posebna izdaja v hrvaščini: poglavje 03 zvezek 022 str. 149 - 165
English
Select your language
Official EU languages:
Access to European Union law
EUR-Lex
Access to European Union law
This document is an excerpt from the EUR-Lex website
Use quotation marks to search for an "exact phrase". Append an asterisk (*) to a search term to find variations of it (transp*, 32019R*). Use a question mark (?) instead of a single character in your search term to find variations of it (ca?e finds case, cane, care).
Need more search options? Use the
Advanced search
Document 32007R0702
Commission Regulation (EC) No 702/2007 of 21 June 2007 amending Commission Regulation (EEC) No 2568/91 on the characteristics of olive oil and olive-residue oil and on the relevant methods of analysis
Uredba Komisije (ES) št. 702/2007 z dne 21. junija 2007 o spremembi Uredbe (EGS) št. 2568/91 o značilnostih oljčnega olja in olja iz oljčnih tropin ter o ustreznih analiznih metodah
Uredba Komisije (ES) št. 702/2007 z dne 21. junija 2007 o spremembi Uredbe (EGS) št. 2568/91 o značilnostih oljčnega olja in olja iz oljčnih tropin ter o ustreznih analiznih metodah
UL L 161, 22.6.2007, p. 11–27
(BG, ES, CS, DA, DE, ET, EL, EN, FR, IT, LV, LT, HU, MT, NL, PL, PT, RO, SK, SL, FI, SV) Dokument je bil objavljen v posebni izdaji.
(HR)
 No longer in force, Date of end of validity: 23/11/2022; implicitno zavrnjeno 32022R2104
No longer in force, Date of end of validity: 23/11/2022; implicitno zavrnjeno 32022R2104
Language
HTML
PDF
Official Journal
|
22.6.2007 |
SL |
Uradni list Evropske unije |
L 161/11 |
UREDBA KOMISIJE (ES) št. 702/2007
z dne 21. junija 2007
o spremembi Uredbe (EGS) št. 2568/91 o značilnostih oljčnega olja in olja iz oljčnih tropin ter o ustreznih analiznih metodah
KOMISIJA EVROPSKIH SKUPNOSTI JE –
ob upoštevanju Pogodbe o ustanovitvi Evropske skupnosti,
ob upoštevanju Uredbe Sveta (ES) št. 865/2004 z dne 29. aprila 2004 o skupni ureditvi trga za oljčno olje in namizne oljke ter o spremembi Uredbe (EGS) št. 827/68 (1) in zlasti člena 5(3) Uredbe,
ob upoštevanju naslednjega:
|
(1) |
Uredba Komisije (EGS) št. 2568/91 (2) opredeljuje fizikalne ter kemične značilnosti oljčnega olja in olja iz oljčnih tropin, določa pa tudi metode za ocenjevanje teh značilnosti. Te metode in mejne vrednosti v zvezi z značilnostmi olja je treba posodobiti ob upoštevanju mnenja kemijskih strokovnjakov in v skladu z opravljenim delom v okviru Mednarodnega sveta za oljkarstvo. |
|
(2) |
Kemijski strokovnjaki so zlasti menili, da je količinska določitev odstotnega deleža 2-gliceril monopalmitata natančnejša za odkrivanje zaestrenega olja. Znižanje mejne vrednosti za stigmastadiena v deviškem oljčnem olju omogoča tudi boljše razlikovanje med deviškim in rafiniranim oljčnim oljem. |
|
(3) |
Da bi zagotovili dovolj časa za prilagoditev novim standardom in uvedbo sredstev, potrebnih za njihovo uporabo, ter se izognili motnjam pri trgovanju, je treba določiti, da se ta uredba uporablja šele po 1. januarju 2008. Zaradi enakih razlogov je treba določiti, da se lahko oljčno olje in olje iz oljčnih tropin, zakonito proizvedena in označena v Skupnosti ali zakonito uvožena v Skupnost ter dana v prosti promet pred tem datumom, tržita do porabe zalog. |
|
(4) |
Ukrepi, predvideni v tej uredbi, so v skladu z mnenjem Upravljalnega odbora za oljčno olje in namizne oljke – |
SPREJELA NASLEDNJO UREDBO:
Člen 1
Uredba (EGS) št. 2568/91 se spremeni:
|
1. |
v členu 2(1) se šesta alinea nadomesti z:
|
|
2. |
priloge se spremenijo, kakor je določeno v Prilogi k tej uredbi. |
Člen 2
Ta uredba začne veljati tretji dan po objavi v Uradnem listu Evropske unije.
Uporablja se od 1. januarja 2008.
Vendar pa se lahko proizvodi, ki so bili zakonito proizvedeni in označeni v Skupnosti ali zakonito uvoženi v Skupnost ter dani v prosti promet pred 1. januarju 2008, tržijo do porabe vseh zalog.
Ta uredba je v celoti zavezujoča in se neposredno uporablja v vseh državah članicah.
V Bruslju, 21. junija 2007
Za Komisijo
Mariann FISCHER BOEL
Članica Komisije
(1) UL L 161, 30.4.2004, str. 97.
(2) UL L 248, 5.9.1991, str. 1. Uredba, kakor je bila nazadnje spremenjena z Uredbo (ES) št. 1989/2003 (UL L 295, 13.11.2003, str. 57).
PRILOGA
Priloge k Uredbi (EGS) št. 2568/91 se spremenijo:
|
1. |
povzetek se spremeni:
|
|
2. |
Priloga I se nadomesti z naslednjim: „PRILOGA I LASTNOSTI OLJČNEGA OLJA Opombe:
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
3. |
Dodatek 1 se spremeni:
|
|
4. |
v Prilogi II se naslov nadomesti z naslednjim: |
|
5. |
Priloga IV se nadomesti z naslednjim: „PRILOGA IV DOLOČEVANJE VSEBNOSTI VOSKOV S KAPILARNO PLINSKO KROMATOGRAFIJO 1. NAMEN Ta metoda opisuje postopek za določitev vsebnosti voskov v oljčnem olju. Voski se ločijo glede na število ogljikovih atomov. Metodo lahko uporabimo zlasti za razlikovanje med oljčnim oljem, pridobljenim s stiskanjem, in oljčnim oljem, pridobljenim z ekstrakcijo (olje iz oljčnih tropin). 2. PRINCIP Maščobo, ki smo ji dodali ustrezen interni standard, na z vodo omočenem silikagelu frakcioniramo s kolonsko kromatografijo; eluirano frakcijo najprej ujamemo pri pogojih preskušanja (katere polarnost je manjša kot pri trigliceridih), nato jo neposredno analiziramo s kapilarno plinsko kromatografijo. 3. OPREMA 3.1 Erlenmajerica prostornine 25 ml. 3.2 Steklena kolona za plinsko kromatografijo, z notranjim premerom 15,0 mm, dolžine 30 do 40 cm in opremljena s petelinčkom. 3.3 Plinski kromatograf, primeren za uporabo s kapilarno kolono in opremljen s sistemom za neposredno injiciranje v kolono, ki obsega: 3.3.1 termostatsko kontrolirano komoro za kolone, s programatorjem temperature; 3.3.2 hladen injektor za injiciranje neposredno v kolono; 3.3.3 plamensko ionizacijski detektor in konverter-ojačevalec; 3.3.4 rekorder-integrator, primeren za uporabo s konverterjem-ojačevalcem (3.3.3), z odzivnim časom, manjšim od 1 sekunde, in nastavljivo hitrostjo pomika papirja. (Uporabimo lahko tudi računalniške sisteme, pri katerih je predvideno zajemanje podatkov plinske kromatografije prek osebnega računalnika.); 3.3.5 Steklena ali kvarčna kapilarna kolona dolžine 8 do 12 m, z notranjim premerom 0,25 do 0,32 mm, znotraj pokrita s tekočo stacionarno fazo in enotne debeline 0,10 do 0,30 μm. (Tekoče stacionarne faze, namenjene uporabi, v prodaji vrste SE 52 ali SE 54.) 3.4 10 μl-mikrobrizgalka s kaljeno iglo in z možnostjo neposrednega injiciranja v kolono. 3.5 Električni vibrator. 3.6 Rotavapor. 3.7 Žarilna peč. 3.8 Analitska tehtnica, ki zagotavlja ± 0,1 mg natančnost. 3.9 Običajna laboratorijska steklovina. 4. REAGENTI 4.1 Silikagel z velikostjo delcev med 60 in 200 μm. Silikagel vsaj 4 ure žarimo pri 500 °C. Nato ga ohladimo in dodamo 2 % vode glede na količino silikagela. Dobro premešamo, da se zmes homogenizira. Pred uporabo hranimo vsaj 12 ur v temi. 4.2 n-heksan, kromatografske čistoče. 4.3 Dietil eter, kromatografske čistoče. 4.4 n-heptan, kromatografske čistoče. 4.5 Standardna raztopina lauril arahidata koncentracije 0,1 % (m/V) v heksanu (interni standard). (Lahko uporabimo tudi -palmitil palmitat ali miristil stearat.) 4.5.1 Sudan 1 (1-fenilazo-2-naftol). 4.6 Nosilni plin: vodik ali helij, čist, plinsko-kromatografske čistoče. 4.7 Pomožni plini:
5. POSTOPEK 5.1 Priprava kromatografske kolone V n-heksanu (4.2) suspendiramo 15 g silikagela (4.1) in napolnimo kolono (3.2). Počakamo, da se posede. Homogenost posedanja, ki poveča homogenost kromatografskih pasov, lahko dosežemo z električnim vibratorjem (3.5). Spiramo s 30 ml n-heksana, da odstranimo vse morebitne nečistoče. S tehtnico (3.8) v erlenmajerico prostornine 25 ml (3.1) natehtamo natanko 500 mg vzorca in dodamo ustrezno količino internega standarda (4.5), ki je odvisna od pričakovane vsebnosti voskov. Na primer, pri oljčnem olju dodamo 0,1 mg, pri olju iz oljčnih tropin pa 0,25 do 0,5 mg lauril arahidata. Tako pripravljen vzorec prenesemo v kromatografsko kolono, in sicer z dvema 2-ml odmerkoma n-heksana (4.2). Topilo izpuščamo iz kolone tako dolgo, da doseže 1 mm nad zgornjim robom absorbenta, zatem spiramo z dodatnimi 70 ml n-heksana, da odstranimo morebitne naravno prisotne n-alkane. Nato začnemo kromatografsko eluiranje, in sicer zberemo 180 ml mešanice n-heksan/etilni eter v razmerju 99:1 in pri pretoku približno 15 kapljic na 10 sekund. Eluiranje vzorca mora potekati pri sobni temperaturi 22 ± 4 °C. Opombe:
Iz tako dobljene frakcije na rotavaporju (3.6) odparimo skoraj vse topilo. Preostala 2 ml topila odstranimo z uporabo blagega toka dušika, nato dodamo 2-4 ml n-heptana. 5.2 Plinsko-kromatografska analiza 5.2.1 Predpriprava Kolono pritrdimo na plinski kromatograf (3.3), in sicer vstopni del na injektor, izhodnega pa na detektor. Izvedemo splošno preverjanje plinskega kromatografa (tesnjenje plinskih povezav, pravilno delovanje detektorja in rekorderja itd.). Če kolono uporabljamo prvič, je priporočljivo, da jo kondicioniramo. Pri majhnem pretoku plina skozi kolono vključimo plinski kromatograf. Postopoma segrevamo, tako da po približno 4 urah dosežemo temperaturo 350 °C. Pri tej temperaturi kolono kondicioniramo vsaj dve uri, nato naravnamo instrument v skladu s pogoji delovanja (naravnamo pretok, prižgemo plamen, povežemo z elektronskim rekorderjem (3.3.4), naravnamo delovno temperaturo peči za kolono, naravnamo detektor itd.). Pri občutljivosti, ki je vsaj dvakrat večja od delovne, posnamemo bazno linijo. Le-ta mora biti linearna, brez kakršnih koli vrhov in odklonov. Negativen premočrtni odklon kaže na slabo tesnjenje med kolono in instrumentom, pozitiven odklon pa na slabo kondicionirano kolono. 5.2.2 Izbira delovnih pogojev Splošni delovni pogoji, ki jih je treba upoštevati, so naslednji:
Te pogoje lahko spremenimo glede na značilnosti kolone in plinskega kromografa, da bi dosegli ločevanje vseh voskov, zadostno resolucijo vrhov (glej sliko) in retencijski čas internega standarda C32, ki mora biti 18 ± 3 minute. Najznačilnejši vrh voskov mora biti izmerjen najmanj 60 % od spodnjega dela skale. Integracijske parametre moramo določiti tako, da dobimo pravilno vrednotenje površin vrhov. Opomba: Glede na visoko končno temperaturo je dovoljen pozitivni odklon, ki pa ne sme biti večji od 10 % od spodnjega dela skale. 5.3 Izvedba analize Z 10-μl mikrobrizgalko odvzamemo 1 μl raztopine; bat izvlečemo, da se igla izprazni. Iglo vbodemo v injektor in po eni ali dveh sekundah hitro vbrizgnemo. Po približno petih sekundah iglo pazljivo izvlečemo. Kromatogram snemamo, dokler ne eluirajo vsi voski. Bazna linija mora ves čas izpolnjevati zahtevane pogoje. 5.4 Identifikacija vrhov Različne vrhove identificiramo na podlagi retencijskih časov in s primerjavo med mešanicami voskov z znanimi retencijskimi časi, ki so bile analizirane pri enakih pogojih. Slika prikazuje kromatogram voskov deviškega oljčnega olja. 5.5 Kvantitativna analiza Z integratorjem izračunamo površine vrhov alifatskih estrov od C40 do C46 in površino vrha internega standarda. Na podlagi naslednje formule izračunamo vsebnost voskov posameznega estra v mg/kg maščobe:
Kjer je:
6. PODAJANJE REZULTATOV Navedemo vsoto vsebnosti posameznih sestavin različnih voskov od C40 do C46, v mg/kg maščobe (ppm). Opomba: Spojine, ki jih je treba količinsko določiti, se nanašajo na vrhove s sodim številom ogljikovih atomov med estri C40 do C46, tako kot je to prikazano na kromatograma voskov oljčnega olja. Če sta za ester C46 dva vrhova, je za njegovo identifikacijo priporočljiva analiza frakcije voskov olja iz oljčnih tropin, kjer je vrh C46 dobro viden, ker je razločno večji. Rezultate je treba izraziti na eno decimalno mesto natančno. Slika Kromatogram voskov v oljčnem olju (9)
Dodatek Določanje linearne hitrosti plina V plinski kromatograf, naravnan na običajne delovne pogoje, injiciramo 1 do 3 μl metana (ali propana). Merimo čas, ki ga plin potrebuje za pot skozi kolono od trenutka vbrizga do trenutka, ko se pojavi vrh (tM). Linearna hitrost v cm/s je izražena s formulo L/tM, kjer je L dolžina kolone v cm, tM pa izmerjeni čas v sekundah. |
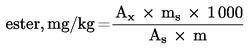
|
6. |
Priloga VII se nadomesti z naslednjim: „PRILOGA VII DOLOČANJE ODSTOTNEGA DELEŽA 2-GLICERIL MONOPALMITATA 1. PREDMET UREJANJA IN PODROČJE UPORABE S to metodo je opisan analitski postopek za določevanje odstotnega deleža palmitinske kisline na položaju 2 v trigliceridih z ovrednotenjem 2-gliceril monopalmitata. To metodo uporabljamo za rastlinska olja, tekoča pri sobni temperaturi (20 °C). 2. PRINCIP Po pripravi pustimo, da na vzorec olja učinkuje pankreatična lipaza: poteče delna hidroliza, specifična za položaja 1 in 3 v molekuli triglicerida, ki povzroči nastanek 2-monoacilglicerolov. Odstotni delež 2-gliceril monopalmitata v monoacilglicerolni frakciji se po sililiranju določi s kapilarno plinsko kromatografijo. 3. APARATURE IN OBIČAJNA LABORATORIJSKA OPREMA 3.1 Erlenmajerica, 25 ml 3.2 Čaše 100, 250 in 300 ml 3.3 Steklena kromatografska kolona z notranjim premerom 21–23 mm, dolžine 400 mm, opremljena s sintrano stekleno ploščico in petelinčkom 3.4 Merilni valji prostornine 10, 50, 100 in 200 ml 3.5 Bučke prostornine 100 in 250 ml 3.6 Rotavapor 3.7 Epruvete za centrifugiranje prostornine 10 ml s koničnim dnom in obrušenim zamaškom 3.8 Centrifuga za epruvete prostornine 10 in 100 ml 3.9 Termostat, ki omogoča vzdrževanje temperature pri 40 °C ± 0,5 °C 3.10 Merilni pipeti prostornine 1 in 2 ml 3.11 Brizgalka prostornine 1 ml 3.12 Mikrobrizgalka prostornine 100 μl 3.13 Lij-ločnik, 1 000 ml 3.14 Plinski kromatograf za kapilarne kolone, opremljen z injicirnim sistemom za hladno injiciranje vzorca neposredno v kolono in pečjo, ki lahko vzdržuje izbrano temperaturo znotraj 1 °C 3.15 Injektor za hladno injiciranje vzorca neposredno v kolono 3.16 Plamensko ionizacijski detektor in elektrometer 3.17 Rekorder-integrator, kompatibilen z elektrometrom, z odzivnim časom, manjšim od 1 sekunde, in nastavljivo hitrostjo pomika papirja 3.18 Steklena ali kvarčna kapilarna kolona dolžine 8 do 12 m, z notranjim premerom 0,25 do 0,32 mm, pokrita z metilpolisiloksanom ali 5-odstotnim fenil metilpolisiloksanom, debeline 0,10–0,30 μm, ki jo je mogoče uporabiti pri 370 °C 3.19 Mikrobrizgalka prostornine 10 μl z nesnemljivo, vsaj 7,5 cm dolgo iglo, za neposredno vbrizganje na začetek kolone 4. REAGENTI 4.1 Silikagel z velikostjo delcev med 0,063 in 0,200 mm (70/280 mesh), pripravljen na naslednji način: silikagel damo v porcelansko posodico, ga pri 160 °C 4 ure sušimo v sušilniku, nato pa pustimo, da se na sobni temperaturi ohladi v eksikatorju. Nato dodamo količino vode, ki ustreza 5 % teže silikagela: v erlenmajerico prostornine 500 ml natehtamo 152 g silikagela in dodamo 8 g destilirane vode, zamašimo ter homogeniziramo. Pred uporabo pustimo mirovati vsaj 12 ur. 4.2 n-heksan (kromatografske čistoče) 4.3 Izopropanol 4.4 Izpropanol, vodna raztopina 1/1 (V/V) 4.5 Pankreatična lipaza. Aktivnost uporabljene lipaze mora biti med 2,0 in 10 lipaznimi enotami na mg (V prodaji so pankreatične lipaze z aktivnostjo med 2 in 10 enotami na mg encima.) 4.6 Pufrska raztopina tris-hidroksi-metilaminometana: 1 M vodna raztopina, ki ji s koncentrirano HCI (1/1 V/V) uravnamo pH na vrednost 8 (preverimo s pH-metrom) 4.7 Natrijev holat, encimske čistosti, 0,1-odstotna vodna raztopina (to raztopino je treba uporabiti v petnajstih dneh po pripravi) 4.8 Kalcijev klorid, 22-odstotna vodna raztopina 4.9 Dietil eter kromatografske čistoče 4.10 Elucijsko topilo: mešanica n-heksana/dietilnega etra (87/13) (V/V) 4.11 Natrijev hidroksid, 12-odstotna raztopina v masnih odstotkih 4.12 Fenolftalein, 1-odstotna raztopina v etanolu 4.13 Nosilni plin: vodik ali helij, za plinsko kromatografijo 4.14 Pomožna plina: vodik, najmanj 99-odstoten, brez vlage in organskih snovi, in zrak, za plinsko kromatografijo in enake čistoče 4.15 Regent za silaniziranje: mešanica piridina, heksametildisilazana, trimetilklorosilana v razmerju 9:3:1 (V/V/V) (V prodaji so raztopine, pripravljene za uporabo. Uporabimo lahko tudi druge reagente za silaniziranje, zlasti bis-trimetilsilil trifluoroacetamid + 1-odstoten trimetilklorosilan, razredčen z enako količino brezvodnega piridina.) 4.16 Referenčni vzorci: čisti monogliceridi ali mešanice monogliceridov, za katere je znano, da imajo podobno odstotkovno sestavo kot vzorec. 5. POSTOPEK 5.1 Priprava vzorca 5.1.1 Olj z deležem prostih kislin, manjšim od 3 %, pred kolonsko kromatografijo ni treba nevtralizirati s silikagelom. Olja, katerih delež prostih kislin je večji od 3 %, je treba nevtralizirati v skladu s točko 5.1.1.1. 5.1.1.1 V lij-ločnik prostornine 1 000 ml (3.13) vlijemo 50 g olja in 200 ml n-heksana. Dodamo 100 ml izopropanola in tako količino 12-odstotne raztopine natrijevega hidroksida (4.11), da ustreza deležu prostih kislin, povečanemu za 5 odstotkov. Eno minuto močno stresamo. Dodamo 100 ml destilirane vode, ponovno pretresemo in pustimo, da se usede. Po ločevanju odstranimo spodnjo plast, ki vsebuje mila. Odstranimo morebitne vmesne plasti (sluz in netopne snovi). Heksansko raztopino nevtraliziranega olja izpiramo z zaporednimi 50- do 60-mililitrskimi odmerki raztopine izopropanola in vode 1/1 (V/V) (4.4) tako dolgo, da je izpiralna faza nevtralna na fenolftalein. Večino heksana odstranimo z destilacijo v vakuumu (uporabimo na primer rotavapor) in olje prelijemo v 100-mililitrsko bučko (3.5). Olje sušimo v vakuumu, dokler se topilo v celoti ne odstrani. Po koncu tega postopka mora biti vsebnost kislin v olju manjša od 0,5 %. 5.1.2 V erlenmajerico prostornine 25 ml damo 1,0 g olja, pripravljenega po spodnjih navodilih (3.1), in ga raztopimo v 10 ml razvijalne mešanice (4.10). Pred kolonsko kromatografijo s silikagelom raztopino pustimo mirovati najmanj 15 minut. Če je raztopina motna, jo centrifugiramo, da zagotovimo optimalne pogoje za kromatografijo. (Uporabimo lahko komercialne 500-mg silikagelne kartuše SPE.) 5.1.3 Priprava kromatografske kolone V kolono (3.3) vlijemo približno 30 m razvijalnega topila (4.10), s stekleno paličko v spodnji del kolone vstavimo košček vate; stisnemo, da odstranimo zrak. V čaši pripravimo raztopino 25 g silikagela (4.1) v približno 80 ml razvijalne raztopine in jo z lijakom prelijemo v kolono. Preverimo, da smo v kolono dali ves silikagel; speremo z elucijskim topilom (4.10), odpremo petelinček in pustimo, da raven tekočine seže približno 2 mm nad zgornjo raven silikagela. 5.1.4 Kolonska kromatografija V erlenmajerico prostornine 25 ml (3.1.) natehtamo natanko 1,0 g vzorca, ki smo ga pripravili v skladu s točko 5.1. Vzorec raztopimo v 10 ml elucijskega topila (4.10). Raztopino prelijemo v kromatografsko kolono, ki smo jo pripravili v skladu s točko 5.1.3. Pazimo, da ne premešamo površine kolone. Odpremo ventil in pustimo raztopino vzorca odtekati, dokler ne doseže ravni silikagela. Eluiramo s 150 ml razvijalnega topila. Količino pretoka nastavimo na 2 ml/min (tako da 150 ml odteče v kolono v približno 60–70 minutah). Eluat zberemo v 250-mililitrsko bučko z znano maso. V vakuumu odparimo topilo in njegove zadnje sledi odstranimo s tokom dušika. Bučko stehtamo in izračunamo pridobljeni ekstrakt (Če uporabljamo silikagelne kartuše SPE, storimo naslednje: v kartuše, ki smo jih predhodno kondicionirali s 3 ml n-heksana, vlijemo 1 ml raztopine (5.1.2). Po filtraciji raztopine razvijemo s 4 ml n-heksana/dietilnega etra 9/1 (V/V). Eluat zberemo v 10-mililitrsko epruveto in ga z uvajanjem toka dušika odparimo do suhega. Na suhem preostanku pustimo učinkovati pankreatično lipazo (5.2). Ključno je, da pred in po uporabi kartuše SPE preverimo sestavo maščobnih kislin. 5.2 Hidroliza s pankreatično lipazo 5.2.1 V epruveto centrifuge natehtamo 0,1 g olja, pripravljenega v skladu s točko 5.1. Dodamo 2 ml pufrske raztopine (4.6), 0,5 ml raztopine natrijevega holata (4.7) in 0,2 ml raztopine kalcijevega klorida, pri čemer po vsakem dodajanju dobro pretresemo. Epruveto zapremo z obrušenim zamaškom in jo namestimo v termostat, naravnan na 40 ± 0,5 °C. 5.2.2 Dodamo 20 mg lipaze, previdno pretresemo (pazimo, da ne zmočimo zamaška) in damo epruveto za natanko 2 minuti v termostat, nato jo damo ven, jo natanko 1 minuto močno stresamo in pustimo, da se ohladi. 5.2.3 Dodamo 1 ml dietilnega etra, zamašimo in močno stresamo, nato centrifugiramo ter raztopino etra z mikrobrizgalko prenesemo v čisto in suho epruveto. 5.3 Priprava silaniziranih derivatov in plinska kromatografija 5.3.1 Z mikrobrizgalko prenesemo 100 μl raztopine (5.2.3) v epruveto prostornine 10 ml s koničastim dnom. 5.3.2 Topilo odstranimo z uvajanjem rahlega toka dušika, dodamo 200 μl reagenta za silaniziranje (4.15), zamašimo epruveto in pustimo mirovati 20 minut. 5.3.3 Po 20 minutah dodamo 1 do 5 ml n-heksana (odvisno od kromatografskih pogojev): raztopina, ki jo dobimo, je nared za plinsko kromatografijo. 5.4 Plinska kromatografija Pogoji za postopek so naslednji:
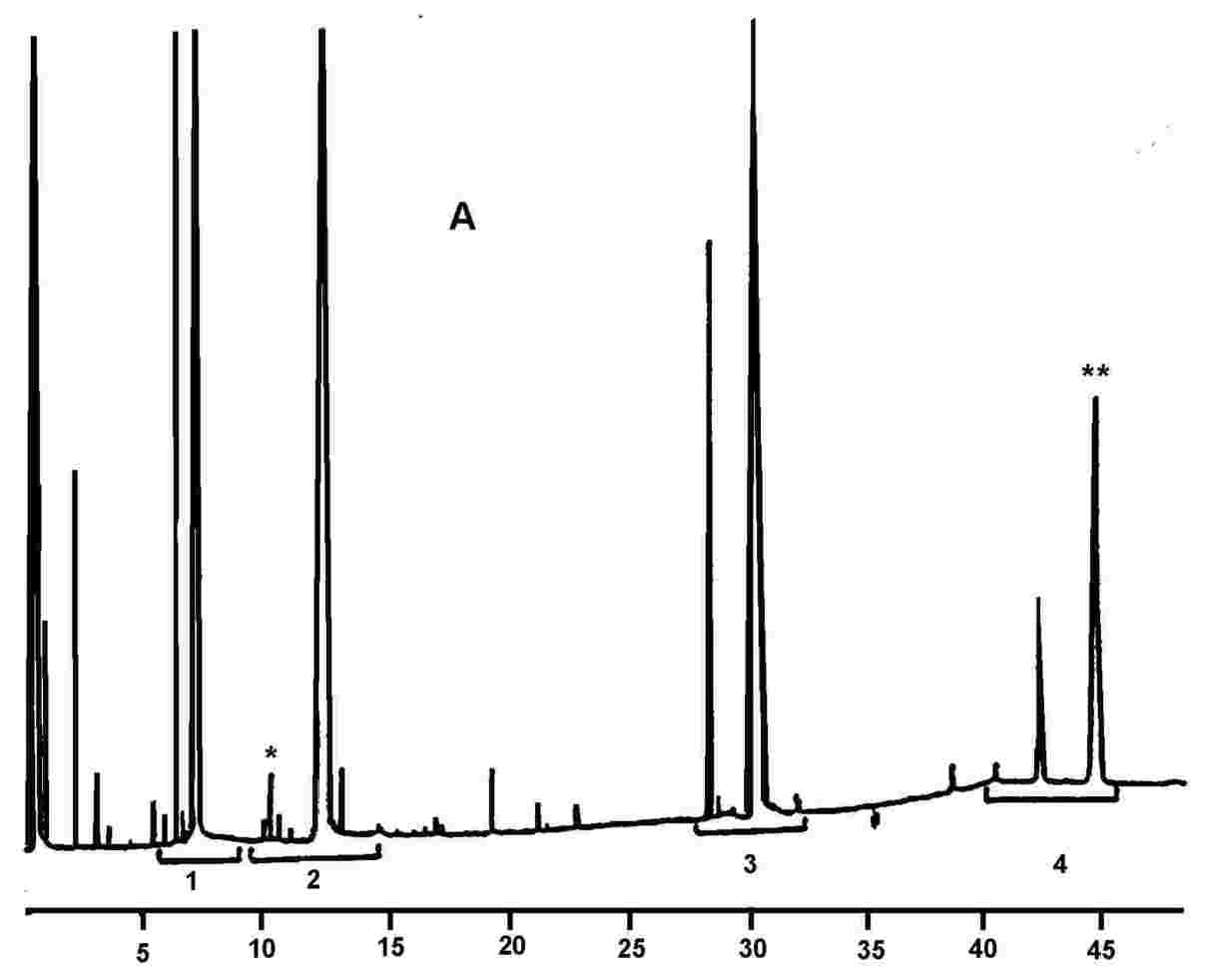
5.4.1 Identifikacija vrhov Posamezne monoacilglicerole identificiramo na podlagi dobljenih retencijskih časov in glede na čase, dobljene za standardne mešanice monogliceridov, analizirane pri enakih pogojih. 5.4.2 Kvantitativna analiza Površina vsakega vrha se izračuna z elektronskim integratorjem. 6. PODAJANJE REZULTATOV Odstotek gliceril monopalmitata izračunamo iz razmerja med ustrezno površino vrha in vsoto površin vrhov vseh monoacilglicerolov (glej Sliko 2), in sicer na podlagi formule: gliceril monopalmitat (%): kjer je:
Rezultat je treba podati na eno decimalko natančno. 7. POROČILO O ANALIZI V poročilu o analizi je treba podrobno navesti:
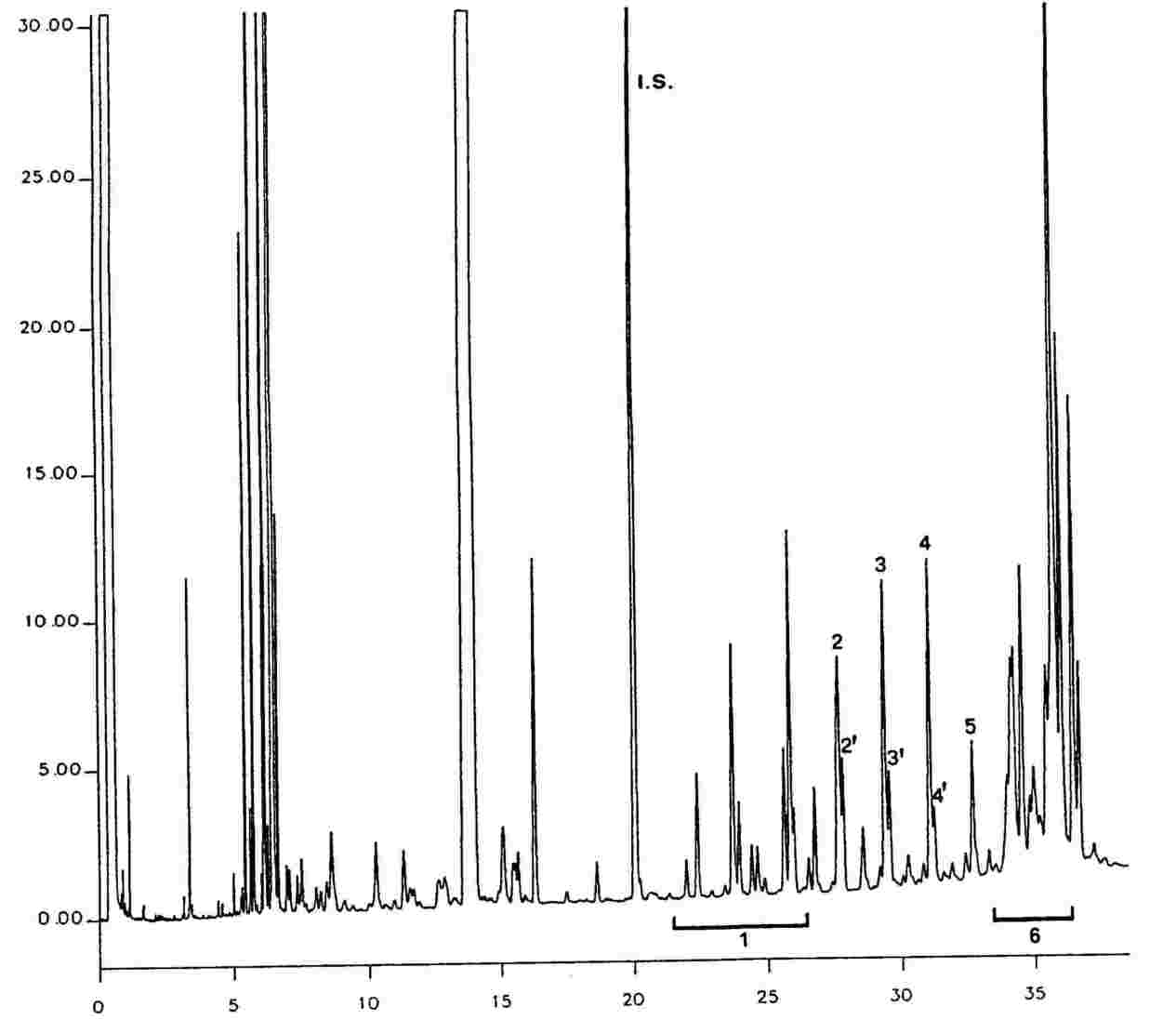
Slika 1 Kromatogram proizvodov reakcije silaniziranja, dobljenih z delovanjem lipaze na rafiniranem oljčnem olju, ki mu je dodanih 20 % 100 % zaestrenega olja
Slika 2 Kromatogram (A) nezaestrenega oljčnega olja po delovanju lipaze, in silaniziranega; pri teh pogojih (kapilarna kolona 8–12 m) frakcija voskov eluira hkrati s frakcijo diacilglicerolov ali malo zatem. Vsebnost triacilglicerolov po lipazi ne sme preseči 15 %.
Kromatogram: (B) zaestrenega olja po delovanju lipaze; po silaniziranju; pri teh pogojih (kapilarna kolona 8–12 m) frakcija voskov eluira hkrati s frakcijo diglicerida ali malo zatem. Vsebnost trigliceridov po lipazi ne sme preseči 15 %
8. OPOMBE Opomba 1. PRIPRAVA LIPAZE Lipaze z zadostno aktivnostjo so komercialno dostopne. Lahko jih pripravimo tudi v laboratoriju, in sicer na naslednji način: 5 kg sveže prašičje trebušne slinavke ohladimo na 0 °C. Odstranimo okoliško trdo maščevje in vezno tkivo ter jo zmeljemo v mlinčku z rezili, da dobimo kašasto tekočino. To tekočino 4 do 6 ur mešamo z 2,5 litra brezvodnega acetona in nato centrifugiramo. Izvleček naredimo še trikrat z enako količino acetona, nato dvakrat z mešanico acetona/dietilnega etra (1/1) (V/V) in dvakrat z dietilnim etrom. Preostanek 48 ur sušimo v vakuumu, da dobimo stabilen prah, ki ga je treba dolgo časa hraniti v hladilniku in pred vlago. Opomba 2. PREVERJANJE AKTIVNOSTI LIPAZE Oljno emulzijo pripravimo na naslednji način: V mešalniku 10 minut mešamo mešanico 165 ml raztopine gumarabikuma (100 g/l), 15 g zdrobljenega ledu in 20 ml predhodno nevtraliziranega oljčnega olja. V čašo prostornine 50 ml zaporedoma damo 10 ml te emulzije, nato 0,3 ml raztopine natrijevega holata (0,2 g/ml) in 20 ml destilirane vode. Čašo damo v termostat, naravnan na 37 °C; namestimo elektrode pH metra in spiralni mešalnik. Z bireto po kapljicah dodajamo raztopino natrijevega hidroksida 0,1 N, dokler ne dobimo pH vrednosti 8,3. Dodamo ustrezno količino v vodi raztopljene lipaze v prahu (0,1 g/ml lipaze). Takoj ko pH meter pokaže pH 8,3, sprožimo štoparico in po kapljah dodajamo raztopino natrijevega hidroksida, in sicer s tako hitrostjo, da ohranimo pH vrednost 8,3. Vsako minuto odčitamo volumen porabljene raztopine. Podatke zabeležimo v sistem koordinatnih osi, in sicer odčitke časa navedemo kot absciso, kot ordinato pa ml alkalne raztopine 0,1 N, ki smo jih porabili za ohranitev konstantnega pH. Dobiti moramo linearen graf. Aktivnost lipaze, izmerjena v lipaznih enotah na mg, je izražena z naslednjo formulo:
kjer je:
Lipazna enota je opredeljena kot količina encima, ki sprosti 10 mikro-ekvivalentov kisline na minuto.“; |




|
7. |
v Prilogi XA se točka 6.2 nadomesti z naslednjim:
|
(1) Vsota skupnih ali posameznih izomerov, ki jih določimo s kapilarno kolono.
(2) Ali če je mediana napake manjša ali enaka 2,5 in je mediana sadežnosti 0.
(3) Olja z vsebnostjo voskov med 300 mg/kg in 350 mg/kg se uvrščajo med lampante oljčna olja, če je vsebnost alifatskih alkoholov manjša ali enaka 350 mg/kg ali če je delež eritrodiola in uvaola manjši ali enak 3,5 %.
(4) Olja z vsebnostjo voskov med 300 mg/kg in 350 mg/kg se uvrščajo med surova olja iz oljčnih tropin, če je skupna vsebnost alifatskih alkoholov večja od 350 mg/kg in če je delež eritrodiola in uvaola večji od 3,5 %.
(5) Vsebnost drugih maščobnih kislin (%): palmitinske: 7,5–20,0; palmitoleinske: 0,3–3,5; heptadekanojske ≤ 0,3; heptadecenojske: ≤ 0,3; stearinske: 0,5–5,0; oleinske: 55,0–83,0; linolenske: 3,5–21,0.
(6) Vsota: delta-5,23-stigmastadienola+klerosterola+beta-sitosterola+sitostanola+delta-5-avenasterola+delta-5,24-stigmastadienola.
(7) Olja z vsebnostjo voskov med 300 mg/kg in 350 mg/kg se uvrščajo med lampante oljčna olja, če je skupna vsebnost alifatskih alkoholov manjša ali enaka 350 mg/kg ali če je delež eritrodiola in uvaola manjši ali enak 3,5 %.
(8) Olja z vsebnostjo voskov med 300 mg/kg in 350 mg/kg se uvrščajo med surova olja iz oljčnih tropin, če je skupna vsebnost alifatskih alkoholov večja od 350 mg/kg in če je delež eritrodiola in uvaola večji od 3,5 %.
(9) Po eluciji sterolnih estrov kromatografska linija ne sme imeti značilnih vrhov (trigliceridi).