Spanish special edition: Chapter 13 Volume 011 P. 203 - 208
Portuguese special edition: Chapter 13 Volume 011 P. 203 - 208
Special edition in Finnish: Chapter 13 Volume 011 P. 140 - 145
Special edition in Swedish: Chapter 13 Volume 011 P. 140 - 145
Special edition in Czech: Chapter 13 Volume 006 P. 154 - 159
Special edition in Estonian: Chapter 13 Volume 006 P. 154 - 159
Special edition in Latvian: Chapter 13 Volume 006 P. 154 - 159
Special edition in Lithuanian: Chapter 13 Volume 006 P. 154 - 159
Special edition in Hungarian Chapter 13 Volume 006 P. 154 - 159
Special edition in Maltese: Chapter 13 Volume 006 P. 154 - 159
Special edition in Polish: Chapter 13 Volume 006 P. 154 - 159
Special edition in Slovak: Chapter 13 Volume 006 P. 154 - 159
Special edition in Slovene: Chapter 13 Volume 006 P. 154 - 159
Special edition in Bulgarian: Chapter 13 Volume 005 P. 194 - 199
Special edition in Romanian: Chapter 13 Volume 005 P. 194 - 199
English
Select your language
Official EU languages:
Access to European Union law
EUR-Lex
Access to European Union law
This document is an excerpt from the EUR-Lex website
Use quotation marks to search for an "exact phrase". Append an asterisk (*) to a search term to find variations of it (transp*, 32019R*). Use a question mark (?) instead of a single character in your search term to find variations of it (ca?e finds case, cane, care).
Need more search options? Use the
Advanced search
Document 31981L0432
Commission Directive 81/432/EEC of 29 April 1981 laying down the Community method of analysis for the official control of vinyl chloride released by materials and articles into foodstuffs
Commission Directive 81/432/EEC of 29 April 1981 laying down the Community method of analysis for the official control of vinyl chloride released by materials and articles into foodstuffs
Commission Directive 81/432/EEC of 29 April 1981 laying down the Community method of analysis for the official control of vinyl chloride released by materials and articles into foodstuffs
OJ L 167, 24/06/1981, p. 6–11
(DA, DE, EL, EN, FR, IT, NL) This document has been published in a special edition(s)
(ES, PT, FI, SV, CS, ET, LV, LT, HU, MT, PL, SK, SL, BG, RO)
 No longer in force, Date of end of validity: 30/04/2011; Repealed by 32011R0010
No longer in force, Date of end of validity: 30/04/2011; Repealed by 32011R0010
- Date of document:
- 29/04/1981
- Date of effect:
- 19/05/1981; Entry into force Date notif.
- Date of notification:
- 19/05/1981
- Date of transposition:
- 01/10/1982; At the latest See Art 2
- Date of end of validity:
- 30/04/2011; Repealed by 32011R0010
- Author:
- European Commission
- Responsible body:
- DG III – Industry, DG03/C/01
- Form:
- Directive
- Addressee:
- The ten Member States: Belgium, Denmark, Germany, Ireland, Greece, France, Italy, Luxembourg, Netherlands, United Kingdom
- Additional information:
- Extended to the EEA by 21994A0103(01)
- Authentic language:
- Danish, German, Greek, English, French, Italian, Dutch, Icelandic, Norwegian
- Treaty:
- Treaty establishing the European Economic Community
- Legal basis:
-
- 31978L0142 - A03
- Link
- Link
- Link
- Select all documents mentioning this document No data available in the table
- Modified by:
-
Relation Act Comment Subdivision concerned From To Repealed by 32011R0010 01/05/2011 - Link
- EUROVOC descriptor:
- Subject matter:
- Directory code:
Language
HTML
PDF
Official Journal
|
24.6.1981 |
EN |
Official Journal of the European Communities |
L 167/6 |
COMMISSION DIRECTIVE
of 29 April 1981
laying down the Community method of analysis for the official control of vinyl chloride released by materials and articles into foodstuffs
(81/432/EEC)
THE COMMISSION OF THE EUROPEAN COMMUNITIES,
Having regard to the Treaty establishing the European Economic Community,
Having regard to Council Directive 78/142/EEC of 30 January 1978 on the approximation of the laws of the Member States relating to materials and articles which contain vinyl chloride monomer and are intended to come into contact with foodstuffs (1), and in particular Article 3 thereof,
Whereas Article 2 of Directive 78/142/EEC lays down that materials and articles must not pass on to the foodstuffs which are in, or have been brought into, contact with such materials and articles any vinyl chloride detectable by a method having a limit of detection of 001 mg/kg, and Article 3 that this limit must be controlled by a Community method of analysis;
Whereas, on the basis of a series of inter-laboratory collaborative trials, the method described in the Annex has proved to be sufficiently accurate and reproducible to be adopted as a Community method;
Whereas the measures provided for in this Directive are in accordance with the opinion of the Standing Committee on Foodstuffs,
HAS ADOPTED THIS DIRECTIVE:
Article 1
The analysis necessary for official control of vinyl chloride released by materials and articles into foodstuffs shall be performed according to the method described in the Annex hereto.
Article 2
The Member States shall bring into force the laws, regulations and administrative provisions necessary to comply with this Directive not later than 1 October 1982. They shall forthwith inform the Commission thereof.
Article 3
This Directive is addressed to the Member States.
Done at Brussels, 29 April 1981.
For the Commission
Karl-Heinz NARJES
Member of the Commission
(1) OJ No L 44, 15.2. 1978, p. 15.
ANNEX
DETERMINATION OF VINYL CHLORIDE RELEASED BY MATERIALS AND ARTICLES INTO FOODSTUFFS
1. SCOPE AND FIELD OF APPLICATION
The method determines the level of vinyl chloride in foodstuffs.
2. PRINCIPLE
The level of vinyl chloride (VC) in foodstuffs is determined by means of gas-chromatography using the ‘headspace’ method.
3. REAGENTS
|
3.1. |
Vinyl chloride (VC), of purity greater than 99.5% (v/v). |
|
3.2. |
N, N-dimethylacetamide (DMA), free from any impurity with the same retention time as VC or as the internal standard (3.3) under the conditions of the test. |
|
3.3. |
Diethyl ether or cis-2-butene, in DMA (3.2) as the internal standard solution. These internal standards must not contain any impurity with the same retention time as VC, under the conditions of the test. |
|
3.4. |
Distilled water or demineralized water of equivalent purity. |
4. APPARATUS
NB:
An instrument or piece of apparatus is mentioned only if it is special, or made to particular specifications. Usual laboratory apparatus is assumed to be available.
|
4.1. |
Gas-chromatograph fitted with automatic headspace sampler or with facilities for manual sample injection. |
|
4.2. |
Flame ionization detector or other detectors mentioned in point 7. |
|
4.3. |
Gas-chromatographic column The column must permit the separation of the peaks of air, of VC and of the internal standard, if used. Furthermore, the combined 4.2 and 4.3 system must allow the signal obtained with a solution containing 0·005 mg VC/litre DMA or 0·005 mg VC/kg DMA to be equal to at least five times the background noise. |
|
4.4. |
Sample phials or flasks fitted with silicon or butyl rubber septa When using manual sampling techniques, the taking of a sample from the headspace with a syringe may cause a partial vacuum to form inside the phial or flask. Hence, for manual techniques where the phials are not pressurized before the sample is taken, the use of large phials is recommended. |
|
4.5. |
Micro-syringes. |
|
4.6. |
Gas-tight syringes for manual headspace sampling. |
|
4.7. |
Analytical balance accurate to 0.1 mg. |
5. PROCEDURE
CAUTION: VC is a hazardous substance and a gas at ambient temperature therefore, the preparation of solutions should be carried out in a well-ventilated fume cupboard.
NB:
Take all the necessary precautions to ensure that no VC or DMA is lost.
When employing manual sampling techniques, an internal standard (3.3) should be used.
When using an internal standard, the same solution must be utilized throughout the procedure.
5.1. Preparation of standard VC solution (solution A)
5.1.1. Concentrated standard VC solution at approximately 2 000 mg/kg
Accurately weigh to the nearest 0.1 mg a suitable glass vessel and place in it a quantity (e.g. 50 ml) of DMA (3.2). Re-weigh. Add to the DMA a quantity (e.g. 0.1 g) of VC (3.1) in liquid or gas form, injecting it slowly onto the DMA. The VC may also be added by bubbling it into the DMA, provided that a device is used which will prevent loss of DMA. Reweigh to the nearest 0.1 mg. Wait two hours to allow equilibrium to be attained. If an internal standard is to be employed, add internal standard so that the concentration of the internal standard in the concentrated standard VC solution is the same as in the internal standard solution prepared under point 3.3. Keep the standard solution in a refrigerator.
5.1.2. Preparation of dilute standard VC solution
Take a weighed amount of concentrated standard solution of VC (5.1.1) and dilute, to a known volume or a known weight, with DMA (3.2) or with internal standard solution (3.3). The concentration of the resultant dilute standard solution (solution A) is expressed as mg/litre or mg/kg respectively.
5.1.3. Preparation of the response curve with solution A
NB:
The curve must comprise at least seven pairs of points.
The repeatability of the responses (1) must be lower than 0.002 mg VC/litre or kg of DMA.
The curve must be calculated from these points by the least squares technique, i.e., the regression line must be calculated using the following equation:
y = a1x + ao
where:
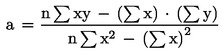
and:

where:
|
y |
= |
the height or area of peaks in any single determination; |
|
x |
= |
the corresponding concentration on the regression line; |
|
n |
= |
number of determinations carried out (n ≥ 14). |
The curve must be linear, i.e., the standard deviation (s) of the differences between the measured responses (yi) and the corresponding value of the responses calculated from the regression line (zi) divided by the mean value (y) of all the measured responses shall not exceed 0·07.
This shall be calculated from:

where:

and:

where:
|
yi |
= |
each individual measured response; |
|
zi |
= |
the corresponding value of the response (yi) on the calculated regression line; |
|
n |
= |
≥ 14. |
Prepare two series of at least seven phials (4.4). Add to each phial volumes of dilute standard VC solution (5.1.2) and DMA (3.2) or internal standard solution in DMA (3.3) such that the final VC concentration of the duplicate solutions will be approximately equal to 0, 0·005, 0·010, 0·020, 0·030, 0·040, 0·050, etc., mg/litre or mg/kg of DMA and that each phial contains the same total volume of solution. The quantity of dilute standard VC solution (5.1.2) must be such that the ratio between the total volume (μl) of added VC solution and quantity (g or ml) of DMA, or internal standard solution (3.3) does not exceed five. Seal the phials and proceed as described under points 5.4.2, 5.4.3 and 5.4.5. Construct a graph in which the ordinate values show the areas (or heights) of the VC peaks of the duplicate solutions, or the ratio of these areas (or heights) to those of the relevant internal standard peaks, and the abscissa values show the VC concentrations of the duplicate solutions.
5.2. Validation of preparation of standard solutions obtained in point 5.1
5.2.1. Preparation of a second standard VC solution (solution B)
Repeat the procedure described under points 5.1.1 and 5.1.2 to obtain a second dilute standard solution with, in this case, a concentration approximately equal to 0·02 mg VC: 1, or 0·02 mg VC/kg of DMA or internal standard solution. Add this solution to two phials (4.4). Seal the phials and proceed as described under points 5.4.2, 5.4.3 and 5.4.5.
5.2.2. Validation of solution A
If the average of two gas-chromatographic determinations relating to the solution B (5.2.1) does not differ by more than 5% from the corresponding point of the response curve obtained in point 5.1.3, the solution A is validated. If the difference is greater than 5%, reject all the solutions obtained in points 5.1 and 5.2 and repeat the procedure from the beginning.
5.3. Preparation of the ‘addition’ curve
NB:
The curve must comprise at least seven pairs of points.
The curve must be calculated from these points by the least squares technique (5.1.3, third indent).
The curve must be linear, i.e., the standard deviation(s) of the differences between the measured responses (yi) and the corresponding value of the responses calculated from the regression line (zi) divided by the mean value ( ) of all the measured responses shall not exceed 0·07 (5.1.3, fourth indent).
) of all the measured responses shall not exceed 0·07 (5.1.3, fourth indent).
5.3.1. Preparation of sample
The sample of foodstuff to be analyzed must be representative of the foodstuff presented for analysis. The foodstuff must, therefore, be mixed or reduced to small pieces and mixed before the sample is taken.
5.3.2. Procedure
Prepare two series of at least seven phials (4.4). Add to each phial a quantity, not less than 5 g, of sample obtained from the foodstuff under investigation (5.3.1). Ensure that an equal quantity is added to each phial. Close the phial immediately. Add to each phial for each gram of sample 1 ml, preferably of distilled water, or demineralized water of at least equivalent purity, or an appropriate solvent if necessary. (Note: for homogeneous foodstuffs, addition of distilled of demineralized water is not necessary.) Add to each phial volumes of dilute standard VC solution (5.1.2), containing the internal standard (3.3), if considered useful, such that concentrations of VC added to the phials equal to 0, 0·005, 0·010, 0·020, 0·030, 0·040 and 0·050, etc., mg/kg of foodstuffs. Ensure that the total volume of DMA or DMA containing internal standard (3.3) in each phial is the same. The quantity of dilute standard VC solution (5.1.2) and additional DMA where used, must be such that the ratio between the total volume (μl) of these solutions and the quantity (g) of foodstuff contained in the phial is as low as possible but not more than five and is the same in all phials. Seal the phials and proceed as described under point 5.4.
5.4. Gas-chromatographic determinations
|
5.4.1. |
Agitate the phials avoiding contact between the contained liquid and the septum (4.4) to obtain a solution or a suspension of the samples of foodstuff as homogeneous as possible. |
|
5.4.2. |
Put all the sealed phials (5.2 and 5.3) in a waterbath for two hours at 60 ± 1 oC to allow equilibrium to be attained. Agitate again, if necessary. |
|
5.4.3. |
Take a sample from the headspace in the phial. When utilizing manual sampling techniques care must be exercized in obtaining a reproducible sample (4.4) in particular the syringe must be pre-warmed to the temperature of the sample. Measure the area (or height) of the peaks relating to the VC and internal standard, if used. |
|
5.4.4. |
Construct a graph in which the ordinate value shows the areas (or heights) of the VC peaks or the ratio of the areas (or heights) of VC peaks to the areas (or heights) of the internal standard peaks and the abscissa values show the quantities of VC added (mg) relating to the quantities of the sample of foodstuff weighed in each phial (kg). Measure the abscissa intercept from the graph. The value so obtained is the concentration of VC.in the sample of the foodstuff under investigation. |
|
5.4.5. |
Remove from the column (4.3) excess DMA using an appropriate method as soon as peaks of DMA appear on the chromatogram. |
6. RESULTS
The VC released by materials and articles into the foodstuff under investigation expressed in mg/kg shall be defined as the average of the two determinations (5.4) provided that the repeatability criterion in point 8 is satisfied.
7. CONFIRMATION OF THE VC
In cases where the VC released by materials and articles into the foodstuffs as calculated under point 6, exceeds the criterion in Article 2 (2) of Council Directive 78/142/EEC of 30 January 1978, the values obtained in each of the two determinations (5.4) must be confirmed in one of three ways:
|
(i) |
by using at least one other column (4.3) having a stationary phase of different polarity. This procedure should continue until a chromatogram is obtained with no evidence of overlap of the VC and/or internal standard peaks with constituents of the sample of foodstuff, |
|
(ii) |
by using other detectors, e.g. the micro-electrolytic conductivity detector (2), |
|
(iii) |
by using mass spectrometry; in this case, if molecular ions with parent masses (m/e) of 62 and 64 are found in the ratio of 3: 1 it may be regarded with high probability as confirming the presence of VC. In case of doubt the total mass spectrum must be checked. |
8. REPEATABILITY
The difference between the results of two determinations (5.4) carried out simultaneously or in rapid succession on the same sample, by the same analyst, under the same conditions, must not exceed 0·003 mg VC/kg of foodstuff.
(1) See recommendation ISO DIS 5725: 1977.
(2) See Journal of Chromatographic Science (volume 12), March 1974, page 152.







All