

EUR-Lex Access to European Union law
This document is an excerpt from the EUR-Lex website
Document 32008R0429
Commission Regulation (EC) No 429/2008 of 25 April 2008 on detailed rules for the implementation of Regulation (EC) No 1831/2003 of the European Parliament and of the Council as regards the preparation and the presentation of applications and the assessment and the authorisation of feed additives (Text with EEA relevance)
Verordening (EG) nr. 429/2008 van de Commissie van 25 april 2008 tot vaststelling van voorschriften ter uitvoering van Verordening (EG) nr. 1831/2003 van het Europees Parlement en de Raad wat betreft de opstelling en indiening van aanvragen en de beoordeling van en de verlening van vergunningen voor toevoegingsmiddelen voor diervoeding (Voor de EER relevante tekst)
Verordening (EG) nr. 429/2008 van de Commissie van 25 april 2008 tot vaststelling van voorschriften ter uitvoering van Verordening (EG) nr. 1831/2003 van het Europees Parlement en de Raad wat betreft de opstelling en indiening van aanvragen en de beoordeling van en de verlening van vergunningen voor toevoegingsmiddelen voor diervoeding (Voor de EER relevante tekst)
OJ L 133, 22.5.2008, p. 1–65
(BG, ES, CS, DA, DE, ET, EL, EN, FR, IT, LV, LT, HU, MT, NL, PL, PT, RO, SK, SL, FI, SV)
Special edition in Croatian: Chapter 03 Volume 035 P. 64 - 128
 In force: This act has been changed. Current consolidated version: 27/03/2021
In force: This act has been changed. Current consolidated version: 27/03/2021
|
22.5.2008 |
NL |
Publicatieblad van de Europese Unie |
L 133/1 |
VERORDENING (EG) Nr. 429/2008 VAN DE COMMISSIE
van 25 april 2008
tot vaststelling van voorschriften ter uitvoering van Verordening (EG) nr. 1831/2003 van het Europees Parlement en de Raad wat betreft de opstelling en indiening van aanvragen en de beoordeling van en de verlening van vergunningen voor toevoegingsmiddelen voor diervoeding
(Voor de EER relevante tekst)
DE COMMISSIE VAN DE EUROPESE GEMEENSCHAPPEN,
Gelet op het Verdrag tot oprichting van de Europese Gemeenschap,
Gelet op Verordening (EG) nr. 1831/2003 van het Europees Parlement en de Raad van 22 september 2003 betreffende toevoegingsmiddelen voor diervoeding (1), en met name op artikel 7, leden 4 en 5,
Na raadpleging van de Europese Autoriteit voor voedselveiligheid overeenkomstig artikel 7, leden 4 en 5, van Verordening (EG) nr. 1831/2003,
Overwegende hetgeen volgt:
|
(1) |
Er moeten uitvoeringsvoorschriften worden vastgesteld voor de procedure voor de verlening van vergunningen voor toevoegingsmiddelen voor diervoeding krachtens Verordening (EG) nr. 1831/2003, waaronder voorschriften voor de opstelling en indiening van de aanvragen en voor de beoordeling van en de verlening van vergunningen voor deze toevoegingsmiddelen. Deze voorschriften moeten in de plaats komen van de bepalingen in de bijlage bij Richtlijn 87/153/EEG van de Raad (2) tot vaststelling van richtsnoeren voor de beoordeling van toevoegingsmiddelen in diervoeding. |
|
(2) |
In deze voorschriften moet worden bepaald aan welke vereisten het bij de aanvraag gevoegde dossier moet voldoen. Met name moet worden bepaald welke wetenschappelijke gegevens ter identificatie en karakterisering van het toevoegingsmiddel in kwestie moeten worden ingediend en welke onderzoeken moeten worden voorgelegd om aan te tonen dat het toevoegingsmiddel werkzaam is en veilig is voor mens, dier en milieu, zodat de Europese Autoriteit voor Voedselveiligheid (hierna „de Autoriteit” genoemd) de vergunningsaanvragen kan controleren en beoordelen. |
|
(3) |
Afhankelijk van de aard van het toevoegingsmiddel of de wijze van gebruik waarvoor een aanvraag wordt ingediend, kunnen min of meer uitgebreide onderzoeken nodig zijn om de eigenschappen of effecten van het toevoegingsmiddel te beoordelen. De bedrijven moet dus enige flexibiliteit worden gegund wat betreft de soort onderzoeken en gegevens die zij moeten indienen om aan te tonen dat het toevoegingsmiddel in kwestie veilig en werkzaam is. Bedrijven die van deze flexibiliteit gebruikmaken, moeten dat in het dossier verantwoorden. |
|
(4) |
De Autoriteit moet de mogelijkheid hebben om zo nodig om aanvullende informatie te verzoeken om te kunnen bepalen of het toevoegingsmiddel voldoet aan de voorwaarden voor vergunningverlening van artikel 5 van Verordening (EG) nr. 1831/2003. |
|
(5) |
Bij de opstelling van dossiers betreffende toevoegingsmiddelen die voor gebruik in voeder of water bestemd zijn, is het essentieel dat passende kwaliteitsnormen worden toegepast zodat er geen betwisting is over de resultaten van de laboratoriumtests. |
|
(6) |
Zo nodig moeten specifieke vereisten worden vastgesteld voor elke categorie toevoegingsmiddelen als bedoeld in artikel 6, lid 1, van Verordening (EG) nr. 1831/2003. |
|
(7) |
Om vergunningaanvragen voor minder gangbare soorten te stimuleren en tegelijk een adequaat veiligheidsniveau te behouden, moeten specifieke voorwaarden worden vastgesteld om het mogelijk te maken de resultaten van onderzoek op gangbare soorten te extrapoleren naar minder gangbare diersoorten. |
|
(8) |
In de uitvoeringsvoorschriften voor vergunningsaanvragen moet ermee rekening worden gehouden dat voor voedselproducerende dieren andere vereisten gelden dan voor niet-voedselproducerende dieren, aangezien de beoordeling van de veiligheid voor de menselijke verbruiker bij deze laatste niet relevant is. |
|
(9) |
Overeenkomstig Richtlijn 86/609/EEG van de Raad van 24 november 1986 inzake de onderlinge aanpassing van de wettelijke en bestuursrechtelijke bepalingen van de lidstaten betreffende de bescherming van dieren die voor experimentele en andere wetenschappelijke doeleinden worden gebruikt (3), moet zo weinig mogelijk gebruik worden gemaakt van procedures waarbij laboratoriumdieren voor experimentele en andere wetenschappelijke doeleinden en dierproeven worden gebruikt. |
|
(10) |
Om te voorkomen dat onderzoeken onnodig worden herhaald, moet worden voorzien in vereenvoudigde procedures voor de verlening van een vergunning voor toevoegingsmiddelen die reeds in levensmiddelen mogen worden gebruikt. |
|
(11) |
Wat betreft toevoegingsmiddelen waarvoor reeds krachtens Richtlijn 70/524/EEG van de Raad (4) een vergunning zonder tijdsbeperking is verleend, moet de aanvrager, indien er geen onderzoeken beschikbaar zijn, zo nodig de mogelijkheid krijgen om de werkzaamheid aan te tonen aan de hand van andere informatie, met name informatie over de lange gebruiksgeschiedenis van het toevoegingsmiddel in kwestie. |
|
(12) |
Er moeten voorschriften worden vastgesteld voor aanvragen voor wijzigingen van vergunningen overeenkomstig artikel 13, lid 3, van Verordening (EG) nr. 1831/2003. |
|
(13) |
Voorts moeten voorschriften worden vastgesteld voor aanvragen voor de verlenging van vergunningen overeenkomstig artikel 14 van Verordening (EG) nr. 1831/2003. |
|
(14) |
In de bepalingen betreffende de onderzoeken naar de veiligheid en werkzaamheid die ter staving van de aanvraag moeten worden verricht, moet worden voorzien in een overgangsperiode gedurende welke de huidige voorschriften van toepassing blijven. Aanvragen die vóór de inwerkingtreding van deze verordening worden ingediend, moeten nog overeenkomstig de bijlage bij Richtlijn 87/153/EEG worden behandeld. Aangezien sommige onderzoeken veel tijd in beslag nemen, moeten aanvragers die hun aanvraag binnen een bepaalde termijn na de inwerkingtreding indienen, de keuze hebben tussen de voorschriften van deze verordening en die van de bijlage bij Richtlijn 87/153/EEG. De uitvoeringsvoorschriften zijn opgesteld op basis van de huidige wetenschappelijke en technische kennis en moeten zo nodig aan nieuwe ontwikkelingen worden aangepast. |
|
(15) |
De in deze verordening vervatte maatregelen zijn in overeenstemming met het advies van het Permanent Comité voor de voedselketen en de diergezondheid, |
HEEFT DE VOLGENDE VERORDENING VASTGESTELD:
Artikel 1
Definities
Voor de toepassing van deze verordening gelden de volgende definities:
|
(1) |
„gezelschapsdieren en andere niet-voedselproducerende dieren”: dieren die behoren tot soorten die door mensen worden gevoederd, gefokt of gehouden, maar niet door mensen worden gegeten, met uitzondering van paarden; |
|
(2) |
„minder gangbare soorten”: andere voedselproducerende dieren dan runderen (melk- en vleesrunderen, met inbegrip van kalveren), schapen (vleesschapen), varkens, kippen (met inbegrip van legkippen), kalkoenen en vissen die tot de zalmenfamilie behoren. |
Artikel 2
Toepassing
1. Een aanvraag voor een vergunning voor een toevoegingsmiddel voor diervoeding, als bedoeld in artikel 7 van Verordening (EG) nr. 1831/2003, wordt ingediend aan de hand van het formulier in bijlage I.
De aanvraag gaat vergezeld van een dossier overeenkomstig artikel 3 (hierna „het dossier” genoemd), dat alle nadere gegevens en documenten als bedoeld in artikel 7, lid 3, van Verordening (EG) nr. 1831/2003 bevat.
2. Indien de aanvrager overeenkomstig artikel 18 van Verordening (EG) nr. 1831/2003 vraagt om bepaalde delen van het in lid 1 bedoelde dossier vertrouwelijk te behandelen, voert hij voor ieder document of ieder deel van een document verifieerbare redenen aan waarom de openbaarmaking van die informatie een aanzienlijke nadelige invloed kan hebben op zijn concurrentiepositie. Vertrouwelijke delen worden van de rest van het dossier gescheiden en worden niet opgenomen in de samenvatting als bedoeld in artikel 7, lid 3, onder h), van Verordening (EG) nr. 1831/2003. De aanvrager zendt de Commissie een kopie van de delen van het dossier die hij vertrouwelijk behandeld wenst te zien en van de daarvoor aangevoerde redenen.
Artikel 3
Dossier
1. In het dossier moet op adequate en toereikende wijze worden aangetoond dat het toevoegingsmiddel voldoet aan de voorwaarden voor vergunningverlening van artikel 5 van Verordening (EG) nr. 1831/2003.
2. De algemene vereisten voor de opstelling en indiening van het dossier zijn vermeld in bijlage II.
De specifieke vereisten waaraan het dossier in voorkomend geval moet voldoen, zijn vermeld in bijlage III.
De minimale duur van langetermijnonderzoeken is vermeld in bijlage IV.
3. In afwijking van lid 2 mag de aanvrager een dossier indienen dat niet aan de in lid 2 genoemde vereisten voldoet, mits hij een geldige reden aanvoert voor ieder element dat niet aan die vereisten voldoet.
Artikel 4
Overgangsmaatregelen
1. Op aanvragen voor een vergunning die vóór de datum van inwerkingtreding van deze verordening worden ingediend, blijft de bijlage bij Richtlijn 87/153/EEG van toepassing.
2. Als aanvragers hun aanvraag voor een vergunning vóór 11 juni 2009 indienen, kunnen zij ervoor opteren dat de secties III en IV van de delen I en II van de bijlage bij Richtlijn 87/153/EEG verder worden toegepast in plaats van de punten 1.3, 1.4, 2.1.3, 2.1.4, 2.2.3, 2.2.4, 3.3, 3.4, 4.1.3, 4.1.4, 4.2.3, 4.2.4, 5.3, 5.4, 6.3, 6.4, 7.3, 7.4, 8.3 en 8.4 van bijlage III en in plaats van de bepalingen in de kolom „Minimale duur van langetermijnonderzoeken naar de werkzaamheid” van de tabellen in bijlage IV.
Artikel 5
Inwerkingtreding
Deze verordening treedt in werking op de twintigste dag volgende op die van haar bekendmaking in het Publicatieblad van de Europese Unie.
Deze verordening is verbindend in al haar onderdelen en is rechtstreeks toepasselijk in elke lidstaat.
Gedaan te Brussel, 25 april 2008.
Voor de Commissie
Androulla VASSILIOU
Lid van de Commissie
(1) PB L 268 van 18.10.2003, blz. 29. Verordening gewijzigd bij Verordening (EG) nr. 378/2005 van de Commissie (PB L 59 van 5.3.2005, blz. 8).
(2) PB L 64 van 7.3.1987, blz. 19. Ingetrokken bij Verordening (EG) nr. 1831/2003.
(3) PB L 358 van 18.12.1986, blz. 1. Richtlijn gewijzigd bij Richtlijn 2003/65/EG van het Europees Parlement en de Raad (PB L 230 van 16.9.2003, blz. 32).
(4) PB L 270 van 14.12.1970, blz. 1. Richtlijn laatstelijk gewijzigd bij Verordening (EG) nr. 1800/2007 van de Commissie (PB L 317 van 16.10.2004, blz. 37).
BIJLAGE I
AANVRAAGFORMULIER ALS BEDOELD IN ARTIKEL 2, LID 1, EN ADMINISTRATIEVE GEGEVENS
1. AANVRAAGFORMULIER
EUROPESE COMMISSIE
DIRECTORAAT-GENERAAL
GEZONDHEID EN CONSUMENTENBESCHERMING
(Adres)
Datum: …
|
Betreft |
: |
Aanvraag voor een vergunning voor een toevoegingsmiddel overeenkomstig Verordening (EG) nr. 1831/2003 |
|
|
Vergunning voor een toevoegingsmiddel of voor een nieuw gebruik van een toevoegingsmiddel (artikel 4, lid 1, van Verordening (EG) nr. 1831/2003) |
|
|
Vergunning voor een bestaand product (artikel 10, lid 2 of 7, van Verordening (EG) nr. 1831/2003) |
|
|
Wijziging van een bestaande vergunning (artikel 13, lid 3, van Verordening (EG) nr. 1831/2003) |
|
|
Verlenging van een vergunning voor een toevoegingsmiddel (artikel 14 van Verordening (EG) nr. 1831/2003) |
|
|
Urgente vergunningverlening (artikel 15 van Verordening (EG) nr. 1831/2003) |
(Gelieve een van de vakjes aan te kruisen)
De aanvrager(s) en/of zijn (hun) vertegenwoordiger(s) in de Gemeenschap (artikel 4, lid 3, van Verordening (EG) nr. 1831/2003) dient (dienen) onder de voorwaarden van artikel 7, lid 3, onder a), van Verordening (EG) nr. 1831/2003 (naam, adres…)
…
…
de volgende aanvraag in voor een vergunning voor het volgende product als toevoegingsmiddel voor diervoeding:
1.1. Identificatie en karakterisering van het toevoegingsmiddel
Naam van het toevoegingsmiddel (karakterisering van de werkzame stof(fen) of het agens (de agentia) zoals gedefinieerd in de punten 2.2.1.1 en 2.2.1.2 van bijlage II):
…
…
Handelsnaam (indien nodig voor de aan de houder gebonden vergunningen):
…
…
In de categorie(ën) en functionele groep(en) toevoegingsmiddelen (1) (lijst):
…
…
Doelsoorten:
…
…
…
Naam van de vergunninghouder (artikel 9, lid 6, van Verordening (EG) nr. 1831/2003):
…
…
Voor dit toevoegingsmiddel is reeds overeenkomstig de voederwetgeving een vergunning verleend krachtens Richtlijn ..…/………/(E)EG of Verordening (EG) nr. ……/…… met nummer …………. als (categorie toevoegingsmiddel)
…
Voor dit toevoegingsmiddel is reeds overeenkomstig de levensmiddelenwetgeving een vergunning verleend krachtens Richtlijn ..…/………(E)EG of Verordening (EG) nr. ……/………met nummer ………… als
…
voor gebruik in
…
Indien het product geheel of gedeeltelijk uit een genetisch gemodificeerd organisme (ggo) bestaat of daarmee is geproduceerd, gelieve de volgende gegevens te verstrekken:
|
|
eenduidig identificatienummer (Verordening (EG) nr. 65/2004 van de Commissie (2)) (in voorkomend geval): … |
|
|
hetzij nadere gegevens over een eventuele overeenkomstig Verordening (EG) nr. 1829/2003 van het Europees Parlement en de Raad (3) verleende vergunning: … |
|
|
hetzij nadere gegevens over een eventuele aanvraag voor een vergunning overeenkomstig Verordening (EG) nr. 1829/2003 die in behandeling is: … |
1.2. Gebruiksvoorwaarden
1.2.1. Gebruik in volledige diervoeders
Diersoort of -categorie:
…
…
Maximumleeftijd of -gewicht:
…
…
Minimumdosis (indien van toepassing): mg of aantal activiteitseenheden (4) of kolonievormende eenheden (CFU) of ml/kg volledig diervoeder met een vochtgehalte van 12 %
…
…
Maximumdosis (indien van toepassing): mg of aantal activiteitseenheden of CFU of ml/kg volledig diervoeder met een vochtgehalte van 12 %
…
…
Bij vloeibaar voeder mogen de minimum- en maximumdoses per liter worden opgegeven.
1.2.2. Gebruik in water
Minimumdosis (indien van toepassing): mg of aantal activiteitseenheden of CFU of ml/l water
…
…
Maximumdosis (indien van toepassing): mg of aantal activiteitseenheden of CFU of ml/l water
…
…
1.2.3. Specifieke gebruiksvoorwaarden (indien van toepassing)
Diersoort of -categorie:
…
…
Maximumleeftijd:
…
…
Minimumdosis (indien van toepassing): mg of aantal activiteitseenheden of CFU of ml/kg aanvullend diervoeder met een vochtgehalte van 12 %
…
…
Maximumdosis (indien van toepassing): mg of aantal activiteitseenheden of CFU of ml/kg aanvullend diervoeder met een vochtgehalte van 12 %
…
…
Bij vloeibaar voeder mogen de minimum- en maximumdoses per liter worden opgegeven.
Gebruiksvoorwaarden of -beperkingen (indien van toepassing):
…
…
…
Specifieke voorwaarden of beperkingen bij het hanteren (indien van toepassing):
…
…
…
…
Maximumresidugehalte (indien van toepassing):
diersoort of -categorie:
…
…
indicatorresidu:
…
…
te onderzoeken weefsels of producten:
…
…
…
Maximumresidugehalte in weefsels of producten (μg/kg):
…
…
…
Wachttijd:
…
1.3. Referentiemonsters
Monsternummer communautair referentielaboratorium (CRL) (indien van toepassing):
…
Nummer of code van de partij:
…
Productiedatum:
…
Vervaldatum:
…
Concentratie:
…
Gewicht:
…
Materiële beschrijving:
…
Beschrijving van de recipiënt:
…
Opslagvoorschriften:
…
1.4. Gevraagde wijziging (indien van toepassing)
…
…
…
…
Een kopie van deze aanvraag is rechtstreeks aan de Autoriteit (met het dossier) en het CRL (met de referentiemonsters) toegezonden.
Handtekening: …
1.5. Bijlagen:
|
|
volledig dossier (alleen voor de Autoriteit); |
|
|
openbare samenvatting van het dossier; |
|
|
gedetailleerde samenvatting van het dossier; |
|
|
lijst van de delen van het dossier waarvoor een vertrouwelijke behandeling wordt gevraagd, en kopie van de delen in kwestie (alleen voor de Commissie en de Autoriteit); |
|
|
kopie van de administratieve gegevens van de aanvrager(s); |
|
|
drie monsters van het toevoegingsmiddel voor het CRL, overeenkomstig artikel 7, lid 3, onder f), van Verordening (EG) nr. 1831/2003 (alleen voor het CRL); |
|
|
veiligheidsinformatieblad (alleen voor het CRL); |
|
|
identiteits- en analysecertificaat (alleen voor het CRL), en |
|
|
bevestiging dat de bijdrage in de kosten aan het CRL is betaald (artikel 4 van Verordening (EG) nr. 378/2005 (5)). |
De delen van het formulier die van toepassing zijn, invullen en de delen die niet van toepassing zijn, doorhalen. Het origineel van het aanvraagformulier (met de gevraagde bijlagen) rechtstreeks aan de Europese Commissie toezenden.
2. ADMINISTRATIEVE GEGEVENS VAN DE AANVRAGER(S)
Contactgegevens voor de indiening van een aanvraag voor een vergunning voor een toevoegingsmiddel overeenkomstig Verordening (EG) nr. 1831/2003
|
1) |
Bedrijf of persoon die de aanvraag indient
|
|
2) |
Contactpersoon (voor alle briefwisseling met de Commissie, de Autoriteit en het CRL)
|
(1) Voor de functionele groep „andere zoötechnische toevoegingsmiddelen” in de categorie „zoötechnische toevoegingsmiddelen” moet duidelijk worden vermeld welke functie het toevoegingsmiddel moet hebben.
(2) PB L 10 van 16.1.2004, blz. 5.
(3) PB L 268 van 18.10.2003, blz. 1. Verordening laatstelijk gewijzigd bij Verordening (EG) nr. 298/2008 (PB L 97 van 9.4.2008, blz. 64).
(4) De „eenheid” moet door de aanvrager worden gedefinieerd.
(5) Verordening (EG) nr. 378/2005 van de Commissie van 4 maart 2005 tot vaststelling van gedetailleerde voorschriften voor de uitvoering van Verordening (EG) nr. 1831/2003 van het Europees Parlement en de Raad, wat betreft de verplichtingen en taken van het communautaire referentielaboratorium betreffende vergunningsaanvragen voor toevoegingsmiddelen voor diervoeding (PB L 59 van 5.3.2005, blz. 8). Verordening gewijzigd bij Verordening (EG) nr. 850/2007 (PB L 188 van 20.7.2007, blz. 3).
BIJLAGE II
ALGEMENE VEREISTEN WAARAAN HET IN ARTIKEL 3 BEDOELDE DOSSIER MOET VOLDOEN
ALGEMEEN
Deze bijlage bevat de vereisten voor de vaststelling van de lijst en de kenmerken van de onderzoeken en informatie over stoffen, micro-organismen en preparaten die overeenkomstig artikel 7 van Verordening (EG) nr. 1831/2003 moeten worden ingediend met dossiers voor:
|
— |
een vergunning voor een nieuw toevoegingsmiddel; |
|
— |
een vergunning voor een nieuw gebruik van een toevoegingsmiddel; |
|
— |
een wijziging van een bestaande vergunning voor een toevoegingsmiddel, of |
|
— |
een verlenging van een vergunning voor een toevoegingsmiddel. |
Aan de hand van de dossiers moet het mogelijk zijn de toevoegingsmiddelen op grond van de meest recente gegevens te beoordelen en na te gaan of zij voldoen aan de fundamentele toelatingsvoorwaarden die zijn vastgesteld in artikel 5 van Verordening (EG) nr. 1831/2003.
Welke onderzoeken moeten worden voorgelegd en hoe uitgebreid deze moeten zijn, hangt af van de aard, de categorie en de functionele groep van het toevoegingsmiddel, het type vergunning (al dan niet aan de vergunninghouder gebonden), de stof zelf, de doeldieren en de gebruiksvoorwaarden. In deze bijlage en in bijlage III wordt aangegeven welke onderzoeken en informatie de aanvrager bij zijn aanvraag moet voegen.
Als in het dossier bepaalde gegevens ontbreken of als anderszins wordt afgeweken van de in deze bijlage of in de bijlagen II, III en IV gevraagde gegevens, moet de aanvrager daar duidelijk de redenen voor opgeven.
Het dossier moet gedetailleerde verslagen bevatten van de verrichte onderzoeken. Deze moeten worden gepresenteerd volgens de nummering in deze bijlage. Het dossier moet de referenties en kopieën van alle aangehaalde wetenschappelijke publicaties bevatten, alsook kopieën van eventuele andere adviezen ter zake die reeds door erkende wetenschappelijke instellingen zijn uitgebracht. Indien deze onderzoeken reeds overeenkomstig de communautaire wetgeving door een Europese wetenschappelijke instantie zijn beoordeeld, volstaat een verwijzing naar de resultaten van die beoordeling. Gegevens uit eerder verrichte en gepubliceerde of collegiaal getoetste onderzoeken moeten duidelijk betrekking hebben op hetzelfde toevoegingsmiddel als dat waarvoor de vergunning wordt aangevraagd.
De onderzoeken, inclusief eerder verrichte en gepubliceerde of collegiaal getoetste onderzoeken moeten worden verricht en gedocumenteerd volgens passende kwaliteitsnormen, bv. goede laboratoriumpraktijken (GLP), zoals vastgesteld bij Richtlijn 2004/10/EG van het Europees Parlement en de Raad van 11 februari 2004 betreffende de onderlinge aanpassing van de wettelijke en bestuursrechtelijke bepalingen inzake de toepassing van de beginselen van goede laboratoriumpraktijken en het toezicht op de toepassing ervan voor tests op chemische stoffen (1) of door de Internationale Organisatie voor Normalisatie (ISO).
Als in-vivo- of in-vitro-onderzoek buiten de Gemeenschap wordt uitgevoerd, moet de aanvrager aantonen dat de onderzoeksfaciliteiten voldoen aan de GLP-beginselen van de Organisatie voor Economische Samenwerking en Ontwikkeling (OESO) of de ISO-normen.
De fysisch-chemische, toxicologische en ecotoxicologische eigenschappen moeten worden bepaald volgens de methoden van Richtlijn 67/548/EEG van de Raad van 27 juni 1967 betreffende de aanpassing van de wettelijke en bestuursrechtelijke bepalingen inzake de indeling, de verpakking en het kenmerken van gevaarlijke stoffen (2), laatstelijk gewijzigd bij Richtlijn 2004/73/EG van de Commissie (3), of volgens geactualiseerde methoden die door internationale wetenschappelijke instanties zijn erkend. Indien andere methoden worden gebruikt, moet dat worden gemotiveerd.
Het gebruik van in-vitromethoden en methoden waardoor de gebruikelijke tests op laboratoriumdieren worden verfijnd of vervangen of waarvoor minder proefdieren nodig zijn, wordt aangemoedigd. Deze methoden moeten dezelfde kwaliteit en zekerheid bieden als de methode die zij vervangen.
De beschrijving van de analysemethoden in diervoeder en water moet in overeenstemming zijn met de GLP-voorschriften van Richtlijn 2004/10/EG en/of EN ISO/IEC 17025. Deze methoden moeten voldoen aan de bepalingen van artikel 11 van Verordening (EG) nr. 882/2004 van het Europees Parlement en de Raad van 29 april 2004 inzake officiële controles op de naleving van de wetgeving inzake diervoeders en levensmiddelen en de voorschriften inzake diergezondheid en dierenwelzijn (4).
Ieder dossier moet een openbare samenvatting en een gedetailleerde wetenschappelijke samenvatting bevatten aan de hand waarvan het toevoegingsmiddel in kwestie kan worden geïdentificeerd en gekarakteriseerd.
Ieder dossier moet een voorstel voor monitoring na het in de handel brengen, voor zover vereist krachtens artikel 7, lid 3, onder g), van Verordening (EG) nr. 1831/2003, en een voorstel voor etiketteringsvoorschriften als bedoeld in artikel 7, lid 3, onder e), van Verordening (EG) nr. 1831/2003 bevatten.
Beoordeling van de veiligheid
Deze is gebaseerd op onderzoeken die beogen aan te tonen dat het gebruik van het toevoegingsmiddel veilig is voor:
|
a) |
de doelsoorten bij de hoogste voorgestelde concentraties in het diervoeder of het water en bij een veelvoud van die concentraties om een veiligheidsmarge vast te stellen; |
|
b) |
consumenten die levensmiddelen consumeren die afkomstig zijn van dieren waarin het toevoegingsmiddel of residuen of metabolieten daarvan aanwezig zijn. In dit geval wordt de veiligheid gewaarborgd door het vaststellen van maximumresidugehalten (MRL's) en wachttijden op basis van een aanvaardbare dagelijkse inname (ADI) of een „tolerable upper intake level” (UL); |
|
c) |
personen die wellicht door inademing of door contact met slijmvliezen, ogen of huid aan het toevoegingsmiddel worden blootgesteld wanneer zij het toevoegingsmiddel hanteren of aan voormengsels of volledige diervoeders toevoegen of wanneer zij voeder of water gebruiken dat het toevoegingsmiddel bevat; |
|
d) |
mens en dier gelet op de selectie en verspreiding van genen voor antimicrobiële resistentie, en |
|
e) |
het milieu, wat betreft het toevoegingsmiddel zelf of daarvan, rechtstreeks en/of door uitscheiding van de dieren, afgeleide producten. |
Als het toevoegingsmiddel uit verscheidene bestanddelen bestaat, mag de veiligheid voor de consument van elk bestanddeel afzonderlijk worden beoordeeld en mag vervolgens het cumulatieve effect in aanmerking worden genomen, mits wordt aangetoond dat er geen wisselwerking tussen de bestanddelen is. Zo niet moet het volledige mengsel worden beoordeeld.
Beoordeling van de werkzaamheid
Deze is gebaseerd op onderzoeken die beogen aan te tonen dat het toevoegingsmiddel werkzaam is met betrekking tot de beoogde gebruiksdoeleinden als omschreven in artikel 6, lid 1, en bijlage I van Verordening (EG) nr. 1831/2003.
1. SECTIE I: SAMENVATTING VAN HET DOSSIER
1.1. Openbare samenvatting overeenkomstig artikel 7, lid 3, onder h), van Verordening (EG) nr. 1831/2003
De aanvrager moet een samenvatting indienen waarin de voornaamste kenmerken van het toevoegingsmiddel worden beschreven. Deze samenvatting bevat geen vertrouwelijke informatie en wordt als volgt ingedeeld:
1.1.1. Inhoud
|
a) |
naam van de aanvrager(s); |
|
b) |
identificatie van het toevoegingsmiddel; |
|
c) |
productiemethode en analysemethode; |
|
d) |
onderzoeken naar de veiligheid en werkzaamheid van het toevoegingsmiddel; |
|
e) |
voorgestelde gebruiksvoorwaarden, en |
|
f) |
voorstel voor monitoring na het in de handel brengen. |
1.1.2. Beschrijving
|
a) |
naam en adres van de aanvrager(s) Deze informatie moet in alle gevallen worden verstrekt, ongeacht het type vergunning (al dan niet aan de vergunninghouder gebonden). Indien het dossier door een groep aanvragers wordt ingediend, moeten de namen van alle aanvragers worden vermeld. |
|
b) |
identificatie van het toevoegingsmiddel De identificatie van het toevoegingsmiddel omvat samenvatting van de volgens bijlage II of III vereiste informatie, afhankelijk van het type vergunning. Het gaat met name om de naam van het toevoegingsmiddel, de voorgestelde indeling naar categorie en functionele groep, de doeldiersoorten of -categorieën en de doses. |
|
c) |
productiemethode en analysemethode Het productieproces moet worden beschreven. Er moet een beschrijving worden gegeven van de algemene procedures van de analysemethoden voor de officiële controles op het toevoegingsmiddel als zodanig in voormengsels en in diervoeders, zoals voorgeschreven in deze bijlage en bijlage III. Op basis van de informatie die overeenkomstig deze bijlage en bijlage III wordt ingediend, moet in voorkomend geval ook de procedure van de methode(n) voor de officiële controles op het toevoegingsmiddel of metabolieten daarvan in voedsel van dierlijke oorsprong worden beschreven. |
|
d) |
onderzoeken naar de veiligheid en werkzaamheid van het toevoegingsmiddel Er moet worden vermeld welke conclusie uit de verschillende verrichte onderzoeken is getrokken met betrekking tot de veiligheid en werkzaamheid van het toevoegingsmiddel. Ter staving van de conclusie van de aanvrager(s) kunnen de resultaten van de onderzoeken in tabelvorm worden vermeld. Alleen onderzoeken overeenkomstig bijlage III moeten in de samenvatting worden vermeld. |
|
e) |
voorgestelde gebruiksvoorwaarden De gebruiksvoorwaarden moeten door de aanvrager(s) worden voorgesteld. Met name moet de aanvrager de te gebruiken concentratie in water of diervoeder aangeven, alsook gedetailleerde voorwaarden voor gebruik in aanvullend diervoeder. Er moet ook informatie worden verstrekt over eventuele andere wijzen van toediening of toevoeging aan diervoeder of water. In voorkomend geval moeten de specifieke gebruiksvoorwaarden (bv. onverenigbaarheid), de specifieke etiketteringsvoorschriften en de diersoorten waarvoor het toevoegingsmiddel bestemd is, worden vermeld. |
|
f) |
voorstel voor monitoring na het in de handel brengen Dit deel heeft alleen betrekking op toevoegingsmiddelen die overeenkomstig artikel 7, lid 3, onder g), van Verordening (EG) nr. 1831/2003 tot een andere categorie behoren dan genoemd in artikel 6, lid 1, onder a) en b), en op toevoegingsmiddelen die vallen onder de communautaire wetgeving inzake het in de handel brengen van producten die geheel of gedeeltelijk uit ggo's bestaan of daarmee zijn geproduceerd. |
1.2. Wetenschappelijke samenvatting van het dossier
Er moet een wetenschappelijke samenvatting worden ingediend met gegevens uit elk deel van de documenten die overeenkomstig deze bijlage en bijlage III ter staving van de aanvraag worden ingediend. Ook de conclusies van de aanvrager(s) moeten worden vermeld.
In de samenvatting moeten, in dezelfde volgorde als in deze bijlage, alle onderdelen ter sprake komen, waarbij naar de desbetreffende bladzijden van het dossier wordt verwezen.
1.3. Lijst van documenten en andere bijzonderheden
De aanvrager moet het nummer en de titel vermelden van de stukken die hij ter staving van de aanvraag indient. Er moet een gedetailleerde index met verwijzing naar de stukken en bladzijden worden bijgevoegd.
1.4. Lijst van de delen van het dossier waarvoor een vertrouwelijke behandeling wordt gevraagd (in voorkomend geval)
In de lijst moet worden verwezen naar de desbetreffende stukken en bladzijden.
2. SECTIE II: IDENTITEIT EN KARAKTERISERING VAN EN GEBRUIKSVOORWAARDEN VOOR HET TOEVOEGINGSMIDDEL; ANALYSEMETHODEN
Het toevoegingsmiddel moet volledig worden geïdentificeerd en gekarakteriseerd.
2.1. Identiteit van het toevoegingsmiddel
2.1.1. Naam van het additief
Voor toevoegingsmiddelen die aan een vergunninghouder gebonden zijn, wordt zo nodig een handelsnaam voorgesteld.
2.1.2. Voorgestelde indeling
Naargelang van de voornaamste functies van het toevoegingsmiddel overeenkomstig artikel 6 en bijlage I van Verordening (EG) nr. 1831/2003 wordt een voorstel gedaan om het in een of meer categorieën en functiegroepen in te delen.
Er moeten ook gegevens over andere bekende aanwendingsmogelijkheden van identieke werkzame stoffen of agentia (bv. in levensmiddelen, in de humane of diergeneeskunde, in landbouw en de industrie) worden verstrekt. Eventuele andere vergunningen voor gebruik als toevoegingsmiddel voor diervoeder, levensmiddelenadditief of diergeneesmiddel of andere soorten vergunningen voor de werkzame stof moeten worden vermeld.
2.1.3. Kwalitatieve en kwantitatieve samenstelling (werkzame stof/agens, overige bestanddelen, onzuiverheden, variatie tussen charges)
De werkzame stof(fen)/het agens (de agentia) en alle overige bestanddelen van het toevoegingsmiddel moeten met het gewichtsaandeel in het eindproduct worden vermeld. De kwalitatieve en kwantitatieve variatie tussen charges van de werkzame stof(fen)/het agens (de agentia) moet worden bepaald.
Bij micro-organismen moet het aantal levensvatbare cellen of sporen, uitgedrukt in CFU per gram, worden bepaald.
Bij enzymen moet elke gedeclareerde (voornaamste) activiteit worden beschreven en moet het aantal eenheden van elke activiteit in het eindproduct worden vermeld. Ook relevante nevenactiviteiten moeten worden vermeld. De activiteitseenheden moeten worden gedefinieerd, bij voorkeur als μmol product dat per minuut uit het substraat vrijkomt, met vermelding van pH en temperatuur.
Als het werkzame bestanddeel van het toevoegingsmiddel een mengsel van werkzame bestanddelen of agentia is, die alle duidelijk (kwalitatief en kwantitatief) kunnen worden bepaald, moeten deze werkzame stoffen/agentia afzonderlijk worden beschreven en moet de mengverhouding worden vermeld.
Andere mengsels, waarvan de bestanddelen niet met één chemische formule kunnen worden beschreven en/of niet allemaal kunnen worden geïdentificeerd, moeten worden gekarakteriseerd door het bestanddeel of de bestanddelen die tot de activiteit van het mengsel bijdragen en/of door een of meer typische hoofdbestanddelen.
Onverminderd eventuele verzoeken van de Autoriteit om aanvullende informatie overeenkomstig artikel 8, lid 2, van Verordening (EG) nr. 1831/2003, hoeft de aanvrager geen beschrijving te geven van overige bestanddelen die geen veiligheidsrisico's inhouden, met uitzondering van werkzame stoffen en agentia voor toevoegingsmiddelen die niet tot de categorieën zoötechnische toevoegingsmiddelen, coccidiostatica en histomonostatica behoren en niet binnen de werkingssfeer van Verordening (EG) nr. 1829/2003 vallen. In ieder geval moeten alle in het dossier vermelde onderzoeken zijn uitgevoerd op hetzelfde toevoegingsmiddel als dat waarvoor de vergunning wordt aangevraagd. Daarnaast mag informatie worden verstrekt over eventuele andere bereidingen die ermee kunnen worden gemaakt. In documenten van derden mag een interne identificatiecode worden gebruikt. In een verklaring moet een lijst van de identificatiecodes worden opgenomen en moet worden bevestigd dat de identificatiecodes verwijzen naar de formuleringen waarvoor de aanvraag wordt ingediend.
2.1.4. Zuiverheid
Chemische en microbiële onzuiverheden en stoffen met toxische of andere ongewenste eigenschappen die niet opzettelijk aan het toevoegingsmiddel worden toegevoegd en niet aan de activiteit ervan bijdragen, moeten door de aanvrager worden geïdentificeerd en gekwantificeerd. Bij gistingsproducten moet de aanvrager ook bevestigen dat er geen productieorganismen in het toevoegingsmiddel aanwezig zijn. Het protocol voor de routinescreening van de productiepartijen op contaminanten en onzuiverheden moet worden beschreven.
Alle verstrekte gegevens moeten het voorstel voor een specificatie van het toevoegingsmiddel ondersteunen.
Afhankelijk van het productieproces zijn krachtens de communautaire wetgeving de onderstaande voorschriften van toepassing.
2.1.4.1. Toevoegingsmiddelen waarvoor de vergunning aan een vergunninghouder gebonden is
In het geval van toevoegingsmiddelen waarvoor de vergunning aan de vergunninghouder gebonden is, moet op basis van de bestaande normen voor andere, soortgelijke doeleinden worden beschreven welk specifiek proces de producent gebruikt. Daarvoor kunnen specificaties van het Gezamenlijk Comité van deskundigen voor levensmiddelenadditieven van de FAO/WHO (JECFA) of specificaties uit EG-vergunningen voor levensmiddelenadditieven worden gebruikt.
2.1.4.2. Toevoegingsmiddelen waarvoor de vergunning niet aan een vergunninghouder gebonden is
In het geval van toevoegingsmiddelen waarvoor de vergunning niet aan een vergunninghouder gebonden is, mogen bestaande normen worden gebruikt die voor andere, soortgelijke doeleinden worden gebruikt of die specificaties uit in de EG toegelaten levensmiddelenadditieven of JECFA-specificaties bevatten. Indien er geen dergelijke normen beschikbaar zijn, of indien dit op het productieproces van toepassing is, moeten ten minste de volgende bijzonderheden worden beschreven en moeten de desbetreffende concentraties worden bepaald:
|
— |
voor micro-organismen: microbiologische verontreiniging, mycotoxinen en zware metalen; |
|
— |
voor gistingsproducten (die geen micro-organismen als werkzame agentia bevatten) gelden dezelfde voorschriften als voor micro-organismen (zie hierboven). Voorts moet worden vermeld in welke mate er afgewerkt groeimedium in het eindproduct aanwezig is; |
|
— |
voor uit planten gewonnen stoffen: microbiologische en botanische verontreiniging (met name ricinus, onkruidzaden en moederkoren), mycotoxinen, pesticidenverontreiniging, maximumgehalten aan oplosmiddelen en, in voorkomend geval, toxicologisch relevante stoffen die in de plant van oorsprong kunnen voorkomen; |
|
— |
voor uit dieren gewonnen stoffen: microbiologische verontreiniging, zware metalen en, in voorkomend geval, maximumgehalten aan oplosmiddelen; |
|
— |
voor minerale stoffen: zware metalen, dioxinen en pcb's; |
|
— |
voor producten die door chemische synthese en processen zijn verkregen: alle chemische stoffen die in de synthetische processen zijn gebruikt, alsook alle tussenproducten die in het eindproduct achterblijven, moeten worden geïdentificeerd, met opgave van de desbetreffende concentraties. |
In voorkomend geval moeten de mycotoxinen voor analysedoeleinden worden geselecteerd volgens de verschillende matrices.
2.1.5. Aggregatietoestand van elke vorm van het product
Wat vaste preparaten betreft, moeten gegevens worden verstrekt over deeltjesgrootteverdeling, deeltjesvorm, dichtheid, bulkdichtheid, stofvorming en het gebruik van processen die de fysische eigenschappen beïnvloeden. Wat vloeibare preparaten betreft, moeten gegevens over viscositeit en oppervlaktespanning worden verstrekt. Als het toevoegingsmiddel bestemd is om in water te worden gebruikt, moet de oplosbaarheid of dispersiegraad worden gedocumenteerd.
2.2. Karakterisering van de werkzame stof(fen)/het agens (de agentia)
2.2.1. Beschrijving
De werkzame stof of het agens moet kwalitatief worden beschreven. Dit omvat de zuiverheid en de oorsprong van de stof of het agens en eventuele andere relevante kenmerken.
2.2.1.1. Chemische stoffen
Chemisch gedefinieerde stoffen moeten worden beschreven met de generische naam, de chemische naam volgens de IUPAC-nomenclatuur, andere internationale generische namen en afkortingen en/of het CAS-nummer. Ook de structuurformule, de molecuulformule en het molecuulgewicht moeten worden vermeld.
Van chemisch gedefinieerde verbindingen die als aroma worden gebruikt, moeten het Flavis-nummer en de desbetreffende chemische groep worden vermeld. In het geval van plantenextracten moeten de fytochemische merkers worden vermeld.
Mengsels waarvan de bestanddelen niet met één chemische formule kunnen worden beschreven en/of niet allemaal kunnen worden geïdentificeerd, moeten worden gekarakteriseerd door het bestanddeel of de bestanddelen die tot de activiteit van het mengsel bijdragen en/of door een of meer typische hoofdbestanddelen. De merkstof moet worden geïdentificeerd om de stabiliteit te kunnen beoordelen en voor de traceerbaarheid.
Wat enzymen en enzympreparaten betreft, moet voor elke gedeclareerde activiteit het nummer en de systematische naam worden vermeld die de International Union of Biochemistry (IUB) voorstelt in de recentste editie van de „Enzyme Nomenclature”. Voor activiteiten die nog niet zijn opgenomen, moet een systematische naam worden gebruikt overeenkomstig de nomenclatuurregels van de IUB. Triviale namen kunnen worden aanvaard voor zover zij ondubbelzinnig zijn, in het hele dossier op consistente wijze worden gebruikt en bij de eerste vermelding duidelijk in verband kunnen worden gebracht met de systematische naam en het IUB-nummer. Van elke enzymactiviteit moet de biologische herkomst worden vermeld.
Van chemische stoffen die door gisting worden geproduceerd, moet ook de microbiële herkomst worden vermeld (zie punt 2.2.1.2. Micro-organismen).
2.2.1.2. Micro-organismen
Van alle micro-organismen, ongeacht of zij als product of als productiestam worden gebruikt, moet de herkomst worden vermeld.
Van micro-organismen die als product of productiestam worden gebruikt, moet in voorkomend geval de wijzigingsgeschiedenis worden vermeld. Van elk micro-organisme moeten de naam en de taxonomische classificatie worden vermeld volgens de recentst gepubliceerde internationale nomenclatuurregels (ICN). Bacteriestammen moeten in een internationaal erkende kweekverzameling (bij voorkeur in de EU) worden gedeponeerd en daar gedurende de toegestane levensduur van het toevoegingsmiddel worden bijgehouden. Er moet een bewijs van deponering worden afgegeven waarop het volgnummer wordt vermeld waaronder de stam wordt bijgehouden. Daarnaast moeten alle relevante morfologische, fysiologische en moleculaire kenmerken worden beschreven die nodig zijn om de stam eenduidig te identificeren, en moet worden beschreven hoe de genetische stabiliteit kan worden bevestigd. In het geval van ggo's moeten de genetische wijzigingen worden beschreven. Van elk ggo moet het eenduidige identificatienummer worden vermeld overeenkomstig Verordening (EG) nr. 65/2004 van de Commissie van 14 januari 2004 tot vaststelling van een systeem voor de ontwikkeling en toekenning van eenduidige identificatienummers voor genetisch gemodificeerde organismen.
2.2.2. Relevante eigenschappen
2.2.2.1. Chemische stoffen
De fysische en chemische eigenschappen moeten worden beschreven. Voor zover van toepassing moeten dissociatieconstante, pKa, elektrostatische eigenschappen, smeltpunt, kookpunt, dichtheid, dampspanning, oplosbaarheid in water en organische oplosmiddelen, Kow en Kd/Koc, massa- en absorptiespectra, NMR-gegevens, mogelijke isomeren en eventuele andere relevante fysische eigenschappen worden vermeld.
Door gisting geproduceerde stoffen moeten vrij zijn van antimicrobiële activiteiten die een rol spelen bij het gebruik van antibiotica bij mens of dier.
2.2.2.2. Micro-organismen
|
— |
toxinen en virulentiefactoren Wat toxinen en virulentiefactoren betreft, moet worden aangetoond dat deze afwezig zijn of geen probleem vormen. Bacteriestammen die behoren tot een taxonomische groep waarvan bekend is dat bepaalde leden toxinen of andere virulentiefactoren kunnen produceren, moeten afdoende worden getest om op moleculair niveau en zo nodig op celniveau aan te tonen dat er geen reden tot zorg is. In het geval van stammen micro-organismen die geen geschiedenis van blijkbaar veilig gebruik hebben en waarvan de biologie relatief onbekend is, is een volledig pakket toxicologische onderzoeken nodig. |
|
— |
antibioticaproductie en antibioticaresistentie Micro-organismen die als toevoegingsmiddel of productiestam worden gebruikt, moeten vrij van antibiotica-activiteit zijn en mogen niet in staat zijn stoffen te produceren die bij mens of dier als antibiotica worden gebruikt. Stammen micro-organismen die bestemd zijn om als toevoegingsmiddel te worden gebruikt, mogen niet verder bijdragen aan het reservoir antibioticaresistente genen dat reeds aanwezig is in de darmflora van dieren en in het milieu. Daarom moeten alle bacteriestammen worden getest op resistentie tegen antibiotica die in de humane geneeskunde en de diergeneeskunde worden gebruikt. Als resistentie wordt vastgesteld, moeten de genetische basis van de resistentie en het risico van overdracht naar andere darmorganismen worden bepaald. Stammen micro-organismen die resistentie hebben ontwikkeld tegen één of meer antimicrobiële stoffen mogen niet als toevoegingsmiddel worden gebruikt, tenzij wordt aangetoond dat de resistentie het gevolg is van één of meer chromosoommutaties en niet overdraagbaar is. |
2.3. Productieproces en eventuele specifieke bewerkingsprocedés
Om na te gaan welke kritieke punten van het proces van invloed kunnen zijn op de zuiverheid van de werkzame stof(fen), het agens (de agentia) of het toevoegingsmiddel, moet het productieproces worden beschreven. Er moet een veiligheidsinformatieblad worden verstrekt betreffende de chemische stoffen die in het productieproces worden gebruikt.
2.3.1. Werkzame stof(fen)/agens (agentia)
Het productieproces (bv. chemische synthese, gisting, kweek, extractie uit organisch materiaal, distillatie) voor de bereiding van het toevoegingsmiddel moet, zo nodig met een flowchart, worden beschreven. De samenstelling van de gistings- of cultuurmedia moet worden vermeld. De zuiveringsmethoden moeten gedetailleerd worden beschreven.
Op genetisch gemodificeerde micro-organismen die als bron van toevoegingsmiddelen worden gebruikt en onder ingeperkte omstandigheden gekweekt worden, is Richtlijn 90/219/EEG (5) van de Raad van toepassing. Het gistingsproces (cultuurmedium, gistingsomstandigheden en verdere verwerking van de gistingsproducten) moet worden beschreven.
2.3.2. Toevoegingsmiddel
Het productieproces van het toevoegingsmiddel moet gedetailleerd worden beschreven. De voornaamste fasen van de bereiding van het toevoegingsmiddel, waaronder het punt (de punten) waarop de werkzame stof(fen) of het agens (de agentia) wordt (worden) toegevoegd, en alle verdere stappen die van invloed zijn op de bereiding, moeten, zo nodig met een flowchart, worden beschreven.
2.4. Fysisch-chemische en technologische eigenschappen van het toevoegingsmiddel
2.4.1. Stabiliteit
De stabiliteit wordt over het algemeen gemeten door een analytische follow-up van de werkzame stof(fen) of het agens (de agentia) of de activiteit of levensvatbaarheid daarvan. Bij enzymen kan de stabiliteit worden gedefinieerd in termen van afname van de katalytische werking, bij micro-organismen in termen van afname van de levensvatbaarheid en bij aroma's in termen van smaakverlies. Bij andere chemische mengsels en extracten kan de stabiliteit worden beoordeeld door een of meer geschikte merkstoffen te monitoren.
Stabiliteit van het toevoegingsmiddel
De stabiliteit van elke formulering van het toevoegingsmiddel bij blootstelling aan verschillende omgevingsfactoren (licht, temperatuur, pH, vocht, zuurstof en verpakkingsmateriaal) moet worden onderzocht. De verwachte houdbaarheid van het toevoegingsmiddel zoals het in de handel wordt gebracht, moet worden gebaseerd op ten minste twee modelsituaties die de diverse te verwachten gebruiksomstandigheden omvatten (bv. 25 oC en 60 % relatieve luchtvochtigheid (HR) en 40 oC en 75 % HR).
Stabiliteit van het toevoegingsmiddel bij gebruik in voormengsels en diervoeder
Bij toevoegingsmiddelen die in voormengsels en diervoeders worden gebruikt, met uitzondering van aromatische stoffen, moet de stabiliteit van elke formulering van het toevoegingsmiddel onder de gewoonlijke productie- en opslagomstandigheden van voormengsels en diervoeder worden onderzocht. De onderzoeken naar de stabiliteit in voormengsels moeten ten minste zes maanden beslaan. De stabiliteit moet bij voorkeur worden getest met voormengsels die sporenelementen bevatten. Zo niet moet op het etiket van het toevoegingsmiddel worden vermeld: „niet mengen met sporenelementen”.
De onderzoeken naar de stabiliteit in diervoeder moeten normaliter ten minste drie maanden beslaan. De algemene stabiliteit moet worden gecontroleerd in slobber en pellets (ook de invloed van de verwerking tot pellets of andere soorten behandeling) voor de voornaamste in de aanvraag genoemde diersoorten.
Bij toevoegingsmiddelen die bestemd zijn om in water te worden gebruikt, moet de stabiliteit van elke formulering in water en onder gesimuleerde gebruiksomstandigheden worden onderzocht.
Als de stabiliteit afneemt, moeten zo nodig de eventuele afbraak- of ontledingsproducten worden gekarakteriseerd.
Er moeten gegevens worden verstrekt van analyses die ten minste één waarneming aan het begin en één waarneming aan het einde van de opslagperiode omvatten.
Zo nodig moet een gedetailleerde beschrijving worden gegeven van de kwalitatieve en kwantitatieve samenstelling van de voormengsels of diervoeders die voor het onderzoek zijn gebruikt.
2.4.2. Homogeniteit
Behalve voor aromastoffen moet worden aangetoond dat het toevoegingsmiddel zich homogeen kan verdelen in voormengsels, diervoeder of water.
2.4.3. Overige kenmerken
Andere kenmerken, zoals stofvorming, elektrostatische eigenschappen en dispergeerbaarheid in vloeistoffen moeten worden beschreven.
2.4.4. Fysisch-chemische onverenigbaarheid of interacties
Te verwachten fysisch-chemische onverenigbaarheid of interacties met diervoeder, dragermateriaal, andere toegelaten toevoegingsmiddelen of geneesmiddelen moeten worden vermeld.
2.5. Gebruiksvoorwaarden voor het toevoegingsmiddel
2.5.1. Voorgestelde gebruikswijze in diervoeder
De diersoort of -categorie en de leeftijdsgroep of het productiestadium van het dier moeten worden aangegeven volgens de categorieën in bijlage IV bij deze verordening. Eventuele contra-indicaties moeten worden vermeld. Het voorgestelde gebruik, in water of diervoeder, moet worden omschreven.
Er moeten nadere gegevens worden verstrekt over de wijze van toediening en de aan voormengsels, diervoeder of drinkwater toe te voegen hoeveelheid. Ook de voorgestelde dosis in het volledige diervoeder, de voorgestelde duur van de toediening en de eventuele wachttijd moeten worden vermeld. Als een bijzonder gebruik van een toevoegingsmiddel in aanvullend diervoeder wordt voorgesteld, moet dat worden gerechtvaardigd.
2.5.2. Informatie over de veiligheid van gebruikers en werknemers
2.5.2.1. Chemische stoffen
Er moet een veiligheidsinformatieblad worden verstrekt overeenkomstig Richtlijn 91/155/EEG van de Commissie van 5 maart 1991 houdende beschrijving en vaststelling van de wijze van uitvoering van het systeem voor specifieke informatie inzake gevaarlijke preparaten krachtens artikel 10 van Richtlijn 88/379/EEG van de Raad (6). Zo nodig moeten bij de productie, hantering, gebruik en verwijdering maatregelen ter preventie van beroepsrisico's en beschermingsmiddelen worden voorgesteld.
2.5.2.2. Micro-organismen
Er moet een classificatie worden verstrekt overeenkomstig Richtlijn 2000/54/EG van het Europees Parlement en de Raad van 18 september 2000 betreffende de bescherming van de werknemers tegen de risico's van blootstelling aan biologische agentia op het werk (zevende bijzondere richtlijn in de zin van artikel 16, lid 1, van Richtlijn 89/391/EEG) (7). Over micro-organismen die overeenkomstig deze richtlijn niet in groep 1 zijn ingedeeld, moeten de afnemers informatie krijgen zodat zij overeenkomstig artikel 3, lid 2, van deze richtlijn de nodige maatregelen ter bescherming van de veiligheid van hun werknemers kunnen treffen.
2.5.2.3. Etiketteringsvoorschriften
Onverminderd de etiketterings- en verpakkingsvoorschriften van artikel 16 van Verordening (EG) nr. 1831/2003 moeten eventuele specifieke etiketteringsvoorschriften en, zo nodig, specifieke gebruiks- en hanteringsvoorwaarden (waaronder bekende onverenigbaarheid en contra-indicaties) en aanwijzingen voor een correct gebruik worden verstrekt.
2.6. Analysemethoden en referentiemonsters
De analysemethoden moeten worden verstrekt volgens de genormaliseerde presentatie die door de ISO wordt aanbevolen (namelijk ISO 78-2).
Overeenkomstig de Verordeningen (EG) nr. 1831/2003 en (EG) nr. 378/2005 worden de in deze rubriek genoemde analysemethoden door het CRL geëvalueerd. Het CRL zendt de Autoriteit een evaluatieverslag waarin het aangeeft of deze methoden geschikt zijn voor officiële controles op het toevoegingsmiddel waarvoor een vergunning wordt aangevraagd. De evaluatie door het CRL betreft vooral de in de punten 2.6.1 en 2.6.2 vermelde methoden.
Indien overeenkomstig Verordening (EG) nr. 2377/90 van de Raad van 26 juni 1990 houdende een communautaire procedure tot vaststelling van maximumwaarden voor residuen van geneesmiddelen voor diergeneeskundig gebruik in levensmiddelen van dierlijke oorsprong (8) een MRL is vastgesteld voor het toevoegingsmiddel waarvoor een vergunning wordt aangevraagd, hoeft punt 2.6.2 niet door het CRL te worden geëvalueerd. Wat punt 2.6.2 betreft, verstrekt de aanvrager dezelfde methode, informatie en bijzonderheden (waaronder desbetreffende updates) die moeten worden verstrekt aan het Europees Geneesmiddelenbureau overeenkomstig bijlage V bij Verordening (EG) nr. 2377/90 en de „Notice to Applicants and Guidelines”, vol. 8 van de reeks „Voorschriften inzake geneesmiddelen in de Europese Gemeenschap”.
Als het CRL, de Autoriteit of de Commissie dat nodig achten, kunnen ook de in punt 2.6.3 beschreven analysemethoden worden geëvalueerd.
Overeenkomstig Verordening (EG) nr. 378/2005 zendt de aanvrager de referentiemonsters vóór de evaluatie van het technische dossier rechtstreeks naar het CRL; vóór de vervaldatum stuurt hij vervangingsmonsters.
De aanvragers volgen de gedetailleerde richtsnoeren die het CRL overeenkomstig artikel 12 van Verordening (EG) nr. 378/2005 verstrekt.
2.6.1. Analysemethoden voor de werkzame stof
Er moet een gedetailleerde karakterisering worden verstrekt van de kwalitatieve en, indien van toepassing, kwantitatieve analysemethode(n) voor de controle op de naleving van de voorgestelde maximum- of minimumgehalten van de werkzame stof(fen)/het agens (de agentia) in het toevoegingsmiddel, voormengsels, diervoeder en, in voorkomend geval, water.
|
2.6.1.1. |
Deze methoden moeten aan dezelfde eisen voldoen als analysemethoden voor officiële controles, overeenkomstig artikel 11 van Verordening (EG) nr. 882/2004. Met name moeten zij aan ten minste een van de volgende eisen voldoen:
|
|
2.6.1.2. |
Bij de gedetailleerde karakterisering van de methode(n) moeten de nodige kenmerken worden beschreven zoals vermeld in bijlage III bij Verordening (EG) nr. 882/2004. |
|
2.6.1.3. |
De prestatiekenmerken van intern gevalideerde methoden moeten worden gecontroleerd door de methode in een tweede, erkend en onafhankelijk laboratorium te laten testen. Samen met de resultaten van deze tests kan eventueel andere informatie worden ingediend waaruit blijkt dat de methode ook door een officieel laboratorium kan worden gebruikt. Indien het tweede laboratorium deel uitmaakt van het consortium van nationale referentielaboratoria (NRL's) dat het CRL overeenkomstig Verordening (EG) nr. 378/2005 bijstaat, moet dit laboratorium, met het oog op de onafhankelijkheid en de betrokkenheid bij de beoordeling van de door de aanvrager ingediende informatie, zodra het CRL de aanvraag ontvangt een belangenverklaring aan het CRL toezenden waarin het zijn werkzaamheden in verband met de aanvraag beschrijft, en mag het niet aan de beoordeling van de aanvraag meewerken. |
|
2.6.1.4. |
In zijn evaluatieverslag voor de Autoriteit kan het CRL passende kenmerken selecteren als genoemd in bijlage III bij Verordening (EG) nr. 882/2004. |
|
2.6.1.5. |
De prestatiecriteria voor methoden voor bepaalde groepen stoffen (bv. enzymen) kunnen worden vastgesteld in de gedetailleerde richtsnoeren die het CRL opstelt overeenkomstig artikel 12 van Verordening (EG) nr. 378/2005. |
2.6.2. Analysemethoden voor de bepaling van het gehalte aan residuen van het toevoegingsmiddel of metabolieten daarvan in voedsel
Er moet een gedetailleerde karakterisering worden verstrekt van de kwalitatieve en kwantitatieve analysemethode(n) voor de bepaling van de indicatorresiduen en/of metabolieten van het toevoegingsmiddel in de te onderzoeken weefsels en dierlijke producten.
|
2.6.2.1. |
Deze methoden moeten aan dezelfde eisen voldoen als analysemethoden voor officiële controles, overeenkomstig artikel 11 van Verordening (EG) nr. 882/2004. Met name moeten zij aan ten minste een van de in punt 2.6.1.1 genoemde eisen voldoen. |
|
2.6.2.2. |
Bij de gedetailleerde karakterisering van de methode(n) moeten de nodige kenmerken worden beschreven zoals vermeld in bijlage III bij Verordening (EG) nr. 882/2004 en moeten de voorschriften van Beschikking 2002/657/EG van de Commissie (10) in acht worden genomen. Zo nodig moeten dezelfde prestatiecriteria worden onderzocht als in de besluiten van de Commissie tot vaststelling van analysemethoden voor het opsporen van bepaalde stoffen en residuen daarvan in producten van levende dieren overeenkomstig Richtlijn 96/23/EG van de Raad. De bepaalbaarheidsgrens van elke methode mag niet meer bedragen dan de helft van de desbetreffende MRL en moet ten minste gevalideerd zijn voor een bereik van de helft van de MRL tot tweemaal de MRL. |
|
2.6.2.3. |
De prestatiekenmerken van intern gevalideerde methoden moeten worden gecontroleerd door de methode in een tweede, erkend en onafhankelijk laboratorium te laten testen. De resultaten van die tests moeten worden verstrekt. Indien het tweede laboratorium deel uitmaakt van het consortium van nationale referentielaboratoria (NRL's) dat het CRL overeenkomstig Verordening (EG) nr. 378/2005 bijstaat, moet dit laboratorium, met het oog op de onafhankelijkheid en de betrokkenheid bij de beoordeling van de door de aanvrager ingediende informatie, zodra het CRL de aanvraag ontvangt een belangenverklaring aan het CRL toezenden waarin het zijn werkzaamheden in verband met de aanvraag beschrijft, en mag het niet aan de beoordeling van de aanvraag meewerken. |
|
2.6.2.4. |
In zijn evaluatieverslag voor de Autoriteit kan het CRL passende kenmerken selecteren uit de in punt 2.6.2.2 genoemde kenmerken. |
|
2.6.2.5. |
De prestatiecriteria voor methoden voor bepaalde groepen stoffen (bv. enzymen) kunnen worden vastgesteld in de gedetailleerde richtsnoeren die het CRL opstelt overeenkomstig artikel 12 van Verordening (EG) nr. 378/2005. |
2.6.3. Analysemethoden voor de identificatie en karakterisering van het toevoegingsmiddel
De aanvrager moet een beschrijving geven van de methoden die zijn gebruikt voor de bepaling van de in de punten 2.1.3, 2.1.4, 2.1.5, 2.2.2, 2.4.1, 2.4.2, 2.4.3 en 2.4.4 genoemde kenmerken.
Overeenkomstig bijlage II bij Verordening (EG) nr. 1831/2003, gewijzigd bij Verordening (EG) nr. 378/2005, kunnen de overeenkomstig dit punt voorgelegde methoden, indien dat nuttig wordt geacht, in het kader van de beoordeling van de aanvraag ook door de Autoriteit of de Commissie worden geëvalueerd.
Het is aan te bevelen dat de overeenkomstig dit punt beschreven methoden internationaal erkend zijn. Methoden die niet internationaal erkend zijn, moeten volledig worden beschreven. In dat geval moeten er onderzoeken worden uitgevoerd door erkende en onafhankelijke laboratoria. Deze moeten volgens passende kwaliteitsnormen (bv. GLP overeenkomstig Richtlijn 2004/10/EG of ISO-normen) worden gedocumenteerd.
De methoden voor de identificatie en karakterisering van het toevoegingsmiddel moeten aan dezelfde eisen voldoen als de analysemethoden die voor officiële controles worden gebruikt, namelijk de eisen van artikel 11 van Verordening (EG) nr. 882/2004, met name als het om wettelijke voorschriften gaat (bv. onzuiverheden en ongewenste stoffen).
3. SECTIE III: ONDERZOEKEN NAAR DE VEILIGHEID VAN HET TOEVOEGINGSMIDDEL
Met de in deze sectie en in de specifieke bijlagen genoemde onderzoeken wordt beoogd te beoordelen:
|
— |
of het toevoegingsmiddel bij de doelsoorten veilig kan worden gebruikt; |
|
— |
of er een risico is op selectie en/of overdracht van antibioticaresistentie en verhoogde persistentie en verspreiding van maag-darmpathogenen; |
|
— |
aan welke risico's de consument kan worden blootgesteld door het consumeren van voedsel afkomstig van dieren die voeder hebben gegeten dat het toevoegingsmiddel bevat of daarmee behandeld is, of door het consumeren van voedsel dat residuen van het toevoegingsmiddel of metabolieten daarvan bevat; |
|
— |
welke risico's er bij inademing of bij contact met slijmvliezen, ogen of huid zijn voor personen die waarschijnlijk in aanraking zullen komen met het toevoegingsmiddel als zodanig of met voormengsels of diervoeder waarin het toevoegingsmiddel is verwerkt, en |
|
— |
wat de risico's zijn op nadelige gevolgen voor het milieu door het toevoegingsmiddel zelf of daarvan afkomstige producten, rechtstreeks en/of door uitscheiding door de dieren. |
3.1. Onderzoek naar de veiligheid van het gebruik van het toevoegingsmiddel voor de doeldieren
Met de in deze sectie genoemde onderzoeken wordt beoogd te beoordelen:
|
— |
of het toevoegingsmiddel als zodanig veilig kan worden gebruikt bij de doelsoorten, en |
|
— |
of er een risico is op selectie en/of overdracht van antibioticaresistentie en verhoogde persistentie en verspreiding van maag-darmpathogenen. |
3.1.1. Tolerantie bij de doeldieren
Doel van het tolerantieonderzoek is een beperkte beoordeling van de toxiciteit op korte termijn van het toevoegingsmiddel bij de doeldieren te maken. Voorts wordt hiermee een veiligheidsmarge vastgesteld voor als het toevoegingsmiddel in hogere doses dan de aanbevolen dosis wordt geconsumeerd. Voor elke diersoort of -categorie waarvoor een aanvraag wordt ingediend, moet een dergelijk tolerantieonderzoek worden verricht om aan te tonen dat het toevoegingsmiddel veilig is. In sommige gevallen is het aanvaardbaar dat een aantal elementen van het tolerantieonderzoek in het kader van een van de onderzoeken naar de werkzaamheid worden onderzocht, mits deze onderzoeken aan onderstaande voorwaarden voldoen. Alle in deze sectie genoemde onderzoeken moeten gebaseerd zijn op het in sectie II beschreven toevoegingsmiddel.
3.1.1.1. De opzet van een tolerantieonderzoek moet minimaal drie groepen omvatten:
|
— |
een groep die geen toevoegingsmiddel krijgt toegediend; |
|
— |
een groep die de hoogste aanbevolen dosis krijgt toegediend, en |
|
— |
een experimentele groep die een veelvoud van de hoogste aanbevolen dosis krijgt toegediend. |
De experimentele groep krijgt gewoonlijk tienmaal de hoogste aanbevolen dosis van het toevoegingsmiddel toegediend. De proefdieren moeten regelmatig worden gecontroleerd op zichtbare tekenen van klinische effecten, prestatiekenmerken, productkwaliteit (indien van toepassing), hematologie, routinebloedchemie en andere parameters die met de biologische eigenschappen van het toevoegingsmiddel verband kunnen houden. Kritische eindpunten die bekend zijn van de toxicologische onderzoeken bij laboratoriumdieren, moeten in overweging worden genomen. Bij het onderzoek naar de werkzaamheid gevonden nadelige effecten moeten hier ook worden vermeld. Onverklaarde sterfgevallen bij het tolerantieonderzoek moeten met een necropsie en zo nodig histologisch worden onderzocht.
Indien wordt aangetoond dat het honderdvoudige van de hoogste aanbevolen dosis getolereerd wordt, zijn geen hematologie of routinebloedchemie nodig. Indien het product slechts bij minder dan tienmaal de hoogste aanbevolen dosis wordt getolereerd, moet het onderzoek zo worden opgezet dat een veiligheidsmarge voor het toevoegingsmiddel kan worden berekend en moeten extra eindpunten worden versterkt (met een necropsie, histologie of zo nodig andere criteria).
Naargelang van hun van toxiciteit, metabolisme of gebruik hoeft voor sommige toevoegingsmiddelen geen tolerantieonderzoek te worden verricht.
Bij de proefopzet moet aandacht worden besteed aan een toereikend statistisch onderscheidingsvermogen.
3.1.1.2. Duur van de tolerantieonderzoeken
Tabel 1
Duur van de tolerantieonderzoeken: varkens
|
Doeldieren |
Duur van de onderzoeken |
Kenmerk van de doeldieren |
|
Speenvarkens |
14 dagen |
Bij voorkeur van 14 dagen tot het spenen |
|
Gespeende biggen |
42 dagen |
Gedurende 42 dagen na het spenen |
|
Mestvarkens |
42 dagen |
Lichaamsgewicht ≤ 35 kg bij het begin van het onderzoek |
|
Fokzeugen |
1 cyclus |
Vanaf de inseminatie totdat de biggen gespeend zijn |
Als de aanvraag wordt ingediend voor speenvarkens en gespeende biggen, wordt een gecombineerd onderzoek (14 dagen bij speenvarkens en 28 dagen bij gespeende biggen) voldoende geacht. Als de tolerantie bij gespeende biggen is aangetoond, is geen afzonderlijk onderzoek bij mestvarkens vereist.
Tabel 2
Duur van de tolerantieonderzoeken: pluimvee
|
Doeldieren |
Duur van de onderzoeken |
Kenmerk van de doeldieren |
|
Mestkippen/opfokleghennen |
35 dagen |
Vanaf het uitkomen |
|
Legkippen |
56 dagen |
Bij voorkeur tijdens het eerste derde deel van de legtijd |
|
Mestkalkoenen |
42 dagen |
Vanaf het uitkomen |
De gegevens over de tolerantie bij mestkippen en mestkalkoenen kunnen worden gebruikt om de tolerantie bij leg- en fokkippen, respectievelijk leg- en fokkalkoenen aan te tonen.
Tabel 3
Duur van de tolerantieonderzoeken: runderen
|
Doeldieren |
Duur van de onderzoeken |
Kenmerk van de doeldieren |
|
Mestkalveren |
28 dagen |
Aanvankelijk lichaamsgewicht ≤ 70 kg |
|
Opfokkalveren, mestrunderen of fokrunderen |
42 dagen |
|
|
Melkkoeien |
56 dagen |
|
Indien een aanvraag wordt ingediend voor opfokkalveren en mestrunderen, wordt een gecombineerd onderzoek (28 dagen voor elke periode) voldoende geacht.
Tabel 4
Duur van de tolerantieonderzoeken: schapen
|
Doeldieren |
Duur van de onderzoeken |
Kenmerk van de doeldieren |
|
Opfok- en mestlammeren |
28 dagen |
|
Tabel 5
Duur van de tolerantieonderzoeken: zalmachtigen en andere vissen
|
Doeldieren |
Duur van de onderzoeken |
Kenmerk van de doeldieren |
|
Zalm en forel |
90 dagen |
|
In plaats van een 90 dagenonderzoek mag ook een onderzoek worden uitgevoerd waarbij het oorspronkelijke lichaamsgewicht van de dieren vanaf het begin van de test minstens verdubbelt.
Indien het toevoegingsmiddel alleen voor broedvissen bestemd is, moet het tolerantieonderzoek zo dicht mogelijk bij de paaitijd worden uitgevoerd. Het tolerantieonderzoek moet minimaal 90 dagen duren en er moet aandacht worden besteed aan de kwaliteit van de eitjes en het aantal eitjes dat in leven blijft.
Tabel 6
Duur van de tolerantieonderzoeken: gezelschapsdieren en andere niet-voedselproducerende dieren
|
Doeldieren |
Duur van de onderzoeken |
Kenmerk van de doeldieren |
|
Honden en katten |
28 dagen |
|
Tabel 7
Duur van de tolerantieonderzoeken: konijnen
|
Doeldieren |
Duur van de onderzoeken |
Kenmerk van de doeldieren |
|
Mestkonijnen |
28 dagen |
|
|
Fokkonijnen |
1 cyclus |
Vanaf de inseminatie totdat de jongen gespeend zijn |
Als de aanvraag wordt ingediend voor konijntjes die gezoogd worden en gespeende konijnen, wordt een periode van 49 dagen (vanaf één week na de geboorte) voldoende geacht. Het moerkonijn moet tot het spenen worden onderzocht.
Indien een toevoegingsmiddel gedurende een specifieke, kortere periode wordt toegediend dan voor de diercategorie vermeld is, moet het volgens de voorgestelde gebruiksvoorwaarden worden toegediend. De observatieperiode mag echter niet minder dan 28 dagen bedragen en betreft de betrokken eindpunten (bv. bij fokzeugen: het aantal levend geboren biggen wat de zwangerschap betreft, en het gewicht van de gespeende biggen wat de lactatieperiode betreft).
3.1.1.3. Omstandigheden waaronder de experimenten hebben plaatsgevonden
Er moet over elk onderzoek afzonderlijk verslag worden uitgebracht, met gedetailleerde gegevens over alle experimentele groepen. Het testprotocol moet zorgvuldig worden opgesteld en algemene beschrijvende gegevens bevatten. In het bijzonder moet het volgende worden opgetekend:
|
1) |
beslag of koppel: plaats en grootte; voederings- en opfokomstandigheden, voedermethode; voor waterdieren: grootte van en aantal bakken of hokken op het bedrijf, lichtomstandigheden en waterkwaliteit, inclusief watertemperatuur en zoutgehalte; |
|
2) |
dieren: diersoort (voor waterdieren bestemd voor menselijke consumptie moet de gewone naam worden vermeld, met tussen haakjes de binomiale Latijnse naam), ras of stam, leeftijd (grootte voor waterdieren), geslacht, identificatieprocedure, fysiologische status en algemene gezondheid; |
|
3) |
data en precieze duur van de tests: datum en aard van de verrichte onderzoeken; |
|
4) |
voeding: beschrijving van de vervaardiging en kwantitatieve samenstelling van de voeding: gebruikte ingrediënten, relevante voedingsstoffen (geanalyseerde waarden) en voedingswaarde. Gegevens over de voederopname; |
|
5) |
de concentratie van de werkzame stof(fen) of het agens (de agentia) (en van eventueel ter vergelijking gebruikte stoffen) in het diervoeder moet worden bepaald door een controleanalyse volgens een adequate, erkende methode: referentienummer(s) van de charges; |
|
6) |
aantal proef- en controlegroepen en het aantal dieren in elke groep: het aantal in de proeven opgenomen dieren moet voldoende zijn voor statistische analyses. Vermeld moet worden welke statistische evaluatiemethoden zijn gebruikt. Het verslag moet betrekking hebben op alle bij de proeven betrokken dieren en/of experimentele eenheden. Gevallen die niet kunnen worden beoordeeld door gebrek aan of verlies van gegevens moeten worden vermeld, waarbij de verdeling binnen de groepen dieren moet worden geclassificeerd; |
|
7) |
ongewenste bijwerkingen van de behandeling bij afzonderlijke dieren of groepen moeten worden gerapporteerd onder vermelding van het moment van optreden en de prevalentie (met bijzonderheden over het in het onderzoek gebruikte observatieprogramma), en |
|
8) |
therapeutische of preventieve behandelingen mogen geen wisselwerking hebben met de voorgestelde wijze van werking van het toevoegingsmiddel en moeten afzonderlijk worden opgetekend. |
3.1.2. Microbieel onderzoek
Er moet onderzoek worden verricht om te bepalen in welke mate het toevoegingsmiddel in staat is kruisresistentie tegen in de humane of diergeneeskunde gebruikte antibiotica te induceren, onder praktijkomstandigheden in de doelsoort op resistente bacteriestammen te selecteren, effect te hebben op opportunistische pathogenen die zich in het spijsverteringskanaal bevinden, en verspreiding of uitscheiding van relevante zoönoseverwekkers te veroorzaken.
Indien de werkzame stof bij de in het voeder gebruikte concentraties een antimicrobiële werking heeft, moet de minimaal remmende concentratie (MIC) voor de desbetreffende soorten bacteriën volgens gestandaardiseerde procedures worden bepaald. Indien een relevante antimicrobiële activiteit wordt aangetoond, moet worden bepaald in welke mate het toevoegingsmiddel in staat is in vitro en in de doelsoort op resistente bacteriestammen te selecteren en om kruisresistentie tegen de desbetreffende antibiotica te induceren (11).
Er moeten tests bij de aanbevolen te gebruiken hoeveelheid worden uitgevoerd voor alle microbiële toevoegingsmiddelen en voor andere toevoegingsmiddelen waarbij een effect op de darmflora kan worden verwacht. Deze onderzoeken moeten aantonen dat het gebruik van het toevoegingsmiddel geen omstandigheden creëert die leiden tot een wildgroei en verspreiding van potentieel ziekteverwekkende micro-organismen.
De keuze van de te controleren micro-organismen hangt af van de doelsoort, maar moet relevante zoönotische soorten omvatten, ongeacht of zij symptomen veroorzaken bij de doeldieren.
3.2. Onderzoek naar de veiligheid van het gebruik van het toevoegingsmiddel voor de consumenten
Doel is de veiligheid van het gebruik van het toevoegingsmiddel voor de consument te beoordelen en te bepalen welke residuen van het toevoegingsmiddel of metabolieten daarvan eventueel kunnen achterblijven in voedsel dat afkomstig is van dieren die water of voeder hebben gekregen dat het toevoegingsmiddel bevatte of daarmee behandeld was.
3.2.1. Metabolisme- en residuonderzoek
De bepaling van het metabolisme van het toevoegingsmiddel in de doelsoorten is een beslissende fase van de identificatie en kwantificatie van de residuen in eetbare weefsels of producten afkomstig van dieren die water of voeder met het toevoegingsmiddel hebben gekregen. Er moeten onderzoeken worden verstrekt over de absorptie, de distributie, het metabolisme en de uitscheiding van de stoffen (en metabolieten daarvan).
De onderzoeken moeten worden uitgevoerd volgens internationaal erkende standaardtestmethoden en overeenkomstig de geldende communautaire wetgeving of de Guidelines for methodological details van de OESO en de GLP-beginselen. Bij het onderzoek moet de communautaire wetgeving inzake dierenwelzijn worden nageleefd. Onderzoeken mogen niet onnodig worden herhaald.
Bij het metabolisme- en residuonderzoek moeten de doeldieren de werkzame stof in het voeder toegediend krijgen (niet via een maagsonde, tenzij dat naar behoren wordt gemotiveerd).
Van metabolieten die meer dan 10 % van de residuen in eetbare weefsels en producten en meer dan 20 % van de totale residuen in de uitscheidingsproducten uitmaken, moet een structurele identificatie worden verricht. Als het metabolisme van de werkzame stof van toxicologisch belang is, moeten ook metabolieten die minder dan bovengenoemde percentages uitmaken, worden geïdentificeerd.
Aan de hand van kinetisch onderzoek van de residuen wordt de blootstelling van de consument berekend en worden zo nodig een wachttijd en MRL's vastgesteld. Er moet een voorstel voor een indicatorresidu worden gedaan.
Naargelang van hun van aard of gebruik hoeft voor sommige toevoegingsmiddelen niet altijd een metabolisme- en residuonderzoek te worden verricht.
3.2.1.1. Metabolismeonderzoek
Doel van het metabolismeonderzoek is de absorptie, de verdeling, de biologische omzetting en de uitscheiding van het toevoegingsmiddel bij de doelsoorten te beoordelen.
De volgende onderzoeken zijn vereist:
|
1) |
metabolismebalans na eenmalige toediening van de werkzame stof bij de voorgestelde gebruiksdosis (totale hoeveelheid gelijk aan de dagelijkse inname) en eventueel (indien gerechtvaardigd) bij een veelvoud van die dosis, teneinde snelheid en mate van absorptie, verdeling (plasma/bloed) en uitscheiding (urine, gal, feces, melk of eieren, uitgeademde lucht, uitscheiding door de kieuwen) te bepalen, zo nodig bij mannelijke en vrouwelijke dieren, en |
|
2) |
de metabole profilering, de identificatie van de metaboliet(en) in uitscheidingsproducten en weefsels en de verdeling in weefsels en producten worden bepaald na herhaaldelijke toediening van de gemerkte verbinding tot een aan de hand van de plasmaniveaus vastgestelde (metabolische) evenwichtstoestand is bereikt. De toegediende dosis moet overeenkomen met de hoogste voorgestelde gebruiksdosis en moet in het voeder worden gemengd. |
3.2.1.2. Residuonderzoek
De hoeveelheid en de aard van de niet-extraheerbare residuen in eetbare weefsels en producten moeten worden onderzocht.
Residuonderzoek is vereist voor alle stoffen waarvoor metabolismeonderzoek vereist is.
Als de stof een natuurlijk bestanddeel van lichaamsvloeistoffen of -weefsels is, of van nature in aanzienlijke hoeveelheden aanwezig is in voedsel of diervoeder, mag het residuonderzoek beperkt blijven tot een vergelijking van het gehalte in weefsels/producten in een onbehandelde groep en in een groep die de hoogste aangegeven dosis heeft gekregen.
Bij gangbare soorten moeten tegelijk de totale residuen van toxicologisch belang worden beoordeeld en het indicatorresidu van de werkzame stof in eetbare weefsels (lever, nieren, spieren, huid, huid met vet) en producten (melk, eieren en honing) worden geïdentificeerd. Het indicatorresidu is de voor het analyse geselecteerde residu waarvan bekend is dat de concentratie verband houdt met het totale residu van toxicologisch belang in de weefsels. Het onderzoek moet ook aantonen hoelang de residuen in de weefsels of producten achterblijven, zodat een passende wachttijd kan worden vastgesteld.
Voor het bepalen van de wachttijd wordt voorgesteld minimaal de volgende aantallen dieren en/of producten op elk gekozen tijdstip te bemonsteren:
|
— |
eetbare weefsels:
|
|
— |
producten:
|
Zo nodig moet een passende verdeling tussen mannelijke en vrouwelijke dieren worden gemaakt.
De residuen moeten worden gemeten bij nul wachttijd (evenwichtstoestand) en op ten minste drie andere bemonsteringstijdstippen.
Er moet een indicatorresidu worden voorgesteld.
Het onderzoek naar absorptie, verdeling en uitscheiding, waaronder de identificatie van de voornaamste metabolieten, moet worden uitgevoerd bij laboratoriumdieren van de soorten waarbij de laagste NOAEL is verkregen of, standaard, bij ratten (mannetjes en vrouwtjes). Aanvullend onderzoek naar bepaalde metabolieten kan nodig zijn als die door de doelsoort worden geproduceerd maar niet in significante mate in het laboratoriumdier worden gevormd.
3.2.1.3. Onderzoek naar metabolisme en dispositie
Er moet een metabolismeonderzoek worden uitgevoerd dat de metabolismebalans, het metabole profiel en de identificatie van de voornaamste metabolieten in urine en feces omvat. Als een andere soort laboratoriumdieren een duidelijk verschil in gevoeligheid vertoont in vergelijking met ratten, moet aanvullende informatie worden verstrekt.
3.2.1.4. Biologische beschikbaarheid van residuen
Bij de evaluatie van het risico voor de consument als gevolg van gebonden residuen in dierlijke producten kan een extra veiligheidsfactor in aanmerking genomen worden op basis van de bepaling van de biologische beschikbaarheid van die residuen met behulp van geschikte laboratoriumdieren en erkende methoden.
3.2.2. Toxicologisch onderzoek
De veiligheid van het toevoegingsmiddel wordt beoordeeld op basis van toxicologisch in-vivo- en in-vitro-onderzoek bij laboratoriumdieren. Daarbij wordt gewoonlijk het volgende gemeten:
|
1) |
acute toxiciteit; |
|
2) |
genotoxiciteit (mutageniteit, clastogeniteit); |
|
3) |
subchronische orale toxiciteit; |
|
4) |
chronische orale toxiciteit/carcinogeniteit; |
|
5) |
reproductietoxiciteit, inclusief teratogeniteit, en |
|
6) |
andere onderzoeken. |
Indien er redenen tot ongerustheid zijn, moet verder onderzoek worden verricht dat de nodige aanvullende informatie oplevert voor de beoordeling van de veiligheid van de werkzame stof en residuen daarvan.
Op basis van de resultaten van deze onderzoeken moet een toxicologische NOAEL worden vastgesteld.
Aanvullend onderzoek naar bepaalde metabolieten kan nodig zijn als die door de doelsoort worden geproduceerd maar niet in significante mate in het laboratoriumdier worden gevormd. Als er gegevens over metabolisch onderzoek bij mensen beschikbaar zijn, moet daarmee rekening worden gehouden om te beslissen welk soort aanvullend onderzoek eventueel nodig is.
Het toxicologische onderzoek moet met de werkzame stof worden uitgevoerd. Als de werkzame stof aanwezig is in een gistingsproduct, moet het gistingsproduct worden getest. Het geteste gistingsproduct moet identiek zijn met het gistingsproduct dat in het commerciële product zal worden gebruikt.
De onderzoeken moeten worden uitgevoerd volgens internationaal erkende standaardtestmethoden en overeenkomstig de geldende communautaire wetgeving of de Guidelines for methodological details van de OESO en de GLP-beginselen. Bij onderzoek met dierproeven moet de communautaire wetgeving inzake dierenwelzijn worden nageleefd. Dergelijk onderzoek mag niet onnodig worden herhaald.
3.2.2.1. Acute toxiciteit
Onderzoek naar de acute toxiciteit is vereist om de toxiciteit van het bestanddeel te classificeren en in beperkte mate te karakteriseren.
Onderzoek naar de acute toxiciteit moet op ten minste twee soorten zoogdieren worden uitgevoerd. Zo nodig mag één soort laboratoriumdier worden vervangen door een doelsoort.
Er hoeft geen exacte LD50 te worden bepaald; het volstaat om de minimale letale dosis bij benadering te bepalen. De maximumdosis mag niet hoger zijn dan 2 000 mg/kg lichaamsgewicht.
Om het aantal en het lijden van de proefdieren te verminderen, worden er voortdurend nieuwe protocollen voor onderzoek naar de toxiciteit van een acute dosis ontwikkeld. Onderzoek dat volgens deze nieuwe procedures wordt uitgevoerd, wordt aanvaard mits het naar behoren gevalideerd is.
De volgende OESO-richtsnoeren moeten worden nageleefd: 402 (acute dermale toxiciteit), 420 (vastedosismethode), 423 (acutetoxiciteitsklasse) en 425 (up-and-downprocedure).
3.2.2.2. Onderzoek naar de genotoxiciteit, met inbegrip van de mutageniteit
Om werkzame stoffen en eventuele metabolieten en afbraakproducten daarvan met mutagene en genotoxische eigenschappen te identificeren, moet een goed gekozen combinatie van verschillende genotoxiciteitstests worden uitgevoerd. Zo nodig moeten de tests zonder en met metabole activering in een zoogdiersysteem worden uitgevoerd en moet rekening worden gehouden met de compatibiliteit van het testmateriaal met het testsysteem.
De basisreeks bestaat uit de volgende tests:
|
1) |
inductie van genmutaties bij bacteriën en/of in zoogdiercellen (bij voorkeur muizenlymfoom tk assay); |
|
2) |
inductie van chromosoomafwijkingen in zoogdiercellen, en |
|
3) |
in-vivotest bij zoogdiersoorten. |
Aanvullende tests kunnen noodzakelijk zijn, afhankelijk van de resultaten van bovengenoemde tests en rekening houdend met het algehele toxiciteitsprofiel en het beoogde gebruik van de stof.
De protocollen moeten in overeenstemming zijn met OESO-richtsnoer 471 (Salmonella typhimurium Reverse Mutation Test), 472 (Escherichia coli Reverse Mutation Test), 473 (in vitro Mammalian Chromosomal Aberration Test), 474 (Mammalian Erythrocyte Micronucleus Test), 475 (Mammalian Bone Marrow Chromosomal Aberration Test), 476 (in vitro Mammalian Cell Gene Mutation Test) of 482 (Unscheduled DNA Synthesis in Mammalian Cells in vitro) en met andere relevante OESO-richtsnoeren voor in-vitro- en in-vivotests.
3.2.2.3. Onderzoek naar subchronische toxiciteit bij herhaalde toediening
Om het subchronische toxische potentieel van de werkzame stof te onderzoeken, moet ten minste één onderzoek van ten minste 90 dagen bij knaagdieren worden uitgevoerd. Als dat nodig wordt geacht, moet een tweede onderzoek bij een andere diersoort dan knaagdieren worden verricht. Naast de controlegroep moet de teststof oraal bij ten minste drie concentraties worden toegediend teneinde een dosis-effectrelatie te bepalen. Normaliter zullen er bij de maximale dosis schadelijke effecten aan het licht komen. Bij de laagste dosering mag normaliter geen toxiciteit blijken.
Het protocol voor dit onderzoek moet in overeenstemming zijn met de OESO-richtsnoer 408 (knaagdieren) of 409 (niet-knaagdieren).
3.2.2.4. Onderzoek naar de chronische orale toxiciteit (inclusief carcinogeniteit)
Om het chronische toxische potentieel en het carcinogene potentieel te onderzoeken, moet gedurende ten minste twaalf maanden een toxiciteitsonderzoek bij ten minste één soort worden uitgevoerd. De meest geschikte soort wordt gekozen op basis van alle beschikbare wetenschappelijke gegevens, waaronder de resultaten van het 90 dagenonderzoek. Standaard wordt voor ratten gekozen. Indien om een tweede onderzoek wordt gevraagd, wordt een knaagdier of een andere zoogdiersoort gebruikt. Naast de controlegroep moet de teststof oraal bij ten minste drie concentraties worden toegediend teneinde een dosis-effectrelatie te bepalen.
Indien het onderzoek naar de chronische toxiciteit wordt gecombineerd met een onderzoek naar de carcinogeniteit, wordt de duur verlengd tot 18 maanden voor muizen en hamsters en tot 24 maanden voor ratten.
Carcinogeniteitsonderzoek is wellicht niet nodig als de werkzame stof en de metabolieten daarvan:
|
1) |
bij de genotoxiciteitstests consequent negatieve resultaten opleveren; |
|
2) |
niet structureel verwant zijn aan bekende carcinogenen, en |
|
3) |
bij chronische toxiciteitstests geen effecten te zien geven die duiden op mogelijke pre(neoplasie). |
De protocollen moeten in overeenstemming zijn met OESO-richtsnoer 452 (onderzoek naar chronische toxiciteit) of 453 (gecombineerd onderzoek naar chronische toxiciteit en carcinogeniteit).
3.2.2.5. Onderzoek naar de reproductietoxiciteit (inclusief prenatale-ontwikkelingstoxiciteit)
Om eventuele aantasting van de mannelijke of vrouwelijke voortplantingsfunctie of schadelijke gevolgen voor de nakomelingen ten gevolge van de toediening van de werkzame stof vast te stellen, moeten de volgende onderzoeken van de voortplantingsfunctie worden uitgevoerd:
|
1) |
onderzoek naar de reproductietoxiciteit over twee generaties, en |
|
2) |
onderzoek naar de prenatale-ontwikkelingstoxiciteit (teratogeniteitsonderzoek). |
Voor nieuwe proeven mogen gevalideerde alternatieve methoden waarvoor minder proefdieren nodig zijn, worden gebruikt.
3.2.2.5.1. Onderzoek naar de reproductietoxiciteit over twee generaties
Er moet bij ten minste één soort, gewoonlijk knaagdieren, onderzoek naar de voortplantingsfunctie worden uitgevoerd dat zich over ten minste twee generaties nakomelingen (F1, F2) uitstrekt. Dit kan gecombineerd worden met onderzoek naar de teratogeniteit. De onderzochte stof moet op een passende tijd vóór de paring oraal worden toegediend aan het mannelijke en het vrouwelijke dier. De toediening moet worden voortgezet tot de F2-generatie gespeend is.
Alle relevante gegevens met betrekking tot vruchtbaarheid, dracht, werpen, moederlijk gedrag, zogen, groei en ontwikkeling van de F1-generatie vanaf de bevruchting tot de volgroeidheid en de ontwikkeling van de F2-generatie tot het spenen, moeten zorgvuldig worden geobserveerd en gerapporteerd. De protocollen voor het onderzoek naar de reproductietoxiciteit moeten in overeenstemming zijn met OESO-richtsnoer 416.
3.2.2.5.2. Onderzoek naar de prenatale-ontwikkelingstoxiciteit (teratogeniteitsonderzoek)
Doel is vast te stellen of de blootstelling, van de implantatie tot het einde van de zwangerschap, schadelijke effecten heeft op het drachtige vrouwtje of op de ontwikkeling van het embryo en de foetus. Mogelijke effecten zijn: hogere toxiciteit in het drachtige vrouwtje, dood van het embryo of de foetus, groeiafwijkingen en structurele afwijkingen bij de foetus.
Gewoonlijk worden voor het eerste onderzoek ratten gebruikt. Als het teratogeniteitsonderzoek een negatief of onduidelijk resultaat oplevert, moet een ander onderzoek naar de ontwikkelingstoxiciteit worden uitgevoerd bij een tweede soort, bij voorkeur konijnen. Als het teratogeniteitsonderzoek bij ratten een positief resultaat oplevert, is een onderzoek bij een tweede soort niet nodig, tenzij uit een overzicht van alle belangrijke onderzoeken blijkt dat de aanvaardbare dagelijkse inname (ADI) op de teratogeniteit bij ratten gebaseerd is. In dat geval is een onderzoek bij een tweede soort nodig om na te gaan wat de gevoeligste soort voor dit eindpunt is. De protocollen moeten in overeenstemming zijn met OESO-richtsnoer 414.
3.2.2.6. Ander specifiek toxicologisch en farmacologisch onderzoek
Indien er redenen tot ongerustheid zijn, moet verder onderzoek worden verricht dat nuttige aanvullende informatie oplevert voor de beoordeling van de veiligheid van de werkzame stof en residuen daarvan. Het kan onder meer gaan om onderzoek naar het farmacologische effect, het effect op jonge (prepuberale) dieren, immunotoxiciteit en neurotoxiciteit.
3.2.2.7. Bepaling van „no observed adverse effect levels” (NOAEL's)
NOAEL's zijn gewoonlijk gebaseerd op het toxicologische effect, maar soms is het farmacologische effect geschikter.
De laagste NOAEL moet worden genomen. Bij het bepalen van de laagste NOAEL, uitgedrukt als mg/kg lichaamsgewicht per dag, moet rekening worden gehouden met alle resultaten van de vorige punten, in combinatie met alle relevante gepubliceerde gegevens (met inbegrip van relevante informatie over de effecten van de werkzame stof op de mens) en, waar van toepassing, informatie over nauw verwante chemische structuren.
3.2.3. Beoordeling van de veiligheid voor de consument
De veiligheid voor de consument wordt beoordeeld door de vastgestelde ADI te vergelijken met de berekende theoretische inname van het toevoegingsmiddel of metabolieten daarvan via voedsel. Voor vitaminen en sporenelementen mag, in plaats van een ADI, een UL („tolerable upper intake level”) worden gebruikt.
3.2.3.1. Voorstel voor een aanvaardbare dagelijkse inname (ADI) voor de werkzame stof(fen)
De ADI (uitgedrukt in mg toevoegingsmiddel of aan het toevoegingsmiddel gerelateerd materiaal per persoon per dag) wordt berekend door de laagste NOAEL (mg per kg lichaamsgewicht) te delen door een geschikte veiligheidsfactor en te vermenigvuldigen met een gemiddeld menselijk lichaamsgewicht van 60 kg.
Indien van toepassing moet een ADI worden voorgesteld. Voor de ADI kan eventueel „geen limiet” worden vastgesteld vanwege de geringe toxiciteit in dierproeven. Als de stof genotoxische of carcinogene eigenschappen vertoont die van belang zijn voor de mens, mag geen ADI worden voorgesteld.
Opdat een ADI kan worden vastgesteld, moet het metabolisme van de werkzame stof hetzelfde zijn in de doeldieren en in de laboratoriumdieren (zie punt 3.2.1.4. Biologische beschikbaarheid van residuen), zodat de consumenten aan dezelfde residuen worden blootgesteld als de proefdieren in het toxicologische onderzoek. Zo niet kan alsnog een ADI worden vastgesteld aan de hand van aanvullend onderzoek bij een tweede soort laboratoriumdieren of met de metabolieten die specifiek zijn voor de doelsoort.
Bij het vaststellen van de veiligheidsfactor voor de vaststelling van de ADI voor een bepaald toevoegingsmiddel moet rekening worden gehouden met de aard van de biologische effecten, de kwaliteit van de gegevens die zijn gebruikt om de NOAEL vast te stellen, het belang van de effecten voor de mens, de omkeerbaarheid van de effecten en eventuele kennis over de rechtstreekse effecten van de residuen op de mens.
Bij de berekening van de ADI moet een veiligheidsfactor van ten minste 100 worden gebruikt (indien een volledig toxicologisch pakket beschikbaar is). Indien er gegevens over het effect van de werkzame stof op mensen beschikbaar zijn, kan een lagere veiligheidsfactor aanvaardbaar zijn. Indien er extra bronnen van onzekerheid zijn of indien de NOAEL wordt vastgesteld op grond van een bepaald kritisch eindpunt, bijvoorbeeld teratogeniteit, kunnen hogere veiligheidsfactoren worden gebruikt.
3.2.3.2. Tolerable upper intake level (UL)
Voor bepaalde toevoegingsmiddelen kan het beter zijn de veiligheidsbeoordeling te baseren op de UL. Dit is de maximale chronische dagelijkse inname van een voedingsstof (uit alle bronnen) waarvan het (door nationale of internationale wetenschappelijke instellingen) onwaarschijnlijk wordt geacht dat het een schadelijk effect op consumenten of bepaalde groepen consumenten heeft.
Het dossier moet gegevens bevatten die aantonen dat het gebruik van het toevoegingsmiddel niet zal leiden tot een situatie waarin de UL kan worden overschreden, rekening houdend met alle mogelijke bronnen van de voedingsstof.
Als het resulterende gehalte aan residuen van het toevoegingsmiddel of metabolieten daarvan in producten van dierlijke oorsprong hoger is dan wat voor deze producten normaal wordt geacht of kan worden verwacht, moet dat duidelijk worden aangegeven.
3.2.3.3. Blootstelling van de consument
De totale inname van het toevoegingsmiddel en/of metabolieten daarvan uit alle bronnen door de consument moet minder bedragen dan de ADI of de UL.
Bij de berekening van de theoretische inname uit voedsel van dierlijke oorsprong wordt rekening gehouden met de concentratie (totale residuen als rekenkundig gemiddelde en hoogste afzonderlijke waarde) die na het stoppen van het gebruik van het toevoegingsmiddel wordt gemeten in weefsels en producten. Zo nodig moeten daarnaast ook de waarden voor de dagelijkse menselijke voedselconsumptie na de verschillende wachttijden worden berekend volgens een worstcasescenario.
In het geval van toevoegingsmiddelen die voor verscheidene soorten bestemd zijn, moet de blootstelling door weefsels afzonderlijk worden berekend voor zoogdieren, vogels en vissen en moet de hoogste waarde worden genomen. Indien van toepassing moet de blootstelling door melk en eieren bij deze waarde worden opgeteld. Als een toevoegingsmiddel bijvoorbeeld wordt gebruikt bij zogende dieren en legkippen, worden de hoogste waarden voor de respectievelijke eetbare weefsels opgeteld bij die voor de consumptie van melk en eieren. Als een toevoegingsmiddel wordt gebruikt bij vissen, legpluimvee en zogende dieren, worden de hoogste waarden voor de respectievelijke eetbare weefsels opgeteld bij die voor de consumptie van eieren en melk. Andere combinaties worden op dezelfde wijze behandeld.
In sommige gevallen (bv. nutritionele en sensoriële toevoegingsmiddelen of toevoegingsmiddelen bestemd voor minder gangbare soorten) kan het passend zijn de beoordeling van de blootstelling van de consument verder te verfijnen aan de hand van meer realistische consumptiecijfers. Daarbij moet echter nog steeds de meest voorzichtige schatting worden genomen. Zo mogelijk moeten daarvoor communautaire gegevens worden gebruikt.
Tabel 1
Theoretische dagelijkse consumptie door de mens (g weefsels of producten)
|
|
Zoogdieren |
Vogels |
Vis |
Overige |
|
Spieren |
300 |
300 |
300 (12) |
|
|
Lever |
100 |
100 |
— |
|
|
Nieren |
50 |
10 |
— |
|
|
Vet |
50 (13) |
90 (14) |
— |
|
|
+ Melk |
1 500 |
— |
— |
|
|
+ Eieren |
— |
100 |
— |
|
|
+ Honing |
|
|
|
20 |
3.2.3.4. Voorstel voor maximumresidugehalten (MRL's)
Het maximumresidugehalte is de maximale concentratie aan residuen (uitgedrukt in μg indicatorresidu per kg eetbaar nat weefsel of product) in voedsel dat de EG als wettelijk toegestaan aanvaardt of als aanvaardbaar erkent. Het is gebaseerd op het type en de hoeveelheid residu die overeenkomstig de ADI wordt geacht geen toxicologisch gevaar in te houden voor de gezondheid van de mens. Zonder ADI kan er geen MRL worden vastgesteld.
Bij de vaststelling van MRL's voor toevoegingsmiddelen moet ook rekening worden gehouden met residuen die uit andere bronnen (bv. voedsel van plantaardige oorsprong) afkomstig zijn. Voorts kan de MRL worden verlaagd om rekening te houden met de gebruiksvoorwaarden van toevoegingsmiddelen en de beschikbaarheid van praktische analysemethoden.
Zo nodig moeten voor verschillende weefsels of producten van de doelsoort afzonderlijke MRL's (uitgedrukt in μg indicatorresidu per kg eetbaar nat weefsel of product) worden vastgesteld. De afzonderlijke MRL's voor de verschillende weefsels of producten moeten afgestemd zijn op de depletiekinetiek en de variabiliteit van de residugehalten in die weefsels of producten bij de diersoorten waarvoor het toevoegingsmiddel bestemd is. De variabiliteit wordt gewoonlijk aangegeven met een 95 %-betrouwbaarheidsinterval van het gemiddelde. Als het betrouwbaarheidsinterval door een laag aantal monsters niet kan worden berekend, wordt de variabiliteit uitgedrukt met de hoogste afzonderlijke waarde.
Onderzoek naar de MRL's voor coccidiostatica en histomonostatica moet worden uitgevoerd volgens de geldende voorschriften voor geneesmiddelen voor diergeneeskundig gebruik (Voorschriften inzake geneesmiddelen in de Europese Gemeenschap, vol. 8, Notice to Applicants and Guidelines. Veterinary medicinal products. Establishment of maximum residue limits (MRLs) for residues of veterinary medicinal products in foodstuffs of animal origin, oktober 2005).
Onderzoek naar de MRL's voor andere toevoegingsmiddelen dan coccidiostatica en histomonostatica moet in voorkomend geval worden uitgevoerd overeenkomstig deze bijlage.
Om de blootstelling van de consument aan de totale residuen (zoals berekend volgens punt 3.2.3.3) te bepalen, moeten de voorgestelde MRL's voor de verschillende weefsels rekening houden met de verhouding tussen het indicatorresidu en het totale residu (tabel 2).
Tabel 2
Definities die worden gebruikt voor het afleiden van een MRL
|
i-j |
Afzonderlijke weefsels/producten (lever, nier, spier, huid + vet, melk, eieren, honing) op verschillende tijdstippen |
|
MRLi-j |
Maximumresidugehalte in weefsels/producten (mg indicatorstof kg-1) |
|
Qti-j |
Dagelijkse consumptie door de mens van afzonderlijke weefsels/producten (kg), vastgesteld volgens tabel 1 of verfijning daarvan |
|
TRCi-j |
Totale concentratie aan residuen in afzonderlijke weefsels/producten (mg kg-1) |
|
MRCi-j |
Concentratie indicatorresidu in afzonderlijke weefsels/producten (mg kg-1) |
|
RMTRi-j |
Verhouding tussen MRCi-j en TRCi-j voor afzonderlijke weefsels/producten |
|
DITRi-j |
Opname via de voeding voor afzonderlijke weefsels/producten, berekend uit totale residuen (mg) DITRi-j = Qti-j × TRCi-j |
|
DITRMRLi-j |
Opname via de voeding, berekend uit de MRL's (mg) voor afzonderlijke weefsels/producten DITRMRLi-j = Qti-j × MRLi-j × RMTRi-j -1 |
De gemeten waarden voor TRC en MRC worden in voorkomend geval in het model in tabel 3 opgenomen. De overige waarden worden berekend. Indien er geen volledige reeks gegevens beschikbaar is doordat sommige waarden onder de aantoonbaarheidsgrens (LOD) liggen, is een extrapolatie van de RMTR aanvaardbaar.
Een MRL kan slechts worden afgeleid indien de som van de afzonderlijke DITR's lager is dan de ADI. Indien de ADI wordt overschreden, kunnen ook gegevens van een langere wachttijd of lagere doses worden gebruikt. Een eerste voorstel voor een MRL kan worden verkregen door de MRC-waarde als leidraad te gebruiken en rekening te houden met de bepaalbaarheidsgrens (LOQ) van de analysemethode. De som van de DITRMRL's die uit de voorgestelde MRL's zijn verkregen, moet onder de ADI en dicht bij de som van de afzonderlijke DITR's liggen. Indien de ADI wordt overschreden, moet een lagere MRL worden voorgesteld en moet de vergelijking worden herhaald.
Sommige toevoegingsmiddelen kunnen aanleiding geven tot residuen beneden de MRL-waarden in melk, eieren of vlees, die niettemin van invloed kunnen zijn op de voedselkwaliteit bij bepaalde verwerkingsprocedés. Voor dergelijke toevoegingsmiddelen kan het aangewezen zijn niet alleen MRL's vast te stellen, maar ook een „maximumresidugehalte voor de verwerking van (levensmiddel)” (MPCR) te overwegen.
Tabel 3
Model voor het afleiden van een voorstel voor een MRL
|
|
Lever |
Nieren |
Spieren |
Huid + vet |
Melk |
Eieren |
Honing |
Totaal |
|
TRC (15) (mg kg-1) |
|
|
|
|
|
|
|
— |
|
MRC (16) (mg kg-1) |
|
|
|
|
|
|
|
— |
|
RMTR (16) |
|
|
|
|
|
|
|
— |
|
DITR (17) (mg) |
|
|
|
|
|
|
|
|
|
MRL proposed (mg kg-1) |
|
|
|
|
|
|
|
— |
|
DITRMRL(mg) |
|
|
|
|
|
|
|
|
3.2.3.5. Voorstel voor een wachttijd
De wachttijd is de tijd die na het staken van de toediening van het toevoegingsmiddel moet verlopen om ervoor te zorgen dat de residugehalten tot onder de MRL's dalen.
3.3. Onderzoek naar de veiligheid van het gebruik van het toevoegingsmiddel voor gebruikers/werknemers
Werknemers worden voornamelijk blootgesteld door inademing of plaatselijke blootstelling bij de vervaardiging, het hanteren of het gebruik van het toevoegingsmiddel. Zo kunnen werknemers op landbouwbedrijven worden blootgesteld bij het hanteren of mengen van het toevoegingsmiddel. Er moet aanvullende informatie worden verstrekt over de wijze waarop de stoffen gehanteerd moeten worden.
Een beoordeling van het risico voor de werknemers moet worden bijgevoegd. In voorkomend geval is de ervaring in de fabriek waar de stof vervaardigd wordt, vaak een belangrijke informatiebron om te beoordelen hoeveel risico de werknemers lopen op blootstelling aan het toevoegingsmiddel zelf door inademing of aanraking. Er moet bijzondere aandacht worden besteed aan toevoegingsmiddelen/met toevoegingsmiddelen behandeld diervoeder en/of uitscheidingsproducten van dieren die bestaan uit of kunnen resulteren in droog poedervormig materiaal, en potentieel allergene toevoegingsmiddelen.
3.3.1. Toxicologische beoordeling van het risico voor de veiligheid van gebruikers/werknemers
De risico's voor werknemers moeten worden beoordeeld aan de hand van een reeks onderzoeken met het toevoegingsmiddel in de vorm waarvoor de aanvraag is ingediend. Er moet acute-inhalatieonderzoek worden verricht, tenzij het onwaarschijnlijk is dat dat product inadembaar stof of een nevel kan vormen. Er moet onderzoek naar huidirritatie worden verricht; indien dat negatieve resultaten oplevert, moet de irritatie van de slijmvliezen (bv. de ogen) worden beoordeeld. Ook de allergene potentie/potentie voor huidsensibilisatie moet worden beoordeeld. De met het oog op de veiligheid voor de consument verkregen toxiciteitsgegevens (zie punt 3.2.2) moeten worden gebruikt om de systemische toxiciteit te beoordelen. Al deze gegevens moeten zo nodig worden beoordeeld door rechtstreekse metingen en specifiek onderzoek.
3.3.1.1. Effect op de ademhalingswegen
Er moet worden aangetoond dat de stof- of nevelconcentraties van het toevoegingsmiddel in de lucht geen gevaar voor de gezondheid van gebruikers of werknemers opleveren. Dit moet zo nodig worden aangetoond met:
|
— |
inhalatieproeven bij laboratoriumdieren; |
|
— |
gepubliceerde epidemiologische gegevens en/of de eigen gegevens van de aanvrager over zijn fabriek en/of irritatie, en |
|
— |
proeven betreffende de sensibilisatie van de ademhalingswegen. |
Indien deeltjes of druppeltjes met een diameter van minder dan 50 μm meer dan 1 gewichtspercent van het product uitmaken, moet onderzoek naar de acute inhalatietoxiciteit worden verricht.
De protocollen voor het onderzoek naar de acute inhalatietoxiciteit moeten in overeenstemming zijn met OESO-richtsnoer 403. Indien subchronischetoxiciteitsonderzoek nodig wordt geacht, moet dat worden uitgevoerd overeenkomstig OESO-richtsnoer 412 (inhalatietoxiciteit bij herhaalde toediening: onderzoek over 28 of 14 dagen) of 413 (subchronische inhalatietoxiciteit: onderzoek over 90 dagen).
3.3.1.2. Effect op de ogen en de huid
De afwezigheid van irritatie en/of sensibilisatie moet worden aangetoond met rechtreeks bewijsmateriaal afkomstig van bekende situaties betreffende mensen, voor zover dat beschikbaar is. Dit moet worden aangevuld met resultaten van gevalideerde dierproeven inzake huid- en oogirritatie en het sensibiliserende vermogen, waarbij het desbetreffende toevoegingsmiddel is gebruikt. Ook de allergene potentie/potentie voor huidsensibilisatie moet worden beoordeeld. De protocollen voor deze onderzoeken moeten in overeenstemming zijn met de OESO-richtsnoeren 404 (huidirritatie/corrosie), 405 (irritatie/corrosie van de ogen), 406 (huidsensibilisatie) en 429 (huidsensibilisatie — lokale lymfkliertest).
Indien er, uit gepubliceerde gegevens of specifieke in-vitrotests, bijtende eigenschappen bekend zijn, hoeven er geen verdere in-vivotests te worden uitgevoerd.
Indien het toevoegingsmiddel toxisch is bij inademing, moet de dermale toxiciteit in overweging worden genomen. De onderzoeken moeten in overeenstemming zijn met OESO-richtsnoer 402 (acute dermale toxiciteit).
3.3.1.3. Systemische toxiciteit
De met het oog op de veiligheid van de consument en andere vereisten verkregen toxiciteitsgegevens (onder meer toxiciteit bij herhaalde toediening, mutageniteit, carcinogeniteit, reproductietoxiciteit en metabolisme) moeten worden gebruikt om de systemische toxiciteit te beoordelen.
3.3.1.4. Beoordeling van de blootstelling
Er moet informatie worden verstrekt over de mogelijke blootstelling door het gebruik van het toevoegingsmiddel, via alle routes (door inademing, via de huid of door inslikken). De informatie moet zo mogelijk een kwantitatieve beoordeling bevatten, bijvoorbeeld de karakteristieke concentratie in de lucht, besmetting van de huid of inslikken. Indien geen kwantitatieve informatie beschikbaar is, moet voldoende informatie worden verstrekt om de blootstelling naar behoren te kunnen beoordelen.
3.3.2. Maatregelen om de blootstelling te beperken
Aan de hand van de informatie van de beoordeling van de toxicologie en de blootstelling moet een conclusie worden getrokken over de risico's voor de gezondheid van de gebruikers/werknemers (inademing, irritatie, sensibilisatie en systemische toxiciteit). Er kunnen voorzorgsmaatregelen worden voorgesteld om de blootstelling te verminderen of te elimineren. Persoonlijke beschermingsmiddelen mogen echter alleen als laatste toevluchtsmiddel worden gebruikt ter bescherming tegen eventuele restrisico's nadat beheersingsmaatregelen zijn genomen. Het verdient de voorkeur om bijvoorbeeld een andere formulering van het product te overwegen.
3.4. Onderzoek naar de veiligheid van het gebruik van het toevoegingsmiddel voor het milieu
Het is belangrijk het milieueffect van toevoegingsmiddelen te onderzoeken omdat toevoegingsmiddelen vaak langdurig en aan grote groepen dieren worden toegediend en omdat de werkzame stof(fen) in belangrijke mate kan (kunnen) worden uitgescheiden in de vorm van de oorspronkelijke stof of metabolieten daarvan.
Het milieueffect van toevoegingsmiddelen moet stapsgewijze worden beoordeeld. In een eerste fase moeten alle toevoegingsmiddelen worden beoordeeld om na te gaan welke toevoegingsmiddelen niet verder hoeven te worden getest. Voor de overige toevoegingsmiddelen is een tweede fase nodig om aanvullende informatie te verkrijgen. Op grond daarvan kan nader onderzoek nodig worden geacht. Dit onderzoek moet overeenkomstig Richtlijn 67/548/EEG worden uitgevoerd.
3.4.1. Fase I-beoordeling
Doel van de fase I-beoordeling is na te gaan of het waarschijnlijk is dat het toevoegingsmiddel of metabolieten daarvan een aanzienlijk milieueffect hebben, en of een fase II-beoordeling noodzakelijk is (zie beslissingsboom).
Indien een van beide onderstaande criteria van toepassing is, kan een fase II-beoordeling achterwege blijven, tenzij wetenschappelijk is aangetoond dat er reden tot bezorgdheid is:
|
a) |
uit de chemische aard en het biologische effect van het toevoegingsmiddel en de gebruiksvoorwaarden blijkt dat het effect te verwaarlozen zal zijn, bijvoorbeeld als het toevoegingsmiddel:
|
|
b) |
de ongunstigste voorspelde concentratie in het milieu („predicted environmental concentration”, PEC) is te gering om een reden van bezorgdheid te vormen. De PEC moet worden beoordeeld voor elk betrokken compartiment (zie verder), waarbij wordt verondersteld dat 100 % van de opgenomen dosis wordt uitgescheiden in de vorm van de oorspronkelijke stof. |
Indien de aanvrager niet kan aantonen dat een van bovenstaande uitzonderingen op het toevoegingsmiddel van toepassing is, is een fase II-beoordeling vereist.
3.4.1.1. Toevoegingsmiddelen voor landzoogdieren
Als uitscheidingsproducten van vee worden uitgereden, kan het gebruik van toevoegingsmiddelen leiden tot verontreiniging van de bodem, het grondwater en het oppervlaktewater (door drainage of wegvloeien).
De ongunstigste PEC voor de bodem (PECbodem) zou zich voordoen als alle uitgescheiden stoffen werden uitgereden. Indien de PECbodem (standaardwaarde: 5 cm diepte) minder dan 10 μg/kg bedraagt, is geen verdere beoordeling vereist.
Als de PEC voor de grondwaterverontreiniging (PECgw) minder dan 0,1 μg/l bedraagt, is geen fase II-beoordeling van het milieueffect van het toevoegingsmiddel op het grondwater vereist.
3.4.1.2. Toevoegingsmiddelen voor waterdieren
Toevoegingsmiddelen in aquacultuur kunnen leiden tot verontreiniging van sediment en water. Bij vissen die in kooien worden gehouden, wordt verondersteld dat het sediment het belangrijkste compartiment is voor de beoordeling van het milieurisico. Bij vis die aan land wordt gekweekt, wordt de uitstroom naar het oppervlaktewater als het voornaamste milieurisico beschouwd.
De ongunstigste PEC voor het sediment (PECsediment) zou zich voordoen als alle uitgescheiden stoffen in het sediment terechtkwamen. Indien de PECsediment (standaardwaarde: 20 cm diepte) minder dan 10 μg/kg nat gewicht bedraagt, is geen verdere beoordeling vereist.
Indien de PEC in het oppervlaktewater (PECow) minder dan 0,1 μg/l bedraagt, is geen verdere beoordeling vereist.
Fase I — Beslissingsboom
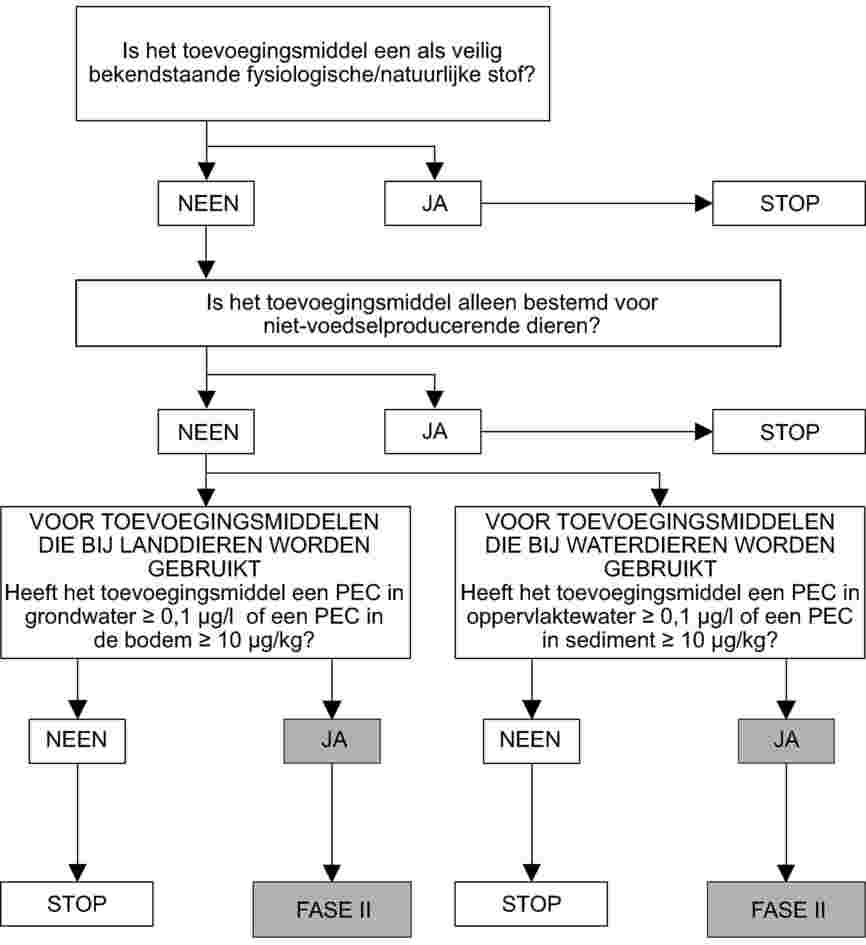
3.4.2. Fase II-beoordeling
Doel van fase II is te beoordelen in welke mate toevoegingsmiddelen niet-doelsoorten in het milieu, waaronder zowel water- als landdieren, kunnen beïnvloeden of in onaanvaardbare concentraties in het grondwater kunnen terechtkomen. Het is niet praktisch om een beoordeling te maken van het effect van het toevoegingsmiddel op elke soort in het milieu die aan het toevoegingsmiddel kan worden blootgesteld nadat het aan de doelsoort is toegediend. De onderzochte taxonomische niveaus zijn bedoeld als surrogaat of indicator voor de diverse soorten die in het milieu aanwezig is.
De fase II-beoordeling wordt verricht op basis van een risicoquotiënt, waarbij voor elk compartiment de berekende PEC wordt vergeleken met de voorspelde concentratie waarbij geen effect optreedt („predicted no effect concentration”, PNEC). De PNEC wordt bepaald aan de hand van proefondervindelijk bepaalde eindpunten, gedeeld door een passende beoordelingsfactor. De PNEC-waarde moet voor elk compartiment worden berekend.
De fase II-beoordeling begint zo mogelijk met een verfijning van de PEC. Het milieurisico wordt in twee stappen beoordeeld.
Tijdens de eerste stap, fase II A, wordt een beperkt aantal onderzoeken naar het milieutraject en -effect gebruikt om een voorzichtige schatting van het risico te maken op grond van de blootstelling en de effecten in het desbetreffende milieucompartiment. Als de verhouding tussen PEC en PNEC kleiner is dan 1, is geen verdere beoordeling vereist, tenzij bioaccumulatie wordt verwacht.
Indien de PEC/PNEC-verhouding op een onaanvaardbaar risico wijst (verhouding groter dan 1) moet de aanvrager overgaan tot fase II B om de beoordeling van het milieurisico te verfijnen.
3.4.2.1. Fase II A
In aanvulling op de in fase I bestudeerde compartimenten moet de PEC voor oppervlaktewater worden berekend, rekening houdend met wegvloeien en drainage.
Op basis van gegevens die nog niet in fase I zijn onderzocht, kan voor elk betrokken milieucompartiment een verfijndere PEC worden berekend. Daarbij moet rekening worden gehouden met:
|
a) |
de concentratie van de betrokken werkzame stof(fen)/metabolieten in mest/vissenfeces na toediening van het toevoegingsmiddel aan dieren in de voorgestelde dosis. Bij deze berekening moet rekening worden gehouden met de doseringen en het volume van de uitscheidingsproducten; |
|
b) |
de mogelijke afbraak van de uitgescheiden betrokken werkzame stof(fen)/metabolieten bij de normale verwerking en opslag van de mest voordat die wordt uitgereden; |
|
c) |
de adsorptie/desorptie van de betrokken werkzame stof(fen)/metabolieten aan/uit de bodem of het sediment bij aquacultuur, bij voorkeur te bepalen door onderzoek van de bodem of het sediment (OESO 106); |
|
d) |
de afbraak in de bodem of watersedimentsystemen (OESO 307, resp. 308), en |
|
e) |
andere factoren zoals fotolyse, hydrolyse, verdamping en verdunning door ploegen. |
Voor de fase II-risicobeoordeling moet voor elk betrokken milieucompartiment de hoogste PEC-waarde worden genomen die uit deze berekeningen voortvloeit.
Indien een hoge persistentie in de bodem/het sediment wordt verwacht, dat wil zeggen als het meer dan één jaar duurt tot de stof tot 90 % van de oorspronkelijke concentratie is afgebroken: DT90 > 1 jaar), moet het accumulatiepotentieel worden onderzocht.
Er moet worden bepaald bij welke concentraties van het toevoegingsmiddel (of metabolieten daarvan) ernstige nadelige kortetermijneffecten optreden voor verschillende trofische niveaus in de betrokken milieucompartimenten. Het gaat meestal om acute tests, waarbij de OESO-richtsnoeren of soortgelijke beproefde richtsnoeren moeten worden gevolgd. Het onderzoek naar het bodemmilieu moet het volgende omvatten: toxiciteit voor regenwormen, drie landplanten en bodemmicro-organismen (bv. effect op de stikstofbinding). Het onderzoek naar het zoetwatermilieu moet het volgende omvatten: toxiciteit voor vissen, Daphnia magna, algen en een sedimentorganisme. In het geval van zeekooien moeten drie verschillende taxa sedimentorganismen worden onderzocht.
Voor elk betrokken compartiment moet de PNEC worden berekend. De PNEC wordt normaliter afgeleid uit de laagste toxiciteitswaarde die bij bovengenoemde tests is waargenomen, die wordt gedeeld door een veiligheidsfactor van minimaal 100, afhankelijk van het eindpunt en het aantal gebruikte testsoorten.
Het bioaccumulatiepotentieel kan worden geraamd aan de hand van de n-octanol/waterverdelingscoëfficiënt, Log Kow. Waarden groter dan of gelijk aan 3 wijzen op mogelijke bioaccumulatie. Om het risico van secundaire vergiftiging te beoordelen, moet worden overwogen om in fase II B een onderzoek naar de bioconcentratiefactor (BCF) uit te voeren.
3.4.2.2. Fase II B (uitvoeriger ecotoxicologisch onderzoek)
Voor toevoegingsmiddelen waarbij na de fase II A-beoordeling een milieurisico niet kan worden uitgesloten, is meer informatie nodig over de effecten op biologische soorten in het (de) milieucompartiment(en) waarin volgens het fase II A-onderzoek problemen zouden kunnen optreden. In dat geval zijn nadere tests nodig om de chronische en specifiekere effecten op de betrokken soorten micro-organismen, dieren en planten te bepalen. Deze aanvullende informatie maakt het mogelijk een lagere veiligheidsfactor toe te passen.
Geschikte aanvullende ecotoxiciteitstests worden in een aantal publicaties beschreven, bijvoorbeeld in de OESO-richtsnoeren. Deze tests moeten zorgvuldig worden gekozen, zodat zij geschikt zijn voor de situatie waarin het toevoegingsmiddel en/of de metabolieten daarvan in het milieu vrijkomen en verspreid worden. De verfijning van de beoordeling van het effect op de bodem (PNECbodem) kan worden gebaseerd op onderzoek naar het chronische effect op regenwormen, aanvullend onderzoek van de bodemmicroflora en een aantal belangrijke plantensoorten, onderzoek op graslandongewervelden (waaronder insecten) en wilde vogels.
De verfijning van de beoordeling van het effect op het water/sediment kan worden gebaseerd op chronische toxiciteitstests op de waterorganismen/benthische organismen die in fase II A het gevoeligst zijn gebleken.
Bioaccumulatieonderzoek moet zo nodig worden uitgevoerd volgens OESO-richtsnoer 305.
4. SECTIE IV: ONDERZOEK NAAR DE WERKZAAMHEID VAN HET TOEVOEGINGSMIDDEL
Er moet met onderzoek worden aangetoond dat het toevoegingsmiddel voor elke voorgestelde toepassing werkzaam is en ten minste een van de in artikel 5, lid 3, van Verordening (EG) nr. 1831/2003 genoemde kenmerken bezit overeenkomstig de categorieën en functionele groepen toevoegingsmiddelen als genoemd in artikel 6 en bijlage I van die verordening. Aan de hand van deze onderzoeken moet de werkzaamheid van het toevoegingsmiddel ook overeenkomstig de veehouderijpraktijk in de EU kunnen worden beoordeeld.
De opzet van het onderzoek moet worden gemotiveerd door het gebruik van het toevoegingsmiddel en de diersoort en -categorie. Indien er dieren worden gebruikt, moeten de tests op dusdanige wijze worden uitgevoerd dat de gezondheids- en houderijomstandigheden van de dieren geen negatief effect hebben op de interpretatie van de resultaten. Voor elk experiment moeten de positieve en negatieve effecten, zowel in technologisch als in biologisch opzicht, worden beschreven. Er moet ook worden aangetoond dat er geen effecten zijn die afbreuk doen aan de bijzondere kenmerken van dierlijke producten. Idealiter moeten de tests voldoen aan de criteria van een erkende kwaliteitsborgingsregeling die aan externe audits wordt onderworpen. Bij gebrek aan een dergelijke regeling moet worden aangetoond dat de werkzaamheden zijn verricht door gekwalificeerd personeel, met passende faciliteiten en uitrusting en onder de verantwoordelijkheid van een met naam genoemde study director.
Het testprotocol wordt zorgvuldig opgesteld door de study director en bevat beschrijvende gegevens zoals de gebruikte methoden, apparaten en materialen, bijzonderheden over soort, ras of stam van de dieren, het aantal dieren en de omstandigheden waaronder zij werden gehuisvest en gevoederd. Bij alle onderzoeken met dieren moeten de testomstandigheden worden beschreven overeenkomstig punt 3.1.1.3. De eindverslagen, de onbewerkte gegevens, de onderzoeksplannen en de naar behoren gekarakteriseerde en geïdentificeerde teststoffen moeten met het oog op latere raadpleging worden gearchiveerd.
Het onderzoek moet de werkzaamheid bij de laagste aanbevolen dosis van het toevoegingsmiddel aantonen aan de hand van gevoelige parameters in vergelijking met een positieve en eventueel ook een negatieve controlegroep. Indien een maximale dosis wordt aanbevolen, moet ook deze worden onderzocht. Om de nodige wetenschappelijke keuzevrijheid te laten voor de opzet en uitvoering van de onderzoeken, wordt geen bepaalde opzet aanbevolen.
Er moet ook aandacht worden besteed aan bekende of potentiële biologische of chemische wisselwerkingen tussen het toevoegingsmiddel en andere toevoegingsmiddelen en/of diergeneesmiddelen en/of voederbestanddelen, voor zover dit van belang is voor de werkzaamheid van het toevoegingsmiddel in kwestie (bv. compatibiliteit van een microbieel toevoegingsmiddel met coccidiostatica en histomonostatica of organisch zuur).
4.1. In-vitro-onderzoek
Voor alle technologische en bepaalde sensoriële toevoegingsmiddelen die de kenmerken van het diervoeder beïnvloeden, moet de werkzaamheid worden aangetoond met laboratoriumonderzoek. Het onderzoek moet aldus worden opgezet dat het een representatief aantal voedermiddelen omvat waaraan het toevoegingsmiddel zal worden toegevoegd. De resultaten moeten bij voorkeur met parametervrije tests worden beoordeeld. De verwachte veranderingen moeten met een p-waarde kleiner dan of gelijk aan 0,05 worden aangetoond.
Voor andere soorten toevoegingsmiddel kan in-vitro-onderzoek, en met name onderzoek dat aspecten van het maag-darmkanaal simuleert, worden gebruikt om de werkzaamheid te staven. Dit onderzoek moet statistisch kunnen worden geëvalueerd.
4.2. Kortetermijnwerkzaamheidsonderzoek bij dieren
Om aan te tonen in welke mate een nieuwe vorm of bron van een voedingsstof of kleurstof een gelijkwaardig, reeds goedgekeurd of bekend toevoegingsmiddel kan vervangen, kan onderzoek naar de biologische beschikbaarheid worden gebruikt.
Om de werkingswijze aan te tonen, kan onderzoek naar de vertering/balans worden gebruikt ter ondersteuning van onderzoek naar de dierlijke productie. In sommige gevallen, met name wat milieuvoordelen betreft, kan de werkzaamheid beter worden aangetoond met balansonderzoek dan met langetermijnonderzoek naar de werkzaamheid. Bij dergelijk onderzoek moeten de aantallen en de soorten/categorieën dieren aangepast zijn aan de voorgestelde gebruiksvoorwaarden.
Zo nodig kunnen andere kortetermijnwerkzaamheidsonderzoeken bij dieren worden voorgesteld, eventueel ter vervanging van langetermijnwerkzaamheidsonderzoeken met dieren, mits dit afdoende gemotiveerd wordt.
4.3. Langetermijnwerkzaamheidsonderzoek bij dieren
De onderzoeken moeten op ten minste twee verschillende plaatsen worden gedaan.
Bij de proefopzet moet aandacht worden besteed aan een toereikend statistisch onderscheidingsvermogen en type 1- en 2-risico's. Het protocol moet voldoende gevoelig zijn om eventuele effecten van het toevoegingsmiddel bij de laagste aanbevolen dosis (risicotype 1 α, p ≤ 0,05 in het algemeen en p ≤ 0,1 voor herkauwers, minder gangbare soorten, gezelschapsdieren en niet-voedselproducerende dieren) op te sporen en voldoende statistisch onderscheidingsvermogen hebben om te garanderen dat het testprotocol aan de doelstelling van het onderzoek voldoet. Het type 2 β-risico moet lager zijn dan of gelijk zijn 20 % in het algemeen en 25 % voor herkauwers, minder gangbare soorten, gezelschapsdieren en niet-voedselproducerende dieren. Daartoe moet het onderscheidingsvermogen (1-β) groter zijn dan of gelijk zijn aan 80 % (75 % voor herkauwers, minder gangbare soorten, gezelschapsdieren en niet-voedselproducerende dieren).
Erkend wordt dat de aard van bepaalde toevoegingsmiddel het moeilijk maakt om te omschrijven welke testomstandigheden optimale resultaten kunnen opleveren. Indien meer dan drie tests beschikbaar zijn, moet daarom een meta-analyse worden overwogen. Daarom moeten voor alle proeven soortgelijke protocollen worden gevolgd, zodat de gegevens later op homogeniteit kunnen worden gecontroleerd en (als de tests dat vereisen) kunnen worden samengevoegd met het oog op statistische evaluatie met p ≤ 0,05.
4.4. Duur van het langetermijnwerkzaamheidsonderzoek bij dieren
In het algemeen komt de duur van het werkzaamheidsonderzoek overeen met de aangegeven toedieningsduur.
Het werkzaamheidsonderzoek moet worden uitgevoerd overeenkomstig de landbouwpraktijken in de EU en gedurende de in bijlage IV vermelde minimumperiode.
Indien een toevoegingsmiddel gedurende een specifieke, kortere periode wordt toegediend dan voor de diercategorie vermeld is, moet het volgens de voorgestelde gebruiksvoorwaarden worden toegediend. De observatieperiode mag echter niet minder dan 28 dagen bedragen en betreft de betrokken eindpunten (bv. bij fokzeugen: het aantal levend geboren biggen wat de zwangerschap betreft en het gewicht van de gespeende biggen wat de lactatieperiode betreft).
Voor andere diersoorten of -categorieën waarvoor in bijlage IV geen minimumduur voor het onderzoek is vastgesteld, wordt een toedieningsduur volgens de voorgestelde gebruiksvoorwaarden in acht genomen.
4.5. Werkzaamheidsvereisten voor categorieën en functionele groepen van toevoegingsmiddelen
Voor alle toevoegingsmiddelen die een effect op dieren beogen te hebben, is in-vitro-onderzoek vereist.
Voor de categorieën zoötechnische toevoegingsmiddelen, coccidiostatica en histomonostatica moet de werkzaamheid met ten minste drie langetermijnonderzoeken naar de werkzaamheid worden aangetoond. Voor sommige zoötechnische toevoegingsmiddelen en andere categorieën toevoegingsmiddelen die een effect op dieren beogen te hebben, kunnen kortermijnonderzoeken naar de werkzaamheid worden aanvaard, mits de werkzaamheid ondubbelzinnig wordt aangetoond.
Voor andere categorieën toevoegingsmiddelen die geen rechtstreeks effect op dieren hebben, moet ten minste één in-vitrowerkzaamheidsonderzoek worden verricht.
4.6. Onderzoek naar de kwaliteit van de dierlijke producten indien dit niet het aangegeven effect is
Om aan te tonen dat het toevoegingsmiddel geen negatief of ander ongewenst effect heeft op de organoleptische en nutritionele (in voorkomend geval hygiënische en technologische) eigenschappen van voedsel afkomstig van dieren die het toevoegingsmiddel toegediend hebben gekregen (voor zover dit niet het beoogde effect is), moeten tijdens een van de werkzaamheidsonderzoeken de nodige monsters worden genomen. Er moeten twee groepen worden onderzocht: een groep die geen toevoegingsmiddel toegediend heeft gekregen en een groep die de hoogste voorgestelde dosis van het toevoegingsmiddel heeft gekregen. De gegevens moeten een statistische evaluatie mogelijk maken. Als deze onderzoeken achterwege worden gelaten, moet dat naar behoren worden gemotiveerd.
5. SECTIE V: PLAN VOOR MONITORING NA HET IN DE HANDEL BRENGEN
Overeenkomstig artikel 7, lid 3, onder g), van Verordening (EG) nr. 1831/2003 moet voor bepaalde categorieën toevoegingsmiddelen een voorstel voor monitoring na het in de handel brengen worden ingediend om eventuele directe of indirecte, onmiddellijk, vertraagd of onverwacht optredende effecten van het gebruik van het toevoegingsmiddel op de gezondheid van mens of dier of het milieu te kunnen traceren en signaleren, overeenkomstig de kenmerken van de betrokken producten.
De opzet van het monitoringplan moet per geval worden uitgewerkt; er moet worden aangegeven wie (bv. de aanvrager, de gebruikers) de verschillende taken in het kader van het monitoringplan moet uitvoeren, wie verantwoordelijk is voor de invoering en de correcte uitvoering van het monitoringplan, en hoe de bevoegde autoriteiten op de hoogte zullen worden gebracht van nieuwe informatie over de gebruiksveiligheid van het toevoegingsmiddel. Indien schadelijke effecten worden vastgesteld, moeten de Commissie en de Autoriteit daarvan in kennis worden gesteld, onverminderd de bepalingen betreffende toezicht van artikel 12 van Verordening (EG) nr. 1831/2003.
Indien het toevoegingsmiddel ook een erkend antibioticum is en indien bij het gebruik van de normale dosis in voeder is gebleken dat het resistente bacteriestammen selecteert, moet in het kader van de monitoring na het in de handel brengen onderzoek in de praktijk worden verricht om de resistentie van bacteriën tegen het toevoegingsmiddel te monitoren.
In het geval van coccidiostatica en histomonostatica moet onderzoek in de praktijk naar de resistentie van Eimeria spp. en Histomonas meleagridis worden verricht, bij voorkeur tijdens het laatste deel van de vergunningstermijn.
(1) PB L 50 van 20.2.2004, blz. 44.
(2) PB 196 van 16.8.1967, blz. 1. Richtlijn laatstelijk gewijzigd bij Richtlijn 2006/121/EG van het Europees Parlement en de Raad (PB L 396 van 30.12.2006, blz. 852; gerectificeerd in PB L 136 van 29.5.2007, blz. 281).
(3) PB L 152 van 30.4.2004, blz. 1; gerectificeerd in PB L 216 van 16.6.2004, blz. 3.
(4) PB L 165 van 30.4.2004; gerectificeerd in PB L 191 van 28.5.2004, blz. 1.
(5) PB L 117 van 8.5.1990, blz. 1. Richtlijn laatstelijk gewijzigd bij Beschikking 2005/174/EG van de Commissie (PB L 59 van 5.3.2005, blz. 20).
(6) PB L 76 van 22.3.1991, blz. 35. Richtlijn laatstelijk gewijzigd bij Richtlijn 2001/58/EG (PB L 212 van 7.8.2001, blz. 24).
(7) PB L 262 van 17.10.2000, blz. 21.
(8) PB L 224 van 18.8.1990, blz. 1. Verordening laatstelijk gewijzigd bij Verordening (EG) nr. 203/2008 van de Commissie (PB L 60 van 5.3.2008, blz. 18).
(9) M. Thompson et al.: Harmonized Guidelines For Single Laboratory Validation Of Methods Of Analysis (IUPAC Technical Report) Pure Appl. Chem., vol. 74, nr. 5, blz. 835-855, 2002.
(10) PB L 221 van 17.8.2002, blz. 8. Beschikking laatstelijk gewijzigd bij Beschikking 2004/25/EG (PB L 6 van 10.1.2004, blz. 38).
(11) Een niet-uitputtende lijst is beschikbaar op www.efsa.europa.eu/en/science/feedap/feedap_opinion/993. html.
(12) Spieren en huid in natuurlijke verhoudingen.
(13) Voor varkens 50 g vet en huid in natuurlijke verhoudingen.
(14) Vet en huid in natuurlijke verhoudingen.
(15) Rekening houdend met de voorgestelde wachttijd.
(16) Idealiter tegelijk met de TRC vastgesteld.
(17) Uit de TRC-waarden berekend.
BIJLAGE III
SPECIFIEKE VEREISTEN WAARAAN HET IN ARTIKEL 3 BEDOELDE DOSSIER MOET VOLDOEN WAT BETREFT BEPAALDE CATEGORIEËN TOEVOEGINGSMIDDELEN OF BEPAALDE BIJZONDERE SITUATIES, OVEREENKOMSTIG ARTIKEL 7, LID 5, VAN VERORDENING (EG) NR. 1831/2003
Verordening (EG) nr. 1831/2003 voorziet waar nodig voor elke categorie toevoegingsmiddelen of voor andere bijzondere doeleinden overeenkomstig artikel 7, lid 5, in aanvullende ondersteuning bij de opstelling van de dossiers.
Lijst van de specifieke vereisten voor de opstelling van dossiers voor:
|
1) |
Technologische toevoegingsmiddelen |
|
2) |
Sensoriële toevoegingsmiddelen |
|
3) |
Nutritionele toevoegingsmiddelen |
|
4) |
Zoötechnische toevoegingsmiddelen |
|
5) |
Coccidiostatica en histomonostatica |
|
6) |
Extrapolatie van gangbare naar minder gangbare diersoorten |
|
7) |
Gezelschapsdieren en andere niet-voedselproducerende dieren |
|
8) |
Toevoegingsmiddelen die reeds als levensmiddelenadditief zijn toegelaten |
|
9) |
Wijziging van vergunningen |
|
10) |
Verlenging van vergunningen |
|
11) |
Herbeoordeling van toevoegingsmiddelen waarvoor reeds bij Richtlijn 70/524/EEG een vergunning is verleend |
Aanvragen mogen overeenkomstig meer dan een van bovengenoemde specifieke vereisten worden ingediend.
Algemene voorwaarden
Als in het dossier gegevens ontbreken waarom in deze secties wordt gevraagd, moet dat worden gemotiveerd.
1. TECHNOLOGISCHE TOEVOEGINGSMIDDELEN
1.1. Sectie I: samenvatting van het dossier
De volledige sectie I van bijlage II is van toepassing.
1.2. Sectie II: identiteit en kenmerken van en gebruiksvoorwaarden voor het toevoegingsmiddel; analysemethoden
Sectie II van bijlage II is als volgt van toepassing:
|
— |
op toevoegingsmiddelen waarvan de vergunning niet aan een specifieke vergunninghouder is verleend, zijn de punten 2.1.2, 2.1.3, 2.1.4, 2.1.4.2, 2.2, 2.3.1, 2.3.2, 2.4.1, 2.4.2, 2.4.4, 2.5 en 2.6 van toepassing; |
|
— |
op andere toevoegingsmiddelen, waarvan de vergunning aan een specifieke vergunninghouder is verleend, is de volledige sectie II van toepassing. |
1.3. Sectie III: onderzoek naar de werkzaamheid van het toevoegingsmiddel
De subsecties 3.1, 3.2 en 3.4 van bijlage II zijn niet van toepassing op inkuiltoevoegingsmiddelen mits wordt aangetoond dat:
|
— |
er geen aantoonbare hoeveelheden van de werkzame stof(fen), van relevante metabolieten daarvan of van het agens (de agentia) in het uiteindelijke diervoeder overleven, of |
|
— |
de werkzame stof(fen) en het agens (de agentia) als normale bestanddelen in kuilvoeder voorkomen en dat het gebruik van het toevoegingsmiddel de concentratie niet wezenlijk verhoogt ten opzichte van kuilvoeder zonder het toevoegingsmiddel (d.w.z. dat de blootstelling niet wezenlijk verandert). |
In andere gevallen is de volledige sectie III van bijlage II is van toepassing.
1.3.1. Onderzoek naar de veiligheid van het gebruik van het toevoegingsmiddel voor de doeldieren
Op lichaamsvreemde (1) stoffen is de volledige subsectie 3.1 van bijlage II van toepassing.
1.3.1.1. Onderzoek naar tolerantie bij de doeldieren
In het geval van inkuiltoevoegingsmiddelen:
|
— |
moet het product aan het basisvoeder worden toegevoegd en moeten de resultaten worden vergeleken met een negatieve controle met hetzelfde voeder. Het basisvoeder mag bestaan uit één bron van kuilvoeder dat zonder toevoegingsmiddel bereid is; |
|
— |
moet de dosis die voor het tolerantieonderzoek wordt gekozen, een veelvoud zijn van de concentratie die in het kuilvoeder aanwezig is op het tijdstip waarop het gewoonlijk wordt gebruikt, voor zover deze met zekerheid kan worden vastgesteld. Er moet bijzondere aandacht worden besteed aan producten die levensvatbare micro-organismen bevatten en aan de mate waarin deze tijdens het inkuilen kunnen overleven en zich kunnen voortplanten. |
Het tolerantieonderzoek mag gewoonlijk beperkt blijven tot een herkauwerssoort, normaliter melkkoeien. Onderzoek bij andere soorten is alleen vereist als de aard van het kuilvoeder het meer geschikt maakt voor niet-herkauwers.
Andere stoffen:
voor de overige stoffen waarvoor een vergunning als technologisch toevoegingsmiddel wordt aangevraagd en waarvoor nog geen vergunning voor gebruik in diervoeder verleend is, moet worden aangetoond dat de hoogste voorgestelde dosis niet schadelijk is voor dieren. Dit bewijs mag beperkt blijven tot één proef bij een van de gevoeligste doelsoorten of bij één soort laboratoriumdieren.
1.3.1.2. Microbieel onderzoek
De volledige subsectie 3.1.2 van bijlage II is van toepassing.
1.3.2. Onderzoek naar de veiligheid van het gebruik van het toevoegingsmiddel voor de consumenten
|
1.3.2.1. |
Metabolisme- en residuonderzoek Metabolisme- en residuonderzoek is niet vereist indien:
Metabolismeonderzoek is evenmin vereist indien de stof van nature in aanzienlijke hoeveelheden in levensmiddelen of diervoeder aanwezig is of als de stof een natuurlijk bestanddeel van lichaamsvloeistoffen of weefsels is. In die gevallen is echter wel residuonderzoek vereist. Dit mag beperkt blijven tot een vergelijking van het gehalte in weefsels/producten in een onbehandelde groep en in een groep die de hoogste aanbevolen dosis heeft gekregen. |
|
1.3.2.2. |
Toxicologisch onderzoek Toxicologisch onderzoek is niet vereist indien:
In het geval van micro-organismen en enzymen waarop geen van bovengenoemde uitzonderingen van toepassing is, moet onderzoek naar de genotoxiciteit (met inbegrip van de mutageniteit) en de subchronische orale toxiciteit worden verricht. Bij het onderzoek naar de genotoxiciteit mogen geen levende cellen aanwezig zijn. Op lichaamsvreemde stoffen waarop geen van bovengenoemde uitzonderingen van toepassing is, is de volledige subsectie 3.2.2 van toepassing. Andere stoffen worden per geval behandeld, rekening houdend met de mate en wijze van blootstelling. |
|
1.3.2.3. |
Beoordeling van de veiligheid voor de consument De volledige subsectie 3.2.3 van bijlage II is van toepassing op toevoegingsmiddelen waarvoor een vergunning voor voedselproducerende dieren is aangevraagd. |
1.3.3. Onderzoek naar de veiligheid van het gebruik van het toevoegingsmiddel voor gebruikers/werknemers
De volledige subsectie 3.3 van bijlage II is van toepassing. Van toevoegingsmiddelen die micro-organismen en enzymen bevatten, wordt verondersteld dat zij de ademhalingswegen sensibiliseren, tenzij het tegendeel wordt aangetoond.
1.3.4. Onderzoek naar de veiligheid van het gebruik van het toevoegingsmiddel voor het milieu
De volledige subsectie 3.4 van bijlage II is van toepassing. In het geval van inkuiltoevoegingsmiddelen moet het effect van het toevoegingsmiddel op de uitstroom uit de hoop of kuil worden onderzocht.
1.4. Sectie IV: onderzoek naar de werkzaamheid van het toevoegingsmiddel
Technologische toevoegingsmiddelen zijn bedoeld om de kenmerken van diervoeder te verbeteren of stabieler te maken, maar hebben meestal geen rechtstreeks biologisch effect op de dierlijke productie. De werkzaamheid van het toevoegingsmiddel moet worden aangetoond aan de hand van relevante criteria zoals aangegeven in erkende aanvaardbare methoden, onder de beoogde praktische gebruiksomstandigheden en in vergelijking met voor controledoeleinden geschikte diervoeders.
De werkzaamheid moet worden beoordeeld door in-vitro-onderzoek, behalve voor stoffen die radionuclidenbesmetting tegengaan. De eindpunten voor de verschillende functionele groepen worden aangegeven in de volgende tabel:
Eindpunten voor verschillende technologische toevoegingsmiddelen
|
Functionele groep |
Eindpunten voor het aantonen van de werkzaamheid |
||||||||||
|
Remming van de groei van micro-organismen, met name biotische organismen en organismen die bederf veroorzaken. Er moet worden aangetoond hoelang het conserverende effect duurt. |
||||||||||
|
Bescherming tegen oxidatieschade aan belangrijke voedingstoffen/bestanddelen bij de verwerking en/of opslag van diervoeders. Er moet worden aangetoond hoelang het beschermende effect duurt. |
||||||||||
|
Vorming/behoud van stabiele emulsies van voedselingrediënten die anders niet of moeilijk kunnen worden gemengd. |
||||||||||
|
Behoud van de fysisch-chemische toestand van diervoeders. |
||||||||||
|
Viscositeit van voedermiddelen of diervoeders. |
||||||||||
|
Vorming van een gel, waardoor de textuur van een diervoeder verandert. |
||||||||||
|
Duurzaamheid van pellets of verbetering van de pelletvorming. |
||||||||||
|
Bewijs van verminderde besmetting van voedsel van dierlijke oorsprong. |
||||||||||
|
Stroombaarheid. Er moet worden aangetoond hoelang het effect duurt. |
||||||||||
|
pH en/of buffercapaciteit van diervoeders. |
||||||||||
|
|
||||||||||
|
Onuitwisbare identificatie van voedermiddelen. |
Inkuiltoevoegingsmiddelen
Om het gewenste effect op het inkuilproces (2) aan te tonen, moeten afzonderlijke tests worden uitgevoerd. Indien het om alle of niet-gespecificeerde voedergewassen gaat, moeten de tests worden uitgevoerd met één exemplaar van elk van de volgende categorieën:
|
— |
gemakkelijk in te kuilen voedergewassen: > 3 % oplosbare koolhydraten in het verse materiaal (bv. hele maisplanten, raaigras, dravik en suikerbietenpulp); |
|
— |
middelmatig moeilijk in te kuilen voedergewassen: 1,5 à 3 % oplosbare koolhydraten in het verse materiaal (bv. veldbeemdgras, zwenkgras en voorgedroogde alfalfa); |
|
— |
moeilijk in te kuilen voedergewassen: < 1,5 % oplosbare koolhydraten in het verse materiaal (bv. kropaar en peulvruchten). |
Indien de aanvraag beperkt is tot bepaalde subcategorieën die in termen van droge stof worden beschreven, moeten de grenzen van het drogestofgehalte uitdrukkelijk worden vermeld. Er moeten drie tests worden uitgevoerd op materiaal dat representatief is voor deze gehalten, zo mogelijk met exemplaren van verschillende botanische oorsprong.
Voor bijzondere soorten diervoeders zijn specifieke tests vereist.
Het onderzoek moet normaliter 90 dagen of meer duren, bij constante temperatuur (aanbevolen temperatuur: tussen 15 en 25 oC). Indien het onderzoek minder lang duurt, moet dat worden gemotiveerd.
In de regel moeten de volgende parameters worden onderzocht in vergelijking met de negatieve controle:
|
— |
droge stof en berekend verlies aan droge stof (gecorrigeerd voor vluchtige bestanddelen); |
|
— |
daling van de pH; |
|
— |
concentratie vluchtige vetzuren (bv. azijnzuur, boterzuur en propionzuur) en melkzuur; |
|
— |
concentratie alcoholen (ethanol); |
|
— |
concentratie ammoniak (g/kg totale stikstof), en |
|
— |
gehalte aan wateroplosbare koolhydraten. |
Daarnaast moeten zo nodig ook andere microbiologische en chemische parameters worden onderzocht om specifieke claims te staven (bv. aantal lactaatassimilerende gisten, aantal Clostridia, aantal Listeria en biogene aminen).
Een beoogde vermindering van de uitstroom wordt beoordeeld ten opzichte van de totale uitstroom over de volledige testperiode, rekening houdend met het te verwachten milieueffect (bv. ecotoxiciteit van de uitstroom of biologisch zuurstofverbruik). De vermindering van de productie van uitstroom moet rechtstreeks worden aangetoond. De voederkuil moet voldoende capaciteit hebben om de uitstroom onder druk te kunnen laten wegvloeien. Het onderzoek moet ten minste 50 dagen duren. Een eventuele andere duur moet worden gemotiveerd.
Een verbetering van de aerobe stabiliteit moet worden aangetoond in vergelijking met een negatieve controle. Stabiliteitsonderzoek moet ten minste zeven dagen na blootstelling aan de lucht en duren. Er moet worden aangetoond dat het toevoegingsmiddel de stabiliteit met ten minste twee dagen verlengt in vergelijking met een onbehandeld controlegewas. Aanbevolen wordt om de test bij een omgevingstemperatuur van 20 oC uit te voeren en een temperatuursstijging van 3 oC of meer ten opzichte van de achtergrondtemperatuur als teken van instabiliteit te beschouwen. In plaats van de temperatuur mag ook de CO2-productie worden gemeten.
1.5. Sectie V: plan voor monitoring na het in de handel brengen
Deze sectie is van toepassing overeenkomstig artikel 7, lid 3, onder g), van Verordening (EG) nr. 1831/2003. Dat wil zeggen dat een plan voor monitoring na het in de handel brengen alleen vereist is voor toevoegingsmiddel die ggo's zijn of daarmee zijn geproduceerd.
2. SENSORIËLE TOEVOEGINGSMIDDELEN
2.1. Kleurstoffen
2.1.1. Sectie I: samenvatting van het dossier
De volledige sectie I van bijlage II is van toepassing.
2.1.2. Sectie II: identiteit en kenmerken van en gebruiksvoorwaarden voor het toevoegingsmiddel; analysemethoden
Sectie II van bijlage II is als volgt van toepassing:
|
— |
op toevoegingsmiddelen waarvan de vergunning niet aan een specifieke vergunninghouder is verleend, zijn de punten 2.1.2, , 2.1.4, 2.1.4.2, 2.2, 2.3.1, 2.3.2, 2.4.1, 2.4.2, 2.4.4, 2.5 en 2.6 van toepassing; |
|
— |
op andere toevoegingsmiddelen, waarvan de vergunning aan een specifieke vergunninghouder is verleend, is de volledige sectie II van toepassing. |
2.1.3. Sectie III: onderzoek naar de veiligheid van het gebruik van het toevoegingsmiddel
De volledige subsectie 3.3 van bijlage II is op alle toevoegingsmiddelen van toepassing.
|
1) |
op stoffen die bij toediening aan dieren aan een levensmiddel van dierlijke oorsprong kleur geven, zijn de volledige subsecties 3.1, 3.2 en 3.4 van sectie III van bijlage II van toepassing; |
|
2) |
voor stoffen die diervoeders kleur geven of daaraan kleur teruggeven, moeten de onderzoeken betreffende subsectie 3.1 van sectie III worden uitgevoerd op dieren die het toevoegingsmiddel bij de aanbevolen dosis toegediend krijgen. Ook verwijzingen naar bestaande wetenschappelijke literatuur kunnen als bewijsmateriaal dienen. De volledige subsecties 3.2 en 3.4 van sectie III van bijlage II zijn van toepassing; |
|
3) |
voor stoffen die een gunstig effect hebben op de kleur van siervissen of -vogels moeten de onderzoeken betreffende subsectie 3.1 van sectie III van bijlage II worden uitgevoerd op dieren die het toevoegingsmiddel bij de aanbevolen dosis toegediend krijgen. Ook verwijzingen naar bestaande wetenschappelijke literatuur kunnen als bewijsmateriaal dienen. De subsecties 3.2 en 3.4 zijn echter niet vereist. |
2.1.4. Sectie IV: onderzoek naar de werkzaamheid van het toevoegingsmiddel
De volledige sectie IV van bijlage II is van toepassing.
|
a) |
Voor stoffen die bij toediening aan dieren aan een levensmiddel van dierlijke oorsprong kleur geven: de kleurverandering van producten afkomstig van dieren die het toevoegingsmiddel bij de aanbevolen dosis toegediend hebben gekregen, moet volgens een geschikte methode worden gemeten. Er moet worden aangetoond dat het gebruik van het toevoegingsmiddel geen negatief effect heeft op de stabiliteit en de organoleptische en nutritionele kwaliteiten van de levensmiddelen. Indien de effecten van een bepaalde stof op de samenstelling/kenmerken van dierlijke producten goed gedocumenteerd is, kunnen andere onderzoeken (bv. naar de biologische beschikbaarheid) in principe voldoende zijn om de werkzaamheid aan te tonen. |
|
b) |
Voor stoffen die aan een diervoeder kleur geven of daaraan kleur teruggeven: de werkzaamheid moet worden aangetoond door passende laboratoriumonderzoeken onder de beoogde gebruiksvoorwaarden en in vergelijking met controlevoeders. |
|
c) |
Voor stoffen die een gunstig effect hebben op de kleur van siervissen of -vogels: het onderzoek ter staving van het (de) effect(en) moet worden uitgevoerd op dieren die het toevoegingsmiddel bij de aanbevolen dosis toegediend krijgen. De kleurverandering moet volgens een passende methode worden gemeten. De werkzaamheid kan ook met andere onderzoeken (bv. naar de biologische beschikbaarheid) of onder verwijzing naar wetenschappelijke literatuur worden aangetoond. |
2.1.5. Sectie V: plan voor monitoring na het in de handel brengen
Deze sectie is van toepassing overeenkomstig artikel 7, lid 3, onder g), van Verordening (EG) nr. 1831/2003. Dat wil zeggen dat een plan voor monitoring na het in de handel brengen alleen vereist is voor toevoegingsmiddel die ggo's zijn of daarmee zijn geproduceerd.
2.2. Aromatische stoffen
2.2.1. Sectie I: samenvatting van het dossier
De volledige sectie I van bijlage II is van toepassing.
2.2.2. Sectie II: identiteit en kenmerken van en gebruiksvoorwaarden voor het toevoegingsmiddel; analysemethoden
Wat de groep „natuurlijke producten” betreft, worden hele planten, dieren en andere organismen en delen en producten daarvan die slechts een zeer beperkte bewerking zoals pletten, malen of drogen hebben ondergaan (bv. talrijke kruiden en specerijen) over het algemeen niet beschouwd als behorend tot de functionele groep aroma's van de categorie sensoriële toevoegingsmiddelen.
Voor de beoordeling van toepassingen van deze producten worden aroma's als volgt ingedeeld:
|
1. |
Natuurlijke producten:
|
|
2. |
Natuurlijke of dienovereenkomstige synthetische, chemisch gedefinieerde aroma's. |
|
3. |
Kunstmatige stoffen. Er moet worden aangegeven tot welke groep het product waartoe waarvoor een vergunning wordt aangevraagd, behoort. Indien het product niet in een van bovengenoemde groepen kan worden ondergebracht, moet dat worden vermeld en gemotiveerd. |
|
2.2.2.1. |
Karakterisering van de werkzame stof(fen)/het agens (de agentia) De volledige subsectie 2.2 van bijlage II is van toepassing. Bovendien geldt het volgende: Voor alle groepen aroma's moeten steeds de desbetreffende identificatienummers (bv. FLAVIS (3), Raad van Europa (4), JECFA, CAS (5) of een andere internationaal erkende nummering) die specifiek voor de identificatie van aroma's in levensmiddelen en diervoeder worden gebruikt, worden vermeld, voor zover deze beschikbaar zijn.
|
|
2.2.2.2. |
Productie- en vervaardigingswijze De volledige subsectie 2.3 van bijlage II is van toepassing. In het geval van niet chemisch gedefinieerde natuurlijke producten — meestal complexe mengsels van talrijke, door extractie verkregen bestanddelen — moet het extractieproces in detail worden beschreven. Aanbevolen wordt om daarvoor de algemeen gebruikte terminologie voor botanische gedefinieerde aroma's te gebruiken, bv. etherische olie, absolue, tinctuur, extract en dergelijke (7). De gebruikte extractiemiddelen, de voorzorgsmaatregelen tegen residuen daarvan en het eventuele gehalte aan onvermijdbare residuen moeten worden vermeld. Bij de karakterisering van het extract mag naar de extractiewijze worden verwezen. |
|
2.2.2.3. |
Analysemethoden
Op alle overige aroma's, zoals natuurlijke extracten die stoffen van toxicologisch belang bevatten, natuurlijke of dienovereenkomstige synthetische, chemisch gedefinieerde aroma's die zelf van toxicologisch belang zijn, en kunstmatige aroma's is de volledige subsectie 2.6 van toepassing. |
2.2.3. Sectie III: onderzoek naar de werkzaamheid van het toevoegingsmiddel
Voor alle aroma's moet de blootstelling van en inname door dieren, zowel door natuurlijke blootstelling als na toevoeging van het aroma aan het diervoeder, worden berekend en vermeld.
Op aroma's die tot de groep kunstmatige stoffen behoren, is de volledige sectie III van bijlage II van toepassing.
|
2.2.3.1. |
Onderzoek naar de veiligheid van het gebruik van het toevoegingsmiddel voor de doeldieren
|
|
2.2.3.2. |
Onderzoek naar de veiligheid van het gebruik van het toevoegingsmiddel voor de consumenten Er moet worden aangetoond dat de metabolieten van het aroma geen accumulatie van producten die van toxicologisch belang zijn voor de mens veroorzaken in het dier. Indien het gebruik van het aroma als toevoegingsmiddel in diervoeder residuen in levensmiddelen van dierlijke oorsprong meebrengt, moet de blootstelling van de consument gedetailleerd worden berekend en vermeld.
|
|
2.2.3.3. |
Onderzoek naar de veiligheid van het gebruik van het toevoegingsmiddel voor gebruikers/werknemers De volledige subsectie 3.3 van bijlage II is van toepassing. |
|
2.2.3.4. |
Onderzoek naar de veiligheid van het gebruik van het toevoegingsmiddel voor het milieu De volledige subsectie 3.4 van bijlage II is van toepassing. |
2.2.4. Sectie IV: onderzoek naar de werkzaamheid van het toevoegingsmiddel
De aromatische eigenschappen moeten worden aangetoond, gewoonlijk met gepubliceerde literatuur. In voorkomend geval mogen zij ook met ervaring met het gebruik in de praktijk worden aangetoond; anders is onderzoek bij dieren vereist.
Indien het product waarvoor een vergunning wordt aangevraagd, naast de functie die onder de definitie van aromastoffen in bijlage I bij Verordening (EG) nr. 1831/2003 valt, ook nog andere functies in diervoeder, dieren of levensmiddelen van dierlijke oorsprong heeft, moet dat grondig worden onderzocht en worden vermeld.
2.2.5. Sectie V: plan voor monitoring na het in de handel brengen
Deze sectie is van toepassing overeenkomstig artikel 7, lid 3, onder g), van Verordening (EG) nr. 1831/2003. Dat wil zeggen dat een plan voor monitoring na het in de handel brengen alleen vereist is voor toevoegingsmiddelen die ggo's zijn of daarmee zijn geproduceerd.
3. NUTRITIONELE TOEVOEGINGSMIDDELEN
3.1. Sectie I: samenvatting van het dossier
De volledige sectie I van bijlage II is van toepassing.
3.2. Sectie II: identiteit en kenmerken van en gebruiksvoorwaarden voor het toevoegingsmiddel; analysemethoden
Sectie II van bijlage II is als volgt van toepassing:
|
— |
op toevoegingsmiddelen waarvan de vergunning niet aan een specifieke vergunninghouder is verleend, zijn de punten 2.1.2, 2.1.3, 2.1.4, 2.1.4.2, 2.2, 2.3.1, 2.3.2, 2.4.1, 2.4.2, 2.4.4, 2.5 en 2.6 van toepassing; |
|
— |
op andere toevoegingsmiddelen, waarvan de vergunning aan een specifieke vergunninghouder is verleend, is de volledige sectie II van toepassing. |
3.3. Sectie III: onderzoek naar de werkzaamheid van het toevoegingsmiddel
3.3.1. Onderzoek naar de veiligheid van het gebruik van het toevoegingsmiddel voor de doelsoorten
|
3.3.1.1. |
Tolerantie bij de doelsoorten
|
|
3.3.1.2. |
Microbieel onderzoek De volledige subsectie 3.1.2 van bijlage II is van toepassing. |
3.3.2. Onderzoek naar de veiligheid van het gebruik van het toevoegingsmiddel voor de consumenten
|
3.3.2.1. |
Metabolisme- en residuonderzoek Metabolisme- en residuonderzoek is doorgaans niet vereist. Het metabolisme van ureumderivaten bij herkauwers moet in het kader van het werkzaamheidsonderzoek worden onderzocht. Onderzoek naar residuen of afzetting is alleen vereist voor toevoegingsmiddelen van de functionele groep „vitaminen, provitaminen en chemisch duidelijk omschreven stoffen met een gelijkaardige werking” die in het lichaam kunnen accumuleren en voor toevoegingsmiddelen van de functionele groep „verbindingen van sporenelementen” waarvan de biologische beschikbaarheid verhoogd is. In dat geval is de procedure van subsectie 3.2.1 van bijlage II niet van toepassing. Het onderzoek mag beperkt blijven tot een vergelijking van het gehalte in weefsels of producten in een groep die de hoogste aangegeven dosis heeft gekregen en een positieve controle (referentiestof). |
|
3.3.2.2. |
Toxicologisch onderzoek Toxicologisch onderzoek is vereist voor gistingsproducten en toevoegingsmiddelen waarvoor nog geen vergunning is verleend. Wat gistingsproducten betreft, moet onderzoek naar de genotoxiciteit en de subchronische toxiciteit worden verricht, tenzij:
Indien het productieorganisme behoort tot een groep waarvan bekend is dat bepaalde stammen toxinen kunnen produceren, moet de aanwezigheid daarvan uitdrukkelijk worden uitgesloten. |
|
3.3.2.3. |
Beoordeling van de veiligheid voor de consument De volledige subsectie 3.2.3 van bijlage II is van toepassing. |
3.3.3. Onderzoek naar de veiligheid van het gebruik van het toevoegingsmiddel voor gebruikers/werknemers
De volledige subsectie 3.3 van bijlage II is van toepassing.
3.3.4. Onderzoek naar de veiligheid van het gebruik van het toevoegingsmiddel voor het milieu
Op nieuwe werkzame stoffen die tot de functionele groep „verbindingen van sporenelementen” behoren, is de volledige subsectie 3.4 van bijlage II van toepassing.
3.4. Sectie IV: onderzoek naar de werkzaamheid van het toevoegingsmiddel
Onderzoek naar de werkzaamheid is niet vereist voor ureum, aminozuren en zouten en analogen daarvan die reeds als toevoegingsmiddel zijn toegelaten, verbindingen van sporenelementen die reeds als toevoegingsmiddel zijn toegelaten en vitaminen, provitaminen en chemisch duidelijk omschreven stoffen met een gelijkaardige werking die reeds als toevoegingsmiddel zijn toegelaten.
Een kortetermijnonderzoek ter staving van de werkzaamheid is vereist voor ureumderivaten, zouten van aminozuren en analogen daarvan die nog niet als toevoegingsmiddel zijn toegelaten, verbindingen van sporenelementen die nog niet toevoegingsmiddel zijn toegelaten en vitaminen, provitaminen en chemisch duidelijk omschreven stoffen met een gelijkaardige werking die nog niet als toevoegingsmiddel zijn toegelaten.
Voor andere stoffen waarmee een nutritioneel effect wordt beoogd, is ten minste een langetermijnonderzoek naar de werkzaamheid overeenkomstig sectie 4 van bijlage II vereist.
Zo nodig moet het onderzoek aantonen dat het toevoegingsmiddel in de nutritionele behoeften van de dieren voorziet. Bij de tests moet een testgroep worden gebruikt waarin de dieren een lagere concentratie van de nutriënt krijgen dan zij nodig hebben. Er mag echter geen gebruik worden gemaakt van een controlegroep met een ernstig tekort. Over het algemeen volstaat het de werkzaamheid aan te tonen bij één diersoort of -categorie, waaronder laboratoriumdieren.
3.5. Sectie V: plan voor monitoring na het in de handel brengen
Deze sectie is van toepassing overeenkomstig artikel 7, lid 3, onder g), van Verordening (EG) nr. 1831/2003.
4. ZOÖTECHNISCHE TOEVOEGINGSMIDDELEN
4.1. Andere zoötechnische toevoegingsmiddelen dan enzymen en micro-organismen
4.1.1. Sectie I: samenvatting van het dossier
De volledige sectie I van bijlage II is van toepassing.
4.1.2. Sectie II: identiteit en kenmerken van en gebruiksvoorwaarden voor het toevoegingsmiddel; analysemethoden
De volledige sectie II van bijlage II is van toepassing.
4.1.3. Sectie III: onderzoek naar de werkzaamheid van het toevoegingsmiddel
|
4.1.3.1. |
Onderzoek naar de veiligheid van het gebruik van het toevoegingsmiddel voor de doeldieren De volledige subsectie 3.1 van bijlage II is van toepassing. |
|
4.1.3.2. |
Onderzoek naar de veiligheid van het gebruik van het toevoegingsmiddel voor de consumenten
|
|
4.1.3.3. |
Onderzoek naar de veiligheid van het toevoegingsmiddel voor gebruikers/werknemers De volledige subsectie 3.3 van bijlage II is van toepassing. |
|
4.1.3.4. |
Onderzoek naar de veiligheid van het toevoegingsmiddel voor het milieu De volledige subsectie 3.4 van bijlage II is van toepassing. |
4.1.4. Sectie IV: onderzoek naar de werkzaamheid van het toevoegingsmiddel
De volledige sectie IV van bijlage II is van toepassing.
|
1) |
Toevoegingsmiddelen die dierlijke productie, prestaties of welzijn gunstig beïnvloeden en functionele groep „andere zoötechnische toevoegingsmiddelen”. De werkzaamheid kan alleen worden aangetoond met betrekking tot elke doelsoort of -categorie. Naargelang van de eigenschappen van het toevoegingsmiddel kunnen de resultaten worden gemeten aan de hand van prestatiekenmerken (bv. voederrendement, gemiddelde dagelijkse gewichtstoename, stijging van de productie van de dieren), de samenstelling van het karkas, de prestaties van het beslag, voortplantingsparameters of het dierenwelzijn. De werkwijze kan worden aangetoond met kortermijnonderzoeken naar de werkzaamheid of laboratoriumonderzoek waarbij relevante eindpunten worden gemeten. |
|
2) |
Toevoegingsmiddelen die het milieueffect van de dierlijke productie gunstig beïnvloeden. Van toevoegingsmiddelen met een gunstig milieueffect (bv. lagere uitscheiding van stikstof of fosfor, lagere methaanproductie, bijsmaken) kan de werkzaamheid bij de doelsoort worden aangetoond door drie kortermijnonderzoeken naar de werkzaamheid bij dieren waarbij duidelijk een gunstig effect blijkt. Bij de onderzoeken moet rekening worden gehouden met een mogelijke aanpassingsreactie op het toevoegingsmiddel. |
4.1.5. Sectie V: plan voor monitoring na het in de handel brengen
Deze sectie is van toepassing overeenkomstig artikel 7, lid 3, onder g), van Verordening (EG) nr. 1831/2003.
4.2. Zoötechnische toevoegingsmiddelen: micro-organismen en enzymen
4.2.1. Sectie I: samenvatting van het dossier
De volledige sectie I van bijlage II is van toepassing.
4.2.2. Sectie II: identiteit en kenmerken van en gebruiksvoorwaarden voor het toevoegingsmiddel; analysemethoden
De volledige sectie II van bijlage II is van toepassing.
4.2.3. Sectie III: onderzoek naar de werkzaamheid van het toevoegingsmiddel
|
4.2.3.1. |
Onderzoek naar de veiligheid van het gebruik van het toevoegingsmiddel voor de doeldieren De volledige subsectie 3.1.1 van bijlage II is van toepassing. De aanvragers worden aangemoedigd om in de experimentele groep zo mogelijk een honderdvoudige overdosis te gebruiken om het aantal vereiste eindpunten dienovereenkomstig te verminderen. Daartoe kan een geconcentreerde vorm van het toevoegingsmiddel worden gebruikt. De concentratie kan worden aangepast door de hoeveelheid draagstof te verminderen, maar de verhouding tussen de actieve stof of het agens en de overige gistingsproducten moet dezelfde blijven als in het eindproduct. In het geval van enzymen moet het voeder een geschikt substraat bevatten. De volledige subsectie 3.1.2 van bijlage II is van toepassing op alle micro-organismen en op enzymen die een rechtstreeks katalytisch effect op micro-organismen hebben of volgens de aanvraag op een andere wijze de darmflora beïnvloeden. Indien er sprake is van nieuwe blootstelling of een aanzienlijk sterkere blootstelling aan micro-organismen, kan aanvullend onderzoek nodig zijn om aan te tonen dat deze geen negatief effect op de commensale darmflora heeft. Bij herkauwers zijn rechtstreekse tellingen van micro-organismen alleen nodig indien er aanwijzingen zijn van een negatief effect op de herkauwfunctie (in vitro gemeten als wijziging van de concentratie vluchtige vetzuren, daling van de propionaatconcentratie of verminderde celluloseafbraak). |
|
4.2.3.2. |
Onderzoek naar de veiligheid van het gebruik van het toevoegingsmiddel voor de consumenten
Enzymen en micro-organismen vormen slechts een deel van het toevoegingsmiddel, dat meestal ook andere, van het gistingsproces afkomstige bestanddelen bevat. Daarom moet worden onderzocht of het toevoegingsmiddel geen mutagene of andere stoffen bevat die schadelijk kunnen zijn voor consumenten die voedsel eten dat afkomstig is van dieren die voeder of water hebben gekregen dat met het toevoegingsmiddel behandeld is. Levensvatbare bacteriën die bestemd zijn om direct of indirect door zoogdieren (waaronder de mens) te worden gegeten, worden echter meestal geselecteerd uit groepen organismen met een geschiedenis van blijkbaar veilig gebruik of met welomschreven toxische risico's. Ook de risico's van de micro-organismen die momenteel voor de productie van enzymen worden gebruikt, zijn meestal goed bekend en worden door moderne productiemethoden aanzienlijk beperkt. Daarom wordt toxiciteitsonderzoek (bv. naar orale toxiciteit of genotoxiciteit) niet nodig geacht voor enzymen uit microbiële bronnen en voor micro-organismen met een geschiedenis van blijkbaar veilig gebruik waarbij de bestanddelen van het gistingsproces welomschreven en bekend zijn. In het geval van levende organismen en organismen die voor de productie van enzymen worden gebruikt, moeten echter altijd de specifieke aandachtspunten van subsectie 2.2.2.2 van bijlage II worden onderzocht. Indien het om een nieuw organisme of een nieuwe toepassing gaat en indien er te weinig over de biologie van het (productie)organisme bekend is om mogelijke productie van toxische metabolieten uit te sluiten, moet onderzoek naar de genotoxiciteit en de orale toxiciteit worden verricht op het toevoegingsmiddel dat levensvatbare micro-organismen of enzymen bevat. In dit geval gaat het meer bepaald om onderzoek naar de genotoxiciteit met inbegrip van de mutageniteit en onderzoek naar de subchronische orale toxiciteit. Aanbevolen wordt om dit onderzoek uit te voeren met de celvrije gistingsbouillon of, in het geval van vastestoffermentatie, met een geschikt extract. |
|
4.2.3.3. |
Onderzoek naar de veiligheid van het toevoegingsmiddel voor gebruikers/werknemers De volledige subsectie 3.3 van bijlage II is van toepassing.
|
|
4.2.3.4. |
Onderzoek naar de veiligheid van het toevoegingsmiddel voor het milieu De volledige subsectie 3.4 van bijlage II is volledig van toepassing op micro-organismen die niet uit de darm afkomstig zijn en niet ubiquitair zijn in het milieu. |
4.2.4. Sectie IV: onderzoek naar de werkzaamheid van het toevoegingsmiddel
De volledige sectie IV van bijlage II is van toepassing.
|
1) |
Toevoegingsmiddelen die de dierlijke productie, prestaties of welzijn gunstig beïnvloeden en functionele groep „andere zoötechnische toevoegingsmiddelen”. De werkzaamheid kan alleen worden aangetoond met betrekking tot elke doelsoort of -categorie. Naargelang van de eigenschappen van het toevoegingsmiddel kunnen de resultaten worden gemeten aan de hand van prestatiekenmerken (bv. voederrendement, gemiddelde dagelijkse gewichtstoename, stijging van de productie van de dieren), de samenstelling van het karkas, de prestaties van het beslag, voortplantingsparameters of het dierenwelzijn. De werkwijze kan worden aangetoond met kortermijnonderzoeken naar de werkzaamheid of laboratoriumonderzoek waarbij relevante eindpunten worden gemeten. |
|
2) |
Toevoegingsmiddelen die het milieueffect van de dierlijke productie gunstig beïnvloeden. Van toevoegingsmiddelen met een gunstig milieueffect (bv. lagere uitscheiding van stikstof of fosfor, lagere methaanproductie, bijsmaken) kan de werkzaamheid bij de doelsoort worden aangetoond door drie kortermijnonderzoeken naar de werkzaamheid bij dieren waarbij duidelijk een gunstig effect blijkt. Bij de onderzoeken moet rekening worden gehouden met een mogelijke aanpassingsreactie op het toevoegingsmiddel. |
4.2.5. Sectie V: plan voor monitoring na het in de handel brengen
Deze sectie is van toepassing overeenkomstig artikel 7, lid 3, onder g), van Verordening (EG) nr. 1831/2003.
5. COCCIDIOSTATICA EN HISTOMONOSTATICA
5.1. Sectie I: samenvatting van het dossier
De volledige sectie I van bijlage II is van toepassing.
5.2. Sectie II: identiteit en kenmerken van en gebruiksvoorwaarden voor het toevoegingsmiddel; analysemethoden
De volledige sectie II van bijlage II is van toepassing.
5.3. Sectie III: onderzoek naar de werkzaamheid van het toevoegingsmiddel
5.3.1. Onderzoek naar de veiligheid van het gebruik van het toevoegingsmiddel voor de doeldieren
De volledige subsectie 3.1 van bijlage II is van toepassing.
5.3.2. Onderzoek naar de veiligheid van het gebruik van het toevoegingsmiddel voor de consumenten
De volledige subsectie 3.2 van bijlage II is van toepassing.
5.3.3. Onderzoek naar de veiligheid van het gebruik van het toevoegingsmiddel voor gebruikers/werknemers
De volledige subsectie 3.3 van bijlage II is van toepassing.
5.3.4. Onderzoek naar de veiligheid van het gebruik van het toevoegingsmiddel voor het milieu
De volledige subsectie 3.4 van bijlage II is van toepassing.
5.4. Sectie IV: onderzoek naar de werkzaamheid van het toevoegingsmiddel
Deze toevoegingsmiddelen beschermen dieren tegen de gevolgen van besmetting met Eimeria spp. of Histomonas meleagridis. Er moet aandacht worden besteed aan het aantonen van de specifieke effecten van het toevoegingsmiddel (bv. desbetreffende soort) en de profylactische eigenschappen (bv. vermindering van morbiditeit, mortaliteit, oöcystentelling en lesiescore). Zo nodig moet informatie over de effecten op de groei en de voederconversie (mestpluimvee, opfokleghennen en konijnen) of het uitkomstpercentage (vermeerderingspluimvee) worden verstrekt.
Om de vereiste gegevens over de werkzaamheid te verkrijgen, moeten drie soorten proeven op doeldieren worden uitgevoerd:
|
— |
kunstmatige enkelvoudige en gecombineerde infecties; |
|
— |
natuurlijke/kunstmatige infectie om gebruiksomstandigheden te simuleren; |
|
— |
reële gebruiksomstandigheden bij praktijkproeven. |
De proeven met kunstmatige enkelvoudige en gecombineerde infecties (bv. pluimvee in batterijkooien) zijn bedoeld om de relatieve werkzaamheid tegen de parasieten aan te tonen en hoeven niet te worden herhaald. Bij onderzoek onder gesimuleerde gebruiksomstandigheden (bv. pluimvee in grondhokken, konijnen in batterijkooien) zijn drie significante resultaten vereist. Er zijn ook drie praktijkproeven met een zekere mate van natuurlijke infectie vereist.
5.5. Sectie V: plan voor monitoring na het in de handel brengen
Deze sectie is van toepassing overeenkomstig artikel 7, lid 3, onder g), van Verordening (EG) nr. 1831/2003.
6. EXTRAPOLATIE VAN GANGBARE NAAR MINDER GANGBARE DIERSOORTEN
Minder gangbare soorten zijn gedefinieerd in artikel 1, lid 2, van deze verordening.
Voor een voorgestelde uitbreiding van het toegelaten gebruik tot een diersoort die fysiologisch weinig verschilt van een diersoort waarvoor het gebruik van het toevoegingsmiddel al is toegelaten, volstaan normaliter beperktere gegevens.
Onderstaande vereisten gelden alleen voor aanvragen voor vergunningen voor minder gangbare soorten voor toevoegingsmiddelen die reeds zijn toegelaten voor gangbare soorten. Op vergunningen voor nieuwe toevoegingsmiddelen die alleen voor minder gangbare soorten worden aangevraagd, zijn alle secties volledig van toepassing, afhankelijk van de categorie of functionele groep van het toevoegingsmiddel (zie de overeenkomstige specifieke vereisten van bijlage III).
6.1. Sectie I: samenvatting van het dossier
De volledige sectie I van bijlage II is van toepassing.
6.2. Sectie II: identiteit en kenmerken van en gebruiksvoorwaarden voor het toevoegingsmiddel; analysemethoden
Sectie II van bijlage II is als volgt van toepassing:
|
— |
op toevoegingsmiddelen waarvan de vergunning aan een specifieke vergunninghouder is verleend, is de volledige sectie II van toepassing; |
|
— |
op andere toevoegingsmiddelen zijn de punten 2.1.2, 2.1.3, 2.1.4, 2.1.4.2, 2.2, 2.3.1, 2.3.2, 2.4.1, 2.4.2, 2.4.4, 2.5 en 2.6 van toepassing. |
6.3. Sectie III: onderzoek naar de veiligheid van het gebruik van het toevoegingsmiddel
6.3.1. Onderzoek naar de veiligheid van het gebruik van het toevoegingsmiddel voor de doeldieren
|
6.3.1.1. |
Tolerantie bij de doelsoorten De vereisten voor de verschillende categorieën en functionele groepen toevoegingsmiddelen zijn van toepassing. In principe is geen tolerantieonderzoek bij minder gangbare soorten vereist indien het toevoegingsmiddel een ruime veiligheidsmarge (ten minste factor tien) heeft bij fysiologisch vergelijkbare gangbare soorten. Indien bij drie gangbare doelsoorten (waaronder niet-herkauwers, herkauwers en pluimvee) een vergelijkbare, ruime veiligheidsmarge wordt vastgesteld, is geen extra tolerantieonderzoek vereist voor fysiologisch niet vergelijkbare minder gangbare soorten (bv. paarden of konijnen). Indien tolerantieonderzoek vereist is, moet het onderzoek bij minder gangbare soorten (met uitzondering van konijnen) ten minste 28 dagen duren bij dieren in de groei en 42 bij volwassen dieren. Voor konijnen geldt de volgende duur: mestkonijnen: 28 dagen, fokkonijnen: één cyclus (vanaf de inseminatie totdat de jongen gespeend zijn). Als de aanvraag wordt ingediend voor konijntjes die gezoogd worden en gespeende konijnen, wordt een periode van 49 dagen (vanaf één week na de geboorte) voldoende geacht. Het moerkonijn moet tot het spenen worden onderzocht. Onderzoek bij vissen (met uitzondering van zalmachtigen) moet 90 dagen duren. |
6.3.2. Onderzoek naar de veiligheid van het gebruik van het toevoegingsmiddel voor de consumenten
|
6.3.2.1. |
Metabolismeonderzoek De vereisten voor de verschillende categorieën en functionele groepen toevoegingsmiddelen zijn van toepassing. Metabolismeonderzoek is niet vereist indien het toevoegingsmiddel reeds mag worden gebruikt bij een soort die fysiologisch vergelijkbaar is met de minder gangbare soort waarvoor de vergunning is aangevraagd. Indien de soorten niet fysiologisch vergelijkbaar zijn, wordt een vergelijking tussen de metabolismeprofielen aan de hand van in-vitro-onderzoek (bv. in hepatocyten met merkstof) voldoende geacht om de metabolische verwantschap te beoordelen. Indien de minder gangbare soort niet fysiologisch vergelijkbaar is met een gangbare soort, moet het metabolisme van het toevoegingsmiddel worden onderzocht bij de minder gangbare soort. |
|
6.3.2.2. |
Residuonderzoek Indien de metabolische verwantschap wordt vermeld of aangetoond, is alleen een kwantificatie van het indicatorresidu in eetbare weefsels en producten vereist. In andere gevallen is de volledige subsectie 3.2.1.2 van bijlage II van toepassing. |
|
6.3.2.3. |
Beoordeling van de veiligheid voor de consument Voorstel voor maximumresidugehalten (MRL's) Bij de vaststelling van MRL's kan ervan worden uitgegaan dat het gehalte aan residuen in eetbare weefsels niet wezenlijk verschilt tussen minder gangbare en gangbare soorten. De MRL's kunnen als volgt binnen klassen dieren worden geëxtrapoleerd:
MRL's voor paarden kunnen worden geëxtrapoleerd als er MRL's voor een gangbare herkauwer en een gangbare niet-herkauwer bestaan. Indien identieke MRL's zijn afgeleid bij gangbare soorten met een verschillend metabolisch vermogen en een verschillende weefselsamenstelling, zoals runderen (of schapen), varkens en kippen (of pluimvee), mogen dezelfde MRL's ook worden vastgesteld voor schapen, paarden en konijnen. Dit betekent dat extrapolatie mogelijk wordt geacht voor alle voedselproducerende dieren behalve vissen. Volgens het richtsnoer van het Comité voor geneesmiddelen voor diergeneeskundig gebruik (10) voor de vaststelling van MRL's voor zalmachtigen en andere vissen mogen MRL's in spierweefsel van een gangbare soort reeds naar zalmachtigen en andere vissen worden geëxtrapoleerd mits de moederstof aanvaardbaar is als indicatorresidu voor de MRL in spierweefsel en huid. De MRL's mogen bijgevolg naar alle voedselproducerende dieren worden geëxtrapoleerd. Er moeten analysemethoden beschikbaar zijn voor de controle op residuen in eetbare weefsels en producten van alle voedselproducerende dieren. |
6.3.3. Onderzoek naar de veiligheid van het gebruik van het toevoegingsmiddel voor gebruikers/werknemers
De volledige subsectie 3.3 van bijlage II is van toepassing.
6.3.4. Onderzoek naar de veiligheid van het gebruik van het toevoegingsmiddel voor het milieu
De beoordeling van het milieurisico mag worden geëxtrapoleerd uit de beoordeling voor de fysiologisch vergelijkbare gangbare soort. Voor toevoegingsmiddelen die voor konijnen bestemd zijn, is de volledige sectie van toepassing, rekening houdend met de vereisten voor de verschillende categorieën en functionele groepen toevoegingsmiddelen.
6.4. Sectie IV: onderzoek naar de werkzaamheid van het toevoegingsmiddel
Indien het toevoegingsmiddel reeds is toegelaten voor een fysiologisch vergelijkbare soort en voor dezelfde functie en indien de werkwijze van het toevoegingsmiddel bekend of aangetoond is, volstaat het dezelfde werkwijze bij de minder gangbare soort aan te tonen om de werkzaamheid aan te tonen. Indien geen dergelijk verband kan worden gelegd, moet de werkzaamheid worden aangetoond volgens de algemene voorschriften van sectie IV van bijlage II. In sommige gevallen kan het aangewezen zijn diersoorten in hetzelfde productiestadium te combineren (bv. geiten en schapen voor melkproductie). De significantie moet in elk onderzoek (p ≤ 0,1) of, indien mogelijk, door een meta-analyse (p ≤ 0,05) worden aangetoond.
Indien de werkzaamheid moet worden aangetoond, moet de duur van het werkzaamheidsonderzoek overeenkomen met de vergelijkbare productiestadia van de fysiologisch vergelijkbare gangbare soort. In andere gevallen moet de minimale duur van het onderzoek voldoen aan de voorschriften van subsectie 4.4 van bijlage II en bijlage IV.
6.5. Sectie V: plan voor monitoring na het in de handel brengen
Deze sectie is van toepassing overeenkomstig artikel 7, lid 3, onder g), van Verordening (EG) nr. 1831/2003.
7. GEZELSCHAPSDIEREN EN ANDERE NIET-VOEDSELPRODUCERENDE DIEREN
Gezelschapsdieren en andere niet-voedselproducerende dieren zijn gedefinieerd in artikel 1, lid 1, van deze verordening.
7.1. Sectie I: samenvatting van het dossier
De volledige sectie I van bijlage II is van toepassing.
7.2. Sectie II: identiteit en kenmerken van en gebruiksvoorwaarden voor het toevoegingsmiddel; analysemethoden
Sectie II van bijlage II is als volgt van toepassing:
|
— |
op toevoegingsmiddelen waarvan de vergunning aan een specifieke vergunninghouder is verleend, is de volledige sectie II van toepassing; |
|
— |
op andere toevoegingsmiddelen zijn de punten 2.1.2, 2.1.3, 2.1.4, 2.1.4.2, 2.2, 2.3.1, 2.3.2, 2.4.1, 2.4.2, 2.4.4, 2.5 en 2.6 van toepassing. |
7.3. Sectie III: onderzoek naar de werkzaamheid van het toevoegingsmiddel
7.3.1. Onderzoek naar de veiligheid van het gebruik van het toevoegingsmiddel voor de doeldieren
De vereisten voor de verschillende categorieën en functionele groepen toevoegingsmiddelen zijn van toepassing. Indien een tolerantieonderzoek vereist is, moet dat minimaal 28 dagen duren.
Een tolerantieonderzoek is niet vereist indien is aangetoond dat het toevoegingsmiddel bij drie gangbare soorten (waaronder niet-herkauwers, herkauwers en pluimvee) een vergelijkbare en ruime veiligheidsmarge heeft.
7.3.2. Onderzoek naar de veiligheid van het gebruik van het toevoegingsmiddel voor de consumenten
Deze subsectie is meestal niet van toepassing. Wel moet er aandacht worden besteed aan de veiligheid van de eigenaar.
7.3.3. Onderzoek naar de veiligheid van het gebruik van het toevoegingsmiddel voor gebruikers/werknemers
De volledige subsectie 3.3 van bijlage II is van toepassing.
7.3.4. Onderzoek naar de veiligheid van het gebruik van het toevoegingsmiddel voor het milieu
Subsectie 3.4 van bijlage II is niet van toepassing.
7.4. Sectie IV: onderzoek naar de werkzaamheid van het toevoegingsmiddel
De vereisten voor de verschillende categorieën en functionele groepen toevoegingsmiddelen zijn van toepassing.
Indien een toevoegingsmiddel waarvoor onderzoek bij dieren vereist is, reeds is toegelaten voor andere, fysiologisch vergelijkbare soorten, hoeft de werkzaamheid niet meer te worden aangetoond, mits het beoogde effect en de werkwijze identiek zijn. Indien het toevoegingsmiddel nog niet toegelaten is of indien het beoogde effect of de werkwijze afwijken van de eerdere vergunning, moet de werkzaamheid worden aangetoond volgens de algemene voorschriften van sectie IV van bijlage II.
Het langetermijnwerkzaamheidsonderzoek moet ten minste 28 dagen duren.
7.5. Sectie V: plan voor monitoring na het in de handel brengen
Deze sectie is van toepassing overeenkomstig artikel 7, lid 3, onder g), van Verordening (EG) nr. 1831/2003.
8. TOEVOEGINGSMIDDELEN DIE REEDS IN LEVENSMIDDELEN MOGEN WORDEN GEBRUIK
8.1. Sectie I: samenvatting van het dossier
De volledige sectie I van bijlage II is van toepassing.
8.2. Sectie II: identiteit en kenmerken van en gebruiksvoorwaarden voor het toevoegingsmiddel; analysemethoden
Sectie II van bijlage II is als volgt van toepassing:
|
— |
op toevoegingsmiddelen waarvan de vergunning aan een specifieke vergunninghouder is verleend, is de volledige sectie II van toepassing; |
|
— |
op andere toevoegingsmiddelen zijn de punten 2.1.2, 2.1.3, 2.1.4, 2.1.4.2, 2.2, 2.3.1, 2.3.2, 2.4.1, 2.4.2, 2.4.4, 2.5 en 2.6 van toepassing. |
8.3. Sectie III: onderzoek naar de werkzaamheid van het toevoegingsmiddel
De recentste formele beoordelingen van de veiligheid van het toevoegingsmiddel moeten worden bijgevoegd, aangevuld met eventuele latere gegevens.
Voor toevoegingsmiddelen die in de EU zonder beperkingen zijn toegelaten als additief in of bestanddeel van levensmiddelen, is onderzoek naar de veiligheid voor consumenten en werknemers gewoonlijk niet nodig.
De subsecties 3.1, 3.2 en 3.3 van bijlage II zijn van toepassing, waarbij rekening moet worden gehouden met de huidige kennis over de veiligheid van het gebruik van deze stoffen in levensmiddelen. Dienovereenkomstig kunnen deze stoffen die ook in levensmiddelen worden gebruikt, als volgt worden ingedeeld:
|
— |
ADI niet gespecificeerd (zonder uitdrukkelijke vermelding van de „upper limit of intake”, voor stoffen met een zeer lage toxiciteit): |
|
— |
ADI of UL vastgesteld, of |
|
— |
geen ADI toegewezen (dit geldt voor stoffen waarvan de veiligheid niet kan worden beoordeeld bij gebrek aan informatie). |
8.3.1. Onderzoek naar de veiligheid van het gebruik van het toevoegingsmiddel voor de doeldieren
Indien in diervoeder en levensmiddelen een vergelijkbare hoeveelheid van het toevoegingsmiddel wordt gebruikt, mag de veiligheid voor de doelsoort worden gebaseerd op de beschikbare toxicologische in-vivogegevens, de chemische structuur en de metabole capaciteit van de doelsoort. Indien in levensmiddelen een aanzienlijk grotere hoeveelheid wordt gebruikt dan in diervoeder, kan, afhankelijk van de soort stof, een tolerantieonderzoek bij de doelsoort vereist zijn.
8.3.2. Onderzoek naar de veiligheid van het gebruik van het toevoegingsmiddel voor de consumenten
Indien het gebruik van het toevoegingsmiddel resulteert in een grotere blootstelling van de consument of een ander metabolietenpatroon dan bij gebruik in levensmiddelen, moeten verdere gegevens over toxicologie en residuen worden verstrekt.
|
8.3.2.1. |
Levensmiddelenadditieven waarvoor geen ADI is gespecificeerd De veiligheid voor de consument hoeft niet te worden beoordeeld, behalve indien het gebruik van het toevoegingsmiddel resulteert in een ander metabolietenpatroon dan bij gebruik in levensmiddelen. |
|
8.3.2.2. |
Levensmiddelenadditieven waarvoor een ADI of UL is vastgesteld De veiligheid voor de consument moet worden beoordeeld, rekening houdend met de extra blootstelling door het gebruik in diervoeder of de specifieke blootstelling door de metabolieten die in de doelsoort ontstaan. Daartoe kunnen literatuurgegevens over residuen worden geëxtrapoleerd. Indien residuonderzoek vereist is, mag dat beperkt blijven tot een vergelijking van het gehalte in weefsels/producten in een onbehandelde groep en in een groep die de hoogste aangegeven dosis heeft gekregen. |
|
8.3.2.3. |
Levensmiddelenadditieven waaraan geen ADI is toegewezen Er moet duidelijk worden vermeld waarom er geen ADI is toegewezen. Indien dat reden tot bezorgdheid geeft en indien het gebruik van het toevoegingsmiddel in diervoeder de blootstelling van de consument aanzienlijk zou doen stijgen, is een volledige toxicologische beoordeling vereist. De extra blootstelling door het gebruik in diervoeder kan worden geëxtrapoleerd uit literatuurgegevens over residuen. Indien residuonderzoek vereist is, mag dat beperkt blijven tot een vergelijking van het gehalte in weefsels/producten in een onbehandelde groep en in een groep die de hoogste aangegeven dosis heeft gekregen. |
8.3.3. Onderzoek naar de veiligheid van het gebruik van het toevoegingsmiddel voor gebruikers/werknemers
De volledige subsectie 3.3 van bijlage II is van toepassing.
Bij de beoordeling van de veiligheid van het toevoegingsmiddel voor de gebruikers moet rekening worden gehouden met de voorzorgsmaatregelen bij het hanteren waarin is voorzien voor het gebruik van dit toevoegingsmiddel in levensmiddelen.
8.3.4. Onderzoek naar de veiligheid van het gebruik van het toevoegingsmiddel voor het milieu
Subsectie 3.4 van bijlage II is van toepassing.
8.4. Sectie IV: onderzoek naar de werkzaamheid van het toevoegingsmiddel
Indien in diervoeder dezelfde functie wordt beoogd als in levensmiddelen, hoeft de werkzaamheid niet meer te worden aangetoond. Zo niet zijn de vereisten met betrekking tot de werkzaamheid van sectie IV van bijlage II van toepassing.
8.5. Sectie V: plan voor monitoring na het in de handel brengen
Deze sectie is van toepassing overeenkomstig artikel 7, lid 3, onder g), van Verordening (EG) nr. 1831/2003.
9. WIJZIGING VAN VERGUNNINGEN
Aangezien gebruik kan worden gemaakt van de beoordeling van de gegevens die voor een eerdere vergunning zijn verstrekt, hoeft een dossier voor een aanvraag overeenkomstig artikel 13, lid 3, van Verordening (EG) nr. 1831/2003 alleen aan de onderstaande vereisten te voldoen.
In een aanvraag voor een wijziging van elementen zoals de identificatie, de karakterisering en de gebruiksvoorwaarden in een bestaande vergunning voor een toevoegingsmiddel moet worden aangetoond dat deze wijziging geen nadelig effect heeft op de doelsoort, de consument, de gebruikers en het milieu. Een toevoegingsmiddel kan in dit opzicht als identiek worden beschouwd als de werkzame stof(fen) of het agens (de agentia) en de gebruiksvoorwaarden dezelfde zijn, als de zuiverheid in wezen gelijk is en als er geen nieuwe bestanddelen aan zijn toegevoegd die een reden tot bezorgdheid kunnen zijn. Voor dergelijke producten mag een verkorte aanvraag worden ingediend omdat het normaliter niet nodig is het onderzoek naar de veiligheid voor de doelsoort, de consument, de gebruikers en het milieu en het werkzaamheidsonderzoek te herhalen.
De aanvraag moet aan de volgende vereisten voldoen:
|
1. |
de volledige bijlage I is van toepassing — dit omvat gedetailleerde gegevens over de aangevraagde wijziging; |
|
2. |
de volledige sectie II van bijlage II is van toepassing; |
|
3. |
er moeten gegevens worden verstrekt waaruit blijkt dat de chemische of biologische kenmerken van het toevoegingsmiddel in wezen gelijk zijn aan die van het reeds toegelaten product; |
|
4. |
zo nodig moet de biologische equivalentie worden aangetoond, hetzij door specificatie, hetzij met gepubliceerde literatuur of specifieke onderzoeken. Indien de biologische equivalentie niet volledig is aangetoond, moet worden aangetoond dat de wachttijd in overeenstemming is met de MRL; |
|
5. |
er moet worden aangetoond dat het toevoegingsmiddel in het licht van de huidige wetenschappelijke kennis onder de goedgekeurde voorwaarden nog steeds veilig is voor de doelsoort, de consument, de gebruikers en het milieu; |
|
6. |
indien in de vergunning monitoringvoorschriften zijn opgenomen, moet een verslag worden ingediend van de resultaten van de monitoring na het in de handel brengen, en |
|
7. |
de specifieke gegevens ter ondersteuning van de aanvraag voor een wijziging moeten worden ingediend overeenkomstig de desbetreffende delen van de secties III, IV en V van bijlage II. |
10. VERLENGING VAN VERGUNNINGEN
Aanvragen voor een verlenging van een vergunning overeenkomstig artikel 14 van Verordening (EG) nr. 1831/2003 moeten aan de volgende vereisten voldoen:
10.1. Sectie I: samenvatting van het dossier
De volledige sectie I van bijlage II is van toepassing. Een kopie van de oorspronkelijke EG-vergunning voor het in de handel brengen of de laatste verlenging van die vergunning moet worden bijgevoegd. Er moet een geactualiseerd dossier worden opgesteld overeenkomstig de recentste vereisten en er moet een lijst worden ingediend van alle wijzigingen sinds de oorspronkelijke vergunning of de laatste verlenging daarvan. De aanvrager moet een samenvatting van het dossier indienen waarin hij het onderwerp van de aanvraag vermeldt en eventuele nieuwe informatie over de identiteit en de veiligheid verstrekt die sinds de vorige vergunning of verlenging beschikbaar is gekomen.
10.2. Sectie II: identiteit en kenmerken van en gebruiksvoorwaarden voor het toevoegingsmiddel; analysemethoden
Sectie II van bijlage II is als volgt van toepassing:
|
— |
op toevoegingsmiddelen waarvan de vergunning aan een specifieke vergunninghouder is verleend, is de volledige sectie II van toepassing; |
|
— |
op andere toevoegingsmiddelen zijn de punten 2.1.2, 2.1.3, 2.1.4, 2.1.4.2, 2.2, 2.3.1, 2.3.2, 2.4.1, 2.4.2, 2.4.4, 2.5 en 2.6 van toepassing. |
Er moet worden aangetoond dat de samenstelling, zuiverheid en activiteit van het toevoegingsmiddel niet wezenlijk zijn gewijzigd of veranderd ten opzichte van het toevoegingsmiddel waarvoor een vergunning is verleend. Veranderingen in het fabricageprocedé moeten worden vermeld.
10.3. Sectie III: onderzoek naar de werkzaamheid van het toevoegingsmiddel
Er moet worden aangetoond dat het toevoegingsmiddel in het licht van de huidige kennis onder de goedgekeurde voorwaarden nog steeds veilig is voor de doelsoort, de consument, de gebruikers en het milieu. Er moet een veiligheidsupdate worden verstrekt voor de periode sinds de oorspronkelijke vergunning of de laatste verlenging daarvan, met informatie over de volgende punten:
|
— |
rapportage over nadelige effecten, waaronder ongelukken (voordien onbekende effecten, ernstige effecten van ongeacht welke aard, verhoogde incidentie van bekende effecten), op de doeldieren, de consumenten, de gebruikers en het milieu. Deze rapportage moet de aard van het effect, het aantal getroffen personen of organismen, de afloop, de gebruiksvoorwaarden en een beoordeling van het oorzakelijk verband omvatten; |
|
— |
rapportage van voordien onbekende interacties en kruisbesmettingen; |
|
— |
indien van toepassing, gegevens over de controle op residuen; |
|
— |
gegevens van epidemiologisch en/of toxicologisch onderzoek; |
|
— |
eventuele andere informatie over de veiligheid van het toevoegingsmiddel en de risico's van het toevoegingsmiddel voor dieren, mensen en het milieu. |
Indien over een of meer van deze punten geen nadere informatie wordt verstrekt, moet dat duidelijk worden gemotiveerd.
Indien in de eerdere vergunning monitoringvoorschriften zijn opgenomen, moeten de resultaten van de monitoring na het in de handel brengen worden gerapporteerd.
Indien de aanvraag tot verlenging van de vergunning overeenkomstig artikel 14, lid 2, onder d), van Verordening (EG) nr. 1831/2003 een voorstel tot wijziging of aanvulling van de voorwaarden van de oorspronkelijke vergunning, onder andere de voorwaarden die verband houden met de toekomstige monitoring, inhoudt, moeten de specifieke gegevens ter ondersteuning van het voorstel tot wijziging worden ingediend overeenkomstig de desbetreffende delen van de secties III, IV en V van bijlage II.
11. HERBEOORDELING VAN TOEVOEGINGSMIDDELEN WAARVOOR REEDS BIJ RICHTLIJN 70/524/EEG EEN VERGUNNING IS VERLEEND
Dit punt 11 is van toepassing op toevoegingsmiddelen waarvoor bij Richtlijn 70/524/EEG een vergunning is verleend en die overeenkomstig artikel 10, lid 2, van Verordening (EG) nr. 1831/2003 herbeoordeeld moeten worden. Het gaat om toevoegingsmiddel van de volgende groepen:
|
— |
antioxidanten; |
|
— |
aromatische en eetlustopwekkende stoffen; |
|
— |
emulgatoren, stabilisatoren, verdikkingsmiddelen en geleermiddelen; |
|
— |
kleurstoffen, met inbegrip van pigmenten; |
|
— |
conserveermiddelen; |
|
— |
vitaminen, provitaminen en chemisch duidelijk omschreven stoffen met een gelijkaardige werking; |
|
— |
sporenelementen; |
|
— |
bindmiddelen, antiklontermiddelen en coagulanten; |
|
— |
zuurteregelaars, en |
|
— |
stoffen die complexen vormen met radionucliden. |
De vereiste grondigheid en kwaliteit van de risicobeoordeling voor deze toevoegingsmiddelen is dezelfde als voor andere toevoegingsmiddelen. Aangezien deze toevoegingsmiddelen reeds lange tijd veilig worden gebruikt, mogen, overeenkomstig de bepalingen van deze verordening, gegevens uit reeds gepubliceerde onderzoeken worden gebruikt om aan te tonen dat het toevoegingsmiddel nog steeds veilig is voor de doelsoort, de consument, de gebruikers en het milieu.
11.1. Sectie I: samenvatting van het dossier
De volledige sectie I van bijlage II is van toepassing.
11.2. Sectie II: identiteit en kenmerken van en gebruiksvoorwaarden voor het toevoegingsmiddel; analysemethoden
Sectie II van bijlage II is als volgt van toepassing:
|
— |
op toevoegingsmiddelen waarvan de vergunning niet aan een specifieke vergunninghouder is verleend, zijn de punten 2.1.2, 2.1.3, 2.1.4, 2.1.4.2, 2.2, 2.3.1, 2.3.2, 2.4.1, 2.4.2, 2.4.4, 2.5 en 2.6 van toepassing; |
|
— |
op andere toevoegingsmiddelen, waarvan de vergunning aan een specifieke vergunninghouder is verleend, is de volledige sectie II van toepassing. |
11.3. Sectie III: onderzoek naar de werkzaamheid van het toevoegingsmiddel
Indien de veiligheid van een toevoegingsmiddel voor de doelsoort, de consument, gebruikers/werknemers en het milieu reeds is beoordeeld, moet een samenvatting van de veiligheidsonderzoeken die voor de eerdere vergunning zijn ingediend, alsook eventuele nieuwe informatie die sindsdien is verkregen, worden ingediend. Indien de veiligheid van het gebruik van het toevoegingsmiddel in diervoeder niet formeel is beoordeeld, mogen onderzoeken en gegevens uit de wetenschappelijke literatuur worden gebruikt, mits deze gelijkwaardig zijn aan de onderzoeken en gegevens die voor een nieuwe aanvraag vereist zijn. Zo niet moet een volledige reeks veiligheidsonderzoeken worden ingediend.
11.4. Sectie IV: onderzoek naar de werkzaamheid van het toevoegingsmiddel
In voorkomend geval mag ander materiaal dan onderzoeken, met name over een lange gebruiksgeschiedenis, worden ingediend om aan te tonen dat aan de vereisten met betrekking tot de werkzaamheid van artikel 5, lid 3, van Verordening (EG) nr. 1831/2003 is voldaan.
11.5. Sectie V: plan voor monitoring na het in de handel brengen
Deze sectie is van toepassing overeenkomstig artikel 7, lid 3, onder g), van Verordening (EG) nr. 1831/2003.
(1) Een lichaamsvreemde stof is een chemische stof die geen natuurlijk bestanddeel is van het organisme dat eraan wordt blootgesteld. Het kan ook gaan om stoffen die in veel hogere concentraties dan gewoonlijk aanwezig zijn.
(2) In deze verordening betekent „inkuilproces” het proces waarbij de aantasting van organisch materiaal wordt beperkt door aanzuring in anaërobe omstandigheden, hetzij door gisting, hetzij door toevoeging van inkuiltoevoegingsmiddelen.
(3) Identificatienummer voor chemisch gedefinieerde aroma's dat wordt gebruikt in FLAVIS (Flavour Information System), de EU-databank die wordt gebruikt in het kader van Verordening (EG) nr. 1565/2000 van de Commissie van 18 juli 2000 (PB L 180 van 19.7.2000, blz. 8) tot vaststelling van de maatregelen die vereist zijn voor de vaststelling van een beoordelingsprogramma in toepassing van Verordening (EG) nr. 2232/96 van het Europees Parlement en de Raad (PB L 299 van 23.11.1996, blz. 1).
(4) CoE-nr.: nummer van de Raad van Europa dat voor botanisch gedefinieerde aromastoffen wordt gebruikt in Verslag nr. 1 van de Raad van Europa over „Natural sources of flavourings”, deel I, Straatsburg, 2000, en de daaropvolgende delen.
(5) CAS-nummer (CAS-nr.): Chemical Abstracts Service Registry Number, uniek identificatienummer voor chemische stoffen dat algemeen wordt gebruikt in lijsten van chemische stoffen.
(6) In deze sectie van deze verordening betekent „stof van toxicologisch belang” een stof met een toelaatbare dagelijkse of wekelijkse inname (TDI of TWI), een ADI, een gebruiksbeperking of een werkzaam bestanddeel zoals gedefinieerd in Richtlijn 88/388/EEG van de Raad van 22 juni 1988 betreffende de onderlinge aanpassing van de wetgevingen der lidstaten inzake aroma's voor gebruik in levensmiddelen en de uitgangsmaterialen voor de bereiding van die aroma's.
(7) Gedefinieerd in aanhangsel 4 bij Verslag nr. 1 van de Raad van Europa over „Natural sources of flavourings”, deel I, Straatsburg, 2000.
(8) JECFA (FAO/WHO, 1996, Food additive series 35, IPCS, WHO Geneva). De overeenkomstige drempel voor de doeldieren moet worden aangepast aan het gewicht van het dier en de voederinname.
(9) PB L 180 van 19.7.2000, blz. 8.
(10) „Note for guidance of the establishment of maximum residue limits for Salmonidae and other fin fish”. Europees Bureau voor de geneesmiddelenbeoordeling, eenheid Evaluatie van geneesmiddelen voor diergeneeskundig gebruik, EMEA/CVMP/153b/97-FINAL.
BIJLAGE IV
Categorieën en definities van doeldieren en minimale duur van de werkzaamheidsonderzoeken
1. Tabel. Diercategorieën: varkens
|
Categorie |
Definitie van de diercategorie |
Duur (ongeveer) (gewicht/leeftijd) |
Minimale duur van langetermijnonderzoeken naar de werkzaamheid |
||
|
Periode/leeftijd |
Leeftijd |
Gewicht |
|||
|
Speenvarkens |
Jonge varkens die worden gezoogd |
Vanaf de geboorte |
Tot 21 à 42 dagen |
Tot 6 à 11 kg |
14 dagen |
|
Gespeende biggen |
Jonge varkens die niet meer worden gezoogd en worden opgefokt tot fok- of vleesvarkens |
Vanaf 21 à 42 dagen |
Tot 120 dagen |
Tot 35 kg |
42 dagen |
|
Speenvarkens en gespeende biggen |
Jonge varkens, vanaf de geboorte, die worden opgefokt tot fok- of vleesvarkens |
Vanaf de geboorte |
Tot 120 dagen |
Tot 35 kg |
58 dagen |
|
Mestvarkens |
Varkens die niet meer worden gezoogd en bestemd zijn voor vleesproductie, tot de dag waarop zij naar het slachthuis worden vervoerd |
Vanaf 60 à 120 dagen |
Tot 120 à 250 dagen (of volgens plaatselijke gewoonte) |
80 à 150 kg (of volgens plaatselijke gewoonte) |
Tot slachtgewicht, maar niet minder dan 70 dagen |
|
Fokzeugen |
Zeugen die minstens eenmaal geïnsemineerd of gedekt zijn |
Vanaf de eerste inseminatie |
|
|
Vanaf de inseminatie totdat de tweede worp gespeend is (twee cycli) |
|
Zeugen, ten behoeve van de biggen |
Vrouwelijke varkens die minstens eenmaal geïnsemineerd of gedekt zijn |
|
|
|
Vanaf ten minste twee weken voor het werpen totdat de biggen gespeend zijn |
2. Tabel. Diercategorieën: pluimvee
|
Categorie |
Definitie van de diercategorie |
Duur (ongeveer) (gewicht/leeftijd) |
Minimale duur van langetermijnonderzoeken naar de werkzaamheid |
||
|
Periode |
Leeftijd |
Gewicht |
|||
|
Mestkippen |
Dieren die worden vetgemest |
Vanaf het uitkomen |
Tot 35 dagen |
Tot ongeveer 1 600 g (maximaal 2 kg) |
35 dagen |
|
Opfokleghennen |
Vrouwelijke dieren die worden opgefokt voor de productie van consumptie-eieren of voor fokdoeleinden |
Vanaf het uitkomen |
Tot ongeveer 16 weken (maximaal 20 weken) |
— |
112 dagen (indien geen gegevens over de werkzaamheid beschikbaar zijn voor mestkippen) |
|
Legkippen |
Productieve vrouwelijke dieren die voor de productie van eieren worden gehouden |
Vanaf 16 à 21 dagen |
Tot ongeveer 13 maanden (maximaal 18 maanden) |
Vanaf 1 200 g (wit)/1 400 g (bruin) |
168 dagen |
|
Mestkalkoenen |
Dieren die worden vetgemest |
Vanaf het uitkomen |
Tot ongeveer 14 weken (maximaal 20 weken) Tot ongeveer 16 weken (maximaal 24 weken) |
Vrouwtjes: tot ongeveer 7 000 g (maximaal 10 000 g) Mannetjes: tot ongeveer 12 000 g (maximaal 20 000 g) |
84 dagen |
|
Fokkalkoenen |
Vrouwelijke en mannelijke dieren die voor fokdoeleinden worden gehouden |
Gehele periode |
Van 30 weken tot ongeveer 60 weken |
Vrouwtjes: vanaf ongeveer 15 000 g Mannetjes: vanaf ongeveer 30 000 g |
Ten minste 6 maanden |
|
Kalkoenen die worden opgefokt voor fokdoeleinden |
Jonge vrouwelijke en mannelijke dieren die worden opgefokt voor fokdoeleinden |
Vanaf het uitkomen |
Tot 30 weken |
Vrouwtjes: tot ongeveer 15 000 g Mannetjes: tot ongeveer 30 000 g |
Gehele periode (indien geen gegevens over de werkzaamheid beschikbaar zijn voor mestkalkoenen) |
3. Tabel. Diercategorieën: runderen (als huisdier gehouden runderen met inbegrip van de soorten Bubalus en Bison)
|
Categorie |
Definitie van de diercategorie |
Duur (ongeveer) (gewicht/leeftijd) |
Minimale duur van langetermijnonderzoeken naar de werkzaamheid |
||
|
Periode |
Leeftijd |
Gewicht |
|||
|
Opfokkalveren |
Kalveren die worden opgefokt voor fokdoeleinden of voor de productie van rundvlees |
Vanaf de geboorte |
Tot 4 maanden |
Tot 60 à 80 kg (maximaal 145 kg) |
56 dagen |
|
Mestkalveren |
Kalveren voor de productie van kalfsvlees |
Vanaf de geboorte |
Tot 6 maanden |
Tot 180 kg (maximaal 250 kg) |
Tot het slachten, maar niet minder dan 84 dagen |
|
Mestrunderen |
Runderen die niet meer worden gezoogd en bestemd zijn voor vleesproductie, tot de dag waarop zij naar het slachthuis worden vervoerd |
Vanaf de volledige ontwikkeling van de herkauwfunctie |
Tot 10 à 36 maanden |
Maximaal 350 à 700 kg |
168 dagen |
|
Melkkoeien voor melkproductie |
Vrouwelijke runderen die ten minste één kalf hebben gekregen |
|
|
|
84 dagen (volledige lactatieperiode rapporteren) |
|
Fokkoeien |
Vrouwelijke runderen die minstens eenmaal geïnsemineerd of gedekt zijn |
Vanaf de eerste inseminatie totdat de tweede worp gespeend is |
|
|
Twee cycli (indien de reproductieparameters vereist zijn) |
4. Tabel. Diercategorieën: schapen
|
Categorie |
Definitie van de diercategorie |
Duur (ongeveer) (gewicht/leeftijd) |
Minimale duur van langetermijnonderzoeken naar de werkzaamheid |
||
|
Periode |
Leeftijd |
Gewicht |
|||
|
Opfoklammeren |
Lammeren die worden opgefokt voor fokdoeleinden |
Vanaf de geboorte |
Tot 3 maanden |
15 à 20 kg |
56 dagen |
|
Mestlammeren |
Lammeren die worden opgefokt voor de productie van lamsvlees |
Vanaf de geboorte |
Tot 6 maanden (of ouder) |
Tot 55 kg |
Tot slachtgewicht, maar niet minder dan 56 dagen |
|
Melkschapen (voor melkproductie) |
Schapen die ten minste één lam hebben gekregen |
|
|
|
84 dagen (volledige lactatieperiode rapporteren) |
|
Fokooien |
Vrouwelijke schapen die minstens eenmaal geïnsemineerd of gedekt zijn |
Vanaf de eerste inseminatie totdat de tweede worp gespeend is |
|
|
Twee cycli (indien de reproductieparameters vereist zijn) |
5. Tabel. Diercategorieën: geiten
|
Categorie |
Definitie van de diercategorie |
Duur (ongeveer) (gewicht/leeftijd) |
Minimale duur van langetermijnonderzoeken naar de werkzaamheid |
||
|
Periode |
Leeftijd |
Gewicht |
|||
|
Opfokgeitenlammeren |
Geitenlammeren die worden opgefokt voor fokdoeleinden |
Vanaf de geboorte |
Tot 3 maanden |
15 à 20 kg |
Ten minste 56 dagen |
|
Mestgeitenlammeren |
Geitenlammeren die worden opgefokt voor de productie van geitenvlees |
Vanaf de geboorte |
Tot 6 maanden |
|
Ten minste 56 dagen |
|
Melkgeiten (voor melkproductie) |
Geiten die ten minste een lam hebben gekregen |
|
|
|
84 dagen (volledige lactatieperiode rapporteren) |
|
Fokgeiten |
Geiten die minstens eenmaal geïnsemineerd of gedekt zijn |
Vanaf de eerste inseminatie totdat de tweede worp gespeend is |
|
|
Twee cycli (indien de reproductieparameters vereist zijn) |
6. Tabel. Diercategorieën: vissen
|
Categorie |
Definitie van de diercategorie |
Duur (ongeveer) (gewicht/leeftijd) |
Minimale duur van langetermijnonderzoeken naar de werkzaamheid |
||
|
Periode |
Leeftijd |
Gewicht |
|||
|
Zalm en forel |
|
|
|
200 à 300 g |
90 dagen of tot het oorspronkelijke lichaamsgewicht is verdubbeld |
|
Zalm en forel |
Broedvissen |
Zo dicht mogelijk bij de paaitijd |
|
|
90 dagen |
7. Tabel. Diercategorieën: konijnen
|
Categorie |
Definitie van de diercategorie |
Duur (ongeveer) (gewicht/leeftijd) |
Minimale duur van langetermijnonderzoeken naar de werkzaamheid |
||
|
Periode |
Leeftijd |
Gewicht |
|||
|
Konijntjes die gezoogd worden en gespeende konijnen |
|
Vanaf één week na de geboorte |
|
|
56 dagen |
|
Mestkonijnen |
Konijnen die worden opgefokt voor de productie van vlees |
Na het spenen |
Tot 8 à 11 weken |
|
42 dagen |
|
Fokkonijnen (voor voortplantingsdoeleinden) |
Vrouwelijke konijnen die minstens eenmaal geïnsemineerd of gedekt zijn |
Vanaf de inseminatie totdat de tweede worp gespeend is |
|
|
Twee cycli (indien de reproductieparameters vereist zijn) |
|
Fokkonijnen (ten behoeve van de jongen) |
Vrouwelijke konijnen die minstens eenmaal geïnsemineerd zijn |
Vanaf de eerste inseminatie |
|
|
Vanaf ten minste twee weken voor het werpen totdat de biggen gespeend zijn (bv. voor micro-organismen) |
8. Tabel. Diercategorieën: paarden
|
Categorie |
Definitie van de diercategorie |
Duur (ongeveer) (gewicht/leeftijd) |
Minimale duur van langetermijnonderzoeken naar de werkzaamheid |
||
|
Periode |
Leeftijd |
Gewicht |
|||
|
Paarden |
Alle categorieën |
|
|
|
56 dagen |