This document is an excerpt from the EUR-Lex website
Document 02009R0152-20170524
Commission Regulation (EC) No 152/2009 of 27 January 2009 laying down the methods of sampling and analysis for the official control of feed (Text with EEA relevance)
Consolidated text: Kommissionens förordning (EG) nr 152/2009 av den 27 januari 2009 om provtagnings- och analysmetoder för offentlig kontroll av foder (Text av betydelse för EES)
Kommissionens förordning (EG) nr 152/2009 av den 27 januari 2009 om provtagnings- och analysmetoder för offentlig kontroll av foder (Text av betydelse för EES)

02009R0152 — SV — 24.05.2017 — 006.001
Den här texten är endast avsedd som ett dokumentationshjälpmedel och har ingen rättslig verkan. EU-institutionerna tar inget ansvar för innehållet. De autentiska versionerna av motsvarande rättsakter, inklusive ingresserna, publiceras i Europeiska unionens officiella tidning och finns i EUR-Lex. De officiella texterna är direkt tillgängliga via länkarna i det här dokumentet
|
KOMMISSIONENS FÖRORDNING (EG) nr 152/2009 av den 27 januari 2009 om provtagnings- och analysmetoder för offentlig kontroll av foder (EGT L 054 26.2.2009, s. 1) |
Ändrad genom:
|
|
|
Officiella tidningen |
||
|
nr |
sida |
datum |
||
|
KOMMISSIONENS FÖRORDNING (EU) nr 278/2012 av den 28 mars 2012 |
L 91 |
8 |
29.3.2012 |
|
|
KOMMISSIONENS FÖRORDNING (EU) nr 51/2013 av den 16 januari 2013 |
L 20 |
33 |
23.1.2013 |
|
|
KOMMISSIONENS FÖRORDNING (EU) nr 691/2013 av den 19 juli 2013 |
L 197 |
1 |
20.7.2013 |
|
|
KOMMISSIONENS FÖRORDNING (EU) nr 709/2014 av den 20 juni 2014 |
L 188 |
1 |
27.6.2014 |
|
|
L 92 |
35 |
6.4.2017 |
||
|
L 115 |
22 |
4.5.2017 |
||
KOMMISSIONENS FÖRORDNING (EG) nr 152/2009
av den 27 januari 2009
om provtagnings- och analysmetoder för offentlig kontroll av foder
(Text av betydelse för EES)
Artikel 1
Provtagning för offentlig kontroll av foder, särskilt när det gäller bestämning av beståndsdelar, inbegripet material som innehåller eller består av eller har framställts av genetiskt modifierade organismer (GMO), fodertillsatser enligt definitionen i Europaparlamentets och rådets förordning (EG) nr 1831/2003 ( 1 ) och främmande ämnen enligt definitionen i Europaparlamentets och rådets direktiv 2002/32/EG ( 2 ) ska utföras enligt de metoder som anges i bilaga I.
Den provtagningsmetod som anges i bilaga I är tillämplig på kontroll av foder vad gäller bestämning av bekämpningsmedelsrester enligt definitionen i Europaparlamentets och rådets förordning (EG) nr 396/2005 ( 3 ) och kontrollen av efterlevnaden av förordning (EU) nr 619/2011.
Artikel 2
Beredning av analysprov och resultatangivelse ska följa metoderna i bilaga II.
Artikel 3
Analyser för offentlig kontroll av foder ska utföras enligt metoderna i bilaga III (Analysmetoder för kontroll av foderråvarors och foderblandningars sammansättning), bilaga IV (Analysmetoder för kontroll av halten godkända fodertillsatser), bilaga V (Analysmetoder för kontroll av främmande ämnen i foder) och bilaga VI (Analysmetoder för bestämning av beståndsdelar av animaliskt ursprung för offentlig kontroll av foder).
Artikel 4
Energivärdet i foderblandningar till fjäderfä ska beräknas i enlighet med bilaga VII.
Artikel 5
De analysmetoder för kontroll av förekomst av otillåtna fodertillsatser som anges i bilaga VIII ska användas som konfirmeringsmetoder.
Artikel 6
Direktiven 71/250/EEG, 71/393/EEG, 72/199/EEG, 73/46/EEG, 76/371/EEG, 76/372/EEG, 78/633/EEG, 81/715/EEG, 84/425/EEG, 86/174/EEG, 93/70/EEG, 93/117/EG, 98/64/EG, 1999/27/EG, 1999/76/EG, 2000/45/EG, 2002/70/EG och 2003/126/EG ska upphöra att gälla.
Hänvisningar till de upphävda direktiven ska anses som hänvisningar till denna förordning och ska läsas i enlighet med jämförelsetabellerna i bilaga IX.
Artikel 7
Denna förordning träder i kraft den tjugonde dagen efter det att den har offentliggjorts i Europeiska unionens officiella tidning.
Den ska tillämpas från och med den 26 augusti 2009.
Denna förordning är till alla delar bindande och direkt tillämplig i alla medlemsstater.
BILAGA I
PROVTAGNINGSMETODER
1. SYFTE OCH TILLÄMPNINGSOMRÅDE
Prov som är avsedda för offentlig kontroll av foder ska tas med de metoder som beskrivs nedan. Prov som har tagits på detta sätt ska anses vara representativa för provmängderna.
Syftet med representativ provtagning är att ta ut en liten fraktion av ett parti på ett sådant sätt att bestämningen av en viss egenskap hos denna fraktion representerar medelvärdet av denna egenskap i partiet. Partiet ska provtas genom att det upprepade gånger tas delprov på olika enskilda positioner i partiet. Dessa delprov ska blandas så att det bildas ett enda samlingsprov, från vilket representativa slutliga prov ska beredas genom representativ delning.
Om det vid en okulärbesiktning visar sig att delar av det foder som ska provtas skiljer sig i kvalitet från resten av fodret från samma parti, ska sådana delar skiljas från resten av fodret och behandlas som ett separat delparti. Om det inte är möjligt att dela upp fodret i separata delpartier ska fodret provtas som ett parti. I sådana fall ska detta nämnas i provtagningsrapporten.
Om det har konstaterats att ett foder som provtagits i enlighet med bestämmelserna i denna förordning inte uppfyller EU-kraven, och detta ingår i ett foderparti av samma kategori eller varuslag, ska man anta att inget foder i detta parti uppfyller kraven, utom om man efter en utförlig bedömning kan konstatera att det inte finns några belägg för att resten av partiet inte uppfyller EU-kraven.
2. DEFINITIONER
|
— |
parti (eller sats) : en identifierad mängd foder som konstaterats ha gemensamma egenskaper som ursprung, sort, förpackningsmetod, förpackare, avsändare eller märkning; när det gäller en produktionsprocess: en produktionsenhet från ett och samma tillverkningsställe som framställts med samma produktionsparametrar, eller ett antal sådana enheter som producerats samtidigt och lagrats tillsammans. |
|
— |
provmängd : ett parti eller en identifierad del av partiet eller delpartiet. |
|
— |
förseglat prov : ett prov som förseglats på sådant sätt att det blir omöjligt att skaffa sig tillträde till provet om inte förseglingen bryts eller avlägsnas. |
|
— |
delprov : en viss mängd som tagits från ett ställe i provmängden. |
|
— |
samlingsprov : en blandning av delprov som tagits från samma provmängd. |
|
— |
reducerat prov : en del av samlingsprovet som erhållits ur detta genom representativ reduktion. |
|
— |
slutligt prov : en del av det reducerade provet eller av det homogeniserade samlingsprovet. |
|
— |
laboratorieprov : ett prov som är avsett för laboratoriet (såsom det tagits emot av laboratoriet) och som kan vara det slutliga provet, det reducerade provet eller samlingsprovet. |
3. ALLMÄNNA BESTÄMMELSER
— Provtagningspersonal: Proven ska tas av personer som den behöriga myndigheten har bemyndigat för detta.
— Provet ska förseglas på sådant sätt att det blir omöjligt att skaffa sig tillträde till provet om inte förseglingen bryts eller avlägsnas. Förseglingens märke bör vara klart identifierbart och synligt. Alternativt kan provet placeras i ett kärl som kan tillslutas på ett sådant sätt att det inte kan öppnas utan att kärlet eller behållaren oåterkalleligt skadas, och återanvändning av kärlet eller behållaren därigenom undviks.
— Identifiering av provet: provet ska vara märkt på ett outplånligt sätt och ska identifieras på ett sådant sätt att det finns en otvetydig koppling till provtagningsrapporten.
— Från varje samlingsprov ska minst två slutliga prov tas: minst ett för kontroll (genomförande) och ett för foderföretagaren (försvar). Slutligen får ett slutligt prov tas som referens. Om hela samlingsprovet homogeniseras, tas de slutliga proven från det homogeniserade samlingsprovet, såvida detta förfarande inte strider mot medlemsstaternas bestämmelser om foderföretagares rättigheter.
4. UTRUSTNING
|
4.1 |
Provtagningsutrustningen ska vara tillverkad av material som inte kan kontaminera de produkter som ska provtas. Utrustning som är avsedd att användas flera gånger ska vara lätt att rengöra för att undvika korskontaminering. |
|
4.2 |
Utrustning som rekommenderas för provtagning på fast foder
4.2.1 Manuell provtagning 4.2.1.1 Flatbottnad skyffel med vertikala sidor
4.2.2 Mekanisk provtagning Lämplig mekanisk utrustning får användas för provtagning på foder i rörelse. Lämplig betyder att prov tas på åtminstone hela sektionen av flödet. Provtagning på foder i rörelse (vid höga flödeshastigheter) kan utföras med automatiska provtagare. 4.2.3 Provdelare Om så är möjligt och lämpligt, bör utrustning avsedd att dela upp provet i ungefär lika stora delar användas för beredning av reducerade prov på ett representativt sätt. |
5. KVANTITATIVA KRAV FÖR ANTALET DELPROV
— De kvantitativa kraven i punkterna 5.1 och 5.2 för antalet delprov är tillämpliga på provmängder i storleken högst 500 ton vilka kan tas på ett representativt sätt. Den beskrivna provtagningsmetoden gäller även större kvantiteter än den föreskrivna maximala provmängdsstorleken, under förutsättning att det högsta antalet delprov som anges i tabellerna nedan lämnas utan avseende och antalet delprov bestäms utifrån den kvadratrotsformel som anges i den relevanta delen av förfarandet (se punkt 5.3) och minimistorleken på samlingsprovet ökas proportionellt. Detta förhindrar inte att ett större parti delas upp i mindre delpartier och att varje delparti provas i enlighet med det förfarande som beskrivs i punkterna 5.1 och 5.2.
— Provmängden ska vara av sådan storlek att prov kan tas i var och en av dess beståndsdelar.
— För mycket stora partier eller delpartier (> 500 ton) och för partier som transporteras eller lagras på sådant sätt att provtagning inte kan ske enligt det provtagningsförfarande som anges i punkt 5.1 och 5.2 i detta kapitel ska det provtagningsförfarande som anges i punkt 5.3 användas.
— Om en foderföretagare enligt lag är skyldig att följa denna förordning inom ramen för ett obligatoriskt övervakningssystem, får foderföretagaren avvika från de kvantitativa krav som föreskrivs i detta kapitel för att ta hänsyn till operativa egenskaper, förutsatt att foderföretagaren på tillfredsställande sätt har visat för den behöriga myndigheten att provtagningsmetoden är likvärdig i fråga om representativitet och efter godkännande av den behöriga myndigheten.
— Om det inte är möjligt att använda den angivna provtagningsmetoden vad gäller de kvantitativa kraven beroende på oacceptabla kommersiella skador på partiet (på grund av förpackningstyper, transportmedel, lagring osv.) kan i undantagsfall en alternativ provtagningsmetod användas, förutsatt att den ger så representativa resultat som möjligt och att den beskrivs och dokumenteras till fullo.
5.1 Kvantitativa krav för delprov vid kontroll av ämnen eller produkter som är jämnt fördelade i fodret
5.1.1 Fast foder i lös vikt
|
Provmängdens storlek |
Minsta antal delprov |
|
≤ 2,5 ton |
7 |
|
> 2,5 ton |
√ 20 gånger det antal ton som utgör provmängden (1), upp till 40 delprov |
|
(*1) Om det erhållna talet inte är ett heltal ska det avrundas till närmaste högre heltal. |
|
5.1.2 Löst flytande foder
|
Provmängdens storlek |
Minsta antal delprov |
|
≤ 2,5 ton eller ≤ 2 500 liter |
4 (1) |
|
> 2,5 ton eller > 2 500 liter |
7 (1) |
|
(*1) Om det inte är möjligt att göra vätskan homogen måste antalet delprov ökas. |
|
5.1.3 Förpackat foder
Foder (fast och flytande) kan förpackas i påsar, säckar, burkar, tunnor etc. som i tabellen anges som enheter. Stora enheter (≥ 500 kg eller liter) ska provtas i enlighet med bestämmelserna för löst foder (se 5.1.1 och 5.1.2.)
|
Provmängdens storlek |
Minsta antal enheter från vilka (minst) ett delprov ska tas (1) |
|
1–20 enheter |
1 enhet (2) |
|
21–150 enheter |
3 enheter (2) |
|
151–400 enheter |
5 enheter (2) |
|
> 400 enheter |
¼ av det √antal enheter som utgör provmängden (3), upp till 40 enheter |
|
(*1) Om öppnande av en enhet kan påverka analysen (t.ex. blötfoder som lätt fördärvas) ska ett delprov utgöras av den oöppnade enheten. (*2) För enheter med ett innehåll på högst 1 kg eller 1 liter ska ett delprov utgöras av innehållet i den ursprungliga enheten. (*3) Om det erhållna talet inte är ett heltal ska det avrundas till närmaste högre heltal. |
|
5.1.4 Foderblock och saltstenar
Prov ska tas på minst ett block eller en sten per provmängd om 25 enheter, som mest fyra block eller stenar.
För block eller stenar på högst 1 kg styck ska ett delprov utgöras av innehållet i ett block eller en sten.
5.1.5 Grovfoder/vallfoder
|
Provmängdens storlek |
Minsta antal delprov (1) |
|
≤ 5 ton |
5 |
|
> 5 ton |
√ 5 gånger det antal ton som utgör provmängden (2), upp till 40 delprov |
|
(*1) Faktum är att i vissa situationer (t.ex. beträffande ensilage) är det inte möjligt att ta de delprov som krävs utan att orsaka oacceptabel skada på partiet. En alternativ provtagningsmetod får användas i sådana situationer och en vägledning för provtagning på sådana partier kommer att utarbetas innan den här förordningen träder i kraft. (*2) Om det erhållna talet inte är ett heltal ska det avrundas till närmaste högre heltal. |
|
5.2 Kvantitativa krav för delprov vid kontroll av beståndsdelar eller ämnen som kan vara ojämnt fördelade i foder
Dessa kvantitativa krav för delprov ska användas i följande situationer:
— Kontroll av aflatoxiner, mjöldryga, andra mykotoxiner och skadliga botaniska orenheter i foderråvaror.
— Kontroll av korskontaminering genom en beståndsdel, inbegripet genetiskt modifierade material eller ett ämne för vilket en ojämn fördelning förväntas i foderråvaror.
Om kontrollmyndigheten har en stark misstanke om att det finns en sådan ojämn fördelning även vid korskontaminering av en beståndsdel eller ett ämne i en foderblandning kan de kvantitativa kraven enligt nedanstående tabell tillämpas.
|
Provmängdens storlek |
Minsta antal delprov |
|
< 80 ton |
Se de kvantitativa kraven i punkt 5.1. Antalet delprov som ska tas ska multipliceras med 2,5. |
|
≥ 80 ton |
100 |
5.3 Kvantitativa krav för delprov i fråga om mycket stora partier
När det gäller stora provmängder (provmängder > 500 ton) är antalet delprov som ska tas = 40 delprov + √ton vid kontroll av ämnen eller produkter som är jämnt fördelade i fodret eller 100 delprov + √ton vid kontroll av beståndsdelar eller ämnen som kan vara ojämnt fördelade i foderråvara.
6. KVANTITATIVA KRAV FÖR SAMLINGSPROV
|
Det krävs ett samlingsprov per provmängd. |
||
|
|
Typ av foder |
|
|
6.1 |
Löst foder |
4 kg |
|
6.2 |
Förpackat foder: |
4 kg (3) |
|
6.3 |
Flytande eller halvflytande foder: |
4 liter |
|
6.4 |
Foderblock eller saltstenar: |
|
|
6.4.1 |
Med en vikt på mer än 1 kg styck |
4 kg |
|
6.4.2 |
Med en vikt på högst 1 kg styck |
vikten av fyra block eller stenar i ursprunglig storlek |
|
6.5 |
Grovfoder/vallfoder |
4 kg (4) |
|
(*1) Om det provtagna fodret har ett högt värde kan en mindre mängd delprov tas, förutsatt att detta beskrivs och dokumenteras i provtagningsrapporten. (*2) Enligt bestämmelserna i kommissionens förordning (EU) nr 619/2011 av den 24 juni 2011 om provtagnings- och analysmetoder för offentlig kontroll av foder vad gäller förekomst av genetiskt modifierat material där godkännandeförfarandet fortfarande pågår eller godkännandet har upphört att gälla (EGT L 166, 25.6.2011, s. 9) ska samlingsprovet för kontroll av förekomst av genetiskt modifierat material innehålla minst 35 000 korn/utsäde. Detta innebär för majs att storleken på samlingsprovet måste vara minst 10,5 kg och för sojabönor 7 kg. För andra utsäden och spannmål som korn, hirs, havre, ris, råg, vete och raps, motsvarar samlingsprovets storlek på 4 kg mer än 35 000 korn/utsäde. (*3) I fråga om förpackat foder är det också möjligt att storleken 4 kg för samlingsprovet inte kan uppnås beroende på de enskilda enheternas storlek. (*4) Om det är fråga om grovfoder eller vallfoder med låg densitet (t.ex. hö och halm) bör samlingsprovet vara minst 1 kg. |
||
7. KVANTITATIVA KRAV FÖR SLUTLIGA PROV
Slutliga prov
Minst ett slutligt prov ska analyseras. Det slutliga prov som analyseras ska vara av minst följande mängd:
|
Fast foder |
|
|
Flytande eller halvflytande foder |
500 ml (1) |
|
(*1) Enligt bestämmelserna i förordning (EU) nr 619/2011 ska det slutliga provet för kontroll av förekomst av genetiskt modifierat material innehålla minst 10 000 korn/utsäde. Detta innebär för majs att storleken på det slutliga provet ska vara minst 3 000 g och för sojabönor 2 000 g. För andra utsäden och spannmål som korn, hirs, havre, ris, råg, vete och raps, motsvarar det slutliga provet storlek på 500 g mer än 10 000 korn/utsäde. (*2) Om samlingsprovet är betydligt mindre än 4 kg eller liter (se fotnoterna under punkt 6) kan också en mindre kvantitet av det slutliga provet tas, förutsatt att detta beskrivs och dokumenteras i provtagningsrapporten. (*3) Vid provtagning på baljväxter, spannmål och trädnötter för bestämning av bekämpningsmedelsrester ska minimistorleken på det slutliga provet vara 1 kg enligt bestämmelserna i kommissionens direktiv 2002/63/EG (EGT L 187, 16.7.2002, s. 30). |
|
8. PROVTAGNINGSMETOD FÖR MYCKET STORA PARTIER ELLER PARTIER SOM LAGRAS OCH TRANSPORTERAS PÅ ETT SÄTT SOM INNEBÄR ATT PROVTAGNING PÅ HELA PARTIET ÄR OMÖJLIG
8.1 Allmänna principer
Om det inte är möjligt att ta delprov på hela partiet på grund av det sätt som partiet transporteras eller lagras på bör provtagning på sådana partier helst ske när partiet är i flöde.
För stora lagerlokaler avsedda för lagring av foder bör företagarna uppmuntras att installera utrustning i lagerlokalen som möjliggör (automatisk) provtagning på hela det lagrade partiet.
Om man använder de provtagningsförfaranden som anges i detta kapitel, ska foderföretagaren eller dennes företrädare informeras om provtagningsförfarandet. Om detta provtagningsförfarande ifrågasätts av foderföretagaren eller dennes företrädare ska foderföretagaren eller dennes företrädare ge den behöriga myndigheten möjlighet att ta prov på hela partiet på dennes bekostnad.
8.2 Stora partier som transporteras med fartyg
8.2.1 Dynamisk provtagning på stora partier som transporteras med fartyg
Provtagning på stora partier på fartyg ska företrädesvis ske när produkten är i flöde (dynamisk provtagning).
Provtagningen ska göras per lastrum (enhet som kan separeras fysiskt). Lastrum töms dock delvis det ena efter det andra, vilket innebär att den ursprungliga fysiska separationen inte längre föreligger efter att lasten överförts till lagringsanläggningar. Provtagning kan därför ske i förhållande till den ursprungliga fysiska separationen eller i förhållande till separationen efter överföring till lagringsanläggningarna.
Lossning av ett fartyg kan pågå i flera dagar. Normalt ska provtagning ske med jämna mellanrum under hela lossningen. Det är dock inte alltid möjligt eller lämpligt att en officiell inspektör är närvarande för provtagning under hela lossningen. Därför får provtagning ske på en del (provmängd) av hela partiet. Antalet delprov bestäms med hänsyn till provmängdens storlek.
Om provtagning utförs på en del av ett foderparti som ingår i samma kategori eller varuslag och det har konstaterats att den delen av partiet inte uppfyller EU-kraven, ska man anta att inget foder i detta parti uppfyller kraven, utom om man efter en utförlig bedömning kan konstatera att det inte finns några belägg för att resten av partiet inte uppfyller EU-kraven.
Även om det officiella provet tas automatiskt är det nödvändigt att en inspektör är närvarande. Om den automatiska provtagningen utförs med förinställda parametrar som inte kan ändras under provtagningen och delproven samlas i ett förseglat kärl, varigenom eventuella bedrägerier förebyggs, krävs det enbart att en inspektör är närvarande i början av provtagningen, varje gång kärlet för proven behöver bytas och i slutet av provtagningen.
8.2.2 Provtagning på partier som transporteras med fartyg genom stationär provtagning
Om provtagningen utförs stationärt ska man tillämpa samma förfarande som föreskrivs för lagringsanläggningar (silor) som kan nås ovanifrån (se punkt 8.4.1).
Provtagningen ska utföras på den tillgängliga delen (ovanifrån) av partiet/rumslasten. Antalet delprov bestäms med hänsyn till provmängdens storlek. Om provtagning utförs på en del av ett foderparti som ingår i samma kategori eller varuslag, och det har konstaterats att den delen av partiet inte uppfyller EU-kraven, ska man anta att inget foder i detta parti uppfyller kraven, utom om man efter en utförlig bedömning kan konstatera att det inte finns några belägg för att resten av partiet inte uppfyller EU-kraven.
8.3 Provtagning på stora partier som förvaras i lager
Provtagningen ska utföras på den åtkomliga delen av partiet. Antalet delprov bestäms med hänsyn till provmängdens storlek. Om provtagning utförs på en del av ett foderparti som ingår i samma kategori eller varuslag, och det har konstaterats att den delen av partiet inte uppfyller EU-kraven, ska man anta att inget foder i detta parti uppfyller kraven, utom om man efter en utförlig bedömning kan konstatera att det inte finns några belägg för att resten av partiet inte uppfyller EU-kraven.
8.4 Provtagning på lagringsanläggningar (silor)
8.4.1 Provtagning på silor som (lätt) kan nås ovanifrån
Provtagningen ska utföras på den åtkomliga delen av partiet. Antalet delprov bestäms med hänsyn till provmängdens storlek. Om provtagning utförs på en del av ett foderparti som ingår i samma kategori eller varuslag, och det har konstaterats att den delen av partiet inte uppfyller EU-kraven, ska man anta att inget foder i detta parti uppfyller kraven, utom om man efter en utförlig bedömning kan konstatera att det inte finns några belägg för att resten av partiet inte uppfyller EU-kraven.
8.4.2 Provtagning på silor som inte kan nås ovanifrån (slutna silor)
8.4.2.1
Prov på foder som lagras i sådana silor kan inte tas statiskt. Om prov måste tas på fodret i silon och det inte finns någon möjlighet att flytta varupartiet ska det därför avtalas med företagaren att han eller hon ska informera inspektören om när silon kommer att tömmas för att göra det möjligt att ta prov på fodret när det är i flöde.
8.4.2.2
Provtagningsförfarandet innebär att en kvantitet av 50–100 kg överförs till ett kärl och att provet tas från detta. Storleken på samlingsprovet motsvarar hela partiet och antalet delprov står i relation till de kvantiteter i silon som överförs till ett kärl för provtagning. Om provtagning utförs på en del av ett foderparti som ingår i samma kategori eller varuslag, och det har konstaterats att den delen av partiet inte uppfyller EU-kraven, ska man anta att inget foder i detta parti uppfyller kraven, utom om man efter en utförlig bedömning kan konstatera att det inte finns några belägg för att resten av partiet inte uppfyller EU-kraven.
8.5 Provtagning på löst foder i stora slutna behållare
Prov på sådana partier kan ofta enbart tas när de lossas. Det är i vissa fall omöjligt att lossa vid importstället eller kontrollpunkten och därför bör provtagningen ske när sådana behållare lossas.
9. ANVISNINGAR OM UTTAGNING, BEREDNING OCH FÖRPACKNING AV PROVEN
9.1 Allmänt
Proven ska tas och beredas utan onödigt dröjsmål med beaktande av de försiktighetsåtgärder som krävs för att produkten varken ska förändras eller kontamineras. Instrument, ytor och behållare som kommer i kontakt med proven ska vara rena och torra.
9.2 Delprov
Delprov ska tas slumpvis i hela provmängden och ha ungefär samma storlek.
Delprovets storlek ska vara minst 100 gram eller, för grovfoder eller vallfoder med låg specifik vikt, 25 gram.
Om färre än 40 delprov ska uttas enligt bestämmelserna om provtagningsförfarande i punkt 8 ska delprovens storlek fastställas i förhållande till den storlek som krävs för samlingsprovet (se punkt 6).
Om det utförs provtagning på små partier av förpackat foder, där det enligt de kvantitativa kraven ska uttas ett begränsat antal delprov, ska ett delprov utgöras av innehållet i en ursprunglig enhet med ett innehåll på högst 1 kg eller 1 liter.
Om det utförs provtagning på förpackat foder som består av små enheter (t.ex. < 250 g) beror delprovets storlek på enhetens storlek.
9.2.1 Löst foder
Provtagningen kan, om så är lämpligt, göras medan provmängden är i rörelse (vid lastning eller lossning).
9.2.2 Förpackat foder
När det antal enheter som krävs för provtagning enligt kapitel 5 har valts ut ska en del av innehållet i varje enhet tas ut med spjut eller skyffel. Vid behov tas proven efter det att enheterna har tömts separat.
9.2.3 Flytande eller halvflytande foder som är homogent eller kan homogeniseras
När det antal enheter som krävs för provtagning enligt kapitel 5 har valts ut ska innehållet vid behov homogeniseras och en mängd tas från varje enhet.
Delproven får tas när innehållet lossas.
9.2.4 Flytande eller halvflytande foder som inte kan homogeniseras
När det antal enheter som krävs för provtagning enligt kapitel 5 har valts ut ska prov tas från olika nivåer.
Proven får också tas när innehållet lossas, men de första fraktionerna ska då kastas.
I båda fallen ska den totala volymen vara minst tio liter.
9.2.5 Foderblock och saltstenar
När det antal block eller stenar som krävs för provtagning enligt kapitel 5 har valts ut får en del av varje block eller sten tas ut. Om det finns en misstanke om att ett block eller en sten inte är homogen får hela blocket eller stenen tas ut som prov.
För block eller stenar på högst 1 kg styck ska ett delprov utgöras av innehållet i ett block eller en sten.
9.3 Beredning av samlingsprov
Delproven ska blandas till ett enda samlingsprov.
9.4 Beredning av slutliga prov
Materialet i samlingsprovet ska blandas omsorgsfullt ( 4 ):
— Varje prov ska placeras i en lämplig behållare/ett lämpligt kärl. Alla nödvändiga försiktighetsåtgärder ska vidtas för att undvika att provets sammansättning förändras eller att provet kontamineras eller förfalskas under transport och lagring.
— Vid kontroll av beståndsdelar eller ämnen som är jämnt fördelade i fodret, får samlingsprovet reduceras representativt till minst 2,0 kg eller 2,0 liter (reducerat prov) ( 5 ), helst med antingen mekanisk eller automatisk delare. Vid kontroll av förekomsten av bekämpningsmedelsrester i ärtväxter, sädeskorn och trädnötter ska det reducerade provet vara minst 3 kg. Om fodrets beskaffenhet inte medger användning av delare eller delaren inte är tillgänglig, får provet reduceras med kvarteringsmetoden. Av de reducerade proven ska de slutliga proven (för kontroll, försvar och referens) därefter beredas av ungefär samma mängd och ska därvid följa de kvantitativa kraven i kapitel 7. Vid kontroll av beståndsdelar, inklusive genetiskt modifierade material eller ämnen som kan vara ojämnt fördelade i foderråvaror, ska samlingsprovet vara:
—
— helt homogeniserat och därefter delat i slutliga prov, eller
— reducerat till minst 2 kg eller 2 liter ( 6 ) med mekanisk eller automatisk delare. Endast om fodrets beskaffenhet inte medger användning av delare får provet vid behov reduceras med kvarteringsmetoden. För kontroll av förekomst av genetiskt modifierat material enligt förordning (EU) nr 619/2011 ska det reducerade provet innehålla minst 35 000 korn/utsäde för att göra det möjligt att erhålla de slutliga proven för genomförande, försvar och referens på minst 10 000 korn/utsäde (se fotnot (**) i kapitel 6 och fotnot (*) i kapitel 7).
9.5 Förpackning av prov
Behållarna eller förpackningarna ska förseglas och märkas på sådant sätt att de inte kan öppnas utan att förseglingen skadas. Hela märkningen ska ingå i förseglingen.
9.6 Sändning av proven till laboratoriet
Provet ska utan onödigt dröjsmål sändas till det utsedda analyslaboratoriet tillsammans med de upplysningar som den som utför analysen behöver.
10. REGISTRERING AV PROV
Alla prov ska registreras så att varje provmängd och dess storlek entydigt kan identifieras.
I registret ska också anges varje avvikelse från det provtagningsförfarande som anges i den här förordningen.
Förutom att registret ska hållas tillgängligt för det officiella kontrollaboratoriet, ska det hållas tillgängligt för foderföretagaren och/eller det laboratorium som utsetts av foderföretagaren.
BILAGA II
ALLMÄNNA BESTÄMMELSER OM ANALYSMETODER FÖR FODER
A. BEREDNING AV ANALYSPROV
1. Syfte
De metoder som beskrivs nedan gäller beredning för analys av prov som skickats till kontrollaboratorierna efter provtagning i enlighet med bilaga I.
Laboratorieproven ska beredas så att den uppvägda mängden enligt analysmetoden är homogen och representativ för de slutliga proven.
2. Försiktighetsåtgärder
Vilken provberedningsmetod som ska användas beror på analysmetoden och de beståndsdelar eller ämnen som ska kontrolleras. Det är därför viktigt att man ser till att provberedningsmetoden lämpar sig för den analysmetod som används och för de beståndsdelar eller ämnen som ska kontrolleras.
Alla nödvändiga moment ska utföras på ett sådant sätt att man i görligaste mån undviker att provet kontamineras eller att dess sammansättning ändras.
Malning, blandning och siktning bör göras utan dröjsmål så att provet i minsta möjliga utsträckning utsätts för luft och ljus. Kvarnar och rivare som avsevärt kan hetta upp provet bör inte användas.
Malning för hand rekommenderas för foder som är särskilt känsliga för värme. Man bör också se till att apparaten inte i sig själv är en källa till kontaminering.
Om provet inte kan beredas utan betydande förändringar av dess vattenhalt ska vattenhalten före och efter beredningen bestämmas med den metod som anges i bilaga III del A.
3. Förfarande
3.1 Allmänt förfarande
Testdelprovet tas från det slutliga provet. Koning och kvartering rekommenderas inte eftersom det kan ge testdelproven större neddelningsfel.
3.1.1
— Blanda det siktade slutliga provet och placera det i en lämplig ren, torr behållare med lufttät förslutning. Blanda provet igen, för att garantera fullständig homogenisering, i direkt samband med invägning för analys (testdelprov).
3.1.2
— Om inte annat anges i metodbeskrivningen torkas det slutliga provet till en vattenhalt på 8–12 % genom den förberedande torkning som görs vid bestämning av vattenhalt enligt bilaga III del A punkt 4.3. Fortsätt sedan enligt anvisningarna i 3.1.1.
3.1.3
— Placera det slutliga provet i en lämplig ren, torr behållare med lufttät förslutning. Blanda provet omsorgsfullt, för att garantera fullständig homogenisering, i direkt anslutning till invägning för analys (testdelprov).
3.1.4
— Slutliga prov som inte kan beredas enligt någon av de föregående metoderna ska behandlas med någon annan metod som garanterar att de provportioner som vägs in för analys (testdelprov) är homogena och representativa för det slutliga provet.
3.2 Särskilt förfarande vid undersökning genom okulärbesiktning eller genom mikroskopi eller om hela samlingsprovet homogeniserats
— Vid en undersökning genom okulärbesiktning (utan att använda mikroskop) används hela laboratorieprovet för undersökningen.
— Vid undersökning i mikroskop får laboratoriet reducera samlingsprovet eller ytterligare reducera det reducerade provet. De slutliga proven för försvars- och eventuellt referensändamål tas enligt ett förfarande som motsvarar förfarandet för det slutliga provet för genomförande.
— Om hela samlingsprovet homogeniseras ska de slutliga proven tas från det homogeniserade samlingsprovet.
4. Provförvaring
Proven ska förvaras vid en temperatur som inte påverkar deras sammansättning. Prov avsedda för analys av vitaminer eller ämnen som är särskilt ljuskänsliga ska förvaras under sådana förhållanden att provet inte påverkas negativt av ljus.
B. BESTÄMMELSER OM REAGENS OCH UTRUSTNING FÖR ANALYS
1. Om inget annat anges för analysmetoden ska alla reagens vara analysrena (p.a.). Vid spåranalys ska reagensens renhet kontrolleras mot ett blankprov. Beroende på resultatet kan det krävas ytterligare rening av reagensen.
2. Om det i metodbeskrivningen inte anges vilken typ av lösnings- eller spädningsmedel som ska användas för beredning av lösningar, spädning, sköljning eller tvättning ska vatten användas. I allmänhet ska vattnet vara demineraliserat eller destillerat. I särskilda fall, som anges i metodbeskrivningarna, ska vattnet behandlas med särskilda reningsmetoder.
3. Eftersom viss utrustning normalt sett förekommer på kontrollaboratorier, nämns i metodbeskrivningarna endast instrument och apparatur som är speciella eller som ska användas på ett speciellt sätt. Instrument och apparatur ska vara rena, särskilt när mycket små mängder ska bestämmas.
C. VAL AV ANALYSMETOD OCH RESULTATANGIVELSE
1. Extraktion
I flera fall anges en specifik extraktionsmetod för en viss analysmetod. I allmänhet kan andra extraktionsmetoder än den som finns i metodbeskrivningen användas om det kan visas att den använda extraktionsmetoden har samma extraktionseffektivitet för den analyserade matrisen som den metod som anges för analysmetoden.
2. Rening
I många fall anges en specifik reningsmetod för en viss analysmetod. I allmänhet kan andra reningsmetoder än den som finns i metodbeskrivningen användas om det kan visas att den använda reningsmetoden ger samma analysresultat för den analyserade matrisen som den metod som anges för analysmetoden.
3. Antal bestämningar
När det gäller analys av främmande ämnen gäller att om resultatet från den första bestämningen är betydligt (> 50 %) lägre än den specifikation som ska kontrolleras så behövs ingen ytterligare bestämning, förutsatt att lämpliga kvalitetsförfaranden används. I andra fall behövs två analyser (en andra bestämning) för att utesluta möjligheten till intern korskontaminering eller oavsiktlig hopblandning av prov. Medelvärdet av de två bestämningarna, där mätosäkerheten beaktas, används för kontroll av att kraven är uppfyllda.
I fråga om kontroll av det deklarerade innehållet av ett ämne eller en beståndsdel gäller att om resultatet från den första bestämningen bekräftar det deklarerade innehållet, dvs. analysresultatet hamnar inom ett intervall för acceptabel variation jämfört med det deklarerade innehållet, behövs ingen ytterligare bestämning, förutsatt att lämpliga kvalitetsförfaranden används. I andra fall behövs två analyser (en andra bestämning) för att utesluta möjligheten till intern korskontaminering eller oavsiktlig hopblandning av prov. Medelvärdet av de två bestämningarna, där mätosäkerheten beaktas, används för kontroll av att kraven är uppfyllda.
I en del fall definieras detta acceptabla variationsintervall, t.ex. i Europaparlamentets och rådets förordning (EG) nr 767/2009 av den 13 juli 2009 om utsläppande på marknaden och användning av foder, om ändring av Europaparlamentets och rådets förordning (EG) nr 1831/2003 och om upphävande av rådets direktiv 79/373/EEG, kommissionens direktiv 80/511/EEG, rådets direktiv 82/471/EEG, 83/228/EEG, 93/74/EEG, 93/113/EG och 96/25/EG samt kommissionens beslut 2004/217/EG ( 7 ).
4. Angivande av analysmetod som använts
I analysrapporten ska anges vilken analysmetod som använts.
5. Rapportering av analysresultat
Analysresultatet ska uttryckas med lämpligt antal värdesiffror på det sätt som anges i metodbeskrivningen och ska vid behov korrigeras för vattenhalten i det slutliga provet före beredning.
6. Mätosäkerhet och utbyte vid analys av främmande ämnen
I fråga om främmande ämnen enligt direktiv 2002/32/EG ska en produkt avsedd som foder inte anses uppfylla kraven på högsta tillåtna halt om analysresultatet, för foder med en vattenhalt på 12 %, bedöms överstiga högsta tillåtna halt med hänsyn tagen till den utvidgade mätosäkerheten och korrigering för utbytet. För att bedöma om kraven uppfylls använder man den analyserade halten, korrigerad för utbyte, minus den utvidgade mätosäkerheten. Denna metod kan bara användas om det går att uppskatta mätosäkerheten och korrigering för utbyte med hjälp av analysmetoden (den kan exempelvis inte användas för mikroskopisk analys).
Analysresultatet ska rapporteras enligt följande (om analysmetoden gör det möjligt att uppskatta mätosäkerhet och utbyte):
a) Korrigerat för utbyte, med uppgift om hur stort utbytet är. Korrigering är inte nödvändig om utbytet är 90–110 %.
b) Som x +/– U, där x är analysresultatet och U den utvidgade mätosäkerheten, beräknad med en täckningsfaktor på 2, vilket ger en konfidensgrad på ca 95 %.
Om analysresultatet är betydligt (> 50 %) lägre än den specifikation som ska kontrolleras, och förutsatt att lämpliga kvalitetsförfaranden använts och att analysen endast syftar till att kontrollera om fodret uppfyller kraven i lagstiftningen, får analysresultatet rapporteras utan korrigering för utbyte. Utbyte och mätosäkerhet behöver inte anges i dessa fall.
BILAGA III
ANALYSMETODER FÖR KONTROLL AV FODERRÅVARORS OCH FODERBLANDNINGARS SAMMANSÄTTNING
A. BESTÄMNING AV VATTENHALT
1. Syfte och tillämpningsområde
Med denna metod kan vattenhalten i foder bestämmas. Det bör framhållas att när det gäller foder som innehåller flyktiga ämnen, exempelvis organiska syror, bestäms också en betydande mängd flyktiga ämnen tillsammans med vatteninnehållet.
Metoden omfattar inte analys av mjölkprodukter som används som foderråvaror, analys av mineralämnen och blandningar som främst består av mineralämnen, analys av animaliska och vegetabiliska fetter och oljor eller analys av oljeväxtfrön och oljehaltiga frukter.
2. Princip
Provet torkas på angivet sätt, alltefter fodrets beskaffenhet. Viktförlusten bestäms genom vägning. När det gäller fast foder med hög vattenhalt är det nödvändigt att göra en förberedande torkning.
3. Utrustning
3.1 Kross av ej vattenabsorberande material som är lätt att rengöra, möjliggör snabb och jämn krossning utan nämnvärd värmeutveckling, så långt möjligt förhindrar kontakt med omgivande luft och som uppfyller kraven i 4.1.1 och 4.1.2 (t.ex. rivkvarnar med variabel malning, vattenkylda kvarnar eller liknande).
3.2 Analysvåg med en noggrannhet av 1 mg.
3.3 Torra behållare av korrosionsfri metall eller av glas och med lufttäta lock. Arbetsytan ska göra det möjligt att sprida ut provet till ca 0,3 g/cm2.
3.4 Elektrisk isotermisk ugn (± 2 oC) med god ventilation och möjlighet till snabb temperaturreglering ( 8 ).
3.5 Reglerbar elektrisk vakuumugn med oljepump och antingen en anordning för tillförsel av varm, torr luft eller ett torkmedel (t.ex. kalciumoxid).
3.6 Exsickator med tjock perforerad metall- eller porslinsplatta och med ett effektivt torkmedel.
4. Utförande
|
Observera: |
De moment som beskrivs i detta avsnitt måste utföras omedelbart efter det att provförpackningarna öppnats. Minst två parallella analyser ska utföras. |
4.1 Beredning
4.1.1
Ta minst 50 g av provet. Krossa eller sönderdela vid behov för att göra provet homogent med avseende på vattenhalt (se 6).
4.1.2
Ta minst 50 g av provet. Mal till partiklar som till minst 50 % passerar genom en sikt med en maskvidd av 0,5 mm och som lämnar högst 10 % återstod på en rundmaskig sikt med en maskvidd av 1 mm.
4.1.3
Ta ca 25 g av provet och väg med en noggrannhet av 10 mg. Tillsätt lämplig mängd vattenfri sand som vägts med en noggrannhet av 10 mg och blanda tills en homogen massa erhålls.
4.2 Torkning
4.2.1
Väg en behållare (3.3) med lock med en noggrannhet av 1 mg. Väg upp ca 5 g av provet med en noggrannhet av 1 mg i den vägda behållaren och fördela det jämnt. Sätt in behållaren utan lock i ugnen som förvärmts till 103 oC. Sätt in behållaren i ugnen så snabbt som möjligt så att ugnstemperaturen inte sjunker alltför mycket. Låt torka i 4 timmar räknat från det att ugnstemperaturen åter stigit till 103 oC. Sätt på locket på behållaren, ta ut denna ur ugnen, låt svalna i 30–45 minuter i exsickatorn (3.6) och väg med en noggrannhet av 1 mg.
Foder som till större delen består av oljor och fetter torkas i ugnen i ytterligare 30 minuter vid 130 oC. Skillnaden i vattenhalt mellan de båda vägningarna får inte överstiga 0,1 %.
4.2.2
Väg en behållare (3.3) med lock med en noggrannhet av 0,5 mg. Väg upp ca 5 g av det krossade provet med en noggrannhet av 1 mg i den vägda behållaren och fördela det jämnt. Sätt in behållaren utan lock i ugnen som förvärmts till 130 oC. Sätt in behållaren i ugnen så snabbt som möjligt så att ugnstemperaturen inte sjunker alltför mycket. Låt torka i två timmar räknat från det att ugnstemperaturen åter stigit till 130 oC. Sätt på locket på behållaren, ta ut denna ur ugnen, låt svalna i 30–45 minuter i exsickatorn (3.6) och väg med en noggrannhet av 1 mg.
4.2.3 Foderblandningar som innehåller mer än 4 % sackaros eller laktos; foderråvaror som t.ex. johannesbrödsfrön, hydrolyserade spannmålsprodukter, malt, torkad betsnitsel, lösliga biprodukter från fisk- eller sockerindustrin; foderblandningar som innehåller mer än 25 % mineralsalter inklusive kristallvatten
Väg en behållare (3.3) med lock med en noggrannhet av 0,5 mg. Väg upp ca 5 g av provet med en noggrannhet av 1 mg i den vägda behållaren och fördela det jämnt. Sätt in behållaren utan lock i vakuumugnen (3.5) som förvärmts till 80–85 oC. Sätt in behållaren i ugnen så snabbt som möjligt så att ugnstemperaturen inte sjunker alltför mycket.
Ställ in trycket på 100 torr och låt torka vid detta tryck i 4 timmar, antingen med en torr varmluftsström eller med användning av torkmedel (ca 300 g till 20 prov). Om det senare alternativet väljs ska vakuumpumpen frånkopplas när det angivna trycket har uppnåtts. Räkna torkningstiden från det att ugnstemperaturen åter har nått 80–85 oC. Låt försiktigt ugnen återgå till atmosfärstryck. Öppna ugnen, sätt omedelbart lock på behållaren, ta ut den ur ugnen, låt svalna i 30–45 minuter i exsickatorn (3.6) och väg med en noggrannhet av 1 mg. Låt torka i ytterligare 30 minuter i vakuumugnen vid 80–85 oC och väg på nytt. Skillnaden i vattenhalt mellan de båda vägningarna får inte överstiga 0,1 %.
4.3 Förberedande torkning
4.3.1
Fasta foder med hög vattenhalt som försvårar krossning måste genomgå en förberedande torkning enligt följande:
Väg upp 50 g okrossat prov med en noggrannhet av 10 mg (pellets och briketter kan vid behov delas grovt) i en lämplig behållare (t.ex. en 20 × 12 cm aluminiumplåt med 0,5 cm upphöjd kant). Låt torka i ugn vid 60–70 oC tills vattenhalten har reducerats till 8–12 %. Ta ut provet ur ugnen, låt svalna utan lock i laboratoriet i 1 timme och väg med en noggrannhet av 10 mg. Krossa provet omedelbart enligt 4.1.1 och torka enligt 4.2.1 eller 4.2.3 beroende på fodrets beskaffenhet.
4.3.2
Spannmål med en vattenhalt över 17 % måste genomgå en förberedande torkning enligt följande:
Väg upp 50 g omalda spannmålskärnor med en noggrannhet av 10 mg i en lämplig behållare (t.ex. en 20 × 12 cm aluminiumplåt med 0,5 cm upphöjd kant). Låt torka i ugn i 5–7 minuter vid 130 oC. Ta ut provet ur ugnen, låt svalna utan lock i laboratoriet i två timmar och väg med en noggrannhet av 10 mg. Mal omedelbart enligt 4.1.2 och torka enligt 4.2.2.
5. Beräkning av resultat
Vattenhalten (X) i procent av provet beräknas med nedanstående formler.
5.1 Torkning utan förberedande torkning
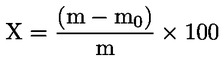
där:
|
m |
= |
provets ursprungliga vikt i gram |
|
m0 |
= |
det torkade provets vikt i gram. |
5.2 Torkning med förberedande torkning

där:
|
m |
= |
provets ursprungliga vikt i gram |
|
m1 |
= |
provets vikt i gram efter förberedande torkning |
|
m2 |
= |
provets vikt i gram efter krossning eller malning |
|
m0 |
= |
det torkade provets vikt i gram. |
5.3 Repeterbarhet
Skillnaden mellan resultaten från två parallella bestämningar som utförs på samma prov får inte överstiga 0,2 % av den absoluta vattenhalten.
6. Anmärkning
Om det är nödvändigt att krossa provet och detta medför en iakttagbar förändring av vattenhalten, måste resultaten av analyserna av fodrets beståndsdelar korrigeras på grundval av provets ursprungliga vattenhalt.
B. BESTÄMNING AV VATTENHALT I ANIMALISKA OCH VEGETABILISKA OLJOR OCH FETTER
1. Syfte och tillämpningsområde
Med denna metod är det möjligt att bestämma halter av vatten och flyktiga ämnen i animaliska och vegetabiliska oljor och fetter.
2. Princip
Provet torkas till konstant vikt (viktförlusten mellan två på varandra följande vägningar får inte överstiga 1 mg) vid 103 oC. Viktförlusten bestäms genom vägning.
3. Utrustning
3.1 Flatbottnad skål av korrosionsresistent material, 8–9 cm i diameter och ca 3 cm hög.
3.2 Termometer med förstärkt kula och expansionsrör i överdelen, graderad från ca 80 oC till minst 110 oC och ca 10 cm lång.
3.3 Sandbad eller elektrisk värmeplatta.
3.4 Exsickator med ett effektivt torkmedel.
3.5 Analysvåg.
4. Utförande
Väg med 1 mg noggrannhet upp ca 20 g av det homogeniserade provet på den torra, vägda skålen (3.1) där termometern (3.2) placerats. Upphetta på sandbadet eller värmeplattan (3.3) under ständig omrörning med termometern så att temperaturen stiger till 90 oC på ca 7 minuter.
Sänk värmen och ge akt på mängden bubblor som stiger från skålens botten. Temperaturen får inte överstiga 105 oC. Fortsätt att röra om och skrapa skålens botten tills det inte längre bildas några bubblor.
För att försäkra sig om att vattnet helt har avlägsnats upphettar man upprepade gånger till 103 ± 2 oC och kyler till 93 oC mellan upphettningarna. Därefter får provet svalna till rumstemperatur i exsickatorn (3.4) varefter det vägs. Detta upprepas tills viktförlusten mellan två på varandra följande vägningar inte längre överstiger 2 mg.
|
Observera: |
Om provets vikt ökar efter flera upphettningar, tyder detta på att fettet har oxiderats. Resultatet beräknas då utifrån den vägning som gjordes omedelbart innan vikten började öka. |
5. Beräkning av resultat
Provets vattenhalt X i procent beräknas med formeln

där:
|
m |
= |
provets vikt i gram |
|
m1 |
= |
skålens vikt (i gram) inklusive innehåll före upphettning |
|
m2 |
= |
skålens vikt (i gram) inklusive innehåll efter upphettning. |
Resultat under 0,05 % ska redovisas som ”under 0,05 %”.
Repeterbarhet
Skillnaden mellan resultaten från två parallella bestämningar som utförs på samma prov får inte överstiga 0,05 % av den absoluta vattenhalten.
C. BESTÄMNING AV RÅPROTEINHALT
1. Syfte och tillämpningsområde
Med denna metod kan råproteinhalten i foder bestämmas på grundval av kvävehalten, bestämd med Kjeldahlmetoden.
2. Princip
Provet uppsluts med svavelsyra i närvaro av en katalysator. Syralösningen görs basisk med natriumhydroxidlösning. Ammoniaken destilleras av och samlas upp i en uppmätt kvantitet svavelsyra varefter syraöverskottet titreras med en standardlösning av natriumhydroxid.
Alternativt destilleras den frigjorda ammoniaken till ett överskott av borsyralösning, som sedan titreras med saltsyra eller svavelsyra.
3. Reagens
3.1 Kaliumsulfat.
3.2 Katalysator: koppar(II)oxid, CuO, eller koppar(II)sulfatpentahydrat, CuSO4 5H2O.
3.3 Zinkgranulat.
3.4 Svavelsyra, ρ20 = 1,84 g/ml.
3.5 Svavelsyra, standardlösning, c(H2SO4) = 0,25 mol/l.
3.6 Svavelsyra, standardlösning, c(H2SO4) = 0,10 mol/l.
3.7 Svavelsyra, standardlösning, c(H2SO4) = 0,05 mol/l.
3.8 Metylrött (indikator): lös 300 mg metylrött i 100 ml etanol, σ = 95–96 % (v/v).
3.9 Natriumhydroxidlösning (teknisk kvalitet kan användas) β = 40 g/100 ml (m/v: 40 %).
3.10 Natriumhydroxid, standardlösning, c(NaOH) = 0,25 mol/l.
3.11 Natriumhydroxid, standardlösning, c(NaOH) = 0,10 mol/l.
3.12 Pimpstensgranulat som saltsyresköljts och glödgats.
3.13 Acetanilid (smältpunkt = 114 oC, N-halt = 10,36 %).
3.14 Sackaros (kvävefri).
3.15 Borsyra (H3BO3).
3.16 Indikatorlösning av metylrött: lös 100 mg metylrött i 100 ml etanol eller metanol.
3.17 Lösning av bromkresolgrönt: lös 100 mg bromkresolgrönt i 100 ml etanol eller metanol.
3.18 Borsyralösning (10–40 g/l beroende på vilken utrustning som används).
Vid kolorimetrisk bestämning av ekvivalenspunkten ska indikatorerna metylrött och bromkresolgrönt tillsättas borsyralösningarna. För beredning av 1 liter borsyralösning tillsätts 7 ml lösning av metylrött (3.16) och 10 ml lösning av bromkresolgrönt (3.17) innan volymen justeras.
Beroende på det vatten som används kan borsyralösningens pH variera mellan olika satser. Det är ofta nödvändigt med en liten tillsats av bas för att få ett positivt blankprov.
|
Anmärkning: |
En tillsats av ca 3–4 ml NaOH (3.11) till 1 liter borsyra (10 g/l) ger vanligen en god justering. Lösningen förvaras i rumstemperatur skyddad från ljus och ammoniakångor. |
3.19 Saltsyra, standardlösning, c(HCl) = 0,10 mol/l.
|
Anmärkning: |
Andra koncentrationer av lösningarna (3.5, 3.6, 3.7, 3.10, 3.11 och 3.19) kan användas om beräkningarna korrigeras för detta. Koncentrationerna bör alltid anges med fyra decimaler. |
4. Utrustning
Lämplig utrustning för uppslutning, destillation och titrering enligt Kjeldahlmetoden.
5. Utförande
5.1 Uppslutning
Väg upp 1 g av provet med 0,001 g noggrannhet och överför detta till uppslutningsapparatens kolv. Tillsätt 15 g kaliumsulfat (3.1), en lämplig mängd katalysator (3.2) (0,3 –0,4 g koppar(II)oxid eller 0,9 –1,2 g koppar(II)sulfatpentahydrat), 25 ml svavelsyra (3.4) och vid behov några korn pimpsten (3.12). Blanda.
Upphetta kolven först måttligt, och rotera den vid behov då och då tills innehållet har förkolnat och skummet försvunnit. Upphetta sedan intensivare tills vätskan kommit i konstant kokning. Värmen är tillräcklig om den kokande syran kondenserar på kolvens väggar. Se till att kolvens väggar inte blir överhettade och att organiska partiklar inte fastnar på dem.
När lösningen har blivit klar och ljusgrön fortsätts kokningen i ytterligare två timmar, varefter lösningen får svalna.
5.2 Destillation
Tillsätt försiktigt tillräckligt med vatten för att sulfaterna ska lösas upp fullständigt. Låt svalna. Tillsätt sedan några korn zinkgranulat (3.3) om så behövs. Fortsätt enligt 5.2.1 eller 5.2.2.
5.2.1
Tillför en exakt uppmätt mängd av 25 ml svavelsyra (3.5) eller (3.7), beroende på den förmodade kvävehalten, till destillationsapparatens uppsamlingskolv. Tillsätt några droppar metylrött (3.8).
Förbind kolven med destillationsapparatens kylare och sänk ned kylarens ände minst 1 cm (se anmärkning 8.3) i vätskan i uppsamlingskolven. Häll långsamt 100 ml natriumhydroxidlösning (3.9) i kolven utan att ammoniak försvinner (se anmärkning 8.1). Upphetta kolven tills ammoniaken har destillerats färdigt.
5.2.2
Om destillatets ammoniakinnehåll titreras manuellt används nedanstående förfarande. Om destillationsenheten är helautomatisk och inkluderar titrering av destillatets ammoniakinnehåll, ska tillverkarens bruksanvisning följas.
Ställ en uppsamlingskolv innehållande 25–30 ml borsyralösning (3.18) under kylarens utlopp på ett sådant sätt att utloppsröret mynnar under ytan i ett överskott av borsyralösning. Ställ in destillationsenheten så att 50 ml natriumhydroxidlösning (3.9) tillsätts. Använd destillationsenheten enligt tillverkarens bruksanvisning och destillera av den ammoniak som frigjorts genom tillsatsen av natriumhydroxidlösning. Samla upp destillatet i borsyralösningen. Mängden destillat (tiden för ångdestillation) beror på mängden kväve i provet. Följ tillverkarens bruksanvisning.
|
Anmärkning: |
I en halvautomatisk destillationsenhet sker såväl tillsats av ett överskott av natriumhydroxid som ångdestillation automatiskt. |
5.3 Titrering
Fortsätt enligt 5.3.1 eller 5.3.2.
5.3.1
Överskottet av svavelsyra titreras i uppsamlingskolven med natriumhydroxidlösning (3.10 eller 3.11), beroende på vilken koncentration av svavelsyra som använts, tills ekvivalenspunkten är nådd.
5.3.2
Uppsamlingskolvens innehåll titreras med standardlösning av saltsyra (3.19) eller med standardlösning av svavelsyra (3.6) med hjälp av en byrett. Den använda mängden titrator avläses.
Vid kolorimetrisk bestämning av ekvivalenspunkten nås denna så snart provet börjar skifta färg till rosa. Avläs byretten med 0,05 ml noggrannhet. En upplyst magnetomrörare eller en fotometer kan göra det lättare att påvisa färgomslaget.
Detta kan också göras automatiskt med hjälp av en ångdestillationsapparat med automatisk titrering.
Följ tillverkarens bruksanvisning för den använda destillationsapparaten eller destillations-/titreringsapparaten.
|
Anmärkning: |
Om automatisk titrering används, inleds titreringen omedelbart efter det att destillationen har börjat, och 1 % borsyralösning (3.18) används. Med en helautomatisk destillationsenhet kan man också göra en potentiometrisk titrering av ammoniak, där ekvivalenspunkten bestäms genom kontinuerlig pH-mätning. I så fall används en automatisk titreringsapparat med en pH-meter som med hjälp av standardmetoder är noggrant kalibrerad i intervallet pH 4–7. Ekvivalenspunkten nås vid pH 4,6 där titrerkurvan är som brantast (inflexionspunkten). |
5.4 Blankprov
För att säkerställa att reagensen är fria från kväve utförs ett blankprov (uppslutning, destillation och titrering) med 1 g sackaros (3.14) i stället för provet.
6. Beräkning av resultat
Beräkningarna görs enligt 6.1 eller 6.2.
6.1 Beräkning för titrering enligt 5.3.1
Råproteinhalten i viktprocent beräknas med formeln

där:
|
V0 |
= |
volymen (ml) NaOH (3.10 eller 3.11) i blankprovet |
|
V1 |
= |
volymen (ml) NaOH (3.10 eller 3.11) vid titreringen av provet |
|
c |
= |
koncentrationen (mol/l) av natriumhydroxid (3.10 eller 3.11) |
|
m |
= |
provets vikt i gram. |
6.2 Beräkning för titrering enligt 5.3.2
6.2.1
Råproteinhalten i viktprocent beräknas med formeln

där:
|
m |
= |
provets vikt i gram |
|
c |
= |
koncentrationen (mol/l) av saltsyra i standardlösningen (3.19) |
|
V0 |
= |
volymen (ml) saltsyra i blankprovet |
|
V1 |
= |
volymen (ml) saltsyra i provet. |
6.2.2
Råproteinhalten i viktprocent beräknas med formeln
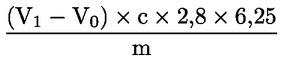
där:
|
m |
= |
provets vikt i gram |
|
c |
= |
koncentrationen (mol/l) av svavelsyra i standardlösningen (3.6) |
|
V0 |
= |
volymen (ml) svavelsyra (3.6) i blankprovet |
|
V1 |
= |
volymen (ml) svavelsyra (3.6) i provet. |
7. Verifiering av metoden
7.1 Repeterbarhet
Skillnaden mellan resultaten från två parallella bestämningar som utförs på samma prov får inte överstiga
— 0,2 % i absoluta tal för råproteinhalter under 20 %,
— 1,0 % av det högre värdet för råproteinhalter på 20–40 %,
— 0,4 % i absoluta tal för råproteinhalter över 40 %.
7.2 Noggrannhet
Utför analysen (uppslutning, destillation och titrering) på 1,5 –2,0 g acetanilid (3.13) i närvaro av 1 g sackaros (3.14). 1 g acetanilid förbrukar 14,80 ml svavelsyra (3.5). Utbytet ska vara minst 99 %.
8. Anmärkningar
8.1 Den använda apparaten kan vara manuell, halvautomatisk eller automatisk. Om apparaten kräver att materialet flyttas mellan uppslutnings- och destillationsstegen måste flyttningen ske utan förlust. Om destillationsapparaturens kolv inte är försedd med en dropptratt ska natriumhydroxiden tillsättas omedelbart innan kolven ansluts till kylaren, varvid vätskan långsamt hälls ned längs kolvens vägg.
8.2 Om det uppslutna materialet tjocknar görs bestämningen om med en större mängd svavelsyra (3.4) än den ovan angivna.
8.3 För produkter med låg kvävehalt kan volymen svavelsyra (3.7) som tillsätts i uppsamlingskolven vid behov minskas till 10–15 ml, varefter vatten fylls på till 25 ml.
8.4 För rutinanalyser kan alternativa metoder för bestämning av råprotein användas, men Kjeldahlmetoden som beskrivs i denna del C är referensmetoden. De resultat som erhålls med den alternativa metoden (t.ex. Dumas) måste för varje matris individuellt visas vara likvärdiga jämfört med referensmetoden. Resultaten kan dock skilja sig något från resultat erhållna med referensmetoden även om likvärdigheten har kontrollerats. Därför måste analysrapporten ange vilken metod som använts för bestämning av råprotein.
D. BESTÄMNING AV UREA
1. Syfte och tillämpningsområde
Med denna metod kan ureahalten i foder bestämmas.
2. Princip
Provet slammas upp i vatten med klarmedel. Suspensionen filtreras. Ureahalten i filtratet bestäms efter tillsats av 4-dimetylaminobensaldehyd (4-DMAB) genom mätning av absorbansen vid våglängden 420 nm.
3. Reagens
3.1 Lösning av 4-dimetylaminobensaldehyd: lös upp 1,6 g 4-DMAB i 100 ml 96 % etanol och tillsätt 10 ml saltsyra (ρ20 = 1,19 g/ml). Denna reagens håller sig i högst två veckor.
3.2 Carrez-lösning I: lös upp 21,9 g zinkacetat, Zn(CH3COO)2 2H2O, och 3 g isättika i vatten. Späd med vatten till 100 ml.
3.3 Carrez-lösning II: lös upp 10,6 g kaliumferrocyanid, K4Fe(CN)6 3H2O, i vatten. Späd med vatten till 100 ml.
3.4 Aktivt kol som inte absorberar urea (ska kontrolleras).
3.5 Urea, 0,1 % lösning (w/v).
4. Utrustning
4.1 Rotationsskakapparat: ca 35–40 varv per minut.
4.2 Provrör: 160 × 16 mm med slipade glasproppar.
4.3 Spektrofotometer.
5. Utförande
5.1 Analys av provet
Väg med en noggrannhet av 1 mg upp 2 g av provet i en 500 ml mätkolv tillsammans med 1 g aktivt kol (3.4). Tillsätt 400 ml vatten och 5 ml Carrez-lösning I (3.2), blanda i ca 30 sekunder och tillsätt 5 ml Carrez-lösning II (3.3). Blanda i 30 minuter på skakapparaten. Späd med vatten till full volym, skaka om och filtrera.
För över 5 ml av de transparenta, färglösa filtraten till provrör med slipade glasproppar, tillsätt 5 ml 4-DMAB-lösning (3.1) och blanda. Ställ provrören i ett vattenbad som håller 20 oC (+/– 4 oC). Mät efter 15 minuter provlösningens absorbans med spektrofotometer vid 420 nm. Jämför med blankprovslösningen av reagensen.
5.2 Kalibreringskurva
För över volymer på 1, 2, 4, 5 och 10 ml av urealösningen (3.5) till 100 ml mätkolvar och späd med vatten till full volym. Avlägsna 5 ml från varje lösning, tillsätt 5 ml 4-DMAB-lösning (3.1) till var och en av dem, homogenisera och mät absorbansen enligt ovan jämfört med en kontrollösning som innehåller 5 ml 4-DMAB-lösning och 5 ml ureafritt vatten. Konstruera kalibreringskurvan.
6. Beräkning av resultat
Bestäm ureamängden i provet med hjälp av kalibreringskurvan.
Uttryck resultatet i procent av provet.
7. Anmärkningar
7.1 Om ureahalten överstiger 3 %, reducera provet till 1 g eller späd den ursprungliga lösningen så att det inte finns mer än 50 mg urea per 500 ml.
7.2 Om ureahalten är låg, öka provmängden så mycket som kan ske med bibehållen transparens och färglöshet hos filtratet.
7.3 Om provet innehåller enkla kväveföreningar som aminosyror ska absorbansen mätas vid 435 nm.
E. BESTÄMNING AV FLYKTIGA KVÄVEFÖRENINGAR
I. GENOM MIKRODIFFUSION
1. Syfte och tillämpningsområde
Med denna metod kan halten flyktiga kväveföreningar, uttryckt som ammoniak, bestämmas i foder.
2. Princip
Provet extraheras med vatten och lösningen klaras och filtreras. De flyktiga kväveföreningarna avskiljs genom mikrodiffusion med kaliumkarbonatlösning, samlas upp i borsyrelösning och titreras med svavelsyra.
3. Reagens
3.1 Triklorättiksyra, lösning 20 % (w/v).
3.2 Indikator: lös 33 mg bromkresolgrönt och 65 mg metylrött i 100 ml etanol 95–96 % (v/v).
3.3 Borsyralösning: lös 10 g borsyra i en 1 000 ml mätkolv med 200 ml etanol 95–96 % (v/v) och 700 ml vatten. Tillsätt 10 ml indikator (3.2). Blanda och justera vid behov lösningens färg till ljusrött genom att tillsätta natriumhydroxidlösning. 1 ml av lösningen fixerar maximalt 300 μg NH3.
3.4 Mättad kaliumkarbonatlösning: lös 100 g kaliumkarbonat i 100 ml kokande vatten. Låt svalna och filtrera.
3.5 Svavelsyra, 0,01 mol/l.
4. Utrustning
4.1 Rotationsskakapparat: ca 35–40 varv per minut.
4.2 Conwayceller av glas eller plast (se diagram).
4.3 Mikrobyretter graderade i 1/100 ml.
5. Utförande
Väg upp 10 g av provet med en noggrannhet av 1 mg och överför det tillsammans med 100 ml vatten till en 200 ml mätkolv. Blanda på skakapparaten i 30 minuter. Tillsätt 50 ml triklorättiksyralösning (3.1), späd med vatten till full volym, skaka kraftigt och filtrera genom ett veckat filtrerpapper.
Tillsätt med pipett 1 ml borsyrelösning (3.3) i den inre delen av Conwaycellen och 1 ml av provfiltratet i cellens krona. Täck delvis med det infettade locket. Droppa snabbt 1 ml mättad kaliumkarbonatlösning (3.4) i kronan och tillslut locket lufttätt. Rotera försiktigt cellen i horisontellt läge så att de två reagensen blandas. Inkubera antingen vid rumstemperatur i minst fyra timmar eller vid 40 oC i en timme.
Titrera med hjälp av en mikrobyrett (4.3) de flyktiga föreningarna i borsyralösningen med svavelsyra (3.5).
Utför ett blankprov med samma metod men utan analysprov.
6. Beräkning av resultat
1 ml 0,01 M H2SO4 motsvarar 0,34 mg ammoniak.
Uttryck resultatet i procent av provet.
Repeterbarhet
Skillnaden mellan resultaten från två parallella bestämningar som utförs på samma prov får inte överstiga
— 10 % av det högre värdet för ammoniakhalter under 1,0 %,
— 0,1 % i absoluta tal för ammoniakhalter på 1,0 % eller högre.
7. Anmärkning
Om provets ammoniakhalt överstiger 0,6 % ska det ursprungliga filtratet spädas ut.
CONWAY CELL
Scale 1/1

II. GENOM DESTILLATION
1. Syfte och tillämpningsområde
Med denna metod kan halten flyktiga kväveföreningar, uttryckt som ammoniak, bestämmas i fiskmjöl som är i det närmaste ureafritt. Den kan endast användas vid ammoniakhalter under 0,25 %.
2. Princip
Provet extraheras med vatten och lösningen klaras och filtreras. De flyktiga kväveföreningarna avskiljs vid kokpunkten genom tillsats av magnesiumoxid och samlas upp i en bestämd mängd svavelsyra. Överskottet av svavelsyra återtitreras med natriumhydroxidlösning.
3. Reagens
3.1 Triklorättiksyra, lösning 20 % (w/v).
3.2 Magnesiumoxid.
3.3 Skumdämpande emulsion (t.ex. silikon).
3.4 Svavelsyra, 0,05 mol/l.
3.5 Natriumhydoxidlösning, 0,1 mol/l.
3.6 Metylröttlösning, 0,3 %, i 95–96 % (v/v) etanol.
4. Utrustning
4.1 Rotationsskakapparat: ca 35–40 varv per minut.
4.2 Destillationsapparat av Kjeldahltyp.
5. Utförande
Väg upp 10 g av provet med en noggrannhet av 1 mg och överför det tillsammans med 100 ml vatten till en 200 ml mätkolv. Blanda på skakapparaten i 30 minuter. Tillsätt 50 ml triklorättiksyralösning (3.1), späd med vatten till full volym, skaka om kraftigt och filtrera genom ett veckat filtrerpapper.
Ta ut klart filtrat i en volym avpassad för den förmodade halten flyktiga kväveföreningar (100 ml brukar vara lämpligt). Späd till 200 ml och tillsätt 2 g magnesiumoxid (3.2) och några droppar skumdämpande emulsion (3.3). Lösningen ska ge basisk reaktion med lackmuspapper; om så inte är fallet tillsätts lite magnesiumoxid (3.2). Fortsätt enligt 5.2 och 5.3 i analysmetoden för bestämning av råproteinhalten (del C i denna bilaga).
Utför ett blankprov med samma metod men utan analysprov.
6. Beräkning av resultat
1 ml 0,05 mol/l H2SO4 motsvarar 1,7 mg ammoniak.
Uttryck resultatet i procent av provet.
Repeterbarhet
Skillnaden mellan resultaten från två parallella bestämningar som utförs på samma prov får inte överstiga 10 % (relativt värde).
F. BESTÄMNING AV AMINOSYROR (UTOM TRYPTOFAN)
1. Syfte och tillämpningsområde
Med denna metod kan fria aminosyror (syntetiska och naturliga) och totalhalten aminosyror (peptidbundna och fria) i foder bestämmas med hjälp av en aminosyraanalysator. Den är tillämplig på följande aminosyror: cyst(e)in, metionin, lysin, treonin, alanin, arginin, asparaginsyra, glutaminsyra, glycin, histidin, isoleucin, leucin, fenylalanin, prolin, serin, tyrosin och valin.
Metoden skiljer inte mellan aminosyrornas salter och kan inte skilja mellan aminosyrornas D- och L-former. Den är inte användbar för bestämning av tryptofan eller hydroxyanaloger av aminosyror.
2. Princip
2.1 Fria aminosyror
De fria aminosyrorna extraheras med utspädd saltsyra. Samextraherade kvävehaltiga makromolekyler fälls ut med sulfosalicylsyra och avlägsnas genom filtrering. Den filtrerade lösningens pH justeras till 2,20 . Aminosyrorna separeras genom jonbyteskromatografi och bestäms fotometriskt vid 570 nm genom reaktion med ninhydrin.
2.2 Totalhalt aminosyror
Valet av förfarande beror på vilka aminosyror som ska bestämmas. Cyst(e)in och metionin måste oxideras till cysteinsyra respektive metioninsulfon före hydrolys. Tyrosin måste bestämmas i hydrolysat av ooxiderade prov. Alla övriga aminosyror som räknas upp i punkt 1 kan bestämmas i antingen det oxiderade eller ooxiderade provet.
Oxidation utförs vid 0 oC med en blandning av permyrsyra och fenol. Överskott av oxidationsreagens avlägsnas genom tillsats av dinatriumdisulfit. Det oxiderade eller ooxiderade provet hydrolyseras med saltsyra (3.20) i 23 timmar. Hydrolysatet justeras till pH 2,20 . Aminosyrorna separeras genom jonbyteskromatografi och bestäms fotometriskt vid 570 nm (440 nm för prolin) genom reaktion med ninhydrin.
3. Reagens
Dubbeldestillerat vatten eller vatten av likvärdig kvalitet ska användas (konduktivitet < 10 μS/cm).
3.1 Väteperoxid, w = 30 % (w/w).
3.2 Myrsyra, w = 98–100 % (w/w).
3.3 Fenol.
3.4 Dinatriumdisulfit
3.5 Natriumhydroxid.
3.6 5-sulfosalicylsyradihydrat.
3.7 Saltsyra, densitet ca 1,18 g/ml.
3.8 Trinatriumcitratdihydrat.
3.9 2,2 '-tiodietanol (tiodiglykol).
3.10 Natriumklorid.
3.11 Ninhydrin.
3.12 Petroleumeter, kokpunktsintervall 40–60 oC.
3.13 Norleucin eller annan förening lämplig som intern standard.
3.14 Kvävgas (< 10 ppm syrgas).
3.15 1-oktanol.
3.16 Aminosyror.
3.16.1 Referenssubstanser enligt listan i punkt 1. Rena substanser, fria från kristallvatten. Torka i vakuum över P2O5 eller H2SO4 i 1 vecka före användning.
3.16.2 Cysteinsyra.
3.16.3 Metioninsulfon.
3.17 Natriumhydroxidlösning, c = 7,5 mol/l:
Lös 300 g NaOH (3.5) i vatten och späd till 1 liter.
3.18 Natriumhydroxidlösning, c = 1 mol/l:
Lös 40 g NaOH (3.5) i vatten och späd till 1 liter.
3.19 Myrsyra-fenollösning:
Blanda 889 g myrsyra (3.2) med 111 g vatten och tillsätt 4,73 g fenol (3.3).
3.20 Hydrolysblandning, c = 6 mol HCl/l innehållande 1 g fenol/l:
Tillsätt 1 g fenol (3.3) till 492 ml HCl (3.7) och späd med vatten till 1 liter.
3.21 Extraktionsblandning, c = 0,1 mol HCl/l innehållande 2 % tiodiglykol: Ta 8,2 ml HCl (3.7), späd med ca 900 ml vatten, tillsätt 20 ml tiodiglykol (3.9) och späd med vatten till 1 liter (blanda inte 3.7 och 3.9 direkt).
3.22 5-sulfosalicylsyra, ß = 6 %:
Lös 60 g 5-sulfosalicylsyra (3.6) i vatten och späd till 1 liter.
3.23 Oxidationsblandning (permyrsyra-fenol):
Blanda 0,5 ml väteperoxid (3.1) med 4,5 ml myrsyra-fenollösning (3.19) i en liten bägare. Inkubera i 20–30 oC i 1 timme för att permyrsyra ska bildas, kyl sedan i isvattenbad (15 min.) före tillsats till provet.
Varning: Undvik hudkontakt, använd skyddskläder.
3.24 Citratbuffert, c = 0,2 mol Na+/l, pH 2,20 :
Lös 19,61 g natriumcitrat (3.8), 5 ml tiodiglykol (3.9), 1 g fenol (3.3) och 16,50 ml HCl (3.7) i ca 800 ml vatten. Justera pH till 2,20 . Späd med vatten till 1 liter.
3.25 Elueringsbuffertar som lämpar sig för den analysator som ska användas (4.9).
3.26 Ninhydrinreagens som lämpar sig för den analysator som ska användas (4.9).
3.27 Standardlösningar av aminosyror. Dessa lösningar ska lagras vid en temperatur under 5 oC.
3.27.1 Stamlösningar av aminosyror (3.16.1).
c = 2,5 μmol/ml av var och en i saltsyra.
Finns att köpa i handeln.
3.27.2 Stamlösning av cysteinsyra och metioninsulfon, c = 1,25 μmol/ml.
Lös 0,2115 g cysteinsyra (3.16.2) och 0,2265 g metioninsulfon (3.16.3) i citratbuffert (3.24) i en 1 000 ml mätkolv och späd till märket med citratbuffert. Kan lagras vid en temperatur under 5 oC i högst tolv månader. Denna lösning används ej om stamlösningen (3.27.1) innehåller cysteinsyra och metioninsulfon.
3.27.3 Stamlösning av intern standard, t.ex. norleucin, c = 20 μmol/ml.
Lös 0,6560 g norleucin (3.13) i citratbuffert (3.24) i en mätkolv och späd till 250 ml med citratbuffert. Kan lagras vid en temperatur under 5 oC i högst 6 månader.
3.27.4 Kalibreringslösning av standardaminosyror för användning med hydrolysater, c = 5 nmol/50 μl för cysteinsyra och metioninsulfon och c = 10 nmol/50 μl för övriga aminosyror. Lös 2,2 g natriumklorid (3.10) i en 100 ml bägare med 30 ml citratbuffert (3.24). Tillsätt 4,00 ml stamlösning av aminosyror (3.27.1), 4,00 ml stamlösning av cysteinsyra och metioninsulfon (3.27.2) och 0,50 ml stamlösning av intern standard (3.27.3) om sådan används. Justera pH till 2,20 med natriumhydroxid (3.18).
Överför kvantitativt till en 50 ml mätkolv, späd till märket med citratbuffert (3.24) och blanda.
Kan lagras vid en temperatur under 5 oC i högst 3 månader.
Se även punkt 9.1.
3.27.5 Kalibreringslösning av standardaminosyror för användning med hydrolysater beredda enligt 5.3.3.1 och för användning med extrakt (5.2). Kalibreringslösningen bereds enligt 3.27.4 men natriumkloriden utelämnas.
Kan lagras vid en temperatur under 5 oC i högst 3 månader.
4. Utrustning
4.1 100 eller 250 ml rundkolv med återloppskylare.
4.2 100 ml borosilikatglasflaska med skruvkork försedd med gummi/teflontätning (t.ex. Duran, Schott) för användning i ugn.
4.3 Ugn med fläktventilation och temperaturreglering med noggrannhet bättre än ± 2 oC.
4.4 pH-meter (tre decimalers noggrannhet).
4.5 Membranfilter (0,22 μm).
4.6 Centrifug.
4.7 Rotationsindunstare.
4.8 Mekanisk skakapparat eller magnetomrörare.
4.9 Aminosyraanlysator eller HPLC-utrustning med jonbytarkolonn, anordning för ninhydrin, derivatisering efter kolonnpassagen och fotometrisk detektor.
Kolonnen fylls med sulfonerad polystyrenharts som kan separera aminosyrorna från varandra och från andra ninhydrinpositiva material. Flödet i buffert- och ninhydrinledningarna ombesörjs av pumpar vars flödesstabilitet är ± 0,5 % under en period som täcker både standardkalibreringskörningen och analysen av provet.
Med vissa aminosyraanalysatorer kan man använda hydrolysmetoder där hydrolysatet har en natriumkoncentration på c = 0,8 mol/l och innehåller all överskottsmyrsyra från oxidationssteget. Andra ger inte en tillfredsställande separation av vissa aminosyror i hydrolysat med ett överskott av myrsyra och/eller hög natriumkoncentration. I detta fall reduceras syravolymen genom indunstning till ca 5 ml efter hydrolysen och före pH-justeringen. Indunstningen ska utföras i vakuum vid högst 40 oC.
5. Utförande
5.1 Provberedning
Provet mals så att det kan passera genom en 0,5 mm sikt. Prov med hög vattenhalt ska antingen lufttorkas vid en temperatur på högst 50 oC eller frystorkas före malningen. Prov med hög fetthalt ska extraheras med petroleumeter (3.12) före malningen.
5.2 Bestämning av fria aminosyror i foder och förblandningar
Väg med 0,2 mg noggrannhet upp en lämplig mängd (1–5 g) av det beredda provet (5.1) i en E-kolv och tillsätt 100,0 ml av extraktionsblandningen (3.21). Blanda i 60 minuter med en mekanisk skakapparat eller magnetomrörare (4.8). Låt sedimentera och pipettera över 10,0 ml av supernatanten till en 100 ml bägare.
Tillsätt 5,0 ml sulfosalicylsyralösning (3.22) under omrörning och fortsätt röra om med hjälp av en magnetomrörare i fem minuter. Filtrera eller centrifugera supernatanten för att avlägsna eventuella fällningar. Överför 10,0 ml av den kvarvarande lösningen till en 100 ml bägare och justera pH till 2,20 med natriumhydroxidlösning (3.18), överför till en mätkolv av lämplig volym med citratbuffert (3.24) och späd till märket med buffertlösningen (3.24).
Om en intern standard används, tillsätt 1,00 ml av denna (3.27.3) för varje 100 ml färdig lösning och späd till märket med buffertlösningen (3.24).
Gå vidare till kromatografin enligt 5.4.
Om extrakten inte kromatograferas samma dag måste de förvaras vid en temperatur under 5 oC.
5.3 Bestämning av totalhalt aminosyror.
5.3.1
Väg med 0,2 mg noggrannhet upp 0,1 –1 g av det beredda provet (5.1) i
— en 100 ml rundkolv (4.1) för öppen hydrolys (5.3.2.3) eller,
— en 250 ml rundkolv (4.1) om en låg natriumkoncentration är nödvändig (5.3.3.1) eller,
— en 100 ml flaska med skruvkork (4.2) för sluten hydrolys (5.3.2.4).
Det invägda provet ska innehålla ca 10 mg kväve, och vattenhalten får inte överstiga 100 mg.
Ställ kolven/flaskan i ett isvattenbad och kyl till 0 oC, tillsätt 5 ml oxidationsblandning (3.23) och blanda med hjälp av en glasspatel med böjd spets. Tillslut kolven/flaskan innehållande spateln med en lufttät folie, ställ isvattenbadet innehållande den tillslutna behållaren i ett kylskåp vid 0 oC och låt stå i 16 timmar. Avlägsna provet ur kylskåpet efter 16 timmar och avlägsna överskottet av oxidationsreagens genom tillsats av 0,84 g dinatriumdisulfit (3.4).
Fortsätt till 5.3.2.1.
5.3.2
5.3.2.1
Tillsätt 25 ml av hydrolysblandningen (3.20) till det oxiderade prov som beretts enligt 5.3.1. Skölj ned eventuella provrester som fastnat på kärlets sidor och på spateln.
Fortsätt enligt 5.3.2.3 eller 5.3.2.4, beroende på vilken hydrolysmetod som används.
5.3.2.2
Väg med 0,2 mg noggrannhet upp 0,1 –1 g av det beredda provet (5.1) i antingen en 100 ml eller 250 ml rundkolv (4.1) eller en 100 ml flaska med skruvkork (4.2). Det invägda provet ska innehålla ca 10 mg kväve. Tillsätt försiktigt 25 ml av hydrolysblandningen (3.20) och blanda med provet. Fortsätt enligt antingen 5.3.2.3 eller 5.3.2.4.
5.3.2.3
Tillsätt tre glaspärlor till blandningen i kolven (beredd enligt 5.3.2.1 eller 5.3.2.2) och återloppskoka i 23 timmar. Efter fullbordad hydrolys, skölj kylaren med 5 ml citratbuffert (3.24). Koppla bort kolven och kyl den i ett isvattenbad.
Fortsätt enligt 5.3.3.
5.3.2.4
Sätt in flaskan med blandningen, beredd enligt 5.3.2.1 eller 5.3.2.2, i en ugn (4.3) vid 110 oC. För att förhindra tryckökning (på grund av gasutveckling) och explosion bör skruvkorken sättas löst på flaskan under den första timmen. Tillslut inte flaskan med skruvkorken. Efter 1 timme dras skruvkorken åt och flaskan med får stå i ugnen (4.3) i 23 timmar. Efter fullbordad hydrolys, ta ut flaskan ur ugnen, öppna försiktigt skruvkorken och ställ flaskan i ett isvattenbad. Låt svalna.
Beroende på metod för pH-justering (5.3.3), överför flaskans innehåll kvantitativt till en 250 ml bägare eller till en 250 ml rundkolv med citratbuffert (3.24).
Fortsätt enligt 5.3.3.
5.3.3
Beroende på aminosyraanalysatorns (4.9) natriumtolerans, fortsätt enligt 5.3.3.1 eller 5.3.3.2 för pH-justeringen.
5.3.3.1
Det är tillrådligt att använda en stamlösning av intern standard (3.27.3) om aminosyraanalysatorn kräver en låg natriumkoncentration (om syravolymen måste reduceras).
Tillsätt i detta fall 2,00 ml stamlösning av intern standard (3.27.3) till hydrolysatet före indunstningen.
Tillsätt 2 droppar 1-oktanol (3.15) till det hydrolysat som erhållits enligt 5.3.2.3 eller 5.3.2.4.
Låt provet indunsta i rotationsindunstare (4.7) till 5–10 ml i vakuum vid 40 oC. Om volymen av någon anledning reduceras till mindre än 5 ml måste hydrolysatet kastas och analysen göras om.
Justera pH till 2,20 med natriumhydroxidlösning (3.18) och fortsätt till 5.3.4.
5.3.3.2
Ta det hydrolysat som erhölls enligt 5.3.2.3 eller 5.3.2.4 och neutralisera det delvis genom att försiktigt och under omrörning tillsätta 17 ml natriumhydroxidlösning (3.17), och se till att temperaturen hålls under 40 oC.
Justera pH till 2,20 vid rumstemperatur med natriumhydroxidlösning (först 3.17 och sedan 3.18). Fortsätt till 5.3.4.
5.3.4
Överför kvantitativt det pH-justerade hydrolysatet (5.3.3.1 eller 5.3.3.2) med citratbuffert (3.24) till en 200 ml mätkolv och späd till märket med buffert (3.24).
Om en intern standard inte redan använts, tillsätt 2,00 ml av denna (3.27.3) och späd till märket med citratbuffert (3.24). Blanda omsorgsfullt.
Gå vidare till kromatografin enligt 5.4.
Om provlösningarna inte kromatograferas samma dag måste de förvaras vid en temperatur under 5 oC.
5.4 Kromatografi
Före kromatografin, låt extraktet (5.2) eller hydrolysatet (5.3.4) anta rumstemperatur. Skaka blandningen och filtrera en lämplig mängd genom ett 0,22 μm membranfilter (4.5). Den klara lösning som erhålls får genomgå jonbyteskromatografi i en aminosyraanalysator (4.9).
Injektionen kan utföras antingen manuellt eller automatiskt. Det är viktigt att samma mängd lösning ± 0,5 % alltid tillsätts kolonnen för analys av standardlösning och prov, utom när en intern standard används, och att kvoten natrium/aminosyra i standard- och provlösning är så lika som möjligt.
Hur ofta kalibreringsprov behöver köras beror i allmänhet på ninhydrinreagensens stabilitet och typen av analyssystem. Standarden eller provet späds med citratbuffert (3.24) för att ge en topparea för standarden på 30–200 % av provets aminosyratopparea.
Kromatografin av aminosyror varierar något beroende på typen av analysator och harts. Det valda systemet måste kunna separera aminosyrorna från varandra och från ninhydrinpositiva material. Kromatografisystemet bör i driftsområdet ge en linjär respons på förändringar i de aminsyramängder som tillförs kolonnen.
Under kromatografisteget gäller de botten/toppkvoter som anges nedan när en ekvimolär lösning (av de aminosyror som mäts) analyseras. Denna ekvimolära lösning måste innehålla minst 30 % av den maximala mängd av varje aminosyra som med noggrannhet kan bestämmas med analyssystemet (4.9).
För separering av treonin-serin bör botten/toppkvoten för den lägre av de två överlappande aminosyrorna på kromatogrammet inte överstiga 2:10. (Om endast cyst(e)in, metionin, treonin och lysin bestäms kommer otillräcklig separation från angränsande toppar att påverka bestämningen). För alla andra aminosyror måste separationen vara bättre än 1:10.
Systemet måste kunna separera lysin från ”lysinartefakter” och ornitin.
6. Beräkning av resultat
Arean för provets och standardens toppar mäts för varje enskild aminosyra, och mängden (X) i gram aminosyra per kg prov beräknas med formeln

Om intern standard används, multiplicera med
![]()
|
A |
= |
topparea, hydrolysat eller extrakt |
|
B |
= |
topparea, kalibreringsstandardlösning |
|
C |
= |
topparea, intern standard i hydrolysat eller extrakt |
|
D |
= |
topparea, intern standard, kalibreringsstandardlösning |
|
M |
= |
molvikt för den aminosyra som bestäms |
|
c |
= |
standardlösningens koncentration i μmol/ml |
|
m |
= |
provets vikt (g) (korrigerad till ursprungsvikten om provet torkats eller avfettats) |
|
V |
= |
ml totalt hydrolysat (5.3.4) eller ml beräknad total spädningsvolym för extraktet (6.1). |
Cystin och cystein bestäms båda som cysteinsyra i hydrolysat av oxiderat prov, men beräknas som cystin (C6H12N2O4S2, molvikt 240,30 g/mol) med hjälp av molvikten 120,15 g/mol (= 0,5 × 240,30 g/mol).
Metionin bestäms som metioninsulfon i hydrolysat av oxiderat prov, men beräknas som metionin med hjälp av molvikten för metionin: 149,21 g/mol.
Tillsatt fritt metionin bestäms efter extraktion som metionin, för beräkningen används samma molvikt.
6.1. Den totala spädningsvolymen för extrakten (F) för bestämning av fria aminosyror (5.2) beräknas med formeln

|
V |
= |
Det färdiga extraktets volym |
7. Utvärderingen av metoden
Metoden testades i en internationell provningsjämförelse 1990 med fyra olika fodertyper (blandat grisfoder, foderblandning för slaktkycklingar, proteinkoncentrat, förblandning). Resultaten, efter eliminering av extremvärden, i form av medelvärden och standardavvikelser redovisas i nedanstående tabeller.
Medelvärden i g/kg
|
Referensmaterial |
Aminosyra |
|||
|
Treonin |
Cyst(e)in |
Metionin |
Lysin |
|
|
Grisfoder |
6,94 n = 15 |
3,01 n = 17 |
3,27 n = 17 |
9,55 n = 13 |
|
Foderblandning för slaktkycklingar |
9,31 n = 16 |
3,92 n = 18 |
5,08 n = 18 |
13,93 n = 16 |
|
Proteinkoncentrat |
22,32 n = 16 |
5,06 n = 17 |
12,01 n = 17 |
47,74 n = 15 |
|
Förblandning |
58,42 n= 16 |
— |
90,21 n = 16 |
98,03 n = 16 |
|
n = Antal deltagande laboratorier. |
||||
7.1 Repeterbarhet
Repeterbarheten uttryckt som ”standardavvikelse inom laboratoriet” i ovannämnda provningsjämförelse redovisas i nedanstående tabell.
Standardavvikelse inom laboratoriet (Sr) i g/kg
|
Referensmaterial |
Aminosyra |
|||
|
Treonin |
Cyst(e)in |
Metionin |
Lysin |
|
|
Grisfoder |
0,13 n = 15 |
0,10 n = 17 |
0,11 n = 17 |
0,26 n = 13 |
|
Foderblandning för slaktkycklingar |
0,20 n = 16 |
0,11 n = 18 |
0,16 n = 18 |
0,28 n = 16 |
|
Proteinkoncentrat |
0,48 n = 16 |
0,13 n = 17 |
0,27 n = 17 |
0,99 n = 15 |
|
Förblandning |
1,30 n= 16 |
— |
2,19 n = 16 |
2,06 n = 16 |
|
n = Antal deltagande laboratorier. |
||||
Variationskoefficient (%) för standardavvikelse inom laboratoriet (Sr)
|
Referensmaterial |
Aminosyra |
|||
|
Treonin |
Cyst(e)in |
Metionin |
Lysin |
|
|
Grisfoder |
1,9 n = 15 |
3,3 n = 17 |
3,4 n = 17 |
2,8 n = 13 |
|
Foderblandning för slaktkycklingar |
2,1 n = 16 |
2,8 n = 18 |
3,1 n = 18 |
2,1 n = 16 |
|
Proteinkoncentrat |
2,7 n = 16 |
2,6 n = 17 |
2,2 n = 17 |
2,4 n = 15 |
|
Förblandning |
2,2 n = 16 |
— |
2,4 n = 16 |
2,1 n = 16 |
|
n = Antal deltagande laboratorier. |
||||
7.2 Reproducerbarhet
Standardavvikelsen mellan laboratorier i ovannämnda provningsjämförelse redovisas i nedanstående tabell.
Standardavvikelse mellan laboratorier (SR) i g/kg
|
Referensmaterial |
Aminosyra |
|||
|
Treonin |
Cyst(e)in |
Metionin |
Lysin |
|
|
Grisfoder |
0,28 n = 15 |
0,30 n = 17 |
0,23 n = 17 |
0,30 n = 13 |
|
Foderblandning för slaktkycklingar |
0,48 n = 16 |
0,34 n = 18 |
0,55 n = 18 |
0,75 n = 16 |
|
Proteinkoncentrat |
0,85 n = 16 |
0,62 n = 17 |
1,57 n = 17 |
1,24 n = 15 |
|
Förblandning |
2,49 n = 16 |
— |
6,20 n = 16 |
6,62 n = 16 |
|
n = Antal deltagande laboratorier. |
||||
Variationskoefficient (%) för standardavvikelse mellan laboratorier (SR)
|
Referensmaterial |
Aminosyra |
|||
|
Treonin |
Cyst(e)in |
Metionin |
Lysin |
|
|
Grisfoder |
4,1 n = 15 |
9,9 n = 17 |
7,0 n = 17 |
3,2 n = 13 |
|
Foderblandning för slaktkycklingar |
5,2 n = 16 |
8,8 n = 18 |
10,9 n = 18 |
5,4 n = 16 |
|
Proteinkoncentrat |
3,8 n = 16 |
12,3 n = 17 |
13,0 n = 17 |
3,0 n = 15 |
|
Förblandning |
4,3 n = 16 |
— |
6,9 n = 16 |
6,7 n = 16 |
|
n = Antal deltagande laboratorier. |
||||
8. Användning av referensmaterial
Korrekt tillämpning av metoden ska verifieras genom analys av certifierade referensmaterial om sådana finns tillgängliga. Kalibrering med godkända kalibreringslösningar för aminosyror rekommenderas.
9. Anmärkningar
9.1 På grund av skillnaderna mellan aminosyraanalysatorer bör de slutliga koncentrationerna hos kalibreringslösningarna för standardaminosyror (se 3.27.4 och 3.27.5) och hos hydrolysatet (se 5.3.4) användas som riktlinje.
Det intervall där sambandet mellan det verkliga värdet och det med apparaten uppmätta värdet är linjärt, måste kontrolleras för alla aminosyror.
Standardlösningen späds med citratbuffert så att man får toppar i mitten av intervallet.
9.2 I de fall HPLC används för att analysera hydrolysater måste försöksbetingelserna optimeras i enlighet med tillverkarens rekommendationer.
9.3 Om denna metod tillämpas på foder som innehåller mer än 1 % klorid (kraftfoder, mineralfoder eller tillskottsfoder) kan den uppmätta halten av metionin bli för låg. För att undvika detta måste provet behandlas på särskilt sätt.
G. BESTÄMNING AV TRYPTOFAN
1. Syfte och tillämpningsområde
Med denna metod kan totalhalten tryptofan och halten fritt tryptofan bestämmas i foder. Ingen åtskillnad görs mellan D- och L-formerna.
2. Princip
För bestämning av totalhalten tryptofan hydrolyseras provet i alkalisk miljö med mättad bariumhydroxidlösning och upphettas till 110 oC i 20 timmar. Efter hydrolysen tillsätts en intern standard.
För bestämning av halten fritt tryptofan extraheras provet i lätt sur miljö i närvaro av den interna standarden.
Halten av tryptofan och intern standard i hydrolysatet eller extraktet bestäms med HPLC med fluorescensdetektion.
3. Reagens
3.1 Dubbeldestillerat vatten eller vatten av likvärdig kvalitet ska användas (konduktivitet < 10 μS/cm).
3.2 Referenssubstans: tryptofan (renhet/halt ≥ 99 %) torkat under vakuum över fosforpentoxid.
3.3 Intern standard: α-metyltryptofan (renhet/halt ≥ 99 %) torkat under vakuum över fosforpentoxid.
3.4 Bariumhydroxidoktahydrat (undvik att exponera Ba(OH)2·8 H2O för större mängder luft, eftersom det då kan bildas BaCO3 som stör bestämningen) (jfr punkt 9.3).
3.5 Natriumhydroxid.
3.6 Ortofosforsyra, w = 85 % (w/w).
3.7 Saltsyra, ρ20 = 1,19 g/ml.
3.8 Metanol, HPLC-kvalitet.
3.9 Petroleumeter, kokpunktsintervall 40–60 oC.
3.10 Natriumhydroxidlösning, c = 1 mol/l:
Lös 40,0 g NaOH (3.5) i vatten och späd till 1 liter med vatten (3.1).
3.11 Saltsyra, c = 6 mol/l:
Späd 492 ml HCl (3.7) med vatten till slutvolymen 1 liter.
3.12 Saltsyra, c = 1 mol/l:
Späd 82 ml HCl (3.7) med vatten till slutvolymen 1 liter.
3.13 Saltsyra, c = 0,1 mol/l:
Späd 8,2 ml HCl (3.7) med vatten till slutvolymen 1 liter.
3.14 Ortofosforsyra, c = 0,5 mol/l:
Späd 34 ml ortofosforsyra (3.6) med vatten (3.1) till slutvolymen 1 liter.
3.15 Koncentrerad tryptofanlösning (3.2), c = 2,50 μmol/ml:
Lös 0,2553 g tryptofan (3.2) i saltsyra (3.13) i en 500 ml mätkolv och späd till märket med saltsyra (3.13). Lösningen kan lagras vid - 18 oC i högst 4 veckor.
3.16 Koncentrerad lösning av intern standard, c = 2,50 μmol/ml:
Lös 0,2728 g α-metyltryptofan (3.3) i saltsyra (3.13) i en 500 ml mätkolv och späd till märket med saltsyra (3.13). Lösningen kan lagras vid - 18 oC i högst 4 veckor.
3.17 Standardkalibreringslösning av tryptofan och intern standard:
Tag 2,00 ml koncentrerad tryptofanlösning (3.15) och 2,00 ml koncentrerad lösning av intern standard (α-metyltryptofan) (3.16). Späd med vatten (3.1) och metanol (3.8) till ungefär samma volym och metanolkoncentration (10–30 %) som det färdiga hydrolysatet.
Denna lösning ska vara nyberedd.
Den ska skyddas från direkt solljus under beredningen.
3.18 Ättiksyra.
3.19 1,1,1-triklor-2-metyl-2-propanol.
3.20 Etanolamin, w > 98 % (w/w).
3.21 Lösning av 1 gram 1,1,1-triklor-2-metyl-2-propanol (3.19) i 100 ml metanol (3.8).
3.22 Mobil fas för HPLC: 3,00 g ättiksyra (3.18) + 900 ml vatten (3.1) + 50,0 ml lösning (3.21) av 1,1,1-triklor-2-metyl-2-propanol (3.19) i metanol (3.8) (1 g/100 ml). Justera pH till 5,00 med etanolamin (3.20). Späd till 1 000 ml med vatten (3.1).
4. Utrustning
4.1 HPLC-utrustning med en spektrofluorometrisk detektor.
4.2 HPLC-kolonn, 125 × 4 mm, C18, 3 μm packning, eller motsvarande.
4.3 pH-meter.
4.4 Polypropylenflaska, 125 ml, med vid hals och skruvkork.
4.5 Membranfilter, 0,45 μm.
4.6 Autoklav, 110 (± 2) oC, 1,4 (±0,1 ) bar.
4.7 Mekanisk skakapparat eller magnetomrörare.
4.8 Vortexblandare.
5. Utförande
5.1 Provberedning
Provet mals så att det kan passera genom en 0,5 mm sikt. Prov med hög vattenhalt ska antingen lufttorkas vid en temperatur på högst 50 oC eller frystorkas före malningen. Prov med hög fetthalt ska extraheras med petroleumeter (3.9) före malning.
5.2 Bestämning av fritt tryptofan (extrakt)
Väg med en noggrannhet av 1 mg upp en lämplig mängd (1–5 g) av det beredda provet (5.1) i en E-kolv. Tillsätt 100,0 ml saltsyra (3.13) och 5,00 ml koncentrerad lösning av intern standard (3.16). Skaka eller blanda i 60 minuter med en mekanisk skakapparat eller magnetomrörare (4.7). Låt sedimentera och pipettera sedan över 10,0 ml av supernatanten till en bägare. Tillsätt 5 ml ortofosforsyra (3.14). Justera pH till 3 med natriumhydroxid (3.10). Tillsätt så mycket metanol (3.8) att koncentrationen blir 10–30 % i slutvolymen. Överför till en mätkolv av lämplig volym och späd med vatten till den volym som behövs för kromatografi (ungefär samma volym som standardkalibreringslösningen [3.17]).
Filtrera några ml av lösningen genom ett 0,45 μm membranfilter (4.5) före injektion i HPLC-kolonnen. Gå vidare till kromatografin enligt 5.4.
Skydda standardlösning och extrakt från direkt solljus. Om extrakten inte kan analyseras samma dag kan de lagras vid 5 oC i högst 3 dagar.
5.3 Bestämning av totalhalt tryptofan (hydrolysat)
Väg med en noggrannhet av 0,2 mg upp 0,1 –1 g av det beredda provet (5.1) i en polypropylenflaska (4.4). Det invägda provet ska innehålla ca 10 mg kväve. Tillsätt 8,4 g bariumhydroxidoktahydrat (3.4) och 10 ml vatten. Blanda på en vortexblandare (4.8) eller med magnetomrörare (4.7). Lämna kvar den teflonbelagda magneten i blandningen. Skölj kärlets väggar med 4 ml vatten. Sätt på skruvkorken och skruva åt den löst. Sätt kärlet i en autoklav (4.6) med kokande vatten och låt ånga i 30–60 minuter. Stäng autoklaven och autoklavera vid 110 (± 2) oC i 20 timmar.
Sänk temperaturen till strax under 100 oC innan autoklaven öppnas. Tillsätt 30 ml rumstempererat vatten till den varma blandningen för att undvika kristallisering av Ba(OH)2·8 H2O. Skaka eller rör försiktigt. Tillsätt 2,00 ml koncentrerad lösning av intern standard (α-metyltryptofan) (3.16). Kyl kärlet i isvattenbad i 15 minuter.
Tillsätt 5 ml ortofosforsyra (3.14). Låt kärlet vara kvar i kylbadet och neutralisera lösningen med saltsyra (3.11) under omrörning. Justera pH till 3,0 med saltsyra (3.12). Tillsätt så mycket metanol att koncentrationen blir 10–30 % i slutvolymen. Överför till en mätkolv av lämplig volym och späd med vatten till den volym som behövs för kromatografi (t.ex. 100 ml). Tillsatsen av metanol bör göras utan att någon fällning bildas.
Filtrera några ml av lösningen genom ett 0,45 μm membranfilter (4.5) före injektion i HPLC-kolonnen. Gå vidare till kromatografin enligt 5.4.
Skydda standardlösning och hydrolysat mot direkt solljus. Om hydrolysaten inte kan analyseras samma dag kan de lagras vid 5 oC i högst tre dagar.
5.4 HPLC-bestämning
Följande betingelser för isokratisk eluering anges som vägledning. Andra betingelser kan väljas om de ger likvärdiga resultat (se även 9.1 och 9.2).
|
HPLC kolonn (4.2): |
125 × 4 mm, C18, 3 μm packning eller motsvarande |
|
Kolonntemperatur: |
Rumstemperatur |
|
Mobil fas (3.22): |
3,00 g ättiksyra (3.18) + 900 ml vatten (3.1) + 50,0 ml lösning (3.21) av 1,1,1-triklor-2-metyl-2-propanol (3.19) i metanol (3.8) (1 g/100 ml). Justera pH till 5,00 med etanolamin (3.20). Späd till 1 000 ml med vatten (3.1) |
|
Flödeshastighet: |
1 ml/min |
|
Total körningstid: |
ca 34 minuter |
|
Detektionsvåglängd: |
excitation: 280 nm, emission: 356 nm. |
|
Injektionsvolym: |
20 μl |
6. Beräkning av resultat
Mängden tryptofan (X) i gram per 100 g prov beräknas med formeln

|
A |
= |
topparea för intern standard, kalibreringsstandardlösning (3.17) |
|
B |
= |
topparea för tryptofan, extrakt (5.2) eller hydrolysat (5.3) |
|
V1 |
= |
volym i ml (2 ml) av koncentrerad tryptofanlösning (3.15) som satts till kalibreringslösningen (3.17) |
|
c |
= |
koncentration i μmol/ml (= 2,50 ) av koncentrerad tryptofanlösning (3.15) som satts till kalibreringslösningen (3.17) |
|
V2 |
= |
volym i ml av koncentrerad lösning av intern standard (3.16) som satts till vid extraktionen (5.2) (= 5,00 ml) eller till hydrolysatet (5.3) (= 2,00 ml) |
|
C |
= |
topparea för intern standard, extrakt (5.2) eller hydrolysat (5.3) |
|
D |
= |
topparea för tryptofan, kalibreringsstandardlösning (3.17) |
|
V3 |
= |
volym i ml (2,00 ml) av koncentrerad lösning av intern standard (3.16) som satts till kalibreringsstandardlösningen (3.17) |
|
m |
= |
provets vikt i gram (korrigerad till den ursprungliga vikten om provet har torkats och/eller avfettats) |
|
M |
= |
molvikt för tryptofan (= 204,23 g/mol). |
7. Repeterbarhet
Skillnaden mellan resultaten från två parallella bestämningar som utförs på samma prov får inte överstiga 10 % av det högre värdet.
8. Resultat av en provningsjämförelse
I en provningsjämförelse inom EU (fjärde jämförelsen) analyserades tre prov av upp till tolv laboratorier för att hydrolysmetoden skulle kunna certifieras. Varje prov analyserades fem gånger. Resultaten redovisas i nedanstående tabell.
|
|
Prov 1 Grisfoder |
Prov 2 Grisfoder med tillsats av L-tryptofan |
Prov 3 Kraftfoder för grisar |
|
L |
12 |
12 |
12 |
|
n |
50 |
55 |
50 |
|
Medelvärde [g/kg] |
2,42 |
3,40 |
4,22 |
|
sr [g/kg] |
0,05 |
0,05 |
0,08 |
|
r [g/kg] |
0,14 |
0,14 |
0,22 |
|
CVr [ %] |
1,9 |
1,6 |
1,9 |
|
SR [g/kg] |
0,15 |
0,20 |
0,09 |
|
R [g/kg] |
0,42 |
0,56 |
0,25 |
|
CVR [ %] |
6,3 |
6,0 |
2,2 |
|
L |
= |
antal laboratorier som lämnade in resultat |
|
n |
= |
antal återstående enskilda resultat efter eliminering av extremvärden (enligt Cochran-Dixons extremvärdestest) |
|
sr |
= |
standardavvikelse för repeterbarhet |
|
SR |
= |
standardavvikelse för reproducerbarhet |
|
r |
= |
repeterbarhet |
|
R |
= |
reproducerbarhet |
|
CVr |
= |
variationskoefficient för repeterbarhet, % |
|
CVR |
= |
variationskoefficient för reproducerbarhet, % |
I en annan provningsjämförelse inom EU (tredje jämförelsen) analyserades två prov av upp till tretton laboratorier för certifiering av extraktionsmetoden för fritt tryptofan. Varje prov analyserades fem gånger. Resultaten redovisas i nedanstående tabell.
|
|
Prov 4 Vete- och sojablandning |
Prov 5 Vete- och sojablandning (= prov 4) med tillsats av tryptofan (0,457 g/kg) |
|
L |
12 |
12 |
|
n |
55 |
60 |
|
Medelvärde [g/kg] |
0,391 |
0,931 |
|
sr [g/kg] |
0,005 |
0,012 |
|
r [g/kg] |
0,014 |
0,034 |
|
CVr [ %] |
1,34 |
1,34 |
|
SR [g/kg] |
0,018 |
0,048 |
|
R [g/kg] |
0,050 |
0,134 |
|
CVR [ %] |
4,71 |
5,11 |
|
L |
= |
antal laboratorier som lämnade in resultat |
|
n |
= |
antalet återstående enskilda resultat efter eliminering av extremvärden (enligt Cochran-Dixons extremvärdestest) |
|
sr |
= |
standardavvikelse för repeterbarhet |
|
SR |
= |
standardavvikelse för reproducerbarhet |
|
r |
= |
repeterbarhet |
|
R |
= |
reproducerbarhet |
|
CVr |
= |
variationskoefficient för repeterbarhet, % |
|
CVR |
= |
variationskoefficient för reproducerbarhet, % |
I ytterligare en provningsjämförelse inom EU analyserades fyra prov av upp till sju laboratorier för certifiering av metoden för tryptofanhydrolys. Resultaten redovisas i nedanstående tabell. Varje prov analyserades fem gånger.
|
|
Prov 1 Blandat grisfoder (CRM 117) |
Prov 2 Fiskmjöl med låg fetthalt (CRM 118) |
Prov 3 Sojamjöl (CRM 119) |
Prov 4 Skummjölkspulver (CRM 120) |
|
L |
7 |
7 |
7 |
7 |
|
n |
25 |
30 |
30 |
30 |
|
Medelvärde [g/kg] |
2,064 |
8,801 |
6,882 |
5,236 |
|
sr [g/kg] |
0,021 |
0,101 |
0,089 |
0,040 |
|
r [g/kg] |
0,059 |
0,283 |
0,249 |
0,112 |
|
CVr [ %] |
1,04 |
1,15 |
1,30 |
0,76 |
|
SR [g/kg] |
0,031 |
0,413 |
0,283 |
0,221 |
|
R [g/kg] |
0,087 |
1,156 |
0,792 |
0,619 |
|
CVR [ %] |
1,48 |
4,69 |
4,11 |
4,22 |
|
L |
= |
antal laboratorier som lämnade in resultat |
|
n |
= |
antalet återstående enskilda resultat efter eliminering av extremvärden (enligt Cochran-Dixons extremvärdestest) |
|
sr |
= |
standardavvikelse för repeterbarhet |
|
SR |
= |
standardavvikelse för reproducerbarhet |
|
r |
= |
repeterbarhet |
|
R |
= |
reproducerbarhet |
|
CVr |
= |
variationskoefficient för repeterbarhet, % |
|
CVR |
= |
variationskoefficient för reproducerbarhet, % |
9. Anmärkningar
9.1 För att få bättre separation mellan tryptofan och α-metyltryptofan kan man prova att använda följande kromatografiska parametrar:
Isokratisk eluering följt av gradientkolonnrening:
|
HPLC-kolonn: |
125 × 4 mm, C18, 5 μm packning eller motsvarande |
||
|
Kolonntemperatur: |
32 oC |
||
|
Mobil fas: |
A: 0,01 mol/l KH2PO4/metanol, 95+5 (v+v) B: metanol |
||
|
Gradientprogram: |
0 min |
100 % A |
0 % B |
|
|
15 min |
100 % A |
0 % B |
|
|
17 min |
60 % A |
40 % B |
|
|
19 min |
60 % A |
40 % B |
|
|
21 min |
100 % A |
0 % B |
|
|
33 min |
100 % A |
0 % B |
|
Flödeshastighet: |
1,2 ml/min |
||
|
Total körningstid: |
ca 33 min |
||
9.2 Kromatografin varierar beroende på typen av HPLC och vilket slags packning som används i kolonnen. Det valda systemet måste klara av att ge baslinjeseparation mellan tryptofan och intern standard. Det är också viktigt att nedbrytningsprodukter tydligt kan separeras från tryptofan och intern standard. Man bör köra hydrolysat utan intern standard för att kunna upptäcka eventuella orenheter på baslinjen under den interna standarden. Det är viktigt att körningstiden är så lång att alla nedbrytningsprodukter hinner elueras ut, annars kan sena elueringstoppar störa efterföljande körningar.
Kromatografisystemet måste ge linjär respons i driftsintervallet. Den linjära responsen ska mätas med en konstant koncentration (den normala) av intern standard och varierande koncentrationer av tryptofan. Det är viktigt att topparna för både tryptofan och intern standard ligger inom det linjära intervallet för HPLC-/fluorescenssystemet. Om topparna för tryptofan och/eller intern standard är för låga eller för höga bör bestämningen göras om med annan provstorlek och/eller slutvolym.
9.3 Bariumhydroxid
Bariumhydroxid blir mer svårlösligt när det åldras. Detta ger en grumlig lösning för HPLC-bestämning, vilket kan ge alltför låga värden för tryptofan.
H. BESTÄMNING AV RÅFETT
1. Syfte och tillämpningsområde
Med denna metod kan råfetthalten i foder bestämmas. Metoden omfattar inte analys av oljeväxtfrön och oljehaltiga frukter.
Beroende på fodrets egenskaper och sammansättning samt bestämningens syfte används en av nedanstående två metoder.
1.1 Metod A – direkt extraherbart råfett
Denna metod är tillämplig på foderråvaror av vegetabiliskt ursprung, utom dem som omfattas av metod B.
1.2 Metod B – total råfetthalt
Denna metod är tillämplig på foderråvaror av animaliskt ursprung och på alla foderblandningar. Den ska användas för alla råvaror från vilka fett inte kan extraheras helt utan föregående hydrolys, till exempel gluten, jäst, potatisprotein och produkter som genomgår processer såsom extrudering, bearbetning till flingor samt uppvärmning.
1.3 Tolkning av resultaten
I samtliga fall där ett högre resultat fås genom metod B än metod A, ska resultatet av metod B räknas som det sanna värdet.
2. Princip
2.1 Metod A
Provet extraheras med petroleumeter. Lösningsmedlet destilleras av och återstoden torkas och vägs.
2.2 Metod B
Provet behandlas under uppvärmning med saltsyra. Blandningen kyls och filtreras. Återstoden sköljs och torkas och bestäms enligt metod A.
3. Reagens
3.1 Petroleumeter, kokpunktsintervall 40–60 oC. Bromtalet måste vara lägre än 1 och återstoden efter förångning mindre än 2 mg/100 ml.
3.2 Vattenfritt natriumsulfat.
3.3 Saltsyra, c = 3 mol/l.
3.4 Filtreringshjälpmedel, till exempel kiselgur, Hyflo-supercel.
4. Utrustning
4.1 Extraktionsutrustning. Om denna är försedd med hävert (Soxhletapparat), ska förångningstakten vara sådan att ca tio kretslopp sker per timme. Om hävert saknas ska förångningstakten vara ca 10 ml per minut.
4.2 Extraktionshylsorna måste vara fria från ämnen som är lösliga i petroleumeter och ha en porositet som överensstämmer med kraven i 4.1.
4.3 Torkugn, antingen en vakuumtorkugn inställd på 75 ± 3 oC eller en varmluftsugn inställd på 100 ± 3 oC.
5. Utförande
5.1 Metod A (se punkt 8.1)
Väg 5 gram av provet med en noggrannhet av 1 mg, överför det till en extraktionshylsa (4.2) och täck med en fettfri bomullstuss.
Placera hylsan i en extraktionsapparat (4.1) och extrahera i sex timmar med petroleumeter (3.1). Samla upp petroleumeterextraktet i en torr, vägd kolv med pimpstensskärvor ( 9 ).
Destillera av lösningsmedlet. Torka återstoden i 1,5 timmar i torkugnen (4.3). Kyl i en exsickator och väg. Torka igen i 30 minuter för att säkerställa att fettets vikt är konstant (viktförlusten mellan två på varandra följande vägningar får inte överstiga 1 mg).
5.2 Metod B
Väg upp 2,5 gram av provet med en noggrannhet av 1 mg (se punkt 8.2) i en 400 ml bägare eller en 300 ml E-kolv. Tillsätt 100 ml saltsyra (3.3) och pimpstensskärvor. Täck över bägaren med ett urglas eller koppla E-kolven till en återloppskylare. Låt blandningen koka sakta i 1 timme över en svag låga eller värmeplatta. Låt inte produkten fastna på behållarens insida.
Kyl och tillsätt så mycket filtreringshjälpmedel (3.4) att inget fett förloras under filtreringen. Filtrera genom ett fuktat, fettfritt, dubbelt filtrerpapper. Skölj återstoden i kallt vatten tills ett neutralt filtrat erhålls. Kontrollera att filtratet inte innehåller något fett. Om så ändå är fallet ska provet extraheras med petroleumeter, enligt metod A, före hydrolys.
Lägg det dubbla filtrerpappret med återstoden på ett urglas och torka i 1,5 timmar i varmluftsugnen (4.3) vid 100 ± 3 oC.
Placera det dubbla filtrerpappret med den torra återstoden i en extraktionshylsa (4.2) och täck med en fettfri bomullstuss. Placera hylsan i en extraktionsapparat (4.1) och följ anvisningarna i punkt 5.1 andra och tredje styckena.
6. Resultatangivelse
Återstodens vikt uttrycks i procent av provet.
7. Repeterbarhet
Skillnaden mellan resultaten av två parallella bestämningar som utförs på samma prov av samma person får inte överstiga
— 0,2 % i absoluta tal för råfetthalter under 5 %,
— 4,0 % av det högre värdet för halter på 5–10 %,
— 0,4 % i absoluta tal för halter över 10 %.
8. Anmärkningar
8.1 För produkter med hög fetthalt, som är svåra att krossa eller från vilka det är svårt att få fram ett homogent reducerat prov, används följande förfarande.
Väg upp 20 gram av provet med en noggrannhet av 1 mg och blanda med minst 10 gram vattenfritt natriumsulfat (3.2). Extrahera med petroleumeter (3.1) enligt punkt 5.1. Späd extraktet med petroleumeter (3.1) till 500 ml och blanda. Överför 50 ml av lösningen till en liten, torr, vägd kolv som innehåller pimpstensskärvor. Destillera bort lösningsmedlet, torka och följ anvisningarna i punkt 5.1 sista stycket.
Avlägsna lösningsmedlet från extraktionsåterstoden i hylsan, krossa återstoden till 1 mm partikelstorlek, återför till extraktionshylsan (tillsätt inte natriumsulfat) och följ anvisningarna i punkt 5.1 andra och tredje styckena.
Provets fetthalt i procent beräknas med formeln
(10m1 + m2) × 5
där:
|
m1 |
= |
återstodens vikt i gram efter första extraktionen (den alikvot av extraktet som används för analys) |
|
m2 |
= |
återstodens vikt i gram efter andra extraktionen. |
8.2 För produkter med låg fetthalt kan provmängden ökas till 5 gram.
8.3 Sällskapsdjursfoder med hög vattenhalt kan behöva blandas med vattenfritt natriumsulfat före hydrolys och extraktion enligt metod B.
8.4 I punkt 5.2 kan det vara effektivare att använda varmt vatten i stället för kallt vatten för att tvätta återstoden efter filtrering.
8.5 Torktiden på 1,5 timmar kan behöva förlängas för vissa foder. För lång torktid bör undvikas eftersom detta kan leda till låga resultat. En mikrovågsugn kan också användas.
8.6 Förextraktion enligt metod A före hydrolys och upprepad extraktion enligt metod B rekommenderas om råfetthalten är högre än 15 %. Detta beror delvis på fodrets egenskaper och på typen av fett i fodret.
I. BESTÄMNING AV VÄXTTRÅD
1. Syfte och tillämpningsområde
Med denna metod är det möjligt att bestämma foders halt av fettfria organiska ämnen som är olösliga i sur och basisk miljö och som vanligen betecknas växttråd.
2. Princip
Provet, som vid behov avfettas, behandlas i tur och ordning med kokande lösningar av svavelsyra och kaliumhydroxid i bestämda koncentrationer. Återstoden separeras genom filtrering på ett sintrat glasfilter och sköljs, torkas, vägs och föraskas vid en temperatur på 475–500 oC. Viktförlusten efter föraskning motsvarar andelen växttråd i analysprovet.
3. Reagens
3.1 Svavelsyra, c = 0,13 mol/l.
3.2 Skumdämpningsmedel (t.ex. n-oktanol).
3.3 Filtreringshjälpmedel (Celite 545 eller motsvarande), upphettat till 500 oC i fyra timmar (8.6).
3.4 Aceton.
3.5 Petroleumeter, kokpunktsintervall 40–60 oC.
3.6 Saltsyra, c = 0,5 mol/l.
3.7 Kaliumhydroxidlösning, c = 0,23 mol/l.
4. Utrustning
4.1 Värmeenhet för uppslutning med svavelsyra och kaliumhydroxidlösning, försedd med en hållare för filterdegeln (4.2) och ett utloppsrör med kran för avtappning av vätska samt anslutning för vakuum och eventuellt tryckluft. Innan enheten tas i bruk varje dag ska den förvärmas med kokande vatten i 5 minuter.
4.2 Glasfilterdegel med insmält filterskiva av sintrat glas, porstorlek 40–90 μm. Innan degeln används första gången, värm den till 500 oC i några minuter och låt den svalna (8.6).
4.3 Cylinder på minst 270 ml med återloppskylare, lämplig för kokning.
4.4 Torkugn med termostat.
4.5 Muffelugn med termostat.
4.6 Extraktionsenhet bestående av en stödplatta för filterdegeln (4.2) och med ett utloppsrör med kran för utsläpp av vakuum och vätska.
4.7 Kopplingsringar för ihopmontering av värmeenheten (4.1), filterdegeln (4.2) och cylindern (4.3) och för anslutning av extraktionsenheten (4.6) och filterdegeln.
5. Utförande
Väg med 1 mg noggrannhet upp 1 g av det beredda provet i filterdegeln (4.2), (jfr punkterna 8.1, 8.2 och 8.3) och tillsätt 1 g filtreringshjälpmedel (3.3).
Montera ihop värmeenheten (4.1) och filterdegeln (4.2) och anslut sedan cylindern (4.3) till filterdegeln. Häll 150 ml kokande svavelsyra (3.1) i cylinder/degeluppställningen och tillsätt vid behov några droppar skumdämpningsmedel (3.2).
Koka upp vätskan inom 5 ± 2 minuter och låt koka kraftigt i exakt 30 minuter.
Öppna kranen till utloppsröret (4.1) och vakuumfiltrera svavelsyran genom filterdegeln. Tvätta återstoden tre gånger med 30 ml kokande vatten per gång och se till att återstoden vakuumfiltreras torr efter varje tvättning.
Stäng utloppskranen och häll 150 ml kokande kaliumhydroxidlösning (3.7) i cylinder/degeluppställningen. Tillsätt några droppar skumdämpningsmedel (3.2). Koka upp vätskan inom 5 ± 2 minuter och låt koka kraftigt i exakt 30 minuter. Filtrera och tvätta på samma sätt som vid behandlingen med svavelsyra.
Efter den sista tvättningen och torkningen kopplas degeln med innehåll loss och ansluts till extraktionsenheten (4.6). Anslut vakuum, tvätta återstoden i degeln tre gånger med 25 ml aceton (3.4) per gång och se till att återstoden vakuumfiltreras torr efter varje tvättning.
Torka degeln till konstant vikt i ugnen vid 130 oC. Låt den svalna i exsickator efter varje torkning och väg snabbt. Sätt in degeln i en muffelugn och föraska till konstant vikt (viktförlusten mellan två på varandra följande vägningar får inte överstiga 2 mg) vid 475–500 oC i minst 30 minuter.
Efter varje upphettning ska degeln först svalna i ugnen och sedan i exsickatorn innan den vägs.
Gör ett blankprov utan provet. Viktförlusten till följd av föraskningen får inte överstiga 4 mg.
6. Beräkning av resultat
Provets växttrådhalt i procent beräknas med formeln

där:
|
m |
= |
provets vikt i gram |
|
m0 |
= |
viktförlust efter föraskning vid bestämningen, i gram |
|
m1 |
= |
viktförlust efter föraskning vid blankprovet, i gram. |
7. Repeterbarhet
Skillnaden mellan resultaten från två parallella bestämningar som utförs på samma prov får inte överstiga
— 0,6 % i absoluta tal om växttrådhalten är mindre än 10 %,
— 6 % av det högre värdet om växttrådhalten uppgår till 10 % eller mer.
8. Anmärkningar
8.1 Foder som innehåller mer än 10 % råfett ska före analys avfettas med petroleumeter (3.5). Anslut filterdegeln (4.2) med innehåll till extraktionsenheten (4.6), anslut vakuum, tvätta återstoden tre gånger med 30 ml petroleumeter och se till att återstoden vakuumfiltreras torr. Anslut degeln med innehåll till värmeenheten (4.1) och fortsätt enligt beskrivningen i punkt 5.
8.2 Foder som innehåller fetter som inte kan extraheras direkt med petroleumeter (3.5) ska avfettas enligt beskrivningen i 8.1 och därefter avfettas ännu en gång efter kokning med syra. Efter kokning med syra och därpå följande tvättning ansluts degeln med innehåll till extraktionsenheten (4.6) och tvättas tre gånger med 30 ml aceton per gång och därefter ytterligare tre gånger med 30 ml petroleumeter per gång. Vakuumfiltrera torrt och fortsätt analysen från och med kaliumhydroxidbehandlingen enligt punkt 5.
8.3 Om fodret innehåller mer än 5 % karbonat, uttryckt som kalciumkarbonat, ansluts degeln (4.2) med det uppvägda provet till värmeenheten (4.1). Tvätta provet tre gånger med 30 ml saltsyra (3.6) per gång. Låt provet stå ca en minut efter varje tillsats innan det filtreras. Tvätta en gång med 30 ml vatten och fortsätt sedan enligt beskrivningen i 5.
8.4 Om den utrustning som används utgörs av ett stativ (flera deglar anslutna till samma värmeenhet) får två enskilda bestämningar av samma analysprov inte ingå i samma provserie.
8.5 Om det efter kokningen är svårt att filtrera de sura och basiska lösningarna blåses tryckluft genom värmeenhetens utloppsrör, varefter filtreringen fullföljes.
8.6 För att förlänga glasfilterdeglarnas livslängd bör temperaturen vid föraskningen inte överstiga 500 oC. Var noga med att undvika alltför hastiga temperaturförändringar vid uppvärmning och avkylning.
J. BESTÄMNING AV SOCKER
1. Syfte och tillämpningsområde
Med denna metod är det möjligt att bestämma halten av reducerande socker och totalhalten socker efter invertering, uttryckt som glukos eller, om så är lämpligt, sackaros med omvandlingsfaktorn 0,95 . Metoden är tillämplig på foderblandningar. För andra typer av foder finns särskilda metoder. Om så krävs ska laktos bestämmas separat och tas med i resultatberäkningen.
2. Princip
Sockerarterna extraheras i utspädd etanol. Lösningen klaras med Carrez-lösning I och II. Efter det att etanolen eliminerats bestäms kvantiteterna före och efter invertering med Luff-Schoorl-metoden.
3. Reagens
3.1 Etanol 40 % (v/v), densitet: 0,948 g/ml vid 20o C, fenolftaleinneutral.
3.2 Carrez-lösning I: lös 21,9 g zinkacetat, Zn(CH3COO)2 2H2O, och 3 g isättika i vatten. Späd med vatten till 100 ml.
3.3 Carrez-lösning II: lös 10,6 g kaliumferrocyanid, K4Fe(CN)6 3H2O, i vatten. Späd med vatten till 100 ml.
3.4 Metylorange, lösning 0,1 % (w/v).
3.5 Saltsyra, 4 mol/l.
3.6 Saltsyra, 0,1 mol/l.
3.7 Natriumhydoxidlösning, 0,1 mol/l.
3.8 Luff-Schoorl-reagens:
Häll citronsyralösningen (3.8.2) i natriumkarbonatlösningen (3.8.3) under noggrann omrörning. Tillsätt kopparsulfatlösningen (3.8.1) och späd med vatten till 1 liter. Låt stå över natten och filtrera.
Kontrollera koncentrationen hos det erhållna reagenset (Cu 0,05 mol/l, Na2CO3 1 mol/l), se punkt 5.4 sista stycket. Lösningens pH ska vara ca 9,4 .
3.8.1 Kopparsulfatlösning: lös 25 g järnfritt kopparsulfat, CuSO4 5H2O, i 100 ml vatten.
3.8.2 Citronsyralösning: lös 50 g citronsyra C6H8O7·H2O i 50 ml vatten.
3.8.3 Natriumkarbonatlösning: lös upp 143,8 g vattenfritt natriumkarbonat i ca 300 ml varmt vatten. Låt svalna.
3.9 Natriumtiosulfatlösning, 0,1 mol/l.
3.10 Stärkelselösning: tillsätt en blandning av 5 g löslig stärkelse i 30 ml vatten till 1 liter kokande vatten. Koka i tre minuter, låt svalna och tillsätt vid behov 10 mg kvicksilverjodid som konserveringsmedel.
3.11 Svavelsyra, 3 mol/l.
3.12 Kaliumjodidlösning, 30 % (w/v).
3.13 Granulerad pimpsten som kokats i saltsyra, sköljts med vatten och torkats.
3.14 3-metylbutan-1-ol.
4. Utrustning
Rotationsskakapparat: ca 35–40 varv per minut.
5. Utförande
5.1 Extraktion av provet
Väg med en noggrannhet av 1 mg upp 2,5 g av provet i en 250 ml mätkolv. Tillsätt 200 ml etanol (3.1) och blanda på skakapparaten i 1 timme. Tillsätt 5 ml Carrez-lösning I (3.2) och rör om i ca 30 sekunder. Tillsätt 5 ml Carrez-lösning II (3.3) och rör om i 1 minut. Späd med etanol (3.1) till full volym, homogenisera och filtrera. Ta undan 200 ml av filtratet och låt indunsta till ungefär halv volym så att det mesta av etanolen elimineras. Överför återstoden efter avdunstningen kvantitativt, med varmt vatten, till en 200 ml mätkolv, kyl, späd med vatten till full volym, homogenisera och filtrera vid behov. Denna lösning används för att bestämma halten av reducerande socker och, efter invertering, totalhalten socker.
5.2 Bestämning av reducerande socker
Pipettera av högst 25 ml av lösningen med ett innehåll av mindre än 60 mg reducerande socker uttryckt som glukos. Späd vid behov med destillerat vatten till 25 ml och bestäm halten reducerande socker med Luff-Schoorl-metoden. Resultatet uttrycks som procent glukos i provet.
5.3 Bestämning av totalhalten socker efter invertering
Pipettera över 50 ml av lösningen till en 100 ml mätkolv. Tillsätt några droppar metylorangelösning (3.4) och därefter – försiktigt och under ständig omrörning – saltsyra (3.5) tills vätskan blir tydligt röd. Tillsätt 15 ml saltsyra (3.6), sänk ned kolven i ett kraftigt kokande vattenbad och låt stå i 30 minuter. Kyl hastigt till ca 20 oC och tillsätt 15 ml natriumhydroxidlösning (3.7). Späd med vatten till 100 ml och homogenisera. Avlägsna högst 25 ml av lösningen med ett innehåll av mindre än 60 mg reducerande socker uttryckt som glukos. Späd vid behov med destillerat vatten till 25 ml och bestäm halten reducerande socker med Luff-Schoorl-metoden. Resultatet uttrycks som procent glukos i provet eller, om så är lämpligt, sackaros, genom att multiplicera med faktorn 0,95 .
5.4 Titrering med Luff-Schoorl-metoden
Pipettera över 25 ml Luff-Schoorl-reagens (3.8) till en 300 ml E-kolv. Tillsätt exakt 25 ml av den klarade sockerlösningen. Tillsätt två korn pimpsten (3.13), hetta upp under omrörning för hand över medelstor öppen låga och låt vätskan koka i cirka två minuter. Ställ omedelbart E-kolven på ett asbestklätt trådnät med ett hål med ca 6 cm diameter under vilket en låga brinner. Lågan ska justeras så att endast E-kolvens botten hettas upp. Anslut en återloppskylare till E-kolven. Låt koka i exakt 10 minuter. Kyl omedelbart i kallt vatten och titrera efter ca 5 minuter på följande sätt:
Tillsätt 10 ml kaliumjodidlösning (3.12) och omedelbart därefter (försiktigt, på grund av risken för kraftig skumbildning) 25 ml svavelsyra (3.11). Titrera med natriumtiosulfatlösning (3.9) tills en matt gul färg uppträder, tillsätt stärkelseindikatorn (3.10) och slutför titreringen.
Utför samma titrering på en noggrant uppmätt blandning av 25 ml Luff-Schoorl-reagens (3.8) och 25 ml vatten efter tillsats av 10 ml kaliumjodidlösning (3.12) och 25 ml svavelsyra (3.11) utan kokning.
6. Beräkning av resultat
Fastställ med hjälp av tabellen nedan den glukosmängd i mg som motsvarar skillnaden mellan resultaten av de båda titreringarna, uttryckt i mg 0,1 M natriumtiosulfat. Uttryck resultatet i procent av provet.
7. Särskilda förfaranden
7.1 För foder som innehåller mycket melass eller som inte är särskilt homogena vägs 20 g upp och överförs med 500 ml vatten till en 1 000 ml mätkolv. Blanda på skakapparaten i en timme. Gör lösningen klar med Carrez-reagens I (3.2) och Carrez-reagens II (3.3) enligt beskrivningen i 5.1 men med fyra gånger större mängd av varje reagens. Späd till full volym med etanol 80 % (v/v).
Homogenisera och filtrera. Avlägsna etanolen enligt beskrivningen i 5.1. Om det inte finns någon dextrinerad stärkelse, tillsätts destillerat vatten till full volym.
7.2 För melass och foderråvaror som innehåller mycket socker och som är nästan stärkelsefria (johannesbröd, torkad betsnitsel osv.), vägs 5 g upp i en 250 ml mätkolv, varefter 200 ml destillerat vatten tillsätts och lösningen blandas på skakapparaten i en timme eller mer om så behövs. Gör lösningen klar med Carrez-reagens I (3.2) och Carrez-reagens II (3.3) enligt beskrivningen i 5.1. Späd med kallt vatten till full volym, homogenisera och filtrera. Bestäm totalhalten socker enligt beskrivningen i 5.3.
8. Anmärkningar
8.1 För att förhindra skumbildning är det lämpligt att (oberoende av volymen) tillsätta ca 1 ml 3-metylbutan-1-ol (3.14) före uppkokning med Luff-Schoorl-reagens.
8.2 Skillnaden mellan totalhalten socker efter invertering, uttryckt som glukos, och halten reducerande socker, uttryckt som glukos, multiplicerad med 0,95 ger sackaroshalten i procent.
8.3 För att bestämma halten reducerande socker med undantag av laktos kan två metoder användas:
8.3.1 För en ungefärlig beräkning multipliceras den laktoshalt som fastställts med en annan analysmetod med 0,675 , och det erhållna resultatet dras från halten reducerande socker.
8.3.2 För en exakt beräkning av reducerande socker med undantag av laktos måste samma prov användas för de två slutliga bestämningarna. Den ena analysen utförs på en del av den lösning som erhållits enligt 5.1 och den andra på en del av den lösning som erhållits vid bestämningen av laktos med den metod som fastställts för detta ändamål (efter det att de andra sockerarterna jästs och lösningen klarats).
I båda fallen bestäms mängden befintligt socker med Luff-Schoorl-metoden och beräknas i mg glukos. Det ena av värdena dras från det andra och skillnaden uttrycks i procent av provet.
Exempel
De två valda volymerna motsvarar vid varje bestämning ett prov på 250 mg.
I det första fallet förbrukas 17 ml 0,1 M natriumtiosulfatlösning, vilket motsvarar 44,2 mg glukos, och i det andra fallet 11 ml, vilket motsvarar 27,6 mg glukos.
Skillnaden är 16,6 mg glukos.
Halten reducerande socker (med undantag av laktos) beräknad som glukos är alltså:

Värdetabell för 25 ml Luff-Schoorl-reagens
ml Na2S2O30,1 mol/l, uppvärmning 2 minuter, kokning 10 minuter
|
Na2S2O3 0,1 mol/l |
Glukos, fruktos, invertsocker C6 H12 O6 |
Laktos C12H22O11 |
Maltos C12H22O11 |
Na2S2O3 0,1 mol/l |
|||
|
ml |
mg |
skillnad |
mg |
skillnad |
mg |
skillnad |
ml |
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
2,4 4,8 7,2 9,7 12,2 14,7 17,2 19,8 22,4 25,0 27,6 30,3 33,0 35,7 38,5 41,3 44,2 47,1 50,0 53,0 56,0 59,1 62,2 |
2,4 2,4 2,5 2,5 2,5 2,5 2,6 2,6 2,6 2,6 2,7 2,7 2,7 2,8 2,8 2,9 2,9 2,9 3,0 3,0 3,1 3,1 |
3,6 7,3 11,0 14,7 18,4 22,1 25,8 29,5 33,2 37,0 40,8 44,6 48,4 52,2 56,0 59,9 63,8 67,7 71,7 75,7 79,8 83,9 88,0 |
3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,8 3,8 3,8 3,8 3,8 3,8 3,9 3,9 3,9 4,0 4,0 4,1 4,1 4,1 |
3,9 7,8 11,7 15,6 19,6 23,5 27,5 31,5 35,5 39,5 43,5 47,5 51,6 55,7 59,8 63,9 68,0 72,2 76,5 80,9 85,4 90,0 94,6 |
3,9 3,9 3,9 4,0 3,9 4,0 4,0 4,0 4,0 4,0 4,0 4,1 4,1 4,1 4,1 4,1 4,2 4,3 4,4 4,5 4,6 4,6 |
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
K. BESTÄMNING AV LAKTOS
1. Syfte och tillämpningsområde
Med denna metod kan laktoshalten bestämmas i foder som innehåller mer än 0,5 % laktos.
2. Princip
Sockerarterna löses upp i vatten. Lösningen fermenteras med jästen Saccharomyces cerevisiae som lämnar laktosen opåverkad. Efter klarning och filtrering bestäms laktoshalten i filtratet med Luff-Schoorl-metoden.
3. Reagens
3.1 Suspension av Saccharomyces cerevisiae: slamma upp 25 g färsk jäst i 100 ml vatten. Suspensionen håller sig högst en vecka i kylskåp.
3.2 Carrez-lösning I: lös 21,9 g zinkacetat, Zn (CH3 COO)2 2H2O, och 3 g isättika i vatten. Späd med vatten till 100 ml.
3.3 Carrez-lösning II: lös 10,6 g kaliumferrocyanid, K4Fe(CN)6 3H2O, i vatten. Späd med vatten till 100 ml.
3.4 Luff-Schoorl-reagens:
Häll citronsyralösningen (3.4.2) i natriumkarbonatlösningen (3.4.3) under noggrann omrörning. Tillsätt kopparsulfatlösningen (3.4.1) och späd med vatten till 1 liter. Låt stå över natten och filtrera. Kontrollera koncentrationen hos det erhållna reagenset (Cu 0,05 mol/l, Na2CO3 1 mol/l). Lösningens pH ska vara ca 9,4 .
3.4.1 Kopparsulfatlösning: lös 25 g järnfritt kopparsulfat, CuSO4 5H2O, i 100 ml vatten.
3.4.2 Citronsyralösning: lös 50 g citronsyra, C6H8O7 · H2O, i 50 ml vatten.
3.4.3 Natriumkarbonatlösning: lös 143,8 g vattenfritt natriumkarbonat i ca 300 ml varmt vatten. Låt svalna.
3.5 Granulerad pimpsten som kokats i saltsyra, sköljts med vatten och torkats.
3.6 Kaliumjodidlösning, 30 % (w/v).
3.7 Svavelsyra, 3 mol/l.
3.8 Natriumtiosulfatlösning, 0,1 mol/l.
3.9 Stärkelselösning: tillsätt en blandning av 5 g löslig stärkelse i 30 ml vatten till 1 liter kokande vatten. Koka i 3 minuter, låt svalna och tillsätt vid behov 10 mg kvicksilverjodid som konserveringsmedel.
4. Utrustning
Vattenbad med termostaten inställd på 38–40 oC.
5. Utförande
Väg med en noggrannhet av 1 mg upp 1 g av provet i en 100 ml mätkolv. Tillsätt 25–30 ml vatten. Låt kolven stå i ett kokande vattenbad i 30 minuter och kyl sedan till ca 35 oC. Tillsätt 5 ml jästsuspension (3.1) och homogenisera. Låt kolven stå i vattenbad i två timmar vid en temperatur av 38–40 oC. Kyl därefter till ca 20 oC.
Tillsätt 2,5 ml Carrez-lösning I (3.2), rör om i 30 sekunder och tillsätt sedan 2,5 ml Carrez-lösning II (3.3) och rör om i ytterligare 30 sekunder. Späd med vatten till 100 ml, blanda och filtrera. Pipettera upp högst 25 ml av filtratet som bör innehålla 40–80 mg laktos och överför detta till en 300 ml E-kolv. Späd vid behov med vatten till 25 ml.
Utför ett blankprov på samma sätt med 5 ml jästsuspension (3.1). Fastställ laktoshalten enligt Luff-Schoorl på följande sätt: tillsätt exakt 25 ml Luff-Schoorl-reagens (3.4) och två korn pimpsten (3.5). Rör om för hand medan innehållet hettas upp över medelstor öppen låga och låt vätskan koka i ca 2 minuter. Ställ omedelbart E-kolven på ett asbestklätt trådnät med ett hål med ca 6 cm diameter under vilket en låga brinner. Lågan ska justeras så att endast E-kolvens botten hettas upp. Anslut en återloppskylare till E-kolven. Låt koka i exakt 10 minuter. Kyl omedelbart i kallt vatten och titrera efter ca 5 minuter på följande sätt:
Tillsätt 10 ml kaliumjodidlösning (3.6) och omedelbart därefter (försiktigt, på grund av risken för kraftig skumbildning) 25 ml svavelsyra (3.7). Titrera med natriumtiosulfatlösning (3.8) tills en matt gul färg uppträder, tillsätt stärkelseindikatorn (3.9) och slutför titreringen.
Utför samma titrering på en noggrant uppmätt blandning av 25 ml Luff-Schoorl-reagens (3.4) och 25 ml vatten efter tillsats av 10 ml kaliumjodidlösning (3.6) och 25 ml svavelsyra (3.7) utan kokning.
6. Beräkning av resultat
Fastställ med hjälp av tabellen nedan den laktosmängd i mg som motsvarar skillnaden mellan resultaten av de båda titreringarna, uttryckt i ml 0,1 M natriumtiosulfat.
Uttryck resultatet som halten vattenfri laktos i procent av provet.
7. Anmärkning
För produkter som innehåller mer än 40 % jäsbart socker används mer än 5 ml jästsuspension (3.1).
Värdetabell för 25 ml Luff-Schoorl-reagens
ml Na2S2O30,1 mol/l, uppvärmning 2 minuter, kokning 10 minuter
|
Na2S2O3 0,1 mol/l |
Glukos, fruktos, invertsocker C6H12O6 |
Laktos C12H22O11 |
Maltos C12H22O11 |
Na2S2O3 0,1 mol/l |
|||
|
ml |
mg |
skillnad |
mg |
skillnad |
mg |
skillnad |
ml |
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
2,4 4,8 7,2 9,7 12,2 14,7 17,2 19,8 22,4 25,0 27,6 30,3 33,0 35,7 38,5 41,3 44,2 47,1 50,0 53,0 56,0 59,1 62,2 |
2,4 2,4 2,5 2,5 2,5 2,5 2,6 2,6 2,6 2,6 2,7 2,7 2,7 2,8 2,8 2,9 2,9 2,9 3,0 3,0 3,1 3,1 |
3,6 7,3 11,0 14,7 18,4 22,1 25,8 29,5 33,2 37,0 40,8 44,6 48,4 52,2 56,0 59,9 63,8 67,7 71,7 75,7 79,8 83,9 88,0 |
3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,8 3,8 3,8 3,8 3,8 3,8 3,9 3,9 3,9 4,0 4,0 4,1 4,1 4,1 |
3,9 7,8 11,7 15,6 19,6 23,5 27,5 31,5 35,5 39,5 43,5 47,5 51,6 55,7 59,8 63,9 68,0 72,2 76,5 80,9 85,4 90,0 94,6 |
3,9 3,9 3,9 4,0 3,9 4,0 4,0 4,0 4,0 4,0 4,0 4,1 4,1 4,1 4,1 4,1 4,2 4,3 4,4 4,5 4,6 4,6 |
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
L. BESTÄMNING AV STÄRKELSE
POLARIMETRISK METOD
1. Syfte och tillämpningsområde
Med denna metod är det möjligt att bestämma halten av stärkelse och nedbrytningsprodukter av stärkelse med hög molekylvikt i foder, i syfte att kontrollera att det deklarerade energivärdet är korrekt (bestämmelser i bilaga VII) och att rådets direktiv 96/25/EG ( 10 ) efterlevs.
2. Princip
Metoden omfattar två bestämningar. I den första behandlas provet med utspädd saltsyra. Efter klarning och filtrering mäts lösningens optiska rotation polarimetriskt.
I den andra extraheras provet med 40 % etanol. Efter att filtratet surgjorts med saltsyra, klarats och filtrerats mäts den optiska rotationen på samma sätt som vid den första bestämningen.
Genom att multiplicera skillnaden mellan de båda mätningarna med en känd faktor erhålls provets stärkelsehalt.
3. Reagens
3.1 Saltsyra, lösning 25 % (w/w), densitet: 1,126 g/ml.
3.2 Saltsyra, lösning 1,13 % (w/v).
Koncentrationen måste kontrolleras genom titrering med 0,1 M natriumhydroxidlösning i närvaro av 0,1 % (w/v) metylrött i 94 % (v/v) etanol. För att neutralisera 10 ml krävs 30,94 ml 0,1 M NaOH.
3.3 Carrez-lösning I: lös 21,9 g zinkacetat, Zn(CH3COO)2 2H2O, och 3 g isättika i vatten. Späd med vatten till 100 ml.
3.4 Carrez-lösning II: lös 10,6 g kaliumferrocyanid, K4Fe(CN)6 3H2O, i vatten. Späd med vatten till 100 ml.
3.5 Etanol 40 % (v/v), densitet: 0,948 g/ml vid 20 oC.
4. Utrustning
4.1 250 ml E-kolv med standardfog av slipat glas och återloppskylare.
4.2 Polarimeter eller sackarimeter.
5. Utförande
5.1 Provberedning
Krossa provet tills det är tillräckligt finfördelat för att helt kunna passera genom en sikt med 0,5 mm runda maskor.
5.2 Bestämning av den totala optiska rotationen (P eller S) (se anmärkning 7.1)
Väg med en noggrannhet av 1 mg upp 2,5 g av det krossade provet i en 100 ml mätkolv. Tillsätt 25 ml saltsyra (3.2), skaka om så att provet fördelas jämnt och tillsätt ytterligare 25 ml saltsyra (3.2). Sänk ned kolven i ett kokande vattenbad under kraftig och jämn omskakning under de tre första minuterna för att förhindra agglomeratbildning. Vattenbadet måste innehålla så mycket vatten att det fortsätter att koka då kolven sänks ned i det. Kolven får inte tas upp ur vattenbadet vid omskakningen. Ta upp kolven ur vattenbadet efter exakt 15 minuter, tillsätt 30 ml kallt vatten och kyl omedelbart till 20 oC.
Tillsätt 5 ml Carrez-lösning I (3.3) och skaka i ca 30 sekunder. Tillsätt sedan 5 ml Carrez-lösning II (3.4) och skaka i ytterligare ca 30 sekunder. Späd med vatten till full volym, blanda och filtrera. Om filtratet inte är fullständigt klart (vilket är sällsynt) upprepas bestämningen med en större kvantitet Carrez-lösning I och II, till exempel 10 ml.
Lösningens optiska rotation mäts i ett 200 mm rör med polarimeter eller sackarimeter.
5.3 Bestämning av den optiska rotationen (P' eller S') hos ämnen som är lösliga i 40 % etanol
Väg med en noggrannhet av 1 mg upp 5 g av provet i en 100 ml mätkolv och tillsätt ca 80 ml etanol (3.5) (se anmärkning 7.2). Låt kolven stå i en timme i rumstemperatur och skaka under denna tid om kraftigt vid sex tillfällen så att provet blandas väl med etanolen. Späd med etanol (3.5) till full volym, blanda och filtrera.
Pipettera över 50 ml av filtratet (motsvarar 2,5 g av provet) till en 250 ml E-kolv, tillsätt 2,1 ml saltsyra (3.1) och skaka kraftigt. Anslut en återloppskylare till E-kolven och sänk därefter ned E-kolven i ett kokande vattenbad. Ta efter exakt 15 minuter upp E-kolven ur vattenbadet, överför innehållet till en 100 ml mätkolv (skölj med lite kallt vatten) och kyl till 20 oC.
Klara provet med Carrez-lösning I (3.3) och II (3.4), späd med vatten till full volym, blanda, filtrera och mät den optiska rotationen på det sätt som anges i 5.2 andra och tredje styckena.
6. Beräkning av resultat
Stärkelsehalten ( %) beräknas på följande sätt:
6.1 Mätning med polarimeter
|
P |
= |
total optisk rotation i vinkelgrader |
|
P' |
= |
optisk rotation i vinkelgrader för ämnen som är lösliga i 40 % (v/v) etanol |
|
|
= |
specifik optisk rotation för ren stärkelse. De konventionella numeriska värdena för denna faktor är
|
6.2 Mätning med sackarimeter
|
S |
= |
total optisk rotation i sackarimetergrader |
|
S' |
= |
optisk rotation i sackarimetergrader för ämnen som är lösliga i 40 % (v/v) etanol |
|
N |
= |
vikt (g) av sackaros i 100 ml vatten som ger en optisk rotation på 100 sackarimetergrader vid mätning med ett 200 mm rör 16,29 g för franska sackarimetrar 26,00 g för tyska sackarimetrar 20,00 g för sackarimetrar av blandtyp
|
6.3 Repeterbarhet
Skillnaden mellan resultaten av två parallella bestämningar som utförs på samma prov får inte överstiga 0,4 i absoluta tal om stärkelsehalten är under 40 %, och 1 % i relativt värde om stärkelsehalten är 40 % eller högre.
7. Anmärkningar
7.1 Om provet innehåller mer än 6 % karbonater, beräknat som kalciumkarbonat, måste dessa avlägsnas genom behandling med en väl avvägd mängd utspädd svavelsyra, innan den totala optiska rotationen bestäms.
7.2 För produkter med hög laktoshalt, som mjölkserum i pulverform och skummjölkspulver, används följande förfarande efter tillsats av 80 ml etanol (3.5): Anslut en återloppskylare till kolven och sänk därefter ned kolven i ett vattenbad vid 50 oC i 30 minuter. Låt svalna och fortsätt analysen enligt anvisningarna i 5.3.
7.3 Följande foderråvaror kan, om de förekommer i betydande mängder i fodret, störa bestämningen av stärkelsehalt med polarimeter och kan därigenom ge upphov till felaktiga resultat:
— (Socker)betsprodukter som (socker)betmassa, (socker)betmelass, melasserad (socker)betmassa, (socker)betvinass, (bet)socker.
— Citruspulpa.
— Linfrön, linfröexpeller, extraherade linfrön.
— Rapsfrö, rapsfröexpeller, extraherade rapsfrön, rapsfröskal.
— Solrosfrön, extraherade solrosfrön, delvis skalade extraherade solrosfrön.
— Kopraexpeller, extraherad kopra.
— Potatispulpa.
— Vattenfri jäst.
— Inulinrika produkter (t.ex. snitsel och mjöl av jordärtskockor).
— Fettgrevar.
M. BESTÄMNING AV RÅASKA
1. Syfte och tillämpningsområde
Med denna metod kan halten råaska i foder bestämmas.
2. Princip
Provet föraskas vid 550 oC. Återstoden vägs.
3. Reagens
Ammoniumnitrat, lösning 20 % (w/v).
4. Utrustning
4.1 Värmeplatta.
4.2 Elektrisk muffelugn med termostat.
4.3 Deglar av kvarts, porslin eller platina; antingen rektangulära (ca 60 × 40 × 25 mm) eller cirkelformade (diameter: 60–75 mm, höjd: 20–40 mm).
5. Utförande
Väg med en noggrannhet av 1 mg upp ca 5 g av provet (2,5 g för produkter som har benägenhet att svälla) i en degel som först har upphettats till 550 oC, svalnat och tarerats. Ställ degeln på värmeplattan och upphetta sakta tills provet förkolnar. Föraska enligt 5.1 eller 5.2.
5.1 Sätt in degeln i den kalibrerade muffelugnen inställd på 550 oC. Håll provet vid denna temperatur tills en vit, ljusgrå eller rödaktig aska bildas som förefaller fri från kolhaltiga partiklar. Ställ degeln i en exsickator, låt svalna och väg omedelbart.
5.2 Sätt in degeln i den kalibrerade muffelugnen inställd på 550 oC. Föraska i 3 timmar. Ställ degeln i en exsickator, låt svalna och väg omedelbart. Föraska på nytt i 30 minuter för att säkerställa att askans vikt är konstant (viktförlusten mellan två på varandra följande vägningar får inte överstiga 1 mg).
6. Beräkning av resultat
Beräkna återstodens vikt med taran fråndragen.
Resultatet uttrycks i procent av provet.
7. Anmärkningar
7.1 Askan av ämnen som är svåra att föraska måste först genomgå en förberedande föraskning i minst 3 timmar, kylas och sedan tillsättas några droppar 20 % ammoniumnitratlösning eller vatten (försiktigt så att inte askan sprids eller klumpar bildas). Fortsätt kalcineringen efter torkning i ugn. Upprepa förfarandet så många gånger som behövs tills föraskningen är fullständig.
7.2 För ämnen som är motståndskraftiga mot den behandling som beskrivs i 7.1 förfars på följande sätt: efter föraskning i tre timmar läggs askan i varmt vatten och filtreras genom ett litet askfritt filter. Föraska filtret och dess innehåll i den ursprungliga degeln. Överför filtratet till den avsvalnade degeln, låt indunsta tills det är torrt, föraska och väg.
7.3 För oljor och fetter: väg noggrant upp ett prov på 25 g i en degel av lämplig storlek. Förkola provet genom att antända den med en remsa askfritt filtrerpapper. Fukta efter förbränning med minsta möjliga mängd vatten. Torka och föraska enligt beskrivningen i punkt 5.
N. BESTÄMNING AV ASKA SOM ÄR OLÖSLIG I SALTSYRA
1. Syfte och tillämpningsområde
Med denna metod kan halten i foder av mineralämnen som är olösliga i saltsyra bestämmas. Två metoder kan användas beroende på typen av prov.
1.1 Metod A: tillämplig på organiska foderråvaror och på de flesta foderblandningar.
1.2 Metod B: tillämplig på mineralfoder och mineralfoderblandningar och på foderblandningar vilkas halt av ämnen som är olösliga i saltsyra överstiger 1 % enligt bestämning med metod A.
2. Princip
2.1 Metod A: provet föraskas, askan kokas i saltsyra och den olösliga återstoden filtreras och vägs.
2.2 Metod B: provet behandlas med saltsyra. Lösningen filtreras varpå återstoden föraskas och den aska som erhålls behandlas enligt metod A.
3. Reagens
3.1 Saltsyra, 3 mol/l.
3.2 Triklorättiksyra, lösning 20 % (w/v).
3.3 Triklorättiksyra, lösning 1 % (w/v).
4. Utrustning
4.1 Värmeplatta.
4.2 Elektrisk muffelugn med termostat.
4.3 Deglar av kvarts, porslin eller platina, rektangulära (ca 60 × 40 × 25 mm) eller cirkelformade (diameter: 60–75 mm, höjd: 20–40 mm).
5. Utförande
5.1 Metod A:
Föraska provet med samma metod som för bestämning av råaska. Aska som erhållits vid den analysen kan också användas.
Överför askan till en 250–400 ml bägare med 75 ml saltsyra (3.1). Koka upp långsamt och låt koka försiktigt i 15 minuter. Filtrera den varma lösningen genom ett askfritt filtrerpapper och skölj återstoden med varmt vatten tills ingen syrareaktion längre är märkbar. Torka filtret med återstoden och föraska i en tarerad degel vid lägst 550 oC och högst 700 oC. Kyl i exsickator och väg.
5.2 Metod B:
Väg med en noggrannhet av 1 mg upp 5 g av provet i en 250–400 ml bägare. Tillsätt 25 ml vatten och därefter 25 ml saltsyra (3.1), blanda och vänta tills gasutvecklingen upphör. Tillsätt ytterligare 50 ml saltsyra (3.1). Vänta tills gasutvecklingen upphör och ställ sedan bägaren i ett kokande vattenbad i 30 minuter eller längre så att eventuell stärkelse hydrolyseras fullständigt. Filtrera den fortfarande varma lösningen genom ett askfritt filter och tvätta filtret i 50 ml varmt vatten (jfr punkt 7). Lägg filtret med återstod i en degel, torka och föraska vid en temperatur av lägst 550 oC och högst 700 oC. Överför askan till en 250–400 ml bägare med 75 ml saltsyra (3.1). Fortsätt enligt beskrivningen i punkt 5.1 andra stycket.
6. Beräkning av resultat
Beräkna återstodens vikt med taran fråndragen. Resultatet uttrycks i procent av provet.
7. Anmärkning
Om filtreringen visar sig svår att genomföra ska analysen påbörjas på nytt, varvid de 50 ml saltsyra (3.1) ersätts med 50 ml 20 % triklorättiksyra (3.2) och filtret sköljs med en varm lösning av 1 % triklorättiksyra (3.3).
O. BESTÄMNING AV KARBONATER
1. Syfte och tillämpningsområde
Med denna metod kan mängden karbonater, konventionellt uttryckt som kalciumkarbonat, bestämmas i de flesta fodertyper.
I vissa fall (t.ex. för järnkarbonat) måste dock en särskild metod användas.
2. Princip
Karbonaterna löses upp i saltsyra. Den koldioxid som frigörs samlas upp i ett mätglas, och dess volym jämförs med den som under samma förhållanden frigörs av en känd mängd kalciumkarbonat.
3. Reagens
3.1 Saltsyra, densitet 1,10 g/ml.
3.2 Kalciumkarbonat.
3.3 Svavelsyra, ca 0,05 mol/l, färgad med metylrött.
4. Utrustning
Scheibler-Dietrichapparat (se figur) eller motsvarande.
5. Utförande
Väg upp en provmängd som beroende på dess karbonathalt ska vara
— 0,5 g för produkter som innehåller 50–100 % karbonater, uttryckt som kalciumkarbonat,
— 1 g för produkter som innehåller 40–50 % karbonater, uttryckt som kalciumkarbonat,
— 2–3 g för övriga produkter.
Placera provet i apparatens specialkolv (4), som är utrustad med ett litet rör av okrossbart material som innehåller 10 ml saltsyra (3.1) och anslut kolven till apparaten. Vrid trevägskranen (5) så att röret (1) öppnas. Ställ in vätskenivån på nollmarkeringen med hjälp av det flyttbara röret (2) som är fyllt med färgad svavelsyra (3.3) och anslutet till mätglaset (1). Vrid kranen (5) så att rören (1) och (3) förbinds och kontrollera att nivån är noll.
Luta kolven (4) så att saltsyran (3.1) sprids långsamt över provet. Jämna ut trycket genom att sänka röret (2). Skaka kolven (4) tills ingen koldioxid längre frigörs.
Återställ trycket genom att justera tillbaka vätskenivån till den ursprungliga i rören (1) och (2). Avläs efter några minuter när gasvolymen har blivit konstant.
Utför ett kontrolltest under samma förhållanden med 0,5 g kalciumkarbonat (3.2).
6. Beräkning av resultat
Karbonathalten, uttryckt som kalciumkarbonat, beräknas med formeln

där:
|
X |
= |
% (w/w) karbonater, uttryckt som kalciumkarbonat, i provet |
|
V |
= |
ml CO2 som frigörs av provet |
|
V1 |
= |
ml CO2 som frigörs av 0,5 g CaCO3 |
|
m |
= |
provets vikt i gram. |
7. Anmärkningar
7.1 Om provet väger mer än 2 g ska först 15 ml destillerat vatten tillsättas i kolven (4) och blandas med provet innan försöket påbörjas. Använd samma volym vatten vid kontrolltestet.
7.2 Om den apparat som används har andra dimensioner än Scheibler-Dietrichapparaten måste man göra motsvarande ändringar av det använda provets och kontrollsubstansens mängd samt vid resultatberäkningen.

P. BESTÄMNING AV TOTALFOSFOR
FOTOMETRISK METOD
1. Syfte och tillämpningsområde
Med denna metod kan totalfosforhalten bestämmas i foder. Metoden är särskilt lämplig för analys av produkter med låg fosforhalt. I vissa fall (fosforrika produkter) kan en gravimetrisk metod användas.
2. Princip
Provet mineraliseras antingen genom torrföraskning (organiska foder) eller genom uppslutning med syra (mineralfoderblandningar och flytande foder) och läggs i en syralösning. Lösningen behandlas med molybdovanadatreagens. Absorbansen hos den gula lösning som då bildas mäts i spektrofotometer vid 430 nm.
3. Reagens
3.1 Kalciumkarbonat.
3.2 Saltsyra, ρ20 = 1,10 g/ml (ca 6 mol/l).
3.3 Salpetersyra, ρ20 = 1,045 g/ml.
3.4 Salpetersyra, ρ20 = 1,38 –1,42 g/ml.
3.5 Svavelsyra, ρ20 = 1,84 g/ml.
3.6 Molybdovanadatreagens: blanda 200 ml ammoniumheptamolybdatlösning (3.6.1), 200 ml ammoniumvanadatlösning (3.6.2) och 134 ml salpetersyra (3.4) i en 1 000 ml mätkolv. Späd med vatten till full volym.
3.6.1 Ammoniumheptamolybdatlösning: lös 100 g ammoniumheptamolybdat (NH4) 6Mo7O24·4H2O, i varmt vatten. Tillsätt 10 ml ammoniak (densitet 0,91 g/ml) och späd med vatten till 1 liter.
3.6.2 Ammoniumvanadatlösning: lös 2,35 g ammoniumvanadat NH4VO3 i 400 ml varmt vatten. Tillsätt långsamt under ständig omrörning 20 ml utspädd salpetersyra (7 ml HNO3 [3.4] + 13 ml vatten) och späd med vatten till 1 liter.
3.7 Standardfosforlösning, 1 mg/ml: lös 4,387 g kaliumdivätefosfat KH2PO4 i vatten. Späd med vatten till 1 liter.
4. Utrustning
4.1 Deglar av kvarts, porslin eller platina.
4.2 Elektrisk muffelugn med termostaten inställd på 550 oC.
4.3 250 ml Kjeldahlkolv.
4.4 Mätkolvar och precisionspipetter.
4.5 Spektrofotometer.
4.6 Provrör, ca 16 mm i diameter, med proppar, 14,5 mm i diameter, volym 25–30 ml.
5. Utförande
5.1 Beredning av lösningen
Lösningen bereds enligt 5.1.1 eller 5.1.2 beroende på typ av prov.
5.1.1
Väg upp 1 g eller mer av provet med en noggrannhet av 1 mg. Placera provet i en Kjeldahlkolv, tillsätt 20 ml svavelsyra (3.5), skaka om så att materialet helt impregneras med syra och så att den inte fastnar på kolvens väggar, upphetta och låt koka i 10 minuter. Låt svalna något, tillsätt 2 ml salpetersyra (3.4), upphetta försiktigt, låt svalna något, tillsätt lite mer salpetersyra (3.4) och upphetta åter till kokpunkten. Upprepa detta förfarande tills en färglös lösning erhålls. Kyl, tillsätt lite vatten, häll över vätskan i en 500 ml mätkolv och skölj ur Kjeldahlkolven med varmt vatten. Låt svalna, späd med vatten till full volym, homogenisera och filtrera.
5.1.2
Väg med en noggrannhet av 1 mg upp ca 2,5 g av provet i en degel. Blanda provet fullständigt med 1 g kalciumkarbonat (3.1). Föraska i ugnen vid 550 oC tills en vit eller grå aska erhålls (visst inslag av träkol har ingen betydelse). Överför askan till en 250 ml bägare. Tillsätt 20 ml vatten och saltsyra (3.2) tills skumningen upphör. Tillsätt ytterligare 10 ml saltsyra (3.2). Ställ bägaren i sandbad och indunsta fullständigt så att kiseldioxiden blir olöslig. Lös åter upp återstoden i 10 ml salpetersyra (3.3) och koka på sandbadet eller på en värmeplatta i 5 minuter utan fullständig indunstning. Häll över vätskan i en 500 ml mätkolv och skölj ur bägaren flera gånger med varmt vatten. Låt svalna, späd med vatten till full volym, homogenisera och filtrera.
5.2 Färgutveckling och mätning av absorbans
Späd ut en alikvot av det filtrat som erhållits enligt 5.1.1 eller 5.1.2 så att en fosforhalt av högst 40 μg/ml erhålls. Överför 10 ml av denna lösning till ett provrör (4.6) och tillsätt 10 ml molybdovanadatreagens (3.6). Homogenisera och låt stå i minst 10 minuter vid 20 oC. Mät absorbansen i spektrofotometer vid 430 nm mot en lösning av 10 ml molybdovanadatreagens (3.6) i 10 ml vatten.
5.3 Kalibreringskurva
Av standardlösningen (3.7) bereds lösningar som innehåller 5, 10, 20, 30 och 40 μg fosfor per ml. Ta 10 ml av var och en av dessa lösningar och tillsätt 10 ml molybdovanadatreagens (3.6). Homogenisera och låt stå i minst 10 minuter vid 20 oC. Mät absorbansen på det sätt som anges i 5.2. Konstruera kalibreringskurvan genom att avsätta absorbansvärdena mot motsvarande kvantiteter fosfor. Vid koncentrationer mellan 0 och 40 μg/ml är kurvan linjär.
6. Beräkning av resultat
Bestäm mängden fosfor i provet med hjälp av kalibreringskurvan.
Resultatet uttrycks i procent av provet.
Repeterbarhet
Skillnaden mellan resultaten från två parallella bestämningar som utförs på samma prov får inte överstiga
— 3 % av det högre värdet för fosforhalter under 5 %,
— 0,15 % i absoluta tal för fosforhalter på 5 % eller mer.
Q. BESTÄMNING AV KLOR I KLORIDER
1. Syfte och tillämpningsområde
Med denna metod kan mängden klor i vattenlösliga klorider, konventionellt uttryckt som natriumklorid, bestämmas. Metoden är tillämplig på alla foder.
2. Princip
Kloriderna löses upp i vatten. Om produkten innehåller organiskt material ska den klaras. Lösningen surgörs något med salpetersyra och kloriderna fälls ut som silverklorid genom tillsats av silvernitratlösning. Överskottet av silvernitrat titreras med ammoniumtiocyanatlösning enligt Volhardmetoden.
3. Reagens
3.1 Ammoniumtiocyanatlösning 0,1 mol/l.
3.2 Silvernitratlösning 0,1 mol/l.
3.3 Mättad lösning av ammoniumjärnsulfat (NH4)Fe(SO4)2.
3.4 Salpetersyra, densitet 1,38 g/ml.
3.5 Dietyleter.
3.6 Aceton.
3.7 Carrez-lösning I: lös 21,9 g zinkacetat, Zn(CH3COO)2·2H2O, och 3 g isättika i vatten. Späd med vatten till 100 ml.
3.8 Carrez-lösning II: lös 10,6 g kaliumferrocyanid, K4Fe(CN)6·3H2O, i vatten. Späd med vatten till 100 ml.
3.9 Aktivt kol, fritt från klorider och inte kloridabsorberande.
4. Utrustning
Rotationsskakapparat: ca 35–40 varv per minut.
5. Utförande
5.1 Beredning av lösningen
Beroende på typ av prov bereds en lösning enligt 5.1.1, 5.1.2 eller 5.1.3.
Utför samtidigt ett blankprov utan det prov som ska analyseras.
5.1.1
Väg med en noggrannhet av 1 mg upp ett prov på högst 10 g som innehåller högst 3 g klor i kloridform. Överför provet och 400 ml vatten till en 500 ml mätkolv vid ca 20 oC. Blanda på skakapparaten i 30 minuter, späd till full volym, homogenisera och filtrera.
5.1.2
Väg med en noggrannhet av 1 mg upp ca 5 g av provet tillsammans med 1 g aktivt kol i en 500 ml mätkolv. Tillsätt 400 ml vatten (ca 20 oC) och 5 ml Carrez-lösning I (3.7), rör om i 30 sekunder och tillsätt därefter 5 ml Carrez-lösning II (3.8). Blanda på skakapparaten i 30 minuter, späd till full volym, homogenisera och filtrera.
5.1.3
Bered lösningen enligt beskrivningen i 5.1.2 men filtrera inte. Dekantera (centrifugera vid behov) och överför 100 ml av supernatanten till en 200 ml mätkolv. Blanda med aceton (3.6) och späd med detta lösningsmedel till full volym, homogenisera och filtrera.
5.2 Titrering
Pipettera över 25–100 ml (beroende på den förmodade klorhalten) av det filtrat som erhållits enligt 5.1.1, 5.1.2 eller 5.1.3 till en E-kolv. Analysprovet får inte innehålla mer än 150 mg klor (Cl). Späd vid behov med vatten till minst 50 ml, tillsätt 5 ml salpetersyra (3.4), 20 ml mättad ammoniumjärnsulfatlösning (3.3) och två droppar ammoniumtiocyanatlösning (3.1) som tillförs med hjälp av en byrett som är fylld till nollmarkeringen. Tillför silvernitratlösningen (3.2) med en byrett så att ett överskott på 5 ml erhålls. Tillsätt 5 ml dietyleter (3.5) och skaka kraftigt så att fällningen koagulerar. Titrera överskottet av silvernitrat med ammoniumtiocyanatlösningen (3.1) tills den rödbruna färgen har bestått i 1 minut.
6. Beräkning av resultat
Mängden klor (X) uttryckt som procent natriumklorid beräknas med formeln

där:
|
V1 |
= |
ml tillsatt silvernitratlösning 0,1 mol/l |
|
V2 |
= |
ml ammoniumtiocyanatlösning 0,1 mol/l som använts vid titreringen |
|
m |
= |
provets vikt. |
Om blankprovet visar att silvernitratlösning, 0,1 mol/l, har förbrukats ska detta värde dras från volymen (V1 – V2).
7. Anmärkningar
7.1. Titreringen kan även utföras potentiometriskt.
7.2. Produkter med mycket hög fetthalt ska först avfettas med dietyleter eller petroleumeter.
7.3. Vid analys av fiskmjöl kan titreringen utföras med Mohrs metod.
BILAGA IV
ANALYSMETODER FÖR KONTROLL AV HALTEN GODKÄNDA FODERTILLSATSER
A. BESTÄMNING AV VITAMIN A
1. Syfte och tillämpningsområde
Med denna metod kan halten av vitamin A (retinol) i foder och förblandningar bestämmas. Med vitamin A avses all-trans-retinylalkohol och dess cis-isomerer som bestäms med denna metod. Halten av vitamin A uttrycks som internationella enheter (IE) per kg, där 1 IE motsvarar aktiviteten hos 0,300 μg all-trans-vitamin A-alkohol eller 0,344 μg all-trans-vitamin A-acetat eller 0,550 μg all-trans-vitamin A-palmitat.
Kvantifieringsgränsen för vitamin A är 2 000 IE/kg.
2. Princip
Provet hydrolyseras med kaliumhydroxid som lösts i etanol, och vitamin A extraheras sedan över till petroleumeter. Lösningsmedlet får därefter avdunsta, och återstoden av provet löses i metanol och späds vid behov till lämplig koncentration. Halten av vitamin A bestäms genom omvänd fas-kromatografi (RP-HPLC) med UV- eller fluorescensdetektor. De kromatografiska parametrarna väljs så att all-trans-vitamin A-alkohol inte separeras från sina cis-isomerer.
3. Reagens
3.1 Etanol, σ = 96 %
3.2 Petroleumeter, kokpunktsintervall 40–60 oC
3.3 Metanol
3.4 Kaliumhydroxidlösning, c = 50 g/100 ml
3.5 Natriumaskorbatlösning, c = 10 g/100 ml (jfr punkt 7.7)
3.6 Natriumsulfid, Na2S· x H20, där x = 7–9
3.6.1 Natriumsulfidlösning, c = 0,5 mol/l i glycerol, ß = 120 g/l för x = 9 (jfr punkt 7.8)
3.7 Fenolftalein som lösts i etanol, c = 2 g/100 ml (3.1)
3.8 2-propanol
3.9 Mobil fas för HPLC: blandning av metanol (3,3 ) och vatten, t.ex. 980 + 20 (v + v). Det exakta förhållandet mellan volymerna anpassas efter kolonnens egenskaper.
3.10 Kvävgas, syrefri
3.11 All-trans-vitamin A-acetat, av extra hög renhetsgrad, med certifierad aktivitet, t.ex. 2,80 × 106 IE/g
3.11.1 Stamlösning av all-trans-vitamin A-acetat: Väg med en noggrannhet av 0,1 mg upp 50 mg vitamin A-acetat (3.11) i en 100 ml mätkolv. Lös i 2-propanol (3.8) och späd till märket med samma lösningsmedel. Den nominella halten av vitamin A i lösningen är 1 400 IE/ml. Den exakta halten ska bestämmas enligt 5.6.3.1.
3.12 All-trans-vitamin A-palmitat, av extra hög renhetsgrad, med certifierad aktivitet, t.ex. 1,80 × 106 IE/g
3.12.1 Stamlösning av all-trans-vitamin A-palmitat: Väg med en noggrannhet av 0,1 mg upp 80 mg vitamin A-palmitat (3.12) i en 100 ml mätkolv. Lös i 2-propanol (3.8) och späd till märket med samma lösningsmedel. Den nominella halten av vitamin A i lösningen är 1 400 IE/ml. Den exakta halten ska bestämmas enligt 5.6.3.2.
3.13 2,6-di-tert-butyl-4-metylfenol (BHT) (jfr punkt 7.5)
4. Utrustning
4.1 Rotationsindunstare
4.2 Bruna glaskärl
4.2.1 Sättkolvar eller E-kolvar, 500 ml, med slipad hals
4.2.2 Smalhalsade mätkolvar, 10, 25, 100 och 500 ml, med slipade proppar
4.2.3 Koniska separertrattar, 1 000 ml, med slipade proppar
4.2.4 Rundkolvar, 250 ml, med slipad hals
4.3 Allihn-kylare, mantellängd 300 mm, med fog av slipat glas och adapter för gasinledningsrör
4.4 Veckat filtrerpapper för fasseparation, diameter 185 mm (t.ex. Schleicher & Schuell 597 HY 1/2)
4.5 HPLC-utrustning med injektionssystem
4.5.1 HPLC-kolonn, 250 × 4 mm, C18, 5 eller 10 μm packning eller motsvarande (prestandakriterium: en enda topp för alla isomerer av retinol vid normala HPLC-förhållanden)
4.5.2 UV- eller fluorescensdetektor, med variabel våglängd
4.6 Spektrofotometer med 10 mm kvartsceller
4.7 Vattenbad med magnetomrörare
4.8 Extraktionsapparat (se figur 1) bestående av:
4.8.1 Glascylinder, 1 liter, med slipad hals och propp.
4.8.2 Adapter av slipat glas med en sidoarm och ett justerbart rör genom mitten. Det justerbara röret bör ha U-formad nederdel och en pip i övre änden så att det övre vätskeskiktet i cylindern kan överföras till en separertratt.
5. Utförande
|
Anmärkning: |
Vitamin A är känsligt för (UV-)ljus och oxidation. Alla arbetsmoment bör därför utföras i skydd från ljus (bruna glaskärl eller glaskärl insvepta i aluminiumfolie) och från syre (spola med kvävgas). Vid extraktionen ersätts luften ovanför vätskan med kvävgas (undvik övertryck genom att lätta på proppen då och då). |
5.1 Provberedning
Mal provet så att det kan passera genom en 1 mm sikt, men undvik friktionsvärme. För att undvika förlust av vitamin A måste malningen göras omedelbart innan provet vägs och förtvålas.
5.2 Förtvålning
Väg med en noggrannhet av 1 mg upp 2–25 g av provet, beroende på halten av vitamin A, i en 500 ml sättkolv eller E-kolv (4.2.1). Tillsätt – i tur och ordning, och samtidigt som flaskan eller kolven roteras – 130 ml etanol (3.1), ca 100 mg BHT (3.13), 2 ml natriumaskorbatlösning (3.5) och 2 ml natriumsulfidlösning (3.6). Flaskan eller kolven kopplas till en kylare (4.3) och ställs därefter i ett vattenbad med magnetomrörare (4.7). Värm till kokpunkten och låt återloppskoka i 5 minuter. Tillsätt 25 ml kaliumhydroxidlösning (3.4) genom kylaren (4.3) och låt återloppskoka i ytterligare 25 minuter under omrörning och i ett långsamt flöde av kvävgas. Skölj sedan kylaren med ca 20 ml vatten och kyl innehållet i flaskan eller kolven till rumstemperatur.
5.3 Extraktion
Överför genom dekantering den förtvålade lösningen kvantitativt till en 1 000 ml separertratt (4.2.3) eller till en extraktionsapparat (4.8) genom att skölja med sammanlagt 250 ml vatten. Skölj ur förtvålningskärlet först med 25 ml etanol (3.1) och sedan med 100 ml petroleumeter (3.2) och överför sköljvätskan till separertratten eller extraktionsapparaten. Förhållandet mellan vatten och etanol i de sammanslagna lösningarna bör vara omkring 2:1. Skaka kraftigt i 2 minuter och låt sedan stå i 2 minuter.
5.3.1
När skikten har separerat (jfr punkt 7.3) överförs skiktet med petroleumeter till en annan separertratt (4.2.3). Upprepa extraktionen två gånger med 100 ml petroleumeter (3.2) och två gånger med 50 ml petroleumeter (3.2).
Tvätta de sammanslagna extrakten i separertratten två gånger genom att tillsätta portioner på 100 ml vatten och sedan försiktigt rotera tratten (för att undvika att emulsioner bildas). Skaka sedan tratten upprepade gånger med portioner på 100 ml vatten, tills vattnet inte längre färgas vid tillsats av fenolftaleinlösning (3.7) (fyra tvättningar brukar räcka). Avlägsna vatten i suspension genom att filtrera det tvättade extraktet genom ett torrt veckat filtrerpapper för fasseparation (4.4) till en 500 ml mätkolv (4.2.2). Skölj ur separertratten och filtret med 50 ml petroleumeter (3.2), späd till märket med petroleumeter (3.2) och blanda omsorgsfullt.
5.3.2
När skikten har separerat (jfr punkt 7.3) ersätts glascylinderns (4.8.1) propp med adaptern av slipat glas (4.8.2). Den U-formade nederdelen av det justerbara röret ska mynna precis ovanför gränsytan mellan skikten. Överför det övre skiktet av petroleumeter till en 1 000 ml separertratt (4.2.3) genom att föra in kvävgas via sidoröret. Tillsätt 100 ml petroleumeter (3.2) till glascylindern, sätt i proppen och skaka kraftigt. Låt skikten separera och överför det övre skiktet till separertratten som ovan. Upprepa extraktionen med ytterligare 100 ml petroleumeter (3.2) och sedan två gånger med 50 ml-portioner av petroleumeter (3.2). Överför petroleumeterskikten till separertratten.
Tvätta de sammanslagna extrakten av petroleumeter enligt 5.3.1 och gå vidare enligt instruktionen i den punkten.
5.4 Beredning av provlösning för HPLC
Pipettera över en alikvot av petroleumeterlösningen (från 5.3.1 eller 5.3.2) till en 250 ml rundkolv (4.2.4). Låt lösningsmedlet avdunsta nästan fullständigt i en rotationsindunstare (4.1) med reducerat tryck och en temperatur på vattenbadet som inte överstiger 40 oC. Återställ atmosfärstryck genom att släppa in kvävgas (3.10) och ta sedan bort kolven från rotationsindunstaren. Avlägsna återstående lösningsmedel med en kvävgasström (3.10) och lös omedelbart upp återstoden av provet i en känd volym (10–100 ml) metanol (3.3). (Halten av vitamin A bör ligga i intervallet 5–30 IE/ml.)
5.5 Bestämning med HPLC
Vitamin A separeras i en kolonn för omvänd fas-kromatografi, C18, (4.5.1) och halten bestäms med en UV-detektor (325 nm) eller fluorescensdetektor (excitation: 325 nm, emission: 475 nm) (4.5.2).
Injicera en alikvot (t.ex. 20 μl) av den metanollösning som erhölls i 5.4 och eluera med den mobila fasen (3.9). Beräkna den genomsnittliga topphöjden (arean) för flera injektioner av samma provlösning och de genomsnittliga topphöjderna (areorna) för flera injektioner av kalibreringslösningarna (5.6.2).
Följande betingelser anges som vägledning. Andra betingelser kan väljas om de ger likvärdiga resultat.
|
HPLC-kolonn (4.5.1): |
250 × 4 mm, C18, 5 eller 10 μm packning, eller motsvarande |
|
Mobil fas (3.9): |
Blandning av metanol (3.3) och vatten, t.ex. 980 + 20 (v + v). |
|
Flödeshastighet: |
1–2 ml/min |
|
Detektor (4.5.2): |
UV-detektor (325 nm) eller fluorescensdetektor (excitation: 325 nm, emission: 475 nm) |
5.6 Kalibrering
5.6.1
Pipettera över 20 ml stamlösning av vitamin A-acetat (3.11.1) eller 20 ml stamlösning av vitamin A-palmitat (3.12.1) till en 500 ml sättkolv eller E-kolv (4.2.1) och hydrolysera enligt 5.2 men utan tillsats av BHT. Extrahera sedan med petroleumeter (3.2) enligt 5.3 och späd till 500 ml med petroleumeter (3.2). Låt 100 ml av extraktet indunsta i en rotationsindunstare (jfr punkt 5.4) tills provet är nästan torrt. Avlägsna resterande lösningsmedel i en kvävgasström (3.10) och lös provet på nytt i 10,0 ml metanol (3.3). Den nominella halten av vitamin A i lösningen är 560 IE/ml. Den exakta halten ska bestämmas enligt 5.6.3.3. Arbetslösningen ska vara nyberedd.
Pipettera över 2,0 ml av arbetslösningen till en 20 ml mätkolv, späd till märket med metanol (3.3) och blanda. Den nominella halten av vitamin A i denna utspädda arbetslösning är 56 IE/ml.
5.6.2
För över 1,0 , 2,0 , 5,0 och 10,0 ml av den utspädda arbetslösningen till ett antal 20 ml mätkolvar, späd till märket med metanol (3.3) och blanda. Den nominella halten av vitamin A i dessa lösningar är 2,8 – 5,6 – 14,0 respektive 28,0 IE/ml.
Injicera upprepade gånger 20 μl av varje kalibreringslösning och bestäm de genomsnittliga topphöjderna (areorna). Använd de genomsnittliga topphöjderna (areorna) för att konstruera en kalibreringskurva med beaktande av resultatet av UV-kontrollen (5.6.3.3).
5.6.3
5.6.3.1
Pipettera över 2,0 ml stamlösning av vitamin A-acetat (3.11.1) till en 50 ml mätkolv (4.2.2) och späd till märket med 2-propanol (3.8). Den nominella halten av vitamin A i denna lösning är 56 IE/ml. Pipettera över 3,0 ml av den utspädda vitamin A-acetatlösningen till en 25 ml mätkolv och späd till märket med 2-propanol (3.8). Den nominella halten av vitamin A i denna lösning är 6,72 IE/ml. Mät UV-spektrumet för lösningen mot 2-propanol (3.8) i en spektrofotometer (4.6) mellan 300 och 400 nm. Extinktionsmaximum ska ligga mellan 325 och 327 nm.
Beräkning av halten av vitamin A:
Halten vitamin A (IE/ml) = E326 × 19,0
(
![]()
för vitamin A-acetat = 1 530 vid 326 nm i 2-propanol)
5.6.3.2
Pipettera över 2,0 ml stamlösning av vitamin A-palmitat (3.12.1) till en 50 ml mätkolv (4.2.2) och späd till märket med 2-propanol (3.8). Den nominella halten av vitamin A i denna lösning är 56 IE/ml. Pipettera över 3,0 ml av den utspädda vitamin A-palmitatlösningen till en 25 ml mätkolv och späd till märket med 2-propanol (3.8). Den nominella halten av vitamin A i denna lösning är 6,72 IE/ml. Mät UV-spektrumet för lösningen mot 2-propanol (3.8) i en spektrofotometer (4.6) mellan 300 och 400 nm. Extinktionsmaximum ska ligga mellan 325 och 327 nm.
Beräkning av halten av vitamin A:
Halten vitamin A (IE/ml) = E326 × 19,0
(
![]()
för vitamin A-palmitat = 957 vid 326 nm i 2-propanol)
5.6.3.3
Pipettera över 3,0 ml av den outspädda arbetslösning av vitamin A som beretts enligt 5.6.1 till en 50 ml mätkolv (4.2.2) och späd till märket med 2-propanol (3.8). Pipettera över 5,0 ml av denna lösning till en 25 ml mätkolv och späd till märket med 2-propanol (3.8). Den nominella halten av vitamin A i denna lösning är 6,72 IE/ml. Mät UV-spektrumet för lösningen mot 2-propanol (3.8) i en spektrofotometer (4.6) mellan 300 och 400 nm. Extinktionsmaximum ska ligga mellan 325 och 327 nm.
Beräkning av halten av vitamin A:
Halten vitamin A (IE/ml) = E325 × 18,3
(
![]()
för vitamin A-alkohol = 1821 vid 325 nm i 2-propanol)
6. Beräkning av resultat
Använd den genomsnittliga topphöjden (arean) för provlösningens vitamin A-toppar för att bestämma halten i IE/ml med hjälp av kalibreringskurvan (5.6.2).
Halten av vitamin A (IE/kg) i provet beräknas med formeln

[UI/kg]
där:
|
c |
= |
halten av vitamin A i provlösningen (5.4) i IE/ml |
|
V1 |
= |
provlösningens volym (5.4) i ml |
|
V2 |
= |
volym för den uttagna alikvoten (5.4) i ml |
|
m |
= |
provets vikt i gram. |
7. Anmärkningar
7.1 För prov med låg halt av vitamin A kan det vara lämpligt att slå ihop petroleumeterextrakten från två förtvålningsomgångar (uppvägd mängd: 25 g) till en enda provlösning för bestämning med HPLC.
7.2 Det prov som ska bestämmas bör inte innehålla mer än 2 g fett.
7.3 Om ingen fasseparation äger rum tillsätts ca 10 ml etanol (3.1) för att få emulsionen att skikta sig.
7.4 För torskleverolja och andra rena fetter bör förtvålningstiden förlängas till 45–60 minuter.
7.5 I stället för BHT kan man använda hydrokinon.
7.6 Retinol-isomerer kan separeras med en kolonn för normal-fas-kromatografi. Topphöjderna (areorna) för alla cis- och trans-isomerer måste då läggas ihop vid beräkningen.
7.7 I stället för natriumaskorbatlösning kan man använda ca 150 mg askorbinsyra.
7.8 I stället för natriumsulfidlösning kan man använda ca 50 mg EDTA.
7.9 Vid bestämning av vitamin A i mjölkersättning måste följande beaktas:
— Vid förtvålning (5.2): Det kan vara nödvändigt att öka mängden kaliumhydroxidlösning (3.4) på grund av mängden fett i provet.
— Vid extraktion (5.3): Det kan vara nödvändigt att anpassa volymförhållandet mellan vatten och etanol (2:1) till följd av emulsioner.
Gör ett utbytesförsök på ett separat prov för att kontrollera att bestämningsmetoden ger tillförlitliga resultat för just denna matris (mjölkersättning). Om utbytet är lägre än 80 % måste resultatet av bestämningen korrigeras för detta.
8. Repeterbarhet
Skillnaden mellan resultaten från två parallella bestämningar som utförs på samma prov får inte överstiga 15 % av det högre värdet.
9. Resultat av en provningsjämförelse ( 11 )
|
|
Förblandning |
Förblandat foder |
Mineralkoncentrat |
Proteinfoder |
Smågrisfoder |
|
L |
13 |
12 |
13 |
12 |
13 |
|
N |
48 |
45 |
47 |
46 |
49 |
|
Medelvärde [IE/kg] |
17,02 × 106 |
1,21 × 106 |
537 100 |
151 800 |
18 070 |
|
sr [IE/kg] |
0,51 × 106 |
0,039 × 106 |
22 080 |
12 280 |
682 |
|
r [IE/kg] |
1,43 × 106 |
0,109 × 106 |
61 824 |
34 384 |
1 910 |
|
CVr [ %] |
3,0 |
3,5 |
4,1 |
8,1 |
3,8 |
|
sR [IE/kg] |
1,36 × 106 |
0,069 × 106 |
46 300 |
23 060 |
3 614 |
|
R [IE/kg] |
3,81 × 106 |
0,193 × 106 |
129 640 |
64 568 |
10 119 |
|
CVR [ %] |
8,0 |
6,2 |
8,6 |
15 |
20 |
|
L: |
: |
antal laboratorier |
|
n |
: |
antal enskilda värden |
|
sr |
: |
standardavvikelse för repeterbarhet |
|
sR |
: |
standardavvikelse för reproducerbarhet |
|
r |
: |
repeterbarhet |
|
R |
: |
reproducerbarhet |
|
CVr |
: |
variationskoefficient för repeterbarhet |
|
CVR |
: |
variationskoefficient för reproducerbarhet |
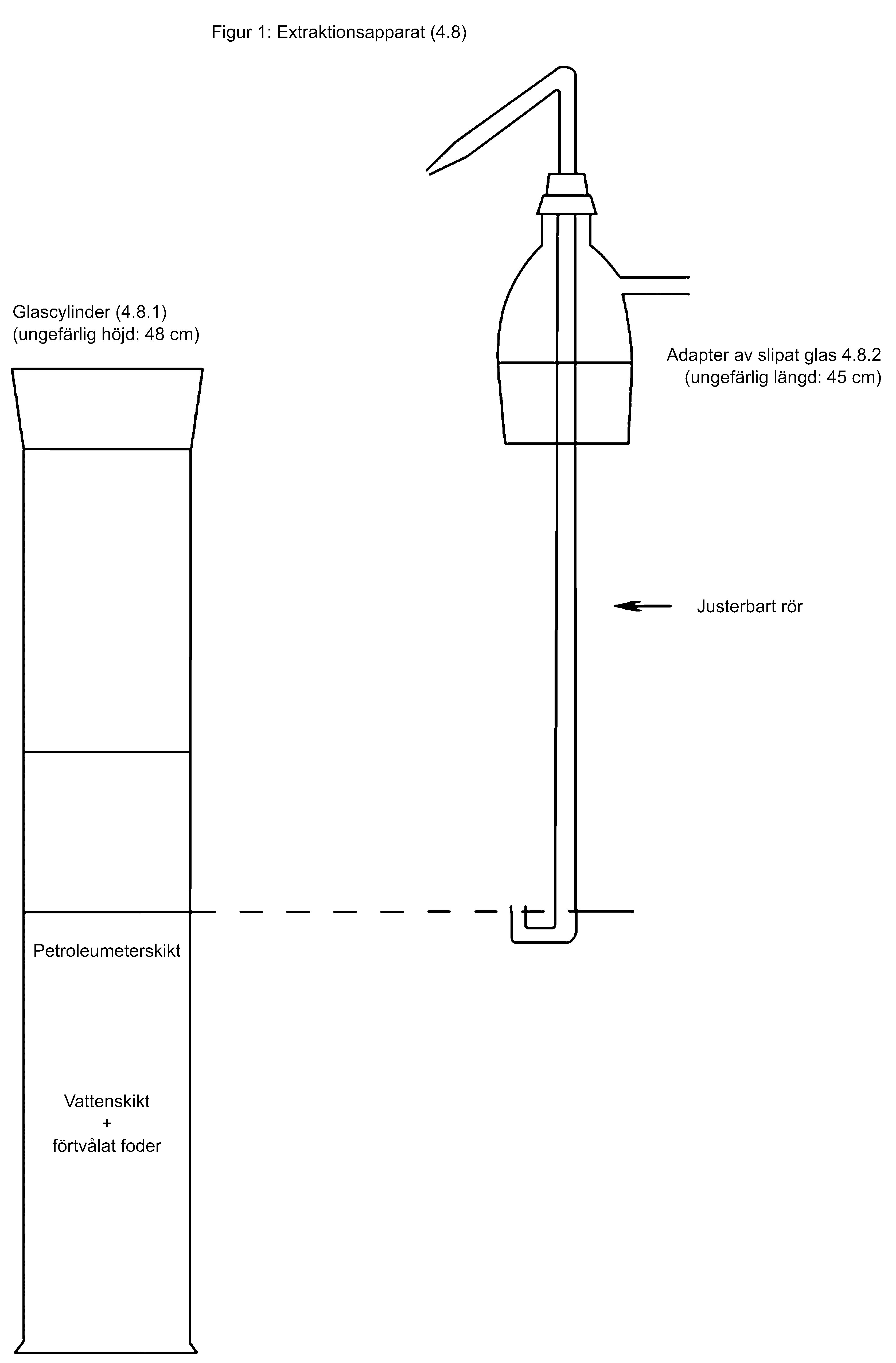
B. BESTÄMNING AV VITAMIN E
1. Syfte och tillämpningsområde
Med denna metod kan halten av vitamin E i foder och förblandningar bestämmas. Halten av vitamin E uttrycks som mg DL-alfa-tokoferolacetat/kg. 1 mg DL-alfa-tokoferolacetat motsvarar 0,91 mg DL-alfa-tokoferol (vitamin E).
Kvantifieringsgränsen för vitamin E är 2 mg/kg. Denna gräns kan bara uppnås med fluorescensdetektor. Med UV-detektor ligger kvantifieringsgränsen på 10 mg/kg.
2. Princip
Provet hydrolyseras med kaliumhydroxid som lösts i etanol, och vitamin E extraheras över till petroleumeter. Lösningsmedlet får därefter avdunsta, och återstoden av provet löses i metanol och späds vid behov till lämplig koncentration. Halten av vitamin E bestäms genom omvänd fas-kromatografi (RP-HPLC) med fluorescens- eller UV-detektor.
3. Reagens
3.1 Etanol, σ = 96 %
3.2 Petroleumeter, kokpunktsintervall 40–60 oC
3.3 Metanol
3.4 Kaliumhydroxidlösning, c = 50 g/100 ml
3.5 Natriumaskorbatlösning, c = 10 g/100 ml (jfr punkt 7.7)
3.6 Natriumsulfid, Na2S· x H20, där x = 7–9
3.6.1 Natriumsulfidlösning, c = 0,5 mol/l i glycerol, β = 120 g/l för x = 9 (jfr punkt 7.8)
3.7 Fenolftalein som lösts i etanol, c = 2 g/100 ml (3.1)
3.8 Mobil fas för HPLC: blandning av metanol (3.3) och vatten, t.ex. 980 + 20 (v + v). Det exakta förhållandet mellan volymerna anpassas efter kolonnens egenskaper.
3.9 Kvävgas, syrefri
3.10 DL-alfa-tokoferolacetat, av extra hög renhetsgrad, med certifierad aktivitet
3.10.1 Stamlösning av DL-alfa-tokoferolacetat: Väg med en noggrannhet av 0,1 mg upp 100 mg DL-alfa-tokoferolacetat (3.10) i en 100 ml mätkolv. Lös i etanol (3.1) och späd till märket med samma lösningsmedel. 1 ml av denna lösning innehåller 1 mg DL-alfa-tokoferolacetat. (För UV-kontroll, se 5.6.1.3; för stabilisering, se 7.4.)
3.11 DL-alfa-tokoferol, av extra hög renhetsgrad, med certifierad aktivitet
3.11.1 Stamlösning av DL-alfa-tokoferol: Väg med en noggrannhet av 0,1 mg upp 100 mg DL-alfa-tokoferol (3.11) i en 100 ml mätkolv. Lös i etanol (3.1) och späd till märket med samma lösningsmedel. 1 ml av denna lösning innehåller 1 mg DL-alfa-tokoferol. (För UV-kontroll, se 5.6.2.3; för stabilisering, se 7.4.)
3.12 2,6-di-tert-butyl-4-metylfenol (BHT) (jfr punkt 7.5)
4. Utrustning
4.1 Rotationsindunstare
4.2 Bruna glaskärl
4.2.1 Sättkolvar eller E-kolvar, 500 ml, med slipad hals.
4.2.2 Smalhalsade mätkolvar, 10, 25, 100 och 500 ml, med slipade proppar.
4.2.3 Koniska separertrattar, 1 000 ml, med slipade proppar.
4.2.4 Rundkolvar, 250 ml, med slipad hals.
4.3 Allihn-kylare, mantellängd 300 mm, med fog av slipat glas och adapter för gasinledningsrör
4.4 Veckat filtrerpapper för fasseparation, diameter 185 mm (t.ex. Schleicher & Schuell 597 HY 1/2)
4.5 HPLC-utrustning med injektionssystem
4.5.1 HPLC-kolonn, 250 × 4 mm, C18, 5 eller 10 μm packning eller motsvarande.
4.5.2 Fluorescens- eller UV-detektor, med variabel våglängd.
4.6 Spektrofotometer med 10 mm kvartsceller
4.7 Vattenbad med magnetomrörare
4.8 Extraktionsapparat (se figur 1) bestående av:
4.8.1 Glascylinder, 1 liter, med slipad hals och propp.
4.8.2 Adapter av slipat glas med en sidoarm och ett justerbart rör genom mitten. Det justerbara röret bör ha U-formad nederdel och en pip i övre änden så att det övre vätskeskiktet i cylindern kan överföras till en separertratt.
5. Utförande
|
Anmärkning: |
Vitamin E är känsligt för (UV-)ljus och oxidation. Alla arbetsmoment bör därför utföras i skydd från ljus (bruna glaskärl eller glaskärl insvepta i aluminiumfolie) och från syre (spola med kvävgas). Vid extraktionen ersätts luften ovanför vätskan med kvävgas (undvik övertryck genom att lätta på proppen då och då). |
5.1 Provberedning
Mal provet så att det kan passera genom en 1 mm sikt, men undvik friktionsvärme. För att undvika förlust av vitamin E måste malningen göras omedelbart innan provet vägs och förtvålas.
5.2 Förtvålning
Väg med en noggrannhet av 0,01 g upp 2–25 g av provet, beroende på halten av vitamin E, i en 500 ml sättkolv eller E-kolv (4.2.1). Tillsätt – i tur och ordning, och samtidigt som flaskan eller kolven roteras – 130 ml etanol (3.1), ca 100 mg BHT (3.12), 2 ml natriumaskorbatlösning (3.5) och 2 ml natriumsulfidlösning (3.6). Flaskan eller kolven kopplas till en kylare (4.3) och ställs därefter i ett vattenbad med magnetomrörare (4.7). Värm till kokpunkten och låt återloppskoka i 5 minuter. Tillsätt därefter 25 ml kaliumhydroxidlösning (3.4) genom kylaren (4.3) och låt återloppskoka i ytterligare 25 minuter under omrörning och ett långsamt flöde av kvävgas. Skölj sedan kylaren med ca 20 ml vatten och kyl innehållet i flaskan eller kolven till rumstemperatur.
5.3 Extraktion
Överför genom dekantering den förtvålade lösningen kvantitativt till en 1 000 ml separertratt (4.2.3) eller till en extraktionsapparat (4.8) genom att skölja med sammanlagt 250 ml vatten. Skölj ur förtvålningskärlet först med 25 ml etanol (3.1) och sedan med 100 ml petroleumeter (3.2) och överför sköljvätskan till separertratten eller extraktionsapparaten. Förhållandet mellan vatten och etanol i de sammanslagna lösningarna bör vara omkring 2:1. Skaka kraftigt i 2 minuter och låt sedan stå i 2 minuter.
5.3.1
När skikten har separerat (se punkt 7.3) överförs skiktet med petroleumeter till en annan separertratt (4.2.3). Upprepa extraktionen två gånger med 100 ml petroleumeter (3.2) och två gånger med 50 ml petroleumeter (3.2).
Tvätta de sammanslagna extrakten i separertratten två gånger genom att tillsätta portioner på 100 ml vatten och sedan försiktigt rotera tratten (för att undvika att emulsioner bildas). Skaka sedan tratten upprepade gånger med portioner på 100 ml vatten, tills vattnet inte längre färgas vid tillsats av fenolftaleinlösning (3.7) (fyra tvättningar brukar räcka). Avlägsna vatten i suspension genom att filtrera det tvättade extraktet genom ett torrt veckat filtrerpapper för fasseparation (4.4) till en 500 ml mätkolv (4.2.2). Skölj ur separertratten och filtret med 50 ml petroleumeter (3.2), späd till märket med petroleumeter (3.2) och blanda omsorgsfullt.
5.3.2
När skikten har separerat (jfr punkt 7.3) ersätts glascylinderns (4.8.1) propp med adaptern av slipat glas (4.8.2). Den U-formade nederdelen av det justerbara röret ska mynna precis ovanför gränsytan mellan skikten. Överför det övre skiktet av petroleumeter till en 1 000 ml separertratt (4.2.3) genom att föra in kvävgas via sidoröret. Tillsätt 100 ml petroleumeter (3.2) till glascylindern, sätt i proppen och skaka kraftigt. Låt skikten separera och överför det övre skiktet till separertratten som ovan. Upprepa extraktionen med ytterligare 100 ml petroleumeter (3.2) och sedan två gånger med 50 ml-portioner av petroleumeter (3.2). Överför petroleumeterskikten till separertratten.
Tvätta de sammanslagna extrakten av petroleumeter enligt 5.3.1 och gå vidare enligt instruktionen i den punkten.
5.4 Beredning av provlösning för HPLC
Pipettera över en alikvot av petroleumeterlösningen (från 5.3.1 eller 5.3.2) till en 250 ml rundkolv (4.2.4). Låt lösningsmedlet avdunsta nästan fullständigt i en rotationsindunstare (4.1) med reducerat tryck och en temperatur på vattenbadet som inte överstiger 40 oC. Återställ atmosfärstryck genom att släppa in kvävgas (3.9) och ta sedan bort kolven från rotationsindunstaren. Avlägsna återstående lösningsmedel med en kvävgasström (3.9) och lös omedelbart upp återstoden av provet i en känd volym (10–100 ml) metanol (3.3). (Halten av DL-alfa-tokoferol bör ligga i intervallet 5–30 μg/ml.)
5.5 Bestämning med HPLC
Vitamin E separeras i en kolonn för omvänd fas-kromatografi, C18, (4.5.1) och halten bestäms med fluorescensdetektor (excitation: 295 nm, emission: 330 nm) eller UV-detektor (292 nm) (4.5.2).
Injicera en alikvot (t.ex. 20 μl) av den metanollösning som erhölls i 5.4 och eluera med den mobila fasen (3.8). Beräkna de genomsnittliga topphöjderna (areorna) för flera injektioner av samma provlösning och de genomsnittliga topphöjderna (areorna) för flera injektioner av kalibreringslösningarna (5.6.2).
Följande betingelser anges som vägledning. Andra betingelser kan väljas om de ger likvärdiga resultat.
|
HPLC-kolonn (4.5.1): |
250 × 4 mm, C18, 5 eller 10 μm packning, eller motsvarande |
|
Mobil fas (3.8): |
Blandning av metanol (3.3) och vatten, t.ex. 980 + 20 (v + v) |
|
Flödeshastighet: |
1–2 ml/min |
|
Detektor (4.5.2) |
Fluorescensdetektor (excitation: 295 nm, emission: 330 nm) eller UV-detektor (292 nm) |
5.6 Kalibrering (DL-alfa-tokoferolacetat eller DL-alfa-tokoferol)
5.6.1
5.6.1.1
Pipettera över 25 ml stamlösning av DL-alfa-tokoferolacetat (3.10.1) till en 500 ml sättkolv eller E-kolv (4.2.1) och hydrolysera enligt 5.2. Extrahera sedan med petroleumeter (3.2) enligt 5.3 och späd till 500 ml med petroleumeter. Låt 25 ml av extraktet indunsta i en rotationsindunstare (jfr punkt 5.4) tills provet är nästan torrt. Avlägsna resterande lösningsmedel i en kvävgasström (3.9) och lös provet på nytt i 25,0 ml metanol (3.3). Den nominella halten av DL-alfa-tokoferol i lösningen är 45,5 μg/ml, vilket motsvarar 50 μg DL-alfa-tokoferolacetat/ml. Arbetslösningen ska vara nyberedd.
5.6.1.2
Överför 1,0 –2,0 –4,0 och 10,0 ml av arbetslösningen till ett antal 20 ml mätkolvar. Späd till märket med metanol (3.3) och blanda. De nominella halterna av DL-alfa-tokoferolacetat i dessa lösningar är 2,5 –5,0 –10,0 och 25,0 μg/ml, vilket motsvarar 2,28 –4,55 –9,10 respektive 22,8 μg DL-alfa-tokoferol/ml.
Injicera upprepade gånger 20 μl av varje kalibreringslösning och bestäm de genomsnittliga topphöjderna (areorna). Använd de genomsnittliga topphöjderna (areorna) för att konstruera en kalibreringskurva.
5.6.1.3
Späd 5,0 ml stamlösning av DL-alfa-tokoferolacetat (3.10.1) till 25,0 ml med etanol och mät UV-spektrumet för denna lösning mot etanol (3.1) i en spektrofotometer (4.6) mellan 250 och 320 nm.
Absorptionsmaximum bör ligga vid 284 nm:
![]()
= 43,6 vid 284 nm i etanol 1 cm
Vid denna spädning bör extinktionskoefficienten ligga mellan 0,84 och 0,88 .
5.6.2
5.6.2.1
Pipettera över 2 ml stamlösning av DL-alfa-tokoferol (3.11.1) till en 50 ml mätkolv. Lös tokoferolet i metanol (3.3) och späd till märket med samma lösningsmedel. Den nominella halten av DL-alfa-tokoferol i lösningen är 40 μg/ml, vilket motsvarar 44,0 μg DL-alfa-tokoferolacetat/ml. Arbetslösningen ska vara nyberedd.
5.6.2.2
Överför 1,0 –2,0 –4,0 och 10,0 ml av arbetslösningen till ett antal 20 ml mätkolvar. Späd till märket med metanol (3.3) och blanda. De nominella halterna av DL-alfa-tokoferol i dessa lösningar är 2,0 –4,0 –8,0 och 20,0 μg/ml, vilket motsvarar 2,20 –4,40 –8,79 respektive 22,0 μg DL-alfa-tokoferolacetat/ml.
Injicera upprepade gånger 20 μl av varje kalibreringslösning och bestäm de genomsnittliga topphöjderna (areorna). Använd de genomsnittliga topphöjderna (areorna) för att konstruera en kalibreringskurva.
5.6.2.3
Späd 2,0 ml stamlösning av DL-alfa-tokoferol (3.11.1) till 25,0 ml med etanol och mät UV-spektrumet för denna lösning mot etanol (3.1) i en spektrofotometer (4.6) mellan 250 och 320 nm. Absorptionsmaximum bör ligga vid 292 nm:
![]()
= 75,8 vid 292 nm i etanol 1 cm
Vid denna spädning bör extinktionskoefficienten ligga på 0,6 .
6. Beräkning av resultat
Använd den genomsnittliga topphöjden (arean) för provlösningens vitamin E-toppar för att bestämma halten i μg/ml (beräknat som alfa-tokoferolacetat) med hjälp av kalibreringskurvan (5.6.1.2 eller 5.6.2.2).
Halten av vitamin E, w (mg/kg) i provet beräknas med formeln

[mg/kg]
där:
|
c |
= |
halten av vitamin E (uttryckt som alfa-tokoferolacetat) i provlösningen (5.4) i μg/ml |
|
V1 |
= |
provlösningens volym (5.4) i ml |
|
V2 |
= |
volym för den uttagna alikvoten (5.4) i ml |
|
m |
= |
provets vikt i gram. |
7. Anmärkningar
7.1 För prov med låg halt av vitamin E kan det vara lämpligt att slå ihop petroleumeterextrakten från två förtvålningsomgångar (uppvägd mängd: 25 g) till en enda provlösning för bestämning med HPLC.
7.2 Det prov som ska bestämmas bör inte innehålla mer än 2 g fett.
7.3 Om ingen fasseparation äger rum tillsätts ca 10 ml etanol (3.1) för att få emulsionen att skikta sig.
7.4 Efter spektrofotometrisk bestämning av lösningen av DL-alfa-tokoferolacetat eller DL-alfa-tokoferol enligt 5.6.1.3 resp. 5.6.2.3, tillsätts ca 10 mg BHT (3.12) till denna (3.10.1 eller 3.10.2). Lösningen förvaras sedan i kylskåp (i högst 4 veckor).
7.5 I stället för BHT kan man använda hydrokinon.
7.6 Med en kolonn för normal-fas-kromatografi går det att separera alfa-, beta-, gamma- och delta-tokoferol.
7.7 I stället för natriumaskorbatlösning kan man använda ca 150 mg askorbinsyra.
7.8 I stället för natriumsulfidlösning kan man använda ca 50 mg EDTA.
7.9 Vitamin E-acetat hydrolyseras mycket snabbt i alkalisk miljö och är därför mycket känsligt för oxidation, särskilt i närvaro av spårelement som järn eller koppar. Vid bestämning av vitamin E i förblandningar som innehåller mer än 5 000 mg/kg finns det alltså en risk att vitamin E bryts ned. Därför bör en HPLC-metod med enzymatisk nedbrytning av vitamin E-beredningen utan någon alkalisk förtvålning användas för att bekräfta resultaten.
8. Repeterbarhet
Skillnaden mellan resultaten från två parallella bestämningar som utförs på samma prov får inte överstiga 15 % av det högre värdet.
9. Resultat av en provningsjämförelse ( 12 )
|
|
Förblandning |
Förblandat foder |
Mineralkoncentrat |
Proteinfoder |
Smågrisfoder |
|
L |
12 |
12 |
12 |
12 |
12 |
|
N |
48 |
48 |
48 |
48 |
48 |
|
Medelvärde [mg/kg] |
17 380 |
1 187 |
926 |
315 |
61,3 |
|
sr [mg/kg] |
384 |
45,3 |
25,2 |
13,0 |
2,3 |
|
r [mg/kg] |
1 075 |
126,8 |
70,6 |
36,4 |
6,4 |
|
CVr [ %] |
2,2 |
3,8 |
2,7 |
4,1 |
3,8 |
|
sR mg/kg] |
830 |
65,0 |
55,5 |
18,9 |
7,8 |
|
R [mg/kg] |
2 324 |
182,0 |
155,4 |
52,9 |
21,8 |
|
CVR [ %] |
4,8 |
5,5 |
6,0 |
6,0 |
12,7 |
|
L |
= |
antal laboratorier |
|
n |
= |
antal enskilda värden |
|
sr |
= |
standardavvikelse för repeterbarhet |
|
sR |
= |
standardavvikelse för reproducerbarhet |
|
r |
= |
repeterbarhet |
|
R |
= |
reproducerbarhet |
|
CVr |
= |
variationskoefficient för repeterbarhet |
|
CVR |
= |
variationskoefficient för reproducerbarhet |

C. BESTÄMNING AV SPÅRELEMENTEN JÄRN, KOPPAR, MANGAN OCH ZINK
1. Syfte och tillämpningsområde
Med denna metod kan halten av spårelementen järn, koppar, mangan och zink i foder bestämmas. Kvantifieringsgränser:
— järn (Fe): 20 mg/kg
— koppar (Cu): 10 mg/kg
— mangan (Mn): 20 mg/kg
— zink (Zn): 20 mg/kg
2. Princip
Provet löses upp i saltsyra efter destruktion av eventuellt organiskt material. Grundämnena järn, koppar, mangan och zink bestäms efter lämplig spädning genom atomabsorptionsspektrometri.
3. Reagens
Inledande anmärkningar
Vid beredning av reagens och analyslösningar ska man använda vatten som är fritt från de katjoner som ska bestämmas, genom att det antingen har dubbeldestillerats i en destillationsapparat av borosilikatglas eller kvartsglas eller har genomgått dubbel behandling med jonbytarharts.
Reagensen måste minst vara av p.a.-kvalitet. Det ska kontrolleras genom ett blankprov att de är fria från det grundämne som ska bestämmas. Om det visar sig nödvändigt måste reagensen renas ytterligare.
I stället för de standardlösningar som beskrivs nedan kan i handeln förekommande standardlösningar användas, förutsatt att de är garanterade och att de kontrolleras före användning.
3.1 Saltsyra, d: 1,19 g/ml.
3.2 Saltsyra, 6 mol/l.
3.3 Saltsyra, 0,5 mol/l.
3.4 38–40 % fluorvätesyra (v/v) med en järnhalt (Fe) under 1 mg/l och en rest efter indunstning på mindre än 10 mg (i form av sulfat)/liter.
3.5 Svavelsyra (d: 1,84 g/ml).
3.6 Väteperoxid (ca 100 volymer syre [30 viktprocent]).
3.7 Standardlösning av järn (1 000 μg Fe/ml) beredd på följande sätt (eller motsvarande kommersiellt tillgängliga lösning): Lös upp 1 g järntråd i 200 ml 6 M saltsyra (3.2), tillsätt 16 ml väteperoxid (3.6) och späd till 1 liter med vatten.
3.7.1 Arbetslösning av järn (100 μg Fe/ml) beredd genom spädning av 1 del standardlösning (3.7) med 9 delar vatten.
3.8 Standardlösning av koppar (1 000 μg Cu/ml) beredd på följande sätt (eller motsvarande kommersiellt tillgängliga lösning):
— Lös 1 g kopparpulver i 25 ml 6 M saltsyra (3.2), tillsätt 5 ml väteperoxid (3.6) och späd till 1 liter med vatten.
3.8.1 Arbetslösning av koppar (10 μg Cu/ml) beredd genom spädning av standardlösningen (3.8) med vatten (1:9) varefter den erhållna lösningen späds med vatten (1:9).
3.9 Standardlösning av mangan (1 000 μg Mn/ml) beredd på följande sätt (eller motsvarande kommersiellt tillgängliga lösning):
— Lös 1 g manganpulver i 25 ml 6 M saltsyra (3.2) och späd till 1 liter med vatten.
3.9.1 Arbetslösning av mangan (10 μg Mn/ml) beredd genom spädning av standardlösningen (3.9) med vatten (1:9) varefter den erhållna lösningen späds med vatten (1:9).
3.10 Standardlösning av zink (1 000 μg Zn/ml) beredd på följande sätt (eller motsvarande kommersiellt tillgängliga lösning):
— Lös 1 g zink i rems- eller bladform i 25 ml 6 M saltsyra (3.2) och späd till 1 liter med vatten.
3.10.1 Arbetslösning av zink (10 μg Zn/ml) beredd genom spädning av standardlösningen (3.10) med vatten (1:9) varefter den erhållna lösningen späds med vatten (1:9).
3.11 Lantankloridlösning: Lös 12 g lantanoxid i 150 ml vatten, tillsätt 100 ml 6 M saltsyra (3.2) och späd till 1 liter med vatten.
4. Utrustning
4.1 Muffelugn med temperaturreglering och helst med skrivare.
4.2 Glaskärlen ska vara av resistent borosilikattyp, och utrustningen bör bara användas för bestämning av spårelement.
4.3 Atomabsorptionsspektrofotometer som motsvarar metodens krav i fråga om känslighet och precision inom det aktuella mätområdet.
5. Utförande ( 13 )
5.1 Prov som innehåller organiskt material
5.1.1 ( 14 )
5.1.1.1 Väg med en noggrannhet av 0,2 mg upp 5–10 g av provet i en degel av kvarts eller platina (jfr anmärkning b), torka i ugn vid 105 oC och sätt in degeln i den kalla muffelugnen (4.1). Stäng ugnen (jfr anmärkning c) och höj temperaturen gradvis till 450–475 oC under ca 90 minuter. Behåll denna temperatur under 4–16 timmar (t.ex. över natten) så att kolhaltigt material avlägsnas. Öppna därefter ugnen och låt den svalna (jfr anmärkning d).
Fukta askan med vatten och för över den till en 250 ml bägare. Tvätta ur degeln med sammanlagt ca 5 ml saltsyra (3.1) och tillför sedan detta långsamt och försiktigt i bägaren. (CO2-bildning kan leda till en kraftig reaktion.) Tillsätt saltsyra (3.1) droppvis under skakning tills all bubbelbildning upphört. Låt provet indunsta fullständigt, under omrörning då och då med glasstav.
Tillsätt 15 ml 6 M saltsyra (3.2) till återstoden, därefter ca 120 ml vatten. Rör med glasstaven, som ska stå kvar i bägaren, och täck över bägaren med ett urglas. Koka upp försiktigt och håll lösningen vid kokpunkten tills askan är fullständigt upplöst. Filtrera genom askfritt filtrerpapper och samla upp filtratet i en 250 ml mätkolv. Skölj bägaren och filtret en gång med 5 ml varm 6 M saltsyra (3.2) och två gånger med kokande vatten. Fyll mätkolven till märket med vatten (HCl-koncentration ca 0,5 M).
5.1.1.2 Om återstoden i filtret är svart (kol) sätts den in i ugnen på nytt och föraskas vid 450–475 oC. Denna föraskning, som bara tar några timmar (ca 3–5 timmar), är klar när askan blivit vit eller nästan vit. Lös upp återstoden i ca 2 ml saltsyra (3.1), låt indunsta fullständigt och tillsätt därefter 5 ml 6 M saltsyra (3.2). Lösningen upphettas och filtreras sedan över till mätkolven. Späd till märket med vatten (HCl-koncentration ca 0,5 M).
Anmärkningar:
a) Vid bestämning av spårelement är det viktigt att vara uppmärksam på riskerna för kontaminering, särskilt av zink, koppar och järn. Den utrustning som används vid provberedningen måste därför vara fri från dessa metaller.
För att minska den allmänna risken för kontaminering ska bestämningen utföras i dammfri miljö med ytterst noggrant rengjord utrustning och omsorgsfullt diskade glaskärl. Bestämning av zink är särskilt känslig för många slag av kontaminering, t.ex. genom glas, reagens och damm.
b) Vikten av det prov som ska föraskas beräknas utifrån fodrets ungefärliga halt av spårelement i förhållande till känsligheten hos den spektrofotometer som används. För vissa foder med låg halt av spårelement kan det vara nödvändigt att utgå från ett prov på 10–20 g och späda den slutliga lösningen till endast 100 ml.
c) Föraskningen ska utföras i en stängd ugn utan tillförsel av luft eller syre.
d) Pyrometern får inte visa högre temperatur än 475o C.
5.1.2
5.1.2.1
Bered, för vart och ett av de spårelement som ska bestämmas, en serie kalibreringslösningar utifrån arbetslösningarna (3.7.1, 3.8.1, 3.9.1 och 3.10.1) med en HCl-koncentration på ca 0,5 M för varje kalibreringslösning och, i fråga om järn, mangan och zink, en lantankloridkoncentration motsvarande 0,1 % La (w/v).
Koncentrationerna av spårelement måste väljas så att de ligger inom känslighetsområdet för den spektrofotometer som används. Följande tabeller ger exempel på sammansättningen hos typiska serier av kalibreringslösningar. Beroende på typen av spektrofotometer och dess känslighet kan det dock vara nödvändigt att välja andra koncentrationer.
|
μg Fe/ml |
0 |
0,5 |
1 |
2 |
3 |
4 |
5 |
|
ml arbetslösning (3.7.1) (1 ml = 100 μg Fe) |
0 |
0,5 |
1 |
2 |
3 |
4 |
5 |
|
ml HCl (3.2) |
7 |
7 |
7 |
7 |
7 |
7 |
7 |
|
+ 10 ml lantankloridlösning (3.11); späd till 100 ml med vatten |
|||||||
|
μg Cu/ml |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
ml arbetslösning (3.8.1) (1 ml = 10 μg Cu) |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
|
ml HCl (3.2) |
8 |
8 |
8 |
8 |
8 |
8 |
8 |
|
μg Mn/ml |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
ml arbetslösning (3.9.1) (1 ml = 10 μg Mn) |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
|
ml HCl (3.2) |
7 |
7 |
7 |
7 |
7 |
7 |
7 |
|
+ 10 ml lantankloridlösning (3.11); späd till 100 ml med vatten |
|||||||
|
μg Zn/ml |
0 |
0,05 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
|
ml arbetslösning (3.10.1) (1 ml = 10 μg Zn) |
0 |
0,5 |
1 |
2 |
4 |
6 |
8 |
|
ml HCl (3.2) |
7 |
7 |
7 |
7 |
7 |
7 |
7 |
|
+ 10 ml lantankloridlösning (3.11); späd till 100 ml med vatten |
|||||||
5.1.2.2
Vid bestämning av koppar kan den lösning som beretts enligt 5.1.1 normalt sett användas direkt. Om det är nödvändigt att justera dess koncentration så att den ligger i intervallet för kalibreringslösningarna pipetteras en alikvot till en 100 ml mätkolv och späds till märket med 0,5 M saltsyra (3.3).
Vid bestämning av järn, mangan och zink pipetteras en alikvot av den lösning som beretts enligt 5.1.1 till en 100 ml mätkolv. Tillsätt 10 ml lantankloridlösning (3.11) och späd till märket med 0,5 M saltsyra (3.3) (jfr punkt 8).
5.1.2.3
Blankprovet måste omfatta samtliga moment som anges i denna metod med den enda skillnaden att analysprovet utesluts. Kalibreringslösningen ”0” får inte användas som blankprov.
5.1.2.4
Mät atomabsorptionen hos kalibreringslösningarna och hos den lösning som ska analyseras med en oxiderande luft-acetylenlåga vid följande våglängder:
Fe: 248,3 nm
Cu: 324,8 nm
Mn: 279,5 nm
Zn: 213,8 nm
Utför varje mätning fyra gånger.
5.2 Mineralfoder
Om provet inte innehåller något organiskt material behövs ingen föregående föraskning. Följ anvisningarna från och med 5.1.1.1 andra stycket. Indunstning med fluorvätesyra kan uteslutas.
6. Beräkning av resultat
Beräkna med hjälp av en kalibreringskurva halten av spårelementet i den lösning som ska analyseras och uttryck resultatet i milligram spårelement per kilogram prov (ppm).
7. Repeterbarhet
Skillnaden mellan resultaten från två parallella bestämningar som utförs på samma prov av samma person får inte överstiga
— 5 mg/kg, i absoluta tal, för halter av spårelementet i fråga på upp till 50 mg/kg,
— 10 % av det högre värdet för halter av spårelementet i fråga på 50–100 mg/kg,
— 10 mg/kg, i absoluta tal, för halter av spårelementet i fråga på 100–200 mg/kg,
— 5 % av det högre värdet för halter av spårelementet i fråga som överstiger 200 mg/kg.
8. Anmärkning
Höga halter av fosfater kan påverka bestämningen av järn, mangan och zink. Sådana interferenser måste korrigeras genom tillsats av lantankloridlösning (3.11). Denna tillsats behöver dock inte göras, vare sig till den lösning som ska analyseras eller till kalibreringslösningarna, om viktförhållandet Ca + Mg/P i provet är > 2.
D. BESTÄMNING AV HALOFUGINON
DL-trans-7-brom-6-klor-3-[3-(3-hydroxi-2-piperidyl)acetonyl]quinazolin-4-(3H)-on-hydrobromid
1. Syfte och tillämpningsområde
Med denna metod kan halten av halofuginon i foder bestämmas. Kvantifieringsgränsen är 1 mg/kg.
2. Princip
Efter behandling med varmt vatten extraheras halofuginon, i form av den fria basen, till etylacetat och extraheras därefter, i form av hydroklorid, till en syralösning. Extraktet renas med jonbyteskromatografi. Halten av halofuginon bestäms genom omvänd fas-kromatografi (HPLC) med UV-detektor.
3. Reagens
3.1 Acetonitril, HPLC-kvalitet
3.2 Amberlite XAD-2-harts
3.3 Ammoniumacetat
3.4 Etylacetat
3.5 Isättika
3.6 Halofuginon referenssubstans (DL-trans-7-brom-6-klor-3-[3-hydroxi-2-piperidyl)acetonyl]quinazolin-4-(3H)-on-hydrobromid, E 764)
3.6.1
Väg med en noggrannhet av 0,1 mg upp 50 mg halofuginon (3.6) i en 500 ml mätkolv. Lös i ammoniumacetatbuffertlösning (3.18) och späd sedan med samma lösning till märket och blanda. Lösningen är stabil i tre veckor om den förvaras mörkt vid 5 oC.
3.6.2
För över 1,0 –2,0 –3,0 –4,0 respektive 6,0 ml stamlösning (3.6.1) till en serie 100 ml mätkolvar. Späd till märket med den mobila fasen (3.21) och blanda. Koncentrationen av halofuginon i dessa lösningar är 1,0 –2,0 –3,0 –4,0 respektive 6,0 μg/ml. Lösningarna ska vara nyberedda.
3.7 Saltsyra (ρ20 ca 1,16 g/ml)
3.8 Metanol
3.9 Silvernitrat
3.10 Natriumaskorbat
3.11 Natriumkarbonat
3.12 Natriumklorid
3.13 EDTA (etylendiamintetraättiksyra, dinatriumsalt)
3.14 Vatten, HPLC-kvalitet
3.15 Natriumkarbonatlösning, c = 10 g/100 ml
3.16 Natriumkloridmättad natriumkarbonatlösning, c = 5 g/100 ml
Lös 50 g natriumkarbonat (3.11) i vatten. Späd till 1 liter och tillsätt natriumklorid (3.12) tills lösningen är mättad.
3.17 Saltsyra, ca 0,1 mol/l
Späd 10 ml HCl (3.7) med vatten till 1 liter.
3.18 Ammoniumacetatbuffertlösning, ca 0,25 mol/l
Lös 19,3 g ammoniumacetat (3.3) och 30 ml ättiksyra (3.5) i vatten (3.14) och späd till 1 liter.
3.19 Förbehandling av Amberlite XAD-2-harts
Skölj en lämplig mängd amberlit (3.2) med vatten tills alla kloridjoner har tvättats ur. Detta bekräftas genom en kontroll med silvernitrat (3.20) på sköljvattnet. Skölj därefter hartsen med 50 ml metanol (3.8). Häll bort metanolen och lagra hartsen i färsk metanol.
3.20 Silvernitratlösning, ca 0,1 mol/l
Lös 0,17 g silvernitrat (3.9) i 10 ml vatten.
3.21 Mobil fas för HPLC
Blanda 500 ml acetonitril (3.1) med 300 ml ammoniumacetatbuffertlösning (3.18) och 1 200 ml vatten (3.14). Justera pH till 4,3 med ättiksyra (3.5). Filtrera lösningen genom ett 0,22 μm filter (4.8) och avlufta den (t.ex. i ultraljudsbad i 10 minuter). Lösningen är stabil i en månad om den förvaras mörkt i ett slutet kärl.
4. Utrustning
4.1 Ultraljudsbad
4.2 Rotationsindunstare
4.3 Centrifug
4.4 HPLC-utrustning med UV-detektor med variabel våglängd eller diod-array-detektor
4.4.1 HPLC-kolonn, 300 × 4 mm, C18, 10 μm packning, eller en motsvarande kolonn
4.5 Glaskolonn (300 × 10 mm) med sintrat glasfilter och avstängningsventil
4.6 Glasfiberfilter, diameter 150 mm
4.7 Membranfilter, 0,45 μm
4.8 Membranfilter, 0,22 μm
5. Utförande
|
Anmärkning: |
Halofuginon som fri bas är instabil i alkaliska lösningar och etylacetatlösningar. Det bör därför inte vara i etylacetat i mer än 30 minuter. |
5.1 Allmänt
5.1.1 Ett blankprov analyseras för att kontrollera att varken halofuginon eller störande ämnen är närvarande.
5.1.2 Gör ett utbytesförsök genom analys av ett blankprov som spikats med ungefär samma mängd halofuginon som finns i provet. För spikning på nivån 3 mg/kg tillsätts 300 μl stamlösning (3.6.1) till 10 g av blankprovet. Blanda och vänta i 10 minuter innan extraktionen (5.2) påbörjas.
|
Anmärkning: |
Blankprovet bör vara av liknande typ som analysprovet. Vid analys ska halofuginon inte kunna påvisas. |
5.2 Extraktion
Väg med en noggrannhet av 0,1 g upp 10 gram av det beredda provet i ett 200 ml centrifugrör. Tillsätt 0,5 g natriumaskorbat (3.10), 0,5 g EDTA (3.13) samt 20 ml vatten och blanda. Ställ röret i vattenbad (80 oC) i 5 minuter. Låt svalna till rumstemperatur. Tillsätt därefter 20 ml natriumkarbonatlösning (3.15) och blanda. Tillsätt omedelbart därefter 100 ml etylacetat (3.4) och skaka röret kraftigt för hand i 15 sekunder. Ställ röret i ultraljudsbadet (4.1) i 3 minuter och lossa proppen. Centrifugera i 2 minuter och dekantera etylacetatfasen genom ett glasfiberfilter (4.6) till en 500 ml separertratt. Upprepa extraktionen av provet med en ny volym på 100 ml etylacetat. Tvätta de kombinerade extrakten i 1 minut med 50 ml natriumkloridmättad natriumkarbonatlösning (3.16). Häll bort vattenskiktet.
Extrahera det organiska skiktet i 1 minut med 50 ml saltsyra (3.17). Det undre syraskiktet tappas av till en 250 ml separertratt. Extrahera det organiska skiktet en gång till i 1,5 minuter med ytterligare 50 ml saltsyra och slå samman med det första extraktet. Tvätta de kombinerade syraextrakten genom att rotera kärlet i ca 10 sekunder med 10 ml etylacetat (3.4).
Överför vattenskiktet kvantitativt till en 250 ml rundkolv och häll bort den organiska fasen. Avlägsna allt resterande etylacetat från syralösningen i en rotationsindunstare (4.2). Vattenbadets temperatur bör inte överstiga 40 oC. Vid ett tryck på ca 25 mbar avdunstar allt resterande etylacetat inom 5 minuter vid 38 oC.
5.3 Rening
5.3.1
Förbered en XAD-2-kolonn för varje provextrakt. För över 10 g förbehandlad amberlit (3.19) till en glaskolonn (4.5) med metanol (3.8). Sätt en liten propp av glasull i toppen av hartskolonnen. Låt metanolen rinna ur kolonnen och skölj hartsen med 100 ml vatten. Stoppa flödet när vätskan når toppen av hartskolonnen. Låt kolonnen komma i jämvikt i 10 minuter före användning. Se till att kolonnen aldrig torkar ut.
5.3.2
För över extraktet (5.2) kvantitativt till toppen av den förberedda amberlitkolonnen (5.3.1) och eluera. Häll bort eluatet. Flödeshastigheten vid elueringen bör inte överskrida 20 ml/minut. Skölj rundkolven med 20 ml saltsyra (3.17) som därefter används för att skölja hartskolonnen. Avlägsna kvarvarande syralösning med en luftström. Häll bort sköljresterna. Tillsätt 100 ml metanol (3.8) i kolonnen och eluera 5–10 ml. Samla upp eluatet i en 250 ml rundkolv. Vänta i 10 minuter så att den återstående metanolen kommer i jämvikt med hartsen. Fortsätt elueringen med en flödeshastighet på högst 20 ml/minut och samla upp eluatet i samma rundkolv. Låt metanolen avdunsta i en rotationsindunstare (4.2). Vattenbadets temperatur bör inte överskrida 40 oC. Överför återstoden kvantitativt till en 10 ml mätkolv genom att använda den mobila fasen (3.21). Späd med den mobila fasen till märket och blanda. En alikvot av lösningen filtreras genom ett membranfilter (4.7). Denna lösning används sedan för HPLC-bestämningen (5.4).
5.4 HPLC-bestämning
5.4.1
Följande betingelser anges som vägledning. Andra betingelser kan väljas om de ger likvärdiga resultat.
HPLC-kolonn (4.4.1)
Mobil fas för HPLC (3.21)
Flödeshastighet: 1,5 –2 ml/minut
Detektionsvåglängd: 243 nm
Injektionsvolym: 40–100 μl.
Kontrollera att det kromatografiska systemet är stabilt genom att upprepade gånger injicera en kalibreringslösning (3.6.2) med koncentrationen 3,0 μg/ml tills topphöjder (areor) och retentionstider är konstanta.
5.4.2
Injicera varje kalibreringslösning (3.6.2) upprepade gånger och mät topphöjderna (areorna) för varje koncentration. Konstruera en kalibreringskurva med kalibreringslösningarnas genomsnittliga topphöjder (areor) längs y-axeln och motsvarande koncentrationerna i μg/ml längs x-axeln.
5.4.3
Provextraktet (5.3.2) injiceras upprepade gånger, varvid samma volym ska användas som för kalibreringslösningarna. Bestäm därefter den genomsnittliga topphöjden (arean) för halofuginon.
6. Beräkning av resultat
Använd den genomsnittliga topphöjden (arean) för provlösningens halofuginontoppar för att bestämma koncentrationen i μg/ml med hjälp av kalibreringskurvan.
Provets halofuginonhalt w (mg/kg) beräknas med formeln

där:
|
c |
: |
provlösningens halofuginonkoncentration i μg/ml |
|
m |
: |
provets vikt i gram. |
7. Validering av resultat
7.1 Identitet
Analytens identitet kan bekräftas genom co-kromatografi eller genom användning av en diod-array-detektor med vars hjälp spektrumen för provextraktet och den kalibreringslösning (3.6.2) som innehåller 6,0 μg/ml jämförs.
7.1.1
Ett provextrakt spikas genom tillsats av en lämplig mängd kalibreringslösning (3.6.2). Mängden tillsatt halofuginon bör motsvara den uppskattade mängden halofuginon i provextraktet.
Endast halofuginontoppens höjd ska öka efter det att hänsyn tagits till både den tillsatta mängden och spädningen av extraktet. Toppens bredd på halva höjden ska ligga inom ± 10 % av den ursprungliga bredden.
7.1.2
Resultaten utvärderas enligt följande kriterier:
a) Våglängderna för prov- och standardspektrumens absorptionsmaximum, i toppens spets på kromatogrammet, ska ligga inom en marginal som beror på detektionssystemets upplösningsförmåga. För diod-array-detektion är denna i allmänhet ± 2 nm.
b) Mellan 225 och 300 nm får prov- och standardspektrumen, i toppens spets på kromatogrammet, inte skilja sig åt för de delar av spektrumet där den relativa absorbansen är 10–100 %. Detta kriterium är uppfyllt om samma maximum observeras och avvikelsen mellan de två spektrumen inte vid någon mätpunkt överstiger 15 % av standardanalytens absorbans.
c) Mellan 225 och 300 nm får spektrumen för den uppåtgående delen, spetsen och den nedåtgående delen av toppen från provextraktet inte skilja sig från varandra för de delar av spektrumet där den relativa absorbansen är 10–100 %. Detta kriterium är uppfyllt om samma maximum observeras och avvikelsen mellan spektrumen inte vid någon mätpunkt överstiger 15 % av absorbansen vid spetsen.
Om något av dessa kriterier inte är uppfyllt har analytens förekomst inte bekräftats.
7.2 Repeterbarhet
Skillnaden mellan resultaten från två parallella bestämningar som utförs på samma prov får inte överstiga 0,5 mg/kg för halofuginonhalter till och med 3 mg/kg.
7.3 Utbyte
För ett spikat blankprov ska utbytet vara minst 80 %.
8. Resultat av en provningsjämförelse
I en provningsjämförelse ( 15 ) analyserades tre prov av åtta laboratorier.
Resultat
|
|
Prov A (blankprov) Vid mottagandet |
Prov B (mjöl) |
Prov C (pellets) |
||
|
|
|
Vid mottagandet |
Efter två månader |
Vid mottagandet |
Efter två månader |
|
Medelvärde [mg/kg] |
ND |
2,80 |
2,42 |
2,89 |
2,45 |
|
SR [mg/kg] |
— |
0,45 |
0,43 |
0,40 |
0,42 |
|
CVR [ %] |
— |
16 |
18 |
14 |
17 |
|
Utbyte [ %] |
|
86 |
74 |
88 |
75 |
|
ND |
= |
analyten har ej påvisats |
|
SR |
= |
standardavvikelse för reproducerbarhet |
|
CVR |
= |
variationskoefficient för reproducerbarhet, % |
E. BESTÄMNING AV ROBENIDIN
1,3-bis[(4-klorbensyliden)amin]guanidinhydroklorid
1. Syfte och tillämpningsområde
Med denna metod kan robenidinhalten i foder bestämmas. Kvantifieringsgränsen är 5 mg/kg.
2. Princip
Provet extraheras med surgjord metanol. Extraktet torkas och en alikvot renas i en kolonn med aluminiumoxid. Robenidinet elueras från kolonnen med metanol och koncentreras. Den erhållna lösningen späds med en lämplig volym av den mobila fasen. Halten av robenidin bestäms genom omvänd fas-kromatografi (HPLC) med UV-detektor.
3. Reagens
3.1 Metanol
3.2 Surgjord metanol
För över 4,0 ml saltsyra (ρ20 = 1,18 g/ml) till en 500 ml mätkolv. Späd med metanol (3.1) till märket och blanda. Denna lösning ska vara nyberedd.
3.3 Acetonitril, HPLC-kvalitet
3.4 Molekylsikt
Typ 3A, 8–12 mesh (1,6 –2,5 mm kulor, kristallint aluminiumsilikat, pordiameter 0,3 nm).
3.5 Aluminiumoxid, sur, aktivitetsgrad I, för kolonnkromatografi
För över 100 g aluminiumoxid till ett lämpligt kärl och tillsätt 2,0 ml vatten. Förslut kärlet och skaka i ca 20 minuter. Förvara sedan i ett väl tillslutet kärl.
3.6 Kaliumdivätefosfatlösning, c = 0,025 mol/l
Lös 3,40 g kaliumdivätefosfat i vatten (HPLC-kvalitet) i en 1 000 ml mätkolv. Späd till märket och blanda.
3.7 Dinatriumvätefosfatlösning, c = 0,025 mol/l
Lös 3,55 g vattenfri (eller 4,45 g dihydrat eller 8,95 g dodekahydrat) dinatriumvätefosfat i vatten (HPLC-kvalitet) i en 1 000 ml mätkolv. Späd till märket och blanda.
3.8 Mobil fas för HPLC
Blanda följande reagens:
650 ml acetonitril (3.3),
250 ml vatten (HPLC-kvalitet),
50 ml kaliumdivätefosfatlösning (3.6),
50 ml dinatriumvätefosfatlösning (3.7).
Filtrera lösningen genom ett 0,22 μm filter (4.6) och avlufta den (t.ex. i ultraljudsbad i 10 minuter).
3.9 Referenssubstans
Rent robenidin: 1,3 -bis[(4-klorbensyliden)amin]guanidin-hydroklorid.
3.9.1
Väg med en noggrannhet av 0,1 mg upp 30 mg referenssubstans av robenidin (3.9). Lös i surgjord metanol (3.2) i en 100 ml mätkolv. Späd till märket med samma lösningsmedel och blanda. Slå in kolven i aluminiumfolie och förvara mörkt.
3.9.2
För över 10,0 ml stamlösning (3.9.1) till en 250 ml mätkolv. Späd till märket med mobil fas (3.8) och blanda. Slå in kolven i aluminiumfolie och förvara mörkt.
3.9.3
För över 5,0 –10,0 –15,0 –20,0 respektive 25,0 ml arbetsstandardlösning (3.9.2) till en serie 50 ml mätkolvar. Späd till märket med den mobila fasen (3.8) och blanda. Koncentrationen av robenidin i dessa lösningar är 1,2 –2,4 –3,6 –4,8 respektive 6,0 μg/ml. Lösningarna ska vara nyberedda.
3.10 Vatten, HPLC-kvalitet
4. Utrustning
4.1 Glaskolonn
Kolonn i brunt glas, med avstängningsventil och en behållare med ca 150 ml volym, invändig diameter 10–15 mm, längd 250 mm.
4.2 Mekanisk skakapparat eller magnetomrörare
4.3 Rotationsindunstare
4.4 HPLC-utrustning med UV-detektor med variabel våglängd eller diod-array-detektor för intervallet 250–400 nm
4.4.1. HPLC-kolonn: 300 × 4 mm, C18, 10 μm packning eller motsvarande
4.5 Glasfiberfilter (Whatman GF/A eller motsvarande)
4.6 Membranfilter, 0,22 μm
4.7 Membranfilter, 0,45 μm
5. Utförande
|
Anmärkning: |
Robenidin är ljuskänsligt. Bruna glaskärl bör därför användas vid alla arbetsmoment. |
5.1 Allmänt
5.1.1 Ett blankprov analyseras för att kontrollera att varken robenidin eller störande ämnen är närvarande.
5.1.2 Gör ett utbytesförsök genom analys av ett blankprov (5.1.1) som spikats med ungefär samma mängd robenidin som finns i provet. För spikning på nivån 60 mg/kg överförs 3,0 ml stamlösning (3.9.1) till en 250 ml E-kolv. Indunsta lösningen till ca 0,5 ml i en kvävgasström. Tillsätt 15 g av blankprovet, blanda och vänta i 10 minuter innan extraktionen (5.2) påbörjas.
|
Anmärkning: |
Blankprovet bör vara av liknande typ som analysprovet. Vid analys ska robenidin inte kunna påvisas. |
5.2 Extraktion
Väg med en noggrannhet av 0,01 g upp ca 15 gram av det beredda provet och för över till en 250 ml E-kolv. Tillsätt 100,0 ml surgjord metanol (3.2), tillslut kolven och låt stå i 1 timme i skakapparat (4.2). Filtrera lösningen genom ett glasfiberfilter (4.5) och samla upp allt filtrat i en 150 ml E-kolv. Tillsätt 7,5 g molekylsikt (3.4), tillslut kolven och skaka i 5 minuter. Filtrera omedelbart genom ett glasfiberfilter. Spara lösningen för reningen (5.3).
5.3 Rening
5.3.1
Sätt en liten propp av glasull i glaskolonnens (4.1) nedre öppning och pressa ihop den med en glasstav. Väg upp 11,0 g av den förbehandlade aluminiumoxiden (3.5) och för över till kolonnen. Försök minimera exponeringen för luft under detta arbetsmoment. Knacka försiktigt på kolonnens nederdel så att aluminiumoxiden sätter sig.
5.3.2
Pipettera över 5,0 ml av det provextrakt som beretts enligt 5.2 till kolonnen. Håll pipettens spets nära kolonnväggen och låt lösningen absorberas av aluminiumoxiden. Eluera robenidinet från kolonnen med 100 ml metanol (3.1), flödeshastighet 2–3 ml/minut, och samla upp eluatet i en 250 ml rundkolv. Låt metanollösningen indunsta fullständigt i rotationsindunstaren (4.3) under reducerat tryck vid 40 oC. Lös upp återstoden i 3–4 ml mobil fas (3.8) och överför kvantitativt till en 10 ml mätkolv. Skölj kolven med flera volymer mobil fas om 1–2 ml och överför sköljvätskan till mätkolven. Späd till märket med samma lösningsmedel och blanda. En alikvot av lösningen filtreras genom ett 0,45 μm membranfilter (4.7). Denna lösning används sedan för HPLC-bestämningen (5.4).
5.4 HPLC-bestämning
5.4.1
Följande betingelser anges som vägledning. Andra betingelser kan väljas om de ger likvärdiga resultat.
HPLC-kolonn (4.4.1),
Mobil fas för HPLC (3.8),
Flödeshastighet: 1,5 –2 ml/minut,
Detektionsvåglängd: 317 nm,
Injektionsvolym: 20–50 μl.
Kontrollera att det kromatografiska systemet är stabilt genom att upprepade gånger injicera en kalibreringslösning (3.9.3) med koncentrationen 3,6 μg/ml tills topphöjder och retentionstider är konstanta.
5.4.2
Injicera varje kalibreringslösning (3.9.3) upprepade gånger och mät topphöjderna (areorna) för varje koncentration. Konstruera en kalibreringskurva med kalibreringslösningarnas genomsnittliga topphöjder (areor) längs y-axeln och motsvarande koncentrationer i μg/ml längs x-axeln.
5.4.3
Provextraktet (5.3.2) injiceras upprepade gånger, varvid samma volym ska användas som för kalibreringslösningarna. Bestäm den genomsnittliga topphöjden (arean) för robenidin.
6. Beräkning av resultat
Använd den genomsnittliga topphöjden (arean) för provlösningens robenidintoppar för att bestämma koncentrationen i μg/ml med hjälp av kalibreringskurvan (5.4.2).
Provets robenidinhalt w (mg/kg) beräknas med formeln
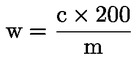
där:
|
c |
= |
provlösningens robenidinkoncentration i μg/ml |
|
m |
= |
provets vikt i gram. |
7. Validering av resultat
7.1 Identitet
Analytens identitet kan bekräftas genom co-kromatografi eller genom användning av en diod-array-detektor med vars hjälp spektrumen för provextraktet och den kalibreringslösning (3.9.3) som innehåller 6 μg/ml jämförs.
7.1.1
Ett provextrakt spikas genom tillsats av en lämplig mängd kalibreringslösning (3.9.3). Mängden tillsatt robenidin bör motsvara den uppskattade mängden robenidin i provextraktet.
Endast robenidintoppens höjd ska öka efter det att hänsyn tagits till både den tillsatta mängden och spädningen av extraktet. Toppens bredd på halva höjden ska ligga inom ± 10 % av den ursprungliga bredden
7.1.2
Resultaten utvärderas enligt följande kriterier:
a) Våglängderna för prov- och standardspektrumens absorptionsmaximum, i toppens spets på kromatogrammet, ska ligga inom en marginal som beror på detektionssystemets upplösningsförmåga. För diod-array-detektion är denna i allmänhet ± 2 nm.
b) Mellan 250 och 400 nm får prov- och standardspektrumen, i toppens spets på kromatogrammet, inte skilja sig åt för de delar av spektrumet där den relativa absorbansen är 10–100 %. Detta kriterium är uppfyllt om samma maximum observeras och avvikelsen mellan de två spektrumen inte vid någon mätpunkt överstiger 15 % av standardanalytens absorbans.
c) Mellan 250 och 400 nm får spektrumen för den uppåtgående delen, spetsen och den nedåtgående delen av toppen från provextraktet inte skilja sig från varandra för de delar av spektrumet där den relativa absorbansen är 10–100 %. Detta kriterium är uppfyllt om samma maximum observeras och avvikelsen mellan spektrumen inte vid någon mätpunkt överstiger 15 % av absorbansen vid spetsen.
Om något av dessa kriterier inte är uppfyllt har analytens förekomst inte bekräftats.
7.2 Repeterbarhet
Skillnaden mellan resultaten från två parallella bestämningar som utförs på samma prov får inte överstiga 10 % av det högre värdet för robenidinhalter över 15 mg/kg.
7.3 Utbyte
För ett spikat blankprov ska utbytet vara minst 85 %.
8. Resultat av en provningsjämförelse
I en provningsjämförelse inom EU fick tolv laboratorier analysera fyra prov av fjäderfä- och kaninfoder i form av mjöl eller pellets. För varje prov gjordes dubbla analyser. Resultaten redovisas i nedanstående tabell:
|
|
Fjäderfä |
Kanin |
||
|
|
Mjöl |
Pellets |
Mjöl |
Pellets |
|
Medelvärde [mg/kg] |
27,00 |
27,99 |
43,6 |
40,1 |
|
sr [mg/kg] |
1,46 |
1,26 |
1,44 |
1,66 |
|
CVr [ %] |
5,4 |
4,5 |
3,3 |
4,1 |
|
SR [mg/kg] |
4,36 |
3,36 |
4,61 |
3,91 |
|
CVR [ %] |
16,1 |
12,0 |
10,6 |
9,7 |
|
Utbyte [ %] |
90,0 |
93,3 |
87,2 |
80,2 |
|
sr |
= |
standardavvikelse för repeterbarhet |
|
CVr |
= |
variationskoefficient för repeterbarhet, % |
|
SR |
= |
standardavvikelse för reproducerbarhet |
|
CVR |
= |
variationskoefficient för reproducerbarhet, % |
F. BESTÄMNING AV DICLAZURIL
(+)-4-klorfenyl [2,6-diklor-4-(2,3,4,5-tetrahydro-3,5-dioxo-1,2,4-triazin-2-yl)fenyl]acetonitril
1. Syfte och tillämpningsområde
Med denna metod kan halten av diclazuril i foder och förblandningar bestämmas. Detektionsgränsen är 0,1 mg/kg och kvantifieringsgränsen 0,5 mg/kg.
2. Princip
Efter tillsats av intern standard extraheras provet med surgjord metanol. För foder renas en alikvot av extraktet på en C18-kolonn för fastfasextraktion. Diclazuril elueras från kolonnen med en blandning av surgjord metanol och vatten. Efter indunstning löses återstoden i DMF/vatten. För förblandningar indunstas extraktet och återstoden löses i DMF/vatten. Diclazurilhalten bestäms med ternär-gradient omvänd fas-kromatografi (HPLC) med UV-detektor.
3. Reagens
3.1 Vatten, HPLC-kvalitet.
3.2 Ammoniumacetat
3.3 Tetrabutylammoniumvätesulfat (TBHS)
3.4 Acetonitril, HPLC-kvalitet
3.5 Metanol, HPLC-kvalitet
3.6 N, N-dimetylformamid (DMF)
3.7 Saltsyra, ρ20 = 1,19 g/ml
3.8 Referenssubstans: diclazuril II-24: (+)-4-klorfenyl [2,6-diklor-4-(2,3,4,5-tetrahydro-3,5-dioxo-1,2,4-triazin-2-yl)fenyl]acetonitril med garanterad renhet, E771
3.8.1
Väg med en noggrannhet av 0,1 mg upp 25 mg av diclazuril referenssubstans (3.8) i en 50 ml mätkolv. Lös i DMF (3.6), späd till märket med DMF (3.6) och blanda. Slå in kolven i aluminiumfolie eller använd en kolv av brunt glas. Förvara i kylskåp. Lösningen är stabil i 1 månad vid högst 4 oC.
3.8.2
Överför 5,00 ml stamlösning (3.8.1) till en 50 ml mätkolv, späd till märket med DMF (3.6) och blanda. Slå in kolven i aluminiumfolie eller använd en kolv av brunt glas. Förvara i kylskåp. Lösningen är stabil i en månad vid högst 4 oC.
3.9 Intern standard: 2,6-diklor-(4-klorfenyl)-4-(4,5-dihydro-3,5-dioxo-1,2,4-triazin-2-(3H)-yl)-metylbensen-acetonitril
3.9.1
Väg med en noggrannhet av 0,1 mg upp 25 mg intern standard (3.9) i en 50 ml mätkolv. Lös i DMF (3.6), späd till märket med DMF (3.6) och blanda. Slå in kolven i aluminiumfolie eller använd en kolv av brunt glas. Förvara i kylskåp. Lösningen är stabil i 1 månad vid högst 4 oC.
3.9.2
Överför 5,00 ml stamlösning av intern standard (3.9.1) till en 50 ml mätkolv, späd till märket med DMF (3.6) och blanda. Slå in kolven i aluminiumfolie eller använd en kolv av brunt glas. Förvara i kylskåp. Lösningen är stabil i 1 månad vid högst 4 oC.
3.9.3
(p = nominell halt av diclazuril i förblandningen i mg/kg)
Väg med en noggrannhet av 0,1 mg upp p/10 mg intern standard i en 100 ml mätkolv, lös i DMF (3.6) i ett ultraljudsbad (4.6), späd till märket med DMF och blanda. Slå in kolven i aluminiumfolie eller använd en kolv av brunt glas. Förvara i kylskåp. Lösningen är stabil i 1 månad vid högst 4 oC.
3.10 Kalibreringslösning, 2 μg/ml.
Pipettera över 2,00 ml diclazurilstandardlösning (3.8.2) och 2,00 ml intern standardlösning (3.9.2) till en 50 ml mätkolv. Tillsätt 16 ml DMF (3.6), späd till märket med vatten och blanda. Denna lösning ska vara nyberedd.
3.11 C18-fastfasextraktionskolonn, t.ex. Bond Elut, storlek: 1 cm3, sorbent: 100 mg.
3.12 Extraktionsmedel: surgjord metanol.
Pipettera 5,0 ml saltsyra (3.7) till 1 000 ml metanol (3.5) och blanda.
3.13 Mobil fas för HPLC
3.13.1 Eluent A: ammoniumacetat–tetrabutylammoniumvätesulfatlösning.
Lös 5 g ammoniumacetat (3.2) och 3,4 g TBHS (3.3) i 1 000 ml vatten (3.1) och blanda.
3.13.2 Eluent B: acetonitril (3.4).
3.13.3 Eluent C: metanol (3.5).
4. Utrustning
4.1 Mekanisk skakapparat.
4.2 Utrustning för ternär-gradient-HPLC
4.2.1 HPLC-kolonn, Hypersil ODS, 3 μm packning, 100 × 4,6 mm, eller motsvarande
4.2.2 UV-detektor med variabel våglängd eller diod-array-detektor.
4.3 Rotationsindunstare
4.4 Membranfilter, 0,45 μm
4.5 Vakuumgrenrör
4.6 Ultraljudsbad
5. Utförande
5.1 Allmänt
5.1.1
Ett blankprov analyseras för att kontrollera att varken diclazuril eller störande ämnen är närvarande. Blankprovet bör vara av liknande typ som analysprovet och varken diclazuril eller störande ämnen ska kunna påvisas i det.
5.1.2
Gör ett utbytesförsök genom analys av ett blankprov som spikats med ungefär samma mängd diclazuril som finns i provet. För spikning på nivån 1 mg/kg, sätt 0,1 ml stamlösning av intern standard (3.8.1) till 50 g av ett blankprov, blanda noga och låt stå i 10 minuter. Blanda sedan flera gånger före extraktionen (5.2).
Om ett blankprov av samma typ som provet inte finns att tillgå (se 5.1.1) kan ett utbytesförsök göras med standardadditionsmetoden. Denna innebär att det prov som ska analyseras spikas med ungefär samma mängd diclazuril som finns i provet. Detta prov analyseras tillsammans med det ospikade provet, och utbytet beräknas sedan genom subtraktion.
5.2 Extraktion
5.2.1
Väg med 0,01 g noggrannhet upp ca 50 g prov. Överför detta till en 500 ml E-kolv, tillsätt 1,00 ml intern standardlösning (3.9.2) och 200 ml extraktionsmedel (3.12) och tillslut kolven. Skaka blandningen i skakapparaten (4.1) över natten. Låt stå i 10 minuter. Överför en 20 ml alikvot av supernatanten till ett lämpligt glaskärl och späd med 20 ml vatten. Överför denna lösning till en extraktionskolonn (3.11) och låt den passera genom kolonnen med hjälp av vakuum (4.5). Tvätta kolonnen med 25 ml av en blandning av extraktionsmedel (3.12) och vatten, 65 + 35 (v + v). Kasta de uppsamlade fraktionerna och eluera ämnena med 25 ml av en blandning av extraktionsmedel (3.12) och vatten, 80 + 20 (v + v). Indunsta denna fraktion i en rotationsindunstare (4.3) vid 60 oC tills den precis har torkat. Lös återstoden i 1,0 ml DMF (3.6), tillsätt 1,5 ml vatten och blanda. Filtrera genom ett membranfilter (4.4). Gå vidare till HPLC-bestämningen (5.3).
5.2.2
Väg med 0,001 g noggrannhet upp ca 1 g prov. Överför detta till en 500 ml E-kolv, tillsätt 1,00 ml intern standardlösning (3.9.3) och 200 ml extraktionsmedel (3.12) och tillslut kolven. Skaka blandningen i skakapparaten (4.1) över natten. Låt stå i 10 minuter. Överför en alikvot på10 000 /p ml (p = nominell halt av diclazuril i förblandningen i mg/kg) av supernatanten till en rundkolv av lämplig storlek. Indunsta i en rotationsindunstare (4.3) under reducerat tryck vid 60 oC tills återstoden precis har torkat. Lös återstoden i 10,0 ml DMF (3.6), tillsätt 15,0 ml vatten (3.1) och blanda. Gå vidare till HPLC-bestämningen (5.3).
5.3 HPLC-bestämning
5.3.1
Följande betingelser anges som vägledning. Andra betingelser kan väljas om de ger likvärdiga resultat.
|
HPLC-kolonn (4.2.1): |
100 × 4,6 mm, Hypersil ODS, 3 μm packning, eller motsvarande |
|
|
Mobil fas : |
Eluent A (3.13.1): |
Vattenlösning av ammonium-acetat och tetrabutylammonium-vätesulfat |
|
|
Eluent B (3.13.2): |
acetonitril |
|
|
Eluent C (3.13.3): |
metanol |
|
Elueringssätt: |
— linjär gradient — utgångsförhållanden: A + B + C = 60 + 20 + 20 (v + v + v) — efter 10 minuters gradienteluering i 30 min. till: A + B + C = 45 + 20 + 35 (v + v + v). Skölj med B i 10 min. |
|
|
Flödeshastighet: |
1,5 –2 ml/min |
|
|
Injektionsvolym: |
20 μl |
|
|
Detektorvåglängd: |
280 nm |
|
Kontrollera det kromatografiska systemets stabilitet genom att upprepade gånger injicera kalibreringslösningen (3.10), som innehåller 2 μg/ml, till dess att topphöjder och retentionstider är konstanta.
5.3.2
Injicera 20 μl av kalibreringslösningen (3.10) upprepade gånger och bestäm den genomsnittliga topphöjden (arean) för diclazuril och den interna standarden.
5.3.3
Injicera 20 μl av provlösningen (5.2.1 eller 5.2.2) upprepade gånger och bestäm den genomsnittliga topphöjden (arean) för diclazuril och den interna standarden.
6. Beräkning av resultat
6.1 Foder
Provets diclazurilhalt w (mg/kg) beräknas med formeln

[mg/kg]
där:
|
hd, s |
= |
topphöjd (area) för diclazuril i provlösningen (5.2.1) |
|
hi, s |
= |
topphöjd (area) för den interna standarden i provlösningen (5.2.1) |
|
hd, c |
= |
topphöjd (area) för diclazuril i kalibreringslösningen (3.10) |
|
hi, c |
= |
topphöjd (area) för den interna standarden i kalibreringslösningen (3.10) |
|
cd, c |
= |
diclazurilkoncentration i kalibreringslösningen i μg/ml (3.10) |
|
m |
= |
provets vikt i gram |
|
V |
= |
provextraktets volym i ml (= 2,5 ml), se 5.2.1. |
6.2 Förblandningar
Provets diclazurilhalt w (mg/kg) beräknas med formeln
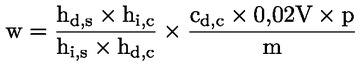
[mg/kg]
där:
|
hd, c |
= |
topphöjd (area) för diclazuril i kalibreringslösningen (3.10) |
|
hi, c |
= |
topphöjd (area) för den interna standarden i kalibreringslösningen (3.10) |
|
hd, s |
= |
topphöjd (area) för diclazuril i provlösningen (5.2.2) |
|
hi, s |
= |
topphöjd (area) för den interna standarden i provlösningen (5.2.2) |
|
cd, c |
= |
diclazurilkoncentration i kalibreringslösningen i μg/ml (3.10) |
|
m |
= |
provets vikt i gram |
|
V |
= |
provextraktets volym i ml (= 25 ml), se 5.2.2 |
|
p |
= |
nominell halt av diclazuril i mg/kg i förblandningen. |
7. Validering av resultaten
7.1 Identitet
Analytens identitet kan bekräftas genom co-kromatografi eller genom användning av en diod-array-detektor med vars hjälp spektrumen för provextraktet (5.2.1 eller 5.2.2) och kalibreringslösningen (3.10) jämförs.
7.1.1
Ett provextrakt (5.2.1 eller 5.2.2) spikas genom tillsats av en lämplig mängd kalibreringslösning (3.10). Mängden tillsatt diclazuril bör vara samma som den mängd diclazuril som finns i provextraktet.
Endast höjden på diclazuriltoppen och på den topp som motsvarar den interna standarden ska öka efter det att hänsyn tagits till både den tillsatta mängden och spädningen av extraktet. Toppens bredd på halva höjden ska ligga inom ± 10 % av den ursprungliga bredden på toppen för diclazuril eller för den interna standarden i det ospikade provextraktet.
7.1.2
Resultaten utvärderas enligt följande kriterier:
a) Våglängderna för prov- och standardspektrumens absorptionsmaximum, i toppens spets på kromatogrammet, ska ligga inom en marginal som beror på detektionssystemets upplösningsförmåga. För diod-array-detektion är denna i allmänhet ± 2 nm.
b) Mellan 230 och 320 nm får prov- och standardspektrumen, i toppens spets på kromatogrammet, inte skilja sig åt för de delar av spektrumet där den relativa absorbansen är 10–100 %. Detta kriterium är uppfyllt om samma maximum observeras och avvikelsen mellan de två spektrumen inte vid någon mätpunkt överstiger 15 % av standardanalytens absorbans.
c) Mellan 230 och 320 nm får spektrumen för den uppåtgående delen, spetsen och den nedåtgående delen av toppen från provextraktet inte skilja sig från varandra för de delar av spektrumet där den relativa absorbansen är 10–100 %. Detta kriterium är uppfyllt om samma maximum observeras och avvikelsen mellan spektrumen inte vid någon mätpunkt överstiger 15 % av absorbansen vid spetsen.
Om något av dessa kriterier inte är uppfyllt har analytens förekomst inte bekräftats.
7.2 Repeterbarhet
Skillnaden mellan resultaten från två parallella bestämningar som utförs på samma prov får inte överstiga
— 30 % av det högre värdet för diclazurilhalter på 0,5 –2,5 mg/kg
— 0,75 mg/kg för diclazurilhalter på 2,5 –5 mg/kg
— 15 % av det högre värdet för diclazurilhalter över 5 mg/kg
7.3 Utbyte
För ett spikat (blank)prov ska utbytet vara minst 80 %.
8. Resultat av en provningsjämförelse
I en provningsjämförelse analyserades fem prov av elva laboratorier. Proven utgjordes av två förblandningar, en blandad med en organisk matris (O 100) och den andra med en oorganisk matris (A 100). Den teoretiska halten är 100 mg diclazuril per kg. De tre foderblandningarna för fjäderfä kom från tre olika företag (NL) (L1/Z1/K1). Den teoretiska halten är 1 mg diclazuril per kg. Laboratorierna instruerades att göra en eller två analyser av varje prov. (Närmare upplysningar om provningsjämförelsen finns i Journal of AOAC International, volym 77, nr 6, 1994, s. 1359–1361). Resultaten redovisas i nedanstående tabell.
|
|
Prov 1 A 100 |
Prov 2 O 100 |
Prov 3 L1 |
Prov 4 Z1 |
Prov 5 K1 |
|
L |
11 |
11 |
11 |
11 |
6 |
|
n |
19 |
18 |
19 |
19 |
12 |
|
Medelvärde |
100,8 |
103,5 |
0,89 |
1,15 |
0,89 |
|
Sr (mg/kg) |
5,88 |
7,64 |
0,15 |
0,02 |
0,03 |
|
CVr ( %) |
5,83 |
7,38 |
17,32 |
1,92 |
3,34 |
|
SR (mg/kg) |
7,59 |
7,64 |
0,17 |
0,11 |
0,12 |
|
CVR ( %) |
7,53 |
7,38 |
18,61 |
9,67 |
13,65 |
|
Nominell halt (mg/kg) |
100 |
100 |
1 |
1 |
1 |
|
L |
= |
antal laboratorier |
|
n |
= |
antal enskilda värden |
|
Sr |
= |
standardavvikelse för repeterbarhet |
|
CVr |
= |
variationskoefficient för repeterbarhet |
|
SR |
= |
standardavvikelse för reproducerbarhet |
|
CVR |
= |
variationskoefficient för reproducerbarhet |
9. Anmärkningar
Diclazurilresponsen måste tidigare ha visats vara linjär i det koncentrationsintervall mätningarna gäller.
G. BESTÄMNING AV NATRIUMLASALOCID
Ett natriumsalt av en polyeter-monokarboxylsyra som produceras av Streptomyces lasaliensis
1. Syfte och tillämpningsområde
Med denna metod kan halten natriumlasalocid bestämmas i foder och förblandningar. Detektionsgränsen är 5 mg/kg och kvantifieringsgränsen 10 mg/kg.
2. Princip
Natriumlasalocid extraheras från provet med surgjord metanol och en bestämning görs med omvänd fas-kromatografi (HPLC) och en spektrofluorometrisk detektor.
3. Reagens
3.1 Kaliumdivätefosfat (KH2PO4)
3.2 Ortofosforsyra, w = 85 % (w/w)
3.3 Lösning av ortofosforsyra, c = 20 %
Späd 23,5 ml ortofosforsyra (3.2) med vatten till 100 ml.
3.4 6-metyl-2-heptylamin (1,5 -dimetylhexylamin), w = 99 % (w/w)
3.5 Metanol, HPLC-kvalitet.
3.6 Saltsyra, densitet = 1,19 g/ml
3.7 Fosfatbuffertlösning, c = 0,01 mol/l
Lös 1,36 gram KH2PO4 (3.1) i 500 ml vatten (3.11). Tillsätt 3,5 ml ortofosforsyra (3.2) och 10,0 ml 6-metyl-2-heptylamin (3.4). Justera pH till 4,0 med ortofosforsyralösningen (3.3) och späd med vatten (3.11) till 1 000 ml.
3.8 Surgjord metanol
För över 5,0 ml saltsyra (3.6) till en 1 000 ml mätkolv. Späd till märket med metanol (3.5) och blanda. Denna lösning ska vara nyberedd.
3.9 Mobil fas för HPLC: lösning av fosfatbuffert och metanol 5 + 95 (v + v)
Blanda 5 ml fosfatbuffertlösning (3.7) med 95 ml metanol (3.5).
3.10 Natriumlasalocid referenssubstans med garanterad renhet, C34H53O8Na (ett natriumsalt av en polyeter-monokarboxylsyra som produceras av Streptomyces lasaliensis), E763
3.10.1
Väg upp 50 mg natriumlasalocid (3.10) med en noggrannhet av 0,1 mg i en 100 ml mätkolv. Lös natriumlasalociden i surgjord metanol (3.8), späd till märket med samma lösningsmedel och blanda. Denna lösning ska vara nyberedd.
3.10.2
Pipettera 10,0 ml stamlösning (3.10.1) till en 100 ml mätkolv. Späd till märket med surgjord metanol (3.8) och blanda. Denna lösning ska vara nyberedd.
3.10.3
Överför 1,0 –2,0 –4,0 –5,0 och 10,0 ml arbetsstandardlösning (3.10.2) till en serie 50 ml mätkolvar. Späd till märket med surgjord metanol (3.8) och blanda. De erhållna lösningarna innehåller 1,0 –2,0 –4,0 –5,0 respektive 10,0 μg natriumlasalocid per ml. Dessa lösningar ska vara nyberedda.
3.11 Vatten, HPLC-kvalitet.
4. Utrustning
4.1 Termostatreglerat ultraljudsbad (eller skakapparat med vattenbad)
4.2 Membranfilter, 0,45 μm
4.3 HPLC-utrustning med injektor, anpassad för volymer på 20 μl
4.3.1 HPLC-kolonn, 125 × 4 mm, omvänd fas, C18, 5 μm packning eller motsvarande
4.3.2 Spektrofluorometer med variabel excitations- och emissionsvåglängd
5. Utförande
5.1 Allmänt
5.1.1
Inför utbytesförsöket (5.1.2) analyseras ett blankprov för att kontrollera att varken natriumlasalocid eller störande ämnen är närvarande. Blankprovet bör vara av liknande typ som analysprovet och varken natriumlasalocid eller störande ämnen ska kunna påvisas i det.
5.1.2
Gör ett utbytesförsök genom analys av ett blankprov som spikats med ungefär samma mängd natriumlasalocid som finns i provet. För spikning på nivån 100 mg/kg överförs 10,0 ml stamlösning (3.10.1) till en 250 ml E-kolv och får indunsta till dess att ca 0,5 ml återstår. Tillsätt 50 g av blankprovet, blanda ordentligt, låt stå i 10 minuter och blanda igen flera gånger före extraktionen (5.2).
Om ett blankprov av liknande typ som provet inte finns att tillgå (se 5.1.1), kan ett utbytesförsök göras med standardadditionsmetoden. Det prov som ska analyseras spikas då med ungefär samma mängd natriumlasalocid som finns i provet. Det erhållna provet analyseras sedan tillsammans med ett prov som inte spikats, och utbytet beräknas genom subtraktion.
5.2 Extraktion
5.2.1
Väg upp 5–10 g av provet med en noggrannhet av 0,01 gram i en 250 ml E-kolv med propp. Tillsätt med pipett 100,0 ml surgjord metanol (3.8). Sätt i proppen löst och rotera kolven för att dispergera provet. Ställ kolven i ett ca 40 oC ultraljudsbad (4.1) i 20 minuter. Ta upp kolven ur badet och låt den svalna till rumstemperatur. Låt stå i ca 1 timme så att de suspenderade partiklarna sjunker till botten. Filtrera en alikvot genom ett 0,45 μm membranfilter (4.2) till ett lämpligt kärl. Gå vidare till HPLC-bestämningen (5.3).
5.2.2
Väg med en noggrannhet av 0,001 gram upp ca 2 gram av den omalda förblandningen i en 250 ml mätkolv. Tillsätt 100,0 ml surgjord metanol (3.8) och rotera kolven för att dispergera provet. Ställ kolven med innehåll i ett ca 40 oC ultraljudsbad (4.1) i 20 minuter. Ta upp kolven ur badet och låt den svalna till rumstemperatur. Späd till märket med surgjord metanol (3.8) och blanda omsorgsfullt. Låt stå i 1 timme så att de suspenderade partiklarna sjunker till botten. Filtrera en alikvot genom ett 0,45 μm membranfilter (4.2). Späd en lämplig volym av klart filtratet med surgjord metanol (3.8) så att en slutlig provlösning erhålls som innehåller ca 4 μg natriumlasalocid/ml. Gå vidare till HPLC-bestämningen (5.3).
5.3 HPLC-bestämning
5.3.1
Följande betingelser anges som vägledning. Andra betingelser kan väljas om de ger likvärdiga resultat.
|
HPLC-kolonn (4.3.1): |
125 × 4 mm, omvänd fas C18, 5 μm packning eller motsvarande |
|
Mobil fas (3.9): |
Blandning av fosfatbuffertlösning (3.7) och metanol (3.5), 5+95 (v+v) |
|
Flödeshastighet: |
1,2 ml/min |
|
Detektionsvåglängder: |
|
|
Excitation: |
310 nm |
|
Emission: |
419 nm |
|
Injektionsvolym: |
20 μl |
Kontrollera att det kromatografiska systemet är stabilt genom att upprepade gånger injicera kalibreringslösningen (3.10.3) med koncentrationen 4,0 μg/ml till dess att topphöjder (areor) och retentionstider är konstanta.
5.3.2
Injicera varje kalibreringslösning (3.10.3) upprepade gånger och bestäm de genomsnittliga topphöjderna (areorna) för varje koncentration. Konstruera en kalibreringskurva med kalibreringslösningarnas genomsnittliga topphöjd (area) längs y-axeln och motsvarande koncentrationer i μg/ml längs x-axeln.
5.3.3
Injicera upprepade gånger de provextrakt som erhållits i 5.2.1 eller 5.2.2. Använd samma volym som för kalibreringslösningen. Bestäm de genomsnittliga topphöjderna (areorna) för natriumlasalocid.
6. Beräkning av resultat
Använd den genomsnittliga topphöjd (area) som erhållits vid injektion av provlösningen (5.3.3) för att bestämma koncentrationen natriumlasalocid (μg/ml) med hjälp av kalibreringskurvan.
6.1 Foder
Halten av natriumlasalocid, w (mg/kg) i provet beräknas med formeln
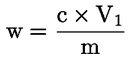
[mg/kg]
där:
|
c |
= |
koncentrationen av natriumlasalocid i provlösningen (5.2.1) i μg/ml |
|
V1 |
= |
provextraktets volym i ml (= 100 ml), se 5.2.1 |
|
m |
= |
provets vikt i gram. |
6.2 Förblandningar
Halten av natriumlasalocid, w (mg/kg) i provet beräknas med formeln

[mg/kg]
där:
|
c |
= |
koncentrationen av natriumlasalocid i provlösningen (5.2.1) i μg/ml |
|
V2 |
= |
provextraktets volym i ml (= 250 ml), se 5.2.2 |
|
f |
= |
spädningsfaktor, se 5.2.2 |
|
m |
= |
provets vikt i gram. |
7. Validering av resultaten
7.1 Identitet
Metoder som grundar sig på spektrofluorometri medför inte samma problem med interferenser som metoder med UV-detektion. Analytens identitet kan bekräftas genom co-kromatografi.
7.1.1
Ett provextrakt (5.2.1 eller 5.2.2) spikas genom tillsats av en lämplig mängd kalibreringslösning (3.10.3). Mängden tillsatt natriumlasalocid bör vara ungefär lika stor som den ursprungliga mängden natriumlasalocid i provextraktet. Endast natriumlasalocidtoppens höjd ska öka i förhållande till mängden tillsatt natriumlasalocid och provextraktets spädning. Toppens bredd på halva höjden får inte avvika mer än ± 10 % från värdet för den ursprungliga topp som motsvarar det ospikade provextraktet.
7.2 Repeterbarhet
Skillnaden mellan resultaten från två parallella bestämningar som utförs på samma prov får inte överstiga
— 15 % av det högre värdet, för halter av natriumlasalocid på 30–100 mg/kg,
— 15 mg/kg, för halter på 100–200 mg/kg,
— 7,5 % av det högre värdet, för halter som överstiger 200 mg/kg.
7.3 Utbyte
För det spikade (blank)provet ska utbytet vara minst 80 %. För de spikade förblandningsproven ska utbytet vara minst 90 %.
8. Resultat av en provningsjämförelse
I en provningsjämförelse ( *1 ) analyserades två förblandningar (prov 1 och 2) och fem fodertyper (prov 3–7) av tolv laboratorier. Varje prov analyserades två gånger. Resultaten redovisas i nedanstående tabell.
|
|
Prov 1 Förblandning för kyckling |
Prov 2 Förblandning för kalkon |
Prov 3 Pellets för kalkon |
Prov 4 Kycklingfoder |
Prov 5 Kalkonfoder |
Prov 6 Fjäderfäfoder A |
Prov 7 Fjäderfäfoder B |
|
L |
12 |
12 |
12 |
12 |
12 |
12 |
12 |
|
n |
23 |
23 |
23 |
23 |
23 |
23 |
23 |
|
Medelvärde [mg/kg] |
5 050 |
16 200 |
76,5 |
78,4 |
92,9 |
48,3 |
32,6 |
|
sr [mg/kg] |
107 |
408 |
1,71 |
2,23 |
2,27 |
1,93 |
1,75 |
|
CVr [ %] |
2,12 |
2,52 |
2,24 |
2,84 |
2,44 |
4,00 |
5,37 |
|
sR [mg/kg] |
286 |
883 |
3,85 |
7,32 |
5,29 |
3,47 |
3,49 |
|
CVR [ %] |
5,66 |
5,45 |
5,03 |
9,34 |
5,69 |
7,18 |
10,70 |
|
Nominell halt [mg/kg] |
5 000 (1) |
16 000 (1) |
80 (1) |
105 (1) |
120 (1) |
50 (2) |
35 (2) |
|
(*1) Halt deklarerad av tillverkaren. (*2) Foder berett i laboratoriet. |
|||||||
|
L |
= |
antal laboratorier |
|
n |
= |
antal enskilda värden |
|
sr |
= |
standardavvikelse för repeterbarhet |
|
sR |
= |
standardavvikelse för reproducerbarhet |
|
CVr |
= |
variationskoefficient för repeterbarhet, % |
|
CVR |
= |
variationskoefficient för reproducerbarhet, % |
BILAGA V
ANALYSMETODER FÖR KONTROLL AV FRÄMMANDE ÄMNEN I FODER
A. BESTÄMNING AV HALT FRI GOSSYPOL OCH TOTALHALT GOSSYPOL
1. Syfte och tillämpningsområde
Med denna metod kan halten fri gossypol, totalhalten gossypol och kemiskt besläktade ämnen bestämmas i bomullsfrö, bomullsfrömjöl och bomullsfrökakor samt i foderblandningar som innehåller dessa foderråvaror, om halten av fri gossypol, totalhalten gossypol och kemiskt besläktade ämnen överstiger 20 mg/kg.
2. Princip
Gossypol extraheras i närvaro av 3-amino-1-propanol, antingen med en blandning av 2-propanol och hexan (vid bestämning av fritt gossypol) eller med dimetylformamid (vid bestämning av totalhalt gossypol). Gossypol omvandlas med anilin till gossypol-dianilin, vars absorbans mäts vid 440 nm.
3. Reagens
3.1 Blandning av 2-propanol och hexan: blanda 60 volymdelar 2-propanol med 40 volymdelar n-hexan.
3.2 Lösningsmedel A: Tillsätt ca 500 ml 2-propanol-hexanblandning (3.1), 2 ml 3-amino-1-propanol, 8 ml isättika och 50 ml vatten i en 1 000 ml mätkolv. Späd med 2-propanol-hexanblandningen (3.1) till full volym. Lösningen är stabil i en vecka.
3.3 Lösningsmedel B: Pipettera över 2 ml 3-amino-1-propanol och 10 ml isättika till en 100 ml mätkolv. Kyl till rumstemperatur och späd med N, N-dimetylformamid till full volym. Lösningen är stabil i en vecka.
3.4 Anilin: Om blankprovets absorbans överstiger 0,022 , destilleras anilinet över zinkpulver och de första och sista tio procenten av destillatet kastas. Om detta reagens kylförvaras i en brun glaskolv med propp är det hållbart i flera månader.
3.5 Standardgossypollösning A: Tillsätt 27,9 mg gossypolacetat i en 250 ml mätkolv. Lös upp och späd med lösningsmedel A (3.2) till full volym. Pipettera över 50 ml av denna lösning till en 250 ml mätkolv och späd med lösningsmedel A till full volym. Gossypolkoncentrationen i denna lösning är 0,02 mg/ml. Låt lösningen stå 1 timme i rumstemperatur innan den används.
3.6 Standardgossypollösning B: Tillsätt 27,9 mg gossypolacetat i en 50 ml mätkolv. Lös upp och späd med lösningsmedel B (3.3) till full volym. Gossypolkoncentrationen i denna lösning är 0,5 mg/ml.
Standardgossypollösningarna A och B är stabila i 24 timmar om de skyddas från ljus.
4. Utrustning
4.1 Rotationsskakapparat: ca 35 varv per minut.
4.2 Spektrofotometer.
5. Utförande
5.1 Analysprov
Analysprovets storlek beror på den antagna gossypolhalten i provet. Det är bäst att arbeta med ett litet analysprov och en relativt stor alikvot av filtratet så att man får en tillräckligt stor mängd gossypol för att kunna göra en exakt fotometrisk mätning. Vid bestämning av fri gossypol i bomullsfrö, bomullsfrömjöl och bomullsfrökakor bör analysprovet väga högst 1 gram. När det gäller foderblandningar kan provet väga upp till 5 g. I de flesta fall är det lämpligt med en alikvot på 10 ml av filtratet som bör innehålla 50–100 μg gossypol. Vid bestämning av totalhalt gossypol bör analysprovet väga 0,5 –5 g så att en alikvot på 2 ml av filtratet innehåller 40–200 μg gossypol.
Analysen bör utföras vid rumstemperatur, ca 20 oC.
5.2 Bestämning av fri gossypol
Placera analysprovet i en 250 ml kolv med slipad hals där kolvens botten täckts med krossat glas. Tillsätt med pipett 50 ml lösningsmedel A (3.2), tillslut kolven och blanda i 1 timme på skakapparaten. Filtrera genom torrt filter och samla upp filtratet i en liten kolv med slipad hals. Täck tratten med urglas under filtreringen.
Pipettera över identiska alikvoter av filtratet med en gossypolhalt på 50–100 μg till två 25 ml mätkolvar (A och B). Späd vid behov med lösningsmedel A (3.2) till 10 ml. Späd därefter innehållet i kolv A med 2-propanol-hexanblandningen (3.1) till full volym. Denna lösning används som referenslösning för provlösningens mätvärden.
Pipettera över 10 ml av lösningsmedel A (3.2) till var och en av två andra 25 ml mätkolvar (C och D). Späd innehållet i kolv C med 2-propanol-hexanblandningen (3.1) till full volym. Denna lösning används som referenslösning för blankprovlösningens mätvärden.
Tillsätt 2 ml anilin (3.4) till kolvarna D och B. Upphetta i 30 minuter på kokande vattenbad så att färgen framträder. Kyl till rumstemperatur, späd med 2-propanol-hexanblandning (3.1) till full volym, homogenisera och låt stå 1 timme.
Bestäm dels blankprovlösningens (D) absorbans genom att jämföra med referenslösningen (C), dels provlösningens (B) absorbans genom att jämföra med referenslösningen (A) i spektrofotometern vid 440 nm med 1 cm glasceller.
Subtrahera blankprovlösningens absorbans från provlösningens (korrigerad absorbans). Beräkna utifrån det erhållna värdet halten fri gossypol enligt anvisningarna i punkt 6.
5.3 Bestämning av totalhalt gossypol
Placera ett analysprov som innehåller 1–5 mg gossypol i en 50 ml mätkolv och tillsätt 10 ml lösningsmedel B (3.3). Bered samtidigt ett blankprov genom att tillföra 10 ml lösningsmedel B (3.3) i en annan 50 ml mätkolv. Upphetta de två kolvarna i 30 minuter på kokande vattenbad. Kyl till rumstemperatur och späd innehållet i båda kolvarna med 2-propanol-hexanblandningen till full volym (3.1). Homogenisera och låt stå i 10–15 minuter. Filtrera därefter och samla upp filtraten i kolvar med slipad hals.
Pipettera över 2 ml av provfiltratet till var och en av två 25 ml mätkolvar, och 2 ml av blankprovfiltratet till två andra 25 ml mätkolvar. Späd innehållet i en kolv från varje serie med 2-propanol-hexanblandningen (3.1) till 25 ml. Dessa lösningar används som referenslösningar.
Tillsätt 2 ml anilin (3.4) till var och en av de andra två kolvarna. Upphetta i 30 minuter på kokande vattenbad så att färgen framträder. Kyl till rumstemperatur, späd med 2-propanol-hexanblandningen (3.1) till 25 ml, homogenisera och låt stå 1 timme.
Bestäm absorbansen enligt anvisningarna i punkt 5.2 för fri gossypol. Beräkna utifrån det erhållna värdet totalhalten gossypol enligt anvisningarna i punkt 6.
6. Beräkning av resultat
Resultaten kan beräknas antingen från den specifika absorbansen (6.1) eller med hjälp av en kalibreringskurva (6.2).
6.1 Från den specifika absorbansen
Den specifika absorbansen blir enligt de beskrivna villkoren följande:
|
Fri gossypol |
|
|
Totalhalt gossypol |
|

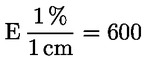
Halten fri gossypol eller totalhalten gossypol beräknas med formeln % gossypol:

där:
|
E |
= |
korrigerad absorbans, bestämd enligt 5.2 |
|
p |
= |
provets vikt i gram |
|
a |
= |
alikvot av filtratet i ml. |
6.2 Från en kalibreringskurva
6.2.1
Ställ i ordning två serier med fem 25 ml mätkolvar. Pipettera över 2,0 –4,0 –6,0 –8,0 respektive 10,0 ml standardgossypollösning A (3.5) till vardera serien av mätkolvar. Späd med lösningsmedel A (3.2) till 10 ml. Komplettera varje serie med en 25 ml mätkolv som endast innehåller 10 ml lösningsmedel A (3.2) (blankprov).
Späd innehållet i den första serien mätkolvar (inklusive den med blankprovet) till 25 ml med 2-propanol-hexanblandningen (3.1) (referensserie).
Tillsätt 2 ml anilin (3.4) till var och en av mätkolvarna i den andra serien (inklusive den med blankprovet). Upphetta i 30 minuter på kokande vattenbad så att färgen framträder. Kyl till rumstemperatur, späd med 2-propanol-hexanblandningen (3.1) till full volym, homogenisera och låt stå 1 timme (standardserie).
Bestäm enligt 5.2 absorbansen för lösningarna i standardserien genom att jämföra med motsvarande lösningar i referensserien. Konstruera kalibreringskurvan genom att avsätta absorbansvärdena mot motsvarande kvantiteter gossypol (i μg).
6.2.2
Ställ i ordning sex stycken 50 ml mätkolvar. Tillsätt 10 ml lösningsmedel B i den första mätkolven (3.3) och 2,0 –4,0 –6,0 –8,0 respektive 10,0 ml standardgossypollösning B i de övriga (3.6). Späd innehållet i varje mätkolv till 10 ml med lösningsmedel B (3.3). Upphetta i 30 minuter på kokande vattenbad. Kyl till rumstemperatur, späd med 2-propanol-hexanblandningen (3.1) till full volym och homogenisera.
Överför 2,0 ml av dessa lösningar till två serier med vardera sex 25 ml mätkolvar. Späd innehållet i den första seriens kolvar till 25 ml med 2-propanol-hexanblandningen (3.1) (referensserie).
Tillsätt 2 ml anilin (3.4) till var och en av kolvarna i den andra serien. Upphetta i 30 minuter på kokande vattenbad. Kyl till rumstemperatur, späd med 2-propanol-hexanblandningen (3.1) till full volym, homogenisera och låt stå 1 timme (standardserie).
Bestäm enligt 5.2 absorbansen för lösningarna i standardserien genom att jämföra med motsvarande lösningar i referensserien. Konstruera kalibreringskurvan genom att avsätta absorbansvärdena mot motsvarande kvantiteter gossypol (i μg).
6.3 Repeterbarhet
Skillnaden mellan resultaten av två parallella bestämningar som utförs på samma prov får inte överstiga
— 15 % av det högre värdet för gossypolhalter under 500 ppm,
— 75 ppm i absoluta tal för halter på 500–750 ppm,
— 10 % av det högre värdet för halter över 750 ppm.
B. BESTÄMNING AV HALTER AV DIOXINER (PCDD/PCDF) OCH PCB
KAPITEL I
Provtagningsmetoder och tolkning av analysresultat
1. Tillämpningsområde och definitioner
Provtagning för offentlig kontroll av halter av polyklorerade dibenso-p-dioxiner (PCDD) och polyklorerade dibensofuraner (PCDF) (nedan kallade PCDD/PCDF), dioxinlika polyklorerade bifenyler (nedan kallade dioxinlika PCB) ( 16 ) och icke-dioxinlika PCB i foder ska göras i enlighet med bilaga I. De kvantitativa kraven i fråga om kontroll av ämnen eller produkter som är jämnt fördelade i fodret enligt punkt 5.1 i bilaga I ska tillämpas. De erhållna samlingsproven ska betraktas som representativa för de partier eller delpartier från vilka de tas. De halter som bestäms i laboratorieproven ska ligga till grund för bedömningen av om proven är överensstämmande med gränsvärdena enligt direktiv 2002/32/EG.
I denna del B ska definitionerna i bilaga I till kommissionens beslut 2002/657/EG ( 17 ) gälla.
Dessutom gäller följande definitioner:
screeningmetoder : metoder för att påvisa prov med halter av PCDD/PCDF och dioxinlika PCB som överskrider gränsvärdena eller åtgärdsgränserna. De ska ge en kostnadseffektiv och hög analyskapacitet och därigenom öka möjligheterna att upptäcka nya incidenter med hög exponering och hälsorisker för konsumenterna. Screeningmetoder ska baseras på bioanalytiska metoder eller GC-MS-metoder. Provresultat som överskrider den använda brytpunkten vid kontroll av överensstämmelsen med gränsvärdet ska kontrolleras genom en fullständig ny analys av det ursprungliga provet med en konfirmeringsmetod.
konfirmeringsmetoder : metoder för att få fram fullständig eller kompletterande information som möjliggör en entydig identifiering och kvantifiering av PCDD/PCDF och dioxinlika PCB vid gränsvärdet, eller vid behov vid åtgärdsgränsen. Sådana metoder bygger på gaskromatografi i kombination med högupplösande masspektrometri (GC-HRMS) eller gaskromatografi i kombination med tandem-masspektrometri (GC-MS/MS).
2. Partiets eller delpartiets överensstämmelse med gränsvärdet
2.1 Icke-dioxinlika PCB
Partiet eller delpartiet är överensstämmande med gränsvärdet om analysresultatet för summan av PCB 28, PCB 52, PCB 101, PCB 138, PCB 153 och PCB 180 (nedan kallade icke-dioxinlika PCB) inte överskrider gränsvärdet enligt direktiv 2002/32/EG, med beaktande av den utvidgade mätosäkerheten ( 18 ). Partiet eller delpartiet är icke-överensstämmande med gränsvärdet enligt direktiv 2002/32/EG om det är ställt utom rimligt tvivel att medelvärdet av två analysresultat avseende den övre koncentrationen ( 19 ) fastställda genom analys av dubbelprov ( 20 ) överskrider gränsvärdet, med beaktande av den utvidgade mätosäkerheten, dvs. att den analyserade koncentrationen efter att den utvidgade mätosäkerheten subtraherats används för bedömning av överensstämmelse.
Den utvidgade mätosäkerheten beräknas med en täckningsfaktor på 2 som ger en konfidensgrad på cirka 95 %. Ett parti eller delparti är icke-överensstämmande om medelvärdet av de uppmätta värdena minus den utvidgade mätosäkerheten för medelvärdet är högre än gränsvärdet.
De bestämmelser som nämns i denna punkt ska tillämpas på de analysresultat som erhålls vid provtagning för offentlig kontroll. När det gäller analys för överklagande eller referensändamål ska nationella regler gälla.
2.2 Dioxiner (PCDD/PCDF) och dioxinlika PCB
Partiet eller delpartiet är överensstämmande med gränsvärdena i följande fall:
— En enda analys med en screeningmetod för vilken andelen prov som felaktigt bedömts överensstämma med gränsvärdet är lägre än 5 % ger ett resultat som visar att halten inte överskrider respektive gränsvärde för PCDD/PCDF och för summan av PCDD/PCDF och dioxinlika PCB enligt direktiv 2002/32/EG.
— En enda analys med en konfirmeringsmetod ger ett resultat som inte överskrider respektive gränsvärde för PCDD/PCDF och för summan av PCDD/PCDF och dioxinlika PCB enligt direktiv 2002/32/EG, med beaktande av den utvidgade mätosäkerheten.
För screeninganalyser ska en brytpunkt fastställas vad gäller bedömningen av om provet/n överensstämmer med gränsvärdet för PCDD/PCDF respektive för summan av PCDD/PCDF och dioxinlika PCB.
Partiet eller delpartiet är icke-överensstämmande med gränsvärdet enligt direktiv 2002/32/EG om det är ställt utom rimligt tvivel att medelvärdet av två analysresultat avseende den övre koncentrationen ( 21 ) fastställda genom analys av dubbelprov ( 22 ) med en konfirmeringsmetod överskrider gränsvärdet, med beaktande av den utvidgade mätosäkerheten, dvs. att den analyserade koncentrationen efter att den utvidgade mätosäkerheten subtraherats används för bedömning av överensstämmelse.
Den utvidgade mätosäkerheten beräknas med en täckningsfaktor på 2 som ger en konfidensgrad på cirka 95 %. Ett parti eller delparti är icke-överensstämmande om medelvärdet av de uppmätta värdena minus den utvidgade mätosäkerheten för medelvärdet är högre än gränsvärdet.
Summan av den skattade utvidgade mätosäkerheten för de enskilda analysresultaten för PCDD/PCDF och dioxinlika PCB ska användas när man anger summan av PCDD/PCDF och dioxinlika PCB.
De bestämmelser som nämns i denna punkt ska tillämpas på de analysresultat som erhålls vid provtagning för offentlig kontroll. När det gäller analys för överklagande eller referensändamål ska nationella regler gälla.
3. Resultat som överskrider de åtgärdsgränser som anges i bilaga II till direktiv 2002/32/EG
Åtgärdsgränserna fungerar som ett redskap för provurval i de fall där det är nödvändigt att identifiera en föroreningskälla och att vidta åtgärder för att minska eller eliminera den. Screeningmetoderna ska fastställa lämpliga brytpunkter för urvalet av dessa prov. Om det behövs betydande insatser för att identifiera en föroreningskälla och minska eller eliminera föroreningen är det lämpligt att bekräfta om åtgärdsgränsen överskridits genom att analysera dubbelprov med en konfirmeringsmetod, med beaktande av den utvidgade mätosäkerheten ( 23 ).
KAPITEL II
Provberedning och krav för analysmetoder för offentlig kontroll av halter av dioxiner (PCDD/PCDF) och dioxinlika PCB i foder
1. Tillämpningsområde
Kraven i detta kapitel ska tillämpas när foder analyseras för offentlig kontroll av halterna av 2,3,7,8-substituerade PCDD/PCDF och dioxinlika PCB och när det gäller provberedning och analyskrav för andra lagstadgade ändamål, inbegripet foderföretagarens kontroller för att säkerställa överensstämmelse med bestämmelserna i Europaparlamentets och rådets förordning (EG) nr 183/2005 ( 24 ).
Övervakningen av förekomsten av PCDD/PCDF och dioxinlika PCB i foder kan utföras med två olika typer av analysmetoder:
a) Screeningmetoder
Syftet med screeningmetoder är att påvisa prov med halter av PCDD/PCDF och dioxinlika PCB som överskrider gränsvärdena eller åtgärdsgränserna. Screeningmetoder ska säkerställa en kostnadseffektiv och hög analyskapacitet och därigenom öka möjligheterna att upptäcka nya incidenter med hög exponering och hälsorisker för konsumenterna. När dessa metoder används ska man sträva efter att undvika resultat som felaktigt bedömts överensstämma. De kan bland annat bestå av bioanalytiska metoder och GC-MS-metoder.
Screeningmetoder jämför analysresultatet med en brytpunkt och ger ett ja/nej-svar för bedömning av om gränsvärdet eller åtgärdsgränsen eventuellt har överskridits. Koncentrationen av PCDD/PCDF och av summan av PCDD/PCDF och dioxinlika PCB i prov som misstänks vara icke-överensstämmande med gränsvärdet ska bestämmas eller bekräftas med en konfirmeringsmetod.
Screeningmetoderna kan dessutom ge en indikation på halterna av de PCDD/PCDF och dioxinlika PCB som förekommer i provet. Om bioanalytiska screeningmetoder används ska resultatet uttryckas som bioanalytiska ekvivalenter (BEQ), medan om fysikalisk-kemiska GC-MS-metoder används ska resultatet uttryckas som toxicitetsekvivalenter (TEQ). Screeningmetodernas resultat i form av numeriska värden är lämpliga för att visa på överensstämmelse eller misstänkt icke-överensstämmelse eller på att åtgärdsgränsen överskridits, och ger en indikation på koncentrationsintervallet vid uppföljning med konfirmeringsmetoder. Däremot är de inte lämpliga för utvärdering av bakgrundshalter, uppskattning av intag, uppföljning av tidstrender avseende halter eller omprövning av åtgärdsgränser och gränsvärden.
b) Konfirmeringsmetoder
Konfirmeringsmetoder möjliggör en entydig identifiering och kvantifiering av de PCDD/PCDF och dioxinlika PCB som förekommer i ett prov och ger fullständig information på kongennivå. Dessa metoder möjliggör därför kontroll av gränsvärden och åtgärdsgränser, inklusive konfirmering av de resultat som erhållits med screeningmetoder. Dessutom kan resultaten användas för andra ändamål, t.ex. bestämning av låga bakgrundshalter vid foderövervakning, uppföljning av tidstrender, bedömning av exponering samt för att bygga upp en databas för eventuell omprövning av åtgärdsgränser och gränsvärden. De är också viktiga för att fastställa kongenmönster så att källan till en eventuell förorening kan identifieras. Sådana metoder bygger på GC-HRMS. För att bekräfta överensstämmelse eller icke-överensstämmelse med gränsvärdet kan även GC-MS/MS användas.
2. Bakgrund
För att beräkna den totala koncentrationen av dioxinlika föreningar i ett prov, den så kallade toxicitetsekvivalenten (TEQ), ska koncentrationen av varje enskilt ämne i ett givet prov multipliceras med respektive ämnes toxicitetsekvivalensfaktor (TEF) (se fotnot 1 i kapitel I) och därefter adderas.
I denna del B avses med den accepterade särskilda kvantifieringsgränsen för en enskild kongen den lägsta analythalten som med rimlig statistisk säkerhet kan mätas och som uppfyller de identifieringskriterier som beskrivs i internationellt erkända standarder, t.ex. standarden EN 16215:2012 (Djurfoder – Bestämning av dioxiner och dioxinlika PCB:er med GC/HRMS och av indikator-PCB:er med GC/HRMS) och/eller i EPA-metoderna 1613 och 1668, med senare revideringar.
Kvantifieringsgränsen för en enskild kongen kan identifieras
a) som den koncentration av en analyt i ett provextrakt som ger en instrumentrespons för de två skilda joner som ska övervakas, med ett S/N-förhållande (signal–brusförhållande) på 3:1 för den mindre känsliga rådatasignalen, eller
b) om beräkningen av signal–brusförhållandet på grund av tekniska skäl inte ger tillförlitliga resultat, som den lägsta koncentrationen på en kalibreringskurva som ger en godtagbar (≤ 30 %) och konsekvent (uppmätt åtminstone i början och slutet av en provserie) avvikelse till den genomsnittliga relativa responsfaktorn som beräknats för alla punkter på kalibreringskurvan för varje provserie. Kvantifieringsgränsen beräknas utifrån den lägsta koncentrationen med beaktande av utbytet för interna standarder och mängden prov.
Bioanalytiska screeningmetoder ger inte resultat på kongennivå utan endast en indikation ( 25 ) på TEQ-värdet, uttryckt som BEQ, på grund av att det i ett provextrakt även kan finnas föreningar som ger en respons i analysen men som inte uppfyller TEQ-principens alla krav.
Screeningmetoder och konfirmeringsmetoder får endast användas för kontroll av en viss matris om metoderna har tillräckligt hög känslighet för att på ett tillförlitligt sätt påvisa halter vid åtgärdsgränsen eller gränsvärdet.
3. Krav för kvalitetssäkring
|
3.1 |
Åtgärder ska vidtas för att undvika korskontaminering under alla moment vid provtagning och analys. |
|
3.2 |
Proven ska lagras och transporteras i behållare av glas, aluminium, polypropen eller polyeten som är lämpade för lagring och inte påverkar halterna av PCDD/PCDF och dioxinlika PCB i proven. Spår av pappersdamm ska avlägsnas från provbehållaren. |
|
3.3 |
Proven ska lagras och transporteras på ett sådant sätt att foderprovet bibehålls i oförändrat tillstånd. |
|
3.4 |
Varje laboratorieprov ska vid behov finmalas och blandas omsorgsfullt enligt en metod som garanterar fullständig homogenisering (t.ex. så att de passerar en 1 mm sikt). Om vattenhalten är för hög ska proven torkas innan de mals. |
|
3.5 |
Reagenser, glasvaror och utrustning ska kontrolleras för om de eventuellt kan påverka de TEQ- eller BEQ-baserade resultaten. |
|
3.6 |
En analys av ett blankprov ska göras genom att man utför hela analysen men utelämnar provet. |
|
3.7 |
För bioanalytiska metoder ska alla glasvaror och lösningsmedel som används analyseras för att garantera att de är fria från ämnen som kan störa påvisandet av målföreningar i mätområdet. Glasvaror ska sköljas med lösningsmedel eller upphettas till de temperaturer som krävs för att avlägsna spår av PCDD/PCDF, dioxinlika föreningar och störande föreningar från dess yta. |
|
3.8 |
Det prov som extraheras ska vara tillräckligt stort för att uppfylla kraven vad gäller ett tillräckligt lågt mätområde som inkluderar gränsvärden eller åtgärdsgränser. |
|
3.9 |
De särskilda provberedningsförfaranden som används för de givna produkterna ska följa internationellt accepterade riktlinjer. |
4. Krav för laboratorier
|
4.1 |
I enlighet med förordning (EG) nr 882/2004 ska laboratorier ackrediteras av ett godkänt ackrediteringsorgan som verkar enligt ISO Guide 58 för att säkerställa att de tillämpar ett system för kvalitetssäkring av analysverksamheten. Laboratorierna ska ackrediteras enligt standarden EN ISO/IEC 17025. Principerna i de tekniska vägledningarna om uppskattning av mätosäkerhet och kvantifieringsgränser vid analys av PCDD/PCDF och PCB ska följas i tillämpliga fall ( 26 ). |
|
4.2 |
Laboratoriekompetensen ska bevisas genom fortlöpande medverkan i provningsjämförelser för bestämning av halten av PCDD/PCDF och dioxinlika PCB i relevanta fodermatriser och koncentrationsintervall. |
|
4.3 |
Laboratorier som använder screeningmetoder vid rutinkontroll av prov ska ha ett nära samarbete med laboratorier som använder konfirmeringsmetoden, både för kvalitetskontroll och för att konfirmera analysresultatet för misstänkta prov. |
5. Grundläggande krav för analysmetoder för dioxiner (PCDD/PCDF) och dioxinlika PCB
5.1 Lågt mätområde och låga kvantifieringsgränser
Eftersom vissa typer av PCDD/PCDF är extremt toxiska ska detektionsgränsen för dessa föreningar vara i storleksordningen femtogram (10– 15 g). För de flesta PCB-kongener räcker det att kvantifieringsgränsen är i storleksordningen nanogram (10– 9 g). Vid mätning av mer toxiska dioxinlika PCB-kongener (särskilt kongener som inte är substituerade på orto-position, s.k. non-orto kongener) ska de lägsta halterna i mätområdet vara i storleksordningen picogram (10– 12 g). För alla andra PCB-kongener räcker det att kvantifieringsgränsen är i storleksordningen nanogram (10– 9 g).
5.2 Hög selektivitet (specificitet)
|
5.2.1 |
PCDD/PCDF och dioxinlika PCB ska kunna skiljas från en stor mängd andra föreningar som extraheras samtidigt och som förekommer i koncentrationer flerfaldigt högre än hos de givna analyterna och som kan störa analysen. GC-MS-metoder ska kunna särskilja mellan olika kongener, exempelvis mellan de toxiska kongenerna (t.ex. de sjutton 2,3,7,8-substituerade PCDD/PCDF och de tolv dioxinlika PCB) och andra kongener. |
|
5.2.2 |
Bioanalytiska metoder ska kunna påvisa målföreningar och ange dem som summan av PCDD/PCDF och/eller dioxinlika PCB. Upprening av prov ska syfta till att avlägsna föreningar som orsakar resultat som felaktigt bedömts vara icke-överensstämmande eller föreningar som kan minska responsen, vilket orsakar resultat som felaktigt bedömts överensstämma. |
5.3 Hög noggrannhet (riktighet och precision, skenbart utbyte för bioassay)
|
5.3.1 |
Bestämning med GC-MS-metoder ska ge en giltig uppskattning av den sanna koncentrationen i ett prov. En hög noggrannhet krävs för att resultatet av en provanalys inte ska avvisas på grund av att det fastställda TEQ-värdet har för dålig tillförlitlighet. Noggrannheten uttrycks som riktighet (skillnaden mellan det medelvärde som har uppmätts för en analyt i ett certifierat material och dess certifierade värde, uttryckt som ett procenttal av detta värde) och precision (RSDR, dvs. den relativa standardavvikelsen beräknad utifrån de resultat som erhållits under reproducerbara förhållanden). |
|
5.3.2 |
För bioanalytiska metoder ska det skenbara utbytet för bioassay bestämmas. Det skenbara utbytet för bioassay motsvaras av det BEQ-värde som beräknas utifrån kalibreringskurvan för TCDD eller PCB 126, korrigeras för blank och därefter delas med det TEQ-värde som fastställs med konfirmeringsmetoden. Syftet är att korrigera för faktorer som till exempel förlusten av PCDD/PCDF och dioxinlika föreningar under extraktions- och uppreningsstegen, föreningar som extraherats samtidigt och ökar eller minskar resultatet (agonistiska och antagonistiska effekter), kvaliteten på kurvpassningen eller skillnader mellan TEF-värden och REP-värden (relativ potensfaktor). Det skenbara utbytet för bioassay beräknas utifrån lämpliga referensprov med representativa kongenmönster kring den givna nivån. |
5.4 Validering i intervallet för gränsvärdet och allmänna åtgärder för kvalitetskontroll
|
5.4.1 |
Laboratorierna ska visa en metods prestanda i intervallet för gränsvärdet, t.ex. 0,5 gång, 1 gång och 2 gånger gränsvärdet med en godtagbar variationskoefficient för upprepade analyser, under valideringsförfarandet och rutinanalyser. |
|
5.4.2 |
Laboratorierna ska regelbundet analysera blankprov och spikade prov eller kontrollprov (helst certifierat referensmaterial om sådant finns) som en åtgärd för intern kvalitetskontroll. Kontrollkort för analyser av blankprov, spikade prov eller kontrollprov ska framställas och kontrolleras för att säkerställa att den analytiska prestandan uppfyller kraven. |
5.5 Kvantifieringsgräns
|
5.5.1 |
För en bioanalytisk screeningmetod är det inte ett absolut krav att kvantifieringsgränsen fastställs, men metoden ska bevisligen kunna särskilja mellan blankprovet och brytpunkten. När ett BEQ-värde anges ska en rapporteringsnivå fastställas för att hantera prov som ger en respons under denna nivå. Rapporteringsnivån ska bevisligen vara skild från metodens blankprov med åtminstone en faktor på tre och ge respons vid halter under mätområdet. Den ska därför beräknas utifrån prov med halter av målföreningarna kring de miniminivåer som krävs och inte utifrån ett S/N-förhållande (signal–brusförhållande) eller ett blankprov. |
|
5.5.2 |
Kvantifieringsgränsen för en konfirmeringsmetod ska motsvara ungefär en femtedel av gränsvärdet. |
5.6 Analyskriterier
För att resultaten från konfirmerings- eller screeningmetoder ska vara tillförlitliga ska följande kriterier vara uppfyllda i intervallet för gränsvärdet för TEQ-värdet respektive BEQ-värdet, vilka kan bestämmas antingen för summan av PCDD/PCDF och dioxinlika PCB (dvs. totalt TEQ eller totalt BEQ) eller separat för PCDD/PCDF och för dioxinlika PCB.
|
|
Screening med bioanalytiska eller fysikalisk-kemiska metoder |
Konfirmeringsmetoder |
|
Andelen prov som felaktigt bedömts vara överensstämmande (1) |
< 5 % |
|
|
Riktighet |
|
– 20 % till + 20 % |
|
Repeterbarhet (RSDr) |
< 20 % |
|
|
Intermediärt precisionsmått (RSDR) |
< 25 % |
< 15 % |
|
(1) Med avseende på gränsvärdena. |
||
5.7 Särskilda krav för screeningmetoder
|
5.7.1 |
Både GC-MS och bioanalytiska metoder får användas för screening. För GC-MS-metoder ska kraven i punkt 6 uppfyllas. För cellbaserade bioanalytiska metoder anges särskilda krav i punkt 7. |
|
5.7.2 |
Laboratorier som använder screeningmetoder vid rutinkontroll av prov ska ha ett nära samarbete med laboratorier som använder konfirmeringsmetoden. |
|
5.7.3 |
Screeningmetodens prestanda ska kontrolleras vid rutinanalyser genom kvalitetskontroll av analysverksamheten och löpande metodvalidering. Det ska finnas ett fortlöpande program för att kontrollera överensstämmande resultat. |
|
5.7.4 |
Kontroll av en eventuell hämning av cellens respons och cytotoxicitet Vid rutinmässig screening ska 20 % av provextrakten analyseras med och utan tillsats av 2,3,7,8-TCDD i en halt som motsvarar gränsvärdet eller åtgärdsgränsen för att kontrollera om responsen eventuellt hämmas av störande ämnen i provextraktet. Den uppmätta koncentrationen i det spikade provet ska jämföras med summan av koncentrationen i det ej spikade provet och den tillsatta koncentrationen. Om den uppmätta koncentrationen är mer än 25 % lägre än den beräknade koncentrationen (summan) tyder detta på att signalen eventuellt hämmas och det berörda provet ska genomgå konfirmeringsanalys med GC-HRMS. Resultaten ska övervakas med kontrollkort. |
|
5.7.5 |
Kvalitetskontroll av överensstämmande prov Ungefär 2–10 % av de överensstämmande proven ska konfirmeras med GC-HRMS, beroende på provmatrisen och laboratoriets erfarenhetsnivå. |
|
5.7.6 |
Kvalitetskontrolluppgifter som grund för bestämning av andelen prov som felaktigt bedömts överensstämma Efter screening av prov vars halter ligger under eller över gränsvärdena eller åtgärdsgränserna ska man bestämma andelen resultat som felaktigt bedömts överensstämma. Den faktiska andelen prov som felaktigt bedömts överensstämma ska vara lägre än 5 %. När man vid kvalitetskontrollen av överensstämmande prov har tillgång till minst 20 konfirmerade resultat per matris eller matrisgrupp ska dessa uppgifter användas som grund för att dra slutsatser om andelen prov som felaktigt bedömts överensstämma. Resultat från prov som analyserats i provningsjämförelser eller vid föroreningsincidenter och som täcker koncentrationsintervall upp till exempelvis två gånger gränsvärdet kan också tas med i de minst 20 resultat som ska ingå i utvärderingen. Proven ska täcka de vanligaste kongenmönstren och komma från olika källor. Även om det främsta syftet med screeningmetoder är att påvisa prov som överskrider åtgärdsgränsen, är gränsvärdet kriteriet för att fastställa andelen som felaktigt bedömts överensstämma, med beaktande av den utvidgade mätosäkerheten för konfirmeringsmetoden. |
|
5.7.7 |
Screeningresultat som eventuellt är icke-överensstämmande ska alltid kontrolleras genom en fullständig ny analys av det ursprungliga provet med en konfirmeringsmetod. Dessa prov kan också användas för att utvärdera andelen resultat som felaktigt bedömts vara icke-överensstämmande. För screeningmetoder motsvaras andelen prov som felaktigt bedömts vara icke-överensstämmande av den fraktion prov som med hjälp av konfirmeringsanalyser konstaterats överensstämma, även om dessa prov hade konstaterats vara eventuellt icke-överensstämmande vid tidigare screening. Utvärderingen av hur väl screeningmetoden fungerar ska göras på grundval av en jämförelse mellan antalet prov som felaktigt bedömts vara icke-överensstämmande och det totala antalet prov som kontrollerats. Detta värde ska vara så lågt att det lönar sig att använda en screeningmetod. |
|
5.7.8 |
Bioanalytiska metoder ska under valideringsbetingelser ge en godtagbar indikation på TEQ-värdet, beräknat och uttryckt som BEQ. Även för bioanalytiska metoder som används under repeterbarhetsbetingelser bör normalt sett den interna repeterbarheten (RSDr) vara lägre än reproducerbarheten (RSDR). |
6. Särskilda krav för GC-MS-metoder för screening eller konfirmering
6.1 Godtagbara skillnader mellan WHO-TEQ-värdena avseende den övre och den lägre koncentrationen
Det får inte skilja mer än 20 % mellan den övre och den lägre koncentrationen vid konfirmering av att gränsvärdet, eller vid behov åtgärdsgränsen, har överskridits.
6.2 Kontroll av utbytet
|
6.2.1 |
För validering av analysmetoden ska 13C-märkta 2,3,7,8-klorsubstituerade interna PCDD/PCDF-standarder och 13C-märkta interna dioxinlika PCB-standarder tillsättas alldeles i början av analysen, t.ex. före extraktionen. Minst en kongen för varje tetra- till oktaklorerad homolog grupp av PCDD/PCDF och minst en kongen för varje homolog grupp av dioxinlika PCB ska tillsättas (alternativt tillsätts minst en kongen för varje masspektrometrisk SIR-funktion [selected ion recording] som används för att övervaka PCDD/PCDF och dioxinlika PCB). För konfirmeringsmetoder ska alla de sjutton 13C-märkta 2,3,7,8-substituerade interna PCDD/PCDF-standarderna och alla de tolv 13C-märkta interna dioxinlika PCB-standarderna användas. |
|
6.2.2 |
Med hjälp av lämpliga kalibreringslösningar ska de relativa responsfaktorerna också bestämmas för de kongener till vilka ingen 13C-märkt analog tillsätts. |
|
6.2.3 |
För foder av vegetabiliskt eller animaliskt ursprung som innehåller mindre än 10 % fett ska interna standarder tillsättas före extraktionen. För foder av animaliskt ursprung som innehåller mer än 10 % fett ska interna standarder tillsättas antingen före eller efter fettextraktionen. Extraktionens effektivitet ska valideras på lämpligt sätt, beroende på när de interna standarderna tillsätts. |
|
6.2.4 |
Innan en GC-MS-analys utförs ska en eller två utbytesstandarder (surrogat) tillsättas. |
|
6.2.5 |
Det krävs en kontroll av utbytet. För konfirmeringsmetoder ska utbytet för de enskilda interna standarderna ligga i intervallet 60–120 %. Lägre eller högre utbytesgrad för enskilda kongener – särskilt för vissa hepta- och oktaklorerade dibenso-p-dioxiner och dibensofuraner – ska godtas under förutsättning att deras bidrag till TEQ-värdet inte överstiger 10 % av det totala TEQ-värdet (baserat på summan av PCDD/PCDF och dioxinlika PCB). För GC-MS-screeningmetoder ska utbytet ligga i intervallet 30–140 %. |
6.3 Avlägsnande av störande ämnen
— PCDD/PCDF ska separeras från störande klorföreningar, såsom icke-dioxinlika PCB och klorerade difenyletrar, med hjälp av lämpliga kromatografiska tekniker (lämpligtvis med en florisil-, aluminiumoxid- och/eller kolkolonn).
— Gaskromatografisk separation av isomerer ska ha mindre än 25 % mellan två toppar för 1,2,3,4,7,8-HxCDF och 1,2,3,6,7,8-HxCDF.
6.4 Kalibrering med standardkurva
Kalibreringskurvan ska omfatta det relevanta intervallet för gränsvärdet eller åtgärdsgränsen.
6.5 Särskilda kriterier för konfirmeringsmetoder
— För GC-HRMS:
—
Vid HRMS ska upplösningen vara större än eller lika med 10 000 i hela massområdet vid 10 % valley.
De ska uppfylla ytterligare kriterier för identifiering och konfirmering som beskrivs i internationellt erkända standarder, t.ex. standarden EN 16215:2012 (Djurfoder – Bestämning av dioxiner och dioxinlika PCB:er med GC/HRMS och av indikator-PCB:er med GC/HRMS) och/eller i EPA-metoderna 1613 och 1668, med senare revideringar.
— För GC-MS/MS:
—
Övervakning av minst två specifika moderjoner, var och en med en specifik motsvarande dotterjon från transitionen, för alla märkta och omärkta analyter som omfattas av analysens tillämpningsområde.
Högsta tillåtna tolerans för relativ jonintensitet på ± 15 % för utvalda dotterjoner från transitionen i jämförelse med beräknade eller uppmätta värden (medelvärde från kalibreringsstandarder) vid identiska MS/MS-betingelser, i synnerhet kollisionsenergi och kollisionsgastryck, för varje transition av en analyt.
Upplösningen för varje kvadrupol ska minst motsvara upplösningen för en massenhet (dvs. tillräcklig upplösning för att separera två toppar som skiljer sig åt med en massenhet) i syfte att minimera risken för att eventuella interferenser stör de givna analyterna.
De ska uppfylla ytterligare kriterier som beskrivs i internationellt erkända standarder, t.ex. standarden EN 16215:2012 (Djurfoder – Bestämning av dioxiner och dioxinlika PCB:er med GC/HRMS och av indikator-PCB:er med GC/HRMS) och/eller i EPA-metoderna 1613 och 1668, med senare revideringar, utom kravet på att använda GC-HRMS.
7. Särskilda krav för bioanalytiska metoder
Bioanalytiska metoder bygger på biologiska principer såsom cellbaserade metoder, receptorbaserade metoder eller immunologiska metoder. I denna punkt 7 fastställs krav för bioanalytiska metoder i allmänhet.
Med en screeningmetod klassificeras enkelt uttryckt ett prov antingen som överensstämmande eller som misstänkt icke-överensstämmande genom en jämförelse mellan det beräknade BEQ-värdet och brytpunkten (se punkt 7.3). Prov under brytpunkten är överensstämmande, medan prov som är lika med eller över brytpunkten är misstänkt icke-överensstämmande och kräver analys med en konfirmeringsmetod. I praktiken kan ett BEQ-värde som motsvarar 2/3 av gränsvärdet användas som brytpunkt, förutsatt att det kan säkerställas att andelen prov som felaktigt bedömts vara överensstämmande är lägre än 5 % och att andelen prov som felaktigt bedömts vara icke-överensstämmande är godtagbar. Om man använder separata gränsvärden för PCDD/PCDF och för summan av PCDD/PCDF och dioxinlika PCB krävs lämpliga brytpunkter för bioassay av PCDD/PCDF för att utan fraktionering kunna kontrollera provens överensstämmelse. För kontroll av prov som överskrider åtgärdsgränserna är en lämplig procentandel av respektive åtgärdsgräns passande som brytpunkt.
Om ett vägledande värde uttrycks i BEQ ska provresultaten ges i mätområdet och överstiga rapporteringsgränsen (se punkt 7.1.1 och 7.1.6).
7.1 Utvärdering av försöksrespons
7.1.1
— När man beräknar koncentrationer utifrån en kalibreringskurva för TCDD kommer de högsta koncentrationerna i kurvan att ha en hög variation (hög variationskoefficient). Mätområdet är det område där variationskoefficienten är mindre än 15 %. Den lägsta koncentrationen i mätområdet (rapporteringsgränsen) ska ligga över metodens blankvärde med åtminstone en faktor på tre. Den högsta koncentrationen i mätområdet motsvaras vanligtvis av EC70-värdet (70 % av den maximalt effektiva koncentrationen), men den är lägre om variationskoefficienten överstiger 15 % i detta område. Mätområdet ska fastställas under valideringen. Brytpunkterna (se punkt 7.3) ska med god marginal ligga inom mätområdet.
— Standardlösningar och provextrakt ska analyseras som trippelprov eller åtminstone som dubbelprov. När dubbelprov analyseras ska en standardlösning eller ett kontrollextrakt som provas i 4–6 brunnar (fördelade över plattan) ge en respons eller en koncentration (endast möjligt i mätområdet) som har en variationskoefficient mindre än 15 %.
7.1.2
7.1.2.1 Kalibrering med standardkurva
— Provhalter ska skattas genom att försöksresponsen jämförs med en kalibreringskurva för TCDD (eller PCB 126 eller en standardblandning med PCDD/PCDF och dioxinlika PCB) utifrån vilken BEQ-värdet kan beräknas i extraktet och därefter i provet.
— Kalibreringskurvor ska bestå av 8–12 koncentrationer (åtminstone som dubbelprov) och ha tillräckligt med koncentrationer i den nedre delen av kurvan (mätområdet). Särskild uppmärksamhet ska ägnas kvaliteten på kurvpassningen i mätområdet. När man ska bedöma anpassningsgraden vid icke-linjär regression är R2-värdet i sig inte viktigt. Man uppnår en bättre kurvpassning genom att minimera skillnaden mellan beräknade och observerade koncentrationer inom kurvans mätområde, t.ex. genom att minimera residualkvadratsumman.
— Den skattade halten i provextraktet korrigeras därefter för BEQ-värdet hos ett blankprov för matrisen eller lösningsmedlet (för att beakta föroreningar från lösningsmedel och andra kemikalier som använts) och för det skenbara utbytet (som beräknats utifrån BEQ-värdet för lämpliga referensprov med representativa kongenmönster kring gränsvärdet eller åtgärdsgränsen). När man korrigerar för ett utbyte ska det skenbara utbytet ligga inom det intervall som krävs (se punkt 7.1.4). Referensprov som används för att korrigera utbytet ska uppfylla de krav som anges i punkt 7.2.
7.1.2.2 Kalibrering med referensprov
Om egenskaperna hos referensprovens matris överensstämmer med dem hos de okända provens matris kan man också använda en kalibreringskurva som genererats från minst fyra referensprov (se punkt 7.2.4: ett blankprov för matrisen och tre referensprov på 0,5 gång, 1 gång och 2 gånger gränsvärdet eller åtgärdsgränsen), varigenom man inte behöver korrigera för blank och utbyte. I detta fall kan den försöksrespons som motsvarar 2/3 av gränsvärdet (se punkt 7.3) beräknas direkt utifrån dessa prov och användas som brytpunkt. För kontroll av prov som överskrider åtgärdsgränserna är en lämplig procentandel av dessa åtgärdsgränser passande som brytpunkt.
7.1.3
Extrakt kan delas upp i fraktioner som innehåller dels PCDD/PCDF, dels dioxinlika PCB, vilket gör att man kan ange separata TEQ-värden för PCDD/PCDF och för dioxinlika PCB (uttryckta som BEQ). Företrädesvis ska man använda en kalibreringskurva för PCB 126-standard för att utvärdera resultaten för den fraktion som innehåller dioxinlika PCB.
7.1.4
Det skenbara utbytet för bioassay ska beräknas utifrån lämpliga referensprov med representativa kongenmönster kring gränsvärdet eller åtgärdsgränsen och uttryckas som en procentandel av BEQ-värdet i jämförelse med TEQ-värdet. Beroende på vilken typ av analysmetod och vilka TEF-värden ( 27 ) som används kan skillnaderna mellan TEF- och REP-faktorerna för dioxinlika PCB ge upphov till låga skenbara utbyten för dioxinlika PCB i jämförelse med för PCDD/PCDF. Om en separat bestämning av PCDD/PCDF och dioxinlika PCB utförs ska därför de skenbara utbytena för bioassay ligga i intervallet 20–60 % för dioxinlika PCB och i intervallet 50–130 % för PCDD/PCDF (intervallen gäller för kalibreringskurvan för TCDD). Bidraget från dioxinlika PCB till summan av PCDD/PCDF och dioxinlika PCB kan variera mellan olika matriser och prov. Dessa variationer återspeglas i de skenbara utbytena för bioassay, vilka ska ligga i intervallet 30–130 % för summan av PCDD/PCDF och dioxinlika PCB. Om TEF-värdena för PCDD/PCDF och dioxinlika PCB i EU-lagstiftningen skulle ändras väsentligt, måste dessa intervall ses över.
7.1.5
Förlusten av föreningar under uppreningen ska kontrolleras under valideringen. Ett blankprov som spikats med en blandning av olika kongener ska genomgå upprening (minst n = 3) och därefter ska utbyte och variabilitet kontrolleras med en konfirmeringsmetod. Utbytet ska ligga i intervallet 60–120 %, särskilt för kongener som i olika blandningar bidrar med mer än 10 % till TEQ-värdet.
7.1.6
Vid rapportering av BEQ-värden ska en rapporteringsgräns bestämmas utifrån relevanta matrisprov som omfattar typiska kongenmönster. Däremot ska inte kalibreringskurvan för standarder användas eftersom precisionen är låg för den nedre delen av kurvan. Även effekter från extraktion och upprening ska beaktas. Rapporteringsgränsen ska ligga över metodens blankvärde med åtminstone en faktor på tre.
7.2 Användning av referensprov
|
7.2.1 |
Referensproven ska representera provmatriser, kongenmönster och koncentrationsintervall för PCDD/PCDF och dioxinlika PCB kring gränsvärdet eller åtgärdsgränsen. |
|
7.2.2 |
Ett blankprov för matrisen, eller när detta inte är möjligt ett blankprov för metoden, och ett referensprov vid gränsvärdet eller åtgärdsgränsen ska ingå i varje försöksserie. Dessa prov ska extraheras och analyseras samtidigt under identiska betingelser. Referensprovet ska ha en klart högre respons än blankprovet eftersom detta säkerställer metodens lämplighet. Dessa prov kan användas för att korrigera för blank och utbyte. |
|
7.2.3 |
De referensprov som valts ut för att korrigera för utbyte ska vara representativa för alla proven, vilket innebär att kongenmönstrena inte ska resultera i att halterna underskattas. |
|
7.2.4 |
För kontroll av gränsvärdet eller åtgärdsgränsen kan ytterligare referensprov på exempelvis 0,5 gång och 2 gånger gränsvärdet eller åtgärdsgränsen tas med i syfte att visa analysens prestanda inom det givna intervallet. Dessa prov kan kombineras för att beräkna BEQ-värdena i proven (se punkt 7.1.2.2). |
7.3 Bestämning av brytpunkter
Förhållandet mellan bioanalytiska resultat angivna som BEQ och resultat från konfirmeringsmetoden angivna som TEQ ska fastställas, till exempel genom matrisanpassade kalibreringsexperiment som omfattar referensprov spikade med 0 gång, 0,5 gång, 1 gång och 2 gånger gränsvärdet och 6 prov per halt (totalt n = 24). Korrektionsfaktorer (blank och utbyte) kan beräknas utifrån detta förhållande, men de ska kontrolleras i enlighet med punkt 7.2.2.
Brytpunkter ska fastställas för bedömning av om prov överensstämmer med gränsvärdena, eller vid behov för kontroll av åtgärdsgränserna, med respektive gränsvärden eller åtgärdsgränser som fastställts för antingen PCDD/PCDF och dioxinlika PCB var för sig, eller för summan av PCDD/PCDF och dioxinlika PCB. De representeras av den nedre gränsen för fördelningen av de bioanalytiska resultaten (korrigerade för blank och utbyte) som motsvarar beslutsgränsen för konfirmeringsmetoden med en konfidensgrad på 95 %, vilket innebär att andelen prov som felaktigt bedömts vara överensstämmande är mindre än 5 % och RSDR är mindre än 25 %. Beslutsgränsen för konfirmeringsmetoden motsvaras av gränsvärdet, med beaktande av den utvidgade mätosäkerheten.
Brytpunkten (uttryckt som BEQ) kan beräknas enligt ett av de sätt som anges i punkt 7.3.1, 7.3.2 och 7.3.3 (se figur 1).
|
7.3.1 |
Med användning av det nedre konfidensbandet i det 95-procentiga prediktionsintervallet för beslutsgränsen för konfirmeringsmetoden och med formeln
där
|
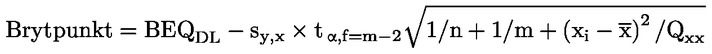

|
7.3.2 |
Beräknat utifrån de bioanalytiska resultaten (korrigerade för blank och utbyte) från flera analyser av prov (n ≥ 6) som förorenats med en halt som motsvarar beslutsgränsen för konfirmeringsmetoden, vilken är den nedre gränsen för fördelningen vid motsvarande medelvärde för BEQ, och med formeln brytpunkt = BEQDL – 1,64 × SDR där
|
|
7.3.3 |
Beräknat som medelvärdet av de bioanalytiska resultaten (uttryckta som BEQ, korrigerade för blank och utbyte) utifrån flera analyser av prov (n ≥ 6) som förorenats med en halt som motsvarar 2/3 av gränsvärdet eller åtgärdsgränsen. Detta grundas på att denna halt kommer att ligga kring den brytpunkt som fastställts enligt punkt 7.3.1 eller 7.3.2. Tillvägagångssätt för beräkning av brytpunkter som bygger på en 95-procentig konfidensgrad, vilket innebär att andelen prov som felaktigt bedömts vara överensstämmande är mindre än 5 %, och RSDR är mindre än 25 %. 1. Utifrån det nedre konfidensbandet i det 95-procentiga prediktionsintervallet för beslutsgränsen för konfirmeringsmetoden. 2. Utifrån flera analyser av prov (n ≥ 6) som förorenats med en halt som motsvarar beslutsgränsen för konfirmeringsmetoden, vilket är den nedre gränsen för fördelningen (en klockformad kurva i figuren) vid motsvarande medelvärde för BEQ.
|

|
7.3.4 |
Begränsningar för brytpunkter De BEQ-baserade brytpunkter som beräknats utifrån den RSDR som fastställts under valideringen med hjälp av ett begränsat antal prov med olika matriser eller kongenmönster kan vara högre än de gränsvärden eller åtgärdsgränser som baserats på TEQ-värden, eftersom en bättre precision kan uppnås vid validering än vid rutinanalyser då ett okänt spektrum av möjliga kongenmönster måste kontrolleras. I sådana fall ska brytpunkterna helst beräknas utifrån en RSDR som motsvarar 25 % eller 2/3 av gränsvärdet eller åtgärdsgränsen. |
7.4 Prestanda
|
7.4.1 |
Eftersom inga interna standarder kan användas i bioanalytiska metoder ska försök som kontrollerar repeterbarheten hos dessa metoder utföras i syfte att bedöma standardavvikelsen inom en försöksserie och mellan försöksserier. Repeterbarheten ska vara under 20 % och reproducerbarheten inom laboratoriet under 25 %. Detta ska grundas på de beräknade halterna uttryckta som BEQ efter korrigering för blank och utbyte. |
|
7.4.2 |
Under valideringsförfarandet ska det visas att metoden kan skilja mellan ett blankprov och ett prov med en halt som motsvarar brytpunkten, så att prov som ligger över den relevanta brytpunkten kan identifieras (se punkt 7.1.2). |
|
7.4.3 |
Målföreningar, möjliga interferenser och högsta godtagbara värden för blankprov ska definieras. |
|
7.4.4 |
Vid analys av ett provextrakt (n = 3) får standardavvikelsen inte överstiga 15 % för en respons eller en koncentration som beräknats utifrån responsen (endast möjligt i mätområdet). |
|
7.4.5 |
De okorrigerade resultaten från referensprov, uttryckta som BEQ (blank och vid gränsvärdet eller åtgärdsgränsen), ska användas vid utvärderingen av den bioanalytiska metodens prestanda under en konstant tidsperiod. |
|
7.4.6 |
Kontrollkort för metodens blankprov och för varje typ av referensprov ska framställas och kontrolleras för att säkerställa att den analytiska prestandan uppfyller kraven, särskilt för metodens blankprov när det gäller kravet på minsta skillnad till de lägsta koncentrationerna i mätområdet och för referensprov när det gäller reproducerbarheten inom laboratoriet. Metodens blankprov ska kontrolleras så att man när blankprovet dras av från provresultatet inte får resultat som felaktigt bedöms vara överensstämmande. |
|
7.4.7 |
Resultaten från analyserna med konfirmeringsmetoderna av misstänkta prov och av 2–10 % av de överensstämmande proven (minst 20 prov per matris) ska samlas in och användas för att utvärdera screeningmetodens prestanda och förhållandet mellan BEQ- och TEQ-värdena. Dessa uppgifter kan användas för omprövning av de brytpunkter som tillämpas på rutinprov för validerade matriser. |
|
7.4.8 |
Att en metod har en bra prestanda kan också visas genom deltagande i provningsjämförelser. Om ett laboratorium kan visa en bra prestanda kan också resultaten från prov som analyserats i provningsjämförelser och som täcker koncentrationsintervallet upp till exempelvis två gånger gränsvärdet tas med i utvärderingen av andelen prov som felaktigt bedömts vara överensstämmande. Proven ska täcka de vanligaste kongenmönstren och komma från olika källor. |
|
7.4.9 |
Vid incidenter kan brytpunkterna omprövas för att återspegla den specifika matrisen och kongenmönstret för denna enda incident. |
8. Rapportering av resultat
8.1 Konfirmeringsmetoder
|
8.1.1 |
Analysresultaten ska innehålla halterna av de enskilda PCDD/PCDF- och dioxinlika PCB-kongenerna och TEQ-värdena angivna som lägre koncentrationer, övre koncentrationer och mellanvärden. Rapporteringen av resultat ska på så sätt ge maximalt med information och därigenom göra det möjligt att tolka resultaten utifrån särskilda krav. |
|
8.1.2 |
Rapporten ska ange den metod som använts för extraktionen av PCDD/PCDF och dioxinlika PCB. |
|
8.1.3 |
Utbytena för de enskilda interna standarderna ska redovisas om de ligger utanför det intervall som anges i punkt 6.2.5 eller när gränsvärdet överskrids (i så fall ska utbytet för ett av de två dubbelproven anges). I övriga fall ska de redovisas på begäran. |
|
8.1.4 |
Eftersom den utvidgade mätosäkerheten ska beaktas vid bedömning av om ett prov är överensstämmande ska denna parameter anges. Analysresultatet ska anges som x ± U, där x är analysresultatet och U den utvidgade mätosäkerheten beräknad med en täckningsfaktor på 2, vilket ger en konfidensgrad på cirka 95 %. Om det har gjorts en separat bestämning av PCDD/PCDF och av dioxinlika PCB ska summan av den skattade utvidgade mätosäkerheten för de enskilda analysresultaten användas när man anger summan av PCDD/PCDF och dioxinlika PCB. |
|
8.1.5 |
Resultaten ska anges i samma enheter och med minst samma antal signifikanta siffror som används för att ange gränsvärden i direktiv 2002/32/EG. |
8.2 Bioanalytiska screeningmetoder
|
8.2.1 |
Resultatet från screeningen ska anges som ”överensstämmande” eller som ”misstänkt icke-överensstämmande”. |
|
8.2.2 |
Dessutom kan ett vägledande resultat för PCDD/PCDF och/eller dioxinlika PCB, uttryckt som BEQ och inte som TEQ, anges. |
|
8.2.3 |
Prov vars respons ligger under rapporteringsgränsen ska anges som ”under rapporteringsgränsen”. Prov vars respons ligger över mätområdet ska anges som ”överskridande mätområdet” och den halt som motsvarar den högsta koncentrationen i mätområdet ska anges som BEQ. |
|
8.2.4 |
För varje typ av provmatris ska rapporten innehålla uppgifter om det gränsvärde eller den åtgärdsgräns som utvärderingen baseras på. |
|
8.2.5 |
Rapporten ska innehålla uppgifter om typ av försök som har gjorts, den grundläggande principen för försöket och typ av kalibrering. |
|
8.2.6 |
Rapporten ska ange den metod som använts för extraktion av PCDD/PCDF och dioxinlika PCB. |
|
8.2.7 |
För prov som misstänks vara icke-överensstämmande ska rapporten innehålla information om de åtgärder som ska vidtas. Koncentrationen av PCDD/PCDF och summan av PCDD/PCDF och dioxinlika PCB i proven med förhöjda halter ska bestämmas/bekräftas med en konfirmeringsmetod. |
|
8.2.8 |
Icke-överensstämmande resultat ska endast rapporteras från konfirmeringsanalys. |
8.3 Fysikalisk-kemiska screeningmetoder
|
8.3.1 |
Resultaten från screeningen ska anges som ”överensstämmande” eller som ”misstänkt icke-överensstämmande”. |
|
8.3.2 |
För varje typ av provmatris ska rapporten innehålla uppgifter om det gränsvärde eller den åtgärdsgräns som utvärderingen baseras på. |
|
8.3.3 |
Halter för enskilda PCDD/PCDF- och/eller dioxinlika PCB-kongener samt TEQ-värden kan dessutom anges angivna som lägre koncentrationer, övre koncentrationer och mellanvärden. Resultaten ska anges i samma enheter och med minst samma antal signifikanta siffror som används för att ange gränsvärden i direktiv 2002/32/EG. |
|
8.3.4 |
Utbytena för de enskilda interna standarderna ska redovisas om de ligger utanför det intervall som anges i punkt 6.2.5 eller när gränsvärdet överskrids (i så fall ska utbytet för ett av de två dubbelproven anges). I övriga fall ska de redovisas på begäran. |
|
8.3.5 |
Rapporten ska ange den GC-MS-metod som använts. |
|
8.3.6 |
Rapporten ska ange den metod som använts för extraktion av PCDD/PCDF och dioxinlika PCB. |
|
8.3.7 |
För prov som misstänks vara icke-överensstämmande ska rapporten innehålla information om de åtgärder som ska vidtas. Koncentrationen av PCDD/PCDF och summan av PCDD/PCDF och dioxinlika PCB i proven med förhöjda halter ska bestämmas/bekräftas med en konfirmeringsmetod. |
|
8.3.8 |
Bedömningen av om ett prov är icke-överensstämmande kan endast göras efter konfirmeringsanalys. |
KAPITEL III
Provberedning och krav för analysmetoder för offentlig kontroll av halter av icke-dioxinlika PCB i foder
1. Tillämpningsområde
Kraven i denna bilaga ska tillämpas när foder analyseras för offentlig kontroll av halterna av icke-dioxinlika PCB och när det gäller provberedning och analyskrav för andra lagstadgade ändamål, inbegripet foderföretagarens kontroller för att säkerställa överensstämmelse med bestämmelserna i förordning (EG) nr 183/2005.
2. Detektionsmetoder som ska användas
Gaskromatografi i kombination med elektroninfångningsdetektion (GC-ECD), GC-LRMS, GC-MS/MS, GC-HRMS eller likvärdiga metoder.
3. Identifiering och konfirmering av givna analyter
|
3.1 |
Relativ retentionstid i förhållande till interna standarder eller referensstandarder (godtagbar avvikelse ± 0,25 %). |
|
3.2 |
Gaskromatografisk separation av icke-dioxinlika PCB från störande ämnen, särskilt andra PCB som elueras samtidigt, i synnerhet om provhalterna ligger i intervallet för lagstadgade gränsvärden och det ska konfirmeras att proven är icke-överensstämmande ( 28 ). |
|
3.3 |
Krav för GC-MS-metoder Övervakning av minst följande antal molekyljoner eller joner karakteristiska för molekylklustret: a) Två specifika joner för HRMS. b) Tre specifika joner för LRMS. c) Två specifika moderjoner, var och en med en specifik motsvarande dotterjon från transitionen, för MS-MS. Högsta tillåtna toleranser för kvoten för förekomsten av utvalda massfragment: Relativ avvikelse mellan kvoten för förekomsten av utvalda massfragment från den teoretiska förekomsten eller kalibreringsstandarden för måljon (den vanligast förekommande jonen som övervakas) och konfirmeringsjon(er): ± 15 %. |
|
3.4 |
Krav för GC-ECD-metoder Resultat som överskrider gränsvärdet ska konfirmeras med hjälp av två GC-kolonner som har stationära faser med olika polaritet. |
4. Bevis för metodens prestanda
Metodens prestanda ska valideras i intervallet för gränsvärdet (0,5–2 gånger gränsvärdet) med en godtagbar variationskoefficient för upprepade analyser (se krav för intermediärt precisionsmått i punkt 9).
5. Kvantifieringsgräns
Summan av kvantifieringsgränserna ( 29 ) för icke-dioxinlika PCB får inte vara högre än 1/3 av gränsvärdet ( 30 ).
6. Kvalitetskontroll
Laboratorierna ska regelbundet kontrollera blankprov, analysera spikade prov och prov för kvalitetskontroll samt medverka i provningsjämförelser med relevanta matriser.
7. Kontroll av utbytet
|
7.1 |
Lämpliga interna standarder med fysikalisk-kemiska egenskaper som är jämförbara med de givna analyternas ska användas. |
|
7.2 |
Tillsats av intern standard Ska tillsättas produkter före extraktion och upprening. |
|
7.3 |
Krav för metoder som använder alla sex isotopmärkta icke-dioxinlika PCB-kongener: a) Resultaten ska korrigeras för de interna standardernas utbyten. b) Utbytet för isotopmärkta interna standarder ska ligga i intervallet 60–120 %. c) Ett lägre eller högre utbyte för enskilda kongener vars bidrag till summan av icke-dioxinlika PCB är mindre än 10 % är godtagbart. |
|
7.4 |
Krav för metoder som inte använder alla sex isotopmärkta interna standarder eller använder andra interna standarder: a) Utbyten för interna standarder ska kontrolleras för varje prov. b) Utbytena för interna standarder ska ligga i intervallet 60–120 %. c) Resultaten ska korrigeras för de interna standardernas utbyten. |
|
7.5 |
Utbytena för omärkta kongener ska kontrolleras genom spikade prov eller prov för kvalitetskontroll med koncentrationer i intervallet för gränsvärdet. Godtagbara utbyten för dessa kongener ska ligga i intervallet 60–120 %. |
8. Krav för laboratorier
I enlighet med förordning (EG) nr 882/2004 ska laboratorier ackrediteras av ett godkänt ackrediteringsorgan som verkar enligt ISO Guide 58 för att säkerställa att de tillämpar ett system för kvalitetssäkring av analysverksamheten. Laboratorier ska ackrediteras enligt standarden EN ISO/IEC 17025. Principerna i vägledningen Technical Guidelines for the estimation of measurement uncertainty and limits of quantification for PCB analysis ska dessutom följas i tillämpliga fall ( 31 ).
9. Prestanda: Kriterier för summan av icke-dioxinlika PCB vid gränsvärdet
|
|
Masspektrometri med isotoputspädning (1) |
Andra metoder |
|
Riktighet |
– 20 % till + 20 % |
– 30 % till + 30 % |
|
Intermediärt precisionsmått (RSD %) |
≤ 15 % |
≤ 20 % |
|
Skillnaden mellan beräkningen av övre och lägre koncentration |
≤ 20 % |
≤ 20 % |
|
(1) Alla sex 13C-märkta analogerna ska användas som interna standarder. |
||
10. Rapportering av resultat
|
10.1 |
Analysresultaten ska innehålla halterna av de enskilda icke-dioxinlika PCB och summan av dessa PCB-kongener angivna som lägre koncentrationer, övre koncentrationer och mellanvärden. Rapporteringen av resultaten ska på så sätt ge maximalt med information och därigenom göra det möjligt att tolka resultaten utifrån särskilda krav. |
|
10.2 |
Rapporten ska ange den metod som använts för extraktion av PCB. |
|
10.3 |
Utbytena för de enskilda interna standarderna ska redovisas om de ligger utanför det intervall som anges i punkt 7 eller när gränsvärdet överskrids. I övriga fall ska de redovisas på begäran. |
|
10.4 |
Eftersom den utvidgade mätosäkerheten ska beaktas vid bedömning av om ett prov är överensstämmande ska också denna parameter anges. Analysresultatet ska anges som x ± U, där x är analysresultatet och U den utvidgade mätosäkerheten beräknad med en täckningsfaktor på 2, vilket ger en konfidensgrad på cirka 95 %. |
|
10.5 |
Resultaten ska anges i samma enheter och med minst samma antal signifikanta siffror som används för att ange gränsvärden i direktiv 2002/32/EG. |
BILAGA VI
ANALYSMETODER FÖR BESTÄMNING AV BESTÅNDSDELAR AV ANIMALISKT URSPRUNG FÖR OFFENTLIG KONTROLL AV FODER
1. SYFTE OCH TILLÄMPNINGSOMRÅDE
Bestämningen av beståndsdelar av animaliskt ursprung i foder ska utföras med ljusmikroskopi eller polymeraskedjereaktion (PCR) i enlighet med bestämmelserna i denna bilaga.
Med hjälp av dessa två metoder är det möjligt att påvisa förekomsten av beståndsdelar av animaliskt ursprung i foderråvaror och foderblandningar. Däremot går det inte att med dessa metoder beräkna mängden av dessa beståndsdelar i foderråvaror och foderblandningar. Båda metoderna har en detektionsgräns under 0,1 % (w/w).
Med PCR är det möjligt att identifiera den taxonomiska gruppen för de beståndsdelar av animaliskt ursprung som finns i foderråvaror och foderblandningar.
Dessa metoder ska användas vid kontroll av att de förbud som fastställs i artikel 7.1 och bilaga IV till förordning (EG) nr 999/2001 och i artikel 11.1 i förordning (EG) nr 1069/2009 tillämpas.
Beroende på vilken typ av foder som testas kan dessa metoder användas inom ett enda protokoll, antingen ensamma eller i kombination, i enlighet med de standardrutiner (SOP) som fastställs av EU:s referenslaboratorium för animaliska proteiner i foder och som offentliggörs på dess webbplats ( 32 ).
2. METODER
2.1 Ljusmikroskopi
2.1.1 Princip
De beståndsdelar av animaliskt ursprung som kan förekomma i de foderråvaror och foderblandningar som sänds för analys identifieras på grundval av typiska egenskaper som kan urskiljas med mikroskop (t.ex. muskelfibrer och andra köttpartiklar, brosk, ben, horn, hår, borst, blod, fjädrar, äggskal, fiskben och fiskfjäll).
2.1.2 Reagens och utrustning
2.1.2.1 Reagens
2.1.2.1.1 Koncentreringsmedel
2.1.2.1.1.1 Tetrakloreten (densitet 1,62).
2.1.2.1.2 Färgreagens
|
2.1.2.1.2.1 |
Alizarinlösning (späd 2,5 ml 1 M saltsyra med 100 ml vatten och tillsätt 200 mg alizarin till denna lösning). |
2.1.2.1.3 Monteringsmedium
2.1.2.1.3.1 Lut (NaOH 2,5 % w/v eller KOH 2,5 % w/v).
2.1.2.1.3.2 Glycerol (outspädd, viskositet: 1 490 cP).
2.1.2.1.3.3 Norland ® Optical Adhesive 65 (viskositet: 1 200 cP) eller ett harts med motsvarande egenskaper för preparering av permanent fixerade preparat på objektglas.
2.1.2.1.4 Monteringsmedium med färgande egenskaper
|
2.1.2.1.4.1 |
Lugols lösning (lös 2 g kaliumjodid i 100 ml vatten och tillsätt 1 g jod under regelbunden omskakning). |
|
2.1.2.1.4.2 |
Cystinreagens (2 g blyacetat, 10 g NaOH/100 ml vatten). |
|
2.1.2.1.4.3 |
Fehlings lösning (före användning blandas lika delar (1:1) av stamlösningarna A och B. Lösning A: lös 6,9 g koppar(II)sulfatpentahydrat i 100 ml vatten. Lösning B: lös 34,6 g kaliumnatriumtartrattetrahydrat och 12 g NaOH i 100 ml vatten). |
|
2.1.2.1.4.4 |
Tetrametylbensidin/väteperoxid (lös 1 g 3,3’,5,5’-tetrametylbensidin (TMB) i 100 ml isättika och 150 ml vatten. Före användning blandas 4 delar TMB-lösning med 1 del 3 % väteperoxid). |
2.1.2.1.5 Sköljmedel
2.1.2.1.5.1 Etanol ≥ 96 % (teknisk kvalitet).
2.1.2.1.5.2 Aceton (teknisk kvalitet).
2.1.2.1.6 Blekmedel
2.1.2.1.6.1 I handeln förekommande natriumhypokloritlösning (9–14 % aktivt klor).
2.1.2.2 Utrustning
2.1.2.2.1 Analysvåg med en noggrannhet av 0,001 g.
2.1.2.2.2 Utrustning för malning: kvarn eller mortel.
2.1.2.2.3 Siktar med kvadratiska maskor med en maskvidd på 0,25 mm och 1 mm.
|
2.1.2.2.4 |
Konisk separertratt i glas med en volym på 250 ml och med en avstängningsventil i teflon eller slipat glas placerad i botten av tratten. Diametern på avstängningsventilen ska vara minst 4 mm. Alternativt får en sedimentbägare med konisk botten användas, förutsatt att laboratoriet har visat att detektionsgränsen är likvärdig med den som uppnås med den koniska separertratten i glas. 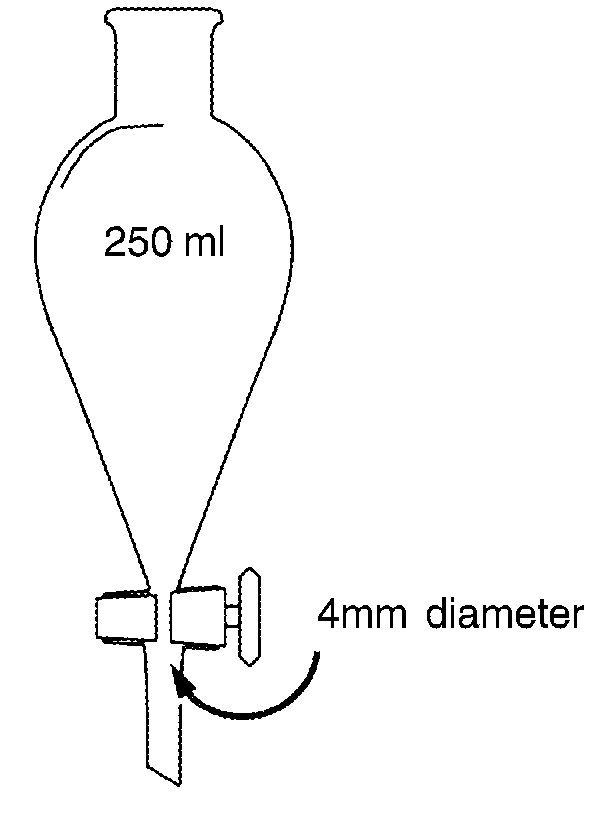
|
|
2.1.2.2.5 |
Stereomikroskop med en förstoringsgrad på minst 6,5× till 40×. |
|
2.1.2.2.6 |
Ljusfältsmikroskop med en förstoringsgrad på minst 100× till 400×. Även polariserad ljusmikroskopi och interferenskontrastmikroskopi får användas. |
|
2.1.2.2.7 |
Laboratorieglas av standardtyp. |
|
2.1.2.2.8 |
Utrustning för preparering av objektglas: vanliga objektglas, objektglas med en fördjupning, täckglas (20 × 20 mm), pincett och liten spatel. |
2.1.3 Provtagning och provberedning
2.1.3.1 Provtagning
Ett representativt prov som tagits i enlighet med bestämmelserna i bilaga I ska användas.
2.1.3.2 Försiktighetsåtgärder
För att undvika korskontaminering i laboratoriet ska all utrustning som används flera gånger rengöras noggrant före användning. Separertrattar ska tas isär före rengöring. Separertrattar och glasvaror ska först diskas för hand och sedan i diskmaskin. Siktar ska rengöras med en borste med styva syntetiska hår. Efter siktningen av fett material såsom fiskmjöl rekommenderas en avslutande rengöring av sikten med aceton och tryckluft.
2.1.3.3 Beredning av andra prov än fett eller olja
|
2.1.3.3.1 |
Torkning av prov: Prov med en vattenhalt över 14 % ska torkas före beredning. |
|
2.1.3.3.2 |
Siktning av prov före beredning: Det rekommenderas att pelleterat foder och palmkärnor siktas genom 1 mm maskor före beredning och att de två fraktionerna därefter bereds och analyseras som separata prov. |
|
2.1.3.3.3 |
Analysprov och malning: Minst 50 g av provet ska delas upp i analysprov och därefter malas. |
|
2.1.3.3.4 |
Extraktion och beredning av sediment: En portion på 10 g (med en noggrannhet av 0,01 g) av det malda analysprovet förs över i en separertratt eller en sedimentbägare med konisk botten och 50 ml tetrakloreten tillsätts. När det gäller fiskmjöl eller andra rena animaliska produkter, mineralämnen eller förblandningar som bildar mer än 10 % sediment ska högst 3 g prov föras över i tratten. Blandningen skakas kraftigt i minst 30 sekunder och ytterligare minst 50 ml tetrakloreten tillsätts försiktigt under det att insidan av tratten sköljs för att avlägsna eventuella vidhäftande partiklar. Den erhållna blandningen ska stå i minst fem minuter innan sedimentet avskiljs genom att avstängningsventilen öppnas. Om en sedimentbägare med konisk botten används ska blandningen omröras kraftigt i minst 15 sekunder och eventuella vidhäftande partiklar på bägarens sidor sköljs noggrant ner med minst 10 ml ren tetrakloreten. Blandningen ska stå i tre minuter och därefter återigen omröras i 15 sekunder och eventuella vidhäftande partiklar på bägarens sidor sköljs noggrant ner med minst 10 ml ren tetrakloreten. Den erhållna blandningen ska stå i minst fem minuter och sedan avlägsnas den flytande fraktionen genom noggrann dekantering så att inget sediment försvinner. Sedimentet torkas och vägs (med en noggrannhet av 0,001 g). Om mer än 5 % av sedimentet består av partiklar större än 0,50 mm ska det siktas genom 0,25 mm maskor och båda fraktionerna ska undersökas. |
|
2.1.3.3.5 |
Extraktion och beredning av flotat: Efter att sedimentet tagits fram enligt metoden ovan bör det finnas två faser i separertratten: en vätskefas som består av tetrakloreten och en fast fas som består av flytande material. Den fasta fasen är flotatet och det erhålls genom att avstängningsventilen öppnas och all tetrakloreten hälls bort. Separertratten vänds upp och ner och flotatet överförs till en stor petriskål och lufttorkas därefter i ett dragskåp. Om mer än 5 % av flotatet består av partiklar större än 0,50 mm ska det siktas genom 0,25 mm maskor och båda fraktionerna ska undersökas. |
|
2.1.3.3.6 |
Beredning av råvara: Minst 5 g av det malda analysprovet ska beredas. Om mer än 5 % av provet består av partiklar större än 0,50 mm ska det siktas genom 0,25 mm maskor och båda fraktionerna ska undersökas. |
2.1.3.4 Beredning av prov bestående av fett eller olja
Följande protokoll ska användas vid beredning av prov bestående av fett eller olja:
— Om fettet är fast värms det i en ugn tills det blir flytande.
— Med pipett överförs 40 ml av fettet från botten av provet till ett centrifugrör.
— Centrifugera i tio minuter vid 4 000 varv per minut.
— Om fettet är fast efter centrifugeringen värms det i en ugn tills det blir flytande.
— Upprepa centrifugeringen i fem minuter vid 4 000 varv per minut.
— Med en liten sked eller spatel placeras hälften av de dekanterade föroreningarna på objektglas för mikroskopisk undersökning. Glycerol rekommenderas som moteringsmedium.
— De återstående föroreningarna används för beredning av sedimentet enligt punkt 2.1.3.3.
2.1.3.5 Användning av färgreagens
För att lättare kunna identifiera beståndsdelar av animaliskt ursprung får färgreagens användas vid provberedningen i enlighet med de riktlinjer som utfärdas av EU:s referenslaboratorium för animaliska proteiner i foder och som offentliggörs på dess webbplats.
Om alizarinlösning används för att färga sedimentet ska följande protokoll användas:
— Det torkade sedimentet överförs till ett provrör i glas och sköljs två gånger med ca 5 ml etanol (varje gång vortexas provet i 30 sekunder, lösningen får stå i ca 1½ minut och därefter hälls lösningsmedlet bort).
— Sedimentet bleks genom att minst 1 ml natriumhypokloritlösning tillsätts. Reaktionen får fortsätta i tio minuter. Provröret fylls med vatten, sedimentet får stå i 2–3 minuter och därefter hälls vattnet och de suspenderade partiklarna försiktigt bort.
— Sedimentet sköljs ytterligare två gånger med ca 10 ml vatten (varje gång vortexas provet i 30 sekunder, lösningen får stå och därefter hälls vattnet bort).
— Till sedimentet tillsätts 2–10 droppar alizarinlösning och blandningen vortexas. Reaktionen får pågå i 30 sekunder och det infärgade sedimentet sköljs två gånger med ca 5 ml etanol och sedan en gång med aceton (varje gång vortexas provet i 30 sekunder, lösningen får stå i ca en minut och därefter hälls lösningsmedlet bort).
— Det infärgade sedimentet torkas.
2.1.4 Undersökning i mikroskop
2.1.4.1 Preparering av objektglas
Objektglasen ska prepareras med sediment och antingen med flotat eller med råvara, enligt laborantens eget val. Om provet har siktats vid provberedningen ska båda fraktionerna (den fina och den grova) prepareras. De prov från fraktionerna som fördelas på objektglasen ska vara representativa för hela fraktionen.
Ett tillräckligt antal objektglas ska prepareras så att hela det undersökningsprotokoll som anges i punkt 2.1.4.2 kan utföras.
Objektglasen ska monteras med ett lämpligt monteringsmedium i enlighet med de standardrutiner som fastställs av EU:s referenslaboratorium för animaliska proteiner i foder och som offentliggörs på dess webbplats. Objektglasen ska täckas med täckglas.
2.1.4.2 Undersökningsprotokoll för att påvisa animaliska partiklar i foderblandningar och foderråvaror
De preparerade objektglasen ska undersökas i mikroskop i enlighet med undersökningsprotokollet i diagram 1 för foderblandningar och andra foderråvaror än rent fiskmjöl, eller med undersökningsprotokollet i diagram 2 för rent fiskmjöl.
Den mikroskopiska undersökningen ska utföras på sediment och antingen på flotat eller på råvara, enligt laborantens eget val. För de grova fraktionerna får utöver det vanliga mikroskopet även stereomikroskop användas. Varje objektglas ska undersökas i sin helhet vid olika förstoringsgrader.
Det minsta antalet objektglas som ska undersökas i varje steg av undersökningsprotokollet ska följas strikt, förutom om mängden fraktionsmaterial inte gör det möjligt att uppnå det föreskrivna antalet objektglas. Högst sex objektglas per bestämning ska undersökas.
För att underlätta identifieringen av partiklarnas typ och ursprung får hjälpmedel användas såsom beslutsstödssystem, bildbibliotek och referensprov.


2.1.4.3 Antal bestämningar
Om man efter en första bestämning, som utförts i enlighet med det relevanta undersökningsprotokollet (diagram 1 eller diagram 2), inte kan påvisa några animaliska partiklar av en viss typ (dvs. landdjur eller fisk) behövs ingen ytterligare bestämning och analysresultatet ska rapporteras med användning av ordalydelsen i punkt 2.1.5.1.
Om man efter en första bestämning, som utförts i enlighet med det relevanta undersökningsprotokollet (diagram 1 eller diagram 2), kan påvisa totalt 1–5 animaliska partiklar av en viss typ (dvs. landdjur eller fisk) ska en andra bestämning utföras med ett nytt analysprov på 50 g. Om man efter denna andra bestämning kan påvisa 0–5 animaliska partiklar av en viss typ ska analysresultatet rapporteras med användning av ordalydelsen i punkt 2.1.5.2, annars ska en tredje bestämning utföras med ett nytt analysprov på 50 g. Om summan av de partiklar av en viss typ som påvisats efter den första och andra bestämningen likväl överstiger 15 behövs ingen ytterligare bestämning och analysresultatet ska rapporteras direkt med användning av ordalydelsen i punkt 2.1.5.3. Om summan av de animaliska partiklarna av en viss typ som påvisats efter tre bestämningar överstiger 15 ska analysresultatet rapporteras med användning av ordalydelsen i punkt 2.1.5.3. I annat fall ska analysresultatet rapporteras med användning av ordalydelsen i punkt 2.1.5.2.
Om man efter en första bestämning, som utförts i enlighet med det relevanta undersökningsprotokollet (diagram 1 eller diagram 2), kan påvisa fler än fem animaliska partiklar av en viss typ (dvs. landdjur eller fisk) ska analysresultatet rapporteras med användning av ordalydelsen i punkt 2.1.5.3.
2.1.5 Redovisning av resultaten
Vid rapportering av resultaten ska laboratoriet ange på vilken typ av material analysen har gjorts (sediment, flotat eller råvara) och hur många bestämningar som utförts.
Laboratorierapporten ska åtminstone innehålla uppgifter om huruvida beståndsdelar från landdjur och fisk förekommer.
De olika fallen ska rapporteras enligt följande:
|
2.1.5.1 |
Inga animaliska partiklar av en viss typ har påvisats — I det inlämnade provet har inga partiklar från landdjur kunnat påvisas med ljusmikroskop. — I det inlämnade provet har inga partiklar från fisk kunnat påvisas med ljusmikroskop. |
|
2.1.5.2 |
I genomsnitt har 1–5 animaliska partiklar av en viss typ påvisats — I det inlämnade provet har i genomsnitt högst fem partiklar från landdjur kunnat påvisas per bestämning med ljusmikroskop. Partiklarna har identifierats som … [ben, brosk, muskelvävnad, hår, horn …]. Antalet partiklar som påvisats är så lågt att det ligger under den mikroskopiska metodens detektionsgräns, vilket innebär att risken för falskt positiva resultat inte kan uteslutas. Eller i förekommande fall: — I det inlämnade provet har i genomsnitt högst fem partiklar från fisk kunnat påvisas per bestämning med ljusmikroskop. Partiklarna har identifierats som … [fiskben, fiskfjäll, brosk, muskelvävnad, otolit, gäl …]. Antalet partiklar som påvisats är så lågt att det ligger under den mikroskopiska metodens detektionsgräns, vilket innebär att risken för falskt positiva resultat inte kan uteslutas. Om provet har siktats före beredningen ska det i laboratorierapporten anges i vilken fraktion (siktad fraktion, pelleterad fraktion eller palmkärnor) de animaliska partiklarna har påvisats. Om animaliska partiklar endast påvisas i den siktade fraktionen kan det tyda på en förorening från omgivningen. |
|
2.1.5.3 |
I genomsnitt har fler än fem animaliska partiklar av en viss typ påvisats — I det inlämnade provet har i genomsnitt fler än fem partiklar från landdjur kunnat påvisas per bestämning med ljusmikroskop. Partiklarna har identifierats som … [ben, brosk, muskelvävnad, hår, horn …]. Eller i förekommande fall: — I det inlämnade provet har i genomsnitt fler än fem partiklar från fisk kunnat påvisas per bestämning med ljusmikroskop. Partiklarna har identifierats som … [fiskben, fiskfjäll, brosk, muskelvävnad, otolit, gäl …]. Om provet har siktats före beredningen ska det i laboratorierapporten anges i vilken fraktion (siktad fraktion, pelleterad fraktion eller palmkärnor) de animaliska partiklarna har påvisats. Om animaliska partiklar endast påvisas i den siktade fraktionen kan det tyda på en förorening från omgivningen. |
2.2 PCR
2.2.1 Princip
Deoxiribonukleinsyrafragment (DNA-fragment) av animaliskt ursprung som kan förekomma i foderråvaror och foderblandningar påvisas med en genetisk amplifieringsteknik (PCR) som inriktas på artspecifika DNA-sekvenser.
För PCR krävs först en DNA-extraktion. Därefter amplifieras det erhållna DNA-extraktet för att påvisa de djurarter som metoden omfattar.
2.2.2 Reagens och utrustning
2.2.2.1 Reagens
2.2.2.1.1 Reagens för DNA-extraktion
Endast reagens som godkänns av EU:s referenslaboratorium för animaliska proteiner i foder och som offentliggörs på dess webbplats får användas.
2.2.2.1.2 Reagens för genetisk amplifiering
2.2.2.1.2.1 Primrar och prober
Endast primrar och prober med oligonukleotidsekvenser som validerats av EU:s referenslaboratorium för animaliska proteiner i foder får användas ( 33 ).
2.2.2.1.2.2 Mastermix
Endast mastermixlösningar som inte innehåller reagens som kan ge falska resultat på grund av att animaliskt DNA förekommer får användas ( 34 ).
2.2.2.1.2.3 Reagens för dekontaminering
2.2.2.1.2.3.1 Saltsyrelösning (0,1 N).
2.2.2.1.2.3.2 Blekmedel (natriumhypokloritlösning med 0,15 % aktivt klor).
2.2.2.1.2.3.3 Icke-frätande reagens för dekontaminering av dyr utrustning såsom analysvågar (t.ex. DNA Erase™ från MP Biomedicals).
2.2.2.2 Utrustning
2.2.2.2.1 Analysvåg med en noggrannhet av 0,001 g.
2.2.2.2.2 Utrustning för malning.
2.2.2.2.3 Termocykler för realtids-PCR.
2.2.2.2.4 Mikrocentrifug för mikrocentrifugrör.
2.2.2.2.5 Uppsättning mikropipetter som täcker volymintervallet 1–1 000 μl.
|
2.2.2.2.6 |
Molekylärbiologiska plastartiklar av standardtyp: mikrocentrifugrör, pipettspetsar med filter för mikropipetter och plattor som passar termocyklern. |
|
2.2.2.2.7 |
Frysar för att lagra prov och reagens. |
2.2.3 Provtagning och provberedning
2.2.3.1 Provtagning
Ett representativt prov som tagits i enlighet med bestämmelserna i bilaga I ska användas.
2.2.3.2 Provberedning
Beredningen av laboratorieprov ska fram till DNA-extraktionen uppfylla kraven i bilaga II. Minst 50 g av provet ska delas upp i analysprov och därefter malas.
Provberedningen ska i enlighet med ISO 24276 utföras i ett annat rum än det som används för DNA-extraktion och genetisk amplifiering.
Två prov på minst 100 mg vardera ska beredas.
2.2.4 DNA-extraktion
DNA ska extraheras ur varje berett prov enligt de standardrutiner som fastställs av EU:s referenslaboratorium för animaliska proteiner i foder och som offentliggörs på dess webbplats.
För varje extraktionsserie ska följande två extraktionskontroller beredas i enlighet med ISO 24276:
— Ett blankprov.
— En positiv extraktionskontroll för DNA.
2.2.5 Genetisk amplifiering
Den genetiska amplifieringen ska utföras enligt de metoder som validerats för varje art som behöver identifieras. Dessa metoder anges i de standardrutiner som fastställs av EU:s referenslaboratorium för animaliska proteiner i foder och som offentliggörs på dess webbplats. Varje DNA-extrakt ska analyseras vid minst två olika koncentrationer för att bedöma inhibering.
För varje målart ska följande två amplifieringskontroller beredas i enlighet med ISO 24276:
— En positiv kontroll för mål-DNA ska användas för varje platta eller serie av PCR-analyser.
— En reagenskontroll för amplifieringen (även kallad reagenskontroll utan templat [no template control]) ska användas för varje platta eller serie av PCR-analyser.
2.2.6 Tolkning och redovisning av resultat
Vid rapportering av resultaten ska laboratoriet åtminstone ange provens vikt, vilken extraktionsmetod som använts, antalet bestämningar som utförts och metodens detektionsgräns.
Resultaten ska inte tolkas och rapporteras om den positiva extraktionskontrollen för DNA och de positiva kontrollerna för mål-DNA inte ger positiva resultat för den berörda analysen och om reagenskontrollen för amplifikationen samtidigt är negativ.
Om resultaten från de två proven inte överensstämmer ska åtminstone den genetiska amplifieringen upprepas. Om laboratoriet misstänker att orsaken till den bristande överensstämmelsen kan vara DNA-extraktet, ska en ny DNA-extraktion och genetisk amplifiering utföras innan resultaten tolkas.
Den slutliga redovisningen av resultaten ska grundas på en kombination och tolkning av resultaten från de två proven i enlighet med de standardrutiner som fastställs av EU:s referenslaboratorium för animaliska proteiner i foder och som offentliggörs på dess webbplats.
2.2.6.1 Negativt resultat
Ett negativt resultat ska rapporteras enligt följande:
I det inlämnade provet har inget DNA från X påvisats (där X är djurarten eller gruppen av djurarter som metoden omfattar).
2.2.6.2 Positivt resultat
Ett positivt resultat ska rapporteras enligt följande:
I det inlämnade provet har DNA från X påvisats (där X är djurarten eller gruppen av djurarter som metoden omfattar).
BILAGA VII
METOD FÖR BERÄKNING AV ENERGIVÄRDET I FODER TILL FJÄDERFÄ
1. Beräkningsmetod och angivelse av energivärde
Energivärdet i foderblandningar till fjäderfä ska beräknas enligt formeln nedan på grundval av visst analyserat innehåll i procent. Värdet ska anges i megajoule (MJ) omsättbar energi (OE), med korrigering för kväve, per kilo foderblandning:
MJ/kg OE = 0,1551 × % råprotein + 0,3431 × % råfett + 0,1669 × % stärkelse + 0,1301 × % totalhalt socker (uttryckt som sackaros).
2. Toleranser som är tillämpliga på deklarerade värden
Om den officiella kontrollen visar en avvikelse (högre eller lägre energivärde i fodret) mellan kontrollresultatet och det deklarerade värdet ska en minimitolerans på 0,4 MJ/kg omsättbar energi tillåtas.
3. Resultatangivelse
Det resultat som fås med hjälp av ovanstående formel ska anges med en decimal.
4. Provtagnings- och analysmetoder
Provtagning av foderblandningen och bestämning av de beståndsdelar som anges i beräkningsmetoden ska utföras enligt gemenskapens metoder för provtagning och analys vid officiell foderkontroll.
Följande metoder ska användas:
— För bestämning av råfetthalt: metod B för bestämning av råfett i bilaga III del H.
— För bestämning av stärkelsehalt: den polarimetriska metod som anges i bilaga III del L.
BILAGA VIII
ANALYSMETODER FÖR KONTROLL AV FÖREKOMST AV OTILLÅTNA FODERTILLSATSER
Viktig anmärkning:
Känsligare analysmetoder än de som anges i denna bilaga får användas för att påvisa otillåten förekomst av fodertillsatser som inte längre är tillåtna.
Analysmetoderna i denna bilaga ska användas som konfirmeringsmetoder.
A. BESTÄMNING AV METYLBENSOKVAT
7-bensyloxi-6-butyl-3-metoxikarbonyl-4-kinolon
1. Syfte och tillämpningsområde
Med denna metod kan halten metylbensokvat i foder bestämmas. Kvantifieringsgränsen är 1 mg/kg.
2. Princip
Metylbensokvat extraheras från provet med en lösning av metansulfonsyra i metanol. Extraktet renas med diklormetan, genom jonbyteskromatografi och därefter åter med diklormetan. Metylbensokvathalten bestäms genom omvänd fas-kromatografi (HPLC) med en UV-detektor.
3. Reagens
3.1 Diklormetan
3.2 Metanol, HPLC-kvalitet
3.3 Mobil fas för HPLC
Blandning av metanol (3.2) och vatten (HPLC-kvalitet) 75 + 25 (v + v).
Filtrera lösningen genom ett 0,22 μm filter (4.5) och avlufta den (t.ex. i ultraljudsbad i 10 minuter).
3.4 Metansulfonsyralösning, c = 2 %
Späd 20,0 ml metansulfonsyra till 1 000 ml med metanol (3.2).
3.5 Saltsyrelösning, c = 10 %
Blanda 100 ml saltsyra (ρ201,18 g/ml) med vatten till 1 000 ml.
3.6 Katjonbytarharts Amberlite CG-120 (Na), 100–200 mesh
Hartset behandlas före användning: slamma upp 100 g harts i 500 ml saltsyrelösning (3.5) och värm under konstant omrörning på en värmeplatta tills det kokar. Låt svalna och dekantera av syran. Vakuumfiltrera genom ett filtrerpapper. Tvätta hartset två gånger med 500 ml vatten per gång och därefter med 250 ml metanol (3.2). Skölj hartset med ytterligare 250 ml metanol och torka genom att lufta filterkakan. Förvara det torkade hartset i en tillsluten flaska.
3.7 Referenssubstans: ren metylbensokvat (7-bensyloxi-6-butyl-3-metoxikarbonyl-4-kinolon)
3.7.1
Väg upp 50 mg metylbensokvat referenssubstans (3.7) med 0,1 mg noggrannhet. Lös upp i metansulfonsyralösning (3.4) i en 100 ml mätkolv. Fyll till märket och blanda.
3.7.2
Överför 5,0 ml stamlösning av metylbensokvat (3.7.1) till en 50 ml mätkolv. Späd till märket med metanol (3.2) och blanda.
3.7.3
Överför 1,0 –2,0 –3,0 –4,0 respektive 5,0 ml arbetsstandardlösning (3.7.2) till en serie 25 ml mätkolvar. Späd till märket med den mobila fasen (3.3) och blanda. De erhållna lösningarna har en metylbensokvatkoncentration på 2,0 –4,0 –6,0 –8,0 respektive 10,0 μg/ml. Dessa lösningar ska vara nyberedda.
4. Utrustning
4.1 Skakapparat.
4.2 Rotationsindunstare.
4.3 Glaskolonn (250 × 15 mm) med avstängningsventil och en behållare med ca 200 ml volym.
4.4 HPLC-apparat med UV-detektor med variabel våglängd eller diod-array-detektor.
4.4.1 HPLC-kolonn: 300 × 4 mm, C18, 10 μm packning eller motsvarande.
4.5 Membranfilter, 0,22 μm.
4.6 Membranfilter, 0,45 μm.
5. Utförande
5.1 Allmänt
5.1.1 Ett blankprov analyseras för att kontrollera att varken metylbensokvat eller störande ämnen är närvarande.
5.1.2 Gör ett utbytesförsök genom analys av ett blankprov som spikats med ungefär samma mängd metylbensokvat som finns i provet. För spikning på nivån 15 mg/kg, tillsätt 600 μl stamlösning (3.7.1) till 20 g av blankprovet, blanda och vänta i 10 minuter före extraktion (5.2).
Anmärkning: För denna metod bör blankprovet vara av samma typ som analysprovet och metylbensokvat ska inte kunna påvisas.
5.2 Extraktion
Väg med en noggrannhet av 0,01 g upp ca 20 gram av det beredda provet och överför till en 250 ml E-kolv. Tillsätt 100,0 ml metansulfonsyralösning (3.4) och skaka i skakapparat (4.1) i 30 minuter. Filtrera lösningen genom ett filtrerpapper och spara filtratet för vätskeextraktionen (5.3).
5.3 Vätskeextraktion
Överför 25,0 ml av filtratet (se 5.2) till en 500 ml separertratt med 100 ml saltsyrelösning (3.5). Tillsätt 100 ml diklormetan (3.1) till separertratten och skaka i 1 minut. Låt skikten separera och tappa av det undre skiktet (diklormetan) i en 500 ml rundkolv. Upprepa extraktionen av vattenfasen med ytterligare två portioner om 40 ml diklormetan och överför dessa till det första extraktet i rundkolven. Indunsta diklormetanextraktet i en rotationsindunstare (4.2) under reducerat tryck vid 40 oC tills det är torrt. Lös upp återstoden i 20–25 ml metanol (3.2), tillslut kolven och spara allt extrakt för jonbyteskromatografin (5.4).
5.4 Jonbyteskromatografi
5.4.1
För in en glasullstuss i glaskolonnens nedre del (4.3). Slamma upp 5,0 g av det behandlade katjonbytarhartset (3.6) med 50 ml saltsyra (3.5), överför till glaskolonnen och låt sedimentera. Tappa av överskottet av syra till precis över hartsytan. Tvätta kolonnen med vatten tills eluatet är lackmusneutralt. Tillsätt 50 ml metanol (3.2) till kolonnen och låt rinna ned till hartsytan.
5.4.2
Pipettera försiktigt över det extrakt som fåtts fram enligt punkt 5.3 till kolonnen. Skölj rundkolven med två portioner på 5–10 ml metanol (3.2) och överför dessa sköljvätskor till kolonnen. Låt extraktet rinna ned till hartsytan och tvätta kolonnen med 50 ml metanol. Se till att flödeshastigheten inte överstiger 5 ml per minut. Häll bort eluatet. Eluera metylbensokvatet från kolonnen med 150 ml metansulfonsyralösning (3.4) och samla upp kolonneluatet i en 250 ml E-kolv.
5.5 Vätskeextraktion
Överför det eluat som erhållits enligt punkt 5.4.2 till en 1 liters separertratt. Skölj E-kolven med 5–10 ml metanol (3.2) och blanda sköljvätskorna med innehållet i separertratten. Tillsätt 300 ml saltsyrelösning (3.5) och 130 ml diklormetan (3.1). Skaka i 1 minut och låt faserna separera. Tappa av det undre skiktet (diklormetan) i en 500 ml rundkolv. Upprepa extraktionen i vattenfasen med ytterligare två portioner på 70 ml diklormetan och blanda dessa extrakt med det första i rundkolven.
Indunsta diklormetanextraktet i rotationsindunstaren (4.2) under reducerat tryck vid 40 oC tills det är torrt. Lös upp återstoden i kolven i ca 5 ml metanol (3.2) och överför denna lösning kvantitativt till en 10 ml mätkolv. Skölj rundkolven med ytterligare två portioner på 1–2 ml metanol och överför dessa till mätkolven. Späd med metanol till märket och blanda. Filtrera en alikvot av lösningen genom ett membranfilter (4.6). Spara denna lösning för HPLC-bestämning (5.6).
5.6 HPLC-bestämning
5.6.1
Följande betingelser anges som vägledning. Andra betingelser kan väljas om de ger likvärdiga resultat.
— HPLC-kolonn (4.4.1).
— Mobil fas för HPLC: metanol-vattenblandning (3.3).
— Flödeshastighet: 1–1,5 ml/minut.
— Detektionsvåglängd: 265 nm.
— Injektionsvolym: 20–50 μl.
Kontrollera att kromatografisystemet är stabilt genom att upprepade gånger injicera kalibreringslösningen (3.7.3) på 4 μg/ml, till dess att topphöjder (areor) och retentionstider är konstanta.
5.6.2
Injicera varje kalibreringslösning (3.7.3) upprepade gånger och mät topphöjderna (areorna) för varje koncentration. Konstruera en kalibreringskurva med kalibreringslösningarnas genomsnittliga topphöjder (areor) längs y-axeln och motsvarande koncentrationer i μg/ml längs x-axeln.
5.6.3
Injicera provextraktet (5.5) upprepade gånger med samma volym som för kalibreringslösningarna, och bestäm den genomsnittliga topphöjden (arean) för metylbensokvat.
6. Beräkning av resultat
Använd den genomsnittliga topphöjden (arean) för provlösningens metylbensokvattoppar för att bestämma koncentrationen i μg/ml med hjälp av kalibreringskurvan (5.6.2).
Provets metylbensokvathalt w (mg/kg) beräknas med formeln

där:
|
c |
= |
provlösningens metylbensokvatkoncentration i μg/ml |
|
m |
= |
provets vikt i gram. |
7. Validering av resultat
7.1 Identitet
Analytens identitet kan bekräftas genom co-kromatografi eller genom användning av en diod-array-detektor med vars hjälp spektrumen för provextraktet och den kalibreringslösning (3.7.3) som innehåller 10 μg/ml jämförs.
7.1.1
Ett provextrakt spikas genom tillsats av lämplig mängd arbetsstandardlösning (3.7.2). Mängden tillsatt metylbensokvat bör vara ungefär lika stor som den uppskattade mängden metylbensokvat i provextraktet.
Endast metylbensokvattoppens höjd ska öka efter det att hänsyn tagits till både den tillsatta mängden och spädningen av extraktet. Toppens bredd på halva höjden ska ligga inom 10 % av den ursprungliga bredden.
7.1.2
Resultaten utvärderas enligt följande kriterier:
a) Våglängderna för prov- och standardspektrumens absorptionsmaximum, i toppens spets på kromatogrammet, ska ligga inom en marginal som beror på detektionssystemets upplösningsförmåga. För diod-array-detektion är denna i allmänhet ± 2 nm.
b) Mellan 220 och 350 nm får prov- och standardspektrumen, i toppens spets på kromatogrammet, inte skilja sig åt för de delar av spektrumet där den relativa absorbansen är 10–100 %. Detta kriterium är uppfyllt om samma maximum observeras och avvikelsen mellan de två spektrumen inte vid någon mätpunkt överstiger 15 % av standardanalytens absorbans.
c) Mellan 220 och 350 nm får spektrumen för den uppåtgående delen, spetsen och den nedåtgående delen av toppen från provextraktet inte skilja sig från varandra för de delar av spektrumet där den relativa absorbansen är 10–100 %. Detta kriterium är uppfyllt om samma maximum observeras och avvikelsen mellan spektrumen inte vid någon mätpunkt överstiger 15 % av absorbansen vid spetsen.
Om något av dessa kriterier inte är uppfyllt har analytens förekomst inte bekräftats.
7.2 Repeterbarhet
Skillnaden mellan resultaten från två parallella bestämningar som utförs på samma prov får inte överstiga 10 % av det högre värdet för metylbensokvathalter på 4–20 mg/kg.
7.3 Utbyte
För ett spikat blankprov ska utbytet vara minst 90 %.
8. Resultat av en provningsjämförelse
Fem prov analyserades av tio laboratorier. Varje prov analyserades två gånger.
|
|
Blankprov |
Mjöl 1 |
Pellet 1 |
Mjöl 2 |
Pellet 2 |
|
Medelvärde [mg/kg] |
ND |
4,50 |
4,50 |
8,90 |
8,70 |
|
sr [mg/kg] |
–– |
0,30 |
0,20 |
0,60 |
0,50 |
|
CVr [ %] |
–– |
6,70 |
4,40 |
6,70 |
5,70 |
|
sR [mg/kg] |
–– |
0,40 |
0,50 |
0,90 |
1,00 |
|
CVR [ %] |
–– |
8,90 |
11,10 |
10,10 |
11,50 |
|
Utbyte [ %] |
–– |
92,00 |
93,00 |
92,00 |
89,00 |
|
ND |
= |
analyten har ej påvisats |
|
sr |
= |
standardavvikelse för repeterbarhet |
|
CVr |
= |
variationskoefficient för repeterbarhet, % |
|
sR |
= |
standardavvikelse för reproducerbarhet |
|
CVR |
= |
variationskoefficient för reproducerbarhet, % |
B. BESTÄMNING AV OLAKINDOX
2-[N-2'-(hydroxietyl)karbamoyl]-3-metylkinoxalin-N1,N4-dioxid
1. Syfte och tillämpningsområde
Med denna metod kan halten olakindox i foder bestämmas. Kvantifieringsgränsen är 5 mg/kg.
2. Princip
Provet extraheras med en vatten-metanolblandning. Olakindoxhalten bestäms genom omvänd fas-kromatografi (HPLC) med en UV-detektor.
3. Reagens
3.1 Metanol.
3.2 Metanol, HPLC-kvalitet.
3.3 Vatten, HPLC-kvalitet.
3.4 Mobil fas för HPLC.
Blandning av vatten (3.3) och metanol (3.2), 900 + 100 (v + v).
3.5 Referenssubstans: ren olakindox 2-[N-2'-(hydroxietyl)karbamoyl]-3-metylkinoxalin-N1,N4-dioxid, E 851.
3.5.1
Väg med 0,1 mg noggrannhet upp 50 mg olakindox (3.5) i en 200 ml mätkolv och tillsätt ca 190 ml vatten. Ställ kolven i ett ultraljudsbad (4.1) i 20 minuter. Låt därefter lösningen anta rumstemperatur, späd till märket med vatten och blanda. Slå in mätkolven i aluminiumfolie och förvara den i kylskåp. Denna lösning ska beredas på nytt varje månad.
3.5.2
Överför 10,0 ml stamlösning (3.5.1) till en 100 ml mätkolv, späd till märket med den mobila fasen (3.4) och blanda. Slå in mätkolven i aluminiumfolie och förvara den i kylskåp. Denna lösning ska beredas på nytt varje dag.
3.5.3
Överför 1,0 –2,0 –5,0 –10,0 –15,0 respektive 20,0 ml arbetsstandardlösning (3.5.2) till ett antal 50 ml mätkolvar. Späd till märket med den mobila fasen (3.4) och blanda. Slå in mätkolvarna i aluminiumfolie. Dessa lösningar har en olakindoxhalt på 0,5 –1,0 –2,5 –5,0 –7,5 respektive 10,0 μg/ml.
Lösningarna ska beredas på nytt varje dag.
4. Utrustning
4.1 Ultraljudsbad.
4.2 Mekanisk skakapparat.
4.3 HPLC-utrustning med UV-detektor med variabel våglängd eller diod-array-detektor.
4.3.1 HPLC-kolonn, 250 × 4 mm, C18, 10 μm packning eller motsvarande.
4.4 Membranfilter, 0,45 μm.
5. Utförande
|
Anmärkning: |
Olakindox är ljuskänsligt. Utför alla moment i dämpad belysning eller använd bruna glaskärl. |
5.1 Allmänt
5.1.1 Ett blankprov analyseras för att kontrollera att varken olakindox eller störande ämnen är närvarande.
5.1.2 Gör ett utbytesförsök genom analys av ett blankprov som spikats med ungefär samma mängd olakindox som finns i provet. För spikning på nivån 50 mg/kg, överför 10,0 ml stamlösning (3.5.1) till en 250 ml E-kolv och indunsta till ca 0,5 ml. Tillsätt 50 g av blankprovet, blanda ordentligt och låt stå i 10 minuter och rör sedan om igen flera gånger före extraktionen (5.2).
|
Anmärkning: |
För denna metod bör blankprovet vara av samma typ som analysprovet och olakindox ska inte kunna påvisas. |
5.2 Extraktion
Väg med 0,01 g noggrannhet upp ca 50 g av provet. Överför materialet till en 1 000 ml E-kolv, tillsätt 100 ml metanol (3.1) och ställ kolven i ett ultraljudsbad (4.1) i 5 minuter. Tillsätt 410 ml vatten och låt kolven stå i badet ytterligare 15 minuter. Ta kolven ur badet, skaka den i 30 minuter i skakapparaten (4.2) och filtrera genom ett veckat filter. Överför 10,0 ml av filtratet till en 20 ml mätkolv, späd med vatten till märket och blanda. Filtrera en alikvot genom ett membranfilter (4.4). (Jfr punkt 9.) Gå vidare till HPLC-bestämningen (5.3).
5.3 HPLC-bestämning
5.3.1
Följande betingelser anges som vägledning. Andra betingelser kan väljas om de ger likvärdiga resultat.
|
Analyskolonn (4.3.1) |
|
|
Mobil fas (3.4): |
blandning av vatten (3.3) och metanol (3.2), 900 + 100 (v + v). |
|
Flödeshastighet: |
1,5 –2 ml/min. |
|
Detektionsvåglängd: |
380 nm. |
|
Injektionsvolym: |
20–100 μl. |
Kontrollera att kromatografisystemet är stabilt genom att upprepade gånger injicera kalibreringslösningen (3.5.3) på 2,5 μg/ml, till dess att topphöjder och retentionstider är konstanta.
5.3.2
Injicera varje kalibreringslösning (3.5.3) upprepade gånger och bestäm de genomsnittliga topphöjderna (areorna) för varje koncentration. Konstruera en kalibreringskurva med kalibreringslösningarnas genomsnittliga topphöjd (area) längs y-axeln och motsvarande koncentrationer i μg/ml längs x-axeln.
5.3.3
Injicera provextraktet (5.2) upprepade gånger med samma volym som för kalibreringslösningarna och bestäm den genomsnittliga topphöjden (arean) för olakindox.
6. Beräkning av resultat
Använd den genomsnittliga topphöjden (arean) för provlösningens olakindoxtoppar för att bestämma koncentrationen i μg/ml med hjälp av kalibreringskurvan.
Provets olakindoxhalt w (mg/kg) beräknas med formeln:
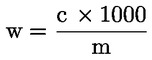
där:
|
c |
= |
provextraktets olakindoxkoncentration (5.2) i μg/ml |
|
m |
= |
provets vikt i g (5.2). |
7. Validering av resultat
7.1 Identitet
Analytens identitet kan bekräftas genom co-kromatografi eller genom användning av en diod-array-detektor med vars hjälp spektrumen för provextraktet (5.2) och den kalibreringslösning (3.5.3) som innehåller 5,0 μg/ml jämförs.
7.1.1
Ett provextrakt (5.2) spikas genom tillsats av lämplig mängd kalibreringslösning (3.5.3). Mängden tillsatt olakindox bör vara ungefär lika stor som mängden olakindox i provextraktet.
Endast olakindoxtoppens höjd ska öka efter det att hänsyn tagits till både den tillsatta mängden och spädningen av extraktet. Toppens bredd på halva höjden ska ligga inom ± 10 % av den ursprungliga bredden på olakindoxtoppen i det ospikade provextraktet.
7.1.2
Resultaten utvärderas enligt följande kriterier:
a) Våglängderna för prov- och standardspektrumens absorptionsmaximum, i toppens spets på kromatogrammet, ska ligga inom en marginal som beror på detektionssystemets upplösningsförmåga. För diod-array-detektion är denna i allmänhet ± 2 nm.
b) Mellan 220 och 400 nm får prov- och standardspektrumen, i toppens spets på kromatogrammet, inte skilja sig åt för de delar av spektrumet där den relativa absorbansen är 10–100 %. Detta kriterium är uppfyllt om samma maximum observeras och avvikelsen mellan de två spektrumen inte vid någon mätpunkt överstiger 15 % av standardanalytens absorbans.
c) Mellan 220 och 400 nm får spektrumen för den uppåtgående delen, spetsen och den nedåtgående delen av toppen från provextraktet inte skilja sig från varandra för de delar av spektrumet där den relativa absorbansen är 10–100 %. Detta kriterium är uppfyllt om samma maximum observeras och avvikelsen mellan de två spektrumen inte vid någon mätpunkt överstiger 15 % av absorbansen vid spetsen.
Om något av dessa kriterier inte är uppfyllt har analytens förekomst inte bekräftats.
7.2 Repeterbarhet
Skillnaden mellan resultaten från två parallella bestämningar som utförs på samma prov får inte överstiga 15 % av det högre värdet för olakindoxhalter på 10–200 mg/kg.
7.3 Utbyte
För ett spikat blankprov ska utbytet vara minst 90 %.
8. Resultat av en provningsjämförelse
I en provningsjämförelse inom EU analyserades fyra prov från smågrisfoder, inklusive ett blankprov, av upp till tretton laboratorier. Resultaten redovisas i nedanstående tabell.
|
|
Prov 1 |
Prov 2 |
Prov 3 |
Prov 4 |
|
L |
13 |
10 |
11 |
11 |
|
n |
40 |
40 |
44 |
44 |
|
Medelvärde [mg/kg] |
— |
14,6 |
48,0 |
95,4 |
|
Sr [mg/kg] |
— |
0,82 |
2,05 |
6,36 |
|
SR [mg/kg] |
— |
1,62 |
4,28 |
8,42 |
|
CVr [ %] |
— |
5,6 |
4,3 |
6,7 |
|
CVR [ %] |
— |
11,1 |
8,9 |
8,8 |
|
Nominell halt |
|
|
|
|
|
[mg/kg] |
— |
15 |
50 |
100 |
|
Utbyte % |
— |
97,3 |
96,0 |
95,4 |
|
L |
= |
antal laboratorier |
|
n |
= |
antal enskilda värden |
|
Sr |
= |
standardavvikelse för repeterbarhet |
|
SR |
= |
standardavvikelse för reproducerbarhet |
|
CVr |
= |
variationskoefficient för repeterbarhet |
|
CVR |
= |
variationskoefficient för reproducerbarhet. |
9. Anmärkning
Även om metoden inte har validerats för foder med en olakindoxhalt över 100 mg/kg, bör man kunna få tillfredsställande resultat genom att ta en mindre mängd prov och/eller späda extraktet (5.2) så att man får en koncentration inom kalibreringskurvans område.
C. BESTÄMNING AV AMPROLIUM
1-[(4-amino-2-propyl-5-pyrimidylmetyl]-2-metyl-pyridiniumkloridhydroklorid
1. Syfte och tillämpningsområde
Med denna metod kan amproliumhalten i foder och förblandningar bestämmas. Detektionsgränsen är 1 mg/kg och kvantifieringsgränsen 5 mg/kg.
2. Princip
Provet extraheras med en metanol-vattenblandning. Efter spädning med den mobila fasen och membranfiltrering bestäms amproliumhalten genom katjonbyteskromatografi (HPLC) med UV-detektor.
3. Reagens
3.1 Metanol.
3.2 Acetonitril, HPLC-kvalitet.
3.3 Vatten, HPLC-kvalitet.
3.4 Natriumdivätefosfatlösning, c = 0,1 mol/l.
Lös 13,80 g natriumdivätefosfatmonohydrat i vatten (3.3) i en 1 000 ml mätkolv, späd till märket med vatten (3.3) och blanda.
3.5 Natriumperkloratlösning, c =1,6 mol/l.
Lös 224,74 g natriumperkloratmonohydrat i vatten (3.3) i en 1 000 ml mätkolv, späd till märket med vatten (3.3) och blanda.
3.6 Mobil fas för HPLC (jfr punkt 9.1).
Blandning av acetonitril (3.2), natriumdivätefosfatlösning (3.4) och natriumperkloratlösning (3.5), 450+450+100 (v+v+v). Filtrera lösningen genom ett 0,22 μm membranfilter (4.3) och avlufta den (t.ex. i ultraljudsbad [4.4] i minst 15 minuter) före användning.
3.7 Referenssubstans: rent amprolium, 1-[(4-amino-2-propylpyrimidin-5-yl)metyl]-2-metyl-pyridiniumkloridhydroklorid, E 750 (jfr punkt 9.2).
3.7.1
Väg med 0,1 mg noggrannhet upp 50 mg amprolium (3.7) i en 100 ml mätkolv, lös upp i 80 ml metanol (3.1) och ställ kolven i ett ultraljudsbad i 10 min (4.4). Låt därefter lösningen anta rumstemperatur, späd till märket med vatten och blanda. Lösningen är stabil i en månad vid högst 4 oC.
3.7.2
Pipettera över 5,0 ml stamlösning (3.7.1) till en 50 ml mätkolv, späd till märket med extraktionsmedlet (3.8) och blanda. Lösningen är stabil i en månad vid högst 4 oC.
3.7.3
Överför 0,5 –1,0 respektive 2,0 ml arbetsstandardlösning (3.7.2) till tre 50 ml mätkolvar. Späd till märket med den mobila fasen (3.6) och blanda. De erhållna lösningarna har en amproliumhalt på 0,5 –1,0 respektive 2,0 μg/ml. Dessa lösningar ska vara nyberedda.
3.8 Extraktionsmedel.
Blandning av metanol (3.1) och vatten 2+1 (v+v).
4. Utrustning
4.1 HPLC-utrustning med injektionssystem avpassad för injektionsvolymer på 100 μl.
4.1.1 HPLC-kolonn 125 × 4 mm, katjonbytare Nucleosil 10 SA, 5 eller 10 μm packning, eller motsvarande.
4.1.2 UV-detektor med variabel våglängd eller diod-array-detektor.
4.2 Membranfilter, PTFE-material, 0,45 μm.
4.3 Membranfilter, 0,22 μm.
4.4 Ultraljudsbad.
4.5 Mekanisk skakapparat eller magnetomrörare.
5. Utförande
5.1 Allmänt
5.1.1
Inför utbytesförsöket (5.1.2) analyseras ett blankprov för att kontrollera att varken amprolium eller störande ämnen är närvarande. Blankprovet bör vara av liknande typ som analysprovet och varken amprolium eller störande ämnen ska kunna påvisas i det.
5.1.2
Gör ett utbytesförsök genom analys av ett blankprov som spikats med ungefär samma mängd amprolium som finns i provet. För spikning på nivån 100 mg/kg, överför 10,0 ml stamlösning (3.7.1) till en 250 ml kolv och indunsta till ca 0,5 ml. Tillsätt 50 g av blankprovet, blanda ordentligt och låt stå i 10 minuter och rör sedan om igen flera gånger före extraktionen (5.2).
Om ett blankprov av liknande typ som provet inte finns att tillgå (se 5.1.1), kan ett utbytesförsök göras med standardadditionsmetoden. Det prov som ska analyseras spikas då med ungefär samma mängd amprolium som finns i provet. Detta prov analyseras tillsammans med det ospikade provet och utbytet kan beräknas genom subtraktion.
5.2 Extraktion
5.2.1
Väg med 0,01 g noggrannhet upp 5–40 g av provet, beroende på amproliumhalten, i en 500 ml E-kolv och tillsätt 200 ml extraktionsmedel (3.8). Ställ kolven i ultraljudsbad (4.4) och låt stå i 15 minuter. Ta kolven ur badet och skaka eller rör om i 1 timme med hjälp av skakapparat eller magnetomrörare (4.5). Späd en alikvot av extraktet med den mobila fasen (3.6) till en amproliumhalt på 0,5 –2 μg/ml och blanda (se anmärkning 9.3). Filtrera 5–10 ml av denna utspädda lösning genom ett membranfilter (4.2). Gå vidare till HPLC-bestämningen (5.3).
5.2.2
Väg med 0,001 g noggrannhet upp 1–4 g av förblandningen, beroende på amproliumhalten, i en 500 ml E-kolv och tillsätt 200 ml extraktionsmedel (3.8). Ställ kolven i ultraljudsbad (4.4) och låt stå i 15 minuter. Ta kolven ur badet och skaka eller rör om i 1 timme med hjälp av skakapparat eller magnetomrörare (4.5). Späd en alikvot av extraktet med den mobila fasen (3.6) till en amproliumhalt på 0,5 –2 μg/ml och blanda. Filtrera 5–10 ml av denna utspädda lösning genom ett membranfilter (4.2). Gå vidare till HPLC-bestämningen (5.3).
5.3 HPLC-bestämning
5.3.1
Följande betingelser anges som vägledning. Andra betingelser kan väljas om de ger likvärdiga resultat.
|
HPLC-kolonn (4.1.1): |
125 × 4 mm, katjonbytare Nucleosil 10 SA, 5 eller 10 μm packning, eller motsvarande. |
|
Mobil fas (3.6): |
Blandning av acetonitril (3.2), natriumdivätefosfatlösning (3.4) och natriumperkloratlösning (3.5), 450+450+100 (v+v+v). |
|
Flödeshastighet: |
0,7 –1 ml/min |
|
Detektionsvåglängd: |
264 nm |
|
Injektionsvolym: |
100 μl |
Kontrollera att kromatografisystemet är stabilt genom att upprepade gånger injicera kalibreringslösningen (3.7.3) på 1,0 μg/ml, till dess att topphöjder och retentionstider är konstanta.
5.3.2
Injicera varje kalibreringslösning (3.7.3) upprepade gånger och bestäm de genomsnittliga topphöjderna (areorna) för varje koncentration. Konstruera en kalibreringskurva med kalibreringslösningarnas genomsnittliga topphöjd (area) längs y-axeln och motsvarande koncentrationer i μg/ml längs x-axeln.
5.3.3
Injicera provextraktet (5.2) upprepade gånger med samma volym som för kalibreringslösningarna och bestäm den genomsnittliga topphöjden (arean) för amprolium.
6. Beräkning av resultat
Använd den genomsnittliga topphöjden (arean) för provlösningens amproliumtoppar för att bestämma koncentrationen i g/ml med hjälp av kalibreringskurvan.
Provets amproliumhalt w (mg/kg) beräknas med formeln

[mg/kg]
där:
|
V |
= |
extraktionsmedlets (3.8) volym i ml enligt 5.2 (dvs. 200 ml) |
|
c |
= |
provextraktets amproliumkoncentration (5.2) i μg/ml |
|
f |
= |
spädningsfaktor enligt 5.2 |
|
m |
= |
provets vikt i gram. |
7. Validering av resultat
7.1 Identitet
Analytens identitet kan bekräftas genom co-kromatografi eller genom användning av en diod-array-detektor med vars hjälp spektrumen för provextraktet (5.2) och den kalibreringslösning (3.7.3) som innehåller 2,0 μg/ml jämförs.
7.1.1
Ett provextrakt (5.2) spikas genom tillsats av en lämplig mängd kalibreringslösning (3.7.3). Mängden tillsatt amprolium bör vara ungefär lika stor som mängden amprolium i provextraktet.
Endast amproliumtoppens höjd ska öka efter det att hänsyn tagits till både den tillsatta mängden och spädningen av extraktet. Toppens bredd ska på halva höjden ligga inom ± 10 % av den ursprungliga bredden på amproliumtoppen i det ospikade provextraktet.
7.1.2
Resultaten utvärderas enligt följande kriterier:
a) Våglängderna för prov- och standardspektrumens absorptionsmaximum, i toppens spets på kromatogrammet, ska ligga inom en marginal som beror på detektionssystemets upplösningsförmåga. För diod-array-detektion är denna i allmänhet ± 2 nm.
b) Mellan 210 och 320 nm får prov- och standardspektrumen, i toppens spets på kromatogrammet, inte skilja sig åt för de delar av spektrumet där den relativa absorbansen är 10–100 %. Detta kriterium är uppfyllt om samma maximum observeras och avvikelsen mellan de två spektrumen inte vid någon mätpunkt överstiger 15 % av standardanalytens absorbans.
c) Mellan 210 och 320 nm får spektrumen för den uppåtgående delen, spetsen och den nedåtgående delen av toppen från provextraktet inte skilja sig från varandra för de delar av spektrumet där den relativa absorbansen är 10–100 %. Detta kriterium är uppfyllt om samma maximum observeras och avvikelsen mellan de två spektrumen inte vid någon mätpunkt överstiger 15 % av absorbansen vid spetsen.
Om något av dessa kriterier inte är uppfyllt har analytens förekomst inte bekräftats.
7.2 Repeterbarhet
Skillnaden mellan resultaten från två parallella bestämningar som utförs på samma prov får inte överstiga
— 15 % av det högre värdet för amproliumhalter på 25–500 mg/kg,
— 75 mg/kg för amproliumhalter på 500–1 000 mg/kg,
— 7,5 % av det högre värdet för amproliumhalter på över 1 000 mg/kg.
7.3 Utbyte
För ett spikat (blank)prov ska utbytet vara minst 90 %.
8. Resultat av en provningsjämförelse
I en provningsjämförelse analyserades tre olika fjäderfäfoder (prov 1–3), ett mineralfoder (prov 4) och en förblandning (prov 5). Resultaten redovisas i nedanstående tabell.
|
|
Prov 1 (blankprov) |
Prov 2 |
Prov 3 |
Prov 4 |
Prov 5 |
|
L |
14 |
14 |
14 |
14 |
15 |
|
n |
56 |
56 |
56 |
56 |
60 |
|
Medelvärde [mg/kg] |
— |
45,5 |
188 |
5 129 |
25 140 |
|
sr [mg/kg] |
— |
2,26 |
3,57 |
178 |
550 |
|
CVr [ %] |
— |
4,95 |
1,90 |
3,46 |
2,20 |
|
sR [mg/kg] |
— |
2,95 |
11,8 |
266 |
760 |
|
CVR [ %] |
— |
6,47 |
6,27 |
5,19 |
3,00 |
|
Nominell halt [mg/kg] |
— |
50 |
200 |
5 000 |
25 000 |
|
L |
= |
antal laboratorier |
|
n |
= |
antal enskilda värden |
|
sr |
= |
standardavvikelse för repeterbarhet |
|
CVr |
= |
variationskoefficient för repeterbarhet |
|
sR |
= |
standardavvikelse för reproducerbarhet |
|
CVR |
= |
variationskoefficient för reproducerbarhet. |
9. Anmärkningar
9.1 Om provet innehåller tiamin kommer tiamintoppen i kromatogrammet att ligga strax före amproliumtoppen. Med denna metod måste amprolium och tiamin separeras. Om amprolium och tiamin inte separeras med den kolonn (4.1.1) som används för den här metoden ska upp till 50 % av acetonitrildelen av den mobila fasen (3.6) ersättas med metanol.
9.2 Enligt den brittiska farmakopén har amproliumlösningens spektrum (c = 0,02 mol/l) i saltsyra (c = 0,1 mol/l) maximum vid 246 och 262 nm. Absorbansen ska vara 0,84 vid 246 nm och 0,80 vid 262 nm.
9.3 Extraktet ska alltid spädas med den mobila fasen, annars kan retentionstiden för amproliumtoppen förändras väsentligt på grund av ändrad jonstyrka.
D. BESTÄMNING AV KARBADOX
Metyl-3-(2-kinoxalinylmetylen)karbazat-N 1 ,N 4 -dioxid
1. Syfte och tillämpningsområde
Med denna metod kan karbadoxhalten i foder, förblandningar och foderberedningar bestämmas. Detektionsgränsen är 1 mg/kg och kvantifieringsgränsen 5 mg/kg.
2. Princip
Provet jämviktas med vatten och extraheras med metanol-acetonitril. För foder renas en alikvot av det filtrerade extraktet på en aluminiumoxidkolonn. För förblandningar och foderberedningar späds en alikvot av det filtrerade extraktet till lämplig koncentration med vatten, metanol och acetonitril. Karbadoxhalten bestäms genom omvänd fas-kromatografi (HPLC) med en UV-detektor.
3. Reagens
3.1 Metanol.
3.2 Acetonitril, HPLC-kvalitet.
3.3 Ättiksyra, w = 100 %.
3.4 Aluminiumoxid: neutral, aktivitetsgrad I.
3.5 Metanol-acetonitril 1 + 1 (v + v).
Blanda 500 ml metanol (3.1) med 500 ml acetonitril (3.2).
3.6 Ättiksyra, σ = 10 %.
Späd 10 ml ättiksyra (3.3) till 100 ml med vatten.
3.7 Natriumacetat.
3.8 Vatten, HPLC-kvalitet.
3.9 Acetatbuffertlösning, c = 0,01 mol/l, pH = 6,0 .
Lös 0,82 g natriumacetat (3.7) i 700 ml vatten (3.8) och justera pH till 6,0 med ättiksyra (3.6). Överför till en 1 000 ml mätkolv, späd med vatten (3.8) till märket och blanda.
3.10 Mobil fas för HPLC.
Blanda 825 ml acetatbuffert (3.9) med 175 ml acetonitril (3.2).
Filtrera lösningen genom ett 0,22 μm filter (4.5) och avlufta den (t.ex. i ultraljudsbad i 10 minuter).
3.11 Referenssubstans.
Ren karbadox: Metyl-3-(2-kinoxalinylmetylen)karbazat-N1,N4-dioxid, E 850.
3.11.1
Väg med 0,1 mg noggrannhet upp 25 mg av karbadox referenssubstans (3.11) i en 250 ml mätkolv. Lös i metanol-acetonitril (3.5) med hjälp av ultraljud (4.7). Låt lösningen därefter anta rumstemperatur, späd till märket med metanol-acetonitril (3.5) och blanda. Slå in kolven i aluminiumfolie eller använd bruna glaskärl. Förvara i kylskåp. Lösningen är stabil i en månad vid högst 4 oC.
3.11.2
Överför 2,0 –5,0 –10,0 respektive 20,0 ml stamlösning (3.11.1) till ett antal 100 ml mätkolvar. Tillsätt 30 ml vatten, späd till märket med metanol-acetonitril (3.5) och blanda. Slå in mätkolvarna i aluminiumfolie. Dessa lösningar har en karbadoxhalt på 2,0 –5,0 –10,0 respektive 20,0 μg/ml.
Kalibreringslösningarna ska vara nyberedda.
|
Anmärkning: |
För bestämning av karbadox i foder som innehåller mindre än 10 mg/kg ska kalibreringslösningar med en koncentration under 2,0 μg/ml användas. |
3.12 Blandning av vatten och [metanol-acetonitril] (3.5), 300 + 700 (v + v).
Blanda 300 ml vatten med 700 ml metanol-acetonitrilblandning (3.5).
4. Utrustning
4.1 Skakapparat eller magnetomrörare.
4.2 Glasfiberfilter (Whatman GF/A eller motsvarande).
4.3 Glaskolonn (längd 300–400 mm, inre diameter ca 10 mm) med sintrat glas och med avtappningskran.
|
Anmärkning: |
En glaskolonn med stängningsventil eller med avsmalnande ände kan också användas. I så fall ska en glasullstuss föras in i kolvens nedre del med hjälp av en glasstav. |
4.4 HPLC-utrustning med injektionssystem, avpassad för injektionsvolymer på 20 μl.
4.4.1 HPLC-kolonn: 300 × 4 mm, C18, 10 μm packning eller motsvarande.
4.4.2 UV-detektor med variabel våglängd eller diod-array-detektor för intervallet 225–400 nm.
4.5 Membranfilter, 0,22 μm.
4.6 Membranfilter, 0,45 μm.
4.7 Ultraljudsbad.
5. Utförande
|
Anmärkning: |
Karbadox är ljuskänsligt. Utför alla moment i dämpad belysning eller använd bruna glaskärl eller glaskärl insvepta i aluminiumfolie. |
5.1 Allmänt
5.1.1
Inför utbytesförsöket (5.1.2) analyseras ett blankprov för att kontrollera att varken karbadox eller störande ämnen är närvarande. Blankprovet bör vara av liknande typ som analysprovet och varken karbadox eller störande ämnen ska kunna påvisas i det.
5.1.2
Gör ett utbytesförsök genom analys av ett blankprov (5.1.1) som spikats med ungefär samma mängd karbadox som finns i provet. För spikning på nivån 50 mg/kg, överför 5,0 ml stamlösning (3.11.1) till en 200 ml E-kolv. Indunsta lösningen till ca 0,5 ml i ett kvävgasflöde. Tillsätt 10 g av blankprovet, blanda och vänta i 10 minuter före extraktion (5.2).
Om ett blankprov av liknande typ som provet inte finns att tillgå (se 5.1.1), kan ett utbytesförsök göras med standardadditionsmetoden. Det prov som ska analyseras spikas då med ungefär samma mängd karbadox som finns i provet. Detta prov analyseras tillsammans med det ospikade provet och utbytet kan beräknas genom subtraktion.
5.2 Extraktion
5.2.1
Väg med 0,01 g noggrannhet upp 10 gram av provet och för över till en 200 ml E-kolv. Tillsätt 15,0 ml vatten, blanda och jämvikta i 5 minuter. Tillsätt 35,0 ml metanol-acetonitrilblandning (3.5), tillslut kolven och skaka eller rör om i 30 minuter med hjälp av skakapparat eller magnetomrörare (4.1). Filtrera lösningen genom ett glasfiberfilter (4.2). Spara lösningen för reningen (5.3).
5.2.2
Väg med 0,001 g noggrannhet upp 1 g omalt prov och överför till en 200 ml E-kolv. Tillsätt 15,0 ml vatten, blanda och jämvikta i 5 minuter. Tillsätt 35,0 ml metanol-acetonitrilblandning (3.5), tillslut kolven och skaka eller rör om i 30 minuter med hjälp av skakapparat eller magnetomrörare (4.1). Filtrera lösningen genom ett glasfiberfilter (4.2).
Pipettera över en alikvot av filtratet till en 50 ml mätkolv. Tillsätt 15,0 ml vatten, fyll till märket med metanol-acetonitril (3.5) och blanda. Karbadoxkoncentrationen i den slutliga lösningen bör vara ca 10 μg/ml. Filtrera en alikvot genom ett 0,45 μm membranfilter (4.6).
Gå vidare till HPLC-bestämningen (5.4).
5.2.3
Väg med 0,001 g noggrannhet upp 0,2 g omalt prov och för över till en 250 ml E-kolv. Tillsätt 45,0 ml vatten, blanda och jämvikta i 5 minuter. Tillsätt 105,0 ml metanol-acetonitrilblandning (3.5), tillslut kolven och homogenisera. Ultraljudsbehandla (4.7) provet i 15 minuter och skaka eller rör om i 15 minuter (4.1). Filtrera lösningen genom ett glasfiberfilter (4.2).
Späd en alikvot av filtratet med vatten-metanol-acetonitrilblandningen (3.12) till en slutlig karbadoxkoncentration på 10–15 μg/ml (för en 10 % lösning blir spädningsfaktorn 10). Filtrera en alikvot genom ett 0,45 μm membranfilter (4.6).
Gå vidare till HPLC-bestämningen (5.4).
5.3 Rening
5.3.1
Väg upp 4 g aluminiumoxid (3.4) och överför det till glaskolonnen (4.3).
5.3.2
Tillsätt 15 ml av det filtrerade extraktet (5.2.1) i aluminiumoxidkolonnen och häll bort de första 2 ml eluat. Samla upp följande 5 ml och filtrera en alikvot genom ett 0,45 m filter (4.6).
Gå vidare till HPLC-bestämningen (5.4).
5.4 HPLC-bestämning
5.4.1
Följande betingelser anges som vägledning. Andra betingelser kan väljas om de ger likvärdiga resultat.
|
HPLC-kolonn (4.4.1): |
300 × 4 mm, C18, 10 μm packning eller motsvarande |
|
Mobil fas (3.10): |
Blandning av acetatbuffertlösning (3.9) och acetonitril (3.2), 825 + 175 (v+v) |
|
Flödeshastighet: |
1,5 –2 ml/min |
|
Detektionsvåglängd: |
365 nm |
|
Injektionsvolym: |
20 μl |
Kontrollera att det kromatografiska systemet är stabilt genom att upprepade gånger injicera kalibreringslösningen (3.11.2) på 5,0 μg/ml till dess att topphöjder (areor) och retentionstider är konstanta.
5.4.2
Injicera varje kalibreringslösning (3.11.2) upprepade gånger och mät topphöjderna (areorna) för varje koncentration. Konstruera en kalibreringskurva med kalibreringslösningarnas genomsnittliga topphöjd (area) längs y-axeln och de motsvarande koncentrationerna i μg/ml längs x-axeln.
5.4.3
Injicera provextraktet (se 5.3.2 för foder, 5.2.2 för förblandningar och 5.2.3 för foderberedningar) upprepade gånger och bestäm den genomsnittliga topphöjden (arean) för karbadox.
6. Beräkning av resultat
Använd den genomsnittliga topphöjden (arean) för provlösningens karbadoxtoppar för att bestämma koncentrationen i μg/ml med hjälp av kalibreringskurvan (5.4.2).
6.1 Foder
Provets karbadoxhalt w (mg/kg) beräknas med formeln
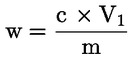
[mg/kg]
där:
|
c |
= |
provextraktets karbadoxkoncentration (5.3.2) i μg/ml |
|
V1 |
= |
extraktionsvolym i ml (dvs. 50) |
|
m |
= |
provets vikt i gram. |
6.2 Förblandningar och beredningar
Provets karbadoxhalt w (mg/kg) beräknas med formeln
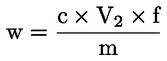
[mg/kg]
där:
|
c |
= |
provextraktets karbadoxkoncentration (5.2.2 eller 5.2.3) i μg/ml |
|
V2 |
= |
extraktionsvolym i ml (dvs. 50 för förblandningar, 150 för beredningar) |
|
f |
= |
spädningsfaktor enligt 5.2.2 (förblandningar) eller 5.2.3 (beredningar) |
|
m |
= |
provets vikt i gram. |
7. Validering av resultat
7.1 Identitet
Analytens identitet kan bekräftas genom co-kromatografi eller genom användning av en diod-array-detektor med vars hjälp spektrumen för provextraktet och den kalibreringslösning (3.11.2) som innehåller 10,0 μg/ml jämförs.
7.1.1
Ett provextrakt spikas genom tillsats av en lämplig mängd kalibreringslösning (3.11.2). Mängden tillsatt karbadox bör vara ungefär lika stor som mängden karbadox i provextraktet.
Endast karbadoxtoppens höjd ska öka efter det att hänsyn tagits till både den tillsatta mängden och spädningen av extraktet. Toppens bredd på halva höjden ska ligga inom 10 % av den ursprungliga bredden.
7.1.2
Resultaten utvärderas enligt följande kriterier:
a) Våglängderna för prov- och standardspektrumens absorptionsmaximum, i toppens spets på kromatogrammet, ska ligga inom en marginal som beror på detektionssystemets upplösningsförmåga. För diod-array-detektion är denna i allmänhet ± 2 nm.
b) Mellan 225 och 400 nm får prov- och standardspektrumen, i toppens spets på kromatogrammet, inte skilja sig åt för de delar av spektrumet där den relativa absorbansen är 10–100 %. Detta kriterium är uppfyllt om samma maximum observeras och avvikelsen mellan de två spektrumen inte vid någon mätpunkt överstiger 15 % av standardanalytens absorbans.
c) Mellan 225 och 400 nm får spektrumen för den uppåtgående delen, spetsen och den nedåtgående delen av toppen från provextraktet inte skilja sig från varandra för de delar av spektrumet där den relativa absorbansen är 10–100 %. Detta kriterium är uppfyllt om samma maximum observeras och avvikelsen mellan spektrumen inte vid någon mätpunkt överstiger 15 % av absorbansen vid spetsen.
Om något av dessa kriterier inte är uppfyllt har analytens förekomst inte bekräftats.
7.2 Repeterbarhet
För innehåll på minst 10 mg/kg får skillnaden mellan resultaten från två parallella bestämningar som utförs på samma prov inte överskrida 15 % av det högre värdet.
7.3 Utbyte
För ett spikat (blank)prov ska utbytet vara minst 90 %.
8. Resultat av en provningsjämförelse
I en provningsjämförelse analyserades sex fodertyper, fyra förblandningar och tre foderberedningar av åtta laboratorier. Varje prov analyserades två gånger. (Närmare uppgifter om provningsjämförelsen finns i Journal of the AOAC, volym 71, 1988, s. 484–490). Resultaten (efter eliminering av extremvärden) redovisas i nedanstående tabell.
Tabell 1
Resultat av provningsjämförelsen för foder
|
|
Prov 1 |
Prov 2 |
Prov 3 |
Prov 4 |
Prov 5 |
Prov6 |
|
L |
8 |
8 |
8 |
8 |
8 |
8 |
|
n |
15 |
14 |
15 |
15 |
15 |
15 |
|
Medelvärde (mg/kg) |
50,0 |
47,6 |
48,2 |
49,7 |
46,9 |
49,7 |
|
Sr (mg/kg) |
2,90 |
2,69 |
1,38 |
1,55 |
1,52 |
2,12 |
|
CVr (%) |
5,8 |
5,6 |
2,9 |
3,1 |
3,2 |
4,3 |
|
SR (mg/kg) |
3,92 |
4,13 |
2,23 |
2,58 |
2,26 |
2,44 |
|
CVR ( %) |
7,8 |
8,7 |
4,6 |
5,2 |
4,8 |
4,9 |
|
Nominell halt (g/kg) |
50,0 |
50,0 |
50,0 |
50,0 |
50,0 |
50,0 |
Tabell 2
Resultat av provningsjämförelsen för förblandningar och foderberedningar
|
|
Förblandningar |
Foderberedningar |
|||||
|
A |
B |
C |
D |
A |
B |
C |
|
|
L |
7 |
7 |
7 |
7 |
8 |
8 |
8 |
|
n |
14 |
14 |
14 |
14 |
16 |
16 |
16 |
|
Medelvärde (g/kg) |
8,89 |
9,29 |
9,21 |
8,76 |
94,6 |
98,1 |
104 |
|
Sr (g/kg) |
0,37 |
0,28 |
0,28 |
0,44 |
4,1 |
5,1 |
7,7 |
|
CVr (%) |
4,2 |
3,0 |
3,0 |
5,0 |
4,3 |
5,2 |
7,4 |
|
SR (g/kg) |
0,37 |
0,28 |
0,40 |
0,55 |
5,4 |
6,4 |
7,7 |
|
CVR (%) |
4,2 |
3,0 |
4,3 |
6,3 |
5,7 |
6,5 |
7,4 |
|
Nominell halt (g/kg) |
10,0 |
10,0 |
10,0 |
10,0 |
100 |
100 |
100 |
|
L |
= |
antal laboratorier |
|
n |
= |
antal enskilda värden |
|
Sr |
= |
standardavvikelse för repeterbarhet |
|
CVr |
= |
variationskoefficient för repeterbarhet |
|
SR |
= |
standardavvikelse för reproducerbarhet |
|
CVR |
= |
variationskoefficient för reproducerbarhet. |
BILAGA IX
JÄMFÖRELSETABELLER SOM AVSES I ARTIKEL 6
1. Direktiv 71/250/EEG
|
Direktiv 71/250/EEG |
Denna förordning |
|
Artikel 1 första stycket |
Artikel 3 |
|
Artikel 1 andra stycket |
Artikel 2 |
|
Artikel 2 |
–– |
|
Artikel 3 |
–– |
|
Bilagan, del 1 |
Bilaga II |
|
Bilagan, del 2 |
–– |
|
Bilagan, del 3 |
–– |
|
Bilagan, del 4 |
Bilaga III, del O |
|
Bilagan, del 5 |
Bilaga III, del M |
|
Bilagan, del 6 |
Bilaga III, del N |
|
Bilagan, del 7 |
Bilaga III, del Q |
|
Bilagan, del 9 |
Bilaga III, del K |
|
Bilagan, del 10 |
–– |
|
Bilagan, del 11 |
–– |
|
Bilagan, del 12 |
Bilaga III, del J |
|
Bilagan, del 14 |
Bilaga III, del D |
|
Bilagan, del 16 |
–– |
2. Direktiv 71/393/EEG
|
Direktiv 71/393/EEG |
Denna förordning |
|
Artikel 1 |
Artikel 3 |
|
Artikel 2 |
–– |
|
Artikel 3 |
–– |
|
Bilagan, del I |
Bilaga III, del A |
|
Bilagan, del II |
Bilaga III, del E |
|
Bilagan, del III |
Bilaga III, del P |
|
Bilagan, del IV |
Bilaga III, del H |
3. Direktiv 72/199/EEG
|
Direktiv 72/199/EEG |
Denna förordning |
|
Artikel 1 |
Artikel 3 |
|
Artikel 2 |
–– |
|
Artikel 3 |
–– |
|
Artikel 4 |
–– |
|
Bilaga I, del 1 |
Bilaga III, del L |
|
Bilaga I, del 2 |
Bilaga III, del C |
|
Bilaga I, del 3 |
–– |
|
Bilaga I, del 4 |
–– |
|
Bilaga I, del 5 |
Bilaga V, del A |
|
Bilaga II |
–– |
4. Direktiv 73/46/EEG
|
Direktiv 73/46/EEG |
Denna förordning |
|
Artikel 1 |
Artikel 3 |
|
Artikel 3 |
–– |
|
Artikel 4 |
–– |
|
Bilaga I, del 1 |
Bilaga III, del B |
|
Bilaga I, del 2 |
–– |
|
Bilaga I, del 3 |
Bilaga III, del I |
5. Direktiv 76/371/EEG
|
Direktiv 76/371/EEG |
Denna förordning |
|
Artikel 1 |
Artikel 1 |
|
Artikel 2 |
–– |
|
Artikel 3 |
–– |
|
Bilagan |
Bilaga I |
6. Direktiv 76/372/EEG
|
Direktiv 76/372/EEG |
Denna förordning |
|
Artikel 1 |
–– |
|
Artikel 2 |
–– |
|
Artikel 3 |
–– |
|
Bilagan |
–– |
7. Direktiv 78/633/EEG
|
Direktiv 78/633/EEG |
Denna förordning |
|
Artikel 1 |
Artikel 3 |
|
Artikel 2 |
–– |
|
Artikel 3 |
–– |
|
Bilagan, del 1 |
–– |
|
Bilagan, del 2 |
–– |
|
Bilagan, del 3 |
Bilaga IV, del C |
8. Direktiv 81/715/EEG
|
Direktiv 81/715/EEG |
Denna förordning |
|
Artikel 1 |
–– |
|
Artikel 2 |
–– |
|
Artikel 3 |
–– |
|
Bilagan |
–– |
9. Direktiv 84/425/EEG
|
Direktiv 84/425/EEG |
Denna förordning |
|
Artikel 1 |
–– |
|
Artikel 2 |
–– |
|
Artikel 3 |
–– |
|
Bilagan |
–– |
10. Direktiv 86/174/EEG
|
Direktiv 86/174/EEG |
Denna förordning |
|
Artikel 1 |
Artikel 4 |
|
Artikel 2 |
–– |
|
Artikel 3 |
–– |
|
Bilagan |
Bilaga VII |
11. Direktiv 93/70/EEG
|
Direktiv 93/70/EEG |
Denna förordning |
|
Artikel 1 |
Artikel 3 |
|
Artikel 2 |
–– |
|
Artikel 3 |
–– |
|
Bilagan |
Bilaga IV, del D |
12. Direktiv 93/117/EG
|
Direktiv 93/117/EG |
Denna förordning |
|
Artikel 1 |
Artiklarna 3 och 5 |
|
Artikel 2 |
–– |
|
Artikel 3 |
–– |
|
Bilagan, del 1 |
Bilaga IV del E |
|
Bilagan, del 2 |
Bilaga VIII, del A |
13. Direktiv 98/64/EG
|
Direktiv 98/64/EG |
Denna förordning |
|
Artikel 1 |
Artiklarna 3 och 5 |
|
Artikel 2 |
–– |
|
Artikel 3 |
–– |
|
Artikel 4 |
–– |
|
Bilagan, del A |
Bilaga III, del F |
|
Bilagan, del C |
Bilaga VIII, del B |
14. Direktiv 1999/27/EG
|
Direktiv 1999/27/EG |
Denna förordning |
|
Artikel 1 |
Artiklarna 3 och 5 |
|
Artikel 2 |
–– |
|
Artikel 3 |
–– |
|
Artikel 4 |
–– |
|
Artikel 5 |
–– |
|
Artikel 6 |
–– |
|
Artikel 7 |
–– |
|
Bilagan, del A |
Bilaga VIII, del C |
|
Bilagan, del B |
Bilaga IV, del F |
|
Bilagan, del C |
Bilaga VIII, del D |
15. Direktiv 1999/76/EG
|
Direktiv 1999/76/EG |
Denna förordning |
|
Artikel 1 |
Artikel 3 |
|
Artikel 2 |
–– |
|
Artikel 3 |
–– |
|
Artikel 4 |
–– |
|
Bilagan |
Bilaga IV, del G |
16. Direktiv 2000/45/EG
|
Direktiv 2000/45/EG |
Denna förordning |
|
Artikel 1 |
Artikel 3 |
|
Artikel 2 |
–– |
|
Artikel 3 |
–– |
|
Artikel 4 |
–– |
|
Bilagan, del A |
Bilaga IV, del A |
|
Bilagan, del B |
Bilaga IV, del B |
|
Bilagan, del C |
Bilaga III, del G |
17. Direktiv 2002/70/EG
|
Direktiv 2002/70/EG |
Denna förordning |
|
Artikel 1 |
Artikel 1 |
|
Artikel 2 |
Artiklarna 2 och 3 |
|
Artikel 3 |
–– |
|
Artikel 4 |
–– |
|
Artikel 5 |
–– |
|
Bilaga I |
Bilaga I och bilaga V del B (I) |
|
Bilaga II |
Bilaga II och bilaga V del B (II) |
18. Direktiv 2003/126/EG
|
Direktiv 2003/126/EG |
Denna förordning |
|
Artikel 1 |
Artikel 3 |
|
Artikel 2 |
–– |
|
Artikel 3 |
–– |
|
Artikel 4 |
–– |
|
Artikel 5 |
–– |
|
Artikel 6 |
–– |
|
Bilagan |
Bilaga VI |
( 1 ) EUT L 268, 18.10.2003, s. 29.
( 2 ) EGT L 140, 30.5.2002, s. 10.
( 3 ) EUT L 70, 16.3.2005, s. 1.
( 4 ) Eventuella klumpar ska brytas upp (om så är nödvändigt genom att de särskiljs från provet och sedan återförs till detta).
( 5 ) Utom i fråga om grovfoder eller vallfoder med låg specifik vikt.
( 6 ) Utom i fråga om grovfoder eller vallfoder med låg specifik vikt.
( 7 ) EUT L 229, 1.9.2009, s. 1.
( 7 ) För torkning av spannmål, mjöl och gröpe måste ugnen ha så hög effekt att den vid en inställning på 131 oC åter uppnår denna temperatur på mindre än 45 minuter efter det att det maximala antalet prov har placerats i den för att torka samtidigt. Ventilationen måste vara så god att resultatet, efter två timmars torkning av så många prov av vanligt vete som ugnen rymmer, skiljer sig från resultatet efter fyra timmars torkning med mindre än 0,15 %.
( 7 ) Om fettet senare ska genomgå kvalitetstester, ersätts pimpstensskärvorna med glaspärlor.
( 7 ) EGT L 125, 23.5.1996, s. 35.
( 7 ) Genomförd av arbetsgruppen för foder inom Verband Deutscher Landwirtschaftlicher Untersuchungs- und Forschungsanstalten (VDLUFA).
( 7 ) Genomförd av arbetsgruppen för foder inom Verband Deutscher Landwirtschaftlicher Untersuchungs- und Forschungsanstalten (VDLUFA).
( 7 ) Andra metoder för uppslutning (t.ex. uppslutning med mikrovågor, under tryck) får användas om man kunnat visa att de ger liknande resultat.
( 7 ) Grönfoder (färskt eller torkat) innehåller ofta stora mängder silikater. Dessa måste avlägsnas, eftersom spårelement annars riskerar att kvarhållas. Prov från sådant foder måste därför behandlas enligt följande modifierade förfarande: Utför steg 5.1.1.1 till och med filtreringen. Skölj filtrerpapperet med den olösliga återstoden två gånger med kokande vatten, och lägg det sedan i en degel av kvarts eller platina. Glödga i muffelugnen (4.1) vid en temperatur lägre än 550 oC tills allt kolhaltigt material har försvunnit. Låt svalna. Tillsätt några droppar vatten, följt av 10–15 ml fluorvätesyra (3.4) och låt vätskan avdunsta fullständigt vid ca 150 oC. Eventuella kvarvarande silikater i återstoden löses upp i några milliliter fluorvätesyra (3.4) som därefter får avdunsta fullständigt. Tillsätt fem droppar svavelsyra (3.5) och upphetta tills ingen vit rök längre avges. Tillsätt 5 ml 6 M saltsyra (3.2) samt ca 30 ml vatten och upphetta på nytt. Filtrera över lösningen till en 250 ml mätkolv och späd till märket med vatten (HCl-koncentration ca 0,5 M). Fortsätt sedan bestämningen enligt punkt 5.1.2.
( 7 ) The Analyst 108, 1983, s. 1252–1256.
( *1 ) Analyst, 1995, 120, 2175–2180.
( 8 ) Tabell över TEF-värden (toxiska ekvivalensfaktorer) för PCDD, PCDF och dioxinlika PCB: Världshälsoorganisationens toxicitetsekvivalensfaktorer (WHO-TEF) för bedömningen av risker för människor på grundval av slutsatserna från WHO:s expertmöte inom det internationella programmet för kemikaliesäkerhet (IPCS) i Genève i juni 2005 (Martin van den Berg et al, ”The 2005 World Health Organization Re-evaluation of Human and Mammalian Toxic Equivalency Factors for Dioxins and Dioxin-like Compounds”, Toxicological Sciences, 2006, 93(2), s. 223–241.
|
Kongen |
TEF-värde |
Kongen |
TEF-värde |
|
Dibenso-p-dioxiner (PCDD) och dibenso-p-furaner (PCDF) |
|
Dioxinlika PCB: non-orto PCB + mono-orto PCB |
|
|
2,3,7,8-TCDD |
1 |
|
|
|
1,2,3,7,8-PeCDD |
1 |
Non-orto PCB |
|
|
1,2,3,4,7,8-HxCDD |
0,1 |
PCB 77 |
0,0001 |
|
1,2,3,6,7,8-HxCDD |
0,1 |
PCB 81 |
0,0003 |
|
1,2,3,7,8,9-HxCDD |
0,1 |
PCB 126 |
0,1 |
|
1,2,3,4,6,7,8-HpCDD |
0,01 |
PCB 169 |
0,03 |
|
OCDD |
0,0003 |
Mono-orto PCB |
|
|
2,3,7,8-TCDF |
0,1 |
PCB 105 |
0,00003 |
|
1,2,3,7,8-PeCDF |
0,03 |
PCB 114 |
0,00003 |
|
2,3,4,7,8-PeCDF |
0,3 |
PCB 118 |
0,00003 |
|
1,2,3,4,7,8-HxCDF |
0,1 |
PCB 123 |
0,00003 |
|
1,2,3,6,7,8-HxCDF |
0,1 |
PCB 156 |
0,00003 |
|
1,2,3,7,8,9-HxCDF |
0,1 |
PCB 157 |
0,00003 |
|
2,3,4,6,7,8-HxCDF |
0,1 |
PCB 167 |
0,00003 |
|
1,2,3,4,6,7,8-HpCDF |
0,01 |
PCB 189 |
0,00003 |
|
1,2,3,4,7,8,9-HpCDF |
0,01 |
|
|
|
OCDF |
0,0003 |
|
|
Förkortningar: ”T” = tetra, ”Pe” = penta, ”Hx” = hexa, ”Hp” = hepta, ”O” = okta, ”CDD” = klordibensodioxin, ”CDF” = klordibensofuran, ”CB” = klorbifenyl.
( 9 ) Kommissionens beslut 2002/657/EG av den 14 augusti 2002 om genomförande av rådets direktiv 96/23/EG avseende analysmetoder och tolkning av resultat (EGT L 221, 17.8.2002, s. 8).
( 10 ) Principerna i vägledningen Guidance Document on Measurement Uncertainty for Laboratories performing PCDD/F and PCB Analysis using Isotope Dilution Mass Spectrometry (http://ec.europa.eu/food/safety/animal-feed_en) ska följas i tillämpliga fall.
( 11 ) Begreppet övre koncentration kräver att man använder kvantifieringsgränsen för bidraget från varje icke-kvantifierad kongen. Begreppet lägre koncentration kräver att man använder noll för bidraget från varje icke-kvantifierad kongen. Begreppet mellanvärde kräver att man använder halva kvantifieringsgränsen vid beräkning av bidraget från varje icke-kvantifierad kongen. Analys av dubbelprov: en separat analys av de givna analyterna med användning av en andra alikvot från samma homogeniserade prov.
( 12 ) I allmänhet gäller de krav för analys av dubbelprov som anges i kapitel C punkt 3 i bilaga II. För metoder med 13C-märkt intern standard för de relevanta analyterna behövs dock analys av dubbelprov endast om resultatet från den första bestämningen inte är överensstämmande. Analys av dubbelprov behövs för att utesluta möjligheten av intern korskontamination eller oavsiktlig hopblandning av prov. När analysen utförs till följd av en föroreningsincident behöver resultatet inte konfirmeras genom analys av dubbelprov om de prov som valts ut för analys kan spåras till föroreningsincidenten och den uppmätta halten är betydligt över gränsvärdet.
( 13 ) Begreppet övre koncentration kräver att man använder kvantifieringsgränsen för bidraget från varje icke-kvantifierad kongen till TEQ-värdet (toxicitetsekvivalenter). Begreppet lägre koncentration kräver att man använder noll för bidraget från varje icke-kvantifierad kongen till TEQ-värdet. Begreppet mellanvärde kräver att man använder halva kvantifieringsgränsen vid beräkning av bidraget från varje icke-kvantifierad kongen till TEQ-värdet.
( 14 ) I allmänhet gäller de krav för analys av dubbelprov som anges i kapitel C punkt 2 i bilaga II. För konfirmeringsmetoder med 13C-märkt intern standard för de relevanta analyterna behövs dock analys av dubbelprov endast om resultatet från den första bestämningen inte är överensstämmande. Analys av dubbelprov behövs för att utesluta möjligheten av intern korskontamination eller oavsiktlig hopblandning av prov. När analysen utförs till följd av en föroreningsincident behöver resultatet inte konfirmeras genom analys av dubbelprov om de prov som valts ut för analys kan spåras till föroreningsincidenten och den uppmätta halten är betydligt över gränsvärdet.
( 15 ) Samma förklaring och krav avseende analys av dubbelprov gäller för kontroll av åtgärdsgränser som för gränsvärden i fotnot 2.
( 16 ) Europaparlamentets och rådets förordning (EG) nr 183/2005 av den 12 januari 2005 om fastställande av krav för foderhygien (EUT L 35, 8.2.2005, s. 1).
( 17 ) Bioanalytiska metoder är inte specifika för de kongener som ingår i TEF-systemet. Även andra strukturellt besläktade föreningar som aktiverar Ah-receptorn kan förekomma i provextraktet och därigenom bidra till den totala responsen. Bioanalytiska resultat kan därför inte vara en uppskattning av, utan snarare en indikation på TEQ-värdet i provet.
( 18 ) Guidance Document on Measurement Uncertainty for Laboratories performing PCDD/F and PCB Analysis using Isotope Dilution Mass Spectrometry (http://ec.europa.eu/food/safety/animal-feed_en) och Guidance Document on the Estimation of LOD and LOQ for Measurements in the Field of Contaminants in Feed and Food (http://ec.europa.eu/food/safety/animal-feed_en).
( 19 ) Nuvarande krav är grundade på de TEF-värden som offentliggjorts i Martin Van den Berg m.fl., Toxicological Sciences, 2006, 93(2), s. 223–241.
( 20 ) Kongener som ofta elueras samtidigt är t.ex. PCB 28/31, PCB 52/69 och PCB 138/163/164. När det gäller GC-MS ska även eventuella interferenser från fragment av mer högklorerade kongener beaktas.
( 21 ) Principerna i vägledningen Guidance Document on the Estimation of LOD and LOQ for Measurements in the Field of Contaminants in Feed and Food (http://ec.europa.eu/food/safety/animal-feed_en) ska följas i tillämpliga fall.
( 22 ) Reagensblankprovet bör lämna ett så litet bidrag som möjligt till halten av en förorening i ett prov. Det är laboratoriets skyldighet att kontrollera hur halterna i blankproven varierar, i synnerhet om blankproven dras av från proven.
( 23 ) Nuvarande krav är grundade på de TEF-värden som offentliggjorts i Martin Van den Berg m.fl., Toxicological Sciences, 2006, 93(2), s. 223–241.
( 24 ) Se http://eurl.craw.eu/
( 25 ) Förteckningen över de primrar och prober för varje djurart som metoden omfattar finns på webbplatsen för EU:s referenslaboratorium för animaliska proteiner i foder.
( 26 ) Exempel på funktionella mastermixar finns på webbplatsen för EU:s referenslaboratorium för animaliska proteiner i foder.