

EUR-Lex Access to European Union law
This document is an excerpt from the EUR-Lex website
Document 02009R0152-20170524
Commission Regulation (EC) No 152/2009 of 27 January 2009 laying down the methods of sampling and analysis for the official control of feed (Text with EEA relevance)
Consolidated text: Règlement (CE) n o 152/2009 de la Commission du 27 janvier 2009 portant fixation des méthodes d'échantillonnage et d'analyse destinées au contrôle officiel des aliments pour animaux (Texte présentant de l'intérêt pour l'EEE)
Règlement (CE) n o 152/2009 de la Commission du 27 janvier 2009 portant fixation des méthodes d'échantillonnage et d'analyse destinées au contrôle officiel des aliments pour animaux (Texte présentant de l'intérêt pour l'EEE)

02009R0152 — FR — 24.05.2017 — 006.001
Ce texte constitue seulement un outil de documentation et n’a aucun effet juridique. Les institutions de l'Union déclinent toute responsabilité quant à son contenu. Les versions faisant foi des actes concernés, y compris leurs préambules, sont celles qui ont été publiées au Journal officiel de l’Union européenne et sont disponibles sur EUR-Lex. Ces textes officiels peuvent être consultés directement en cliquant sur les liens qui figurent dans ce document
|
RÈGLEMENT (CE) No 152/2009 DE LA COMMISSION du 27 janvier 2009 portant fixation des méthodes d'échantillonnage et d'analyse destinées au contrôle officiel des aliments pour animaux (Texte présentant de l'intérêt pour l'EEE) (JO L 054 du 26.2.2009, p. 1) |
Modifié par:
|
|
|
Journal officiel |
||
|
n° |
page |
date |
||
|
L 91 |
8 |
29.3.2012 |
||
|
RÈGLEMENT (UE) No 51/2013 DE LA COMMISSION du 16 janvier 2013 |
L 20 |
33 |
23.1.2013 |
|
|
RÈGLEMENT (UE) No 691/2013 DE LA COMMISSION du 19 juillet 2013 |
L 197 |
1 |
20.7.2013 |
|
|
L 188 |
1 |
27.6.2014 |
||
|
L 92 |
35 |
6.4.2017 |
||
|
L 115 |
22 |
4.5.2017 |
||
RÈGLEMENT (CE) No 152/2009 DE LA COMMISSION
du 27 janvier 2009
portant fixation des méthodes d'échantillonnage et d'analyse destinées au contrôle officiel des aliments pour animaux
(Texte présentant de l'intérêt pour l'EEE)
Article premier
Les prélèvements d’échantillons destinés au contrôle officiel des aliments pour animaux sont effectués conformément aux méthodes décrites à l’annexe I, notamment pour ce qui concerne la détermination des constituants – y compris les matières premières contenant des organismes génétiquement modifiés (OGM), consistant en de tels organismes ou produites à partir de ceux-ci –, des additifs pour l’alimentation animale tels que définis par le règlement (CE) no 1831/2003 du Parlement européen et du Conseil ( 1 ) et des substances indésirables telles que définies par la directive 2002/32/CE du Parlement européen et du Conseil ( 2 ).
La méthode d’échantillonnage établie à l’annexe I est applicable pour le contrôle des aliments pour animaux en ce qui concerne la détermination des résidus de pesticides tels que définis par le règlement (CE) no 396/2005 du Parlement européen et du Conseil ( 3 ) et la conformité au règlement (UE) no 619/2011.
Article 2
La préparation des échantillons en vue de l'analyse et l'expression des résultats sont effectuées conformément aux méthodes décrites à l'annexe II.
Article 3
L'analyse réalisée aux fins du contrôle officiel des aliments pour animaux est effectuée conformément aux méthodes décrites à l'annexe III (méthodes d'analyse relatives au contrôle de la composition des matières premières pour aliments des animaux et des aliments composés), à l'annexe IV (méthodes d'analyse relatives au contrôle des teneurs en additifs autorisés des aliments pour animaux), à l'annexe V (méthodes d'analyse relatives au contrôle des substances indésirables dans les aliments pour animaux) et à l'annexe VI (méthodes d'analyse applicables en matière d'identification des constituants d'origine animale pour le contrôle officiel des aliments pour animaux).
Article 4
La valeur énergétique des aliments composés destinés à la volaille est calculée conformément à la méthode décrite à l'annexe VII.
Article 5
Les méthodes d'analyse relatives à la détection d'additifs dont la présence n'est plus autorisée dans les aliments pour animaux, décrites à l'annexe VIII, sont appliquées à des fins de confirmation.
Article 6
Les directives 71/250/CEE, 71/393/CEE, 72/199/CEE, 73/46/CEE, 76/371/CEE, 76/372/CEE, 78/633/CEE, 81/715/CEE, 84/425/CEE, 86/174/CEE, 93/70/CEE, 93/117/CE, 98/64/CE, 1999/27/CE, 1999/76/CE, 2000/45/CE, 2002/70/CE et 2003/126/CE sont abrogées.
Les références faites aux directives abrogées s'entendent comme faites au présent règlement et sont à lire selon le tableau de correspondance figurant à l'annexe IX.
Article 7
Le présent règlement entre en vigueur le vingtième jour suivant celui de sa publication au Journal officiel de l'Union européenne.
Il s'applique à partir du 26 août 2009
Le présent règlement est obligatoire dans tous ses éléments et directement applicable dans tout État membre.
ANNEXE I
MÉTHODES D’ÉCHANTILLONNAGE
1. OBJET ET CHAMP D’APPLICATION
Les échantillons destinés au contrôle officiel des aliments pour animaux sont prélevés conformément aux modalités indiquées ci-après. Les échantillons ainsi obtenus sont considérés comme étant représentatifs des portions échantillonnées.
Le but de l’échantillonnage représentatif est d’obtenir une petite fraction d’un lot de façon que la détermination d’une caractéristique particulière de cette fraction représente la valeur moyenne de la caractéristique considérée pour l’ensemble du lot. Le lot est échantillonné par le prélèvement répété d’échantillons élémentaires en différents points du lot. Ces échantillons élémentaires sont combinés par mélange pour former un échantillon global dont sont tirés, par division représentative, des échantillons finals représentatifs.
Si un examen visuel révèle que la qualité de certaines portions de l’aliment pour animaux devant être soumis à l’échantillonnage diffère de celle du reste du même lot, ces portions sont séparées du reste et traitées comme un sous-lot distinct. S’il n’est pas possible de le diviser en sous-lots distincts, l’aliment pour animaux est échantillonné en tant que lot unique. Dans de tels cas, il en est fait mention dans le rapport d’échantillonnage.
Lorsqu’il est constaté que des aliments pour animaux soumis à un échantillonnage conformément aux dispositions du présent règlement, et qui font partie d’un lot de la même catégorie ou correspondant à la même description, ne satisfont pas aux exigences de l’Union européenne, il est présumé que la totalité des aliments de ce lot est défectueuse, sauf si une évaluation détaillée n’a pas fourni d’éléments indiquant que le reste du lot ne répond pas aux exigences de l’Union européenne.
2. DÉFINITIONS
|
— |
Lot : quantité identifiée d’aliment pour animaux dont il est établi qu’elle présente des caractéristiques communes, telles que l’origine, la variété, le type d’emballage, l’emballeur, l’expéditeur ou l’étiquetage, et, dans le cas d’un processus de production, une quantité de produit fabriquée dans une seule usine en utilisant des paramètres de production uniformes ou plusieurs de ces quantités lorsqu’elles sont produites en ordre continu et entreposées ensemble. |
|
— |
Portion échantillonnée : lot ou partie identifiée du lot ou du sous-lot. |
|
— |
Échantillon scellé : échantillon scellé de manière à ce qu’il soit impossible d’accéder à l’échantillon sans briser ou retirer le scellé. |
|
— |
Échantillon élémentaire : quantité prélevée en un point de la portion échantillonnée. |
|
— |
Échantillon global : ensemble d’échantillons élémentaires prélevés sur la même portion échantillonnée. |
|
— |
Échantillon réduit : partie de l’échantillon global, obtenue par réduction représentative de celui-ci. |
|
— |
Échantillon final : partie de l’échantillon réduit ou de l’échantillon global homogénéisé. |
|
— |
Échantillon de laboratoire : échantillon destiné au laboratoire (tel que reçu par le laboratoire) qui peut être l’échantillon final, l’échantillon réduit ou l’échantillon global. |
3. DISPOSITIONS GÉNÉRALES
— Personnel chargé du prélèvement des échantillons: les prélèvements sont effectués par des personnes mandatées à cet effet par l’autorité compétente.
— L’échantillon doit être scellé de sorte qu’il soit impossible d’y accéder sans briser ou retirer le scellé. La marque du scellé doit être clairement identifiable et visible. L’échantillon peut également être placé dans un récipient pouvant être fermé de manière à ce qu’il soit impossible de l’ouvrir sans endommager irréversiblement le réceptacle ou le contenant, afin d’éviter sa réutilisation.
— Identification de l’échantillon: l’échantillon doit être pourvu d’une marque indélébile et être identifié de façon à établir un lien non équivoque avec le rapport d’échantillonnage.
— Au moins deux échantillons finals sont prélevés sur chaque échantillon global: au moins un à des fins de contrôle (surveillance du respect de la loi) et un autre destiné à l’exploitant du secteur de l’alimentation animale (à des fins de recours). Un échantillon final peut éventuellement être prélevé à des fins de référence. Si l’échantillon global complet est homogénéisé, les échantillons finals sont prélevés sur celui-ci, à moins que cette procédure ne soit contraire à la législation des États membres concernant le droit des exploitants du secteur de l’alimentation animale.
4. APPAREILLAGE
|
4.1. |
Les appareils destinés aux prélèvements doivent être construits en matériaux qui ne contaminent pas les produits à prélever. Les appareils destinés à être utilisés à plusieurs reprises doivent être faciles à nettoyer pour éviter toute contamination croisée. |
|
4.2. |
Appareils recommandés pour le prélèvement d’échantillons d’aliments pour animaux solides
4.2.1. Prélèvement manuel 4.2.1.1. Pelle à fond plat et à bords verticaux
4.2.2. Prélèvement mécanique Des appareils mécaniques appropriés peuvent être utilisés pour prélever des échantillons d’aliments en mouvement. «Appropriés» signifie que ces appareils permettent de prélever au moins une section complète du flot. Le prélèvement d’aliments pour animaux en mouvement (à des débits élevés) peut être effectué au moyen d’échantillonneurs automatiques. 4.2.3. Diviseur Le cas échéant, des appareils destinés à diviser l’échantillon en parts approximativement égales devraient être utilisés pour la préparation d’échantillons réduits représentatifs. |
5. EXIGENCES QUANTITATIVES CONCERNANT LE NOMBRE D’ÉCHANTILLONS ÉLÉMENTAIRES
— Les exigences quantitatives relatives au nombre d’échantillons élémentaires figurant aux points 5.1 et 5.2 s’appliquent à des portions échantillonnées n’excédant pas 500 tonnes et qui peuvent faire l’objet d’un échantillonnage représentatif. La procédure d’échantillonnage décrite est aussi valable pour des quantités supérieures à la taille maximale prescrite pour la portion échantillonnée, à condition de ne pas tenir compte du nombre maximal d’échantillons élémentaires indiqué dans les tableaux ci-après – le nombre d’échantillons élémentaires étant déterminé grâce à la formule mathématique indiquée dans la partie pertinente de la procédure (voir le point 5.3) – et d’augmenter proportionnellement la taille minimale de l’échantillon global. Il est néanmoins possible de diviser un lot de grande taille en sous-lots plus petits et d’échantillonner chaque sous-lot conformément à la procédure décrite aux points 5.1 et 5.2.
— La dimension de la portion échantillonnée doit être telle que toutes les parties qui la composent puissent être échantillonnées.
— Pour les lots ou sous-lots de très grande taille (> 500 tonnes) et les lots qui sont transportés ou stockés d’une manière qui ne permet pas d’effectuer l’échantillonnage conformément à la procédure prévue aux points 5.1 et 5.2 du présent chapitre, la procédure d’échantillonnage prévue au point 5.3 doit être appliquée.
— Si la législation impose à l’exploitant du secteur de l’alimentation animale de se conformer au présent règlement dans le cadre d’un système de contrôle obligatoire, l’exploitant peut s’écarter des exigences quantitatives prévues par le présent chapitre afin de tenir compte de caractéristiques opérationnelles, à la condition qu’il ait démontré, à la satisfaction de l’autorité compétente, l’équivalence de la procédure d’échantillonnage du point de vue de la représentativité, et après avoir obtenu l’autorisation de l’autorité compétente.
— Dans des cas exceptionnels, s’il n’est pas possible d’appliquer la méthode d’échantillonnage décrite en ce qui concerne les exigences quantitatives, parce qu’elle entraînerait une dégradation inacceptable de la valeur commerciale du lot (à cause des formes d’emballage, des moyens de transport, du mode de stockage, etc.), un autre mode de prélèvement peut être appliqué, à condition qu’il soit aussi représentatif que possible et qu’il soit décrit en détail et bien documenté.
5.1. Exigences quantitatives concernant les échantillons élémentaires en rapport avec le contrôle des substances ou produits répartis uniformément dans les aliments pour animaux
5.1.1. Aliments solides en vrac
|
Taille de la portion échantillonnée |
Nombre minimal d’échantillons élémentaires |
|
≤ 2,5 tonnes |
7 |
|
> 2,5 tonnes |
√ 20 fois le nombre de tonnes constituant la portion échantillonnée (1), jusqu’à 40 échantillons élémentaires |
|
(*1) Lorsque le résultat obtenu est un nombre décimal, il doit être arrondi au nombre entier immédiatement supérieur. |
|
5.1.2. Aliments liquides en vrac
|
Taille de la portion échantillonnée |
Nombre minimal d’échantillons élémentaires |
|
≤ 2,5 tonnes ou ≤ 2 500 litres |
4 (1) |
|
> 2,5 tonnes ou > 2 500 litres |
7 (1) |
|
(*1) S’il n’est pas possible d’homogénéiser le liquide, le nombre d’échantillons élémentaires doit être augmenté. |
|
5.1.3. Aliments emballés
Les aliments pour animaux (solides et liquides) peuvent être emballés dans des sachets, des sacs, des boîtes, des barils, etc., qui sont désignés comme des unités dans le tableau ci-après. Les unités de grande taille (≥ 500 kg ou litres) doivent être échantillonnées conformément aux dispositions prévues pour les aliments pour animaux en vrac (voir points 5.1.1 et 5.1.2).
|
Taille de la portion échantillonnée |
Nombre minimal d’unités sur lesquelles (au moins) un échantillon élémentaire doit être prélevé (1) |
|
De 1 à 20 unités |
1 unité (2) |
|
De 21 à 150 unités |
3 unités (2) |
|
De 151 à 400 unités |
5 unités (2) |
|
> 400 unités |
¼ du √ nombre d’unités constituant la portion échantillonnée (3), jusqu’à 40 unités |
|
(*1) Si l’ouverture d’une unité est susceptible d’avoir une incidence sur l’analyse (par exemple s’il s’agit d’aliments pour animaux humides et périssables), l’unité non ouverte constitue un échantillon élémentaire. (*2) S’agissant des unités dont le contenu n’excède pas 1 kg ou 1 litre, le contenu d’une unité d’origine constitue un échantillon élémentaire. (*3) Lorsque le résultat obtenu est un nombre décimal, il doit être arrondi au nombre entier immédiatement supérieur. |
|
5.1.4. Aliments en briques et pierres à lécher
Au minimum une brique ou pierre à prélever par portion échantillonnée de 25 unités, jusqu’à quatre briques ou pierres au maximum.
Pour les briques ou pierres à lécher d’un poids unitaire n’excédant pas 1 kg, le contenu d’une brique ou d’une pierre constitue un échantillon élémentaire.
5.1.5. Fourrages grossiers/fourrage
|
Taille de la portion échantillonnée |
Nombre minimal d’échantillons élémentaires (1) |
|
≤ 5 tonnes |
5 |
|
> 5 tonnes |
√ 5 fois le nombre de tonnes constituant la portion échantillonnée (2), jusqu’à 40 échantillons élémentaires |
|
(*1) Il est admis que dans certains cas (par exemple, celui des ensilages), il n’est pas possible de prélever les échantillons élémentaires requis sans endommager le lot de façon inacceptable. Dans de telles situations, une autre méthode d’échantillonnage peut être appliquée; des orientations pour l’échantillonnage de ce type de lots seront élaborées avant l’entrée en application du présent règlement. (*2) Lorsque le résultat obtenu est un nombre décimal, il doit être arrondi au nombre entier immédiatement supérieur. |
|
5.2. Exigences quantitatives concernant les échantillons élémentaires en rapport avec le contrôle des constituants ou substances susceptibles d’être répartis non uniformément dans les aliments pour animaux
Ces exigences quantitatives concernant les échantillons élémentaires s’appliquent dans les situations suivantes:
— contrôle de la présence d’aflatoxines, d’ergot de seigle, d’autres mycotoxines et impuretés botaniques nuisibles dans les matières premières des aliments pour animaux,
— contrôle de la contamination croisée par un constituant, y compris le matériel génétiquement modifié, ou une substance susceptibles d’être répartis non uniformément dans les matières premières des aliments pour animaux.
Si l’autorité de contrôle suspecte fortement qu’une telle répartition non uniforme existe également dans le cas d’une contamination croisée par un constituant ou une substance dans un aliment composé pour animaux, les exigences quantitatives figurant dans le tableau ci-dessous peuvent être appliquées.
|
Taille de la portion échantillonnée |
Nombre minimal d’échantillons élémentaires |
|
< 80 tonnes |
Voir les exigences quantitatives au point 5.1. Le nombre d’échantillons élémentaires à prélever doit être multiplié par 2,5. |
|
≥ 80 tonnes |
100 |
5.3. Exigences quantitatives concernant les échantillons élémentaires dans les cas de lots de très grande taille
Dans le cas de portions échantillonnées de grande taille (> 500 tonnes), le nombre d’échantillons élémentaires à prélever = 40 échantillons élémentaires + √ tonnes pour le contrôle des substances ou produits répartis uniformément dans les aliments pour animaux, ou 100 échantillons élémentaires + √ tonnes pour le contrôle des constituants ou substances susceptibles d’être répartis non uniformément dans les matières premières pour aliments des animaux.
6. EXIGENCES QUANTITATIVES CONCERNANT LES ÉCHANTILLONS GLOBAUX
|
Un seul échantillon global par portion échantillonnée est requis. |
||
|
|
Nature des aliments pour animaux |
|
|
6.1. |
Aliments en vrac |
4 kg |
|
6.2. |
Aliments emballés |
4 kg (3) |
|
6.3. |
Aliments liquides ou semi-liquides |
4 litres |
|
6.4. |
Aliments en briques ou pierres à lécher: |
|
|
6.4.1. |
d’un poids unitaire excédant 1 kg |
4 kg |
|
6.4.2. |
d’un poids unitaire n’excédant pas 1 kg |
poids de quatre briques ou pierres d’origine |
|
6.5. |
Fourrage/fourrage grossier |
4 kg (4) |
|
(*1) Si la valeur des aliments pour animaux est élevée, il est possible de prélever une plus petite quantité pour l’échantillon global, pour autant que cela soit décrit et documenté dans le rapport d’échantillonnage. (*2) Conformément aux dispositions du règlement (UE) no 619/2011 de la Commission du 24 juin 2011 fixant les méthodes d’échantillonnage et d’analyse du contrôle officiel des aliments pour animaux en vue de la détection de matériel génétiquement modifié faisant l’objet d’une procédure d’autorisation ou dont l’autorisation a expiré (JO L 166 du 25.6.2011, p. 9), l’échantillon global pour le contrôle de la présence de matériel génétiquement modifié doit contenir au moins 35 000 graines/semences. Cela signifie que l’échantillon global doit peser au moins 10,5 kg pour le maïs et 7 kg pour le soja. Pour d’autres graines ou semences telles que l’orge, le millet, l’avoine, le riz, le seigle, le blé et le colza, un échantillon global de 4 kg correspond à plus de 35 000 graines. (*3) Dans le cas des aliments pour animaux emballés, il peut également s’avérer impossible de constituer un échantillon global de 4 kg, en fonction de la taille des unités individuelles. (*4) S’il s’agit de fourrage grossier ou de fourrage de faible densité relative (par exemple, de foin ou de paille), l’échantillon global doit peser au moins 1 kg. |
||
7. EXIGENCES QUANTITATIVES CONCERNANT LES ÉCHANTILLONS FINALS
Échantillons finals
L’analyse d’au moins un échantillon final est requise. La quantité de l’échantillon final destinée à l’analyse ne peut être inférieure aux valeurs suivantes:
|
Aliments solides |
|
|
Aliments liquides ou semi-liquides |
500 ml (1) |
|
(*1) Conformément aux dispositions du règlement (UE) no 619/2011, l’échantillon final pour le contrôle de la présence de matériel génétiquement modifié doit contenir au moins 10 000 graines/semences. Cela signifie que l’échantillon final doit peser au moins 3 000 g pour le maïs et 2 000 g pour le soja. Pour d’autres graines ou semences telles que l’orge, le millet, l’avoine, le riz, le seigle, le blé et le colza, un échantillon final de 500 g correspond à plus de 10 000 graines. (*2) Si la taille de l’échantillon global est sensiblement inférieure à 4 kg ou 4 litres (voir notes de bas de page du point 6), il est possible de prélever une plus petite quantité pour l’échantillon final, pour autant que cela soit décrit et documenté dans le rapport d’échantillonnage. (*3) En cas d’échantillonnage de légumineuses, de grains de céréales et de fruits à coque visant à déterminer la présence de résidus de pesticides, l’échantillon final doit peser au moins 1 kg conformément aux dispositions de la directive 2002/63/CE de la Commission (JO L 187 du 16.7.2002, p. 30). |
|
8. MÉTHODE D’ÉCHANTILLONNAGE POUR LES LOTS DE TRÈS GRANDE TAILLE OU LES LOTS STOCKÉS OU TRANSPORTÉS DE TELLE MANIÈRE QU’UN ÉCHANTILLONNAGE DANS L’ENSEMBLE DU LOT N’EST PAS POSSIBLE
8.1. Principes généraux
Si le mode de transport ou de stockage d’un lot ne permet pas de prélever des échantillons élémentaires dans l’ensemble du lot, l’échantillonnage doit de préférence être effectué lorsque ce lot est en mouvement.
Dans le cas des grands entrepôts destinés au stockage d’aliments pour animaux, il convient d’encourager les opérateurs à installer dans l’entrepôt des équipements permettant de prélever (automatiquement) des échantillons dans l’ensemble du lot stocké.
Si les procédures d’échantillonnage prévues dans le présent chapitre sont appliquées, l’exploitant du secteur de l’alimentation animale ou son représentant en sont informés. Si l’exploitant du secteur de l’alimentation animale ou son représentant remettent en cause la procédure d’échantillonnage, ils autorisent l’autorité compétente à prélever des échantillons dans l’ensemble du lot, à leurs propres frais.
8.2. Lots de grande taille transportés par bateau
8.2.1. Échantillonnage dynamique de lots de grande taille transportés par bateau
L’échantillonnage de lots de grande taille transportés par bateau est effectué de préférence lorsque le produit est en mouvement (échantillonnage dynamique).
L’échantillonnage doit être fait par cale (entité qui peut être physiquement séparée). Cependant, les cales sont vidées partiellement l’une après l’autre, de sorte que la séparation physique initiale n’existe plus après le transfert dans les installations de stockage. L’échantillonnage peut donc être réalisé en fonction de la séparation physique initiale ou en fonction de la séparation après transfert dans les installations de stockage.
Le déchargement d’un bateau peut durer plusieurs jours. Normalement, l’échantillonnage doit être effectué à intervalles réguliers tout au long du déchargement. Toutefois, la présence d’un inspecteur officiel chargé de l’échantillonnage pendant toute l’opération de déchargement n’est pas toujours possible ou appropriée. Par conséquent, la réalisation d’un échantillonnage sur une partie (portion échantillonnée) du lot est autorisée. Le nombre d’échantillons élémentaires est déterminé en tenant compte de la taille de la portion échantillonnée.
Si l’échantillonnage d’une partie d’un lot d’aliments pour animaux de la même catégorie ou correspondant à la même description révèle que cette partie du lot ne satisfait pas aux exigences de l’Union européenne, il est présumé que la totalité des aliments de ce lot est défectueuse, sauf si une évaluation détaillée n’a pas fourni d’éléments indiquant que le reste du lot ne répond pas aux exigences de l’Union européenne.
Même si l’échantillon officiel est prélevé automatiquement, la présence d’un inspecteur est nécessaire. Cependant, si l’échantillonnage automatique est effectué selon des paramètres préétablis qui ne peuvent être modifiés durant l’échantillonnage et si les échantillons élémentaires sont collectés dans un réceptacle scellé prévenant toute fraude, la présence d’un inspecteur n’est requise qu’au début de l’échantillonnage, chaque fois que le réceptacle contenant les échantillons doit être changé, ainsi qu’à la fin de l’échantillonnage.
8.2.2. Échantillonnage statique de lots transportés par bateau
Si l’échantillonnage est effectué de façon statique, la procédure prévue pour les installations de stockage (silos) accessibles par le haut doit être appliquée (voir le point 8.4.1).
L’échantillonnage doit être effectué sur la partie accessible (par le haut) du lot/de la cale. Le nombre d’échantillons élémentaires est déterminé en tenant compte de la taille de la portion échantillonnée. Si l’échantillonnage d’une partie d’un lot d’aliments pour animaux de la même catégorie ou correspondant à la même description révèle que cette partie du lot ne satisfait pas aux exigences de l’Union européenne, il est présumé que la totalité des aliments de ce lot est défectueuse, sauf si une évaluation détaillée n’a pas fourni d’éléments indiquant que le reste du lot ne répond pas aux exigences de l’Union européenne.
8.3. Échantillonnage de lots de grande taille stockés dans des entrepôts
L’échantillonnage doit être effectué sur la partie accessible du lot. Le nombre d’échantillons élémentaires est déterminé en tenant compte de la taille de la portion échantillonnée. Si l’échantillonnage d’une partie d’un lot d’aliments pour animaux de la même catégorie ou correspondant à la même description révèle que cette partie du lot ne satisfait pas aux exigences de l’Union européenne, il est présumé que la totalité des aliments de ce lot est défectueuse, sauf si une évaluation détaillée n’a pas fourni d’éléments indiquant que le reste du lot ne répond pas aux exigences de l’Union européenne.
8.4. Échantillonnage dans des installations de stockage (silos)
8.4.1. Échantillonnage dans des silos (facilement) accessibles par le haut
L’échantillonnage doit être effectué sur la partie accessible du lot. Le nombre d’échantillons élémentaires est déterminé en tenant compte de la taille de la portion échantillonnée. Si l’échantillonnage d’une partie d’un lot d’aliments pour animaux de la même catégorie ou correspondant à la même description révèle que cette partie du lot ne satisfait pas aux exigences de l’Union européenne, il est présumé que la totalité des aliments de ce lot est défectueuse, sauf si une évaluation détaillée n’a pas fourni d’éléments indiquant que le reste du lot ne répond pas aux exigences de l’Union européenne.
8.4.2. Échantillonnage dans des silos inaccessibles par le haut (silos fermés)
8.4.2.1.
Les aliments pour animaux entreposés dans des silos de ce type ne peuvent faire l’objet d’un échantillonnage statique. Par conséquent, si les aliments pour animaux entreposés dans le silo doivent faire l’objet d’un échantillonnage et s’il n’est pas possible de déplacer le lot, il doit être convenu avec l’opérateur qu’il informe l’inspecteur du moment où le silo sera déchargé afin de permettre l’échantillonnage des aliments en mouvement.
8.4.2.2.
La procédure d’échantillonnage consiste à transférer dans un réceptacle une quantité comprise entre 50 et 100 kg et à y prélever l’échantillon. La taille de l’échantillon global correspond au lot entier et le nombre d’échantillons élémentaires dépend de la quantité transférée du silo vers le réceptacle en vue de l’échantillonnage. Si l’échantillonnage d’une partie d’un lot d’aliments pour animaux de la même catégorie ou correspondant à la même description révèle que cette partie du lot ne satisfait pas aux exigences de l’Union européenne, il est présumé que la totalité des aliments de ce lot est défectueuse, sauf si une évaluation détaillée n’a pas fourni d’éléments indiquant que le reste du lot ne répond pas aux exigences de l’Union européenne.
8.5. Échantillonnage d’aliments pour animaux en vrac dans des conteneurs fermés de grande taille
Il est fréquent que de tels lots ne puissent faire l’objet d’un échantillonnage que quand ils sont déchargés. Dans certains cas, il est impossible de décharger ce type de conteneurs au point d’importation ou de contrôle; c’est pourquoi l’échantillonnage devrait avoir lieu lorsqu’ils sont déchargés.
9. INSTRUCTIONS CONCERNANT LE PRÉLÈVEMENT, LA PRÉPARATION ET LE CONDITIONNEMENT DES ÉCHANTILLONS
9.1. Généralités
Prélever et préparer les échantillons sans délai indu en tenant compte des précautions requises pour éviter que le produit ne soit altéré ou contaminé. Les instruments ainsi que les surfaces et les récipients destinés à recevoir les échantillons doivent être propres et secs.
9.2. Échantillons élémentaires
Les échantillons élémentaires doivent être prélevés au hasard dans l’ensemble de la portion échantillonnée et être approximativement égaux en poids.
La taille minimale de l’échantillon élémentaire est de 100 g ou de 25 g dans le cas de fourrage grossier ou fourrage de faible densité relative.
Si, conformément aux règles de la procédure d’échantillonnage établie au point 8, moins de 40 échantillons élémentaires doivent être prélevés, la taille des échantillons élémentaires doit être déterminée en fonction de la taille requise pour l’échantillon global à constituer (voir le point 6).
En cas d’échantillonnage de petits lots d’aliments pour animaux emballés sur lesquels, conformément aux exigences quantitatives, un nombre limité d’échantillons élémentaires doit être prélevé, le contenu d’une unité d’origine n’excédant pas 1 kg ou 1 litre constitue un échantillon élémentaire.
En cas d’échantillonnage d’aliments pour animaux emballés composés de petites unités (par exemple < 250 g), la taille de l’échantillon élémentaire dépend de la taille de l’unité.
9.2.1. Aliments en vrac
Selon le cas, l’échantillonnage peut avoir lieu lors de la mise en mouvement de la portion échantillonnée (chargement ou déchargement).
9.2.2. Aliments emballés
Le nombre requis d’unités à échantillonner étant délimité comme indiqué au chapitre 5, prélever une partie du contenu de chaque unité au moyen d’une sonde ou d’une pelle. Si nécessaire, prélever les échantillons après avoir vidé séparément les unités.
9.2.3. Aliments liquides ou semi-liquides homogènes ou homogénéisables
Le nombre requis d’unités à échantillonner étant délimité comme indiqué au chapitre 5, effectuer un prélèvement dans chaque unité après en avoir homogénéisé le contenu, si nécessaire.
Les échantillons élémentaires peuvent être prélevés lors du soutirage du contenu.
9.2.4. Aliments liquides ou semi-liquides non homogénéisables
Le nombre requis d’unités à échantillonner étant délimité comme indiqué au chapitre 5, prélever les échantillons à différents niveaux.
Les échantillons peuvent également être prélevés lors du soutirage du contenu, après élimination des premières fractions.
Dans les deux cas, le volume total prélevé ne peut être inférieur à 10 litres.
9.2.5. Aliments en briques et pierres à lécher
Le nombre requis de briques ou de pierres à échantillonner étant délimité comme indiqué au chapitre 5, une partie de chaque brique ou pierre peut être prélevée. En cas de suspicion de non-homogénéité d’une brique ou d’une pierre, l’intégralité de cette dernière peut faire office d’échantillon.
Pour les briques ou pierres à lécher dont le poids unitaire n’excède pas 1 kg, une brique ou une pierre constituent un échantillon élémentaire.
9.3. Préparation des échantillons globaux
Mélanger les échantillons élémentaires pour constituer un seul échantillon global.
9.4. Préparation des échantillons finals
Mélanger soigneusement l’échantillon global ( 4 ).
— Introduire chaque échantillon dans un récipient/réceptacle approprié. Prendre toutes les précautions nécessaires pour éviter toute modification de la composition de l’échantillon ou toute contamination ou altération pouvant survenir au cours du transport ou du stockage.
— Pour le contrôle des constituants ou substances répartis uniformément dans les aliments pour animaux, l’échantillon global peut être réduit de façon représentative à 2 kg ou 2 litres au moins (échantillon réduit) ( 5 ), de préférence au moyen d’un diviseur mécanique ou automatique. Pour le contrôle de la présence de résidus de pesticides dans les légumineuses, les grains de céréales et les fruits à coque, la taille minimale de l’échantillon réduit est de 3 kg. Si la nature des aliments pour animaux ne permet pas d’utiliser un diviseur ou si ce dernier n’est pas disponible, l’échantillon peut être réduit par la méthode des quartiers. À partir des échantillons réduits, préparer ensuite des échantillons finals (à des fins de contrôle, de recours et de référence) de quantité approximativement égale et conformes aux exigences quantitatives figurant au chapitre 7. En cas de contrôle des constituants, y compris le matériel génétiquement modifié, ou substances susceptibles d’être répartis non uniformément dans les matières premières pour aliments des animaux, l’échantillon global est:
—
— complètement homogénéisé puis divisé en échantillons finals, ou
— réduit à 2 kg ou 2 litres au moins ( 6 ) au moyen d’un diviseur mécanique ou automatique. Si la nature des aliments pour animaux ne permet pas d’utiliser un diviseur, et dans ce cas uniquement, l’échantillon peut, si nécessaire, être réduit par la méthode des quartiers. Pour le contrôle de la présence de matériel génétiquement modifié dans le cadre du règlement (UE) no 619/2011, l’échantillon réduit doit contenir au moins 35 000 graines/semences pour permettre d’obtenir des échantillons finals à des fins de contrôle, de recours et de référence comportant au moins 10 000 graines/semences [voir la note de bas de page (**) au chapitre 6 et la note de bas de page (*) au chapitre 7].
9.5. Conditionnement des échantillons
Sceller et étiqueter les récipients ou emballages de façon à ce qu’il soit impossible de les ouvrir sans briser le scellé. L’étiquette générale doit être incorporée dans le scellé.
9.6. Envoi des échantillons au laboratoire
Transmettre l’échantillon sans délai indu au laboratoire d’analyse désigné, avec les indications nécessaires à l’analyse.
10. PROCÈS-VERBAL D’ÉCHANTILLONNAGE
Pour chaque échantillon, établir un procès-verbal d’échantillonnage permettant d’identifier sans ambiguïté la portion échantillonnée et sa taille.
Mentionner également dans le procès-verbal tout écart par rapport à la procédure d’échantillonnage prévue par le présent règlement.
Mettre le procès-verbal à la disposition du laboratoire de contrôle officiel ainsi que de l’exploitant du secteur de l’alimentation animale et/ou du laboratoire désigné par ce dernier.
ANNEXE II
DISPOSITIONS GÉNÉRALES CONCERNANT LES MÉTHODES D’ANALYSE DES ALIMENTS POUR ANIMAUX
A. PRÉPARATION DES ÉCHANTILLONS EN VUE DE L’ANALYSE
1. Objet
Les procédures décrites ci-après portent sur la préparation, en vue de leur analyse, des échantillons envoyés aux laboratoires de contrôle après leur prélèvement conformément aux dispositions de l’annexe I.
La préparation des échantillons de laboratoire doit permettre que les prises d’essais prévues dans les méthodes d’analyse soient homogènes et représentatives des échantillons finals.
2. Précautions à prendre
La procédure à suivre pour préparer des échantillons dépend des méthodes d’analyse à appliquer et des constituants ou substances à contrôler. Il est donc essentiel de veiller à ce que la procédure suivie en la matière soit adaptée à la méthode d’analyse appliquée ainsi qu’aux constituants ou substances à contrôler.
Effectuer toutes les opérations de façon à éviter autant que possible une contamination de l’échantillon ou des modifications de sa composition.
Effectuer les broyages, les mélanges et les tamisages sans délai, en exposant au minimum l’échantillon à l’air et à la lumière. Éviter l’utilisation de moulins ou de broyeurs susceptibles de produire un échauffement notable de l’échantillon.
Le broyage manuel est recommandé pour les aliments particulièrement sensibles à la chaleur. Veiller, en outre, à ce que l’appareillage même ne soit pas une source de contamination.
Si la préparation entraîne inévitablement une modification significative de la teneur en humidité de l’échantillon, déterminer sa teneur en humidité avant et après la préparation selon la méthode prévue à l’annexe III, point A.
3. Procédure
3.1. Procédure générale
L’aliquote de test est prélevée sur l’échantillon final. La méthode des quartiers opposés n’est pas recommandée, car elle peut aboutir à des aliquotes de test présentant une erreur de division élevée.
3.1.1.
— Mélanger l’échantillon final tamisé et le recueillir dans un récipient approprié, propre et sec, muni d’une fermeture hermétique. Mélanger de nouveau pour garantir une homogénéisation complète, immédiatement avant de prélever la prise d’essai (aliquote de test).
3.1.2.
— Sauf indication spécifique dans les méthodes d’analyse, dessécher l’échantillon final, de façon à ramener sa teneur en humidité à un niveau compris entre 8 et 12 %, en appliquant le procédé de prédessiccation décrit au point 4.3 de la méthode de dosage de l’humidité mentionnée à l’annexe III, point A. Procéder ensuite comme indiqué au point 3.1.1.
3.1.3.
— Recueillir l’échantillon final dans un récipient approprié, propre et sec, muni d’une fermeture hermétique. Mélanger soigneusement pour garantir une homogénéisation complète, immédiatement avant de prélever la prise d’essai (aliquote de test).
3.1.4.
— Si l’échantillon final ne peut être préparé selon l’un des procédés indiqués ci-dessus, appliquer tout autre procédé de préparation approprié permettant d’obtenir des prises d’essai (aliquotes de test) homogènes et représentatives des échantillons finals.
3.2. Procédure spécifique en cas d’examen visuel ou microscopique ou dans les cas où l’échantillon global est entièrement homogénéisé
— En cas d’examen visuel (sans microscope), l’échantillon de laboratoire est examiné dans son intégralité.
— En cas d’examen au microscope, le laboratoire peut réduire l’échantillon global ou réduire encore davantage l’échantillon réduit. Les échantillons finals destinés à des fins de recours et, éventuellement, à des fins de référence sont prélevés selon une procédure équivalente à la procédure appliquée pour prélever l’échantillon final destiné à des fins de contrôle.
— Si l’échantillon global est entièrement homogénéisé, les échantillons finals sont prélevés sur l’échantillon global homogénéisé.
4. Conservation et stockage des échantillons
Conserver les échantillons à une température ne pouvant modifier leur composition. Conserver les échantillons destinés à l’analyse de vitamines ou de substances particulièrement sensibles à la lumière dans des conditions telles qu’ils ne soient pas altérés par la lumière.
B. DISPOSITIONS CONCERNANT LES RÉACTIFS ET L’APPAREILLAGE UTILISÉS DANS LES MÉTHODES D’ANALYSE
1. Sauf indication spécifique dans les méthodes d’analyse, tous les réactifs doivent être de qualité «pour analyse» (p.a.). Pour l’analyse des oligoéléments, la pureté des réactifs doit être contrôlée par un essai à blanc. Selon le résultat obtenu, une purification supplémentaire des réactifs peut être requise.
2. Les opérations de mise en solution, de dilution, de rinçage ou de lavage mentionnées dans les méthodes d’analyse sans indication quant à la nature du solvant ou du diluant impliquent qu’il faut utiliser de l’eau. En règle générale, l’eau doit être déminéralisée ou distillée. Dans des cas particuliers, indiqués dans les méthodes d’analyse, elle doit être soumise à des procédés spécifiques de purification.
3. Compte tenu de l’équipement usuel des laboratoires de contrôle, seuls les instruments et appareils spéciaux ou devant répondre à des conditions spécifiques sont mentionnés dans les méthodes d’analyse. Ce matériel doit être propre, tout particulièrement pour les déterminations de très faibles quantités de substances.
C. APPLICATION DES MÉTHODES D’ANALYSE ET EXPRESSION DES RÉSULTATS
1. Procédé d’extraction
Plusieurs méthodes déterminent un procédé d’extraction spécifique. En règle générale, des procédés d’extraction autres que celui visé dans la méthode peuvent être appliqués à condition que le procédé d’extraction appliqué ait une efficacité d’extraction équivalente avérée, pour la matrice analysée, à celle du procédé mentionné dans la méthode.
2. Procédé de purification
Plusieurs méthodes déterminent un procédé de purification spécifique. En règle générale, des procédés de purification autres que celui visé dans la méthode peuvent être appliqués, à condition qu’il soit prouvé que le procédé de purification appliqué donne des résultats d’analyse équivalents, pour la matrice analysée, à ceux que donne le procédé mentionné dans la méthode.
3. Nombre de déterminations
Si, lors de l’analyse de substances indésirables, le résultat de la première détermination est nettement (> 50 %) inférieur à la spécification à contrôler, il n’est pas nécessaire de procéder à une détermination supplémentaire, à condition que les procédures appropriées en matière de qualité aient été suivies. Dans les autres cas, une double analyse (deuxième détermination) est nécessaire pour exclure la possibilité d’une contamination croisée interne ou un mélange accidentel des échantillons. La moyenne des deux déterminations, compte tenu de l’incertitude de mesure, sert à vérifier la conformité.
Si, lors du contrôle de la teneur déclarée en une substance ou un ingrédient donné, le résultat de la première détermination confirme l’exactitude de la teneur déclarée (ce qui signifie que l’écart entre le résultat de l’analyse et la teneur déclarée se situe dans la plage admissible), il n’est pas nécessaire de procéder à des déterminations supplémentaires, à condition que les procédures appropriées en matière de qualité aient été suivies. Dans les autres cas, une double analyse (deuxième détermination) est nécessaire pour exclure la possibilité d’une contamination croisée interne ou un mélange accidentel des échantillons. La moyenne des deux déterminations, compte tenu de l’incertitude de mesure, sert à vérifier la conformité.
Dans certains cas, l’écart admissible est défini par la législation, comme dans le règlement (CE) no 767/2009 du Parlement européen et du Conseil du 13 juillet 2009 concernant la mise sur le marché et l’utilisation des aliments pour animaux, modifiant le règlement (CE) no 1831/2003 du Parlement européen et du Conseil et abrogeant la directive 79/373/CEE du Conseil, la directive 80/511/CEE de la Commission, les directives 82/471/CEE, 83/228/CEE, 93/74/CEE, 93/113/CE et 96/25/CE du Conseil, ainsi que la décision 2004/217/CE de la Commission ( 7 ).
4. Mention de la méthode d’analyse appliquée
Le bulletin d’analyse doit mentionner la méthode d’analyse appliquée.
5. Indication du résultat de l’analyse
Le résultat de l’analyse doit être exprimé conformément aux indications données dans la méthode d’analyse avec un nombre approprié de chiffres significatifs, et être corrigé, si nécessaire, en fonction de la teneur en humidité de l’échantillon final avant sa préparation.
6. Incertitude de mesure et taux de récupération en cas d’analyse de substances indésirables
En ce qui concerne les substances indésirables au sens de la directive 2002/32/CE, un produit destiné à l’alimentation animale est considéré comme ne satisfaisant pas à la teneur maximale fixée lorsque le résultat de l’analyse, rapporté à un aliment d’une teneur en humidité de 12 %, est jugé supérieur à la teneur maximale, compte tenu de l’incertitude de mesure élargie et de la correction de la récupération. La concentration analysée, corrigée au titre de la récupération et après soustraction de l’incertitude de mesure élargie, est utilisée pour l’évaluation de la conformité. Ce procédé est applicable uniquement dans les cas où la méthode d’analyse autorise l’estimation de l’incertitude de mesure et de la correction de la récupération (ce qui n’est pas possible, par exemple, dans le cas d’une analyse microscopique).
Le résultat de l’analyse est rapporté comme suit (lorsque la méthode d’analyse appliquée permet d’estimer l’incertitude de mesure et le taux de récupération):
a) corrigé au titre de la récupération, le taux de récupération étant indiqué. La correction de la récupération n’est pas nécessaire lorsque le taux de récupération est compris entre 90 et 110 %;
b) sous la forme «x +/– U», où x est le résultat de l’analyse et U l’incertitude de mesure élargie, calculée à l’aide d’un coefficient de couverture 2 qui donne un niveau de confiance d’environ 95 %.
Néanmoins, si le résultat de l’analyse est nettement (> 50 %) inférieur à la spécification à contrôler, et à condition que les procédures appropriées en matière de qualité aient été suivies et que l’analyse vise uniquement à contrôler si les dispositions légales sont respectées, ce résultat peut être mentionné sans correction de la récupération, et la mention du taux de récupération et de l’incertitude de mesure peut être omise dans ce cas.
ANNEXE III
MÉTHODES D'ANALYSE RELATIVES AU CONTRÔLE DE LA COMPOSITION DES MATIÈRES PREMIÈRES POUR ALIMENTS DES ANIMAUX ET DES ALIMENTS COMPOSÉS
A. DOSAGE DE L'HUMIDITÉ
1. Objet et champ d'application
La méthode permet de déterminer la teneur en humidité des aliments pour animaux. Dans le cas d'aliments pour animaux contenant des substances volatiles telles que des acides organiques, il a été observé qu'une quantité significative de substances volatiles était dosée en même temps que la teneur en humidité.
Elle ne concerne pas l'analyse des produits laitiers en tant que matières premières pour aliments des animaux, l'analyse des substances minérales et des mélanges essentiellement composés de substances minérales, l'analyse des graisses et des huiles animales et végétales, ni l'analyse des graines et des fruits oléagineux.
2. Principe
L'échantillon est soumis à la dessiccation dans des conditions définies, variant en fonction de la nature de l'aliment pour animaux. La perte de poids est déterminée par pesée. Il est nécessaire de procéder à une prédessiccation lorsqu'il s'agit d'aliments solides qui ont une teneur en humidité élevée.
3. Appareillage
3.1. Broyeur construit en matériau n'absorbant pas l'humidité, facile à nettoyer, permettant un broyage rapide et uniforme sans provoquer d'échauffement sensible, évitant au maximum le contact avec l'air extérieur et permettant de satisfaire aux exigences énoncées aux points 4.1.1 et 4.1.2 (par exemple, microbroyeurs à marteaux ou à refroidissement à eau, moulins à cônes démontables, broyeurs à mouvement lent ou à disques dentés).
3.2. Balance d'analyse d'une précision de 1 mg.
3.3. Récipients secs en métal inoxydable ou en verre munis d'un couvercle hermétique; surface utile permettant d'obtenir une répartition de la prise d'essai de 0,3 g par cm2.
3.4. Étuve isotherme (± 2 oC) à chauffage électrique, assurant une régulation rapide de la température et convenablement ventilée ( 8 ).
3.5. Étuve à vide, à chauffage électrique réglable, munie d'une pompe à huile et soit d'un dispositif à introduction d'air chaud déshydraté, soit d'un déshydratant (par exemple, de l'oxyde de calcium).
3.6. Dessiccateur à plaque en métal ou en porcelaine, épaisse, perforée, contenant un déshydratant efficace.
4. Mode opératoire
|
NB: |
Les opérations décrites au présent point doivent être effectuées immédiatement après l'ouverture des emballages contenant les échantillons. Les analyses doivent être effectuées au moins en double. |
4.1. Préparation
4.1.1.
Prélever au moins 50 g de l'échantillon. Si nécessaire, broyer ou diviser de façon appropriée pour éviter toute variation de la teneur en humidité (voir point 6).
4.1.2.
Prélever au moins 50 g de l'échantillon. Moudre en particules dont au moins 50 % passent par un tamis à mailles de 0,5 mm et ne laissent pas plus de 10 % de refus sur le tamis à mailles rondes de 1 mm.
4.1.3.
Prélever et peser, à 10 mg près, 25 g environ de l'échantillon, y ajouter une quantité appropriée de sable anhydre, pesée à 10 mg près, et mélanger jusqu'à obtention d'un produit homogène.
4.2. Dessiccation
4.2.1.
Tarer, à 1 mg près, un récipient (3.3) muni de son couvercle. Peser, à 1 mg près, dans le récipient taré 5 g environ de l'échantillon et répartir uniformément la prise d'essai. Placer le récipient, sans son couvercle, dans l'étuve préchauffée à 103 oC. Pour éviter que la température de l'étuve ne descende trop, introduire le récipient en un minimum de temps. Laisser sécher durant quatre heures à partir du moment où l'étuve a de nouveau atteint la température de 103 oC. Remettre le couvercle sur le récipient, retirer celui-ci de l'étuve, laisser refroidir 30 à 45 minutes dans le dessiccateur (3.6) et peser à 1 mg près.
Dans le cas des aliments constitués essentiellement de matières grasses, effectuer une dessiccation complémentaire de 30 minutes dans l'étuve à 130 oC. L'écart entre les deux pesées ne peut excéder 0,1 % d'humidité.
4.2.2.
Tarer, à 0,5 mg près, un récipient (3.3) muni de son couvercle. Peser, à 1 mg près, dans le récipient taré 5 g environ de l'échantillon broyé et répartir uniformément la prise d'essai. Placer le récipient, sans son couvercle, dans l'étuve préchauffée à 130 oC. Pour éviter que la température de l'étuve ne descende trop, introduire le récipient en un minimum de temps. Laisser sécher durant deux heures à partir du moment où l'étuve a de nouveau atteint la température de 130 oC. Remettre le couvercle sur le récipient, retirer celui-ci de l'étuve, laisser refroidir 30 à 45 minutes dans le dessiccateur (3.6) et peser à 1 mg près.
4.2.3. Aliments composés pour animaux contenant plus de 4 % de saccharose ou de lactose: matières premières pour aliments des animaux tels que caroube, produits céréaliers hydrolysés, germes de malt, cossettes de betteraves, solubles de poisson et sucres; aliments composés pour animaux contenant plus de 25 % de sels minéraux renfermant de l'eau de cristallisation
Tarer, à 0,5 mg près, un récipient (3.3) muni de son couvercle. Peser, à 1 mg près, dans le récipient taré 5 g environ de l'échantillon et répartir uniformément la prise d'essai. Placer le récipient, sans son couvercle, dans l'étuve à vide (3.5) préchauffée à une température comprise entre 80 et 85 oC. Pour éviter que la température de l'étuve ne descende trop, introduire le récipient en un minimum de temps.
Amener la pression à 100 torrs et laisser sécher à cette pression durant quatre heures, soit sous un courant d'air sec et chaud, soit à l'aide d'un déshydratant (300 g environ pour 20 échantillons). Dans ce dernier cas, couper la connexion avec la pompe à vide lorsque la pression prescrite est atteinte. Compter la durée de séchage à partir du moment où l'étuve a de nouveau atteint la température de 80 à 85 oC. Ramener ensuite avec précaution l'étuve à la pression atmosphérique. Ouvrir l'étuve, couvrir immédiatement le récipient de son couvercle, retirer le récipient de l'étuve, laisser refroidir durant 30 à 45 minutes dans le dessiccateur (3.6) et peser à 1 mg près. Procéder à une dessiccation complémentaire de 30 minutes dans l'étuve à vide à la température de 80 à 85 oC et peser de nouveau. L'écart entre les deux pesées ne peut excéder 0,1 % d'humidité.
4.3. Prédessiccation
4.3.1.
Les aliments solides, dont la teneur en humidité est élevée et rend le broyage difficile, doivent être prédesséchés comme suit.
Peser, à 10 mg près, 50 g de l'échantillon non broyé (une division grossière peut être effectuée, si nécessaire, dans le cas des aliments comprimés ou agglomérés) dans un récipient approprié (par exemple, une plaque en aluminium de 20 × 12 cm munie d'un bord de 0,5 cm). Laisser sécher dans une étuve à la température de 60 à 70 oC, jusqu'à ce que la teneur en humidité soit ramenée à une valeur comprise entre 8 et 12 %. Retirer de l'étuve, laisser refroidir à découvert dans le laboratoire durant une heure et peser à 10 mg près. Broyer immédiatement après comme indiqué au point 4.1.1 et effectuer la dessiccation comme indiqué au point 4.2.1 ou 4.2.3, selon la nature de l'aliment.
4.3.2.
Les grains dont la teneur en humidité est supérieure à 17 % doivent être prédesséchés comme suit.
Peser, à 10 mg près, 50 g du grain non moulu dans un récipient approprié (par exemple, une plaque en aluminium de 20 × 12 cm munie d'un bord de 0,5 cm). Laisser sécher dans une étuve pendant 5 à 7 minutes à la température de 130 oC. Retirer de l'étuve, laisser refroidir à découvert dans le laboratoire durant deux heures et peser à 10 mg près. Moudre immédiatement après, comme indiqué au point 4.1.2 et effectuer la dessiccation comme indiqué au point 4.2.2.
5. Calcul des résultats
La teneur en humidité (X), exprimée en pourcentage de l'échantillon, est donnée par les formules suivantes.
5.1. Dessiccation sans prédessiccation

où:
|
m |
= |
poids initial, en grammes, de la prise d'essai, |
|
m0 |
= |
poids, en grammes, de la prise d'essai sèche. |
5.2. Dessiccation avec prédessiccation

où:
|
m |
= |
poids initial, en grammes, de la prise d'essai, |
|
m1 |
= |
poids, en grammes, de la prise d'essai après prédessiccation, |
|
m2 |
= |
poids, en grammes, de la prise d'essai après broyage ou mouture, |
|
m0 |
= |
poids, en grammes, de la prise d'essai sèche. |
5.3. Répétabilité
La différence entre les résultats de deux dosages parallèles effectués sur un même échantillon ne peut dépasser 0,2 % de l'humidité (valeur absolue).
6. Observation
Si un broyage se révèle nécessaire et s'il s'avère que celui-ci entraîne une variation de la teneur en humidité du produit, les résultats de l'analyse des composants de l'aliment doivent être corrigés en fonction de la teneur en humidité de l'échantillon initial.
B. DOSAGE DE L'HUMIDITÉ DANS LES GRAISSES ET LES HUILES ANIMALES ET VÉGÉTALES
1. Objet et champ d'application
La méthode permet de déterminer la teneur en humidité (eau et autres matières volatiles) des graisses et des huiles animales et végétales.
2. Principe
L'échantillon est soumis à la dessiccation à 103 oC jusqu'à cessation de la diminution de la masse (la perte de masse entre deux pesées successives doit être inférieure ou égale à 1 mg). La perte de poids est déterminée par pesée.
3. Appareillage
3.1. Récipient à fond plat, en matériau résistant à la corrosion, d'un diamètre de 8 à 9 cm et d'une hauteur de 3 cm environ.
3.2. Thermomètre, avec bulbe renforcé, et chambre de dilatation à l'extrémité supérieure, gradué de 80 oC environ à 110 oC au moins, d'une longueur de 10 cm environ.
3.3. Bain de sable ou plaque chauffante électrique.
3.4. Dessiccateur, contenant un déshydratant efficace.
3.5. Balance d'analyse.
4. Mode opératoire
Peser, à 1 mg près, 20 g de l'échantillon homogénéisé dans le récipient (3.1) sec et taré contenant le thermomètre (3.2). Chauffer sur le bain de sable ou la plaque chauffante (3.3), en agitant constamment à l'aide du thermomètre, de façon à ce que la température atteigne 90 oC en 7 minutes environ.
Réduire l'intensité du chauffage en suivant la fréquence avec laquelle les bulles montent du fond du récipient. La température ne peut dépasser 105 oC. Continuer à agiter en raclant le fond du récipient jusqu'à cessation de la formation de bulles.
Pour assurer l'élimination complète de l'humidité, répéter à plusieurs reprises le chauffage à 103 oC ± 2 oC, en refroidissant à 93 oC entre les chauffages successifs. Laisser refroidir ensuite dans le dessiccateur (3.4) jusqu'à température ambiante et peser. Répéter cette opération jusqu'à ce que la perte de poids entre deux pesées successives n'excède plus 2 mg.
|
NB: |
Une augmentation de poids de l'échantillon après chauffage répété indique une oxydation de la graisse. Si cela se produit, calculer le résultat à partir de la pesée effectuée immédiatement avant le début de l'augmentation de poids. |
5. Calcul des résultats
La teneur en humidité (X), exprimée en pourcentage de l'échantillon, est donnée par la formule suivante.

où:
|
m |
= |
poids, en grammes, de la prise d'essai, |
|
m1 |
= |
poids, en grammes, du récipient avec son contenu, avant le chauffage, |
|
m2 |
= |
poids, en grammes, du récipient avec son contenu, après le chauffage. |
Les résultats inférieurs à 0,05 % doivent être rapportés par la mention «moins de 0,05 %».
Répétabilité
La différence entre les résultats de deux dosages parallèles effectués sur un même échantillon ne peut dépasser 0,05 % en valeur absolue.
C. DÉTERMINATION DE LA TENEUR EN PROTÉINES BRUTES
1. Objet et champ d'application
La méthode permet de déterminer la teneur en protéines brutes des aliments pour animaux à partir de la teneur en azote dosée selon la méthode de Kjeldahl.
2. Principe
L'échantillon est minéralisé à l'acide sulfurique en présence d'un catalyseur. La solution acide est alcalinisée par une solution d'hydroxyde de sodium. L'ammoniac est entraîné par distillation et recueilli dans une quantité déterminée d'acide sulfurique dont l'excès est titré par une solution étalon d'hydroxyde de sodium.
Autre possibilité: l'ammoniac libéré est distillé dans un excès de solution d'acide borique, après quoi l'ammoniac combiné avec l'acide borique est titré par une solution d'acide chlorhydrique ou d'acide sulfurique.
3. Réactifs
3.1. Sulfate de potassium.
3.2. Catalyseur: oxyde de cuivre (II) CuO ou sulfate de cuivre (II) pentahydraté CuSO45H2O.
3.3. Zinc en granulés.
3.4. Acide sulfurique, ρ20 = 1,84 g/ml.
3.5. Acide sulfurique, solution étalon volumétrique, c(H2SO4) = 0,25 mol/l.
3.6. Acide sulfurique, solution étalon volumétrique, c(H2SO4) = 0,10 mol/l.
3.7. Acide sulfurique, solution étalon volumétrique, c(H2SO4) = 0,05 mol/l.
3.8. Rouge de méthyle (indicateur): dissoudre 300 g de rouge de méthyle dans 100 ml d'éthanol, σ = 95-96 % (v/v).
3.9. Solution d'hydroxyde de sodium (utilisation possible de la qualité technique) β = 40 g/100 ml (m/v: 40 %).
3.10. Hydroxyde de sodium, solution étalon volumétrique, c(NaOH) = 0,25 mol/l.
3.11. Hydroxyde de sodium, solution étalon volumétrique, c(NaOH) = 0,10 mol/l.
3.12. Granulés de pierre ponce lavés à l'acide chlorhydrique et calcinés.
3.13. Acétanilide (point de fusion = 114 oC, teneur N = 10,36 %).
3.14. Saccharose (exempt d'azote).
3.15. Acide borique (H3BO3).
3.16. Solution d'indicateur de rouge de méthyle: dissoudre 100 mg de rouge de méthyle dans 100 ml d'éthanol ou de méthanol.
3.17. Solution de vert de bromocrésol: dissoudre 100 mg de vert de bromocrésol dans 100 ml d'éthanol ou de méthanol.
3.18. Solution d'acide borique (de 10 g/l à 40 g/l en fonction de l'appareillage utilisé).
En cas de détection colorimétrique au point d'équivalence, les indicateurs (rouge de méthyle et vert de bromocrésol) doivent être ajoutés aux solutions d'acide borique. Si 1 l de solution d'acide borique est préparé, avant ajustement du volume, ajouter 7 ml de solution d'indicateur de rouge de méthyle (3.16) et 10 ml de solution de vert de bromocrésol (3.17).
En fonction de l'eau utilisée, le pH de la solution d'acide borique peut varier d'un lot à l'autre. Il est fréquent qu'il faille opérer un ajustement au moyen d'un petit volume d'alcali pour obtenir un blanc positif.
|
NB: |
L'ajout d'environ 3 à 4 ml de NaOH (3.11) dans 1 l de solution d'acide borique à 10 g/l donne généralement de bons ajustements. Conserver la solution à température ambiante et la protéger de la lumière et des sources de vapeurs d'ammoniac pendant sa conversation. |
3.19. Acide chlorhydrique, solution étalon volumétrique, c(HCL) = 0,10 mol/l.
|
NB: |
Il est permis de modifier les concentrations des solutions volumétriques (points 3.5, 3.6, 3.7, 3.10, 3.11, et 3.19), à condition d'apporter les corrections nécessaires dans les calculs. Les concentrations devraient toujours être exprimées par des chiffres à quatre décimales. |
4. Appareillage
Appareils permettant de réaliser les opérations de minéralisation, de distillation et de titrage selon la méthode de Kjeldahl.
5. Mode opératoire
5.1. Minéralisation
Peser, à 0,001 g près, 1 g de l'échantillon et introduire la prise d'essai dans le ballon de l'appareil de minéralisation. Ajouter 15 g de sulfate de potassium (3.1), une quantité appropriée de catalyseur (3.2) [0,3 à 0,4 g d'oxyde de cuivre (II) ou 0,9 à 1,2 g de sulfate de cuivre (II) pentahydraté], 25 ml d'acide sulfurique (3.4) et, si nécessaire, quelques grains de pierre ponce (3.12) et mélanger.
Chauffer le ballon, avec modération dans un premier temps, en agitant de temps en temps, si nécessaire, jusqu'à carbonisation de la masse et disparition de l'écume; chauffer ensuite plus intensément jusqu'à ébullition régulière du liquide. Le chauffage est approprié si l'acide en ébullition se condense sur la paroi du ballon. Éviter la surchauffe des parois et l'adhérence de particules organiques.
Lorsque la solution apparaît limpide et vert clair, maintenir l'ébullition durant 2 heures puis laisser refroidir.
5.2. Distillation
Ajouter avec précaution une quantité suffisante d'eau pour garantir une dissolution complète des sulfates. Laisser refroidir et ajouter, s'il le faut, quelques grains de zinc (3.3). Procéder conformément au point 5.2.1 ou 5.2.2.
5.2.1.
Introduire, dans le flacon collecteur de l'appareil à distiller, 25 ml exactement mesurés d'acide sulfurique 3.5 ou 3.7, selon la teneur présumée en azote. Introduire quelques gouttes d'indicateur au rouge de méthyle (3.8).
Connecter le ballon de minéralisation au réfrigérant de l'appareil à distiller et plonger l'extrémité du réfrigérant sur une hauteur de 1 cm au moins dans le liquide du flacon collecteur (voir observation 8.3). Verser lentement 100 ml de solution d'hydroxyde de sodium (3.9) dans le ballon de minéralisation sans provoquer de perte d'ammoniac (voir observation 8.1). Chauffer le ballon jusqu'à distillation complète de l'ammoniac.
5.2.2.
Lorsque le titrage de la teneur en ammoniac du distillat est effectué manuellement, le mode opératoire décrit ci-après est applicable. Lorsque l'unité de distillation est entièrement automatisée, y compris pour ce qui concerne le titrage de la teneur en ammoniac du distillat, suivre les instructions d'utilisation de l'unité de distillation fournies par le fabricant.
Placer un flacon collecteur contenant de 25 à 30 ml de solution d'acide borique (3.18) en sortie du réfrigérant de manière telle que le tube d'évacuation soit plongé dans l'excès de solution d'acide borique. Régler l'unité de distillation pour obtenir 50 ml de solution d'hydroxyde de sodium (3.9). Manier l'unité de distillation conformément aux instructions du fabricant et éliminer l'ammoniac libéré par distillation en ajoutant la solution d'hydroxyde de sodium. Recueillir le distillat dans la solution d'acide borique. La quantité de distillat (durée de distillation à la vapeur) dépend de la quantité d'azote dans l'échantillon. Suivre les instructions du fabricant.
|
NB: |
Une unité de distillation semi-automatique effectue automatiquement l'addition d'excès d'hydroxyde de sodium et la distillation à la vapeur. |
5.3. Titrage
Procéder conformément au point 5.3.1 ou 5.3.2.
5.3.1.
Titrer, dans le flacon collecteur, l'excès d'acide sulfurique par la solution d'hydroxyde de sodium (3.10 ou 3.11), selon la concentration de l'acide sulfurique utilisé, jusqu'à atteindre le point d'équivalence.
5.3.2.
Titrer le contenu du flacon collecteur par la solution étalon volumétrique d'acide chlorhydrique (3.19) ou par la solution étalon volumétrique d'acide sulfurique (3.6) au moyen d'une burette et lire la quantité de solution titrée utilisée.
En cas de détection colorimétrique au point d'équivalence, le point d'équivalence est atteint dès la première trace de couleur rose dans le contenu. Estimer le contenu de la burette à 0,05 ml près. Un agitateur magnétique illuminé ou un détecteur photométrique peut faciliter la visualisation du point d'équivalence.
Cette étape peut être réalisée automatiquement au moyen d'une unité de distillation à titrage automatique.
Suivre les instructions du fabricant lors de l'utilisation d'une unité de distillation ou d'une unité de distillation avec titreur.
|
NB: |
En cas d'utilisation d'un système de titrage automatique, le titrage débute immédiatement après le démarrage de la distillation et la solution d'acide borique à 1 % (3.18) est utilisée. En cas d'utilisation d'une unité de distillation entièrement automatique, le titrage automatique de l'ammoniac peut se faire par détection au point d'équivalence au moyen d'un système potentiométrique (pH). Dans ce cas, un titreur automatique à pH-mètre est utilisé. Le pH-mètre doit être correctement étalonné dans une plage allant de pH 4 à pH 7 conformément aux procédures d'étalonnage habituelles. Le point d'équivalence du titrage est atteint à pH 4,6 , qui est le point d'inflexion de la courbe de titrage. |
5.4. Essai à blanc
Pour confirmer que les réactifs sont exempts d'azote, effectuer un essai à blanc (minéralisation, distillation et titrage) en utilisant 1 g de saccharose (3.14) en lieu en place de l'échantillon.
6. Calcul des résultats
Les calculs sont effectués conformément au point 6.1 ou 6.2.
6.1. Calcul pour le titrage conformément au point 5.3.1
La teneur en protéines brutes, exprimée sous forme de pourcentage en poids, se calcule selon la formule suivante:

où:
|
V0 |
= |
volume (ml) de NaOH (3.10 ou 3.11) utilisé dans l'essai à blanc, |
|
V1 |
= |
volume (ml) de NaOH (3.10 ou 3.11) utilisé pour le titrage de l'échantillon, |
|
c |
= |
concentration (mol/l) de l'hydroxyde de sodium (3.10 ou 3.11), |
|
m |
= |
poids (g) d'échantillon. |
6.2. Calcul pour le titrage conformément au point 5.3.2
6.2.1.
La teneur en protéines brutes, exprimée sous forme de pourcentage en poids, se calcule selon la formule suivante:

où:
|
m |
= |
poids (g) de la prise d'essai, |
|
c |
= |
concentration (mol/l) de la solution étalon volumétrique d'acide chlorhydrique (3.19), |
|
V0 |
= |
volume (ml) d'acide chlorhydrique utilisé dans l'essai à blanc, |
|
V1 |
= |
volume (ml) d'acide chlorhydrique utilisé dans la prise d'essai. |
6.2.2.
La teneur en protéines brutes, exprimée sous forme de pourcentage en poids, se calcule selon la formule suivante:
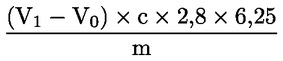
où:
|
m |
= |
poids (g) de la prise d'essai, |
|
c |
= |
concentration (mol/l) de la solution étalon volumétrique d'acide sulfurique (3.6), |
|
V0 |
= |
volume (ml) d'acide sulfurique (3.6) utilisé dans l'essai à blanc, |
|
V1 |
= |
volume (ml) d'acide sulfurique (3.6) utilisé dans la prise d'essai. |
7. Vérification de la méthode
7.1. Répétabilité
La différence entre les résultats de deux déterminations parallèles effectuées sur le même échantillon ne peut dépasser:
— 0,2 % en valeur absolue pour les teneurs en protéines brutes inférieures à 20 %,
— 1,0 % par rapport au résultat le plus élevé, pour les teneurs en protéines brutes comprises entre 20 et 40 %,
— 0,4 % en valeur absolue pour les teneurs supérieures à 40 %.
7.2. Exactitude
Effectuer l'analyse (minéralisation, distillation et titrage) à l'aide de 1,5 à 2,0 g d'acétanilide (3.13) en présence de 1 g de saccharose (3.14); 1 g d'acétanilide consomme 14,80 ml d'acide sulfurique (3.5). La récupération doit être de 99 % au moins.
8. Observations
8.1. L'appareil peut être de type manuel, semi-automatique ou automatique. Si l'appareil nécessite un transfert entre les étapes de minéralisation et de distillation, ce transfert doit être effectué sans perte. Si le ballon de l'appareil à distiller n'est pas équipé d'un entonnoir à robinet, ajouter l'hydroxyde de sodium immédiatement avant la connexion du ballon au réfrigérant en versant lentement le liquide le long de la paroi.
8.2. Si le produit minéralisé se solidifie, recommencer la détermination en utilisant une quantité d'acide sulfurique (3.4) plus grande que celle indiquée ci-dessus.
8.3. Pour les produits à faible teneur en azote, le volume d'acide sulfurique (3.7) à mettre dans le ballon collecteur peut être réduit, si nécessaire, à 10 ou à 15 ml et porté à 25 ml par addition d'eau.
8.4. Pour les analyses de routine, d'autres méthodes d'analyse peuvent être appliquées pour le dosage des protéines brutes, mais la méthode de Kjeldahl décrite au présent point C est la méthode de référence. L'équivalence entre les résultats de la méthode de substitution (par exemple, la méthode de Dumas) et ceux de la méthode de référence doit être démontrée pour chaque matrice séparément. Étant donné que, même après vérification de l'équivalence, les résultats obtenus au moyen d'une méthode de substitution peuvent s'écarter légèrement des résultats qui auraient été obtenus au moyen de la méthode de référence, il est nécessaire de mentionner sur le bulletin d'analyse la méthode d'analyse appliquée pour le dosage des protéines brutes.
D. DOSAGE DE L'URÉE
1. Objet et champ d'application
La méthode permet de déterminer la teneur en urée des aliments pour animaux.
2. Principe
L'échantillon est mis en suspension dans de l'eau en présence d'un défécant. La suspension est filtrée. La teneur en urée du filtrat est déterminée, après addition de 4-diméthylaminobenzaldéhyde (4-DMAB), par mesure de la densité optique à la longueur d'onde de 420 mm.
3. Réactifs
3.1. Solution de 4-diméthylaminobenzaldéhyde: dissoudre 1,6 g de 4-DMAB dans 100 ml d'éthanol à 96 % et ajouter 10 ml d'acide chlorhydrique (ρ201,19 g/ml). Ce réactif se conserve au maximum deux semaines.
3.2. Solution de Carrez I: dissoudre dans l'eau 21,9 g d'acétate de zinc Zn(CH3COO)2 2H2O, et 3 g d'acide acétique glacial. Ajuster à 100 ml avec de l'eau.
3.3. Solution de Carrez II: dissoudre dans l'eau 10,6 g de ferrocyanure de potassium K4 Fe (CN)6 3 H2O. Ajuster à 100 ml avec de l'eau.
3.4. Charbon actif n'adsorbant pas l'urée (à contrôler).
3.5. Urée, solution à 0,1 % (p/v).
4. Appareillage
4.1. Mélangeur (culbuteur): environ 35 à 40 retournements par minute.
4.2. Tubes à essais: 160 × 16 mm, à bouchon rodé.
4.3. Spectrophotomètre.
5. Mode opératoire
5.1. Analyse de l'échantillon
Peser, à 1 mg près, 2 g de l'échantillon et les introduire avec 1 g de charbon actif (3.4) dans un flacon jaugé de 500 ml. Ajouter 400 ml d'eau et 5 ml de solution de Carrez I (3.2), agiter pendant environ 30 secondes et ajouter ensuite 5 ml de solution de Carrez II (3.3). Mélanger durant trente minutes dans le culbuteur. Ajuster au trait de jauge avec de l'eau, agiter et filtrer.
Prélever 5 ml des filtrats limpides et incolores, les introduire dans les tubes à essais à bouchon rodé, ajouter 5 ml de solution de 4-DMAB (3.1) et mélanger. Placer les tubes dans un bain d'eau à 20 oC (+/- 4 oC). Après quinze minutes, mesurer la densité optique de la solution d'échantillon au spectrophotomètre à 420 nm par comparaison avec la solution de l'essai à blanc des réactifs.
5.2. Courbe d'étalonnage
Prélever des volumes de 1, 2, 4, 5 et 10 ml de la solution d'urée (3.5), les introduire dans des fioles jaugées de 100 ml et ajuster au trait de jauge avec de l'eau. Prélever 5 ml de chaque solution, y ajouter chaque fois 5 ml de solution de 4-DMAB (3.1), homogénéiser et mesurer la densité optique comme indiqué plus haut par comparaison avec une solution de référence contenant 5 ml de 4-DMAB et 5 ml d'eau exempte d'urée. Tracer la courbe d'étalonnage.
6. Calcul des résultats
Déterminer la quantité d'urée de la prise d'essai en se référant à la courbe d'étalonnage.
Exprimer le résultat sous forme de pourcentage de l'échantillon.
7. Observations
7.1. Pour les teneurs en urée supérieures à 3 %, réduire la prise d'essai à 1 g ou diluer la solution initiale pour ne pas avoir plus de 50 mg d'urée dans 500 ml.
7.2. Pour les faibles teneurs en urée, augmenter la prise d'essai pour autant que le filtrat reste limpide et incolore.
7.3. Si l'échantillon contient des composés azotés simples, tels que les acides aminés, il convient d'effectuer la mesure de la densité optique à 435 nm.
E. DOSAGE DES BASES AZOTÉES VOLATILES
I. PAR MICRODIFFUSION
1. Objet et champ d'application
La méthode permet de déterminer la teneur en bases azotées volatiles, exprimées en ammoniac, des aliments pour animaux.
2. Principe
L'échantillon est extrait par l'eau et la solution est déféquée et filtrée. Les bases azotées volatiles sont déplacées à l'aide d'une solution de carbonate de potassium par microdiffusion, recueillies dans une solution d'acide borique et titrées par l'acide sulfurique.
3. Réactifs
3.1. Acide trichloracétique, solution à 20 % (p/v).
3.2. Indicateur: dissoudre 33 mg de vert de bromocrésol et 65 mg de rouge de méthyle dans 100 ml d'éthanol à 95-96 % (v/v).
3.3. Solution d'acide borique: dans une fiole jaugée de 1 l, dissoudre 10 g d'acide borique dans 200 ml d'éthanol à 95-96 % (v/v) et 700 ml d'eau. Ajouter 10 ml d'indicateur (3.2). Mélanger et ajuster si nécessaire la coloration de la solution au rouge clair par addition d'une solution d'hydroxyde de sodium. 1 ml de cette solution permet de fixer au maximum 300 μg de NH3.
3.4. Solution saturée de carbonate de potassium: dissoudre 100 g de carbonate de potassium dans 100 ml d'eau en ébullition. Laisser refroidir et filtrer.
3.5. Acide sulfurique 0,01 mol/l.
4. Appareillage
4.1. Mélangeur (culbuteur): environ 35 à 40 retournements par minute.
4.2. Cellules de Conway (voir schéma), en verre ou en plastique.
4.3. Microburettes, graduées au 1/100 ml.
5. Mode opératoire
Peser, à 1 mg près, 10 g de l'échantillon et les introduire avec 100 ml d'eau dans une fiole jaugée de 200 ml. Mélanger ou agiter durant 30 minutes dans le culbuteur. Ajouter 50 ml de solution d'acide trichloracétique (3.1), ajuster au trait de jauge avec de l'eau, agiter vigoureusement et filtrer sur un filtre à plis.
Introduire à la pipette, dans la partie centrale de la cellule de Conway, 1 ml de solution d'acide borique (3.3) et dans la couronne de la cellule 1 ml du filtrat de l'échantillon. Couvrir partiellement à l'aide du couvercle graissé. Laisser tomber rapidement dans la couronne 1 ml de solution saturée de carbonate de potassium (3.4) et fermer le couvercle de façon hermétique. Remuer avec précaution la cellule en lui donnant un mouvement de rotation dans un plan horizontal, afin d'assurer le mélange des deux réactifs. Laisser incuber soit durant quatre heures au moins à température ambiante, soit durant une heure à 40 oC.
Titrer les bases volatiles dans la solution d'acide borique par l'acide sulfurique (3.5) en utilisant une microburette (4.3).
Effectuer un essai à blanc en appliquant le même mode opératoire, en l'absence d'échantillon à analyser.
6. Calcul des résultats
1 ml de H2SO40,01 mol/l correspond à 0,34 mg d'ammoniac.
Exprimer le résultat sous forme de pourcentage de l'échantillon.
Répétabilité
La différence entre les résultats de deux dosages parallèles effectués sur le même échantillon ne peut dépasser:
— 10 % en valeur relative pour les teneurs en ammoniac inférieures à 1,0 %,
— 0,1 % en valeur absolue pour les teneurs en ammoniac égales ou supérieures à 1,0 %.
7. Observation
Si la teneur en ammoniac de l'échantillon est supérieure à 0,6 %, diluer le filtrat initial.
CONWAY CELL
Scale 1/1

II. PAR DISTILLATION
1. Objet et champ d'application
La méthode permet de déterminer la teneur en bases azotées volatiles, exprimées en ammoniac, des farines de poisson ne contenant presque pas d'urée. Elle n'est applicable que pour des teneurs en ammoniac inférieures à 0,25 %.
2. Principe
L'échantillon est extrait par l'eau et la solution est déféquée et filtrée. Les bases azotées volatiles sont déplacées à l'ébullition par addition d'oxyde de magnésium et recueillies dans une quantité déterminée d'acide sulfurique dont l'excès est titré en retour par une solution d'hydroxyde de sodium.
3. Réactifs
3.1. Acide trichloracétique, solution à 20 % (p/v).
3.2. Oxyde de magnésium.
3.3. Émulsion antimousse (silicone, par exemple).
3.4. Acide sulfurique 0,05 mol/l.
3.5. Solution d'hydroxyde de sodium 0,1 mol/l.
3.6. Solution à 0,3 % de rouge de méthyle dans l'éthanol à 95-96 % (v/v).
4. Appareillage
4.1. Mélangeur (culbuteur): environ 35 à 40 retournements par minute.
4.2. Appareil à distiller de type Kjeldahl.
5. Mode opératoire
Peser, à 1 mg près, 10 g de l'échantillon et les introduire avec 100 ml d'eau dans une fiole jaugée de 200 ml. Mélanger ou agiter durant 30 minutes dans le culbuteur. Ajouter 50 ml de solution d'acide trichloracétique (3.1), ajuster au trait de jauge avec de l'eau, agiter vigoureusement et filtrer sur un filtre à plis.
Prélever une quantité de filtrat limpide approprié à la teneur présumée en bases azotées volatiles (100 ml conviennent en général). Diluer à 200 ml et ajouter 2 g d'oxyde de magnésium (point 3.2) et quelques gouttes d'émulsion antimousse (3.3). La solution doit être alcaline au papier de tournesol; si elle ne l'est pas, ajouter de l'oxyde de magnésium (3.2). Procéder conformément aux points 5.2 et 5.3 de la méthode de détermination de la teneur en protéines brutes (point C de la présente annexe).
Effectuer un essai à blanc en appliquant le même mode opératoire, en l'absence d'échantillon à analyser.
6. Calcul des résultats
1 ml de H2SO40,05 mol/l correspond à 1,7 mg d'ammoniac.
Exprimer le résultat sous forme de pourcentage de l'échantillon.
Répétabilité
La différence entre les résultats de deux dosages parallèles effectués sur un même échantillon ne peut dépasser, en valeur relative, 10 % d'ammoniac.
F. DOSAGE DES ACIDES AMINÉS (À L'EXCEPTION DU TRYPTOPHANE)
1. Objet et champ d'application
La méthode permet de doser les acides aminés libres (de synthèse et naturels) et totaux (liés dans des peptides et libres) dans les aliments pour animaux au moyen d'un analyseur d'acides aminés. Elle s'applique aux acides aminés suivants: cyst(é)ine, méthionine, lysine, thréonine, alanine, arginine, acide aspartique, acide glutamique, glycine, histidine, isoleucine, leucine, phénylalanine, proline, serine, tyrosine et valine.
La méthode ne fait pas de distinction entre les sels des acides aminés et ne permet pas de différencier les formes D et L des acides aminés. Elle ne convient pas pour le dosage du tryptophane et des analogues hydroxylés des acides aminés.
2. Principe
2.1. Acides aminés libres
Les acides aminés libres sont extraits à l'aide d'acide chlorhydrique dilué. Les macromolécules azotées coextraites sont précipitées à l'aide d'acide sulfosalicylique et éliminées par filtration. La solution filtrée est ajustée à pH 2,20 . Les acides aminés sont séparés par chromatographie par échange d'ions et dosés par réaction avec la ninhydrine et détection photométrique à 570 nm.
2.2. Acides aminés totaux
Le mode opératoire dépend des acides aminés à examiner. La cyst(é)ine et la méthionine doivent être oxydées pour devenir respectivement de l'acide cystéique et de la méthionine sulfone avant hydrolyse. La tyrosine doit être dosée dans l'hydrolysat d'échantillons non oxydés. Tous les autres acides aminés mentionnés au point 1 peuvent être dosés soit dans un échantillon oxydé, soit dans un échantillon non oxydé.
L'oxydation s'effectue à 0 oC à l'aide d'un mélange d'acide performique et de phénol. L'excédent de réactif d'oxydation est décomposé à l'aide de disulfite de sodium. L'échantillon oxydé ou non oxydé est hydrolysé à l'aide d'acide chlorhydrique (3.20) pendant 23 heures. L'hydrolysat est ajusté à pH 2,20 . Les acides aminés sont séparés par chromatographie par échange d'ions et dosés par réaction à la ninhydrine et détection photométrique à 570 nm (440 nm pour la proline).
3. Réactifs
Utiliser de l'eau bidistillée ou de l'eau de qualité équivalente (conductivité < 10 μS)
3.1. Peroxyde d'hydrogène, p (p/p) = 30 %.
3.2. Acide formique, p (p/p) = 98-100 %.
3.3. Phénol.
3.4. Disulfite de disodium.
3.5. Hydroxyde de sodium.
3.6. Acide 5-sulfosalicylique dihydraté.
3.7. Acide chlorhydrique, densité approximative de 1,18 g/ml.
3.8. Citrate de tri-sodium dihydraté.
3.9. 2,2 '-Thiodiéthanol (thiodiglycol).
3.10. Chlorure de sodium.
3.11. Ninhydrine.
3.12. Éther de pétrole, intervalle d'ébullition: 40-60 oC.
3.13. Norleucine ou autre composé pouvant être utilisé comme étalon interne.
3.14. Azote gazeux (< 10 ppm d'oxygène).
3.15. 1-Octanol.
3.16. Acides aminés.
3.16.1. Substances étalons visées au point 1. Composés purs ne contenant pas d'eau de cristallisation. Sécher sous vide sur P2O5 ou H2SO4 pendant une semaine avant utilisation.
3.16.2. Acide cystéique.
3.16.3. Méthionine sulfone.
3.17. Solution d'hydroxyde de sodium, c = 7,5 mol/l:
dissoudre 300 g de NaOH (3.5) dans de l'eau et ajuster à 1 l.
3.18. Solution d'hydroxyde de sodium, c = 1 mol/l:
dissoudre 40 g de NaOH (3.5) dans de l'eau et ajuster à 1 l.
3.19. Solution d'acide formique — phénol:
mélanger 889 g d'acide formique (3.2) et 111 g d'eau en ajoutant 4,73 g de phénol (3.3).
3.20. Mélange à hydrolyser, c = 6 mol HCl/l contenant 1 g de phénol/l:
ajouter 1 g de phénol (3.3) à 492 ml de HCl (3.7) et ajuster à 1 l avec de l'eau.
3.21. Mélange d'extraction, c = 0,1 mol HCl/l contenant 2 % de thiodiglycol: prendre 8,2 ml de HCl (3.7), diluer dans environ 900 ml d'eau, ajouter 20 ml de thiodiglycol (3.9) et ajuster à 1 l avec de l'eau (ne pas mélanger directement 3.7 et 3.9).
3.22. Acide 5-sulfosalicylique, ß = 6 %:
dissoudre 60 g d'acide 5-sulfosalicylique (3.6) dans de l'eau et ajuster à 1 l avec de l'eau.
3.23. Mélange d'oxydation (acide performique-phénol):
mélanger 0,5 ml de peroxyde d'hydrogène (3.1) avec 4,5 ml d'une solution de phénol et d'acide formique (3.19) dans un petit bécher. Incuber à une température de 20 à 30 oC pendant une heure pour obtenir de l'acide performique, laisser refroidir ensuite sur un bain d'eau glacée (15 minutes) avant de l'ajouter à l'échantillon.
Attention: éviter tout contact avec la peau et porter des vêtements de protection.
3.24. Tampon au citrate, c = 0,2 mol de Na+/l, pH= 2,20 :
dissoudre 19,61 g de citrate de sodium (3.8), 5 ml de thiodiglycol (3.9), 1 g de phénol (3.3) et 16,50 ml de HCl (3.7) dans environ 800 ml d'eau. Ajuster le pH à 2,20 . Ajuster à 1 l avec de l'eau.
3.25. Tampons d'élution préparés conformément aux prescriptions prévues pour l'analyseur (4.9).
3.26. Réactif à la ninhydrine préparé conformément aux prescriptions prévues pour l'analyseur (4.9).
3.27. Solutions étalons d'acides aminés. Conserver ces solutions à une température inférieure à 5 oC.
3.27.1. Solution mère étalon d'acides aminés (3.16.1).
c = 2,5 μmol/ml de chacun dans l'acide chlorhydrique.
S'obtient dans le commerce.
3.27.2. Solution mère étalon d'acide cystéique et de méthionine sulfone, c = 1,25 μmol/ml.
Dissoudre 0,2115 g d'acide cystéique (3.16.2) et 0,2265 g de méthionine sulfone (3.16.3) dans du tampon au citrate (3.24) dans une fiole jaugée de 1 l et ajuster au trait de jauge avec du tampon au citrate. Conserver à une température inférieure à 5 oC pendant 12 mois au maximum. Cette solution n'est pas utilisée si la solution mère de l'étalon (3.27.1) contient de l'acide cystéique et de la méthionine sulfone.
3.27.3. Solution de l'étalon interne, par exemple norleucine. c = 20 μmol/ml.
Dissoudre 0,6560 g de norleucine (3.13) dans du tampon au citrate (3.24) dans une fiole jaugée et ajuster à 250 ml à l'aide du tampon au citrate. Conserver à une température inférieure à 5 oC pendant 6 mois au maximum.
3.27.4. Solution d'étalonnage des acides aminés à utiliser avec des hydrolysats, c = 5 nmol/50 μl d'acide cystéique et de méthionine sulfone et c = 10 nmol/50 μl des autres acides aminés. Dissoudre 2,2 g de chlorure de sodium (3.10) dans un bécher de 100 ml avec 30 ml de tampon au citrate (3.24). Ajouter 4,00 ml de la solution étalon d'acides aminés (3.27.1), 4,00 ml de solution étalon d'acide cystéique et de méthionine de sulfone (3.27.2) et 0,50 ml de solution de l'étalon interne (3.27.3) le cas échéant. Ajuster le pH à 2,20 avec de l'hydroxyde de sodium (3.18).
Transférer dans une fiole jaugée de 50 ml, ajuster au trait de jauge de jauge avec du tampon au citrate (3.24) et mélanger.
Conserver à une température inférieure à 5 oC pendant 3 mois au maximum.
Voir aussi les observations du point 9.1.
3.27.5. Solution d'étalonnage des acides aminés à utiliser avec des hydrolysats préparés conformément au point 5.3.3.1 et destinés à être utilisés avec des extraits (5.2). Préparer la solution d'étalonnage conformément au point 3.27.4, mais sans chlorure de sodium.
Conserver à une température inférieure à 5 oC pendant 3 mois au maximum.
4. Appareillage
4.1. Ballon sphérique de 100 ou de 250 ml pourvu d'un réfrigérant à reflux.
4.2. Flacon en verre de borosilicate de 100 ml avec bouchon à vis avec garniture de caoutchouc/téflon (par exemple Duran, Schott) pouvant être utilisé dans une étuve.
4.3. Étuve à ventilation forcée et régulation de la température d'une précision supérieure à ± 2 oC.
4.4. pH-mètre (à graduation à trois décimales).
4.5. Filtre à membrane (0,22 μm).
4.6. Centrifugeuse.
4.7. Évaporateur rotatif sous vide.
4.8. Agitateur mécanique ou magnétique.
4.9. Analyseur d'acides aminés ou équipement pour CLHP avec colonne échangeuse d'ions, dispositif pour ninhydrine, dérivatisation postcolonne et détecteur photométrique.
La colonne est remplie avec des résines de polystyrène sulfonées capables de séparer les différents acides aminés les uns des autres et d'autres produits réagissant à la ninhydrine. Le flux du tampon et du réactif de ninhydrine est assuré par des pompes dont le degré de stabilité du flux est de ±0,5 % dans la période couvrant l'analyse de la solution d'étalonnage et l'analyse de l'échantillon.
Avec certains analyseurs d'acides aminés, on peut utiliser des méthodes d'hydrolyse dans lesquelles l'hydrolysat a une concentration de sodium de c = 0,8 mol/l et contient tout l'acide formique résiduel provenant de l'oxydation. D'autres appareils ne donnent pas une séparation satisfaisante de certains acides aminés si l'hydrolysat contient trop d'acide formique et/ou présente des concentrations élevées d'ions sodium. Dans ce cas, le volume de l'acide est réduit par évaporation à environ 5 ml après l'hydrolyse et avant l'ajustement du pH. L'évaporation doit être faite sous vide à 40 oC au maximum.
5. Mode opératoire
5.1. Préparation de l'échantillon
Broyer l'échantillon pour le réduire à une granulométrie de 0,5 mm. Les échantillons à forte teneur en humidité doivent être séchés à l'air à une température de 50 oC au maximum ou par lyophilisation avant broyage. Les échantillons à forte teneur en matières grasses doivent être extraits par l'éther de pétrole (3.12) avant broyage.
5.2. Dosage des acides aminés libres dans les aliments pour animaux et les prémélanges
Peser, à 0,2 mg près, une quantité adéquate (1-5 g) de l'échantillon préparé (5.1) dans un erlenmeyer et ajouter 100,0 ml du mélange d'extraction (3.21). Agiter le mélange pendant 60 min à l'aide d'un agitateur mécanique ou magnétique (4.8). Laisser décanter le sédiment et introduire à la pipette 10,0 ml de la solution surnageante dans un bécher de 100 ml.
Ajouter 5,0 ml de la solution d'acide sulfosalicylique (3.22) tout en remuant et poursuivre l'agitation pendant 5 min à l'aide de l'agitateur magnétique. Filtrer ou centrifuger la phase surnageante en vue d'en séparer le précipitat éventuel. Verser 10,0 ml de la solution obtenue dans un bécher de 100 ml, ajuster le pH à 2,20 à l'aide d'une solution d'hydroxyde de sodium (3.18), transférer le tout dans une fiole jaugée d'un volume approprié à l'aide du tampon au citrate (3.24) et ajuster au trait de jauge avec la solution tampon (3.24).
Si un étalon interne est utilisé, ajouter 1,00 ml de l'étalon interne (3.27.3) par fraction de 100 ml de la solution finale et ajuster au trait de jauge avec la solution tampon (3.24).
Passer à la chromatographie conformément au point 5.4.
Si les extraits ne sont pas chromatographiés le même jour, les conserver à une température inférieure à 5 oC.
5.3. Dosage des acides aminés totaux
5.3.1.
Peser, à 0,2 mg près, entre 0,1 et 1 g de l'échantillon préparé (5.1) dans:
— un ballon sphérique de 100 ml (4.1) pour hydrolyse ouverte (5.3.2.3), ou
— un ballon sphérique de 250 ml (4.1) si l'on a besoin d'une faible concentration de sodium (5.3.3.1), ou
— un flacon de 100 ml à bouchon à vis (4.2) (pour hydrolyse fermée 5.3.2.4).
La portion d'échantillon pesée doit avoir une teneur en azote d'environ 10 mg et une teneur en humidité de 100 mg au plus.
Placer le ballon sphérique ou le flacon dans un bain d'eau glacée et refroidir à 0 oC; ajouter 5 ml du mélange à oxyder (3.23) et mélanger à l'aide d'une spatule de verre à bout courbé. Fermer hermétiquement le ballon/le flacon contenant la spatule à l'aide d'un film hermétique à l'air, placer le bain d'eau glacée avec le récipient ainsi fermé dans un réfrigérateur à 0 oC et l'y laisser séjourner pendant 16 heures. Enlever ensuite le produit du réfrigérateur et décomposer l'excès de réactif d'oxydation par addition de 0,84 g de disulfite de sodium (3.4).
Passer à l'opération du point 5.3.2.1.
5.3.2.
5.3.2.1.
À l'échantillon oxydé préparé conformément au point 5.3.1, ajouter 25 ml du mélange d'hydrolyse (3.20) en prenant soin d'éliminer par rinçage tout résidu de l'échantillon adhérant aux parois du récipient et à la spatule.
Selon la méthode d'hydrolyse utilisée, appliquer la méthode visée au point 5.3.2.3 ou 5.3.2.4.
5.3.2.2.
Peser dans un ballon sphérique de 100 ml ou de 250 ml (4.1) ou dans un tube de 100 ml pourvu d'un bouchon à vis (4.2), à 0,2 mg près, de 0,1 à 1 g de l'échantillon préparé (5.1). La portion d'échantillon pesée doit avoir une teneur en azote d'environ 10 mg. Ajouter avec précaution 25 ml du mélange à hydrolyser (3.20) et mélanger avec l'échantillon. Procéder conformément au point 5.3.2.3 ou 5.3.2.4.
5.3.2.3.
Ajouter 3 billes de verre au mélange contenu dans le ballon (préparé conformément au point 5.3.2.1 ou 5.3.2.2) et faire bouillir sous reflux en bouillonnement constant pendant 23 heures. Après l'hydrolyse, rincer le réfrigérant au moyen de 5 ml du tampon au citrate (3.24). Déconnecter le ballon et le faire refroidir dans un bain glacé.
Poursuivre conformément au point 5.3.3.
5.3.2.4.
Placer le flacon contenant le mélange préparé conformément au point 5.3.2.1 ou 5.3.2.2 dans une étuve (4.3) chauffée à 110 oC. Pendant la première heure, placer le bouchon à vis sur le récipient de manière à éviter une élévation de la pression (due à l'évolution des substances gazeuses) et une explosion. Ne pas fermer le récipient en vissant le bouchon. Après 1 heure, fermer le récipient à l'aide du bouchon à vis et laisser reposer dans l'étuve (4.3) pendant 23 heures. Après l'hydrolyse, enlever le flacon de l'étuve, ouvrir avec précaution le bouchon et placer le flacon dans un bain d'eau glacée. Laisser refroidir.
Selon la procédure adoptée pour l'ajustement du pH (5.3.3), transférer quantitativement le contenu du flacon dans un bécher de 250 ml ou dans un ballon sphérique de 250 ml à l'aide du tampon au citrate (3.24).
Poursuivre conformément au point 5.3.3.
5.3.3.
En fonction de la tolérance en sodium de l'analyseur d'acides aminés (4.9), procéder conformément aux points 5.3.3.1 ou 5.3.3.2 pour l'ajustement du pH.
5.3.3.1.
Il est recommandé d'utiliser un étalon interne (3.27.3) si on utilise des analyseurs d'acides aminés demandant une faible concentration de sodium (lorsque le volume d'acide doit être réduit).
Dans ce cas, ajouter 2,00 ml de la solution de l'étalon interne (3.27.3) à l'hydrolysat avant évaporation.
Ajouter 2 gouttes de 1-Octanol (3.15) à l'hydrolysat obtenu conformément à la procédure du point 5.3.2.3 ou 5.3.2.4.
À l'aide d'un évaporateur rotatif (4.7), réduire sous vide à 40 oC le volume à un niveau compris entre 5 et 10 ml. Si le volume est accidentellement réduit à moins de 5 ml, l'hydrolysat doit être rejeté et l'analyse recommencée.
Ajuster le pH à 2,20 à l'aide d'une solution d'hydroxyde de sodium (3.18) et poursuivre conformément au point 5.3.4.
5.3.3.2.
Recueillir les hydrolysats obtenus conformément au point 5.3.2.3 ou 5.3.2.4 et les neutraliser partiellement en ajoutant avec précaution, tout en agitant, 17 ml d'une solution d'hydroxyde de sodium (3.17), la température restant inférieure à 40 oC.
L'ajustement du pH final à 2,20 s'effectue à température ambiante au moyen d'une solution d'hydroxyde de sodium conforme au point 3.17 et, enfin, d'une solution d'hydroxyde de sodium conforme au point 3.18. Poursuivre conformément au point 5.3.4.
5.3.4.
Transférer quantitativement l'hydrolysat à pH ajusté (5.3.3.1 ou 5.3.3.2) avec le tampon au citrate (3.24) dans une fiole jaugée de 200 ml et ajuster au trait de jauge à l'aide du tampon (3.24).
Si l'étalon interne (3.27.3) n'a pas encore été utilisé, en ajouter 2,00 ml et ajuster au trait de jauge avec du tampon au citrate (3.24). Mélanger soigneusement.
Poursuivre par la chromatographie (5.4).
Si les solutions des échantillons ne sont pas chromatographiées le même jour, les conserver à une température inférieure à 5 oC.
5.4. Chromatographie
Avant la chromatographie, porter l'extrait (5.2) ou l'hydrolysat (5.3.4) à température ambiante. Agiter le mélange et filtrer une partie appropriée à travers un filtre à membrane de 0,22 μm (4.5). La solution claire obtenue est soumise à une chromatographie par échange d'ions au moyen d'un analyseur d'acides aminés (4.9).
L'injection peut être opérée manuellement ou automatiquement. Il importe que la même quantité de solution (± 0,5 %) soit toujours ajoutée à la colonne pour l'analyse des étalons et des échantillons, sauf lorsqu'un étalon interne est utilisé, et que les rapports sodium/acide aminé dans les solutions étalons et les solutions des échantillons soient aussi identiques que possible.
En général, la fréquence des injections de la solution d'étalonnage dépend de la stabilité du réactif ninhydrine et du système d'analyse. La solution étalon ou d'échantillon est diluée avec un tampon au citrate (3.24) pour donner une surface de pic de la solution étalon de 30 à 200 % de la surface de pic de l'acide aminé dans l'échantillon.
La chromatographie des acides aminés varie légèrement selon le type d'analyseur employé et de résine utilisée. Le système retenu doit permettre de séparer les acides aminés les uns des autres et des substances réagissant à la ninhydrine. Au cours de l'opération, le système chromatographique doit donner une réponse linéaire à des variations des quantités d'acides aminés ajoutés dans la colonne.
Pendant la phase de chromatographie, les rapports de hauteur entre creux et sommets de pics mentionnés ci-dessous sont applicables lorsqu'une solution équimolaire des acides aminés est analysée. Cette solution équimolaire doit contenir au moins 30 % de la charge maximale de chaque acide aminé qui peut être déterminée avec précision à l'aide du système analyseur d'acides aminés (4.9).
Pour la séparation thréonine-sérine, le rapport de hauteur entre la vallée et le sommet du plus bas des deux acides aminés chevauchant, dans le chromatogramme, ne doit pas dépasser 2:10 [si le dosage porte seulement sur la cyst(é)ine, la méthionine, la thréonine et la lysine, une séparation insuffisante des pics voisins aura une influence défavorable sur le dosage]. Pour tous les autres acides aminés, la séparation doit être meilleure que 1:10.
Le système doit garantir que la lysine est séparée des «artefacts de lysine» et de l'ornithine.
6. Calcul des résultats
La surface des pics de l'échantillon et de la solution étalon est mesurée pour chaque acide aminé particulier et la quantité (X), exprimée en grammes d'acide aminé par kg d'échantillon, est calculée selon la formule suivante:
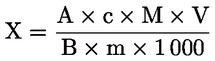
Si un étalon interne est utilisé, multiplier par
![]()
|
A |
= |
surface de pic, hydrolysat ou extrait, |
|
B |
= |
surface de pic, solution d'étalonnage, |
|
C |
= |
surface de pic, étalon interne dans l'hydrolysat ou l'extrait, |
|
D |
= |
surface de pic, étalon interne, solution d'étalonnage, |
|
M |
= |
poids molaire de l'acide aminé dosé, |
|
c |
= |
concentration de l'étalon en μmol/ml, |
|
m |
= |
poids en grammes de l'échantillon (corrigé pour obtenir le poids initial si le produit est séché et/ou dégraissé), |
|
V |
= |
ml d'hydrolysat total (5.3.4) ou ml du volume de l'extrait dilué total calculé (6.1). |
La cystine et la cystéine sont dosées toutes deux sous forme d'acide cystéique dans les hydrolysats de l'échantillon oxydé mais calculées sous forme de cystine (C6H12N2O4S2, M 240,30 g/mol) en utilisant un M de 120,15 g/mol (= 0,5 × 240,30 g/mol).
La méthionine est dosée sous forme de méthionine sulfone dans les hydrolysats de l'échantillon oxydé, mais calculée sous forme de méthionine en utilisant un M de 149,21 g/mol.
La méthionine libre ajoutée est dosée après extraction sous forme de méthionine; pour le calcul, appliquer la même valeur à M.
6.1. Le volume de la dilution totale des extraits (F) pour le dosage des acides aminés libres (5.2) est calculé comme suit:

|
V |
= |
volume de l'extrait final. |
7. Évaluation de la méthode
La méthode a été testée dans une comparaison interlaboratoire internationale faite en 1990 sur la base de quatre aliments pour animaux différents (aliment composé pour porcs, aliment composé pour poulets de chair, concentré protéique, prémélange). La moyenne et l'écart type des résultats, après élimination des aberrants, sont mentionnés dans les tableaux ci-après.
Moyennes en g/kg
|
Matériau de référence |
Acide aminé |
|||
|
Thréonine |
Cyst(é)ine |
Méthionine |
Lysine |
|
|
Aliment composé pour porcs |
6,94 n = 15 |
3,01 n = 17 |
3,27 n = 17 |
9,55 n = 13 |
|
Aliment composé pour poulets de chair |
9,31 n = 16 |
3,92 n = 18 |
5,08 n = 18 |
13,93 n = 16 |
|
Concentré protéique |
22,32 n = 16 |
5,06 n = 17 |
12,01 n = 17 |
47,74 n = 15 |
|
Prémélange |
58,42 n = 16 |
— |
90,21 n = 16 |
98,03 n = 16 |
|
n = nombre de laboratoires participants. |
||||
7.1. Répétabilité
La répétabilité, exprimée sous forme d'«écart type à l'intérieur du laboratoire» de la comparaison interlaboratoire susvisée, est indiquée dans les tableaux ci-après.
Écart type à l'intérieur du laboratoire (Sr) en g/kg
|
Matériau de référence |
Acide aminé |
|||
|
Thréonine |
Cyst(é)ine |
Méthionine |
Lysine |
|
|
Aliment composé pour porcs |
0,13 n = 15 |
0,10 n = 17 |
0,11 n = 17 |
0,26 n = 13 |
|
Aliment composé pour poulets de chair |
0,20 n = 16 |
0,11 n = 18 |
0,16 n = 18 |
0,28 n = 16 |
|
Concentré protéique |
0,48 n = 16 |
0,13 n = 17 |
0,27 n = 17 |
0,99 n = 15 |
|
Prémélange |
1,30 n = 16 |
— |
2,19 n = 16 |
2,06 n = 16 |
|
n = nombre de laboratoires participants. |
||||
Coefficient de variation (%) pour l'écart type à l'intérieur du laboratoire (Sr)
|
Matériau de référence |
Acide aminé |
|||
|
Thréonine |
Cyst(é)ine |
Méthionine |
Lysine |
|
|
Aliment composé pour porcs |
1,9 n = 15 |
3,3 n = 17 |
3,4 n = 17 |
2,8 n = 13 |
|
Aliment composé pour poulets de chair |
2,1 n = 16 |
2,8 n = 18 |
3,1 n = 18 |
2,1 n = 16 |
|
Concentré protéique |
2,7 n = 16 |
2,6 n = 17 |
2,2 n = 17 |
2,4 n = 15 |
|
Prémélange |
2,2 n = 16 |
— |
2,4 n = 16 |
2,1 n = 16 |
|
n = nombre de laboratoires participants. |
||||
7.2 Reproductibilité
Les résultats relatifs à l'écart type entre laboratoires pour la comparaison interlaboratoire mentionnée ci-dessus figurent au tableau ci-dessous.
Écart type entre laboratoires (SR) en g/kg
|
Matériau de référence |
Acide aminé |
|||
|
Thréonine |
Cyst(é)ine |
Méthionine |
Lysine |
|
|
Aliment composé pour porcs |
0,28 n = 15 |
0,30 n = 17 |
0,23 n = 17 |
0,30 n = 13 |
|
Aliment composé pour poulets de chair |
0,48 n = 16 |
0,34 n = 18 |
0,55 n = 18 |
0,75 n = 16 |
|
Concentré protéique |
0,85 n = 16 |
0,62 n = 17 |
1,57 n = 17 |
1,24 n = 15 |
|
Prémélange |
2,49 n = 16 |
— |
6,20 n = 16 |
6,62 n = 16 |
|
n = nombre de laboratoires participants. |
||||
Coefficient de variation (%) pour l'écart type entre laboratoires (SR)
|
Matériau de référence |
Acide aminé |
|||
|
Thréonine |
Cyst(é)ine |
Méthionine |
Lysine |
|
|
Aliment composé pour porcs |
4,1 n = 15 |
9,9 n = 17 |
7,0 n = 17 |
3,2 n = 13 |
|
Aliment composé pour poulets de chair |
5,2 n = 16 |
8,8 n = 18 |
10,9 n = 18 |
5,4 n = 16 |
|
Concentré protéique |
3,8 n = 16 |
12,3 n = 17 |
13,0 n = 17 |
3,0 n = 15 |
|
Prémélange |
4,3 n = 16 |
— |
6,9 n = 16 |
6,7 n = 16 |
|
n = nombre de laboratoires participants. |
||||
8. Utilisation de matériaux de référence
L'application correcte de la méthode est vérifiée par répétition des mesures des matériaux de référence certifiés éventuellement disponibles. L'étalonnage au moyen d'une solution d'étalonnage certifiée d'acides aminés est recommandée.
9. Observations
9.1. Par suite des différences entre appareils analyseurs d'acides aminés, les concentrations finales des solutions d'étalonnage des acides aminés (voir 3.27.4 et 3.27.5) et de l'hydrolysat (voir 5.3.4) ont un caractère indicatif.
Le champ de la réponse linéaire de l'appareillage doit être vérifié pour tous les acides aminés.
La solution étalon est diluée avec un tampon au citrate pour donner des surfaces de pic se situant au milieu du champ.
9.2. Dans les cas où un appareillage de chromatographie liquide à haute performance est utilisé pour analyser les hydrolysats, les conditions d'essai doivent être optimisées conformément aux recommandations du fabricant.
9.3. L'application de la méthode aux aliments pour animaux contenant plus de 1 % de chlorure (concentré, aliments minéraux, aliments complémentaires) pourrait entraîner une sous-estimation de la méthionine, et un traitement spécial est à prévoir.
G. DOSAGE DU TRYPTOPHANE
1. Objet et champ d'application
La méthode permet de doser le tryptophane total et libre dans les aliments pour animaux. Elle ne fait pas de distinction entre les formes D et L.
2. Principe
Pour le dosage du tryptophane total, l'échantillon est hydrolysé en milieu alcalin avec une solution d'hydroxyde de baryum saturée et il est chauffé à 110 oC pendant vingt heures. Après l'hydrolyse, l'étalon interne est ajouté.
Pour le dosage du tryptophane libre, l'échantillon est extrait dans des conditions d'acidité modérée en présence de l'étalon interne.
Le tryptophane et l'étalon interne dans l'hydrolysat ou dans l'extrait sont dosés par CLHP à l'aide d'un détecteur fluorimétrique.
3. Réactifs
3.1. Utiliser de l'eau bidistillée ou de l'eau de qualité équivalente (conductivité < 10 μS/cm).
3.2. Substance étalon: tryptophane (pureté/teneur ≥ 99 %) séché sous vide sur pentoxyde de phosphore.
3.3. Étalon interne: α-méthyl-tryptophane (pureté/teneur ≥ 99 %) séché sous vide sur pentoxyde de phosphore.
3.4. Hydroxyde de baryum octahydraté (veiller à ne pas exposer excessivement le Ba(OH)2·8H2O à l'air afin d'éviter la formation de BaCO3 qui pourrait perturber le dosage) (voir l'observation figurant au point 9.3).
3.5. Hydroxyde de sodium.
3.6. Acide orthophosphorique, p (p/p) = 85 %.
3.7. Acide chlorhydrique, ρ201,19 g/ml.
3.8. Méthanol, de qualité CLHP.
3.9. Éther de pétrole, intervalle d'ébullition: 40-60 oC.
3.10. Solution d'hydroxyde de sodium, c = 1 mol/l:
dissoudre 40,0 g de NaOH (3.5) dans de l'eau et ajuster au litre avec de l'eau (3.1).
3.11. Acide chlorhydrique, c = 6 mol/l:
prendre 492 ml de HCl (3.7) et ajuster au litre avec de l'eau.
3.12. Acide chlorhydrique, c = 1 mol/l:
prendre 82 ml de HCl (3.7) et ajuster au litre avec de l'eau.
3.13. Acide chlorhydrique, c = 0,1 mol/l:
prendre 8,2 ml de HCl (3.7) et ajuster au litre avec de l'eau.
3.14. Acide orthophosphorique, c = 0,5 mol/l:
prendre 34 ml d'acide orthophosphorique (3.6) et ajuster au litre avec de l'eau (3.1).
3.15. Solution concentrée de tryptophane (3.2), c = 2,50 μmol/ml:
dans une fiole jaugée de 500 ml, dissoudre 0,2553 g de tryptophane (3.2) dans de l'acide chlorhydrique (3.13) et ajuster au trait de jauge avec de l'acide chlorhydrique (3.13). Conserver à - 18 oC pendant quatre semaines au maximum.
3.16. Solution concentrée de l'étalon interne, c = 2,50 μmol/ml:
dans une fiole jaugée de 500 ml, dissoudre 0,2728 g de α-méthyl-tryptophane (3.3) dans de l'acide chlorhydrique (3.13) et ajuster au trait de jauge avec de l'acide chlorhydrique (3.13). Conserver à - 18 oC pendant quatre semaines au maximum.
3.17. Solution d'étalonnage de tryptophane et étalon interne:
prendre 2,00 ml de solution concentrée de tryptophane (3.15) et 2,00 ml de solution concentrée de l'étalon interne (α-méthyl-tryptophane) (3.16). Diluer avec de l'eau (3.1) et du méthanol (3.8) approximativement au même volume et approximativement à la même concentration de méthanol (10-30 %) que l'hydrolysat fini.
Cette solution doit être préparée peu avant d'être utilisée.
Se protéger de la lumière directe du soleil pendant la préparation.
3.18. Acide acétique.
3.19. Trichloro-1,1,1-méthyl-2-propanol-2.
3.20. Éthanolamine p (p/p) > 98 %.
3.21. Solution de 1 g de trichloro-1,1,1-méthyl-2-propanol-2 (3.19) dans 100 ml de méthanol (3.8).
3.22. Phase mobile pour CLHP: 3,00 g d'acide acétique (3.18) + 900 ml d'eau (3.1) +50,0 ml de solution (3.21) de trichloro-1,1,1-méthyl-2-propanol-2 (3.19) dans du méthanol (3.8) (1 g/100 ml). Ajuster le pH à 5,00 à l'aide d'éthanolamine (3.20). Ajuster à 1 000 ml avec de l'eau (3.1).
4. Appareillage
4.1. Équipement pour CLHP avec détection spectrofluorimétrique.
4.2. Colonne de chromatographie liquide, 125 mm × 4 mm, C18, particules de 3 μm, ou équivalent.
4.3. pH-mètre.
4.4. Fiole en polypropylène, capacité 125 ml, à large col et bouchon à vis.
4.5. Filtre à membrane, 0,45 μm.
4.6. Autoclave, 110 (± 2) oC, 1,4 (±0,1 ) bar.
4.7. Agitateur mécanique ou magnétique.
4.8. Agitateur Vortex.
5. Mode opératoire
5.1. Préparation des échantillons
Broyer l'échantillon pour le réduire à une granulométrie de 0,5 mm. Les échantillons à forte teneur en humidité doivent être séchés à l'air à une température de 50 oC au maximum ou par lyophilisation avant broyage. Les échantillons à forte teneur en matières grasses doivent être extraits par éther de pétrole (3.9) avant broyage.
5.2. Dosage du tryptophane libre (extrait)
Peser, à 1 mg près, une quantité adéquate (1-5 g) de l'échantillon préparé (5.1) dans une fiole erlenmeyer. Ajouter 100,0 ml d'acide chlorhydrique (3.13) et 5,00 ml de solution concentrée de l'étalon interne (3.16). Agiter ou mélanger pendant 60 minutes à l'aide d'un agitateur mécanique ou magnétique (4.7). Laisser décanter le sédiment et introduire à la pipette 10,0 ml de la solution surnageante dans un bécher. Ajouter 5 ml d'acide orthophosphorique (3.14). Ajuster le pH à 3 avec de l'hydroxyde de sodium (3.10). Ajouter suffisamment de méthanol (3.8) pour obtenir une concentration comprise entre 10 et 30 % de méthanol dans le volume final. Transférer dans une fiole jaugée de volume approprié et diluer avec de l'eau au volume nécessaire pour la chromatographie [environ le même volume que la solution étalon de calibration (3.17)].
Filtrer quelques millilitres de la solution au travers d'un filtre à membrane de 0,45 μm (4.5) avant d'injecter sur la colonne CLHP. Passer à la chromatographie conformément au point 5.4.
Protéger la solution étalon et les extraits de la lumière directe du soleil. Si les extraits ne peuvent pas être analysés le jour même, ils doivent être conservés à une température de 5 oC pendant trois jours au maximum.
5.3. Dosage du tryptophane total (hydrolysat)
Peser, à 0,2 mg près, entre 0,1 et 1 g de l'échantillon préparé (5.1) dans la fiole en polypropylène (4.4). La portion d'échantillon pesée doit avoir une teneur en azote d'environ 10 mg. Ajouter 8,4 g d'hydroxyde de baryum octahydraté (3.4) et 10 ml d'eau. Mélanger avec un agitateur Vortex (4.8) ou un agitateur magnétique (4.7). Laisser l'aimant enrobé de téflon dans le mélange. Laver les parois du récipient avec 4 ml d'eau. Poser le bouchon à vis et fermer la fiole sans serrer. Transférer dans un autoclave (4.6) avec de l'eau bouillante et de la vapeur d'eau pendant 30 à 60 minutes. Fermer l'autoclave et le mettre en marche à 110 (± 2) oC pendant vingt heures.
Avant d'ouvrir l'autoclave, réduire la température à un peu moins de 100 oC. Pour éviter la cristallisation de Ba(OH)2·8H2O, ajouter au mélange chaud 30 ml d'eau à la température ambiante. Agiter ou remuer doucement. Ajouter 2,00 ml de la solution concentrée de l'étalon interne (α-méthyl-tryptophane) (3.16). Refroidir les récipients dans un bain d'eau/de glace pendant 15 minutes.
Ajouter ensuite 5 ml d'acide orthophosphorique (3.14). Maintenir le récipient dans le bain réfrigérant et neutraliser au HCl (3.11) tout en remuant, puis ajuster le pH à 3,0 à l'aide de HCl (3.12). Ajouter suffisamment de méthanol pour obtenir une concentration comprise entre 10 et 30 % de méthanol dans le volume final. Transférer dans une fiole jaugée de volume approprié et diluer avec de l'eau au volume nécessaire pour la chromatographie (par exemple 100 ml). L'addition de méthanol ne doit pas provoquer de précipitation.
Filtrer quelques millilitres de la solution au travers d'un filtre à membrane de 0,45 μm (4.5) avant d'injecter sur la colonne CLHP. Passer à la chromatographie conformément au point 5.4.
Protéger la solution étalon et les hydrolysats de la lumière directe du soleil. Si les hydrolysats ne peuvent pas être analysés le jour même, ils doivent être conservés à une température de 5 oC pendant trois jours au maximum.
5.4. Dosage par CLHP
Les conditions suivantes de l'élution isocratique sont proposées à titre indicatif; d'autres conditions peuvent être appliquées, pourvu qu'elles donnent des résultats équivalents (voir également les observations figurant aux points 9.1 et 9.2).
|
Colonne de chromatographie liquide (4.2): |
125 mm × 4 mm, C18, particules de 3 μm, ou équivalent |
|
Température de la colonne: |
température ambiante |
|
Phase mobile (3.22): |
3,00 g d'acide acétique (3.18) + 900 ml d'eau (3.1) +50,0 ml de solution (3.21) de trichloro-1,1,1-méthyl-2-propanol-2 (3.19) dans du méthanol (3.8) (1 g/100 ml). Ajuster le pH à 5,00 à l'aide d'éthanolamine (3.20). Ajuster à 1 000 ml avec de l'eau (3.1). |
|
Débit: |
1 ml/min |
|
Temps d'élution total: |
environ 34 minutes |
|
Longueur d'onde de détection: |
excitation: 280 nm; émission: 356 nm |
|
Volume d'injection: |
20 μl |
6. Calcul des résultats
La quantité de tryptophane (X), exprimée en g par 100 g d'échantillon, est calculée à l'aide de la formule suivante:
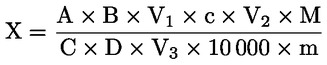
|
A |
= |
surface du pic de l'étalon interne, solution étalon de calibration (3.17), |
|
B |
= |
surface du pic de tryptophane, extrait (5.2) ou hydrolysat (5.3), |
|
V1 |
= |
volume en ml (2 ml) de solution concentrée de tryptophane (3.15) ajoutée à la solution d'étalonnage (3.17), |
|
c |
= |
concentration en μmol/ml (= 2,50 ) de solution concentrée de tryptophane (3.15) ajoutée à la solution d'étalonnage (3.17), |
|
V2 |
= |
volume en ml de la solution concentrée de l'étalon interne (3.16) ajoutée à l'extraction (5.2) (= 5,00 ml) ou à l'hydrolysat (5.3) (= 2,00 ml), |
|
C |
= |
surface du pic de l'étalon interne, extrait (5.2) ou hydrolysat (5.3), |
|
D |
= |
surface du pic de tryptophane, solution étalon de calibration (3.17), |
|
V3 |
= |
volume en ml (= 2,00 ml) de la solution concentrée de l'étalon interne (3.16) ajoutée à la solution étalon de calibration (3.17), |
|
m |
= |
poids de l'échantillon en g (corrigé pour obtenir le poids initial si le produit est séché et/ou dégraissé), |
|
M |
= |
poids molaire du tryptophane (= 204,23 g/mol). |
7. Répétabilité
La différence entre les résultats de deux dosages parallèles effectués sur le même échantillon ne peut dépasser 10 % par rapport au résultat le plus élevé.
8. Résultats d'une étude collaborative
Une étude collaborative communautaire (quatrième comparaison interlaboratoire) a été réalisée: douze laboratoires ont analysé trois échantillons pour certifier la méthode par hydrolyse. Chaque échantillon a été soumis à cinq analyses. Les résultats figurent dans le tableau ci-après.
|
|
Échantillon 1 Aliment pour porcs |
Échantillon 2 Aliment pour porcs complété par du L-tryptophane |
Échantillon 3 Aliment concentré pour porcs |
|
L |
12 |
12 |
12 |
|
n |
50 |
55 |
50 |
|
Moyenne [g/kg] |
2,42 |
3,40 |
4,22 |
|
sr [g/kg] |
0,05 |
0,05 |
0,08 |
|
r [g/kg] |
0,14 |
0,14 |
0,22 |
|
CVr [%] |
1,9 |
1,6 |
1,9 |
|
SR [g/kg] |
0,15 |
0,20 |
0,09 |
|
R [g/kg] |
0,42 |
0,56 |
0,25 |
|
CVR [%] |
6,3 |
6,0 |
2,2 |
|
L |
= |
nombre de laboratoires ayant présenté des résultats |
|
n |
= |
nombre de résultats individuels retenus une fois les résultats aberrants éliminés (identifiés selon les tests de Cochran-Dixon) |
|
sr |
= |
écart type de répétabilité |
|
SR |
= |
écart type de reproductibilité |
|
r |
= |
répétabilité |
|
R |
= |
reproductibilité |
|
CVr |
= |
coefficient de variation de la répétabilité, en % |
|
CVR |
= |
coefficient de variation de la reproductibilité, en % |
Une autre étude collaborative communautaire (troisième comparaison interlaboratoire) a été réalisée: jusqu'à treize laboratoires ont analysé deux échantillons pour certifier la méthode par extraction du tryptophane libre. Chaque échantillon a été soumis à cinq analyses. Les résultats figurent dans le tableau ci-après.
|
|
Échantillon 4 Mélange de blé et de soja |
Échantillon 5 Mélange de blé et de soja (= échantillon 4) avec addition de tryptophane (0,457 g/kg) |
|
L |
12 |
12 |
|
n |
55 |
60 |
|
Moyenne [g/kg] |
0,391 |
0,931 |
|
sr [g/kg] |
0,005 |
0,012 |
|
r [g/kg] |
0,014 |
0,034 |
|
CVr [%] |
1,34 |
1,34 |
|
SR [g/kg] |
0,018 |
0,048 |
|
R [g/kg] |
0,050 |
0,134 |
|
CVR [%] |
4,71 |
5,11 |
|
L |
= |
nombre de laboratoires ayant présenté des résultats |
|
n |
= |
nombre de résultats individuels retenus une fois les résultats aberrants éliminés (identifiés selon les tests de Cochran-Dixon) |
|
sr |
= |
écart type de répétabilité |
|
SR |
= |
écart type de reproductibilité |
|
r |
= |
répétabilité |
|
R |
= |
reproductibilité |
|
CVr |
= |
coefficient de variation de la répétabilité, en % |
|
CVR |
= |
coefficient de variation de la reproductibilité, en % |
Une autre étude interlaboratoire communautaire a été réalisée: sept laboratoires ont analysé quatre échantillons pour certifier la méthode par hydrolyse. Chaque échantillon a été soumis à cinq analyses. Les résultats figurent dans le tableau ci-après.
|
|
Échantillon 1 Aliment composé pour porcs (CRM117) |
Échantillon 2 Farine de poisson à faible teneur en matières grasses (CRM 118) |
Échantillon 3 Farine de soja (CRM 119) |
Échantillon 4 Lait écrémé en poudre (CRM 120) |
|
L |
7 |
7 |
7 |
7 |
|
n |
25 |
30 |
30 |
30 |
|
Moyenne [g/kg] |
2,064 |
8,801 |
6,882 |
5,236 |
|
sr [g/kg] |
0,021 |
0,101 |
0,089 |
0,040 |
|
r [g/kg] |
0,059 |
0,283 |
0,249 |
0,112 |
|
CVr [%] |
1,04 |
1,15 |
1,30 |
0,76 |
|
SR [g/kg] |
0,031 |
0,413 |
0,283 |
0,221 |
|
R [g/kg] |
0,087 |
1,156 |
0,792 |
0,619 |
|
CVR [%] |
1,48 |
4,69 |
4,11 |
4,22 |
|
L |
= |
nombre de laboratoires ayant présenté des résultats |
|
n |
= |
nombre de résultats individuels retenus une fois les résultats aberrants éliminés (identifiés selon les tests de Cochran-Dixon) |
|
sr |
= |
écart type de répétabilité |
|
SR |
= |
écart type de reproductibilité |
|
r |
= |
répétabilité |
|
R |
= |
reproductibilité |
|
CVr |
= |
coefficient de variation de la répétabilité, en % |
|
CVR |
= |
coefficient de variation de la reproductibilité, en % |
9. Observations
9.1. Les conditions de chromatographie spéciales suivantes peuvent donner une meilleure séparation entre le tryptophane et le α-méthyl-tryptophane.
Élution isocratique suivie d'un nettoyage de la colonne par gradient:
|
Colonne de chromatographie liquide: |
125 mm × 4 mm, C18, particules de 5 μm, ou équivalent |
||
|
Température de la colonne: |
32 oC |
||
|
Phase mobile: |
A: 0,01 mol/l KH2PO4/méthanol, 95+5 (V + V). B: méthanol |
||
|
Programme du gradient: |
0 min |
100 % A |
0 % B |
|
|
15 min |
100 % A |
0 % B |
|
|
17 min |
60 % A |
40 % B |
|
|
19 min |
60 % A |
40 % B |
|
|
21 min |
100 % A |
0 % B |
|
|
33 min |
100 % A |
0 % B |
|
Débit: |
1,2 ml/min |
||
|
Temps d'élution total: |
environ 33 minutes. |
||
9.2. La chromatographie varie selon le type de CLHP et selon le remplissage de la colonne. Le système retenu doit donner un retour à la ligne de base entre les pics du tryptophane et de l'étalon interne. De plus, il est important que les produits de décomposition soient bien séparés du tryptophane et de l'étalon interne. Il faut réaliser un essai sans étalon interne de façon à vérifier l'absence d'impuretés sur la ligne de base au niveau de l'étalon interne. Il est important que le temps d'élution soit suffisamment long pour permettre l'élution de tous les produits de décomposition, faute de quoi des pics d'élution tardifs peuvent interférer avec les opérations de chromatographie ultérieures.
Dans la gamme des opérations, le système chromatographique doit donner une réponse linéaire. Cette réponse doit être mesurée à une concentration constante (la concentration normale) de l'étalon interne et à différentes concentrations de tryptophane. Il est important que la hauteur des pics du tryptophane et de l'étalon interne se situe dans la gamme linéaire du système CLHP/fluorescence. Si le ou les pics du tryptophane et/ou de l'étalon interne sont trop petits ou trop grands, l'analyse doit être répétée avec un échantillon d'une autre dimension et/ou un volume final modifié.
9.3. Hydroxyde de baryum
Avec le temps, l'hydroxyde de baryum devient plus difficile à dissoudre. Cela donne une solution trouble pour le dosage par CLHP, ce qui peut entraîner de faibles résultats pour le tryptophane.
H. DOSAGE DES MATIÈRES GRASSES BRUTES
1. Objet et champ d'application
La méthode est destinée à déterminer la teneur en matières grasses brutes des aliments pour animaux. Elle ne concerne pas l'analyse des graines et des fruits oléagineux.
L'application des deux procédés décrits ci-après dépend de la nature et de la composition des aliments pour animaux et de la raison pour laquelle l'analyse est effectuée.
1.1. Procédé A — matières grasses directement extractibles
Ce procédé est applicable aux matières premières pour aliments des animaux d'origine végétale, à l'exception de celles auxquelles s'applique le procédé B.
1.2. Procédé B — matières grasses brutes totales
Ce procédé est applicable aux matières premières pour aliments des animaux d'origine animale et à tous les aliments composés. Il doit être utilisé pour toutes les matières premières dont les matières grasses ne sont pas complètement extractibles sans hydrolyse préalable, par exemple les glutens, les levures, les protéines de pomme de terre et les produits soumis à des procédés tels que l'extrusion, la floconnisation et le chauffage.
1.3. Interprétation des résultats
Dans tous les cas où l'application du procédé B donne un résultat supérieur à celui obtenu grâce à l'application du procédé A, le résultat obtenu au moyen du procédé B est accepté comme valeur réelle.
2. Principe
2.1. Procédé A
L'échantillon est extrait par de l'éther de pétrole. Le solvant est éliminé par distillation et le résidu est séché et pesé.
2.2. Procédé B
L'échantillon est traité à chaud par de l'acide chlorhydrique. Le mélange est refroidi et filtré. Après avoir été lavé et séché, le résidu est soumis à l'analyse selon le procédé A.
3. Réactifs
3.1. Éther de pétrole, intervalle d'ébullition: 40 à 60 oC. L'indice de brome doit être inférieur à 1 et le résidu après évaporation inférieur à 2 mg/100 ml.
3.2. Sulfate de sodium, anhydre.
3.3. Acide chlorhydrique, c = 3 mol/l.
3.4. Adjuvant de filtration, par exemple terre de diatomées, Hyflo-supercel.
4. Appareillage
4.1. Extracteur. Si l'appareil est muni d'un siphon (appareil de Soxhlet), le débit du reflux doit être réglé de façon à obtenir environ dix cycles par heure; s'il s'agit d'un appareil sans siphon, le débit de liquide reflué doit être de 10 ml environ par minute.
4.2. Cartouches d'extraction, exemptes de matière soluble dans l'éther de pétrole, dont la porosité est compatible avec les exigences du point 4.1.
4.3. Étuve à dessiccation, soit par le vide à 75 oC ± 3 oC, soit à pression atmosphérique à 100 oC ± 3 oC.
5. Mode opératoire
5.1. Procédé A (voir observation 8.1)
Peser, à 1 mg près, 5 g de l'échantillon, les introduire dans une cartouche d'extraction (4.2) et les recouvrir d'un tampon de coton dégraissé.
Placer la cartouche dans un extracteur (4.1) et extraire durant 6 heures par de l'éther de pétrole (3.1). Recueillir l'extrait dans une fiole sèche, tarée et contenant des fragments de pierre ponce ( 9 ).
Éliminer le solvant par distillation. Sécher le résidu en plaçant la fiole pendant une heure et demie dans l'étuve à dessiccation (4.3). Laisser refroidir dans un dessiccateur et peser. Sécher de nouveau pendant 30 minutes pour s'assurer que le poids des matières grasses reste constant (la perte de poids entre deux pesées successives doit être inférieure à 1 mg).
5.2. Procédé B
Peser, à 1 mg près, 2,5 g de l'échantillon (voir observation 8.2), les introduire dans un bécher de 400 ml ou une fiole conique de 300 ml et ajouter 100 ml d'acide chlorhydrique (3.3) et des fragments de pierre ponce. Recouvrir le bécher d'un verre de montre ou munir la fiole conique d'un réfrigérant à reflux. Amener le mélange à ébullition douce, à l'aide d'une petite flamme ou d'une plaque chauffante, et l'y maintenir durant une heure. Éviter que le produit adhère aux parois du récipient.
Refroidir et ajouter une quantité d'adjuvant de filtration (3.4) suffisante pour éviter toute perte de matières grasses lors de la filtration. Filtrer sur un double papier filtre mouillé, exempt de matières grasses. Laver le résidu à l'eau froide jusqu'à neutralité du filtrat. Vérifier que le filtrat ne contient pas de matières grasses. La présence de celles-ci indique qu'une extraction de l'échantillon par l'éther de pétrole, selon le procédé A, doit être effectuée préalablement à l'hydrolyse.
Placer le double papier filtre contenant le résidu sur un verre de montre et sécher durant une heure et demie dans l'étuve à pression atmosphérique (4.3) à 100 oC ± 3 oC.
Introduire le double papier filtre contenant le résidu sec dans une cartouche à extraction (4.2) et le recouvrir d'un tampon de coton dégraissé. Placer la cartouche dans un extracteur (4.1) et poursuivre le mode opératoire comme indiqué au point 5.1, deuxième et troisième alinéas.
6. Expression du résultat
Exprimer le poids du résidu sous forme de pourcentage de l'échantillon.
7. Répétabilité
La différence entre les résultats de deux dosages parallèles effectués sur un même échantillon par le même analyste ne peut dépasser:
— 0,2 %, en valeur absolue, pour les teneurs en matières grasses brutes inférieures à 5 %,
— 4,0 % du résultat le plus élevé pour les teneurs de 5 à 10 %,
— 0,4 %, en valeur absolue, pour les teneurs supérieures à 10 %.
8. Observations
8.1. Pour les produits à teneur élevée en matières grasses qui sont difficiles à broyer ou non appropriés au prélèvement d'une prise d'essai réduite homogène, procéder de la manière suivante.
Peser, à 1 mg près, 20 g de l'échantillon et les mélanger à 10 g ou plus de sulfate de sodium anhydre (3.2). Extraire par l'éther de pétrole (3.1) comme indiqué au point 5.1. Ajuster l'extrait obtenu à 500 ml avec de l'éther de pétrole (3.1) et mélanger. Introduire 50 ml de la solution dans une petite fiole sèche, tarée et contenant des fragments de pierre ponce. Éliminer le solvant par distillation, sécher et poursuivre le mode opératoire comme indiqué au point 5.1, dernier alinéa.
Éliminer le solvant du résidu d'extraction se trouvant dans la cartouche, broyer le résidu à la finesse de 1 mm, le replacer dans la cartouche (ne pas ajouter de sulfate de sodium) et poursuivre le mode opératoire comme indiqué au point 5.1, deuxième et troisième alinéas.
Calculer la teneur en matières grasses, exprimée en pourcentage de l'échantillon, au moyen de la formule suivante:
(10m1 + m2) × 5
où:
|
m1 |
= |
poids, en grammes, du résidu de la première extraction (partie aliquote de l'extrait), |
|
m2 |
= |
poids, en grammes, du résidu de la seconde extraction. |
8.2. La prise d'essai des produits pauvres en matières grasses peut être portée à 5 g.
8.3. Les aliments pour animaux de compagnie ayant une teneur élevée en eau peuvent devoir être mélangés avec du sulfate de sodium anhydre préalablement à l'hydrolyse et à l'extraction selon le procédé B.
8.4. Au point 5.2, il peut être plus efficace d'utiliser de l'eau chaude au lieu de l'eau froide pour laver le résidu après filtration.
8.5. Il se peut que le temps de séchage (1 h 30) doive être allongé pour certains aliments pour animaux. Un temps de séchage excessif doit être évité dans la mesure où il peut donner de faibles résultats. Un four à micro-ondes peut également être utilisé.
8.6. Une première extraction selon le procédé A préalablement à l'hydrolyse et une deuxième extraction selon le procédé B sont recommandées si la teneur en matières grasses brutes est supérieure à 15 %. Cela dépend dans une certaine mesure de la nature des aliments pour animaux et de la nature des matières grasses contenues dans ces aliments.
I. DOSAGE DE LA CELLULOSE BRUTE
1. Objet et champ d'application
La méthode permet de doser, dans les aliments pour animaux, les matières organiques exemptes de graisses et insolubles en milieu acide et en milieu alcalin, conventionnellement désignées sous le nom de cellulose brute.
2. Principe
L'échantillon, dégraissé si nécessaire, est traité successivement par des solutions bouillantes d'acide sulfurique et d'hydroxyde de potassium de concentrations déterminées. Le résidu est séparé par filtration sur un filtre en verre fritté, lavé, séché, pesé puis incinéré entre 475 et 500 oC. La perte de poids résultant de l'incinération correspond à la cellulose brute de la prise d'essai.
3. Réactifs
3.1. Acide sulfurique, c = 0,13 mol/l.
3.2. Antimousse (n-octanol, par exemple).
3.3. Adjuvant de filtration (Celite 545 ou équivalent) chauffé à 500 oC pendant quatre heures (8.6).
3.4. Acétone.
3.5. Éther de pétrole, intervalle d'ébullition: 40-60 oC.
3.6. Acide chlorhydrique, c = 0,5 mol/l.
3.7. Solution d'hydroxyde de potassium, c = 0,23 mol/l.
4. Appareillage
4.1. Unité de chauffage pour la digestion par des solutions d'acide sulfurique ou d'hydroxyde de potassium. Cette unité est munie d'un support à creuset filtrant (4.2) et pourvue d'un tuyau avec vanne vers le vide et la vidange. Elle est éventuellement pourvue d'air comprimé. Avant chaque utilisation journalière, préchauffer l'unité avec de l'eau bouillante pendant cinq minutes.
4.2. Creuset filtrant en verre muni d'un filtre en verre fritté de porosité 40-90 μm. Avant la première utilisation, chauffer à 500 oC pendant quelques minutes et refroidir (8.6).
4.3. Cylindre d'au moins 270 ml avec un réfrigérant à reflux, approprié à l'ébullition.
4.4. Étuve avec thermostat.
4.5. Four à moufle avec thermostat.
4.6. Unité d'extraction comprenant un support pour le creuset filtrant (4.2) et un tuyau d'évacuation avec vanne vers le vide et la vidange.
4.7. Joints pour assembler l'unité de chauffage (4.1), le creuset (4.2) et le cylindre (4.3), et pour connecter l'unité d'extraction (4.6) à froid et le creuset.
5. Mode opératoire
Peser, à 1 mg près, 1 g de l'échantillon préparé, le mettre dans le creuset (4.2) (voir observations 8.1, 8.2, 8.3) et ajouter 1 g d'adjuvant de filtration (3.3).
Assembler l'unité de chauffage (4.1) et le creuset filtrant (4.2), puis attacher le cylindre (4.3) au creuset. Remplir de 150 ml d'acide sulfurique bouillant (3.1) l'ensemble cylindre-creuset et, si nécessaire, ajouter quelques gouttes d'antimousse (3.2).
Porter le liquide à ébullition en 5 ± 2 minutes et bouillir vivement pendant 30 minutes exactement.
Positionner la vanne vers le tuyau de vidange (4.1) et, sous vide, filtrer l'acide sulfurique à travers le creuset filtrant et laver le résidu avec trois fois 30 ml d'eau bouillante, en veillant à ce que le résidu aille à sec après chaque lavage.
Fermer la vanne de vidange et verser 150 ml de solution bouillante d'hydroxyde de potassium (3.7) dans l'ensemble cylindre-creuset et ajouter quelques gouttes d'antimousse (3.2). Porter le liquide à ébullition en 5 ± 2 minutes et bouillir vivement pendant exactement 30 minutes. Filtrer et répéter la procédure de lavage comme pour l'acide sulfurique.
Après le lavage final et le séchage, disjoindre le creuset et son contenu et le reconnecter à l'unité d'extraction à froid (4.6). Mettre le vide et laver le résidu dans le creuset avec trois portions successives de 25 ml d'acétone (3.4) en veillant à ce que le résidu aille à sec après chaque lavage.
Sécher le creuset jusqu'à poids constant à l'étuve à 130 oC. Après chaque séchage, refroidir dans le dessiccateur et peser rapidement. Placer le creuset dans un four à moufle et incinérer jusqu'à poids constant (la perte de poids entre deux pesées successives doit être inférieure ou égale à 2 mg) entre 475 et 500 oC pendant au moins 30 minutes.
Après chaque chauffage, refroidir d'abord dans le four puis dans le dessiccateur avant de peser.
Effectuer un essai à blanc sans l'échantillon. La perte de poids résultant de l'incinération ne peut excéder 4 mg.
6. Calcul des résultats
La teneur en cellulose brute exprimée sous forme de pourcentage de l'échantillon est donnée par la formule suivante:

où:
|
m |
= |
poids de l'échantillon (en g), |
|
m0 |
= |
perte de poids (en g) après incinération, lors du dosage, |
|
m1 |
= |
perte de poids (en g) après incinération, lors de l'essai à blanc. |
7. Répétabilité
La différence entre les résultats de deux dosages parallèles effectués sur le même échantillon ne peut dépasser:
— 0,6 % en valeur absolue, pour les teneurs en cellulose brute inférieures à 10 %,
— 6 % par rapport au résultat le plus élevé, pour les teneurs en cellulose brute égales ou supérieures à 10 %.
8. Observations
8.1. Les aliments pour animaux contenant plus de 10 % de matières grasses brutes doivent être dégraissés avant l'analyse au moyen d'éther de pétrole (3.5). Connecter le creuset filtrant (4.2) et son contenu à l'unité d'extraction à froid (4.6), appliquer le vide et laver le résidu avec trois fois 30 ml d'éther de pétrole. S'assurer que le résidu est sec. Connecter le creuset et son contenu à l'unité de chauffage (4.1) et continuer comme décrit au point 5.
8.2. Les aliments pour animaux contenant des matières grasses qui ne peuvent être directement extraites par l'éther de pétrole (3.5) doivent être dégraissés comme indiqué au point 8.1 et dégraissés de nouveau après ébullition avec l'acide. Après l'ébullition avec l'acide et les lavages qui suivent, connecter le creuset et son contenu à l'unité d'extraction à froid (4.6) et laver trois fois avec 30 ml d'acétone et ensuite trois fois avec 30 ml d'éther de pétrole. Filtrer sous vide jusqu'au séchage et continuer l'analyse telle qu'elle est décrite au point 5 en commençant au niveau du traitement par l'hydroxyde de potassium.
8.3. Si l'aliment pour animaux contient plus de 5 % de carbonates, exprimés en carbonate de calcium, connecter le creuset (4.2) avec l'échantillon pesé à l'unité de chauffage (4.1). Laver l'échantillon avec trois fois 30 ml d'acide chlorhydrique (3.6). Après chaque addition, attendre environ 1 minute avant de filtrer. Laver une fois avec 30 ml d'eau et continuer ensuite comme décrit au point 5.
8.4. Si un appareil en forme de dressoir est utilisé (plusieurs creusets attachés à la même unité de chauffage), ne pas effectuer deux essais du même échantillon dans la même série.
8.5. Si, après ébullition, il est difficile de filtrer les solutions acide et basique, utiliser l'air comprimé par le tuyau de vidange de l'unité de chauffage, puis continuer la filtration.
8.6. Il convient que la température d'incinération ne dépasse pas 500 oC, de façon à allonger la durée de vie des creusets filtrants en verre. Prendre soin d'éviter les chocs thermiques excessifs pendant les cycles de chauffage et de refroidissement.
J. DOSAGE DES SUCRES
1. Objet et champ d'application
La méthode permet de doser les sucres réducteurs et les sucres totaux après inversion, exprimés en glucose ou, le cas échéant, en saccharose, par conversion à l'aide du facteur 0,95 . Elle est applicable aux aliments composés. Des modalités particulières sont prévues pour d'autres aliments. Il convient, si nécessaire, de doser séparément le lactose et d'en tenir compte dans le calcul des résultats.
2. Principe
Les sucres sont dissous dans l'éthanol dilué; la solution est déféquée au moyen des solutions de Carrez I et II. Après élimination de l'éthanol, les dosages sont effectués avant et après inversion, selon la méthode de Luff-Schoorl.
3. Réactifs
3.1. Solution d'éthanol à 40 % (v/v), densité: 0,948 g/ml à 20 oC, amené au point de virage de la phénolphtaléine.
3.2. Solution de Carrez I: dissoudre dans l'eau 21,9 g d'acétate de zinc Zn(CH3COO)2 2H2O et 3 g d'acide acétique glacial. Ajuster à 100 ml avec de l'eau.
3.3. Solution de Carrez II: dissoudre dans l'eau 10,6 g de ferrocyanure de potassium K4Fe (CN)6 3 H2O. Ajuster à 100 ml avec de l'eau.
3.4. Solution à 0,1 % (p/v) de méthylorange.
3.5. Acide chlorhydrique à 4 mol/l.
3.6. Acide chlorhydrique à 0,1 mol/l.
3.7. Solution d'hydroxyde de sodium 0,1 mol/l.
3.8. Réactif selon Luff-Schoorl
Verser, tout en agitant prudemment, la solution d'acide citrique (3.8.2) dans la solution de carbonate de sodium (3.8.3). Ajouter ensuite la solution de sulfate de cuivre (3.8.1) et ajuster à 1 l avec de l'eau. Laisser reposer une nuit et filtrer.
Contrôler la concentration du réactif ainsi obtenu (Cu 0,05 mol/l; Na2 CO3 1 mol/l), voir point 5.4, dernier alinéa. Le pH de la solution doit être de 9,4 environ.
3.8.1. Solution de sulfate de cuivre: dissoudre 25 g de sulfate de cuivre, CuSO4 5H2O, exempt de fer, dans 100 ml d'eau.
3.8.2. Solution d'acide citrique: dissoudre 50 g d'acide citrique, C6H8O7·H2O dans 50 ml d'eau.
3.8.3. Solution de carbonate de sodium: dissoudre 143,8 g de carbonate de sodium anhydre dans environ 300 ml d'eau chaude. Laisser refroidir.
3.9. Solution de thiosulfate de sodium 0,1 mol/l.
3.10. Solution d'amidon: ajouter un mélange de 5 g d'amidon soluble dans 30 ml d'eau à 1 l d'eau bouillante. Faire bouillir durant trois minutes, laisser refroidir, ajouter au besoin 10 mg d'iodure mercurique comme agent conservateur.
3.11. Acide sulfurique 3 mol/l.
3.12. Solution à 30 % (p/v) d'iodure de potassium.
3.13. Granulés de pierre ponce bouillis dans l'acide chlorhydrique, lavés à l'eau et séchés.
3.14. 3-méthylbutan-l-ol.
4. Appareillage
Mélangeur (culbuteur): environ 35 à 40 retournements par minute.
5. Mode opératoire
5.1. Mise en solution
Peser, à 1 mg près, 2,5 g de l'échantillon et les introduire dans une fiole jaugée de 250 ml. Ajouter 200 ml d'éthanol (3.1) et mélanger pendant une heure dans le culbuteur. Ajouter 5 ml de solution de Carrez I (3.2) et agiter pendant environ 30 secondes. Ajouter 5 ml de solution de Carrez II (3.3) et agiter de nouveau pendant une minute. Ajuster au trait de jauge avec de l'éthanol (3.1), homogénéiser et filtrer. Prélever 200 ml du filtrat et évaporer environ la moitié du volume de manière à éliminer la majeure partie de l'éthanol. Transvaser quantitativement le résidu d'évaporation, à l'aide d'eau chaude, dans une fiole jaugée de 200 ml, refroidir, ajuster au trait de jauge avec de l'eau, homogénéiser et filtrer, si nécessaire. Cette solution sera utilisée pour le dosage des sucres réducteurs et, après inversion, pour le dosage des sucres totaux.
5.2. Dosage des sucres réducteurs
Prélever à la pipette une quantité de solution n'excédant pas 25 ml et contenant moins de 60 mg de sucres réducteurs, exprimés en glucose. Si nécessaire, ajuster à 25 ml avec de l'eau distillée et déterminer la teneur en sucres réducteurs au moyen de la méthode de Luff-Schoorl. Le résultat est exprimé sous forme de pourcentage de glucose dans l'échantillon.
5.3. Dosage des sucres totaux après inversion
Prélever à la pipette 50 ml de solution et les porter dans une fiole jaugée de 100 ml. Ajouter quelques gouttes de solution de méthylorange (3.4), puis, prudemment et tout en agitant, de l'acide chlorhydrique (3.5) jusqu'à virage net au rouge. Ajouter 15 ml d'acide chlorhydrique (3.6), plonger la fiole dans un bain d'eau à forte ébullition et l'y maintenir durant trente minutes. Refroidir rapidement à 20 oC environ et ajouter 15 ml de solution d'hydroxyde de sodium (3.7). Ajuster à 100 ml avec de l'eau et homogénéiser. Prélever une quantité n'excédant pas 25 ml et contenant moins de 60 mg de sucres réducteurs, exprimés en glucose. Si nécessaire, ajuster à 25 ml avec de l'eau distillée et déterminer la teneur en sucres réducteurs au moyen de la méthode de Luff-Schoorl. Le résultat est exprimé sous forme de pourcentage de glucose ou, le cas échéant, de saccharose, en multipliant par le facteur 0,95 .
5.4. Titrage selon la méthode de Luff-Schoorl
Prélever à la pipette 25 ml du réactif selon Luff-Schoorl (3.8) et les porter dans un erlenmeyer de 300 ml; ajouter 25 ml, exactement mesurés, de la solution déféquée de sucres. Ajouter deux granulés de pierre ponce (3.13), chauffer, en agitant à la main, sur une flamme libre de hauteur moyenne et porter le liquide à ébullition en deux minutes environ. Placer immédiatement l'erlenmeyer sur une toile métallique pourvue d'un écran d'amiante muni d'un trou de 6 cm environ de diamètre, sous laquelle on a préalablement allumé une flamme. Celle-ci est réglée de façon à ce que seul le fond de l'erlenmeyer soit chauffé. Adapter ensuite un réfrigérant à reflux sur l'erlenmeyer. A partir de ce moment, faire bouillir pendant dix minutes exactement. Refroidir immédiatement dans l'eau froide et après cinq minutes environ, titrer comme suit:
Ajouter 10 ml de solution d'iodure de potassium (3.12) et, immédiatement après et avec prudence (en raison du risque de formation d'une mousse abondante), 25 ml d'acide sulfurique (3.11). Titrer ensuite par la solution de thiosulfate de sodium (3.9) jusqu'à apparition d'une coloration jaune terne, ajouter l'indicateur à l'amidon (3.10) et achever le titrage.
Effectuer le même titrage sur un mélange exactement mesuré de 25 ml de réactif selon Luff-Schoorl (3.8) et 25 ml d'eau, après avoir ajouté 10 ml de solution d'iodure de potassium (3.12) et 25 ml d'acide sulfurique (3.11), sans porter à ébullition.
6. Calcul des résultats
Déterminer, à l'aide de la table, la quantité de glucose (en mg) qui correspond à la différence entre les valeurs des deux titrages, exprimées en mg de thiosulfate de sodium 0,1 mol/l. Exprimer le résultat sous forme de pourcentage de l'échantillon.
7. Modes opératoires particuliers
7.1. Pour les aliments très riches en mélasse et d'autres aliments peu homogènes, peser 20 g et les introduire dans une fiole jaugée de 1 l avec 500 ml d'eau. Mélanger pendant une heure dans le culbuteur. Déféquer au moyen des réactifs de Carrez I (3.2) et II (3.3) comme décrit au point 5.1 en utilisant, toutefois, une quantité quatre fois plus élevée de chaque réactif. Ajuster au trait de jauge avec de l'éthanol à 80 % (v/v).
Homogénéiser et filtrer. Éliminer l'éthanol comme décrit au point 5.1. En l'absence d'amidon dextrinisé, ajuster au trait de jauge avec de l'eau distillée.
7.2. Pour les mélasses et les matières premières pour aliments des animaux riches en sucres et presque exempts d'amidon (caroubes, cossettes séchées de betteraves, etc.), peser 5 g, les introduire dans une fiole jaugée de 250 ml, ajouter 200 ml d'eau distillée et mélanger pendant une heure, ou plus si nécessaire, dans le culbuteur. Déféquer ensuite au moyen des réactifs de Carrez I (3.2) et II (3.3), comme décrit au point 5.1. Ajuster au trait de jauge avec de l'eau froide, homogénéiser et filtrer. Pour doser les sucres totaux, poursuivre comme décrit au point 5.3.
8. Observations
8.1. Il est recommandé d'ajouter environ 1 ml de 3-méthylbutan-l-ol (3.14) (sans tenir compte du volume), avant l'ébullition avec le réactif de Luff-Schoorl, afin d'éviter la formation de mousse.
8.2. La différence entre la teneur en sucres totaux après inversion, exprimés en glucose, et la teneur en sucres réducteurs, exprimés en glucose, multipliée par 0,95 , donne la teneur en saccharose pour cent.
8.3. Pour déterminer la teneur en sucres réducteurs, à l'exclusion du lactose, deux voies peuvent être adoptées.
8.3.1. Pour un calcul approximatif, on multiplie par 0,675 la teneur en lactose déterminée par une méthode d'analyse différente et on retranche le résultat obtenu de la teneur en sucres réducteurs.
8.3.2. Pour un calcul précis des sucres réducteurs, à l'exclusion du lactose, il est nécessaire de partir du même échantillon pour les deux dosages finals. L'une des analyses est effectuée sur une partie de la solution obtenue en application du point 5.1, l'autre sur une partie de la solution obtenue lors du dosage du lactose selon la méthode prévue à cet effet (après fermentation des autres espèces de sucres et défécation).
Dans les deux cas, la quantité de sucre présente est déterminée selon la méthode de Luff-Schoorl et calculée en mg de glucose. L'une des deux valeurs est retranchée de l'autre et la différence est exprimée en pourcentage de l'échantillon.
Exemple:
Les deux volumes prélevés correspondent, pour chaque dosage, à un échantillon de 250 mg.
On consomme 17 ml de solution de thiosulfate de sodium 0,1 mol/l, ce qui correspond à 44,2 mg de glucose, dans le premier cas, et 11 ml de la solution, ce qui correspond à 27,6 mg de glucose, dans le second cas.
La différence s'élève à 16,6 mg de glucose.
La teneur en sucres réducteurs (lactose excepté), calculée en glucose est donc de:

Table des valeurs pour 25 ml de réactif selon Luff-Schoorl
ml de Na2 S2 O30,1 mol/l, deux minutes de chauffage, dix minutes d'ébullition
|
Na2 S2 O3 0,1 mol/l |
Glucose, fructose, sucres invertis C6 H12 O6 |
Lactose C12 H22 O11 |
Maltose C12 H22 O11 |
Na2 S2 O3 0,1 mol/l |
|||
|
ml |
mg |
différence |
mg |
différence |
mg |
différence |
ml |
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
2,4 4,8 7,2 9,7 12,2 14,7 17,2 19,8 22,4 25,0 27,6 30,3 33,0 35,7 38,5 41,3 44,2 47,1 50,0 53,0 56,0 59,1 62,2 |
2,4 2,4 2,5 2,5 2,5 2,5 2,6 2,6 2,6 2,6 2,7 2,7 2,7 2,8 2,8 2,9 2,9 2,9 3,0 3,0 3,1 3,1 |
3,6 7,3 11,0 14,7 18,4 22,1 25,8 29,5 33,2 37,0 40,8 44,6 48,4 52,2 56,0 59,9 63,8 67,7 71,7 75,7 79,8 83,9 88,0 |
3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,8 3,8 3,8 3,8 3,8 3,8 3,9 3,9 3,9 4,0 4,0 4,1 4,1 4,1 |
3,9 7,8 11,7 15,6 19,6 23,5 27,5 31,5 35,5 39,5 43,5 47,5 51,6 55,7 59,8 63,9 68,0 72,2 76,5 80,9 85,4 90,0 94,6 |
3,9 3,9 3,9 4,0 3,9 4,0 4,0 4,0 4,0 4,0 4,0 4,1 4,1 4,1 4,1 4,1 4,2 4,3 4,4 4,5 4,6 4,6 |
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
K. DOSAGE DU LACTOSE
1. Objet et champ d'application
La méthode permet de déterminer la teneur en lactose des aliments pour animaux qui en contiennent plus de 0,5 %.
2. Principe
Les sucres sont dissous dans l'eau. La solution est soumise à la fermentation par la levure Saccharomyces cerevisiae qui laisse le lactose intact. Après défécation et filtration, la teneur en lactose du filtrat est déterminée par la méthode de Luff-Schoorl.
3. Réactifs
3.1. Suspension de Saccharomyces cerevisiae: mettre en suspension 25 g de levure fraîche dans 100 ml d'eau. La suspension se conserve une semaine au maximum au réfrigérateur.
3.2. Solution de Carrez I: dissoudre dans l'eau 21,9 g d'acétate de zinc Zn (CH3COO)2 2H2O et 3 g d'acide acétique glacial. Ajuster à 100 ml avec de l'eau.
3.3. Solution de Carrez II: dissoudre dans l'eau 10,6 g de ferrocyanure de potassium K4Fe (CN)6 3 H2O. Ajuster à 100 ml avec de l'eau.
3.4. Réactif selon Luff-Schoorl:
verser, tout en agitant prudemment, la solution d'acide citrique (3.4.2) dans la solution de carbonate de sodium (3.4.3). Ajouter la solution de sulfate de cuivre (3.4.1) et ajuster à 1 l avec de l'eau. Laisser reposer une nuit et filtrer. Contrôler la concentration du réactif ainsi obtenu (Cu 0,05 mol/l; Na2 CO3 1 mol/l). Le pH de la solution doit être de 9,4 environ.
3.4.1. Solution de sulfate de cuivre: dissoudre 25 g de sulfate de cuivre Cu SO4 5H2O, exempt de fer, dans 100 ml d'eau.
3.4.2. Solution d'acide citrique: dissoudre 50 g d'acide citrique C6H8O7·H2O dans 50 ml d'eau.
3.4.3. Solution de carbonate de sodium: dissoudre 143,8 g de carbonate de sodium anhydre dans environ 300 ml d'eau chaude. Laisser refroidir.
3.5. Granulés de pierre ponce bouillis dans l'acide chlorhydrique, lavés à l'eau et séchés.
3.6. Solution à 30 % (p/v) d'iodure de potassium.
3.7. Acide sulfurique 3 mol/l.
3.8. Solution de thiosulfate de sodium 0,1 mol/l.
3.9. Solution d'amidon: ajouter un mélange de 5 g d'amidon soluble dans 30 ml d'eau à 1 l d'eau bouillante. Faire bouillir durant trois minutes, laisser refroidir et ajouter, si nécessaire, 10 mg d'iodure mercurique comme agent conservateur.
4. Appareillage
Bain d'eau muni d'un thermostat réglé à 38-40 oC.
5. Mode opératoire
Peser, à 1 mg près, 1 g de l'échantillon et introduire cette prise d'essai dans une fiole jaugée de 100 ml. Ajouter 25 à 30 ml d'eau. Placer la fiole pendant trente minutes dans un bain d'eau bouillante et refroidir ensuite à 35 oC environ. Ajouter 5 ml de suspension de levure (3.1) et homogénéiser. Laisser reposer la fiole durant deux heures dans un bain d'eau, à la température de 38 à 40 oC. Refroidir jusqu'à 20 oC environ.
Ajouter 2,5 ml de solution de Carrez I (3.2) et agiter pendant trente secondes; ajouter ensuite 2,5 ml de solution de Carrez II (3.3) et agiter de nouveau pendant trente secondes. Ajuster à 100 ml avec de l'eau, mélanger et filtrer. Prélever à la pipette une quantité de filtrat n'excédant pas 25 ml et contenant de préférence 40 à 80 mg de lactose et introduire celle-ci dans un erlenmeyer de 300 ml. Si nécessaire, ajuster à 25 ml avec de l'eau.
Procéder de la même façon à un essai à blanc avec 5 ml de suspension de levure (3.1). Déterminer comme suit la teneur en lactose selon Luff-Schoorl: ajouter 25 ml exactement du réactif selon Luff-Schoorl (3.4) et deux granulés de pierre ponce (3.5). Chauffer, en agitant à la main, sur une flamme libre de hauteur moyenne et porter le liquide à ébullition en deux minutes environ. Placer immédiatement l'erlenmeyer sur une toile métallique pourvue d'un écran d'amiante muni d'un trou de 6 cm environ de diamètre, sous laquelle on a préalablement allumé une flamme. Celle-ci est réglée de façon à ce que seul le fond de l'erlenmeyer soit chauffé. Adapter ensuite un réfrigérant à reflux sur l'erlenmeyer. À partir de ce moment, faire bouillir pendant dix minutes exactement. Refroidir immédiatement dans l'eau froide et après cinq minutes environ, titrer comme suit:
Ajouter 10 ml de solution d'iodure de potassium (3.6) et, immédiatement après et avec prudence (en raison du risque de formation d'une mousse abondante), 25 ml d'acide sulfurique (3.7). Titrer ensuite par la solution de thiosulfate de sodium (3.8) jusqu'à apparition d'une coloration jaune terne, ajouter l'indicateur à l'amidon (3.9) et achever le titrage.
Effectuer le même titrage sur un mélange exactement mesuré de 25 ml de réactif selon Luff-Schoorl (3.4) et 25 ml d'eau, après avoir ajouté 10 ml de solution d'iodure de potassium (3.6) et 25 ml d'acide sulfurique (3.7), sans porter à ébullition.
6. Calcul des résultats
Déterminer, à l'aide de la table jointe, la quantité de lactose (en mg) qui correspond à la différence entre les résultats des deux titrages, exprimés en ml de thiosulfate de sodium 0,1 mol/l.
Exprimer le résultat sous forme de pourcentage de lactose anhydre dans l'échantillon.
7. Observation
Pour les produits contenant plus de 40 % de sucres fermentescibles, utiliser plus de 5 ml de suspension de levure (3.1).
Table des valeurs pour 25 ml de réactif selon Luff-Schoorl
ml de Na2 S2 O30,1 mol/l, deux minutes de chauffage, dix minutes d'ébullition
|
Na2 S2 O3 0,1 mol/l |
Glucose, fructose, sucres invertis C6 H12 O6 |
Lactose C12 H22 O11 |
Maltose C12 H22 O11 |
Na2 S2 O3 0,1 mol/l |
|||
|
ml |
mg |
différence |
mg |
différence |
mg |
différence |
ml |
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
2,4 4,8 7,2 9,7 12,2 14,7 17,2 19,8 22,4 25,0 27,6 30,3 33,0 35,7 38,5 41,3 44,2 47,1 50,0 53,0 56,0 59,1 62,2 |
2,4 2,4 2,5 2,5 2,5 2,5 2,6 2,6 2,6 2,6 2,7 2,7 2,7 2,8 2,8 2,9 2,9 2,9 3,0 3,0 3,1 3,1 |
3,6 7,3 11,0 14,7 18,4 22,1 25,8 29,5 33,2 37,0 40,8 44,6 48,4 52,2 56,0 59,9 63,8 67,7 71,7 75,7 79,8 83,9 88,0 |
3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,8 3,8 3,8 3,8 3,8 3,8 3,9 3,9 3,9 4,0 4,0 4,1 4,1 4,1 |
3,9 7,8 11,7 15,6 19,6 23,5 27,5 31,5 35,5 39,5 43,5 47,5 51,6 55,7 59,8 63,9 68,0 72,2 76,5 80,9 85,4 90,0 94,6 |
3,9 3,9 3,9 4,0 3,9 4,0 4,0 4,0 4,0 4,0 4,0 4,1 4,1 4,1 4,1 4,1 4,2 4,3 4,4 4,5 4,6 4,6 |
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
L. DOSAGE DE L'AMIDON
MÉTHODE POLARIMÉTRIQUE
1. Objet et champ d'application
La méthode permet de déterminer la teneur en amidon et en produits de dégradation à haut poids moléculaire de l'amidon des aliments pour animaux, en vue de contrôler le respect de la valeur énergétique déclarée (dispositions de l'annexe VII) et de la directive 96/25/CE du Conseil ( 10 ).
2. Principe
La méthode comprend un double dosage. Lors du premier, l'échantillon est traité au moyen d'acide chlorhydrique dilué. Après défécation et filtration, on mesure par polarimétrie le pouvoir rotatoire de la solution.
Lors du second, l'échantillon est extrait au moyen d'éthanol à 40 %. Après acidification du filtrat par l'acide chlorhydrique, défécation et filtration, on mesure le pouvoir rotatoire dans les mêmes conditions que lors du premier dosage.
La différence entre les deux, multipliée par un facteur connu, donne la teneur en amidon de l'échantillon.
3. Réactifs
3.1. Acide chlorhydrique, solution à 25 % (p/p), densité: 1,126 g/ml.
3.2. Acide chlorhydrique, solution à 1,13 % (p/v).
La concentration doit être vérifiée par titrage à l'aide d'une solution d'hydroxyde de sodium 0,1 mol/l en présence de rouge de méthyle à 0,1 % (p/v) dans l'éthanol à 94 % (v/v). 30,94 ml de NaOH 0,1 mol/l sont nécessaires pour la neutralisation de 10 ml.
3.3. Solution de Carrez I: dissoudre dans l'eau 21,9 g d'acétate de zinc Zn(CH3COO)2 2H2O et 3 g d'acide acétique glacial. Ajuster à 100 ml avec de l'eau.
3.4. Solution de Carrez II: dissoudre dans l'eau 10,6 g de ferrocyanure de potassium K4 Fe (CN)6 3H2O. Ajuster à 100 ml avec de l'eau.
3.5. Éthanol, solution à 40 % (v/v), densité: 0,948 g/ml à 20 oC.
4. Appareillage
4.1. Erlenmeyer de 250 ml à rodage normalisé, avec réfrigérant à reflux.
4.2. Polarimètre ou saccharimètre.
5. Mode opératoire
5.1. Préparation de l'échantillon
Broyer l'échantillon de façon à ce qu'il passe en totalité au travers d'un tamis à mailles rondes de 0,5 mm de diamètre.
5.2. Détermination du pouvoir rotatoire total (P ou S) (voir observation 7.1)
Peser, à 1 mg près, 2,5 g de l'échantillon broyé et les introduire dans une fiole jaugée de 100 ml. Ajouter 25 ml d'acide chlorhydrique (3.2), agiter pour obtenir une bonne répartition de la prise d'essai et ajouter de nouveau 25 ml d'acide chlorhydrique (3.2). Plonger la fiole dans un bain d'eau bouillante et, durant les 3 premières minutes qui suivent, agiter énergiquement et régulièrement pour éviter la formation d'agglomérats. La quantité d'eau du bain doit être suffisante pour permettre de maintenir le bain en ébullition lorsque la fiole y est plongée. Celle-ci ne peut être retirée du bain au cours de l'agitation. Après 15 minutes exactement, retirer la fiole du bain, y ajouter 30 ml d'eau froide et refroidir immédiatement jusqu'à 20 oC.
Ajouter 5 ml de solution de Carrez I (3.3) et agiter pendant environ 30 secondes. Ajouter ensuite 5 ml de solution de Carrez II (3.4) et agiter de nouveau pendant 30 secondes environ. Ajuster au trait de jauge avec de l'eau, mélanger et filtrer. Si le filtrat n'est pas parfaitement limpide (ce qui est rare), recommencer l'analyse en utilisant une plus grande quantité des solutions de Carrez I et II, par exemple 10 ml.
Mesurer le pouvoir rotatoire de la solution dans un tube de 200 mm au polarimètre ou au saccharimètre.
5.3. Détermination du pouvoir rotatoire (P' ou S') des substances solubles dans de l'éthanol à 40 %
Peser, à 1 mg près, 5 g de l'échantillon, les introduire dans une fiole jaugée de 100 ml et ajouter 80 ml environ d'éthanol (3.5) (voir observation 7.2). Laisser la fiole reposer durant 1 h à température ambiante; au cours de ce laps de temps, procéder six fois à une agitation énergique de façon à ce que la prise d'essai soit bien mélangée à l'éthanol. Ajuster au trait de jauge avec de l'éthanol (3.5), mélanger et filtrer.
Introduire à la pipette 50 ml du filtrat (= 2,5 g de l'échantillon) dans un erlenmeyer de 250 ml, ajouter 2,1 ml d'acide chlorhydrique (3.1) et agiter énergiquement. Ajuster un réfrigérant à reflux à l'erlenmeyer et plonger celui-ci dans un bain d'eau bouillante. Après 15 minutes exactement, retirer l'erlenmeyer du bain, transvaser son contenu dans une fiole jaugée de 100 ml, en rinçant avec un peu d'eau froide, et refroidir jusqu'à 20 oC.
Déféquer ensuite à l'aide des solutions de Carrez I (3.3) et II (3.4), ajuster au trait de jauge avec de l'eau, mélanger, filtrer et mesurer le pouvoir rotatoire comme indiqué au point 5.2, deuxième et troisième alinéas.
6. Calcul des résultats
La teneur en amidon (%) est calculée de l'une des manières suivantes.
6.1. Mesures effectuées au polarimètre

|
P |
= |
pouvoir rotatoire total en degrés d'angle, |
|
P' |
= |
pouvoir rotatoire en degrés d'angle des substances solubles dans l'éthanol à 40 % (v/v), |
|
|
= |
pouvoir rotatoire spécifique de l'amidon pur. Les valeurs numériques D habituellement acceptées pour ce facteur sont les suivantes:
|
6.2. Mesures effectuées au saccharimètre
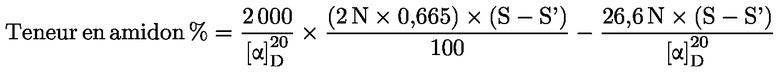
|
S |
= |
pouvoir rotatoire total en degrés de saccharimètre, |
|
S' |
= |
pouvoir rotatoire en degrés de saccharimètre des substances solubles dans l'éthanol à 40 % (v/v), |
|
N |
= |
poids (g) du saccharose dans 100 ml d'eau produisant une rotation optique de 100 degrés de saccharimètre mesurés à l'aide d'un tube de 200 mm: 16,29 g pour les saccharimètres français, 26,00 g pour les saccharimètres allemands, 20,00 g pour les saccharimètres mixtes. |
|
|
= |
pouvoir rotatoire spécifique de l'amidon pur (voir 6.1). |
6.3. Répétabilité
La différence entre les résultats de deux dosages parallèles effectués sur un même échantillon ne peut dépasser 0,4 en valeur absolue pour les teneurs en amidon inférieures à 40 % et 1 % en valeur relative pour les teneurs en amidon égales ou supérieures à 40 %.
7. Observations
7.1. Lorsque l'échantillon contient plus de 6 % de carbonates, calculés en carbonate de calcium, ceux-ci doivent être détruits par un traitement à l'aide d'une quantité exactement appropriée d'acide sulfurique dilué, avant la détermination du pouvoir rotatoire total.
7.2. Dans le cas des produits à forte teneur en lactose, tels que les poudres de lactosérum ou de lait écrémé, procéder comme suit après l'addition de 80 ml d'éthanol (3.5). Ajuster à la fiole un réfrigérant à reflux, plonger la fiole durant 30 minutes dans un bain d'eau à 50 oC. Laisser refroidir et poursuivre l'analyse comme indiqué au point 5.3.
7.3. Les matières premières suivantes, lorsqu'elles sont présentes dans des proportions significatives dans les aliments pour animaux, sont réputées donner lieu à des interférences lors de la détermination de la teneur en amidon par la méthode polarimétrique, de sorte que les résultats obtenus pourraient être faussés:
— sous-produits de betterave (sucrière) tels que la pulpe de betterave (sucrière), la mélasse de betterave (sucrière), la pulpe de betterave (sucrière) mélassée, la vinasse de betterave (sucrière), le sucre de betterave,
— pulpe d'agrumes,
— graines de lin; tourteau de pression de graines de lin; tourteau d'extraction de graines de lin,
— graines de colza; tourteau de pression de colza; tourteau d'extraction de colza; pellicules de colza,
— graines de tournesol; tourteau d'extraction de tournesol; tourteau d'extraction de tournesol partiellement décortiqué,
— tourteau de pression de coprah; tourteau d'extraction de coprah,
— pulpe de pommes de terre,
— levures déshydratées,
— produits riches en inuline (par exemple, cossettes et farine de topinambour),
— cretons.
M. DOSAGE DES CENDRES BRUTES
1. Objet et champ d'application
La méthode permet de déterminer la teneur en cendres brutes des aliments pour animaux.
2. Principe
L'échantillon est incinéré à 550 oC; le résidu est pesé.
3. Réactifs
Nitrate d'ammonium, solution à 20 % (p/v).
4. Appareillage
4.1. Plaque chauffante.
4.2. Four à moufle électrique, avec thermostat.
4.3. Creusets à incinération en silice, porcelaine ou platine rectangulaires (environ 60 × 40 × 25 mm) ou ronds (diamètre: 60 à 75 mm, hauteur: 20 à 40 mm).
5. Mode opératoire
Peser, à 1 mg près, 5 g environ de l'échantillon (2,5 g pour les produits ayant tendance à gonfler) dans un creuset à incinération préalablement chauffé à 550 oC, refroidi et taré. Placer le creuset sur la plaque chauffante et chauffer progressivement jusqu'à carbonisation de la matière. Incinérer conformément au point 5.1 ou 5.2.
5.1. Introduire le creuset dans le four à moufle réglé à 550 oC. Maintenir à cette température jusqu'à obtention de cendres blanches, gris clair ou rougeâtres, apparemment dépourvues de particules charbonneuses. Placer le creuset dans un dessiccateur, laisser refroidir et peser immédiatement.
5.2. Introduire le creuset dans le four à moufle réglé à 550 oC. Incinérer pendant 3 heures. Placer le creuset dans un dessiccateur, laisser refroidir et peser immédiatement. Incinérer de nouveau pendant 30 minutes pour s'assurer que le poids des cendres reste constant (la perte de poids entre deux pesées successives doit être inférieure à 1 mg).
6. Calcul des résultats
Calculer le poids du résidu en déduisant la tare.
Exprimer le résultat sous forme de pourcentage de l'échantillon.
7. Observations
7.1. Les cendres des matières difficiles à incinérer doivent être soumises à une première incinération de trois heures au moins, refroidies et additionnées de quelques gouttes d'une solution à 20 % de nitrate d'ammonium ou d'eau (prudemment, pour éviter la dispersion ou le collage des cendres). Poursuivre la calcination après dessiccation à l'étuve. Répéter si nécessaire l'opération jusqu'à incinération complète.
7.2. Pour les matières qui résistent au traitement indiqué au point 7.1, opérer comme suit: après une incinération de trois heures, reprendre les cendres par de l'eau chaude et filtrer sur un petit filtre sans cendres. Incinérer le filtre et son contenu dans le creuset initial. Amener le filtrat dans le creuset refroidi, évaporer à sec, incinérer et peser.
7.3. Dans le cas des matières grasses, peser avec exactitude une prise d'essai de 25 g dans un creuset de capacité appropriée. Carboniser en enflammant la matière au moyen d'une mèche de papier filtre sans cendres. Après combustion, humecter par le minimum nécessaire d'eau. Sécher et incinérer comme indiqué au point 5.
N. DOSAGE DES CENDRES INSOLUBLES DANS L'ACIDE CHLORHYDRIQUE
1. Objet et champ d'application
La méthode permet de déterminer la teneur en matières minérales insolubles dans l'acide chlorhydrique des aliments pour animaux. Deux procédés sont prévus en fonction de la nature de l'échantillon.
1.1. Procédé A: applicable aux matières premières organiques pour aliments des animaux et à la plupart des aliments composés.
1.2. Procédé B: applicable aux composés et aux mélanges minéraux ainsi qu'aux aliments composés dont la teneur en matières insolubles dans l'acide chlorhydrique, déterminée selon le procédé A, est supérieure à 1 %.
2. Principe
2.1. Procédé A: l'échantillon est incinéré, les cendres sont traitées à ébullition par l'acide chlorhydrique et le résidu insoluble est filtré et pesé.
2.2. Procédé B: l'échantillon est traité par l'acide chlorhydrique. La solution est filtrée, le résidu est incinéré et les cendres obtenues sont traitées comme dans le procédé A.
3. Réactifs
3.1. Acide chlorhydrique à 3 mol/l.
3.2. Acide trichloracétique, solution à 20 % (p/v).
3.3. Acide trichloracétique, solution à 1 % (p/v).
4. Appareillage
4.1. Plaque chauffante.
4.2. Four à moufle électrique, avec thermostat.
4.3. Creusets à incinération en silice, porcelaine ou platine rectangulaires (environ 60 × 40 × 25 mm) ou ronds (diamètre: 60 à 75 mm, hauteur: 20 à 40 mm).
5. Mode opératoire
5.1. Procédé A
Incinérer la prise d'essai selon le mode opératoire décrit pour le dosage des cendres brutes. Les cendres obtenues lors de ce dosage peuvent également être utilisées.
Introduire les cendres dans un bécher de 250 à 400 ml à l'aide de 75 ml d'acide chlorhydrique (3.1). Porter prudemment le liquide à ébullition douce et maintenir celle-ci pendant quinze minutes. Filtrer la solution chaude sur un papier filtre sans cendres et laver le résidu avec de l'eau chaude jusqu'à disparition de réaction acide. Sécher le filtre contenant le résidu et incinérer dans un creuset taré à une température de 550 oC au moins et de 700 oC au plus. Refroidir dans un dessiccateur et peser.
5.2. Procédé B
Peser, à 1 mg près, 5 g de l'échantillon et les introduire dans un bécher de 250 à 400 ml. Ajouter successivement 25 ml d'eau et 25 ml d'acide chlorhydrique (3.1), mélanger et attendre la fin de l'effervescence. Ajouter encore 50 ml d'acide chlorhydrique (3.1). Attendre la fin d'un éventuel dégagement gazeux, placer ensuite le bécher dans un bain d'eau bouillante et l'y maintenir pendant trente minutes ou plus, si nécessaire, afin d'hydrolyser complètement l'amidon éventuellement présent. Filtrer à chaud sur filtre sans cendres et laver le filtre à l'aide de 50 ml d'eau chaude (voir observation 7). Placer le filtre contenant le résidu dans un creuset à incinération, sécher et incinérer à une température de 550 oC au moins et de 700 oC au plus. Introduire les cendres dans un bécher de 250 à 400 ml à l'aide de 75 ml d'acide chlorhydrique (3.1); poursuivre comme indiqué au point 5.1, deuxième alinéa.
6. Calcul des résultats
Calculer le poids du résidu en déduisant la tare. Exprimer le résultat sous forme de pourcentage de l'échantillon.
7. Observation
Si la filtration se révèle difficile, recommencer le dosage en remplaçant les 50 ml d'acide chlorhydrique (3.1) par 50 ml d'acide trichloracétique à 20 % (3.2) et en lavant le filtre à l'aide d'une solution chaude d'acide trichloracétique à 1 % (3.3).
O. DOSAGE DES CARBONATES
1. Objet et champ d'application
La méthode permet de doser les carbonates, conventionnellement exprimés en carbonate de calcium, dans la plupart des aliments pour animaux.
Il faut néanmoins utiliser une méthode particulière dans certains cas (carbonate de fer, par exemple).
2. Principe
Les carbonates sont décomposés par l'acide chlorhydrique; le gaz carbonique libéré est recueilli dans un tube gradué et son volume est comparé à celui dégagé, dans les mêmes conditions, par une quantité connue de carbonate de calcium.
3. Réactifs
3.1. Acide chlorhydrique, densité 1,10 g/ml.
3.2. Carbonate de calcium.
3.3. Acide sulfurique 0,05 mol/l environ, coloré par du rouge de méthyle.
4. Appareillage
Appareil selon Scheibler-Dietrich (voir schéma) ou appareil équivalent.
5. Mode opératoire
Selon la teneur en carbonates de l'échantillon, peser une prise d'essai comme indiqué ci-après:
— 0,5 g pour les produits contenant de 50 à 100 % de carbonates, exprimés en carbonate de calcium,
— 1 g pour les produits contenant de 40 à 50 % de carbonates, exprimés en carbonate de calcium,
— 2 à 3 g pour les autres produits.
Introduire la prise d'essai dans le flacon spécial (4) de l'appareil, muni d'un petit tube en matière incassable contenant 10 ml d'acide chlorhydrique (3.1), et raccorder le flacon à l'appareil. Tourner le robinet à trois voies (5) de façon à ce que le tube (1) communique avec l'extérieur. A l'aide du tube mobile (2), qui est rempli d'acide sulfurique coloré (3.3) et relié au tube gradué (1), amener le niveau du liquide à la graduation zéro. Tourner le robinet (5), de façon à faire communiquer les tubes (1) et (3), et vérifier le niveau zéro.
Laisser couler lentement l'acide chlorhydrique (3.1) sur la prise d'essai en inclinant le flacon (4). Égaliser la pression en abaissant le tube (2). Agiter le flacon (4) jusqu'à cessation complète du dégagement de gaz carbonique.
Rétablir la pression en ramenant le liquide au même niveau dans les tubes (1) et (2). Faire la lecture après quelques minutes, lorsque le volume gazeux est devenu constant.
Effectuer dans les mêmes conditions un essai de comparaison sur 0,5 g de carbonate de calcium (3.2).
6. Calcul des résultats
La teneur en carbonates, exprimés en carbonate de calcium, est donnée par la formule suivante:

où:
|
X |
= |
% (p/p) de carbonates dans l'échantillon, exprimés en carbonate de calcium, |
|
V |
= |
ml de CO2 dégagés par la prise d'essai, |
|
V1 |
= |
ml de CO2 dégagés par 0,5 g de CaCO3, |
|
m |
= |
poids (en g) de la prise d'essai. |
7. Observations
7.1. Lorsque la prise d'essai est supérieure à 2 g, introduire préalablement 15 ml d'eau distillée dans le flacon (4) et mélanger avant de commencer l'essai. Employer le même volume d'eau pour l'essai de comparaison.
7.2. Si l'on utilise un appareil d'un volume différent de celui de Scheibler-Dietrich, il faut y adapter la prise d'essai de l'échantillon et de la substance de comparaison ainsi que le calcul des résultats.

P. DOSAGE DU PHOSPHORE TOTAL
MÉTHODE PHOTOMÉTRIQUE
1. Objet et champ d'application
La méthode permet de déterminer la teneur en phosphore total des aliments pour animaux. Elle est particulièrement indiquée pour l'analyse des produits pauvres en phosphore. Dans certains cas (produits riches en phosphore), une méthode gravimétrique peut être appliquée.
2. Principe
L'échantillon est minéralisé, soit par voie sèche (pour les aliments organiques), soit par voie humide (pour les composés minéraux et les aliments liquides) et mis en solution acide. La solution est traitée par le réactif vanadomolybdique. La densité optique de la solution jaune ainsi formée est mesurée au spectrophotomètre à 430 nm.
3. Réactifs
3.1. Carbonate de calcium.
3.2. Acide chlorhydrique, ρ20 = 1,10 g/ml (environ 6 mol/l).
3.3. Acide nitrique, ρ20 = 1,045 g/ml.
3.4. Acide nitrique, ρ20 = 1,38 à 1,42 g/ml.
3.5. Acide sulfurique, ρ20 = 1,84 g/ml.
3.6. Réactif vanadomolybdique: mélanger 200 ml de solution d'heptamolybdate d'ammonium (3.6.1), 200 ml de solution de monovanadate d'ammonium (3.6.2) et 134 ml d'acide nitrique (3.4) dans une fiole jaugée de 1 l. Ajuster au trait de jauge avec de l'eau.
3.6.1. Solution d'heptamolybdate d'ammonium: dissoudre dans l'eau chaude 100 g d'heptamolybdate d'ammonium (NH4) 6Mo7O24.4H2O. Ajouter 10 ml d'ammoniaque (densité 0,91 g/ml) et ajuster à 1 l avec de l'eau.
3.6.2. Solution de monovanadate d'ammonium: dissoudre dans 400 ml d'eau chaude 2,35 g de monovanadate d'ammonium NH4VO3. Ajouter lentement et tout en agitant 20 ml d'acide nitrique dilué [7 ml de HNO3 (3.4) + 13 ml de H2O] et ajuster à 1 l avec de l'eau.
3.7. Solution étalon à 1 mg de phosphore par ml: dissoudre dans l'eau 4,387 g de dihydrogénophosphate de potassium KH2PO4. Ajuster à 1 l avec de l'eau.
4. Appareillage
4.1. Creusets à incinération en silice, en porcelaine ou en platine.
4.2. Four à moufle électrique, muni d'un thermostat réglé à 550 oC.
4.3. Matras de Kjeldahl, 250 ml.
4.4. Fioles jaugées et pipettes de précision.
4.5. Spectrophotomètre.
4.6. Tubes à essai d'un diamètre de 16 mm environ, à rodage normalisé 14,5 mm; capacité: 25 à 30 ml.
5. Mode opératoire
5.1. Préparation de la solution
Selon la nature de l'échantillon, préparer une solution comme indiqué au point 5.1.1 ou 5.1.2.
5.1.1.
Peser, à 1 mg près, 1 g ou plus de l'échantillon. Introduire la prise d'essai dans un matras de Kjeldahl, ajouter 20 ml d'acide sulfurique (3.5), agiter pour imprégner complètement la matière d'acide et éviter qu'elle n'adhère aux parois du ballon, chauffer et maintenir pendant 10 minutes à ébullition. Laisser refroidir légèrement, ajouter 2 ml d'acide nitrique (3.4), chauffer doucement, laisser refroidir légèrement, ajouter à nouveau un peu d'acide nitrique (3.4) et porter à ébullition. Répéter ces opérations jusqu'à obtention d'une solution incolore. Refroidir, ajouter un peu d'eau, transvaser le liquide dans une fiole jaugée de 500 ml en rinçant le matras de Kjeldahl à l'eau chaude. Laisser refroidir, ajuster au trait de jauge avec de l'eau, homogénéiser et filtrer.
5.1.2.
Peser, à 1 mg près, 2,5 g environ de l'échantillon dans un creuset à incinération. Mélanger intimement la prise d'essai à 1 g de carbonate de calcium (3.1). Incinérer au four à 550 oC jusqu'à obtention de cendres blanches ou grises (une petite quantité de charbon ne gêne pas). Transvaser les cendres dans un bécher de 250 ml. Ajouter 20 ml d'eau et de l'acide chlorhydrique (3.2) jusqu'à cessation de l'effervescence. Ajouter encore 10 ml d'acide chlorhydrique (3.2). Porter le bécher sur un bain de sable et évaporer à sec pour insolubiliser la silice. Reprendre le résidu par 10 ml d'acide nitrique (3.3) et faire bouillir pendant 5 minutes sur le bain de sable ou la plaque chauffante, sans évaporer à sec. Transvaser le liquide dans une fiole jaugée de 500 ml en rinçant le bécher à plusieurs reprises à l'eau chaude. Laisser refroidir, ajuster au trait de jauge avec de l'eau, homogénéiser et filtrer.
5.2. Développement de la coloration et mesure de la densité optique
Diluer une partie aliquote du filtrat obtenu conformément au point 5.1.1 ou 5.1.2 pour obtenir une concentration de phosphore atteignant au maximum 40 μg/ml. Introduire 10 ml de cette solution dans un tube à essai (4.6) et y ajouter 10 ml du réactif vanadomolybdique (3.6). Homogénéiser et laisser reposer 10 minutes au moins à la température de 20 oC. Mesurer la densité optique au spectrophotomètre à 430 nm par comparaison avec une solution obtenue par addition de 10 ml du réactif vanadomolybdique (3.6) à 10 ml d'eau.
5.3. Courbe d'étalonnage
Préparer à partir de la solution étalon (3.7) des solutions contenant respectivement 5, 10, 20, 30 et 40 μg de phosphore par ml. Prélever 10 ml de chacune de ces solutions et y ajouter 10 ml du réactif vanadomolybdique (3.6). Homogénéiser et laisser reposer 10 minutes au moins à la température de 20 oC. Mesurer la densité optique comme indiqué au point 5.2. Tracer la courbe d'étalonnage en portant en ordonnée les valeurs de la densité optique et en abscisse les quantités correspondantes de phosphore. La courbe est linéaire pour les concentrations comprises entre 0 et 40 μg/ml.
6. Calcul des résultats
Déterminer la quantité de phosphore de la prise d'essai en se référant à la courbe d'étalonnage.
Exprimer le résultat sous forme de pourcentage de l'échantillon.
Répétabilité
La différence entre les résultats de deux dosages parallèles effectués sur le même échantillon ne peut dépasser:
— 3 % par rapport au résultat le plus élevé, pour les teneurs en phosphore inférieures à 5 %,
— 0,15 % en valeur absolue pour les teneurs en phosphore égales ou supérieures à 5 %.
Q. DOSAGE DU CHLORE DES CHLORURES
1. Objet et champ d'application
La méthode permet de doser le chlore des chlorures solubles dans l'eau, conventionnellement exprimé en chlorure de sodium. Elle est applicable à tous les aliments pour animaux.
2. Principe
Les chlorures sont dissous dans l'eau. Si le produit contient des matières organiques, on procède à une défécation. La solution est légèrement acidifiée par l'acide nitrique et les chlorures sont précipités sous forme de chlorure d'argent à l'aide d'une solution de nitrate d'argent. L'excès de nitrate d'argent est titré par une solution de thiocyanate d'ammonium, selon la méthode de Volhard.
3. Réactifs
3.1. Solution de thiocyanate d'ammonium 0,1 mol/l.
3.2. Solution de nitrate d'argent 0,1 mol/l.
3.3. Solution saturée de sulfate d'ammonium ferrique (NH4)Fe(SO4)2.
3.4. Acide nitrique, densité 1,38 g/ml.
3.5. Éther diéthylique.
3.6. Acétone.
3.7. Solution de Carrez I: dissoudre dans l'eau 21,9 g d'acétate de zinc Zn (CH3COO)2·2H2O et 3 g d'acide acétique glacial. Ajuster à 100 ml avec de l'eau.
3.8. Solution de Carrez II: dissoudre dans l'eau 10,6 g de ferrocyanure de potassium K4Fe(CN)6·3 H2O. Ajuster à 100 ml avec de l'eau.
3.9. Charbon actif exempt de chlorures et n'en adsorbant pas.
4. Appareillage
Mélangeur (culbuteur): environ 35 à 40 retournements par minute.
5. Mode opératoire
5.1. Préparation de la solution
Selon la nature de l'échantillon, préparer une solution comme indiqué au point 5.1.1, 5.1.2 ou 5.1.3.
Effectuer, en parallèle, un essai à blanc exempt d'échantillon à analyser.
5.1.1.
Peser, à 1 mg près, une prise d'essai de 10 g au maximum ne contenant pas plus de 3 g de chlore sous forme de chlorures et l'introduire dans un flacon jaugé de 500 ml avec 400 ml d'eau à 20 oC environ. Mélanger durant trente minutes dans le culbuteur, ajuster au trait de jauge, homogénéiser et filtrer.
5.1.2.
Peser, à 1 mg près, 5 g de l'échantillon et les introduire avec 1 g de charbon actif dans un flacon jaugé de 500 ml. Ajouter 400 ml d'eau à 20 oC environ et 5 ml de solution de Carrez I (3.7), agiter pendant 30 secondes et ajouter ensuite 5 ml de solution de Carrez II (3.8). Mélanger durant trente minutes dans le culbuteur, ajuster au trait de jauge, homogénéiser et filtrer.
5.1.3.
Préparer la solution comme indiqué au point 5.1.2 mais ne pas filtrer. Décanter (si nécessaire, centrifuger), prélever 100 ml du liquide surnageant et les introduire dans une fiole jaugée de 200 ml. Mélanger avec de l'acétone (3.6) et ajuster au trait de jauge avec ce solvant, homogénéiser et filtrer.
5.2. Titrage
Introduire à la pipette, dans un erlenmeyer, 25 à 100 ml du filtrat (selon la teneur présumée en chlore) obtenu conformément au point 5.1.1, 5.1.2 ou 5.1.3. La portion aliquote ne peut contenir plus de 150 mg de chlore (Cl). Diluer, si nécessaire, à 50 ml au moins avec de l'eau, ajouter 5 ml d'acide nitrique (3.4), 20 ml de solution saturée de sulfate d'ammonium ferrique (3.3) et 2 gouttes de solution de thiocyanate d'ammonium (3.1) débitées à l'aide d'une burette remplie jusqu'au trait de jauge zéro. Débiter ensuite à l'aide d'une burette la solution de nitrate d'argent (3.2) de façon à obtenir un excès de 5 ml. Ajouter 5 ml d'éther diéthylique (3.5) et agiter fortement pour rassembler le précipité. Titrer l'excès de nitrate d'argent par la solution de thiocyanate d'ammonium (3.1) jusqu'à ce que le virage au rouge-brun persiste pendant une minute.
6. Calcul des résultats
La quantité de chlore (X), exprimée en chlorure de sodium sous forme de pourcentage, est donnée par la formule suivante:

où:
|
V1 |
= |
ml de solution de nitrate d'argent 0,1 mol/l ajoutés, |
|
V2 |
= |
ml de solution de thiocyanate d'ammonium 0,1 mol/l utilisés lors du titrage, |
|
m |
= |
poids de l'échantillon. |
Si l'essai à blanc indique une consommation de solution de nitrate d'argent 0,1 mol/l, retrancher cette valeur du volume (V1 - V2).
7. Observations
7.1. Le titrage peut également être réalisé par potentiométrie.
7.2. Pour les produits très riches en matières grasses, procéder à un dégraissage préalable par l'éther diéthylique ou l'éther de pétrole.
7.3. Dans le cas des farines de poisson, le titrage peut être effectué selon la méthode de Mohr.
ANNEXE IV
MÉTHODES D'ANALYSE RELATIVES AU CONTRÔLE DES TENEURS EN ADDITIFS AUTORISÉS DES ALIMENTS POUR ANIMAUX
A. DOSAGE DE LA VITAMINE A
1. Objet et champ d'application
La méthode permet de déterminer la teneur en vitamine A (rétinol) des aliments pour animaux et des prémélanges. La vitamine A comprend le rétinol-all-trans et ses isomères-cis, qui sont déterminés par cette méthode. La teneur en vitamine A est exprimée en unités internationales (UI) par kg. Une UI correspond à l'activité de 0,300 μg de vitamine A alcool-all-trans ou à 0,344 μg de vitamine A acétate all-trans ou de 0,550 μg de vitamine A palmitate all-trans.
La limite de quantification est de 2 000 UI de vitamine A/kg.
2. Principe
L'échantillon est hydrolysé avec une solution d'hydroxyde de potassium éthanolique, et la vitamine A est extraite dans de l'éther de pétrole. Le solvant est enlevé par évaporation; le résidu est dissous dans du méthanol et, si nécessaire, dilué à la concentration requise. La teneur en vitamine A est déterminée par chromatographie liquide haute performance en phase inverse (CLHP-PI) à l'aide d'un détecteur UV ou fluorimétrique. Les paramètres de la chromatographie sont choisis de telle sorte qu'il n'y ait pas de séparation entre la vitamine A alcool-all-trans et ses isomères-cis.
3. Réactifs
3.1. Éthanol, σ = 96 %.
3.2. Éther de pétrole, intervalle d'ébullition 40 oC-60 oC.
3.3. Méthanol.
3.4. Solution d'hydroxyde de potassium, c = 50 g/100 ml.
3.5. Solution d'ascorbate de sodium, c = 10 g/100 ml (voir l'observation figurant au point 7.7).
3.6. Sulfure de sodium, Na2S · x H2O (x = 7-9).
3.6.1. Solution de sulfure de sodium, c = 0,5 mol/l dans du glycérol, β = 120 g/l (pour x = 9) (voir l'observation figurant au point 7.8).
3.7. Solution de phénolphtaléine, c = 2 g/100 ml dans de l'éthanol (3.1).
3.8. Propanol-2.
3.9. Phase mobile pour CLHP: mélange de méthanol (3,3 ) et d'eau, par exemple: 980 + 20 (v + v). Les proportions exactes sont déterminées par les caractéristiques de la colonne employée.
3.10. Azote, libre d'oxygène.
3.11. Vitamine A acétate all-trans, extra-pure, d'activité garantie, par exemple: 2,80 × 106 UI/g.
3.11.1. Solution mère de vitamine A acétate all-trans: peser, à 0,1 mg près, 50 mg de vitamine A acétate (3.11) dans une fiole jaugée de 100 ml. Dissoudre dans du propanol-2 (3.8) et ajuster au trait de jauge avec le même solvant. La concentration nominale de cette solution est de 1 400 UI de vitamine A par ml. La teneur exacte doit être déterminée selon le point 5.6.3.1.
3.12. Vitamine A palmitate all-trans, extra-pure, d'activité garantie, par exemple: 1,80 × 106 UI/g.
3.12.1. Solution mère de vitamine A palmitate all-trans: peser, à 0,1 mg près, 80 mg de vitamine A palmitate (3.12) dans une fiole jaugée de 100 ml. Dissoudre dans du propanol-2 (3.8) et ajuster au trait de jauge avec le même solvant. La concentration nominale de cette solution est de 1 400 UI de vitamine A par ml. La teneur exacte doit être déterminée selon le point 5.6.3.2.
3.13. BHT (di-tert-butyl-2,6-méthyl-4-phénol) (voir l'observation figurant au point 7.5).
4. Appareillage
4.1. Évaporateur rotatif sous vide.
4.2. Verrerie ambrée.
4.2.1. Ballons à fond plat ou coniques, 500 ml, avec col en verre rodé.
4.2.2. Fioles jaugées à bouchon en verre rodé, col étroit, de 10, 25, 100 et 500 ml.
4.2.3. Ampoules à décanter coniques, 1 000 ml, à bouchon en verre rodé.
4.2.4. Ballons piriformes, 250 ml, avec col en verre rodé.
4.3. Réfrigérant d'Allihn, 300 mm de longueur utile, avec joint en verre rodé et adaptateur pour tuyau d'alimentation en gaz.
4.4. Papier filtre plissé pour la séparation des phases, d'un diamètre de 185 mm (par exemple: Schleicher & Schuell 597 HY 1/2).
4.5. Équipement CLHP avec système à injection.
4.5.1. Colonne de chromatographie liquide de 250 mm × 4 mm, C18, particules de 5 ou 10 μm, ou équivalent (critère de performance: un seul pic pour tous les isomères de rétinol dans les conditions CLHP).
4.5.2. Détecteur UV ou fluorimétrique, de longueur d'onde variable.
4.6. Spectrophotomètre avec cellules de quartz de 10 mm.
4.7. Bain-marie avec agitateur magnétique.
4.8. Appareil d'extraction (voir la figure 1) se composant:
4.8.1. d'une éprouvette d'une capacité de 1 l, à col et à bouchon en verre rodé;
4.8.2. d'une pièce à rodage mâle munie d'une tige latérale et d'un tube réglable passant en son centre. Ce tube doit avoir une partie inférieure en U et un bec gicleur à son extrémité opposée, de telle sorte que la couche liquide supérieure dans l'éprouvette puisse être transférée dans une ampoule à décanter.
5. Mode opératoire
|
NB |
: |
La vitamine A est sensible à la lumière (UV) et à l'oxydation. Toutes les opérations doivent être réalisées en l'absence de lumière (dans du verre ambré ou protégé d'une feuille d'aluminium) et en l'absence d'oxygène (éliminé avec de l'azote). Pendant l'extraction, l'air au-dessus du liquide doit être remplacé par de l'azote (éviter l'excès de pression en desserrant le bouchon de temps en temps). |
5.1. Préparation de l'échantillon
Moudre l'échantillon pour qu'il passe par un tamis à mailles de 1 mm, en évitant la production de chaleur. Le broyage doit avoir lieu immédiatement avant la pesée et la saponification, sinon il peut y avoir des pertes en vitamine A.
5.2. Saponification
Selon la teneur en poids de la vitamine A, peser, à 1 mg près, de 2 à 25 g de l'échantillon dans un ballon à fond plat ou conique de 500 ml (4.2.1). Ajouter successivement, tout en remuant, 130 ml d'éthanol (3.1), environ 100 mg de BHT (3.13), 2 ml de solution d'ascorbate de sodium (3.5) et 2 ml de solution de sulfure de sodium (3.6). Adapter un réfrigérant (4.3) au ballon et immerger celui-ci dans un bain-marie avec agitateur magnétique (4.7). Chauffer jusqu'à ébullition et laisser refluer pendant 5 minutes. Ajouter alors 25 ml de solution d'hydroxyde de potassium (3.4) par le réfrigérant (4.3) et laisser refluer de nouveau pendant 25 min, tout en agitant sous un faible courant d'azote. Rincer alors le réfrigérant avec environ 20 ml d'eau et laisser refroidir le contenu du ballon à la température ambiante.
5.3. Extraction
Transférer quantitativement par décantation la solution de saponification en rinçant avec un volume total de 250 ml d'eau dans une ampoule à décanter de 1 000 ml (4.2.3) ou dans l'appareil d'extraction (4.8). Rincer successivement le ballon de saponification avec 25 ml d'éthanol (3.1) et 100 ml d'éther de pétrole (3.2) et transférer le liquide de rinçage dans l'ampoule à décanter ou dans l'appareil d'extraction. La proportion d'eau et d'éthanol dans les solutions combinées doit être d'environ 2:1. Secouer énergiquement pendant 2 minutes et laisser reposer pendant 2 minutes.
5.3.1.
Lorsque les couches sont séparées (voir l'observation figurant au point 7.3), transférer la couche d'éther de pétrole dans une autre ampoule à décanter (4.2.3). Répéter deux fois l'opération avec 100 ml d'éther de pétrole (3.2), puis deux fois avec 50 ml d'éther de pétrole (3.2).
Laver deux fois les extraits combinés dans l'ampoule à décanter en remuant doucement (afin d'éviter la formation d'émulsions) avec des portions de 100 ml d'eau et de nouveau en secouant avec d'autres portions de 100 ml d'eau jusqu'à ce que l'eau demeure incolore après addition de solution de phénolphtaléine (3.7) (quatre lavages sont généralement suffisants). Filtrer l'extrait lavé sur un filtre plissé sec pour la séparation des phases (4.4) afin d'éliminer l'eau résiduelle et transférer dans une fiole jaugée de 500 ml (4.2.2). Rincer l'ampoule à décanter et le filtre avec 50 ml d'éther de pétrole (3.2), ajuster au trait de jauge avec de l'éther de pétrole (3.2) et bien mélanger.
5.3.2.
Lorsque les couches sont séparées (voir l'observation figurant au point 7.3), remettre le bouchon de l'éprouvette (4.8.1) sur la pièce à rodage mâle (4.8.2) et placer l'extrémité inférieure en forme de U du tube réglable de telle sorte qu'elle se trouve juste au-dessus du niveau de l'interface. En exerçant une pression d'azote par la tige latérale, transférer la couche supérieure d'éther de pétrole dans une ampoule à décanter de 1 000 ml (4.2.3). Ajouter 100 ml d'éther de pétrole (3.2) dans le cylindre en verre, boucher et bien secouer. Laisser les couches se séparer et transférer la couche supérieure dans l'ampoule à décanter comme précédemment. Répéter la procédure d'extraction avec de nouveau 100 ml d'éther de pétrole (3.2), puis deux fois avec 50 ml d'éther de pétrole (3.2) et ajouter les couches d'éther de pétrole dans l'ampoule à décanter.
Laver les extraits combinés d'éther de pétrole selon la procédure décrite au point 5.3.1 et procéder selon ce point.
5.4. Préparation de la solution d'échantillon pour CLHP
Introduire à la pipette une portion aliquote de la solution d'éther de pétrole (issue de 5.3.1 ou de 5.3.2) dans un ballon piriforme de 250 ml (4.2.4). Laisser évaporer le solvant presque entièrement dans l'évaporateur rotatif (4.1) sous une pression réduite, à une température de bain ne dépassant pas 40 oC. Restaurer la pression atmosphérique en laissant entrer l'azote (3.10) et enlever le ballon de l'évaporateur rotatif. Supprimer le reste du solvant dans un courant d'azote (3.10) et dissoudre immédiatement le résidu dans un volume connu (10-100 ml) de méthanol (3.3) (la concentration de vitamine A doit être de l'ordre de 5 UI/ml à 30 UI/ml).
5.5. Dosage par CLHP
La vitamine A est séparée sur une colonne C18 à phase inverse (4.5.1), et la concentration est mesurée à l'aide d'un détecteur UV (325 nm) ou d'un détecteur fluorimétrique (excitation: 325 nm; émission: 475 nm) (4.5.2).
Injecter une portion aliquote (par exemple: 20 μl) de la solution méthanolique obtenue sous 5.4 et éluer avec la phase mobile (3.9). Calculer la hauteur moyenne du pic (surface) de plusieurs injections de la même solution d'échantillon et les hauteurs moyennes des pics (surfaces) de plusieurs injections des solutions d'étalonnage (5.6.2).
Les conditions suivantes sont proposées à titre indicatif; d'autres conditions peuvent être appliquées pourvu qu'elles donnent des résultats équivalents.
|
Colonne de chromatographie liquide (point 4.5.1): |
250 mm × 4 mm, C18, particules de 5 ou de 10 μm, ou équivalent |
|
Phase mobile (3.9): |
mélange de méthanol (3.3) et d'eau, par exemple 980 + 20 (v + v). |
|
Débit: |
1-2 ml/min |
|
Détecteur (4.5.2): |
détecteur UV (325 nm) ou détecteur fluorimétrique (excitation: 325 nm/émission: 475 nm) |
5.6. Étalonnage
5.6.1.
Introduire à la pipette 20 ml de la solution mère de vitamine A acétate (3.11.1) ou 20 ml de la solution mère de vitamine A palmitate (3.12.1) dans un ballon à fond plat ou conique de 500 ml (4.2.1) et hydrolyser comme décrit au point 5.2, mais sans ajouter de BHT. Procéder ensuite à l'extraction avec de l'éther de pétrole (3.2) conformément au point 5.3 et ajuster à 500 ml avec de l'éther de pétrole (3.2). Laisser évaporer 100 ml de cet extrait presque entièrement sur l'évaporateur rotatif (voir 5.4), enlever le solvant résiduel dans un courant d'azote (3.10) et redissoudre le résidu dans 10,0 ml de méthanol (3.3). La concentration nominale de cette solution est de 560 UI de vitamine A par ml. La teneur exacte doit être déterminée conformément au point 5.6.3.3. La solution étalon de travail doit être préparée peu avant d'être utilisée.
Introduire à la pipette 2,0 ml de cette solution étalon de travail dans une fiole jaugée de 20 ml, ajuster au trait de jauge avec du méthanol (3.3) et mélanger. La concentration nominale de cette solution étalon de travail diluée est de 56 UI de vitamine A par ml.
5.6.2.
Transférer 1,0 , 2,0 , 5,0 et 10,0 ml de la solution étalon de travail diluée dans une série de fioles jaugées de 20 ml, ajuster au trait de jauge avec du méthanol (3.3) et mélanger. Les concentrations nominales de ces solutions sont de 2,8 , 5,6 , 14,0 et 28,0 UI de vitamine A par ml.
Injecter plusieurs fois 20 μl de chaque solution d'étalonnage et déterminer les hauteurs moyennes des pics (surfaces). D'après les hauteurs moyennes des pics (surfaces), tracer une courbe d'étalonnage tenant compte des résultats du contrôle UV (5.6.3.3).
5.6.3.
5.6.3.1.
Introduire à la pipette 2,0 ml de la solution mère de vitamine A acétate (3.11.1) dans une fiole jaugée de 50 ml (4.2.2) et ajuster au trait de jauge avec du propanol-2 (3.8). La concentration nominale de cette solution est de 56 UI de vitamine A par ml. Introduire à la pipette 3,0 ml de cette solution de vitamine A acétate diluée dans une fiole jaugée de 25 ml et ajuster au trait de jauge avec du propanol-2 (3.8). La concentration nominale de cette solution est de 6,72 UI de vitamine A par ml. Mesurer le spectre UV de cette solution contre du propanol-2 (3.8) dans le spectrophotomètre (4.6) entre 300 nm et 400 nm. L'extinction maximale doit se situer entre 325 nm et 327 nm.
Calcul de la teneur en vitamine A:
UI de vitamine A/ml = E326 × 19,0
(
![]()
de la vitamine A acétate = 1 530 à 326 nm dans du propanol-2)
5.6.3.2.
Introduire à la pipette 2,0 ml de la solution mère de vitamine A palmitate (3.12.1) dans une fiole jaugée de 50 ml (4.2.2) et ajuster au trait de jauge avec du propanol-2 (3.8). La concentration nominale de cette solution est de 56 UI de vitamine A par ml. Introduire à la pipette 3,0 ml de cette solution de vitamine A de palmitate diluée dans une fiole jaugée de 25 ml et ajuster au trait de jauge avec du propanol-2 (3.8). La concentration nominale de cette solution est de 6,72 UI de vitamine A par ml. Mesurer le spectre UV de cette solution contre du propanol-2 (3.8) dans le spectrophotomètre (4.6) entre 300 nm et 400 nm. L'extinction maximale doit se situer entre 325 nm et 327 nm.
Calcul de la teneur en vitamine A:
UI de vitamine A/ml = E326 × 19,0
(
![]()
de la vitamine A palmitate = 957 à 326 nm dans du propanol-2)
5.6.3.3.
Introduire à la pipette 3,0 ml de la solution étalon de travail de vitamine A non diluée, préparée conformément au point 5.6.1, dans une fiole jaugée de 50 ml (4.2.2) et ajuster au trait de jauge avec du propanol-2 (3.8). Introduire à la pipette 5,0 ml de cette solution dans une fiole jaugée de 25 ml et ajuster au trait de jauge avec du propanol-2 (3.8). La concentration nominale de cette solution est de 6,72 UI de vitamine A par ml. Mesurer le spectre UV de cette solution contre du propanol-2 (3.8) dans le spectrophotomètre (4.6) entre 300 nm et 400 nm. L'extinction maximale doit se situer entre 325 nm et 327 nm.
Calcul de la teneur en vitamine A:
UI de vitamine A/ml = E325 × 18,3
(
![]()
de la vitamine A alcool = 1 821 à 325 nm dans du propanol-2)
6. Calcul des résultats
À partir de la hauteur moyenne (surface) des pics de vitamine A de la solution d'échantillon, déterminer la concentration de vitamine A dans cette solution, en UI/ml, par référence à la courbe d'étalonnage (5.6.2).
La teneur w en vitamine A de l'échantillon, exprimée en UI/kg, est donnée par la formule suivante:

[UI/kg]
où:
|
c |
= |
concentration de vitamine A dans la solution d'échantillon (5.4) en UI/ml, |
|
V1 |
= |
volume de la solution d'échantillon (5.4) en ml, |
|
V2 |
= |
volume de la portion aliquote prélevée sous 5.4 en ml, |
|
m |
= |
poids de la prise d'essai, en grammes. |
7. Observations
7.1. Pour les échantillons ayant une faible concentration de vitamine A, il peut être utile de rassembler les extraits dans l'éther de pétrole issus de deux saponifications (quantité pesée: 25 g) à une solution d'échantillon pour dosage par CLHP.
7.2. L'échantillon prélevé pour l'analyse ne doit pas contenir plus de 2 g de matières grasses.
7.3. S'il n'y a pas séparation des phases, ajouter environ 10 ml d'éthanol (3.1) pour briser l'émulsion.
7.4. Avec de l'huile de foie de morue et d'autres matières grasses pures, le temps de saponification doit être porté à 45-60 minutes.
7.5. Le BHT peut être remplacé par de l'hydroquinone.
7.6. En utilisant une colonne en phase directe, il est possible de séparer les isomères de rétinol. Dans ce cas, les hauteurs (surfaces) de tous les pics des isomères cis et trans doivent toutefois être additionnés en vue du calcul des résultats.
7.7. La solution d'ascorbate de sodium peut être remplacée par environ 150 mg d'acide ascorbique.
7.8. La solution de sulfure de sodium peut être remplacée par environ 50 mg de EDTA.
7.9. En cas d'analyse de la vitamine A dans des aliments d'allaitement, une attention particulière doit être accordée:
— à la saponification (5.2): en raison de la quantité de graisse présente dans l'échantillon, il peut être nécessaire d'augmenter la quantité de solution d'hydroxyde de potassium (3.4),
— à l'extraction (5.3): en raison de la présence d'émulsions, il peut être nécessaire d'adapter le ratio (2:1) eau/éthanol.
Il convient, pour vérifier si la méthode d'analyse appliquée donne des résultats fiables sur cette matrice spécifique (aliments d'allaitement), d'effectuer un test de récupération sur une prise d'essai supplémentaire. Si le taux de récupération est inférieur à 80 %, le résultat de l'analyse doit être corrigé au titre de la récupération.
8. Répétabilité
La différence entre les résultats de deux dosages parallèles effectués sur le même échantillon ne peut dépasser 15 % par rapport au résultat le plus élevé.
9. Résultats d'une étude collaborative ( 11 )
|
|
Prémélange |
Aliment prémélangé |
Concentré minéral |
Concentré protéique |
Aliment pour porcelets |
|
L |
13 |
12 |
13 |
12 |
13 |
|
n |
48 |
45 |
47 |
46 |
49 |
|
moyenne [UI/kg] |
17,02 × 106 |
1,21 × 106 |
537 100 |
151 800 |
18 070 |
|
sr [UI/kg] |
0,51 × 106 |
0,039 × 106 |
22 080 |
12 280 |
682 |
|
r [UI/kg] |
1,43 × 106 |
0,109 × 106 |
61 824 |
34 384 |
1 910 |
|
CVr [%] |
3,0 |
3,5 |
4,1 |
8,1 |
3,8 |
|
sR [UI/kg] |
1,36 × 106 |
0,069 × 106 |
46 300 |
23 060 |
3 614 |
|
R [UI/kg] |
3,81 × 106 |
0,193 × 106 |
129 640 |
64 568 |
10 119 |
|
CVR [%] |
8,0 |
6,2 |
8,6 |
15 |
20 |
|
L |
= |
nombre de laboratoires |
|
n |
= |
nombre de valeurs individuelles |
|
sr |
= |
écart type de répétabilité |
|
sR |
= |
écart type de reproductibilité |
|
r |
= |
répétabilité |
|
R |
= |
reproductibilité |
|
CVr |
= |
coefficient de variation de la répétabilité |
|
CVR |
= |
coefficient de variation de la reproductibilité |
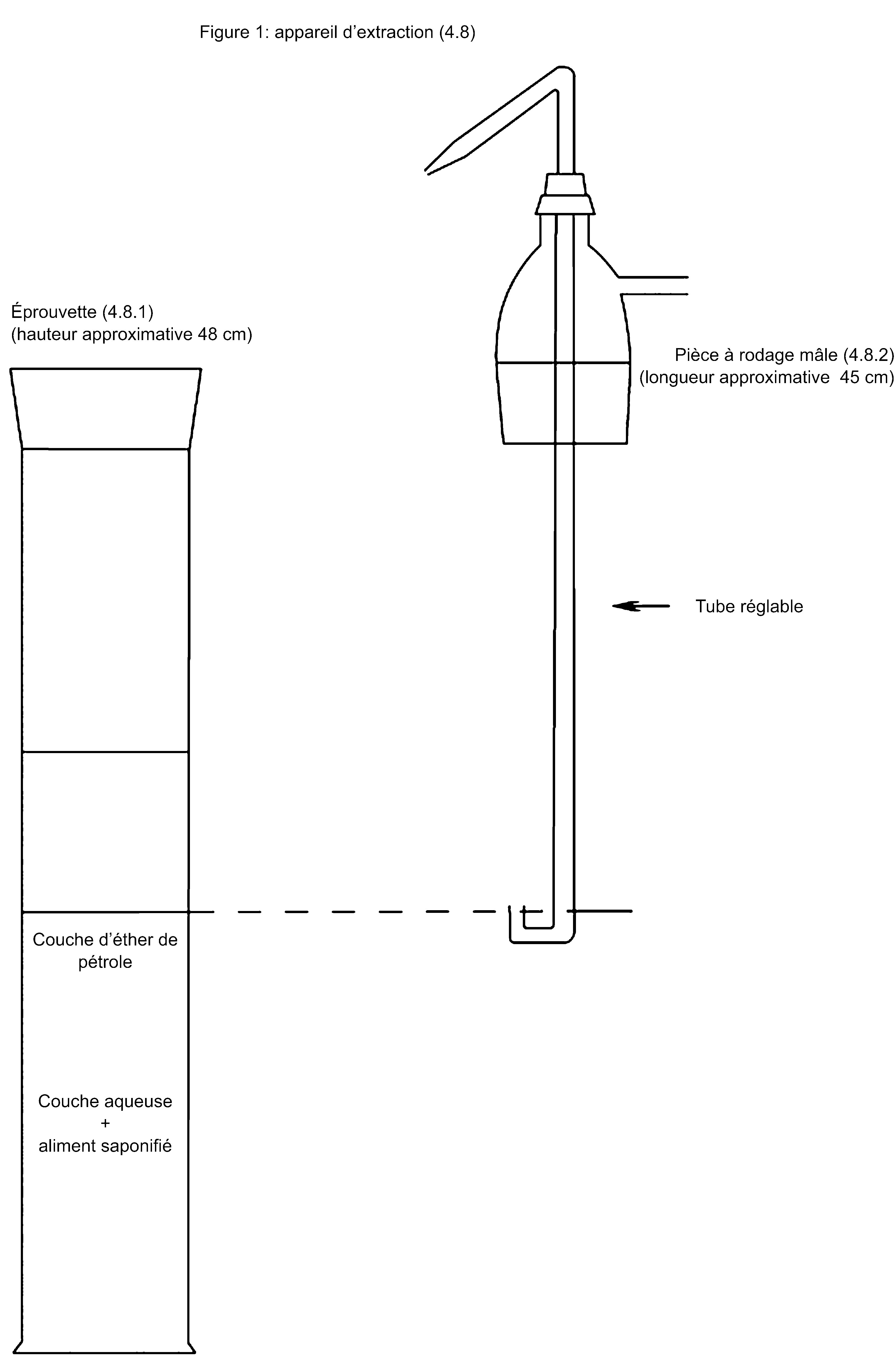
B. DOSAGE DE LA VITAMINE E
1. Objet et champ d'application
La méthode permet de déterminer la teneur en vitamine E des aliments pour animaux et des prémélanges. La teneur en vitamine E est exprimée en mg d'acétate de DL-α-tocophérol par kg. 1 mg d'acétate de DL-α-tocophérol correspond à 0,91 mg de DL-α-tocophérol (vitamine E).
La limite de quantification est de 2 mg de vitamine E/kg. Cette limite de quantification ne peut être atteinte qu'avec un détecteur fluorimétrique. La limite de quantification est fixée à 10 mg/kg en cas d'utilisation d'un détecteur UV.
2. Principe
L'échantillon est hydrolysé avec une solution d'hydroxyde de potassium éthanolique et la vitamine E est extraite dans de l'éther de pétrole. Le solvant est enlevé par évaporation; le résidu est dissous dans du méthanol et, si nécessaire, dilué à la concentration requise. La teneur en vitamine E est déterminée par chromatographie liquide haute performance en phase inverse (CLHP-PI) à l'aide d'un détecteur UV ou fluorimétrique.
3. Réactifs
3.1. Éthanol, σ = 96 %.
3.2. Éther de pétrole, intervalle d'ébullition 40 oC-60 oC.
3.3. Méthanol.
3.4. Solution d'hydroxyde de potassium, c = 50 g/100 ml.
3.5. Solution d'ascorbate de sodium, c = 10 g/100 ml (voir l'observation figurant au point 7.7).
3.6. Sulfure de sodium, Na2S · x H2O (x = 7-9).
3.6.1. Solution de sulfure de sodium, c = 0,5 mol/l dans du glycérol, β = 120 g/l (pour x = 9) (voir l'observation figurant au point 7.8).
3.7. Solution de phénolphtaléine, c = 2 g/100 ml dans de l'éthanol (3.1).
3.8. Phase mobile pour CLHP: mélange de méthanol (3.3) et d'eau, par exemple: 980 + 20 (v + v). Les proportions exactes sont déterminées par les caractéristiques de la colonne employée.
3.9. Azote, libre d'oxygène.
3.10. Acétate de DL-α-tocophérol, extra-pur, d'activité garantie.
3.10.1. Solution mère d'acétate de DL-α-tocophérol: peser, à 0,1 mg près, 100 mg d'acétate de DL-α-tocophérol (3.10) dans une fiole jaugée de 100 ml. Dissoudre dans de l'éthanol (3.1) et ajuster au trait de jauge avec le même solvant. 1 ml de cette solution contient 1 mg d'acétate de DL-α-tocophérol (pour le contrôle UV: voir point 5.6.1.3, pour la stabilisation: voir l'observation figurant au point 7.4).
3.11. DL-α-tocophérol, extra-pur, d'activité garantie.
3.11.1. Solution mère de DL-α-tocophérol: peser, à 0,1 mg près, 100 mg de DL-α-tocophérol (3.11) dans une fiole jaugée de 100 ml. Dissoudre dans de l'éthanol (3.1) et ajuster au trait de jauge avec le même solvant. 1 ml de cette solution contient 1 mg de DL-α-tocophérol (pour le contrôle UV: voir point 5.6.2.3, pour la stabilisation: voir l'observation figurant au point 7.4).
3.12. BHT (di-tert-butyl-2,6-méthyl-4-phénol) (voir l'observation figurant au point 7.5).
4. Appareillage
4.1. Évaporateur rotatif.
4.2. Verrerie ambrée.
4.2.1. Ballons à fond plat ou coniques, 500 ml, avec col en verre rodé.
4.2.2. Fioles jaugées à bouchon en verre rodé, col étroit, de 10, 25, 100 et 500 ml.
4.2.3. Ampoules à décanter coniques, 1 000 ml, à bouchon en verre rodé.
4.2.4. Ballons piriformes, 250 ml, avec col en verre rodé.
4.3. Réfrigérant d'Allihn, 300 mm de longueur utile, avec joint en verre rodé et adaptateur pour tuyau d'alimentation en gaz.
4.4. Papier filtre plissé pour la séparation des phases, d'un diamètre de 185 mm (par exemple, Schleicher & Schuell 597 HY 1/2).
4.5. Équipement CLHP avec système à injection.
4.5.1. Colonne de chromatographie liquide de 250 mm × 4 mm, C18, particules de 5 ou 10 μm, ou équivalent.
4.5.2. Détecteur UV ou fluorimétrique, de longueur d'onde variable.
4.6. Spectrophotomètre avec cellules de quartz de 10 mm.
4.7. Bain-marie avec agitateur magnétique.
4.8. Appareil d'extraction (voir la figure 1) se composant:
4.8.1. d'une éprouvette d'une capacité de 1 l, à col et bouchon en verre rodé;
4.8.2. d'une pièce à rodage mâle munie d'une tige latérale et d'un tube réglable passant en son centre. Ce tube doit avoir une partie inférieure en U et un bec gicleur à son extrémité opposée, de telle sorte que la couche liquide supérieure dans l'éprouvette puisse être transférée dans une ampoule à décanter.
5. Mode opératoire
|
NB |
: |
La vitamine E est sensible à la lumière (UV) et à l'oxydation. Toutes les opérations doivent être réalisées en l'absence de lumière (dans du verre ambré ou protégé d'une feuille d'aluminium) et en l'absence d'oxygène (éliminé avec de l'azote). Pendant l'extraction, l'air au-dessus du liquide doit être remplacé par de l'azote (éviter l'excès de pression en desserrant le bouchon de temps en temps). |
5.1. Préparation de l'échantillon
Moudre l'échantillon pour qu'il passe par un tamis à mailles de 1 mm, en évitant la production de chaleur. Le broyage doit avoir lieu immédiatement avant la pesée et la saponification, sinon il peut y avoir des pertes en vitamine E.
5.2. Saponification
Selon la teneur en poids de la vitamine E, peser, à 0,01 g près, de 2 à 25 g de l'échantillon dans un ballon à fond plat ou conique de 500 ml (4.2.1). Ajouter successivement, tout en remuant, 130 ml d'éthanol (3.1), environ 100 mg de BHT (3.13), 2 ml de solution d'ascorbate de sodium (3.5) et 2 ml de solution de sulfure de sodium (3.6). Adapter le réfrigérant (4.3) au ballon et immerger celui-ci dans un bain-marie avec agitateur magnétique (4.7). Chauffer jusqu'à ébullition et laisser refluer pendant 5 minutes. Ajouter alors 25 ml de solution d'hydroxyde de potassium (3.4) par le réfrigérant (4.3) et laisser refluer de nouveau pendant 25 min, tout en agitant sous un faible courant d'azote. Rincer alors le réfrigérant avec environ 20 ml d'eau et laisser refroidir le contenu du ballon à la température ambiante.
5.3. Extraction
Transférer quantitativement par décantation la solution de saponification en rinçant avec un volume total de 250 ml d'eau dans une ampoule à décanter de 1 000 ml (4.2.3) ou dans l'appareil d'extraction (4.8). Rincer successivement le ballon de saponification avec 25 ml d'éthanol (3.1) et 100 ml d'éther de pétrole (3.2) et transférer le liquide de rinçage dans l'ampoule à décanter ou dans l'appareil d'extraction. La proportion d'eau et d'éthanol dans les solutions combinées doit être d'environ 2:1. Secouer énergiquement pendant 2 minutes et laisser reposer pendant 2 minutes.
5.3.1.
Lorsque les couches sont séparées (voir l'observation figurant au point 7.3), transférer la couche d'éther de pétrole dans une autre ampoule à décanter (4.2.3). Répéter deux fois l'opération avec 100 ml d'éther de pétrole (3.2), puis deux fois avec 50 ml d'éther de pétrole (3.2).
Laver deux fois les extraits combinés dans l'ampoule à décanter en remuant doucement (afin d'éviter la formation d'émulsions) avec des portions de 100 ml d'eau et de nouveau en secouant avec d'autres portions de 100 ml d'eau jusqu'à ce que l'eau demeure incolore après addition de solution de phénolphtaléine (3.7) (quatre lavages sont généralement suffisants). Filtrer l'extrait lavé sur un filtre plissé sec pour la séparation des phases (4.4) afin d'éliminer l'eau résiduelle et transférer dans une fiole jaugée de 500 ml (4.2.2). Rincer l'ampoule à décanter et le filtre avec 50 ml d'éther de pétrole (3.2), ajuster au trait de jauge avec de l'éther de pétrole (3.2) et bien mélanger.
5.3.2.
Lorsque les couches sont séparées (voir l'observation figurant au point 7.3), remettre le bouchon de l'éprouvette (4.8.1) sur la pièce à rodage mâle (4.8.2) et placer l'extrémité inférieure en forme de U du tube réglable de telle sorte qu'elle se trouve juste au-dessus du niveau de l'interface. En exerçant une pression d'azote par la tige latérale, transférer la couche supérieure d'éther de pétrole dans une ampoule à décanter de 1 000 ml (4.2.3). Ajouter 100 ml d'éther de pétrole (3.2) dans le cylindre en verre, boucher et bien secouer. Laisser les couches se séparer et transférer la couche supérieure dans l'ampoule à décanter comme précédemment. Répéter la procédure d'extraction avec de nouveau 100 ml d'éther de pétrole (3.2), puis deux fois avec 50 ml d'éther de pétrole (3.2) et ajouter les couches d'éther de pétrole dans l'ampoule à décanter.
Laver les extraits combinés d'éther de pétrole selon la procédure décrite au point 5.3.1 et procéder selon ce point.
5.4. Préparation de la solution d'échantillon pour CLHP
Introduire à la pipette une portion aliquote de la solution d'éther de pétrole (issue de 5.3.1 ou de 5.3.2) dans un ballon piriforme de 250 ml (4.2.4). Laisser évaporer le solvant presque entièrement dans l'évaporateur rotatif (4.1) sous une pression réduite, à une température de bain ne dépassant pas 40 oC. Restaurer la pression atmosphérique en laissant entrer l'azote (3.9) et enlever le ballon de l'évaporateur. Supprimer le reste du solvant dans un courant d'azote (3.9) et dissoudre immédiatement le résidu dans un volume connu (10-100 ml) de méthanol (3.3) (la concentration de DL-α-tocophérol doit être de l'ordre de 5 μg/ml à 30 μg/ml).
5.5. Dosage par CLHP
La vitamine E est séparée sur une colonne C18 à phase inverse (4.5.1) et la concentration est mesurée à l'aide d'un détecteur fluorimétrique (excitation: 295 nm; émission: 330 nm) ou d'un détecteur UV (292 nm) (4.5.2).
Injecter une portion aliquote (par exemple, 20 μl) de la solution méthanolique obtenue sous 5.4 et éluer avec la phase mobile (3.8). Calculer les hauteurs moyennes des pics (surfaces) de plusieurs injections de la même solution d'échantillon et les hauteurs moyennes des pics (surfaces) de plusieurs injections des solutions d'étalonnage (5.6.2).
Les conditions suivantes sont proposées à titre indicatif; d'autres conditions peuvent être appliquées, pourvu qu'elles donnent des résultats équivalents.
|
Colonne de chromatographie liquide (point 4.5.1): |
250 mm × 4 mm, C18, particules de 5 ou de 10 μm, ou équivalent |
|
Phase mobile (3.8): |
mélange de méthanol (3.3) et d'eau, par exemple 980 + 20 (v+v) |
|
Débit: |
1-2 ml/min |
|
Détecteur (4.5.2): |
détecteur fluorimétrique (excitation: 295 nm/émission: 330 nm) ou détecteur UV (292 nm) |
5.6. Étalonnage (acétate de DL-α-tocophérol ou DL-α-tocophérol)
5.6.1.
5.6.1.1.
Introduire à la pipette 25 ml de la solution mère d'acétate de DL-α-tocophérol (3.10.1) dans un ballon à fond plat ou conique de 500 ml (4.2.1) et hydrolyser comme décrit au point 5.2. Procéder ensuite à l'extraction avec de l'éther de pétrole (3.2) conformément au point 5.3 et ajuster à 500 ml avec de l'éther de pétrole. Laisser évaporer 25 ml de cet extrait presque entièrement sur l'évaporateur rotatif (voir le point 5.4), enlever le solvant résiduel dans un courant d'azote (3.9) et redissoudre le résidu dans 25,0 ml de méthanol (3.3). La concentration nominale de cette solution est de 45,5 μg de DL-α-tocophérol par ml, équivalant à 50 μg d'acétate de DL-α-tocophérol par ml. La solution étalon de travail doit être préparée peu avant d'être utilisée.
5.6.1.2.
Transférer 1,0 , 2,0 , 4,0 et 10,0 ml de la solution étalon de travail dans une série de fioles jaugées de 20 ml, ajuster au trait de jauge avec du méthanol (3.3) et mélanger. Les concentrations nominales de ces solutions sont de 2,5 , 5,0 , 10,0 et 25,0 μg/ml d'acétate de DL-α-tocophérol, soit 2,28 , 4,55 , 9,10 et 22,8 μg/ml de DL-α-tocophérol.
Injecter plusieurs fois 20 μl de chaque solution d'étalonnage et déterminer les hauteurs moyennes des pics (surfaces). D'après les hauteurs moyennes des pics (surfaces), tracer une courbe d'étalonnage.
5.6.1.3.
Diluer 5,0 ml de la solution mère d'acétate de DL-α-tocophérol (3.10.1), ajuster à 25,0 ml dans de l'éthanol et mesurer le spectre UV de cette solution contre de l'éthanol (3.1) dans le spectrophotomètre (4.6) entre 250 nm et 320 nm.
L'absorption maximale doit être de 284 nm:
![]()
= 43,6 à 284 nm dans de l'éthanol.
À cette dilution, une valeur d'extinction de 0,84 à 0,88 doit être obtenue.
5.6.2.
5.6.2.1.
Introduire à la pipette 2 ml de la solution étalon de DL-α-tocophérol (3.11.1) dans une fiole jaugée de 50 ml, dissoudre dans du méthanol (3.3) et ajuster au trait de jauge avec du méthanol. La concentration nominale de cette solution est de 40 μg de DL-α-tocophérol par ml, équivalant à 44,0 μg d'acétate de DL-α-tocophérol par ml. La solution étalon de travail doit être préparée peu avant d'être utilisée.
5.6.2.2.
Transférer 1,0 , 2,0 , 4,0 et 10,0 ml de la solution étalon de travail dans une série de fioles jaugées de 20 ml, ajuster au trait de jauge avec du méthanol (3.3) et mélanger. Les concentrations nominales de ces solutions sont de 2,0 , 4,0 , 8,0 et 20,0 μg/ml de DL-α-tocophérol, soit 2,20 , 4,40 , 8,79 et 22,0 μg/ml d'acétate de DL-α-tocophérol.
Injecter plusieurs fois 20 μl de chaque solution d'étalonnage et déterminer les hauteurs moyennes des pics (surfaces). D'après les hauteurs moyennes des pics (surfaces), tracer une courbe d'étalonnage.
5.6.2.3.
Diluer 2,0 ml de la solution mère de DL-α-tocophérol (3.11.1), ajuster à 25,0 ml dans de l'éthanol et mesurer le spectre UV de cette solution contre de l'éthanol (3.1) dans le spectrophotomètre (4.6) entre 250 nm et 320 nm. L'absorption maximale doit être de 292 nm:
![]()
= 75,8 à 292 nm dans de l'éthanol.
À cette dilution, une valeur d'extinction de 0,6 doit être obtenue.
6. Calcul des résultats
À partir de la hauteur moyenne (surface) des pics de vitamine E de la solution d'échantillon, déterminer la concentration de vitamine E dans cette solution, en μg/ml, (exprimée en acétate d'alphatocophérol) par référence à la courbe d'étalonnage (5.6.1.2 ou 5.6.2.2).
La teneur w en vitamine E de l'échantillon, exprimée en mg/kg, est donnée par la formule suivante:

[mg/kg]
où:
|
c |
= |
concentration de vitamine E (calculée en acétate d'alphatocophérol) dans la solution d'échantillon (5.4) en μg/ml, |
|
V1 |
= |
volume de la solution d'échantillon (5.4) en ml, |
|
V2 |
= |
volume de la portion aliquote prélevée sous 5.4 en ml, |
|
m |
= |
poids de la prise d'essai, en grammes. |
7. Observations
7.1. Pour les échantillons ayant une faible concentration de vitamine E, il peut être utile de rassembler les extraits dans l'éther de pétrole issus de deux saponifications (quantité pesée: 25 g) à une solution d'échantillon pour dosage par CLHP.
7.2. L'échantillon prélevé pour l'analyse ne doit pas contenir plus de 2 g de matières grasses.
7.3. S'il n'y a pas séparation des phases, ajouter environ 10 ml d'éthanol (3.1) pour briser l'émulsion.
7.4. Une fois la mesure spectrophotométrique de la solution d'acétate de DL-α-tocophérol ou de DL-α-tocophérol effectuée, respectivement selon le point 5.6.1.3 ou 5.6.2.3, ajouter environ 10 mg de BHT (3.12) à la solution (3.10.1 ou 3.10.2) et conserver cette solution au réfrigérateur (durée maximale de conservation: quatre semaines).
7.5. Le BHT peut être remplacé par de l'hydroquinone.
7.6. Il est possible d'utiliser une colonne en phase directe pour séparer les tocophérols α, β, γ et δ.
7.7. La solution d'ascorbate de sodium peut être remplacée par environ 150 mg d'acide ascorbique.
7.8. La solution de sulfure de sodium peut être remplacée par environ 50 mg d'EDTA.
7.9. La vitamine E acétate s'hydrolyse très rapidement en milieu alcalin et est dès lors très sensible à l'oxydation, particulièrement en présence d'oligoéléments tels que le fer et le cuivre. Le dosage de la vitamine E dans des prémélanges présentant des teneurs supérieures à 5 000 mg/kg pourrait entraîner une dégradation de la vitamine E. C'est pourquoi il est recommandé d'utiliser, aux fins de la confirmation, une méthode de dosage CLHP comprenant une digestion enzymatique de la vitamine E sans saponification alcaline.
8. Répétabilité
La différence entre les résultats de deux dosages parallèles effectués sur le même échantillon ne peut dépasser 15 % par rapport au résultat le plus élevé.
9. Résultats d'une étude collaborative ( 12 )
|
|
Prémélange |
Aliment prémélangé |
Concentré minéral |
Concentré protéique |
Aliment pour porcelets |
|
L |
12 |
12 |
12 |
12 |
12 |
|
n |
48 |
48 |
48 |
48 |
48 |
|
Moyenne [mg/kg] |
17 380 |
1 187 |
926 |
315 |
61,3 |
|
sr [mg/kg] |
384 |
45,3 |
25,2 |
13,0 |
2,3 |
|
r [mg/kg] |
1 075 |
126,8 |
70,6 |
36,4 |
6,4 |
|
CVr [%] |
2,2 |
3,8 |
2,7 |
4,1 |
3,8 |
|
sR mg/kg] |
830 |
65,0 |
55,5 |
18,9 |
7,8 |
|
R [mg/kg] |
2 324 |
182,0 |
155,4 |
52,9 |
21,8 |
|
CVR [%] |
4,8 |
5,5 |
6,0 |
6,0 |
12,7 |
|
L |
= |
nombre de laboratoires |
|
n |
= |
nombre de valeurs individuelles |
|
sr |
= |
écart type de répétabilité |
|
sR |
= |
écart type de reproductibilité |
|
r |
= |
répétabilité |
|
R |
= |
reproductibilité |
|
CVr |
= |
coefficient de variation de la répétabilité |
|
CVR |
= |
coefficient de variation de la reproductibilité |
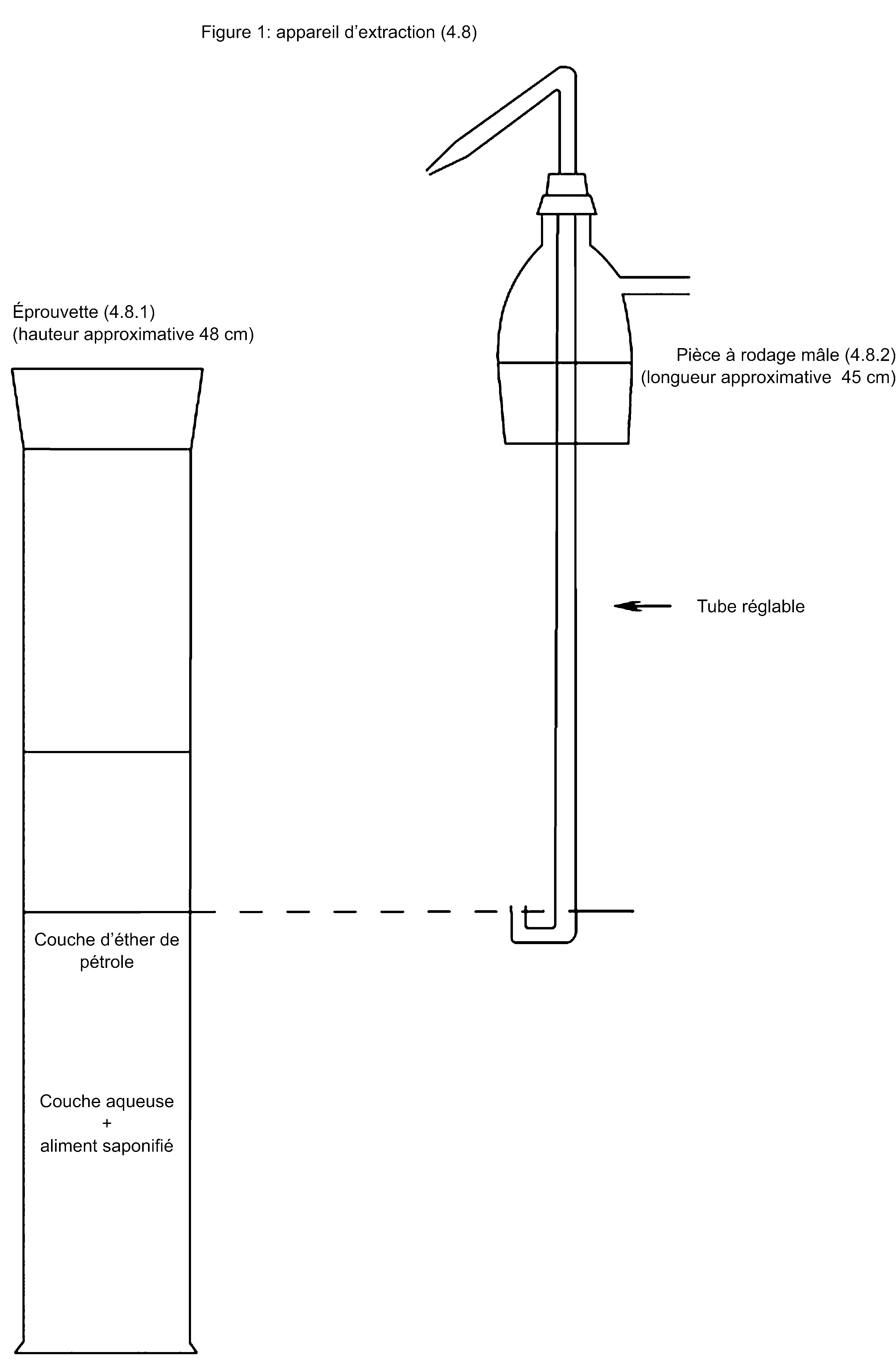
C. DOSAGE DES OLIGOÉLÉMENTS FER, CUIVRE, MANGANÈSE ET ZINC
1. Objet et champ d'application
La méthode permet de doser les oligoéléments fer, cuivre, manganèse et zinc dans les aliments pour animaux. Les limites de quantification sont les suivantes:
— fer (Fe): 20 mg/kg,
— cuivre (Cu): 10 mg/kg,
— manganèse (Mn): 20 mg/kg,
— zinc (Zn): 20 mg/kg.
2. Principe
L'échantillon est mis en solution dans l'acide chlorhydrique après destruction éventuelle des matières organiques. Les éléments fer, cuivre, manganèse et zinc sont dosés, après dilution appropriée, par spectrométrie d'absorption atomique.
3. Réactifs
Remarques préliminaires
L'eau utilisée pour la préparation des réactifs et des solutions requises au cours de l'analyse doit être exempte des cations à déterminer. Elle est obtenue soit par double distillation de l'eau dans un appareil en borosilicate ou en quartz, soit par double permutation sur résine échangeuse d'ions.
Les réactifs doivent être au moins de qualité «pour analyse». L'absence de l'élément à déterminer doit être contrôlée par un essai à blanc. Si nécessaire, les réactifs doivent être soumis à une purification plus poussée.
Les solutions étalons décrites ci-après peuvent être remplacées par des solutions étalons commerciales, à condition que celles-ci soient garanties et contrôlées avant l'emploi.
3.1. Acide chlorhydrique (d:1,19 g/ml).
3.2. Acide chlorhydrique (6 mol/l).
3.3. Acide chlorhydrique (0,5 mol/l).
3.4. Acide fluorhydrique à 38-40 % (v/v), ayant une teneur en fer (Fe) inférieure à 1 mg/l et dont le résidu d'évaporation est inférieur à 10 mg (exprimés en sulfates)/l.
3.5. Acide sulfurique (d: 1,84 g/ml).
3.6. Peroxyde d'hydrogène [à environ 100 volumes d'oxygène (30 % en poids)].
3.7. Solution étalon de fer (1 000 μg Fe/ml) préparée comme suit, ou solution commerciale équivalente: dissoudre 1 g de fer en fil dans 200 ml d'acide chlorhydrique 6 mol/l (3.2), ajouter 16 ml de peroxyde d'hydrogène (3.6.) et ajuster à 1 l avec de l'eau.
3.7.1. Solution étalon de travail de fer (100 μg Fe/ml): diluer une part de la solution étalon (3.7) dans neuf parts d'eau.
3.8. Solution étalon de cuivre (1 000 μg Cu/ml) préparée comme suit, ou solution commerciale équivalente:
— dissoudre 1 g de cuivre en poudre dans 25 ml d'acide chlorhydrique 6 mol/l (3.2), ajouter 5 ml de peroxyde d'hydrogène (3.6) et ajuster à 1 l avec de l'eau.
3.8.1. Solution étalon de travail de cuivre (10 μg Cu/ml): diluer une part de la solution étalon (3.8) dans neuf parts d'eau; diluer ensuite une part de la solution obtenue dans neuf parts d'eau.
3.9. Solution étalon de manganèse (1 000 μg Mn/ml) préparée comme suit, ou solution commerciale équivalente:
— dissoudre 1 g de manganèse en poudre dans 25 ml d'acide chlorhydrique 6 mol/l (3.2) et ajuster à 1 l avec de l'eau.
3.9.1. Solution étalon de travail de manganèse (10 μg Mn/ml): diluer une part de la solution étalon (3.9) dans neuf parts d'eau; diluer ensuite une part de la solution obtenue dans neuf parts d'eau.
3.10. Solution étalon de zinc (1 000 μg Zn/ml) préparée comme suit, ou solution commerciale équivalente:
— dissoudre 1 g de zinc en ruban ou en plaque dans 25 ml d'acide chlorhydrique 6 mol/l (3.2) et ajuster à 1 l avec de l'eau.
3.10.1. Solution étalon de travail de zinc (10 μg Zn/ml): diluer une part de la solution étalon (3.10) dans neuf parts d'eau; diluer ensuite une part de la solution obtenue dans neuf parts d'eau.
3.11. Solution de chlorure de lanthane: dissoudre 12 g d'oxyde de lanthane dans 150 ml d'eau, ajouter 100 ml d'acide chlorhydrique 6 mol/l (3.2) et ajuster à 1 l avec de l'eau.
4. Appareillage
4.1. Four à moufle, à température réglable et, de préférence, contrôlée.
4.2. Verrerie en borosilicate, résistante. Il est recommandé d'utiliser un matériel servant exclusivement aux dosages des oligoéléments.
4.3. Spectrophotomètre d'absorption atomique, répondant aux exigences de la méthode en ce qui concerne la sensibilité et la précision dans la gamme des mesures utiles.
5. Mode opératoire ( 13 )
5.1. Échantillons contenant des composés organiques
5.1.1. ( 14 )
5.1.1.1. Placer 5 à 10 g de l'échantillon, pesés à 0,2 mg près, dans un creuset en quartz ou en platine [voir note b)], sécher à l'étuve à 105 oC et introduire le creuset dans le four à moufle (4.1) froid. Fermer le four [voir note c)] et élever progressivement sa température pour atteindre 450 à 475 oC en 90 minutes environ. Maintenir cette température durant 4 à 16 h (par exemple, toute une nuit) de façon à éliminer la matière charbonneuse, ouvrir ensuite le four et laisser refroidir [voir note d)].
Humidifier les cendres avec de l'eau et les transférer dans un bécher de 250 ml. Rincer le creuset à l'aide de 5 ml d'acide chlorhydrique (3.1) et transvaser lentement et avec précaution la solution de rinçage dans le bécher (une réaction violente peut se produire par formation de CO2). Ajouter ensuite goutte à goutte de l'acide chlorhydrique (3.1), tout en remuant le contenu du bécher, jusqu'à cessation de l'effervescence. Évaporer à sec en remuant périodiquement à l'aide d'une tige de verre.
Ajouter au résidu 15 ml d'acide chlorhydrique 6 mol/l (3.2) et ensuite environ 120 ml d'eau. Agiter avec la tige de verre, qu'il importe de laisser dans le bécher, et couvrir ce dernier avec un verre de montre. Porter le liquide à ébullition douce et maintenir l'ébullition jusqu'à ce que les cendres ne se dissolvent apparemment plus. Filtrer sur un papier filtre sans cendres et recueillir le filtrat dans une fiole jaugée de 250 ml. Laver le bécher et le filtre avec 5 ml d'acide chlorhydrique 6 mol/l (3.2) chaud et à deux reprises avec de l'eau bouillante. Ajuster au trait de jauge avec de l'eau (la concentration de HCl est d'environ 0,5 mol/l).
5.1.1.2. Si le résidu se trouvant dans le filtre apparaît noir (charbonneux), le replacer dans le four et incinérer de nouveau à 450-475 oC. Cette incinération, qui requiert seulement quelques heures (3 à 5 heures environ), est complétée lorsque les cendres apparaissent blanches ou presque blanches. Dissoudre le résidu dans environ 2 ml d'acide chlorhydrique (3.1), évaporer à sec et ajouter 5 ml d'acide chlorhydrique 6 mol/l (3.2). Chauffer, filtrer la solution dans la fiole jaugée et ajuster au trait de jauge avec de l'eau (la concentration de HCl est d'environ 0,5 mol/l).
Notes:
a) il est important, lors du dosage des oligoéléments, d'être attentif aux risques de contamination, notamment par le zinc, le cuivre et le fer. Les instruments utilisés pour la préparation des échantillons doivent par conséquent être exempts de ces métaux.
Pour réduire les risques de contamination, il convient de travailler en atmosphère exempte de poussières avec un matériel rigoureusement propre et une verrerie soigneusement lavée. Le dosage du zinc est particulièrement sensible aux nombreux types de contaminations dues, par exemple, à la verrerie, aux réactifs et à la poussière;
b) calculer le poids de l'échantillon à incinérer en fonction de la teneur approximative de l'aliment en oligo-élément à doser et de la sensibilité du spectrophotomètre utilisé. Pour certains aliments pauvres en oligo-éléments, il peut être nécessaire de prélever un échantillon de 10 à 20 g et de limiter le volume de la solution finale à 100 ml;
c) incinérer dans un four fermé sans injection d'air ou d'oxygène;
d) la température indiquée par le pyromètre ne peut dépasser 475 oC.
5.1.2.
5.1.2.1.
Préparer, pour chaque oligoélément à doser, une gamme de solutions d'étalonnage à partir des solutions étalons de travail 3.7.1, 3.8.1, 3.9.1 et 3.10.1, de façon à ce que chaque solution d'étalonnage ait une concentration de HCl d'environ 0,5 mol/l et, dans le cas du fer, du manganèse et du zinc, une concentration de chlorure de lanthane correspondant à 0,1 % de lanthane (p/v).
Les concentrations choisies d'oligoéléments doivent se trouver dans la zone de sensibilité du spectrophotomètre utilisé. Les tableaux ci-après donnent, à titre d'exemple, des types de composition de solution d'étalonnage; selon le type et la sensibilité du spectrophotomètre utilisé, il peut être nécessaire de choisir d'autres concentrations.
|
μg Fe/ml |
0 |
0,5 |
1 |
2 |
3 |
4 |
5 |
|
ml de solution étalon de travail (3.7.1) (1 ml = 100 μg Fe) |
0 |
0,5 |
1 |
2 |
3 |
4 |
5 |
|
ml HCl (3.2) |
7 |
7 |
7 |
7 |
7 |
7 |
7 |
|
+ 10 ml de solution de chlorure de lanthane (3.11); ajuster à 100 ml avec de l'eau. |
|||||||
|
μg Cu/ml |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
ml de solution étalon de travail (3.8.1) (1 ml = 10 μg Cu) |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
|
ml HCl (3.2) |
8 |
8 |
8 |
8 |
8 |
8 |
8 |
|
μg Mn/ml |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
ml de solution étalon de travail (3.9.1) (1 ml = 10 μg Mn) |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
|
ml HCl (3.2) |
7 |
7 |
7 |
7 |
7 |
7 |
7 |
|
+ 10 ml de solution de chlorure de lanthane (3.11); ajuster à 100 ml avec de l'eau. |
|||||||
|
μg Zn/ml |
0 |
0,05 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
|
ml de solution étalon de travail (3.10.1) (1 ml = 10 μg Zn) |
0 |
0,5 |
1 |
2 |
4 |
6 |
8 |
|
ml HCl (3.2) |
7 |
7 |
7 |
7 |
7 |
7 |
7 |
|
+ 10 ml de solution de chlorure de lanthane (3.11); ajuster à 100 ml avec de l'eau. |
|||||||
5.1.2.2.
Pour le dosage du cuivre, la solution préparée conformément au point 5.1.1 peut, en règle générale, être utilisée directement. S'il est nécessaire d'amener sa concentration dans la gamme des concentrations des solutions d'étalonnage, une partie aliquote peut être introduite à la pipette dans une fiole jaugée de 100 ml et ajustée au trait de jauge avec de l'acide chlorhydrique 0,5 mol/l (3.3).
Pour le dosage du fer, du manganèse et du zinc, introduire à la pipette une portion aliquote de la solution préparée conformément au point 5.1.1 dans une fiole jaugée de 100 ml; ajouter 10 ml de solution de chlorure de lanthane (3.11) et ajuster au trait de jauge avec de l'acide chlorhydrique 0,5 mol/l (3.3) (voir aussi point 8 «Observation»).
5.1.2.3.
Effectuer un essai à blanc comportant toutes les étapes prescrites du mode opératoire, en excluant la présence de l'échantillon. La solution d'étalonnage «0» ne peut être utilisée comme «blanc».
5.1.2.4.
Mesurer l'absorption atomique des solutions d'étalonnage et de la solution à analyser, en utilisant une flamme oxydante air-acétylène, aux longueurs d'onde ci-après:
Fe: 248,3 nm;
Cu: 324,8 nm;
Mn: 279,5 nm;
Zn: 213,8 nm.
Effectuer chaque mesure quatre fois.
5.2. Aliments minéraux
En l'absence de matière organique, l'incinération préalable est inutile. Appliquer le mode opératoire à partir du deuxième alinéa du point 5.1.1.1. L'évaporation en présence d'acide fluorhydrique peut être omise.
6. Calcul des résultats
Calculer la concentration d'oligoéléments dans la solution à analyser à l'aide d'une courbe d'étalonnage et exprimer le résultat en mg d'oligoélément par kg d'échantillon (ppm).
7. Répétabilité
La différence entre les résultats de deux dosages parallèles effectués sur un même échantillon par le même analyste ne peut dépasser:
— 5 mg/kg, en valeur absolue, pour les teneurs en oligoélément concerné allant jusqu'à 50 mg/kg,
— 10 % du résultat le plus élevé pour les teneurs en oligoélément comprises entre 50 et 100 mg/kg,
— 10 mg/kg, en valeur absolue, pour les teneurs en oligoélément concerné comprises entre 100 et 200 mg/kg,
— 5 % du résultat le plus élevé pour les teneurs en oligoélément concerné supérieures à 200 mg/kg.
8. Observation
La présence de grandes quantités de phosphates peut interférer dans le dosage du fer, du manganèse et du zinc. Cette interférence doit être corrigée par addition de solution de chlorure de lanthane (3.11). Néanmoins, si l'échantillon a un rapport pondéral Ca + Mg/P > 2, l'addition de solution de chlorure de lanthane (3.11) à la solution à analyser et aux solutions d'étalonnage peut être omise.
D. DOSAGE DE L'HALOFUGINONE
DL-trans-7-bromo-6-chloro-3-[3-(3-hydroxy-2-piperidyl) acétonyl]-quinazoline-4-(3H)-one bromhydrate
1. Objet et champ d'application
La méthode permet de déterminer la teneur en halofuginone des aliments pour animaux. La limite de quantification est de 1 mg/kg.
2. Principe
Après traitement à l'eau chaude, l'halofuginone est extrait sous forme de base libre dans de l'acétate d'éthyle et ensuite séparé sous forme de chlorhydrate dans une solution acide aqueuse. L'extrait est purifié par chromatographie sur résines échangeuses d'ions. La teneur en halofuginone est déterminée par chromatographie liquide à haute performance (CLHP) en phase inverse à l'aide d'un détecteur d'UV.
3. Réactifs
3.1. Acétonitrile, de qualité CLHP.
3.2. Résine d'Amberlite XAD-2.
3.3. Acétate d'ammonium.
3.4. Acétate d'éthyle.
3.5. Acide acétique, glacial.
3.6. Halofuginone étalon (DL-trans-7-bromo-6-chloro-3-[3-(3-hydroxy-2-piperidyl) acétonyl]-quinazoline-4-(3H)-one bromhydrate, E 764).
3.6.1.
Peser, à 0,1 mg près, 50 mg d'halofuginone (3.6) dans une fiole jaugée de 500 ml, dissoudre dans une solution tampon d'acétate d'ammonium (3.18), ajuster au trait de jauge avec la solution tampon et mélanger. Cette solution est stable pendant trois semaines à 5 oC si elle est conservée à l'abri de la lumière.
3.6.2.
Dans une série de fioles jaugées de 100 ml, transvaser 1,0 , 2,0 , 3,0 , 4,0 et 6,0 ml de la solution mère de l'étalon (3.6.1). Ajuster au trait de jauge avec la phase mobile (3.21) et mélanger. Ces solutions ont des concentrations respectives de 1,0 , 2,0 , 3,0 , 4,0 et 6,0 μg/ml d'halofuginone. Les solutions doivent être préparées peu avant d'être utilisées.
3.7. Acide chlorhydrique (ρ20 environ 1,16 g/ml).
3.8. Méthanol.
3.9. Nitrate d'argent.
3.10. Ascorbate de sodium.
3.11. Carbonate de sodium.
3.12. Chlorure de sodium.
3.13. EDTA (acide éthylènediamine tétracétique, sel disodique).
3.14. Eau, de qualité CLHP.
3.15. Solution de carbonate de sodium, c = 10 g/100 ml.
3.16. Solution de carbonate de sodium saturée au chlorure de sodium, c = 5 g/100 ml.
Dissoudre 50 g de carbonate de sodium (3.11) dans de l'eau, ajuster à 1 l et ajouter du chlorure de sodium (3.12) jusqu'à saturation de la solution.
3.17. Acide chlorhydrique, environ 0,1 mol/l.
Diluer avec de l'eau 10 ml d'acide chlorhydrique (3.7) jusqu'à 1 l.
3.18. Solution tampon d'acétate d'ammonium, environ 0,25 mol/l.
Dissoudre 19,3 g d'acétate d'ammonium (3.3) et 30 ml d'acide acétique (3.5) dans de l'eau (3.14) et ajuster à 1 l.
3.19. Préparation de la résine d'Amberlite XAD-2.
Laver une quantité appropriée d'Amberlite (3.2) avec de l'eau jusqu'à l'élimination de tous les ions chlorure, indiquée par un essai au nitrate d'argent (3.20) réalisé sur la phase aqueuse éliminée. Laver ensuite la résine avec 50 ml de méthanol (3.8), écarter le méthanol et conserver la résine sous méthanol frais.
3.20. Solution de nitrate d'argent, environ 0,1 mol/l.
Dissoudre 0,17 g de nitrate d'argent (3.9) dans 10 ml d'eau.
3.21. Phase mobile de la CLHP
Mélanger 500 ml d'acétonitrile (3.1) avec 300 ml de solution tampon d'acétate d'ammonium (3.18) et 1 200 ml d'eau (3.14). Ajuster au moyen d'acide acétique (3.5) à pH 4,3 . Filtrer à travers un filtre de 0,22 μm (4.8) et dégazer la solution (par exemple, par traitement aux ultrasons pendant 10 minutes). Cette solution est stable pendant un mois si elle est conservée à l'abri de la lumière dans un récipient fermé.
4. Appareillage
4.1. Bain ultrasonique.
4.2. Évaporateur rotatif.
4.3. Centrifugeuse.
4.4. Équipement pour CLHP avec détecteur d'ultraviolets à longueur d'onde variable ou détecteur à barrettes de diodes.
4.4.1. Colonne de chromatographie liquide, 300 mm × 4 mm, C18, particules de 10 μm, ou équivalent.
4.5. Colonne de verre (300 mm × 10 mm), munie d'un filtre de verre fritté et d'un robinet.
4.6. Filtres en fibre de verre, de 150 mm de diamètre.
4.7. Filtres à membrane, 0,45 μm.
4.8. Filtres à membrane, 0,22 μm.
5. Mode opératoire
|
NB |
: |
L'halofuginone, en tant que base libre, est instable dans des solutions alcalines et d'acétate d'éthyle. Il ne doit pas rester dans l'acétate d'éthyle pendant plus de 30 minutes. |
5.1. Généralités
5.1.1. Un aliment blanc doit être analysé afin de vérifier l'absence d'halofuginone ou de substances interférentes.
5.1.2. Il convient d'effectuer un test de récupération en analysant l'aliment blanc après l'avoir supplémenté par ajout d'une quantité d'halofuginone similaire à celle présente dans l'échantillon. Pour parvenir à une teneur de 3 mg/kg, ajouter 300 μl de la solution mère de l'étalon (3.6.1) à 10 g d'aliment blanc, mélanger et attendre 10 minutes avant de procéder à l'extraction (5.2).
|
NB |
: |
Dans le cadre de cette méthode, l'aliment blanc doit être d'un type similaire dans sa composition à celui de l'échantillon et, lors de son analyse, il ne doit pas être détecté d'halofuginone. |
5.2. Extraction
Peser, à 0,1 g près, 10 g de l'échantillon préparé dans un tube à centrifuger de 200 ml, ajouter 0,5 g d'ascorbate de sodium (3.10), 0,5 g d'EDTA (3.13) et 20 ml d'eau, puis mélanger. Placer le tube pendant 5 minutes au bain-marie (80 oC). Après refroidissement à température ambiante, ajouter 20 ml de solution de carbonate de sodium (3.15) et mélanger. Ajouter immédiatement 100 ml d'acétate d'éthyle (3.4) et agiter vigoureusement à la main pendant 15 secondes. Placer ensuite le tube pendant 3 minutes dans le bain ultrasonique (4.1) en dévissant le bouchon. Centrifuger pendant 2 minutes et décanter la phase d'acétate d'éthyle à travers un filtre en fibre de verre (4.6) dans une ampoule à décanter de 500 ml. Répéter l'extraction de l'échantillon avec une seconde portion de 100 ml d'acétate d'éthyle. Laver les extraits combinés pendant 1 minute avec 50 ml de la solution de carbonate de sodium saturée au chlorure de sodium (3.16) et éliminer la phase aqueuse.
Extraire la couche organique pendant 1 minute avec 50 ml d'acide chlorhydrique (3.17). Faire passer la couche acide inférieure dans une ampoule à décanter de 250 ml. Réextraire la couche organique pendant 1,5 minute avec 50 ml d'acide chlorhydrique supplémentaires et combiner avec le premier extrait. Laver les extraits d'acide combinés par agitation pendant environ 10 secondes avec 10 ml d'acétate d'éthyle (3.4).
Transvaser quantitativement la couche aqueuse dans un ballon à fond rond de 250 ml et éliminer la phase organique. À l'aide d'un évaporateur rotatif (4.2), évaporer totalement le reste d'acétate d'éthyle de la solution acide. La température du bain-marie ne doit pas dépasser 40 oC. Sous un vide d'environ 25 mbar, la totalité de l'acétate d'éthyle résiduel sera éliminée en 5 minutes à 38 oC.
5.3. Purification
5.3.1.
Pour chaque extrait d'échantillon, préparer une colonne XAD-2. À l'aide de méthanol (3.8), transvaser 10 g d'Amberlite préparée (3.19) dans une colonne de verre (4.5). Ajouter un petit tampon de laine de verre au sommet du lit de la résine. Laisser le méthanol s'écouler de la colonne et laver la résine avec 100 ml d'eau, en arrêtant le débit lorsque le liquide atteint le sommet du lit de la résine. Laisser la colonne se stabiliser pendant 10 minutes avant de l'utiliser. Ne jamais laisser la colonne aller à sec.
5.3.2.
Transvaser l'extrait (5.2) quantitativement au sommet de la colonne d'Amberlite préparée (5.3.1) et éluer, en écartant l'éluat. La vitesse d'élution ne doit pas dépasser 20 ml/min. Rincer le ballon à fond rond avec 20 ml d'acide chlorhydrique (3.17) et utiliser cette solution pour laver la colonne de résine. Faire passer un courant d'air pour éliminer la solution acide restante. Éliminer les liquides du lavage. Ajouter 100 ml de méthanol (3.8) à la colonne et laisser éluer 5 à 10 ml en recueillant l'éluat dans un ballon à fond rond de 250 ml. Laisser le reste de méthanol se stabiliser pendant 10 minutes avec la résine et poursuivre l'élution à une vitesse ne dépassant pas 20 ml/min, en recueillant l'éluat dans le même ballon à fond rond. À l'aide d'un évaporateur rotatif (4.2), évaporer le méthanol, en veillant à ce que la température du bain-marie ne dépasse pas 40 oC. Transvaser le résidu quantitativement dans une fiole jaugée de 10 ml à l'aide de la phase mobile (3.21). Ajuster au trait de jauge avec la phase mobile et mélanger. Une partie aliquote est filtrée à travers un filtre à membrane (4.7). Réserver cette solution pour l'analyse par CLHP (5.4).
5.4. Dosage par CLHP
5.4.1.
Les conditions ci-après sont proposées à titre indicatif; d'autres conditions peuvent être appliquées si elles donnent des résultats équivalents.
Colonne de chromatographie liquide (4.4.1).
Phase mobile de la CLHP (3.21).
Débit: 1,5 à 2 ml/min.
Longueur d'onde de détection: 243 nm.
Volume d'injection: 40 à 100 μl.
Vérifier la stabilité du système chromatographique, en injectant plusieurs fois la solution d'étalonnage (3.6.2) à 3,0 μg/ml, jusqu'à l'obtention de hauteurs (surfaces) de pics et de temps de rétention constants.
5.4.2.
Injecter plusieurs fois chaque solution d'étalonnage (3.6.2) et mesurer les hauteurs (surfaces) des pics pour chaque concentration. Tracer la courbe d'étalonnage en portant les moyennes des hauteurs ou surfaces de pics des solutions d'étalonnage en ordonnée et les concentrations correspondantes en μg/ml en abscisse.
5.4.3.
Injecter plusieurs fois l'extrait d'échantillon (5.3.2), en utilisant le même volume que pour les solutions d'étalonnage, et déterminer la hauteur (surface) de pic moyenne de l'halofuginone.
6. Calcul des résultats
À partir de la hauteur (surface) moyenne des pics d'halofuginone de la solution d'échantillon, déterminer la concentration d'halofuginone dans cette solution, en μg/ml, par référence à la courbe d'étalonnage (5.4.2).
La teneur en halofuginone w (mg/kg) de l'échantillon est donnée par la formule suivante:

où:
|
c |
= |
concentration d'halofuginone dans la solution d'échantillon, en μg/ml, |
|
m |
= |
poids de la prise d'essai, en grammes. |
7. Validation des résultats
7.1. Identité
L'identité de l'analyte peut être confirmée par cochromatographie ou par l'utilisation d'un détecteur à barrettes de diodes permettant de comparer les spectres de l'extrait d'échantillon et de la solution d'étalonnage (3.6.2) à 6,0 μg/ml d'halofuginone.
7.1.1.
Un extrait d'échantillon est supplémenté par ajout d'une quantité appropriée de solution d'étalonnage (3.6.2). La quantité d'halofuginone ajoutée doit être similaire à la quantité estimée d'halofuginone trouvée dans l'extrait d'échantillon.
Seule la hauteur du pic d'halofuginone doit être augmentée, compte tenu à la fois de la quantité ajoutée et de la dilution de l'extrait. La largeur du pic, à la moitié de sa hauteur, doit être d'environ 10 % de sa largeur initiale.
7.1.2.
Évaluer les résultats conformément aux critères suivants:
a) les longueurs d'onde d'absorption maximale des spectres de l'échantillon et de l'étalon, enregistrées au sommet des pics sur le chromatogramme, doivent être identiques, dans une marge déterminée par le pouvoir de résolution du système de détection. Dans le cadre d'une détection par barrettes de diodes, elle est généralement d'environ 2 nm;
b) entre 225 et 300 nm, les spectres de l'échantillon et de l'étalon, enregistrés au sommet des pics sur le chromatogramme, ne peuvent être différents pour les parties du spectre comprises entre 10 et 100 % d'absorbance relative. Ce critère est rempli lorsque les mêmes maxima sont présents et que l'écart observé entre les deux spectres ne dépasse nulle part 15 % de l'absorbance de l'analyte étalon;
c) entre 225 et 300 nm, les spectres de la courbe ascendante, du sommet et de la courbe descendante du pic fourni par l'extrait d'échantillon ne peuvent être visuellement différents les uns des autres pour les parties du spectre comprises entre 10 et 100 % d'absorbance relative. Ce critère est rempli lorsque les mêmes maxima sont présents et que l'écart observé entre les spectres ne dépasse nulle part 15 % de l'absorbance du spectre au sommet du pic.
Si l'un de ces critères n'est pas rempli, la présence de l'analyte n'est pas confirmée.
7.2. Répétabilité
La différence entre les résultats des deux analyses parallèles effectuées sur le même échantillon ne peut dépasser 0,5 mg/kg pour des teneurs en halofuginone allant jusqu'à 3 mg/kg.
7.3. Récupération
En ce qui concerne l'échantillon blanc supplémenté, la récupération doit être de 80 % au minimum.
8. Résultats d'une étude collaborative
Dans le cadre d'une étude collaborative ( 15 ), trois échantillons ont été analysés par huit laboratoires.
Résultats
|
|
Échantillon A (blanc) À la réception |
Échantillon B (farine) |
Échantillon C (granulés) |
||
|
|
|
À la réception |
Après deux mois |
À la réception |
Après deux mois |
|
Moyenne [mg/kg] |
ND |
2,80 |
2,42 |
2,89 |
2,45 |
|
SR [mg/kg] |
— |
0,45 |
0,43 |
0,40 |
0,42 |
|
CVR [%] |
— |
16 |
18 |
14 |
17 |
|
Réc. [%] |
|
86 |
74 |
88 |
75 |
|
ND |
= |
non détecté |
|
SR |
= |
écart type de reproductibilité |
|
CVR |
= |
coefficient de variation de la reproductibilité (%) |
|
Réc. |
= |
récupération (%) |
E. DOSAGE DE LA ROBÉNIDINE
Chlorhydrate de 1,3 bis [(4-chlorobenzylidène) amino] guanidine
1. Objet et champ d'application
La méthode permet de déterminer la teneur en robénidine des aliments pour animaux. la limite de quantification est de 5 mg/kg.
2. Principe
L'échantillon est extrait à l'aide de méthanol acidifié. L'extrait est séché et une partie aliquote purifiée dans une colonne d'oxyde d'aluminium. La robénidine est éluée de la colonne à l'aide de méthanol, concentrée et portée à un volume adéquat avec la phase mobile. La teneur en robénidine est déterminée par chromatographie liquide haute performance (CLHP) en phase inverse, à l'aide d'un détecteur d'ultraviolets.
3. Réactifs
3.1. Méthanol.
3.2. Méthanol acidifié.
Transvaser 4,0 ml d'acide chlorhydrique (ρ20 = 1,18 g/ml) dans une fiole jaugée de 500 ml, ajuster au trait de jauge avec du méthanol (3.1) et mélanger. Cette solution doit être préparée peu avant d'être utilisée.
3.3. Acétonitrile, de qualité CLHP.
3.4. Tamis moléculaire.
Type 3A, perles de 8 à 12 mesh (perles de 1,6 à 2,5 mm, aluminosilicate cristallin, diamètre des pores de 0,3 mm).
3.5. Oxyde d'aluminium: acide, degré d'activité I pour chromatographie sur colonne.
Transvaser 100 g d'oxyde d'aluminium dans un récipient approprié et ajouter 2,0 ml d'eau. Boucher et agiter pendant environ 20 minutes. Conserver dans un récipient bien fermé.
3.6. Solution de dihydrogénophosphate de potassium, c = 0,025 mol/l.
Dissoudre 3,40 g de dihydrogénophosphate de potassium dans de l'eau (qualité CLHP) dans une fiole jaugée de 1 000 ml, ajuster au trait de jauge et mélanger.
3.7. Solution de monohydrogénophosphate disodique, c = 0,025 mol/l.
Dissoudre 3,55 g de monohydrogénophosphate disodique anhydre (ou 4,45 g de dihydrate ou 8,95 g de dodécahydrate) dans de l'eau (de qualité CLHP), dans une fiole jaugée de 1 L, ajuster au trait de jauge et mélanger.
3.8. Phase mobile de la CLHP.
Mélanger les réactifs suivants:
650 ml d'acétonitrile (3.3),
250 ml d'eau (de qualité CLHP),
50 ml de solution de dihydrogénophosphate de potassium (3.6),
50 ml de solution de monohydrogénophosphate disodique (3.7).
Filtrer à travers un filtre de 0,22 μm (4.6) et dégazer la solution (par exemple, par traitement aux ultrasons pendant 10 minutes).
3.9. Substance étalon.
Robénidine pure: chlorhydrate de 1,3 -bis [(4-chlorobenzylidène)amino]guanidine.
3.9.1.
Peser, à 0,1 mg près, 30 mg de substance étalon de robénidine (3.9). Dissoudre dans du méthanol acidifié (3.2), dans une fiole jaugée de 100 ml, ajuster au trait de jauge avec le même solvant et mélanger. Envelopper la fiole dans une feuille d'aluminium et conserver à l'abri de la lumière.
3.9.2.
Transvaser 10,0 ml de la solution mère de l'étalon (3.9.1) dans une fiole jaugée de 250 ml, ajuster au trait de jauge avec la phase mobile (3.8) et mélanger. Envelopper la fiole dans une feuille d'aluminium et conserver à l'abri de la lumière.
3.9.3.
Dans une série de fioles jaugées de 50 ml, transvaser 5,0 , 10,0 , 15,0 , 20,0 et 25,0 ml de la solution étalon intermédiaire (3.9.2). Ajuster au trait de jauge avec la phase mobile (3.8) et mélanger. Ces solutions ont des concentrations respectives de 1,2 , 2,4 , 3,6 , 4,8 et 6,0 μg/ml de robénidine. Les solutions doivent être préparées peu avant d'être utilisées.
3.10. Eau de qualité CLHP.
4. Appareillage
4.1. Colonne de verre.
Colonne construite en verre ambré, munie d'un robinet et d'un réservoir d'une capacité d'environ 150 ml, d'un diamètre intérieur de 10-15 mm, d'une longueur de 250 mm.
4.2. Agitateur mécanique ou magnétique.
4.3. Évaporateur rotatif.
4.4. Équipement pour CLHP avec détecteur d'ultraviolets à longueur d'onde variable ou détecteur à barrettes de diodes fonctionnant dans l'intervalle de 250-400 nm.
4.4.1. Colonne de chromatographie liquide: 300 mm × 4 mm, C18, particules de 10 μm, ou équivalent.
4.5. Papier filtre en fibre de verre (Whatman GF/A ou équivalent).
4.6. Filtres à membrane, 0,22 μm.
4.7. Filtres à membrane, 0,45 μm.
5. Mode opératoire
|
NB |
: |
La robénidine est photosensible. L'emploi de verrerie ambrée est recommandé pour toutes les opérations. |
5.1. Généralités
5.1.1. Il est recommandé d'analyser un aliment blanc pour vérifier l'absence de robénidine et de substances interférentes.
5.1.2. Il convient d'effectuer un test de récupération en analysant l'aliment blanc (5.1.1) après l'avoir supplémenté par ajout d'une quantité de robénidine similaire à celle présente dans l'échantillon. Pour parvenir à une teneur de 60 mg/kg, transvaser 3,0 ml de la solution mère de l'étalon (3.9.1) dans une fiole conique de 250 ml. Ramener la solution à environ 0,5 ml par évaporation dans un courant d'azote. Ajouter 15 g de l'aliment blanc, mélanger et laisser reposer 10 minutes avant de procéder à l'extraction (5.2).
|
NB |
: |
Dans le cadre de cette méthode, l'aliment blanc doit être d'un type similaire à celui de l'échantillon et, lors de son analyse, il ne doit pas être détecté de robénidine. |
5.2. Extraction
Peser, à 0,01 g près, 15 g de l'échantillon préparé. Transvaser dans une fiole conique de 250 ml et ajouter 100,0 ml de méthanol acidifié (3.2), boucher le récipient et agiter pendant une heure avec l'agitateur (4.2). Filtrer la solution à travers un papier filtre en fibre de verre (4.5) et recueillir la totalité du filtrat dans une fiole conique de 150 ml. Ajouter 7,5 g de tamis moléculaire (3.4), boucher le récipient et agiter pendant 5 minutes. Filtrer immédiatement à travers un papier filtre en fibre de verre. Conserver cette solution pour la purification (5.3).
5.3. Purification
5.3.1.
Munir l'extrémité inférieure d'une colonne de verre d'un petit tampon de laine de verre (4.1) et tasser à l'aide d'une baguette de verre. Peser et transvaser dans la colonne 11,0 g d'oxyde d'aluminium préparé (3.5). Il convient de réduire l'exposition à l'air au cours de cette opération. Tapoter l'extrémité inférieure de la colonne remplie pour décanter l'oxyde d'aluminium.
5.3.2.
À l'aide d'une pipette, transférer sur la colonne 5,0 ml de l'extrait d'échantillon préparé (5.2). Maintenir l'embout de la pipette contre la paroi de la colonne et laisser l'oxyde d'aluminium absorber la solution. Éluer la robénidine de la colonne à l'aide de 100 ml de méthanol (3.1), à un débit de 2-3 ml/min, et recueillir l'éluat dans un ballon à fond rond de 250 ml. Sécher la solution de méthanol par évaporation, à pression réduite, à 40 oC, à l'aide d'un évaporateur rotatif (4.3). Redissoudre le résidu dans 3-4 ml de la phase mobile (3.8) et transvaser quantitativement dans une fiole jaugée de 10 ml. Rincer la fiole avec plusieurs fractions de 1-2 ml de la phase mobile et transvaser ces rinçages dans la fiole jaugée. Ajuster au trait de jauge avec le même solvant et mélanger. Une partie aliquote est filtrée à travers un filtre à membrane de 0,45 μm (4.7). Réserver cette solution pour l'analyse par CLHP (5.4).
5.4. Dosage par CLHP
5.4.1.
Les conditions ci-après sont proposées à titre indicatif; d'autres conditions peuvent être appliquées si elles donnent des résultats équivalents.
Colonne de chromatographie liquide (4.4.1).
Phase mobile de la CLHP (3.8).
Débit: 1,5 à 2 ml/minute.
Longueur d'onde de détection: 317 nm.
Volume d'injection: 20 à 50 μl.
Vérifier la stabilité du système chromatographique en injectant plusieurs fois la solution d'étalonnage (3.9.3) à 3,6 μg/ml, jusqu'à obtention de hauteurs (surfaces) de pics et de temps de rétention constants.
5.4.2.
Injecter plusieurs fois chaque solution d'étalonnage (3.9.3) et mesurer les hauteurs (surfaces) des pics pour chaque concentration. Tracer la courbe d'étalonnage en portant les moyennes des hauteurs ou surfaces de pics des solutions d'étalonnage en ordonnée et les concentrations correspondantes en μg/ml en abscisse.
5.4.3.
Injecter plusieurs fois l'extrait d'échantillon (5.3.2) en utilisant le même volume que pour les solutions d'étalonnage et déterminer la hauteur (surface) moyenne des pics de robénidine.
6. Calcul des résultats
À partir de la hauteur (surface) moyenne des pics de robénidine de la solution d'échantillon, déterminer la concentration de robénidine dans cette solution, en μg/ml, par référence à la courbe d'étalonnage (5.4.2).
La teneur en robénidine w (mg/kg) de l'échantillon est donnée par la formule suivante:
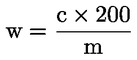
où:
|
c |
= |
concentration de robénidine dans la solution d'échantillon, en μg/ml, |
|
m |
= |
poids de la prise d'essai, en grammes. |
7. Validation des résultats
7.1. Identité
L'identité de l'analyte peut être confirmée par cochromatographie ou à l'aide d'un détecteur à barrettes de diodes permettant de comparer les spectres de l'extrait d'échantillon et de la solution d'étalonnage (3.9.3) contenant 6 μg/ml de robénidine.
7.1.1.
Un extrait d'échantillon est supplémenté par ajout d'une quantité appropriée de solution d'étalonnage (3.9.3). La quantité de robénidine ajoutée doit être similaire à la quantité estimée de robénidine trouvée dans l'extrait d'échantillon.
Seule la hauteur du pic de robénidine doit être augmentée, compte tenu à la fois de la quantité ajoutée et de la dilution de l'extrait. La largeur du pic, à la moitié de sa hauteur, doit être d'environ 10 % de sa largeur initiale.
7.1.2.
Évaluer les résultats conformément aux critères suivants:
a) les longueurs d'onde d'absorption maximale des spectres de l'échantillon et de l'étalon, enregistrées au sommet des pics sur le chromatogramme, doivent être identiques, dans une marge déterminée par le pouvoir de résolution du système de détection. Dans le cadre d'une détection par barrettes de diodes, elle est généralement de plus ou moins 2 nm;
b) entre 250 et 400 nm, les spectres de l'échantillon et de l'étalon, enregistrés au sommet des pics sur le chromatogramme, ne peuvent être différents pour les parties du spectre comprises entre 10 et 100 % d'absorbance relative. Ce critère est rempli lorsque les mêmes maxima sont présents et que l'écart observé entre les deux spectres ne dépasse nulle part 15 % de l'absorbance de l'analyte étalon;
c) entre 250 et 400 nm, les spectres de la courbe ascendante, du sommet et de la courbe descendante du pic fourni par l'extrait d'échantillon ne peuvent être visuellement différents les uns des autres pour les parties du spectre comprises entre 10 et 100 % d'absorbance relative. Ce critère est rempli lorsque les mêmes maxima sont présents et que l'écart observé entre les spectres ne dépasse nulle part 15 % de l'absorbance du spectre au sommet du pic.
Si l'un de ces critères n'est pas rempli, la présence de l'analyte n'est pas confirmée.
7.2. Répétabilité
La différence entre les résultats de deux dosages parallèles effectués sur le même échantillon ne peut dépasser 10 % du résultat le plus élevé pour une teneur en robénidine supérieure à 15 mg/kg.
7.3. Récupération
En ce qui concerne l'échantillon blanc supplémenté, la récupération doit être de 85 % au minimum.
8. Résultats d'une étude collaborative
Dans le cadre d'une étude collaborative CE, quatre échantillons d'aliments pour volailles et lapins, sous forme de farine ou de granulés, ont été analysés par douze laboratoires. Pour chaque échantillon, les analyses ont été effectuées en double. Les résultats sont indiqués dans le tableau ci-dessous:
|
|
Volailles |
Lapins |
||
|
|
Farine |
Granulés |
Farine |
Granulés |
|
Moyenne [mg/kg] |
27,00 |
27,99 |
43,6 |
40,1 |
|
sr [mg/kg] |
1,46 |
1,26 |
1,44 |
1,66 |
|
CVr [%] |
5,4 |
4,5 |
3,3 |
4,1 |
|
SR [mg/kg] |
4,36 |
3,36 |
4,61 |
3,91 |
|
CVR [%] |
16,1 |
12,0 |
10,6 |
9,7 |
|
Récupération [%] |
90,0 |
93,3 |
87,2 |
80,2 |
|
sr |
= |
écart type de répétabilité |
|
CVr |
= |
coefficient de variation de la répétabilité (%) |
|
SR |
= |
écart type de reproductibilité |
|
CVR |
= |
coefficient de variation de la reproductibilité (%) |
F. DOSAGE DU DICLAZURIL
(+)-4-chlorophényl[2,6-dichloro-4-(2,3,4,5-tétrahydro-3,5-dioxo-1,2,4-triazin-2-yl)phényl]acétonitrile
1. Objet et champ d'application
La méthode permet de déterminer la teneur en diclazuril des aliments pour animaux et des prémélanges. La limite de détection est de 0,1 mg/kg; la limite de quantification est de 0,5 mg/kg.
2. Principe
Après l'addition d'un étalon interne, l'échantillon est extrait à l'aide de méthanol acidifié. Pour les aliments pour animaux, une partie aliquote de l'extrait est purifiée sur une cartouche C18 pour extraction en phase solide. Le diclazuril est élué de la cartouche à l'aide d'un mélange de méthanol acidifié et d'eau. Après évaporation, le résidu est dissous dans un mélange DMF/eau. Pour les prémélanges, l'extrait est évaporé et le résidu est dissous dans un mélange DMF/eau. La teneur en diclazuril est déterminée par chromatographie en phase liquide à haute performance (CLHP) à gradients ternaires et en phase inverse, à l'aide d'un détecteur UV.
3. Réactifs
3.1. Eau, de qualité CLHP.
3.2. Acétate d'ammonium.
3.3. Hydrogénosulfate de tétrabutylammonium (TBHS).
3.4. Acétonitrile, de qualité CLHP.
3.5. Méthanol, de qualité CLHP.
3.6. N, N-diméthylformamide (DMF).
3.7. Acide chlorhydrique, ρ20 = 1,19 g/ml.
3.8. Substance étalon: diclazuril II-24: (+)-4-chlorophényl[2,6-dichloro-4-(2,3,4,5-tétrahydro-3,5-dioxo-1,2,4-triazin-2-yl)phényl]acétonitrile de pureté garantie, E771.
3.8.1.
Peser, à 0,1 mg près, 25 mg de substance étalon de diclazuril (3.8) dans une fiole jaugée de 50 ml. Dissoudre dans du DMF (3.6), ajuster au trait de jauge avec du DMF (3.6) et mélanger. Envelopper le flacon dans une feuille d'aluminium ou utiliser un flacon ambré et le mettre au réfrigérateur. À une température de ≤ 4 oC, la solution est stable pendant un mois.
3.8.2.
Transférer 5,00 ml de la solution mère de l'étalon (3.8.1) dans une fiole jaugée de 50 ml, ajuster au trait de jauge avec du DMF (3.6) et mélanger. Envelopper le flacon dans une feuille d'aluminium ou utiliser un flacon ambré et le mettre au réfrigérateur. À une température de ≤ 4 oC, la solution est stable pendant un mois.
3.9. Étalon interne: 2,6 dichloro-a-(4-chlorophényl)-4-(4,5 dihydro-3,5-dioxo-1,2,4-triazine-2 (3H)-yl) α-méthylbenzène-acétonitrile.
3.9.1.
Peser, à 0,1 mg près, 25 mg de substance étalon interne (3.9) dans une fiole jaugée de 50 ml. Dissoudre dans du DMF (3.6), ajuster au trait de jauge avec du DMF (3.6) et mélanger. Envelopper le flacon dans une feuille d'aluminium ou utiliser un flacon ambré et le mettre au réfrigérateur. À une température de ≤ 4 oC, la solution est stable pendant un mois.
3.9.2.
Transférer 5,00 ml de la solution mère de l'étalon interne (3.9.1) dans une fiole jaugée de 50 ml, ajuster au trait de jauge avec du DMF (3.6) et mélanger. Envelopper le flacon dans une feuille d'aluminium ou utiliser un flacon ambré et le mettre au réfrigérateur. À une température de ≤ 4 oC, la solution est stable pendant un mois.
3.9.3.
(p = teneur nominale en diclazuril du prémélange en mg/kg)
Peser, à 0,1 mg près, p/10 mg de l'étalon interne dans une fiole jaugée de 100 ml, dissoudre dans du DMF (3.6) dans un bain ultrasonique (4.6), ajuster au trait de jauge avec du DMF et mélanger. Envelopper le flacon dans une feuille d'aluminium ou utiliser un flacon ambré et le mettre au réfrigérateur. À une température de ≤ 4 oC, la solution est stable pendant un mois.
3.10. Solution d'étalonnage, 2 μg/ml
Introduire à la pipette 2,00 ml de solution étalon de diclazuril (3.8.2) et 2,00 ml de solution de l'étalon interne (3.9.2) dans une fiole jaugée de 50 ml. Ajouter 16 ml de DMF (3.6), ajuster au trait de jauge avec de l'eau et mélanger. Cette solution doit être préparée peu avant d'être utilisée.
3.11. Cartouche C18 pour extraction en phase solide, par exemple, Bond Elut, taille: 1 cc, masse de sorption: 100 mg.
3.12. Solvant d'extraction: méthanol acidifié.
Introduire à la pipette 5,0 ml d'acide chlorhydrique (3.7) dans 1 000 ml de méthanol (3.5) et mélanger.
3.13. Phase mobile pour CLHP
3.13.1. Éluant A: acétate d'ammonium — solution d'hydrogénosulfate de tétrabutylammonium.
Dissoudre 5 g d'acétate d'ammonium (3.2) et 3,4 g de TBHS (3.3) dans 1 000 ml d'eau (3.1) et mélanger.
3.13.2. Éluant B: acétonitrile (3.4).
3.13.3. Éluant C: méthanol (3.5).
4. Appareillage
4.1. Agitateur mécanique.
4.2. Équipement pour CLHP à gradients ternaires.
4.2.1. Colonne pour chromatographie liquide, remplie d'Hypersil ODS de 3 μm, 100 mm × 4,6 mm, ou équivalent.
4.2.2. Détecteur UV de longueur d'onde variable ou détecteur à barrettes de diodes.
4.3. Évaporateur rotatif.
4.4. Filtre à membrane de 0,45 μm.
4.5. Distributeur à vide.
4.6. Bain ultrasonique.
5. Mode opératoire
5.1. Généralités
5.1.1.
Analyser un aliment blanc pour vérifier l'absence de diclazuril ou de substances interférentes. L'aliment blanc doit être du même type que celui de l'échantillon; il ne doit être détecté ni diclazuril ni substances interférentes.
5.1.2.
Effectuer un test de récupération par analyse de l'aliment blanc auquel a été ajoutée une quantité de diclazuril similaire à celle présente dans l'échantillon. Pour obtenir une concentration de 1 mg/kg, ajouter 0,1 ml de la solution mère de l'étalon (3.8.1) à 50 g de l'aliment blanc, mélanger soigneusement et laisser reposer 10 minutes, mélanger de nouveau plusieurs fois avant de procéder à l'extraction (5.2).
En l'absence d'aliment blanc de même type que celui de l'échantillon (voir 5.1.1), le test de récupération peut être effectué selon la méthode par addition de l'étalon. Dans ce cas, l'échantillon à analyser est supplémenté d'une quantité de diclazuril semblable à celle déjà présente dans l'échantillon. Celui-ci est analysé avec l'échantillon non supplémenté et la récupération peut être calculée par différence.
5.2. Extraction
5.2.1.
Peser, à 0,01 g près, environ 50 g de l'échantillon. Transférer dans une fiole erlenmeyer de 500 ml, ajouter 1,00 ml de la solution de l'étalon interne (3.9.2), 200 ml du solvant d'extraction (3.12) et boucher la fiole. Secouer le mélange sur l'agitateur (4.1) pendant une nuit. Laisser déposer pendant 10 minutes. Transférer une partie aliquote de 20 ml du surnageant dans un récipient en verre approprié et diluer dans 20 ml d'eau. Transférer cette solution dans une cartouche à extraction (3.11) et filtrer sous vide (4.5). Laver la cartouche à l'aide de 25 ml du mélange de solvant d'extraction (3.12) et d'eau, 65 + 35 (V + V). Éliminer les fractions collectées et éluer les composés à l'aide de 25 ml d'un mélange de solvant d'extraction (3.12) et d'eau, 80 + 20 (V + V). Évaporer cette fraction jusqu'à ce qu'elle commence à sécher au moyen d'un évaporateur rotatif (4.3) à 60 oC. Dissoudre le résidu dans 1,0 ml de DMF (3.6), ajouter 1,5 ml d'eau (3.1) et mélanger. Faire passer dans un filtre à membrane (4.4). Procéder au dosage par CLHP (5.3).
5.2.2.
Peser, à 0,001 g près, environ 1 g de l'échantillon. Transférer dans une fiole erlenmeyer de 500 ml, ajouter 1,00 ml de solution de l'étalon interne (3.9.3) et 200 ml de solvant d'extraction (3.12), puis boucher la fiole. Passer le mélange à l'agitateur (4.1) pendant une nuit. Laisser déposer pendant 10 minutes. Transférer une partie aliquote de 10 000 /p ml (p = teneur nominale du prémélange en diclazuril en mg/kg) du surnageant dans un ballon à fond rond de dimension appropriée. Évaporer ce mélange jusqu'à ce qu'il commence à sécher, sous une pression réduite et à 60 oC au moyen d'un évaporateur rotatif (4.3). Redissoudre le résidu dans 10,0 ml de DMF (3.6), ajouter 15,0 ml d'eau (3.1) et mélanger. Procéder au dosage par CLHP (5.3).
5.3. Dosage par CLHP
5.3.1.
Les conditions suivantes sont proposées à titre indicatif, d'autres conditions peuvent être appliquées si elles donnent des résultats équivalents.
|
Colonne de chromatographie liquide (4.2.1): |
100 mm × 4,6 mm, remplie d'Hypersil ODS de 3 μm, ou équivalent. |
|
|
Phase mobile: |
Éluant A (3.13.2): |
solution aqueuse d'acétate d'ammonium et d'hydrogénosulfate de tétrabutylammonium |
|
|
Éluant B (3.13.2): |
acétonitrile |
|
|
Éluant C (3.13.3): |
méthanol |
|
Mode d'élution: |
— gradient linéaire — conditions initiales: A + B + C = 60 + 20 + 20 (v + v + v) — au bout de 10 minutes, élution par gradient pendant 30 min jusqu'à: A + B + C = 45 + 20 + 35 (v + v + v) Rincer avec B pendant 10 minutes |
|
|
Débit: |
1,5 -2 ml/min |
|
|
Volume d'injection: |
20 μl |
|
|
Longueur d'onde de détection: |
280 nm. |
|
Vérifier la stabilité du système chromatographique en injectant plusieurs fois la solution d'étalonnage (3.10) contenant 2 μg/ml jusqu'à obtention de hauteurs de pic et de temps de rétention constants.
5.3.2.
Injecter 20 μl de la solution d'étalonnage (3.10) plusieurs fois et déterminer la hauteur (surface) moyenne des pics du diclazuril et de l'étalon interne.
5.3.3.
Injecter 20 μl de la solution d'échantillon (5.2.1 ou 5.2.2) plusieurs fois et déterminer la hauteur (surface) moyenne des pics du diclazuril et de l'étalon interne.
6. Calcul des résultats
6.1. Aliments pour animaux
La teneur en diclazuril w (en mg/kg) de l'échantillon est donnée par la formule suivante:

[mg/kg]
où:
|
hd,s |
= |
hauteur (surface) du pic de diclazuril dans la solution d'échantillon (5.2.1), |
|
hi,s |
= |
hauteur (surface) du pic de l'étalon interne dans la solution d'échantillon (5.2.1), |
|
hd,c |
= |
hauteur (surface) du pic de diclazuril dans la solution d'étalonnage (3.10), |
|
hi,c |
= |
hauteur (surface) du pic de l'étalon interne dans la solution d'étalonnage (3.10), |
|
cd,c |
= |
concentration de diclazuril dans la solution d'étalonnage en μg/ml (3.10), |
|
m |
= |
poids de la prise d'essai, en grammes, |
|
V |
= |
volume de l'extrait d'échantillon selon 5.2.1 (c'est-à-dire 2,5 ml). |
6.2. Prémélanges
La teneur en diclazuril w (en mg/kg) de l'échantillon est donnée par la formule suivante:

[mg/kg]
où:
|
hd,c |
= |
hauteur (surface) du pic de diclazuril dans la solution d'étalonnage (3.10), |
|
hi,c |
= |
hauteur (surface) du pic de l'étalon interne dans la solution d'étalonnage (3.10), |
|
hd,s |
= |
hauteur (surface) du pic de diclazuril dans la solution d'échantillon (5.2.2), |
|
hi,s |
= |
hauteur (surface) du pic de l'étalon interne dans la solution d'échantillon (5.2.2), |
|
cd,c |
= |
concentration de diclazuril dans la solution d'étalonnage en μg/ml (3.10), |
|
m |
= |
poids de la prise d'essai, en grammes, |
|
V |
= |
volume de l'extrait d'échantillon selon 5.2.2 (c'est-à-dire 25 ml), |
|
p |
= |
teneur nominale en diclazuril du prémélange, en mg/kg). |
7. Validation des résultats
7.1. Identité
L'identité de l'analyte peut être confirmée par cochromatographie ou à l'aide d'un détecteur à barrettes de diodes qui permet de comparer les spectres de l'extrait d'échantillon (5.2.1 ou 5.2.2) et de la solution d'étalonnage (3.10).
7.1.1.
Un extrait d'échantillon (5.2.1 ou 5.2.2) est additionné d'une quantité appropriée de la solution d'étalonnage (3.10). La quantité de diclazuril ajoutée doit être semblable à la quantité de diclazuril constatée dans l'extrait d'échantillon.
Seule la hauteur du pic du diclazuril et du pic de l'étalon interne doit être augmentée, compte tenu à la fois de la quantité ajoutée et de la dilution de l'extrait. La largeur du pic à mi-hauteur doit se situer à ± 10 % de la largeur initiale du pic du diclazuril ou du pic de l'étalon interne de l'extrait d'échantillon non supplémenté.
7.1.2.
Évaluer les résultats conformément aux critères suivants:
a) les longueurs d'onde d'absorption maximale des spectres de l'échantillon et de l'étalon, enregistrées au sommet des pics sur le chromatogramme, doivent être identiques, dans une marge déterminée par le pouvoir de résolution du système de détection. Dans le cadre d'une détection par barrettes de diodes, elle est généralement de ± 2 nm;
b) entre 230 et 320 nm, les spectres de l'échantillon et de l'étalon enregistrés au sommet du pic sur le chromatogramme ne doivent pas être différents pour les parties du spectre situées entre 10 et 100 % de l'absorbance relative. Ce critère est rempli lorsque les mêmes maxima sont présents et que, en aucun point, l'écart observé entre les deux spectres ne dépasse 15 % de l'absorbance de l'étalon de l'analyte;
c) entre 230 en 320 nm, les spectres de la courbe ascendante, du sommet et de la courbe descendante du pic produits par l'extrait d'échantillon ne doivent pas être différents les uns des autres pour les parties du spectre situées entre 10 et 100 % de l'absorbance relative. Ce critère est rempli lorsque les mêmes maxima sont présents et que, en aucun point, l'écart observé entre les spectres ne dépasse 15 % de l'absorbance du spectre au sommet du pic.
Si l'un de ces critères n'est pas rempli, la présence de l'analyte n'est pas confirmée.
7.2. Répétabilité
La différence entre les résultats de deux dosages parallèles effectués sur le même échantillon ne peut dépasser:
— 30 % du résultat supérieur pour les teneurs en diclazuril comprises entre 0,5 et 2,5 mg/kg,
— 0,75 mg/kg pour les teneurs en diclazuril comprises entre 2,5 et 5 mg/kg,
— 15 % du résultat supérieur pour les teneurs en diclazuril supérieures à 5 mg/kg.
7.3. Récupération
Pour un échantillon (blanc) supplémenté, la récupération doit être de 80 % au moins.
8. Résultats d'une étude collaborative
Dans le cadre d'une étude collaborative, cinq échantillons ont été analysés par onze laboratoires. Ces échantillons consistaient en deux prémélanges: l'un était mélangé à une matrice organique (O 100) et l'autre à une matrice inorganique (A 100). La teneur théorique est de 100 mg de diclazuril par kg. Les trois aliments pour volaille mélangés étaient fabriqués par trois producteurs différents (NL) (L1/Z1/K1). La teneur théorique est de 1 mg de diclazuril par kg. Les laboratoires avaient pour instruction d'analyser chacun des échantillons une seule fois ou en double (des informations plus détaillées sur cette étude collaborative figurent dans le Journal of AOAC International, Volume 77, no 6, 1994, pp. 1359-1361). Les résultats de l'étude figurent ci-après:
|
|
Échantillon 1 A 100 |
Échantillon 2 O 100 |
Échantillon 3 L1 |
Échantillon 4 Z1 |
Échantillon 5 K1 |
|
L |
11 |
11 |
11 |
11 |
6 |
|
n |
19 |
18 |
19 |
19 |
12 |
|
Moyenne |
100,8 |
103,5 |
0,89 |
1,15 |
0,89 |
|
Sr (mg/kg) |
5,88 |
7,64 |
0,15 |
0,02 |
0,03 |
|
CVr (%) |
5,83 |
7,38 |
17,32 |
1,92 |
3,34 |
|
SR (mg/kg) |
7,59 |
7,64 |
0,17 |
0,11 |
0,12 |
|
CVR (%) |
7,53 |
7,38 |
18,61 |
9,67 |
13,65 |
|
Teneur nominale (mg/kg) |
100 |
100 |
1 |
1 |
1 |
|
L |
= |
nombre de laboratoires |
|
n |
= |
nombre de valeurs individuelles |
|
Sr |
= |
écart type de répétabilité |
|
CVr |
= |
coefficient de variation de la répétabilité |
|
SR |
= |
écart type de reproductibilité |
|
CVR |
= |
coefficient de variation de la reproductibilité |
9. Observation
Il doit être préalablement démontré que la réaction du diclazuril est linéaire sur toute la gamme des concentrations mesurées.
G. DOSAGE DU LASALOCIDE-SODIUM
Sel sodique de polyéther de l'acide monocarboxylique, produit par Streptomyces lasaliensis
1. Objet et champ d'application
La méthode permet de déterminer la teneur en lasalocide-sodium des aliments pour animaux et des prémélanges. La limite de détection est de 5 mg/kg; la limite de quantification est de 10 mg/kg.
2. Principe
Le lasalocide-sodium est extrait d'échantillon par du méthanol acidifié et dosé par chromatographie liquide à haute performance (CLHP) en phase inverse à l'aide d'un détecteur spectrofluorimétrique.
3. Réactifs
3.1. Phosphate monopotassique (KH2P04).
3.2. Acide orthophosphorique, p (p/p) = 85 %.
3.3. Solution d'acide orthophosphorique, c = 20 %
Diluer 23,5 ml d'acide orthophosphorique (3.2) dans de l'eau et ajuster à 100 ml.
3.4. 6-méthyl-2-heptylamine (1,5 -diméthylhexylamine), p (p/p) = 99 %.
3.5. Méthanol, de qualité CLHP.
3.6. Acide chlorhydrique, densité = 1,19 g/ml.
3.7. Solution tampon de phosphate, c = 0,01 mol/l.
Dissoudre 1,36 g de KH2PO4 (3.1) dans 500 ml d'eau (3.11), ajouter 3,5 ml d'acide orthophosphorique (3.2) et 10,0 ml de 6-méthyl-2-heptylamine (3.4). Ajuster le pH à 4,0 à l'aide de la solution d'acide orthophosphorique (3.3), diluer à l'eau et ajuster à 1 000 ml (3.11).
3.8. Méthanol acidifié
Transférer 5,0 ml d'acide chlorhydrique (3.6) dans une fiole jaugée de 1 000 ml, ajuster au trait de jauge avec du méthanol (3.5) et mélanger. Cette solution doit être préparée peu avant d'être utilisée.
3.9. Phase mobile pour CLHP, solution tampon de phosphate et méthanol 5 + 95 (v + v)
Mélanger 5 ml de solution tampon de phosphate (3.7) avec 95 ml de méthanol (3.5).
3.10. Substance étalon: lasalocide-sodium garanti pur, C34H53O8Na (sel sodique de polyéther de l'acide monocarboxylique, produit par Streptomyces lasaliensis), E763.
3.10.1.
Peser, à 0,1 mg près, 50 mg de lasalocide-sodium (3.10) dans une fiole jaugée de 100 ml, dissoudre dans du méthanol acidifié (3.8), ajuster au trait de jauge avec le même solvant et mélanger. Cette solution doit être préparée peu avant d'être utilisée.
3.10.2.
Introduire à la pipette 10,0 ml de la solution mère de l'étalon (3.10.1) dans une fiole jaugée de 100 ml, ajuster au trait de jauge avec du méthanol acidifié (3.8) et mélanger. Cette solution doit être préparée peu avant d'être utilisée.
3.10.3.
Transférer 1,0 , 2,0 , 4,0 , 5,0 et 10,0 ml de la solution étalon intermédiaire (3.10.2) dans une série de fioles jaugées de 50 ml. Ajuster au trait de jauge avec du méthanol acidifié (3.8) et mélanger. Ces solutions correspondent respectivement à 1,0 , 2,0 , 4,0 , 5,0 et 10,0 μg de lasalocide-sodium par ml et doivent être préparées peu avant d'être utilisées.
3.11. Eau, de qualité CLHP.
4. Appareillage
4.1. Bain ultrasonique (ou bain-marie vibrant) avec réglage de température.
4.2. Filtres à membrane de 0,45 μm.
4.3. Équipement CLHP avec système à injection permettant d'injecter des volumes de 20 μl.
4.3.1. Colonne de chromatographie liquide, 125 mm × 4 mm, en phase inverse C18, particules de 5 μm, ou équivalent.
4.3.2. Spectrofluorimètre avec correction des longueurs d'onde variables (sollicitation et émission).
5. Mode opératoire
5.1. Généralités
5.1.1.
Pour procéder au test de récupération (5.1.2), analyser un aliment blanc pour vérifier l'absence de lasalocide-sodium ou de substances interférentes. L'aliment blanc doit être du même type que celui de l'échantillon; il ne doit être détecté ni lasalocide-sodium ni substances interférentes.
5.1.2.
Effectuer un test de récupération par analyse de l'aliment blanc auquel a été ajoutée une quantité de lasalocide-sodium similaire à celle présente dans l'échantillon. Pour obtenir une concentration de 100 mg/kg, transférer 10,0 ml de la solution mère de l'étalon (3.10.1) dans une fiole erlenmeyer de 250 ml et concentrer la solution par évaporation à environ 0,5 ml. Ajouter 50 g de l'aliment blanc, mélanger soigneusement et laisser reposer 10 minutes tout en mélangeant de nouveau plusieurs fois avant de procéder à l'extraction (5.2).
En l'absence d'aliment blanc de même type que celui de l'échantillon (voir 5.1.1), le test de récupération peut être effectué selon la méthode par addition de l'étalon. Dans ce cas, l'échantillon à analyser est supplémenté par ajout d'une quantité de lasalocide-sodium semblable à celle déjà présente dans l'échantillon. Celui-ci est analysé avec l'échantillon non supplémenté et la récupération peut être calculée par différence.
5.2. Extraction
5.2.1.
Peser, à 0,01 g près, de 5 g à 10 g de l'échantillon dans une fiole erlenmeyer de 250 ml avec bouchon. Y introduire à la pipette 100,0 ml de méthanol acidifié (3.8). Boucher légèrement et agiter pour obtenir une dispersion. Placer la fiole dans le bain ultrasonique (4.1) à 40 oC environ pendant 20 minutes, l'enlever et laisser refroidir à température ambiante. Laisser reposer pendant une heure environ, jusqu'à décantation des matières en suspension, et filtrer une partie aliquote sur un filtre à membrane de 0,45 μm (4.2) dans un récipient approprié. Procéder au dosage par CLHP (5.3).
5.2.2.
Peser, à 0,001 g près, 2 g de prémélange non broyé dans une fiole jaugée de 250 ml. Ajouter 100,0 ml de méthanol acidifié (3.8) et agiter pour obtenir une dispersion. Placer la fiole dans le bain ultrasonique (4.1) à 40 oC environ pendant 20 minutes, l'enlever et laisser refroidir à température ambiante. Diluer jusqu'au trait de jauge avec du méthanol acidifié (3.8) et mélanger soigneusement. Laisser reposer pendant une heure, jusqu'à décantation des matières en suspension et filtrer une partie aliquote sur un filtre à membrane de 0,45 μm (4.2). Diluer un volume adéquat de filtrat clair avec du méthanol acidifié (3.8), de manière à obtenir une solution d'essai finale contenant environ 4 μg/ml de lasalocide-sodium. Procéder au dosage par CLHP (5.3).
5.3. Dosage par CLHP
5.3.1.
Les conditions suivantes sont proposées à titre indicatif; d'autres conditions peuvent être appliquées si elles donnent des résultats équivalents:
|
Colonne de chromatographie liquide (4.3.1): |
125 mm × 4 mm, en phase inverse, C18, particules de 5 μm, ou équivalent |
|
Phase mobile (3.9): |
Mélange de solution tampon de phosphate (3.7) et de méthanol (3.5), 5 + 95 (V + V) |
|
Débit: |
1,2 ml/min |
|
Longueurs d'onde de détection: |
|
|
Sollicitation: |
310 nm |
|
Émission: |
419 nm |
|
Volume d'injection: |
20 μl |
Vérifier la stabilité du système chromatographique en injectant plusieurs fois la solution d'étalonnage (3.10.3) à 4,0 μg/ml jusqu'à obtention de hauteurs (surfaces) de pics et de temps de rétention constants.
5.3.2.
Injecter chaque solution d'étalonnage (3.10.3) plusieurs fois et déterminer les hauteurs (surfaces) moyennes des pics pour chaque concentration. Établir une courbe d'étalonnage en utilisant les hauteurs (surfaces) moyennes des pics des solutions d'étalonnage comme ordonnées et les concentrations correspondantes en μg/ml comme abscisses.
5.3.3.
Injecter l'extrait d'échantillon (obtenu conformément au point 5.2.1 ou 5.2.2) plusieurs fois en utilisant le même volume que celui retenu pour la solution d'étalonnage et déterminer la hauteur (surface) moyenne des pics du lasalocide-sodium.
6. Calcul des résultats
À partir de la hauteur (surface) moyenne des pics du lasalocide-sodium de la solution d'échantillon (5.3.3), déterminer la concentration de lasalocide-sodium (μg/ml) par référence à la courbe d'étalonnage.
6.1. Aliments pour animaux
La teneur w en lasalocide-sodium, exprimée en mg/kg de l'échantillon, est donnée par la formule suivante:

[mg/kg]
où:
|
c |
= |
concentration de lasalocide-sodium dans la solution d'échantillon (5.2.1) en μg/ml, |
|
V1 |
= |
volume de l'extrait d'échantillon selon 5.2.1 en ml (soit 100), |
|
m |
= |
poids de la prise d'essai, en grammes. |
6.2. Prémélanges
La teneur w en lasalocide-sodium, exprimée en mg/kg de l'échantillon, est donnée par la formule suivante:

[mg/kg]
où:
|
c |
= |
concentration de lasalocide-sodium dans la solution d'échantillon (5.2.2) en μg/ml, |
|
V2 |
= |
volume de l'extrait d'échantillon selon 5.2.2 en ml (soit 250), |
|
f |
= |
facteur de dilution selon 5.2.2, |
|
m |
= |
poids de la prise d'essai, en grammes. |
7. Validation des résultats
7.1. Identité
Les méthodes fondées sur la spectrofluorimétrie sont moins sujettes aux interférences que celles qui utilisent un détecteur UV. L'identité de l'analyte peut être confirmée par cochromatographie.
7.1.1.
Un extrait d'échantillon (5.2.1 ou 5.2.2) est additionné d'une quantité appropriée de la solution d'étalonnage (3.10.3). La quantité de lasalocide-sodium ajoutée doit être semblable à la quantité de lasalocide-sodium constatée dans l'extrait d'échantillon. Seule la hauteur du pic du lasalocide-sodium doit être augmentée, compte tenu de la quantité ajoutée et de la dilution de l'extrait. La largeur du pic à mi-hauteur doit se situer à ± 10 % de la largeur initiale du pic de lasalocide-sodium de l'extrait d'échantillon non supplémenté.
7.2. Répétabilité
La différence entre les résultats de deux dosages parallèles effectués sur le même échantillon ne peut dépasser:
— 15 % du résultat supérieur pour les teneurs en lasalocide-sodium comprises entre 30 mg/kg et 100 mg/kg,
— 15 mg/kg pour les teneurs en lasalocide-sodium comprises entre 100 mg/kg et 200 mg/kg,
— 7,5 % du résultat supérieur pour les teneurs en lasalocide-sodium supérieures à 200 mg/kg.
7.3. Récupération
Pour les échantillons d'aliments pour animaux (blancs) supplémentés, la récupération doit être de 80 % au minimum. Pour les échantillons de prémélanges supplémentés, la récupération doit être de 90 % au minimum.
8. Résultats d'une étude collaborative
Dans le cadre d'une étude collaborative ( *1 ), 2 prémélanges (échantillons 1 et 2) et 5 aliments (échantillons 3-7) ont été analysés par 12 laboratoires. Pour chaque échantillon, les analyses ont été effectuées en double. Les résultats figurent dans le tableau ci-après.
|
|
Échantillon 1 Prémé-lange poulets |
Échantillon 2 Prémé-lange dindes |
Échantillon 3 Granulés dindes |
Échantillon 4 Miettes poulets |
Échantillon 5 Aliment dindes |
Échantillon 6 Aliment volaille A |
Échantillon 7 Aliment volaille B |
|
L |
12 |
12 |
12 |
12 |
12 |
12 |
12 |
|
n |
23 |
23 |
23 |
23 |
23 |
23 |
23 |
|
Moyenne [mg/kg] |
5 050 |
16 200 |
76,5 |
78,4 |
92,9 |
48,3 |
32,6 |
|
sr [mg/kg] |
107 |
408 |
1,71 |
2,23 |
2,27 |
1,93 |
1,75 |
|
CVr [%] |
2,12 |
2,52 |
2,24 |
2,84 |
2,44 |
4,00 |
5,37 |
|
sR [mg/kg] |
286 |
883 |
3,85 |
7,32 |
5,29 |
3,47 |
3,49 |
|
CVR [%] |
5,66 |
5,45 |
5,03 |
9,34 |
5,69 |
7,18 |
10,70 |
|
Teneur nominale [mg/kg] |
5 000 (1) |
16 000 (1) |
80 (1) |
105 (1) |
120 (1) |
50 (2) |
35 (2) |
|
(*1) Teneur déclarée par le fabricant. (*2) Aliment préparé au laboratoire. |
|||||||
|
L |
= |
nombre de laboratoires |
|
n |
= |
nombre de valeurs individuelles |
|
sr |
= |
écart type de répétabilité |
|
sR |
= |
écart type de reproductibilité |
|
CVr |
= |
coefficient de variation de la répétabilité (%) |
|
CVR |
= |
coefficient de variation de la reproductibilité (%) |
ANNEXE V
MÉTHODES D'ANALYSE RELATIVES AU CONTRÔLE DE SUBSTANCES INDÉSIRABLES DANS LES ALIMENTS POUR ANIMAUX
A. DOSAGE DU GOSSYPOL LIBRE ET TOTAL
1. Objet et champ d'application
La méthode permet de déterminer les teneurs en gossypol libre, en gossypol total et en substances chimiquement apparentées des graines, des farines et des tourteaux de coton ainsi que des aliments composés contenant ces matières premières pour aliments des animaux, lorsque la présence de gossypol libre, de gossypol total et de substances chimiquement apparentées dépasse 20 mg/kg.
2. Principe
Le gossypol est extrait en présence de 3-amino-l-propanol, soit par un mélange d'isopropanol et d'hexane pour le dosage du gossypol libre, soit par la diméthylformamide pour le dosage du gossypol total. Le gossypol est transformé au moyen d'aniline en gossypol-dianiline, dont la densité optique est mesurée à 440 nm.
3. Réactifs
3.1. Mélange isopropanol-hexane: mélanger 60 parts en volume d'isopropanol à 40 parts en volume de n-hexane.
3.2. Solvant A: porter, dans une fiole jaugée de 1 l, 500 ml environ du mélange isopropanol-hexane (3.1), 2 ml de 3-amino-l-propanol, 8 ml d'acide acétique glacial et 50 ml d'eau. Ajuster au trait de jauge avec le mélange isopropanol-hexane (3.1). Ce réactif est stable durant une semaine.
3.3. Solvant B: porter à la pipette, dans une fiole jaugée de 100 ml, 2 ml de 3-amino-l-propanol et 10 ml d'acide acétique glacial. Refroidir à la température ambiante et ajuster au trait de jauge avec la N, N-diméthylformamide. Ce réactif est stable durant une semaine.
3.4. Aniline: si la densité optique de l'essai à blanc excède 0,022 , distiller l'aniline sur poudre de zinc en éliminant les première et dernière fractions de 10 % du distillat. En flacon bouché de verre brun et au réfrigérateur, ce réactif se conserve plusieurs mois.
3.5. Solution étalon A de gossypol: porter, dans une fiole jaugée de 250 ml, 27,9 mg d'acétate de gossypol. Dissoudre et ajuster au trait de jauge avec le solvant A (3.2). Introduire à la pipette 50 ml de cette solution dans une fiole jaugée de 250 ml et ajuster au trait de jauge avec le solvant A. La concentration de gossypol dans cette solution est de 0,02 mg/ml. Laisser reposer durant 1 h à température ambiante avant l'emploi.
3.6. Solution étalon B de gossypol: porter, dans une fiole jaugée de 50 ml, 27,9 mg d'acétate de gossypol. Dissoudre et ajuster au trait de jauge avec le solvant B (3.3). La concentration de gossypol dans cette solution est de 0,5 mg/ml.
Conservées à l'abri de la lumière, les solutions étalon A et B de gossypol sont stables durant 24 h.
4. Appareillage
4.1. Mélangeur (culbuteur): environ 35 retournements par minute.
4.2. Spectrophotomètre.
5. Mode opératoire
5.1. Prise d'essai
La prise d'essai est en rapport avec la teneur présumée en gossypol de l'échantillon. Il est préférable d'opérer sur une petite prise d'essai et sur une partie aliquote du filtrat relativement importante, de façon à obtenir une quantité de gossypol suffisante pour effectuer une mesure photométrique précise. Pour le dosage du gossypol libre dans les graines, les farines et les tourteaux de coton, la prise d'essai ne doit pas excéder 1 g; pour les aliments composés, elle pourra atteindre 5 g. Une partie aliquote de 10 ml de filtrat convient dans la plupart des cas; elle doit contenir de 50 à 100 μg de gossypol. Pour le dosage du gossypol total, la prise d'essai pourra varier de 0,5 à 5 g afin qu'une partie aliquote de 2 ml de filtrat contienne de 40 à 200 μg de gossypol.
L'analyse devrait être effectuée à une température ambiante proche de 20 oC.
5.2. Dosage du gossypol libre
Introduire la prise d'essai dans une fiole à col rodé de 250 ml, dont le fond est recouvert de verre pilé. Ajouter à la pipette 50 ml de solvant A (3.2), boucher la fiole et mélanger durant 1 h dans le mélangeur. Filtrer sur filtre sec et recueillir le filtrat dans une petite fiole à col rodé. Au cours de la filtration, recouvrir l'entonnoir d'un verre de montre.
Introduire à la pipette dans deux fioles jaugées de 25 ml (A et B) des parties aliquotes identiques de filtrat contenant de 50 à 100 μg de gossypol. Si nécessaire, ajuster le volume à 10 ml avec du solvant A (3.2). Ajuster ensuite au trait de jauge le contenu de la fiole (A) avec le mélange isopropanol-hexane (3.1). Cette solution sera utilisée comme solution de référence pour la mesure de la solution d'échantillon.
Introduire à la pipette 10 ml du solvant A (3.2) dans deux autres fioles jaugées de 25 ml (C et D). Ajuster au trait de jauge le contenu de la fiole (C) avec le mélange isopropanol-hexane (3.1). Cette solution sera utilisée comme solution de référence pour la mesure de la solution de l'essai à blanc.
Ajouter 2 ml d'aniline (3.4) dans les fioles (D) et (B). Chauffer durant 30 minutes sur un bain d'eau bouillante pour développer la coloration. Refroidir à la température ambiante, ajuster au trait de jauge avec le mélange isopropanol-hexane (3.1), homogénéiser et laisser reposer durant 1 h.
Déterminer au spectrophotomètre à 440 nm, dans des cuvettes en verre de 1 cm, la densité optique de la solution de l'essai à blanc (D) par comparaison avec la solution de référence (C) et la densité optique de la solution d'échantillon (B) par comparaison avec la solution de référence (A).
Soustraire la densité optique de la solution de l'essai à blanc de celle de la solution d'échantillon (= densité optique corrigée). Calculer à partir de cette valeur la teneur en gossypol libre comme indiqué au point 6.
5.3. Dosage du gossypol total
Introduire une prise d'essai contenant de 1 à 5 mg de gossypol dans une fiole jaugée de 50 ml et ajouter 10 ml de solvant B (3.3). Préparer simultanément un essai à blanc, en introduisant 10 ml de solvant B (3.3), dans une autre fiole jaugée de 50 ml. Chauffer les deux fioles durant 30 minutes sur un bain d'eau bouillante. Refroidir à la température ambiante et ajuster au trait de jauge le contenu de chaque fiole avec le mélange isopropanol-hexane (3.1). Homogénéiser et laisser déposer pendant 10 à 15 minutes, filtrer ensuite et recueillir les filtrats dans des fioles à col rodé.
Introduire à la pipette 2 ml du filtrat de l'échantillon dans deux fioles jaugées de 25 ml et 2 ml du filtrat de l'essai à blanc dans deux autres fioles de 25 ml. Ajuster le contenu d'une fiole de chaque série à 25 ml avec le mélange isopropanol-hexane (3.1). Ces solutions seront utilisées comme solutions de référence.
Ajouter 2 ml d'aniline (3.4) dans les deux autres fioles. Chauffer durant 30 minutes sur un bain d'eau bouillante pour développer la coloration. Refroidir à la température ambiante, ajuster à 25 ml avec le mélange isopropanol-hexane (3.1), homogénéiser et laisser reposer durant 1 h.
Déterminer la densité optique comme indiqué au point 5.2 pour le gossypol libre. Calculer à partir de cette valeur la teneur en gossypol total comme indiqué au point 6.
6. Calcul des résultats
Le calcul des résultats peut se faire soit à partir de la densité optique spécifique (6.1), soit par référence à une courbe d'étalonnage (6.2).
6.1. À partir de la densité optique spécifique
Dans les conditions décrites, les densités optiques spécifiques sont les suivantes:
|
Gossypol libre |
|
|
Gossypol total |
|
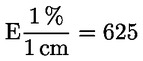
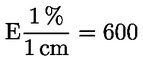
La teneur en gossypol libre ou total de l'échantillon est donnée par la formule suivante:

où:
|
E |
= |
densité optique corrigée, déterminée comme indiqué au point 5.2, |
|
p |
= |
prise d'essai en grammes, |
|
a |
= |
partie aliquote du filtrat en ml. |
6.2. À partir d'une courbe d'étalonnage
6.2.1.
Préparer deux séries de cinq fioles jaugées de 25 ml. Dans chaque série, introduire à la pipette dans les fioles des volumes respectifs de 2,0 , 4,0 , 6,0 , 8,0 et 10,0 ml de la solution étalon A de gossypol (3.5). Ajuster les volumes à 10 ml avec le solvant A (3.2). Compléter chaque série par une fiole jaugée de 25 ml contenant uniquement 10 ml de solvant A (3.2) (essai à blanc).
Ajuster à 25 ml le volume des fioles de la première série (y compris la fiole destinée à l'essai à blanc) avec le mélange isopropanol-hexane (3.1) (série de référence).
Ajouter 2 ml d'aniline (3.4) dans chaque fiole de la seconde série (y compris la fiole destinée à l'essai à blanc). Chauffer durant 30 minutes sur un bain d'eau bouillante pour développer la coloration. Refroidir à la température ambiante, ajuster au trait de jauge avec le mélange isopropanol-hexane (3.1), homogénéiser et laisser reposer durant 1 h (série étalon).
Déterminer dans les conditions indiquées au point 5.2 la densité optique des solutions de la série étalon par comparaison avec les solutions correspondantes de la série de référence. Tracer graphiquement la courbe d'étalonnage en portant les densités optiques en regard des quantités de gossypol (en μg).
6.2.2.
Préparer 6 fioles jaugées de 50 ml. Introduire dans la première fiole 10 ml de solvant B (3.3) et, dans les autres, respectivement 2,0 , 4,0 , 6,0 , 8,0 et 10,0 ml de la solution étalon B de gossypol (3.6). Ajuster le contenu de chaque fiole à 10 ml avec le solvant B (3.3). Chauffer durant 30 minutes sur un bain d'eau bouillante. Refroidir à la température ambiante, ajuster au trait de jauge avec le mélange isopropanol-hexane (3.1) et homogénéiser.
Introduire 2,0 ml de ces solutions dans deux séries de 6 fioles jaugées de 25 ml. Ajuster à 25 ml le contenu des fioles de la première série avec le mélange isopropanol-hexane (3.1) (série de référence).
Ajouter 2 ml d'aniline (3.4) dans chaque fiole de la seconde série. Chauffer durant 30 minutes sur un bain d'eau bouillante. Refroidir à la température ambiante, ajuster au trait de jauge avec le mélange isopropanol-hexane (3.1), homogénéiser et laisser reposer durant 1 h (série étalon).
Déterminer, dans les conditions indiquées au point 5.2, la densité optique des solutions de la série étalon par comparaison avec les solutions correspondantes de la série de référence. Tracer graphiquement la courbe d'étalonnage en portant les densités optiques en regard des quantités de gossypol (en μg).
6.3. Répétabilité
La différence entre les résultats de deux dosages parallèles effectués sur le même échantillon ne peut dépasser:
— 15 %, en valeur relative, du résultat supérieur pour les teneurs en gossypol inférieures à 500 ppm,
— 75 ppm, en valeur absolue, pour les teneurs comprises entre 500 et 750 ppm,
— 10 %, en valeur relative, du résultat supérieur pour les teneurs dépassant 750 ppm.
B. DÉTERMINATION DES TENEURS EN DIOXINES (PCDD/PCDF) ET EN PCB
CHAPITRE I
Méthodes de prélèvement d'échantillons et interprétation des résultats d'analyse
1. Champ d'application et définitions
Les échantillons destinés au contrôle officiel des teneurs en dibenzo-p-dioxines polychlorées (PCDD), en dibenzofuranes polychlorés (PCDF), en polychlorobiphényles (PCB) ( 16 ) de type dioxine et en PCB autres que ceux de type dioxine des aliments pour animaux sont prélevés conformément aux dispositions de l'annexe I. Les exigences quantitatives concernant le contrôle des substances ou des produits répartis uniformément dans les aliments pour animaux prévues au point 5.1 de l'annexe I sont appliquées. Les échantillons globaux ainsi obtenus sont considérés comme représentatifs des lots ou sous-lots sur lesquels ils sont prélevés. Le respect des teneurs maximales fixées dans la directive 2002/32/CE est établi sur la base des teneurs déterminées dans les échantillons de laboratoire.
Aux fins du présent règlement, les définitions figurant à l'annexe I de la décision 2002/657/CE de la Commission ( 17 ) s'appliquent.
Outre ces définitions, les définitions ci-après s'appliquent aux fins de la présente partie B:
«méthodes de dépistage» : méthodes servant à sélectionner les échantillons dont les teneurs en PCDD, PCDF et PCB de type dioxine dépassent les teneurs maximales ou les seuils d'intervention. Elles offrent une grande capacité de traitement d'échantillons à la fois efficace et économique et augmentent la probabilité de découvrir de nouveaux cas d'exposition élevée susceptibles de mettre la santé des consommateurs en péril. Les méthodes de dépistage sont fondées sur des méthodes de bioanalyse ou sur des méthodes CG-SM. Les résultats des échantillons dépassant la valeur seuil utilisée aux fins du contrôle du respect de la teneur maximale, sont vérifiés; à cette fin, l'échantillon initial est soumis à une nouvelle analyse complète, réalisée au moyen d'une méthode de confirmation;
«méthodes de confirmation» : méthodes qui fournissent des informations complètes ou complémentaires permettant l'identification et la quantification univoque des PCDD, PCDF et des PCB de type dioxine à la teneur maximale ou, en cas de besoin, au seuil d'intervention. Ces méthodes recourent à la chromatographie en phase gazeuse couplée à la spectrométrie de masse haute résolution (CG-SMHR) ou à la chromatographie en phase gazeuse couplée à la spectrométrie de masse en tandem (CG-SM/SM).
2. Conformité du lot ou sous-lot avec la teneur maximale
2.1. Spécifications relatives aux PCB autres que ceux de type dioxine
Le lot ou sous-lot est conforme à la teneur maximale si le résultat d'analyse pour la somme des PCB 28, PCB 52, PCB 101, PCB 138, PCB 153 et PCB 180 (ci-après les «PCB autres que ceux de type dioxine») ne dépasse pas la teneur maximale fixée dans la directive 2002/32/CE, compte tenu de l'incertitude de mesure élargie ( 18 ). Le lot ou sous-lot n'est pas conforme à la teneur maximale fixée dans la directive 2002/32/CE si la moyenne des deux résultats d'analyse supérieurs ( 19 ) obtenus lors d'une double analyse ( 20 ), dépasse la teneur maximale avec une quasi-certitude, compte tenu de l'incertitude de mesure élargie; la concentration analysée sert ainsi, après déduction de l'incertitude de mesure élargie, à évaluer la conformité.
L'incertitude de mesure élargie est calculée au moyen d'un facteur d'élargissement de 2, qui donne un niveau de confiance d'environ 95 %. Un lot ou sous-lot est non conforme si la moyenne des valeurs mesurées, diminuée de l'incertitude élargie de la moyenne, est supérieure à la teneur maximale fixée.
Les règles énoncées aux alinéas précédents du présent point s'appliquent aux résultats d'analyse des échantillons destinés au contrôle officiel. En cas d'analyse à des fins de recours ou d'arbitrage, les règles nationales s'appliquent.
2.2. Spécifications relatives aux PCDD/PCDF et aux PCB de type dioxine
Le lot ou sous-lot est conforme à la teneur maximale si le résultat d'une seule analyse,
— effectuée selon une méthode de dépistage avec un taux de faux conformes inférieur à 5 %, indique que la teneur ne dépasse pas les limites maximales respectives fixées pour les PCDD/PCDF et la somme des PCDD/PCDF et des PCB de type dioxine dans la directive 2002/32/CE,
— effectuée selon une méthode de confirmation, ne dépasse pas les teneurs maximales respectives fixées pour les PCDD/PCDF et la somme des PCDD/PCDF et des PCB de type dioxine dans la directive 2002/32/CE, compte tenu de l'incertitude de mesure élargie.
Pour les analyses de dépistage, une valeur seuil doit être établie pour déterminer la conformité aux teneurs maximales respectives fixées pour les PCDD/PCDF ou pour la somme des PCDD/PCDF et des PCB de type dioxine.
Le lot ou sous-lot n'est pas conforme à la teneur maximale fixée dans la directive 2002/32/CE si la moyenne des deux résultats d'analyse supérieurs ( 21 ) obtenus lors d'une double analyse ( 22 ) à l'aide d'une méthode de confirmation, dépasse la teneur maximale avec une quasi-certitude, compte tenu de l'incertitude de mesure élargie; la concentration analysée sert ainsi, après déduction de l'incertitude de mesure élargie, à évaluer la conformité.
L'incertitude de mesure élargie est calculée au moyen d'un facteur d'élargissement de 2, qui donne un niveau de confiance d'environ 95 %. Un lot ou sous-lot est non conforme si la moyenne des valeurs mesurées, diminuée de l'incertitude élargie de la moyenne, est supérieure à la teneur maximale fixée.
La somme des estimations des incertitudes élargies des résultats d'analyse distincts concernant les PCDD/PCDF et les PCB de type dioxine doit être utilisée pour la somme des PCDD/PCDF et des PCB de type dioxine.
Les règles énoncées aux alinéas précédents du présent point s'appliquent aux résultats d'analyse des échantillons destinés au contrôle officiel. En cas d'analyse à des fins de recours ou d'arbitrage, les règles nationales s'appliquent.
3. Résultats dépassant les seuils d'intervention prévus à l'annexe II de la directive 2002/32/CE
Les seuils d'intervention sont utilisés aux fins de la sélection d'échantillons dans les cas où il est nécessaire de déterminer une source de contamination et de prendre des mesures de réduction ou d'élimination de celle-ci. Les méthodes de dépistage établissent des valeurs seuil appropriées aux fins de la sélection de ces échantillons. S'il est nécessaire d'accomplir des efforts considérables pour déterminer une source et pour réduire ou éliminer la contamination, il convient de confirmer le dépassement des seuils d'intervention en réalisant une double analyse au moyen d'une méthode de confirmation et en tenant compte de l'incertitude de mesure élargie ( 23 ).
CHAPITRE II
Préparation des échantillons et prescriptions applicables aux méthodes d'analyse à utiliser pour le contrôle officiel des teneurs en dioxines (PCDD/PCDF) et en PCB de type dioxine des aliments pour animaux
1. Champ d'application
Les prescriptions du présent chapitre s'appliquent aux analyses d'aliments pour animaux effectuées aux fins du contrôle officiel des teneurs en PCDD/PCDF substitués en 2,3,7,8 et en PCB de type dioxine, ainsi qu'à d'autres fins réglementaires, y compris les contrôles effectués par l'exploitant du secteur de l'alimentation animale pour veiller au respect des dispositions de l'article 4 du règlement (CE) no 183/2005 du Parlement européen et du Conseil ( 24 ).
La présence de PCDD/PCDF et de PCB de type dioxine dans les aliments pour animaux peut être contrôlée au moyen des deux types de méthodes d'analyse décrites ci-après.
a) Méthodes de dépistage
Les méthodes de dépistage servent à sélectionner les échantillons dont les teneurs en PCDD/PCDF et en PCB de type dioxine dépassent les teneurs maximales ou les seuils d'intervention. Elles doivent offrir une grande capacité de traitement d'échantillons à la fois efficace et économique et augmenter la probabilité de découvrir de nouveaux cas d'exposition élevée des consommateurs et de risques pour leur santé. Leur application doit permettre d'éviter les faux conformes. Elles peuvent comprendre des méthodes de bioanalyse et des méthodes de CG-SM.
Elles permettent de comparer le résultat d'une analyse avec une valeur seuil et de confirmer ou d'infirmer (réponse positive ou négative) l'éventuel dépassement de la teneur maximale ou du seuil d'intervention. La concentration de PCDD/PCDF et la somme des PCDD/PCDF et des PCB de type dioxine dans les échantillons suspectés de non-conformité à la teneur maximale doivent être déterminées ou confirmées au moyen d'une méthode de confirmation.
En outre, les méthodes de dépistage peuvent donner une indication des teneurs en PCDD/PCDF et en PCB de type dioxine des échantillons. En cas d'application de méthodes bioanalytiques de dépistage, le résultat est exprimé en équivalents de bioanalyse (BEQ), alors qu'en cas d'application de méthodes CG-SM physico-chimiques, il est exprimé en équivalents toxiques (TEQ). Les résultats numériques obtenus au moyen de méthodes de dépistage permettent de démontrer la conformité, la suspicion de non-conformité ou le dépassement des seuils d'intervention et donnent une indication sur la plage des teneurs en cas de suivi au moyen de méthodes de confirmation. Ils ne permettent pas, par exemple, d'évaluer les niveaux de bruit de fond, d'estimer l'ingestion, de suivre l'évolution chronologique des teneurs ou de réévaluer les seuils d'intervention et les teneurs maximales.
b) Méthodes de confirmation
Les méthodes de confirmation permettent l'identification et la quantification univoques des PCDD/PCDF et des PCB de type dioxine présents dans un échantillon et de fournir des informations complètes à l'échelle du congénère. Par conséquent, ces méthodes permettent de contrôler les teneurs maximales et les seuils d'intervention et de confirmer les résultats obtenus au moyen des méthodes de dépistage. En outre, les résultats peuvent servir à d'autres fins, telles que la détermination des faibles niveaux de bruit de fond dans le contrôle des aliments pour animaux, le suivi de l'évolution chronologique, l'évaluation de l'exposition et la création d'une base de données en vue de la réévaluation éventuelle des seuils d'intervention et des teneurs maximales. Ils sont aussi importants pour établir les profils de congénères aux fins de la détermination de la source d'une contamination éventuelle. Ces méthodes reposent sur la chromatographie en phase gazeuse couplée à la spectrométrie de masse haute résolution (CG-SMHR). La chromatographie en phase gazeuse couplée à la spectrométrie de masse en tandem (CG-SM/SM) peut également être utilisée pour confirmer la conformité ou la non-conformité à la teneur maximale.
2. Contexte
Pour le calcul des concentrations d'équivalents toxiques (TEQ), les concentrations des différentes substances dans un échantillon donné sont multipliées par leurs facteurs d'équivalence toxique (TEF) respectifs (voir note de bas de page 1 au chapitre I), puis elles sont additionnées de façon à donner la concentration totale de composés de type dioxine, exprimée en TEQ.
Aux fins de la présente partie B, la limite de quantification spécifique acceptée d'un congénère est la teneur minimale en analyte mesurable avec une certitude statistique raisonnable, satisfaisant aux critères d'identification décrits dans des normes internationalement reconnues, telles que la norme EN 16215:2012 (Aliments des animaux — Dosage des dioxines et des PCB de type dioxine par CG-SMHR et des PCB indicateurs par CG-SMHR) et/ou les méthodes EPA 1613 et 1668 révisées.
La limite de quantification d'un congénère peut être:
a) la concentration d'un analyte dans l'extrait d'un échantillon qui produit une réponse instrumentale à deux ions suivis avec un rapport S/B (signal/bruit) de 3:1 pour le signal de données brut le moins intense; ou
b) si, pour des raisons techniques, le calcul du rapport signal/bruit ne donne pas des résultats fiables, le point de concentration minimal sur une courbe d'étalonnage qui donne un écart acceptable (≤ 30 %) et cohérent (mesuré au moins au début et à la fin d'une série analytique d'échantillons) par rapport au facteur de réponse relatif moyen calculé pour tous les points de la courbe d'étalonnage de chaque série d'échantillons. La limite de quantification est calculée à partir du point de concentration le plus bas, compte tenu du taux de récupération des étalons internes et du prélèvement d'échantillons.
Les méthodes bioanalytiques de dépistage ne donnent pas de résultats à l'échelle du congénère mais une simple indication ( 25 ) de la valeur TEQ, exprimée en équivalents de bioanalyse (BEQ) compte tenu du fait qu'il est possible que tous les composés présents dans un extrait d'échantillon produisant une réponse lors de l'essai ne remplissent pas l'ensemble des conditions du principe du TEQ.
Les méthodes de dépistage et de confirmation ne peuvent être appliquées aux fins du contrôle d'une matrice donnée que si les méthodes sont suffisamment sensibles pour déceler les teneurs de manière fiable au niveau du seuil d'intervention ou de la teneur maximale.
3. Prescriptions d'assurance qualité
|
3.1. |
Des mesures doivent être prises pour que soit évitée toute contamination croisée à chaque étape de la procédure d'échantillonnage et d'analyse. |
|
3.2. |
Les échantillons doivent être conservés et transportés dans des récipients en verre, en aluminium, en polypropylène ou en polyéthylène adaptés à la conservation, préservant les teneurs en PCDD/PCDF et en PCB de type dioxine dans les échantillons de la moindre influence. Toute trace de poussière de papier doit être enlevée du contenant de l'échantillon. |
|
3.3. |
La conservation et le transport de l'échantillon doivent être effectués d'une façon telle que l'intégrité de l'échantillon d'aliment pour animaux est préservée. |
|
3.4. |
Si nécessaire, chaque échantillon pour laboratoire doit être broyé finement et soigneusement mélangé, selon une méthode garantissant une homogénéisation complète (par exemple de façon à pouvoir passer au travers d'un tamis à mailles de 1 mm). Les échantillons doivent être séchés avant le broyage si leur teneur en humidité est trop élevée. |
|
3.5. |
Il y a lieu de contrôler les réactifs, la verrerie et l'équipement en vue de déceler toute influence des résultats exprimés en TEQ ou en BEQ. |
|
3.6. |
Un essai à blanc est réalisé, en suivant tout le procédé d'analyse, mais sans l'échantillon. |
|
3.7. |
Pour les méthodes de bioanalyse, l'ensemble de la verrerie et des solvants utilisés dans l'analyse doivent faire l'objet d'un dépistage de composés interférant avec la détection des composés cibles dans la plage de travail. La verrerie est rincée à l'aide de solvants ou chauffée à des températures permettant d'éliminer de sa surface les traces de PCDD/PCDF, de composés de type dioxine et de composés interférents. |
|
3.8. |
La quantité de l'extrait doit être suffisamment élevée, de façon à répondre aux prescriptions en ce qui concerne une plage de travail suffisamment basse comprenant les concentrations des teneurs maximales ou du seuil d'intervention. |
|
3.9. |
Les procédures spécifiques de préparation des échantillons utilisées pour les produits considérés doivent être conformes aux lignes directrices internationalement acceptées. |
4. Prescriptions applicables aux laboratoires
|
4.1. |
Conformément aux dispositions du règlement (CE) no 882/2004, les laboratoires doivent être accrédités par un organisme habilité qui se conforme au guide ISO 58, de manière à garantir qu'ils appliquent les procédures d'assurance qualité à leurs analyses. Les laboratoires doivent être accrédités selon la norme EN ISO/CEI 17025. Les principes énoncés dans les directives techniques pour l'estimation de l'incertitude de mesure et les limites de quantification pour l'analyse des PCDD/PCDF et des PCB doivent être suivis lorsqu'ils sont applicables ( 26 ). |
|
4.2. |
L'aptitude des laboratoires doit être prouvée par la participation continue et réussie à des études interlaboratoires sur le dosage des PCDD/PCDF et des PCB de type dioxine dans les matrices d'aliments pour animaux et les plages de concentration correspondantes. |
|
4.3. |
Les laboratoires appliquant les méthodes de dépistage pour les contrôles de routine des échantillons coopèrent étroitement avec les laboratoires appliquant la méthode de confirmation, tant pour le contrôle qualité que pour la confirmation du résultat de l'analyse des échantillons suspects. |
5. Prescriptions fondamentales applicables aux procédés d'analyse relatifs aux dioxines (PCDD/PCDF) et aux PCB de type dioxine
5.1. Sensibilité élevée et limite de quantification basse
En ce qui concerne les PCDD/PCDF, étant donné la toxicité extrêmement élevée de certains de ces composés, les seuils de détection doivent être de l'ordre de quelques femtogrammes (10– 15 g). Pour la plupart des congénères PCB, une limite de quantification de l'ordre du nanogramme (10– 9 g) est déjà suffisante. Pour la mesure des congénères PCB de type dioxine plus toxiques (en particulier les congénères non ortho substitués), la limite inférieure de la plage de travail doit être sous le picogramme (10– 12 g). Pour tous les autres congénères PCB, une limite de quantification de l'ordre du nanogramme (10– 9 g) est suffisante.
5.2. Grande sélectivité (spécificité)
|
5.2.1. |
Il est nécessaire de distinguer les PCDD/PCDF et les PCB de type dioxine d'une multitude d'autres composés extraits simultanément de l'échantillon qui sont susceptibles d'interférer et peuvent être présents à des concentrations supérieures, jusqu'à plusieurs ordres de grandeur, à celles des analytes à doser. Pour les méthodes de CG-SM, il est nécessaire d'établir une distinction entre les congénères, notamment entre les congénères toxiques (par exemple, les dix-sept PCDD/PCDF substitués en 2,3,7,8 et les douze PCB de type dioxine) et les autres congénères. |
|
5.2.2. |
Les méthodes de bioanalyse doivent permettre la détection des composés cibles en tant que somme des PCDD/PCDF et/ou des PCB de type dioxine. La purification des échantillons est destinée à éliminer les composés à l'origine de faux non conformes ou les composés susceptibles d'atténuer la réponse et de donner des faux conformes. |
5.3. Grande exactitude (justesse et fidélité, taux de récupération apparent du bioessai)
|
5.3.1. |
Pour les méthodes de CG-SM, le dosage doit fournir une estimation juste de la concentration réelle dans un échantillon. Une grande exactitude est nécessaire pour empêcher que le résultat d'une analyse d'échantillon ne soit écarté en raison du manque de fiabilité de la valeur TEQ déterminée. L'exactitude est une expression de la justesse (la différence entre la valeur moyenne mesurée pour un analyte dans un matériau certifié et sa valeur certifiée, exprimée en pourcentage de cette valeur) et de la fidélité (RSDR est l'écart type relatif calculé à partir des résultats obtenus dans des conditions de reproductibilité). |
|
5.3.2. |
Pour les méthodes de bioanalyse, le taux de récupération apparent du bioessai doit être déterminé. Le taux de récupération apparent du bioessai désigne la valeur BEQ calculée à partir de la courbe d'étalonnage de la TCDD ou du PCB 126 corrigée du blanc, puis divisée par la valeur TEQ déterminée par la méthode de confirmation. Elle vise à corriger des facteurs tels que la perte de PCDD/PCDF et de composés de type dioxine durant les phases d'extraction et de purification, la coextraction de composés qui augmentent ou atténuent la réponse (effets agonistes et antagonistes), la qualité de l'ajustement de la courbe ou les différences entre les valeurs TEF (facteur d'équivalence toxique) et REP (potentiel relatif). Le taux de récupération apparent du bioessai est calculé à partir d'échantillons de référence appropriés avec des profils de congénères représentatifs proches du niveau considéré. |
5.4. Validation dans la plage de la teneur maximale et mesures générales de contrôle qualité
|
5.4.1. |
Les laboratoires doivent démontrer la validité de la méthode dans une certaine plage proche de la teneur maximale, par exemple à des niveaux correspondant à 0,5 fois, 1 fois et 2 fois la teneur maximale, avec un coefficient de variation acceptable pour les analyses répétées, durant la procédure de validation et l'analyse de routine. |
|
5.4.2. |
Des essais à blanc et des expériences avec enrichissement ou des analyses sur des échantillons témoins (si possible, des matériaux de référence certifiés) sont effectués régulièrement dans le cadre des mesures internes de contrôle qualité. Il est nécessaire de réaliser et de vérifier des cartes de contrôle qualité pour les essais à blanc, les expériences avec enrichissement ou l'analyse des échantillons témoins afin de garantir que les performances analytiques sont conformes aux prescriptions. |
5.5. Limite de quantification
|
5.5.1. |
Pour une méthode bioanalytique de dépistage, l'établissement de la limite de quantification n'est pas indispensable, mais il doit être démontré que la méthode permet de distinguer la valeur de blanc de la valeur seuil. En cas de transmission d'une valeur BEQ, il est nécessaire d'établir un seuil d'inscription permettant de savoir que faire des échantillons produisant une réponse au-dessous de ce seuil. Le seuil d'inscription doit présenter une différence avérée d'un facteur de trois au moins par rapport aux échantillons du blanc de procédure produisant une réponse au-dessous de la plage de travail. Il est donc calculé à partir d'échantillons contenant les composés cibles proches de la teneur minimale requise et non à partir d'un rapport S/B ou d'un blanc d'essai. |
|
5.5.2. |
La limite de quantification pour une méthode de confirmation est de l'ordre d'un cinquième de la teneur maximale. |
5.6. Critères d'analyse
La fiabilité des résultats des méthodes de confirmation ou de dépistage impose le respect des critères ci-après dans la plage de la teneur maximale pour la valeur TEQ ou la valeur BEQ, qu'elle soit exprimée en TEQ totaux ou BEQ totaux (somme des PCDD/PCDF et des PCB de type dioxine) ou séparément pour les PCDD/PCDF et les PCB de type dioxine.
|
|
Dépistage au moyen de méthodes de bioanalyse ou de méthodes physico-chimiques |
Méthodes de confirmation |
|
Taux de faux conformes (1) |
< 5 % |
|
|
Justesse |
|
De – 20 % à + 20 % |
|
Répétabilité (RSDr) |
< 20 % |
|
|
Fidélité intermédiaire (RSDR) |
< 25 % |
< 15 % |
|
(1) Au regard des teneurs maximales. |
||
5.7. Prescriptions spécifiques applicables aux méthodes de dépistage
|
5.7.1. |
Le dépistage peut être effectué au moyen de méthodes de CG-SM et de méthodes de bioanalyse. Pour les méthodes de CG-SM, les prescriptions établies au point 6 doivent être appliquées. Pour les méthodes de bioanalyse cellulaire, des prescriptions spécifiques sont établies au point 7. |
|
5.7.2. |
Les laboratoires appliquant les méthodes de dépistage pour les contrôles de routine d'échantillons coopèrent étroitement avec les laboratoires appliquant la méthode de confirmation. |
|
5.7.3. |
Les performances de la méthode de dépistage doivent être vérifiées durant l'analyse de routine par un contrôle qualité des analyses et par la validation continue de la méthode. Il doit exister un programme continu de contrôle des résultats conformes. |
|
5.7.4. |
Contrôle de l'atténuation éventuelle de la réponse cellulaire et cytotoxicité: vingt pour cent des extraits d'échantillons doivent être mesurés par dépistage de routine sans et avec ajout de la 2,3,7,8-TCDD à la teneur maximale ou au seuil d'intervention, pour vérifier si la réponse est éventuellement atténuée par des substances interférentes présentes dans l'extrait d'échantillon. La concentration mesurée dans l'échantillon enrichi doit être comparée à la somme de la concentration de l'extrait non enrichi et de la concentration de l'enrichissement. Si cette concentration mesurée est inférieure de plus de 25 % à la concentration (somme) calculée, cela indique la possibilité d'atténuation du signal et l'échantillon en question doit faire l'objet de l'analyse de confirmation par CG-SMHR. Les résultats sont contrôlés à l'aide de cartes de contrôle de la qualité. |
|
5.7.5. |
Contrôle de la qualité sur des échantillons conformes: environ 2 à 10 % des échantillons conformes, en fonction de la matrice et de l'expérience du laboratoire, doivent être confirmés par CG-SMHR. |
|
5.7.6. |
Détermination des taux de faux conformes à partir des données du contrôle qualité: le taux de faux conformes résultant du dépistage des échantillons au-dessous et au-dessus de la teneur maximale ou du seuil d'intervention doit être déterminé. Les taux réels de faux conformes doivent être inférieurs à 5 %. Dès lors que le contrôle qualité des échantillons conformes fait apparaître au moins vingt résultats confirmés par matrice/groupe de matrices, des conclusions sur le taux de faux conformes doivent être tirées à partir de cette base de données. Les résultats des échantillons analysés au moyen d'essais interlaboratoires ou durant des cas de contamination, jusqu'à concurrence d'une concentration de deux fois la teneur maximale, par exemple, peuvent figurer parmi les vingt résultats à atteindre pour déterminer le taux de faux conformes. Les échantillons couvrent les profils de congénères les plus fréquents, représentant différentes sources. Bien que les bioanalyses de dépistage servent avant tout à révéler les échantillons dépassant le seuil d'intervention, le critère appliqué pour la détermination des taux de faux conformes est la teneur maximale, compte tenu de l'incertitude de mesure élargie de la méthode de confirmation. |
|
5.7.7. |
Les résultats du dépistage indiquant que des échantillons pourraient être non conformes doivent toujours être vérifiés; à cette fin, l'échantillon initial est soumis à une nouvelle analyse complète, réalisée au moyen d'une méthode de confirmation. Ces échantillons peuvent également servir à l'évaluation du taux de faux non conformes. Pour les méthodes de dépistage, le taux de faux non conformes est la fraction des résultats dont la conformité est confirmée par une analyse de confirmation, alors que lors du dépistage précédent, les échantillons avaient été déclarés potentiellement non conformes. L'évaluation des avantages de la méthode de dépistage doit se fonder sur la comparaison du nombre d'échantillons faussement non conformes et du nombre total d'échantillons contrôlés. Ce taux doit être suffisamment bas pour rendre l'utilisation de la méthode de dépistage avantageuse. |
|
5.7.8. |
Dans des conditions de validation, les méthodes de bioanalyse doivent fournir une indication juste de la valeur TEQ, calculée et exprimée en BEQ. De plus, pour les méthodes de bioanalyse appliquées dans des conditions de répétabilité, le RSDr intralaboratoire sera généralement inférieur au RSDR (reproductibilité). |
6. Prescriptions spécifiques applicables aux méthodes de CG-SM à respecter à des fins de dépistage ou de confirmation
6.1. Écarts acceptables entre l'estimation supérieure et l'estimation inférieure des OMS-TEQ
L'écart entre l'estimation supérieure et l'estimation inférieure ne peut dépasser 20 % pour la confirmation du dépassement des teneurs maximales ou, en cas de besoin, des seuils d'intervention.
6.2. Mesure des taux de récupération
|
6.2.1. |
Des étalons internes de PCDD/PCDF substitués en 2,3,7,8 marqués au 13C et des étalons internes de PCB de type dioxine marqués au 13C doivent être ajoutés au tout début de la méthode d'analyse, par exemple avant la phase d'extraction, afin de valider le procédé d'analyse. Il est nécessaire d'ajouter au moins un congénère pour chacun des groupes d'isomères tétrachlorés à octachlorés des PCDD/PCDF et au moins un congénère pour chaque groupe d'isomères des PCB de type dioxine (une autre méthode consiste à ajouter au moins un congénère pour chaque fenêtre d'acquisition spectrométrique utilisée pour le contrôle des PCDD/PCDF et des PCB de type dioxine). Pour les méthodes de confirmation, il y a lieu d'utiliser l'ensemble des dix-sept étalons internes de PCDD/PCDF substitués en 2,3,7,8 marqués au 13C ainsi que la totalité des douze étalons internes de PCB de type dioxine marqués au 13C. |
|
6.2.2. |
Des facteurs de réponse relatifs doivent également être déterminés dans le cas des congénères pour lesquels aucun analogue marqué au 13C n'est ajouté, en utilisant des solutions d'étalonnage appropriées. |
|
6.2.3. |
Pour les aliments pour animaux d'origine végétale et les aliments pour animaux d'origine animale contenant moins de 10 % de graisses, il est obligatoire d'ajouter les étalons internes avant la phase d'extraction. Pour les aliments pour animaux d'origine animale contenant plus de 10 % de graisses, les étalons internes sont ajoutés avant ou après l'extraction des graisses. L'efficacité de l'extraction doit faire l'objet d'une validation appropriée, eu égard à la phase au cours de laquelle les étalons internes sont introduits. |
|
6.2.4. |
Avant l'analyse CG-SM, un ou deux étalons de récupération (substitution) doivent être ajoutés. |
|
6.2.5. |
Le taux de récupération doit être mesuré. Dans le cas des méthodes de confirmation, les taux de récupération des étalons internes doivent se situer dans une plage comprise entre 60 et 120 %. Pour des congénères individuels, en particulier pour certains dibenzo-p-dioxines et dibenzofuranes heptachlorés et octachlorés, des taux de récupération inférieurs ou supérieurs sont acceptables, à condition que leur contribution à la valeur TEQ ne dépasse pas 10 % de la valeur TEQ totale (sur la base de la somme des PCDD/PCDF et des PCB de type dioxine). Dans le cas des méthodes de dépistage par CG-SM, les taux de récupération doivent se situer dans une plage comprise entre 30 et 140 %. |
6.3. Élimination des substances interférentes
— Les PCDD/PCDF sont séparés des composés chlorés interférents, tels que les PCB autres que ceux de type dioxine et les diphényléthers chlorés, au moyen de techniques chromatographiques appropriées (de préférence au moyen d'une colonne de florisil, d'alumine et/ou de charbon),
— La séparation des isomères par chromatographie en phase gazeuse est inférieure à 25 % de pic à pic entre 1,2,3,4,7,8-HxCDF et 1,2,3,6,7,8-HxCDF.
6.4. Étalonnage avec courbe étalon
La plage de la courbe d'étalonnage doit couvrir la plage correspondante des teneurs maximales ou des seuils d'intervention.
6.5. Critères spécifiques applicables aux méthodes de confirmation
— CG-SMHR:
—
En SMHR, la résolution caractéristique doit être égale ou supérieure à 10 000 pour toute la plage de masse avec une vallée de 10 %.
Les autres critères d'identification et de confirmation sont remplis conformément à des normes internationalement reconnues, telles que la norme EN 16215:2012 (Aliments des animaux — Dosage des dioxines, des PCB de type dioxine et des PCB indicateurs par CG-SMHR) et/ou les méthodes EPA 1613 et 1668 révisées.
— CG-SM/SM:
—
Il s'agit de suivre au moins deux ions précurseurs spécifiques et, pour chacun d'eux, un ion de fragmentation spécifique produit pour tous les analytes marqués et non marqués dans le champ d'application de l'analyse.
La tolérance autorisée maximale des intensités relatives des ions est de ± 15 % pour les ions de fragmentation sélectionnés par rapport aux valeurs calculées ou mesurées (moyenne des étalons), dans des conditions SM/SM identiques (notamment en ce qui concerne l'énergie de collision et la pression du gaz de collision), pour chaque transition d'un analyte.
La résolution de chaque quadrupôle doit être égale ou supérieure à la résolution massique unitaire (résolution massique unitaire: résolution suffisante pour séparer deux pics d'une unité massique) de manière à minimiser les éventuelles interférences avec les analytes considérés.
Les autres critères sont remplis conformément à des normes internationalement reconnues, telles que la norme EN 16215:2012 (Aliments des animaux — Dosage des dioxines, des PCB de type dioxine et des PCB indicateurs par CG-SMHR) et/ou les méthodes EPA 1613 et 1668 révisées, sauf pour ce qui concerne l'obligation de recourir à la CG-SMHR.
7. Prescriptions spécifiques applicables aux méthodes de bioanalyse
Les méthodes de bioanalyse sont des méthodes fondées sur le recours aux principes biologiques tels que les bioessais cellulaires, les tests d'interaction récepteurs ou les immuno-essais. Le présent point 7 énonce les prescriptions applicables aux méthodes de bioanalyse en général.
Dans une méthode de dépistage, l'échantillon est en principe déclaré conforme ou est déclaré suspecté d'être non conforme. À cette fin, la valeur BEQ calculée est comparée à la valeur seuil (voir point 7.3). Les échantillons au-dessous de la valeur seuil sont déclarés conformes, et ceux à la valeur seuil ou au-dessus de celle-ci sont déclarés suspectés d'être non conformes et nécessitent une analyse au moyen d'une méthode de confirmation. Dans la pratique, une valeur BEQ équivalant aux deux tiers de la teneur maximale peut servir de valeur seuil, à condition qu'un taux de faux conformes inférieur à 5 % et un taux acceptable de faux non conformes soient garantis. Avec des teneurs maximales distinctes pour les PCDD/PCDF et pour la somme des PCDD/PCDF et des PCB de type dioxine, le contrôle de la conformité des échantillons sans fractionnement requiert des valeurs seuil de bioessai appropriées pour les PCDD/PCDF. Pour la vérification des échantillons dépassant les seuils d'intervention, la valeur seuil doit être un pourcentage approprié des seuils d'intervention respectifs.
Si une valeur indicative est exprimée en BEQ, les résultats des échantillons doivent se situer dans la plage de travail et dépasser le seuil d'inscription (voir points 7.1.1 et 7.1.6).
7.1. Évaluation de la réponse à l'essai
7.1.1.
— Le calcul des concentrations à partir d'une courbe d'étalonnage de la TCDD fera apparaître une variation importante (coefficient de variation élevé — CV) des valeurs à l'extrémité supérieure de la courbe. La plage de travail est la zone où ce CV est inférieur à 15 %. L'extrémité inférieure de la plage de travail (seuil d'inscription) doit par ailleurs être établie à un niveau supérieur d'un facteur de trois au moins aux blancs de procédure. L'extrémité supérieure de la plage de travail est habituellement représentée par la valeur EC70 (70 % de la concentration effective maximale), mais elle se situe à un niveau inférieur si le CV est supérieur à 15 % dans cette plage. La plage de travail est établie pendant la validation. Les valeurs seuil (point 7.3) doivent se situer dans la plage de travail,
— Les solutions étalon et les extraits d'échantillon doivent être analysés en triple ou au moins en double. En cas d'utilisation de doubles, une solution étalon ou un extrait témoin analysé dans quatre à six puits répartis sur la plaque produisent une réponse ou une concentration (possible uniquement dans la plage de travail) sur la base d'un CV inférieur à 15 %.
7.1.2.
7.1.2.1. Étalonnage avec courbe étalon
— Les teneurs dans les échantillons doivent être estimées par comparaison de la réponse à l'essai avec une courbe d'étalonnage de la TCDD (ou du PCB 126 ou d'un mélange type de PCDD/PCDF/PCB de type dioxine) pour calculer la valeur BEQ dans l'extrait et, par la suite, dans l'échantillon,
— Les courbes d'étalonnage doivent contenir de huit à douze concentrations (au moins en double), la concentration dans la partie inférieure de la courbe (plage de travail) devant être suffisante. Une attention particulière doit être accordée à la qualité de l'ajustement de la courbe dans la plage de travail. La valeur R2 a peu ou n'a pas de valeur en tant que telle pour l'appréciation de la justesse de l'ajustement en régression non linéaire. La réduction de l'écart entre les valeurs calculées et les valeurs observées dans la plage de travail de la courbe améliorera l'ajustement (la réduction de la somme des résidus au carré, par exemple),
— Le niveau estimatif dans l'extrait d'échantillon doit ensuite être corrigé de la valeur BEQ calculée pour un échantillon blanc de matrice/de solvant (pour tenir compte des impuretés provenant des solvants et substances chimiques utilisés) et du taux de récupération apparent (calculé à partir de la valeur BEQ d'échantillons de référence adéquats avec des profils de congénères représentatifs proches de la teneur maximale ou du seuil d'intervention). Pour la correction par le taux de récupération, le taux de récupération apparent doit se situer dans les limites de la plage requise (voir point 7.1.4). Les échantillons de référence utilisés pour corriger du taux de récupération doivent respecter les prescriptions énoncées au point 7.2.
7.1.2.2. Étalonnage avec des échantillons de référence
Une autre solution consiste à utiliser une courbe d'étalonnage élaborée à partir d'au moins quatre échantillons de référence (voir point 7.2.4): une matrice blanche et trois échantillons de référence à 0,5 fois, 1 fois et 2 fois la teneur maximale ou le seuil d'intervention, ce qui permet de se passer de la correction par le blanc et le taux de récupération si les propriétés de la matrice des échantillons de référence correspondent à celles des échantillons inconnus. Dans ce cas, la réponse à l'essai correspondant aux deux tiers de la teneur maximale (voir point 7.3) peut être calculée directement à partir de ces échantillons et servir de valeur seuil. Pour la vérification des échantillons dépassant les seuils d'intervention, la valeur seuil doit être un pourcentage approprié de ces seuils d'intervention.
7.1.3.
Les extraits peuvent être séparés en fractions contenant des PCDD/PCDF et des PCB de type dioxine, permettant une indication distincte des teneurs en PCDD/PCDF et en PCB de type dioxine (en BEQ). Il convient d'utiliser de préférence une courbe d'étalonnage du PCB 126 pour évaluer les résultats de la fraction contenant les PCB de type dioxine.
7.1.4.
Le «taux de récupération apparent du bioessai» doit être calculé à partir d'échantillons de référence appropriés avec des profils de congénères représentatifs qui sont proches de la teneur maximale ou du seuil d'intervention et être exprimé en pourcentage de la valeur BEQ par rapport à la valeur TEQ. En fonction du type de bioessai et des TEF ( 27 ) utilisés, les écarts entre les facteurs TEF et REP pour les PCB de type dioxine peuvent entraîner des taux de récupération apparents faibles pour les PCB de type dioxine par rapport aux PCDD/PCDF. Par conséquent, en cas de dosage distinct des PCDD/PCDF et des PCB de type dioxine, les taux de récupération apparents du bioessai doivent être de 20 à 60 % pour les PCB de type dioxine et de 50 à 130 % pour les PCDD/PCDF (les plages s'appliquent pour la courbe d'étalonnage de la TCDD). Comme la contribution des PCB de type dioxine à la somme des PCDD/PCDF et des PCB de type dioxine peut varier en fonction des matrices et des échantillons, les taux de récupération apparents du bioessai pour la somme des PCDD/PCDF et des PCB de type dioxine reflètent ces plages et doivent se situer entre 30 et 130 %. Toute modification substantielle des valeurs TEF aux fins de la législation de l'Union relative aux PCDD/PCDF et aux PCB de type dioxine requiert la révision de ces plages.
7.1.5.
La perte de composés durant la purification doit être vérifiée pendant la validation. Un échantillon blanc enrichi d'un mélange des différents congénères doit faire l'objet d'une purification (n = 3 au moins) et la récupération et la variabilité doivent être vérifiées au moyen d'une méthode de confirmation. Le taux de récupération doit se situer entre 60 et 120 %, surtout pour les congénères contribuant à hauteur de plus de 10 % à la valeur TEQ dans différents mélanges.
7.1.6.
S'agissant de l'inscription des valeurs BEQ dans un rapport, un seuil d'inscription est déterminé à partir des échantillons de matrice considérés associant des profils de congénères types, mais pas à partir de la courbe d'étalonnage des étalons, la précision de la plage inférieure de la courbe n'étant pas suffisante. Les effets de l'extraction et de la purification doivent être pris en compte. Le seuil d'inscription doit être établi à un niveau supérieur d'un facteur de trois au moins aux blancs de procédure.
7.2. Utilisation d'échantillons de référence
|
7.2.1. |
Les échantillons de référence représentent la matrice de prélèvement, les profils de congénères et les plages de concentration des PCDD/PCDF et des PCB de type dioxine proches de la teneur maximale ou du seuil d'intervention. |
|
7.2.2. |
Chaque série d'essais doit comporter une matrice blanche ou, à défaut, un blanc de procédure et un échantillon de référence à la teneur maximale ou au seuil d'intervention. Ces échantillons doivent être extraits et analysés au même moment et dans les mêmes conditions. La réponse de l'échantillon de référence doit être nettement plus élevée que celle de l'échantillon blanc, garantissant ainsi la validité de l'essai. Ces échantillons peuvent être utilisés pour corriger du blanc et du taux de récupération. |
|
7.2.3. |
Les échantillons de référence choisis pour corriger du taux de récupération doivent être représentatifs des échantillons de l'essai, ce qui signifie que les profils de congénères ne peuvent pas conduire à une surestimation des teneurs. |
|
7.2.4. |
Des échantillons de référence supplémentaires, d'une concentration égale à 0,5 fois et 2 fois la teneur maximale ou le seuil d'intervention, par exemple, peuvent être inclus pour démontrer l'efficacité de l'essai dans la plage pertinente pour le contrôle de la teneur maximale ou du seuil d'intervention. Agrégés, ces échantillons peuvent servir au calcul des valeurs BEQ dans les échantillons d'essai (voir point 7.1.2.2). |
7.3. Détermination de valeurs seuil
Le lien entre les résultats de bioanalyse en BEQ et les résultats des méthodes de confirmation en TEQ doit être établi, par exemple par des expériences d'étalonnage avec adaptation matricielle, à l'aide d'échantillons de référence enrichis pour atteindre 0, 0,5 fois, 1 fois et 2 fois la teneur maximale, avec six répétitions sur chaque teneur (n = 24). Les facteurs de correction (blanc et taux de récupération) peuvent être estimés à partir de ce lien mais doivent être contrôlés conformément au point 7.2.2.
Des valeurs seuil sont établies pour déterminer la conformité de l'échantillon avec les teneurs maximales ou pour vérifier que les seuils d'intervention, s'ils sont considérés, sont conformes aux teneurs maximales ou seuils d'intervention respectifs établis pour les PCDD/PCDF et pour les PCB de type dioxine pris isolément, ou pour la somme des PCDD/PCDF et des PCB de type dioxine. Elles sont représentées par l'extrémité inférieure de la répartition des résultats de bioanalyse (corrigés du blanc et du taux de récupération) correspondant à la limite de décision de la méthode de confirmation fondée sur un niveau de confiance de 95 %, soit un taux de faux conformes inférieur à 5 %, et sur un RSDR inférieur à 25 %. La limite de décision de la méthode de confirmation est la teneur maximale, compte tenu de l'incertitude de mesure élargie.
La valeur seuil (en BEQ) peut être calculée selon l'une des formules énoncées aux points 7.3.1, 7.3.2 et 7.3.3 (voir graphique 1).
|
7.3.1. |
Utilisation de la plage inférieure de l'intervalle de prédiction de 95 % à la limite de décision de la méthode de confirmation.
où
|

|
7.3.2. |
Calcul à partir des résultats de bioanalyse (corrigés du blanc et du taux de récupération) de multiples analyses d'échantillons (n ≥ 6) contaminés à hauteur de la limite de décision de la méthode de confirmation, en tant qu'extrémité inférieure de la répartition des données à la valeur BEQ moyenne correspondante: Valeur seuil = BEQDL – 1,64 × SDR où
|
|
7.3.3. |
Mesure en tant que valeur moyenne des résultats de bioanalyse (en BEQ, corrigée du blanc et du taux de récupération) à partir de l'analyse multiple d'échantillons (n ≥ 6) contaminés aux deux tiers de la teneur maximale ou du seuil d'intervention, sachant que ce niveau sera proche de la valeur seuil déterminée conformément au point 7.3.1 ou 7.3.2: Calcul des valeurs seuil sur la base d'un niveau de confiance de 95 %, soit un taux de faux conformes inférieur à 5 %, et d'un RSDR inférieur à 25 %: 1) à partir de la plage inférieure de l'intervalle de prédiction de 95 % à la limite de décision de la méthode de confirmation. 2) à partir de l'analyse multiple d'échantillons (n ≥ 6) contaminés à hauteur de la limite de décision de la méthode de confirmation en tant qu'extrémité inférieure de la répartition des données (représentée dans le graphique par une courbe en cloche) à la valeur BEQ moyenne correspondante.
|

|
7.3.4. |
Restriction aux valeurs seuil Les valeurs seuil fondées sur la valeur BEQ calculées à partir du RSDR atteint durant la validation à l'aide d'un nombre limité d'échantillons de matrices/profils de congénères différents peuvent être supérieures aux teneurs maximales ou aux seuils d'intervention fondés sur la valeur TEQ en raison d'une plus grande fiabilité que celle qu'il est possible d'atteindre dans les analyses de routine lorsqu'un spectre inconnu de profils de congénères possibles doit être contrôlé. Dans de tels cas, les valeurs seuil sont calculées à partir d'un RSDR égal à 25 %, ou, de préférence, aux deux tiers de la teneur maximale ou du seuil d'intervention. |
7.4. Caractéristiques de performance
|
7.4.1. |
Étant donné qu'aucun étalon interne ne peut être utilisé dans les méthodes de bioanalyse, des tests de répétabilité de ces méthodes doivent être effectués pour obtenir des données sur l'écart type au sein des séries d'essais et entre elles. La répétabilité doit être inférieure à 20 % et la reproductibilité intralaboratoire inférieure à 25 %. Ce calcul doit être fondé sur les valeurs calculées en BEQ après correction par le blanc et le taux de récupération. |
|
7.4.2. |
Dans le cadre de la procédure de validation, l'essai doit permettre de distinguer un échantillon blanc d'une teneur à la valeur seuil, permettant ainsi l'identification des échantillons au-dessus de la valeur seuil correspondante (voir point 7.1.2). |
|
7.4.3. |
Les composés cibles, les interférences potentielles et les valeurs maximales tolérées pour le blanc doivent être définis. |
|
7.4.4. |
L'écart type relatif de la concentration calculée à partir des réponses (possible uniquement dans la plage de travail) d'un triple dosage d'un extrait d'échantillon ne peut être supérieur à 15 %. |
|
7.4.5. |
Les résultats non corrigés du ou des échantillons de référence exprimés en BEQ (blanc et teneur maximale ou seuil d'intervention) doivent être utilisés pour évaluer les performances de la méthode de bioanalyse dans un intervalle de temps constant. |
|
7.4.6. |
Il convient de réaliser et de vérifier des cartes de contrôle qualité pour les blancs de procédure et chaque type d'échantillon de référence afin de s'assurer que la performance analytique est conforme aux prescriptions, notamment pour les blancs de procédure en ce qui concerne la différence minimale requise par rapport à l'extrémité inférieure de la plage de travail et pour les échantillons de référence en ce qui concerne la reproductibilité intralaboratoire. Les blancs de procédure doivent être bien contrôlés en vue d'éviter les faux conformes lorsqu'ils sont retranchés. |
|
7.4.7. |
Les résultats, obtenus au moyen des méthodes de confirmation, des échantillons suspects et de 2 à 10 % des échantillons conformes (au minimum vingt échantillons par matrice) sont collectés et utilisés pour l'évaluation des performances de la méthode de dépistage et du lien entre les valeurs BEQ et TEQ. Cette base de données peut être utilisée aux fins de la réévaluation des valeurs seuil applicables aux échantillons de routine pour les matrices validées. |
|
7.4.8. |
Les bonnes performances des méthodes peuvent également être démontrées à l'aide d'essais interlaboratoires. Les résultats des échantillons analysés dans des essais interlaboratoires, couvrant une concentration atteignant, par exemple, jusqu'à deux fois la teneur maximale, peuvent également faire partie de l'évaluation du taux de faux conformes, si un laboratoire est en mesure de démontrer ses bonnes performances. Les échantillons couvrent les profils de congénères les plus fréquents, représentant différentes sources. |
|
7.4.9. |
En cas de crise, les valeurs seuil peuvent être réévaluées, reflétant mieux la matrice et les profils de congénères particuliers de ce cas précis. |
8. Inscription des résultats dans un rapport
8.1. Méthodes de confirmation
|
8.1.1. |
Les résultats d'analyse comprennent les teneurs en congénères individuels des PCDD/PCDF et des PCB de type dioxine et les valeurs TEQ sont indiquées en estimation inférieure, estimation supérieure et estimation intermédiaire, afin que soit consigné un maximum de données, ce qui permet une interprétation des résultats en fonction de prescriptions spécifiques. |
|
8.1.2. |
Le rapport doit mentionner la méthode utilisée pour extraire les PCDD/PCDF et les PCB de type dioxine. |
|
8.1.3. |
Les taux de récupération des étalons internes individuels doivent être mentionnés s'ils se situent en dehors de la plage mentionnée au point 6.2.5 ou si la teneur maximale est dépassée (dans ce cas, les taux de récupération doivent être fournis pour l'une des deux analyses faites en double). Dans tous les autres cas, ils doivent être communiqués sur demande. |
|
8.1.4. |
L'incertitude de mesure élargie doit également être mentionnée dans le rapport, car ce paramètre doit être pris en compte lorsqu'il s'agit de déterminer la conformité d'un échantillon. Par conséquent, les résultats de l'analyse sont consignés sous la forme x +/– U, où x est le résultat de l'analyse et U l'incertitude de mesure élargie calculée au moyen d'un facteur d'élargissement de 2 qui donne un niveau de confiance d'environ 95 %. En cas de dosage distinct des PCDD/PCDF et des PCB de type dioxine, la somme des estimations de l'incertitude élargie des résultats d'analyse distincts concernant les PCDD/PCDF et les PCB de type dioxine doit être utilisée pour la somme des PCDD/PCDF et des PCB de type dioxine. |
|
8.1.5. |
Les résultats doivent être exprimés dans les mêmes unités et par au moins le même nombre de chiffres significatifs que les teneurs maximales établies dans la directive 2002/32/CE. |
8.2. Méthodes bioanalytiques de dépistage
|
8.2.1. |
Le dépistage donne un résultat qui doit être énoncé comme étant «conforme» ou «suspecté d'être non conforme» («suspect») dans le rapport. |
|
8.2.2. |
Le dépistage peut aussi donner, pour les PCDD/PCDF et/ou les PCB de type dioxine, un résultat exprimé en BEQ, et non en TEQ. |
|
8.2.3. |
Les échantillons dont la réponse est au-dessous du seuil d'inscription doivent être énoncés comme étant «sous le seuil d'inscription». Les échantillons dont la réponse est au-dessus de la plage de travail doivent être déclarés comme étant «au-dessus de la plage de travail» et le niveau correspondant à l'extrémité supérieure de la plage de travail doit être indiqué en BEQ. |
|
8.2.4. |
Pour chaque type de matrice de prélèvement, le rapport doit mentionner la teneur maximale ou le seuil d'intervention sur lequel repose l'évaluation. |
|
8.2.5. |
Le rapport doit mentionner le type d'essai appliqué, le principe de base de l'essai et le type d'étalonnage. |
|
8.2.6. |
Le rapport doit mentionner la méthode utilisée pour extraire les PCDD/PCDF et les PCB de type dioxine. |
|
8.2.7. |
Dans le cas d'échantillons suspectés d'être non conformes, le rapport doit comprendre une note sur les mesures à prendre. La concentration de PCDD/PCDF et la somme des PCDD/PCDF et des PCB de type dioxine dans ces échantillons présentant des teneurs élevées doivent être déterminées/confirmées au moyen d'une méthode de confirmation. |
|
8.2.8. |
Les résultats non conformes ne sont mentionnés dans le rapport que sur la base de l'analyse de confirmation. |
8.3. Méthodes physico-chimiques de dépistage
|
8.3.1. |
Le dépistage donne un résultat qui doit être énoncé comme étant «conforme» ou «suspecté d'être non conforme» («suspect») dans le rapport. |
|
8.3.2. |
Pour chaque type de matrice de prélèvement, le rapport doit mentionner la teneur maximale ou le seuil d'intervention sur lequel repose l'évaluation. |
|
8.3.3. |
En outre, les teneurs en congénères individuels des PCDD/PCDF et des PCB de type dioxine et les valeurs TEQ indiquées en estimation inférieure, estimation supérieure et estimation intermédiaire peuvent être données. Les résultats doivent être exprimés dans les mêmes unités et par au moins le même nombre de chiffres significatifs que les teneurs maximales établies dans la directive 2002/32/CE. |
|
8.3.4. |
Les taux de récupération des étalons internes individuels doivent être mentionnés s'ils se situent en dehors de la plage mentionnée au point 6.2.5 ou si la teneur maximale est dépassée (dans ce cas, les taux de récupération doivent être fournis pour l'une des deux analyses faites en double). Dans tous les autres cas, ils doivent être communiqués sur demande. |
|
8.3.5. |
Le rapport doit mentionner la méthode CG-SM appliquée. |
|
8.3.6. |
Le rapport doit mentionner la méthode utilisée pour extraire les PCDD/PCDF et les PCB de type dioxine. |
|
8.3.7. |
Dans le cas d'échantillons suspectés d'être non conformes, le rapport doit comprendre une note sur les mesures à prendre. La concentration de PCDD/PCDF et la somme des PCDD/PCDF et des PCB de type dioxine dans ces échantillons présentant des teneurs élevées doivent être déterminées/confirmées au moyen d'une méthode de confirmation. |
|
8.3.8. |
La non-conformité ne peut être décidée qu'après une analyse de confirmation. |
CHAPITRE III
Préparation des échantillons et prescriptions applicables aux méthodes d'analyse à utiliser pour le contrôle officiel des teneurs en PCB autres que ceux de type dioxine des aliments pour animaux
1. Champ d'application
Les prescriptions du présent chapitre s'appliquent aux analyses d'aliments pour animaux effectuées aux fins du contrôle officiel des teneurs en PCB autres que ceux de type dioxine, ainsi qu'à d'autres fins réglementaires, y compris les contrôles effectués par l'exploitant du secteur de l'alimentation animale pour veiller au respect des dispositions du règlement (CE) no 183/2005.
2. Méthodes de détection applicables
Chromatographie en phase gazeuse/détection à capture d'électrons (CG-DCE), CG-SMBR, CG-SM/SM, CG-SMHR ou méthodes équivalentes.
3. Identification et confirmation des analytes considérés
|
3.1. |
Temps de rétention relatif par rapport aux étalons internes ou aux étalons de référence (écart admissible de ± 0,25 %). |
|
3.2. |
Séparation par chromatographie en phase gazeuse des PCB autres que ceux de type dioxine des substances interférentes, surtout les PCB coélués, notamment si les teneurs des échantillons sont dans les limites légales et si la non-conformité doit être confirmée ( 28 ). |
|
3.3. |
Prescriptions applicables aux techniques de CG-SM Il s'agit de suivre au moins le nombre suivant d'ions moléculaires ou d'ions caractéristiques du cluster moléculaire: a) deux ions spécifiques pour la SMHR; b) trois ions spécifiques pour la SMBR; c) deux ions précurseurs spécifiques et, pour chacun d'eux, un ion de fragmentation spécifique produit pour la SM-SM. Tolérances maximales admises applicables aux rapports isotopiques des fragments de masse sélectionnés: écart relatif entre le rapport de l'ion cible (l'ion recherché le plus abondant) et du ou des ions qualificateurs et le rapport théorique de ces ions ou celui déterminé grâce à un standard d'étalonnage: ± 15 %. |
|
3.4. |
Prescriptions applicables aux techniques de CG-DCE Les résultats dépassant la teneur maximale doivent être confirmés avec deux colonnes de CG présentant des phases stationnaires de polarité différente. |
4. Démonstration des performances de la méthode
Les performances de la méthode doivent être validées dans la plage autour de la teneur maximale (0,5 à 2 fois la teneur maximale) avec un coefficient de variation acceptable pour les analyses répétées (voir prescriptions relatives à la fidélité intermédiaire au point 9).
5. Limite de quantification
La somme des limites de quantification ( 29 ) des PCB autres que ceux de type dioxine ne peut être supérieure à un tiers de la teneur maximale ( 30 ).
6. Contrôle qualité
Essais à blanc, analyse d'échantillons enrichis, échantillons servant au contrôle qualité, participation à des études interlaboratoires sur les matrices pertinentes à intervalles réguliers.
7. Mesure des taux de récupération
|
7.1. |
Des étalons internes appropriés présentant des propriétés physico-chimiques comparables à celles des analytes considérés doivent être utilisés. |
|
7.2. |
Ajout d'étalons internes: ajout dans les échantillons (avant l'extraction et la purification). |
|
7.3. |
Prescriptions applicables aux méthodes recourant aux six congénères des PCB autres que ceux de type dioxine marqués d'un isotope: a) les résultats doivent être corrigés des taux de récupération des étalons internes; b) les taux de récupération des étalons internes marqués d'un isotope doivent se situer entre 60 et 120 %; c) les taux de récupération inférieurs ou supérieurs pour les congénères individuels avec une contribution à la somme des PCB autres que ceux de type dioxine inférieure à 10 % sont acceptables. |
|
7.4. |
Prescriptions applicables aux méthodes ne recourant pas à l'ensemble des six étalons internes marqués d'un isotope ou recourant à d'autres étalons internes: a) le taux de récupération du ou des étalons internes doit être mesuré pour chaque échantillon; b) les taux de récupération du ou des étalons internes doivent se situer entre 60 et 120 %; c) les résultats doivent être corrigés des taux de récupération des étalons internes; |
|
7.5. |
Les taux de récupération des congénères non marqués doivent être vérifiés à l'aide d'échantillons enrichis ou d'échantillons de contrôle qualité présentant des concentrations de l'ordre de la teneur maximale. Les taux de récupération pour ces congénères sont réputés acceptables s'ils se situent entre 60 et 120 %. |
8. Prescriptions applicables aux laboratoires
Conformément aux dispositions du règlement (CE) no 882/2004, les laboratoires doivent être accrédités par un organisme habilité qui se conforme au guide ISO 58, de manière à garantir qu'ils appliquent les procédures d'assurance qualité à leurs analyses. Les laboratoires doivent être accrédités selon la norme EN ISO/CEI 17025. En outre, les principes énoncés dans les directives techniques pour l'estimation de l'incertitude de mesure et les limites de quantification pour l'analyse des PCB doivent être suivis lorsqu'ils sont applicables ( 31 ).
9. Caractéristiques des performances: critères afférents à la somme des PCB autres que ceux de type dioxine à la teneur maximale
|
|
Spectrométrie de masse par dilution isotopique (1) |
Autres techniques |
|
Justesse |
De – 20 % à + 20 % |
De – 30 % à + 30 % |
|
Fidélité intermédiaire (RSD %) |
≤ 15 % |
≤ 20 % |
|
Différence entre l'estimation supérieure et l'estimation inférieure |
≤ 20 % |
≤ 20 % |
|
(1) Les six analogues marqués au 13C doivent être utilisés comme étalons internes. |
||
10. Inscription des résultats dans un rapport
|
10.1. |
Les résultats d'analyse comprennent les teneurs en congénères individuels des PCB autres que ceux de type dioxine et la somme des PCB autres que ceux de type dioxine et sont indiqués en estimation inférieure, estimation supérieure et estimation intermédiaire, afin que soit consigné un maximum de données, ce qui permet une interprétation des résultats en fonction de prescriptions spécifiques. |
|
10.2. |
Le rapport mentionne la méthode utilisée pour extraire les PCB. |
|
10.3. |
Les taux de récupération des étalons internes individuels doivent être fournis s'ils se situent en dehors de la plage mentionnée au point 7 ou si la teneur maximale est dépassée. Dans tous les autres cas, ils doivent être fournis sur demande. |
|
10.4. |
L'incertitude de mesure élargie doit également être inscrite dans le rapport, car ce paramètre doit être pris en compte lorsqu'il s'agit de déterminer la conformité d'un échantillon. Par conséquent, les résultats de l'analyse sont consignés sous la forme x +/– U, où x est le résultat de l'analyse et U l'incertitude de mesure élargie calculée au moyen d'un facteur d'élargissement de 2 qui donne un niveau de confiance d'environ 95 %. |
|
10.5. |
Les résultats doivent être exprimés dans les mêmes unités et par au moins le même nombre de chiffres significatifs que les teneurs maximales établies dans la directive 2002/32/CE. |
ANNEXE VI
MÉTHODES D’ANALYSE APPLICABLES EN MATIÈRE D’IDENTIFICATION DES CONSTITUANTS D’ORIGINE ANIMALE POUR LE CONTRÔLE OFFICIEL DES ALIMENTS POUR ANIMAUX
1. OBJET ET CHAMP D’APPLICATION
L’identification des constituants d’origine animale dans les aliments pour animaux doit être effectuée à l’aide de la microscopie optique ou d’une réaction d’amplification en chaîne par polymérase (PCR), conformément aux dispositions prévues dans la présente annexe.
Ces deux méthodes permettent de détecter la présence de constituants d’origine animale dans les matières premières pour aliments des animaux et dans les aliments composés pour animaux. Toutefois, elles ne permettent pas de calculer la quantité de ces constituants dans les matières premières pour aliments des animaux et dans les aliments composés pour animaux. La limite de détection des deux méthodes est inférieure à 0,1 % (p/p).
L’amplification en chaîne par polymérase permet d’identifier le groupe taxonomique des constituants d’origine animale présents dans les matières premières pour aliments des animaux et dans les aliments composés pour animaux.
Ces méthodes doivent être appliquées pour le contrôle du respect des interdictions prévues à l’article 7, paragraphe 1, et à l’annexe IV du règlement (CE) no 999/2001, ainsi qu’à l’article 11, paragraphe 1, du règlement (CE) no 1069/2009.
En fonction du type d’aliment pour animaux soumis aux essais, ces méthodes peuvent être utilisées, suivant un protocole opérationnel unique, individuellement ou conjointement conformément aux procédures opérationnelles normalisées établies par le laboratoire de référence de l’Union européenne pour la détection de protéines animales dans les aliments pour animaux (EURL-AP) et publiées sur son site internet ( 32 ).
2. MÉTHODES
2.1. Microscopie optique
2.1.1. Principe
Les constituants d’origine animale susceptibles d’être présents dans les matières premières pour aliments des animaux et dans les aliments composés pour animaux envoyés pour analyse sont identifiés sur la base de caractéristiques typiques et identifiables au microscope telles que les fibres musculaires et autres particules de viande, les cartilages, les os, la corne, les poils, les soies, le sang, les plumes, les coquilles d’œuf, les arêtes et les écailles de poisson.
2.1.2. Réactifs et appareillage
2.1.2.1. Réactifs
2.1.2.1.1. Agent de concentration
2.1.2.1.1.1. Tétrachloréthylène (densité relative 1,62)
2.1.2.1.2. Réactif de coloration
|
2.1.2.1.2.1. |
Solution de rouge d’alizarine (diluer 2,5 ml d’acide chlorhydrique 1 M dans 100 ml d’eau et ajouter 200 mg de rouge d’alizarine à cette solution) |
2.1.2.1.3. Milieux de montage
2.1.2.1.3.1. Lessive de soude et de potasse (NaOH à 2,5 % p/v ou KOH à 2,5 % p/v)
2.1.2.1.3.2. Glycérol (non dilué, viscosité: 1 490 cP)
2.1.2.1.3.3. Norland® Optical Adhesive 65 (viscosité: 1 200 cP) ou résine ayant des propriétés équivalentes pour la préparation de lames permanentes
2.1.2.1.4. Milieux de montage avec propriétés de coloration
|
2.1.2.1.4.1. |
Solution de lugol (dissoudre 2 g d’iodure de potassium dans 100 ml d’eau et ajouter 1 g d’iode en agitant fréquemment) |
|
2.1.2.1.4.2. |
Réactif cystinique (2 g d’acétate de plomb, 10 g NaOH/100 ml d’eau) |
|
2.1.2.1.4.3. |
Liqueur de Fehling [préparée avant l’utilisation à partir de parts égales (1/1) de deux solutions-mères A et B. Solution A: dissoudre 6,9 g de sulfate de cuivre (II) pentahydraté dans 100 ml d’eau. Solution B: dissoudre 34,6 g de tartrate double de sodium et de potassium tétrahydraté et 12 g de NaOH dans 100 ml d’eau] |
|
2.1.2.1.4.4. |
Tétraméthylbenzidine/Peroxyde d’hydrogène [dissoudre 1 g de 3,3’,5,5’-tétraméthylbenzidine (TMB) dans 100 ml d’acide acétique glacial et 150 ml d’eau. Avant l’utilisation, mélanger quatre parts de cette solution de TMB et une part de peroxyde d’hydrogène à 3 %] |
2.1.2.1.5. Agents de rinçage
2.1.2.1.5.1. Éthanol ≥ 96 % (qualité technique)
2.1.2.1.5.2. Acétone (qualité technique)
2.1.2.1.6. Réactif de blanchiment
2.1.2.1.6.1. Solution commerciale d’hypochlorite de sodium (9-14 % de chlore actif)
2.1.2.2. Appareillage
2.1.2.2.1. Balance de précision à 0,001 g
2.1.2.2.2. Équipement de broyage: broyeur ou mortier
2.1.2.2.3. Tamis à mailles carrées de 0,25 mm et 1 mm de largeur
|
2.1.2.2.4. |
Ampoule à décanter conique en verre d’une capacité de 250 ml munie d’un robinet en téflon ou en verre rodé à la base du cône. Le diamètre de l’ouverture du robinet doit être supérieur ou égal à 4 mm. Un bécher de décantation à fond conique peut également être utilisé, à condition que le laboratoire ait démontré que les niveaux de détection sont équivalents à ceux obtenus en utilisant l’ampoule à décanter conique en verre. 
|
|
2.1.2.2.5. |
Microscope stéréoscopique couvrant une plage de grossissement final allant de 6,5 à 40 fois au minimum |
|
2.1.2.2.6. |
Microscope composé à fond clair par éclairage à transmission couvrant une plage de grossissement final allant de 100 à 400 fois au minimum. Un microscope en lumière polarisée ou à contraste interférentiel différentiel peut également être utilisé |
|
2.1.2.2.7. |
Verrerie courante de laboratoire |
|
2.1.2.2.8. |
Matériel pour la préparation sur lame: lames de microscope classiques, lames creuses, lamelles (20 × 20 mm), brucelles, spatule fine |
2.1.3. Échantillonnage et préparation des échantillons
2.1.3.1. Échantillonnage
Utiliser un échantillon représentatif prélevé conformément aux dispositions fixées à l’annexe I
2.1.3.2. Précautions à prendre
Afin d’éviter une contamination croisée en laboratoire, tous les équipements recyclables doivent être soigneusement nettoyés avant l’emploi. Les éléments de l’ampoule à décanter doivent être démontés avant le nettoyage. Les éléments de l’ampoule à décanter et la verrerie doivent être prélavés manuellement puis lavés en machine. Les tamis doivent être nettoyés à l’aide d’une brosse à poils synthétiques durs. Un dernier nettoyage des tamis avec de l’acétone et de l’air comprimé est recommandé après le tamisage de matières grasses telles que des farines de poisson.
2.1.3.3. Préparation des échantillons autres que les matières grasses ou les huiles
|
2.1.3.3.1. |
Séchage d’échantillons: les échantillons présentant une teneur en humidité supérieure à 14 % doivent être séchés avant le traitement. |
|
2.1.3.3.2. |
Prétamisage des échantillons: il est recommandé de prétamiser les aliments pour animaux en granulés et les bouchons à l’aide d’un tamis à mailles de 1 mm puis de préparer et d’analyser les deux fractions obtenues comme deux échantillons distincts. |
|
2.1.3.3.3. |
Sous-échantillonnage et broyage: au moins 50 g de l’échantillon doivent être séparés en sous-échantillons destinés à être analysés puis broyés. |
|
2.1.3.3.4. |
Extraction et préparation du résidu: transvaser une portion de 10 g (exactitude de 0,01 g) du sous-échantillon broyé dans l’ampoule à décanter ou le bécher à décantation à fond conique et ajouter 50 ml de tétrachloréthylène. La portion transvasée dans l’ampoule est limitée à 3 g dans le cas des farines de poissons ou d’autres produits d’origine animale purs, d’ingrédients minéraux ou de prémélanges générant plus de 10 % de résidus. Agiter vigoureusement le mélange pendant au moins 30 secondes et ajouter avec précaution au moins 50 ml de tétrachloréthylène en lavant la surface intérieure de l’ampoule pour éliminer les particules adhérentes. Laisser le mélange obtenu se décanter pendant au moins 5 minutes avant de séparer le résidu en ouvrant le robinet. Si un bécher de décantation à fond conique est utilisé, agiter vigoureusement le mélange pendant au moins 15 secondes et laver soigneusement la surface intérieure avec au moins 10 ml de tétrachloréthylène propre pour éliminer les particules adhérant aux parois du bécher. Laisser le mélange se décanter pendant 3 minutes et agiter à nouveau pendant 15 secondes puis laver soigneusement la surface intérieure avec au moins 10 ml de tétrachloréthylène propre pour éliminer les particules adhérant aux parois du bécher. Laisser le mélange obtenu se décanter pendant au moins 5 minutes puis retirer la fraction liquide, l’éliminer en la laissant soigneusement se décanter et en prenant soin de ne rien perdre du résidu. Le résidu total doit être séché puis pesé (exactitude de 0,001 g). Si le résidu est constitué de plus de 5 % de particules supérieures à 0,5 mm, il doit être passé à travers un tamis de 0,25 mm, et les deux fractions obtenues doivent être examinées. |
|
2.1.3.3.5. |
Extraction et préparation des matières flottantes: après la récupération du résidu par la méthode décrite ci-dessus, deux phases devraient rester dans l’ampoule à décanter: une phase liquide constituée de tétrachloréthylène et une phase solide composée de matières flottantes, laquelle est récupérée par le versement complet du tétrachloréthylène hors de l’ampoule en ouvrant le robinet. L’ampoule à décanter doit être retournée, et les matières flottantes doivent être transvasées dans une grande boîte de Pétri et séchées à l’air dans une hotte de laboratoire. Si les matières flottantes sont constituées de plus de 5 % de particules supérieures à 0,5 mm, elles doivent être passées à travers un tamis de 0,25 mm et les deux fractions obtenues doivent être examinées. |
|
2.1.3.3.6. |
Préparation des matières premières: préparer une portion d’au moins 5 g du sous-échantillon broyé. Si la matière est constituée de plus de 5 % de particules supérieures à 0,5 mm, elle doit être passée à travers un tamis de 0,25 mm et les deux fractions obtenues doivent être examinées. |
2.1.3.4. Préparation des échantillons constitués de matières grasses ou d’huiles
Le protocole suivant doit être respecté pour la préparation des échantillons constitués de matières grasses ou d’huiles:
— s’il s’agit de graisse solide, chauffer celle-ci dans un four jusqu’à ce qu’elle devienne liquide,
— au moyen d’une pipette, transvaser 40 ml de graisse ou d’huile du fond de l’échantillon dans un tube de centrifugation,
— centrifuger pendant 10 minutes à 4 000 tours/minute,
— si la graisse s’est solidifiée pendant la centrifugation, la réchauffer au four jusqu’à ce qu’elle redevienne liquide,
— centrifuger une nouvelle fois pendant 5 minutes à 4 000 tours/minute,
— à l’aide d’une petite cuillère ou d’une spatule, transvaser une moitié des impuretés obtenues sur des lames microscopiques pour examen; il est recommandé d’utiliser du glycérol comme milieu de montage,
— utiliser les impuretés restantes pour la préparation du résidu, comme décrit au point 2.1.3.3.
2.1.3.5. Utilisation de réactifs de coloration
Pour faciliter l’identification correcte des constituants d’origine animale, l’opérateur peut utiliser des réactifs de coloration au cours de la préparation de l’échantillon, conformément aux orientations formulées par le laboratoire de référence de l’Union européenne pour la détection de protéines animales dans les aliments pour animaux et publiées sur son site internet.
Si la solution de rouge d’alizarine est utilisée pour la coloration du résidu, le protocole suivant doit être appliqué:
— transvaser le résidu séché dans une éprouvette en verre et le rincer deux fois avec environ 5 ml d’éthanol (agiter chaque fois au vortex pendant 30 secondes, laisser le solvant se décanter pendant environ 1 minute 30 puis l’éliminer),
— blanchir le résidu avec au moins 1 ml de solution d’hypochlorite de sodium; laisser réagir pendant 10 minutes; remplir l’éprouvette d’eau, laisser le résidu se décanter pendant 2 à 3 minutes puis éliminer doucement l’eau et les particules en suspension,
— rincer deux fois le résidu avec environ 10 ml d’eau (agiter au vortex pendant 30 secondes, laisser se décanter et, chaque fois, éliminer l’eau),
— ajouter 2 à 10 gouttes de solution de rouge d’alizarine et agiter le mélange au vortex; laisser réagir pendant 30 secondes et rincer deux fois le résidu coloré avec environ 5 ml d’éthanol, puis une nouvelle fois avec de l’acétone (agiter chaque fois au vortex pendant 30 secondes, laisser le solvant se décanter environ une minute puis l’éliminer);
— sécher le résidu.
2.1.4. Examen au microscope
2.1.4.1. Préparation des lames
Les lames microscopiques sont préparées à partir du résidu et, selon le choix de l’opérateur, à partir des matières flottantes ou de la matière première. Si un tamisage a été effectué au cours de la préparation de l’échantillon, les deux fractions obtenues (la fraction fine et la fraction grossière) doivent être préparées. Les prises d’essai des fractions étalées sur les lames doivent être représentatives de la fraction entière.
Un nombre suffisant de lames doit être préparé afin de garantir la réalisation d’un protocole d’examen complet, tel que prévu au point 2.1.4.2.
Les lames microscopiques doivent être montées avec le milieu de montage adéquat, conformément aux procédures opérationnelles normalisées établies par le laboratoire de référence de l’Union européenne pour la détection de protéines animales dans les aliments pour animaux et publiées sur son site internet. Elles sont recouvertes de lamelles.
2.1.4.2. Protocoles d’observation pour la détection de particules animales dans les aliments composés pour animaux et les matières premières pour aliments des animaux
Les lames microscopiques sont observées conformément au protocole d’observation figurant dans le schéma 1 pour les aliments composés pour animaux et les matières premières pour aliments des animaux autres que les farines de poisson pures, ou dans le schéma 2 pour les farines de poisson pures.
Le résidu et, selon le choix de l’opérateur, les matières flottantes ou la matière première, doivent être observés au microscope composé. Les grosses fractions peuvent en outre être examinées au microscope stéréoscopique. Chaque lame doit être observée entièrement à différents grossissements.
Le nombre minimal de lames à observer à chaque étape du protocole d’observation doit être rigoureusement respecté, à moins que la totalité du matériau de la fraction ne permette pas d’atteindre le nombre de lames prévu. Six lames au plus doivent être observées pour chaque détermination.
Afin de déterminer plus facilement la nature et l’origine des particules, l’opérateur peut utiliser des outils d’aide tels que les systèmes d’aide à la décision, les bibliothèques d’images et les échantillons de référence.

2.1.4.3. Nombre de déterminations
Si, à la suite d’une première détermination effectuée conformément au protocole d’observation prévu dans le schéma 1 ou 2, selon le cas, aucune particule animale d’une nature donnée (c’est-à-dire dérivée d’animaux terrestres ou de poissons) n’est détectée, aucune détermination supplémentaire n’est nécessaire, et le résultat de l’analyse doit être rapporté selon les libellés prévus au point 2.1.5.1.
Si, à la suite d’une première détermination effectuée conformément au protocole d’observation prévu dans le schéma 1 ou 2, selon le cas, le nombre total de particules animales d’une nature donnée (c’est-à-dire dérivées d’animaux terrestres ou de poissons) qui sont détectées varie de 1 à 5, une deuxième détermination doit être effectuée à partir d’un nouveau sous-échantillon de 50 g. Si, à la suite de cette deuxième détermination, le nombre de particules animales de cette même nature qui sont détectées varie de 0 à 5, le résultat de l’analyse doit être rapporté selon les libellés prévus au point 2.1.5.2, sinon une troisième détermination doit être effectuée à partir d’un nouveau sous-échantillon de 50 g. Toutefois, si, à la suite des deux premières déterminations, la somme des particules d’une nature donnée qui sont détectées sur l’ensemble des deux déterminations est supérieure à 15, aucune détermination supplémentaire n’est nécessaire et le résultat de l’analyse doit être directement rapporté selon les libellés prévus au point 2.1.5.3. Si, à l’issue de la troisième détermination, la somme des particules animales d’une nature donnée qui sont détectées sur l’ensemble des trois déterminations est supérieure à 15, le résultat de l’analyse doit être rapporté selon les libellés prévus au point 2.1.5.3. Dans tout autre cas, le résultat de l’analyse doit être rapporté selon les libellés prévus au point 2.1.5.2.
Si, à la suite d’une première détermination effectuée conformément au protocole d’observation prévu dans le schéma 1 ou 2, selon le cas, plus de cinq particules animales d’une nature donnée (c’est-à-dire dérivées d’animaux terrestres ou de poissons) sont détectées, le résultat de l’analyse doit être communiqué selon les libellés prévus au point 2.1.5.3.
2.1.5. Expression des résultats
Lorsqu’il rapporte les résultats, le laboratoire doit indiquer le type de matériel sur lequel l’analyse a été conduite (résidu, matières flottantes ou matière première) et le nombre de déterminations effectuées.
Le rapport du laboratoire doit contenir au minimum des informations concernant la présence de constituants dérivés d’animaux terrestres et de poissons.
Les différents cas doivent être présentés de la façon suivante:
|
2.1.5.1. |
Aucune particule animale d’une nature donnée n’est détectée: — l’échantillon soumis à l’analyse ne contient aucune particule dérivée d’un animal terrestre détectable au microscope optique, — l’échantillon soumis à l’analyse ne contient aucune particule dérivée d’un poisson détectable au microscope optique. |
|
2.1.5.2. |
Entre une et cinq particules animales d’une nature donnée sont détectées en moyenne: — l’échantillon soumis à l’analyse ne contient, en moyenne, par détermination, pas plus de cinq particules dérivées d’animaux terrestres détectables au microscope optique. Les particules ont été identifiées comme étant… [de l’os, du cartilage, des muscles, des poils, de la corne, etc.]. Cette faible présence, inférieure à la limite de détection de la méthode de l’examen microscopique, signifie qu’un risque de faux résultat positif ne peut être exclu. Ou, le cas échéant, — l’échantillon soumis à l’analyse ne contient, en moyenne, par détermination, pas plus de cinq particules dérivées de poissons détectables au microscope optique. Les particules ont été identifiées comme étant… [de l’os, des écailles, du cartilage, des muscles, des otolithes, des branchies, etc.]. Cette faible présence, inférieure à la limite de détection de la méthode de l’examen microscopique, signifie qu’un risque de faux résultat positif ne peut être exclu. En cas de prétamisage de l’échantillon de laboratoire, le rapport de laboratoire mentionne la fraction (fraction tamisée, fraction de granulés ou de bouchons) dans laquelle les particules animales ont été détectées, dans la mesure où seule la détection de particules animales dans la fraction tamisée peut être le signe d’une contamination de l’environnement. |
|
2.1.5.3. |
Plus de cinq particules animales d’une nature donnée sont détectées en moyenne: — l’échantillon soumis à l’analyse contient, en moyenne, par détermination, plus de cinq particules dérivées d’animaux terrestres détectables au microscope optique. Les particules ont été identifiées comme étant… [de l’os, du cartilage, des muscles, des poils, de la corne, etc.]. Ou, le cas échéant, — l’échantillon soumis à l’analyse contient, en moyenne, par détermination, plus de cinq particules dérivées de poissons détectables au microscope optique. Les particules ont été identifiées comme étant … [de l’os, des écailles, du cartilage, des muscles, des otolithes, des branchies, etc.]. En cas de prétamisage de l’échantillon de laboratoire, le rapport de laboratoire mentionne la fraction (fraction tamisée, fraction de granulés ou de bouchons) dans laquelle les particules animales ont été détectées, dans la mesure où seule la détection de particules animales dans la fraction tamisée peut être le signe d’une contamination de l’environnement. |
2.2. Amplification en chaîne par polymérase
2.2.1. Principe
Des fragments d’acide désoxyribonucléique (ADN) d’origine animale susceptibles d’être présents dans les matières premières pour aliments des animaux et les aliments composés pour animaux sont détectés selon une technique d’amplification génique (amplification en chaîne par polymérase, PCR), en ciblant des séquences d’ADN spécifiques par espèce.
L’amplification en chaîne par polymérase nécessite tout d’abord une étape d’extraction de l’ADN. L’extrait d’ADN ainsi obtenu est ensuite soumis à l’étape d’amplification, afin de détecter l’espèce animale visée par l’essai.
2.2.2. Réactifs et appareillage
2.2.2.1. Réactifs
2.2.2.1.1. Réactifs pour l’étape d’extraction de l’ADN
Seuls les réactifs approuvés par le laboratoire de référence de l’Union européenne pour la détection de protéines animales dans les aliments pour animaux et publiés sur son site internet doivent être utilisés.
2.2.2.1.2. Réactifs pour l’étape d’amplification génique
2.2.2.1.2.1. Amorces et sondes
Seules les amorces et les sondes présentant des séquences d’oligonucléotides validées par le laboratoire de référence de l’Union européenne pour la détection de protéines animales dans les aliments pour animaux doivent être utilisées ( 33 ).
2.2.2.1.2.2. Mélange maître
Seules des solutions de mélanges maîtres ne contenant pas de réactifs susceptibles de fausser les résultats en raison de la présence d’ADN animal doivent être utilisées ( 34 ).
2.2.2.1.2.3. Réactifs de décontamination
2.2.2.1.2.3.1. Solution d’acide chlorhydrique (0,1 N)
2.2.2.1.2.3.2. Eau de Javel (solution d’hypochlorite de sodium à 0,15 % de chlore actif)
2.2.2.1.2.3.3. Réactifs de décontamination non corrosifs pour des dispositifs coûteux tels que des balances de précision (exemple: DNA Erase™ de MP Biomedicals)
2.2.2.2. Appareillage
2.2.2.2.1. Balance de précision à 0,001 g
2.2.2.2.2. Équipement de broyage
2.2.2.2.3. Thermocycleur permettant la PCR en temps réel
2.2.2.2.4. Microcentrifugeuse pour tubes de microcentrifugation
2.2.2.2.5. Ensemble de micropipettes permettant d’introduire à la pipette de 1 μl à 1 000 μl
|
2.2.2.2.6. |
Matériel en plastique standard de biologie moléculaire: tubes de microcentrifugation, embouts à filtres pour micropipettes, microplaques adaptées au thermocycleur. |
|
2.2.2.2.7. |
Congélateurs pour la conservation des échantillons et des réactifs |
2.2.3. Échantillonnage et préparation des échantillons
2.2.3.1. Échantillonnage
Utiliser un échantillon représentatif prélevé conformément aux dispositions fixées à l’annexe I.
2.2.3.2. Préparation de l’échantillon
Effectuer la préparation d’échantillons de laboratoire précédant l’extraction de l’ADN dans le respect des exigences fixées à l’annexe II. Au moins 50 g de l’échantillon doivent être séparés en sous-échantillons destinés à être analysés puis broyés.
Conformément à la norme ISO 24276, l’échantillon doit être préparé dans une salle différente de celles qui sont utilisées pour l’extraction de l’ADN et les réactions d’amplification génique.
Préparer deux prises d’essai d’au moins 100 mg chacune.
2.2.4. Extraction de l’ADN
L’extraction de l’ADN doit être effectuée sur chaque prise d’essai préparée suivant les procédures opérationnelles normalisées établies par le laboratoire de référence de l’Union européenne pour la détection de protéines animales dans les aliments pour animaux et publiées sur son site internet.
Conformément à la norme ISO 24276, deux témoins d’extraction doivent être préparés pour chaque série d’extractions:
— un témoin d’extraction à blanc,
— un témoin positif d’extraction de l’ADN.
2.2.5. Amplification génique
L’amplification génique doit être effectuée suivant les méthodes validées pour chaque espèce devant être identifiée. Ces méthodes sont fixées dans les procédures opérationnelles normalisées établies par le laboratoire de l’Union européenne pour la détection de protéines animales dans les aliments pour animaux et publiées sur son site internet. Chaque extrait d’ADN doit être analysé au moins à deux dilutions différentes afin d’évaluer l’inhibition.
Conformément à la norme ISO 24276, deux témoins d’amplification doivent être préparés pour chaque espèce cible:
— un témoin positif de l’ADN cible doit être utilisé pour chaque plaque ou série d’épreuves PCR,
— un témoin du réactif d’amplification (également appelé «témoin négatif») doit être utilisé pour chaque plaque ou série d’épreuves PCR.
2.2.6. Interprétation et expression des résultats
Lorsqu’il rapporte les résultats, le laboratoire doit indiquer au moins la masse des prises d’essai utilisées, la technique d’extraction utilisée, le nombre de déterminations effectuées et la limite de détection de la méthode.
Les résultats ne doivent pas être interprétés et rapportés si le témoin positif d’extraction de l’ADN et le témoin positif de l’ADN cible ne donnent pas de résultats positifs pour la cible faisant l’objet des essais alors que le témoin du réactif d’amplification est négatif.
Si les résultats des deux prises d’essai ne sont pas cohérents, renouveler au moins la phase d’amplification génique. Si le laboratoire soupçonne que les extraits d’ADN peuvent être la cause de l’incohérence des résultats, une nouvelle extraction d’ADN suivie d’une amplification génique doit être effectuée avant l’interprétation des résultats.
L’expression définitive des résultats repose sur l’intégration et l’interprétation des résultats des deux prises d’essai, conformément aux procédures opérationnelles normalisées établies par le laboratoire de référence de l’Union européenne pour la détection de protéines animales dans les aliments pour animaux et publiées sur son site internet.
2.2.6.1. Résultat négatif
Un résultat négatif est rapporté comme suit:
Aucun ADN de X n’a été détecté dans l’échantillon soumis à l’analyse (X étant l’espèce animale ou le groupe d’espèces animales visé par l’essai).
2.2.6.2. Résultats positifs
Un résultat positif est rapporté comme suit:
De l’ADN de X a été détecté dans l’échantillon soumis à l’analyse (X étant l’espèce animale ou le groupe d’espèces animales visé par l’essai).
ANNEXE VII
MÉTHODE DE CALCUL DE LA VALEUR ÉNERGÉTIQUE DES ALIMENTS COMPOSÉS DESTINÉS À LA VOLAILLE
1. Mode de calcul et expression de la valeur énergétique
La valeur énergétique des aliments composés destinés à la volaille est calculée selon la formule ci-après, à partir des pourcentages de certains composants analytiques des aliments; cette valeur est exprimée en mégajoules (MJ), d'énergie métabolisable (EM), corrigée en azote, par kilogramme d'aliment composé, tel quel:
MJ/kg d'EM = 0,1551 × % protéine brute + 0,3431 × % matières grasses brutes + 0,1669 × % amidon + 0,1301 × % sucres totaux (exprimés en saccharose).
2. Tolérances applicables aux valeurs déclarées
Si, à la suite des contrôles officiels, on constate un écart entre le résultat du contrôle et la valeur énergétique déclarée constituant une augmentation ou une diminution de la valeur énergétique de l'aliment, une tolérance de 0,4 MJ/kg d'énergie métabolisable est au moins appliquée.
3. Expression du résultat
Le résultat obtenu, après application de la formule ci-avant, est indiqué à une décimale près.
4. Méthodes d'échantillonnage et d'analyse
Le prélèvement de l'échantillon de l'aliment composé et la détermination des teneurs en composants analytiques indiquées dans la méthode de calcul sont réalisés respectivement selon les méthodes d'échantillonnage et d'analyse communautaires destinées au contrôle officiel des aliments pour animaux.
Doivent être appliqués:
— pour le dosage des matières grasses brutes: le procédé B de la méthode de dosage des matières grasses brutes, prévu à l'annexe III, partie H,
— pour le dosage de l'amidon: la méthode polarimétrique, prévue à l'annexe III, partie L.
ANNEXE VIII
MÉTHODES D'ANALYSE RELATIVES À LA DÉTECTION D'ADDITIFS DONT LA PRÉSENCE N'EST PLUS AUTORISÉE DANS LES ALIMENTS POUR ANIMAUX
Remarques importantes:
Il est permis d'appliquer des méthodes d'analyse plus sensibles que les méthodes d'analyse prévues dans la présente annexe pour détecter des additifs dont la présence n'est plus autorisée dans les aliments pour animaux.
Les méthodes d'analyse décrites dans la présente annexe doivent être appliquées à des fins de confirmation.
A. DOSAGE DU MÉTHYL-BENZOQUATE
7-Benzyloxy-6-butyl-3-méthoxycarbonyl-4-quinolone
1. Objet et champ d'application
La méthode permet de déterminer la teneur en méthyl-benzoquate des aliments pour animaux. La limite de quantification est de 1 mg/kg.
2. Principe
Le méthyl-benzoquate est extrait d'échantillon dans une solution méthanolique d'acide méthanesulfonique. L'extrait est purifié au dichlorométhane, par chromatographie sur résines échangeuses d'ions, puis à nouveau au dichlorométhane. La teneur en méthyl-benzoquate est déterminée par chromatographie liquide haute performance (CLHP) en phase inverse à l'aide d'un détecteur d'ultraviolets.
3. Réactifs
3.1. Dichlorométhane.
3.2. Méthanol, de qualité CLHP.
3.3. Phase mobile de la CLHP
mélange de méthanol (3.2) et d'eau (qualité CLHP) 75 + 25 (v + v).
Filtrer à travers un filtre de 0,22 μm (4.5) et dégazer la solution (par exemple, par traitement aux ultrasons pendant 10 minutes).
3.4. Solution d'acide méthanesulfonique, c = 2 %
Porter 20,0 ml d'acide méthanesulfonique à 1 000 ml par dilution dans du méthanol (3.2).
3.5. Solution d'acide chlorhydrique, c = 10 %
Porter 100 ml d'acide chlorhydrique (ρ201,18 g/ml) à 1 000 ml par dilution dans de l'eau.
3.6. Résine d'Amberlite échangeuse de cations CG-120 (Na), de 100 à 200 mesh
La résine est prétraitée avant l'emploi. Mélanger 100 g de résine avec 500 ml de solution d'acide chlorhydrique (3.5) et porter le mélange à ébullition sur une plaque chauffante, sans cesser d'agiter. Laisser refroidir et décanter l'acide. Filtrer sous vide à travers un filtre en papier. Laver la résine à deux reprises avec 500 ml d'eau, puis avec 250 ml de méthanol (3.2). Rincer la résine avec de nouveau 250 ml de méthanol et sécher par courant d'air à travers le gâteau de filtration. Conserver la résine séchée dans un flacon bouché.
3.7. Substance étalon: méthyl-benzoquate pur (7-benzyloxy-6-butyl-3-méthoxycarbonyl-4-quinolone)
3.7.1.
Peser, à 0,1 mg près, 50 mg de substance étalon (3.7), dissoudre dans une solution d'acide méthanesulfonique (3.4) dans une fiole jaugée de 100 ml, ajuster au trait de jauge et mélanger.
3.7.2.
Transvaser 5,0 ml de solution mère de l'étalon (3.7.1) dans une fiole jaugée de 50 ml, ajuster au trait de jauge avec du méthanol (3.2) et mélanger.
3.7.3.
Dans une série de fioles jaugées de 25 ml, transvaser 1,0 , 2,0 , 3,0 , 4,0 et 5,0 ml de la solution étalon intermédiaire de méthyl-benzoquate (3.7.2). Ajuster au trait de jauge avec la phase mobile (3.3) et mélanger. Ces solutions ont des concentrations respectives de 2,0 , 4,0 , 6,0 , 8,0 et 10,0 μg/ml de méthyl-benzoquate. Les solutions doivent être préparées peu avant d'être utilisées.
4. Appareillage
4.1. Agitateur.
4.2. Évaporateur rotatif.
4.3. Colonne de verre (250 mm × 15 mm) munie d'un robinet et d'un réservoir d'environ 200 ml.
4.4. Équipement pour CLHP avec détecteur d'ultraviolets à longueur d'onde variable ou détecteur à barrettes de diodes.
4.4.1. Colonne de chromatographie liquide: 300 mm × 4 mm, C18, particules de 10 μm, ou équivalent.
4.5. Filtres à membrane, 0,22 μm.
4.6. Filtres à membrane, 0,45 μm.
5. Mode opératoire
5.1. Généralités
5.1.1. Analyser un aliment blanc afin de vérifier l'absence de méthyl-benzoquate ou de substances interférentes.
5.1.2. Un test de récupération doit être effectué en analysant un aliment blanc qui a été supplémenté par ajout d'une quantité de méthyl-benzoquate similaire à celle présente dans l'échantillon. Pour parvenir à une teneur de 15 mg/kg, ajouter 600 μl de la solution mère de l'étalon (3.7.1) à 20 g d'aliment blanc, mélanger et attendre 10 minutes avant de procéder à l'extraction (5.2).
Dans le cadre de cette méthode, l'aliment blanc doit être d'un type similaire dans sa composition à celui de l'échantillon et, lors de son analyse, il ne doit pas être détecté de méthyl-benzoquate.
5.2. Extraction
Peser, à 0,01 g près, 20 g de l'échantillon préparé et transférer dans une fiole conique de 250 ml. Ajouter 100,0 ml d'acide méthanesulfonique (3.4) et agiter mécaniquement (4.1) pendant 30 minutes. Filtrer la solution à travers un filtre en papier et conserver le filtrat pour la séparation liquide-liquide (5.3).
5.3. Séparation liquide-liquide
Transvaser 25 ml du filtrat (5.2) dans une ampoule à décanter de 500 ml contenant 100 ml de solution d'acide chlorhydrique (3.5). Ajouter 100 ml de dichlorométhane (3.1) dans l'ampoule et agiter pendant 1 minute. Après séparation des phases, laisser la phase inférieure (dichlorométhane) s'écouler dans un ballon à fond rond de 500 ml. Répéter l'extraction de la phase aqueuse avec deux autres portions de dichlorométhane de 40 ml et combiner celles-ci avec le premier extrait dans le ballon rond. Évaporer totalement l'extrait de dichlorométhane au moyen de l'évaporateur rotatif (4.2), qui doit assurer une température de 40 oC et fonctionner à pression réduite. Dissoudre le résidu dans 20 à 25 ml de méthanol (3.2), boucher le ballon et conserver la totalité de l'extrait pour la chromatographie par échange d'ions (5.4).
5.4. Chromatographie par échange d'ions
5.4.1.
Introduire un petit tampon de laine de verre dans l'extrémité inférieure d'une colonne de verre (4.3). Préparer une boue de 5,0 g de résine échangeuse de cations traitée (3.6) et de 50 ml d'acide chlorhydrique (3.5), verser dans la colonne et laisser reposer. Laisser l'acide s'écouler de manière à établir son niveau juste au-dessus de la surface de la résine et laver la colonne à l'eau jusqu'à ce que l'effluent soit neutre au papier de tournesol. Transvaser 50 ml de méthanol (3.2) dans la colonne et le laisser s'écouler jusqu'à ce qu'il atteigne la surface de la résine.
5.4.2.
Au moyen d'une pipette, transvaser soigneusement l'extrait de l'opération 5.3 dans la colonne. Rincer le ballon à fond rond avec deux portions de 5 à 10 ml de méthanol (3.2) et transvaser ces liquides de lavage dans la colonne. Laisser l'extrait s'écouler jusqu'à la surface de la résine et laver la colonne avec 50 ml de méthanol en veillant à ce que le débit ne dépasse pas 5 ml/min. Éliminer l'effluent. Éluer le méthyl-benzoquate de la colonne au moyen de 150 ml de solution d'acide méthanesulfonique (3.4) et recueillir l'éluat de la colonne dans une fiole conique de 250 ml.
5.5. Séparation liquide-liquide
Transvaser l'éluat de l'opération 5.4.2 dans une ampoule à décanter de 1 l. Rincer la fiole conique avec 5 à 10 ml de méthanol (3.2) et combiner le liquide de lavage avec le contenu de l'ampoule à décantation. Ajouter 300 ml de solution d'acide chlorhydrique (3.5) et 130 ml de dichlorométhane (3.1). Agiter pendant 1 minute et laisser les phases se séparer. Laisser s'écouler la phase inférieure (dichlorométhane) dans un ballon à fond rond de 500 ml. Répéter l'extraction de la phase aqueuse avec deux nouvelles portions de 70 ml de dichlorométhane et combiner ces extraits avec le premier dans le ballon à fond rond.
Évaporer totalement l'extrait de dichlorométhane au moyen de l'évaporateur rotatif (4.2), qui doit assurer une température de 40 oC et fonctionner à pression réduite. Dissoudre le résidu dans le ballon dans environ 5 ml de méthanol (3.2) et transvaser quantitativement cette solution dans une fiole graduée de 10 ml. Rincer le ballon à fond rond avec deux nouvelles portions de 1 à 2 ml de méthanol et transvaser le liquide dans la fiole graduée. Ajuster au trait de jauge avec du méthanol et mélanger. Filtrer une portion aliquote à travers un filtre à membrane (4.6). Réserver cette solution pour l'analyse par CLHP (5.6).
5.6. Dosage par CLHP
5.6.1.
Les conditions suivantes sont données à titre indicatif. D'autres paramètres peuvent être utilisés à condition de produire des résultats équivalents:
— colonne de chromatographie liquide (4.4.1),
— phase mobile de la CLHP: mélange méthanol-eau (3.3),
— débit: 1 à 1,5 ml/min,
— longueur d'onde de détection: 265 nm,
— volume injecté: 20 à 50 μl.
Contrôler la stabilité du système chromatographique, en injectant plusieurs fois la solution d'étalonnage (3.7.3) à 4 μg/ml, jusqu'à l'obtention de surfaces ou de hauteurs de pics et de temps de rétention constants.
5.6.2.
Injecter plusieurs fois chaque solution d'étalonnage (3.7.3) et mesurer les hauteurs (surfaces) des pics pour chaque concentration. Tracer la courbe d'étalonnage en portant les moyennes des hauteurs ou des surfaces de pics des solutions d'étalonnage en ordonnée et les concentrations correspondantes en μg/ml en abscisse.
5.6.3.
Injecter plusieurs fois l'extrait d'échantillon (5.5), en utilisant le même volume que pour les solutions d'étalonnage, et déterminer la hauteur (surface) de pic moyenne du méthyl-benzoquate.
6. Calcul des résultats
À partir de la hauteur (surface) moyenne des pics du méthyl-benzoquate de la solution d'échantillon, déterminer la concentration de méthyl-benzoquate dans cette solution, en μg/ml, par référence à la courbe d'étalonnage (5.6.2).
La teneur en méthyl-benzoquate (w en mg/kg) de l'échantillon est donnée par la formule suivante:

où:
|
c |
= |
concentration de méthyl-benzoquate dans la solution d'échantillon, en μg/ml, |
|
m |
= |
poids de la prise d'essai, en grammes. |
7. Validation des résultats
7.1. Identité
L'identité de l'analyte peut être confirmée par cochromatographie ou par l'utilisation d'un détecteur à barrettes de diodes, qui permet de comparer les spectres de l'extrait d'échantillon et de la solution d'étalonnage (3.7.3) à 10 μg/ml de méthyl-benzoquate.
7.1.1.
Un extrait d'échantillon est supplémenté par addition d'une quantité appropriée de solution étalon intermédiaire (3.7.2). La quantité de méthyl-benzoquate ajoutée doit être similaire à la quantité estimée de méthyl-benzoquate trouvée dans l'extrait d'échantillon.
Seule la hauteur du pic de méthyl-benzoquate doit être augmentée, compte tenu à la fois de la quantité ajoutée et de la dilution de l'extrait. La largeur du pic, à la moitié de sa hauteur, doit être d'environ 10 % de sa largeur initiale.
7.1.2.
Évaluer les résultats conformément aux critères suivants:
a) les longueurs d'onde d'absorption maximale des spectres de l'échantillon et de l'étalon, enregistrées au sommet des pics sur le chromatogramme, doivent être identiques, dans une marge déterminée par le pouvoir de résolution du système de détection. Dans le cadre d'une détection par barrettes de diodes, elle est généralement de plus ou moins 2 nm;
b) entre 220 et 350 nm, les spectres de l'échantillon et de l'étalon, enregistrés au sommet du pic sur le chromatogramme, ne doivent pas être différents pour les parties du spectre comprises entre 10 et 100 % de densité optique relative. Ce critère est rempli lorsque les mêmes maxima sont présents et que l'écart observé entre les deux spectres ne dépasse nulle part 15 % de densité optique de l'analyte étalon;
c) entre 220 et 350 nm, les spectres de la courbe ascendante, du sommet et de la courbe descendante du pic fourni par l'extrait d'échantillon ne peuvent être visuellement différents les uns des autres pour les parties du spectre comprises entre 10 et 100 % de densité optique relative. Ce critère est rempli lorsque les mêmes maxima sont présents et que l'écart observé entre les spectres ne dépasse nulle part 15 % de la densité optique du spectre au sommet du pic.
Si l'un de ces critères n'est pas rempli, la présence de l'analyte n'est pas confirmée.
7.2. Répétabilité
La différence entre les résultats de deux dosages parallèles effectués sur le même échantillon ne peut dépasser: 10 % du résultat le plus élevé pour des teneurs en méthyl-benzoquate comprises entre 4 et 20 mg/kg.
7.3. Récupération
En ce qui concerne l'échantillon blanc supplémenté, la récupération doit être de 90 % au minimum.
8. Résultats d'une étude collaborative
Cinq échantillons ont été analysés par dix laboratoires. Pour chaque échantillon, les analyses ont été effectuées en double.
|
|
Blanc |
Farine 1 |
Granulés 1 |
Farine 2 |
Granulés 2 |
|
Moyenne [mg/kg] |
ND |
4,50 |
4,50 |
8,90 |
8,70 |
|
sr [mg/kg] |
— |
0,30 |
0,20 |
0,60 |
0,50 |
|
CVr [%] |
— |
6,70 |
4,40 |
6,70 |
5,70 |
|
sR [mg/kg] |
— |
0,40 |
0,50 |
0,90 |
1,00 |
|
CVR [%] |
— |
8,90 |
11,10 |
10,10 |
11,50 |
|
Récupération [%] |
— |
92,00 |
93,00 |
92,00 |
89,00 |
|
ND |
= |
non détecté |
|
sr |
= |
écart type de répétabilité |
|
CVr |
= |
coefficient de variation de la répétabilité (%) |
|
sR |
= |
écart type de reproductibilité |
|
CVR |
= |
coefficient de variation de la reproductibilité (%) |
B. DOSAGE DE L'OLAQUINDOX
2-[N -2'-(hydroxyéthyl )carbamoyl]-3- méthylquinoxaline-N 1 ,N 4 -dioxyde)
1. Objet et champ d'application
La méthode permet de déterminer la teneur en olaquindox des aliments pour animaux. La limite de quantification est de 5 mg/kg.
2. Principe
L'échantillon est soumis à une extraction par un mélange eau/méthanol. La teneur en olaquindox est déterminée par chromatographie liquide haute performance (CLHP) en phase inverse, avec détection UV.
3. Réactifs
3.1. Méthanol.
3.2. Méthanol, de qualité CLHP.
3.3. Eau, de qualité CLHP.
3.4. Phase mobile pour CLHP
Mélange eau (3.3)-méthanol (3.2), 900 + 100 (v + v).
3.5. Substance étalon: olaquindox pur, 2-[N-2'-(hydroxyéthyl)carbamoyl]-3-méthylquinoxaline-N1,N4-dioxyde, E 851.
3.5.1.
Peser, à 0,1 mg près, 50 mg d'olaquindox (3.5) dans une fiole jaugée de 200 ml et ajouter environ 190 ml d'eau. Placer la fiole pendant 20 minutes dans un bain ultrasonique (4.1). Après traitement ultrasonique, porter la solution à température ambiante, l'ajuster au trait de jauge avec de l'eau et mélanger. Envelopper la fiole d'une feuille d'aluminium et la placer au réfrigérateur. Renouveler la solution chaque mois.
3.5.2.
Transférer 10,0 ml de solution mère de l'étalon (3.5.1) dans une fiole jaugée de 100 ml, ajuster au trait de jauge avec la phase mobile (3.4) et mélanger. Envelopper la fiole d'une feuille d'aluminium et la placer au réfrigérateur. Renouveler la solution chaque jour.
3.5.3.
Transférer 1,0 , 2,0 , 5,0 , 10,0 , 15,0 et 20,0 ml de solution étalon intermédiaire (3.5.2) dans une série de fioles graduées de 50 ml. Ajuster au trait de jauge avec la phase mobile (3.4) et mélanger. Envelopper les fioles avec une feuille d'aluminium. Ces solutions correspondent respectivement à 0,5 , 1,0 , 2,5 , 5,0 , 7,5 et 10,0 μg d'olaquindox par ml.
Renouveler les solutions chaque jour.
4. Appareillage
4.1. Bain ultrasonique.
4.2. Agitateur mécanique.
4.3. Équipement CLHP à détecteur UV de longueur d'onde variable ou détecteur à barrettes de diodes.
4.3.1. Colonne de chromatographie liquide de 250 mm × 4 mm, C18, particules de 10 μm, ou équivalent.
4.4. Filtres à membrane, 0,45 μm.
5. Mode opératoire
|
NB |
: |
L'olaquindox est photosensible. Effectuer toutes les opérations sous lumière diffuse ou utiliser du verre ambré. |
5.1. Généralités
5.1.1. Analyser un aliment blanc pour vérifier l'absence d'olaquindox ou de substances interférentes.
5.1.2. Effectuer un test de récupération par analyse d'un aliment blanc auquel il a été ajouté une quantité d'olaquindox similaire à celle présente dans l'échantillon. Pour obtenir une concentration de 50 mg/kg, transférer10,0 ml de la solution mère de l'étalon (3.5.1) dans une fiole erlenmeyer de 250 ml et concentrer la solution par évaporation à environ 0,5 ml. Ajouter 50 g de l'aliment blanc, mélanger soigneusement et attendre 10 minutes tout en mélangeant de nouveau plusieurs fois avant de procéder à l'extraction (5.2).
|
NB |
: |
Aux fins de la présente méthode, l'aliment blanc doit être d'un type similaire à celui de l'échantillon et aucune présence d'olaquindox ne doit y être détectée. |
5.2. Extraction
Peser, à 0,01 g près, 50 g de l'échantillon. Transférer dans une fiole erlenmeyer de 1 000 ml, ajouter 100 ml de méthanol (3.1) et placer la fiole pendant 5 min dans un bain ultrasonique (4.1). Ajouter 410 ml d'eau et laisser dans le bain ultrasonique pendant 15 min de plus. Enlever la fiole du bain ultrasonique, l'agiter pendant 30 min sur l'agitateur (4.2) et filtrer à travers un filtre plissé. Transvaser 10,0 ml du filtrat dans une fiole jaugée de 20 ml, ajuster au trait de jauge avec de l'eau et mélanger. Une partie aliquote est filtrée à travers un filtre à membrane (4.4) (voir point 9 Observation). Procéder au dosage par CLHP (5.3).
5.3. Dosage par CLHP
5.3.1.
Les conditions suivantes sont proposées à titre indicatif, d'autres conditions peuvent être appliquées si elles donnent des résultats équivalents.
|
Colonne d'analyse (4.3.1) |
|
|
Phase mobile (3.4): |
mélange d'eau (3.3) et de méthanol (3.2), 900 + 100 (v + v) |
|
Débit: |
1,5 -2 ml/min |
|
Longueur d'onde de détection: |
380 nm |
|
Volume d'injection: |
20 μl-100 μl |
Vérifier la stabilité du système chromatographique en injectant plusieurs fois la solution d'étalonnage (3.5.3) contenant 2,5 μg/ml jusqu'à obtention de hauteurs de pic et de temps de rétention constants.
5.3.2.
Injecter chaque solution d'étalonnage (3.5.3) plusieurs fois et déterminer les hauteurs (surfaces) moyennes des pics pour chaque concentration. Tracer la courbe d'étalonnage en portant les hauteurs (surfaces) de pic moyennes des solutions d'étalonnage en ordonnée et les concentrations correspondantes en μg/ml en abscisse.
5.3.3.
Injecter l'extrait d'échantillon (5.2) plusieurs fois en utilisant le même volume que celui retenu pour les solutions d'étalonnage et déterminer la hauteur (surface) moyenne des pics de l'olaquindox.
6. Calcul des résultats
À partir de la hauteur (surface) moyenne des pics de l'olaquindox de la solution d'échantillon, déterminer la concentration d'olaquindox dans cette solution, en μg/ml, par référence à la courbe d'étalonnage (5.3.2).
La teneur (w) en olaquindox, exprimée en mg/kg de l'échantillon, est donnée par la formule suivante:

où:
|
c |
= |
concentration d'olaquindox dans l'extrait d'échantillon (5.2) en μg/ml, |
|
m |
= |
poids de la prise d'essai, en g (5.2). |
7. Validation des résultats
7.1. Identité
L'identité de l'analyte peut être confirmée par cochromatographie ou à l'aide d'un détecteur à barrettes de diodes qui permet de comparer les spectres de l'extrait d'échantillon (5.2) et de la solution d'étalonnage (3.5.3) contenant 5,0 μg/ml.
7.1.1.
Un extrait d'échantillon (5.2) est additionné d'une quantité appropriée de la solution d'étalonnage (3.5.3). La quantité d'olaquindox ajoutée doit être semblable à la quantité d'olaquindox constatée dans l'extrait d'échantillon.
Seule la hauteur du pic de l'olaquindox doit être augmentée, compte tenu de la quantité ajoutée et de la dissolution de l'extrait. La largeur du pic à mi-hauteur doit se situer à ± 10 % de la largeur initiale du pic d'olaquindox de l'extrait d'échantillon non supplémenté.
7.1.2.
Évaluer les résultats conformément aux critères suivants:
a) les longueurs d'onde d'absorption maximale des spectres de l'échantillon et de l'étalon, enregistrées au sommet des pics sur le chromatogramme, doivent être identiques, dans une marge déterminée par le pouvoir de résolution du système de détection. Dans le cadre d'une détection par barrettes de diodes, elle est généralement de ± 2 nm;
b) entre 220 et 400 nm, les spectres de l'échantillon et de l'étalon enregistrés au sommet du pic sur le chromatogramme ne doivent pas être différents pour les parties du spectre situées entre 10 et 100 % de l'absorbance relative. Ce critère est rempli lorsque les mêmes maxima sont présents et que, en aucun point, l'écart observé entre les deux spectres ne dépasse 15 % de l'absorbance de l'analyte étalon;
c) entre 220 en 400 nm, les spectres de la courbe ascendante, du sommet et de la courbe descendante du pic produits par l'extrait d'échantillon ne doivent pas être différents les uns des autres pour les parties du spectre situées entre 10 et 100 % de l'absorbance relative. Ce critère est rempli lorsque les mêmes maxima sont présents et que, en aucun point, l'écart observé entre les spectres ne dépasse 15 % de l'absorbance du spectre au sommet du pic.
Si l'un de ces critères n'est pas rempli, la présence de l'analyte n'est pas confirmée.
7.2. Répétabilité
La différence entre les résultats de deux dosages parallèles effectués sur le même échantillon ne peut dépasser 15 % du résultat supérieur pour les teneurs en olaquindox comprises entre 10 et 200 mg/kg.
7.3. Récupération
En ce qui concerne l'échantillon blanc supplémenté, la récupération doit être de 90 % au minimum.
8. Résultats d'une étude collaborative
Une étude collaborative communautaire a été organisée: jusqu'à treize laboratoires ont analysé quatre échantillons d'aliments pour porcelets, y compris un aliment blanc. Les résultats de l'étude figurent ci-après.
|
|
Échantillon 1 |
Échantillon 2 |
Échantillon 3 |
Échantillon 4 |
|
L |
13 |
10 |
11 |
11 |
|
n |
40 |
40 |
44 |
44 |
|
Moyenne [mg/kg] |
— |
14,6 |
48,0 |
95,4 |
|
Sr [mg/kg] |
— |
0,82 |
2,05 |
6,36 |
|
SR [mg/kg] |
— |
1,62 |
4,28 |
8,42 |
|
CVr [%] |
— |
5,6 |
4,3 |
6,7 |
|
CVR [%] |
— |
11,1 |
8,9 |
8,8 |
|
Teneur nominale |
|
|
|
|
|
[mg/kg] |
— |
15 |
50 |
100 |
|
Récupération % |
— |
97,3 |
96,0 |
95,4 |
|
L |
= |
nombre de laboratoires |
|
n |
= |
nombre de valeurs individuelles |
|
Sr |
= |
écart type de répétabilité |
|
SR |
= |
écart type de reproductibilité |
|
CVr |
= |
coefficient de variation de la répétabilité |
|
CVR |
= |
coefficient de variation de la reproductibilité |
9. Observation
Bien que la méthode n'ait pas été validée pour les aliments contenant plus de 100 mg/kg d'olaquindox, on peut obtenir des résultats satisfaisants en utilisant une prise d'essai plus faible et/ou en diluant l'extrait (5.2) de façon telle que sa concentration se situe dans la zone de la courbe d'étalonnage (5.3.2).
C. DOSAGE DE L'AMPROLIUM
Chlorhydrate du chlorure de 1-[(4-amino-2-propylpirimidin-5-yl)méthyl]-2-méthyl-pyridinium
1. Objet et champ d'application
La méthode permet de déterminer la teneur en amprolium des aliments pour animaux et des prémélanges. La limite de détection est de 1 mg/kg et la limite de quantification de 5 mg/kg.
2. Principe
L'échantillon est soumis à une extraction par un mélange méthanol/eau. Après dilution dans une phase mobile et filtration sur membrane, la teneur en amprolium est déterminée par chromatographie liquide haute performance (CLHP) avec échange de cations à l'aide d'un détecteur UV.
3. Réactifs
3.1. Méthanol.
3.2. Acétonitrile, de qualité CLHP.
3.3. Eau, de qualité CLHP.
3.4. Solution de phosphate monosodique, c = 0,1 mol/l
Dissoudre 13,80 g de phosphate monosodique monohydraté dans de l'eau (3.3) dans une fiole jaugée de 1 000 ml, ajuster au trait de jauge avec de l'eau (3.3) et mélanger.
3.5. Solution de perchlorate de sodium, c = 1,6 mol/l
Dissoudre 224,74 g de perchlorate de sodium monohydraté dans de l'eau (3.3) dans une fiole jaugée de 1 000 ml, ajuster au trait de jauge avec de l'eau (3.3) et mélanger.
3.6. Phase mobile pour CLHP (voir point 9.1)
Mélange d'acétonitrile (3.2), de solution de phosphate monosodique (3.4) et de solution de perchlorate de sodium (3.5), 450 + 450 + 100 (v + v + v). Avant l'emploi, faire passer dans un filtre à membrane de 0,22 μm (4.3) et dégazer la solution [par exemple dans un bain ultrasonique (4.4) pendant au moins 15 minutes].
3.7. Substance étalon: amprolium pur, chlorhydrate chlorure de 1-[(4-amino-2-propylpirimidin-5-yl)méthyl]-2-méthyl-pyridinium, E 750 (voir 9.2).
3.7.1.
Peser, à 0,1 mg près, 50 mg d'amprolium (3.7) dans une fiole jaugée de 100 ml, dissoudre dans 80 ml de méthanol (3.1) et placer la fiole pendant 10 min dans un bain ultrasonique (4.4). Après traitement ultrasonique, porter la solution à température ambiante, l'ajuster au trait de jauge avec de l'eau et mélanger. À une température ≤ 4 oC, la solution est stable pendant un mois.
3.7.2.
Transférer, au moyen d'une pipette, 5,0 ml de la solution mère de l'étalon (3.7.1) dans une fiole jaugée de 50 ml, ajuster au trait de jauge avec le solvant d'extraction (3.8) et mélanger. À une température ≤ 4 oC, la solution est stable pendant un mois.
3.7.3.
Transférer 0,5 , 1,0 et 2,0 ml de la solution étalon intermédiaire (3.7.2) dans une série de fioles jaugées de 50 ml. Ajuster au trait de jauge avec la phase mobile (3.6) et mélanger. Ces solutions correspondent respectivement à 0,5 , 1,0 et 2,0 μg d'amprolium par ml. Ces solutions doivent être préparées peu avant d'être utilisées.
3.8. Solvant d'extraction
Mélange de méthanol (3.1) et d'eau 2 + 1 (v + v).
4. Appareillage
4.1. Équipement CLHP avec système à injection permettant d'injecter des volumes de 100 μl.
4.1.1. Colonne pour chromatographie liquide de 125 mm × 4 mm à échange de cations remplie de Nucleosil 10 SA de 5 ou 10 μm, ou équivalent.
4.1.2. Détecteur UV de longueur d'onde variable ou détecteur à barrettes de diodes.
4.2. Filtre à membrane en PTFE de 0,45 μm.
4.3. Filtre à membrane de 0,22 μm.
4.4. Bain ultrasonique.
4.5. Agitateur mécanique ou magnétique.
5. Mode opératoire
5.1. Généralités
5.1.1.
Pour procéder au test de récupération (5.1.2), analyser un aliment blanc pour vérifier l'absence d'amprolium ou de substances interférentes. L'aliment blanc doit être du même type que celui de l'échantillon; il ne doit être détecté ni amprolium ni substances interférentes.
5.1.2
Effectuer un test de récupération par analyse de l'aliment blanc auquel a été ajoutée une quantité d'amprolium similaire à celle présente dans l'échantillon. Pour obtenir une concentration de 100 mg/kg, transférer 10,0 ml de la solution mère de l'étalon (3.7.1) dans une fiole erlenmeyer de 250 ml et concentrer la solution par évaporation à environ 0,5 ml. Ajouter 50 g de l'aliment blanc, mélanger soigneusement et attendre 10 min tout en mélangeant de nouveau plusieurs fois avant de procéder à l'extraction (5.2).
En l'absence d'aliment blanc de même type que celui de l'échantillon (voir 5.1.1), le test de récupération peut être effectué selon la méthode par addition de l'étalon. Dans ce cas, l'échantillon à analyser est supplémenté d'une quantité d'amprolium semblable à celle déjà présente dans l'échantillon. Celui-ci est analysé avec l'échantillon non supplémenté et la récupération peut être calculée par différence.
5.2. Extraction
5.2.1.
Peser, à 0,01 g près, de 5 à 40 g de l'échantillon selon sa teneur en amprolium dans une fiole erlenmeyer de 500 ml et ajouter 200 ml de solvant d'extraction (3.8). Placer la fiole dans le bain ultrasonique (4.4) pendant 15 minutes. Enlever la fiole du bain ultrasonique et la soumettre pendant 1 heure à une agitation mécanique ou magnétique (4.5). Diluer une partie aliquote de l'extrait avec la phase mobile (3.6) pour obtenir une teneur en amprolium de 0,5 à 2 μg/ml et mélanger (voir observation 9.3). Filtrer 5 à 10 ml de cette solution diluée sur un filtre à membrane (4.2). Procéder au dosage par CLHP (5.3).
5.2.2.
Peser, à 0,001 g près, de 1 à 4 g du prémélange selon sa teneur en amprolium dans une fiole erlenmeyer de 500 ml et ajouter 200 ml de solvant d'extraction (3.8). Placer la fiole dans le bain ultrasonique (4.4) pendant 15 minutes. Enlever la fiole du bain ultrasonique et la soumettre pendant 1 heure à une agitation mécanique ou magnétique (4.5). Diluer une partie aliquote de l'extrait avec la phase mobile (3.6) pour obtenir une teneur en amprolium de 0,5 à 2 μg/ml et mélanger. Filtrer 5 à 10 ml de cette solution diluée sur un filtre à membrane (4.2). Procéder au dosage par CLHP (5.3).
5.3. Dosage par CLHP
5.3.1.
Les conditions suivantes sont proposées à titre indicatif, d'autres conditions peuvent être appliquées si elles donnent des résultats équivalents.
|
Colonne de chromatographie |
|
|
liquide (4.1.1): |
125 mm × 4 mm avec échange de cations avec Nucleosil 10 SA de 5 ou de 10 μm, ou équivalent |
|
Phase mobile (3.6): |
mélange d'acétonitrile (3.2), de solution de phosphate monosodique (3.4) et de solution de perchlorate de sodium (3.5), 450 + 450 + 100 (v + v + v) |
|
Débit: |
0,7 -1 ml/min |
|
Longueur d'onde de détection: |
264 nm |
|
Volume d'injection: |
100 μl |
Vérifier la stabilité du système chromatographique en injectant plusieurs fois la solution d'étalonnage (3.7.3) contenant 1,0 μg/ml jusqu'à obtention de hauteurs de pic et de temps de rétention constants.
5.3.2.
Injecter chaque solution d'étalonnage (3.7.3) plusieurs fois et déterminer les hauteurs (surfaces) moyennes des pics pour chaque concentration. Tracer la courbe d'étalonnage en portant les hauteurs (surfaces) de pic moyennes des solutions d'étalonnage en ordonnée et les concentrations correspondantes en μg/ml en abscisse.
5.3.3.
Injecter l'extrait d'échantillon (5.2) plusieurs fois en utilisant le même volume que celui retenu pour les solutions d'étalonnage et déterminer la hauteur (surface) moyenne des pics de l'amprolium.
6. Calcul des résultats
À partir de la hauteur (surface) moyenne des pics de l'amprolium de la solution d'échantillon, déterminer la concentration d'amprolium dans cette solution, en μg/ml, par référence à la courbe d'étalonnage (5.3.2).
La teneur w en amprolium, exprimée en mg/kg de l'échantillon, est donnée par la formule suivante:

[mg/kg]
où:
|
V |
= |
volume du solvant d'extraction (3.8) en ml selon 5.2 (c'est-à-dire 200 ml), |
|
c |
= |
concentration d'amprolium dans l'extrait d'échantillon (5.2) en μg/ml, |
|
f |
= |
facteur de dilution selon 5.2, |
|
m |
= |
poids de la prise d'essai, en grammes. |
7. Validation des résultats
7.1. Identité
L'identité de l'analyte peut être confirmée par cochromatographie ou à l'aide d'un détecteur à barrettes de diodes qui permet de comparer les spectres de l'extrait d'échantillon (5.2) et de la solution d'étalonnage (3.7.3) contenant 2,0 μg/ml.
7.1.1.
Un extrait d'échantillon (5.2) est supplémenté par ajout d'une quantité appropriée de la solution d'étalonnage (3.7.3). La quantité d'amprolium ajoutée doit être semblable à la quantité d'amprolium constatée dans l'extrait d'échantillon.
Seule la hauteur du pic de l'amprolium doit être augmentée, compte tenu de la quantité ajoutée et de la dilution de l'extrait. La largeur du pic à mi-hauteur doit se situer à ± 10 % de la largeur initiale du pic d'amprolium de l'extrait d'échantillon non supplémenté.
7.1.2.
Évaluer les résultats conformément aux critères suivants:
a) les longueurs d'onde d'absorption maximale des spectres de l'échantillon et de l'étalon, enregistrées au sommet des pics sur le chromatogramme, doivent être identiques, dans une marge déterminée par le pouvoir de résolution du système de détection. Dans le cadre d'une détection par barrettes de diodes, elle est généralement de ± 2 nm;
b) entre 210 et 320 nm, les spectres de l'échantillon et de l'étalon enregistrés au sommet du pic sur le chromatogramme ne doivent pas être différents pour les parties du spectre situées entre 10 et 100 % de l'absorbance relative. Ce critère est rempli lorsque les mêmes maxima sont présents et que, en aucun point, l'écart observé entre les deux spectres ne dépasse 15 % de l'absorbance de l'analyte étalon;
c) entre 210 et 320 nm, les spectres de la courbe ascendante, du sommet et de la courbe descendante du pic produits par l'extrait d'échantillon ne doivent pas être différents les uns des autres pour les parties du spectre situées entre 10 et 100 % de l'absorbance relative. Ce critère est rempli lorsque les mêmes maxima sont présents et que, en aucun point, l'écart observé entre les spectres ne dépasse 15 % de l'absorbance du spectre au sommet du pic.
Si l'un de ces critères n'est pas rempli, la présence de l'analyte n'est pas confirmée.
7.2. Répétabilité
La différence entre les résultats de deux dosages parallèles effectués sur le même échantillon ne peut dépasser:
— 15 % du résultat supérieur pour les teneurs en amprolium comprises entre 25 et 500 mg/kg,
— 75 mg/kg pour les teneurs en amprolium comprises entre 500 et 1 000 mg/kg,
— 7,5 % du résultat supérieur pour les teneurs en amprolium supérieures à 1 000 mg/kg.
7.3. Récupération
Pour un échantillon (blanc) supplémenté, la récupération doit être de 90 % au moins.
8. Résultats d'une étude collaborative
Dans le cadre d'une étude collaborative, trois aliments pour volaille (échantillons 1-3), un aliment minéral (échantillon 4) et un prémélange (échantillon 5) ont été analysés. Les résultats de l'étude figurent dans le tableau ci-après.
|
|
Échantillon 1 (aliment blanc) |
Échantillon 2 |
Échantillon 3 |
Échantillon 4 |
Échantillon 5 |
|
L |
14 |
14 |
14 |
14 |
15 |
|
n |
56 |
56 |
56 |
56 |
60 |
|
Moyenne [mg/kg] |
— |
45,5 |
188 |
5 129 |
25 140 |
|
sr [mg/kg] |
— |
2,26 |
3,57 |
178 |
550 |
|
CVr [%] |
— |
4,95 |
1,90 |
3,46 |
2,20 |
|
sR [mg/kg] |
— |
2,95 |
11,8 |
266 |
760 |
|
CVR [%] |
— |
6,47 |
6,27 |
5,19 |
3,00 |
|
Teneur nominale [mg/kg] |
— |
50 |
200 |
5 000 |
25 000 |
|
L |
= |
nombre de laboratoires |
|
n |
= |
nombre de valeurs individuelles |
|
sr |
= |
écart type de répétabilité |
|
CVr |
= |
coefficient de variation de la répétabilité |
|
sR |
= |
écart type de reproductibilité |
|
CVR |
= |
coefficient de variation de la reproductibilité |
9. Observations
9.1. Si l'échantillon contient de la thiamine, le pic de la thiamine dans le chromatogramme apparaît peu avant le pic de l'amprolium. D'après cette méthode, l'amprolium et la thiamine doivent être séparés. Si l'amprolium et la thiamine ne sont pas séparés par la colonne (4.1.1) utilisée dans cette méthode, remplacer jusqu'à 50 % de la portion d'acétonitrile de la phase mobile (3.6) par du méthanol.
9.2. Selon la pharmacopée britannique, le spectre de la solution d'amprolium (c = 0,02 mol/l) dans l'acide chlorhydrique (c = 0,1 mol/l) présente des maxima à 246 nm et à 262 nm. L'absorbance sera de 0,84 à 246 nm et de 0,80 à 262 nm.
9.3. L'extrait doit toujours être dilué avec la phase mobile, faute de quoi le temps de rétention du pic d'amprolium peut changer de façon significative en raison des variations de la force ionique.
D. DOSAGE DU CARBADOX
Méthyl 3-(2-quinoxalinylméthylène)carbazate N 1 ,N 4 -dioxyde
1. Objet et champ d'application
La méthode permet de déterminer la teneur en carbadox des aliments pour animaux, des prémélanges et des préparations. La limite de détection est de 1 mg/kg et la limite de quantification de 5 mg/kg.
2. Principe
L'échantillon est équilibré avec de l'eau et soumis à une extraction par un mélange méthanol/acétonitrile. Pour les aliments pour animaux, une partie aliquote de l'extrait filtré est nettoyée sur une colonne d'oxyde d'aluminium. Pour les prémélanges et les préparations, une partie aliquote de l'extrait filtré est diluée à une concentration appropriée avec de l'eau, du méthanol et de l'acétonitrile. La teneur en carbadox est déterminée par chromatographie liquide haute performance (CLHP) en phase inverse, avec détection UV.
3. Réactifs
3.1. Méthanol.
3.2. Acétonitrile, de qualité CLHP.
3.3. Acide acétique, w = 100 %.
3.4. Oxyde d'aluminium: neutre, degré d'activité I.
3.5. Méthanol-acétonitrile 1 + 1 (v + v)
Mélanger 500 ml de méthanol (3.1) à 500 ml d'acétonitrile (3.2).
3.6. Acide acétique, σ = 10 %
Diluer 10 ml d'acide acétique (3.3) dans 100 ml d'eau.
3.7. Acétate de sodium.
3.8. Eau, de qualité CLHP.
3.9. Solution tampon à l'acétate, c = 0,01 mol/l, pH = 6,0
Dissoudre 0,82 g d'acétate de sodium (3.7) dans 700 ml d'eau (3.8) et ajuster le pH à 6,0 avec de l'acide acétique (3.6). Transférer dans une fiole jaugée de 1 000 ml, ajuster au trait de jauge avec de l'eau (3.8) et mélanger.
3.10. Phase mobile pour CLHP.
Mélanger 825 ml de solution tampon à l'acétate (3.9) à 175 ml d'acétonitrile (3.2).
Filtrer à travers un filtre de 0,22 μm (4.5) et dégazer la solution (par exemple, par traitement aux ultrasons pendant 10 minutes).
3.11. Substance étalon.
Carbadox pur: méthyl 3-(2-quinoxalinylméthylène)carbazate N1,N4-dioxyde, E 850.
3.11.1.
Peser, à 0,1 mg près, 25 mg de substance étalon de carbadox (3.11) dans une fiole jaugée de 250 ml. Dissoudre dans un mélange de méthanol-acétonitrile (3.5) par traitement aux ultrasons (4.7). Après le traitement ultrasonique, porter la solution à la température ambiante, ajuster au trait de jauge avec du méthanol-acétonitrile (3.5) et mélanger. Envelopper la fiole dans une feuille d'aluminium ou utiliser un récipient en verre ambré et le mettre au réfrigérateur. À une température ≤ 4 oC, la solution est stable pendant un mois.
3.11.2.
Transférer 2,0 , 5,0 , 10,0 et 20,0 ml de la solution mère de l'étalon (3.11.1) dans une série de fioles jaugées de 100 ml. Ajouter 30 ml d'eau, ajuster au trait de jauge avec du méthanol-acétonitrile (3.5) et mélanger. Envelopper les fioles dans une feuille d'aluminium. Ces solutions correspondent respectivement à 2,0 , 5,0 , 10,0 et 20,0 μg/ml de carbadox.
Elles doivent être préparées peu avant d'être utilisées.
|
N.B. |
Pour doser le carbadox dans des aliments pour animaux contenant moins de 10 mg/kg, il faut préparer des solutions d'étalonnage à des concentrations inférieures à 2,0 μg/ml. |
3.12. Mélange eau-[méthanol-acétonitrile] (3.5), 300 + 700 (v + v)
Mélanger 300 ml d'eau à 700 ml du mélange de méthanol-acétonitrile (3.5).
4. Appareillage
4.1. Agitateur de laboratoire ou mélangeur magnétique.
4.2. Papier filtre en fibre de verre (Whatman GF/A ou équivalent).
4.3. Colonne de verre (longueur 300 à 400 mm, diamètre interne environ 10 mm), avec cloison en verre fritté et valve d'évacuation.
|
NB |
: |
Il est également possible d'utiliser une colonne de verre munie d'un robinet ou une colonne de verre avec extrémité effilée; dans ce cas, un petit tampon de laine de verre est inséré à l'extrémité inférieure et enfoncé à l'aide d'une baguette de verre. |
4.4. Équipement CLHP avec système à injection permettant d'injecter des volumes de 20 μl.
4.4.1. Colonne de chromatographie liquide: 300 mm × 4 mm, C18, particules de 10 μm, ou équivalent.
4.4.2. Détecteur UV de longueur d'onde variable ou détecteur à barrettes de diodes opérant entre 225 et 400 nm.
4.5. Filtre à membrane de 0,22 μm.
4.6. Filtre à membrane de 0,45 μm.
4.7. Bain ultrasonique.
5. Mode opératoire
|
NB |
: |
Le carbadox est photosensible. Opérer toujours sous lumière tamisée, ou utiliser des récipients en verre ambré ou enveloppés dans une feuille d'aluminium. |
5.1. Généralités
5.1.1.
Pour procéder au test de récupération (5.1.2), analyser un aliment blanc pour vérifier l'absence de carbadox ou de substances interférentes. L'aliment blanc doit être du même type que celui de l'échantillon; il ne doit être détecté ni carbadox ni substances interférentes.
5.1.2.
Effectuer un test de récupération par analyse de l'aliment blanc (5.1.1) auquel a été ajoutée une quantité de carbadox similaire à celle présente dans l'échantillon. Pour obtenir une concentration de 50 mg/kg, transférer 5,0 ml de la solution mère de l'étalon (3.11.1) dans une fiole erlenmeyer de 200 ml. Concentrer la solution par évaporation à environ 0,5 ml dans un courant d'azote. Ajouter 10 g de l'aliment blanc, mélanger et laisser reposer 10 minutes avant de procéder à l'extraction (5.2).
En l'absence d'aliment blanc de même type que celui de l'échantillon (voir 5.1.1), le test de récupération peut être effectué selon la méthode par addition de l'étalon. Dans ce cas, l'échantillon à analyser est supplémenté d'une quantité de carbadox semblable à celle déjà présente dans l'échantillon. Cet échantillon est analysé avec l'échantillon non supplémenté et la récupération peut être calculée par différence.
5.2. Extraction
5.2.1.
Peser, à 0,01 g près, 10 g de l'échantillon et transférer dans une fiole erlenmeyer de 200 ml. Ajouter 15,0 ml d'eau, mélanger et équilibrer pendant 5 min. Ajouter 35,0 ml de méthanol-acétonitrile (3.5), boucher et soumettre pendant 30 min à agitation mécanique ou magnétique (4.1). Faire passer la solution par un papier filtre en fibre de verre (4.2). Conserver cette solution pour la purification (5.3).
5.2.2.
Peser, à 0,001 g près, 1 g de l'échantillon non broyé et transférer dans une fiole erlenmeyer de 200 ml. Ajouter 15,0 ml d'eau, mélanger et équilibrer pendant 5 min. Ajouter 35,0 ml de méthanol-acétonitrile (3.5), boucher et soumettre pendant 30 min à agitation mécanique ou magnétique (4.1). Faire passer la solution par un papier filtre en fibre de verre (4.2).
Introduire à la pipette une partie aliquote du filtrat dans une fiole jaugée de 50 ml. Ajouter 15,0 ml d'eau, ajuster au trait de jauge avec du méthanol-acétonitrile (3.5) et mélanger. La concentration de carbadox de la solution finale doit être d'environ 10 μg/ml. Une partie aliquote est passée par un filtre de 0,45 μm (4.6).
Procéder au dosage par CLHP (5.4).
5.2.3.
Peser, à 0,001 g près, 0,2 g de l'échantillon non broyé et transférer dans une fiole erlenmeyer de 250 ml. Ajouter 45,0 ml d'eau, mélanger et équilibrer pendant 5 min. Ajouter 105,0 ml de méthanol-acétonitrile (3.5), boucher et homogénéiser. Soumettre l'échantillon aux ultrasons (4.7) pendant 15 min, puis à une agitation mécanique ou magnétique pendant 15 min (4.1). Faire passer la solution par un papier filtre en fibre de verre (4.2).
Diluer une partie aliquote du filtrat avec le mélange eau-méthanol-acétonitrile (3.12) jusqu'à une concentration finale de carbadox de 10 à 15 μg/ml (pour une préparation à 10 %, le facteur de dilution est de 10). Une partie aliquote est passée par un filtre de 0,45 μm (4.6).
Procéder au dosage par CLHP (5.4).
5.3. Purification
5.3.1.
Peser 4 g d'oxyde d'aluminium (3.4) et transférer dans la colonne de verre (4.3).
5.3.2.
Faire passer 15 ml de l'extrait filtré (5.2.1) dans la colonne d'oxyde d'aluminium et éliminer les deux premiers millilitres de l'éluat. Collecter les 5 ml suivants et faire passer une partie aliquote par un filtre de 0,45 μm (4.6).
Procéder au dosage par CLHP (5.4).
5.4. Dosage par CLHP
5.4.1.
Les conditions ci-après sont proposées à titre indicatif; d'autres conditions peuvent être appliquées si elles donnent des résultats équivalents.
|
Colonne de chromatographie liquide (4.4.1): |
|
|
|
300 mm × 4 mm, C18, particules de 10 μm ou équivalent |
|
Phase mobile (3.10): |
Mélange de solution tampon à l'acétate (3.9) et d'acétonitrile (3.2), 825 + 175 (v + v) |
|
Débit: |
1,5 -2 ml/min |
|
Longueur d'onde de détection: |
365 nm |
|
Volume d'injection: |
20 μl |
Vérifier la stabilité du système chromatographique en injectant plusieurs fois la solution d'étalonnage (3.11.2) contenant 5,0 μg/ml, jusqu'à obtention de hauteurs (surfaces) de pics et de temps de rétention constants.
5.4.2.
Injecter chaque solution d'étalonnage (3.11.2) plusieurs fois et déterminer les hauteurs (surfaces) des pics pour chaque concentration. Établir une courbe d'étalonnage en utilisant les hauteurs ou surfaces moyennes des pics des solutions d'étalonnage comme ordonnées et les concentrations correspondantes en μg/ml comme abscisses.
5.4.3.
Injecter l'extrait d'échantillon [(5.3.2) pour les aliments pour animaux, (5.2.2) pour les prémélanges et (5.2.3) pour les préparations] plusieurs fois et déterminer la hauteur (surface) moyenne des pics du carbadox.
6. Calcul des résultats
À partir de la hauteur (surface) moyenne des pics du carbadox de la solution d'échantillon, déterminer la concentration de carbadox dans cette solution, en μg/ml, par référence à la courbe d'étalonnage (5.4.2).
6.1. Aliments pour animaux
La teneur w en carbadox, exprimée en mg/kg, de l'échantillon est donnée par la formule suivante:

[mg/kg]
où:
|
c |
= |
concentration de carbadox dans l'extrait d'échantillon (5.3.2) en μg/ml, |
|
V1 |
= |
volume d'extraction en ml (c'est-à-dire 50), |
|
m |
= |
poids de la prise d'essai, en grammes. |
6.2. Prémélanges et préparations
La teneur w en carbadox, exprimée en mg/kg, de l'échantillon est donnée par la formule suivante:

[mg/kg]
où:
|
c |
= |
concentration de carbadox dans l'extrait d'échantillon (5.2.2. ou 5.2.3) en μg/ml, |
|
V2 |
= |
volume d'extraction en ml (c'est-à-dire 50 pour les prémélanges et 150 pour les préparations), |
|
f |
= |
facteur de dilution selon 5.2.2 (prémélanges) ou 5.2.3 (préparations), |
|
m |
= |
poids de la prise d'essai, en grammes. |
7. Validation des résultats
7.1. Identité
L'identité de l'analyte peut être confirmée par cochromatographie ou à l'aide d'un détecteur à barrettes de diodes qui permet de comparer les spectres de l'extrait d'échantillon et de la solution d'étalonnage (3.11.2) contenant 10,0 μg/ml.
7.1.1.
Un extrait d'échantillon est supplémenté par ajout d'une quantité appropriée de la solution d'étalonnage (3.11.2). La quantité de carbadox ajoutée doit être semblable à la quantité de carbadox évaluée dans l'extrait d'échantillon.
Seule la hauteur du pic du carbadox doit être augmentée, compte tenu à la fois de la quantité ajoutée et de la dilution de l'extrait. La largeur du pic, à la moitié de sa hauteur, doit être d'environ 10 % de sa largeur initiale.
7.1.2.
Évaluer les résultats conformément aux critères suivants:
a) les longueurs d'onde d'absorption maximale des spectres de l'échantillon et de l'étalon, enregistrées au sommet des pics sur le chromatogramme, doivent être identiques, dans une marge déterminée par le pouvoir de résolution du système de détection. Pour la détection par barrettes de diodes, elle est généralement de ± 2 nm;
b) entre 225 et 400 nm, les spectres de l'échantillon et de l'étalon, enregistrés au sommet des pics sur le chromatogramme, ne peuvent être différents pour les parties du spectre comprises entre 10 et 100 % d'absorbance relative. Ce critère est rempli lorsque les mêmes maxima sont présents et que l'écart observé entre les deux spectres ne dépasse nulle part 15 % de l'absorbance de l'analyte étalon;
c) entre 225 en 400 nm, les spectres de la courbe ascendante, du sommet et de la courbe descendante du pic produits par l'extrait d'échantillon ne doivent pas être différents les uns des autres pour les parties du spectre situées entre 10 et 100 % de l'absorbance relative. Ce critère est rempli lorsque les mêmes maxima sont présents et que l'écart observé entre les spectres ne dépasse nulle part 15 % de l'absorbance du spectre au sommet du pic.
Si l'un de ces critères n'est pas rempli, la présence de l'analyte n'est pas confirmée.
7.2. Répétabilité
Pour les teneurs d'au moins 10 mg/kg, la différence entre les résultats de deux dosages parallèles effectués sur le même échantillon ne peut dépasser 15 % du résultat supérieur.
7.3. Récupération
Pour un échantillon (blanc) supplémenté, la récupération doit être de 90 % au moins.
8. Résultats d'une étude collaborative
Dans le cadre d'une étude collaborative, six aliments pour animaux, quatre prémélanges et trois préparations ont été analysés par huit laboratoires. Pour chaque échantillon, les analyses ont été effectuées en double (des informations plus détaillées sur cette étude collaborative figurent dans le Journal of AOAC International, volume 71, 1988, pp. 484-490.) Les résultats de l'étude (hormis les valeurs aberrantes) figurent ci-après:
Tableau 1
Résultats de l'étude collaborative sur les aliments pour animaux
|
|
Échantillon 1 |
Échantillon 2 |
Échantillon 3 |
Échantillon 4 |
Échantillon 5 |
Échantillon 6 |
|
L |
8 |
8 |
8 |
8 |
8 |
8 |
|
n |
15 |
14 |
15 |
15 |
15 |
15 |
|
Moyenne (mg/kg) |
50,0 |
47,6 |
48,2 |
49,7 |
46,9 |
49,7 |
|
Sr (mg/kg) |
2,90 |
2,69 |
1,38 |
1,55 |
1,52 |
2,12 |
|
CVr (%) |
5,8 |
5,6 |
2,9 |
3,1 |
3,2 |
4,3 |
|
SR (mg/kg) |
3,92 |
4,13 |
2,23 |
2,58 |
2,26 |
2,44 |
|
CVR (%) |
7,8 |
8,7 |
4,6 |
5,2 |
4,8 |
4,9 |
|
Teneur nominale (mg/kg) |
50,0 |
50,0 |
50,0 |
50,0 |
50,0 |
50,0 |
Tableau 2
Résultats de l'étude collaborative sur les prémélanges et les préparations
|
|
Prémélanges |
Préparations |
|||||
|
A |
B |
C |
D |
A |
B |
C |
|
|
L |
7 |
7 |
7 |
7 |
8 |
8 |
8 |
|
n |
14 |
14 |
14 |
14 |
16 |
16 |
16 |
|
Moyenne (g/kg) |
8,89 |
9,29 |
9,21 |
8,76 |
94,6 |
98,1 |
104 |
|
Sr (g/kg) |
0,37 |
0,28 |
0,28 |
0,44 |
4,1 |
5,1 |
7,7 |
|
CVr (%) |
4,2 |
3,0 |
3,0 |
5,0 |
4,3 |
5,2 |
7,4 |
|
SR (g/kg) |
0,37 |
0,28 |
0,40 |
0,55 |
5,4 |
6,4 |
7,7 |
|
CVR (%) |
4,2 |
3,0 |
4,3 |
6,3 |
5,7 |
6,5 |
7,4 |
|
Teneur nominale (g/kg) |
10,0 |
10,0 |
10,0 |
10,0 |
100 |
100 |
100 |
|
L |
= |
nombre de laboratoires |
|
n |
= |
nombre de valeurs individuelles |
|
Sr |
= |
écart type de répétabilité |
|
CVr |
= |
coefficient de variation de la répétabilité |
|
SR |
= |
écart type de reproductibilité |
|
CVR |
= |
coefficient de variation de la reproductibilité |
ANNEXE IX
TABLEAUX DE CORRESPONDANCE VISÉS À L'ARTICLE 6
1. Directive 71/250/CEE
|
Directive 71/250/CEE |
Le présent règlement |
|
Article 1er, premier alinéa |
Article 3 |
|
Article 1er, deuxième alinéa |
Article 2 |
|
Article 2 |
— |
|
Article 3 |
— |
|
Annexe, point 1 |
Annexe II |
|
Annexe, point 2 |
— |
|
Annexe, point 3 |
— |
|
Annexe, point 4 |
Annexe III, point O |
|
Annexe, point 5 |
Annexe III, point M |
|
Annexe, point 6 |
Annexe III, point N |
|
Annexe, point 7 |
Annexe III, point Q |
|
Annexe, point 9 |
Annexe III, point K |
|
Annexe, point 10 |
— |
|
Annexe, point 11 |
— |
|
Annexe, point 12 |
Annexe III, point J |
|
Annexe, point 14 |
Annexe III, point D |
|
Annexe, point 16 |
— |
2. Directive 71/393/CEE
|
Directive 71/393/CEE |
Le présent règlement |
|
Article 1er |
Article 3 |
|
Article 2 |
— |
|
Article 3 |
— |
|
Annexe, point 1 |
Annexe III, point A |
|
Annexe, point 2 |
Annexe III, point E |
|
Annexe, point 3 |
Annexe III, point P |
|
Annexe, point 4 |
Annexe III, point H |
3. Directive 72/199/CEE
|
Directive 72/199/CEE |
Le présent règlement |
|
Article 1er |
Article 3 |
|
Article 2 |
— |
|
Article 3 |
— |
|
Article 4 |
— |
|
Annexe I, point 1 |
Annexe III, point L |
|
Annexe I, point 2 |
Annexe III, point C |
|
Annexe I, point 3 |
— |
|
Annexe I, point 4 |
— |
|
Annexe I, point 5 |
Annexe V, point A |
|
Annexe II |
— |
4. Directive 73/46/CEE
|
Directive 73/46/CEE |
Le présent règlement |
|
Article 1er |
Article 3 |
|
Article 3 |
— |
|
Article 4 |
— |
|
Annexe I, point 1 |
Annexe III, point B |
|
Annexe I, point 2 |
— |
|
Annexe I, point 3 |
Annexe III, point I |
5. Directive 76/371/CEE
|
Directive 76/371/CEE |
Le présent règlement |
|
Article 1er |
Article 1er |
|
Article 2 |
— |
|
Article 3 |
— |
|
Annexe |
Annexe I |
6. Directive 76/372/CEE
|
Directive 76/372/CEE |
Le présent règlement |
|
Article 1er |
— |
|
Article 2 |
— |
|
Article 3 |
— |
|
Annexe |
— |
7. Directive 78/633/CEE
|
Directive 78/633/CEE |
Le présent règlement |
|
Article 1er |
Article 3 |
|
Article 2 |
— |
|
Article 3 |
— |
|
Annexe, point 1 |
— |
|
Annexe, point 2 |
— |
|
Annexe, point 3 |
Annexe IV, point C |
8. Directive 81/715/CEE
|
Directive 81/715/CEE |
Le présent règlement |
|
Article 1er |
— |
|
Article 2 |
— |
|
Article 3 |
— |
|
Annexe |
— |
9. Directive 84/425/CEE
|
Directive 84/425/CEE |
Le présent règlement |
|
Article 1er |
— |
|
Article 2 |
— |
|
Article 3 |
— |
|
Annexe |
— |
10. Directive 86/174/CEE
|
Directive 86/174/CEE |
Le présent règlement |
|
Article 1er |
Article 4 |
|
Article 2 |
— |
|
Article 3 |
— |
|
Annexe |
Annexe VII |
11. Directive 93/70/CEE
|
Directive 93/70/CEE |
Le présent règlement |
|
Article 1er |
Article 3 |
|
Article 2 |
— |
|
Article 3 |
— |
|
Annexe |
Annexe IV, point D |
12. Directive 93/117/CE
|
Directive 93/117/CE |
Le présent règlement |
|
Article 1er |
Articles 3 et 5 |
|
Article 2 |
— |
|
Article 3 |
— |
|
Annexe, point 1 |
Annexe IV, point E |
|
Annexe, point 2 |
Annexe VIII, point A |
13. Directive 98/64/CE
|
Directive 98/64/CE |
Le présent règlement |
|
Article 1er |
Articles 3 et 5 |
|
Article 2 |
— |
|
Article 3 |
— |
|
Article 4 |
— |
|
Annexe, partie A |
Annexe III, point F |
|
Annexe, partie C |
Annexe VIII, point B |
14. Directive 1999/27/CE
|
Directive 1999/27/CE |
Le présent règlement |
|
Article 1er |
Articles 3 et 5 |
|
Article 2 |
— |
|
Article 3 |
— |
|
Article 4 |
— |
|
Article 5 |
— |
|
Article 6 |
— |
|
Article 7 |
— |
|
Annexe, partie A |
Annexe VIII, point C |
|
Annexe, partie B |
Annexe IV, point F |
|
Annexe, partie C |
Annexe VIII, point D |
15. Directive 1999/76/CE
|
Directive 1999/76/CE |
Le présent règlement |
|
Article 1er |
Article 3 |
|
Article 2 |
— |
|
Article 3 |
— |
|
Article 4 |
— |
|
Annexe |
Annexe IV, point G |
16. Directive 2000/45/CE
|
Directive 2000/45/CE |
Le présent règlement |
|
Article 1er |
Article 3 |
|
Article 2 |
— |
|
Article 3 |
— |
|
Article 4 |
— |
|
Annexe, partie A |
Annexe IV, point A |
|
Annexe, partie B |
Annexe IV, point B |
|
Annexe, partie C |
Annexe III, point G |
17. Directive 2002/70/CE
|
Directive 2002/70/CE |
Le présent règlement |
|
Article 1er |
Article 1er |
|
Article 2 |
Articles 2 et 3 |
|
Article 3 |
— |
|
Article 4 |
— |
|
Article 5 |
— |
|
Annexe I |
Annexe I et Annexe V, point B.I |
|
Annexe II |
Annexe II et Annexe V, point B.II |
18. Directive 2003/126/CE
|
Directive 2003/126/CE |
Le présent règlement |
|
Article 1er |
Article 3 |
|
Article 2 |
— |
|
Article 3 |
— |
|
Article 4 |
— |
|
Article 5 |
— |
|
Article 6 |
— |
|
Annexe |
Annexe VI |
( 1 ) JO L 268 du 18.10.2003, p. 29.
( 2 ) JO L 140 du 30.5.2002, p. 10.
( 3 ) JO L 70 du 16.3.2005, p. 1.
( 4 ) Écraser tous les agrégats (si nécessaire, en les séparant de la masse et en réunissant ensuite le tout).
( 5 ) Sauf pour le fourrage grossier ou fourrage de faible densité relative.
( 6 ) Sauf pour le fourrage grossier ou fourrage de faible densité relative.
( 7 ) JO L 229 du 1.9.2009, p. 1.
( 7 ) Pour la dessiccation des céréales, des farines, des gruaux et des semoules, l'étuve doit avoir une capacité calorifique telle que, réglée préalablement à une température de 131 oC, elle puisse de nouveau atteindre cette température moins de 45 minutes après la mise en place du nombre maximal de prises d'essai à sécher simultanément. Elle doit avoir une ventilation telle que, en séchant pendant deux heures toutes les prises d'essai de froment tendre qu'elle peut contenir, les résultats présentent une différence inférieure à 0,15 % par rapport aux résultats obtenus après quatre heures de séchage.
( 7 ) Remplacer les fragments de pierre ponce par des perles de verre lorsque la matière grasse doit faire l'objet d'examens qualitatifs ultérieurs.
( 7 ) JO L 125 du 23.5.1996, p. 35.
( 7 ) Étude menée par le groupe de travail sur les aliments pour animaux de la Verband Deutscher Landwirtschaftlicher Untersuchungs- und Forschungsanstalten (VDLUFA).
( 7 ) Étude menée par le groupe de travail sur les aliments pour animaux de la Verband Deutscher Landwirtschaftlicher Untersuchungs- und Forschungsanstalten (VDLUFA).
( 7 ) D'autres méthodes de minéralisation peuvent être utilisées, à condition qu'il ait été démontré qu'elles donnaient des résultats similaires (telle la minéralisation sous pression par micro-ondes).
( 7 ) Les fourrages verts (frais ou déshydratés) sont susceptibles de contenir de grandes quantités de silice végétale pouvant retenir des oligoéléments et qui doivent être éliminés. Les échantillons de ces aliments pour animaux doivent par conséquent être soumis au traitement suivant. Effectuer l'opération 5.1.1.1. jusqu'au stade de la filtration. Laver le papier filtre contenant le résidu insoluble à deux reprises avec de l'eau bouillante et le placer dans un creuset en quartz ou en platine. Incinérer dans le four à moufle (4.1) à une température inférieure à 550 oC jusqu'à ce que toute la matière charbonneuse ait complètement disparu. Laisser refroidir, ajouter quelques gouttes d'eau puis 10 à 15 ml d'acide fluorhydrique (3.4) et évaporer à sec à 150 oC environ. Si le résidu contient encore de la silice, dissoudre celle-ci dans quelques ml d'acide fluorhydrique (3.4) et évaporer à sec. Ajouter 5 gouttes d'acide sulfurique (3.5) et chauffer jusqu'à disparition des fumées blanches. Ajouter 5 ml d'acide chlorhydrique 6 mol/l (3.2) et environ 30 ml d'eau, chauffer, filtrer la solution dans une fiole jaugée de 250 ml et ajuster au trait de jauge avec de l'eau (la concentration en HCl est de 0,5 mol/l environ). Poursuivre le procédé à partir du point 5.1.2.
( 7 ) The Analyst 108, 1983, pp. 1252-1256.
( *1 ) Analyst, 1995, 120, 2175-2180.
( 8 ) Tableau des facteurs d'équivalence toxique (TEF) pour les PCDD, PCDF et PCB de type dioxine: TEF-OMS pour l'évaluation des risques pour l'homme, fondés sur les conclusions de la réunion des experts du Programme international sur la sécurité des substances chimiques (PISSC) de l'Organisation mondiale de la santé (OMS), qui s'est tenue à Genève en juin 2005 [Martin van den Berg et al., The 2005 World Health Organization Re-evaluation of Human and Mammalian Toxic Equivalency Factors for Dioxins and Dioxin-like Compounds, Toxicological Sciences 93(2), 223–241 (2006)].
|
Congénère |
Valeur TEF |
Congénère |
Valeur TEF |
|
Dibenzo-p-dioxines («PCDD») et Dibenzo-p-furanes («PCDF») |
|
PCB «de type dioxine» PCB non-ortho + PCB mono-ortho |
|
|
2,3,7,8-TCDD |
1 |
|
|
|
1,2,3,7,8-PeCDD |
1 |
PCB non ortho |
|
|
1,2,3,4,7,8-HxCDD |
0,1 |
PCB 77 |
0,0001 |
|
1,2,3,6,7,8-HxCDD |
0,1 |
PCB 81 |
0,0003 |
|
1,2,3,7,8,9-HxCDD |
0,1 |
PCB 126 |
0,1 |
|
1,2,3,4,6,7,8-HpCDD |
0,01 |
PCB 169 |
0,03 |
|
OCDD |
0,0003 |
PCB mono ortho |
|
|
2,3,7,8-TCDF |
0,1 |
PCB 105 |
0,00003 |
|
1,2,3,7,8-PeCDF |
0,03 |
PCB 114 |
0,00003 |
|
2,3,4,7,8-PeCDF |
0,3 |
PCB 118 |
0,00003 |
|
1,2,3,4,7,8-HxCDF |
0,1 |
PCB 123 |
0,00003 |
|
1,2,3,6,7,8-HxCDF |
0,1 |
PCB 156 |
0,00003 |
|
1,2,3,7,8,9-HxCDF |
0,1 |
PCB 157 |
0,00003 |
|
2,3,4,6,7,8-HxCDF |
0,1 |
PCB 167 |
0,00003 |
|
1,2,3,4,6,7,8-HpCDF |
0,01 |
PCB 189 |
0,00003 |
|
1,2,3,4,7,8,9-HpCDF |
0,01 |
|
|
|
OCDF |
0,0003 |
|
|
Abréviations utilisées: «T» = tétra; «Pe» = penta; «Hx» = hexa; «Hp» = hepta; «O» = octa; «CDD» = chlorodibenzodioxine; «CDF» = chlorodibenzofurane; «CB» = chlorobiphényle.
( 9 ) Décision 2002/657/CE de la Commission du 14 août 2002 portant modalités d'application de la directive 96/23/CE du Conseil en ce qui concerne les performances des méthodes d'analyse et l'interprétation des résultats (JO L 221 du 17.8.2002, p. 8).
( 10 ) Les principes énoncés dans le document «Guidance Document on Measurement Uncertainty for Laboratories performing PCDD/PCDF and PCB Analysis using Isotope Dilution Mass Spectrometry» (http://ec.europa.eu/food/safety/animal-feed_en) doivent être suivis lorsqu'ils sont applicables.
( 11 ) L'«estimation supérieure» est une valeur calculée sur la base d'une contribution de chaque congénère non quantifié égale à la limite de quantification. L'«estimation inférieure» est une valeur calculée sur la base d'une contribution de chaque congénère non quantifié égale à zéro. L'«estimation intermédiaire» est une valeur calculée sur la base d'une contribution de chaque congénère non quantifié égale à la moitié de la limite de quantification.
( 12 ) La «double analyse» est l'analyse séparée des analytes visés au moyen d'une deuxième partie aliquote du même échantillon homogénéisé. En général, les exigences en matière de double analyse prévues à l'annexe II, chapitre C, point 3, s'appliquent. Néanmoins, pour les méthodes qui prévoient des étalons internes marqués au 13C pour les analytes considérés, la double analyse n'est nécessaire que si le résultat de la première détermination n'est pas conforme. La double analyse est nécessaire pour exclure la possibilité d'une contamination croisée interne ou un mélange accidentel des échantillons. Si l'analyse est effectuée lors d'une contamination, la confirmation par double analyse peut être omise lorsque la traçabilité permet d'établir le lien entre les échantillons prélevés en vue de l'analyse et le cas de contamination et lorsque la teneur constatée est considérablement supérieure à la teneur maximale.
( 13 ) L'«estimation supérieure» est une valeur calculée sur la base d'une contribution de chaque congénère non quantifié à l'équivalent toxique (TEQ) égale à la limite de quantification. L'«estimation inférieure» est une valeur calculée sur la base d'une contribution de chaque congénère non quantifié au TEQ égale à zéro. L'«estimation intermédiaire» est une valeur calculée sur la base d'une contribution de chaque congénère non quantifié au TEQ égale à la moitié de la limite de quantification.
( 14 ) En général, les exigences en matière de double analyse prévues à l'annexe II, chapitre C, point 2, s'appliquent. Néanmoins, pour les méthodes de confirmation qui prévoient des étalons internes marqués au 13C pour les analytes considérés, la double analyse n'est nécessaire que si le résultat de la première détermination n'est pas conforme. La double analyse est nécessaire pour exclure la possibilité d'une contamination croisée interne ou un mélange accidentel des échantillons. Si l'analyse est effectuée lors d'une contamination, la confirmation par double analyse peut être omise lorsque la traçabilité permet d'établir le lien entre les échantillons prélevés en vue de l'analyse et le cas de contamination et lorsque la teneur constatée est considérablement supérieure à la teneur maximale.
( 15 ) Les explications et les prescriptions concernant la double analyse applicable aux teneurs maximales figurant à la note de bas de page 2 valent également pour la mesure des seuils d'intervention.
( 16 ) Règlement (CE) no 183/2005 du Parlement européen et du Conseil du 12 janvier 2005 établissant des exigences en matière d'hygiène des aliments pour animaux (JO L 35 du 8.2.2005, p. 1).
( 17 ) Les méthodes de bioanalyse ne sont pas spécifiques aux congénères inclus dans le système des TEF. D'autres composés structurellement proches activant le récepteur aryl hydrocarbone (AhR) et susceptibles d'être présents dans l'extrait d'échantillon contribuent à la réponse générale. Aussi les résultats d'une bioanalyse ne sauraient-ils être une estimation et constituent plutôt une indication de la valeur TEQ dans l'échantillon.
( 18 ) «Guidance Document on Measurement Uncertainty for Laboratories performing PCDD/F and PCB Analysis using Isotope Dilution Mass Spectrometry» (http://ec.europa.eu/food/safety/animal-feed_en), «Guidance Document on the Estimation of LOD and LOQ for Measurements in the Field of Contaminants in Feed and Food» (http://ec.europa.eu/food/safety/animal-feed_en)
( 19 ) Les prescriptions actuelles sont fondées sur les TEF publiés dans M. Van den Berg et al., Toxicological Sciences 93(2), p. 223 à 241 (2006).
( 20 ) Les congénères qui coéluent souvent sont, par exemple, les PCB 28/31, les PCB 52/69 et les PCB 138/163/164. Pour la CG-SM, il faut tenir compte aussi des interférences possibles de fragments de congénères plus fortement chlorés.
( 21 ) Les principes décrits dans le document «Guidance Document on the Estimation of LOD and LOQ for Measurements in the Field of Contaminants in Feed and Food» (http://ec.europa.eu/food/safety/animal-feed_en) doivent être suivis lorsqu'ils sont applicables.
( 22 ) Il est fortement recommandé d'avoir une contribution de la valeur de blanc de réactif inférieure à la teneur en un contaminant d'un échantillon. Il incombe au laboratoire de contrôler la variation des valeurs de blanc, notamment si ces valeurs sont soustraites.
( 23 ) Les prescriptions actuelles sont fondées sur les TEF publiés dans M. Van den Berg et al., Toxicological Sciences 93(2), p. 223 à 241 (2006).
( 24 ) http://eurl.craw.eu/
( 25 ) La liste des amorces et sondes pour chaque espèce animale ciblée par l’essai est disponible sur le site web du laboratoire de référence de l’Union européenne pour la détection de protéines animales dans les aliments pour animaux.
( 26 ) Des exemples de mélanges maîtres utilisables sont disponibles sur le site web du laboratoire de référence de l’Union européenne pour la détection de protéines animales dans les aliments pour animaux.