This document is an excerpt from the EUR-Lex website
Document 52023XC0508(01)
Commission Guidance on the content and structure of the summary of the clinical investigation report (Text with EEA relevance) 2023/C 163/06
Commission Guidance on the content and structure of the summary of the clinical investigation report (Text with EEA relevance) 2023/C 163/06
Commission Guidance on the content and structure of the summary of the clinical investigation report (Text with EEA relevance) 2023/C 163/06
C/2023/2622
OJ C 163, 8.5.2023, p. 7–13
(BG, ES, CS, DA, DE, ET, EL, EN, FR, GA, HR, IT, LV, LT, HU, MT, NL, PL, PT, RO, SK, SL, FI, SV)
|
8.5.2023 |
EN |
Official Journal of the European Union |
C 163/7 |
COMMISSION GUIDANCE
on the content and structure of the summary of the clinical investigation report
(Text with EEA relevance)
(2023/C 163/06)
Table of Contents
|
1. |
Introduction | 8 |
|
2. |
The summary of the clinical investigation report | 8 |
|
3. |
Revision clause | 12 |
|
4. |
Glossary and abbreviations | 12 |
|
5. |
References | 12 |
1. Introduction
This document is intended to provide Commission guidance, in accordance with Article 77(6) of Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices (hereafter: the MDR), for the content and structure of the summary of the clinical investigation report.
This guidance aims to ensure that the summary of the clinical investigation report presents information about the design, conduct, analysis and results of the clinical investigation in terms and in a format that are easily understandable to the intended user of the medical device.1
According to Article 77 (5) of the MDR, the sponsor of a clinical investigation shall submit a report of the clinical investigation within one year of the end of the clinical investigation or within three months of the early termination and this report shall be accompanied by a summary. The minimum requirements of the clinical investigation report are outlined in Section 7, Chapter III of Annex XV of the MDR. Section 7, Chapter III of Annex XV of the MDR also outlines what will be covered by the summary, namely:
|
— |
Title of the clinical investigation |
|
— |
Purpose of the clinical investigation |
|
— |
Description of the investigation, investigational design and methods used |
|
— |
Results of the investigation |
|
— |
Conclusion of the investigation |
According to Article 77(5) of the MDR, the report and summary shall be submitted to Member States in which the clinical investigation was conducted by means of the electronic system referred to in Article 73 of the MDR. According to Article 77(7) of the MDR, the report and the summary shall become publicly accessible through the electronic system referred to in Article 73 of the MDR, at the latest when the device is registered in accordance with Article 29 of the MDR and before it is placed on the market. In cases of early termination or temporary halt, the summary and the report shall become publicly accessible immediately after submission.
2. The summary of the clinical investigation report
Explanatory note
|
— |
This is a summary document. Only relevant information should be supplied. Where a ‘brief description’ is requested be as concise as possible. Avoid simply copying bodies of text from the full clinical investigation report. |
|
— |
The language used must be appropriate for the intended user(s) of the device. Their levels of health literacy and numeracy should be considered at all times when developing this summary document. |
|
— |
Ensure no promotional content is included in this document. |
2.1. Cover page
|
Date of summary: |
|
|
Title of clinical investigation: |
|
|
Name and contact details of the entity sponsoring the study: |
|
|
Name of the entity funding the study2: |
|
|
Single identification number: |
Prior to full functioning of EUDAMED, this will be the CIV-ID, which is obtained from the authorising competent authority. Once EUDAMED is functional, this refers to the single identification number as referred to in Article 70(1) of MDR |
|
Clinical investigation plan number: |
|
2.2. Content and structure of the summary of the clinical investigation report
|
Title of the clinical investigation - summary information4 |
|
|
Brief Study Title |
|
|
Full study title3 |
A brief description of the design, the experimental medical device, comparator (when relevant), care provider (when relevant), and population of the clinical investigation. |
|
Dates of investigation3 |
Start (the first act of recruitment in the clinical investigation) and end (the last visit of the last subject in the clinical investigation) dates of the clinical investigation5. Please see MDCG document 2021-6 for further description of these terms. |
|
Location(s) |
Where the investigation was conducted, including site location and country. |
|
If relevant, reason for temporary halt or early termination3 |
Where relevant. Reasons may include positive active findings, positive control findings, safety findings, futility, slow recruitment, external evidence, etc.2 |
|
Purpose of the clinical investigation4 |
||||||||
|
Brief explanation of the rationale for the clinical investigation, including:
Depending on the context of the clinical investigation, please describe:
|
|
Description of the investigational device, clinical investigation, and methods used4 |
|
|
Do not include any results, analysis, conclusions or discussion points in this section. |
|
|
Description of participants4 |
Description of the eligibility criteria for participants and the settings. Where applicable, a description of the eligibility criteria for centres involved in the investigation and those performing the interventions. |
|
Description of the device and comparator6 |
Should include a description of the investigational device, its version/variant and intended purpose including the different components required for the medical intervention(s) involving the device under investigation (e.g. the pre and post-operative care, medical/surgical intervention, etc.). For comparative studies, descriptions of both the experimental intervention and comparator should be provided. |
|
Description of procedures to use the device9 |
Brief description of the procedures and methods necessary for use of the device in the clinical investigation. |
|
Study Design4 |
Description and justification of the chosen study design, i.e. randomised controlled trial (parallel, cluster, crossover, factorial, non-inferiority), non-randomised comparative study, non-comparative study, other. |
|
Objectives and endpoints 4 |
Brief description clearly stating the primary and secondary objectives of the investigation as well as hypotheses that were tested. Primary and secondary endpoints should also be clearly defined. |
|
Sample size 7 |
Please include the power and sample size estimations. Include adjustments made in this calculation for dropout rate/lost to follow up, where relevant.10 |
|
Randomisation and blinding8 |
Where relevant, a description of the methods of intervention allocation and blinding methods used (if any). |
|
Follow up duration4 |
Length of time participants were followed in the study. Also indicate the intended lifetime of the device under investigation. |
|
Concomitant treatments4 |
Description of any treatments that were necessary for all subjects receiving the medical device in this clinical investigation. Please explain how this may differ from usual standard of care for this medical device, if relevant. |
|
Statistical analysis methods4 |
Brief description of the statistical methods used to provide outcomes’ estimates, compare groups for the primary outcome, to perform additional analyses, to adjust for biases, to deal with missing data. |
|
Substantial modifications11 |
Where relevant, a table describing any substantial modifications to the clinical investigation plan, relevant versions of the clinical investigation plan and dates of these modifications. Please confirm approval from an ethics committee has been received for these modifications. |
|
Results of the investigation4 |
|
|
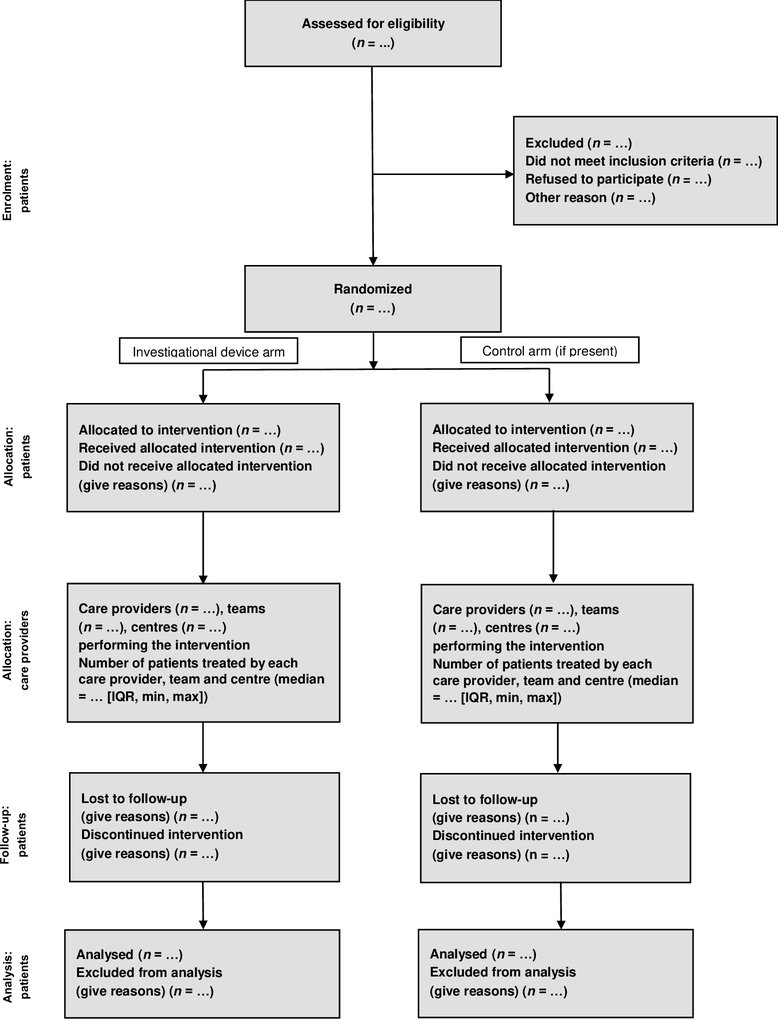
Participant flow12 |
Number of participants screened, enrolled, allocated and followed up to each intervention. The use of this or similar flowchart is strongly recommended13. Please note, the below flowchart is for randomised two arm studies. The flowchart will need to be adjusted for different study designs.
|
|
Baseline demographic and clinical characteristics4 |
If available, description of age, sex, ethnic background, country, stage of disease, relevant co-morbidities and any other disease-influencing factors in participants. When applicable, description of care providers (case volume, qualification, expertise, etc.) and centres (volume) in each group. |
|
Outcome of the intervention4 |
Results on primary endpoint including in absolute numbers when feasible (e.g. 10/20, not 50 %), estimated effect size and the precision of the result. Please include whether an analysis is intention-to-treat or per protocol. Results of secondary endpoints or additional analyses can be reported, but the dangers of drawing conclusions from these results should be discussed. |
|
Safety outcomes 14 |
Description of adverse events15, adverse device effects16 and device deficiencies15Listed table in order of most to least frequent, in numerical absolute numbers (X out of YX subjects) and percentage (X% of subjects) and the nature of these events/effects (expected/unexpected). Only aggregated information related to these events/effects should be presented.5List of any subject deaths. Number of subjects withdrawn from the investigation and reasons. |
|
Deviations to clinical investigation plan5 |
Where relevant, a description of any deviations to the clinical investigation plan which occurred during the clinical investigation |
|
Conclusion of the clinical investigation4 |
|
|
What the results mean17 |
Brief description of the results of this investigation: overall assessment of benefits vs risks of the intervention in light of the investigation What does this clinical investigation add to the clinical data related to the safety and performance of the device? |
|
What do the results add to the current scientific knowledge17 |
Brief description of the results of this investigation in the context of current evidence: overall benefits vs risks assessment of the investigational medical device in the context of all other evidence available, and the implications of the results in clinical practice. |
|
Limitations18 |
Any limitations of the investigation, such as biases in the investigation, uncertainties which remain following the investigation or limitations in the applicability of the results to real world settings |
|
Potential for future studies18 |
|
3. Revision clause
Based on the experience gained through the application of these guidelines, the Commission may consider revising this document.
4. Glossary and abbreviations
|
CIV-ID |
Clinical Investigation Identification (number) |
|
EUDAMED |
European Database for Medical Devices |
|
MDR |
Medical Device Regulation |
5. References
|
1. |
Regulation (EU) 2017/745 Article 77 (5) |
|
2. |
The funding entity is the entity which provides financing for the clinical investigation, as per the reference to finance in Regulation (EU) 745/2017 Annex XV, Chapter II, Section 3.1.4 |
|
3. |
Regulation (EU) 2017/745 Article 77 |
|
4. |
Regulation (EU) 2017/745 Annex XV, Chapter III, Section 7 |
|
5. |
MDCG guidance document 2021-6: Questions & Answers regarding clinical investigation |
|
6. |
Regulation (EU) 2017/745 Annex XV, Chapter II, Section 3.6.2 |
|
7. |
Regulation (EU) 2017/745 Annex XV, Chapter II, Section 3.6.3 |
|
8. |
Regulation (EU) 2017/745 Annex XV, Chapter II, Section 3.6.4 |
|
9. |
Regulation (EU) 2017/745 Annex XV, Chapter II, Section 3.6.5 |
|
10. |
Regulation (EU) 2017/745 Annex XV, Chapter II, Section 3.7 |
|
11. |
‘Substantial modification’ as described in MDCG guidance document 2021-6 Regulation (EU) 2017/745 – Questions & Answers regarding clinical investigation |
|
12. |
Adapted from Regulation (EU) 2017/745 Annex XV, Chapter III, Section 7 and International standard EN ISO 14155:2020 Annex D.7 |
|
13. |
Modified CONSORT flow diagram for individual randomized, controlled trials of nonpharmacologic treatment. Isabelle Boutron, MD, PhD; et al for the CONSORT Group: Extending the CONSORT Statement to Randomized Trials of Nonpharmacologic Treatment: Explanation and Elaboration. Ann Intern Med. 2008;148:295-309. |
|
14. |
Regulation (EU) 2017/745 Article 80 |
|
15. |
Regulation (EU) 2017/745 Article 2 |
|
16. |
Regulation (EU) 2017/745 Annex XV, Chapter II, Section 2.5 |
|
17. |
Adapted from Regulation (EU) 2017/745 Annex XV, Chapter III, Section 7 and International standard ISO 14155:2020 Annex D.8 |
|
18. |
International standard ISO 14155:2020 Annex D.8 |