This document is an excerpt from the EUR-Lex website
Document 01991R2568-20160804
Commission Regulation (EEC) No 2568/91 of 11 July 1991 on the characteristics of olive oil and olive-residue oil and on the relevant methods of analysis
Consolidated text: Commission Regulation (EEC) No 2568/91 of 11 July 1991 on the characteristics of olive oil and olive-residue oil and on the relevant methods of analysis
Commission Regulation (EEC) No 2568/91 of 11 July 1991 on the characteristics of olive oil and olive-residue oil and on the relevant methods of analysis

1991R2568 — EN — 04.08.2016 — 029.001
This text is meant purely as a documentation tool and has no legal effect. The Union's institutions do not assume any liability for its contents. The authentic versions of the relevant acts, including their preambles, are those published in the Official Journal of the European Union and available in EUR-Lex. Those official texts are directly accessible through the links embedded in this document
|
COMMISSION REGULATION (EEC) No 2568/91 of 11 July 1991 on the characteristics of olive oil and olive-residue oil and on the relevant methods of analysis (OJ L 248 5.9.1991, p. 1) |
Amended by:
|
|
|
Official Journal |
||
|
No |
page |
date |
||
|
L 349 |
36 |
18.12.1991 |
||
|
L 150 |
17 |
2.6.1992 |
||
|
L 176 |
27 |
30.6.1992 |
||
|
L 199 |
18 |
18.7.1992 |
||
|
L 327 |
28 |
13.11.1992 |
||
|
L 22 |
58 |
30.1.1993 |
||
|
Amended by: COMMISSION REGULATION (EEC) No 826/93 of 6 April 1993 |
L 87 |
6 |
7.4.1993 |
|
|
L 66 |
29 |
18.3.1993 |
||
|
L 24 |
33 |
29.1.1994 |
||
|
L 280 |
43 |
29.10.1994 |
||
|
L 69 |
1 |
29.3.1995 |
||
|
L 258 |
49 |
28.10.1995 |
||
|
L 341 |
25 |
12.12.1997 |
||
|
L 28 |
5 |
4.2.1998 |
||
|
L 282 |
55 |
20.10.1998 |
||
|
L 46 |
15 |
20.2.1999 |
||
|
L 65 |
9 |
7.3.2001 |
||
|
L 276 |
8 |
19.10.2001 |
||
|
L 128 |
8 |
15.5.2002 |
||
|
L 295 |
57 |
13.11.2003 |
||
|
L 161 |
11 |
22.6.2007 |
||
|
L 178 |
11 |
5.7.2008 |
||
|
L 23 |
1 |
27.1.2011 |
||
|
COMMISSION IMPLEMENTING REGULATION (EU) No 661/2012 of 19 July 2012 |
L 192 |
3 |
20.7.2012 |
|
|
COMMISSION IMPLEMENTING REGULATION (EU) No 299/2013 of 26 March 2013 |
L 90 |
52 |
28.3.2013 |
|
|
COMMISSION IMPLEMENTING REGULATION (EU) No 1348/2013 of 16 December 2013 |
L 338 |
31 |
17.12.2013 |
|
|
COMMISSION DELEGATED REGULATION (EU) 2015/1830 of 8 July 2015 |
L 266 |
9 |
13.10.2015 |
|
|
COMMISSION IMPLEMENTING REGULATION (EU) 2015/1833 of 12 October 2015 |
L 266 |
29 |
13.10.2015 |
|
|
COMMISSION IMPLEMENTING REGULATION (EU) 2016/1227 of 27 July 2016 |
L 202 |
7 |
28.7.2016 |
|
Corrected by:
COMMISSION REGULATION (EEC) No 2568/91
of 11 July 1991
on the characteristics of olive oil and olive-residue oil and on the relevant methods of analysis
Article 1
1. Oils, the characteristics of which comply with those set out in points 1 and 2 of Annex I to this Regulation, shall be deemed to be virgin olive oils within the meaning of point 1(a) and (b) of the Annex to Regulation No 136/66/EEC.
2. Oil, the characteristics of which comply with those set out in point 3 of Annex I to this Regulation, shall be deemed to be lampante olive oil within the meaning of point 1(c) of the Annex to Regulation No 136/66/EEC.
3. Oil, the characteristics of which comply with those set out in point 4 of Annex I to this Regulation, shall be deemed to be refined olive oil within the meaning of point 2 of the Annex to Regulation No 136/66/EEC.
4. Oil, the characteristics of which comply with those set out in point 5 of Annex I to this Regulation, shall be deemed to be olive oil composed of refined olive oils and virgin olive oils within the meaning of point 3 of the Annex to Regulation No 136/66/EEC.
5. Oil, the characteristics of which comply with those set out in point 6 of Annex I to this Regulation, shall be deemed to be crude olive-pomace oil within the meaning of point 4 of the Annex to Regulation No 136/66/EEC.
6. Oil, the characteristics of which comply with those set out in point 7 of Annex I to this Regulation, shall be deemed to be refined olive-pomace oil within the meaning of point 5 of the Annex to Regulation No 136/66/EEC.
7. Oil, the characteristics of which comply with those set out in point 8 of Annex I to this Regulation, shall be deemed to be olive-pomace oil within the meaning of point 6 of the Annex to Regulation No 136/66/EEC.
Article 2
1. The characteristics of oils laid down in Annex I shall be determined in accordance with the following methods of analysis:
(a) for the determination of the free fatty acids, expressed as the percentage of oleic acid, the method set out in Annex II;
(b) for the determination of the peroxide index, the method set out in Annex III;
(c) for determination of the wax content, the method set out in Annex IV;
(d) for the determination of the composition and content of sterols and triterpene dialcohols by capillary-column gas chromatography, the method set out in Annex V;
(e) for the determination of the percentage of 2- glyceryl monopalmitate, the method set out in Annex VII;
(f) for spectrophotometric analysis, the method set out in Annex IX;
(g) for the determination of the fatty acid composition, the method set out in Annex X;
(h) for the determination of the volatile halogenated solvents, the method set out in Annex XI;
(i) for the evaluation of the organoleptic characteristics of virgin olive oil, the method set out in Annex XII;
(j) for the determination of stigmastadienes, the method set out in Annex XVII;
(k) for determining the content of triglycerides with ECN42, the method set out in Annex XVIII;
(l) for the determination of the aliphatic and triterpenic alcohols content, the method set out in Annex XIX;
(m) for the determination of the content of waxes, fatty acid methyl esters and fatty acid ethyl esters, the method set out in Annex XX.
▼M28 —————
2. Verification by national authorities or their representatives of the organoleptic characteristics of virgin oils shall be effected by tasting panels approved by the Member States.
The organoleptic characteristics of an oil as referred to in the first subparagraph shall be deemed consonant with the category declared if a panel approved by the Member State confirms the grading.
Should the panel not confirm the category declared as regards the organoleptic characteristics, at the interested party's request, the national authorities or their representatives shall have carried out without delay two counter-assessments by other approved panels, at least one by a panel approved by the producer Member State concerned. The characteristics concerned shall be deemed consonant with the characteristics declared if at least two of the counter-assessments confirm the declared grade. If that is not the case, the interested party shall be responsible for the cost of the counter-assessments.
3. When the national authorities or their representatives verify the characteristics of the oil as provided for in paragraph 1, samples shall be taken in accordance with international standards EN ISO 661 on the preparation of test samples and EN ISO 5555 on sampling. However, notwithstanding point 6.8 of standard EN ISO 5555, in case of batches of such oils in immediate packaging, the sample shall be taken in accordance with Annex Ia to this Regulation. In case of bulk oils for which the sampling cannot be performed according to EN ISO 5555, the sampling shall be performed in accordance with instructions provided by the competent authority of the Member State.
Without prejudice to standard EN ISO 5555 and Chapter 6 of standard EN ISO 661, the samples taken shall be put in a dark place away from strong heat as quickly as possible and sent to the laboratory for analysis no later than the fifth working day after they are taken, otherwise the samples shall be kept in such a way that they will not be degraded or damaged during transport or storage before being sent to the laboratory.
4. For the purposes of the verification provided for in paragraph 3, the analyses referred to in Annexes II, III, IX, XII and XX and, where applicable, any counter-analyses required under national law, shall be carried out before the minimum durability date in case of packaged products. In case of sampling of bulk oils, those analyses shall be carried out no later than the sixth month after the month in which the sample was taken.
No time limit shall apply to the other analyses provided for in this Regulation.
Unless the sample was taken less than two months before the minimum durability date, if the results of the analyses do not match the characteristics of the category of olive oil or olive-pomace oil declared, the party concerned shall be notified no later than one month before the end of the period laid down in the first subparagraph.
5. For the purpose of determining the characteristics of olive oils by the methods provided for in the first subparagraph of paragraph 1, the analysis results shall be directly compared with the limits laid down in this Regulation.
Article 2a
1. For the purpose of this Article, ‘olive oil marketed’ means total quantity of olive oil and olive pomace oil of a relevant Member State that is consumed in that Member State or exported from that Member State.
2. Member States shall ensure that conformity checks are carried out selectively, based on a risk analysis, and with appropriate frequency, so as to ensure that the olive oil marketed is consistent with the category declared.
3. The criteria to assess the risk may include:
(a) the category of oil, the period of production, the price of oils in relation to other vegetable oils, the blending and packing operations, the storage facilities and conditions, the country of origin, the country of destination, the means of transport or the volume of the lot;
(b) the position of the operators in the marketing chain, the volume and/or value marketed by them, the range of oil categories they market, the type of business carried out such as milling, storage, refining, blending, packaging or retail sale;
(c) findings made during previous checks including the number and type of defects found, the usual quality of oils marketed, the performance of technical equipment used;
(d) the reliability of operators’ quality assurance systems or self-checking systems related to the conformity to marketing standards;
(e) the place where the check is carried out, in particular if it is the first point of entry into the Union, the last point of exit from the Union or the place where the oils are produced, packaged, loaded or sold to the final consumer;
(f) any other information that might indicate a risk of non-compliance.
4. Member States shall lay down in advance:
(a) the criteria for assessing the risk of non-conformity of lots;
(b) on the basis of a risk analysis for each risk category, the minimum number of operators or lots and/or quantities which will be subject to a conformity check.
At least one conformity check per thousand tonnes of olive oil marketed in the Member State shall be carried out per year.
5. Member States shall verify compliance by:
(a) carrying out, in any order, the analyses set out in Annex I; or
(b) following the order set out in Annex Ib on the decision tree until one of the decisions appearing in the decision tree is reached.
▼M19 —————
Article 3
Where it is found that an oil does not correspond to its category description, the Member State concerned shall, without prejudice to any other penalties, apply effective, proportionate and dissuasive penalties to be determined in the light of the seriousness of the irregularity detected.
Where checks reveal significant irregularities, Member States shall increase the frequency of checks in relation to marketing stage, oil category, origin, or other criteria.
Article 4
1. The Member States may approve assessment panels so that national authorities or their representatives can assess and verify organoleptic characteristics.
The terms of approval shall be set by Member States and ensure that:
— the requirements of Annex XII.4 are met,
— the panel head is given training recognised for this purpose by the Member State,
— continued approval depends on performance in annual checks arranged by the Member State.
Member States shall notify to the Commission a list of approved panels and the action taken under this paragraph.
2. Where Member States encounter difficulties in setting up tasting panels in their territory, they may call on a tasting panel approved in another Member State.
3. Each Member State draw up a list of tasting panels set up by professional or inter-branch organizations in accordance with the conditions laid down in paragraph 1 and shall ensure that those conditions are complied with.
▼M19 —————
Article 6
1. The oil content of oil cake and other residues resulting from the extraction of olive oil (CN codes 2306 90 11 and 2306 90 19 ) shall be determined using the method set out in Annex XV.
2. The oil content referred to in paragraph 1 shall be expressed as a percentage of the weight of oil to the weight of dry matter.
Article 7
The Community provisions concerning the presence of contaminants shall apply.
As regards halogenated solvents, the limits for all categories of olive oils are as follows:
— maximum content of each halogenated solvent detected: 0,1 mg/kg,
— maximum total content of halogenated solvents detected: 0,2 mg/kg.
Article 7a
Natural or legal persons and groups of persons who hold olive oil and olive pomace oil from the extraction at the mill up to the bottling stage included, for whatever professional or commercial purposes, shall be required to keep entry and withdrawal registers for each category of such oils.
Member State shall ensure that the obligation laid down in the first paragraph is duly complied with.
Article 8
1. Member States shall notify the Commission of the measures implementing this Regulation. They shall inform the Commission of any subsequent amendments.
2. No later than 31 May of each year, Member States shall transmit to the Commission a report on the implementation of this Regulation during the previous calendar year. The report shall contain at least the results of the conformity checks carried out on olive oils as per the templates set out in Annex XXI.
3. The notifications referred to in this Regulation shall be made in accordance with Commission Regulation (EC) No 792/2009 ( 1 ).
Article 9
Regulation (EEC) No 1058/77 is hereby repealed.
Article 10
1. This Regulation shall enter into force on the third day following its publication in the Official Journal of the European Communities.
However, the method set out in Annex XII shall apply from ►M1 1 November 1992 ◄ , except in so far as operations relating to the intervention system are concerned.
That method shall not apply to virgin olive oil prepared for the market prior to 1 November 1992.
2. This Regulation shall not apply to olive oil and olive-residue oil packaged before the entry into force of this Regulation and marketed up to 31 October 1992.
This Regulation shall be binding in its entirety and directly applicable in all Member States.
ANNEXES
Summary
|
Annex I: |
Olive oil characteristics |
|
Annex Ia: |
Sampling of olive oil or olive-pomace oil delivered in immediate packaging |
|
Annexe Ib: |
Decision tree for verifying whether an olive oil sample is consistent with the category declared |
|
Annex II: |
Determination of free fatty acids, cold method |
|
Annex III: |
Determination of the peroxide value |
|
Annex IV: |
Determination of wax content by capillary column gas chromatography |
|
Annex V: |
Determination of the composition and content of sterols and triterpenes dialcohols by capillary-column gas chromatography |
|
Annex VII: |
►M21 Determination of the percentage of 2-glyceryl monopalmitate ◄ |
|
▼M20 ————— |
|
|
Annex IX: |
Spectrophotometric investigation in the ultraviolet |
|
Annex X: |
Determination of fatty acid methyl esters by gas chromatography |
|
Annex XI: |
Determination of the volatile halogenated solvents of olive oil |
|
Annex XII: |
The international olive council’s method for the organoleptic assessment of virgin olive oil |
|
▼M20 ————— |
|
|
▼M19 ————— |
|
|
Annex XV: |
Oil content of olive residue |
|
Annex XVI: |
Determination of iodine value |
|
Annex XVII: |
Method for the determination of stigmastadienes in vegetable oils |
|
Annex XVIII: |
Determination of the difference between actual and theoretical content of triacylglycerols with ECN 42 |
|
Annex XIX: |
►M28 Determination of aliphatic and triterpenic alcohols content by capillary gas chromatography ◄ |
|
Annex XX: |
Method for the determination of the content of waxes, fatty acid methyl esters and fatty acid ethyl esters by capillary gas chromatography |
|
▼M28 ————— |
|
|
ANNEX XXI: |
Results of conformity checks carried out on olive oils referred to in Article 8(2) |
ANNEX I
OLIVE OIL CHARACTERISTICS
|
Category |
Fatty acid ethyl esters (FAEEs) (*) |
Acidity (%) (*) |
Peroxide index mEq O2/kg (*) |
Waxes mg/kg (**) |
2-glyceril monopalmitate (%) |
Stigmastadienes mg/kg (1) |
Difference: ECN42 (HPLC) and ECN42 (theoretical calculation) |
K232 (*) |
K268 or K270 (*) |
Delta-K (*) |
Organoleptic evaluation Median defect (Md) (*) |
Organoleptic evaluation Fruity median (Mf) (*) |
|
1. Extra virgin olive oil |
FAEEs ≤ 40 mg/kg (2013-2014 crop year) (2) FAEEs ≤ 35 mg/kg (2014-2016 crop year) FAEEs ≤ 30 mg/kg (after 2016 crop years) |
≤ 0,8 |
≤ 20 |
C42 + C44 + C46 ≤ 150 |
≤ 0,9 if total palmitic acid % ≤ 14 % |
≤ 0,05 |
≤ |0,2| |
≤ 2,50 |
≤ 0,22 |
≤ 0,01 |
Md = 0 |
Mf > 0 |
|
≤ 1,0 if total palmitic acid % > 14 % |
||||||||||||
|
2. Virgin olive oil |
— |
≤ 2,0 |
≤ 20 |
C42 + C44 + C46 ≤ 150 |
≤ 0,9 if total palmitic acid % ≤ 14 % |
≤ 0,05 |
≤ |0,2| |
≤ 2,60 |
≤ 0,25 |
≤ 0,01 |
Md ≤ 3,5 |
Mf > 0 |
|
≤ 1,0 if total palmitic acid % > 14 % |
||||||||||||
|
3. Lampante olive oil |
— |
> 2,0 |
— |
C40 + C42 + C44 + C46 ≤ 300 (3) |
≤ 0,9 if total palmitic acid % ≤ 14 % |
≤ 0,50 |
≤ |0,3| |
— |
— |
— |
Md > 3,5 (4) |
— |
|
≤ 1,1 if total palmitic acid % > 14 % |
||||||||||||
|
4. Refined olive oil |
— |
≤ 0,3 |
≤ 5 |
C40 + C42 + C44 + C46 ≤ 350 |
≤ 0,9 if total palmitic acid % ≤ 14 % |
— |
≤ |0,3| |
— |
≤ 1,10 |
≤ 0,16 |
— |
— |
|
≤ 1,1 if total palmitic acid % > 14 % |
||||||||||||
|
5. Olive oil composed of refined and virgin olive oils |
— |
≤ 1,0 |
≤ 15 |
C40 + C42 + C44 + C46 ≤ 350 |
≤ 0,9 if total palmitic acid % ≤ 14 % |
— |
≤ |0,3| |
— |
≤ 0,90 |
≤ 0,15 |
— |
— |
|
≤ 1,0 if total palmitic acid % > 14 % |
||||||||||||
|
6. Crude olive-pomace oil |
— |
— |
— |
C40 + C42 + C44 + C46 > 350 (5) |
≤ 1,4 |
— |
≤ |0,6| |
— |
— |
— |
— |
— |
|
7. Refined olive-pomace oil |
— |
≤ 0,3 |
≤ 5 |
C40 + C42 + C44 + C46 > 350 |
≤ 1,4 |
— |
≤ |0,5| |
— |
≤ 2,00 |
≤ 0,20 |
— |
— |
|
8. Olive-pomace oil |
— |
≤ 1,0 |
≤ 15 |
C40 + C42 + C44 + C46 > 350 |
≤ 1,2 |
— |
≤ |0,5| |
— |
≤ 1,70 |
≤ 0,18 |
— |
— |
|
(1) Total isomers which could (or could not) be separated by capillary column. (2) The limit applies to olive oils produced as from 1 March 2014. (3) Oils with a wax content of between 300 mg/kg and 350 mg/kg are considered to be lampante olive oil if the total aliphatic alcohol content is less than or equal to 350 mg/kg or if the erythrodiol and uvaol content is less than or equal to 3,5 %. (4) The median defect may be less than or equal to 3,5 and the fruity median equal to 0. (5) Oils with a wax content of between 300 mg/kg and 350 mg/kg are considered to be crude olive-pomace oil if the total aliphatic alcohol content is above 350 mg/kg and if the erythrodiol and uvaol content is greater than 3,5 %. |
||||||||||||
|
Category |
Fatty acid composition (1) |
Total transoleic isomers (%) |
Total translinoleic + translinolenic isomers (%) |
Sterols composition |
Total sterols (mg/kg) |
Erythrodiol and uvaol (%) (**) |
||||||||||
|
Myristic (%) |
Linolenic (%) |
Arachidic (%) |
Eicosenoic (%) |
Behenic (%) |
Lignoceric (%) |
Cholesterol (%) |
Brassicasterol (%) |
Campesterol (2) (%) |
Stigmasterol (%) |
App β – sitosterol (3) (%) (**) |
Delta-7-stigmastenol (2) (%) |
|||||
|
1. Extra virgin olive oil |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,40 |
≤ 0,20 |
≤ 0,20 |
≤ 0,05 |
≤ 0,05 |
≤ 0,5 |
≤ 0,1 |
≤ 4,0 |
< Camp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 000 |
≤ 4,5 |
|
2. Virgin olive oil |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,40 |
≤ 0,20 |
≤ 0,20 |
≤ 0,05 |
≤ 0,05 |
≤ 0,5 |
≤ 0,1 |
≤ 4,0 |
< Camp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 000 |
≤ 4,5 |
|
3. Lampante olive oil |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,40 |
≤ 0,20 |
≤ 0,20 |
≤ 0,10 |
≤ 0,10 |
≤ 0,5 |
≤ 0,1 |
≤ 4,0 |
— |
≥ 93,0 |
≤ 0,5 |
≥ 1 000 |
≤ 4,5 (4) |
|
4. Refined olive oil |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,40 |
≤ 0,20 |
≤ 0,20 |
≤ 0,20 |
≤ 0,30 |
≤ 0,5 |
≤ 0,1 |
≤ 4,0 |
< Camp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 000 |
≤ 4,5 |
|
5. Olive oil composed of refined and virgin olive oils |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,40 |
≤ 0,20 |
≤ 0,20 |
≤ 0,20 |
≤ 0,30 |
≤ 0,5 |
≤ 0,1 |
≤ 4,0 |
< Camp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 000 |
≤ 4,5 |
|
6. Crude olive-pomace oil |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,40 |
≤ 0,30 |
≤ 0,20 |
≤ 0,20 |
≤ 0,10 |
≤ 0,5 |
≤ 0,2 |
≤ 4,0 |
— |
≥ 93,0 |
≤ 0,5 |
≥ 2 500 |
> 4,5 (5) |
|
7. Refined olive-pomace oil |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,40 |
≤ 0,30 |
≤ 0,20 |
≤ 0,40 |
≤ 0,35 |
≤ 0,5 |
≤ 0,2 |
≤ 4,0 |
< Camp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 800 |
> 4,5 |
|
8. Olive-pomace oil |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,40 |
≤ 0,30 |
≤ 0,20 |
≤ 0,40 |
≤ 0,35 |
≤ 0,5 |
≤ 0,2 |
≤ 4,0 |
< Camp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 600 |
> 4,5 |
|
(1) Other fatty acids content (%): palmitic: 7,50-20,00; palmitoleic: 0,30-3,50; heptadecanoic: ≤ 0,30; heptadecenoic: ≤ 0,30; stearic: 0,50-5,00; oleic: 55,00-83,00; linoleic: 2,50-21,00. (2) See the Appendix to this Annex. (3) App β-sitosterol: Delta-5,23-stigmastadienol + chlerosterol + beta-sitosterol+sitostanol + delta-5-avenasterol + delta-5,24-stigmastadienol. (4) Oils with a wax content of between 300 mg/kg and 350 mg/kg are considered to be lampante olive oil if the total aliphatic alcohol content is less than or equal to 350 mg/kg or if the erythrodiol and uvaol content is less than or equal to 3,5 %. (5) Oils with a wax content of between 300 mg/kg and 350 mg/kg are considered to be crude olive-pomace oil if the total aliphatic alcohol content is above 350 mg/kg or if the erythrodiol and uvaol content is greater than 3,5 %. |
||||||||||||||||
Notes:
(a) The results of the analyses must be expressed to the same number of decimal places as used for each characteristic. The last digit must be increased by one unit if the following digit is greater than 4.
(b) If just a single characteristic does not match the values stated, the category of an oil can be changed or the oil declared impure for the purposes of this Regulation.
(c) If a characteristic is marked with an asterisk (*), referring to the quality of the oil, this means the following: — for lampante olive oil, it is possible for both the relevant limits to be different from the stated values at the same time, — for virgin olive oils, if at least one of these limits is different from the stated values, the category of the oil will be changed, although they will still be classified in one of the categories of virgin olive oil.
(d) If a characteristic is marked with two asterisks (**), this means that for all types of olive-pomace oil, it is possible for both the relevant limits to be different from the stated values at the same time.
Appendix
DECISION TREE
Campesterol decision tree for virgin and extra virgin olive oils:
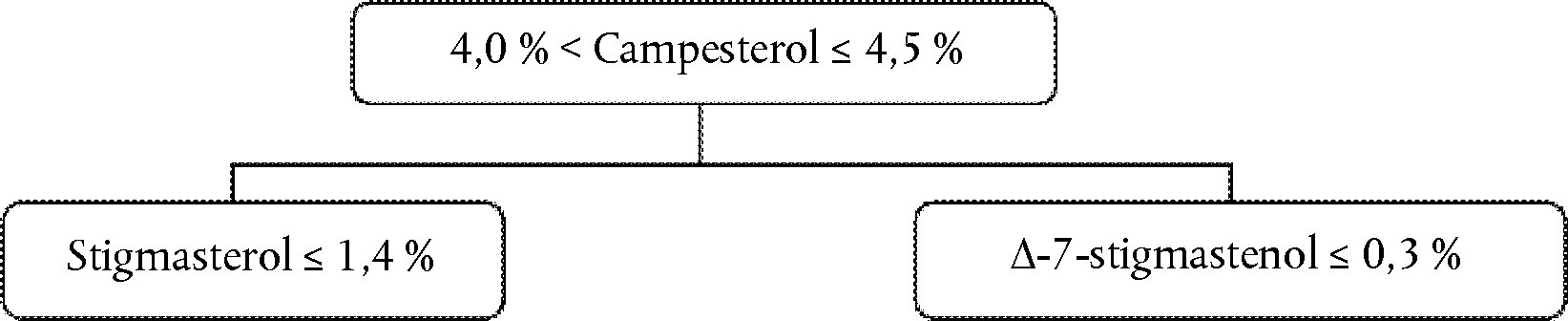
The other parameters shall comply with the limits fixed in this Regulation.
Delta-7-stigmastenol decision tree for:
— Extra virgin and virgin olive oils
—
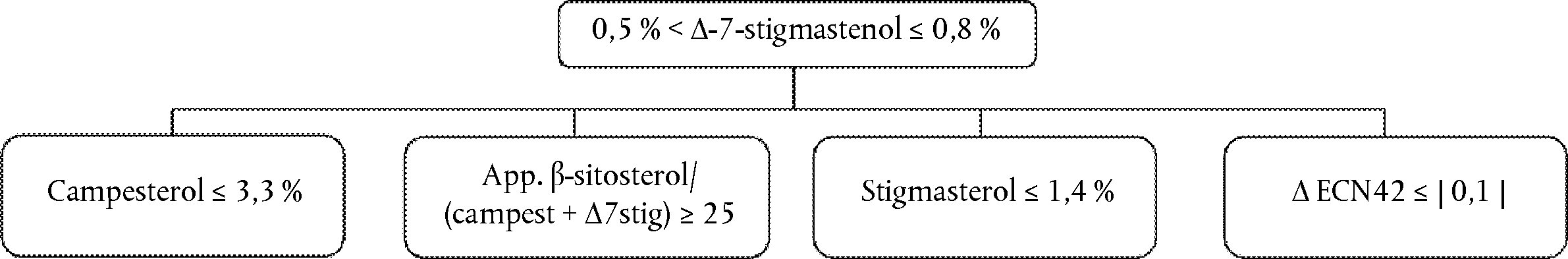
The other parameters shall comply with the limits fixed in this Regulation.
— Olive-pomace oils (crude and refined)
—

ANNEX Ia
SAMPLING OF OLIVE OIL OR OLIVE-POMACE OIL DELIVERED IN IMMEDIATE PACKAGING
This method of sampling is applied to batches of olive oil or olive-pomace oil put up in immediate packaging. Different sampling methods apply, depending on whether the immediate packaging exceeds 5 litres or not.
‘Batch’ shall mean a set of sales units which are produced, manufactured and packed in circumstances such that the oil contained in each sales unit is considered to be homogenous in terms of all analytical characteristics. The individuation of a batch must be done in accordance with Directive 2011/91/EU of the European Parliament and of the Council ( 2 ).
‘Increment’ shall mean the quantity of oil contained in an immediate package and taken from a random point of the batch.
1. CONTENT OF PRIMARY SAMPLE
1.1. Immediate packaging not exceeding 5 litres
‘Primary Sample’ for immediate packaging not exceeding 5 litres shall mean the number of increments taken from a batch and in agreement with Table 1.
Table 1
Primary sample minimum size must comprise the following
|
Where the immediate packaging has a capacity of |
The primary sample must comprise the oil from |
|
(a) 1 litre or more |
(a) 1 immediate pack; |
|
(b) less than 1 litre |
(b) the minimum number of packs with a total capacity of at least 1,0 litre |
The number of packs referred to in Table 1, which shall constitute a primary sample, can be increased by each Member State, according to their own needs (for example organoleptic assessment by a different laboratory from that which performed the chemical analyses, counter-analysis, etc.).
1.2. Immediate packaging exceeding 5 litres
‘Primary Sample’ for immediate packaging exceeding 5 litres shall mean a representative part of the total increments, obtained by a process of reduction and in agreement with Table 2. The primary sample must be composed of various examples.
‘Example’ of a primary sample shall mean each of the packages making up the primary sample.
Table 2
Minimum number of increments to be selected
|
Number of packages in the lot |
Minimum number of increments to be selected |
|
Up to 10 |
1 |
|
From … 11 to 150 |
2 |
|
From … 151 to 500 |
3 |
|
From … 501 to 1 500 |
4 |
|
From … 1 501 to 2 500 |
5 |
|
> 2 500 per 1 000 packages |
1 extra increment |
In order to reduce the volume of the sampling immediate packs, the content of the sampling increments is homogenised for the preparation of the primary sample. The portions of the different increments are poured into a common container for homogenisation by stirring, so that it will be best protected from air.
The content of the primary sample must be poured into a series of packages of the minimum capacity of 1,0 liter, each one of which constitutes an example of the primary sample.
The number of primary samples can be increased by each Member State, according to their own necessity (for example organoleptic assessment by a different laboratory from the one that performed the chemical analyses, counter-analysis, etc).
Each package must be filled in a way to minimise the air layer on top and then suitably closed and sealed to ensure the product is tamper-proof.
These examples must be labeled to ensure correct identification.
2. ANALYSES AND RESULTS
2.1. Each primary sample must be subdivided into laboratory samples, in accordance with point 2.5 of standard EN ISO 5555, and analysed according to the order shown in the decision tree set out in Annex Ib or in any other random order.
2.2. Where all the results of the analyses comply with the characteristics of the category of oil declared, the whole batch is to be declared to comply.
If a single result of the analyses does not comply with the characteristics of the category of oil declared, the whole batch is to be declared non compliant.
3. VERIFICATION OF THE CATEGORY OF BATCH
3.1. In order to verify the batch category, the competent authority may increase the number of primary samples taken at different points of the batch according to the following table:
Table 3
Number of primary samples determined by the size of batch
|
Size of batch (litres) |
Number of primary samples |
|
Less than 7 500 |
2 |
|
From 7 500 to less than 25 000 |
3 |
|
From 25 000 to less than 75 000 |
4 |
|
From 75 000 to less than 125 000 |
5 |
|
Equal to and more than 125 000 |
6 + 1 each 50 000 litres more |
Each increment constituting a primary sample must be taken from a continuous place in the batch; it is necessary to take note of the location of each primary sample and to identify it unambiguously.
The formation of each primary sample must be carried out according to the procedures referred to in points 1.1 and 1.2.
Each primary sample is then subjected to the analyses referred to in Article 2(1).
3.2. When one of the results of the analyses referred to in Article 2(1) of at least one primary sample does not comply with the characteristics of the declared category of oil, the whole sampling batch shall be declared non compliant.
ANNEX Ib
DECISION TREE FOR VERIFYING WHETHER AN OLIVE OIL SAMPLE IS CONSISTENT WITH THE CATEGORY DECLARED
Table 1

Table 2

Table 3

Appendix 1
Table of equivalence between the Annexes to this Regulation and the analyses specified in the decision tree
|
— Acidity |
Annex II |
Determination of free fatty acids, cold method |
|
— Peroxide value |
Annex III |
Determination of peroxide value |
|
— UV spectrometry |
Annex IX |
Spectrophotometric analysis |
|
— Organoleptic assessment |
Annex XII |
Organoleptic assessment of virgin olive oil |
|
— Ethyl esters |
Annex XX |
Method for the determination of the content of waxes, fatty acid methyl esters and fatty acid ethyl esters by capillary gas chromatography |
|
— 3,5-Stigmastadienes |
Annex XVII |
Method for the determination of stigmastadienes in vegetable oils |
|
— Trans isomers of fatty acids |
Annex X |
Determination of fatty acid methyl esters by gas chromatography |
|
— Fatty acids content |
Annex X |
Determination of fatty acid methyl esters by gas chromatography |
|
— ΔECN42 |
Annex XVIII |
Determination of the composition of triglycerides with ECN42 (difference between the HPLC data and theoretical content) |
|
— Sterols composition and total sterols — Erythrodiol and Uvaol |
Annex V |
Determination of the composition and content of sterols and triterpene dialcohols by capillary-column gas chromatography |
|
— Waxes |
Annex IV |
Determination of wax content by capillary column gas chromatography |
|
— Aliphatic and triterpenic alcohols |
Annex XIX |
Determination of aliphatic and triterpenic alcohols content by capillary gas chromatography |
|
— Saturated fatty acids in position 2 |
Annex VII |
Determination of the percentage of 2-glyceryl monopalmitate |
ANNEX II
DETERMINATION OF FREE FATTY ACIDS, COLD METHOD
1. SCOPE AND FIELD OF APPLICATION
This method describes the determination of free fatty acids in olive oils and olive pomace oils. The content of free fatty acids is expressed as acidity calculated as the percentage of oleic acid.
2. PRINCIPLE
A sample is dissolved in a mixture of solvents and the free fatty acids present titrated using a potassium hydroxide or sodium hydroxide solution.
3. REAGENTS
All the reagents should be of recognized analytical quality and the water used either distilled or of equivalent purity.
|
3.1 |
Diethyl ether; 95 % ethanol (v/v), mixture of equal parts by volume. Neutralize precisely at the moment of use with the potassium hydroxide solution (3.2), with the addition of 0,3 ml of the phenolphthalein solution (3.3) per 100 ml of mixture. Note 1: Diethyl ether is highly inflammable and may form explosive peroxides. Special care should be taken in its use. Note 2: If it is not possible to use diethyl ether, a mixture of solvents containing ethanol and toluene may be used. If necessary, ethanol may be replaced by propanol-2. |
|
3.2 |
Potassium hydroxide or sodium hydroxide, titrated ethanolic or aqueous solution, c(KOH) [or c(NaOH)] about 0,1 mol/l or, if necessary, c(KOH) [or c(NaOH)] about 0,5 mol/l. Commercial solutions are available. The exact concentration of potassium hydroxide solution (or sodium hydroxide solution) must be known and checked prior to use. Use a solution prepared at least five days before use and decanted into a brown glass bottle with a rubber stopper. The solution should be colourless or straw coloured. If phase separation is observed when using aqueous solution of potassium hydroxide (or sodium hydroxide), replace the aqueous solution by an ethanolic solution. Note 3: A stable colourless solution of potassium hydroxide (or sodium hydroxide) may be prepared as follows. Bring to the boil 1 000 ml of ethanol or water with 8 g of potassium hydroxide (or sodium hydroxide) and 0,5 g of aluminium shavings and continue boiling under reflux for one hour. Distil immediately. Dissolve in the distillate the required quantity of potassium hydroxide (or sodium hydroxide). Leave for several days and decant the clear supernatant liquid from the precipitate of potassium carbonate (or sodium carbonate). The solution may also be prepared without distillation as follows: to 1 000 ml of ethanol (or water) add 4 ml of aluminium butylate and leave the mixture for several days. Decant the supernatant liquid and dissolve the required quantity of potassium hydroxide (or sodium hydroxide). The solution is ready for use. |
|
3.3 |
Phenolphthalein, 10 g/l solution in 95 to 96 % ethanol (v/v) or alkali blue 6B or thymolphthalein, 20 g/l solution in 95 to 96 % ethanol (v/v). In the case of strongly coloured oils, alkali blue or thymolphthalein shall be used. |
4. APPARATUS
Usual laboratory equipment including:
4.1 Analytical balance;
4.2 250 ml conical flask;
4.3 10 ml burette class A, graduated in 0,05 ml, or equivalent automatic burette.
5. PROCEDURE
5.1 Preparation of the test sample
When the sample is cloudy, it should be filtered.
5.2 Test portion
Take a sample depending on the presumed acidity in accordance with the following table:
|
Expected acidity (oleic acidity g/100 g) |
Mass of sample (g) |
Weighing accuracy (g) |
|
0 to 2 |
10 |
0,02 |
|
> 2 to 7,5 |
2,5 |
0,01 |
|
> 7,5 |
0,5 |
0,001 |
Weigh the sample in the conical flask (4.2).
5.3 Determination
Dissolve the sample (5.2) in 50 to 100 ml of the previously neutralized mixture of diethyl ether and ethanol (3.1).
Titrate while stirring with the 0,1 mol/l solution of potassium hydroxide (or sodium hydroxide) (3.2) (see Note 4) until the indicator changes (the colour of the coloured indicator persists for at least 10 seconds).
Note 4: If the quantity of 0,1 mol/l potassium hydroxide (or sodium hydroxide) solution required exceeds 10 ml, use the 0,5 mol/l solution or change the sample mass according to the expected free acidity and the proposed table.
Note 5: If the solution becomes cloudy during titration, add enough of the solvents (3.1) to give a clear solution.
Carry out a second determination only if the first result is higher than the specified limit for the category of the oil.
6. EXPRESSION OF RESULTS
Acidity as a percentage of oleic acid by weight is equal to:

where:
|
V |
= |
the volume of titrated potassium hydroxide solution (or sodium hydroxide) used, in millilitres; |
|
c |
= |
the exact concentration in moles per litre of the titrated solution of potassium hydroxide (or sodium hydroxide) used; |
|
M |
= |
282 g/mol, the molar mass in grams per mole of oleic acid; |
|
m |
= |
the mass of the sample, in grams. |
Oleic acidity is reported as follows:
(a) to two decimal places for values from 0 up to and including 1;
(b) to one decimal place for values from 1 up to and including 100.
ANNEX III
DETERMINATION OF PEROXIDE VALUE
1. SCOPE
This Standard describes a method for the determination of the peroxide value of oils and fats.
2. FIELD OF APPLICATION
This Standard is applicable to animal and vegetable oils and fats.
3. DEFINITION
The peroxide value is the quantity of those substances in the sample, expressed in terms of milliequivalents of active oxygen per kilogram, which oxidize potassium iodide under the operating conditions described.
4. PRINCIPLE
Treatment of the test portion, in solution in acetic acid and chloroform, by a solution of potassium iodide. Titration of the liberated iodine with standardized sodium thiosulphate solution.
5. APPARATUS
All the equipment used shall be free from reducing or oxidizing substances.
Note:
Do not grease ground surfaces.
|
5.1. |
3 ml glass scoop. |
|
5.2. |
Flasks, with ground necks and stoppers, of about 250 ml capacity, dried beforehand and filled with a pure, dry inert gas (nitrogen or, preferably, carbon dioxide). |
|
5.3. |
25- or 50-ml burette, graduated in 0,1 ml. |
6. REAGENTS
|
6.1. |
Chloroform, analytical reagent quality, freed from oxygen by bubbling a current of pure, dry inert gas through it. |
|
6.2. |
Glacial acetic acid, analytical reagent quality, freed from oxygen by bubbling a current of pure, ►C1 dry inert gas ◄ through it. |
|
6.3. |
Potassium iodide, saturated aqueous solution, recently prepared, free from iodine and iodates. |
|
6.4. |
Sodium thiosulphate, 0,01 or 0,002 ►C1 Mol/L accurately ◄ standardized aqueous solution, standardized just before use. |
|
6.5. |
Starch solution, 10 g/l aqueous dispersion, recently prepared from natural soluble starch. |
7. SAMPLE
Take care that the sample is taken and stored away from the light, kept cold and contained in completely filled glass containers, hermetically sealed with ground-glass or cork stoppers.
8. PROCEDURE
The test shall be carried out in diffuse daylight or in artificial light. Weigh in a glass scoop (5.1) or, failing this, in a flask (5.2), to the nearest 0,001 g, a mass of the sample in accordance with the following table, according to the expected peroxide value:
|
Expected peroxide value (meq) |
Weight of test portion (g) |
|
0 to 12 |
5,0 to 2,0 |
|
12 to 20 |
2,0 to 1,2 |
|
20 to 30 |
1,2 to 0,8 |
|
30 to 50 |
0,8 to 0,5 |
|
50 to 90 |
0,5 to 0,3 |
Unstopper a flask (5.2) and introduce the glass scoop containing the test portion. Add 10 ml of chloroform (6.1). Dissolve the test portion rapidly by stirring. Add 15 ml of acetic acid (6.2), then 1 ml of potassium iodide solution (6.3). Insert the stopper quickly, shake for one minute, and leave for exactly five minutes away from the light at a temperature from 15 to 25 °C.
Add about 75 ml of distilled water. Titrate the liberated iodine with the sodium thiosulphate solution ►C1 (6.4) (0,002 Mol/L solution for expected values less than 12, and 0,01 Mol/L solution ◄ for expected values above 12) shaking vigorously, using starch solution (6.5) as indicator.
Carry out two determinations on the same test sample.
Carry out simultaneously a blank test. If the result of the blank exceeds 0,05 ml of ►C1 0,01 mol/L sodium ◄ thiosulphate solution (6.4), replace the impure reagents.
9. EXPRESSION OF RESULTS
The peroxide value (PV), expressed in milliequivalents of active oxygen per kilogram, is given by the formula:

where:
V = the number of ml of the standardized sodium thiosulphate solution (6.4) used for the test, corrected to take into account the blank test;
T = ►C1 the exact molarity ◄ of the sodium thiosulphate solution (6.4) used;
m = the weight in g, of the test portion.
Take as the result the arithmetic mean of the two determinations carried out.
ANNEX IV
DETERMINATION OF WAX CONTENT BY CAPILLARY COLUMN GAS CHROMATOGRAPHY
1. SUBJECT
This method describes a process for determining the wax content of olive oils. Waxes are separated according to the number of their carbon atoms. The method may be used in particular to distinguish between olive oil obtained by pressing and that obtained by extraction (olive-residue oil).
2. PRINCIPLE
Addition of a suitable internal standard to the fat or oil, then fractionation by chromatography on a hydrated silica gel column. Recovery under the test conditions of the fraction eluted first (the polarity of which is less than that of the triglycerides), then direct analysis by capillary column gas chromatography.
3. EQUIPMENT
|
3.1. |
25 ml Erlenmeyer flask. |
|
3.2. |
Glass column for gas chromatography, internal diameter 15,0 mm, length 30 to 40 cm, fitted with a stopcock. |
|
3.3. |
Suitable gas chromatograph with a capillary column, equipped with a system for direct introduction into the column comprising the following:
|
|
3.4. |
10 μl microsyringe for on-column injection, equipped with a hardened needle. |
|
3.5. |
Electrovibrator. |
|
3.6. |
Rotary evaporator. |
|
3.7. |
Muffle furnace. |
|
3.8. |
Analytical balance with guaranteed precision of ± 0,1 mg. |
|
3.9. |
Normal laboratory glassware. |
4. REAGENTS
|
4.1. |
Silica gel with a granule size of between 60 and 200 μm. Place the gel in the furnace at 500 °C for at least four hours. After cooling, add 2 % water in relation to the quantity of sampled silica gel. Shake well to homogenise the slurry. Keep in darkness for at least 12 hours prior to use. |
|
4.2. |
n-hexane, for chromatography. |
|
4.3. |
Ethyl ether, for chromatography. |
|
4.4. |
n-heptane, for chromatography. |
|
4.5. |
Standard solution of lauryl arachidate, at 0,1 % (m/v) in hexane (internal standard). (It is also possible to use palmityl palmitate or myristyl stearate.)
|
|
4.6. |
Carrier gas: hydrogen or helium, gas-chromatographic purity. |
|
4.7. |
Auxiliary gases: — pure hydrogen for gas chromatography, — pure air for gas chromatography. |
5. PROCEDURE
5.1. Preparation of the chromatographic column.
Suspend 15 g of silica gel (4.1) in the n-hexane (4.2) and introduce it into the column (3.2). Allow to settle spontaneously. Complete settling with the aid of an electrovibrator (3.5) to make the chromatographic layer more homogeneous. Percolate 30 ml of n-hexane to remove any impurities. Using the balance (3.8) weigh exactly 500 mg of the sample into the 25 ml Erlenmeyer flask (3.1), add the appropriate quantity of the internal standard (4.5) according to the presumed wax content. For example, add 0,1 mg of lauryl arachidate for olive oil, and 0,25 to 0,5 mg for olive-residue oil. Transfer the prepared sample to the chromotography column using two 2 ml portions of n-hexane (4.2).
Allow the solvent to flow away until it reaches 1 mm above the upper level of the absorbant then percolate a further 70 ml of n-hexane in order to eliminate the n-alkanes naturally present. Then start the chromatographic elution by collecting 180 ml of the mixture of n-hexane/ethyl ether (ratio 99:1), keeping a rate of flow of approximately 15 drops every 10 seconds. Elution of the sample must be carried out at a room temperature of 22 ± 4 °C.
NB:
— The n-hexane/ethyl ether mixture (99:1) must be prepared every day.
— For a visual check on the correct elution of the waxes 100 μl of 1 % Sudan in the elution mixture can be added to the sample in solution. Since the colourant has an intermediate retention, between waxes and triglycerides, when the coloration has reached the bottom of the column the elution should be suspended because all the waxes will have been eluted.
Dry the fraction thus obtained in a rotary evaporator (3.6.) until virtually all the solvent has been eliminated. Eliminate the final 2 ml of solvent with the aid of a weak current of nitrogen; then add 2-4 ml n-heptane.
5.2. Analysis by gas chromatography
5.2.1. Preparatory work
Fit the column to the gas chromatograph (3.3) by connecting the inlet port to the on-column system and the outlet port to the detector. Perform a general check on the GC apparatus (operation of gas circuits, detector and recorder efficiency, etc.).
If the column is being used for the first time it should be conditioned first. Pass a little gas through the column, then turn on the GC apparatus. Heat gradually until 350 °C is reached after about four hours. Maintain that temperature for at least two hours then regulate the apparatus to operating conditions (set gas flow, light flame, connect to the electronic recorder (3.3.4), set temperature of column chamber, detector, etc.) and record the signal at a sensitivity at least twice as high as that required for the analysis. The baseline must be linear, with no peaks of any kind, and must not show any deviation.
A negative straight-line drift indicates that the column connections are not tight; a positive drift that the column has not been sufficiently conditioned.
5.2.2. Choice of operating conditions
The operating conditions are generally as follows:
— column temperature:
—
|
|
20 °C/minute |
|
5 °C/minute |
|
20 °C/minute |
|
|
Initially 80 °C (1′) |
→ |
240 °C |
→ |
325 °C (6′) |
→ |
340 °C (10′) |
— detector temperature: 350 °C;
— quantity of substance injected: 1 μl of the n-heptane solution (2-4 ml);
— carrier gas: helium or hydrogen at the correct linear velocity for the gas selected (see Appendix);
— instrument sensitivity: suitable for the following conditions:
The conditions may be modified according to the characteristics of the column and the GC apparatus to obtain separation of all the waxes and a satisfactory peak resolution (see figure); the internal standard C32 retention time must be 18 ± 3 minutes. The most representative wax peak must be at least 60 % of the full scale.
The peak integration parameters must be established so as to obtain a correct evaluation of the areas of the peaks in question.
NB: Given the high final temperature, a positive drift of no more than 10 % of the full scale is permitted.
5.3. Performance of the analysis
Sample 1 μl of the solution using the 10 μl microsyringe; withdraw the syringe plunger so that the needle is empty. Place the needle in the injector and after 1-2 seconds inject quickly; remove the needle slowly after about five seconds.
Record until the waxes are completely eluted.
The base line must always satisfy the required conditions.
5.4. Identification of peaks
Identification of the different peaks should be based on retention time by comparison with wax mixtures of known retention times analysed under the same conditions.
The figure is a chromatogram of the waxes of a virgin olive oil.
5.5. Evaluation of quantity
Calculate the areas of the peaks of the internal standard and the aliphatic esters of C40 to C46 using the integrator.
Calculate the wax content of each of the esters in mg/kg fat using the formula:

where:
|
Ax |
= |
area of each ester’s peak, in square millimetres; |
|
As |
= |
area of the internal standard’s peak, in square millimetres; |
|
ms |
= |
mass of added internal standard, in milligrams; |
|
m |
= |
mass of sample for analysis, in grams. |
6. EXPRESSION OF RESULTS
Indicate the total of the contents of the various C40 to C46 waxes in mg/kg fat (ppm).
NB: The components to be quantified refer to the peaks with carbon pair numbers between esters C40 and C46, using the example of the olive oil wax chromatogram shown in the figure below. If ester C46 appears twice, it is recommended that to identify it the fraction of the waxes of an olive-residue oil should be analysed where the C46 peak is easy to identify because it is in the clear majority.
The results should be expressed to one decimal place.
Figure
Chromatogram of the waxes of an olive oil ( 3 )
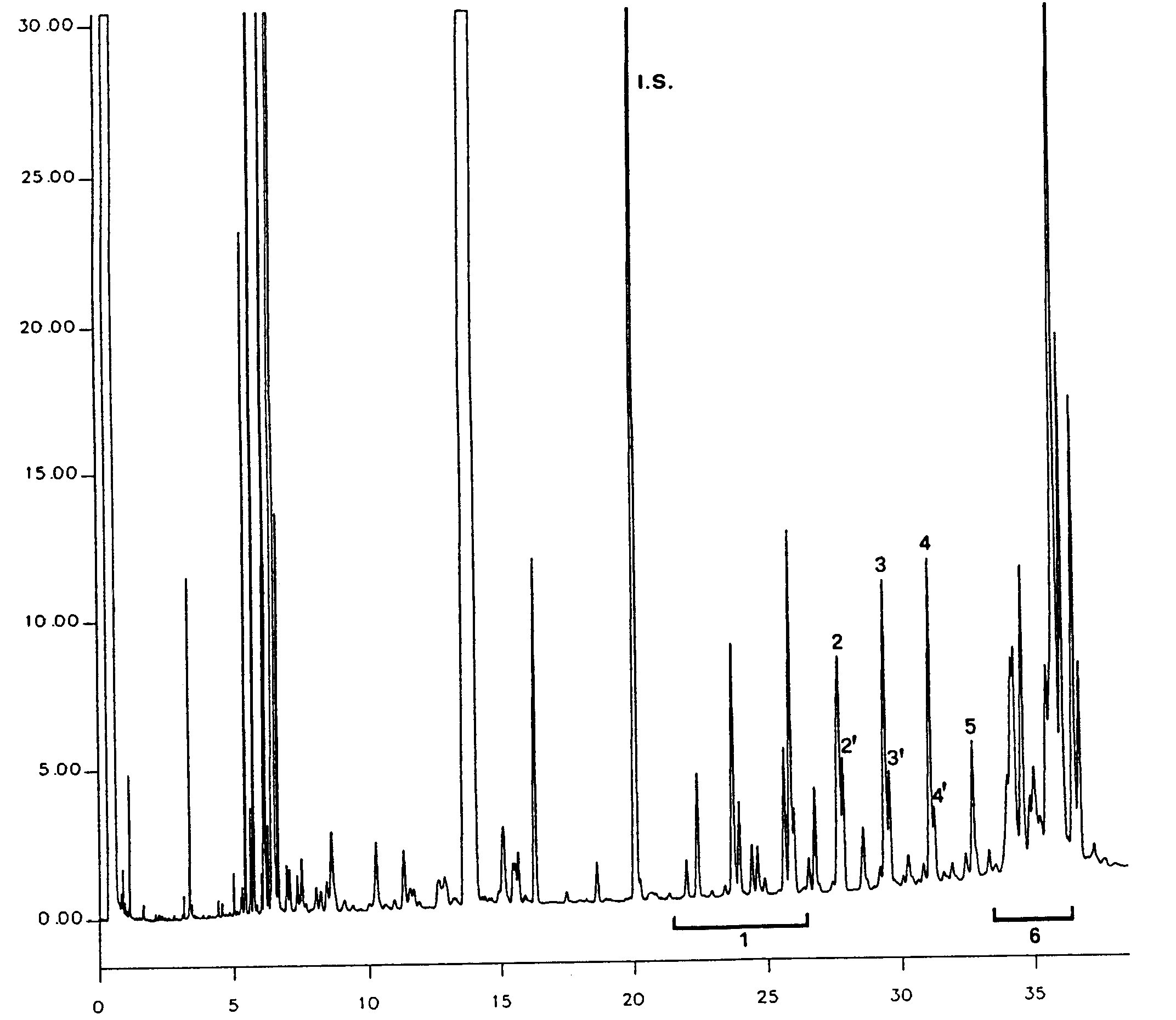
Key:
|
I.S. |
= |
Lauryl arachidate |
|
1. |
= |
Diterpenic esters |
|
2 + 2′ |
= |
C40 esters |
|
3 + 3′ |
= |
C42 esters |
|
4 + 4′ |
= |
Esters C44 |
|
5. |
= |
C46 esters |
|
6. |
= |
Sterol esters and triterpenic alcohol |
Appendix
Determination of the linear velocity of the gas
Inject 1-3 μl methane (or propane) into the GC apparatus after it has been regulated to normal operating conditions. Measure the time it takes for the gas to flow through the column from the time it is injected to the time the peak appears (tM).
The linear velocity in cm/s is given by the formula L/tM, where L is the length of the column in cm and tM the time measured in seconds.
ANNEX V
DETERMINATION OF THE COMPOSITION AND CONTENT OF STEROLS AND TRITERPENES DIALCOHOLS BY CAPILLARY-COLUMN GAS CHROMATOGRAPHY
1. SCOPE
The method describes a procedure for determining the individual and total sterols and triterpene dialcohols content of olive oils and olive-pomace oils.
2. PRINCIPLE
The oil, with added α-cholestanol as an internal standard, is saponified with potassium hydroxide in ethanolic solution and the unsaponifiable matter is then extracted with ethyl ether.
The sterols and triterpene dialcohols fraction is separated from the unsaponifiable matter by thin-layer chromatography on a basic silica gel plate. The fractions recovered from the silica gel are transformed into trimethylsilyl ethers and then analysed by capillary column gas chromatography.
3. APPARATUS
The usual laboratory equipment and in particular the following:
3.1. 250 ml flask fitted with a reflux condenser with ground-glass joints.
3.2. 500 ml separating funnel.
3.3. 250 ml flasks.
3.4. Complete apparatus for analysis by thin-layer chromatography using 20 x 20 cm glass plates.
3.5. Ultraviolet lamp with a wavelength of 254 or 366 nm.
3.6. 100 μl and 500 μl microsyringes.
3.7. Cylindrical filter funnel with a G3 porous septum (porosity 15-40 μm) of diameter approximately 2 cm and a depth of 5 cm, suitable for filtration under vacuum with male ground-glass joint.
3.8. 50 ml vacuum conical flask with ground-glass female joint, which can be fitted to the filter funnel (point 3.7).
3.9. 10 ml test tube with a tapering bottom and a sealing glass stopper.
3.10. Gas chromatograph suitable for use with a capillary column with split injection system, consisting of:
3.10.1. A thermostatic chamber for columns capable of maintaining the desired temperature with an accuracy of ± 1°C;
3.10.2. A temperature-adjustable injection unit with a persilanised glass vaporising element and split system;
3.10.3. A flame ionisation detector (FID);
3.10.4. Data acquisition system suitable for use with the FID detector (point 3.10.3.), capable of manual integration.
3.11. Fused-silica capillary column of length 20 to 30 m, internal diameter 0,25 to 0,32 mm, coated with 5 % diphenyl - 95 % dimethylpolysiloxane (SE-52 or SE-54 stationary phase or equivalent), to a uniform thickness between 0,10 and 0,30 μm.
3.12. Microsyringe, of 10 ml capacity, for gas chromatography, with cemented needle suitable for split injection.
3.13. Calcium dichloride desiccator
4. REAGENTS
4.1. Potassium hydroxide minimum titre 85 %.
4.2. Potassium hydroxide ethanolic solution, approximately 2 N.
Dissolve 130 g of potassium hydroxide (point 4.1) with cooling in 200 ml of distilled water and then make up to one litre with ethanol (point 4.10). Keep the solution in well-stoppered dark glass bottles and stored maximum two days.
4.3. Ethyl ether, for analysis quality.
4.4. Potassium hydroxide ethanolic solution, approximately 0,2 N.
Dissolve 13 g of potassium hydroxide (point 4.1) in 20 ml of distilled water and make up to one litre with ethanol (point 4.10).
4.5. Anhydrous sodium sulphate, for analysis quality.
4.6. Glass plates (20x20 cm) coated with silica gel, without fluorescence indicator, thickness 0,25 mm (commercially available ready for use).
4.7. Toluene, for chromatography quality.
4.8. Acetone, for chromatography quality.
4.9. n-Hexane, for chromatography quality.
4.10. Ethyl ether, for chromatography quality.
4.11. Ethanol of analytical quality.
4.12. Ethyl acetate of analytical quality.
4.13. Reference solution for thin-layer chromatography: cholesterol or phytosterols, and erythrodiol 5 % solution in ethyl acetate (point 4.11).
4.14. 2,7-dichlorofluorescein, 0,2 % in ethanolic solution. Make slightly basic by adding a few drops of 2 N alcoholic potassium hydroxide solution (point 4.2).
4.15. Anhydrous pyridine, for chromatography quality (see Note 5).
4.16. Hexamethyl disilazane of analytical quality.
4.17. Trimethylchlorosilane of analytical quality.
4.18. Sample solutions of sterol trimethylsilyl ethers.
To be prepared at the time of use from sterols and erythrodiol obtained from oils containing them.
4.19. α-cholestanol, purity more than 99 % (purity must be checked by GC analysis).
4.20. α-cholestanol internal standard solution, 0,2 % solution (m/V) in ethyl acetate (point 4.11).
4.21. Phenolphthalein solution, 10 g/L in ethanol (point 4.10).
4.22. Carrier gas: hydrogen or helium, gas-chromatographic purity.
4.23. Auxiliary gases: hydrogen, helium, nitrogen and air, of gas-chromatographic purity.
4.24. n-Hexane (point 4.9)/ethyl ether (point 4.10) mixture 65:35 (V/V).
4.25. Silylation reagent, consisting of a 9:3:1 (V/V/V) mixture of pyridine/hexamethyl disilazane/trimethylchlorosilane.
5. PROCEDURE
|
5.1. |
Preparation of the unsaponifiable matter.
|
|
5.2. |
Separation of the sterol and triterpene dialcohols fraction (erythrodiol + uvaol)
|
|
5.3. |
Preparation of the trimethylsilyl ethers.
|
|
5.4. |
Gas chromatographic analysis.
|
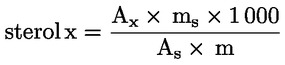
6. EXPRESSION OF THE RESULTS
6.1. Report individual sterol concentrations as mg/kg of fatty material and their sum as ‘total sterols’.
The composition of each of the individual sterols and of the erythrodiol and uvaol should be expressed to one decimal point.
Total sterol composition must be expressed without any decimal point.
6.2. Calculate the percentage of each individual sterol from the ratio of the relevant peak area to the total peak area for sterols:

where:
|
Ax |
= |
peak area for x; |
|
ΣA |
= |
total peak area for sterols. |
6.3. Apparent β-sitosterol: Δ5-23-stigmastadienol + clerosterol + β-sitosterol + sitostanol + Δ5-avenasterol + Δ5-24-stigmastadienol.
6.4. Calculate the percentage of erythrodiol and uvaol:

where
|
ΣA |
= |
sum area for sterol in computing system counts; |
|
Er |
= |
area of Erythrodiol in computing system counts; |
|
Uv |
= |
area of Uvaol in computing system counts; |
Appendix
Determination of the linear speed of the gas
With the gas chromatograph set to normal operating conditions, inject 1 to 3 μl of methane (or propane) and measure the time taken by the gas to pass through the column, from the time of injection to the time at which the peak appears (tM).
The linear speed in cm/s is given by L/tM, where L is the length of the column in centimetres and tM is the measured time, in seconds.
Table 1
Relative retention times for sterols
|
Peak |
Identification |
Relative retention time |
||
|
SE 54 column |
SE 52 column |
|||
|
1 |
Cholesterol |
Δ-5-cholesten-3ß-ol |
0,67 |
0,63 |
|
2 |
Cholestanol |
5α-cholestan-3ß-ol |
0,68 |
0,64 |
|
3 |
Brassicasterol |
[24S]-24-methyl-Δ-5,22-cholestadien-3ß-ol |
0,73 |
0,71 |
|
* |
Ergosterol |
[24S] 24 methy Δ5-7-22 cholestatrien 3ß-ol |
0,78 |
0,76 |
|
4 |
24-methylene-cholesterol |
24-methylene-Δ-5,24-cholestadien-3ß-o1 |
0,82 |
0,80 |
|
5 |
Campesterol |
(24R)-24-methyl-Δ-5-cholesten-3ß-ol |
0,83 |
0,81 |
|
6 |
Campestanol |
(24R)-24-methyl-cholestan-3ß-ol |
0,85 |
0,82 |
|
7 |
Stigmasterol |
(24S)-24-ethyl-Δ-5,22-cholestadien-3ß-ol |
0,88 |
0,87 |
|
8 |
Δ-7-campesterol |
(24R)-24-methyl-Δ-7-cholesten-3ß-ol |
0,93 |
0,92 |
|
9 |
Δ-5,23-stigmastadienol |
(24R,S)-24-ethyl-Δ-5,23-choIestadien-3ß-ol |
0,95 |
0,95 |
|
10 |
Clerosterol |
(24S)-24-ethyl-Δ-5,25-cholestadien-3ß-ol |
0,96 |
0,96 |
|
11 |
ß-sitosterol |
(24R)-24-ethyl-Δ-5-cholesten-3ß-ol |
1,00 |
1,00 |
|
12 |
Sitostanol |
24-ethyl-cholestan-3ß-ol |
1,02 |
1,02 |
|
13 |
Δ-5-avenasterol |
(24Z)-24-ethylidene-Δ-cholesten-3ß-ol |
1,03 |
1,03 |
|
14 |
Δ-5-24-stigmastadienol |
(24R,S)-24-ethyl-Δ-5,24-cholestadien-3ß-ol |
1,08 |
1,08 |
|
15 |
Δ-7-stigmastenol |
(24R,S)-24-ethyl-Δ-7-cholesten-3ß-ol |
1,12 |
1,12 |
|
16 |
Δ-7-avenasterol |
(24Z)-24-ethylidene-Δ-7-cholesten-3ß-ol |
1,16 |
1,16 |
|
17 |
Erythrodiol |
5α olean-12en-3ß28 diol |
1,41 |
1,41 |
|
18 |
Uvaol |
Δ12-ursen-3ß28 diol |
1,52 |
1,52 |
Figure 1
Gas chromatogram of the sterol and triterpene dialchols fraction of a lampante olive oil (spiked with internal standard)

Figure 2
Gas chromatogram of the sterol and triterpene dialchols fraction of a refined olive oil (spiked with internal standard)

Figure 3
TLC plate olive-pomace oil with the zone that must be scraped for sterols and triterpenic dialcohols determination

1 – Squalene
2 – Triterpene and Aliphatic alcohols
3 – Sterols and Triterpenic dialcohols
4 – Start and free fatty acids
▼M26 —————
ANNEX VII
DETERMINATION OF THE PERCENTAGE OF 2-GLYCERYL MONOPALMITATE
1. PURPOSE AND SCOPE
This method describes the analysis procedure for determining the percentage of palmitic acid in position 2 of the triglycerides by evaluating 2-glyceryl monopalmitate.
This method can be applied to liquid vegetable oils at ambient temperature (20 °C).
2. PRINCIPLE
After preparation the oil sample is subjected to the action of pancreatic lipase: partial and specific hydrolysis in positions 1 and 3 of the triglyceride molecule causes monoglycerides to appear in position 2. The percentage of 2-glyceryl monopalmitate in the monoglyceride fraction is determined after silylation by capillary-column gas chromatography.
3. APPARATUS AND MATERIALS
|
3.1. |
25 ml Erlenmeyer flask |
|
3.2. |
100, 250 and 300 ml beakers |
|
3.3. |
Glass chromatograph column, internal diameter 21-23 mm, length 400 mm, fitted with a sintered glass disc and a stopcock |
|
3.4. |
10, 50, 100 and 200 ml measuring cylinders |
|
3.5. |
100 and 250 ml flasks |
|
3.6. |
Rotary evaporator |
|
3.7. |
10 ml conical-bottomed centrifuge tubes with groundglass stopper |
|
3.8. |
Centrifuge for 10 and 100 ml tubes |
|
3.9. |
Thermostat permitting a stable temperature of 40 ± 0,5 °C |
|
3.10. |
1 and 2 ml graduated pipettes |
|
3.11. |
1 ml hypodermic syringe |
|
3.12. |
100 μl microsyringe |
|
3.13. |
1 000 ml funnel |
|
3.14. |
Capillary gas chromatograph with an on-column cold injector for direct injection of the sample into the column and a furnace able to maintain the selected temperature to approximately 1 °C |
|
3.15. |
On-column cold injector for direct injection of the sample into the column |
|
3.16. |
Flame ionisation detector and electrometer |
|
3.17. |
Recorder-integrator adapted to the electrometer with a response rate no greater than 1 sec and a variable paper roll rate |
|
3.18. |
Capillary column made of glass or fused silica 8-12 metres long, 0,25-0,32 mm internal diameter, covered with methylpolysiloxane or phenyl methylpolysiloxane 5 %, 0,10-0,30 μm thick, useable at 370 °C |
|
3.19. |
10 μl microsyringe fitted with a hardened needle, at least 7,5 cm long for direct on-column injection. |
4. REAGENTS
|
4.1. |
Silica gel with a grain size of between 0,063 and 0,200 mm (70/280 mesh) prepared as follows: Place the silica gel in a porcelain capsule, dry in an incubator at 160 °C for four hours, then leave to cool at room temperature in a desiccator. Add water equivalent to 5 % of the mass of the silica gel as follows: Weigh 152 g silica gel into an Erlenmeyer flask then add 8 g of distilled water, stopper and shake gently to distribute the water evenly. Leave to stand for at least 12 hours before use. |
|
4.2. |
n-hexane (for chromatography) |
|
4.3. |
Isopropanol |
|
4.4. |
Isopropanol, 1/1 (v/v) aqueous solution |
|
4.5. |
Pancreatic lipase. It must have an activity of between 2,0 and 10 lipase units per mg. (Pancreatic lipases with an activity of between 2 and 10 units per mg enzyme are commercially available.) |
|
4.6. |
Buffer solution of trishydroxymethylaminomethane: 1 M aqueous solution adjusted to pH 8 (potentiometric control) by conc. HCl (1/1 v/v) |
|
4.7. |
Enzyme-quality sodium cholate, 0,1 % aqueous solution (this solution must be used within two weeks of its preparation) |
|
4.8. |
Calcium chloride, 22 % aqueous solution |
|
4.9. |
Diethyl ether for chromatography |
|
4.10. |
Developer solvent: mixture of n-hexane/diethyl ether (87:13 v:v) |
|
4.11. |
Sodium hydroxide, 12 % by weight solution |
|
4.12. |
Phenolphthalein, 1 % solution in ethanol |
|
4.13. |
Carrier gas: hydrogen or helium, for gas chromatography |
|
4.14. |
Auxiliary gases: hydrogen, 99 % minimum purity, free from moisture and organic substances, and air, for gas chromatography, of the same purity |
|
4.15. |
Silanisation reagent: mixture of pyridine/hexamethyldisilazane, trimethylchlorosilane 9/3/1 (v/v/v). (Ready-to-use solutions are commercially available. Other silylation reagents may be used, particularly bis-trimethylsilyl trifluoracetamide + 1 % trimethylchlorosilane, diluted with an identical volume of anhydrous pyridine.) |
|
4.16. |
Reference samples: pure monoglycerides or monoglyceride mixtures with a known percentage composition similar to that of the sample. |
5. METHOD
5.1. Sample preparation
|
5.1.1. |
Oils with a free acidity of less than 3 % do not need to be neutralised before chromatography on a silica gel column. Oils with a free acidity of more than 3 % must be neutralised as per point 5.1.1.1.
|
|
5.1.2. |
Put 1,0 g of the oil prepared as above into a 25 ml Erlenmeyer flask (3.1) and dissolve in 10 ml of developer mixture (4.10). Leave the solution to stand for at least 15 minutes before silica gel column chromatography. If the solution is cloudy centrifuge it to ensure optimum conditions for chromatography. (Ready-to-use 500 mg silica gel SPE cartridges can be used). |
|
5.1.3. |
Preparation of the chromatography column Pour about 30 ml of the developer solvent (4.10) into the column (3.3), insert a piece of cotton into the bottom part of the column using a glass rod; press to eliminate the air. In a beaker prepare a suspension of 25 g of silica gel (4.1) in about 80 ml of developer solvent and pour it into the column using a funnel. Check that all the silica gel is in the column; wash with developer solvent (4.10), open the stopcock and allow the liquid to reach a level about 2 mm above the level of the silica gel. |
|
5.1.4. |
Column chromatography Weigh accurately 1,0 g of sample prepared as in point 5.1 into a 25 ml Erlenmeyer flask (3.1). Dissolve the sample in 10 ml of developer solvent (4.10). Pour the solution into the chromatography column prepared as in point 5.1.3. Avoid disturbing the surface of the column. Open the stopcock and pour the sample solution until it reaches the level of the silica gel. Develop with 150 ml of the developer solvent. Adjust the flow rate to 2 ml/min (so that 150 ml enters the column in about 60-70 minutes). Recover the eluate in a previously weighed 250 ml flask. Evaporate the solvent under vacuum and remove the final traces of the solvent under a nitrogen current. Weigh the flask and calculate the recovered extract. (If ready-to-use silica gel SPE cartridges are used use the following method: Put 1 ml of solution (5.1.2) into the prepared cartridges with 3 ml of n-hexane. After percolating the solution develop with 4 ml of n-hexane/diethyl ether 9/1 (v/v). Recover the eluate in a 10 ml tube and evaporate to dry in a nitrogen current. Expose the dry residue to pancreatic lipase (5.2). (It is essential to check the fatty acid composition before and after crossing the SPE cartridge.) |
5.2. Hydrolysis by pancreatic lipase
|
5.2.1. |
Weigh into the centrifuge tube 0.1 g of the oil prepared as in point 5.1. Add 2 ml of buffer solution (4.6), 0,5 ml of the sodium cholate solution (4.7) and 0,2 ml of the calcium chloride solution, stirring well after each addition. Close the tube with the groundglass stopper and place in the thermostat at 40 + 0,5 °C. |
|
5.2.2. |
Add 20 mg of lipase, shake carefully (avoid wetting the stopper) and place the tube in the thermostat for exactly two minutes. Then remove it, shake vigorously for exactly 1 minute and leave to cool. |
|
5.2.3. |
Add 1 ml of diethyl ether, stopper and shake vigorously, then centrifuge and transfer the ether solution into a clean, dry tube using a microsyringe. |
5.3. Preparation of the silanised derivatives and gas chromatography
|
5.3.1. |
With a microsyringe insert 100 μl of solution (5.2.3) into a 10 ml conical-bottomed tube. |
|
5.3.2. |
Remove the solvent under a slight nitrogen current, add 200 μl of silanisation reagent (4.15), stopper the tube and leave to stand for 20 minutes. |
|
5.3.3. |
After 20 minutes, add 1 to 5 ml of n-hexane (depending on the chromatography conditions): the resulting solution is ready for gas chromatography. |
5.4. Gas chromatography
Operating conditions:
— Injector temperature (on-column injector) lower than solvent boiling point (68 °C);
— Detector temperature: 350 °C;
— Column temperature: programming of furnace temperature: 60 °C for 1 minute, increasing by 15 °C per minute up to 180 °C, then by 5 °C per minute up to 340 °C, then 340 °C for 13 minutes;
— Carrier gas: hydrogen or helium, set at a linear velocity sufficient to obtain the resolution reflected in Figure 1. The retention time of the C54 triglyceride must be 40 ± 5 minutes (see Figure 2). (The operating conditions indicated above are indicative. Operators will have to optimise them to obtain the desired resolution. The peak corresponding to 2-glyceryl monopalmitate must have a minimum height equal to 10 % of the recorder scale.)
— Quantity of substance injected: 0,5-1 μl of the n-hexane solution (5 ml) (5.3.3).
5.4.1. Identification of the peaks
The individual monoglycerides are identified from their retention times and by comparison with those obtained for standard monoglyceride mixtures under the same conditions.
5.4.2. Quantitative evaluation
The area of each peak is calculated using an electronic integrator.
6. EXPRESSION OF RESULTS
The percentage of glyceryl monopalmitate is calculated from the ratio between the area of the corresponding peak and the areas of the peaks of all the monoglycerides (see Figure 2) using the formula:
glyceryl monopalmitate (%):

where:
|
Ax |
= |
area of the peak corresponding to glyceryl monopalmitate |
|
ΣA |
= |
sum of the areas of all the monoglyceride peaks |
The result must be to one decimal place.
7. ANALYSIS REPORT
The analysis report must specify:
— reference to this method,
— all the information needed for a full identification of the sample,
— the analysis result,
— any deviation from the method, whether as the result of a decision by the parties concerned or for another reason,
— details to identify the laboratory, the date of the analysis and the signatures of those responsible for the analysis.
Figure 1
Chromatogram of the products of the silanisation reaction obtained by the action of lipase on a refined olive oil with 20 % esterified oil added (100 %)

Key: ‘acides gras libres’ = free fatty acids; ‘Huile d’olive raffinée + 20 % huile estérifiée’ = refined olive oil + 20 % esterified oil; ‘1-2 monopalmitoléine’ = 1-2 monopalmitolein; ‘1-2 mono C18 insat.’ = unsaturated 1-2 mono C18
Figure 2
Chromatogram of:
|
(A) |
unesterified olive oil, after lipase; after silanisation; under these conditions (8-12 m capillary column) the wax fraction is eluted at the same time as the diglyceride fraction or slightly afterwards. After lipase, the triglyceride content should not exceed 15 % 
Key:
|
Chromatogram of:
|
(B) |
unesterified oil after lipase; after silanisation; under these conditions (8-12 m capillary column) the wax fraction is eluted at the same time as the diglyceride fraction or slightly afterwards. After lipase, the triglyceride content should not exceed 15 %. 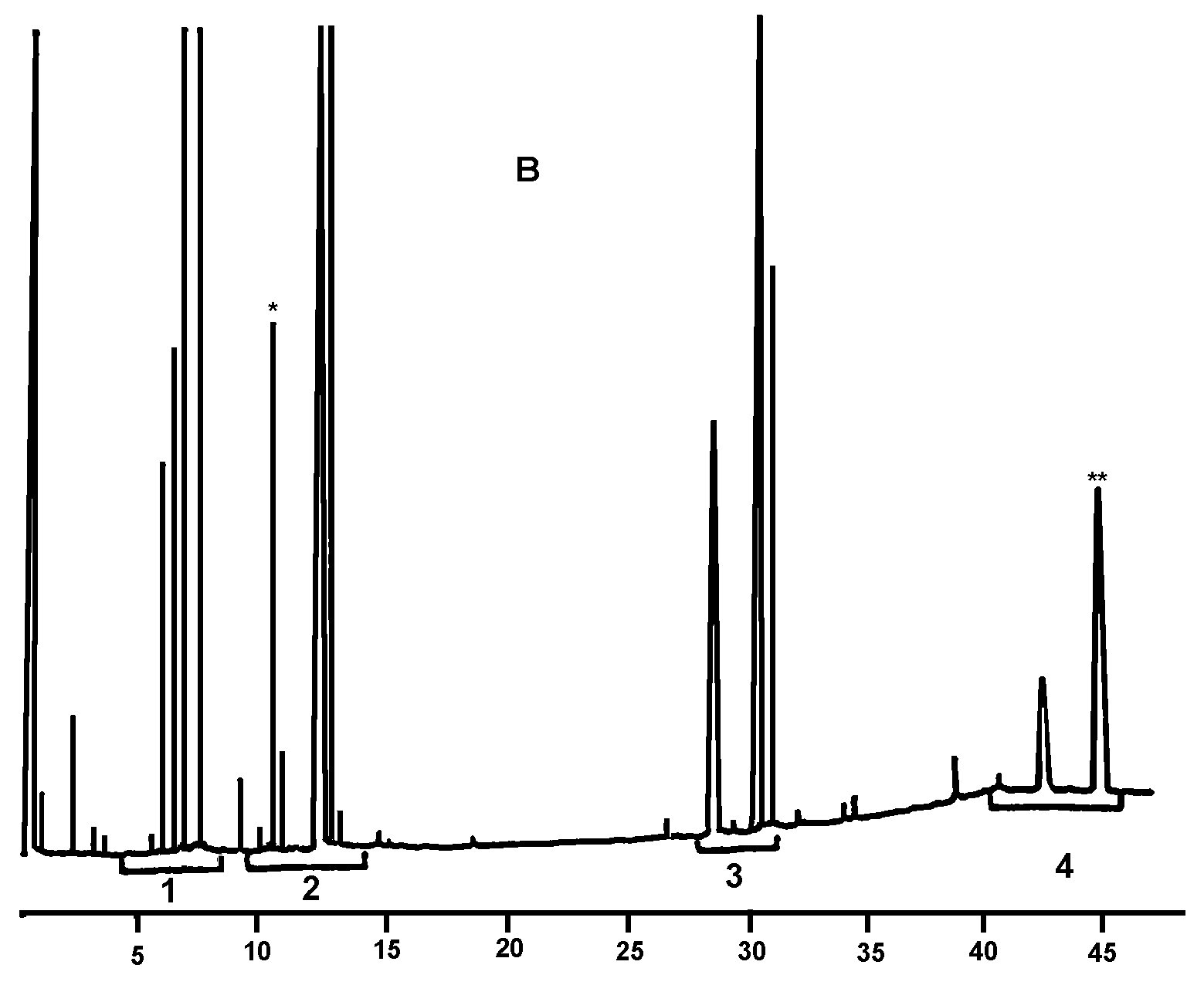
Key:
|
8. NOTES
PREPARATION OF THE LIPASE
Lipases with satisfactory activity are commercially available. They can also be prepared in the laboratory in the following manner:
Cool to 0 °C 5 kg of fresh pig’s pancreas. Remove the surrounding solid fat and the connective tissue and grind to a liquid paste in a blender. Stir the paste with 2,5 litres of anhydrous acetone for 4-6 hours, then centrifuge. Extract the residue three more times with the same volume of anhydrous acetone, then twice with an acetone/diethyl ether mixture (1/1 v/v) and twice with diethyl ether.
Vacuum-dry the residue for 48 hours to obtain a stable powder which can be stored for a long time in a refrigerator away from moisture.
MONITORING LIPASE ACTIVITY
Prepare an olive oil emulsion as follows:
In a mixer stir for 10 minutes a mixture of 165 ml of a 100 g/l gum arabic solution, 15 g of crushed ice and 20 ml of a previously neutralised olive oil.
Pour 10 ml of the emulsion into a 50 ml beaker, then 0,3 ml of a 0,2 g/ml sodium cholate solution and then 20 ml of distilled water.
Put the beaker in a thermostat set at 37 °C; introduce the electrodes of the pH meter and the screw agitator.
Using a burette, add a 0,1 N sodium hydroxide solution drop by drop until a pH of 8,3 is obtained.
Add an aliquot of the lipase powder suspension in water (0,1 g/ml of lipase). As soon as the pH meter reads 8,3, start the chronometer and add the sodium hydroxide solution drop by drop at a rate which maintains the pH at 8,3. Note every minute the volume of solution consumed.
Record the data on an x/y graph with the time on the x-axis and millilitres of 0,1 N alkaline solution consumed to keep a constant pH on the y-axis. A linear graph should be obtained.
Lipase activity, expressed in lipase units per mg, is given by the following formula:

where:
|
A |
is activity in lipase units/mg |
|
V |
is the number of millilitres of 0,1 N sodium hydroxide solution per minute (calculated on the basis of the graph) |
|
N |
is the titre of the sodium hydroxide solution |
|
m |
is the mass in mg of the test lipase. |
A lipase unit is defined as the quantity of enzyme which releases 10 micro-equivalents of acid per minute.
▼M20 —————
ANNEX IX
SPECTROPHOTOMETRIC INVESTIGATION IN THE ULTRAVIOLET
FOREWORD
Spectrophotometric examination in the ultraviolet can provide information on the quality of a fat, its state of preservation and changes brought about by technological processes. The absorption at the wavelengths specified in the method is due to the presence of conjugated diene and triene systems resulting from oxidation processes and/or refining practices. These absorptions are expressed as specific extinctions
![]()
(the extinction of 1 % w/v solution of the fat in the specified solvent, in a 10 mm cell) conventionally indicated by K (also referred to as ‘extinction coefficient’).
1. SCOPE
This Annex describes the procedure for performing a spectrophotometric examination of olive oil in the ultraviolet region.
2. PRINCIPLE OF THE METHOD
A sample is dissolved in the required solvent and the absorbance of the solution is measured at the specified wavelengths with reference to pure solvent.
The specific extinctions at 232 nm and 268 nm in iso-octane or 232 nm and 270 nm in cyclohexane are calculated for a concentration of 1 % w/v in a 10 mm cell.
3. EQUIPMENT
|
3.1. |
A spectrophotometer suitable for measurements at ultraviolet wavelengths (220 nm to 360 nm), with the capability of reading individual nanometric units. A regular check is recommended for the accuracy and reproducibility of the absorbance and wavelength scales as well as for stray light.
|
|
3.2. |
Rectangular quartz cuvettes, with covers, suitable for measurements at the ultraviolet wavelengths (220 to 360 nm) having an optical path-length of 10 mm. When filled with water or other suitable solvent the cuvettes should not show differences between them of more than 0,01 extinction units. |
|
3.3. |
One- mark volumetric flasks, capacity 25 ml, class A. |
|
3.4. |
Analytical balance, capable of being read to the nearest 0,0001 g |
4. REAGENTS
During the analysis, unless otherwise stated, use only reagents of recognised analytical grade and distilled or demineralised water or water of equivalent purity.
Solvent: Iso-octane (2,2,4 trimethylpentane) for the measurements at 232 nm and 268 nm and cyclohexane for the measurements at 232 nm and 270 nm, having an absorbance less than 0,12 at 232 nm and less than 0,05 at 270 nm against distilled water, measured in a 10 mm cell.
5. PROCEDURE
|
5.1. |
The sample must be perfectly homogeneous and without suspended impurities. If not, it must be filtered through paper at a temperature of approximately 30 °C. |
|
5.2. |
Weigh accurately approximately 0,25 g (to the nearest 1 mg) of the sample so prepared into a 25 ml graduated flask, make up to the mark with the specified solvent and homogenise. The resulting solution must be perfectly clear. If opalescence or turbidity is present, filter quickly through paper. NOTE: Generally, a mass of 0,25 to 0,30 g is sufficient for absorbance measurements of virgin and extra virgin olive oils at 268 nm and 270 nm. For measurements at 232 nm, 0,05 g of sample are usually required, so two distinct solutions are usually prepared. For absorbance measurements of olive pomace oils, refined olive oils and adulterated olive oils, a smaller portion of sample, e.g. 0,1 g is usually needed due to their higher absorbance. |
|
5.3. |
If necessary, correct the baseline (220-290 nm) with solvent in both quartz cells (sample and reference), then fill the sample quartz cell with the test solution and measure the extinctions at 232, 268 or 270 nm against the solvent used as a reference. The extinction values recorded must lie within the range 0,1 to 0,8 or within the range of linearity of the spectrophotometer which should be verified. If not, the measurements must be repeated using more concentrated or more dilute solutions as appropriate. |
|
5.4. |
After measuring the absorbance at 268 or 270 nm, measure the absorbance at λmax, λmax + 4 and λmax – 4. These absorbance values are used to determine the variation in the specific extinction (ΔΚ). NOTE: λmax is considered to be 268 nm for isooctane used as solvent and 270 nm for cyclohexane. |
6. EXPRESSION OF THE RESULTS
|
6.1. |
Record the specific extinctions (extinction coefficients) at the various wavelengths calculated as follows:
where:
expressed to two decimal places. |
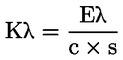
|
6.2. |
Variation of the specific extinction (ΔΚ) The variation of the absolute value of the extinction (ΔΚ) is given by:
where Km is the specific extinction at the wavelength for maximum absorption at 270 nm and 268 nm depending on the solvent used. The results should be expressed to two decimal places. |
ANNEX X
DETERMINATION OF FATTY ACID METHYL ESTERS BY GAS CHROMATOGRAPHY
1. SCOPE
This Annex gives guidance on the gas chromatographic determination of free and bound fatty acids in vegetable fats and oils following their conversion into fatty acid methyl esters (FAME).
The bound fatty acids of the triacylglycerols (TAGs) and, depending on the esterification method, the free fatty acids (FFA), are converted into fatty acid methyl esters (FAME), which are determined by capillary gas chromatography.
The method described in this Annex allows the determination of FAME from C12 to C24, including saturated, cis- and transmonounsaturated and cis- and trans-polyunsaturated fatty acid methyl esters.
2. PRINCIPLE
Gas chromatography (GC) is used for the quantitative analysis of FAME. The FAME are prepared according to Part A. They are then injected into and vaporised within the injector. The separation of FAME is performed on analytical columns of specific polarity and length. A Flame Ionisation Detector (FID) is used for the detection of the FAME. The conditions of analysis are given in Part B.
Hydrogen or helium may be used as the carrier gas (mobile phase) in the gas chromatography of FAME with FID. Hydrogen speeds up separation and gives sharper peaks. The stationary phase is a microscopic layer of a thin liquid film on an inert solid surface made of fused silica.
As they pass through the capillary column the volatilised compounds being analysed interact with the stationary phase coating the inner surface of the column. Due to this different interaction of different compounds, they elute at a different time, which is called the retention time of the compound for a given set of analysis parameters. The comparison of the retention times is used for the identification of the different compounds.
PART A
PREPARATION OF THE FATTY ACID METHYL ESTERS FROM OLIVE OIL AND OLIVE-POMACE OIL
1. SCOPE
This part specifies the preparation of the methyl esters of fatty acids. It includes methods for preparing fatty acid methyl esters from olive and olive-pomace oils.
2. FIELD OF APPLICATION
The preparation of the fatty acid methyl esters from olive oils and olive-pomace oils are performed by transesterification with methanolic solution of potassium hydroxide at room temperature. The necessity of purification of the sample prior to the trans-esterification depends on the sample's free fatty acids content and the analytical parameter to be determined, it can be chosen according to the following table:
|
Category of oil |
Method |
|
Virgin olive oil with acidity ≤ 2,0 % |
1. Fatty acids 2. trans-Fatty acids 3. ΔECN42 (after purification with silica-gel SPE) |
|
Refined olive oil |
|
|
Olive oil composed of refined olive oil and virgin olive oils |
|
|
Refined olive pomace oil |
|
|
Olive pomace oil |
|
|
Virgin olive oil with acidity > 2,0 % Crude olive pomace oil |
1. Fatty acids (after purification with silica-gel SPE) 2. trans-Fatty acids (after purification with silica-gel SPE) 3. ΔECN42 (after purification with silica-gel SPE) |
3. METHODOLOGY
3.1. Trans-esterification with methanolic solution of potassium hydroxide at room temperature
3.1.1. Principle
Methyl esters are formed by trans-esterification with methanolic potassium hydroxide as an intermediate stage before saponification takes place.
3.1.2. Reagents
|
3.1.2.1. |
Methanol containing not more than 0,5 % (m/m) water. |
|
3.1.2.2. |
Hexane, chromatographic quality. |
|
3.1.2.3. |
Heptane, chromatographic quality. |
|
3.1.2.4. |
Diethyl ether, stabilised for analysis. |
|
3.1.2.5. |
Acetone, chromatographic quality. |
|
3.1.2.6. |
Elution solvent for purifying the oil by column/SPE chromatography, mixture hexane/diethyl ether 87/13 (v/v). |
|
3.1.2.7. |
Potassium hydroxide, approximately 2M methanolic solution: dissolve 11,2 g of potassium hydroxide in 100 ml of methanol. |
|
3.1.2.8. |
Silica gel cartridges, 1 g (6 ml), for solid phase extraction. |
3.1.3. Apparatus
|
3.1.3.1. |
Screw-top test tubes (5 ml volume) with cap fitted with a PTFE joint. |
|
3.1.3.2. |
Graduated or automatic pipettes, 2 ml and 0,2 ml. |
3.1.4. Purification of oil samples
When necessary, the samples will be purified by passing the oil through a silica gel solid-phase extraction cartridge. A silica gel cartridge (3.1.2.8) is placed in a vacuum elution apparatus and washed with 6 ml of hexane (3.1.2.2); washing is performed without vacuum. Then a solution of the oil (0,12 g approximately) in 0,5 ml of hexane (3.1.2.2) is loaded onto the column. The solution is pulled down and then eluted with 10 ml of hexane/diethyl ether (87:13 v/v) (3.1.2.6). The combined eluates are homogenised and divided in two similar volumes. An aliquot is evaporated to dryness in a rotary evaporator under reduced pressure at room temperature. The residue is dissolved in 1 ml of heptane and the solution is ready for fatty acid analysis by GC. The second aliquot is evaporated and the residue is dissolved in 1 ml of acetone for triglyceride analysis by HPLC, if necessary.
3.1.5. Procedure
In a 5 ml screw-top test tube (3.1.3.1) weigh approximately 0,1 g of the oil sample. Add 2 ml of heptane (3.1.2.2), and shake. Add 0,2 ml of the methanolic potassium hydroxide solution (3.1.2.7), put on the cap fitted with a PTFE joint, tighten the cap, and shake vigorously for 30 seconds. Leave to stratify until the upper solution becomes clear. Decant the upper layer containing the methyl esters. The heptane solution is ready for injection into the gas chromatograph. It is advisable to keep the solution in the refrigerator until gas chromatographic analysis. Storage of the solution for more than 12 hours is not recommended.
PART B
ANALYSIS OF FATTY ACID METHYL ESTERS BY GAS CHROMATOGRAPHY
1. SCOPE
This part gives general guidance for the application of capillary column gas chromatography to determine the qualitative and quantitative composition of a mixture of fatty acid methyl esters obtained in accordance with the method specified in Part A.
The part is not applicable to polymerised fatty acids.
2. REAGENTS
2.1. Carrier gas
Inert gas (helium or hydrogen), thoroughly dried and with an oxygen content of less than 10 mg/kg.
Note 1: Hydrogen can double the speed of analysis but is hazardous. Safety devices are available.
2.2. Auxiliary gases
|
2.2.1. |
Hydrogen (purity ≥ 99,9 %), free from organic impurities. |
|
2.2.2. |
Air or oxygen, free from organic impurities. |
|
2.2.3. |
Nitrogen (purity > 99 %). |
2.3. Reference standard
Mixture of methyl esters of pure fatty acids, or the methyl esters of a fat of known composition, preferably similar to that of the fatty matter to be analysed. Cis and trans isomers of octadecenoic, octadecadienoic and octadecatrienoic methyl esters are useful for the identification of trans isomers of unsaturated acids.
Care should be taken to prevent the oxidation of polyunsaturated fatty acids.
3. APPARATUS
The instructions given are for the usual equipment used for gas chromatography, employing capillary columns and a flame-ionisation detector.
3.1. Gas chromatograph
The gas chromatograph shall comprise the following elements.
3.1.1. Injection system
Use an injection system with capillary columns, in which case the injection system should be specially designed for use with such columns. It may be of the split type or the splitless on-column injector type.
3.1.2. Oven
The oven shall be capable of heating the capillary column to a temperature of at least 260 °C and of maintaining the desired temperature to within 0,1 °C. The last requirement is particularly important when a fused silica tube is used.
The use of temperature-programmed heating is recommended in all cases, and in particular for fatty acids with less than 16 carbon atoms.
3.1.3. Capillary column
|
3.1.3.1. |
Tube, made of a material inert to the substances to be analysed (usually glass or fused silica). The internal diameter shall be between 0,20 to 0,32 mm. The internal surface shall undergo an appropriate treatment (e.g. surface preparation, inactivation) before receiving the stationary phase coating. A length of 60 m is sufficient for fatty acid and cis and trans isomers of fatty acids. |
|
3.1.3.2. |
Stationary phase, polar polysiloxane (cyanopropylsilicone) bonded (cross-linked) columns are suitable. Note 2: There is a risk that polar polysiloxanes may give rise to difficulties in the identification and separation of linolenic acid and C20 acids. The coatings shall be thin, i.e. 0,1 to 0,2 μm. |
|
3.1.3.3. |
Assembly and conditioning of the column Observe the normal precautions for assembling capillary columns, i.e. arrangement of the column in the oven (support), choice and assembly of joints (leak tightness), positioning of the ends of the column in the injector and the detector (reduction of dead-spaces). Place the column under a flow of carrier gas (e.g. 0,3 bar (30 kPa) for a column of length 25 m and internal diameter 0,3 mm). Condition the column by temperature programming of the oven at 3 °C/min from ambient temperature to a temperature 10 °C below the decomposition limit of the stationary phase. Maintain the oven at this temperature for one hour until stabilisation of the baseline. Return it to 180 °C to work under isothermal conditions. Note 3: Suitably pre-conditioned columns are available commercially. |
3.1.4. Flame ionisation detector and converter-amplifier
3.2. Syringe
The syringe shall have a maximum capacity of 10 μl, graduated in 0,1 μl divisions.
3.3. Data acquisition system
Data acquisition system connected online with the detectors and employed with a software program suitable for peak integration and normalisation.
4. PROCEDURE
The operations described in 4.1 to 4.3 are for the use of a flame-ionisation detector.
4.1. Test conditions
4.1.1. Selection of optimum operating conditions for capillary columns
Owing to the efficiency and permeability of capillary columns, the separation of the constituents and the duration of the analysis are largely dependent on the flow-rate of the carrier gas in the column. It will therefore be necessary to optimise the operating conditions by adjusting this parameter (or simply column head loss) depending on whether the aim is to improve separation or speed up analysis.
The following conditions have proved to be suitable for the separation of FAMEs (C4 to C26). Examples of chromatograms are shown in Appendix B:
|
Injector temperature: |
250 °C |
|
Detector temperature: |
250 °C |
|
Oven temperature: |
165 °C (8 min) to 210 °C at 2 °C/min |
|
Carrier gas hydrogen: |
column head pressure, 179 kPa |
|
Total flow: |
154,0 ml/min; |
|
Split ratio: |
1:100 |
|
Injection volume: |
1 μl |
4.1.2. Determination of the resolution (see Appendix A)
Calculate the resolution, R, of two neighbouring peaks I and II, using the formula:
R = 2 × ((ddr(II) – dr(I) )/(ω(I) + ω(II))) or R = 2 × ((tr(II) – tr(I) )/(ω(I) + ω(II))) (USP) (United States Pharmacopeia),
or
R = 1,18 × ((tr(II) – tr(I))/(ω0,5(I) + ω0,5(II))) (EP, BP, JP, DAB), (JP (Japanese Pharmacopeia), EP (Pharmacopée Européenne), BP (British Pharmacopeia))
where:
d r(I) is the retention distance of peak I;
d r(II) is the retention distance of peak II;
t r(I) is the retention time of peak I;
t r(II) is the retention time of peak II;
ω(I) is the width of the base of peak I;
ω(II) is the width of the base of peak II;
ω0,5 is the peak width of the specified compound, at mid-height of the peak;
If ω(I) ≈ ω(II), calculate R using the following formulas:
R = (dr(II) – d r(I))/ω = (dr(II) – d r(I))/4σ
where:
σ is the standard deviation (see Appendix A, Figure 1).
If the distance dr between the two peaks d r(II) - d r(I) is equal to 4σ, the resolution factor R = 1.
If two peaks are not separated completely, the tangents to the inflection points of the two peaks intersect at point C. In order to completely separate the two peaks, the distance between the two peaks must be equal to:
d r(II) - d r(I) = 6 σ from where R = 1,5 (see Appendix A, Figure 3).
5. EXPRESSION OF RESULTS
5.1. Qualitative analysis
Identify the methyl ester peaks of the sample from the chromatogram in Appendix B, figure 1, if necessary by interpolation, or by comparison with those of the methyl esters reference mixtures (as indicated at point 2.3).
5.2. Quantitative analysis
5.2.1. Determination of the composition
Calculate the mass fraction w i of the individual fatty acid methyl esters, expressed as a percentage by mass of methyl esters, as follows:
5.2.2. Method of calculation
5.2.2.1. General case
Calculate the content of a given component i, expressed as a percentage by mass of methyl esters, by determining the percentage represented by the area of the corresponding peak relative to the sum of the areas of all the peaks, using the following formula:
wi = (Ai/ΣA) × 100
where:
Ai is the area under the peak of the individual fatty acid methyl ester i;
ΣA is the sum of the areas under all the peaks of all the individual fatty acid methyl esters.
The results are expressed to two decimal places.
Note 4: For fats and oils, the mass fraction of the fatty acid methyl esters is equal to the mass fraction of the triacylglycerols in grams per 100 g. For cases in which this assumption is not allowed, see 5.2.2.2.
5.2.2.2. Use of correction factors
In certain cases, for example in the presence of fatty acids with less than eight carbon atoms or of acids with secondary groups, the areas shall be corrected with specific correction factors (Fci). These factors shall be determined for each single instrument. For this purpose suitable reference materials with certified fatty acid composition in the corresponding range shall be used.
Note 5: These correction factors are not identical to the theoretical FID correction factors, which are given in Appendix A, as they also include the performance of the injection system etc. However, in the case of bigger differences, the whole system should be checked for performance.
For this reference mixture, the mass percentage of the FAME i is given by the formula:
wi = (mi /Σm) × 100
where
m i is the mass of the FAME i in the reference mixture;
Σm is the total of the masses of the various components as FAMEs of the reference mixture.
From the chromatogram of the reference mixture, calculate the percentage by area for the FAME i as follows:
wi = (Ai/ΣA) × 100
where:
Ai is the area of the FAME i in the reference mixture;
ΣA is the sum of all the areas of all the FAMEs of the reference mixture.
The correction factor Fc is then
Fc = (mi × ΣA)/(Ai/Σm)
For the sample, the percentage by mass of each FAME i is:
wi = (Fi × Ai)/Σ (Fi × Ai)
The results are expressed to two decimal places.
Note 6: The calculated value corresponds to the percentage of mass of the individual fatty acid calculated as triacylglycerols per 100 g fat.
5.2.2.3. Use of an internal standard
In certain analyses (for example where not all of the fatty acids are quantified, such as when acids with four and six carbons are present alongside acids with 16 and 18 carbons, or when it is necessary to determine the absolute amount of a fatty acid in a sample) it is necessary to use an Internal Standard. Fatty acids with 5, 15 or 17 carbons are frequently used. The correction factor (if any) for the Internal Standard should be determined.
The percentage by mass of component i, expressed as methyl esters, is then given by the formula:
wi = (mIS × Fi × Ai)/(m × FIS × AIS)
where:
A i is the area the FAME i;
A IS is the area of the internal standard;
F i is the correction factor of the fatty acid i, expressed as FAME;
F IS is the correction factor of the internal standard;
m is the mass of the test portion, in milligrams
m IS is the mass of the internal standard, in milligrams.
The results are expressed to two decimal places.
6. TEST REPORT
The test report shall specify the methods used for the preparation of the methyl esters and for the gas chromatographic analysis. It shall also mention all operating details not specified in this Standard Method, or regarded as optional, together with details of any incidents which may have influenced the results.
The test report shall include all the information necessary for complete identification of the sample.
7. PRECISION
7.1. Results of interlaboratory test
Details of an interlaboratory test on the precision of the method are set out in Annex C to standard IOC/T.20/Doc. No 33. The values derived from this interlaboratory test may not be applicable to concentration ranges and matrices other than those given.
7.2. Repeatability
The absolute difference between two independent single test results, obtained using the same method on identical test material in the same laboratory by the same operator using the same equipment within a short interval of time, will in not more than 5 % of cases be greater than r given in tables 1 to 14 in Annex C to standard IOC/T.20/Doc. No 33.
7.3. Reproducibility
The absolute difference between two single test results, obtained using the same method on identical test material in different laboratories with different operators using different equipment, will in not more than 5 % of cases be greater than R given in tables 1 to 14 in Annex C to standard IOC/T.20/Doc. No 33.
Appendix A
Figure 1

ω0,5 width at half height of the triangle (ABC) and b width at half height of the triangle (NPM).
|
Figure 2 |
Figure 3 |
|
|
|

Appendix B
Figure 1
Gas chromatographic profile obtained by the cold methylation method from olive-pomace oil

The chromatographic peaks correspond to the methyl and ethyl esters except where otherwise indicated.
ANNEX XI
DETERMINATION OF VOLATILE HALOGENATED SOLVENTS CONTENT OF OLIVE OIL
1. METHOD
Analysis by gas chromatography using the head space technique.
2. EQUIPMENT
|
2.1. |
Gas chromatography apparatus fitted with an electron capture detector (ECD). |
|
2.2. |
Head space apparatus. |
|
2.3. |
Gas chromatography column, of glass, 2 m long and 2 mm in diameter, stationary phase. OV101 10 % or equivalent, impregnating a calcined diatomaceous earth, acid washed and silanised and of a particle size of 80 to 100 mesh. |
|
2.4. |
Carrier and auxiliary gas: nitrogen for gas chromatography, suitable for detection by electron capture. |
|
2.5. |
Glass flasks, 10 to 15 ml, with teflon coating and aluminium stopper with fitment for entry of syringe. |
|
2.6. |
Hermetically sealing clamps. |
|
2.7. |
Gas syringe 0,5 to 2 ml. |
3. REAGENTS
Standard: halogenated solvents of a degree of purity suitable for gas chromatography.
4. PROCEDURE
|
4.1. |
Exactly weigh around 3 g of oil in a glass flask (not to be reused); hermetically seal it. Place it in a thermostat at 70 °C for one hour. Using a syringe carefully remove 0,2 to 0,5 ml of the head space. Inject this into the column of the gas chromatography apparatus regulated as follows: — injector temperature: 150 °C, — column temperature: 70 to 80 °C, — detector temperature: 200 to 250 °C. Other temperatures may also be used provided the results remain equivalent. |
|
4.2. |
Reference solutions: prepare standard solutions using refined olive oil with no trace of solvents with concentrations ranging from 0,05 to 1 ppm (mg/kg) and corresponding to the presumed content of the sample. The halogenated solvents may be diluted using pentane. |
|
4.3. |
Quantitative assessment: correlate the surfaces or the elevations of the peaks of the sample and of the standard solution of the concentration presumed closest. If the deviation is greater than 10 % the analysis must be repeated in comparison with another standard solution until the deviation is within 10 %. The content is determined on the basis of the average of the elementary injections. |
|
4.4. |
Expression of results: in ppm (mg/kg). The detection limit for the method is 0,01 mg/kg. |
ANNEX XII
THE INTERNATIONAL OLIVE COUNCIL’S METHOD FOR THE ORGANOLEPTIC ASSESSMENT OF VIRGIN OLIVE OIL
1. PURPOSE AND SCOPE
The purpose of the international method described in this Annex is to determine the procedure for assessing the organoleptic characteristics of virgin olive oil within the meaning of point 1 of Part VIII of Annex VII to Regulation (EU) No 1308/2013 of the European Parliament and of the Council ( 4 ) and to establish the method for its classification on the basis of those characteristics. It also provides indications for optional labelling.
The method described is applicable only to virgin olive oils and to the classification or labelling of such oils according to the intensity of the defects perceived and of the fruitiness, as determined by a group of tasters selected, trained and monitored as a panel.
The IOC standards mentioned in this Annex are used in their last available version.
2. GENERAL BASIC VOCABULARY FOR SENSORY ANALYSIS
Refer to the standard IOC/T.20/Doc. No 4 "Sensory Analysis: General Basic Vocabulary"
3. SPECIFIC VOCABULARY
3.1. Negative attributes
Fusty/muddy sediment: Characteristic flavour of oil obtained from olives piled or stored in such conditions as to have undergone an advanced stage of anaerobic fermentation, or of oil which has been left in contact with the sediment that settles in underground tanks and vats and which has also undergone a process of anaerobic fermentation.
Musty-humid-earthy: Characteristic flavour of oils obtained from fruit in which large numbers of fungi and yeasts have developed as a result of its being stored in humid conditions for several days or of oil obtained from olives that have been collected with earth or mud on them and which have not been washed.
Winey-vinegary-acid-sour: Characteristic flavour of certain oils reminiscent of wine or vinegar. This flavour is mainly due to a process of aerobic fermentation in the olives or in olive paste left on pressing mats which have not been properly cleaned and leads to the formation of acetic acid, ethyl acetate and ethanol.
Rancid: Flavour of oils which have undergone an intense process of oxidation.
Frostbitten olives (wet wood): Characteristic flavour of oils extracted from olives which have been injured by frost while on the tree.
3.1.1. Other negative attributes
|
Heated or Burnt |
Characteristic flavour of oils caused by excessive and/or prolonged heating during processing, particularly when the paste is thermally mixed, if this is done under unsuitable thermal conditions. |
|
Hay-wood |
Characteristic flavour of certain oils produced from olives that have dried out. |
|
Rough |
Thick, pasty mouthfeel sensation produced by certain old oils. |
|
Greasy |
Flavour of oil reminiscent of that of diesel oil, grease or mineral oil. |
|
Vegetable water |
Flavour acquired by the oil as a result of prolonged contact with vegetable water which has undergone fermentation processes. |
|
Brine |
Flavour of oil extracted from olives which have been preserved in brine. |
|
Metallic |
Flavour that is reminiscent of metals. It is characteristic of oil which has been in prolonged contact with metallic surfaces during crushing, mixing, pressing or storage. |
|
Esparto |
Characteristic flavour of oil obtained from olives pressed in new esparto mats. The flavour may differ depending on whether the mats are made of green esparto or dried esparto. |
|
Grubby |
Flavour of oil obtained from olives which have been heavily attacked by the grubs of the olive fly (Bactrocera oleae). |
|
Cucumber |
Flavour produced when an oil is hermetically packed for too long, particularly in tin containers, and which is attributed to the formation of 2,6 nonadienal. |
3.2. Positive attributes
|
Fruity |
Set of olfactory sensations characteristic of the oil which depends on the variety and comes from sound, fresh olives, either ripe or unripe. It is perceived directly and/or through the back of the nose. |
|
Bitter |
Characteristic primary taste of oil obtained from green olives or olives turning colour. It is perceived in the circumvallate papillae on the ‘V’ region of the tongue. |
|
Pungent |
Biting tactile sensation characteristic of oils produced at the start of the crop year, primarily from olives that are still unripe. It can be perceived throughout the whole of the mouth cavity, particularly in the throat. |
3.3. Optional terminology for labelling purposes
Upon request, the panel leader may certify that the oils which have been assessed comply with the definitions and ranges corresponding solely to the following terms according to the intensity and perception of the attributes.
Positive attributes (fruity, bitter and pungent): According to the intensity of perception:
— Robust, when the median of the attribute is more than 6;
— Medium, when the median of the attribute is between 3 and 6;
— Delicate, when the median of the attribute is less than 3.
|
Fruitiness |
Set of olfactory sensations characteristic of the oil which depends on the variety of olive and comes from sound, fresh olives in which neither green nor ripe fruitiness predominates. It is perceived directly and/or through the back of the nose. |
|
Green fruitiness |
Set of olfactory sensations characteristic of the oil which is reminiscent of green fruit, depends on the variety of olive and comes from green, sound, fresh olives. It is perceived directly and/or through the back of the nose. |
|
Ripe fruitiness |
Set of olfactory sensations characteristic of the oil which is reminiscent of ripe fruit, depends on the variety of olive and comes from sound, fresh olives. It is perceived directly and/or through the back of the nose. |
|
Well balanced |
Oil which does not display a lack of balance, by which is meant the olfactory- gustatory and tactile sensation where the median of the bitter attribute and the median of the pungent attribute are not more than 2 points above the median of the fruitiness. |
|
Mild oil |
Oil for which the median of the bitter and pungent attributes is 2 or less. |
List of terms according to the intensity of perception:
|
Terms subject to production of an organoleptic test certificate |
Median of the attribute |
|
Fruitiness |
— |
|
Ripe fruitiness |
— |
|
Green fruitiness |
— |
|
Delicate fruitiness |
Less than 3 |
|
Medium fruitiness |
Between 3 and 6 |
|
Robust fruitiness |
More than 6 |
|
Delicate ripe fruitiness |
Less than 3 |
|
Medium ripe fruitiness |
Between 3 and 6 |
|
Robust ripe fruitiness |
More than 6 |
|
Delicate green fruitiness |
Less than 3 |
|
Medium green fruitiness |
Between 3 and 6 |
|
Robust green fruitiness |
More than 6 |
|
Delicate bitterness |
Less than 3 |
|
Medium bitterness |
Between 3 and 6 |
|
Robust bitterness |
More than 6 |
|
Delicate pungency |
Less than 3 |
|
Medium pungency |
Between 3 and 6 |
|
Robust pungency |
More than 6 |
|
Well balanced oil |
The median of the bitter attribute and the median of the pungent attribute are not more than 2 points above the median of the fruitiness |
|
Mild oil |
The median of the bitter attribute and the median of the pungent attribute are not more than 2 |
4. GLASS FOR OIL TASTING
Refer to the standard IOC/T.20/Doc. No 5, "Glass for Oil Tasting".
5. TEST ROOM
Refer to the standard IOC/T.20/Doc. No 6, "Guide for the Installation of a Test Room".
6. ACCESSORIES
The following accessories, which are required by tasters to perform their task properly, must be supplied in each booth and must be within easy reach:
— glasses (standardised) containing the samples, code numbered, covered with a watch-glass and kept at 28 °C ± 2 °C;
— profile sheet (see Figure 1) on hard copy, or on soft copy provided that the conditions of the profile sheet are met, together with the instructions for its use if necessary
— pen or indelible ink
— trays with slices of apple and/or water, carbonated water and/or rusks
— glass of water at ambient temperature
— sheet recalling the general rules listed in sections 8.4 and 9.1.1
— spittoons.
7. PANEL LEADER AND TASTERS
7.1. Panel leader
The panel leader must be a suitably trained person with an expert knowledge of the kinds of oils which he or she will come across in the course of their work. They are the key figure in the panel and responsible for its organisation and running.
The work of the panel leader calls for basic training in the tools of sensory analysis, sensory skill, meticulousness in the preparation, organisation and performance of the tests and skill and patience to plan and execute the tests in a scientific manner.
They are the sole person responsible for selecting, training and monitoring the tasters in order to ascertain their level of aptitude. They are thus responsible for the appraisal of the tasters, which must always be objective and for which they must develop specific procedures based on tests and solid acceptance and rejection criteria. See standard IOC/T.20/Doc. No 14, "Guide for the selection, training and monitoring of skilled virgin olive oil tasters".
Panel leaders are responsible for the performance of the panel and hence for its evaluation, of which they must give reliable, objective proof. In any case, they must demonstrate at all times that the method and tasters are under control. Periodic calibration of the panel is recommended (IOC/T.20/Doc. No 14, § 5).
They hold ultimate responsibility for keeping the records of the panel. These records must always be traceable. They must comply with the assurance and quality requirements laid down in international sensory analysis standards and ensure the anonymity of the samples at all times.
They shall be responsible for inventorying and ensuring that the apparatus and equipment needed to comply with the specifications of this method is properly cleaned and maintained and shall keep written proof thereof, as well as of the compliance with the test conditions.
They shall be in charge of the reception and storage of the samples upon their arrival at the laboratory as well as of their storage after being tested. When doing so, they shall ensure at all times that the samples remain anonymous and are properly stored, for which purpose they must develop written procedures in order to ensure that the entire process is traceable and affords guarantees.
In addition, they are responsible for preparing, coding and presenting the samples to the tasters according to an appropriate experimental design in line with pre-established protocols, as well as for assembling and statistically processing the data obtained by the tasters.
They shall be in charge of developing and drafting any other procedures that might be necessary to complement this standard and to ensure that the panel functions properly.
They must seek ways of comparing the results of the panel with those obtained by other panels undertaking the analysis of virgin olive oil in order to ascertain whether the panel is working properly.
It is the duty of the panel leader to motivate the panel members by encouraging interest, curiosity and a competitive spirit among them. To do so, they are strongly recommended to ensure a smooth two-way flow of information with the panel members by keeping them informed about all the tasks they carry out and the results obtained. In addition, they shall ensure that their opinion is not known and shall prevent possible leaders from asserting their criteria over the other tasters.
They shall summon the tasters sufficiently in advance and shall answer any queries regarding the performance of the tests, but shall refrain from suggesting any opinion to them on the sample.
7.1.1. Deputy panel leader
The panel leader may, on justified grounds, be replaced by a deputy panel leader who may stand in for duties regarding the performance of the tests. This substitute must have all the necessary skills required of a panel leader.
7.2. Tasters
The people acting as tasters in organoleptic tests carried out on olive oils must do so voluntarily. It is therefore advisable for candidates to submit an application in writing. Candidates shall be selected, trained and monitored by the panel leader in accordance with their skills in distinguishing between similar samples; it should be borne in mind that their accuracy will improve with training.
Tasters must act like real sensory observers, setting aside their personal tastes and solely reporting the sensations they perceive. To do so, they must always work in silence, in a relaxed, unhurried manner, paying the fullest possible sensory attention to the sample they are tasting.
Between 8 and 12 tasters are required for each test, although it is wise to keep some extra tasters in reserve to cover possible absences.
8. TEST CONDITIONS
8.1. Presentation of the sample
The oil sample for analysis shall be presented in standardised tasting glasses conforming to the standard IOC/T.20/Doc. No 5 ‘Glass for oil tasting’.
The glass shall contain 14–16 ml of oil, or between 12,8 and 14,6 g if the samples are to be weighed, and shall be covered with a watch-glass.
Each glass shall be marked with a code made up of digits or a combination of letters and digits chosen at random. The code will be marked by means of an odourfree system.
8.2. Test and sample temperature
The oil samples intended for tasting shall be kept in the glasses at 28 °C ± 2 °C throughout the test. This temperature has been chosen because it makes it easier to observe organoleptic differences than at ambient temperature and because at lower temperatures the aromatic compounds peculiar to these oils volatilise poorly while higher temperatures lead to the formation of volatile compounds peculiar to heated oils. See the standard IOC/T.20/Doc. No 5 ‘Glass for Oil Tasting’ for the method which has to be used for heating the samples when in the glass.
The test room must be at a temperature between 20 ° and 25 °C (see IOC/T.20/Doc. No 6).
8.3. Test times
The morning is the best time for tasting oils. It has been proved that there are optimum perception periods as regards taste and smell during the day. Meals are preceded by a period in which olfactory–gustatory sensitivity increases, whereas afterwards this perception decreases.
However, this criterion should not be taken to the extreme where hunger may distract the tasters, thus decreasing their discriminatory capacity; therefore, it is recommended to hold the tasting sessions between 10.00 in the morning and 12 noon.
8.4. Tasters: general rules of conduct
The following recommendations apply to the conduct of the tasters during their work.
When called by the panel leader to participate in an organoleptic test, tasters should be able to attend at the time set beforehand and shall observe the following:
— They shall not smoke or drink coffee at least 30 minutes before the time set for the test.
— They must not have used any fragrance, cosmetic or soap whose smell could linger until the time of the test. They must use an unperfumed soap to wash their hands which they shall then rinse and dry as often as necessary to eliminate any smell.
— They shall fast at least one hour before the tasting is carried out.
— Should they feel physically unwell, and in particular if their sense of smell or taste is affected, or if they are under any psychological effect that prevents them from concentrating on their work, the tasters shall refrain from tasting and shall inform the panel leader accordingly.
— When they have complied with the above, the tasters shall take up their place in the booth allotted to them in an orderly, quiet manner.
— They shall carefully read the instructions given on the profile sheet and shall not begin to examine the sample until fully prepared for the task they have to perform (relaxed and unhurried). If any doubts should arise, they should consult the panel leader in private.
— They must remain silent while performing their tasks.
— They must keep their mobile phone switched off at all times to avoid interfering with the concentration and work of their colleagues.
9. PROCEDURE FOR THE ORGANOLEPTIC ASSESSMENT AND CLASSIFICATION OF VIRGIN OLIVE OIL
9.1. Tasting technique
9.1.1. The tasters shall pick up the glass, keeping it covered with the watch-glass, and shall bend it gently; they shall then rotate the glass fully in this position so as to wet the inside as much as possible. Once this stage is completed, they shall remove the watch-glass and smell the sample, taking slow deep breaths to evaluate the oil. Smelling should not exceed 30 seconds. If no conclusion has been reached during this time, they shall take a short rest before trying again.
When the olfactory test has been performed, the tasters shall then evaluate the buccal sensations (overall retronasal olfactory, gustatory and tactile sensations). To do so, they shall take a small sip of approximately 3 ml of oil. It is very important to distribute the oil throughout the whole of the mouth cavity, from the front part of the mouth and tongue along the sides to the back part and to the palate support and throat, since it is a known fact that the perception of tastes and tactile sensations varies in intensity depending on the area of the tongue, palate and throat.
It should be stressed that it is essential for a sufficient amount of the oil to be spread very slowly over the back of the tongue towards the palate support and throat while the taster concentrates on the order in which the bitter and pungent stimuli appear. If this is not done, both of these stimuli may escape notice in some oils or else the bitter stimulus may be obscured by the pungent stimulus.
Taking short, successive breaths, drawing in air through the mouth, enables the taster not only to spread the sample extensively over the whole of the mouth but also to perceive the volatile aromatic compounds via the back of the nose by forcing the use of this channel.
NB: When the tasters do not perceive fruitiness in a sample and the intensity of the classifying negative attribute is 3,5 or less the panel leader may decide to arrange for the tasters to analyse the sample again at ambient temperature (COI/T.20/Doc. No 6/Rev. 1, September 2007, section 3 — General specifications for installation of a test room) while specifying the context and concept of ambient temperature. When the sample reaches room temperature, the tasters should re-assess it to check solely whether fruitiness is perceived. If it is, they should mark the intensity on the scale.
The tactile sensation of pungency should be taken into consideration. For this purpose it is advisable to ingest the oil.
9.1.2. When organoleptically assessing a virgin olive oil, it is recommended that FOUR SAMPLES at the most be evaluated in each session with a maximum of three sessions per day, to avoid the contrast effect that could be produced by immediately tasting other samples.
As successive tastings produce fatigue or loss of sensitivity caused by the preceding samples, it is necessary to use a product that can eliminate the remains of the oil from the preceding tasting from the mouth.
The use of a small slice of apple is recommended which, after being chewed, can be disposed of in the spittoon. Then rinse out the mouth with a little water at ambient temperature. At least 15 minutes shall lapse between the end of one session and the start of the next.
9.2. Use of the profile sheet by tasters
The profile sheet intended for use by tasters is detailed in Figure 1 of this Annex.
Each taster on the panel shall smell and then taste ( 5 ) the oil under consideration. They shall then enter the intensity with which they perceive each of the negative and positive attributes on the 10-cm scale shown in the profile sheet provided.
Should the tasters perceive any negative attributes not listed in section 4, they shall record them under the "others" heading, using the term or terms that most accurately describes the attributes.
9.3. Use of the data by the panel leaders
The panel leader shall collect the profile sheets completed by each taster and shall review the intensities assigned to the different attributes. Should they find any anomaly, they shall invite the taster to revise his or her profile sheet and, if necessary, to repeat the test.
The panel leader shall enter the assessment data of each panel member in a computer program like that provided by the standard IOC/T.20/Doc. No 15 with a view to statistically calculating the results of the analysis, based on the calculation of their median. See point 9.4 and the Appendix to this Annex. The data for a given sample shall be entered with the aid of a matrix comprising 9 columns representing the 9 sensory attributes and n lines representing the n panel members used.
When a defect is perceived and entered under the ‘others’ heading by at least 50 % of the panel, the panel leader shall calculate the median of the defect and shall arrive at the corresponding classification.
The value of the robust coefficient of variation which defines classification (defect with the strongest intensity and fruity attribute) must be no greater than 20 %.
If the opposite is the case, the panel leader must repeat the evaluation of the specific sample in another tasting session.
If this situation arises often, the panel leader is recommended to give the tasters specific additional training (IOC/T.20/Doc. No 14, § 5) and to use the repeatability index and deviation index to check taster performance (IOC/T.20/Doc. No 14, § 6).
9.4. Classification of the oil
The oil is graded as follows in line with the median of the defects and the median for the fruity attribute. The median of the defects is defined as the median of the defect perceived with the greatest intensity. The median of the defects and the median of the fruity attribute are expressed to one decimal place.
The oil is graded by comparing the median value of the defects and the median of the fruity attribute with the reference ranges given below. The error of the method has been taken into account when establishing the limits of these ranges, which are therefore considered to be absolute. The software packages allow the grading to be displayed as a table of statistics or a graph.
(a) Extra virgin olive oil: the median of the defects is 0 and the median of the fruity attribute is above 0;
(b) Virgin olive oil: the median of the defects is above 0 but not more than 3,5 and the median of the fruity attribute is above 0;
(c) Lampante virgin olive oil: the median of the defects is above 3,5 or the median of the defects is less than or equal to 3,5 and the fruity median is equal to 0.
Note 1: When the median of the bitter and/or pungent attribute is more than 5,0, the panel leader shall state so on the test certificate.
For assessments intended to monitor compliance, one test shall be carried out. In the case of counter assessments, the analysis must be carried out in duplicate in different tasting sessions. The results of the duplicate analysis must be statistically homogenous. (See point 9.5). If not, the sample must be reanalysed twice again. The final value of the median of the classification attributes will be calculated using the average of both medians.
9.5 Criteria for the acceptance and rejection of duplicates
The normalised error, defined below, shall be used to determine whether the two results of a duplicate analysis are homogenous or statistically acceptable:

Where Me1 and Me2 are the medians of the two duplicates (respectively first and second analysis) and U1 and U2 are the expanded uncertainties obtained for the two values, calculated as follows as specified in Appendix:
U
1 = c × s* and
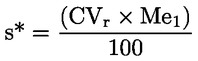
For the expanded uncertainty, c = 1,96; hence:
U1 = 0,0196 × CVr × Me1
where CVr is the robust coefficient of variation.
For it to be stated that the two values obtained are not statistically different, En must be equal to or less than 1,0.
Appendix
Method for calculating the median and the confidence intervals
Median
![]()
The median is defined as the real number Xm characterised by the fact that the probability (p) that the distribution values (X) are below this number (Xm), is less than and equal to 0,5 and that simultaneously the probability (p) that the distribution values (X) are below or equal to Xm is greater than and equal to 0,5. A more practical definition is that the median is the 50th percentile of a distribution of numbers arranged in increasing order. In simpler terms, it is the midpoint of an ordered set of odd numbers, or the mean of two midpoints of an ordered set of even numbers.
Robust standard deviation
In order to arrive at a reliable estimate of the variability around the mean it is necessary to refer to the robust standard deviation as estimated according to Stuart and Kendall (4). The formula gives the asymptotic robust standard deviation, i.e. the robust estimate of the variability of the data considered where N is the number of observations and IQR is the interquartile range which encompasses exactly 50% of the cases of a given probability distribution:

The interquartile range is calculated by calculating the magnitude of the difference between the 75th and 25th percentile.
![]()
Where the percentile is the value Xpc characterised by the fact that the probability (p) that the distribution values are less than Xpc is less than and equal to a specific hundreth and that simultaneously the probability (p) that the distribution values are less than or equal to Xpc is greater than and equal to that specific hundredth. The hundredth indicates the distribution fractile chosen. In the case of the median it is equal to 50/100.

For practical purposes, the percentile is the distribution value corresponding to a specific area subtended from the distribution or density curve. To give an example, the 25th percentile represents the distribution value corresponding to an area equal to 0,25 or 25/100.
In this method percentiles are computed on the basis of the real values which appear in the data matrix (percentiles computing procedure).
Robust coefficient of variation (%)
The CVr% represents a pure number which indicates the percentage variability of the set of numbers analysed. For this reason it is very useful for checking the reliability of the panel assessors.
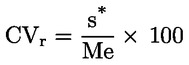
Confidence intervals of the median at 95%
The confidence intervals at 95% (value of the error of the first kind equal to 0,05 or 5%) represent the interval within which the value of the median could vary if it were possible to repeat an experiment an infinite number of times. In practice, it indicates the interval of variability of the test in the operating conditions adopted starting from the assumption that it is possible to repeat it many times. As with the CVr%, the interval helps to assess the reliability of the test.

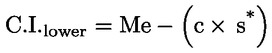
where C = 1,96 for the confidence interval at the 95% level.
An example of the calculation sheet is presented in Annex I to the standard IOC/T 20/Doc. No 15.
References
(1) Wilkinson, L. 1990. Systat: The system for statistics. Evanston, IL.SYSTAT Inc.
(2) Cicchitelli, G. 1984. Probabilità e Statistica. Maggioli Editore, Rimini.
(3) Massart, D.L.; Vandeginste, B.G.M.; Deming, Y.; Michotte, L. 1988. Chemometrics. A textbook. Elsevier. Amsterdam.
(4) Kendall, M.G.; Stuart, A. 1967. The advanced theory of statistics. Vol. 1. Hafner Publishing Co.
(5) McGill, R.; Tukey, J.W.; Larsen, W.A. 1978. Variation of Box Plots. The American Statistician, 32, (2), 12-16.
(6) IOC/T.28/Doc. No 1 September 2007, Guidelines for the accreditation of sensory testing laboratories with particular reference to virgin olive oil according to standard ISO/IEC 17025:2005.
(7) IOC/T.20/Doc. No 14.
(8) IOC/T.20/Doc. No 15.
(9) ISO/IEC 17025:05.
▼M20 —————
▼M19 —————
ANNEX XV
1. OIL CONTENT OF OLIVE RESIDUE
1.1. Apparatus
— suitable extraction apparatus fitted with a 200 to 250 ml round-bottomed flask,
— electrically heated bath (e.g., sand bath, water bath) or hotplate,
— analytical balance,
— oven regulated to a maximum of 80° C,
— electrically heated oven fitted with a thermostatic device regulated to 103 ± 2° C and one that can be swept with a stream of air or operated at reduced pressure,
— mechanical mill, easy to clean, and one that allows the olive residues to be ground without a rise in their temperature or any appreciable alteration in their content of moisture, volatile matter or substances extractable with hexane,
— extraction thimble and cotton wool or filter paper from which substances extractable with hexane have already been removed,
— dessicator,
— sieve with 1 mm diameter apertures,
— small particles of previously dried pumice stone.
1.2. Reagent
Normal hexane, technical grade, which must leave a residue of less than 0,002 g per 100 ml, on complete evaporation.
2. PROCEDURE
2.1. Preparation of the test sample
If necessary, use the mechanical mill, which has previously been properly cleaned, to grind the laboratory sample in order to reduce it to particles that can pass completely through the sieve.
Use about one twentieth of the sample to complete the process of cleaning the mill, discard the ground material, grind the remainder and collect, mix carefully and analyze without delay.
2.2. Test portion
As soon as the grinding operation has been completed, weigh out about 10 g of the sample to the nearest 0,01 g for testing.
2.3. Preparation of the extraction thimble
Place the test portion in the thimble and plug with cotton wool. If a filter paper is used, envelope the test portion in it.
2.4. Peliminary drying
If the olive residues are very moist (i.e., moisture and volatile matter content more than 10 %), carry out preliminary drying by placing the loaded thimble (or filter paper) in the oven heated for an appropriate time at not more than 80° C in order to reduce the moisture and volatile matter content to less than 10 %.
2.5. Preparation of the round-bottomed flask
Weigh to the nearest 1 mg the flask containing one or two particles of pumice stone, previously dried in the stove at 103 ± 2° C and then cooled in a dessicator for not less than one hour.
2.6. Initial extraction
Into the extraction apparatus insert the thimble (or filter paper) containing the test portion. Pour into the flask the requisite quantity of hexane. Fit the flask to the extraction apparatus and place the whole on the electrically heated bath. Adjust the rate of heating in such a way that the reflux rate is not less than three drops per second (moderate, not violent boiling). After four hours extraction, allow to cool. Remove the thimble from the extraction apparatus and place it in a stream of air in order to drive off most of the impregnating solvent.
2.7. Second extraction
Tip the contents of the thimble into the micro-grinder and grind as finely as possible. Return the ground mixture to the thimble without loss and place it back in the extraction apparatus.
Continue the extraction for a further two hours using the same round-bottomed flask containing the initial extract.
The resultant solution in the extraction flask must be clear. If not, filter it through a filter paper and wash the original flask and the filter paper several times with hexane. Collect the filtrate and the washing solvent in a second round-bottomed flask which has been dried and tared to the nearest 1 mg.
2.8. Removal of solvent and weighing of extract
Remove the greater part of the solvent by distillation on an electrically heated bath. Remove the last traces of solvent by heating the flask in the oven at 103 ± 2° C for 20 minutes. Assist the elimination process either by blowing in air, or preferably an inert gas, at intervals or by using reduced pressure.
Leave the flask in a dessicator to cool for at least one hour and weigh to the nearest 1 mg.
Heat again for 10 minutes under the same conditions, cool in a dessicator and reweigh.
The difference between the two weighings shall not exceed 10 mg. If it does, heat again for periods of 10 minutes followed by cooling and weighing until the weight difference is 10 mg or less. Note the last weight of the flask.
Carry out duplicate determinations on the test sample.
3. EXPRESSION OF RESULTS
3.1. Method of calculation and formula
(a) The extract expressed as a percentage by mass of the product as received is equal to:
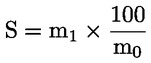
|
where: |
S = is the percentage by mass of extract of the product as received, m0 = is the mass, in grams, of the test portion, m1 = is the mass, in grams, of the extract after drying. |
Take as the result the arithmetic mean of the duplicate determinations, providing the repeatability conditions are satisfied.
Express the result to the first decimal place.
(b) The extract is expressed on a dry matter basis by using the formula:

where:
S = is the percentage of extract by means of the product as received (see (a)),
U = is its moisture and volatile matter content.
3.2. Repeatability
The difference between the duplicate determinations carried out simultaneously or in rapid sucession by the same analyst shall not exceed 0,2 g of hexane extract per 100 g of sample.
If this condition is not satisfied, repeat the analysis on two other test portions. If, in this case too, the difference exceeds 0,2 g, take as the result the arithmetic mean of the four determinations.
ANNEX XVI
DETERMINATION OF IODINE VALUE
1. SCOPE
This International Standard specifies a method for the determination of the iodine value of animal and vegetable fats and oils, referred to hereafter as fats.
2. DEFINITION
For the purposes of this International Standard, the following definition applies:
|
2.1. |
iodine value. The mass of iodine absorbed by the sample under the operating conditions specified in this International Standard. The iodine value is expressed as grams of iodine per 100 g of sample. |
3. PRINCIPLE
Dissolution of a test portion in solvent and addition of Wijs reagent. After a specified time, addition of potassium iodide solution and water, and titration of the liberated iodine with sodium thiosulfate solution.
4. REAGENTS
All reagents shall be of recognized analytical grade:
|
4.1. |
water, complying with the requirements of ISO 3696, Grade 3. |
|
4.2. |
potassium iodide, 100 g/l solution, not containing iodate or free iodine. |
|
4.3. |
starch, solution. Mix 5 g of soluble starch in 30 ml of water, add this mixture to 1 000 ml of boiling water, boil for three minutes and allow to cool. |
|
4.4. |
sodium thiosulfate, standard volumetric solution c (Na2S2O3.5H2O) = 0,1 mol/l, standardized not more than seven days before use. |
|
4.5. |
solvent, prepared by mixing equal volumes of cyclohexane and acetic acid. |
|
4.6. |
Wijs reagent, containing iodine monochloride in acetic acid. Commercially available Wijs reagent shall be used. |
5. APPARATUS
Usual laboratory apparatus and, in particular, the following:
|
5.1. |
glass weighing scoops, suitable for the test portion and for inserting into the flasks (6.2). |
|
5.2. |
conical flasks, of 500 ml capacity, fitted with ground glass stoppers and completely dry. |
6. PREPARATION OF THE TEST SAMPLE
The homogenized sample is dried over sodium sulphate and filtered.
7. PROCEDURE
|
7.1. |
Test portion The mass of the test portion varies according to its expected iodine value as shown in Table 1.
Table 1
Weigh the test portion to the nearest 0,1 mg in a glass weighing scoop (5.1). |
|
7.2. |
Determination Place the test portion in a 500 ml flask (6.2). Add 20 ml of the solvent (4.5) to dissolve the fat. Add exactly 25 ml of the Wijs reagent (4.6), insert the stopper, swirl the contents and place the flask in the dark. Do not use a mouth pipette for the Wijs reagent. Similarily, prepare a blank with the solvent and the reagent but omitting the test portion. For samples having an iodine valve below 150, leave the flasks in the dark for one hour; for those with an iodine value above 150 and for polymerized products or products oxidized to a considerable extent, leave for two hours. At the end of the time, add 20 ml of the potassium iodide solution (4.2) and 150 ml of water (4.1) to each of the flasks. Titrate with the standard volumetric sodium thiosulfate solution (4.4) until the yellow colour due to iodine has almost disappeared. Add a few drops of the starch solution (4.3) and continue the titration until the blue colour just disappears after very vigorous shaking. Note: Potentiometric determination of the end point is permissible. |
|
7.3. |
Number of determinations Carry out two determinations on the same test sample. |
8. EXPRESSION OF RESULTS
The iodine value is given by the expression

where:
c = is the numerical value of the exact concentration, in moles per litre, of the standard volumetric sodium thiosulfate solution (4.4) used;
V1 = is the numerical value of the volume, in millilitres, of the standard volumetric sodium thiosulfate solution (4.4) used for the blank test;
V2 = is the numerical value of the volume, in millilitres, of the standard volumetric sodium thiosulfate solution (4.4) used for the determination;
m = is the numerical value of the mass, in grams, of the test portion (7.1).
Take as the result the arithmetic mean of the two determinations, provided that the requirement for repeatability (9.2) is satisfied.
ANNEX XVII
METHOD FOR THE DETERMINATION OF STIGMASTADIENES IN VEGETABLE OILS
1. PURPOSE
Determination of stigmastadienes in vegetable oils containing low concentrations of these hydrocarbons, particularly in virgin olive oil and crude olive-residue oil.
2. SCOPE
The standard may be applied to all vegetable oils although measurements are reliable only where the content of these hydrocarbons lies between 0,01 and 4,0 mg/kg. The method is particularly suited to detecting the presence of refined vegetable oils (olive, olive residue, sunflower, palm, etc.) in virgin olive oil since refined oils contained stigmastadienes and virgin oils do not.
3. PRINCIPLE
Isolation of unsaponifiable matter. Separation of steroidal hydrocarbon fraction by column chromatography on silica gel and analysis by capillary gas chromatography.
4. APPARATUS
|
4.1. |
250 ml flasks suitable for use with a reflux condenser. |
|
4.2. |
Separating funnels of 500 ml capacity. |
|
4.3. |
100 ml round-bottom flasks. |
|
4.4. |
Rotary evaporator. |
|
4.5. |
Glass chromatography column (1,5 to 2,0 cm internal diameter by 50 cm length) with Teflon tap and a plug of glass wool fibre or sintered glass disc at the bottom. To prepare silica gel column, pour hexane into the chromatography column to a depth of approximately 5 cm and then fill with a slurry of silica gel in hexane (15 g in 40 ml) with the help of hexane portions. Allow to settle and finish settling by applying slight vibration. Add anhydrous sodium sulphate to a height of approximately 0,5 cm, finally elute the excess hexane. |
|
4.6. |
Gas chromatograph with flame ionization detector, split or cold on-column injector and oven programmable to within ± 1 °C. |
|
4.7. |
Fused silica capillary column for gas chromatography (0,25 or 0,32 mm internal diameter by 25 m length) coated with 5 %-phenylmethylsilicone phase, 0,25 mm film thickness. Note 1: Other columns of similar or lower polarity can be used. |
|
4.8. |
Integrator-recorder with possibility of valley-valley integration mode. |
|
4.9. |
5 to 10 ml microsyringe for gas chromatography with cemented needle. |
|
4.10. |
Electrical heating mantle or hot place. |
5. REAGENTS
All reagents should be of analytical grade unless otherwise specified. The water used should be distilled water, or water of at least equivalent purity.
|
5.1. |
Hexane or mixture of alkanes of bp interval 65 to 70 °C, distilled with rectifying column. Note 2: The solvent must be distilled to remove impurities. |
|
5.2. |
96 v/v ethanol. |
|
5.3. |
Anhydrous sodium sulphate. |
|
5.4. |
Alcoholic potassium hydroxide solution at 10 %. Add 10 ml of water to 50 g potassium hydroxide, stir, and then dissolve the mixture in ethanol to 500 ml. Note 3: Alcoholic potash turns brown on standing. It should be prepared freshly each day and kept in well stoppered dark glass bottles. |
|
5.5. |
Silica gel 60 for column chromatography, 70 to 230 mesh, (Merck, reference 7734 or similar). Note 4: Usually, silica gel can be used directly from the container without any treatment. However, some batches of silica gel may show low activity resulting in bad chromatographic separations. Under this circumstance, the silica gel should be treated in the following way: Activate the silica gel by heating for a minimum of four hours at 550 °C. After heating, place the silica gel in a desiccator while the gel is cooling and then transfer the silica gel to a stoppered flask. Add 2 % of water and shake until no lumps can be seen and the powder flows freely. If batches of silica gel result in chromatograms with interfering peaks, the silica gel should be treated as above. An alternative could be the use of extra pure silica gel 60 (Merck, reference 7754). |
|
5.6. |
Stock solution (200 ppm) of cholesta-3,5-diene (Sigma, 99 % purity) in hexane (10 mg in 50 ml). |
|
5.7. |
Standard solution of cholesta-3,5-diene hexane at concentration of 20 ppm, obtained by dilution of above solution. Note 5: The solutions 5.6 and 5.7 are stable for a period of at least four months if kept at less than 4 °C. |
|
5.8. |
Solution of n-nonacosane in hexane at concentration of approximately 100 ppm. |
|
5.9. |
Carrier gas for chromatography: helium or hydrogen of 99,9990 % purity. |
|
5.10. |
Auxiliary gases for flame ionization detector: hydrogen of 99,9990 % purity and purified air. |
6. PROCEDURE
6.1. Preparation of unsaponifiable matter
|
6.1.1. |
Weigh 20 ± 0,1 g of oil into a 250-ml flask (4.1), add 1 ml of the standard solution of cholesta-3,5-diene (20μg) and 75 ml of alcoholic potash at 10 %, fit reflux condenser, and heat to slight boiling for 30 minutes, Remove the flask containing the sample from the heat and allow the solution to cool slightly (do not allow to cool completely as the sample will set). Add 100 ml of water and transfer the solution to a separating funnel (4.2) with the aid of 100 ml of hexane. Shake the mixture vigorously for 30 seconds and allow the separate. Note 6: If an emulsion is produced which does not rapidly disappear, add small quantities of ethanol. |
|
6.1.2. |
Transfer the aqueous phase beneath to a second separating funnel and extract again with 100 ml of hexane. Once more run off the lower phase and wash the hexane extracts (combined in another separating funnel) three times with 100 ml each time of a mixture of ethanol-water (1: 1) until neutral pH is reached. |
|
6.1.3. |
Pass the hexane solution through anhydrous sodium sulphate (50 g), wash with 20 ml hexane and evaporate in a rotary evaporator at 30 °C under reduced pressure until dryness. |
6.2. Separation of steroidal hydrocarbon fraction
|
6.2.1. |
Take the residue to the fractioning column with the aid of two 1-ml portions of hexane, run the sample onto the column by allowing the solution level to drop to the top of the sodium sulphate and start the chromatographic elution with hexane at a flow rate of 1 ml/min approximately. Discard the first 25 to 30 ml of eluate and then collect the following 40 ml fraction. After collection, transfer this fraction to a 100-ml round bottomed flask (4.3). Note 7: The first fraction contains saturated hydrocarbons (Figure 1 a) and the second fraction the steroidal ones. Further elution provides squalene and related compounds. To achieve a good separation between saturated and steroidal hydrocarbons, the optimization of fraction volumes is required. For this, the volume of the first fraction should be adjusted so that when the second fraction is analysed the peaks representing the saturated hydrocarbons are low (see Figure 1 c); if they do not appear but the intensity of the standard peak is low, the volume should be reduced. Anyway, a complete separation between the components of the first and second fractions is unnecessary; as there is no overlapping of peaks during GC analysis if GC conditions are ajusted as indicated in 6.3.1. The optimization of the volume of the second fraction if generally not needed as a good separation exists with the further components. Nevertheless, the presence of a large peak at approximately 1,5 minutes lower retention time than the standard is due to squalene, and it is indicative of a bad separation. |
|
6.2.2. |
Evaporate the second fraction in a rotary evaporator at 30 °C under reduced pressure until dryness, and immediately dissolve the residue in 0,2 ml of hexane. Keep the solution in the refrigerator until analysis. Note 8: Residues 6.1.3 and 6.2.2 should not be kept dry and at room temperature. As soon as they are obtained, the solvent should be added and the solutions should be kept in the refrigerator. |
6.3. Gas chromatography
|
6.3.1. |
Working conditions for split injection: — injector temperature: 300 °C, — detector temperature: 320 °C, — integrator-recorder: the parameters for integration should be fixed so as to give a correct assessment of the areas. Valley-valley integration mode is recommended, — sensitivity: about 16 times the minimum attenuation, — amount of solution injected: 1μl, — oven programming temperatures: initial 235 °C for six minutes and then rising at 2 °C/minute up to 285 °C, — injector with 1: 15 flow divider, — carrier: helium or hydrogen at about 120 kPa pressure. These conditions may be adjusted in accordance with the characteristics of the chromatograph and the column to give chromatograms meeting the following requirements: internal standard peak within approximately five minutes of the time given in 6.3.2; the internal standard peak should be at least 80 % of the full scale. The gas chromatographic system must be checked injecting a mixture of the stock solution of cholestadiene (5.6) and n-nonacosane solution (5.8). The cholesta-3,5-diene peak must appear before the n-nonacosane (Figure 1c); if it does not occur two actions can be undertaken: reduce the oven temperature and/or use a less polar column. |
|
6.3.2. |
Peak identification The internal standard peak appears at approximately 19 minutes and the 3,5-stigmastadiene at a relative retention time of approximately 1,29 (see Figure 1b). The 3,5-stigmastadiene occurs with small quantities of an isomer, and usually, both elute together as a single chromatographic peak. Nevertheless, if the column is too polar or shows a high resolving power, the isomer can appear as a small peak before and close to that of stigmasta-3,5-diene (Figure 2). In order to ensure that the stigmastadienes are eluted as one peak, it is advisable to replace the column by one which is either less polar or has a wider internal diameter. Note 9: Stigmastadienes for reference can be obtained from the analysis of a refined vegetable oil by using less amount of sample (1 to 2 g). Stigmastadienes originate a prominent and easily identifiable peak. |
|
6.3.3. |
Quantitative analysis The stigmastadienes content is determined according to the formula: mg/kg of stigmastadienes =
Detection limit: about 0,01 mg/kg. |

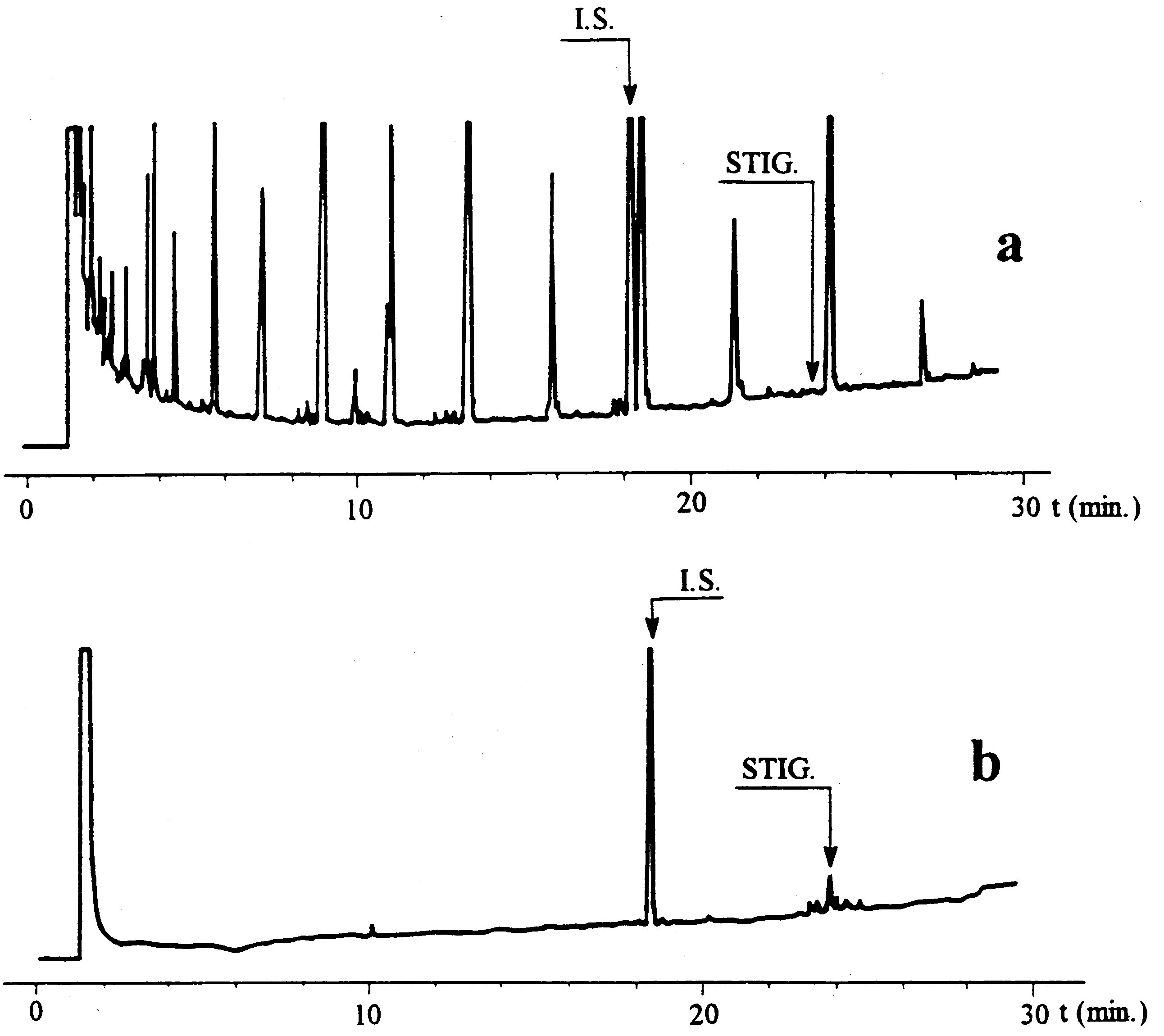

Figure 1
Gas chromatograms obtained from olive oil samples analysed on a fused silica capillary column (0,25 mm internal diameter by 25 m) coated with 5 %-phenylmethylsilicone, 0,25 μm film thickness.
(a) First fraction (30 ml) from a virgin oil, spiked with standard.
(b) Second fraction (40 ml) from an olive oil containing 0,10 mg/kg of stigmastadienes.
(c) Second fraction (40 ml) containing a small proportion of the first fraction.
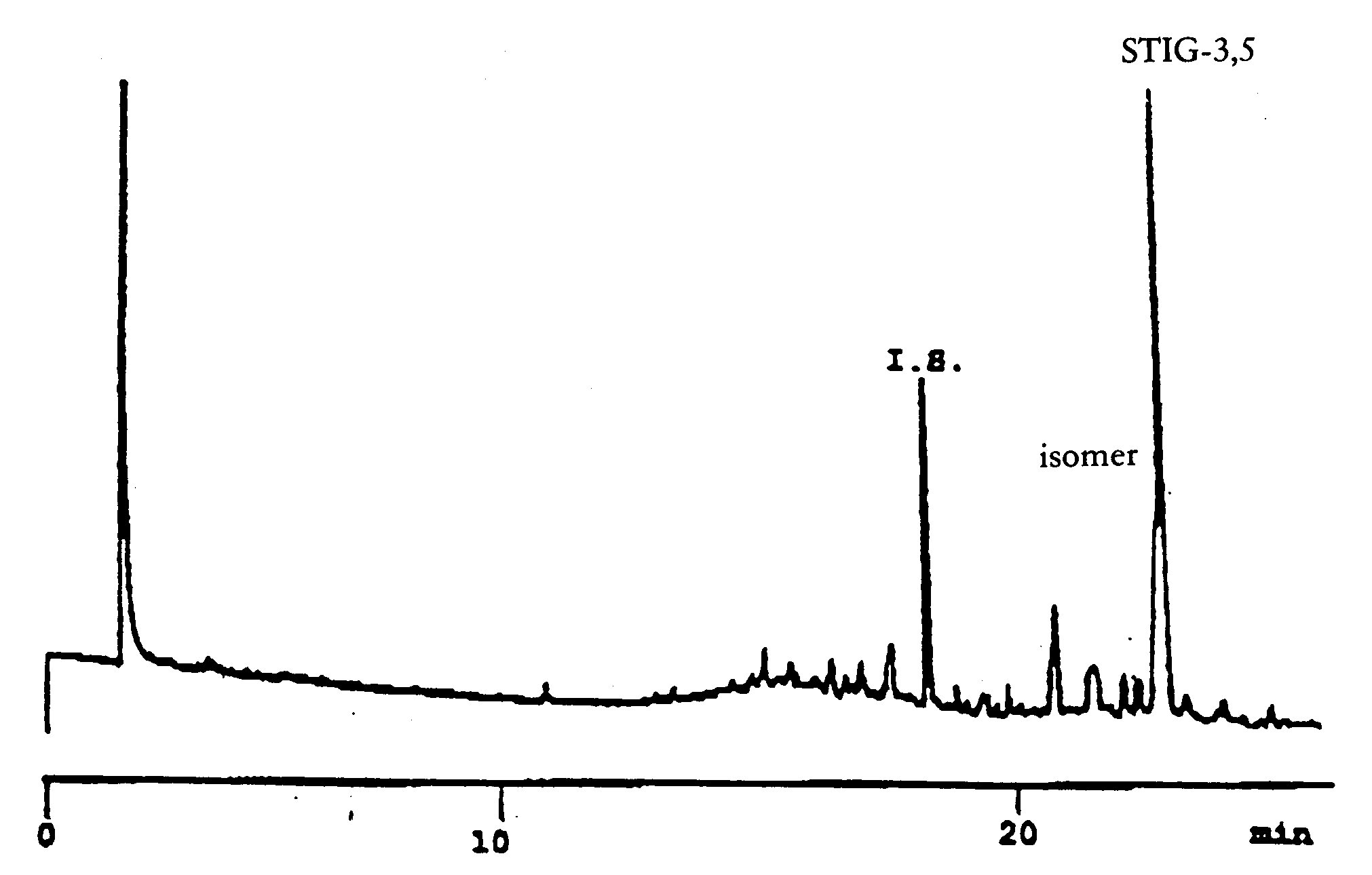
Figure 2
Gas chromatogram obtained from a refined olive oil sample analysed on DB-5 column showing the isomer of 3,5-stigmastadiene.
ANNEX XVIII
DETERMINATION OF THE DIFFERENCE BETWEEN ACTUAL AND THEORETICAL CONTENT OF TRIACYLGLYCEROLS WITH ECN 42
1. SCOPE
Determination of the absolute difference between the experimental values of triacylglycerols (TAGs) with equivalent carbon number 42 (ECN42HPLC) obtained by determination in the oil by high performance liquid chromatography and the theoretical value of TAGs with an equivalent carbon number of 42 (ECN 42theoretical) calculated from the fatty acid composition.
2. FIELD OF APPLICATION
The standard is applicable to olive oils. The method is applicable to the detection of the presence of small amounts of seed oils (rich in linoleic acid) in every class of olive oils.
3. PRINCIPLE
The content of triacylglycerols with ECN 42 determined by HPLC analysis and the theoretical content of triacylglycerols with ECN 42 (calculated on the basis of GLC determination of fatty acid composition) correspond within a certain limit for genuine olive oils. A difference larger than the values adopted for each type of oil points out that the oil contains seed oils.
4. METHOD
The method for the calculation of the theoretical content of triacylglycerols with ECN 42 and of the difference with respect to the HPLC data is essentially made by the coordination of analytical data obtained by means of other methods. It is possible to distinguish three phases: determination of fatty acid composition by capillary gas chromatography, calculation of theoretical composition of triacylglycerols with ECN 42, HPLC determination of ECN 42 triacylglycerols.
4.1. Apparatus
|
4.1.1. |
Round-bottomed flasks, 250 and 500 ml. |
|
4.1.2. |
Beakers 100 ml. |
|
4.1.3. |
Glass chromatographic column, 21 mm internal diameter, 450 mm length, with cock and normalised cone (female) at the top. |
|
4.1.4. |
Separating funnels, 250 ml, with normalised cone (male) at the bottom, suitable for connection to the top of the column. |
|
4.1.5. |
Glass rod, 600 mm length. |
|
4.1.6. |
Glass funnel, 80 mm diameter. |
|
4.1.7. |
Volumetric flasks, 50 ml. |
|
4.1.8. |
Volumetric flasks, 20 ml. |
|
4.1.9. |
Rotary evaporator. |
|
4.1.10. |
High performance liquid chromatograph, allowing thermostatic control of column temperature. |
|
4.1.11. |
Injection units for 10 μl delivery. |
|
4.1.12. |
Detector: differential refractometer. The full scale sensitivity should be at least 10–4 units of refractive index. |
|
4.1.13. |
Column: stainless steel tube 250 mm length x 4,5 mm internal diameter packed with 5 μm diameter particles of silica with 22 to 23 % carbon in the form of octadecylsilane. |
|
4.1.14. |
Data processing software. |
|
4.1.15. |
Vials, of about 2 ml volumes, with Teflon-layered septa and screw caps. |
4.2. Reagents
The reagents should be of analytical purity. Elution solvents should be de-gassed, and may be recycled several times without effect on the separations.
|
4.2.1. |
Petroleum ether 40– 60 °C chromatographic grade or hexane. |
|
4.2.2. |
Ethyl ether, peroxide-free, freshly distilled. |
|
4.2.3. |
Elution solvent for purifying the oil by column chromatography mixture petroleum ether/ethyl ether 87/13 (v/v). |
|
4.2.4. |
Silica gel, 70-230 mesh, type Merck 7734, with water content standardised at 5 % (w/w/). |
|
4.2.5. |
Glass wool. |
|
4.2.6. |
Acetone for HPLC. |
|
4.2.7. |
Acetonitrile or propionitrile for HPLC. |
|
4.2.8. |
HPLC elution solvent: acetonitrile + acetone (proportions to be adjusted to obtain the desired separation; begin with 50:50 mixture) or propionitrile. |
|
4.2.9. |
Solubilisation solvent: acetone. |
|
4.2.10. |
Reference triglycerides: commercial triglycerides (tripalmitin, triolein, etc.) may be used and the retention times then plotted in accordance with the equivalent carbon number, or alternatively reference chromatograms obtained from soya oil, mixture 30:70 soya oil — olive oil and pure olive oil (see notes 1 and 2 and figures 1 to 4). |
|
4.2.11. |
Solid phase extraction column with silica phase 1 g, 6 ml. |
4.3. Sample preparation
As a number of interfering substances can give rise to false positive results, the sample must always be purified according to IUPAC method 2.507, used for the determination of polar compounds in frying fats.
4.3.1. Chromatographic column preparation
Fill the column (4.1.3) with about 30 ml of elution solvent (4.2.3), then introduce inside the column some glass wool (4.2.5) pushing it to the bottom of the column by means of the glass rod (4.1.5).
In a 100 ml beaker, suspend 25 g of silica gel (4.2.4) in 80 ml of elution mixture (4.2.3), then transfer it to the column by means of a glass funnel (4.1.6).
To ensure the complete transfer of the silica gel to the column, wash the beaker with the elution mixture and transfer the washing portions to the column too.
Open the cock and let the solvent elute from the column until its level is about 1 cm over the silica gel.
4.3.2. Column chromatography
Weigh with the accuracy of 0,001 g, 2,5 ± 0,1 g of oil, previously filtered, homogenised and anhydrified, if necessary, in a 50 ml volumetric flask (4.1.7).
Dissolve it in about 20 ml of elution solvent (4.2.3). If necessary, slightly heat it to make the dissolution easily. Cool at room temperature and adjust the volume with elution solvent.
By means of a volumetric pipette, introduce 20 ml of solution inside the column prepared according to 4.3.1, open the cock and let the solvent elute to the silica gel layer level.
Then elute with 150 ml of elution solvent (4.2.3), adjusting the solvent rate at about 2 ml/min (150 ml will take about 60-70 minutes to pass through the column).
The eluate is recovered in a 250 ml round-bottomed flask (4.1.1) previously tared in an oven and exactly weighed. Eliminate the solvent at reduced pressure in a rotary evaporator (4.1.9) and weigh the residue that will be used to prepare the solution for HPLC analysis and for methyl ester preparation.
The sample recovery from the column must be 90 % at least for the extra virgin, virgin, ordinary, refined and olive oil categories, and a minimum of 80 % for lampante and olive-pomace oils.
4.3.3. SPE purification
Silica SPE column is activated by passing 6 ml of hexane (4.2.3) under vacuum, avoiding dryness.
Weigh to an accuracy of 0,001 g, 0,12 g in a 2 ml vial (4.1.15) and dissolve with 0,5 ml of hexane (4.2.3).
Load the SPE column with the solution and elute with 10 ml of hexane-diethyl ether (87:13 v/v) (4.2.3) under vacuum.
The collected fraction is evaporated to dryness in a rotary evaporator (4.1.9) under reduced pressure at room temperature. The residue is dissolved in 2 ml of acetone (4.2.6) for triacylglycerol (TAG) analysis.
4.4. HPLC analysis
4.4.1. Preparation of the samples for chromatographic analysis
A 5 % solution of the sample to be analysed is prepared by weighing 0,5 ± 0,001 g of the sample into a 10 ml graduated flask and making up to 10 ml with the solubilisation solvent (4.2.9).
4.4.2. Procedure
Set up the chromatographic system. Pump elution solvent (4.2.8) at a rate of 1,5 ml/min to purge the entire system. Wait until a stable base line is obtained.
Inject 10 μl of the sample prepared as in point 4.3.
4.4.3. Calculation and expression of results
Use the area normalisation method, i.e. assume that the sum of the areas of the peaks corresponding to TAGs from ECN 42 up to ECN 52 is equal to 100 %.
Calculate the relative percentage of each triglyceride using the formula:
![]()
.
The results should be given to at least two decimal places.
See notes 1 to 4.
4.5. Calculation of triacylglycerols composition (moles %) from fatty acid composition data (area %)
4.5.1. Determination of fatty acid composition
Fatty acid composition is determined by ISO 5508 by means of a capillary column. The methyl esters are prepared according to COI/T.20/Doc. No 24.
4.5.2. Fatty acids for calculation
Glycerides are grouped by their Equivalent Carbon Number (ECN), taking into account the following equivalencies between ECN and fatty acids. Only fatty acids with 16 and 18 carbon atoms were taken into consideration, because only these are important for olive oil. The fatty acids should be normalised to 100 %.
|
Fatty acid (FA) |
Abbreviation |
Molecular weight (MW) |
ECN |
|
Palmitic acid |
P |
256,4 |
16 |
|
Palmitoleic acid |
Po |
254,4 |
14 |
|
Stearic acid |
S |
284,5 |
18 |
|
Oleic acid |
O |
282,5 |
16 |
|
Linoleic acid |
L |
280,4 |
14 |
|
Linolenic acid |
Ln |
278,4 |
12 |
4.5.3. Conversion of area % into moles for all fatty acids (1)
|
|
|
|
|
|
|
|


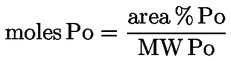
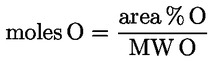


4.5.4. Normalisation of fatty acid moles to 100 % (2)

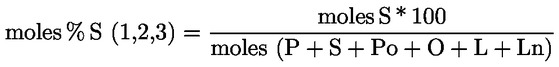

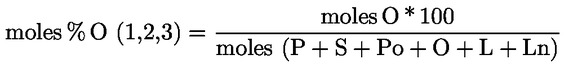

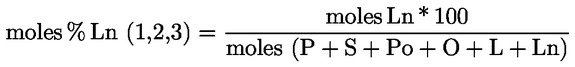
The result gives the percentage of each fatty acid in moles % in the overall (1, 2, 3–) position of the TAGs.
Then the sum of the saturated fatty acids P and S (SFA) and the unsaturated fatty acids Po, O, L and Ln (UFA) are calculated (3):
![]()
![]()
4.5.5. Calculation of the fatty acid composition in 2- and 1, 3- positions of TAGs
The fatty acids are distributed to three pools as follows: one for 2- position and two identical for 1- and 3- positions, with different coefficients for the saturated (P and S) and unsaturated acids (Po, O, L and Ln).
4.5.5.1. Saturated fatty acids in 2-position [P(2) and S(2)] (4):
![]()
![]()
4.5.5.2. Unsaturated fatty acids in 2-position [Po(2), O(2), L(2) and Ln(2)] (5):




4.5.5.3. Fatty acids in 1,3-positions [P(1,3), S(1,3), Po(1,3), O(1,3), L(1,3) and Ln(1,3)] (6):


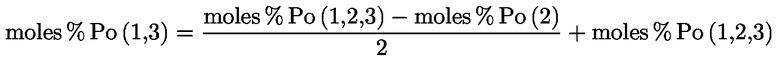



4.5.6. Calculation of triacylglycerols
4.5.6.1. TAGs with one fatty acid (AAA, here LLL, PoPoPo) (7)
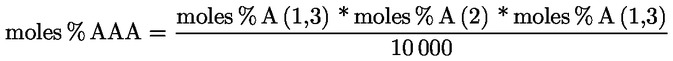
4.5.6.2. TAGs with two fatty acids (AAB, here PoPoL, PoLL) (8)


4.5.6.3. TAGs with three different fatty acids (ABC, here OLLn, PLLn, PoOLn, PPoLn) (9)
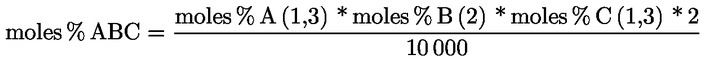

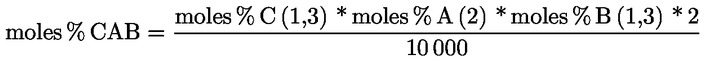
4.5.6.4. Triacylglycerols with ECN42
The triacylglycerols with ECN42 are calculated according to equations 7, 8 and 9 and are then given in order of expected elution in HPLC (normally only three peaks).
LLL
PoLL and the positional isomer LPoL
OLLn and the positional isomers OLnL and LnOL
PoPoL and the positional isomer PoLPo
PoOLn and the positional isomers OPoLn and OLnPo
PLLn and the positional isomers LLnP and LnPL
PoPoPo
SLnLn and the positional isomer LnSLn
PPoLn and the positional isomers PLnPo and PoPLn
The triacylglycerols with ECN42 are given by the sum of the nine triacylglycerols including their positional isomers. The results should be given to at least two decimal places.
5. EVALUATION OF THE RESULTS
The calculated theoretical content and the content determined by the HPLC analysis are compared. If the difference in the absolute value of the HPLC data minus the theoretical data is greater than the values stated for the appropriate oil category in the standard, the sample contains seed oil.
Results are given to two decimal figures.
6. EXAMPLE (THE NUMBERS REFER TO THE SECTIONS IN THE TEXT OF THE METHOD)
— 4.5.1. Calculation of moles % fatty acids from GLC data (normalised area %)
The following data are obtained for the fatty acid composition by GLC:
|
FA |
P |
S |
Po |
O |
L |
Ln |
|
MW |
256,4 |
284,5 |
254,4 |
282,5 |
280,4 |
278,4 |
|
Area % |
10,0 |
3,0 |
1,0 |
75,0 |
10,0 |
1,0 |
— 4.5.3 Conversion of area % into moles for all fatty acids (see formula (1))
|
moles P |
= |
|

|
moles S |
= |
|
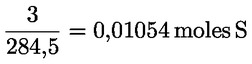
|
moles Po |
= |
|
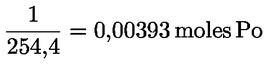
|
moles O |
= |
|

|
moles L |
= |
|
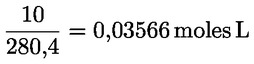
|
moles Ln |
= |
|

|
Total |
= |
0,35821 moles TAGs |
— 4.5.4 Normalisation of fatty acid moles to 100 % (see formula (2))
|
moles % P(1,2,3) |
= |
|
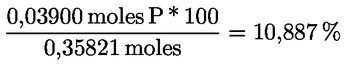
|
moles % S(1,2,3) |
= |
|

|
moles % Po(1,2,3) |
= |
|

|
moles % O(1,2,3) |
= |
|

|
moles % L(1,2,3) |
= |
|

|
moles % Ln(1,2,3) |
= |
|
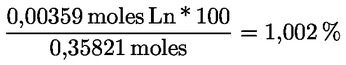
|
Total moles % |
= |
100 % |
Sum of the saturated and unsaturated fatty acids in the 1,2,3-position of TAGs (see formula (3)):
![]()
![]()
— 4.5.5 Calculation of the fatty acid composition in 2- and 1,3-positions of the TAGs
— 4.5.5.1 Saturated fatty acids in 2-position [P(2) and S(2)] (see formula (4))
![]()
![]()
— 4.5.5.2 Unsaturated fatty acids in 2-position [Po(1,3), O(1,3), L(1,3) and Ln(1,3)] (see formula (5))

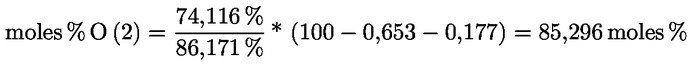

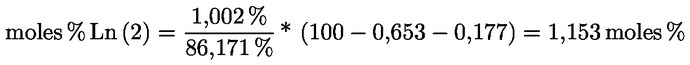
— 4.5.5.3 Fatty acids in 1,3-positions [P(1,3), S(1,3), Po(1,3), O(1,3), L(1,3) and Ln(1,3)] (see formula (6))
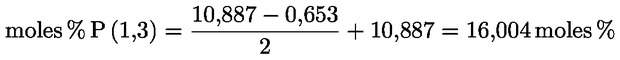
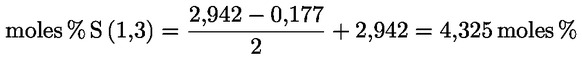




— 4.5.6. Calculation of triacylglycerols
From the calculated fatty acid composition in sn-2- and sn-1,3-positions:
|
FA in |
1,3-pos |
2-pos |
|
P |
16,004 % |
0,653 % |
|
S |
4,325 % |
0,177 % |
|
Po |
1,015 % |
1,262 % |
|
O |
68,526 % |
85,296 % |
|
L |
9,204 % |
11,457 % |
|
Ln |
0,927 % |
1,153 % |
|
Sum |
100,0 % |
100,0 % |
the following triacylglycerols are calculated:
LLL
PoPoPo
PoLL with 1 positional isomer
SLnLn with 1 positional isomer
PoPoL with 1 positional isomer
PPoLn with 2 positional isomers
OLLn with 2 positional isomers
PLLn with 2 positional isomers
PoOLn with 2 positional isomers
— 4.5.6.1. TAGs with one fatty acid (LLL, PoPoPo) (see formula (7))
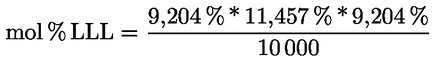 , = 0,09706 mol LLL
, = 0,09706 mol LLL
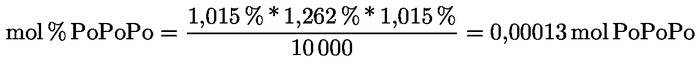
— 4.5.6.2 TAGs with two fatty acids (PoLL, SLnLn, PoPoL) (see formula (8))
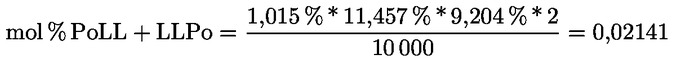
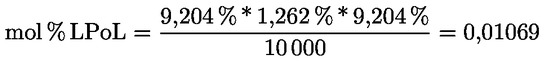
0,03210 mol PoLL


0,00094 mol SLnLn


0,00354 mol PoPoL
— 4.5.6.3 TAGs with three different fatty acids (PoPLn, OLLn, PLLn, PoOLn) See formula (9)
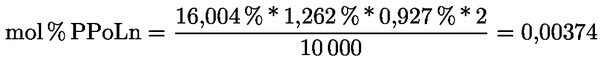


0,00761 mol PPoLn


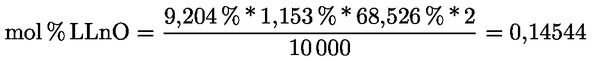
0,43655 mol OLLn


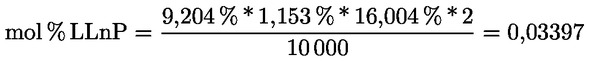
0,06907 mol PLLn



0,04812 mol PoOLn
ECN42 = 0,69512 mol TAGs
Note 1: The elution order can be determined by calculating the equivalent carbon numbers, often defined by the relation ![]() , where CN is the carbon number and n is the number of double bonds; it can be calculated more precisely by taking into account the origin of the double bond. If no, nl and nln are the numbers of double bonds attributed to oleic, linoleic and linolenic acids respectively, the equivalent carbon number can be calculated by means of the relation of the formula:
, where CN is the carbon number and n is the number of double bonds; it can be calculated more precisely by taking into account the origin of the double bond. If no, nl and nln are the numbers of double bonds attributed to oleic, linoleic and linolenic acids respectively, the equivalent carbon number can be calculated by means of the relation of the formula:
![]()
where the coefficient do, dl and dln can be calculated by means of the reference triglycerides. Under the conditions specified in this method, the relation obtained will be close to:
![]()
Note 2: With several reference triglycerides, it is also possible to calculate the resolution with respect to triolein:
![]()
trioleinby use of the reduced retention time
![]()
The graph of log α against f (number of double bonds) enables the retention values to be determined for all the triglycerides of fatty acids contained in the reference triglycerides — see Figure 1.
Note 3: The efficiency of the column should permit clear separation of the peak of trilinolein from the peaks of the triglycerides with an adjacent RT. The elution is carried out up to ECN 52 peak.
Note 4: A correct measure of the areas of all peaks of interest for the present determination is ensured if the second peak corresponding to ECN 50 is 50 % of full scale of the recorder.
Figure 1
Graph of log α against f (number of double bonds)
Number of double bonds
La: lauric acid; My: myristic acid; P: palmitic acid; S: stearic acid; O: oleic acid; L: linoleic acid; Ln: linolenic acid
Figure 2
Low linoleic olive oil
(a)

With solvent: Acetone/Acetonitrile.
PROFILE a: Main components of chromatographic peaks: ECN42: (1) LLL + PoLL; (2) OLLn + PoOLn; (3) PLLn; ECN44: (4) OLL + PoOL; (5) OOLn + PLL; (6) POLn + PPoPo; (7) OOL + PoOO; ECN46: (8) OOL + LnPP; (9) PoOO; (10) SLL + PLO; (11) PoOP + SPoL + SOLn + SPoPo; (12) PLP; ECN48: (13) OOO + PoPP; (14 + 15) SOL + POO; (16) POP; ECN50: (17) SOO; (18) POS + SLS.
(b)

With solvent: Propionitrile
PROFILE b: Main components of chromatographic peaks: ECN42: (1) LLL; (2) OLLn + PoLL; (3) PLLn; ECN44: (4) OLL; (5) OOLn + PoOL; (6) PLL + PoPoO; (7) POLn + PPoPo + PPoL; ECN46: (8) OOL + LnPP; (9) PoOO; (10) SLL + PLO; (11) PoOP + SPoL + SOLn + SPoPo; (12) PLP; ECN48: (13) OOO + PoPP; (14) SOL; (15) POO; (16) POP; ECN50: (17) SOO; (18) POS + SLS
Figure 3
High linoleic olive oil
(a)
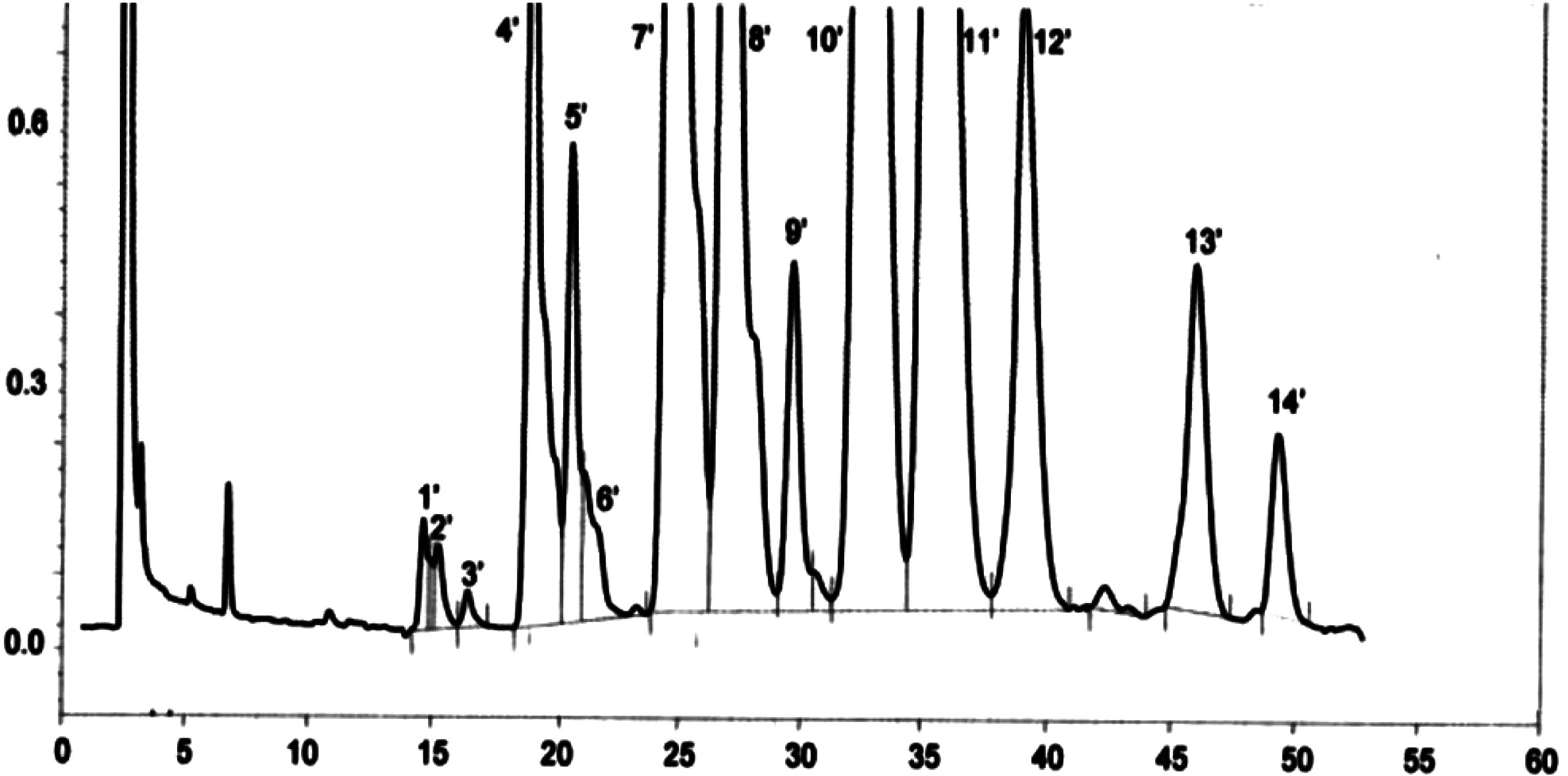
With solvent: Acetone/Acetonitrile (50:50).
Profile a: Main components of chromatographic peaks: ECN42: (1’) LLL + PoLL; (2’) OLLn + PoOLn; (3’) PLLn; ECN44: (4’) OLL + PoOL; (5’) OOLn + PLL; (6’) POLn + PPoPo; ECN46: (7’) OOL + PoOO; (8’) PLO + SLL + PoOP; (9’) PLP + PoPP; ECN48: (10’) OOO; (11’) POO + SLL + PPoO; (12’) POP + PLS; ECN50: (13’) SOO; (14’) POS + SLS
(b)
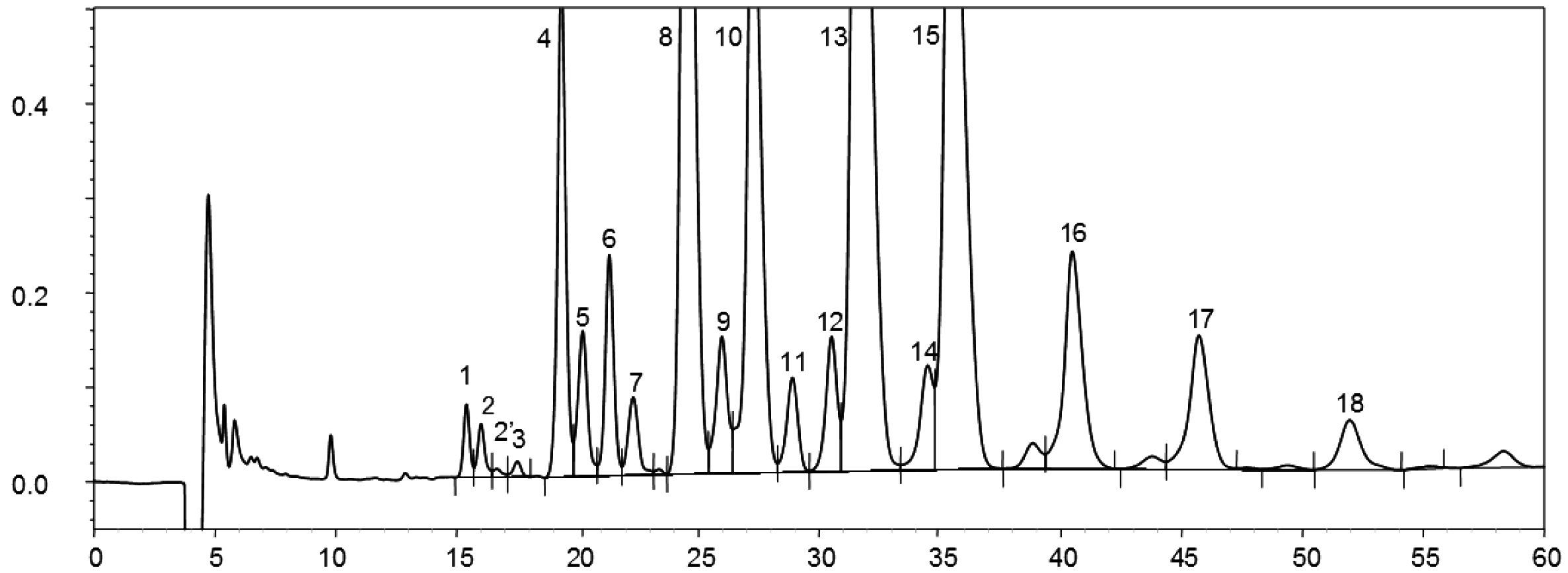
With solvent: Propionitrile.
Profile b: Main components of chromatographic peaks: ECN42: (1) LLL; (2 + 2’) OLLn + PoLL; (3) PLLn; ECN44: (4) OLL; (5) OOLn + PoOL; (6) PLL + PoPoO; (7) POLn + PPoPo + PPoL; ECN46: (8) OOL + LnPP; (9) PoOO; (10) SLL + PLO; (11) PoOP + SPoL + SOLn + SPoPo; ECN48: (12) PLP; (13) OOO + PoPP; (14) SOL; (15) POO; (16) POP; ECN50: (17) SOO; (18) POS + SLS; ECN52: (19) AOO.
ANNEX XIX
DETERMINATION OF ALIPHATIC AND TRITERPENIC ALCOHOLS CONTENT BY CAPILLARY GAS CHROMATOGRAPHY
1. SUBJECT MATTER
This Annex describes a method for the determination of aliphatic and triterpenic alcohols content in oils and fats.
2. PRINCIPLE OF THE METHOD
The fatty substance, with 1-eicosanol added as internal standard, is saponified with ethanolic potassium hydroxide and then the unsaponifiable matter extracted with ethyl ether. The alcoholic fraction is separated from the unsaponifiable matter by chromatography on a basic silica gel plate; the alcohols recovered from the silica gel are transformed into trimethylsilyl ethers and analysed by capillary gas chromatography.
3. EQUIPMENT
3.1. 250 ml round-bottomed flask fitted with a reflux condenser having ground-glass joints.
3.2. 500 ml separating funnel.
3.3. 250 ml round-bottomed flasks.
3.4. Chromatographic tank for thin-layer chromatographic analysis, for glass plates of dimensions 20 x 20 cm.
3.5. Ultraviolet lamp having a wavelength of 366 or 254 nm.
3.6. 100 μl and 500 μl microsyringes.
3.7. A cylindrical filter funnel with a G3 porous septum (porosity 15 to 40 μm) of diameter approximately 2 cm and a depth of some 5 cm, with an attachment suitable for filtration under vacuum and a 12/21 male ground glass joint.
3.8. 50 ml vacuum conical flask with a 12/21 ground-glass female joint which can be fitted to the filter funnel (3.7).
3.9. A 10 ml test tube with a tapering bottom and a sealing stopper.
3.10. Gas chromatograph for use with a capillary column, and provided with a splitting system composed of:
3.10.1. Thermostatic chamber for columns (column oven) to hold the temperature desired with a precision of ± 1 °C.
3.10.2. A temperature-adjustable injection unit with a persilanised glass vaporising element.
3.10.3. A flame ionisation detector and converter-amplifier.
3.10.4. Recorder-integrator for operation with the converter-amplifier (3.10.3), with response time not exceeding one second and with variable paper-speed.
3.11. Glass or fused silica capillary column, of length 20 to 30 m, internal diameter 0,25 to 0,32 mm, with SE-52 or SE-54 liquid phase or equivalent, with a film thickness between 0,10 and 0,30 μm.
3.12. Microsyringe for gas chromatography, of 10 μl capacity with hardened needle.
3.13. Analytical balance sensitive to 1 mg (with 0,1 mg display).
4. REAGENTS
4.1. Potassium hydroxide, approximately 2 N ethanolic solution: 130 g potassium hydroxide (minimum concentration 85 %) is dissolved, with cooling, in 200 ml distilled water and then made up to one litre with ethanol. The solution should be stored in a well-stoppered opaque glass bottle.
4.2. Ethyl ether, pure for analysis.
4.3. Anhydrous sodium sulphate, analytical purity.
4.4. Glass plates coated with silica gel, without fluorescence indicator, thickness 0,25 mm (commercially available ready for use).
4.5. Potassium hydroxide, approximately 0,2 N ethanolic solution; 13 g of potassium hydroxide are dissolved in 20 ml of distilled water and made up to one litre with ethanol.
4.6. Benzene, for chromatography (see 5.2.2).
4.7. Acetone, for chromatography (See 5.2.2).
4.8. Hexane, for chromatography (see 5.2.2).
4.9. Ethyl ether, for chromatography (see 5.2.2).
4.10. Chloroform, for chromatography.
4.11. Reference solution for thin-layer chromatography: C20-C28 alcohols 0,5 % in chloroform, or a fraction of alcohols obtained as indicated in point 5.2 from the unsaponifiable matter of an olive-pomace oil.
4.12. 0,2 % solution of 2', 7'-dichlorofluorescein in ethanol. Make slightly basic by adding a few drops of 2 N alcoholic potassium hydroxide solution.
4.13. Anhydrous pyridine, for chromatography.
4.14. Hexamethyl disilazane.
4.15. Trimethylchlorosilane.
4.16. Standard solutions of trimethylsilyl ethers of aliphatic alcohols from C20 to C28. They may be prepared from mixtures of pure alcohols at the time they are required for use.
4.17. A 0,1 % (m/v) solution of 1-eicosanol in chloroform (internal standard).
4.18. Carrier gas: hydrogen or helium, gas-chromatographic purity.
4.19. Auxiliary gas: nitrogen, gas-chromatographic purity.
5. PROCEDURE
5.1. Preparation of the unsaponifiables
|
5.1.1. |
Using a 500 μl microsyringe place, into a 250 ml round-bottom flask, a volume of 0,1 % 1-eicosanol solution in chloroform (4.17) containing a quantity of 1-eicosanol approximately equal to 10 % of the aliphatic alcohols content in that portion of sample to be taken for analysis. For example, to 5 g of sample add 250 μl of the 0,1 % 1-eicosanol solution if olive oil and 1 500 μl if olive pomace oil. Evaporate to dryness in current of nitrogen and then weigh accurately 5 g of the dry filtered sample into the same flask. |
|
5.1.2. |
Add 50 ml of 2 N potassium hydroxide ethanolic solution, fit the reflux condenser and heat the apparatus to slight boiling on a steam bath, stirring continuously throughout the heating process until saponification has taken place (the solution becomes clear). Continue heating for a further 20 minutes and then add 50 ml of distilled water through the condenser. The condenser is then disconnected and the flask cooled to approximately 30 °C. |
|
5.1.3. |
The contents of the flask are quantitatively transferred to a separating funnel of 500 ml capacity by adding distilled water several times, using a total of around 50 ml distilled water. Add approximately 80 ml of ethyl ether, shake vigorously for approximately 30 seconds and allow to settle (Note 1). Separate off the lower aqueous phase collecting it in a second separating funnel. Two further extractions are effected on the aqueous phase, in the same manner, using each time 60 to 70 ml ethyl ether. Note 1: Emulsions may be eliminated by adding, using as a spray, small quantities of ethyl alcohol or methyl alcohol. |
|
5.1.4. |
The ethyl ether extracts are combined in a separating funnel and washed with distilled water (50 ml at a time) until the washing water gives a neutral reaction. Discard the washing water, dry with anhydrous sodium sulphate and filter, into a flask of 250 ml capacity which has been weighed beforehand, the funnel and filter being washed with small quantities of ethyl ether which are added to the total. |
|
5.1.5. |
Distil the ether down to a few ml, then bring to dryness under a slight vacuum or in a current of nitrogen, completing drying in an oven at 100 °C for approximately a quarter of an hour, and then weigh after cooling in a desiccator. |
5.2. Separation of alcoholic fractions
|
5.2.1. |
Preparation of basic TLC plates: the silica gel plates (4.4) are immersed completely, in 0,2 N potassium hydroxide solution (4.5) for 10 seconds, and then left to dry for two hours under an extractor hood and finally placed in an oven at 100 °C for one hour. Remove from the oven and keep in a calcium chloride desiccator until required for use (plates treated in this way must be used within 15 days). Note 2: When basic silica gel plates are used to separate the alcoholic fraction there is no need to treat the unsaponifiables with alumina. It follows that all acid compounds (fatty acids and others) are retained at the origin thereby obtaining both aliphatic alcohol and terpenic alcohol bands which are both separated distinctly from the sterol band. |
|
5.2.2. |
Place a 65/35 by volume hexane/ethyl ether mixture in the plate-developing chamber to a depth of approximately 1 cm ( 6 ). Close the chamber using an appropriate cover and leave for half an hour to allow equilibration between vapour and liquid. Strips of filter paper dipping into the eluent may be affixed to the inside surfaces of the tank to reduce the development time by approximately one third and obtain more uniform, regular elution of the components. Note 3: The developing solution must be replaced for each analysis in order to obtain reproducible developing conditions. |
|
5.2.3. |
An approximately 5 % solution of unsaponifiable matter (5.1.5) in chloroform is prepared and 0,3 ml of the solution is streaked as a uniform strip of minimum thickness, using the 100 μl microsyringe, on a TLC plate at approximately 2 cm from the bottom of the TLC plate. Aligned with the origin, 2 to 3 μl of the aliphatic alcohols reference solution (4.11) are spotted for the identification of the aliphatic alcohols band after development has been completed. |
|
5.2.4. |
Place the plate inside the development tank as stated in 5.2.2. The ambient temperature should be maintained between 15 and 20 °C. Immediately close the chamber with the cover and allow to elute until the solvent front reaches approximately 1 cm from the upper edge of the plate. The plate is then removed from the development chamber and the solvent evaporated under a hot air current or the plate is left for a while under the extractor hood. |
|
5.2.5. |
The plate is sprayed lightly and evenly with the solution of 2′, 7′-dichlorofluorescein when the plate is observed under ultra violet light. The aliphatic alcohols band can be identified through being aligned with the stain obtained from the reference solution: mark the limits of the band with a black pencil; outlining the band of aliphatic alcohols and the band immediately above that, which is the terpenic alcohols band, together (Note 4). Note 4: The aliphatic alcohols band and the terpenic alcohols band are to be grouped together in view of the possible migration of some aliphatic alcohols into the triterpenic alcohols band. An example of the TLC separation in given in Figure 1 of the Appendix. |
|
5.2.6. |
Using a metal spatula scrape off the silica gel in the marked area. Place the finely comminuted material removed into the filter funnel (3.7). Add 10 ml of hot chloroform, mix carefully with the metal spatula and filter under vacuum, collecting the filtrate in the conical flask (3.8) attached to the filter funnel. Wash the silica gel in the flask three times with ethyl ether (approximately 10 ml each time) collecting the filtrate in the same flask attached to the funnel. Evaporate the filtrate to a volume of 4 to 5 ml, transfer the residual solution to the previously weighed 10 ml test tube (3.9), evaporate to dryness by mild heating in a gentle flow of nitrogen, make up again using a few drops of acetone, evaporate again to dryness, place in an oven at 105 °C for approximately 10 minutes and then allow to cool in a desiccator and weigh. The residue inside the test tube is composed of the alcoholic fraction. |
5.3. Preparation of the trimethylsilyl ethers
|
5.3.1. |
The reagent for silylation, consisting of a mixture of 9:3:1 by volume (Note 5) of pyridine-hexamethyldisilazane-trimethylchlorosilane in the proportion of 50 μl for each milligram of aliphatic alcohols, is added to the test tube containing the alcoholic fraction, avoiding all absorption of moisture (Note 6). Note 5: Solutions which are ready for use are available commercially. Other silanising reagents such as, for example, bis-trimethylsilyl, trifluor acetamide + 1 % trimethyl chlorosilane, which has to be diluted with an equal volume of anhydrous pyridine, are also available. Note 6: The slight opalescence which may form is normal and does not cause any interference. The formation of a white floc or the appearance of a pink colour are indicative of the presence of moisture or deterioration of the reagent. If these occur the test must be repeated. |
|
5.3.2. |
Stopper the test tube, shake carefully (without overturning) until the aliphatic alcohols are completely dissolved. Stand for at least 15 minutes at ambient temperature and then centrifuge for a few minutes. The clear solution is ready for gas chromatographic analysis. |
5.4. Gas chromatography analysis
5.4.1. Preliminary operations, column packing
|
5.4.1.1. |
Fit the column in the gas chromatograph, attaching the inlet end to the injector connected to the splitting system and the outlet end to the detector. Carry out a general check of the gas chromatography assembly (tightness of gas fittings, efficiency of the detector, efficiency of the splitting system and of the recording system, etc.). |
|
5.4.1.2. |
If the column is being used for the first time it is recommended that it should be subjected to conditioning. A little carrier gas is passed through the capillary column and then the gas chromatography assembly is switched on and gradually heated until a temperature not less than 20 °C above the operating temperature (see Note 7) is attained. That temperature is held for not less than two hours and then the assembly is brought to the operating conditions (regulation of gas flow, split flame ignition, connection to the electronic recorder, adjustment of the temperature of the capillary column oven, the detector and the injector, etc.) and the signal is adjusted to a sensitivity not less than twice the highest level contemplated for the execution of the analysis. The course of the base line must be linear, without peaks of any kind, and must not drift. A negative straight-line drift indicates leakage from the column connections; a positive drift indicates inadequate conditioning of the column. Note 7: The conditioning temperature shall be at least 20 °C less than the maximum temperature contemplated for the liquid phase employed. |
5.4.2. Choice of operating conditions
5.4.2.1. The guideline operating conditions are as follows:
— column temperature: the initial isotherm is set at 180 °C for eight minutes and then programmed at 5 °C/minute to 260 °C and a further 15 minutes at 260 °C,
— temperature of evaporator: 280 °C,
— temperature of detector: 290 °C,
— linear velocity of carrier gas: helium 20 to 35 cm/s, hydrogen 30 to 50 cm/s,
— splitting ratio: 1:50 to 1:100,
— sensitivity of instrument: 4 to 16 times the minimum attenuation,
— sensitivity of recording: 1 to 2 mV fs,
— paper speed: 30 to 60 cm/h,
— quantity of substance injected: 0,5 to 1 μl of TMSE solution.
The above conditions may be modified according to the characteristics of the column and of the gas chromatograph to obtain chromatograms satisfying the following conditions:
— alcohol C26 retention time shall be 18 ± 5 minutes,
— the alcohol C22 peak shall be 80 ± 20 % of the full scale value for olive oil and 40 ± 20 % of the full scale value for seed oil.
5.4.2.2. The above requirements are checked by repeated injection of the standard TMSE mixture of alcohols and the operating conditions are adjusted to yield the best possible results.
5.4.2.3. The parameters for the integration of peaks shall be set so that a correct appraisal of the areas of the peaks considered is obtained.
5.4.3. Analytical procedure
5.4.3.1. Using the microsyringe of 10 μl capacity draw in 1 μl of hexane followed by 0,5 μl of air and subsequently 0,5 to 1 μl of the sample solution; raise the plunger of the syringe further so the needle is emptied. Push the needle through the membrane of the injection unit and after one to two seconds inject rapidly, then slowly remove the needle after some five seconds.
5.4.3.2. Recording is effected until the TMSE of the aliphatic alcohols present have been eluted completely. The base line shall always correspond to the requirements of 5.4.1.2.
5.4.4. Peak identification.
The identification of individual peaks is effected according to the retention times and by comparison with the standard TMSE mixture, analysed under the same conditions.
Examples of chromatogram of the alcoholic fraction of a refined olive oil is shown in Figures 2 and 3 of the Appendix.
5.4.5. Quantitative evaluation
5.4.5.1. The peak areas of 1-eicosanol and of the aliphatic alcohols C22, C24, C26 and C28 are calculated by electronic integration.
5.4.5.2. The contents of each aliphatic alcohol, expressed in mg/
1 000
g fatty substance, are calculated as follows:
![]()
where:
Ax = area of the alcohol peak x
As = area of 1-eicosanol
ms = mass of 1-eicosanol in milligrams
m = mass of sample drawn for determination, in grams.
6. EXPRESSION OF THE RESULTS
The contents of the individual aliphatic alcohols in mg/1 000 g of fatty substance and the sum of the ‘total aliphatic alcohols’ are reported.
Appendix
TLC separation example and chromatogram examples
Figure 1
Thin-layer chromatography plate of the unsaponifiable fraction from olive oil eluted with hexane/ethyl ether (65/35)

|
1 |
Alcohol C26 |
A |
Sterols |
|
2 |
Alcohol C30 |
B |
Aliphatic alcohols |
|
3 |
Alcohol C20 |
C |
Triterpenic alcohols |
|
4 |
Mix Alcohols C20-22-26-30 |
D |
Squalene |
|
5 |
Extra virgin unsaponifiable |
|
|
Figure 2
Chromatogram of the alcoholic fraction of a refined olive oil

|
1 = Phytol |
5 = Alcohol C24 |
9 = 24-Methylen-cycloartenol |
|
2 = Geranyl geraniol |
6 = Alcohol C26 |
10 = Citrostadienol |
|
3 = Alcohol C20 (IS) |
7 = Alcohol C28 |
11 = Cyclobranol |
|
4 = Alcohol C22 |
8 = Cycloartenol |
|
Figure 3
Aliphatic and triterpenic alcohols of a refined olive oil and a second centrifugation olive oil

|
1 = Phytol |
5 = Alcohol C24 |
9 = 24-Methylen-cycloartenol (24MeCA) |
|
2 = Geranyl geraniol (CX) |
6 = Alcohol C26 |
10 = Citrostadienol |
|
3 = Alcohol C20 |
7 = Alcohol C28 |
11 = Cyclobranol |
|
4 = Alcohol C22 |
8 = Cycloartenol (CA) |
|
ANNEX XX
Method for the determination of the content of waxes, fatty acid methyl esters and fatty acid ethyl esters by capillary gas chromatography
1. PURPOSE
This method is for the determination of the content of waxes, fatty acid methyl and ethyl esters in olive oils. The individual waxes and alkyl esters are separated according to the number of carbon atoms. The method is recommended as a tool for distinguishing between olive oil and olive-pomace oil and as a quality parameter for extra virgin olive oils enabling the detection of fraudulent mixtures of extra virgin olive oils with lower quality oils whether they are virgin, lampante or some deodorised oils.
2. PRINCIPLE
Addition of suitable internal standards to the oil and fractionation by chromatography on a hydrated silica gel column. Recovery of the fraction eluted under the test conditions (with a lower polarity than that of the triacylglycerols) and direct analysis by capillary gas chromatography.
3. APPARATUS
|
3.1. |
Erlenmeyer flask, 25 ml. |
|
3.2. |
Glass column for liquid chromatography, internal diameter 15 mm, length 30-40 cm, fitted with a suitable stopcock. |
|
3.3. |
Gas chromatograph suitable for use with a capillary column, equipped with a system for direct, on-column injection comprising:
|
|
3.4. |
Microsyringe, 10 μl, with hardened needle, for direct on-column injection. |
|
3.5. |
Electric shaker. |
|
3.6. |
Rotary evaporator. |
|
3.7. |
Muffle oven. |
|
3.8. |
Analytical balance for weighing to an accuracy of ± 0,1 mg. |
|
3.9. |
Usual laboratory glassware. |
4. REAGENTS
|
4.1. |
Silica gel, 60-200 μm mesh. Place the silica gel in the muffle oven at 500 °C for at least 4 h. Allow to cool and then add 2 % water in relation to the quantity of silica gel used. Shake well to homogenise slurry and keep in the desiccator for at least 12 h prior to use. |
|
4.2. |
n-hexane, chromatography grade or residue grade (the purity must by checked). WARNING – Fumes may ignite. Keep away from sources of heat, sparks or naked flames. Make sure the bottles are always properly closed. Ensure proper ventilation during usage. Avoid build-up of fumes and remove any possible fire risk, such as heaters or electric apparatus not manufactured from non-inflammable material. Pernicious if inhaled, because it may cause nerve cell damage. Avoid breathing in the fumes. Use a suitable respiratory apparatus if necessary. Avoid contact with eyes and skin. |
|
4.3. |
Ethyl ether, chromatography grade
WARNING – Highly inflammable and moderately toxic. Irritates the skin. Pernicious if inhaled. May cause damage to eyes. Effects may be delayed. It can form explosive peroxides. Fumes may ignite. Keep away from sources of heat, sparks or naked flames. Make sure the bottles are always properly closed. Ensure proper ventilation during usage. Avoid build-up of fumes and remove any possible fire risk, such as heaters or electric apparatus not manufactured from non-inflammable material. Do not evaporate to dryness or near dryness. The addition of water or an appropriate reducing agent can reduce peroxide formation. Do not drink. Avoid breathing in the fumes. Avoid prolonged or repeated contact with skin. |
|
4.4. |
n-heptane, chromatography grade, or iso-octane WARNING – Inflammable. Pernicious if inhaled. Keep away from sources of heat, sparks or naked flames. Make sure the bottles are always properly closed. Ensure proper ventilation during usage. Avoid breathing in the fumes. Avoid prolonged or repeated contact with skin. |
|
4.5. |
Standard solution of lauryl arachidate (Note 3), at 0,05 % (m/V) in heptane (internal standard for waxes). Note 3: Palmityl palmitate, myristyl stearate or arachidyl laureate may also be used. |
|
4.6. |
Standard solution of methyl heptadecanoate, at 0,02 % (m/V) in heptane (internal standard for methyl and ethyl esters). |
|
4.7. |
Sudan 1 (1-phenylazo-2-naphthol). |
|
4.8. |
Carrier gas: hydrogen or helium, pure, gas chromatography grade. WARNING Hydrogen. Highly inflammable, under pressure. Keep away from sources of heat, sparks, naked flames or electric apparatus not manufactured from non-inflammable material. Make sure the bottle valve is shut when not in use. Always use with a pressure reducer. Release the tension of the reducer spring before opening the bottle valve. Do not stand in front of the bottle outlet when opening the valve. Ensure proper ventilation during usage. Do not transfer hydrogen from one bottle to another. Do not mix gas in the bottle. Make sure the bottles cannot be knocked over. Keep them away from sunlight and sources of heat. Store in a corrosive-free environment. Do not use damaged or unlabelled bottles. Helium. Compressed gas at high pressure. It reduces the amount of oxygen available for breathing. Keep the bottle shut. Ensure proper ventilation during usage. Do not enter storage areas unless they are properly ventilated. Always use with a pressure reducer. Release the tension of the reducer spring before opening the bottle valve. Do not transfer gas from one bottle to another. Make sure the bottles cannot be knocked over. Do not stand in front of the bottle outlet when opening the valve. Keep them away from sunlight and sources of heat. Store in a corrosive-free environment. Do not use damaged or unlabelled bottles. Do not inhale. Use solely for technical purposes. |
|
4.9. |
Auxiliary gases: — Hydrogen, pure, gas chromatography grade. — Air, pure, gas chromatography grade. WARNING Air. Compressed gas at high pressure. Use with caution in the presence of combustible substances as the self-ignition temperature of most of the organic compounds in the air is considerably lower under high pressure. Make sure the bottle valve is shut when not in use. Always use a pressure reducer. Release the tension of the reducer spring before opening the bottle valve. Do not stand in front of the bottle outlet when opening the valve. Do not transfer gas from one bottle to another. Do not mix gas in the bottle. Make sure the bottles cannot be knocked over. Keep them away from sunlight and sources of heat. Store in a corrosive-free environment. Do not use damaged or unlabelled bottles. Air intended for technical purposes must not be used for inhaling or respiratory apparatus. |
5. PROCEDURE
5.1. Preparation of the chromatography column
Suspend 15 g of silica gel (point 4.1) in n-hexane (point 4.2) and introduce into the column (point 3.2). Allow to settle spontaneously. Complete settling with the aid of an electric shaker to make the chromatographic bed more homogeneous. Percolate 30 ml of n-hexane to remove any impurities. Weigh exactly about 500 mg of the sample into the 25-ml flask (point 3.1), using the analytical balance (point 3.8), and add a suitable amount of internal standard (point 4.5), depending on the assumed wax content, e.g. add 0,1 mg of lauryl arachidate in the case of olive oil, 0,25-0,50 mg in the case of olive-pomace oil and 0,05 mg of methyl heptadecanoate for olive oils (point 4.6).
Transfer the prepared sample to the chromatography column with the aid of two 2-ml portions of n-hexane (point 4.2).
Allow the solvent to flow to 1 mm above the upper level of the absorbent. Percolate a further of n-hexane/ethyl ether (99:1) and collect 220 ml at a flow of about 15 drops every 10 seconds. (This fraction contains the methyl and ethyl esters and waxes). (Note 4) (Note 5).
Note 4: The n-hexane/ethyl ether (99:1) mixture should be freshly prepared every day
Note 5: 100 μl of Sudan I dye at 1 % in the elution mixture can be added to the sample solution to check visually that the waxes are eluted properly.
The retention time of the dye lies in between that of the waxes and triacylglycerols. Hence, when the dye reaches the bottom of the chromatography column, elution has to be suspended because all the waxes have been eluted.
Evaporate the resultant fractions in a rotary evaporator until the solvent is almost removed. Remove the last 2 ml under a weak current of nitrogen. Collect the fraction containing the methyl and ethyl esters is diluted with 2-4 ml of n-heptane or iso-octane.
5.2. Gas chromatography analysis
5.2.1. Preliminary procedure
Fit the column to the gas chromatograph (point 3.3), connecting the inlet port to the on-column system and the outlet port to the detector. Check the gas chromatography apparatus (operation of gas loops, efficiency of detector and recorder system, etc.).
If the column is being used for the first time, it is advisable to condition it. Run a light flow of gas through the column, then switch on the gas chromatography apparatus. Gradually heat until a temperature of 350 °C is reached after approximately 4 h.
Maintain this temperature for at least 2 h, then regulate the apparatus to the operating conditions (regulate gas flow, light flame, connect to electronic recorder (point 3.3.4), regulate oven temperature for column, regulate detector, etc.). Record the signal at a sensitivity at least twice as high as that required for the analysis. The base line should be linear, with no peaks of any kind, and must not have any drift.
Negative straight-line drift indicates that the column connections are not correct while positive drift indicates that the column has not been properly conditioned.
5.2.2. Choice of operating conditions for waxes and methyl and ethyl esters (Note 6).
The operating conditions are generally as follows:
|
— |
Column temperature : 20 °C/min 5 °C/min 80 °C at first (1′) |
|
— |
Detector temperature : 350 °C. |
|
— |
Amount injected : 1 μl of n-heptane solution (2-4 ml). |
|
— |
Carrier gas : helium or hydrogen at the optimal linear speed for the gas chosen (see Appendix A). |
|
— |
Instrument sensitivity : suitable for fulfilling the above conditions. |
Note 6: Due to the high final temperature, positive drift is allowed but may not exceed more than 10 % of the full-scale value.
These conditions may be modified to suit the characteristics of the column and the gas chromatograph in order to separate all the waxes and fatty acid methyl and ethyl esters and to obtain satisfactory peak separation (see Figures 2, 3 and 4) and a retention time of 18 ± 3 minutes for the lauryl arachidate internal standard. The most representative peak of the waxes must be over 60 % of the full-scale value while the methyl heptadecanoate internal standard for the methyl and ethyl esters must reach the full-scale value.
The peak integration parameters should be determined in such a way as to obtain a correct evaluation of the peak areas considered.
5.3. Performance of the analysis
Take up 10 μl of the solution with the aid of the 10 μl micro-syringe, drawing back the plunger until the needle is empty. Introduce the needle into the injection system and inject quickly after 1–2 s. After about 5 s, gently extract the needle.
Perform the recording until the waxes or stigmastadienes are completely eluted, depending on the fraction being analysed.
The base line must always meet the required conditions.
5.4. Peak identification
Identify the peaks from the retention times by comparing them with mixtures of waxes with known retention times, analysed under the same conditions. The alkyl esters are identified from mixtures of methyl and ethyl esters of the chief fatty acids in olive oils (palmitic and oleic).
Figure 1 provides a chromatogram of the waxes in a virgin olive oil. Figures 2 and 3 show the chromatograms of two retail extra virgin olive oils, one with methyl and ethyl esters and the other without them. Figure 4 gives the chromatograms for a top-quality extra virgin olive oil and the same oil spiked with 20 % deodorised oil.
5.5. Quantitative analysis of the waxes
Determine the area of the peaks corresponding to the lauryl arachidate internal standard and the aliphatic esters from C40 to C46 with the aid of the integrator.
Determine the total waxes content by adding each individual wax, in mg/kg of fat, as follows:

where:
|
Ax |
= |
area corresponding to the peak for the individual ester, in computer counts |
|
As |
= |
area corresponding to the peak for the lauryl arachidate internal standard, in computer counts |
|
ms |
= |
mass of the lauryl arachidate internal standard added, in milligrams; |
|
m |
= |
mass of the sample taken for determination, in grams. |
5.5.1. Quantitative analysis of the methyl and ethyl esters
With the aid of the integrator, determine the areas of the peaks corresponding to the methyl heptadecanoate internal standard, the methyl esters of the C16 and C18 fatty acids and the ethyl esters of the C16 and C18 fatty acids.
Determine the content of each alkyl ester, in mg/kg of fat, as follows:
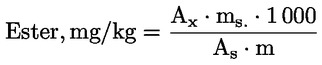
where:
|
Ax |
= |
area corresponding to the peak for the individual C16 and C18 ester, in computer counts |
|
As |
= |
area corresponding to the peak for the methyl heptadecanoate internal standard, in computer counts |
|
ms |
= |
mass of the methyl heptadecanoate internal standard added, in milligrams; |
|
m |
= |
mass of the sample taken for determination, in grams. |
6. EXPRESSION OF RESULTS
Report the sum of the contents of the different waxes from C40 to C46 (Note 7) in milligrams per kilograms of fat.
Report the sum of the contents of the methyl esters and ethyl esters from C16 to C18 and the total of the two.
Results should be expressed to the nearest mg/kg.
Note 7: The components for quantification refer to the peaks with even carbon numbers amongst the C40 - C46 esters, according to the specimen chromatogram of the waxes in olive oil provided in the attached figure. For identification purposes, if the C46 ester is split, it is recommended to analyse the wax fraction of an olive-pomace oil where the C46 peak is distinguishable because it is clearly predominant.
Report the ratio between ethyl esters and methyl esters
Figure 1
Example of a gas chromatogram of the wax fraction of an olive oil ( 7 )

Peaks with a retention time from 5 to 8 min of the fatty acid methyl and ethyl esters
Keys:
|
I.S. |
= |
Lauryl arachidate |
|
1 |
= |
Diterpenic esters |
|
2+2′ |
= |
C40 esters |
|
3+3′ |
= |
C42 esters |
|
4+4′ |
= |
C44 esters |
|
5 |
= |
C46 esters |
|
6 |
= |
Sterol esters and triterpene alcohols |
Figure 2
Methyl esters, ethyl esters and waxes in a virgin olive oil

Keys:
|
1 |
– |
Methyl C16 |
|
2 |
– |
Ethyl C16 |
|
3 |
– |
Methyl heptadecanoate I.S. |
|
4 |
– |
Methyl C18 |
|
5 |
– |
Ethyl C18 |
|
6 |
– |
Squalene |
|
7 |
– |
Lauryl arachidate I.S. |
|
A |
– |
Diterpenic esters |
|
B |
– |
Waxes |
|
C |
– |
Sterol esters and triterpenic esters |
Figure 3
Methyl esters, ethyl esters and waxes in an extra virgin olive oil

Keys:
|
1 |
– |
Methyl heptadecanoate I.S. |
|
2 |
– |
Methyl C18 |
|
3 |
– |
Ethyl C18 |
|
4 |
– |
Squalene |
|
5 |
– |
Lauryl arachidate I.S. |
|
A |
– |
Diterpenic esters |
|
B |
– |
Waxes |
|
C |
– |
Sterol esters and triterpenic esters |
Figure 4
Part of a chromatogram of an extra virgin olive oil and the same oil spiked with deodorised oil

Keys:
|
1 |
– |
Methyl myristate I.S. |
|
2 |
– |
Methyl palmitate |
|
3 |
– |
Ethyl palmitate |
|
4 |
– |
Methyl heptadecanoate I.S. |
|
5 |
– |
Methyl linoleate |
|
6 |
– |
Methyl oleate |
|
7 |
– |
Methyl stearate |
|
8 |
– |
Ethyl linoleate |
|
9 |
– |
Ethyl oleate |
|
10 |
– |
Ethyl stearate |
Appendix A
Determination of linear gas speed
Inject 1:3 μl of methane (or propane) into the gas chromatograph after adjusting it to the normal operating conditions. Measure the time the gas takes to run through the column from the moment it is injected until the peak emerges (tM).
The linear speed in cm/s is given by L/tM where L is the length of the column, in cm, and tM is the time measured in s.
▼M28 —————
ANNEX XXI
Results of conformity checks carried out on olive oils referred to in Article 8(2)
|
|
Labelling |
Chemical parameters |
Organoleptic characteristics (4) |
Final conclusion |
|||||||||||||
|
Sample |
Category |
Country of origin |
Place of inspection (1) |
Legal name |
Designation of origin |
Storage conditions |
Erroneous information |
Legibility |
C/NC (3) |
Parameters out of limit Y/N |
If so, please indicate which one(s) (2) |
C/NC (3) |
Median defect |
Fruity Median |
C/NC (3) |
Required action |
Sanction |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(1) Internal market (mill, bottlers, retail stage), export, import. (2) Each characteristic of olive oil set out in Annex I shall have a code. (3) Conform/not conform. (4) Not required for olive oil and pomace-oil. |
|||||||||||||||||
( 1 ) OJ L 228, 1.9.2009, p. 3.
( 2 ) Directive 2011/91/EU of the European Parliament and of the Council of 13 December 2011 on indications or marks identifying the lot to which a foodstuff belongs (OJ L 334, 16.12.2011, p. 1).
( 3 ) After elution of the sterol esters the chromatogram trace must not show any significant peaks (triglycerides).
( 4 ) Regulation (EU) No 1308/2013 of the European Parliament and of the Council of 17 December 2013 establishing a common organisation of the markets in agricultural products and repealing Council Regulations (EEC) No 922/72, (EEC) No 234/79, (EC) No 1037/2001 and (EC) No 1234/2007 (OJ L 347, 20.12.2013, p. 671).
( 5 ) They may refrain from tasting an oil when they notice any extremely intense negative attribute by direct olfactory means, in which case they shall record this exceptional circumstance in the profile sheet.
( 6 ) In these cases in particular, a 95/5 by volume benzene/acetone eluent mixture must be used to obtain distinct band separation.
( 7 ) After elution of the sterol esters, the chromatogram should not show any significant peaks (triacylglycerols).