

EUR-Lex Access to European Union law
This document is an excerpt from the EUR-Lex website
Document 01991R2568-20160804
Commission Regulation (EEC) No 2568/91 of 11 July 1991 on the characteristics of olive oil and olive-residue oil and on the relevant methods of analysis
Consolidated text: Regolamento (CEE) n . 2568/91 della Commissione dell'11 luglio 1991 relativo alle caratteristiche degli oli d'oliva e degli oli di sansa d'oliva nonché ai metodi ad essi attinenti
Regolamento (CEE) n . 2568/91 della Commissione dell'11 luglio 1991 relativo alle caratteristiche degli oli d'oliva e degli oli di sansa d'oliva nonché ai metodi ad essi attinenti

1991R2568 — IT — 04.08.2016 — 029.001
Il presente testo è un semplice strumento di documentazione e non produce alcun effetto giuridico. Le istituzioni dell’Unione non assumono alcuna responsabilità per i suoi contenuti. Le versioni facenti fede degli atti pertinenti, compresi i loro preamboli, sono quelle pubblicate nella Gazzetta ufficiale dell’Unione europea e disponibili in EUR-Lex. Tali testi ufficiali sono direttamente accessibili attraverso i link inseriti nel presente documento
|
REGOLAMENTO (CEE) N. 2568/91 DELLA COMMISSIONE dell'11 luglio 1991 (GU L 248 dell'5.9.1991, pag. 1) |
Modificato da:
Rettificato da:
REGOLAMENTO (CEE) N. 2568/91 DELLA COMMISSIONE
dell'11 luglio 1991
relativo alle caratteristiche degli oli d'oliva e degli oli di sansa d'oliva nonché ai metodi ad essi attinenti
Articolo 1
1. Sono considerati oli di oliva vergini ai sensi del punto 1, lettere a) e b), dell'allegato del regolamento n. 136/66/CEE gli oli le cui caratteristiche sono conformi a quelle indicate rispettivamente nei punti 1 e 2 dell'allegato I del presente regolamento.
2. È considerato olio di oliva lampante ai sensi del punto 1, lettera c), dell'allegato del regolamento n. 136/66/CEE, l'olio le cui caratteristiche sono conformi a quelle indicate nell'allegato I, punto 3, del presente regolamento.
3. È considerato olio di oliva raffinato ai sensi del punto 2 dell'allegato del regolamento n. 136/66/CEE, l'olio le cui caratteristiche sono conformi a quelle indicate nell'allegato I, punto 4, del presente regolamento.
4. È considerato olio di oliva composto di oli di oliva raffinati e di oli di oliva vergini ai sensi del punto 3 dell'allegato del regolamento n. 136/66/CEE, l'olio le cui caratteristiche sono conformi a quelle indicate nell'allegato I, punto 5, del presente regolamento.
5. È considerato olio di sansa di oliva greggio ai sensi del punto 4 dell'allegato del regolamento n. 136/66/CEE, l'olio le cui caratteristiche sono conformi a quelle indicate nell'allegato I, punto 6, del presente regolamento.
6. È considerato olio di sansa di oliva raffinato ai sensi del punto 5 dell'allegato del regolamento n. 136/66/CEE, l'olio le cui caratteristiche sono conformi a quelle indicate nell'allegato I, punto 7, del presente regolamento.
7. È considerato olio di sansa di oliva ai sensi del punto 6 dell'allegato del regolamento n. 136/66/CEE, l'olio le cui caratteristiche sono conformi a quelle indicate nell'allegato I, punto 8, del presente regolamento.
Articolo 2
1. Le caratteristiche degli oli figuranti nell'allegato I sono determinate in base ai seguenti metodi di analisi:
(a) per la determinazione degli acidi grassi liberi, espressi in percentuale di acido oleico, il metodo di cui all'allegato II;
(b) per la determinazione dell'indice di perossidi, il metodo di cui all'allegato III;
(c) per la determinazione del contenuto di cere, il metodo di cui all'allegato IV;
(d) per la determinazione della composizione e del contenuto di steroli e dialcoli triterpenici mediante gascromatografia con colonna capillare, il metodo di cui all'allegato V;
(e) per la determinazione della percentuale di 2-gliceril monopalmitato, il metodo di cui all’allegato VII;
(f) per l'analisi spettrofotometrica, il metodo di cui all'allegato IX;
(g) per la determinazione della composizione di acidi grassi, il metodo di cui all'allegato X;
(h) per la determinazione dei solventi alogenati volatili, il metodo di cui all'allegato XI;
(i) per la valutazione delle caratteristiche organolettiche degli oli di oliva vergini, il metodo di cui all'allegato XII;
(j) per la determinazione degli stigmastadieni, il metodo di cui all'allegato XVII;
(k) per la determinazione dei trigliceridi con ECN42, il metodo di cui all'allegato XVIII;
(l) per la determinazione del contenuto di alcoli alifatici e triterpenici, il metodo di cui all'allegato XIX;
(m) per la determinazione del contenuto di cere e metil ed etil esteri degli acidi grassi, il metodo di cui all’allegato XX.
▼M28 —————
2. La verifica delle caratteristiche organolettiche degli oli di oliva vergini da parte delle autorità nazionali o dei loro rappresentanti è effettuata da panel di assaggiatori riconosciuti dagli Stati membri.
Le caratteristiche organolettiche di un olio, ai sensi del primo comma, si considerano conformi alla categoria di olio di oliva dichiarata se il panel di assaggiatori riconosciuto dallo Stato membro ne conferma la classificazione.
Qualora il panel non confermi la categoria dichiarata, sotto il profilo delle sue caratteristiche organolettiche, a richiesta dell'interessato le autorità nazionali o i loro rappresentanti incaricano altri panel riconosciuti di effettuare quanto prima due controanalisi, di cui almeno una deve essere effettuata da un panel riconosciuto dallo Stato membro di produzione dell'olio. Le caratteristiche in questione sono considerate conformi a quelle dichiarate se le due controanalisi confermano la classificazione dichiarata. In caso contrario il costo delle controanalisi è a carico dell'interessato.
3. Per quanto riguarda la verifica delle caratteristiche degli oli da parte delle autorità nazionali o di loro rappresentanti, prevista al paragrafo 1, il prelievo dei campioni si effettua secondo le norme internazionali EN ISO 661 relativa alla preparazione dei campioni per le prove e EN ISO 5555 relativa al campionamento. Tuttavia, in deroga al punto 6.8 della norma EN ISO 5555, per le partite di oli in imballaggi immediati il prelievo del campione si effettua conformemente all'allegato I bis del presente regolamento. Nel caso degli oli sfusi per i quali il campionamento non può essere eseguito conformemente alla norma EN ISO 5555, i campioni sono prelevati secondo le istruzioni impartite dall'autorità competente dello Stato membro.
Fatte salve le disposizioni della norma EN ISO 5555 e del capitolo 6 della norma EN ISO 661, i campioni prelevati sono messi quanto prima al riparo dalla luce e da fonti di calore elevato e sono inviati al laboratorio per le analisi entro il quinto giorno lavorativo successivo a quello del prelievo; altrimenti i campioni sono conservati in modo da evitarne il degrado o il danneggiamento durante il trasporto o lo stoccaggio in attesa di essere inviati al laboratorio.
4. Ai fini della verifica prevista al paragrafo 3, le analisi di cui agli allegati II, III, IX, XII e XX nonché, eventualmente, le controanalisi previste dalla legislazione nazionale, sono effettuate anteriormente alla data di durata minima per quanto riguarda i prodotti condizionati. In caso di campionamento di oli sfusi, tali analisi sono effettuate entro il sesto mese successivo a quello del prelievo del campione.
Per le altre analisi previste dal presente regolamento non si applica nessun termine.
Salvo se il campione sia stato prelevato meno di due mesi prima della data di durata minima, nel caso in cui i risultati delle analisi non corrispondano alle caratteristiche della categoria di olio di oliva o di olio di sansa di oliva dichiarata, l'interessato ne viene informato al più tardi un mese prima dello scadere del termine di cui al primo comma.
5. Ai fini della determinazione delle caratteristiche degli oli di oliva secondo i metodi di cui al paragrafo 1, primo comma, i risultati delle analisi sono direttamente confrontati con i limiti fissati dal presente regolamento.
Articolo 2 bis
1. Ai fini del presente articolo, si intende per«olio d’olivacommercializzato» ilquantitativo totale di olio d’oliva e di olio di sansa di uno Stato membro cheè consumato in tale Stato membro oppure esportato da tale Statomembro.
2. Gli Stati membri fanno in modo che i controlli di conformitàsiano effettuati selettivamente, in base ad un’analisi di rischio e conadeguata frequenza, onde garantire che l’olio d’oliva immesso in commerciocorrisponda alla categoria dichiarata.
3. I criteri di valutazione del rischio possono includere:
a) la categoria dell’olio, il periodo di produzione, il prezzo degli oli rispetto a quello di altri oli vegetali, le operazioni di miscelazione e confezionamento, gli impianti e le condizioni di stoccaggio, il paese d’origine, il paese di destinazione, il mezzo di trasporto o il volume della partita;
b) la posizione degli operatori nella catena di commercializzazione, il volume e/o il valore nonché la gamma di categorie di oli che commercializzano, il tipo di attività economica svolta quali la molitura, l’immagazzinamento, la raffinazione, la miscelazione, il confezionamento e la vendita al minuto;
c) le risultanze che emergono da controlli precedenti, segnatamente per quanto riguarda il numero e il tipo di carenze accertate, la qualità abituale degli oli commercializzati e il livello di prestazione delle attrezzature tecniche adoperate;
d) l’affidabilità dei sistemi di assicurazione della qualità degli operatori o dei loro sistemi di autocontrollo rispetto alla conformità alle norme di commercializzazione;
e) il luogo in cui il controllo viene effettuato, in particolare se si tratta del primo punto di ingresso nell’Unione, dell’ultimo punto di uscita dall’Unione o del luogo in cui gli oli sono prodotti, confezionati, caricati o venduti al consumatore finale;
f) qualsiasi altra informazione da cui si possa evincere un rischio di non conformità.
4. Gli Stati membri stabiliscono in anticipo:
a) i criteri di valutazione del rischio di non conformità delle partite;
b) sulla base di un’analisi del rischio per ogni singola categoria di rischio, il numero minimo di operatori o di partite e/o di quantitativi minimi che saranno soggetti ad un controllo di conformità.
Almeno un controllo annuale di conformità è effettuato permille tonnellate di olio d’oliva commercializzato annualmente nello Statomembro.
5. Gli Stati membri verificano la conformità:
a) procedendo, in un ordine qualsiasi, alle analisi di cui all’allegato I; o
b) nell’ordine previsto dall’albero decisionale di cui all’allegato I ter, fino a raggiungere una delle decisioni figuranti nel suddetto albero decisionale.
▼M19 —————
Articolo 3
Qualora si constati che un olio non corrisponde alla descrizione della sua categoria, lo Stato membro interessato applica sanzioni effettive, proporzionate e dissuasive stabilite in base alla gravità dell’irregolarità accertata, ferme restando altre sanzioni eventuali.
Se dai controlli emergono irregolarità sostanziali, gli Stati membri aumentano la frequenza dei controlli relativi alla fase di commercializzazione, alla categoria dell’olio, all’origine o ad altri criteri.
Articolo 4
1. Ai fini della valutazione e del controllo delle caratteristiche organolettiche da parte delle autorità nazionali o dei loro rappresentanti, gli Stati membri possono procedere al riconoscimento di panel di assaggiatori.
Le condizioni del riconoscimento sono stabilite dallo Stato membro in particolare in modo da:
— rispondere alle condizioni di cui all'allegato XII, punto 4,
— garantire che la formazione del capo del panel si compia presso un organismo riconosciuto e alle condizioni a tal fine stabilite dallo Stato membro,
— subordinare la validità del riconoscimento ai risultati ottenuti nell'ambito di un sistema di controllo annuale istituito dallo Stato membro.
Ogni Stato membro comunica alla Commissione l'elenco dei panel riconosciuti e le misure adottate conformemente al presente paragrafo.
2. Lo Stato membro che incontri difficoltà per la costituzione di un comitato di assaggio sul proprio territorio può ricorrere ad un comitato di assaggio riconosciuto da un altro Stato membro.
3. Ogni Stato membro compila l'elenco dei comitati di assaggio istituiti da associazioni professionali o interprofessionali in conformità del paragrafo 1 e controlla il rispetto di dette norme.
▼M19 —————
Articolo 6
1. Il tenore in olio delle sanse e degli altri residui dell'estrazione dell'olio (codice NC 2306 90 11 e 2306 90 19 ) è determinato conformemente al metodo che figura nell'allegato XV.
2. Il tenore in olio di cui al paragrafo 1 è espresso in percentuale del suo peso rispetto a quello della sostanza secca.
Articolo 7
Si applicano le disposizioni comunitarie relative alla presenza di contaminanti.
Per quanto riguarda il tenore di solventi alogenati, i limiti per tutte le categorie di oli di oliva sono i seguenti:
— tenore massimo di ciascun solvente alogenato rilevato: 0,1 mg/kg
— tenore massimo della somma dei solventi alogenati rilevati: 0,2 mg/kg.
Articolo 7 bis
Le persone e i gruppi di persone fisiche o giuridiche che detengono, ai fini dell’esercizio della loro professione o a fini commerciali, olio d’oliva ed olio di sansa, dalla fase dell’estrazione al frantoio fino all’imbottigliamento incluso, hanno l’obbligo di tenere registri di entrata e di uscita per ogni categoria di questi oli.
Gli Stati membri assicurano che l’obbligo di cui al primo paragrafo sia debitamente rispettato.
Articolo 8
1. Ogni Stato membro comunica alla Commissione le misureadottate per l’applicazione del presente regolamento. Lo Stato membro comunicaalla Commissione qualsiasi ulteriore modifica di dette misure.
2. Entro il 31 maggio di ogni anno, lo Stato membro trasmette alla Commissione una relazione sull’applicazione del presente regolamento nel corso dell’anno civile precedente. La relazione contiene almeno i risultati dei controlli di conformità effettuati sugli oli d’oliva, presentati in base ai modelli figuranti nell’allegato XXI.
3. Le notifiche di cui al presente regolamento sono effettuate anorma del regolamento (CE) n. 792/2009 della Commissione ( 1 )
Articolo 9
Il regolamento (CEE) n. 1058/77 è abrogato.
Articolo 10
1. Il presente regolamento entra in vigore il terzo giorno successivo a quello della pubblicazione nella Gazzetta ufficiale delle Comunità europee.
Tuttavia, il metodo che figura nell'allegato XII viene applicato a decorrere dal ►M1 1o novembre 1992 ◄ , salvo per quanto riguarda le operazioni legate all'intervento.
Questo metodo non si applica all'olio d'oliva vergine condizionato anteriormente al 1o novembre 1992.
2. Il presente regolamento non si applica agli oli d'oliva e agli oli di sansa d'oliva condizionati anteriormente all'entrata in vigore del presente regolamento e commercializzati fino al 31 ottobre 1992.
Il presente regolamento è obbligatorio in tutti i suoi elementi e direttamente applicabile in ciascuno degli Stati membri.
ALLEGATI
Sommario
|
Allegato I: |
Caratteristiche degli oli di oliva |
|
Allegato I bis: |
Campionatura delle partite di olio di oliva o di olio di sansa di oliva consegnate in imballaggi immediati |
|
Allegato I ter: |
Schema decisionale per la verifica della conformità di un campione di olio di oliva alla categoria dichiarata |
|
Allegato II: |
Determinazione degli acidi grassi liberi, metodo a freddo |
|
Allegato III: |
Determinazione del numero di perossidi |
|
Allegato IV: |
Determinazione del contenuto di cere mediante gascromatografia con colonna capillare |
|
Allegato V: |
Determinazione della composizione e del contenuto di steroli e dialcoli triterpenici mediante gascromatografia con colonna capillare |
|
Allegato VII: |
►M21 Determinazione della percentuale di 2-gliceril monopalmitato ◄ |
|
▼M20 ————— |
|
|
Allegato IX: |
Analisi spettrofotometrica nell’ultravioletto |
|
Allegato X: |
Determinazione degli esteri metilici degli acidi grassi mediante gascromatografia |
|
Allegato XI: |
Determinazione del tenore dei solventi alogenati |
|
Allegato XII: |
Metodo del consiglio oleicolo internazionale per la valutazione organolettica degli oli di oliva vergini |
|
▼M20 ————— |
|
|
▼M19 ————— |
|
|
Allegato XV: |
Metodo di determinazione del tenore in olio d'oliva delle sanse |
|
Allegato XVI: |
Determinazione del numero di iodio |
|
Allegato XVII: |
Metodo di determinazione degli stigmastadieni negli oli vegetali |
|
Allegato XVIII: |
Determinazione della differenza tra il contenuto effettivo e il contenuto teorico di triacilgliceroli con ECN 42 |
|
Allegato XIX: |
►M28 Determinazione del contenuto di alcoli alifatici e triterpenici mediante gascromatografia con colonna capillare ◄ |
|
Allegato XX: |
Metodo per la determinazione del contenuto di cere e metil ed etil esteri degli acidi grassi mediante gascromatografia con colonna capillare |
|
▼M28 ————— |
|
|
Allegato XXI: |
Risultati dei controlli di conformità eseguiti sugli oli di oliva di cui al paragrafo 2 dell’articolo 8 |
ALLEGATO I
CARATTERISTICHE DEGLI OLI DI OLIVA
|
Categoria |
Etil esteri degli acidi grassi (EEAG) (*) |
Acidità (%) (*) |
Numero dei perossidi mEq O2/kg (*) |
Cere mg/kg (**) |
2 gliceril monopalmitato (%) |
Stigmastadieni mg/kg (1) |
Differenza: ECN42 (HPLC) e ECN42 (calcolo teorico) |
K232 (*) |
K268 o K270 (*) |
Delta-K (*) |
Valutazione organolettica Mediana del difetto (Md) (*) |
Valutazione organolettica Mediana del fruttato (Mf) (*) |
|
1. Olio extra vergine di oliva |
EEAG ≤ 40 mg/kg (campagna 2013-2014) (2) EEAG ≤ 35 mg/kg (campagna 2014-2016) EEAG ≤ 30 mg/kg (campagne successive al 2016) |
≤ 0,8 |
≤ 20 |
C42 + C44 + C46 ≤ 150 |
≤ 0,9 se % acido palmitico totale ≤ 14 % |
≤ 0,05 |
≤ |0,2| |
≤ 2,50 |
≤ 0,22 |
≤ 0,01 |
Md = 0 |
Mf > 0 |
|
≤ 1,0 se % acido palmitico totale > 14 % |
||||||||||||
|
2. Olio di oliva vergine |
— |
≤ 2,0 |
≤ 20 |
C42 + C44 + C46 ≤ 150 |
≤ 0,9 se % acido palmitico totale ≤ 14 % |
≤ 0,05 |
≤ |0,2| |
≤ 2,60 |
≤ 0,25 |
≤ 0,01 |
Md ≤ 3,5 |
Mf > 0 |
|
≤ 1,0 se % acido palmitico totale > 14 % |
||||||||||||
|
3. Olio di oliva lampante |
— |
> 2,0 |
— |
C40 + C42 + C44 + C46 ≤ 300 (3) |
≤ 0,9 se % acido palmitico totale ≤ 14 % |
≤ 0,50 |
≤ |0,3| |
— |
— |
— |
Md > 3,5 (4) |
— |
|
≤ 1,1 se % acido palmitico totale > 14 % |
||||||||||||
|
4. Olio di oliva raffinato |
— |
≤ 0,3 |
≤ 5 |
C40 + C42 + C44 + C46 ≤ 350 |
≤ 0,9 se % acido palmitico totale ≤ 14 % |
— |
≤ |0,3| |
— |
≤ 1,10 |
≤ 0,16 |
— |
— |
|
≤ 1,1 se % acido palmitico totale > 14 % |
||||||||||||
|
5. Olio di oliva composto di oli di oliva raffinati e di oli di oliva vergini |
— |
≤ 1,0 |
≤ 15 |
C40 + C42 + C44 + C46 ≤ 350 |
≤ 0,9 se % acido palmitico totale ≤ 14 % |
— |
≤ |0,3| |
— |
≤ 0,90 |
≤ 0,15 |
— |
— |
|
≤ 1,0 se % acido palmitico totale > 14 % |
||||||||||||
|
6. Olio di sansa di oliva greggio |
— |
— |
— |
C40 + C42 + C44 + C46 > 350 (5) |
≤ 1,4 |
— |
≤ |0,6| |
— |
— |
— |
— |
— |
|
7. Olio di sansa di oliva raffinato |
— |
≤ 0,3 |
≤ 5 |
C40 + C42 + C44 + C46 > 350 |
≤ 1,4 |
— |
≤ |0,5| |
— |
≤ 2,00 |
≤ 0,20 |
— |
— |
|
8. Olio di sansa di oliva |
— |
≤ 1,0 |
≤ 15 |
C40 + C42 + C44 + C46 > 350 |
≤ 1,2 |
— |
≤ |0,5| |
— |
≤ 1,70 |
≤ 0,18 |
— |
— |
|
(1) Somma degli isomeri che potrebbero (o non potrebbero) essere separati mediante colonna capillare. (2) Questo limite si applica agli oli di oliva prodotti a decorrere dal 1o marzo 2014. (3) Gli oli con un tenore di cera compreso tra 300 mg/kg e 350 mg/kg sono considerati olio di oliva lampante se gli alcoli alifatici totali sono pari o inferiori a 350 mg/kg o se la percentuale di eritrodiolo e uvaolo è pari o inferiore a 3,5 %. (4) La mediana del difetto può essere pari o inferiore a 3,5 e la mediana del fruttato uguale a 0. (5) Gli oli con un tenore di cera compreso tra 300 mg/kg e 350 mg/kg sono considerati olio di sansa di oliva greggio se gli alcoli alifatici totali sono superiori a 350 mg/kg e se la percentuale di eritrodiolo e uvaolo è superiore a 3,5 %. |
||||||||||||
|
Categoria |
Composizione in acidi grassi (1) |
Somma degli isomeri transoleici (%) |
Somma degli isomeri translinoleici + translinolenici (%) |
Composizione in steroli |
Steroli totali (mg/kg) |
Eritrodiolo e uvaolo (%) (**) |
||||||||||
|
Miristico (%) |
Linolenico (%) |
Arachico (%) |
Eicosenoico (%) |
Beenico (%) |
Lignocerico (%) |
Colesterolo (%) |
Brassicasterolo (%) |
Campesterolo (2) (%) |
Stigmasterolo (%) |
β — sitosterolo apparente (3) (%) (**) |
Delta-7-stigmastenolo (2) (%) |
|||||
|
1. Olio extra vergine di oliva |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,40 |
≤ 0,20 |
≤ 0,20 |
≤ 0,05 |
≤ 0,05 |
≤ 0,5 |
≤ 0,1 |
≤ 4,0 |
< Camp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 000 |
≤ 4,5 |
|
2. Olio di oliva vergine |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,40 |
≤ 0,20 |
≤ 0,20 |
≤ 0,05 |
≤ 0,05 |
≤ 0,5 |
≤ 0,1 |
≤ 4,0 |
< Camp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 000 |
≤ 4,5 |
|
3. Olio di oliva lampante |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,40 |
≤ 0,20 |
≤ 0,20 |
≤ 0,10 |
≤ 0,10 |
≤ 0,5 |
≤ 0,1 |
≤ 4,0 |
— |
≥ 93,0 |
≤ 0,5 |
≥ 1 000 |
≤ 4,5 (4) |
|
4. Olio di oliva raffinato |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,40 |
≤ 0,20 |
≤ 0,20 |
≤ 0,20 |
≤ 0,30 |
≤ 0,5 |
≤ 0,1 |
≤ 4,0 |
< Camp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 000 |
≤ 4,5 |
|
5. Olio di oliva composto di oli di oliva raffinati e di oli di oliva vergini |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,40 |
≤ 0,20 |
≤ 0,20 |
≤ 0,20 |
≤ 0,30 |
≤ 0,5 |
≤ 0,1 |
≤ 4,0 |
< Camp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 000 |
≤ 4,5 |
|
6. Olio di sansa di oliva greggio |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,40 |
≤ 0,30 |
≤ 0,20 |
≤ 0,20 |
≤ 0,10 |
≤ 0,5 |
≤ 0,2 |
≤ 4,0 |
— |
≥ 93,0 |
≤ 0,5 |
≥ 2 500 |
> 4,5 (5) |
|
7. Olio di sansa di oliva raffinato |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,40 |
≤ 0,30 |
≤ 0,20 |
≤ 0,40 |
≤ 0,35 |
≤ 0,5 |
≤ 0,2 |
≤ 4,0 |
< Camp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 800 |
> 4,5 |
|
8. Olio di sansa di oliva |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,40 |
≤ 0,30 |
≤ 0,20 |
≤ 0,40 |
≤ 0,35 |
≤ 0,5 |
≤ 0,2 |
≤ 4,0 |
< Camp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 600 |
> 4,5 |
|
(1) Tenore di altri acidi grassi (%): palmitico: 7,50-20,00; palmitoleico: 0,30-3,50; eptadecanoico: ≤ 0,30; eptadecenoico: ≤ 0,30; stearico: 0,50-5,00; oleico: 55,00-83,00; linoleico: 2,50-21,00. (2) V. Appendice del presente allegato. (3) β-sitosterolo apparente: delta-5,23-stigmastadienolo + clerosterolo + beta-sitosterolo + sitostanolo + delta-5-avenasterolo + delta-5,24-stigmastadienolo. (4) Gli oli con un tenore di cera compreso tra 300 mg/kg e 350 mg/kg sono considerati olio di oliva lampante se gli alcoli alifatici totali sono pari o inferiori a 350 mg/kg o se la percentuale di eritrodiolo e uvaolo è pari o inferiore a 3,5 %. (5) Gli oli con un tenore di cera compreso tra 300 mg/kg e 350 mg/kg sono considerati olio di sansa di oliva greggio se gli alcoli alifatici totali sono superiori a 350 mg/kg o se la percentuale di eritrodiolo e uvaolo è superiore a 3,5 %. |
||||||||||||||||
Note:
a) I risultati delle analisi devono essere espressi con un numero di decimali uguale a quello previsto per ogni caratteristica. L'ultima cifra deve essere aumentata di una unità se la cifra successiva è superiore a 4.
b) È sufficiente che una sola caratteristica non sia conforme ai valori indicati perché l'olio venga cambiato di categoria o dichiarato non conforme riguardo alla sua purezza ai fini del presente regolamento.
c) Le caratteristiche contrassegnate con un asterisco (*) e riguardanti le qualità dell'olio implicano che: — per l'olio di oliva lampante, i due corrispondenti valori limite possono non essere rispettati simultaneamente, — per gli oli di oliva vergini, l'inosservanza di almeno uno di questi valori limite comporta il cambiamento di categoria, pur rimanendo classificati in una delle categorie degli oli di oliva vergini.
d) Le caratteristiche contrassegnate con due asterischi (**) implicano che per tutti gli oli di sansa di oliva i due corrispondenti valori limite possono non essere rispettati simultaneamente.
Appendice
Schema decisionale
Schema decisionale per il campesterolo nell'olio di oliva vergine e nell'olio extra vergine di oliva.

Gli altri parametri devono rispettare i limiti fissati dal presente regolamento.
Schema decisionale per il delta-7-stigmastenolo.
— Nell'olio di oliva vergine e nell'olio extra vergine di oliva
—

Gli altri parametri devono rispettare i limiti fissati dal presente regolamento.
— Negli oli di sansa di oliva (greggio e raffinato)
—

ALLEGATO I bis
CAMPIONATURA DELLE PARTITE DI OLIO DI OLIVA O DI OLIO DI SANSA DI OLIVA CONSEGNATE IN IMBALLAGGI IMMEDIATI
Il presente metodo di campionatura si applica alle partite di olio di oliva o di olio di sansa di oliva condizionate in imballaggi immediati. A seconda che la capacità dell'imballaggio immediato sia o no superiore a 5 litri si applicano metodi di campionatura diversi.
Si intende per «partita» un insieme di unità di vendita prodotte, fabbricate e condizionate in circostanze tali che l'olio contenuto in ciascuna di queste unità di vendita è considerato omogeneo per tutte le caratteristiche analitiche. La partita deve essere identificata conformemente alla direttiva 2011/91/UE del Parlamento europeo e del Consiglio ( 2 ).
Si intende per «incremento» la quantità di olio contenuta in un imballaggio immediato prelevata da un punto a caso della partita.
1. CONTENUTO DEL CAMPIONE ELEMENTARE
1.1. Imballaggi immediati di capacità non superiore a 5 litri
Nel caso degli imballaggi immediati di capacità non superiore a 5 litri si intende per «campione elementare» il numero di incrementi prelevati da una partita in conformità alla tabella 1.
Tabella 1
Dimensione minima del campione elementare
|
In caso di imballaggi immediati aventi una capacità |
Il campione elementare deve essere costituito dall'olio proveniente da |
|
a) superiore o uguale a 1 litro |
a) 1 imballaggio immediato |
|
b) inferiore a 1 litro |
b) dal numero minimo di imballaggi la cui capacità totale è almeno pari a 1,0 litro |
Il numero di imballaggi di cui alla tabella 1, che costituiscono un campione elementare, può essere aumentato dai singoli Stati membri, secondo le rispettive esigenze (ad esempio, valutazione organolettica da parte di un laboratorio diverso da quello che ha eseguito le analisi chimiche, controanalisi ecc.).
1.2. Imballaggi immediati di capacità superiore a 5 litri
Nel caso degli imballaggi immediati di capacità superiore a 5 litri si intende per «campione elementare» una parte rappresentativa del totale degli incrementi, ottenuta mediante un processo di riduzione in conformità alla tabella 2. Il campione elementare deve essere costituito da più esemplari.
Si intende per «esemplare» di un campione elementare ciascuno degli imballaggi che costituiscono il campione elementare.
Tabella 2
Numero minimo di incrementi da selezionare
|
Numero di imballaggi del lotto |
Numero minimo di incrementi da selezionare |
|
Fino a 10 |
1 |
|
Da …. 11 a 150 |
2 |
|
Da …. 151 a 500 |
3 |
|
Da …. 501 a 1 500 |
4 |
|
Da …. 1 501 a 2 500 |
5 |
|
> 2 500 per 1 000 imballaggi |
1 incremento supplementare |
Per ridurre il volume degli imballaggi immediati da sottoporre a campionamento, il contenuto degli incrementi è omogeneizzato ai fini della preparazione del campione elementare. Le porzioni dei diversi incrementi sono versate in un contenitore comune per essere omogeneizzati mediante agitazione, in modo da essere protetti al meglio dall'aria.
Il contenuto del campione elementare deve essere versato in una serie di imballaggi della capacità minima di 1,0 litro, ciascuno dei quali costituisce un esemplare del campione elementare.
Il numero di campioni elementari può essere aumentato dai singoli Stati membri, secondo le rispettive esigenze (ad esempio, valutazione organolettica da parte di un laboratorio diverso da quello che ha eseguito le analisi chimiche, controanalisi ecc.).
Ciascun imballaggio deve essere riempito in modo tale ridurre il più possibile lo strato d'aria sovrastante e quindi idoneamente chiuso e sigillato per garantire che il prodotto non possa essere manomesso.
Gli esemplari in questione devono essere etichettati per identificarli con precisione.
2. ANALISI E RISULTATI
2.1. Ciascun campione elementare deve essere suddiviso in campioni di laboratorio, conformemente al punto 2.5 della norma EN ISO 5555, e sottoposto alle analisi nell'ordine indicato nello schema decisionale di cui all'allegato I ter o in qualunque altro ordine casuale.
2.2. Qualora tutti i risultati delle analisi siano conformi alle caratteristiche della categoria di olio dichiarata, l'intera partita in questione è dichiarata conforme.
Qualora anche uno solo dei risultati non sia conforme alle caratteristiche della categoria di olio dichiarata, l'intera partita in questione è dichiarata non conforme.
3. VERIFICA DELLA CATEGORIA DELLA PARTITA
3.1. Per verificare la categoria della partita, l'autorità competente può aumentare il numero di campioni elementari effettuati in punti diversi della partita secondo la tabella seguente:
Tabella 3
Numero di campioni elementari determinati dalle dimensioni della partita
|
Dimensione della partita (litri) |
Numero di campioni elementari |
|
Meno di 7 500 |
2 |
|
Da 7 500 a meno di 25 000 |
3 |
|
Da 25 000 a meno di 75 000 |
4 |
|
Da 75 000 a meno di 125 000 |
5 |
|
125 000 o più |
6 + 1 ogni 50 000 litri supplementari |
Ciascun incremento che costituisce un campione elementare deve essere prelevato da punti contigui della partita; è necessario annotare l'ubicazione di ciascun campione elementare e identificarlo inequivocabilmente.
La costituzione di ciascun campione elementare deve rispettare le procedure di cui ai punti 1.1 e 1.2.
Ciascun campione elementare è quindi sottoposto alle analisi di cui all'articolo 2, paragrafo 1.
3.2. Qualora, per almeno uno dei campioni elementari, uno dei risultati delle analisi di cui all'articolo 2, paragrafo 1, non sia conforme alle caratteristiche della categoria di olio dichiarata, l'intera partita è dichiarata non conforme.
ALLEGATO I ter
SCHEMA DECISIONALE PER LA VERIFICA DELLA CONFORMITÀ DI UN CAMPIONE DI OLIO DI OLIVA ALLA CATEGORIA DICHIARATA
Tabella 1

Tabella 2

Tabella 3
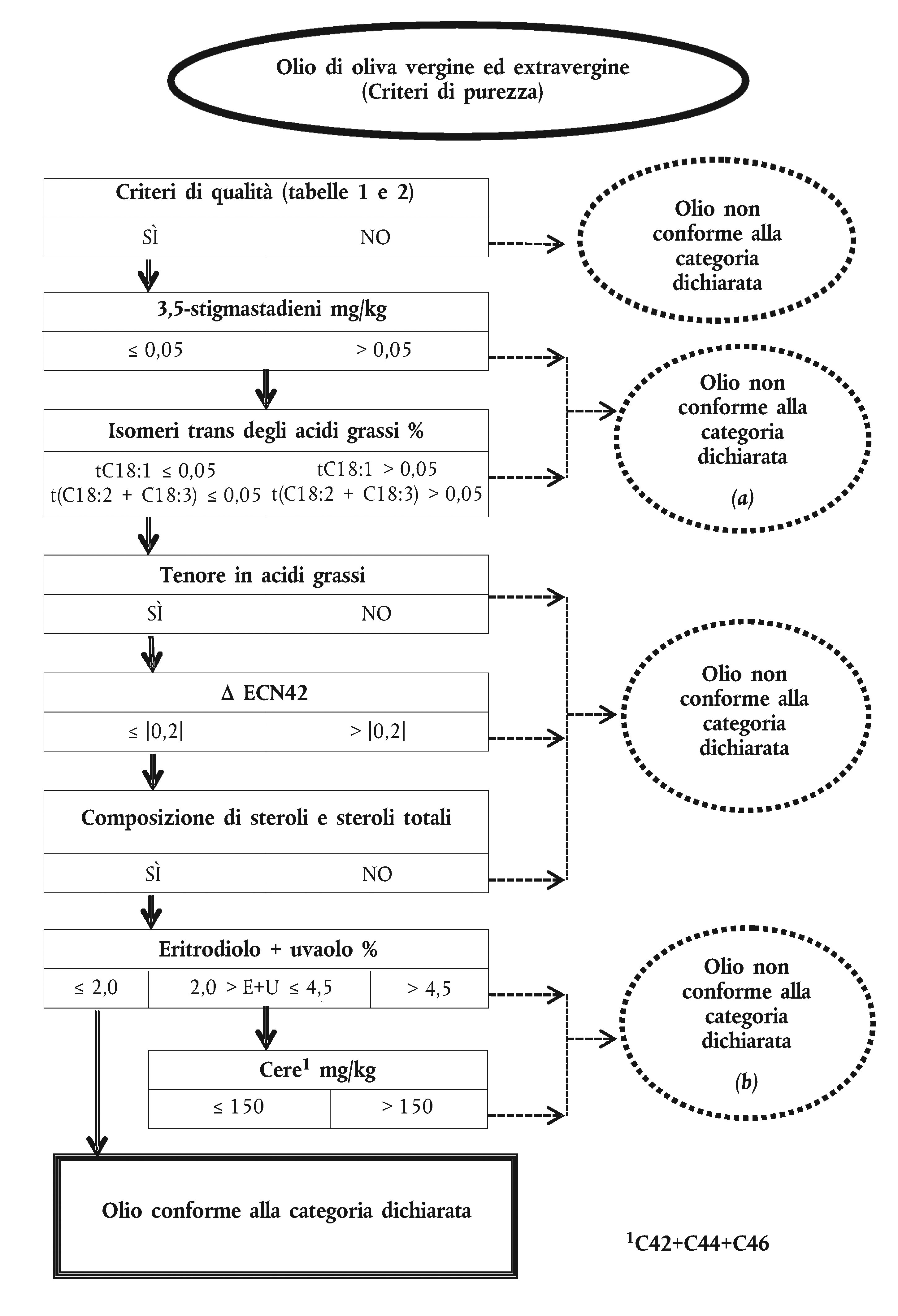
Appendice 1
Corrispondenza tra gli allegati del presente regolamento e le analisi previste nello schema decisionale
|
— Acidità |
Allegato II |
Determinazione degli acidi grassi liberi, metodo a freddo |
|
— Indice di perossidi |
Allegato III |
Determinazione del numero di perossidi |
|
— Spettrometria a raggi ultravioletti |
Allegato IX |
Analisi spettrofotometrica |
|
— Valutazione organolettica |
Allegato XII |
Valutazione organolettica degli oli di oliva vergini |
|
— Etil esteri |
Allegato XX |
Metodo per la determinazione del contenuto di cere e metil ed etil esteri degli acidi grassi mediante gascromatografia con colonna capillare |
|
— Stigmasta-3,5-diene |
Allegato XVII |
Metodo di determinazione degli stigmastadieni negli oli vegetali |
|
— Isomeri trans degli acidi grassi |
Allegato X |
Determinazione degli esteri metilici degli acidi grassi mediante gascromatografia |
|
— Composizione di acidi grassi |
Allegato X |
Determinazione degli esteri metilici degli acidi grassi mediante gascromatografia |
|
— ΔECN42 |
Allegato XVIII |
Determinazione dei trigliceridi con ECN42 (differenze fra i dati HPLC e il contenuto teorico) |
|
— Composizione di steroli e steroli totali — Eritrodiolo e uvaolo |
Allegato V |
Determinazione della composizione e del contenuto di steroli e dialcoli triterpenici mediante gascromatografia con colonna capillare |
|
— Cere |
Allegato IV |
Determinazione del contenuto di cere mediante gascromatografia con colonna capillare |
|
— Alcoli alifatici e triterpenici |
Allegato XIX |
Determinazione del contenuto di alcoli alifatici e triterpenici mediante gascromatografia con colonna capillare |
|
— Acidi grassi saturi in posizione 2 |
Allegato VII |
Determinazione della percentuale di 2- gliceril monopalmitato |
ALLEGATO II
DETERMINAZIONE DEGLI ACIDI GRASSI LIBERI, METODO A FREDDO
1. OGGETTO E CAMPO D'APPLICAZIONE
Il presente metodo descrive la determinazione degli acidi grassi liberi negli oli d'oliva e negli oli di sansa d'oliva. Il tenore in acidi grassi liberi viene espresso mediante l'acidità calcolata come percentuale di acido oleico.
2. PRINCIPIO
Dissoluzione di una aliquota della sostanza da analizzare in una miscela di solventi, poi titolazione degli acidi grassi liberi presenti mediante una soluzione di idrossido di potassio o idrossido di sodio.
3. REAGENTI
Tutti i reagenti devono essere di qualità analitica riconosciuta; l'acqua impiegata dev'essere acqua distillata o di purezza equivalente.
|
3.1 |
Etere etilico; etanolo al 95 % (V/V), miscela 1 — 1 in volume. Neutralizzare esattamente al momento dell'impiego con la soluzione di idrossido di potassio (3.2) in presenza di 0,3 ml della soluzione di fenolftaleina (3.3) per 100 ml di miscela. Nota 1: l'etere etilico è molto infiammabile e può formare perossidi esplosivi. Esso deve pertanto essere usato con precauzioni particolari. Nota 2: se non è possibile usare l'etere etilico, si può ricorrere a una miscela di solventi costituita da etanolo e da toluene. Se necessario, l'etanolo può essere sostituito dal 2-propanolo. |
|
3.2 |
Idrossido di potassio o idrossido di sodio, soluzione etanolica o acquosa titolata c(KOH) o [c(NaOH)] all'incirca 0,1 mol/l oppure, se necessario, c(KOH) [o c(NaOH)] 0,5 mol/l circa. Esistono in commercio soluzioni pronte per l'uso. La concentrazione esatta della soluzione di idrossido di potassio (o di idrossido di sodio) deve essere nota e verificata prima dell'uso. Impiegare una soluzione preparata almeno 5 giorni prima dell'uso e decantata in un flacone di vetro bruno chiuso con un tappo di gomma. La soluzione deve essere incolore o giallo pallida. Qualora si riscontri una separazione delle fasi utilizzando una soluzione acquosa di idrossido di potassio (o di idrossido di sodio), questa dev'essere sostituita con una soluzione etanolica. Nota 3: una soluzione incolore stabile di idrossido di potassio (o di idrossido di sodio) può essere preparata come segue. Portare e mantenere per un'ora all'ebollizione a ricadere 1 000 ml di etanolo o acqua con 8 g di idrossido di potassio (o di idrossido di sodio) e 0,5 g di trucioli di alluminio. Distillare immediatamente. Sciogliere nel distillato il quantitativo necessario di idrossido di potassio (o di idrossido di sodio). Lasciar riposare per parecchi giorni e decantare il liquido chiaro sopranatante del precipitato di carbonato di potassio (o di carbonato di sodio). La soluzione può essere preparata altresì senza distillazione, come segue. A 1 000 ml di etanolo (o acqua) aggiungere 4 ml di butilato di alluminio e lasciar riposare la miscela per qualche giorno. Decantare il liquido sopranatante e sciogliervi il quantitativo necessario di idrossido di potassio (o di idrossido di sodio). Questa soluzione è pronta per l'uso. |
|
3.3 |
Fenolftaleina, soluzione di 10 g/l in etanolo al 95-96 % (V/V) o blu alcalino 6B o timolftaleina, soluzione di 20 g/l in etanolo al 95-96 % (V/V). Nel caso di oli fortemente colorati, dev'essere utilizzato il blu alcalino o la timolftaleina. |
4. APPARECCHIATURA
Materiale corrente da laboratorio, in particolare:
4.1 bilancia analitica;
4.2 beuta, avente una capacità di 250 ml;
4.3 buretta da 10 ml, classe A, graduata in divisioni di 0,05 ml, o buretta automatica equivalente.
5. PROCEDIMENTO
5.1 Preparazione del campione per l'analisi
Quando il campione è torbido, occorre filtrarlo.
5.2 Sostanza da analizzare
Prelevare un'aliquota della sostanza da analizzare, a seconda dell'acidità presunta, secondo le indicazioni della seguente tabella.
|
Acidità prevista (acidità in acido oleico g/100 g) |
Massa della sostanza da analizzare (g) |
Precisione della pesata della sostanza da analizzare (g) |
|
da 0 a 2 |
10 |
0,02 |
|
da > 2 a 7,5 |
2,5 |
0,01 |
|
> 7,5 |
0,5 |
0,001 |
Pesare la sostanza da analizzare nella beuta (4.2).
5.3 Determinazione
Sciogliere l'aliquota di sostanza da analizzare (5.2) in 50-100 ml della miscela etere etilico/etanolo (3.1) precedentemente neutralizzata.
Titolare, agitando, con la soluzione di idrossido di potassio (o di idrossido di sodio) di 0,1 mol/l (3.2) (cfr. nota 4) fino a viraggio dell'indicatore (colorazione dell'indicatore colorato persistente per almeno 10 s).
Nota 4: se il quantitativo necessario di soluzione di idrossido di potassio (o di idrossido di sodio) di 0,1 mol/l supera 10 ml, usare una soluzione di 0,5 mol/l o modificare la massa della sostanza da analizzare a seconda dell'acidità libera prevista e alla tabella proposta.
Nota 5: se la soluzione diventa torbida durante la titolazione, aggiungere un quantitativo sufficiente della miscela di solventi (3.1) per ottenere una soluzione chiara.
Effettuare una seconda determinazione soltanto nel caso in cui il primo risultato sia superiore al limite specificato per la categoria dell'olio in questione.
6. ESPRESSIONE DEI RISULTATI
L'acidità, espressa come percentuale di acido oleico, è pari a:
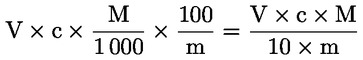
dove:
|
V |
è il volume, in millilitri, della soluzione titolata di idrossido di potassio (o di idrossido di sodio) usata; |
|
c |
è la concentrazione esatta, in moli per litro, della soluzione titolata di idrossido di potassio (o di idrossido di sodio) usata; |
|
M |
è pari a 282 g/mol, la massa molare in g per mole di acido oleico; |
|
m |
è la massa in g del campione. |
L'acidità oleica è indicata come segue:
a) con due cifre decimali per i valori da 0 fino a 1 compreso;
b) con una cifra decimale per i valori da 1 fino a 100 compreso.
ALLEGATO III
DETERMINAZIONE DEL NUMERO DI PEROSSIDI
1. OGGETTO
Si tratta di un metodo per la determinazione del numero di perossidi in oli e grassi.
2. CAMPO D'APPLICAZIONE
Oli e grassi animali e vegetali.
3. DEFINIZIONE
Il numero di perossidi è il quantitativo delle sostanze presenti nel campione, espresse in milliequivalenti di ossigeno attivo per kg, che ossidano lo ioduro di potassio nelle condizioni che vengono descritte.
4. PRINCIPIO
Trattamento della sostanza in esame, sciolta in acido acetico e cloroformio, con una soluzione di ioduro di potassio. Titolazione dello iodio liberato con soluzione di tiosolfato di sodio standardizzata.
5. APPARECCHIATURA
Tutta l'apparecchiatura usata dev'essere esente da sostanze riducenti od ossidanti.
Nota:
Non ungere le superfici smerigliate.
|
5.1. |
Ditale di vetro da 3 ml. |
|
5.2. |
►C2 Palloni a collo e tappo smerigliato ◄ , aventi una capacità di circa 250 ml, previamente asciugati e riempiti di gas puro, secco inerte (azoto o, di preferenza, anidride carbonica). |
|
5.3. |
Buretta da 25 o 50 ml, graduata in 0,1 ml. |
6. REAGENTI
|
6.1. |
Cloroformio, di qualità per reagente analitico, liberato dall'ossigeno facendovi gorgogliare una corrente di gas inerte puro e secco. |
|
6.2. |
Acido acetico glaciale, di qualità per analisi, liberato dall'ossigeno facendovi gorgogliare una corrente di gas puro e secco. |
|
6.3. |
Ioduro di potassio, soluzione acquosa satura, di recente preparazione, esente da iodio e da iodati. |
|
6.4. |
Tiosolfato di sodio, 0,01 o 0,02 N, soluzione acquosa accuratamente standardizzata immediatamente prima dell'uso. |
|
6.5. |
Soluzione di amido, dispersione acquosa di 10 g/l, di recente preparazione da amido naturale solubile. |
7. CAMPIONE
Prelevare il campione e conservarlo al riparo dalla luce, tenendolo al fresco e mettendolo in contenitori di vetro completamente riempiti, sigillati ermeticamente con tappi a smeriglio o di sughero.
8. PROCEDIMENTO
La prova dev'essere effettuata alla luce del giorno diffusa oppure alla luce artificiale. Pesare in un ditale di vetro (5.1) oppure, in mancanza, in un pallone (5.2) con l'approssimazione di 0,001 g, una massa del campione conformemente alla seguente tabella e al numero di perossidi previsto:
|
Numero di perossidi previsto (meq) |
Peso della sostanza da analizzare (in g) |
|
0 — 12 |
5,0 — 2,0 |
|
12 — 20 |
2,0 — 1,2 |
|
20 — 30 |
1,2 — 0,8 |
|
30 — 50 |
0,8 — 0,5 |
|
50 — 90 |
0,5 — 0,3 |
Stappare un pallone (5.2) ed introdurre il ditale di vetro contenente la sostanza da analizzare. Aggiungere 10 ml di cloroformio (6.1). Sciogliere la sostanza da analizzare rapidamente, agitando. Aggiungere 15 ml di acido acetico (6.2), quindi 1 ml di soluzione di ioduro di potassio (6.3). Ritappare rapidamente, agitare per 1 minuto e lasciar riposare per 5 minuti esatti al riparo dalla luce, ad una temperatura compresa tra 15 e 25 °C.
Aggiungere circa 75 ml di acqua distillata. Titolare lo iodio liberato con una soluzione di tiosolfato di sodio (6.4) (soluzione 0,002 N per valori previsti inferiori a 12 e soluzione 0,01 N per valori previsti superiori a 12) agitando vigorosamente, usando la soluzione di amido (6.5) come indicatore.
Eseguire due determinazioni sullo stesso campione di sostanza.
Eseguire contemporaneamente una prova in bianco. Se il risultato del bianco supera 0,05 ml di soluzione 0,01 N di tiosolfato di sodio (6.4), sostituire i reagenti impuri.
9. ESPRESSIONE DEI RISULTATI
Il numero di perossidi (P.V.), espresso in milliequivalenti di ossigeno attivo per kg, viene dato dalla formula:

dove:
V = è il numero di ml della soluzione standardizzata di tiosolfato di sodio (6.4) usata per la prova, corretto in modo da tener conto della prova in bianco.
T = è la normalità esatta della soluzione di tiosolfato di sodio (6.4) usata.
m = è il peso in g della sostanza da analizzare.
Considerare come risultato la media aritmetica delle due determinazioni eseguite.
ALLEGATO IV
DETERMINAZIONE DEL CONTENUTO DI CERE MEDIANTE GASCROMATOGRAFIA CON COLONNA CAPILLARE
1. OGGETTO
Il metodo descrive un procedimento per la determinazione del contenuto di cere negli oli d’oliva. Le cere sono separate in funzione del numero di atomi di carbonio. Il metodo può essere impiegato in particolare per differenziare l’olio d’oliva di pressione da quello di estrazione (olio di sansa).
2. PRINCIPIO
La sostanza grassa, addizionata di opportuno standard interno, viene frazionata mediante cromatografia su colonna di gel di silice idratato; la frazione eluita per prima nelle condizioni di prova (avente polarità inferiore a quella dei trigliceridi) viene recuperata e quindi analizzata direttamente mediante gascromatografia in colonna capillare.
3. APPARECCHIATURA
|
3.1. |
Beuta da 25 ml. |
|
3.2. |
Colonna in vetro per gascromatografia con diametro interno 15,0 mm e altezza 30-40 cm, provvista di rubinetto. |
|
3.3. |
Gascromatografo idoneo per il funzionamento con colonna capillare, dotato di sistema di introduzione diretta in colonna costituito da:
|
|
3.4. |
Microsiringa per introduzione diretta in colonna da 10 μl con ago cementato. |
|
3.5. |
Vibratore elettrico. |
|
3.6. |
Evaporatore rotante. |
|
3.7. |
Forno a muffola. |
|
3.8. |
Bilancia analitica in grado di garantire una precisione di misura di + 0,1 mg. |
|
3.9. |
Normale vetreria da laboratorio. |
4. REAGENTI
|
4.1. |
Gel di silice di granulometria compresa tra 60 e 200 μm. Il gel di silice è posto in muffola a 500 °C per almeno 4 ore. Dopo raffreddamento è addizionato del 2 % di acqua rispetto alla quantità di gel di silice prelevata. Si agita bene allo scopo di omogeneizzare la massa. Si conserva al buio per almeno 12 ore prima dell’uso. |
|
4.2. |
n-esano per cromatografia. |
|
4.3. |
Etere etilico per cromatografia. |
|
4.4. |
n-eptano per cromatografia. |
|
4.5. |
Soluzione campione di lauril arachidato, allo 0,1 % (v/m) in esano (standard interno). (Si può utilizzare anche palmitil palmitato o miristil stearato).
|
|
4.6. |
Gas vettore: idrogeno o elio puro per gascromatografia. |
|
4.7. |
Gas ausiliari: — idrogeno puro per gascromatografia, — aria pura per gascromatografia. |
5. PROCEDIMENTO
5.1. Preparazione della colonna cromatografica
Mettere in sospensione 15 g di gel di silice (4.1) in n-esano (4.2) e introdurla in colonna (3.2). Dopo assestamento spontaneo, completare lo stesso con l’ausilio di un vibratore elettrico per rendere più omogeneo il letto cromatografico. Percolare 30 ml di n-esano allo scopo di allontanare le eventuali impurezze. Pesare esattamente, nella beuta da 25 ml (3.1), circa 500 mg di campione con la bilancia (3.8), addizionare l’opportuna quantità di standard interno (4.5) in funzione del presunto contenuto di cere. Ad esempio aggiungere 0,1 mg di lauril arachidato nel caso di olio di oliva, e da 0,25 a 0,50 mg nel caso di olio di sansa. Immettere il campione così preparato nella colonna cromatografica con l’ausilio di due porzioni da 2 ml ciascuna di n-esano (4.2).
Lasciar fluire il solvente fino a 1 mm sopra l’assorbente, quindi percolare altri 70 ml di n-esano allo scopo di eliminare gli n-alcani naturalmente presenti. Iniziare quindi l’eluizione cromatografica raccogliendo 180 ml di miscela n-esano/etere etilico in rapporto 99:1, rispettando un flusso di circa 15 gocce ogni 10 secondi. L’eluizione del campione deve essere effettuata a una temperatura ambiente di 22 °C ± 4.
NB:
— La miscela n-esano/etere etilico (99:1) deve essere preparata ogni giorno.
— Per controllare visivamente la corretta eluizione delle cere, è possibile aggiungere al campione in soluzione 100 μl di sudan 1 all’1 % nella miscela di eluizione. Il colorante ha una ritenzione intermedia tra le cere e i trigliceridi, pertanto quando la colorazione raggiunge il fondo della colonna cromatografica bisogna sospendere l’eluizione, in quanto tutte le cere sono state eluite.
Essiccare la frazione così ottenuta mediante evaporatore rotante (3.6) fino ad eliminazione quasi completa del solvente. Eliminare gli ultimi 2 ml di solvente mediante un debole flusso di azoto; aggiungere quindi 2-4 ml di n-eptano.
5.2. Analisi gascromatografica
5.2.1. Operazioni preliminari
Installare la colonna nel gascromatografo (3.3), collegando il terminale di ingresso al sistema «on column» e il terminale di uscita al rivelatore. Eseguire i controlli generali del complesso gascromatografico (tenuta dei circuiti dei gas, efficienza del rivelatore e del sistema di registrazione, ecc.).
Se la colonna è messa in uso per la prima volta è consigliabile procedere al suo condizionamento. Far fluire un leggero flusso di gas attraverso la colonna, quindi avviare il complesso gascromatografico. Riscaldare gradualmente fino a raggiungere, dopo circa 4 ore, la temperatura di 350 °C. Mantenere tale temperatura per almeno 2 ore, quindi portare il complesso alle condizioni di funzionamento (regolazione del flusso dei gas, accensione della fiamma, collegamento con il registratore elettronico (3.3.4), regolazione della temperatura della camera per colonna, del rivelatore, ecc.) e registrare il segnale ad una sensibilità almeno due volte superiore a quella prevista per l’esecuzione dell’analisi. Il tracciato della linea di base deve risultare lineare, esente da picchi di qualsiasi natura, e non deve presentare deriva.
Una deriva rettilinea negativa indica imperfetta tenuta delle connessioni della colonna, una deriva positiva indica un insufficiente condizionamento della colonna.
5.2.2. Scelta delle condizioni operative
Le condizioni operative di massima sono le seguenti:
— temperatura della colonna:
—
|
|
20 °C/min |
|
5 °C/min |
|
20 °C/min |
|
|
inizio a 80 °C (1′) |
→ |
240 °C |
→ |
325 °C (6′) |
→ |
340 °C (10′) |
— temperatura del rivelatore: 350 °C,
— quantità di sostanza iniettata: 1 μl della soluzione (2-4 ml) di n-eptano,
— gas vettore: elio o idrogeno alla velocità lineare ottimale per il gas prescelto (cfr. appendice),
— sensibilità strumentale: idonea a soddisfare le sottostanti condizioni.
Tali condizioni possono essere variate in funzione delle caratteristiche della colonna e del gascromatografo in modo da avere una separazione di tutte le cere, una risoluzione soddisfacente dei picchi (cfr. figura) e una ritenzione dello standard interno C32 di 18 ± 3 minuti. Il picco delle cere più rappresentativo deve avere un’altezza superiore al 60 % del fondo scala.
I parametri di integrazione dei picchi devono essere impostati in modo da ottenere una corretta valutazione delle aree dei picchi che vengono presi in considerazione.
NB:Vista l’elevata temperatura finale, è ammessa una deriva positiva che non deve superare il 10 % del fondo scala.
5.3. Esecuzione dell’analisi
Prelevare 1 μl di soluzione con la microsiringa da 10 μl; estrarre lo stantuffo della siringa in modo che l’ago resti vuoto. Introdurre l’ago attraverso il dispositivo di iniezione e, dopo 1-2 secondi, iniettare rapidamente; estrarre quindi lentamente l’ago dopo circa 5 secondi.
Effettuare la registrazione fino a completa eluizione delle cere.
La linea di base deve rispondere sempre ai requisiti richiesti.
5.4. Identificazione dei picchi
L’identificazione dei singoli picchi viene effettuata in base ai tempi di ritenzione e in confronto a miscele di cere a tempi di ritenzione noti, analizzate nelle medesime condizioni.
Nella figura è riportato un cromatogramma delle cere di un olio di oliva vergine.
5.5. Valutazione quantitativa
Procedere al calcolo delle aree dei picchi dello standard interno e degli esteri alifatici da C40 a C46 per mezzo dell’integratore.
Calcolare il contenuto di cere in ogni singolo estere, in mg/kg di sostanza grassa, secondo la formula seguente:

in cui:
|
Ax |
= |
area del picco del singolo estere, in millimetri quadrati |
|
As |
= |
area del picco dello standard interno, in millimetri quadrati |
|
ms |
= |
massa di standard interno aggiunta, in milligrammi |
|
m |
= |
massa di campione prelevato per la determinazione, in grammi. |
6. ESPRESSIONE DI RISULTATI
Riportare la somma dei contenuti delle singole cere da C40 a C46 in mg/kg di sostanza grassa (ppm).
NB:I componenti da quantificare si riferiscono ai picchi a numero di carbonio pari compresi tra gli esteri C40 e C46, secondo l’esempio di cromatogramma delle cere dell’olio di oliva riportato nella figura seguente. Se l’estere C46 risulta sdoppiato, si consiglia, ai fini della sua identificazione, di analizzare la frazione cerosa di un olio di sansa dove il picco C46 risulta facilmente individuabile in quanto nettamente maggioritario.
I risultati si esprimono con due cifre decimali.
Figura
Cromatogramma delle cere di un olio d’oliva ( 3 )
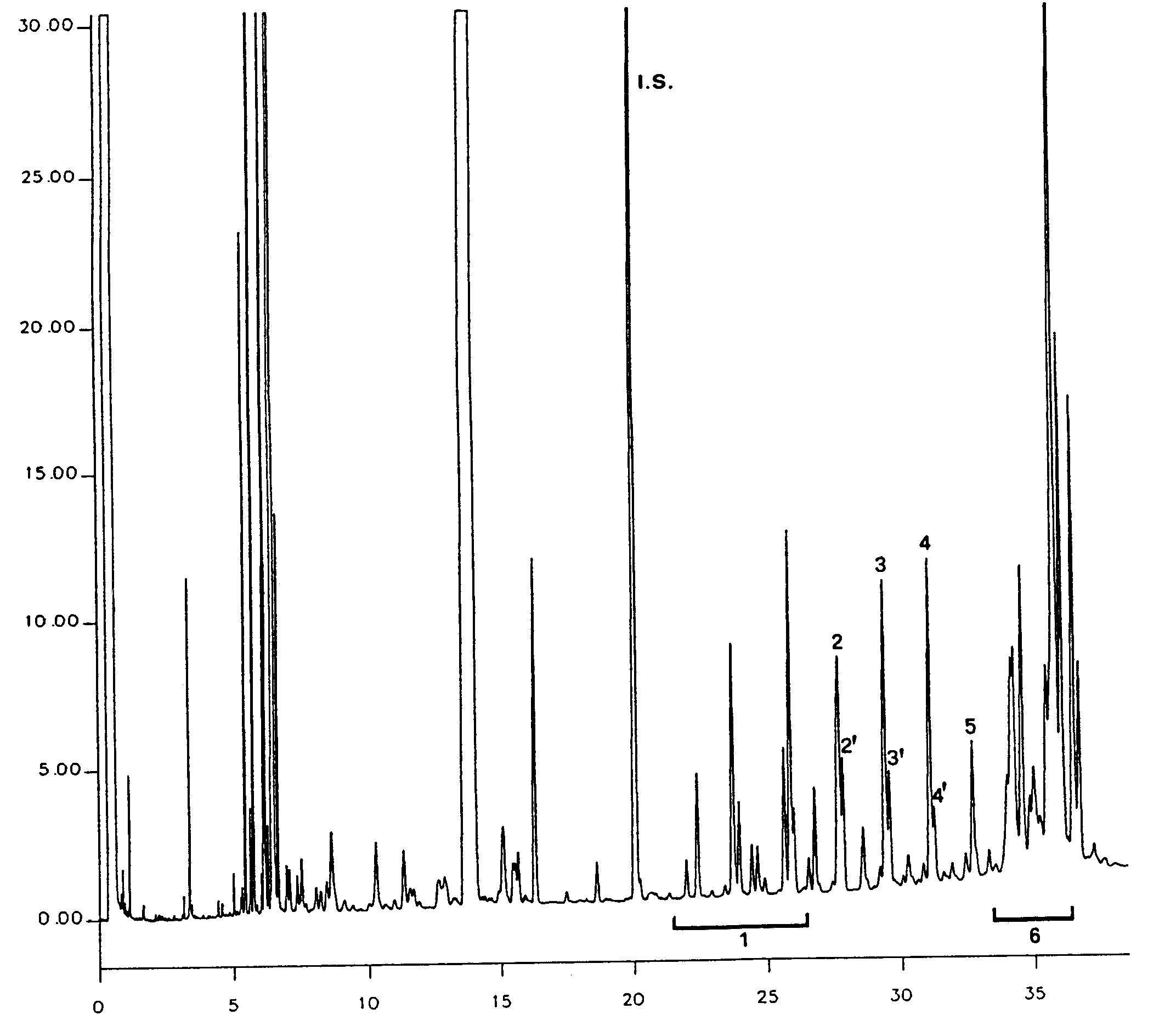
Legenda:
|
I.S. |
= |
Lauril arachidato |
|
1. |
= |
Esteri diterpenici |
|
2 + 2′ |
= |
Esteri C40 |
|
3 + 3′ |
= |
Esteri C42 |
|
4 + 4′ |
= |
Esteri C44 |
|
5. |
= |
Esteri C46 |
|
6. |
= |
Esteri steroli e alcoli triterpenici. |
Appendice
Determinazione della velocità lineare del gas
Iniettare nel gascromatografo, regolato alle normali condizioni operative, da 1 a 3 μl di metano (o propano). Cronometrare il tempo che il gas impiega a percorrere la colonna, dal momento dell’iniezione al momento dell’uscita del picco (tM).
La velocità lineare in cm/s è data da L/tM in cui L è la lunghezza della colonna in centimetri e tM è il tempo cronometrato in secondi.
ALLEGATO V
DETERMINAZIONE DELLA COMPOSIZIONE E DEL CONTENUTO DI STEROLI E DIALCOLI TRITERPENICI MEDIANTE GASCROMATOGRAFIA CON COLONNA CAPILLARE
1. OGGETTO
Il metodo descrive un procedimento per la determinazione del contenuto di steroli e dialcoli triterpenici, singoli e totali, degli oli di oliva e degli oli di sansa di oliva.
2. PRINCIPIO
L'olio, addizionato di α-colestanolo quale standard interno, è saponificato con idrossido di potassio in soluzione etanolica, quindi l'insaponificabile viene estratto con etere etilico.
La frazione costituita da steroli e dialcoli triterpenici è separata dall'insaponificabile mediante cromatografia su strato sottile su una placca di gel di silice basica. Le frazioni recuperate dal gel di silice vengono trasformate in trimetilsilileteri e quindi analizzate mediante gascromatografia in colonna capillare.
3. APPARECCHIATURA
La normale apparecchiatura di laboratorio e in particolare quanto segue.
3.1. Matraccio da 250 ml, munito di refrigerante a ricadere con giunti a smeriglio.
3.2. Imbuto separatore da 500 ml.
3.3. Matracci da 250 ml.
3.4. Attrezzatura completa per analisi cromatografica su strato sottile, per lastre di vetro 20 × 20 cm.
3.5. Lampada a luce ultravioletta, con lunghezza d'onda 254 o 366 nm.
3.6. Microsiringhe da 100 μl e 500 μl.
3.7. Imbuto cilindrico filtrante a setto poroso G 3 (porosità 15-40 μm) di diametro circa 2 cm e altezza circa 5 cm, idoneo per filtrazione sotto vuoto con giunto smerigliato maschio.
3.8. Beuta per vuoto da 50 ml con giunto femmina smerigliato, adattabile all'imbuto filtrante (punto 3.7.).
3.9. Provetta da 10 ml a fondo conico con tappo di vetro a tenuta.
3.10. Gascromatografo idoneo per il funzionamento con colonna capillare, dotato di dispositivo di iniezione di tipo split, costituito da:
3.10.1. camera termostatica per le colonne, in grado di mantenere la temperatura desiderata con la precisione di ± 1 °C;
3.10.2. complesso di iniezione termoregolabile con elemento vaporizzatore in vetro persilanizzato e sistema split;
3.10.3. rivelatore a ionizzazione di fiamma;
3.10.4. sistema di acquisizione di dati idoneo per il funzionamento con il rivelatore a ionizzazione di fiamma (punto 3.10.3.), integrabile manualmente.
3.11. Colonna capillare in silice fusa, lunga 20 - 30 m, diametro interno 0,25 - 0,32 mm, internamente ricoperta di una miscela costituita da 5% difenil - 95% dimetilpolisilossano (fase stazionaria SE-52 o SE-54 o equivalenti), fino a uno spessore uniforme compreso fra 0,10 e 0,30 μm.
3.12. Microsiringa per gascromatografia da 10 μl con ago cementato idonea all'iniezione split.
3.13. Essiccatore a cloruro di calcio.
4. REAGENTI
4.1. Idrossido di potassio, titolo minimo 85%.
4.2. Idrossido di potassio, soluzione etanolica circa 2 N:
si sciolgono, sotto raffreddamento, 130 g di idrossido di potassio (punto 4.1) in 200 ml di acqua distillata, quindi si porta ad 1 litro con etanolo (punto 4.10). Si conserva la soluzione in bottiglie di vetro scuro ben tappate, per un massimo di due giorni.
4.3. Etere etilico, puro per analisi.
4.4. Idrossido di potassio, soluzione etanolica circa 0,2 N:
si sciolgono 13 g di idrossido di potassio (punto 4.1) in 20 ml di acqua distillata e si porta a 1 litro con etanolo (punto 4.10).
4.5. Sodio solfato anidro, puro per analisi.
4.6. Lastre di vetro (20x20 cm) stratificate con gel di silice, senza indicatore di fluorescenza, spessore 0,25 mm (sono reperibili in commercio già pronte per l'uso).
4.7. Toluene, per cromatografia.
4.8. Acetone, per cromatografia.
4.9. n-esano, per cromatografia.
4.10. Etere etilico, per cromatografia.
4.11. Etanolo per analisi.
4.12. Acetato di etile per analisi.
4.13. Soluzione di riferimento per la cromatografia su strato sottile: colesterolo o fitosteroli, e soluzione di eritrodiolo al 5% in acetato di etile (punto 4.11).
4.14. 2,7-diclorofluoresceina, soluzione etanolica allo 0,2%. Si rende leggermente basica aggiungendo qualche goccia di soluzione alcolica 2 N di idrossido di potassio (punto 4.2).
4.15. Piridina anidra, per cromatografia (v. Nota 5).
4.16. Esametildisilazano per analisi.
4.17. Trimetilclorosilano per analisi.
4.18. Soluzioni campione di trimetilsilileteri degli steroli:
si preparano al momento dell'impiego partendo da steroli ed eritrodiolo ottenuti da oli che li contengano.
4.19. α-colestanolo, puro ad oltre il 99% (la purezza deve essere verificata mediante analisi gascromatografica).
4.20. α-colestanolo, soluzione di standard interno allo 0,2% (m/V) in acetato di etile (punto 4.11).
4.21. Fenolftaleina, soluzione di 10 g/l in etanolo (punto 4.10).
4.22. Gas vettore: idrogeno o elio, puri per gascromatografia.
4.23. Gas ausiliari: idrogeno, elio, azoto e aria, puri per gascromatografia.
4.24. Miscela n-esano (punto 4.9)/etere etilico (punto 4.10) in rapporto 65:35 (V/V).
4.25. Reagente per la sililazione costituito da una miscela 9:3:1 (V/V/V) di piridina-esametildisilazano- trimetilclorosilano.
5. PROCEDIMENTO
|
5.1. |
Preparazione dell'insaponificabile.
|
|
5.2. |
Separazione della frazione costituita da steroli e dialcoli triterpenici (eritrodiolo + uvaolo)
|
|
5.3. |
Preparazione dei trimetilsilileteri.
|
|
5.4. |
Analisi gascromatografica.
|

6. ESPRESSIONE DEI RISULTATI
6.1. Si riportano le concentrazioni dei singoli steroli, in mg/kg di sostanza grassa e, come steroli totali, la loro somma.
Il contenuto di ogni singolo sterolo, dell'eritrodiolo e dell'uvaolo deve essere espresso con una cifra decimale.
Il contenuto totale di steroli deve essere espresso senza decimali.
6.2. Si calcola il contenuto percentuale di ogni singolo sterolo dal rapporto fra l'area del picco corrispondente e la somma delle aree dei picchi degli steroli:

in cui:
|
Ax |
= |
area del picco x; |
|
ΣA |
= |
somma delle aree di tutti i picchi degli steroli. |
6.3. β-sitosterolo apparente: Δ5-23-stigmastadienolo + clerosterolo + β-sitosterolo + sitostanolo + Δ5-avenasterolo + Δ5-24-stigmastadienolo.
6.4 Si calcola il contenuto percentuale di eritrodiolo e uvaolo:

in cui:
|
ΣA |
= |
somma delle aree degli steroli in unità di calcolo dell'integratore; |
|
Er |
= |
area dell'eritrodiolo in unità di calcolo dell'integratore; |
|
Uv |
= |
area dell'uvaolo in unità di calcolo dell'integratore. |
Appendice
Determinazione della velocità lineare del gas
Nel gascromatografo, regolato alle normali condizioni operative, si iniettano 1 - 3 μl di metano (o propano) e si cronometra il tempo che il gas impiega a percorrere la colonna, dal momento dell'iniezione al momento della comparsa del picco (tM).
La velocità lineare in cm/s è data da L/tM, in cui L è la lunghezza della colonna in centimetri e tM è il tempo cronometrato in secondi.
Tabella 1
Tempi di ritenzione relativi degli steroli
|
Picco |
Identificazione |
Tempo di ritenzione relativo |
||
|
Colonna SE 54 |
Colonna SE 52 |
|||
|
1 |
Colesterolo |
Δ-5-colesten-3ß-olo |
0,67 |
0,63 |
|
2 |
Colestanolo |
5α-colestan-3ß-olo |
0,68 |
0,64 |
|
3 |
Brassicasterolo |
[24S]-24-metil-Δ-5,22-colestadien-3ß-olo |
0,73 |
0,71 |
|
* |
Ergosterolo |
[24S] 24 metil Δ5-7-22 colestatrien 3ß-olo |
0,78 |
0,76 |
|
4 |
24-metilencolesterolo |
24-metilen-Δ-5,24-colestadien-3ß-o1o |
0,82 |
0,80 |
|
5 |
Campesterolo |
(24R)-24-metil-Δ-5-colesten-3ß-olo |
0,83 |
0,81 |
|
6 |
Campestanolo |
(24R)-24-metil-colestan-3ß-olo |
0,85 |
0,82 |
|
7 |
Stigmasterolo |
(24S)-24-etil-Δ-5,22-colestadien-3ß-olo |
0,88 |
0,87 |
|
8 |
Δ-7-campesterolo |
(24R)-24-metil-Δ-7-colesten-3ß-olo |
0,93 |
0,92 |
|
9 |
Δ-5,23-stigmastadienolo |
(24R,S)-24-etil-Δ-5,23-colestadien-3ß-olo |
0,95 |
0,95 |
|
10 |
Clerosterolo |
(24S)-24-etil-Δ-5,25-colestadien-3ß-olo |
0,96 |
0,96 |
|
11 |
ß-sitosterolo |
(24R)-24-etil-Δ-5-colesten-3ß-olo |
1,00 |
1,00 |
|
12 |
Sitostanolo |
24-etil-colestan-3ß-olo |
1,02 |
1,02 |
|
13 |
Δ-5-avenasterolo |
(24Z)-24-etiliden-Δ-colesten-3ß-olo |
1,03 |
1,03 |
|
14 |
Δ-5-24-stigmastadienolo |
(24R,S)-24-etil-Δ-5,24-colestadien-3ß-olo |
1,08 |
1,08 |
|
15 |
Δ-7-stigmastenolo |
(24R,S)-24-etil-Δ-7-colesten-3ß-olo |
1,12 |
1,12 |
|
16 |
Δ-7-avenasterolo |
(24Z)-24-etiliden-Δ-7-colesten-3ß-olo |
1,16 |
1,16 |
|
17 |
Eritrodiolo |
5α olean-12en-3ß28 diolo |
1,41 |
1,41 |
|
18 |
Uvaolo |
Δ12-ursen-3ß28 diolo |
1,52 |
1,52 |
Figura 1
Gascromatogramma della frazione costituita da steroli e dialcoli triterpenici di un olio di oliva lampante (addizionato di standard interno)

Figura 2
Gascromatogramma della frazione costituita da steroli e dialcoli triterpenici di un olio di oliva raffinato (addizionato di standard interno)

Figura 3
Placca cromatografica di un olio di sansa di oliva con la zona da raschiare per la determinazione degli steroli e dei dialcoli triterpenici

1 – Squalene
2 – Alcoli triterpenici e alifatici
3 – Steroli e dialcoli triterpenici
4 – Acidi grassi iniziali e liberi
▼M26 —————
ALLEGATO VII
DETERMINAZIONE DELLA PERCENTUALE DI 2 GLICERIL MONOPALMITATO
1. OGGETTO E CAMPO DI APPLICAZIONE
Il metodo descrive il procedimento analitico per la determinazione della percentuale di acido palmitico in posizione 2 nei trigliceridi mediante valutazione del 2-gliceril monopalmitato.
Esso si applica agli oli vegetali liquidi a temperatura ambiente (20 °C).
2. PRINCIPIO
Una volta preparato, il campione di olio è sottoposto all’azione della lipasi pancreatica: un’idrolisi parziale e specifica nelle posizioni 1 e 3 della molecola di trigliceride determina la comparsa dei monogliceridi in posizione 2. La percentuale di 2-gliceril monopalmitato nella frazione monogliceridica è determinata, previa sililazione, mediante gascromatografia in colonna capillare.
3. APPARECCHIATURA E MATERIALE
|
3.1. |
Beuta da 25 ml. |
|
3.2. |
Beakers da 100, 250 e 300 ml. |
|
3.3. |
Colonna di vetro per cromatografia, con diametro interno 21-23 mm e lunghezza 400 mm, provvista di disco di vetro sinterizzato e di rubinetto. |
|
3.4. |
Provette tarate da 10, 50, 100 e 200 ml. |
|
3.5. |
Matracci da 100 e 250 ml. |
|
3.6. |
Evaporatore rotante. |
|
3.7. |
Provette da centrifuga a fondo conico da 10 ml con tappo smerigliato. |
|
3.8. |
Centrifuga per provette da 10 e 100 ml. |
|
3.9. |
Termostato in grado di mantenere una temperatura di 40 °C + 0,5 °C. |
|
3.10. |
Pipette tarate da 1 e 2 ml. |
|
3.11. |
Siringa ipodermica da 1 ml. |
|
3.12. |
Microsiringa da 100 μl. |
|
3.13. |
Imbuto da 1 000 ml. |
|
3.14. |
Gascromatografo idoneo per il funzionamento con colonna capillare, dotato di dispositivo di iniezione «on column» a freddo per l’introduzione diretta del campione nella colonna e di stufa in grado di mantenere la temperatura desiderata con l’approssimazione di 1 °C. |
|
3.15. |
Iniettore a freddo «on column» per introduzione diretta in colonna. |
|
3.16. |
Rivelatore a ionizzazione di fiamma ed elettrometro. |
|
3.17. |
Registratore-integratore adatto all’elettrometro, con tempo di risposta non superiore a 1 secondo e con velocità della carta variabile. |
|
3.18. |
Colonna capillare in vetro o silice fusa, lunga 8-12 m, diametro interno 0,25-0,32 mm, ricoperta di metilpolisilossano o di fenilmetilpolisilossano al 5 %, di spessore compreso fra 0,10 e 0,30 μm, resistente a una temperatura di 370 °C. |
|
3.19. |
Microsiringa da 10 μl con ago cementato lungo almeno 7,5 cm, per iniezione diretta in colonna. |
4. REAGENTI
|
4.1. |
Gel di silice di granulometria compresa tra 0,063 e 0,200 mm (70/280 mesh), preparato nel modo seguente: mettere il gel di silice in una capsula di porcellana, essiccare nella stufa a 160 °C per 4 ore, quindi lasciar raffreddare a temperatura ambiente in essiccatore. Aggiungere un volume d’acqua equivalente al 5 % del peso del gel di silice, procedendo come segue: in una beuta da 500 ml pesare 152 g di gel di silice e aggiungere 8 g di acqua distillata, tappare e agitare delicatamente per ottenere una ripartizione uniforme dell’acqua. Lasciare a riposo per almeno 12 ore prima dell’uso. |
|
4.2. |
n-esano per cromatografia. |
|
4.3. |
Isopropanolo. |
|
4.4. |
Isopropanolo in soluzione acquosa 1:1 (v/v). |
|
4.5. |
Lipasi pancreatica avente un’attività compresa tra 2,0 e 10 unità di lipasi per mg (esistono in commercio lipasi pancreatiche con attività compresa tra 2 e 10 unità per mg di enzima). |
|
4.6. |
Soluzione tampone di tris-idrossimetilamminometano: soluzione acquosa 1 M portata a pH 8 (controllare con il potenziometro) mediante aggiunta di acido cloridrico concentrato (1:1 v/v). |
|
4.7. |
Colato di sodio (qualità enzimatica), soluzione acquosa allo 0,1 % (la soluzione deve essere utilizzata entro i 15 giorni successivi alla preparazione). |
|
4.8. |
Cloruro di calcio, soluzione acquosa al 22 %. |
|
4.9. |
Etere etilico per cromatografia. |
|
4.10. |
Solvente di sviluppo: miscela n-esano/etere etilico (87/13) (v/v). |
|
4.11. |
Idrossido di sodio, soluzione al 12 % in peso. |
|
4.12. |
Fenolftaleina, soluzione etanolica all’1 %. |
|
4.13. |
Gas vettore: idrogeno o elio per gascromatografia. |
|
4.14. |
Gas ausiliari: idrogeno (minimo 99 %), esente da umidità e sostanze organiche, e aria per gascromatografia della stessa purezza. |
|
4.15. |
Reagente di silanizzazione: miscela di piridina-esametildisilazano-trimetilclorosilano 9/3/1 (v/v/v) (Esistono in commercio soluzioni pronte per l’uso. Possono essere utilizzati anche altri reagenti silanizzanti, quali ad esempio il bis-trimetiltrifluorolacetammide + 1 % trimetilclorosilano da diluire con uno stesso volume di piridina anidra). |
|
4.16. |
Campioni di riferimento: monogliceridi puri o miscele di monogliceridi a composizione percentuale nota, simile a quella del campione. |
5. PROCEDIMENTO
5.1. Preparazione del campione
|
5.1.1. |
Gli oli la cui acidità libera è inferiore al 3 % non devono essere neutralizzati prima della cromatografia su colonna di gel di silice. Gli oli la cui acidità libera è superiore al 3 % devono essere sottoposti a neutralizzazione secondo il procedimento descritto al punto 5.1.1.1.
|
|
5.1.2. |
Introdurre nella beuta da 25 ml (3.1) 1,0 g di olio preparato nel modo sopra indicato e scioglierlo in 10 ml di solvente di sviluppo (4.10). Lasciare a riposo la soluzione per almeno 15 minuti prima di procedere alla cromatografia su colonna di gel di silice. Se la soluzione è torbida, centrifugarla per garantire condizioni ottimali per la cromatografia. (Si possono utilizzare cartucce di gel di silice SPE da 500 mg pronte per l’uso). |
|
5.1.3. |
Preparazione della colonna cromatografica Versare nella colonna (3.3) circa 30 ml di solvente di sviluppo (4.10); introdurre un batuffolo di cotone nella parte inferiore della colonna con l’ausilio di una bacchetta di vetro; comprimere per far uscire l’aria. Preparare in un beaker una sospensione di 25 g di gel di silice (4.1) in circa 80 ml di solvente di sviluppo e versarla nella colonna attraverso un imbuto. Verificare che tutto il gel di silice sia stato immesso nella colonna; lavare con solvente di sviluppo (4.8), aprire il rubinetto e lasciar fluire il liquido fino a circa 2 mm sopra il livello superiore del gel di silice. |
|
5.1.4. |
Cromatografia su colonna Nella beuta da 25 ml (3.1.) pesare esattamente 1,0 g di campione preparato secondo il procedimento descritto al punto 5.1. Sciogliere il campione in 10 ml di solvente di sviluppo (4.10). Versare la soluzione nella colonna cromatografica preparata nel modo indicato al punto 5.1.3. Evitare di agitare la superficie della colonna. Aprire il rubinetto e lasciar fluire la soluzione campione fino al livello del gel di silice. Sviluppare con 150 ml di solvente di sviluppo. Regolare il flusso a 2 ml/min (in modo che i 150 ml passino attraverso la colonna in 60-70 minuti circa). Recuperare l’eluato in un matraccio da 250 ml previamente tarato. Far evaporare il solvente sotto vuoto ed eliminarne le ultime tracce con una corrente di azoto. Pesare il matraccio e calcolare l’estratto raccolto. [Se si utilizzano cartucce di silice SPE pronte per l’uso, procedere come segue: introdurre 1 ml di soluzione (5.1.2) nelle cartucce previamente preparate con 3 ml di n-esano. Una volta percolata la soluzione, sviluppare con 4 ml di n-esano/etere etilico 9:1 (v/v). Recuperare l’eluato in una provetta da 10 ml e sottoporlo a evaporazione in corrente di azoto fino a essiccazione completa. Sottoporre il residuo secco all’azione della lipasi pancreatica (5.2). È essenziale che la composizione in acidi grassi sia verificata prima e dopo passaggio su cartuccia SPE]. |
5.2. Idrolisi con lipasi pancreatica
|
5.2.1. |
Pesare nella provetta della centrifuga 0,1 g di olio preparato nel modo descritto al punto 5.1. Aggiungere 2 ml di soluzione tampone (4.6), 0,5 ml della soluzione di colato di sodio (4.7) e 0,2 ml della soluzione di cloruro di calcio, agitando bene dopo ogni aggiunta. Chiudere la provetta con il tappo smerigliato e inserirla nel termostato a 40 + 0,5 °C. |
|
5.2.2. |
Aggiungere 20 mg di lipasi, agitare accuratamente (evitando di bagnare il tappo), mettere la provetta nel termostato per esattamente 2 minuti, quindi ritirarla, agitare vigorosamente per esattamente 1 minuto e lasciar raffreddare. |
|
5.2.3. |
Aggiungere 1 ml di etere etilico, tappare e agitare vigorosamente, quindi centrifugare e travasare la soluzione in una provetta pulita e asciutta con la microsiringa. |
5.3. Preparazione dei derivati silanizzati e della gascromatografia
|
5.3.1. |
Introdurre con la microsiringa 100 μl di soluzione (5.2.3) in una provetta da 10 ml a fondo conico. |
|
5.3.2. |
Eliminare il solvente con una leggera corrente di azoto, aggiungere 200 μl di reagente di silanizzazione (4.15), tappare la provetta e lasciare a riposo per 20 minuti. |
|
5.3.3. |
Dopo 20 minuti, aggiungere da 1 a 5 ml di n-esano (in funzione delle condizioni cromatografiche): la soluzione così ottenuta è pronta per la gascromatografia. |
5.4. Gascromatografia
Le condizioni operative sono le seguenti:
— temperatura dell’iniettore (iniettore «on column») inferiore alla temperatura di ebollizione del solvente (68 °C),
— temperatura del rivelatore: 350 °C,
— temperatura della colonna: temperatura della stufa programmata a 60 °C per 1 minuto, quindi incrementata di 15 °C al minuto fino a 180 °C, poi di 5 °C al minuto fino a 340 °C e mantenuta a 340 °C per 13 minuti,
— gas vettore: idrogeno o elio regolato alla velocità lineare adeguata per ottenere la risoluzione rappresentata nella figura 1; il tempo di ritenzione del trigliceride C54 deve essere di 40 + 5 minuti (cfr. figura 2). (Le condizioni operative sopra indicate sono proposte a titolo indicativo. L’operatore provvederà ad ottimizzarle per ottenere la risoluzione desiderata. Il picco corrispondente al 2-gliceril monopalmitato deve avere un’altezza minima pari al 10 % della scala del registratore.),
— quantità di sostanza iniettata: 0,5-1 μl della soluzione (5 ml) di n-esano (5.3.3).
5.4.1. Identificazione dei picchi
I singoli monogliceridi sono identificati in base ai tempi di ritenzione, in confronto a quelli ottenuti con miscele standard di monogliceridi analizzate nelle medesime condizioni.
5.4.2. Valutazione quantitativa
L’area dei picchi è calcolata mediante un integratore elettronico.
6. ESPRESSIONE DEI RISULTATI
La percentuale di 2-gliceril monopalmitato è calcolata in base al rapporto tra l’area del picco corrispondente e la somma delle aree dei picchi di tutti i monogliceridi (cfr. figura 2), applicando la seguente formula:
gliceril monopalmitato (%):

in cui:
|
Ax |
= |
area del picco corrispondente al gliceril monopalmitato |
|
ΣA |
= |
somma delle aree di tutti i picchi dei monogliceridi. |
Il risultato si esprime con una cifra decimale.
7. RAPPORTO DI ANALISI
Il rapporto di analisi deve recare:
— il riferimento al metodo descritto,
— ogni indicazione utile per la completa identificazione del campione,
— il risultato dell’analisi,
— ogni deviazione dal metodo indicato, sia essa dovuta ad una decisione delle parti interessate o a qualsiasi altro motivo,
— gli estremi per l’identificazione del laboratorio, la data dell’analisi e la firma dei responsabili.
Figura 1
Cromatogramma dei prodotti della reazione di silanizzazione ottenuti dall’azione della lipasi su un olio d’oliva raffinato addizionato del 20 % di olio esterificato (100 %).

Legenda: «acides gras libres» = acidi grassi liberi; «Huile d’olive raffinée + 20 % huile estérifiée» = olio d’oliva raffinato + 20 % olio esterificato; «1-2 monopalmitoléine» = 1-2 monopalmitoleina; «1-2 mono C18 insat.» = 1-2 mono C18 insaturi
Figura 2
Cromatogramma di:
|
(A) |
olio d’oliva non esterificato dopo lipasi; dopo silanizzazione; in queste condizioni (colonna capillare 8-12 m), la frazione cerosa viene eluita contemporaneamente alla frazione di digliceride o poco dopo. Dopo lipasi, il tenore di trigliceridi non dovrebbe superare il 15 %. 
Legenda:
|
Cromatogramma di:
|
(B) |
olio esterificato dopo lipasi; dopo silanizzazione; in queste condizioni (colonna capillare 8-12 m), la frazione cerosa viene eluita contemporaneamente alla frazione di digliceride o poco dopo. Dopo lipasi, il tenore di trigliceridi non dovrebbe superare il 15 %. 
Legenda:
|
8. NOTE
PREPARAZIONE DELLA LIPASI
Esistono in commercio lipasi con attività soddisfacente. Si possono anche preparare in laboratorio con il seguente procedimento.
Raffreddare 5 kg di pancreas suino fresco a 0 °C. Rimuovere il grasso solido e il tessuto connettivo circostanti e triturare in un mescolatore in modo da ottenere un fluido pastoso. Agitare questa pasta per 4-6 ore con 2,5 l di acetone anidro e centrifugare. Estrarre il residuo altre tre volte con lo stesso volume di acetone, poi due volte con una miscela 1:1 (v/v) di acetone e di etere etilico e due volte con etere etilico.
Essiccare il residuo sotto vuoto per 48 ore in modo da ottenere una polvere stabile, da conservare in frigorifero al riparo dall’umidità.
CONTROLLO DELL’ATTIVITÀ LIPASICA
Preparare un’emulsione di olio d’oliva nel modo seguente:
Agitare per 10 minuti in un mescolatore una miscela di 165 ml di soluzione di gomma arabica a 100g/l, 15 g di ghiaccio tritato e 20 ml di un olio d’oliva neutralizzato.
In un beaker da 50 ml versare 10 ml di questa emulsione, quindi 0,3 ml di soluzione di colato di sodio a 0,2 g/ml e 20 ml di acqua distillata.
Mettere il beaker in un termostato regolato a 37 °C; introdurre gli elettrodi del pHmetro e l’agitatore ad elica.
Mediante una buretta aggiungere goccia a goccia una soluzione di idrossido di sodio 0,1 N fino ad ottenere un pH 8,3.
Aggiungere un volume di soluzione acquosa di polvere di lipasi (0,1 g/ml di lipasi). Non appena il pHmetro indica un pH 8,3, avviare il cronometro e aggiungere goccia a goccia la soluzione di idrossido di sodio in modo da mantenere il pH a 8,3. Annotare il volume di soluzione consumato ogni minuto.
Riportare i dati in un diagramma indicando le letture di tempo nelle ascisse e i millilitri di soluzione alcalina 0,1 N consumati per mantenere costante il pH nelle ordinate. Si deve ottenere un grafico lineare.
L’attività della lipasi, misurata in unità di lipasi per mg, è data dalla formula seguente:

in cui:
|
A |
è l’attività in unità di lipasi/mg |
|
V |
è il numero di ml di soluzione di idrossido di sodio 0,1 N/min, desunto dal diagramma |
|
N |
è la normalità della soluzione di idrossido di sodio |
|
m |
è la massa in mg della lipasi utilizzata per la prova. |
L’unità di lipasi è definita come la quantità di enzima che libera 10 microequivalenti di acido al minuto.
▼M20 —————
ALLEGATO IX
ANALISI SPETTROFOTOMETRICA NELL'ULTRAVIOLETTO
PREMESSA
L'analisi spettrofotometrica nell'ultravioletto può fornire informazioni sulla qualità di una sostanza grassa, sul suo stato di conservazione e sulle modificazioni indotte da processi tecnologici. L'assorbimento alle lunghezze d'onda specificate nel metodo è dovuto alla presenza di sistemi dienici e trienici coniugati derivanti da processi di ossidazione e/o da pratiche di raffinazione. I valori di tale assorbimento sono espressi come estinzione specifica
![]()
(estinzione di una soluzione della sostanza grassa all'1 % (p/v) nel solvente prescritto, in una cuvetta di 10 mm) convenzionalmente indicata con K (detto anche «coefficiente di estinzione»).
1. OGGETTO
Il presente allegato descrive il procedimento per l'esecuzione dell'analisi spettrofotometrica nell'ultravioletto dell'olio d'oliva.
2. PRINCIPIO DEL METODO
Un campione viene disciolto nel solvente stabilito e l'assorbanza della soluzione è misurata alle lunghezze d'onda prescritte, in riferimento al solvente puro.
Si determinano i valori dell'estinzione specifica alle lunghezze d'onda di 232 e 268 nm nell'isoottano o di 232 e 270 nm nel cicloesano, per una concentrazione dell'1 % p/v in una cuvetta di 10 mm.
3. APPARECCHIATURA
|
3.1. |
Spettrofotometro per misurazioni a lunghezze d'onda dell'ultravioletto (da 220 a 360 nm), con possibilità di lettura per ogni unità nanometrica. Si raccomanda l'esecuzione di controlli regolari in relazione all'accuratezza e alla riproducibilità delle scale di lunghezza d'onda e di assorbanza, nonché alla luce parassita.
|
|
3.2. |
Cuvette in quarzo rettangolari, con coperchio, adatte per misurazioni a lunghezze d'onda dell'ultravioletto (da 220 a 360 nm), con un percorso ottico di 10 mm. Riempite di acqua o con altro solvente idoneo, le cuvette non devono presentare fra loro differenze superiori a 0,01 unità di estinzione. |
|
3.3. |
Matracci volumetrici tarati da 25 ml, classe A. |
|
3.4. |
Bilancia analitica, idonea a fornire una lettura con l'approssimazione di 0,0001 g. |
4. REAGENTI
Salvo indicazione contraria, durante l'analisi utilizzare soltanto reagenti di qualità analitica riconosciuta e acqua distillata o demineralizzata o acqua di purezza equivalente.
Solvente: Isoottano (2,2,4 trimetilpentano) per misurazioni a 232 nm e 268 nm o cicloesano per misurazioni a 232 nm e 270 nm, con assorbanza inferiore a 0,12 a 232 nm e a 0,05 a 270 nm rispetto all'acqua distillata, misurata in una cuvetta di 10 mm.
5. PROCEDIMENTO
|
5.1. |
Il campione deve essere perfettamente omogeneo ed esente da impurezze sospese. In caso contrario deve essere filtrato su carta alla temperatura di circa 30 °C. |
|
5.2. |
Del campione così preparato si versano circa 0,25 g (con un'approssimazione di 1 mg) in un matraccio tarato da 25 ml, si porta a volume con il solvente prescritto e si omogeneizza. La soluzione risultante deve essere perfettamente limpida. Qualora si riscontri opalescenza o torbidità si filtra rapidamente con carta. NOTA: in generale, è sufficiente una massa di 0,25-0,30 g per misurare l'assorbenza di oli di oliva vergini ed extra vergini a 268 nm e 270 nm. Per misurazioni a 232 nm occorrono di norma 0,05 g di campione; in tal caso si preparano generalmente due soluzioni distinte. Per misurare l'assorbanza di oli di sansa d'oliva, oli d'oliva raffinati e oli d'oliva adulterati, che presentano un'assorbanza più elevata, è generalmente sufficiente un campione di quantità inferiore (ad esempio 0,1 g). |
|
5.3. |
Se necessario, si corregge la linea di base (220-290 nm) con solvente in entrambe le cuvette di quarzo (campione e riferimento), quindi si riempie con la soluzione di prova la cuvetta di quarzo del campione e si misurano le estinzioni a 232, 268 o 270 nm rispetto al solvente utilizzato come riferimento. I valori di estinzione registrati devono essere compresi tra 0,1 e 0,8 o nell'intervallo di linearità dello spettrofotometro (che deve essere verificato). In caso contrario è necessario ripetere le misurazioni utilizzando soluzioni più concentrate o più diluite, a seconda del caso. |
|
5.4. |
Dopo aver misurato l'assorbanza a 268 o 270 nm, si misura l'assorbanza a λmax, λmax + 4 e λmax – 4. Questi valori di assorbanza sono utilizzati per determinare la variazione dell'estinzione specifica (ΔΚ). NOTA: il valore di λmax è fissato a 268 nm per l'isoottano utilizzato come solvente e a 270 nm per il cicloesano. |
6. ESPRESSIONE DEI RISULTATI
|
6.1. |
Si riportano le estinzioni specifiche (coefficienti di estinzione) alle varie lunghezze d'onda calcolate come segue:
in cui:
il valore è espresso con due cifre decimali. |

|
6.2. |
Variazione dell'estinzione specifica (ΔΚ) La variazione del valore assoluto dell'estinzione (ΔΚ) è data da:
in cui Km è l'estinzione specifica alla lunghezza d'onda di massimo assorbimento a 270 nm e 268 nm, a seconda del solvente utilizzato. Il valore è espresso con due cifre decimali. |
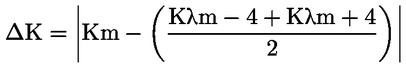
ALLEGATO X
DETERMINAZIONE DEGLI ESTERI METILICI DEGLI ACIDI GRASSI MEDIANTE GASCROMATOGRAFIA
1. OGGETTO
Il presente allegato fornisce orientamenti per la determinazione mediante gascromatografia degli acidi grassi liberi e legati nei grassi e negli oli vegetali dopo la loro conversione in esteri metilici di acidi grassi (FAME).
Gli acidi grassi legati dei triacilgliceroli (TAG) e, a seconda del metodo di esterificazione, gli acidi grassi liberi (FFA), sono convertiti in esteri metilici di acidi grassi (FAME), determinati mediante gascromatografia con colonna capillare.
Il metodo descritto nel presente allegato consente di determinare gli esteri metilici degli acidi grassi da C12 a C24, compresi quelli saturi, monoinsaturi cis e trans e polinsaturi cis e trans.
2. PRINCIPIO
La gascromatografia (GC) è utilizzata per l'analisi quantitativa dei FAME. I FAME sono preparati conformemente alla parte A. Essi sono successivamente iniettati e vaporizzati nell'iniettore. La separazione dei FAME viene eseguita su colonne analitiche di polarità e lunghezza specifiche. Un rivelatore a ionizzazione di fiamma (FID) è utilizzato per il rilevamento dei FAME. Le condizioni di analisi figurano nella parte B.
L'idrogeno o l'elio possono essere utilizzati come gas vettore (fase mobile) nella gascromatografia dei FAME con FID. L'idrogeno accelera la separazione e produce picchi più marcati. La fase stazionaria consiste in un microscopico strato di una sottile pellicola liquida su una superficie solida inerte di silice fusa.
Mentre passano attraverso la colonna capillare i composti volatili in esame interagiscono con la fase stazionaria che riveste la superficie interna della colonna. A causa di tale differenza di interazione dei diversi composti, la loro eluizione avviene in un momento diverso, detto «tempo di ritenzione» del composto per un determinato insieme di parametri analitici. La comparazione dei tempi di ritenzione serve a identificare i diversi composti.
PARTE A
PREPARAZIONE DEGLI ESTERI METILICI DI ACIDI GRASSI DA OLIO DI OLIVA E DA OLIO DI SANSA DI OLIVA
1. OGGETTO
La presente parte illustra la preparazione degli esteri metilici degli acidi grassi e include metodi di preparazione degli esteri metilici di acidi grassi da oli di oliva e di oli di sansa di oliva.
2. CAMPO DI APPLICAZIONE
La preparazione degli esteri metilici di acidi grassi da oli di oliva e oli di sansa di oliva avviene mediante transesterificazione con soluzione metanolica di idrossido di potassio a temperatura ambiente. La necessità di depurare il campione prima della transesterificazione dipende dal tenore di acidi grassi liberi del campione e dal parametro analitico da determinare; il metodo può essere scelto secondo la seguente tabella:
|
Categoria di olio |
Metodo |
|
Olio di oliva vergine di acidità ≤ 2,0 %, |
1. Acidi grassi 2. Acidi grassi trans 3. ΔECN 42 (dopo purificazione tramite estrazione su fase solida (SPE) con gel di silice) |
|
Olio di oliva raffinato |
|
|
Olio di oliva composto da oli d'oliva raffinati e da oli d'oliva vergini |
|
|
Olio di sansa di oliva raffinato |
|
|
Olio di sansa di oliva |
|
|
Olio di oliva vergine con acidità > 2,0 % Olio di sansa di oliva greggio |
1. Acidi grassi (dopo purificazione tramite estrazione su fase solida (SPE) con gel di silice) 2. Acidi grassi trans (dopo purificazione tramite estrazione su fase solida (SPE) con gel di silice) 3. ΔECN 42 (dopo purificazione tramite estrazione su fase solida (SPE) con gel di silice) |
3. METODOLOGIA
3.1. Transesterificazione in soluzione metanolica di idrossido di potassio a temperatura ambiente
3.1.1. Principio
Gli esteri metilici si formano per transesterificazione in una soluzione metanolica di idrossido di potassio come fase intermedia prima della saponificazione.
3.1.2. Reagenti
|
3.1.2.1. |
Metanolo con tenore di acqua non superiore allo 0,5 % (m/m). |
|
3.1.2.2. |
Esano per cromatografia. |
|
3.1.2.3. |
Eptano per cromatografia. |
|
3.1.2.4. |
Etere dietilico, stabilizzato per analisi. |
|
3.1.2.5. |
Acetone per cromatografia. |
|
3.1.2.6. |
Solvente di eluizione per la purificazione dell'olio mediante cromatografia su colonna/estrazione su fase solida (SPE); miscela di esano/etere dietilico in proporzioni 87:13(v/v). |
|
3.1.2.7. |
Idrossido di potassio, soluzione metanolica di circa 2M: sciogliere 11,2 g di idrossido di potassio in 100 ml di metanolo. |
|
3.1.2.8. |
Cartucce di gel di silice, 1 g (6 ml), per l'estrazione in fase solida (SPE). |
3.1.3. Apparecchiatura
|
3.1.3.1. |
Provette con tappo a vite (volume 5 ml) munito di guarnizione in PTFE. |
|
3.1.3.2. |
Pipette graduate o automatiche da 2 ml e 0,2 ml. |
3.1.4. Purificazione dei campioni di olio
All'occorrenza i campioni verranno purificati facendo passare l'olio su una cartuccia di gel di silice per estrazione in fase solida. Inserire una cartuccia di gel di silice (3.1.2.8) in un apparecchio di eluizione sotto vuoto e lavare con 6 ml di esano (3.1.2.2); effettuare il lavaggio senza vuoto. Quindi immettere nella colonna una soluzione d'olio (0,12 g circa) in 0,5 ml di esano (3.1.2.2). Far scendere la soluzione per eluizione con 10 ml di esano/etere dietilico (87:13 v/v) (3.1.2.6). Omogeneizzare tutti gli eluati e dividerli in due volumi simili. Fare evaporare un'aliquota fino ad essiccamento in un evaporatore rotante, a pressione ridotta e temperatura ambiente. Dissolvere il residuo in 1 ml di eptano: si ottiene una soluzione pronta per l'analisi degli acidi grassi mediante GC. Far evaporare la seconda aliquota e dissolvere il residuo in 1 ml di acetone per l'analisi dei trigliceridi mediante HPLC, se necessario.
3.1.5. Procedura
Pesare circa 0,1 g del campione di olio in una provetta da 5 ml con tappo a vite (3.1.3.1). Aggiungere 2 ml di eptano e mescolare (3.1.2.2). Aggiungere 0,2 ml di soluzione metanolica di idrossido di potassio (3.1.2.7), chiudere con il tappo munito di guarnizione in PTFE, stringere bene il tappo e agitare energicamente per 30 secondi. Lasciare depositare finché la parte superiore della soluzione diventa chiara. Far decantare lo strato superiore che contiene gli esteri metilici. La soluzione di eptano ottenuta è pronta per essere iniettata nel gascromatografo. Si consiglia di conservare la soluzione in frigorifero fino al momento dell'analisi gascromatografica. Non si consiglia di conservare la soluzione per un periodo superiore alle 12 ore.
PARTE B
ANALISI DEGLI ESTERI METILICI DEGLI ACIDI GRASSI MEDIANTE GASCROMATOGRAFIA
1. OGGETTO
La presente parte fornisce un orientamento generale per l'applicazione della gascromatografia con colonne capillari ai fini della determinazione della composizione qualitativa e quantitativa di una miscela di esteri metilici degli acidi grassi ottenuta con il metodo specificato nella parte A.
La presente parte non è applicabile agli acidi grassi polimerizzati.
2. REAGENTI
2.1. Gas vettore
Gas inerte (elio o idrogeno) completamente essiccato ed avente un tenore di ossigeno inferiore a 10 mg/kg.
Nota 1: L'idrogeno può raddoppiare la velocità dell'analisi, ma è pericoloso. Sono disponibili dispositivi di sicurezza.
2.2. Gas ausiliari
|
2.2.1. |
Idrogeno (purezza ≥ 99,9 %), esente da impurezze organiche. |
|
2.2.2. |
Aria od ossigeno, esente da impurezze organiche. |
|
2.2.3. |
Azoto (purezza > 99 %). |
2.3. Standard di riferimento
Miscela di esteri metilici di acidi grassi puri oppure esteri metilici di un grasso di composizione nota, di preferenza analogo a quello della sostanza grassa da analizzare. Gli isomeri cis e trans di esteri metilici ottadecadienoici, ottadecenoici e ottadecatrienoici sono utili per l'identificazione degli isomeri trans degli acidi insaturi.
Si deve prestare attenzione a prevenire l'ossidazione degli acidi grassi polinsaturi.
3. APPARECCHIATURA
Le istruzioni precisano che deve essere usata la consueta apparecchiatura per gascromatografia, facendo uso di colonne capillari e di un rivelatore a ionizzazione di fiamma.
3.1. Gascromatografo
Il gascromatografo deve comprendere i seguenti elementi.
3.1.1. Sistema d'iniezione
Utilizzare un sistema d'iniezione con colonne capillari, nel qual caso il sistema di iniezione deve essere appositamente progettato per l'utilizzo delle colonne. L'iniezione può essere del tipo split o splitless (iniezione on-column).
3.1.2. Forno
Il forno deve poter scaldare la colonna capillare ad almeno 260 °C e mantenere la temperatura desiderata con l'approssimazione di ± 0,1 °C. Quest'ultimo requisito è particolarmente importante se si usa una provetta in silice fusa.
L'utilizzo di un apparecchio dotato di un programmatore di temperatura è raccomandato in tutti i casi e in particolare per gli acidi grassi con meno di 16 atomi di carbonio.
3.1.3. Colonna capillare
|
3.1.3.1. |
Tubo costituito di materiale inerte rispetto alle sostanze da analizzare (di solito vetro o silice fusa). Il diametro interno deve essere compreso tra 0,20 mm e 0,32 mm. La superficie interna deve esser sottoposta a un opportuno trattamento (ad es. preparazione della superficie, inattivazione) prima di ricevere la pellicola della fase stazionaria. Una lunghezza di 60 m è sufficiente per gli acidi grassi e per gli isomeri cis e trans degli acidi grassi. |
|
3.1.3.2. |
Fase stazionaria, polisilossano polare (cianopropilsilicone). Le colonne a fase legata (a legami reticolari) sono adeguate. Nota 2: Vi è il rischio che i polisilossani polari creino difficoltà nell'identificazione e separazione dell'acido linolenico e degli acidi a C20. Lo spessore delle pellicole deve essere sottile, ad esempio 0,1 μm-0,2 μm. |
|
3.1.3.3. |
Montaggio e condizionamento della colonna Osservare le normali precauzioni necessarie per il montaggio delle colonne capillari, ovvero sistemazione della colonna nel forno (supporto), scelta e collegamento dei giunti (a tenuta stagna), sistemazione delle estremità della colonna nell'iniettore e nel rivelatore (riduzione degli spazi morti). Far fluire attraverso la colonna un flusso di gas vettore [ad es. 0,3 bar (30 kPa) per una colonna di 25 m di lunghezza e di 0,3 mm di diametro interno]. Condizionare la colonna programmando la temperatura del forno a 3 °C/min dalla temperatura ambiente a una temperatura di 10 °C inferiore al limite di decomposizione della fase stazionaria. Mantenere il forno a questa temperatura per 1 h fino a stabilizzazione della linea di base. Riportarla a 180 °C in modo da lavorare in condizioni isotermiche. Nota 3: Sono disponibili in commercio adeguate colonne precondizionate. |
3.1.4. Rivelatore a ionizzazione di fiamma e convertitore-amplificatore
3.2. Siringa
La siringa deve avere una capacità massima di 10 μl ed essere graduata in divisioni di 0,1 μl.
3.3. Sistema di acquisizione dei dati
Sistema di acquisizione dei dati collegato online con i rilevatori e utilizzato con un software adeguato per l'integrazione e la normalizzazione dei picchi.
4. PROCEDURA
Le operazioni descritte dal paragrafo 4.1 al paragrafo 4.3 riguardano l'utilizzo di un rivelatore a ionizzazione di fiamma.
4.1. Condizioni di prova
4.1.1. Selezione delle condizioni operative ottimali per colonne capillari
In considerazione delle caratteristiche di efficienza e di permeabilità delle colonne capillari, la separazione dei costituenti e la durata dell'analisi sono ampiamente dipendenti dal flusso del gas vettore nella colonna. Sarà pertanto necessario ottimizzare le condizioni operative adeguando questo parametro (o semplicemente la perdita di carico in testa alla colonna), a seconda che si voglia migliorare la separazione o accelerare l'analisi.
Le seguenti condizioni si sono rivelate adatte per la separazione dei FAME (da C4 a C26). Esempi di cromatogrammi figurano nell'appendice B:
|
Temperatura dell'iniettore: |
250 °C |
|
Temperatura del rivelatore: |
250 °C |
|
Temperatura del forno: |
da 165 °C (8 min) a 210 °C a 2 °C/min |
|
Gas vettore idrogeno: pressione in testa alla colonna: |
179 kPa |
|
Flusso totale: |
154,0 ml/min; |
|
Rapporto di partizione (split ratio): |
1:100 |
|
Volume di iniezione: |
1 μl |
4.1.2. Determinazione della risoluzione (cfr. appendice A)
Calcolare la risoluzione, R, di due picchi vicini I e II, utilizzando la seguente formula:
R = 2 × ((dr(II) – d r(I))/(ω(I) + ω(II))) or R = 2 × ((tr(II) – t r(I))/(ω(I) + ω(II))) (USP) (United States Pharmacopeia),
Oppure
R = 1,18 × ((tr(II) – tr(I))/(ω0,5(I) + ω0,5(II))) (EP, BP, JP, DAB), (JP (Japanese Pharmacopeia), EP (Pharmacopée Européenne), (BP (British Pharmacopeia))
in cui:
d r(I) è la distanza di ritenzione del picco I;
d r(II) è la distanza di ritenzione del picco II;
t r(I) è il tempo di ritenzione del picco I;
t r(II) è il tempo di ritenzione del picco II;
ω(I) è la larghezza della base del picco I;
ω(II) è la larghezza della base del picco II;
ω0,5 è la larghezza del picco dello specifico composto, a metà altezza del picco;
Se ω(I) ≈ ω(II), calcolare R applicando le seguenti formule:
R = (dr(II) – d r(I))/ω = (dr(II) – d r(I))/4σ
in cui:
σ è la deviazione standard (cfr. appendice A, figura 1).
Se la distanza dr tra due picchi d r(II) – d r(I) è uguale a 4σ, il fattore di risoluzione R = 1.
Se due picchi non sono completamente separati, le tangenti ai punti di flesso dei due picchi si intersecano al punto C. Per separare completamente i due picchi, la distanza tra i due picchi deve essere pari a:
d r(II) – d r(I) = 6 σ da cui R = 1,5 (cfr. appendice A, figura 3).
5. ESPRESSIONE DEI RISULTATI
5.1. Analisi qualitativa
Identificare i picchi dell'estere metilico del campione dal cromatogramma nell'appendice B, figura 1, se necessario per interpolazione, o dal raffronto con quelli delle miscele degli esteri metilici di riferimento (come indicato al punto 2.3).
5.2. Analisi quantitativa
5.2.1. Determinazione della composizione
Calcolare la frazione di massa wi dei singoli esteri metilici di acidi grassi, espressa come percentuale in massa degli esteri metilici, come segue:
5.2.2. Metodo di calcolo
5.2.2.1. Caso generale
Calcolare il contenuto di un dato componente i, espresso come percentuale in massa degli esteri metilici, determinando la percentuale rappresentata dal rapporto tra l'area del picco corrispondente e la somma delle aree di tutti i picchi, usando la formula seguente:
wi = (Ai/ΣA) × 100
in cui:
Ai è l'area del picco del singolo estere metilico di acidi grassi i;
ΣA è la somma delle aree dei picchi di tutti i singoli esteri metilici degli acidi grassi.
I risultati sono espressi con due cifre decimali.
Nota 4: Per oli e grassi, la frazione in massa degli esteri metilici di acidi grassi è pari alla frazione di massa dei triacilgliceroli in grammi per 100 g. Per i casi in cui tale ipotesi non è ammessa, cfr. 5.2.2.2.
5.2.2.2. Uso di fattori correttivi
In taluni casi, ad esempio in presenza di acidi grassi con meno di otto atomi di carbonio oppure di acidi con gruppi secondari, le aree devono essere corrette con specifici fattori di correzione (Fci). Questi fattori sono determinati per ogni singolo strumento. A tal fine vanno utilizzati materiali di riferimento appropriati con una composizione in acidi grassi certificata nel corrispondente intervallo.
Nota 5: Questi fattori correttivi non sono identici ai fattori di correzione teorici FID, riportati nell'appendice A, poiché comprendono anche le prestazioni del sistema di iniezione ecc. Tuttavia, in caso di differenze maggiori, l'intero sistema dovrebbe essere controllato per verificarne le prestazioni.
Per questa miscela di riferimento, la percentuale in massa del FAME i è data dalla formula:
wi = (mi /Σm) × 100
in cui:
m i è la massa del FAME i nella miscela di riferimento;
Σm è il totale delle masse dei vari componenti come i FAME della miscela di riferimento.
Dal cromatogramma della miscela di riferimento calcolare la percentuale in area del FAME i come segue:
wi = (Ai/ΣA) × 100
in cui:
Ai è l'area del FAME i nella miscela di riferimento;
ΣA è la somma delle aree di tutti i FAME della miscela di riferimento.
Il fattore di correzione Fc è quindi
Fc = (mi × ΣA)/(Ai/Σm)
Per il campione, la percentuale in massa di ciascun FAME i è:
wi = (Fi × Ai)/Σ (Fi × Ai)
I risultati sono espressi con due cifre decimali.
Nota 6: Il valore calcolato corrisponde alla percentuale in massa dei singoli acidi grassi calcolata come triacilgliceroli per 100 g di grassi.
5.2.2.3. Uso di uno standard interno
In alcune analisi (ad es. quando non tutti gli acidi grassi sono quantificati e quando sono presenti acidi con 4 e 6 atomi di carbonio accanto ad acidi con 16 e 18 atomi di carbonio, oppure quando è necessario determinare il quantitativo assoluto di un acido grasso in un campione) è necessario ricorrere a uno standard interno. Vengono spesso usati acidi grassi con 5, 15 o 17 atomi di carbonio. Deve essere determinato l'eventuale fattore di correzione per lo standard interno.
La percentuale in massa del componente i, espressa in esteri metilici, è data dalla formula:
wi = (mIS × Fi × Ai)/(m × FIS × AIS)
in cui:
A i è l'area del FAME i;
A IS è l'area dello standard interno;
F i è il fattore di correzione dell'acido grasso i, espressa come FAME;
F IS è il fattore di correzione dello standard interno;
m è la massa in milligrammi della quantità di sostanza prelevata per l'analisi,
m IS è la massa in milligrammi dello standard interno.
I risultati sono espressi con due cifre decimali.
6. RELAZIONE SULLA PROVA
La relazione sulla prova deve specificare i metodi usati per la preparazione degli esteri metilici e per l'analisi gascromatografica. Essa deve citare altresì tutti i dettagli operativi non specificati nel presente metodo standard oppure considerati facoltativi, nonché i particolari di qualsiasi evento che possa avere influenzato i risultati.
La relazione sulla prova deve comprendere tutti i dati necessari per la completa identificazione del campione.
7. PRECISIONE DEL METODO
7.1. Risultati del test interlaboratorio
Maggiori dettagli di un test interlaboratorio per verificare la precisione del metodo sono stabiliti nell'allegato C della norma COI/T.20/Doc. n. 33. I valori tratti da questa prova interlaboratorio potrebbero non essere applicabili a intervalli di concentrazioni e a matrici diverse da quelle date.
7.2. Ripetibilità
La differenza assoluta tra i risultati di due prove indipendenti, eseguite dallo stesso operatore entro un breve lasso di tempo, utilizzando la stessa attrezzatura e lo stesso metodo sul medesimo materiale di prova, nello stesso laboratorio, eccede in non più del 5 % dei casi il valore di ripetibilità (r) riportato nelle tabelle 1-14 dell'allegato C della norma COI/T.20/Doc. n. 33.
7.3. Riproducibilità
La differenza assoluta tra i risultati di due prove, eseguite da diversi operatori utilizzando lo stesso metodo su materiale di prova identico in laboratori diversi con diverse attrezzature, eccede in non più del 5 % dei casi il valore di riproducibilità (R) riportato nelle tabelle 1-14 dell'allegato C della norma COI/T.20/Doc. n. 33.
Appendice A
Figura 1

Con larghezza di ω0,5 a metà altezza del triangolo (ABC) e larghezza b a metà altezza del triangolo (NPM).
|
Figura 2 |
Figura 3 |
|
|
|


Appendice B
Figura 1
Profilo gascromatografico di un olio di sansa di oliva, ottenuto con il metodo della metilazione a freddo
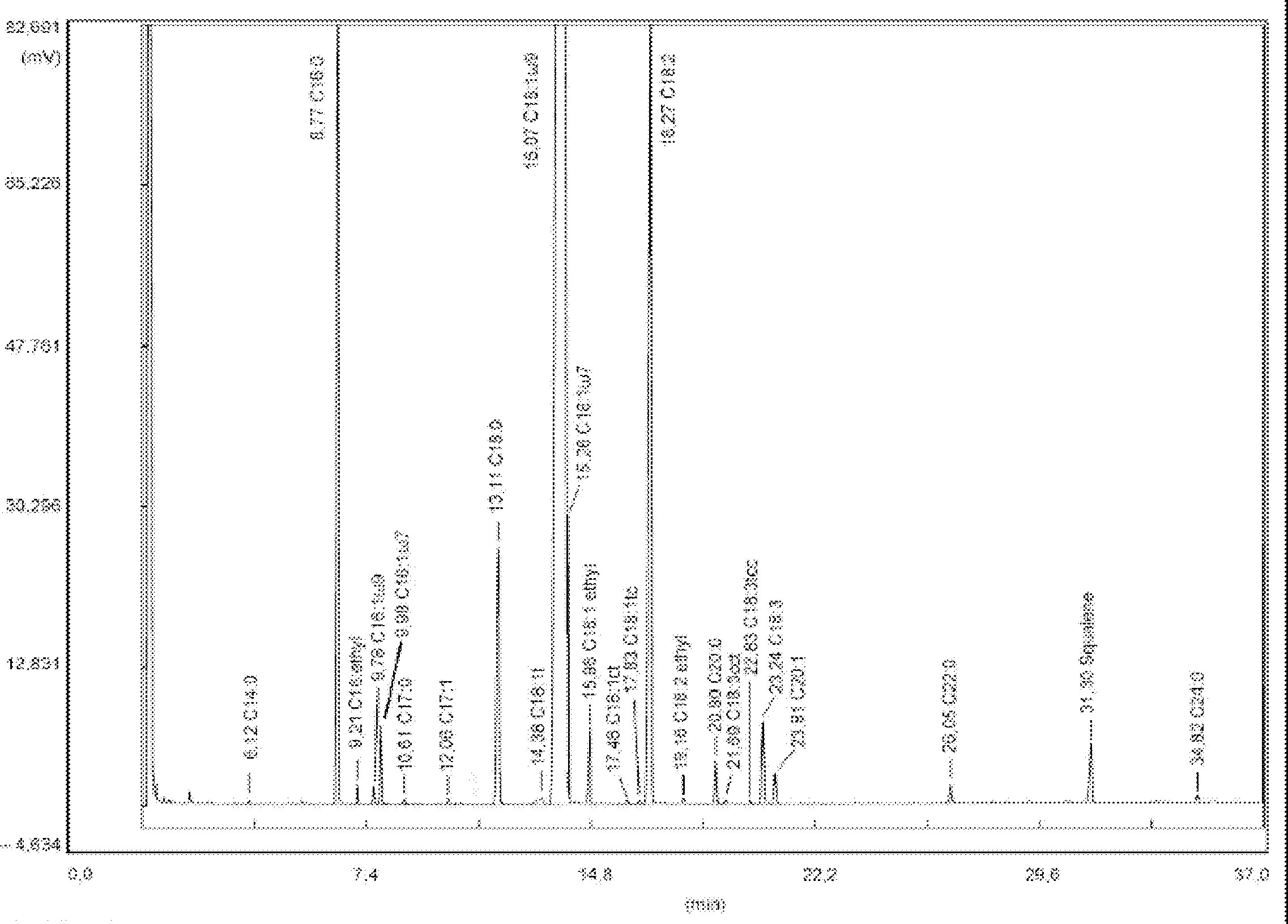
I picchi cromatografici corrispondono agli esteri metilici ed etilici, salvo altre indicazioni.
ALLEGATO XI
DETERMINAZIONE DEL TENORE DI SOLVENTI ALOGENATI VOLATILI NELL'OLIO DI OLIVA
1. PRINCIPIO
Analisi mediante cromatografia in fase gassosa secondo la tecnica dello spazio di testa (head space).
2. APPARECCHIATURA
|
2.1. |
Apparecchio di cromatografia in fase gassosa, munito di rivelatore a cattura di elettroni. |
|
2.2. |
Apparecchiatura per spazio di testa. |
|
2.3. |
Colonna per cromatografia in fase gassosa, in vetro, di 2 m di lunghezza e 2 mm di diametro, fase stazionaria. OV101 al 10 % o equivalente, impregnato su una terra di diatomee calcinata, lavata con acidi e silanizzata, di granulometria 80-100 Mesh. |
|
2.4. |
Gas vettore e gas ausiliario; azoto per cromatografia in fase gassosa, idoneo alla rivelazione per cattura di elettroni. |
|
2.5. |
Bottiglie in vetro da 10 a 15 ml, munite di una guarnizione in teflon e di un tappo d'alluminio provvisto di un orifizio per prelievo a mezzo siringa. |
|
2.6. |
Pinze a chiusura ermetica. |
|
2.7. |
Siringa per gas da 0,5 a 2 ml. |
3. REATTIVI
Solventi alogenati volatili di purezza idonea all'impiego per cromatografia in fase gassosa.
4. PROCEDIMENTO
|
4.1. |
Pesare con precisione 3 g d'olio circa in una bottiglia di vetro (da non riutilizzare) e chiudere la bottiglia ermeticamente. Riporre la bottiglia in un termostato a 70 °C per un'ora. Prelevare con precisione con la siringa un volume da 0,2 a 0,5 ml dallo spazio di testa ed iniettarlo nella colonna del cromatografo in fase gassosa regolato come segue: — temperatura dell'iniettore: 150 °C — temperatura della colonna: 70-80 °C — temperatura del rivelatore: 200-250 °C Si possono usare temperature diverse purché i risultati siano equivalenti. |
|
4.2. |
Soluzioni di riferimento: preparare soluzioni standard, usando olio d'oliva, raffinato senza traccia di solventi, a concentrazioni variabili tra 0,05 e 1 mg/kg ed in relazione con il tenore presunto del campione. L'eventuale diluizione deve essere fatta con pentano. |
|
4.3. |
Valutazione quantitativa: fare il rapporto tra le superfici o le altezze dei picchi del campione e della soluzione standard corrispondente alla concentrazione presunta più vicina. Se lo scarto relativo supera il 10 % occorre ripetere l'analisi comparato con una nuova soluzione standard, fino a che la sua concentrazione rientri nel suddetto scarto relativo. Il tenore è determinato in base ad una media di iniezioni elementari. |
|
4.4. |
Espressione dei risultati: i risultati sono espressi in mg/kg (ppm). Il limite di rivelazione del metodo è di 0,01 mg/kg. |
ALLEGATO XII
METODO DEL CONSIGLIO OLEICOLO INTERNAZIONALE PER LA VALUTAZIONE ORGANOLETTICA DEGLI OLI DI OLIVA VERGINI
1. OGGETTO E CAMPO D'APPLICAZIONE
Il presente metodo internazionale stabilisce una procedura che consente di valutare le caratteristiche organolettiche degli oli di oliva vergini ai sensi dell'allegato VII, parte VIII, punto 1, del regolamento (UE) n. 1308/2013 del Parlamento europeo e del Consiglio ( 4 ) e di classificarli in base a tali caratteristiche. Il metodo contiene inoltre indicazioni per un'etichettatura facoltativa.
Il metodo descritto è applicabile soltanto agli oli di oliva vergini e alla loro classificazione o etichettatura in funzione dell'intensità dei difetti percepiti e del flavor fruttato, determinata da un gruppo di assaggiatori selezionati, addestrati e controllati, costituito in panel.
Le norme COI citate nel presente allegato si intendono nell'ultima versione disponibile.
2. VOCABOLARIO GENERALE DI BASE PER L'ANALISI SENSORIALE
V. norma COI/T.20/Doc. n. 4 "Analisi sensoriale: vocabolario generale"
3. VOCABOLARIO SPECIFICO
3.1. Attributi negativi
Riscaldo/Morchia Flavor caratteristico dell'olio ottenuto a partire da olive ammassate o depositate in condizioni che hanno favorito un forte sviluppo della fermentazione anaerobica, o flavor dell'olio rimasto in contatto con fanghi di decantazione in serbatoi o vasche, che abbiano anch'essi subito processi di fermentazione anaerobica.
Muffa-umidità-terra Flavor caratteristico dell'olio ottenuto da frutti nei quali si sono sviluppati abbondanti funghi e lieviti per essere rimasti ammassati in ambienti umidi per molti giorni o dell'olio ottenuto da olive raccolte con terra o infangate e non lavate.
Avvinato-inacetito-acido-agro Flavor caratteristico di alcuni oli che ricorda quello del vino o dell'aceto. Esso è dovuto essenzialmente a un processo di fermentazione aerobica delle olive o dei resti di pasta di olive in fiscoli non lavati correttamente, che porta alla formazione di acido acetico, acetato di etile ed etanolo.
Rancido Flavor degli oli che hanno subito un processo ossidativo intenso.
Olive gelate (legno umido) Flavor caratteristico dell'olio estratto da olive che hanno subito una gelata sull'albero.
3.1.1. Altri attributi negativi
|
Cotto o stracotto |
Flavor caratteristico dell'olio dovuto a eccessivo e/o prolungato riscaldamento, che si verifica in particolare durante la termo-gramolatura, se realizzata in condizioni termiche inadeguate. |
|
Fieno — Legno |
Flavor caratteristico di alcuni oli provenienti da olive secche. |
|
Grossolano |
Sensazione orale/tattile densa e pastosa prodotta da alcuni oli vecchi. |
|
Lubrificanti |
Flavor dell'olio che ricorda il gasolio, il grasso o l'olio minerale. |
|
Acqua di vegetazione |
Flavor acquisito dall'olio a causa di un contatto prolungato con le acque di vegetazione che hanno subito un processo di fermentazione. |
|
Salamoia |
Flavor dell'olio estratto da olive conservate in salamoia. |
|
Metallico |
Flavor che ricorda il metallo. È caratteristico dell'olio mantenuto a lungo in contatto con superfici metalliche durante i procedimenti di frangitura, gramolatura, pressione o stoccaggio. |
|
Sparto |
Flavor caratteristico dell'olio ottenuto da olive pressate in fiscoli di sparto nuovi. Può presentare caratteristiche diverse a seconda dello sparto utilizzato per costruire i fiscoli (sparto verde o secco). |
|
Verme |
Flavor dell'olio ottenuto da olive fortemente colpite da larve di mosca dell'olivo (Bactrocera oleae). |
|
Cetriolo |
Flavor dell'olio che ha subito un condizionamento ermetico eccessivamente prolungato, particolarmente in lattine, e che viene attribuito alla formazione di 2-6 nonadienale. |
3.2. Attributi positivi
|
Fruttato |
Insieme delle sensazioni olfattive, che dipendono dalla varietà delle olive, caratteristiche dell'olio ottenuto da frutti sani e freschi, verdi o maturi, percepite per via diretta e/o retronasale. |
|
Amaro |
Sapore elementare caratteristico dell'olio ottenuto da olive verdi o invaiate, percepito dalle papille caliciformi che formano la V linguale. |
|
Piccante |
Sensazione tattile di pizzicore caratteristica degli oli prodotti all'inizio della campagna, principalmente da olive ancora verdi, che può essere percepita in tutta la cavità orale, in particolare in gola. |
3.3. Terminologia facoltativa ai fini dell'etichettatura
Su richiesta, il capo panel può certificare che gli oli valutati corrispondono alle definizioni e agli intervalli relativi esclusivamente alle diciture di seguito elencate, in funzione dell'intensità e della percezione degli attributi.
Attributi positivi (fruttato, amaro e piccante): in funzione dell'intensità della percezione:
— robusto, quando la mediana dell'attributo è superiore a 6;
— medio, quando la mediana dell'attributo è compresa fra 3 e 6;
— delicato, quando la mediana dell'attributo è inferiore a 3.
|
Fruttato |
Insieme delle sensazioni olfattive, che dipendono dalla varietà delle olive, caratteristiche dell'olio ottenuto da frutti sani e freschi senza predominanza del fruttato verde o del fruttato maturo, percepite per via diretta e/o retronasale. |
|
Fruttato verde |
Insieme delle sensazioni olfattive che ricordano i frutti verdi, dipendono dalla varietà delle olive e sono caratteristiche dell'olio ottenuto da frutti verdi, sani e freschi, percepite per via diretta e/o retronasale. |
|
Fruttato maturo |
Insieme delle sensazioni olfattive che ricordano i frutti maturi, dipendono dalla varietà delle olive e sono caratteristiche dell'olio ottenuto da frutti sani e freschi, percepite per via diretta e/o retronasale. |
|
Equilibrato |
Olio che non presenta squilibrio. Per squilibrio si intende la sensazione olfatto-gustativa e tattile dell'olio in cui la mediana dell'attributo amaro e/o quella dell'attributo piccante non superano di 2 punti la mediana del fruttato. |
|
Olio dolce |
Olio in cui la mediana dell'attributo amaro e quella dell'attributo piccante sono inferiori o uguali a 2. |
Elenco delle diciture in funzione dell'intensità della percezione:
|
Diciture soggette alla presentazione di un certificato delle prove organolettiche |
Mediana dell'attributo |
|
Fruttato |
— |
|
Fruttato maturo |
— |
|
Fruttato verde |
— |
|
Fruttato delicato |
Meno di 3 |
|
Fruttato medio |
Compreso tra 3 e 6 |
|
Fruttato robusto |
Più di 6 |
|
Fruttato maturo delicato |
Meno di 3 |
|
Fruttato maturo medio |
Compreso tra 3 e 6 |
|
Fruttato maturo robusto |
Più di 6 |
|
Fruttato verde delicato |
Meno di 3 |
|
Fruttato verde medio |
Compreso tra 3 e 6 |
|
Fruttato verde robusto |
Più di 6 |
|
Amaro delicato |
Meno di 3 |
|
Amaro medio |
Compreso tra 3 e 6 |
|
Amaro robusto |
Più di 6 |
|
Piccante delicato |
Meno di 3 |
|
Piccante medio |
Compreso tra 3 e 6 |
|
Piccante robusto |
Più di 6 |
|
Olio equilibrato |
La mediana dell'attributo amaro e quella dell'attributo piccante non superano di 2 punti la mediana del fruttato |
|
Olio dolce |
La mediana dell'attributo amaro e quella dell'attributo piccante non superano 2 |
4. BICCHIERE PER L'ASSAGGIO DI OLI
V. norma COI/T.20/Doc. n. 5 "Bicchiere per l'assaggio di oli".
5. SALA DI ASSAGGIO
V. norma COI/T.20/Doc. n. 6 "Guida per l'allestimento di una sala di assaggio".
6. ACCESSORI
Affinché gli assaggiatori possano svolgere correttamente il loro compito, ogni cabina deve essere attrezzata con i seguenti accessori:
— bicchieri (normalizzati) contenenti i campioni, contrassegnati in chiave, coperti da vetri di orologio e mantenuti a 28 °C ± 2 °C;
— scheda di profilo (v. figura 1) su carta, o in formato elettronico che riproduca le caratteristiche della scheda di profilo, eventualmente corredata da istruzioni per l'uso;
— penna a sfera o inchiostro indelebile;
— piattini con fettine di mela e/o acqua, acqua gassata e/o fette biscottate;
— bicchiere d'acqua a temperatura ambiente;
— un documento che riassume le norme generali citate ai punti 8.4 e 9.1.1;
— sputacchiere.
7. IL CAPO PANEL E GLI ASSAGGIATORI
7.1. Il capo panel
Il capo panel dovrà essere una persona sufficientemente formata, intenditrice ed esperta nei tipi di olio che troverà nel suo lavoro. È la figura chiave del panel e il responsabile della sua organizzazione e del suo funzionamento.
Per poter svolgere il suo compito il capo del panel deve possedere una formazione di base in analisi sensoriale e conoscerne gli strumenti. Il suo lavoro richiede abilità sensoria, meticolosità nella preparazione, nell'organizzazione e nell'esecuzione delle prove, nonché abilità e pazienza per pianificare ed eseguire le prove con rigore scientifico.
Il capo panel seleziona gli assaggiatori e provvede al loro addestramento e al controllo del loro operato in modo da garantire il mantenimento di un adeguato livello attitudinale. La qualificazione degli assaggiatori rientra pertanto nelle sue responsabilità. La valutazione della qualificazione deve essere obiettiva e a tal fine il capo panel prevederà procedure specifiche, basate su prove e criteri di inclusione/esclusione ben definiti. V. norma COI/T.20/Doc. n. 14 "Guida per la selezione, l'addestramento e il controllo degli assaggiatori qualificati di olio di oliva extra vergine".
Il capo panel è responsabile della prestazione del panel ed è pertanto chiamato a effettuare la valutazione del panel, che andrà accreditata in modo fedele e obiettivo. In ogni caso, il capo panel deve essere sempre in grado di dimostrare che ha il pieno controllo del metodo e degli assaggiatori. Si raccomanda la calibrazione periodica del panel (COI/T.20/Doc. n. 14, § 5).
È il primo responsabile per quanto riguarda la tenuta e la conservazione dei registri del panel. I registri saranno sempre rintracciabili e conformi alle esigenze di garanzia e qualità previste dalla norme internazionali sull'analisi sensoriale e garantiranno in ogni momento l'anonimato dei campioni.
Il capo panel è responsabile delle attrezzature e del materiale da utilizzare in conformità con le specificazioni del presente metodo, ne assicura l'inventario, la perfetta pulizia e conservazione. Redige un rendiconto relativo agli aspetti sopra citati, in cui dichiara che la prova si è svolta nel rispetto delle condizioni previste.
Il ricevimento e lo stoccaggio dei campioni al loro arrivo in laboratorio e la conservazione dei campioni dopo analisi avviene sotto la sua responsabilità; il capo panel assicura in ogni momento l'anonimato e l'adeguata conservazione dei campioni. A tal fine formulerà procedure scritte che consentano di assicurare la tracciabilità del processo.
Si svolgono sotto la sua responsabilità anche le operazioni di preparazione e codificazione dei campioni, la presentazione dei campioni agli assaggiatori secondo il protocollo sperimentale fissato, la raccolta e l'elaborazione statistica dei dati ricevuti dagli assaggiatori.
Il capo panel ha inoltre il compito di definire tutte le procedure eventualmente necessarie al completamento della presente norma e al buon funzionamento del panel.
Cercherà formule che permettono di raffrontare i risultati del panel con quelli di altri panel che svolgono l'analisi dell'olio di oliva vergine, per verificare il buon funzionamento del suo panel.
Il capo panel ha inoltre il compito di motivare i membri del gruppo, suscitandone l'interesse, la curiosità e lo spirito di emulazione. Per questo motivo si raccomanda vivamente di curare una buona comunicazione con i membri del gruppo, che devono sentirsi partecipi del lavoro che svolgono e dei risultati ottenuti. Deve d'altra parte evitare di far conoscere la sua opinione e impedire che i criteri di possibili leader si impongano agli altri assaggiatori.
Convocherà gli assaggiatori con sufficiente anticipo e chiarirà loro qualsiasi dubbio sulla realizzazione delle prove, pur astenendosi dal suggerire qualsiasi opinione sul campione.
7.1.1. Vicecapo panel
Per motivi giustificati, il capo panel può essere sostituito nei suoi compiti concernenti la realizzazione delle prove da un vicecapo. Il vicecapo deve possedere tutte le competenze necessarie per la funzione di capo panel.
7.2. Assaggiatori
Le persone che intervengono come assaggiatori nelle prove organolettiche di oli di oliva devono farlo a titolo volontario. Si raccomanda pertanto di richiedere ai candidati la presentazione di una domanda scritta. I candidati sono selezionati, addestrati ed esaminati dal capo panel in base alla capacità di distinguere campioni simili; occorre ricordare che la precisione dell'assaggiatore migliora con l'addestramento.
L'assaggiatore deve comportarsi come un vero osservatore sensoriale e riferire esclusivamente le sensazioni percepite, senza tener conto dei gusti personali. Svolgerà il suo lavoro in silenzio, con animo disteso e senza fretta, prestando la massima attenzione al campione che sta analizzando.
Per ciascuna prova occorrono da 8 a 12 assaggiatori. È bene prevedere alcuni assaggiatori di riserva, per supplire a eventuali assenze.
8. CONDIZIONI DELLA PROVA
8.1. Presentazione del campione
Il campione di olio di oliva da analizzare sarà presentato in bicchieri per l'assaggio standardizzati, conformemente alla norma COI/T.20/Doc. n. 5 «Bicchiere per l'assaggio di oli».
Il bicchiere conterrà 14-16 ml di olio o un quantitativo compreso tra 12,8 e 14,6 g se i campioni sono pesati, e sarà coperto da un vetro d'orologio.
Ogni bicchiere sarà contraddistinto, mediante un sistema di scrittura inodore, da un codice composto da cifre, o da cifre e lettere, scelto aleatoriamente.
8.2. Temperatura del campione e della sala
I campioni di olio oggetto della prova sensoriale devono essere mantenuti a una temperatura di 28 °C ± 2 °C per tutta la durata della prova. Questa temperatura è stata scelta perché, a differenza della temperatura ambiente, consente di rilevare più facilmente le differenze organolettiche. A temperature inferiori si ha infatti una scarsa volatilizzazione dei composti aromatici propri degli oli, mentre temperature superiori portano alla formazione dei composti volatili propri degli oli riscaldati. V. norma COI/T.20/Doc. n. 5 «Bicchiere per l'assaggio di oli» per quanto riguarda il sistema di riscaldamento dei campioni, da usare quando i campioni sono stati introdotti nei bicchieri.
La temperatura della sala di assaggio deve essere compresa tra 20 ° e 25 °C (v. COI/T. 20/Doc. n. 6).
8.3. Orario delle prove
Le ore di lavoro più idonee all'assaggio di oli sono quelle del mattino: è stato dimostrato che durante la giornata si hanno dei momenti in cui la percezione di gusti e odori è ottimale. I pasti sono preceduti da un periodo di aumento della sensibilità olfatto-gustativa, e seguiti da una diminuzione.
Tale criterio tuttavia non va applicato in modo troppo radicale e occorre evitare che gli assaggiatori siano distratti dalla fame, con la conseguente riduzione della capacità di discriminazione. Le prove di assaggio dovrebbero pertanto essere organizzate tra le 10 e le 12 del mattino.
8.4. Norme generali di condotta per gli assaggiatori
Si indicano di seguito alcune raccomandazioni circa il comportamento richiesto agli assaggiatori durante la prova.
Ricevuto da parte del responsabile del panel l'invito a partecipare alla prova organolettica, l'assaggiatore si dispone a effettuarla all'ora indicata, attenendosi a quanto segue.
— Non fumare e non bere caffè per almeno 30 minuti prima dell'ora fissata per la prova.
— Non aver usato profumi, cosmetici o saponi il cui odore può persistere al momento della prova. Per lavarsi le mani, servirsi di un sapone non profumato, poi sciacquarle e asciugarle in modo da eliminare ogni traccia di odore.
— Non mangiare nulla per almeno un'ora prima della prova.
— Se le sue condizioni fisiologiche sono compromesse, specie in caso di alterazione del senso dell'olfatto o del gusto, o se risente di qualsiasi effetto psicologico che può impedirgli di concentrarsi sul suo lavoro, dovrà astenersi dall'assaggio e comunicarlo al capo panel.
— L'assaggiatore, rispettate le norme precedenti, occuperà il suo posto nella cabina assegnatagli, nella maniera più ordinata e silenziosa possibile.
— Leggerà con attenzione le istruzioni contenute nella scheda di profilo e non inizierà l'esame del campione finché non sarà totalmente pronto a svolgere il suo compito (rilassato e non affrettato). In caso di dubbio, si consulterà in privato con il capo del panel.
— Realizzerà il suo lavoro in silenzio.
— Manterrà spento il telefono cellulare, per salvaguardare la concentrazione e il lavoro dei suoi colleghi.
9. PROCEDURA DI VALUTAZIONE ORGANOLETTICA E CLASSIFICAZIONE DELL'OLIO DI OLIVA VERGINE
9.1. Tecnica di assaggio
9.1.1. L'assaggiatore prenderà il bicchiere tenendolo coperto con il vetro di orologio, lo inclinerà leggermente e in questa posizione lo girerà completamente per bagnare il più possibile la superficie interna. Fatto ciò, separerà il vetro d'orologio e odorerà il campione, facendo inspirazioni lente e profonde, al fine di valutarlo. Il periodo di odorazione non deve eccedere i 30 secondi. Se in questo periodo non è giunto a nessuna conclusione, l'assaggiatore farà una pausa e procederà a un nuovo tentativo.
Conclusa la prova olfattiva, procederà alla valutazione delle sensazioni orali (sensazione congiunta olfatto-gustativa per via retronasale e tattile). Prenderà un sorso d'olio di circa 3 ml. È importante ripartire l'olio per tutta la cavità orale, dalla parte anteriore e dalla lingua, passando sulle parti laterali e la parte posteriore, fino al velo palatino e alla gola, in quanto, come è noto, la percezione dei sapori e delle sensazioni tattili varia d'intensità secondo le zone della lingua, del palato e della gola.
Si deve insistere sulla necessità che l'olio si spanda in quantità sufficiente e molto lentamente dalla parte posteriore della lingua verso il velo palatino e la gola, concentrando l'attenzione sull'ordine di apparizione degli stimoli amaro e piccante; in caso contrario, per alcuni oli i due stimoli potrebbero passare inavvertiti o l'amaro potrebbe essere coperto dal piccante.
Aspirazioni corte e successive, attraverso la bocca, permettono sia di estendere il campione nella cavità orale sia di percepire i componenti volatili aromatici mediante il passaggio forzato per la via retronasale.
NB: Qualora l'assaggiatore non percepisca il fruttato in un campione e l'intensità dell'attributo negativo di classificazione sia pari o inferiore a 3,5 il capo panel può decidere di far analizzare nuovamente all'assaggiatore il campione a temperatura ambiente (COI/T.20/Doc. n. 6/Rev. 1, settembre 2007, punto 3 — Guida per l'allestimento di una sala di assaggio), specificando il contesto e il concetto di temperatura ambiente. Quando il campione raggiunge la temperatura ambiente, l'assaggiatore deve procedere a una nuova valutazione per esaminare esclusivamente la presenza di fruttato. In caso affermativo, deve contrassegnare l'intensità sulla scala.
Occorre tener conto anche della sensazione tattile del piccante ed è pertanto opportuno che l'olio venga inghiottito.
9.1.2. Per gli oli vergini si raccomanda di effettuare la valutazione organolettica su un numero di campioni non superiore a QUATTRO per seduta, con un massimo di 3 sedute al giorno, per evitare l'effetto di contrasto che potrebbe produrre l'assaggio immediato di altri campioni.
Poiché gli assaggi successivi sono alterati dalla fatica o dalla perdita di sensibilità dovuta ai precedenti, sarà necessario servirsi di un prodotto capace di eliminare dalla bocca i resti d'olio dell'assaggio precedente.
Si raccomanda l'uso di un pezzettino di mela che, una volta masticato, può essere sputato; sciacquarsi poi con un poco d'acqua a temperatura ambiente. Tra un assaggio e l'altro devono passare almeno 15 minuti.
9.2. Uso della scheda di profilo da parte dell'assaggiatore
La scheda di profilo ad uso dell'assaggiatore è oggetto della figura 1 del presente Allegato.
Ogni assaggiatore membro del panel deve odorare l'olio sottoposto ad esame, e poi passare all'assaggio ( 5 ). In seguito appunterà sulla scala di 10 cm della scheda di profilo a sua disposizione l'intensità alla quale percepisce ciascuno degli attributi negativi e positivi.
Nel caso in cui fossero percepiti attributi negativi non enumerati al punto 4, questi devono essere indicati alla voce "altri" impiegando il o i termini che li descrivono con la maggior precisione.
9.3. Uso dei dati da parte del capo panel
Il capo panel raccoglie le schede di profilo compilate dagli assaggiatori e controlla le intensità assegnate ai diversi attributi; se constata un'anomalia, chiede all'assaggiatore di rivedere la sua scheda di profilo e, se necessario, di ripetere la prova.
Il capo panel introduce i dati della valutazione di ogni componente del panel in un programma informatico come quello previsto dalla norma COI/T.20/Doc. n. 15 e procede a calcolare statisticamente i risultati dell'analisi, basandosi sul calcolo della mediana. V. punto 9.4 e Appendice del presente Allegato. L'inserimento dei dati per un campione va effettuato servendosi di una matrice composta di 9 colonne corrispondenti ai 9 attributi sensoriali e di 9 righe corrispondenti agli n componenti del panel impiegati.
Quando un difetto percepito è riportato alla voce «altri» da almeno il 50 % del panel, il capo panel deve procedere al calcolo della mediana del difetto in questione e alla corrispondente classificazione.
Il valore del coefficiente di variazione robusto che definisce la classificazione (difetto con l'intensità più alta e attributo fruttato) deve essere inferiore o pari al 20 %.
Altrimenti, il capo panel deve ripetere la valutazione del campione in questione in una seduta di assaggio distinta.
Se tale situazione si verifica frequentemente, si raccomanda al capo panel di fornire agli assaggiatori un ulteriore addestramento specifico (COI/T.20/Doc. n. 14, § 5) e di controllare le prestazioni dell'assaggiatore avvalendosi dell'indice di ripetibilità e di deviazione (COI/T.20/Doc. n. 14, § 6).
9.4. Classificazione dell'olio di oliva
L'olio è classificato nelle categorie sotto riportate in funzione della mediana dei difetti e della mediana dell'attributo fruttato. Per mediana dei difetti si intende la mediana del difetto percepito con l'intensità più alta. La mediana dei difetti e la mediana del fruttato sono espresse con una sola cifra decimale.
La classificazione dell'olio avviene confrontando il valore della mediana dei difetti e della mediana del fruttato con gli intervalli di riferimento indicati di seguito. Poiché i limiti di questi intervalli sono stati stabiliti tenendo conto del margine di errore del metodo, sono considerati assoluti. I programmi informatici consentono di visualizzare la classificazione su una tabella di dati statistici o un grafico.
a) Olio extravergine di oliva: la mediana dei difetti è pari a 0 e la mediana del fruttato è superiore a 0;
b) olio di oliva vergine: la mediana dei difetti è superiore a 0 e inferiore o pari a 3,5 e la mediana del fruttato è superiore a 0;
c) olio di oliva vergine lampante: la mediana dei difetti è superiore a 3,5 oppure la mediana dei difetti è inferiore o pari a 3,5 e la mediana del fruttato è pari a 0.
Nota 1: quando la mediana dell'amaro e/o piccante è superiore a 5,0, il capo panel lo segnalerà nel certificato di analisi dell'olio.
Per le analisi eseguite ai fini del controllo di conformità, si effettua una unica prova. Nel caso di analisi contraddittorie dev'essere effettuata l'analisi in doppio in sessioni di assaggio distinte. I risultati dell'analisi in doppio devono essere statisticamente omogenei. (Cfr. punto 9.5) In caso negativo, il campione deve essere analizzato ancora due volte. Il valore finale della mediana degli attributi di classificazione sarà calcolato sulla base della media di entrambe le mediane.
9.5 Criteri di accettazione e rifiuto di duplicati
L'errore normalizzato, definito di seguito, è usato per determinare se i due risultati dell'analisi in doppio sono omogenei o statisticamente accettabili:

Se Me1 e Me2 sono le mediane dei due duplicati (rispettivamente della prima e della seconda analisi) e U1 e U2 sono le incertezze estese ottenute per i due valori, calcolate come segue come specificato nell'appendice:
U
1 = c × s* and

Per l'incertezza estesa, c = 1,96; da cui
U1 = 0,0196 × CVr × Me1
dove CVr è il coefficiente di variazione robusto.
Per dichiarare che i due valori ottenuti non sono statisticamente differenti, En deve essere pari o inferiore a 1,0.
Appendice
Metodo di calcolo della mediana e degli intervalli di confidenza
Mediana
![]()
La mediana è definita come il numero reale Xm caratterizzato dal fatto che la probabilità (p) che i valori della distribuzione (X) siano minori di questo numero (Xm) è minore o uguale a 0,5 e che, contemporaneamente, la probabilità (p) che i valori della distribuzione (X) siano minori o uguali a Xm è maggiore o uguale a 0,5. Una definizione più operativa è quella che definisce la mediana come il 50o percentile di una distribuzione di numeri ordinata in modo crescente. In termini più semplici, essa rappresenta il valore centrale di una serie ordinata di numeri dispari, oppure la media dei due valori centrali di una serie ordinata di numeri pari.
Deviazione standard robusta
Per avere una stima attendibile della variabilità intorno alla mediana ci si rifà alla stima della deviazione standard robusta secondo Stuart e Kendall (4). La formula indica la deviazione standard robusta asintotica, ossia la stima robusta della variabilità dei dati considerati, in cui N è il numero di osservazioni e IQR l'intervallo interquartile, che racchiude esattamente il 50% dei casi di una data distribuzione probabilistica:
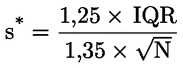
Il calcolo dell'intervallo interquartile si esegue calcolando la grandezza dello scarto tra il 75o e il 25o percentile.
![]()
in cui il percentile è quel valore Xpc caratterizzato dal fatto che la probabilità (p) che i valori della distribuzione siano minori di Xpc è minore o uguale a un determinato centesimo e che, contemporaneamente, la probabilità (p) che i valori della distribuzione siano minori o uguali a Xpc è maggiore o uguale a quel determinato centesimo. Il centesimo indica la frazione di distribuzione scelta. Nel caso della mediana questa è pari a 50/100.

Operativamente, il percentile è quel valore di distribuzione che corrisponde a una determinata area sottesa dalla curva di distribuzione o di densità. Ad esempio, il 25o percentile rappresenta il valore di distribuzione corrispondente a un'area pari a 0,25 o 25/100.
Questo metodo prevede il computo dei percentili sulla base dei valori reali figuranti nella matrice dei dati (procedura di computo dei percentili).
Coefficiente di variazione robusto (in %)
Il CVr% rappresenta un numero puro che indica la percentuale di variabilità della serie di numeri analizzata; per questo motivo risulta molto informativo sull'attendibilità dei giudici del panel.
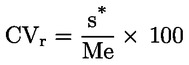
Intervalli di confidenza della mediana al 95%
Gli intervalli di confidenza al 95% (valore dell'errore del primo tipo pari a 0,05 o 5%) rappresentano l'intervallo entro il quale il valore della mediana potrebbe variare se fosse possibile ripetere infinite volte un esperimento. In pratica indicano l'intervallo di variabilità della prova nelle condizioni operative adottate qualora si potesse ripeterla parecchie volte. L'intervallo aiuta a valutare, come con il CVr%, l'attendibilità della prova.

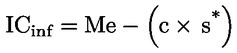
in cui C = 1,96 per l'intervallo di confidenza al 95%.
Un esempio di foglio di calcolo è riportato nell'allegato I della norma COI/T 20/Doc. n. 15.
Bibliografia
(1) Wilkinson, L. 1990. Systat: The system for statistics. Evanston, IL.SYSTAT Inc.
(2) Cicchitelli, G. 1984. Probabilità e Statistica. Maggioli Editore, Rimini.
(3) Massart, D.L.; Vandeginste, B.G.M.; Deming, Y.; Michotte, L. 1988. Chemometrics. A textbook. Elsevier. Amsterdam.
(4) Kendall, M.G.; Stuart, A. 1967. The advanced theory of statistics. Vol. 1. Hafner Publishing Co.
(5) McGill, R.; Tukey, J.W.; Larsen, W.A. 1978. Variation of Box Plots. The American Statistician, 32, (2), 12-16.
(6) COI/T.28/Doc. n. 1 Settembre 2007, Linee guida per l'accreditamento dei laboratori di analisi sensoriale con particolare riguardo all'olio vergine di oliva secondo la norma ISO/IEC 17025:2005.
(7) COI/T.20/Doc. n. 14.
(8) COI /T.20/Doc. n. 15.
(9) ISO/IEC 17025:05.
▼M20 —————
▼M19 —————
ALLEGATO XV
1. METODO DI DETERMINAZIONE DEL TENORE IN OLIO D'OLIVA DELLE SANSE
1.1. Materiale
— apparecchio da estrazione appropriato, munito di un pallone da 200-250 ml,
— bagno a riscaldamento elettrico (bagno a sabbia, bagno ad acqua, ecc.) o piastra riscaldante,
— bilancia analitica,
— stufa regolata su un massimo di 80 °C,
— stufa a riscaldamento elettrico, provvista di un dispositivo di termoregolazione regolato su 103 ± 2 °C e tale da consentire una insufflazione d'aria o una depressione,
— frantoio meccanico facile da pulire, che permette la frantumazione dei noccioli senza riscaldamento e senza modificazione sensibile del loro tenore in acqua e in olio,
— ditale da estrazione e cotone idrofilo o carta da filtro, esenti da sostanze estraibili con esano,
— essiccatore,
— setaccio a maglie da 1 mm di diametro,
— pietra pomice in granuli, previamente essiccata.
1.2. Reattivo
n-esano tecnico, il cui residuo all'evaporazione completa dev'essere inferiore a 0,002 g/100 ml.
2. MODO DI OPERARE
2.1. Preparazione del campione per l'analisi
Frantumare il campione contrattuale, se necessario, nel frantoio meccanico ben pulito in precedenza, allo scopo di ridurlo in particelle che attraversino completamente il setaccio.
Utilizzare 1/20 circa del campione per completare la pulizia del frantoio, scartare il prodotto di questa macinazione, frantumare il resto, raccoglierlo, mescolarlo con cura e analizzarlo immediatamente.
2.2. Quantità di sostanza da analizzare
Immediatamente dopo la fine della frantumazione, pesare con l'approssimazione di 0,01 g circa 10 g del campione.
2.3. Preparazione del ditale da estrazione
Porre la sostanza destinata all'analisi nella cartuccia, che va tappata con il tampone di cotone idrofilo. Nel caso che si utilizzi una carta da filtro, impacchettare le sanse frantumate in tale carta.
2.4. Preessiccazione
Se la sansa è molto umida (tenore in acqua ed in sostanze volatili superiore al 10 %), effettuare un'essiccazione preliminare ponendo per un tempo conveniente il ditale riempito (o la carta da filtro) nella stufa riscaldata ad un massimo di 80 °C, per ricondurre il tenore in acqua ed in materie volatili al di sotto del 10 %.
2.5. Preparazione del pallone
Pesare con l'approssimazione di 1 mg il pallone contenente 1-2 granuli di pomice, previamente essiccato in stufa a 103 ± 2 °C e poi raffreddato per almeno un'ora in essiccatore.
2.6. Prima estrazione
Porre nell'apparecchio da estrazione il ditale (o la carta da filtro) contenente la sostanza da analizzare. Versare nel pallone la quantità necessaria di esano. Adattare il pallone all'apparecchio da estrazione e porre il tutto sul bagno a riscaldamento elettrico. Effettuare il riscaldamento in condizioni tali che la portata del riflusso sia di almeno tre gocce al secondo (ebollizione moderata, non tumultuosa).
Dopo quattro ore di estrazione, lasciar raffreddare. Togliere il ditale dall'apparecchio di estrazione e porlo in una corrente d'aria, al fine di eliminare la maggior parte del solvente che lo impregna.
2.7. Seconda estrazione
Vuotare il ditale nel microfrantoio e macinare il più finemente possibile. Reintrodurre quantitativamente la miscela nel ditale e rimettere questo nell'apparecchio da estrazione.
Ricominciare l'estrazione per altre due ore, utilizzando lo stesso pallone che contiene la prima sostanza estratta.
La soluzione ottenuta nel pallone da estrazione dev'essere limpida. Se così non fosse, filtrarla su carta, lavando più volte il primo pallone e la carta da filtro con esano. Raccogliere filtrato e solvente di lavaggio in un secondo pallone, previamente essiccato e tarato con l'approssimazione di 1 mg.
2.8. Eliminazione del solvente e pesata dell'estratto
Eliminare la maggior parte del solvente distillato su bagno a riscaldamento elettrico. Eliminare le ultime tracce di solvente riscaldando il pallone in stufa a 103 ± 2 °C per 20 minuti. Facilitare questa eliminazione sia insufflando ogni tanto dell'aria o, preferibilmente, del gas inerte, sia operando sotto pressione ridotta.
Lasciar raffreddare il pallone in essiccatore per almeno un'ora, poi pesarlo con l'approssimazione di 1 mg.
Riscaldare di nuovo per 10 minuti nelle stesse condizioni, poi raffreddare in essiccatore e pesare.
La differenza fra i risultati di queste due pesate dev'essere inferiore od uguale a 10 mg. In caso contrario, riscaldare di nuovo per periodi di 10 minuti, seguiti da raffreddamento e pesata, finché la differenza di massa sia uguale tutt'al più a 10 mg. Adottare per il calcolo il valore dato dall'ultima pesata.
Effettuare due determinazioni sullo stesso campione.
3. ESPRESSIONE DEI RISULTATI
3.1. Modo di calcolo e formula
a) L'estratto, espresso come massa percentuale sul prodotto tal quale, è dato dalla formula:

nella quale:
S = percentuale in massa sul prodotto tal quale,
m0 = massa in grammi della quantità di sostanza prelevata per l'analisi,
m1 = massa in grammi dell'estratto dopo essiccazione.
Prendere come risultato la media aritmetica delle due determinazioni, se le condizioni di ripetibilità sono adempiute.
Esprimere il risultato con una sola cifra decimale.
b) L'estratto viene riferito alla sostanza secca utilizzando la formula seguente:

dove:
S = percentuale in massa di estratto sul prodotto tal quale (vedi lettera a),
U = suo tenore in acqua e in sostanze volatili.
3.2. Ripetibilità
La differenza fra i risultati di due determinazioni effettuate simultaneamente o in rapida successione dallo stesso analista non deve eccedere i 0,2 g di estratto ottenuto con l'esano per ogni 100 g di campione.
In caso contrario, ripetere l'analisi su due altri quantitativi di sostanza. Se ancora questa volta la differenza eccede gli 0,2 g, assumere come risultato la media aritmetica delle quattro determinazioni effettuate.
ALLEGATO XVI
DETERMINAZIONE DEL NUMERO DI IODIO
1. OGGETTO
La presente norma internazionale definisce un metodo per la determinazione del numero di iodio nei grassi e negli oli di origine animale e vegetale, qui di seguito denominati «grassi».
2. DEFINIZIONE
Ai fini della presente norma internazionale, si applica la seguente definizione.
|
2.1. |
Numero di iodio: la massa di iodio assorbita dal campione nelle condizioni specificate nella presente norma internazionale. Il numero di iodio viene espresso in grammi di iodio per 100 g di campione. |
3. PRINCIPIO
Scioglimento della sostanza da analizzare nel solvente ed aggiunta di reagente di Wijs. Trascorso un certo lasso di tempo, aggiunta di soluzione di ioduro di potassio e di acqua e titolazione dello iodio liberato con soluzione di tiosolfato di sodio.
4. REAGENTI
Tutti i reagenti devono essere di grado analitico riconosciuto.
|
4.1. |
Ioduro di potassio, soluzione di 100 g/l, non contenente iodato o iodio libero. |
|
4.2. |
Amido, soluzione. Versare 5 g di amido solubile in 30 ml d'acqua, aggiungere questa miscela a 1 000 ml di acqua bollente, fare bollire per 3 minuti e lasciar raffreddare. |
|
4.3. |
Tiosolfato di sodio, soluzione volumetrica standard c (Na2S2O3.5H2O) = 0,1 mol/l, standardizzato non oltre 7 giorni prima dell'uso. |
|
4.4. |
Solvente, preparato miscelando volumi eguali di cicloesano e di acido acetico. |
|
4.5. |
Reagente di Wijs, contenente monocloruro di iodio in acido acetico. È opportuno usare il reagente di Wijs disponibile in commercio. Nota: Il reagente contiene 9 g di ICl3 + 9 g di I in acido acetico. |
5. APPARECCHIATURA
La consueta apparecchiatura di laboratorio e in particolare quanto segue.
|
5.1. |
Ditali da pesata, in vetro, idonei per la sostanza da analizzare e per l'inserimento nelle beute (5.2). |
|
5.2. |
Beute, aventi una capacità di 500 ml, provviste di tappi in vetro smerigliato e completamente asciutte. |
6. PREPARAZIONE DEL CAMPIONE DI SOSTANZA DA ANALIZZARE
Il campione omogeneizzato è seccato su solfato sodico e filtrato.
7. PROCEDIMENTO
|
7.1. |
Sostanza da analizzare Il peso della sostanza da analizzare varia a seconda del numero di iodio che si prevede, come indicato nella tabella 1.
Tabella 1
Pesare la sostanza da analizzare con l'approssimazione di 0,1 mg in un ditale da pesata in vetro (5.1). |
|
7.2. |
Determinazione Versare la sostanza da analizzare in una beuta da 500 ml (5.2). Aggiungere 20 ml del solvente (4.4) in modo da sciogliere li grasso. Aggiungere esattamente 25 ml del reagente di Wijs (4.5), inserire il tappo, agitare il contenuto e riporre la beuta al buio. Per il reagente di Wijs non usare una pipetta a bocca. Analogamente preparare un bianco col solvente ed il reagente, ma tralasciando la sostanza da analizzare. Per le sostanze aventi un numero di iodio inferiore a 150, mantenere le beute al buio per un'ora; per quelle aventi un numero di iodio superiore a 150 nonché per i prodotti polimerizzati oppure per i prodotti notevolmente ossidati, lasciar riposare per due ore. Trascorso il periodo necessario, aggiungere 20 ml della soluzione di ioduro di potassio (4.1) e 150 ml di acqua a ciascuna delle beute. Titolare con la soluzione volumetrica standard di tiosolfato di sodio (4.3) finché la colorazione gialla dovuta allo iodio non sia quasi scomparsa. Aggiungere alcune gocce della soluzione di amido (4.2) e continuare la titolazione finché la colorazione blu sia appena scomparsa a seguito di agitazione molto vigorosa. Nota — È consentita la determinazione potenziometrica del punto finale. |
|
7.3. |
Numero di determinazioni Effettuare due determinazioni sullo stesso campione. |
8. ESPRESSIONE DEI RISULTATI
Il numero di iodio viene dato dalla seguente espressione:

Dove:
c = è il numerico della concentrazione esatta, in moli per litro, della soluzione di Tiosolfato di sodio Titolata (5.4) utilizzata;
V1 = è il valore numerico del volume, in ml, della soluzione di Tiosolfato di sodio Titolata (5.4) utilizzata;
V2 = è il valore numerico del volume, in ml, delle soluzioni di Tiosolfato di sodio (5.4) utilizzato per la determinazione;
m = è il valore numerico del peso, in g, della sostanza da analizzare (7.1).
Prendere come risultato la media aritmetica di due determinazioni.
ALLEGATO XVII
METODO DI DETERMINAZIONE DEGLI STIGMASTADIENI NEGLI OLI VEGETALI
1. SCOPO
Determinazione degli stigmastadieni negli oli vegetali contenenti basse concentrazioni di questi idrocarburi, soprattutto oli d'oliva vergini e oli di sansa d'oliva grezzi.
2. OGGETTO
Il metodo può essere applicato a tutti gli oli vegetali, ma è attendibile soltanto se il tenore di questi idrocarburi è compreso tra 0,01 e 4,0 mg/kg. Esso è particolarmente adatto a rivelare la presenza di oli vegetali raffinati (oliva, sansa, girasole, palma, ecc.) nell'olio di oliva vergine, dato che gli oli raffinati contengono stigmastadieni, mentre gli oli vergini non li contengono.
3. PRINCIPIO
Isolamento dell'insaponificabile. Separazione della frazione costituita dagli steroidi a carattere di idrocarburi mediante cromatografia su colonna di gel di silice e analisi mediante gascromatografia su capillare.
4. APPARECCHIATURA
|
4.1. |
Palloni idonei da 250 ml, con condensatore a riflusso. |
|
4.2. |
Imbuti separatori da 500 ml. |
|
4.3. |
Palloni a fondo rotondo da 100 ml. |
|
4.4. |
Evaporatore rotante. |
|
4.5. |
Colonna per cromatografia in vetro (1,5-2,0 cm di diametro interno, della lunghezza di 50 cm) provvista di rubinetto in teflon e di tappo in fibra di lana di vetro o disco di vetro sinterizzato all'estremità inferiore. Per preparare la colonna di gel di silice versare l'esano nella colonna cromatografica fino a raggiungere uno spessore di circa 5 cm e riempire quindi con un impasto di gel di silice in esano (15 g in 40 ml) aiutandosi con porzioni di esano. Lasciar depositare completando poi il deposito con leggere vibrazioni. Aggiungere solfato di sodio anidro fino all'ottenimento di uno spessore di circa 0,5 cm ed infine eluire l'esano in eccesso. |
|
4.6. |
Gascromatografo provvisto di rilevatore a ionizzazione di fiamma, iniettore a separazione o a freddo, lungo la colonna e stufa programmabile con l'approssimazione di ± 1 °C. |
|
4.7. |
Colonna capillare di silice fusa per gascromatografia (0,25 o 0,30 mm di diametro interno, della lunghezza di 25 m) ricoperte di fase di fenilmetilsilicone al 5 %, spessore 0,25 mm. Nota 1. Possono essere usate altre colonne di polarità equivalente o inferiore. |
|
4.8. |
Registratore-integratore con possibilità d'integrazione da valle a valle. |
|
4.9. |
Microsiringa per gascromatografia da 5-10 ml con ago cementato. |
|
4.10. |
Camicia di riscaldamento o piastra termica, elettrica. |
5. REAGENTI
Tutti i reagenti debbono essere puri per analisi se non specificato diversamente. L'acqua usata dev'essere distillata oppure di purezza per lo meno equivalente.
|
5.1. |
Esano o miscela di alcani con intervallo di ebollizione a 65-70 °C, distillato su colonna di rettificazione. Nota 2. Il solvente dev'essere distillato in modo da eliminare le impurezze. |
|
5.2. |
Etanolo al 96 % v/v. |
|
5.3. |
Solfato di sodio anidro. |
|
5.4. |
Soluzione alcolica di idrossido di potassio al 10 %. Aggiungere 10 ml d'acqua a 50 g di idrossido di potassio, agitare e sciogliere quindi la miscela in etanolo fino a 500 ml. Nota 3. La potassa alcolica vira al bruno se lasciata riposare. Dev'essere preparata di fresco ogni giorno e tenuta in bottiglie di vetro scure ben tappate. |
|
5.5. |
Gel di silice 60 per cromatografia su colonna 70-230 mesh (Merck, ref. 7734 o simili). Nota 4. Di regola il gel di silice può essere usato prelevandolo direttamente dal contenitore senza alcun trattamento preliminare. Tuttavia alcune partite di silice sono scarsamente attive e determinano separazioni cromatografiche di cattiva qualità. In casi del genere, il gel di silice dev'essere disattivato scaldandolo per almeno quattro ore a 550 °C; successivamente, sistemarlo in un essiccatore fino a raffreddamento e trasferirlo quindi in un pallone provvisto di tappo. Aggiungere il 2 % di acqua e agitare fino a scomparsa dei grumi e libero flusso della polvere. Il gel di silice dev'essere trattato come sopra se le partite di gel di silice danno cromatogrammi con picchi di interferenza. Come alternativa, può essere usato gel di silice 60 extra puro (Merck, ref. 7754). |
|
5.6. |
Soluzione madre (200 ppm) di colesta-3,5-diene (Sigma, purezza 99 %) in esano (10 mg in 50 ml). |
|
5.7. |
Soluzione standard di colesta-3,5-diene in esano alla concentrazione di 20 ppm, ottenuta diluendo la soluzione di cui sopra. Nota 5. Le soluzioni 5.6 e 5.7 non si deteriorano per almeno 4 mesi se conservate a una temperatura inferiore ai 4 °C. |
|
5.8. |
Soluzione di n-nonacosano in esano ad una concentrazione di circa 100 ppm. |
|
5.9. |
Gas vettore per cromatografia: idrogeno o elio puro al 99,9990 %. |
|
5.10. |
Gas ausiliari per il rivelatore a ionizzazione di fiamma: idrogeno puro al 99,9990 % ed aria purificata. |
6. PROCEDIMENTO
6.1. Preparazione dell'insaponificabile:
|
6.1.1. |
Pesare 20 g, con l'approssimazione di ± 0,1 di olio in un pallone da 250 ml (4.1), aggiungere 1 ml della soluzione standard di colesta-3,5-diene (20mg) e 75 ml di potassa alcolica al 10 %, preparare il condensatore a riflusso e portare a leggera ebollizione per 30 minuti. Allontanare il pallone contenente il campione dalla fonte di calore e lasciare raffreddare leggermente la soluzione (non far raffreddare completamente, altrimenti il campione si depositerebbe). Aggiungere 100 ml d'acqua e trasferire la soluzione in un imbuto a decantazione (4.2) con l'ausilio di 100 ml di esano. Agitare la miscela vigorosamente per 30 secondi e lasciar stratificare. Nota 6. Se si forma un'emulsione che non scompare rapidamente, aggiungere piccoli quantitativi di etanolo. |
|
6.1.2. |
Trasferire la fase acquosa inferiore in un secondo imbuto separatore ed estrarre nuovamente con 100 ml di esano. Eliminare ancora la fase inferiore e lavare gli estratti di esano (raccolti in un altro imbuto separatore) tre volte con tre porzioni, di 100 ml ciascuna, di una miscela etanolo-acqua (1: 1) fino a raggiungimento di pH neutro. |
|
6.1.3. |
Far passare la soluzione di esano attraverso del solfato di sodio anidro (50 g), lavare con 20 ml di esano a far evaporare in evaporatore rotante a 30 °C e bassa pressione fino a secchezza. |
6.2. Separazione della frazione di idrocarburo steroidico:
|
6.2.1. |
Trasferire il residuo nella colonna di frazionamento con l'ausilio di due porzioni di esano da 1 ml, far passare il campione attraverso la colonna lasciando che la soluzione scenda fino alla somità del solfato di sodio e avviare l'eluizione cromatografica con esano ad una velocità di efflusso di 1 ml/min. circa. Eliminare i primi 25-30 ml dell'eluizione e raccogliere quindi la rimanente frazione di 40 ml. Dopo averla raccolta, trasferirla in un pallone a fondo rotondo da 100 ml (4.3). Nota 7. La prima frazione contiene idrocarburi saturi (figura 1a), la seconda quelli steroidici. Continuando l'eluizione si ottiene squalene e composti connessi. Ai fini di una buona separazione tra idrocarburi saturi e steroidici, è necessario ottimalizzare le frazioni di volume. A questo scopo il volume della prima frazione dev'essere regolato in modo che, quando viene analizzata la seconda frazione, i picchi che rappresentano gli idrocarburi saturi siano bassi (vedasi figura 1c); se essi non compaiono, ma l'intensità del picco standard è bassa, il volume dev'essere ridotto. In ogni caso non è necessaria una separazione completa dei componenti della prima e seconda frazione, dato che durante l'analisi gascromatografica non vi è sovrapposizione di picchi, se detta analisi viene eseguita nelle condizioni precisate al paragrafo 6.3.1. In generale non è necessaria l'ottimalizzazione della seconda frazione in volume, data la buona separazione degli altri componenti. Tuttavia la presenza di una grosso picco per un tempo di ritenzione inferiore di circa 1,5 minuti rispetto allo standard è dovuta allo squalene e sta ad indicare una cattiva separazione. |
|
6.2.2. |
Evaporare la seconda frazione in un evaporatore a 30 °C e bassa pressione fino a secchezza e sciogliere immediatamente il residuo in 0,2 ml di esano. Conservare la soluzione in frigorifero fino all'analisi. Nota 8. I residui 6.1.3 e 6.2.2 non devono essere lasciati asciugare, né tenuti a temperatura ambiente. Non appena essi vengono ottenuti, è necessario aggiungere il solvente e conservare le soluzioni in frigorifero. |
6.3. Gascromatografia:
|
6.3.1. |
Condizioni operative per l'iniezione a separazione: — Temperatura dell'iniettore: 300 °C. — Temperatura del rivelatore: 320 °C. — Registratore-integratore: i parametri di integrazione devono essere fissati in modo che forniscano una corretta valutazione delle aree. Si raccomanda un'integrazione da valle a valle. — Sensibilità: circa 16 volte l'attenuazione minima. — Quantitativo di soluzione iniettato: 1ml. — Temperature di programmazione della stufa: inizialmente 235 °C per 6 min. e successivamente aumento di 2 °C/min. fino a 285 °C. — Iniettore provvisto di separatore di flusso a 1: 15. — Vettore: elio o idrogeno a una pressione di circa 120 kPa. Queste condizioni possono essere adeguate alle caratteristiche del cromatografo e della colonna in modo da ottenere cromatogrammi che rispettino i seguenti requisiti: picco dello standard interno entro 5 min. circa dei tempi definiti al paragrafo 6.3.2; detto picco dev'essere pari ad almeno l'80 % della scala completa. Il sistema gascromatografico deve essere verificato iniettando una miscela della soluzione madre di colestadiene (5.6) con la soluzione di n-nonacosano (5.8). Il picco del colesta-3,5-diene deve comparire prima di quello dell'n-nonacosano (figura 1c); se ciò non succede, si hanno due possibilità: abbassare la temperatura della stufa e/o usare una colonna meno polare. |
|
6.3.2. |
Identificazione del picco Il picco dello standard interno compare dopo circa 19 min. e lo stigmasta-3,5-diene, a un tempo di ritenzione relativo di circa 1,29 (cfr. figura 1b). Lo stigma-3,5-diene è associato a piccoli quantitativi di un isomero e di solito entrambi danno origine a un unico picco cromatografico. Tuttavia, se la colonna è troppo polare oppure mostra un forte potere risolvente, l'isomero può comparire sotto forma di piccolo picco prima e accanto a quello dello stigmasta-3,5-diene (Figura 2). Per essere certi che gli stigmastadieni vengano eluiti in un picco unico, è consigliabile sostituire la colonna con una meno polare oppure con una di diametro interno superiore. Nota 9. Gli stigmastadieni di riferimento possono essere ottenuti dall'analisi di un olio vegetale raffinato usando un quantitativo inferiore di campione (1-2 g). Gli stigmastadieni danno un picco significativo e facilmente identificabile. |
|
6.3.3. |
Analisi quantitativa Il tenore di stigmastadieni viene determinato con la formula seguente: mg/kg di stigmastadieni =
Limite di rivelazione: circa 0,01 mg/kg. |
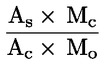


Figura 1
Gascromatogrammi ottenuti da campioni di olio d'oliva analizzati su colonna capillare di silice fusa (0,25 mm di diametro interno, della lunghezza di 25 m) ricoperti di fenilmetilsilicone al 5 %, con uno spessore 0,25 mm.
a) Prima frazione (30 ml) di olio vergine, addizionata intenzionalmente con lo standard.
b) Seconda frazione (40 ml) di olio d'oliva contenente 0,10 mg/kg di stigmastadieni.
c) Seconda frazione (40 ml) contenente una piccola proporzione della prima frazione.

Figura 2
Gascromatogramma ottenuto da un campione di olio di oliva raffinato analizzato su colonna DB-5 che mostra l'isomero dello stigmasta-3,5-diene.
ALLEGATO XVIII
DETERMINAZIONE DELLA DIFFERENZA TRA IL CONTENUTO EFFETTIVO E IL CONTENUTO TEORICO DI TRIACILGLICEROLI CON ECN 42
1. OGGETTO
Determinazione della differenza assoluta tra i valori sperimentali dei triacilgliceroli (TAG) con numero di carbonio equivalente pari a 42 (ECN 42HPLC) ottenuti mediante determinazione nell’olio per cromatografia liquida ad alta prestazione (HPLC) e il valore teorico dei TAG con un numero di carbonio equivalente pari a 42 (ECN 42teorico), calcolato a partire dalla composizione degli acidi grassi.
2. CAMPO DI APPLICAZIONE
Il metodo è applicabile agli oli d’oliva. Mira a individuare la presenza di piccole quantità di oli di semi (ricchi in acido linoleico) in qualunque categoria di olio d’oliva.
3. PRINCIPIO
Per gli oli puri, il contenuto in triacilgliceroli con ECN 42, determinato mediante HPLC, corrisponde all’incirca al contenuto teorico di triacilgliceroli con ECN 42 (calcolato in base alla determinazione mediante GLC (gascromatografia gas-liquido) della composizione in acidi grassi). Una differenza superiore ai valori adottati per ciascun tipo di olio indica che l’olio contiene oli di semi.
4. METODO
Il metodo per il calcolo del contenuto teorico di triacilgliceroli con ECN 42 e della differenza rispetto ai dati HPLC si basa essenzialmente sulla coordinazione dei dati analitici ottenuti mediante altri metodi. È possibile distinguere tre fasi: determinazione della composizione in acidi grassi per cromatografia in fase gassosa su colonna capillare, calcolo della composizione teorica dei triacilgliceroli con ECN 42 e determinazione HPLC dei triacilgliceroli con ECN 42.
4.1. Apparecchiatura
|
4.1.1. |
Matracci a fondo arrotondato (palloni) da 250 e 500 ml. |
|
4.1.2. |
Becher da 100 ml. |
|
4.1.3. |
Colonna cromatografica in vetro (diametro interno: 21 mm,lunghezza 450 mm), provvista di rubinetto e cono normalizzato (femmina) allasommità. |
|
4.1.4. |
Imbuti separatori da 250 ml con cono normalizzato (maschio)alla base, che si può adattare alla la sommità della colonna. |
|
4.1.5. |
Bacchetta di vetro, lunghezza 600 mm. |
|
4.1.6. |
Imbuto di vetro, diametro 80 mm. |
|
4.1.7. |
Matracci volumetrici, 50 ml. |
|
4.1.8. |
Matracci volumetrici, 20 ml. |
|
4.1.9. |
Evaporatore rotante. |
|
4.1.10. |
Cromatografo in fase liquida ad alta prestazione, conregolazione termostatica della temperatura della colonna. |
|
4.1.11. |
Unità di iniezione di 10 μl. |
|
4.1.12. |
Rivelatore: rifrattometro differenziale. La sensibilità sututta la scala deve essere pari almeno a 10–4 unitàdell’indice refrattivo. |
|
4.1.13. |
Colonna: tubo in acciaio inossidabile della lunghezza di250 mm e del diametro interno di 4,5 mm, riempito con particelle di silice deldiametro di 5 μm, contenente il 22-23 % di carbonio sotto forma diottadecilsilano. |
|
4.1.14. |
Software di elaborazione dati. |
|
4.1.15. |
Fialette da circa 2 ml, con setti in teflon e cappucci avite. |
4.2. Reagenti
I reagenti devono evidenziare una purezza per analisi. I solventi di eluizione devono essere degassati e possono essere riciclati più volte senza ripercussioni sulle separazioni.
|
4.2.1. |
Etere di petrolio 40-60 °C, qualità per cromatografia oesano. |
|
4.2.2. |
Etere etilico, esente da perossidi, distillato da poco. |
|
4.2.3. |
Solvente per eluizione per purificare l’olio mediantecromatografia su colonna: miscela di etere di petrolio/etere etilico 87:13(v/v). |
|
4.2.4. |
Gel di silice, 70-230, tipo Merck 7734, con un tenore diacqua normalizzato al 5 % (m/m). |
|
4.2.5. |
Lana di vetro. |
|
4.2.6. |
Acetone per HPLC. |
|
4.2.7. |
Acetonitrile o propionitrile per HPLC. |
|
4.2.8. |
Solvente di eluzione per HPLC: acetonitrile + acetone(proporzioni da regolare in modo da ottenere la separazione desiderata:cominciare con una miscela 50:50), o propionitrile. |
|
4.2.9. |
Solvente di solubilizzazione: acetone. |
|
4.2.10. |
Trigliceridi di riferimento: si possono usare i trigliceridiche si trovano in commercio (tripalmitina, trioleina ecc.), riportando su ungrafico i rispettivi tempi di ritenzione in funzione del numero di carbonioequivalente, oppure utilizzare cromatogrammi di riferimento ottenuti impiegandoolio di soia, miscela 30:70 olio di soia - olio di oliva e olio di oliva puro(cfr. note 1 e 2 e figure 1, 2, 3 e 4). |
|
4.2.11. |
Colonna di estrazione in fase solida (SPE) con gel disilice, 1 g, 6 ml. |
4.3. Preparazione del campione
Poiché alcune sostanze interferenti possono dar luogo a risultati falsamente positivi, il campione deve essere sempre purificato secondo il metodo IUPAC 2.507, impiegato per la determinazione dei composti polari nei grassi di frittura.
4.3.1. Preparazione della colonnacromatografica
Riempire la colonna (4.1.3) con 30 ml circa di solvente dieluizione (4.2.3), introducendo poi nella colonna un tampone di lana di vetro(4.2.5) spingendolo sul fondo della colonna con la bacchetta di vetro(4.1.5).
In un becher da 100 ml, sospendere 25 g di gel di silice(4.2.4) in 80 ml di miscela di eluizione (4.2.3), trasferendola quindi nellacolonna mediante un imbuto di vetro (4.1.6).
Per assicurare il completo trasferimento del gel di silicenella colonna, lavare il becher con la miscela di eluizione e trasferire nellacolonna anche le porzioni di lavaggio.
Aprire il rubinetto e lasciar eluire il solvente dallacolonna fin a quando il suo livello supera di circa 1 cm quello del gel disilice.
4.3.2. Cromatografia sucolonna
In un matraccio tarato da 50 ml (4.1.7), pesare, con laprecisione di 0,001 g, 2,5 ± 0,1 g di olio previamente filtrato, omogeneizzatoe se necessario disidratato.
Diluirlo in 20 ml circa di solvente di eluizione (4.2.3), senecessario riscaldandolo leggermente per facilitare la dissoluzione.Raffreddare a temperatura ambiente e portare a volume con solvente dieluizione.
Con una pipetta volumetrica, introdurre 20 ml di soluzioneall’interno della colonna preparata come indicato al punto 4.3.1, aprire ilrubinetto e lasciar defluire il solvente fino al livello dello strato di gel disilice.
Eluire in seguito con 150 ml di solvente di eluizione(4.2.3), regolando il flusso del solvente a circa 2 ml/min (per passareattraverso la colonna, 150 ml impiegheranno circa 60-70 minuti).
Recuperare l’eluato in un matraccio a fondo arrotondato da250 ml (4.1.1) precedentemente tarato in una stufa e pesato con accuratezza.Eliminare il solvente a pressione ridotta in un evaporatore rotante (4.1.9) epesare il residuo che sarà impiegato per preparare la soluzione per l’analisiHPLC e per la preparazione dell’estere metilico.
Il campione deve essere recuperato almeno al 90 % per lecategorie olio extra vergine, olio vergine, olio raffinato e olio d’oliva, eall’80 % per l’olio lampante e per l’olio di sansa.
4.3.3. Purificazione medianteSPE
Attivare la colonna SPE con assorbente siliceo eluendo 6 mldi esano (4.2.3) sotto vuoto, evitandone l’essicazione.
Pesare con un’accuratezza dello 0,001 g, 0,12 g in unapipetta da 2 ml (4.1.15) e dissolvere in 0,5 ml di esano (4.2.3).
Versare nella colonna SPE la soluzione ed eluire con 10 mldi esano-etere dietilico (87:13 v/v) (4.2.3) sotto vuoto.
La frazione ottenuta è fatta evaporare completamente in unevaporatore rotante (4.1.9) a pressione ridotta e a temperatura ambiente. Ilresiduo è disciolto in 2 ml di acetone (4.2.6) per l’analisi deitriacilgliceroli (TAG).
4.4. Analisi HPLC
4.4.1. Preparazione dei campioni perl’analisi cromatografica
Preparare una soluzione al 5 % del campione da analizzarepesando 0,5 g (con uno scarto massimo di ± 0,001 g) del campione in unmatraccio tarato da 10 ml e portare a volume (10 ml) con il solvente disolubilizzazione (4.2.9).
4.4.2. Procedimento
Predisporre il sistema cromatografico. Far defluire ilsolvente di eluizione (4.2.8) ad un flusso di 1,5 ml/min, in modo da spurgarel’intero sistema. Attendere finché la linea di base è diventata stabile.
Iniettare 10 μl del campione preparato come indicato alpunto 4.3.
4.4.3. Calcolo ed espressione deirisultati
Impiegare il metodo della normalizzazione interna, ossiapartendo dal presupposto che la somma delle aree dei picchi corrispondenti aiTAG da ECN 42 a ECN 52 è uguale al 100 %.
Calcolare la percentuale relativa di ciascun triglicerideapplicando la formula:
![]()
I risultati devono essere espressi con almeno due cifredecimali.
Vedere le note 1, 2, 3 e 4.
4.5. Calcolo della composizione dei triacilgliceroli (% di moli) a partire dai dati relativi alla composizione in acidi grassi (% dell’area)
4.5.1. Determinazione della composizionein acidi grassi
La composizione in acidi grassi è determinata conformementealla norma ISO 5508, mediante una colonna capillare. La preparazione degliesteri metilici è eseguita conformemente al metodo COI/T.20/Doc. n. 24.
4.5.2. Acidi grassi considerati per ilcalcolo
I gliceridi sono raggruppati secondo i loro numeri dicarbonio equivalente (ECN), tenendo conto delle equivalenze fra ECN e acidigrassi riportate qui di seguito. Sono stati presi in considerazione soltantogli acidi grassi con 16 e 18 atomi di carbonio, perché sono i soli ad avereimportanza per l’olio di oliva. Gli acidi grassi devono essere riproporzionatial 100 %.
|
Acidi grassi (FA) |
Abbreviazione |
Peso molecolare (PM) |
ECN |
|
Acido palmitico |
P |
256,4 |
16 |
|
Acido palmitoleico |
Po |
254,4 |
14 |
|
Acido stearico |
S |
284,5 |
18 |
|
Acido oleico |
O |
282,5 |
16 |
|
Acido linoleico |
L |
280,4 |
14 |
|
Acido linolenico |
Ln |
278,4 |
12 |
4.5.3. Trasformazione in moli della %dell’area per tutti gli acidi grassi (1)
|
|
|
|
|
|
|
|
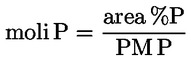

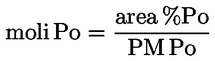
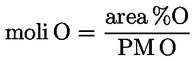

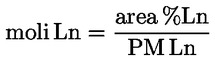
4.5.4. Riproporzionamento degli acidigrassi al 100 % (2)


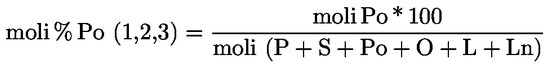



Il risultato esprime la percentuale di ciascun acido grassoin moli % sulla posizione complessiva (1, 2, 3) dei TAG.
Si calcola quindi la somma degli acidi grassi saturi P e S(SFA) e degli acidi grassi insaturi Po, O, L e Ln (UFA) (3):
![]()
![]()
4.5.5. Calcolo della composizione degliacidi grassi nelle posizioni 2, e 1, 3 dei TAG:
Gli acidi grassi sono distribuiti in tre gruppi, nel modoseguente: uno per la posizione 2 e due identici per le posizioni 1 e 3, concoefficienti diversi per gli acidi saturi (P e S) e gli acidi insaturi (Po, O,L e Ln).
4.5.5.1. Acidi grassi saturi nella posizione 2 [P(2) e S(2)] (4)
![]()
![]()
4.5.5.2. Acidi grassi insaturi nella posizione 2 [Po(2), O(2), L(2) e Ln(2)] (5):

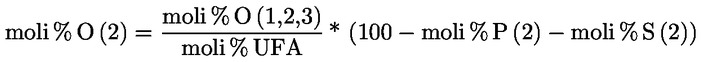
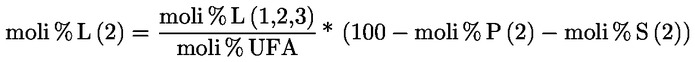

4.5.5.3. Acidi grassi nelle posizioni 1e 3 [P(1,3), S(1,3), Po(1,3), O(1,3), L(1,3) e Ln(1,3)] (6):



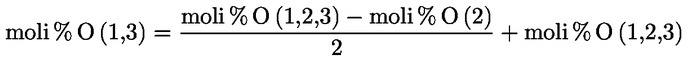


4.5.6. Calcolo deitriacilgliceroli
4.5.6.1. TAG con un solo acido grasso (AAA, qui LLL, PoPoPo) (7)

4.5.6.2. TAG con due acidi grassi (AAB, qui PoPoL, PoLL) (8)

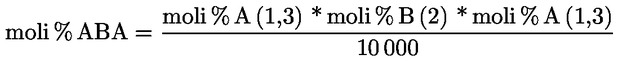
4.5.6.3. TAG con tre diversi acidi grassi (ABC, qui OLLn, PLLn, PoOLn, PPoLn) (9)
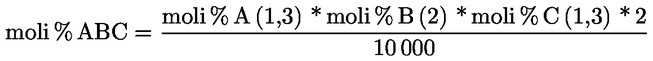
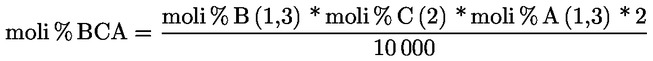

4.5.6.4. Triacilgliceroli con ECN 42
I triagliceroli ECN 42 sono calcolati secondo le equazioni 7, 8 e 9 e sono espressi nell’ordine previsto di eluizione in HPLC (normalmente soltanto tre picchi).
LLL
PoLL e isomero di posizione LPoL
OLLn e isomeri di posizione OLnL e LnOL
PoPoL e isomero di posizione PoLPo
PoOLn e isomeri di posizione OPoLn e OLnPo
PLLn e isomeri di posizione LLnP e LnPL
PoPoPo
SLnLn e isomero di posizione LnSLn
PPoLn e isomeri di posizione PLnPo e PoPLn
I triacilgliceroli ECN 42 sono espressi dalla somma dei nove triacilgliceroli, compresi i loro isomeri di posizione. I risultati devono essere espressi con almeno due cifre decimali.
5. VALUTAZIONE DEI RISULTATI
Si confrontano il contenuto teorico calcolato e il contenuto determinato mediante HPLC. Se la differenza nel valore assoluto tra i dati HPLC e i dati teorici è superiore ai valori riportati nella norma per la pertinente categoria di olio, il campione contiene olio di semi.
I risultati vanno espressi con due cifre decimali.
6. ESEMPIO (I NUMERI SI RIFERISCONO ALLE SEZIONI NEL TESTO DEL METODO)
— 4.5.1. Calcolo della % di moli degli acidi grassi a partire dai dati della GLC (% normalizzata dell’area)
Per la composizione in acidi grassi si ottengono mediante GLC i dati seguenti:
|
FA |
P |
S |
Po |
O |
L |
Ln |
|
PM |
256,4 |
284,5 |
254,4 |
282,5 |
280,4 |
278,4 |
|
Area % |
10,0 |
3,0 |
1,0 |
75,0 |
10,0 |
1,0 |
— 4.5.3 Conversione, in moli, della % dell’area per tutti gli acidi grassi (cfr. formula (1)]
|
moli P |
= |
|

|
moli S |
= |
|
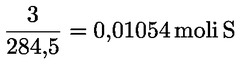
|
moli Po |
= |
|

|
moli O |
= |
|

|
moli L |
= |
|

|
moli Ln |
= |
|

|
Somma |
= |
0,35821 moli TAG |
— 4.5.4 Normalizzazione al 100 % delle moli di acidi grassi (cfr. formula (2)]
|
moli % P(1,2,3) |
= |
|

|
moli % S(1,2,3) |
= |
|

|
moli % Po(1,2,3) |
= |
|
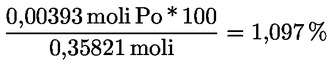
|
moli % O(1,2,3) |
= |
|

|
moli % L(1,2,3) |
= |
|

|
moli % Ln(1,2,3) |
= |
|

|
Totale moli % |
= |
100 % |
Somma degli acidi grassi saturi e insaturi nella posizione 1,2,3- dei TAG (cfr. formula (3)]:
![]()
![]()
— 4.5.5 Calcolo della composizione di acidi grassi nelle posizioni 2 e 1, 3 dei TAG
— 4.5.5.1 Acidi grassi saturi nella posizione 2 [P(2) e S(2)](cfr. formula (4)]
![]()
![]()
— 4.5.5.2 Acidi grassi insaturi nella posizione 2 [Po(1,3),O(1,3), L(1,3) e Ln(1,3)] (cfr. formula (5)]


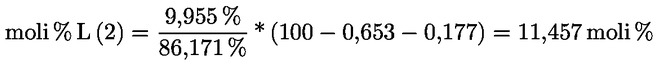

— 4.5.5.3 Acidi grassi nelle posizioni 1, 3 [P(1,3), S(1,3),Po(1,3), O(1,3), L(1,3) e Ln(1,3)] (cfr. formula (6)]






— 4.5.6. Calcolo dei triacilgliceroli
Dalla composizione calcolata di acidi grassi nelle posizioni 2 e 1e 3:
|
AG in |
posiz.1,3- |
posiz. 2- |
|
P |
16,004 % |
0,653 % |
|
S |
4,325 % |
0,177 % |
|
Po |
1,015 % |
1,262 % |
|
O |
68,526 % |
85,296 % |
|
L |
9,204 % |
11,457 % |
|
Ln |
0,927 % |
1,153 % |
|
Somma |
100,0 % |
100,0 % |
si calcolano i seguenti triacilgliceroli:
LLL
PoPoPo
PoLL con 1 isomero di posizione
SLnLn con 1 isomero di posizione
PoPoL con 1 isomero di posizione
PPoLn con 2 isomeri di posizione
OLLn con 2 isomeri di posizione
PLLn con 2 isomeri di posizione
PoOLn con 2 isomeri di posizione
— 4.5.6.1. TAG con un unico acido grasso (LLL, PoPoPo) (cfr.formula (7)]
 =0,09706 moli LLL
=0,09706 moli LLL

— 4.5.6.2 TAG con due acidi grassi (PoLL, SLnLn, PoPoL) (cfr.formula (8)]


0,03210 moli PoLL


0,00094 moli SLnLn
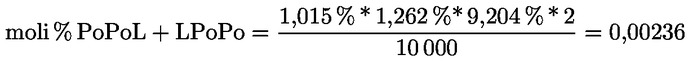

0,00354 moli PoPoL
— 4.5.6.3 TAG con tre diversi acidi grassi (PoPLn, OLLn, PLLn,PoOLn) Cfr. formula (9)



0,00761 moli PPoLn



0,43655 moli OLLn
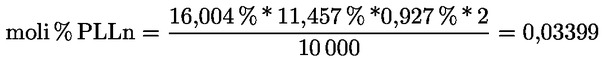


0,06907 moli PLLn

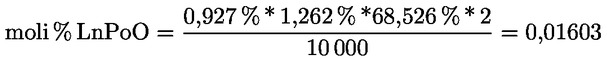
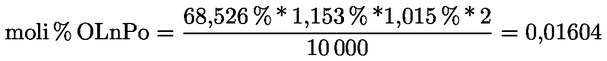
0,04812 di moli PoOLn
ECN42 = 0,69512 % di moli TAG
Nota 1: L’ordine di eluizione può essere determinato calcolando i numeri di carbonio equivalente, spesso definiti dalla relazione ![]() , dove CN è il numero di carbonio ed n è il numero di doppi legami; esso può essere calcolato precisamente tenendo conto dell’origine del doppio legame. Se no, nl e nln sono i numeri di doppi legami attribuiti rispettivamente agli acidi oleico, linoleico e linolenico, il numero di carbonio equivalente può essere calcolato mediante la relazione data dalla formula:
, dove CN è il numero di carbonio ed n è il numero di doppi legami; esso può essere calcolato precisamente tenendo conto dell’origine del doppio legame. Se no, nl e nln sono i numeri di doppi legami attribuiti rispettivamente agli acidi oleico, linoleico e linolenico, il numero di carbonio equivalente può essere calcolato mediante la relazione data dalla formula:
![]()
dove i coefficienti do, dl e dln possono essere calcolati mediante i trigliceridi di riferimento. Nelle condizioni specificate nel presente metodo, la relazione ottenuta sarà vicina a:
![]()
Nota 2: Con diversi trigliceridi di riferimento, è anche possibile calcolare la risoluzione rispetto alla trioleina:
![]()
trioleinaimpiegando il tempo corretto di ritenzione RT
![]()
Il grafico di log α in funzione di f (numero di doppi legami) consente di determinare i valori di ritenzione per tutti i trigliceridi degli acidi grassi contenuti nei trigliceridi di riferimento - cfr. figura 1.
Nota 3: l’efficienza della colonna dovrebbe permettere una chiara separazione del picco della trilinoleina dai picchi dei trigliceridi con un RT adiacente. L’eluizione viene effettuata fino al picco corrispondente a ECN 52.
Nota 4: una misura corretta delle aree di tutti i picchi rilevanti per la presente determinazione è garantita quando l’altezza del secondo picco della serie ECN 50 è pari al 50 % del massimo della scala di registrazione.
Figura 1
Grafico di log α in funzione di f (numero didoppi legami)
Numero di doppi legami
La: acido laurico; My: acido miristico; P: acido palmitico; S: acido stearico; O: acido oleico; L: acido linoleico; Ln: acido linolenico
Figura 2
Olio di oliva a basso tenore in acidolinoleico
a

Con solvente: acetone/acetonitrile.
PROFILO a: componenti principali dei picchi cromatografici: ECN42: (1) LLL + PoLL; (2) OLLn + PoOLn; (3) PLLn; ECN44: (4) OLL + PoOL; (5) OOLn + PLL; (6) POLn + PPoPo; (7) OOL + PoOO; ECN46: (8) OOL + LnPP; (9) PoOO; (10) SLL + PLO; (11) PoOP + SPoL + SOLn + SPoPo; (12) PLP; ECN48: (13) OOO + PoPP; (14 + 15) SOL + POO; (16) POP; ECN50: (17) SOO; (18) POS + SLS.
b

Con solvente: propionitrile
PROFILO b: componenti principali dei picchi cromatografici: ECN42: (1) LLL; (2) OLLn + PoLL; (3) PLLn; ECN44: (4) OLL; (5) OOLn + PoOL; (6) PLL + PoPoO; (7) POLn + PPoPo + PPoL; ECN46: (8) OOL + LnPP; (9) PoOO; (10) SLL + PLO; (11) PoOP + SPoL + SOLn + SPoPo; (12) PLP; ECN48: (13) OOO + PoPP; (14) SOL; (15) POO; (16) POP; ECN50: (17) SOO; (18) POS + SLS
Figura 3
Olio di oliva ad alto tenore in acidolinoleico
a

Con solvente: Acetone/Acetonitrile (50:50).
Profilo a: componenti principali dei picchi cromatografici: ECN42: (1’) LLL + PoLL; (2’) OLLn + PoOLn; (3’) PLLn; ECN44: (4’) OLL + PoOL; (5’) OOLn + PLL; (6’) POLn + PPoPo; ECN46: (7’) OOL + PoOO; (8’) PLO + SLL + PoOP; (9’) PLP + PoPP; ECN48: (10’) OOO; (11’) POO + SLL + PPoO; (12’) POP + PLS; ECN50: (13’) SOO; (14’) POS + SLS
b
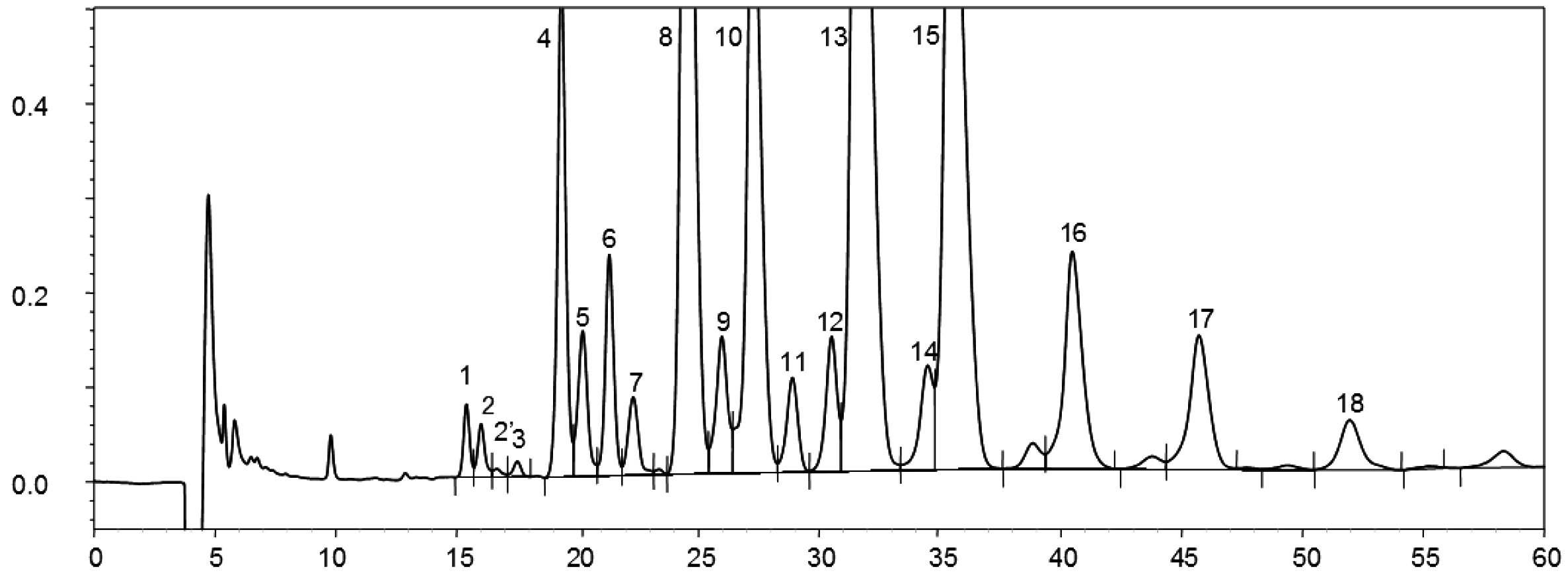
Con solvente: propionitrile.
Profilo b: componenti principali dei picchi cromatografici: ECN42: (1) LLL; (2 + 2’) OLLn + PoLL; (3) PLLn; ECN44: (4) OLL; (5) OOLn + PoOL; (6) PLL + PoPoO; (7) POLn + PPoPo + PPoL; ECN46: (8) OOL + LnPP; (9) PoOO; (10) SLL + PLO; (11) PoOP + SPoL + SOLn + SPoPo; ECN48: (12) PLP; (13) OOO + PoPP; (14) SOL; (15) POO; (16) POP; ECN50: (17) SOO; (18) POS + SLS; ECN52: (19) AOO.
ALLEGATO XIX
DETERMINAZIONE DEL CONTENUTO DI ALCOLI ALIFATICI E TRITERPENICI MEDIANTE GASCROMATOGRAFIA CON COLONNA CAPILLARE
1. OGGETTO
Il presente allegato descrive un procedimento per la determinazione del contenuto di alcoli alifatici e triterpenici negli oli e nelle sostanze grasse.
2. PRINCIPIO DEL METODO
La sostanza grassa, addizionata di 1-eicosanolo quale standard interno, è saponificata con idrossido di potassio in soluzione etanolica, quindi l'insaponificabile viene estratto con etere etilico. Dall'insaponificabile estratto è separata la frazione degli alcoli mediante cromatografia su placca di gel di silice basica; gli alcoli recuperati dal gel di silice vengono trasformati in trimetilsilileteri ed analizzati mediante gascromatografia in colonna capillare.
3. APPARECCHIATURA
3.1. Matraccio da 250 ml, munito di refrigerante a ricadere con giunti a smeriglio.
3.2. Imbuto separatore da 500 ml.
3.3. Matracci da 250 ml.
3.4. Attrezzatura completa per analisi cromatografica su strato sottile, per lastre di vetro 20 × 20 cm.
3.5. Lampada a luce ultravioletta, con lunghezza d'onda 366 o 254 nm.
3.6. Microsiringhe da 100 e 500 microlitri.
3.7. Imbuto cilindrico filtrante a setto poroso G 3 (porosità 15-40 micrometri) di diametro circa 2 cm e altezza circa 5 cm, con attacco idoneo per filtrazione sotto vuoto e giunto smerigliato maschio 12/21.
3.8. Beuta per vuoto da 50 ml con giunto femmina smerigliato 12/21 adattabile all'imbuto filtrante (3.7).
3.9. Provetta da 10 ml a fondo conico con tappo a tenuta.
3.10. Gascromatografo idoneo per il funzionamento con colonna capillare, dotato di sistema di splittaggio, costituito da:
3.10.1. Camera termostatica per la colonna, idonea a mantenere la temperatura desiderata con la precisione di circa 1 °C.
3.10.2. Complesso di iniezione termoregolabile con elemento vaporizzante in vetro persilanizzato.
3.10.3. Rivelatore a ionizzazione di fiamma e convertitore-amplificatore.
3.10.4. Registratore-integratore idoneo per il funzionamento con il convertitore-amplificatore (3.10.3), con tempo di risposta non superiore a 1 secondo e con velocità della carta variabile.
3.11. Colonna capillare in vetro o silice fusa, lunga 20 + 30 m, diametro interno 0,25 + 0,32 mm, internamente ricoperta con liquido SE-52 o SE-54 o equivalenti, con spessore uniforme compreso fra 0,10 e 0,30 micrometri.
3.12. Microsiringa per gascromatografia da 10 microlitri con ago cementato.
3.13. Bilancia di previsione con sensibilità di 1 mg (con indicazione 0,1 mg).
4. REATTIVI
4.1. Potassio idrossido, soluzione metanolica circa 2 N: si sciolgono, sotto raffreddamento, 130 g di idrossido di potassio (titolo minimo 85 %) in 200 ml di acqua distillata, quindi si porta ad 1 litro con etanolo. La soluzione si conserva in bottiglie di vetro scuro ben tappate.
4.2. Etere etilico, puro per analisi.
4.3. Sodio solfato anidro, puro per analisi.
4.4. Lastre di vetro stratificate con gel di silice, senza indicatore di fluorescenza, spessore 0,25 mm (sono reperibili in commercio già pronte per l'uso).
4.5. Potassio idrossido, soluzione etanolica circa 0,2 N: si sciolgono 13 g di idrossido di potassio in 20 ml di acqua distillata e si porta a 1 litro con etanolo.
4.6. Benzene, per cromatografia (cfr. 5.2.2).
4.7. Acetone, per cromatografia (5.2.2).
4.8. Esano, per cromatografia (cfr. 5.2.2).
4.9. Etere etilico, per cromatografia (cfr. 5.2.2).
4.10. Cloroformio, puro per analisi.
4.11. Soluzione di riferimento per la cromatografia su strato sottile: alcoli C20-C28 allo 0,5 % in cloroformio, o una frazione di alcoli ottenuti come indicato al punto 5.2 dall'insaponificabile di un olio di sansa di oliva.
4.12. 2,7-diclorofluoresceina, soluzione etanolica allo 0,2 %. Si rende leggermente basica aggiungendo qualche goccia di soluzione alcolica 2 N di idrossido di potassio.
4.13. Piridina anidra, per cromatografia.
4.14. Esametildisilazano.
4.15. Trimetilclorosilano.
4.16. Soluzione etanolo di trimetilsilileteri degli alcoli alifatici da C20 a C28. Si preparano al momento dell'impiego a partire da miscele di alcoli puri.
4.17. 1-eicosanolo, soluzione allo 0,1 % (m/v) in cloroformio (standard interno).
4.18. Gas vettore: idrogeno o elio, puri per gascromatografia.
4.19. Gas ausiliare: azoto puro per gascromatografia.
5. PROCEDIMENTO
5.1. Preparazione dell'insaponificabile
|
5.1.1. |
Nel matraccio da 250 ml si introduce, impiegando la microsiringa da 500 microlitri, un volume di soluzione di 1-eicosanolo allo 0,1 % in cloroformio (4.17) che contenga una quantità di 1-eicosanolo corrispondente a circa il 10 % del contenuto di alcoli alifatici nell'aliquota di campione da prelevare per la determinazione. Ad esempio per 5 g di campione si aggiungano 250 microlitri della soluzione di 1-eicosanolo allo 0,1 % se trattasi di oli di oliva e 1 500 microlitri se trattasi di olio di sansa di oliva. Si evapora il cloroformio in corrente di azoto fino a secchezza, quindi nello stesso matraccio si pesano esattamente circa 5 g di campione secco e filtrato. |
|
5.1.2. |
Si aggiungono 50 ml di soluzione etanolica di idrossido di potassio 2 N, si applica il refrigerante a ricadere e si scalda a leggera ebollizione su bagnomaria sotto continua energica agitazione, fino a saponificazione avvenuta (la soluzione diviene limpida). Si continua il riscaldamento ancora per 20 minuti, quindi si aggiungono 50 ml di acqua distillata facendoli scendere dall'alto del refrigerante, si stacca il refrigerante e si raffredda il matraccio a circa 30 °C. |
|
5.1.3. |
Si travasa il contenuto del matraccio quantitativamente, in un imbuto separatore da 500 ml, aiutandosi con acqua distillata, a più riprese, impiegandone complessivamente circa 50 ml. Si aggiungono circa 80 ml di etere etilico, si agita energicamente per circa 30 secondi e si lascia stratificare (nota 1). Si separa la fase acquosa sottostante raccogliendola in un secondo imbuto separatore. Sulla fase acquosa si effettuano ancora due estrazioni, con le stesse modalità impiegando ogni volta 60-70 ml di etere etilico. Nota 1: Eventuali emulsioni possono essere eliminate aggiungendo, mediante spruzzetta, piccole quantità di alcool etilico o metilico. |
|
5.1.4. |
Si riuniscono gli estratti eterei in un unico imbuto separatore e si lavano con acqua distillata (50 ml per volta) fino a reazione neutra delle acque di lavaggio. Eliminata l'acqua di lavaggio, si essicca con solfato di sodio anidro e si filtra, su solfato sodico anidro, in un matraccio da 250 ml previamente pesato, lavando imbuto e filtro con piccole quantità di etere etilico. |
|
5.1.5. |
Si distilla l'etere fino a pochi ml, quindi si porta a secco sotto leggero vuoto o in corrente di azoto, si completa l'essiccamento in stufa a 100 °C per un quarto d'ora circa e, dopo raffreddamento in essiccatore, si pesa. |
5.2. Separazione della frazione degli alcoli
|
5.2.1. |
Preparazione delle lastre basiche: si immergono le lastre al gel di silice (4.4), completamente, nella soluzione etanolica 0,2 N di idrossido di potassio (4.5) per 10 secondi, si lasciano quindi asciugare sotto cappa per 2 ore ed infine si pongono in stufa a 100 °C per 1 ora. Si tolgono dalla stufa e si conservano in essiccatore a cloruro di calcio fino al momento dell'impiego (le placche così trattate devono essere impiegate entro 15 giorni). Nota 2: Impiegando per la separazione della frazione alcolica delle lastre di gel di silice basiche si elimina la necessità del trattamento dell'insaponificabile con allumina. In tal modo vengono trattenuti sulla linea di caricamento tutti i composti di natura acida (acidi grassi ed altro) ottenendosi così le bande degli alcoli alifatici e terpenici nettamente separate dalla banda degli steroli. |
|
5.2.2. |
Nella camera di sviluppo delle lastre si introduce una miscela esano-etere etilico 65/35 (V/V) fino all'altezza di circa 1 cm ( 6 ). Si chiude la camera con l'apposito coperchio e si lascia così per almeno mezz'ora in modo che si stabilisca l'equilibrio liquido-vapore. Sulle superfici interne della camera possono essere fissate delle strisce di carta da filtro che peschino nell'eluente: questo accorgimento permette di ridurre di circa 1/3 il tempo di sviluppo e di ottenere una più uniforme e regolare eluizione dei componenti. Nota 3: Al fine di ottenere condizioni di eluizione perfettamente riproducibili la miscela di sviluppo deve essere sostituita ad ogni prova. |
|
5.2.3. |
Si prepara una soluzione al 5 % circa di insaponificabile (5.1.5) in cloroformio e, con la microsiringa da 100 microlitri si depositano su una placca cromatografica (5.2.1) a 2 cm circa da una estremità, 0,3 ml di detta soluzione, in striscia il più possibile sottile ed uniforme. In allineamento con la linea di caricamento, ad un'estremità della lastra si depositano 2-3 microlitri della soluzione di riferimento degli alcoli (4.11), allo scopo di identificare, a sviluppo ultimato, la banda degli alcoli alifatici. |
|
5.2.4. |
Si pone la placca nella camera di sviluppo preparata come detto in 5.2.2. La temperatura dovrà essere mantenuta fra 15 e 20 °C. Si chiude subito la camera col coperchio e si lascia eluire fino a che il fronte del solvente sia arrivato a circa 1 cm dal bordo superiore della placca. Si rimuove quindi la placca dalla camera di sviluppo e si evapora il solvente in corrente di aria calda oppure lasciando la placca ad asciugare per un po' di tempo sotto cappa. |
|
5.2.5. |
Si spruzza la placca debolmente e uniformemente con la soluzione di 2,7-diclorofluoresceina. Osservando la placca alla luce ultravioletta si individua la banda degli alcoli alifatici per allineamento con la macchia ottenuta con la soluzione di riferimento e si delimita con una matita nera l'insieme della banda degli alcoli alifatici e della banda immediatamente superiore corrispondente agli alcoli terpenici (nota 4). Nota 4: La banda degli alcoli alifatici e la banda degli alcoli terpenici devono essere raggruppate a motivo della possibile migrazione di alcuni alcoli alifatici nella banda degli alcoli triterpenici. Un esempio di separazione cromatografica su strato sottile è riportata nella figura 1 dell'appendice. |
|
5.2.6. |
Con una spatola metallica si raschia il gel di silice compreso nell'area delimitata. Il materiale asportato, finemente sminuzzato, viene introdotto nell'imbuto filtrante (3.7); si aggiungono 10 ml di cloroformio caldo, si mescola accuratamente con la spatola metallica e si filtra aiutandosi con il vuoto, raccogliendo il filtrato nella beuta (3.8), collegata all'imbuto filtrante. Si lava il gel di silice nell'imbuto per tre volte con etere etilico (circa 10 ml per volta) raccogliendo sempre il filtrato nella stessa beuta adattata all'imbuto. Si evapora il filtrato fino ad un volume di circa 4-5 ml, si trasferisce la soluzione residua nella provetta da 10 ml (3.9) previamente pesata, si porta a secco con blando riscaldamento in leggera corrente di azoto, si riprende con qualche goccia di acetone, si riporta ancora a secco, si pone 10 minuti circa in stufa a 105 °C, indi si lascia raffreddare in essiccatore e si pesa. Il residuo contenuto nella provetta è costituito dalla frazione alcolica. |
5.3. Preparazione dei trimetilsilileteri
|
5.3.1. |
Nella provetta contenente la frazione alcolica si aggiunge il reattivo per la sililazione, costituito da una miscela di piridina-esametildisilazanotrimetilclorosilano 9:3:1 (v/v/v) (nota 5) in ragione di 50 microlitri per ogni milligrammo di alcoli alifatici, evitando ogni assorbimento di umidità (nota 6). Nota 5: Esistono in commercio soluzioni già pronte per l'uso; sono inoltre disponibili altri reagenti silanizzanti, quali ad esempio il bis-trimetiltrifluorolacetammide + 1 % trimetilclorosilano da diluire con uno stesso volume di piridina anidra. Nota 6: L'eventuale formazione di una leggera opalescenza è normale e non è causa di alcun disturbo. La formazione di un flocculato bianco o la comparsa di una colorazione rosa sono indizio della presenza di umidità o di alterazione del reattivo. In questo caso la prova dovrà essere ripetuta. |
|
5.3.2. |
Si tappa la provetta, si agita cautamente (senza capovolgere) fino a completa solubilizzazione degli alcoli alifatici. Si lascia a sé per almeno 15 minuti a temperatura ambiente, quindi si centrifuga per alcuni minuti: la soluzione limpida è pronta per l'analisi gascromatografica. |
5.4. Analisi gascromatografica
5.4.1. Operazioni preliminari, condizionamento della colonna
|
5.4.1.1. |
Si installa nel gascromatografo la colonna, collegando il terminale di ingresso all'iniettore connesso col sistema di splittaggio, e il terminale di uscita al rivelatore. Si eseguono i controlli generali del complesso gascromatografico (tenuta dei circuiti dei gas, efficienza del rivelatore, efficienza del sistema di splittaggio e del sistema di registrazione, ecc.). |
|
5.4.1.2. |
Se la colonna è messa in uso per la prima volta è consigliabile procedere al suo condizionamento. Si fa fluire un leggero flusso di gas attraverso la colonna stessa, quindi si accende il complesso gascromatografico e si inizia un riscaldamento graduale fino a raggiungere una temperatura di almeno 20 °C superiore a quella di esercizio (nota 7). Si mantiene tale temperatura per almeno 2 ore, quindi si porta il complesso alle condizioni di funzionamento (regolazione del flusso dei gas e dello splittaggio, accensione della fiamma, collegamento con il registratore elettronico, regolazione della temperatura della camera per la colonna, del rivelatore e dell'iniettore, ecc.) e si registra il segnale ad una sensibilità almeno 2 volte superiore a quella prevista per l'esecuzione dell'analisi. Il tracciato della linea di base deve risultare lineare, esente da picchi di qualsiasi natura, e non deve presentare deriva. Una deriva rettilinea negativa indica imperfetta tenuta delle connessioni della colonna, una deriva positiva indica un insufficiente condizionamento della colonna. Nota 7: La temperatura di condizionamento deve in ogni caso essere inferiore di almeno 20 °C alla temperatura massima prevista per il liquido di ripartizione impiegato. |
5.4.2. Scelta delle condizioni operative
5.4.2.1. Condizioni operative di massima sono le seguenti:
— temperatura della colonna: inizio isoterma 8 minuti a 180 °C, quindi programma 5 °C/minuto fino a 260 °C e ancora 15 minuti a 260 °C,
— temperatura dell'evaporatore: 280 °C,
— temperatura del rivelatore: 290 °C,
— velocità lineare del gas vettore: elio da 20 a 35 cm/s; idrogeno da 30 a 50 cm/s,
— rapporto di splittaggio: da 1/50 a 1/100,
— sensibilità strumentale: da 4 a 16 volte l'attenuazione minima,
— sensibilità di registrazione: da 1 a 2 mV su fondo scala,
— velocità della carta: da 30 a 60 cm/ora,
— quantità di sostanza iniettata: da 0,5 a 1 microlitri di soluzione di TMSE.
Tali condizioni possono essere modificate in funzione delle caratteristiche della colonna e del gascromatografo in modo da ottenere cromatogrammi che soddisfino le condizioni seguenti:
— il tempo di ritenzione dell'alcol C26 deve essere di 18 ± 5 minuti,
— il picco dell'alcol C22 deve essere per l'olio di oliva 80 ± 20 % del fondo scala e per gli oli di semi 40 ± 20 % del fondo scala.
5.4.2.2. Per verificare i suddetti requisiti si effettuano ripetute iniezioni con le miscele campione di TMSE degli alcoli e si ritoccano le condizioni operative fino a raggiungere i migliori risultati.
5.4.2.3. I parametri di integrazione dei picchi dovranno essere impostati in modo da ottenere una corretta valutazione delle aree dei picchi che vengono presi in considerazione.
5.4.3. Esecuzione dell'analisi
5.4.3.1. Con la microsiringa da 10 microlitri si preleva 1 ml di esano, si aspirano 0,5 microlitri di aria e successivamente da 0,5 a 1 microlitri della soluzione del campione; si alza ancora lo stantuffo della siringa in modo che l'ago sia vuoto. Si introduce l'ago attraverso la membrana del complesso di iniezione e dopo 1-2 secondi si inietta rapidamente e si estrae quindi lentamente l'ago dopo circa 5 secondi.
5.4.3.2. Si effettua la registrazione fino a completa eluizione dei TMSE degli alcoli presenti. La linea di base deve essere sempre corrispondente ai requisiti richiesti (5.4.1.2).
5.4.4. Identificazione dei picchi.
L'identificazione dei singoli picchi viene effettuata in base ai tempi di ritenzione e per paragone con miscele di TMSE degli alcoli, analizzate nelle medesime condizioni.
Nelle figure 2 e 3 dell'appendice sono riportati esempi di cromatogramma della frazione alcolica di un olio di oliva raffinato.
5.4.5. Valutazione quantitativa
5.4.5.1. Si procede al calcolo con l'integratore, delle aree dei picchi dell'1-eicosanolo e degli alcoli alifatici da C22 C24, C26, C28.
5.4.5.2. Si calcola il contenuto di ogni singolo alcool alifatico, in mg/
1 000
g di sostanza grassa come segue:

in cui:
Ax = area del picco dell'alcool x
As = area del picco dell'1-eicosanolo
ms = peso di 1-eicosanolo aggiunto, in milligrammi
m = peso del campione prelevato per la determinazione, in grammi.
6. ESPRESSIONE DEI RISULTATI
Si riportano i contenuti dei singoli alcoli alifatici, in mg/1 000 g di sostanza grassa e, come «alcoli alifatici totali», la loro somma.
Appendice
Esempio di separazione cromatografica su strato sottile ed esempi di cromatogramma
Figura 1
Cromatografia su strato sottile della frazione insaponificabile dall'olio di oliva eluito in esano/etere etilico (65/35)

|
1 |
Alcol C26 |
A |
Steroli |
|
2 |
Alcol C30 |
B |
Alcoli alifatici |
|
3 |
Alcol C20 |
C |
Alcoli triterpenici |
|
4 |
Alcoli misti C20-22-26-30 |
D |
Squalene |
|
5 |
Extra vergine insaponificabile |
|
|
Figura 2
Cromatogramma della frazione alcolica di un olio di oliva raffinato

|
1 = Fitolo |
5 = Alcol C24 |
9 = 24-Metilen-cicloartenolo |
|
2 = Geranil geraniolo |
6 = Alcol C26 |
10 = Citrostadienolo |
|
3 = Alcol C20 (IS) |
7 = Alcol C28 |
11 = Ciclobranolo |
|
4 = Alcol C22 |
8 = Cicloartenolo |
|
Figura 3
Alcoli alifatici e triterpenici di un olio di oliva raffinato e un olio di oliva di seconda centrifugazione
|
1 = Fitolo |
5 = Alcol C24 |
9 = 24-Metilen-cicloartenolo (24MeCA) |
|
2 = Geranil geraniolo (CX) |
6 = Alcol C26 |
10 = Citrostadienolo |
|
3 = Alcol C20 |
7 = Alcol C28 |
11 = Ciclobranolo |
|
4 = Alcol C22 |
8 = Cicloartenolo (CA) |
|
ALLEGATO XX
Metodo per la determinazione del contenuto di cere e metil ed etil esteri degli acidi grassi mediante gascromatografia con colonna capillare
1. OGGETTO
Il presente metodo permette di determinare il contenuto di cere e metil ed etil esteri degli acidi grassi negli oli di oliva. Le singole cere e gli alchil esteri sono separati in funzione del numero di atomi di carbonio. L’impiego del metodo viene consigliato come mezzo atto a differenziare l’olio di oliva dall’olio di sansa di oliva e come parametro di qualità per gli oli extra vergini, in quanto permette di individuare false miscele di oli extra vergini di oliva e oli di bassa qualità e di capire se si tratta di oli vergini, lampanti o deodorati.
2. PRINCIPIO
La sostanza grassa, addizionata di opportuni standard interni, viene frazionata mediante cromatografia su colonna di gel di silice idratato; la frazione eluita nelle condizioni di prova (con polarità minore di quella dei trigliceridi) viene recuperata e analizzata direttamente mediante gascromatografia in colonna capillare.
3. APPARECCHIATURA
|
3.1. |
Beuta da 25 ml. |
|
3.2. |
Colonna in vetro per cromatografia liquida avente diametro interno di 15 mm e altezza da 30 a 40 cm con opportuno rubinetto. |
|
3.3. |
Gascromatografo idoneo per il funzionamento con colonna capillare, dotato di sistema di introduzione diretta in colonna costituito da:
|
|
3.4. |
Microsiringa da 10 μl con ago cementato, idonea per iniezione diretta in colonna. |
|
3.5. |
Vibratore elettrico |
|
3.6. |
Evaporatore rotante |
|
3.7. |
Muffola |
|
3.8. |
Bilancia analitica in grado di garantire un’accuratezza della misura di ± 0,1 mg |
|
3.9. |
Normale vetreria da laboratorio |
4. REAGENTI
|
4.1. |
Gel di silice con granulometria compresa tra 60 e 200 μm. Porre il gel di silice in muffola a 500 °C per almeno 4 h. Dopo il raffreddamento addizionare il 2 % di acqua riferito alla quantità di gel di silice prelevata. Agitare bene allo scopo di omogeneizzare la massa e conservare nell’essiccatore almeno per 12 h prima dell’impiego. |
|
4.2. |
n-Esano per cromatografia o analisi dei residui (verificare la purezza). AVVERTENZA: i vapori possono incendiarsi. Tenere lontano da sorgenti di calore, scintille o fiamme libere. Tenere i contenitori ben chiusi. Usare con ventilazione adeguata. Evitare l’accumulo di vapori ed eliminare ogni possibile causa di incendio, quali riscaldatori o apparecchi elettrici non antideflagranti. Nocivo per inalazione: può causare danni alle cellule del sistema nervoso. Evitare di respirare i vapori, usare se necessario un apparecchio respiratorio adatto. Evitare il contatto con gli occhi e la pelle. |
|
4.3. |
Etere etilico, per cromatografia
AVVERTENZA: prodotto altamente infiammabile. Moderatamente tossico. Irritante per la pelle. Nocivo per inalazione. Può causare danni agli occhi. Gli effetti possono essere differiti. Può formare perossidi esplosivi. I vapori possono incendiarsi. Tenere lontano da sorgenti di calore, scintille o fiamme libere. Tenere i contenitori ben chiusi. Usare con ventilazione adeguata. Evitare l’accumulo di vapori ed eliminare ogni possibile causa di incendio, quali riscaldatori o apparecchi elettrici non antideflagranti. Non evaporare a secchezza o quasi-secchezza. L’aggiunta di acqua o di un agente riducente appropriato può ridurre la formazione di perossidi. Non ingerire. Evitare di respirare i vapori. Evitare il contatto prolungato o ripetuto con la pelle. |
|
4.4. |
n-Eptano per cromatografia o Isottano AVVERTENZA: prodotto infiammabile. Nocivo per inalazione. Tenere lontano da sorgenti di calore, scintille o fiamme libere. Tenere i contenitori ben chiusi. Usare con ventilazione adeguata. Evitare di respirare i vapori. Evitare il contatto prolungato o ripetuto con la pelle. |
|
4.5. |
Soluzione campione di lauril arachidato (Nota 3), allo 0,05 % (m/V) in eptano (standard interno per cere). Nota 3: È possibile utilizzare anche palmitil palmitato o miristil stearato o arachidil laurato. |
|
4.6. |
Soluzione campione di metileptadecanoato, allo 0,02 % (m/V) in eptano (standard interno metil ed etil esteri). |
|
4.7. |
Sudan 1 (1-fenilazo-2-naftolo) |
|
4.8. |
Gas vettore: idrogeno o elio puri per gascromatografia AVVERTENZA Idrogeno. Altamente infiammabile, sotto pressione. Tenere lontano da fonti di calore, scintille o fiamme libere o apparecchi elettrici non antideflagranti. Tenere sempre la valvola della bombola chiusa quando non si usa. Usare sempre un regolatore di pressione. Togliere la tensione alla molla del riduttore prima di aprire la valvola della bombola. Non sostare davanti al foro di uscita della bombola quando si apre la valvola. Usare con ventilazione adeguata. Non trasferire l’idrogeno da una bombola a un’altra. Non miscelare gas nella bombola. Tenere sempre ben assicurate le bombole affinché non possano cadere. Tenere le bombole lontane dal sole o da sorgenti di calore. Non tenere in ambienti corrosivi. Non usare bombole danneggiate o senza etichetta. Elio. Gas compresso sotto alta pressione. Riduce l’ossigeno disponibile per la respirazione, tenere il contenitore chiuso. Usare con ventilazione adeguata. Non entrare nei locali di conservazione se non sono adeguatamente ventilati. Usare sempre un regolatore di pressione. Togliere la tensione alla molla del riduttore prima di aprire la valvola della bombola. Non trasferire il gas da una bombola a un’altra. Tenere sempre ben assicurate le bombole affinché non possano cadere. Non sostare davanti al foro di uscita della bombola quando si apre la valvola. Tenere le bombole lontane dal sole o da sorgenti di calore. Non tenere in ambienti corrosivi. Non usare bombole danneggiate o senza etichetta. Non usare per inalazione e farne solo uso tecnico. |
|
4.9. |
Gas ausiliari: — idrogeno, puro per gascromatografia — aria, pura per gascromatografia AVVERTENZA Aria. Gas compresso sotto alta pressione. Usare con cautela in presenza di sostanze combustibili in quanto la temperatura di autoaccensione della maggior parte dei composti organici nell’aria si abbassa notevolmente ad alta pressione. Tenere sempre la valvola della bombola chiusa quando non si usa. Usare sempre un regolatore di pressione. Togliere la tensione alla molla del riduttore prima di aprire la valvola della bombola. Non sostare davanti al foro di uscita della bombola quando si apre la valvola. Non trasferire il gas da una bombola a un’altra. Non miscelare gas nella bombola. Tenere sempre ben assicurate le bombole affinché non possano cadere. Tenere le bombole lontane dal sole o da sorgenti di calore. Non tenere in ambienti corrosivi. Non usare bombole danneggiate o senza etichetta. Non usare per inalazione o per apparecchi respiratori l’aria destinata a usi tecnici. |
5. PROCEDIMENTO
5.1. Preparazione della colonna cromatografica
Mettere in sospensione 15 g di gel di silice (4.1) in n-esano (4.2) e introdurla in colonna (3.2). A sedimentazione avvenuta completare l’assestamento mediante l’uso di un vibratore elettrico al fine di rendere più omogeneo il letto cromatografico. Percolare 30 ml di n-esano allo scopo di allontanare le eventuali impurezze. Pesare esattamente, nella beuta da 25 ml (3.1), circa 500 mg di campione con la bilancia analitica (3.8). Addizionare l’opportuna quantità di campione di riferimento (4.5) in funzione del presunto contenuto di cere. Ad esempio aggiungere 0,1 mg di lauril arachidato nel caso di olio di oliva, da 0,25 a 0,50 mg nel caso di olio di sansa e 0,05 mg di metileptadecanoato per gli oli di oliva (4.6).
Trasferire il campione così preparato nella colonna cromatografica aiutandosi con due porzioni da 2 ml ciascuna di n-esano (4.2).
Lasciare fluire il solvente fino a un battente di 1 mm, quindi far percolare altro n-esano/etere etilico (99:1) e raccogliere 220 ml, rispettando un flusso di circa 15 gocce ogni 10 secondi. (Questa frazione contiene i metil ed etil esteri e le cere). (Nota 4) (Nota 5).
Nota 4: La miscela n-esano/etere etilico (99:1) dovrà essere preparata ogni giorno.
Nota 5: Onde controllare visivamente la corretta eluizione delle cere, è possibile aggiungere al campione in soluzione 100 μl di Sudan I all’1 % nella miscela di eluizione.
Il colorante ha un tempo di ritenzione intermedio tra le cere e i trigliceridi, pertanto quando la colorazione raggiunge il fondo della colonna cromatografica bisogna sospendere l’eluizione, in quanto tutte le cere sono state eluite.
Evaporare la frazione così ottenuta mediante evaporatore rotante fino ad allontanamento quasi completo del solvente, eliminare gli ultimi 2 ml con l’aiuto di un debole flusso di azoto e riprendere la frazione che contiene i metil ed etil esteri e che va diluita con 2-4 ml di n-eptano o isottano.
5.2. Analisi gascromatografica
5.2.1. Operazioni preliminari
Installare nel gascromatografo (3.3) la colonna, collegando il terminale di ingresso connesso al sistema in colonna e il terminale di uscita al rilevatore. Eseguire i controlli generali del complesso gascromatografico (tenuta dei circuiti dei gas, efficienza del rivelatore e del sistema di registrazione, ecc.).
Se la colonna è messa in uso per la prima volta è consigliabile procedere al suo condizionamento: far fluire un leggero flusso di gas attraverso la colonna quindi avviare il complesso gascromatografico e iniziare un riscaldamento graduale fino a raggiungere, dopo circa 4 h, la temperatura di 350 °C.
Mantenere tale temperatura per almeno 2 h, quindi portare il complesso alle condizioni di funzionamento (regolazione del flusso del gas, accensione della fiamma, collegamento con il registratore elettronico (3.3.4), regolazione della temperatura della camera per colonna, del rivelatore, ecc.) e registrare il segnale a una sensibilità almeno due volte superiore a quella prevista per l’esecuzione dell’analisi. Il tracciato della linea di base deve risultare lineare, esente da picchi di qualsiasi natura e non deve presentare deriva.
Una deriva rettilinea negativa indica imperfetta tenuta delle connessioni della colonna, una deriva positiva indica un insufficiente condizionamento della colonna.
5.2.2. Scelta delle condizioni operative per le cere ed i metil ed etil esteri (Nota 6)
Le condizioni operative di massima sono le seguenti:
|
— |
Temperatura della colonna : 20 °C/min 5 °C/min inizio a 80 °C (1′) |
|
— |
Temperatura del rivelatore : 350 °C. |
|
— |
Quantità di sostanza iniettata : 1 μl della soluzione (2-4 ml) di n-eptano. |
|
— |
Gas vettore : elio o idrogeno alla velocità lineare ottimale per il gas prescelto (vedere Appendice A). |
|
— |
Sensibilità strumentale : idonea a soddisfare le condizioni di cui sopra. |
Nota 6: Vista l’elevata temperatura finale è ammessa una deriva positiva che non deve superare il 10 % del fondo scala.
Tali condizioni possono essere modificate in funzione delle caratteristiche della colonna e del gascromatografo in modo da avere una separazione di tutte le cere e dei metil ed etil esteri degli acidi grassi e una risoluzione soddisfacente dei picchi (vedi figure 2, 3 e 4) e un tempo di ritenzione dello standard interno del lauril arachidato di 18 ± 3 minuti. Il picco delle cere più rappresentativo deve superare di oltre il 60 % il fondo scala, mentre lo standard interno del metileptadecanoato dei metil ed etil esteri deve raggiungere il valore del fondo scala.
I parametri di integrazione dei picchi dovranno essere impostati in modo da ottenere una corretta valutazione delle aree dei picchi presi in considerazione.
5.3. Esecuzione dell’analisi
Con la microsiringa da 10 μl si prelevano 10 μl di soluzione; si alza lo stantuffo della siringa in modo che l’ago sia vuoto. Si introduce l’ago attraverso il dispositivo di iniezione e dopo 1-2 secondi si inietta rapidamente e si estrae quindi lentamente l’ago dopo circa 5 secondi.
Si effettua la registrazione fino a completa eluizione delle cere e degli stigmastadieni a seconda della frazione che viene analizzata.
La linea di base deve essere sempre corrispondente ai requisiti richiesti.
5.4. Identificazione dei picchi
L’identificazione dei singoli picchi viene effettuata in base ai tempi di ritenzione e per paragone con miscele di cere a tempi di ritenzione noti, analizzate nelle medesime condizioni. Per gli alchil esteri l’identificazione viene effettuata mediante miscele di metil ed etil esteri dei principali acidi grassi degli oli di oliva (palmitico e oleico).
Nella figura 1 è riportato un cromatogramma delle cere presenti in un olio di oliva vergine. Nelle figure 2 e 3 sono riportati i cromatogrammi di due oli extra vergini di oliva del commercio, uno con metil ed etil esteri e un altro senza. Nella figura 4 vengono riportati i cromatogrammi relativi a un olio extra vergine di ottima qualità e dello stesso olio addizionato del 20 % di olio deodorato.
5.5. Valutazione quantitativa delle cere
Si procede al calcolo delle aree dei picchi corrispondente allo standard interno del lauril arachidato e agli esteri alifatici da C 40 a C 46 mediante un integratore.
Si calcola il contenuto totale in cere aggiungendo ogni singola cera, in mg/kg di sostanza grassa, come segue:

dove:
|
Ax |
= |
area del picco del singolo estere, in unità di calcolo dell’integratore |
|
As |
= |
area del picco dello standard interno del lauril arachidato, in unità di calcolo dell’integratore |
|
ms |
= |
massa di standard interno del lauril arachidato aggiunto, in milligrammi |
|
m |
= |
massa di campione prelevato per la determinazione, in grammi |
5.5.1. Valutazione quantitativa dei metil ed etil esteri
Si procede al calcolo delle aree dei picchi corrispondenti allo standard interno del metileptadecanoato, ai metil esteri degli acidi grassi a C16 e C18 e agli etil esteri degli acidi grassi C16 e C18 mediante un integratore.
Si calcola il contenuto di ogni singolo alchil estere, in mg/kg di sostanza grassa, come segue:
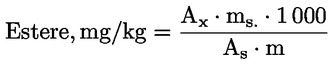
dove:
|
Ax |
= |
area del picco del singolo estere C16 e C18, in unità di calcolo dell’integratore |
|
As |
= |
area del picco dello standard interno del metileptadecanoato, in unità di calcolo dell’integratore |
|
ms |
= |
massa di standard interno del metileptadecanoato aggiunto, in milligrammi |
|
m |
= |
massa di campione prelevato per la determinazione, in grammi |
6. ESPRESSIONE DEI RISULTATI
Riportare la somma dei contenuti delle singole cere da C40 a C46 (Nota 7) in milligrammi per chilogrammo di sostanza grassa.
Riportare la somma dei singoli contenuti dei metil ed etil esteri da C16 a C18 e la loro somma.
I risultati vengono espressi arrotondando al mg/kg più vicino.
Nota 7: I componenti da quantificare si riferiscono ai picchi a numero di carbonio pari compresi tra gli esteri C40 e C46, secondo l’esempio di cromatogramma delle cere dell’olio di oliva riportato nella figura allegata. Qualora l’estere C46 risulti sdoppiato, si consiglia, ai fini della sua corretta identificazione, di analizzare la frazione cerosa di un olio di sansa dove il picco C46 risulta individuabile in quanto nettamente maggioritario.
Riportare il rapporto tra etil esteri e metil esteri
Figura 1
Esempio di cromatogramma della frazione cere di un olio di oliva ( 7 )
Picchi con un tempo di ritenzione da 5 a 8 minuti dei metil ed etil esteri degli acidi grassi
Legenda:
|
Standard interno |
= |
lauril arachidato |
|
1 |
= |
Esteri diterpenici |
|
2+2′ |
= |
Esteri C40 |
|
3+3′ |
= |
Esteri C42 |
|
4+4′ |
= |
Esteri C44 |
|
5 |
= |
Esteri C46 |
|
6 |
= |
Esteri steroli e alcoli triterpenici |
Figura 2
Metil ed etil esteri e cere di un olio vergine di oliva

Legenda:
|
1 |
– |
Metile C16 |
|
2 |
– |
Etile C16 |
|
3 |
– |
Standard interno metileptadecanoato |
|
4 |
– |
Metile C18 |
|
5 |
– |
Etile C18 |
|
6 |
– |
Squalene |
|
7 |
– |
Standard interno lauril arachidato |
|
A |
– |
Esteri diterpenici |
|
B |
– |
Cere |
|
C |
– |
Esteri steroli e alcoli triterpenici |
Figura 3
Metil ed etil esteri e cere di un olio extra vergine di oliva

Legenda:
|
1 |
– |
Standard interno metileptadecanoato |
|
2 |
– |
Metile C18 |
|
3 |
– |
Etile C18 |
|
4 |
– |
Squalene |
|
5 |
– |
Standard interno lauril arachidato |
|
A |
– |
Esteri diterpenici |
|
B |
– |
Cere |
|
C |
– |
Esteri steroli e alcoli triterpenici |
Figura 4
Parte di cromatogramma relativa a un olio extra vergine di oliva e allo stesso olio addizionato di olio deodorato

Legenda:
|
1 |
– |
Standard interno metil miristato |
|
2 |
– |
Metil palmitato |
|
3 |
– |
Etil palmitato |
|
4 |
– |
Standard interno metileptadecanoato |
|
5 |
– |
Metil linoleato |
|
6 |
– |
Metil oleato |
|
7 |
– |
Metil stearato |
|
8 |
– |
Etil linoleato |
|
9 |
– |
Etil oleato |
|
10 |
– |
Etil stearato |
Appendice A
Determinazione della velocità lineare del gas
Nel cromatografo, regolato alle normali condizioni operative, si iniettano 1:3 μl di metano (o propano) e si cronometra il tempo che il gas impiega a percorrere la colonna, dal momento dell’iniezione al momento dell’uscita del picco (tM).
La velocità lineare in cm/s è data da L/tM in cui L è la lunghezza della colonna in centimetri e tM è il tempo cronometrato in secondi.
▼M28 —————
ALLEGATO XXI
Risultati dei controlli di conformità eseguiti sugli oli di oliva di cui al paragrafo 2 dell’articolo 8
|
|
Etichettatura |
Parametri chimici |
Caratteristiche organolettiche (4) |
Conclusione finale |
|||||||||||||
|
Campione |
Categoria |
Paese di origine |
Luogo di ispezione (1) |
Denominazione legale |
Denominazione di origine |
Condizioni di conservazione |
Informazione erronea |
Leggibilità |
C/NC (3) |
Parametri fuori limite SÌ/NO |
Se sì, indicare quale/quali (2) |
C/NC (3) |
Mediana dei difetti |
MEDIA-na del fruttato |
C/NC (3) |
Azione richiesta |
Sanzione |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(1) Mercato interno (frantoi, imbottigliatori, fase di vendita al dettaglio), esportazione, importazione (2) Ciascuna caratteristica dell’olio di oliva di cui all’allegato I deve avere un codice (3) Conforme/non conforme (4) Non richieste per l’olio di oliva e per l’olio di sansa |
|||||||||||||||||
( 1 ) GU L 228 dell’1.9.2009, pag. 3.
( 2 ) Direttiva 2011/91/UE del Parlamento europeo e del Consiglio, del 13 dicembre 2011, relativa alle diciture o marche che consentono di identificare la partita alla quale appartiene una derrata alimentare (GU L 334 del 16.12.2011, pag. 1).
( 3 ) Dopo l’eluizione degli esteri degli steroli, il tracciato cromatografico non deve presentare picchi significativi (trigliceridi).
( 4 ) Regolamento (UE) n. 1308/2013 del Parlamento europeo e del Consiglio, del 17 dicembre 2013, recante organizzazione comune dei mercati dei prodotti agricoli e che abroga i regolamenti (CEE) n. 922/72, (CEE) n. 234/79, (CE) n. 1037/2001 e (CE) n. 1234/2007 del Consiglio (GU L 347 del 20.12.2013, pag. 671).
( 5 ) Qualora osservi per via olfattiva diretta attributi negativi estremamente intensi, l'assaggiatore, in via eccezionale, potrà astenersi dall'assaggio. Indicherà l'accaduto sulla scheda di profilo.
( 6 ) In questi casi, in particolare, si deve utilizzare la miscela eluente benzene-acetone 95/5 (v/v) per ottenere una buona separazione delle bande.
( 7 ) Dopo l’eluzione degli esteri degli steroli il cromatogramma non deve presentare picchi significativi (trigliceridi).