This document is an excerpt from the EUR-Lex website
Document 02011R1007-20180215
Regulation (EU) No 1007/2011 of the European Parliament and of the Council of 27 September 2011 on textile fibre names and related labelling and marking of the fibre composition of textile products and repealing Council Directive 73/44/EEC and Directives 96/73/EC and 2008/121/EC of the European Parliament and of the Council (Text with EEA relevance)
Consolidated text: Verordnung (EU) Nr. 1007/2011 des Europäischen Parlaments und des Rates vom 27. September 2011 über die Bezeichnungen von Textilfasern und die damit zusammenhängende Etikettierung und Kennzeichnung der Faserzusammensetzung von Textilerzeugnissen und zur Aufhebung der Richtlinie 73/44/EWG des Rates und der Richtlinien 96/73/EG und 2008/121/EG des Europäischen Parlaments und des Rates (Text von Bedeutung für den EWR)
Verordnung (EU) Nr. 1007/2011 des Europäischen Parlaments und des Rates vom 27. September 2011 über die Bezeichnungen von Textilfasern und die damit zusammenhängende Etikettierung und Kennzeichnung der Faserzusammensetzung von Textilerzeugnissen und zur Aufhebung der Richtlinie 73/44/EWG des Rates und der Richtlinien 96/73/EG und 2008/121/EG des Europäischen Parlaments und des Rates (Text von Bedeutung für den EWR)

- Date of document:
- 15/02/2018
- Date of effect:
- 15/02/2018
- Author:
- Not available
- Form:
- Konsolidierter Text
- Additional information:
- LASTMODIN 32011R1007R(05)
- Link
- Link
- Link
- Select all documents mentioning this document
- Consolidation: basic act:
- 32011R1007
02011R1007 — DE — 15.02.2018 — 004.001
Dieser Text dient lediglich zu Informationszwecken und hat keine Rechtswirkung. Die EU-Organe übernehmen keine Haftung für seinen Inhalt. Verbindliche Fassungen der betreffenden Rechtsakte einschließlich ihrer Präambeln sind nur die im Amtsblatt der Europäischen Union veröffentlichten und auf EUR-Lex verfügbaren Texte. Diese amtlichen Texte sind über die Links in diesem Dokument unmittelbar zugänglich
|
VERORDNUNG (EU) Nr. 1007/2011 DES EUROPÄISCHEN PARLAMENTS UND DES RATES vom 27. September 2011 über die Bezeichnungen von Textilfasern und die damit zusammenhängende Etikettierung und Kennzeichnung der Faserzusammensetzung von Textilerzeugnissen und zur Aufhebung der Richtlinie 73/44/EWG des Rates und der Richtlinien 96/73/EG und 2008/121/EG des Europäischen Parlaments und des Rates (Text von Bedeutung für den EWR) (ABl. L 272 vom 18.10.2011, S. 1) |
Geändert durch:
|
|
|
Amtsblatt |
||
|
Nr. |
Seite |
Datum |
||
|
L 338 |
1 |
21.12.2011 |
||
|
DELEGIERTE VERORDNUNG (EU) Nr. 286/2012 DER KOMMISSION vom 27. Januar 2012 |
L 95 |
1 |
31.3.2012 |
|
|
L 158 |
1 |
10.6.2013 |
||
|
DELEGIERTE VERORDNUNG (EU) 2018/122 DER KOMMISSION vom 20. Oktober 2017 |
L 22 |
3 |
26.1.2018 |
|
Berichtigt durch:
VERORDNUNG (EU) Nr. 1007/2011 DES EUROPÄISCHEN PARLAMENTS UND DES RATES
vom 27. September 2011
über die Bezeichnungen von Textilfasern und die damit zusammenhängende Etikettierung und Kennzeichnung der Faserzusammensetzung von Textilerzeugnissen und zur Aufhebung der Richtlinie 73/44/EWG des Rates und der Richtlinien 96/73/EG und 2008/121/EG des Europäischen Parlaments und des Rates
(Text von Bedeutung für den EWR)
KAPITEL 1
ALLGEMEINE BESTIMMUNGEN
Artikel 1
Gegenstand
In dieser Verordnung sind die Vorschriften für die Verwendung von Bezeichnungen von Textilfasern und die damit zusammenhängende Etikettierung und Kennzeichnung der Faserzusammensetzung von Textilerzeugnissen, Vorschriften über die Etikettierung oder Kennzeichnung nichttextiler Teile tierischen Ursprungs und Vorschriften über die Bestimmung der Faserzusammensetzung von Textilerzeugnissen durch quantitative Analyse von binären und ternären Textilfasergemischen festgelegt, um das Funktionieren des Binnenmarkts zu verbessern und den Verbrauchern zutreffende Informationen zur Verfügung zu stellen.
Artikel 2
Geltungsbereich
(1) Diese Verordnung gilt für Textilerzeugnisse, wenn sie auf dem Unionsmarkt bereitgestellt werden, und für die in Absatz 2 genannten Erzeugnisse.
(2) Für die Zwecke dieser Verordnung werden die folgenden Erzeugnisse in der gleichen Weise wie Textilerzeugnisse behandelt:
a) Erzeugnisse mit einem Gewichtsanteil an Textilfasern von mindestens 80 %;
b) Bezugsmaterial für Möbel, Regen- und Sonnenschirme mit einem Gewichtsanteil an Textilkomponenten von mindestens 80 %;
c) die Textilkomponenten
i) der oberen Schicht mehrschichtiger Fußbodenbeläge,
ii) von Matratzenbezügen,
iii) von Bezügen von Campingartikeln,
sofern diese Textilkomponenten einen Gewichtsanteil von mindestens 80 %dieser oberen Schichten oder Bezüge ausmachen;
d) Textilien, die in andere Waren eingearbeitet sind und zu deren Bestandteil werden, sofern ihre Zusammensetzung angegeben ist.
(3) Diese Verordnung gilt nicht für Textilerzeugnisse, die ohne Übereignung an Heimarbeiter oder selbständige Unternehmen zur Weiterverarbeitung übergeben werden.
(4) Diese Verordnung gilt nicht für maßgeschneiderte Textilerzeugnisse, die von selbständigen Schneidern hergestellt wurden.
Artikel 3
Begriffsbestimmungen
(1) Im Sinne dieser Verordnung bezeichnet der Ausdruck
a) „Textilerzeugnis“ ein Erzeugnis, das im rohen, halbbearbeiteten, bearbeiteten, halbverarbeiteten, verarbeiteten, halbkonfektionierten oder konfektionierten Zustand ausschließlich Textilfasern enthält, unabhängig von dem zur Mischung oder Verbindung angewandten Verfahren;
b) „Textilfaser“
i) ein Erzeugnis, das durch seine Flexibilität, seine Feinheit und seine große Länge im Verhältnis zum Höchstquerschnitt gekennzeichnet ist und sich somit zur Herstellung von Textilerzeugnissen eignet, oder
ii) ein flexibles Band oder ein Schlauch mit einer Normalbreite von höchstens 5 mm, einschließlich der Bänder, die von breiteren Bändern oder Bahnen abgeschnitten werden, hergestellt auf der Grundlage der zur Herstellung der in Anhang I Tabelle 2 aufgeführten Fasern dienenden Stoffe und geeignet zur Herstellung von Textilerzeugnissen;
c) „Normalbreite“ die Breite des Bandes oder des Schlauches in gefalteter, abgeflachter, gepresster oder gedrehter Form, oder bei nicht einheitlicher Breite die Durchschnittsbreite;
d) „Textilkomponente“ einen Teil eines Textilerzeugnisses mit einem feststellbaren Fasergehalt;
e) „Fremdfasern“ Fasern, die nicht auf dem Etikett oder der Kennzeichnung angegeben sind;
f) „Futter“ eine separate Komponente, die bei der Fertigung von Kleidungsstücken und anderen Erzeugnissen verwendet wird und aus ein- oder mehrschichtigem Textilmaterial besteht, das an einem oder mehreren Säumen befestigt ist;
g) „Etikettierung“ die Angabe der erforderlichen Informationen auf dem Textilerzeugnis durch die Anbringung eines Etiketts;
h) „Kennzeichnung“ die unmittelbare Angabe der erforderlichen Informationen auf dem Textilerzeugnis durch Aufnähen, Aufsticken, Drucken, Prägen oder jede andere Technik des Anbringens;
i) „globale Etikettierung“ die Verwendung eines einzigen Etiketts für mehrere Textilerzeugnisse oder -komponenten;
j) „Einwegartikel“ ein Textilerzeugnis, das dazu konzipiert ist, nur einmal oder kurzfristig verwendet zu werden und dessen normale Verwendung nicht zu einer späteren Verwendung zum gleichen Zweck oder zu einem ähnlichen Zweck dient;
k) „vereinbarter Zuschlag“ den Wert der Feuchtigkeitsaufnahme, der bei der Berechnung des Prozentsatzes der Faserkomponenten auf der Basis des Trockengewichts der reinen Fasern nach Anwendung der vereinbarten Zuschläge zu verwenden ist.
(2) Für die Zwecke dieser Verordnung gelten die in Artikel 2 der Verordnung (EG) Nr. 765/2008 festgelegten Definitionen der Begriffe „Bereitstellung auf dem Markt“, „Inverkehrbringen“, „Hersteller“, „Einführer“, „Händler“, „Wirtschaftsakteure“, „harmonisierte Norm“, „Marktüberwachung“ und „Marktüberwachungsbehörde“.
Artikel 4
Allgemeine Voraussetzung für die Bereitstellung von Textilerzeugnissen auf dem Markt
Textilerzeugnisse dürfen nur dann auf dem Markt bereitgestellt werden, wenn sie etikettiert oder gekennzeichnet sind oder ihnen Handelsdokumente im Einklang mit dieser Verordnung beiliegen.
KAPITEL 2
BEZEICHNUNGEN VON TEXTILFASERN UND DAMIT ZUSAMMENHÄNGENDE ETIKETTIERUNGS- UND KENNZEICHNUNGSVORSCHRIFTEN
Artikel 5
Bezeichnungen von Textilfasern
(1) Für die Beschreibung der Faserzusammensetzungen auf Etiketten und Kennzeichnungen von Textilerzeugnissen dürfen nur die Textilfaserbezeichnungen nach Anhang I verwendet werden.
(2) Die Verwendung der Bezeichnungen nach Anhang I ist jenen Textilfasern vorbehalten, die in ihren Eigenschaften der Beschreibung nach diesem Anhang entsprechen.
Die Bezeichnungen nach Anhang I dürfen weder alleinstehend noch in Wortverbindungen oder als Eigenschaftswort für andere Fasern verwendet werden.
Die Verwendung des Begriffs „Seide“ ist zur Angabe der Form oder besonderen Aufmachung von Textilfasern als Endlosfasern nicht zulässig.
Artikel 6
Anträge auf Einfügung neuer Bezeichnungen von Textilfasern
Jeder Hersteller oder jede für den Hersteller handelnde Person kann beantragen, dass die Kommission eine neue Textilfaserbezeichnung in die Liste in Anhang I aufnimmt.
Der Antrag muss ein technisches Dossier enthalten, das gemäß Anhang II zusammengestellt wurde.
Artikel 7
Reine Textilerzeugnisse
(1) Nur Textilerzeugnisse, die ausschließlich aus einer Faser bestehen, dürfen den Zusatz „100 %“, „rein“ oder „ganz“ auf dem Etikett oder der Kennzeichnung tragen.
Für andere Textilerzeugnisse dürfen diese oder ähnliche Formulierungen nicht verwendet werden.
(2) Unbeschadet des Artikels 8 Absatz 3 kann auch ein Textilerzeugnis, das einen Gewichtsanteil an Fremdfasern von nicht mehr als 2 % enthält, als ausschließlich aus einer Faser bestehend behandelt werden, sofern dieser Anteil dadurch gerechtfertigt ist, dass er bei guter Herstellungspraxis technisch unvermeidbar und nicht Ergebnis einer systematischen Hinzufügung ist.
Ein im Streichverfahren gewonnenes Textilerzeugnis kann auch als ausschließlich aus einer Faser bestehend behandelt werden, wenn es einen Gewichtsanteil an Fremdfasern von nicht mehr als 5 % enthält, sofern dieser Anteil dadurch gerechtfertigt ist, dass er bei guter Herstellungspraxis technisch unvermeidbar und nicht Ergebnis einer systematischen Hinzufügung ist.
Artikel 8
Erzeugnisse aus Schurwolle
(1) Ein Wollerzeugnis darf nur dann mit einer der in Anhang III aufgeführten Bezeichnungen etikettiert oder gekennzeichnet werden, wenn es ausschließlich aus einer Wollfaser besteht, die niemals in einem Fertigerzeugnis enthalten war und die weder einem anderen als dem zur Herstellung des Erzeugnisses erforderlichen Spinn- und/oder Filzprozess unterlegen hat noch einer faserschädigenden Behandlung oder Benutzung ausgesetzt wurde.
(2) Abweichend von Absatz 1 dürfen die in Anhang III genannten Bezeichnungen für die in einem Textilfasergemisch enthaltene Wolle verwendet werden, wenn alle folgenden Bedingungen erfüllt sind:
a) die gesamte in dem Gemisch enthaltene Wolle entspricht den Voraussetzungen des Absatzes 1;
b) der Anteil dieser Wolle am Gesamtgewicht des Gemischs beträgt nicht weniger als 25 %;
c) die Wolle ist im Fall eines intimen Fasergemischs nur mit einer einzigen anderen Faser gemischt.
Die vollständige prozentuale Zusammensetzung eines solchen Gemischs ist anzugeben.
(3) Der Gewichtsanteil von Fremdfasern bei den in den Absätzen 1 und 2 bezeichneten Erzeugnissen, einschließlich im Streichverfahren gewonnener Wollerzeugnisse, darf 0,3 % nicht überschreiten und muss dadurch gerechtfertigt sein, dass er bei guter Herstellungspraxis technisch unvermeidbar und nicht Ergebnis einer systematischen Hinzufügung ist.
Artikel 9
Multifaser-Textilerzeugnisse
(1) Auf dem Etikett oder der Kennzeichnung von Textilerzeugnissen werden die Bezeichnung und der Gewichtsanteil aller im Erzeugnis enthaltenen Fasern in absteigender Reihenfolge angegeben.
(2) Abweichend von Absatz 1 und unbeschadet des Artikels 7 Absatz 2 dürfen Fasern, deren Anteil am Gesamtgewicht des Textilerzeugnisses bis zu 5 % beträgt, oder Fasern, deren Anteil am Gesamtgewicht des Textilerzeugnisses zusammen bis zu 15 % beträgt, als „sonstige Fasern“ bezeichnet werden, wobei ihr Gewichtsanteil unmittelbar davor oder dahinter anzugeben ist, vorausgesetzt, dass sie zum Zeitpunkt der Herstellung schwierig zu bestimmen sind.
(3) Erzeugnisse mit einer Kette aus reiner Baumwolle und einem Schuss aus reinem Leinen, bei denen der Hundertsatz des Leinens mindestens 40 % des Gesamtgewichts des entschlichteten Gewebes ausmacht, können als „Halbleinen“ bezeichnet werden, wobei die Angabe der Zusammensetzung „Kette reine Baumwolle — Schuss reiner Flachs (bzw. Leinen)“ hinzugefügt werden muss.
(4) Unbeschadet des Artikels 5 Absatz 1 dürfen für Textilerzeugnisse, deren Zusammensetzung zum Zeitpunkt ihrer Herstellung schwierig zu bestimmen ist, die Bezeichnungen „diverse Faserarten“ oder „Erzeugnis unbestimmter Zusammensetzung“ auf dem Etikett oder der Kennzeichnung verwendet werden.
(5) Abweichend von Absatz 1 dieses Artikels können Fasern, die nicht in Anhang I aufgeführt sind, als „sonstige Fasern“ bezeichnet werden, wobei ihr Gewichtsanteil unmittelbar davor oder dahinter anzugeben ist.
Artikel 10
Fasern mit dekorativer Wirkung und Fasern mit antistatischer Wirkung
(1) Sichtbare und isolierbare Fasern, mit denen eine rein dekorative Wirkung erzielt werden soll und die nicht mehr als 7 % des Gewichts des Fertigerzeugnisses ausmachen, müssen nicht in den in den Artikeln 7 und 9 vorgesehenen Faserzusammensetzungen berücksichtigt werden.
(2) Metallfasern und andere Fasern, die zur Erzielung einer antistatischen Wirkung zugesetzt werden und die nicht mehr als 2 % des Gewichts des Fertigerzeugnisses ausmachen, müssen nicht in den in den Artikeln 7 und 9 vorgesehenen Faserzusammensetzungen berücksichtigt werden.
►C2 (3) Im Fall der in Artikel 9 Absatz 3 genannten Erzeugnisse ◄ werden die in den Absätzen 1 und 2 des vorliegenden Artikels vorgesehenen Prozentsätze getrennt für das Gewicht der Schussfäden und der Kettfäden berechnet.
Artikel 11
Mehrkomponenten-Textilerzeugnisse
(1) Jedes Textilerzeugnis, das aus zwei oder mehr Textilkomponenten besteht, die nicht denselben Textilfasergehalt haben, ist mit einem Etikett oder einer Kennzeichnung zu versehen, das bzw. die für jede Komponente den Textilfasergehalt angibt.
(2) Die in Absatz 1 vorgesehene Etikettierung oder Kennzeichnung ist nicht erforderlich für Textilkomponenten, die die beiden folgenden Bedingungen erfüllen:
a) Sie sind nicht die Hauptfutterstoffe und
b) sie machen weniger als 30 % des Gesamtgewichts des Textilerzeugnisses aus.
(3) Zwei oder mehrere Textilerzeugnisse mit demselben Fasergehalt, die nach den Gepflogenheiten ein einheitliches Ganzes bilden, brauchen nur mit einem Etikett oder einer Kennzeichnung versehen zu werden.
Artikel 12
Textilerzeugnisse, die nichttextile Teile tierischen Ursprungs enthalten
(1) Nichttextile Teile tierischen Ursprungs in Textilerzeugnissen sind unter Verwendung des Hinweises „Enthält nichttextile Teile tierischen Ursprungs“ bei der Etikettierung oder Kennzeichnung von Erzeugnissen, die solche Teile enthalten, anzugeben, wenn sie auf dem Markt bereitgestellt werden.
(2) Die Etikettierung oder Kennzeichnung darf nicht irreführend sein und muss so erfolgen, dass sie vom Verbraucher ohne Schwierigkeiten verstanden werden kann.
Artikel 13
Etikettierung und Kennzeichnung von in Anhang IV aufgeführten Textilerzeugnissen
Die Faserzusammensetzung der in Anhang IV aufgeführten Textilerzeugnisse ist nach den dort festgelegten Etikettierungs- und Kennzeichnungsbestimmungen anzugeben.
Artikel 14
Etiketten und Kennzeichnungen
(1) Textilerzeugnisse werden zur Angabe ihrer Faserzusammensetzung etikettiert oder gekennzeichnet, wenn sie auf dem Markt bereitgestellt werden.
Die Etikettierung und Kennzeichnung von Textilerzeugnissen muss dauerhaft, leicht lesbar, sichtbar und zugänglich und — im Falle eines Etiketts — fest angebracht sein.
(2) Unbeschadet des Absatzes 1 können diese Etiketten oder Kennzeichnungen durch Begleitpapiere (Handelsdokumente) ersetzt oder ergänzt werden, wenn die Erzeugnisse Wirtschaftsakteuren in der Lieferkette oder zur Erfüllung eines Auftrags eines öffentlichen Auftraggebers im Sinne von Artikel 1 der Richtlinie 2004/18/EG des Europäischen Parlaments und des Rates vom 31. März 2004 über die Koordinierung der Verfahren zur Vergabe öffentlicher Bauaufträge, Lieferaufträge und Dienstleistungsaufträge ( 1 ) geliefert werden.
(3) Die in den Artikeln 5, 7, 8 und 9 genannten Bezeichnungen von Textilfasern und Beschreibungen der Faserzusammensetzung sind in den in Absatz 2 dieses Artikels genannten Begleitpapieren (Handelsdokumenten) deutlich anzugeben.
Die Verwendung von Abkürzungen ist nicht zulässig; eine Ausnahme gilt für Lochkartenschlüssel oder für Abkürzungen, die in internationalen Normen definiert sind, sofern ihre Bedeutung in demselben Handelsdokument erläutert wird.
Artikel 15
Verpflichtung zur Etikettierung oder Kennzeichnung
(1) Bringt ein Hersteller ein Textilerzeugnis in Verkehr, so stellt er die Etikettierung oder Kennzeichnung und die Richtigkeit der darin enthaltenen Informationen sicher. Ist der Hersteller nicht in der Union niedergelassen, so stellt der Einführer die Etikettierung oder Kennzeichnung und die Richtigkeit der darin enthaltenen Informationen sicher.
(2) Ein Händler gilt für die Zwecke dieser Verordnung als Hersteller, wenn er ein Erzeugnis unter seinem Namen oder seiner Handelsmarke in Verkehr bringt, das Etikett selbst anbringt oder den Inhalt des Etiketts ändert.
(3) Stellt ein Händler ein Textilerzeugnis auf dem Markt bereit, so stellt er sicher, dass es die entsprechende Etikettierung oder Kennzeichnung gemäß dieser Verordnung trägt.
(4) Die in den Absätzen 1, 2 und 3 genannten Wirtschaftsakteure stellen sicher, dass die Informationen, die vorgelegt werden, wenn Textilerzeugnisse auf dem Markt bereitgestellt werden, nicht mit den in dieser Verordnung vorgesehenen Bezeichnungen von Textilfasern und den Beschreibungen der Faserzusammensetzung verwechselt werden können.
Artikel 16
Verwendung der Bezeichnungen von Textilfasern und der Beschreibungen der Faserzusammensetzung
(1) Wird ein Textilerzeugnis auf dem Markt bereitgestellt, so werden die in den Artikeln 5, 7, 8 und 9 genannten Beschreibungen der Textilfaserzusammensetzung in Katalogen, in Prospekten, auf Verpackungen, Etiketten und Kennzeichnungen in einer Weise angegeben, dass sie leicht lesbar, sichtbar und deutlich erkennbar sind, sowie in einem Schriftbild, das in Bezug auf Schriftgröße, Stil und Schriftart einheitlich ist. Diese Informationen müssen für Verbraucher vor dem Kauf deutlich sichtbar sein; dies gilt auch für Fälle, in denen der Kauf auf elektronischem Wege erfolgt.
(2) Markenzeichen oder Firmenbezeichnungen dürfen den in den Artikeln 5, 7, 8 und 9 genannten Beschreibungen der Textilfaserzusammensetzung unmittelbar voran- oder nachgestellt werden.
Enthält jedoch ein Markenzeichen oder eine Firmenbezeichnung allein stehend oder in Wortverbindungen oder als Eigenschaftswort eine der in Anhang I aufgeführten Bezeichnungen von Textilfasern oder eine Bezeichnung, die leicht damit zu verwechseln ist, so ist solch ein Markenzeichen oder solch eine Firmenbezeichnung den in Artikel 5, 7, 8 und 9 genannten Beschreibungen der Textilfaserzusammensetzung unmittelbar voran- oder nachzustellen.
Andere Informationen werden stets getrennt davon aufgeführt.
(3) Die Etikettierung oder Kennzeichnung erfolgt in der Amtssprache oder den Amtssprachen des Mitgliedstaats, in dessen Hoheitsgebiet die Textilerzeugnisse dem Verbraucher bereitgestellt werden, es sei denn der betreffende Mitgliedstaat schreibt etwas anderes vor.
Bei Nähgarn, Stopfgarn oder Stickgarn, das auf Spulen, Fadenrollen, in Strähnen, Knäueln oder sonstigen kleinen Einheiten angeboten wird, gilt Unterabsatz 1 für die globale Etikettierung gemäß Artikel 17 Absatz 3. Werden solche Erzeugnisse einzeln verkauft, so können sie in einer beliebigen Amtssprache der Organe der Union etikettiert oder gekennzeichnet sein, sofern sie auch eine globale Etikettierung aufweisen.
Artikel 17
Ausnahmeregelungen
(1) Die Vorschriften der Artikel 11, 14, 15 und 16 gelten vorbehaltlich der in den Absätzen 2, 3 und 4 des vorliegenden Artikels geregelten Ausnahmen.
(2) Die Angabe der Bezeichnungen von Textilfasern oder der Faserzusammensetzung in der Etikettierung und Kennzeichnung der in Anhang V aufgeführten Textilerzeugnisse ist nicht vorgeschrieben.
Enthält jedoch ein Markenzeichen oder eine Firmenbezeichnung eine der in Anhang I aufgeführten Bezeichnungen oder eine damit verwechselbare Bezeichnung allein stehend oder in Wortverbindungen oder als Eigenschaftswort, so gelten die Artikel 11, 14, 15 und 16.
(3) Sind in Anhang VI aufgeführte Textilerzeugnisse von der gleichen Art und weisen sie die gleiche Faserzusammensetzung auf, so dürfen sie mit einer globalen Etikettierung zusammen auf dem Markt bereitgestellt werden.
(4) Die Faserzusammensetzung von Textilerzeugnissen, die als Meterware verkauft werden, kann auf dem Stück oder auf der Rolle, das bzw. die auf dem Markt bereitgestellt wird, angegeben werden.
(5) Die in den Absätzen 3 und 4 genannten Textilerzeugnisse werden so auf dem Markt bereitgestellt, dass ihre Faserzusammensetzung jedem Käufer in der Lieferkette bis hin zum Verbraucher bekannt ist.
KAPITEL 3
MARKTÜBERWACHUNG
Artikel 18
Überprüfungen zur Marktüberwachung
Die Marktüberwachungsbehörden überprüfen, ob die Faserzusammensetzung der Textilerzeugnisse mit der angegebenen Faserzusammensetzung dieser Erzeugnisse nach Maßgabe dieser Verordnung übereinstimmt.
Artikel 19
Bestimmung der Faserzusammensetzung
(1) Zum Zwecke der Bestimmung der Faserzusammensetzung von Textilerzeugnissen erfolgen die Überprüfungen nach Artikel 18 gemäß den Methoden des Anhangs VIII oder mit in diesen Anhang aufzunehmenden harmonisierten Normen.
(2) Bei der Bestimmung der Faserzusammensetzungen gemäß den Artikeln 7, 8 und 9 sind die in Anhang VII aufgeführten Artikel nicht zu berücksichtigen.
(3) Die in den Artikeln 7, 8 und 9 genannte Faserzusammensetzung wird unter Anwendung des in Anhang IX vorgesehenen vereinbarten Zuschlags auf die Trockenmasse jeder Faser berechnet, nachdem die in Anhang VII aufgeführten Artikel ausgesondert wurden.
(4) Die Labore, die für den Test von Textilgemischen verantwortlich sind, für die es kein einheitliches Analyseverfahren auf Unionsebene gibt, bestimmen die Faserzusammensetzung dieser Gemische, wobei im Analysebericht die erzielten Ergebnisse, das angewandte Verfahren und dessen Genauigkeit anzugeben sind.
Artikel 20
Toleranzen
(1) Für die Zwecke der Ermittlung der Faserzusammensetzung von Textilerzeugnissen gelten die Toleranzen gemäß den Absätzen 2, 3 und 4.
(2) Unbeschadet des Artikels 8 Absatz 3 entfällt die Angabe von Fremdfasern in der Faserzusammensetzung gemäß Artikel 9, wenn der prozentuale Anteil dieser Fasern folgende Werte nicht erreicht:
a) 2 % des Gesamtgewichts des Textilerzeugnisses, sofern dieser Anteil dadurch gerechtfertigt ist, dass er bei guter Herstellungspraxis technisch unvermeidbar und nicht Ergebnis einer systematischen Hinzufügung ist, oder
b) 5 % des Gesamtgewichts bei Textilerzeugnissen, die im Streichverfahren gewonnen wurden, sofern dieser Anteil dadurch gerechtfertigt ist, dass er bei guter Herstellungspraxis technisch unvermeidbar und nicht Ergebnis einer systematischen Hinzufügung ist.
(3) Eine Herstellungstoleranz von 3 % zwischen der gemäß Artikel 9 angegebenen Faserzusammensetzung und den Anteilen, die mit der gemäß Artikel 19 durchgeführten Analyse ermittelt werden, bezogen auf das Gesamtgewicht der auf dem Etikett oder in der Kennzeichnung angegebenen Fasern, ist zulässig. Diese Toleranz gilt auch für
a) Fasern, die gemäß Artikel 9 mit den Worten „sonstige Fasern“ bezeichnet werden dürfen,
b) den Hundertteil von Wolle gemäß Artikel 8 Absatz 2 Buchstabe b.
Für die Zwecke der Analyse werden die Toleranzen einzeln berechnet. Das für die Berechnung der Toleranz nach diesem Absatz heranzuziehende Gesamtgewicht ist das Gewicht der Fasern des Fertigerzeugnisses, abzüglich des Gewichts von Fremdfasern, die bei der Anwendung der Toleranz gemäß Absatz 2 des vorliegenden Artikels möglicherweise festgestellt wurden.
(4) Die Kumulierung der Toleranzen nach den Absätzen 2 und 3 ist nur zulässig, wenn sich herausstellt, dass die bei der Anwendung der Toleranz nach Absatz 2 durch die Analyse möglicherweise festgestellten Fremdfasern von der gleichen chemischen Art sind wie eine oder mehrere der auf dem Etikett oder in der Kennzeichnung angegebenen Fasern.
(5) Für besondere Textilerzeugnisse, deren Herstellungsverfahren höhere Toleranzen erfordert als die in den Absätzen 2 und 3 festgelegten Toleranzen, kann die Kommission höhere Toleranzen zulassen.
Vor dem Inverkehrbringen des Textilerzeugnisses stellt der Hersteller einen Antrag auf Genehmigung durch die Kommission, in dem er ausreichende Gründe für die außergewöhnlichen Umstände bei der Herstellung nennt und diese außergewöhnlichen Umstände nachweist. Die Genehmigung kann nur in außergewöhnlichen Fällen und bei entsprechender Rechtfertigung seitens des Herstellers gewährt werden.
Die Kommission erlässt gegebenenfalls durch delegierte Rechtsakte gemäß Artikel 22 technische Kriterien und Verfahrensvorschriften für die Anwendung dieses Absatzes.
KAPITEL 4
SCHLUSSBESTIMMUNGEN
Artikel 21
Delegierte Rechtsakte
(1) Der Kommission wird die Befugnis übertragen, delegierte Rechtsakte gemäß Artikel 22 in Bezug auf technische Kriterien und Verfahrensvorschriften zur Anwendung von Artikel 20 Absatz 5, Änderungen der Anhänge II, IV, V, VI, VII, VIII und IX, die erforderlich sind, um dem technischen Fortschritt Rechnung zu tragen, und Änderungen des Anhangs I, um gemäß Artikel 6 neue Textilfasernamen in die in diesem Anhang genannte Liste aufzunehmen, zu erlassen.
(2) Beim Erlass dieser delegierten Rechtsakte handelt die Kommission nach den Bestimmungen dieser Verordnung.
Artikel 22
Ausübung der Befugnisübertragung
(1) Die Befugnis zum Erlass delegierter Rechtsakte wird der Kommission unter den in diesem Artikel festgelegten Bedingungen übertragen.
(2) Die Befugnis zum Erlass delegierter Rechtsakte gemäß Artikel 20 Absatz 5 und Artikel 21 wird der Kommission für einen Zeitraum von fünf Jahren ab dem 7. November 2011 übertragen. Die Kommission erstellt spätestens neun Monate vor Ablauf des Zeitraums von fünf Jahren einen Bericht über die Befugnisübertragung. Die Befugnisübertragung verlängert sich stillschweigend um Zeiträume gleicher Länge, es sei denn, das Europäische Parlament oder der Rat widersprechen einer solcher Verlängerung spätestens drei Monate vor Ablauf des jeweiligen Zeitraums.
(3) Die Befugnisübertragung gemäß Artikel 20 Absatz 5 und Artikel 21 kann vom Europäischen Parlament oder vom Rat jederzeit widerrufen werden. Der Beschluss über den Widerruf beendet die Übertragung der in diesem Beschluss angegebenen Befugnis. Er wird am Tag nach seiner Veröffentlichung im Amtsblatt der Europäischen Union oder zu einem darin angegebenen späteren Zeitpunkt wirksam. Die Gültigkeit von delegierten Rechtsakten, die bereits in Kraft sind, wird von dem Beschluss über den Widerruf nicht berührt.
(4) Sobald die Kommission einen delegierten Rechtsakt erlässt, übermittelt sie ihn gleichzeitig dem Europäischen Parlament und dem Rat.
(5) Ein delegierter Rechtsakt, der gemäß Artikel 20 Absatz 5 und Artikel 21 erlassen worden ist, tritt nur in Kraft, wenn weder das Europäische Parlament noch der Rat innerhalb einer Frist von zwei Monaten nach Übermittlung dieses Rechtsakts an das Europäische Parlament und den Rat Einwände erhoben haben oder wenn vor Ablauf dieser Frist das Europäische Parlament und der Rat beide der Kommission mitgeteilt haben, dass sie keine Einwände erheben werden. Auf Initiative des Europäischen Parlaments oder des Rates wird diese Frist um zwei Monate verlängert.
Artikel 23
Berichterstattung
Bis zum 8. November 2014 legt die Kommission dem Europäischen Parlament und dem Rat einen Bericht über die Anwendung dieser Verordnung vor, wobei ein Schwerpunkt auf den Anträgen auf neue Bezeichnungen von Textilfasern und deren Annahme liegt, gegebenenfalls zusammen mit einem Gesetzgebungsvorschlag.
Artikel 24
Überprüfung
(1) Bis zum 30. September 2013 legt die Kommission dem Europäischen Parlament und dem Rat einen Bericht über mögliche neue Etikettierungsvorschriften vor, die auf Unionsebene eingeführt werden sollten, damit den Verbrauchern genaue, sachdienliche, verständliche und vergleichbare Informationen über die Merkmale von Textilerzeugnissen zur Verfügung gestellt werden.
(2) Der Bericht beruht auf einer Konsultation aller Beteiligten und berücksichtigt die geltenden einschlägigen europäischen und internationalen Normen.
(3) Dem Bericht werden gegebenenfalls Gesetzgebungsvorschläge beigefügt, und es werden darin unter anderem folgende Aspekte geprüft:
a) ein Ursprungsetikettierungssystem, mit dem den Verbrauchern zutreffende Informationen über das Ursprungsland und zusätzliche Informationen, mit denen die lückenlose Rückverfolgbarkeit von Textilerzeugnissen gewährleistet wird, zur Verfügung gestellt werden sollen, unter Berücksichtigung der Ergebnisse der Entwicklungen im Zusammenhang mit potenziellen horizontalen Ursprungsland-Vorschriften;
b) ein vereinheitlichtes Etikettierungssystem für die Pflege;
c) ein unionsweit einheitliches Etikettierungssystem für Größen für einschlägige Textilerzeugnisse;
d) eine Angabe allergener Stoffe;
e) elektronische Etikettierung und andere neue Technologien und die Verwendung sprachunabhängiger Symbole oder Codes zur Ermittlung von Fasern.
Artikel 25
Studie zu gefährlichen Stoffen
Bis zum 30. September 2013 führt die Kommission eine Studie durch, in der bewertet wird, ob ein Kausalzusammenhang zwischen allergischen Reaktionen und in Textilerzeugnissen verwendeten chemischen Stoffen oder Gemischen besteht. Auf der Grundlage dieser Studie legt die Kommission gegebenenfalls Gesetzgebungsvorschläge im Rahmen der geltenden Vorschriften der Union vor.
Artikel 26
Übergangsbestimmung
Textilerzeugnisse, die der Richtlinie 2008/121/EG entsprechen und vor dem 8. Mai 2012 in Verkehr gebracht werden, können bis zum 9. November 2014 weiterhin auf dem Markt bereitgestellt werden.
Artikel 27
Aufhebung
Die Richtlinien 73/44/EWG, 96/73/EG und 2008/121/EG werden mit Wirkung vom 8. Mai 2012 aufgehoben.
Bezugnahmen auf die aufgehobenen Richtlinien gelten als Bezugnahmen auf diese Verordnung und sind nach Maßgabe der Entsprechungstabellen in Anhang X zu lesen.
Artikel 28
Inkrafttreten
Diese Verordnung tritt am zwanzigsten Tag nach ihrer Veröffentlichung im Amtsblatt der Europäischen Union in Kraft.
Sie gilt ab dem 8. Mai 2012.
Diese Verordnung ist in allen ihren Teilen verbindlich und gilt unmittelbar in jedem Mitgliedstaat.
ANHANG I
Liste der Bezeichnungen von Textilfasern
(gemäß Artikel 5)
Tabelle 1
|
Nummer |
Bezeichnung |
Beschreibung der Fasern |
|
1 |
Wolle |
Faser vom Fell des Schafes (Ovis aries) oder ein Gemisch aus Fasern von der Schafschur und aus Haaren der unter Nummer 2 genannten Tiere |
|
2 |
Alpaka, Lama, Kamel, Kaschmir, Mohair, Angora (-kanin), Vikunja, Yak, Guanako, Kaschgora, Biber, Fischotter, mit oder ohne zusätzliche Bezeichnung „Wolle“ oder „Tierhaar“ |
Haare nachstehender Tiere: Alpaka, Lama, Kamel, Kaschmirziege, Angoraziege, Angorakanin, Vikunja, Yak, Guanako, Kaschgoraziege, Biber, Fischotter |
|
3 |
Tierhaar, mit oder ohne Angabe der Tiergattung (z. B. Rinderhaar, Hausziegenhaar, Rosshaar) |
Haare von verschiedenen Tieren, soweit diese nicht unter den Nummern 1 und 2 genannt sind |
|
4 |
Seide |
Faser, die ausschließlich aus Kokons seidenspinnender Insekten gewonnen wird |
|
5 |
Baumwolle |
Faser aus den Samen der Baumwollpflanze (Gossypium) |
|
6 |
Kapok |
Faser aus dem Fruchtinneren des Kapok (Ceiba pentandra) |
|
7 |
Flachs bzw. Leinen |
Bastfaser aus den Stängeln des Flachses (Linum usitatissimum) |
|
8 |
Hanf |
Bastfaser aus den Stängeln des Hanfes (Cannabis sativa) |
|
9 |
Jute |
Bastfaser aus den Stängeln des Corchorus olitorius und Corchorus capsulatis. Im Sinne dieser Verordnung sind der Jute gleichgestellt: Fasern aus Hibiscus cannabinus, Hibiscus sabdariffa, Abutilon avicennae, Urena lobata, Urena sinuata |
|
10 |
Manila |
Faser aus den Blattscheiden der Musa textilis |
|
11 |
Alfa |
Faser aus den Blättern der Stipa tenacissima |
|
12 |
Kokos |
Faser aus der Frucht der Cocos nucifera |
|
13 |
Ginster |
Bastfaser aus den Stängeln des Cytisus scoparius und/oder des Spartium junceum |
|
14 |
Ramie |
Faser aus dem Bast der Boehmeria nivea und der Boehmeria tenacissima |
|
15 |
Sisal |
Faser aus den Blättern der Agave sisalana |
|
16 |
Sunn |
Faser aus dem Bast der Crotalaria juncea |
|
17 |
Henequen |
Faser aus dem Bast der Agave fourcroydes |
|
18 |
Maguey |
Faser aus dem Bast der Agave cantala |
Tabelle 2
|
Nummer |
Bezeichnung |
Beschreibung der Fasern |
|
19 |
Acetat |
Faser aus Zellulose-Acetat mit weniger als 92 %, jedoch mindestens 74 % acetylierter Hydroxylgruppen |
|
20 |
Alginat |
Faser aus den Metallsalzen der Alginsäure |
|
21 |
Cupro |
Regenerierte Zellulosefaser nach dem Kupfer-Ammoniak-Verfahren |
|
22 |
Modal |
Nach einem geänderten Viskoseverfahren hergestellte regenerierte Zellulosefaser mit hoher Reißkraft und hohem Modul in feuchtem Zustand. Die Reißkraft (BC) in aufgemachtem Zustand und die Kraft (BM), die erforderlich ist, um in feuchtem Zustand eine Dehnung von 5 % zu erzielen, sind Folgende: BC (Zentinewton) ≥ 1,3 BM (Zentinewton) ≥ 0,5 wobei T die mittlere längenbezogene Masse in Dezitex ist. |
|
23 |
Regenerierte Proteinfaser |
Faser aus regeneriertem und durch chemische Agenzien stabilisiertem Eiweiß |
|
24 |
Triacetat |
Aus Zellulose-Acetat hergestellte Faser, bei der mindestens 92 % der Hydroxylgruppen acetyliert sind |
|
25 |
Viskose |
Bei Endlosfasern und Spinnfasern nach dem Viskoseverfahren hergestellte regenerierte Zellulosefaser |
|
26 |
►C1 Polyacryl ◄ |
Faser aus linearen Makromolekülen, deren Kette aus mindestens 85 Gewichtsprozent Acrylnitril aufgebaut wird |
|
27 |
Polychlorid |
Faser aus linearen Makromolekülen, deren Kette aus mehr als 50 Gewichtsprozent chloriertem Olefin (z. B. Vinylchlorid, Vinylidenchlorid) aufgebaut wird |
|
28 |
Fluorfaser |
Faser aus linearen Makromolekülen, die aus aliphatischen Fluor-Kohlenstoff-Monomeren gewonnen werden |
|
29 |
Modacryl |
Faser aus linearen Makromolekülen, deren Kette aus mehr als 50 und weniger als 85 Gewichtsprozent Acrylnitril aufgebaut wird |
|
30 |
Polyamid oder Nylon |
Faser aus synthetischen linearen Makromolekülen, deren Kette sich wiederholende Amidbindungen aufweist, von denen mindestens 85 % an lineare aliphatische oder zykloaliphatische Einheiten gebunden sind |
|
31 |
Aramid |
Fasern aus linearen synthetischen Makromolekülen mit aromatischen Gruppen, deren Kette aus Amid- oder Imidbindungen besteht, von denen mindestens 85 % direkt an zwei aromatische Kerne gebunden sind und deren Imidbindungen, wenn vorhanden, die Anzahl der Amidbindungen nicht übersteigen darf |
|
32 |
Polyimid |
Faser aus synthetischen linearen Makromolekülen, deren Kette sich wiederholende Imideinheiten aufweist |
|
33 |
Lyocell |
Durch Auflösungs- und Spinnverfahren in organischem Lösungsmittel (Gemisch aus organischen Chemikalien und Wasser) hergestellte regenerierte Zellulosefaser ohne Bildung von Derivaten |
|
34 |
Polylactid |
Faser aus linearen Makromolekülen, deren Kette zu mindestens 85 Masseprozent aus Milchsäureestereinheiten besteht, die aus natürlich vorkommenden Zuckern gewonnen werden, und deren Schmelzpunkt bei mindestens 135 °C liegt |
|
35 |
Polyester |
Faser aus linearen Makromolekülen, deren Kette zu mindestens 85 Gewichtsprozent aus dem Ester eines Diols mit Terephtalsäure besteht |
|
36 |
Polyethylen |
Faser aus gesättigten linearen Makromolekülen nicht substituierter aliphatischer Kohlenwasserstoffe |
|
37 |
Polypropylen |
Faser aus linearen gesättigten aliphatischen Kohlenwasserstoffen, in denen jeder zweite Kohlenstoff eine Methylgruppe in isotaktischer Anordnung trägt, ohne weitere Substitution |
|
38 |
Polyharnstoff |
Faser aus linearen Makromolekülen, deren Kette eine Wiederkehr der funktionellen Harnstoffgruppe (NH-CO-NH) aufweist |
|
39 |
Polyurethan |
Faser aus linearen Makromolekülen, deren Kette eine Wiederkehr der funktionellen Urethangruppen aufweist |
|
40 |
Vinylal |
Faser aus linearen Makromolekülen, deren Kette aus Polyvinylalkohol mit variablem Acetalisierungsgrad aufgebaut wird |
|
41 |
Trivinyl |
Faser aus drei verschiedenen Vinylmonomeren, die sich aus Acrylnitril, aus einem chlorierten Vinylmonomer und aus einem dritten Vinylmonomer zusammensetzt, von denen keines 50 % der Gewichtsanteile aufweist |
|
42 |
Elastodien |
Elastische Faser, die aus natürlichem oder synthetischem Polyisopren besteht, entweder aus einem oder mehreren polymerisierten Dienen, mit oder ohne einem oder mehreren Vinylmonomeren, und die, unter Einwirkung einer Zugkraft um die dreifache ursprüngliche Länge gedehnt, nach Entlastung sofort wieder nahezu in ihre Ausgangslage zurückkehrt |
|
43 |
Elasthan |
Elastische Faser, die aus mindestens 85 Gewichtsprozent von segmentiertem Polyurethan besteht, und die, unter Einwirkung einer Zugkraft um die dreifache ursprüngliche Länge gedehnt, nach Entlastung sofort wieder nahezu in ihre Ausgangslage zurückkehrt |
|
44 |
Glasfaser |
Faser aus Glas |
|
45 |
Elastomultiester |
Faser, die durch die Interaktion von zwei oder mehr chemisch verschiedenen linearen Makromolekülen in zwei oder mehr verschiedenen Phasen entsteht (von denen keine 85 % Gewichtsprozent übersteigt), die als wichtigste funktionale Einheit Estergruppen enthält (zu mindestens 85 %) und die nach geeigneter Behandlung um die anderthalbfache ursprüngliche Länge gedehnt, nach Entlastung sofort wieder nahezu in ihre Ausgangslage zurückkehrt |
|
46 |
Elastolefin |
Für Fasern aus mindestens 95 Gewichtsprozent Makromolekülen, zum Teil quervernetzt, zusammengesetzt aus Ethylen und wenigstens einem anderen Olefin, und die, unter Einwirkung einer Zugkraft um die anderthalbfache ursprüngliche Länge gedehnt, nach Entlastung sofort wieder nahezu in ihre Ausgangslage zurückkehren |
|
47 |
Melamin |
Faser, die zu mindestens 85 Gewichtsprozent aus quervernetzten, aus Melaminderivaten bestehenden Makromolekülen aufgebaut ist |
|
48 |
Bezeichnung entsprechend dem Stoff, aus dem sich die Fasern zusammensetzen, z. B. Metall (metallisch, metallisiert), Asbest, Papier, mit oder ohne Zusatz „Faser“ oder „Garn“ |
Fasern aus verschiedenen oder neuartigen Stoffen, die vorstehend nicht aufgeführt sind |
|
49 |
Polypropylen/Polyamid-Bikomponentenfaser |
Bikomponentenfaser, die zu 10 bis 25 Gewichtsprozent aus in eine Polypropylenmatrix eingebetteten Polyamidfibrillen besteht |
|
50 |
Polyacrylat |
Faser aus quervernetzten Makromolekülen, die aus mehr als 35 Gewichtsprozent Acrylatgruppen (Säure, Leichtmetallsalze oder Ester) und weniger als 10 Gewichtsprozent Acrylnitrilgruppen in der Kette und bis zu 15 Gewichtsprozent Stickstoff in der Quervernetzung aufgebaut wird |
ANHANG II
Mindestanforderungen an ein technisches Dossier, das in den Antrag für eine neue Bezeichnung von Textilfasern aufzunehmen ist
(gemäß Artikel 6)
Ein technisches Dossier, das einem Antrag auf Aufnahme einer neuen Bezeichnung von Textilfasern in die Liste in Anhang I gemäß Artikel 6 beizufügen ist, muss mindestens die folgenden Angaben enthalten:
1. Vorgeschlagene Bezeichnung der Textilfaser:
Die vorgeschlagene Bezeichnung muss sich auf die chemische Zusammensetzung beziehen und gegebenenfalls Aufschluss über die Eigenschaften der Faser geben. Die vorgeschlagene Bezeichnung darf nicht mit Rechten des geistigen Eigentums verbunden sein und sie darf nicht mit dem Hersteller in Zusammenhang stehen.
2. Vorgeschlagene Definition der Textilfaser:
Die vorgeschlagene Definition muss die Faserzusammensetzung beschreiben. Die Eigenschaften, die in der Definition der neuen Textilfaser genannt werden, wie beispielsweise Elastizität, müssen durch Standardtestverfahren überprüfbar sein, die im technischen Dossier zusammen mit den Versuchsergebnissen der Analysen vorzulegen sind.
3. Identifizierung der Textilfaser: chemische Formel, Unterschiede zu bereits vorhandenen Textilfasern, FTIR-Spektrum gegebenenfalls in Verbindung mit einzelnen Daten wie Schmelzpunkt, Dichte, Refraktionsindex und Brandverhalten.
4. Vorgeschlagener vereinbarter Zuschlag für die Berechnung der Faserzusammensetzung.
5. Vorgeschlagene Identifizierungs- und Quantifizierungsverfahren, einschließlich Versuchsdaten:
Der Antragsteller muss beurteilen, ob sich die in Anhang VIII dieser Verordnung aufgeführten Methoden oder die in jenen Anhang aufzunehmenden harmonisierten Normen einsetzen lassen, um die wahrscheinlichsten im Handel zu erwartenden Gemische der neuen Textilfaser mit anderen Textilfasern zu analysieren, und wenigstens eine dieser Methoden vorschlagen. Im Falle von Methoden oder harmonisierten Normen, bei denen die Textilfaser als unlöslicher Bestandteil gelten kann, muss der Antragsteller die Faktoren „d“ angeben, welche den Massenkorrekturfaktoren entsprechen, die bei den Berechnungen (zur Berücksichtigung des bei der Analyse bekanntlich auftretenden Massenverlustes) der neuen Textilfaser anzuwenden sind.
Sind die in dieser Verordnung aufgeführten Methoden ungeeignet, muss der Antragsteller dies angemessen begründen und eine oder mehrere neue Methoden vorschlagen. Für die vorgeschlagenen neuen Methoden sind jeweils der Anwendungsbereich (einschließlich der Fasergemische), das Prinzip (vor allem das chemische Verfahren und seine Schritte), die Geräte und Reagenzien, der Analysengang, die Berechnung und Ergebnisdarstellung (einschließlich des Werts der Faktoren „d“) und die Genauigkeit (Konfidenzintervall der Versuchsergebnisse) zu beschreiben.
Der Antrag muss alle Versuchsdaten, insbesondere zu den Fasermerkmalen, für die vorgeschlagenen Identifizierungs- und Quantifizierungsmethoden enthalten. Mit dem Dossier müssen auch Daten über die Genauigkeit, Robustheit und Reproduzierbarkeit der Methoden vorgelegt werden.
6. Verfügbare Informationen über mögliche allergische Reaktionen und andere schädliche Auswirkungen der neuen Textilfaser auf die menschliche Gesundheit, einschließlich der Ergebnisse von Tests, die zu diesem Zweck in Einklang mit den einschlägigen Rechtsvorschriften der Union durchgeführt wurden.
7. Zur Begründung des Antrags zusätzlich vorzulegende Angaben zu den Herstellungsverfahren und der Bedeutung für den Verbraucher:
Das technische Dossier muss zumindest Angaben zu der Anzahl der Hersteller, dem Ort der Produktionsstätten und der erwarteten Marktverfügbarkeit der neuen Faser bzw. der aus ihr hergestellten Erzeugnisse enthalten.
8. Bereitstellung von Proben:
Der Hersteller oder eine für den Hersteller handelnde Person stellt repräsentative Proben der neuen, reinen Textilfaser und der relevanten Textilfasergemische, die benötigt werden, um die Genauigkeit, Robustheit und Reproduzierbarkeit der vorgeschlagenen Identifizierungs- und Quantifizierungsmethoden zu überprüfen, bereit. Die Kommission kann beim Hersteller oder bei der für den Hersteller handelnden Person zusätzliche Proben der relevanten Fasergemische anfordern.
Der Hersteller oder eine für den Hersteller handelnde Person stellt repräsentative Proben der neuen, reinen Textilfaser und der relevanten Textilfasergemische, die benötigt werden, um eine Validierung der vorgeschlagenen Identifizierungs- und Quantifizierungsmethoden vorzunehmen, bereit. Die Kommission kann beim Hersteller oder der für den Hersteller handelnden Person zusätzliche Proben der relevanten Fasergemische anfordern.
ANHANG III
Bezeichnungen gemäß Artikel 8 Absatz 1
|
— |
Bulgarisch : „необработена вълна“ |
|
— |
Spanisch : „lana virgen“ oder „lana de esquilado“ |
|
— |
Tschechisch : „střižní vlna“ |
|
— |
Dänisch : „ren, ny uld“ |
|
— |
Deutsch : „Schurwolle“ |
|
— |
Estnisch : „uus vill“ |
|
— |
Griechisch : „παρθένο μαλλί“ |
|
— |
Englisch : „fleece wool“ oder „virgin wool“ |
|
— |
Französisch : „laine vierge“ oder „laine de tonte“ |
|
— |
Kroatisch : „runska vuna“ |
|
— |
Irisch : „olann lomra“ |
|
— |
Italienisch : „lana vergine“ oder „lana di tosa“ |
|
— |
Lettisch : „pirmlietojuma vilna“ oder „cirptā vilna“ |
|
— |
Litauisch : „natūralioji vilna“ |
|
— |
Ungarisch : „élőgyapjú“ |
|
— |
Maltesisch : „suf verġni“ |
|
— |
Niederländisch : „scheerwol“ |
|
— |
Polnisch : „żywa wełna“ |
|
— |
Portugiesisch : „lã virgem“ |
|
— |
Rumänisch : „lână virgină“ |
|
— |
Slowakisch : „strižná vlna“ |
|
— |
Slowenisch : „runska volna“ |
|
— |
Finnisch : „uusi villa“ |
|
— |
Schwedisch : „ny ull“ |
ANHANG IV
Besondere Etikettierungs- und Kennzeichnungsvorschriften für bestimmte Textilerzeugnisse
(gemäß Artikel 13)
|
Erzeugnisse |
Etikettierungs- und Kennzeichnungsvorschriften |
|
1. Folgende Miederwaren: |
►C3 Die Faserzusammensetzung ist auf dem Etikett und der Kennzeichnung entweder durch Angabe der Zusammensetzung des gesamten Erzeugnisses oder, global oder getrennt, durch Angabe der Zusammensetzung der einzelnen Teile dieser Artikel anzugeben: ◄ |
|
a) Büstenhalter |
►C3 äußerer und innerer Stoff der Oberfläche der Schalen und des Rückenteils; ◄ |
|
b) Korsetts und Hüfthalter |
Vorderteil, Rückenteil und Seitenteile; |
|
c) Korseletts |
►C3 äußerer und innerer Stoff der Oberfläche der Schalen, der verstärkten Vorderteile, der verstärkten Rückenteile und der Seitenteile ◄ |
|
2. Andere, oben nicht aufgeführte Miederwaren: |
►C3 Die Faserzusammensetzung ist entweder durch Angabe der Zusammensetzung des gesamten Erzeugnisses oder, global oder getrennt, durch Angabe der Zusammensetzung der verschiedenen Teile dieser Artikel anzugeben. Diese Etikettierung ist für Teile, die weniger als 10 % des Gesamtgewichts des Erzeugnisses ausmachen, nicht vorgeschrieben ◄ |
|
3. Alle Miederwaren: |
Die getrennte Etikettierung und Kennzeichnung der verschiedenen Teile der Miederwaren hat so zu erfolgen, dass für den Verbraucher ohne Schwierigkeiten erkennbar ist, auf welchen Teil des Erzeugnisses sich die auf dem Etikett oder der Kennzeichnung angegebenen Hinweise beziehen |
|
4. Ausgebrannte Textilerzeugnisse: |
Die Faserzusammensetzung ist für das Gesamterzeugnis anzugeben; sie kann durch getrennte Nennung der Zusammensetzung des Grundmaterials und der der Ausbrennung unterworfenen Teile angegeben werden. Diese beiden Bestandteile sind ausdrücklich zu nennen |
|
5. Stickerei-Textilerzeugnisse: |
Die Faserzusammensetzung ist für das Gesamterzeugnis anzugeben; sie kann durch getrennte Nennung der Zusammensetzung des Grundmaterials und der Stickereifäden angegeben werden. Diese beiden Bestandteile sind ausdrücklich zu nennen. Diese Etikettierung oder Kennzeichnung ist nur für bestickte Teile vorgeschrieben, die mindestens 10 % der Oberfläche des Erzeugnisses ausmachen |
|
6. Garn mit einem Kern und einer Umspinnung aus verschiedenen Faserarten, das als solches dem Verbraucher auf dem Markt bereitgestellt wird: |
Die Zusammensetzung ist für das Gesamterzeugnis anzugeben; sie kann durch getrennte Nennung der Zusammensetzung des Kerns und der Umspinnung angegeben werden. Diese beiden Bestandteile sind ausdrücklich zu nennen |
|
7. Textilerzeugnisse aus Samt oder Plüsch oder ähnlichen Stoffen: |
Hier ist die Faserzusammensetzung für das Gesamterzeugnis anzugeben; sie kann, wenn diese Erzeugnisse aus einer Grundschicht und einer unterschiedlichen Nutzschicht bestehen und aus verschiedenen Fasern zusammengesetzt sind, getrennt für diese Bestandteile angegeben werden. Diese beiden Bestandteile sind ausdrücklich zu nennen |
|
8. Bodenbeläge und Teppiche, bei denen die Grundschicht und die Nutzschicht aus verschiedenen Fasern bestehen: |
Die Faserzusammensetzung braucht nur für die Nutzschicht angegeben zu werden. Die Nutzschicht ist ausdrücklich zu nennen |
ANHANG V
Textilerzeugnisse, für die keine Etikettierung oder Kennzeichnung vorgeschrieben ist
(gemäß Artikel 17 Absatz 2)
1. Hemdsärmelhalter
2. Armbänder für Uhren, aus Spinnstoffen
3. Etiketten und Abzeichen
4. Polstergriffe, aus Spinnstoffen
5. Kaffeewärmer
6. Teewärmer
7. Schutzärmel
8. Muffe, nicht aus Plüsch
9. Künstliche Blumen
10. Nadelkissen
11. Bemalte Leinwand
12. Textilerzeugnisse für Grundmaterial, Grundschichten, Verstärkungen und Versteifungen
13. Gebrauchte, konfektionierte Textilerzeugnisse, sofern sie ausdrücklich als solche bezeichnet sind
14. Gamaschen
15. Verpackungsmaterial, nicht neu und als solches verkauft
16. Täschner- und Sattlerwaren, aus Spinnstoffen
17. Reiseartikel, aus Spinnstoffen
18. Fertige oder noch fertigzustellende handgestickte Tapisserien und Material zu ihrer Herstellung, einschliesslich Handstickgarne, die getrennt vom Grundmaterial zum Verkauf angeboten werden und speziell zur Verwendung für solche Tapisserien aufgemacht sind
19. Reißverschlüsse
20. Mit Textilien überzogene Knöpfe und Schnallen
21. Buchhüllen aus Spinnstoffen
22. Spielzeug
23. Textile Teile von Schuhwaren
24. Deckchen aus mehreren Bestandteilen mit einer Oberfläche von weniger als 500 cm2
25. Topflappen und Topfhandschuhe
26. Eierwärmer
27. Kosmetiktäschchen
28. Tabakbeutel aus Stoff
29. Futterale bzw. Etuis für Brillen, Zigaretten und Zigarren, Feuerzeuge und Kämme, aus Stoff
30. Hüllen für Mobiltelefone und tragbare Medienabspielgeräte mit einer Oberfläche von höchstens 160 cm2
31. Schutzartikel für den Sport, ausgenommen Handschuhe
32. Toilettenbeutel
33. Schuhputzbeutel
34. Bestattungsartikel
35. Einwegerzeugnisse, ausgenommen Watte
36. Den Vorschriften des Europäischen Arzneibuchs unterliegende Textilerzeugnisse, für die ein entsprechender Vermerk aufgenommen wurde, wieder verwendbare medizinische und orthopädische Binden und allgemeines orthopädisches Textilmaterial
37. Textilerzeugnisse, einschließlich Seile, Taue und Bindfäden (vorbehaltlich Anhang VI Nummer 12), die normalerweise bestimmt sind:
a) zur Verwendung als Werkzeug bei der Herstellung und der Verarbeitung von Gütern;
b) zum Einbau in Maschinen, Anlagen (für Heizung, Klimatisierung, Beleuchtung usw.), Haushaltsgeräte und andere Geräte, Fahrzeuge und andere Transportmittel oder zum Betrieb, zur Wartung oder zur Ausrüstung dieser Geräte, mit Ausnahme von Planen und Textilzubehör für Kraftfahrzeuge, das getrennt von den Fahrzeugen verkauft wird
38. Textilerzeugnisse für den Schutz und die Sicherheit, wie z. B. Sicherheitsgurte, Fallschirme, Schwimmwesten, Notrutschen, Brandschutzvorrichtungen, kugelsichere Westen, besondere Schutzanzüge (z. B. Feuerschutz, Schutz vor Chemikalien oder anderen Sicherheitsrisiken)
39. Ballonhallen (Sport-, Ausstellungs-, Lagerhallen usw.), sofern Angaben über die Leistungen und technischen Einzelheiten dieser Erzeugnisse mitgeliefert werden
40. Segel
41. Textilwaren für Tiere
42. Fahnen und Banner
ANHANG VI
Textilerzeugnisse, für die eine globale Etikettierung ausreicht
(gemäß Artikel 17 Absatz 3)
1. Scheuertücher
2. Putztücher
3. Bordüren und Besatz
4. Borten
5. Gürtel
6. Hosenträger
7. Strumpf- und Sockenhalter
8. Schnürsenkel
9. Bänder
10. Gummielastische Bänder
11. Verpackungsmaterial, neu und als solches verkauft
12. Schnüre für Verpackungen und landwirtschaftliche Verwendungszwecke; Schnüre, Seile und Taue, die nicht unter Anhang V Nummer 37 fallen ( *1 )
13. Deckchen
14. Taschentücher und Ziertaschentücher
15. Haarnetze
16. Krawatten und Fliegen für Kinder
17. Lätzchen, Seiflappen und Waschhandschuhe
18. Nähgarne, Stopfgarne und Stickgarne, die für den Einzelverkauf aufgemacht sind
19. Gurte für Vorhänge und Jalousien
ANHANG VII
Bei der Bestimmung der Faserzusammensetzung nicht zu berücksichtigende Artikel
(gemäß Artikel 19 Absatz 2)
|
Erzeugnisse |
Ausgenommene Artikel |
|
a) Alle Textilerzeugnisse: |
i) nicht textile Teile, Webkanten, Etiketten und Abzeichen, Bordüren und Besatz, die nicht Bestandteil des Erzeugnisses sind, mit Textilien überzogene Knöpfe und Schnallen, Zubehör, Schmuckbesatz, nichtelastische Bänder, an bestimmten, eng begrenzten Stellen eingearbeitete elastische Fäden und Bänder sowie, unter den in Artikel 10 genannten Bedingungen, sichtbare und isolierbare Fasern rein dekorativer Funktion und Fasern mit antistatischer Wirkung ii) Fettstoffe, Bindemittel, Beschwerungen, Appreturen, Imprägniermittel, zusätzliche Färbe- und Druckhilfsmittel sowie sonstige Textilbearbeitungserzeugnisse |
|
b) Fußbodenbeläge und Teppiche: |
sämtliche Teile außer der Nutzschicht |
|
c) Polstergewebe: |
Binde- und Füllketten sowie Binde- und Füllschüsse, die nicht Teil der Nutzschicht sind |
|
d) Vorhänge, Gardinen und Übergardinen: |
Binde- und Füllketten sowie Binde- und Füllschüsse, die nicht Teil der Vorderseite des Stoffes sind |
|
e) Socken: |
Zusätzliches Elastikgarn im Bündchen sowie Verstärkungsgarn an Zehen und Ferse |
|
f) Strumpfhosen: |
Zusätzliches Elastikgarn im Bündchen sowie Verstärkungsgarn an Zehen und Ferse |
|
g) Andere als die unter den Buchstaben b bis f genannten Textilerzeugnisse: |
►C3
|
ANHANG VIII
Methoden für die quantitative Analyse von binären und ternären Textilfasergemischen
(gemäß Artikel 19 Absatz 1)
KAPITEL 1
I. Vorbereitung der Vorproben und der Analyseproben zur Bestimmung der Zusammensetzung von Textilerzeugnissen
1. ANWENDUNGSBEREICH
Dieses Kapitel enthält allgemeine Hinweise für die Herstellung von Vorproben geeigneter Größe (d. h. nicht über 100 g) zur Vorbehandlung für quantitative Analysen aus Laboratoriumssammelproben sowie für die Auswahl von Analyseproben aus Vorproben, aus denen die nichtfaserigen Bestandteile in einer Vorbehandlung entfernt worden sind ( 2 ).
2. DEFINITIONEN
2.1. „Prüfgut“
Diejenige Materialmenge, die aufgrund einer Reihe von Untersuchungsergebnissen beurteilt werden soll. Dazu kann zum Beispiel das gesamte Material einer Stofflieferung gehören, das gesamte von einer bestimmten Maschine gewebte Gewebe, eine Sendung Garne oder ein Ballen bzw. eine aus mehreren Ballen bestehende Lieferung.
2.2. „Laboratoriumssammelprobe“
Teil des Prüfguts, der als repräsentativ aus der Gesamtmenge entnommen worden ist und an das Laboratorium eingesandt wird. Größe und Art der Laboratoriumssammelprobe sind so zu wählen, dass sie die Schwankungen innerhalb der Lieferung richtig wiedergibt und im Laboratorium leicht zu handhaben ist ( 3 ).
2.3. „Vorprobe“
Teil der Laboratoriumssammelprobe, aus dem in einer Vorbehandlung die nichtfaserigen Bestandteile entfernt und anschließend die Analyseproben entnommen werden. Größe und Art der Vorprobe sind so zu wählen, dass sie die Schwankungen innerhalb der Laboratoriumssammelprobe richtig wiedergibt ( 4 ).
2.4. „Analyseprobe“
Teil der Vorprobe, der für die quantitative Einzelanalyse erforderlich ist.
3. PRINZIP
Die Vorprobe wird so ausgewählt, dass sie die Laboratoriumssammelprobe repräsentiert.
Die Analyseproben werden aus der Vorprobe so ausgewählt, dass sie die letztere repräsentieren.
4. PROBENAHME AUS LOSEN FASERN
4.1. Ungerichtete Fasern
Die Vorprobe wird aus zufallsbedingt der Laboratoriumssammelprobe entnommenen Büscheln zusammengestellt. Die gesamte Vorprobe wird gründlich mit Hilfe einer Laboratoriumskrempel gemischt ( 5 ). Der Krempelflor sowie die noch in der Krempel hängenden Fasern und der aus der Krempel herausgefallene Faserbruch werden vorbehandelt. Anschließend werden die Analyseproben im jeweiligen Gewichtsverhältnis aus dem Krempelflor, den anhängenden Fasern und den herausgefallenen Fasern entnommen.
Bleibt die Form des Krempelflors durch die Vorbehandlung im Wesentlichen unverändert, so werden die Analyseproben in der unter Nummer 4.2 beschriebenen Weise entnommen. Wird der Krempelflor durch die Vorbehandlung zerstört, so werden für die Restproben nach dem Zufallsprinzip mindestens 16 kleine Büschel von geeigneter und unter sich möglichst gleicher Größe aus der vorbehandelten Probe herausgenommen und zu einer Probe zusammengefasst.
4.2. Gerichtete Fasern (Krempelflor, Kammzug, Vorgarne)
Aus zufällig ausgewählten Teilen der Laboratoriumssammelprobe werden mindestens 10 Schnittproben zu je etwa 1 g hergestellt. Die auf diese Weise erhaltene Vorprobe wird vorbehandelt. Dann werden die Schnittproben Kante an Kante aneinandergelegt und daraus die Analyseproben in der Weise hergestellt, dass jeweils ein Schnitt so durch die Muster gelegt wird, dass von jeder der 10 Längen ein Teil erfasst wird.
5. PROBENAHME AUS GARNEN
5.1. Garne auf Hülsen oder in Strängen
Es müssen alle Hülsen oder Stränge der Laboratoriumssammelprobe verwendet werden.
Man entnimmt jeder Hülse oder jedem Strang entsprechend zusammenhängende Fäden gleicher Länge, indem man Stränge gleicher Zahl auf eine Haspel ( 6 ) aufwindet, oder auf andere Weise. Die einzelnen Längen werden zu einem einzigen Strang oder Kabel zusammengelegt, wobei darauf zu achten ist, dass in jedem Strang oder Kabel immer gleiche Fadenlängen von jeder Hülse oder jedem Strang vorhanden sind.
Die auf diese Weise erhaltene Vorprobe wird vorbehandelt.
Zur Entnahme von Analyseproben aus der vorbehandelten Vorprobe werden Fadenabschnitte gleicher Menge aus dem Strang oder Kabel herausgenommen; dabei ist darauf zu achten, dass keiner der darin enthaltenen Fäden ausgelassen wird.
Ist t die Feinheit in „Tex“ und n die Anzahl der Hülsen oder Stränge der Laboratoriumssammelprobe, so beträgt die Fadenlänge von jeder Hülse oder jedem Strang, die eine Vorprobe von 10 g ergibt, 106/nt cm.
Ist nt sehr hoch, zum Beispiel über 2 000 , so wird ein schwerer Fadenstrang aufgewunden und in zwei Teile zerschnitten, um einen Strang von geeignetem Gewicht herzustellen. Die Enden einer Probe in Strangform sind vor Beginn der Vorbehandlung sorgfältig zusammenzubinden; die Analyseproben sind an einer Stelle zu entnehmen, die von dem abgebundenen Ende genügend weit entfernt ist.
5.2. Kettfäden
Die Vorprobe wird in der Weise entnommen, dass man vom Ende der Kette ein Stück abschneidet, das mindestens 20 cm lang ist und alle Kettfäden mit Ausnahme der Webkante enthält, die verworfen wird. Ist die Probe zu schwer, um im Ganzen vorbehandelt zu werden, so wird sie in zwei oder mehr Teile unterteilt, wobei jedes Teil vor der Vorbehandlung zusammengebunden wird. Die einzelnen Teile werden getrennt vorbehandelt und danach wieder zusammengefasst. Von der Vorprobe wird eine Analyseprobe von passender Länge in genügendem Abstand von der Bündelung abgeschnitten, wobei darauf zu achten ist, dass keiner der Kettfäden ausgelassen wird. Bei einer Kette mit n Fäden der Feinheit t in „Tex“ beträgt die Länge einer Probe von 1 g Gewicht 105/nt cm.
6. PROBENAHME AUS TEXTILEN FLÄCHENGEBILDEN
6.1. Laboratoriumssammelprobe bestehend aus einem einzigen repräsentativen Abschnitt
Man schneidet einen diagonalen Streifen von Ecke zu Ecke und entfernt die Webkanten. Dieser Streifen ist die Vorprobe. Für eine Vorprobe von x g beträgt die Fläche des Streifens x104/G cm2. G = Flächengewicht des Gewebes in g/m2.
Nach der Vorbehandlung zerschneidet man den Streifen quer zur Länge in vier gleiche Teile und legt diese übereinander. Man entnimmt Analyseproben aus einem beliebigen Teil des übereinander geschichteten Materials, indem man alle Lagen in der Weise durchschneidet, dass man von jeder Lage eine Analyseprobe mit gleicher Länge erhält.
Enthält der Stoff ein gewebtes Muster, so darf die Breite der Vorprobe, parallel zur Richtung der Kette gemessen, nicht kleiner sein als eine Wiederholung des Musters in der Kette. Ist unter diesen Bedingungen die Vorprobe zu groß, um im Ganzen vorbehandelt zu werden, so muss sie in gleiche Teile zerschnitten werden, die getrennt vorzubehandeln sind; diese Teile sind vor der Herstellung der Analyseprobe übereinanderzulegen, doch ist darauf zu achten, dass entsprechende Teile des Musters nicht zusammenfallen.
6.2. Laboratoriumssammelproben, die aus mehreren Stoffabschnitten bestehen
Jeder Abschnitt wird gemäß 6.1 analysiert; die Ergebnisse werden getrennt angegeben.
7. PROBENAHME VON KONFEKTIONSARTIKELN UND ENDERZEUGNISSEN
Die Laboratoriumssammelprobe besteht in der Regel aus einem ganzen Konfektionsartikel bzw. Enderzeugnis oder aus einem repräsentativen Teilstück desselben.
Gegebenenfalls ist der Prozentsatz der verschiedenen Teile zu bestimmen, die nicht denselben Fasergehalt haben, damit festgestellt werden kann, ob Artikel 11 eingehalten ist.
Dem Teil des Konfektionsartikels bzw. des Enderzeugnisses, dessen Zusammensetzung durch ein Etikett gekennzeichnet werden muss, ist eine repräsentative Vorprobe zu entnehmen. Trägt das Erzeugnis mehrere Etiketten, so müssen repräsentative Vorproben aus jedem Teil, der durch ein Etikett gekennzeichnet wird, entnommen werden.
Ist das Erzeugnis, dessen Zusammensetzung bestimmt werden soll, nicht homogen, so kann es erforderlich sein, aus jedem Teil des Erzeugnisses Vorproben zu entnehmen und den verhältnismäßigen Anteil der einzelnen Teile, bezogen auf das ganze Erzeugnis, zu bestimmen.
Die Prozentsätze werden dann unter Berücksichtigung der verhältnismäßigen Anteile der untersuchten Teile errechnet.
Diese Vorproben werden vorbehandelt.
Anschließend werden den vorbehandelten Vorproben repräsentative Analyseproben entnommen.
II. Einführung in die Methoden für die quantitative Analyse von Textilfasergemischen
Die quantitativen Analysemethoden bei Textilfasergemischen stützen sich hauptsächlich auf zwei Verfahren, nämlich auf das der manuellen Trennung und das der chemischen Trennung der Fasern.
Dem manuellen Trennverfahren sollte nach Möglichkeit der Vorzug gegeben werden, denn im Allgemeinen führt es zu genaueren Ergebnissen als das chemische Verfahren. Das manuelle Verfahren lässt sich auf alle Textilerzeugnisse, bei denen die dieses Erzeugnis bildenden Fasern kein untrennbares Gemisch darstellen, anwenden, z. B. auf die aus mehreren Bestandteilen zusammengesetzten Garne, bei denen die einzelnen Bestandteile aus einer einzigen Faserart gebildet werden, sowie auf Gewebe, bei denen die Kette aus einer anderen Faser als der Schuss besteht, oder auf Gewirke, die aus verschiedenartigen Garnen zusammengesetzt sind.
In der Regel stützt sich die quantitative chemische Analysemethode bei Textilfasergemischen auf die selektive Auflösbarkeit der Einzelbestandteile des Gemischs. Nach Entfernung eines Bestandteils wird der unlösliche Rückstand gewogen, und der Anteil des löslichen Bestandteils wird unter Zugrundelegung des Gewichtsverlusts berechnet. In diesem ersten Teil des Anhangs werden die Angaben, die sich generell bei einer Analyse nach diesem Verfahren ergeben und für die im vorliegenden Anhang genannten Fasergemische jeder Zusammensetzung gelten, zusammengestellt. Er muss daher in Verbindung mit den nachfolgenden einzelnen Abschnitten des Anhangs benutzt werden, die ausführliche Verfahren für besondere Fasergemische enthalten. Es kann vorkommen, dass bestimmte chemische Analysen auf anderen Prinzipien als dem der selektiven Auflösbarkeit beruhen. In diesem Fall werden in dem entsprechenden Teil der einschlägigen Methode ausführliche Angaben hierüber gemacht.
Die während der Herstellung der Textilerzeugnisse benutzten Fasergemische und in geringerem Maße auch die Fasergemische in den fertigen Textilien können oftmals als natürliche Beimengungen oder zur Erleichterung des Verarbeitungsvorgangs Fette, Wachse oder Hilfsstoffe bzw. wasserlösliche Stoffe enthalten. Diese nicht zur Faser gehörenden Stoffe müssen vor der Analyse ausgesondert werden. Aus diesem Grund wird ebenfalls eine Methode der Vorbehandlung zwecks Entfernung von Ölen, Fetten, Wachsen und wasserlöslichen Stoffen angegeben.
Textilien können ferner Harze oder andere Stoffe enthalten, die ihnen bestimmte Eigenschaften verleihen sollen. Diese Stoffe, zu denen in Ausnahmefällen auch Farbstoffe gehören, können die Einwirkung des Reagenzes auf die löslichen Bestandteile beeinträchtigen bzw. teilweise oder vollständig durch das Reagenz beseitigt werden. Diese Zusatzstoffe können somit Fehler hervorrufen und müssen vor der Analyse der Probe ausgeschieden werden. Ist dies unmöglich, so sind die in diesem Anhang beschriebenen Methoden für die quantitative Analyse nicht mehr anwendbar.
Farbstoffe in gefärbten Fasern werden als fester Bestandteil der Faser angesehen und nicht entfernt.
Bei diesen Analysen wird von der Trockenmasse ausgegangen, weshalb ein Verfahren zur Bestimmung der Trockenmasse angegeben wird.
Bei der Ergebnisdarstellung werden zur Trockenmasse der einzelnen Fasern die in Anhang IX angegebenen Zuschläge zugerechnet.
Die in dem Gemisch enthaltenen Fasern sind vor der Analyse zu identifizieren. Bei bestimmten Methoden kann der unlösliche Bestandteil von Gemischen teilweise in dem Reagenz aufgelöst werden, das zur Auflösung des löslichen Bestandteils bzw. der löslichen Bestandteile verwendet wird.
Nach Möglichkeit wurden die Reagenzien so gewählt, dass sie nur einen geringen oder überhaupt keinen Einfluss auf die unlöslichen Fasern haben. Ist bei der Analyse mit einem Gewichtsverlust zu rechnen, so müssen die Ergebnisse entsprechend korrigiert werden; Korrekturfaktoren hierfür sind angegeben. Diese wurden in mehreren Laboratorien dadurch bestimmt, dass durch Vorbehandlung gereinigte Fasern mit dem entsprechenden Reagenz unter Befolgung der Analysemethode behandelt wurden.
Diese Korrekturfaktoren gelten nur für normale Fasern, und weitere Korrekturfaktoren können erforderlich sein, wenn die Fasern vor oder während der Verarbeitung nicht intakt geblieben sind. Die angegebenen chemischen Analysemethoden gelten für Einzelanalysen.
Es muss sowohl beim manuellen Trennungsverfahren als auch beim chemischen Trennungsverfahren mindestens an zwei getrennten Analyseproben je eine Analyse durchgeführt werden.
In Zweifelsfällen muss, sofern dies nicht technisch unmöglich ist, eine weitere Analyse durchgeführt werden, und zwar nach einem Verfahren, mit dem sich die bei dem ersten Verfahren als Rückstand gebildete Faser auflösen lässt.
KAPITEL 2
METHODEN FÜR DIE QUANTITATIVE ANALYSE VON BESTIMMTEN BINÄREN TEXTILFASERGEMISCHEN
I. Allgemeine Angaben zu den Methoden für die quantitative chemische Analyse von Textilfasergemischen
I.1. ANWENDUNGSBEREICH
Unter dem „Anwendungsbereich“ jeder Methode werden die Fasern aufgeführt, auf die sie anzuwenden ist.
I.2. PRINZIP
Nach Identifizierung der einzelnen Bestandteile eines Fasergemischs werden zunächst durch eine entsprechende Vorbehandlung die nichtfaserigen Bestandteile entfernt, sodann wird einer der Bestandteile entfernt, und zwar in der Regel durch selektive Auflösung ( 7 ). Der unlösliche Rückstand wird gewogen und der Anteil des löslichen Bestandteils wird aus dem Gewichtsverlust berechnet. Außer bei technischen Schwierigkeiten ist vorzugsweise die in größerer Menge vorhandene Faser aufzulösen, damit man die in geringerer Menge vorhandene Faser als Rückstand erhält.
I.3. ERFORDERLICHES MATERIAL
I.3.1. Geräte
|
I.3.1.1. |
Filtertiegel und Wägegläser zum Einsetzen von Tiegeln oder andere gleichwertige Geräte |
|
I.3.1.2. |
Absaugflasche |
|
I.3.1.3. |
Exsikkator mit gefärbtem Kieselgel als Feuchtigkeitsindikator |
|
I.3.1.4. |
Trockenofen mit Ventilator zur Trocknung der Analyseproben bei 105 ± 3 °C |
|
I.3.1.5. |
Analysenwaage, Empfindlichkeit 0,0002 g |
|
I.3.1.6. |
Soxhlet-Extraktionsapparat oder gleichwertige Apparatur |
|
I.3.2. |
Reagenzien
Alle Reagenzien müssen chemisch rein sein. |
I.4. KONDITIONIERUNGS- UND ANALYSEATMOSPHÄRE
Da die Trockenmasse bestimmt wird, ist weder eine Konditionierung der Probe noch eine Untersuchung in klimatisierter Atmosphäre erforderlich.
I.5. VORPROBE
Es wird eine für die Laboratoriumsprobe repräsentative Vorprobe gewählt, die für sämtliche erforderlichen Analyseproben von jeweils mindestens 1 g ausreicht.
I.6. VORBEHANDLUNG DER VORPROBE ( 8 )
Ist einer der bei der Berechnung der Prozentsätze nicht zu berücksichtigenden Bestandteile vorhanden (siehe Artikel 19), so ist dieser zunächst durch eine geeignete Methode zu entfernen, die jedoch keinen der Faserbestandteile angreifen darf.
Zu diesem Zweck werden die mit Hilfe von Petrolether und Wasser extrahierbaren nichtfaserigen Bestandteile entfernt, indem die Vorprobe im Soxhlet-Apparat mit Petrolether während einer Stunde und mit mindestens sechs Umläufen pro Stunde behandelt wird. Anschließend wird der Petrolether der Vorprobe verdampft; danach wird die Vorprobe durch Direktbehandlung extrahiert, das heißt durch einstündiges Eintauchen in Wasser bei Zimmertemperatur mit darauf folgendem einstündigen Eintauchen in Wasser bei 65 °C ± 5 °C unter zeitweiligem Schütteln, Flottenverhältnis 1:100. Danach wird das überschüssige Wasser durch Ausquetschen, Absaugen oder Zentrifugieren entfernt, bis die Probe lufttrocken ist.
Bei Elastolefin oder Fasergemischen, die Elastolefin und andere Fasern enthalten (Wolle, Tierhaare, Seide, Baumwolle, Flachs bzw. Leinen, Hanf, Jute, Manila, Alfa, Kokos, Ginster, Ramie, Sisal, Cupro, Modal, regenerierte Proteinfasern, Viskose, Polyacryl, Polyamid oder Nylon, Polyester, Elastomultiester), ist das oben beschriebene Verfahren dahingehend leicht abzuändern, dass Petrolether durch Aceton ersetzt wird.
Bei binären Fasergemischen, die Elastolefin und Acetat enthalten, ist folgende Vorbehandlung durchzuführen. Die Vorprobe wird 10 Minuten lang bei 80 °C mit einer Lösung extrahiert, die 25 g/l 50 %ige Orthophosphorsäure und 50 g/l Harnstoff enthält, Flottenverhältnis 1:100. Die Vorprobe wird in Wasser gewaschen, danach lässt man sie abtropfen und wäscht sie in einer 0,1 %igen Natriumbicarbonat-Lösung sowie abschließend vorsichtig in Wasser.
Falls die nichtfaserigen Bestandteile nicht mit Hilfe von Petrolether und Wasser extrahiert werden können, so müssen sie anstatt mit Wasser, wie oben beschrieben, mit einem geeigneten Stoff entfernt werden, der keinen der Faserbestandteile wesentlich verändert. Bei einigen ungebleichten natürlichen Pflanzenfasern (wie zum Beispiel Jute- oder Kokosfasern) ist zu beachten, dass durch die normale Vorbehandlung mit Petrolether und Wasser nicht alle natürlichen nichtfaserigen Bestandteile beseitigt werden. Trotzdem werden keine weiteren Vorbehandlungen vorgenommen, soweit die Probe keine in Petrolether und in Wasser unlöslichen Appreturen enthält.
In den Analysenberichten müssen die gewählten Vorbehandlungsmethoden eingehend geschildert werden.
I.7. ANALYSENGANG
I.7.1. Allgemeine Anweisungen
I.7.1.1. Trocknung
Alle Trocknungsvorgänge sind mindestens 4 Stunden, jedoch nicht mehr als 16 Stunden bei 105 °C ± 3 °C in einem belüfteten Ofen bei geschlossener Ofentür durchzuführen. Beträgt die Trocknungsdauer weniger als 14 Stunden, muss überprüft werden, ob ein konstantes Gewicht erreicht wurde. Dieses Gewicht kann als erreicht gelten, wenn der Gewichtsunterschied nach einer neuen Trocknung von 60 Minuten weniger als 0,05 % beträgt.
Die Filtertiegel und Wägegläser sowie die Proben oder die Rückstände sollen während des Trocknungs-, Abkühlungs- und Wägevorgangs nicht mit bloßen Händen berührt werden.
Die Analyseproben werden in einem Wägeglas mit abgenommenem Stopfen getrocknet. Nach der Trocknung wird das Wägeglas vor Herausnahme aus dem Ofen geschlossen und so schnell wie möglich in den Exsikkator gebracht.
Der Filtertiegel wird in einem Wägeglas zusammen mit dessen abgenommenen Stopfen im Ofen getrocknet. Nach der Trocknung wird das Wägeglas verschlossen und so schnell wie möglich in den Exsikkator gestellt.
Wird ein anderes Gerät als der Filtertiegel verwendet, so trocknet man im Trockenofen, um das Trockengewicht der Fasern ohne Verlust zu bestimmen.
I.7.1.2. Kühlung
Alle Kühlvorgänge werden in dem neben der Waage aufgestellten Exsikkator ausreichend lange durchgeführt, um ein völliges Abkühlen der Wägegläser zu erreichen, wobei die Abkühldauer mindestens 2 Stunden beträgt.
I.7.1.3. Wägung
Nach dem Kühlen wird das Wägeglas innerhalb von zwei Minuten nach Herausnahme aus dem Exsikkator gewogen. Wägegenauigkeit 0,0002 g.
I.7.2. Analysengang
Man entnimmt aus der vorbehandelten Vorprobe eine Analyseprobe von mindestens 1 g. Das Garn oder die Gewebe werden in Längen von etwa 10 mm ausgeschnitten und soweit wie möglich zerlegt (zerschnitten). Die Analyseprobe wird in einem Wägeglas getrocknet, im Exsikkator gekühlt und gewogen. Die Probe wird in ein Glasgefäß gegeben, das im entsprechenden Teil der Unionsmethode beschrieben ist, anschließend wird das Wägeglas sofort wieder gewogen und das Trockengewicht der Probe durch Differenzbildung ermittelt. Die Analyse wird gemäß den Angaben in dem entsprechenden Teil der Methode zu Ende geführt. Der Rückstand wird mikroskopisch geprüft, um festzustellen, ob durch die Behandlung die lösliche Faser völlig ausgesondert worden ist.
I.8. BERECHNUNG UND ERGEBNISDARSTELLUNG
Das Gewicht des unlöslichen Bestandteils wird als Prozentsatz des Gesamtgewichts der im Gemisch enthaltenen Fasern ausgedrückt. Der Prozentsatz des löslichen Faseranteils ergibt sich aus der Differenz. Die Ergebnisberechnung erfolgt auf der Basis der Trockengewichte der reinen Fasern, wobei zum einen vereinbarte Zuschläge, zum anderen Korrekturfaktoren zur Berücksichtigung der Verluste bei der Vorbehandlung und der Analyse angewendet werden. Diese Berechnungen erfolgen nach der unter Nummer I.8.2 angegebenen Formel.
|
I.8.1. |
Berechnung des prozentualen Gewichtsanteils der trockenen und reinen unlöslichen Bestandteile ohne Berücksichtigung des Gewichtsverlusts der Fasern bei der Vorbehandlung
Dabei ist:
Selbstverständlich gelten die im Normalfall anwendbaren „d“-Werte nicht für chemisch angegriffene Fasern. |

|
I.8.2. |
Berechnung des Prozentsatzes der unlöslichen Komponente nach Anwendung der vereinbarten Zuschläge und etwaiger Korrekturfaktoren zur Berücksichtigung des Gewichtsverlusts durch die Vorbehandlung
Dabei ist:
Der Prozentsatz der zweiten Komponente (P2A %) beträgt 100 – P1A %. Bei Anwendung einer besonderen Vorbehandlung müssen die Werte b1 und b2 nach Möglichkeit dadurch bestimmt werden, dass alle reinen Faserbestandteile der bei der Analyse angewandten Vorbehandlung unterworfen werden. Als reine Fasern gelten die Fasern, die frei von jeglichen nichtfaserhaltigen Stoffen sind, mit Ausnahme derjenigen Stoffe, die sie normalerweise (aufgrund ihrer Beschaffenheit und des Herstellungsprozesses) in dem Zustand (ungebleicht, gebleicht) enthalten, in dem sich die zu analysierende Ware befindet. Verfügt man nicht getrennt über reine Faserbestandteile, die zur Herstellung der zu analysierenden Ware gedient haben, so sind für b1 und b2 Durchschnittswerte zugrunde zu legen, die aus der Prüfung ähnlicher Fasern ermittelt wurden, wie sie das untersuchte Gemisch enthält. Wird die normale Vorbehandlung durch Extraktion mit Petrolether und mit Wasser durchgeführt, so kann man im Allgemeinen auf die Korrekturfaktoren b1 und b2 verzichten, außer im Fall von ungebleichter Baumwolle, ungebleichtem Flachs bzw. Leinen und ungebleichtem Hanf, für die ein durch die Vorbehandlung bedingter Gewichtsverlust von 4 %, im Fall von Polypropylen ein solcher von 1 %, konventionell festgelegt ist. Im Fall anderer Fasern wird konventionell festgelegt, dass der durch die Vorbehandlung bedingte Gewichtsverlust für die Berechnung unberücksichtigt bleibt. |
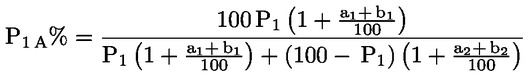
II. Methode der quantitativen Analyse durch manuelle Trennung
II.1. ANWENDUNGSBEREICH
Die Methode lässt sich auf Fasergemische beliebiger Beschaffenheit anwenden, vorausgesetzt, dass sie kein untrennbares Gemisch darstellen und dass sie sich manuell trennen lassen.
II.2. PRINZIP
Nach Identifizierung der einzelnen Bestandteile der Fasergemische werden zunächst die nicht faserhaltigen Bestandteile durch eine geeignete Vorbehandlung ausgesondert, anschließend die Fasern von Hand getrennt, getrocknet und zwecks Berechnung des Anteils der einzelnen Faserarten am Gemisch gewogen.
II.3. GERÄTE
|
II.3.1. |
Wägeglas bzw. andere Geräte, die gleichartige Ergebnisse liefern |
|
II.3.2. |
Exsikkator mit gefärbtem Kieselgel als Feuchtigkeitsindikator |
|
II.3.3. |
Trockenofen mit Ventilator zur Trocknung der Analyseproben bei 105 ± 3 °C |
|
II.3.4. |
Analysenwaage, Empfindlichkeit 0,0002 g |
|
II.3.5. |
Soxhlet-Extraktionsapparat oder andere Geräte, die ein gleichartiges Ergebnis liefern |
|
II.3.6. |
Nadel |
|
II.3.7. |
Garndrehungszähler oder gleichwertige Apparatur |
II.4. Reagenzien
|
II.4.1. |
Petrolether, nachdestilliert, Siedebereich 40 bis 60 °C |
|
II.4.2. |
Destilliertes oder entionisiertes Wasser |
|
II.4.3. |
Aceton |
|
II.4.4. |
Orthophosphorsäure |
|
II.4.5. |
Harnstoff |
|
II.4.6. |
Natriumbicarbonat |
Alle Reagenzien müssen chemisch rein sein.
II.5. KONDITIONIERUNGS- UND ANALYSENATMOSPHÄRE
Siehe Nummer I.4.
II.6. VORPROBE
Siehe Nummer I.5.
II.7. VORBEHANDLUNG DER VORPROBE
Siehe Nummer I.6.
II.8. ANALYSENGANG
II.8.1. Analyse von Garnen
Eine Analyseprobe von mindestens 1 g wird aus einer vorbehandelten Probe entnommen; Bei sehr feinen Garnen kann die Analyse ungeachtet des Gewichts auf einer Mindestlänge von 30 m durchgeführt werden.
Die Garne sind in Stücke von geeigneter Länge zu schneiden; aus diesen sind mit Hilfe einer Präpariernadel und, falls erforderlich, mit Hilfe des Garndrehungszählers die einzelnen Elemente herauszutrennen. Die auf diese Weise herausgetrennten Elemente werden dann in ein tariertes Wägeglas gegeben und bei 105 °C ± 3 °C getrocknet, bis ein konstantes Gewicht gemäß I.7.1 und I.7.2 erreicht ist.
II.8.2. Analyse eines Gewebes
Eine Analyseprobe von mindestens 1 g wird aus einer vorbehandelten Vorprobe entnommen, die Analyseprobe wird so ausgeschnitten, dass sie außerhalb der Webkante liegt, exakt geschnittene Ränder ohne Kräuselung aufweist und parallel zu Schuss und Kette bzw. bei Gewirken gleichlaufend längs und quer zu den Maschenreihen geschnitten ist. Die einzelnen Garne werden getrennt und in tarierten Wägegläsern gesammelt; dann wird wie unter Nummer II.8.1 vorgegangen.
II.9. BERECHNUNG UND ERGEBNISDARSTELLUNG
Das Gewicht jedes Bestandteils wird als Prozentsatz des Gesamtgewichts der im Gemisch enthaltenen Fasern ausgedrückt. Die Berechnung erfolgt auf der Basis des Trockengewichts der reinen Fasern unter Anwendung von vereinbarten Zuschlägen sowie von Korrekturfaktoren zur Berücksichtigung der während der Vorbehandlung aufgetretenen Gewichtsverluste.
|
II.9.1. |
Berechnung des Prozentsatzes der reinen Trockengewichte ohne Berücksichtigung des Gewichtsverlusts der Fasern durch die Vorbehandlung:
|
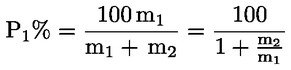
|
II.9.2. |
Berechnung des Prozentsatzes jeder einzelnen Komponente nach Anwendung der vereinbarten Zuschläge und etwaiger Korrekturfaktoren zur Berücksichtigung des Gewichtsverlustes durch die Vorbehandlung: siehe Nummer I.8.2. |
III.1. GENAUIGKEIT DER METHODEN
Die für jede Methode angegebene Genauigkeit bezieht sich auf die Reproduzierbarkeit (Wiederholstreubereich).
Die Reproduzierbarkeit ist der Zuverlässigkeitsgrad, d. h. die Einengung der Übereinstimmung zwischen den Versuchsergebnissen, die in verschiedenen Laboratorien oder zu verschiedenen Zeiten erzielt werden, wenn dabei jeweils nach derselben Methode und an demselben homogenen Prüfgut Einzelergebnisse ermittelt werden.
Die Reproduzierbarkeit wird durch das Konfidenzintervall der Versuchsergebnisse bei einem Konfidenzniveau von 95 % ausgedrückt.
Somit würde die Abweichung zwischen den Ergebnissen einer in verschiedenen Laboratorien durchgeführten Analysenreihe bei richtiger und normaler Anwendung der Methode auf eine gleichartige homogene Mischung nur in fünf von 100 Fällen das Konfidenzintervall überschreiten.
III.2. ANALYSENBERICHT
|
III.2.1. |
Angabe, ob die Analyse nach der hier beschriebenen Methode durchgeführt worden ist. |
|
III.2.2. |
Detaillierte Angaben über etwaige Spezial-Vorbehandlungen (siehe Nummer I.6). |
|
III.2.3. |
Angabe der Einzelergebnisse sowie des arithmetischen Mittels auf eine Dezimalstelle genau. |
IV. Besondere Methoden
Übersichtstabelle
|
Methode |
Anwendungsbereich (1) |
Reagenz |
|
|
Löslicher Bestandteil |
Unlöslicher Bestandteil |
||
|
1. |
Acetat |
Bestimmte andere Fasern |
Aceton |
|
2. |
Bestimmte Eiweißfasern |
Bestimmte andere Fasern |
Hypochlorit |
|
3. |
Viskose, Cupro und bestimmte Modalarten |
Bestimmte andere Fasern |
Ameisensäure-Zinkchlorid |
|
4. |
Polyamid oder Nylon |
Bestimmte andere Fasern |
80 %ige Ameisensäure |
|
5. |
Acetat |
Bestimmte andere Fasern |
Benzylalkohol |
|
6. |
Triacetat oder Polylactid |
Bestimmte andere Fasern |
Dichlormethan |
|
7. |
Bestimmte Zellulosefasern |
Bestimmte andere Fasern |
75 %ige Schwefelsäure m/m |
|
8. |
Polyacrylfasern, bestimmte Modacrylfasern oder bestimmte Polychloridfasern |
Bestimmte andere Fasern |
Dimethylformamid |
|
9. |
Bestimmte Polychloridfasern |
Bestimmte andere Fasern |
Schwefelkohlenstoff/Aceton (55,5/44,5 %) v/v |
|
10. |
Acetat |
Bestimmte andere Fasern |
Essigsäure |
|
11. |
Seide, Polyamid oder Nylon |
Bestimmte andere Fasern |
75 %ige Schwefelsäure m/m |
|
12. |
Jute |
Bestimmte Fasern tierischen Ursprungs |
Stickstoffbestimmungsverfahren |
|
13. |
Polypropylen |
Bestimmte andere Fasern |
Xylol |
|
14. |
Bestimmte Fasern |
Bestimmte andere Fasern |
Konzentrierte Schwefelsäure |
|
15. |
Polychloridfasern, bestimmte Modacryle, bestimmte Elastane, Acetate, Triacetate |
Bestimmte andere Fasern |
Cyclohexanon |
|
16. |
Melamin |
Bestimmte andere Fasern |
90 %ige heiße Ameisensäure |
|
17. |
Polyester |
Bestimmte andere Fasern |
Trichloressigsäure und Chloroform |
|
(1) Die Fasern sind unter der jeweiligen Methode genau aufgelistet. |
|||
METHODE Nr. 1
ACETAT- UND BESTIMMTE ANDERE FASERN
(Aceton-Verfahren)
1. ANWENDUNGSBEREICH
Dieses Verfahren gilt nach Entfernung der Nichtfaserstoffe für binäre Fasergemische von
1. Acetat (19)
mit
2. Wolle (1), Tierhaaren (2 und 3), Seide (4), Baumwolle (5), Flachs (7) Hanf (8), Jute (9), Manila (10), ALFA (11), Kokosfasern (12), Ginster (13), Ramie (14), Sisal (15), Cupro (21), Modal (22), regenerierten Proteinfasern (23), Viskose (25), Polyacryl (26), Polyamid oder Nylon (30), Polyester (35), Polypropylen (37), Elastomultiester (45), Elastolefin (46), Melamin (47), Polypropylen/Polyamid-Bikomponentenfasern (49) und Polyacrylat (50).
Dieses Verfahren ist unter keinen Umständen auf oberflächen-entacetylierte Acetatfasern anwendbar.
2. PRINZIP
Die Acetatfasern werden mittels Aceton aus einer bekannten Trockenmasse herausgelöst. Der Rückstand wird gesammelt, gewaschen, getrocknet und gewogen; seine — erforderlichenfalls berichtigte — Masse wird in Prozentsätzen der Trockenmasse der Mischung ausgedrückt. Der Anteil an trockenen Acetatfasern wird durch Differenzbildung ermittelt.
3. GERÄTE UND REAGENZIEN (neben den im allgemeinen Teil genannten)
3.1. Geräte
Mindestens 200 ml fassender Erlenmeyerkolben mit Glasschliffstopfen.
3.2. Reagenz
Aceton.
4. ANALYSENGANG
Es ist der im allgemeinen Teil beschriebene Analysengang zu befolgen und folgendermaßen vorzugehen:
Der Analyseprobe, die sich in einem mindestens 200 ml fassenden Erlenmeyerkolben mit Glasschliffstopfen befindet, werden 100 ml Aceton je Gramm Analyseprobe zugegeben; der Kolben wird geschüttelt und 30 Minuten lang bei Raumtemperatur unter zeitweiligem Schütteln stehengelassen; anschließend wird die Flüssigkeit über einen gewogenen Glasfiltertiegel dekantiert.
Diese Behandlung wird noch zweimal wiederholt (insgesamt drei Extraktionen), doch jeweils nur 15 Minuten lang, so dass die Gesamtdauer der Acetonbehandlung eine Stunde beträgt. Der Rückstand wird in einen Glasfiltertiegel überführt und danach mit Aceton unter Absaugen ausgewaschen. Der Tiegel wird erneut mit Aceton gefüllt, das man ohne Absaugen von selbst ablaufen lässt.
Zum Schluss wird der Tiegel durch Absaugen geleert, zusammen mit dem Rückstand getrocknet, gekühlt und gewogen.
5. BERECHNUNG UND ERGEBNISDARSTELLUNG
Die Ergebnisse werden nach dem im allgemeinen Teil angegebenen Berechnungsverfahren ermittelt. Der Korrekturfaktor „d“ beträgt 1,00, für Melamin und Polyacrylat 1,01.
6. GENAUIGKEIT DES VERFAHRENS
Bei homogenen Textilgemischen liegt das Konfidenzintervall der Ergebnisse dieses Verfahrens bei höchstens ± 1, wobei das Konfidenzniveau 95 % beträgt.
METHODE Nr. 2
BESTIMMTE EIWEISSFASERN UND BESTIMMTE ANDERE FASERN
(Hypochlorit-Verfahren)
1. ANWENDUNGSBEREICH
Dieses Verfahren gilt nach Entfernung der Nichtfaserstoffe für binäre Fasergemische von
1. bestimmten Eiweißfasern wie: Wolle (1), Tierhaaren (2 und 3), Seide (4) und regenerierte Proteinfasern (23)
mit
2. Baumwolle (5), Cupro (21), Viskose (25), Polyacryl (26), Polychlorid (27), Polyamid oder Nylon (30), Polyester (35), Polypropylen (37), Elastan (43), Glasfasern (44), Elastomultiester (45), Elastolefin (46), Melamin (47) und Polypropylen/Polyamid-Bikomponentenfasern (49).
Sind unterschiedliche Eiweißfasern vorhanden, so liefert das Verfahren deren Gesamtmenge, jedoch nicht die prozentualen Anteile.
2. PRINZIP
Die Eiweißfasern werden mit einer Hypochloritlösung aus einer bekannten Trockenmasse herausgelöst. Der Rückstand wird gesammelt, gewaschen, getrocknet und gewogen; seine — erforderlichenfalls berichtigte — Masse wird in Prozentsätzen der Trockenmasse der Mischung ausgedrückt. Der Anteil an trockenen Eiweißfasern wird durch Differenzbildung ermittelt.
Für die Herstellung der Hypochloritlösung kann Lithiumhypochlorit oder Natriumhypochlorit verwendet werden.
Lithiumhypochlorit empfiehlt sich dann, wenn die Zahl der Analysen gering ist oder Analysen in größeren zeitlichen Abständen durchgeführt werden. Der Grund liegt darin, dass festes Lithiumhypochlorit gegenüber Natriumhypochlorit einen nahezu konstanten Hypochloritanteil enthält. Ist dieser Hypochloritanteil bekannt, muss nicht bei jeder Analyse der Hypochloritgehalt jodometrisch überprüft werden, es kann vielmehr mit konstanter Einwaage an Lithiumhypochlorit gearbeitet werden.
3. GERÄTE UND REAGENZIEN (neben den im allgemeinen Teil genannten)
3.1. Geräte
a) Mindestens 250 ml fassender Erlenmeyerkolben mit Glasschliffstopfen;
b) Thermostat, einstellbar auf 20 °C ± 2 °C.
3.2. Reagenzien
a) Hypochloritreagenz
i) Lithiumhypochloritlösung
Diese besteht aus einer frisch zubereiteten Lösung mit 35 g/l ± 2 g/l) aktivem Chlor (etwa 1 M), der 5 g/l ± 0,5 g/l vorher gelöstes Natriumhydroxid zugegeben wurde. Hierzu werden 100 g Lithiumhypochlorit mit 35 % aktivem Chlor (bzw. 115 g mit 30 % aktivem Chlor) in etwa 700 ml destilliertem Wasser gelöst, 5 g in etwa 200 ml destilliertem Wasser gelöstes Natriumhydroxid hinzugefügt und mit destilliertem Wasser auf 1 Liter aufgefüllt. Die frisch hergestellte Lösung braucht nicht jodometrisch überprüft zu werden.
ii) Natriumhypochloritlösung
Diese besteht aus einer frisch zubereiteten Lösung mit 35 g/l ± 2 g/l aktivem Chlor (etwa 1 M), der 5 g/l ± 0,5 g/l vorher gelöstes Natriumhydroxid zugegeben wurde.
Vor jeder Analyse ist der Gehalt der Lösung an aktivem Chlor jodometrisch zu überprüfen.
b) Verdünnte Essigsäure
5 ml Eisessig werden mit Wasser auf 1 Liter aufgefüllt.
4. ANALYSENGANG
Es ist der im allgemeinen Teil beschriebene Analysengang zu befolgen und folgendermaßen vorzugehen: Etwa 1 g der Analyseprobe wird in dem 250-ml-Kolben mit etwa 100 ml der Hypochloritlösung (Lithium- oder Natriumhypochlorit) versetzt und gut geschüttelt, um die Analyseprobe zu benetzen.
Anschließend wird der Kolben 40 Minuten in einem Thermostat bei 20 °C erwärmt und dabei kontinuierlich oder zumindest in regelmäßigen Abständen geschüttelt. Da die Lösung der Wolle exotherm verläuft, ist die Reaktionswärme durch diese Arbeitsweise zu verteilen und abzuführen. Andernfalls können größere Fehler durch das Anlösen der unlöslichen Fasern entstehen.
Nach 40 Minuten wird der Inhalt des Kolbens durch einen gewogenen Glasfiltertiegel filtriert; etwa zurückgebliebene Fasern werden durch Auswaschen des Kolbens mit etwas Hypochloritreagenz in den Filtertiegel gespült. Der Filtertiegel wird mittels Unterdruck entleert und der Rückstand nacheinander mit Wasser, verdünnter Essigsäure und wieder mit Wasser gewaschen, wobei der Tiegel nach jeder Flüssigkeitszugabe unter Absaugen entleert wird, jedoch erst, nachdem die Flüssigkeit ohne Absaugen durchgelaufen ist.
Zum Schluss wird der Tiegel durch Absaugen geleert, zusammen mit dem Rückstand getrocknet, abgekühlt und gewogen.
5. BERECHNUNG UND ERGEBNISDARSTELLUNG
Die Ergebnisse werden nach dem im allgemeinen Teil angegebenen Berechnungsverfahren ermittelt. Der Korrekturfaktor „d“ hat den Wert 1,00, für Baumwolle, Viskose, Modal und Melamin den Wert 1,01 und für ungebleichte Baumwolle den Wert 1,03.
6. GENAUIGKEIT DES VERFAHRENS
Bei homogenen Textilfasergemischen liegt das Konfidenzintervall der nach diesem Verfahren ermittelten Ergebnisse bei maximal ± 1 mit einem Konfidenzniveau von 95 %.
METHODE Nr. 3
VISKOSE, CUPRO ODER BESTIMMTE MODALARTEN UND BESTIMMTE ANDERE FASERN
(Methode mit Ameisensäuren und Zinkchlorid)
1. ANWENDUNGSBEREICH
Dieses Verfahren gilt nach Entfernung der Nichtfaserstoffe für binäre Fasergemische von
1. Viskose (25) oder Cupro (21) sowie bestimmten Modalarten (22)
mit
2. Baumwolle (5), Polypropylen (37), Elastolefin (46) und Melamin (47).
Wird die Anwesenheit von Modalfasern festgestellt, so ist ein Vorversuch auszuführen, um zu untersuchen, ob diese im Reagenz löslich sind.
Die Methode gilt nicht für Gemische, bei denen die Baumwolle durch übermäßigen chemischen Angriff verändert worden ist oder die Viskose- oder Cuprofasern durch das Vorhandensein bestimmter Farbstoffe, Reagenzien oder Appreturmittel, die nicht vollständig entfernt werden können, nicht mehr vollständig löslich sind.
2. PRINZIP
Die Viskose-, Cupro- oder Modalfasern werden aus einer bekannten Trockenmasse mit einem Reagenz aus Ameisensäure und Zinkchlorid herausgelöst. Der Rückstand wird gesammelt, gewaschen, getrocknet und gewogen; seine berichtigte Masse wird in Prozentsätzen der Trockenmasse des Gemischs ausgedrückt. Der Anteil an trockenen Viskose-, Cupro- oder Modalfasern wird durch Differenzbildung ermittelt.
3. GERÄTE UND REAGENZIEN (neben den im allgemeinen Teil genannten)
3.1. Geräte
a) Mindestens 200 ml fassender Erlenmeyerkolben mit Glasschliffstopfen;
b) Einrichtung zur Erwärmung des Erlenmeyerkolbens auf 40 °C ± 2 °C.
3.2. Reagenzien
a) Lösung aus 20 g geschmolzenem wasserfreiem Zinkchlorid und 68 g wasserfreier Ameisensäure, mit Wasser auf 100 g aufgefüllt (d. h. aus 20 Gewichtsteilen geschmolzenem wasserfreiem Zinkchlorid in 80 Gewichtsteilen Ameisensäure, 85 Gewichtsprozent).
In diesem Zusammenhang wird auf Nummer I.3.2.2 hingewiesen, der vorschreibt, dass alle verwendeten Reagenzien chemisch rein sein müssen; außerdem darf ausschließlich geschmolzenes wasserfreies Zinkchlorid verwendet werden.
b) Ammoniumhydroxydlösung: 20 ml einer konzentrierten Ammoniaklösung (relative Dichte bei 20°C: 0,880) werden mit Wasser auf 1 Liter aufgefüllt.
4. ANALYSENGANG
Es ist der im allgemeinen Teil beschriebene Analysengang zu befolgen und folgendermaßen vorzugehen: Die Probe wird sofort in einen auf 40 °C vorgewärmten Erlenmeyerkolben gegeben und mit 100 ml der auf 40 °C vorgewärmten Ameisensäure-Zinkchloridlösung je Gramm Probe versetzt. Der Kolben wird verschlossen und kräftig geschüttelt. Der Kolben und sein Inhalt werden 2½ Stunden lang bei 40 °C stehengelassen und während dieser Zeit zweimal in Intervallen von je 1 Stunde geschüttelt.
Der Inhalt des Kolbens wird über einen gewogenen Glasfiltertiegel filtriert; dabei werden etwa am Kolben haftende Fasern mit Reagenzlösung in den Filtertiegel gespült. Mit 20 ml Reagenz, die auf 40 °C vorgewärmt ist, nachspülen.
Filtertiegel und Rückstand werden gründlich mit Wasser von 40 °C gewaschen. Danach wird der Faserrückstand mit ca. 100 ml kalter Ammoniaklösung (3.2. b) gespült, wobei sichergestellt werden muss, dass dieser Rückstand 10 Minuten lang vollständig in der Lösung eingetaucht bleibt ( 9 ); danach wird gründlich mit kaltem Wasser gespült.
jedoch erst, nachdem die Flüssigkeit ohne Absaugen durchgelaufen ist.
Zum Schluss wird der noch verbleibende Flüssigkeitsüberschuss durch Absaugen entfernt und Tiegel und Rückstand getrocknet, abgekühlt und gewogen.
5. BERECHNUNG UND ERGEBNISDARSTELLUNG
Die Ergebnisse werden nach dem im allgemeinen Teil angegebenen Berechnungsverfahren ermittelt. Der Korrekturfaktor „d“ beträgt 1,00; nur für Baumwolle beträgt er 1,02 und für Melamin 1,01.
6. GENAUIGKEIT DES VERFAHRENS
Bei homogenen Textilgemischen liegt das Konfidenzintervall der Ergebnisse dieses Verfahrens bei höchstens ± 2, wobei das Konfidenzniveau 95 % beträgt.
METHODE Nr. 4
POLYAMID ODER NYLON UND BESTIMMTE ANDERE FASERN
(Verfahren mit 80 %iger Ameisensäure)
1. ANWENDUNGSBEREICH
Dieses Verfahren gilt nach Entfernung der Nichtfaserstoffe für binäre Fasergemische von
1. Polyamid oder Nylon (30)
mit
2. Wolle (1), Tierhaaren (2 und 3), Baumwolle (5), Cupro (21), Modal (22), Viskose (25), Polyacryl (26), Polychlorid (27), Polyester (35), Polypropylen (37), Glasfasern (44), Elastomultiester (45), Elastolefin (46) und Melamin (47).
Das Verfahren gilt wie vorstehend angegeben für wollhaltige Gemische, doch ist bei einem Wollgehalt von über 25 % die Methode Nr. 2 anzuwenden, d. h. Auflösung der Wolle in einer alkalischen Natriumhypochlorit- oder Lithiumhypochloritlösung.
2. PRINZIP
Das Polyamid oder Nylon wird aus einer bekannten Trockenmasse mittels Ameisensäure herausgelöst. Der Rückstand wird gesammelt, gewaschen, getrocknet und gewogen; seine - erforderlichenfalls berichtigte - Masse wird in Prozentsätzen der Trockenmasse der Mischung ausgedrückt. Der Anteil an trockenem Polyamid oder Nylon wird durch Differenzbildung ermittelt.
3. GERÄTE UND REAGENZIEN (neben den im allgemeinen Teil genannten)
3.1. Geräte
Mindestens 200 ml fassender Erlenmeyerkolben mit Glasschliffstopfen.
3.2. Reagenzien
a) Ameisensäure zu 80 Gewichtsprozent, relative Dichte bei 20 °C: 1,186). 880 ml Ameisensäure zu 90 Gewichtsprozent (relative Dichte bei 20 °C: 1,204) werden mit Wasser auf 1 Liter aufgefüllt. Alternativ dazu werden 780 ml Ameisensäure zu 98 bis 100 Gewichtsprozent (relative Dichte bei 20 °C: 1,220) werden mit Wasser auf 1 Liter aufgefüllt.
Zwischen 77 und 83 Gewichtsprozent Ameisensäure ist die Konzentration nicht kritisch.
b) Verdünntes Ammoniak: 80 ml konzentriertes Ammoniak (relative Dichte bei 20 °C: 0,880) werden mit Wasser auf 1 Liter aufgefüllt.
4. ANALYSENGANG
Es ist der im allgemeinen Teil beschriebene Analysengang zu befolgen und folgendermaßen vorzugehen: Die Probe wird in einem mindestens 200 ml fassenden Erlenmeyerkolben mit 100 ml Ameisensäure je Gramm Probe versetzt, der Kolben wird verschlossen und geschüttelt, um die Probe vollständig zu benetzen; 15 Minuten bei Raumtemperatur unter zeitweiligem Schütteln stehen lassen. Der Inhalt des Erlenmeyerkolbens wird durch einen gewogenen Glasfiltertiegel filtriert; etwa zurückbleibende Fasern werden durch Auswaschen des Kolbens mit etwas Ameisensäurelösung in den Filtertiegel überführt.
Der Filtertiegel wird unter Absaugen entleert und der Rückstand nacheinander mit Ameisensäure, warmem Wasser, verdünntem Ammoniak und schließlich mit kaltem Wasser gewaschen, wobei der Tiegel nach jeder Flüssigkeitszugabe unter Absaugen entleert wird, jedoch erst, nachdem die Flüssigkeit ohne Absaugen durchgelaufen ist.
Zum Schluss wird der Tiegel durch Absaugen geleert, zusammen mit dem Rückstand getrocknet, gekühlt und gewogen.
5. BERECHNUNG UND ERGEBNISDARSTELLUNG
Die Ergebnisse werden nach dem im allgemeinen Teil angegebenen Berechnungsverfahren ermittelt. Der Korrekturfaktor „d“ beträgt 1,00, für Melamin 1,01.
6. GENAUIGKEIT DES VERFAHRENS
Bei homogenen Textilgemischen liegt das Konfidenzintervall der Ergebnisse dieses Verfahrens bei höchstens ± 1, wobei das Konfidenzniveau 95 % beträgt.
METHODE Nr. 5
ACETAT UND BESTIMMTE ANDERE FASERN
(Benzylalkohol-Verfahren)
1. ANWENDUNGSBEREICH
Dieses Verfahren gilt nach Entfernung der Nichtfaserstoffe für binäre Fasergemische von
1. Acetat (19)
mit
2. Triacetatfasern (24), Polypropylen (37), Elastolefin (46), Melamin (47), Polypropylen/Polyamid-Bikomponentenfasern (49) und Polyacrylat (50).
2. PRINZIP
Die Acetatfasern werden aus einer bekannten Trockenmasse des Gemischs mit Hilfe von Benzylalkohol von 52 °C ± 2 °C herausgelöst.
Der Rückstand wird gesammelt, gewaschen, getrocknet und gewogen; seine Masse wird in Prozentsätzen der Trockenmasse des Gemischs ausgedrückt. Der Anteil an trockenen Acetatfasern wird durch Differenzbildung ermittelt.
3. GERÄTE UND REAGENZIEN (neben den im allgemeinen Teil genannten)
3.1. Geräte
a) Mindestens 200 ml fassender Erlenmeyerkolben mit Glasschliffstopfen;
b) mechanischer Schüttler.
c) Thermostat oder anderes Gerät, das den Kolben einer Temperatur von 52 °C ± 2 °C aussetzen kann.
3.2. Reagenzien
a) Benzylalkohol;
b) Ethanol.
4. ANALYSENGANG
Es ist der im allgemeinen Teil beschriebene Analysengang zu befolgen und folgendermaßen vorzugehen:
Der im Erlenmeyerkolben befindlichen Probe werden 100 ml Benzylalkohol je Gramm Probe zugegeben. Der Kolben wird mit einem Stopfen verschlossen und so auf einem Schüttler befestigt, dass er in ein Wasserbad von 52 °C ± 2 °C taucht, wo er mindestens 20 Minuten bei dieser Temperatur geschüttelt wird.
(Unter Umständen kann der mechanische Schüttler durch kräftiges Schütteln mit der Hand ersetzt werden.)
Die Flüssigkeit wird über einen gewogenen Glasfiltertiegel dekantiert. Eine neue Portion Benzylalkohol wird hinzugefügt und der Kolben nochmals 20 Minuten lang bei 52 °C ± 2 °C geschüttelt.
Dann wird über den Filtertiegel dekantiert. Diese Behandlung wird ein drittes Mal wiederholt.
Danach werden die Flüssigkeit und der Rückstand in den Filtertiegel gegossen. Etwa im Kolben zurückbleibende Fasern werden durch Hinzufügung einer zusätzlichen Menge Benzylalkohol von 52 °C ± 2 °C übergespült. Der Tiegel wird nun vollständig trockengeschleudert. Die Fasern werden in einen Kolben überführt;
Ethanol wird zum Spülen beigefügt. Nach kräftigem Schütteln mit der Hand wird über den Filtertiegel dekantiert.
Dieser Spülvorgang ist zwei- oder dreimal zu wiederholen. Der Rückstand wird in den Tiegel überführt und vollständig trockengeschleudert. Filtertiegel und Rückstand werden getrocknet und nach Abkühlung gewogen.
5. BERECHNUNG UND ERGEBNISDARSTELLUNG
Die Ergebnisse werden nach dem im allgemeinen Teil angegebenen Berechnungsverfahren ermittelt. Der Korrekturfaktor „d“ beträgt 1,00, für Melamin 1,01.
6. GENAUIGKEIT DES VERFAHRENS
Bei homogenen Textilgemischen liegt das Konfidenzintervall der Ergebnisse dieses Verfahrens bei höchstens ± 1, wobei das Konfidenzniveau 95 % beträgt.
METHODE Nr. 6
TRIACETAT- ODER POLYLACTIDFASERN UND BESTIMMTE ANDERE FASERN
(Dichlormethan-Verfahren)
1. ANWENDUNGSBEREICH
Dieses Verfahren gilt nach Entfernung der Nichtfaserstoffe für binäre Fasergemische von
1. Triacetat (24) oder Polylactid (34)
mit
2. Wolle (1), Tierhaaren (2 und 3), Seide (4), Baumwolle (5), Cupro (21), Modal (22), Viskose (25), Polyacryl (26), Polyamid oder Nylon (30), Polyester (35), Polypropylen (37), Glasfasern (44), Elastomultiester (45), Elastolefin (46), Melamin (47), Polypropylen/Polyamid-Bikomponentenfasern (49) und Polyacrylat (50).
Triacetatfasern, die durch besondere Behandlung partiell verseift sind, sind im Reagenz nicht mehr voll löslich. In diesem Fall ist die Methode nicht anwendbar.
2. PRINZIP
Die Triacetat- oder Polylactidfasern werden aus einer bekannten Trockenmasse mit Hilfe von Dichlormethan herausgelöst. Der Rückstand wird gesammelt, gewaschen, getrocknet und gewogen; seine — erforderlichenfalls berichtigte — Masse wird in Prozentsätzen der Trockenmasse der Mischung ausgedrückt. Der Anteil an trockenem Triacetat oder Polylactid wird durch Differenzbildung ermittelt.
3. GERÄTE UND REAGENZIEN (neben den im allgemeinen Teil genannten)
3.1. Geräte
Mindestens 200 ml fassender Erlenmeyerkolben mit Glasschliffstopfen.
3.2. Reagenz
Dichlormethan.
4. ANALYSENGANG
Es ist der im allgemeinen Teil beschriebene Analysengang zu befolgen und folgendermaßen vorzugehen:
Die in einem 200-ml-Erlenmeyerkolben mit Glasschliffstopfen befindliche Analyseprobe wird mit 100 ml Dichlormethan je Gramm Analyseprobe versetzt, der Kolben wird mit dem Stopfen verschlossen, geschüttelt zwecks vollständiger Benetzung der Analyseprobe und 30 Minuten bei Raumtemperatur unter Schütteln in Abständen von 10 Minuten stehengelassen. Die Flüssigkeit wird über einen gewogenen Glasfiltertiegel dekantiert. Es werden 60 ml Dichlormethan in den Kolben mit dem Rückstand gegeben, von Hand geschüttelt und der Inhalt des Kolbens über den Glasfiltertiegel filtriert. Etwa zurückbleibende Fasern werden durch Spülen mit einer kleinen zusätzlichen Menge von Dichlormethan in den Tiegel überführt. Der Tiegel wird unter Absaugen entleert, dann erneut mit Dichlormethan gefüllt, das man vollständig ablaufen lässt.
Schließlich wird der Flüssigkeitsüberschuss unter Absaugen entfernt, der Rückstand zur gänzlichen Entfernung des Lösungsmittels mit kochendem Wasser behandelt, abgesaugt und der Tiegel mit Rückstand getrocknet, abgekühlt und gewogen.
5. BERECHNUNG UND ERGEBNISDARSTELLUNG
Die Ergebnisse werden nach dem im allgemeinen Teil angegebenen Berechnungsverfahren ermittelt. Der Korrekturfaktor „d“ beträgt 1,00 für Polyester, Elastomultiester, Elastolefin und Melamin 1,01.
6. GENAUIGKEIT DES VERFAHRENS
Bei homogenen Textilgemischen liegt das Konfidenzintervall der Ergebnisse dieses Verfahrens bei höchstens ± 1, wobei das Konfidenzniveau 95 % beträgt.
METHODE Nr. 7
BESTIMMTE ZELLULOSEFASERN UND BESTIMMTE ANDERE FASERN
(Verfahren mit 75 %iger Schwefelsäure)
1. ANWENDUNGSBEREICH
Dieses Verfahren gilt nach Entfernung der Nichtfaserstoffe für binäre Fasergemische von
1. Baumwolle (5), Flachs (oder Leinen) (7), Hanf (8), Ramie (14), Cupro (21), Modal (22) und Viskose (25)
mit
2. Polyester (35), Polypropylen (37), Elastomultiester (45), Elastolefin (46) und Polypropylen/Polyamid-Bikomponentenfasern (49).
2. PRINZIP
Die Zellulosefasern werden aus einer bekannten Trockenmasse mit Hilfe 75 %iger Schwefelsäure herausgelöst. Der Rückstand wird gesammelt, gewaschen, getrocknet und gewogen; seine Masse wird in Prozentsätzen der Trockenmasse des Gemischs ausgedrückt. Der Anteil an trockenen Zellulosefasern wird durch Differenzbildung ermittelt.
3. GERÄTE UND REAGENZIEN (neben den im allgemeinen Teil genannten)
3.1. Geräte
a) Mindestens 500 ml fassender Erlenmeyerkolben mit Glasschliffstopfen.
b) Thermostat oder anderes Gerät zur Erwärmung des Erlenmeyerkolbens auf 50 °C ± 5 °C.
3.2. Reagenzien
a) Schwefelsäure zu 75 Gewichtsprozent (± 2 %):
Herstellung in der Weise, dass 700 ml Schwefelsäure (relative Dichte bei 20 °C: 1,84) unter Kühlung vorsichtig zu 350 ml destilliertem Wasser zugesetzt werden.
Nach Abkühlung auf Raumtemperatur mit Wasser auf 1 l auffüllen.
b) Verdünnte Ammoniaklösung:
80 ml Ammoniaklösung (relative Dichte bei 20 °C: 0,880) werden mit Wasser auf 1 Liter aufgefüllt.
4. ANALYSENGANG
Es ist der im allgemeinen Teil beschriebene Analysengang zu befolgen und folgendermaßen vorzugehen:
Die in einem mindestens 500 ml fassenden Erlenmeyerkolben mit Glasschliffstopfen befindliche Probe wird mit 200 ml 75 %iger Schwefelsäure je Gramm Probe versetzt, der Kolben wird mit dem Stopfen verschlossen und vorsichtig geschüttelt, um die Probe vollständig zu benetzen.
Der Kolben wird eine Stunde lang auf einer Temperatur von 50 °C ± 5 °C gehalten und in Abständen von etwa 10 Minuten geschüttelt. Danach wird der Inhalt des Kolbens unter Absaugen durch einen Filtertiegel filtriert. Etwa zurückgebliebene Fasern werden durch Spülen des Kolbens mit etwas 75 %iger Schwefelsäure in den Glasfiltertiegel überführt. Der Glasfiltertiegel wird durch Absaugen geleert und der auf dem Filter verbliebene Rückstand durch erneute Zugabe von Schwefelsäure ein erstes Mal ausgewaschen. Es ist erst abzusaugen, nachdem die Flüssigkeit ohne Absaugen hindurchgelaufen ist.
Der Rückstand wird nacheinander mehrmals mit kaltem Wasser, zweimal mit verdünnter Ammoniaklösung und dann gründlich mit kaltem Wasser gewaschen, wobei nach jeder Flüssigkeitszugabe abzusaugen ist, jedoch jedes Mal erst, nachdem die Flüssigkeit ohne Absaugen hindurchgelaufen ist. Schließlich wird der Tiegel durch Absaugen entleert, zusammen mit dem Rückstand getrocknet, abgekühlt und gewogen.
5. BERECHNUNG UND ERGEBNISDARSTELLUNG
Die Ergebnisse werden nach dem im allgemeinen Teil angegebenen Berechnungsverfahren ermittelt. Der Korrekturfaktor „d“ beträgt 1,00; nur für Polypropylen/Polyamid-Bikomponentenfasern beträgt er 1,01.
6. GENAUIGKEIT DES VERFAHRENS
Bei homogenen Textilgemischen liegt das Konfidenzintervall der Ergebnisse dieses Verfahrens bei höchstens ± 1, wobei das Konfidenzniveau 95 % beträgt.
METHODE Nr. 8
POLYACRYLFASERN, BESTIMMTE MODACRYLFASERN BESTIMMTE POLYCHLORIDFASERN UND BESTIMMTE ANDERE FASERN
(Dimethylformamid-Verfahren)
1. ANWENDUNGSBEREICH
Dieses Verfahren gilt nach Entfernung der Nichtfaserstoffe für binäre Fasergemische von
1. Acrylfasern (26), bestimmten Modacrylfasern (29) oder bestimmten Polychloridfasern (27) ( 10 )
mit
2. Wolle (1), Tierhaaren (2 und 3), Seide (4), Baumwolle (5), Cupro (21), Modal (22), Viskose (25), Polyamid oder Nylon (30), Polyester (35), Polypropylen (37), Elastomultiester (45), Elastolefin (46), Melamin (47), Polypropylen/Polyamid-Bikomponentenfasern (49) und Polyacrylat (50).
Es gilt ferner für Polyacryl- und bestimmte Modacrylfasern, die mit vormetallisierten Farbstoffen, jedoch nicht mit Nachchromierfarbstoffen behandelt sind.
2. PRINZIP
Die Polyacrylfasern, bestimmte Modacrylfasern oder bestimmte Polychloridfasern werden aus einer bekannten Trockenmasse mittels Dimethylformamid im kochenden Wasserbad herausgelöst. Der Rückstand wird gesammelt, gewaschen, getrocknet und gewogen; seine — erforderlichenfalls berichtigte — Masse wird in Prozentsätzen der Trockenmasse des Gemischs ausgedrückt. Der Anteil an trockenen Polyacrylfasern, Modacrylfasern oder Polychloridfasern wird durch Differenzbildung ermittelt.
3. GERÄTE UND REAGENZIEN (neben den im allgemeinen Teil genannten)
3.1. Geräte
a) Mindestens 200 ml fassender Erlenmeyerkolben mit Glasschliffstopfen;
b) kochendes Wasserbad.
3.2. Reagenz
Dimethylformamid (Siedepunkt 153 °C ± 1 °C) mit nicht mehr als 0,1 % Wasser.
Da das Reagenz giftig ist, wird empfohlen, unter dem Abzug zu arbeiten.
4. ANALYSENGANG
Es ist der im allgemeinen Teil beschriebene Analysengang zu befolgen und folgendermaßen vorzugehen:
Die in einem mindestens 200 ml fassenden Erlenmeyerkolben mit Glasschliffstopfen befindliche Probe wird mit 80 ml im kochenden Wasserbad vorgewärmtem Dimethylformamid je Gramm Probe versetzt, der Kolben wird mit dem Stopfen verschlossen, geschüttelt zwecks vollständiger Benetzung der Probe und eine Stunde lang im kochenden Wasserbad belassen. Während dieser Zeit werden der Kolben und sein Inhalt fünfmal vorsichtig von Hand geschüttelt.
Die Flüssigkeit wird durch einen gewogenen Glasfiltertiegel dekantiert, wobei die Fasern im Kolben zurückgehalten werden. Es werden erneut 60 ml Dimethylformamid in den Kolben gegeben und wiederum 30 Minuten im kochenden Wasserbad erwärmt, wobei der Kolben und sein Inhalt zweimal vorsichtig von Hand geschüttelt werden.
Der Inhalt des Kolbens wird durch einen Glasfiltertiegel unter Absaugen filtriert.
Die zurückbleibenden Fasern werden durch Ausspülen des Kolbens mit Dimethylformamid in den Glasfiltertiegel überführt. Der Tiegel wird unter Absaugen entleert. Die zurückbleibenden Fasern werden mit etwa 1 Liter Wasser von 70-80 °C Temperatur gewaschen, wobei der Tiegel jedes Mal mit Wasser gefüllt wird.
Nach jeder Wasserzugabe wird kurz abgesaugt, allerdings erst dann, wenn das Wasser abgelaufen ist. Läuft das Waschwasser zu langsam durch den Tiegel, kann kurz abgesaugt werden.
Zum Schluss wird der Tiegel mit dem Rückstand getrocknet, abgekühlt und gewogen.
5. BERECHNUNG UND ERGEBNISDARSTELLUNG
Die Ergebnisse werden nach dem im allgemeinen Teil angegebenen Berechnungsverfahren ermittelt. Der Korrekturfaktor „d“ beträgt 1,00, jedoch für Wolle, Baumwolle, Cupro, Modal, Polyester, Elastomultiester, Melamin und Polyacrylat 1,01.
6. GENAUIGKEIT DES VERFAHRENS
Bei homogenen Textilgemischen liegt das Konfidenzintervall der Ergebnisse dieses Verfahrens bei höchstens ± 1, wobei das Konfidenzniveau 95 % beträgt.
METHODE Nr. 9
BESTIMMTE POLYCHLORIDFASERN UND BESTIMMTE ANDERE FASERN
(Verfahren mit Schwefelkohlenstoff/Aceton 55,5/44,5 % v/v)
1. ANWENDUNGSBEREICH
Dieses Verfahren gilt nach Entfernung der Nichtfaserstoffe für binäre Fasergemische von
1. bestimmten Polychloridfasern (27), d. h. bestimmten, auch nachchlorierten Polyvinylchloridfasern ( 11 )
mit
2. wolle (1), Tierhaaren (2 und 3), Seide (4), Baumwolle (5), Cupro (21), Modal (22), Viskose (25), Polyacryl (26), Polyamid oder Nylon (30), Polyester (35), Polypropylen (37), Glasfasern (44), Elastomultiester (45), Melamin (47), Polypropylen/Polyamid-Bikomponentenfasern (49) und Polyacrylat (50).
Ist der Gehalt an Wolle oder Seide im Gemisch größer als 25 %, so ist Methode Nr. 2 anzuwenden.
Ist der Gehalt an Polyamid oder Nylon im Gemisch größer als 25 %, so ist die Methode Nr. 4 anzuwenden.
2. PRINZIP
Die Polychloridfasern werden aus einer bekannten Trockenmasse mit Hilfe einer azeotropischen Mischung von Schwefelkohlenstoff und Aceton herausgelöst. Der Rückstand wird gesammelt, gewaschen, getrocknet und gewogen; seine - erforderlichenfalls berichtigte - Masse wird in Prozentsätzen der Trockenmasse der Mischung ausgedrückt. Der Anteil an trockenen Polyvinylchloridfasern wird durch Differenzbildung ermittelt.
3. GERÄTE UND REAGENZIEN (neben den im allgemeinen Teil genannten)
3.1. Geräte
a) Mindestens 200 ml fassender Erlenmeyerkolben mit Glasschliffstopfen;
b) mechanischer Schüttler.
3.2. Reagenzien
a) Azeotropische Mischung von Schwefelkohlenstoff und Aceton (55,5 Volumenprozent Schwefelkohlenstoff und 44,5 Volumenprozent Aceton). Da das Reagenz giftig ist, wird empfohlen, unter dem Abzug zu arbeiten.
b) Ethanol (92 Volumenprozent) oder Methanol.
4. ANALYSENGANG
Es ist der im allgemeinen Teil beschriebene Analysengang zu befolgen und folgendermaßen vorzugehen:
Die in einem mindestens 200 ml fassender Erlenmeyerkolben mit Glasschliffstopfen befindliche Probe wird mit 100 ml azeotropischer Mischung je Gramm Probe versetzt. Der Kolben wird sorgfältig verschlossen und 20 Minuten lang mit dem mechanischen Schüttler oder von Hand bei Raumtemperatur kräftig geschüttelt.
Die Flüssigkeit wird durch den gewogenen Glasfiltertiegel dekantiert.
Die Behandlung wird mit 100 ml frischem Lösungsmittel wiederholt. Diese Behandlung wird so oft fortgesetzt, bis ein Tropfen Lösungsmittel nach Verdunstung auf einem Uhrglas keinerlei Polymerniederschlag hinterlässt. Der Rückstand wird mit Hilfe einer zusätzlichen Menge Lösungsmittel in den Filtertiegel überführt, der Filtertiegel durch Sauganwendung entleert; Tiegel und Rückstand werden mit 20 ml Alkohol gespült und anschließend dreimal mit Wasser nachgespült. Vor dem Absaugen muss die Spülflüssigkeit vollständig durchgelaufen sein. Tiegel und Rückstand werden getrocknet, abgekühlt und gewogen.
Die Proben bestimmter Gemische mit hohem Gehalt an Polychlorid schrumpfen beim Trocknen beträchtlich, was die Beseitigung des Polychlorids durch das Lösungsmittel erschwert.
Jedoch verhindert diese Schrumpfung nicht die vollständige Auflösung des Polychlorids.
5. BERECHNUNG UND ERGEBNISDARSTELLUNG
Die Ergebnisse werden nach dem im allgemeinen Teil angegebenen Berechnungsverfahren ermittelt. Der Korrekturfaktor „d“ beträgt 1,00, für Melamin und Polyacrylat 1,01.
6. GENAUIGKEIT DES VERFAHRENS
Bei homogenen Textilfasergemischen liegt das Konfidenzintervall der nach diesem Verfahren ermittelten Ergebnisse bei maximal ± 1 mit einem Konfidenzniveau von 95 %.
METHODE Nr. 10
ACETATFASERN UND BESTIMMTE ANDERE FASERN
(Essigsäure-Verfahren)
1. ANWENDUNGSBEREICH
Dieses Verfahren gilt nach Entfernung der Nichtfaserstoffe für binäre Fasergemische von
1. Acetat (19)
mit
2. bestimmten Polychloridfasern (27), und zwar Polyvinylchloridfasern, auch nachchloriert, sowie Polypropylen (37), Elastolefin (46), Melamin (47) und Polypropylen/Polyamid-Bikomponentenfasern (49).
2. PRINZIP
Die Acetatfasern werden aus einer bekannten Trockenmasse mittels Essigsäure aufgelöst. Der Rückstand wird gesammelt, gewaschen, getrocknet und gewogen; seine — erforderlichenfalls berichtigte — Masse wird in Prozentsätzen der Trockenmasse der Mischung ausgedrückt. Der Anteil an trockenen Acetatfasern wird durch Differenzbildung ermittelt.
3. GERÄTE UND REAGENZIEN (neben den im allgemeinen Teil genannten)
3.1. Geräte
a) Mindestens 200 ml fassender Erlenmeyerkolben mit Glasschliffstopfen;
b) mechanischer Schüttler.
3.2. Reagenz
Essigsäure (mehr als 99 %). Dieses Reagenz ist sehr ätzend, bei seiner Verwendung ist daher Vorsicht geboten.
4. ANALYSENGANG
Es ist der im allgemeinen Teil beschriebene Analysengang zu befolgen und folgendermaßen vorzugehen:
Die in einem mindestens 200 ml fassenden Erlenmeyerkolben mit Glasschliffstopfen befindliche Probe wird mit 100 ml Essigsäure je Gramm Probe versetzt. Der Kolben wird sorgfältig verschlossen und bei Raumtemperatur 20 Minuten mit einem mechanischen Schüttler oder von Hand kräftig geschüttelt. Die Flüssigkeit wird durch den gewogenen Glasfiltertiegel dekantiert. Diese Behandlung wird zweimal unter Verwendung von jeweils 100 ml frischem Lösungsmittel wiederholt, so dass insgesamt drei Extraktionen ausgeführt werden. Der Rückstand wird in den Filtertiegel überführt, der Flüssigkeitsrückstand unter Sauganwendung entleert;
Tiegel und Rückstand werden mit 50 ml Essigsäure sowie anschließend dreimal mit Wasser nachgespült. Nach jedem Auswaschen muss die Flüssigkeit von selbst durchlaufen, bevor abgesaugt wird. Tiegel und Rückstand trocknen, abkühlen und wägen.
5. BERECHNUNG UND ERGEBNISDARSTELLUNG
Die Ergebnisse werden nach dem im allgemeinen Teil angegebenen Berechnungsverfahren ermittelt. Der Korrekturfaktor „d“ beträgt 1,00.
6. GENAUIGKEIT DES VERFAHRENS
Bei homogenen Textilfasergemischen liegt das Konfidenzintervall der nach diesem Verfahren ermittelten Ergebnisse bei maximal ± 1 mit einem Konfidenzniveau von 95 %.
METHODE Nr. 11
SEIDE ODER POLYAMID UND BESTIMMTE ANDERE FASERN
(Verfahren mit 75 %iger Schwefelsäure)
1. ANWENDUNGSBEREICH
Diese Methode gilt nach Entfernung der nichtfaserigen Bestandteile für binäre Gemische von:
1. Seide (4) oder Polyamid oder Nylon (30)
mit
2. Wolle (1), Tierhaaren (2 und 3), Polypropylen (37), Elastolefin (46), Melamin (47) und Polypropylen/Polyamid-Bikomponentenfasern (49).
2. PRINZIP
Die Seidenfasern bzw. Polyamid- oder Nylonfasern werden aus einer bekannten Trockenmasse mit 75-gewichtsprozentiger Schwefelsäure herausgelöst ( 12 ).
Der Rückstand wird gesammelt, gewaschen, getrocknet und gewogen. Seine — erforderlichenfalls berichtigte — Masse wird in Prozentsätzen der Trockenmasse des Gemischs ausgedrückt. Der Anteil an trockener Seide bzw. trockenem Polyamid oder Nylon wird durch Differenzbildung ermittelt.
3. GERÄTE UND REAGENZIEN (neben den im allgemeinen Teil genannten)
3.1. Geräte
Mindestens 200 ml fassender Erlenmeyerkolben mit Glasschliffstopfen;
3.2. Reagenzien
a) 75 %ige Schwefelsäure (± 2 %) Herstellungsweise:
700 ml Schwefelsäure (relative Dichte 1,84 bei 20 °C) werden vorsichtig unter Abkühlung zu 350 ml destilliertem Wasser zugesetzt.
Nach Abkühlung auf Raumtemperatur mit Wasser auf 1 l auffüllen.
b) Verdünnte Schwefelsäure: 100 ml konzentrierte Schwefelsäure (relative Dichte 1,84 bei 20 °C) werden langsam zu 1 900 ml destilliertem Wasser zugesetzt.
c) Verdünntes Ammoniak: 200 ml konzentriertes Ammoniak (relative Dichte 0,880 bei 20 °C) werden mit Wasser auf 1 Liter aufgefüllt.
4. ANALYSENGANG
Es ist der im allgemeinen Teil beschriebene Analysengang zu befolgen und folgendermaßen vorzugehen:
Die in einem mindestens 200 ml fassenden Erlenmeyerkolben mit Schliffstopfen befindliche Probe wird mit 100 ml 75 %iger Schwefelsäure je Gramm Probe versetzt. Der Kolben wird verschlossen, kräftig geschüttelt und 30 Minuten bei Raumtemperatur stehen gelassen. Erneut schütteln, 30 Minuten stehen lassen. Ein letztes Mal schütteln und den Inhalt des Kolbens in den gewogenen Glasfiltertiegel überführen. Etwa im Kolben zurückbleibende Fasern werden mit 75 %iger Schwefelsäure nachgespült. Der Rückstand wird im Filtertiegel nacheinander mit 50 ml verdünnter Schwefelsäure, 50 ml Wasser und 50 ml verdünntem Ammoniak ausgewaschen. Die Fasern müssen jedes Mal etwa 10 Minuten lang mit der Flüssigkeit in Kontakt bleiben, bevor abgesaugt wird. Schließlich wird mit Wasser nachgewaschen, wobei die Fasern 30 Minuten im Wasser verbleiben. Die Restflüssigkeit wird unter Absaugen entfernt, Tiegel und Rückstand werden getrocknet, abgekühlt und gewogen.
Im Fall von binären Gemischen von Polyamid mit Polypropylen/Polyamid-Bikomponentenfasern ist nach dem Überführen der Fasern in den gewogenen Glasfiltertiegel und vor Anwendung des beschriebenen Waschverfahrens der Rückstand im Filtertiegel zweimal jeweils mit 50 ml 75 %iger Schwefelsäure auszuwaschen.
5. BERECHNUNG UND ERGEBNISDARSTELLUNG
Die Ergebnisse werden nach dem im allgemeinen Teil angegebenen Berechnungsverfahren ermittelt. Der Korrekturfaktor „d“ beträgt 1,00; nur für Wolle beträgt er 0,985, für Polypropylen/Polyamid-Bikomponentenfasern 1,005 und für Melamin 1,01.
6. GENAUIGKEIT DES VERFAHRENS
Bei homogenen Textilgemischen liegt das Konfidenzintervall der Ergebnisse dieses Verfahrens bei höchstens ± 1, wobei das Konfidenzniveau 95 % beträgt; nur bei binären Gemischen von Polyamid mit Polypropylen/Polyamid-Bikomponentenfasern liegt das Konfidenzintervall bei maximal ± 2.
METHODE Nr. 12
JUTE UND BESTIMMTE FASERN TIERISCHEN URSPRUNGS
(Verfahren mittels Stickstoffbestimmung)
1. ANWENDUNGSBEREICH
Dieses Verfahren gilt nach Entfernung der Nichtfaserstoffe für binäre Fasergemische von
1. Jute (9)
mit
2. bestimmten Fasern tierischen Ursprungs.
Letztere können aus Tierhaaren (2 und 3) oder aus Wolle (1) oder einer Mischung von Tierhaaren und Wolle bestehen. Das Verfahren eignet sich nicht für Textilfasergemische, die nichtfaserige Bestandteile (Farbstoffe, Appreturmittel usw.) auf Stickstoffbasis enthalten.
2. PRINZIP
Der Stickstoffgehalt der Mischung wird bestimmt und hieraus sowie aus dem bekannten Stickstoffgehalt der beiden Bestandteile der Mischung wird deren proportionaler Anteil berechnet.
3. GERÄTE UND REAGENZIEN (neben den im allgemeinen Teil genannten)
3.1. Geräte
a) Kjeldahl-Zersetzungskolben von 200 bis 300 ml;
b) Kjeldahl-Destillationskolben mit Dampferzeuger;
c) Titriergerät, mit einer Genauigkeit bis zu 0,05 ml.
3.2. Reagenzien
a) Toluol;
b) Methanol;
c) Schwefelsäure, relative Dichte bei 20°C: 1,84 ( 13 );
d) Kaliumsulfat (13) ;
e) Selendioxyd (13) ;
f) Natriumhydroxydlösung (400 g/l). 400 g Natriumhydroxyd werden in 400 bis 500 ml Wasser aufgelöst und die Flüssigkeit mit Wasser auf 1 Liter verdünnt;
g) Indikatormischung. 0,1 g Methylrot werden in 95 ml Ethanol und 5 ml Wasser gelöst; diese Lösung wird mit 0,5 g Bromkresolgrün, das in 475 ml Ethanol und 25 ml Wasser aufgelöst ist, vermischt;
h) Borsäurelösung. 20 g Borsäure werden in 1 Liter Wasser gelöst;
i) Schwefelsäure 0,02 N (Normvolumenlösung).
4. VORBEHANDLUNG DER VORPROBE
Anstelle der im allgemeinen Teil beschriebenen Vorbehandlung wird wie folgt vorgegangen:
Die lufttrockene Vorprobe wird in einem Soxhlet-Apparat mit einer Mischung von einem Volumenteil Toluol und drei Volumenteilen Methanol 4 Stunden lang bei mindestens 5 Umläufen pro Stunde extrahiert. Man lässt die Lösungsmittel aus der Probe in Luft verdampfen und entfernt die letzten Spuren in einem Wärmeschrank bei 105 °C ± 3 °C. Sodann wird die Probe im Wasser (50 ml/g Probemenge) durch Sieden mit Rückkühlung 30 Minuten lang extrahiert. Nach dem Filtrieren wird die Probe in den Kolben zurückgegeben und die Extraktion mit einem gleichen Volumen frischen Wassers wiederholt. Nach erneutem Filtrieren wird das überschüssige Wasser durch Ausdrücken, Absaugen oder Zentrifugieren aus der Probe entfernt, die man dann an der Luft trocknen lässt.
Toluol und Methanol sind giftig; deshalb sind bei ihrer Verwendung alle erforderlichen Vorsichtsmaßnahmen zu treffen.
5. ANALYSENGANG
5.1. Allgemeine Anweisungen
Hinsichtlich Entnahme, Trocknung und Wägung der Probe sind die im allgemeinen Teil gemachten Angaben zu befolgen.
5.2. Ausführliche Anleitung
Zu der Probe von mindestens 1 g im Kjeldahl-Kolben werden der Reihe nach 2,5 g Kaliumsulfat, 0,1 bis 0,2 g Selendioxyd und 10 ml Schwefelsäure (relative Dichte bei 20 °C: 1,84) gegeben. Der Kolben wird zunächst schwach erhitzt, bis das ganze Fasermaterial zersetzt ist, sodann wird kräftiger erhitzt, bis die Lösung klar und nahezu farblos geworden ist. Die Erhitzung wird 15 Minuten lang fortgesetzt. Dann lässt man den Kolben abkühlen, verdünnt den Inhalt vorsichtig mit 10 bis 20 ml Wasser, kühlt ab, überführt den Inhalt quantitativ in einen 200-ml-Maßkolben und füllt das Volumen mit Wasser auf, um die Analysenlösung herzustellen. In einen 100-ml-Erlenmeyerkolben werden etwa 20 ml Borsäurelösung gegeben; der Kolben wird unter dem Kondensator des Kjeldahl-Destilliergeräts so aufgestellt, dass das Ablaufrohr gerade unter den Spiegel der Borsäurelösung eintaucht. Es werden genau 10 ml Analysenlösung in den Destillierkolben überführt und mindestens 5 ml Natriumhydroxydlösung in den Trichter gegeben; man hebt den Stopfen leicht an und lässt die Natriumhydroxydlösung langsam in den Kolben fließen. Wenn die Analysenlösung und die Natriumhydroxydlösung zwei getrennte Schichten bilden, so sind sie durch vorsichtiges Schütteln zu vermischen. Der Destillierkolben wird leicht erwärmt und gleichzeitig Dampf aus dem Dampferzeuger in ihn eingeführt. Es werden etwa 20 ml Destillat gesammelt und das Auffanggefäß so weit gesenkt, dass sich das Ende des Ablaufrohrs etwa 20 mm über dem Spiegel der Flüssigkeit befindet, danach wird eine Minute lang weiter destilliert. Das Ende des Ablaufrohrs wird mit Wasser ausgespült und das Reinigungswasser im Erlenmeyerkolben aufgefangen. Der Erlenmeyerkolben wird sodann entfernt und durch einen zweiten Erlenmeyerkolben ersetzt, der etwa 10 ml Borsäurelösung enthält; darin werden etwa 10 ml Destillat aufgesammelt.
Die beiden Destillate werden getrennt mit 0,02-N-Schwefelsäure unter Verwendung der Indikatormischung titriert. Die Ergebnisse für die beiden Destillate werden notiert. Wenn das Bestimmungsergebnis des zweiten Destillats über 0,2 ml liegt, so ist die Bestimmung zu wiederholen; es muss also nochmals mit einem anderen aliquoten Teil der Analysenlösung destilliert werden.
Es wird ein Blindversuch durchgeführt, wobei die Zersetzung und Destillation lediglich unter Verwendung der Reagenzien erfolgt.
6. BERECHNUNG UND ERGEBNISDARSTELLUNG
6.1. Der prozentuale Anteil des Stickstoffs in der trockenen Probe wird folgendermaßen berechnet:

Dabei ist:
|
A |
= |
Prozentsatz Stickstoff im reinen Trockengewicht der Probe; |
|
V |
= |
Gesamtvolumen in ml der Standardschwefelsäure für die Bestimmung; |
|
b |
= |
Gesamtvolumen in ml der Standardschwefelsäure beim Blindversuch; |
|
N |
= |
tatsächlicher Titer der Standardschwefelsäure; |
|
W |
= |
Trockenmasse (g) der Probe. |
6.2. Bei Verwendung des Wertes 0,22 % für den Stickstoffgehalt der Jute bzw. 16,2 % für den Stickstoffgehalt der tierischen Fasern — beide Werte auf die Trockenmasse bezogen — berechnet sich die Zusammensetzung der Mischung nach folgender Formel:

Dabei sind:
|
PA% |
= |
Prozentsatz der tierischen Faser im reinen Trockengewicht der Probe. |
7. GENAUIGKEIT DES VERFAHRENS
Bei homogenen Textilgemischen liegt das Konfidenzintervall der Ergebnisse dieses Verfahrens bei höchstens ± 1, wobei das Konfidenzniveau 95 % beträgt.
METHODE Nr. 13
POLYPROPYLEN UND BESTIMMTE ANDERE FASERN
(Xylol-Verfahren)
1. ANWENDUNGSBEREICH
Dieses Verfahren gilt nach Entfernung der Nichtfaserstoffe für binäre Fasergemische von
1. Polypropylen (37)
mit
2. Wolle (1), Tierhaaren (2 und 3), Seide (4), Baumwolle (5), Acetat (19), Cupro (21), Modal (22), Triacetat (24), Viskose (25), Polyacrylfasern (26), Polyamid oder Nylon (30), Polyester (35), Glasfasern (44), Elastomultiester (45), Melamin (47) und Polyacrylat (50).
2. PRINZIP
Die Polypropylenfaser wird aus einer bekannten Menge der Trockenmasse durch Lösung mit kochendem Xylol abgetrennt. Der Rückstand wird gesammelt, gewaschen, getrocknet und gewogen; seine — erforderlichenfalls berichtigte — Masse wird in Prozentsätzen der Trockenmasse der Mischung ausgedrückt. Das Mengenverhältnis des Polypropylen wird aus der Gewichtsdifferenz berechnet.
3. GERÄTE UND REAGENZIEN (neben den im allgemeinen Teil genannten)
3.1. Geräte
a) Mindestens 200 ml fassender Erlenmeyerkolben mit Glasschliffstopfen;
b) Rückflusskühler (geeignet für Flüssigkeiten mit erhöhtem Kochpunkt) mit zum Aufsetzen auf den Erlenmeyerkolben (a) angepasstem geschliffenem Übergangsteil;
c) auf die Temperatur des Siedepunkts von Xylol erhitzte Heizhaube.
3.2. Reagenz
Xylol, Siedebereich zwischen 137 °C und 142 °C.
Xylol ist sehr leicht entflammbar und entwickelt giftige Dämpfe. Beim Gebrauch sind entsprechende Schutzvorkehrungen zu treffen.
4. ANALYSENGANG
Es ist der im allgemeinen Teil beschriebene Analysengang zu befolgen und folgendermaßen vorzugehen:
Zu der in den Erlenmeyerkolben (3.1.a) eingegebenen Probe werden 100 ml Xylol (3.2) pro Gramm Probe hinzugegeben. Dann wird der Kühler (3.1.b) aufgesetzt und der Kolbeninhalt zum Kochen gebracht; Kochdauer 3 Minuten.
Die heiße Flüssigkeit wird danach sofort über den tarierten Glasfiltertiegel (siehe Anmerkung 1) dekantiert. Dieser Vorgang wird zweimal unter Verwendung von jeweils 50 ml frischem Lösungsmittel wiederholt.
Der im Kolben zurückgebliebene Rückstand wird nacheinander zweimal mit 30 ml kochendem Xylol, dann zweimal mit je 75 ml Petrolether (I.3.2.1 Allgemeiner Teil) ausgewaschen. Nach dem zweiten Waschen mit Petrolether wird der Inhalt des Kolbens durch den Tiegel filtriert; die zurückgebliebenen Fasern werden mittels einer kleinen zusätzlichen Menge Petrolether in den Tiegel überführt. Den Tiegel und den Rückstand trocknen, abkühlen und wiegen.
1. Der Filtertiegel, über den das Xylol dekantiert wird, muss vorgewärmt werden.
2. Nach den Verfahrensschritten mit dem kochenden Xylol darauf achten, dass der den Rückstand enthaltende Erlenmeyerkolben ausreichend abgekühlt wird, bevor der Petrolether dazugegeben wird.
3. Um die Gefährdung der Laboranten durch die Entflammbarkeit und Giftigkeit möglichst niedrig zu halten, können Heißextraktionsapparaturen und geeignete Verfahren, die gleichartige Ergebnisse erbringen, angewendet werden ( 14 ).
5. BERECHNUNG UND ERGEBNISDARSTELLUNG
Die Ergebnisse werden nach dem im allgemeinen Teil angegebenen Berechnungsverfahren ermittelt. Der Korrekturfaktor „d“ beträgt 1,00, für Melamin und Polyacrylat 1,01.
6. GENAUIGKEIT DES VERFAHRENS
Bei homogenen Textilgemischen liegt das Konfidenzintervall der Ergebnisse dieses Verfahrens bei höchstens ± 1, wobei das Konfidenzniveau 95 % beträgt.
METHODE Nr. 14
BESTIMMTE FASERN UND BESTIMMTE ANDERE FASERN
(Verfahren mit konzentrierter Schwefelsäure)
1. ANWENDUNGSBEREICH
Dieses Verfahren gilt nach Entfernung der Nichtfaserstoffe für binäre Fasergemische von
1. Baumwolle (5), Acetat (19), Cupro (21), Modal (22), Triacetat (24), Viskose (25), bestimmten Polyacrylfasern (26) und Modacrylfasern (29), Polyamid oder Nylon (30), Polyester (35) und Elastomultiester (45)
mit
2. Polychloridfasern (27) auf Homopolymerbasis von Vinylchlorid, auch nachchloriert, sowie Polypropylen (37), Elastolefin (46), Melamin (47) und Polypropylen/Polyamid-Bikomponentenfasern (49).
Die betreffenden Modacrylfasern sind diejenigen, die bei Behandlung in konzentrierter Schwefelsäure (relative Dichte bei 20 °C: 1,84) eine klare Lösung ergeben.
Dieses Verfahren kann insbesondere anstelle der Methoden Nr. 8 und 9 angewendet werden.
2. PRINZIP
Alle Bestandteile außer Polychloridfasern, Polypropylen, Elastolefin, Melamin oder Polypropylen/Polyamid-Bikomponentenfasern (d. h. die unter 1.1 genannten Fasern) werden aus einer bekannten Trockenmasse durch Auflösen in konzentrierter Schwefelsäure (relative Dichte bei 20 °C: 1,84) herausgelöst. Der aus Polychloridfasern, Polypropylen, Elastolefin, Melamin oder Polypropylen/Polyamid-Bikomponentenfasern bestehende Rückstand wird gesammelt, gewaschen, getrocknet und gewogen; seine — erforderlichenfalls berichtigte — Masse wird in Prozentsätzen der Trockenmasse der Mischung ausgedrückt. Der Anteil der anderen Bestandteile wird durch Differenzbildung ermittelt.
3. GERÄTE UND REAGENZIEN (neben den im allgemeinen Teil genannten)
3.1. Geräte
a) Mindestens 200 ml fassender Erlenmeyerkolben mit Glasschliffstopfen;
b) Glasstab mit abgeflachtem Ende.
3.2. Reagenzien
a) Schwefelsäure (relative Dichte bei 20 °C: 1,84);
b) Schwefelsäure, wässrige Lösung etwa 50 % (m/m) Schwefelsäure.
Herstellung in der Weise, dass 400 ml Schwefelsäure (relative Dichte bei 20 °C: 1,84) zu 500 ml destilliertem oder entionisiertem Wasser hinzugegeben. Wenn die Lösung auf Zimmertemperatur abgekühlt ist, wird mit Wasser auf 1 Liter angefüllt.
c) Ammoniak, verdünnte Lösung
60 ml einer konzentrierten Ammoniaklösung (relative Dichte bei 20 °C: 0,880) werden mit destilliertem Wasser zu 1 l Lösung verdünnt.
4. ANALYSENGANG
Es ist der im allgemeinen Teil beschriebene Analysengang zu befolgen und folgendermaßen vorzugehen:
Zu der in den Erlenmeyerkolben (3.1.a) eingegebenen Analyseprobe werden 100 ml Schwefelsäure (3.2.a) pro Gramm Probe hinzugegeben.
10 Minuten lang bei Zimmertemperatur unter häufigerem Umrühren der Probenlösung mit dem Glasstab stehen lassen. Wenn es sich um ein Gewebe oder um gewirkte Ware handelt, so drückt man das Probestück leicht gegen die Kolbenwand, um auf diese Weise die durch die Schwefelsäure ausgelösten Bestandteile abzutrennen.
Die Flüssigkeit wird über einen gewogenen Glasfiltertiegel dekantiert. Dann werden erneut 100 ml Schwefelsäure (3.2. a) in den Kolben gegeben und der Vorgang wiederholt. Den Inhalt des Kolbens über dem Tiegel entleeren und hierbei den faserigen Rückstand mit dem Glasstab abtrennen. Erforderlichenfalls etwas konzentrierte Schwefelsäure (3.2. a) in den Kolben nachgeben, um die an den Wänden hängenbleibenden Faserreste zu entfernen. Den Tiegel durch Absaugen entleeren; das Filtrat des Kolbens völlig entfernen oder den Kolben auswechseln, dann den Rückstand im Tiegel nacheinander mit der 50 %igen Schwefelsäure (3.2. b) destilliertem oder entionisiertem Wasser (I.3.2.3 Allgemeiner Teil) und der Ammoniaklösung (3.2. c) und schließlich gründlich mit destilliertem oder entionisiertem Wasser auswaschen, wobei der Tiegel durch Absaugen nach jeder Zugabe vollständig entleert wird (während des Waschvorgangs ist nicht abzusaugen, sondern erst, nachdem die Flüssigkeit durch ihr Eigengewicht abgelaufen ist). Den Tiegel und den Rückstand trocknen, abkühlen und wiegen.
5. BERECHNUNG UND ERGEBNISDARSTELLUNG
Die Ergebnisse werden nach dem im allgemeinen Teil angegebenen Berechnungsverfahren ermittelt. Der Korrekturfaktor „d“ beträgt 1,00; nur für Melamin und Polypropylen/Polyamid-Bikomponentenfasern beträgt er 1,01.
6. GENAUIGKEIT DES VERFAHRENS
Bei homogenen Textilgemischen liegt das Konfidenzintervall der Ergebnisse dieses Verfahrens bei höchstens ± 1, wobei das Konfidenzniveau 95 % beträgt.
METHODE Nr. 15
POLYCHLORIDFASERN, BESTIMMTE MODACRYLE, BESTIMMTE ELASTHANE, ACETAT, TRIACETAT UND BESTIMMTE ANDERE FASERN
(Cyclohexanonverfahren)
1. ANWENDUNGSBEREICH
Dieses Verfahren gilt nach Entfernung der Nichtfaserstoffe für binäre Fasergemische von
1. Acetat (19), Triacetat (24), Polychlorid (27), bestimmten Modacrylen (29) oder bestimmten Elasthanen (43)
mit
2. Wolle (1), Tierhaaren (2 und 3), Seide (4), Baumwolle (5), Cupro (21), Modal (22), Viskose (25), Polyacrylfasern (26), Polyamid oder Nylon (30), Glasfasern (44), Melamin (47) und Polyacrylat (50).
Sind Modacryl- oder Elasthanfasern vorhanden, so ist ein Vorversuch notwendig, um festzustellen, ob die Fasern in dem Reagenz vollständig löslich sind.
Zur Bestimmung von Gemischen mit Polychloridfasern sind auch die Methoden Nr. 9 oder 14 anwendbar.
2. PRINZIP
Ausgehend von einer bekannten Trockenmasse des Gemischs werden Fasern von Acetat, Triacetat, Polychlorid, bestimmten Modacrylen und bestimmten Elasthanen mit Cyclohexanon bei annähernder Siedetemperatur aufgelöst. Der Rückstand wird gesammelt, gewaschen, getrocknet und gewogen; seine - erforderlichenfalls berichtigte - Masse wird in Prozentsätzen der Trockenmasse der Mischung ausgedrückt. Der Anteil an Polychlorid-, Modacryl-, Elasthan-, Acetat- und Triacetatfasern in Prozent wird durch Differenzbildung ermittelt.
3. GERÄTE UND REAGENZIEN (neben den im allgemeinen Teil genannten)
3.1. Geräte
a) Heißextraktionsgerät, mit dem nach dem in Nummer 4 beschriebenen Verfahren gearbeitet werden kann. (siehe Abbildung, die eine Variante des in „Melliand Textilberichte“ 56 (1975), S. 643-645, beschriebenen Geräts darstellt.)
b) Filtertiegel zur Aufnahme der Analyseprobe;
c) poröse Scheidewand, Porosität 1;
d) für den Destillationskolben geeigneter Rückflusskühler;
e) Heizgerät.
3.2. Reagenzien
a) Cyclohexanon, Siedepunkt 156 °C;
b) Ethanol, verdünnt auf 50 Volumenprozent.
Cyclohexanon ist brennbar und toxisch. Beim Gebrauch sind entsprechende Schutzvorkehrungen zu treffen.
4. ANALYSENGANG
Es ist nach den im allgemeinen Teil angegebenen Anweisungen zu arbeiten und folgendermaßen vorzugehen:
100 ml Cyclohexanon je Gramm Probe in den Destillationskolben geben, das Extraktionsgefäß ein- bzw. aufsetzen und den Filtertiegel mit der Probe einführen. Auf den Filtertiegel die leicht geneigte poröse Trennwand legen, danach den Rückflusskühler aufsetzen. Das Cyclohexanon bis zum Siedepunkt erwärmen, 60 Minuten bei einer Mindestgeschwindigkeit von etwa 12 Zyklen pro Stunde extrahieren.
Nach Extraktion und Abkühlung den Filtertiegel aus dem Extraktionsgefäß nehmen und die poröse Scheidewand entfernen. Den Inhalt des Filtertiegels 3- bis 4-mal mit auf etwa 60 °C erwärmtem 50 %igem Ethanol und anschließend mit 1 Liter Wasser bei 60 °C waschen.
Während und zwischen den Waschvorgängen zunächst keinen Unterdruck anwenden, sondern die Flüssigkeit normal ablaufen lassen und erst dann absaugen.
Zum Schluss wird der Tiegel mit dem Rückstand getrocknet, abgekühlt und gewogen.
5. BERECHNUNG UND ERGEBNISDARSTELLUNG
Die Ergebnisse werden nach dem im allgemeinen Teil angegebenen Berechnungsverfahren ermittelt. Der Korrekturfaktor „d“ hat den Wert 1,00, jedoch bei Polyacrylat den Wert 1,02, bei Seide und Melamin den Wert 1,01 und bei Polyacrylfasern den Wert 0,98.
6. GENAUIGKEIT DES VERFAHRENS
Bei homogenen Textilgemischen liegt das Konfidenzintervall der Ergebnisse dieses Verfahrens bei höchstens ± 1, wobei das Konfidenzniveau 95 % beträgt.

METHODE Nr. 16
MELAMIN UND BESTIMMTE ANDERE FASERN
(Verfahren mit heißer Ameisensäure)
1. ANWENDUNGSBEREICH
Dieses Verfahren gilt nach Entfernung der Nichtfaserstoffe für binäre Fasergemische von
1. Melamin (47)
mit
2. Baumwolle (5), Aramid (31) und Polypropylen (37).
2. PRINZIP
Das Melamin wird mittels erhitzter Ameisensäure (90 % m/m) aus einer bekannten Trockenmasse herausgelöst.
Der Rückstand wird gesammelt, gewaschen, getrocknet und gewogen; seine — erforderlichenfalls berichtigte — Masse wird in Prozentsätzen der Trockenmasse der Mischung ausgedrückt. Der prozentuale Anteil der zweiten Bestandteile wird aus der Gewichtsdifferenz berechnet.
Der empfohlene Temperaturbereich ist unbedingt einzuhalten, weil die Löslichkeit von Melamin äußerst temperaturabhängig ist.
3. GERÄTE UND REAGENZIEN (neben den im allgemeinen Teil genannten)
3.1. Geräte
a) Mindestens 200 ml fassender Erlenmeyerkolben mit Glasschliffstopfen;
b) Wasserbadschüttler oder anderes Gerät zum Schütteln und zur Erwärmung des Kolbens auf konstante 90 ± 2 °C.
3.2. Reagenzien
a) Ameisensäure zu 90 Gewichtsprozent, relative Dichte bei 20 °C: 1,204. 890 ml Ameisensäure zu 98 bis 100 Gewichtsprozent (relative Dichte bei 20 °C: 1,220) werden mit Wasser auf 1 Liter aufgefüllt.
Heiße Ameisensäure ist äußerst korrosiv, bei ihrer Verwendung ist daher Vorsicht geboten.
b) Verdünntes Ammoniak: 80 ml konzentriertes Ammoniak (relative Dichte bei 20 °C: 0,880) werden mit Wasser auf 1 Liter aufgefüllt.
4. ANALYSENGANG
Es ist der im allgemeinen Teil beschriebene Analysengang zu befolgen und folgendermaßen vorzugehen:
Die in einem mindestens 200 ml fassender Erlenmeyerkolben mit Glasschliffstopfen befindliche Analyseprobe wird mit 100 ml Ameisensäure je Gramm Probe versetzt. Der Kolben wird verschlossen und geschüttelt, um die Probe vollständig zu benetzen. Der Kolben wird so auf dem Schüttler befestigt, dass er in ein Wasserbad von 90 °C ± 2 °C taucht, wo er eine Stunde lang kräftig geschüttelt wird. Nach Abkühlung des Kolbens auf Raumtemperatur wird die Flüssigkeit über den tarierten Glasfiltertiegel dekantiert. Man gibt 50 ml Ameisensäure in den Kolben mit dem Rückstand, schüttelt von Hand und filtriert den Inhalt des Kolbens über den Glasfiltertiegel. Etwa zurückbleibende Fasern werden durch Auswaschen des Kolbens mit etwas Ameisensäurelösung in den Tiegel überführt. Den Tiegel durch Absaugen entleeren, dann den Rückstand mit Ameisensäurelösung, heißem Wasser, verdünntem Ammoniak und schließlich kaltem Wasser auswaschen, wobei der Tiegel nach jeder Flüssigkeitszugabe durch Absaugen vollständig entleert wird. jedoch erst, nachdem die Flüssigkeit ohne Absaugen durchgelaufen ist. Zum Schluss wird der Tiegel durch Absaugen geleert, zusammen mit dem Rückstand getrocknet, gekühlt und gewogen.
5. BERECHNUNG UND ERGEBNISDARSTELLUNG
Die Ergebnisse werden nach dem im allgemeinen Teil angegebenen Berechnungsverfahren ermittelt. Der Korrekturfaktor „d“ beträgt 1,02.
6. GENAUIGKEIT DES VERFAHRENS
Bei homogenen Textilgemischen liegt das Konfidenzintervall der Ergebnisse dieses Verfahrens bei höchstens ± 2, wobei das Konfidenzniveau 95 % beträgt.
METHODE Nr. 17
POLYESTER UND BESTIMMTE ANDERE FASERN
(Verfahren mit Trichloressigsäure und Chloroform)
1. ANWENDUNGSBEREICH
Dieses Verfahren gilt nach Entfernung der Nichtfaserstoffe für binäre Fasergemische von
1. Polyester (35)
mit
2. Polyacrylat (50)
2. ALLGEMEINE INFORMATIONEN
Das Prinzip, die Geräte und Reagenzien, der Analysengang, die Berechnung und Ergebnisdarstellung, die auf binäre Fasergemische aus Polyester und Polyacrylat anzuwenden sind, entsprechen den in der Norm EN ISO 1833-25:2013 beschriebenen. Der Korrekturfaktor „d“ beträgt 1,01.
KAPITEL 3
QUANTITATIVE ANALYSE TERNÄRER TEXTILFASERGEMISCHE
EINLEITUNG
In der Regel beruht das Verfahren der quantitativen chemischen Analyse auf der selektiven Auflösbarkeit der Einzelbestandteile. Für dieses Verfahren ergeben sich vier mögliche Varianten:
1. Es wird mit zwei verschiedenen Analyseproben gearbeitet, wobei aus der ersten Probe ein Bestandteil (a) und aus der zweiten Probe ein weiterer Bestandteil (b) herausgelöst werden. Die unlöslichen Rückstände jeder Probe werden gewogen und aus dem Massenverlust wird der Prozentsatz jedes der beiden löslichen Bestandteile errechnet. Der Prozentsatz des dritten Bestandteils (c) wird durch Differenzbildung ermittelt.
2. Es wird mit zwei verschiedenen Analyseproben gearbeitet, wobei aus der ersten Probe ein Bestandteil (a) und aus der zweiten Probe zwei Bestandteile (a und b) herausgelöst werden. Der unlösliche Rückstand der ersten Analyseprobe wird gewogen und aus dem Massenverlust wird der Prozentsatz des Bestandteils (a) errechnet. Der unlösliche Rückstand der zweiten Analyseprobe wird gewogen; er entspricht dem Bestandteil (c). Der Prozentsatz des dritten Bestandteils (b) wird durch Differenzbildung ermittelt.
3. Es wird mit zwei verschiedenen Analyseproben gearbeitet, wobei aus der ersten Probe zwei Bestandteile (a und b) und aus der zweiten Probe zwei Bestandteile (b und c) herausgelöst werden. Die unlöslichen Rückstände entsprechen jeweils den beiden Bestandteilen (c) und (a). Der Prozentsatz des dritten Bestandteils (b) wird durch Differenzbildung ermittelt.
4. Es wird mit nur einer Analyseprobe gearbeitet, wobei nach Entfernung eines der Bestandteile der von den beiden anderen Fasern gebildete, unlösliche Rückstand gewogen und aus dem Massenverlust der Prozentsatz des löslichen Bestandteils errechnet wird. Aus dem Rückstand wird eine der beiden Fasern herausgelöst, der unlösliche Bestandteil wird gewogen und der Prozentsatz des zweiten löslichen Bestandteils wird aus dem Massenverlust errechnet.
Besteht eine Wahlmöglichkeit, wird zur Verwendung einer der drei ersten Varianten geraten.
Der mit der Analyse beschäftigte Sachverständige muss jedoch darauf achten, dass bei der chemischen Analyse Methoden gewählt werden, die Lösungsmittel vorschreiben, die nur eine bestimmte Faser bzw. bestimmte Fasern auflösen, die andere bzw. anderen dagegen nicht auflösen.
Als Beispiele sind in Abschnitt V in einer Tabelle eine Reihe von ternären Fasergemischen aufgeführt sowie Analysenmethoden für binäre Fasergemische, die grundsätzlich auch für die Analyse dieser ternären Fasergemische verwendet werden können.
Um die Fehleranfälligkeit zu minimieren, wird empfohlen, die chemische Analyse, soweit möglich, anhand von wenigstens zwei der vier oben beschriebenen Varianten vorzunehmen.
Die in dem Gemisch enthaltenen Fasern sind vor der Analyse zu identifizieren. Bei bestimmten chemischen Methoden kann der unlösliche Bestandteil von Gemischen teilweise in dem Reagenz aufgelöst werden, das zur Auflösung des löslichen Bestandteils bzw. der löslichen Bestandteile verwendet wird. Nach Möglichkeit wurden die Reagenzien so gewählt, dass sie nur einen geringen oder überhaupt keinen Einfluss auf die unlöslichen Fasern haben. Ist bei der Analyse mit einem Massenverlust zu rechnen, so müssen die Ergebnisse entsprechend korrigiert werden. Korrekturfaktoren hierfür sind angegeben. Diese wurden in mehreren Laboratorien dadurch bestimmt, dass durch Vorbehandlung gereinigte Fasern mit dem entsprechenden Reagenz unter Befolgung der Analysenmethode behandelt wurden. Sie gelten nur für normale Fasern. Weitere Korrekturfaktoren können erforderlich sein, wenn die Fasern vor oder während der Verarbeitung nicht intakt geblieben sind. Wenn die vierte Variante angewandt werden muss, bei der eine Textilfaser der aufeinanderfolgenden Einwirkung von zwei Lösungsmitteln ausgesetzt ist, so ist es notwendig, die etwaigen Massenverluste auf Grund dieser doppelten Behandlung durch Korrekturfaktoren zu berücksichtigen. Sowohl beim manuellen Trennungsverfahren als auch beim chemischen Trennungsverfahren müssen mindestens Doppelbestimmungen durchgeführt werden.
I. Allgemeines über die Methoden der quantitativen chemischen Analyse von ternären Textilfasergemischen
Allgemeine Angaben zu den Verfahren der quantitativen chemischen Analyse von ternären Textilfasergemischen.
I.1. ANWENDUNGSBEREICH
Unter dem Anwendungsbereich wird bei jeder Analysenmethode für binäre Fasergemische aufgeführt, für welche Fasern sie anzuwenden ist. (siehe Kapitel 2 betreffend Methoden der quantitativen Analyse bestimmter binärer Textilfasergemische.)
I.2. PRINZIP
Nach Identifizierung der einzelnen Bestandteile der Fasergemische werden zunächst durch eine entsprechende Vorbehandlung die nichtfaserigen Bestandteile entfernt, sodann werden eine oder mehrere der in der Einleitung beschriebenen vier Varianten des Verfahrens der selektiven Auflösung angewendet. Vorzugsweise sind, sofern keine technischen Schwierigkeiten auftreten, die in größerer Menge vorhandenen Fasern aufzulösen, damit man die in geringster Menge vorhandene Faser als Endrückstand erhält.
I.3. GERÄTE UND REAGENZIEN
I.3.1. Geräte
|
I.3.1.1. |
Filtertiegel und Wägegläser zum Einsetzen von Tiegeln oder andere gleichwertige Geräte |
|
I.3.1.2. |
Absaugflasche |
|
I.3.1.3. |
Exsikkator mit gefärbtem Kieselgel als Feuchtigkeitsindikator |
|
I.3.1.4. |
Trockenofen mit Ventilator zur Trocknung der Analyseproben bei 105 ± 3 °C |
|
I.3.1.5. |
Analysenwaage, Empfindlichkeit 0,0002 g |
|
I.3.1.6. |
Soxhlet-Extraktionsapparat oder gleichwertige Apparatur |
I.3.2. Reagenzien
|
I.3.2.1. |
Petrolether, nachdestilliert, Siedebereich 40 bis 60 °C |
|
I.3.2.2. |
Sonstige Reagenzien sind in den entsprechenden Teilen der Methode angegeben. |
|
I.3.2.3. |
Destilliertes oder entionisiertes Wasser |
|
I.3.2.4. |
Aceton. |
|
I.3.2.5. |
Orthophosphorsäure |
|
I.3.2.6. |
Harnstoff |
|
I.3.2.7. |
Natriumbicarbonat |
Alle Reagenzien müssen chemisch rein sein.
I.4. KONDITIONIERUNGS- UND ANALYSEATMOSPHÄRE
Da die Trockenmasse bestimmt wird, ist weder eine Konditionierung der Probe noch eine Untersuchung in klimatisierter Atmosphäre erforderlich.
I.5. VORPROBE
Es wird eine für die Laboratoriumsprobe repräsentative Vorprobe gewählt, die für sämtliche erforderlichen Analyseproben von jeweils mindestens 1 g ausreicht.
I.6. VORBEHANDLUNG DER VORPROBE ( 15 )
Ist einer der bei der Berechnung der Prozentsätze nicht zu berücksichtigenden Bestandteile vorhanden (siehe Artikel 19), so ist dieser zunächst durch eine geeignete Methode zu entfernen, die jedoch keinen der Faserbestandteile angreifen darf.
Zu diesem Zweck werden die mit Hilfe von Petrolether und Wasser extrahierbaren nichtfaserigen Bestandteile entfernt, indem die Vorprobe im Soxhlet-Apparat mit Petrolether während einer Stunde und mit mindestens sechs Umläufen pro Stunde behandelt wird. Anschließend wird der Petrolether der Vorprobe verdampft; danach wird die Vorprobe durch Direktbehandlung extrahiert, das heißt durch einstündiges Eintauchen in Wasser bei Zimmertemperatur mit darauf folgendem einstündigen Eintauchen in Wasser bei 65 °C ± 5 °C unter zeitweiligem Schütteln, Flottenverhältnis 1:100. Danach wird das überschüssige Wasser durch Ausquetschen, Absaugen oder Zentrifugieren entfernt, bis die Vorprobe lufttrocken ist.
Bei Elastolefin oder Fasergemischen, die Elastolefin und andere Fasern enthalten (Wolle, Tierhaare, Seide, Baumwolle, Flachs bzw. Leinen, Hanf, Jute, Manila, Alfa, Kokos, Ginster, Ramie, Sisal, Cupro, Modal, regenerierte Proteinfasern, Viskose, Polyacryl, Polyamid oder Nylon, Polyester oder Elastomultiester), ist das oben beschriebene Verfahren dahingehend leicht abzuändern, dass Petrolether durch Aceton ersetzt wird.
Falls die nichtfaserigen Bestandteile nicht mit Hilfe von Petrolether und Wasser extrahiert werden können, so müssen sie anstatt mit Wasser, wie oben beschrieben, mit einem geeigneten Stoff entfernt werden, der keinen der Faserbestandteile wesentlich verändert. Bei einigen ungebleichten natürlichen Pflanzenfasern (wie zum Beispiel Jute- oder Kokosfasern) ist zu beachten, dass durch die normale Vorbehandlung mit Petrolether und Wasser nicht alle natürlichen nichtfaserigen Bestandteile beseitigt werden. Trotzdem werden keine weiteren Vorbehandlungen vorgenommen, soweit die Probe keine in Petrolether und in Wasser unlöslichen Appreturen enthält.
In den Analysenberichten müssen die gewählten Vorbehandlungsmethoden eingehend geschildert werden.
I.7. ANALYSENGANG
I.7.1. Allgemeine Anweisungen
I.7.1.1. Trocknung
Alle Trockenoperationen sind mindestens 4 Stunden, jedoch nicht mehr als 16 Stunden lang bei 105 ± 3 °C in einem belüfteten Ofen bei geschlossener Ofentür durchzuführen. Beträgt die Trocknungsdauer weniger als 14 Stunden, muss überprüft werden, ob eine konstante Masse erreicht wurde. Diese kann als erreicht gelten, wenn der Massenunterschied nach einer neuen Trocknung von 60 Minuten weniger als 0,05 % beträgt
Die Filtertiegel und Wägegläser sowie die Proben oder die Rückstände sollen während des Trocknungs-, Abkühlungs- und Wägevorgangs nicht mit bloßen Händen berührt werden.
Die Analyseproben werden in einem Wägeglas mit abgenommenem Stopfen getrocknet. Nach der Trocknung wird das Wägeglas vor Herausnahme aus dem Ofen geschlossen und so schnell wie möglich in den Exsikkator gebracht.
Der Filtertiegel wird in einem Wägeglas zusammen mit dessen abgenommenen Stopfen im Ofen getrocknet. Nach der Trocknung wird das Wägeglas verschlossen und so schnell wie möglich in den Exsikkator gestellt.
Wird ein anderes Gerät als der Filtertiegel verwendet, so wird im Trockenofen getrocknet, um die Trockenmasse der Fasern ohne Verlust zu bestimmen.
I.7.1.2. Kühlung
Alle Kühlvorgänge werden in dem neben der Waage aufgestellten Exsikkator ausreichend lange durchgeführt, um ein völliges Abkühlen der Wägegläser zu erreichen, wobei die Abkühldauer mindestens 2 Stunden beträgt.
I.7.1.3. Wägung
Nach dem Abkühlen wird das Wägeglas innerhalb von zwei Minuten nach Herausnahme aus dem Exsikkator gewogen; Wägegenauigkeit 0,0002 g.
I.7.2. Verfahren
Aus der vorbehandelten Vorprobe wird eine Analyseprobe von mindestens 1 g Masse entnommen. Das Garn oder die Gewebe werden in Längen von etwa 10 mm ausgeschnitten und soweit wie möglich zerlegt (zerschnitten). Die Analyseprobe wird in einem Wägeglas getrocknet, im Exsikkator gekühlt und gewogen. Die Probe wird in ein Glasgefäß gegeben, das im entsprechenden Teil der Unionsmethode beschrieben ist, anschließend wird das Wägeglas sofort wieder gewogen und die Trockenmasse der Probe durch Differenzbildung ermittelt. Die Analyse wird gemäß den Angaben in dem entsprechenden Teil des Verfahrens zu Ende geführt. Der Rückstand wird mikroskopisch geprüft, um festzustellen, ob durch die Behandlung die lösliche Faser völlig ausgesondert worden ist.
I.8. BERECHNUNG UND ERGEBNISDARSTELLUNG
Die Masse jedes Bestandteils wird als Prozentsatz der Gesamtmasse der im Gemisch enthaltenen Fasern ausgedrückt. Die Ergebnisberechnung erfolgt auf der Basis der Trockenmasse der reinen Fasern unter Anwendung a) der vereinbarten Zuschläge sowie b) von Korrekturfaktoren zur Berücksichtigung der Verluste nichtfaseriger Bestandteile während Vorbehandlung und Analyse.
|
I.8.1. |
Berechnung des prozentualen Massenanteils der reinen trockenen Fasern ohne Berücksichtigung des Massenverlusts der Fasern durch die Vorbehandlung I.8.1.1. VARIANTE 1 Formeln, die dann anzuwenden sind, wenn eine Komponente des Gemischs aus einer Probe und eine andere Komponente aus einer zweiten Probe herausgelöst werden:
P 3% = 100 – (P 1% + P 2%)
I.8.1.2. VARIANTE 2 Formeln, die anzuwenden sind, wenn aus der ersten Analyseprobe eine Komponente (a) mit den beiden anderen Komponenten (b + c) als Rückstand und anschließend zwei Komponenten (a + b) mit der dritten Komponente (c) als Rückstand herausgelöst werden. P 1% = 100 – (P 2% + P 3%)
I.8.1.3. VARIANTE 3 Formeln, die dann anzuwenden sind, wenn zwei Komponenten (a + b) einer Probe mit der dritten Komponente (c) als Rückstand und anschließend zwei Komponenten (b + c) mit der ersten Komponente (a) als Rückstand herausgelöst werden:
P 2% = 100 – (P 1% + P 3%)
I.8.1.4. VARIANTE 4 Formeln, die anzuwenden sind, wenn nacheinander zwei Komponenten des Fasergemischs aus der gleichen Probe herausgelöst werden: P 1% = 100 – (P 2% + P 3%)
|
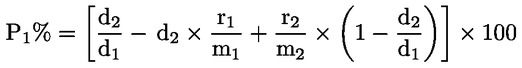



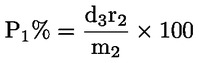


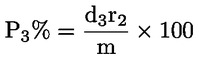
|
I.8.2. |
Berechnung des Prozentsatzes jeder einzelnen Komponente nach Anwendung der vereinbarten Zuschläge und etwaiger Korrekturfaktoren zur Berücksichtigung des Massenverlustes durch die Vorbehandlung: Ist
dann ist
Bei Anwendung einer Spezialvorbehandlung müssen die Größen b1, b2 und b3 nach Möglichkeit dadurch bestimmt werden, dass alle reinen Faserbestandteile der bei der Analyse angewandten Vorbehandlung unterworfen werden. Als reine Fasern gelten die Fasern, die frei von allen nichtfaserhaltigen Stoffen sind, mit Ausnahme derjenigen Stoffe, die sie normalerweise (auf Grund ihrer Beschaffenheit oder des Herstellungsprozesses) in dem Zustand (roh, gebleicht) enthalten, in dem sie sich in der zu analysierenden Ware vorfinden. Sind keine getrennten und reinen Faserbestandteile vorhanden, die zur Herstellung der zu analysierenden Ware gedient haben, so sind für b1, b2 und b3 Durchschnittswerte anzunehmen, die sich aus Prüfungen von ähnlichen wie in der untersuchten Mischung enthaltenen reinen Fasern ergeben. Wird die normale Vorbehandlung durch Extraktion mit Petrolether und mit Wasser durchgeführt, so kann man im allgemeinen auf die Korrekturfaktoren b1, b2 und b3 verzichten, außer im Fall von ungebleichter Baumwolle, ungebleichtem Flachs bzw. ungebleichtem Leinen und ungebleichtem Hanf, bei denen vereinbarungsgemäß ein durch die Vorbehandlung bedingter Verlust von 4 %, bei Polypropylen von 1 %, zugestanden wird. Im Fall anderer Fasern bleibt der Verlust vereinbarungsgemäß für die Berechnung unberücksichtigt. |
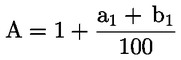
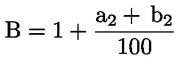


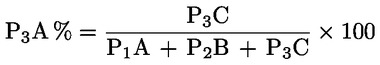
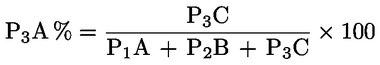
|
I.8.3. |
Anmerkung Berechnungsbeispiele finden sich in Abschnitt IV. |
II. Verfahren der quantitativen Analyse von ternären Textilfasergemischen durch manuelle Trennung
II.1. ANWENDUNGSBEREICH
Die Methode lässt sich auf Fasergemische beliebiger Beschaffenheit anwenden, vorausgesetzt, dass sie kein untrennbares Gemisch darstellen und dass sie sich manuell trennen lassen.
II.2. PRINZIP
Nach Identifizierung der einzelnen Bestandteile der Fasergemische werden zunächst die nichtfaserhaltigen Bestandteile durch eine geeignete Vorbehandlung ausgesondert, anschließend die Fasern von Hand getrennt, getrocknet und zwecks Berechnung des Anteils der einzelnen Faserarten am Gemisch gewogen.
II.3. GERÄTE
|
II.3.1. |
Wägeglas bzw. andere Geräte, die gleichartige Ergebnisse liefern |
|
II.3.2. |
Exsikkator mit gefärbtem Kieselgel als Feuchtigkeitsindikator |
|
II.3.3. |
Trockenofen mit Ventilator zur Trocknung der Analyseproben bei 105 ± 3 °C |
|
II.3.4. |
Analysewaage, Empfindlichkeit 0,0002 g |
|
II.3.5. |
Soxhlet-Extraktionsapparat oder gleichwertige Apparatur |
|
II.3.6. |
Nadel |
|
II.3.7. |
Garndrehungszähler oder gleichwertige Apparatur |
II.4. REAGENZIEN
|
II.4.1. |
Petrolether, nachdestilliert, Siedebereich 40 bis 60 °C |
|
II.4.2. |
Destilliertes oder entionisiertes Wasser |
II.5. KONDITIONIERUNGS- UND ANALYSEATMOSPHÄRE
Siehe Nummer I.4.
II.6. VORPROBE
Siehe Nummer I.5.
II.7. VORBEHANDLUNG DER VORPROBE
Siehe Nummer I.6.
II.8. VERFAHREN
II.8.1. Analyse von Garnen
Eine Analyseprobe mit einer Masse von mindestens 1 g wird aus einer vorbehandelten Vorprobe entnommen. Bei sehr feinen Garnen kann die Analyse ungeachtet der Masse auf einer Mindestlänge von 30 m durchgeführt werden.
Die Garne sind in Stücke von geeigneter Länge zu schneiden; aus diesen sind mit Hilfe einer Präpariernadel und, falls erforderlich, mit Hilfe des Garndrehungszählers die einzelnen Elemente herauszutrennen. Die auf diese Weise herausgetrennten Elemente werden dann in ein tariertes Wägeglas gegeben und bei 105 ± 3 °C getrocknet, bis eine konstante Masse gemäß Nummer I.7.1 und I.7.2 erreicht ist.
II.8.2. Analyse eines Gewebes
Eine Analyseprobe von mindestens 1 g wird aus einer vorbehandelten Probe entnommen; die Analyseprobe wird so ausgeschnitten, dass sie außerhalb der Webkante liegt, exakt geschnittene Ränder ohne Kräuselung aufweist und parallel zu Schuss und Kette bzw. bei Gewirken gleichlaufend längs und quer zu den Maschenreihen geschnitten ist. Die einzelnen Elemente werden getrennt und in tarierten Wägegläsern gesammelt; dann wird wie unter Nummer II.8.1 vorgegangen.
II.9. BERECHNUNG UND ERGEBNISDARSTELLUNG
Die Masse jedes Bestandteils wird als Prozentsatz der Gesamtmasse der im Gemisch enthaltenen Fasern ausgedrückt. Die Berechnung erfolgt auf der Basis des Trockengewichts der reinen Fasern unter Anwendung a) der vereinbarten Zuschläge und b) der erforderlichen Korrekturfaktoren zur Berücksichtigung der während der Vorbehandlung aufgetretenen Massenverluste.
|
II.9.1. |
Berechnung des Prozentsatzes der reinen Trockenmasse ohne Berücksichtigung des Massenverlustes der Fasern durch die Vorbehandlung:
P 3% = 100 – (P 1% + P 2%)
|
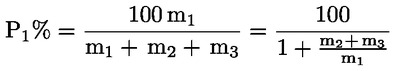
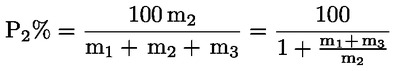
|
II.9.2. |
Berechnung des Prozentsatzes jeder einzelnen Komponente nach Anwendung der vereinbarten Zuschläge und etwaiger Korrekturfaktoren zur Berücksichtigung des Gewichtsverlustes durch die Vorbehandlung: siehe Nummer I.8.2. |
III. Verfahren der quantitativen Analyse von ternären Fasergemischen mit kombinierter manueller und chemischer Trennung
Die Trennung muss, soweit möglich, manuell vorgenommen werden. Hierbei ist der Prozentsatz der getrennten Teile zu berücksichtigen, bevor von den einzelnen Teilen eine Analyse auf chemischem Wege vorgenommen wird.
III.1. GENAUIGKEIT DER VERFAHREN
Die für jedes Verfahren der Analyse binärer Fasergemische angegebene Genauigkeit bezieht sich auf die Reproduzierbarkeit (siehe Kapitel 2 betreffend Methoden der quantitativen Analyse bestimmter binärer Textilfasergemische).
Die Reproduzierbarkeit ist der Zuverlässigkeitsgrad, d. h. der Grad der Übereinstimmung zwischen den Versuchsergebnissen, wenn bei diesen Versuchen in verschiedenen Laboratorien oder zu verschiedenen Zeitpunkten nach demselben Verfahren und an Proben aus demselben homogenen Prüfgut jeweils unterschiedliche Ergebnisse erzielt werden.
Die Reproduzierbarkeit wird durch das Konfidenzintervall der Versuchsergebnisse bei einem Konfidenzniveau von 95 % ausgedrückt.
Dies besagt, dass die Abweichung zwischen zwei Ergebnissen einer in verschiedenen Laboratorien durchgeführten Analysenreihe bei richtiger und normaler Anwendung des Verfahrens auf eine gleichartige homogene Mischung nur in fünf von hundert Fällen das Konfidenzintervall überschreiten darf.
Um die Genauigkeit der Ergebnisse der Analyse eines ternären Fasergemischs zu bestimmen, sind in der Regel in der üblichen Weise diejenigen Werte zugrunde zulegen, die bei den Methoden für binäre Fasergemische angegeben sind, welche für die Analyse des ternären Fasergemischs benutzt wurden.
Da für die vier Varianten der quantitativen chemischen Analyse von ternären Fasergemischen jeweils die Auflösung von zwei Komponenten vorgesehen ist (aus zwei verschiedenen Proben bei den ersten drei Varianten und aus derselben Probe bei der vierten Variante), berechnet sich, wenn man die Genauigkeit der beiden benutzten Methoden für binäre Fasergemische mit E1 und E2 bezeichnet, die Genauigkeit der Ergebnisse für jede Komponente nach folgender Tabelle:
|
Komponente des Gemischs |
Varianten |
||
|
1 |
2 und 3 |
4 |
|
|
a |
E1 |
E1 |
E1 |
|
b |
E2 |
E1 + E2 |
E1 + E2 |
|
c |
E1 + E2 |
E2 |
E1 + E2 |
Bei Anwendung der vierten Variante kann sich auf Grund einer eventuellen, schwer bestimmbaren Wirkung des ersten Reagenz auf den aus den Komponenten b und c bestehenden Probenrückstand eine geringere Genauigkeit ergeben, als nach dem obigen Verfahren berechnet.
III.2. ANALYSENBERICHT
|
III.2.1. |
Angabe der zur Analyse verwendeten Varianten sowie der verwendeten Verfahren, Reagenzien und Korrekturfaktoren. |
|
III.2.2. |
Detaillierte Angaben über etwaige Spezialvorbehandlungen (siehe Nummer I.6). |
|
III.2.3. |
Angabe der Einzelergebnisse sowie des arithmetischen Mittels auf eine Dezimalstelle genau. |
|
III.2.4. |
Nach Möglichkeit sollte die Genauigkeit der Methode für jede Komponente angegeben werden, berechnet nach der Tabelle in Abschnitt III.1. |
IV. Beispiele für die Berechnung der prozentualen Anteile der Komponenten bestimmter ternärer Fasergemische unter Benutzung unter Nummer I.8.1 beschriebener Varianten
Gegeben sei der Fall eines Fasergemischs, bei dem die qualitative Analyse der Zusammensetzung des Rohmaterials folgende Bestandteile ergeben hat: 1. Wolle (Streichgarn); 2. Nylon (Polyamid); 3. ungebleichte Baumwolle.
VARIANTE 1
Arbeitet man mit dieser Variante, d. h. mit zwei verschiedenen Analyseproben, wobei ein Bestandteil (a = Wolle) aus der ersten Probe und ein zweiter Bestandteil (b = Polyamid) aus der zweiten Probe herausgelöst wird, so erhält man folgende Ergebnisse:
1. Trockenmasse der ersten Probe nach der Vorbehandlung (m1) = 1,6000 g.
2. Trockenmasse des Rückstands nach Behandlung mit alkalischem Natriumhypochlorit (Polyamid + Baumwolle) (r1) = 1,4166 g.
3. Trockenmasse der zweiten Probe nach der Vorbehandlung (m2) = 1,8000 g.
4. Trockenmasse des Rückstands nach Behandlung mit Ameisensäure (Wolle + Baumwolle) (r2) = 0,9000 g.
Die Behandlung mit alkalischem Natriumhypochlorit verursacht keinerlei Massenverlust bei Polyamid, während ungebleichte Baumwolle 3 % verliert, so dass d1 = 1,00 und d2 = 1,03 ist.
Die Behandlung mit Ameisensäure verursacht keinerlei Massenverlust bei Wolle und ungebleichter Baumwolle, so dass d3 und d4 = 1,00 ist.
Setzt man in der Formel unter Nummer I.8.1.1 die durch chemische Analyse erzielten Werte und die Korrekturfaktoren ein, so erhält man
P1% (Wolle) = [1,03/1,00 – 1,03 × 1,4166/1,6000 + (0,9000/1,8000) × (1 – 1,03/1,00)] ×100 = 10,30
P2% (Polyamid) = [1,00/1,00 – 1,00 × 0,9000/1,8000 + (1,4166/1,6000) × (1 – 1,00/1,00)] ×100 = 50,00
P3% (Baumwolle) = 100 – (10,30 + 50,00) = 39,70
Die prozentualen Anteile der verschiedenen getrockneten und gereinigten Fasern des Gemischs sind folgende:
|
Wolle |
10,30 % |
|
Polyamid |
50,00 % |
|
Baumwolle |
39,70 % |
Diese Prozentsätze müssen nach den Formeln unter Nummer I.8.2 korrigiert werden, um auch die vereinbarten Zuschläge sowie die Korrekturfaktoren für die nach der Vorbehandlung etwa eingetretenen Massenverluste zu berücksichtigen.
Nach Anhang IX sind die vereinbarten Zuschläge folgende: Wolle (Streichgarn) 17,00 %, Polyamid 6,25 %, Baumwolle 8,50 %. Außerdem erfährt ungebleichte Baumwolle einen Massenverlust von 4 % nach Vorbehandlung durch Petrolether und Wasser.
Man erhält infolgedessen
P1A% (Wolle) = 10,30 × [1+(17,00+0,0)/100] / [10,30 × (1 + (17,00+0,0)/100) + 50,00 × (1 + (6,25+0,0)/100) + 39,70 × (1+(8,50+4,0)/100)] × 100 = 10,97
P2A% (Polyamid) = 50,0 × [(1+ (6,25+0,0)/100)/109,8385] × 100 = 48,37
P3A% (Baumwolle) = 100 – (10,97 + 48,37) = 40,66
Die Rohstoffzusammensetzung des Garns ist infolgedessen
|
Polyamid |
48,4 % |
|
Baumwolle |
40,6 % |
|
Wolle |
11,0 % |
|
|
100,0 % |
VARIANTE 4
Gegeben sei der Fall eines Fasergemischs, dessen qualitative Analyse folgende Bestandteile ergeben hat: Wolle (Streichgarn), Viskose, ungebleichte Baumwolle.
Angenommen, dass unter Benutzung der Variante 4, d. h. durch aufeinanderfolgendes Auflösen zweier Bestandteile des Gemischs derselben Analyseprobe, folgende Ergebnisse erhalten wurden:
1. Trockenmasse der Probe nach der Vorbehandlung (m) = 1,6000 g.
2. Trockenmasse des Rückstands nach Behandlung mit alkalischem Natriumhypochlorit (Viskose + Baumwolle) (r1) = 1,4166 g.
3. Trockenmasse des Rückstands nach der zweiten Behandlung des Rückstands r1 mit Ameisensäure/Zinkchlorid (Baumwolle) (r2) = 0,6630 g.
Die Behandlung mit alkalischem Natriumhypochlorit verursacht keinerlei Massenverlust bei Viskose, während die ungebleichte Baumwolle 3 % verliert, so dass d1 = 1,00 und d2 = 1,03 ist.
Durch die Behandlung mit Ameisensäure/Zinkchlorid erhöht sich die Masse der Baumwolle um 4 %, so dass d3 = 1,03×0,96 = 0,9888 ≈ 0,99 ist (es wird daran erinnert, dass d3 der Korrekturfaktor ist, der den Verlust bzw. die Zunahme der Masse des dritten Bestandteils im ersten bzw. zweiten Reagenz berücksichtigt).
Setzt man in der Formel unter Nummer I.8.1.4. die durch chemische Analyse erzielten Werte und die Korrekturfaktoren ein, so erhält man
P2% (Viskose) = 1,00 × (1,4166/1,6000) × 100 – (1,00/1,03) × 41,02=48,71 %
P3% (Baumwolle) = 0,99 × (0,6630/1,6000) × 100= 41,02 %
P1% (Wolle) = 100 - (48,71 + 41,02) = 10,27 %
Wie bereits für Variante 1 angegeben, sind diese Prozentsätze nach den unter Nummer I.8.2 angegebenen Formeln zu korrigieren.
P1A% (Wolle) = 10,27 × [1 + (17,0+0,0)/100)]/[10,27 × (1+ (17,00+0,0)/100) +48,71 × (1+(13+0,0) / 100) + 41,02 × (1 + (8,5+4,0)/100)] × 100 = 10,61 %
P2A% (Viskose) = 48,71× [1 + (13+0,0)/100]/113,2057 × 100 = 48,62 %
P3A% (Baumwolle) = 100 – (10,61 + 48,62) = 40,77 %
Die Rohstoffzusammensetzung des Gemischs ist infolgedessen
|
Viskose |
48,6 % |
|
Baumwolle |
40,8 % |
|
Wolle |
10,6 % |
|
|
— |
|
|
100,0 % |
V. Tabelle mit typischen ternären Fasergemischen, die mit Hilfe der Analysemethoden der Union für binäre Fasergemische analysiert werden können (als Beispiel)
|
Gemisch Nr. |
Faserbestandteile |
Varianten |
Nr. der verwendeten Methode für binäre Fasergemische mit Angabe der Reagenzien |
||
|
Erster Bestandteil |
Zweiter Bestandteil |
Dritter Bestandteil |
|||
|
1. |
Wolle oder Tierhaare |
Viskose, Cupro oder bestimmte Modalarten |
Baumwolle |
1 und/oder 4 |
2 (Hypochlorit) und 3 (Zinkchlorid/Ameisensäure) |
|
2. |
Wolle oder Tierhaare |
Polyamid oder Nylon |
Baumwolle, Viskose, Cupro oder Modal |
1 und/oder 4 |
2 (Hypochlorit) und 4 (80 %ige Ameisensäure m/m) |
|
3. |
Wolle, Tierhaare oder Seide |
Bestimmte andere Fasern |
Viskose, Cupro, Modal oder Baumwolle |
1 und/oder 4 |
2 (Hypochlorit) und 9 (Schwefelkohlenstoff/Aceton 55,5/44,5% v/v) |
|
4. |
Wolle oder Tierhaare |
Polyamid oder Nylon |
Polyester, Polypropylen/Polyacrylfasern oder Glasfasern |
1 und/oder 4 |
2 (Hypochlorit) und 4 (80 %ige Ameisensäure m/m) |
|
5. |
Wolle, Tierhaare oder Seide |
Bestimmte andere Fasern |
Polyester, Polyacrylfasern, Polyamid oder Nylon oder Glasfasern |
1 und/oder 4 |
2 (Hypochlorit) und 9 (Schwefelkohlenstoff/Aceton 55,5/44,5 % v/v) |
|
6. |
Seide |
Wolle oder Tierhaare |
Polyester |
2 |
11 (75 %-ige Schwefelsäure m/m) und 2 (Hypochlorit) |
|
7. |
Polyamid oder Nylon |
Polyacrylfasern oder bestimmte andere Fasern |
Baumwolle, Viskose, Cupro oder Modal |
1 und/oder 4 |
4 (80 %-ige Ameisensäure m/m) und 8 (Dimethylformamid) |
|
8. |
Bestimmte Polychloridfasern |
Polyamid oder Nylon |
Baumwolle, Viskose, Cupro oder Modal |
1 und/oder 4 |
8 (Dimethylformamid) und 4 (80 %-ige Ameisensäure m/m) oder 9 (Schwefelkohlenstoff/Aceton 55,5/44,5 % v/v) und 4 (80 %-ige Ameisensäure m/m) |
|
9. |
Seide |
Polyamid oder Nylon |
Polyester |
1 und/oder 4 |
8 (Dimethylformamid) und 4 (80 %-ige Ameisensäure m/m) |
|
10. |
Acetat |
Polyamid oder Nylon oder bestimmte andere Fasern |
Viskose, Baumwolle, Cupro oder Modal |
4 |
1 (Aceton) und 4 (80 %-ige Ameisensäure m/m) |
|
11. |
Bestimmte Polychloridfasern |
Polyacrylfasern oder bestimmte andere Fasern |
Polyamid oder Nylon |
2 und/oder 4 |
9 (Schwefelkohlenstoff/Aceton 55,5/44,5 % v/v) und 8 (Dimethylformamid) |
|
12. |
Bestimmte Polychloridfasern |
Polyamid oder Nylon |
Seide |
1 und/oder 4 |
9 (Schwefelkohlenstoff/Aceton 55,5/44,5 % v/v und 4 (80 %-ige Ameisensäure m/m) |
|
13. |
Polyamid oder Nylon |
Viskose, Cupro, Modal oder Baumwolle |
Polyester |
4 |
4 (80 %-ige Ameisensäure m/m) und 7 (75 %-ige Schwefelsäure m/m) |
|
14. |
Acetat |
Viskose, Cupro, Modal oder Baumwolle |
Polyester |
4 |
1 (Aceton) und 7 (75 %-ige Schwefelsäure m/m) |
|
15. |
Seide |
Viskose, Cupro, Modal oder Baumwolle |
Polyester |
4 |
8. (Dimethylformamid) und 7 (75 %-ige Schwefelsäure m/m) |
|
16. |
Acetat |
Wolle, Tierhaare oder Seide |
Baumwolle, Viskose, Cupro, Modal, Polyamid oder Nylon, Polyester, Polyacrylfasern |
4 |
1 (Aceton) und 2 (Hypochlorit) |
|
17. |
Triacetat |
Wolle, Tierhaare oder Seide |
Baumwolle, Viskose, Cupro, Modal, Polyamid oder Nylon, Polyester, Polyacrylfasern |
4 |
6 (Dichlormethan) und 2 (Hypochlorit) |
|
18. |
Seide |
Wolle, Tierhaare oder Seide |
Polyester |
1 und/oder 4 |
8 (Dimethylformamid) und 2 (Hypochlorit) Polyacrylfasern |
|
19. |
Seide |
Seide |
Wolle oder Tierhaare |
4 |
8 (Dimethylformamid) und 11 (75 %-ige Schwefelsäure m/m) |
|
20. |
Seide |
Wolle, Tierhaare oder Seide |
Baumwolle, Viskose, Cupro oder Modal |
1 und/oder 4 |
8 (Dimethylformamid) und 2 (Hypochlorit) |
|
21. |
Wolle, Tierhaare oder Seide |
Baumwolle, Viskose, Modal, Cupro |
Polyester |
4 |
2 (Hypochlorit) und 7 (75 %-ige Schwefelsäure m/m) |
|
22. |
Viskose, Cupro oder bestimmte Modalarten |
Baumwolle |
Polyester |
2 und/oder 4 |
3 (Zinkchlorid/Ameisensäure) und 7 (75 %-ige Schwefelsäure m/m) |
|
23. |
Seide |
Viskose, Cupro oder bestimmte Modalarten |
Baumwolle |
4 |
8 (Dimethylformamid) und 3 (Zinkchlorid/Ameisensäure) |
|
24. |
Bestimmte Polychloridfasern |
Viskose, Cupro oder bestimmte Modalarten |
Baumwolle |
1 und/oder 4 |
9 (Schwefelkohlenstoff/Aceton 55,5/44,5 % v/v) und 3 (Zinkchlorid/Ameisensäure) oder 8 (Dimethylformamid) und 3 (Zinkchlorid/Ameisensäure) |
|
25. |
Acetat |
Viskose, Cupro oder bestimmte Modalarten |
Baumwolle |
4 |
1 (Aceton) und 3 (Zinkchlorid/Ameisensäure) |
|
26. |
Triacetat |
Viskose, Cupro oder bestimmte Modalarten |
Baumwolle |
4 |
6 (Dichlormethan) und 3 (Zinkchlorid/Ameisensäure) |
|
27. |
Acetat |
Seide |
Wolle oder Tierhaare |
4 |
1 (Aceton) und 11 (75 %-ige Schwefelsäure m/m) |
|
28. |
Triacetat |
Seide |
Wolle oder Tierhaare |
4 |
6 (Dichlormethan) und 11 (75 %-ige Schwefelsäure m/m) |
|
29. |
Acetat |
Seide |
Baumwolle, Viskose, Cupro oder Modal |
4 |
1 (Aceton) und 8 (Dimethylformamid) |
|
30. |
Triacetat |
Seide |
Baumwolle, Viskose, Cupro oder Modal |
4 |
6 (Dichlormethan) und 8 (Dimethylformamid) |
|
31. |
Triacetat |
Polyamid oder Nylon |
Baumwolle, Viskose, Cupro oder Modal |
4 |
6 (Dichlormethan) und 4 (80 %-ige Ameisensäure m/m) |
|
32. |
Triacetat |
Baumwolle, Viskose, Cupro oder Modal |
Polyester |
4 |
6 (Dichlormethan) und 7 (75 %-ige Schwefelsäure m/m) |
|
33. |
Acetat |
Polyamid oder Nylon |
Polyester oder Polyacrylfasern |
4 |
1 (Aceton) und 4 (80 %-ige Ameisensäure m/m) |
|
34. |
Acetat |
Seide |
Polyester |
4 |
1 (Aceton) und 8 (Dimethylformamid) |
|
35. |
Bestimmte Polychlorid-fasern |
Baumwolle, Viskose, Cupro oder Modal |
Polyester |
4 |
8 (Dimethylformamid) und 7 (75 %ige Schwefelsäure m/m) oder 9 (Schwefelkohlenstoff/Aceton, 55,5/44,5 % v/v) und 7 (75 %-ige Schwefelsäure m/m) |
|
36. |
Baumwolle |
Polyester |
Elastolefin |
2 und/oder 4 |
7 (75 %-ige Schwefelsäure m/m) und 14 (konzentrierte Schwefelsäure) |
|
37. |
Bestimmte Modacryl-fasern |
Polyester |
Melamin |
2 und/oder 4 |
8 (Dimethylformamid) und 14 (konzentrierte Schwefelsäure) |
ANHANG IX
Vereinbarte Zuschläge, die zur Berechnung des Gewichts der in einem Textilerzeugnis enthaltenen Fasern verwendet werden müssen
(gemäß Artikel 19 Absatz 3)
|
Faser Nr. |
Fasern |
Prozent |
|
1-2 |
Wolle und Tierhaare: |
|
|
gekämmte Fasern |
18,25 |
|
|
gekrempelte Fasern |
17,00 (1) |
|
|
3 |
Tierhaare: |
|
|
gekämmte Fasern |
18,25 |
|
|
gekrempelte Fasern |
17,00 (1) |
|
|
Schweif- und Mähnenhaare: |
|
|
|
gekämmte Fasern |
16,00 |
|
|
gekrempelte Fasern |
15,00 |
|
|
4 |
Seide |
11,00 |
|
5 |
Baumwolle: |
|
|
übliche Fasern |
8,50 |
|
|
merzerisierte Fasern |
10,50 |
|
|
6 |
Kapok |
10,90 |
|
7 |
Flachs bzw. Leinen |
12,00 |
|
8 |
Hanf |
12,00 |
|
9 |
Jute |
17,00 |
|
10 |
Manila |
14,00 |
|
11 |
Alfa |
14,00 |
|
12 |
Kokos |
13,00 |
|
13 |
Ginster |
14,00 |
|
14 |
Ramie (entfettete Fasern) |
8,50 |
|
15 |
Sisal |
14,00 |
|
16 |
Sunn |
12,00 |
|
17 |
Henequen |
14,00 |
|
18 |
Maguey |
14,00 |
|
19 |
Acetat |
9,00 |
|
20 |
Alginat |
20,00 |
|
21 |
Cupro |
13,00 |
|
22 |
Modal |
13,00 |
|
23 |
Protein |
17,00 |
|
24 |
Triacetat |
7,00 |
|
25 |
Viskose |
13,00 |
|
26 |
Polyacryl |
2,00 |
|
27 |
Polychlorid |
2,00 |
|
28 |
Fluorfaser |
0,00 |
|
29 |
Modacryl |
2,00 |
|
30 |
Polyamid oder Nylon: |
|
|
Spinnfaser |
6,25 |
|
|
Endlosfaser |
5,75 |
|
|
31 |
Aramid |
8,00 |
|
32 |
Polyimid |
3,50 |
|
33 |
Lyocell |
13,00 |
|
34 |
Polylactid |
1,50 |
|
35 |
Polyester |
1,50 |
|
36 |
Polyethylen |
1,50 |
|
37 |
Polypropylen |
2,00 |
|
38 |
Polyharnstoff |
2,00 |
|
39 |
Polyurethan: |
|
|
Spinnfaser |
3,50 |
|
|
Endlosfaser |
3,00 |
|
|
40 |
Vinylal |
5,00 |
|
41 |
Trivinyl |
3,00 |
|
42 |
Elastodien |
1,00 |
|
43 |
Elasthan |
1,50 |
|
44 |
Glasfaser: |
|
|
mit einem Durchmesser von über 5 μm |
2,00 |
|
|
mit einem Durchmesser von 5 μm oder weniger |
3,00 |
|
|
45 |
Elastomultiester |
1,50 |
|
46 |
Elastolefin |
1,50 |
|
47 |
Melamin |
7,00 |
|
48 |
Metallfaser |
2,00 |
|
metallisierte Faser |
2,00 |
|
|
Asbest |
2,00 |
|
|
Papiergarn |
13,75 |
|
|
49 |
Polypropylen/Polyamid-Bikomponentenfaser |
1,00 |
|
50 |
Polyacrylat |
30,00 |
|
(1) Der Zuschlag von 17,00 % wird auch angewendet, wenn es nicht möglich ist festzustellen, ob das Textilerzeugnis, das Wolle und/oder Tierhaare enthält, aus gekämmten oder gekrempelten Fasern besteht. |
||
ANHANG X
Entsprechungstabellen
|
Richtlinie 2008/121/EG |
Vorliegende Verordnung |
|
Artikel 1 Absatz 1 |
Artikel 4 |
|
Artikel 1 Absatz 2 Buchstaben a-c |
— |
|
Artikel 1 Absatz 2 Buchstabe d |
Artikel 2 Absatz 3 |
|
Artikel 2 Absatz 1 |
Artikel 3 Absatz 1 |
|
Artikel 2 Absatz 2 Einleitung |
Artikel 2 Absatz 2 Einleitung |
|
Artikel 2 Absatz 2 Buchstabe a |
Artikel 2 Absatz 2 Buchstabe a |
|
Artikel 2 Absatz 2 Buchstabe b |
Artikel 2 Absatz 2 Buchstaben b und c |
|
Artikel 2 Absatz 2 Buchstabe c |
Artikel 2 Absatz 2 Buchstabe d |
|
Artikel 3 |
Artikel 5 |
|
Artikel 4 |
Artikel 7 |
|
Artikel 5 |
Artikel 8 |
|
Artikel 6 Absätze 1 und 2 |
— |
|
Artikel 6 Absatz 3 |
Artikel 9 Absatz 3 |
|
Artikel 6 Absatz 4 |
Artikel 9 Absatz 4 |
|
Artikel 6 Absatz 5 |
Artikel 20 |
|
Artikel 7 |
Artikel 10 |
|
Artikel 8 Absatz 1 Satz 1 |
Artikel 14 Absatz 1 |
|
Artikel 8 Absatz 1 Satz 2 |
Artikel 14 Absatz 2 |
|
Artikel 8 Absatz 2 |
Artikel 14 Absatz 3 |
|
Artikel 8 Absatz 3 Unterabsatz 1 |
Artikel 16 Absatz 1 |
|
Artikel 8 Absatz 3 Unterabsätze 2 und 3 |
Artikel 16 Absatz 2 |
|
Artikel 8 Absatz 4 |
Artikel 16 Absatz 3 |
|
Artikel 8 Absatz 5 |
— |
|
Artikel 9 Absatz 1 |
Artikel 11 Absätze 1 und 2 |
|
Artikel 9 Absatz 2 |
Artikel 11 Absatz 3 |
|
Artikel 9 Absatz 3 |
Artikel 13 und Anhang IV |
|
Artikel 10 Absatz 1 Buchstabe a |
Artikel 17 Absatz 2 |
|
Artikel 10 Absatz 1 Buchstabe b |
Artikel 17 Absatz 3 |
|
Artikel 10 Absatz 1 Buchstabe c |
Artikel 17 Absatz 4 |
|
Artikel 10 Absatz 2 |
Artikel 17 Absatz 5 |
|
Artikel 11 |
Artikel 15 Absatz 4 |
|
Artikel 12 |
Artikel 19 Absatz 2 und Anhang VII |
|
Artikel 13 Absatz 1 |
Artikel 19 Absatz 1 |
|
Artikel 13 Absatz 2 |
— |
|
Artikel 14 Absatz 1 |
— |
|
Artikel 14 Absatz 2 |
— |
|
Artikel 15 |
Artikel 21 |
|
Artikel 16 |
— |
|
Artikel 17 |
— |
|
Artikel 18 |
— |
|
Artikel 19 |
— |
|
Artikel 20 |
— |
|
Anhang I |
Anhang I |
|
Anhang II |
Anhang III |
|
Anhang III |
Anhang V |
|
Anhang III Nummer 36 |
Artikel 3 Absatz 1 Buchstabe j |
|
Anhang IV |
Anhang VI |
|
Anhang V |
Anhang IX |
|
Anhang VI |
— |
|
Anhang VII |
— |
|
Richtlinie 96/73/EG |
Vorliegende Verordnung |
|
Artikel 1 |
Artikel 1 |
|
Artikel 2 |
Anhang VIII Kapitel 1 Abschnitt I Nummer 2 |
|
Artikel 3 |
Artikel 19 Absatz 1 |
|
Artikel 4 |
Artikel 19 Absatz 4 |
|
Artikel 5 |
Artikel 21 |
|
Artikel 6 |
— |
|
Artikel 7 |
— |
|
Artikel 8 |
— |
|
Artikel 9 |
— |
|
Anhang I |
Anhang VIII Kapitel 1 Abschnitt I |
|
Anhang II |
Anhang VIII Kapitel 1 Abschnitt II und Kapitel 2 |
|
Anhang III |
— |
|
Anhang IV |
— |
|
Richtlinie 73/44/EWG |
Vorliegende Verordnung |
|
Artikel 1 |
Artikel 1 |
|
Artikel 2 |
Anhang VIII Kapitel 1 Abschnitt I |
|
Artikel 3 |
Artikel 19 Absatz 1 |
|
Artikel 4 |
Artikel 19 Absatz 4 |
|
Artikel 5 |
Artikel 21 |
|
Artikel 6 |
— |
|
Artikel 7 |
— |
|
Anhang I |
Anhang VIII Kapitel 3 Einleitung und Abschnitte I bis III |
|
Anhang II |
Anhang VIII Kapitel 3 Abschnitt IV |
|
Anhang III |
Anhang VIII Kapitel 3 Abschnitt V |
ERKLÄRUNG DES EUROPÄISCHEN PARLAMENTS UND DES RATES
Das Europäische Parlament und der Rat sind sich bewusst, wie wichtig es ist, den Verbrauchern zutreffende Informationen zur Verfügung zu stellen, insbesondere wenn Erzeugnisse mit einer Ursprungsangabe gekennzeichnet werden, um sie vor betrügerischen, unzutreffenden oder irreführenden Angaben zu schützen. Der Einsatz neuer Technologien wie der elektronischen Etikettierung, einschließlich der Radiofrequenz-Identifikation, kann ein nützliches Mittel für die Bereitstellung solcher Informationen sein, wobei gleichzeitig auch mit den technischen Entwicklungen Schritt gehalten wird. Das Europäische Parlament und der Rat ersuchen die Kommission, bei der Ausarbeitung des Berichts gemäß Artikel 24 der Verordnung zu prüfen, wie sich diese Technologien auf mögliche neue Etikettierungsvorschriften auswirken würden, auch im Hinblick auf die Verbesserung der Rückverfolgbarkeit von Textilerzeugnissen.
( 1 ) ABl. L 134 vom 30.4.2004, S. 114.
( *1 ) Für Erzeugnisse gemäß dieser Nummer, die als Schnittstücke verkauft werden, ist die globale Etikettierung diejenige der Rolle. Zu Seilen und Tauen gemäß dieser Nummer zählen insbesondere Seile und Taue für den Alpinismus und den Wassersport.
( 2 ) Gegebenenfalls ist die Analyseprobe direkt vorzubehandeln.
( 3 ) Für Enderzeugnisse und Konfektionsartikel siehe Nummer 7.
( 4 ) Siehe Nummer 1.
( 5 ) Statt mit einer Laboratoriumskrempel kann auch mit einem Fasermischer gearbeitet oder das Verfahren der „ausgekämmten Büschel“ (Hecheln des Doublierens, Teilens und anteiliges Verwerfen) angewendet werden.
( 6 ) Bei Verwendung in einer geeigneten Haspel können mehrere Hülsen gleichzeitig aufgewunden werden.
( 7 ) Methode Nr. 12 bildet eine Ausnahme. Sie geht von der Bestimmung eines wesentlichen Faktors eines der beiden Bestandteile aus.
( 8 ) Siehe Kapitel 1.1.
( 9 ) Um die Einwirkungsdauer der Ammoniaklösung auf den Faserrückstand während 10 Minuten sicherzustellen, kann man z. B. den Filtertiegel mit einem Vorstoß mit Hahn versehen, der den Abfluss der Ammoniaklösung zu regeln gestattet.
( 10 ) Die Löslichkeit dieser Modacryl- oder Polychloridfasern im Reagenz ist vor der Analyse zu prüfen.
( 11 ) Die Löslichkeit der Polyvinylchloridfasern im Reagenz ist vor der Analyse zu prüfen.
( 12 ) Wildseiden, wie zum Beispiel Tussahseide, werden mit 75 %iger Schwefelsäure nicht vollständig herausgelöst.
( 13 ) Diese Reagenzien müssen stickstofffrei sein.
( 14 ) Diesbezüglich sei auf die in „Melliand Textilberichte“ 56 (1975), S. 643-645, beschriebene Apparatur hingewiesen.
( 15 ) Siehe Kapitel 1 Nummer 1.
( 16 ) Die Werte für d sind in Kapitel 2 dieses Anhangs betreffend die verschiedenen Methoden der Analyse binärer Gemische angegeben.
( 17 ) d3 sollte möglichst vorab durch Versuchsverfahren festgelegt werden.






