

EUR-Lex Access to European Union law
This document is an excerpt from the EUR-Lex website
Document L:2009:054:FULL
Official Journal of the European Union, L 54, 26 February 2009
Официален вестник на Европейския съюз, L 54, 26 февруари 2009г.
Официален вестник на Европейския съюз, L 54, 26 февруари 2009г.
Display all documents published in this Official Journal
|
ISSN 1830-3617 |
||
|
Официален вестник на Европейския съюз |
L 54 |
|

|
||
|
Издание на български език |
Законодателство |
Година 52 |
|
Съдържание |
|
I Актове, приети по силата на Договорите за ЕО/Евратом, чието публикуване е задължително |
Страница |
|
|
|
РЕГЛАМЕНТИ |
|
|
|
* |
Регламент (ЕО) № 152/2009 на Комисията от 27 януари 2009 година за определяне на методите за вземане на проби и анализ за целите на официалния контрол на фуражите ( 1 ) |
|
|
|
||
|
|
* |
|
|
|
|
|
(1) Текст от значение за ЕИП |
|
BG |
Актовете, чиито заглавия се отпечатват с нормален шрифт, са актове по текущо управление на селскостопанската политика и имат кратък срок на действие. Заглавията на всички останали актове се отпечатват с удебелен шрифт и се предшестват от звезда. |
I Актове, приети по силата на Договорите за ЕО/Евратом, чието публикуване е задължително
РЕГЛАМЕНТИ
|
26.2.2009 |
BG |
Официален вестник на Европейския съюз |
L 54/1 |
РЕГЛАМЕНТ (ЕО) № 152/2009 НА КОМИСИЯТА
от 27 януари 2009 година
за определяне на методите за вземане на проби и анализ за целите на официалния контрол на фуражите
(текст от значение за ЕИП)
КОМИСИЯТА НА ЕВРОПЕЙСКИТЕ ОБЩНОСТИ,
като взе предвид Договора за създаване на Европейската общност,
като взе предвид Регламент (EО) № 882/2004 на Европейския парламент и на Съвета от 29 април 2004 година относно официалния контрол, провеждан с цел осигуряване на проверка на съответствието със законодателството в областта на фуражите и храните и правилата за опазване здравето на животните и хуманното отношение към животните (1), и по-специално член 11, параграф 4, букви а), б) и в) от него,
като има предвид, че:
|
(1) |
За прилагането на Директива 70/373/ЕИО в съответствие с член 61, параграф 2 от Регламент (ЕО) № 822/2004 са били приети и остават в сила следните актове:
|
|
(2) |
Тъй като Директива 70/373/ЕИО беше заменена с Регламент (ЕО) № 882/2004, целесъобразно е да се заменят актовете за прилагане на посочената директива с един регламент. Същевременно, методите следва да бъдат адаптирани в светлината на развитието на научните и технологичните знания. Методите, които вече не са пригодни за целите, за които са създадени, следва да бъдат заличени. Предвижда се своевременно да се актуализират разпоредбите за вземане на проби, за да бъдат взети под внимание въведените напоследък подобрения в начина на производство, съхранение, транспорт и продажба на фуражите, но въпреки това е целесъобразно засега да бъдат запазени съществуващите разпоредби за вземането на проби. |
|
(3) |
Следователно е необходимо директиви 71/250/ЕИО, 71/393/ЕИО, 72/199/ЕИО, 73/46/ЕИО, 76/371/ЕИО, 76/372/ЕИО, 78/633/ЕИО, 81/715/ЕИО, 84/425/ЕИО, 86/174/ЕИО, 93/70/ЕИО, 93/117/ЕО, 98/64/ЕО, 1999/27/ЕО, 1999/76/ЕО, 2000/45/ЕО, 2002/70/ЕО и 2003/126/ЕО да бъдат отменени. |
|
(4) |
Мерките, предвидени в настоящия регламент, са в съответствие със становището на Постоянния комитет по хранителната верига и здравето на животните, |
ПРИЕ НАСТОЯЩИЯ РЕГЛАМЕНТ:
Член 1
Вземането на проби за целите на официалния контрол на фуражите с оглед определянето на съставките, добавките и нежеланите вещества, с изключение на остатъчните вещества от пестициди и микроорганизми, се извършват в съответствие с методите, определени в приложение I.
Член 2
Подготовката на проби за анализ и представянето на резултатите се извършва в съответствие с методите, посочени в приложение II.
Член 3
Анализът на фуражите за целите на официалния контрол се провежда, като се използват методите, посочени в приложение III (Методи за анализ с цел контролиране на състава на фуражните съставки и на комбинирания фураж), приложение IV (Методи за анализ с цел контролиране на равнището на позволени добавки във фуражите), приложение V (Методи за анализ с цел контролиране на нежеланите вещества във фуражите) и приложение VI (Методи за анализ за определяне на съставките от животински произход за целите на официалния контрол на фуражите).
Член 4
Енергийната стойност на комбинираните фуражи за домашни птици се изчислява в съответствие с приложение VII.
Член 5
Посочените в приложение VIII методи за анализ с цел контролиране на непозволено наличие на добавки, чието ползване във фуражи вече не е разрешено, се използват за потвърждение.
Член 6
Директиви 71/250/ЕИО, 71/393/ЕИО, 72/199/ЕИО, 73/46/ЕИО, 76/371/ЕИО, 76/372/ЕИО, 78/633/ЕИО, 81/715/ЕИО, 84/425/ЕИО, 86/174/ЕИО, 93/70/ЕИО, 93/117/ЕО, 98/64/ЕО, 1999/27/ЕО, 1999/76/ЕО, 2000/45/ЕО, 2002/70/ЕО и 2003/126/ЕО се отменят.
Позоваванията на отменените директиви се считат за позовавания на настоящия регламент и се четат съгласно таблиците на съответствието в приложение IX.
Член 7
Настоящият регламент влиза в сила на двадесетия ден след публикуването му в Официален вестник на Европейския съюз.
Той се прилага от 26 август 2009 г.
Настоящият регламент е задължителен в своята цялост и се прилага пряко във всички държави-членки.
Съставено в Брюксел на 27 януари 2009 година.
За Комисията
Androulla VASSILIOU
Член на Комисията
(1) ОВ L 165, 30.4.2004 г., стр. 1.
(2) ОВ L 155, 12.7.1971 г., стр. 13.
(3) ОВ L 279, 20.12.1971 г., стр. 7.
(4) ОВ L 123, 29.5.1972 г., стр. 6.
(5) ОВ L 83, 30.3.1973 г., стр. 21.
(6) ОВ L 102, 15.4.1976 г., стр. 1.
(7) ОВ L 102, 15.4.1976 г., стр. 8.
(8) ОВ L 206, 29.7.1978, стр. 43.
(9) ОВ L 257, 10.9.1981 г., стр. 38.
(10) ОВ L 238, 6.9.1984 г., стр. 34.
(11) ОВ L 130, 16.5.1986 г., стр. 53.
(12) ОВ L 234, 17.9.1993 г., стр. 17.
(13) ОВ L 329, 30.12.1993 г., стр. 54.
(14) ОВ L 257, 19.9.1998 г., стр. 14.
(15) ОВ L 118, 6.5.1999 г., стр. 36.
(16) ОВ L 207, 6.8.1999 г., стр. 13.
(17) ОВ L 174, 13.7.2000 г., стр. 32.
(18) ОВ L 209, 6.8.2002 г., стр. 15.
(19) ОВ L 339, 24.12.2003 г., стр. 78.
ПРИЛОЖЕНИЕ I
МЕТОДИ ЗА ВЗЕМАНЕ НА ПРОБИ
1. ЦЕЛ И ОБХВАТ
Пробите, предназначени за официалния контрол на фуражите, се вземат по описаните по-долу методи. Така получените проби се смятат за представителни за изследваните партиди.
2. ПЕРСОНАЛ, КОЙТО ВЗИМА ПРОБИТЕ
Вземането на проби се извършва от лица, упълномощени за тази цел от държавите-членки.
3. ОПРЕДЕЛЕНИЯ
Изследвана партида: количество от продукта, което представлява единица с предполагаеми еднакви характеристики.
Точкова проба: количество, взето от едно място на изследваната партида.
Съставна проба: съвкупността от всички точкови проби, взети от една и съща изследвана партида.
Редуцирана проба: представителна част от съставната проба, получена от последната чрез редуциране.
Крайна проба: част от редуцираната проба или от хомогенизираната съставна проба.
4. АПАРАТУРА
|
4.1. |
Апаратурата за вземане на проби трябва да е изработени от материали, които не могат да замърсят взетите за проба продукти. Тази апаратура може да бъде официално одобрена от държавите членки. |
4.2. Препоръчвана апаратура за вземане на проби от твърди фуражи
4.2.1. Ръчно вземане на проби
|
4.2.1.1. |
Плоска лопатка с вертикални страни |
|
4.2.1.2. |
Сонда с дълъг прорез или камери. Размерите на сондата за вземане на проби трябва да съответстват на характеристиките на изследваната партида (дълбочина на контейнера, размери на чувалите и т.н.) и на големината на частиците на фуража. |
4.2.2. Механично вземане на проби
Може да се използва одобрен механичен инструмент за вземане на проби от фураж от конвейрна лента.
4.2.3. Устройства за разделяне
Устройства, предназначени да разделят пробата на приблизително равни части, могат да се използват както за вземането на точкови проби, така и за подготовката на редуцирани и крайни проби.
5. КОЛИЧЕСТВЕНИ ИЗИСКВАНИЯ
|
5.А. |
Във връзка с контрола на веществата или продуктите, разпределени равномерно във фуражите |
|
|
5.А.1. |
Изследвана партида Размерът на изследваната партида трябва да дава възможност за вземане на проба от всяка от съставните ѝ части. |
|
|
5.А.2. |
Точкови проби |
|
|
5.А.2.1. |
Насипен фураж |
Минимален брой точкови проби: |
|
5.А.2.1.1 |
изследвани партиди, които не надвишават 2,5 метрични тона |
седем |
|
5.А.2.1.2. |
изследвани партиди, които надвишават 2,5 метрични тона: |
√ 20 пъти броят на метричните тонове на изследваната партида (1), до най-много 40 точкови проби |
|
5.А.2.2. |
Опакован фураж; |
Минимален брой опаковки, от които се взима проба (2): |
|
5.А.2.2.1. |
Опаковки по-тежки от 1 kg |
|
|
5.А.2.2.1.1. |
изследвани партиди от една до четири опаковки: |
всички опаковки |
|
5.А.2.2.1.2. |
изследвани партиди от пет до 16 опаковки: |
четири |
|
5.А.2.2.1.3. |
изследвани партиди от над 16 опаковки: |
√ броят на опаковките, от които се състои изследваната партида (1), до най-много 20 опаковки |
|
5.А.2.2.2. |
Опаковки, които не надвишават 1 kg |
четири |
|
5.А.2.3. |
Течни или полутечни фуражи: |
Минимален брой контейнери, от които се взима проба (2): |
|
5.А.2.3.1. |
Контейнери с обем над 1 литър |
|
|
5.А.2.3.1.1. |
изследвани партиди от един до четири контейнера |
всички контейнери |
|
5.А.2.3.1.2. |
изследвани партиди от пет до 16 контейнера |
четири |
|
5.А.2.3.1.3. |
изследвани партиди от над 16 контейнера |
√ броят на контейнерите, от които се състои изследваната партида (1), до най-много 20 контейнера |
|
5.А.2.3.2. |
Контейнери с обем не повече от един литър |
Четири |
|
5.А.2.4. |
Фуражни блокчета и минерални буци за близане |
Минимален брой фуражни блокчета или буци за близане, от които се взима проба (2): едно блокче или една буца на изследвана партида от 25 единици, до най-много четири блокчета или буци |
|
5.А.3. |
Съставна проба Изисква се една съставна проба за всяка изследвана партида. Общата маса на точковите проби, съставляващи съставната проба, не трябва да бъде по-малка от следното: |
|
|
5.А.3.1. |
Насипен фураж |
4 kg |
|
5.А.3.2. |
Пакетиран фураж: |
|
|
5.А.3.2.1. |
опаковки, по-тежки от 1 kg |
4 kg |
|
5.А.3.2.2. |
опаковки, не по-тежки от 1 kg |
теглото на съдържанието на четири оригинални опаковки |
|
5.А.3.3. |
Течни или полутечни фуражи |
|
|
5.А.3.3.1. |
контейнери с обем над един литър |
четири литра |
|
5.А.3.3.2. |
контейнери с обем не повече от един литър |
съдържанието на четири оригинални контейнера |
|
5.А.3.4. |
Фуражни блокчета или минерални буци за близане: |
|
|
5.А.3.4.1 |
всяко(а) с тегло над 1 kg |
4 kg |
|
5.А.3.4.2. |
всяко(а) с тегло не повече от 1 kg |
теглото на четири оригинални блокчета или буци |
|
5.А.4. |
Крайни проби Когато е необходимо, от съставната проба чрез редуциране се получава крайната проба. Изисква се анализ на поне една крайна проба. Масата на крайната проба за анализ не трябва да бъде по-малка от следното: |
|
|
|
Твърди фуражи |
500 g |
|
|
Течни или полутечни фуражи |
500 ml |
|
5.Б. |
Във връзка с контрола на нежелани вещества или вещества, които могат да са разпределени неравномерно във фуражите, като например афлатоксини, мораво рогче, рицин и crotalaria във фуражните суровини (3) |
|
|
5.Б.1. |
Изследвана партида: вж. 5.А.1. |
|
|
5.Б.2. |
Точкови проби |
|
|
5.Б.2.1. |
Насипни фуражи вж. 5.А.2.1. |
|
|
5.Б.2.2. |
Опаковани фуражи: |
Минимален брой опаковки, от които се взима проба: |
|
5.Б.2.2.1. |
изследвани партиди от една до четири опаковки: |
всички опаковки |
|
5.Б.2.2.2. |
изследвани партиди от пет до 16 опаковки |
четири |
|
5.Б.2.2.3. |
изследвани партиди от над 16 опаковки |
√ броят на опаковките, от които се състои изследваната партида (1), до най-много 40 опаковки |
|
5.Б.3. |
Съставни проби Броят на съставните проби се изменя в зависимост от размера на изследваната партида. Минималният брой на съставните проби на изследвана партида е посочен по-долу. Общата маса на точковите проби, съставляващи съставната проба, не трябва да бъде под 4 kg |
|
|
5.Б.3.1. |
Насипен фураж |
|
|
|
Тегло на изследваната партида в метрични тонове: |
Минимален брой съставни проби на изследвана партида: |
|
|
до 1 тон |
1 |
|
|
от 1 до 10 тона |
2 |
|
|
от 10 до 40 тона |
3 |
|
|
над 40 тона |
4 |
|
5.Б.3.2. |
Опакован фураж |
|
|
|
Големина на изследваната партида, изразена чрез броя опаковки: |
Минимален брой съставни проби на изследвана партида: |
|
|
1 до 16 |
1 |
|
|
17 до 200 |
2 |
|
|
201 до 800 |
3 |
|
|
над 800 |
4 |
|
5.Б.4. |
Крайни проби От всяка съставна проба чрез редуциране се получава крайната проба Изисква се анализ на поне една крайна проба за всяка съставна проба. Масата на крайната проба за анализ не трябва да е под 500 g. |
|
6. ИНСТРУКЦИИ ОТНОСНО ВЗИМАНЕТО, ПОДГОТОВКАТА И ОПАКОВАНЕТО НА ПРОБИТЕ
6.1. Общи положения
Пробите се взимат и се подготвят възможно най-бързо, като се спазват необходимите предпазни мерки, за да се избегне промяна или замърсяване на продукта. Инструментите, както и повърхностите и съдовете за поставяне на пробите трябва да са почистени и сухи.
6.2. Точкови проби
6.2.А. Във връзка с контрола на веществата или продуктите, разпределени равномерно във фуражите
Точковите проби трябва да се взимат произволно от цялата изследвана партида. Размерите им трябва да са приблизително еднакви.
6.2.А.1.
Изследваната партида се разделя мислено на приблизително равни части. Избира се произволно определен брой части, съответстващи на броя на точковите проби, предвидени в т. 5.А.2 и от всяка част се взема най-малко една проба.
При необходимост пробите се взимат при преместване на изследваната партида (товарене или разтоварване).
6.2.А.2.
След като е избран необходимият брой опаковки за вземане на проби, както е посочено в т. 5.А.2, със сонда или лопатка се взема част от съдържанието на всяка опаковка. В случаите, когато това е необходимо, пробите се взимат след като опаковките се изпразнят поотделно. Всякакви буци следва да се раздробят, като преди това се отделят от общата маса, и после се върнат обратно в пробата, като операцията се повтаря за всяка съставна проба.
6.2.А.3.
След като се избере необходимият брой контейнери за вземане на проби, както е посочено в т. 5.А.2, съдържанието им се хомогенизира, ако е необходимо и от всеки контейнер се взима определено количество.
Точковите проби могат да се вземат при източване на съдържанието.
6.2.А.4.
След като бъде избран необходимият брой контейнери за вземане на проби, съгласно т. 5.А.2., се взимат проби от различни нива.
Пробите могат да се вземат също така при източване на съдържанието, след отстраняване на първите количества.
И в двата случая общият обем на взетите проби не трябва да бъде под 10 литра.
6.2.А.5.
След като бъде избран необходимият брой блокчета или буци за вземане на проби съгласно т. 5.А.2., се взима част от всяко блокче или буца.
6.2.Б. Във връзка с контрола на нежелани вещества или продукти, които могат да са разпределени неравномерно във фуражите, като например афлатоксини, мораво рогче, рицин и crotalaria във фуражните суровини
Изследваната партида се разделя мислено на определен брой приблизително равни части, съответстващи на броя на предвидените в 5.Б.3. съставни проби. Ако частите са повече от една, цялото количество точкови проби, предвидени в 5.Б.2., се разпределя приблизително равномерно между различните части. След това се взимат проби с приблизително еднаква маса (4), така че общата маса на пробите от всяка част да не е по-малка от минималното количество от 4 kg, изисквано за всяка съставна проба. Не трябва да се събират заедно точковите проби, взети от различни части.
6.3. Подготовка на съставните проби
6.3.А. Във връзка с контрола на веществата или продукти, разпределени равномерно във фуражите
Точковите проби се смесват в една съставна проба.
6.3.Б. Във връзка с контрола на нежелани вещества или продукти, които могат да са разпределени неравномерно във фуражите, като например афлатоксини, мораво рогче, рицин и crotalaria във фуражните суровини
Точковите проби от всяка част на изследваната партида се смесват и се образуват толкова съставни проби, колкото е посочено в т. 5.Б.3., като се отбелязва произходът на всяка съставна проба.
6.4. Подготовка на крайните проби
Материалът във всяка съставна проба се смесва грижливо, за да се получи хомогенна проба (5). Ако е необходимо, за тази цел съставната проба се редуцира с механично или автоматично устройство за разделяне или чрез квартуване най-малко до 2 kg или два литра (съкратена проба).
След това се приготвят най-малко три крайни проби с приблизително еднакви маса или обем, отговарящи на количествените изисквания в т. 5.А.4 или т. 5.Б.4. Всяка проба се поставя в подходящ съд. Вземат се всички необходими предпазни мерки, за да се избегнат промени в състава на пробата или замърсяването и увреждането ѝ по време на транспортиране или складиране.
6.5. Опаковане на крайните проби
Съдовете или опаковките се запечатват с пломба и им се поставят етикети (етикетът трябва да е захванат от пломбата), така че да няма възможност за отваряне, без да се увреди пломбата.
7. ПРОТОКОЛ ЗА ВЗИМАНЕ НА ПРОБИТЕ
За всяко вземане на проба се изготвя протокол, по който се идентифицира недвусмислено изследваната партида.
8. ИЗПРАЩАНЕ НА ПРОБИТЕ
За всяка съставна проба се изпраща по най-бързия начин до упълномощената лаборатория за анализи най-малко една крайна проба заедно с необходимите за анализа данни.
(1) Ако полученото число е дробно, то се закръглява до следващото цяло число.
(2) За опаковки или контейнери, чието съдържание не надвишава 1 kg или един литър и за фуражни блокчета или минерални буци за близане, не по-тежки от 1 kg всяко(а), за точкова проба се приема съдържанието на една оригинална опаковка или контейнер, блокче или буца.
(3) Методите, посочени в 5.А, се използват при контрола на афлатоксини, мораво рогче, рицин и crotalaria в пълноценни и в допълващи фуражи.
(4) При опаковани фуражи част от съдържанието на опаковките, от които трябва да се вземат проби, следва да бъде извадено, като се използва сонда или лопатка, след като опаковките бъдат поотделно изпразнени, когато това е необходимо.
(5) Буците се смачват (ако е необходимо, те се отделят от общата маса, а след това се връщат в пробата) поотделно за всяка обща проба.
ПРИЛОЖЕНИЕ II
ОБЩИ РАЗПОРЕДБИ ОТНОСНО МЕТОДИТЕ ЗА АНАЛИЗ НА ФУРАЖИТЕ
А. ПОДГОТОВКА НА ПРОБИТЕ ЗА АНАЛИЗ
1. Цел
Описаните по-долу процедури се отнасят до подготовката за анализ на крайните проби, изпратени на лабораториите за контрол след вземане на проби, проведено в съответствие с разпоредбите в приложение I.
Посочените проби трябва да бъдат подготвени по такъв начин, че претеглените количества, предвидени в методите за анализ, да са хомогенни и представителни за крайните проби.
2. Предпазни мерки, които следва да се вземат
Процедурата, която трябва да се следва за подготовка на пробите, зависи от използваните методи за анализ. Следователно е от изключително значение да се гарантира, че следваната процедура за подготовка на пробите е подходяща за използвания метод за анализ.
Всички необходими действия трябва да се извършват по такъв начин, че да се избегне, доколкото е възможно, замърсяване на пробата или промени в нейния състав.
Стриването, разбъркването и пресяването се извършват възможно най-бързо, с минимално излагане на пробата на въздух и светлина. Не се използва оборудване за смилане и стриване, което може да предизвика значително загряване на пробата.
За фуражите, които са особено чувствителни към загряване, се препоръчва ръчно смилане. Следи се също самият апарат да не бъде източник на замърсяване с микроелементи.
Ако подготовката не може да се проведе без значими промени в съдържанието на влага на пробата, следва да се определи съдържанието на влага преди и след подготовката в съответствие с метода, описан в част А от приложение III.
3. Процедура
Пробата се разделя на подходящи части, предназначени за анализ и за еталони, като се използват подходящи техники за разделяне като редуващо се фракционно отбиране с лопата и стационарно или ротационно сепариране. Методът на конуса и квартуването не се препоръчват, защото с тях може да се получат проби с голяма грешка при разделяне. Еталонната част от пробата се съхранява в подходящ чист и сух съд, снабден с херметична запушалка, а другата част, тежаща най-малко 100 g, се подготвя за анализ, както е посочено по-долу.
3.1. Фуражи, които могат да бъдат смлени във вида, в който са
Освен ако не е посочено друго в методите за анализ, след смилането ѝ цялата проба се пресява през сито с квадратни отвори със страна 1 mm (в съответствие с препоръка на ISO R565), ако това е необходимо. Прекалено ситното смилане следва да се избягва.
Пресятата проба се разбърква и се прибира в подходящ чист и сух съд, снабден с херметична запушалка. Разбърква се отново, непосредствено преди да се вземе претегленото количество за анализ.
3.2. Фуражи, които могат да бъдат смляни след изсушаване
Освен ако не е посочено друго в методите за анализ, пробата се изсушава по такъв начин, че съдържанието на влага в нея да се сведе до 8—12 %, като се прилага процедурата за предварително изсушаване, описана в т. 4.3 от метода за определяне на влагата, споменат в част А от приложение III. След това се процедира, както е посочено в т. 3.1.
3.3. Течни или полутечни фуражи
Пробата се взима в подходящ чист и сух съд, снабден с херметична запушалка. Разбърква се добре непосредствено преди да се вземе претегленото количество за анализ.
3.4. Други фуражи
Към проби, които не могат да бъдат подготвени по някоя от посочените по-горе процедури, се прилага всяка друга подходяща процедура, с която може да се гарантира, че претеглените количества за анализ са хомогенни и представителни за крайните проби.
4. Съхранение на пробите
Пробите се съхраняват при температура, при която не се променя техният състав. Пробите, предназначени за анализ на витамини или вещества, които са особено чувствителни към светлина, се съхраняват в съдове от кафяво стъкло.
Б. РАЗПОРЕДБИ ЗА РЕАКТИВИТЕ И АПАРАТУРАТА, ИЗПОЛЗВАНИ ПРИ МЕТОДИТЕ ЗА АНАЛИЗ
|
1. |
Освен ако не е посочено друго в методите за анализ, всички аналитични реактиви трябва да са чисти за анализ (ч.з.а.). Когато се прави анализ за определяне на микроелементи, чистотата на реактивите трябва да се проверява чрез празна проба. В зависимост от получените резултати, може да се наложи по-нататъшно пречистване на реактивите. |
|
2. |
Всяка дейност, която включва приготвянето на разтвори, разреждане, изплакване или измиване, спомената в методите на анализ без указание за естеството на употребявания разтворител или разредител, предполага, че трябва да се използва вода. По принцип се използва деминерализирана или дестилирана вода. В особени случаи, които са посочени в методите за анализ, тя трябва да бъде подложена на специално пречистване. |
|
3. |
Като се има предвид обичайната апаратура в контролните лаборатории, в методите за анализ се споменават само онези инструменти и апарати, които са специални или изискват специфичен начин на използване. Те трябва да са чисти, особено когато трябва да се определят много малки количества от веществата. |
В. ПРИЛАГАНЕ НА МЕТОДИТЕ ЗА АНАЛИЗ И ИЗРАЗЯВАНЕ НА РЕЗУЛТАТИТЕ
1. Процедура за екстракция
Различни методи определят конкретната процедура за екстракция. По принцип, могат да бъдат приложени други процедури за екстракция, освен посочената в метода процедура, при условие, че е доказано, че използваната процедура за екстракция има еквивалентна екстракционна ефективност за анализираната матрица, каквато има и упоменатата в метода процедура.
2. Процедура за очистване
Различни методи определят конкретната процедура за очистване. По принцип, могат да бъдат приложени други процедури за очистване, освен посочената в метода процедура, при условие, че е доказано, че използваната процедура за очистване има еквивалентна очиствателна ефективност за анализираната матрица, каквато има и упоменатата в метода процедура.
3. Протокол за използвания метод за анализ
По принцип, за определяне на всяко вещество във фуражите е установен един метод за анализ. Когато са дадени няколко метода, конкретният използван метод в контролната лаборатория трябва да се посочи в протокола за анализ.
4. Брой на определянията
Резултатът, даден в протокола за анализ, е средната стойност, получена от поне две определяния, извършени върху отделни части от пробата и със задоволителна повторяемост.
При анализ на нежелани вещества обаче, ако резултатът от първото определяне е значително (> 50 %) по-нисък от стойността, която подлежи на контрол, не са необходими допълнителни определяния, при условие че са приложени подходящите процедури за осигуряване на качеството.
При контрол на декларирано съдържание на вещество или съставка, ако резултатът от първото определяне потвърди декларираното съдържание, т.е., ако резултатът от анализа попада в интервала на допустимото отклонение за декларираното съдържание, не са необходими допълнителни определяния, при условие че са приложени подходящи процедури за осигуряване на качеството.
В някои случаи въпросният интервал на допустимото отклонение е определен от законодателството, както в Директива 79/373/ЕИО (1).
5. Протокол за аналитичния резултат
Аналитичният резултат се изразява по начина, посочен в метода за анализ, със съответен брой значещи цифри, и, ако е необходимо, се коригира в зависимост от съдържанието на влага в крайната проба преди подготовката.
6. Неопределеност на измерване и степен на възстановяване при анализ на нежелани вещества
По отношение на нежеланите вещества по смисъла на Директива 2002/32/ЕО, включително диоксини и диоксиноподбни РСВ, продукт, предназначен за консумация от животните, се разглежда като неотговарящ на изискването за определеното максимално съдържание, ако се прецени, че аналитичният резултат надвишава максималното съдържание, като се вземе предвид разширената неопределеност на измерване и корекцията за възстановяване. За да се оцени степента на съответствие с изискванията, се използва анализираната концентрация, след като се коригира със стойността на възстановяването и след изваждане на разширената неопределеност на измерване. Посочената процедура се прилага единствено в случаи, когато методът за анализ позволява оценка на неопределеността на измерване и на корекцията за възстановяване (тя е невъзможна например при микроскопски анализ).
Аналитичният резултат се отчита, както следва (доколкото използваният метод за анализ позволява да се оцени неопределеността на измерване и степента на възстановяване):
|
а) |
с корекция за възстановяването, като се посочва равнището на възстановяване. Корекцията за възстановяване не е необходима, ако степента на възстановяване е между 90 и 110 %. |
|
б) |
като „х +/- U“, където х е аналитичният резултат, а U е разширената неопределеност на измерването, като се използва фактор на покриване 2, който дава ниво на достоверност от около 95 %. |
Ако обаче стойността на аналитичния резултат е значително (> 50 %) по-ниска от стойността, която подлежи на контрол, и при условие че са приложени подходящи процедури за осигуряване на качеството и ако анализът се прави само за да се провери съответствието със законовите разпоредби, аналитичният резултат може да се отчете без да се прави корекция за възстановяване и в тези случаи отчитането на степента на възстановяване и на неопределеността при измерване може да бъде пропуснато.
ПРИЛОЖЕНИЕ III
МЕТОДИ ЗА АНАЛИЗ С ЦЕЛ КОНТРОЛ НА СЪСТАВА НА ФУРАЖНИТЕ СУРОВИНИ И НА КОМБИНИРАНИТЕ ФУРАЖИ
А. ОПРЕДЕЛЯНЕ НА СЪДЪРЖАНИЕТО НА ВЛАГА
1. Цел и обхват
Настоящият метод дава възможност да се определи съдържанието на влага във фуражи. В случай, че фуражът съдържа летливи вещества, като например органични киселини, трябва да се отбележи, че заедно със съдържанието на влага се откриват и значителни количества от тези летливи вещества.
Методът не обхваща анализа на млечните продукти в качеството им на фуражни суровини, анализа на неорганичните вещества и смесите, съставени предимно от неорганични вещества, анализа на животинските и растителните мазнини и масла или анализа на маслодайните семена и плодове.
2. Принцип
Пробата се изсушава при точно определени условия, които варират в зависимост от вида фураж. Загубата на тегло се определя посредством претегляне. При работа с твърди фуражи, които имат високо съдържание на влага, е необходимо да се извърши предварително сушене.
3. Апаратура
|
3.1. |
Мелница от неабсорбиращ влага материал, която е лесна за почистване, позволява бързо, равномерно раздробяване без забележимо загряване, предотвратява контакта с външния въздух, доколкото това е възможно, и отговаря на изискванията, определени в т. 4.1.1 и т. 4.1.2 (напр. чукови микромелници или микромелници с водно охлаждане, сглобяеми конусни мелници, нискооборотни мелници или мелници със зъбни колела). |
|
3.2. |
Аналитични везни за измерване с точност до 1 mg. |
|
3.3. |
Сухи съдове от некородиращ метал или от стъкло, с капаци, осигуряващи херметично затваряне; работна повърхност, позволяваща разстилане на изследваната проба с плътност около 0,3 g/cm2. |
|
3.4. |
Електрическа изотермична пещ (± 2 oC) с подходяща вентилация, която осигурява бързо регулиране на температурата (1). |
|
3.5. |
Регулируема електрическа вакуумна пещ, оборудвана с маслена помпа и с механизъм за вкарване на горещ сух въздух или с изсушаващ агент (напр. калциев оксид). |
|
3.6. |
Ексикатор с дебела перфорирана метална или порцеланова плоча, съдържащ ефикасен изсушаващ агент. |
4. Процедура
|
N.B.: |
Описаните в този раздел операции следва да се извършват незабавно след отварянето на опаковките с проби. Провежда се най-малко двукратен анализ. |
4.1. Подготовка
4.1.1.
Вземат се най-малко 50 g от пробата. При необходимост това количество се раздробява или разделя така, че да се избегне промяна в съдържанието на влага (вж. т. 6).
4.1.2.
Вземат се най-малко 50 g от пробата. Смилат се на ситни частици, от които поне 50 % да могат да преминат през сито с големина на отворите 0,5 mm и от които най-много 10 % да не могат да преминат през сито с кръгли отвори с размер 1 mm.
4.1.3.
Вземат се около 25 g от пробата, претеглят се с точност до 10 mg, добавя се съответното количество безводен пясък, претеглено с точност до 10 mg, и се разбърква до получаването на хомогенен продукт.
4.2. Сушене
4.2.1.
Съдът (т. 3.3) се претегля заедно с капака си с точност до 1 mg. В претегления съд се измерват с точност до 1 mg около 5 g от пробата и се разстилат равномерно. Съдът, без капака, се поставя в предварително загрятата до 103 oC пещ. За да се предотврати нежелателното спадане на температурата, съдът се поставя в пещта възможно най-бързо. Пробата се оставя да съхне четири часа, считано от времето на възстановяване на температурата от 103 oC. Капакът се поставя обратно върху съда, последният се изважда от пещта, оставя се да изстива 30—45 min в ексикатора (т. 3.6) и се претегля с точност до 1 mg.
За фуражи, състоящи се предимно от масла и мазнини, пробите се сушат 30 min по-дълго при температура 130 oC. Разликата между стойностите за съдържанието на влага, получени от двете претегляния, не трябва да надвишава 0,1 %.
4.2.2.
Съдът (т. 3.3) се претегля заедно с капака с точност до 0,5 mg. В претегления съд се измерват с точност до 1 mg около 5 g от смляната проба и се разстилат равномерно. Съдът, без капака, се поставя в предварително загрятата до 130 oC пещ. За да се предотврати нежелателното спадане на температурата, съдът се поставя в пещта възможно най-бързо. Пробата се оставя да съхне четири часа, считано от времето на възстановяване на температурата от 130 oC. Капакът се поставя обратно върху съда, последният се изважда от пещта, оставя се да изстива 30—45 min в ексикатора (т. 3.6) и се претегля с точност до 1 mg.
|
4.2.3. |
Комбинирани фуражи, съдържащи повече от 4 % захароза или лактоза: фуражни суровини, като например плодове на рожков, хидролизирани зърнени продукти, малцови семена, парченца изсушено цвекло, рибни и захарни разтворими компоненти; комбинирани фуражи, съдържащи повече от 25 % минерални соли, включително кристализационна вода. Съдът (т. 3.3) се претегля заедно с капака с точност до 0,5 mg. В претегления съд се измерват с точност до 1 mg около 5 g от пробата и се разстилат равномерно. Съдът, без капака, се поставя в предварително загрятата от 80 oС до 85 oC вакуумна пещ (т. 3.5). За да се предотврати нежелателното спадане на температурата, съдът се поставя в пещта възможно най-бързо. Налягането се увеличава до 100 Torr и пробата се оставя да се суши четири часа при това налягане на поток горещ сух въздух или посредством изсушаващ агент (около 300 g за 20 проби). Във втория случай вакуумната помпа се изключва, когато се достигне предписаното налягане. Времето на сушене се отчита от момента на възстановяване на температурата от 80 oС—85 oC в пещта. Налягането в пещта внимателно се понижава и изравнява с атмосферното. Пещта се отваря, капакът се поставя веднага върху съда, съдът се изважда от пещта, оставя се да се охлади за 30—45 min в ексикатора (т. 3.6) и се претегля с точност до 1 mg. Суши се още 30 min във вакумната пещ на 80 oC—85 oC и се претегля отново. Разликата между стойностите за съдържанието на влага, получени от двете претегляния, не трябва да надвишава 0,1 %. |
4.3. Предварително сушене
4.3.1.
Твърдите фуражи с високо съдържание на влага, което затруднява раздробяването, трябва да се подлагат на предварително сушене по следния начин:
Претеглят се 50 g от проба от нераздробен фураж се с точност до 10 mg (пресовани или слепени фуражи при необходимост могат да бъдат грубо разделени) в подходящ съд (напр. алуминиева плоча с размери 20 × 12 cm и височина на ръба 0,5 cm). Пробата се оставя да съхне в пещ при температура 60 oС—70 oC, докато съдържанието на влага спадне до 8 %—12 %. Пробата се изважда от пещта, оставя се да изстине без капак в лабораторията в продължение на един час и се претегля с точност до 10 mg. Незабавно се раздробява, както е указано в т. 4.1.1 и се суши, както е указано в т. 4.2.1 или т. 4.2.3 в зависимост от свойствата на фуража.
4.3.2.
Зърно със съдържание на влага над 17 % трябва да се подложи на предварително сушене по следния начин:
Претеглят се 50 g несмляно зърно с точност до 10 mg в подходящ съд (напр. алуминиева плоча с размери 20 × 12 cm и височина на ръба 0,5 cm). Оставя се да се суши за 5—7 min в пещ при температура 130 oC. Пробата се изважда от пещта, оставя се да изстине без капак в лабораторията в продължение на два часа и се претегля с точност до 10 mg. Незабавно се смила, както е указано в т. 4.1.2 и се суши, както е указано в т. 4.2.2.
5. Изчисляване на резултатите
Съдържанието на влага (Х) като процент от пробата се изчислява по следните формули:
5.1. Изсушаване без предварително сушене

където:
|
m |
= |
първоначално тегло в грамове на пробата за анализ, |
|
m0 |
= |
тегло в грамове на сухата проба за анализ. |
5.2. Изсушаване с предварително сушене

където:
|
m |
= |
първоначално тегло в грамове на пробата за анализ, |
|
m1 |
= |
тегло в грамове на пробата за анализ след предварителното сушене, |
|
m2 |
= |
тегло в грамове на пробата за анализ след раздробяване или смилане, |
|
m0 |
= |
тегло в грамове на сухата проба за анализ. |
5.3. Повторяемост
Разликата между резултатите от две паралелни определяния, проведени върху една и съща проба, не трябва да надвишава 0,2 % от абсолютната стойност на влагата.
6. Забележки
В случай, че се наложи раздробяване, и ако това промени съдържанието на влага в продукта, резултатите от анализа на съставките на фуража трябва да бъдат коригирани въз основа на съдържанието на влага на пробата в първоначалното ѝ състояние.
Б. ОПРЕДЕЛЯНЕ НА ВЛАГА В ЖИВОТИНСКИ И РАСТИТЕЛНИ МАЗНИНИ И МАСЛА
1. Цел и обхват
Настоящият метод дава възможност да се определи съдържанието на вода и летливи вещества, съдържащи се в животински и растителни мазнини и масла.
2. Принцип
Пробата се суши, докато теглото ѝ стане постоянно (намаляването на теглото при две последователни претегляния трябва да е по-малко или равно на 1 mg), при 103 oC. Загубата на тегло се определя посредством претегляне.
3. Апаратура
|
3.1. |
Плоскодънно блюдо от корозионно устойчив материал с диаметър от 8 до 9 cm и с приблизителна височина 3 cm. |
|
3.2. |
Термометър с усилен резервоар и разширение в горната част на капиляра, градуиран от около 80 oC до най-малко 110 oС и дълъг приблизително 10 cm. |
|
3.3. |
Пясъчна баня или електрически котлон. |
|
3.4. |
Ексикатор, съдържащ ефикасен изсушаващ агент. |
|
3.5. |
Аналитични везни. |
4. Процедура
В сухото претеглено блюдо (т. 3.1), в което е поставен термометърът (т. 3.2) се претеглят с точност до 1 mg около 20 g от хомогенизираната проба. Пробата се нагрява върху пясъчната баня или котлона (т. 3.3), като се разбърква непрекъснато с термометъра така, че да се достигне температура 90 oC за около 7 min.
Нагряването се намалява, като се наблюдава честотата, с която се появяват мехурчета от дъното на блюдото. Температурата не трябва да надвишава 105 oC. Разбъркването продължава, като се стърже дъното на блюдото, докато престанат да се образуват мехури.
За да се осигури напълно отстраняване на влагата, нагряването се повтаря няколко пъти до 103 oC ±2 oC с охлаждане до 93 oC между нагряванията. След това се оставя да се охлади до стайна температура в ексикатора (т. 3.4) и се претегля. Тази операция се повтаря, докато загубата на тегло между две последователни претегляния спре да надвишава 2 mg.
|
N.B. |
Увеличаване на теглото на пробата след повторно нагряване е индикация за окисляване на мазнината. В този случай резултатът се пресмята, като за основа се взема претеглянето, проведено непосредствено преди теглото да започне да нараства. |
5. Изчисляване на резултатите
Съдържанието на влага (Х) като процент от пробата се пресмята по следната формула:

където:
|
m |
= |
теглото в грамове на пробата за анализ; |
|
m1 |
= |
теглото в грамове на блюдото със съдържанието му преди нагряване; |
|
m2 |
= |
теглото в грамове на блюдото със съдържанието му след нагряване; |
Резултати по-ниски от 0,05 % трябва да бъдат записани като „по-ниски от 0,05 %“.
Повтаряемост
Разликата във влажността между резултатите в две паралелни определяния, проведени върху една и съща проба, не трябва да надвишава 0,05 % като абсолютна стойност.
В. ОПРЕДЕЛЯНЕ НА СУРОВ ПРОТЕИН
1. Цел и обхват
Настоящият метод дава възможност да се определи общото количество суров протеин в храните за животни на базата на съдържанието на азот, определяно по метода на Келдал.
2. Принцип
Пробата се изварява със сярна киселина в присъствие на катализатор. Киселинният разтвор се алкализира с разтвор на натриев хидроксид. Амонякът се дестилира и събира в определено количество сярна киселина, излишъкът от която се титрува със стандартен разтвор на натриев хидроксид.
Като алтернатива, отделеният амоняк се дистилира в излишък от разтвор на борна киселина, след което се титрува с разтвор на солна или сярна киселина.
3. Реагенти
|
3.1. |
Калиев сулфат. |
|
3.2. |
Катализатор: меден (II) оксид CuO или меден (II) сулфат пентахидрат, CuSO4 5H2O |
|
3.3. |
Цинк на гранули. |
|
3.4. |
Сярна киселина, ρ20 = 1,84 g/ml. |
|
3.5. |
Сярна киселина, стандартен обемен разтвор, c(H2SO4) = 0,25 mol/l. |
|
3.6. |
Сярна киселина, стандартен обемен разтвор, c(H2SO4) = 0,10 mol/l. |
|
3.7. |
Сярна киселина, стандартен обемен разтвор, c(H2SO4) = 0,05 mol/l. |
|
3.8. |
Индикатор метилово червено; разтварят се 300 mg метилово червено в 100 ml етанол, σ = 95—96 % (v/v). |
|
3.9. |
Разтвор на натриев хидроксид (може да се използва технически чист разтвор) β = 40 g/100 ml (m/v: 40 %). |
|
3.10. |
Натриев хидрооксид, стандартен обемен разтвор, c(NaOH) = 0,25 mol/l. |
|
3.11. |
Натриев хидрооксид, стандартен обемен разтвор, c(NaOH) = 0,10 mol/l. |
|
3.12. |
Пемза на гранули, измита в солна киселина и запалена |
|
3.13. |
Ацетанилид (точка на топене = 114 oC, съдържание на N = 10,36 %). |
|
3.14. |
Захароза (без азот). |
|
3.15. |
Борна киселина (H3BO3). |
|
3.16. |
Индикаторен разтвор на метилово червено: разтварят се 100 mg метилово червено в 100 ml етанол или метанол. |
|
3.17. |
Разтвор на бромокрезолово зелено: разтварят се 100 mg бромокрезолово зелено в 100 ml етанол или метанол. |
|
3.18. |
Разтвор на борна киселина (10 g/l—40 g/l в зависимост от използваната апаратура). Когато еквивалентният пункт се определя по колориметричен метод, индикаторните разтвори с метилово червено и бромокрезолно зелено се добавят към разтворите на борна киселина. Ако е приготвен 1 литър разтвор на борна киселина, преди коригирането на обема се добавят 7 ml индикаторен разтвор на метилово червено (т. 3.16) и 10 ml разтвор на бромокрезолно зелено (т. 3.17). В зависимост от използваната вода, pH на разтвора на борна киселина може да се различава в различните партиди. Често е необходимо да направи корекция с малък обем алкално вещество, за да се получи надеждна празна проба
|
|
3.19 |
Солна киселина, стандартен разтвор, c(HCl) = 0,10 mol/l.
|
4. Апаратура
Апаратурата следва да бъде подходяща за изваряване, дестилация и титруване по процедурата на Келдал.
5. Процедура
5.1. Изваряване
Отмерва се 1 g от пробата с точност до 0,001 g и пробата се прехвърля в колбата на апарата за изваряване. Прибавят се 15 g калиев сулфат (т. 3.1), подходящо количество катализатор (т. 3.2) (от 0,3 до 0,4 меден (II) оксид или от 0,9 до 1,2 g меден (II) сулфат пентахидрат), 25 ml сярна киселина (т. 3.4) и ако е необходимо, няколко гранули пемза (т. 3.12) и се разбърква.
Колбата се нагрява първоначално бавно, като от време на време се завърта, докато съдържанието се овъгли и пяната изчезне; след това се нагрява по-интензивно, докато течността започне непрекъснато да кипи. Нагряването е подходящо, ако врящата киселина се кондензира по стените на колбата. Следи се стените да не прегреят и към тях да не прилепват органични частици.
Когато разтворът се избистри и стане бледозелен, продължава да се вари още два часа, след което се оставя да се охлади.
5.2. Дестилация
Внимателно се добавя достатъчно вода, за да се осигури пълно разтваряне на сулфатите. Оставя се да се охлади и след това се прибавят няколко гранули цинк (т. 3.3), ако е необходимо. Действа се според т. 5.2.1 или т. 5.2.2.
5.2.1.
В събирателната колба на апарата за дестилация се поставя точно измерено количество от 25 ml сярна киселина (т. 3.5) или (т. 3.7) в зависимост от предполагаемото съдържание на азот. Добавят се няколко капки индикатор метилово червено (т. 3.8).
Колбата, в която е проведено изваряването, се свързва към хладника на дестилационния апарат и изходът на хладника се потапя в течността в събирателната колба на дълбочина поне 1 cm (вж. забележка в т. 8.3). Бавно се изсипват 100 ml разтвор на натриев хидроксид (т. 3.9) в колбата за изваряване, без загуба на амоняк (вж. забележка в т. 8.1). Колбата се нагрява, докато амонякът се дестилира.
5.2.2.
Когато титруването на съдържанието на амоняк в дестилата се осъществява ръчно, се прилага описаната по-долу процедура. Когато дестилационният модул е напълно автоматизиран и включва титруването на съдържанието на амоняк в дестилата, се следват инструкциите на производителя за работата на дестилационния модул.
Поставя се събирателна колба, в която има 25 ml—30 ml разтвор на борна киселина (т. 3.18), под изхода на хладника по такъв начин, че изходната тръба да е под равнището на излишното количество разтвор на борна киселина. Настройва се дестилационният модул да подава 50 ml разтвор на натриев хидрооксид (т. 3.9). Дестилационният модул се пуска в действие в съответствие с инструкциите на производителя и се дестилира амонякът, отделен при добавянето на разтвора на натриев хидрооксид. Дестилатът се събира в приемащия разтвор на борна киселина. Количеството на дестилата (времето на дестилация на парите) зависи от количеството азот в пробата. Следват се инструкциите на производителя.
|
Забележка: |
В полуавтоматичните дестилационни модули добавянето на излишъка от натриев хидрооксид и дестилацията на парите се извършват автоматично. |
5.3. Титруване
Действа се според т. 5.3.1 или т. 5.3.2.
5.3.1.
Излишното количество сярна киселина в събирателната колба се титрува с разтвор от натриев хидроксид (т. 3.10 или т. 3.11) в зависимост от концентрацията на използваната сярна киселина, докато се достигне еквивалентния пункт.
5.3.2.
Съдържанието на събирателната колба се титрува със стандартен обемен разтвор на солна киселина (т. 3.19) или със стандартен обемен разтвор на сярна киселина (т. 3.6), като се използва бюрета и се отчита количеството на използвания титрационен агент.
Когато се използва колориметричен метод за определяна на еквивалентния пункт, крайната точка е достигната, когато в съдържанието се появи първата розова следа. Показанията от бюретата се отчитат с точност до 0,05 ml. Може да се подпомогне визуализацията на еквивалентния пункт чрез използването на осветена магнитна бъркалка или фотометричен детектор.
Този процес може да се проведе автоматично, като се използва дестилатор на парите с автоматично титруване.
При работа с конкретен дестилатор или дестилатор/титратор се следват указанията на производителя.
|
Забележка: |
Когато се използва автоматизирана система за титруване, титруването започва веднага след началото на дестилацията и се използва разтвор на борна киселина (т. 3.18) с концентрация 1 %. Когато се използва изцяло автоматизирана дестилация, автоматичното титруване на амоняка може да се осъществи също така чрез определяне на еквивалентния пункт като се използва система за потенциометрично определяне на pH. В този случай се използва автоматичен титратор с рН-метър. pH-метърът се калибрира правилно в обхвата от pH 4 до pH 7, като се следват обичайните лабораторни процедури за калибриране на pH. Еквивалентният пункт на титруването на pH е достигнат, когато pH достигне стойност 4,6, като това е наи-високата точка на кривата на титруване (инфлексна точка). |
5.4. Тест с празна проба
За да се потвърди, че реагентите не съдържат азот, се извършва контролен тест с празна проба (изваряване, дестилация и титруване) с 1 g захароза (т. 3.14) вместо с пробата.
6. Изчисляване на резултатите
Изчисляването се извършва в съответствие с т. 6.1 или т. 6.2.
6.1. Пресмятане на титруването в съотвестствие с т. 5.3.1.
Съдържанието на суров протеин, изразено като процент от теглото на пробата, се пресмята по следната формула:

Където:
|
V0 |
= |
е обемът (в ml) на NаОН (т. 3.10 или т. 3.11), използван в теста с празната проба. |
|
V1 |
= |
е обемът (в ml) на NаОН (т. 3.10 или т. 3.11), използван при титруването на пробата. |
|
c |
= |
е концентрацията (в mol/l) на натриев хидроксид (т. 3.10 или т. 3.11). |
|
m |
= |
е теглото (в g) на пробата. |
6.2 Изчисляване на титруването в съответствие с т. 5.3.2.
6.2.1.
Съдържанието на суров протеин, изразено като процент от теглото на пробата, се пресмята по следната формула:

където:
|
m |
= |
е теглото (в g) на частта от пробата за анализ; |
|
c |
= |
е концентарацията (в mol/l) на стандартния обемен разтвор на солна киселина (точка 3.19); |
|
V0 |
= |
е обемът (в ml) на солната киселина, използвана при анализа на празната проба; |
|
V1 |
= |
е обемът (в ml) на солната киселина, използвана при анализа на частта от пробата за анализ. |
6.2.2.
Съдържанието на суров протеин като процент от теглото на пробата се пресмята по следната формула:

където:
|
m |
= |
е теглото (в g) на частта от пробата за анализ; |
|
c |
= |
е концентрацията (в mol/l) на стандартния разтвор на сярна киселина (т. 3.6); |
|
V0 |
= |
е обемът (в ml) на сярната киселина (т. 3.6), използвана при анализа на контролната проба; |
|
V1 |
= |
е обемът (в ml) на сярната киселина (т. 3.6), използвана при анализа на частта от пробата за анализ. |
7. Проверка на метода
7.1. Повторяемост
Разликата между резултатите от две паралелни определяния, проведени върху една и съща проба не трябва да превишава:
|
— |
0,2 % като абсолютна стойност за съдържание на суров протеин под 20 %; |
|
— |
1,0 %, отнесен към по-високата стойност, за съдържание на суров протеин от 20 % до 40 %; |
|
— |
0,4 % като абсолютна стойност за съдържание на суров протеин над 40 %. |
7.2. Точност
Анализът (изваряване, дестилация и титруване) се извършва с 1,5 до 2,0 g ацетанилид (т. 3.13) в присъствието на 1 g захароза (т. 3.14); 1 g ацеталинид консумира 14,80 ml сярна киселина (т. 3.5). Възстановяването трябва да бъде поне 99 %.
8. Забележки
|
8.1. |
Апаратурата трябва да бъде от ръчен, полуавтоматичен или автоматичен тип. Ако апаратурата изисква прехвърляне между етапите на изваряване и дестилация, прехвърлянето трябва да се извърши без загуба. Ако колбата на дестилационния апарат не е снабдена с делителна фуния, натриевият хидроксид се добавя веднага преди свързването на колбата с хладника, като течността се изсипва бавно по стената. |
|
8.2. |
Ако изваряваното вещество се втвърди, определянето се повтаря с по-големи от указаните по-горе количества сярна киселина (т. 3.4). |
|
8.3. |
За продукти с ниско съдържание на азот обемът на сярната киселина (т. 3.7), който трябва да се постави в събирателната колба, може да се намали, ако е необходимо, до 10 или 15 ml и да се допълни до 25 ml с вода. |
|
8.4. |
За рутинни анализи могат да се използват алтернативни методи за определяне на съдържанието на суров протеин, но референтен е описаният в настоящата част В метод на Келдал. Еквивалентността между резултатите, получени с алтернативния метод (напр. DUMAS), и резултатите, получени с референтния метод, трябва да бъде доказвана поотделно за всяка матрица. Тъй като резултатите, получени с алтернативния метод, дори и след проверка на еквивалентността, могат леко да се различават от резултатите, получени с референтния метод, необходимо е да се посочи в протокола от анализа кой метод за определяне на суров протеин е използван. |
Г. ОПРЕДЕЛЯНЕ НА КАРБАМИД
1. Цел и обхват
Настоящият метод позволява определянето на съдържанието на карбамид във фуражите.
2. Принцип
Пробата се суспендира във вода с избистрящ агент. Суспензията се филтрира. Съдържанието на карбамид във филтрата се определя след добавяне на 4-диметиламинобензалдехид (4-DMAB) чрез измерване на оптичната плътност при дължина на вълната 420 nm.
3. Реагенти
|
3.1. |
Разтвор на 4-диметиламинобензалдехид: разтваря се 1,6 g 4-DMAB в 100 ml 96 % етанол и се добавят 10 ml солна киселина (ρ201,19 g/ml). Този реактив се съхранява най-много две седмици. |
|
3.2. |
Разтвор на Карез I: разтварят се във вода 21,9 g цинков ацетат Zn(CH3COO)2 2H2O и 3 g безводна оцетна киселина. Допълва се до 100 ml с вода. |
|
3.3. |
Разтвор на Карез II: разтварят се във вода 10,6 g калиев фероцианид K4 Fe (CN)6 3H2O. Допълва се до 100 ml с вода. |
|
3.4. |
Активен въглен, който не абсорбира карбамид (да се провери). |
|
3.5. |
Разтвор на карбамид с концентрация 0,1 % (т/об). |
4. Апаратура
|
4.1. |
Смесител (барабанен): приблизително 35 до 40 rpm. |
|
4.2. |
Епруветки: 160 × 16 mm, снабдени с шлифовани стъклени запушалки. |
|
4.3. |
Спектрофотометър. |
5. Процедура
5.1. Анализ на пробата
Претеглят се с точност до 1 mg 2 g от пробата и заедно с 1 g активен въглен (т. 3.4) се поставят в мерителна колба с вместимост 500 ml. Добавят се 400 ml вода и 5 ml разтвор на Карез I (т. 3.2), разбъркват се в продължение на около 30 секунди и след което се добавят 5 ml разтвор на Карез II (т. 3.3). Размесват се тридесет минути в барабанния смесител. Допълва се до марката с вода, разклаща се и се филтрира.
Взимат се 5 ml от прозрачните безцветни филтрати, поставят се в епруветки с шлифовани стъклени запушалки, добавя се 5 ml разтвор на 4-DMAB (т. 3.1) и се размесва. Поставят се епруветките във водна баня при 20 oС (+/– 4 oС). След петнадесет минути със спектрофотометър при 420 nm се измерва оптичната плътност на пробния разтвор. Сравнява се с контролния разтвор на реагентите.
5.2. Калибрационна крива
Взимат се обеми от 1, 2, 4, 5 и 10 ml от карбамидния разтвор (т. 3.5), поставят се в мерителни колби с вместимост 100 ml и се допълват до марката с вода. Взимат се по 5 ml от всеки разтвор, добавят се 5 ml разтвор на 4-DМAB (т. 3.1) към всеки от тях, хомогенизират се и се измерва оптичната плътност, както е указано по-горе, като се сравняват с контролен разтвор, съдържащ 5 ml 4-DMAB и 5 ml вода, която не съдържа карбамид. Построява се калибрационната крива.
6. Изчисляване на резултатите
Определя се количеството карбамид в пробата, като се използва калибрационната крива.
Резултатът се изразява като процент от пробата.
7. Забележки
|
7.1. |
В случай на съдържание карбамид над 3 %, пробата се намалява до 1 g или първоначалният разтвор се разрежда така, че да не съдържа повече от 50 mg карбамид в 500 ml. |
|
7.2. |
В случай на ниско съдържание на карбамид, пробата се увеличава дотогава, докато се получи прозрачен и безцветен филтрат. |
|
7.3. |
Ако пробата съдържа прости азотни съединения, като например аминокиселини, оптичната плътност се измерва при 435 nm. |
Д. ОПРЕДЕЛЯНЕ СЪДЪРЖАНИЕТО НА ЛЕТЛИВИ АЗОТНИ ОСНОВИ
I. ЧРЕЗ МИКРОДИФУЗИЯ
1. Цел и обхват
Настоящият метод дава възможност да се определи съдържанието на летливи азотни основи, изразени като амоняк, във фуражите.
2. Принцип
Пробата се извлича с вода и разтворът се избистря и филтрира. Летливите азотни основи се извличат чрез микродифузия, като се използва калиев карбонат, събират се в разтвор на борна киселина и се титруват със сярна киселина.
3. Реагенти
|
3.1. |
Разтвор на трихлороцетна киселина с концентрация 20 % (w/v). |
|
3.2. |
Индикатор: 33 mg бромокрезолово зелено и 65 mg метилово червено се разтварят в 100 ml 95 %—96 % (о/о) етанол. |
|
3.3. |
Разтвор на борна киселина: в градуирана колба от 1 l се разтварят 10 g борна киселина в 200 ml 95 %—96 % (о/о) етанол и 700 ml вода. Добавят се 10 ml индикатор (т. 3.2). Разбърква се и ако е необходимо, цветът на разтвора се коригира до получаване на светло червено, като са добавя разтвор на натриев хидроксид. 1 ml от този разтвор свързва максимум 300 μg NH3. |
|
3.4. |
Наситен разтвор на калиев карбонат: 100 g калиев карбонат се разтварят в 100 ml кипяща вода. Оставя се да изстине и се филтрира. |
|
3.5. |
Сярна киселина, 0,01 mol/l. |
4. Апаратура
|
4.1. |
Смесител (барабанен): приблизително 35—40 r.p.m. |
|
4.2. |
Стъклени или пластмасови клетки на Конуей (вж. диаграмата). |
|
4.3. |
Микробюрети, градуирани в 1/100 ml. |
5. Процедура
Претеглят се 10 g от пробата с точност до 1 mg и заедно със 100 ml вода се поставят в градуирана колба от 200 ml. Разбъркват се или се разклащат в смесителя в продължение на около 30 min. Добавят се 50 ml разтвор на трихлорооцетна киселина (т. 3.1), допълва се до пълния обем с вода, съдържанието силно се разклаща и се филтрира през нагънат филтър.
С помощта на пипета 1 ml разтвор на борна киселина (т. 3.3) се вкарва в централната част на клетката на Конуей, а 1 ml от филтрата на пробата се вкарва във венеца на клетката. Частично се покрива със смазания капак. Капва се бързо 1 ml от наситения разтвор на калиев карбонат (т. 3.4) във венеца и съдът се затваря херметически с капака. След това той се завърта върху хоризонтална плоскост така, че двата реагента да се смесят. Оставя се да инкубира най-малко четири часа при стайна температура или един час при температура 40 oC.
С помощта на микробюрета (т. 4.3) летливите основи в разтвора на борна киселина се титруват със сярна киселина (т. 3.5).
Провежда се контролен опит по същата процедура, но без проба за анализ.
6. Изчисляване на резултатите
1 ml H2SO40,01 mol/l съответства на 0,34 mg амоняк.
Резултатът се изразява като процент от пробата.
Повторяемост
Разликата между резултатите от две паралелни определяния, проведени върху една и съща проба, не трябва да надвишава:
|
— |
10 % в относителна стойност за съдържание на амоняк под 1,0 %, |
|
— |
0,1 % в абсолютна стойност за съдържание на амоняк 1,0 % или повече. |
7. Забележки
При условие че съдържанието на амоняк в пробата надвишава 0,6 %, първоначалният филтрат се разрежда.
CONWAY CELL
Scale 1/1

II. ЧРЕЗ ДЕСТИЛАЦИЯ
1. Цел и обхват
Настоящият метод дава възможност да се определи съдържанието на летливи азотни основи, изразени като амоняк, в рибено брашно, което на практика не съдържа карбамид. Той е приложим само тогава, когато съдържанието на амоняк е по-ниско от 0,25 %.
2. Принцип
Пробата се извлича с вода и разтворът се избистря и филтрира. Летливите азотни основи се извличат в точката на кипене чрез добавяне на магнезиев оксид и се събират в точно определено количество сярна киселина, излишъкът от която се ретитрува с разтвор на натриев хидроксид.
3. Реагенти
|
3.1. |
Разтвор на трихлороцетна киселина с концентрация 20 % (т/об). |
|
3.2. |
Магнезиев оксид |
|
3.3. |
Eмулсия срещу образуване на пяна (напр. силикон). |
|
3.4. |
Сярна киселина, 0,05 mol/l. |
|
3.5. |
Разтвор на натриев хидроксид, 0,1 mol/l. |
|
3.6. |
Разтвор на 0,3 % метилово червено в 95 %—96 % (о/о) етанол. |
4. Апаратура
|
4.1. |
Смесител (барабанен): приблизително 35—40 r.p.m. |
|
4.2. |
Апарат за дестилация тип Келдал. |
5. Процедура
Претеглят се 10 g от пробата с точност до 1 mg и заедно със 100 ml вода се поставят в градуирана колба от 200 ml. Разбъркват се или се разклащат в смесителя в продължение на 30 min. Добавят се 50 ml разтвор на трихлорооцетна киселина (т. 3.1), допълва се до пълния обем с вода, съдържанието силно се разклаща и се филтрира през нагънат филтър.
Взема се известно количество избистрен филтрат, подходящо за предполагаемото съдържание на летливи азотни основи (100 ml обикновено са достатъчни). Разрежда се до 200 ml и се добавят 2 g магнезиев оксид (т. 3.2) и няколко капки емулсия срещу образуване на пяна (т. 3.3). Разтворът трябва да бъде алкален при проба с лакмусова хартия; в противен случай се добавя още магнезиев оксид (т. 3.2). Процедира се в съответствие с т. 5.2 и т. 5.3 от метода за анализ за определяне на съдържанието на суров протеин (част В от настоящото приложение).
Провежда се контролен опит по същата процедура, но без проба за анализ.
6. Изчисляване на резултатите
1 ml H2SO40,05 mol/l съответства на 1,7 mg амоняк.
Резултатът се изразява като процент от пробата.
Повторяемост
Разликата между резултатите от две паралелни определяния, проведени върху една и съща проба, не трябва да надвишава в относителна стойност 10 % амоняк.
Е. ОПРЕДЕЛЯНЕ СЪДЪРЖАНИЕТО НА АМИНОКИСЕЛИНИ (БЕЗ ТРИПТОФАН)
1. Цел и обхват
Настоящият метод служи за определяне с помощта на анализатор за аминокиселини на съдържанието на свободни (синтетични и естествени) аминокиселини и на общото съдържание на аминокиселини (участващи в пептидна връзка и свободни) във фуражите. Методът е приложим за следните аминокиселини: цист(е)ин, метионин, лизин, треонин, аланин, аргинин, аспарагинова киселина, глутаминова киселина, глицин, хистидин, изолевцин, левцин, фенилаланин, пролин, серин, тирозин и валин.
Методът не позволява да се разграничат солите на аминокиселините и да се различават D- и L-формите на аминокиселините. Той не важи за определяне на съдържанието на триптофан или на хидрокси аналози на аминокиселините.
2. Принцип
2.1. Свободни аминокиселини
Свободните аминокиселини се извличат с разредена солна киселина. Извлечените паралелно с тях азотисти макромолекули се утаяват със сяро-салицилова киселина и отстраняват посредством филтриране. Филтрираният разтвор се коригира до pH 2,20. Аминокиселините се отделят посредством йонообменна хроматография и съдържанието им се определя посредством нинхидринова реакция с фотометрично отчитане при 570 nm.
2.2. Общо съдържание на аминокиселини
Процедурата, която ще бъде избрана, зависи от вида на аминокиселините, които се изследват. Цист(е)инът и метионинът трябва да бъдат окислени съответно до цистеинова киселина и метионин сулфон преди хидролизата. Тирозинът трябва да се определя в продукти от хидролизата в неокислени проби. Всички останали аминокиселини, изброени в т. 1, могат да бъдат определени както в окислените, така и в неокислените проби.
Окислението се извършва при 0 oC със смес от пероксимравчена киселина и фенол. Излишъкът от окислителния реагент се разгражда с натриев дисулфит. Пробата, независимо дали е окислена или неокислена, се подлага на хидролиза със солна киселина (т. 3.20) в продължение на 23 часа. Продуктът на хидролизата се коригира до pH 2,20. Аминокиселините се отделят посредством йонообменна хроматография и се определят посредством нинхидринова реакция с фотометрично отчитане при 570 nm (440 nm за пролина).
3. Реагенти
Да се използва двойно дестилирана вода или вода с равностойно качество (проводимост < 10 μS)
|
3.1. |
Водороден прекис, w (w/w) = 30 %. |
|
3.2. |
Мравчена киселина, w (w/w) = 98—100 %. |
|
3.3. |
Фенол. |
|
3.4. |
Натриев дисулфит. |
|
3.5. |
Натриев хидроксид. |
|
3.6. |
Дихидрат на 5-сулфосалициловата киселина. |
|
3.7. |
Солна киселина, плътност приблизително 1,18 g/ml. |
|
3.8. |
Тринатриев цитрат дихидрат. |
|
3.9. |
2,2'-тиодиетанол (тиодигликол). |
|
3.10. |
Натриев хлорид. |
|
3.11. |
Нинхидрин. |
|
3.12. |
Петролен етер, температура на кипене 40 oС—60 oС. |
|
3.13. |
Норлевцин или друго съединение, подходящо за използване като вътрешен стандарт. |
|
3.14. |
Газообразен азот (<10 ppm кислород) |
|
3.15. |
1-октанол |
|
3.16. |
Аминокиселини. |
|
3.16.1. |
Стандартни вещества, посочени в точка 1. Химически чисти съединения, които не съдържат кристализационна вода. Изсушват се във вакуум над P205 или H2SO4 в продължение на 1 седмица преди употреба |
|
3.16.2. |
Цистеинова киселина. |
|
3.16.3. |
Метионин сулфон. |
|
3.17. |
Разтвор на натриев хидроксид, с = 7,5 mol/l: Разтварят се 300 g NaOH (т. 3.5) във вода и се допълва с вода до 1 литър. |
|
3.18. |
Разтвор на натриев хидроксид, с = 1 mol/l: Разтварят се 40 g NaOH (т. 3.5) във вода и се допълва с вода до 1 литър. |
|
3.19. |
Разтвор на мравчена киселина и фенол: Смесват се 889 g мравчена киселина (т. 3.2) със 111 g вода и се добавят 4,73 g фенол (т. 3.3). |
|
3.20. |
Смес за хидролиза, c = 6 mol HCl/l, съдържаща 1 g фенол на литър: Добавя се 1 g фенол (т. 3.3) към 492 ml HCl (т. 3.7) и се допълва с вода до 1 литър. |
|
3.21. |
Смес за екстракция, c = 0,1 mol HCl/l, съдържаща 2 % тиодигликол: вземат се 8,2 ml HCl (т. 3.7), разреждат се с около 900 ml вода, добавят се 20 ml тиодигликол (т. 3.9) и се допълва с вода до 1 литър (да не се смесват директно веществата в т. 3.7 и т. 3.9). |
|
3.22. |
5-сулфосалицилова киселина ß = 6 %: Разтварят се 60 g 5-сулфосалицилова киселина (т. 3.6) във вода и се допълва с вода до 1 литър |
|
3.23. |
Смес за окисляване (пероксимравчена киселина и фенол): Смесват се 0,5 ml водороден прекис (т. 3.1) с 4,5 ml разтвор от мравчена киселина и фенол (т. 3.19) в малка бехерова чаша. Инкубира се при температура 20 oC—30 oC в продължение на 1 час, за да се образува пероксимравчена киселина, след което се охлажда в ледена водна баня (15 min) преди да се добави към пробата. Внимание: Да се избягва контакт с кожата и да се носи защитно облекло. |
|
3.24. |
Цитратен буферен разтвор, с = 0,2 mol Na+/l; pH 2,20: Разтварят се 19,61 g натриев цитрат (т. 3.8), 5 ml тиодигликол (т. 3.9), 1 g фенол (т. 3.3) и 16,50 ml HCl в приблизително 800 ml вода. Стойността на рН се регулира на 2,20. Допълва се до 1 литър с вода. |
|
3.25. |
Буферни разтвори за отмиване, приготвени в съответствие с указанията за използвания уред за анализ (т. 4.9). |
|
3.26. |
Нинхидринов реагент, приготвен в съответствие с указанията за използвания уред за анализ (т. 4.9). |
|
3.27. |
Стандартни разтвори на аминокиселини. Тези разтвори се съхраняват при температура под 5 oC. |
|
3.27.1. |
Основен стандартен разтвор на аминокиселини (т. 3.16.1). c = 2,5 μmol/ml от всяка в солна киселина. Могат да бъдат купени в търговската мрежа. |
|
3.27.2. |
Основен стандартен разтвор на цистеинова киселина и на метионин сулфон, c = 1,25 μmol/ml. Разтварят се 0,2115 g цистеинова киселина (т. 3.16.2) и 0,2265 g метионин сулфон (т. 3.16.3) в цитратния буферен разтвор (т. 3.24) в градуирана колба от 1 литър и се допълва до този обем с цитратен буферен разтвор. Да се съхранява под 5 oC не повече от 12 месеца. Този разтвор не се използва, ако базовият стандартен разтвор (т. 3.27.1) съдържа цистеинова киселина и метионин сулфон. |
|
3.27.3. |
Базов стандартен разтвор на вътрешния стандарт, т.е. норлевцин, c = 20 μmol/ml. Разтварят се 0,6560 g норлевцин (т. 3.13) в цитратния буферен разтвор (3.24) в градуирана колба и се допълва до 250 ml с цитратен буферен разтвор. Да се съхранява под 5 oC не повече от 6 месеца. |
|
3.27.4. |
Калибрационен разтвор на стандартни аминокиселини за употреба с хидролизатите, c = 5 nmol/50 μl цистеинова киселина и метионин сулфон и c = 10 nmol/50 μl за другите аминикиселини. Разтварят се 2,2 g натриев хлорид в бехерова чаша от 100 ml в 30 ml цитратен буферен разтвор (т. 3.24) Добавят се 4,00 ml базов стандартен разтвор на аминокиселини (т. 3.27.1), 4,00 ml базов стандартен разтвор на цистеинова киселина и метионин сулфон (т. 3.27.2) и 0,50 ml базов стандартен разтвор на вътрешния стандарт (т. 3.27.3), ако се използва такъв. Коригира се до рН 2,20 с натриев хидроксид (т. 3.18). Прехвърля се в градуирана колба от 50 ml, допълва се до марката с цитратен буферен разтвор (т. 3.24) и се разбърква. Да се съхранява под 5 oC не повече от 3 месеца. Вж. също забележка 9.1. |
|
3.27.5. |
Калибрационен разтвор на стандартни аминокиселини за употреба с приготвени в съответствие с точка 5.3.3.1 хидролизати и за употреба с екстракти (т. 5.2). Калибрационният разтвор се приготвя в съответствие с т. 3.27.4, но не се добавя натриев хлорид. Да се съхранява под 5 oC не повече от 3 месеца. |
4. Апаратура
|
4.1. |
Облодънна колба с обем 100 или 250 ml, снабдена с обратен хладник. |
|
4.2. |
Шише от боросиликатно стъкло от 100 ml с винтова капачка с гумена/тефлонова гарнитура (напр. Duran, Schott), което може да се използва в пещ. |
|
4.3. |
Пещ с принудителна вентилация и температурен регулатор с точност по-висока от ± 2 oC. |
|
4.4. |
pH-метър (три знака след десетичната запетая). |
|
4.5. |
Мембранен филтър, 0,22 μm. |
|
4.6. |
Центрофуга. |
|
4.7. |
Ротационен вакуумен изпарител. |
|
4.8. |
Механична клатачна машина или магнитна бъркалка. |
|
4.9. |
Уред за анализ на аминокиселини или уред за HPLC с йонобменна колона, устройство за нинхидрин, следколонна дериватизация и фотометричен детектор. Колоната се запълва със сулфонирани полистиренови смоли, които могат да отделят аминокиселините една от друга и от други вещества, реагиращи положително на нинхидрин. Циркулацията на буферния разтвор и на нинхидриновия реагент се осигурява от помпи с устойчивост на потока от ±0,5 % за целия период, включващ както стандартното времетраене на калибрирането, така и извършване на анализа на пробата. При някои уреди за анализ на аминокиселини може да се използва хидролизна процедура, при която хидролизатът е с концентрация на натрий с = 0,8 mol/l и съдържа цялата остатъчна мравчена киселина от етапа на окисляването. Други уреди не осигуряват задоволително отделяне на някои определени аминокиселини, ако хидролизатът съдържа излишък от мравчена киселина и/или високи концентрации на натриеви йони. В този случай количеството на киселината се намалява чрез изпарение до приблизително 5 ml след хидролизата и преди коригиране на киселинността. Изпарението се извършва във вакуумна среда при максимум 40 oС. |
5. Процедура
5.1. Подготовка на пробата
Пробата се смила, така че да може да премине през сито с отвори 0,5 mm. Пробите с голяма влажност трябва да се изсушат на въздух при температура най-много 50 oС или чрез лиофилизация преди смилането. Пробите с високо съдържание на мастни вещества се екстрахират с петролен етер (т. 3.12) преди смилането.
5.2. Определяне на свободни аминокиселини във фуражи и премикси
Претегля се с точност до 0,2 mg подходящо количество (1—5 g) от подготвената проба (т. 5.1) в конусовидна колба и се добавят 100,0 ml от сместа за екстракция (т. 3.21). Разбърква се или се размесва в продължение на 60 min с механична клатачна машина или с магнитна бъркалка (точка 4.8). Оставя се седиментът да се утаи и 10,0 ml от супернатанта се слага с пипета в бехерова чаша 100 ml.
Като се разбърква, се добавят 5,0 ml разтвор на сулфосалицилова киселина (т. 3.22), и с помощта на магнитната бъркалка разбъркването продължава още 5 min. Супернатантът се филтрира или центрофугира, така че да не остане никаква утайка. Поставят се 10,0 ml от получения разтвор в бехерова чаша 100 ml и с натриев хидроксид (т. 3.18) се коригира pH до 2,20, прехвърля се в мерителна колба с подходящ обем, като се използва цитратен буферен разтвор (т. 3.24) и се допълва до пълния обем с буферния разтвор (т. 3.24).
Ако се използва вътрешен стандарт, се добавя 1,00 ml от вътрешния стандарт (т. 3.27.3) за всеки 100 ml от крайния разтвор и се допълва до пълния обем с буферния разтвор (т. 3.24).
Пристъпва се към етапа на хроматографията в съответствие с точка 5.4.
Ако екстрактите няма да се използват през същия ден, те трябва да се съхраняват при температура под 5 oC.
5.3. Определяне на общото количество аминокиселини
5.3.1.
Претеглят се с точност до 0,2 mg между 0,1 и 1 g от подготвената проба (т. 5.1) във:
|
— |
облодънна колба с обем от 100 ml (т. 4.1) за октрита хидролиза (т. 5.3.2.3), или |
|
— |
облодънна колба с обем 250 ml (т. 4.1), ако се изисква ниска концентрация на натрий (т. 5.3.3.1), или |
|
— |
шише от 100 ml, снабдено с винтова капачка (точка 4.2) (за закрита хидролиза, т. 5.3.2.4). |
Претеглената част от пробата трябва да съдържа около 10 mg азот и не повече от 100 mg влага.
Колбата/шишето се слага в ледена водна баня и се охлажда до 0 oC, добавят се 5 ml от сместа за окисляване (т. 3.23) и се разбърква с помощта на стъклена шпатула със закривен връх. Колбата/шишето, в която/което се намира шпатулата, се запечатва херметично с филм, водната баня със запечатания съд се слага в хладилник при 0 oC и се оставя за 16 часа. След 16 часа се изважда от хладилника и излишният окислителен реагент се разлага чрез добавяне на 0,84 g натриев дисулфит (т. 3.4).
Пристъпва се към процедурата в т. 5.3.2.1
5.3.2.
5.3.2.1.
Към окислената проба, подготвена в съответствие с т. 5.3.1 се добавят 25 ml от сместа за хидролизиране (т. 3.20), като се измиват всякакви остатъци от пробата, полепнали по стените на съда и шпатулата.
Според използваната процедура за хидролиза се процедира в съответствие с процедурата в т. 5.3.2.3 или в т. 5.3.2.4.
5.3.2.2.
В облодънна колба с обем 100 ml или от 250 ml (т. 4.1) или в шише от 100 ml с винтoва капачка (т. 4.2) се претеглят с точност до 0,2 mg между 0,1 и 1 g от подготвената проба (т. 5.1). Претеглената част от пробата трябва да съдържа около 10 mg азот. Внимателно се добавят 25 ml от сместа за хидролиза (т. 3.20) и се смесват с пробата. Процедира се в съответствие с процедурата в т. 5.3.2.3 или в т. 5.3.2.4.
5.3.2.3.
Добавят се 3 стъклени топчета към сместа в колбата (приготвена в съответствие с процедурата в т. 5.3.2.1 или в т. 5.3.2.2) и се вари с обратех хладник и при постоянно отделяне на мехурчета в продължение на 23 часа. След приключване на хидролизата, хладникът се изплаква с 5 ml цитратен буферен разтвор (т. 3.24). Колбата се откача и се охлажда в ледена баня.
Процедира се в съответствие с т. 5.3.3.
5.3.2.4.
Шишето с приготвената съгласно т. 5.3.2.1 или т. 5.3.2.2 смес се поставя в сушилня (т. 4.3) при 110 oC. За да се избегне покачване на налягането (поради отделянето на газообразни вещества) и за да се предотврати избухване, през първия час винтовата капачка се поставя върху гърлото на съда без да се затваря. Да не се затваря съдът с капачката. След един час съдът се затваря с капачката и се оставя в сушилнята (т. 4.3) за 23 часа. След приключване на хидролизата, шишето се изважда от сушилнята, внимателно се отваря капачката на шишето и то се слага в ледена водна баня. Оставя се да се охлади.
В зависимост от процедурата за коригиране на pH (т. 5.3.3), съдържанието на шишето количествено се прехвърля в бехерова чаша от 250 ml или в облодънна колба от 250 ml, като се използва цитратен буферен разтвор (т. 3.24).
Процедира се в съответствие с т. 5.3.3.
5.3.3.
В зависимост от толерантността на уреда за анализ на аминокиселини към натрий, коригирането на pH се извършва в съответствие с т. 5.3.31 или с т. 5.3.3.2.
5.3.3.1.
Препоръчва се да се използва вътрешен основен стандартен разтвор (т. 3.27.3), когато се използват уреди за анализ на аминокиселини, изискващи ниски концентрации на натрий (когато трябва да се намали обемът на киселината).
В този случай преди изпарението към хидролизата се добавят 2,00 ml от вътрешния основен стандартен разтвор (т. 3.2.7.3).
Добавят се 2 капки 1-октанол (т. 3.15) към получения в съответствие с т. 5.3.2.3 или т. 5.3.2.4 хидролизат.
Като се използва ротационен изпарител (т. 4.7), обемът се намалява до 5—10 ml във вакуум при 40 oC. Ако случайно обемът е намален на по-малко от 5 ml, хидролизатът трябва да се изхвърли и анализът да се повтори.
Коригира се рН до 2,20 с разтвор на натриев хидроксид (т. 3.18) и се пристъпва към изпълнението на процедурата в т. 5.3.4.
5.3.3.2.
Вземат се получените в съответствие с т. 5.3.2.3 или т. 5.3.2.4 хидролизати и частично се неутрализират, като внимателно се добавят при непрекъснато бъркане 17 ml разтвор на натриев хидроксид (т. 3.17), като се гарантира, че температурата не се покачва над 40 oС.
Коригира се рН до 2,20 при стайна температура, като се използва разтвор на натриев хидроксид (т. 3.17) и накрая разтвор на натриев хидроксид (т. 3.18). Пристъпва се към процедурата в т. 5.3.4
5.3.4.
Хидролизатът с коригирано рН (т. 5.3.3.1 или т. 5.3.3.2) се прехвърля количествено с помощта на цитратен буферен разтвор (т. 3.24) в градуирана колба от 200 ml и се допълва до маркировката с буферен разтвор (т. 3.24).
Ако не е бил използван вътрешен стандарт, се добавят 2,00 ml от вътрешния стандарт (т. 3.27.3) и се допълва до пълния обем с цитратен буферен разтвор (т. 3.24). Старателно се размесва.
Пристъпва се към определяне чрез хроматография (т. 5.4).
Ако разтворите от пробата няма да се изследват през същия ден, те трябва да се съхраняват при температура под 5 oC.
5.4. Хроматографско изследване
Преди започване на хроматографското изследване екстрактът (т. 5.2) или хидролизатът (т. 5.3.4) се затопля до стайна температура. Сместа се разклаща и подходящо количество от нея се филтрира през мембранен филтър с размер на отворите 0,22 μm (т. 4.5). Полученият бистър разтвор се подлага след това на йонообменна храматография, като се използва уред за анализ на аминокиселини (т. 4.9).
Впръскването може да се извърши ръчно или автоматично. Важно е в колоната за анализ да се поставя еднакво — с точност до ±0,5 % — количество разтвор от стандарта и от пробата, освен когато се използва вътрешен стандарт, и съотношението между натрия и аминокиселини в разтворите на стандарта и на пробата да е толкова близко, колкото е възможно.
По принцип, честотата на калибровъчните операции зависи от стабилността на нинхидриновия реагент и от аналичната система. Стандартът или изследваната проба се разреждат с цитратен буферен разтвор (т. 3.24), така че пиковата повърхност на стандарта да бъде от 30 до 200 % от пиковата повърхност на аминокиселината на пробата.
Хроматографията на аминокиселините варира леко според типа на уреда за анализ и според използваната смола. Използваната система трябва да е в състояние да отдели аминокиселините една от друга и от вещества, реагиращи положително на нинхидрин. В работния диапазон на хроматографската система трябва да се наблюдава линейна зависимост спрямо промяната на количествата аминокиселини, които се въвеждат в колоната.
При хроматографския етап на анализа се прилагат съотношенията между най-ниските и най-високите стойности, споменати по-долу, при условие че се анализира еквимоларен разтвор (на определяните аминокиселини). Еквимоларният разтвор трябва да съдържа най-малко 30 % от максималното количество от всяка аминокиселина, която може да да бъде прецизно измерена със системата за анализ на аминокиселини (т. 4.9).
За разграничаването на треонин от серин съотношението между най-ниската и най-високата стойност върху хроматограмата за по-слабо представената от двете застъпващи се аминокиселини не трябва да надвишава 2:10. (ако се определят само цист(е)ин, метионин, треонин и лизин, недостатъчното разграничаване на придружаващите пикови стойности ще повлияе отрицателно върху определянето). За всички останали аминокиселини разграничаването трябва да бъде по-добро от 1:10.
Системата трябва да гарантира, че лизинът се разграничава от „лизиновите артефакти“ и от орнитин.
6. Изчисляване на резултатите
Пиковата повърхност на пробата и на стандарта се измерва за всяка отделна аминокиселина и се пресмята количеството Х в грамове аминокиселина на килограм проба.
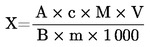
Ако се използва вътрешен стандарт, се умножава по: 
|
A |
= |
пикова повърхност на хидролизат или екстракт |
|
B |
= |
пикова площ на стандартния калибрационен разтвор |
|
C |
= |
пикова повърхност на вътрешния стандарт в хидролизат или екстракт |
|
D |
= |
пикова площ, вътрешен стандарт, калибрационен стандартен разтвор |
|
M |
= |
моларно тегло на определяната амонокиселина |
|
c |
= |
концентрация на стандарта в μmol/ml |
|
m |
= |
тегло на пробата (g) (коригирано, за да се получи началното тегло, ако продуктът е изсушен или обезмаслен) |
|
V |
= |
ml общ хидролизат (т. 5.3.4) или ml изчислен общ разтворен обем на екстракта (т. 6.1) |
Цистинът и цистеинът се определят като цистеинова киселина в продукти на хидролизата на окислена проба, но се изчисляват като цистин (C6H12N2O4S2, M 240,30 g/mol), като се използва M 120,15 g/mol (= 0,5 × 240,30 g/mol).
Метионинът се определя като метионин сулфон в продуктите на хидролизата на окислената проба, но се изчислява като метионин, като се използва M на метионина: 149,21 g/mol.
Добавеният свободен метионин се определя след екстракция като метионин, като за изчисленията се използва същото M.
|
6.1. |
Общият разтворен обем на екстрактите (F) за определянето на аминокиселините (т. 5.2) се изчислява, както следва: 
|
7. Оценка на метода
Методът е изпробван чрез взаимно сравнение, направено на международно ниво през 1990 година, като за целта са използвани четири различни вида фураж (фуражна смеска за свине, смеска за бройлери, протеинов концентрат, премикс). Резултатите за средните стойности и за стандартното отклонение след отстраняване на отклоняващите се стойности са дадени в таблицата в настоящата точка:
Средни стойности в g/kg
|
Еталонен материал |
Аминокиселина |
||||||
|
Треонин |
Цистин/цистеин |
Метионин |
Лизин |
||||
|
Фуражна смеска за свине |
6,94 n = 15 |
3,01 n = 17 |
3,27 n = 17 |
9,55 n = 13 |
|||
|
Смеска за бройлери |
9,31 n = 16 |
3,92 n = 18 |
5,08 n = 18 |
13,93 n = 16 |
|||
|
Протеинов концентрат |
22,32 n = 16 |
5,06 n = 17 |
12,01 n = 17 |
47,74 n = 15 |
|||
|
Премикс |
58,42 n = 16 |
— |
90,21 n = 16 |
98,03 n = 16 |
|||
|
|||||||
7.1. Повторяемост
Повторяемостта на гореспоменатото сравнително изпитване, изразена като „стандартно лабораторно отклонение“, е дадена в таблиците по-долу:
Вътрешно лабораторно стандартно отклонение (Sr) в g/kg
|
Еталонен материал |
Аминокиселина |
||||||
|
Треонин |
Цистин/цистеин |
Метионин |
Лизин |
||||
|
Фуражна смеска за свине |
0,13 n = 15 |
0,10 n = 17 |
0,11 n = 17 |
0,26 n = 13 |
|||
|
Смеска за бройлери |
0,20 n = 16 |
0,11 n = 18 |
0,16 n = 18 |
0,28 n = 16 |
|||
|
Протеинов концентрат |
0,48 n = 16 |
0,13 n = 17 |
0,27 n = 17 |
0,99 n = 15 |
|||
|
Премикс |
1,30 n = 16 |
— |
2,19 n = 16 |
2,06 n = 16 |
|||
|
|||||||
Коефициент на вариация (%) за вътрешно лабораторното стандартно отклонение (Sr)
|
Еталонен материал |
Аминокиселина |
||||||
|
Треонин |
Цистин/цистеин |
Метионин |
Лизин |
||||
|
Фуражна смеска за свине |
1,9 n = 15 |
3,3 n = 17 |
3,4 n = 17 |
2,8 n = 13 |
|||
|
Смеска за бройлери |
2,1 n = 16 |
2,8 n = 18 |
3,1 n = 18 |
2,1 n = 16 |
|||
|
Протеинов концентрат |
2,7 n = 16 |
2,6 n = 17 |
2,2 n = 17 |
2,4 n = 15 |
|||
|
Премикс |
2,2 n = 16 |
— |
2,4 n = 16 |
2,1 n = 16 |
|||
|
|||||||
7.2. Възпроизводимост
В таблицата по-долу са дадени резултатите за стандартното отклонение между лабораториите при гореспоменатото сравнение:
Стандартно отклонение (SR) между лабораториите в g/kg
|
Еталонен материал |
Аминокиселина |
||||||
|
Треонин |
Цистин/цистеин |
Метионин |
Лизин |
||||
|
Фуражна смеска за свине |
0,28 n = 15 |
0,30 n = 17 |
0,23 n = 17 |
0,30 n = 13 |
|||
|
Смеска за бройлери |
0,48 n = 16 |
0,34 n = 18 |
0,55 n = 18 |
0,75 n = 16 |
|||
|
Протеинов концентрат |
0,85 n = 16 |
0,62 n = 17 |
1,57 n = 17 |
1,24 n = 15 |
|||
|
Премикс |
2,49 n = 16 |
— |
6,20 n = 16 |
6,62 n = 16 |
|||
|
|||||||
Коефициент на вариация (%) за стандартното отклонение между лабораториите (SR)
|
Еталонен материал |
Аминокиселина |
||||||
|
Треонин |
Цистин/цистеин |
Метионин |
Лизин |
||||
|
Фуражна смеска за свине |
4,1 n = 15 |
9,9 n = 17 |
7,0 n = 17 |
3,2 n = 13 |
|||
|
Смеска за бройлери |
5,2 n = 16 |
8,8 n = 18 |
10,9 n = 18 |
5,4 n = 16 |
|||
|
Протеинов концентрат |
3,8 n = 16 |
12,3 n = 17 |
13,0 n = 17 |
3,0 n = 15 |
|||
|
Премикс |
4,3 n = 16 |
— |
6,9 n = 16 |
6,7 n = 16 |
|||
|
|||||||
8. Използване на еталонни материали
Точното прилагане на метода се проверява с повтаряне на измерванията върху сертифицирани еталонни материали при наличие на такива. Препоръчва се калибриране със сертифициран калибрационен разтвор на аминокиселини.
9. Забележки
|
9.1. |
Поради различията между уредите за анализ на аминокиселини, за ръководни се считат крайните концентрации на калибрационните разтвори на стандартни аминокиселини (вж. т. 3.27.4 и т. 3.27.5) и на продукта на хидролизата (вж. т. 5.3.4). Диапазонът на линейна чувствителност на апарата трябва да се провери за всички аминокиселини. Стандартният разтвор се разрежда с цитратен буфер, за да може пиковите повърхности да съвпаднат със средата на диапазона. |
|
9.2. |
Когато за анализиране на хидролизатите се използва оборудване за високоефективна течна хроматография, експерименталните условия трябва да бъдат оптимизирани според препоръките на производителя. |
|
9.3. |
При прилагането на метода за фуражи, съдържащи повече от 1 % хлорид (концентрати, минерални фуражи, хранителни добавки) е възможно да се получат твърде ниски стойности за метионина и затова се налага специална обработка. |
Ж. ОПРЕДЕЛЯНЕ НА ТРИПТОФАН
1. Цел и обхват
Методът дава възможност да се определят общият и свободният триптофан във фуражите. Методът не прави разлика между D- и L-формите.
2. Принцип
За да се определи общият триптофан, пробата се хидролизира в алкална среда с наситен разтвор на бариев хидроксид и се загрява до 110 oС в продължение на 20 часа. След хидролизата се прибавя вътрешен стандарт.
За да се определи свободният триптофан, пробата се подлага на екстракция в умерено кисела среда при наличие на вътрешен стандарт.
Триптофанът и вътрешният стандарт в хидролизата или в екстракта се определят чрез HPLC с флуоресцентен детектор.
3. Реагенти
|
3.1. |
Трябва да се използва двойно дестилирана вода или вода с равностойно качество (проводимост < 10 μS/cm) |
|
3.2. |
Стандартно вещество: триптофан (чистота/съдържание ≥ 99 %), изсушен във вакуум над фосфорен пентоксид. |
|
3.3. |
Вътрешен стандарт: α-метилтриптофан (чистота/съдържание ≥ 99 %), изсушен във вакуум над фосфорен пентоксид. |
|
3.4. |
Бариев хидроксид октахидрат (внимава се Ва(ОН)2 . 8Н2О да не се излага прекалено дълго на въздух, за да се избегне образуването на ВаСО3, който може да попречи на определянето) (вж. забележка в т. 9.3). |
|
3.5. |
Натриев хидроксид. |
|
3.6. |
Ортофосфорна киселина, w (w/w) = 85 % |
|
3.7. |
Солна киселина ρ201,19 g/ml |
|
3.8. |
метанол с чистота за HPLC |
|
3.9. |
Петролен етер, диапазон на кипене: 40—60 oС |
|
3.10. |
Разтвор на натриев хидроксид, с = 1 mol/l: Разтварят се 40,0 g NaOH (т. 3.5) във вода и се долива вода (т. 3.1) до 1 литър. |
|
3.11. |
Солна киселина, с = 6 mol/l: Взимат се 492 ml НСl (т. 3.7) и се долива вода до 1 литър. |
|
3.12. |
Солна киселина, с = 1 mol/l: Взимат се 82 ml НСl (т. 3.7) и се долива вода до 1 литър. |
|
3.13. |
Солна киселина, с = 0,1 mol/l: Взимат се 8,2 ml НСl (т. 3.7) и се долива вода до 1 литър. |
|
3.14. |
Ортофосфорна киселина, с = 0,5 mol/l: Взимат се 34 ml ортофосфорна киселина (т. 3.6) и се долива вода (т. 3.1) до 1 литър. |
|
3.15. |
Концентриран разтвор на триптофан (т. 3.2), с = 2,50 μmol/ml: В мерителна колба от 500 ml се разтварят 0,2553 g триптофан (т. 3.2) в солна киселина (т. 3.13) и се допълва до пълния обем със солна киселина (т. 3.13). Разтворът се съхранява при –18 oС в продължение най-много на 4 седмици. |
|
3.16. |
Концентриран разтвор на вътрешния стандарт, с = 2,50 μmol/ml В мерителна колба от 500 ml се разтварят 0,2728 g α-метил-триптофан (т. 3.3) в солна киселина (т. 3.13) и се допълва до пълния обем със солна киселина (т. 3.13). Разтворът се съхранява при –18 oС в продължение най-много на 4 седмици. |
|
3.17. |
Стандартен калибрационен разтвор на триптофан и вътрешен стандарт: Взимат се 2,00 ml концентриран разтвор на триптофан (т. 3.15) и 2,00 ml концентриран разтвор на вътрешния стандарт (α-метил-триптофан) (т. 3.16). Разреждат се с вода (т. 3.1) и метанол (т. 3.8) приблизително до същия обем и приблизително до същата концентрация на метанол (10—30 %) като тези на готовия хидролизат. Разтворът трябва да бъде приготвен непосредствено преди употреба. Да се осигури защита срещу пряка слънчева светлина по време на приготвянето на разтвора. |
|
3.18. |
Оцетна киселина |
|
3.19. |
1,1,1-трихлоро-2-метил-2-пропанол. |
|
3.20. |
Етаноламин w (w/w) > 98 % |
|
3.21. |
Разтвор от 1 g 1,1,1-трихлоро-2-метил-2-пропанол (т. 3.19) в 100 ml метанол (т. 3.8). |
|
3.22. |
Подвижна фаза за HPLC: 3,00 g оцетна киселина (т. 3.18) + 900 ml вода (т. 3.1) + 50 ml разтвор (т. 3.21) на 1,1,1-трихлоро-2-метил-2-пропанол- (т. 3.19) в метанол (т. 3.8) (1 g/100 ml). Коригира се рН с етаноламин (т. 3.20) до достигане на стойност 5,00. Долива се вода (т. 3.1) до 1 000 ml. |
4. Апаратура
|
4.1. |
Оборудване за HPLC със спектрофлуорометричен детектор |
|
4.2. |
Колона за течна хроматография, 125 mm × 4 mm, С18, пълнеж от 3 μm или еквивалентен |
|
4.3. |
рН-метър. |
|
4.4. |
Полипропиленова колба с обем 125 ml, с широко гърло и винтова капачка. |
|
4.5. |
Мембранен филтър, 0,45 μm. |
|
4.6. |
Автоклав, 110 (±2) oС, 1,4 (±0,1) бара |
|
4.7. |
Механичен клатачна машина или магнитна бъркалка. |
|
4.8. |
Миксер Vortex |
5. Процедура
5.1. Подготовка на пробите
Пробата се смила, така че да може да премине през сито с отвори 0,5 mm. Пробите с голяма влажност трябва да се изсушат на въздух при температура най-много 50 oС или чрез лиофилизация преди смилането. Пробите с високо съдържание на мастни вещества се подлагат на екстракция с петролен етер (т. 3.9) преди смилането.
5.2. Определяне на свободния триптофан (екстракт)
Претегля се с точност до 1 mg подходящо количество (1—5 g) от подготвената проба (т. 5.1) в конусовидна колба. Прибавят се 100,0 ml солна киселина (т. 3.13) и 5,00 ml концентриран разтвор на вътрешния стандарт (т. 3.16). Разбърква се или се размесва в продължение на 60 min с механична клатачна машина или или с магнитната бъркалка (т. 4.7). Оставя се седиментът да се утаи и с пипета се прехвърлят 10,0 ml от супернатанта в бехерова чаша. Прибавят се 5 ml ортофосфорна киселина (т. 3.14). Коригира се рН до 3, като се използва натриев хидроксид (т. 3.10). Прибавя се достатъчно количество метанол (т. 3.8), за да се постигне концентрация между 10 и 30 % метанол в крайния обем. Сместа се прехвърля в мерителна колба с подходящ обем и се разрежда с вода до достигане на необходимия за хроматографията обем (приблизително еднакъв с този на калибрационния стандартен разтвор (т. 3.17).
Няколко милилитра от разтвора се фитрират през мембранен филтър от 0,45 μm (т. 4.5), преди да бъдат впръскани в колоната за HPLC. Пристъпва се към етапа на хроматографията в съответствие с точка 5.4.
Стандартният разтвор и екстрактите следва да се пазят от пряка слънчева светлина. Ако не е възможно да се анализират в същия ден, екстрактите могат да се съхраняват при температура 5 oС за най-много 3 дни.
5.3. Определяне на общия триптофан (хидролизат)
Претеглят се с точност до 0,2 mg между 0,1 и 1 g от подготвената проба (т. 5.1) в полипропиленова колба (т. 4.4). Претеглената част от пробата трябва да съдържа около 10 mg азот. Добавят се 8,4 g. бариев хидроксид октахидрат (т. 3,4) и 10 ml вода. Съставките се размесват с миксер Vortex (т. 4.8) или с магнитна бъркалка (т. 4.7). Покритият с тефлон магнит се оставя в сместа. Стените на съда се изплакват с 4 ml вода. Поставя се винтовата капачка и колбата се затваря, без да се затяга капачката. Прехвърля се в автоклав (т. 4.6) с вряща вода и пара за 30—60 min. Автоклавът се затваря и се включва на температура 110 oС (± 2) в продължение на 20 часа.
Преди отварянето на автоклава температурата се намалява до малко под 100 oC. За да се избегне кристализация на Ва(ОН)2 . 8Н2О, към загрятата смес се прибавят 30 ml вода със стайна температура. Леко се разклаща или разбърква. Прибавят се 2,00 ml от концентрирания разтвор на вътрешния стандарт (α-метил-триптофан) (т. 3.16). Съдовете се охлаждат във вода/ледена вана в продължение на 15 min.
След това се прибавят 5 ml ортофосфорна киселина (т. 3.14). Съдът се държи в охлаждащата вана и съдържанието се неутрализира със HCl (т. 3.11), като се бърка непркъснато и рН се коригира до 3,0 с помощта на HCl (т. 3.12). Прибавя се достатъчно количество метанол, за да се достигне концентрация между 10 и 30 % метанол в крайния обем. Прехвърля се в мерителна колба с подходящ обем и се разрежда с вода до достигане на обема, необходим за хроматографията (например 100 ml). Добавянето на метанол не трябва да предизвика утаяване.
Няколко милилитра от разтвора се фитрират през мембранен филтър от 0,45 μm (т. 4.5) преди да бъдат впръскани в колоната за HPLC. Пристъпва се към етапа на хроматографията в съответствие с точка 5.4.
Стандартният разтвор и хидролизатите трябва да се пазят от пряка слънчева светлина. Ако не е възможно да се анализират в същия ден, хидролизатите могат да се съхраняват при температура 5 oС в продължение най-много на 3 дни.
5.4. Определяне с помощта на HPLC
За ориентиране се предлагат следните условия за изократно елуиране; може да се използват и други, при условие, че дават равностойни резултати (вж. също и забележките т. 9.1 и т. 9.2):
|
Колона за течна хроматография (т. 4.2): |
125 mm × 4 mm, С18, пълнеж от 3 μm или еквивалентна |
|
Температура на колоната: |
стайна температура |
|
Подвижна фаза (т. 3.22): |
3,00 g оцетна киселина (т. 3.18) + 900 ml вода (т. 3.1) +50,0 ml разтвор (т. 3.21) на 1,1,1-трихлоро-2-метил-2-пропанол (т. 3.19) в метанол (точка 3.8) (1g/100ml). Стойността на рН се коригира на 5,00 етаноламин (т. 3.20). Долива се до 1 000 ml с вода (т. 3.1). |
|
Скорост на изтичане: |
(1ml/min). |
|
Общо време: |
приблизително 34 min |
|
Дължина на вълната за определяне: |
възбуждане 280 nm, излъчване: 356 nm. |
|
Обем на впръскване |
20 μl |
6. Изчисляване на резултатите
Количеството триптофан (Х) се изчислява в g на 100 g проба.
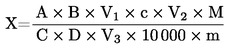
|
A |
= |
пикова площ на вътрешния стандарт, разтвор на стандарта за калибриране (т. 3.17) |
|
B |
= |
пикова площ на триптофана, ексракт (т. 5.2) или хидролизат (т. 5.3) |
|
V1 |
= |
обем в ml (2 ml) на концентрирания разтвор на триптофан (т. 3.15), прибавен към калибрационния разтвор (т. 3.17) |
|
c |
= |
концентрация в μmol/ml (= 2,50) на концентрирания разтвор на триптофан (т. 3.15), прибавен към калибрационния разтвор (т. 3.17) |
|
V2 |
= |
обем в ml на разтвора на вътрешния стандарт (т. 3.16), прибавен към екстракта (т. 5.2) (= 5,00 ml) или към хидролизата (т. 5.3) (= 2,00 ml) |
|
C |
= |
пикова повърхност на вътрешния стандарт, екстракт (т. 5.2) или хидролизат (т. 5.3) |
|
D |
= |
пикова площ на триптофана, калибрационен стандартен разтвор (т. 3.17) |
|
V3 |
= |
обем в ml (= 2,00 ml) от концентрирания разтвор на вътрешния стандарт (т. 3.16), прибавен към калибрационния стандартен разтвор (т. 3.17) |
|
m |
= |
тегло на пробата в g (коригирано, за да се получи началното тегло, ако продуктът е изсушен и/или обезмаслен) |
|
M |
= |
молекулно тегло на триптофана (= 204,23 g/mol) |
7. Повторяемост
Разликата в резултатите от две паралелни определяния на една и съща проба не трябва да надхвърля 10 % по отношение на най-високия резултат.
8. Резултати от съвместно изследване
Беше организирано съвместно изследване (4-то сравнение) в рамките на Общността, при което в 12 лаборатории бяха анализирани три проби, за да се сертифицира методът за изследване чрез хидролиза. Бяха направени 5 еднакви анализа на всяка проба. Резултатите са представени в таблицата по-долу:
|
|
Проба 1 Фураж за свине |
Проба 2 Фураж за свине с добавен L-триптофан |
Проба 3 Концентриран фураж за свине |
|
L |
12 |
12 |
12 |
|
n |
50 |
55 |
50 |
|
Средна стойност в g/kg |
2,42 |
3,40 |
4,22 |
|
sr [g/kg] |
0,05 |
0,05 |
0,08 |
|
r [g/kg] |
0,14 |
0,14 |
0,22 |
|
CVr [ %] |
1,9 |
1,6 |
1,9 |
|
SR [g/kg] |
0,15 |
0,20 |
0,09 |
|
R [g/kg] |
0,42 |
0,56 |
0,25 |
|
CVR [ %] |
6,3 |
6,0 |
2,2 |
|
L |
= |
брой на лабораториите, предали резултати |
|
n |
= |
брой на запазените индивидуални резултати след елиминиране на отклоняващите се стойности (определни чрез тестовете на Cochran и Dixon) |
|
sr |
= |
стандартно отклонение на повторяемостта |
|
SR |
= |
стандартно отклонение на възпроизводимостта |
|
r |
= |
повторяемост |
|
R |
= |
възпроизводимост |
|
CVr |
= |
коефициент на изменение на повторяемостта, % |
|
CVR |
= |
коефициент на изменение на възпроизводимостта, % |
Беше организирано друго съвместно изследване (3-то сравнение) в рамките на Общността, при което в 13 лаборатории бяха анализирани две проби, за да се сертифицира методът за екстракция на свободен триптофан. Бяха направени 5 еднакви анализа на всяка проба. Резултатите са представени в таблицата по-долу:
|
|
Проба 4 Смес от пшеница и соя |
Проба 5 Смес от пшеница и соя (= проба 4) с добавен триптофан (0,457g/kg) |
|
L |
12 |
12 |
|
n |
55 |
60 |
|
Средна стойност в g/kg |
0,391 |
0,931 |
|
sr [g/kg] |
0,005 |
0,012 |
|
r [g/kg] |
0,014 |
0,034 |
|
CVr [ %] |
1,34 |
1,34 |
|
SR [g/kg] |
0,018 |
0,048 |
|
R [g/kg] |
0,050 |
0,134 |
|
CVR [ %] |
4,71 |
5,11 |
|
L |
= |
брой на лабораториите, предали резултати |
|
n |
= |
брой на запазените индивидуални резултати след елиминиране на отклоняващите се стойности (определни чрез тестовете на Cochran и Dixon) |
|
sr |
= |
стандартно отклонение на повторяемостта |
|
SR |
= |
стандартно отклонение на възпроизводимостта |
|
r |
= |
повторяемост |
|
R |
= |
възпроизводимост |
|
CVr |
= |
коефициент на изменение на повторяемостта, % |
|
CVR |
= |
коефициент на изменение на възпроизводимостта, % |
Беше организирано друго паралелно проучване в рамките на Общността, при което в 7 лаборатории бяха анализирани четири проби, за да се сертифицира методът за определяне на триптофан чрез хидролиза. Резултатите са представени по-долу. Бяха направени 5 еднакви анализа на всяка проба.
|
|
Проба 1 Комбиниран фураж за свине (CRM 117) |
Проба 2 Рибно брашно с ниско съдържание на мазнини (CRM 118) |
Проба 3 Соево брашно (CRM 119) |
Проба 4 Обезмаслено мляко на прах (CRM 120) |
|
L |
7 |
7 |
7 |
7 |
|
n |
25 |
30 |
30 |
30 |
|
Средна стойност в g/kg |
2,064 |
8,801 |
6,882 |
5,236 |
|
sr [g/kg] |
0,021 |
0,101 |
0,089 |
0,040 |
|
r [g/kg] |
0,059 |
0,283 |
0,249 |
0,112 |
|
CVr [ %] |
1,04 |
1,15 |
1,30 |
0,76 |
|
SR [g/kg] |
0,031 |
0,413 |
0,283 |
0,221 |
|
R [g/kg] |
0,087 |
1,156 |
0,792 |
0,619 |
|
CVR [ %] |
1,48 |
4,69 |
4,11 |
4,22 |
|
L |
= |
брой на лабораториите, предали резултати |
|
n |
= |
брой на запазените индивидуални резултати след елиминиране на отклоняващите се стойности (определени чрез тестовете на Cochran и Dixon) |
|
sr |
= |
стандартно отклонение на повторяемостта |
|
SR |
= |
стандартно отклонение на възпроизводимостта |
|
r |
= |
повторяемост |
|
R |
= |
възпроизводимост |
|
CVr |
= |
коефициент на изменение на повторяемостта, % |
|
CVR |
= |
коефициент на изменение на възпроизводимостта, % |
9. Забележки
|
9.1. |
Следните специални условия за хроматография могат да доведат до по-добро разграничаване на триптофана и α-метил-триптофана. Изократно елуиране с последващо градиентно почистване на колоната:
|
||||||||||||||||||||||||||||||||||||||||||||
|
9.2. |
Хроматографията варира според типа HPLC и според материала, използван за запълването на колоната. Избраната система трябва да може да разграничи триптофана от вътешния стандарт на равнището на базовата линия. Освен това е важно продуктите на разлагането да се разделят добре от триптофана и вътрешния стандарт. Изследват се хидролизати без вътрешен стандарт, за да се провери базовата линия за примеси при наличие на вътрешен стандарт. Важно е също така е времето за елуиране да е достатъчно продължително, за да има възможност за елуиране на всички продукти на разлагането, в противен случай късните пикове, породени от елуирането, могат да интерферират със следващите хроматографски операции. В работния диапазон хроматографската система трябва да покаже линейна реакция. Линейната реакция се измерва при постоянна концентрация (т.е. нормална концентрация) на вътрешния стандарт и при различни концентрации на триптофана. Важно е височината на пиковете на триптофана и на вътрешния стандарт да се намира в линейния диапазон на системата HPLC/флуоресцентната система. Ако пикът или пиковете на триптофана и/или на вътрешния стандарт са прекалено ниски или прекалено високи, анализът се повтаря с проба с различна големина и/или изменен краен обем. |
|
9.3. |
Бариев хидроксид
С течение на времето разтварянето на бариевият хидроксид става все по-трудно. Това води до мътен разтвор при определяне с HPLC, което може да доведе до слаби резултати за триптофана. |
З. ОПРЕДЕЛЯНЕ СЪДЪРЖАНИЕТО НА СУРОВИ МАСЛА И МАЗНИНИ
1. Цел и обхват
Настоящият методът служи за определяне на съдържанието на сурови масла и мазнини във фуражите. Той не обхваща анализа на маслодайните семена и плодове.
Използването на двете разгледани по-долу процедури зависи от характера и състава на фуража и причината за извършване на анализа.
1.1. Процедура А — Директно екстахируеми сурови масла и мазнини
Настоящият метод е приложим за фуражни суровини от растителен произход, с изключение на включените в обхвата на процедура Б.
1.2. Процедура Б — Общо съдържание на сурови масла и мазнини
Настоящият метод е приложим за фуражни суровини от животински произход и за всички комбинирани фуражи. Той трябва да се използва за всички суровини, от които маслата и мазнините не могат да бъдат екстрахирани напълно, без преди това да се осъществи хидролиза (напр. глутени, мая, картофени протеини и продукти, подлежащи на преработка като екструдиране, сплескване и нагряване).
1.3. Интерпретация на резултатите
Във всички случаи, когато резултатът, получен при използване на процедура Б, е по-висок от получения по процедура А, за действителен се приема резултатът от процедура Б.
2. Принцип
2.1. Процедура А
Пробата се подлага на екстракция с петролен етер. Разтворителят се дестилира, след което остатъкът се изсушава и претегля.
2.2. Процедура Б
Пробата се обработва със солна киселина под действието на топлина. Сместа се охлажда и флитрира. Остатъкът се измива, изсушава и се подлага на определяне на съдържанието в съответствие с процедура А.
3. Реагенти
|
3.1. |
Петролен етер с диапазон на кипене: 40—60 oC. Бромното число трябва да бъде под 1, а остатъкът при изпарение — под 2 mg/100 ml. |
|
3.2. |
Натриев сулфат, безводен. |
|
3.3. |
Солна киселина, с = 3 mol HCl/l |
|
3.4. |
Спомагателно средство за филтриране, напр. инфузорна пръст, Hyflo-supercel. |
4. Апаратура
|
4.1. |
Екстракционен апарат. Когато екстракционният апарат е снабден със сифон (апарат на Сокслет), скоростта на обратния поток трябва да бъде такава, че да се получават около 10 цикъла на час; когато апаратът не е от сифонен тип, скоростта на обратния поток трябва да бъде около 10 ml в минута. |
|
4.2. |
Екстракционни гилзи, незамърсени с вещества, разтворими в петролен етер и с пропускливост, която отговаря на изискванията в т. 4.1. |
|
4.3. |
Сушилня или вакуумна сушилня, настроена на 75 ± 3 oС или въздушна пещ, настроена на 100 ± 3oС. |
5. Процедура
5.1. Процедура А (вж. т. 8.1)
Претеглят се 5 g от пробата с точност до 1 mg, прехвърлят се в екстракционна гилза (т. 4.2), която се закрива с памучен тампон, по който не трябва да има мазнини.
Гилзата се поставя в екстрактора (т. 4.1) и се провежда екстракция в продължение на шест часа с петролен етер (т. 3.1). Екстрактът от петролния етер се събира в суха претеглена колба, в която са поставени парченца пемза (2).
Рзтворителят се отстранява чрез дестилация. Остатъкът се изсушава, като се остави колбата за час и половина в сушилнята (т. 4.3). Оставя се да се охлади в ексикатор и се претегля. Изсушава се отново в продължение на 30 min, за да се гарантира, че теглото на маслата и мазнините ще остане постоянно (загубата на тегло между две последователни претегляния трябва да бъде по-малка или равна на 1 mg).
5.2. Процедура Б
Претеглят се 2,5 g от пробата с точност до 1 mg (вж. т. 8.2), поставят се в бехерова чаша от 400 ml или конусообразна колба от 300 ml и се добавят 100 ml солна киселина (т. 3.3) и парченца пемза. Бехеровата чаша се покрива със стъкло за наблюдение или на конусообразната колба се поставя обратен хладник. Сместа се оставя да заври леко над слаб пламък или котлон и се задръжа така един час. Не трябва да се позволява на продукта да полепва по стените на съда.
Съдържанието се охлажда, добавя се толкова филтратор (т. 3.4), колкото е достатъчно, за да се предотврати загубата на масло или мазнина по време на филтрирането. Сместа се филтрира през навлажнена, чиста от мазнини двойна филтърна хартия. Остатъкът се измива в студена вода до получаване на неутрален филтрат. Проверява се дали филтратът не съдържа масла или мазнини. Наличието им показва, че преди хидролизата пробата трябва да бъде екстрахирана с петролен етер, като се използва процедура А.
Двойната филтърна хартия с остатъка се поставя върху стъкло за наблюдение и се суши в продължение на час и половина в сушилня (т. 4.3) при 100 oС ± 3o С.
Двойната филтърна хартия със сухия остатък се поставя в екстракционна гилза (т. 4.2) и се покрива с чист от мазнини памучен тампон. Гилзата се поставя в екстрактора (т. 4.1) и се процедира, както е посочено във втори и трети параграф от точка 5.1.
6. Изразяване на резултата
Теглото на остатъка се изразява като процент от теглото на пробата.
7. Повторяемост
Разликата между резултатите от две паралелни определяния извършени на една и съща проба от един и същ аналитик, не трябва да бъдат повече от:
|
— |
0,2 % като абсолютна стойност, за съдържание на сурови масла и мазнини под от 5 %, |
|
— |
4,0 % спрямо най-високия резултат за съдържание от 5 до 10 %, |
|
— |
0,4 % като абсолютна стойност, за съдържание над 10 %. |
8. Забележки
|
8.1. |
При продукти с високо съдържание на масла и мазнини, които трудно се трошат или не са подходящи за получаване на хомогенна редуцирана проба за анализ, се процедира както следва. Претегелят се 20 g от пробата с точност до 1 mg и се смесват с 10 g или повече безводен натриев сулфат (т. 3.2). Провежда се екстрахиране с петролен етер (т. 3.1), както е указано в точка 5.1. Към получения екстракт се добавя петролен етер (т. 3.1) до 500 ml и се размесва. Вземат се 50 ml от разтвора и се поставят в малка суха претеглена колба, която съдържа парченцата пемза. Разтворителят се отстранява чрез дестилация, съдържанието на колбата се изсушава и се процедира, както е указано в последния параграф на точка 5.1. От останалия в гилзата остатък от екстракцията се отстранява разтвотителят, остатъкът се натрошава до размер на чстиците от 1 mm, връща се в екстракционната гилза (да не се добавя натриев сулфат) и се процедира, както е указано във втория и третия параграф на точка 5.1. Съдържанието на масла и мазнини се изчислява като процент от пробата, като се използва следната формула: (10m1 + m2) × 5 където:
|
|
8.2. |
За продукти с ниско съдържание на масла и мазнини масата на пробата за анализ може да се увеличи на 5 g. |
|
8.3. |
Може да е необходимо храните за домашни любимци с високо водно съдържание да се смесят с безводен натриев сулфат преди хидролизата и екстракцията по процедура Б. |
|
8.4. |
При описаната в т. 5.2 процедура за измиването на остатъка след филтрирането може да е по-ефикасно да се използва топла вода вместо студена. |
|
8.5. |
Може да се наложи времето за сушене от 1,5 часа да бъде увеличено за някои видове фуражи. Избягва се прекаленото изсушаване, тъй като то може да доведе до занижени резултати. Може да се използва също и микровълнова фурна. |
|
8.6. |
Когато съдържанието на сурови масла/мазнини е по-голямо от 15 %, се порепоръчва преди хидролизата да се направи предварителна екстракция чрез процедура А и повторна екстракция чрез процедура Б. Това донякъде зависи от свойствата на фуражите и свойствата на маслата/мазнините във фуражите. |
И. ОПРЕДЕЛЯНЕ НА СУРОВИ ВЛАКНА
1. Цел и обхват
Настоящият метод дава възможност за определяне във фуражите на неразтворими в кисели и в алкални среди органични вещества, които не съдържат мазнини, обикновено означавани като сурови влакна.
2. Принцип
Пробата, обезмаслена, когато е необходимо, се обработва последователно с кипящи разтвори на сярна киселина и калиев хидроксид с определени концентрации. Остатъкът се отделя чрез филтриране с филтър от синтеровано стъкло, измива се, изсушава се, претегля се и се опепелява при температура от 475 до 500 oC. Загубата на тегло в резултат на опепеляването отговаря на суровите влакна, присъстващи в пробата за анализ.
3. Реагенти
|
3.1. |
Сярна киселина, c = 0,13 mol/l. |
|
3.2. |
Средство срещу образуване на пяна (напр. n-октанол). |
|
3.3. |
Помощно средство за филтриране (Celite 545 или еквивалентно), нагрявано при тепмература 500 oC в продължение на 4 часа (т. 8.6). |
|
3.4. |
Ацетон |
|
3.5. |
Петролен етер, диапазон на кипене от 40 до 60 oC. |
|
3.6. |
Солна киселина, с = 0,5 mol/l. |
|
3.7. |
Разтвор на натриев хидроксид, с = 0,23 mol/l. |
4. Апаратура
|
4.1. |
Нагревателен модул за изваряване с разтвор на сярна киселина и калиев хидроксид, снабден с основа за филтрувален тигел (т. 4.2) и с отвеждаща тръба с кранче, свързана с резервоар за изходна течност и с вакуумна помпа, евентуално снабден с източник на сгъстен въздух. Преди употреба всеки ден модулът се нагрява в продължение на пет минути с вряла вода. |
|
4.2. |
Стъклен филтрувален тигел със запоен синтерован плосък стъклен филтър с размер на порите 40—90 μm. Преди първото използване филтърът се загрява до 500 oC в продължение на няколко минути и се охлажда (т. 8.6). |
|
4.3. |
Цилиндър с вместимост най-малко 270 ml с обратен хладник, подходящ за кипване на вода. |
|
4.4. |
Сушилня с термостат. |
|
4.5. |
Муфелна пещ с термостат |
|
4.6. |
Модул за екстракция, състоящ се от основа за филтрувалния тигел (т. 4.2) и тръба с кранче към вакуумната помпа и резервоара за течност. |
|
4.7. |
Съединителни пръстени за сглобяване на нагревателния модул (т. 4.1), тигела (т. 4.2) и цилиндъра (т. 4.3) и за свързване на модула за студена ектракция (т. 4.6) и тигела. |
5. Процедура
Претегля се 1 g от пробата с точност до 1 mg и се слага в тигела (т. 4.2) (вж. забележки в т. 8.1, т. 8.2 и т. 8.3) и се добавя 1 g помощно средство за филтриране (т. 3.3).
Сглобяват се модулът за нагряване (т. 4.1) и филтрувалният тигел (т. 4.2), след което цилиндърът (т. 4.3) се свързва към тигела. Наливат се 150 ml вряща сярна киселина (т. 3.1) в сглобените цилиндър и тигел и ако е необходимо, се добавят няколко капки средство срещу образуване на пяна (т. 3.2).
Течността се нагрява до кипене за 5 ± 2 min и се оставя да ври при силно кипене точно 30 min.
Отваря се кранчето към отвеждащата тръба (т. 4.1) и под вакуум се филтрира сярната киселина през филтрувалния тигел, като остатъкът се измива три пъти с по 30 ml вряща вода, като се следи след всяко измиване остатъкът да е филтриран до сухо.
Затваря се изходното кранче и се наливат 150 ml врящ разтвор на калиев хидроксид (т. 3.7) в сглобените цилиндър и тигел и се добавят няколко капки средство срещу образуване на пяна (т. 3.2). Течността се нагрява до точката на кипене за 5 ± 2 min и се оставя да ври при силно кипене точно 30 min. Филтрира се и се повтаря процедурата на измиване, която е използвана при етапа на обработка със сярна киселина.
След последното измиване и изсушаване тигелът с неговото съдържание се отделя и се свързва с модула за студена екстракция (т. 4.6). Прилага се вакуумът и остатъкът във филтрувалния тигел се измива три пъти с по 25 ml ацетон (т. 3.4), като се следи след всяко измиване остатъкът да е фултруван до сухо.
Тигелът се изсушава, докато теглото му остане постоянно, в сушилня при температура 130 oC. След всяко сушене той се охлажда в ексикатор и бързо се претегля. Тигелът се поставя в муфелна пещ и се подлага на опепеляване, докато теглото му остане постоянно (загубата на тегло между две последователни претегляния трябва да бъде по-малка или равна на 2 mg) при 475 oC—500 oC в продължение най-малко на 30 min.
След всяко нагряване тигелът се охлажда първо в пещта, а после в ексикатора, преди да бъде претеглен.
Прави се празна проба. Загубата на тегло в резултат от опепеляването не трябва да надвишава 4 mg.
6. Изчисляване на резултатите
Съдържанието на сурови влакна като процент от пробата се дава от израза:

където:
|
m |
= |
тегло в g на пробата; |
|
m0 |
= |
загуба на тегло в g след опепеляване по време на определянето; |
|
m1 |
= |
загуба на тегло в g след опепеляване при празната проба; |
7. Повторяемост
Разликата между резултатите от две определяния, проведени върху една и съща проба, не трябва да превишава:
|
— |
0,6 % като абсолютна стойност за съдържание на сурови влакна под 10 %; |
|
— |
6 % по отношение на най-високата стойност за съдържание на сурови влакна, равно или по-голямо от 10 %. |
8. Забележки
|
8.1. |
Фуражите, съдържащи повече от 10 % сурови мазнини, трябва да бъдат обезмаслени с петролен етер (т. 3.5) преди анализа. Филтрувалният тигел (т. 4.2) със съдържанието си се свързва с модула за студена екстракция (т. 4.6), прилага се вакуумът и остатъкът се измива три пъти с по 30 ml петролен етер, като се следи остатъкът да е филтриран до сухо. Тигелът със съдържанието си се свързва модула за нагряване (т. 4.1) и след това се процедира, както е описано в т. 5. |
|
8.2. |
Фуражите, които не могат да бъдат подложени пряко на екстракция с петролен етер (т. 3.5), следва да бъдат обезмаслени, както е указано в т. 8.1, и след изваряване в киселина да бъдат обезмаслени още веднъж. След изваряването в киселина и последващото измиване тигелът със съдържанието си се свързва към модула за студена екстракция (т. 4.6) и се измива три пъти с по 30 ml ацетон, след което се измива още три пъти с по 30 ml петролен етер. Филтрира се до сухо под вакуум и анализът се продължава, както е описано в т. 5, като се започне с третирането с калиев хидроксид. |
|
8.3. |
Ако фуражът съдържа повече от 5 % карбонати, изразени като калциев карбонат, тигелът (т. 4.2) с претеглената проба се свързва към модула за нагряване (т. 4.1) Пробата се измива три пъти с по 30 ml солна киселина (т. 3.6). След всяко добавяне на киселина се изчаква около минута преди да се започне филтрирането. Пробата се измива един път с 30 ml вода, след което се процедира, както е описано в т. 5. |
|
8.4. |
Ако се използва уред във формата на статив (няколко тигела, свързани към един модул за нагряване), не може в рамките на една и съща серия да се провеждат две отделни определяния върху една и съща проба за анализ. |
|
8.5. |
Ако след варенето филтрирането на киселинния или основния разтвор е затруднено, се използва сгъстен въздух, подаван през отвеждащата тръба на модула за нагряване, след което филтрирането продължава. |
|
8.6. |
Температурата на опепеляване не трябва да надвишава 500 oC, за да се удължи срокът на ползване на стъклените филтрувални тигели. Трябва да се избягват твърде големи температурни разлики при циклите на нагряване и охлаждане. |
Й. ОПРЕДЕЛЯНЕ НА ЗАХАР
1. Цел и обхват
Настоящият метод позволява определянето на съдържанието на редуциращи захари и общи захари след инверсия, изразени като глюкоза или, когато е целесъобразно, като захароза, при преобразуване с коефициент 0,95. Той е приложим за комбинирани фуражи. За другите фуражи са предвидени специални методи. Когато е необходимо, лактозата се измерва отделно и се отчита при изчисляването на резултатите.
2. Принцип
Захарите се екстрахират в разреден етанол; разтворът се избистря с разтвори на Карез I и II. След отстраняване на етанола количествата преди и след инверсията се определят по метода на Луф-Шорл.
3. Реагенти
|
3.1. |
Етанол 40 % (v/v) с плътност: 0,948 g/ml при 20 oС, неутрализиран спрямо фенолфталеин. |
|
3.2. |
Разтвор на Карез I: 21,9 g цинков ацетат Zn(CH3COO)2 2H2O и 3 g безводна оцетна киселина се разтварят във вода. Допълва се до 100 ml с вода. |
|
3.3. |
Разтвор на Карез II: 10,6 g калиев фероцианид K4 Fe (CN)6 3H2O се разтварят във вода. Допълва се до 100 ml с вода. |
|
3.4. |
Разтвор на метилово оранжево с концентрация 0,1 % (w/v). |
|
3.5. |
Солна киселина, 4 mol/l. |
|
3.6. |
Солна киселина, 0,1 mol/l. |
|
3.7. |
Разтвор на натриев хидроксид, 0,1 mol/l. |
|
3.8. |
Реагент на Луф-Шорл: При внимателно разбъркване, разтворът на лимонена киселина (т. 3.8.2) се добавя към разтвора на натриев карбонат (т. 3.8.3). Добавя се разтворът на меден сулфат (т. 3.8.1) и се допълва до 1 литър с вода. Оставя се да престои една нощ и се филтрира. Проверява се концентрацията на така получения реагент (Cu 0,05 mol/l; Na2 CO3 1 mol/l), вж. (т. 5.4), последния параграф. Стойността на рН на разтвора е приблизително 9,4. |
|
3.8.1. |
Разтвор на меден сулфат: 25 g меден сулфат CuSO4,5H2O, несъдържащ желязо, се разтварят в 100 ml вода. |
|
3.8.2. |
Разтвор на лимонена киселина: 50 g лимонена киселина, C6H8O7·H2O се разтварят в 50 ml вода. |
|
3.8.3. |
Разтвор на натриев карбонат: 143,8 g безводен натриев карбонат се разтварят в около 300 ml топла вода. Оставя се да се охлади. |
|
3.9. |
Разтвор на натриев тиосулфат 0,1 mol/l. |
|
3.10. |
Разтвор на нишесте: смес от 5 g разтворимо нишесте в 30 ml вода се прибавя към 1 литър кипяща вода. Оставя се да кипи три минути, оставя се да се охлади и ако е необходимо, се добавят 10 mg живачен йодид като консервант. |
|
3.11. |
Сярна киселина, 3 mol/l. |
|
3.12. |
Разтвор на калиев йодид, 30 % (w/v). |
|
3.13. |
Гранулирана пемза, кипната в солна киселина, промита с вода и изсушена. |
|
3.14. |
3-метилбутан-1-ол |
4. Апаратура
Смесител (барабанен): приблизително 35 до 40 r.p.m.
5. Процедура
5.1. Екстрахиране на пробата
Претеглят се 2,5 g от пробата с точност до 1 mg и се изсипват в мерителна колба от 250 ml. Добавят се 200 ml етанол (т. 3.1) и пробата се размесва един час в барабанния смесител. Добавят се 5 ml разтвор на Карез I (т. 3.2) и се бърка около 30 секунди. Добавят се 5 ml разтвор на Карез II (т. 3.3) и се бърка още една минута. Допълва се до пълния обем с етанол (т. 3.1), хомогенизира се и се филтрира. Отделят се 200 ml от филтрата, който се сгъстява чрез изпаряване, докато обемът му намалее приблизително наполовина, за да се отстрани по-голямата част от етанола. Остатъкът след изпаряването се прибавя количествено в 200 ml мерителна колба с помощта на гореща вода, охлажда се, допълва се с вода до пълния обем, хомогенизира се и, ако е необходимо, се филтрира. Този разтвор се използва за определянето на количеството редуциращи захари и, след инверсия, на общите захари.
5.2. Определяне на редуциращи захари
С помощта на пипета се взимат не повече от 25 ml от разтвора, съдържащ не повече от 60 mg редукторни захари, изразени като глюкоза. Ако е необходимо, обемът се коригира до 25 ml с дестилирана вода и се определя съдържанието на редуциращи захари по метода на Луф-Шорл. Резултатът се изразява като процентно съдържание на глюкоза в пробата.
5.3. Определяне на общи захари след инверсия
С помощта на пипета се взимат 50 ml от разтвора и се прехвърлят в мерителна колба от 100 ml. Добавят се няколко капки разтвор на метилово оранжево (т. 3.4), след това внимателно и при непрекъснато разбъркване се добавя солна киселина (т. 3.5), докато течността придобие определено червен цвят. Добавят се 15 ml солна киселина (т. 3.6), колбата се потапя в силно кипяща вода и се оставя там в продължение на тридесет минути. Охлажда се бързо до около 20 oС и се добавят 15 ml разтвор на натриев хидроксид (т. 3.7). Допълва се до 100 ml с вода и се хомогенизира. Взимат се не повече от 25 ml, които съдържат не повече от 60 mg редуциращи захари, изразени като глюкоза. Ако е необходимо, обемът се коригира до 25 ml с дестилирана вода и се определя съдържанието на редуциращи захари по метода на Луф-Шорл. Резултатът се изразява като проценти глюкоза или, когато е целесъобразно, като захароза, чрез умножаване по коефициент 0,95.
5.4. Титруване по метода на Луф-Шорл
С помощта на пипета се вземат 25 ml от реагента на Луф-Шорл (т. 3.8) и се прехвърлят в ерленмайерова колба от 300 ml; добавят се точно 25 ml от избистрения захарен разтвор. Добавят се 2 гранули пемза (т. 3.13), нагряват се с бъркане на ръка на открит пламък със средна височина и течността се довежда до кипене за около две минути. Ерленмайеровата колба се поставя веднага върху покрита с азбест телена мрежа с отвор с диаметър около 6 cm, под която е запален пламък. Пламъкът следва да се регулира така, че само основата на ерленмайеровата колба да се нагрява. Ерленмайеровата колба се свързва към обратен хладник. Оставя се да кипи точно десет минути. Охлажда се веднага в студена вода и след около пет минути се титрува, както следва:
Добавя се 10 ml разтвор на калиев йодид (т. 3.12) и веднага след това (внимателно, тъй като има опасност от обилно образуване на пяна) се добавят 25 ml сярна киселина (т. 3.11). Титрува се с разтвор на натриев тиосулфат (т. 3.9) до появяване на матовожълт цвят, добавя се нишестеният индикатор (т. 3.10) и се завършва титруването.
Същото титруване се провежда с точно измерена смес от 25 ml реагент на Луф-Шорл (т. 3.8) и 25 ml вода след добавяне на 10 ml разтвор на калиев йодид (т. 3.12) и 25 ml сярна киселина (т. 3.11) без кипене.
6. Изчисляване на резултатите
С помощта на таблицата се определя количеството глюкоза в mg, което съответства на разликата между стойностите на двете титрувания, изразена в mg натриев тиосулфат 0,1 mol/l. Резултатът се изразява като процент от теглото на пробата.
7. Специални процедури
|
7.1. |
Ако се изследват богати на меласа фуражи и други слабо хомогенни фуражи, 20 g от пробата се претеглят и се поставят в 500 ml вода в 1-литрова мерителна колба. Размесват се един час в барабанен смесител. Избистря се с разтвори на Карез I и II (т. 3.2 и т. 3.3), както е описано в т. 5.1, като обаче се използват четирикратни количества от всеки реагент. Допълва се до пълния обем с 80 % етанол (v/v). Хомогенизира се и се филтрира. Етанолът се отстранява, както е описано в т. 5.1. Ако няма декстринизирано нишесте, съдържанието на колбата се допълва до пълния обем с дестилирана вода. |
|
7.2. |
Ако се изследва меласа и суровини за фуражи без примеси, които са богати на захар и почти не съдържат нишесте (рожкови, сушено цвекло и др.), се претеглят 5 g, поставят се в мерителна колба от 250 ml, добавят се 200 ml дестилирана вода и се размесват в барабанния смесител един час или повече, ако е необходимо. Избистря се, като се използва разтвор на Карез I (т. 3.2) и разтвор на Карез II (т. 3.3), както е описано в т. 5.1. Допълва се до пълния обем със студена вода, хомогенизира се и се филтрира. За да се определи количеството общи захари, се продължава, както е описано в т. 5.3. |
8. Забележки
|
8.1. |
За да се избегне образуването на пяна, препоръчва се да се добави (независимо от обема) приблизително 1 ml 3-метилбутан-1-ол (т. 3.14) преди кипването с реагента на Луф-Шорл. |
|
8.2. |
Разликата между съдържанието на общи захари след инверсия, изразени като глюкоза, и съдържанието на редуциращи захари, изразени като глюкоза, умножена по 0,95, дава процентното съдържание на захароза. |
|
8.3. |
Могат да се използват два метода за определяне на количеството на редуциращи захари с изключение на лактоза: |
|
8.3.1. |
За приблизително изчисляване, установеното с друг аналитичен метод съдържание на лактоза се умножава по 0,675 и полученият резултат се изважда от съдържанието на редуциращите захари. |
|
8.3.2. |
За точно изчисляване на количеството редуциращи захари, с изключение на лактоза, трябва да се използва една и съща проба за двете крайни определяния: единият анализ се прави с част от разтвора, получен по процедурата в т. 5.1, а другият — с част от разтвора, получен при определянето на лактозата по метода, посочен за тази цел (след ферментиране на другите видове захар и избистряне). И в двата случая количеството налична захар се определя по метода на Луф-Шорл и се изчислява в mg глюкоза. Едната от стойностите се изважда от другата и разликата се изразява като процент от теглото на пробата. Пример Двата обема, взети за всяко определение, отговарят на проба от 250 mg. В първия случай се изразходват 17 ml разтвор на натриев тиосулфат от 0,1 mol/l, съответстващи на 44,2 mg глюкоза; във втория — 11 ml, съответстващи на 27,6 mg глюкоза. Разликата е 16,6 mg глюкоза. Съдържанието на редуциращи захари (с изключение на лактоза), изчислено като глюкоза, е съответно:
|

Таблица със стойности за 25 ml реагент на Луф-Шорл
ml Na2S2O30,1 mol/l загряване две минути, варене десет минути
|
Na2 S2 O3 0,1 mol/l |
Глюкоза, фруктоза, инвертни захари C6 H12 O6 |
Лактоза C12 H22 O11 |
Малтоза C12 H22 O11 |
Na2 S2 O3 0,1 mol/l |
|||
|
ml |
mg |
разлика |
mg |
разлика |
mg |
разлика |
ml |
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
2,4 4,8 7,2 9,7 12,2 14,7 17,2 19,8 22,4 25,0 27,6 30,3 33,0 35,7 38,5 41,3 44,2 47,1 50,0 53,0 56,0 59,1 62,2 |
2,4 2,4 2,5 2,5 2,5 2,5 2,6 2,6 2,6 2,6 2,7 2,7 2,7 2,8 2,8 2,9 2,9 2,9 3,0 3,0 3,1 3,1 |
3,6 7,3 11,0 14,7 18,4 22,1 25,8 29,5 33,2 37,0 40,8 44,6 48,4 52,2 56,0 59,9 63,8 67,7 71,7 75,7 79,8 83,9 88,0 |
3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,8 3,8 3,8 3,8 3,8 3,8 3,9 3,9 3,9 4,0 4,0 4,1 4,1 4,1 |
3,9 7,8 11,7 15,6 19,6 23,5 27,5 31,5 35,5 39,5 43,5 47,5 51,6 55,7 59,8 63,9 68,0 72,2 76,5 80,9 85,4 90,0 94,6 |
3,9 3,9 3,9 4,0 3,9 4,0 4,0 4,0 4,0 4,0 4,0 4,1 4,1 4,1 4,1 4,1 4,2 4,3 4,4 4,5 4,6 4,6 |
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
К. ОПРЕДЕЛЯНЕ НА ЛАКТОЗА
1. Цел и обхват
Настоящият метод позволява да се определи съдържанието на лактоза във фуражи, съдържащи повече от 0,5 % лактоза.
2. Принцип
Захарите се разтварят във вода. Разтворът се подлага на ферментация под въздействие на дрождите Saccharomyces cerevisiae, които не засягат лактозата. След избистряне и филтриране съдържанието на лактоза във филтрата се определя по метода на Луф-Шорл.
3. Реагенти
|
3.1. |
Суспензия на Saccharomyces cerevisiae: 25 g пресни дрожди се смесват със 100 ml вода до получаването на суспензия. Суспензията може да се запази най-много една седмица в хладилник. |
|
3.2. |
Разтвор на Карез I: 21,9 g цинков ацетат Zn (CH3 COO)2 2H2O и 3 g безводна оцетна киселина се разтварят във вода. Допълва се до 100 ml с вода. |
|
3.3. |
Разтвор на Карез II: 10,6 g калиев фероцианид K4 Fe (CN)6 3H2O се разтварят във вода. Допълва се до 100 ml с вода. |
|
3.4. |
Реагент на Луф-Шорл: При внимателно разбъркване, разтворът на лимонена киселина (точка 3.4.2) се добавя към разтвора на натриев карбонат (3.4.3). Добавя се разтворът на меден сулфат (3.4.1) и се допълва до 1 литър с вода. Оставя се да престои една нощ и се филтрира. Проверява се концентрацията на така получения реактив (Cu 0,05 mol/l; Na2 CO3 1 mol/l). Стойността на рН на разтвора е приблизително 9,4. |
|
3.4.1. |
Разтвор на меден сулфат: 25 g меден сулфат CuSO4 5H2O, несъдържащ желязо, се разтварят в 100 ml вода. |
|
3.4.2. |
Разтвор на лимонена киселина: 50 g лимонена киселина, C6H8O7·H2O се разтварят в 50 ml вода. |
|
3.4.3. |
Разтвор на натриев карбонат: 143,8 g безводен натриев карбонат се разтварят в приблизително 300 ml топла вода. Оставя се да се охлади. |
|
3.5. |
Гранулирана пемза, кипната в солна киселина, промита с вода и изсушена. |
|
3.6. |
Разтвор на калиев йодид, 30 % (w/v). |
|
3.7. |
Сярна киселина, 3 mol/l. |
|
3.8. |
Разтвор на натриев тиосулфат 0,1 mol/l. |
|
3.9. |
Разтвор на нишесте: смес от 5 g разтворимо нишесте в 30 ml вода се прибавя към 1 литър кипяща вода. Оставя се да кипи 3 минути, оставя се да се охлади и, ако е необходимо, се добавят 10 mg живачен йодид като консервант. |
4. Апаратура
Водна баня с термостат, настроен на 38—40 oС.
5. Процедура
Претегля се 1 g от пробата с точност до 1 mg и тази част от пробата се поставя в мерителна колба от 100 ml. Добавят се 25—30 ml вода. Колбата се поставя в кипяща водна баня за тридесет минути и след това се охлажда до приблизително 35 oС. Добавят се 5 ml от дрождената суспензия (т. 3.1) и сместа се хомогенизира. Колбата се оставя да престои два часа на водна баня при температура 38—40 oС. Охлажда се до приблизително 20 oC.
Добавят се 2,5 ml разтвор на Карез I (т. 3.2), бърка се тридесет секунди, след това се добавят 2,5 ml разтвор на Карез II (т. 3.3) и отново се бърка тридесет секунди. Допълва се до 100 ml с вода, разбърква се и се филтрира. С помощта на пипета се взима част от филтрата — не повече от 25 ml, по възможност със съдържание на лактоза от 40 до 80 mg — и се прехвърля в ерленмайерова колба от 300 ml. Ако е необходимо, се допълва до 25 ml с вода.
Прави се празна проба по същия начин с 5 ml дрождена суспензия (3.1). Съдържанието на лактозата се определя по метода на Луф-Шорл, както следва: прибавят се точно 25 ml реагент на Луф-Шорл (3.4) и две гранули пемза (т. 3.5). Бърка се на ръка, като едновременно се нагрява на открит пламък със средна височина, и течността се довежда до кипене за около две минути. Ерленмайеровата колба се поставя веднага върху покрита с азбест телена мрежа с отвор с диаметър около 6 cm, под която е запален пламък. Пламъкът следва да се регулира така, че само основата на ерленмайеровата колба да се нагрява. Ерленмайеровата колба се свързва към обратен хладник. Оставя се да кипи точно десет минути. Охлажда се веднага в студена вода и след около пет минути се титрува, както следва:
Добавят се 10 ml разтвор на калиев йодид (т. 3.6) и веднага след това (внимателно, защото има опасност от обилно образуване на пяна) се добавят 25 ml сярна киселина (т. 3.7). Титрува се с разтвор на натриев тиосулфат (т. 3.8) до получаване на матово-жълт цвят, добавя се нишестеният индикатор (т. 3.9) и се завършва титруването.
Същото титруване се провежда върху точно измерена смес от 25 ml реагент на Луф-Шорл (т. 3.4) и 25 ml вода след добавяне на 10 ml разтвор на калиев йодид (т. 3.6) и 25 ml сярна киселина (т. 3.7) без кипене.
6. Изчисляване на резултатите
Като се използва приложената таблица, се установява количеството лактоза в mg, което отговаря на разликата между резултатите от двете титрувания, изразено в ml натриев тиосулфат 0,1 mol/l.
Резултатът се изразява като процент безводна лактоза от пробата.
7. Забележки
За продукти, съдържащи повече от 40 % ферментируема захар, се използват повече от 5 ml дрождена суспензия (т. 3.1).
Таблица на стойностите за 25 ml реагент на Луф-Шорл
ml Na2S2O30,1 mol/l загряване две минути, кипене десет минути
|
Na2 S2 O3 0,1 mol/l |
Глюкоза, фруктоза, инвертни захари C6 H12 O6 |
Лактоза C12 H22 O11 |
Малтоза C12 H22 O11 |
Na2 S2 O3 0,1 mol/l |
|||
|
ml |
mg |
разлика |
mg |
разлика |
mg |
разлика |
ml |
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
2,4 4,8 7,2 9,7 12,2 14,7 17,2 19,8 22,4 25,0 27,6 30,3 33,0 35,7 38,5 41,3 44,2 47,1 50,0 53,0 56,0 59,1 62,2 |
2,4 2,4 2,5 2,5 2,5 2,5 2,6 2,6 2,6 2,6 2,7 2,7 2,7 2,8 2,8 2,9 2,9 2,9 3,0 3,0 3,1 3,1 |
3,6 7,3 11,0 14,7 18,4 22,1 25,8 29,5 33,2 37,0 40,8 44,6 48,4 52,2 56,0 59,9 63,8 67,7 71,7 75,7 79,8 83,9 88,0 |
3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,8 3,8 3,8 3,8 3,8 3,8 3,9 3,9 3,9 4,0 4,0 4,1 4,1 4,1 |
3,9 7,8 11,7 15,6 19,6 23,5 27,5 31,5 35,5 39,5 43,5 47,5 51,6 55,7 59,8 63,9 68,0 72,2 76,5 80,9 85,4 90,0 94,6 |
3,9 3,9 3,9 4,0 3,9 4,0 4,0 4,0 4,0 4,0 4,0 4,1 4,1 4,1 4,1 4,1 4,2 4,3 4,4 4,5 4,6 4,6 |
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
Л. ОПРЕДЕЛЯНЕ НА НИШЕСТЕ
ПОЛЯРИМЕТРИЧЕН МЕТОД
1. Цел и обхват
Настоящият метод дава възможност да се определят нивата на нишесте и на продукти от разграждането на нишесте с високо молекулно тегло във фуражите, с цел проверка на съответствието с обявената енергийна стойност (разпоредби в приложение VII) и с Директива 96/25/ЕО на Съвета (3).
2. Принцип
Методът включва две определяния. При първото пробата се третира с разредена солна киселина. След избистряне и филтриране се измерва въртенето на плоскостта на поляризацията на разтвора чрез поляриметрия.
При второто пробата се подлага на екстракция с 40 % етанол. След подкисляване на филтрата със солна киселина, избистряне и филтриране се измерва въртенето на плоскостта на поляризацията, както при първото определяне.
Като се умножи разликата между двете измервания с коефициент, който е известен, се получава съдържанието на нишесте в пробата.
3. Реагенти
|
3.1. |
Разтвор на солна киселина, 25 % (w/w) с плътност: 1,126 g/ml. |
|
3.2. |
Разтвор на солна киселина с концентрация 1,13 % (w/v). Kонцентрацията трябва да се провери чрез титруване, като се използва разтвор на натриев хидроксид 0,1 mol/l, в присъствие на 0,1 % (w/v) метилово червено в 94 % (v/v) етанол. За неутрализацията на 10 ml са необходими 30,94 ml NaOH 0,1 mol/l. |
|
3.3. |
Разтвор на Карез I: 21,9 g цинков ацетат Zn(CH3COO)22H2O и 3 g безводна оцетна киселина се разтварят във вода. Допълва се до 100 ml с вода. |
|
3.4. |
Разтвор на Карез II: разтварят се 10,6 g калиев фероцианид, K4Fe(CN)6 3H2O във вода. Допълва се до 100 ml с вода. |
|
3.5. |
Етанол 40 % (v/v) с плътност: 0,948 g/ml при 20 oС. |
4. Апаратура
|
4.1. |
Ерленмайерова колба от 250 ml, със стандартен стъклен шлиф и с обратен хладник. |
|
4.2. |
Поляриметър или захарометър. |
5. Процедура
5.1. Подготовка на пробата
Пробата се разтрошава до получаване на достатъчно дребни частици, за да може цялата проба да премине през сито с кръгли отвори с големина 0,5 mm.
5.2. Определяне на общото въртене на плоскостта на поляризацията (P или S) (вж. Забележка в т. 7.1)
Претеглят се точност до един mg 2,5 g от натрошената проба и се поставят в градуирана колба от 100 ml. Прибавят се 25 ml солна киселина (т. 3.2), разклаща се до получаване на равномерно разпределение на пробата, и се добавят още 25 ml солна киселина (т. 3.2). Колбата се потапя във водна баня с вряща вода, като енергично се разклаща през първите три минути, за да се предотврати образуването на агломерати. Количеството вода във водната баня трябва да е достатъчно, за да може при поставяне на колбата в нея кипенето на продължи. Колбата не трябва да се изважда от банята докато трае разклащането. Точно след 15 min колбата се изважда от водната баня, добавят се 30 ml студена вода и незабавно се охлажда до 20 oС.
Добавят се 5 ml разтвор на Карез I (т. 3.3) и се разбърква около 30 секунди. Добавят се 5 ml разтвор на Карез II (т. 3.4) и се разбърква още около 30 секунди. Допълва се с вода до запълване на обема, разбърква се и се филтрира. Ако филтратът не е идеално бистър (което рядко се случва), определянето се повтаря, като се използва по-голямо количество разтвор на Карез I и II, например 10 ml.
С поляриметър или захарометър се измерва въртенето на плоскостта на поляризацията на разтвора в епруветка от 200 mm.
5.3. Определяне на въртенето на плоскостта на поляризацията (P' или S') на вещества, разтворими в 40 % етанол
Претеглят се 5 g от пробата с точност до един mg, поставят се в градуирана колба от 100 ml и се прибавят 80 ml етанол (т. 3.5) (вж. забележка 7.2). Колбата се оставя е в продължение на 1 час при стайна температура; през това време тя шесткратно се разклаща енергично, така че пробата за анализ напълно да се смеси с етанола. Допълва се с етанол (т. 3.5) до запълване на на обема, разбърква се и се филтрира.
Отмерват се с пипета 50 ml от филтрата (отговаря на 2,5 g от пробата) и се поставят в ерленмайерова колба от 250 ml, прибавят се 2,1 ml солна киселина (т. 3.1) и се разклаща енергично. Обратният хладник се монтира към ерленмайеровата колба и тя се потапя в баня с вряща вода. Точно след 15 минути ерленмайеровата колба се изважда от водната баня, съдържанието ѝ се прехвърля в градуирана колба от 100 ml, като колбата се изплаква с малко студена вода и се охлажда до 20 oС.
Съдържанието се избистря с разтвор на Карез I (т. 3.3) и II (т. 3.4), допълва се с вода до пълния обем, разбърква се, филтрира се и се измерва въртенето на плоскостта на поляризацията, както е посочено в параграфи 2 и 3 от точка 5.2.
6. Изчисляване на резултатите
Съдържанието на нишесте (в %) се изчислява, както следва:
6.1. Измерване с поляриметър

|
P |
= |
общо въртене на плоскостта на поляризацията в градуси |
||||||||||||||
|
P' |
= |
въртене на плоскостта на поляризацията в градуси на веществата, разтворими в 40 % (v/v) етанол |
||||||||||||||
|
|
= |
Специфично въртене на плоскостта на поляризацията за чисто нишесте. Числовите стойности, които обикновено се приемат за този параметър, са следните:
|
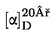
6.2. Измерване със захарометър
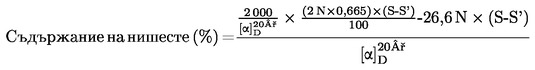
|
S |
= |
Общо въртене на плоскостта на поляризацията в градуси на захарометъра |
|
S' |
= |
Въртене на плоскостта на поляризацията в градуси на захарометъра на веществата, разтворими в 40 % (v/v) етанол |
|
N |
= |
тегло (в g) на разтворена в 100 ml вода захароза, която предизвиква въртене на плоскостта на поляризацията от 100 градуса по скалата на захарометъра, измерени при използване на епруветка от 200 mm 16,29 g при френските захарометри 26,00 g при немските захарометри 20,00 g при смесени захарометри. |
|
|
= |
специфично въртене плоскостта на поляризацията на чистото нишесте (вж. т. 6.1) |

6.3. Повторяемост
Разликата в резултатите от две паралелни определяния, проведени върху една и съща проба, не трябва да превишава 0,4 като абсолютна стойност при съдържание на нишесте по-ниско от 40 %, и 1 % като относителна стойност при съдържание на нишесте от 40 % или повече.
7. Забележки
|
7.1. |
Ако пробата съдържа повече от 6 % карбонати, изчислени като калциев карбонат, те трябва да се неутрализират, като се обработят с точно определено количество разредена сярна киселина преди определянето на общото въртене на плоскостта на поляризацията. |
|
7.2. |
В случай на продукти с високо съдържание на лактоза, като серум от сухо мляко или сухо обезмаслено мляко, след като се добавят 80 ml етанол (т. 3.5), се процедира, както е указано по-долу. Свързва се обратният хладник към колбата и тя се потапя за 30 минути във водна баня при 50 oС. Оставя се да изстине и анализът продължава, както е указано в т. 5.3. |
|
7.3. |
Известно е, че следните фуражни суровини, когато присъстват във фуражите в значителни количества, предизвикват смущения при определяне на съдържанието на нишесте по поляриметричния метод, което би могло да доведе до неточни резултати:
|
М. ОПРЕДЕЛЯНЕ НА СУРОВА ПЕПЕЛ
1. Цел и обхват
Настоящият метод позволява определянето на съдържанието на сурова пепел във фуражите.
2. Принцип
Пробата се опепелява при 550 oС; остатъкът се претегля.
3. Реагенти
Разтвор на амониев нитрат, 20 % (w/v).
4. Апаратура
|
4.1. |
Нагревателна плоча |
|
4.2. |
Електрическа муфелна пещ с термостат |
|
4.3. |
Кварцови, порцеланови или платинени тигли за опепеляване, правоъгълни (прибл. 60 × 40 × 25 mm) или кръгли (диаметър 60—75 mm, височина 20—40 mm) |
5. Процедура
Претеглят се с точност до един mg около 5 g от пробата (2,5 g при продукти, които имат свойството да набъбват) и се поставя в тигел за опепеляване, който е бил предварително нагрят до 550 oC, охладен и тариран. Тигелът се поставя върху нагревателната плоча и се нагрява постепенно, докато веществото се овъгли. Опепеляването се извършва в съответствие с процедурата в т. 5.1 или т. 5.2.
|
5.1. |
Тигелът се поставя в калибрирана муфелна пещ, настроена на 550 oC. Държи се при тази температура, докато се получи бяла, светло сива или червеникава пепел, която видимо не съдържа въгленови частици. Тигелът се поставя в ексикатор, оставя се да се охлади и се претегля незабавно. |
|
5.2. |
Тигелът се поставя в калибрирана муфелна пещ, настроена на 550 oС. Оставя се да се опепелява 3 часа. Тигелът се поставя в ексикатор, оставя се да се охлади и се претегля незабавно. Опепелява се отново в продължение на 30 минути, за да се гарантира, че теглото на пепелта ще остане постоянно (загубата на тегло между две последователни претегляния трябва да бъде по-малка или равна на 1 mg). |
6. Изчисляване на резултатите
Теглото на остатъка се изчислява, като се изважда тарираното тегло.
Резултатът се изразява като процент от пробата.
7. Забележки
|
7.1. |
Пепелта на субстанции, които се опепеляват трудно, трябва да бъде подложена на първоначално опепеляване в продължение на най-малко три часа, да бъде охладена и след това да се добавят няколко капки 20 % разтвор на амониев нитрат или вода (внимателно, за да се избегне разпрашаването на пепелта или образуването на бучици). След изсушаване в пещта калцинирането се продължава. Повтаря се операцията колкото пъти се налага до постигане на пълно опепеляване. |
|
7.2. |
При субстанции, устойчиви към обработката, описана в т. 7.1, се процедира, както следва: след опепеляване в продължение на три часа, пепелта се поставя в гореща вода и се филтрира през малък, несъдържащ пепели филтър. Филтърът и съдържанието му се опепеляват в първоначалния тигел. Филтратът се поставя в охладен тигел, изпарява се до сухо, опепелява се и се претегля. |
|
7.3. |
Когато се изследват масла и мазнини, се претегля точно проба от 25 g в подходящ по размери тигел. Субстанцията се овъглява, като се запали чрез поднасяне на горяща лента от филтърна хартия, която не съдържа пепели. След изгарянето се навлажнява с колкото е възможно по-малко вода, изсушава се и се опепелява, както е описано в точка 5. |
Н. ОПРЕДЕЛЯНЕ НА НЕРАЗТВОРИМА В СОЛНА КИСЕЛИНА ПЕПЕЛ
1. Цел и обхват
Настоящият метод позволява да се определи количеството на неразтворимите в солна киселина минерални вещества във фуражите. В зависимост от естеството на пробата могат да се приложат два метода.
|
1.1. |
Метод А: приложим към органични суровини за фуражи и към повечето комбинирани фуражи. |
|
1.2. |
Метод Б: приложим към минерални вещества и смеси и към комбинирани фуражи, чието съдържание на неразтворими в солна киселина вещества, определено по метод А, е по-голямо от 1 %. |
2. Принцип
|
2.1. |
Метод А: пробата се опепелява, пепелта се кипва в солна киселина и неразтворимият остатък се филтрира и претегля. |
|
2.2. |
Метод Б: пробата се обработва със солна киселина. Разтворът се филтрира, остатъкът се опепелява и така получената пепел се обработва по метод А. |
3. Реагенти
|
3.1. |
Солна киселина, 3 mol/l. |
|
3.2. |
Разтвор на трихлороцетна киселина с концентрация 20 % (w/v). |
|
3.3. |
Разтвор на трихлороцетна киселина с концентрация 1 % (w/v). |
4. Апаратура
|
4.1. |
Нагревателна плоча. |
|
4.2. |
Електрическа муфелна пещ с термостат. |
|
4.3. |
Кварцови, порцеланови или платинени тигли за опепеляване, правоъгълни (прибл. 60 × 40 × 25 mm) или кръгли (диаметър 60—75 mm, височина 20—40 mm) |
5. Процедура
5.1. Метод А:
Пробата се опепелява по метода, описан за определянето на сурова пепел. Може да се използва и пепелта, получена при посочения анализ.
Пепелта се поставя в бехерова чаша с вместимост от 250—400 ml, като се използват 75 ml солна киселина (точка 3.1). Довежда се бавно до кипене и се оставя да кипи умерено в продължение на петнадесет минути. Горещият разтвор се филтрира през филтърна хартия, несъдържаща пепели, и остатъкът се промива с гореща вода до изчезване на киселинната реакция. Филтърът с остатъка се изсушава и се опепелява в тариран тигел при температура, не по-ниска от 550 oС и не по-висока от 700 oС. Охлажда се в ексикатор и се претегля.
5.2. Метод Б:
Претеглят се 5 g от пробата с точност до един mg и се поставят в бехерова чаша с вместимост 250—400 ml. Добавят се последователно 25 ml вода и 25 ml солна киселина (т. 3.1), разбърква се и се изчаква да спре кипенето. Добавят се още 50 ml солна киселина (точка 3.1). Изчаква се да спре отделянето на газ, след което бехеровата чаша се поставя във вана с вряща вода и се държи там тридесет минути или повече, ако е необходимо, за да се хидролизира изцяло нишестето, което евентуално се съдържа в пробата. Докато е горещо, съдържанието се филтрира през филтър, несъдържащ пепел, и филтърът се промива с 50 ml топла вода (вж. забележка в т. 7). Филтърът се поставя с остатъка в тигел за опепеляване, изсушава се и се опепелява при температура, не по-ниска от 550 oС и не по-висока от 700 oС. Пепелта се поставя в бехерова чаша с вместимост 250—400 ml, като се добавят 75 ml солна киселина (точка 3.1); продължава се, както е описано във втория абзац на точка 5.1.
6. Изчисляване на резултатите
Теглото на остатъка се изчислява, като се изважда теглото на тарата. Резултатът се изразява като процент от пробата.
7. Забележки
Ако филтрирането се окаже трудно, анализът се повтаря, като се заместват 50-те ml солна киселина (точка 3.1) с 50 ml 20 % трихлороцетна киселина (точка 3.2) и се промива филтърът с топъл 1 % разтвор на трихлороцетна киселина (точка 3.3).
О. ОПРЕДЕЛЯНЕ НА КАРБОНАТИ
1. Цел и обхват
Настоящият метод позволява количественото определяне на съдържанието на карбонати, обикновено изразявани като калциев карбонат, в повечето фуражи.
В някои случаи обаче (напр. при железен карбонат) трябва да се използва специален метод.
2. Принцип
Карбонатите се разлагат в солна киселина; освободеният въглероден диоксид се събира в градуирана епруветка и обемът му се сравнява с този, освободен при същите условия от известно количество калциев карбонат.
3. Реагенти
|
3.1. |
Солна киселина, плътност 1,10 g/ml. |
|
3.2. |
Калциев карбонат |
|
3.3. |
Сярна киселина, приблизително 0,05 mol/l, оцветена с метилово червено. |
4. Апаратура
Апарат на Шайблер-Дитрих (вж. схемата) или еквивалентен на него апарат.
5. Процедура
В зависимост от съдържанието на карбонати в пробата, претеглят се част от пробата, както е посочено по-долу:
|
— |
0,5 g за продукти, съдържащи от 50 до 100 % карбонати, изразени като калциев карбонат; |
|
— |
1 g за продукти, съдържащи от 40 до 50 % карбонати, изразени като калциев карбонат; |
|
— |
2 до 3 g за останалите продукти. |
Частта от пробата се поставя в специалната колба (4) на апарата, снабдена с малка тръбичка от нечуплив материал, съдържаща 10 ml солна киселина (т. 3.1), и колбата се свързва с апарата. Трипътният кран (5) се завърта така, че тръбата (1) да е свързана с външната среда. Като се използват подвижната тръба (2), напълнена с оцветена сярна киселина (точка 3.3) и свързана към градуираната тръба (1), се допълва течноста до нивото на нулевата марка. Завърта се кранът (5) така, че да се свързва тръби (1) и (3) и се проверява нивото да е на нула.
Солната киселина (точка 3.1) се прелива бавно върху частта от пробата, като колбата се накланя (4). Изравнява се налягането, като се сваля надолу тръбата (2). Разклаща се колбата (4) дотогава, докато спре напълно отделянето на въглероден диоксид.
Налягането се възстановява, като се изравнява наново течността до едно и също ниво в тръби (1) и (2). След няколко минути, когато обемът газ стане постоянен, се прави отчитането.
Прави се контролна проба при същите условия с 0,5 g калциев карбонат (точка 3.2).
6. Изчисляване на резултатите
Съдържанието на карбонати, изразено като калциев карбонат, се изчислява по формулата:
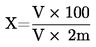
където:
|
X |
= |
% (w/w) карбонати в пробата, изразено като калциев карбонат |
|
V |
= |
обем на СО2 в ml, отделен от частта от пробата. |
|
V1 |
= |
обем CO2 в ml, отделен от 0,5 g CaCO3. |
|
m |
= |
тегло в g на частта от пробата |
7. Забележки
|
7.1. |
Когато частта от пробата тежи повече от 2 g, първо се добавят 15 ml дестилирана вода в колбата (4) и се размесва преди началото на анализа. Използва се същият обем вода за контролната проба. |
|
7.2. |
Ако използваният апарат има обем, различен от този на апарата Шайблер-Дитрих, частта, взета от пробата и от контролното вещество, както и изчисляването на резултатите трябва да се пригодят съответно.
|

П. ОПРЕДЕЛЯНЕ НА ОБЩОТО КОЛИЧЕСТВО ФОСФОР
ФОТОМЕТРИЧЕН МЕТОД
1. Цел и обхват
Настоящият метод дава възможност да се определи общото съдържание на фосфор във фуражите. Той е особено подходящ за анализ на продукти с ниско съдържание на фосфор. В някои случаи (при богати на фосфор продукти) може да се използва гравиметричен метод.
2. Принцип
Пробата се минерализира чрез сухо изгаряне (за органични фуражи) или чрез киселинно изваряване (за минерални смеси и течни фуражи), след което се поставя в разтвор на киселина. Разтворът се обработва с молибдованадатен реагент. Оптичната плътност на така образувания жълт разтвор се измерва със спектрофотометър при 430 nm.
3. Реагенти
|
3.1. |
Калциев карбонат. |
|
3.2. |
Солна киселина ρ20 = 1,10 g/ml (приблизително 6 mol/l). |
|
3.3. |
Азотна киселина ρ20 = 1,045 g/ml. |
|
3.4. |
Азотна киселина ρ20 = 1,38—1,42 g/ml. |
|
3.5. |
Сярна киселина ρ20 = 1,84 g/ml |
|
3.6. |
Молибдованадатен реагент: в градуирана колба от 1 l се смесват 200 ml разтвор на амониев хептамолибдат (точка 3.6.1), 200 ml разтвор на амониев монованадат (точка 3.6.2) и 134 ml азотна киселина (точка 3.4). Допълва се до пълния обем с вода. |
|
3.6.1. |
Разтвор на амониев хептамолибдат: в гореща вода се разтварят 100 g амониев хептамолибдат (NH4) 6Mo7O24.4H2O. Добавят се 10 ml амоняк (с плътност 0,91 g/ml) и обемът се допълва до 1 l с вода. |
|
3.6.2. |
Разтвор на амониев монованадат: 2,35 g амониев монованадат NH4VO3 се разтварят в 400 ml гореща вода. При постоянно бъркане бавно се добавят 20 ml разредена азотна киселина (7 ml HNO3 (т. 3.4) + 13 ml H2O) и обемът се допълва до 1 l с вода. |
|
3.7. |
Стандартен разтвор от 1 mg фосфор на ml: разтварят се 4,387 g калиев дихидроген фосфат KH2PO4 във вода. Допълва се до 1 l с вода. |
4. Апаратура
|
4.1. |
Кварцови, порцеланови или платинени тигли за опепеляване. |
|
4.2. |
Електрическа муфелна пещ с термостат, настроен на 550 oC. |
|
4.3. |
Колба тип Келдал с обем 250 ml. |
|
4.4. |
Градуирани колби и прецизни пипети. |
|
4.5. |
Спектрофотометър. |
|
4.6. |
Епруветки с приблизителен диаметър 16 mm и запушалки, калибровани за диаметър 14,5 mm; вместимост: 25—30 ml. |
5. Процедура
5.1. Приготвяне на разтвора
Според вида на пробата разтворът се приготвя по начина, указан в точка 5.1.1 или в точка 5.1.2.
5.1.1.
Претегля се 1 g или повече от пробата с точност до 1 mg. Пробата за анализ се поставя в колба тип Келдал, добавят се 20 ml сярна киселина (т. 3.5), колбата се разклаща с цел веществото напълно да се импрегнира с киселина и да се предотврати полепването му по стените ѝ, след което се нагрява и се поддържа в точката на кипене в продължение на 10 минути; Оставя се да изстине леко, добавят се 2 ml азотна киселина (точка 3.4), леко се нагрява, оставя се да изстине леко, добавя се още малко азотна киселина (точка 3.4) и отново се довежда до точката на кипене. Тази процедура се повтаря до получаване на безцветен разтвор. Последният се охлажда, добавя се малко вода, течността се прелива в градуирана колба с обем 500 ml, като колбата Келдал се изплаква с гореща вода. Оставя се да изстине, след което обемът се допълва с вода и течността се хомогенизира и филтрира.
5.1.2.
Около 2,5 g от пробата се претеглят с точност до 1 mg в тигел за опепеляване. Пробата за анализ се разбърква до пълно смесване с 1 g калциев карбонат (т. 3.1). Сместа се опепелява в пещта при температура 550 oC до получаване на бяла или сива пепел (малко количество въглен не е от значение). Пепелта се пресипва в бехерова чаша с обем 250 ml. Добавят се 20 ml вода и солна киселина (т. 3.2), докато престане отделянето на газ. Добавят се още 10 ml солна киселина (т. 3.2). Бехеровата чаша се поставя на пясъчна баня и се оставя съдържанието ѝ да се сгъстява чрез изпарение, докато изсъхне, така че силициевият диоксид да стане неразтворим. Остатъкът се разтваря отново в 10 ml азотна киселина (т. 3.3) и се оставя да кипи на пясъчната баня или на нагревателна плоча 5 минути, без да се изпарява до изсъхване. Течността се прелива в градуирана колба с обем 500 ml, като бехеровата чаша се изплаква няколко пъти с гореща вода. Оставя се да изстине, след което обемът се допълва с вода и течността се хомогенизира и филтрира.
5.2. Получаване на оцветяването и измерване на оптичната плътност
Аликвотна част от получения в съответствие с процедурата в т. 5.1.1 или т. 5.1.2 филтрат се разрежда до получаване на концентрация на фосфор не по-висока от 40 μg/ml. Поставят се 10 ml от този разтвор в епруветка (т. 4.6) и се добавят 10 ml от молибдованадатния реагент (т. 3.6). Сместа се хомогенизира и се оставя да престои най-малко 10 минути при температура 20 oC. Оптичната плътност се измерва в спектрофотометър при 430 nm спрямо разтвор, получен чрез добавяне на 10 ml от молибдованадатния реагент (т. 3.6) към 10 ml вода.
5.3. Калибрационна крива
От стандартния разтвор (т. 3.7) се приготвят разтвори, със съдърание на фосфор съответно 5, 10, 20, 30 и 40 μg/ml. Вземат се по 10 ml от всеки от разтворите и към тях се прибавят по 10 ml от молибдованадатния реагент (т. 3.6) Смесите се хомогенизират и се оставят да престоят най-малко 10 минути при температура 20 oC. Измерва се оптичната плътност, както е указано в точка 5.2. Калибрационната крива се очертава, като различните стойности на оптичната плътност се нанасят спрямо съответните количества фосфор. За концентрации между 0 и 40 μg/ml графиката е линейна.
6. Изчисляване на резултатите
Количеството фосфор в пробата за анализ се определя с помощта на калибрационната крива.
Резултатът се изразява като процент от пробата.
Повторяемост
Разликата между резултатите от две паралелни определяния, проведени върху една и съща проба, не трябва да надвишава:
|
— |
3 % отнесени към най-високата стойност за съдържание на фосфор под 5 %, |
|
— |
0,15 % като абсолютна стойност за съдържание на фосфор от 5 % или повече. |
Р. ОПРЕДЕЛЯНЕ НА ХЛОР ОТ ХЛОРИДИ
1. Цел и обхват
Настоящият метод позволява определянето на съдържанието на хлор в разтворими във вода хлориди, обикновено изразявани като натриев хлорид. Той е приложим за всички фуражи.
2. Принцип
Хлоридите се разтварят във вода. Ако продуктът съдържа органични вещества, той се подлага на избистряне. Разтворът се подкислява леко с азотна киселина и хлоридите се утаяват под формата на сребърен хлорид с помощта на разтвор от сребърен нитрат. Излишният сребърен нитрат се титрува с разтвор на амониев тиоцианат по метода на Фолхард.
3. Реагенти
|
3.1. |
Разтвор на амониев тиоцианат 0,1 mol/l. |
|
3.2. |
Разтвор на сребърен нитрат 0,1 mol/l. |
|
3.3. |
Наситен разтвор на амониев ферисулфат (NH4)Fe(SO4)2. |
|
3.4. |
Азотна киселина, плътност: 1,38 g/ml. |
|
3.5. |
Диетилов етер. |
|
3.6. |
Ацетон. |
|
3.7. |
Разтвор на Карез I: 21,9 g цинков ацетат Zn(CH3COO)2 2H2O и 3 g безводна оцетна киселина се разтварят във вода. Допълва се до 100 ml с вода. |
|
3.8. |
Разтвор на Карез II: 10,6 g калиев фероцианид K4 Fe (CN)6 3H2O се разтварят във вода. Допълва се до 100 ml с вода. |
|
3.9. |
Активен въглен, който не съдържа хлориди и не абсорбира хлориди. |
4. Апаратура
Смесител (барабанен): приблизително 35—40 r.p.m.
5. Процедура
5.1. Приготвяне на разтвора
Според естеството на пробата, се приготвя разтвор, както е посочено в т. 5.1.1, т. 5.1.2 или т. 5.1.3.
Същевременно се прави празна проба без подлежащото на анализ вещество.
5.1.1.
Претегля се с точност до един mg проба от не повече от 10 g, която съдържа не повече от 3 g хлор под формата на хлориди. Претегленото количество се слага заедно с 400 ml вода в мерителна колба от 500 ml при температура от около 20 oC. Сместа се размесва тридесет минути в барабанния смесител, допълва се до пълния обем, хомогенизира се и се филтрира.
5.1.2.
Претеглят се около 5 g от пробата с точност до един mg и това количество се поставя с 1 g активен въглен в мерителна колба от 500 ml. Добавят се 400 ml вода с температура около 20 oС и 5 ml разтвор на Карез I (т. 3.7), разбърква се 30 секунди и после се добавят 5 ml разтвор на Карез II (т. 3.8). Сместа се размесва тридесет минути в барабанния смесител, допълва се до пълния обем, хомогенизира се и се филтрира.
5.1.3.
Разтворът се приготвя, както е описано в т. 5.1.2, но не се филтрира. Декантира се (ако се налага — се центрофугира), отливат се 100 ml от супернатанта, които се прехвърлят в мерителна колба от 200ml. Смесва се с ацетон (т. 3.6) и се допълва до пълния обем с този разтворител, хомогенизира се и се филтрира.
5.2. Титруване
С помощта на пипета в ерленмайерова колба се прехвърлят 25 ml—100 ml от филтрата (в зависимост от предполагаемото хлорно съдържание), получен както е описано в т. 5.1.1, т. 5.1.2 или т. 5.1.3. Аликвотната част не трябва да съдържа повече от 150 mg хлор (Cl). Разрежда се, ако е необходимо, до не по-малко от 50 ml с вода, добавят се 5 ml азотна киселина (т. 3.4), 20 ml наситен разтвор на амониев ферисулфат (т. 3.3) и две капки разтвор на амониев тиоцианат (т. 3.1), прехвърлен с помощта на бюрета, допълнена до нулевото деление. Като се използва бюрета, разтворът на сребърен нитрат (т. 3.2) се прехвърля по такъв начин, че да се получи излишък от 5 ml. Добавят се 5 ml диетилов етер (т. 3.5) и се разклаща енергично, за да коагулира утайката. Излишният сребърен нитрат се титрува с разтвора на амониев тиоцианат (т. 3.1) дотогава, докато червено-кафеникавото оцветяване се задържи една минута.
6. Изчисляване на резултатите
Съдържанието на хлор (Х), изразено като процентно съдържание на натриев хлорид, се изчислява по следната формула:
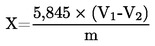
където:
|
V1 |
= |
обем (в ml) на добавения разтвор на сребърен нитрат 0,1 mol/l |
|
V2 |
= |
обем (в ml) на разтвора на амониев тиоцианат 0,1 mol/l, използван за титруване |
|
m |
= |
тегло на пробата. |
В случай че празната проба покаже изразходване на разтвора на сребърен нитрат с концентрация 0,1 mol/l, тази стойност се изважда от обема (V1 - V2).
7. Забележки
|
7.1. |
Титруването може да проведе и чрез потенциометрия. |
|
7.2. |
При продукти, които са много богати на масла и мазнини, първо се извършва обезмасляване с диетилов етер или петролен етер. |
|
7.3. |
При рибено брашно титруването може да се извърши по метода на Мор. |
(1) За изсушаването на зърнени култури, брашно, булгур и едросмляно брашно, фурната трябва да има такъв топлинен капацитет, че при предварително загряване до 131 oC, да достигне отново тази температура за по-малко от 45 минути след като в нея е било сложено максималното количество проби за едновременно сушене. Вентилацията трябва да е такава, че когато толкова проби от обикновено жито, колкото може да събере, се сушат в продължение на два часа, резултатите се различават от онези, които се получават след четиричасово сушене, с по-малко от 0,15 %.
(2) Когато маслото или мазнината трябва след това да бъдат подложени на изпитания за качество, вместо парченца пемза се използват стъклени топчета.
ПРИЛОЖЕНИЕ IV
МЕТОДИ ЗА АНАЛИЗ С ЦЕЛ КОНТРОЛ НА КОЛИЧЕСТВАТА РАЗРЕШЕНИ ДОБАВКИ ВЪВ ФУРАЖИТЕ
А. ОПРЕДЕЛЯНЕ НА ВИТАМИН А
1. Цел и обхват
Настоящият метод дава възможност за определяне на количеството на витамин А (ретинол) във фуражи и премикси. Витамин А включва изцяло транс ретинилов алкохол и неговите цис-изомери, които се определят по този метод. Съдържанието на виамин А се изразява в международни единици (IU) на kg. Една IU отговаря на активността на 0,300 μg изцяло транс-витамин А алкохол или на 0,344 μg изцяло транс-витамин А ацетат, или на 0,550 μg изцяло транс-витамин А палмитат.
Прагът на значимост е 2 000 UI витамин А/kg.
2. Принцип
Пробата се хидролизира с етанолов разтвор на калиев хидроксид и витамин А се извлича в петролен етер. Разтворителят се елиминира чрез изпаряване; остатъкът се разтваря в метанол и, ако е необходимо, се разрежда до изискваната концентрация. Съдържанието на витамин А се определя чрез високоефективна течна хроматография с обратна фаза (RP-HPLC) с помощта на UV-детектор или флуоресцентен детектор. Параметрите на хроматографията се подбират така, че да не се разделя изцяло транс-витамин А алкохол от неговите цис-изомери.
3. Реагенти
|
3.1. |
Етанол, σ = 96 % |
|
3.2. |
Петролен етер, интервал на температурата на кипене 40 oС—60 oС |
|
3.3. |
Метанол |
|
3.4. |
Разтвор на калиев хидроксид, c = 50 g/100 ml |
|
3.5. |
Разтвор на натриев аскорбат, c = 10 g/100 ml (вж. забележката в точка 7.7) |
|
3.6. |
Натриев сулфид, Na2S x H2O(х = 7—9) |
|
3.6.1. |
Разтвор на натриев сулфид, с = 0,5 mol/l в глицерин ß = 120 g/l (за x = 9) (вж. забележките в точка 7.8). |
|
3.7. |
Разтвор на фенолфталеин, c = 2 g/100 ml в етанол (точка 3.1) |
|
3.8. |
2-пропанол |
|
3.9. |
Подвижна фаза за HPLC: смес от метанол (точка 3,3) и вода, например: 980 + 20 (v + v). Точните пропорции се определят от характеристиките на използваната колона. |
|
3.10. |
Азот, незамърсен с кислород. |
|
3.11. |
Изцяло-trans-витамин А ацетат, с изключителна чистота, с гарантирана активност, например 2,80 × 106 IU/g |
|
3.11.1. |
Основен разтвор на изцяло транс-витамин А ацетат: Претеглят се 50 mg витамин А ацетат (т. 3.11) с точност до 0,1 mg в мерителна колба от 100 ml. Разтварят се в 2-пропанол (т. 3.8) и се допълва до марката със същия разтворител. Номиналната концентрация на този разтвор е 1 400 IU витамин А на ml. Точната концентрация следва да се определи в съответствие с точка 5.6.3.1. |
|
3.12. |
Изцяло транс витамин А палмитат, с изключителна чистота, с гарантирана активност, например 1,80 × 106 IU/g. |
|
3.12.1. |
Основен разтвор на all-trans-витамин А палмитат: претеглят се 80 mg витамин А палмитат (т. 3.12) с точност до 0,1 mg в мерителна колба от 100 ml. Разтварят се в 2-пропанол (т. 3.8) и се допълва до марката със същия разтворител. Номиналната концентрация на този разтвор е 1 400 IU витамин А на ml. Точната концентрация следва да се определи в съответствие с точка 5.6.3.2. |
|
3.13. |
2,6-ди-терт-бутил-4-метилфенол (BHT, бутилхидрокситолуол) (вж. забележката в т. 7.5) |
4. Апаратура
|
4.1. |
Вакуумен ротационен изпарител. |
|
4.2. |
Лабораторни съдове от тъмно стъкло |
|
4.2.1. |
Плоскодънни или конусовидни колби от 500 ml, с шлиф |
|
4.2.2. |
Градуирани колби с шлифована стъклена запушалка, тясно гърло, 10, 25, 100 и 500 ml. |
|
4.2.3. |
Конусовидни делителни фунии, 1 000 ml, с шлифована стъклена запушалка |
|
4.2.4. |
Крушообразни колби, 250 ml, с шлиф |
|
4.3. |
Хладник на Алин, дължина на кожуха 300 mm, с шлифован стъклен шлиф, с адаптер за тръба за захранване с газ |
|
4.4. |
Нагъната филтърна хартия за сепарация на фазите, диаметър 185 mm (например Schleicher & Schuell 597 HY 1/2) |
|
4.5. |
Оборудване за HPLC със система за впръскване |
|
4.5.1. |
Колона за течна хроматография, 250 mm × 4 mm, С18, пълнеж от 5 или 10 μm, или еквивалентна (експлоатационен критерий: при првеждане на HPLC трябва да се получи само един пик за всички изомери на ретинола). |
|
4.5.2. |
UV-детектор или флуоресцентен детектор с настройка на дължината на вълната |
|
4.6. |
Спектрофотометър с 10-милиметрови кварцови елементи |
|
4.7. |
Вана за водна баня с магнитна бъркалка |
|
4.8. |
Екстрактор (вж. фиг.1), състоящ се от: |
|
4.8.1. |
Стъклен цилиндър с обем 1 l, с шлифовани стъклени гърло и запушалка |
|
4.8.2. |
Шлифована стъклена вложка, снабдена със странична тръбичка и регулируема тръба, която минава през центъра ѝ. Долната част на регулируемата тръба е с U-образна форма и с дюза в противоположния край, така че горният течен слой в цилиндъра да може да се прехвърли в делителна фуния |
5. Процедура
|
Забележка: |
Витамин А е чувствителен към (УВ) светлина и податлив на окисляване. Всички манипулации се извършват на тъмно (в стъклени съдове от тъмно стъкло или съдове, покрити с алуминиево фолио) и в отсъствие на кислород (елиминира се с азот). По време на екстракцията въздухът над течността се заменя с азот (за да се избягва прекалено голямото налягане, от време на време запушалката трябва да се отваря). |
5.1. Подготовка на пробата
Пробата се смила, така че да може да премине през сито с отвори 1 mm, като се избягва нагряване. Смилането трябва да стане непосредствено преди претеглянето и осапуняването, в противен случай съществува риск от загуби на витамин А.
5.2. Осапуняване
Според тегловното съдържание на витамин А, в плоскодънна или конусовидна колба от 500 ml (т. 4.2.1.) се претеглят с точност до 1mg 2—25 g от пробата. Последователно се прибавят при постоянно разбъркване 130 ml етанол (т. 3.1), около 100 mg BHT (т. 3.13), 2 ml разтвор на натриев аскорбат (т. 3.5) и 2 ml разтвор на натриев сулфид (т. 3.6). Монтира се хладникът (т. 4.3) на колбата и последната се потапя във вана за водна баня с магнитна бъркалка (т. 4.7). Нагрява се до кипене и в продължение на 5 min се оставят парите да преминават през хладника. След това се прибавят 25 ml разтвор на калиев хидроксид (т. 3.4) през хладника (т. 4.3) и отново се оставят парите да преминават през хладника в продължение на 25 min, като се бърка при слаб приток на азот. След това хладникът се изплаква с приблизително 20 ml вода и се оставя съдържанието на колбата да изстине до достигане на стайна температура.
5.3. Екстракция
Чрез декантация осапуняващият разтвор се прехвърля количествено, като колбата се изплаква с общо 250 ml вода, в делителна фуния от 1 000 ml (точка 4.2.3) или в екстрактора (точка 4.8). Колбата за осапуняване се изплаква последователно с 25 ml етанол (т. 3.1) и 100 ml петролен етер (т. 3.2) и течността от изплакването се прехвърля в делителната фуния или в екстрактора. Съотношението между водата и етанола при така комбинираните разтвори трябва да е около 2:1. Разклаща се енергично в продължение на 2 min и се оставя в покой за 2 min.
5.3.1.
Когато слоевете се отделят (вж. забележката в т. 7.3), слоят от петролен етер се прехвърля в друга делителна фуния (т. 4.2.3). Екстракцията се повтаря два пъти със 100 ml петролен етер (т. 3.2), а след това два пъти с 50 ml петролен етер (т. 3.2).
Комбинираните екстракти се промиват два пъти в делителната фуния при леко разклащане (за да се избегне образуването на емулсия) с порции по 100 ml вода и след това с повторно разклащане с нови порции по 100 ml вода, докато последната остане безцветна след добавянето на разтвор на фенолфталеин (т. 3.7) (обикновено са достатъчни четири промивания). Промитият екстракт се филтрира със сух нагънат филтър за отделяне на фазите (т. 4.4), с цел да се отстрани всякаква суспендирана вода, и се прехвърля в градуирана колба от 500 ml (т. 4.2.2). Делителната фуния и филтърът се изплакват с 50 ml петролен етер (т. 3.2), допълва се до пълния обем с петролен етер (т. 3.2) и се разбърква добре.
5.3.2.
Когато слоевете се отделят (вж. забележката в т. 7.3), запушалката на стъкления цилиндър (т. 4.8.1) се заменя с шлифованата стъклена вложка (т. 4.8.2), като долният U-образен край на регулируемата тръба се поставя така, че да се окаже точно над нивото на граничната повърхност. Чрез подаване на азот през страничната тръбичка се прилага налягане и се прехвърля горният слой петролен етер в делителна фуния от 1 000 ml (т. 4.2.3). Прибавят се 100 ml петролен етер (т. 3.2) в стъкления цилиндър, поставя се запушалката и силно се разклаща. Оставят се слоевете да се отделят и горният слой се прехвърля в делителната фуния, както е посочено по-горе. Процедурата на екстракция се повтаря с нови 100 ml петролен етер (т. 3.2), след това още два пъти с по 50 ml петролен етер (т. 3.2) и слоевете петролен етер се добавят в делителната фуния.
Събраните екстракти от петролен етер се промиват съгласно процедурата, описана в точка 5.3.1, и се процедира съгласно указаното в посочената точка.
5.4. Подготовка на разтвора на пробата за HPLC
Аликвотна част от разтвора на петролен етер (т. 5.3.1 или т. 5.3.2) се прехвърля с пипета в крушообразна колба от 250 ml (т. 4.2.4). Оставя се разтворителят да се изпари почти изцяло в ротационния изпарител (т. 4.1) при понижено налягане, при температура на ваната не по-висока от 40 oС. Възстановява се атмосферното налягане, като се вкара азот (точка 3.10), и колбата се изважда от ротационния изпарител. Останалото количество разтворител се отстранява в поток азот (т. 3.10) и остатъкът незабавно се разтваря в известен обем (10—100 ml) метанол (т. 3.3) (концентрацията на витамин А трябва да е в интервала от 5 IU/ml до 30 IU/ml).
5.5. Определяне чрез HPLC
Витамин А се отделя в колона С18 с обратна фаза (т. 4.5.1) и концентрацията му се измерва с помощта на UV-детектор (325 nm) или флуоресцентен детектор (възбуждане: 325 nm, излъчване: 475 nm. (т. 4.5.2).
Впръсква се аликвотна част (например 20 μl) от получения метанолов разтвор (вж. т. 5.4) и се елуира с мобилната фаза (т. 3.9). Изчислява се средната височина на пика (площ) на няколко впръсквания със същия разтвор на пробата и средните височини на пиковете (площи) на няколко впръсквания с калибрационните разтвори (т. 5.6.2).
Дават се следните указания за условията, като могат да се прилагат и други условия, стига те да дават еквивалентни резултати.
|
Колона за течна хроматография (т. 4.5.1): |
250 mm × 4 mm, С18, пълнеж от 5 μm или10 μm или еквивалентна |
|
Подвижна фаза (т. 3.9): |
Смес от метанол (т. 3.3) и вода, например: 980 + 20 (v + v). |
|
Скорост на изтичане: |
1—2 ml/min; |
|
Детектор (т. 4.5.2): |
UV-детектор (325 nm) или флуоресцентен детектор (възбуждане: 325 nm, излъчване: 475 nm) |
5.6. Калибриране
5.6.1.
Отмерват се с пипета 20 ml от основния разтвор на витамин А ацетат (т. 3.11.1) или 20 ml от основния разтвор на витамин А палмитат (т. 3.12.1) в колба с плоско дъно или конусообразна колба с обем 500 ml (т. 4.2.1) и се хидролизира, както е описано в т. 5.2, но без да се прибавя BHT. След това се пристъпва към екстракция с петролен етер (т. 3.2) съгласно процедурата в т. 5.3 и се допълва до 500 ml с петролен етер (т. 3.2). В ротативния изпарител (вж. т. 5.4) 100 ml от този екстракт се подлагат на почти пълно изпаряване, отстранява се останалото количество разтворител в поток от азот (т. 3.10) и остатъкът отново се разтваря в 10,0 ml метанол (т. 3.3). Номиналната концентрация на този разтвор е 560 IU витамин А на ml. Точната концентрация следва да се определи в съответствие с т. 5.6.3.3. Работният стандартен разтвор се приготвя непосредствено преди употреба.
Отмерват се с пипета 2,0 ml от работния стандартен разтвор в градуирана колба от 20 ml, допълва се до пълния обем с метанол (т. 3.3) и се размесва. Номиналната концентрация на този разреден работен стандартен разтвор е 56 IU витамин А на ml.
5.6.2.
Прехвърлят се 1,0, 2,0, 5,0 и 10,0 ml от разредения работен стандартен разтвор в градуирани колби от 20 ml, допълва се до пълния обем с метанол (т. 3.3) и се размесва. Номиналните концентрации на тези разтвори са 2,8, 5,6, 14,0 и 28,0 IU витамин А на ml.
Неколкократно се впръскват 20 μl от всеки калибрационен разтвор и се определят средните височини на пиковете (площите). Според средните височини на пиковете (площите) се начертава калибрационна крива, като се вземат предвид резултатите от UV-контрола (т. 5.6.3.3).
5.6.3.
5.6.3.1.
Отмерват се с пипета 2,0 ml от основния разтвор на витамин А ацетат (т. 3.11.1) в градуирана колба от 50 ml (т. 4.2.2) и се допълва до пълния обем с 2-пропанол (т. 3.8). Номиналната концентрация на този разтвор е 56 IU витамин А на ml. Отмерват се пипета 3,0 ml от този разтвор в градуирана колба от 25 ml и се допълва до пълния обем с с 2-пропанол (т. 3.8). Номиналната концентрация на този разтвор е 6,72 IU витамин А на ml. Ултравиолетовият (UV) спектър на разтвора се сравнява с този на 2-пропанол (т. 3.8) в спектрофотометъра (т. 4.6) между 300 nm и 400 nm. Максимумът на екстинкция трябва да бъде между 325 nm и 327 nm.
Изчисляване на съдържанието на витамин А:
IU витамин A/ml = E326 × 19,0
( за витамин А ацетат = 1 530 при 326 nm в 2-пропанол)
за витамин А ацетат = 1 530 при 326 nm в 2-пропанол)
5.6.3.2.
Отмерват се с пипета 2,0 ml от основния разтвор на витамин А палмитат (т. 3.12.1) в градуирана колба от 50 ml (т. 4.2.2) и се допълва до пълния обем с пропанол-2 (т. 3.8). Номиналната концентрация на този разтвор е 56 IU витамин А на ml. Отмерват се пипета 3,0 ml от този разреден разтвор наа витамин А в градуирана колба от 25 ml и се допълва до пълния обем с 2-пропанол (т. 3.8). Номиналната концентрация на този разтвор е 6,72 IU витамин А на ml. Ултравиолетовият (UV) спектър на разтвора се сравнява с този на 2-пропанол (т. 3.8) в спектрофотометърa (т. 4.6) между 300 nm и 400 nm. Максимумът на екстинкция трябва да бъде между 325 nm и 327 nm.
Изчисляване на съдържанието на витамин А:
IU витамин A/ml = E326 × 19,0
( за витамин А палмитат = 957 при 326 nm в 2-пропанол)
за витамин А палмитат = 957 при 326 nm в 2-пропанол)
5.6.3.3.
Отмерват се с пипета 3,0 ml неразреден работен стандартен разтвор на витамин А, приготвен по процедурата в т. 5.6.1, в градуирана колба от 50 ml (т. 4.2.2) и се допълва до пълния обем с 2-пропанол (т. 3.8). Отмерват се пипета 5,0 ml от този разтвор в градуирана колба от 25 ml и се допълва до пълния обем с 2-пропанол (т. 3.8). Номиналната концентрация на този разтвор е 6,72 IU витамин А на ml. Ултравиолетовият (UV) спектър на разтвора се сравнява с този на 2-пропанол (т. 3.8) в спектрофотометъра (т. 4.6) между 300 nm и 400 nm. Максимумът на екстинкция трябва да бъде между 325 nm и 327 nm.
Изчисляване на съдържанието на витамин А:
IU витамин A/ml = E325 × 18,3
( за витамин А алкохол = 1 821 при 325 nm в 2-пропанол)
за витамин А алкохол = 1 821 при 325 nm в 2-пропанол)
6. Изчисляване на резултатите
Като се изхожда от средната височина (площ) на пиковете на витамин А на разтвора на пробата, се определя концентрацията на този разтвор в IU/ml чрез сравняване с калибрационната крива (т. 5.6.2).
Съдържанието w на витамин А в пробата, изразено в IU/kg, се получава по следната формула:
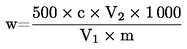 [UI/kg]
[UI/kg]
където:
|
c |
= |
концентрация на витамин А в разтвора на пробата (т. 5.4) в IU/ml |
|
V1 |
= |
обем на разтвора на пробата (т. 5.4) в ml |
|
V2 |
= |
обем на взетата аликвотна част по т. 5.4 в ml |
|
m |
= |
тегло на частта от пробата за анализ в g. |
7. Забележки
|
7.1. |
При проби с ниско съдържание на витамин А, може да се окаже полезно да се обединят получените от две осапунявания екстракти в петролен етер (претеглено количество: 25 g) в един разтвор на пробата за определяне с HPLC. |
|
7.2. |
В теглото на взетата за анализ проба не трябва да се съдържат повече от 2 g мазнини. |
|
7.3. |
Ако не се получи разделяне на фазите, се прибавят около 10 ml етанол (т. 3.1), за да се пресече емулсията. |
|
7.4. |
При рибено масло и други чисти мазнини времето на осапуняване се удължава до 45—60 min. |
|
7.5. |
ВНТ може да се замени с хидрохинон. |
|
7.6. |
Разделянето на ретиноловите изомери е възможно при използване на нормалнофазова колона. В този случай обаче при изчисленията височината (площта) на пиковете на транс- и цисизомерите трябва да се сумира. |
|
7.7. |
Разтворът на натриев аскорбат може да се замени с около 150 ml аскорбинова киселина. |
|
7.8. |
Разтворът на натриев сулфид може да се замени с около 50 ml EDTA. |
|
7.9. |
При анализ на витамин А в заместители на млякото трябва да се обърне специално внимание на:
За да се провери дали прилаганият метод за анализ дава надеждни резултати за конкретната матрица (заместител на мляко), се провежда тест за възстановяване върху още една част от пробата. Ако степента на възстановяване е по-ниска от 80 %, аналитичният резултат следва да се коригира с оглед на възстановяването. |
8. Повторяемост
Разликата в резултатите от две паралелни определяния на една и съща проба не трябва да надхвърля 15 % по отношение на най-високия резултат.
9. Резултати от съвместно изследване (1)
|
|
Премикс |
Премикс фураж |
Минерален концентрат |
Протеинов фураж |
Прасенца |
|
L |
13 |
12 |
13 |
12 |
13 |
|
n |
48 |
45 |
47 |
46 |
49 |
|
Средна стойност в IU/kg |
17,02 × 106 |
1,21 × 106 |
537 100 |
151 800 |
18 070 |
|
sr [IU/kg] |
0,51 × 106 |
0,039 × 106 |
22 080 |
12 280 |
682 |
|
r [IU/kg] |
1,43 × 106 |
0,109 × 106 |
61 824 |
34 384 |
1 910 |
|
CVr [ %] |
3,0 |
3,5 |
4,1 |
8,1 |
3,8 |
|
sR [IU/kg] |
1,36 × 106 |
0,069 × 106 |
46 300 |
23 060 |
3 614 |
|
R [IU/kg] |
3,81 × 106 |
0,193 × 106 |
129 640 |
64 568 |
10 119 |
|
CVR [ %] |
8,0 |
6,2 |
8,6 |
15 |
20 |
|
L |
= |
брой на лабораториите |
|
n |
= |
брой на единичните стойности |
|
sr |
= |
стандартно отклонение на повторяемостта |
|
sR |
= |
стандартно отклонение на възпроизводимостта |
|
r |
= |
повторяемост |
|
R |
= |
възпроизводимост |
|
CVr |
= |
коефициент на вариация на повторяемостта |
|
CVR |
= |
коефициент на вариация на възпроизводимостта |

Б. ОПРЕДЕЛЯНЕ НА ВИТАМИН Е
1. Цел и обхват
Настоящият метод дава възможност за определяне на количеството на витамин Е във фуражи и премикси. Съдържанието на витамин Е се изразява в mg DL-α-токоферол ацетат на kg. 1 mg DL-α-токоферол ацетат отговаря на 0,91 mg DL-α-токоферол (витамин Е).
Прагът на количествено определяне е 2 mg витамин Е/kg. Достигането на този праг е възможно само с флуоресцентен детектор. При използване на UV-детектор прагът на количествено определяне е 10 mg/kg.
2. Принцип
Пробата се хидролизира с етанолов разтвор на калиев хидроксид и витамин Е се извлича в петролен етер. Разтворителят се елиминира чрез изпаряване, остатъкът се разтваря в метанол и, ако е необходимо, се разрежда до изискваната концентрация. Съдържанието на витамин Е се определя чрез високоефективна течна хроматография с обратна фаза с помощта на флуоресцентен детектор или UV-детектор.
3. Реагенти
|
3.1. |
Етанол, σ = 96 % |
|
3.2. |
Петролен етер, интервал на температура на кипене 40 oС—60 oС |
|
3.3. |
Метанол |
|
3.4. |
Разтвор на калиев хидроксид, с = 50 g/100 ml |
|
3.5. |
Разтвор на натриев аскорбат, c = 10 g/100 ml (вж. забележката в т. 7.7.) |
|
3.6. |
Натриев сулфид, Na2S x H2O (х = 7—9) |
|
3.6.1. |
Разтвор на натриев сулфид, с = 0,5 mol/l в глицерин β = 120 g/l (за x = 9) (вж. забележките в т. 7.8). |
|
3.7. |
Разтвор на фенолфталеин, c = 2 g/100 ml в етанол (т. 3.1) |
|
3.8. |
Подвижна фаза за HPLC: смес от метанол (т. 3.3) и вода, например: 980 + 20 (v + v). Точното съотношение се определя от характеристиките на използваната колона. |
|
3.9. |
Азот, незамърсен с кислород. |
|
3.10. |
DL-α-токоферол ацетат, с изключителна чистота, с гарантирана активност. |
|
3.10.1. |
Основен разтвор на DL-α-токоферол ацетат: претеглят се с точност до 0,1 mg 100 mg DL-α-токоферол ацетат (т. 3.10) в градуирана колба от 100 ml. Разтварят се в етанол (т. 3.1) и се допълва до пълния обем със същия разтворител. 1 ml от този разтвор съдържа 1 mg DL-α-токоферол ацетат. (за контрола с ултравиолетови лъчи вж. т. 5.6.1.3; за стабилизирането вж. забележките в т. 7.4). |
|
3.11. |
DL-α-токоферол, с изключителна чистота, с гарантирана активност |
|
3.11.1. |
Основен разтвор на DL-α-токоферол. Претеглят се с точност до 0,1 mg 100 mg DL-α-токоферол (т. 3.11) в градуирана колба от 100 ml. Разтварят се в етанол (т. 3.1) и се допълва до пълния обем със същия разтворител. 1 ml от този разтвор съдържа 1 mg DL-α-токоферол. (за контрола с ултравиолетови лъчи вж. т. 5.6.2.3; за стабилизирането вж. забележките в т. 7.4). |
|
3.12. |
2,6-ди-терт-бутил-4-метилфенол (BHT, бутилхидрокситолуол) (вж. забележката в т. 7.5) |
4. Апаратура
|
4.1. |
Ротационен тънкослоен изпарител. |
|
4.2. |
Лабораторни съдове от тъмно стъкло |
|
4.2.1. |
Плоскодънни или конусовидни колби от 500 ml, с шлиф |
|
4.2.2. |
Градуирани колби с шлифовани стъклени запушалки, тясно гърло, от 10, 25, 100 и 500 ml. |
|
4.2.3. |
Конусовидни делителни фунии, 1 000 ml, с шлифовани стъклени запушалки |
|
4.2.4. |
Крушообразни колби, 250 ml, с шлиф |
|
4.3. |
Хладник на Алин, дължина на кожуха 300 mm, с шлифован стъклен шлиф, с адаптер за тръба за захранване с газ |
|
4.4. |
Нагъната филтърна хартия за сепарация на фазите, диаметър 185 mm (например Schleicher & Schuell 597 HY 1/2) |
|
4.5. |
Оборудване за HPLC със система за впръскване |
|
4.5.1. |
Колона за течна хроматография, 250 mm × 4 mm, С18, пълнеж от 5 или 10 μm, или еквивалентна |
|
4.5.2. |
Флуоресцентен детектор или UV-детектор с настройка на дължината на вълната |
|
4.6. |
Спектрофотометър с 10-милиметрови кварцови елементи |
|
4.7. |
Вана за водна баня с магнитна бъркалка |
|
4.8. |
Екстрактор (вж. фиг.1), състоящ се от: |
|
4.8.1. |
Стъклен цилиндър с обем 1 l, с шлифовани стъклени гърло и запушалка. |
|
4.8.2. |
Шлифована стъклена вложка, снабдена със странична тръбичка и регулируема тръба, която минава през центъра ѝ. Долната част на регулиращата се тръба следва да бъде с U-образна форма и с дюза в противоположния край, така че горният течен слой в цилиндъра да може да се прехвърли в делителна фуния |
5. Процедура
|
Забележка: |
Витамин Е е чувствителен към (UV-) светлина и податлив на окисляване. Всички манипулации трябва да се извършват на тъмно (в съдове от тъмно или покрито с алуминиево фолио стъкло) и в отсъствие на кислород (елиминира се с азот). По време на екстракцията въздухът над течността се заменя с азот (за да се избягва прекалено голямото налягане, от време на време трябва да се отваря запушалката). |
5.1. Подготовка на пробата
Пробата се смила, така че да може да премине през сито с отвори 1 mm, като се избягва нагряване. Смилането трябва да стане непосредствено преди претеглянето и осапуняването, в противен случай съществува риск от загуби на витамин Е.
5.2. Осапуняване
Според тегловното съдържание на витамин Е, от 2 до 25 g от пробата се претеглят с точност до 0,01 g в плоскодънна или конусовидна колба от 500 ml (т. 4.2.1.). Последователно се прибавят при постоянно разклащане 130 ml етанол (т. 3.1), около 100 mg BHT (т. 3.12), 2 ml разтвор на натриев аскорбат (т. 3.5) и 2 ml разтвор на натриев сулфид (точка 3.6). Монтира се хладникът (т. 4.3) на колбата и последната се потапя във вана за водна баня с магнитна бъркалка (т. 4.7). Нагрява се до кипене и в продължение на 5 min се оставят парите да преминават през хладника. След това се прибавят 25 ml разтвор на калиев хидроксид (т. 3.4) през хладника (т. 4.3) и отново се оставят парите да преминават през хладника в продължение на 25 min, като се бърка при слаб приток на азот. Изплаква се хладникът с около 20 ml вода и се оставя съдържанието на колбата да изстине до достигането на стайна температура.
5.3. Екстракция
Чрез декантация осапуняващият разтвор се прехвърля количествено, като колбата се изплаква с общо 250 ml вода, в делителна фуния от 1 000 ml (т. 4.2.3) или в екстрактора (т. 4.8). Колбата за осапуняване се изплаква последователно с 25 ml етанол (т. 3.1) и 100 ml петролен етер (т. 3.2) и течността от изплакването се прехвърля в делителната фуния или в екстрактора. Съотношението между водата и етанола в комбинираните разтвори трябва да е около 2:1. Разклаща се енергично в продължение на 2 min и се оставя в покой за 2 min.
5.3.1.
Когато слоевете се отделят (вж. забележката в т. 7.3), слоят от петролен етер се прехвърля в друга делителна фуния (т. 4.2.3). Екстракцията се повтаря два пъти с 100 ml петролен етер (т. 3.2), а след това два пъти с 50 ml петролен етер (т. 3.2).
Събраните екстракти се промиват два пъти в делителната фуния при леко разклащане (за да се избегне образуването на емулсия) с порции от по 100 ml вода и след това с повторно разклащане с нови порции от по 100 ml вода, докато последната остане безцветна след добавянето на разтвор на фенолфталеин (т. 3.7) (обикновено са достатъчни четири промивания). Промитият екстракт се филтрира със сух нагънат филтър за отделяне на фазите (т. 4.4), за да се отстрани всякаква суспендирана вода, и се прехвърля в градуирана колба от 500 ml (т. 4.2.2). Делителната фуния и филтърът се изплакват с 50 ml петролен етер (т. 3.2), долива се до пълния обем с петролен етер (т. 3.2) и се разбърква добре.
5.3.2.
Когато слоевете се отделят (вж. забележката в т. 7.3), запушалката на стъкления цилиндър (т. 4.8.1) се заменя с шлифованата стъклена вложка (т. 4.8.2), като долният U-образен край на регулиращата се тръба се поставя така, че да се окаже точно над нивото на разделителната повърхност. Чрез подаване на азот през страничната тръбичка се прилага налягане и се прехвърля горният слой петролен етер в делителна фуния от 1 000 ml (т. 4.2.3). Прибавят се 100 ml петролен етер (т. 3.2) в стъкления цилиндър, затваря се с запушалката и силно се разклаща. Оставят се слоевете да се отделят и горният слой се прехвърля в делителната фуния, както е посочено по-горе. Процедурата на екстракция се повтаря с нови 100 ml петролен етер (т. 3.2), след това още два пъти с по 50 ml петролен етер (т. 3.2) и слоевете петролен етер се добавят в делителната фуния.
Събраните екстракти от петролен етер се промиват съгласно процедурата, описана в т. 5.3.1, и се изпълняват процедурата, описана в посочената точка.
5.4. Подготовка на разтвора на пробата за HPLC
Аликвотна част от разтвора на петролен етер (т. 5.3.1 или т. 5.3.2) се прехвърлят с пипета в крушообразна колба от 250 ml (т. 4.2.4). Оставя се разтворителят да се изпари почти изцяло в ротационния изпарител (т. 4.1) при понижено налягане, при температура на ваната не по-висока от 40 oС. Възстановява се атмосферното налягане, като се вкара азот (точка 3.9), и колбата се изважда от ротационния изпарител. Отстранява се останалото количество разтворител в поток азот (т. 3.9) и остатъкът незабавно се разтваря в известен обем (10—100 ml) метанол (т. 3.3) (концентрацията на DL-α-токоферол трябва да е в интервала от 5 μg/ml до 30 μg/ml).
5.5. Определяне чрез HPLC
Витамин Е се отделя в обратнофазова колона С18 (т. 4.5.1) и концентрацията му се измерва с помощта на флуоресцентен детектор (възбуждане: 295 nm, излъчване: 330 nm) или с UV-детектор (292 nm) (т. 4.5.2).
Впръсква се аликвотна част (например 20 μl) от получения в съответствие с точка 5.4 метанолов разтвор и се елуира с подвижната фаза (т. 3.8). Изчисляват се средните височини на пиковете (площите) на няколко впръсквания със същия разтвор на пробата и средните височини на пиковете (площи) на няколко впръсквания с калибрационните разтвори (т. 5.6.2).
Дават се следните указания за условията: могат да се прилагат и други условия, при условие че те дават еквивалентни резултати:
|
Колона за течна хроматография (т. 4.5.1): |
250 mm × 4 mm, С18, пълнеж от 5 μm или от 10 μm или еквивалентна |
|
Подвижна фаза (т. 3.8): |
Смес от метанол (т. 3.3) и вода, например: 980 + 20 (v + v). |
|
Скорост на изтичане: |
1—2 ml/min; |
|
Детектор (т. 4.5.2): |
Флуоресцентен индикатор (възбуждане: 295 nm/излъчване: 330 nm) или UV детектор (292 nm) |
5.6. Калибриране (DL-α-токоферол ацетат или DL-α-токоферол)
5.6.1.
5.6.1.1.
Прехвърлят се с пипета 25 ml от основния разтвор на DL-α-токоферол ацетат (т. 3.10.1) в плоскодъннаили конусообразна колба с обем 500 ml (т. 4.2.1) и се хидролизират, както е описано в точка 5.2. След това се пристъпва към екстракция с петролен етер (т. 3.2) в съответствие с процедурата в т. 5.3 и се допълва до 500 ml с петролен етер. Взимат се 25 ml от този екстракт и се подлагат на изпаряване в ротационния изпарител (т. 5.4) до почти пълно изпаряване на разтворителя, отстранява се останалото количество разтворител в поток от азот (т. 3.9) и остатъкът се разтваря отновов 25,0 ml метанол (т. 3.3). Номиналната концентрация на този разтвор е 45,5 μg DL-α-токоферол на ml, еквивалентен на 50 μg DL-α-токоферол ацетат на ml. Работният стандартен разтвор следва да е приготвен непосредствено преди употреба.
5.6.1.2.
Прехвърлят се 1,0, 2,0, 4,0 и 10,0 ml от работния стандартен разтвор в няколко градуирани колби от 20 ml, допълват се до пълния обем с метанол (т. 3.3) и съдържанието се разбърква. Номиналните концентрации на тези разтвори са 2,5, 5,0, 10,0 и 25,0 μg/ml DL-α-токоферол ацетат, или 2,28, 4,55, 9,10 и 22,8 μg DL-α-токоферол.
Неколкократно се впръскват 20 μl от всеки калибрационен разтвор и се определят средните височини на пиковете (площите). Като се използват средните височини на пиковете (площите), се начертава калибрационната крива.
5.6.1.3.
Разреждат се 5,0 ml от основния разтвор на DL-α-токоферол ацетат (т. 3.10.1) с етанол до 25,0 ml и ултравиолетовият спектър на този разтвор се сравнява със спектъра на етанол (т. 3.1) в спектрален фотометър (т. 4.6) при дължина на вълната между 250 nm и 320 nm.
Максималната абсорбция трябва да е при 284 nm:
 = 43,6 при 284 nm в етанол
= 43,6 при 284 nm в етанол
При това разреждане трябва да се получи стойност на екстинкция от 0,84 до 0,88.
5.6.2.
5.6.2.1.
Прехвърлят се с пипета 2 ml от основния разтвор на DL-α-токоферол (т. 3.11.1) в градуирана колба от 50 ml, разтваря се в метанол (т. 3.3) и се допълва до пълния обем с метанол. Номиналната концентрация на този разтвор е 40 μg DL-α-токоферол на ml, еквивалентен на 44,0 μg DL-α-токоферол ацетат на ml. Работният стандартен разтвор следва да бъде приготвен непосредствено преди употреба.
5.6.2.2.
Прехвърлят се 1,0, 2,0, 4,0 и 10,0 ml от работния стандартен разтвор в няколко градуирани колби от 20 ml, допълват се до пълния обем с метанол (т. 3.3) и съдържанието се разбърква. Номиналните концентрации на тези разтвори са 2,0, 4,0, 8,0 и 20,0 μg/ml DL-α-токоферол, или 2,20, 4,40, 8,79 и 22,0 μg/ml DL-α-токоферол ацетат.
Неколкократно се впръскват 20 μl от всеки калибрационен разтвор и се определят средните височини на пиковете (площите). Като се използват средните височини на пиковете (площите), се построява калибрационната крива.
5.6.2.3.
Разреждат се 2,0 ml от основния разтвор на DL-α-токоферол (т. 3.11.1) с етанол до 25,0 ml и ултравиолетовият спектър на този разтвор се сравнява със спектъра на етанол (т. 3.1) в спектрофотометър (т. 4.6) при дължина на вълната между 250 nm и 320 nm. Максималното поглъщане се наблюдава при 292 nm:
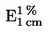 = 75,8 при 292 nm в етанол
= 75,8 при 292 nm в етанол
При това разреждане трябва да се получи стойност на екстинкция равна на 0,6.
6. Изчисляване на резултатите
Като се изхожда от средната височина (повърхността) на пиковете на витамин Е на разтвора на пробата, концентрацията на този разтвор в μg/ml (изчислена като α-токоферол ацетат) се определя чрез сравнение с калибрационната крива (т. 5.6.1.2 или т. 5.6.2.2.).
Съдържанието w на витамин Е в пробата, изразено в mg/kg, се пресмята по следната формула:
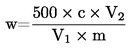 [mg/kg]
[mg/kg]
където:
|
c |
= |
концентрация на витамин Е (като α-токоферол ацетат) в разтвора на пробата (т. 5.4) в μg/ml |
|
V1 |
= |
обем на разтвора на пробата (т. 5.4) в ml |
|
V2 |
= |
обем на взетата аликвотната част (т. 5.4) в ml |
|
m |
= |
тегло на частта от пробата за анализ в g. |
7. Забележки
|
7.1. |
При проби с ниска концентрация на витамин Е, може да се окаже полезно да се обединят направените в петролен етер екстракти, които са получени от две осапунявания (претеглено количество: 25 g) в един разтвор на пробата за определяне чрез HPLC. |
|
7.2. |
Взетата за анализ проба не трябва да съдържа повече от 2 g мазнини. |
|
7.3. |
Ако не се получи разделяне на фазите, се прибавят около 10 ml етанол (т. 3.1), за да се пресече емулсията. |
|
7.4. |
След спектрофотометричното измерване на разтвора на DL-α-токоферол ацетат или на DL-α-токоферол съгласно т. 5.6.1.3 или т. 5.6.2.3 съответно, към разтвора (т. 3.10.1 или т. 3.10.2) се прибавят около 10 mg BHT (т. 3.12) и разтворът се съхранява хладилник (максимален срок на съхранение: 4 седмици). |
|
7.5. |
BHT може да се замени с хидрохинон. |
|
7.6. |
Като се използва нормалнофазова колона, е възможно отделянето на α-, β-, γ- и δ- токоферол. |
|
7.7. |
Разтворът на натриев аскорбат може да се замени с приблизително 150 ml аскорбинова киселина. |
|
7.8. |
Разтворът на натриев сулфид може да се замени с около 50 ml EDTA. |
|
7.9. |
Витамин Е много лесно се хидролизира в алкална среда и поради това е много чувствителен към окисляване, особено в присъствието на микроелементи като желязо или мед. При определяне на витамин Е в премикси при равнища по-високи от 5 000 mg/kg, може да се стигне до разграждане на витамин Е. Поради това за потвърждаване се препоръчва метод с използване на високоефективна течна хроматография, който включва ензимно разграждане на препарата, съдържащ витамин Е, но без етапа на осапуняване в алкална среда. |
8. Повторяемост
Разликата в резултатите от две паралелни определяния, проведени върху една и съща проба, не трябва да надхвърля 15 % по отношение на по-високия резултат.
9. Резултати от съвместно изследване (2)
|
|
Премикс |
Премикс фураж |
Минерален концентрат |
Протеинов фураж |
Прасенца |
|
L |
12 |
12 |
12 |
12 |
12 |
|
n |
48 |
48 |
48 |
48 |
48 |
|
Средна стойност в mg/kg |
17 380 |
1 187 |
926 |
315 |
61,3 |
|
sr [mg/kg] |
384 |
45,3 |
25,2 |
13,0 |
2,3 |
|
r [mg/kg] |
1 075 |
126,8 |
70,6 |
36,4 |
6,4 |
|
CVr [ %] |
2,2 |
3,8 |
2,7 |
4,1 |
3,8 |
|
sR mg/kg] |
830 |
65,0 |
55,5 |
18,9 |
7,8 |
|
R [mg/kg] |
2 324 |
182,0 |
155,4 |
52,9 |
21,8 |
|
CVR [ %] |
4,8 |
5,5 |
6,0 |
6,0 |
12,7 |
|
L |
= |
брой на лабораториите |
|
n |
= |
брой на единичните стойности |
|
sr |
= |
стандартно отклонение на повторяемостта |
|
sR |
= |
стандартно отклонение на възпроизводимостта |
|
r |
= |
повторяемост |
|
R |
= |
възпроизводимост |
|
CVr |
= |
коефициент на вариация на повторяемостта |
|
CVR |
= |
коефициент на вариация на възпроизводимостта |
В. ОПРЕДЕЛЯНЕ НА СЛЕДИ ОТ МИКРОЕЛЕМЕНТИ — ЖЕЛЯЗО, МЕД, МАНГАН И ЦИНК
1. Цел и обхват
Настоящият метод позволява да се определят микроелементите желязо, мед, манган и цинк в храните. Праговете за количествено определяне са следните:
|
— |
желязо (Fе): 20 mg/kg |
|
— |
мед (Cu): 10 mg/kg |
|
— |
манган (Mn): 20 mg/kg |
|
— |
цинк (Zn): 20 mg/kg |
2. Принцип
Пробата се поставя в разтвор на солна киселина след разрушаване на органичните вещества, ако има такива. Елементите желязо, мед, манган и цинк се определят с атомноабсорбционна спектрометрия след подходящо разтваряне.
3. Реагенти
Уводен коментар
За приготвянето на реагентите и аналитичните разтвори се използва вода, която не съдържа подлежащите на определяне катиони, получена чрез двойно дестилиране в боросиликатно или кварцово стъкло или чрез двойно третиране с йонообменна смола.
Реагентите трябва да бъдат поне с чистота за анализ. Отсъствието на елемента за определяне трябва да се проверява чрез експеримент с празна проба. Ако е необходимо, реагентите трябва допълнително да се пречистят.
Вместо описаните по-долу стандартни разтвори, могат да се използват търговски стандартни разтвори, при условие че те са с гарантирано качество и са проверени преди употреба.
|
3.1. |
Солна киселина (d: 1,19 g/ml). |
|
3.2. |
Солна киселина (6 mol/l). |
|
3.3. |
Солна киселина (0,5 mol/l). |
|
3.4. |
Флуороводородна киселина 38—40 % (v/v) със съдържание на желязо (Fe) под 1 mg/l и остатък след изпаряване под 10 mg (като сулфат) на литър. |
|
3.5. |
Сярна киселина (d: 1,84 g/ml). |
|
3.6. |
Водороден прекис (приблизително 100 обема кислород (30 тегловни %). |
|
3.7. |
Стандартен разтвор, съдържащ желязо (1 000 μg Fe/ml), приготвен, както следва, или еквивалентен търговски достъпен разтвор: разтваря се 1 g желязна тел в 200 ml солна киселина с концентрация 6 mol/l (т. 3.2), добавят се 16 ml водороден прекис (т. 3.6) и се допълва до един литър с вода. |
|
3.7.1. |
Работен стандартен разтвор, съдържащ желязо (100 μg Fe/ml), приготвен чрез разреждане на една част от стандартния разтвор (т. 3.7) с 9 части вода. |
|
3.8. |
Стандартен разтвор, съдържащ мед (1 000 μg Cu/ml), приготвен, както следва, или еквивалентен търговски достъпен разтвор:
|
|
3.8.1. |
Работен стандартен разтвор, съдържащ мед (10 μg Сu/ml), приготвен чрез разреждане на една част от стандартния разтвор (т. 3.8) с 9 части вода и след това разреждане на една част от получения разтвор с 9 части вода. |
|
3.9. |
Стандартен разтвор, съдържащ манган (1 000 μg Mn/ml), приготвен, както следва, или еквивалентен търговски достъпен разтвор:
|
|
3.9.1. |
Работен стандартен разтвор, съдържащ манган (10 μg Mn/ml), приготвен чрез разреждане на една част от стандартния разтвор (т. 3.9) с 9 части вода и след това разреждане на една част от получения разтвор с 9 части вода. |
|
3.10. |
Стандартен разтвор, съдържащ цинк (1 000 μg Zn/ml), приготвен, както следва, или еквивалентен търговски достъпен разтвор:
|
|
3.10.1. |
Работен стандартен разтвор, съдържащ цинк (10 μg Zn/ml), приготвен чрез разреждане на една част от стандартния разтвор (т. 3.10) с 9 части вода и след това разреждане на една част от получения разтвор с 9 части вода. |
|
3.11. |
Разтвор на лантанов трихлорид: разтварят се 12 g лантанов оксид в 150 ml вода, добавят се 100 ml солна киселина с концентрация 6 mol/l (т. 3.2) и се допълва до един литър с вода. |
4. Апаратура
|
4.1. |
Муфелна пещ с регулиране на температурата и по възможност със записващо устройство. |
|
4.2. |
Съдовете трябва да бъдат от устойчиво боросиликатно стъкло и се препоръчва да се използва апарат, който е предназначен само за определяне на микроелементи. |
|
4.3. |
Атомнoабсорбционен спектрометър, съответстващ на изискванията на метода, с оглед на чувствителността и прецизността в необходимия обхват. |
5. Процедура (3)
5.1. Проби, съдържащи органични вещества
5.1.1. (4)
|
5.1.1.1 |
Поставят се 5—10 g от пробата, премерена с точност до 0,2 mg, в кварцов или платинен тигел (вж. забележка б)), изсушават се в пещ при 105 oС и тигелът се поставя в студената муфелна пещ (т. 4.1). Пещта се затваря (вж. забележка в) и температурата постепенно се повишава до 450—475 oС за около 90 минути. Тази температура се поддържа 4—16 часа (напр. през нощта) за отделяне на саждите, след това пещта се отваря, за да се охлади (вж. забележка г)). Пепелта се навлажнява с вода и се прехвърля в бехерова чаша от 250 ml. Тигелът се измива с около 5 ml солна киселина (т. 3.1), след което последната бавно и внимателно се добавя към бехерова чаша (може да има бурна реакция поради образуването на СО2). Капка по капка се добавя солна киселина (т. 3.1) като се разбърква докато престане образуването на газови мехурчета. Изпарява се до сухо, като от време навреме се разбърква със стъклена пръчка. Добавят се 15 ml солна киселина с концентрация 6 mol/l (т. 3.2) към остатъка, след което се добавят и 120 ml вода. Разбърква се със стъклена пръчка, която се оставя в бехеровата чаша, и последната се покрива с часовниково стъкло. Бавно се нагрява до кипене и се поддържа тази температура докато спре разтварянето на пепелта. Филтрира се през чиста от пепел филтърна хартия и филтратът се събира в мерителна колба от 250 ml. Бехеровата чаша и филтърът се измиват с 5 ml гореща солна киселина с концентрация 6 mol/l (т. 3.2) и два пъти с вряла вода. Напълва се мерителната колба до марката с вода (концентрация на солната киселина около 0,5 mol/l). |
|
5.1.1.2 |
Ако остатъкът във филтъра изглежда черен (въглерод), той се връща в пещта и се опепелява отново при 450—475 oС. Това опепеляване, което изисква само няколко часа (около 3—5 часа), е завършено, когато пепелта изглежда бяла или почти бяла. Остатъкът се разтваря с около 2 ml солна киселина (т. 3.1), изпарява се до сухо и се добавят 5 ml солна киселина (т. 3.2) с концентрация 6 mol/l. Нагрява се, разтворът в мерителната колба се филтрира и се допълва до пълния обем с вода (концентрация на HCl около 0,5 mol/l). Забележки:
|
5.1.2.
5.1.2.1.
За всеки от елементите, които ще се определят, от работните стандартни разтвори, посочени в точки 3.7.1, 3.8.1, 3.9.1 и 3.10.1, се приготвя серия от калибрационни разтвори, като всеки има концентрация на HCl около 0,5 mol/l (и (за желязото, мангана и цинка) концентрация на лантанoв трихлорид, еквивалентна на 0,1 % Lа (w/v).
Подбраните концентрации на микроелементи трябва да бъдат в рамките на чувствителността на използвания спектрометър. Таблиците по-долу дават примери за състава на типични серии от калибрационни разтвори; в зависимост обаче от вида и чувствителността на използвания спектрофотометър, може да се наложи да се изберат други концентрации.
Желязо
|
μg Fe/ml |
0 |
0,5 |
1 |
2 |
3 |
4 |
5 |
|
ml работен стандартен разтвор (т. 3.7.1) (1 ml = 100 μg Fe) |
0 |
0,5 |
1 |
2 |
3 |
4 |
5 |
|
ml HCl (т. 3.2) |
7 |
7 |
7 |
7 |
7 |
7 |
7 |
|
+ 10 ml разтвор на лантанов трихлорид (т. 3.11) и се долива с вода до 100 ml |
|||||||
Мед
|
μg Cu/ml |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
ml работен стандартен разтвор (т. 3.8.1) (1 ml = 10 μg Cu) |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
|
ml HCl (т. 3.2) |
8 |
8 |
8 |
8 |
8 |
8 |
8 |
Манган
|
μg Mn/ml |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
ml работен стандартен разтвор (т. 3.9.1) (1 ml = 10 μg Mn) |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
|
ml HCl (т. 3.2) |
7 |
7 |
7 |
7 |
7 |
7 |
7 |
|
+ 10 ml разтвор на лантанов трихлорид (т. 3.11) и се долива с вода до 100 ml |
|||||||
Цинк
|
μg Zn/ml |
0 |
0,05 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
|
ml работен стандартен разтвор (т. 3.10.1) (1 ml = 10 μg Zn) |
0 |
0,5 |
1 |
2 |
4 |
6 |
8 |
|
ml HCl (т. 3.2) |
7 |
7 |
7 |
7 |
7 |
7 |
7 |
|
+ 10 ml разтвор на лантанов трихлорид (т. 3.11) и се долива с вода до 100 ml |
|||||||
5.1.2.2.
За определянето на медта разтворът, приготвен както е указано в т. 5.1.1, може да се използва директно. Ако е необходимо концентрацията му да се сведе до рамките на калибрационните разтвори, може да се прехвърли аликвотна част от разтвора с пипета в мерителна колба от 100 ml и да се допълни до марката със солна киселина с концентрация 0,5 mol/l (т. 3.3).
За определянето на желязото, мангана и цинка се прехвърля с пипета аликвотна част от разтвора, приготвен, както е указано в т. 5.1.1, в мерителна колба от 100 ml, добавят се 10 ml разтвор от лантанов трихлорид (т. 3.11) и се допълва до марката със солна киселина с концентрация 0,5 mol/l (т. 3.3).
5.1.2.3.
Контролният експеримент трябва да включва всички указани стъпки на процедурата, с изключение на това, че се пропуска добавянето на пробния материал. Калибрационен разтвор „0“ не трябва да се използва като контролен разтвор.
5.1.2.4.
Измерва се атомната абсорбция на калибрационните разтвори и на разтвора, който ще се анализира, чрез използване на окисляващ въздушноацетиленов пламък при следните дължини на вълната:
|
|
Fe: 248,3 nm |
|
|
Cu: 324,8 nm |
|
|
Mn: 279,5 nm |
|
|
Zn: 213,8 nm |
Всички измервания се извършат четири пъти.
5.2. Минерални фуражи
Ако пробата не съдържа органични вещества, не е необходимо да се извършва предварително опепеляване. Процедира се, както е указано в точка 5.1.1.1, като се започне от втория параграф. Може да се пропусне изпаряването с флуороводородна киселина.
6. Изчисляване на резултатите
С помощта на калибрационна крива се изчисляват концентрациите на микроелементите в разтвора за анализ и резултатът се изразява в милиграми микроелемент на килограм проба (ppm, части на милион).
7. Повторяемост
Разликата между резултатите от две паралелни определяния, извършени с една и съща проба от един и същ аналитик, не трябва да бъдe повече от:
|
— |
5 mg/kg в абсолютна стойност за съдържание на въпросните микроелементи до 50 mg/kg; |
|
— |
10 % от по-високия резултат за съдържание на въпросния микроелемент от 50 до 100 mg/kg; |
|
— |
10 mg/kg в абсолютна стойност за съдържание на въпросните микроелементи от 100 до 200 mg/kg; |
|
— |
5 % от по-високия резултат за съдържание на въпросния микроелемент над 200 mg/kg. |
8. Забележка
Наличието на големи количества фосфати може да окаже неблагоприятно влияние върху определянето на желязо, манган и цинк. Подобно влияние трябва да бъде коригирано с добавянето на разтвор на лантанов трихлорид (т. 3.11). Ако обаче тегловното съотношение Ca + Mg/P > 2, добавянето на разтвор на лантанов трихлорид (т. 3.11) към анализирания разтвор и към калибрационните разтвори може да се пропусне.
Г. ОПРЕДЕЛЯНЕ НА ХАЛОФУГИНОН
DL-транс-7-бромо-6-хлоро-3-[3-(3-хидрокси-2-пиперидил)ацетонил]-хиназолин-4-(3Н)-он хидробромид.
1. Цел и обхват
Методът позволява определянето на съдържанието на халофугинон във фуражите. Прагът на количествено определяне е 1 mg/kg.
2. Принцип
След третиране с гореща вода, халофугинонът се екстрахира като свободна основа в етилов ацетат и след това се разделя като хидрохлорид във воден киселинен разтвор. Екстрактът се очиства посредством йоннообменна хроматография. Съдържанието на халофугинон се определя с помошта на високоефективна течна хроматография с обратна фаза (HPLC), като се използва UV-детектор.
3. Реагенти
|
3.1. |
Ацетонитрил с чистота за HPLC |
|
3.2. |
Смола амберлит XAD-2 |
|
3.3. |
Амониев ацетат |
|
3.4. |
Етилов ацетат |
|
3.5. |
Оцетна киселина, кристална |
|
3.6. |
Халофугинон — стандартно вещество (DL-транс-7-бромо-6-хлоро-3-[3-(хидрокси-2-пиперидил)ацетонил]-хиназолин-4-(3Н)-он хидробромид, Е 764) |
|
3.6.1. |
Претеглят се с точност до 0,1 mg 50 mg халофугинон (т. 3.6) в градуирана колба с вместимост 500 ml, разтварят се в буферен разтвор на амониев ацетат (т. 3.18), допълва се до пълния обем с буферен разтвор и се разбърква. Този разтвор остава стабилен в продължение на три седмици при температура 5 oС, ако се съхранява на тъмно. |
|
3.6.2. |
В поредица от градуирани колби с вместимост 100 ml се прехвърлят 1,0, 2,0, 3,0, 4,0 и 6,0 ml от основния стандартен разтвор на халофугинон (т. 3.6.1). Допълват се до пълния обем с подвижната фаза (т. 3.21) и се разбъркват. Разтворите ще имат съответно концентрации на халофугинон 1,0, 2,0, 3,0, 4,0 и 6,0 μg/ml. Разтворите се приготвят непосредствено преди употреба. |
|
3.7. |
Солна киселина (ρ20 е приблизително 1,16 g/ml) |
|
3.8. |
Метанол |
|
3.9. |
Сребърен нитрат |
|
3.10. |
Натриев аскорбат |
|
3.11. |
Натриев карбонат |
|
3.12. |
Натриев хлорид |
|
3.13. |
EDTA (етилен-диамин-тетра-оцетна киселина, двунатриева сол) |
|
3.14. |
Вода с чистота за HPLC |
|
3.15. |
Разтвор на натриев карбонат, c = 10 g/100 ml |
|
3.16. |
Натриев хлорид — наситен разтвор на натриев карбонат, c = 5 g/100 ml Разтварят се 50 g натриев карбонат (т. 3.11) във вода, разрежда се до 1 литър и се добавя натриев хлорид (т. 3.12), докато се получи наситен разтвор. |
|
3.17. |
Солна киселина, приблизително 0,1 mol/l. Разтварят се 10 ml HCl (т. 3.7) във вода, докато се получи 1 l. |
|
3.18. |
Буферен разтвор на амониев ацетат, приблизително 0,25 mol/l Разтварят се 19,3 g амониев ацетат (т. 3.3) и 30 ml оцетна киселина (т. 3.5) във вода (т. 3.14) и разтворът се разрежда до 1 l. |
|
3.19. |
Подготвяне на смола амберлит XAD-2 Подходящо количество амберлит (т. 3.2) се промива с вода, докато бъдат отстранени всички хлорни йони, като това се установява чрез тест със сребърен нитрат (т. 3.20), който се извършва върху промивната вода. След това смолата се промива с 50 ml метанол (3.8), отстранява се метанолът и се смолата се съхранява под чист метанол. |
|
3.20. |
Разтвор на сребърен нитрат, приблизително 0,1 mol/l. Разтварят се 0,17 g сребърен нитрат (т. 3.9) в 10 ml вода. |
|
3.21. |
Подвижна фаза за HPLC Смесват се 500 ml ацетонитрил (т. 3.1) с 300 ml буферен разтвор на амониев ацетат (т. 3.18) и 1 200 ml вода (т. 3.14). Киселинността се коригира до рН = 4,3, като се използва оцетна киселина (т. 3.5). Разтворът се филтрира през филтър с размер на порите 0,22 μm (т. 4.8) и се обезгазява (напр.чрез подлагане на въздействието на ултразвук в продължение на 10 минути). Този разтвор остава стабилен в продължение на един месец, при условие че се съхранява на тъмно място в затворен съд. |
4. Апаратура
|
4.1. |
Ултразвукова баня |
|
4.2. |
Ротационен тънкослоен изпарител |
|
4.3. |
Центрофуга |
|
4.4. |
Апарат за HPLC с UV-детектор с настройка на дължината на вълната или с детектор с диодна матрица |
|
4.4.1. |
Колона за течна хроматография, 300 mm × 4 mm, С18, пълнеж от 10 μm, или еквивалентна колона |
|
4.5. |
Стъклена колона (300 mm × 10 mm), снабдена с филтър от синтеровано стъкло и спирачно кранче. |
|
4.6. |
Филтри от стъкловлакно, с диаметър 150 mm |
|
4.7. |
Мембранни филтри, 0,45 μm. |
|
4.8. |
Мембранни филтри, 0,22 μm. |
5. Процедура
|
Забележка |
: |
Халофугинонът като свободна основа е нестабилен в алкални разтвори и в разтвор на етилов ацетат. Халофугинонът не трябва да остава в етилов ацетат повече от 30 min. |
5.1. Общи положения
|
5.1.1. |
Анализира се фураж без добавки, за да се провери дали не присъстват халофугинон или други замърсяващи вещества. |
|
5.1.2. |
Провежда се тест за възстановяване, като се анализира фураж без добавки, обогатен чрез добавяне на известно количество халофугинон, подобно на това, което присъства в пробата. За да се обогати фуражът до равнище 3 mg/kg, се добавят 300 μl от основния стандартен разтвор (т. 3.6.1) към 10 g от фуража без добавки, смесва се и се изчаква 10 min, преди да се премине към етапа на екстрахиране (т. 5.2).
|
5.2. Екстракция
Претеглят се с точност до 0,1 g 10 g от подготвената проба в епруветка за центрофугиране с вместимост 200 ml, добавят се 0,5 g натриев аскорбат (т. 3.10), 0,5 g EDTA (т. 3.13) и 20 ml вода и се разбърква. Епруветката се поставя за 5 min във водна баня (80 oС). След охлаждане до стайна температура се добавят 20 ml разтвор на натриев карбонат (т. 3.15) и се разбърква. Незабавно се добавят 100 ml етилов ацетат (т. 3.4) и се разклаща силно с ръка в продължение на 15 секунди. След това епруветката се поставя за три минути в ултразвукова баня (т. 4.1) и се разхлабва запушалката. Центрофугира се в продължение на и се прелива фазата на етиловия ацетат през филтър от стъкловлакно (т. 4.6) в делителна фуния с вместимост 500 ml. Екстрахирането на пробата се повтаря с втора порция от 100 ml етилов ацетат. Събраните на едно място екстракти се промиват в продължение на с 50 ml наситен разтвор на натриев хлорид в разтвор на натриев карбонат (т. 3.16) и се отстранява водният слой.
Екстрахира се органичният слой в продължение на 1 min с 50 ml солна киселина (т. 3.17). Прехвърля се долният киселинен слой в делителна фуния с вместимост 250 ml. Повторно се екстрахира органичният слой в продължение на минута и половина с нови 50 ml разтвор на солна киселина и се прибавя към първия екстракт. Събраните заедно киселинни екстракти се промиват чрез въртене в продължение на приблизително 10 s с 10 ml етилов ацетат (т. 3.4).
Водният слой се прехвърля количествено в облодънна колба от 250 ml, като се отстранява органичната фаза. Изпарява се целият останал етилов ацетат от киселинния разтвор, като се използва ротационен тънкослоен изпарител (т. 4.2). Температурата на водата не трябва да надвишава 40 oC. Под вакуум от приблизително 25 mbar целият остатъчен етилов ацетат се отстранява в рамките на 5 min при температура 38 oС.
5.3. Почистване
5.3.1.
За всеки екстракт на пробата се подготвя по една колона XAD-2. 10 g от подготвения амберлит (т. 3.19) се прехвърлят в стъклена колона (т. 4.5) с метанол (т. 3.8). Добавя се малка запушалка от стъклен памук в горната част на коритото от смола. Метанолът се източва от колоната и смолата се промива със 100 ml вода, като потокът се спира веднага, щом течността достигне горната част на коритото от смола. Оставя се колоната да се уравновеси за 10 min преди употреба. Не се допуска колоната да изсъхва.
5.3.2.
Екстрактът (т. 5.2) се прехвърля количествено в горната част на подготвената колона от амберлит (т. 5.3.1) и се елуюира, като се изхвърля елуатът. Скоростта на потока за елуиране не трябва да надвишава 20 ml/min. Облодънната колба се изплаква с 20 ml солна киселина (т. 3.17), която се използва и за да се измие колоната със смола. Евентуално останалият киселинен разтвор се отстранява с въздушен поток. Промивните течности се отстраняват. Добавят се 100 ml метанол (т. 3.8) към колоната и се оставя процесът на елуиране да продължи от 5 до 10 min, като елуатът се събира в облодънна колба с вместимост 250 ml. Оставя се останалият метанол за 10 min, за да влезе в равновесие със смолата, и се продължава елуирането със скорост 20 ml/min, като елуатът се събира в същата облодънна колба. Изпарява се метанолът върху ротационния тънкослоен изпарител (т. 4.2), като температурата на водната баня не трябва да превишава 40 oС. Остатъкът се прехвърля количествено в мерителна колба от 10 ml, като се използва подвижната фаза (т. 3.21). Долива се до номиналния обем с подвижната фаза и се разбърква. Една аликвотна част се филтрира през мембранен филтър (т. 4.7). Този разтвор се запазва за определяне с помощта на HPLC (т. 5.4).
5.4. Определяне с помощта на HPLC
5.4.1.
Следните указания за параметрите са ориентировъчни, като могат да се използват и други параметри, при условие че с тях се получават еквивалентни резултати:
Колона за течна хроматография (т. 4.4.1);
Подвижна фаза за HPLC (т. 3.21);
Скорост на изтичане: 1,5 до 2 ml/min.
Дължина на вълната за откриване: 243 nm
Обем за впръскване: от 40 до 100 μl.
Проверява се стабилността на хроматографската система, като калибрационният разтвор (т. 3.6.2), съдържащ 3 μg/ml, се впръсква няколко пъти, докато височината на пиковете (или площите) и времето на задържане останат постоянни.
5.4.2.
Всеки калибрационен разтвор (т. 3.6.2) се впръсква няколко пъти и се измерват височините на пиковете (площите) за всяка концентрация. Построява се калибрационна крива, като за ординати се използват средните височини на пиковете (площите) на калибрационните разтвори, а за абсциси — съответните концентрации в μg/ml.
5.4.3.
Екстрактът на пробата (т. 5.3.2) се впръсква няколко пъти, като се използва същият обем, който е взет за калибрационните разтвори, и се определя средната височина на пиковете (площи) за пиковете за халофугинона.
6. Изчисляване на резултатите
Концентрацията на разтвора на пробата в μg/ml се определя от средната височина (площ) на пиковете за халофугинона от разтвора на пробата, като се сравнява с калибрационната крива (т. 5.4.2).
Съдържанието w (mg/kg) на халофугинон в пробата се получава със следната формула:

където:
|
c |
: |
концентрация на халофугинон в разтвора на пробата в μg/ml, |
|
m |
: |
тегло на частта от пробата за анализ в грамове. |
7. Потвърждаване на резултатите
7.1. Идентичност
Идентичността на аналита може да се потвърди с помощта на кохроматография или като се използва детектор с диодна матрица, с който се сравняват спектрите на екстракта на пробата и на калибрационния разтвор (т. 3.6.2), съдържащ 6,0 μg/ml.
7.1.1.
Екстракт на пробата се обогатява като се добавя целесъобразно количество калибрационен разтвор (т. 3.6.2). Количеството добавен халофугинон трябва да е сходно с предполагаемото количество халофугинон, намерен в екстракта на пробата.
Увеличава се само височината на пика на халофугинона, след като се вземе под внимание както добавеното количество, така и разреждането на екстракта. Широчината на пика, на половината от неговата максимална височина, трябва да бъде в рамките на ± 10 % от първоначалната широчина.
7.1.2.
Резултатите се оценяват според следните критерии:
|
а) |
дължината на вълната на максимална абсорбция на пробата трябва да същата като тази на спектрите на стандарта, регистрирана в най-високата точка на хроматограмата, с отконение, определено от разделителната способност на детекторната система. За детектирането с диодна матрица, отклонението обикновено е в рамките на ± 2 nm; |
|
б) |
между 225 и 300 nm спектрите на пробата и на стандарта, отчетени в най-високата точка на хроматограмата, не трябва да се различават помежду си за тези части от спектъра, които са в интервала между 10 до 100 % от относителната екстинкция. Критерият е изпълнен тогава, когато са налице едни и същи максимуми и не е регистрирана точка, в която отклонението между двата спектъра да превишава 15 % от екстинцията на стандартния аналит; |
|
в) |
между 225 и 300 nm спектрите на възходящата линия, максимума и низходящата линия на пика, получени при анализа на екстракта на пробата, не трябва да се различават един от друг в тези части от спектъра, които попадат в интервала между 10 до 100 % относителна екстинкция. Критерият е изпълнен тогава, когато са налице едни и същи максимуми и когато във всички регистрирани точки отклонението между спектрите не надминава 15 % от екстинкцията на спектъра на най-високата точка. |
Ако някой от критериите не е изпълнен, наличието на аналита не се потвърждава.
7.2. Повторяемост
Разликата между резултатите от две паралелни определяния, проведени върху една и съща проба, не трябва да надвишава 0,5 mg/kg за съдържание на халофугинон до 3 mg/kg.
7.3. Възстановяване
За обогатената празна проба възстановяването трябва да е минимум 80 %.
8. Резултати от съвместно изследване
Беше проведено съвместно изследване (5), при което три проби са били подложени на анализ от осем лаборатории.
Резултати
|
|
Проба А (празна) При получаване |
Проба Б (брашно) |
Проба В (пелети) |
||
|
|
|
При получаване |
След два месеца |
При получаване |
След два месеца |
|
Средна стойност в mg/kg |
НО |
2,80 |
2,42 |
2,89 |
2,45 |
|
SR [mg/kg] |
— |
0,45 |
0,43 |
0,40 |
0,42 |
|
CVR [ %] |
— |
16 |
18 |
14 |
17 |
|
Възст. [ %] |
|
86 |
74 |
88 |
75 |
|
НО |
= |
не е открито |
|
SR |
= |
стандартно отклонение на възпроизводимостта |
|
CVR |
= |
коефициент на вариация на възпроизводимостта ( %) |
|
Възст. |
= |
възстановяване ( %) |
Д. ОПРЕДЕЛЯНЕ НА РОБЕНИДИН
1,3-bis[(4-хлоробензилиден)амино]гванидин хидрохлорид
1. Цел и обхват
Настоящият метод позволява определянето на съдържанието на робенидин във фуражите. Прагът на количествено определяне е 5 mg/kg.
2. Принцип
Пробата се екстрахира с помощта на подкиселен метанол. Екстрактът се изсушава, след което аликвотна част от него се подлага на пречистване в колона с алуминиев оксид. Робенидинът се елуира от колоната с метанол, концентрира се и се разрежда до желания обем с подвижна фаза. Съдържанието на робенидин се определя с помощта на високоефективна течна хроматография с обратна фаза (HPLC), като се използва UV-детектор.
3. Реагенти
|
3.1. |
Метанол |
|
3.2. |
Подкиселен метанол Поставят се 4,0 ml солна киселина (ρ20 = 1,18 g/ml) в градуирана колба от 500 ml, допълва се с метанол до пълния обем (т. 3.1), и се разбърква. Разтворът се приготвя непосредствено преди употреба. |
|
3.3. |
Ацетонитрил, с чистота за HPLC |
|
3.4. |
Молекулно сито Тип 3А, частици с меш от 8 до 12 (частици от 1,6—2,5 mm, кристален алуминосиликат, диаметър на порите 0,3 mm) |
|
3.5. |
Алуминиев оксид със степен на киселинна активност I за колонна хроматография В подходящ съд се поставят 100 g алуминиев оксид и се прибавят 2,0 ml вода. Запушва се и се разклаща за приблизително 20 min. Така полученият разтвор се съхранява в добре запушен съд. |
|
3.6. |
Разтвор на калиев дихидроген фосфат, с = 0,025 mol/l В градуирана колба от 1 000 ml се разтварят 3,40 g калиев дихидроген фосфат във вода (чиста за HPLC), допълва се до пълния обем и се разбърква. |
|
3.7. |
Разтвор на динатриев хидроген фосфат, с = 0,025 mol/l В градуирана колба от 1 литър се Разтварят 3,55 g безводен динатриев хидроген фосфат (или 4,45 g дихидрат или 8,95 g додекахидрат) във вода (с чистота за HPLC), допълва се до пълния обем и се разбърква. |
|
3.8. |
Подвижна фаза за HPLC Смесват се следните реагенти:
Разтворът се филтрира през филтър с размер на порите 0,22 μm (т. 4.6) и се обезгазява (напр. чрез подлагане на въздействието на ултразвук в продължение на 10 минути). |
|
3.9. |
Стандартно вещество Чист робенидин: 1,3-bis[(4-хлоробензилиден)амино] гуанидин хидрохлорид. |
|
3.9.1. |
С точност до 0,1 mg се отмерват 30 mg от стандартното вещество робенидин (т. 3.9). Това количество се разтваря в подкиселен метанол (т. 3.2) в градуирана колба от 100 ml, допълва се до пълния обем със същия разтворител и се разбърква. Колбата се увива с алуминиево фолио и се съхранява на тъмно място. |
|
3.9.2. |
Прехвърлят се 10,0 ml от основния стандартен разтвор (т. 3.9.1) в градуирана колба от 250 ml, долива се с подвижната фаза (т. 3.8) до пълния обем и се разбърква. Колбата се увива с алуминиево фолио и се съхранява на тъмно място. |
|
3.9.3. |
В мерителни колби от 50 ml се поставят съответно 5,0, 10,0, 15,0, 20,0 и 25,0 ml от междинния стандартен разтвор (т. 3.9.2). Колбите се допълват до пълния им обем с подвижната фаза (т. 3.8) и се течността се разбърква. Разтворите отговарят съответно на 1,2; 2,4; 3,6; 4,8 и 6,0 μg/ml робенидин. Разтворите се приготвят непосредствено преди употреба. |
|
3.10. |
Вода с чистота за HPLC |
4. Апаратура
|
4.1. |
Стъклена колона Направена от непрозрачно стъкло и снабдена със спирателно кранче, с резервоар с обем приблизително 150 ml, вътрешен диаметър от 10 до 15 mm и дължина 250 mm. |
|
4.2. |
Механична клатачна машина или магнитна бъркалка |
|
4.3. |
Ротационен тънкослоен изпарител. |
|
4.4. |
Апарат за HPLC с UV-детектор с настройка на дължината на вълната или с диодна матрица, която работи в обхвата 250—400 nm. |
|
4.4.1. |
Колона за течна хроматаграфия: 300 mm ×4 mm, С18, пълнеж от 10 μm или еквивалентна |
|
4.5. |
Филтрова хартия от стъклени нишки (Whatman GF/A или еквивалентна) |
|
4.6. |
Мембранни филтри, 0,22 μm. |
|
4.7. |
Мембранни филтри, 0,45 μm. |
5. Процедура
|
Забележка |
: |
Робенидинът е чувствителен към светлина. Трябва да се използват непрозрачни лабораторни съдове при всички операции. |
5.1. Общи положения
|
5.1.1. |
Необходимо е да се анализира фураж без добавки с цел да се провери наличието на робенидин или вщества, които пречат на анализа. |
|
5.1.2. |
Необходимо е да се проведе тест за възстановяване, като се анализира проба от фураж без робенидин (т. 5.1.1.), обогатена чрез добавяне на известно количество робенидин, сходно с това в пробата. За да се постигне обогатяване до равнище 60 mg/kg, в конична колба от 250 ml се прехвърлят 3,0 ml от основния стандартен разтвор (т. 3.9.1). Разтворът се изпарява до прибл. 0,5 ml под приток на азот. Прибавят се 15 g от фуража без добавки, разбърква се добре и се изчаква 10 min, преди да се продължи с етапа на екстракция (т. 5.2).
|
5.2. Екстракция
Отмерват се с точност до 0,01 g приблизително 15 g от приготвената проба. Прехвърлят се в конична колба от 250 ml и се добавят 100,0 ml подкиселен метанол (т. 3.2), съдът се запушва добре и се разклаща в продължение на 1 h в клатачна машина (т. 4.2). Разтворът се филтрира през филтрова хартия от стъклени нишки (т. 4.5) и целият филтрат се събира в конична колба от 150 ml. Прибавят се 7,5 g молекулно сито (т. 3.4), съдът се запушва добре и се разклаща в продължение на 5 min. Незабавно се филтрира през филтрова хартия от стъклени нишки. Разтворът се запазва за етапа на пречистване (т. 5.3).
5.3. Пречистване
5.3.1.
Долният край на стъклената колона (т. 4.1) се запушвас тапа от стъклен памук, която се вкарва с помощта на стъклена пръчка. Отмерват се 11 g от приготвения алуминиев оксид (т. 3.5) и се прехвърлят в колоната. По време на тази манипулация излагането на въздействието на обкръжаващия въздух се свежда до минимум. Напълнената колона се почуква леко в долния край, за да улегне алуминиевият оксид.
5.3.2.
С пипета се прехвърлят в колоната 5 ml от екстракта на пробата, приготвен съгласно точка 5.2. Краят на пипетата се поставя близо до стената на колоната и се оставя разтворът да се абсорбира от алуминиевия оксид. Робенидинът от колоната се елуира, като се използват 100 ml метанол (т. 3.1) при скорост на изтичане 2—3 ml/min, а елуатът се събира в облодънна колба с вместимост 250 ml. Метаноловият разтвор се подлага на изпаряване до изсъхване при намалено налягане при температура 40 oC с помощта на ротационен тънкослоен изпарител (т. 4.3). Остатъкът се разтваря отново в 3—4 ml от подвижната фаза (т. 3.8) и се прехвърля количествено в градуирана колба от 10 ml. Колбата се изплаква на няколко пъти с порции от 1—2 ml от подвижната фаза и изплакнатите количества се прехвърлят в градуираната колба. Допълва се до пълния обем със същия разтворител и се разбърква. Една аликвотна част се филтрира през мембранен филтър от 0,45 μm (т. 4.7). Разтворът се запазва за определяне чрез HPLC (т. 5.4).
5.4. Определяне с помощта на HPLC
5.4.1.
Следните указания за параметрите са ориентировъчни, като могат да се използват и други параметри, при условие че с тях се получават еквивалентни резултати:
Колона за течна хроматография (т. 4.4.1),
подвижна фаза за HPLC (т. 3.8),
Скорост на изтичане: 1,5 до 2 ml/min,
Дължина на вълната, на която е настроен детекторът: 317 nm
обем за впръскване 20—50 μl.
Проверява се стабилността на хроматографската система, като на няколко пъти се впръсква калибрационен разтвор (т. 3.9.3), съдържащ 3,6 μg/ml, докато се постигнат постоянни височини на пика и постоянни времена на задържане.
5.4.2.
Всеки от калибрационните разтвори (т. 3.9.3), се впръсква няколко пъти и се измерват височините (площите) за всяка концентрация. Построява се калибрационна крива, като за ординати се използват средните височини на пиковете или площите на калибрационните разтвори, а за абсциси — съответните концентрации в μg/ml.
5.4.3.
Разтворът на пробата (т. 5.3.2) се впръсква няколко пъти, като се използва същият обем, който е взет за калибрационните разтвори, и се определя средната височина на пиковете (площ) за пиковете на робенидина.
6. Изчисляване на резултатите
Концентрацията на разтвора на пробата в μg/ml се определя от средната височина (площ) на пиковете за робенидина, като се сравнява с калибрационната крива (т. 5.4.2).
Съдържанието w (mg/kg) на робенидин в пробата се пресмята по следната формула:

където:
|
c |
= |
концентрация на робенидин в разтвора на пробата в μg/ml, |
|
m |
= |
тегло на частта от пробата за анализ в грамове. |
7. Потвърждаване на резултатите
7.1. Идентичност
Идентичността на аналита може да се потвърди с помощта на кохроматография или като се използва детектор с диодна матрица, с който се сравняват спектрите на екстракта на пробата и на калибрационния разтвор (т. 3.9.3), съдържащ 6 μg/ml.
7.1.1.
Екстратът на пробата се обогатява, като към него се прибави подходящо количество от калибрационния разтвор (т. 3.9.3). Количеството добавен робенидин трябва да е сходно с предполагаемото количество робенидин, намерен в екстракта на пробата.
Увеличава се само височината на пика на робенидина, след като се вземе под внимание както добавеното количество, така и степента на разреждане на екстракта. Широчината на пика на половината от неговата максимална височина трябва да бъде в рамките на около 10 % от първоначалната широчина.
7.1.2.
Резултатите се оценяват според следните критерии:
|
а) |
дължината на вълната на максимална на пробата трябва да същата като тази на спектрите на стандарта, регистрирана в най-високата точка на хроматограмата и при отклонение, определено от разделителната способност на детекторната система. За детектирането с диодна матрица, отклонението обикновено е в рамките на ± 2 nm; |
|
б) |
между 250 и 400 nm спектрите на пробата и на стандарта, отчетени в най-високата точка на хроматограмата, не трябва да се различават помежду си в тези части от спектъра в интервала между 10 до 100 % относителна екстинкция. Критерият е изпълнен тогава, когато са налице едни и същи максимуми и не е регистрирана точка, в която отклонението между двата спектра да превишава 15 % от екстинцията на стандартния аналит; |
|
в) |
между 250 и 400 nm спектрите на възходящата линия, максимума и низходящата линия на пика, получени при анализа на екстракта на пробата, не трябва да се различават един от друг в тези части от спектъра, които попадат в интервала между 10 до 100 % относителна екстинкция. Критерият е изпълнен тогава, когато са налице едни и същи максимуми и когато във всички регистрирани точки отклонението между спектрите не надминава 15 % от екстинкцията на спектъра на най-високата точка. |
Ако някой от критериите не е изпълнен, наличието на аналита не се потвърждава.
7.2. Повторяемост
Разликата между резултатите от две паралелни определяния, проведени върху една и съща проба, не трябва да надвишава 10 % от по-високия резултат при съдържание на робенидин по-високо от 15 mg/kg.
7.3. Възстановяване
Възстановяването за обогатена проба от фураж без добавки трябва да бъде поне 85 %.
8. Резултати от съвместно изследване
Беше организирано съвместно изследване в рамките на ЕО, при което четири проби от фураж за домашни птици и зайци под формата на брашно или пелети бе анализирано от 12 лаборатории. За всяка проба са проведени по два анализа. Резултатите от изследването са представени в следната таблица:
|
|
Домашни птици |
Зайци |
||
|
|
Брашно |
Пелети |
Брашно |
Пелети |
|
Средна стойност в mg/kg |
27,00 |
27,99 |
43,6 |
40,1 |
|
sr [mg/kg] |
1,46 |
1,26 |
1,44 |
1,66 |
|
CVr [%] |
5,4 |
4,5 |
3,3 |
4,1 |
|
SR [mg/kg] |
4,36 |
3,36 |
4,61 |
3,91 |
|
CVR [%] |
16,1 |
12,0 |
10,6 |
9,7 |
|
Възстановяване (%) |
90,0 |
93,3 |
87,2 |
80,2 |
|
sr |
= |
стандартно отклонение на повторяемостта |
|
CVr |
= |
коефициент на вариация на повторяемостта |
|
SR |
= |
стандартно отклонение на възпроизводимостта |
|
CVR |
= |
коефициент на вариация на възпроизводимостта, % |
Е. ОПРЕДЕЛЯНЕ НА ДИКЛАЗУРИЛ
(+)-4-хлорфенил [2,6-дихлоро-4-(2,3,4,5-тетрахидро-3,5-диоксо-1,2,4-триазин-2-ил)фенил]-ацетонитрил.
1. Цели и обхват
Настоящият метод дава възможност за определяне на равнището на диклазурил във фуражи и премикси. Границата на откриване е 0,1 mg/kg, а границата на определяне е 0,5 mg/kg.
2. Принцип
След прибавяне на вътрешен стандарт, пробата се екстрахира с подкислен метанол. При фуражите, една аликвотна част от екстракта се пречиства в патрон за екстракция на твърда фаза С18. Диклазурилът се елуира от патрона със смес от подкислен метанол и вода. След изпарение, остатъкът се разтваря в DMF/вода. При премиксите екстрактът се изпарява и остатъкът се разтваря в DMF/вода. Съдържанието на диклазурил се определя чрез високоефективна триградиентна течна хроматография (HPLC) с обратна фаза, с помощта на UV детектор.
3. Реагенти
|
3.1. |
Вода с чистота за HPLC |
|
3.2. |
Амониев ацетат |
|
3.3. |
Тетрабутиламониeв хидроген сулфат (TBHS) |
|
3.4. |
Ацетонитрил, с чистота за HPLC |
|
3.5. |
Метанол, с чистота за HPLC |
|
3.6. |
N,N-диметилформамид (DMF) |
|
3.7. |
Солна киселина, ρ20 = 1,19 g/ml |
|
3.8. |
Стандартно вещество: диклазурил II-24: (+)-4-хлорфенил [2,6-дихлоро-4-(2,3,4,5-тетрахидро-3,5-диоксо-1,2,4-триазин-2-ил)фенил]-ацетонитрил с гарантирана чистота, Е771. |
|
3.8.1. |
Претеглят се 25 mg стандартно вещество диклазурил (т. 3.8) с точност до 0,1 mg в градуирана колба от 50 ml. Разтварят се в DMF (т. 3.6), допълва се до пълния обем с DMF (3.6) и се разбърква. Колбата се обвива с алуминиево фолио или се използва колба от тъмно стъкло и се поставя за съхранение в хладилник. При температура ≤ 4 oС, разтворът е стабилен в продължение на 1 месец. |
|
3.8.2. |
Прехвърлят се 5,00 ml от основния стандартен разтвор (т. 3.8.1) в градуирана колба от 50 ml, допълва се до пълния обем с DMF (т. 3.6) и се разбърква. Колбата се обвива с алуминиево фолио или се използва колба от тъмно стъкло и се поставя за съхранение в хладилник. При температура ≤ 4 oС, разтворът е стабилен в продължение на 1 месец. |
|
3.9. |
Вещество, използвано като вътрешен стандарт: 2,6 дихлоро-α-(4-хлорофенил)-4-(4,5 дихидро-3,5-диоксо-1,2,4-триазин-2(3Н)-ил) α-метилбензен-ацетонитрил. |
|
3.9.1. |
Претеглят се 25 mg от веществото за вътрешен стандарт (т. 3.9) с точност до 0,1 mg в градуирана колба от 50 ml. Това количество се разтваря в DMF (т. 3.6), разтворът се допълва до пълния обем с DMF (3.6) и се разбърква. Колбата се обвива с алуминиево фолио или се използва колба от тъмно стъкло и се поставя за съхранение в хладилник. При температура ≤ 4 oС, разтворът е стабилен в продължение на 1 месец. |
|
3.9.2. |
Прехвърлят се 5,00 ml от вътрешния основен стандартен разтвор (т. 3.9.1) в градуирана колба от 50 ml, допълва се до пълния обем с DMF (т. 3.6) и се разбърква. Колбата се обвива с алуминиево фолио или се използва колба от тъмно стъкло и се поставя за съхранение в хладилник. При температура ≤ 4 oС, разтворът е стабилен в продължение на 1 месец. |
|
3.9.3. |
(р = номинално съдържание на диклазурил в mg/kg в премикса) Отмерват се с точност до 0,1 mg p/10 mg от веществото, използвано за вътрешен стандарт, в градуирана колба от 100 ml, разтварят се в DMF (т. 3.6) в ултразвукова вана (т. 4.6), допълва се до пълния обем с DMF и се разбърква. Колбата се обвива с алуминиево фолио или се използва колба от тъмно стъкло и се поставя за съхранение в хладилник. При температура ≤ 4 oС, разтворът е стабилен в продължение на 1 месец. |
|
3.10. |
Калибрационен разтвор 2 μg/ml. Отмерват се с пипета 2,00 ml от стандартния разтвор на диклазурил (т. 3.8.2) и 2 ml от вътрешния стандартен разтвор в градуирана колба от 50 ml. Прибавят се 16 ml DMF (т. 3.6), допълва се до пълния обем с вода и се разбърква. Разтворът трябва да бъде приготвен непосредствено преди употреба. |
|
3.11. |
Патрон за екстракция на твърда фаза С18, напр. Bond Elut, размер: 1 cm3, тегло на сорбента: 100 mg |
|
3.12. |
Екстракционен разтворител: подкислен метанол. Отмерват се с пипета 5,0 ml солна киселина (т. 3.7) и се смесват с 1 000 ml метанол (т. 3.5). |
|
3.13. |
Подвижна фаза за HPLC |
|
3.13.1. |
Елуиращ агент А: разтвор на амониев ацетат — тетрабутиламониев хидроген сулфат. Разтварят се 5 g амониев ацетат (т. 3.2) и 3,4 g TBHS (т. 3.3) в 1 000 ml вода (т. 3.1) и се разбърква. |
|
3.13.2. |
Елуиращ агент Б: ацетонитрил (т. 3.4). |
|
3.13.3. |
Елуиращ агент В: метанол (т. 3.5). |
4. Апаратура
|
4.1. |
Механична клатачна машина |
|
4.2. |
Оборудване за тройно-градиентна HPLC |
|
4.2.1. |
Колона за течна хроматография, Hypersil ODS, пълнеж от 3 μm, 100 mm ×4,6 mm, или еквивалентна. |
|
4.2.2. |
UV детектор с настройка на дължината на вълната или детектор с диодна матрица. |
|
4.3. |
Ротационен тънкослоен изпарител. |
|
4.4. |
Мембранен филтър, 0,45 μm. |
|
4.5. |
Вакуумен колектор. |
|
4.6. |
Ултразвукова баня. |
5. Процедура
5.1. Общи положения
5.1.1.
Анализира се фураж без добавки, за да се провери дали не той не е замърсен с диклазурил или други вещества. Фуражът без добавки трябва да е аналогичен по вид на този от пробата и при анализа да не се открие диклазурил или други нежелани вещества.
5.1.2.
Провежда се тест за възстановяване, като се анализира проба от фураж без добавки, обогатена чрез прибавяне на близко до наличното в пробата за изследване количество диклазурил. За да се обогати фуражът до равнище 1 mg/kg, се добавят 0,1 ml от основния стандартен разтвор (т. 3.8.1) към 50 g от фуража без добавки, смесва се и се изчаква 10 min, като се рамесва няколко пъти, преди да се премине към следващия етап (т. 5.2).
Като алтернатива, ако не е наличен фураж, подобен на този, от който е взета пробата (вж. 5.1.1), тестът за възстановяване може да се извърши чрез стандартия метод на добавяне. В този случай пробата, която ще се анализира, се обогатява с такова количество диклазурил, което е близко до вече съдържащото се в пробата. Тази проба се анализира заедно с необогатена проба, като степента на възстановяване се пресмята чрез изваждане.
5.2. Екстракция
5.2.1.
Претеглят се точност до 0,01 g около 50 g от пробата. Прехвърлят се в конична колба от 500 ml, прибавя се 1,00 ml разтвор на вътрешния стандарт (т. 3.9.2), 200 ml разтворител за екстракция (т. 3.12) и колбата се запушва. Сместа се поставя в клатачна машина (т. 4.1) и се оставя в нея до следващия ден. Сместа се оставя да се утаява в продължение на 10 минути. Прехвърлят се 20 ml аликвотна част от супернатанта в подходящ стъклен съд и се разрежда с 20 ml вода. Този разтвор се прехвърля в патрон за екстракция (т. 3.11) и се прекарва през последния чрез подаване на вакуум (т. 4.5). Патронът се промива с 25 ml смес от екстракционния разтворител (т. 3.12) и вода, 65 + 35 (V + V). Събраните фракции се отстраняват и съединенията се елуират с 25 ml смес от екстракционния разтворител (т. 3.12) и вода, 80 + 20 (V + V). Тази фракция се изпарява до сухо, като се използва ротационният изпарител (т. 4.3) при 60 oС. Остатъкът се разтваря в 1,0 ml DMF (т. 3.6), добавят се 1,5 ml вода (т. 3.1) и се разбърква. Филтрира се през мембранен филтър (т. 4.4). Пристъпва се към определяне чрез HPLC (т. 5.3).
5.2.2.
Претегля се с точност до 0,001 g около 1 g от пробата. Претегленото количество се прехвърля в конична колба от 500 ml, прибавя се 1,00 ml от разтвора на вътрешния стандарт (т. 3.9.3), 200 ml екстракционен разтворител (т. 3.12) и колбата се запушва. Сместа се поставя в клатачна машина (т. 4.1) и се оставя в нея до следващия ден. Сместа се оставя да се утаява в продължение на 10 минути. Аликвотна част с обем 10,000/р ml (р = номинално съдържание на диклазурил в премикса в mg/kg) от супернатанта се прехвърля в облодънна колба с подходящ размер. Течността се изпарява при понижено налягане и при 60 oC до сухо, като се използва ротационният изпарител (т. 4.3). Остатъкът се разтваря отново в 10,0 ml DMF (т. 3.6), прибавят се 15,0 ml вода (т. 3.1) и се разбърква. Пристъпва се към определяне чрез HPLC (т. 5.3).
5.3. Определяне с помощта на HPLC
5.3.1.
Следните условия са дадени като указание; може да се използват други, при условие, че дават равностойни резултати:
|
Колона за течна хроматография (т. 4.2.1): |
100 mm × 4,6 mm, Hypersil ODS, пълнеж от 3 μm, или еквивалентна |
|
||||||
|
подвижна фаза: |
Елуиращ агент А (т. 3.13.1): |
Воден разтвор на амониев ацетат и тетрабутиламониев хидроген сулфат; |
||||||
|
|
Елуиращ агент Б (т. 3.13.2): |
ацетонитрил |
||||||
|
|
Елуиращ агент В (т. 3.13.3): |
метанол |
||||||
|
Начин на елуиране |
Промива се с Б в продължение на 10 min |
|||||||
|
Скорост на изтичане: |
1,5—2 ml/min; |
|
||||||
|
обем за впръскване: |
20 μl |
|
||||||
|
Дължина на вълната, на която е настроен детекторът: |
280 nm |
|
||||||
Стабилността на хроматографската система се проверява, като се впръска няколко пъти калибрационен разтвор (т. 3.10), съдържащ 2 μg/ml, докато се постигнат постоянни височини на пиковете и времена на задържане.
5.3.2.
Впръскват се 20 μl от калибрационния разтвор (т. 3.10) няколко пъти и се определя средната височина (площ) на пика на диклазурила и на пиковете на вътрешния стандарт.
5.3.3.
Впръскват се 20 μl от разтвора на пробата (т. 5.2.1 или т. 5.2.2.) няколко пъти и се определя средната височина (площ) на пика на диклазурила и пиковете на вътрешния стандарт.
6. Изчисляване на резултатите
6.1. Фуражи
Съдържанието на диклазурил w (mg/kg) в пробата се пресмята по следната формула:
 [mg/kg]
[mg/kg]
където:
|
hd, s |
= |
височина (площ) на пика на диклазурил в разтвора на пробата (т. 5.2.1) |
|
hi, s |
= |
височина (площ) на пика на вътрешния стандарт в разтвора на пробата (т. 5.2.1) |
|
hd, c |
= |
височина (площ) на пика на диклазурил в калибрационния разтвор (т. 3.10) |
|
hi, c |
= |
височина (площ) на пика на вътрешния стандарт в калибрационния разтвор (т. 3.10) |
|
cd, c |
= |
концентрация на диклазурил в калибрационния разтвор в μg/ml (т. 3.10) |
|
m |
= |
тегло в g на частта от пробата за анализ. |
|
V |
= |
обем на екстракта от пробата, съгласно т. 5.2.1 (т.е. 2,5 ml) |
6.2. Премикси
Съдържанието на диклазурил w (mg/kg) в пробата се пресмята по следната формула:
 [mg/kg]
[mg/kg]
където:
|
hd,с |
= |
височина (площ) на пика на диклазурил в калибрационния разтвор (т. 3.10) |
|
hi, c |
= |
височина (площ) на пика на вътрешния стандарт в калибрационния разтвор (т. 3.10) |
|
hd, s |
= |
височина (площ) на пика на диклазурил в разтвора на пробата (т. 5.2.2) |
|
hi, s |
= |
височина (площ) на пика на вътрешния стандарт в разтвора на пробата (т. 5.2.2) |
|
cd, c |
= |
концентрация на диклазурил в калибрационния разтвор в μg/ml (т. 3.10) |
|
m |
= |
тегло в g на частта от пробата за анализ. |
|
V |
= |
обем на екстракта от пробата, съгласно т. 5.2.1 (т.е. 25 ml) |
|
p |
= |
номинално съдържание на диклазурил в mg/kg в премикса |
7. Потвърждаване на резултатите
7.1. Идентичност
Идентичността на аналита може да бъде потвърдена чрез кохроматография или като се използва детектор с диодна матрица, с помощта на който се сравняват спектрите на екстракта на пробата (т. 5.2.1 или т. 5.2.2) и на калибрационния разтвор (т. 3.10).
7.1.1.
Обогатява се екстракт от пробата (т. 5.2.1 или т. 5.2.2), като се добави подходящо количество калибрационен разтвор (т. 3.10). Количеството добавен диклазурил трябва да бъде близко до откритото в екстракта от пробата.
След като се вземат под внимание както добавеното количество, така и разреждането на екстракта, увеличават се само височината на пика на диклазурила и тази на пика на вътрешния стандарт. Широчината на пика при половината от височината му трябва да бъде в границите на ± 10 % от оригиналната широчина на пика на диклазурила или на пика на вътрешния стандарт на необогатения екстракт от пробата.
7.1.2.
Резултатите се оценяват според следните критерии:
|
а) |
Дължината на вълната на максимална абсорбция на пробата трябва да бъде същата като тази на спектрите на стандарта, регистрирана в най-високата точка на хроматограмата, с отклонение, определено от разделителната способност на детектиращата система. За отчитане с диодна матрица отклонението обикновено е в рамките на ± 2 nm. |
|
б) |
Между 230 и 320 nm спектрите на пробата и на стандарта, отчетени в най-високата точка на хроматограмата, не трябва да се различават помежду си за онези части от спектъра, които попадат в интервала между 10 до 100 % относителна екстинкция. Критерият е изпълнен тогава, когато са налице едни и същи максимуми и не е регистрирана точка, в която отклонението между двата спектра да превишава 15 % от екстинцията на стандартния аналит; |
|
в) |
между 230 и 320 nm спектрите на възходящата линия, максимума и низходящата линия на пика, получени при анализа на екстракта на пробата, не трябва да се различават един от друг в тези части от спектъра, които попадат в интервала между 10 до 100 % относителна екстинкция. Критерият е изпълнен тогава, когато са налице едни и същи максимуми и когато във всички регистрирани точки отклонението между спектрите не превишава 15 % от екстинкцията на спектъра на най-високата точка. |
Ако някой от критериите не е изпълнен, наличието на аналита не се потвърждава.
7.2. Повторяемост
Разликата между резултатите от две паралелни определяния, проведени върху една и съща проба, не трябва да превишава:
|
— |
30 % по отношение на по-високата стойност при съдържание на диклазурил от 0,5 mg/kg до 2,5 mg/kg; |
|
— |
0,75 mg/kg при съдържание на диклазурил между 2,5 mg/kg и 5 mg/kg; |
|
— |
15 % по отношение на по-високата стойност при съдържание на диклазурил над 5 mg/kg. |
7.3. Възстановяване
При обогатена (празна) проба, извличането трябва да бъде най-малко 80 %.
8. Резултати от съвместно изследване
Проведено беше съвместно изследване, при което 11 лаборатории анализираха 5 проби. Пробите се състояха от два премикса; единият беше смесен с органична матрица (О 100), а другият — с неорганична матрица (А 100). Теоретичното съдържание на диклазурил беше 100 mg/kg. Трите смесени фуража за домашни птици бяха произведени от три различни производителя (NL) (L1/Z1/K1). Теоретичното съдържание на диклазурил беше 1 mg/kg. Лабораториите бяха инструктирани да анализират всяка от пробите един или два пъти. (По-подробна информация за това съвместно изследване може да се открие в Journal of AOAC International, том 77, бр. 6, 1994 г., стр. 1359—1361). Резултатите са представени в следната таблица:
|
|
Проба 1 А 100 |
Проба 2 O 100 |
Проба 3 L1 |
Проба 4 Z1 |
Проба 5 K1 |
|
L |
11 |
11 |
11 |
11 |
6 |
|
n |
19 |
18 |
19 |
19 |
12 |
|
Средно |
100,8 |
103,5 |
0,89 |
1,15 |
0,89 |
|
Sr (mg/kg) |
5,88 |
7,64 |
0,15 |
0,02 |
0,03 |
|
CVr (%) |
5,83 |
7,38 |
17,32 |
1,92 |
3,34 |
|
SR (mg/kg) |
7,59 |
7,64 |
0,17 |
0,11 |
0,12 |
|
CVR (%) |
7,53 |
7,38 |
18,61 |
9,67 |
13,65 |
|
Номинално съдържание (mg/kg) |
100 |
100 |
1 |
1 |
1 |
|
L |
= |
брой на лабораториите |
|
n |
= |
брой на единичните стойности |
|
Sr |
= |
стандартно отклонение на повторяемостта |
|
CVr |
= |
коефициент на вариация на повторяемостта |
|
SR |
= |
стандартно отклонение на възпроизводимостта |
|
CVR |
= |
коефициент на вариация на възпроизводимостта |
9. Забележки
Необходимо е предваретелно да е доказано, че реакцията спрямо диклазурил е линейна в интервала на измерваните стойности.
Ж. ОПРЕДЕЛЯНЕ НА ЛАЗАЛОЦИД-НАТРИЙ
Натриева сол на полиетерна монокарбонова киселина, получена от Streptomyces lasaliensis
1. Цел и обхват
Настоящията метод дава възможност за определяне на равнището на лазалоцид-натрий във фуражи и премикси. Границата на откриване е 5 mg/kg, а границата на количествено определяне е 10 mg/kg.
2. Принцип
Лазалоцид натрий се извлича от пробата в подкислен метанол и се определя чрез високоефективна течна хроматография (HPLC) с обратна фаза, с помощта на спектрофлуориметричен детектор.
3. Реагенти
|
3.1. |
Калиев дихидроген фосфат (KH2PO4) |
|
3.2. |
Ортофосфорна киселина, w (w/w) = 85 % |
|
3.3. |
Разтвор на ортофосфорна киселина, c = 20 % Разредждат се 23,5 ml ортофосфорна киселина (т. 3.2) до 100 ml с вода. |
|
3.4. |
6-Метил-2-хептиламин (1,5-диметилхексиламин), w (w/w) = 99 % |
|
3.5. |
Метанол, с чистота за HPLC |
|
3.6. |
Солна киселина, плътност = 1,19 g/ml |
|
3.7. |
Фосфатен буферен разтвор, с = 0,01 mol/l Разтварят се 1,36 g KH2PO4 (т. 3.1) в 500 ml вода (т. 3.11), добавят се 3,5 ml ортофосфорна киселина (т. 3.2) и 10,0 ml 6-метил-2-хептиламин (т. 3.4). Стойността на рН се коригира до 4,0 чрез прибавяне на разтвор на ортофосфорна киселина (т. 3.3) и разтворът се разрежда до 1 000 ml с вода (т. 3.11). |
|
3.8. |
Подкиселен метанол Прехвърлят се 5,0 ml солна киселина (т. 3.6) в градуирана колба от 1 000 ml, допълва се с метанол (т. 3.5) до пълния обем и се разбърква. Разтворът трябва да бъде приготвен непосредствено преди употреба. |
|
3.9. |
Подвижна фаза за HPLC, фосфатен буферно-метанолов разтвор 5 + 95 (V + V) Смесват се 5 ml от фосфатния буферен разтвор (т. 3.7) с 95 ml метанол (т. 3.5). |
|
3.10. |
Стандартно вещество лазалоцид натрий с гарантирана чистота, C34H53O8Na (натриева сол на полиетерна монокарбонова киселина, получена от Streptomyces lasaliensis), Е763 |
|
3.10.1. |
Претеглят се с точност до 0,1 mg 50 mg лазалоцид натрий (т. 3.10) и се поставят в градуирана колба от 100 ml, разтварят се в подкислен метанол (т. 3.8), допълва се със същия разтворител до пълния обем, и се разбърква. Разтворът трябва да бъде приготвен непосредствено преди употреба. |
|
3.10.2. |
Отмерват се с пипета 10,0 ml от основния стандартен разтвор (т. 3.10.1) в градуирана колба от 100 ml, допълва се с подкислен метанол (т. 3.8) до пълния обем и се разбърква. Разтворът трябва да бъде приготвен непосредствено преди употреба. |
|
3.10.3. |
В няколко колби от 50 ml се прехвърлят 1,0, 2,0, 4,0, 5,0 и 10,0 ml от междинния стандартен разтвор (т. 3.10.2). Допълва се с подкислен метанол (т. 3.8) до пълния обем и се разбърква. Тези разтвори съдържат съответно 1,0, 2,0, 4,0, 5,0 и 10,0 μg/ml лазалоцид натрий. Разтворите трябва да бъдат приготвени непосредствено преди употреба. |
|
3.11. |
Вода с чистота за HPLC. |
4. Апаратура
|
4.1. |
Ултразвукова вана (или вибрираща водна баня) с регулатор на температурата |
|
4.2. |
Мембранен филтър, 0,45 μm. |
|
4.3. |
Оборудване за HPLC със система за впръскване, подходяща за впръскване на обеми от 20 μl. |
|
4.3.1. |
Колона за течна хроматография 125 mm × 4 mm, обратна фаза С18, пълнеж от 5 μm, или еквивалентна |
|
4.3.2. |
Спектрофлуорометър с регулатор на дължината на вълната на възбуждане и излъчване |
5. Процедура
5.1. Общи положения
5.1.1.
За провеждането на теста за възстановяване (т. 5.1.2), се анализира фураж без добавки, за да се провери дали не е замърсен с лазалоцид натрий или с други вещества. Фуражът без добавки трябва да е подобен по вид на този от изследваната проба и в него не трябва да се откриват лазалоцид натрий или нежелани вещества.
5.1.2.
Тестът за възстановяване се извършва, като се анализира пробата от фуража без добавки, обогатена чрез прибавяне на близко до наличното в пробата количество лазалоцид натрий. За да се обогати пробата до ниво до 100 mg/kg, 10,0 ml от стандартния разтвор (т. 3.10.1) се прехвърлят в конична колба от 250 ml и се подлагат на изпарение, докато количеството на разтвора намалее до около 0,5 ml. Добавят се 50 g от фуража без добавки, разбърква се старателно и се оставя за 10 min, като се разбърква неколкократно, преди да се премине към етапа на екстракция (т. 5.2).
Като алтернатива, ако не е наличен фураж без добавки, подобен на този, от който е взета пробата (вж. т. 5.1.1), тестът за възстановяване може да се извърши чрез стандартия метод на добавяне. В този случай пробата, която ще се анализира, се обогатява с такова количество лазалоцид натрий, което е близко до наличното в пробата. Тази проба се анализира заедно с необогатената проба, като извличането се пресмята чрез изваждане.
5.2. Екстракция
5.2.1.
Претеглят се 5 g до 10 g от пробата с точност до 0,01 g и се поставят в конична колба от 250 ml със запушалка. Прибавят се с пипета 100,0 ml подкислен метанол (т. 3.8). Запушва се хлабаво със запушалката и се разклаща, за да може съдържанието да се диспергира. Колбата се поставя в ултразвукова вана (т. 4.1) при температура приблизително 40 oС за 20 минути, след което се изважда и охлажда до стайна температура. Оставя се да престои около 1 час до утаяване на суспензията, след което през мембранен филтър от 0,45 μm (т. 4.2) се филтрира в подходящ съд една аликвотна част. Пристъпва се към определяне чрез HPLC (т. 5.3).
5.2.2.
Около 2 g несмлян премикс се претеглят с точност до 0,001 g и се поставят в градуирана колба от 250 ml. Прибавят се 100,0 ml подкислен метанол (т. 3.8) и се разклаща, за да може съдържанието да се диспергира. Колбата със съдържанието си се поставя в ултразвукова вана (т. 4.1) при температура приблизително 40 oС за 20 минути, след което се изважда и охлажда до стайна температура. Разрежда се с подкислен метанол (т. 3.8) до пълния обем и се разбърква старателно. Оставя се да престои около 1 час до утаяване на суспензията, след което през мембранен филтър от 0,45 μm (т. 4.2) се филтрира една аликвотна част. Подходящ обем от чистия филтрат се разрежда с подкислен метанол (т. 3.8), за да се получи окончателен разтвор за анализ, който съдържа около 4 μg/ml лазалоцид натрий. Пристъпва се към определяне чрез HPLC (т. 5.3).
5.3. Определяне с помощта на HPLC
5.3.1.
За ориентиране се предлагат следните условия; може да се използват и други условия, ако дават равностойни резултати:
|
Колона за течна хроматография (4.3.1): |
125 mm × 4 mm, с обратна фаза С18, пълнеж от 5 μm, или еквивалентна |
|
Подвижна фаза (т. 3.9): |
Смес от разтвор на фосфатен буфер (т. 3.7) и метанол (т. 3.5), 5 + 95 (V+V) |
|
Скорост на изтичане: |
1,2 ml/min |
|
Дължина на вълната за откриване: |
|
|
възбуждане: |
310 nm |
|
излъчване: |
419 nm |
|
обем за впръскване: |
20 μl |
Стабилността на хроматографската система се проверява, като се впръсква няколко пъти калибрационен разтвор (т. 3.10.3), съдържащ 4,0 μg/ml, докато бъдат постигнати постоянни пикови височини (или площи) и времена на задържане.
5.3.2.
Всеки калибрационен разтвор се впръсква (3.10.3) по няколко пъти и се определят средните пикови височини (площи) за всяка концентрация. Построява се калибрационна крива, като за ординати се използват средните височини на пиковете (площите), а за абсциси — съответните концентрации в μg/ml.
5.3.3.
Впръскват се няколко пъти получените в т. 5.2.1. или т. 5.2.2 екстракти от изследваната проба, като се взима същият обем като при калибрационния разтвор, и се определят средните пикови височини (площи) на пиковете на лазалоцид натрий.
6. Изчисляване на резултатите
Като се използва средната пикова височина (площ), получена при впръскване на разтвора на пробата (т. 5.3.3), чрез сравняване с калибрационната крива се определя концентрацията на лазалоцид натрий (μg/ml).
6.1. Фураж
Съдържанието на лазалоцид натрий w (mg/kg) в пробата се пресмята по следната формула:
 [mg/kg]
[mg/kg]
където:
|
c |
= |
концентрация на лазалоцид натрий в разтвора на пробата (т. 5.2.1) в μg/ml |
|
V1 |
= |
обем на екстракта от пробата, съгласно т. 5.2.1 в ml (т.е. 100) |
|
m |
= |
тегло в g на частта от пробата за анализ. |
6.2. Премикси
Съдържанието на лазалоцид натрий w (mg/kg) в пробата се пресмята по следната формула:
 [mg/kg]
[mg/kg]
където:
|
c |
= |
концентрация на лазалоцид натрий в разтвора на пробата (т. 5.2.2) в μg/ml |
|
V2 |
= |
обем на екстракта от пробата, съгласно т. 5.2.2 в ml (т.е. 250) |
|
f |
= |
фактор на разреждане, съгласно т. 5.2.2 |
|
m |
= |
тегло в g на частта от пробата за анализ. |
7. Потвърждаване на резултатите
7.1. Идентичност
Методите, които се базират на спектрофлуориметрията, са по-малко податливи на смущения, отколкото тези, при които се използва UV детекция. Идентичността на аналита може да се потвърди чрез ко-хроматография.
7.1.1.
Обогатява се екстракт от пробата (т. 5.2.1 или т. 5.2.2), като се добави подходящо количество калибрационен разтвор (т. 3.10.3). Количеството добавен лазалоцид натрий трябва да бъде близко до откритото в екстракта от пробата. Увеличава се само височината на пика на лазалоцид натрий, като се вземе предвид добавеното количество лазалоцид натрий и степента на разреждане на екстракта. Широчината на пика на половината от височината му трябва да бъде в рамките на ± 10 % от първоначалната широчина, получена за екстракта от необогатената проба.
7.2. Повторяемост
Разликата между резултатите от две паралелни определяния, проведени върху една и съща проба, не трябва да превишава:
|
— |
15 % по отношение на по-високата стойност при съдържание на лазалоцид натрий от 30 mg/kg до 100 mg/kg; |
|
— |
15 mg/kg при съдържание на лазалоцид натрий от 100 mg/kg до 200 mg/kg; |
|
— |
7,5 % по отношение на по-високата стойност при съдържание на лазалоцид натрий над 200 mg/kg. |
7.3. Възстановяване
За обогатената проба от фуража, възстановяването трябва да бъде най-малко 80 %. При обогатените проби от премикси, възстановяването трябва да бъде най-малко 90 %.
8. Резултати от съвместно изследване
Проведено беше съвместно изследване (6), при което бяха анализирани 2 премикса (проби 1 и 2) и 5 вида фураж (проби 3—7) от 12 лаборатории. За всяка проба са проведени по два анализа. Резултатите са представени в таблицата по-долу:
|
|
Проба 1 Премикс за пилета |
Проба 2 Премикс за пуйки |
Проба 3 Пелети за пуйки |
Проба 4 Трохи за пилета |
Проба 5 Фураж за пуйки |
Проба 6 Фураж за дом. птици А |
Проба 7 Фураж за дом. птици Б |
|
L |
12 |
12 |
12 |
12 |
12 |
12 |
12 |
|
n |
23 |
23 |
23 |
23 |
23 |
23 |
23 |
|
Среднa стойност, [mg/kg] |
5 050 |
16 200 |
76,5 |
78,4 |
92,9 |
48,3 |
32,6 |
|
sr [mg/kg] |
107 |
408 |
1,71 |
2,23 |
2,27 |
1,93 |
1,75 |
|
CVr [ %] |
2,12 |
2,52 |
2,24 |
2,84 |
2,44 |
4,00 |
5,37 |
|
sR [mg/kg] |
286 |
883 |
3,85 |
7,32 |
5,29 |
3,47 |
3,49 |
|
CVR [ %] |
5,66 |
5,45 |
5,03 |
9,34 |
5,69 |
7,18 |
10,70 |
|
Номинално съдържание, [mg/kg] |
5 000 (7) |
16 000 (7) |
80 (7) |
105 (7) |
120 (7) |
50 (8) |
35 (8) |
|
L |
= |
брой на лабораториите |
|
n |
= |
брой на единичните резултати |
|
sr |
= |
стандартно отклонение на повторяемостта |
|
sR |
= |
стандартно отклонение на възпроизводимостта |
|
CVr |
= |
коефициент на вариация на повторяемостта, % |
|
CVR |
= |
коефициент на вариация на възпроизводимостта, % |
(1) Проведено от Работната група по фуража на Verband Deutscher Landwirtschaftlicher Untersuchungs- und Forschungsanstalten (VDLUFA)
(2) Проведено от Работната група по фуража на Verband Deutscher Landwirtschaftlicher Untersuchungs- und Forschungsanstalten (VDLUFA).
(3) Могат да се използват и други методи за изваряване, при условие, че е доказано, че те дават сходни резултати (напр. изваряване под налягане с микровълни).
(4) Зелените фуражи (пресни или изсушени) е възможно да съдържат значително количество силициев диоксид от растителен произход, който може да задържи микроелементите, и който трябва да да бъде отстранен. Поради това, за проби от тези фуражи трябва да се следват следните изменени процедури. Операция 5.1.1.1. се провежда едновременно с филтрирането. Филтърната хартия с неразтворимия остатък се измива два пъти с вряла вода и се поставя в кварцов или платинен тигел. В муфелната пещ (т. 4.1) филтърната хартия се изгаря при температура под 550 oC, докато всички саждени частици изчезнат напълно. Оставя се да изстине, добавят се няколко капки вода и 10—15 ml флуороводородна киселина (т. 3.4) и се подлага на изпаряване до сухо при около 150 oC. Ако има остатъци от силициев диоксид в остатъка, те се разтварят повторно в няколко милилитра флуороводородна киселина (3.4) и се подлагат на изпаряване до сухо. Добавят се няколко капки сярна киселина (т. 3.5) и се нагрява, докато престане да се образува бял дим. След като се добавят около 5 ml солна киселина с концентрация 6 mol/l (т. 3.2) и около 30 ml вода, разтворът се нагрява, филтрира се в мерителна колба от 250 ml и се допълва до пълния обем с вода (концентрация на HCl около 0,5 mol/l). След това се пристъпва към определянето в съответствие с точка 5.1.2
(5) The Analyst 108, 1983 г., стр. 1 252—1 256.
(6) Analyst, 1995, 120, 2175—2180
(7) Заявено от производителя съдържание.
(8) Фураж, подготвен в лабораторията.
ПРИЛОЖЕНИЕ V
МЕТОДИ ЗА АНАЛИЗ С ЦЕЛ КОНТРОЛ НА НЕЖЕЛАНИ ВЕЩЕСТВА ВЪВ ФУРАЖИТЕ
А. ОПРЕДЕЛЯНЕ НА СВОБОДЕН И ОБЩ ГОСИПОЛ
1. Цел и обхват
Настоящият метод дава възможност да се определи равнището на свободния госипол, общия госипол и химически сродни вещества в памучно семе, брашно от памучно семе, в кюспе от памучно семе и в комбинирани фуражи, съдържащи тези фуражни суровини, когато съдържанието на свободен госипол, общ госипол и химически сродни вещества надвишава 20 mg/kg.
2. Принцип
Госиполът се екстрахира в присъствие на 3-аминопропан-1-ол — или със смес от пропан-2-ол и хексан — за определяне на свободния госипол, или с диметилформамид за определяне на общия госипол. Под въздействието на анилин госиполът се превръща в госипол-дианилин, чиято оптична плътност се измерва при 440 nm.
3. Реагенти
|
3.1. |
Смес от пропан-2-ол и хексан: 60 обемни части пропан-2-ол се смесват с 40 обемни части n-хексан. |
|
3.2. |
Разтворител А: в градуирана колба от 1 l се смесват около 500 ml от сместа от пропан-2-ол и хексан (т. 3.1), 2 ml 3-аминопропан-1-ол, 8 ml безводна оцетна киселина и 50 ml вода. Допълва се до пълния обем със смес от пропан-2-ол и хексан (т. 3.1). Реагентът е стабилен в продължение на една седмица. |
|
3.3. |
Разтворител B: в градуирана колба от 100 ml се поставят с пипета 2 ml 3-аминопропан-1-ол и 10 ml безводна оцетна киселина. Охлаждат се до стайна температура и колбата се допълва до пълния обем с N,N-диметилформамид. Реагентът е стабилен в продължение на една седмица. |
|
3.4. |
Анилин: Ако оптичната плътност на празната проба надвишава 0,022, анилинът се дестилира над цинков прах, като се отстраняват първите и последните 10 % фракции от дестилата. Охладен и съхраняван в кафява бутилка със стъклена запушалка, този реагент се съхранява няколко месеца. |
|
3.5. |
Стандартен разтвор А на госипол: В градуирана колба от 250 ml се поставят 27,9 mg госипол ацетат. Разтварят се и колбата се долива до пълния обем с разтворител А (т. 3.2). С пипета 50 ml от този разтвор се прехвърлят в градуирана колба от 250 ml, която се допълва до пълния обем с разтворител А. Концентрацията на госипол в този разтвор е 0,02 mg/ml. Разтворът се оставя на стайна температура за един час преди упортеба. |
|
3.6. |
Стандартен разтвор Б на госипол: В градуирана колба от 50 ml се поставят 27,9 mg госипол ацетат, разтварят се и се долива разтворител Б (т. 3.3) до пълния обем. Концентрацията на госипол в този разтвор е 0,5 mg/ml. Стандартните разтвори А и Б на госипол са стабилни в продължение на 24 часа, ако се съхраняват на тъмно. |
4. Апаратура
|
4.1. |
Смесител (барабанен): около 35 r.p.m. |
|
4.2. |
Спектрофотометър. |
5. Процедура
5.1. Проба за изследване
Големината на пробата зависи от предполагаемото съдържание на госипол в нея. За предпочитане е да се работи с малка проба и относително голяма аликвотна част от филтрата, така че да се получи достатъчно количество госипол за прецизно спектрофотометрично измерване. При определяне на свободния госипол в памучно семе, брашно от памучно семе и в кюспе от памучно семе изследваната проба не трябва да надвишава 1 g; при комбиниран фураж, тя може да достигне и 5 g. За повечето случаи е подходящо да се използва аликвотна част от филтрата с обем 10 ml; тя трябва да съдържа от 50 до 100 μg госипол. При определяне на общия госипол пробата трябва да бъде от 0,5 до 5 g, така че в 2 ml от аликвотната част от филтрата да се съдържат от 40 до 200 μg госипол.
Анализът се провежда при стайна температура от около 20 oC.
5.2. Определяне на свободен госипол.
Пробата се поставя в колба от 250 ml с шлифовано гърло, като дъното на колбата трябва да е покрито с натрошено стъкло. Добавят се с пипета 50 ml от разтворител А (т. 3.2), колбата се запушва и се разбърква в продължение на един час в смесителя. Филтрува се през сух филтър и филтратът се събира в малка колба с шлифовано гърло. По време на филтруването фунията се покрива с часовниково стъкло.
Равни аликвотни части от филтрата, съдържащ от 50 до 100 μg госипол, се прехвърлят с пипета в две градуирани колби (А и В) от по 25 ml. Ако е необходимо, обемът се допълва до 10 ml с разтворител А (т. 3.2). Съдържанието на колба А се допълва до пълния обем със сместа от пропан-2-ол и хексан (т. 3.1). Този разтвор се използва като еталонен разтвор и спрямо него се измерва разтворът на пробата.
В две други градуирани колби от 25 ml (В и Г) се прехвърлят с пипета по 10 ml от разтворител А (т. 3.2). Съдържанието на колба В се допълва до пълния обем със сместа от пропан-2-ол и хексан (т. 3.1). Този разтвор се използва като еталонен и спрямо него се измерват разтворите на празната проба.
Във всяка от колбите Г и Б се добавят по 2 ml анилин (т. 3.4). Колбите се нагряват в продължение на 30 минути над кипяща водна баня, за да се развие оцветяването. Охлаждат се до стайна температура, допълват се до пълния обем със сместа от пропан-2-ол и хексан (т. 3.1), хомогенизират се и се оставят да престоят един час.
Оптическата плътност на празната проба Г се определя чрез сравняване с еталонния разтвор В. Оптическата плътност на пробата Б се определя чрез сравняване със стандартния разтвор А. Измерването се провежда на спектрофотометър при 440 nm, като се използват стъклени кювети с дебелина 1 cm.
Оптическата плътност на празната проба се изважда от тази на изследваната проба (= коригирана оптична плътност). Като се вземе тази стойност, се изчислява количеството свободен госипол, както е описано в точка 6.
5.3. Определяне на общ госипол
В градуирана колба от 50 ml се поставя проба, която съдържа от 1 до 5 mg госипол и се добавят 10 ml разтворител Б (т. 3.3). Едновременно с това се приготвя и празна проба, като 10 ml от разтворител Б (т. 3.3) се прехвърлят в друга градуирана колба от 50 ml. Двете колби се нагряват в продължение на 30 минути над кипяща водна баня. Охлаждат се до стайна температура и съдържанието на всяка колба се допълва до пълния обем със сместа от пропан-2-ол и хексан (т. 3.1). Съдържанието на колбите се хомогенизира и се оставя да се утаи в продължение на 10—15 минути, след което се филтрира и филтратите се събират в колби с шлифовани гърла.
С пипета се поставят по 2 ml от филтрата на пробата в две градуирани колби от 25 ml, а по 2 ml от филтрата на празната проба се слагат в други две градуирани колби от 25 ml. Съдържанието на колбите от всяка група се допълва до 25 ml със сместа от пропан-2-ол и хексан (т. 3.1). Тези разтвори се използват като еталонни разтвори.
Във всяка от другите две колби се добавят по 2 ml анилин (т. 3.4). Колбите се нагряват в продължение на 30 минути над кипяща водна баня, за да се развие оцветяването. Охлаждат се до стайна температура, допълват се до 25 ml със сместа от пропан-2-ол и хексан (т. 3.1), съдържанието им се хомогенизира се и се оставят да престоят един час.
Оптичната плътност се определя, както е указано в т. 5.2 за свободния госипол. Като се вземе тази стойност, се изчислява количеството на общия госипол, както е указано в точка 6.
6. Изчисляване на резултатите
Резултатите могат да се изчислят както чрез специфичната оптична плътност (т. 6.1), така и чрез сравняване с калибрационната крива (т. 6.2).
6.1. Определяне чрез специфичната оптична плътност
Очакваните стойности на специфичните оптични плътности при описаните условия са следните:
|
Свободен госипол: |
|
|
Общ госипол: |
|


Съдържанието на свободен или общ госипол в пробата се изчислява по следната формула:

където:
|
Е |
= |
коригираната оптична плътност, определена както е указано в т 5.2. |
|
p |
= |
масата в g на пробата; |
|
a |
= |
аликвотна част от филтрата в ml. |
6.2. Чрез използване на калибрационната крива
6.2.1.
Подготвят се 2 серии от пет градуирани колби от 25 ml. Във всяка серия колби с пипета се прехвърлят по 2,0, 4,0, 6,0, 8,0 и 10,0 ml аликвотни части от стандартния разтвор А на госипол (т. 3.5). Допълват се до 10 ml с разтворител А (т. 3.2). Към всяка серия се добавя градуирана колба от 25 ml, в която има само 10 ml разтворител А (т. 3.2) (празна проба).
Съдържанието на колбите от първата серия (включително колбата с празната проба) се допълва до 25 ml със сместа от пропан-2-ол и хексан (т. 3.1) (еталонна серия).
Във всяка от колбите от втората серия (включително в колбата с празната проба) се добавят по 2 ml анилин (т. 3.4). Колбите се нагряват в продължение на 30 минути над кипяща водна баня, за да се развие оцветяването. Охлаждат се до стайна температура, допълват се до пълния обем със сместа от пропан-2-ол и хексан (т. 3.1), хомогенизира се съдържанието им и се оставят да престоят един час (стандартна серия).
В съответствие с т. 5.2 се определя оптичната плътност на разтворите в стандартната серия, като се сравняват последните със съответните им разтвори от еталонната серия. Построява се калибрационната крива, като срещу количествата госипол (в μg) се нанася оптичната плътност.
6.2.2.
Подготват се шест градуирани колби от 50 ml. В първата колба се поставят 10 ml от разтворител Б (т. 3.3), а в другите — съответно 2,0, 4,0, 6,0, 8,0 и 10,0 ml от стандартния разтвор Б на госипол (т. 3.6) Всяка колба се допълва до 10 ml с разтворител Б (т. 3.3). Колбите се нагряват в продължение на 30 минути над кипяща водна баня. Охлаждат се до стайна температура, допълват се до пълния обем със сместа от пропан-2-ол и хексан (т. 3.1), и съдържанието им се хомогенизира.
Поставят се 2 ml от тези разтвори в две серии от шест градуирани колби от 25 ml. Съдържанието на колбите от първата серия се допълва до 25 ml със сместа от пропан-2-ол и хексан (т. 3.1) (еталонна серия).
Във всяка от колбите от втората серия се добавят по 2 ml анилин (т. 3.4). Колбите се нагряват в продължение на 30 минути над кипяща водна баня. Охлаждат се до стайна температура, допълват се до пълния обем със сместа от пропан-2-ол и хексан (т. 3.1), хомогенизира се съдържанието им и се оставят да престоят един час (стандартна серия).
В съответствие с т. 5.2 се определя оптичната плътност на разтворите в стандартната серия, като се сравняват последните със съответните им разтвори от еталонната серия. Построява се калибрационната крива, като срещу количествата госипол (в μg) се нанася оптичната плътност.
6.3. Повторяемост
Разликата между резултатите от две паралелни определяния, проведени върху една и съща проба, не трябва да превишава:
|
— |
15 % по отношение на по-високата стойност за съдържание на госипол по-ниско от 500 ppm, |
|
— |
75 ppm в абсолютна стойност за съдржание на госипол не по-малко от 500 ppm и не по-високо от 750 ppm, |
|
— |
10 % по отношение на по-високата стойност при съдържание на госипол над 750 ppm. |
Б. ОПРЕДЕЛЯНЕ НА НИВАТА НА ДИОКСИНИ (PCDD/PCDF) И ДИОКСИНОПОДОБНИ РСВ
I. МЕТОДИ ЗА ВЗИМАНЕ НА ПРОБИ И ТЪЛКУВАНЕ НА РЕЗУЛТАТИТЕ ОТ АНАЛИЗА
1. Цел и обхват
Пробите, предназначени за официалния контрол на нивата на диоксини (полихлорирани дибензо-р-диоксини (PCDD) и полихлорирани дибензофурани (PCDF) и на диоксиноподобни полихлорирани бифенили (PCB) (1) във фуражите се взимат в съответствие с разпоредбите на приложение I. Следва да се прилагат количествените изисквания във връзка с контрола на равномерно разпределени във фуражите вещества, посочени в точка 5.А на приложение I. Така получените сборни проби се приемат за представителни за партидите и подпартидите, от които са взети. Съответствието с максимално допустимите граници, определени в Директива 2002/32/ЕО на Европейския парламент и на Съвета (2), се установява въз основа на равнищата, определени при анализа на лабораторните проби.
2. Съответствие на партидата или подпартидата на спецификацията
Партидата се приема, ако резултатът от един отделен анализ не превишава съответната допустима максимална граница, определена в Директива 2002/32/ЕО, като се взима предвид и евентуалната грешка при измерване.
Партидата не съответства на разпоредбите за максимално допустими граници, определени в Директива 2002/32/ЕО, ако горната граница (3) на аналитичния резултат, потвърден от повторен анализ (4), несъмнено превишава максималната граница, като се взима предвид и евентуалната грешка при измерване.
Грешката при измерване може да се вземе предвид по един от следните начини:
|
— |
като се изчисли разширената неопределеност с помощта на фактор на покриване 2, който дава ниво на сигурност около 95 %. Партидата не съответства на разпоредбите, ако измерената стойност след изваждане на U e над максималната граница. В случай на разделно определяне на диоксини и на диоксиноподобни PCB, сборът от изчислената разширена неопределеност на отделните резултати от анализа на диоксините и от този на диоксиноподобните PCB трябва да се използва за стойност на сбора за диоксини и диоксиноподобни PCB. |
|
— |
като се определи критичната граница (ССα) в съответствие с разпоредбите на Решение 2002/657/ЕО на Комисията (5) (точка 3.1.2.5 от приложението — в случай на вещества, за които е определена разрешена граница). Партидата не съответства на разпоредбите, ако измерената стойност е равна или по-висока от CCα |
Така определените правила на тълкуване са приложими за резултатите от анализа на пробите, предназначени за целите на официалния контрол. Те не накърняват правото на държавите-членки да прилагат национални разпоредби към анализи, провеждани с цел защита или арбитраж.
II. ПОДГОТОВКА НА ПРОБИТЕ И ИЗИСКВАНИЯ КЪМ МЕТОДИТЕ ЗА АНАЛИЗ, ИЗПОЛЗВАНИ ПРИ ОФИЦИАЛЕН КОНТРОЛ НА НИВАТА НА ДИОКСИНИ (PCDD/PCDF) И ДИОКСИНОПОДОБНИ РСВ
1. Цел и приложно поле
Настоящите изисквания се прилагат, когато се анализират фуражни суровини и фураж с цел определяне на диоксини (полихлорирани дибензо-р-диоксини (РСDD) и полихлорирани дибензофурани (PCDF)) и диоксиноподобни полихлорирани бифенили (PCB).
Контролът на съдържанието на диоксини във фуражите може да се осъществи чрез стратегия, основана на скринингов метод, който позволява да се подберат онези проби, в които съдържанието на диоксин или на диоксиноподобни PCB е по-ниско от нивото на значение, но с не повече от 25 %, или надхвърля това ниво. Концентрацията на диоксини в пробите, където равнищата му са високи, трябва да бъде определена/потвърдена чрез метод за потвърждение.
Скрининговите методите са методи, които се използват, за да се открият диоксини и РСВ на на нивото от значение. Те имат висока производителност, и се използват за пресяване на голям брой проби, с цел да бъдат намерени потенциално позитивните. Те са специално разработени, за да се избегнат псевдоотрицателни резултати.
Методите за потвърждение предоставят пълни или допълнителни сведения, позволяващи еднозначна идентификация и количествено определяне на диоксините и на диоксиноподобните PCB на нивото от значение.
2. Контекст
Тъй като екологичните и биологичните проби (включително проби от фуражни суровини/фуражи) по принцип съдържат сложни смеси от различни диоксини и диоксинови конгенери, за да се улесни оценката на риска, е създадено понятието коефициент на токсична еквивалентност (КТЕ). КТЕ са създадени, за да може концентрациите на смеси от 2,3,7,8-заместени PCDD и PCDF и на някои не-орто- и моно-орто- хлор-заместени PCB, които притежават диоксиноподобна активност, да бъдат изразявани в токсични еквиваленти (ТЕ) на 2,3,7,8-TCDD. Концентрациите на отделните вещества в дадена проба се умножават по съответния им КТЕ и след това се сумират, за да дадат общата концентрация на диоксиноподобни съединения, изразена в TE.
Единствено за целите на настоящия регламент приетата специфична граница за количествено определяне на отделен конгенер е концентрацията на аналит в екстракта от пробата, която предизвиква реакция от страна на техническото устройство за два различни йона, която трябва да се следи при съотношение С/Ш (сигнал/шум), равно на 3:1 за по-малко чувствителния сигнал, и която отговаря на основните изисквания като време на задържане и изотопно съотношение, съгласно процедурата за определяне, описана в метод EPA 1613, ревизия Б.
3. Изисквания за осигуряване на качество при подготовката на пробите
Прилагат се общите разпоредби за подготовка на пробите за анализ, посочени в приложение II.
Освен това, следва да се спазват следните изисквания:
|
— |
Пробите трябва да се съхраняват и транспортират в контейнери от стъкло, алуминий, полипропилен или полиетилен. Контейнерът за съхраняване на пробата трябва да бъде почистен от всякакви следи от хартиени частици. Лабораторните стъклени съдове трябва да бъдат изплакнати с разтворители, предварително проверени за наличие на диоксини. |
|
— |
Трябва да се направи анализ с празна проба, като се изпълни цялата аналитична процедура, като единствената разлика е отсъствието на пробата. |
|
— |
Теглото на използваната за екстракция проба трябва да бъде достатъчно, за да отговаря на изискванията по отношение на чувствителността. |
4. Изисквания за лабораториите
|
— |
Лабораториите трябва да докажат качествата на метода в интервала около нивото от значение, например на нива 0,5, 1 или 2 пъти по-високи от това ниво с приемлив коефициент на вариация за повторните анализи. За повече яснота относно критериите за валидност вж. точка 5. |
|
— |
Границата за количествено определяне за метода за потвърдждаване следва да бъде в интервала около една пета от нивото от значение, за да е сигурно, че допустимите коефициенти на вариация попадат в обхвата на нивото от значение. |
|
— |
Следва да се провеждат редовни контролни анализи с празни проби и експерименти с внесена добавка или анализ на контролни проби (за предпочитане сертифициран еталонен материал) като вътрешни мерки за контрол на качеството. |
|
— |
Най-добрият начин да се докаже способността на дадена лаборатория да извършва специфични анализи са завършилите с успех участия в междулабораторни изследвания, които оценяват компетентността на лабораторията. Въпреки това, успешното участие в междулабораторни изследвания на почвени проби или проби от отпадни води например не е достатъчно доказателство за компетентност при анализ на проби от храни за консумация от човека или от фуражи, които са с по-ниски нива на замърсяване. Ето защо непрекъснатото участие в междулабораторни изследвания за определянето на съдържание на диоксини и на диоксиноподобни PCB в съответните матрици от фуражи и храни за консумация от човека е задължително. |
|
— |
Лабораториите се акредитират от признат орган, действащ съгласно Ръководство ISO 58, за да има гаранция, че те прилагат мерки за осигуряване на качеството на анализите. Лабораториите се акредитират съгласно стандарта ISO/IEC/17025. |
5. Изисквания към процедурите за анализ на диоксини и диоксиноподобни РСВ
Основни изисквания за приемане на процедурите за анализ:
|
— |
Повишена чувствителност и ниски граници на откриване За PCDD и при PCDF границите на откриване на трябва да бъдат от порядъка на един пикограм (10-12 g) ТЕ, поради изключително високата токсичност на някои от тези съединения. Известно е, че РСВ се намират в по-високи концентрации от PCDD и PCDF. За повечето конгенери на РСВ е достатъчна чувствителност в нанограмовия(10-9 g) интервал. Въпреки това за измерването на по-токсичните диоксиноподобни конгенери на РСВ (особено не орто- заместените) е нужно да бъде достигната същата чувствителност на отчитане както при PCDD и PCDF. |
|
— |
Висока селективност (специфичност). Необходимо е PCDD, PCDF и диоксиноподобните PCB да бъдат различавани от множество други съединения, екстрахирани едновременно с тях и евентуално интерфериращи с тях, чиито концентрации са с няколко степени по-високи от тези на търсените аналити. За методите на газова хроматография/масспектрометрия (GC/MS) е необходимо диференциране между различните конгенери, както и между токсичните (например седемнадесетте 2,3,7,8-заместени PCDD и PCDF и диоксиноподобните РСВ) и другите конгенери. Биологичните анализи трябва да позволяват да се определят селективно стойностите на ТЕ като сбор от PCDD, PCDF и диоксиноподобните РСВ. |
|
— |
Висока точност (достоверност и прецизност). Определянето трябва да даде валидна и достоверна оценка за действителната концентрация в пробата. Високата точност (точност на измерването: висока степен на съгласуване между резултата от измерването с истинската или очакваната стойност на измерваната величина) е необходима, за да се избегне отхвърляне на резултата от анализа на пробата поради ниска достоверност на оценката на TE. Точността се изразява като достоверност (разлика между средната стойност, измерена за даден аналит в сертифициран материал и неговата сертифицирана стойност, изразена като процент от тази стойност) и прецизност (RSDR, относително стандартно отклонение, което се изчислява на базата на резултатите, получени при възпроизводими условия). |
Методите за скрининг могат да включват биологични анализи и GC/MS методи; методите за потвърждаване са газовата хроматография с високa разделителна способност/масспектрометрията с висока разделителна способност (HRGC/HRMS).
За общата стойност, изразена в ТЕ, трябва да бъдат изпълнени следните критерии:
|
|
Скринингови методи |
Методи за потвърждаване |
|
Псевдоотрицателни резултати |
< 1 % |
|
|
Достоверност |
|
- 20 % до + 20 % |
|
Прецизност, определена чрез RSDR |
< 30 % |
< 15 % |
6. Специфични изисквания за методите gc/ms, които следва да се спазват за целите на скрининга и потвърждаването
|
— |
За да е валидна аналитичната процедура, още в самото начало или при започването на метода за анализ, например преди етапа на екстракция, трябва да бъдат добавени вътрешни стандарти на 2,3,7,8 xлорзаместени PCDD/F, маркирани с 13С и вътрешни стандарти на диоксиноподобни РСВ, маркирани с 13С. Трябва да се добави най-малко един конгенер за всяка от тетра- до октахлорираните хомоложни групи за PSDD/F и най-малко един конгенер за всяка от хомоложните групи за диоксиноподобните РСВ, (или най-малко един конгенер за всяка избрана йонозаписваща функция при масспектрометрията за мониторинг на PSDD/F и диоксиноподобни РСВ). За предпочитане е, особено при методите за потвърждаване, да се използват всички 17 2,3,7,8-заместени вътрешни стандарти за PSDD/F, маркирани с 13C, и всички 12 вътрешни стандарти за диоксиноподобни РСВ, маркирани с 13C. |
|
— |
Като се използват подходящи калибрационни разтвори, трябва също да бъдат определени относителни фактори на отговор за конгенерите, за които не е добавен маркиран с 13C аналог. |
|
— |
За фуражи от растителен произход и фуражи от животински произход, съдържащи по-малко от 10 % мазнини, е задължително вътрешните стандарти да бъдат добавени преди екстракцията. При фуражи от растителен произход, съдържащи повече от 10 % мазнини, вътрешните стандарти могат да бъдат прибавени както преди етапа на екстракция на мазнините, така и след етапа на екстракция на мазнините. Трябва да се извърши подходяща проверка на ефективността на екстракцията в зависимост от това на кой етап са въведени вътрешните стандарти и в зависимост от това дали резултатите са изразени на база продукт, или на база мазнина. |
|
— |
Преди анализа с GC/MS трябва да бъдат прибавени един или два стандарта за субституция. |
|
— |
Необходим е контрол на възстановяването. При методите за потвърждаване възстановяването на oтделните вътрешни стандарти трябва да бъде в интервала от 60 % до 120 %. Допустими са по-ниска или по-висока степен на възстановяване за отделните конгенери, по-специално за някои хепта- и октахлорирани дибензодиоксини и дибензофурани, при условие че техният принос към стойността на ТЕ не надхвърля 10 % от общата стойност на ТЕ (на базата на сбора от PCDD/F и диоксиноподобните РСВ). При скрининговите методи възстановяването трябва да бъде в интервала от 30 % до 140 %. |
|
— |
Отделянето на диоксините от интерфериращи хлорирани съединения като недиоксиноподобни РСВ и хлорирани дифенилни етери се извършва с подходящи хроматографски техники (за предпочитане в колони с флоризил, диалуминиев триоксид и/или въглен). |
|
— |
Разделянето на изомерите чрез газова хроматография трябва да бъде достатъчно (< 25 % от пик до пик между 1,2,3,4,7,8-HxCDF и 1,2,3,6,7,8-HxCDF). |
|
— |
Определянето се извършва съгласно метод EPA 1613 ревизия Б: Тетра- до октахлорирани диоксини и фурани чрез HRGC/HRMS с изотопно разреждане или друг метод със същите критерии за ефикасност. |
|
— |
Разликата между равнищата на горната и долната граница не трябва да надхвърля 20 % за фуражите, замърсени с диоксин, в интервала до максималната граница или над нея. За фуражи с равнища на замърсяване доста под максималното разликата може да варира в интервал от 25 % до 40 %. |
7. Скринингови аналитични методи
7.1. Въведение
При използване на скринингов метод могат се изпълнят различни аналитични подходи: чисто скринингов подход и количествен подход.
Реакцията по отношение на пробите се сравнява с тази по отношение на еталонна проба на нивото от значение. Пробите, при които реакцията е по-ниска от тази при еталонната проба, се обявяват за отрицателни, а тези, при които реакция е по-висока от тази на сравнителната проба, се смятат за положителни. Изисквания:
|
— |
Във всяка серия опити трябва да бъдат подложени на екстракция и анализ по същото време и при същите условия, както и останалите проби, и една или повече празни и еталонни проби. Реакцията по отношение на еталонната проба трябва да бъде значително по-висока от тази при празната проба. |
|
— |
включват се допълнителни еталонни проби с концентрации съответно равни на умножената по коефициент 0,5 и 2 стойност на ниво от значение, за да се покажат същинските характеристики на анализа в интервала от значение с цел контрол на нивото от значение. |
|
— |
в случай, че се изследват други матрици, пригодността на еталонната/ите проба/и трябва да бъде доказана, за предпочитане чрез включване на проби, при които стойността на ТЕ, установена чрез HRGC/HRMS, е сравнима с тази на сравнителната проба или на празна проба, обогатена така, че бъде достигнато същото равнище. |
|
— |
като се има предвид, че при биологичните анализи не може да бъде използван никакъв вътрешен стандарт, тестовете за повторяемост са от голямо значение за получаването на данни за стандартното отклонение в рамките на една серия опити. Коефициентът на вариация трябва да е по-малък от 30 %. |
|
— |
при биологичните анализи трябва да се дефинират целевите съединения, възможните интерференции и максималните допустими граници в празната проба. |
При количествения подход се изисква използване на серии със стандартно разреждане, двойно и тройно пречистване и измерване, както и контролни анализи на празни проби и анализи за определяне на възстановяването. Резултатът може да се изрази чрез ТЕ, което предполага, че съединенията, предизвикали сигнала, отговарят на принципа на ТЕ. За тази цел може да се използва TCDD (или стандартна смес от диоксини/фурани/диоксиноподобни PCB) за построяване на калибрационната крива, която позволява да се изчисли стойността на ТЕ в екстракта, а следователно и в пробата. След това тя се коригира с равнище на TE, изчислено за празната проба (за да се вземат предвид онечистванията от използваните разтворители и химикали) и за възстановяването (изчислено от равнището на TE на проба за контрол на качеството, което е близко до границата на нивото от значение). Важно е да се отбележи, че част от привидното намаляване на възстановяването може да се дължи на матрични ефекти и/или на разликите в стойностите на КТЕ, използвани при биологичните анализи и официалните стойности, определени от СЗО.
7.2. Изисквания за аналитичните методи, използвани при скрининговите изследвания
|
— |
Скрининговите изследвания могат да се извършат посредством методи за анализ GC/MS и биологични анализи. За методите GC/MS трябва да се изпълнят изискванията, изложени в точка 6. За биологичните анализи върхуклетки се прилагат специфичните изисквания, посочени в т. 7.3, а за готовите комплекти за биологични анализи — в т. 7.4. |
|
— |
Необходимо е да се сравнят данните за броя на псевдоположителните и на псевдоотрицателните резултати на голям брой проби под и над максималното равнище или такива на равнището на предприемане на действие със стойността на ТЕ, определена посредством аналитичен метод за потвърждаване. Действителните псевдоотрицателни резултати трябва да бъдат под 1 %. Процентът на псевдоположителните резултати трябва да бъде достатъчно нисък, за да бъде полезно използването на скрининговия метод. |
|
— |
Положителните резултати трябва винаги да бъдат потвърждавани посредством аналитичен метод за потвърждаване (HRGC/HRMS). Освен това проби, които се намират в широк интервал на ТЕ, трябва да бъдат потвърдени чрез HRGC/HRMS (приблизително 2 %—10 % от отрицателните проби). Трябва да има информация за съответствието между биологичния анализ и резултата от HRGC/HRMS. |
7.3. Специфични изисквания за биологични анализи върху клетки
|
— |
При провеждане на биологичен анализ изпълнението на всеки анализ изисква поредици от еталонни концентрации на TCDD или смес от диоксин/фуран (цялостна крива „доза/реакция“ с R2 > 0,95). За целите на скрининга обаче за анализ на проби с ниско съдържание може да се използва по-подробна крива за ниските концентрации. |
|
— |
за резултатите от биологичния анализ за постоянен период от време се използва еталонна концентрация на TCDD (около 3 пъти стойността на границата на определяне) върху формуляр за контрол на качеството. Може да се използва и относителната реакция по отношение на еталонна проба, сравнена с калибрационната крива на TCDD, като се има предвид, че реакцията на клетките може да зависи от голям брой фактори. |
|
— |
Изготвят се и се проверяват диаграми за контрол на качеството на всеки вид еталонен материал, за да се гарантира, че резултатът съответства на предоставените насоки. |
|
— |
Индукцията на разреждането на използваната проба трябва да е в линейната част на кривата на реакция, особено при количествените изчисления. Пробите, които се намират отвъд тази линейна част, трябва да бъдат разредени и повторно анализирани. Ето защо се препоръчва да се анализират едновременно поне 3 разреждания. |
|
— |
Процентното стандартно отклонение не трябва да бъде повече от 15 % при трикратно определяне за всяко разреждане на пробата и не повече от 30 % между три независими експеримента. |
|
— |
За граница на откриването може да бъде определена стойност, 3 пъти по-голяма от стандартното отклонение на реакцията на чистия разтворител или на фона. Друг подход е приемането на реакция, която е над реакцията на фона (фактор на индукция 5 пъти по-голям от реакцията на чистия разтворител), изчислена въз основа на калибрационната крива за деня. Като граница на количественото определяне може да бъде избрана стойност, от 5 до 6 пъти по-висока от стандартното отклонение на реакцията на чистия разтворител или на тази на фона, или да се възприеме реакция, която е очевидно над фона (фактор на индукция 10 пъти по-голям от реакцията на чистия разтворител), изчислена въз основа на калибрационната крива за деня. |
7.4. Специфични изисквания за готови комплекти за биологични анализи
|
— |
Следва да се гарантира, че готовите комплекти биологичните анализи са достатъчно чувствителни и надеждни, за да бъдат приложени върху фуражи. |
|
— |
Трябва да се спазват инструкциите на производителя за подготовката на пробите и за анализите. |
|
— |
Комплектите за анализи не се използват след изтичането на срока им на годност. |
|
— |
Не се използват материали или компоненти, предвидени за други комплекти. |
|
— |
Температурата на съхранение на комплектите се поддържа в определения температурен интервал и те се използват при посочената работна температура. |
|
— |
Границата на откриване за имуноанализите се определя като сумата от средното и 3 пъти стандартното отклонение, основаващо се на десетократно повторен анализ на празна проба, се дели на стойността на наклона в уравнението на линейната регресия. |
|
— |
Използват се еталонни стандарти за сравнение при лабораторните опити, за да се гарантира, че реакцията по отношение на стандарта попада в допустим интервал. |
8. Отчитане на резултатите
Доколкото използваната аналитична процедура позволява това, в аналитичните резултати трябва да се посочват съдържанията на отделните конгенери на PCDD/F и PCB, и аналитичните резултати да бъдат отчитани като ниски, високи и междинни, за да се включат възможно най-много данни при отчитането на резултатите, което да позволи тъкуване на резултатите в зависимост от специфичните изисквания.
В доклада също така се включва съдържанието на мазнини в пробата, както и методът, използван за екстракция на мазнините.
Нивата на възстановяване на отделните вътрешни стандарти трябва да бъдат съобщени, ако се намират извън интервала, посочен в т. 6, или ако надвишават максималното ниво, а в останалите случаи те трябва да бъдат предоставени при поискване.
Тъй като неопределеността при измерване трябва да бъде отчетена при взимане на решение за годността на дадена проба, този параметър също се съобщава. Така например, аналитичните резултати се отчитат като х +/- U, където х е аналитичният резултат, а U — разширената неопределеност при измерване, като се използва фактор на покриване 2, който дава ниво на сигурност от около 95 %. В случай на разделно определяне на диоксини и на диоксиноподобни PCB, сборът от изчислената разширена неопределеност на отделните резултати от анализа на диоксините и от този на диоксиноподобните PCB трябва да се използва за стойност на сбора за диоксини и диоксиноподобни PCB.
Ако се отчита несигурността на измерването, като се прилага CCα (както е описано в т. I.2 от настоящата част Б), този параметър също се съобщава.
(1) Таблица за КТЕ (= коефициенти на токсична еквивалентност) на диоксините, фураните и диоксиноподобните PCB:
|
Конгенер |
Стойност на КТЕ |
Конгенер |
Стойност на КТЕ |
|
Дибензо-p-диоксини („PCDD“) |
|
Диоксиноподобните PCB (полихлорирани бифенили): |
|
|
2,3,7,8-TCDD |
1 |
|
|
|
1,2,3,7,8-PeCDD |
1 |
PCB, различни от орто PCB) |
|
|
1,2,3,4,7,8-HxCDD |
0,1 |
PCB 77 |
0,0001 |
|
1,2,3,6,7,8-HxCDD |
0,1 |
PCB 81 |
0,0001 |
|
1,2,3,7,8,9-HxCDD |
0,1 |
PCB 126 |
0,1 |
|
1,2,3,4,6,7,8-HpCDD |
0,01 |
PCB 169 |
0,01 |
|
OCDD |
0,0001 |
Моно-орто PCB |
|
|
|
|
PCB 105 |
0,0001 |
|
Дибензофурани („PCDF“) |
|
PCB 114 |
0,0005 |
|
2,3,7,8-TCDF |
0,1 |
PCB 118 |
0,0001 |
|
1,2,3,7,8-PeCDF |
0,05 |
PCB 123 |
0,0001 |
|
2,3,4,7,8-PeCDF |
0,5 |
PCB 156 |
0,0005 |
|
1,2,3,4,7,8-HxCDF |
0,1 |
PCB 157 |
0,0005 |
|
1,2,3,6,7,8-HxCDF |
0,1 |
PCB 167 |
0,00001 |
|
1,2,3,7,8,9-HxCDF |
0,1 |
PCB 189 |
0,0001 |
|
2,3,4,6,7,8-HxCDF |
0,1 |
|
|
|
1,2,3,4,6,7,8-HpCDF |
0,01 |
|
|
|
1,2,3,4,7,8,9-HpCDF |
0,01 |
|
|
|
OCDF |
0,0001 |
|
|
|
Използвани съкращения: „T“ = тетра; „Pe“ = пента; „Hx“ = хекса; „Hp“ = хепта; „О“ = окта; „CDD“ = хлородибензо-p-диоксин; „CDF“ = хлородибензофуран; „CB“ = хлоробифенил. |
|||
(2) ОВ L 140, 30.5.2002 г., стр. 10.
(3) Понятието „горна граница“ налага да се използва стойността на границата на количествено определяне като стойност на дела на всеки конгенер, чието количество не е определено, в КТЕ.
Понятието „долна граница“ налага в КТЕ да се използва нула като стойност на дела на всеки конгенер, чието количество не е определено. Понятието „средно ниво“ налага в КТЕ.
да се използва половината от стойността на границата на количествено определяне като стойност на дела на всеки конгенер, чието количество не е определено.
(4) Повторният анализ е необходим, за да се изключи възможността от вътрешно замърсяване на една проба от друга или от случайно объркване на пробите. Първият анализ се използва за проверка на съответствието с разпоредбите, като се отчита грешката при измерване.
В случай че анализът се провежда в контекста на инцидент, при който има замърсяване с диоксин, потвърждението чрез повторен анализ може да бъде пропуснато, ако избраните за анализ проби могат да бъдат проследени и свързани с инцидента, при който е станало замърсяването.
ПРИЛОЖЕНИЕ VI
МЕТОДИ ЗА АНАЛИЗ ЗА ОПРЕДЕЛЯНЕТО НА СЪСТАВКИ ОТ ЖИВОТИНСКИ ПРОИЗХОД ЗА ЦЕЛИТЕ НА ОФИЦИАЛНИЯ КОНТРОЛ НА ФУРАЖИТЕ
Условия, които се прилагат при откриването, определянето или оценката на съставките от животински произход във фуражите чрез микроскопски анализ
1. Цел и приложно поле
Настоящите условия се прилагат при откриването на съставки от животински произход (определени като продукти от преработени трупове или части от бозайници, птици и риби) във фуражите чрез микроскопско изследване, в рамките на координираната програма за контрол в областта на храненето на животните съгласно Регламент (ЕО) № 882/2004 на Европейския парламент и на Съвета (1). При условие че методите, описани в настоящото приложение, се използват при всички официални анализи, може да бъде извършен и втори анализ с използване на вариант на анализа или друг вид анализ, с цел да се подобри точността при откриването на някои типове съставки от животински произход или да се определи по-точно произходът на съставките. Освен това при изследването на някои специфични съставки от животински произход, като плазмата или костите в лойта (вж. също т. 9) може да се използва и вариант на настоящия протокол, при условие че тези анализи се провеждат в допълнение на анализите, предвидени в координираната програма за контрол.
2. Чувствителност
В зависимост от естеството на съставките от животински произход, във фуражите могат да бъдат откривани много малки количества от тях (< 0,1 %).
3. Принцип
За идентификацията се използва взета в съответствие с разпоредбите, изложени в приложение I, представителна проба, която е била подготвена по подходящ начин. Описаният по-долу протокол е подходящ за третирането на фуражи с ниска влажност. Фуражите с влажност над 14 % се изсушават (кондензират) преди третирането им. Някои специални фуражи или фуражни суровини (напр. мазнини, масла), изискват специално третиране (вж. т. 9). Съставките от животински произход се идентифицират на базата на типични характеристики, които се установяват под микроскоп (а именно мускулни влакна и други месни частици, хрущяли, кости, рога, косми, четина, кръв, пера, черупки от яйца, рибешки кости, люспи). Идентификацията се отнася както за пресятата фракция (т. 6.1), така и за концентрираната утайка (т. 6.2) от пробата.
4. Реагенти
4.1. Фиксатори
|
4.1.1. |
Хлоралхидрат (във воден разтвор, 60 % w/v) |
|
4.1.2. |
Основа (2,5 % w/v NaOH или 2,5 % w/v KOH) за пресетите фракции |
|
4.1.3. |
Парафиново масло или глицерин (вискозитет: 68—81) за изследване под микроскоп на утайката |
4.2. Промиващи средства
|
4.2.1. |
Алкохол, 96 %. |
|
4.2.2. |
Ацетон. |
4.3. Концентриращи средства
|
4.3.1. |
Тетрахлороетилен (плътност 1,62). |
4.4. Оцветяващи реагенти
|
4.4.1. |
Разтвор на йод/калиев йодид (разтварят се 2 g калиев йодид в 100 ml вода и се прибавя 1 g йод при често разклащане). |
|
4.4.2. |
Ализариново червено (в 100 ml вода се разтварят 2,5 ml солна киселина 1М и към този разтвор се прибавят 200 mg ализариново червено). |
|
4.4.3. |
Цистинов реактив (2 g оловен ацетат, 10 g NaOH на 100 ml H2O). |
|
4.4.4. |
Разтвор на йод/калиев йодид (разтворени в 70 % разтвор на етанол). |
4.5. Избелващ реагент
|
4.5.1. |
Готов разтвор на натриев хипохлорид (9,6 % активен хлор). |
5. Апаратура и помощни средства
|
5.1. |
Аналитична везна (точност до 0,01 g, с изключение за концентрираната утайка: 0,001 g). |
|
5.2. |
Инструмент за стриване (мелничка или хаванче, по-специално за фуражи с мастно съдържание над 15 % към момента на анализа). |
|
5.3. |
Сито с квадратни отвори с размер на отвора най-много 0,50 mm. |
|
5.4. |
Делителна фуния или бехерова чаша с конично дъно за утаяване. |
|
5.5. |
Стереомикроскоп (увеличение най-малко 40 пъти). |
|
5.6. |
Съставен микроскоп (увеличение най-малко 400 пъти) с преминаваща или поляризирана светлина. |
|
5.7. |
Стандартна лабораторна стъклария. Цялото оборудване се почиства старателно. Делителните фунии и стъкларията трябва да се измият машинно. Ситата се почистват с помощта на четка с твърд косъм. |
6. Процедура
Гранулираните фуражи могат да бъдат предварително пресяти, ако двете фракции се анализират като отделни образци.
Третират се най-малко 50 g от пробата (стрити старателно, ако е необходимо, с помощта на подходящ инструмент за смилане (т. 5.2), с цел да се получи подходяща структура). От смления материал се вземат две представителни части, едната за пресятата фракция (най-малко 5 g) (т. 6.1), а другата за концентрираната утайка (най-малко 5 g) (т. 6.2). Оцветяването с реагенти (т. 6.3) може да бъде използвано и за идентификационни цели.
С цел да се посочи видът на животинските протеини и произходът на частиците, може да се използва устройство за подпомагане на взимането на решения, като например ARIES, както и еталонни проби.
6.1. Идентификация на съставки от животински произход в пресетите фракции
Пресяват се през сито (т. 5.3) най-малко 5 g от пробата в две фракции.
Пресятата/ите фракция/и, съдържаща/и едрите частици (или представителна част от фракцията), се разстила върху подходящ носител, така че да образува фин слой и методично се изследва под стереомикроскоп (т. 5.5) при различни увеличения, за откриване на съставките от животински произход.
Предметните стъкла с пресятата/ите фракция/и, които съдържат фини частици, се изследват методично под съставен микроскоп (т. 5.6) при различни увеличения, за откриване на съставките от животински произход.
6.2. Идентификация на съставки от животински произход в концентрираната утайка
Най-малко 5 g (с точност до 0,01 g) от пробата се пресипват в делителна фуния или бехерова чаша с конично дъно за утаяване и се обработват с най-малко 50 ml тетрахлоретилен (т. 4.3.1). Сместа се разклаща или разбърква неколкократно.
|
— |
Ако се използва затворена делителна фуния, утайката трябва да се остави да престои достатъчно дълго време, преди да се отдели (най-малко 3 минути). Разклащането се повтаря и се изчаква отново 3 минути, за да се утаи утайката. Утайката се се отделя отново. |
|
— |
Ако се използва отворена бехерова чаша, утайката трябва да се остави да се утаява най-малко 5 минути, преди да се отдели утайката. |
Цялата утайка се изсушава и след това се претегля (с точност до 0,001 g). Претеглянето се извършва само ако се изисква количествена оценка. Ако утайката се състои от много едри частици може да се пресее през сито (т. 5.3) в две фракции. Изсушената утайка се изследва със стереомикроскоп (т. 5.5) и със съставен микроскоп (т. 5.6) за установяване на съставки от кости.
6.3. Използване на фиксатори и оцветяващи реагенти
Идентифицирането чрез микроскоп на съставките от животински произход може да се улесни чрез използването на специални фиксатори и оцветители.
Хлоралхидрат (т. 4.1.1): С помощта на внимателно загряване могат да бъдат видяни по-ясно клетъчните структури, защото зрънцата нишесте желират и нежеланите съставки на клетката се отстраняват.
Основа (т. 4.1.2): натриевият хидроксид или калиевият хидроксид почиства съставките на фуража, като подпомага разпознаването на мускулни влакна, косми и други кератинови структури.
Парафиново масло и глицерин (т. 4.1.3): В тези фиксатори могат да бъдат добре идентифицирани костните съставки, защото повечето пори остават запълнени с въздух и се виждат като черни дупки с големина 5—15 μm.
Разтвор на йод/калиев йодид (т. 4.4.1): използва се за откриване на нишесте (синьо-виолетово оцветяване) и протеин (жълто-оранжево оцветяване). Разтворите могат да бъдат разредени, ако се налага.
Разтвор на ализариново червено (т. 4.4.2): Червено/розово оцветяване на кости, кости от риба и люспи. Преди изсушаването на утайката (вж. раздел 6.2) цялото количество утайка се премества в стъклена епруветка и се изплаква два пъти с около 5 ml алкохол (т. 4.2.1) (всеки път се използва вортекс, разтворителят се оставя да се утаи за около една минута, след което се излива). Преди използването на оцветяващия реагент утайката се избелва чрез добавяне на най-малко 1 ml разтвор на натриев хипохлорид (т. 4.5.1). Реакцията се оставя да продължи 10 минути. Епруветката се допълва с вода, оставя се 2—3 минути, докато утайката се утаи, водата и плуващите в нея частици се отстраняват. Утайката се промива още два пъти с около 10 ml вода (използва се вортекс, изчаква се утаяването, водата се излива всеки път). Добавят се от две до 10 или повече капки (в зависимост от количеството остатък) от разтвора на ализариново червено. Сместа се разклаща и се изчаква няколко секунди, за да се прояви реакцията. Оцветената утайка се промива два пъти с по около 5 ml алкохол (т. 4.2.1) и веднъж с ацетон (т. 4.2.2) (всеки път се използва вортекс, а разтворителят се оставя да се утаи за около една минута, след което се излива). Утайката е готова за изсушаване.
Цистинов реагент (т. 4.4.3): Като се прилага внимателно загряване, съставките, които съдържат цистин (косми, пера и др.) се оцветяват в черно-кафяво.
6.4. Изследване на фуражи, които биха могли да съдържат рибно брашно
Под съставен микроскоп се изследва минимум едно предметно стъкло с пресята фина фракция и с фина фракция на утайката (вж. раздели 6.1 и 6.2)
Ако на етикета е посочено наличие на рибно брашно в съставките или ако има съмнение, или бъде открито наличие на рибно брашно при първоначалния преглед, се изследват най-малко две допълнителни предметни стъкла с пресята фина фракция на оригиналната проба и с фракция от общата утайка.
7. Изчисляване и оценка
Държавите-членки гарантират спазването на процедурите, описани в настоящата точка, при всеки официален анализ, целящ количествена оценка (не само наличие) на съставки от животински произход.
Изчислението може да бъде направено само ако съставките от животински произход включват парчета от кости.
Парчетата от кости от сухоземни топлокръвни видове животни (т.е. бозайници и птици) се различат от различните типове рибни кости върху микроскопско предметно стъкло по типичните пори. Съотношението на отделните съставки от животински произход в пробата се оценява, като се отчита:
|
— |
очакваният дял (тегловни проценти) на костите в концентрираната утайка и |
|
— |
делът (тегловни проценти) на костите в съставките от животински произход. |
Оценката трябва да се базира на изследването на най-малко три предметни стъкла (ако е възможно) и на най-малко пет полета за всяко стъкло. В комбинираните фуражи концентрираната утайка като правило съдържа не само парчета от кости на сухоземни животни и рибни кости, но и други частици с високо специфично тегло, например минерали, пясък, части от вдървесени растения и др.
7.1. Очаквана стойност на процентното съдържание на парчета от кости
Процентно съдържание на парчета кости от сухоземни животни = (S × c)/W
Процентно съдържание на парчета от рибни кости и люспи = (S × d)/W
|
S = |
тегло на утайката (mg), с = коефициент на корекция ( %) за разглежданата част кости от сухоземни животни в утайката, d = коефициент на корекция за разглежданата част парчета от рибни кости и люспи в утайката, W = тегло на материала от пробата, от който произлиза утайката (mg). |
7.2. Очаквана стойност на съставките от животински произход
Делът на костите в продуктите от животински произход може да варира значително. (Процентното съдържание на кости е от порядъка на 50—60 % за костните брашна и от порядъка на 20—30 % за месните брашна; при рибните брашна, съдържанието на кости и люспи варира в зависимост от категорията и произхода на рибното брашно, но обикновено е в рамките на 10—20 %).
Ако е известен типът на брашното в пробата, възможно е да се направят следните оценки за съдържанието:
Очаквано съдържание на съставките от продукти от сухоземни животни ( %) = (S × c)/(W × f) × 100
Очаквано съдържание на съставки от продукти на рибна основа ( %) = (S × d)/(W × f) × 100
|
(S = |
тегло на утайката (mg), с = коефициент на корекция ( %) за разглежданата част кости от сухоземни животни в утайката, d = коефициент на корекция за разглежданата част парчета от рибни кости и люспи в утайката, f = коефициент на корекция за дела на костите в съставките от животински произход в изследваната проба, W = тегло на материала от пробата, от който произлиза утайката (mg). |
8. Изразяване на резултата от изследването
Докладът съдържа най-малкото информация относно наличието на съставки, получени от сухоземни животни и рибно брашно. Отделните случаи се представят по следния начин:
|
8.1. |
По отношение на наличието на съставки, получени от сухоземни животни:
|
|
8.2. |
И, по отношение на наличието на рибни брашна:
Ако бъдат открити съставки, получени от риба или сухоземни животни, при необходимост, докладът от изследванията може да дава количествена оценка на откритите съставки (х %, < 0,1 %, между 0,1 и 0,5 %, между 0,5 и 5 % или > 5 %) и да уточнява типа сухоземни животни (ако е възможно) и идентифицираните съставки от животински произход (мускулни влакна, хрущяли, кости, рога, косми, четина, кръв, пера, яйчни черупки, рибни кости, люспи). Ако се прави количествена оценка на съставките от животински произход, то използваният коефициент на корекция f също се упоменава. Когато се установи наличието на кости от сухоземни животни, в доклада се добавя и следната клауза: „Не се изключва възможността гореизброените съставки да произхождат от бозайници“. Тази допълнителна клауза не е необходима, ако парчетата кости на сухоземни животни са определени като парчета кости от птици или бозайници. |
9. Незадължителен протокол за анализа на мазнини или масла
При анализа на мазнини или масла може да се използва следният протокол:
|
— |
Ако мазнината е твърда, тя трябва да се загрее до втечняването ѝ, например в микровълнова печка. |
|
— |
Взимат се с пипета 40 ml мазнина от дъното на пробата и се прехвърлят в епруветка за центрофугиране. |
|
— |
Пробата се подлага на центрофугиране в продължение на 10 минути при 4 000 rpm. |
|
— |
Ако мазнината се втвърди при центрофугирането, тя се загрява още веднъж в печката, докато отново се върне в течно състояние. Центрофугира се отново в продължение на 5 минути при 4 000 rpm. |
|
— |
С малка лъжичка или шпатула половината от отделените примеси се прехвърлят в малко блюдо на Петри или върху микроскопско предметно стъкло, с цел да се открие под микроскоп евентуалното наличие на съставки от животински произход (мускулни влакна, пера, парчета от кости и др.). Препоръчително е използването на парафиново масло или глицерин като фиксатор за микроскопския анализ. |
|
— |
Останалите примеси се използват за подготвянето на утайка по начина, описан в т. 6.2. |
ПРИЛОЖЕНИЕ VII
МЕТОД ЗА ИЗЧИСЛЯВАНЕ НА ЕНЕРГИЙНАТА СТОЙНОСТ НА ФУРАЖИТЕ ЗА ДОМАШНИ ПТИЦИ
1. Метод за изчисляване и изразяване на енергийната стойност
Енергийната стойност на комбинираните фуражи за домашни птици трябва да бъде изчислявана в съответствие с посочената по-долу формула въз основа на процентното съдържание на определени аналитични компоненти във фуража. Тази стойност трябва да бъде изразена в мегаджаули (MJ) обменна енергия (ОЕ) с коригиран азот, на килограм от хранителната смес:
MJ/kg ОЕ = 0,1551 × % суров протеин +0,3431 × % сурови мазнини +0,1669 × % нишесте +0,1301 × % общо захари (изразени като захароза)
2. Допустими отклонения от декларираните стойности
Ако официалната проверка установи несъответствие (по-висока или по-ниска енергийна стойност на фуража), позволеното минимално отклонение между резултата от проверката и декларираната енергийна стойност е 0,4 MJ/kg ОЕ.
3. Изразяване на резултата
След прилагането на горепосочената формула, полученият резултат трябва да бъде отчетен до първия знак след десетичната запетая.
4. Методи за вземане на проби и анализ
Вземането на проби от комбинирания фураж и определянето на съдържанието на компонентите за анализ, посочени в метода за изчисление, трябва да бъде извършено в съответствие с общностните методи за вземане на проби и за анализ, използвани при официалния контрол на фуражите.
Трябва да се прилагат следните методи:
|
— |
за определяне на съдържанието на сурови мазнини: процедура Б от метода за определяне на сурови масла и мазнини, изложена в част З от приложение III. |
|
— |
за определяне на съдържанието на нишесте: полариметричния метод, изложен в част Л от приложение III. |
ПРИЛОЖЕНИЕ VIII
МЕТОДИ ЗА АНАЛИЗ С ЦЕЛ КОНТРОЛ НА НЕЗАКОННОТО НАЛИЧИЕ ВЪВ ФУРАЖИТЕ НА ДОБАВКИ, ЧИЕТО ПОЛЗВАНЕ ВЕЧЕ НЕ Е РАЗРЕШЕНО
Важно указание:За откриване на незаконно наличие във фуражите на добавки, чието ползване вече не е разрешено, могат да се използват методи за анализ, по-чувствителни от упоменатите в настоящото приложение методиМетодите за анализ, упоменати в настоящото приложение, се използват за потвърждаване
А. ОПРЕДЕЛЯНЕ НА METHYL BENZOQUATE
(7-бензокси-6-бутил-3-метоксикарбонил-4-хинолон)
1. Цел и обхват
Настоящият метод позволява определянето на съдържанието на methyl benzoquate във фуражите. Прагът на количествено определяне е 1 mg/kg.
2. Принцип
Methyl benzoquate се екстрахира от пробата с помощта на разтвор на метанол и метансулфонова киселина. Екстрактът се пречиства с дихлорметан чрез йоннообменна хроматография и след това отново с дихлорометан. Съдържанието на methyl benzoquate се определя чрез високоефективна течна хроматография (HPLC) с обратна фаза с помощта на UV-детектор.
3. Реагенти
|
3.1. |
Дихлорметан |
|
3.2. |
Метанол, с чистота за HPLC |
|
3.3. |
Подвижна фаза HPLC Смес от метанол (т. 3.2) и вода (с чистота за HPLC) 75 + 25 (v + v). Разтворът се филтрира през филтър с размер на порите 0,22 μm (т. 4.5) и се обезгазява (напр. чрез подлагане на въздействието на ултразвук в продължение на 10 минути). |
|
3.4. |
Разтвор на метансулфонова киселина, c = 2 % Разреждат се с метанолът 20,0 ml метансулфонова киселина до 1 000 ml (т. 3.2). |
|
3.5. |
Разтвор на солна киселина, с = 10 %: Разреждат се с вода 100 ml солна киселина (ρ201,18 g/ml) до 1 000 ml. |
|
3.6. |
Катионнообменна смола Amberlite CG-120 (Na), меш 100 до 200 Смолата се подлага на предварителна подготовка: суспендират се 100 g от смолата в 500 ml разтвор на солна киселина (т. 3.5), сместа се нагрява на нагревателна плоча до завиране, като непрекъснато се разбърква. Оставя се да се охлади и киселината се източва. Прецежда се през филтърна хартия във вакуум. Смолата се промива два пъти с по 500 ml вода, а след това с 250 ml метанол (т. 3.2). Смолата се промива с още 250 ml метанол и се изсушава, като се пуска въздух през филтрата. Изсушената смола се съхранява в запушено шише. |
|
3.7. |
Стандартно вещество: чист methyl benzoquate (7-benzyloxy-6-butyl-3-methoxycarbonyl-4-quinolone) |
|
3.7.1. |
Отмерерват се 50 mg от стандартното вещество (т. 3.7) с точност до 0,1 mg и се разтварят в разтвор на метаносулфонова киселина (т. 3.4) в градуирана колба от 100 ml, допълва се до пълния обем и се разбърква. |
|
3.7.2. |
Прехвърлят се 5,0 ml от основния стандартен разтвор на methyl benzoquate (т. 3.7.1) в градуирана колба от 50 ml, допълва се до пълния обем с метанол (т. 3.2) и се разбърква. |
|
3.7.3. |
Прехвърлят се 1,0, 2,0, 3,0, 4,0 и 5,0 ml от междинния стандартен разтвор на methyl benzoquate (т. 3.7.2) в няколко градуирани колби от 25 ml. Допълва се до пълния обем с подвижната фаза (т. 3.3) и се разбърква. Концентрацията на methyl benzoquate в тези разтвори е съответно 2,0, 4,0, 6,0, 8,0 и 10,0 μg/ml. Разтворите трябва да се приготвят непосредствено преди употреба. |
4. Апаратура
|
4.1. |
Лабораторна клатачна машина |
|
4.2. |
Ротационен тънкослоен изпарител |
|
4.3. |
Стъклена колона (250 mm × 15 mm), снабдена със запорен кран и резервоар с капацитет от приблизително 200 ml. |
|
4.4. |
Апарат за HPLC с UV-детектор с настройка на дължината на вълната или с детектор с диодна матрица |
|
4.4.1. |
Колона за течна хроматаграфия: 300 mm × 4 mm, С18, пълнеж от 10 μm, или еквивалентна |
|
4.5. |
Мембранни филтри, 0,22 μm. |
|
4.6. |
Мембранни филтри, 0,45 μm. |
5. Процедура
5.1. Общи положения
|
5.1.1. |
Анализира се празна фуражна проба с цел да се провери отсъствието както на methyl benzoquate, така и на интерфериращи вещества. |
|
5.1.2. |
Провежда се тест за възстановяване, като се анализира празната проба, която предварително е била обогатена чрез прибавяне на допълнително количество methyl benzoquate, сходно с количеството в пробата. За да се обогати фуражът до равнище 15 mg/kg, се добавят 600 μl от основния стандартен разтвор (т. 3.7.1) към 20 g от фуража без добавки, смесва се и се изчаква 10 min, преди да се премине към етапа на екстракция (т. 5.2). Следва да се отбележи, че за целите на метода празната проба трябва да бъде сходна по вид с изследваната проба и при анализа ѝ не трябва да се открие наличие на methyl benzoquate. |
5.2. Екстракция
Отмерват се приблизително 20 g от приготвената проба с точност до 0,01 g и се прехвърлят в конична колба от 250 ml. Прибавят се 100 ml разтвор на метансулфонова киселина (т. 3.4) и се разклаща с механично приспособление (т. 4.1) в продължение на 30 min. Филтрира се през филтърна хартия и филтратът се запазва за етапа на разделяне течност-течност (т. 5.3).
5.3. Разделяне течност-течност
В делителна фуния от 500 ml, съдъражаща 100 ml разтвор на солна киселина (т. 3.5), се прехвърлят 25,0 ml от филтрата, получен съгласно процедурата в т. 5.2. Прибавят се 100 ml дихлорметан (т. 3.1) във фунията и се разклаща в продължение на около една минута. Оставят се пластовете да се разделят и се източва долният (дихлорметанов) пласт в облодънна колба от 500 ml. Екстракцията на водната фаза се повтаря с още две дози от 40 ml дихлорметан и тези екстракти се смесват с първия екстракт в облодънната колба. Дихлорметановият екстракт се подлага на изпаряване до сухо в ротационния тънкослоен изпарител (т. 4.2) при понижено налягане и температура от 40 oC. Остатъкът се разтваря в 20—25 ml метанол (т. 3.2), колбата се запушва и се запазва целият екстракт за йоннообменната хроматография (т. 5.4).
5.4. Йоннообменна хроматография
5.4.1.
Запушва се долният край на стъклената колона (т. 4.3) с тапа от стъклен памук. Приготвя се суспензия с 5,0 g от обработената катионнообменна смола (т. 3.6) и 50 ml солна киселина (т. 3.5), налива се в стъклената колона и се оставя да се утаи. Източва се излишното количество киселина до нивото на повърхността на смолата и колоната се промива с вода, докато източваната течност не покаже неутрална реакция спрямо лакмус. Прехвърлят се 50 ml метанол (т. 3.2) в колоната и се оставя да се отцеди до нивото на повърхността на смолата.
5.4.2.
С помощта на пипета внимателно се прехвърля в колоната екстрактът, получен в съотвтствие с процедурата в т. 5.3. Облодънната колба се изплаква с две дози (5—10 ml) метанол (т. 3.2) и течността от изплакванията се прехвърля в колоната. Екстрактът се източва до нивото на смолата и колоната се промива с 50 ml метанол, като скоростта на изтичане е не по-голяма от 5 ml/min. Източеното количество се изхвърля. Като се използват 150 ml разтвор на метансулфонова киселина (т. 3.4), се елуира methyl benzoquate от колоната, а елуатът от колоната се поставя в конична колба от 250 ml.
5.5. Разделяне течност-течност
Елуатът, получен с процедурата в т. 5.4.2, се прехвърля в делителна фуния от 1 l. Коничната колба се изплаква с 5—10 ml метанол (т. 3.2) и течността от изплакванията се смесва със съдържанието на делителната фуния. Прибавят се 300 ml разтвор на солна киселина (т. 3.5) и 130 ml дихлорметан (т. 3.1). Разклаща се за около 1 min и се оставя фазите да се разделят. Долният (дихлорметанов) слой се източва в облодънна колба от 500 ml. Екстракцията на водната фаза се повтаря с още две дози по 70 ml дихлорметан и новополучените екстракти се смесват с първия екстракт в облодънната колба.
Дихлорметановият екстракт се подлага на изпаряване до сухо в ротационния тънкослоен изпарител (т. 4.2) при понижено налягане и температура от 40 oC. Остатъкът в колбата се разтваря в 5 ml метанол (т. 3.2) и този разтвор се прехвърля количествено в градуирана колба от 10 ml. Облодънната колба се изплаква с още две дози метанол (1—2 ml) и съдържанието се прехвърля в градуираната колба. Допълва се с метанол до пълния обем и се разбърква. Една аликвотна част се филтрира през мембранен филтър (т. 4.6). Разтворът се запазва за определяне с HPLC (т. 5.6).
5.6. Определяне с помощта на HPLC
5.6.1.
Дават се следните указания за условията за провеждане на анализа. Могат да бъдат използвани и други условия, ако дават равностойни резултати:
|
— |
колона за течна хроматография (т. 4.4.1); |
|
— |
подвижна фаза за HPLC: смес от метанол и вода (т. 3.3), |
|
— |
скорост на изтичане: 1 до 1,5 ml/min, |
|
— |
дължина на вълната за откриване: 265 nm |
|
— |
обем за впръскване: от 20 до 50 μl. |
Проверява се стабилността на хроматографичната система, като на няколко пъти се впръсква калибрационният разтвор (т. 3.7.3), съдържащ 4 μg/ml, докато се постигнат постоянни височини на пика и постоянни времена на задържане.
5.6.2.
Всеки от калибрационните разтвори (3.7.3) се впръсква няколко пъти и се измерват височините (площите) на пика за всяка концентрация. Построява се калибрационна крива, като за ординати се използват средните височини на пиковете (площите) на калибрационните разтвори, а за абсциси — съответните концентрации в μg/ml.
5.6.3.
Разтворът на пробата (т. 5.5) се впръсква няколко пъти, като се използва същият обем като този за калибрационните разтвори и се определя средната височина (площ) на пиковете на methyl benzoquate.
6. Изчисляване на резултатите
Чрез сравнение с калибрационната крива (т. 5.6.2) и като се използва средната височина (площ) на пиковете на methyl benzoquate за разтвора на пробата, се определя концентрацията на разтвора на пробата в μg/ml.
Съдържанието на methyl benzoquate w (mg/kg) в пробата се изчислява по следната формула:

където:
|
c |
= |
концентрация на methyl benzoquate в разтвора на пробата в μg/ml, |
|
m |
= |
тегло на частта от пробата за анализ в грамове. |
7. Потвърждаване на резултатите
7.1. Идентичност
Идентичността на аналита може да се потвърди чрез кохроматография или чрез използване на детектор с диодна матрица, с помощта на който могат да се сравнят спекрите на екстракта на пробата и на калибрационния разтвор (т. 3.7.3), съдържащ 10 μg/ml.
7.1.1.
Екстрактът от пробата се обогатява, като към него се прибави подходящо количество от междинния стандартен разтвор (т. 3.7.2). Количеството добавен methyl benzoquate трябва да бъде сходно с количество methyl benzoquate, което се предполага, че се съдържа в екстракта от пробата.
Увеличава се само височината на пика на methyl benzoquate, след като се вземат предвид както добавеното количество, така и степента на разреждане на екстракта. Широчината на пика на половината от неговата максимална височина трябва да бъде в рамките на около 10 % от първоначалната широчина.
7.1.2.
Резултатите се оценяват според следните критерии:
|
а) |
дължината на вълната на максимална абсорбция на пробата трябва да съвпада с тази на спектрите на стандарта, регистрирана в най-високата точка на хроматограмата и при отконение, определено от разделителната способност на детектиращата система. При детектиране с диодна матрица, отклонението обикновено е в рамките на 2 nm; |
|
б) |
в диапазона между 220 и 350 nm спектрите на пробата и на стандарта, отчетени в най-високата точка на хроматограмата, не трябва да се различават помежду си в тези части от спектъра в интервала между 10 до 100 % относителна екстинкция. Критерият е изпълнен тогава, когато са налице едни и същи максимуми и не е регистрирана точка, в която отклонението между двата спектъра да превишава 15 % от екстинцията на стандартния аналит; |
|
в) |
между 220 и 350 nm спектрите на възходящата линия, максимума и низходящата линия на пика, получен при анализа на екстракта на пробата, не трябва да се различават един от друг в тези части от спектъра, които попадат в интервала между 10 до 100 % относителна екстинкция. Критерият е изпълнен тогава, когато са налице същите максимуми и когато в нито една регистрирана точка отклонението между спектрите не надвишава 15 % от екстинкцията на спектъра на най-високата точка. |
Ако някой от критериите не е изпълнен, наличието на аналита не се потвърждава.
7.2. Повторяемост
Разликата между резултатите от две паралелни определяния, проведени върху една и съща проба, не трябва да превишава: 10 % спрямо по-високата стойност за съдържание на methyl benzoquate между 4 и 20 mg/kg.
7.3. Възстановяване
Възстановяването за обогатена празна проба трябва да бъде най-малко 90 %.
8. Резултати от съвместно изследване
Пет проби бяха подложени на анализ в 10 лаборатории. За всяка проба са проведени по два анализа.
|
|
Без добавки |
Брашно 1 |
Пелети 1 |
Брашно 2 |
Пелети 2 |
|
Среднa стойност [mg/kg] |
НО |
4,50 |
4,50 |
8,90 |
8,70 |
|
sr [mg/kg] |
— |
0,30 |
0,20 |
0,60 |
0,50 |
|
CVr [ %] |
— |
6,70 |
4,40 |
6,70 |
5,70 |
|
sR [mg/kg] |
— |
0,40 |
0,50 |
0,90 |
1,00 |
|
CVR [ %] |
— |
8,90 |
11,10 |
10,10 |
11,50 |
|
Възстановяване ( %) |
— |
92,00 |
93,00 |
92,00 |
89,00 |
|
НО |
= |
не е открито |
|
sr |
= |
стандартно отклонение на повторяемостта |
|
CVr |
= |
коефициент на вариация на повторяемостта, % |
|
sR |
= |
стандартно отклонение на възпроизводимостта |
|
CVR |
= |
коефициент на вариация на възпроизводимостта, % |
Б. ОПРЕДЕЛЯНЕ НА ОЛАКВИНДОКС
2-[N-2'-(хидроксиетил)карбамоил]-3-метилхиноксалин-N 1 ,N 4 -диоксид
1. Цел и обхват
Настоящият метод позволява определянето на съдържанието на олавкиндокс във фуражите. Прагът на количествено определяне е 5 mg/kg.
2. Принцип
Пробата се подлага на екстракция със смес от метанол и вода. Съдържанието на олквиндокс се определя с помощта на високоефективна течна хроматография с обратна фаза (HPLC), като се използва UV-детектор.
3. Реагенти
|
3.1. |
Метанол |
|
3.2. |
Метанол, с чистота за HPLC |
|
3.3. |
Вода, с чистота за HPLC |
|
3.4. |
Подвижна фаза за HPLC Смес от вода (т. 3.3) и метанол (т. 3.2), 900 +100 (V + V). |
|
3.5. |
Стандартно вещество: чист олаквиндокс 2-[N-2'-(хидроксиетил)карбамоил]-3-метилхиноксалин-N1,N4-диоксид, Е 851. |
|
3.5.1. |
Претеглят се с точност до 0,1 mg 50 mg олаквиндокс (т. 3.5) в градуирана колба от 200 ml и се добавят 190 ml вода. След това колбата се поставя за 20 min в ултразвукова вана (т. 4.1). След обработката с ултразвук, разтворът се охлажда до стайна температура, допълва се с вода до пълния обем и се разбърква. Колбата се завива в алуминиево фолио и се съхранява в хладилник. Този разтвор трябва да се приготвя ежемесечно. |
|
3.5.2. |
Прехвърлят се 10,0 ml от основния стандартен разтвор (т. 3.5.1) в градуирана колба от 100 ml, долива се по пълния обем с подвижната фаза (т. 3.4) и се разбърква. Колбата се завива в алуминиево фолио и се съхранява в хладилник. Този разтвор трябва да се приготвя ежедневно. |
|
3.5.3. |
В поредица градуирани колби от 50 ml се прехвърлят 1,0, 2,0, 5,0, 10,0, 15,0 и 20,0 ml от междинния стандартен разтвор (т. 3.5.2). Колбите се допълват до пълния обем с подвижната фаза (т. 3.4) и съдържанието им се разбърква. Колбите се завиват в алуминиево фолио. Тези разтвори отговарят съответно на 0,5, 1,0, 2,5, 5,0, 7,5 и 10,0 μg олаквиндокс на ml. Тези разтвори трябва да се приготвят ежедневно. |
4. Апаратура
|
4.1. |
Ултразвукова баня |
|
4.2. |
Механична клатачна машина |
|
4.3. |
Оборудване за HPLC с UV-детектор с настройка на дължината на вълната или детектор с диодна матрица |
|
4.3.1. |
Колона за течна хроматография, 250 mm × 4 mm, С18, пълнеж от 10 μm, или еквивалентна |
|
4.4. |
Мембранни филтри, 0,45 μm. |
5. Процедура
|
Забележка |
: |
Олаквиндоксът е чувствителен към светлина. Всички процедури се извършват при приглушена светлина или се използва лабораторна стъклария от тъмно стъкло. |
5.1. Общи положения
|
5.1.1. |
Анализира се проба от фураж без добавки, за да се провери дали в нея няма олаквиндокс или други интерфериращи вещества. |
|
5.1.2. |
Следва да се проведе се тест за възстановяване, като се анализира празната проба, обогатена чрез добавянето на известно количество олаквиндокс, сходно с количество, което се намира в анализираната проба. За да се обогати пробата до равнище 50 mg/kg, 10 ml от основния стандартен разтвор (т. 3.5.1) се прехвърлят в конична колба от 250 ml и се подлагат на изпарение, докато количеството на разтвора намалее до около 0,5 ml. Добавят се 50 g от фуража без добавки, разбърква се старателно и се оставя за 10 min, като се разбърква неколкократно, преди да се премине към етапа на екстракция (т. 5.2).
|
5.2. Екстракция
Претеглят се с точност 0,01 g приблизително 50 g от пробата. Прехвърлят се в коничнаа колба от 1 000 ml, добавят се 100 ml метанол (т. 3.1) и колбата се поставя в ултарзвукова баня (т. 4.1) за 5 минути. Добавят се 410 ml вода и се оставя в ултразвуковата вана за още 15 min. Колбата се изважда от ултразвуковата вана, разклаща се в продължение на 30 минути с клатачната машина (т. 4.2) и се филтрира през нагънат филтър. Прехвърлят се 10,0 ml от филтрата в градуирана колба от 20 ml, долива се вода до пълния обем и се разбърква. Една аликвотна част се филтрираа през мембранен филтър (т. 4.4). (вж. забележката в т. 9) Преминава се към определяне с помощта на HPLC (т. 5.3).
5.3. Определяне с помощта на HPLC
5.3.1.
Следните условия са дадени като указание; може да се използват и други, при условие, че дават равностойни резултати:
|
Аналитична колона (т. 4.3.1): |
|
|
Подвижна фаза (т. 3.4): |
смес от вода (т. 3.3) и метанол (т. 3.2), 900 + 100 (V + V) |
|
Скорост на изтичане: |
1,5—2 ml/min; |
|
Дължина на вълната за откриване: |
380 nm |
|
Обем на впръскване: |
20—100 μl. |
Проверява се стабилността на хроматографската система като няколко пъти се впръсква калибрационен разтвор (т. 3.5.3), в който се съдържат 2,5 μg/ml, докато се постигнат постоянни височини на пиковете и постоянни времена на задържане.
5.3.2.
Впръсква се от всеки калибрационен разтвор (3.5.3) по няколко пъти и се определят средните пикови височини (площи) за всяка концентрация. Построява се калибрационна крива, като средните височини на пиковете (площите) на калибрационните разтвори се използват за ординати, а съответстващите им концентрации в μg/ml за абсциси.
5.3.3.
Екстрактът от пробата (т. 5.2) се впръсква няколко пъти, като се използва същият обем, както за калибриращите разтвори, и се определя средната височина (площта) на пиковете на олаквиндокс.
6. Изчисляване на резултатите
От средната височина (площ) на пиковете на олаквиндокс в разтвора на пробата чрез сравняване с калибрационната крива (т. 5.3.2) се определя концентрацията на разтвора на пробата в μg/ml.
Съдържанието на олаквиндокс w в mg/kg в пробата се определя по формулата:

където:
|
c |
= |
концентрация на олаквиндокс в екстракта на пробата (т. 5.2) в μg/ml |
|
m |
= |
тегло в g на частта от пробата за анализ (т. 5.2) |
7. Потвърждаване на резултатите
7.1. Идентичност
Идентичността на аналита може да се потвърди с помощта на кохроматография или като се използва детектор с диодна матрица, с помощта на които се сравняват спектрите на екстракта на пробата (т. 5.2) и на калибрационния разтвор (т. 3.5.3), съдържащ 5,0 μg/ml.
7.1.1.
Обогатява се екстракт от пробата (т. 5.2), като се добавя подходящо количество калибрационен разтвор (т. 3.5.3). Количество добавен олаквиндокс трябва да бъде сходно с количеството олаквиндокс в екстракта от пробата.
Увеличава се само височината на пика на олаквиндокс след като се вземат предвид както добавеното количество, така и степента на разреждане на екстракта от пробата. Широчината на пика на по средата на височината му трябва да бъде в рамките на ± 10 % от първоначалната широчина на пика на олаквиндокс в необогатения екстракт на пробата.
7.1.2.
Резултатите се оценяват според следните критерии:
|
а) |
Дължината на вълната на максимална абсорбция на пробата трябва да бъде същата като тази на спектрите на стандарта, регистрирана в най-високата точка на хроматограмата и при отконение, определено от разделителната способност на детектиращата система. За детекция с диодна матрица отклонението обикновено е в рамките на ± 2 nm. |
|
б) |
Между 220 и 400 nm спектрите на пробата и на стандарта, отчетени в най-високата точка на хроматограмата, не трябва да се различават помежду си за онези части от спектъра, които попадат в интервала между 10 до 100 % относителна екстинкция. Критерият е изпълнен тогава, когато са налице едни и същи максимуми и не е регистрирана точка, в която отклонението между двата спектъра превишава 15 % от екстинцията на стандартния аналит; |
|
в) |
между 220 и 400 nm спектрите на възходящата линия, максимума и низходящата линия на пика, получен при анализа на екстракта на пробата, не трябва да се различават един от друг в онези части от спектъра, които попадат в интервала между 10 до 100 % относителна екстинкция. Критерият е изпълнен тогава, когато са налице едни и същи максимуми и когато във всички регистрирани точки отклонението между спектрите не надминава 15 % от екстинкцията на спектъра на най-високата точка. |
Ако някой от критериите не е изпълнен, наличието на аналита не се потвърждава.
7.2. Повторяемост
Разликата между резултатите от две паралелни определяния, извършени върху една и съща проба, не трябва да надвишава 15 % спрямо по-високия резултат за съдържание на олаквиндокс между 10 и 200 mg/kg.
7.3. Възстановяване
Възстановяването за обогатена празна проба трябва да бъде най-малко 90 %.
8. Резултати от съвместно изследване
Организирано бе съвместно изследване в ЕО, при което 13 лаборатории анализираха четири проби от фураж за прасенца, включително една проба с необогатен фураж. Резултатите са представени по-долу:
|
|
Проба 1 |
Проба 2 |
Проба 3 |
Проба 4 |
|
L |
13 |
10 |
11 |
11 |
|
n |
40 |
40 |
44 |
44 |
|
Средна стойност в [mg/kg] |
— |
14,6 |
48,0 |
95,4 |
|
sr [mg/kg] |
— |
0,82 |
2,05 |
6,36 |
|
SR [mg/kg] |
— |
1,62 |
4,28 |
8,42 |
|
CVr [ %] |
— |
5,6 |
4,3 |
6,7 |
|
CVR [ %] |
— |
11,1 |
8,9 |
8,8 |
|
Номинално съдържание |
|
|
|
|
|
[mg/kg] |
— |
15 |
50 |
100 |
|
Възстановяване % |
— |
97,3 |
96,0 |
95,4 |
|
L |
= |
брой на лабораториите |
|
n |
= |
брой на единичните стойности |
|
Sr |
= |
стандартно отклонение на повторяемостта |
|
SR |
= |
стандартно отклонение на възпроизводимостта |
|
CVr |
= |
коефициент на вариация на повторяемостта |
|
CVR |
= |
коефициент на вариация на възпроизводимостта. |
9. Забележки
Въпреки че методът още не проверен за фуражи със съдържание на олаквиндокс над 100 mg/kg, възможно е да се постигнат задоволителни резултати, като се вземе проба с по-малко тегло и/или екстрактът (т. 5.2) се разтвори до постигане на концентрация в рамките на диапазона на калибрационната крива (т. 5.3.2).
В. ОПРЕДЕЛЯНЕ НА АМПРОЛИУМ
1-[(4-амино-2-пропилпиримидин-5-ил)метил]-2-метил-пиридин хлорид хидрохлорид
1. Цел и обхват
Настоящият метод дава възможност за определяне на съдържанието на ампролиум във фуражи и премикси. Границата на откриване е 1 mg/kg, а границата на количествено определяне е 5 mg/kg.
2. Принцип
Пробата се подлага на екстракция със смес от метанол и вода. След разреждане с подвижната фаза и филтрация с мембранен филтър, съдържанието на ампролиум се определя чрез високоефективна течна хроматография (HPLC) с катионен обмен, с използване на UV детектор.
3. Реагенти
|
3.1. |
Метанол. |
|
3.2. |
Ацетонитрил, с чистота за HPLC. |
|
3.3. |
Вода, с чистота за HPLC. |
|
3.4. |
Разтвор на натриев дихидроген фосфат, с = 0,1 mol/l. Разтварят се във вода (т. 3.3) 13,80 g натриев дихидроген фосфат монохидрат в градуирана колба от 1 000 ml, допълва се до пълния обем с вода (т. 3.3) и се разбърква. |
|
3.5. |
Разтвор на натриев перхлорат, с = 1,6 mol/l Разтварят се 224,74 g натриев перхлорат монохидрат във вода (т. 3.3) в градуирана колба от 1 000 ml, допълва се с вода (т. 3.3) до пълния обем и се разбърква. |
|
3.6. |
Подвижна фаза за HPLC (вж. забележка 9.1). Смес от ацетонитрил (т. 3.2), разтвор на натриев дихидроген фосфат (т. 3.4) и разтвор на натриев перхлорат (т. 3.5), 450 + 450 + 100 (v + v + v). Преди употреба, разтворът се филтрира през мембранен филтър от 0,22 μm (т. 4.3) и обезгазява (напр.в ултразвукова вана (т. 4.4) в продължение най-малко на 15 минути). |
|
3.7. |
Стандартно вещество: чист ампролиум, 1-[(4-амино-2-пропилпиримидин-5-ил)метил]-2-метил-пиридин хлорид хидрохлорид, Е 750 (вж. 9.2). |
|
3.7.1. |
Претеглят се 50 mg ампролиум (т. 3.7) с точност до 0,1 mg в градуирана колба от 100 ml, това количество се разтваря в 80 ml метанол (т. 3.1) и колбата се поставя в ултразвукова вана (т. 4.4) за 10 минути. След обработката с ултразвук, разтворът се охлажда до стайна температура, допълва се с вода до пълния обем и се разбърква. При температура ≤ 4 oС разтворът е стабилен в продължение на 1 месец. |
|
3.7.2. |
Отмерват се с пипета 5,0 ml от основния стандартен разтвор (т. 3.7.1) в градуирана колба от 50 ml, допълва се до пълния обем с екстракционен разтворител (т. 3.8) и се разбърква. При температура ≤ 4 oС разтворът е стабилен в продължение на 1 месец. |
|
3.7.3. |
Прехвърлят се съответно 0,5, 1,0 и 2,0 ml от междинния стандартен разтвор (т. 3.7.2) в поредица от градуирани колби от 50 ml. Колбите се допълват до пълния обем с подвижната фаза (т. 3.6) и съдържанието им се разбърква. Тези разтвори отговарят съответно на 0,5, 1,0 и 2 μg ампролиум на ml. Разтворите трябва да бъдат приготвени непосредствено преди употреба. |
|
3.8. |
Екстракционен разтворител Смес от метанол (т. 3.1) и вода, 2 + 1 (v + v). |
4. Апаратура
|
4.1. |
Оборудване за HPLC със система за впръскване, подходяща за впръскване на обеми от 100 μl. |
|
4.1.1. |
Колона за течна хроматография 125 mm × 4 mm, пълна с Nucleosil 10 SA за катионен обмен от 5 или 10 μm, или еквивалентна. |
|
4.1.2. |
UV детектор с настройка дължината на вълната или детектор с диодна матрица. |
|
4.2. |
Мембранен филтър от материал PTFE, 0,45 μm. |
|
4.3. |
Мембранен филтър, 0,22 μm. |
|
4.4. |
Ултразвукова вана. |
|
4.5. |
Механична клатачна машина или магнитна бъркалка. |
5. Процедура
5.1. Общи положения
5.1.1.
За изпълнението на теста за възстановяване (т. 5.1.2), се анализира празна проба фураж, за да се провери за наличие на ампролиум и интерфериращи вещества. Фуражът от празната проба трябва да е подобен по вид на този от изследваната проба и в него не трябва да бъдат открити ампролиум или интерфериращи вещества.
5.1.2
Извършва се тест за възстановяване, като се анализира пробата от фураж без добавки, обогатена чрез прибавяне на известно количество ампролиум, сходно с наличното в изследваната проба. За да се обогати пробата до равнище 100 mg/kg, 10,0 ml от основния стандартен разтвор (т. 3.7.1) се прехвърлят в конична колба от 250 ml и се подлагат на изпарение, докато количеството на разтвора намалее до около 0,5 ml. Добавят се 50 g от фуража без добавки, разбърква се старателно и се оставя за 10 min, като се разбърква неколкократно, преди да се премине към етапа на екстракция (т. 5.2).
Като алтернатива, ако не е наличен фураж, подобен на този, от който е взета пробата (вж. т. 5.1.1), тестът за възстановяване може да се извърши чрез стандартия метод на добавяне. В този случай пробата, която ще се анализира, се обогатява с такова количество ампролиум, което е близко до вече съдържащото се в пробата. Тази проба се анализира заедно с необогатената проба, като възстановяването се пресмята чрез изваждане.
5.2. Екстракция
5.2.1.
Претеглят се с точност до 0,01 mg от 5 g до 40 g от пробата, в зависимост от съдържанието на ампролиум, в конична колба от 500 ml, и се добавят 200 ml екстракционен разтворител (т. 3.8). Колбата се поставя в ултразвукова вана (т. 4.4) за 15 min. Колбата се изважда от ултразвуковата вана, разклаща се в продължение на 1 час с клатачна машина или се разбърква с магнитна бъркалка (т. 4.5). Една аликвотна част от екстракта се разрежда с подвижната фаза (т. 3.6) до получаване на съдържание на ампролиум от 0,5 до 2 μg/ml и се разбърква (вж. забележка 9.3). Филтрират се 5 до 10 ml от този разреден разтвор през мембранен филтър (т. 4.2). Пристъпва се към определяне чрез HPLC (т. 5.3).
5.2.2.
Претеглят се точност до 0,001 g от 1 до 4 g премикс, в зависимост от съдържанието на ампролиум, в конична колба от 500 ml, и се добавят 200 ml екстракционен разтворител (т. 3.8). Колбата се поставя в ултразвукова вана (т. 4.4) за 15 min. Колбата се изважда от ултразвуковата вана, разклаща се в продължение на 1 час с клатачна машина или се разбърква с магнитна бъркалка (т. 4.5). Една аликвотна част от екстракта се разрежда с подвижната фаза (т. 3.6) до получаване на съдържание на ампролиум от 0,5 до 2 μг/ml и се разбърква. Филтрират се 5 до 10 ml от този разреден разтвор през мембранен филтър (т. 4.2). Пристъпва се към определяне чрез HPLC (т. 5.3).
5.3. Определяне с помощта на HPLC
5.3.1.
Следните условия са дадени като указание; може да се използват и други, при условие, че дават равностойни резултати:
|
Колона за течна |
|
|
хроматография (т. 4.1.1): |
125 mm × 4 mm, за катионен обмен Nucleosil 10 SA, пълнеж от 5 или 10 μm, или еквивалентна (т. 4.2) |
|
Подвижна фаза (т. 3.6): |
смес от ацетонитрил (т. 3.2), разтвор на натриев дихидроген фосфат (т. 3.4) и разтвор на натриев перхлорат (т. 3.5), 450 + 450 + 100 (v + v + v). |
|
Скорост на изтичане: |
0,7—1 ml/min; |
|
Дължина на вълната за откриване: |
264 nm |
|
Обем на впръскване: |
100 μl |
Проверява се стабилността на хроматографската система като няколко пъти се впръсква калибрационен разтвор (т. 3.7.3), в който се съдържат 1,0 μg/ml, докато се постигнат постоянни височини на пиковете и постоянни времена на задържане.
5.3.2.
Впръсква се от всеки калибрационен разтвор (т. 3.7.3) по няколко пъти и се определят средните височини на пиковете (площите) за всяка концентрация. Построява се калибрационна крива, като средните височини на пиковете (площите) на калибрационните разтвори се използват за ординати, а съответстващите им концентрации в μg/ml — за абсциси.
5.3.3.
Екстрактът от пробата за изследване (5.2) се впръсква няколко пъти, като се използва същият обем като при калибрационните разтвори, и се определя средната височина (площ) на пиковете на ампролиум.
6. Изчисляване на резултатите
От средната височина (площ) на пиковете на ампролиум в разтвора на пробата чрез сравняване с калибрационната крива (т. 5.3.2) се определя концентрацията на разтвора на пробата в μg/ml.
Съдържанието на ампролиум w в mg/kg в пробата се определя по формулата:
 [mg/kg]
[mg/kg]
където:
|
V |
= |
обем на екстракционния разтворител (т. 3.8) в ml, съгласно т. 5.2 (т.е. 200 ml) |
|
c |
= |
концентрация на ампролиум в екстракта на пробата (т. 5.2) в μg/ml |
|
f |
= |
фактор на разреждане, съгласно т. 5.2. |
|
m |
= |
тегло в g на частта от пробата за анализ. |
7. Потвърждаване на резултатите
7.1. Идентичност
Идентичността на аналита може да се потвърди чрез ко-хроматография или чрез използване на детектор с диодна матрица, с помощта на които могат да се сравнят спекрите на екстракта на пробата и на калибрационния разтвор (т. 3.7.3), съдържащ 2,0 μg/ml.
7.1.1.
Обогатява се екстракт от пробата (т. 5.2), като се добави подходящо количество калибрационен разтвор (т. 3.7.3). Количеството добавен ампролиум трябва да бъде близко до това, открито в екстракта на пробата.
Увеличава се само височината на пика на ампролиум, след като се вземат под внимание както добавеното количество, така и степента на разреждане на екстракта. Широчината на пика при половината от височината му трябва да бъде в границите на ± 10 % от първоначалната широчина на пика на ампролиум при необогатения екстракт от пробата.
7.1.2.
Резултатите се оценяват според следните критерии:
|
а) |
Дължината на вълната на максимална абсорбция на пробата трябва да бъде същата като тази на спектрите на стандарта, регистрирана в най-високата точка на хроматограмата и при отконение, определено от разделителната способност на детектиращата система. При детектиране с диодна матрица отклонението обикновено е в рамките на ± 2 nm. |
|
б) |
Между 210 и 320 nm спектрите на пробата и на стандарта, отчетени в най-високата точка на хроматограмата, не трябва да се различават помежду си за онези части от спектъра, които попадат в интервала между 10 до 100 % относителна екстинкция. Критерият е изпълнен тогава, когато са налице едни и същи максимуми и не е регистрирана точка, в която отклонението между двата спектра превишава 15 % от екстинцията на стандартния аналит; |
|
в) |
между 210 и 320 nm спектрите на възходящата линия, максимума и низходящата линия на пика, получен при анализа на екстракта на пробата, не трябва да се различават един от друг в онези части от спектъра, които попадат в интервала между 10 до 100 % относителна екстинкция. Критерият е изпълнен тогава, когато са налице едни и същи максимуми и когато във всички регистрирани точки отклонението между спектрите не надминава 15 % от екстинкцията на спектъра на най-високата точка. |
Ако един от тези критерии не е спазен, наличието на аналита не е потвърдено.
7.2. Повторяемост
Разликата между резултатите от две паралелни определяния, проведени върху същата проба, не трябва да превишава:
|
— |
15 % спрямо по-високата стойност при съдържание на ампролиум от 25 mg/kg до 500 mg/kg; |
|
— |
75 mg/kg при съдържание на ампролиум между 500 mg/kg и 1 000 mg/kg; |
|
— |
7,5 % спрямо по-високата стойност при съдържание на ампролиум над 1 000 mg/kg. |
7.3. Възстановяване
При обогатена (празна) проба, възстановяването трябва да бъде най-малко 90 %.
8. Резултати от съвместно изследване
Проведено беше паралелно изследване, при което бяха анализирани три фуража за домашни птици (проби 1—3), един минерален фураж (проба 4) и един премикс (проба 5). Резултатите са представени в следната таблица:
|
|
Проба 1 Празна проба фураж |
Проба 2 |
Проба 3 |
Проба 4 |
Проба 5 |
|
L |
14 |
14 |
14 |
14 |
15 |
|
n |
56 |
56 |
56 |
56 |
60 |
|
Средна стойност в [mg/kg] |
— |
45,5 |
188 |
5 129 |
25 140 |
|
sr [mg/kg] |
— |
2,26 |
3,57 |
178 |
550 |
|
CVr [ %] |
— |
4,95 |
1,90 |
3,46 |
2,20 |
|
sR [mg/kg] |
— |
2,95 |
11,8 |
266 |
760 |
|
CVR [ %] |
— |
6,47 |
6,27 |
5,19 |
3,00 |
|
Номинално съдържание [mg/kg] |
— |
50 |
200 |
5 000 |
25 000 |
|
L |
= |
брой на лабораториите |
|
n |
= |
брой на единичните стойности |
|
sr |
= |
стандартно отклонение на повторяемостта |
|
CVr |
= |
коефициент на вариация на повторяемостта |
|
sR |
= |
стандартно отклонение на възпроизводимостта |
|
CVR |
= |
коефициент на вариация на възпроизводимостта |
9. Забележки
|
9.1. |
Ако пробата съдържа тиамин, неговият пик в хроматограмата се появява малко преди пика на ампролиума. Ако се следва този метод, ампролиумът и тиаминът трябва да се разделят. Ако те не се разделят с помощта на колоната (т. 4.1.1), използвана в настоящия метод, следва да се заменят до 50 % от ацетонитрила в подвижната фаза (т. 3.6) с метанол. |
|
9.2. |
Съгласно Британската фармакопея, спектърът на разтвор на ампролиум (с = 0,02 mol/l) в солна киселина (с = 0,1 mol/l) показва максимуми при 246 nm и 262 nm. Екстинкцията достига 0,84 при 246 nm и 0,80 при 262 nm. |
|
9.3. |
Екстрактът винаги трябва да бъде разреден с подвижната фаза фаза, тъй като в противен случай времето на задържане на пика на ампролиума може значително да се измени, поради промени в йонната сила на разтвора. |
Г. ОПРЕДЕЛЯНЕ НА КАРБАДОКС
Метил 3-(2-квиноксалинилметилен)карбазат N 1 ,N 4 -диоксид
1. Цел и обхват
Настоящият метод дава възможност за определяне на съдържанието на карбадокс във фуражи, премикси и ветеринарномедицински препарати. Границата на откриване е 1 mg/kg, а границата на количествено определяне е 5 mg/kg.
2. Принцип
Пробата се уравновесява с вода и се подлага на екстракция със смес метанол-ацетонитрил. При фуражите, една аликвотна част от филтрирания екстракт се подлага на очистване в колона с диалуминиев триоксид. При премиксите и ветеринарномедицинските препарати, една аликвотна част от филтрирания екстракт се разрежда с вода, метанол и ацетонитрил до получаване на подходяща концентрация. Съдържанието на карбадокс се определя чрез високоефективна течна хроматография (HPLC) с обратна фаза, с използване на UV детектор.
3. Реагенти
|
3.1. |
Метанол. |
|
3.2. |
Ацетонитрил, с чистота за HPLC. |
|
3.3. |
Оцетна киселина, w = 100 %. |
|
3.4. |
Диалуминиев триоксид: неутрален, клас на активност I. |
|
3.5. |
Метанол-ацетонитрил 1 + 1 (v + v). Смесват се 500 ml метанол (т. 3.1) с 500 ml ацетонитрил (т. 3.2). |
|
3.6. |
Оцетна киселина, σ = 10 %. Разреждат се 10 ml оцетна киселина (т. 3.3) до 100 ml с вода. |
|
3.7. |
Натриев ацетат. |
|
3.8. |
Вода, с чистота за HPLC. |
|
3.9. |
Ацетатен буферен разтвор, с = 0,01 mol/l, pH = 6,0. Разтварят се 0,82 g натриев ацетат (т. 3.7) в 700 ml вода (т. 3.8), като с помощта на оцетна киселина (т. 3.6) се регулира стойността на рН до 6,0. Прехвърля се в градуирана колба от 1 000 ml, допълва се до пълния обем с вода (т. 3.8) и се разбърква. |
|
3.10. |
Подвижна фаза за HPLC Смесват се 825 ml ацетатен буферен разтвор (т. 3.9) с 175 ml ацетонитрил (т. 3.2). Разтворът се филтрира през филтър с размер на порите 0,22 μm (т. 4.5) и се обезгазява (напр. чрез подлагане на въздействието на ултразвук в продължение на 10 минути). |
|
3.11. |
Стандартно вещество Чист карбадокс: Метил 3-(2-квиноксалинилметилен)карбазат N1,N4-диоксид, Е 850. |
|
3.11.1. |
Претеглят се 25 mg стандартна вещество карбадокс (т. 3.11) с точност до 0,1 mg в градуирана колба от 250 ml. Разтварят се в смес метанол-ацетонитрил (т. 3.5) с помощта на ултразвук (т. 4.7). След ултразвуковата обработка, разтворът се оставя да достигне стайна температура, допълва се по пълния обемс с метанол-ацетонитрил (т. 3.5), и се разбърква. Колбата се обвива с алуминиево фолио или се използва съд от непрозрачно стъкло, и се съхранява в хладилник. При температура ≤ 4 oС, разтворът е стабилен в продължение на 1 месец. |
|
3.11.2. |
Прехвърлят се съответно 2,0, 5,0, 10,0 и 20,0 ml от основния стандартен разтвор (т. 3.11.1) в поредица от градуирани колби от 100 ml. Прибавят се 30 ml вода, допълва се метанол-ацетонитрил (т. 3.5) до пълния обем и се разбърква. Колбите се завиват в алуминиево фолио. Разтворите отговарят съответно на 2,0; 5,0; 10,0; и 20,0 μg/ml карбадокс. Калибрационните разтвори трябва да бъдат приготвени непосредствено преди употреба.
|
|
3.12. |
Смес вода-[метанол-ацетонитрил] (т. 3.5), 300 + 700 (v + v) Смесват се 300 ml вода със 700 ml смес на метанол-ацетонитрил (т. 3.5). |
4. Апаратура
|
4.1. |
Клатачна машина или магнитна бъркалка. |
|
4.2. |
Филтър от стъклени нишки (Whatman GF/A или еквивалентен). |
|
4.3. |
Стъклена колона (дължина от 300 до 400 mm, вътрешен диаметър приблизително 10 mm) с преграда от синтеровано стъкло и източващ клапан.
|
|
4.4. |
Оборудване за HPLC със система за впръскване, подходяща за впръскване на обеми от 20 μl. |
|
4.4.1. |
Колона за течна хроматаграфия: 300 mm × 4 mm, С18, пълнеж от 10 μm, или еквивалентна. |
|
4.4.2. |
UV детектор с настройка на дължината на вълната или детектор с диодна матрица, работещ в диапазона от 225 nm до 400 nm. |
|
4.5. |
Мембранен филтър, 0,22 μm. |
|
4.6. |
Мембранен филтър, 0,45 μm. |
|
4.7. |
Ултразвукова вана. |
5. Процедура
|
Забележка |
: |
Карбадоксът е чувствителен към светлина. Всички процедури се извършват на приглушена светлина или се използват стъклени съдове от тъмно стъкло или такива, обвити в алуминиево фолио. |
5.1. Общи положения
5.1.1.
За изпълнението на теста за възстановяване (т. 5.1.2) се анализира празна проба фураж без добавки, за да се провери за наличието на карбадокс и интерфериращи субстанции. Фуражът от празната проба трябва да е подобен по вид на този от изследваната проба, и в него не трябва да бъдат открити карбадокс или интерфериращи вещества.
5.1.2.
Провежда се тест за възстановяване, като се анализира празната проба фураж (т. 5.1.1), обогатена чрез прибавяне на известно количество карбадокс, близко до наличното в изследваната проба. За да се обогати пробата до ниво 50 mg/kg, се пресипват 5,0 ml от основния стандартен разтвор (т. 3.11.1) в конична колба от 200 ml. Разтворът се подлага на изпаряване в струя азот, докато се обемът му се намали приблизително до 0,5 ml. Прибавят се 10 g от празната проба, смесва се добре и се изчакват 10 min, преди да се продължи с етапа на екстракция (т. 5.2).
Като алтернатива, ако не е наличен фураж, подобен на този, от който е взета пробата (вж. т. 5.1.1), тестът за възстановяване може да се извърши чрез стандартия метод на добавяне. В този случай пробата, която ще се анализира, се обогатява с такова количество карбадокс, което е близко до вече съдържащото се в пробата. Тази проба се анализира заедно с необогатената проба, като степента на възстановяване се пресмята чрез изваждане.
5.2. Екстракция
5.2.1.
Претеглят се 10 g от изследваната проба с точност до 0,01 g и се прехвърлят в конична колба от 200 ml. Прибавят се 15,0 ml вода, разбърква се и се уравновесява в продължение на 5 min. Добавят се 35,0 ml метанол-ацетонитрил (т. 3.5), колбата се запушва и се разклаща в продължение на 30 минути в клатачна машина или се разбърква с магнитната бъркалка (т. 4.1). Разтворът се филтрира през филтър от стъклени нишки (т. 4.2). Разтворът се запазва за етапа на пречистване (т. 5.3).
5.2.2.
Претегля се 1 g несмляна проба с точност до 0,001 mg и се прехвърля в конична колба от 200 ml. Прибавят се 15,0 ml вода, разбърква се и се уравновесява в продължение на 5 min. Добавят се 35,0 ml метанол-ацетонитрил (т. 3.5), колбата се запушва и се разклаща в продължение на 30 минути в клатачна машина или се разбърква с магнитната бъркалка (т. 4.1). Разтворът се филтрира през филтър от стъклени нишки (т. 4.2).
Отмерва се пипета една аликвотна част от филтрата в мерителна колба от 50 ml. Прибавят се 15,0 ml вода, допълва се с метанол-ацетонитрил (т. 3.5) до пълния обем и се разбърква. Концентрацията на карбадокс в крайния разтвор трябва да бъде приблизително 10 μg/ml. Една аликвотна част от разтвора се филтрира през филтър с размер на порите 0,45 μm (т. 4.6).
Пристъпва се към определяне чрез HPLC (т. 5.4).
5.2.3.
Претеглят се 0,2 g несмляна проба с точност до 0,001 mg и се прехвърлят в конична колба от 250 ml. Прибавят се 45,0 ml вода, разбърква се и се уравновесява за 5 min. Добавят се 105,0 ml метанол-ацетонитрил (т. 3.5), колбата се запушва и съдържанието и се хомогенизира. Пробата се поставя в ултразвукова вана (т. 4.7) за 15 минути, след което се разклаща или разбърква в продължение на 15 минути (т. 4.1). Разтворът се филтрира през филтър от стъклени нишки (т. 4.2).
Една аликвотна част от филтрата се разрежда със сместа от вода, метанол и ацетонитрил (т. 3.12) до получаване на крайна концентрация на карбадокс от 10—15 μg/ml (за препарат с концентрация 10 %, факторът на разреждане е 10). Една аликвотна част се филтрира през филтър с размер на порите 0,45 μm (т. 4.6).
Пристъпва се към определяне чрез HPLC (т. 5.4).
5.3. Пречистване
5.3.1.
Претеглят се 4 g диалуминев триоксид (т. 3.4) и се прехвърлят в стъклената колона (т. 4.3).
5.3.2.
Прилагат се 15 ml от филтрирания екстракт (т. 5.2.1) към колоната с диалуминиевия оксид и първите 2 ml от елуат се отстраняват. Следващите 5 ml се събират и една аликтвотна част се филтрира през филтър с размер на порите 0,45 μm (т. 4.6).
Пристъпва се към определяне чрез HPLC (т. 5.4).
5.4. Определяне с помощта на HPLC
5.4.1.
Дават се следните указания за параметрите, могат да се използват и други параметри, при условие че с тях се получават еквивалентни резултати:
|
Колона за течна |
|
|
хроматография (т. 4.4.1): |
300 mm × 4 mm, С18, пълнеж от 10 μm или еквивалентна |
|
Подвижна фаза (т. 3.10): |
Смес от ацетатен буфер (т. 3.9) и ацетонитрил (т. 3.2), 825 + 175 (v + v) |
|
Скорост на изтичане: |
1,5—2 ml/min; |
|
Дължина на вълната за откриване: |
365 nm |
|
Обем на впръскване: |
20 μl |
Проверява се стабилността на хроматографичната система, като на няколко пъти се впръсква калибрационният разтвор (т. 3.11.2), съдържащ 5,0 μg/ml, докато се постигнат постоянни височини на пика и постоянни времена на задържане.
5.4.2.
Всеки калибрационен разтвор (3.11.2) се впръсква няколко пъти и се определят средните височини на пиковете (площите) за всяка концентрация. Построява се калибрационна крива, като се използват средните пикови височини или площи на калибрационните разтвори за ординати, а съответните им концентрации в μg/ml за абсциси.
5.4.3.
Екстрактът от пробата [(т. 5.3.2) за фуражи, (т. 5.2.2) за премикси и (т. 5.2.3) за ветеринарномедицинските препарати] се впръсква няколко пъти, и се определя средната пикова височина (площ) на пиковете на карбадокс.
6. Изчисляване на резултатите
Концентрацията на карбадокс в разтвора на пробата в μg/ml се определя от средната височина (площ) на пиковете на карбадокс на разтвора на пробата, като се сравнява с калибрационната крива (т. 5.4.2).
6.1. Фураж
Съдържанието w (mg/kg) на карбадокс в пробата се пресмята по следната формула:
 [mg/kg]
[mg/kg]
където:
|
c |
= |
концентрация на карбадокс в екстракта на пробата (т. 5.3.2) в μg/ml |
|
V1 |
= |
обем на екстракта в ml (т.е. 50) |
|
m |
= |
тегло в g на частта от пробата за анализ. |
6.2. Премикси и препарати
Съдържанието w (mg/kg) на карбадокс (mg/kg) в пробата се пресмята по следната формула:
 [mg/kg]
[mg/kg]
където:
|
c |
= |
концентрация на карбадокс в екстракта на пробата (т. 5.2.2 или 5.2.3) в μg/ml |
|
V2 |
= |
обем на екстракта в ml (т.е. 50 за премикси; 150 за препарати) |
|
f |
= |
фактор на разреждане съгласно т. 5.2.2 (премикси) или 5.2.3 (препарати) |
|
m |
= |
тегло в g на частта от пробата за анализ. |
7. Потвърждаване на резултатите
7.1. Идентичност
Идентичността на аналита може да се потвърди с помощта на кохроматография или като се използва детектор с диодна матрица, с които се сравняват спектрите на екстракта на пробата и на калибрационния разтвор (т. 3.11.2), съдържащ 10,0 μg/ml.
7.1.1.
Обогатява се екстракт от пробата, като се прибави подходящо количество калибрационен разтвор (3.11.2). Количеството добавен карбадокс трябва да бъде близко до това, което се очаква да бъде открито в екстракта на пробата.
Увеличава се само височината на пика на карбадокс след като се вземат под внимание както добавеното количество, така и степента на разреждане на екстракта. Широчината на пика на половината от неговата максимална височина трябва да бъде в рамките на около 10 % от първоначалната широчина.
7.1.2.
Резултатите се оценяват според следните критерии:
|
а) |
Дължината на вълната на максимална абсорбция на пробата трябва да бъде същата като тази на спектрите на стандарта, регистрирана в най-високата точка на хроматограмата и при отконение, определено от разделителната способност на детектиращата система. При детектиране с диодна матрица, тя е обикновено в границите на ± 2 nm; |
|
б) |
между 225 и 400 nm спектрите на пробата и на стандарта, отчетени в най-високата точка на хроматограмата, не трябва да се различават помежду си за тези части от спектъра в интервала между 10 до 100 % относителна екстинкция.;. Критерият е изпълнен тогава, когато са налице едни и същи максимуми и не е регистрирана точка, в която отклонението между двата спектра превишава 15 % от екстинцията на стандартния аналит; |
|
в) |
между от 225 до 400 nm, спектрите на възходящата линия, максимума и низходящата линия на пика, получен от екстракта на пробата, не трябва да се различават един от друг в онези части на спектъра в интервала между 10 до 100 % относителна екстинкция. Критерият е изпълнен тогава, когато налице са едни и същи максимуми и когато във всички регистрирани точки отклонението между спектрите не надминава 15 % от екстинкцията на спектъра на най-високата точка. |
Ако някой от критериите не е изпълнен, наличието на аналита не се потвърждава.
7.2. Повторяемост
При съдържание 10 mg/kg и по-високо, разликата между резултатите от две паралелни определяния, извършени върху същата проба, не трябва да превишава 15 % спрямо по-високия резултат.
7.3. Възстановяване
При обогатена (празна) проба, възстановяването трябва да бъде най-малко 90 %.
8. Резултати от съвместно изследване
Проведено беше съвместно изследване, при което бяха анализирани 6 фуража, 4 премикса и 3 препарата в 8 лаборатории. За всяка проба са проведени по два анализа. (По-подробна информация за това съвместно изследване може да се открие в Journal of AOAC International, том 71, 1988 г., стр. 484—490). Резултатите (с изключение на рязко отличаващите се стойности) са представени по-долу:
Таблица 1
Резултати от съвместно изследванена фуражи
|
|
Проба 1 |
Проба 2 |
Проба 3 |
Проба 4 |
Проба 5 |
Проба 6 |
|
L |
8 |
8 |
8 |
8 |
8 |
8 |
|
n |
15 |
14 |
15 |
15 |
15 |
15 |
|
Среднa стойност, [mg/kg] |
50,0 |
47,6 |
48,2 |
49,7 |
46,9 |
49,7 |
|
Sr (mg/kg) |
2,90 |
2,69 |
1,38 |
1,55 |
1,52 |
2,12 |
|
CVr ( %) |
5,8 |
5,6 |
2,9 |
3,1 |
3,2 |
4,3 |
|
SR (mg/kg) |
3,92 |
4,13 |
2,23 |
2,58 |
2,26 |
2,44 |
|
CVR ( %) |
7,8 |
8,7 |
4,6 |
5,2 |
4,8 |
4,9 |
|
Номинална стойност, (mg/kg) |
50,0 |
50,0 |
50,0 |
50,0 |
50,0 |
50,0 |
Таблица 2
Резултати от съвместно изследване на премикси и препарати
|
|
Премикси |
Препарати |
|||||
|
А |
Б |
В |
Г |
А |
Б |
В |
|
|
L |
7 |
7 |
7 |
7 |
8 |
8 |
8 |
|
n |
14 |
14 |
14 |
14 |
16 |
16 |
16 |
|
Средна стойност (g/kg) |
8,89 |
9,29 |
9,21 |
8,76 |
94,6 |
98,1 |
104 |
|
Sr (mg/kg) |
0,37 |
0,28 |
0,28 |
0,44 |
4,1 |
5,1 |
7,7 |
|
CVr ( %) |
4,2 |
3,0 |
3,0 |
5,0 |
4,3 |
5,2 |
7,4 |
|
SR (g/kg) |
0,37 |
0,28 |
0,40 |
0,55 |
5,4 |
6,4 |
7,7 |
|
CVR ( %) |
4,2 |
3,0 |
4,3 |
6,3 |
5,7 |
6,5 |
7,4 |
|
Номинално съдържание (g/kg) |
10,0 |
10,0 |
10,0 |
10,0 |
100 |
100 |
100 |
|
L |
= |
брой на лабораториите |
|
n |
= |
брой на единичните стойности |
|
Sr |
= |
стандартно отклонение на повторяемостта |
|
CVr |
= |
коефициент на вариация на повторяемостта |
|
SR |
= |
стандартно отклонение на възпроизводимостта |
|
CVR |
= |
коефициент на вариация на възпроизводимостта |
ПРИЛОЖЕНИЕ IX
ТАБЛИЦИ НА СЪОТВЕТСТВИЕТО, ПОСОЧЕНИ В ЧЛЕН 6
1. Директива 71/250/EИО
|
Директива 71/250/EИО |
Настоящият регламент |
|
Член 1, първа алинея |
Член 3 |
|
Член 1, втора алинея |
Член 2 |
|
Член 2 |
— |
|
Член 3 |
— |
|
Приложение, част 1 |
Приложение II |
|
Приложение, част 2 |
— |
|
Приложение, част 3 |
— |
|
Приложение, част 4 |
Приложение III, част О |
|
Приложение, част 5 |
Приложение III, част М |
|
Приложение, част 6 |
Приложение III, част Н |
|
Приложение, част 7 |
Приложение III, част Р |
|
Приложение, част 9 |
Приложение III, част К |
|
Приложение, част 10 |
— |
|
Приложение, част 11 |
— |
|
Приложение, част 12 |
Приложение III, част Й |
|
Приложение, част 14 |
Приложение III, част Г |
|
Приложение, част 16 |
— |
2. Директива 71/393/EИО
|
Директива 71/393/EИО |
Настоящият регламент |
|
Член 1 |
Член 3 |
|
Член 2 |
— |
|
Член 3 |
— |
|
Приложение, част I |
Приложение III, част A |
|
Приложение, част II |
Приложение III, част Д |
|
Приложение, част III |
Приложение III, част П |
|
Приложение, част IV |
Приложение III, част З |
3. Директива 72/199/EИО
|
Директива 72/199/EИО |
Настоящият регламент |
|
Член 1 |
Член 3 |
|
Член 2 |
— |
|
Член 3 |
— |
|
Член 4 |
— |
|
Приложение I, част 1 |
Приложение III, част Л |
|
Приложение I, част 2 |
Приложение III, част В |
|
Приложение I, част 3 |
— |
|
Приложение I, част 4 |
— |
|
Приложение I, част 5 |
Приложение V, част A |
|
Приложение II |
— |
4. Директива 73/46/EИО
|
Директива 73/46/EИО |
Настоящият регламент |
|
Член 1 |
Член 3 |
|
Член 3 |
— |
|
Член 4 |
— |
|
Приложение I, част 1 |
Приложение III, част Б |
|
Приложение I, част 2 |
— |
|
Приложение I, част 3 |
Приложение III, част И |
5. Директива 76/371/EИО
|
Директива 76/371/EИО |
Настоящият регламент |
|
Член 1 |
Член 1 |
|
Член 2 |
— |
|
Член 3 |
— |
|
Приложение |
Приложение I |
6. Директива 76/372/EИО
|
Директива 76/372/EИО |
Настоящият регламент |
|
Член 1 |
— |
|
Член 2 |
— |
|
Член 3 |
— |
|
Приложение |
— |
7. Директива 78/633/EИО
|
Директива 78/633/EИО |
Настоящият регламент |
|
Член 1 |
Член 3 |
|
Член 2 |
— |
|
Член 3 |
— |
|
Приложение, част 1 |
— |
|
Приложение, част 2 |
— |
|
Приложение, част 3 |
Приложение IV, част В |
8. Директива 81/715/EИО
|
Директива 81/715/EИО |
Настоящият регламент |
|
Член 1 |
— |
|
Член 2 |
— |
|
Член 3 |
— |
|
Приложение |
— |
9. Директива 84/425/EИО
|
Директива 84/425/EИО |
Настоящият регламент |
|
Член 1 |
— |
|
Член 2 |
— |
|
Член 3 |
— |
|
Приложение |
— |
10. Директива 86/174/EИО
|
Директива 86/174/EИО |
Настоящият регламент |
|
Член 1 |
Член 4 |
|
Член 2 |
— |
|
Член 3 |
— |
|
Приложение |
Приложение VII |
11. Директива 93/70/EИО
|
Директива 93/70/EИО |
Настоящият регламент |
|
Член 1 |
Член 3 |
|
Член 2 |
— |
|
Член 3 |
— |
|
Приложение |
Приложение IV, част Г |
12. Директива 93/117/EО
|
Директива 93/117/EО |
Настоящият регламент |
|
Член 1 |
Членове 3 и 5 |
|
Член 2 |
— |
|
Член 3 |
— |
|
Приложение, част 1 |
Приложение IV, част Д |
|
Приложение, част 2 |
Приложение VIII, част A |
13. Директива 98/64/EО
|
Директива 98/64/EО |
Настоящият регламент |
|
Член 1 |
Членове 3 и 5 |
|
Член 2 |
— |
|
Член 3 |
— |
|
Член 4 |
— |
|
Приложение, част A |
Приложение III, част Е |
|
Приложение, част В |
Приложение VIII, част Б |
14. Директива 1999/27/EО
|
Директива 1999/27/EО |
Настоящият регламент |
|
Член 1 |
Членове 3 и 5 |
|
Член 2 |
— |
|
Член 3 |
— |
|
Член 4 |
— |
|
Член 5 |
— |
|
Член 6 |
— |
|
Член 7 |
— |
|
Приложение, част A |
Приложение VIII, част В |
|
Приложение, част Б |
Приложение IV, част Е |
|
Приложение част В |
Приложение VIII, част Г |
15. Директива 1999/76/EО
|
Директива 1999/76/EО |
Настоящият регламент |
|
Член 1 |
Член 3 |
|
Член 2 |
— |
|
Член 3 |
— |
|
Член 4 |
— |
|
Приложение |
Приложение IV, част Ж |
16. Директива 2000/45/EО
|
Директива 2000/45/EО |
Настоящият регламент |
|
Член 1 |
Член 3 |
|
Член 2 |
— |
|
Член 3 |
— |
|
Член 4 |
— |
|
Приложение, част A |
Приложение IV, част A |
|
Приложение, част Б |
Приложение IV, част Б |
|
Приложение част C |
Приложение III, част Ж |
17. Директива 2002/70/EО
|
Директива 2002/70/EО |
Настоящият регламент |
|
Член 1 |
Член 1 |
|
Член 2 |
Членове 2 и 3 |
|
Член 3 |
— |
|
Член 4 |
— |
|
Член 5 |
— |
|
Приложение I |
Приложение I и приложение V, част Б (I) |
|
Приложение II |
Приложение II и приложение V, част Б (II) |
18. Директива 2003/126/EО
|
Директива 2003/126/EО |
Настоящият регламент |
|
Член 1 |
Член 3 |
|
Член 2 |
— |
|
Член 3 |
— |
|
Член 4 |
— |
|
Член 5 |
— |
|
Член 6 |
— |
|
Приложение |
Приложение VI |
|
26.2.2009 |
BG |
Официален вестник на Европейския съюз |
L 54/s3 |
СЪОБЩЕНИЕ ЗА ЧИТАТЕЛИТЕИнституциите решиха, че занапред в техните текстове няма да се съдържа позоваване на последното изменение на цитираните актове.Освен ако не е посочено друго, позоваванията на актове в публикуваните тук текстове се отнасят към актуалната версия на съответния акт.