edizione speciale in lingua bulgare: capitolo 15 tomo 017 pag. 150 - 168
edizione speciale in lingua romena: capitolo 15 tomo 017 pag. 150 - 168
edizione speciale in lingua croata: capitolo 15 tomo 015 pag. 31 - 49
English
Select your language
Official EU languages:
Access to European Union law
EUR-Lex
Access to European Union law
This document is an excerpt from the EUR-Lex website
Use quotation marks to search for an "exact phrase". Append an asterisk (*) to a search term to find variations of it (transp*, 32019R*). Use a question mark (?) instead of a single character in your search term to find variations of it (ca?e finds case, cane, care).
Need more search options? Use the
Advanced search
Document 32006L0086
Commission Directive 2006/86/EC of 24 October 2006 implementing Directive 2004/23/EC of the European Parliament and of the Council as regards traceability requirements, notification of serious adverse reactions and events and certain technical requirements for the coding, processing, preservation, storage and distribution of human tissues and cells (Text with EEA relevance)
Direttiva 2006/86/CE della Commissione, del 24 ottobre 2006 , che attua la direttiva 2004/23/CE del Parlamento europeo e del Consiglio per quanto riguarda le prescrizioni in tema di rintracciabilità, la notifica di reazioni ed eventi avversi gravi e determinate prescrizioni tecniche per la codifica, la lavorazione, la conservazione, lo stoccaggio e la distribuzione di tessuti e cellule umani (Testo rilevante ai fini del SEE)
Direttiva 2006/86/CE della Commissione, del 24 ottobre 2006 , che attua la direttiva 2004/23/CE del Parlamento europeo e del Consiglio per quanto riguarda le prescrizioni in tema di rintracciabilità, la notifica di reazioni ed eventi avversi gravi e determinate prescrizioni tecniche per la codifica, la lavorazione, la conservazione, lo stoccaggio e la distribuzione di tessuti e cellule umani (Testo rilevante ai fini del SEE)
GU L 294 del 25.10.2006, p. 32–50
(ES, CS, DA, DE, ET, EL, EN, FR, IT, LV, LT, HU, NL, PL, PT, SK, SL, FI, SV) Questo documento è stato pubblicato in edizioni speciali
(BG, RO, HR)
GU L 314M del 1.12.2007, p. 272–290
(MT)
 In force: This act has been changed. Current consolidated version: 29/04/2015
In force: This act has been changed. Current consolidated version: 29/04/2015
Language
HTML
PDF
Official Journal
|
25.10.2006 |
IT |
Gazzetta ufficiale dell'Unione europea |
L 294/32 |
DIRETTIVA 2006/86/CE DELLA COMMISSIONE
del 24 ottobre 2006
che attua la direttiva 2004/23/CE del Parlamento europeo e del Consiglio per quanto riguarda le prescrizioni in tema di rintracciabilità, la notifica di reazioni ed eventi avversi gravi e determinate prescrizioni tecniche per la codifica, la lavorazione, la conservazione, lo stoccaggio e la distribuzione di tessuti e cellule umani
(Testo rilevante ai fini del SEE)
LA COMMISSIONE DELLE COMUNITÀ EUROPEE,
visto il trattato che istituisce la Comunità europea,
vista la direttiva 2004/23/CE del Parlamento europeo e del Consiglio, del 31 marzo 2004, sulla definizione di norme di qualità e di sicurezza per la donazione, l’approvvigiornamento, il controllo, la lavorazione, la conservazione, lo stoccaggio e la distribuzione di tessuti e cellule umani (1), in particolare l’articolo 8, l’articolo 11, paragrafo 4, e l’articolo 28, lettere a), c), g) e h),
considerando quanto segue:
|
(1) |
La direttiva 2004/23/CE stabilisce norme di qualità e di sicurezza per la donazione, l’approvvigionamento, il controllo, la lavorazione, la conservazione, lo stoccaggio e la distribuzione di tessuti e cellule umani destinati ad applicazioni sull’uomo e di prodotti fabbricati derivanti da tessuti e cellule umani destinati ad applicazioni sull’uomo, al fine di assicurare un elevato livello di protezione della salute umana. |
|
(2) |
Per prevenire la trasmissione di malattie attraverso tessuti e cellule umani destinati ad applicazioni sull’uomo e garantire un livello equivalente di qualità e sicurezza, la direttiva 2004/23/CE prevede la fissazione di prescrizioni tecniche specifiche per ciascuna delle fasi del procedimento di applicazione di tessuti e cellule umani, comprese le norme e le specifiche relative a un sistema di qualità per gli istituti dei tessuti. |
|
(3) |
Conformemente alla direttiva 2004/23/CE gli Stati membri istituiscono un sistema di accreditamento, designazione, autorizzazione o rilascio di licenza per gli istituti dei tessuti e per i procedimenti di preparazione degli istituti dei tessuti, al fine di assicurare un elevato livello di protezione della salute umana. Occorre definire le prescrizioni tecniche per tale sistema. |
|
(4) |
Le prescrizioni per l’accreditamento, la designazione, l’autorizzazione o il rilascio di licenza degli istituti dei tessuti si estendono all’organizzazione e alla gestione, al personale, alle attrezzature e ai materiali, ai servizi/locali, alla documentazione, alle registrazioni e alla verifica della qualità. Gli istituti dei tessuti accreditati, designati, autorizzati o titolari di licenza ottemperano alle ulteriori prescrizioni relative alle specifiche attività da essi svolte. |
|
(5) |
I parametri di qualità dell’aria durante la lavorazione di tessuti e cellule sono un fattore fondamentale che può influire sul rischio di contaminazione di tessuti o cellule. Viene di norma richiesta una qualità dell’aria con numeri di particelle e numeri di colonie microbiche equivalenti a quelli di grado A di cui alla Guida europea alle buone pratiche di fabbricazione, allegato 1, e alla direttiva 2003/94/CE della Commissione (2). Tuttavia in determinate situazioni non sono prescritti parametri di qualità dell’aria con numeri di particelle e numeri di colonie microbiche equivalenti a quelli di grado A. In questi casi occorre dimostrare e documentare che l’ambiente prescelto corrisponde alla qualità e sicurezza richieste per il tipo di tessuto e cellule, di procedimento e applicazione sull’uomo di cui si tratta. |
|
(6) |
Il campo d’applicazione della direttiva deve estendersi alla qualità e sicurezza di tessuti e cellule umani durante la codifica, la lavorazione, la conservazione, lo stoccaggio e la distribuzione al centro sanitario in cui verranno applicati al corpo umano. Non deve tuttavia estendersi alle applicazioni di questi tessuti e cellule sull’uomo (con atti chirurgici, perfusione, inseminazione o trasferimento di embrioni). Le disposizioni della direttiva in materia di rintracciabilità e di notifica di reazioni ed eventi avversi gravi si applicano anche alla donazione, all’approvvigionamento e al controllo di tessuti e cellule umani di cui alla direttiva 2006/17/CE della Commissione (3). |
|
(7) |
L’uso di tessuti e cellule per applicazioni sull’uomo comporta una rischio di trasmissione di malattie e di altri potenziali effetti avversi ai riceventi. Al fine di sorvegliare e ridurre tali effetti occorre definire prescrizioni specifiche in materia di rintracciabilità e una procedura comunitaria di notifica di reazioni ed eventi avversi gravi. |
|
(8) |
Presunte reazioni avverse gravi, nel donatore o nel ricevente, ed eventi avversi gravi dalla donazione alla distribuzione di tessuti e cellule, che possono influire sulla qualità e la sicurezza di tessuti e cellule e possono essere attribuiti all’approvvigionamento (compresa la valutazione e la selezione del donatore), al controllo, alla lavorazione, alla conservazione, allo stoccaggio e alla distribuzione di tessuti e cellule umani devono essere tempestivamente notificati all’autorità competente. |
|
(9) |
Reazioni avverse gravi possono essere individuate durante o dopo l’approvvigionamento in donatori viventi o durante o dopo l’applicazione sull’uomo. Occorre che siano riferite al relativo istituto dei tessuti per successive indagini e notifica all’autorità competente. Se lo desiderano tuttavia, l’organizzazione di approvvigionamento o l’organizzazione responsabile dell’applicazione sull’uomo possono anche inviarne notifica diretta all’autorità competente. Occorre che la direttiva definisca i dati minimi necessari per la notifica all’autorità competente, lasciando impregiudicata la facoltà degli Stati membri di mantenere o adottare sul proprio territorio provvedimenti di protezione più rigorosi in conformità delle disposizioni del trattato. |
|
(10) |
Per minimizzare i costi di trasmissione, evitare sovrapposizioni ed accrescere l’efficienza amministrativa occorre ricorrere alle tecnologie moderne e ai metodi di gestione elettronica per lo svolgimento dei compiti di trasmissione e trattamento delle informazioni. Tali tecnologie devono basarsi su un modello standard di scambio dotato di un sistema idoneo di gestione dei dati di riferimento. |
|
(11) |
Per agevolare la rintracciabilità e l’accesso alle informazioni sulle caratteristiche e proprietà fondamentali di tessuti e cellule occorre definire i dati di base da includere in un codice unico europeo. |
|
(12) |
La presente direttiva rispetta i diritti fondamentali e osserva i principi specificamente sanciti dalla Carta dei diritti fondamentali dell’Unione europea. |
|
(13) |
Le disposizioni di cui alla presente direttiva sono conformi al parere del comitato istituito a norma dell’articolo 29 della direttiva 2004/23/CE, |
HA ADOTTATO LA PRESENTE DIRETTIVA:
Articolo 1
Campo d’applicazione
1. La presente direttiva si applica alla codifica, alla lavorazione, alla conservazione, allo stoccaggio e alla distribuzione di:
|
a) |
tessuti e cellule umani destinati ad applicazioni sull’uomo; |
|
b) |
prodotti fabbricati derivati da tessuti e cellule umani destinati ad applicazioni sull’uomo, qualora tali prodotti non siano disciplinati da altre direttive. |
2. Le disposizioni degli articoli da 5 a 9 della presente direttiva, relative alla rintracciabilità e alla notifica di reazioni ed eventi avversi gravi, si applicano anche alla donazione, all’approvvigionamento e al controllo di tessuti e cellule umani.
Articolo 2
Definizioni
Ai fini della presente direttiva si intendono per:
|
a) |
«cellule riproduttive»: tutti i tessuti e le cellule destinati ad essere utilizzati ai fini della riproduzione assistita; |
|
b) |
«donazione da parte di un partner»: la donazione di cellule riproduttive tra un uomo e una donna che dichiarano di avere rapporti fisici; |
|
c) |
«sistema di qualità»: la struttura organizzativa, le responsabilità, le procedure, i procedimenti e le risorse destinati ad attuare la gestione della qualità, comprese tutte le attività che direttamente o indirettamente contribuiscono alla qualità; |
|
d) |
«gestione della qualità»: le attività coordinate per dirigere e controllare un’organizzazione sul piano della qualità; |
|
e) |
«procedure operative standard (POS)»: istruzioni scritte che descrivono le fasi di un determinato procedimento nonché i materiali e i metodi da utilizzare e il prodotto finale previsto; |
|
f) |
«convalida» (o «qualifica» in caso di attrezzature o ambienti): la produzione di prove documentate, in grado di garantire con un elevato livello di certezza che determinati procedimenti, attrezzature o ambienti diano luogo a un prodotto conforme alle specifiche e alle caratteristiche qualitative prestabilite; un procedimento è convalidato al fine di valutare se un sistema funziona efficacemente in rapporto all’impiego previsto; |
|
g) |
«rintracciabilità»: la facoltà di reperire e identificare i tessuti o le cellule in ogni fase dell’approvvigionamento, della lavorazione, del controllo e dello stoccaggio fino alla distribuzione al ricevente o allo smaltimento, compresa la capacità di identificare il donatore e l’istituto dei tessuti o il centro di produzione che ricevono, lavorano o stoccano i tessuti o le cellule e, a livello di servizi medici, la capacità di identificare i responsabili che applicano i tessuti o le cellule sui riceventi; la rintracciabilità comporta inoltre la facoltà di reperire e identificare tutti i dati pertinenti relativi ai prodotti e ai materiali che vengono a contatto con tali tessuti o cellule; |
|
h) |
«critico»: che ha potenzialmente effetto sulla qualità e/o la sicurezza di cellule e tessuti o è a contatto con cellule e tessuti; |
|
i) |
«organizzazione di approvvigionamento»: un centro sanitario, un’unità ospedaliera o un altro ente in cui si prelevano tessuti e cellule umani che può non essere accreditato, designato, autorizzato o titolare di licenza come istituto dei tessuti; |
|
j) |
«organizzazione responsabile dell’applicazione sull’uomo»: un centro sanitario, un’unità ospedaliera o un altro ente che esegue applicazioni sull’uomo di tessuti e cellule umani. |
Articolo 3
Prescrizioni per l’accreditamento, la designazione, l’autorizzazione o il rilascio di licenza agli istituti dei tessuti
Un istituto dei tessuti ottempera alle prescrizioni di cui all’allegato I.
Articolo 4
Prescrizioni per l’accreditamento, la designazione, l’autorizzazione, il rilascio di licenza ai procedimenti di preparazione di tessuti e cellule
Negli istituti dei tessuti i procedimenti di preparazione ottemperano alle prescrizioni di cui all’allegato II.
Articolo 5
Notifica di reazioni avverse gravi
1. Gli Stati membri garantiscono che:
|
a) |
le organizzazioni di approvvigionamento abbiano predisposto procedure per conservare le registrazioni dei tessuti e cellule prelevati e per notificare tempestivamente agli istituti dei tessuti ogni reazione avversa grave nel donatore vivente che possa influire sulla qualità e sicurezza di tessuti e cellule; |
|
b) |
le organizzazioni responsabili dell’applicazione sull’uomo di tessuti e cellule abbiano predisposto procedure per conservare le registrazioni dei tessuti e cellule applicati e per notificare tempestivamente agli istituti dei tessuti ogni reazione avversa grave osservata nel corso o a seguito dell’applicazione clinica, che possa essere in rapporto con la qualità e la sicurezza dei tessuti e delle cellule; |
|
c) |
gli istituti dei tessuti che distribuiscono tessuti e cellule per applicazioni sull’uomo forniscano all’organizzazione responsabile dell’applicazione sull’uomo di tessuti e cellule informazioni sulle modalità per notificare le reazioni avverse gravi di cui alla lettera b). |
2. Gli Stati membri garantiscono che gli istituti dei tessuti:
|
a) |
abbiano predisposto procedure per comunicare tempestivamente all’autorità competente tutte le informazioni disponibili pertinenti alle presunte reazioni avverse gravi di cui al paragrafo 1, lettere a) e b); |
|
b) |
abbiano predisposto procedure per comunicare tempestivamente all’autorità competente le conclusioni dell’indagine per analizzare le cause e il conseguente esito. |
3. Gli Stati membri garantiscono che:
|
a) |
la persona responsabile di cui all’articolo 17 della direttiva 2004/23/CE notifichi all’autorità competente le informazioni incluse nella notifica di cui alla parte A dell’allegato III; |
|
b) |
gli istituti dei tessuti notifichino all’autorità competente i provvedimenti adottati per quanto riguarda altri tessuti e cellule interessati, distribuiti a fini di applicazioni sull’uomo; |
|
c) |
gli istituti dei tessuti notifichino all’autorità competente le conclusioni dell’indagine, fornendo almeno le informazioni di cui alla parte B dell’allegato III. |
Articolo 6
Notifica di eventi avversi gravi
1. Gli Stati membri garantiscono che:
|
a) |
le organizzazioni di approvvigionamento e gli istituti dei tessuti abbiano predisposto procedure per conservare le registrazioni e per notificare tempestivamente ogni evento avverso grave che si verifichi durante l’approvvigionamento e possa influire sulla qualità e/o sicurezza dei tessuti e cellule; |
|
b) |
le organizzazioni responsabili dell’applicazione sull’uomo di tessuti e cellule abbiano predisposto procedure per notificare tempestivamente agli istituti dei tessuti ogni evento avverso grave che possa influire sulla qualità e sicurezza dei tessuti e cellule; |
|
c) |
gli istituti dei tessuti forniscano all’organizzazione responsabile dell’applicazione sull’uomo informazioni sulle modalità per notificare loro eventi avversi gravi che possano influire sulla qualità e sicurezza dei tessuti e cellule. |
2. In materia di riproduzione assistita si considera evento avverso grave ogni tipo di errore d’identificazione o di confusione di gameti o embrioni. Tutte le persone o le organizzazioni di approvvigionamento o le organizzazioni responsabili dell’applicazione sull’uomo riferiscono tali eventi agli istituti dei tessuti fornitori per indagine e notifica all’autorità competente.
3. Gli Stati membri garantiscono che gli istituti dei tessuti:
|
a) |
abbiano predisposto procedure per comunicare tempestivamente all’autorità competente tutte le informazioni disponibili pertinenti ai presunti eventi avversi gravi di cui al paragrafo 1, lettere a) e b); |
|
b) |
abbiano predisposto procedure per comunicare tempestivamente all’autorità competente le conclusioni dell’indagine per analizzare le cause e il conseguente esito. |
4. Gli Stati membri garantiscono che:
|
a) |
la persona responsabile di cui all’articolo 17 della direttiva 2004/23/CE notifichi all’autorità competente le informazioni incluse nella notifica di cui alla parte A dell’allegato IV; |
|
b) |
gli istituti dei tessuti valutino gli eventi avversi gravi per individuarne le cause evitabili nell’ambito del procedimento; |
|
c) |
gli istituti dei tessuti notifichino all’autorità competente le conclusioni dell’indagine, fornendo almeno le informazioni di cui alla parte B dell’allegato IV. |
Articolo 7
Relazioni annuali
1. Una relazione annuale sulle notifiche delle reazioni e degli eventi avversi gravi ricevute dall’autorità competente viene presentata dagli Stati membri alla Commissione entro il 30 giugno dell’anno successivo. La Commissione presenta alle autorità competenti degli Stati membri una sintesi delle relazioni ricevute. L’autorità competente mette tale relazione a disposizione degli istituti dei tessuti.
2. La trasmissione dei dati si attiene alle specifiche del modello di scambio di dati di cui all’allegato V, parti A e B, e fornisce tutte le informazioni necessarie ad identificare il mittente e a conservare i suoi dati di riferimento.
Articolo 8
Comunicazione di informazioni fra le autorità competenti e alla Commissione
Gli Stati membri garantiscono la comunicazione tra le rispettive autorità competenti e alla Commissione delle opportune informazioni riguardanti reazioni ed eventi avversi gravi, al fine di assicurare che vengano adottati provvedimenti adeguati.
Articolo 9
Rintracciabilità
1. Gli istituti dei tessuti dispongono di sistemi efficaci ed accurati per identificare ed etichettare individualmente cellule/tessuti ricevuti e distribuiti.
2. Gli istituti dei tessuti e le organizzazioni responsabili dell’applicazione sull’uomo conservano per almeno 30 anni i dati di cui all’allegato VI avvalendosi di un sistema di memorizzazione adeguato e leggibile.
Articolo 10
Sistema europeo di codifica
1. Un codice unico europeo d’identificazione viene attribuito a tutti i materiali donati all’istituto dei tessuti, al fine di garantire un’adeguata identificazione del donatore e la rintracciabilità di tutti i materiali donati, nonché di fornire informazioni sulle caratteristiche e proprietà fondamentali dei tessuti e cellule. Il codice comprende almeno le informazioni di cui all’allegato VII.
2. Il paragrafo 1 non si applica alla donazione di cellule riproduttive dal partner.
Articolo 11
Recepimento
1. Gli Stati membri adottano le disposizioni legislative, regolamentari e amministrative necessarie a conformarsi alla presente direttiva entro il 1o settembre 2007. Essi comunicano immediatamente alla Commissione il testo di tali disposizioni nonché una tavola di concordanza tra queste ultime e la presente direttiva.
Gli Stati membri adottano le disposizioni legislative, regolamentari e amministrative necessarie a conformarsi all’articolo 10 della presente direttiva entro il 1o settembre 2008.
Quando gli Stati membri adottano tali disposizioni, queste contengono un riferimento alla presente direttiva o sono corredate da tale riferimento all’atto della pubblicazione ufficiale. Le modalità del riferimento sono decise dagli Stati membri.
2. Gli Stati membri comunicano alla Commissione il testo delle disposizioni essenziali di diritto interno adottate nella materia disciplinata dalla presente direttiva.
Articolo 12
Entrata in vigore
La presente direttiva entra in vigore il ventesimo giorno successivo alla pubblicazione nella Gazzetta ufficiale dell’Unione europea.
Articolo 13
Destinatari
Gli Stati membri sono destinatari della presente direttiva.
Fatto a Bruxelles, il 24 ottobre 2006.
Per la Commissione
Markos KYPRIANOU
Membro della Commissione
(1) GU L 102 del 7.4.2004, pag. 48.
(2) http://pharmacos.eudra.org/F2/eudralex/vol-4/home.htm e GU L 262 del 14.10.2003, pag. 22.
(3) GU L 38 del 9.2.2006, pag. 40.
ALLEGATO I
Prescrizioni per l’accreditamento, la designazione, l’autorizzazione o il rilascio di licenza agli istituti dei tessuti di cui all’articolo 3
A. ORGANIZZAZIONE E GESTIONE
|
1. |
Occorre designare una persona responsabile che abbia le qualifiche e le responsabilità di cui all’articolo 17 della direttiva 2004/23/CE. |
|
2. |
Un istituto dei tessuti deve avere una struttura organizzativa e procedure operative adeguate alle attività per le quali si chiede l’accreditamento/la designazione/l’autorizzazione/il rilascio di licenza; occorre un organigramma che definisca chiaramente i rapporti in materia di responsabilità e di obblighi di riferire. |
|
3. |
Ogni istituto dei tessuti deve potersi avvalere di un medico riconosciuto designato per prestare consulenze e supervisionare le attività mediche dell’istituto, quali la selezione dei donatori, la verifica degli esiti clinici dei tessuti e cellule applicati o le eventuali interazioni con gli utenti clinici. |
|
4. |
Occorre che un sistema documentato di gestione della qualità sia applicato alle attività per le quali si richiede l’accreditamento/la designazione/l’autorizzazione o il rilascio di licenza, in conformità dei parametri di cui alla presente direttiva. |
|
5. |
Occorre garantire che i rischi inerenti all’uso e alla manipolazione di materiale biologico vengano individuati e minimizzati, coerentemente con il mantenimento di qualità e sicurezza adeguate alla destinazione prevista di tessuti e cellule. Sono compresi i rischi specificamente concernenti le procedure, l’ambiente e lo stato di salute del personale dell’istituto dei tessuti. |
|
6. |
Gli accordi tra istituti dei tessuti e terzi devono ottemperare all’articolo 24 della direttiva 2004/23/CE. Gli accordi con terzi devono specificare i termini del rapporto e le responsabilità, nonché i protocolli da seguire per corrispondere alle specifiche di funzionamento richieste. |
|
7. |
Occorre predisporre un sistema documentato, con la supervisione della persona responsabile, per confermare la conformità di tessuti e/o cellule ad adeguate specifiche di sicurezza e qualità per il rilascio e la distribuzione. |
|
8. |
In caso di cessazione delle attività gli accordi conclusi e le procedure adottate in conformità dell’articolo 21, paragrafo 5, della direttiva 2004/23/CE comprendono i dati sulla rintracciabilità e i materiali relativi alla qualità e alla sicurezza di cellule e tessuti. |
|
9. |
Occorre predisporre un sistema documentato che garantisca l’identificazione di ciascuna unità di tessuto o cellule in tutte le fasi delle attività per le quali si chiede l’accreditamento/la designazione/l’autorizzazione/il rilascio di licenza. |
B. PERSONALE
|
1. |
Il personale degli istituti dei tessuti dev’essere sufficiente di numero e qualificato per i compiti da svolgere. La competenza del personale dev’essere valutata ad intervalli adeguati precisati nel sistema di qualità. |
|
2. |
I mansionari di tutto il personale devono essere chiari, documentati e aggiornati. I relativi compiti, competenze e responsabilità devono essere ben documentati e compresi. |
|
3. |
Al personale occorre garantire una formazione iniziale/di base, gli aggiornamenti necessari quando cambiano le procedure o si sviluppano le conoscenze scientifiche, nonché adeguate possibilità di corrispondente crescita professionale. Il programma di formazione deve garantire e documentare che ciascun soggetto:
|
C. ATTREZZATURE E MATERIALI
|
1. |
La progettazione e la manutenzione di tutte le attrezzature e i materiali devono corrispondere alle loro destinazioni previste e minimizzare ogni rischio per i riceventi e/o il personale. |
|
2. |
Tutte le attrezzature e i dispositivi tecnici critici devono essere identificati e convalidati, periodicamente ispezionati e preventivamente sottoposti a manutenzione conformemente alle istruzioni del fabbricante. Le attrezzature o i materiali che incidono su parametri critici di lavorazione o stoccaggio (ad esempio temperatura, pressione, numero di particelle, livello di contaminazione microbica) devono essere identificati ed eventualmente sottoposti a osservazioni, vigilanza, allarmi e interventi correttivi adeguati per individuarne le disfunzioni e i difetti e per garantire che i parametri critici rimangano costantemente al di sotto dei limiti accettabili. Tutte le attrezzature che dispongono di una funzione di misurazione critica devono essere tarate su un parametro di riferimento reperibile, qualora esista. |
|
3. |
Le attrezzature nuove e riparate devono essere controllate al momento dell’installazione e convalidate prima dell’uso. I risultati dei controlli devono essere documentati. |
|
4. |
Occorre procedere periodicamente alla manutenzione, alla pulizia, alla disinfezione e all’igienizzazione di tutte le attrezzature critiche e alle relative registrazioni. |
|
5. |
Occorre disporre di norme di funzionamento per ogni attrezzatura critica, con indicazioni dettagliate di come intervenire in caso di disfunzioni o guasti. |
|
6. |
Le norme per le attività di cui si chiede l’accreditamento/la designazione/l’autorizzazione/il rilascio di licenza devono indicare dettagliatamente le specifiche di tutti i materiali e i reagenti critici. Devono essere in particolare definite le specifiche per gli additivi (ad esempio soluzioni) e i materiali d’imballaggio. I reagenti e i materiali critici devono corrispondere alle prescrizioni e alle specifiche documentate e, se del caso, alle prescrizioni della direttiva 93/42/CEE del Consiglio, del 14 giugno 1993, concernente i dispositivi medici (1) e della direttiva 98/79/CE del Parlamento europeo e del Consiglio, del 27 ottobre 1998, relativa ai dispositivi medico-diagnostici in vitro (2). |
D. SERVIZI/LOCALI
|
1. |
Un istituto dei tessuti deve avere servizi adeguati allo svolgimento di attività per le quali si chiede l’accreditamento/la designazione/l’autorizzazione o il rilascio di licenza, in conformità dei parametri di cui alla presente direttiva. |
|
2. |
Quando tali attività comprendono la lavorazione di tessuti e cellule a contatto con l’ambiente, essa deve svolgersi in un ambiente di specifica qualità e pulizia dell’aria al fine di minimizzare i rischi di contaminazione, compresa la contaminazione incrociata tra donazioni. L’efficacia di questi provvedimenti dev’essere convalidata e controllata. |
|
3. |
Fatte salve diverse disposizioni di cui al punto 4, se i tessuti o le cellule vengono a contatto con l’ambiente durante la lavorazione senza essere poi sottoposti a un procedimento di inattivazione microbica, occorre una qualità dell’aria con numeri di particelle e numeri di colonie microbiche equivalenti a quelli di grado A di cui alla Guida europea alle buone pratiche di fabbricazione (Good Manufacturing Practice: GMP), allegato 1 e alla direttiva 2003/94/CE, con un ambiente di fondo adeguato alla lavorazione dei tessuti/cellule interessati, ma almeno equivalente a GMP di grado D in termini di numeri di particelle e di colonie microbiche. |
|
4. |
Condizioni ambientali meno rigorose di quelle specificate al punto 3 possono essere accettabili qualora:
|
|
5. |
Il punto 4, lettere a), b), c) e d) prevede precisazioni sull’ambiente. Occorre dimostrare e documentare che l’ambiente prescelto corrisponde alla qualità e sicurezza richieste, prendendo almeno in considerazione la destinazione prevista, le modalità di applicazione e lo stato immunitario del ricevente. In ogni corrispondente reparto dell’istituto dei tessuti devono essere disponibili indumenti e attrezzature adeguati per la protezione e l’igiene personali, unitamente a istruzioni scritte relative all’igiene e all’obbligo del camice. |
|
6. |
Quando le attività per le quali si chiede l’accreditamento/la designazione/l’autorizzazione o il rilascio di licenza comportano lo stoccaggio di tessuti e cellule, occorre definire le condizioni di stoccaggio necessarie per mantenere le proprietà richieste per i tessuti e cellule, compresi i corrispondenti parametri relativi a temperatura, umidità o qualità dell’aria. |
|
7. |
Occorre controllare, sorvegliare e registrare i parametri critici (ad esempio temperatura, umidità, qualità dell’aria) per dimostrarne la corrispondenza con le specifiche condizioni di stoccaggio. |
|
8. |
Occorre predisporre servizi di stoccaggio che separino e distinguano nettamente i tessuti e le cellule precedenti al rilascio/in quarantena da quelli rilasciati e da quelli scartati, al fine di prevenirne la confusione e la contaminazione incrociata. Nei locali di stoccaggio di tessuti e cellule sia in quarantena che rilasciati occorre predisporre zone o dispositivi di stoccaggio fisicamente separati o isolamenti di sicurezza all’interno del dispositivo per la tenuta di determinati tessuti e cellule prelevati conformemente a criteri speciali. |
|
9. |
L’istituto dei tessuti deve disporre di politiche e procedure scritte per l’accesso controllato, la pulizia, la manutenzione e lo smaltimento dei rifiuti, nonché per garantire la riorganizzazione della prestazione dei servizi in situazioni di emergenza. |
E. DOCUMENTAZIONE E REGISTRAZIONI
|
1. |
Occorre predisporre un sistema che fornisca una documentazione chiaramente definita ed efficace, registrazioni e schede corrette nonché procedure operative standard (POS) per le attività di cui si chiede l’accreditamento/la designazione/l’autorizzazione/il rilascio di licenza. I documenti devono essere periodicamente verificati ed essere conformi ai parametri di cui alla presente direttiva. Il sistema deve garantire la standardizzazione dell’attività svolta e la rintraccialibilità di tutte le sue fasi, cioè codifica, idoneità di donatori, approvvigionamento, lavorazione, conservazione, stoccaggio, trasporto, distribuzione o smaltimento, compresi gli aspetti relativi al controllo di qualità e alla garanzia di qualità. |
|
2. |
Il materiale, le attrezzature e il personale coinvolti in ogni attività critica devono essere identificati e registrati. |
|
3. |
Negli istituti dei tessuti tutte le modifiche dei documenti devono essere verificate, datate, approvate, documentate ed eseguite puntualmente da personale autorizzato. |
|
4. |
Occorre istituire una procedura di controllo dei documenti che fornisca la storia delle verifiche e delle modifiche dei documenti e che garantisca che ne venga utilizzata solo la versione in corso. |
|
5. |
Occorre dimostrare che le registrazioni sono attendibili e che rappresentano correttamente i risultati. |
|
6. |
Le registrazioni devono essere leggibili e indelebili e possono essere manoscritte oppure avvalersi di un altro sistema convalidato, come il supporto elettronico o il microfilm. |
|
7. |
Fatto salvo quanto disposto all’articolo 9, paragrafo 2, tutte le registrazioni, dati grezzi compresi, che sono critiche per la sicurezza e la qualità dei tessuti e cellule vanno conservate per garantirne l’accessibilità per almeno 10 anni dopo la data di scadenza, l’uso clinico o lo smaltimento. |
|
8. |
Le registazioni devono ottemperare alle prescrizioni di riservatezza di cui all’articolo 14 della direttiva 2004/23/CE. L’accesso alla documentazione e ai dati dev’essere limitato ai soggetti autorizzati dalla persona responsabile, nonché all’autorità competente a fini di applicazione di misure d’ispezione e di controllo. |
F. VERIFICA DELLA QUALITÀ
|
1. |
Occorre predisporre un sistema di verifica delle attività per le quali si richiede l’accreditamento/la designazione/l’autorizzazione/il rilascio di licenza. Le verifiche devono essere eseguite in modo autonomo da persone qualificate e competenti almeno ogni due anni, al fine di accertare l’osservanza dei protocolli approvati e delle prescrizioni normative. I risultati e gli interventi correttivi devono essere documentati. |
|
2. |
Gli scostamenti rispetto ai parametri di qualità e sicurezza richiesti devono essere oggetto di indagini documentate, comprendenti anche decisioni relative ad eventuali interventi correttivi e preventivi. La sorte dei tessuti e delle cellule non conformi dev’essere decisa seguendo procedure scritte con la supervisione della persona responsabile e successivamente registrata. Occorre identificare tutti i tessuti e le cellule interessati e renderne conto. |
|
3. |
Gli interventi correttivi devono essere documentati, avviati e completati con puntualità ed efficacia. L’efficacia degli interventi preventivi e correttivi va valutata dopo l’attuazione. |
|
4. |
Occorre che l’istituto dei tessuti predisponga procedimenti di verifica del funzionamento del sistema di gestione della qualità per garantirne il progresso costante e sistematico. |
(1) GU L 169 del 12.7.1993, pag. 1. Direttiva modificata da ultimo dal regolamento (CE) n. 1882/2003 del Parlamento europeo e del Consiglio (GU L 284 del 31.10.2003, pag. 1).
(2) GU L 331 del 7.12.1998, pag. 1. Direttiva modificata dal regolamento (CE) n. 1882/2003.
ALLEGATO II
Prescrizioni per l’autorizzazione di procedimenti di preparazione di tessuti e cellule negli istituti di tessuti di cui all’articolo 4
L’autorità competente autorizza ogni procedimento di preparazione di tessuti e cellule dopo aver valutato i criteri di selezione del donatore e le procedure di approvvigionamento, i protocolli relativi a ciascuna fase del procedimento, i criteri di gestione della qualità e i criteri quantitativi e qualitativi definitivi per i tessuti e cellule. Tale valutazione deve almeno attenersi alle prescrizioni di cui al presente allegato.
A. RICEVIMENTO ALL’ISTITUTO DEI TESSUTI
Quando l’istituto dei tessuti riceve i tessuti e cellule prelevati, questi devono corrispondere alle prescrizioni di cui alla direttiva 2006/17/CE.
B. LAVORAZIONE
Quando le attività per le quali si chiede l’accreditamento/la designazione/l’autorizzazione/il rilascio di licenza comprendono la lavorazione di tessuti e cellule, le procedure dell’istituto dei tessuti devono attenersi ai seguenti criteri.
|
1. |
Le procedure di lavorazione critiche devono essere convalidate e non devono rendere i tessuti o le cellule clinicamente inefficaci o nocivi per il ricevente. La convalida può basarsi su studi eseguiti dall’istituto medesimo, o su dati di studi pubblicati, o — per procedure di lavorazione pienamente affermate — sulla valutazione retrospettiva dei risultati clinici relativi ai tessuti forniti dall’istituto. |
|
2. |
Occorre dimostrare che il procedimento di convalida può essere svolto in modo coerente ed efficace nell’ambito dello stabilimento dei tessuti ad opera del suo personale. |
|
3. |
Le procedure devono essere documentate nelle POS, che devono attenersi al metodo convalidato e ai parametri di cui alla presente direttiva, conformemente all’allegato I, parte E, punti da 1 a 4. |
|
4. |
Occorre garantire che tutti i procedimenti si svolgano in conformità delle POS approvate. |
|
5. |
Qualora ai tessuti o cellule venga applicato un procedimento d’inattivazione microbica, esso va specificato, documentato e convalidato. |
|
6. |
Prima di ogni modifica significativa della lavorazione, il procedimento modificato dev’essere convalidato e documentato. |
|
7. |
Le procedure di lavorazione vanno periodicamente sottoposte a valutazione critica, per garantire che continuino a conseguire i risultati previsti. |
|
8. |
Le procedure per scartare tessuti e cellule devono impedire la contaminazione di altri tessuti e cellule, dell’ambiente di lavorazione o del personale. Tali procedure devono attenersi alle normative nazionali. |
C. STOCCAGGIO E RILASCIO DI PRODOTTI
Quando le attività per le quali si chiede l’accreditamento/la designazione/l’autorizzazione/il rilascio di licenza comprendono lo stoccaggio e il rilascio di tessuti e cellule, le procedure autorizzate dall’istituto dei tessuti devono attenersi ai seguenti criteri.
|
1. |
Per ogni tipo di condizione di stoccaggio dev’essere precisato un tempo massimo. Il periodo prescelto deve tra l’altro tener conto dell’eventuale deterioramento delle proprietà richieste per tessuti e cellule. |
|
2. |
Occorre un sistema di tenuta d’inventario per i tessuti e/o le cellule per garantire che non vengano rilasciati prima che siano state rispettate tutte le prescrizioni di cui alla presente direttiva. Occorre una procedura operativa standard che precisi le circostanze, le responsabilità e le procedure inerenti al rilascio di tessuti e cellule per la distribuzione. |
|
3. |
Un sistema per l’identificazione di tessuti e cellule in ogni fase di lavorazione nell’istituto dei tessuti deve distinguere nettamente i prodotti rilasciati da quelli non rilasciati (in quarantena) e da quelli scartati. |
|
4. |
Le registrazioni devono dimostrare che prima del rilascio di tessuti e cellule sono state rispettate tutte le corrispondenti specifiche; che in particolare tutti i moduli di dichiarazione in uso, le cartelle mediche pertinenti, le registrazioni di lavorazione e i risultati dei controlli sono stati verificati in base a una procedura scritta da un soggetto autorizzato a questo scopo dalla persona responsabile di cui all’articolo 17 della direttiva 2004/23/CE. Se per comunicare i risultati di laboratorio si usa un computer, una pista di controllo deve indicare il responsabile del loro rilascio. |
|
5. |
Occorre eseguire una valutazione dei rischi documentata, approvata dalla persona responsabile di cui all’articolo 17 della direttiva 2004/23/CE, per decidere la sorte di tutti i tessuti e cellule stoccati dopo l’introduzione di nuovi criteri di selezione o controllo dei donatori o di notevoli modifiche di fasi di lavorazione, al fine di rafforzare la sicurezza o la qualità. |
D. DISTRIBUZIONE E RITIRO
Quando le attività per le quali si chiede l’accreditamento/la designazione/l’autorizzazione/il rilascio di licenza comprendono la distribuzione di tessuti e cellule, le procedure autorizzate dell’istituto dei tessuti devono attenersi ai seguenti criteri.
|
1. |
Occorre definire le condizioni di trasporto critiche, quali la temperatura e le scadenze temporali, per il mantenimento delle proprietà richieste per tessuti e cellule. |
|
2. |
Il contenitore/imballaggio dev’essere sicuro e garantire la conservazione di tessuti e cellule nelle condizioni precisate. Tutti i contenitori e gli imballaggi devono essere convalidati come idonei allo scopo. |
|
3. |
Se la distribuzione viene affidata a terzi, occorre predisporre un accordo documentato che garantisca il mantenimento delle condizioni richieste. |
|
4. |
L’istituto dei tessuti deve disporre di personale autorizzato per valutare le eventuali esigenze di ritiro e per avviare e coordinare le azioni necessarie. |
|
5. |
Occorre predisporre un’efficace procedura di ritiro, che includa una descrizione delle responsabilità e delle azioni da intraprendere, compresa la notifica all’autorità competente. |
|
6. |
Le azioni devono essere intraprese entro un periodo predefinito, devono prevedere che vengano rintracciati i tessuti e cellule interessati e, se del caso, includere una ricostruzione del percorso. L’indagine ha lo scopo di identificare ogni donatore che possa aver contribuito a causare la reazione nel ricevente, di recuperare i tessuti e cellule provenienti da tale donatore e di informare destinatari e riceventi dei tessuti e cellule prelevati da questo donatore dell’eventuale rischio a cui possono essere esposti. |
|
7. |
Occorre predisporre procedure per il trattamento delle richieste di tessuti e cellule. Le norme per l’assegnazione di tessuti e cellule a determinati pazienti o centri sanitari devono essere documentate e loro comunicate a richiesta. |
|
8. |
Occorre predisporre un sistema documentato per il trattamento dei prodotti restituiti, comprendente, se del caso, i criteri per la loro iscrizione nell’inventario. |
E. ETICHETTATURA FINALE PER LA DISTRIBUZIONE
|
1. |
Il contenitore primario di tessuti/cellule deve indicare:
Se alcune delle informazioni di cui alle lettere d) ed e) non possono essere incluse nell’etichetta del contenitore primario, devono essere fornite su un foglio separato ad esso allegato. Il foglio dev’essere imballato insieme al contenitore primario in modo da garantire che rimangano uniti. |
|
2. |
Le seguenti informazioni devono essere riportate sull’etichetta o nella documentazione di accompagnamento:
|
F. ETICHETTATURA ESTERNA DEL CONTENITORE PER LA SPEDIZIONE
A fini di trasporto il contenitore primario dev’essere collocato in un contenitore per la spedizione, la cui etichetta deve contenere almeno le seguenti informazioni:
|
a) |
identificazione dell’istituto dei tessuti d’origine, compresi indirizzo e numero telefonico; |
|
b) |
identificazione dell’organizzazione responsabile dell’applicazione sull’uomo destinatario, compresi indirizzo e numero telefonico; |
|
c) |
indicazione che l’imballaggio contiene tessuti/cellule umani con la dicitura MANIPOLARE CON CAUTELA; |
|
d) |
se ai fini del trapianto occorrono cellule viventi, quali cellule staminali, gameti e embrioni, va aggiunta la dicitura NON IRRADIARE; |
|
e) |
condizioni di trasporto raccomandate (ad esempio conservare al fresco, in posizione verticale, ecc.); |
|
f) |
istruzioni per la sicurezza/metodo di raffreddamento (se del caso). |
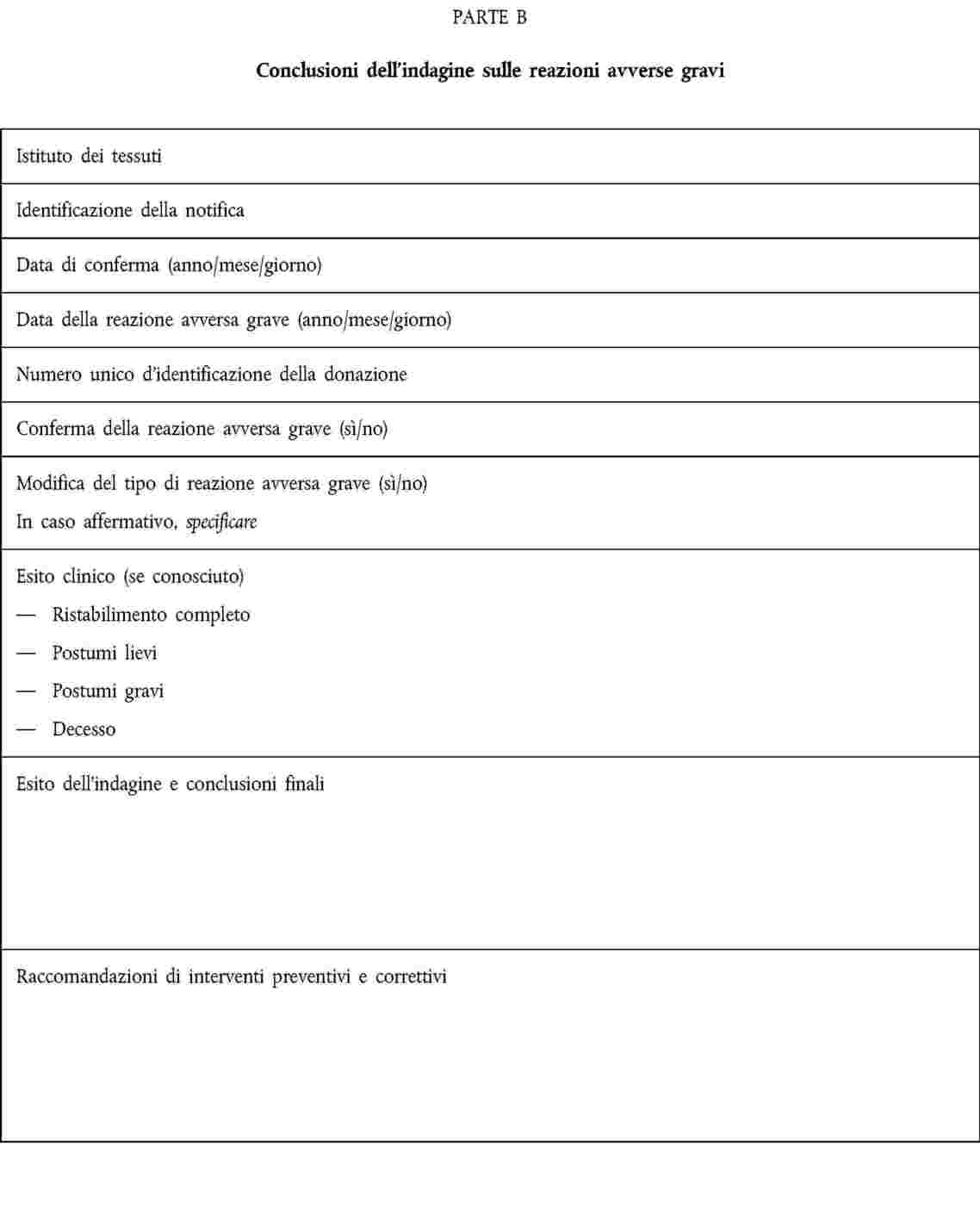
ALLEGATO III
NOTIFICA DI REAZIONI AVVERSE GRAVI

ALLEGATO IV
NOTIFICA DI EVENTI AVVERSI GRAVI

ALLEGATO V
MODELLO DI NOTIFICA ANNUALE


ALLEGATO VI
Informazioni sui dati minimi relativi al donatore/ricevente da conservare a norma dell’articolo 9
A. PER ISTITUTI DEI TESSUTI
Identificazione del donatore
Identificazione della donazione che deve almeno comprendere:
|
— |
l’identificazione dell’organizzazione di approvvigionamento o dell’istituto dei tessuti, |
|
— |
il numero unico d’identificazione della donazione, |
|
— |
la data dell’approvvigionamento, |
|
— |
il luogo dell’approvvigionamento, |
|
— |
il tipo di donazione (ad esempio di un tessuto unico/di più tessuti, autologa/allogenica, da vivente/da deceduto). |
Identificazione del prodotto che deve almeno comprendere:
|
— |
l’identificazione dell’istituto dei tessuti, |
|
— |
il tipo di prodotto di tessuti e cellule (nomenclatura di base), |
|
— |
il numero aggregato delle partite (se del caso), |
|
— |
il numero specifico della sottopartita (se del caso), |
|
— |
la data di scadenza, |
|
— |
lo stato dei tessuti/cellule (in quarantena, idonei all’uso, ecc.), |
|
— |
la descrizione e l’origine dei prodotti, delle fasi di lavorazione in uso, dei materiali e degli additivi che vengono a contatto con tessuti e cellule e hanno effetto sulla loro qualità e/o sicurezza, |
|
— |
l’identificazione del servizio responsabile dell’etichettatura finale. |
Identificazione dell’applicazione che deve almeno comprendere:
|
— |
la data di distribuzione/smaltimento, |
|
— |
l’identificazione del clinico o dell’utente finale/del servizio. |
B. PER ORGANIZZAZIONI RESPONSABILI DELL’APPLICAZIONE SULL’UOMO
|
a) |
Identificazione dell’istituto dei tessuti fornitore |
|
b) |
Identificazione del clinico o dell’utente/del servizio finale |
|
c) |
Tipo di tessuti e cellule |
|
d) |
Identificazione del prodotto |
|
e) |
Identificazione del ricevente |
|
f) |
Data dell’applicazione |
ALLEGATO VII
Informazioni contenute nel sistema di codifica europeo
|
a) |
Identificazione della donazione:
|
|
b) |
Identificazione del prodotto:
|