

EUR-Lex Access to European Union law
This document is an excerpt from the EUR-Lex website
Document 32018R0150
Commission Implementing Regulation (EU) 2018/150 of 30 January 2018 amending Implementing Regulation (EU) 2016/1240 as regards methods for the analysis and quality evaluation of milk and milk products eligible for public intervention and aid for private storage
Komisjoni rakendusmäärus (EL) 2018/150, 30. jaanuar 2018, millega muudetakse rakendusmäärust (EL) 2016/1240 seoses riikliku sekkumise ja eraladustamistoetuse jaoks kõlbliku piima ja kõlblike piimatoodete analüüsimise ning kvaliteedi hindamise meetoditega
Komisjoni rakendusmäärus (EL) 2018/150, 30. jaanuar 2018, millega muudetakse rakendusmäärust (EL) 2016/1240 seoses riikliku sekkumise ja eraladustamistoetuse jaoks kõlbliku piima ja kõlblike piimatoodete analüüsimise ning kvaliteedi hindamise meetoditega
C/2018/0480
OJ L 26, 31.1.2018, p. 14–47
(BG, ES, CS, DA, DE, ET, EL, EN, FR, HR, IT, LV, LT, HU, MT, NL, PL, PT, RO, SK, SL, FI, SV)
 In force
In force
|
31.1.2018 |
ET |
Euroopa Liidu Teataja |
L 26/14 |
KOMISJONI RAKENDUSMÄÄRUS (EL) 2018/150,
30. jaanuar 2018,
millega muudetakse rakendusmäärust (EL) 2016/1240 seoses riikliku sekkumise ja eraladustamistoetuse jaoks kõlbliku piima ja kõlblike piimatoodete analüüsimise ning kvaliteedi hindamise meetoditega
EUROOPA KOMISJON,
võttes arvesse Euroopa Liidu toimimise lepingut,
võttes arvesse Euroopa Parlamendi ja nõukogu 17. detsembri 2013. aasta määrust (EL) nr 1306/2013 ühise põllumajanduspoliitika rahastamise, haldamise ja seire kohta ning millega tunnistatakse kehtetuks nõukogu määrused (EMÜ) nr 352/78, (EÜ) nr 165/94, (EÜ) nr 2799/98, (EÜ) nr 814/2000, (EÜ) nr 1290/2005 ja (EÜ) nr 485/2008, (1) eriti selle artikli 62 lõike 2 punkti i,
ning arvestades järgmist:
|
(1) |
Komisjoni delegeeritud määruses (EL) 2016/1238 (2) ja komisjoni rakendusmääruses (EL) 2016/1240 (3) on sätestatud riikliku sekkumise ja eraladustamistoetuse eeskirjad. Komisjoni määruses (EÜ) nr 273/2008 (4) on sätestatud meetodid, mida kasutatakse, et hinnata piima ja piimatoodete vastavust kõnealustes määrustes sätestatud kõlblikkusnõuetele seoses riikliku sekkumise ja eraladustamistoetusega. |
|
(2) |
Võttes arvesse piima ja piimatoodete analüüsi- ja kvaliteedihindamismetoodika tehnilist arengut, tuleks teha sisulisi muutusi, et kõnealust metoodikat lihtsustada ja ajakohastada viited ISO standarditele. Selguse ja tõhususe huvides ning võttes arvesse määruse (EÜ) nr 273/2008 sätete muutmise ulatust ja tehnilist iseloomu, tuleks kõnealuse määruse asjaomased sätted lisada rakendusmäärusesse (EL) 2016/1240. |
|
(3) |
Et tagada uute standardite ja meetodite ühtne järgimine kõigis liikmesriikides, tuleks laboritele anda piisavalt aega menetluste läbivaatamiseks ja ajakohastatud meetodite rakendamiseks. |
|
(4) |
Seepärast tuleks rakendusmäärust (EL) 2016/1240 vastavalt muuta. |
|
(5) |
Õiguskindluse huvides tuleks määrus (EÜ) nr 273/2008 kehtetuks tunnistada. |
|
(6) |
Käesoleva määrusega ettenähtud meetmed on kooskõlas põllumajandusturgude ühise korralduse komitee arvamusega, |
ON VASTU VÕTNUD KÄESOLEVA MÄÄRUSE:
Artikkel 1
Rakendusmäärust (EL) 2016/1240 muudetakse järgmiselt.
|
1) |
Artiklit 4 muudetakse järgmiselt:
|
|
2) |
Lisatakse artikkel 60a: „Artikkel 60a Riikliku sekkumise ja eraladustamistoetusega seotud piima ja piimatoodete kontrollide erisätted 1. Või, lõssipulbri ja juustu kõlblikkus eraladustamistoetuse saamiseks tehakse kindlaks vastavalt VI, VII ja VIII lisaga kehtestatud meetoditele. Kõnealused meetodid peavad vastama asjaomaste Euroopa või rahvusvaheliste standardite viimastele versioonidele, mis on kehtinud vähemalt kuus kuud enne riikliku sekkumise ajavahemiku esimest päeva, nagu määratletud määruse (EL) nr 1308/2013 artiklis 12. 2. Käesoleva määrusega sätestatud meetodite abil läbi viidud kontrollide tulemusi hinnatakse vastavalt IX lisale.“ |
|
3) |
Lisasid muudetakse vastavalt käesoleva määruse lisale. |
Artikkel 2
Määrus (EÜ) nr 273/2008 tunnistatakse kehtetuks.
Artikkel 3
Käesolev määrus jõustub seitsmendal päeval pärast selle avaldamist Euroopa Liidu Teatajas.
Käesolev määrus on tervikuna siduv ja vahetult kohaldatav kõikides liikmesriikides.
Brüssel, 30. jaanuar 2018
Komisjoni nimel
president
Jean-Claude JUNCKER
(1) ELT L 347, 20.12.2013, lk 549.
(2) Komisjoni 18. mai 2016. aasta delegeeritud määrus (EL) 2016/1238, millega täiendatakse Euroopa Parlamendi ja nõukogu määrust (EL) nr 1308/2013 riikliku sekkumise ja eraladustamistoetuse osas (ELT L 206, 30.7.2016, lk 15).
(3) Komisjoni 18. mai 2016. aasta rakendusmäärus (EL) 2016/1240, millega kehtestatakse Euroopa Parlamendi ja nõukogu määruse (EL) nr 1308/2013 rakenduseeskirjad riikliku sekkumise ja eraladustamistoetuse osas (ELT L 206, 30.7.2016, lk 71).
(4) Komisjoni 5. märtsi 2008. aasta määrus (EÜ) nr 273/2008, millega kehtestatakse nõukogu määruse (EÜ) nr 1255/1999 üksikasjalikud rakenduseeskirjad piima ja piimatoodete analüüsimise ning kvaliteedi hindamise meetodite kohta (ELT L 88, 29.3.2008, lk 1).
LISA
Rakendusmääruse (EL) 2016/1240 lisasid muudetakse järgmiselt.
|
1) |
IV lisa muudetakse järgmiselt:
|
||||||||||||||||||||||||||||||||||||||||||||||||||
|
2) |
V lisas lisatakse järgmine Ia osa: „IA OSA Lõssipulbri analüüsimeetodid seoses riikliku sekkumisega
I liide LÕSSIPULBER: FOSFATIDÜÜLSERIINI JA FOSFATIDÜÜLETANOOLAMIINI KVANTITATIIVNE MÄÄRAMINE Meetod: pöördfaasiline kõrgsurvevedelikkromatograafia (HPLC) 1. EESMÄRK JA RAKENDUSALA Käesoleva meetodiga on võimalik määrata fosfatidüülseriini (PS) ja fosfatidüületanoolamiini (PE) kogus lõssipulbris ja sellega saab tuvastada petipiimapulbri lisandit lõssipulbris. 2. MÄÄRATLUS PS + PE sisaldus: aine massiosa, mis on määratud siinkirjeldatud meetodi abil. Tulemus väljendatakse dipalmitoüülfosfatidüületanoolamiini (PEDP) milligrammides 100 g pulbri kohta. 3. MEETODI PÕHIMÕTE Taastatud piimapulbrist ekstraheeritakse metanooliga aminofosfolipiidid. PS ja PE määratakse o-ftaaldialdehüüdderivaatidena (OPA) pöördfaasilise HPLC ja fluorestsentsdetektori abil. PS ja PE sisaldus uuritavas proovis määratakse kvantitatiivselt standardproovi suhtes, mis sisaldab teadaolevat kogust PEDPd. 4. REAKTIIVID Kõik reaktiivid peavad olema analüütiliselt puhtad. Kasutada tuleb destilleeritud või vähemalt samaväärse puhtusastmega vett, kui ei ole ette nähtud teisiti. 4.1. Standardaine: vähemalt 99 % puhtusastmega PEDP Märkus: standardainet tuleb säilitada – 18 °C juures. 4.2. Reaktiivid standardproovide ja uuritavate proovide valmistamiseks 4.2.1. HPLC jaoks sobiva puhtusastmega metanool 4.2.2. HPLC jaoks sobiva puhtusastmega kloroform 4.2.3. Trüptamiinmonohüdrokloriid 4.3. Reaktiivid o-ftaaldialdehüüdi derivaatide valmistamiseks 4.3.1. Naatriumhüdroksiidi 12 M vesilahus 4.3.2. Boorhappe 0,4 M vesilahus, pH naatriumhüdroksiidiga (4.3.1) reguleeritud 10,0-ni 4.3.3. 2-merkaptoetanool 4.3.4. o-ftaaldialdehüüd (OPA) 4.4. HPLC elueerimislahustid 4.4.1. Elueerimislahustid peavad olema valmistatud HPLC jaoks sobiva puhtusastmega reaktiividest. 4.4.2. HPLC jaoks sobiva puhtusastmega vesi 4.4.3. Metanool, mille puhtus on fluorimeetriliselt kontrollitud 4.4.4. Tetrahüdrofuraan 4.4.5. Naatriumdivesinikfosfaat 4.4.6. Naatriumatsetaat 4.4.7. Äädikhape 5. APARATUUR 5.1. Analüütilised kaalud, mõõtetäpsus 1 mg, jaotise väärtus 0,1 mg. 5.2. Keeduklaasid ruumalaga 25 ja 100 ml 5.3. Pipetid, millega saab lisada 1 ja 10 ml 5.4. Magnetsegur 5.5. Mõõtepipetid, millega saab lisada 0,2, 0,5 ja 5 ml 5.6. Mõõtekolvid ruumalaga 10, 50 ja 100 ml 5.7. Süstlad ruumalaga 20 ja 100 μl. 5.8. Ultrahelivann 5.9. Tsentrifuug, mis võimaldab kiirendusi 27 000 × g 5.10. Klaasviaalid ruumalaga ligikaudu 5 ml 5.11. Mõõtesilinder ruumalaga 25 ml 5.12. pH-meeter täpsusega kuni 0,1 pH ühikut 5.13. HPLC seadmed 5.13.1. Gradientpumbasüsteem, töökiirus 1,0 ml/min 200-baarise rõhu juures 5.13.2. Automaatproovisisesti, mille abil saab valmistada derivaate 5.13.3. Kolonnisoojendi, mis võimaldab hoida kolonni temperatuuril 30 ± 1 °C 5.13.4. Fluorestsentsdetektor, reguleeritud ergastuslainepikkusele 330 nm ja kiirguslainepikkusele 440 nm 5.13.5. Integraator või andmetöötlustarkvara, mis võimaldab mõõta piikide pindala 5.13.6. Kolonn LiChrospher® – 100 (250 × 4,6 mm) või samaväärne kolonn, mis on täidetud oktadetsüülsilaaniga (C 18), osakeste suurus 5 μm. 6. PROOVIVÕTT Proovid tuleb võtta vastavalt ISO standardile 707. 7. TÖÖ KÄIK 7.1. Sisestandardlahuse valmistamine 7.1.1. Kaaluda 100 ml mõõtekolbi (5.6) 30,0 ± 0,1 mg trüptamiinmonohüdrokloriidi (4.2.3) kaalutis ning täita kolb metanooliga (4.2.1) kuni märgini. 7.1.2. Pipettida 1 ml (5.3) saadud lahust 10 ml mõõtekolbi (5.6) ning täita kolb metanooliga (4.2.1) kuni märgini, et saavutada trüptamiini kontsentratsioon 0,15 mM. 7.2. Uuritava proovi lahuse valmistamine 7.2.1. Kaaluda 25 ml keeduklaasi (5.2) 1,000 ± 0,001 g lõssipulbri proovi. Lisada pipetiga (5.3) 10 ml destilleeritud vett temperatuuriga 40 ± 1 °C ja segada magnetseguriga (5.4) 30 minutit, kuni kõik tükid on lahustunud. 7.2.2. Pipettida 0,2 ml (5.5) taastatud piima 10 ml mõõtekolbi (5.6), lisada süstlaga (5.7) 100 μl 0,15 mM trüptamiinilahust (7.1) ja täiendada metanooliga (4.2.1) märgini. Segada hoolikalt, pöörates kolbi korduvalt ümber, ja töödelda ultraheliga (5.8) 15 minutit. 7.2.3. Tsentrifuugida (5.9) 27 000 × g juures 10 minutit ja koguda supernatant klaasviaali (5.10). Märkus: uuritava proovi lahust tuleks HPLC analüüsi tegemiseni säilitada 4 °C juures. 7.3. Välisstandardlahuse valmistamine 7.3.1. Kaaluda 50 ml mõõtekolbi (5.6) 55,4 mg PEDPd (4.1) ning lisada mõõtesilindriga (5.11) ligikaudu 25 ml kloroformi (4.2.2). Kuumutada korgiga suletud kolb temperatuurini 50 ± 1 °C ja segada hoolikalt, kuni PEDP on lahustunud. Jahutada kolb temperatuurini 20 °C, täiendada metanooliga (4.2.1) märgini ja segada kolbi ümber pöörates. 7.3.2. Pipettida 1 ml (5.3) saadud lahust 100 ml mõõtekolbi (5.6) ning täiendada metanooliga (4.2.1) märgini. Pipettida 1 ml (5.3) saadud lahust 10 ml mõõtekolbi (5.6), lisada 100 μl (5.7) 0,15 mM trüptamiinilahust (7.1) ja täiendada metanooliga (4.2.1) märgini. Segada, pöörates kolbi korduvalt ümber. Märkus: standardproovi lahust tuleks HPLC-analüüsi tegemiseni säilitada 4 °C juures. 7.4. Derivaatimisreaktiivi valmistamine Kaaluda 10 ml mõõtekolbi (5.6) 25,0 ± 0,1 mg OPAd (4.3.4), lisada 0,5 ml (5.5) metanooli (4.2.1) ja segada hoolikalt, et OPA lahustuks. Täiendada märgini boorhappe lahusega (4.3.2) ning lisada süstlaga (5.7) 20 μl 2-merkaptoetanooli (4.3.3). Märkus: derivaatimisreaktiivi tuleb säilitada tumedas klaasviaalis 4 °C juures ning see säilib üks nädal. 7.5. Määramine HPLC-ga 7.5.1. Elueerimislahustid (4.4) Lahusti A: 0,3 mM naatriumdivesinikfosfaadi ja 3 mM naatriumatsetaadi lahus (pH reguleeritud äädikhappega väärtuseni 6,5 ± 0,1): metanool: tetrahüdrofuraan = 558:440:2 (mahusuhe) Lahusti B: metanool 7.5.2. Soovitatav elueerimisgradient
Märkus: Joonisel 1 näidatud lahutusvõime saavutamiseks võib olla vaja elueerimisgradienti natuke muuta. Kolonni temperatuur: 30 °C. 7.5.3. Sisestusruumala: 50 μl derivaatimisreaktiivi ja 50 μl proovilahust 7.5.4. Kolonni tasakaalustamine Süsteemi tavalisel igapäevasel käivitamisel pesta kolonni 15 minutit 100 % lahustiga B, seejärel segada A:B suhtes 40:60 ja tasakaalustada 15 minutit voolutuskiirusel 1 ml/min. Teha pimekatse, sisestades metanooli (4.2.1). Märkus: enne pikaajalist hoiustamist pesta kolonni metanooli ja kloroformi seguga (mahusuhe 80:20) 30 minutit. 7.5.5. PS + PE sisalduse määramine uuritavas proovis 7.5.6. Teha järjestikused kromatograafilised analüüsid, hoides nende läbiviimise vahelise aja konstantsena, et saada konstantseid retentsiooniaegu. Tundlikkusteguri arvutamiseks sisestada iga 5–10 uuritava proovilahuse järel välisstandardlahust (7.3). Märkus: pärast iga 20–25 töötsüklit tuleb kolonni puhastada, pestes vähemalt 30 minutit 100 % lahustiga B (7.5.1). 7.6. Integreerimismeetod 7.6.1. PEDP piik PEDP elueeritakse ühe piigina. Piigi pindala määratakse miinimumist miinimumini integreerimisega. 7.6.2. Trüptamiinipiik Trüptamiin elueeritakse ühe piigina (joonis 1). Piigi pindala määratakse miinimumist miinimumini integreerimisega. 7.6.3. PS ja PE piigirühmad Kirjeldatud tingimustel (joonis 1) elueerub PS kahe peamise osaliselt lahknemata piigina, millele eelneb väiksem piik. PE elueerub kolme peamise osaliselt lahknemata piigina. Iga piigirühma kogupindala määramiseks pannakse nulljoon paika joonisel 1 esitatud viisil. 8. TULEMUSTE ARVUTAMINE JA ESITAMINE PS ja PE sisaldus uuritavas proovis arvutatakse järgmiselt: C = 55,36 × ((A2)/(A1)) × ((T1)/(T2)) kus:
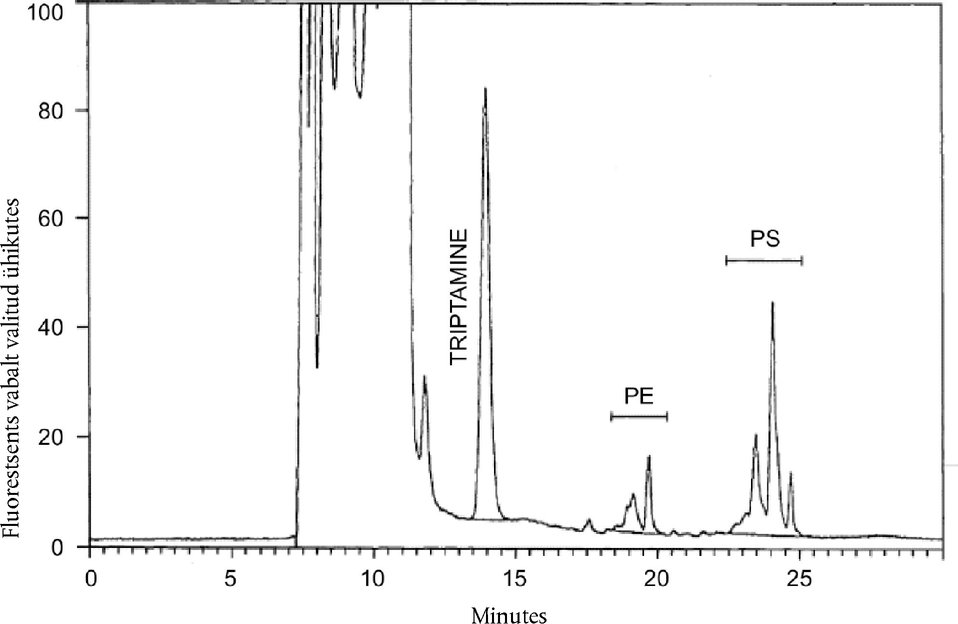
9. MEETODI TÄPSUS Märkus: Korduvuse väärtused on arvutatud IDFi rahvusvahelise standardi (*) kohaselt. 9.1. Korduvus Korduvuse suhteline standardhälve, mis väljendab sõltumatute analüüsitulemuste varieeruvust ning mis on lühikese aja jooksul saadud ühe ja sama analüüsija poolt samades tingimustes sama seadme ja prooviga, ei tohi ületada 2 %. Kui nendes tingimustes on saadud kaks tulemust, ei tohi kahe tulemuse suhteline erinevus olla suurem kui 6 % tulemuste aritmeetilisest keskmisest. 9.2. Korratavus Kui eri laborite analüüsijad on ühe ja sama proovi analüüsimisel saanud kaks erinevat tulemust, kasutades eri seadmeid eri tingimustes, ei tohi kahe tulemuse suhteline erinevus olla suurem kui 11 % nende tulemuste aritmeetilisest keskmisest. 10. KIRJANDUS 10.1. Resmini P., Pellegrino L., Hogenboom J. A., Sadini V., Rampilli M., „Detection of buttermilk solids in skimmilk powder by HPLC quantification of aminophospholipids.“ Sci. Tecn. Latt.-Cas., 39, 395 (1988). Joonis 1 Fosfatidüülseriini (PS) ja fosfatidüületanoolamiini (PE) OPA-derivaatide piigid pulbrist taastatud lõssi metanooliekstrakti HPLC-kromatogrammil. Näidatud on PS, PE ja trüptamiini (sisestandard) piikide integreerimine.
II liide RIIKLIKUKS LADUSTAMISEKS ETTE NÄHTUD LÕSSIPULBRIS ESINEVA LAABIVADAKU TUVASTAMINE KASEIINI MAKROPEPTIIDIDE MÄÄRAMISE TEEL KÕRGSURVEVEDELIKKROMATOGRAAFIA (HPLC) ABIL 1. RAKENDATAVUS JA RAKENDUSALA Käesoleva meetodiga on võimalik kaseiini makropeptiidide määramise teel tuvastada laabivadaku lisand riiklikuks ladustamiseks ette nähtud lõssipulbris. 2. VIIDE Rahvusvaheline standard ISO 707 – Piim ja piimatooted – Proovivõtujuhend. 3. MÄÄRATLUS Tahke laabivadaku sisaldus massiprotsentides leitakse kaseiini makropeptiidide sisalduse kaudu kirjeldatud meetodit kasutades. 4. PÕHIMÕTE
5. REAKTIIVID Kõik reaktiivid peavad olema analüütiliselt puhtad. Kasutada tuleb destilleeritud või vähemalt samaväärse puhtusastmega vett. 5.1. Trikloroäädikhappe lahus Lahustada vees 240 g trikloroäädikhapet (CCl3COOH) ja täiendada ruumalani 1 000 ml. Lahus peab olema puhas ja värvitu. 5.2. Eluentlahus, pH 6,0 Lahustada 1,74 g dikaaliumvesinikfosfaati (K2HPO4), 12,37 g kaaliumdivesinikfosfaati (KH2PO4) ja 21,41 g naatriumsulfaati (Na2SO4) umbes 700 ml vees. Vajaduse korral reguleerida pH 6,0-le, kasutades fosforhappe või kaaliumhüdroksiidi lahust. Täiendada veega 1 000 ml märgini ja homogeniseerida. Märkus: eluendi koostist võib ajakohastada, et see vastaks etalonide sertifikaadile või kolonni täitematerjali tootja soovitustele. Enne kasutamist filtrida eluentlahus läbi membraanfiltri, mille pooride läbimõõt on 0,45 μm. 5.3. Pesulahus Segada üks mahuosa atsetonitriili (CH3CN) üheksa mahuosa veega. Enne kasutamist filtrida segu läbi membraanfiltri, mille pooride läbimõõt on 0,45 μm. Märkus: võib kasutada mõnda muud baktereid tapva toimega pesulahust, mis ei vähenda kolonnide lahutusvõimet. 5.4. Standardproovid 5.4.1. Lõssipulber, mis vastab käesoleva määruse nõuetele (s.t [0]) 5.4.2. Sama lõssipulber, millele on lisatud 5 massiprotsenti standardkoostisega laabivadakupulbrit (s.t [5]) 6. APARATUUR 6.1. Analüütilised kaalud 6.2. Tsentrifuug, mis võimaldab saavutada tsentrifuugimiskiirenduse 2 200 g ja on varustatud umbes 50 ml suletavate tsentrifuugianumatega 6.3. Mehhaaniline loksuti 6.4. Magnetsegur 6.5. Umbes 7 cm läbimõõduga klaaslehtrid 6.6. Umbes 12,5 cm läbimõõduga keskmise tihedusega paberfiltrid 6.7. Klaasist filtrimisseade, mille membraanfiltri pooride läbimõõt on 0,45 μm 6.8. Mõõtepipetid, mis võimaldavad mõõta 10 ml (ISO 648, klass A või ISO/R 835), või doseerimisseade, millega saab mõõta 10,0 ml kahe minuti jooksul 6.9. Doseerimisseade, mis võimaldab mõõta 20,0 ml vett umbes 50 °C juures 6.10. Termostaadiga vesivann, reguleeritud temperatuurile 25 ± 0,5 °C 6.11. HPLC-seade, mille komplekti kuulub:
7. PROOVIVÕTT 7.1. Proove võetakse vastavalt rahvusvahelise standardiga ISO 707 sätestatud korrale. Liikmesriigid võivad kasutada muud proovide võtmise meetodit, kui see on kooskõlas eespool nimetatud standardi põhimõtetega. 7.2. Proovi hoitakse tingimustes, mis välistavad selle riknemise või koostise muutumise. 8. TÖÖ KÄIK 8.1. Uuritava proovi ettevalmistamine Panna lõssipulber õhukindla kaanega anumasse, mille maht on lõssipulbri mahust umbes kaks korda suurem. Sulgeda anum kohe. Segada lõssipulber hästi läbi, anumat korduvalt ümber pöörates. 8.2. Katsekogus Kaaluda tsentrifuugianumasse (6.2) või sobivasse suletavasse anumasse (50 ml) 2,000 ± 0,001 g uuritavat proovi. 8.3. Rasva ja valkude eemaldamine 8.3.1. Lisada katsekogusele 20,0 ml kuuma vett (50 °C). Lahustada pulber, loksutades segu mehaanilisel loksutil (6.3) viis minutit. Asetada anum vesivanni (6.10) ja tasakaalustada 25 °C juures. 8.3.2. Magnetseguriga (6.4) tugevasti segades lisada kahe minuti jooksul ühtlase kiirusega 10,0 ml trikloroäädikhapet (5.1), mille temperatuur on 25 °C. Seejärel asetada tops 60 minutiks vesivanni (6.10). 8.3.3. Tsentrifuugida (6.2) 10 minutit 2 200 g juures või filtrida läbi paberi (6.6), visates filtraadist ära esimesed 5 ml. 8.4. Kromatograafiline määramine 8.4.1. Süstida HPLC-seadmesse (6.11), milles eluentlahuse (5.2) voolukiirus on 1,0 ml/min, täpselt mõõdetud 15–30 μl supernatanti või filtraati (8.3.3). Märkus 1. Võib kasutada muud voolukiirust sõltuvalt kasutatava kolonni sisediameetrist või kolonni tootja soovitustest. Märkus 2. Iga töökatkestuse ajal tuleb kolonne veega pesta. Kolonnidesse ei tohi jätta eluentlahust (5.2). Enne töö katkestamist enam kui 24 tunniks tuleb kolonnid veega läbi pesta ja seejärel pesta vähemalt kolme tunni vältel lahusega (5.3) voolukiirusel 0,2 ml/min. 8.4.2. Uuritava proovi [E] kromatograafilise analüüsi tulemused saadakse kromatogrammina, millel iga piik identifitseeritakse selle retentsiooniaja (RT) järgi järgmiselt.
Kolonni(de) valik võib oluliselt mõjutada üksikpiikide retentsiooniaega. Integraator (6.11.6) arvutab automaatselt välja iga piigi pindala A.
Enne kvantitatiivset tõlgendamist tuleb kontrollida iga kromatogrammi kuju, et avastada võimalikud kõrvalekalded, mis on tingitud seadme või kolonnide puudulikust tööst või analüüsitava proovi päritolust ja omadustest. Kahtluse korral tuleb analüüsi korrata. 8.5. Tundlikkustegurite leidmine 8.5.1. Kasutada täpselt sama meetodit, mida on kirjeldatud standardproovide (5.4) puhul punktides 8.2–8.4.2. Kasutada värskelt valmistatud lahuseid, sest kaseiini makropeptiidid lagunevad 8 % trikloroäädikhappe keskkonnas. Hinnanguline kadu 30 °C juures on 0,2 % tunnis. 8.5.2. Enne proovide kromatograafilist analüüsi tuleb kolonnid tasakaalustada, süstides korduvalt standardproovi (5.4.2) lahust (8.5.1), kuni kaseiini makropeptiidile vastava piigi pindala ja retentsiooniaeg jääb konstantseks. 8.5.3. Määrata tundlikkustegurid R, süstides sama palju filtraate (8.5.1) nagu uuritavate proovide puhul. 9. TULEMUSTE VÄLJENDAMINE 9.1. Arvutusmeetod ja -valemid 9.1.1. Tundlikkusteguri R arvutamine
kus:
kus:
9.1.2. Proovi [E] piikide suhtelise pindala arvutamine
kus:
9.1.3. Proovi [E] piigi III suhtelise retentsiooniaja arvutamine: RRTIII[E] = (RTIII[E])/(RTIII[5]) kus:
9.1.4. Katsed on näidanud lineaarse seose olemasolu III piigi suhtelise retentsiooniaja (s.t RRTIII [E]) ja lisatud vadakupulbri sisalduse vahel kuni 10 % sisalduseni.
RRTIII väärtuste lubatud kõikumine on ± 0,002. Tavaliselt ei erine RRTIII [0] väärtus palju 1,034st. Sõltuvalt kolonnide olukorrast võib väärtus läheneda väärtusele 1,000, kuid peab alati olema sellest suurem. 9.2. Proovis sisalduva laabivadakupulbri protsendi arvutamine W = SIII[E] – [1,3 + (SIII[0] – 0, 9)] kus:
9.3. Meetodi täpsus 9.3.1. Korduvus Ühel ja samal ajal või kohe üksteise järel samade seadmetega ühe ja sama analüüsija poolt identse katsematerjaliga läbi viidud kahe määramise tulemuste vaheline erinevus ei tohi olla suurem kui 0,2 massiprotsenti. 9.3.2. Korratavus Erinevus kahe üksiktulemuse vahel, mis on saadud kahes eri laboris sama katsematerjaliga, ei tohi olla suurem kui 0,4 massiprotsenti. 9.4. Tõlgendamine 9.4.1. Vadak puudub, kui III piigi suhteline pindala SIII [E], väljendatuna laabivadaku grammides 100 g toote kohta, on ≤ 2,0 + (SIII[0] – 0,9), kus:
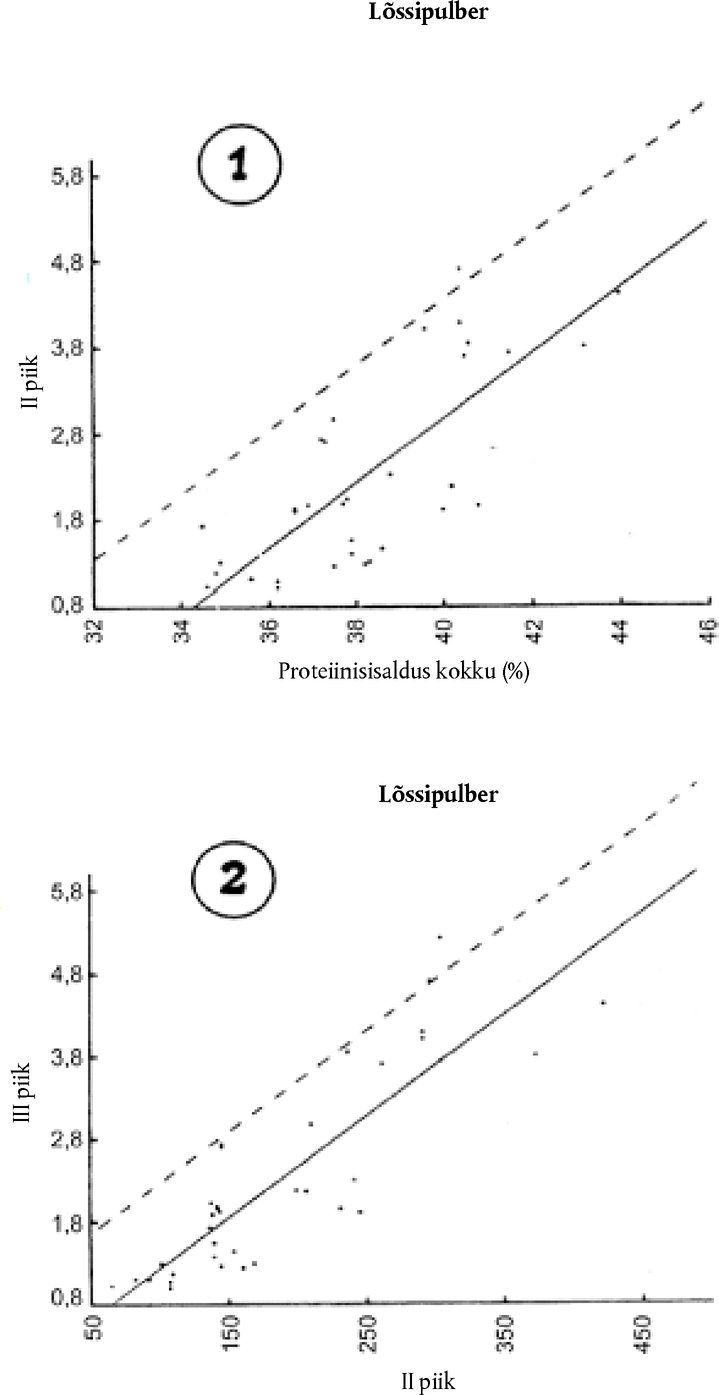
9.4.2. Kui III piigi suhteline pindala SIII[E] > 2,0 + (SIII[0] – 0,9) ja II piigi suhteline pindala SII [E] ≤ 160, määrata laabivadakusisaldus vastavalt punktis 9.2 osutatule. 9.4.3. Kui III piigi suhteline pindala SIII [E] on > 2,0 + (SIII[0] – 0,9) ja II piigi suhteline pindala SII [E] ≤ 160, määrata üldine valgusisaldus (P %), seejärel uurida jooniseid 1 ja 2. 9.4.3.1. Suure valgusisaldusega võltsimata koostisega lõssipulbri proovide analüüsimisel saadud teave on kokku võetud joonistel 1 ja 2. Pidev joon kujutab lineaarset regressiooni, mille koefitsiendid arvutatakse vähimruutude meetodil. Sirge katkendjoon fikseerib III piigi suhtelise pindala ülemmäära, kusjuures on tõenäoline, et 90 % juhtudest seda ei ületata. Jooniste 1 ja 2 katkendjoonte võrrandid:
kus:
Need võrrandid on võrdsed punktis 9.2 nimetatud arvuga 1,3. Lahknevus (T1 ja T2) leitud suhtelise pindala SIII [E] ja suhtelise pindala SIII vahel saadakse järgmiselt: T1 = SIII[E] – [(0,376 P% – 10,7) + (SIII[0] – 0,9)]; T2 = SIII[E] – [(0,0123 SII[E] + 0,93) + (SIII[0] – 0,9)]
Laabivadaku sisaldus arvutatakse vastavalt järgmisele võrrandile: W = T2 + 0,91 kus: 0,91 on pidevjoone ja katkendjoone vaheline kaugus vertikaalteljel.
III liide LAABIVADAKUPULBRI MÄÄRAMINE LÕSSIPULBRIS 1. EESMÄRK: LÕSSIPULBRILE LISATUD LAABIVADAKUPULBRI TUVASTAMINE 2. KIRJANDUS: RAHVUSVAHELINE STANDARD ISO 707 3. MÄÄRATLUS Laabivadakupulbri sisalduse leidmiseks määratakse kaseiini makropeptiidide sisaldus massiprotsentides järgnevalt kirjeldatud meetodil. 4. PÕHIMÕTE Proove analüüsitakse kaseiini makropeptiidi A tuvastamiseks pöördfaasilise kõrgsurvevedelikkromatograafia (HPLC) abil. Saadud tulemusi võrreldakse lõssipulbri standardproovidega, millest osale on lisatud teatav kogus vadakupulbrit. Kui tulemus on suurem kui 1 massiprotsent, näitab see, et proov sisaldab laabivadakupulbrit. 5. REAKTIIVID Kõik reaktiivid peavad olema analüütiliselt puhtad. Kasutada tuleb destilleeritud või vähemalt samaväärse puhtusastmega vett. Atsetonitriili kvaliteet peab vastama spektroskoopia või HPLC nõuetele. 5.1. Trikloroäädikhappe lahus Lahustada vees 240 g trikloroäädikhapet (CCl3COOH) ja täiendada ruumalani 1 000 ml. Lahus peab olema puhas ja värvitu. 5.2. Eluendid A ja B Eluent A: panna 1 000 ml mõõtekolbi 150 ml atsetonitriili (CH3CN), 20 ml isopropanooli (CH3CHOHCH3) ja 1,00 ml trifluoroäädikhapet (TFA, CF3CHOOH). Täita veega ruumalani 1 000 ml. Eluent B: panna 1 000 ml mõõtekolbi 550 ml atsetonitriili, 20 ml isopropanooli ja 1,00 ml TFAd. Täita veega ruumalani 1 000 ml. Filtrida eluentlahus enne kasutamist läbi membraanfiltri, mille pooride läbimõõt on 0,45 μm. 5.3. Kolonni konserveerimine Pärast analüüse pestakse kolonni eluendiga B (gradiendi järgi) ja seejärel 30 minuti jooksul atsetonitriiliga (gradiendi järgi). Kolonni säilitatakse atsetonitriilis. 5.4. Standardproovid 5.4.1. Riikliku ladustamise nõuetele vastav lõssipulber (s.t [0]) 5.4.2. Sama lõssipulber, millele on lisatud 5 massiprotsenti standardkoostisega laabivadakupulbrit (s.t [5]) 5.4.3. Sama lõssipulber, millele on lisatud 50 massiprotsenti standardkoostisega laabivadakupulbrit (s.t [50]) 6. APARATUUR 6.1. Analüütilised kaalud 6.2. Tsentrifuug, mis võimaldab saavutada tsentrifuugimiskiirenduse 2 200 g ja on varustatud umbes 50 ml suletavate tsentrifuugianumatega 6.3. Mehhaaniline loksuti 6.4. Magnetsegur 6.5. Umbes 7 cm läbimõõduga klaaslehtrid 6.6. Umbes 12,5 cm läbimõõduga keskmise tihedusega paberfiltrid 6.7. Klaasist filtrimisseade, mille membraanfiltri pooride läbimõõt on 0,45 μm 6.8. Mõõtepipetid, mis võimaldavad mõõta 10 ml (ISO 648, klass A või ISO/R 835), või doseerimisseade, millega saab mõõta 10,0 ml kahe minuti jooksul. 6.9. Doseerimisseade, mis võimaldab mõõta 20,0 ml vett umbes 50 °C juures 6.10. Termostaadiga vesivann, reguleeritud temperatuurile 25 ± 0,5 °C 6.11. HPLC-seade, mis koosneb järgmistest osadest.
7. PROOVIVÕTT 7.1. Proove võetakse vastavalt rahvusvahelise standardiga ISO 707 sätestatud korrale. Liikmesriigid võivad kasutada muud proovide võtmise meetodit, kui see on kooskõlas eespool nimetatud standardi põhimõtetega. 7.2. Proovi hoitakse tingimustes, mis välistavad selle riknemise või koostise muutumise. 8. TÖÖ KÄIK 8.1. Uuritava proovi ettevalmistamine Panna lõssipulber õhukindla kaanega anumasse, mille maht on lõssipulbri mahust umbes kaks korda suurem. Sulgeda anum kohe. Segada lõssipulber hästi läbi, anumat korduvalt ümber pöörates. 8.2. Katsekogus Kaaluda tsentrifuugianumasse (6.2) või sobivasse suletavasse anumasse (50 ml) 2,00 ± 0 001 g uuritavat proovi. Märkus: segu puhul kasutada uuritava proovi kaalutist, mis pärast rasvast vabastamist vastab kogusele 2,00 g. 8.3. Rasva ja valkude eemaldamine 8.3.1. Lisada katsekogusele 20,0 ml kuuma vett (50 °C). Lahustada pulber, loksutades segu mehaanilisel loksutil (6.3) viis minutit. Asetada anum vesivanni (6.10) ja tasakaalustada 25 °C juures. 8.3.2. Magnetseguriga (6.4) tugevasti segades lisada kahe minuti jooksul ühtlase kiirusega 10,0 ml 25 °C trikloroäädikhapet (5.1). Seejärel asetada anum 60 minutiks vesivanni (6.10). 8.3.3. Tsentrifuugida (6.2) 10 minutit 2 200 g juures või filtrida läbi paberi (6.6), visates filtraadist ära esimesed 5 ml. 8.4. Kromatograafiline määramine 8.4.1. Pöördfaasiline HPLC välistab valepositiivsete tulemuste saamise hapukoorepeti pulbri sisalduse tõttu. 8.4.2. Enne pöördfaasilise HPLC analüüsi läbiviimist tuleb optimeerida gradiendi tingimused. Umbes 6 ml surnud ruumalaga (ruumala lahuste ühinemispunktist kuni injektori silmuseni, viimane kaasa arvatud) gradientsüsteemide puhul on optimaalne kaseiini makropeptiidi A retentsiooniaeg 26 ± 2 minutit. Väiksema surnud ruumalaga (näiteks 2 ml) gradientsüsteemide puhul on optimaalne retentsiooniaeg 22 minutit. Võtta standardproovide (5.4) lahuseid, millest osa sisaldab 50 % laabivadakut. Sisestada HPLC-seadmesse 100 μl supernatanti või filtraati (8.3.3) tabelis 1 esitatud proovigradiendi tingimustel. Tabel 1 Proovigradiendi tingimused kromatograafilise analüüsi optimeerimiseks
Kahe kromatogrammi võrdlus peab andma kaseiini makropeptiidi A piigi asukoha. Järgmist valemit kasutades saab välja arvutada normaalgradiendi puhul kasutatava esialgse lahuse koostise (vt 8.4.3): % B = 10 – 2,5 + (13,5 + (RTcmpA – 26) / 6) × 30 / 27 % B = 7,5 + (13,5 + (RTcmpA – 26) / 6) × 1,11 kus
8.4.3. Võtta uuritavate proovide lahused. Süstida HPLC-seadmesse täpselt mõõdetult 100 μl supernatanti või filtraati (8.3.3) eluendi (5.2) voolukiirusel 1,0 ml/minutis. Eluendi koostis analüüsi alguses saadakse punkti 8.4.2 kohaselt. Harilikult on see ligikaudu A:B = 76:24 (5.2). Kohe pärast sisestamist alustada lineaargradienti, mille tulemuseks on 5 % võrra suurem B kogus 27 minuti pärast. Seejärel alustada lineaargradienti, mille tulemusel on eluendis viie minuti pärast 90 % B. Seda eluendi koostist hoida viis minutit ja pärast seda taastada lineaargradiendiga viie minuti jooksul esialgne koostis. Sõltuvalt pumbasüsteemi mahutavusest võib järgmist analüüsi alustada 15 minutit pärast algtingimuste saavutamist. Märkus 1. Kaseiini makropeptiidi A retentsiooniaeg peab olema 26 ± 2 minutit. Seda on võimalik saavutada, muutes esimese gradiendi alg- ja lõpptingimusi. % B erinevus esimese gradiendi alg- ja lõpptingimustel peab siiski olema 5 % B. Märkus 2. Eluentidest tuleb õhk piisavalt hästi eemaldada ja need peavad sellisena püsima. See on oluline gradientpumbasüsteemi tõrgeteta toimimiseks. Kaseiini makropeptiidi A piigi retentsiooniaja standardhälve peab olema alla 0,1 minuti (n = 10). Märkus 3. Iga viienda proovi järel tuleb sisestada standardproov [5] ja arvutada selle alusel uus tundlikkustegur R (9.1.1). 8.4.4. Uuritava proovi (E) kromatograafilise analüüsi tulemused saadakse kromatogrammina, millel kaseiini makropeptiidi A piik tehakse kindlaks 26minutilise retentsiooniaja alusel. Integraator (6.11.6) arvutab automaatselt kaseiini makropeptiidi A piigi kõrguse H. Kontrollida tuleb iga kromatogrammi nulljoone asukohta. Kui nulljoone asukoht on vale, tuleb analüüsi või integreerimist korrata. Märkus: kui kaseiini makropeptiidi A piik on muudest piikidest piisavalt hästi eraldunud, tuleb nulljoon kindlaks määrata miinimumist miinimumini, muudel juhtudel kasutada vertikaalide tõmbamist ühise nulljooneni, mille alguspunkt peab olema kaseiini makropeptiidi A piigi läheduses (seega mitte ajahetkel t = 0 min!). Standardi ja proovide puhul tuleb kasutada sama tüüpi integreerimist ning ühise nulljoone puhul tuleb kontrollida selle sobivust proovidele ja standardile. Enne kvantitatiivset interpreteerimist tuleb kontrollida iga kromatogrammi kuju, et avastada võimalikud kõrvalekalded, mis on tingitud seadme või kolonni puudulikust tööst või uuritava proovi päritolust ja omadustest. Kahtluse korral tuleb analüüsi korrata. 8.5. Tundlikkustegurite leidmine 8.5.1. Kasutada täpselt sama meetodit, mida on kirjeldatud standardproovide (5.4.1–5.4.2) puhul punktides 8.2–8.4.4. Kasutada värskelt valmistatud lahuseid, sest kaseiini makropeptiid laguneb toatemperatuuril 8 % trikloroäädikhappe keskkonnas. Temperatuuril 4 °C püsib lahus stabiilsena 24 tundi. Pikkade analüüsiseeriate korral on automaatsisesti puhul soovitatav kasutada jahutatud proovialust. Märkus: Punkti 8.4.2 võib vahele jätta, kui eelmistest analüüsidest on teada algtingimustele vastav % B. Standardproovi [5] kromatogramm peab vastama joonisele 1. Sellel joonisel eelneb kaseiini makropeptiidi A piigile kaks väikest piiki. Piigid peavad eristuma samal määral. 8.5.2. Enne proovide kromatograafilist analüüsi sisestada 100 μl standardproovi, mis ei sisalda laabivadakut [0] (5.4.1). Kromatogrammil ei tohi olla kaseiini makropeptiidi A retentsiooniajale vastavat piiki. 8.5.3. Määrata tundlikkustegurid R, sisestades samas mahus filtraati (8.5.1) nagu uuritavate proovide puhul. 9. TULEMUSTE VÄLJENDAMINE 9.1. Arvutusmeetod ja -valemid 9.1.1. Tundlikkusteguri R arvutamine Kaseiini makropeptiidi A piik: R = W/H kus
9.2. Proovis sisalduva laabivadakupulbri protsendi arvutamine W(E) = R × H(E), kus
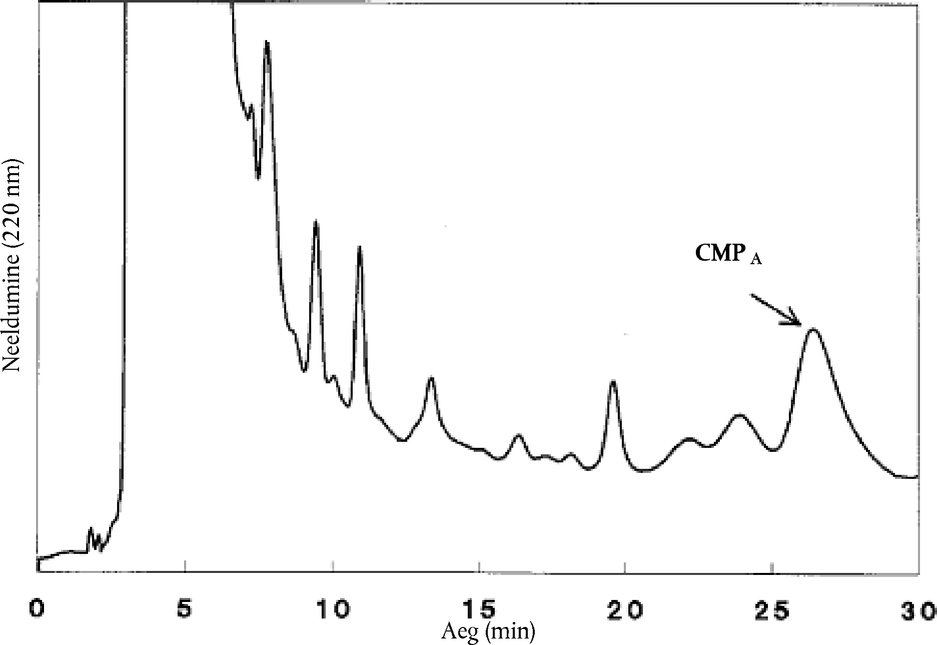
Kui W(E) on suurem kui 1 % ning kui retentsiooniaegade erinevus proovi ja standardproovi [5] puhul on väiksem kui 0,2 minutit, näitab see, et proov sisaldab laabivadakupulbrit. 9.3. Meetodi täpsus 9.3.1. Korduvus Ühel ja samal ajal või kohe üksteise järel samade seadmetega ühe ja sama analüüsija poolt identse katsematerjaliga läbi viidud kahe määramise tulemuste vaheline erinevus ei tohi olla suurem kui 0,2 massiprotsenti. 9.3.2. Korratavus Ei ole määratud. 9.3.3. Lineaarsus Laabivadaku kontsentratsioonide vahemikus 0–16 % peavad tulemused olema lineaarses seoses korrelatsioonikoefitsiendiga > 0,99. 9.4. Tõlgendamine 1 % piir hõlmab korratavusest tulenevalt määramatust. Joonis 1 Ni–4.6 standard
|
|
3) |
Lisatakse järgmised lisad. „ VII LISA Eraladustuslepingute alusel ladustatud lõssipulbri analüüsi meetodid
VII LISA Eraladustuslepingute alusel ladustatud lõssipulbri analüüsi meetodid
VIII LISA Eraladustuslepingute alusel ladustatud juustu analüüsi meetodid
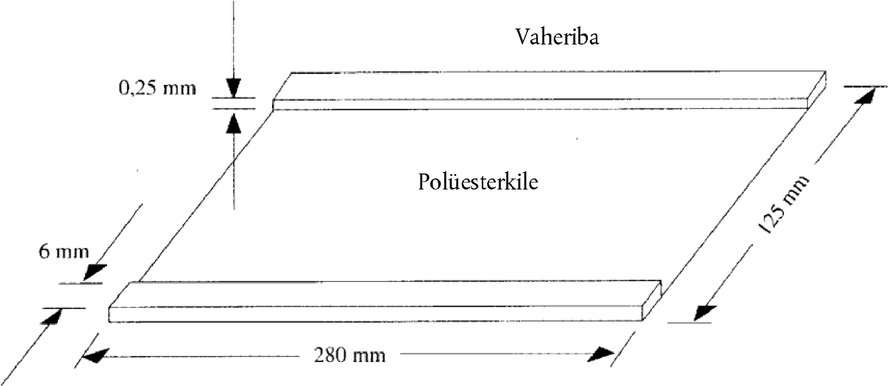
Liide MEETOD LEHMAPIIMA JA LEHMAPIIMA KASEINAADI MÄÄRAMISEKS JUUSTUS, MIS ON VALMISTATUD LAMBA-, KITSE- VÕI PÜHVLIPIIMAST VÕI LAMBA-, KITSE- JA PÜHVLIPIIMA SEGUST 1. RAKENDATAVUS Lehmapiima ja lehmapiima kaseinaadi määramine lamba-, kitse- või pühvlipiimast või lamba-, kitse- ja pühvlipiima segust valmistatud juustus γ-kaseiinide isoelektrilise fookustamise abil pärast lüüsimist plasmiiniga. 2. RAKENDUSALA Käesolev tundlik ja spetsiifiline meetod sobib töötlemata ja kuumtöödeldud lehmapiima ning lehmapiima kaseinaadi tuvastamiseks laagerdumata ja laagerdunud juustus, mis on valmistatud lamba-, kitse- või pühvlipiimast või lamba-, kitse- ja pühvlipiima segust. See meetod ei sobi võltsimise tuvastamiseks, kui piima või juustu on võltsitud lehmapiimast eraldatud kuumtöödeldud vadakuvalgu kontsentraadiga. 3. MEETODI PÕHIMÕTE 3.1. Kaseiinide eraldamine juustust ja etalonainetest 3.2. Eraldatud kaseiinide lahustamine ja lõhustamine plasmiiniga (EC.3.4.21.7) 3.3. Plasmiiniga töödeldud kaseiinide isoelektriline fookustamine karbamiidi juuresolekul ja valkude värvimine 3.4. Värviga töödeldud γ3- ja γ2-kaseiini mustrite (tõend lehmapiima olemasolust) võrdlemine mustritega, mis on saadud katsetes 0 % ja 1 % lehmapiima sisaldavate etalonainetega samas geelis. 4. REAKTIIVID Kui ei ole öeldud teisiti, tuleb kasutada analüütiliselt puhtaid kemikaale. Vesi peab olema kahekordselt destilleeritud või samaväärse puhtusastmega. Märkus: järgnev eeskiri kehtib laboris valmistatud karbamiidisisaldusega polüakrüülamiidgeelide puhul mõõtmetega 265 × 125 × 0,25 mm. Kui kasutatakse muud tüüpi ja teistsuguse suurusega geele, tuleb lahutustingimusi vastavalt kohandada. Isoelektriline fookustamine 4.1. Reaktiivid karbamiidi sisaldavate polüakrüülamiidgeelide valmistamiseks 4.1.1. Geeli põhilahus Lahustada vees:
täiendada ruumalani 100 ml ning säilitada külmikus tumedas klaaspudelis. Märkus: neurotoksilise akrüülamiidi määratletud kaalutiste asemel on parem kasutada müügil olevat akrüülamiidi ja BISi eelvalmistatud lahust. Kui valmislahus sisaldab 30 % massikontsentratsioonis akrüülamiidi ja 0,8 % massikontsentratsioonis BISi, tuleb eespool nimetatud segu valmistamiseks võtta nimetatud kaalutiste asemel 16,2 ml valmislahust. Põhilahuse säilivusaeg on kuni 10 päeva; kui põhilahuse juhtivus on suurem kui 5 μS, tuleb see deioniseerida, segades seda 2 g ioonvahetiga Amberlite MB-3 kolmekümne minuti jooksul ja filtreerides seejärel läbi 0,45 m membraani. 4.1.2. Geeli lahus Segada geeli põhilahus (vt 4.1.1) lisandite ja amfolüütidega (*):
Segada geeli lahus ning degaseerida see ultrahelivannis või vaakumis kahe kuni kolme minuti jooksul. Märkus: geeli lahus valmistada vahetult enne selle valamist kattekilele (vt 6.2). 4.1.3. Katalüsaatori lahused 4.1.3.1. N,N,N′,N′-tetrametüületüleendiamiin (TEMED) 4.1.3.2. 40 % massikontsentratsiooniga ammooniumpersulfaadi lahus (PER): lahustada 800 mg PERi vees ja täiendada ruumalani 2 ml. Märkus: alati tuleb kasutada värskelt valmistatud PERi lahust. 4.2. Kontaktvedelik Petrool või vedel parafiin 4.3. Anoodilahus Lahustada 5,77 g 85massiprotsendilist fosforhapet vees ja täiendada ruumalani 100 ml. 4.4. Katoodilahus Lahustada 2,00 g naatriumhüdroksiidi vees ja täiendada veega ruumalani 100 ml. Proovi ettevalmistamine 4.5. Reaktiivid valgu eraldamiseks 4.5.1. Lahjendatud äädikhape (täiendada 25,0 ml jää-äädikhappe ruumala veega ruumalani 100 ml) 4.5.2. Diklorometaan 4.5.3. Atsetoon 4.6. Valke lahustav puhver Lahustada vees:
ja täiendada ruumalani 50 ml. Märkus: säilitada külmikus; säilimisaeg kuni üks nädal. 4.7. Reaktiivid kaseiinide lõhustamiseks plasmiiniga 4.7.1. Ammooniumkarbonaatpuhver Tiitrida 0,2 mol/l ammooniumvesinikkarbonaadi lahus (1,58 g/100 ml vees, mis sisaldab 0,05 mol/l etüleendiamiintetraatsetaati (EDTA, 1,46 g/100 ml vees), 0,2 mol/l ammooniumkarbonaadi lahusega (1,92 g/100 ml vees, sisaldab 0,05 mol/l EDTAd) kuni pH-ni 8. 4.7.2. Veise plasmiin (EC. 3.4.21.7) aktiivsusega vähemalt 5 U/ml 4.7.3. ε-aminokaproonhappe lahus ensüümi inhibeerimiseks Lahustada 2,624 g ε-aminokaproonhapet (6-amino-n-heksaanhapet) 100 ml 40mahuprotsendilises etanoolis. 4.8. Standardained 4.8.1. Laabiga kalgendatud lamba- ja kitsepiimalõssi segust saadud sertifitseeritud etalonaineid, mis sisaldavad 0 % ja 1 % lehmapiima, on võimalik hankida Komisjoni Etalonainete ja Mõõtmiste Instituudist (Commission's Institute for Reference Materials and Measurements, B-2440 Geel, Belgia). 4.8.2. 0 % ja 1 % lehmapiima sisaldavate laboratoorsete vaheetalonainete valmistamine laabiga kalgendatud pühvlipiimast Lõssi saamiseks tsentrifuugida kas pühvli või veise toorpiima 37 °C ja 2 500 g juures 20 minutit. Pärast tsentrifuugianuma ja selle sisu kiiret jahutamist temperatuurini 6–8 °C kõrvaldada ülemine rasvakiht täielikult. 1 %-lise etalonsegu valmistamiseks panna 1 l keeduklaasi 495 ml pühvlipiimalõssi, lisada 5,00 ml lehmapiimalõssi ja reguleerida piimhapet (massikontsentratsioon 10 %) lisades pH-le 6,4. Temperatuur reguleerida väärtusele 35 °C, lisada 100 μl vasika laapi (aktiivsus 1:10 000, ligikaudu 3 000 U/ml), segada üks minut ja jätta keeduklaas alumiiniumfooliumiga kaetuna 35 °C juures üheks tunniks seisma, et lõss kalgenduks. Kui kalgend on moodustunud, külmkuivatada kogu kalgendunud mass, seda eelnevalt homogeniseerimata ja vadakut eraldamata. Jahvatada külmkuivatatud mass peeneks ühtlaseks pulbriks. Nullprotsendiline etalonaine valmistatakse sama meetodi abil puhtast pühvlipiimalõssist. Etalonaineid tuleb säilitada – 20 °C juures. Märkus: enne etalonainete valmistamist on soovitatav pühvlipiimalõssi puhtust kontrollida plasmiiniga töödeldud kaseiinide isoelektrilise fookustamise teel. Reaktiivid valgu eraldamiseks 4.9. Kinnisti Lahustada 150 g trikloroäädikhapet vees ja täiendada ruumalani 1 000 ml. 4.10. Värvi väljapesemise lahus Täiendada 500 ml metanooli ja 200 ml jää-äädikhappe segu destilleeritud veega ruumalani 2 000 ml. Märkus: iga päev tuleb valmistada värske värvi väljapesemise lahus. Seda saab valmistada, segades 50mahuprotsendilise metanooli ja 20mahuprotsendilise jää-äädikhappe põhilahuseid võrdsed ruumalad. 4.11. Värvimislahused 4.11.1. Värvimislahus (värvi põhilahus 1) Lahustada magnetseguri abil 3,0 g Coomassie-briljantsinist G-250 (C.I. 42655) 1 000 ml 90mahuprotsendilises metanoolis umbes 45 minuti jooksul ja filtreerida läbi kahe keskmise kiirusega toimiva kurdfiltri. 4.11.2. Värvimislahus (värvi põhilahus 2) Lahustada 5,0 g vasksulfaatpentahüdraati 1 000 ml 20mahuprotsendilises äädikhappes. 4.11.3. Värvimislahus (töölahus) Segada vahetult enne värvimist kokku 125 ml kumbagi värvi põhilahust (4.11.1 ja 4.11.2). Märkus: värvimislahus tuleb valmistada kasutamise päeval. 5. SEADMED 5.1. Klaasplaadid (265 × 125 × 4 mm), kummirull (laius 15 cm), nivelleerimislaud 5.2. Geeli kandekile (265 × 125 mm) 5.3. Kattekile (280 × 125 mm). Pikematele servadele panna riba kleeplinti (280 × 6 × 0,25 mm) (vt joonis 1). 5.4. Elektrofookustamiskamber jahutusplaadiga (nt 265 × 125 mm) ja sobiva pingeallikaga (≥ 2,5 kV) või automaatne elektroforeesiseade 5.5. Tsirkulatsioonkrüostaat, mis võimaldab termostaadiga hoida temperatuuri 12 ± 0,5 °C 5.6. Tsentrifuug, mis võimaldab töötada 3 000 g juures 5.7. Elektroodribad (≥ 265 mm pikkused) 5.8. Plastist tilgapudelid anoodi- ja katoodilahustele 5.9. Proovi aplikaatorid (10 × 5 mm, viskoosist või valke väheadsorbeerivast filterpaberist) 5.10. Roostevabast terasest või klaasist anumad värvimiseks ja värvi väljapesemiseks (nt 280 × 150 mm instrumendialused) 5.12. Reguleeritav varrashomogenisaator (varda diameeter 10 mm, 8 000 – 20 000 pööret minutis) 5.13. Magnetsegur 5.14. Ultrahelivann 5.15. Kilekeevitusaparaat 5.16. 25 μl mikropipetid 5.17. Vaakumtsentrifuug või külmkuivati 5.18. Temperatuuridele 35 ja 40 ± 1 °C reguleeritav loksutiga ja termostaadiga vesivann 5.19. Densitomeeter, mis võimaldab töötada lainepikkusel λ = 634 nm 6. TÖÖ KÄIK 6.1. Proovi ettevalmistamine 6.1.1. Kaseiinide eraldamine Panna 100 ml tsentrifuugianumasse juustu või etalonaine kaalutis, mis on ekvivalentne 5 g kuivmassiga, lisada 60 ml destilleeritud vett ja homogeenida varrashomogenisaatori abil (8 000 – 10 000 pööret minutis). Reguleerida pH lahjendatud äädikhappega (4.5.1) väärtusele 4,6 ja tsentrifuugida (5 minutit, 3 000 g). Rasv ja vadak dekanteerida, jääk homogeenida kiirusel 20 000 pööret minutis 40 ml destilleeritud vees, mille pH on lahjendatud äädikhappega (4.5.1) reguleeritud väärtusele 4,5, lisada 20 ml diklorometaani (4.5.2), homogeenida uuesti ja tsentrifuugida (5 minutit, 3 000 g). Eraldada spaatliga kaseiinikiht, mis jääb vee- ja orgaanilise faasi vahele (vt joonis 2), ning kõrvaldada mõlemad faasid dekanteerimise teel. Homogeenida kaseiin uuesti 40 ml destilleeritud vees (vt eespool) ja 20 ml diklorometaanis (4.5.2) ning tsentrifuugida. Seda protseduuri korrata seni (kaks või kolm korda), kuni mõlemad ekstraheerimisfaasid jäävad värvusetuks. Homogeenida valgujääk 50 ml atsetoonis (4.5.3) ja filtrida läbi keskmise kiirusega toimiva kurdpaberfiltri. Filtrile kogutud jääki pesta kahe eraldi 25 ml atsetoonikogusega, kuivatada õhu käes või lämmastikuvoolus ja hõõruda uhmris peeneks. Märkus: kuivi kaseiinipreparaate tuleb säilitada – 20 °C juures. 6.1.2. β-kaseiinide lõhustamine plasmiini abil γ-kaseiinide intensiivistamiseks Dispergeerida 25 mg kaseiinipreparaati (6.1.1) 0,5 ml ammooniumkarbonaatpuhvris (4.7.1) ja homogeenida 20 minutit näiteks ultraheliga töötlemise teel. Soojendada temperatuurini 40 °C, lisada 10 μl plasmiini (4.7.2), segada ja inkubeerida 40 °C juures ühe tunni jooksul pidevalt loksutades. Ensüümi inhibeerimiseks lisada 20 μl ε-aminokaproonhappe lahust (4.7.3), seejärel lisada 200 mg tahket karbamiidi ja 2 mg ditiotreitooli. Märkus: et fookustatud kaseiiniribad tuleksid sümmeetrilisemad, on soovitatav pärast ε-aminokaproonhappe lisamist lahus külmkuivatada ja kuivjääk lahustada 0,5 ml valke lahustavas puhvris (4.6). 6.2. Karbamiidi sisaldavate polüakrüülamiidgeelide valmistamine Niisutada geeli kandekilet (5.2) mõne tilga veega ja rullida see klaasplaadile (5.1), kõrvaldades eralduva vee paberrätiku või lapiga. Vaheribadega (0,25 mm) kattekile (5.3) rullida teisele klaasplaadile samal viisil. Asetada plaat nivelleerimislauale horisontaalasendisse. Ettevalmistatud deaereeritud geelilahusele (4.1.2) lisada 10 μl TEMEDi (4.1.3.1), segada, lisada 10 μl PERi lahust (4.1.3.2), segada hoolikalt ja valada kohe kattekile keskele ühtlaselt laiali. Asetada geeli kandeplaadi üks serv, kilepind allpool, kattekilega plaadile ja lasta kandeplaat aeglaselt alla, nii et kahe kile vahel moodustub ühtlane mullideta geelikile (joonis 3). Lasta geeli kandeplaat õhukese spaatli abil ettevaatlikult lõpuni alla ja asetada selle peale raskuseks veel kolm klaasplaati. Umbes 60 minuti pärast, kui polümerisatsioon on lõppenud, eemaldada kergelt klaasplaate koputades kandekilele polümeriseerunud geel koos kattekilega. Kandekile väline pind puhastada hoolikalt geelijääkidest ja karbamiidist. Kande- ja kattekile vahel olev geel keevitada kiletorusse ja säilitada külmikus (säilivusaeg kuni kuus nädalat). Märkus: kleepribadega kattekilet võib kasutada korduvalt. Kui proove on vähe või kasutatakse automaatset elektroforeesiseadet, võib polüakrüülamiidgeeli lõigata väiksemateks tükkideks (kaks geeli 4,5 × 5 cm). 6.3. Isoelektriline fookustamine Reguleerida jahutustermostaat temperatuurile 12 °C. Geeli kandekile väline pind puhastada petrooliga, lasta jahutusploki keskele mõned tilgad petrooli (4.2). Seejärel rullida geel selle peal lahti nii, et kandekile jääks allapoole ega tekiks mulle. Pühkida liigne petrool ära ja eemaldada kattekile. Niisutada elektroodribad elektroodilahustega (4.3, 4.4), lõigata geeliga ühepikkuseks ja asetada kohale (elektroodide vahekaugus 9,5 cm). Isoelektrilise fookustamise tingimused 6.3.1. Geeli suurus 265 × 125 × 0,25 mm
Märkus: kui geeli paksust või laiust muudetakse, tuleb voolutugevust ja võimsust reguleerida sellele vastavalt (nt 265 × 125 × 0,5 mm geeli puhul tuleb voolutugevust ja võimsust kahekordistada). 6.3.2. Automaatse elektroforeesiseadme pingeprogrammi näide (kaks 5,0 × 4,5 cm geeli; elektroodid on geelile asetatud vahetult, ilma elektroodribadeta)
Proovi aplikaator asetada kohale 2. etapis, kui protsess on kestnud 0 volttundi. Proovi aplikaator eemaldada 2. etapis, kui protsess on kestnud 30 volttundi. 6.4. Valkude värvimine 6.4.1. Valkude kinnistamine Pärast pinge väljalülitamist eemaldada viivitamatult elektroodribad ja asetada geel kohe värvimis-/pesemisküvetti, millesse on valatud 200 ml kinnistit (4.9). Hoida geeli kinnistis 15 minutit, loksutada pidevalt. 6.4.2. Geeli pesemine ja värvimine Nõrutada kogu kinnisti välja ja pesta geeli kaks korda 100 ml värvi väljapesemise lahusega (4.10), iga kord 30 sekundit. Valada värvi väljapesemise lahus ära ja asendada 250 ml värvimislahusega (4.11.3). Lasta geelil värvuda 45 minutit, lahust ettevaatlikult loksutades. 6.4.3. Värvi väljapesemine geelist Valada värvimislahus välja, pesta geeli kaks korda 100 ml värvi väljapesemise lahusega (4.10), seejärel loksutada 200 ml värvi väljapesemise lahusega 15 minutit, korrata värvi väljapesemisetappi vähemalt 2–3 korda, kuni saadakse selge värvitu foon. Seejärel loputada geeli destilleeritud vees (2 × 2 minutit) ja lasta sellel õhu käes kuivada (2–3 tundi) või kuivatada fööniga (10–15 minutit). Märkus 1: kinnistada, pesta, värvida ja värvi välja pesta tuleb 20 °C juures. Kõrgemat temperatuuri mitte kasutada. Märkus 2: kui eelistatakse värvimist tundlikuma hõbedaga (nt Silver Staining Kit, Protein, Pharmacia Biotech, Code nr 17-1150-01), tuleb plasmiiniga töödeldud kaseiiniproove lahjendada kuni kontsentratsioonini 5 mg/ml. 7. HINDAMINE Hindamiseks võrreldakse uuritava proovi valgumustreid etalonainete mustriga samal geelil. Lehmapiima olemasolu juustus, mis on valmistatud lamba-, kitse- või pühvlipiimast või lamba-, kitse- ja pühvlipiima segust, tehakse kindlaks γ2- ja γ3-kaseiini järgi, mille isoelektrilised punktid on pH 6,5 ja pH 7,5 vahel (joonised 4a, 4b ja 5). Avastamispiir on alla 0,5 %. 7.1. Visuaalne hindamine Lehmapiima koguse visuaalseks hindamiseks on soovitav reguleerida proovide ja etalonainete kontsentratsioonid selliseks, et lamba-, kitse- ja/või pühvlipiima γ2- ja γ3-kaseiini ribade intensiivsus oleks ühesugune (vt γ2 E, G, B ja γ3 E, G, B joonistel 4a, 4b ja 5). Sel juhul saab uuritava proovi lehmapiimasisaldust (mis võib olla suurem või väiksem kui 1 % või võrdne 1 %ga) hinnata otse, võrreldes lehmapiima γ3- ja γ2-kaseiini ribade intensiivsusi (vt γ3 C ja γ2 C joonistel 4a, 4b ja 5) 0- ja 1-protsendiliste etalonainete (lammas, kits) või laboratoorse vaheetalonaine (pühvel) vastavate intensiivsustega. 7.2. Densitomeetriline hindamine Võimaluse korral tuleks lehmapiima γ2- ja γ3-kaseiinile vastavate piikide pindalade suhted lamba-, kitse- ja pühvlipiima vastavate piikide pindaladesse (joonis 5) määrata densitomeetriliselt (5.19). Neid suhteid võrreldakse γ2- ja γ3-kaseiinile vastavate piikide pindalade suhetega samal geelil analüüsitud 1-protsendiliste etalonainete (lammas, kits) või laboratoorse vaheetalonaine (pühvel) puhul. Märkus: meetod toimib rahuldavalt, kui 1-protsendilise etalonaine puhul saadakse nii lehma γ2- kui ka γ3-kaseiini olemasolu kohta selge kinnitus, kuid 0-protsendilise etalonaine puhul seda ei saada. Kui selliseid tulemusi ei saada, tuleb määramisprotsessi optimeerida, pidades täpselt kinni meetodi tingimustest kõigis üksikasjades. Proov loetakse positiivseks, kui nii lehma γ2- kui ka γ3-kaseiini ribade intensiivsused või piikide pindalad on samaväärsed või suuremad kui 1-protsendilise etalonaine vastavad näitajad. 8. KIRJANDUS Addeo, F., Moio, L., Chianese, L., Stingo, C., Resmini, P., Berner, I, Krause, I., Di Luccia, A., Bocca A. Use of plasmin to increase the sensitivity of the detection of bovine milk in ovine and/or caprine cheese by gel isoelectric focusing of γ2-caseins. Milchwissenschaft, 45, 708–711 (1990). Addeo, F., Nicolai, M.A., Chianese, L., Moio, L., Spagna Musso, S., Bocca, A., Del Giovine, L. A control method to detect bovine milk in ewe and water buffalo cheese using immunoblotting. Milchwissenschaft, 50, 83–85 (1995). Krause, I., Berner, I., Klostermeyer, H. Sensitive detection of cow milk in ewe and goat milk and cheese by carrier ampholyte – and carrier ampholyte/immobilized pH gradient – isoelectric focusing of γ-caseins using plasmin as signal amplifier. Electrophoresis-Forum, 89 (B. J. Radola, ed.) lk 389-393, Bode-Verlag, München (1989). Krause, Ι., Belitz, H.-D., Kaiser, K.-P. Nachweis von Kuhmilch in Schaf and Ziegenmilch bzw. -käse durch isoelektrische Fokussierung in harnstoffhaltigen Polyacrylamidgelen. Z. Lebensm. Unters. Forsch., 174, 195–199 (1982). Radola, B.J. Ultrathin-layer isoelectric focusing in 50–100 μm polyacrylamide gels on silanised glass plates or polyester films. Electrophoresis, 1, 43–56 (1980). Joonis 1 Kattekile skeem
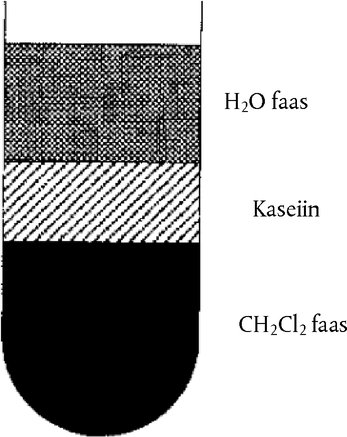
Joonis 2 Kaseiinikiht, mis jääb pärast tsentrifuugimist ujuma vee- ja orgaanilise faasi vahele
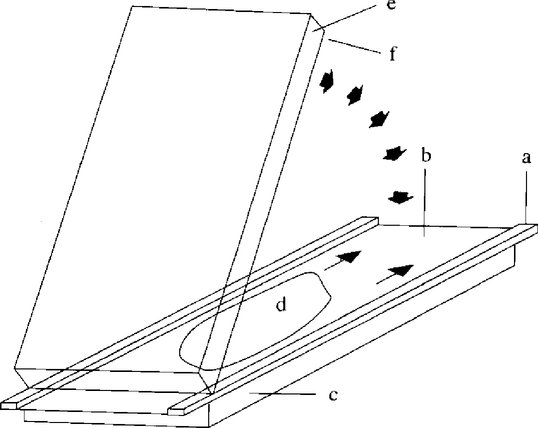
Joonis 3 Üliõhukese polüakrüülamiidgeeli valmistamine pressimismeetodil
a = vaheriba (0,25 mm); b = kattekile (5.3); c, e = klaasplaadid (5.1); d = geelilahus (4.1.2); f = geeli kandekile (5.2) Joonis 4a Erineva lehmapiimasisaldusega lamba- ja kitsepiimajuustust eraldatud ja plasmiiniga töödeldud kaseiinide isoelektriline fookustamine
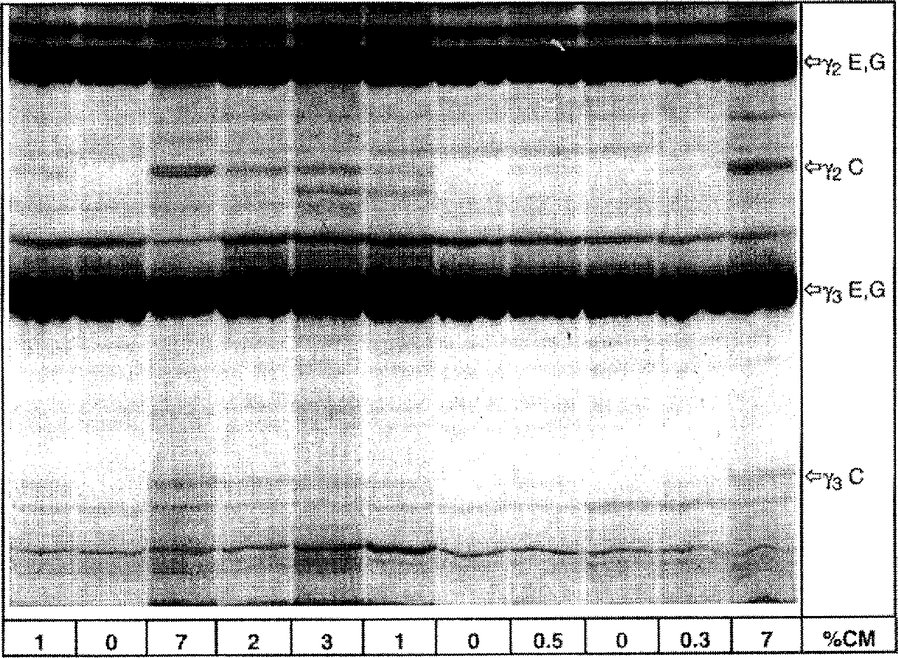
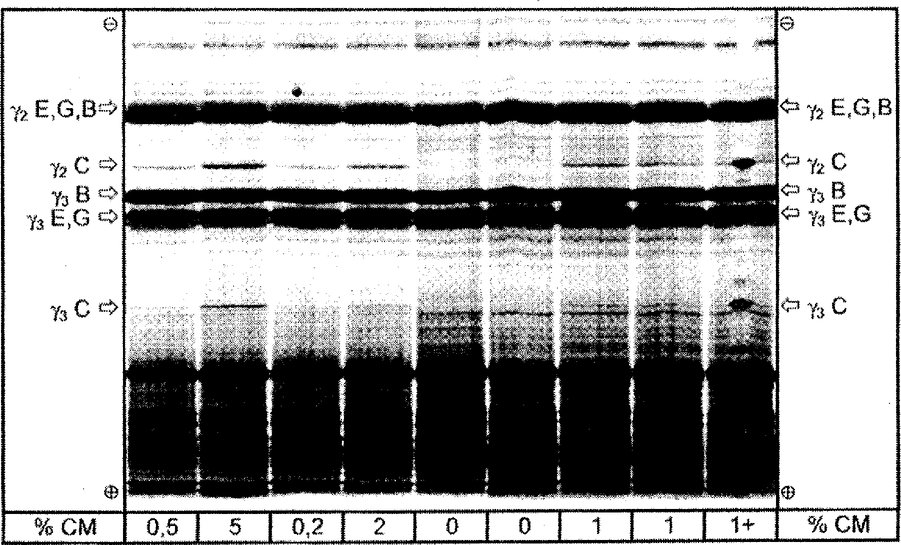
% CM = lehmapiima protsent, C = lehm, E = lammas, G = kits Kujutatud on isoelektriliselt fookustatud geeli ülemist poolt. Joonis 4b Erineva lehmapiimasisaldusega lamba-, kitse- ja pühvlipiimasegu juustust eraldatud ja plasmiiniga töödeldud kaseiinide isoelektriline fookustamine
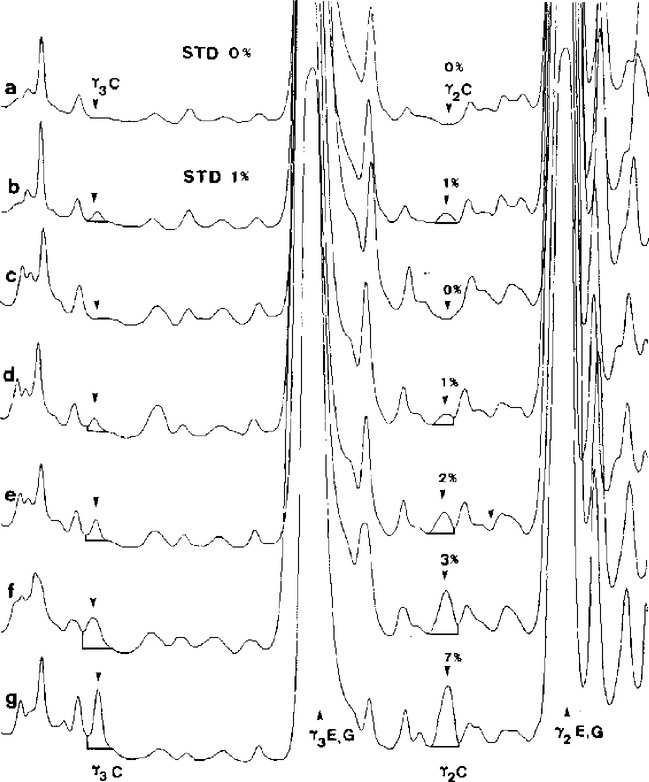
% CM = lehmapiima protsent; 1+ = proov, mis sisaldab 1 % lehmapiima ja millele on lisatud puhast veise kaseiini (spektri keskosas). C = lehm, E = lammas, G = kits, B = pühvel. Kujutatud on isoelektriliselt fookustatud geel kogu lahutusala ulatuses. Joonis 5 Etalonainete (STD) ning lamba- ja kitsepiima segust valmistatud juustu proovide densitogrammide superpositsioon pärast isoelektrilist fookustamist.
a,b = standardained, mis sisaldavad 0 ja 1 % lehmapiima; c-g = juustuproovid, mis sisaldavad 0, 1, 2, 3 ja 7 % lehmapiima. C = lehm, E = lammas, G = kits. Isoelektriliselt fookustatud geeli ülemist poolt skanniti lainepikkusel λ = 634 nm. IX LISA Analüüsi hindamine 1. Kvaliteedi tagamine Analüüsid tehakse vastavalt määruse (EÜ) nr 882/2004 (**) artiklile 12 määratud või liikmesriikide pädevate asutuste määratud laborites. 2. Proovi võtmine ja analüüsitulemuste vaidlustamise
Liide Tootepartii piirnormile vastavuse hindamine 1. Põhimõte Kui riikliku sekkumise ja eraladustusabi käsitlevates õigusaktides on sätestatud üksikasjalik proovi võtmise kord, siis seda korda järgitakse. Kõikidel muudel juhtudel kontrollitakse esitatud tootepartiid vähemalt 3 juhuslikult valitud prooviühikust koosneva prooviga. Võib teha liitproovi. Saadud tulemuse võrdlemiseks sätestatud piirnormidega arvutatakse 95 % usaldusvahemik välja kahekordse standardhälbena, kusjuures asjakohane standardhälve sõltub sellest, kas 1) meetod on väärtuste σr ja σR osas rahvusvaheliselt valideeritud või 2) laborisisesel valideerimisel on välja arvutatud sisemine korratavus. See usaldusvahemik võrdsustatakse tulemuse mõõtemääramatusega. 2. Meetod on rahvusvaheliselt valideeritud Sel juhul on korduvuse standardhälve σr ja korratavuse standardhälve σR kindlaks määratud ning labor saab tõendada vastavust valideeritud meetodi sobivuskriteeriumidele. Arvutatakse korratud mõõtmiste arvu n aritmeetiline keskmine
Kui mõõtmise lõpptulemuse x arvutamiseks kasutatakse ühte järgmistest valemitest: x = y 1 + y 2, x = y 1 – y 2, x = y 1 · y 2 või x = y 1/y 2 , tuleb standardhälbed ühendada tavapäraste meetoditega. Tootepartii loetakse sätestatud ülemisele piirnormile UL mittevastavaks, kui
muudel juhtudel loetakse see piirnormile UL vastavaks. Tootepartii loetakse sätestatud alumisele piirnormile LL mittevastavaks, kui
muudel juhtudel loetakse see piirnormile LL vastavaks. 3. Laborisisene valideerimine laborisisese korratavuse standardhälbe väljaarvutamisega Kui kasutatakse käesolevas määruses määratlemata meetodeid ja täpsuspiirid ei ole kindlaks määratud, tuleb läbi viia laborisisene valideerimine. Selleks tuleb laiendmääramatuse U arvutamise valemis kasutada σr ja σ R asemel laborisisese korduvuse standardhälvet sir ja laborisisese korduvuse standardhälvet si R . Piirnormile vastavuse hindamisel järgitavad eeskirjad on sätestatud punktis 1. Kui leitakse, et tootepartii ei vasta sätestatud piirnormile, tuleb mõõtmist korrata käesolevas määruses kirjeldatud meetodi abil ning hinnata tulemust vastavalt punktile 1.
“ |
 .
. Laiendmääramatus (
Laiendmääramatus (
 ;
;(1) Kasutatava meetodi kiidab heaks makseasutus.
(2) Kõrbenud osakeste analüüse võib teha süstemaatiliselt. Kõnealuseid analüüse tuleb siiski teha alati, kui organoleptilist kontrolli ei tehta.
(3) Kasutatava meetodi (ühe või mõlemad) kiidab heaks makseasutus.
(4) Kasutatava meetodi kiidab heaks makseasutus.
(5) Organoleptilised kontrollid tehakse juhul, kui makseasutus seda pärast riskipõhist analüüsi vajalikuks peab.
(6) Kasutatava meetodi kiidab heaks makseasutus.
(7) Proovi pealekandmine: pärast eelfookustamist (1. etapp) pipettida 18 μl proovi ja standardlahust proovi aplikaatoritele (10 × 5 mm), asetada need 1 mm vahedega geelile, pikisuunas 5 mm kaugusele anoodist, ja vajutada kergelt. Fookustada eelpool nimetatud tingimustel ja eemaldada proovi aplikaatorid ettevaatlikult pärast 60minutilist proovi fookustamist.