

EUR-Lex Access to European Union law
This document is an excerpt from the EUR-Lex website
Document 32014R0900
Commission Regulation (EU) No 900/2014 of 15 July 2014 amending, for the purpose of its adaptation to technical progress, Regulation (EC) No 440/2008 laying down test methods pursuant to Regulation (EC) No 1907/2006 of the European Parliament and of the Council on the Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH) Text with EEA relevance
Verordnung (EU) Nr. 900/2014 der Kommission vom 15. Juli 2014 zur Änderung der Verordnung (EG) Nr. 440/2008 zur Festlegung von Prüfmethoden gemäß der Verordnung (EG) Nr. 1907/2006 des Europäischen Parlaments und des Rates zur Registrierung, Bewertung, Zulassung und Beschränkung chemischer Stoffe (REACH) zwecks Anpassung an den technischen Fortschritt Text von Bedeutung für den EWR
Verordnung (EU) Nr. 900/2014 der Kommission vom 15. Juli 2014 zur Änderung der Verordnung (EG) Nr. 440/2008 zur Festlegung von Prüfmethoden gemäß der Verordnung (EG) Nr. 1907/2006 des Europäischen Parlaments und des Rates zur Registrierung, Bewertung, Zulassung und Beschränkung chemischer Stoffe (REACH) zwecks Anpassung an den technischen Fortschritt Text von Bedeutung für den EWR
ABl. L 247 vom 21.8.2014, p. 1–111
(BG, ES, CS, DA, DE, ET, EL, EN, FR, HR, IT, LV, LT, HU, MT, NL, PL, PT, RO, SK, SL, FI, SV)
 In force
In force
|
21.8.2014 |
DE |
Amtsblatt der Europäischen Union |
L 247/1 |
VERORDNUNG (EU) Nr. 900/2014 DER KOMMISSION
vom 15. Juli 2014
zur Änderung der Verordnung (EG) Nr. 440/2008 zur Festlegung von Prüfmethoden gemäß der Verordnung (EG) Nr. 1907/2006 des Europäischen Parlaments und des Rates zur Registrierung, Bewertung, Zulassung und Beschränkung chemischer Stoffe (REACH) zwecks Anpassung an den technischen Fortschritt
(Text von Bedeutung für den EWR)
DIE EUROPÄISCHE KOMMISSION —
gestützt auf den Vertrag über die Arbeitsweise der Europäischen Union,
gestützt auf die Verordnung (EG) Nr. 1907/2006 des Europäischen Parlaments und des Rates vom 18. Dezember 2006 zur Registrierung, Bewertung, Zulassung und Beschränkung chemischer Stoffe (REACH), zur Schaffung einer Europäischen Agentur für chemische Stoffe, zur Änderung der Richtlinie 1999/45/EG und zur Aufhebung der Verordnung (EWG) Nr. 793/93 des Rates, der Verordnung (EG) Nr. 1488/94 der Kommission, der Richtlinie 76/769/EWG des Rates sowie der Richtlinien 91/155/EWG, 93/67/EWG, 93/105/EG und 2000/21/EG der Kommission (1), insbesondere Artikel 13 Absatz 2,
in Erwägung nachstehender Gründe:
|
(1) |
In der Verordnung (EG) Nr. 440/2008 der Kommission (2) sind die in der Verordnung (EG) Nr. 1907/2006 vorgesehenen Prüfmethoden zur Bestimmung der physikalisch-chemischen Eigenschaften, der Toxizität und der Ökotoxizität von Stoffen festgelegt. |
|
(2) |
Es ist angezeigt, die Verordnung (EG) Nr. 440/2008 zu aktualisieren und vorrangig um neue und aktualisierte Prüfmethoden zu ergänzen, welche die OECD kürzlich angenommen hat, um dem technischen Fortschritt Rechnung zu tragen, und dafür zu sorgen, dass die Anzahl der verwendeten Versuchstiere im Einklang mit der Richtlinie 2010/63/EU des Europäischen Parlaments und des Rates (3) reduziert wird. Die Interessenträger wurden zu dem vorliegenden Entwurf konsultiert. |
|
(3) |
Diese Anpassung an den technischen Fortschritt beinhaltet sechs neue Prüfmethoden für die Bestimmung der Toxizität und anderer Gesundheitsauswirkungen, darunter eine Prüfung auf Entwicklungsneurotoxizität, eine erweiterte Ein-Generationen-Studie zur Reproduktionstoxizität, ein In-vivo-Genmutationsassay an transgenen Nagetieren, ein In-vitro-Test zur Bewertung der Auswirkungen auf die Synthese von Steroidhormonen sowie zwei In-vivo-Verfahren zur Beurteilung östrogener und (anti-)androgener Wirkungen. |
|
(4) |
Die Verordnung (EG) Nr. 440/2008 sollte daher entsprechend geändert werden. |
|
(5) |
Die Maßnahmen dieser Verordnung entsprechen der Stellungnahme des mit Artikel 133 der Verordnung (EG) Nr. 1907/2006 eingesetzten Ausschusses — |
HAT FOLGENDE VERORDNUNG ERLASSEN:
Artikel 1
Der Anhang der Verordnung (EG) Nr. 440/2008 wird nach Maßgabe des Anhangs der vorliegenden Verordnung geändert.
Artikel 2
Diese Verordnung tritt am dritten Tag nach ihrer Veröffentlichung im Amtsblatt der Europäischen Union in Kraft.
Diese Verordnung ist in allen ihren Teilen verbindlich und gilt unmittelbar in jedem Mitgliedstaat.
Brüssel, den 15. Juli 2014
Für die Kommission
Der Präsident
José Manuel BARROSO
(1) ABl. L 396 vom 30.12.2006, S. 1.
(2) Verordnung (EG) Nr. 440/2008 der Kommission vom 30. Mai 2008 zur Festlegung von Prüfmethoden gemäß der Verordnung (EG) Nr. 1907/2006 des Europäischen Parlaments und des Rates zur Registrierung, Bewertung, Zulassung und Beschränkung chemischer Stoffe (REACH) (ABl. L 142 vom 31.5.2008, S. 1).
(3) Richtlinie 2010/63/EU des Europäischen Parlaments und des Rates vom 22. September 2010 zum Schutz der für wissenschaftliche Zwecke verwendeten Tiere (ABl. L 276 vom 20.10.2010, S. 33).
ANHANG
Der Anhang der Verordnung (EG) Nr. 440/2008 wird wie folgt geändert:
Die Kapitel B.53, B.54, B.55, B.56, B.57 und B.58 werden eingefügt:
„B.53. PRÜFUNG AUF ENTWICKLUNGSNEUROTOXIZITÄT
EINLEITUNG
1. Diese Prüfmethode entspricht der OECD-Prüfrichtlinie (TG) 426 (2007). Im Juni 1995 traf sich eine OECD-Arbeitsgruppe aus dem Bereich Reproduktions- und Entwicklungstoxizität in Kopenhagen, um zu erörtern, ob eine Aktualisierung der bestehenden diesbezüglichen OECD-Prüfrichtlinien sowie die Ausarbeitung neuer Leitlinien für bislang noch nicht abgedeckte Endpunkte erforderlich ist (1). Die Arbeitsgruppe empfahl, auf der Grundlage einer inzwischen überarbeiteten Richtlinie der amerikanischen Umweltschutzbehörde (EPA) (2) eine Prüfrichtlinie für Entwicklungsneurotoxizität auszuarbeiten. Im Juni 1996 fand ein zweites Beratungstreffen in Kopenhagen statt, bei dem das Sekretariat über den Entwurf einer neuen Prüfrichtlinie zur Entwicklungsneurotoxizität einschließlich der wichtigsten Eckpunkte mit Angaben zur Auswahl der Versuchstierarten, zur Dosierungsphase, zur Prüfphase, zu den zu bewertenden Endpunkten und zu den Kriterien für die Auswertung der Ergebnisse informiert werden sollte. 1998 wurde eine US-amerikanische Richtlinie für die Bewertung des Neurotoxizitätsrisikos veröffentlicht (3). Im Oktober 2000 wurden nacheinander ein Beratungstreffen von OECD-Sachverständigen und ein Workshop des ILSI Risk Science Institute abgehalten. 2005 fand in Tokio ein Sachverständigen-Beratungstreffen statt. Ziel dieser Veranstaltungen war die Erörterung der wissenschaftlichen und technischen Fragen im Zusammenhang mit der aktuellen Prüfrichtlinie, und die Empfehlungen aus diesen Treffen (4)(5)(6)(7) wurden bei der Ausarbeitung der vorliegenden Prüfmethode berücksichtigt. Weitere Informationen zu Durchführung, Interpretation und Terminologie dieser Prüfmethode finden sich in den Guidance Documents der OECD Nr. 43 (‚Reproductive Toxicity Testing and Assessment‘) (8) und Nr. 20 (‚Neurotoxicity Testing‘) (9).
AUSGANGSÜBERLEGUNGEN
2. Viele Chemikalien schädigen bekanntermaßen beim Menschen und anderen Arten das in der Entwicklung befindliche Nervensystem (10)(11)(12)(13). Für die Bewertung und Beurteilung der toxischen Eigenschaften einer Chemikalie ist unter Umständen eine Bestimmung ihres entwicklungsneurotoxischen Potenzials erforderlich. Anhand von Prüfungen auf Entwicklungsneurotoxizität sollen Daten — einschließlich einer Charakterisierung der Dosis-Wirkungs-Beziehung — zu den potenziellen funktionalen und morphologischen Wirkungen auf das in der Entwicklung befindliche Nervensystem der Nachkommen gewonnen werden, die möglicherweise aus der Exposition in utero und während der frühen Lebensphasen resultieren.
3. Eine Entwicklungsneurotoxizitätsstudie kann als Einzelstudie durchgeführt werden, im Rahmen einer Reproduktionstoxizitätsstudie und/oder einer Neurotoxizitätsstudie am adulten Tier erfolgen (z. B. Prüfmethoden B.34 (14), B.35 (15), B.43 (16)) oder mit einer Studie zur Prüfung auf pränatale Entwicklungstoxizität gekoppelt werden (z. B. Prüfmethode B.31 (17)). Wenn die Entwicklungsneurotoxizitätsstudie im Rahmen einer anderen Studie oder gekoppelt mit einer anderen Studie durchgeführt wird, ist unbedingt darauf zu achten, dass die Integrität beider Studienarten gewahrt bleibt. Bei sämtlichen Prüfungen sind die geltenden Rechtsvorschriften sowie staatliche und institutionelle Leitlinien für die Verwendung von Versuchstieren zu Forschungszwecken einzuhalten (z. B. 18).
4. Das Prüflabor muss sich vor der Durchführung der Studie umfassend über alle zu der Prüfsubstanz vorliegenden Daten informieren. Dazu gehören die Identität und chemische Struktur der Chemikalie, ihre physikalisch-chemischen Eigenschaften, die Ergebnisse jeglicher anderer mit der Chemikalie durchgeführten In-vitro- oder In-vivo-Toxizitätsprüfungen, toxikologische Daten zu strukturverwandten Chemikalien und die vorgesehene(n) Verwendung(en) der Chemikalie. Diese Angaben sind notwendig, um für alle betroffenen Kreise sicherzustellen, dass die Prüfung für den Schutz der menschlichen Gesundheit maßgeblich ist, und sie erleichtern die Auswahl einer geeigneten Anfangsdosis.
PRINZIP DER PRÜFMETHODE
5. Die Prüfsubstanz wird den Tieren während der Trächtigkeit und der Laktationsperiode verabreicht. Anhand der Prüfung der Muttertiere können die Wirkungen auf trächtige und laktierende Weibchen bewertet und vergleichende Informationen (Muttertier versus Nachkommen) gewonnen werden. Die Nachkommen der einzelnen Würfe werden im randomisierten Verfahren für die Neurotoxizitätsbeurteilung ausgewählt. Die Versuchstiere werden bei der Beurteilung auf schwere neurologische und Verhaltensanomalien hin beobachtet. Dies schließt die Bewertung der physischen Entwicklung, der Verhaltensontogenese, der motorischen Aktivität, der motorischen und sensorischen Funktionen sowie der Lernfähigkeit und der Gedächtnisleistung ebenso ein wie die Beurteilung des Hirngewichts und neuropathologischer Veränderungen während der postnatalen Entwicklung und am adulten Tier.
6. Wenn die Prüfmethode als Einzelstudie durchgeführt wird, können zusätzliche verfügbare Tiere der einzelnen Gruppen für spezifische neuropathologische, neurochemische oder elektrophysiologische Verfahren oder Verfahren zur Erforschung des neurologisch bedingten Verhaltens verwendet werden, anhand deren die Daten der gemäß vorliegender Prüfmethode empfohlenen Prüfungen ergänzt werden können (16)(19)(20)(21). Die Zusatzverfahren können insbesondere dann sinnvoll sein, wenn die empirische Beobachtung, die erwarteten Wirkungen oder der Wirkmechanismus/die Wirkungsweise auf eine bestimmte Art von Neurotoxizität hindeuten. Diese Zusatzverfahren können sowohl auf die Muttertiere als auch die Jungtiere angewendet werden. Des Weiteren können Ex-vivo- oder In-vitro-Verfahren angewendet werden, sofern dadurch die Integrität der In-vivo-Verfahren nicht verändert wird.
VORBEREITUNGEN FÜR DIE PRÜFUNG
Auswahl der Versuchstierarten
7. Bevorzugtes Versuchstier ist die Ratte; gegebenenfalls können andere Versuchstierarten verwendet werden. Dabei ist jedoch zu beachten, dass die für diese Prüfmethode angegebenen Gestations- und postnatalen Tage für typischerweise verwendete Rattenstämme spezifisch sind. Im Falle der Verwendung anderer Arten oder eines ungewöhnlichen Stamms sind vergleichbare Tage zu wählen. Die Verwendung anderer Arten ist auf der Grundlage toxikologischer, pharmakokinetischer und/oder anderer Daten zu begründen. Bei der Begründung sollten unter anderem möglicherweise vorliegende artenspezifische postnatale neuropathologische Bewertungen und Bewertungen zum neurologisch bedingten Verhalten berücksichtigt werden. Wenn bei einem früheren Test Bedenken aufgetreten sind, sollte die Art/der Stamm, der die Bedenken hervorgerufen hat, in Betracht gezogen werden. Da verschiedene Rattenstämme unterschiedliche Eigenschaften und Merkmale haben, sollte der ausgewählte Stamm nachweislich über eine adäquate Fekundität und Empfindlichkeit verfügen. Inwiefern andere Arten aufgrund ihrer Empfindlichkeit geeignet sind, eine Entwicklungsneurotoxizität zuverlässig nachzuweisen, sollte dokumentiert werden.
Haltungs- und Fütterungsbedingungen
8. Die Temperatur im Versuchstierraum sollte 22 °C ± 3 °C betragen. Die relative Luftfeuchtigkeit sollte mindestens 30 % betragen und — außer beim Reinigen des Raums — 70 % nicht überschreiten, anzustreben ist ein Wert von 50-60 %. Die Beleuchtung sollte künstlich sein und die Hell- und Dunkelphasen sollten sich im Abstand von 12 Stunden abwechseln. Der Hell-Dunkel-Zyklus kann auch vor der Paarung und für den Verlauf der Studie umgekehrt werden, um die Bewertung der funktionalen und verhaltensbezogenen Endpunkte während der Dunkelphase (unter Rotlicht), also wenn die Tiere normalerweise aktiv sind (22), durchzuführen. Bei jeglicher Änderung des Hell-Dunkel-Zyklus ist ausreichend Zeit für die Akklimatisierung der Tiere einzuplanen, damit diese sich an den neuen Zyklus gewöhnen können. An die Versuchstiere kann herkömmliches Laborfutter verfüttert werden, und eine unbegrenzte Trinkwasserversorgung ist zu gewährleisten. Die Art des Futters und Wassers ist zu protokollieren und jeweils auf den Schadstoffgehalt hin zu analysieren.
9. Die Tiere können entweder einzeln oder in kleinen gleichgeschlechtlichen Gruppen in Käfigen untergebracht werden. Die Verpaarung erfolgt in für diese Zwecke geeigneten Käfigen. Wenn die Paarung nachweislich stattgefunden hat oder spätestens an Tag 15 der Trächtigkeit sind die verpaarten Tiere einzeln in Wurfkäfigen oder speziellen Käfigen für Muttertiere zu halten. Die Käfige sollten so angeordnet werden, dass etwaige Einflüsse der Käfigplatzierung minimiert werden. Verpaarte Weibchen sind mit bestimmten geeigneten Nestmaterialien zu versorgen, wenn die Geburt bevorsteht. Bekanntermaßen können sich eine unangemessene Handhabung oder Stress während der Gravidität nachteilig auswirken, da dies unter anderem zu pränatalen Verlusten und einer veränderten fetalen und postnatalen Entwicklung führen kann. Zum Schutz vor pränatalen Verlusten durch nicht behandlungsbedingte Faktoren ist bei der Handhabung der trächtigen Tiere Sorgfalt angebracht, und Stress infolge von äußeren Faktoren, wie übermäßiger Lärm, ist zu vermeiden.
Vorbereitung der Tiere
10. Es sind gesunde Tiere zu verwenden, die an die Laborbedingungen gewöhnt und zuvor nicht für andere Experimente verwendet wurden, außer wenn die Studie Teil einer anderen Studie ist (siehe Nummer 3). Art, Stamm, Herkunft, Geschlecht, Gewicht und Alter der Versuchstiere sind anzugeben. Jedes Versuchstier wird zur sicheren Identifizierung durch eine eigene Nummer gekennzeichnet. Die Tiere der einzelnen Testgruppen sollten so weit wie möglich ein einheitliches Gewicht und Alter haben und sich im normalen Schwankungsbereich der untersuchten Tierart/des Tierstamms bewegen. Für jede Dosisstufe sind junge adulte Weibchen zu verwenden, die noch nicht geworfen haben. Es ist sicherzustellen, dass Geschwister nicht verpaart werden. Tag 0 der Gravidität ist der Tag, an dem ein Vaginalpfropf und/oder Sperma beobachtet wird. Wenn terminiert trächtige Tiere von einem Lieferanten gekauft werden, ist eine angemessene Akklimatisierungszeit (z. B. 2-3 Tage) einzuplanen. Verpaarte Weibchen sind nach dem Zufallsprinzip den Kontroll- und Behandlungsgruppen zuzuteilen und möglichst gleichmäßig auf die Gruppen zu verteilen (für die gleichmäßige Verteilung auf alle Gruppen wird z. B. eine stratifizierte Randomisierung, beispielsweise auf der Grundlage des Körpergewichts, empfohlen). Vom selben Männchen besamte Weibchen sind gleichmäßig auf die Gruppen zu verteilen.
VERFAHREN
Zahl und Geschlecht der Versuchstiere
11. In jeder Prüf- und Kontrollgruppe sollte sich eine ausreichende Zahl trächtiger Weibchen befinden, die der Prüfsubstanz ausgesetzt werden, damit sichergestellt ist, dass eine angemessene Zahl an Nachkommen für die Neurotoxizitätsbeurteilung zur Verfügung steht. Es wird empfohlen, insgesamt 20 Würfe auf jeder Dosisstufe zu verwenden. Wiederholungsgaben und zeitversetzte Dosierungsschemata sind zulässig, wenn die Gesamtzahlen der Würfe pro Gruppe erreicht und geeignete statistische Modelle zur Berücksichtigung der Wiederholungsgaben verwendet werden.
12. Spätestens am postnatalen Tag 4 (PND 4) (der Tag der Geburt ist PND 0) ist die Größe der einzelnen Würfe durch Eliminierung zufällig ausgewählter überzähliger Jungtiere anzupassen, damit alle Würfe gleich groß sind (23). Die Wurfgröße sollte die durchschnittliche Wurfgröße des verwendeten Nagerstamms nicht überschreiten (8-12). In dem Wurf sollten sich möglichst gleich viele männliche und weibliche Jungtiere befinden. Eine selektive Eliminierung der Jungtiere, z. B. auf der Grundlage des Körpergewichts, ist nicht angebracht. Nach Vereinheitlichung der Würfe (Auslese) und vor der weiteren Prüfung funktionaler Endpunkte sollten einzelne Jungtiere, die für Tests vor bzw. nach dem Absetzen bestimmt sind, eindeutig gekennzeichnet werden, wobei eine beliebige geeignete humane Methode für die Jungtierkennzeichnung anzuwenden ist (z. B. 24).
Zuteilung von Tieren zu funktionalen Prüfungen, Verhaltensprüfungen, zur Bestimmung des Hirngewichts und zu neuropathologischen Beurteilungen
13. Im Rahmen der Prüfmethode gibt es mehrere Ansätze für die Zuteilung der in utero oder während des Säugens exponierten Tiere zu den funktionalen Prüfungen, den Verhaltensprüfungen, der Beurteilung der Geschlechtsreife, der Bestimmung des Hirngewichts und der neuropathologischen Beurteilung (25). Im Einzelfall können zusätzlich noch weitere Prüfungen zum neurologisch bedingten Verhalten (z. B. Sozialverhalten), zur Neurochemie oder Neuropathologie durchgeführt werden, sofern die Integrität der ursprünglich erforderlichen Prüfungen dadurch nicht gefährdet wird.
14. Die Jungtiere werden frühestens am PND 4 aus jeder der einzelnen Dosisgruppen ausgewählt und Endpunktbewertungen zugewiesen. Die Jungtiere sollten möglichst so ausgewählt werden, dass bei sämtlichen Prüfungen Jungtiere beider Geschlechter aus allen Würfen und aus jeder einzelnen Dosisgruppe zu gleichen Teilen vertreten sind. Bei der Prüfung der motorischen Aktivität sind dieselben Paare männlicher und weiblicher Jungtiere in sämtlichen Entwicklungsphasen vor dem Absetzen zu prüfen (siehe Nummer 35). Bei allen anderen Prüfungen können dieselben oder andere Paare männlicher und weiblicher Tiere verschiedenen Verhaltensprüfungen zugeteilt werden. Bei der Prüfung der kognitiven Funktionen müssen für die Prüfungen abgesetzter Jungtiere gegenüber adulten Tieren möglicherweise unterschiedliche Jungtiere zugeteilt werden, um Störeinflüsse aufgrund des Alters und durch Lerneffekte zu vermeiden (26)(27). Beim Absetzen (PND 21) können die nicht für die Prüfungen ausgewählten Jungtiere auf humane Art und Weise entsorgt werden. Alle Änderungen bei der Jungtierzuteilung sind festzuhalten. Die statistische Maßeinheit ist der Wurf (oder das Muttertier), nicht das Jungtier.
15. Die Zuteilung der Jungtiere zu den Untersuchungen vor dem Absetzen, den Untersuchungen nach dem Absetzen, den kognitiven Prüfungen, pathologischen Untersuchungen usw. kann auf verschiedene Weise erfolgen (siehe Abbildung 1 für die allgemeine Planung und Anlage 1 für Zuteilungsbeispiele). Die empfohlenen Mindestanzahlen an Tieren in jeder einzelnen Dosisgruppe lauten jeweils für Untersuchungen vor bzw. nach dem Absetzen wie folgt:
|
Klinische Beobachtungen und Körpergewicht |
Alle Tiere |
|
Ausführliche klinische Beobachtungen |
20/Geschlecht (1/Geschlecht/Wurf) |
|
Hirngewicht (nach Fixierung) PND 11-22 |
10/Geschlecht (1/Wurf) |
|
Hirngewicht (unfixiert) ~ PND 70 |
10/Geschlecht (1/Wurf) |
|
Neuropathologie (Immersions- oder Perfusionsfixierung) PND 11-22 |
10/Geschlecht (1/Wurf) |
|
Neuropathologie (Perfusionsfixierung) ~ PND 70 |
10/Geschlecht (1/Wurf) |
|
Geschlechtsreife |
20/Geschlecht (1/Geschlecht/Wurf) |
|
Andere Entwicklungsparameter (optional) |
Alle Tiere |
|
Verhaltensontogenese |
20/Geschlecht (1/Geschlecht/Wurf) |
|
Motorische Aktivität |
20/Geschlecht (1/Geschlecht/Wurf) |
|
Motorische und sensorische Funktionen |
20/Geschlecht (1/Geschlecht/Wurf) |
|
Lernen und Gedächtnis |
10/Geschlecht (1) (1/Wurf) |
Dosierung
16. Es sollten mindestens drei Dosisstufen und eine gleichzeitige Kontrolle verwendet werden. Die Dosisstufen sollten so gewählt werden, dass es zu einer graduellen Abstufung der toxischen Wirkungen kommt. Außer bei Beschränkungen aufgrund der physikalisch-chemischen oder der biologischen Eigenschaften der Chemikalie sollte die höchste Dosierungsstufe so gewählt werden, dass dadurch irgend eine Form von maternaler Toxizität auftritt (z. B. klinische Anzeichen, verminderte Körpergewichtszunahme (maximal 10 %) und/oder eine nachweisliche dosislimitierende Toxizität in einem Zielorgan). Die Hochdosis kann mit einigen Ausnahmen auf 1 000 mg/kg/Tag Körpergewicht begrenzt werden. Zum Beispiel kann aufgrund der erwarteten menschlichen Exposition die Verwendung einer höheren Dosisstufe erforderlich sein. Alternativ sollten Pilotstudien oder Vorstudien zur Dosisfindung durchgeführt werden, um die höchste einzusetzende Dosis zu bestimmen, bei der es zu minimaler maternaler Toxizität kommt. Wenn sich entweder in einer standardmäßigen Entwicklungstoxizitätsstudie oder in einer Pilotstudie herausgestellt hat, dass die Prüfsubstanz entwicklungstoxisch wirkt, sollte die höchste Dosisstufe diejenige Maximaldosis sein, bei der keine übermäßige Toxizität bei den Nachkommen auftritt bzw. bei der in utero oder am Neugeborenen weder Tod noch Missbildungen in einem Maße feststellbar sind, dass eine Bewertung der Neurotoxizität behindert würde. Bei der niedrigsten Dosisstufe sollte möglichst keine maternale Toxizität oder Entwicklungstoxizität einschließlich Neurotoxizität nachweisbar sein. Es ist eine absteigende Folge von Dosisstufen zu wählen, um dosisabhängige Wirkungen und die niedrigste Dosisstufe ohne zu beobachtende unerwünschte Wirkungen (NOAEL) nachzuweisen oder Dosen in der Nähe der Nachweisgrenze zu bestimmen, anhand deren die Festlegung einer Benchmark-Dosis (BMD) erfolgen kann. Zwei- bis vierfache Abstände erweisen sich häufig als optimale Dosisabstufungen, und meist ist eine zusätzliche vierte Dosisgruppe der Verwendung von sehr großen Dosisabständen (z. B. um mehr als den Faktor 10) vorzuziehen.
17. Bei der Wahl der Dosisstufen sind sämtliche für die Prüfsubstanz oder verwandte Stoffe vorliegenden Daten zur Toxizität sowie zusätzliche Informationen zu Verstoffwechselung und Toxikokinetik zu berücksichtigen. Diese Angaben sind gegebenenfalls für den Nachweis der Angemessenheit des Dosierungsplans hilfreich. Eine direkte Verabreichung an die Jungtiere sollte unter Berücksichtigung der Daten zu Exposition und Pharmakokinetik erfolgen (28)(29). Vor der Durchführung von Studien mit direkter Verabreichung sind die Vor- und Nachteile sorgfältig gegeneinander abzuwägen (30).
18. Die gleichzeitige Kontrollgruppe sollte eine scheinbehandelte Gruppe oder eine Vehikelkontrollgruppe sein, sofern ein Vehikel zur Verabreichung der Prüfsubstanz verwendet wird. Allen Tieren soll normalerweise dieselbe Menge der Prüfsubstanz oder des Vehikels entsprechend ihrem Körpergewicht verabreicht werden. Wird ein Vehikel oder ein anderes Additiv zur Erleichterung der Dosierung verwendet, sollten folgende Aspekte berücksichtigt werden: Auswirkungen auf die Resorption, die Verteilung, die Verstoffwechselung oder die Retention der Prüfsubstanz, Auswirkungen auf die chemischen Eigenschaften der Prüfsubstanz, die deren toxische Eigenschaften verändern können, und ferner Auswirkungen auf die Futter- oder Wasseraufnahme oder den Ernährungszustand der Versuchstiere. Das Vehikel sollte weder Wirkungen verursachen, die die Auswertung der Studie beeinflussen könnten, noch darf es toxisch auf das neurologisch bedingte Verhalten wirken oder zu einer Beeinflussung von Reproduktion oder Entwicklung führen. Bei neuen Vehikeln ist zusätzlich zu einer Vehikelkontrollgruppe eine scheinbehandelte Kontrollgruppe zu verwenden. Die Tiere der Kontrollgruppe(n) sind genauso zu behandeln wie die Tiere der Prüfgruppe(n).
Verabreichung der Dosen
19. Der Verabreichungsweg der Prüfsubstanz bzw. des Vehikels richtet sich nach der vorherrschenden Art der Exposition beim Menschen und den vorliegenden Daten zu Verstoffwechselung und Verteilung bei den Versuchstieren. Die Verabreichung erfolgt üblicherweise auf oralem Weg (z. B. Schlundsonde, Nahrung, Trinkwasser), andere Wege (z. B. dermal, inhalativ) sind jedoch je nach Eigenschaften und den voraussichtlichen oder bekannten menschlichen Expositionswegen ebenfalls möglich (weitere Informationen dazu im Guidance Document Nr. 43(8)). Der gewählte Verabreichungsweg ist zu begründen. Die Prüfsubstanz sollte täglich ungefähr zur selben Zeit verabreicht werden.
20. Die den einzelnen Tieren verabreichte Dosis orientiert sich normalerweise an der letzten Bestimmung des jeweiligen Körpergewichts. Im letzten Graviditätsdrittel ist bei der Anpassung der Dosen jedoch Vorsicht geboten. Wenn bei den behandelten Muttertieren übermäßige Toxizität festgestellt wird, sind die Tiere auf humane Art und Weise zu töten.
21. Die Prüfsubstanz bzw. das Vehikel ist den verpaarten Weibchen ab dem Implantationszeitpunkt (Tag 6 der Gravidität — GD 6) und während der gesamten Säugephase (PND 21) mindestens einmal täglich zu verabreichen, sodass die Jungtiere der Prüfsubstanz während ihrer prä- und postnatalen neurologischen Entwicklung ausgesetzt sind. Das Alter, ab dem die Verabreichung beginnt, und die Verabreichungsdauer und -häufigkeit können angepasst werden, wenn sich herausstellt, dass ein anderer Versuchsplan für die menschliche Exposition maßgeblicher ist. Die Dauer der Dosisgaben ist bei anderen Arten so anzupassen, dass eine Exposition während aller frühen Phasen der Hirnentwicklung sichergestellt ist (d. h. entsprechend dem pränatalen und frühen postnatalen Hirnwachstum beim Menschen). Mit der Verabreichung kann bereits bei Beginn der Trächtigkeit (GD 0) begonnen werden. Es gilt jedoch zu bedenken, dass die Prüfsubstanz potenziell zu einem Abgang vor der Einnistung führen kann. Dieses Risiko ließe sich zwar durch eine Verabreichung erst ab GD 6 vermeiden, doch dann würden die Entwicklungsphasen zwischen Tag GD 0 und Tag GD 6 nicht in die Behandlung einbezogen. Wenn ein Labor bereits geplant verpaarte Tiere erwirbt, ist ein Verabreichungsbeginn an Tag GD 0 gar nicht möglich, sodass der Tag GD 6 für den Verabreichungsbeginn gut geeignet wäre. Das Prüflabor sollte sich bei der Festlegung des Dosierungsplans an einschlägigen Informationen zu den Wirkungsweisen der Prüfsubstanz, früheren Erfahrungen und logistischen Überlegungen orientieren; dazu gehört gegebenenfalls auch eine Verlängerung der Dosisgabe bis nach dem Absetzen. Die Verabreichung ist am Tag der Geburt auszusetzen, wenn das gebärende Tier noch nicht alle Nachkommen geboren hat. Im Allgemeinen wird davon ausgegangen, dass die Exposition der Jungtiere über die Muttermilch erfolgt, eine Direktverabreichung bei den Jungtieren ist jedoch in denjenigen Fällen in Betracht zu ziehen, wenn der Nachweis für eine kontinuierliche Exposition der Nachkommen fehlt. Der Nachweis für eine kontinuierliche Exposition kann z. B. pharmakokinetischen Daten, Toxizitätszeichen bei den Nachkommen oder veränderten Biomarkern entnommen werden (28).
BEOBACHTUNGEN
Beobachtungen an Muttertieren
22. Sämtliche Muttertiere sind mindestens einmal täglich sorgfältig auf ihren Gesundheitszustand einschließlich Anzeichen für Morbidität und Mortalität zu beobachten.
23. Während der Behandlungs- und Beobachtungsphasen sind regelmäßig unter Einbeziehung von mindestens zehn Muttertieren pro Dosisstufe eingehendere klinische Beobachtungen durchzuführen (mindestens zweimal während der Verabreichung in der Graviditätsphase und zweimal während der Verabreichung in der Säugephase). Die Tiere sind außerhalb des Käfigs, in dem sie gehalten werden, von qualifiziertem technischem Personal zu beobachten, denen die Behandlung des jeweiligen Tieres nicht bekannt ist. Dabei sind standardisierte Verfahren anzuwenden, um den Stress des Tieres zu minimieren, die Voreingenommenheit des Beobachters so gering wie möglich zu halten und die Zuverlässigkeit bei Beobachtungen durch mehrere Personen zu maximieren. Die Beobachtungen innerhalb einer bestimmten Studie sollten möglichst durch ein und denselben Untersucher erfolgen.
24. Die bei der Beobachtung festgestellten Anzeichen sind zu dokumentieren. Wenn irgend möglich, ist darüber hinaus die Größenordnung der beobachteten Anzeichen festzuhalten. Bei den klinischen Beobachtungen ist insbesondere auf Veränderungen an Haut, Fell, Augen, Schleimhäuten, auf Sekrete sowie autonome Körperfunktionen (z. B. Tränensekretion, Piloerektion, Pupillengröße, ungewöhnliche Atemmuster und/oder Mundatmung sowie ungewöhnliche Harnentleerung oder Defäkation) zu achten.
25. Ungewöhnliche Reaktionen hinsichtlich Körperhaltung, Aktivitätsgrad (z. B. verminderte oder verstärkte Erkundung der üblichen Untersuchungsumgebung) und Bewegungskoordination sind ebenfalls zu vermerken. Veränderungen des Gangs (z. B. watschelnder Gang, Ataxie), der Haltung (z. B. Buckelhaltung) und des Reaktionsverhaltens beim Aufnehmen oder Umsetzen des Versuchstiers oder anderen Umgebungsstimuli sowie Auftreten von klonischen oder tonischen Bewegungen, Krämpfen oder Tremors, Stereotypien (z. B. übermäßige Fellpflege, ungewöhnliche Kopfbewegungen, repetitives Laufen im Kreis) oder auffälliges Verhalten (z. B. Beißen oder übermäßiges Lecken, Selbstverstümmelung, Rückwärtslaufen, Lautäußerungen) oder Aggressionen sollen protokolliert werden.
26. Sämtliche Toxizitätszeichen sind mit Tag des Einsetzens, Tageszeit, Schweregrad und Dauer zu notieren.
27. Die Tiere sind zum Zeitpunkt der Dosierung mindestens einmal wöchentlich während der Dauer der Studie und am oder um den Tag der Geburt herum sowie am PND 21 (Absetzen) zu wiegen. In Studien mit Substanzverabreichung per Schlundsonde sind die Muttertiere mindestens zweimal wöchentlich zu wiegen. Die Dosis ist jeweils nach jeder Bestimmung des Körpergewichts entsprechend anzupassen. Die Futteraufnahme ist wöchentlich mindestens während der Trächtigkeit und der Säugeperiode zu messen. Die Wasseraufnahme ist mindestens wöchentlich zu messen, wenn die Exposition über die Wasserversorgung erfolgt.
Beobachtungen an Nachkommen
28. Sämtliche Nachkommen sind mindestens einmal täglich sorgfältig auf Toxizitätszeichen sowie auf Morbidität und Mortalität hin zu beobachten.
29. Während der Behandlungs- und Beobachtungsphasen sollten eingehendere klinische Beobachtungen an den Nachkommen durchgeführt werden. Die Nachkommen (mindestens ein Jungtier pro Geschlecht und Wurf) sind von medizinisch-technischen Assistenten (MTA) zu beobachten, denen die Behandlung des jeweiligen Tieres nicht bekannt ist. Dabei sind standardisierte Verfahren anzuwenden, um die Voreingenommenheit des Beobachters so gering wie möglich zu halten und die Zuverlässigkeit bei Beobachtungen durch mehrere Personen zu maximieren. Die Beobachtungen sollten möglichst durch ein- und denselben Untersucher erfolgen. Es sind entsprechend dem untersuchten Entwicklungsstadium mindestens die unter den Nummern 24 und 25 beschriebenen Endpunkte zu überwachen.
30. Sämtliche bei den Nachkommen auftretenden Toxizitätszeichen sind mit Tag des Einsetzens, Tageszeit, Schweregrad und Dauer zu notieren.
Physische Merkmale und Entwicklungsparameter
31. Veränderungen bei Entwicklungsparametern vor dem Absetzen (z. B. Ohrmuschelentfaltung, Augenöffnung, Schneidezahndurchbruch) korrelieren eng mit dem Körpergewicht (30)(31). Das Körpergewicht ist möglicherweise der beste Indikator für die physische Entwicklung. Eine Messung von Entwicklungsparametern empfiehlt sich daher nur dann, wenn bereits im Vorfeld nachgewiesen ist, dass diese Endpunkte zusätzliche Informationen liefern werden. Die zeitliche Planung für die Bewertung dieser Parameter ist Tabelle 1 zu entnehmen. In Abhängigkeit von den erwarteten Wirkungen und den Ergebnissen der ersten Messungen sollten gegebenenfalls weitere Zeitpunkte hinzugefügt oder die Messungen in anderen Entwicklungsstadien durchgeführt werden.
32. Für die Bewertung der physischen Entwicklung sollte eher das postkoitale Alter als das postnatale Alter zugrunde gelegt werden (33). Wenn die Jungtiere am Tag des Absetzens getestet werden, sollte die Prüfung vor dem eigentlichen Absetzen erfolgen, um die Ergebnisse nicht durch den Stress zu verzerren, der beim Absetzen entsteht. Prüfungen, die nach dem Absetzen an den Jungtieren erfolgen, sollten zudem nicht innerhalb der ersten zwei Tage nach dem Absetzen durchgeführt werden.
Tabelle 1
Zeitliche Planung für die Bewertung der physischen Merkmale und Entwicklungsparameter und der funktionalen Endpunkte/Verhaltensendpunkte (2) .
|
Altersphasen Endpunkte |
Vor Absetzen (3) |
Adoleszenz (3) |
Junge adulte Tiere (3) |
|
Physische Merkmale und Entwicklungsparameter |
|||
|
Körpergewicht und klinische Beobachtungen |
Wöchentlich (4) |
mindestens vierzehntägig |
mindestens vierzehntägig |
|
Hirngewicht |
PND 22 (5) |
bei Beendigung |
|
|
Neuropathologie |
PND 22 (5) |
bei Beendigung |
|
|
Geschlechtsreife |
— |
zu gegebener Zeit |
— |
|
Andere Entwicklungsparameter (6) |
gegebenenfalls |
— |
— |
|
Funktionale Endpunkte/Verhaltensendpunkte |
|||
|
Verhaltensontogenese |
Mindestens zwei Messungen |
|
|
|
Motorische Aktivität (einschließlich Habituation) |
1-3 Mal (7) |
— |
einmal |
|
Motorische und sensorische Funktionen |
— |
einmal |
einmal |
|
Lernen und Gedächtnis |
— |
einmal |
einmal |
33. Die lebenden Jungtiere sind zu zählen und ihr Geschlecht ist zu bestimmen, z. B. durch Sichtkontrolle oder Messung des anogenitalen Abstands (34)(35); jedes Jungtier eines Wurfs ist einzeln zu wiegen, und zwar so bald wie möglich nach der Geburt, danach wöchentlich während der Laktation und im Anschluss daran mindestens vierzehntägig. Bei der Beurteilung der Geschlechtsreife sollten pro Wurf bei mindestens einem Weibchen und einem Männchen Alter und Körpergewicht bestimmt werden, wenn die Öffnung der Vagina (36) bzw. die Präputialseparation (37) beobachtet werden.
Verhaltensontogenese
34. Die Ontogenese ausgewählter Verhaltensweisen sollte bei mindestens einem Jungtier pro Geschlecht und Wurf während der entsprechenden Altersphase gemessen werden, wobei an allen Prüftagen und bei jedem bewerteten Verhalten jeweils dieselben Jungtiere zu verwenden sind. Die Messtage sind gleichmäßig über diesen Zeitraum zu verteilen, um entweder die normale oder die behandlungsbedingte Veränderung der Ontogenese des jeweiligen Verhaltens bestimmen zu können (38). Nachfolgend sind einige Beispiele für Verhalten aufgeführt, deren Ontogenese bewertet werden kann: Aufrichtungsreflex, negative Geotaxis und motorische Aktivität (38)(39)(40).
Motorische Aktivität
35. Die motorische Aktivität ist in den Lebensphasen vor dem Absetzen und in späteren Phasen beim adulten Tier zu beobachten (41)(42)(43)(44)(45). Bezüglich Prüfungen vor dem Absetzen siehe Nummer 32. Die Prüfsitzung sollte so lange dauern, dass bei unbehandelten Kontrolltieren eine Habituation innerhalb der Sitzung nachgewiesen werden kann. Es wird dringend empfohlen, bei der Bewertung der Verhaltensontogenese die motorische Aktivität zu berücksichtigen. Wenn bei einem Test die Verhaltensontogenese geprüft werden soll, sollten für alle vor dem Absetzen stattfindenden Prüfsitzungen dieselben Tiere eingesetzt werden. Die Prüfungen sollten so häufig stattfinden, dass die Ontogenese einer Habituation innerhalb einer Sitzung bewertet werden kann (44). Dafür sind gegebenenfalls drei oder mehr Prüfzeitpunkte vor dem Tag des Absetzens und einschließlich dieses Tags erforderlich (z. B. PND 13, 17 und 21). Dieselben Tiere oder Wurfgeschwister sollten darüber hinaus als adulte Tiere gegen Ende der Studie geprüft werden (z. B. PND 60-70). Nach Bedarf können die Prüfungen an weiteren Zusatztagen durchgeführt werden. Die motorische Aktivität sollte mittels eines automatischen Aktivitätsmessgeräts beobachtet werden, das in der Lage ist, sowohl ein Ansteigen als auch ein Abfallen der Aktivität zu erfassen (d. h. die vom Gerät gemessene Basisaktivität sollte weder so niedrig sein, dass dadurch die Erfassung eines Abfalls verhindert wird, noch so hoch, dass dadurch die Erfassung von Aktivitätsanstiegen unmöglich gemacht wird). Jedes der Geräte sollte im Rahmen standardisierter Verfahren geprüft worden sein, um bei Betrieb mehrerer Geräte an mehreren Tagen eine möglichst hohe Betriebssicherheit zu gewährleisten. Die Behandlungsgruppen sollten soweit wie möglich gleichmäßig auf die Geräte verteilt werden. Jedes Tier ist einzeln zu testen. Bei der Beobachtung der Behandlungsgruppen über die Prüfzeiträume hinweg ist auf Ausgewogenheit zu achten, um Störeinflüsse aufgrund des zirkadianen Aktivitätsrhythmus zu vermeiden. Es ist nach Möglichkeit sicherzustellen, dass die Prüfbedingungen nur geringfügig variieren und diese Variationen nicht systematisch mit der Behandlung zusammenhängen. Zu den Variablen, die die Messung zahlreicher Verhaltensweisen, einschließlich der motorischen Aktivität, beeinflussen können, gehören Geräuschpegel, Testkäfigform und -größe, Temperatur, relative Luftfeuchte, Lichtverhältnisse, Gerüche, Verwendung des vertrauten Haltungskäfigs oder eines neuen Testkäfigs und Ablenkungen aus der Umgebung.
Motorische und sensorische Funktionen
36. Die motorischen und sensorischen Funktionen sind mindestens einmal während der Adoleszenzphase und einmal beim jungen adulten Tier eingehend zu untersuchen (z. B. PND 60-70). Bezüglich Prüfungen zum Zeitpunkt des Absetzens siehe Nummer 32. Es sollte eine ausreichende Anzahl an Tests durchgeführt werden, um eine aussagekräftige Stichprobenmenge zu den sensorischen Modalitäten (z. B. somatosensorisch, vestibulär) und den motorischen Funktionen (z. B. Kraft, Koordination) sicherzustellen. Die motorischen und sensorischen Funktionen können unter anderem mittels des generalisierten Streckreflexes (extensor-thrust) (46), des Aufrichtungsreflexes (47)(48), der akustischen Schreckreflex-Habituation (40)(49)(50)(51)(52)(53)(54) und evozierter Potenziale (55) untersucht werden.
Lern- und Gedächtnisprüfungen
37. Nach dem Absetzen (beispielsweise an Tag 25 ± 2 Tage) und beim jungen adulten Tier (PND 60 oder älter) sollte ein Test für assoziatives Lernen und Gedächtnis durchgeführt werden. Bezüglich Prüfungen vor dem Absetzen siehe Nummer 32. Bei diesen beiden Entwicklungsphasen können derselbe Test oder andere Tests verwendet werden. Bei der Auswahl der Prüfung(en), anhand deren die Lern- und Gedächtnisfähigkeit bei abgesetzten Tieren und adulten Ratten untersucht werden kann, besteht ein gewisser Handlungsspielraum. Die Prüfungen sollten jedoch so aufgebaut sein, dass sie die folgenden beiden Kriterien erfüllen: Erstens sollte die Lernfähigkeit entweder als Veränderung über mehrere wiederholte Lernexperimente oder -sitzungen hinweg oder, in Prüfungen mit nur einem Versuch, durch Verwendung einer Versuchsbedingung, die nichtassoziative Wirkungen der Trainingserfahrung kontrolliert, beurteilt werden. Zweitens sollten die Prüfungen eine Gedächtnismessung (Kurzzeit- oder Langzeitgedächtnis) zusätzlich zum ursprünglichen Lernen (Aneignung) beinhalten, doch diese Gedächtnismessung ist nur dann dokumentierbar, wenn innerhalb derselben Prüfung auch eine Messung der Aneignung stattgefunden hat. Wenn die Lern- und Gedächtnisprüfungen eine Wirkung der Prüfsubstanz ergeben, sind gegebenenfalls Zusatztests in Erwägung zu ziehen, um auszuschließen, dass das Versuchsergebnis auf alternative Ursachen, zum Beispiel geänderte sensorische, motivationale und/oder motorische Fähigkeiten, zurückzuführen ist. Zusätzlich zu den beiden oben genannten Kriterien wird empfohlen, die Lern- und Gedächtnistests danach auszuwählen, ob bei ihnen in Bezug auf die zu untersuchende Chemikalienklasse nachweislich eine Prüfempfindlichkeit vorliegt, sofern sich entsprechende Hinweise in der Fachliteratur finden. Falls sich in der Literatur keine Hinweise finden, können unter anderem folgende Prüfungen unter Einhaltung der oben genannten Kriterien durchgeführt werden: passive Vermeidung (43)(56)(57), Delayed-matching-to-Position-Aufgaben für die adulte Ratte (58) und die infantile Ratte (59), olfaktorische Konditionierung (43)(60), Morris-Wasserlabyrinth (61)(62)(63), Biel-Maze-Test oder Cincinnati-Maze-Test (64)(65), Radial Arm-Maze (66), T-Maze (43) und Aneignung und Beibehaltung von zeitplangesteuertem Verhalten (26)(67)(68). In der Literatur sind zusätzliche Tests für abgesetzte Jungtiere (26)(27) und adulte Ratten (19)(20) beschrieben.
Nekropsie
38. Die Muttertiere können nach dem Absetzen der Nachkommen getötet werden.
39. Die neuropathologische Beurteilung der Nachkommen wird unter Verwendung der Gewebe von auf humane Art am Tag PND 22 oder zu einem früheren Zeitpunkt zwischen PND 11 und PND 22 getöteten Nachkommen sowie am Ende der Studie durchgeführt. Bei am PND 22 getöteten Nachkommen sind die Hirngewebe zu untersuchen; bei am Ende der Prüfung getöteten Tieren sind sowohl die Gewebe des Zentralnervensystems (ZNS) als auch die des peripheren Nervensystems (PNS) zu beurteilen. Tiere, die am PND 22 oder früher getötet werden, können durch Immersion oder Perfusion fixiert werden. Am Ende der Prüfung getötete Tiere sind durch Perfusion zu fixieren. Bei allen die Gewebepräparation betreffenden Aspekten sollte auf Ausgewogenheit geachtet werden, d. h. von der Perfusion der Tiere über das Schneiden der Gewebeproben und die Probenvorbereitung bis hin zur Färbung der Objektträger sollte jeder Satz repräsentative Proben jeder Dosisgruppe enthalten. Eine weiterführende neuropathologische Anleitung findet sich im OECD Guidance Document Nr. 20 (9), siehe auch (103).
Probenvorbereitung
40. Alle zum Zeitpunkt der Nekropsie offenkundigen schwerwiegenden Abnormitäten sind zu dokumentieren. Es sind Gewebeproben von allen wichtigen Regionen des Nervensystems zu entnehmen. Die Gewebeproben sind in einem geeigneten Fixativ aufzubewahren und im Einklang mit allgemein bekannten histologischen Standardprotokollen (69)(70)(71)(103) aufzubereiten. Für ZNS- und PNS-Gewebe ist die Paraffineinbettung geeignet, doch wenn ein höherer Auflösungsgrad erforderlich ist (z. B. bei peripheren Nerven bei Verdacht auf periphere Neuropathie und/oder bei der morphometrischen Analyse peripherer Nerven), ist gegebenenfalls eine Nachfixierung mit Osmium in Kombination mit Epoxydeinbettung angezeigt. Für die morphometrische Analyse gewonnenes Hirngewebe ist bei allen Dosisstufen zur gleichen Zeit in ein geeignetes Medium einzubetten, um ein Schrumpfen der Prüfgegenstände zu vermeiden, das bei zu langer Aufbewahrung im Fixativ auftreten kann (6).
Neuropathologische Untersuchung
41. Bei der qualitativen Untersuchung sollen
|
i) |
Regionen innerhalb des Nervensystems ermittelt werden, die Anzeichen für neuropathologische Veränderungen zeigen, |
|
ii) |
diejenigen Arten neuropathologischer Veränderungen ermittelt werden, die auf die Exposition gegenüber der Prüfsubstanz zurückzuführen sind, und |
|
iii) |
der Schweregrad der neuropathologischen Veränderungen bestimmt werden. |
Repräsentative histologische Schnitte der Gewebeproben sind von einem entsprechend geschulten Pathologen mikroskopisch auf das Vorliegen neuropathologischer Veränderungen zu untersuchen. Alle neuropathologischen Veränderungen sind subjektiv nach Schweregrad einzuteilen. Eine Hämatoxylin-Eosin-Färbung ist für die Hirnschnitt-Beurteilung von Tieren, die spätestens am PND 22 auf humane Weise getötet wurden, gegebenenfalls ausreichend. Bei ZNS- und PNS-Geweben von am Ende der Prüfung getöteten Tieren sollte jedoch eine Myelinfärbung (z. B. Luxol Fast Blue/Kresylviolett) und eine Silberfärbung (z. B. nach Bielschowsky oder Bodian) durchgeführt werden. Je nach professioneller Einschätzung des Pathologen und in Abhängigkeit von den beobachteten Veränderungen können auch andere Färbungen zur Identifizierung und Charakterisierung bestimmter Veränderungstypen in Betracht kommen (z. B. histochemischer Nachweis des sauren Gliafaserproteins (Glial fibrillary acidic protein, GFAP) oder Lektinhistochemie zur Bewertung glialer und mikroglialer Veränderungen (72), Fluoro-Jade-Färbung zur Feststellung von Nekrosen (73)(74) oder Silberfärbungen speziell für neuronale Degenerationen (75)).
42. Es sollte eine morphometrische (quantitative) Bewertung durchgeführt werden, da diese Daten für den Nachweis einer behandlungsbedingten Wirkung relevant sein können und hilfreich sind für die Beurteilung der behandlungsbedingten Unterschiede bei Hirngewicht und Morphologie (76)(77). Von den Nervengeweben sind Proben herzustellen und für die morphometrische Untersuchung aufzubereiten. Die morphometrischen Bewertungen können z. B. lineare oder flächige Messungen bestimmter Hirnregionen umfassen (78). Bei den linearen oder flächigen Messungen müssen gleichwertige, sorgfältig auf der Grundlage zuverlässiger mikroskopischer Messpunkte ausgewählte Schnitte verwendet werden (6). Mithilfe der Stereologie können behandlungsbedingte Wirkungen auf bestimmte Parameter wie Volumen oder Zellzahl für bestimmte neuroanatomische Regionen festgestellt werden (79)(80)(81)(82)(83)(84).
43. Die Gehirne sind auf Hinweise für behandlungsbedingte neuropathologische Veränderungen hin zu untersuchen, und von allen wichtigen Hirnregionen sind geeignete Proben zu nehmen (z. B. Riechkolben, Großhirnrinde (Cortex cerebri), Hippocampus, Basalganglien, Thalamus, Hypothalamus, Mittelhirn (Tectum, Tegmentum und Pedunculus cerebri), Pons, Medulla oblongata, Kleinhirn), um eine gründliche Untersuchung zu gewährleisten. Die Schnitte müssen unbedingt bei allen Tieren in derselben Ebene entnommen werden. Bei adulten Tieren, die am Ende der Prüfung auf humane Art getötet werden, sind repräsentative Schnitte des Rückenmarks und des PNS als Proben zu entnehmen. Die untersuchten Bereiche sollten das Auge mit Sehnerv und Netzhaut, das Rückenmark an den zervikalen und lumbalen Verdickungen, die dorsalen und ventralen Nervenwurzelfasern, den proximalen Ischiasnerv, den proximalen Schienbeinnerv (am Knie) und die Wadenmuskel-Äste des Schienbeinnervs umfassen. Die Gewebeschnitte durch Rückenmark und peripheres Nervengewebe sollen sowohl Quer- als auch Längsschnitte umfassen.
44. Bei der neuropathologischen Beurteilung sollte zusätzlich zu Zellveränderungen (z. B. neuronale Vakuolisierung, Degeneration, Nekrose) und Gewebeveränderungen (z. B. Gliose, Leukozyteninfiltration, Zystenbildung) auch nach Hinweisen auf Entwicklungsschäden am Nervensystem gesucht werden (6)(85)(86)(87)(88)(89). Diesbezüglich sind behandlungsbedingte Wirkungen unbedingt von normalen Entwicklungsereignissen zu unterscheiden, die üblicherweise in der mit dem Tötungszeitpunkt zusammenfallenden Entwicklungsphase auftreten (90). Beispiele für signifikante Veränderungen, die auf eine Entwicklungsschädigung hindeuten, sind unter anderem:
|
— |
Veränderungen der Größe oder Form bei Riechkolben, Großhirn oder Kleinhirn, |
|
— |
Veränderungen der relativen Größe verschiedener Hirnregionen einschließlich Vergrößerungen oder Verkleinerungen von Regionen, die aus dem Verlust oder der Fortdauer von normalerweise kurzlebigen Zellpopulationen oder axonalen Projektionen resultieren (z. B. externes Keimlager des Kleinhirns, Gehirnbalken), |
|
— |
Veränderungen der Proliferation, Migration und Differenzierung, wie sie in Bereichen mit ausgeprägter Apoptose oder Nekrose vorkommen, Cluster oder zerstreute Populationen ektopischer, fehlplatzierter oder fehlgebildeter Neuronen oder Änderungen an der relativen Größe verschiedener Schichten kortikaler Strukturen, |
|
— |
Veränderungen beim Entstehungsmuster der Myelinhülle einschließlich einer globalen Größenverminderung oder veränderte Färbung der myelinisierten Strukturen, |
|
— |
Feststellung eines Wasserkopfs, insbesondere Erweiterung der Ventrikel, Stenose des Aquaeductus cerebri und Verdünnung der Großhirnhemisphären. |
Analyse der Dosis-Wirkungs-Beziehung neuropathologischer Veränderungen
45. Das folgende, schrittweise aufgebaute Verfahren wird für die qualitative und quantitative neuropathologische Analyse empfohlen. Als erstes werden die Schnitte der Gruppe mit der höchsten Dosis mit denjenigen der Kontrollgruppe verglichen. Wenn bei den Tieren der Gruppe, die die höchste Dosis erhalten hat, keine neuropathologischen Veränderungen feststellbar sind, ist keine weitere Analyse erforderlich. Wenn neuropathologische Veränderungen bei der Hochdosisgruppe nachweisbar sind, werden auch die Tiere der mittleren und der niedrigsten Dosisgruppen untersucht. Wenn die Hochdosisgruppe aufgrund einer anderen konfundierenden Toxizität oder des Todes der Tiere abgeschlossen ist, sind die Gruppen mit hoher und mittlerer Dosis auf neuropathologische Veränderungen hin zu analysieren. Wenn es in den niedrigeren Dosisgruppen Hinweise auf Neurotoxizität gibt, ist bei diesen Gruppen eine neuropathologische Analyse durchzuführen. Wenn bei der qualitativen oder quantitativen Untersuchung behandlungsbedingte neuropathologische Veränderungen gefunden werden, sind die Dosisabhängigkeit des Auftretens, die Häufigkeit und der Schweregrad der Läsionen oder der morphometrischen Veränderungen auf der Grundlage einer Beurteilung aller Tiere aus sämtlichen Dosisgruppen zu bestimmen. In die Beurteilung sind alle Hirnregionen einzubeziehen, bei denen sich neuropathologische Veränderungen gleich welcher Art finden. Für jede Läsionsart sind die zur Festlegung der einzelnen Schweregrade verwendeten Eigenschaften zu beschreiben und die zur Unterscheidung der Schweregrade verwendeten Merkmale anzugeben. Anhand der Häufigkeit der einzelnen Läsionsarten und ihres Schweregrads ist eine statistische Analyse zur Beurteilung der Art der Dosis-Wirkungs-Beziehung durchzuführen. Es wird empfohlen, codierte Objektträger zu verwenden (91).
DATEN UND BERICHTERSTATTUNG
Daten
46. Die Daten sind sowohl einzeln zu protokollieren als auch in tabellarischer Form zusammenzufassen, wobei ersichtlich sein muss, bei welcher der einzelnen Prüfgruppen welche Arten von Veränderungen aufgetreten sind und bei wie vielen Muttertieren, nach Geschlecht gegliederten Nachkommen und Würfen diese Veränderungen feststellbar waren. Wenn eine direkte postnatale Exposition der Nachkommen erfolgt ist, sind Verabreichungsweg, -dauer und -zeitraum in den Bericht aufzunehmen.
Beurteilung und Auswertung der Ergebnisse
47. Eine Prüfung auf Entwicklungsneurotoxizität liefert Informationen darüber, wie sich eine Chemikalie bei wiederholter Exposition in utero und während der frühen postnatalen Entwicklung auswirkt. Da der Schwerpunkt sowohl auf die allgemeine Toxizität als auch auf entwicklungsneurotoxische Endpunkte gelegt wird, lassen die Prüfungsergebnisse eine Unterscheidung zu zwischen den Wirkungen auf die neurologische Entwicklung, die ohne allgemeine maternale Toxizität auftreten, und Veränderungen, die nur bei Dosen auftreten, die auch beim Muttertier toxisch wirken. Da die Wechselbeziehung zwischen Prüfungsdesign, statistischer Analyse und biologischer Signifikanz der Daten äußerst komplex ist, können die Entwicklungsneurotoxizitätsdaten nur unter Hinzuziehung eines Experten korrekt ausgewertet werden (107)(109). Bei der Auswertung der Testergebnisse sollte ein WoE-Ansatz angewendet werden (20)(92)(93)(94). Die Muster der verhaltensbedingten oder morphologischen Befunde sollten, sofern vorhanden, ebenso erörtert werden wie eine nachweisliche Dosis-Wirkungs-Beziehung. In diese Analyse sollten Daten aus allen Studien einfließen, die für die Beurteilung der Entwicklungsneurotoxizität relevant sind, dazu gehören unter anderem epidemiologische Studien oder Fallberichte am Menschen sowie tierexperimentelle Studien (z. B. toxikokinetische Daten, Daten zur Struktur-Wirkungs-Beziehung, Daten aus anderen Toxizitätsstudien). Dies schließt auch die Beziehung zwischen den Dosen der Prüfsubstanz und dem Auftreten/Nichtauftreten neurotoxischer Wirkungen bzw. deren Häufigkeit und Ausprägung bei beiden Geschlechtern mit ein (20)(95).
48. Die Datenauswertung sollte ferner eine Diskussion sowohl der biologischen als auch der statistischen Signifikanz beinhalten. Die statistische Analyse sollte bei der Datenauswertung unterstützend herangezogen werden, aber nicht allein bestimmend dafür sein. Allein aus dem Fehlen einer statistischen Signifikanz sollte ebenso wenig automatisch geschlossen werden, dass keine behandlungsbedingten Wirkungen vorliegen, wie das Vorhandensein einer statistischen Signifikanz nicht als einzige Begründung für das Vorliegen einer behandlungsbedingten Wirkung herangezogen werden darf. Um mögliche falsch-negative Ergebnisse zu vermeiden und den Schwierigkeiten vorzubeugen, die naturgemäß mit einem ‚negativen Beweis‘ verbunden sind, sollten vorhandene positive und historische Kontrolldaten in die Diskussion einbezogen werden, insbesondere dann, wenn keine behandlungsbedingten Wirkungen gefunden werden (102)(106). Die Wahrscheinlichkeit falsch-positiver Ergebnisse sollte vor dem Hintergrund der statistischen Gesamtauswertung der Daten erörtert werden (96). Sofern ein Zusammenhang zwischen neuropathologischen Veränderungen und Verhaltensänderungen beobachtet wurde, sollte dieser in die Beurteilung einbezogen werden.
49. Sämtliche Ergebnisse sind mithilfe zum Prüfungsdesign passender statistischer Modelle zu analysieren (108). Die Entscheidung für eine parametrische bzw. eine parameterfreie Analyse ist anhand bestimmter Faktoren wie zum Beispiel der Art der Daten (umgewandelte/nicht umgewandelte Daten) und deren Verteilung sowie der relativen Robustheit der gewählten statistischen Analyse zu begründen. Bei der Auswahl der statistischen Analysemethoden sollten Prüfungszweck und Prüfungsdesign als Orientierungshilfe dienen, um das Auftreten von Typ-I-Fehlern (falsch-positive Ergebnisse) und Typ-II-Fehlern (falsch-negative Ergebnisse) so gering wie möglich zu halten (96)(97)(104)(105). Bei Entwicklungsstudien, bei denen jeweils mehrere Jungtiere desselben Wurfs geprüft werden, sollte der Wurf als solcher in das statistische Modell einbezogen werden, um aufgeblähte Typ-I-Fehlerquoten zu vermeiden (98)(99)(100)(101). Die statistische Maßeinheit sollte der Wurf sein, nicht das Jungtier. Die Experimente sollten so ausgestaltet sein, dass Wurfgeschwister nicht als unabhängige Beobachtungsobjekte behandelt werden. Jeder am selben Versuchstier wiederholt gemessene Endpunkt sollte anhand statistischer Modelle analysiert werden, die der Tatsache Rechnung tragen, dass diese Messungen nicht unabhängig sind.
Prüfbericht
50. Der Prüfbericht sollte folgende Angaben enthalten:
Prüfsubstanz:
|
— |
physikalische Beschaffenheit und gegebenenfalls physikalisch-chemische Eigenschaften, |
|
— |
Identifizierungsdaten einschließlich Quelle und |
|
— |
Reinheit der Zubereitung und bekannte und/oder erwartete Verunreinigungen. |
Vehikel (falls verwendet):
|
— |
Begründung für die Wahl des Vehikels (falls nicht Wasser oder physiologische Kochsalzlösung). |
Versuchstiere:
|
— |
Art und Stamm und Begründung für die Verwendung einer anderen Art als der Ratte, |
|
— |
Anbieter, von dem die Versuchstiere stammen, |
|
— |
Zahl, Alter bei Beginn und Geschlecht der Versuchstiere, |
|
— |
Herkunft, Haltungsbedingungen, Nahrung, Wasser usw. und |
|
— |
Gewicht der einzelnen Tiere bei Versuchsbeginn. |
Prüfbedingungen:
|
— |
Begründung der gewählten Dosisstufen, |
|
— |
Begründung des Verabreichungswegs und -zeitraums, |
|
— |
Angabe der verabreichten Dosen einschließlich Einzelheiten zu Vehikel, Volumen und physikalischer Form des verabreichten Stoffs, |
|
— |
Angaben zur Zubereitungsform der Prüfsubstanz/des Futters, der erreichten Konzentration, Stabilität und Homogenität der Zubereitung, |
|
— |
Methode zur eindeutigen Kennzeichnung der Muttertiere und der Nachkommen, |
|
— |
genaue Beschreibung der Randomisierungsmethode(n) für die Zuteilung der Muttertiere zu Behandlungsgruppen, für die Auswahl von Jungtieren zwecks Auslese und für die Zuteilung von Jungtieren zu Prüfgruppen, |
|
— |
Einzelheiten zur Verabreichung der Prüfsubstanz, |
|
— |
gegebenenfalls Angaben zur Umrechnung der Konzentration der Prüfsubstanz im Futter/Wasser bzw. im Inhalat (ppm) in die entsprechende Dosis (mg pro kg Körpergewicht und Tag), |
|
— |
Umgebungsbedingungen |
|
— |
Einzelheiten zu Futter- und Wasserqualität (z. B. Leitungswasser, destilliertes Wasser) und |
|
— |
Datum des Beginns und des Endes der Prüfung. |
Beobachtungen und Prüfverfahren:
|
— |
ausführliche Beschreibung der zur Standardisierung von Beobachtungen und Verfahren angewendeten Vorgehensweise sowie operative Festlegungen für die Auswertung der Beobachtungen, |
|
— |
Verzeichnis aller verwendeten Prüfverfahren und Begründung für deren Anwendung, |
|
— |
ausführliche Beschreibung der verhaltensbezogenen/funktionalen, pathologischen, neurochemischen oder elektrophysiologischen Verfahren einschließlich detaillierter Angaben zu automatischen Geräten, |
|
— |
Kalibrierungsverfahren und Verfahren zur Gewährleistung der Gleichwertigkeit der verwendeten Geräte und der ausgewogenen Zusammensetzung der Prüfgruppen während der Prüfverfahren und |
|
— |
kurze Begründung, warum einzelne Entscheidungen die Hinzuziehung eines Sachverständigen erfordern. |
Ergebnisse (Einzelergebnisse und Zusammenfassung, gegebenenfalls einschließlich Durchschnittswert und Varianz):
|
— |
Zahl der Tiere zu Beginn und am Ende der Prüfung, |
|
— |
Zahl der bei den einzelnen Prüfmethoden eingesetzten Tiere und Würfe, |
|
— |
Kennzeichnungsnummern der einzelnen Tiere und der Würfe, aus denen sie stammen, |
|
— |
Wurfgröße und mittleres Geburtsgewicht nach Geschlecht, |
|
— |
Körpergewicht und Daten zur Änderung des Körpergewichts einschließlich des terminalen Körpergewichts der Muttertiere und der Nachkommen |
|
— |
Nahrungsaufnahmedaten und gegebenenfalls Wasseraufnahmedaten (z. B. wenn die Prüfsubstanz mit dem Wasser verabreicht wird), |
|
— |
Daten für toxische Reaktion, aufgeschlüsselt nach Geschlecht und Dosisstufe, einschließlich Zeichen für Toxizität oder Mortalität, gegebenenfalls einschließlich Todeszeitpunkt und -ursache, |
|
— |
Art, Schweregrad, Dauer, Tag des Einsetzens, Tageszeit und weiterer Verlauf der ausführlichen klinischen Beobachtungen, |
|
— |
Bewertung der einzelnen Entwicklungsparameter (Gewicht, Geschlechtsreife und Verhaltensontogenese) zu jedem Beobachtungzeitpunkt, |
|
— |
ausführliche Beschreibung der nach Geschlecht aufgeschlüsselten verhaltensbezogenen, funktionalen, neuropathologischen, neurochemischen und elektrophysiologischen Befunde einschließlich Zunahmen und Abnahmen im Vergleich zu Kontrollgruppen, |
|
— |
Ergebnisse der Nekropsie, |
|
— |
Hirngewichte, |
|
— |
sämtliche Diagnosen aus neurologischen Anzeichen und Läsionen einschließlich natürlicherweise aufgetretener Krankheiten oder Zustände, |
|
— |
Abbildungen von Beispielbefunden, |
|
— |
niedrig aufgelöste Bilder zur Bewertung der Homologie der für die Morphometrie verwendeten Schnitte, |
|
— |
Resorptions- und Metabolismusdaten einschließlich ergänzender Daten aus einer separaten Toxikokinesestudie (falls vorhanden), |
|
— |
statistische Aufarbeitung der Ergebnisse einschließlich statistischer Modelle für die Daten- und Ergebnisanalyse, und zwar unabhängig davon, ob diese signifikant waren oder nicht, und |
|
— |
Verzeichnis der an der Prüfung beteiligten Personen einschließlich ihrer Berufsausbildung. |
Diskussion der Ergebnisse:
|
— |
Dosis-Wirkungs-Beziehungen, aufgeschlüsselt nach Geschlecht und Gruppe, |
|
— |
Einfließen etwaiger anderer toxischer Wirkungen in die Schlussfolgerung bezüglich des neurotoxischen Potenzials der Prüfsubstanz, aufgeschlüsselt nach Geschlecht und Gruppe, |
|
— |
Auswirkung etwaiger toxikokinetischer Informationen auf die Schlussfolgerung, |
|
— |
Abgleich mit anderen bekannten Neurotoxinen auf ähnliche Wirkungen, |
|
— |
Daten zur Untermauerung der Zuverlässigkeit und Empfindlichkeit der Prüfmethode (d. h. positive Daten und historische Kontrolldaten), |
|
— |
Zusammenhänge zwischen neuropathologischen und funktionalen Wirkungen (sofern diese vorliegen) und |
|
— |
NOAEL oder Benchmark-Dosis für Muttertiere und Nachkommen, aufgeschlüsselt nach Geschlecht und Gruppe. |
Schlussfolgerungen:
|
— |
Diskussion der Gesamtauswertung der Daten auf Grundlage der Ergebnisse, einschließlich der Bestimmung des NOAEL und einer Schlussfolgerung darüber, ob die Prüfsubstanz eine entwicklungsneurotoxische Wirkung hat oder nicht. |
LITERATUR
|
(1) |
OECD (1995). Draft Report of the OECD Ad Hoc Working Group on Reproduction and Developmental Toxicity. Copenhagen, Denmark, 13.-14. Juni 1995. |
|
(2) |
US EPA (1998). U.S. Environmental Protection Agency Health Effects Test Guidelines. OPPTS 870.6300. Developmental Neurotoxicity Study. US EPA 712-C-98-239. Abrufbar unter [http://www.epa.gov/opptsfrs/OPPTS_Harmonized/870_Health_Effects_Test_Guidelines/Series/]. |
|
(3) |
US EPA (1998). Guidelines for Neurotoxicity Risk Assessment. US EPA 630/R-95/001F. Abrufbar unter [http://cfpub.epa.gov/ncea/cfm/recordisplay.cfm?PrintVersion=True&deid=12479]. |
|
(4) |
Cory-Slechta, D.A., Crofton, K.M., Foran, J.A., Ross, J.F., Sheets, L.P., Weiss, B., Mileson, B. (2001). Methods to identify and characterize developmental neurotoxicity for human health risk assessment: I. Behavioral effects. Environ. Health Perspect., 109:79-91. |
|
(5) |
Dorman, D.C., Allen, S.L., Byczkowski, J.Z., Claudio, L., Fisher, J.E. Jr., Fisher, J.W., Harry, G.J., Li, A.A., Makris, S.L., Padilla, S., Sultatos, L.G., Mileson, B.E. (2001). Methods to identify and characterize developmental neurotoxicity for human health risk assessment: III. Pharmacokinetic and pharmacodynamic considerations. Environ. Health Perspect., 109:101-111. |
|
(6) |
Garman, R.H., Fix,A.S., Jortner, B.S., Jensen, K.F., Hardisty, J.F., Claudio, L., Ferenc, S. (2001). Methods to identify and characterize developmental neurotoxicity for human health risk assessment: II. Neuropathology. Environ. Health Perspect., 109:93-100. |
|
(7) |
OECD (2003). Report of the OECD Expert Consultation Meeting on Developmental Neurotoxicity Testing. Washington D.C., US, 23.-25. Oktober 2000. |
|
(8) |
OECD (2008). OECD Environment, Health and Safety Publications Series on Testing and Assessment No. 43. Guidance Document on Mammalian Reproductive Toxicity Testing and Assessment. Environment Directorate, OECD, Paris. July 2008. Abrufbar unter [http://search.oecd.org/officialdocuments/displaydocumentpdf/?cote=env/jm/mono(2008)16&doclanguage=en]. |
|
(9) |
OECD (2003). OECD Environment, Health and Safety Publications Series on Testing and Assessment No. 20. Guidance Document for Neurotoxicity Testing. Environment Directorate, OECD, Paris, September 2003. Abrufbar unter [http://www.oecd.org/document/22/0,2340,en_2649_34377_1916054_1_1_1_1,00.html]. |
|
(10) |
Kimmel, C.A., Rees, D.C., Francis, E.Z. (1990) Qualitative and quantitative comparability of human and animal developmental neurotoxicity. Neurotoxicol. Teratol., 12: 173-292. |
|
(11) |
Spencer, P.S., Schaumburg, H.H., Ludolph, A.C. (2000) Experimental and Clinical Neurotoxicology, 2nd Edition, ISBN 0195084772, Oxford University Press, New York. |
|
(12) |
Mendola, P., Selevan, S.G., Gutter, S., Rice, D. (2002) Environmental factors associated with a spectrum of neurodevelopmental deficits. Ment. Retard. Dev. Disabil. Res. Rev. 8:188-197. |
|
(13) |
Slikker, W.B., Chang, L.W. (1998) Handbook of Developmental Neurotoxicology, 1 st Edition, ISBN 0126488606, Academic Press, New York. |
|
(14) |
Kapitel B.34 dieses Anhangs, Prüfung auf Reproduktionstoxizität während einer Generation. |
|
(15) |
Kapitel B.35 dieses Anhangs, Zweigenerationenstudie zur Prüfung auf Reproduktionstoxizität. |
|
(16) |
Kapitel B.43 dieses Anhangs, Prüfung auf Neurotoxizität bei Nagetieren. |
|
(17) |
Kapitel B.31 diese Anhangs, Studie zur Prüfung auf pränatale Entwicklungstoxizität. |
|
(18) |
Richtlinie 2010/63/EU des Europäischen Parlaments und des Rates vom 22. September 2010 zum Schutz der für wissenschaftliche Zwecke verwendeten Tiere (ABl. L 276 vom 20.10.2010, S. 33). |
|
(19) |
WHO (1986) Principles and Methods for the Assessment of Neurotoxicity Associated with Exposure to Chemicals, (Environmental Health Criteria 60), Albany, New York: World Health Organization Publications Center, USA. Abrufbar unter [http://www.inchem.org/documents/ehc/ehc/ehc060.htm]. |
|
(20) |
WHO (2001) Neurotoxicity Risk Assessment for Human Health: Principles and Approaches, (Environmental Health Criteria 223), World Health Organization Publications, Geneva. Abrufbar unter [http://www.intox.org/databank/documents/supplem/supp/ehc223.htm]. |
|
(21) |
Chang, L.W., Slikker, W. (1995) Neurotoxicology: Approaches and Methods, 1st Edition, ISBN 012168055X, Academic Press, New York. |
|
(22) |
De Cabo, C., Viveros, M.P. (1997) Effects of neonatal naltrexone on neurological and somatic development in rats of both genders. Neurotoxicol. Teratol., 19:499-509. |
|
(23) |
Agnish, N.D., Keller, K.A. (1997) The rationale for culling of rodent litters. Fundam. Appl. Toxicol., 38:2-6. |
|
(24) |
Avery, D.L., Spyker, J.M. (1977) Foot tattoo of neonatal mice. Lab. Animal Sci., 27:110-112. |
|
(25) |
Wier, P.J., Guerriero, F.J., Walker, R.F. (1989) Implementation of a primary screen for developmental neurotoxicity. Fundam. Appl. Toxicol., 13:118-136. |
|
(26) |
Spear, N.E., Campbell, B.A. (1979) Ontogeny of Learning and Memory. ISBN 0470268492, Erlbaum Associates, New Jersey. |
|
(27) |
Krasnegor, N.A., Blass, E.M., Hofer, M.A., Smotherman, W. (1987) Perinatal Development: A Psychobiological Perspective. Academic Press, Orlando. |
|
(28) |
Zoetis, T., Walls, I. (2003) Principles and Practices for Direct Dosing of Pre-Weaning Mammals in Toxicity Testing and Research. ILSI Press, Washington, DC. |
|
(29) |
Moser, V., Walls, I., Zoetis, T. (2005) Direct dosing of preweaning rodents in toxicity testing and research: Deliberations of an ILSI RSI expert working group. Int. J. Toxicol., 24:87-94. |
|
(30) |
Conolly, R.B., Beck, B.D., Goodman, J.I. (1999) Stimulating research to improve the scientific basis of risk assessment. Toxicol. Sci., 49:1-4. |
|
(31) |
ICH (1993) ICH Harmonised Tripartite Guideline: Detection of Toxicity to Reproduction for Medical Products (S5A). International Conference on Harmonisation of Technical Requirements for Registration of Phamaceuticals for Human Use. |
|
(32) |
Lochry, E.A. (1987) Concurrent use of behavioral/functional testing in existing reproductive and developmental toxicity screens: Practical considerations. J. Am. Coll. Toxicol., 6:433-439. |
|
(33) |
Tachibana, T., Narita, H., Ogawa, T., Tanimura, T. (1998) Using postnatal age to determine test dates leads to misinterpretation when treatments alter gestation length, results from a collaborative behavioral teratology study in Japan. Neurotoxicol. Teratol., 20:449-457. |
|
(34) |
Gallavan, R.H. Jr., Holson, J.F., Stump, D.G., Knapp, J.F., Reynolds, V.L. (1999) Interpreting the toxicologic significance of alterations in anogenital distance: potential for confounding effects of progeny body weights. Reprod. Toxicol., 13:383-390. |
|
(35) |
Gray, L.E. Jr., Ostby, J., Furr, J., Price, M., Veeramachaneni, D.N., Parks, L. (2000) Perinatal exposure to the phthalates DEHP, BBP, and DINP, but not DEP, DMP, or DOTP, alters sexual differentiation of the male rat. Toxicol. Sci., 58:350-365. |
|
(36) |
Adams, J., Buelke-Sam, J., Kimmel, C.A., Nelson, C.J., Reiter, L.W., Sobotka, T.J., Tilson, H.A., Nelson, B.K. (1985) Collaborative behavioral teratology study: Protocol design and testing procedure. Neurobehav. Toxicol. Teratol., 7:579-586. |
|
(37) |
Korenbrot, C.C., Huhtaniemi, I.T., Weiner, R.W. (1977) Preputial separation as an external sign of pubertal development in the male rat. Biol. Reprod., 17:298-303. |
|
(38) |
Spear, L.P. (1990) Neurobehavioral assessment during the early postnatal period. Neurotoxicol. Teratol., 12:489-95. |
|
(39) |
Altman, J., Sudarshan, K. (1975) Postnatal development of locomotion in the laboratory rat. Anim. Behav., 23:896-920. |
|
(40) |
Adams, J. (1986) Methods in Behavioral Teratology. In: Handbook of Behavioral Teratology. Riley, E.P., Vorhees, C.V. (eds.) Plenum Press, New York, S. 67-100. |
|
(41) |
Reiter, L.W., MacPhail, R.C. (1979) Motor activity: A survey of methods with potential use in toxicity testing. Neurobehav. Toxicol., 1:53-66. |
|
(42) |
Robbins, T.W. (1977) A critique of the methods available for the measurement of spontaneous motor activity, Handbook of Psychopharmacology, Vol. 7, Iverson, L.L., Iverson, D.S., Snyder, S.H., (eds.) Plenum Press, New York, S. 37-82. |
|
(43) |
Crofton, K.M., Peele, D.B., Stanton, M.E. (1993) Developmental neurotoxicity following neonatal exposure to 3,3′-iminodipropionitrile in the rat. Neurotoxicol. Teratol., 15:117-129. |
|
(44) |
Ruppert, P.H., Dean, K.F., Reiter, L.W. (1985) Development of locomotor activity of rat pups in figure-eight mazes. Dev. Psychobiol., 18:247-260. |
|
(45) |
Crofton, K.M., Howard, J.L., Moser, V.C., Gill, M.W., Reiter, L.W., Tilson, H.A., MacPhail, R.C. (1991) Interlaboratory comparison of motor activity experiments: Implications for neurotoxicological assessments. Neurotoxicol. Teratol., 13:599-609. |
|
(46) |
Ross, J. F., Handley, D. E., Fix, A. S., Lawhorn, G. T., Carr, G. J. (1997) Quantification of the hind-limb extensor thrust response in rats. Neurotoxicol. Teratol., 19:1997. 405-411. |
|
(47) |
Handley, D.E., Ross, J.F., Carr, G.J. (1998) A force plate system for measuring low-magnitude reaction forces in small laboratory animals.Physiol. Behav., 64:661-669. |
|
(48) |
Edwards, P.M., Parker, V.H. (1977) A simple, sensitive, and objective method for early assessment of acrylamide neuropathy in rats. Toxicol. Appl. Pharmacol., 40:589-591. |
|
(49) |
Davis, M. (1984) The mammalian startle response. In: Neural Mechanisms of Startle Behavior, Eaton, R.C. (ed.), Plenum Press, New York, S. 287-351 |
|
(50) |
Koch, M. (1999) The neurobiology of startle. Prog. Neurobiol., 59:107-128. |
|
(51) |
Crofton, K.M. (1992) Reflex modification and the assessment of sensory dysfunction. In Target Organ Toxicology Series: Neurotoxicology, Tilson, H., Mitchell, C. (eds.). Raven Press, New York, S. 181-211. |
|
(52) |
Crofton, K.M., Sheets, L.P. (1989) Evaluation of sensory system function using reflex modification of the startle response. J. Am. Coll. Toxicol., 8:199-211. |
|
(53) |
Crofton, K.M, Lassiter, T.L, Rebert, C.S. (1994) Solvent-induced ototoxicity in rats: An atypical selective mid-frequency hearing deficit. Hear. Res.,80:25-30. |
|
(54) |
Ison, J.R. (1984) Reflex modification as an objective test for sensory processing following toxicant exposure. Neurobehav. Toxicol. Teratol., 6:437-445. |
|
(55) |
Mattsson, J.L., Boyes, W.K., Ross, J.F. (1992) Incorporating evoked potentials into neurotoxicity test schemes. In: Target Organ Toxicology Series: Neurotoxicity, Tilson, H., Mitchell, C., (eds.), Raven Press, New York, S. 125-145. |
|
(56) |
Peele, D.B., Allison, S.D., Crofton, K.M. (1990) Learning and memory deficits in rats following exposure to 3,3′-iminopropionitrile. Toxicol. Appl. Pharmacol., 105:321-332. |
|
(57) |
Bammer, G. (1982) Pharmacological investigations of neurotransmitter involvement in passive avoidance responding: A review and some new results. Neurosci. Behav. Rev., 6:247-296. |
|
(58) |
Bushnell, P.J. (1988) Effects of delay, intertrial interval, delay behavior and trimethyltin on spatial delayed response in rats. Neurotoxicol. Teratol., 10:237-244. |
|
(59) |
Green, R.J., Stanton, M.E. (1989) Differential ontogeny of working memory and reference memory in the rat. Behav. Neurosci., 103:98-105. |
|
(60) |
Kucharski, D., Spear, N.E. (1984) Conditioning of aversion to an odor paired with peripheral shock in the developing rat. Develop. Psychobiol., 17:465-479. |
|
(61) |
Morris, R. (1984) Developments of a water-maze procedure for studying spatial learning in the rat. J. Neurosci. Methods, 11:47-60. |
|
(62) |
Brandeis, R., Brandys, Y., Yehuda, S. (1989) The use of the Morris water maze in the study of memory and learning. Int. J. Neurosci., 48:29-69. |
|
(63) |
D'Hooge, R., De Deyn, P.P. (2001) Applications of the Morris water maze in the study of learning and memory. Brain Res. Rev, 36:60-90. |
|
(64) |
Vorhees, C.V. (1987) Maze learning in rats: A comparison of performance in two water mazes in progeny prenatally exposed to different doses of phenytoin. Neurotoxicol. Teratol., 9:235-241. |
|
(65) |
Vorhees, C.V. (1997) Methods for detecting long-term CNS dysfunction after prenatal exposure to neurotoxins. Drug Chem. Toxicol., 20:387-399. |
|
(66) |
Akaike, M., Tanaka, K., Goto, M., Sakaguchi, T. (1988) Impaired Biel and Radial arm maze learning in rats with methyl-nitrosurea induced microcephaly. Neurotoxicol. Teratol., 10:327-332. |
|
(67) |
Cory-Slechta, D.A., Weiss, B., Cox, C. (1983) Delayed behavioral toxicity of lead with increasing exposure concentration. Toxicol. Appl. Pharmacol., 71:342-352. |
|
(68) |
Campbell, B.A., Haroutunian, V. (1981) Effects of age on long-term memory: Retention of fixed interval responding. J. Gerontol., 36:338-341. |
|
(69) |
Fix, A.S, Garman, R.H. (2000) Practical aspects of neuropathology: A technical guide for working with the nervous system. Toxicol. Pathol., 28:122-131. |
|
(70) |
Prophet, E.B., Mills, B., Arrington, J.B., Sobin, L.H. (1994) Laboratory Methods in Histotechnology, American Registry of Pathology, Washington, DC, S. 84-107. |
|
(71) |
Bancroft, J.D., Gamble, M. (2002) Theory and Practice of Histological Techniques, 5th edition, Churchill Livingstone, London. |
|
(72) |
Fix, A.S., Ross, J.F., Stitzel, S.R., Switzer, R.C. (1996) Integrated evaluation of central nervous system lesions: stains for neurons, astrocytes, and microglia reveal the spatial and temporal features of MK-801-induced neuronal necrosis in the rat cerebral cortex. Toxicol. Pathol., 24: 291-304. |
|
(73) |
Schmued, L.C., Hopkins, K.J. (2000) Fluoro-Jade B: A high affinity tracer for the localization of neuronal degeneration. Brain Res., 874:123-130. |
|
(74) |
Krinke, G.J., Classen, W., Vidotto, N., Suter, E., Wurmlin, C.H. (2001) Detecting necrotic neurons with fluoro-jade stain. Exp. Toxic. Pathol., 53:365-372. |
|
(75) |
De Olmos, I.S., Beltramino, C.A., and de Olmos de Lorenzo, S. (1994) Use of an amino-cupric-silver technique for the detection of early and semiacute neuronal degeneration caused by neurotoxicants, hypoxia and physical trauma. Neurotoxicol. Teratol., 16, 545-561. |
|
(76) |
De Groot, D.M.G., Bos-Kuijpers, M.H.M., Kaufmann, W.S.H., Lammers, J.H.C.M., O'Callaghan, J.P., Pakkenberg, B., Pelgrim, M.T.M., Waalkens-Berendsen, I.D.H., Waanders, M.M., Gundersen, H.J. (2005a) Regulatory developmental neurotoxicity testing: A model study focusing on conventional neuropathology endpoints and other perspectives. Environ. Toxicol. Pharmacol., 19:745-755. |
|
(77) |
De Groot, D.M.G., Hartgring, S., van de Horst, L., Moerkens, M., Otto, M., Bos-Kuijpers, M.H.M., Kaufmann, W.S.H., Lammers, J.H.C.M., O'Callaghan, J.P., Waalkens-Berendsen, I.D.H., Pakkenberg, B., Gundersen, H.J. (2005b) 2D and 3D assessment of neuropathology in rat brain after prenatal exposure to methylazoxymethanol, a model for developmental neurotoxicity. Reprod. Toxicol., 20:417-432. |
|
(78) |
Rodier, P.M., Gramann, W.J. (1979) Morphologic effects of interference with cell proliferation in the early fetal period. Neurobehav. Toxicol., 1:129-135. |
|
(79) |
Howard, C.V., Reed, M.G. (1998) Unbiased Stereology: Three-Dimensional Measurement in Microscopy, Springer-Verlag, New York. |
|
(80) |
Hyman, B.T., Gomez-Isla, T., Irizarry, M.C. (1998) Stereology: A practical primer for neuropathology. J. Neuropathol. Exp. Neurol., 57:305-310. |
|
(81) |
Korbo, L., Andersen, B.B., Ladefoged, O., Møller, A. (1993) Total numbers of various cell types in rat cerebellar cortex estimated using an unbiased stereological method. Brain Res., 609:262-268. |
|
(82) |
Schmitz, C. (1997) Towards more readily comprehensible procedures in disector stereology. J. Neurocytol., 26:707-710. |
|
(83) |
West, M.J. (1999) Stereological methods for estimating the total number of neurons and synapses: Issues of precision and bias. Trends Neurosci., 22:51-61. |
|
(84) |
Schmitz, C., Hof, P.R. (2005) Design-based stereology in neuroscience. Neuroscience, 130:813-831. |
|
(85) |
Gavin, C.E., Kates, B., Gerken, L.A., Rodier, P.M. (1994) Patterns of growth deficiency in rats exposed in utero to undernutrition, ethanol, or the neuroteratogen methylazoxymethanol (MAM). Teratology, 49:113-121. |
|
(86) |
Ohno, M., Aotani, H., Shimada, M. (1995) Glial responses to hypoxic/ischemic encephalopathy in neonatal rat cerebrum. Develop. Brain Res., 84:294-298. |
|
(87) |
Jensen KF, Catalano SM. (1998) Brain morphogenesis and developmental neurotoxicology. In: Handbook of Developmental Neurotoxicology, Slikker, Jr. W., Chang, L.W. (eds.) Academic Press, New York, S. 3-41. |
|
(88) |
Ikonomidou, C., Bosch, F., Miksa, M., Bittigau, P., Vöckler, J., Dikranian, K., Tenkova, T.I., Stefovska, V., Turski, L., Olney, J.W. (1999) Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science, 283:70-74. |
|
(89) |
Ikonomidou, C., Bittigau, P., Ishimaru, M.J., Wozniak, D.F., Koch, C., Genz, K., Price, M.T., Sefovska, V., Hörster, F., Tenkova, T., Dikranian, K., Olney, J.W. (2000) Ethanol-induced apoptotic degeneration and fetal alcohol syndrome. Science, 287:1056-1060. |
|
(90) |
Friede, R. L. (1989) Developmental Neuropathology. Second edition. Springer-Verlag, Berlin. |
|
(91) |
House, D.E., Berman, E., Seeley, J.C., Simmons, J.E. (1992) Comparison of open and blind histopathologic evaluation of hepatic lesions. Toxicol. Let., 63:127-133. |
|
(92) |
Tilson, H.A., MacPhail, R.C., Crofton, K.M. (1996) Setting exposure standards: a decision process. Environ. Health Perspect., 104:401-405. |
|
(93) |
US EPA (2005) Guidelines for Carcinogen Risk Assessment. US EPA NCEA-F-0644A. |
|
(94) |
US EPA (1996) Guidelines for Reproductive Toxicity Risk Assessment, Federal Register 61(212): 56274-56322. |
|
(95) |
Danish Environmental Protection Agency (1995) Neurotoxicology. Review of Definitions, Methodology, and Criteria. Miljøprojekt nr. 282. Ladefoged, O., Lam, H.R., Østergaard, G., Nielsen, E., Arlien-Søborg, P. |
|
(96) |
Muller, K.E., Barton, C.N., Benignus, V.A. (1984). Recommendations for appropriate statistical practice in toxicologic experiments. Neurotoxicology, 5:113-126. |
|
(97) |
Gad, S.C. (1989) Principles of screening in toxicology with special emphasis on applications to Neurotoxicology. J. Am. Coll. Toxicol., 8:21-27. |
|
(98) |
Abby, H., Howard, E. (1973) Statistical procedures in developmental studies on a species with multiple offspring. Dev. Psychobiol., 6:329-335. |
|
(99) |
Haseman, J.K., Hogan, M.D. (1975) Selection of the experimental unit in teratology studies. Teratology, 12:165-172. |
|
(100) |
Holson, R.R., Pearce, B. (1992) Principles and pitfalls in the analysis of prenatal treatment effects in multiparous species. Neurotoxicol. Teratol., 14:221-228. |
|
(101) |
Nelson, C.J., Felton, R.P., Kimmel, C.A., Buelke-Sam, J., Adams, J. (1985) Collaborative Behavioral Teratology Study: Statistical approach. Neurobehav. Toxicol. Teratol., 7:587-90. |
|
(102) |
Crofton, K.M., Makris, S.L., Sette, W.F., Mendez, E., Raffaele, K.C. (2004) A qualitative retrospective analysis of positive control data in developmental neurotoxicity studies. Neurotoxicol. Teratol., 26:345-352. |
|
(103) |
Bolon, B., Garman, R., Jensen, K., Krinke, G., Stuart, B., and an ad hoc working group of the STP Scientific and Regulatory Policy Committee. (2006) A ‚best practices‘ approach to neuropathological assessment in developmental neurotoxicity testing — for today. Toxicol. Pathol. 34:296-313. |
|
(104) |
Tamura, R.N., Buelke-Sam, J. (1992) The use of repeated measures analysis in developmental toxicology studies. Neurotoxicol. Teratol., 14(3):205-210. |
|
(105) |
Tukey, J.W., Ciminera, J.L., Heyse, J.F. (1985) Testing the statistical certainty of a response to increasing doses of a drug. Biometrics, 41:295-301. |
|
(106) |
Crofton, K.M., Foss, J.A., Haas, U., Jensen, K., Levin, E.D., and Parker, S.P. (2008) Undertaking positive control studies as part of developmental neurotoxicity testing: report from the ILSI Research Foundation/Risk Science Institute expert working group on neurodevelopmental endpoints. Neurotoxicology and Teratology, 30(4):266-287. |
|
(107) |
Raffaele, K.C., Fisher, E., Hancock, S., Hazelden, K., and Sobrian, S.K. (2008) Determining normal variability in a developmental neurotoxicity test: report from the ILSI Research Foundation/Risk Science Institute expert working group on neurodevelopmental endpoints. Neurotoxicology and Teratology, 30(4):288-325. |
|
(108) |
Holson, R.R., Freshwater, L., Maurissen, J.P.J., Moser, V.C., and Phang, W. (2008) Statistical issues and techniques appropriate for developmental neurotoxicity testing: a report from the ILSI Research Foundation/Risk Science Institute expert working group on neurodevelopmental endpoints. Neurotoxicology and Teratology, 30(4):326-348. |
|
(109) |
Tyl, R.W., Crofton, K.M., Moretto, A., Moser, V.C., Sheets, L.P., and Sobotka, T.J. (2008) Identification and interpretation of developmental neurotoxicity effects: a report from the ILSI Research Foundation/Risk Science Institute expert working group on neurodevelopmental endpoints Neurotoxicology and Teratology, 30(4):349-381. |
Abbildung 1
Allgemeines Prüfschema für funktionale Prüfungen/Verhaltensprüfungen, neuropathologische Beurteilung und Hirngewichte. Dieses Diagramm basiert auf der Beschreibung unter den Nummern 13, 14 und 15 (PND = postnataler Tag). Beispiele für die Zuteilung der Versuchstiere sind Anlage 1 zu entnehmen.
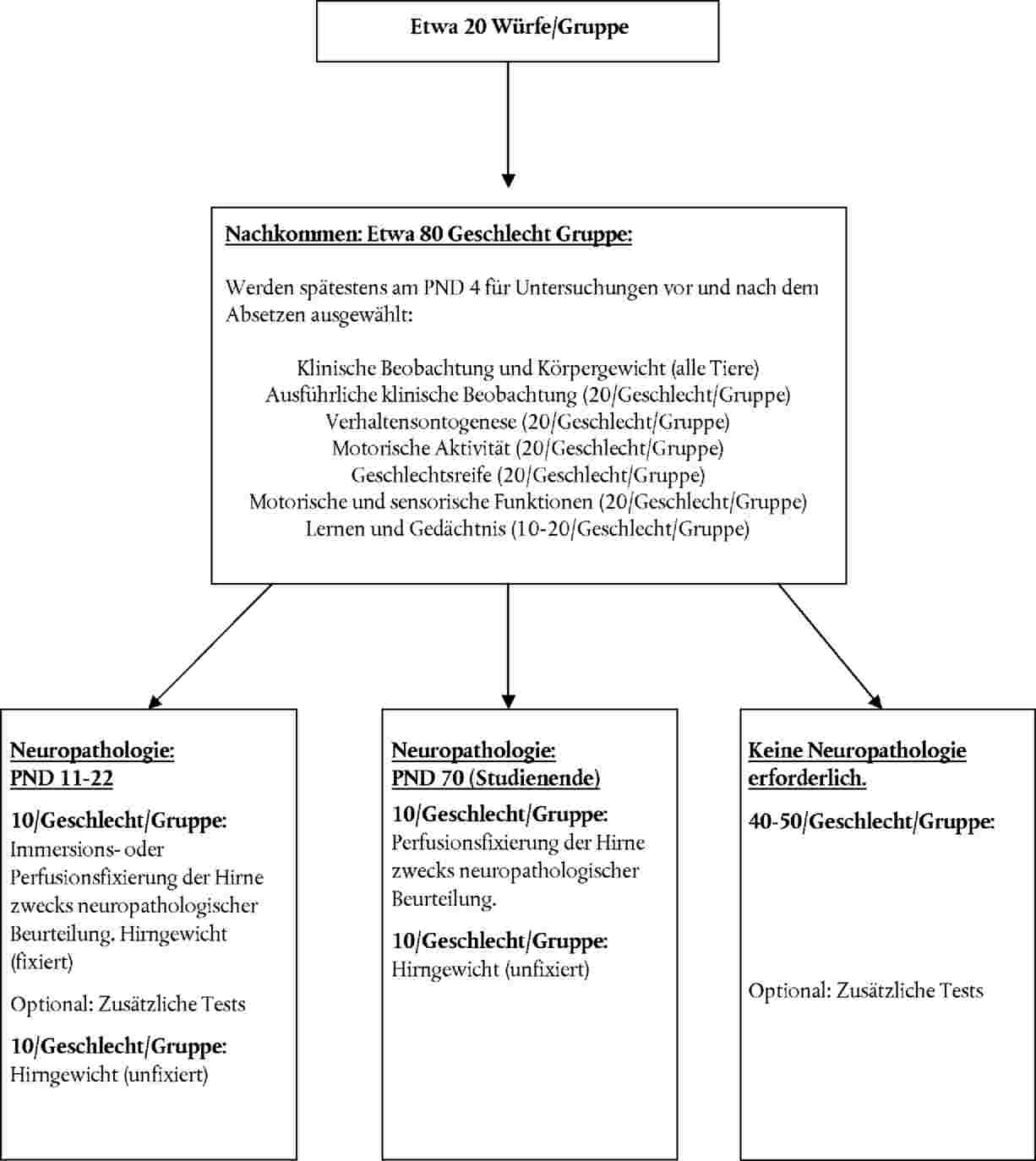
Anlage 1
|
1. |
Beispiele für mögliche Zuteilungen sind in nachstehender Tabelle beschrieben. Diese Beispiele sollen veranschaulichen, dass die Versuchstiere den verschiedenen Prüfparadigmen auf unterschiedliche Art und Weise zugeteilt werden können. |
Beispiel 1
|
2. |
Bei einem Satz von 20 Jungtieren pro Geschlecht und Dosisstufe (ein Männchen und ein Weibchen pro Wurf) wird die Verhaltensontogenese vor dem Absetzen geprüft. Von diesen Tieren werden 10 Jungtiere pro Geschlecht und Dosisstufe (ein Männchen oder ein Weibchen pro Wurf) am PND 22 auf humane Weise getötet. Die Gehirne werden entfernt, gewogen und für die histopathologische Beurteilung aufbereitet. Zusätzlich werden die am unfixierten Hirn erhobenen Hirngewichtsdaten der verbleibenden 10 Männchen und 10 Weibchen pro Dosisstufe gesammelt. |
|
3. |
Bei einem weiteren Satz von 20 Jungtieren pro Geschlecht und Dosisstufe (ein Männchen und ein Weibchen pro Wurf) werden funktionale und Verhaltenstests nach dem Absetzen durchgeführt (ausführliche klinische Beobachtungen und Prüfung der motorischen Aktivität, der akustischen Schreckreaktion und der kognitiven Funktionen am adoleszenten Tier), und das Alter der Geschlechtsreife wird bewertet. Von diesen Tieren werden 10 Jungtiere pro Geschlecht und Dosisstufe (ein Männchen oder ein Weibchen pro Wurf) bei Studienende betäubt und per Perfusion fixiert (etwa PND 70). Die Gehirne werden, nachdem sie zusätzlich in situ fixiert wurden, entfernt und für die neuropathologische Beurteilung aufbereitet. |
|
4. |
Für die kognitiven Funktionsprüfungen an jungen adulten Tieren (z. B. PND 60-70) wird ein dritter Satz von 20 Jungtieren pro Geschlecht und Dosisstufe verwendet (ein Männchen und ein Weibchen pro Wurf). Von diesen Tieren werden 10 Jungtiere pro Geschlecht und Gruppe (ein Männchen oder ein Weibchen pro Wurf) bei Studienende getötet und die Gehirne entfernt und gewogen. |
|
5. |
Die verbleibenden 20 Tiere pro Geschlecht und Gruppe werden für möglicherweise durchzuführende zusätzliche Prüfungen zurückbehalten. Tabelle 1
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Beispiel 2
|
6. |
Bei einem Satz von 20 Jungtieren pro Geschlecht und Dosisstufe (ein Männchen und ein Weibchen pro Wurf) wird die Verhaltensontogenese vor dem Absetzen geprüft. Von diesen Tieren werden 10 Jungtiere pro Geschlecht und Dosisstufe (ein Männchen oder ein Weibchen pro Wurf) am PND 11 auf humane Weise getötet. Die Gehirne werden entfernt, gewogen und für die histopathologische Beurteilung aufbereitet. |
|
7. |
Bei einem weiteren Satz von 20 Jungtieren pro Geschlecht und Dosisstufe (ein Männchen und ein Weibchen pro Wurf) werden Untersuchungen nach dem Absetzen durchgeführt (ausführliche klinische Beobachtungen, Prüfung der motorischen Aktivität, Bewertung des Alters der Geschlechtsreife und Prüfung der motorischen und sensorischen Funktionen). Von diesen Tieren werden 10 Jungtiere pro Geschlecht und Dosisstufe (ein Männchen oder ein Weibchen pro Wurf) bei Studienende betäubt und per Perfusion fixiert (etwa PND 70). Die Gehirne werden, nachdem sie zusätzlich in situ fixiert wurden, entfernt, gewogen und für die neuropathologische Beurteilung aufbereitet. |
|
8. |
Für kognitive Funktionsprüfungen an adoleszenten und jungen adulten Tieren werden 10 Jungtiere pro Geschlecht und Dosisstufe verwendet (ein Männchen oder ein Weibchen pro Wurf). Bei den kognitiven Funktionsprüfungen werden für die Prüfungen am PND 23 und an jungen adulten Tieren unterschiedliche Tiere verwendet. Nach Beendigung werden die für die Prüfungen am adulten Tier verwendeten 10 Tiere pro Geschlecht und Gruppe getötet. Ihre Gehirne werden entfernt und gewogen. |
|
9. |
Die verbleibenden, nicht für die Prüfungen ausgewählten 20 Tiere pro Geschlecht und Gruppe werden beim Absetzen getötet und entsorgt. Tabelle 2
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Beispiel 3
|
10. |
Bei einem Satz von 20 Jungtieren pro Geschlecht und Dosisstufe (ein Männchen und ein Weibchen pro Wurf) wird am PND 11 das Gehirngewicht bestimmt und eine neuropathologische Bewertung durchgeführt. Von diesen Tieren werden 10 Jungtiere pro Geschlecht und Dosisstufe (ein Männchen oder ein Weibchen pro Wurf) am PND 11 auf humane Weise getötet, und die Gehirne werden entfernt, gewogen und für die histopathologische Beurteilung aufbereitet. Zusätzlich werden die am unfixierten Hirn erhobenen Hirngewichtsdaten der verbleibenden 10 Männchen und 10 Weibchen pro Dosisstufe gesammelt. |
|
11. |
Bei einem weiteren Satz von 20 Jungtieren pro Geschlecht und Dosisstufe (ein Männchen und ein Weibchen pro Wurf) wird die Verhaltensontogenese (motorische Aktivität) beurteilt, es werden Untersuchungen nach dem Absetzen durchgeführt (motorische Aktivität und Bewertung des Alters der Geschlechtsreife), und an den adoleszenten Tieren werden kognitive Funktionsprüfungen durchgeführt. |
|
12. |
Bei einem weiteren Satz von 20 Tieren pro Geschlecht und Dosisstufe (ein Männchen und ein Weibchen pro Wurf) werden die motorischen und sensorischen Funktionen (akustische Schreckreaktion) geprüft und ausführliche klinische Beobachtungen vorgenommen. Von diesen Tieren werden 10 Jungtiere pro Geschlecht und Dosisstufe (ein Männchen oder ein Weibchen pro Wurf) bei Studienende betäubt und per Perfusion fixiert (etwa PND 70). Die Gehirne werden, nachdem sie zusätzlich in situ fixiert wurden, entfernt, gewogen und für die neuropathologische Beurteilung aufbereitet. |
|
13. |
Bei einem weiteren Satz von 20 Jungtieren pro Geschlecht und Dosisstufe (ein Männchen und ein Weibchen pro Wurf) werden kognitive Funktionsprüfungen an den jungen adulten Tieren durchgeführt. Von diesen Tieren werden 10 Jungtiere pro Geschlecht und Gruppe (ein Männchen oder ein Weibchen pro Wurf) bei Studienende getötet und die Gehirne entfernt und gewogen. Tabelle 3
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Anlage 2
Begriffsbestimmungen
Chemikalie: ein Stoff oder eine Mischung.
Prüfsubstanz: jede(r) mittels dieser Prüfmethode getestete Stoff bzw. Mischung
B.54. UTEROTROPHER BIOASSAY MIT NAGERN: EIN KURZZEIT-SCREENING-TEST AUF ÖSTROGENE EIGENSCHAFTEN
EINLEITUNG
1. Diese Prüfmethode entspricht der OECD-Prüfrichtlinie (TG) 440 (2007). Die OECD setzte sich 1998 zur Priorität, bestehende Prüfrichtlinien für Screening und Testung potenzieller endokriner Disruptoren zu überarbeiten und neue Richtlinien zu entwickeln (1). Ein Aspekt dieser Maßnahme war die Ausarbeitung einer Prüfrichtlinie für die In-vivo-Uterusgewichtsprüfung (Uterotrophic Bioassay) mit Nagern. Der uterotrophe Bioassay mit Nagern wurde nachfolgend einem umfassenden Validierungsprogramm unterzogen, in dessen Rahmen unter anderem ein ausführliches Hintergrunddokument erstellt (2)(3) und umfassende Intra- und Interlaborstudien durchgeführt wurden, um die Relevanz und Reproduzierbarkeit der In-vivo-Prüfung anhand eines wirksamen Referenzöstrogens, schwacher Östrogen-Rezeptoragonisten, eines starken Östrogen-Rezeptorantagonisten und einer negativen Referenzchemikalie (4)(5)(6)(7)(8)(9) nachzuweisen. Die vorliegende Prüfmethode B.54 ist das Resultat der bei der Validierung des Prüfprogramms gewonnenen Erfahrungen und Ergebnisse im Bereich Östrogenagonisten.
2. Der uterotrophe Bioassay ist ein Kurzzeit-Screening-Test, der auf die 1930er-Jahre zurückgeht (27)(28) und durch einen Fachausschuss erstmals 1962 für Screeningzwecke standardisiert wurde (32)(35). Der Test basiert auf einer Zunahme des Uterusgewichts, die auch als uterotrophe Reaktion bezeichnet wird (vgl. Literaturhinweis 29). Dabei wird untersucht, ob eine Chemikalie biologische Abläufe auslöst, die der Wirkung von Agonisten oder Antagonisten natürlicher Östrogene (z. B. 17β-Östradiol) entsprechen; die Methode wird allerdings wesentlich häufiger für den Nachweis von Agonisten als für den Nachweis von Antagonisten eingesetzt. Der Uterus reagiert auf zweierlei Art auf Östrogen: Zunächst erhöht sich das Gewicht aufgrund von Wassereinlagerung. Darauf folgt eine Gewichtszunahme aufgrund von Gewebewachstum (30). Die Uterusreaktionen sind bei Ratten und Mäusen qualitativ vergleichbar.
3. Dieser Bioassay wird als In-vivo-Screening-Test durchgeführt und sollte im Zusammenhang mit dem Rahmenkonzept der OECD ‚Conceptual Framework for the Testing and Assessment of Endocrine Disrupting Chemicals‘ (Testung und Bewertung endokrin wirksamer Stoffe) (Anlage 2) angewendet werden. In diesem Rahmenkonzept ist der uterotrophe Bioassay ein In-vivo-Test der Stufe 3, mit dem Daten über einen einzelnen endokrinen Mechanismus, nämlich die östrogene Wirkung, gewonnen werden sollen.
4. Der uterotrophe Bioassay ist als Teil einer Reihe von In-vitro- und In-vivo-Tests zur Bestimmung von Chemikalien, die mit dem endokrinen System in Wechselwirkung treten können, konzipiert und soll letztendlich der Bewertung des Risikos für die Umwelt und die menschliche Gesundheit dienen. Im Rahmen des Validierungsprogramms der OECD wurde sowohl anhand von starken als auch von schwachen Östrogenagonisten untersucht, wie gut sich die Prüfung zur Erkennung von Chemikalien mit östrogener Wirkung eignet (4)(5)(6)(7)(8). Dabei konnte die Empfindlichkeit des Prüfverfahrens bezüglich Östrogenagonisten ebenso fundiert nachgewiesen werden wie eine gute Intra- und Interlabor-Reproduzierbarkeit.
5. Was negative Verbindungen anbelangt, wurde nur eine ‚negative‘ Referenzchemikalie (deren Wirkungslosigkeit bereits im Rahmen von Uterusgewichtsprüfungen sowie bei in vitro durchgeführten Rezeptorbindungs- und Rezeptorprüfungen belegt worden war) in das Validierungsprogramm aufgenommen, aber es wurden zusätzliche, vom Validierungsprogramm der OECD unabhängige Prüfdaten ausgewertet, die die Spezifizität des uterotrophen Bioassays beim Screening von Östrogenagonisten weiter untermauern (16).
AUSGANGSÜBERLEGUNGEN UND BEGRENZUNGEN
6. Östrogenagonisten und -antagonisten können an die Östrogenrezeptoren a und b binden und die transkriptionelle Aktivität der Rezeptoren aktivieren bzw. hemmen. Dies ist potenziell mit gesundheitlichen Risiken verbunden und schließt auch negative Auswirkungen auf Reproduktion und Entwicklung mit ein. Aus diesem Grund müssen Chemikalien möglichst schnell daraufhin geprüft und beurteilt werden können, ob es sich um potenzielle Östrogenagonisten oder -antagonisten handelt. Die in vitro bestimmte Affinität eines Liganden für einen Östrogenrezeptor oder die transkriptionelle Aktivierung von Reportergenen liefert zwar wertvolle Informationen, ist aber nur eine von mehreren Determinanten für das Gefahrenpotenzial. Andere Determinanten können die metabolische Aktivierung und Deaktivierung beim Eintritt in den Körper, die Verteilung auf die Zielgewebe und die Ausscheidung aus dem Körper sein, was zumindest teilweise vom Verabreichungsweg und von der geprüften Chemikalie abhängig ist. Daraus ergibt sich die Notwendigkeit, die potenzielle Wirkung einer Chemikalie in vivo unter einschlägigen Bedingungen zu prüfen, sofern die Eigenschaften der Chemikalie hinsichtlich Resorption, Verteilung, Metabolismus und Ausscheidung (ADME) nicht bereits entsprechende Informationen liefern. Die Uterusgewebe reagieren mit starkem und raschem Wachstum auf die Stimulation mit Östrogenen, was insbesondere für Labornager gilt, deren Östruszyklus etwa vier Tage dauert. Die Verwendung von Nagerarten, insbesondere Ratten, ist zudem bei Toxizitätsstudien zur Beurteilung des Gefahrenpotenzials weit verbreitet. Aus diesem Grund ist der Nageruterus das geeignete Zielorgan für das In-vivo-Screening von Östrogenagonisten und -antagonisten.
7. Der vorliegenden Prüfmethode liegen die Prüfpläne der OECD-Validierungsstudie zugrunde, die sich in Intra- und Interlaborstudien als zuverlässig und wiederholbar erwiesen haben (5)(7). Derzeit sind zwei Methoden verfügbar, und zwar die Methode am adulten ovarektomierten Weibchen (Methode mit adulten OVX-Tieren) und die Methode am unreifen, nicht ovarektomierten Weibchen (Methode mit unreifen Tieren). Das Validierungsprogramm der OECD hat ergeben, dass die Empfindlichkeit und Reproduzierbarkeit beider Methoden vergleichbar ist. Die Methode mit unreifen Ratten, deren Hypothalamus-Hypophysen-Gonaden-Achse (HPG-Achse) intakt ist, ist in gewissen Punkten weniger spezifisch, deckt dafür aber eine größere Bandbreite an Untersuchungsmöglichkeiten ab als die am ovarektomierten Tier, weil sie für Chemikalien empfindlich ist, die auch mit der HPG-Achse interagieren und nicht nur mit dem Östrogenrezeptor. Die HGP-Achse ist etwa ab dem 15. Lebenstag funktionsfähig. Vorher kann die Pubertät nicht — zum Beispiel durch Behandlung mit GnRH — beschleunigt werden. Beim Eintritt in die Pubertät haben die Weibchen vor der Vaginalöffnung mehrere stumme Zyklen, die nicht zu einer Vaginalöffnung oder Ovulation führen, bei denen es jedoch zu einigen hormonellen Schwankungen kommt. Wenn die HPG-Achse direkt oder indirekt durch eine Chemikalie stimuliert wird, hat das eine frühzeitige Pubertät und Ovulation sowie eine beschleunigte Vaginalöffnung zur Folge. Dies wird nicht nur durch Chemikalien hervorgerufen, die auf die HPG-Achse wirken; auch bestimmte Nahrung mit einem höheren Niveau metabolisierbarer Energie stimuliert das Wachstum und beschleunigt die Vaginalöffnung, ohne dass eine östrogene Wirkung vorliegt. Solche Chemikalien würden bei adulten OVX-Tieren keine uterotrophe Reaktion hervorrufen, weil deren HPG-Achse nicht funktionsfähig ist.
8. Aus Tierschutzgründen sollte der Methode, bei der unreife Ratten verwendet werden, der Vorzug eingeräumt werden, weil dadurch eine chirurgische Vorbehandlung der Tiere entfällt und eine mögliche Nichtverwendung derjenigen Tiere vermieden wird, die Anzeichen für einen beginnenden Östruszyklus aufweisen (siehe Nummer 30).
9. Eine Zunahme des Uterusgewichts ist nicht ausschließlich östrogenen Ursprungs, d. h. auch Chemikalien, die keine Östrogenagonisten oder -antagonisten sind, können eine uterotrophe Reaktion hervorrufen. Beispielsweise können auch relativ hohe Dosen von Progesteron, Testosteron oder verschiedenen synthetischen Progestinen ebenfalls stimulierend wirken (30). Jede Reaktion kann histologisch auf Keratinisation und Kornifikation der Vagina analysiert werden (30). Ein positives Ergebnis eines uterotrophen Bioassays sollte unabhängig vom möglichen Ursprung der Reaktion weitere Abklärungsmaßnahmen nach sich ziehen. Weitere Nachweise für die östrogene Wirkung könnten durch In-vitro-Prüfungen, wie zum Beispiel ER-Bindungstests und Tests auf transkriptionelle Aktivierung oder andere In-vivo-Prüfungen, wie zum Beispiel die Prüfung an pubertären Weibchen, gewonnen werden.
10. Unter Berücksichtigung der Tatsache, dass der uterotrophe Bioassay ein In-vivo-Screening-Test ist, wurde der gewählte Validierungsansatz sowohl Tierschutzbelangen als auch einer mehrstufigen Prüfstrategie gerecht. Zu diesem Zweck lag der Schwerpunkt darauf, die beiden Hauptaspekte zu validieren, auf die es bei zahlreichen Chemikalien ankommt, nämlich die Reproduzierbarkeit und die Empfindlichkeit für die östrogene Wirkung, während der Antiöstrogenwirkungskomponente der Prüfung dagegen nur wenig Aufmerksamkeit gewidmet wurde. Es wurde nur ein Antiöstrogen mit starker Wirkung getestet, da die Anzahl an Chemikalien mit eindeutig antiöstrogenem Profil (bei denen die antiöstrogene Wirkung nicht durch eine teilweise vorhandene östrogene Wirkung verschleiert wird) ausgesprochen begrenzt ist. Der vorliegenden Prüfmethode liegt folglich der Prüfplan für den Nachweis der östrogenen Wirkung zugrunde, während der Prüfplan für die Prüfung auf Antagonisten in einem Guidance Document der OECD (37) enthalten ist. Die Reproduzierbarkeit und Empfindlichkeit der Prüfung von Chemikalien mit rein antiöstrogener Wirkung werden zu einem späteren Zeitpunkt klarer definiert, wenn das Prüfverfahren eine Zeit lang routinemäßig im Einsatz war und mehr Chemikalien mit dieser Wirkungsart ermittelt worden sind.
11. Es wird darauf hingewiesen, dass bei allen tierexperimentellen Verfahren die örtlichen Standards der Versuchstierpflege einzuhalten sind. Die nachfolgend ausgeführten Pflege- und Behandlungsbeschreibungen stellen Mindestanforderungen dar; Vorrang vor diesen Standards haben lokale Bestimmungen, zum Beispiel die Richtlinie 2010/63/EU des Europäischen Parlaments und des Rates vom 22. September 2010 zum Schutz der für wissenschaftliche Zwecke verwendeten Tiere (38). Eine weiterführende Anleitung für die tierschutzgerechte Behandlung von Versuchstieren ist in dem OECD Guidance Document (25) enthalten.
12. Wie bei allen Prüfungen, bei denen lebende Tiere verwendet werden, ist vor Testbeginn sicherzustellen, dass die Daten wirklich benötigt werden. Zum Beispiel werden die Daten möglicherweise zur weiteren Abklärung der folgenden beiden Szenarien gebraucht:
|
— |
wenn ein hohes Expositionspotenzial (Stufe 1 des Rahmenkonzepts, Anlage 2) oder Anzeichen für eine östrogene Wirkung (Stufe 2) vorliegen, um zu untersuchen, ob solche Wirkungen in vivo vorkommen können; |
|
— |
wenn die Ergebnisse der In-vivo-Tests der Stufen 4 und 5 auf eine östrogene Wirkung hindeuten, um zu untermauern, dass diese Ergebnisse mit einem Östrogenmechanismus zusammenhängen, der nicht mit einem In-vitro-Test erklärt werden kann. |
13. Die für die Zwecke dieser Prüfmethode verwendeten Begriffe sind in Anlage 1 definiert.
PRINZIP DER PRÜFMETHODE
14. Die Empfindlichkeit des uterotrophen Bioassays beruht auf einer Versuchsanordnung, bei der die Hypothalamus-Hypophysen-Eierstock-Achse nicht funktionsfähig ist, was zu einem niedrigen endogenen Östrogenspiegel führt. Dadurch werden ein niedriges Ausgangsgewicht des Uterus zu Anfang der Prüfung und das größtmögliche Spektrum an Reaktionen auf die verabreichten Östrogene sichergestellt. Diese Anforderung ist bei weiblichen Nagern in zwei Fällen erfüllt:
|
i) |
bei unreifen Weibchen nach dem Absetzen und vor Einsetzen der Pubertät und |
|
ii) |
bei jungen, adulten, ovarektomierten Weibchen mit ausreichend Zeit für die Rückbildung des Uterusgewebes. |
15. Die Prüfsubstanz wird täglich oral per Schlundsonde oder durch subkutane Injektion verabreicht. Die abgestuften Dosen der Prüfsubstanz werden mindestens zwei Versuchsgruppen (vgl. Nummer 33 für weitere Informationen) verabreicht, wobei pro Gruppe eine Dosisstufe gegeben wird und der Verabreichungszeitraum bei der Methode mit unreifen Tieren drei aufeinanderfolgende Tage und bei der Methode mit unreifen OVX-Tieren mindestens drei aufeinanderfolgende Tage beträgt. Die Tiere werden etwa 24 Stunden nach der letzten Dosisgabe seziert. Bei Östrogenagonisten wird das mittlere Uterusgewicht der behandelten Versuchstiergruppen im Vergleich zur Vehikelgruppe auf eine statistisch signifikante Erhöhung hin bewertet. Eine statistisch signifikante Erhöhung des mittleren Uterusgewichts einer Prüfgruppe deutet auf eine positive Reaktion in diesem Bioassay hin.
BESCHREIBUNG DER METHODE
Auswahl der Versuchstierarten
16. Es können die üblichen Labornagerstämme verwendet werden. Bei der Validierung wurden beispielsweise Stämme der Sprague-Dawley- und der Wistar-Ratte eingesetzt. Es sollten keine Stämme verwendet werden, von denen ein weniger gutes Ansprechen der Uteri bekannt ist oder vermutet wird. Das Labor sollte die Empfindlichkeit des verwendeten Stamms wie unter den Nummern 26 und 27 beschrieben nachweisen.
17. Ratten und Mäuse werden seit den 1930er-Jahren routinemäßig für den uterotrophen Bioassay verwendet. Die Validierungsprüfungen der OECD wurden nur anhand von Ratten durchgeführt, und zwar aufgrund der Überlegung, dass bei zwei aller Voraussicht nach gleichwertigen Spezies eine Art für eine weltweit gültige Validierung ausreichen sollte und so Ressourcen und Tiere eingespart werden können. Die Ratte ist bei den meisten Prüfungen auf Reproduktions- und Entwicklungstoxizität die Spezies der Wahl. Da es bereits eine umfangreiche Sammlung historischer Daten zu Mäusen gibt, wurde eine Follow-up-Validierungsstudie mit einer begrenzten Anzahl an Mäusen durchgeführt (16), um den Anwendungsbereich der Prüfmethode des uterotrophen Bioassays mit Nagern auf die Verwendung von Mäusen als Prüfspezies auszudehnen. Getreu der ursprünglichen Absicht, Ressourcen und Tiere einzusparen, wurde der Ansatz einer Überbrückungsstudie mit einer begrenzten Anzahl an Prüfsubstanzen und teilnehmenden Laboratorien und ohne codierte Probentestung gewählt. Diese Überbrückungsstudie ergab für den uterotrophen Bioassay mit jungen adulten, ovarektomierten Mäusen, dass die an Ratten und Mäusen gewonnenen Daten sich sowohl qualitativ als auch quantitativ gut decken. Wenn das Ergebnis des uterotrophen Bioassays gegebenenfalls als Vorstudie für eine Langzeitstudie verwendet wird, können somit Tiere desselben Stamms und derselben Herkunft für beide Studienarten verwendet werden. Der Überbrückungsansatz war auf OVX-Mäuse beschränkt, und der Bericht enthält keine robusten Daten, die eine Validierung des Modells mit unreifen Tieren erlauben würden. Daher fällt das Tiermodell mit unreifen Mäusen nicht in den Anwendungsbereich der aktuellen Prüfmethode.
18. Demnach können in bestimmten Fällen Mäuse anstelle von Ratten verwendet werden. Die Wahl dieser Versuchstierart ist auf der Grundlage toxikologischer, pharmakokinetischer und/oder anderer Kriterien zu begründen. Bei Mäusen muss gegebenenfalls der Prüfplan geändert werden. Zum Beispiel ist die Nahrungsaufnahme im Verhältnis zum Körpergewicht bei Mäusen höher als bei Ratten, und der Phytoöstrogengehalt der Nahrung sollte daher bei Mäusen niedriger sein als bei Ratten (9)(20)(22).
Haltungs- und Fütterungsbedingungen
19. Bei allen Verfahren sind die örtlichen Standards der Versuchstierpflege einzuhalten. Bei diesen Pflege- und Behandlungsbeschreibungen handelt es sich um Mindestanforderungen; Vorrang haben lokale Rechtsvorschriften, wie zum Beispiel die Richtlinie 2010/63/EU des Europäischen Parlaments und des Rates vom 22. September 2010 zum Schutz der für wissenschaftliche Zwecke verwendeten Tiere (38). Die Temperatur im Versuchstierraum sollte 22 °C (± 3 °C) betragen. Die relative Luftfeuchtigkeit sollte mindestens 30 % betragen und — außer beim Reinigen des Raums — 70 % nicht überschreiten. Angestrebt werden sollte eine Luftfeuchtigkeit von 50-60 %. Die Beleuchtung sollte künstlich sein. Die Hell- und Dunkelphasen sollten sich im Abstand von 12 Stunden abwechseln.
20. Labornahrung und Trinkwasser sind ad libitum bereitzustellen. Die jungen adulten Tiere können entweder einzeln oder in Gruppen von bis zu drei Tieren pro Käfig untergebracht werden. Da die unreifen Tiere noch sehr jung sind, ist die Unterbringung in der Gruppe aus sozialen Gründen empfehlenswert.
21. Ein hoher Gehalt an Phytoöstrogenen in der Labornahrung führt bei Nagern erwiesenermaßen zu einer Erhöhung des Uterusgewichts, die für eine Verfälschung der Ergebnisse des uterotrophen Bioassays ausreichend ist (13)(14)(15). Hohe Gehalte an Phytoöstrogenen und ein hohes Niveau metabolisierbarer Energie in der Labornahrung können ferner bei unreifen Tieren einen vorzeitigen Pubertätseintritt zur Folge haben. Phytoöstrogene in der Nahrung sind vorwiegend auf Soja- und Alfalfaprodukte als Zutaten im Laborfutter zurückzuführen, wobei die Phytoöstrogenkonzentrationen in der Standard-Labornahrung zwischen den einzelnen Chargen variieren (23). Das Körpergewicht ist eine wichtige Variable, da die aufgenommene Futtermenge in Relation zum Körpergewicht steht. Die tatsächlich aus demselben Futter aufgenommene Phytoöstrogendosis kann daher zwischen verschiedenen Tierarten und, innerhalb einer Art, je nach Alter abweichen (9). Bei unreifen Rattenweibchen kann die Futteraufnahme in Relation zum Körpergewicht ungefähr doppelt so hoch ausfallen wie bei ovarektomierten jungen adulten Weibchen. Bei jungen adulten Mäusen kann die Futteraufnahme in Relation zum Körpergewicht ungefähr viermal so hoch ausfallen wie bei ovarektomierten jungen adulten Rattenweibchen.
22. Die Ergebnisse anderer uterotropher Bioassays (9)(17)(18)(19) haben jedoch gezeigt, dass begrenzte Mengen an Phytoöstrogenen in der Nahrung akzeptabel sind und sich nicht negativ auf die Aussagekraft des Assays auswirken. Zur Orientierung: Der Phytoöstrogengehalt sollte 350 μg Genistein-Äquivalente pro Gramm Labornahrung bei unreifen Sprague Dawley- und Wistar-Rattenweibchen nicht überschreiten (6)(9). Diese Nahrung sollte auch dann geeignet sein, wenn junge adulte ovarektomierte Ratten getestet werden, weil die Nahrungsaufnahme in Relation zum Körpergewicht bei jungen adulten Ratten geringer ist als bei unreifen Tieren. Wenn adulte ovarektomierte Mäuse oder stärker auf Phytoöstrogene reagierende Ratten verwendet werden sollen, muss eine proportionale Reduzierung der Phytoöstrogengehalte in der Nahrung in Erwägung gezogen werden (20). Ferner können Unterschiede bei der verfügbaren verstoffwechselbaren Energie bei verschiedenen Arten von Labornahrung das Einsetzen der Pubertät beeinflussen (21)(22).
23. Vor Beginn des Versuchs ist bei der Auswahl der Labornahrung sorgfältig darauf zu achten, dass diese keinen erhöhten Gehalt an Phytoöstrogenen (siehe (6) und (9)) oder an metabolisierbarer Energie aufweist, denn beides kann zu einer Verzerrung der Ergebnisse führen (15)(17)(19)(22)(36). Die Gewährleistung der ordnungsgemäßen Durchführung des vom Labor verwendeten Prüfsystems gemäß den Nummern 26 und 27 stellt eine wichtige Kontrolle dieser beiden Faktoren dar. Sicherheitshalber sollten im Einklang mit der Guten Laborpraxis (GLP) repräsentative Stichproben von allen im Laufe der Prüfung verabreichten Nahrungschargen genommen werden, um bei Bedarf den Phytoöstrogengehalt zu analysieren (z. B. wenn im Vergleich zu historischen Kontrolldaten ein hohes Uterus-Kontrollgewicht vorliegt oder bei ungenügendem Ansprechen auf das Referenzöstrogen 17α-Ethinylestradiol). Die Aliquote sind im Rahmen der Prüfung zu analysieren oder so aufzubewahren, dass sich die Probe nicht vor der Analyse zersetzen kann (zum Beispiel durch Einfrieren bei – 20 °C).
24. Einige Arten von Einstreu können natürlicherweise Chemikalien mit östrogener oder antiöstrogener Wirkung enthalten (zum Beispiel beeinflusst Maiseinstreu bekanntermaßen den Zyklus von Ratten und hat vermutlich antiöstrogene Eigenschaften). Der Phytoöstrogengehalt der gewählten Einstreu sollte so gering wie möglich sein.
Vorbereitung der Tiere
25. Die Versuchstiere, die keine Krankheitszeichen oder physischen Abnormitäten aufweisen, werden randomisiert auf die Kontroll- und Behandlungsgruppen aufgeteilt. Die Käfige sollten so angeordnet werden, dass etwaige Einflüsse der Käfigplatzierung minimiert werden. Jedes Tier ist eindeutig zu kennzeichnen. Unreife Tiere sind bis zum Absetzen möglichst gemeinsam mit Muttertieren oder Ammen zu halten, bis sie sich akklimatisiert haben. Die Akklimatisierungszeit vor Versuchsbeginn sollte sowohl für junge adulte Tiere als auch für unreife, mit Muttertieren oder Ammen gelieferte Tiere etwa fünf Tage betragen. Wenn gerade abgesetzte, unreife Tiere ohne Muttertiere erworben werden, ist gegebenenfalls eine Verkürzung der Akklimatisierungszeit notwendig, da mit der Dosierung sofort nach dem Absetzen begonnen werden sollte (vgl. Nummer 29).
VERFAHREN
Eignungsprüfung des Prüflabors
26. Für die Eignungsprüfung des Prüflabors gibt es zwei Optionen:
|
— |
Regelmäßige Verifizierung durch Abgleich mit einer zu Anfang durchgeführten positiven Kontrollstudie/Baselinestudie (vgl. Nummer 27). Mindestens alle sechs Monate sowie immer dann, wenn eine Veränderung eingetreten ist, die die Prüfleistung beeinflussen könnte (z. B. neue Futterzubereitung, Änderung des Sektionspersonals, Änderung des Tierstamms, geänderter Lieferant usw.), ist die Empfindlichkeit des Prüfsystems (das Tiermodell) anhand einer (mit Hilfe der unter Nummer 27 beschriebenen positiven Kontrollstudie/Baselinestudie bestimmten) geeigneten Dosis des Referenzöstrogens 17a-Ethinylestradiol (CAS Nr. 57-63-6) (EE) zu verifizieren. |
|
— |
Verwendung gleichzeitiger Kontrollgruppen, indem bei jeder Prüfung eine Gruppe hinzugefügt wird, die eine geeignete Dosis des Referenzöstrogens erhält. |
Wenn das Tiermodell nicht wie erwartet reagiert, sind die Versuchsbedingungen zu überprüfen und entsprechend zu ändern. Die bei jedem der Ansätze verwendete Dosis des Referenzöstrogens sollte etwa die ED70 bis 80 sein.
27. Baselinestudie/positive Kontrollstudie — Bevor ein Labor anhand der vorliegenden Prüfmethode erstmals eine Studie durchführt, sollte seine Eignung nachgewiesen werden, und zwar durch Prüfung der Empfindlichkeit des Tiermodells und Festlegung der Dosis-Wirkungs-Beziehung für das Referenzöstrogen 17a-Ethinylestradiol (CAS Nr. 57-63-6) (EE) bei einem Minimum von vier Dosen. Die Reaktion des Uterusgewichts wird mit bekannten historischen Daten abgeglichen (vgl. Literaturhinweis (5)). Wenn diese Baseline- bzw. positive Kontrollstudie nicht zu den erwarteten Ergebnissen führt, sind die Versuchsbedingungen zu überprüfen und entsprechend zu modifizieren.
Zahl und Zustand der Versuchstiere
28. Jede Behandlungs- und Kontrollgruppe sollte aus mindestens sechs Tieren zusammengesetzt sein (sowohl beim Prüfplan für die Methode mit unreifen Tieren als auch bei dem für die Methode mit adulten OVX-Tieren).
Alter der unreifen Tiere
29. Beim uterotrophen Bioassay mit unreifen Tieren ist der Tag der Geburt anzugeben. Mit der Dosierung sollte so früh begonnen werden, dass sichergestellt ist, dass der mit der Pubertät assoziierte natürliche Anstieg der endogenen Östrogene am Ende der Prüfsubstanzverabreichung noch nicht stattgefunden hat. Andererseits gibt es Hinweise darauf, dass sehr junge Tiere möglicherweise weniger empfindlich reagieren. Zur Festlegung des optimalen Alters sollte jedes Labor seine eigenen Hintergrunddaten zur Geschlechtsreife heranziehen.
Als Faustregel kann mit der Dosierung bei Ratten unmittelbar nach einem frühen Absetzen am postnatalen Tag 18 (PND 18) begonnen werden (PND 0 ist der Tag der Geburt). Die Dosierung sollte bei Ratten möglichst am PND 21 abgeschlossen sein, spätestens jedoch vor PND 25, weil nach diesem Alter die Hypothalamus-Hypophysen-Eierstock-Achse funktionsfähig wird und die endogenen Östrogenspiegel unter Umständen zu steigen beginnen, was mit einer Erhöhung der mittleren Baseline-Uterusgewichte und einem Ansteigen der Gruppen-Standardabweichungen einhergeht (2)(3)(10)(11)(12).
Ovarektomieverfahren
30. Bei Prüfungen mit ovarektomierten Ratten- und Mäuseweibchen (Behandlungs- und Kontrollgruppen) ist die Ovarektomie im Alter zwischen sechs und acht Wochen durchzuführen. Bei Ratten sollte zwischen der Ovarektomie und dem ersten Verabreichungstag mindestens ein Zeitraum von 14 Tagen liegen, damit der Uterus sich wieder auf eine stabile Mindestgröße (die Ausgangsgröße für die Prüfung) zurückbilden kann. Bei Mäusen sollten mindestens sieben Tage zwischen der Ovarektomie und dem ersten Verabreichungstag liegen. Da bereits kleine Mengen Ovarialgewebe für eine signifikante Erhöhung der Östrogenspiegel ausreichen (3), sollten die Tiere vor ihrer Verwendung mittels Beobachtung der bei Vaginalabstrichen an fünf aufeinanderfolgenden Tagen (z. B. Tage 10-14 nach der Ovarektomie bei Ratten) gewonnenen Epithelzellen getestet werden. Tiere, die Anzeichen für einen beginnenden Östruszyklus zeigen, sollten nicht verwendet werden. Später, bei der Nekropsie, sind die Ovarialstümpfe auf Reste von Ovarialgewebe zu untersuchen. Wird Ovarialgewebe gefunden, sollte das Tier nicht in die Berechnungen mit einbezogen werden (3).
31. Zu Beginn der Ovarektomie liegt das ordnungsgemäß narkotisierte Tier in Bauchlage. Der Längsschnitt zur Eröffnung der dorsolateralen Bauchwand sollte etwa 1 cm betragen und in der Mitte zwischen unterem Rippenbogenrand und Beckenkamm sowie einige Millimeter neben dem lateralen Rand des Lendenmuskels verlaufen. Das Ovar wird der Bauchhöhle entnommen und auf einem sterilen Feld abgelegt. Dann wird es an der Verbindungsstelle von Eileiter und Gebärmutterkörper abgetrennt. Wenn sichergestellt ist, dass keine starke Blutung auftritt, ist die Bauchwand zuzunähen und die Haut mit Klammern oder einer entsprechenden Naht zu schließen. Die Ligaturpunkte sind schematisch in Abbildung 1 dargestellt. In Absprache mit einem auf Nager spezialisierten Veterinärmediziner sollte eine geeignete postoperative Schmerzlinderung erfolgen.
Körpergewicht
32. Bei der Methode mit adulten OVX-Tieren korreliert das Uterusgewicht nicht mit dem Körpergewicht, weil das Uterusgewicht durch Hormone, zum Beispiel Östrogene, nicht aber durch Wachstumsfaktoren, die die Körpergröße regulieren, beeinflusst wird. Beim Modell mit unreifen Tieren dagegen ist ein Zusammenhang zwischen Uterusgewicht und Körpergewicht gegeben, so lange das Tier noch nicht voll entwickelt ist (34). Zu Beginn des Versuchs sollten die Gewichtsunterschiede bei den unreifen Tieren möglichst gering sein und ± 20 % des Durchschnittsgewichts nicht überschreiten. Das bedeutet, dass die Wurfgröße durch den Züchter standardisiert werden sollte, sodass sichergestellt ist, dass Nachkommen verschiedener Muttertiere ungefähr gleich gefüttert werden. Die Tiere werden durch randomisierte Gewichtsverteilung so den Kontroll- und Behandlungsgruppen zugewiesen, dass das durchschnittliche Körpergewicht der einzelnen Gruppen sich statistisch nicht von dem der anderen Gruppen unterscheidet. Eine Zuteilung von Wurfgeschwistern zur selben Behandlungsgruppe ist zu vermeiden, sofern das ohne Erhöhung der Anzahl der für die Untersuchung verwendeten Würfe möglich ist.
Dosierung
33. Um festzustellen, ob eine Prüfsubstanz in vivo eine östrogene Wirkung entfalten kann, sind normalerweise zwei Dosisgruppen und eine Kontrollgruppe ausreichend, weshalb dieser Versuchsanordnung aus Tierschutzgründen der Vorrang eingeräumt werden sollte. Wenn der Zweck des Versuchs entweder die Erstellung einer Dosis-Wirkungs-Kurve oder die Umrechnung auf niedrigere Dosen ist, sind mindestens drei Dosisgruppen erforderlich. Wenn Informationen gewonnen werden sollen, die über das Vorhandensein einer östrogenen Aktivität hinausgehen (wie zum Beispiel eine Einschätzung der Wirkstärke), sollte ein anderer Dosierungsplan in Betracht gezogen werden. Abgesehen von der Behandlung mit der Prüfsubstanz sollten die Tiere der Kontrollgruppe genauso behandelt werden wie die Tiere in den Prüfgruppen. Wird die Prüfsubstanz mit einem Vehikel verabreicht, muss die Kontrollgruppe dieselbe Menge des Vehikels erhalten wie die behandelten Gruppen (bzw. das höchste verwendete Volumen, wenn die verschiedenen Gruppen unterschiedliche Volumina erhalten).
34. Das Ziel des uterotrophen Bioassays ist die Auswahl von Dosen, bei denen die Tiere auf jeden Fall überleben und die nach drei aufeinanderfolgenden Tagen mit einer Chemikaliengabe von maximal 1 000 mg pro Kilogramm und Tag nicht mit signifikanter Toxizität oder einem Leiden der Tiere einhergehen. Bei der Auswahl sämtlicher Dosisstufen sind alle zu der Prüfsubstanz oder verwandten Stoffen verfügbaren toxikologischen und (toxiko-)kinetischen Daten zu berücksichtigen. Bei der Festlegung der Höchstdosis sollten insbesondere die LD50 und/oder Angaben zur akuten Toxizität berücksichtigt werden, um den Tod der Tiere oder starkes Leiden und Qualen zu vermeiden (24)(25)(26). Die höchste Dosis sollte die maximal verträgliche Dosis (MTD) darstellen; eine auf einer Dosisstufe, die eine positive uterotrophe Reaktion hervorgerufen hat, durchgeführte Studie wäre ebenfalls akzeptabel. Für Screeningzwecke sind große Intervalle (z. B. eine halbe logarithmische Einheit, entsprechend einer Dosissteigerung um den Faktor 3,2, oder sogar bis zu einer vollen logarithmischen Einheit) zwischen den Dosisstufen allgemein akzeptabel. Liegen keine geeigneten Daten vor, kann eine Dosisfindungsstudie zur Bestimmung der zu verwendenden Dosen durchgeführt werden.
35. Wenn die Stärke der östrogenen Wirkung eines Agonisten aufgrund von In-vitro- (oder In-silico-)Daten beurteilt werden kann, können alternativ auch diese Daten für die Dosiswahl herangezogen werden. Zum Beispiel wird die Menge der Prüfsubstanz, die uterotrophe Reaktionen vergleichbar mit denjenigen des Referenzagonisten (Ethinylestradiol) hervorrufen würde, anhand ihrer In-vitro-Wirkstärke im Verhältnis zu Ethinylestradiol abgeschätzt. Die höchste Prüfdosis ergibt sich aus der Multiplikation dieser Äquivalenzdosis mit einem geeigneten Faktor, zum Beispiel 10 oder 100.
Ausführungen zur Dosisfindung
36. Falls notwendig, kann eine Vorstudie zur Dosisfindung mit wenigen Tieren durchgeführt werden. Diesbezüglich kann das OECD Guidance Document Nr. 19 (25) konsultiert werden, in dem klinische Anzeichen für Toxizität oder Leiden der Tiere festgelegt sind. Sofern es im Rahmen dieser Dosisfindungsstudie nach drei Verabreichungstagen praktikabel ist, können die Uteri etwa 24 Stunden nach der letzten Dosisgabe operativ entfernt und gewogen werden. Diese Daten können dann zur Planung der Hauptstudie herangezogen werden (Auswahl der akzeptablen Höchstdosis und der niedrigeren Dosen sowie Empfehlung für die Anzahl an Dosisgruppen).
Verabreichung der Dosen
37. Die Prüfsubstanz wird oral per Schlundsonde oder durch subkutane Injektion verabreicht. Bei der Wahl des Verabreichungswegs sind Tierschutzaspekte sowie toxikologische Aspekte wie die Art der Humanexposition gegenüber der Chemikalie (z. B. Schlundsonde, um eine Exposition über die Nahrungsaufnahme zu simulieren; subkutane Injektion, um Einatmen oder Aufnahme über die Haut am Modell zu testen), die physikalisch-chemischen Eigenschaften der Prüfsubstanz und insbesondere verfügbare toxikologische Angaben und Daten zu Verstoffwechselung und Kinetik (z. B. die Notwendigkeit, eine First-pass-Metabolisierung zu vermeiden; bessere Effizienz über einen bestimmten Verabreichungsweg) mit einzubeziehen.
38. Als Erstes sollte möglichst immer die Verwendung einer wässrigen Lösung/Suspension in Erwägung gezogen werden. Da aber die meisten Östrogenliganden beziehungsweise ihre metabolischen Vorläufer eher hydrophob sind, ist der gängigste Ansatz die Verwendung einer Lösung/Suspension auf Ölbasis (z. B. Mais-, Erdnuss-, Sesam- oder Olivenöl). Diese Öle unterscheiden sich jedoch in ihrem Energie- und Fettgehalt, sodass das Vehikel die Menge der insgesamt aufgenommenen metabolisierbaren Energie (ME) beeinflussen kann, was sich wiederum — insbesondere bei der Methode mit unreifen Tieren — auf die gemessenen Endpunkte, beispielsweise das Uterusgewicht, auswirken kann (33). Aus diesem Grund sollte vor dem Versuch jedes geplante Vehikel mit Kontrollen ohne Vehikel verglichen werden. Die Prüfsubstanzen können in einer sehr kleinen Menge von 95 %igem Ethanol oder anderen geeigneten Lösungsmitteln gelöst und im Prüfvehikel auf ihre endgültigen Arbeitskonzentrationen verdünnt werden. Die toxischen Eigenschaften des Lösungsmittels müssen bekannt sein und sollten in einer separaten Kontrollgruppe, in der nur das Lösungsmittel geprüft wird, getestet werden. Wenn die Prüfsubstanz als stabil erachtet wird, kann der Lösungsvorgang durch schonende Erwärmung und kräftige mechanische Einwirkung unterstützt werden. Die Stabilität der Prüfsubstanz im Vehikel sollte bestimmt werden. Ist die Prüfsubstanz für die Dauer des Versuchs stabil, kann eine Start-Aliquote der Substanz vorbereitet werden und die spezifizierten Dosierungsverdünnungen können täglich zubereitet werden.
39. Die zeitliche Planung der Dosisgaben ist abhängig vom verwendeten Tiermodell (vgl. Nummer 29 für das Modell mit unreifen Tieren und Nummer 30 für das Modell mit adulten OVX-Tieren). Unreife Rattenweibchen erhalten täglich an drei aufeinanderfolgenden Tagen eine Gabe der Prüfsubstanz. Auch für ovarektomierte Rattenweibchen wird eine dreitägige Behandlungsdauer empfohlen, aber eine längere Exposition ist akzeptabel und kann den Nachweis schwach aktiver Chemikalien erleichtern. Im Falle von ovarektomierten Mäuseweibchen ist bei starken Östrogen-Agonisten eine Verabreichungsdauer von drei Tagen im Allgemeinen ausreichend und eine Verlängerung auf sieben Tage nicht mit signifikanten Vorteilen verbunden; für schwache Östrogene wurde diese Beziehung allerdings in der Validierungsstudie (16) nicht nachgewiesen, weshalb eine verlängerte Dosisgabe von sieben aufeinanderfolgenden Tagen bei adulten OVX-Mäusen angeraten ist.Die Dosis sollte täglich ungefähr zur selben Zeit verabreicht werden. Die Dosisgaben sind bei Bedarf anzupassen, um eine konstante Dosierung in Relation zum Körpergewicht des Tiers (z. B. mg Prüfsubstanz pro kg Körpergewicht und Tag) sicherzustellen. Das Prüfvolumen sollte im Verhältnis zum Körpergewicht möglichst konstant gehalten werden, indem die Konzentration der Lösung, die die Dosierung enthält, angepasst wird, um so bei allen Dosisstufen und Verabreichungswegen ein im Verhältnis zum Körpergewicht konstantes Volumen sicherzustellen.
40. Wird die Prüfsubstanz über eine Sonde verabreicht, so sollte dies in einer täglichen Einmaldosis unter Verwendung einer Schlundsonde oder einer geeigneten Intubationskanüle erfolgen. Das maximale Flüssigkeitsvolumen, das jeweils verabreicht werden kann, hängt von der Größe des Versuchstiers ab. Es sind die örtlichen Tierschutzrichtlinien zu befolgen, das Volumen sollte jedoch 5 ml pro kg Körpergewicht nicht überschreiten, außer bei wässrigen Lösungen, von denen 10 ml pro kg Körpergewicht gegeben werden können.
41. Wird die Prüfsubstanz durch subkutane Injektion verabreicht, so sollte dies über eine tägliche Einmaldosis geschehen. Die Dosen sind mittels steriler Nadel (z. B. 23 oder 25 Gauge) und Tuberkulinspritze dorsal im Bereich der Schulterblätter oder in die Lendenregion zu injizieren. Die Injektionsstelle kann rasiert werden. Wenn Injektionsflüssigkeit verloren geht, an der Injektionsstelle ausläuft oder unvollständig verabreicht wird, ist dies zu protokollieren. Das Gesamtvolumen, das pro Ratte und Tag injiziert wird, sollte 5 ml pro kg Körpergewicht, aufgeteilt auf zwei Injektionsstellen, nicht überschreiten; dies gilt nicht für wässrige Lösungen, von denen 10 ml pro kg Körpergewicht gegeben werden können.
Beobachtungen
Allgemeine und klinische Beobachtungen
42. Allgemeine klinische Beobachtungen sollten mindestens einmal täglich durchgeführt werden, bei Anzeichen für Toxizität häufiger. Die Beobachtungen sind möglichst immer zur selben Tageszeit/zu denselben Tageszeiten und unter Berücksichtigung des Zeitraums nach der Verabreichung, in dem die Wirkungsgipfel zu erwarten sind, durchzuführen. Alle Tiere sind auf Anzeichen von Mortalität, Morbidität und allgemeine klinische Zeichen wie Verhaltensänderungen, Änderungen an Haut, Fell, Augen und Schleimhäuten, Sekrete und Exkrete sowie autonome Körperfunktionen (z. B. Tränensekretion, Piloerektion, Pupillengröße, ungewöhnliche Atemmuster) zu beobachten.
Körpergewicht und Futteraufnahme
43. Alle Tiere sind täglich auf 0,1 g genau zu wiegen, wobei unmittelbar vor Behandlungsbeginn, also wenn die Tiere den Gruppen zugeteilt werden, mit dem Wiegen zu beginnen ist. Optional kann die aufgenommene Futtermenge während des Behandlungszeitraums pro Käfig durch Wiegen der Futterspender bestimmt werden. Die Daten zur Futteraufnahme sind in Gramm pro Ratte und Tag auszudrücken.
Sektion und Bestimmung des Uterusgewichts
44. Die Ratten sind 24 Stunden nach der letzten Behandlung auf humane Weise zu töten. Idealerweise werden die Nekropsien über die Gruppen hinweg randomisiert durchgeführt, um ein unmittelbares Abarbeiten der Dosisgruppen nach oben oder unten zu vermeiden, da dies die Daten geringfügig beeinflussen könnte. Ziel des Bioassays ist die Bestimmung sowohl des Uterusfeuchtgewichts als auch des geblotteten Uterusgewichts. Das Feuchtgewicht beinhaltet den Uterus und den Gehalt an Uterusflüssigkeit. Das geblottete Gewicht wird bestimmt, nachdem die Uterusflüssigkeit ausgedrückt und beseitigt wurde.
45. Vor der Sektion wird die Vagina bei unreifen Tieren auf ihren Öffnungsstatus hin untersucht. Die Sektion beginnt mit der Eröffnung der Bauchwand von der Schambeinfuge an. Anschließend werden das Uterushorn und die Ovarien, sofern vorhanden, von der dorsalen Bauchwand gelöst. Dann werden Harnblase und Harnleiter von der ventralen und lateralen Seite des Uterus und der Vagina entfernt. Die fibröse Adhäsion zwischen Rektum und Vagina wird gelöst, bis die Verbindung zwischen Vaginalöffnung und Perinealhaut erkennbar ist. Uterus und Vagina werden vom Körper gelöst, indem die Vaginalwand genau oberhalb der Verbindung zur Perinealhaut abgeschnitten wird (siehe Abbildung 2). Der Uterus sollte durch vorsichtiges Abtrennen des Mesometriums an der Stelle, an der es jeweils dorsolateral mit der Längsseite eines Uterushorns verbunden ist, von der Körperwand gelöst werden. Sobald der Uterus aus dem Körper entfernt ist, sollten die weiteren Arbeiten rasch genug erfolgen, um ein Austrocknen der Gewebe zu vermeiden. Ein Gewichtsverlust durch Austrocknen fällt bei kleinen Geweben wie dem Uterus stärker ins Gewicht (23). Wenn Ovarien vorhanden sind, werden sie so am Ovidukt entfernt, dass keine Gebärmutterflüssigkeit aus dem Uterushorn verloren geht. Bei ovarektomierten Tieren sind die Ovarialstümpfe auf Reste von Ovarialgewebe zu untersuchen. Überschüssiges Fett- und Bindegewebe ist abzuschaben. Die Vagina wird genau unterhalb des Gebärmutterhalses vom Uterus getrennt, sodass der Gebärmutterhals mit dem Uteruskörper verbunden bleibt (siehe Abbildung 2).
46. Die Uteri werden einzeln in eindeutig gekennzeichneten und gewogenen Behälter verbracht (z. B. Petrischale oder Wägeschiffchen aus Kunststoff) und kontinuierlich gegen Austrocknung vor dem Wiegen geschützt (beispielsweise indem ein leicht mit Kochsalzlösung angefeuchtetes Filterpapier in den Behälter gelegt wird). Der Uterus wird mit der Uterusflüssigkeit auf 0,1 mg genau gewogen (Uterusfeuchtgewicht).
47. Anschließend wird jeder Uterus einzeln weiterverarbeitet, um die Gebärmutterflüssigkeit zu entfernen. Beide Uterushörner werden durchstochen oder längs aufgeschnitten. Der Uterus wird auf leicht angefeuchtetes Filterpapier (z. B. Whatman Nr. 3) gelegt und vorsichtig mit einem zweiten Stück leicht angefeuchteten Filterpapiers ausgedrückt, bis die Uterusflüssigkeit vollständig entfernt ist (Blotten). Der Uterus wird ohne die Gebärmutterflüssigkeit auf 0,1 mg genau gewogen (Uterusgewicht nach Blotten, nachfolgend Uterustrockengewicht).
48. Am Uterusgewicht bei Studienende lässt sich ablesen, ob das geeignete Alter bei der unreifen intakten Ratte tatsächlich nicht überschritten wurde, ausschlaggebend sind in dieser Hinsicht jedoch die historischen Daten des vom Labor verwendeten Rattenstamms (vgl. Nummer 56 bezüglich der Interpretation der Ergebnisse).
Optionale Untersuchungen
49. Nach dem Wiegen können die Uteri in 10 % neutral gepuffertem Formalin fixiert und nach Hämatoxylin-Eosin-Färbung (HE-Färbung) histopathologisch untersucht werden. Die Vagina kann ebenfalls entsprechend untersucht werden (siehe Nummer 9). Für einen quantitativen Vergleich kann ferner eine morphometrische Untersuchung des Epithelgewebes der Gebärmutterschleimhaut durchgeführt werden.
DATEN UND BERICHTERSTATTUNG
Daten
50. Folgende Daten über die Prüfung sind anzugeben:
|
— |
die Anzahl der Tiere zu Beginn des Assays, |
|
— |
die Anzahl und Identität der während der Prüfung tot aufgefundenen oder aus humanen Gründen getöteten Tiere sowie Datum und Uhrzeit des Todes oder der Tötung der Tiere, |
|
— |
die Anzahl und Identität der Tiere, die Toxizitätszeichen aufweisen, und eine Beschreibung der beobachteten Toxizitätszeichen einschließlich Zeitpunkt des Einsetzens, Dauer und Schweregrad der toxischen Wirkungen und |
|
— |
die Anzahl und Identität der Tiere, bei denen Läsionen festgestellt werden, und eine Beschreibung der Art der Läsionen. |
51. Zu den einzelnen Tieren sollten die Daten bezüglich Körpergewicht, Uterusfeuchtgewicht und Uterustrockengewicht protokolliert werden. Um festzustellen, ob die Verabreichung der Prüfsubstanz zu einer statistisch signifikanten (p < 0,05) Erhöhung des Uterusgewichts geführt hat, sollten einseitige statistische Analysen für die Agonisten durchgeführt werden. Es sind geeignete statistische Analysen durchzuführen, um die Trocken- und Feuchtgewichte der Uteri auf behandlungsbedingte Veränderungen zu überprüfen. Zum Beispiel können die Daten anhand einer Kovarianzanalyse mit dem Körpergewicht bei Nekropsie als Kovariate beurteilt werden. Vor der Datenanalyse kann eine varianzstabilisierende logarithmische Transformation der Uterusdaten durchgeführt werden. Der Dunnett-Test und der Hsu-Test sind für paarweise Vergleiche jeder Dosisgruppe mit Vehikelkontrollgruppen und die Berechnung der Konfidenzintervalle geeignet. Es können studentisierte Residuenplots zur Erfassung möglicher Ausreißer und zur Bewertung der Homogenität der Varianzen eingesetzt werden. Diese Verfahren kamen im Rahmen des Validierungsprogramms der OECD unter Verwendung von PROC GLM (GLM-Verfahren) im Statistik-Softwaresystem SAS (Statistisches Analysesystem, SAS Institute, Cary, NC), Version 8 (6)(7) zum Einsatz.
52. Der Abschlussbericht muss Folgendes beinhalten:
Prüfeinrichtung:
|
— |
verantwortliches Personal und Zuständigkeiten während der Studie, |
|
— |
Daten der Baselinestudie/positiven Kontrollstudie und in regelmäßigen Abständen gewonnene positive Kontrolldaten (vgl. Nummern 26 und 27). |
Prüfsubstanz:
|
— |
Beschreibung der Prüfsubstanzen, |
|
— |
physikalische Beschaffenheit und gegebenenfalls physikalisch-chemische Eigenschaften, |
|
— |
Zubereitungsverfahren von Verdünnungen und Häufigkeit der Zubereitung, |
|
— |
etwaige Daten über die Stabilität, |
|
— |
etwaige Analysen der Dosierungslösungen. |
Vehikel:
|
— |
Beschreibung des Prüfvehikels (Art, Lieferant und Charge), |
|
— |
Begründung der Wahl des Vehikels (sofern anders als Wasser). |
Versuchstiere:
|
— |
Tierart und -stamm und Begründung der getroffenen Wahl, |
|
— |
Lieferant und genaue Angabe der Einrichtung, aus der die Tiere stammen, |
|
— |
Alter bei Lieferung und Geburtsdatum, |
|
— |
bei unreifen Tieren, Angabe, ob diese mit Mutter- oder Ammentier geliefert werden, und Datum des Absetzens, |
|
— |
Angaben zum Akklimatisierungsverfahren für die Tiere, |
|
— |
Anzahl der Tiere pro Käfig, |
|
— |
Angaben/Methode zur eindeutigen Kennzeichnung der einzelnen Tiere und der Gruppe. |
Prüfbedingungen:
|
— |
Angaben zum Randomisierungsverfahren (angewendete Methode), |
|
— |
Begründung der Dosiswahl, |
|
— |
Angaben zur Formulierung der Prüfsubstanz, ihrer erreichten Konzentration, Stabilität und Homogenität, |
|
— |
Angaben zur Verabreichung der Prüfsubstanz und Begründung des gewählten Expositionswegs, |
|
— |
Futter (Bezeichnung, Art, Lieferant, Inhalt und, sofern bekannt, Phytoöstrogengehalte), |
|
— |
Trinkwasserversorgung (z. B. Leitungswasser oder gefiltertes Wasser) und Art der Tränkung (Großbehälter mit Trinkschläuchen, Flaschentränkung usw.), |
|
— |
Einstreu (Bezeichnung, Art, Lieferant, Inhalt), |
|
— |
Protokoll der Haltungsbedingungen, Beleuchtungsintervalle, Raumtemperatur, Luftfeuchtigkeit, Raumreinigung, |
|
— |
ausführliche Beschreibung der Nekropsie und der Uterusgewichtsbestimmung, |
|
— |
Beschreibung der statistischen Verfahren. |
Ergebnisse
Bezüglich der einzelnen Tiere:
|
— |
sämtliche Angaben zum Körpergewicht pro Tier und Tag (von der Gruppenzuteilung bis zur Nekropsie) (auf 0,1 g genau), |
|
— |
Alter jedes einzelnen Tiers (ausgedrückt in Tagen; Tag der Geburt = Tag 0) ab der Verabreichung der Prüfsubstanz, |
|
— |
Datum und Uhrzeit der einzelnen Dosisgaben, |
|
— |
berechnetes Volumen und verabreichte Dosierung sowie beobachtete Dosierungsverluste während oder nach der Verabreichung, |
|
— |
täglicher Bericht zum Zustand der Tiere einschließlich einschlägiger Symptome und Beobachtungen, |
|
— |
vermutete Todesursache (bei während der Studie in moribundem Zustand oder tot aufgefundenen Tieren), |
|
— |
Datum und Uhrzeit der schmerzlosen Tötung einschließlich des Zeitabstands zur letzten Dosisgabe, |
|
— |
Uterusfeuchtgewicht (auf 0,1 mg genau) und etwaige Beobachtungen zu Uterusflüssigkeitsverlusten während der Sektion und Vorbereitung zum Wiegen, |
|
— |
Uterustrockengewicht (auf 0,1 mg) genau. |
Bezüglich der einzelnen Tiergruppen:
|
— |
mittleres Körpergewicht pro Tag (auf 0,1 g genau) und Standardabweichungen (von der Gruppenzuteilung bis zur Nekropsie), |
|
— |
mittleres Uterusfeuchtgewicht und mittleres Uterustrockengewicht pro Gruppe (auf 0,1 mg genau) und Standardabweichungen, |
|
— |
Futterverbrauch pro Tag, falls gemessen (berechnet in Gramm verzehrtes Futter pro Tier), |
|
— |
Ergebnisse der statistischen Analysen zum Vergleich der Uterusfeucht- und -trockengewichte der Behandlungsgruppen mit denselben Maßnahmen in den Vehikelkontrollgruppen, |
|
— |
Ergebnisse der statistischen Analysen zum Vergleich des Körpergewichts insgesamt und der Körpergewichtszunahme der Behandlungsgruppen mit denselben Maßnahmen in den Vehikelkontrollgruppen. |
53. Zusammenfassung der wichtigsten Elemente der Prüfmethode
|
|
Ratte |
Maus |
|
Versuchstiere |
||
|
Stamm |
Allgemein üblicher Labornagerstamm |
|
|
Anzahl der Tiere |
Mindestens sechs Tiere pro Dosisgruppe |
|
|
Anzahl der Gruppen |
Mindestens zwei Prüfgruppen (für weitere Hinweise siehe Nummer 33) und eine negative Kontrollgruppe Für Hinweise zu positiven Kontrollgruppen siehe Nummern 26 und 27 |
|
|
Haltungs- und Fütterungsbedingungen |
||
|
Temperatur im Versuchstierraum |
22 °C ± 3 °C |
|
|
Relative Luftfeuchtigkeit |
50-60 %, mind. 30 %, max. 70 % |
|
|
Tägliche Beleuchtungsintervalle |
12 h hell/12 h dunkel |
|
|
Nahrung und Trinkwasser |
Ad libitum |
|
|
Unterbringung |
Einzeln oder in Gruppen von bis zu drei Tieren (unreife Tiere sollten aus sozialen Gründen in der Gruppe untergebracht werden) |
|
|
Futter und Einstreu |
Futter und Einstreu sollten möglichst niedrige Phytoöstrogengehalte aufweisen |
|
|
Prüfplan |
||
|
Methode |
Methode mit unreifen, nicht ovarektomierten Tieren (bevorzugtes Verfahren) Methode mit ovarektomierten adulten Weibchen |
Methode mit ovarektomierten adulten Weibchen |
|
Alter der unreifen Tiere bei Dosisgabe |
Beginn frühestens PND 18; letzte Dosisgabe vor PND 25 |
Bei dieser Prüfmethode nicht relevant |
|
Alter bei Ovarektomie |
Zwischen der 6. und 8. Lebenswoche |
|
|
Alter der ovarektomierten Tiere bei Dosisgabe |
Es sollten mindestens 14 Tage zwischen der Ovarektomie und dem ersten Verabreichungstag liegen. |
Es sollten mindestens sieben Tage zwischen der Ovarektomie und dem ersten Verabreichungstag liegen. |
|
Körpergewicht |
Das Körpergewicht sollte nur minimal variieren und nicht mehr als ± 20 % vom Durchschnittsgewicht abweichen. |
|
|
Dosierung |
||
|
Verabreichungsweg |
Schlundsonde oder subkutane Injektion |
|
|
Häufigkeit der Verabreichung |
Tägliche Einmaldosis |
|
|
Flüssigkeitsvolumen bei Schlundsonde und Injektion |
≤ 5 ml/kg Körpergewicht (bzw. bis zu 10 ml/kg Körpergewicht bei wässrigen Lösungen) (bei subkutaner Gabe an zwei Injektionsstellen) |
|
|
Verabreichungsdauer |
Drei aufeinanderfolgende Tage beim Modell mit unreifen Tieren Mindestens drei aufeinanderfolgende Tage beim OVX-Modell |
Sieben aufeinanderfolgende Tage beim OVX-Modell |
|
Zeitpunkt der Nekropsie |
Etwa 24 Stunden nach der letzten Dosisgabe |
|
|
Ergebnisse |
||
|
Positive Reaktion |
Statistisch signifikante Erhöhung des durchschnittlichen Uterusgewichts (Feuchtgewicht und/oder Trockengewicht) |
|
|
Referenzöstrogen |
17α-Ethinylestradiol |
|
LEITFADEN FÜR DIE INTERPRETATION UND AKZEPTANZ DER ERGEBNISSE
54. Im Allgemeinen ist das Ergebnis einer Prüfung auf östrogene Wirkung dann als positiv anzusehen, wenn zumindest bei der höchsten Dosisstufe im Vergleich zu der nur mit dem Lösungsmittel behandelten Kontrollgruppe eine statistisch signifikante Erhöhung des Uterusgewichts (p < 0,05) zu verzeichnen ist. Ein positives Testergebnis wird ferner durch den Nachweis einer biologisch plausiblen Beziehung zwischen Dosis und Wirkstärke untermauert, wobei zu beachten ist, dass eine Prüfsubstanz sowohl östrogene als auch antiöstrogene Wirkungen haben kann, die sich gegenseitig überlagern, was sich unter Umständen auf den Verlauf der Dosis-Wirkungs-Kurve auswirkt.
55. Es ist darauf zu achten, dass die maximal verträgliche Dosis nicht überschritten wird, weil die Interpretation der Daten sonst nicht aussagekräftig ist. Vor diesem Hintergrund sind ein verringertes Körpergewicht, klinische Anzeichen und andere Befunde sorgfältig zu prüfen.
56. Ein wesentliches Kriterium für die Akzeptanz der bei dem uterotrophen Bioassay gewonnenen Daten sind die Uterusgewichte der Vehikelkontrollgruppe. Hohe Kontrollwerte können die Empfindlichkeit des Bioassays und die Fähigkeit zur Feststellung sehr schwacher Östrogenagonisten beeinträchtigen. Die Auswertung der Fachliteratur und die im Laufe der Validierung des uterotrophen Bioassays erhobenen Daten legen die Vermutung nahe, dass hohe Durchschnittswerte bei Kontrollgruppen, insbesondere wenn unreife Tiere verwendet werden, spontan vorkommen können (2)(3)(6)(9). Da das Uterusgewicht bei unreifen Ratten von zahlreichen Variablen abhängig ist — wie zum Beispiel von Stamm und Körpergewicht —, kann keine allgemeingültige Obergrenze für das Uterusgewicht angegeben werden. Als Faustregel gilt, dass Uterustrockengewichte bei unreifen Kontrollratten zwischen 40 und 45 mg als verdächtig einzustufen sind, und dass Uterusgewichte über 45 mg Anlass zu einer Wiederholung der Prüfung geben können. Dies muss jedoch von Fall zu Fall entschieden werden (3)(6)(8). Wenn bei adulten Ratten die Ovarektomie nicht vollständig durchgeführt wurde, führt das ovariale Restgewebe unter Umständen zu einer endogenen Östrogenproduktion, was eine Verzögerung bei der Rückbildung des Uterusgewichts zur Folge hat.
57. Bei der Vehikelkontrollgruppe scheint ein Uterustrockengewicht von unter 0,09 % des Körpergewichts unreifer Rattenweibchen und von unter 0,04 % des Körpergewichts ovarektomierter junger adulter Weibchen akzeptable Ergebnisse zu liefern [siehe Tabelle 31 (2)]. Liegen die Uterusgewichte der Kontrollgruppe über diesen Werten, sind verschiedene Faktoren, darunter das Alter der Tiere, die vollständige Entfernung der Ovarien, Phytoöstrogene im Futter usw. zu überprüfen, und negative Prüfergebnisse (also kein Anzeichen für östrogene Wirkung) mit Vorsicht zu verwenden.
58. Historische Daten der Vehikelkontrollgruppen sollten vom Labor in dessen Datenbestand aufgezeichnet werden. Die historischen Daten zur Wirkung positiver Referenzöstrogene, zum Beispiel 17a-Ethinylestradiol, sind ebenfalls vom Labor einzupflegen. Die Labore können ferner die Reaktion auf bekannte schwache Östrogenagonisten testen. Alle diese Daten können mit Daten aus der Fachliteratur (2)(3)(4)(5)(6)(7)(8) abgeglichen werden. So kann sichergestellt werden, dass die Methoden des Labors in ausreichendem Maße empfindlich sind.
59. Die Uterustrockengewichte zeigten im Laufe der Validierungsprüfung der OECD eine geringere Variabilität als die Uterusfeuchtgewichte (6)(7). Eine signifikante Reaktion bei einer der Bestimmungsarten würde allerdings auf ein positives Testergebnis, also eine Östrogenwirkung der Prüfsubstanz, hindeuten.
60. Die uterotrophe Reaktion ist nicht ausschließlich östrogenen Ursprungs. Ein positives Testergebnis des uterotrophen Bioassays sollte jedoch allgemein als Nachweis für ein östrogenes Potenzial im lebenden Organismus gewertet werden und normalerweise den Anstoß für weitere Abklärungsmaßnahmen liefern (siehe Nummer 9 und OECD Conceptual Framework for the Testing and Assessment of Endocrine Disrupting Chemicals, Anlage 2).
Abbildung 1
Schematische Darstellung der operativen Entfernung der Ovarien

Die Operation beginnt mit der Eröffnung der dorsolateralen Bauchwand in der Mitte zwischen unterem Rippenbogenrand und Beckenkamm sowie einige Millimeter neben dem lateralen Rand des Lendenmuskels. Die Ovarien sind innerhalb der Bauchhöhle zu lokalisieren. Anschließend werden die Ovarien jeweils auf einem sterilen Feld aus der Bauchhöhle vorgelagert, der Bereich zwischen Ovar und Uterus wird zur Blutstillung ligiert, und die Ovarien werden jeweils oberhalb der Ligatur an der Verbindungsstelle zwischen Ovidukt und Uterushorn abgeschnitten. Wenn sichergestellt ist, dass keine starke Blutung mehr besteht, ist die Bauchhöhle zuzunähen und die Haut mit Klammern oder einer Naht zu schließen. Die Tiere sollten sich, bevor sie verwendet werden, mindestens 14 Tage lang erholen können; in dieser Zeit kann sich auch der Uterus zurückbilden.
Abbildung 2
Entfernung und Vorbereitung der Uterusgewebe für die Gewichtsbestimmung

Die Sektion beginnt mit der Eröffnung der Bauchwand ab der Schambeinfuge. Anschließend werden die Ovarien (sofern vorhanden) und die Uterushörner von der dorsalen Bauchwand gelöst. Harnblase und Harnleiter werden von der ventralen und lateralen Seite des Uterus und der Vagina entfernt. Die fibröse Adhäsion zwischen Rektum und Vagina wird gelöst, bis die Verbindung zwischen Vaginalöffnung und Perinealhaut erkennbar ist. Uterus und Vagina werden vom Körper getrennt, indem die Vaginalwand genau oberhalb der Verbindung zur Perinealhaut abgeschnitten wird (siehe Abbildung). Der Uterus sollte durch vorsichtiges Abtrennen des Mesometriums an der Stelle, an der es jeweils dorsolateral mit der Längsseite eines Uterushorns verbunden ist, von der Körperwand gelöst werden. Nach Entfernung aus dem Körper ist überschüssiges Fett- und Bindegewebe abzuschaben. Wenn Ovarien vorhanden sind, werden sie so am Ovidukt entfernt, dass keine Gebärmutterflüssigkeit aus dem Uterushorn verloren geht. Bei ovarektomierten Tieren sind die Ovarialstümpfe auf Reste von Ovarialgewebe zu untersuchen. Die Vagina wird genau unterhalb des Gebärmutterhalses vom Uterus getrennt, sodass der Gebärmutterhals mit dem Uteruskörper verbunden bleibt. Der Uterus kann nun gewogen werden.
Anlage 1
BEGRIFFSBESTIMMUNGEN
Antiöstrogene Wirkung : die Fähigkeit einer Chemikalie, die Wirkung von Östradiol 17ß in einem Säugetierorganismus zu unterdrücken.
Chemikalie : ein Stoff oder eine Mischung.
Dosierung : ein allgemeiner Begriff, der die Dosis, ihre Häufigkeit und die Dauer der Verabreichung umfasst.
Dosis : die Menge der verabreichten Prüfsubstanz. Für die Zwecke des uterotrophen Bioassays wird die Dosis ausgedrückt als Masse der Prüfsubstanz je Einheit Körpergewicht des Versuchstiers pro Tag (z. B. mg pro kg Körpergewicht und Tag).
Empfindlichkeit : der Anteil aller positiven/wirkenden Chemikalien, die durch den Test korrekt eingestuft werden. Die Empfindlichkeit ist ein Maß für die Genauigkeit einer Prüfmethode mit kategorialen Ergebnissen und ein wichtiger Aspekt bei der Bewertung ihrer Relevanz.
Maximal verträgliche Dosis (MTD) : die höchstmögliche Dosis der Prüfsubstanz, die nach Einbringen in den Körper nicht zum Tod von Versuchstieren führt (bezeichnet als LD0) (IUPAC, 1993).
Östrogene Wirkung : die Fähigkeit einer Chemikalie, in einem Säugetierorganismus wie Östradiol 17ß zu wirken.
Postnataler Tag X : der x-te Lebenstag nach der Geburt.
Prüfsubstanz : jede(r) mittels dieser Prüfmethode getestete Stoff bzw. Mischung.
Spezifizität : der Anteil aller negativen/wirkungslosen Stoffe, die durch den Test korrekt eingestuft werden. Die Spezifizität ist ein Maß für die Genauigkeit einer Prüfmethode mit kategorialen Ergebnissen und ein wichtiger Aspekt bei der Bewertung ihrer Relevanz.
Tag der Geburt : der postnatale Tag 0.
Uterotroph : Begriff zur Beschreibung einer positiven Wirkung auf das Gewicht der Uterusgewebe.
Validierung : wissenschaftlicher Prozess zur Beschreibung der operationellen Anforderungen und Grenzen einer Prüfmethode und zum Nachweis ihrer Zuverlässigkeit und Eignung für einen bestimmten Zweck.
Anlage 2

ANMERKUNGEN ZUM RAHMENKONZEPT
|
Anmerkung 1: |
Ein Einstieg in das Rahmenkonzept und ein Ausstieg sind auf allen Stufen möglich und abhängig davon, welche Informationen für Gefahren- und Risikobeurteilungszwecke benötigt werden. |
|
Anmerkung 2: |
Bei Stufe 5 sollte der Aspekt der Ökotoxikologie Endpunkte beinhalten, die Hinweise auf schädliche Wirkmechanismen und eine potenzielle Schädigung der Population liefern. |
|
Anmerkung 3: |
Wenn ein multimodales Modell mehrere Assays mit nur einem Endpunkt (Single-Endpoint-Assays) umfasst, ersetzt dieses Modell die Durchführung der Single-Endpoint-Assays. |
|
Anmerkung 4: |
Die Bewertung der einzelnen Chemikalien sollte von Fall zu Fall und unter Berücksichtigung aller verfügbaren Daten sowie vor dem Hintergrund des Zwecks der Stufen des Rahmenkonzepts erfolgen. |
|
Anmerkung 5: |
Das Rahmenkonzept ist derzeit noch nicht als vollständig anzusehen. Auf Stufe 3, 4 und 5 beinhaltet es Assays, die entweder schon verfügbar sind oder sich gerade in der Validierungsphase befinden. Bei Letzteren ist die Aufnahme in das Rahmenkonzept noch vorläufig. Sobald ihre Entwicklung und Validierung abgeschlossen ist, werden sie offizieller Bestandteil des Konzepts. |
|
Anmerkung 6: |
Stufe 5 ist nicht so zu verstehen, dass es sich bei den darin enthaltenen Tests nur um endgültige Prüfungen handelt. Die Prüfungen dieser Stufe sollen vielmehr einen Beitrag zur allgemeinen Gefahren- und Risikobewertung leisten. |
LITERATUR
|
(1) |
OECD (1998). Report of the First Meeting of the OECD Endocrine Disrupter Testing and Assessment (EDTA) Task Force, 10.-11. März 1998, ENV/MC/CHEM/RA(98)5. |
|
(2) |
OECD (2003). Detailed Background Review of the Uterotrophic Bioassay: Summary of the Available Literature in Support of the Project of the OECD Task Force on Endocrine Disrupters Testing and Assessment (EDTA) to Standardise and Validate the Uterotrophic Bioassay. OECD Environmental Health and Safety Publication Series on Testing and Assessment No. 38. ENV/JM/MONO(2003)1. |
|
(3) |
Owens JW, Ashby J. (2002). Critical Review and Evaluation of the Uterotrophic Bioassay for the Identification of Possible Estrogen Agonists and Antagonists: In Support of the Validation of the OECD Uterotrophic Protocols for the Laboratory Rodent. Crit. Rev. Toxicol. 32:445-520. |
|
(4) |
OECD (2006). OECD Report of the Initial Work Towards the Validation of the Rodent Uterotrophic Assay — Phase 1. OECD Environmental Health and Safety Publication Series on Testing and Assessment No. 65. ENV/JM/MONO(2006)33. |
|
(5) |
Kanno, J, Onyon L, Haseman J, Fenner-Crisp P, Ashby J, Owens W. (2001). The OECD program to validate the rat uterotrophic bioassay to screen compounds for in vivo estrogenic responses: Phase 1. Environ Health Perspect. 109:785-94. |
|
(6) |
OECD (2006). OECD Report of the Validation of the Rodent Uterotrophic Bioassay: Phase 2 — Testing of Potent and Weak Oestrogen Agonists by Multiple Laboratories. OECD Environmental Health and Safety Publication Series on Testing and Assessment No. 66. ENV/JM/MONO(2006)34. |
|
(7) |
Kanno J, Onyon L, Peddada S, Ashby J, Jacob E, Owens W. (2003). The OECD program to validate the rat uterotrophic bioassay: Phase Two — Dose Response Studies. Environ. Health Persp.111:1530-1549 |
|
(8) |
Kanno J, Onyon L, Peddada S, Ashby J, Jacob E, Owens W. (2003). The OECD program to validate the rat uterotrophic bioassay: Phase Two — Coded Single Dose Studies. Environ. Health Persp.111:1550-1558. |
|
(9) |
Owens W, Ashby J, Odum J, Onyon L. (2003). The OECD program to validate the rat uterotrophic bioassay: Phase Two — Dietary phytoestrogen analyses. Environ. Health Persp. 111:1559-1567. |
|
(10) |
Ogasawara Y, Okamoto S, Kitamura Y, Matsumoto K. (1983). Proliferative pattern of uterine cells from birth to adulthood in intact, neonatally castrated, and/or adrenalectomized mice assayed by incorporation of [I125]iododeoxyuridine. Endocrinology 113:582-587. |
|
(11) |
Branham WS, Sheehan DM, Zehr DR, Ridlon E, Nelson CJ. (1985). The postnatal ontogeny of rat uterine glands and age-related effects of 17b-estradiol. Endocrinology 117:2229-2237. |
|
(12) |
Schlumpf M, Berger L, Cotton B, Conscience-Egli M, Durrer S, Fleischmann I, Haller V, Maerkel K, Lichtensteiger W. (2001). Estrogen active UV screens. SÖFW-J. 127:10-15. |
|
(13) |
Zarrow MX, Lazo-Wasem EA, Shoger RL. (1953). Estrogenic activity in a commercial animal ration. Science, 118:650-651. |
|
(14) |
Drane HM, Patterson DSP, Roberts BA, Saba N. (1975). The chance discovery of oestrogenic activity in laboratory rat cake. Fd. Cosmet. Toxicol. 13:425-427. |
|
(15) |
Boettger-Tong H, Murphy L, Chiappetta C, Kirkland JL, Goodwin B, Adlercreutz H, Stancel GM, Makela S. (1998). A case of a laboratory animal feed with high estrogenic activity and its impact on in vivo responses to exogenously administered estrogens. Environ. Health Perspec.106:369-373. |
|
(16) |
OECD (2007). Additional data supporting the Test Guideline on the Uterotrophic Bioassay in rodents. OECD Environmental Health and Safety Publication Series on Testing and Assessment No. 67. |
|
(17) |
Degen GH, Janning P, Diel P, Bolt HM. (2002). Estrogenic isoflavones in rodent diets. Toxicol. Lett. 128:145-157. |
|
(18) |
Wade MG, Lee A, McMahon A, Cooke G, Curran I. (2003). The influence of dietary isoflavone on the uterotrophic response in juvenile rats. Food Chem. Toxicol. 41:1517-1525. |
|
(19) |
Yamasaki K, Sawaki M, Noda S, Wada T, Hara T, Takatsuki M. (2002). Immature uterotrophic assay of estrogenic compounds in rats given different phytoestrogen content diets and the ovarian changes in the immature rat uterotrophic of estrogenic compounds with ICI 182,780 or antide. Arch. Toxicol. 76:613-620. |
|
(20) |
Thigpen JE, Haseman JK, Saunders HE, Setchell KDR, Grant MF, Forsythe D. (2003). Dietary phytoestrogens accelerate the time of vaginal opening in immature CD-1 mice. Comp. Med. 53:477-485. |
|
(21) |
Ashby J, Tinwell H, Odum J, Kimber I, Brooks AN, Pate I, Boyle CC. (2000). Diet and the aetiology of temporal advances in human and rodent sexual development. J. Appl. Toxicol.20:343-347. |
|
(22) |
Thigpen JE, Lockear J, Haseman J, Saunders HE, Caviness G, Grant MF, Forsythe DB. (2002). Dietary factors affecting uterine weights of immature CD-1 mice used in uterotrophic bioassays. Cancer Detect. Prev. 26:381-393. |
|
(23) |
Thigpen JE, Li L-A, Richter CB, Lebetkin EH, Jameson CW. (1987). The mouse bioassay for the detection of estrogenic activity in rodent diets: I. A standardized method for conducting the mouse bioassay. Lab. Anim. Sci.37:596-601. |
|
(24) |
OECD (2008). Acute oral toxicity — up-and-down procedure. OECD Guideline for the testing of chemicals No 425. |
|
(25) |
OECD (2000). Guidance document on the recognition, assessment and use of clinical signs as humane endpoints for experimental animals used in safety evaluation. Environmental Health and Safety Monograph Series on Testing and Assessment No 19. ENV/JM/MONO(2000)7. |
|
(26) |
OECD (2001). Guidance document on acute oral toxicity. Environmental Health and Safety Monograph Series on Testing and Assessment No 24. ENV/JM/MONO(2001)4. |
|
(27) |
Bulbring, E., and Burn, J.H. (1935). The estimation of oestrin and of male hormone in oily solution. J. Physiol. 85:320-333. |
|
(28) |
Dorfman, R.I., Gallagher, T.F. and Koch, F.C (1936). The nature of the estrogenic substance in human male urine and bull testis. Endocrinology 19:33-41. |
|
(29) |
Reel, J.R., Lamb IV, J.C. and Neal, B.H. (1996). Survey and assessment of mammalian estrogen biological assays for hazard characterization. Fundam. Appl. Toxicol. 34:288-305. |
|
(30) |
Jones, R.C. and Edgren, R.A. (1973). The effects of various steroid on the vaginal histology in the rat. Fertil. Steril. 24:284-291. |
|
(31) |
OECD (1982). Organization for Economic Co-operation and Development — Principles of Good Laboratory Practice, ISBN 92-64-12367-9, Paris. |
|
(32) |
Dorfman R.I. (1962). Methods in Hormone Research, Vol. II, Part IV: Standard Methods Adopted by Official Organization. New York, Academic Press. |
|
(33) |
Thigpen J.E. et al. (2004). Selecting the appropriate rodent diet for endocrine disruptor research and testing studies. ILAR J 45(4):401-416. |
|
(34) |
Gray L.E. and Ostby J. (1998). Effects of pesticides and toxic substances on behavioral and morphological reproductive development: endocrine versus non-endocrine mechanism. Toxicol Ind Health. 14(1-2):159-184. |
|
(35) |
Booth AN, Bickoff EM and Kohler GO. (1960). Estrogen-like activity in vegetable oils and mill by-products. Science, 131:1807-1808. |
|
(36) |
Kato H, Iwata T, Katsu Y, Watanabe H, Ohta Y, Iguchi T (2004). Evaluation of estrogenic activity in diets for experimental animals using in vitro assay. J. Agric Food Chem. 52, 1410-1414. |
|
(37) |
OECD (2007). Guidance Document on the Uterotrophic Bioassay Procedure to Test for Antioestrogenicity. Series on Testing and Assessment. No. 71. |
|
(38) |
Richtlinie 2010/63/EU des Europäischen Parlaments und des Rates vom 22. September 2010 zum Schutz der für wissenschaftliche Zwecke verwendeten Tiere (ABl. L 276 vom 20.10.2010, S. 33). |
B.55. HERSHBERGER-BIOASSAY MIT RATTEN: EIN KURZZEIT-SCREENING-TEST AUF (ANTI-)ANDROGENE EIGENSCHAFTEN
EINLEITUNG
1. Diese Prüfmethode entspricht der OECD-Prüfrichtlinie (TG) 441 (2009). Die OECD setzte sich 1998 zur Priorität, bestehende Prüfrichtlinien für Screening und Testung potenzieller endokriner Disruptoren zu überarbeiten und neue Richtlinien zu entwickeln (1). Ein Aspekt der Maßnahme war die Ausarbeitung einer Prüfrichtlinie für den Hershberger-Bioassay (Hershberger-Test) mit Ratten. Diese Prüfung wurde, nachdem sie über mehrere Jahrzehnte hinweg von der pharmazeutischen Industrie angewendet worden war, erstmals 1962 durch einen offiziellen Fachausschuss als Screening-Instrument für androgene Chemikalien standardisiert (2). Im Zeitraum 2001-2007 wurde der Hershberger-Test mit Ratten einem umfassenden Validierungsprogramm unterzogen, in dessen Rahmen ein Hintergrunddokument (23), ein ausführliches Methodikpapier (3) und ein Sektionsleitfaden (21) erstellt sowie umfassende Intra- und Interlaborstudien durchgeführt wurden, um die Zuverlässigkeit und Reproduzierbarkeit des Bioassays nachzuweisen. Diese Validierungsstudien wurden mit einem starken Referenzandrogen (Testosteronpropionat, TP), zwei starken synthetischen Androgenen (Trenbolonacetat und Methyltestosteron), einem stark antiandrogenen Arzneistoff (Flutamid), einem starken Inhibitor (Finasterid) der Synthese des natürlichen Androgens (Dihydrotestosteron, DHT), mehreren schwach antiandrogenen Pestiziden (Linuron, Vinclozolin, Procymidon, p,p′-DDE), einem starken Inhibitor der 5α-Reduktase (Finasterid) und zwei bekannt negativen Chemikalien (Dinitrophenol und Nonylphenol) (4)(5)(6)(7)(8) durchgeführt. Die vorliegende Prüfmethode ist das Resultat der jahrzehntelangen Anwendung des Bioassays in der Praxis und der bei der Validierung des Prüfprogramms gewonnenen Erfahrungen und Ergebnisse auf diesem Gebiet.
2. Der Hershberger-Test ist ein in vivo durchgeführter Kurzzeit-Screening-Test, bei dem akzessorische Gewebe des männlichen Reproduktionstrakts untersucht werden. Der Test wurde erstmals in den 1930er-Jahren durchgeführt und in den 1940er-Jahren um die Anwendung auf Muskeln des männlichen Fortpflanzungstrakts erweitert, die auf Androgene reagieren (2)(9-15). In den 1960er-Jahren wurden über 700 potenzielle Androgene anhand eines standardisierten Prüfplans (2)(14) beurteilt; die Verwendung des Tests galt damals sowohl für Androgene als auch für Antiandrogene als Standardmethode (2)(15). Bei der vorliegenden In-vivo-Prüfung werden die Gewichtsveränderungen bei fünf Arten androgenabhängiger Gewebe an kastrierten, peripubertären Rattenmännchen untersucht. Es wird beurteilt, ob eine Chemikalie biologische Abläufe auslöst, die der Wirkung von Androgenagonisten, Androgenantagonisten oder 5α-Reduktaseinhibitoren entsprechen. Bei den fünf androgenabhängigen Zielgeweben der vorliegenden Prüfmethode handelt es sich um die ventrale Prostata (VP), das Samenbläschen (SB) (einschließlich Flüssigkeiten und Koagulationsdrüse), den Muskelkomplex Musculus levator ani und bulbospongiosus (LABC), die paarigen Cowperschen Drüsen (COW) und die Glans penis (GP). Bei der kastrierten, peripubertären männlichen Ratte reagieren alle diese fünf Gewebe mit einer Erhöhung ihres absoluten Gewichts auf Androgene. Wird bei denselben fünf Geweben eine Gewichtszunahme durch Verabreichung eines starken Referenzandrogens stimuliert, so reagieren diese auf die Gabe von Antiandrogenen mit einer Verringerung ihres absoluten Gewichts. Das primäre Modell für den Hershberger-Test, das in den Phasen 1, 2 und 3 des Hershberger-Validierungsprogramms validiert wurde, war das chirurgisch kastrierte, peripubertäre Rattenmännchen.
3. Der Hershberger-Bioassay wird als mechanistischer In-vivo-Screening-Test auf Androgenagonisten, Androgenantagonisten und 5a-Reduktaseinhibitoren eingesetzt und ist im Kontext des Rahmenkonzepts der OECD ‚Conceptual Framework for the Testing and Assessment of Endocrine Disrupting Chemicals‘ (Testung und Bewertung endokrin wirksamer Substanzen (endokrine Disruptoren) (Anlage 2) zu sehen. In diesem Rahmenkonzept ist der Hershberger-Test ein In-vivo-Test der Stufe 3, mit dem Daten über einen einzelnen endokrinen Mechanismus gewonnen werden sollen, nämlich die (anti-)androgene Wirkung. Die Prüfung ist als Teil einer Reihe von In-vitro- und In-vivo-Tests zur Bestimmung von Chemikalien konzipiert, die das Potenzial haben, mit dem endokrinen System in Wechselwirkung zu treten, und soll letztendlich der Bewertung des Risikos für die Umwelt und die menschliche Gesundheit dienen.
4. Da aus Tierschutzgründen Bedenken gegen das Kastrationsverfahren bestanden, wurde als alternatives Modell für den Hershberger-Test die Verwendung des intakten (unkastrierten) abgesetzten, stimulierten männlichen Jungtiers angestrebt, damit die Kastration vermieden werden kann. Die Prüfmethode mit dem stimulierten, abgesetzten Tier wurde einer Validierung unterzogen (24), bei der sich allerdings herausstellte, dass diese Variante des Hershberger-Tests offenbar nicht geeignet ist, die Auswirkungen schwach wirksamer Antiandrogene in den geprüften Dosierungen auf das Gewicht androgenabhängiger Organe zuverlässig nachzuweisen. Daher wurde sie nicht in die vorliegende Prüfmethode aufgenommen. Da diese Prüfvariante jedoch nicht nur unter Tierschutzgesichtspunkten sinnvoll ist, sondern möglicherweise auch Aufschluss über andere Wirkungsweisen geben kann, ist sie im OECD Guidance Document Nr. 115 (25) beschrieben.
AUSGANGSÜBERLEGUNGEN UND BEGRENZUNGEN
5. Androgenagonisten und -antagonisten können an die Androgenrezeptoren binden und die vom Rezeptor gesteuerte Gentranskription aktivieren bzw. hemmen. Darüber hinaus hemmen manche Chemikalien bei einigen androgenempfindlichen Zielgeweben die Umwandlung von Testosteron in das stärker wirksame natürliche Androgen Dihydrotestosteron (5a-Reduktasehemmer). Solche Chemikalien sind potenziell gesundheitsschädlich und können unter anderem negative Auswirkungen auf Reproduktion und Entwicklung haben. Aus diesem Grund müssen Chemikalien möglichst schnell daraufhin geprüft und beurteilt werden können, ob es sich um potenzielle Androgenagonisten, -antagonisten oder 5a-Reduktaseinhibitoren handelt. Die Affinität eines Liganden für einen Androgenrezeptor, die in vitro durch Rezeptorbindungstests oder transkriptionelle Aktivierung von Reportergenen bestimmt werden kann, liefert zwar wertvolle Informationen, ist aber nur eine von mehreren Determinanten für das Gefahrenpotenzial. Andere Determinanten können die metabolische Aktivierung und Deaktivierung beim Eintritt in den Körper, die Verteilung auf die Zielgewebe und die Ausscheidung aus dem Körper sein. Daraus ergibt sich die Notwendigkeit, die potenzielle Wirkung einer Chemikalie in vivo unter einschlägigen Bedingungen und bei entsprechender Exposition zu prüfen. Die In-vivo-Beurteilung ist weniger kritisch, wenn die Eigenschaften der Chemikalie hinsichtlich Resorption, Verteilung, Metabolismus und Ausscheidung (ADME) bekannt sind. Androgenabhängige Gewebe reagieren mit schnellem und starkem Wachstum auf eine Stimulation mit Androgenen, was insbesondere für kastrierte, peripubertäre Rattenmännchen gilt. Die Verwendung von Nagerarten, insbesondere Ratten, ist zudem bei Toxizitätsstudien zur Beurteilung des Gefahrenpotenzials weit verbreitet. Daher ist die vorliegende Variante der Prüfmethode, bei der die fünf genannten Zielgewebe an kastrierten, peripubertären Ratten untersucht werden, für das In-vivo-Screening von Androgenagonisten, Androgenantagonisten und 5a-Reduktaseinhibitoren geeignet.
6. Dieser Prüfmethode liegen die Prüfpläne der OECD-Validierungsstudie zugrunde, sich in Intra- und Interlaborstudien als zuverlässig und wiederholbar erwiesen haben (4)(5)(6)(7)(8). Die Prüfmethode umfasst Nachweisverfahren sowohl für eine androgene als auch für eine antiandrogene Wirkung.
7. Obgleich die verschiedenen Laboratorien, die den Hershberger-Bioassay im Rahmen des Validierungsprogramms der OECD durchgeführt haben, relativ unterschiedliche TP-Dosen für den Nachweis von Antiandrogenen verwendeten (0,2 versus 0,4 mg pro kg und Tag, subkutane Injektion), unterschieden sich diese beiden Prüfplanvarianten in Bezug auf die Eignung für den Nachweis schwacher oder starker antiandrogener Wirkungen nur geringfügig. Selbstverständlich darf jedoch die TP-Dosis weder so hoch sein, dass die Wirkungen von schwachen Androgenrezeptor-Antagonisten (AR-Antagonisten) blockiert werden, noch so niedrig, dass die androgenen Gewebe selbst dann kaum Wachstum zeigen, wenn nicht gleichzeitig Antiandrogene verabreicht werden.
8. Die Wachstumsreaktion der einzelnen androgenabhängigen Gewebe ist nicht ausschließlich androgenen Ursprungs, d. h. auch Chemikalien, die keine Androgenagonisten sind, können das Gewicht bestimmter Gewebe beeinflussen. Wenn mehrere Gewebe gleichzeitig mit Wachstum reagieren, erhärtet das jedoch den Verdacht, dass ein überwiegend androgenspezifischer Mechanismus vorliegt. Beispielsweise können hohe Dosen starker Östrogene zu einer Gewichtszunahme der Samenbläschen führen; die anderen androgenabhängigen Gewebe des Tests reagieren jedoch nicht in ähnlicher Weise. Antiandrogene Chemikalien können entweder als Androgenrezeptorantagonisten oder als 5a-Reduktaseinhibitoren wirken. 5a-Reduktaseinhibitoren wirken unterschiedlich, weil die Umwandlung in das wirksamere Dihydrotestosteron von Gewebe zu Gewebe variiert. Antiandrogene, die die 5a-Reduktase hemmen, wie zum Beispiel Finasterid, wirken sich im Vergleich zu einem starken AR-Antagonisten wie Flutamid stärker auf die ventrale Prostata aus als auf andere Gewebe. Diese unterschiedliche Gewebereaktion kann für eine Differenzierung zwischen AR-vermittelter und 5a-Reduktase-vermittelter Wirkungsweise herangezogen werden. Ferner ist der Androgenrezeptor evolutionsbedingt mit den Rezeptoren anderer Steroidhormone verwandt, und einige andere Hormone können in hohen, supraphysiologischen Dosen die wachstumsfördernden Effekte von TP binden und ihnen entgegenwirken (13). Des Weiteren leuchtet es ein, dass ein verbesserter Steroidmetabolismus und eine darauf folgende Senkung des Testosterons im Serum das Wachstum androgenabhängiger Gewebe verringern kann. Jedes positive Ergebnis des Hershberger-Tests sollte daher normalerweise anhand eines WoE-Ansatzes beurteilt werden; dazu gehören In-vitro-Tests wie die AR- und ER-Bindungstests (ER — Östrogenrezeptor) und entsprechende Tests zur transkriptionellen Aktivierung oder die Ergebnisse anderer In-vivo-Tests, bei denen ähnliche androgene Zielgewebe untersucht werden, zum Beispiel der Test am pubertären Männchen, die 15-Tage-Untersuchung am intakten Männchen oder Studien mit wiederholter Gabe über 28 oder 90 Tage.
9. Die Erfahrung hat gezeigt, dass xenobiotische Androgene seltener vorkommen als xenobiotische Antiandrogene. Es ist daher davon auszugehen, dass der Hershberger-Bioassay überwiegend für das Screening von Antiandrogenen eingesetzt werden wird. Die Prüfung auf androgene Wirkungen wäre dennoch für steroidale oder steroidähnliche Chemikalien oder auch für Chemikalien zu empfehlen, bei denen die Prüfungen der Stufe 1 oder 2 des Rahmenkonzepts (Anlage 2) Hinweise auf ein androgenes Potenzial ergeben haben. Ebenso können bei Prüfungen der Stufe 5 unerwünschte Wirkungen beobachtet werden, die mit (anti-)androgenen Profilen assoziiert sind, sodass bewertet werden muss, ob eine Chemikalie eine endokrine Wirkungsweise hat.
10. Es wird darauf hingewiesen, dass bei allen tierexperimentellen Verfahren die örtlichen Standards der Versuchstierpflege einzuhalten sind; die folgenden Pflege- und Behandlungsbeschreibungen sind Mindestanforderungen; Vorrang vor diesen Standards haben lokale Bestimmungen, zum Beispiel die Richtlinie 2010/63/EU des Europäischen Parlaments und des Rates vom 22. September 2010 zum Schutz der für wissenschaftliche Zwecke verwendeten Tiere (26). Eine weiterführende Anleitung für tierschutzgerechte Behandlung von Versuchstieren ist in dem OECD Guidance Document (17) enthalten.
11. Wie bei allen Bioassays mit Versuchstieren ist sorgfältig abzuwägen, ob die Durchführung der Studie notwendig ist. Grundsätzlich sprechen die folgenden beiden Gründe für eine Durchführung:
|
— |
Es liegt ein hohes Expositionspotenzial (Stufe 1 des Rahmenkonzepts) vor, oder bei In-vitro-Tests (Stufe 2) werden Anzeichen für eine (anti-)androgene Wirkung festgestellt, aufgrund deren abgeklärt werden muss, ob solche Effekte auch in vivo vorkommen können. |
|
— |
Bei den In-vivo-Tests der Stufe 4 oder 5 werden Effekte festgestellt, die einer (anti-)androgenen Wirkung entsprechen; in diesem Fall sollte bei weiteren Untersuchungen die spezifische Wirkungsweise untersucht bzw. bestimmt werden, ob die Effekte auf einen (anti-)androgenen Mechanismus zurückzuführen sind. |
12. Die für die Zwecke dieser Prüfmethode verwendeten Begriffe sind in Anlage 1 definiert.
PRINZIP DER PRÜFMETHODE
13. Die Empfindlichkeit des Hershberger-Bioassays beruht auf der Verwendung männlicher Tiere mit minimaler endogener Androgenproduktion. Dies wird durch den Einsatz kastrierter Männchen erreicht, denen nach der Kastration ausreichend Zeit eingeräumt wurde, damit die Zielgewebe sich wieder auf eine einheitliche Mindestgröße, die Baseline- bzw. Ausgangsgröße des Versuchs, zurückbilden können. Dadurch ist beim Screening auf ein androgenes Potenzial der Spiegel an zirkulierenden endogenen Androgenen niedrig, die Hypothalamus-Hypophysen-Gonaden-Achse kann dies nicht über Feedbackmechanismen kompensieren, die Reaktionsfähigkeit der Gewebe ist maximiert, und das Gewebegewicht bei Studienbeginn schwankt so wenig wie möglich. Wenn das antiandrogene Potenzial untersucht werden soll, kann durch die Stimulation mit einem Referenzandrogen eine konsistentere Gewebegewichtszunahme erreicht werden. Folglich sind beim Hershberger-Bioassay nur sechs Tiere pro Dosisgruppe erforderlich, während bei anderen Tests mit intakten pubertären oder adulten Männchen 15 Tiere pro Dosisgruppe verwendet werden sollten.
14. Die peripubertären Rattenmännchen werden in geeigneter Weise unter Verwendung zugelassener Narkosemittel und steriler Arbeitstechniken kastriert. In den ersten Tagen nach dem Eingriff sollten zur Vermeidung postoperativer Beschwerden Analgetika verabreicht werden. Durch eine Kastration wird die Genauigkeit der Prüfung verbessert: schwache Androgene und Antiandrogene sind leichter nachweisbar, weil kompensatorische endokrine Feedbackmechanismen ausgeschaltet werden, die beim intakten Tier die Wirkungen der verabreichten Androgene und Antiandrogene abschwächen können, und weil die starken Schwankungen der Serumtestosteronspiegel zwischen den einzelnen Tieren beseitigt werden. Demzufolge wird durch die Kastration die Anzahl der Tiere verringert, die für das Screening dieser endokrinen Wirkungen benötigt werden.
15. Beim Screening auf eine potenzielle androgene Wirkung ist die Prüfsubstanz täglich über einen Zeitraum von zehn aufeinanderfolgenden Tagen per Schlundsonde oder durch subkutane Injektion zu verabreichen. Es sind mindestens zwei Versuchstiergruppen mit den Prüfsubstanzen zu behandeln, wobei pro Gruppe eine Dosisstufe getestet wird. Etwa 24 Stunden nach der letzten Dosisgabe werden die Tiere seziert. Eine statistisch signifikante Gewichtszunahme bei mindestens zwei Zielorganen der Prüfsubstanzgruppen im Vergleich zur Vehikelkontrollgruppe ist ein Hinweis darauf, dass die Prüfsubstanz potenziell eine androgene Wirkung hat (vgl. Nummer 60). Androgene wie Trenbolon, die nicht durch die 5a-Reduktase umgewandelt werden können, haben ausgeprägtere Wirkungen auf den LABC-Komplex und die GP als TP, doch sollte bei allen Geweben eine Gewichtszunahme zu verzeichnen sein.
16. Beim Screening auf eine potenzielle antiandrogene Wirkung ist die Prüfsubstanz täglich über einen Zeitraum von zehn aufeinanderfolgenden Tagen per Schlundsonde oder durch subkutane Injektion zu verabreichen, und zwar bei gleichzeitiger subkutaner Gabe von TP (0,2 bzw. 0,4 mg pro kg und Tag). Im Laufe des Validierungsverfahrens wurde festgelegt, dass entweder 0,2 oder 0,4 mg TP pro kg und Tag gegeben werden können, da Antiandrogene mit beiden Dosen wirksam nachgewiesen werden konnten und deshalb nur eine der beiden Dosen zur Verwendung bei der Prüfung ausgewählt werden sollte. Die abgestuften Dosen der Prüfsubstanz werden mindestens drei Behandlungsgruppen verabreicht, wobei pro Gruppe eine Dosisstufe getestet wird. Etwa 24 Stunden nach der letzten Dosisgabe werden die Tiere seziert. Eine statistisch signifikante Gewichtsabnahme bei mindestens zwei Zielorganen der Gruppen, die die Prüfsubstanz plus TP erhalten haben, im Vergleich zur Kontrollgruppe, die nur TP erhalten hat, ist ein Hinweis darauf, dass die Prüfsubstanz potenziell eine antiandrogene Wirkung hat (vgl. Nummer 61).
BESCHREIBUNG DER METHODE
Auswahl der Tierart und des Stamms
17. Ratten werden seit den 1930er-Jahren routinemäßig für den Hershberger-Bioassay verwendet. Obwohl aus biologischer Sicht plausibel ist, dass Ratten und Mäuse ähnliche Reaktionen zeigen würden, ist die Ratte nach 70 Jahren Erfahrung mit dem Rattenmodell die Spezies der Wahl für den Hershberger-Test. Da die anhand des Hershberger-Tests erhobenen Daten gegebenenfalls später als Vorstudie für eine mehrere Generationen umfassende Langzeitstudie dienen, können somit ferner Tiere derselben Art, desselben Stamms und derselben Herkunft für beide Studienarten verwendet werden.
18. Laut dem vorliegenden Prüfplan können die Labore den für die Prüfung zu verwendenden Rattenstamm selbst auswählen, wobei es sich im Allgemeinen um den bereits in der Vergangenheit üblicherweise von dem teilnehmenden Labor verwendeten Stamm handeln sollte. Es können üblicherweise verwendete Laborrattenstämme eingesetzt werden; allerdings ist von der Verwendung von Stämmen abzusehen, bei denen die Geschlechtsreife wesentlich später als am 42. Lebenstag einsetzt, da die Kastration dieser Männchen am 42. Lebenstag die Gewichtsbestimmung der Glans Penis, die nur nach der Separation des Präputiums vom Penisschaft erfolgen kann, unmöglich machen kann. Aus diesem Grund sollten, außer in Ausnahmefällen, keine von der Fisher-344-Ratte abstammenden Stämme eingesetzt werden. Bei der Fisher-344-Ratte hat die sexuelle Entwicklung einen anderen zeitlichen Verlauf als bei anderen häufiger verwendeten Stämmen wie zum Beispiel Sprague Dawley oder Wistar (16). Wenn ein solcher Stamm verwendet werden soll, sollte das Labor die Tiere zu einem etwas späteren Zeitpunkt kastrieren und es sollte die Empfindlichkeit des eingesetzten Stamms belegen können. Das Labor muss die Wahl des Rattenstamms klar begründen. Dient der Screening-Test gegebenenfalls als Vorstudie zu einer Prüfung mit wiederholter oraler Gabe, einer Prüfung auf Reproduktions- und Entwicklungstoxizität oder einer Langzeitstudie, sollten für alle Prüfungen möglichst Tiere desselben Stamms und derselben Herkunft verwendet werden.
Haltungs- und Fütterungsbedingungen
19. Bei allen Verfahren sind die örtlichen Standards der Versuchstierpflege einzuhalten. Bei diesen Pflege- und Behandlungsbeschreibungen handelt es sich um Mindestanforderungen; Vorrang haben strengere lokale Rechtsvorschriften, wie zum Beispiel die Richtlinie 2010/63/EU des Europäischen Parlaments und des Rates vom 22. September 2010 zum Schutz der für wissenschaftliche Zwecke verwendeten Tiere (26). Die Temperatur im Versuchstierraum sollte 22 °C (± 3 °C) betragen. Die relative Luftfeuchtigkeit sollte mindestens 30 % betragen und — außer beim Reinigen des Raums — 70 % nicht überschreiten. Angestrebt werden sollte eine Luftfeuchtigkeit von 50-60 %. Die Beleuchtung sollte künstlich sein. Die Hell- und Dunkelphasen sollten sich im Abstand von 12 Stunden abwechseln.
20. Eine Unterbringung in der Gruppe ist aufgrund des geringen Alters der Tiere und der Tatsache, dass Ratten soziale Wesen sind, einer Haltung im Einzelkäfig vorzuziehen. Durch eine Unterbringung von zwei oder drei Tieren pro Käfig werden Enge und damit verbundener Stress vermieden, der sich wiederum auf die hormonelle Steuerung der Entwicklung der akzessorischen Gewebe des Reproduktionstrakts auswirken kann. Die Käfige sollten gründlich gereinigt werden, um etwaige Schad- und Schmutzstoffe zu beseitigen, und so angeordnet werden, dass etwaige Einflüsse der Käfigplatzierung minimiert werden. Eine Überbelegung ist durch angemessen große Käfige (~ 2 000 cm2) zu vermeiden.
21. Die Tiere sind einzeln und auf möglichst schmerzlose Weise zu kennzeichnen (z. B. durch eine Ohrmarke). Die Kennzeichnungsmethode ist zu protokollieren.
22. Labornahrung und Trinkwasser sind ad libitum bereitzustellen. Laboratorien, die den Hershberger-Bioassay durchführen, sollten die Labornahrung verfüttern, die sie üblicherweise bei Chemikalienprüfungen verwenden. Im Laufe der Validierungsstudien zu dem Bioassay traten keine Wirkungen oder Schwankungen auf, die der Nahrung zugeschrieben werden konnten. Die verwendete Nahrung ist zu protokollieren, und eine Probe des Laborfutters ist für potenzielle zukünftige Analysezwecke aufzubewahren.
Leistungskriterien für androgenabhängige Organgewichte
23. Während der Validierungsstudie gab es keinerlei Anzeichen dafür, dass sich eine Verringerung des Körpergewichts auf die Erhöhung oder Verringerung der Gewichte der Zielgewebe (also der im Laufe des Tests zu wiegenden Gewebe) auswirkt.
24. Bei den verschiedenen, im Rahmen des Validierungsprogramms erfolgreich eingesetzten Rattenstämmen waren die Gewichte der androgenabhängigen Organe bei den schwereren Rattenstämmen höher als bei den leichteren Stämmen. Daher beinhalten die Leistungskriterien des Hershberger-Bioassays keine absoluten erwarteten Organgewichte für die positiven und negativen Kontrollen.
25. Da der Variationskoeffizient (VK) für ein Gewebe umgekehrt proportional zur Teststärke ist, basieren die Leistungskriterien des Hershberger-Bioassays auf maximalen VK-Werten für jedes Gewebe (Tabelle 1). Die VK-Werte stammen aus den Validierungsstudien der OECD. Bei negativen Ergebnissen sollten die Laboratorien die VK der Kontrollgruppe und der Behandlungsgruppe mit der höchsten Dosis prüfen, um feststellen zu können, ob die maximalen VK-Leistungskriterien überschritten wurden.
26. Die Studie sollte wiederholt werden, wenn 1) mindestens drei der zehn möglichen Einzel-VK in der Kontroll- und in der Hochdosisgruppe die für Prüfungen auf Agonisten und Antagonisten in Tabelle 1 festgelegten Höchstwerte überschreiten und 2) wenn mindestens zwei Zielgewebe marginal nicht signifikant sind (d. h. r-Werte zwischen 0,05 und 0,10).
Tabelle 1
Maximal zulässige VK für akzessorische Zielgewebe des Reproduktionstrakts im Modell mit kastrierten Tieren laut OECD-Validierungsstudien (12)
|
Gewebe |
Antiandrogene Wirkungen |
Androgene Wirkungen |
|
Samenbläschen |
40 % |
40 % |
|
Ventrale Prostata |
40 % |
45 % |
|
LABC |
20 % |
30 % |
|
Cowpersche Drüsen |
35 % |
55 % |
|
Glans penis |
17 % |
22 % |
VERFAHREN
Konformität mit Vorschriften und Eignungsprüfung des Prüflaboratoriums
27. Anders als bei dem Uterotrophie-Test (Kapitel B.54 dieses Anhangs) ist ein Nachweis der Laborkompetenz vor Studienbeginn beim Hershberger-Test nicht notwendig, weil gleichzeitige Positivkontrollen (mit Testosteronpropionat und Flutamid) und Negativkontrollen integraler Bestandteil der Prüfung sind.
Zahl und Zustand der Versuchstiere
28. Die Behandlungs- und Kontrollgruppen bestehen jeweils aus mindestens sechs Tieren. Dies gilt sowohl für den Prüfplan zur Untersuchung androgener Wirkungen als auch für den zur Untersuchung antiandrogener Wirkungen.
Kastration
29. Den Tieren sollte nach ihrer Ankunft mehrere Tage Zeit gegeben werden, sich zu akklimatisieren, um sicherzugehen, dass sie gesund sind und gut gedeihen. Da Tiere, die vor dem 42. Lebenstag bzw. dem postnatalen Tag 42 (PND 42) kastriert werden, gegebenenfalls noch keine Präputialseparation zeigen, sollte die Kastration frühestens am PND 42 durchgeführt werden, nicht vorher. Die Tiere werden unter Narkose durch Aufschneiden des Skrotums und Entfernung beider Hoden und Nebenhoden kastriert; die Blutgefäße und Samenleiter werden ligiert. Wenn sichergestellt ist, dass keine Blutung besteht, sollte das Skrotum vernäht oder mit chirurgischen Klammern geschlossen werden. In den ersten Tagen nach dem Eingriff sollten die Tiere Analgetika erhalten, um die postoperativen Beschwerden zu lindern. Wenn bereits kastrierte Tiere von einem Versuchstieranbieter erworben werden, sind Alter und Stadium der Geschlechtsreife vom Anbieter zu bescheinigen.
Akklimatisierung nach der Kastration
30. Die Anpassung der Tiere an die Laborbedingungen sollte nach der Kastration mindestens sieben Tage lang fortgesetzt werden, damit sich das Gewicht der Zielorgane zurückbilden kann. Die Tiere sollten täglich beobachtet werden, und Tiere, die Anzeichen für eine Krankheit oder physische Abnormitäten zeigen, sind aus der Studie zu nehmen. Mit der Behandlung bzw. der ersten Dosisgabe (der Studie) kann somit zwischen PND 49 (frühester Termin) und PND 60 (spätester Termin) begonnen werden. Die Nekropsie sollte spätestens am Tag PND 70 durchgeführt werden. Diese Flexibilität lässt den Laboratorien genügend Raum für eine effiziente Versuchsplanung.
Körpergewicht und Randomisierung der Gruppen
31. Unterschiede beim individuellen Körpergewicht tragen zu Schwankungen der Gewebegewichte bei, und zwar sowohl innerhalb der Tiergruppen als auch zwischen den Gruppen. Verstärkte Gewebegewichtsschwankungen führen zu einem höheren Variationskoeffizienten (VK) und verringern die (manchmal auch als Testempfindlichkeit bezeichnete) Teststärke der Prüfung. Daher sollten Schwankungen des Körpergewichts sowohl experimentell als auch statistisch begrenzt werden.
32. Auf experimenteller Ebene sollte angestrebt werden, die Schwankungen der Körpergewichte sowohl innerhalb der Prüfgruppen als auch zwischen ihnen zu verringern. Erstens ist die Verwendung ungewöhnlich kleiner oder großer Tiere zu vermeiden. Diese sollten nicht in die Studienkohorte aufgenommen werden. Zu Beginn der Studie sollte das Gewicht der Versuchstiere nicht um mehr als ± 20 % des Durchschnittsgewichts schwanken (z. B. 175 g ± 35 g bei kastrierten, peripubertären Ratten). Zweitens sollten die Tiere durch randomisierte Gewichtsverteilung den Kontroll- und Behandlungsgruppen zugewiesen werden, sodass das durchschnittliche Körpergewicht der einzelnen Gruppen sich statistisch nicht von dem der anderen Gruppen unterscheidet. Das angewendete Block-Randomisierungsverfahren ist zu protokollieren.
33. Da Toxizität zu einer Abnahme des Körpergewichts bei den Behandlungsgruppen im Vergleich zur Kontrollgruppe führen kann, könnte statt des Körpergewichts bei Nekropsie das Körpergewicht am ersten Verabreichungstag der Prüfsubstanz als statistische Kovariate herangezogen werden.
Dosierung
34. Um festzustellen, ob eine Prüfsubstanz in vivo eine androgene Wirkung entfalten kann, sind normalerweise zwei Dosisgruppen mit Prüfsubstanz plus positive Kontrollgruppe und Vehikelkontrollgruppe (negative Kontrollgruppe) (siehe Nummer 43) ausreichend, weshalb dieser Versuchsanordnung aus Tierschutzgründen der Vorrang eingeräumt werden sollte. Wenn der Zweck des Versuchs entweder die Erstellung einer Dosis-Wirkungs-Kurve oder die Umrechnung auf niedrigere Dosen ist, sind mindestens drei Dosisgruppen erforderlich. Wenn Informationen gewonnen werden sollen, die über das Vorhandensein einer androgenen Wirkung hinausgehen (wie zum Beispiel eine Einschätzung der Wirkstärke), sollte ein anderer Dosierungsplan in Betracht gezogen werden. Bei einem Test auf antiandrogene Eigenschaften wird die Prüfsubstanz zusammen mit einem Referenz-Androgenagonisten verabreicht. Es sind mindestens drei Prüfgruppen mit verschiedenen Dosen der Prüfsubstanz sowie eine positive und eine negative Kontrollgruppe (vgl. Nummer 44) zu verwenden. Abgesehen von der Behandlung mit der Prüfsubstanz sollten die Tiere der Kontrollgruppe genauso behandelt werden wie die Tiere in den Prüfgruppen. Wird die Prüfsubstanz mit einem Vehikel verabreicht, erhält die Kontrollgruppe das Vehikel im höchsten bei den Prüfgruppen verwendeten Volumen.
35. Bei der Wahl der Dosisstufen sind sämtliche für die Prüfsubstanz oder verwandte Stoffe vorliegenden Daten zur Toxizität und (Toxiko-)Kinetik zu berücksichtigen. Bei der Festlegung der höchsten Dosisstufe sollten erstens der LD50-Wert und/oder Angaben zu einer akuten Toxizität berücksichtigt werden, um den Tod oder starkes Leiden und Qualen der Tiere zu vermeiden (17)(18)(19)(20), und zweitens sind Daten zu Prüfungen auf subchronische und chronische Toxizität heranzuziehen. Im Allgemeinen sollte die höchste Dosis nicht zu einer Verringerung des finalen Körpergewichts der Tiere um mehr als 10 % im Vergleich zur Kontrollgruppe führen. Die höchste Dosis sollte entweder 1) die höchste Dosis sein, bei der die Tiere auf jeden Fall überleben und nach Verabreichung an zehn aufeinanderfolgenden Tagen von bis zu 1 000 mg pro kg und Tag (Höchstdosis) (vgl. Nummer 36) keine signifikanten Toxizitäts- oder Leidenszeichen aufweisen, oder 2) eine Dosis, die (anti-)androgene Wirkungen hervorruft, je nachdem, welche der beiden Dosen niedriger ist. Für Screeningzwecke sind große Intervalle, z. B. eine halbe logarithmische Einheit (entsprechend einer Dosissteigerung um den Faktor 3,2) oder sogar eine volle logarithmische Einheit zwischen den Dosierungen akzeptabel. Liegen keine geeigneten Daten vor, kann unterstützend eine Dosisfindungsstudie (siehe Nummer 37) zur Bestimmung der zu verwendenden Dosen durchgeführt werden.
Grenzdosisstufe
36. Treten bei der Grenzdosis von 1 000 mg pro kg Körpergewicht und Tag und bei einer niedrigeren Dosis unter Anwendung der für diese Studie beschriebenen Verfahren keine statistisch signifikanten Änderungen bei den Gewichten der Reproduktionsorgane auf, so kann auf zusätzliche Dosisstufen gegebenenfalls verzichtet werden. Vom Konzept der Grenzdosis ist nur dann abzuweichen, wenn die Daten zur menschlichen Exposition darauf hindeuten, dass eine höhere Dosisstufe notwendig ist.
Ausführungen zur Dosisfindung
37. Sofern erforderlich können die geeigneten Dosisgruppen in einer Dosisfindungsstudie mit wenigen Tieren ausgewählt [unter Anwendung der Methoden für die Prüfung auf akute Toxizität (Kapitel B.1 bis und B.1 tris des vorliegenden Anhangs (27), OECD TG 425 (19))]. Das Ziel beim Hershberger-Test ist die Auswahl von Dosen, bei denen die Tiere auf jeden Fall überleben und nach zehn aufeinanderfolgenden Tagen mit einer Chemikaliengabe von maximal 1 000 mg pro Kilogramm und Tag (siehe Nummern 35 und 36) keine signifikante Toxizität oder sonstiges Leiden aufweisen. Für eine Definition der klinischen Toxizitäts- oder Leidenszeichen bei den Tieren wird auf das OECD Guidance Document (17) verwiesen. Sofern es im Rahmen dieser Dosisfindungsstudie nach zehn Verabreichungstagen praktikabel ist, können die Zielgewebe etwa 24 Stunden nach der letzten Dosisgabe operativ entfernt und gewogen werden. Diese Daten können dann bei der Dosiswahl für die Hauptstudie unterstützend herangezogen werden.
Referenzchemikalien und Vehikel
38. Als Referenzandrogenagonist sollte Testosteronpropionat (TP), CAS Nr. 57-82-5, dienen. Die TP-Dosierungsmenge kann entweder 0,2 mg pro kg Körpergewicht und Tag oder 0,4 mg pro kg Körpergewicht und Tag betragen. Als Referenzandrogenantagonist sollte Flutamid (FT), CAS Nr. 1311-84-7, verwendet werden. Die FT-Dosierungsmenge sollte 3 mg pro kg Körpergewicht und Tag betragen, und FT sollte gleichzeitig mit der TP-Dosierung verabreicht werden.
39. Als Erstes sollte möglichst immer die Verwendung einer wässrigen Lösung/Suspension in Erwägung gezogen werden. Da aber viele Androgenliganden beziehungsweise ihre metabolischen Vorläufern eher hydrophob sind, ist der gängigste Ansatz die Verwendung einer Lösung/Suspension auf Ölbasis (z. B. Mais-, Erdnuss-, Sesam- oder Olivenöl). Die Prüfsubstanzen können in einer sehr kleinen Menge 95 %igem Ethanol oder anderen geeigneten Lösungsmitteln gelöst und im Prüfvehikel auf ihre endgültigen Arbeitskonzentrationen verdünnt werden. Die toxischen Eigenschaften des Lösungsmittels sollten bekannt sein und in einer separaten Kontrollgruppe, in der nur das Lösungsmittel geprüft wird, getestet werden. Wenn die Prüfsubstanz als stabil erachtet wird, kann der Lösungsvorgang durch schonende Erwärmung und kräftige mechanische Einwirkung unterstützt werden. Die Stabilität der Prüfsubstanz im Vehikel sollte bestimmt werden. Ist die Prüfsubstanz für die Dauer der Studie stabil, kann eine Start-Aliquote der Substanz vorbereitet werden und die spezifizierten Dosierungsverdünnungen können täglich zubereitet werden, wobei darauf zu achten ist, dass die Proben nicht verunreinigt werden.
Verabreichung der Dosen
40. TP sollte durch subkutane Injektion verabreicht werden, FT über eine Schlundsonde.
41. Die Prüfsubstanz wird oral per Schlundsonde oder durch subkutane Injektion verabreicht. Bei der Wahl des Verabreichungswegs sind Tierschutzaspekte und die physikalisch-chemischen Eigenschaften der Prüfsubstanz zu berücksichtigen. Zusätzlich sind toxikologische Aspekte wie die Art der Humanexposition gegenüber der Chemikalie (z. B. Schlundsonde, um eine Aufnahme mit der Nahrung zu simulieren; subkutane Injektion, um Einatmen oder Aufnahme über die Haut am Modell zu testen) und die in der Fachliteratur genannten toxikologischen Angaben und Daten zu Verstoffwechselung und Kinetik (z. B. die Notwendigkeit, eine First-pass-Metabolisierung zu vermeiden; bessere Effizienz über einen bestimmten Verabreichungsweg) in die Planung mit einzubeziehen, bevor umfassende Langzeitprüfungen durchgeführt werden, wenn durch Injektion positive Ergebnisse erzielt werden.
42. Die Tiere sollten die Dosierung an zehn aufeinanderfolgenden Tagen jeweils auf die gleiche Art und Weise, in der gleichen zeitlichen Abfolge und im Abstand von jeweils etwa 24 Stunden erhalten. Die Dosierungsstufe ist täglich auf der Grundlage des ebenfalls täglich bestimmten Körpergewichts anzupassen. Für jeden Expositionstag sind Dosisvolumen und Verabreichungszeit zu notieren. Es ist sorgfältig darauf zu achten, dass die unter Nummer 35 genannte Höchstdosis nicht überschritten wird, weil die Interpretation der Daten sonst nicht aussagekräftig ist. Vor diesem Hintergrund sind ein verringertes Körpergewicht, klinische Anzeichen und andere Befunde sorgfältig zu prüfen. Wird die Prüfsubstanz über eine Sonde verabreicht, sollte eine Schlundsonde oder eine geeignete Intubationskanüle verwendet werden. Das maximale Flüssigkeitsvolumen, das jeweils verabreicht werden kann, hängt von der Größe des Versuchstiers ab. Es sind die örtlichen Tierschutzrichtlinien zu befolgen, das Volumen sollte jedoch 5 ml pro kg Körpergewicht nicht überschreiten, außer bei wässrigen Lösungen, von denen 10 ml pro kg Körpergewicht gegeben werden können. Die subkutanen Injektionen sind mittels steriler Nadel (z. B. 23 oder 25 Gauge) und Tuberkulinspritze dorsal im Bereich der Schulterblätter oder in die Lendenregion zu applizieren. Die Injektionsstelle kann rasiert werden. Wenn Injektionsflüssigkeit verloren geht, an der Injektionsstelle ausläuft oder unvollständig verabreicht wird, ist dies zu protokollieren. Das insgesamt pro Ratte und Tag injizierte Volumen sollte 0,5 ml pro kg Körpergewicht nicht überschreiten.
Spezifische Vorgehensweise für Androgenagonisten
43. Bei der Prüfung auf Androgenagonisten ist die Vehikelkontrollgruppe die negative Kontrollgruppe und die mit TP behandelte Gruppe die positive Kontrollgruppe. Für die Untersuchung auf biologische Abläufe, die der Wirkung von Androgenagonisten entsprechen, werden den Behandlungsgruppen an zehn aufeinanderfolgenden Tagen ausgewählte Dosen einer Prüfsubstanz verabreicht wird. Die Gewichte der fünf akzessorischen Gewebe des Reproduktionstrakts der Tiere der Prüfgruppen werden mit denen der Vehikelgruppe verglichen, um zu untersuchen, ob statistisch signifikante Gewichtserhöhungen feststellbar sind.
Spezifische Vorgehensweise für Androgenantagonisten und 5α-Reduktaseinhibitoren
44. Bei der Prüfung auf Androgenantagonisten und 5α-Reduktaseinhibitoren ist die mit TP behandelte Gruppe die negative Kontrollgruppe und die Gruppe, die gleichzeitig Referenzdosen von TP und FT erhält, ist die positive Kontrollgruppe. Für die Untersuchung auf biologische Abläufe, die der Wirkung von Androgenantagonisten und 5a-Reduktaseinhibitoren entsprechen, werden den Behandlungsgruppen an zehn aufeinanderfolgenden Tagen ausgewählte Dosen einer TP-Referenzdosis und einer Prüfsubstanz verabreicht. Die Gewichte der fünf akzessorischen Gewebe des Reproduktionstrakts der Tiere der Prüfgruppen, die TP plus die Prüfsubstanz erhalten haben, werden mit denen der Referenzgruppe verglichen, die nur TP erhalten hat, um zu untersuchen, ob statistisch signifikante Gewichtsverringerungen feststellbar sind.
BEOBACHTUNGEN
Klinische Beobachtungen
45. Allgemeine klinische Beobachtungen sollten mindestens einmal täglich durchgeführt werden, bei Anzeichen für Toxizität häufiger. Die Beobachtungen sind möglichst immer zur selben Tageszeit/zu denselben Tageszeiten und unter Berücksichtigung des Zeitraums nach der Verabreichung, in dem voraussichtlich die Wirkungsgipfel zu erwarten sind, durchzuführen. Alle Tiere sind auf Anzeichen von Mortalität, Morbidität und allgemeine klinische Zeichen wie Verhaltensänderungen, Änderungen an Haut, Fell, Augen und Schleimhäuten, Sekrete und Exkrete sowie autonome Körperfunktionen (z. B. Tränensekretion, Piloerektion, Pupillengröße, ungewöhnliche Atemmuster) zu beobachten.
46. Tot aufgefundene Tiere sind zu entfernen und ohne weitere Datenanalyse zu beseitigen. Vor der Nekropsie verendete Tiere sind mit den offenkundigen Todesursachen zu protokollieren. Moribunde Tiere sind auf humane Weise zu töten. Vor der Nekropsie in moribundem Zustand aufgefundene und anschließend getötete Tiere sind mit den offenkundigen Krankheitsursachen zu protokollieren.
Körpergewicht und Futteraufnahme
47. Alle Tiere sind täglich auf 0,1 g genau zu wiegen, wobei unmittelbar vor Behandlungsbeginn, also wenn die Tiere den Gruppen zugeteilt werden, mit dem Wiegen zu beginnen ist. Optional kann die aufgenommene Futtermenge während des Behandlungszeitraums pro Käfig durch Wiegen der Futterspender bestimmt werden. Die Daten zur Futteraufnahme sind in Gramm pro Ratte und Tag auszudrücken.
Sektion und Bestimmung der Gewebe- und Organgewichte
48. Die Ratten sollten etwa 24 Stunden nach der letzten Prüfsubstanzgabe nach der üblichen Vorgehensweise des durchführenden Labors getötet und ausgeblutet und anschließend seziert werden. Die humane Tötungsmethode ist im Laborbericht zu protokollieren.
49. Idealerweise werden die Nekropsien über die Gruppen hinweg randomisiert durchgeführt, um ein unmittelbares Abarbeiten der Dosisgruppen nach oben oder unten zu vermeiden, da dies die Daten geringfügig beeinflussen könnte. Alle Nekropsiebefunde, d. h. pathologische Veränderungen/sichtbare Läsionen, sind zu notieren und zu protokollieren.
50. Die fünf androgenabhängigen Gewebe (VP, SB, LABC, COW, GP) sind zu wiegen. Diese Gewebe sollten herausgeschnitten, sorgfältig von überschüssigem anhaftendem Gewebe und Fett befreit und ihr frisches (unfixiertes) Gewicht einzeln bestimmt werden. Jedes Gewebe ist mit besonderer Umsicht zu behandeln, um Flüssigkeitsverluste zu vermeiden und so ein Austrocknen zu verhindern, da dies zu niedrigeren Gewichten und damit zu signifikanten Fehlern und Schwankungen der dokumentierten Werte führen kann. Bestimmte Gewebe können sehr klein oder schwierig zu sezieren sein, was zwangsläufig zu Schwankungen führt. Daher sollte die Sektion der akzessorischen Gewebe des Reproduktionstrakts unbedingt von Personen durchgeführt werden, die mit den Standard-Sektionsverfahren für diese Gewebe vertraut sind. Eine Standardarbeitsanweisung (SOP) für Sektionen ist in Form eines Handbuchs bei der OECD erhältlich (21). Mit einer gründlichen Schulung nach diesem SOP-Handbuch lässt sich diese potenzielle Schwankungsquelle der Studie minimieren. Um eine unterschiedliche Aufbereitung der Gewebe zu vermeiden, sollte eine bestimmte Gewebeart immer von demselben Prosektor bearbeitet werden. Ist dies nicht möglich, sollte die Nekropsie so geplant werden, dass jeder Prosektor bei allen Behandlungsgruppen ein bestimmtes Gewebe seziert, und nicht so, dass eine Person alle Gewebe einer Kontrollgruppe seziert, während eine andere Person für die Gewebe der behandelten Gruppen zuständig ist. Jedes akzessorische Gewebe des Reproduktionstrakts ist ohne vorheriges Blotten auf 0,1 mg genau zu wiegen, und das Gewicht ist für jedes Tier einzeln zu dokumentieren.
51. Bestimmte Gewebe können sehr klein oder schwierig zu sezieren sein, was zwangsläufig zu Schwankungen führt. Frühere Arbeiten haben einen Bereich von Variationskoeffizienten (VK) ergeben, der anscheinend je nach Befähigung des Labors unterschiedlich ist. In einigen wenigen Fällen wurden große Unterschiede bei absoluten Gewebegewichten, zum Beispiel der VP und der COW, innerhalb ein und desselben Labors beobachtet.
52. Optional können die Gewichte von Leber, paarigen Nieren und paarigen Nebennieren bestimmt werden. Auch hier sind die Gewebe von anhaftendem Bindegewebe und Fett zu befreien. Die Leber ist zu wiegen und der Messwert auf 0,1 g genau zu dokumentieren; die paarigen Nieren und paarigen Nebennieren sind jeweils zu wiegen und die Messwerte auf 0,1 mg genau zu dokumentieren. Leber, Nieren und Nebennieren werden nicht nur durch Androgene beeinflusst, sie liefern auch nützliche Hinweise auf systemische Toxizität.
53. Eine Bestimmung des luteinisierenden Hormons (LH), des follikelstimulierenden Hormons (FSH) und des Testosterons (T) im Serum ist optional. Mithilfe der T-Spiegel im Serum kann bestimmt werden, ob die Prüfsubstanz eine Metabolisierung des Testosterons durch die Leber induziert, was zu niedrigeren Serumspiegeln führt. Ohne die T-Daten kann ein solcher Effekt auf einen antiandrogenen Mechanismus zurückzuführen sein. Die LH-Spiegel liefern Informationen darüber, ob ein Antiandrogen nicht nur in der Lage ist, Organgewichte zu verringern, sondern auch die hypothalamisch-hypophysäre Funktion zu beeinflussen, was in Langzeitstudien zu Hodentumoren führen kann. FSH ist ein wichtiges Hormon für die Spermatogenese. Bestimmungen von T4 und T3 im Serum sind ebenfalls optionale Messungen, die nützliche Zusatzhinweise über die Fähigkeit zur Störung der Schilddrüsenhormon-Homöostase liefern können. Wenn Hormonwerte bestimmt werden sollen, ist den Ratten vor der Nekropsie unter Narkose per Herzpunktion Blut zu entnehmen, wobei bei der Wahl der Narkosemethode darauf zu achten ist, dass sie die Hormonmessung nicht beeinflusst. Die Methode der Serumgewinnung, die Herkunft von Kits für Radioimmunassays (RIA) oder andere Messverfahren, die Analyseverfahren und die Ergebnisse sind zu dokumentieren. Die Serumspiegel von LH und T sind als ng/ml Serum zu dokumentieren.
54. Die nachstehend beschriebene Sektion der Gewebe basiert auf einem ausführlichen Sektionsleitfaden mit Fotos, der im Rahmen des Validierungsprogramms als zusätzliches Informationsmaterial veröffentlicht wurde (21). Darüber hinaus ist ein Video zur Sektion über die Website der koreanischen Nahrungs- und Arzneimittelbehörde erhältlich (22).
|
— |
Bei dem mit der ventralen Seite nach oben gelagerten Tier wird bestimmt, ob sich das Präputium des Penis von der Glans penis getrennt hat. Wenn das der Fall ist, wird das Präputium zurückgeschoben und die Glans penis entfernt, auf 0,1 mg genau gewogen und das Gewicht aufgezeichnet. |
|
— |
Die abdominale Haut und Bauchwand werden eröffnet und die Eingeweide freigelegt. Wenn die optionalen Organe gewogen werden sollen, ist die Leber zu entnehmen und auf 0,1 g genau zu wiegen; Magen und Eingeweide sind zu entnehmen, dann sind die paarigen Nieren und die paarigen Nebennieren zu entnehmen und auf 0,1 mg genau zu wiegen. Nach diesen Sektionsschritten ist die Blase freigelegt und die Sektion der akzessorischen männlichen Zielgewebe beginnt. |
|
— |
Für die Sektion der VP ist die Blase durch Schneiden des Bindegewebes entlang der Mittellinie von der ventralen Muskelschicht zu trennen. Die Blase ist nach anterior in Richtung der Samenbläschen (SB) zu verlagern, sodass der linke und der rechte Lappen der ventralen Prostata (die von einer Fettschicht bedeckt sind) freigelegt werden. Das Fett wird vorsichtig vom rechten und linken Lappen der VP gelöst. Der rechte Lappen der VP wird vorsichtig von der Harnröhre weggelagert und von der Harnröhre abgetrennt. Während der rechte Lappen der VP noch gehalten wird, wird vorsichtig der linke Lappen der VP von der Harnröhre weggelagert und abgeschnitten; das Gewicht wird auf 0,1 mg genau bestimmt und dokumentiert. |
|
— |
Um Samenbläschen und Koagulationsdrüse (SBKD) zu sezieren, wird die Blase nach kaudal verlagert und so der Samenleiter sowie der rechte und linke Lappen der Samenbläschen plus Koagulationsdrüsen freigelegt. Um Flüssigkeitsverluste zu vermeiden, ist an der Basis der SBKD, an der Stelle, an der der Samenleiter auf die Harnröhre trifft, eine Gefäßklemme zu befestigen. Die SBKD sind vorsichtig abzuschneiden, und Fett und Adnexe sind bei noch befestigter Gefäßklemme abzuschaben und in ein tariertes Wägeschiffchen zu legen. Anschließend wird die Gefäßklemme abgenommen, das Gewicht auf 0,1 mg genau bestimmt und das Ergebnis dokumentiert. |
|
— |
Um den Komplex Musculus levator ani und bulbospongiosus (LABC) zu sezieren, werden die Muskeln und die Penisbasis freigelegt. Die LA-Muskeln umschlingen das Kolon, während die anterioren LA- und BC-Muskeln mit dem Bulbus penis verbunden sind. Die Haut und Adnexe der Perianalregion, die sich von der Penisbasis zum anterioren Ende des Anus erstreckt, werden entfernt. Die BC-Muskeln werden nach und nach vom Bulbus penis und den Geweben freigelegt. Das Kolon wird durchgeschnitten, dann können die vollständigen LABC-Muskeln seziert und entnommen werden. Die LABC-Muskeln sind von Fett und Adnexen zu befreien, auf 0,1 mg genau zu wiegen, und die Ergebnisse sind festzuhalten. |
|
— |
Nach Entfernung der LABC-Muskeln sind die runden Cowperschen oder Bulbourethraldrüsen (COW) an der Basis und leicht dorsal des Bulbus penis sichtbar. Sie sind vorsichtig zu sezieren, um eine Beschädigung der dünnen Kapsel und damit einen Flüssigkeitsverlust zu vermeiden. Die paarigen COW sind auf 0,1 mg genau zu wiegen, und das Ergebnis ist festzuhalten. |
|
— |
Wenn während der Nekropsie bzw. des Sezierens Flüssigkeitsverluste aus einer Drüse auftreten, sind diese ebenfalls zu dokumentieren. |
55. Wenn zur Beurteilung der einzelnen Chemikalien mehr Tiere seziert werden müssen als pro Tag angemessen wäre, kann der Studienbeginn auf zwei aufeinanderfolgende Tage verteilt werden, sodass sowohl die Nekropsie als auch die damit zusammenhängenden Arbeiten auf zwei Tage verteilt werden. Bei einer derart gestaffelten Vorgehensweise ist pro Tag jeweils die Hälfte der Tiere pro Behandlungsgruppe zu verwenden.
56. Die Tierkörper sind nach der Nekropsie auf geeignete Weise zu entsorgen.
BERICHTERSTATTUNG
Daten
57. Die Daten sind einzeln zu protokollieren (d. h. Körpergewicht, Gewichte der akzessorischen Gewebe des Reproduktionstrakts, optionale Messungen sowie andere Wirkungen und Beobachtungen) und für jede Tiergruppe (Durchschnittswerte und Standardabweichungen aller Messungen) zu dokumentieren. Die Daten sind in tabellarischer Form zusammenzufassen. Aus den Daten muss Folgendes hervorgehen: Anzahl der Tiere zu Beginn der Prüfung, Anzahl der Tiere, die während der Prüfung tot aufgefunden werden oder die Toxizitätszeichen aufweisen, und eine Beschreibung der beobachteten Toxizitätszeichen einschließlich Zeitpunkt des Einsetzens, Dauer und Schweregrad.
58. Der Abschlussbericht sollte Folgendes enthalten:
Prüfeinrichtung
|
— |
Name und Ort der Einrichtung, |
|
— |
Prüfungsleiter und anderes Personal sowie Zuständigkeiten während der Prüfung, |
|
— |
Beginn und Ende der Prüfung, d. h. Datum der ersten Verabreichung der Prüfsubstanz, und Datum der letzten Nekropsie. |
Prüfsubstanz
|
— |
Herkunft, Los-/Chargennummer, Identität, Reinheit, vollständige Anschrift des Lieferanten und Eigenschaften der Prüfsubstanz(en), |
|
— |
physikalische Beschaffenheit und, sofern zutreffend, physikalisch-chemische Eigenschaften, |
|
— |
Lagerungsbedingungen und Methode und Häufigkeit der Verdünnungszubereitung, |
|
— |
etwaige Daten über die Stabilität, |
|
— |
etwaige Analysen der Dosierungslösungen/Suspensionen. |
Vehikel
|
— |
Beschreibung des Vehikels (Art, Lieferant und Chargennr.), |
|
— |
Begründung der Wahl des Vehikels (sofern anders als Wasser). |
Versuchstiere und Tierhaltungsverfahren
|
— |
Tierart und -stamm und Begründung der getroffenen Wahl, |
|
— |
Herkunft oder Lieferant der Versuchstiere mit vollständiger Anschrift, |
|
— |
Anzahl und Alter der gelieferten Tiere, |
|
— |
Haltungsbedingungen (Temperatur, Beleuchtung usw.) |
|
— |
Futter (Bezeichnung, Art, Lieferant, Chargennr., Inhalt und, sofern bekannt, Phytoöstrogengehalte), |
|
— |
Einstreu (Bezeichnung, Art, Lieferant, Inhalt), |
|
— |
Käfighaltungsbedingungen und Anzahl der Tiere pro Käfig. |
Prüfbedingungen
|
— |
Alter bei Kastration und Dauer der Akklimatisierung nach Kastration, |
|
— |
Gewicht der einzelnen Tiere bei Prüfungsbeginn (auf 0,1 g genau), |
|
— |
Randomisierungsverfahren und Protokoll der Zuteilung zu Vehikel-, Referenzchemikalien- und Prüfsubstanzgruppen und Käfigen, |
|
— |
Durchschnittswerte und Standardabweichungen der Körpergewichte der einzelnen Gruppen pro Wägetag während der Prüfung, |
|
— |
Begründung der Dosiswahl, |
|
— |
Art und Weise der Verabreichung der Prüfsubstanz und Begründung des gewählten Expositionswegs, |
|
— |
Falls es sich um eine Prüfung auf antiandrogene Wirkungen handelt, die Behandlung mit TP (Dosis und Volumen), |
|
— |
Behandlung mit der Prüfsubstanz (Dosis und Volumen), |
|
— |
Zeitpunkt der Dosierung, |
|
— |
Nekropsieverfahren einschließlich Angaben zu Entblutung und Narkosemitteln, |
|
— |
wenn Serumanalysen durchgeführt werden, sind Angaben zum Analyseverfahren zu machen. Wenn zum Beispiel RIA angewendet wird, sind das RIA-Verfahren, die Herkunft der RIA-Kits, die Ablaufdaten der Kits, das Szintillationszählungsverfahren und das Standardisierungsverfahren zu protokollieren. |
Ergebnisse
|
— |
tägliche Beobachtungen für jedes Tier während der Dosierung einschließlich: |
|
— |
Körpergewicht (auf 0,1 g genau), |
|
— |
klinische Zeichen (falls vorhanden), |
|
— |
etwaige Messungen oder Aufzeichnungen zur Nahrungsaufnahme. |
|
— |
Nekropsiebefunde für jedes Tier einschließlich: |
|
— |
Datum der Nekropsie, |
|
— |
Behandlungsgruppe, |
|
— |
Kennziffer des Tiers, |
|
— |
Prosektor, |
|
— |
Uhrzeit der Nekropsie/Sektion, |
|
— |
Alter des Tiers, |
|
— |
finales Körpergewicht bei Nekropsie, wobei etwaige statistisch signifikante Erhöhungen oder Verringerungen zu vermerken sind, |
|
— |
Reihenfolge des Entblutens und Sezierens der einzelnen Tiere bei der Nekropsie, |
|
— |
Gewichte der fünf androgenabhängigen Zielgewebe: |
|
— |
ventrale Prostata (auf 0,1 mg genau), |
|
— |
Samenbläschen plus Koagulationsdrüsen einschließlich Flüssigkeit (paarig, auf 0,1 mg genau), |
|
— |
Muskelkomplex Levator ani/bulbospongiosus (auf 0,1 mg genau), |
|
— |
Cowpersche Drüsen (Frischgewicht — paarig, auf 0,1 mg genau), |
|
— |
Glans penis (Frischgewicht, auf 0,1 mg genau), |
|
— |
Gewichte der optionalen Gewebe, falls bestimmt: |
|
— |
Leber (auf 0,1 g genau), |
|
— |
Niere (paarig, auf 0,1 mg genau), |
|
— |
Nebenniere (paarig, auf 0,1 mg genau), |
|
— |
Allgemeine Bemerkungen und Kommentare, |
|
— |
Analyse von Serumhormonen, falls bestimmt
|
|
— |
Allgemeine Bemerkungen und Kommentare. |
Datenzusammenfassung
Die Daten sind in tabellarischer Form zusammenzufassen, und zwar mit Stichprobengröße für jede Gruppe, Mittelwert und Standardfehler des Mittelwerts oder Standardabweichung. Ebenfalls in tabellarischer Form aufgeführt sein müssen die Körpergewichte bei Nekropsie, Körpergewichtsänderungen ab Dosierungsbeginn bis Nekropsie, Gewichte der akzessorischen Zielgewebe des Reproduktionstrakts und optional bestimmte Organgewichte.
Diskussion der Ergebnisse
Analyse der Ergebnisse
59. Das Körpergewicht und das Gewicht der einzelnen Organe bei Nekropsie sind statistisch auf Parameter wie die Varianzhomogenität mit entsprechender bedarfsgerechter Datentransformation zu analysieren. Die Behandlungsgruppen sind anhand einer Varianzanalyse oder ähnlicher Analysetechniken mit einer Kontrollgruppe zu vergleichen, gefolgt von paarweisen Vergleichen (z. B. dem einseitigen Test nach Dunnett) und dem Kriterium der statistischen Differenz, z. B. p ≤ 0,05. Es ist zu ermitteln, welche Gruppen eine statistische Signifikanz aufweisen. ‚Relative Organgewichte‘ sind jedoch aufgrund der dieser Datenbearbeitung zugrunde liegenden ungültigen statistischen Hypothesen zu vermeiden.
60. Bei der Prüfung auf Androgenagonismus sollte die Prüfgruppe, die nur das Vehikel erhält, die Kontrollgruppe sein. Die Parameter der Wirkungsweise einer Prüfsubstanz können bei den Zielgeweben zu unterschiedlichen relativen Reaktionen führen; so kann z. B. Trenbolon, das nicht durch die 5α-Reduktase umgewandelt werden kann, eine stärkere Wirkung auf den LABC-Komplex und die GP haben als TP. Eine statistisch signifikante Gewichtserhöhung (p ≤ 0,05) bei mindestens zwei der fünf androgenabhängigen Zielgewebe (VP, LABC, GP, KD und SBKD) ist als positives Ergebnis im Sinne einer Wirkung als Androgenagonist zu werten; ferner sollte bei allen Zielgeweben ein gewisses erhöhtes Wachstum zu beobachten sein. Eine kombinierte Beurteilung der Reaktionen aller akzessorischen Gewebe des Reproduktionstrakts kann mittels geeigneter multivariater Datenanalysemethoden erfolgen. Dies kann zu besseren Analyseergebnissen führen, insbesondere dann, wenn nur bei einem einzigen Gewebe eine statistisch signifikante Reaktion feststellbar ist.
61. Bei der Prüfung auf Androgenantagonismus sollte die Prüfgruppe, die das Referenzandrogen (also nur Testosteronpropionat) erhält, die Kontrollgruppe sein. Die Parameter der Wirkungsweise einer Prüfsubstanz können bei den Zielgeweben zu unterschiedlichen relativen Reaktionen führen; so haben z. B. 5α-Reduktaseinhibitoren wie Finasterid im Vergleich zu wirksamen AR-Antagonisten wie Flutamid eine stärkere Wirkung auf die ventrale Prostata als auf andere Gewebe. Eine statistisch signifikante Gewichtsverringerung (p ≤ 0,05) bei mindestens zwei der fünf androgenabhängigen Zielgewebe (VP, LABC, GP, KD und SBKD) im Verhältnis zu der nur mit TP behandelten Gruppe ist als positives Ergebnis im Sinne einer Wirkung als Androgenantagonist zu werten; ferner sollte bei allen Zielgeweben eine gewisse Wachstumsverringerung zu beobachten sein. Eine kombinierte Beurteilung der Reaktionen aller akzessorischen Gewebe des Reproduktionstrakts kann mittels geeigneter multivariater Datenanalysemethoden erfolgen. Dies kann zu besseren Analyseergebnissen führen, insbesondere dann, wenn nur bei einem einzigen Gewebe eine statistisch signifikante Reaktion feststellbar ist.
62. Die Daten sind in tabellarischer Form zusammenzufassen, und zwar mit Mittelwert, Standardfehler des Mittelwerts (Standardabweichung wäre ebenfalls akzeptabel) und Stichprobengröße für jede Gruppe. Es sollten auch Tabellen mit Einzeldaten beigefügt werden. Die Einzelwerte, Mittelwerte und die Werte für Standardfehler (Standardabweichung) und Variationskoeffizienten für die Kontrolldaten sind daraufhin zu prüfen, ob sie die akzeptablen Kriterien für die Konsistenz mit erwarteten historischen Werten erfüllen. Übersteigen die Variationskoeffizienten (VK) die Werte in Tabelle 1 (vgl. Nummern 25 und 26) für die einzelnen Organgewichte, so ist zu prüfen, ob Fehler bei der Datenerfassung oder -eingabe vorliegen oder ob das Labor die Sektion der androgenabhängigen Gewebe noch nicht exakt beherrscht, sodass weitere Schulungsmaßnahmen/praktische Erfahrung gerechtfertigt sind. Im Allgemeinen sind die VK (die Standardabweichung geteilt durch das mittlere Organgewicht) von Labor zu Labor und von Prüfung zu Prüfung reproduzierbar. Die vorgelegten Daten sollten mindestens umfassen: ventrale Prostata, Samenbläschen, Musculus levator ani plus bulbospongiosus, Cowpersche Drüsen, Glans penis, Leber und Körpergewichte sowie Körpergewichtsänderungen von Dosierungsbeginn bis Nekropsie. Die Daten können auch nach Kovarianzanpassung für das Körpergewicht eingereicht werden, doch dies sollte eine Einreichung der nicht angepassten Daten nicht ersetzen. Zusätzlich sollte die Präputialseparation, wenn sie bei einer der Gruppen nicht erfolgt ist, bei ihrem Eintreten erfasst und statistisch anhand des exakten Tests nach Fisher mit der Kontrollgruppe verglichen werden.
63. Wenn beim Vergleich der Computerdateneinträge mit den ursprünglichen Datenblättern Organgewichtswerte auffallen, die nicht biologisch plausibel sind oder um mehr als drei Standardabweichungen vom Behandlungsgruppendurchschnitt abweichen, sollten diese sorgfältig auf ihre Richtigkeit überprüft und gegebenenfalls verworfen werden, da es sich wahrscheinlich um Erfassungsfehler handelt.
64. Ein Vergleich der Prüfungsergebnisse mit den VK-Werten der OECD (siehe Tabelle 1) ist häufig ein wichtiger Schritt bei der Interpretation der Validität der Prüfungsergebnisse. Die historischen Daten der Vehikelkontrollgruppen sollten vom Labor in dessen Datenbestand eingepflegt werden. Die historischen Daten zur Wirkung positiver Referenzchemikalien, zum Beispiel TP und FT, sind ebenfalls vom Labor einzupflegen. Die Laboratorien können ferner die Reaktion auf bekannt schwache Androgenagonisten und -antagonisten in regelmäßigen Abständen testen und diese Daten einpflegen. Diese Daten können mit den verfügbaren OECD-Daten verglichen werden, um sicherzustellen, dass mit den Methoden des Labors eine ausreichende statistische Präzision und Aussagekraft erzielt wird.
Anlage 1
BEGRIFFSBESTIMMUNGEN
Androgen (Adj.) : Begriff zur Beschreibung einer positiven Wirkung auf das Wachstum androgenabhängiger Gewebe.
Antiandrogen (Adj.) : die Fähigkeit einer Chemikalie, die Wirkung von TP in einem Säugetierorganismus zu unterdrücken.
Chemikalie : ein Stoff oder eine Mischung.
Dosierung : ein allgemeiner Begriff, der die Dosis, ihre Häufigkeit und die Dauer der Verabreichung umfasst.
Dosis : die Menge der verabreichten Prüfsubstanz. Für die Zwecke des Hershberger-Bioassays wird die Dosis ausgedrückt als Masse der Prüfsubstanz je Einheit Körpergewicht des Versuchstiers pro Tag (z. B. mg pro kg Körpergewicht und Tag).
Empfindlichkeit : die Fähigkeit einer Prüfmethode, Chemikalien korrekt anhand derjenigen Eigenschaft zu erkennen, auf die diese getestet werden.
Moribund (Adj.) : Begriff zur Beschreibung des Zustands eines sterbenden, todgeweihten Tiers.
Postnataler Tag X : der x-te Lebenstag nach der Geburt.
Prüfsubstanz : jede(r) mittels dieser Prüfmethode getestete Stoff bzw. Mischung.
Spezifizität : die Fähigkeit einer Prüfmethode, Chemikalien korrekt anhand des Fehlens derjenigen Eigenschaft zu erkennen, auf die diese getestet werden.
Tag der Geburt : der postnatale Tag 0.
Validierung : wissenschaftlicher Prozess zur Beschreibung der operationellen Anforderungen und Grenzen einer Prüfmethode und zum Nachweis ihrer Zuverlässigkeit und Eignung für einen bestimmten Zweck.
Anlage 2

ANMERKUNGEN ZUM RAHMENKONZEPT
|
Anmerkung 1: |
Ein Einstieg in das Rahmenkonzept und ein Ausstieg sind auf allen Stufen möglich und abhängig davon, welche Informationen für Gefahren- und Risikobeurteilungszwecke benötigt werden. |
|
Anmerkung 2: |
Bei Stufe 5 sollte der Aspekt der Ökotoxikologie Endpunkte beinhalten, die Hinweise auf schädliche Wirkmechanismen und eine potenzielle Schädigung der Population liefern. |
|
Anmerkung 3: |
Wenn ein multimodales Modell mehrere Assays mit nur einem Endpunkt (Single-Endpoint-Assays) umfasst, ersetzt dieses Modell die Durchführung der Single-Endpoint-Assays. |
|
Anmerkung 4: |
Die Bewertung der einzelnen Chemikalien sollte von Fall zu Fall und unter Berücksichtigung aller verfügbaren Daten sowie vor dem Hintergrund des Zwecks der Stufen des Rahmenkonzepts erfolgen. |
|
Anmerkung 5: |
Das Rahmenkonzept ist derzeit noch nicht als vollständig anzusehen. Auf Stufe 3, 4 und 5 beinhaltet es Assays, die entweder schon verfügbar sind oder sich gerade in der Validierungsphase befinden. Bei Letzteren ist die Aufnahme in das Rahmenkonzept noch vorläufig. Sobald ihre Entwicklung und Validierung abgeschlossen ist, werden sie offizieller Bestandteil des Konzepts. |
|
Anmerkung 6: |
Stufe 5 ist nicht so zu verstehen, dass es sich bei den darin enthaltenen Tests nur um endgültige Prüfungen handelt. Die Prüfungen dieser Stufe sollen vielmehr einen Beitrag zur allgemeinen Gefahren- und Risikobewertung leisten. |
LITERATUR
|
(1) |
OECD (1998). Report of the First Meeting of the OECD Endocrine Disrupter Testing and Assessment (EDTA) Task Force, 10.-11. März 1998, ENV/MC/CHEM/RA(98)5. |
|
(2) |
Dorfman RI (1962). Standard methods adopted by official organization. Academic Press, NY. |
|
(3) |
Gray LE Jr, Furr J and Ostby JS (2005). Hershberger assay to investigate the effects of endocrine disrupting compounds with androgenic and antiandrogenic activity in castrate-immature male rats. In: Current Protocols in Toxicology 16.9.1-16.9.15. J Wiley and Sons Inc. |
|
(4) |
OECD (2006). Final OECD report of the initial work towards the validation of the rat Hershberger assay. Phase 1. Androgenic response to testosterone propionate and anti-androgenic effects of flutamide. Environmental Health and Safety, Monograph Series on Testing and Assessment No 62. ENV/JM/MONO(2006)30. |
|
(5) |
OECD (2008). Report of the OECD Validation of the Rat Hershberger Bioassay: Phase 2: Testing of Androgen Agonists, Androgen Antagonists and a 5a-Reductase Inhibitor in Dose Response Studies by Multiple Laboratories. Environmental Health and Safety, Monograph Series on Testing and Assessment No 86. ENV/JM/MONO(2008)3. |
|
(6) |
OECD (2007). Report of the Validation of the Rat Hershberger Assay: Phase 3: Coded Testing of Androgen Agonists, Androgen Antagonists and Negative Reference Chemicals by Multiple Laboratories. Surgical Castrate Model Protocol. Environmental Health and Safety, Monograph Series on Testing and Assessment No 73. ENV/JM/MONO(2007)20. |
|
(7) |
Owens, W, Zeiger E, Walker M, Ashby J, Onyon L, Gray, Jr, LE (2006). The OECD programme to validate the rat Hershberger bioassay to screen compounds for in vivo androgen and antiandrogen responses. Phase 1: Use of a potent agonist and a potent antagonist to test the standardized protocol. Env. Health Persp. 114:1265-1269. |
|
(8) |
Owens W, Gray LE, Zeiger E, Walker M, Yamasaki K, Ashby J, Jacob E (2007). The OECD program to validate the rat Hershberger bioassay to screen compounds for in vivo androgen and antiandrogen responses: phase 2 dose-response studies. Environ Health Perspect. 115(5):671-8. |
|
(9) |
Korenchevsky V (1932). The assay of testicular hormone preparations. Biochem J 26:413-422. |
|
(10) |
Korenchevsky V, Dennison M, Schalit R (1932). The response of castrated male rats to the injection of the testicular hormone. Biochem J26:1306-1314. |
|
(11) |
Eisenberg E, Gordan GS (1950). The levator ani muscle of the rat as an index of myotrophic activity of steroidal hormones. J Pharmacol Exp Therap 99:38-44. |
|
(12) |
Eisenberg E, Gordan GS, Elliott HW (1949). Testosterone and tissue respiration of the castrate male rat with a possible test for mytrophic activity. Endocrinology 45:113-119. |
|
(13) |
Hershberger L, Shipley E, Meyer R (1953). Myotrophic activity of 19-nortestosterone and other steroids determined by modified levator ani muscle method. Proc Soc Exp Biol Med 83:175-180. |
|
(14) |
Hilgar AG, Vollmer EP (1964). Endocrine bioassay data: Androgenic and myogenic. Washington DC: United States Public Health Service. |
|
(15) |
Dorfman RI (1969). Androgens and anabolic agents. In: Methods in Hormone Research, volume IIA. (Dorfman RI, ed.) New York:Academic Press, 151-220. |
|
(16) |
Massaro EJ (2002). Handbook of Neurotoxicology, volume I. New York: Humana Press, S. 38. |
|
(17) |
OECD (2000). Guidance document on the recognition, assessment and use of clinical signs as humane endpoints for experimental animals used in safety evaluation. Environmental Health and Safety Monograph Series on Testing and Assessment No 19. ENV/JM/MONO(2000)7. |
|
(18) |
OECD (1982). Organization for Economic Co-operation and Development — Principles of Good Laboratory Practice, ISBN 92-64-12367-9, Paris. |
|
(19) |
OECD (2008). Acute oral toxicity — up-and-down procedure. OECD Guideline for the testing of chemicals No 425. |
|
(20) |
OECD (2001). Guidance document on acute oral toxicity. Environmental Health and Safety Monograph Series on Testing and Assessment No 24. ENV/JM/MONO(2001)4. |
|
(21) |
Supplemental materials for Owens et al. (2006). The OECD programme to validate the rat Hershberger bioassay to screen compounds for in vivo androgen and antiandrogen responses. Phase 1: Use of a potent agonist and a potent antagonist to test the standardized protocol. Env. Health Persp. 114:1265-1269. Siehe Abschnitt II, Sektionsleitfaden für die Laboratorien, abrufbar unter: [http://www.ehponline.org/docs/2006/8751/suppl.pdf]. |
|
(22) |
Koreanische Nahrungs- und Arzneimittelbehörde. Visueller Referenzleitfaden zur Verfahrensweise beim Hershberger-Test einschließlich Sektionsvideo. [http://rndmoa.kfda.go.kr/endocrine/reference/education_fr.html]. |
|
(23) |
OECD (2008). Background Review Document on the Rodent Hershberger Bioassay. Environmental Health and Safety Monograph Series on Testing and Assessment No 90. ENV/JM/MONO(2008)17. |
|
(24) |
OECD (2008). Draft Validation report of the Intact, Stimulated, Weanling Male Rat Version of the Hershberger Bioassay. |
|
(25) |
OECD (2009). Guidance Document on the Weanling Hershberger Bioassay in rats: A shortterm screening assay for (anti)androgenic properties. Series on Testing and Assessment, Number 115. |
|
(26) |
Richtlinie 2010/63/EU des Europäischen Parlaments und des Rates vom 22. September 2010 zum Schutz der für wissenschaftliche Zwecke verwendeten Tiere (ABl. L 276 vom 20.10.2010, S. 33). |
|
(27) |
Die folgenden Kapitel des vorliegenden Anhangs:
|
B.56 ERWEITERTE EIN-GENERATIONEN-PRÜFUNG AUF REPRODUKTIONSTOXIZITÄT
EINLEITUNG
1. Diese Prüfmethode entspricht der OECD-Prüfrichtlinie (TG) 443 (2012). Sie basiert auf dem Vorschlag des Fachausschusses Agricultural Chemical Safety Assessment (ACSA) des ILSI Health and Environmental Sciences Institute (HESI) für eine in Bezug die Lebensphase F1 erweiterte Ein-Generationen-Prüfung auf Reproduktionstoxizität (EOGRTS), wie in Cooper et al., 2006 (1) veröffentlicht. Der Prüfplan wurde in einigen Punkten verbessert und präzisiert, um Flexibilität zu gewährleisten und deutlich zu machen, dass auf vorhandenem Wissen aufgebaut wird, gleichzeitig jedoch Beobachtungen am lebenden Tier genutzt werden, um den Prüfungsablauf zu steuern und zu maßschneidern. Die nachstehende Prüfmethode beschreibt die Verfahrensschritte einer EOGRTS-Prüfung im Einzelnen. Sie basiert auf drei Kohorten von F1-Tieren:
Kohorte 1: Bewertung reproduktionstoxischer/entwicklungstoxischer Endpunkte; diese Kohorte kann auf eine F2-Generation erweitert werden.
Kohorte 2: Bewertung der potenziellen Auswirkung der Exposition gegenüber der Prüfsubstanz auf das sich entwickelnde Nervensystem.
Kohorte 3: Bewertung der potenziellen Auswirkung der Exposition gegenüber der Prüfsubstanz auf das sich entwickelnde Immunsystem.
2. Bei Entscheidungen darüber, ob auch die zweite Generation bewertet und die Kohorten für Entwicklungsneurotoxizität und/oder Entwicklungsimmuntoxizität ausgelassen werden sollten, sollte vorhandenes Wissen über die Prüfsubstanz ebenso berücksichtigt werden wie die Bedürfnisse der verschiedenen Regulierungsbehörden. Zweck der Prüfmethode ist es, die möglichen Verfahrenschritte für die Bewertung der einzelnen Kohorten im Detail zu beschreiben.
3. Für Regulierungsbehörden, die zur Produktion einer zweiten Generation interne Auslöser (trigger) verwenden, enthält das OECD Guidance Document Nr. 117 (39) entsprechende Verfahrensvorschriften.
AUSGANGSÜBERLEGUNGEN UND ZIELE
4. Hauptziel der EOGRTS-Prüfung ist es, bestimmte Lebensphasen zu evaluieren, die von anderen Arten von Toxizitätsstudien nicht abgedeckt werden, und nach etwaigen Wirkungen einer prä- und postnatalen Chemikalienexposition zu suchen. Zur Bestimmung der reproduktionstoxischen Endpunkte ist vorgesehen, in einem ersten Schritt — und soweit sie vorliegen — Informationen aus Prüfungen auf Toxizität bei wiederholter Verabreichung einer Prüfsubstanz (einschließlich Screeningstests auf Reproduktionstoxizität, z. B. OECD TG 422 (32)), oder kurzfristige Screeningtests auf endokrine Disruptoren, (z. B. Uterotropher Bioassay — Prüfmethode B.54 (36); und Hershberger-Test — Prüfmethode B.55 (37)) heranzuziehen, um Auswirkungen auf die Fortpflanzungsorgane von männlichen und weiblichen Tieren festzustellen. Diese könnten bei männlichen Tieren Daten über die Spermatogenese (Testikular-Histopathologie) und bei weiblichen Tieren Daten über Östruszyklen, Follikelzahlen/Eizellenreifung und die Integrität der Eierstöcke (Histopathologie) umfassen. Anschließend werden durch die EOGRTS-Prüfung die reproduktionstoxischen Endpunkte untersucht, die die Interaktion von männlichen mit weiblichen Tieren, von weiblichen Tieren mit befruchteten Eizellen (Conceptus) und von weiblichen Tieren mit ihren Nachkommen und der F1-Generation bis nach der Geschlechtsreife voraussetzen (siehe OECD Guidance Document Nr. 151, das diese Prüfmethode (40) unterstützt).
5. Der Prüfplan ermöglicht die Bewertung der prä- und postnatalen Auswirkungen von Chemikalien auf die Entwicklung sowie eine detaillierte Bewertung der systemischen Toxizität bei trächtigen und laktierenden weiblichen Tieren sowie jungen und adulten Nachkommen. Durch ausführliche Untersuchung der wichtigsten entwicklungstoxischen Endpunkte (wie Lebensfähigkeit der Nachkommen, Gesundheit der Neugeborenen, Entwicklungsstand bei der Geburt und körperliche sowie funktionale Entwicklung bis zum Erwachsenenalter) sollen spezifische Zielorgane bei den Nachkommen ermittelt werden. Die Prüfung liefert und/oder bestätigt außerdem Informationen über die Auswirkungen einer Prüfsubstanz auf die Integrität und Leistungsfähigkeit der Fortpflanzungsorgane adulter männlicher und weiblicher Tiere. Es werden speziell — aber nicht ausschließlich — die folgenden Parameter geprüft: Gonadenfunktion, Östruszyklus, epididymale Spermienreifung, Paarungsverhalten, Empfängnis, Trächtigkeit, Geburt und Laktation. Außerdem werden die Erkenntnisse aus den Untersuchungen der Entwicklungsneurotoxizität und Immuntoxizität potenzielle Wirkungen in diesen Systemen charakterisieren. Die aus diesen Prüfungen gewonnenen Daten dürften die Bestimmung der NOAEL-Werte (No Observed Adverse Effect Level), der LOAEL-Werte (Lowest Observed Adverse Effect Level) und/oder Benchmark-Dosen für die verschiedenen Endpunkte ermöglichen und/oder sollten zur Charakterisierung der in früheren Untersuchungen der Toxizität bei wiederholter Verabreichung (repeat-dose studies) festgestellten Wirkungen verwendet werden und/oder als Richtwerte für spätere Tests dienen.
6. Ein Schema des Protokolls ist in Abbildung 1 gegeben. Die Prüfsubstanz wird in abgestuften Dosen kontinuierlich an mehrere Gruppen geschlechtsreifer männlicher und weiblicher Versuchstiere verabreicht. Diese Parentalgeneration (P) erhält die Dosen vor der Paarung während eines vorgegebenen Expositionszeitraums (ausgewählt auf Basis der für die Prüfsubstanz vorliegenden Informationen; mindestens aber zwei Wochen) und während eines zweiwöchigen Paarungszeitraums. P-Männchen werden mindestens bis zum Entwöhnen der F1-Nachkommen weiterbehandelt. Die Behandlung sollte mindestens zehn Wochen dauern. Sie können auch länger behandelt werden, falls Auswirkungen auf die Fortpflanzung unschlüssig sind. Die Behandlung der P-Weibchen wird während Trächtigkeit und Laktation bis nach der Entwöhnung ihrer Würfe fortgesetzt (d. h. acht- bis zehnwöchige Behandlung). Die F1-Nachkommen werden vom Zeitpunkt ihrer Entwöhnung bis zum Erwachsenenalter mit der Prüfsubstanz weiterbehandelt. Wird eine zweite Generation untersucht (siehe OECD Guidance Document Nr. 117 (39)), so werden die F1-Nachkommen so lange behandelt, bis die F2-Generation entwöhnt ist bzw. bis die Prüfung abgeschlossen ist.
7. Alle Tiere werden klinisch und pathologisch auf Anzeichen von Toxizität untersucht, wobei insbesondere auf die Integrität und Leistungsfähigkeit der Fortpflanzungsorgane der männlichen und der weiblichen adulten Tiere sowie auf Gesundheit, Wachstum, Entwicklung und Fortpflanzungsfähigkeit der Nachkommen geachtet wird. Am Tag des Absetzens werden ausgewählte Nachkommen für weitere Untersuchungen, einschließlich Untersuchungen der Geschlechtsreife, der Integrität und Funktionsfähigkeit der Fortpflanzungsorgane, der neurologischen Endpunkte und der Verhaltensendpunkte sowie der Immunfunktionen in bestimmte Untergruppen (Kohorten 1-3, siehe die Nummern 33 und 34 sowie Abbildung 1) eingeteilt.
8. Bei der Prüfung sollten die im OECD Guidance Document Nr. 19 on the recognition, assessment, and use of clinical signs as humane endpoints for experimental animals used in safety evaluation (34) genannten Grundsätze und Erwägungen beachtet werden.
9. Wurden genügend Prüfungen durchgeführt, um über die Eignung dieses neuen Prüfplans zu befinden, wird die Prüfmethode überprüft und auf der anhand der gewonnenen Erfahrungen gegebenenfalls überarbeitet.
Abbildung 1
Schema der erweiterten Ein-Generationen-Prüfung auf Reproduktionstoxizität

BESCHREIBUNG DER PRÜFMETHODE/VORBEREITUNGEN FÜR DEN TEST
Versuchstiere
Auswahl von Versuchstierart und -stamm
10. Die Art der Versuchstiere für die Prüfung auf Reproduktionstoxizität sollte unter Berücksichtigung aller verfügbaren Informationen sorgfältig ausgewählt werden. Wegen der Fülle der vorliegenden Hintergrundinformationen und der Vergleichbarkeit mit allgemeinen Toxizitätsprüfungen ist die Ratte in der Regel das Versuchstier der Wahl, und die in dieser Prüfmethode angegebenen Kriterien und Empfehlungen beziehen sich auf diese Tierart. Falls eine andere Tierart verwendet wird, ist dies zu begründen, und das Protokoll ist entsprechend abzuändern. Stämme mit geringer Fruchtbarkeit oder allgemein anerkanntem spontanem Vorkommen von Entwicklungsstörungen sollten nicht verwendet werden.
Alter, Körpergewicht und Aufnahmekriterien
11. Es sind gesunde Elterntiere zu verwenden, die zuvor nicht in anderen Versuchen verwendet wurden. Es sind sowohl männliche als auch weibliche Tiere zu untersuchen, wobei die weiblichen Tiere nicht geworfen haben noch trächtig sein dürfen. Die P-Tiere sollten geschlechtsreif sein, bei der ersten Dosisverabreichung über ein ähnliches Gewicht (innerhalb des Geschlechts) verfügen, bei der Paarung etwa gleich alt (ungefähr 90 Tage) und für die Prüfart und den Prüfstamm repräsentativ sein. Die Tiere sind nach ihrem Eintreffen im Labor mindestens fünf Tage lang einzugewöhnen. Sie werden nach dem Zufallsprinzip so auf die Kontroll- und Behandlungsgruppen verteilt, dass die verschiedenen Gruppen vergleichbare mittlere Körpergewichtswerte aufweisen (d. h. ± 20 % des Mittelwerts).
Unterbringungs- und Fütterungsbedingungen
12. Die Temperatur im Versuchstierraum sollte 22 °C (± 3°) betragen. Die relative Luftfeuchtigkeit sollte bei 30-70 % liegen, wobei eine Luftfeuchtigkeit von 50-60 % ideal ist. Die Hell- und Dunkelphasen der künstlichen Beleuchtung sollten jeweils 12 Stunden dauern. Es kann herkömmliches Laborfutter verfüttert werden, die Tiere sollten jedoch unbegrenzten Zugang zu Trinkwasser haben. Es ist besonders auf den Phytoöstrogengehalt des Futters zu achten, da einige reproduktionstoxische Endpunkte durch einen zu hohen Phytoöstrogengehalt im Futter beeinträchtigt werden könnten. Es wird standardisiertes Futter mit offener Rezeptur empfohlen, dessen Gehalt an östrogen wirkenden chemischen Substanzen reduziert wurde (2) (30). Die Auswahl des Futters kann eventuell dadurch beeinflusst werden, dass eine geeignete Menge Prüfsubstanz beigemischt werden muss, wenn die Prüfsubstanz über das Futter verabreicht werden soll. Der Gehalt, die Homogenität und die Stabilität der Prüfsubstanz in den Futterrationen sind zu überprüfen. Futter und Trinkwasser sind regelmäßig auf Schadstoffe zu analysieren. Proben jeder im Laufe des Versuchs verwendeten Futtercharge sind bis zur Fertigstellung des Abschlussberichts unter geeigneten Bedingungen (z. B. durch Einfrieren bei – 20 °C) aufzubewahren, falls die Ergebnisse eine weitere Analyse der Futterbestandteile erfordern.
13. Die Tiere sollten in kleinen Gruppen gleichen Geschlechts und gleicher Behandlung untergebracht werden. Sie können auch einzeln untergebracht werden, um potenzielle Verletzungen zu vermeiden (z. B. männliche Tiere nach der Paarungszeit). Die Paarung sollte in zweckbestimmten Käfigen erfolgen. Nach dem nachweislich erfolgten Deckakt sind weibliche Tiere, von denen angenommen wird, dass sie trächtig sind, in separaten Käfigen speziell für trächtige Tiere bzw. in Wurfkäfigen zu halten, in denen ihnen vorgegebene und geeignete Nistmaterialien zur Verfügung stehen. Würfe werden bis zur Entwöhnung bei ihren Müttern gehalten. F1-Tiere sollten ab dem Tag ihres Absetzens bis zum Versuchsende in kleinen Gruppen gleichen Geschlechts und gleicher Behandlung gehalten werden. Sie können auch einzeln gehalten werden, soweit dies wissenschaftlich gerechtfertigt ist. Der Phytoöstrogengehalt der gewählten Einstreu sollte so gering wie möglich sein.
Zahl und Kennzeichnung der Versuchstiere
14. Normalerweise sollte sich in jeder Prüf- und Kontrollgruppe eine ausreichende Menge Begattungspaare befinden, um pro Dosisgruppe mindestens 20 trächtige Weibchen zu ergeben. Das Ziel besteht darin, so viele trächtige Weibchen zu erhalten, dass eine aussagekräftige Bewertung des Potenzials der Prüfsubstanz, die Fruchtbarkeit, die Trächtigkeit und das Verhalten der Muttertiere der P-Generation sowie Wachstum und Entwicklung der F1-Nachkommen von der Befruchtung bis hin zur Geschlechtsreife zu beeinträchtigen, gewährleistet ist. Wird die gewünschte Zahl trächtiger Tiere nicht erzielt, so wird die Studie dadurch nicht zwangsläufig unbrauchbar. Hier ist der jeweilige Einzelfall zu bewerten, wobei zu prüfen ist, ob eine mögliche kausale Beziehung zur Prüfsubstanz hergestellt werden kann.
15. Jedes P-Tier erhält eine individuelle Kennnummer, bevor mit der Dosisverabreichung begonnen wird. Wenn historische Labordaten darauf hindeuten, dass ein erheblicher Teil der weiblichen Tiere möglicherweise keinen regelmäßigen (4- oder 5-tägigen) Östruszyklus aufweist, wird empfohlen, die Östruszyklen vor Beginn der Behandlung zu untersuchen. Alternativ kann die Gruppe vergrößert werden, um sicherzustellen, dass zu Beginn der Behandlung mindestens 20 weibliche Tiere in jeder Gruppe einen regelmäßigen (4- oder 5-tägigen) Zyklus aufweisen. Alle F1-Nachkommen werden bei der ersten Untersuchung der Neugeborenen am Tag der Geburt (Tag 0) oder am Tag 1 nach der Geburt (postnatal day, PND) einzeln gekennzeichnet. Aufzeichnungen, aus denen der Wurf, dem sie entstammen, hervorgeht, sind für alle F1-Tiere und gegebenenfalls auch für alle F2-Tiere während der gesamten Versuchsdauer aufzubewahren.
Prüfsubstanz
Verfügbare Informationen über die Prüfsubstanz
16. Die Überprüfung vorliegender Informationen ist für die Wahl des Verabreichungswegs, des Vehikels der Tierart und der Dosisabstufungen sowie für potenzielle Änderungen des Verabreichungszeitplans wichtig. Daher sollten bei der Planung der EOGRTS-Prüfung alle über die Prüfsubstanz vorliegenden sachdienlichen Informationen wie physikalisch-chemische Eigenschaften, toxikokinetische Eigenschaften (einschließlich des artenspezifischen Stoffwechsels), toxikodynamische Eigenschaften, Struktur-Wirkungs-Beziehungen (SAR), In-vitro-Stoffwechselprozesse, Ergebnisse früherer Toxizitätsstudien und sachdienliche Informationen über strukturelle Analogien berücksichtigt werden. Vorläufige Informationen über Resorption, Verteilung, Metabolismus und Elimination (ADME) sowie Bioakkumulation können aus der chemischen Struktur, physikalisch-chemischen Daten, dem Umfang der Plasmaproteinbindung oder toxikokinetischen Studien abgeleitet werden, während Ergebnisse aus Toxizitätsstudien zusätzliche Informationen, z. B. über den NOAEL-Wert, den Stoffwechsel oder eine Stoffwechselinduktion liefern.
Berücksichtigung toxikokinetischer Daten
17. Obwohl nicht erforderlich, können toxikokinetische Daten (TK-Daten) aus früheren Dosisfindungs- oder anderen Studien bei der Erstellung des Prüfplans, der Dosisabstufung und der Auswertung der Ergebnisse äußerst nützlich sein. Ganz besonders zweckdienlich sind dabei Daten, die 1) die Exposition von Föten und Jungtieren gegenüber der Prüfsubstanz (oder relevanten Metaboliten) bestätigen, 2) zur Ableitung einer internen Dosimetrie beitragen und 3) eine potenzielle dosisabhängige Sättigung kinetischer Prozesse bewerten. Falls weitere TK-Daten vorliegen, beispielsweise Metabolitprofile, Konzentrations-Zeit-Kurven usw. sind diese ebenfalls zu berücksichtigen. Ergänzende TK-Daten können auch während des Hauptversuchs erhoben werden, vorausgesetzt die Erhebung und Interpretation der wichtigsten Versuchsendpunkte wird dadurch nicht beeinträchtigt.
Grundsätzlich wären folgende TK-Datensätze bei der Planung der EOGRTS-Prüfung nützlich:
|
— |
späte Trächtigkeitsphase (z. B. Trächtigkeitstag (GD) 20) — mütterliches und fetales Blut, |
|
— |
Mitt-Laktation (PND 10) — mütterliches Blut, Jungtierblut und/oder Milch, |
|
— |
kurz nach der Entwöhnung (z. B. PND 28) — Blutproben der entwöhnten Jungtiere. |
Die Bestimmung der spezifischen Analyten (wie Ausgangssubstanz und/oder Metaboliten) und des Probenahmeplans sollte flexibel gehandhabt werden. Beispielsweise hängen Zahl und Zeitpunkt der Probenahmen an einem beliebigen Entnahmetag vom Expositionsweg und der Vorabkenntnis der toxikokinetischen Merkmale nicht trächtiger Tiere ab. Bei Versuchen mit Verabreichung über das Futter sind Probenahmen stets zum selben Zeitpunkt an jedem dieser Tage ausreichend, wohingegen bei Verabreichungen über eine Schlundsonde unter Umständen zusätzliche Probenahmen erforderlich sind, um einen besseren Schätzwert für den Bereich der internen Dosen zu erhalten. Eine vollständige Konzentrations-Zeit-Kurve muss jedoch nicht für jeden Probenahmetag erstellt werden. Erforderlichenfalls können Blutproben nach Geschlechtern innerhalb der Würfe für fetale und neonatale Analysen gepoolt werden.
Verabreichungsweg
18. Bei der Wahl des Verabreichungswegs sollten die Routen berücksichtigt werden, die für die menschliche Exposition am relevantesten sind. Obwohl das Protokoll für die Verabreichung der Prüfsubstanz über das Futter ausgelegt ist, kann es für eine Verabreichung über andere Wege (Trinkwasser, Schlundsonde, Inhalation, dermal) je nach Eigenschaften der Prüfsubstanz und den erforderlichen Informationen abgeändert werden.
Wahl des Vehikels
19. Bei Bedarf wird die Prüfsubstanz in einem geeigneten Vehikel gelöst oder suspendiert. Es wird empfohlen, nach Möglichkeit zunächst die Verwendung einer wässrigen Lösung/Suspension und erst dann eine Lösung/Emulsion in Öl (z. B. Maisöl) in Betracht zu ziehen. Bei anderen Vehikeln als Wasser sollten deren toxische Merkmale bekannt sein. Die Verwendung von Vehikeln mit einer potenziellen intrinsischen Toxizität (z. B. Aceton, DMSO) ist zu vermeiden. Die Stabilität der Prüfsubstanz im Vehikel sollte bestimmt werden. Wird ein Vehikel oder ein anderes Additiv zur Erleichterung der Dosierung verwendet, so sollten folgende Aspekte berücksichtigt werden: die Auswirkungen auf die Resorption, die Verteilung, die Verstoffwechselung oder die Retention der Prüfsubstanz, Auswirkungen auf die chemischen Eigenschaften der Prüfsubstanz, die deren toxische Eigenschaften verändern können, und ferner Auswirkungen auf die Futter- oder Wasseraufnahme oder den Ernährungszustand der Versuchstiere.
Wahl der Dosis
20. Normalerweise sollten im Versuch mindestens drei Dosisstufen und eine gleichzeitige Kontrolle verwendet werden. Bei der Wahl der geeigneten Dosisstufen sollte der Prüfer alle vorliegenden Informationen berücksichtigen, einschließlich der Dosierungsinformationen aus früheren Versuchen, TK-Daten zu trächtigen und nicht trächtigen Tieren, das Ausmaß der Übertragung über die Muttermilch und die Schätzwerte für die menschliche Exposition. Falls toxikokinetische Daten vorliegen, die auf eine dosisbedingte Sättigung der toxikokinetischen Prozesse hindeuten, ist darauf zu achten, dass hohe Dosisstufen vermieden werden, die eindeutig eine hohe Sättigung aufweisen, natürlich nur unter der Voraussetzung, dass menschliche Expositionen möglichst deutlich unter dem Sättigungspunkt liegen. In solchen Fällen sollte die höchste Dosisstufe auf oder knapp über dem Wendepunkt zum Übergang zu nichtlinearem toxikokinetischem Verhalten liegen.
21. Sind keine relevanten toxikokinetischen Daten verfügbar, sollten die Dosisstufen auf der Grundlage der toxischen Wirkungen gewählt werden, es sei denn, es bestehen Beschränkungen aufgrund der physikalisch-chemischen Beschaffenheit der Prüfsubstanz. Werden die Dosisstufen auf der Grundlage der Toxizität gewählt, ist die höchste Dosisstufe so anzusetzen, dass bei den Tieren zwar toxische Wirkungen, aber keine Todesfälle oder schwere Leiden hervorgerufen werden.
22. Die Dosisstufen sind in absteigender Reihenfolge zu wählen, um etwaige dosisabhängige Wirkungen nachzuweisen und NOAEL-Werte oder Dosen an der Nachweisgrenze zu ermitteln, über die eine Benchmark-Dosis (BMD) für die empfindlichsten Endpunkte abgeleitet werden kann. Zwei- bis vierfache Abstände erweisen sich häufig als optimale Dosisabstufungen, um große Dosisstufenabstände zwischen NOAEL- und LOAEL-Werten zu vermeiden. Außerdem ist es oft besser, eine vierte Prüfgruppe einzurichten, statt zwischen den Dosen sehr große Abstände (z. B. mehr als × 10) zu verwenden.
23. Bis auf die Behandlung mit der Prüfsubstanz sind die Tiere in der Kontrollgruppe genauso zu behandeln wie die Tiere in den Prüfgruppen. Diese Gruppe sollte eine unbehandelte oder scheinbehandelte Gruppe oder eine Vehikelkontrollgruppe sein, sofern ein Vehikel zur Verabreichung der Prüfsubstanz verwendet wird. Wird ein Vehikel verwendet, erhält die Kontrollgruppe das Vehikel im höchsten verwendeten Volumen.
Limit-Test
24. Wenn in Versuchen mit wiederholter Verabreichung bei einer Dosis von mindestens 1 000 mg/kg Körpergewicht/Tag keine Anzeichen von Toxizität zu erkennen sind oder wenn aufgrund von Daten über struktur- und/oder stoffwechselverwandte Stoffe, die auf ähnliche In-vivo/In-vitro-Stoffwechseleigenschaften hindeuten, keine Toxizität zu erwarten ist, kann möglicherweise auf einen Versuch mit mehreren Dosisstufen verzichtet werden. In solchen Fällen könnte die EOGRTS-Prüfung mit einer Kontrollgruppe und mit einer einzelnen Dosis von mindestens 1 000 mg/kg Körpergewicht/Tag durchgeführt werden. Sollten jedoch bei dieser Grenzdosis Nachweise einer Reproduktions- oder Entwicklungstoxizität festgestellt werden, sind zur Ermittlung eines NOAEL-Wertes weitere Versuche bei niedrigeren Dosisstufen erforderlich. Das Kriterium des Limit-Tests findet jedoch nur Anwendung, wenn das Niveau der menschlichen Exposition keine höhere Dosisstufe erfordert.
VERFAHREN
Exposition der Nachkommen
25. Die bevorzugte Verabreichungsmethode ist die Aufnahme über die Nahrung. Falls die Untersuchungen mit Schlundsonden durchgeführt werden, wird darauf hingewiesen, dass die Jungtiere die Prüfsubstanz in der Regel nur indirekt über die Milch erhalten, bis nach der Entwöhnung auch für sie die Direktverabreichung beginnt. Bei Futter- oder Trinkwasserversuchen erhalten die Jungtiere die Prüfsubstanz zusätzlich direkt, sobald sie während der letzten Woche der Laktationszeit beginnen, selbst zu fressen. Wenn zu wenig Prüfsubstanz in die Muttermilch übergeht und nicht nachwiesen werden kann, dass die Nachkommen kontinuierlich exponiert sind, sollte eine Änderung des Prüfplans in Betracht gezogen werden. In diesem Fall sollte auf der Grundlage vorliegender toxikokinetischer Erkenntnisse, der Toxizität bei den Nachkommen oder veränderter Biomarker bei Jungtieren während der Laktationszeit eine Direktverabreichung in Betracht gezogen werden (3) (4). Vor der Durchführung von Direktverabreichungsversuchen mit säugenden Jungtieren sind die Vor- und Nachteile sorgfältig gegeneinander abzuwägen (5).
Verabreichungszeitplan und Verabfolgung der Dosen
26. Unter Umständen liegen aus früheren ausreichend langen Prüfungen mit wiederholter Verabreichung Informationen zum Östruszyklus, zur Histopathologie des männlichen und weiblichen Fortpflanzungsapparats und zur Analyse testikulärer/epididymaler Spermien vor. Daher soll bei der EOGRTS-Prüfung die Dauer der Behandlung vor der Paarung die Feststellung von Auswirkungen auf funktionale Veränderungen ermöglichen, die das Paarungsverhalten und die Fruchtbarkeit beeinträchtigen können. Die Behandlung vor der Paarung sollte so lange dauern, bis bei P-Männchen und P-Weibchen ein Expositionsgleichgewicht (Steady-State) erreicht ist. Eine zweiwöchige Behandlung vor der Paarung wird in den meisten Fällen für beide Geschlechter als angemessen angesehen. Bei Weibchen werden damit drei bis vier vollständige Östruszyklen abgedeckt, was genügen sollte, um etwaige Auswirkungen auf den Zyklus festzustellen. Bei männlichen Tieren entspricht dies der Zeit, die für den epididymalen Übergang der reifenden Spermien erforderlich ist; dieser Zeitraum sollte für die Feststellung posttestikulärer Auswirkungen auf Spermien (während der Endphasen der Spermiation und der epididymalen Spermienreifung) bei der Paarung ausreichen. Am Ende des Versuchs, d. h. der Zeitpunkt, für den die testikuläre und epididymale Histopathologie und die Analyse der Spermienparameter anberaumt sind, waren die P- und F1-Männchen zumindest für die Dauer eines vollständigen Spermatogeneseprozesses exponiert ((6) (7) (8) (9) und OECD Guidance Document Nr. 151 (40)).
27. Die Expositionsszenarien für Männchen vor der Paarung könnten angepasst werden, wenn in früheren Untersuchungen nachweislich testikuläre Toxizität (Beeinträchtigung der Spermatogenese) oder Beeinträchtigungen der Integrität und Funktionsfähigkeit der Spermien festgestellt wurden. Gleichermaßen können auch bei Weibchen vor der Paarung andere Expositionsszenarien gerechtfertigt sein, wenn Auswirkungen der Prüfsubstanz auf den Östruszyklus und damit auf die Empfänglichkeit bekannt sind. In besonderen Fällen kann es unter Umständen akzeptabel sein, dass mit der Behandlung der P-Weibchen erst nach einem spermienpositiven Abstrich begonnen wird (siehe OECD Guidance Document Nr. 151 (40)).
28. Sobald die Phase der Verabreichung vor der Paarung feststeht, sollten die Tiere bis zur Nekropsie nach einem 7 Tage/Woche-Schema mit der Prüfsubstanz behandelt werden. Allen Tieren sollte die Prüfsubstanz nach derselben Methode verabreicht werden. Die Verabreichung sollte während der zweiwöchigen Paarungszeit und — bei P-Weibchen — während der gesamten Trächtigkeitsdauer und Laktationsperiode bis zu ihrer Tötung am Tag nach dem Absetzen fortgeführt werden. Männchen sollten bis zu ihrer Tötung am Tag der Absetzung der F1-Tiere in derselben Weise zu behandeln. Bei der Nekropsie ist weiblichen Tieren Priorität einzuräumen; sie sollten am selben/einem ähnlichen Laktationstag seziert werden. Die Nekropsie der männlichen Tiere kann sich je nach Verfügbarkeit der Laboreinrichtungen über mehrere Tage erstrecken. Sofern mit der direkten Verabreichung der Prüfsubstanz nicht bereits während der Laktationsperiode begonnen wurde, sollte die Direktverabfolgung an ausgewählte F1-Männchen und F1-Weibchen am Tag des Absetzens beginnen und bis zur geplanten Nekropsie (je nach in Kohorteneinteilung) fortgesetzt werden.
29. Bei über das Futter oder das Trinkwasser verabreichten Prüfsubstanzen ist unbedingt sicherzustellen, dass die Mengen der jeweiligen Prüfsubstanz den normalen Nährstoff- oder Wasserhaushalt nicht beeinträchtigen. Wird die Prüfsubstanz über die Nahrung verabreicht, kann entweder eine konstante Konzentration im Futter/Wasser (ppm) oder eine konstante Dosis, bezogen auf das Körpergewicht des Tieres, verwendet werden. Dabei ist anzugeben, welche Option gewählt wurde.
30. Wird die Prüfsubstanz über eine Schlundsonde verabreicht, sollte die Menge der jeweils verabreichten Flüssigkeit grundsätzlich 1 ml/100 g Körpergewicht nicht übersteigen (0,4 ml/100 g Körpergewicht ist der Höchstwert für Öl, z. B. Maisöl). Außer bei reizenden oder ätzenden Stoffen, die in der Regel in höheren Konzentrationen eine verstärkte Wirkung hervorrufen, sollten Abweichungen bei den verabreichten Prüfsubstanzmengen minimiert werden, indem die Konzentration so angepasst wird, dass auf allen Dosisstufen ein konstantes Volumen gewährleistet ist. Die Behandlung sollte jeden Tag etwa zur gleichen Uhrzeit erfolgen. Die Dosis für jedes Tier sollte sich auf das aktuelle Körpergewichtstützen und ist bei adulten Männchen und nicht-trächtigen adulten Weibchen mindestens wöchentlich und bei trächtigen Weibchen und F1-Tieren alle zwei Tage anzupassen, wenn die Verabreichung vor dem Absetzen und während der zwei Wochen nach der Entwöhnung erfolgt. Falls toxikokinetische Daten auf eine schwache plazentare Übertragung der Prüfsubstanz hindeuten, muss die in der der letzten Trächtigkeitswoche über die Schlundsonde verabreichte Dosis unter Umständen angepasst werden, um die Verabreichung einer zu giftigen Dosis an das Muttertier zu verhindern. Weibliche Tiere sollten am Wurftag nicht mit einer Schlundsonde oder auf andere Weise behandelt werden, die eine Hantierung der Tiere erfordert; es ist besser, am Wurftag keine Prüfsubstanz zu verabreichen, als den Geburtsvorgang zu beeinträchtigen.
Paarung
31. Jedes P-Weibchen sollte so lange einem einzigen nach dem Zufallsprinzip ausgewählten nicht verwandten Männchen aus derselben Dosisgruppe zugeführt werden (1:1-Paarung), bis die Kopulation nachweislich stattgefunden hat oder zwei Wochen abgelaufen sind. Falls nicht genügend Männchen vorhanden sind, weil männliche Tiere beispielsweise noch vor der Paarung gestorben sind, können Männchen, die bereits ein Weibchen gedeckt haben (1:1) mit einem zweiten Weibchen angepaart werden, damit alle Weibchen gedeckt werden. Tag 0 der Trächtigkeit wird als der Tag festgelegt, an dem die Deckung (durch Vaginalpfropf oder Spermaspuren) nachgewiesen werden kann. Die Tiere sind nach der nachweislichen Kopulation so bald wie möglich zu trennen. Falls nach zwei Wochen keine Paarung stattgefunden hat, sollten die Tiere ohne weitere Paarungsmöglichkeit getrennt werden. Begattungspaare sind in den Aufzeichnungen als solche zu vermerken.
Wurfgröße
32. An PND 4 kann die Größe eines jeden Wurfs angepasst werden, indem überschüssige Jungtiere nach dem Zufallsprinzip aussortiert werden, um pro Wurf nach Möglichkeit fünf männliche und fünf weibliche Tiere zu erhalten. Die selektive Eliminierung von Jungtieren, z. B. auf der Grundlage des Körpergewichts, wird nicht empfohlen. Wenn es wegen der Anzahl männlicher bzw. weiblicher Jungtiere nicht möglich ist, pro Wurf jeweils fünf Jungtiere eines jeden Geschlechts zu erhalten, ist auch eine grobe Anpassung (beispielsweise sechs Männchen und vier Weibchen) akzeptabel.
Auswahl von Jungtieren für Untersuchungen nach der Entwöhnung (siehe Abbildung 1)
33. Zum Zeitpunkt der Entwöhnung (um PND 21) werden aus allen verfügbaren Würfen bis zu 20 Jungtiere pro Dosis- und Kontrollgruppe für weitere Untersuchungen ausgewählt und bis zur Geschlechtsreife gehalten (sofern keine früheren Tests erforderlich sind). Die Jungtiere werden nach dem Zufallsprinzip ausgewählt, wobei offensichtliche Kümmerlinge (Tiere mit mehr als zwei Standardabweichungen unter dem mittleren Jungtiergewicht des betreffenden Wurfes) grundsätzlich nicht einbezogen werden, da sie für die Behandlungsgruppe kaum repräsentativ sind.
An PND 21 werden die ausgewählten F1-Jungtiere nach dem Zufallsprinzip in eine der drei folgenden Tierkohorten eingeteilt:
Kohorte 1 (1A und 1B)= Testung auf Reproduktions-/Entwicklungstoxizität,
Kohorte 2 (2A und 2B)= Testung auf Entwicklungsneurotoxizität,
Kohorte 3= Testung auf Entwicklungsimmuntoxizität.
Kohorte 1A: Ein Männchen und ein Weibchen pro Wurf/Gruppe (20 pro Geschlecht/Gruppe): prioritäre Auswahl für die primäre Untersuchung der Auswirkungen auf die Fortpflanzungsorgane und der allgemeinen Toxizität.
Kohorte 1B: Ein Männchen und ein Weibchen pro Wurf/Gruppe (20 pro Geschlecht/Gruppe): prioritäre Auswahl für Folgeuntersuchungen der Reproduktionsleistung durch Paarung von F1-Tieren (siehe OECD Guidance Document Nr. 117 (39)) und zur Generierung zusätzlicher histopathologischer Daten in Fällen mutmaßlich reproduktionstoxischer oder endokrin wirksamer Stoffe oder wenn Ergebnisse für Kohorte 1A unschlüssig sind.
Kohorte 2A: Insgesamt 20 Jungtiere pro Gruppe (10 Männchen und 10 Weibchen pro Gruppe; ein Männchen oder ein Weibchen pro Wurf), eingeteilt für Untersuchungen neurologisch bedingten Verhaltens mit anschließender neurohistopathologischer Untersuchung im adulten Alter.
Kohorte 2B: Insgesamt 20 Jungtiere pro Gruppe (10 Männchen und 10 Weibchen pro Gruppe; ein Männchen oder ein Weibchen pro Wurf), eingeteilt für neurohistopathologische Untersuchungen am Tag des Absetzens (PND 21 oder 22). Wenn nicht genügend Tiere vorhanden sind, sollten Tiere vorrangig der Kohorte 2A zugewiesen werden.
Kohorte 3: Insgesamt 20 Jungtiere pro Gruppe (10 Männchen und 10 Weibchen pro Gruppe; ein Männchen oder ein Weibchen pro Wurf, soweit möglich). Unter Umständen werden zusätzliche Jungtiere aus der Kontrollgruppe benötigt, die im TDAR-Assay (T-Zell-abhängige Antikörperantwort) an PND 56 ± 3 Tage als positive Kontrolltiere dienen.
34. Sollte ein Wurf nicht genügend Tieren umfassen, um alle Kohorten zu bedienen, so wird Kohorte 1 Vorrang eingeräumt, da sie zur Produktion einer F2-Generation erweitert werden kann. Zusätzliche Jungtiere können bei speziellem Bedarf, z. B. wenn es sich bei einer Chemikalie möglicherweise um ein Neurotoxin, ein Immuntoxin oder ein Reproduktionstoxin handelt, jeder beliebigen Kohorte zugewiesen werden. Diese Jungtiere können für Untersuchungen zu anderen Zeitpunkten oder ergänzender Endpunkte verwendet werden. Jungtiere, die keinen Kohorten zugeteilt werden, werden klinisch und biochemisch (Nummer 55) untersucht und seziert (Nummer 68) untersucht.
Zweite Paarung von P-Tieren
35. Eine zweite Paarung wird für P-Tiere in der Regel nicht empfohlen, denn sie geht mit dem Verlust wichtiger Informationen über die Zahl der Implantationsstellen (und somit Angaben über Abgänge nach der Implantation und über perinatale Abgängen — Indikatoren eines teratogenen Potenzials) für den ersten Wurf einher. Wirkungen in exponierten weiblichen Tieren ließen sich durch eine Erweiterung der Prüfung durch Paarung der F1-Generation leichter bestätigen oder klären. Eine zweite Paarung der P-Männchen mit unbehandelten weiblichen Tieren ist jedoch stets eine Möglichkeit, um unschlüssige Ergebnisse zu klären oder um bei der ersten Paarung festgestellte Auswirkungen auf die Fruchtbarkeit näher zu charakterisieren.
BEOBACHTUNGEN AM LEBENDEN TIER
Klinische Beobachtungen
36. Bei den P-Tieren und den ausgewählten F1-Tieren wird einmal täglich eine allgemeine klinische Untersuchung vorgenommen. Bei Verabreichung der Prüfsubstanz über eine Schlundsonde sind die klinischen Beobachtungen (auf mögliche Toxizitätsanzeichen, die Plasmakonzentrationsspitzen zugeordnet werden) vor und nach der Verabreichung vorzunehmen. Pertinente Verhaltensänderungen, Anzeichen für eine schwierige oder langwierige Geburt und alle Toxizitätsanzeichen werden festgehalten. Zweimal täglich — am Wochenende einmal täglich — werden alle Tiere auf Anzeichen von schwerer Toxizität, Morbidität und Mortalität untersucht.
37. Darüber hinaus werden alle P- und F1 -Tiere (nach dem Entwöhnen) einmal wöchentlich eingehender untersucht; diese Untersuchung könnte praktischerweise durchgeführt werden, wenn das Tier gewogen wird, um den umgangsbedingten Stress auf ein Mindestmaß zu reduzieren. Die Untersuchungen sind sorgfältig durchzuführen und nach einer speziell vom Prüflabor entwickelten Bewertungsskala zu dokumentieren. Die Testbedingungen sollten möglichst konstant sein. Die festzuhaltenden Anzeichen umfassen, ohne darauf beschränkt zu sein, u. a. Veränderungen an Haut, Fell, Augen, Schleimhäuten sowie Sekrete und Exkretionen und autonome Körperfunktionen (wie Tränensekretion, Piloerektion, Pupillenveränderung, ungewöhnliche Atmungsmuster). Gang- und Haltungsstörungen, Reaktionen auf Berührungen sowie etwaige klonisch-tonische Anfälle, stereotype Verhaltensweisen (z. B. übermäßiges Putzen, wiederholte Kreisbewegungen) oder abnormes Verhalten (z. B. Selbstverstümmelung, Rückwärtsgehen) sollten ebenfalls dokumentiert werden.
Körpergewicht und Futter-/Trinkwasseraufnahme
38. P-Tiere werden am ersten Tag der Verabreichung der Prüfsubstanz und danach mindestens einmal wöchentlich gewogen. Darüber hinaus werden P-Weibchen während der Laktation an denselben Tagen gewogen wie die Jungtiere in ihren Würfen (siehe Nummer 44). Alle F1-Tiere werden am Tag des Absetzens (PND 21) und danach mindestens einmal wöchentlich einzeln gewogen. Das Körpergewicht wird auch an dem Tag aufgezeichnet, an dem die Pubertät eintritt (Abschluss der Präputialseparation oder Öffnung der Vagina). Alle Tiere werden bei der Tötung gewogen.
39. Im Laufe des Versuchs werden Futter- und Wasseraufnahme (bei Verabreichung der Prüfsubstanz im Trinkwasser) mindestens einmal wöchentlich stets an denselben Tagen gemessen und erfasst, an denen auch das Körpergewicht gemessen und dokumentiert wird (außer während der Kohabitation). Die Futteraufnahme der F1-Tiere jedes Käfigs wird ab der Einteilung in eine Kohorte einmal wöchentlich dokumentiert.
Östruszyklen
40. Es existieren möglicherweise bereits vorläufige Informationen über die Auswirkungen der Prüfsubstanz auf den Östruszyklus aus früheren Prüfungen auf Toxizität bei wiederholter Verabreichung, die zur Festlegung eines prüfsubstanzspezifischen Protokolls für die EOGRTS-Prüfung herangezogen werden können. In der Regel beginnt die Bewertung der weiblichen Zyklizität (durch Vaginalzytologie) zu Beginn des Behandlungszeitraums und wird bis zur Paarungsbestätigung bzw. bis zum Ende der zweiwöchigen Paarungszeit fortgesetzt. Falls weibliche Tiere vor der Behandlung auf einen normalen Östruszyklus hin untersucht wurden, ist es zweckdienlich, die Abstriche auch nach Beginn der Behandlung fortzusetzen; wenn jedoch zu Beginn der Behandlung Bedenken in Bezug auf unspezifische Auswirkungen bestehen (beispielsweise eine beginnende ausgeprägte Futterverweigerung), kann den Tieren bis zu zwei Wochen Zeit gelassen werden, um sich der Behandlung anzupasen, bevor die zweiwöchige Abstrichperiode anläuft, die der Paarung vorausgeht. Wenn die Behandlungszeit für die weiblichen Tiere auf diese Art und Weise (d. h. auf eine vierwöchige Behandlung vor der Paarung) verlängert wird, sollte der Erwerb jüngerer Tiere und die Verlängerung des Behandlungszeitraums für männliche Tiere vor der Paarung in Erwägung gezogen werden. Bei der Entnahme vaginaler/zervikaler Zellen ist sorgfältig darauf zu achten, dass die Schleimhaut nicht gereizt und infolgedessen eine Pseudogravidität eingeleitet wird (10) (11).
41. Vaginalabstriche sind bei allen F1-Weibchen in der Kohorte 1A nach beginnender Öffnung der Vagina bis zur Erfassung des ersten verhornten Abstrichs täglich zu untersuchen, um den zeitlichen Abstand zwischen diesen beiden Ereignissen zu bestimmen. Außerdem sollten die Östruszyklen bei allen F1-Weibchen in Kohorte 1A über einen Zeitraum von zwei Wochen, etwa ab PND 75, überwacht werden. Sollte sich die Paarung der F1-Generation als nötig erweisen, wird die Vaginalzytologie in Kohorte 1B vom Zeitpunkt der Paarung an bis zur nachweislichen Deckung überwacht.
Deckung und Gravidität
42. Zusätzlich zu den Standardendpunkten (z. B. Körpergewicht, Futteraufnahme, klinische Beobachtungen, einschließlich Mortalitäts-/Morbiditätskontrollen) werden die Zeitpunkte der Paarung, der Befruchtung und der Geburt aufgezeichnet und das präkoitale Intervall (Paarung bis Befruchtung) sowie die Dauer der Gravidität (Befruchtung bis Geburt) berechnet. Die P-Weibchen sind zum Zeitpunkt des voraussichtlichen Partus sorgfältig auf Anzeichen von Dystokie zu untersuchen. Alle Abnormitäten beim Nistverhalten oder bei der Säugeleistung sind zu dokumentieren.
43. Der Wurftag ist für das Muttertier Laktationstag (LD) 0 und für die Nachkommen der postnatale Tag (PND) 0. Alternativ dazu können auch die Tage nach der Kopulation herangezogen werden, um Fehler bei den Daten über die postnatale Entwicklung zu vermeiden, die auf Unterschiede bei der Trächtigkeitsdauer zurückzuführen sind; die Zeit nach der Geburt ist jedoch ebenfalls zu dokumentieren. Dies ist vor allem wichtig, wenn sich die Prüfsubstanz auf die Trächtigkeitsdauer auswirkt.
Parameter für die Nachkommen
44. Jeder Wurf ist so bald wie möglich nach der Geburt (PND 0 oder 1) zu untersuchen, um die Anzahl und Geschlecht der Jungtiere, Totgeburten, Lebendgeburten und etwaige grobe Anomalien (extern sichtbare Abnormalitäten, einschließlich Gaumenspalten; subkutane Hämorrhagien; anormale Hautfarbe oder Hautbeschaffenheit; Vorhandensein der Nabelschnur; Abwesenheit von Milch im Magen; Präsenz von trockener Absonderungen) festzustellen. Darüber hinaus sollte die erste klinische Untersuchung der Neugeborenen auch eine qualitative Beurteilung der Körpertemperatur, des Bewegungsmusters und der Reaktion auf Berührungen beinhalten. Jungtiere, die am PND 0 oder zu einem späteren Zeitpunkt tot aufgefunden werden, sind auf mögliche Defekte und auf die Todesursache hin zu untersuchen. Lebende Jungtiere werden am PND 0 oder 1 gezählt und danach regelmäßig, mindestens jedoch an PND 4, 7, 14 und 21, einzeln gewogen. Klinische Untersuchungen, die je nach dem Alter der Tiere durchzuführen sind, sollten wiederholt werden, wenn die Nachkommen gewogen werden, oder auch öfter, wenn bei ihrer Geburt fallspezifische Befunde festgestellt wurden. Zu achten ist insbesondere auf Veränderungen an Haut, Fell, Augen und Schleimhäuten, auf Absonderungen und Ausscheidungen sowie autonome Körperfunktionen. Gang- und Haltungsstörungen sowie Reaktionen auf Berührungen und etwaige klonisch-tonische Anfälle, stereotype oder bizarre Verhaltensweisen sind ebenfalls zu dokumentieren.
45. Der anogenitale Abstand (AGD) sollte bei jedem Jungtier mindestens einmal (zwischen PND 0 und PND 4) gemessen werden. Das Körpergewicht des Jungtiers wird am Tag der Messung des AGD erfasst, der auf Jungtiergröße — vorzugsweise die Quadratwurzel des Körpergewichts — genormt sein sollte (12). Das Vorhandensein von Brustwarzen/Warzenhöfen bei männlichen Jungtieren ist am PND 12 oder 13 zu kontrollieren.
46. Alle ausgewählten F1-Tiere werden täglich auf Vorhaut-Eichel-Trennung bei männlichen Tieren bzw. Öffnung der Vagina bei weiblichen Tieren untersucht, und zwar vor dem Tag, an dem das Erreichen dieser Endpunkte erwartet wird, um festzustellen, ob die Geschlechtsreife eventuell früh eintritt. Alle Abnormalitäten der Geschlechtsorgane (wie persistente Vaginalfilamente, Hypospadie oder Spaltpenis) sind festzuhalten. Die Geschlechtsreife der F1-Tiere wird mit der körperlichen Entwicklung verglichen, indem Alter und Körpergewicht zum Zeitpunkt der Vorhaut-Eichel-Trennung bei männlichen Tieren bzw. der Öffnung der Vagina bei weiblichen Tieren bestimmt werden (13).
Bewertung der potenziellen Entwicklungsneurotoxizität (Kohorten 2A und 2B)
47. Für Bewertungen der Neurotoxizität sind aus jeder Behandlungsgruppe zehn männliche und zehn weibliche Tiere der Kohorte 2A sowie zehn männliche und zehn weibliche Tiere der Kohorte 2B zu verwenden (für jede Kohorte: ein männliches oder ein weibliches Tier pro Wurf; alle Würfe müssen durch mindestens ein nach dem Zufallsprinzip ausgewähltes Jungtier vertreten sein). Tiere der Kohorte 2A sind FOB-Tests (Functional Observational Battery) sowie Untersuchungen auf akustische Schreckreaktion und motorische Aktivität (siehe Nummern 48-50) sowie neuropathologischen Tests (siehe Nummern 74-75) zu unterziehen. Dabei ist nach Möglichkeit sicherzustellen, dass die Prüfbedingungen möglichst wenig variieren und dass diese Variationen nicht systematisch mit der Behandlung zusammenhängen. Zu den Variablen, die das Verhalten beeinflussen können, gehören Geräuschpegel (beispielsweise intermittierender Lärm), Temperatur, Feuchtigkeit, Beleuchtung, Gerüche, Tageszeit und Ablenkungen aus der Umgebung. Die Ergebnisse der Neurotoxizitätstests sind bezogen auf entsprechende historische Kontrollreferenzbereiche auszulegen. Tiere der Kohorte 2B sollten an PND 21 oder 22 neuropathologisch untersucht werden (siehe Nummern 74-75).
48. Ein Test auf akustische Schreckreaktion sollte am PND 24 (± 1 Tag) mit Tieren der Kohorte 2A durchgeführt werden. Die Untersuchung der Behandlungsgruppe und der Kontrollgruppe ist ausgewogen über den Tag zu verteilen. Jede Testreihe umfasst 50 Prüfungen. Bei diesen Tests wird die mittlere Reaktionsamplitude für jeden Block von 10 Prüfungen (5 Blöcke mit je 10 Prüfungen) berechnet, dessen Prüfbedingungen optimiert wurden, um während der Testreihe Habituation zu erzeugen. Diese Verfahren sollten mit der Prüfmethode B.53 (35) übereinstimmen.
49. Zu einem geeigneten Zeitpunkt zwischen PND 63 und PND 75 werden die Tiere der Kohorte 2A einem FOB-Test und einem automatisierten Motoriktest unterzogen. Diese Verfahren sollten mit den Prüfmethoden B.43 (33) und B.53 (35) übereinstimmen. Der FOB-Test beinhaltet eine ausführliche Beschreibung des äußeren Erscheinungsbildes, des Verhaltens und der funktionalen Integrität des Versuchstiers. Diese Bewertungen beruhen auf Beobachtungen im Haltungskäfig, in einem Standardgehege (offenes Feld), in dem sich das Tier frei bewegen kann, sowie durch Manipulationstests. Die Untersuchungen sollten in aufsteigender Reihenfolge der Interaktivität durchgeführt werden. Anhang 1 enthält eine Liste von Maßnahmen. Alle Tiere sollten von geschulten Beobachtern untersucht werden, denen die Behandlungsphase des jeweiligen Tieres nicht bekannt ist. Dabei sind standardisierte Verfahren anzuwenden, um beobachterbedingte Abweichungen auf ein Mindestmaß zu begrenzen. Nach Möglichkeit sollten die Tiere in einem bestimmten Test stets durch denselben Beobachter untersucht werden. Falls dies nicht möglich ist, ist die Zuverlässigkeit bei verschiedenen Beobachtern auf andere Weise nachzuweisen. Für jeden Parameter der Verhaltensprüfbatterie sind Skalen und Bewertungskriterien zu verwenden, deren Anwendung genau festgelegt ist. Nach Möglichkeit sind für Beobachtungsendpunkte, die subjektive Einstufungen beinhalten, objektive quantitative Messungen durchzuführen. In Bezug auf die Motorik wird jedes Tier einzeln getestet. Die Testreihe sollte so lange dauern, bis bei Kontrollen eine Habituation innerhalb der Testreihe nachgewiesen werden kann. Die Motorik sollte mithilfe eines automatischen Bewegungsmessgeräts untersucht werden, das in der Lage ist, sowohl eine Zunahme als auch ein Nachlassen der Bewegung zu erfassen (d. h., die vom Gerät gemessene Basisbewegung sollte weder so schwach sein, dass dadurch die Erfassung eines Nachlassen der Bewegung unmöglich wird, noch so stark, dass Bewegungszunahmen nicht erfasst werden können). Jedes einzelne Gerät sollte im Rahmen standardisierter Verfahren geprüft worden sein, um bei Einsatz mehrerer Geräte über mehrere Tage eine möglichst hohe Betriebssicherheit zu gewährleisten. Die Behandlungsgruppen sollten so weit wie möglich gleichmäßig auf die Geräte verteilt werden. Die Untersuchung Beobachtung der Behandlungsgruppen sollte über den Tag verteilt werden, um zirkadianen Aktivitätsrhythmus zu berücksichtigen.
50. Falls vorhandene Informationen auf die Notwendigkeit hindeuten, weitere funktionelle Tests durchzuführen (z. B. sensorische, soziale oder kognitive Tests), sind diese zu berücksichtigen, ohne die Integrität der anderen durchgeführten Untersuchungen zu beeinträchtigen. Falls diese Tests an denselben Tieren durchgeführt werden, die auch für den Standardtest auf akustische Schreckreaktion, den FOB-Test und den Motoriktest verwendet wurden, sind andere Tests einzuplanen, um das Risiko, dass die Integrität dieser Tests beeinträchtigt wird, auf ein Mindestmaß zu begrenzen. Zusätzliche Prüfverfahren können insbesondere dann sinnvoll sein, wenn die empirische Beobachtung, die erwarteten Wirkungen oder der Wirkmechanismus/die Wirkungsweise auf eine bestimmte Art von Neurotoxizität hindeuten.
Bewertung einer potenziellen Entwicklungsimmuntoxizität (Kohorte 3)
51. Am postnatalen PND 56 (± 3 Tage) sollten aus jeder Behandlungsgruppe zehn männliche und zehn weibliche Tiere der Kohorte 3 (ein männliches oder ein weibliches Tier pro Wurf; alle Würfe müssen durch mindestens ein nach dem Zufallsprinzip ausgewähltes Jungtier vertreten sein) im Einklang mit den aktuellen Immuntoxizitätstestverfahren (14) (15) einem Test auf T-Zell-abhängige Antikörperantwort (d. h. auf primäre IgM-Antikörperantwort auf ein T-Zell-abhängiges Antigen, wie zum Beispiel rote Blutkörperchen von Schafen oder Schlitzschnecken-Hämocyanin (KLH)) unterzogen werden. Die Reaktion kann bestimmt werden durch Zählung spezifischer plaquebildender Zellen (PFC) in der Milz oder durch Bestimmung des Titerwertes für SRBC- oder KLH-spezifische IgM-Antikörper im Serum mittels ELISA-Test auf dem Höhepunkt der Reaktion. Die Maximalreaktionen lassen sich in der Regel vier (PFC-Reaktion) oder fünf (ELISA-Test) Tage nach der intravenösen Beimpfung festzustellen. Wird die primäre Antikörperreaktion durch Auszählen der plaquebildenden Zellen bestimmt, so ist die Bewertung von Untergruppen von Tieren an getrennten Tagen zulässig, sofern die Immunisierung der Untergruppe und die Tötung der Tiere zeitlich so geplant sind, dass die plaquebildenden Zellen zum Zeitpunkt der Höchstreaktion gezählt werden, die Untergruppen aus ebenso vielen männlichen wie weiblichen Nachkommen aller Dosisgruppen, einschließlich Kontrolltieren, bestehen und die Tiere der Untergruppen ungefähr im selben postnatalen Alter untersucht werden.Die Exposition gegenüber der Prüfsubstanz wird bis zum Tag vor der Entnahme der Milz zur Bestimmung der PFC-Reaktion oder des Serums für den ELISA-Test fortgesetzt.
Folgeuntersuchung auf potenzielle Reproduktionstoxizität (Kohorte 1B)
52. Tiere der Kohorte 1B können gegebenenfalls auch nach PND 90 weiterbehandelt und gezüchtet werden, um erforderlichenfalls eine F2-Generation zu produzieren. Männliche und weibliche Tiere derselben Dosisgruppen sind ab oder nach PND 90 bis zu zwei Wochen lang, allerdings nicht über PND 120 hinaus, zusammenzuführen (wobei die Paarung von Geschwistern zu vermeiden ist). Es sollten genau so vorgegangen werden wie bei den P-Tieren. Sofern dies nachgewiesen weren kann, reicht es jedoch unter Umständen jedoch aus, die Würfe an PND 4 zu töten, statt sie bis zur Entwöhnung oder darüber hinaus weiter zu untersuchen.
ABSCHLIESSENDE BEOBACHTUNGEN
Klinisch-biochemische/Hämatologische Untersuchungen
53. Systemische Wirkungen in P-Tieren sollten überwacht werden. An einer vorgegebenen Stelle werden von zehn nach dem Zufallsprinzip ausgewählten P-Männchen und -Weibchen pro Dosisgruppe am Versuchsende Nüchternblutproben entenommen, unter angemessenen Bedingungen gelagert und teilweise oder vollständig hämatologischen, klinischen, bio-chemischen Untersuchungen, einer T4- und TSH-Analyse oder anderen Tests unterzogen, die aufgrund des bekannten Wirkungsprofils der Prüfsubstanz naheliegen (siehe OECD Guidance Document Nr. 151 (40)). Die folgenden hämatologischen Parameter sollten dabei untersucht werden: Hämatokrit, Hämoglobinkonzentration, Erythrozytenzahl, Gesamt- und Differential-Leukozytenzahl, Thrombozytenzahl und Blutgerinnungszeit/-fähigkeit. Die Plasma- oder Serumuntersuchungen sollten Folgendes umfassen: Glucose, Gesamtcholesterin, Harnstoff, Kreatinin, Gesamtprotein und Albumin sowie mindestens zwei Enzyme, die auf hepatozelluläre Wirkungen schließen lassen (wie Alanin-Aminotransferase, Aspartat-Aminotransferase, alkalische Phosphatase, γ-Glutamyltranspeptidase und Glutamatdehydrogenase). Die Bestimmung weiterer Enzyme und Gallensäuren kann unter bestimmten Umständen ebenfalls wertvolle Hinweise liefern. Darüber hinaus können Blutproben von allen Tieren entnommen und für eine spätere Analyse aufbewahrt werden, um unschlüssige Wirkungsergebnisse zu klären oder interne Expositionsdaten zu generieren. Ist keine zweite Paarung der P-Tiere beabsichtigt, werden die Blutproben unmittelbar vor oder bei der geplanten Tötung der Tiere gezogen. Falls Tiere behalten werden, sind die Blutproben einige Tage vor der zweiten Paarung der Tiere zu ziehen. Sofern aus vorliegenden Daten aus Untersuchungen mit wiederholter Verabreichung nicht hervorgeht, dass der Parameter nicht durch die Prüfsubstanz beeinträchtigt wird, sollten vor Abschluss der Studie eine Urinuntersuchung durchgeführt und die folgenden Parameter bewertet werden: Aussehen, Volumen, Osmolalität oder Dichte, pH-Wert, Protein, Glucose, Blut und Blutzellen, Zelltrümmer. Urin kann auch gesammelt werden um die Ausscheidung der Prüfsubstanz und/oder Metaboliten zu überwachen.
54. Systemische Wirkungen sind auch in F1-Tieren zu überwachen. Von zehn nach dem Zufallsprinzip ausgewählten Männchen und Weibchen der Kohorte 1A pro Dosisgruppe werden am Versuchsende an einer vorgegebenen Stelle Nüchternblutproben entnommen, unter angemessenen Bedingungen gelagert und klinischen bio-chemischen Standarduntersuchungen, einschließlich einer Bewertung der Serumspiegel auf Schilddrüsenhormone (T4- und TSH), hämatologischen Untersuchungen (Gesamt- und Differenzialleukozytenzahl) sowie Urinuntersuchungen unterzogen.
55. Die an PND 4 überzähligen Jungtiere werden makroskopisch untersucht, wobei erwogen werden kann, die Konzentration der Schilddrüsenhormone (T4) im Serum zu bestimmen. Erforderlichenfalls können nach Würfen Blutproben Neugeborener (PND 4) für biochemische Analysen und zur Bestimmung der Konzentration der Schilddrüsenhormone gepoolt werden. Blutproben für die T4- und TSH-Analyse wird außerdem von gerade entwöhnten Tieren gezogen, die am PND 22 makroskopisch untersucht werden (F1-Jungtiere, die nicht für Kohorten ausgewählt werden).
Spermienparameter
56. Spermienparameter sollten in allen männlichen Tieren der P-Generation gemessen werden, sofern keine Daten vorliegen, die nachweisen, dass Spermienparameter in einem 90-Tage-Versuch nicht beeinträchtigt werden. Die Untersuchung der Spermienparameter sollte bei allen männlichen Tieren der Kohorte 1A durchgeführt werden.
57. Bei Versuchsabschluss wird für alle männlichen P- und F1-Tieren (Kohorte 1A) das Gewicht der Hoden und Nebenhoden aufgezeichnet. Mindestens ein Hoden und ein Nebenhoden werden für die histopathologische Untersuchung konserviert. Der verbleibende Nebenhoden wird zur Auszählung von Spermienreserven im Nebenhodenschwanz (Cauda epididymis) (16) (17) verwendet. Darüber hinaus werden Spermien aus dem Nebenhodenschwanz bzw. aus dem Samenleiter (Vas deferens) mithilfe von Methoden gewonnen, die Schäden für die Bewertung der Motilität und Morphologie der Spermien auf ein Mindestmaß begrenzen (18).
58. Die Spermienmotilität kann entweder unverzüglich nach der Tötung bewertet oder für eine spätere Analyse aufgezeichnet werden. Der Anteil der progressiv beweglichen Spermien könnte entweder subjektiv oder mithilfe einer computergestützten Bewegungsanalyse objektiv bestimmt werden (19) (20) (21) (22) (23) (24). Für die Bewertung der Morphologie der Spermien sollte eine Spermienprobe aus dem Nebenhoden (oder aus dem Samenleiter) als Fest- oder Feuchtpräparat (25) untersucht werden, wobei mindestens 200 Spermien pro Probe entweder als normal (sowohl Kopf als auch Mittelstück/Schwanz erscheinen normal) oder abnormal einzustufen sind. Morphologische Abnormalitäten der Spermien wären beispielsweise die Verschmelzung von Köpfen, isolierte Köpfe und Kopf- und/oder Schwanzmissbildungen (26). Missgebildete oder große Spermienköpfe können auf Störungen bei der Spermiation hindeuten.
59. Werden zum Zeitpunkt der Sektion Spermienproben eingefroren, Abstriche fixiert und Bilder zur Analyse der Spermienmotilität dokumentiert (27), kann die anschließende Analyse auf männliche Tiere der Kontrollgruppe und der Hochdosisgruppe beschränkt werden. Werden jedoch behandlungsbezogene Wirkungen beobachtet, sind auch die niedrigeren Dosisgruppen zu bewerten.
Makroskopische Untersuchung
60. Bei Versuchsende oder bei vorzeitigem Tod werden alle P- und F1-Tiere seziert und makroskopisch auf etwaige strukturelle Abnormalitäten oder pathologische Veränderungen hin untersucht. Dabei ist besonders auf die Organe des Fortpflanzungssystems zu achten. Jungtiere, die in moribundem Zustand auf humane Weise getötet werden, und tote Jungtiere sind zu dokumentieren und sollten — wenn sie nicht mazeriert werden — auf mögliche Defekte und/oder die Ursache des Todes untersucht und konserviert werden.
61. Bei adulten P- und F1-Weibchen ist am Tag der Sektion ein Vaginalabstrich zu untersuchen, um das Stadium des Östruszyklus zu bestimmen und eine Korrelation zur histopathologischen Untersuchung der Fortpflanzungsorgane zu ermöglichen. Die Uteri aller P-Weibchen (und gegebenenfalls auch aller F1-Weibchen) sind so auf Vorhandensein und Anzahl von Implantationsstellen zu untersuchen, dass die histopathologische Bewertung nicht beeinträchtigt wird.
Wiegen der Organe und Konservation von Gewebe -P-Tiere und adulte F1 -Tiere
62. Bei Versuchsende werden von allen P-Tieren und von allen adulten F1-Tieren der relevanten (nachstehend angeführten) Kohorten möglichst bald nach der Sektion Körpergewicht und Nassgewicht der nachstehend angeführten Organe bestimmt, um Austrocknen zu vermeiden. Die genannten Organe sind anschließend unter geeigneten Bedingungen zu konservieren. Sofern keine anderslautenden Vorgaben vorliegen, können paarige Organe einzeln oder zusammen entsprechend der üblichen Praxis des ausführenden Labors gewogen werden.
|
— |
Uterus (mit Ovidukten und Zervix), Ovarien; |
|
— |
Hoden, Nebenhoden (Gesamt und Nebenhodenschwanz für die zur Spermienauszählung verwendeten Proben); |
|
— |
Prostata (dorsolaterale und ventrale Teile zusammen). Bei der Entfernung der anhaftenden Gewebe von der Prostata ist sorgfältig darauf zu achten, dass die mit Flüssigkeit gefüllten Samenbläschen nicht perforiert werden. Wirkte sich die Behandlung auf das Gesamtgewicht der Prostata aus, sind die dorsolateralen und ventralen Segmente nach ihrer Fixierung sorgfältig zu sezieren und separat zu wiegen; |
|
— |
Samenbläschen mit Koagulationsdrüsen und ihre Flüssigkeiten (als eine Einheit); |
|
— |
Gehirn, Leber, Nieren, Herz, Milz, Thymusdrüse, Hypophyse, Schilddrüse (nach Fixierung), Nebennieren sowie bekannte Zielorgane oder -gewebe. |
63. Zusätzlich zu den oben angeführten Organen sollten auch Proben des peripheren Nervengewebes, der Muskeln, des Rückenmarks, des Auges mit Sehnerv, des Magen-Darm-Trakts, der Harnblase, der Lunge, der Trachea (mit anhaftender Schilddrüse und Nebenschilddrüse), des Knochenmarks, des Samenleiters (bei männlichen Tieren), der Brustdrüse (bei männlichen und weiblichen Tieren) und der Vagina unter geeigneten Bedingungen zu konservieren.
64. Bei Tieren der Kohorte 1A sind alle Organe zu wiegen und für die histopathologische Untersuchung zu konservieren.
65. Für die Ermittlung prä- und postnatal induzierter immuntoxischer Wirkungen sind zehn männliche und zehn weibliche Tiere der Kohorte 1A einer jeden Behandlungsgruppe (ein Männchen oder ein Weibchen pro Wurf; alle Würfe werden durch mindestens ein nach dem Zufallsprinzip ausgewählten Jungtier repräsentiert) bei Versuchsende den folgenden Untersuchungen zu unterziehen:
|
— |
Wiegen der (regionären) Lymphknoten entlang des Expositionswegs und der überregionären Lymphknoten (zusätzlich zum Gewicht der Nebennieren, des Thymus und der Milz, das bereits für alle Tiere der Kohorte 1A bestimmt wurde); |
|
— |
Untersuchung einer Hälfte der Milz auf Lymphozytensubpopulation in der Milz (T-Lymphozyten CD4+ und CD8+, B-Lymphozyten und natürliche Killerzellen); die andere Milzhälfte wird für die histopathologische Auswertung konserviert. |
Die Analyse der Lymphozytensubpopulationen in der Milz nicht immunisierter Tiere (Kohorte 1A) zeigt, ob die Exposition mit einer Veränderung der immunologischen Steady-State-Verteilung von ‚Helferzellen‘ (CD4+) oder von zytotoxischen T-Lymphozyten (CD8+) oder von natürlichen Killerzellen (NK) (schnelle Reaktionen auf neoplastische Zellen und Pathogene) zusammenhängt.
66. Bei Tieren der Kohorte 1B sollten die folgenden Organe gewogen und die entsprechenden Gewebe zum Blockstadium umgewandelt werden:
|
— |
Vagina (nicht gewogen), |
|
— |
Uterus mit Zervix, |
|
— |
Ovarien, |
|
— |
Hoden (zumindest einer), |
|
— |
Nebenhoden, |
|
— |
Samenbläschen und Koagulationsdrüsen, |
|
— |
Prostata, |
|
— |
Hypophyse, |
|
— |
vorgesehene Zielorgane. |
Histopathologische Untersuchungen von Kohorte 1B sollten dann durchgeführt werden, wenn die Ergebnisse der Kohorte 1A unschlüssig sind oder ein Verdacht auf Reproduktionstoxine oder endokrine Disruptoren besteht.
67. Kohorten 2A und 2B: Tests auf Entwicklungsneurotoxizität (PND 21 oder PND 22 sowie adulte Nachkommen). Tiere der Kohorte 2A werden nach den Verhaltenstests getötet, das Gewicht ihres Gehirns wird aufgezeichnet und sie werden zur Bewertung der Neurotoxizität vollständig neurohistopathologisch untersucht. Tiere der Kohorte 2B werden am PND 21 oder PND 22 getötet; das Gewicht ihres Gehirns wird aufgezeichnet und das Gehirn wird zur Bewertung der Neurotoxizität mikroskopisch untersucht. Für Tiere der Kohorte 2A und optional für Tiere der Kohorte 2B ist eine Perfusionsfixierung im Sinne der Prüfmethode B.53 (35) erforderlich.
Wiegen der Organe und Konservierung von Gewebe — entwöhnte F1-Tiere
68. Nicht für die Kohorten ausgewählte Jungtiere, einschließlich Kümmerlinge, werden nach dem Absetzen an PND 22 getötet, sofern die Ergebnisse nicht darauf hindeuten, dass weitere Beobachtungen an lebenden Tieren erforderlich sind. Getötete Jungtiere werden seziert und ihre Fortpflanzungsorgane werden, wie unter den Nummern 62 und 63 beschrieben, untersucht. Bei bis zu zehn Jungtieren pro Geschlecht und Gruppe aus möglichst vielen Würfen sollten Gehirn, Milz und Thymusdrüse gewogen und unter geeigneten Bedingungen konserviert werden. Darüber hinaus kann von diesen männlichen und weiblichen Jungtieren Brustdrüsengewebe für eine weitere mikroskopische Analyse (13) (siehe OECD Guidance Document Nr. 151 (40)) konserviert werden. Massive Abnormalitäten und Zielgewebe sollten für eine etwaige spätere histologische Untersuchungen sichergestellt werden.
Histopathologische Untersuchung — P-Tiere
69. Eine vollständige histopathologische Untersuchung der unter den Nummern 62 und 63 genannten Organe wird bei allen P-Tieren der Hochdosisgruppen und der Kontrollgruppen durchgeführt. Organe, die behandlungsbedingte Veränderungen aufweisen, sollten auch bei allen Tieren in Niedrigdosisgruppen untersucht werden, um die Bestimmung eines NOAEL-Werts zu unterstützen. Darüber hinaus sollten die Fortpflanzungsorgane aller Tiere mit mutmaßlich verringerter Fruchtbarkeit histopathologisch zu untersuchen, wie Tiere, die sich nicht gepaart haben, die nicht empfangen haben, nicht gedeckt wurden, keine gesunden Nachkommen geboren haben oder bei denen der Östruszyklus oder die Zahl, die Motilität oder die Morphologie der Spermien beeinträchtigt waren, sowie alle makroskopischen Läsionen histopathologisch untersucht werden.
Histopathologische Untersuchung — F1-Tiere
Tiere der Kohorte 1
70. Eine vollständige histopathologische Untersuchung der unter den Nummern 62 und 63 genannten Organe wird bei allen adulten Tieren der Hochdosisgruppen und der Kontrollgruppen der Kohorte 1A durchgeführt. Alle Würfe sollten durch mindestens ein Jungtier pro Geschlecht repräsentiert sein. Organe und Gewebe, die behandlungsbedingte Veränderungen aufweisen, sind auch von allen Tieren in Niedrigdosisgruppen zu untersuchen, um die Bestimmung eines NOAEL-Werts zu unterstützen. Für die Bewertung der prä- und postnatal induzierter Wirkungen auf Lymphorgane sollten neben einer bereits in allen 1A-Tieren durchgeführten histopathologischen Bewertung der Thymusdrüse, der Milz und der Nebennieren auch die gesammelten Lymphknoten und das Knochenmark von zehn männlichen und zehn weiblichen Tieren der Kohorte 1A histopathologisch untersucht werden.
71. Bei mutmaßlichen Reproduktionstoxinen oder endokrinen Disruptoren sollten nach der Beschreibung unter Nummer 66 zu Blockstadien umgewandeltes Gewebe der Fortpflanzungsorgane und des endokrinen Gewebes von allen Tieren der Kohorte 1B histopathologisch untersucht werden. Falls die Ergebnisse der Kohorte 1A unschlüssig sind, sollte auch die Kohorte 1B histologisch untersucht werden.
72. Ovarien adulter Weibchen sollten Primordialfollikel und heranreifende Follikel sowie Gelbkörper enthalten; daher sollte eine histopathologische Untersuchung darauf abzielen, Primordialfollikel und kleine heranreifende Follikel sowie Gelbkörper in F1-Weibchen quantitativ zu bestimmen; die Zahl der Tiere, die Wahl der Ovarsektion und der Umfang der Stichprobe für die Sektion sollten für das angewandte Bewertungsverfahren statistisch aussagekräftig sein. Zunächst können die Follikel bei Tieren der Kontrollgruppe und der Hochdosisgruppe gezählt werden; wenn bei letzteren eine Schadwirkung festgestellt wird, sind auch Tiere der Niedrigdosisgruppen zu untersuchen. Die Untersuchung sollte auch die Zählung der Primordialfollikel umfassen, die mit der Zählung der kleinen heranwachsenden Follikel kombiniert werden kann, um Eierstöcke behandelter Tiere mit Eierstöcken unbehandelter Tiere vergleichen zu können (siehe OECD Guidance Document Nr. 151 (40)). Die Gelbkörperuntersuchung sollte parallel zur Untersuchung der Östruszyklizität stattfinden, damit das Zyklusstadium bei der Bewertung berücksichtigt werden kann. Ovidukt, Uterus und Vagina sind auf eine angemessene organtypische Entwicklung hin zu untersuchen.
73. Eine eingehende histopathologische Untersuchung der Hoden wird bei männlichen F1-Tieren durchgeführt, um behandlungsbedingte Wirkungen auf die Entwicklung der Hoden (Differenzen) sowie auf die Spermatogenese festzustellen (38). Nach Möglichkeit sind Sektionen des Rete testis zu untersuchen. Caput, Corpus und Cauda des Nebenhodens und der Samenleiter werden auf angemessene organtypische Entwicklung sowie auf die für die P-Männchen erforderlichen Parameter hin untersucht.
Tiere der Kohorte 2
74. Nach dem Abschluss der Untersuchungen neurologisch bedingter Verhaltensweisen (nach PND 75 aber vor PND 90) werden alle Tiere der Hochdosisgruppen und der Kontrollgruppen der Kohorte 2A nach Geschlecht neurohistopathologisch untersucht. Das Gehirn aller Tiere der Hochdosisgruppen und der Kontrollgruppen der Kohorte 2B wird an PND 21 oder 22 nach Geschlecht histopathologisch untersucht. Organe oder Gewebe, die behandlungsbedingte Veränderungen aufweisen, sollten auch bei Tieren in den Niedrigdosisgruppen untersucht werden, um die Bestimmung eines NOAEL-Wertes zu unterstützen. Bei Tieren der Kohorten 2A und 2B werden multiple Sektionen des Gehirns geprüft, um eine Untersuchung von Riechkolben, Großhirnrinde (Cortex cerebri), Hippocampus, Basalganglien, Thalamus, Hypothalamus, Mittelhirn (Tectum, Tegmentum und Pedunculus cerebri), Pons, Medulla oblongata, Kleinhirn) zu gewährleisten. Nur bei Tieren der Kohorte 2A werden die Augen (Netzhaut und Sehnerv) sowie Proben des peripheren Nervengewebes, der Muskeln und des Rückenmarks untersucht. Alle neurohistologischen Verfahren sollten mit der Prüfmethode B.53 (35) übereinstimmen.
75. Morphometrische (quantitative) Bewertungen sollten an repräsentativen Regionen des Gehirns (homologe und sorgfältig auf der Grundlage zuverlässiger mikroskopischer Messpunkte ausgewählte Sektionen) durchgeführt werden und können auch lineare und/oder areale Messungen der spezifischen Gehirnregionen umfassen. An jedem Orientierungspunkt (Ebene) sind mindestens drei konsekutive Schnitte vorzunehmen, damit der einheitlichste und repräsentativste Schnitt für die spezifische Hirnregion bewertet werden kann. Der Neuropathologe sollte mit angemessenem Urteilsvermögen bewerten, ob die für die Messung präparierten Schnitte mit den anderen Schnitten in der Probenreihe homolog sind und sich daher für die Einbeziehung eignen, da sich insbesondere lineare Messungen über einen relativ kurzen Abstand ändern können (28). Nicht homologe Schnitte sollten nicht verwendet werden. Das Ziel besteht zwar darin, Proben von allen diesem Zweck vorbehaltenen Tieren (10 je Geschlecht und Dosisstufe) zu entnehmen, es kann aber auch eine kleinere Anzahl an Proben angemessen sein. Proben von weniger als 6 Tieren je Geschlecht und Dosisstufe gelten für die Zwecke der vorliegenden Prüfmethode in der Regel jedoch nicht als ausreichend. Mithilfe der Stereologie können behandlungsbedingte Wirkungen auf bestimmte Parameter wie Volumen oder Zellzahl für bestimmte neuroanatomische Regionen festgestellt werden. Bei allen die Gewebepräparation betreffenden Aspekten sollte auf Ausgewogenheit geachtet werden, d. h. von der Gewebefixierung über das Schneiden der Gewebeproben und die Probenvorbereitung bis hin zur Färbung der Objektträger sollte jeder Satz repräsentative Proben einer jeden Dosisgruppe enthalten. Bei morphometrischen oder stereologischen Analysen sollte Hirngewebe bei allen Dosisstufen zur gleichen Zeit in ein geeignetes Medium eingebettet werden, um ein Schrumpfen der Prüfgegenstände zu vermeiden, was bei zu langer Aufbewahrung im Fixativ auftreten kann.
BERICHTERSTATTUNG
Daten
76. Die Daten sind sowohl einzeln zu protokollieren als auch in tabellarischer Form zusammenzufassen. Gegebenenfalls sind für jede Prüfgruppe und für jede Generation die folgenden Angaben aufzuzeichnen: die Zahl der Tiere zu Beginn der Prüfung und die Zahl der während der Prüfung tot aufgefundenen oder aus Tierschutzgründen getöteten Tiere, ferner der Zeitpunkt des Todes oder der Tötung, die Zahl der fruchtbaren Tiere, die Zahl der trächtigen Weibchen, die Zahl der Weibchen, die Jungtiere werfen, und die Zahl der Tiere, die Toxizitätszeichen aufweisen, sowie eine Beschreibung der beobachteten Toxizität, einschließlich des Zeitpunkts, zu dem die toxischen Wirkungen erstmalig aufgetreten sind, ihrer Dauer und ihres Schweregrads.
77. Die numerischen Daten sollten nach einem geeigneten statistischen Verfahren ausgewertet werden. Die Statistikmethoden sollten Teil des Prüfplans sowie geeignet sein, um Nichtnormaldaten (z. B. Zähldaten), zensierte Daten (z. B. eingeschränkte Beobachtungszeit), Unabhängigkeit (z. B. Wirkungen der Würfe und wiederholte Messungen) sowie ungleiche Varianzen zu bewältigen. Allgemeingültige lineare gemischte Modelle und Dosis-Wirkungs-Modelle decken ein breites Spektrum an Analysetools ab, die für die im Rahmen dieser Prüfmethode erzeugten Daten geeignet sein können. Der Bericht sollte ausreichende Informationen über das angewandte Analyseverfahren und Computerprogramm enthalten, damit ein unabhängiger Überprüfer/Statistiker die Analyse bewerten und nachvollziehen kann.
Auswertung der Ergebnisse
78. Die Befunde sind im Hinblick auf die beobachteten Wirkungen, einschließlich der Befunde der makroskopischen und mikroskopischen Untersuchungen, zu bewerten. Ausgewertet werden u. a. die Beziehung oder die fehlende Beziehung zwischen der Dosis und dem Vorliegen, dem Auftreten und dem Schweregrad von Abnormalitäten, einschließlich makroskopischer Veränderungen. Zielorgane, Fruchtbarkeit, klinische Abnormalitäten, Reproduktionsleistung und Wurfleistung, Veränderungen des Körpergewichts, Mortalität und alle sonstigen toxischen Auswirkungen und Auswirkungen auf die Entwicklung sind ebenfalls zu bewerten. Besonderes Augenmerk gilt dabei geschlechtsspezifischen Veränderungen. Bei der Auswertung der Testergebnisse sind die physikalisch-chemischen Eigenschaften der Prüfsubstanz und — falls verfügbar — toxikokinetische Daten, einschließlich plazentarer Übertragung und Milchausscheidung, zu berücksichtigen.
Prüfbericht
79. Der Prüfbericht sollte die folgenden, zu den in dieser Prüfung untersuchten P-, F1-Tieren und gegebenenfalls F2-Tieren generierten Daten enthalten:
Prüfsubstanz:
|
— |
Alle vorliegenden relevanten Informationen über die Prüfsubstanz und toxikokinetische und toxikodynamische Eigenschaften der Prüfsubstanz, |
|
— |
Kenndaten, |
|
— |
Reinheit. |
Vehikel (falls verwendet):
|
— |
Begründung der Wahl des Vehikels, sofern anders als Wasser. |
Versuchstiere:
|
— |
Tierart/Stamm, |
|
— |
Anzahl, Alter und Geschlecht der Tiere, |
|
— |
Herkunft, Unterbringungsbedingungen, Ernährung, Nistmaterial usw., |
|
— |
Gewicht der einzelnen Tiere bei Versuchsbeginn, |
|
— |
Vaginalabstrichdaten für P-Weibchen vor Beginn der Behandlung (falls zu diesem Zeitpunkt Daten erhoben wurden), |
|
— |
Paarungsdaten zur P-Generation mit Angabe der männlichen und weiblichen Paarungspartner und des Paarungsergebnisses, |
|
— |
Aufzeichnungen über den Herkunftswurf bei adulten Tieren der Generation F1. |
Prüfungsbedingungen:
|
— |
Begründung der gewählten Dosisstufen, |
|
— |
Einzelheiten zur chemischen Formulierung der Prüfsubstanz/Futterzubereitung und zu den erreichten Konzentrationen, |
|
— |
Stabilität und Homogenität des Präparats im Vehikel oder in der Trägersubstanz (z. B. Futter, Trinkwasser), im Blut und/oder in der Milch unter den Verwendungs- und Lagerbedingungen zwischen den Verwendungen, |
|
— |
Einzelheiten der Verabreichung der Prüfsubstanz, |
|
— |
gegebenenfalls Angaben zur Umrechnung der Konzentration der Prüfsubstanz im Futter/Wasser (ppm) in die entsprechende Dosis (mg/kg Körpergewicht/Tag), |
|
— |
Angaben zu Futter- und Wasserqualität (einschließlich Futterzusammensetzung, sofern verfügbar), |
|
— |
genaue Beschreibung der Verfahren für die Zufallsauswahl von Jungtieren zwecks Tötung und für die Einteilung von Jungtieren in die Prüfgruppen, |
|
— |
Umgebungsbedingungen, |
|
— |
Verzeichnis der am Versuch Studie beteiligten Personen, einschließlich fachlicher Schulung. |
Ergebnisse (Sammel- und Einzeldaten, aufgeschlüsseltnach Geschlecht und Dosis):
|
— |
Futteraufnahme, Wasseraufnahme (falls erfasst), Futtereffizienz (Körpergewichtszunahme pro Gramm aufgenommenen Futters, ausgenommen während der Kohabitation und Laktation) und bei P- und F1-Tieren Aufnahme der Prüfsubstanz (bei Verabreichung über das Futter/Trinkwasser), |
|
— |
Angaben zur Resorption (falls vorhanden), |
|
— |
bei P-Tieren: Angaben zum Körpergewicht, |
|
— |
bei ausgewählten F1-Tieren nach der Entwöhnung: Angaben zum Körpergewicht, |
|
— |
Zeitpunkt des Todes, falls während des Versuchs eingetreten, oder Angabe, ob Tiere bis zum Versuchsende überlebt haben, |
|
— |
Art, Schweregrad und Dauer der klinischen Beobachtungen (mit Angaben zur Reversibilität), |
|
— |
Angaben zu hämatologischen Untersuchungen und Urinuntersuchungen sowie klinisch-chemische Daten, einschließlich TSH und T4, |
|
— |
Phänotypanalyse der Milzzellen (T-, B-, NK-Zellen), |
|
— |
Knochenmarkzellularität, |
|
— |
Daten zur toxischen Reaktion, |
|
— |
Zahl weiblicher P- und F1-Tiere mit normalem oder abnormalem Östruszyklus sowie Zyklusdauer, |
|
— |
Zeit bis zur Paarung (präkoitales Intervall, Anzahl Tage zwischen Paarung und Deckung), |
|
— |
toxische oder andere Wirkungen auf die Fortpflanzung, einschließlich Zahl und Anteil der Tiere, die sich gepaart haben, trächtig wurden, geworfen und laktiert haben, sowie der männlichen Tiere, die gezeugt haben, und der weiblichen Tiere mit Anzeichen von Dystokie/langwieriger oder schwieriger Geburt, |
|
— |
Dauer der Gravidität und gegebenenfalls des Partus, |
|
— |
Anzahl an Implantationen, Wurfgröße und Anteil der männlichen Jungtiere, |
|
— |
Anzahl und Anteil der Abgänge nach der Implantation, der Lebendgeburten und der Totgeburten, |
|
— |
Angaben zum Gewicht des Wurfs und der Jungtiere (Männchen und Weibchen, einzeln und zusammen), Anzahl der Kümmerlinge, falls diese bestimmt werden, |
|
— |
Anzahl Jungtiere mit deutlich sichtbaren Abnormalitäten, |
|
— |
Toxische oder andere Wirkungen in Nachkommen, postnatales Wachstum, Lebensfähigkeit usw., |
|
— |
Angaben zu physischen Merkmalen bei Jungtieren und anderen postnatalen Entwicklungsparametern, |
|
— |
Daten zur Geschlechtsreife von F1-Tieren, |
|
— |
gegebenenfalls Daten zu funktionellen Beobachtungen bei Jungtieren und adulten Tieren, |
|
— |
Körpergewicht bei der Tötung sowie Angaben zum absoluten und relativen Organgewicht bei P- und adulten F1-Tieren, |
|
— |
Sektionsbefunde, |
|
— |
ausführliche Beschreibung aller histopathologischen Befunde, |
|
— |
für männliche P- und F1-Tiere: Gesamtzahl der Spermien aus dem Nebenhodenschwanz, Anteil der progressiv beweglichen Spermien, Anteil der morphologisch normalen Spermien und Anteil der Spermien, der der jeweils festgestellten Abnormalität entspricht, |
|
— |
für P- und F1-Weibchen: gegebenenfalls Anzahl und Reifestadium der Follikel in den Eierstöcken, |
|
— |
Zählung der Gelbkörper in den Ovarien der F1-Weibchen, |
|
— |
nach Möglichkeit statistische Auswertung der Ergebnisse. |
Parameter für Kohorte 2:
|
— |
Ausführliche Beschreibung der zur Standardisierung von Beobachtungen und Verfahren angewendeten Vorgehensweise sowie Vorgaben für die Auswertung der Ergebnisse, |
|
— |
Auflistung aller angewandten Prüfverfahren mit Begründung, |
|
— |
ausführliche Beschreibung der angewandten verhaltensbezogenen/funktionalen, neuropathologischen und morphometrischen Verfahren, einschließlich detaillierter Angaben zu automatischen Vorgängen, |
|
— |
Kalibrierungsverfahren und Verfahren zur Gewährleistung der Gleichwertigkeit der verwendeten Geräte und der Ausgewognenheit der Prüfgruppen während der Prüfung, |
|
— |
kurze Begründung, warum bestimmte Entscheidungen die Hinzuziehung eines Sachverständigen erfordern, |
|
— |
ausführliche Beschreibung der nach Geschlecht und Dosisgruppe aufgeschlüsselten verhaltensbezogenen/funktionalen, neuropathologischen und morphometrischen Befunde, einschließlich Erhöhung und Verringerung der Kontrollhäufigkeit, |
|
— |
Hirngewicht, |
|
— |
etwaige Diagnosen aufgrund neurologischer Anzeichen und Läsionen, einschließlich natürlich aufgetretener Krankheiten oder Zustände, |
|
— |
Abbildungen von Beispielbefunden, |
|
— |
Low-Power-Imaging zur Bewertung der Homologie der für die Morphometrie verwendeten Sektionen, |
|
— |
statistische Auswertung der Ergebnisse, einschließlich statistischer Modelle für die Daten- und Ergebnisanalyse, und zwar unabhängig davon, ob diese Daten oder Ergebnisse signifikant waren oder nicht, |
|
— |
Beitrag etwaiger anderer toxischer Wirkungen zur Schlussfolgerung bezüglich des neurotoxischen Potenzials der Prüfsubstanz, aufgeschlüsselt nach Geschlechtern und Dosisgruppen, |
|
— |
Auswirkung etwaiger toxikokinetischer Informationen auf die Schlussfolgerungen, |
|
— |
Daten zur Untermauerung der Zuverlässigkeit und Empfindlichkeit der Prüfmethode (d. h. positive Daten und historische Kontrolldaten), |
|
— |
Zusammenhänge zwischen neuropathologischen und funktionalen Wirkungen (sofern diese vorliegen), |
|
— |
NOAEL-Wert oder Benchmark-Dosis für Muttertiere und Nachkommen, aufgeschlüsselt nach Geschlechtern und Dosisgruppen, |
|
— |
Diskussion der Gesamtauswertung der Daten auf Grundlage der Ergebnisse, einschließlich einer Schlussfolgerung darüber, ob die Prüfsubstanz eine entwicklungsneurotoxische Wirkung hat oder nicht, und des NOAEL-Werts. |
Parameter für die Kohorte 3:
|
— |
IgM-Antikörper-Titer im Serum (Sensibilisierung für SRBC oder KLH) oder Einheiten IgM-plaquebildender Zellen in der Milz (Sensibilisierung für SRBC), |
|
— |
die Leistung der TDAR-Methode sollte als Teil des Optimierungsprozesses durch das Labor, das den Test erstmals einrichtet, und regelmäßig (beispielsweise jährlich) durch alle Labore bestätigt werden, |
|
— |
Diskussion der Gesamtauswertung der Daten auf Grundlage der Ergebnisse, einschließlich einer Schlussfolgerung darüber, ob die Prüfsubstanz eine entwicklungsimmuntoxische Wirkung hat oder nicht, und des NOAEL-Wertes. |
Diskussion der Ergebnisse
Schlussfolgerungen, einschließlich der NOAEL-Werte für Wirkungen bei Elterntieren und deren Nachkommen
Alle Informationen, die nicht während des Versuchs generiert wurden, für die Auswertung der Ergebnisse jedoch zweckdienlich sind (z. B. Ähnlichkeiten der Wirkungen mit bekannten Neurotoxinen) sind ebenfalls anzuführen.
Auswertung der Ergebnisse
80. Eine erweiterte Ein-Generationen-Prüfung auf Reproduktionstoxizität (EOGRTS) generiert Daten über die Wirkungen wiederholter Verabreichungen einer Chemikalie während aller Phasen des Fortpflanzungszyklus. Sie gibt insbesondere Auskunft über das Fortpflanzungssystem sowie über Entwicklung, Wachstum, Überleben und funktionale Endpunkte der Nachkommen bis zum postnatalen Tag (PND) 90.
81. Bei der Auswertung der Prüfungsergebnisse sollten alle verfügbaren Daten über die Prüfsubstanz, einschließlich physikalisch-chemischer, toxikokinetischer und toxikodynamischer Eigenschaften, sowie verfügbare relevante Informationen über strukturelle Analogien und Ergebnisse vorausgegangener Toxizitätsstudien mit der Prüfsubstanz (z. B. akute Toxizität, Toxizität bei wiederholter Verabreichung, mechanistische Studien und Studien, in denen bewertet wird, ob bei den In-vivo-/In-vitro-Stoffwechseleigenschaften erhebliche qualitative und quantitative artspezifische Unterschiede vorliegen) berücksichtigt werden. Die Ergebnisse der makroskopischen Untersuchung (Nekropsie) und die Organgewichte sollten nach Möglichkeit im Kontext der Beobachtungen bewertet werden, die bereits in anderen Versuchen mit wiederholter Verabreichung gemacht wurden. Wachstumsverlangsamung bei Nachkommen könnte auf den Einfluss der Prüfsubstanz auf die Milchzusammensetzung zurückgeführt werden (29).
Kohorte 2 (Entwicklungsneurotoxizität)
82. Neurologisch bedingtes Verhalten und neuropathologische Ergebnisse sollten unter Berücksichtigung aller Befunde von Fachleuten nach einem Weight-of-evidence-Ansatz ausgewertet werden. Die Muster verhaltensbedingter oder morphologischer Befunde sollten, sofern vorhanden, ebenso erörtert werden wie eine nachweisliche Dosis-Wirkungs-Beziehung. In diese Charakterisierung sollte die Beurteilung der Entwicklungsneurotoxizität, einschließlich epidemiologischer Untersuchungen beim Menschen oder Fallberichte sowie tierexperimentelle Studien (z. B. toxikokinetische Daten, Daten zur Struktur-Wirkungs-Beziehung, Daten aus anderen Toxizitätsstudien) einfließen. Die Datenauswertung sollte ferner eine Diskussion sowohl der biologischen als auch der statistischen Signifikanz beinhalten. Sofern ein Zusammenhang zwischen neuropathologischen Veränderungen und Verhaltensänderungen beobachtet wurde, sollte dieser in die Beurteilung einbezogen werden. Leitlinien zur Auswertung der entwicklungsneurotoxischen Ergebnisse finden sich in der Prüfmethode B.53 (35) und in Tyl et al., 2008 (31).
Kohorte 3 (Entwicklungsimmuntoxizität)
83. Die mithilfe des TDAR-Assay (T-Zell-abhängigen Antikörperantwort) ermittelte Unterdrückung oder Verstärkung der Immunfunktion sollte im Kontext aller festgestellten Beobachtungen bewertet werden. Die Signifikanz des TDAR-Ergebnisses kann durch andere Wirkungen auf immunologisch verwandte Indikatoren (z. B. Knochenmarkzellularität, Gewicht und histopathologische Untersuchung des Lymphdrüsengewebes, Lymphozyten-Teilmengenverteilung) untermauert werden. Die im TDAR ermittelten Wirkungen sind möglicherweise weniger aussagekräftig bei anderen Toxizitäten, die bei niedrigeren Expositionskonzentrationen festgestellt werden.
84. Als Hilfe bei der Auswertung der reproduktionstoxischen und neurotoxischen Ergebnisse ist das OECD Guidance Document Nr. 43 zu Rate zu ziehen (26).
LITERATUR
|
(1) |
Cooper, R.L., J.C. Lamb, S.M. Barlow, K. Bentley, A.M. Brady, N. Doerr, D.L. Eisenbrandt, P.A. Fenner-Crisp, R.N. Hines, L.F.H. Irvine, C.A. Kimmel, H. Koeter, A.A. Li, S.L. Makris, L.P. Sheets, G.J.A. Speijers and K.E. Whitby (2006). ‚A Tiered Approach to Life Stages Testing for Agricultural Chemical Safety Assessment‘, Critical Reviews in Toxicology, 36, S. 69-98. |
|
(2) |
Thigpen, J.E., K.D.R. Setchell, K.B. Ahlmark, J. Locklear, T. Spahr, G.F. Leviness, M.F. Goelz, J.K. Haseman, R.R. Newbold, and D.B. Forsythe (1999). ‚Phytoestrogen Content of Purified Open and Closed Formula Laboratory Animal Diets‘, Lab. Anim. Sci., 49, S. 530-536. |
|
(3) |
Zoetis, T. and I. Walls (2003). Principles and Practices for Direct Dosing of Pre-Weaning Mammals in Toxicity Testing and Research, ILSI Press, Washington, DC. |
|
(4) |
Moser, V.C., I. Walls and T. Zoetis (2005). ‚Direct Dosing of Preweaning Rodents in Toxicity Testing and Research: Deliberations of an ILSI RSI Expert Working Group‘, International Journal of Toxicology, 24, S. 87-94. |
|
(5) |
Conolly, R.B., B.D. Beck, and J.I. Goodman (1999). ‚Stimulating Research to Improve the Scientific Basis of Risk Assessment‘, Toxicological Sciences, 49, S. 1-4. |
|
(6) |
Ulbrich, B. and A.K. Palmer (1995). ‚Detection of Effects on Male Reproduction — a Literature Survey‘, Journal of the American College of Toxicologists, 14, S. 293-327. |
|
(7) |
Mangelsdorf, I., J. Buschmann and B. Orthen (2003). ‚Some Aspects Relating to the Evaluation of the Effects of Chemicals on Male Fertility‘, Regulatory Toxicology and Pharmacology, 37, S. 356-369. |
|
(8) |
Sakai, T., M. Takahashi, K. Mitsumori, K. Yasuhara, K. Kawashima, H. Mayahara und Y. Ohno (2000). ‚Collaborative work to evaluate toxicity on male reproductive organs by repeated dose studies in rats — overview of the studies‘, Journal of Toxicological Sciences, 25, S. 1-21. |
|
(9) |
Creasy, D.M. (2003). ‚Evaluation of Testicular Toxicology: A Synopsis and Discussion of the Recommendations Proposed by the Society of Toxicologic Pathology‘, Birth Defects Research, Part B, 68, S. 408-415. |
|
(10) |
Goldman, J.M., A.S. Murr, A.R. Buckalew, J.M. Ferrell and R.L. Cooper (2007). ‚The Rodent Estrous Cycle: Characterization of Vaginal Cytology and its Utility in Toxicological Studies‘, Birth Defects Research, Part B, 80 (2), S. 84-97. |
|
(11) |
Sadleir, R.M.F.S. (1979). ‚Cycles and Seasons‘, in C.R. Auston and R.V. Short (eds.), Reproduction in Mammals: I. Germ Cells and Fertilization, Cambridge, New York. |
|
(12) |
Gallavan, R.H. Jr, J.F. Holson, D.G. Stump, J.F. Knapp and V.L. Reynolds (1999). ‚Interpreting the Toxicologic Significance of Alterations in Anogenital Distance: Potential for Confounding Effects of Progeny Body Weights‘, Reproductive Toxicology, 13: S. 383-390. |
|
(13) |
Korenbrot, C.C., I.T. Huhtaniemi and R.I. Weiner (1977). ‚Preputial Separation as an External Sign of Pubertal Development in the Male Rat‘, Biological Reproduction, 17, S. 298-303. |
|
(14) |
Ladics, G.S. (2007). ‚Use of SRBC Antibody Responses for Immunotoxicity Testing‘, Methods, 41, S. 9-19. |
|
(15) |
Gore, E.R., J. Gower, E. Kurali, J.L. Sui, J. Bynum, D. Ennulat and D.J. Herzyk (2004). ‚Primary Antibody Response to Keyhole Limpet Hemocyanin in Rat as a Model for Immunotoxicity Evaluation‘, Toxicology, 197, S. 23-35. |
|
(16) |
Gray, L.E., J. Ostby, J. Ferrell, G. Rehnberg, R. Linder, R. Cooper, J. Goldman, V. Slott and J. Laskey (1989). ‚A Dose-Response Analysis of Methoxychlor-Induced Alterations of Reproductive Development and Function in the Rat‘, Fundamental and Applied Toxicology, 12, S. 92-108. |
|
(17) |
Robb, G.W., R.P. Amann and G.J. Killian (1978). ‚Daily Sperm Production and Epididymal Sperm Reserves of Pubertal and Adult Rates‘, Journal of Reproduction and Fertility,54, S. 103-107. |
|
(18) |
Klinefelter, G.R., L.E. Jr Gray and J.D. Suarez (1991). ‚The Method of Sperm Collection Significantly Influences Sperm Motion Parameters Following Ethane Dimethanesulfonate Administration in the Rat‘. Reproductive Toxicology,5, S. 39-44. |
|
(19) |
Seed, J., R.E. Chapin, E.D. Clegg., L.A. Dostal, R.H. Foote, M.E. Hurtt, G.R. Klinefelter, S.L. Makris, S.D. Perreault, S. Schrader, D. Seyler, R. Sprando, K.A. Treinen, D.N. Veeramachaneni and L.D. Wise (1996). ‚Methods for Assessing Sperm Motility, Morphology, and Counts in the Rat, Rabbit, and Dog: a Consensus Report‘, Reproductive Toxicology, 10, S. 237-244. |
|
(20) |
Chapin, R.E., R.S. Filler, D. Gulati, J.J. Heindel, D.F. Katz, C.A. Mebus, F. Obasaju, S.D. Perreault, S.R. Russell and S. Schrader (1992). ‚Methods for Assessing Rat Sperm Motility‘, Reproductive Toxicology, 6, S. 267-273. |
|
(21) |
Klinefelter, G.R., N.L. Roberts and J.D. Suarez (1992). ‚Direct Effects of Ethane Dimethanesulphonate on Epididymal Function in Adult Rates: an In Vitro Demonstration‘, Journal of Andrology, 13, S. 409-421. |
|
(22) |
Slott, V.L., J.D. Suarez and S.D. Perreault (1991). ‚Rat Sperm Motility Analysis: Methodologic Considerations‘, Reproductive Toxicology, 5, S. 449-458. |
|
(23) |
Slott, V.L., and S.D. Perreault (1993). ‚Computer-Assisted Sperm Analysis of Rodent Epididymal Sperm Motility Using the Hamilton-Thorn Motility Analyzer‘, Methods in Toxicology, Part A, Academic, Orlando, Florida, S. 319-333. |
|
(24) |
Toth, G.P., J.A. Stober, E.J. Read, H. Zenick and M.K. Smith (1989). ‚The Automated Analysis of Rat Sperm Motility Following Subchronic Epichlorhydrin Administration: Methodologic and Statistical Considerations‘, Journal of Andrology, 10, S. 401-415. |
|
(25) |
Linder, R.E., L.F. Strader, V.L. Slott and J.D. Suarez (1992). ‚Endpoints of Spermatoxicity in the Rat After Short Duration Exposures to Fourteen Reproductive Toxicants‘, Reproductive Toxicology, 6, S. 491-505. |
|
(26) |
OECD (2008). Guidance Document on Mammalian Reproductive Toxicity Testing and Assessment, Series on Testing and Assessment, Nr. 43, ENV/JM/MONO(2008)16, OECD, Paris. |
|
(27) |
Working, P.K., M. Hurtt (1987). ‚Computerized Videomicrographic Analysis of Rat Sperm Motility‘, Journal of Andrology, 8, S. 330-337. |
|
(28) |
Bolin, B., R. Garman, K. Jensen, G. Krinke, B. Stuart, and an ad Hoc Working Group of the STP Scientific and Regulatory Policy Committee (2006). ‚A ‚Best Practices‘ Approach to Neuropathologic Assessment in Developmental Neurotoxicity Testing — for Today‘, Toxicological Pathology, 34, S. 296-313. |
|
(29) |
Stütz, N., B. Bongiovanni, M. Rassetto, A. Ferri, A.M. Evangelista de Duffard, and R. Duffard (2006). ‚Detection of 2,4-dichlorophenoxyacetic Acid in Rat Milk of Dams Exposed During Lactation and Milk Analysis of their Major Components‘, Food Chemicals Toxicology, 44, S. 8-16. |
|
(30) |
Thigpen, JE, K.D.R. Setchell, J.K. Haseman, H.E. Saunders, G.F. Caviness, G.E. Kissling, M.G. Grant and D.B. Forsythe (2007). ‚Variations in Phytoestrogen Content between Different Mill Dates of the Same Diet Produces Significant Differences in the Time of Vaginal Opening in CD-1 Mice and F344 Rates but not in CD Sprague Dawley Rates‘, Environmental health perspectives, 115(12), S. 1717-1726. |
|
(31) |
Tyl, R.W., K. Crofton, A. Moretto, V. Moser, L.P. Sheets and T.J. Sobotka (2008). ‚Identification and Interpretation of Developmental Neurotoxicity Effects: a Report from the ILSI Research Foundation/Risk Science Institute Expert Working Group on Neurodevelopmental Endpoints‘, Neurotoxicology and Teratology, 30: S. 349-381. |
|
(32) |
OECD (1996). Combined Repeated Dose Toxicity Study with the Reproduction/Developmental Toxicity Screening Test, OECD Guideline for Testing of Chemicals, Nr. 422, OECD, Paris. |
|
(33) |
Kapitel B.43 dieses Anhangs, Prüfung auf Neurotoxizität bei Nagetieren. |
|
(34) |
OECD (2000). Guidance Document on the recognition, assessment, and use of clinical signs as humane endpoints for experimental animals used in safety evaluations, Series on Testing and Assessment, Nr. 19, ENV/JM/MONO(2000)7, OECD, Paris. |
|
(35) |
Kapitel B.53 dieses Anhangs, Prüfung auf Entwicklungsneurotoxizität. |
|
(36) |
Kapitel B.54 dieses Anhangs: Uterotropher Bioassay an Nagetieren: Ein Kurzzeit-Screening-Test auf östrogene Eigenschaften. |
|
(37) |
Kapitel B.55 dieses Anhangs: Hershberger-Bioassay an Ratten: Ein Kurzzeit-Screening-Test auf (anti-)androgene Eigenschaften. |
|
(38) |
OECD (2009). Guidance Document for Histologic Evalution of Endocrine and Reproductive Test in Rodents, Series on Testing and Assessment, Nr. 106, OECD, Paris. |
|
(39) |
OECD (2011). Guidance Document on the Current Implementation of Internal Triggers in the Extended One Generation Reproductive Toxicity Study in the United States and Canada, Series on Testing and Assessment, Nr. 117, ENV/JM/MONO(2011)21, OECD, Paris. |
|
(40) |
OECD (2013). Guidance Document supporting TG 443: Extended One Generation Reproductive Toxicity Study, Series on Testing and Assessment, Nr. 151, OECD, Paris. |
Anlage 1
Maßnahmen und Beobachtungen im Rahmen der FOB (Functional Observational Battery) (Kohorte 2A)
|
Käfig & offenes Gehege |
Manipulation |
Physiologie |
|
Haltung |
Leicht zu greifen |
Temperatur |
|
Unfreiwillige klonische und tonische Bewegungen |
Leicht zu hantieren |
Körpergewicht |
|
Schließung der Augenlider |
Muskeltonus |
Pupillenreaktion |
|
Piloerektion |
Reaktion bei Annäherung |
Pupillengröße |
|
Salivation |
Reaktion auf Berühren |
|
|
Tränensekretion |
Akustische Reaktion |
|
|
Lautäußerungen |
Reaktion bei Schwanzkneifen |
|
|
Aufbäumen |
Aufrichtungsreaktion |
|
|
Anomaler Gang |
Spreizung des Landefußes |
|
|
Erregung |
Greifkraft der Vordergliedmaßen |
|
|
Stereotypie |
Greifkraft der Hintergliedmaßen |
|
|
Bizarres Verhalten |
|
|
|
Färbungen |
|
|
|
Atmungsstörung |
|
|
Anlage 2
BEGRIFFSBESTIMMUNGEN
Chemikalie : ein Stoff oder eine Mischung.
Prüfsubstanz : jede(r) mittels dieser Prüfmethode getestete Stoff bzw. Mischung.
B.57. H295R-STEROIDGENESE-ASSAY
EINLEITUNG
1. Diese Prüfmethode entspricht der OECD-Prüfrichtlinie (TG) 456 (2011). Die OECD setzte sich 1998 zur Priorität, bestehende Prüfrichtlinien für Screening und Testung potenziell endokriner Disruptoren zu überarbeiten und neue Richtlinien zu entwickeln. Das Rahmenkonzept der OECD für die Testung und Bewertung endokriner Disruptoren von 2002 umfasst fünf Stufen, wobei jede Stufe einem anderen Grad der biologischen Komplexität entspricht (1). Bei dem in der vorliegenden Prüfmethode beschriebenen In-vitro Testsystem — dem H295R-Steroidgenese-Assay — wird eine menschliche Adenokarzinom-Zelllinie (NCI-H295R-Zellen) verwendet. Der Assay stellt einen ‚In-vitro-Assay der Stufe 2 dar, und liefert mechanistische Daten‘, die zum Screening und für Priorisierungszwecke zu verwenden sind. Die Ausarbeitung und Standardisierung des Assays als Screeningmethode für chemische Auswirkungen auf die Steroidgenese, insbesondere auf die Produktion von 17β-Östradiol (E2) und Testosteron (T), erfolgte in mehreren Schritten. Der H295R-Assay ist optimiert und validiert worden (2) (3) (4) (5).
2. Das Ziel des H295R-Steroidgenese-Assays besteht darin, Chemikalien nachzuweisen, durch die die Produktion von E2 und T beeinflusst wird. Mit dem H295R-Assay sollen Xenobiotika ermittelt werden, die diejenigen endogenen Bestandteile als Zielstelle(n) haben, die den intrazellularen biochemischen Pfad bilden, der mit Cholesterol beginnt und bis zur Produktion von E2 und/oder T führt. Mit dem H295R-Assay sollen keine Chemikalien bestimmt werden, die die Steroidgenese aufgrund von Auswirkungen auf die Hypothalamus-Hypophysen-Gonaden-Achse (HPG-Achse) beeinträchtigen. Der Assay zielt vielmehr darauf ab, im Hinblick auf das Potenzial einer Chemikalie, die Produktion von T und E2 zu induzieren oder zu hemmen, eine JA/NEIN-Antwort zu liefern; in einigen Fällen können jedoch auch quantitative Ergebnisse erzielt werden (siehe Nummern 53 und 54). Die Ergebnisse des Assays werden als relative Veränderungen in der Hormonproduktion im Vergleich zu den Lösungsmittelkontrollen (LK) angegeben. Der Assay zielt nicht darauf ab, spezifische mechanistische Informationen über die Interaktion der Prüfsubstanz mit dem endokrinen System zu liefern. Anhand der Zelllinie wurden Forschungen durchgeführt, um Auswirkungen auf bestimmte Enzyme und Zwischenhormone, wie zum Beispiel Progesteron, zu bestimmen (2).
3. Die in der vorliegenden Prüfmethode verwendeten Begriffe und Abkürzungen sind in der Anlage beschrieben. Ein detailliertes Protokoll mit Anweisungen zur Herstellung von Lösungen, Kultivierung von Zellen und Durchführung verschiedener Aspekte der Prüfung ist als Anhang I-III des OECD-Dokuments ‚Multi-Laboratory Validation of the H295R Steroidogenesis Assay to Identify Modulators of Testosterone and Estradiol Production‘ verfügbar (4).
AUSGANGSÜBERLEGUNGEN UND GRENZEN
4. An der Biosynthese sexueller Steroidhormone sind fünf verschiedene Enzyme, die sechs unterschiedliche Reaktionen katalysieren, beteiligt. Die enzymatische Umwandlung von Cholesterin in Pregnenolon durch die Cytochrom P450-abhängige Cholesterin-Monooxygenase (CYP11A) ist der erste Schritt in einer Reihe biochemischer Reaktionen, die in der Synthese steroider Endprodukte gipfeln. Je nach Reihenfolge der beiden nächsten Reaktionen teilt sich der Pfad der Steroidgenese in zwei Pfade auf: den Δ5-Hydroxysteroid-Pfad und den Δ4-Ketosteroid-Pfad, die in der Produktion von Androstenedion (Abbildung 1) zusammenlaufen.
5. Androstenedion wird durch 17β-Hydroxysteroid Dehydrogenase (17β-HSD) in Testosteron (T) umgewandelt. Testosteron ist sowohl ein Zwischen- als auch ein Endhormonprodukt. Im männlichen Organismus kann T durch 5α-Reduktase, die in Zellmembranen, Kernhülle und im Retikulum von Zielgewebe mit androgener Wirkung, wie zum Beispiel Prostata und Samenbläschen, gefunden wird, in Dihydrotestosteron (DHT) umgewandelt werden. DHT ist als Androgen erheblich wirksamer als T und gilt ebenfalls als Endprodukthormon. Der H295R-Assay misst kein DHT (siehe Nummer 10).
6. Das Enzym im Pfad der Steroidgenese, das androgene Chemikalien in östrogene Chemikalien umwandelt, ist Aromatase (CYP19). CYP19 wandelt T in 17β-Östradiol (E2) und Androstenedion in Östron um. E2 und T gelten als Endprodukthormone des Steroidgenesepfads.
7. Die Spezifizität der Lyaseaktivität von CYP17 ist bei den verschiedenen Intermediaten von Tierart zu Tierart unterschiedlich. Im Menschen bevorzugt das Enzym Substrate des Δ5-Hydroxysteroid-Pfads (Pregnenolon); dagegen werden in der Ratte Substrate im Δ4-Ketosteroid-Pfad (Progesteron) begünstigt (19). Solche Unterschiede in der CYP17-Lyaseaktivität können einige artspezifische Unterschiede in der Reaktion auf Chemikalien erklären, die die Steroidgenese in vivo verändern (6). Es hat sich erwiesen, dass die H295-Zellen die Expression des humanen adulten Nebennierenenzyms und das Muster der Steroidproduktion am genauesten widerspiegeln (20), aber auch dafür bekannt sind, Enzyme sowohl für den Δ5-Hydroxysteroid als auch für den Δ4-Ketosteroid-Pfad für Androgensynthese zu exprimieren (7) (11) (13) (15).
Abbildung 1
Pfad der Steroidgenese in H295R-Zellen

Anmerkung:
Enzyme sind kursiv gedruckt, Hormone sind fett gedruckt, und Pfeile zeigen die Richtung der Synthese an. Ein grauer Hintergrund zeigt Corticosteroidpfade/-produkte an. Sexuelle Steroidhormonpfade/-produkte sind eingekreist. CYP = Cytochrom P450; HSD = Hydroxysteroid-Dehydrogenase; DHEA = Dehydroepiandrosteron.
8. Die menschliche H295R-Adenokarzinom-Zelllinie ist ein nützliches In-vitro-Modell für die Ermittlung der Auswirkungen auf die Synthese von Steroidhormonen (2) (7) (8) (9) (10). Die H295R-Zelllinie exprimiert Gene, die alle wichtigen Enzyme für die oben genannte Steroidgenese verschlüsseln (11) (15) (Abbildung 1). Das ist eine einzigartige Eigenschaft, weil die In-vivo-Expression dieser Gene gewebe- und entwicklungsstadiumspezifisch ist, d. h., kein Gewebe- oder Entwicklungsstadium exprimiert alle an der Steroidgenese beteiligten Gene (2). H295R-Zellen weisen physiologische Eigenschaften zonal undifferenzierter Nebennierenzellen menschlicher Föten auf (11). Die Zellen stellen ein einzigartiges In-vitro-System dar, da sie die Fähigkeit besitzen, alle in der adulten Nebennierenrinde und den Gonaden gefundenen Steroidhormone zu produzieren. Mit ihnen können Auswirkungen sowohl auf die Corticosteroidsynthese als auch auf die Produktion von sexuellen Steroidhormonen, wie zum Beispiel Androgene und Östrogene, geprüft werden, auch wenn der Assay nur für den Nachweis von T und E2 validiert wurde. Die durch das Prüfsystem erfassten Änderungen in Form einer Veränderung der Produktion von T und E2 können das Ergebnis einer Vielzahl unterschiedlicher Interaktionen der Prüfsubstanzen mit Steroidgenesefunktionen sein, die durch die H295R-Zellen exprimiert werden. Dazu gehört die Modulation der Expression, die Synthese oder die Funktion von bei der Produktion, Umwandlung oder Eliminierung von Steroidhormonen beteiligten Enzymen (12) (13) (14). Die Hemmung der Hormonproduktion kann auf eine direkte kompetitive Bindung an ein Enzym, einen Einfluss auf Ko-Faktoren, wie zum Beispiel NADPH (Nicotinamidadenindinukleotidphosphat) und cAMP (zyklisches Adenosinmonophosphat), und/oder eine Steroidstoffwechselerhöhung oder die Reprimierung der Genexpression bestimmter Enzyme im Steroidgenesepfad zurückgeführt werden. Während die Hemmung sowohl von direkten als auch indirekten an der Hormonproduktion beteiligten Prozessen abhängig sein kann, erfolgt die Induktion in der Regel indirekt, beispielsweise durch beeinflussende Ko-Faktoren wie NADPH und cAMP (wie etwa bei Forskolin), einen sinkenden Steroidstoffwechsel (13) und/oder eine Hochregulierung der Genexpression der Steroidgenese.
9. Der H295R-Assay weist mehrere Vorteile auf:
|
— |
Er ermöglicht den Nachweis von Erhöhungen und Rückgängen bei der Produktion von T und E2; |
|
— |
er ermöglicht die direkte Bewertung der potenziellen Auswirkung einer Chemikalie auf die Zellviabilität/Zytotoxizität. Dies ist ein wichtiges Element, weil dadurch Wirkungen, die auf die Zytotoxizität zurückgehen, von Wirkungen, die auf die direkte Interaktion der Chemikalien mit steroidogenen Pfaden zurückgehen, unterschieden werden können, was in Gewebeexplantatsystemen, die aus einer Vielzahl von Zelltypen unterschiedlicher Empfindlichkeiten und Funktionalitäten bestehen, nicht möglich ist; |
|
— |
er erfordert keine Versuchstiere; |
|
— |
die H295R-Zelllinie ist im Handel erhältlich. |
10. Die wichtigsten Einschränkungen des Assays sind folgende:
|
— |
Seine metabolische Kapazität ist nicht bekannt, wahrscheinlich aber eher begrenzt; daher würden Chemikalien, die metabolisch aktiviert werden müssen, in diesem Assay wahrscheinlich übersehen. |
|
— |
Da H295R von Nebennierengewebe abgeleitet wird, besitzt es die Enzyme, die Gluco- und Mineralocorticoide ebenso erzeugen können wie Geschlechtshormone; daher könnten die Auswirkungen auf die Produktion von Gluco- und Mineralocorticoiden die in dem Assay festgestellten T- und E2-Spiegel beeinflussen. |
|
— |
Der Assay misst kein DHT. Aus diesem Grund ist auch nicht davon auszugehen, dass mit ihm Chemikalien festgestellt werden können, die die 5α-Reduktase hemmen; für diesen Zweck kann der Hershberger-Assay (16) verwendet werden. |
|
— |
Mit dem H295R-Assay werden keine Chemikalien festgestellt, die die Steroidgenese dadurch stören, dass sie die Hypothalamus-Hypophysen-Gonaden-Achse (HPG-Achse) beeinträchtigen, da dies nur in intakten Tieren untersucht werden kann. |
PRINZIP DER PRÜFMETHODE
11. Zweck des Assays ist der Nachweis von Chemikalien, die die Produktion von T und E2 beeinträchtigen. T ist außerdem ein Zwischenprodukt im Produktionspfad von E2. Mit dem Assay können Chemikalien nachgewiesen werden, die die Enzyme des Steroidgenesepfades in der Regel hemmen oder auslösen.
12. Der Assay wird in der Regel unter Standardzellkulturbedingungen in 24-Mulden-Kulturplatten durchgeführt. Alternativ dazu können andere Plattengrößen für die Durchführung des Assays verwendet werden; die Saat- und Versuchsbedingungen sind dabei jedoch so anzupassen, dass die Leistungskriterien erfüllt sind.
13. Nach einer Akklimatisierungsphase von 24 Stunden in Multiwellplatten werden die Zellen 48 Stunden lang mindestens dreifach sieben Konzentrationen der Prüfsubstanz ausgesetzt. Als Negativ- und Positivkontrollen dienen Testreihen mit Lösungsmittel und jeweils einem bekannten Stoff, der die Hormonproduktion hemmt bzw. induziert, in einer festgelegten Konzentration. Am Ende der Expositionszeit wird das Medium aus jeder Mulde entfernt. Unmittelbar nach Entfernen des Mediums wird in jeder Mulde die Zellviabilität untersucht. Die Hormonkonzentrationen im Medium können mit einer Vielzahl von Methoden gemessen werden, wozu auch im Handel erhältliche Kits zur Hormonmessung und/oder Gerätetechniken wie die Flüssigchromatographie-Massenspektrometrie gehören. Die Daten werden als ‚fold change‘ (x-fache Änderung) bezüglich der Lösungsmittelkontrolle und der niedrigsten Konzentration mit messbarer Wirkung (Lowest-Observed-Effect-Concentration, LOEC) angegeben. Falls der Assay negativ ist, wird die höchste geprüfte Konzentration als geprüfte Konzentration ohne messbare schädliche Wirkung (No-Observed-Effect-Concentration, NOEC) dokumentiert. Schlussfolgerungen bezüglich der Fähigkeit einer Chemikalie, die Steroidgenese zu beeinträchtigen, sollten sich auf mindestens zwei unabhängige Testreihen stützen. Die erste Testreihe kann zur Dosisfindung, gegebenenfalls mit anschließender Anpassung der Konzentrationen für die Testreihen 2 und 3, dienen, wenn Probleme mit der Löslichkeit oder der Zytotoxizität auftreten oder die Aktivität der Chemikalie am Ende des geprüften Konzentrationsbereichs zu liegen scheint.
KULTURTECHNIK
Zelllinie
14. Die NCI-H295R-Zellen sind nach Unterzeichnung einer Materialübertragungsvereinbarung (Material Transfer Agreement, MTA) (14) im Handel von der American Type Culture Collections (ATCC) erhältlich.
Einleitung
15. Wegen Änderungen in der E2-Produktionskapazität der Zellen mit zunehmendem Alter/steigender Passagenzahl (2) sollten die Zellen vor ihrer Verwendung unter Befolgung eines speziellen Protokolls gezüchtet werden, und die Zahl der Passagen nach dem Auftauen der Zellen sollte ebenso dokumentiert werden wie die Zahl der Passagen, bei denen die Zellen eingefroren und in flüssigem Stickstoff gelagert wurden. Die erste Zahl gibt die tatsächliche Anzahl Zellpassagen an und die zweite Zahl beschreibt die Anzahl Passagen, bei denen die Zellen eingefroren und eingelagert wurden. So würden beispielsweise Zellen, die nach Passage fünf eingefroren und aufgetaut und anschließend dreimal geteilt wurden (vier Passagen, wenn die frisch aufgetauten Zellen als Passage 1 gezählt werden), nachdem sie wieder gezüchtet wurden, als Passage 4.5 gekennzeichnet. Ein Beispiel für ein Nummerierungsschema ist in Anhang 1 des Validierungsberichts angeführt (4).
16. Als Basis für angereichertes Medium und Einfriermedium wird Stammmedium verwendet. Angereichertes Medium ist zur Züchtung von Zellen ein notwendiger Bestandteil. Einfriermedium wurde speziell für ein auswirkungsfreies Einfrieren von Zellen für die langfristige Lagerung konzipiert. Vor der Verwendung sollte Nu-Serum (oder ein vergleichbares Serum mit gleichen Eigenschaften, das nachgewiesenermaßen Daten erzeugt, die die Anforderungen an die Prüfleistung und die Qualitätskontrolle erfüllen), das ein Bestandteil des angereicherten Mediums ist, auf Hintergrundkonzentrationen von T und E2 untersucht werden. Die Zubereitung dieser Lösungen wird im Anhang II des Validierungsberichts beschrieben (4).
17. Nach Ansetzen einer H295R-Zellkultur aus einer ursprünglichen ATCC-Charge sind die Zellen über fünf Passagen zu vermehren (d. h.,die Zellen werden vier Mal geteilt). Passage-Fünf-Zellen werden dann in flüssigem Stickstoff zur Lagerung eingefroren. Vor dem Einfrieren der Zellen wird eine Probe der vorangegangenen Passage-Vier-Zellen in einer Qualitätskontrollplatte getestet (siehe die Nummern 36 und 37), um zu überprüfen, ob die Basalhormonproduktion und die Reaktion auf positive Kontrollchemikalien die in Tabelle 5 festgelegten Qualitätskontrollkriterien für den Assay erfüllen.
18. H295R-Zellen müssen gezüchtet, eingefroren und in flüssigem Stickstoff gelagert werden, um sicherzustellen, dass für die Kultivierung und die Verwendung stets Zellen der richtigen Passage/des richtigen Alters verfügbar sind. Die maximale Anzahl Passagen nach Übernahme einer neuen (15) oder gefrorenen (16) Zellcharge in die Kultur, die zur Verwendung im H295R-Assay akzeptabel ist, sollte 10 nicht übersteigen. Akzeptable Passagen für Kulturen von Zellen aus einer bei Passage 5 eingefrorenen Charge wären beispielsweise 4.5 bis 10.5. Bei Zellen, mit denen von diesen eingefrorenen Chargen begonnen wurde, ist das in Nummer 19 beschriebene Verfahren zu befolgen. Diese Zellen sind in mindestens vier (4) zusätzlichen Passagen (Passage 4.5) zu züchten, bevor sie in Tests eingesetzt werden können.
Begin einer neuer Zellkultur mit Zellen aus der Gefrierlagerung
19. Das Verfahren mit zum Start einer neuen Zellkultur mit Zellen aus der Gefrierlagerung ist anzuwenden, wenn eine neue Charge Zellen aus der Lagerung in flüssigem Stickstoff zu Zucht- und Testzwecken entnommen wird. Dieses Verfahren ist in Anhang III des Validierungsberichts ausführlich beschrieben (4). Die Zellen werden aus der Kryokonservierung entnommen, rasch aufgetaut, in angereichertem Medium in ein Zentrifugenröhrchen überführt, bei Zimmertemperatur abzentrifugiert, in angereichertem Medium resuspendiert und in eine Kulturflasche übertragen. Das Medium sollte am nächsten Tag gewechselt werden. Die H295R-Zellen werden in einem Inkubator bei 37 °C in einer mit 5 % CO2-angereicherten Luftatmosphäre kultiviert und das Medium wird 2-3 Mal pro Woche gewechselt. Wenn die Zellen zu ungefähr 85-90 % konfluent sind, sollten sie aufgeteilt werden. Die Zellen müssen aufgeteilt werden, um die Lebensfähigkeit und das Wachstum der Zellen sicherzustellen und um ausreichend Zellen für die Durchführung von Bioassays zur Verfügung zu haben. Die Zellen werden drei Mal mit Phosphat-gepufferter Salzlösung (PBS, ohne Ca2+ Mg2+.) ausgewaschen und durch Zugabe eines geeigneten Enzyms, z. B. Trypsin, in PBS (ohne Ca2+ Mg2+) aus der Kulturflasche herausgelöst. Die Reaktion sollte nach Ablösen der Zellen von der Kulturflasche durch Zugabe eines dreifachen Volumens an angereichertem Medium, im Verhältnis zu dem für die Enzymbehandlung verwendeten Volumen, gestoppt werden. Die Zellen werden in ein Zentrifugenröhrchen überführt, bei Zimmertemperatur zentrifugiert; der Überstand wird entfernt und das Pellet in angereichertem Medium resuspendiert. Eine entsprechende Menge Zellsuspension wird in die neue Kulturflasche gegeben. Die Menge an Zellsuspension sollte sollte so gewählt werden, dass die Zellen innerhalb von fünf bis sieben Tagen Konfluenz erreichen. Das empfohlene Subkultivierungsverhältnis beträgt 1:3 bis 1:4. Die Platte ist sorgfältig zu beschriften. Die Zellen sind jetzt für die Verwendung im Assay bereit. Überschüssige Zellen sollten in flüssigem Stickstoff eingelagert werden so wie in Paragraph 20 beschrieben.
Einfrieren von H295R-Zellen (Vorbereitung von Zellen für die Kryokonservierung)
20. Um H295R-Zellen zum Einfrieren vorzubereiten, ist das oben beschriebene Verfahren für die Aufteilung von Zellen bis zu dem Schritt der Resuspendierung des Zellenpellets am Boden des Zentrifugenröhrchens zu befolgen. Hier wird das Zellenpellet in Einfriermedium resuspendiert. Die Lösung wird in ein entsprechend etikettiertes Kryogenfläschchen übertragen und bei – 80 °C 24 Stunden lang eingefroren. Danach wird das Kryogenfläschchen zur Lagerung in flüssigen Stickstoff gegeben. Dieses Verfahren ist in Anhang III des Validierungsberichts ausführlich beschrieben (4).
Plattierung und Vorinkubation von Zellen für die Durchführung der Tests
21. Die benötigte Anzahl an nach den Angaben in Absatz 19 vorbereiteten 24-Muldenplatten hängt von der Zahl der zu prüfenden Chemikalien und der Konfluenz der Zellen in den Kulturschalen ab. In der Regel bietet eine Kulturflasche (75 cm2) mit 80-90 % konfluenten Zellen genügend Zellen für eine bis eineinhalb Platten (24-Mulden) mit einer angestrebten Dichte von 200 000 bis 300 000 Zellen pro ml Medium, was innerhalb von 24 Stunden zu ungefähr 50-60 % Konfluenz in den Mulden führt (Abbildung 2). Dies ist typischerweise die optimale Zelldichte für die Hormonproduktion im Assay. Bei höheren Dichten verändern sich sowohl die T- als auch die Muster der T- als auch der E2-Produktion. Bevor der Assay das erste Mal durchgeführt wird, empfiehlt es sich, unterschiedliche Einsaatdichten zwischen 200 000 und 300 000 Zellen pro ml zu prüfen und die Dichte, die sich bei 50-60 % Konfluenz in der Mulde nach 24 Stunden ergibt, für weitere Versuche auszuwählen.
Abbildung 2
Mikrofotografie von H295R-Zellen bei einer Einsaatdichte von 50 % in einer 24-Mulden-Kulturplatte nach 24 Stunden, aufgenommen am Rand (A) und in der Mitte (B) einer Mulde.

22. Das Medium wird aus der Kulturflasche abpipettiert und die Zellen werden drei Mal mit steriler PBS (ohne Ca2+Mg2+) gespült. Es wird eine Enzymlösung (in PBS) zugegeben, um die Zellen von der Kulturflasche abzulösen. Nachdem ein angemessener Zeitraum zur Ablösung der Zellen verstrichen ist, sollte die Enzymwirkung durch Zugabe eines angereicherten Mediums im dreifachen Verhältnis zu dem für die Enzymbehandlung verwendeten Volumen gestoppt werden. Die Zellen werden in ein Zentrifugenröhrchen überführt und bei Zimmertemperatur zentrifugiert; der Überstand wird entfernt und das Zellpellet in angereichertem Medium resuspendiert. Anschließend wird die Zelldichte bestimmt, z. B. mittels einer Zählkammer oder eines Zellzählers. Die Zelllösung sollte entsprechend der gewünschten Dichte für das Ausplattieren verdünnt und gründlich gemischt werden, um eine homogene Zelldichte sicherzustellen. Die Zellen sollten mit 1 ml Zelllösung/Mulde plattiert und die Platten und Mulden beschriftet werden. Die ausgesäten Platten werden 24 Stunden lang bei 37 °C und 5 % CO2 in Luft inkubiert, damit die Zellen an den Mulden anwachsen können.
ANFORDERUNGEN AN DIE QUALITÄTSKONTROLLE
23. Es ist wichtig, bei der Dosierung exakte Volumina der Lösungen und Proben in die Mulden zu geben, weil diese Volumina die Konzentrationen bestimmen, die in den Berechnungen der Assay-Ergebnisse verwendet werden.
24. Vor dem Ansetzen einer Zellkultur und der späteren Durchführung von Tests, hat jedes Labor die Empfindlichkeit seines Hormonmesssystems nachzuweisen (Nummern 29-31).
25. Werden antikörperbasierte Hormonmessungsassays verwendet, sind die Prüfsubstanzen vor Beginn der Tests, wie unter Nummer 32 beschrieben, darauf zu prüfen, ob sie das für die quantitative Bestimmung von T und E2 verwendete Bestimmungssystem unter Umständen beeinträchtigen können.
26. Für den Assay wird DMSO als Lösungsmittel empfohlen. Falls ein alternatives Lösungsmittel verwendet wird, ist Folgendes zu bestimmen:
|
— |
die Löslichkeit der Prüfsubstanz, Forskolin und Prochloraz im Lösungsmittel und |
|
— |
die Zytotoxizität in Abhängigkeit von der Konzentration des Lösungsmittels. |
Es wird empfohlen, dass die maximal zulässige Lösungsmittelkonzentration eine 10-fache Verdünnung der am wenigsten zytotoxischen Konzentration des Lösungsmittels nicht übersteigen sollte.
27. Bevor die Tests zum ersten Mal durchgeführt werden, hat das Labor einen Eignungsversuch durchzuführen und nachzuweisen, dass es die entsprechende Zellkultur und die Versuchsbedingungen, die für die chemischen Prüfungen laut den Beschreibungen in den Absätzen 33-35 erforderlich sind, erzielen und aufrechterhalten kann.
28. Wenn Testreihen begonnen werden, bei denen eine neue Charge verwendet wird, ist vor der Verwendung einer neuen Zellcharge eine Testreihe mit einer Kontrollplatte durchzuführen, um die Leistungsfähigkeit der Zellen, wie unter den Nummern 36 und 37 beschrieben, zu bewerten.
Leistung des Hormonmesssystems
Methodenempfindlichkeit, -genauigkeit, -präzision und Kreuzreaktivität mit der Probenmatrix
29. Jedes Labor kann für die Analyse der Produktion von T und E2 durch H295R-Zellen ein Hormonmesssystem seiner Wahl verwenden, so lange dieses die Leistungskriterien, einschließlich der Quantifizierungsgrenze (Limit of Quantification, LOQ) erfüllt. Nominal liegt die Quantifizierungsgrenze bei 100 pg/ml für T bzw. bei 10 pg/ml für E2. Diese Grenzen basieren auf den in den Validierungsstudien beobachteten Basalhormonspiegeln. In Abhängigkeit zu den im durchführenden Labor erzielten Basalhormonspiegeln können jedoch auch höhere oder niedrigere Werte angemessen sein. Vor dem Ansetzen einer Qualitätskontrollplatte und der Einleitung von Testreihen hat das Labor nachzuweisen, dass der Hormonassay, der verwendet werden soll, Hormonkonzentrationen in angereichertem Medium so genau und präzise bestimmen kann, dass die in den Tabellen 1 und 5 angegebenen Qualitätskontrollkriterien erfüllt werden. Dieser Nachweis erfolgt durch die Analyse eines mit einer internen Hormonkontrolle versetzten angereicherten Mediums. Das angereicherte Medium ist mit mindestens drei Konzentrationen eines jeden Hormons zu versetzen (z. B. 100, 500 und 2 500 pg/ml von T; 10, 50 und 250 pg/ml von E2; oder für die niedrigsten Spikekonzentrationen für T und E2 können die auf den Nachweisgrenzen des gewählten Hormonmesssystems gestützten niedrigstmöglichen Konzentrationen verwendet werden) und zu analysieren. Die gemessenen Hormonkonzentrationen der nicht extrahierten Proben sollten innerhalb von 30 % der Nominalkonzentrationen liegen und die Variation zwischen wiederholten Messungen derselben Probe sollte 25 % nicht übersteigen (weitere Kriterien für die Qualitätskontrolle finden sich auch in Tabelle 8). Werden diese Qualitätskontrollkriterien erfüllt, wird davon ausgegangen, dass der ausgewählte Hormonassay ausreichend genau und präzise ist und nicht zu Kreuzreaktionen mit Mediumsbestandteilen (Probenmatrix) führt, zumindest nicht in einem derartigen Ausmaß, dass von einer deutlichen Beeinflussung des Assayergebnisses auszugehen ist. In einem solchen Fall ist keine Extraktion von Proben vor der Messung der Hormone erforderlich.
30. Für den Fall, dass die in den Tabellen 1 und 8 angeführten Qualitätskontrollkriterien nicht erfüllt werden, kann es zu einer erheblichen Matrixwirkung kommen und es ist ein Versuch mit extrahiertem und mit Spike (T oder E2) versetztem Medium durchzuführen. Ein Beispiel für ein Extraktionsverfahren ist in Anhang 1 des Validierungsberichts angeführt (4). Die Messungen der Hormonkonzentrationen in den extrahierten Proben sind dreifach durchzuführen (17). Wenn gezeigt werden kann, dass die Bestandteile des Mediums nach der Extraktion die Hormonbestimmungsmethode nach den Festlegungen durch die Qualitätskontrollkriterien nicht beeinträchtigen, sind alle weiteren Versuche mithilfe extrahierter Proben durchzuführen. Falls die Qualitätskontrollkriterien nach der Extraktion nicht erfüllt werden können, ist das verwendete Hormonmesssystem für den Zweck des H295R Steroidgenese-Assay nicht geeignet und es ist eine alternative Hormonbestimmungsmethode zu verwenden.
Standardkurve
31. Die Hormonkonzentrationen der Lösungsmittelkontrollen (LK) sollten innerhalb des linearen Teils der Standardkurve liegen. Die Werte der Lösungsmittelkontrollen sollten vorzugsweise nahe an den Mittelpunkt des linearen Anteils fallen, um sicherzustellen, dass die Stimulation und die Hemmung der Hormonsynthese gemessen werden kann. Die zu messenden Verdünnungen des Mediums (oder Extrakte) sind entsprechend auszuwählen. Die lineare Beziehung ist durch einen geeigneten statistischen Ansatz zu bestimmen.
Test auf chemische Interferenz
32. Sollen zur Messung der Hormone antikörperbasierte Assays, wie zum Beispiel Enzyme-Linked Immunosorbent-Assays (ELISA) oder Radio-Immuno-Assays (RIA) verwendet werden, ist jede Chemikalie vor Beginn der tatsächlichen Prüfung der Chemikalien darauf zu prüfen, ob sie das einzusetzende Hormonmesssystem möglicherweise beeinträchtigt (Anhang III des Validierungsberichts (4)), da diese Tests durch einige Chemikalien beeinträchtigt werden können (17). Wenn aus der Bestimmung der Hormonanalyse hervorgeht, dass eine Beeinflussung ≥ 20 % der Basalhormonproduktion für T und/oder E2 erfolgt, ist der Test auf chemische Beeinflussung des Hormonassay (wie im Anhang III des Validierungsberichts (4) Abschnitt 5.0 beschrieben) für alle Stammlösungsverdünnungen der Prüfsubstanz durchzuführen, um die Schwellendosis zu ermitteln, bei der eine signifikante Beeinflussung (≥ 20 %) erfolgt. Wenn die Beeinflussung geringer ist als 30 % können die Ergebnisse um die Beeinflussung korrigiert werden. Wenn die Beeinflussung größer ist als 30 %, sind die Daten ungültig und die Daten bei diesen Konzentrationen sind zu verwerfen. Wenn es bei mehr als einer nicht zytotoxischen Konzentration zu einer signifikanten Beeinflussung des Hormonmesssystems durch die Prüfsubstanz kommt, muss ein anderes Hormonmesssystem verwendet werden. Um Beeinflussungen von kontaminierenden Chemikalien zu vermeiden, wird empfohlen, dass Hormone aus dem Medium mithilfe eines geeigneten Lösungsmittels extrahiert werden. Geeignete Methoden finden sich im Validierungsbericht (4).
Tabelle 1
Leistungskriterien für Hormonmesssysteme
|
Parameter |
Kriterium |
|
Empfindlichkeit der Messmethode |
Quantifizierungsgrenze (LOQ) T: 100 pg/ml; E2: 10 pg/ml (18) |
|
Hormonextraktionseffizienz (nur, wenn Extraktion nötig ist) |
Die durchschnittlichen Wiederfindungsraten (gestützt auf dreifache Messungen) für die versetzten Hormonmengen sollten nicht mehr als 30 % von der Menge abweichen, die zugegeben wurde. |
|
Chemische Beeinflussung (nur antikörperbasierte Systeme) |
Es sollte zu keiner wesentlichen (≥ 30 % der Basalhormonproduktion des entsprechenden Hormons) Kreuzreaktion mit einem der von den Zellen produzierten Hormone kommen (19) (20) |
Eignungsprüfung des Prüflabors
33. Bevor unbekannte Chemikalien geprüft werden, hat ein Labor nachzuweisen, dass es die Kompetenz besitzt, die entsprechenden Zellkultur- und die Versuchsbedingungen, die für die erfolgreiche Durchführung des Assays erforderlich sind, herzustellen und aufrechtzuerhalten. Dieser Nachweis erfolgt im Rahmen einer Eignungsprüfung. Da die Leistung eines Assays unmittelbar mit den durchführenden Labortechnikern zusammenhängt, sollten diese Verfahren teilweise wiederholt werden, wenn es zu einem Wechsel des Laborpersonals kommt.
34. Diese Eignungsprüfung wird unter denselben Bedingungen durchgeführt, wie sie unter den Nummern 38 bis 40 beschrieben sind, d. h. Zellen werden sieben zunehmenden Konzentrationen starker, mittlerstarker und schwacher Induktoren und Inhibitoren sowie einer negativen Chemikalie ausgesetzt werden (siehe Tabelle 2). Zu den zu prüfenden Chemikalien gehören im Einzelnen der starke Induktor Forskolin (CAS-Nr. 66575-29-9), der starke Inhibitor Prochloraz (CAS-Nr. 67747-09-5), der mittelstarke Induktor Atrazin (CAS-Nr. 1912-24-9), der mittelstarke Inhibitor Aminoglutethimid (CAS-Nr. 125-84-8), der schwache Induktor (E2-Produktion) und der schwache Inhibitor (T-Produktion) Bisphenol A (CAS-Nr. 80-05-7) sowie die negative Chemikalie Human-Choriongonadotropin (hCG) (CAS-Nr. 9002-61-3), wie in Tabelle 2 dargestellt. Testreihen mit separaten Platten werden für alle Chemikalien durchgeführt; dabei ist das in Tabelle 6 angegebene Format zu verwenden. Eine Qualitätskontrollplatte (Tabelle 4, Nummern 36-37) ist bei den täglichen Testreihen für die Eignungsprüfungschemikalien einzubeziehen.
Tabelle 2
Eignungsprüfungschemikalien und Expositionskonzentrationen
|
Chemikalie |
Prüfkonzentrationen [μM] |
|
Prochloraz |
0 (21), 0,01, 0,03, 0,1, 0,3, 1, 3, 10 |
|
Forskolin |
0 (21), 0,03, 0,1, 0,3, 1, 3, 10, 30 |
|
Atrazin |
0 (21), 0,03, 0,1, 1, 3, 10, 30, 100 |
|
Aminoglutethimid |
0 (21), 0,03, 0,1, 1, 3, 10, 30, 100 |
|
Bisphenol A |
0 (21), 0,03, 0,1, 1, 3, 10, 30, 100 |
|
HCG |
0 (21), 0,03, 0,1, 1, 3, 10, 30, 100 |
Die Exposition von H295R-Zellen gegenüber Eignungsprüfungschemikalien sollte bei der Eignungsprüfung des Labors in 24-Mulden-Platten durchgeführt werden. Die Dosierung erfolgt für alle Prüfsubstanzdosen in μM. Die Dosen sind in DMSO bei 0,1 % v/v pro Mulde zu verabreichen. Alle Prüfkonzentrationen sind in Triplikaten zu testen (Tabelle 6). Für jede Chemikalie werden separate Platten verwendet. Eine Qualitätskontrollplatte wird in alle täglichen Testreihen einbezogen.
35. Zellviabilitäts- und Hormonanalysen sind nach den Vorgaben der Nummern 42 bis 46 durchzuführen. Der Schwellenwert [niedrigste Konzentration mit messbarer Wirkung (Lowest-Observed-Effect-Concentration, LOEC)] und das Einstufungsergebnis sind zu dokumentieren und mit den Werten in Tabelle 3 zu vergleichen. Die Daten gelten als akzeptabel, wenn sie die Bedingungen für den LOEC-Wert und das Einstufungsergebnis in Tabelle 3 erfüllen.
Tabelle 3
Schwellenwerte (LOEC-Werte) und Einstufungergebnisse für Eignungsprüfungschemikalien
|
|
CAS-Nummer |
LOEC [μM] |
Entscheidungsergebnis |
||
|
T |
E2 |
T |
E2 |
||
|
Prochloraz |
67747-09-5 |
≤ 0,1 |
≤ 1,0 |
+ (22) (Inhibition) |
+ (Inhibition) |
|
Forskolin |
66575-29-9 |
≤ 10 |
≤ 0,1 |
+ (Induktion) |
+ (Induktion) |
|
Atrazin |
1912-24-9 |
≤ 100 |
≤ 10 |
+ (Induktion) |
+ (Induktion) |
|
Aminoglutethimid |
125-84-8 |
≤ 100 |
≤ 100 |
+ (Inhibition) |
+ (Inhibition) |
|
Bisphenol A |
80-05-7 |
≤ 10 |
≤ 10 |
+ (Inhibition) |
+ (Induktion) |
|
HCG |
9002-61-3 |
entfällt |
entfällt |
Negativ |
Negativ |
Qualitätskontrollplatte
36. Die Qualitätskontrollplatte (QK-Platte) wird zur Überprüfung der Leistung der H295R-Zellen unter Standardkulturbedingungen und zur Errichtung einer historischen Datenbasis für frühere Daten über Hormonkonzentrationen in Lösungsmittelkontrollen und Positiv- und Negativkontrollen sowie zur Festlegung anderer im Laufe der Zeit durchzuführenden Qualitätskontrollmaßnahmen verwendet.
|
— |
Die Leistung der H295R-Zellen ist mithilfe einer QK-Platte für jede neue ATCC-Charge oder nach der erstmaligen Verwendung zuvor eingefrorener Zellen zu bewerten, sofern die Eignungsprüfung des Labors (Nummern 32-34) nicht mit dieser Zellcharge durchgeführt worden ist. |
|
— |
Eine QK-Platte gewährleistet bei der Prüfung von Chemikalien eine vollständige Bewertung der Assay-Bedingungen (z. B. Zellviabilität, Lösungsmittelkontrollen, Negativ- und Positivkontrollen sowie Intra- und Interassay-Variabilität) und sollte Bestandteil jeder Testreihe sein. |
37. Der Qualitätskontrolltest wird in einer 24-Muldenplatte durchgeführt und folgt für die Inkubation, die Dosierung, die Zellviabilität/Zytotoxizität, die Hormonextraktion und die Hormonanalyse denselben Verfahren, die sie bereits unter den Nummern 38 bis 46 für Chemikalientestungen beschrieben wurden. Die Qualitätskontrollplatte enthält Blindproben, Lösungsmittelkontrollen und zwei Konzentrationen eines bekannten Induktors (Forskolin, 1, 10 μM) und Inhibitors (Prochloraz, 0,1, 1 μM) für die E2- und T-Synthese. Darüber hinaus wird in ausgewählten Mulden als Positivkontrolle für den Viabilitäts-/Zytotoxizitäts-Assay MeOH verwendet. Eine ausführliche Beschreibung der Plattenanordnung findet sich in Tabelle 4. Die Kriterien, die bei der QK-Platte erfüllt sein müssen, sind in Tabelle 5 aufgeführt. Die Mindestwerte für die Basalhormonproduktion für T und E2 sind sowohl bei den Mulden mit der Lösungsmittelkontrolle als auch bei den Blindproben einzuhalten.
Tabelle 4
Anordnung der Qualitätskontrollplatte für die Prüfung der Leistung nicht exponierter H295R-Zellen und Zellen, die gegenüber einem bekannten Inhibitor (PRO = Prochloraz) und Induktor (FOR = Forskolin) der E2- und T-Produktion exponiert wurden. Nach abgeschlossenem Expositionsversuch und der Entfernung des Mediums wird eine 70 %ige Methanollösung in alle MeOH-Mulden gegeben, die als Positivkontrolle für die Zytotoxizität dienen (siehe Zytotoxizitäts-Assay in Anhang III des Validierungsberichts (4)).
|
|
1 |
2 |
3 |
4 |
5 |
6 |
|
A |
Blindprobe (23) |
Blindprobe (23) |
Blindprobe (23) |
Blindprobe (23) (+ MeOH) (24) |
Blindprobe (23) (+ MeOH) (24) |
Blindprobe (23) (+ MeOH) (24) |
|
B |
DMSO (25) 1 μl |
DMSO (25) 1 μl |
DMSO (25) 1 μl |
DMSO (25) 1 μl (+ MeOH) (24) |
DMSO (25) 1 μl (+ MeOH) (24) |
DMSO (25) 1 μl (+ MeOH) (24) |
|
C |
FOR 1 μM |
FOR 1 μM |
FOR 1 μM |
PRO 0,1 μM |
PRO 0,1 μM |
PRO 0,1 μM |
|
D |
FOR 10 μM |
FOR 10 μM |
FOR 10 μM |
PRO 1 μM |
PRO 1 μM |
PRO 1 μM |
Tabelle 5
Leistungskriterien für die Qualitätskontrollplatte
|
|
T |
E2 |
|
Basalhormonproduktion in der Lösungsmittelkontrolle (LK) |
≥ 5-fache LOQ |
≥ 2,5-fache LOQ |
|
Induktion (10 μM Forskolin) |
≥ 1,5-fache LK |
≥ 7,5-fache LK |
|
Inhibition (1 μM Prochloraz) |
≤ 0,5-fache LK |
≤ 0,5-fache LK |
VERFAHREN DER CHEMIKALIENEXPOSITION
38. Die vorinkubierten Zellen werden aus dem Inkubator entnommen (Nummer 21) und vor der Zudosierung unter einem Mikroskop kontrolliert, um sicherzustellen, dass sie sich in gutem Zustand befinden (Anhaftung, Morphologie).
39. Die Zellen werden in eine Biosicherheitswerkbank platziert und das angereicherte Medium wird entfernt und durch ein neues angereichertes Medium ersetzt (1 ml/Mulde). Für diese Prüfmethode wird DMSO als Lösungsmittel empfohlen. Sollten jedoch Gründe für die Verwendung anderer Lösungsmittel vorliegen, ist dies wissenschaftlich zu begründen. Die Zellen werden durch Zugabe von 1 μl der geeigneten Stammlösung in DMSO (siehe Anhang II des Validierungsberichts (4)) pro 1 ml angereichertes Medium (Muldenvolumen) gegenüber der Prüfsubstanz exponiert. Dadurch ergibt sich eine Endkonzentration von 0,1 % DMSO in den Mulden. Um eine angemessene Vermischung sicherzustellen, wird im Allgemeinen empfohlen, die geeignete Stammlösung der Prüfsubstanz in DMSO mit angereichertem Medium zu mischen, um so für jede Dosis die gewünschte Endkonzentration zu erhalten, und die Mischung unverzüglich nach Entfernen des alten Mediums in die Mulden zu geben. Bei einer solchen Vorgehensweise sollte die Konzentration von DMSO (0,1 %) in allen Mulden gleich bleiben. Die Mulden mit den beiden höchsten Konzentrationen werden mithilfe eines Stereomikroskops visuell auf Bildung von Niederschlag oder Trübung (Hinweis auf die unvollständige Löslichkeit der Prüfsubstanz) untersucht. Werden solche Bedingungen (Trübung, Niederschlagsbildung) beobachtet, sind die Mulden mit den nächstniedrigeren Konzentrationen ebenfalls zu untersuchen (und so weiter), und Konzentrationen, die sich nicht vollständig gelöst haben, sind aus der weiteren Bewertung und Analyse auszuschließen. Die Platte wird bei 37 oC und 5 % CO2 in der Luftatmosphäre 48 Stunden lang in den Inkubator zurückgestellt. Die Anordnung der Prüfsubstanzplatte ist in Tabelle 6 dargelegt. Die Stämme 1-7 zeigen Bestückung mit zunehmenden Dosen Prüfsubstanz.
Tabelle 6
Dosierungsschema für die Exposition von H295R-Zellen gegenüber Prüfsubstanzen in einer 24-Muldenplatte
|
|
1 |
2 |
3 |
4 |
5 |
6 |
|
A |
DMSO |
DMSO |
DMSO |
Stamm 4 |
Stamm 4 |
Stamm 4 |
|
B |
Stamm 1 |
Stamm 1 |
Stamm 1 |
Stamm 5 |
Stamm 5 |
Stamm 5 |
|
C |
Stamm 2 |
Stamm 2 |
Stamm 2 |
Stamm 6 |
Stamm 6 |
Stamm 6 |
|
D |
Stamm 3 |
Stamm 3 |
Stamm 3 |
Stamm 7 |
Stamm 7 |
Stamm 7 |
40. Nach 48 Stunden werden die Expositionsplatten aus dem Inkubator genommen und jede Mulde wird unter dem Mikroskop auf den Zustand der Zellen (Anhaftung, Morphologie, Grad der Konfluenz) und Anzeichen von Zytotoxizität untersucht. Das Medium aus jeder Mulde wird in zwei gleiche Mengen (von jeweils ungefähr 490 μl) unterteilt und in zwei separate, entsprechend gekennzeichnete Fläschchen gegeben (d. h. ein Aliquot als Ersatzprobe für jede Mulde). Um zu verhindern, dass die Zellen austrocknen, wird jeweils immer nur aus einer Reihe oder einer Kolonne Medium entfernt und durch das Medium für die Zellviabilitäts-/Zytotoxizitätsprüfung ersetzt. Wenn die Zellviabilität/Zytotoxizität nicht unverzüglich bestimmt wird, werden in jede Mulde 200 μl PBS mit Ca2+ und Mg2+ zugegeben. Die Medien werden bis zur weiteren Analyse der Hormonkonzentrationen (siehe Nummern 44-46) bei — 80 oC eingefroren. Obwohl in Medium bei — 80 oC aufbewahrtes T und E2 in der Regel zwar mindestens drei Monate lang stabil bleiben, sollte die Hormonstabilität während der Lagerung in jedem Labor dennoch dokumentiert werden.
41. Unmittelbar nachdem das Medium entfernt wurde, wird für jede Expositionsplatte die Zellviabilität/Zytotoxizität bestimmt.
Bestimmung der Zellviabilität
42. Zur Bestimmung der potenziellen Auswirkung der Prüfsubstanz auf die Zellviabilität/Zytotoxizität kann ein beliebiger Zellviabilitäts-/Zytotoxizitäts-Assay durchgeführt werden. Der Assay sollte eine reale Messung des Anteils der in einer Mulde präsenten lebensfähigen Zellen gewährleisten oder es sollte nachgewiesen werden, dass er direkt mit dem (einer linearen Funktion des) Live/Dead®-Assay vergleichbar ist (siehe Anhang III des Validierungsberichts (4)). Ein alternativer Assay, der sich ebenfalls bewährt hat, ist der MTT-Test [3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazoliumbromid] (18). Die Bewertung der Zellviabilität nach den vorgenannten Methoden entspricht einer relativen Messung, die nicht unbedingt lineare Beziehungen zur absoluten Zahl der Zellen in einer Mulde aufweist. Daher sollte der Prüfer parallel dazu jede Mulde einer subjektiven visuellen Beurteilung unterziehen und Digitalfotos der Lösungsmittelkontrollen und der beiden höchsten nicht zytotoxischen Konzentrationen aufnehmen und archivieren, um so eine spätere Beurteilung der genauen Zelldichte zu ermöglichen, sollte eine solche erforderlich werden. Sollten Sichtkontrolle oder Viabilitäts-/Zytotoxizitäts-Assay auf eine Erhöhung der Zellenanzahl hindeuten, so muss diese überprüft werden. Wird die Erhöhung der Zellenanzahl bestätigt, ist dies im Prüfbericht anzugeben. Die Zellviabilität wird bezogen auf die durchschnittliche Reaktion in den Lösungsmittelkontrollen ausgedrückt (bei der davon ausgegangen wird, dass sie zu 100 % lebensfähigen Zellen entspricht) und in einer für den jeweils verwendeten Zellviabilitäts-/Zytotoxizitäts-Assay geeigneten Weise berechnet. Für den MTT-Assay kann die folgende Formel verwendet werden:
% lebensfähige Zellen = (Reaktion in der Mulde – durchschnittliche Reaktion in mit MeOH [= 100 % tote Zellen] behandelten Mulden) ÷ (durchschnittliche Reaktion in Mulden mit Lösungsmittelkontrolle – durchschnittliche Reaktion in mit MeOH [= 100 % tote Zellen] behandelten Mulden)
43. Mulden mit einer niedrigeren Viabilität als 80 % im Verhältnis zu der durchschnittlichen Viabilität in den Lösungsmittelkontrollen (= 100 % Viabilität) sollten in die endgültige Datenanalyse nicht einbezogen werden. Kommt es bei einer Zytotoxizität von beinahe 20 % zur Hemmung der Steroidgenese, muss sich der Prüfer vergewissern, dass die Ursache für die Hemmung nicht die Zytotoxizität ist.
Hormonanalyse
44. Jedes Labor kann für die Analyse von T und E2 ein Hormonmesssystem seiner Wahl verwenden. Überschüssige Aliquote von Medium aus den einzelnen Behandlungsgruppen können für die Zubereitung von Verdünnungen verwendet werden, um die Konzentration in den linearen Teil der Standardkurve zu bringen. Gemäß Nummer 29 sollte jedes Labor nachweisen, dass sein Hormonmesssystem (z. B. ELISA, RIA, LC-MS, LC-MS/MS) den Qualitätskontrollkriterien entspricht. Dieser Nachweis erfolgt durch die Analyse eines mit einer internen Hormonkontrolle versetzten angereicherten Mediums, bevor Qualitätskontrolltestreihen durchgeführt oder Chemikalien geprüft werden. Um sicherzustellen, dass die Bestandteile des Prüfsystems die Hormonmessung nicht beeinträchtigen, müssen die Hormone vor ihrer Messung möglicherweise aus dem Medium extrahiert werden (für die Bedingungen, unter denen eine Extraktion erforderlich oder nicht erforderlich ist, siehe Nummer 30). Es wird empfohlen, bei der Extraktion die in Anhang III des Validierungsberichts angegebenen Verfahrensvorschriften zu befolgen (4).
45. Wird ein im Handel erhältliches Testkit für die Messung der Hormonproduktion verwendet, sollte die Hormonanalyse nach den Vorgaben in den Bedienungsanleitungen des jeweiligen Herstellers durchgeführt werden. Die meisten Hersteller verfügen über ein spezielles Verfahren für die Durchführung von Hormonanalysen. Verdünnungen von Proben müssen so angepasst werden, dass die erwarteten Hormonkonzentrationen für die Lösungsmittelkontrollen in die Mitte des linearen Bereichs der Standardkurve des einzelnen Assays fallen (Anhang III des Validierungsberichts (4)). Werte außerhalb des linearen Bereichs der Standardkurve sind zu verwerfen.
46. Die endgültigen Hormonkonzentrationen werden folgendermaßen berechnet:
Beispiel:
|
Extrahiert: |
450 μl Medium |
|
Rekonstituiert in: |
250 μl Assay-Puffer |
|
Verdünnung im Assay: |
1:10 (um die Probe in den linearen Bereich der Standardkurve zu bringen) |
|
Hormonkonzentration im Assay: |
150 pg/ml (bereits an die Konzentration pro ml getesteter Probe angepasst) |
|
Wiederfindung: |
89 % |
|
Endgültige Hormonkonzentration = |
(Hormonkonzentration (pro ml) ÷ Wiederfindung) (Verdünnungsfaktor) |
|
Endgültige Hormonkonzentration = |
(150 pg/ml) ÷ (0,89) × (250 μl/450 μl) × 10 = 936,3 pg/ml |
Wahl der Prüfkonzentrationen
47. Es sind mindestens zwei unabhängige Assay-Testreihen durchzuführen. Sofern für die Wahl der Prüfkonzentrationen nicht bereits Informationen (wie Angaben zu Löslichkeitsgrenzen oder zur Zytotoxizität) vorliegen, wird empfohlen, die Prüfkonzentrationen für die erste Testreihe in log10-Abständen festzulegen, wobei 10-3 M die Höchstkonzentration darstellt. Ist die Chemikalie löslich und bei keiner der geprüften Konzentrationen zytotoxisch und war die erste Testreihe bei allen Konzentrationen negativ, so ist dies in einer weiteren Testreihe, die unter denselben Bedingungen wie die erste Testreihe durchgeführt wird, zu bestätigen (Tabelle 7). Sind die Ergebnisse der ersten Testreihe unschlüssig (d. h., der fold change [x-fache Änderung] ist im Vergleich zur Lösungsmittelkontrolle nur für eine einzige Konzentration statistisch signifikant) oder positiv (d. h., der fold change ist für zwei oder mehr nebeneinanderliegende Konzentrationen statistisch signifikant), so sollte der Test, wie in Tabelle 7 angegeben, mit verfeinerten Prüfkonzentrationen wiederholt werden. Die Prüfkonzentrationen in den Testreihen zwei und drei (falls zutreffend) sind auf der Grundlage der Ergebnisse aus der ersten Testreihe anzupassen, indem die Bracketing-Konzentrationen, die eine Wirkung hervorgerufen haben, mit in 1/2-log-Abständen verfeinert werden (wenn z. B. die ursprüngliche Testreihe mit 0,001, 0,01, 0,1, 1, 10, 100, 1000 μM zu Induktionen bei 1 und 10 μM geführt hat, sollten für die zweite Testreihe die Konzentrationen 0,1, 0,3, 1, 3, 10, 30, 100 μM verwendet werden), sofern keine niedrigeren Konzentrationen eingesetzt werden müssen, um einen LOEC-Wert zu erhalten. Im letztgenannten Fall sollten in der zweiten Testreihe mindestens fünf Konzentrationen unter der in der Testreihe geprüften niedrigsten Konzentration mit 1/2-log-Abständen verwendet werden. Wird die erste Testreihe durch die zweite Testreihe nicht bestätigt, (d. h., die zuvor positiv getestete Konzentration ± 1 Konzentrationssteigerung ergibt keine statistische Signifikanz), muss ein dritter Versuch unter den anfänglichen Testbedingungen durchgeführt werden. Unschlüssige Ergebnisse in der ersten Testreihe werden als negative Ergebnisse angesehen, wenn die gemessene Wirkung in keiner der beiden folgenden Testreihen bestätigt werden konnte. Unschlüssige Ergebnisse aus der ersten Testreihe werden als positive Reaktionen (Wirkungen) gewertet, wenn die Reaktion in mindestens einer weiteren Testreihe innerhalb einer Konzentrationssteigerung von ± 1 bestätigt werden kann (das Verfahren zur Datenauswertung findet sich in Abschnitt 55).
Tabelle 7
Entscheidungsmatrix für mögliche Ergebnisszenarien
|
Testreihe 1 |
Testreihe 2 |
Testreihe 3 |
Entscheidung |
|||
|
Szenario |
Entscheidung |
Szenario |
Entscheidung |
Szenario |
positiv |
negativ |
|
negativ |
Bestätigen (26) |
negativ |
Aufhören |
|
|
X |
|
negativ |
Bestätigen (26) |
positiv |
Verfeinern (27) |
negativ |
|
X |
|
unschlüssig (28) |
Verfeinern (27) |
negativ |
Bestätigen (26) |
negativ |
|
X |
|
unschlüssig (28) |
Verfeinern (27) |
negativ |
Bestätigen (26) |
positiv |
X |
|
|
unschlüssig (28) |
Verfeinern (27) |
positiv |
|
|
X |
|
|
positiv |
Verfeinern (27) |
negativ |
Bestätigen (26) |
positiv |
X |
|
|
negativ |
Bestätigen (26) |
positiv |
Verfeinern (27) |
positiv |
X |
|
|
positiv |
Verfeinern (27) |
positiv |
Aufhören |
|
X |
|
Qualitätskontrolle der Testplatte
48. Neben den Qualitätskriterien für die Qualitätskontrollplatte sind weitere Qualitätskriterien einzuhalten, die in Tabelle 8 dargelegt sind und zulässige Abweichungen zwischen Replikatmulden, Wiederholungsversuche, die Linearität und Empfindlichkeit der Hormonmesssysteme, die Variabilität zwischen Wiederholungs- Hormonmessungen derselben Probe und die prozentuale Wiederfindung gespikter Hormone nach der Mediumextraktion (falls zutreffend; die Anforderungen an die Extraktion finden sich in Nummer 30) betreffen. Die Daten sollten innerhalb der Bereiche liegen, die für jeden Parameter, der für die weitere Auswertung zu berücksichtigen ist, festgelegt wurden. Werden diese Kriterien nicht erfüllt, ist auf dem Arbeitsblatt anzugeben, dass die Qualitätskontrollkriterien bei der fraglichen Probe nicht eingehalten wurden, und die Probe ist entweder erneut zu analysieren oder aus dem Datensatz zu streichen.
Tabelle 8
Akzeptanzbereiche und/oder Variation (in %) für die H295R-Assay-Testplattenparameter
(LOQ: Quantifizierungsgrenze des Hormonmesssystems. VK: Variationskoeffizient; LK: Lösungsmittelkontrolle; DPM: Disintegrationen pro Minute)
|
|
Vergleich zwischen |
T |
E2 |
|
Basalhormonproduktion in LKs |
x-fach größer als LOQ |
≥ 5-fach |
≥ 2,5-fach |
|
Expositionsversuche — Innerhalb des Platten-VK für LKs (Replikatmulden) |
Absolute Konzentrationen |
≤ 30 % |
≤ 30 % |
|
Expositionsversuche — Zwischen Platten-VKs für LKs (Wiederholungsversuche) |
Fold Change (x-fache Änderung) |
≤ 30 % |
≤ 30 % |
|
Hormonmesssystem — Empfindlichkeit |
Feststellbare x-fache Abnahme im Vergleich zur LK |
≥ 5-fach |
≥ 2,5-fach |
|
Hormonmesssystem — Wiederholungs- messung des VK für LK (29) |
Absolute Konzentrationen |
≤ 25 % |
≤ 25 % |
|
Mediumextraktion — Wiederfindung des internen 3H-Standards (falls zutreffend) |
DPM |
≥ 65 % Nominal |
|
DATENANALYSE UND BERICHTERSTATTUNG
Datenanalyse
49. Zur Bewertung der relativen Zunahme/Abnahme der chemisch veränderten Hormonproduktion, sind die Ergebnisse auf den mittleren Lösungsmittelkontrollwert jeder Testplatte zu standardisieren und als Änderungen bezogen auf die Lösungsmittelkontrolle in jeder Testplatte anzugeben. Alle Daten sind als Mittelwert ± 1 Standardabweichung anzugeben.
50. Nur Hormondaten für Mulden, bei denen die Zytotoxizität niedriger war als 20 %, sind in die Datenanalyse aufzunehmen. Relative Änderungen sind folgendermaßen zu berechnen:
Relative Veränderung = (Hormonkonzentration in jeder Mulde) ÷ (Mittlere Hormonkonzentration in allen Mulden mit Lösungsmittelkontrolle).
51. Sollte die Sichtkontrolle der Mulde oder der unter Nummer 42 beschriebene Viabilitäts-/Zytotoxizitäts-Assay auf eine Erhöhung der Zellanzahl hindeuten, muss diese offensichtliche Erhöhung überprüft werden. Wird die Erhöhung der Zellanzahl bestätigt, ist dies im Prüfbericht anzugeben.
52. Bevor statistische Analysen durchgeführt werden, sind die Normalitäts- und Varianzhomogenitätshypothesen zu bewerten. Die Normalität ist mittels Normalwahrscheinlichkeitsplot (Standard-Probability-Plot)oder nach anderen geeigneten statistischen Methoden (z. B. Shapiro-Wilk's Test) zu bewerten. Sind die Daten (Fold Changes) nicht normal verteilt, sollte versucht werden, die Daten umzuwandeln, um eine approximative Normalverteilung zu erhalten. Wenn die Daten normalverteilt oder approximativ normalverteilt sind, sollten die Unterschiede zwischen den verschiedenen Gruppen chemischer Konzentrationen und den Lösungsmittelkontrollen anhand eines parametrischen Tests (z. B. Dunnett's Test) analysiert werden, wobei die Konzentration die unabhängige und die Reaktion (Fold Change) die abhängige Variable darstellt. Sind die Daten nicht normalverteilt, ist ein geeigneter nichtparametrischer Test zu verwenden (z. B. Kruskal-Wallis-Test, Steel's Many-One Rank Test). Unterschiede gelten als signifikant bei p ≤ 0,05. Die statistischen Bewertungen erfolgen auf Grundlage der durchschnittlichen Werte der Mulden, die unabhängige Wiederholungsdatenpunkte darstellen. Es wird davon ausgegangen, dass es aufgrund der großen Dosisstufenabstände in der ersten Testreihe (log10-Staffelung) nicht möglich sein wird, eindeutige Konzentration-Wirkungs-Beziehungen zu beschreiben, bei denen die beiden höchsten Dosen im linearen Bereich der Sigmoidkurve liegen. Daher finden bei der ersten Testreihe oder etwaigen anderen Datensätzen, bei denen dieser Fall eintritt (z. B. wenn keine maximale Wirksamkeit geschätzt werden kann), statistische Daten mit fester Variable des Typs I Anwendung.
53. Wenn mehr als zwei Datenpunkte im linearen Bereich der Kurve liegen und soweit die maximalen Wirksamkeiten berechnet werden können (wie dies für bestimmte zweite Testreihen mit 1/2-log-Abstand zwischen den Expositionskonzentrationen erwartet wird), ist ein Probit-, Logit- oder ein anderes geeignetes Regressionsmodell für die Berechnung der wirksamen Konzentrationen zu verwenden (z. B. EC50 und EC20).
54. Die Ergebnisse sind sowohl in grafischer Form (Balkendiagramme, die Mittelwert ± 1 Standardabweichung darstellen) als auch in tabellarischer Form (LOEC-/NOEC-Wert, Wirkungsrichtung und Stärke der maximalen Reaktion, die Teil des Dosis-Wirkung-Teils der Daten ist) vorzulegen (siehe Beispiel in Abbildung 3). Die Datenauswertung wird nur dann als gültig angesehen, wenn sie auf mindestens zwei unabhängig durchgeführten Testreihen basiert. Ein Versuch oder eine Testreihe gilt als unabhängig, wenn er/sie an einem anderen Tag mit neuen Lösungen und Kontrollen durchgeführt wurde. Der für die Testreihen 2 und 3 (falls erforderlich) verwendete Konzentrationsbereich kann auf der Grundlage der Ergebnisse aus der ersten Testreihe angepasst werden, um den Dosis-Wirkungs-Bereich mit der niedrigsten Konzentration mit messbarer Wirkung (LOEC) (siehe Nummer 47) besser bestimmen zu können.
Abbildung 3
Beispiel für die Präsentation und Bewertung der bei der Durchführung des H295R-Assays generierten Daten in grafischer und tabellarischer Form
(Sternchen zeigen statistisch signifikante Unterschiede zur Lösungsmittelkontrolle (p< 0,05) an. LOEC: niedrigste Konzentration mit gemessener Wirkung; Max. Änderung: Maximale Stärke der bei den einzelnen Konzentration gemessenen Reaktion, bezogen auf die durchschnittlichen Reaktion der Lösunsgsmittelkontrolle (= 1))

|
Chemikalie |
LOEC |
Max. Änderung |
|
Forskolin |
0,01 |
0,15-fach |
|
Letrozol |
0,001 |
29-fach |
Verfahren für die Datenauswertung
55. Eine Prüfsubstanz gilt als positiv, wenn die x-fache Induktion bei zwei nebeneinander liegenden Konzentrationen in mindestens zwei unabhängigen Testreihen (Tabelle 7) statistisch gesehen von der Lösungsmittelkontrolle abweicht (p ≤ 0,05). Eine Prüfsubstanz gilt nach zwei unabhängigen negativen Testreihen oder nach drei Testreihen, von denen zwei negative Ergebnisse und einer entweder unschlüssige oder positive Ergebnisse aufweisen, als negativ. Wenn die in drei unabhängigen Versuchen generierten Daten die in Tabelle 7 angeführten Entscheidungskriterien nicht erfüllen, können die Versuchsergebnisse nicht ausgewertet werden. Ergebnisse bei Konzentrationen, die über den Löslichkeitsgrenzen liegen, oder bei zytotoxischen Konzentrationen sollten nicht in die Datenauswertung einfließen.
Prüfbericht
56. Der Prüfbericht sollte folgende Angaben enthalten:
Prüfanstalt
|
— |
Name und Standort, |
|
— |
Versuchsleiter und sonstiges Personal und ihre jeweiligen Zuständigkeiten, |
|
— |
Versuch: Start- und Enddatum. |
Prüfsubstanz, Reagenzien und Kontrollen,
|
— |
Identität (Name/gegebenenfalls CAS-Nummer), Quelle, Nummer der Partie/Charge, Reinheit, Lieferant und Charakterisierung von Prüfsubstanz, Reagenzien und Kontrollen, |
|
— |
physikalische Beschaffenheit und gegebenenfalls physikalisch-chemische Eigenschaften der Prüfsubstanz, |
|
— |
Lagerbedingungen und Methode sowie Häufigkeit der Zubereitung der Prüfsubstanzen, Reagenzien und Kontrollen, |
|
— |
Stabilität der Prüfsubstanz. |
Zellen
|
— |
Herkunft und Art der Zellen, |
|
— |
Zahl der Passagen (Zellpassagen-Identifikator) der im Test verwendeten Zellen, |
|
— |
Beschreibung der Verfahren für die Pflege der Zellkulturen. |
Vorbedingungen für den Test (falls zutreffend),
|
— |
Beschreibung und Ergebnisse des Testsystems zum Nachweis der Interferenz von Chemikalien mit der Hormonproduktion, |
|
— |
Beschreibung und Ergebnisse der Messungen der Wirksamkeit der Hormonextraktion, |
|
— |
Standard- und Kalibrierungskurven für alle durchzuführenden analytischen Assays, |
|
— |
Nachweisgrenzen für die gewählten analytischen Assays. |
Prüfbedingungen
|
— |
Zusammensetzungen der Medien, |
|
— |
Konzentration der Prüfsubstanz, |
|
— |
Zelldichte (geschätzte oder gemessene Zellkonzentrationen nach 24 Stunden und nach 48 Stunden), |
|
— |
Löslichkeit der Prüfsubstanz (Löslichkeitsgrenze, falls diese bestimmt wird), |
|
— |
Inkubationszeit und -bedingungen. |
Prüfergebnisse
|
— |
Rohdaten (Kontrollen und Prüfsubstanzen) für jede Mulde — jede wiederholte Messung in Form der ursprünglichen Daten, die von dem für die Messung der Hormonproduktion verwendeten Gerät gemessen wurden (z. B. OD, Fluoreszenzeinheiten, DPM usw.), |
|
— |
Validierung der Normalität oder Erläuterung der Datenumwandlung, |
|
— |
mittlere Reaktionen ± 1 Standardabweichung für jede gemessene Mulde, |
|
— |
Zytotoxizitätsdaten (Prüfkonzentrationen, die die Zytotoxizität verursacht haben), |
|
— |
Bestätigung, dass die Qualitätskontrollanforderungen eingehalten wurden, |
|
— |
relative Änderung im Vergleich zur Lösungsmittelkontrolle, zur berücksichtigung der Zytotoxizität, |
|
— |
Balkendiagramm, das die relative x-fache Änderung (Fold Change bei jeder Konzentration, die Standardabweichung und die statistische Signifikanz, wie unter den Nummern 49-54 beschrieben, aufzeigt. |
Datenauswertung
|
— |
Die Verfahrensvorschriften für die Datenauswertung gelten für Ergebnisse und Befunde. |
Diskussion
|
— |
Liefert der Versuch Anhaltspunkte dafür, dass die T-/E2-Daten durch indirekte Wirkungen auf die Gluko- und Mineralocorticoid-Pfade beeinflusst werden könnten? |
Schlussfolgerungen
LITERATUR
|
(1) |
OECD (2002). Rahmenkonzept der OECD für die Testung und Bewertung endokrin wirksamer Substanzen (endokrine Disruptoren) (‚OECD Conceptual Framework for the Testing and Assessment of Endocrine Disrupting Chemicals‘), in Kapitel B.54 Anlage 2 dieses Anhangs. |
|
(2) |
Hecker, M., Newsted, J.L., Murphy, M.B., Higley, E.B., Jones, P.D., Wu, R. and Giesy, J.P. (2006). Human adrenocarcinoma (H295R) cells for rapid in vitro determination of effects on steroidogenesis: Hormone production. Toxicol. Appl. Pharmacol., 217, 114-124. |
|
(3) |
Hecker, M., Hollert, H., Cooper, R., Vinggaard, A.-M., Akahori, Y., Murphy, M., Nellemann, C., Higley, E., Newsted, J., Wu, R., Lam, P., Laskey, J., Buckalew, A., Grund, S., Nakai, M., Timm, G., and Giesy, J. P. (2007). The OECD validation program of the H295R steroidgenesis assay for the identification of in vitro inhibitors or inducers of testosterone and estradiol production, Phase 2: inter laboratory pre-validation studies. Env. Sci. Pollut. Res., 14, 23-30. |
|
(4) |
OECD (2010). Multi-Laboratory Validation of the H295R Steroidogenesis Assay to Identify Modulators of Testosterone and Estradiol Production. OECD Series of Testing and Assessment No. 132, ENV/JM/MONO(2010)31, Paris. Abrufbar unter [http://www.oecd.org/document/30/0,3746,en_2649_34377_1916638_1_1_1_1,00.html]. |
|
(5) |
OECD (2010). Peer Review Report of the H295R Cell-Based Assay for Steroidogenesis. OECD Series of Testing and Assessment No. 133, ENV/JM/MONO(2010)32, Paris. Abrufbar unter [http://www.oecd.org/document/30/0,3746,en_2649_34377_1916638_1_1_1_1,00.html]. |
|
(6) |
Battelle (2005). Detailed Review Paper on Steroidogenesis. Abrufbar unter [http://www.epa.gov/endo/pubs/edmvs/steroidogenesis_drp_final_3_29_05.pdf]. |
|
(7) |
Hilscherova, K., Jones, P. D., Gracia, T., Newsted, J. L., Zhang, X., Sanderson, J. T., Yu, R. M. K., Wu, R. S. S. and Giesy, J. P. (2004). Assessment of the Effects of Chemicals on the Expression of Ten Steroidogenic Genes in the H295R Cell Line Using Real-Time PCR, Toxicol. Sci., 81, 78-89. |
|
(8) |
Sanderson, J. T., Boerma, J., Lansbergen, G. and Van den Berg, M. (2002). Induction and inhibition of aromatase (CYP19) activity by various classes of pesticides in H295R human adrenocortical carcinoma cells, Toxicol. Appl. Pharmacol., 182, 44-54. |
|
(9) |
Breen, M.S., Breen, M., Terasaki, N., Yamazaki, M. and Conolly, R.B. (2010). Computational model of steroidogenesis in human H295R cells to predict biochemical response to endocrine-active chemicals: Model development for metyrapone. Environ. Health Perspect., 118: 265-272. |
|
(10) |
Higley, E.B., Newsted, J.L., Zhang, X., Giesy, J.P. and Hecker, M. (2010). Assessment of chemical effects on aromatase activity using the H295R cell line, Environ. Sci. Poll. Res., 17:1137-1148. |
|
(11) |
Gazdar, A. F., Oie, H. K., Shackleton, C. H., Chen, T. R., Triche, T. J., Myers, C. E., Chrousos, G. P., Brennan, M. F., Stein, C. A. and La Rocca, R. V. (1990). Establishment and characterization of a human adrenocortical carcinoma cell line that expresses Multiple pathways of steroid biosynthesis. Cancer Res., 50, 5488-5496. |
|
(12) |
He, Y.H., Wiseman, S.B., Zhang, X.W., Hecker, M., Jones, P.D., El-Din, M.G., Martin, J.W. and Giesy, J.P. (2010). Ozonation attenuates the steroidogenic disruptive effects of sediment free oil sands process water in the H295R cell line. Chemosphere, 80:578-584. |
|
(13) |
Zhang, X.W., Yu, R.M.K., Jones, P.D., Lam, G.K.W., Newsted, J.L., Gracia, T., Hecker, M., Hilscherova, K., Sanderson, J.T., Wu, R.S.S. and Giesy, J.P. (2005). Quantitative RT-PCR methods for evaluating toxicant-induced effects on steroidogenesis using the H295R cell line. Environ. Sci. Technol., 39:2777-2785. |
|
(14) |
Higley, E.B., Newsted, J.L., Zhang, X., Giesy, J.P. and Hecker, M. (2010). Differential assessment of chemical effects on aromatase activity, and E2 and T production using the H295R cell line. Environ. Sci. Pol. Res., 17:1137-1148. |
|
(15) |
Rainey, W. E., Bird, I. M., Sawetawan, C., Hanley, N. A., Mccarthy, J. L., Mcgee, E. A., Wester, R. and Mason, J. I. (1993). Regulation of human adrenal carcinoma cell (NCI-H295) production of C19 steroids. J. Clin. Endocrinol. Metab., 77, 731-737. |
|
(16) |
Kapitel B.55 dieses Anhangs: Hershberger Bioassay mit Ratten: Ein Kurzzeit Screening-Test auf (anti-)androgene Eigenschaften. |
|
(17) |
Shapiro, R., and Page, L.B. (1976). Interference by 2,3-dimercapto-1-propanol (BAL) in angiotensin I radioimmunoassay, J. Lab. Clin. Med., 2, 222-231. |
|
(18) |
Mosmann, T. (1983). Rapid colorimetric assay for growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods., 65, 55-63. |
|
(19) |
Brock, B.J., Waterman, M.R. (1999). Biochemical differences between rat and human cytochrome P450c17 support the different steroidogenic needs of these two species. Biochemistry. 38:1598-1606. |
|
(20) |
Oskarsson, A., Ulleras, E., Plant, K., Hinson, J. Goldfarb, P.S., (2006). Steroidogenic gene expression in H295R cells and the human adrenal gland: adrenotoxic effects of lindane in vitro. J. Appl. Toxicol., 26:484-492. |
Anlage
BEGRIFFSBESTIMMUNGEN
Angereichertes Medium : Stammmedium plus BD Nu-Serum und ITS+ Premium Mix (siehe Anhang II des Validierungsberichts) (4).
Chemikalie : ein Stoff oder eine Mischung.
CYP : Cytochrom-P450-Monooxygenase, eine Familie von Genen und den daraus hervorgegangenen Enzymen, die bei der Katalyse vielfältiger biochemischer Reaktionen, einschließlich Synthese und Verstoffwechselung von Steroidhormonen, beteiligt sind.
DPM : Disintegration (Zerfall) pro Minute, Anzahl Atome in einer bestimmten Menge radioaktiven Materials, deren Zerfall minütlich nachgewiesen wird.
E2 : 17β-Estradiol; wichtigstes Östrogen in Säugetiersystemen.
Einfriermedium : Mediumzum Einfrieren von Zellen und zur Lagerung gefrorener Zellen, bestehend aus Kulturmedium plus BD NuSerum und Dimethylsulfoxid.
H295R-Zellen : humane Adenokarzinom-Zellen, die die physiologischen Merkmale zonal undifferenzierter Nebennierenzellen menschlicher Föten aufweisen und die alle Enzyme des steroidogenen Pfades exprimieren. Sie sind bei der ATCC erhältlich.
Konfluenz : Bedeckung der Oberfläche eines Kulturmediums mit Zellen oder Proliferation von Zellen im Kulturmedium.
Linearer Bereich : Bereich innerhalb der Standardkurve eines Hormonmesssystems mit Ergebnissen proportional zur Konzentration des in der Probe präsenten Analyts.
LOEC (Lowest Observed Effect Concentration): die niedrigste Konzentrationstufe, bei der die Assay-Reaktion statistisch gesehen von der Reaktion der Lösungsmittelkontrolle abweicht.
LOQ (Limit of Quantification): Quantifizierungsgrenze; kleinste Menge einer Chemikalie, die innerhalb eines bestimmten Konfidenzintervalls im Gegensatz zur Leermessung (Blindwert) gerade noch nachgewiesen werden kann. Für die Zwecke der vorliegenden Methode und falls nicht anders angegeben, wird der LOQ-Wert in der Regel vom Hersteller der Testsysteme vorgegeben.
NOEC (No Observed Effect Concentration): die höchste getestete Konzentration, bei der im Test keine signifikante Wirkung festgestellt wird.
Passage : die Anzahl Zellteilungen nach dem Ansetzen einer Kultur aus gefrorenen Zellen, wobei der Ausgangspassage der gefrorenen Zellen die Nummer eins (1) zugeordnet wird. Zellen, die einmal gesplittet wurden, werden als Passage 2 bezeichnet, usw.
PBS (Phosphate Buffered Saline): Dulbeccos phosphatgepufferte Salzlösung.
Prüfsubstanz : ein(e) nach dieser Prüfmethode geprüfter Stoff/geprüfte Mischung.
Qualitätskontrolle (QK) : die Maßnahmen, die nötig sind, um aussagekräftige Daten zu gewährleisten.
Qualitätskontrollplatte : eine 24-Muldenplatte mit zwei Konzentrationen der positiven und negativen Kontrollen zur Überwachung der Leistung einer neuen Charge Zellen oder zur Bereitstellung der positiven Kontrollen für den Assay, wenn Chemikalien geprüft werden.
Stammmedium : die Basis für die Zubereitung anderer Reagenzien, bestehend aus einer 1:1-Mischung von Dulbecco's Modified Eagle's Medium (DMEM) und Ham's F-12 Nutrient Mixture (DMEM/F12) in 15 mM-HEPES-Puffer ohne Phenolrot oder Natriumhydrogenkarbonat. Natriumhydrogenkarbonat wird als Puffer zugegeben (siehe Anhang II des Validierungsberichts) (4).
Steroidgenese : Synthesepfad vom Cholesterol zu verschiedenen Steroidhormonen. Bestimmte Zwischenprodukte auf dem steroiden Synthesepfad, wie Progesteron und Testosteron, sind eigenständige wichtige Hormone, dienen aber auch als Vorläufer für nachgeschaltete Hormone.
T : Testosteron, eines der beiden wichtigsten Androgene bei Säugern.
Testplatte : Platte, auf der H295R-Zellen der Prüfsubstanz ausgesetzt werden. Testplatten enthalten die Lösungsmittelkontrolle und die Prüfsubstanz in sieben Konzentrationsstufen in Dreifachmulden.
Testreihe : ein unabhängiger Versuch, gekennzeichnet durch einen neuen Satz Lösungen und Kontrollen.
Trypsin 1X : eine verdünnte Lösung des Enzyms Trypsin, einer pankreatischen Serin-Protease, die zur Ablösung von Zellen von einer Zellkulturplatte verwendet wird (siehe Anhang III des Validierungsberichts) (4).
VK : Variationskoeffizient, definiert als Verhältnis der Standardabweichung einer Verteilung zu ihrem arithmetischen Mittelwert.
B.58 GENMUTATIONS-ASSAYS AN SOMATISCHEN ZELLEN UND KEIMZELLEN TRANSGENER NAGETIERE
EINLEITUNG
1. Diese Prüfmethode entspricht der OECD-Prüfrichtlinie (TG) 488 (2013). EU-Prüfmethoden stehen für eine Vielzahl von In-vitro-Assays zum Nachweis von Chromosomen- und/oder Genmutationen zur Verfügung. Es gibt Prüfmethoden für In-vivo-Endpunkte (d. h. Chromosomenaberrationen und nicht geplante DNA-Synthesen), die jedoch keine Genmutationen messen. Mutationsassays an transgenen Nagetieren (transgenic rodents, TGR) decken den Bedarf an praktischen und zugänglichen In-vivo-Genmutationstests.
2. Die TGR-Mutationsassays sind gründlich überprüft worden (24) (33). Für diese Tests werden transgene Ratten und Mäuse verwendet, die mehrere Kopien chromosomal integrierten Plasmids oder Phagen-Shuttle-Vektoren enthalten. Die Transgene enthalten Reporter-Gene zum Nachweis verschiedener Arten von Mutationen, die in vivo durch Prüfsubstanzen induziert wurden.
3. Mutationen bei Nagetieren werden bewertet, indem das Transgen wiedergefunden und der Phänotyp des Reporter-Gens in einer bakteriellen Wirtszelle ohne Reporter-Gen analysiert wird. Mit TGR-Mutationsassays werden Mutationen gemessen, die in genetisch neutralen Genen induziert wurden, die sich in praktisch jedem Gewebe des Nagetiers wiederfinden. Mit diesen Assays können somit viele der bestehenden Grenzen, die die In-vivo-Untersuchung von Genmutationen in endogenen Genen derzeit noch behindern (z. B. begrenzte Menge geeigneten Gewebes für die Analyse, Negativ-/Positivselektion im Vergleich zu den Mutationen).
4. Die vorliegenden Daten deuten darauf hin, dass Transgene in ähnlicher Weise wie endogene Gene auf Mutagene reagieren, insbesondere was den Nachweis des Austauschs einer Base gegen eine andere, Rasterschubmutationen und kleine Deletionen und Insertionen betrifft(24).
5. Die Internationalen Workshops für Genotoxizitätsprüfungen (International Workshop on Genotoxicity Testing, IWGT) haben die Berücksichtigung von TGR-Mutationsassays für den In-vivo-Nachweis von Genmutationen befürwortet und ein Durchführungsprotokoll empfohlen (15) (29). Die vorliegende Prüfmethode stützt sich auf diese Empfehlungen. Eine weitergehende Analyse, die die Verwendung dieses Protokolls unterstützt, findet sich unter (16).
6. Es wird davon ausgegangen, dass es in Zukunft möglich sein wird, TGR-Mutationsassays mit einer Prüfung auf Toxizität bei wiederholten Dosisverabreichungen (Kapitel B.7 dieses Anhangs) zu kombinieren. Es sind jedoch weitere Daten erforderlich, um sicherzustellen, dass die Empfindlichkeit der TGR-Mutationsassays durch den kürzeren — eintägigen — Zeitabstand zwischen dem Ende des Verabreichungszeitraums und dem Zeitpunkt der Probenahme (wie sie bei Prüfungen auf Toxizität bei wiederholter Verabreichung üblich ist) im Vergleich zu dem bei TGR-Mutationsassays üblichen Drei-Tage-Abstand nicht beeinträchtigt wird. Es sind auch Daten erforderlich, die belegen, dass die Leistung der Assays mit wiederholter Verabreichung durch die Verwendung eines transgenen Nagerstamms anstelle der traditionellen Nagerstämme nicht beeinträchtigt wird. Sobald diese Daten vorliegen, wird die vorliegende Prüfmethode aktualisiert.
7. Die wichtigsten Begriffe sind im Anhang bestimmt.
AUSGANGSÜBERLEGUNGEN
8. Für die nachstehend angeführten TGR-Mutationsassays liegen ausreichende Daten vor, die ihre Verwendung im Rahmen der vorliegenden Prüfmethode, soweit sie unter Standardbedingungen durchgeführt werden, unterstützen: lacZ Bakteriophage Maus (Muta™Mouse); lacZ Plasmid Maus; gpt Delta (gpt und Spi– ) Maus und Ratte; lacI Maus und Ratte (Big Blue®). Außerdem kann der cII-Positivselektionsassay zur Bewertung von Mutationen in den Modellen Big Blue® und Muta™Mouse verwendet werden. Die Mutagenese in den TGR-Modellen wird in der Regel als Mutantenhäufigkeit bewertet; erforderlichenfalls kann eine Molekularanalyse der Mutationen aber zusätzliche Informationen liefern (siehe Nummer 24).
9. Diese In-vivo-Genmutationstests an Nagetieren sind insbesondere für die Bewertung des mutagenen Risikos von Relevanz, da die Assay-Reaktionen vom In-vivo-Stoffwechsel, von der Pharmakokinetik, den DNA-Reparaturprozessen und der Transläsionssynthese abhängen, auch wenn diese bei den einzelnen Tierarten, Geweben und Arten der DNA-Schädigungen unterschiedlich sein können. Ein In-vivo-Genmutationsassay ist auch für die weitere Untersuchung einer bei einer In-vitro-Prüfung festgestellten mutagenen Wirkung und zur weiteren Berücksichtigung von Testergebnissen von Nutzen, bei denen andere In-vivo-Endpunkte verwendet werden (24). Abgesehen davon, dass zwischen Genmutationen und der Auslösung von Krebs ein kausaler Zusammenhang besteht, ist die Genmutation ein relevanter Endpunkt für die Prognose anderer mutationsbedingter Erkrankungen somatischer Gewebe als Krebs (12) (13) und von über die Keimbahn übertragenen Krankheiten.
10. Wenn Anzeichen dafür bestehen, dass die Prüfsubstanz oder ein reaktiver Metabolit keines der Zielgewebe erreicht, ist die Durchführung eines TGR-Mutationsassays nicht sinnvoll.
PRINZIP DER PRÜFMETHODE
11. In den unter Nummer 8 beschriebenen Assays ist das Zielgen bakterieller oder bakteriophager Herkunft, und seine Wiederfindung anhand der genomischen Nagetier-DNA erfolgt durch Einführung des Transgens in einen λ-Bakteriophagen oder Plasmid-Shuttle-Vektor. Bei dem Verfahren wird aus dem zu untersuchenden Nagetiergewebe genomische DNA extrahiert; diese genomische DNA wird in vitro verarbeitet (d. h. die λ-Vektoren werden verpackt, bzw. die Plasmiden werden ligiert und elektroporiert, um den Shuttle-Vektor wiederzufinden), und anschließend werden unter geeigneten Bedingungen Mutationen in bakteriellen Wirtszellen nachgewiesen. Bei den Assays werden neutrale Transgene eingesetzt, die sich ohne weiteres in den meisten Geweben wiederfinden lassen.
12. Bei dem grundlegenden TGR-Mutationsversuch wird das Nagetier über einen bestimmten Zeitraum mit einer Chemikalie behandelt. Die Chemikalien können in jeder geeigneten Form, auch durch Implantation, verabreicht werden (z. B. Prüfung von Medizinprodukten). Der Gesamtdauer, während der die Chemikalie einem Tier verabreicht wird, wird Verabreichungszeitraum genannt. Auf die Verabreichung folgt — vor der Tötung des Tieres — in der Regel ein Zeitraum, in dem die Chemikalie nicht verabreicht wird und in dem nicht reparierte DNA-Läsionen in stabile Mutationen fixiert werden. In der Literatur wird dieser Zeitraum unterschiedlich entweder als Manifestationszeit, Fixierungszeit oder Expressionszeit bezeichnet; das Ende dieses Zeitraums markiert den Probenahmezeitpunkt (15) (29). Nach dem Töten des Tieres wird die genomische DNA aus dem zu untersuchenden Gewebe isoliert und gereinigt.
13. Daten zu jeweils ein und demselben Gewebe/Tier aus mehreren Verpackungen/Ligationen werden normalerweise aggregiert und die Mutantenhäufigkeit wird in der Regel mithilfe von insgesamt 105 bis 107 plaquebildenden oder kolonienbildenden Einheiten bewertet. Bei Anwendung von Positivselektionsmethoden wird die Gesamtzahl der plaquebildenden Einheiten mit einem separaten Satz nicht-selektiver Platten bestimmt.
14. Es wurden Positivselektionsmethoden entwickelt, um den Nachweis von Mutationen sowohl im gpt-Gen[gpt Delta Maus und Ratte, gpt – Phänotyp (20) (22) (28)] als auch im lacZ-Gen [Muta™Maus oder lacZ-Plasmid Maus (3) (10) (11) (30)] zu erleichtern; hingegen werden Mutationen des lacI-Gens in Big Blue®-Tieren über eine nicht-selektive Methode nachgewiesen, bei der Mutanten durch die Erzeugung (blau) gefärbter Plaques gekennzeichnet werden. Eine Positivselektionsmethode liegt auch für den Nachweis von Punktmutationen vor, zu denen es im cII-Gen des λ-Bakteriophagen-Shuttle-Vektors kommt [Big Blue® Maus oder Ratte und Muta™Maus (17)] sowie von Deletionen in den λ rot und gam [Spi– Auswahl in gpt Delta Maus und Ratte (21) (22) (28)]. Die Mutantenhäufigkeitwird berechnet, indem die Zahl der Plaques/Plasmiden mit Mutationen im Transgen durch die Gesamtzahl der aus derselben DNA-Probe gewonnenen Plaques/Plasmiden geteilt wird. In TGR-Genmutationsstudien ist die Mutantenhäufigkeit der gemessene Parameter. Darüber hinaus kann eine Mutationshäufigkeit als Anteil der Zellen mit unabhängigen Mutationen bestimmt werden; bei dieser Berechnung muss die klonale Expansiondurch Sequenzierung der wiedergefundenen Mutanten korrigiert werden (24).
15. Die in den lacI-, lacZ-, cII- und gpt- Punktmutationsassays gezählten Mutationen bestehen hauptsächlich aus Substitutionsmutationen (Basenpaare), Rasterschubmutationen und kleinen Insertionen/Deletionen. Der relative Anteil dieser Mutationstypen unter spontanen Mutationen ist vergleichbar mit dem Anteil, der im endogenen Hprt-Gen gemessen wird. Große Deletionen werden nur mit der Spi–-Auswahl und den lacZ-Plasmidassays nachgewiesen (24). Zu untersuchende Mutationen sind In-vivo-Mutationen, zu denen es in der Maus oder in der Ratte kommt. In-vitro und Ex-vivo-Mutationen, zu denen es bei der Phagen-/Plasmidwiederfindung, Replikation oder Reparatur kommen kann, sind relativ selten und können in einigen Systemen durch die bakterielle Wirtszelle/das System der positiven Selektion eindeutig bestimmt oder ausgeschlossen werden.
BESCHREIBUNG DER METHODE
Vorbereitungen
Auswahl der Versuchstierarten
16. Derzeit ist eine Vielzahl von Modellen für den Nachweis von Genmutationen bei transgenen Mäusen erhältlich und diese Systeme sind gängiger als Modelle für transgene Ratten. Wenn die Ratte eindeutig ein geeigneteres Modell ist als die Maus (beispielsweise wenn der Mechanismus der Karzinogenese bei einem nur in Ratten vorkommenden Tumor untersucht wird, um einen Bezug zu einer Rattentoxizitätsstudie herzustellen, oder wenn bekannt ist, dass der Stoffwechsel der Ratte für den menschlichen Stoffwechsel repräsentativer ist) ist die Verwendung von Modellen für den Nachweis von Genmutationen bei transgenen Ratten in Erwägung zu ziehen.
Haltungs- und Fütterungsbedingungen
17. Die Temperatur im Versuchstierraum sollte idealerweise 22 °C (± 3 °C) betragen. Auch wenn die relative Luftfeuchtigkeit mindestens 30 % betragen und — außer beim Reinigen des Raums — 70 % nicht überschreiten sollte, ist eine Luftfeuchtigkeit von 50-60 % anzustreben. Die Beleuchtung sollte künstlich sein, und die täglichen Hell- und Dunkelphasen sollten sich im Abstand von 12 Stunden abwechseln. An die Versuchstiere kann herkömmliches Laborfutter verfüttert werden, und eine unbegrenzte Trinkwasserversorgung ist zu gewährleisten. Die Auswahl des Futters wird eventuell dadurch beeinflusst, dass eine geeignete Beimischung der Prüfsubstanz sichergestellt werden muss, wenn die Prüfsubstanz auf diese Art verabreicht werden soll. Die Tiere sollten in kleinen gleichgeschlechtlichen Gruppen (nicht mehr als fünf Tiere) untergebracht werden, wenn kein aggressives Verhalten erwartet wird. Sie können auch einzeln gehalten werden, wenn dies wissenschaftlich gerechtfertigt ist.
Vorbereitung der Tiere
18. Gesunde, jung-adulte geschlechtsreife Tiere (acht bis zwölf Wochen alt zu Beginn der Behandlung) werden nach dem Zufallsprinzip in die Kontroll- bzw. Behandlungsgruppen eingeteilt. Die Tiere werden individuell gekennzeichnet. Sie werden über einen Zeitraum von mindestens fünf Tagen unter Laborbedingungen eingewöhnt. Die Käfige sollten so angeordnet werden, dass etwaige Wirkungen aufgrund des Käfigstandorts minimiert werden. Bei Versuchsbeginn sollten die Gewichtsunterschiede der Tiere möglichst gering sein und ± 20 % des geschlechtsspezifischen Durchschnittsgewichts nicht überschreiten.
Herstellung der Dosen
19. Feste Prüfsubstanzen sollten vor der Verabreichung an die Tiere in geeigneten Lösungsmitteln oder Vehikeln gelöst oder suspendiert oder in das Futter bzw. das Trinkwasser gegeben werden. Flüssige Prüfsubstanzen können direkt verabreicht oder zuvor verdünnt werden. Für inhalative Expositionen können die Prüfsubstanzen in Abhängigkeit zu ihren physikalisch-chemischen Eigenschaften als Gas, Dampf oder festes/flüssiges Aerosol verabreicht werden. Es sind frische Zubereitungen der Prüfsubstanz zu verwenden, es sei denn, die Stabilität der Substanz bei Lagerung ist erwiesen.
Prüfbedingungen
Lösungsmittel/Vehikel
20. Das Lösungsmittel/Vehikel sollte in der verwendeten Dosierung keine toxischen Wirkungen hervorrufen und nicht im Verdacht stehen, mit der Prüfsubstanz eine chemische Reaktion einzugehen. Werden keine allgemein anerkannten Lösungsmittel/Vehikel verwendet, so sollte ihre Einbeziehung durch Kompatibilitätsdaten untermauert werden. Es empfiehlt sich, als erste Wahl möglichst die Verwendung eines wässrigen Lösungsmittels/Vehikels in Erwägung zu ziehen.
Positivkontrollen
21. In der Regel sind parallel Positivkontrolltiere zu verwenden. Bei Laboren, die ihre Kompetenz unter Beweis gestellt haben (siehe Nummer 23) und diese Assays routinemäßig verwenden, kann DNA von zuvor behandelten Positivkontrolltieren in jeden Versuch einbezogen werden, um den Erfolg der Methode zu bestätigen. Derartige DNA aus vorausgegangenen Versuchen ist von derselben Tierart und denselben Zielgeweben zu gewinnen und ordnungsgemäß zu lagern (siehe Nummer 36). Werden parallel Positivkontrollen verwendet, müssen diese nicht auf dem gleichen Verabreichungsweg gegeben werden wie die Prüfsubstanz; von den Positivkontrollen sollte jedoch bekannt sein, dass sie Mutationen in einem oder mehreren für die Prüfsubstanz relevanten Geweben hervorrufen. Die Dosen der Chemikalien für die Positivkontrolle sind so auszuwählen, dass sie schwache oder moderate Wirkungen hervorrufen, mit denen die Leistung und Empfindlichkeit des Assays kritisch bewertet werden können. Beispiele für derartige Chemikalien und bestimmte Zielgewebe finden sich in Tabelle 1.
Tabelle 1
Beispiele für Positivkontrollchemikalien und bestimmte Zielgewebe
|
Positivkontroll-chemikalie und CAS-Nummer |
Einecs-Name und Einecs-Nummer |
Charakterisierung |
Mutationszielgewebe |
|
|
Ratte |
Maus |
|||
|
N-Ethyl-N-Nitroso-Harnstoff [CAS-Nr. 759-73-9] |
N-Ethyl-N-Nitroso-Harnstoff [212-072-2] |
Direkt wirkendes Mutagen |
Leber, Lunge |
Knochenmark, Kolon, Kolonepithel, Darm, Leber, Lunge, Milz, Niere, Granulosazellen der Ovarien, männliche Keimzellen |
|
Ethyl carbamat (Urethan) [CAS-Nr. 51-79-6] |
Urethan [200-123-1] |
Mutagen, erfordert Stoffwechsel, ruft aber nur schwache Wirkungen hervor |
|
Knochenmark, Vormagen, Dünndarm, Leber, Lunge, Milz |
|
2,4-Diaminotoluen [CAS-Nummer 95-80-7] |
4-Methyl-m-phenylenediamin [202-453-1] |
Mutagen, erfordert Stoffwechsel, ebenfalls positiv in dem Spi–Assay |
Leber |
Leber |
|
Benzo[a]pyren [CAS-Nr. 50-32-8] |
Benzo[def]chrysen [200-028-5] |
Mutagen, erfordert Stoffwechsel |
Leber, Omenta, |
Knochenmark, Brust, Kolon, Vormagen, Drüsenmagen, Herz, Leber, Lunge, männliche Keimzellen |
Negativkontrollen
22. In jede Probenahme sind Negativkontrolltiere einzubeziehen, die nur ein Lösungsmittel oder ein Vehikel erhalten und ansonsten in derselben Weise behandelt werden wie die Behandlungsgruppen. Um die Eignung der Vehikelkontrolle festzustellen, sollten darüber hinaus in jede Probenahme auch unbehandelte Kontrolltiere einbezogen werden, soweit keine historischen oder veröffentlichten Kontrolldaten vorliegen, aus denen hervorgeht, dass das gewählte Lösungsmittel/Vehikel keine schädlichen oder mutagenen Wirkungen hervorruft.
Eignungsprüfung des Prüflaboratoriums
23. Das Labor sollte seine Kompetenz unter Beweis stellen, erwartete Ergebnisse aus veröffentlichten Daten (24) in folgenden Bereichen zu reproduzieren: 1) Mutantenhäufigkeiten bei Positivkontrollchemikalien (einschließlich schwacher Reaktionen), wie den in Tabelle 1 angeführten Chemikalien, Nicht-Mutagenen und Vehikelkontrollen; und 2) die Wiederfindung von Transgenen aus genomischer DNA (z. B. Verpackungseffizienz).
Sequenzierung von Mutanten
24. Für Zulassungsanträge ist keine DNA-Sequenzierung von Mutanten erforderlich, insbesondere, wenn ein eindeutiges positives oder negatives Ergebnis erzielt wird. Sequenzierungsdaten können sich jedoch als nützlich erweisen, wenn eine hohe interindividuelle Streuung festgestellt wird. In solchen Fällen können durch Sequenzierung ‚Jackpot‘-Mutationen oder klonale Ereignisse ausgeschlossen werden, indem der Anteil eines bestimmten Gewebes an eindeutigen Mutanten ermittelt wird. Die Sequenzierung von ungefähr zehn Mutanten pro Gewebe und Tier sollte für die einfache Bestimmung, ob klonale Mutanten zur Mutantenhäufigkeit beitragen, ausreichen; um die Mutantenhäufigkeit mathematisch um die Klonalität zu berichtigen, kann es sich als notwendig erweisen, bis zu 25 Mutanten zu sequenzieren. Die Sequenzierung von Mutanten kann auch in Betracht gezogen werden, wenn kleinere Zunahmen der Mutantenhäufigkeit (d. h. Zunahmen knapp über den Werten der unbehandelten Kontrolltiere) festgestellt werden. Unterschiede im Mutantenspektrum zwischen den Mutantenkolonien aus behandelten und unbehandelten Tieren können eine mutagene Wirkung unterstützen (29). Mutationsspektra können außerdem für die Aufstellung mechanistischer Hypothesen nützlich sein. Wenn Sequenzierung als fester Bestandteil in das Versuchsprotokoll aufgenommen werden soll, ist der Versuch besonders sorgfältig zu planen, insbesondere im Hinblick auf die Anzahl der pro Probe sequenzierten Mutanten, um entsprechend dem verwendeten statistischen Modell eine angemessene Aussagekraft zu erzielen (siehe Nummer 43).
VERFAHREN
Zahl und Geschlecht der Versuchstiere
25. Es sollten genügend Tiere pro Gruppe vorgesehen sein, damit die statistische Aussagekraft gewährleistet ist, die nötig ist, um mindestens eine Verdopplung der Mutantenhäufigkeit nachzuweisen. Die Gruppen bestehen aus mindestens fünf Tieren; falls die statistische Aussagekraft jedoch zu gering ist, ist die Zahl der Tiere gegebenenfalls zu erhöhen. In der Regel sind männliche Tiere zu verwenden. In einigen Fällen kann die Durchführung von Tests ausschließlich an weiblichen Tieren gerechtfertigt sein; beispielsweise wenn frauenspezifische Humanmedikamente getestet werden oder wenn spezielle Untersuchungen zum weiblichen Stoffwechsel durchgeführt werden. Liegen im Hinblick auf Toxizität oder Stoffwechsel signifikante Unterschiede zwischen den Geschlechtern vor, sind für die Tests sowohl männliche als auch weibliche Tiere erforderlich.
Verabreichungszeitraum
26. Gestützt auf die Beobachtung, dass sich Mutationen mit jeder Behandlung steigern, ist ein Schema für wiederholte Verabreichungen erforderlich, wobei über einen Zeitraum von 28 Tagen täglich behandelt wird. Dies wird allgemein sowohl für das Hervorrufen einer ausreichenden Akkumulation von Mutationen durch schwache Mutagene als auch für die Bereitstellung einer angemessenen Expositionsdauer für den Nachweis von Mutationen in langsam proliferierenden Organen als akzeptabel angesehen. Bei einigen Bewertungen mögen alternative Behandlungsschemata angebracht sein. Diese alternativen Dosierungspläne sind im Protokoll wissenschaftlich zu begründen. Die Behandlungen sollten nicht kürzer sein als die Zeit, die für die vollständige Induktion aller relevanten metabolisierender Enzyme erforderlich ist, und kürzere Behandlungen erfordern möglicherweise mehrere Probenahmen, die sich für Organe mit unterschiedlicher Proliferation eignen. In jedem Fall sind zur Begründung eines Protokolls alle vorliegenden Informationen (z. B. zur allgemeinen Toxizität bzw. zum Stoffwechsel und zur Pharmakokinetik) zu verwenden, vor allem, wenn von den vorgenannten Standardempfehlungen abgewichen wird. Länger als acht Wochen andauernde Behandlungszeiten können zwar die Empfindlichkeit erhöhen, müssen aber klar erläutert und begründet werden, weil lange Behandlungszeiten durch klonale Expansion zu einem scheinbaren Anstieg der Mutantenhäufigkeit führen können (29).
Dosisstufen
27. Dosisstufen sollten sich auf die Ergebnisse einer Dosisfindungsstudie stützen, bei der die allgemeine Toxizität gemessen und die über den gleichen Expositionsweg durchgeführt wurde oder aber auf bereits vorliegende Ergebnisse aus Untersuchungen der subakuten Toxizität. Zur Bestimmung der Dosisbereiche können nicht transgene Tiere desselben Nagetierstamms verwendet werden. Der Haupttest sollte eine vollständige Untersuchung einer Negativkontrollgruppe beinhalten, um Informationen über Dosisreaktionen zu gewinnen (siehe Nummer 22) und mindestens drei angemessen abgestufte Dosierungen umfassen, außer bei Verwendung der Grenzdosis (siehe Nummer 28). Die höchste Dosis sollte die maximal verträgliche Dosis (maximum tolerated dose, MTD) sein. Die MTD wird als die Dosis festgelegt, die Toxizitätsanzeichen in einem Ausmaß hervorruft, das darauf schließen lässt, dass nach demselben Dosierungsplan verabreichte höhere Dosen voraussichtlich zum Tod des Tieres führen würden. Chemikalien mit spezifischen biologischen Aktivitäten bei niedrigen, nicht toxischen Dosen (wie Hormone und Mitogene) und Chemikalien mit gesättigten toxikokinetischen Eigenschaften können als Ausnahmen von den Kriterien zur Dosisfestsetzung angesehen werden und sind auf Fallbasis zu bewerten. Die verwendeten Dosisstufen sollten von der Dosis mit der höchsten Toxizität bis zur Dosis mit der geringsten oder überhaupt keiner Toxizität reichen.
Limit-Test
28. Falls Dosisfindungsversuche oder vorliegende Daten aus verwandten Nagetierstämmen darauf hinweisen, dass ein Behandlungsplan, der zumindest die Grenzdosis vorsieht (siehe unten), keine messbaren toxischen Wirkungen hervorruft,und wenn aufgrund von Daten über strukturverwandte Chemikalien keine Genotoxizität erwartet wird, kann unter Umständen auf eine vollständige Untersuchung mit drei Dosisstufen verzichtet werden. Für einen Verabreichungszeitraum von 28 Tagen (d. h. 28 tägliche Behandlungen) beträgt die Grenzdosis 1 000 mg/kg Körpergewicht/Tag. Für Verabreichungszeiträume von 14 Tagen oder weniger beträgt die Grenzdosis 2 000 mg/kg/Körpergewicht/Tag (von 28 täglichen Behandlungen abweichende Dosierungspläne sind im Protokoll wissenschaftlich zu begründen; siehe Nummer 26).
Verabreichung der Dosen
29. In der Regel wird die Prüfsubstanz über eine Schlundsonde oder eine geeignete Intubationskanüle verabreicht. Bei der Planung eines Assays ist grundsätzlich die Art der Humanexposition zu berücksichtigen. Folglich sind unter Umständen auch andere Expositionswege (subkutan, intravenös, topisch, inhalativ oder intratracheal oder über das Trinkwasser/Futter oder Implantation) akzeptabel, soweit sie begründet werden können. Eine Intraperitonealinjektion wird nicht empfohlen, weil sie beim Menschen keinen physiologisch relevanten Expositionsweg darstellt. Das maximale Flüssigkeitsvolumen, das einem Versuchstier über eine Schlundsonde oder über eine Injektion jeweils verabreicht werden kann, hängt von der Größe des Versuchstiers ab. Dieses Volumen sollte 2 ml/100 g Körpergewicht nicht überschreiten. Die Verwendung höherer Volumina muss gerechtfertigt werden. Abgesehen von reizenden oder ätzenden Stoffen, die in der Regel bei höheren Konzentrationen eine verstärkte Wirkung hervorrufen, sollte die Variabilität des Prüfvolumens auf ein Mindestmaß begrenzt werden, indem die Konzentration so angepasst wird, dass auf allen Dosisstufen ein konstantes Volumen gewährleistet ist.
Probenahmezeitpunkt
Somatische Zellen
30. Der Probenahmezeitpunkt stellt einen kritischen Parameter dar, weil er von dem für die Fixierung der Mutationen erforderlichen Zeitraum abhängt. Dieser Zeitraum ist gewebespezifisch und hängt offensichtlich mit der Turnover-Zeit der Zellpopulation zusammen, wobei Knochenmark und Darm schnell und die Leber erheblich langsamer reagieren. Ein angemessener Kompromiss für die Messung der Mutantenhäufigkeiten sowohl bei schnell als auch bei langsam proliferierenden Geweben wären tägliche Behandlungen an 28 aufeinanderfolgenden Tagen (wie unter Nummer 26 angegeben) mit Probenahme drei Tage nach der letzten Behandlung; dabei kann es vorkommen, dass die maximale Mutantenhäufigkeit in langsam proliferierendem Gewebe unter diesen Bedingungen nicht sichtbar wird. Wenn langsam proliferierende Gewebe von ausschlaggebender Bedeutung sind, ist eine spätere Probenahme 28 Tage nach dem 28-tägigen Verabreichungszeitraum möglicherweise angemessener (16) (29). In solchen Fällen würde die Probenahme drei Tage nach der letzten Behandlung durch die spätere Probenahme ersetzt und bedürfte einer wissenschaftlichen Begründung.
Keimzellen
31. TGR-Assays sind für die Untersuchung von Genmutationen in männlichen Keimzellen (7) (8) (27), für die der Zeitpunkt und die Kinetik der Spermatogenese genau bestimmt wurden (27), gut geeignet. Die geringe Zahl der — selbst nach einer Superovulation — für die Analyse verfügbaren Eizellen und mangelnde DNA-Synthese in der Eizelle schließen die Bestimmung von Mutationen in weiblichen Keimzellen in Studien mit transgenen Tieren von vorne herein aus (31).
32. Der Probenahmezeitpunkt ist bei männlichen Keimzellen so auszuwählen, dass Proben aus dem während der gesamten Keimzellenentwicklung exponierten Zelltypbereich entnommen werden und die Phase, auf die die Probenahme abzielt, ausreichend exponiert wurde. Die Entwicklungszeit der Keimzellen von spermatogonialen Stammzellen zu reifen Spermien, die den Samenleiter/Nebenhodenschwanz erreichen,beträgt ~ 49 Tage bei der Maus (36) und ~ 70 Tage bei der Ratte (34) (35). Nach einer 28-tägigen Exposition und einer anschließenden dreitägigen Probenahme repräsentieren die aus dem Samenleiter/Nebenhodenschwanz (7) (8) entnommenen Spermien eine Population von Zellen, die ungefähr in der zweiten Hälfte der Spermatogenese exponiert wurden; in diesen Zeitraum fallen auch die meiotische und die postmeiotische Phase, nicht aber die spermatogoniale oder Stammzellenphase. Um adäquate Proben von Zellen aus dem Samenleiter/Nebenhodenschwanz entnehmen zu können, die während des Expositionszeitraums spermatogoniale Stammzellen waren, ist frühestens sieben Wochen (Mäuse) oder zehn Wochen (Ratten) nach Ende der Behandlung eine zusätzliche Probenahme erforderlich.
33. Zellen, die nach dem Schema ‚28 + 3‘-Tage aus den Hodenkanälchen herausgepresst wurden, bilden eine für alle Entwicklungsphasen der Keimzellen angereicherte Mischpopulation (7) (8). Die Phasen, in denen Keimzellmutationen induziert werden, lassen sich durch Beprobung derartiger Zellen zum Nachweis von Genmutationen weniger präzise bewerten als durch die Beprobung von Spermatozoen aus dem Samenleiter/Nebenhodenschwanz (weil die Proben aus den Kanälchen aus einer ganzen Reihe von Keimzelltypen bestehen und diese Zellpopulation in einem gewissen Maß mit somatischen Zellen kontaminiert ist). Bei der Beprobung von Zellen aus Hodenkanälchen UND von Spermatozoen aus dem Samenleiter/Nebenhodenschwanz nach nur ‚28 + 3‘-Probenahmetagen würden in gewissem Umfang jedoch auch Zellen erfasst, die in fast allen Keimzellentwicklungsphasen exponiert wurden, was für den Nachweis bestimmter Keimzellmutagene von Nutzen sein könnte.
Beobachtungen
34. Allgemeine klinische Beobachtungen sollten mindestens einmal täglich erfolgen, vorzugsweise jeweils zur gleichen Zeit und unter Berücksichtigung des Zeitraums nach der Verabreichung, in dem die Wirkungsspitzen erwartet werden. Der Gesundheitszustand der Tiere ist zu dokumentieren. Mindestens zweimal täglich sind alle Tiere auf Morbiditäts- und Mortalitätsanzeichen zu überprüfen. Alle Tiere sind mindestens einmal wöchentlich sowie zum Zeitpunkt ihrer Tötung zu wiegen. Die Futteraufnahme wird mindestens wöchentlich gemessen. Wenn die Prüfsubstanz über das Trinkwasser verabreicht wird, wird die Wasseraufnahme bei jedem Wasserwechsel und mindestens einmal wöchentlich gemessen. Tiere, die nichtletale Anzeichen einer übermäßigen Toxizität aufweisen, sind vor Testende zu euthanasieren (23).
Gewebeentnahme
35. Die Argumente für eine Gewebeentnahme sollten fundiert sein. Da Mutationsinduktionen in praktisch jedem beliebigen Gewebe untersucht werden können, sollten die zu entnehmenden Gewebe nach dem Versuchsmotiv und unter Berücksichtigung aller vorliegenden Daten zur Mutagenität, Karzinogenität oder Toxizität der Prüfsubstanz ausgewählt werden. Zu den wichtigen zu berücksichtigenden Faktoren sollten auch der Verabreichungsweg (gestützt auf den (die) wahrscheinlichen Expositionsweg(e) beim Menschen), die voraussichtliche Gewebeverteilung und der mögliche Wirkungsmechanismus gehören. Liegen keinerlei Hintergrundinformationen vor, sind einige somatische Gewebe zu entnehmen, die für die Untersuchung von Interesse sind. Bei diesem Gewebe sollte es sich um schnell und langsam proliferierendes Gewebe und um Kontaktstellengewebe handeln. Darüber hinaus sind (nach der Beschreibung unter den Nummern 32 und 33) Spermatozoen aus dem Samenleiter/Nebenhodenschwanz und in der Entwicklung befindliche Keimzellen aus den Hodenkanälchen zu entnehmen und für den Fall, dass eine künftige Analyse der Keimzellenmutagenität erforderlich wird, aufzubewahren. Die Organe sind zu wiegen, und bei größeren Organen sollten von allen Tieren Gewebe aus demselben Organbereich entnommen werden.
Lagerung von Gewebe und DNA
36. Gewebe (oder Gewebehomogenate) sind bei oder unter einer Temperatur von – 70 °C zu lagern und für die DNA-Isolierung innerhalb von fünf Jahren zu verwenden. Bei einer Temperatur von 4 °C in einem geeigneten Puffer kühl gelagerte isolierte DNA sollte idealerweise innerhalb eines Jahres für Mutationsanalysen verwendet werden.
Gewebeauswahl für Mutantenanalysen
37. Die Gewebe sind unter anderem nach folgenden Krierien auszuwählen: 1) Verabreichungsweg oder Stelle des Erstkontakts (z. B. Drüsenmagen, wenn die Verabreichung oral erfolgt; Lunge, wenn die Verabreichung inhalativ erfolgt, oder Haut bei einer topischen Anwendung), und 2) in allgemeinen Toxizitätsstudien gemessene pharmakokinetische Parameter, die die Gewebedisposition, -retention oder -akkumulation oder Zielorgane für die Toxizität anzeigen. Wenn Versuche aufgrund von Ergebnissen aus Karzinogenitätsuntersuchungen durchgeführt werden, sind Zielgewebe für die Karzinogenität in Erwägung zu ziehen. Die für die Analyse ausgewählten Gewebe sollten den Nachweis von Chemikalien maximieren, die in vitro direkt mutagen wirken, schnell verstoffwechselt werden, hochreaktiv sind oder schlecht absorbiert werden, oder von Chemikalien, bei denen das Zielgewebe über den Verabreichungsweg bestimmt wird (6).
38. In Ermangelung von Hintergrundinformationen und unter Berücksichtigung der durch den Verabreichungsweg bedingten Kontaktstelle sind die Leber und mindestens ein sich schnell teilendes Gewebe (z. B. Drüsenmagen, Knochenmark) auf Mutagenität zu untersuchen. In den meisten Fällen kann den oben angeführten Anforderungen durch die Analyse zweier sorgfältig ausgewählter Gewebe genüge getan werden. In einigen Fällen sind möglicherweise drei oder mehr Gewebe nötig.Wenn es spezifische Besorgnis hinsichtlich Keimzellmutationen gibt, auch aufgrund positiver Befunde in Somazellen, sollten Keimzellgewebe auf Mutationen untersucht werden.
Messmethoden
39. Für die empfohlenen transgenen Modelle zum Nachweis von Mutanten stehen folgende Standardlabormethoden bzw. veröffentlichte Methoden zur Verfügung: lacZ-Bakteriophage lambda und Plasmid (30); lacI Maus (2) (18); gpt Delta Maus (22); gpt Delta Ratte (28); cII (17). Änderungen sind zu begründen und ordnungsgemäß zu dokumentieren. Daten aus multiplen Verpackungen können zusammengefasst und zum Erzielen einer angemessenen Anzahl Plaques oder Kolonien verwendet werden. Wird eine große Anzahl Verpackungsreaktionen benötigt, um die geeignete Anzahl Plaques zu erzielen, kann dies jedoch ein Hinweis auf schlechte DNA-Qualität sein. In solchen Fällen sind die Daten mit Vorsicht zu behandeln, weil sie unzuverlässig sein könnten. Die optimale Gesamtzahl an Plaques oder Kolonien pro DNA-Probe richtet sich nach der statistischen Wahrscheinlichkeit des Nachweises einer ausreichenden Anzahl Mutanten bei einer gegebenen Häufigkeit spontaner Mutanten. In der Regel sind mindestens 125 000 bis 300 000 Plaques erforderlich, wenn die Häufigkeit spontaner Mutanten bei 3 × 10–5 (15) liegt. Beim Big Blue® lacI Assay muss unbedingt gewährleistet sein, dass durch Einbeziehung geeigneter Farbkontrollen parallel zu allen Ausstrichen der gesamte Bereich der Mutanten-Farbphänotypen nachgewiesen werden kann. Gewebe und die resultierenden Proben (Items) sind nach einem Blockschema zu bearbeiten und zu analysieren, bei dem Items aus der Vehikel-/Lösungsmittel-Kontrollgruppe, der Positivkontrollgruppe (falls verwendet) oder der DNA-Positivkontrolle (falls zutreffend) und jede Behandlungsgruppe zusammen verarbeitet werden.
DATEN UND BERICHTERSTATTUNG
Auswertung der Ergebnisse
40. Die Daten zu den einzelnen Tieren sind in tabellarischer Form vorzulegen. Die Versuchseinheit ist das Tier. Der Bericht sollte folgende Angaben enthalten: Gesamtzahl der plaquebildenden Einheiten (Plaque-Forming Units, PFU) bzw. der koloniebildenden Einheiten (Colony-Forming Units, CFU), Anzahl Mutanten und Mutantenhäufigkeit für jedes Gewebe und Tier. Bei multiplen Verpackungs-/Rettungsreaktionen ist die Zahl der Reaktionen pro DNA-Probe anzugeben. Auch wenn es sich empfiehlt, die Daten über jede einzelne Reaktion aufzubewahren, müssen lediglich die Gesamt-PFU bzw. -CFU schriftlich festgehalten werden. Daten zur Toxizität und klinische Anzeichen gemäß Nummer 34 sind zu dokumentieren. Sequenzierungsergebnisse sind für jeden einzelnen analysierten Mutanten vorzulegen, wobei die entsprechend durchgeführten Mutationshäufigkeits-berechnungen für jedes Tier und jedes Gewebe anzugeben sind.
Statistische Auswertung und Interpretation der Ergebnisse
41. Es gibt mehrere Kriterien für die Entscheidung über ein positives Ergebnis, wie eine dosisabhängigee Zunahme der Mutantenhäufigkeit oder eine deutliche Zunahme der Mutantenhäufigkeit in einer einzigen Dosisgruppe im Vergleich zur Lösungsmittel-/Vehikelkontrollgruppe. Es sind mindestens drei behandelte Dosisgruppen zu analysieren, um ausreichende Daten für Dosis-Wirkungs-Analysen zu liefern. Das Hauptaugenmerk sollte auf der biologischen Relevanz der Ergebnisse liegen, wobei geeignete statistische Methoden als Hilfsmittel für die Auswertung der Testergebnisse verwendet werden können (4) (14) (15) (25) (26). Die verwendeten statistischen Tests müssen das Tier als Versuchseinheit berücksichtigen.
42. Eine Prüfsubstanz, bei der die Ergebnisse die oben angeführten Kriterien in keinem Gewebe erfüllen, gilt im vorliegenden Test als nicht mutagen. Für die biologische Relevanz eines negativen Ergebnisses ist die Gewebeexposition zu bestätigen.
43. Für DNA-Sequenzierungsanalysen stehen eine Reihe statistischer Konzepte zur Erleichterung der Auswertung der Ergebnisse zur Verfügung (1) (5) (9) (19).
44. Indem ermittelt wird, ob die gemessenen Werte innerhalb oder außerhalb des historischen Kontrollbereichs liegen, lassen sich Leitlinien für die Bewertung der biologischen Signifikanz der Reaktion herleiten (32).
Prüfbericht
45. Der Prüfbericht sollte folgende Angaben enthalten:
Prüfsubstanz:
|
— |
Angaben zur Identifikation und CAS-Nummer, falls bekannt, |
|
— |
Herkunft, Chargennummer, falls verfügbar, |
|
— |
physikalische Beschaffenheit und Reinheit, |
|
— |
physikalisch-chemische Eigenschaften, die für die Durchführung des Versuchs von Belang sind, |
|
— |
Stabilität der Prüfsubstanz, falls bekannt. |
Lösungsmittel/Vehikel:
|
— |
Begründung der Wahl des Vehikels, |
|
— |
Löslichkeit und Stabilität der Prüfsubstanz im Lösungsmittel/Vehikel, falls bekannt, |
|
— |
Zubereitung des Futters, des Trinkwassers oder der Inhalationsrezepturen, |
|
— |
analytische Bestimmungen zu Rezepturen (z. B. Stabilität, Homogenität, Nennkonzentrationen). |
Versuchstiere:
|
— |
Art/Stamm und Begründung der getroffenen Wahl, |
|
— |
Anzahl, Alter und Geschlecht der Tiere, |
|
— |
Herkunft, Unterbringungsbedingungen, Futter usw., |
|
— |
Gewicht der einzelnen Tiere bei Versuchsbeginn, einschließlich Körpergewichtsspanne, Mittelwert und Standardabweichung für jede Gruppe. |
Prüfbedingungen:
|
— |
Daten zu den Positiv- und Negativkontrollen (Vehikel/Lösungsmittel), |
|
— |
Daten aus der Dosisfindungsstudie, |
|
— |
Begründung der gewählten Dosisstufen, |
|
— |
Angaben zur Prüfsubstanzzubereitung, |
|
— |
Einzelheiten der Verabreichung der Prüfsubstanz, |
|
— |
Begründung des Verabreichungswegs, |
|
— |
Methoden zur Toxizitätsmessung beim Tier, einschließlich, soweit verfügbar, histopathologischer oder hämatologischer Analysen, sowie Angaben zur Häufigkeit, mit der die Tiere beobachtet und ihre Körpergewichte gemessen wurden, |
|
— |
Methoden zum Nachweis, dass die Prüfsubstanz das Zielgewebe oder den allgemeinen Kreislauf erreicht hat, falls negative Ergebnisse erzielt werden, |
|
— |
(gegebenenfalls) tatsächliche Dosis (in mg/kg je Körpergewicht und Tag), berechnet auf Basis der Konzentration (ppm) der Prüfsubstanz im Futter/Wasser und ihrer Aufnahme, |
|
— |
Angaben zur Futter- und Wasserqualität, |
|
— |
detaillierte Beschreibung der Behandlungs- und Probenahmepläne und Begründung der jeweils getroffenen Wahl, |
|
— |
Tötungsmethode, |
|
— |
Verfahren zur Isolation und Aufbewahrung von Gewebe, |
|
— |
Methoden zur Isolierung der genomischen DNA von Nagetieren, Rettung des Transgens aus der genomischen DNA und Übertragung der transgenen DNA auf eine bakterielle Wirtszelle, |
|
— |
Herkunft und Chargennummern aller Zellen, Laborkits und Reagenzien (soweit zutreffend), |
|
— |
Methoden zur Auszählung von Mutanten, |
|
— |
Methoden für die Molekularanalyse von Mutanten und deren Verwendung bei der Berichtigung um die Klonalität und/oder bei der Berechnung von Mutationshäufigkeiten, falls zutreffend. |
Ergebnisse:
|
— |
Gesundheitszustand des Tieres vor und während des Versuchszeitraums, einschließlich Toxizitätsanzeichen, |
|
— |
Körper- und Organgewicht bei der Tötung, |
|
— |
für jedes Gewebe/Tier: Anzahl Mutanten, Anzahl bewerteter Plaques bzw. Kolonien und Mutantenhäufigkeit, |
|
— |
für jede Gewebe-/Tiergruppe: Anzahl Verpackungsreaktionen pro DNA-Probe, Gesamtzahl der Mutanten, mittlere Mutantenhäufigkeit, Standardabweichung, |
|
— |
nach Möglichkeit Dosis-Wirkungs-Beziehung, |
|
— |
für jedes Gewebe/Tier: Anzahl unabhängiger Mutanten und mittlere Mutationshäufigkeit, soweit eine Molekularanalyse der Mutationen durchgeführt wurde, |
|
— |
Daten über Negativkontrolltiere, die gleichzeitig in den Versuch einbezogen waren, und historische Kontrolldaten aus Negativkontrollen mit Angaben zu Bereichen, Mittelwerten und Standardabweichungen, |
|
— |
Daten über Positivkontrolltiere, die gleichzeitig in den Versuch einbezogen waren (oder Daten zur DNA von Positivkontrolltieren aus früheren Versuchen), |
|
— |
analytische Bestimmungen, falls verfügbar (z. B. für die Verpackung verwendete DNA-Konzentrationen, DNA-Sequenzierungsdaten), |
|
— |
verwendete statistische Analysen und Methoden. |
Diskussion der Ergebnisse
Schlussfolgerung
LITERATUR
|
(1) |
Adams, W.T. and T.R. Skopek (1987). ‚Statistical Test for the Comparison of Samples from Mutational Spectra‘. J. Mol. Biol., 194: 391-396. |
|
(2) |
Bielas, J.H. (2002). ‚A more Efficient Big Blue® Protocol Improves Transgene Rescue and Accuracy in an Adduct and Mutation Measurement‘. Mutation Res., 518: 107-112. |
|
(3) |
Boerrigter, M.E., M.E. Dollé, H.-J. Martus, J.A. Gossen and J. Vijg (1995). ‚Plasmid-based Transgenic Mouse Model for Studying in vivo Mutations‘Nature, 377(6550): 657-659 |
|
(4) |
Carr, G.J. and N.J. Gorelick (1995). ‚Statistical Design and Analysis of Mutation Studies in Transgenic Mice‘, Environ. Mol. Mutagen, 25(3): 246-255. |
|
(5) |
Carr, G.J. and N.J. Gorelick (1996). ‚Mutational Spectra in Transgenic Animal Research: Data Analysis and Study Design Based upon the Mutant or Mutation Frequency‘, Environ. Mol. Mutagen, 28: 405-413. |
|
(6) |
Dean, S.W., T.M. Brooks, B. Burlinson, J. Mirsalis, B. Myhr, L. Recio and V. Thybaud (1999). ‚Transgenic Mouse Mutation Assay Systems can Play an important Role in Regulatory Mutagenicity Testing in vivo for the Detection of Site-of-contact Mutagens‘, Mutagenesis, 14(1): 141-151. |
|
(7) |
Douglas, G.R., J. Jiao, J.D. Gingerich, J.A. Gossen and L.M. Soper(1995). ‚Temporal and Molecular Characteristics of Mutations Induced by Ethylnitrosourea in Germ Cells Isolated from Seminiferous Tubules and in Spermatozoa of lacZ Transgenic Mice‘, Proc. Natl. Acad. Sci. USA, 92: 7485-7489. |
|
(8) |
Douglas, G.R., J.D. Gingerich, L.M. Soper and J. Jiao (1997). ‚Toward an Understanding of the Use of Transgenic Mice for the Detection of Gene Mutations in Germ Cells‘, Mutation Res., 388(2-3): 197-212. |
|
(9) |
Dunson, D.B. and K.R. Tindall (2000). ‚Bayesian Analysis of Mutational Spectra‘, Genetics, 156: 1411-1418. |
|
(10) |
Gossen, J.A., W.J. de Leeuw, C.H. Tan, E.C. Zwarthoff, F. Berends, P.H. Lohman, D.L. Knook and J. Vijg(1989). ‚Efficient Rescue of Integrated Shuttle Vectors from Transgenic Mice: a Model for Studying Mutations in vivo‘, Proc. Natl. Acad. Sci. USA, 86(20): 7971-7975. |
|
(11) |
Gossen, J.A. and J. Vijg (1993), ‚A Selective System for lacZ-Phage using a Galactose-sensitive E. coli Host‘, Biotechniques, 14(3): 326, 330. |
|
(12) |
Erikson, R.P. (2003). ‚Somatic Gene Mutation and Human Disease other than Cancer‘, Mutation Res., 543: 125-136. |
|
(13) |
Erikson, R.P. (2010). ‚Somatic Gene Mutation and Human Disease other than Cancer: an Update‘, Mutation Res., 705: 96-106. |
|
(14) |
Fung, K.Y., G.R. Douglas and D. Krewski (1998). ‚Statistical Analysis of lacZ Mutant Frequency Data from Muta™Mouse Mutagenicity Assays‘, Mutagenesis, 13(3): 249-255. |
|
(15) |
Heddle, J.A., S. Dean, T. Nohmi, M. Boerrigter, D. Casciano, G.R. Douglas, B.W. Glickman, N.J. Gorelick, J.C. Mirsalis, H.-J Martus, T.R. Skopek, V. Thybaud, K.R.Tindall and N. Yajima (2000). ‚In vivo Transgenic Mutation Assays‘, Environ. Mol. Mutagen., 35: 253-259. |
|
(16) |
Heddle, J.A., H.-J. Martus and G.R. Douglas (2003). ‚Treatment and Sampling Protocols for Transgenic Mutation Assays‘, Environ. Mol. Mutagen., 41: 1-6. |
|
(17) |
Jakubczak, J.L., G. Merlino, J.E. French, W.J. Muller, B. Paul, S. Adhya and S. Garges (1996). ‚Analysis of Genetic Instability during Mammary Tumor Progression using a novel Selection-based Assay for in vivo Mutations in a Bacteriophage λ Transgene Target‘, Proc. Natl. Acad. Sci. USA, 93(17): 9073-9078. |
|
(18) |
Kohler, S.W., G.S. Provost, P.L. Kretz, A. Fieck, J.A. Sorge and J.M. Short (1990). ‚The Use of Transgenic Mice for Short-term, in vivo Mutagenicity Testing‘, Genet. Anal. Tech. Appl., 7(8): 212–218. |
|
(19) |
Lewis P.D., B. Manshian, M.N. Routledge, G.B. Scott and P.A. Burns (2008). ‚Comparison of Induced and Cancer-associated Mutational Spectra using Multivariate Data Analysis‘, Carcinogenesis, 29(4): 772-778. |
|
(20) |
Nohmi, T., M. Katoh, H. Suzuki, M. Matsui, M. Yamada, M. Watanabe, M. Suzuki, N. Horiya, O. Ueda, T. Shibuya, H. Ikeda and T. Sofuni (1996). ‚A new Transgenic Mouse Mutagenesis Test System using Spi — and 6-thioguanine Selections‘, Environ. Mol. Mutagen., 28(4): 465-470. |
|
(21) |
Nohmi, T., M. Suzuki, K. Masumura, M. Yamada, K. Matsui, O. Ueda, H. Suzuki, M. Katoh, H. Ikeda and T. Sofuni (1999). ‚Spi– Selection: an Efficient Method to Detect γ-ray-induced Deletions in Transgenic Mice‘, Environ. Mol. Mutagen., 34(1): 9-15. |
|
(22) |
Nohmi, T., T. Suzuki and K.I. Masumura (2000). ‚Recent Advances in the Protocols of Transgenic Mouse Mutation Assays‘, Mutation Res., 455(1-2): 191-215. |
|
(23) |
OECD (2000). Guidance Document on the recognition, assessment, and use of clinical signs as humane endpoints for experimental animals used in safety evaluations, Series on Testing and Assessment, Nr. 19, ENV/JM/MONO(2000)7, OECD, Paris. |
|
(24) |
OECD (2009). Detailed Review Paper on Transgenic Rodent Mutation Assays, Series on Testing and Assessment, No 103, ENV/JM/MONO(2009)7, OECD, Paris. |
|
(25) |
Piegorsch, W.W., B.H. Margolin, M.D. Shelby, A. Johnson, J.E. French, R.W. Tennant and K.R. Tindall (1995). ‚Study Design and Sample Sizes for a lacI Transgenic Mouse Mutation Assay‘, Environ. Mol. Mutagen., 25(3): 231-245. |
|
(26) |
Piegorsch, W.W., A.C. Lockhart, G.J. Carr, B.H. Margolin, T. Brooks, … G.R. Douglas, U.M. Liegibel, T. Suzuki, V. Thybaud, J.H. van Delft and N.J. Gorelick (1997). ‚Sources of Variability in Data from a Positive Selection lacZ Transgenic Mouse Mutation Assay: an Interlaboratory Study‘, Mutation. Res., 388(2-3): 249-289. |
|
(27) |
Singer, T.M., I.B. Lambert, A. Williams, G.R. Douglas and C.L. Yauk(2006). ‚Detection of Induced Male Germline Mutation: Correlations and Comparisons between Traditional Germline Mutation Assays, Transgenic Rodent Assays and Expanded Simple Tandem Repeat Instability Assays‘, Mutation. Res., 598: 164-193. |
|
(28) |
Toyoda-Hokaiwado, N., T. Inoue, K. Masumura, H. Hayashi, Y. Kawamura, Y. Kurata, M. Takamune, M. Yamada, H. Sanada, T. Umemura, A. Nishikawa and T. Nohmi (2010). ‚Integration of in vivo Genotoxicity and Short-term Carcinogenicity Assays using F344 gpt delta Transgenic Rates: in vivo Mutagenicity of 2,4-diaminotoluene and 2,6-diaminotoluene Structural Isomers‘, Toxicol. Sci., 114(1): 71-78. |
|
(29) |
Thybaud, V., S. Dean, T. Nohmi, J. de Boer, G.R. Douglas, B.W. Glickman, N.J. Gorelick, J.A. Heddle, R.H. Heflich, I. Lambert, H.-J. Martus, J.C. Mirsalis, T. Suzuki and N. Yajima (2003). ‚In vivo Transgenic Mutation Assays‘, Mutation Res., 540: 141-151. |
|
(30) |
Vijg, J. and G.R. Douglas (1996), ‚Bacteriophage λ and Plasmid lacZ Transgenic Mice for studying Mutations in vivo‘ in: G. Pfeifer (ed.), Technologies for Detection of DNA Damage and Mutations, Part II, Plenum Press, New York, NY, USA, pp. 391-410. |
|
(31) |
Yauk, C.L., J.D. Gingerich, L. Soper, A. MacMahon, W.G. Foster and G.R. Douglas (2005). ‚A lacZ Transgenic Mouse Assay for the Detection of Mutations in Follicular Granulosa Cells‘, Mutation Res., 578(1-2): 117-123. |
|
(32) |
Hayashi, M., K. Dearfield, P. Kasper, D. Lovell, H.-J. Martus, V. Thybaud (2011). ‚Compilation and Use of Genetic Toxicity Historical Control Data‘, Mutation Res., doi:10.1016/j.mrgentox.2010.09.007. |
|
(33) |
OECD (2011). Retrospective Performance Assessment of OECD Test Guideline on Transgenic Rodent Somatic and Germ Cell Gene Mutation Assays, Series on Testing and Assessment, No 145, ENV/JM/MONO(2011)20, OECD, Paris. |
|
(34) |
Clermont, Y. (1972). ‚Kinetics of spermatogenesis in mammals seminiferous epithelium cycle and spermatogonial renewal‘. Physiol. Rev. 52: 198-236. |
|
(35) |
Robaire, B., Hinton, B.T., and Oregbin-Crist, M.-C. (2006). ‚The Epididymis‘, in Neil, J.D., Pfaff, D.W., Chalis, J.R.G., de Kretser, D.M., Richards, J.S., and P. M, Wassarman (eds.), Physiology of Reproduction, Elsevier, the Netherlands, pp. 1071-1148. |
|
(36) |
Russell, L.B. (2004). ‚Effects of male germ-cell stage on the frequency, nature, and spectrum of induced specific-locus mutations in the mouse‘, Genetica, 122: 25–36. |
Anlage
BEGRIFFSBESTIMMUNGEN
Basenpaarsubstitution : eine Art Mutation, die dazu führt, dass eine einzelne DNA-Nukleotidbase gegen eine andere DNA-Nukleotidbase ausgetauscht wird.
Chemikalie : ein Stoff oder eine Mischung.
Cos-Stelle : ein 12-Nukleotidsegment einer Einzelstrang-DNA, das an beiden Enden des Doppelstrang-Genoms des Bakteriophagen vorkommt.
Deletion : eine Mutation, bei der ein oder mehrere (sequenzielle) Nukleotiden vom Genom verloren werden.
Elektroporation : Anwendung elektrischer Impulse zur Erhöhung der Durchlässigkeit von Zellmembranen.
Endogenes Gen : ein Gen, das aus dem Genom selbst stammt.
Extra-binomiale Variation : größere Variabilität bei Wiederholungsschätzungen eines Populationsanteils als bei einer Binomialverteilung der Population erwartet würde.
Große Deletionen : Deletionen mehrerer Kilobasen in der DNA (die durch Spi – Auswahl und die lacZ-Plasmidassays gezielt nachgewiesen werden).
Insertion : Hinzufügen eines oder mehrerer Nukleotidenbasenpaare in eine DNA-Sequenz.
Jackpot : große Anzahl Mutanten, durch klonale Expansion aus einer einzigen Mutation entstanden.
Kapsid : die ein Viruspartikel umschließende Proteinhülle.
Klonale Expansion : die Erzeugung vieler Zellen aus einer einzigen (Mutanten-)Zelle.
Koloniebildende Einheit (Colony-Forming Unit, CFU) : ein Größe zur Quantifizierung lebensfähiger Bakterien.
Konkatamer : eine Wiederholungssequenz einer DNA-Kette (Biomolekül).
Ligation : die kovalente Anbindung von zwei DNA-Molekülenden durch DNA-Ligase.
Mitogen : eine Chemikalie, die eine Zelle zur Zellteilung anregt und Mitose auslöst (d. h. Zellteilung).
Neutrales Gen : ein Gen, das weder von positivem, noch von negativem Selektionsdruck beeinflusst wird.
Plaquebildende Einheit (Plaque Forming Unit, PFU) : eine Größe zur Quantifizierung lebensfähiger Bakteriophagen.
Positivselektion : eine Methode, bei der nur Mutanten überleben.
Probenahmezeitpunkt : das Ende des Zeitraums vor der Tötung des Tieres, in dem die Chemikalie nicht verabreicht wird und in dem nicht reparierte DNA-Läsionen sich in Form stabiler Mutationen fixieren.
Prüfsubstanz : jede(r) nach dieser Prüfmethode getesteter Stoff bzw. Mischung.
Punktmutation : allgemeiner Begriff für eine Mutation, die nur eine kleine Sequenz der DNA betrifft, einschließlich kleiner Insertionen, Deletionen und Basenpaarsubstitutionen.
Rasterschubmutation : eine genetische Mutation innerhalb einer ein Protein/Peptid codierenden DNA-Sequenz, verursacht durch Insertionen oder Deletionen einer Reihe von Nukleotiden, die nicht gleichmäßig durch drei geteilt werden können.
Reporter-Gen : ein Gen, dessen Produkt (mutantes Gen) einfach nachzuweisen ist.
Shuttle-Vektor : ein Vektor, der so strukturiert ist, dass er sich in zwei verschiedenen Wirtsarten vermehren kann; entsprechend kann in einen Shuttle-Vektor eingebrachte DNA in zwei unterschiedlichen Zelltypen oder zwei unterschiedlichen Organismen geprüft oder manipuliert werden.
Transgen : bezogen auf einen Organismus oder der Organismus selbst, dessen Genom durch die Übertragung eines oder mehrerer Gene einer anderen Spezies verändert worden ist.
Verabreichungszeitraum : die Gesamtdauer, während der einem Tier Prüfsubstanz verabreicht wird.
Verpackung : die Synthese infektiöser Phagenpartikel aus der Zubereitung von Phagenkapsid und Schwanzproteinen und einem Konkatamer von Phagen-DNA-Molekülen. Wird gemeinhin zur Verpackung von auf einen Lambda-Vektor geklonter DNA (die durch Cos-Stellen getrennt ist) in infektiöse Lambdapartikel verwendet.
Verpackungseffizienz : die Effizienz, mit der verpackte Bakteriophagen in Wirtsbakterien wiedergefunden werden.
(1) Je nach Empfindlichkeit der kognitiven Funktionsprüfungen sollte die Untersuchung einer wesentlich größeren Anzahl an Tieren in Erwägung gezogen werden, z. B. bis zu ein Männchen und ein Weibchen pro Wurf (bezüglich Tierzuteilung siehe Anlage 1) (für weiterführende Hinweise zur Stichprobengröße siehe OECD Guidance Document Nr. 43 (8)).
(2) Dieser Tabelle ist zu entnehmen, wie oft die Messungen mindestens durchgeführt werden sollten. In Abhängigkeit von den erwarteten Wirkungen und den Ergebnissen der ersten Messungen sollten gegebenenfalls weitere Zeitpunkte (z. B. alte Tiere) hinzugefügt oder die Messungen in anderen Entwicklungsstadien durchgeführt werden.
(3) Es wird empfohlen, die Jungtiere nicht in den ersten zwei Tagen nach dem Absetzen zu testen (vgl. Nummer 32). Das empfohlene Lebensalter für die Prüfung der adoleszenten Tiere ist jeweils: Lernen und Gedächtnis = PND 25 ± 2; motorische und sensorische Funktionen = PND 25 ± 2. Das empfohlene Lebensalter für die Prüfung junger adulter Tiere ist PND 60-70.
(4) Wenn die Prüfsubstanz den Jungtieren direkt verabreicht wird, sollte das Körpergewicht mindestens zweimal wöchentlich bestimmt werden, um die Dosis in Zeiten rascher Gewichtszunahme anpassen zu können.
(5) Hirngewicht und Neuropathologie können gegebenenfalls zu einem früheren Zeitpunkt bewertet werden (z. B. PND 11) (vgl. Nummer 39).
(6) Gegebenenfalls sind neben dem Körpergewicht auch andere Parameter der Entwicklung (z. B. Augenöffnung) zu dokumentieren (vgl. Nummer 31).
(7) Vgl. Nummer 35.
(8) Für dieses Beispiel werden die Würfe auf vier Männchen und vier Weibchen verkleinert; die männlichen Jungtiere werden von eins bis vier durchnummeriert, die weiblichen von fünf bis acht.
(9) Für dieses Beispiel werden die Würfe auf vier Männchen und vier Weibchen verkleinert; die männlichen Jungtiere werden von eins bis vier durchnummeriert, die weiblichen von fünf bis acht.
(10) Bei der Prüfung der kognitiven Fähigkeiten sind für die Tests am PND 23 und die Tests an jungen adulten Tieren unterschiedliche Tiere zu verwenden (z. B. gerade/ungerade Würfe von insgesamt 20 Jungtieren).
(11) Für dieses Beispiel werden die Würfe auf vier Männchen und vier Weibchen verkleinert; die männlichen Jungtiere werden von eins bis vier durchnummeriert, die weiblichen von fünf bis acht.
(12) Der Schwellenwert des Variationskoeffizienten für ein gegebenes Gewebe wurde anhand einer (fortlaufend vom kleinsten zum größten Wert aufgebauten) VK-Kurve ermittelt, in der alle Durchschnittswerte aller Versuche der Validierungsprüfung unter Verwendung eines spezifischen Modells (Agonisten oder Antagonisten) enthalten waren. Der VK-Schwellenwert wurde vom ‚Bruchpunkt‘ abgeleitet, also dem Punkt, ab dem die Abstände zu den nächsthöheren VK in der Reihe erheblich größer sind als zu den vorhergehenden VK. Es ist anzumerken, dass mit dieser Analyse zwar relativ zuverlässige ‚Bruchpunkte‘ für das antagonistische Prüfmodell ermittelt werden konnten, dass die VK-Kurven für die Prüfung auf Agonisten allerdings einen einheitlicheren Anstieg zeigten, was dazu führte, dass die Festlegung eines Schwellen-VK mittels dieser Methode in gewisser Weise willkürlich erfolgte.
(13) Die Forschung hat gezeigt, dass die Brustdrüse, insbesondere in ihrer frühen Entwicklungsphase, einen empfindlichen Endpunkt für die Östrogenwirkung darstellt. Endpunkte, die Brustdrüsen von Jungtieren beiderlei Geschlechts betreffen, sollten deshalb, sobald sie validiert wurden, in diese Prüfmethode einbezogen werden.
(14) ATCC CRL-2128; ATCC, Manassas, VA, USA, [http://www.lgcstandards-atcc.org/].
(15) ‚Neue Charge‘ bezieht sich auf eine frische Charge Zellen, die von ATCC bezogen wurde.
(16) ‚Eingefrorene Charge‘ bezieht sich auf Zellen, die zuvor gezüchtet und anschließend in einem anderen Labor als ATCC eingefroren wurden.
Anmerkung:Wenn die Extraktion erforderlich ist, werden für jeden Extrakt drei wiederholte Messungen durchgeführt. Jede Probe wird nur einmal extrahiert.
Anmerkung: Die Messgrenzen der Methode basieren auf den in Tabelle 5 angeführten Basalhormonproduktionswerten und sind leistungsgestützt. Wenn eine höhere Basalhormonproduktion erzielt werden kann, kann der Grenzwert auch höher sein.
(19) Einige T- und E2-Antikörper können bei einem höheren Anteil unter Umständen zu Kreuzreaktionen mit Androstendion bzw. Östron führen. In solchen Fällen ist es nicht möglich, die Wirkungen auf 17β-HSD genau zu bestimmen. Die Daten können dennoch nützliche Auskünfte über die Wirkungen auf die Östrogen- oder Androgenproduktion im Allgemeinen geben. In solchen Fällen sind die Angaben als Androgen-/Östrogenreaktionen und nicht als E2 und T auszuweisen.
(20) Dazu gehören: Cholesterin, Pregnenolon, Progesteron, 11-Deoxykortikosteron, Kortikosteron, Aldosteron, 17α-Pregnenolon, 17α-Progesteron, Deoxykortison, Kortison, DHEA, Androstenedion, Östron.
(21) Lösungsmittel (DMSO) Kontrolle (0), 1 μl DMSO/Mulde.
(22) +, positiv
entfällt: keine Angabe, weil nach der Exposition gegenüber nicht-zytotoxischen Konzentrationen der negativen Kontrolle keine Änderungen eintreten sollten.
(23) Zellen in Blindprobenmulden erhalten nur Medium (d. h. kein Lösungsmittel).
(24) Methanol (MeOH) wird nach abgeschlossener Exposition und nach Entfernung des Mediums aus diesen Mulden zugegeben.
(25) DMSO-Lösungsmittelkontrolle (1 μl/Mulde).
(26) Vorangegangene Testreihe nach demselben Prüfplan bestätigen.
(27) Assay mit einem 1/2-log-Abstand zwischen den Konzentrationen erneut durchführen (mit Bracketing der Konzentration, die im vorangegangenen Versuch zu einem signifikant abweichenden Testergebnis geführt hat).
(28) Ein Fold Change (x-fache Veränderung) bei einer Konzentration weicht statistisch signifikant von der Lösungsmittelkontrolle ab.
(29) Bezogen auf Wiederholungsmessungen ein und derselben Probe.