

EUR-Lex Access to European Union law
This document is an excerpt from the EUR-Lex website
Document 32008R0440
Council Regulation (EC) No 440/2008 of 30 May 2008 laying down test methods pursuant to Regulation (EC) No 1907/2006 of the European Parliament and of the Council on the Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH) (Text with EEA relevance)
Kommissionens förordning (EG) nr 440/2008 av den 30 maj 2008 om testmetoder enligt Europaparlamentets och rådets förordning (EG) nr 1907/2006 om registrering, utvärdering, godkännande och begränsning av kemikalier (Reach) (Text av betydelse för EES)
Kommissionens förordning (EG) nr 440/2008 av den 30 maj 2008 om testmetoder enligt Europaparlamentets och rådets förordning (EG) nr 1907/2006 om registrering, utvärdering, godkännande och begränsning av kemikalier (Reach) (Text av betydelse för EES)
OJ L 142, 31.5.2008, p. 1–739
(BG, ES, CS, DA, DE, ET, EL, EN, FR, IT, LV, LT, HU, MT, NL, PL, PT, RO, SK, SL, FI, SV)
Special edition in Croatian: Chapter 13 Volume 033 P. 3 - 741
 In force: This act has been changed. Current consolidated version: 26/03/2023
In force: This act has been changed. Current consolidated version: 26/03/2023
- Date of document:
- 30/05/2008
- Date of effect:
- 01/06/2008; ikraftträdande off.gör.dag + 1 se art. 4
- Date of end of validity:
- No end date
- Author:
- Europeiska kommissionen
- Form:
- Förordning
- Additional information:
- av betydelse för EES
- Treaty:
- Fördraget om upprättandet av Europeiska gemenskapen
- Legal basis:
-
- 32006R1907 - A13P3
- Link
- Link
- Link
- Select all documents mentioning this document No data available in the table
- Modified by:
-
Relation Act Comment Subdivision concerned From To Corrected by 32008R0440R(01) (EN) Corrected by 32008R0440R(02) (FI) Corrected by 32008R0440R(03) (LT) Corrected by 32008R0440R(04) (IT) Corrected by 32008R0440R(05) (LT) Modified by 32009R0761 tillägg bilaga 1 punkt A kapitel A.22 27/08/2009 Modified by 32009R0761 ersätter bilaga 1 punkt C kapitel C.3 27/08/2009 Modified by 32009R0761 tillägg bilaga 1 punkt B kapitel B.46 27/08/2009 Modified by 32009R0761 tillägg bilaga 1 punkt C kapitel C.26 27/08/2009 Modified by 32009R0761 tillägg bilaga 1 punkt C kapitel C.25 27/08/2009 Modified by 32009R0761 ersätter bilaga 1 punkt A kapitel A.4 27/08/2009 Modified by 32010R1152 tillägg bilaga B 12/12/2010 Modified by 32012R0640 bilaga 23/07/2012 Modified by 32014R0260 TXT 22/03/2014 Modified by 32014R0900 tillägg bilaga kapitel B.53 24/08/2014 Modified by 32014R0900 tillägg bilaga kapitel B.57 24/08/2014 Modified by 32014R0900 tillägg bilaga kapitel B.55 24/08/2014 Modified by 32014R0900 tillägg bilaga kapitel B.58 24/08/2014 Modified by 32014R0900 tillägg bilaga kapitel B.56 24/08/2014 Modified by 32014R0900 tillägg bilaga kapitel B.54 24/08/2014 Modified by 32016R0266 ersätter bilaga kapitel C.26 04/03/2016 Modified by 32016R0266 tillägg bilaga kapitel C.32 04/03/2016 Modified by 32016R0266 tillägg bilaga kapitel C.37 04/03/2016 Modified by 32016R0266 tillägg bilaga kapitel C.38 04/03/2016 Modified by 32016R0266 tillägg bilaga kapitel C.41 04/03/2016 Modified by 32016R0266 tillägg bilaga kapitel C.44 04/03/2016 Modified by 32016R0266 tillägg bilaga kapitel C.45 04/03/2016 Modified by 32016R0266 tillägg bilaga kapitel C.43 04/03/2016 Modified by 32016R0266 tillägg bilaga kapitel C.40 04/03/2016 Modified by 32016R0266 tillägg bilaga kapitel C.46 04/03/2016 Modified by 32016R0266 tillägg bilaga kapitel C.35 04/03/2016 Modified by 32016R0266 tillägg bilaga skäl text 04/03/2016 Modified by 32016R0266 tillägg bilaga kapitel C.39 04/03/2016 Modified by 32016R0266 tillägg bilaga kapitel C.31 04/03/2016 Modified by 32016R0266 tillägg bilaga kapitel A.24 04/03/2016 Modified by 32016R0266 tillägg bilaga kapitel C.33 04/03/2016 Modified by 32016R0266 tillägg bilaga kapitel C.36 04/03/2016 Modified by 32016R0266 ersätter bilaga kapitel C.3 04/03/2016 Modified by 32016R0266 tillägg bilaga kapitel C.42 04/03/2016 Modified by 32016R0266 ersätter bilaga kapitel C.11 04/03/2016 Modified by 32016R0266 tillägg bilaga kapitel C.34 04/03/2016 Modified by 32017R0735 tillägg bilaga s. C kapitel C.51 18/05/2017 Modified by 32017R0735 tillägg bilaga s. B kapitel B.62 18/05/2017 Modified by 32017R0735 tillägg bilaga s. C kapitel C.48 18/05/2017 Modified by 32017R0735 tillägg bilaga s. B kapitel B.61 18/05/2017 Modified by 32017R0735 ersätter bilaga s. C kapitel C.20 18/05/2017 Modified by 32017R0735 ersätter bilaga s. B kapitel B.11 18/05/2017 Modified by 32017R0735 tillägg bilaga s. C kapitel C.49 18/05/2017 Modified by 32017R0735 tillägg bilaga s. A kapitel A.25 18/05/2017 Modified by 32017R0735 ersätter bilaga s. C kapitel C.13 18/05/2017 Modified by 32017R0735 upphävande bilaga s. B kapitel B.18 18/05/2017 Modified by 32017R0735 upphävande bilaga s. B kapitel B.16 18/05/2017 Modified by 32017R0735 tillägg bilaga s. C kapitel C.47 18/05/2017 Modified by 32017R0735 ersätter bilaga s. B kapitel B.12 18/05/2017 Modified by 32017R0735 ersätter bilaga s. C kapitel C.29 P 66 18/05/2017 Modified by 32017R0735 upphävande bilaga s. B kapitel B.15 18/05/2017 Modified by 32017R0735 upphävande bilaga s. B kapitel B.24 18/05/2017 Modified by 32017R0735 ersätter bilaga s. B kapitel B.10 18/05/2017 Modified by 32017R0735 tillägg bilaga s. B kapitel B.59 18/05/2017 Modified by 32017R0735 ersätter bilaga s. B kapitel B.47 18/05/2017 Modified by 32017R0735 tillägg bilaga s. B kapitel B.60 18/05/2017 Modified by 32017R0735 ersätter bilaga s. B kapitel B.5 18/05/2017 Modified by 32017R0735 ersätter bilaga s. B kapitel B.49 18/05/2017 Modified by 32017R0735 upphävande bilaga s. B kapitel B.20 18/05/2017 Modified by 32017R0735 upphävande bilaga s. B kapitel B.19 18/05/2017 Modified by 32017R0735 ersätter bilaga s. B kapitel B.48 18/05/2017 Modified by 32017R0735 tillägg bilaga s. C kapitel C.50 18/05/2017 Modified by 32019R1390 tillägg bilaga del B kapitel B.63 16/10/2019 Modified by 32019R1390 tillägg bilaga del C kapitel C.52 16/10/2019 Modified by 32019R1390 ersätter bilaga del B kapitel B.46 16/10/2019 Modified by 32019R1390 tillägg bilaga del B kapitel B.69 16/10/2019 Modified by 32019R1390 ersätter bilaga del B kapitel B.40a 16/10/2019 Modified by 32019R1390 tillägg bilaga del B kapitel B.70 16/10/2019 Modified by 32019R1390 tillägg bilaga del C kapitel C.53 16/10/2019 Modified by 32019R1390 tillägg bilaga del B kapitel B.67 16/10/2019 Modified by 32019R1390 ersätter bilaga del B kapitel B.23 16/10/2019 Modified by 32019R1390 ersätter bilaga del B kapitel B.22 16/10/2019 Modified by 32019R1390 tillägg bilaga del B kapitel B.71 16/10/2019 Modified by 32019R1390 ersätter bilaga del B kapitel B.4 16/10/2019 Modified by 32019R1390 tillägg bilaga del B kapitel B.66 16/10/2019 Modified by 32019R1390 ersätter bilaga del B kapitel B.17 16/10/2019 Modified by 32019R1390 tillägg bilaga del B kapitel B.65 16/10/2019 Modified by 32019R1390 tillägg bilaga del B kapitel B.64 16/10/2019 Modified by 32019R1390 ersätter bilaga del B kapitel B.40 16/10/2019 Modified by 32019R1390 tillägg bilaga del B kapitel B.68 16/10/2019 Modified by 32023R0464 ersätter bilaga del B kapitel B.59 text 26/03/2023 Modified by 32023R0464 ersätter bilaga del B kapitel B.40bis text 26/03/2023 Modified by 32023R0464 ersätter bilaga del C kapitel C.39 text 26/03/2023 Modified by 32023R0464 ersätter bilaga del A kapitel A.4 text 26/03/2023 Modified by 32023R0464 ersätter bilaga del A kapitel A.12 text 26/03/2023 Modified by 32023R0464 ersätter bilaga del B kapitel B.8 text 26/03/2023 Modified by 32023R0464 ersätter bilaga del B kapitel B.29 text 26/03/2023 Modified by 32023R0464 ersätter bilaga del B kapitel B.71 text 26/03/2023 Modified by 32023R0464 ersätter bilaga del A kapitel A.15 text 26/03/2023 Modified by 32023R0464 ersätter bilaga del C kapitel C.33 text 26/03/2023 Modified by 32023R0464 ersätter bilaga del C kapitel C.36 text 26/03/2023 Modified by 32023R0464 ersätter bilaga del B kapitel B.31 text 26/03/2023 Modified by 32023R0464 ersätter bilaga del B kapitel B.48 text 26/03/2023 Modified by 32023R0464 ersätter bilaga del B kapitel B.47 text 26/03/2023 Modified by 32023R0464 ersätter bilaga del B kapitel B.66 text 26/03/2023 Modified by 32023R0464 ersätter bilaga del B kapitel B.39 text 26/03/2023 Modified by 32023R0464 ersätter bilaga del B kapitel B.41 text 26/03/2023 Modified by 32023R0464 ersätter bilaga del A kapitel A.20 text 26/03/2023 Modified by 32023R0464 ersätter bilaga del C kapitel C.15 text 26/03/2023 Modified by 32023R0464 ersätter bilaga del A kapitel A.8 text 26/03/2023 Modified by 32023R0464 tillägg bilaga del 0 26/03/2023 Modified by 32023R0464 ersätter bilaga del A kapitel A.21 text 26/03/2023 Modified by 32023R0464 ersätter bilaga del B kapitel B.60 text 26/03/2023 Modified by 32023R0464 ersätter bilaga del B kapitel B.26 text 26/03/2023 Modified by 32023R0464 ersätter bilaga del B kapitel B.51 text 26/03/2023 Modified by 32023R0464 ersätter bilaga del A kapitel A.9 text 26/03/2023 Modified by 32023R0464 ersätter bilaga del A kapitel A.16 text 26/03/2023 Modified by 32023R0464 ersätter bilaga del C kapitel C.9 text 26/03/2023 Modified by 32023R0464 ersätter bilaga del C kapitel C.32 text 26/03/2023 Modified by 32023R0464 ersätter bilaga del B kapitel B.46 text 26/03/2023 Modified by 32023R0464 ersätter bilaga del B kapitel B.61 text 26/03/2023 Modified by 32023R0464 ersätter bilaga del B kapitel B.5 text 26/03/2023 Modified by 32023R0464 ersätter bilaga del B kapitel B.69 text 26/03/2023 Modified by 32023R0464 ersätter bilaga del B kapitel B.30 text 26/03/2023 Modified by 32023R0464 ersätter bilaga del B kapitel B.25 text 26/03/2023 Modified by 32023R0464 ersätter bilaga del B kapitel B.13/14 text 26/03/2023 Modified by 32023R0464 ersätter bilaga del A kapitel A.11 text 26/03/2023 Modified by 32023R0464 ersätter bilaga del B kapitel B.6 text 26/03/2023 Modified by 32023R0464 ersätter bilaga del B kapitel B.17 text 26/03/2023 Modified by 32023R0464 ersätter bilaga del A kapitel A.10 text 26/03/2023 Modified by 32023R0464 ersätter bilaga del C kapitel C.1 text 26/03/2023 Modified by 32023R0464 ersätter bilaga del B kapitel B.33 text 26/03/2023 Modified by 32023R0464 ersätter bilaga del B kapitel B.32 text 26/03/2023 Modified by 32023R0464 ersätter bilaga del A kapitel A.17 text 26/03/2023 Modified by 32023R0464 ersätter bilaga del A kapitel A.3 text 26/03/2023 Modified by 32023R0464 ersätter bilaga del B kapitel B.3 text 26/03/2023 Modified by 32023R0464 ersätter bilaga del B kapitel B.68 text 26/03/2023 Modified by 32023R0464 ersätter bilaga del B kapitel B.34 text 26/03/2023 Modified by 32023R0464 ersätter bilaga del B kapitel B.56 text 26/03/2023 Modified by 32023R0464 ersätter bilaga del B kapitel B.70 text 26/03/2023 Modified by 32023R0464 ersätter bilaga del B kapitel B.35 text 26/03/2023 Modified by 32023R0464 ersätter bilaga del B kapitel B.22 text 26/03/2023 Modified by 32023R0464 ersätter bilaga del B kapitel B.58 text 26/03/2023 - Affected by case:
-
- Interpreted by 62017CJ0487
- Instruments cited:
- Link
- EUROVOC descriptor:
- Subject matter:
- Directory code:
-
- 05.20.20.10 Fri rörlighet för arbetstagare och socialpolitik / Socialpolitik / Arbetsförhållanden / Säkerhet på arbetsplatsen
- 13.30.18.00 Industripolitik och den inre marknaden / Den inre marknaden: tillnärmning av lagstiftning / Farliga ämnen
- 15.10.20.50 Miljö, konsument- och hälsoskydd / Miljö / Föroreningar / Kemikalier, industriella risker och bioteknik
|
31.5.2008 |
SV |
Europeiska unionens officiella tidning |
L 142/1 |
KOMMISSIONENS FÖRORDNING (EG) nr 440/2008
av den 30 maj 2008
om testmetoder enligt Europaparlamentets och rådets förordning (EG) nr 1907/2006 om registrering, utvärdering, godkännande och begränsning av kemikalier (Reach)
(Text av betydelse för EES)
EUROPEISKA GEMENSKAPERNAS KOMMISSION HAR ANTAGIT DENNA FÖRORDNING
med beaktande av fördraget om upprättandet av Europeiska gemenskapen,
med beaktande av Europaparlamentets och rådets förordning nr 1907/2006 av den 18 december 2006 om registrering, utvärdering, godkännande och begränsning av kemikalier (Reach), inrättande av en europeisk kemikaliemyndighet, ändring av direktiv 1999/45/EG och upphävande av rådets förordning (EEG) nr 793/93 och kommissionens förordning (EG) nr 1488/94 samt rådets direktiv 76/769/EEG och kommissionens direktiv 91/155/EEG, 93/67/EEG, 93/105/EG och 2000/21/EG (1), särskilt artikel 13.3, och
av följande skäl:
|
(1) |
Enligt förordning (EG) nr 1907/2006 ska testmetoder antas på gemenskapsnivå för ämnestestsyften om sådana tester krävs för att generera information om ämnenas inneboende egenskaper. |
|
(2) |
I bilaga V till rådets direktiv 67/548/EEG av den 27 juni 1967 om tillnärmning av lagar och andra författningar om klassificering, förpackning och märkning av farliga ämnen (2) fastställs metoder för ämnens och preparats (beredningars) fysikalisk-kemikaliska egenskaper, toxicitet och ekotoxicitet. Bilaga V till direktiv 67/548/EEG har upphävts genom direktiv 2006/121/EG, med verkan från och med den 1 juni 2008. |
|
(3) |
De testmetoder som ingår i bilaga V till direktiv 67/548/EEG bör tas med i denna förordning. |
|
(4) |
Denna förordning hindrar inte användning av andra testmetoder, förutsatt att användningen av dessa är i enlighet med artikel 13(3) i förordning (EG) nr 1907/2006. |
|
(5) |
Principerna för att ersätta, minska och förbättra användningen av djur i provningsförfaranden bör till fullo beaktas när testmetoder utarbetas, särskilt när lämpliga validerade metoder för att ersätta, minska eller förbättra djurförsök blir tillgängliga. |
|
(6) |
De åtgärder som föreskrivs i denna förordning är förenliga med yttrandet från den kommitté som inrättats genom artikel 133 i förordning (EG) nr 1907/2006. |
HÄRIGENOM FÖRESKRIVS FÖLJANDE.
Artikel 1
De testmetoder som ska tillämpas för de syften som avses i förordning (EG) nr 1907/2006 fastställs i bilagan till denna förordning.
Artikel 2
Kommissionen ska där så är lämpligt se över de testmetoder som ingår i denna förordning i syfte att ersätta, minska eller förfina testning på ryggradsdjur.
Artikel 3
Hänvisningar till bilaga V till direktiv 67/548/EEG ska läsas som hänvisningar till denna förordning.
Artikel 4
Denna förordning träder i kraft dagen efter det att den har offentliggjorts i Europeiska unionens officiella tidning.
Den ska tillämpas från och med den 1 juni 2008.
Utfärdad i Bryssel den 30 maj 2008.
På kommissionens vägnar
Stavros DIMAS
Ledamot av kommissionen
(1) EUT L 396, 30.12.2006, s. 1. Rättad i EUT L 136, 29.5.2007, s. 3.
(2) EGT 196, 16.8.1967, s. 1. Direktivet senast ändrat genom Europaparlamentets och rådets direktiv 2006/121/EG (EUT L 396, 30.12.2006, s. 850. Rättat i EUT L 136, 29.5.2007, s. 281) – uppdateras med lämpliga referenser så snart den 30:e anpassningen offentliggjorts.
BILAGA
DEL A: METODER FÖR BESTÄMNING AV FYSIKALISK-KEMISKA EGENSKAPER
INNEHÅLLSFÖRTECKNING
|
A.1 |
SMÄLT-/FRYSPUNKT |
|
A.2 |
KOKPUNKT |
|
A.3 |
RELATIV DENSITET |
|
A.4 |
ÅNGTRYCK |
|
A.5 |
YTSPÄNNING |
|
A.6 |
VATTENLÖSLIGHET |
|
A.8 |
FÖRDELNINGSKOEFFICIENT |
|
A.9 |
FLAMPUNKT |
|
A.10 |
ANTÄNDLIGHET (FASTA ÄMNEN) |
|
A.11 |
ANTÄNDLIGHET (GASER) |
|
A.12 |
ANTÄNDLIGHET (KONTAKT MED VATTEN) |
|
A.13 |
PYROFORISKA EGENSKAPER HOS FASTA OCH FLYTANDE ÄMNEN |
|
A.14 |
EXPLOSIVA GASER |
|
A.15 |
SJALVANTÄNDNINGSTEMPERATUR (VÄTSKOR OCH GASER) |
|
A.16 |
RELATIV SJALVANTÄNDNINGSTEMPERATUR (FASTA ÄMNEN) |
|
A.17 |
OXIDERANDE EGENSKAPER (FASTA ÄMNEN) |
|
A.18 |
MOLEKYLÄR MEDEL VIKT OCH MOLEKYLÄR VIKTFÖRDELNING AV POLYMERER |
|
A.19 |
INNEHÅLL AV LÅGMOLEKYLVIKTSPOLYMERER |
|
A.20 |
POLYMERERS BETEENDE I VATTEN VID LÖSNING/EXTRAKTION |
|
A.21 |
OXIDERANDE EGENSKAPER (VÄTSKOR) |
A.1 SMÄLT-/FRYSPUNKT
1. METOD
De flesta av de beskrivna metoderna är baserade på OECD:s riktlinjer för undersökning av kemikalier (se hänvisning 1). Grundprinciperna anges i hänvisningarna 2 och 3.
1.1 INLEDNING
De beskrivna metoderna och apparaterna kan användas för bestämning av ämnens smältpunkt, utan någon begränsning beträffande deras renhetsgrad.
Valet av metod beror på arten av det ämne som ska undersökas. Det är följaktligen avgörande om ämnet lätt, med svårighet eller inte alls kan pulvriseras.
För vissa ämnen är det mer ändamålsenligt att bestämma frys- eller stelningspunkten, och i denna metod inbegrips standarder för sådana bestämningar.
Om det till följd av ämnets särskilda egenskaper inte går att korrekt bestämma ovan nämnda parametrar kan det vara ändamålsenligt att bestämma den lägsta flytpunkten.
1.2 DEFINITIONER OCH ENHETER
Smältpunkten definieras som den temperatur vid vilken fasövergången från fast till flytande tillstånd sker vid normalt atmosfäriskt tryck, och denna temperatur motsvarar idealt fryspunkten.
Eftersom fasövergången för många ämnen sker under ett punktintervall beskrivs den ofta som smäkintervallet.
Omvandling av enheter (K till oC):
t = T – 273,15
|
t |
: |
temperaturen i grader Celsius ( oC) |
|
T |
: |
termodynamiska temperaturen Kelvin (K) |
1.3 REFERENSÄMNEN
Man behöver inte använda referensämnen varje gång ett nytt ämne ska undersökas. Referensämnen ska först och främst användas för att då och då kontrollera metoden och för jämförelse med resultat som uppnåtts med andra metoder.
Vissa referensämnen anges i hänvisningarna (se hänvisning 4).
1.4 TESTMETODSPRINCIP
Temperaturen (temperarurintervallet) för fasövergången från fast till flytande tillstånd eller från flytande till fast tillstånd bestäms. I praktiken bestäms temperaturen vid begynnande smältning/frysning och vid slutlig smältning/frysning under uppvärmning/avkylning av ett prov av testämnet vid atmosfäriskt tryck. Fem typer av metoder beskrivs, nämligen kapillärmetoder, varmebordsmetoder, fryspunktsbestämning, metoder för termisk analys och bestämning av lägsta flytpunkt (utvecklad för petroleumoljor).
I vissa fall kan det vara praktiskt att mäta fryspunkten i stället för smältpunkten.
1.4.1 Kapillärmetoder
1.4.1.1 Smältpunktsanordningar med vätskebad
Ett kapillärrör fylls med en liten, tätt packad mängd av det finmalda ämnet. Röret uppvärms tillsammans med en termometer och temperaturstegringen ställs in till mindre än ca 1 K/min under själva smältningen. Temperaturen vid begynnande och slutlig smältning bestäms.
1.4.1.2 Srnältpunktsanordningar med metallblock
Enligt beskrivningen under 1.4.1.1, bortsett från att kapillärröret och termometern är placerade i ett uppvärmt metallblock och kan iakttas genom hål i blocket.
1.4.1.3 Bestämmng med fotocell
Provet i kapillärröret uppvärms automatiskt i en metallcylinder. En ljusstråle riktas genom ämnet via ett hål i cylindern på en exakt inställd fotocell. Vid smältningen ändras de flesta ämnens optiska egenskaper från ogenomskinligt till genomskinligt. Ljusstyrkan som når fotocellen ökar, varvid en stoppsignal sänds till en digital indikator som visar temperaturen hos en platinamotståndstermometer som är placerad i värmekammaren. Denna metod kan inte användas för vissa starkt färgade ämnen.
1.4.2 Värmebord
1.4.2.1 Koflers värmebänk
Koflers värmebänk består av två elektriskt uppvärmda metallstycken med olika värmeledningsförmåga och är så utformad att dess temperaturgradient i längdriktningen nästan är linjär. Värmebänkens temperaturområde kan spänna från 283 K till 543 K och är försedd med en särskild anordning för temperaturavläsning som består av en löpare med visare och en flagga, och som är särskilt utformad för den aktuella bänken. Smältpunkten bestäms genom att det aktuella ämnet läggs i ett tunt lager direkt på värmebänkens ovansida. Efter några sekunder utvecklas en skarp skiljelinje mellan den flytande och den fasta fasen. Skiljelinjens temperatur avläses genom att visaren ställs in på linjen.
1.4.2.2 Smältmikroskop
Flera värmebord med mikroskop används vid bestämning av smältpunkten på mycket små provmängder. I de flesta värmeborden mäts temperaturen med ett känsligt termoelement, men ibland används även kvicksilvertermometrar. En typisk smältpunktsanordning, bestående av värmebord med mikroskop, är utrustad med en värmekammare som innehåller en metallplatta på vilken proven placeras på ett objektglas. I mitten av metallplattan finns ett hål genom vilket ljus kan sändas från mikroskopets belysningsspegel. När anordningen används är kammaren stängd med en glasplatta för att förhindra tillträde av luft till provet.
Uppvärmningen av provet regleras med en reostat. Om det krävs mycket exakta mätningar kan polariserat ljus användas för optiskt anisotropa ämnen.
1.4.2.3 Meniskmetoden
Denna metod används endast för polyamiden
Den temperatur vid vilken en silikonoljemenisk, som är innesluten mellan ett värmebord och ett täckglas som vilar på polyamidprovämnet, förskjuts iakttas visuellt.
1.4.3 Metod för bestämning av fryspunkt
Provet placeras i ett särskilt provrör och sätts i en apparat för bestämning av fryspunkten. Provet avkyls under försiktig och oavbruten omrörning och temperaturen mäts med passande mellanrum. När temperaturen är konstant vid flera avläsningar registreras denna temperatur (korrigerad för termometerfel) som fryspunkten.
Underkylning ska undvikas genom att man upprättar jämvikt mellan den fasta fasen och vätskefasen.
1.4.4 Termisk analys
1.4.4.1 Differentiell termisk analys (DTA)
Genom denna metod registreras temperaturskillnaden mellan ämnet och ett referensämne som en funktion av temperaturen medan ämnet och referensämnet utsätts för samma kontrollerade temperaturprogram. En fasövergång i provet som åtföljs av en entalpiändring visar sig genom en endoterm (smältning) eller exoterm (frysning) avvikelse från den registrerade baslinjen.
1.4.4.2 Differentiell scanningkalorimetri (DSC)
Genom denna metod registreras skillnaden i den energimängd som tillförs ett ämne och ett referensmaterial som en funktion av temperaturen medan ämnet och referensämnet utsätts för samma kontrollerade temperaturprogram. Denna skillnad motsvarar den energi som är nödvändig för att upprätthålla en temperaturskillnad på noll mellan ämnet och referensmaterialet. En fasövergång i provet ledsagad av en entalpiändring visar sig genom en endoterm (smältning) eller exoterm (frysning) avvikelse från den registrerade baslinjen.
1.4.5 Flytpunkt
Denna metod har utvecklats för petroleumoljor och är lämplig för oljeaktiga ämnen med låg smältpunkt.
Efter att provet först har värmts upp kyls det ned med en bestämd hastighet, och dess flytegenskaper undersöks med 3K:s mellanrum. Den lägsta temperaturen vid vilken rörelse av ämnet kan iakttas registreras som flytpunkten.
1.5 KVALITETSKRITERIER
I följande tabell anges de olika metodernas lämplighet och noggrannhet vid bestämning av smältpunkt/smältpunktsintervall.
TABELL: METODERNAS LÄMPLIGHET
A. Kapillärmetoder
|
Mätmetod |
Ämnen som kan pulvriseras |
Ämnen som inte lätt pulvriseras |
Temperatur område |
Uppskattad exakthet (1) |
Befintlig standard |
|
Smältpunktsanordning med vätskebad |
ja |
några få |
273 till 573 K |
±0,3 K |
JIS K 0064 |
|
Smältpunkts-anordning med metallblock |
ja |
några få |
293 till >573K |
±0,5 K |
ISO 1218 (E) |
|
Bestämning med fotocell |
ja |
flera med hjälp av särskild utrustning |
253 till 573 K |
±0,5 K |
|
B. Värmebord och frysmetoder
|
Mätmetod |
Ämnen som kan pulvriseras |
Ämnen som inte latt pulvriseras |
Temperaturområde |
Uppskattad exakthet (2) |
Befintlig standard |
|
Koflers värmebank |
ja |
nej |
283 till > 573 K |
±1,0 K |
ANSI/ASTM D 3451-76 |
|
Smältmikroskop |
ja |
några få |
273 till > 573 K |
±0,5 K |
DIN 53736 |
|
Meniskmetod |
nej |
endast för polyamider |
293 till > 573 K |
±0,5 K |
ISO 1218 (E) |
|
Fryspunkt-metoder |
ja |
ja |
223 till 573 K |
±0,5 K |
t.ex. BS 4695 |
C. Termisk analys
|
Mätmetod |
Ämnen som kan pulvriseras |
Ämnen som inte lätt pulvriseras |
Temperaturområde |
Uppskattad cxakthet (3) |
Befintlig standard |
|
Differentiell termisk analys |
ja |
nej |
173 till 1 273 K |
upp till 600 K ±0,5 K upp till 1 273 K ±2,0 K |
ASTM E 537-76 |
|
Differentiell kalorimetri |
ja |
ja |
173 till 1 273 K |
upp till 600 K ±0,5 K upp till 1 273 K ±2,0 K |
ASTM E 537-76 |
D. Flytpunkt
|
Matmetod |
Ämnen som kan pulvriseras |
Ämnen som inte lätt pulvriseras |
Temperaturområde |
Uppskattad exakthet (4) |
Befintlig standard |
|
Flytpunkt |
för petroleum- oljor och oljeaktiga ämnen |
för petroleum- oljor och oljeaktiga ämnen |
223 till 323 K |
±3,0 K |
ASTM D 97-66 |
1.6 METODBESKRIVNING
Tillvägagångssättet för nästan alla testmetoderna beskrivs i internationella och nationella standarder (se tillägg 1).
1.6.1 Kapillärrörsmetoder
Fint pulvriserade ämnen uppvisar vid långsam uppvärmning vanligtvis ett smältningsförlopp enligt figur 1.
Figur 1
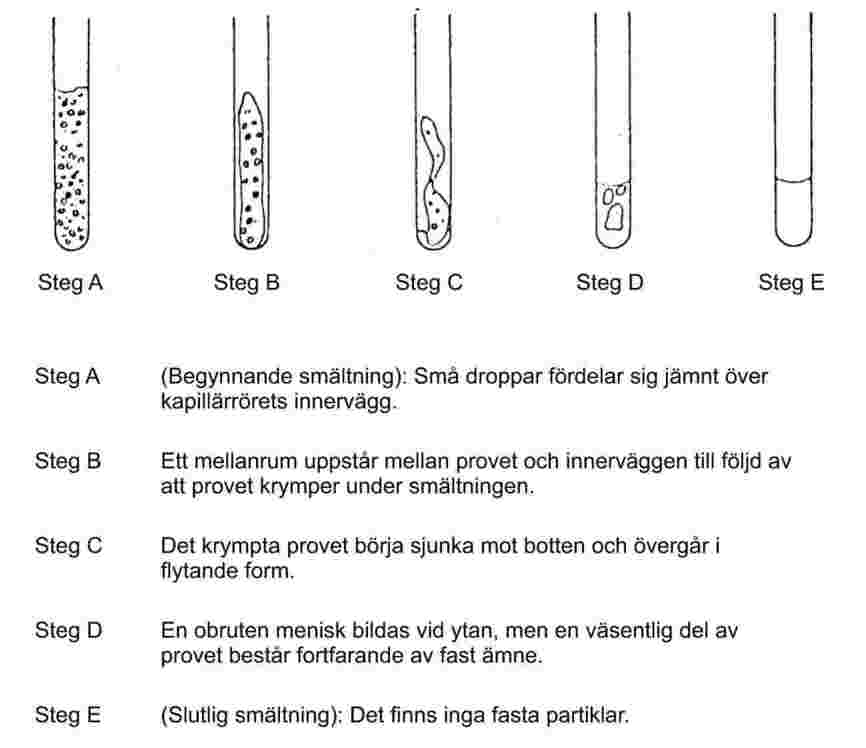
Vid bestämning av smältpunkten antecknas temperaturerna vid begynnande och slutlig smältning.
1.6.1.1 Smältpunktsanordningar med vätskebad
Figur 2 visar ett exempel på en standardiserad smältpunktsapparat (JIS K 0064) av glas. Alla mått är angivna i mm.
Figur 2
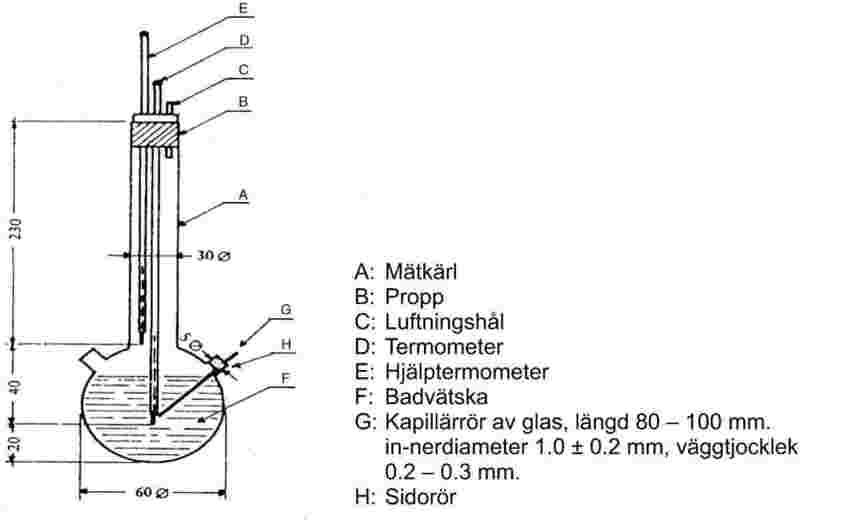
Badvätska:
En lämplig vätska väljs. Valet beror på den smältpunkt som ska bestämmas, t.ex. paraffinolja för smältpunkter upp till 473 K och silikonolja för smältpunkter upp till 573 K.
För smältpunkter över 523 K kan en blandning bestående av 3 delar svavelsyra och 2 delar kaliumsulfat (viktförhållande) användas. Vid användning av denna blandning ska lämpliga försiktighetsåtgärder vidtas.
Termometer:
Endast termometrar som uppfyller kraven i följande eller likvärdiga standarder bör användas:
ASTM E 1-71, DIN 12770, JIS K 8001.
Förfarande:
Det torra ämnet pulvriseras fint i en mortel och fylls i kapillärröret, som är tillslutet i den ena änden, till en höjd av ungefär 3 mm efter tät packning. För att få ett jämnt packat prov låter man kapillärröret falla från en höjd av ungefär 700 mm genom ett lodrätt glasrör ned på ett urglas.
Det fyllda kapillärröret placeras i badet så att den mittersta delen av termometerns kvicksilverkula berör den del av kapillärröret där provet finns. Normalt placeras kapillärröret i apparaten när badets temperatur är ungefär 10 K under smältpunkten.
Badvätskan uppvärms med en temperaturstegring av ungefar 3 K/min under omrörning av vätskan. När temperaturen är ungefär 10 K under den förväntade smältpunkten ställs temperaturstegringen in till högst 1 K/min.
Beräkning:
Smältpunkten beräknas enligt följande:
T-TD+0,00016(TD – TE)n
där:
|
T |
= |
korrigerad smältpunkt i K |
|
TD |
= |
temperaturavläsning av termometer D i K |
|
TE |
= |
temperaturavläsning av termometer E i K |
|
n |
= |
antal gradindelningar av den utstående delen av termometer D:s kvicksilverpelare. |
1.6.1.2 Smältpunktsanordning med metallblock
Apparatur:
Denna består av
|
— |
ett cylindriskt metallblock, vars översta del är ihålig och bildar en kammare (se figur 3), |
|
— |
en metallpropp med två eller flera hål som gör det möjligt att föra in rör i metallblocket, |
|
— |
ett uppvärmningssystem för metallblocket, t.ex. i form av ett elektriskt motstånd inneslutet i blocket, |
|
— |
en reostat för reglering av energitillförseln, om elektrisk uppvärmning används, |
|
— |
fyra fönster av värmebeständigt glas på kammarens sidovägg, placerade diametralt och vinkelrätt mot varandra. Framför ett av dessa fönster finns en lupp monterad för iakttagelse av kapillärröret. De tre andra fönstren används för att belysa kammarens insida med hjälp av lampor, |
|
— |
ett kapillärrör av värmebeständigt glas, tillslutet i ena änden (se 1.6.1.1). |
Termometer:
Se standarderna i punkt 1.6.1.1. Även termoelektriska instrument med jämförbar noggrannhet kan användas.
Figur 3

1.6.1.3 Bestämning med fotocell
Apparatur och förfarande:
Apparaturen består av en metallkammare med ett automatiskt uppvärmningssystem. Tre kapillärrör fylls enligt 1.6.1.1 och placeras i ugnen.
Flera linjära temperaturstegringar kan användas för att kalibrera apparaturen och lämplig temperaturstegring justeras elektriskt med en förvald konstant och linjär hastighet. Skrivare visar den faktiska ugnstemperaturen och temperaturen för ämnet i kapillärrören.
1.6.2 Värmebord
1.6.2.1 Koflers värmebänk
Se tillägg.
1.6.2.2 Smältmikroskop
Se tillägg.
1.6.2.3 Meniskmetoden (polyamider)
Se tillägg.
Uppvärmningshastigheten vid smältpunkten ska vara lägre än 1 K/min.
1.6.3 Metoder för bestämning av fryspunkt
Se tillägg.
1.6.4 Termisk analys
1.6.4.1 Differentiell termisk analys
Se tillägg.
1.6.4.2 Differentiell scanningkalorimetri
Se tillägg.
1.6.5 Bestämning av flytpunkt
Se tillägg.
2. DATA
I vissa fall är termometerkorrektion nödvändigt.
3. RAPPORTERING
Testrapporten ska, om möjligt, innehålla följande uppgifter:
|
— |
Använd testmetod. |
|
— |
Noggrann beskrivning av substansen (beteckning och orenheter) och eventuella inledande reningsåtgärder. |
|
— |
Den uppskattade noggrannheten. |
Som smältpunkt anges medelvärdet av minst två mätningar, som ligger inom gränserna för den uppskattade noggrannheten (se tabellerna).
Om skillnaden mellan temperaturen vid begynnande och slutlig smältning ligger inom gränsvardena för metodens noggrannhet, anges temperaturen vid den slutliga smältningen som smältpunkten. I motsatt fall anges båda temperaturerna.
Om ämnet upplöses eller sublimeras innan smältpunkten nås anges den temperatur där detta fenomen iakttas.
Alla uppgifter och upplysningar av betydelse för tolkningen av resultaten måste anges, i synnerhet vad gäller orenheter och ämnets fysiska tillstånd.
4. HÄNVISNINGAR
|
1. |
OECD, Paris, ”Test Guideline 102”, rådets beslut K(81) 30 slutligt, 1981. |
|
2. |
IUPAC, B. Le Neindre, B. Vodar, eds, ”Experimental thermodynamics”, Butterworths, London1975, vol. II, 803–834. |
|
3. |
R. Weissberg ed., ”Technique of organic Chemistry, Physical Methods of Organic Chemistry”, 3rd ed., Interscience Publ., New York, 1959,, vol. I del I kapitel VII. |
|
4. |
IUPAC, ”Physiochemical measurements: Catalogue of reference materials from national laboratories, Pure and applied chemistry”, vol. 48, 1976, 505–515. |
Tillägg
Kompletterande tekniska upplysningar finns i följande standarder:
1. Kapillärmetoder
1.1 Smältpunktsanordningar med vätskebad
|
ASTM E 324-69 |
Standard test method for relative initial and final melting points and the melting range of organic chemicals |
|
BS 4634 |
Method for determination of melting point and/or melting range |
|
DIN 53181 |
Bestimmung des Schmelzintervalles von Harzen nach Kapillarverfahren |
|
JIS K 00-64 |
Testing methods for melting point of chemical products |
1.2 Smältpunktsanordningar med metallblock
|
DIN 53736 |
Visuelle Bestimmung der Schmelztemperatur von teilkristallinen Kunststoffen |
|
ISO 1218 (E) |
Plastics – polyamides – determination of ”melting point” |
2. Värmebord
2.1 Koflers värmebänk
|
ANSI/ASTM D 3451 |
76 Standard recommended practices for testing polymeric powder coatings |
2.2 Smältmikroskop
|
DIN 53736 |
Visuelle Bestimmung der Schmelztemperatur von teilkristallinen Kunststoffen |
2.3 Meniskmetoden (polyamider)
|
ISO 1218 (E) |
Plastics – polyamides – determination of ”melting point” |
|
ANSI/ASTM D 2133-66 |
Standard specification for acetal resin injection moulding and extrusion materials |
|
NF T 51-050 |
Résines de polyamides. Determination du ”point de fusion” Methode du ménisque |
3. Metoder för bestämning av fryspunkt
|
BS 4633 |
Method for the determination of crystallizing point |
|
BS 4695 |
Method for Determination of Melting Point of petroleum wax (Cooling Curve) |
|
DIN 51421 |
Bestimmung des Gefrierpunktes von Flugkraftstoffen, Ottokraftstoffen und Motorenbenzolen |
|
ISO 2207 |
Cires de pétrole: détermination de la température de figeage |
|
DIN 53175 |
Bestimmung des Erstarrungspunktes von Fettsäuren |
|
NF T 60-114 |
Point de fusion des paraffines |
|
NF T 20-051 |
Méthode de détermination du point de cristallisation (point de congélation) |
|
ISO 1392 |
Method for the determination of the freezing point |
4. Termisk analys
4.1 Differentiell termisk analys
|
ASTM E 537-76 |
Standard method for asseying the thermal stability of chemicals by methods of differential thermal analysis |
|
ASTM E 473-85 |
Standard definitions of terms relating to thermal analysis |
|
ASTM E 472-86 |
Standard practice for reporting thermoanalytical data |
|
DIN 51005 |
Thermisches Analyse, Begriffe |
4.2 Differentiell scanningkalorimetri
|
ASTM E 537-76 |
Standard method for asseying the thermal stability of chemicals by methods of differential thermal analysis |
|
ASTM E 473-85 |
Standard definitions of terms relating to thermal analysis |
|
ASTM E 472-86 |
Standard practice for reporting thermoanalytical data |
|
DIN 51005 |
Thermisches Analyse, Begriffe |
5. Bestämning av flytpunkt
|
NBN 52014 |
Echantillonnage et analyse des produits du pétrole: Point de trouble et point d'écoulement limite – Monsterneming en ontleding van aardolieproducten: Troebelingspunt en vloeipunt |
|
ASTM D 97-66 |
Standard test method for pour point of petroleum oils |
|
ISO 3016 |
Petroleum oils – Determination of pour point. |
A.2 KOKPUNKT
1. METOD
De flesta av de beskrivna metoderna är baserade på OECD:s riktlinjer för undersökning av kemikalier (se hänvisning 1). Grundprinciperna anges i hänvisningarna 2 och 3.
1.1. INLEDNING
De metoder och anordningar som beskrivs här kan användas för alla vätskor och ämnen med låg smältpunkt, förutsatt att det inte inträffar någon kemisk reaktion under kokpunkten (t.ex. autooxidation, omordning, nedbrytning). Metoderna kan användas för alla vätskor, oavsett renhetsgrad.
De metoder där man använder bestämning med fotocell och de som är baserade på termisk analys framhävs särskilt eftersom de kan användas för bestämning av både smält- och kokpunkter. Dessutom kan dessa mätningar utföras automatiskt.
Den ”dynamiska metoden” har den fördelen att den också kan användas för bestämning av ångtryck, och det är inte nödvändigt att korrigera kokpunkten till standardtryck (101,325 kPa) eftersom standardtrycket kan ställas in med en manostat under mätningen.
Anmärkningar:
Orenheters inflytande på kokpunktsbestämningen beror i hög grad på arten av orenhet. Om provet innehåller flyktiga orenheter, som kan påverka resultaten, kan ämnet eventuellt lenas.
1.2. DEFINITIONER OCH ENHETER
Standardkokpunkten definieras som den temperatur vid vilken en vätskas ångtryck är 101,325 kPa.
Om kokpunkten inte mäts vid normalt atmosfäriskt tryck kan ångtryckets beroende av temperaturen beskrivas genom Clausius-Clapeyrons ekvation:

där:
|
p |
= |
ämnets ångtryck i pascal |
|
A Hv |
= |
ämnets förångningsvärme i J mol-1 |
|
R |
= |
den universella gaskonstanten = 8,314 J mol-1 K-1 |
|
T |
= |
termodynamisk temperatur i K |
Kokpunkten anges i förhållande till det aktuella trycket under mätningen.
Omvandling
Tryck (enhet: kPa)
|
100 kPa |
= |
bar = 0,1 MPa (enheten ”bar” är fortfarande tillåten men rekommenderas inte). |
|
133 Pa |
= |
1 mm Hg = 1 Torr (enheterna ”mm Hg” och ”Torr” är inte tillåtna). |
|
1 atm |
= |
standardatmosfär = 101 325 Pa (enheten ”atm” är inte tillåten). |
Temperatur (enhet: K)
t = T – 273,15
|
t |
: |
temperatur i grader Celsius ( oC) |
|
T |
: |
termodynamisk temperatur i kelvin (K) |
1.3 REFERENSÄMNEN
Man behöver inte använda referensämnen varje gång ett nytt ämne ska undersökas. Referensämnen ska först och främst användas för att då och då kontrollera metoden och för jämförelse med resultat som uppnåtts med andra metoder.
I de metoder som anges i tillägget anges vissa referensämnen.
1.4 TESTMETODSPRINCIP
Det finns fem metoder för bestämning av kokpunkten (kokpunktsintervallet) som är baserade på mätning av den temperatur vid vilken proven kokar, och två metoder som är baserade på termisk analys.
1.4.1 Bestämning med hjälp av ebulliometer
Ebulliometern utvecklades ursprungligen för molekylviktsbestämning via kokpunktshöjningar, men de lämpar sig även för exakt bestämning av kokpunkter. En mycket enkel apparat beskrivs i ASTM D 1120-72 (se tillägg). I denna apparat uppvärms vätskan under jämviktsbetingelser i atmosfäriskt tryck tills den kokar.
1.4.2 Dynamisk metod
Genom denna metod mäts ångans förtätningstemperatur med hjälp av en lämplig termometer i återloppet vid kokning. Med denna metod kan trycket varieras.
1.4.3 Destillationsmetod för bestämning av kokpunkt
Genom denna metod destilleras vätskan, och ångans förtätningstemperatur och destillatmängden mäts.
1.4.4 Siwoloboffs metod
Ett prov uppvärms i ett provrör som är nedsänkt i vätska i ett värmebad. Ett sammangjutet kapillärrör med en luftbubbla i den nedre änden sänks ned i provröret.
1.4.5 Bestämning med fotocell
Vid användning av Siwoloboffs princip sker automatisk fotoelektrisk mätning av uppstigande bubblor.
1.4.6 Differentiell termisk analys
Genom denna metod registrerar temperaturskillnaden mellan ämnet och ett referensämne som en funktion av temperaturen medan ämnet och referensämnet utsätts för samma kontrollerade temperaturprogram. En fasöver-gång i provet som åtföljs av en entalpiändring visar sig genom en endoterm (smältning) eller exoterm (frysning) avvikelse från den registrerade baslinjen.
1.4.7 Differentiell scanningkalorimetri
Genom denna metod registreras skillnaden i den energimängd som tillförs ett ämne och ett referensmaterial som en funktion av temperaturen medan ämnet och referensämnet utsätts för samma kontrollerade temperaturprogram. Denna skillnad motsvarar den energi som är nödvändig för att upprätthålla en temperaturskillnad på noll mellan ämnet och referensmaterialet. En fasövergång i provet som åtföljs av en entalpiändring visar sig genom en endoterm (smältning) eller exoterm (frysning) avvikelse från den registrerade baslinjen.
1.5 KVALITETSKRITERIER
I tabell 1 anges de olika metodernas lämplighet och noggrannhet vid bestämning av kokpunkt/kokpunktsintervall.
Tabell 1
Jämförelse av metoderna
|
Mätmetod |
Uppskattad noggrannhet |
Befintliga standarder |
|
Ebulliometer |
ASTM D 1120-72 (5) |
|
|
Dynamisk metod |
±0,5 K (upp till 600 K) (6) |
|
|
Destillationsprocess (kokpunktsintervall) |
±0,5 K (upp till 600 K) |
ISO/R 918, DIN 53171, BS 4591/71 |
|
Siwoloboffs metod |
± 2 K (upp till 600 K) (6) |
|
|
Bestämning med fotocell |
±0,3 K (vid 373 K) (6) |
|
|
Differentiell termisk kalorimetri |
±0,5 K (upp till 600 K) ±2,0 K (upp till 1 273 K) |
ASTM E 537-76 |
|
Differentiell scanningkalorimetn |
±0,5 K (upp till 600 K) ±2,0 K (upp till 1 273 K) |
ASTM E 537-76 |
1.6 METODBESKRIVNING
Tillvägagångssättet för vissa metoder beskrivs i internationella och nationella standarder (se tillägg).
1.6.1 Ebulliometer
Se tillägg.
1.6.2 Dynamisk metod
Se testmetod A.4 för bestämning av ångtrycket.
Den kokpunkt, som iakttas vid ett tryck av 101,325 kPa, antecknas.
1.6.3 Destillationsförlopp (kokpunktsintervall)
Se tillägg.
1.6.4 Siwoloboffs metod
Provet uppvärms i en smältpunktsapparat i ett provrör med en diameter på ungefär 5 mm (se figur 1).
Figur 1 visar en typ av en standardiserad smält- och kokpunktsapparat (JIS K 0064) (gjord av glas, alla mått i mm).
Figur 1
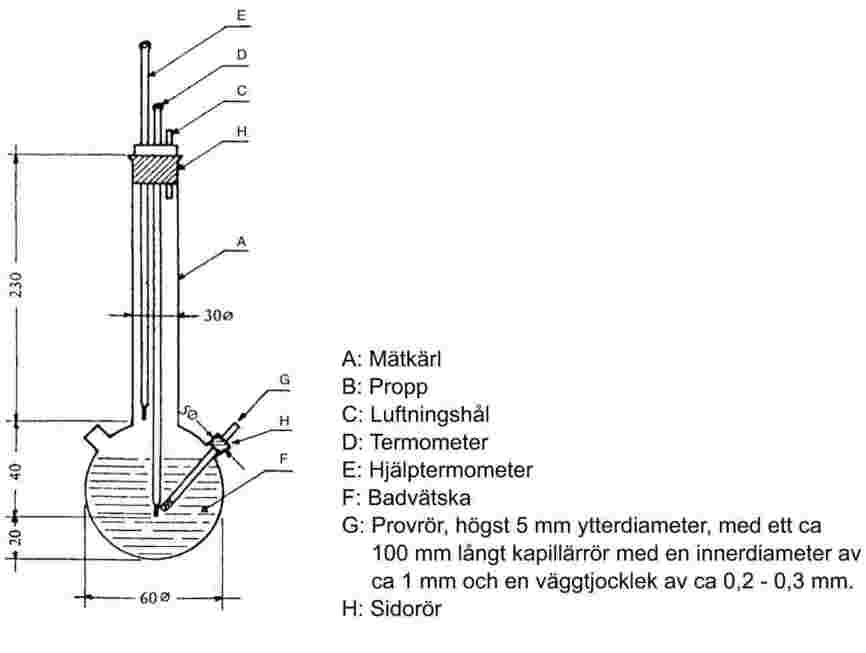
Ett kapillärrör, som är tillslutet ungefär 1 cm ovanför den nedre änden, placeras i provröret. Provet fylls så högt upp att den tillslutna delen av kapillärröret befinner sig under vattenytan. Provröret med kapillärröret fästs vid termometern med ett gummiband eller med ett stöd från sidan (se figur 2).
|
Figur 2 Siwoloboffs princip |
Figur 3 Modifierad princip |
|
|
|


Badvätskan väljs med hänsyn till kokpunkten. Vid temperaturer upp till 573 K kan silikonolja användas. Paraf-finolja kan endast användas upp till 473 K. Uppvärmningen av badvätskan ska i början regleras till en temperaturstegring av 3 K/min, under omrörning. Vid ungefär 10 K under den förväntade kokpunkten sänks värmetillförseln så att temperaturstegringen blir mindre än 1 K/min. När temperaturen närmar sig kokpunkten börjar bubblor strömma ut från kapillärröret.
Kokpunkten är den temperatur då bubbelströmmen under en kortvarig temperaturminskning upphör och vätskan börjar stiga upp i kapillärröret. Termometeravläsningen visar ämnets kokpunkt.
I den modifierade principen (se figur 3) bestäms kokpunkten i ett kapillärrör avsett för smältpunktsbestämning. Det sträcks till en finspetsig tråd, ungefär 2 cm lång (a), och en liten provmängd sugs upp. Den öppna änden av det fina kapillärröret tillsluts genom smältning på så vis att en liten luftbubbla blir kvar i änden. Vid uppvärmning i smältpunktsapparaten (b) utvidgar sig luftbubblan. Kokpunkten är den temperatur vid vilken ämnesproppen når upp till ytan av badvätskan.
1.6.5 Bestämning med fotocell
Provet uppvärms i ett kapillärrör i ett uppvärmt metallblock.
En ljusstråle riktas via lämpliga hål i blocket genom ämnet mot en exakt inställd fotocell.
Medan provets temperatur stiger utvecklas enstaka luftbubblor från kapillärröret. När kokpunkten är nådd ökar antalet bubblor kraftigt. Detta leder till att styrkan ändras på det ljus som är riktat mot fotocellen och en stoppsignal sänds till en indikator som visar temperaturen på en motståndstermometer av platina som är placerad i blocket.
Denna metod är särskilt användbar eftersom den kan användas för bestämningar under rumstemperatur ned till 253,15 K (- 20 oC) utan ändringar av apparaturen. Instrumentet ska endast placeras i ett kylbad.
1.6.6 Termisk analys
1.6.6.1 Differentiell termisk analys
Se tillägg.
1.6.6.2 Differentiell scanningkalorimetri
Se tillägg.
2. DATA
Vid små avvikelser från normalt tryck (högst ± 5 kPa) korrigeras kokpunktstemperaturer till Tn med användning av följande Sidney Young ekvation:
Tn = T + (fT x Δp)
där:
|
Δ p |
= |
(101,325 – p) [observera förtecknet] |
|
P |
= |
ptrycket mätt i kPa |
|
fT |
= |
kokpunktens ändring med trycket i K/kPa |
|
T |
= |
uppmätt kokpunkt i K |
|
Tn |
= |
kokpunkt korrigerad till normalt tryck i K |
I ovannämnda internationella och nationella standarder finns temperaturkorrektionsfaktorer fT för ett flertal ämnen och ekvationer för approximation av dem.
Exempelvis anges i DIN 53171 följande grova korrektioner för lösningsmedel som ingår i färger:
Tabell 2:
Temperaturkorrektionsfaktorer ft
|
Temperatur T (K) |
Korrektionsfaktor fT (K/kPa) |
|
323,15 |
0,26 |
|
348,15 |
0,28 |
|
373,15 |
0,31 |
|
398,15 |
0,33 |
|
423,15 |
0,35 |
|
448,15 |
0,37 |
|
473,15 |
0,39 |
|
498,15 |
0,41 |
|
523,15 |
0,44 |
|
548,15 |
0,45 |
|
573,15 |
0,47 |
3. RAPPORTERING
Testrapporten ska, om möjligt, innehålla följande uppgifter:
|
— |
Använd testmetod. |
|
— |
Noggrann beskrivning av substansen (beteckning och orenheter) och eventuella inledande reningsåtgärder. |
|
— |
Den uppskattade noggrannheten. |
Som kokpunkt anges medelvärdet av minst två mätningar, som ligger mom gränserna för den uppskattade noggrannheten (se tabell 1).
De uppmätta kokpunkterna och medelvärdet av dessa ska anges, och det tryck vid vilket mätningarna gjordes ska anges i kPa. Trycket bör helst ligga i närheten av normalt atmosfäriskt tryck.
Alla uppgifter och upplysningar av betydelse för tolkningen av resultaten ska anges, i synnerhet vad gäller orenheter och ämnets fysiska tillstånd.
4. HÄNVISNINGAR
|
1. |
OECD, Paris, 1981, Test Guideline 103, rådets beslut K(81) 30 slutligt. |
|
2. |
IUPAC, B. Le Neindre, B. Vodar, eds. Experimental thermodynamics, Butterworths, London 1975, vol. II. |
|
3. |
R. Weissberg ed.: Technique of organic Chemistry, Physical Methods of Organic Chemistry, 3rd ed., Inter-science Publ., New York, 1959, vol. I, del I, kapitel VII. |
Tillägg
Kompletterande tekniska uppgifter finns, i följande standarder:
1. Ebulliometer
|
ASTM D 1120-72 |
Standard test method for boiling point of engine anti-freezes |
2. Destillationsprocess (kokpunktsintervall)
|
ISO/R 918 |
Test Method for Distillation (Distillation Field and Distillation Range) |
|
BS 4349/68 |
Method for determination of distillation of petroleum products |
|
BS 4591/71 |
Method for the determination of distillation characteristics |
|
DIN 53171 |
Lösungsmittel fur Anstrichstoffe, Bestimmung des Siederverlaufes |
|
NF T 20-608 |
Distillation: determination du rendement et de l'intervalle de distillation |
3. Differentiell termisk analys och differentiell scanningkalorimetri
|
ASTM E 537-76 |
Standard method for assessine the thermal stability of chemicals by methods or differential thermal analysis |
|
ASTM E 473-85 |
Standard definitions of terms relating to thermal analysis |
|
ASTM E 472-86 |
Standard practice for reporting thermoanalytical data |
|
DIN 51005 |
Thermisches Analyse, Begriffe |
A.3 RELATIV DENSITET
1. METOD
De flesta av de beskrivna metoderna är baserade på OECD:s riktlinjer för undersökning av kemikalier (se hänvisning 1). Grundprinciperna anges i hänvisning 2.
1.1 INLEDNING
De beskrivna metoderna för bestämning av relativ densitet kan användas för fasta ämnen och vätskor, oavsett renhetsgrad. De olika metoder som får användas anges i tabell 1.
1.2 DEFINITIONER OCH ENHETER
Fasta ämnens och vätskors relativa densitet (D4 20) är förhållandet mellan det undersökta ämnets vikt per volymenhet, bestämd vid 20 oC och vikten per samma volymenhet vatten, bestämd vid 4 oC. Den relativa densiteten har ingen dimension.
Ett ämnes densitet, Q, är kvoten av vikten, m, och dess volym, v.
Densiteten, Q, anges, i SI-enheter, i kg/m3.
1.3 REFERENSÄMNEN (SE HÄNVISNING 1 OCH 3)
Referensämnen behöver inte användas varje gång ett nytt ämne ska undersökas. De ska först och främst användas för att då och då kontrollera metoden och för jämförelse med resultat som uppnåtts med andra metoder.
1.4 TESTMETODSPRINCIP
Fyra metoder används.
1.4.1 Flottörmetoder
1.4.1.1 Hydrometer (för vätskor)
Tillräckligt noggranna och snabba densitetsbestämningar kan göras med flytande hydrometrar, med vilka en vätskas densitet kan härledas från nedsänkningsgraden som avläses på en skala.
1.4.1.2 Hydrostatisk balans (för vätskor och fasta ämnen)
Skillnaden mellan provets vikt mätt i luft och i en lämplig vätska (t.ex. vatten) kan användas för att bestämma dess densitet.
För fasta ämnen är den mätta densiteten endast representativ för just det prov som har använts vid bestämningen. När densiteten hos vätskor ska bestämmas vägs först en kropp med känd volym, v, i luft och sedan i vätskan.
1.4.1.3 Metod med nedsänkt kula (för vätskor) (se hänvisning 4)
Med denna metod bestäms en vätskas densitet utifrån skillnaden mellan vätskans vikt före och efter nedsänkning av en kula med känd volym i testvätskan.
1.4.2 Pyknometermetoder
Pyknometrar av olika utformning och med kända volymer kan användas till fasta ämnen och vätskor. Densiteten beräknas utifrån viktskillnaden mellan den fulla och den tomma pyknometern och dess kända volym.
1.4.3 Luftjämförelsepyknometer (för fasta ämnen)
Densiteten hos ett fast ämne, oavsett form, kan bestämmas vid rumstemperatur med en gasjämförelsepyknometer. Ämnets volym mäts i luft eller en inert gas i en cylinder med varierbar kalibrerad volym. Densiteten beräknas genom att vikten mäts efter volymbestämningen.
1.4.4 Oscillerande densitometer (se hänvisning 5, 6 och 7)
En vätskas densitet kan bestämmas med en oscillerande densitometer. En mekanisk oscillator i form av ett U-rör sätts i vibration vid en viss frekvens som är beroende på oscillatorns vikt. När ett prov införs ändras oscillatorns resonansfrekvens. Apparaturen ska kalibreras med två vätskor med känd densitet. Dessa vätskor ska helst väljas så att deras densiteter spänner över hela det område som ska mätas.
1.5 KVALITETSKRITERIER
I tabellen anges de olika metodernas lämplighet för bestämning av densitet.
1.6 METODBESKRIVNING
Tillägget innehåller en lista över några standarder, från vilka ytterligare tekniska uppgifter kan inhämtas.
Mätningarna ska utföras vid 20 oC, och minst två mätningar ska göras.
2. DATA
Se standarder.
3. RAPPORTERING
Testrapporten ska, om möjligt, innehålla följande uppgifter:
|
— |
Använd testmetod. |
|
— |
Exakt specificering av ämnet (beteckning och orenheter) och eventuella inledande reningsåtgärder. |
Den relativa densiteten  ska anges enligt definitionen i 1.2 tillsammans med det undersökta ämnets fysiska tillstånd.
ska anges enligt definitionen i 1.2 tillsammans med det undersökta ämnets fysiska tillstånd.
Alla uppgifter och anmärkningar av betydelse för tolkningen av resultaten ska rapporteras, i synnerhet vad gäller orenheter och ämnets fysiska tillstånd.
Tabell
Metodernas lämplighet
|
Mätmetod |
Densitet |
Maximal dynamisk viskositet |
Befintliga standarder |
|||
|
Fasta |
Vätskor |
|||||
|
|
ja |
5 Pa s |
ISO 387, ISO 649-2, NF T 20-050 |
||
|
|
|
|
|
||
|
ja |
|
|
ISO 1183 (A) |
||
|
|
ja |
5 Pa s |
ISO 901 and 758 |
||
|
|
ja |
20 Pa s |
DIN 53217 |
||
|
|
|
|
ISO 3507 |
||
|
ja |
|
|
ISO 1183(B), NF T 20-053 |
||
|
|
ja |
500 Pa s |
ISO 758 |
||
|
ja |
|
|
DIN 55990 Teil 3, DIN 53243 |
||
|
|
yes |
5 Pa s |
|
||
4. HÄNVISNINGAR
|
1. |
OECD, Paris, 1981, Test Guideline 109, rådets beslut K(81) 30 slutligt. |
|
2. |
R. Weissberger ed., Technique of Organic Chemistry, Physical Methods of Organic Chemistry, 3rd ed., kapitel IV, Interscience Publ., New York, 1959, vol. I, del 1. |
|
3. |
IUPAC, Recommended reference materials for realization of physico-chemical properties, Pure and applied chemistry, 1976, vol. 48, s. 508. |
|
4. |
Wagenbreth, H., Die Tauchkugel zur Bestimmung der Dichte von Fliüssigkeiten, Technisches Messen tm, 1979, vol. 11, s. 427–430. |
|
5. |
Leopold, H., Die digitale Messung von Flussigkeiten, Elektronik, 1970, vol. 19, s. 297–302. |
|
6. |
Baumgarten, D., Füllmengenkontrolle bei vorgepackten Erzeugnissen – Verfahren zur Dichtebestimmung bei fliüssigen Produkten und ihre praktische Anwendung, Die Pharmazeutische Industrie, 1975, vol. 37, s. 717–726. |
|
7. |
Riemann, J., Der Einsatz der digitalen Dichtemessung im Brauereilaboratorium, Brauwissenschaft, 1976, vol. 9, s. 253–255. |
Tillägg
Kompletterande tekniska uppgifter finns i följande standarder:
1. Flottörmetoder
1.1 Hydrometer
|
DIN 12790, ISO 3S7 |
Hydrometer, general instructions |
|
DIN 12791 |
Part I: Density hydrometers; construction, admjustment and use; Part II: Density hydrometers; standardized sizes, designation Part III: Use and test |
|
ISO 649-2 |
Laboratory glassware: Density hydrometers for general purpose |
|
NF T 20-050 |
Chemical products for industrial use – Determination of density of liquids Aerometric method |
|
DIN 12793 |
Laboratory glassware: range find hydrometers |
1.2 Hydrostatisk balans
För fasta ämnen
|
ISO 1183 |
Method A: Methods för determining the density and relative density of plastics excluding cellular plastics |
|
NF T 20-049 |
Chemical products for industrial use – Determination of the density of solids other than powders and cellular products – Hydrostatic balance method |
|
ASTM-D-792 |
Specific gravity and density of plastics by displacement |
|
DIN 53479 |
Testing of plastics and elastometers; determination of density |
För vätskor
|
ISO 901 |
ISO 758 |
|
DIN 51757 |
Testing of mineral oils and related materials; determination of density |
|
ASTM D 941-55, ASTM D 1296-67 och ASTM D 1481-62 |
|
|
ASTM D 1298 |
Density, specific gravity or API gravity of crude petroleum and liquid petroleum products by hydrometer method |
|
BS 4714 |
Density, specific gravity or API gravity of crude petroleum and liquid petro-leum products by hydrometer method |
1.3 Metod med nedsänkt kula
|
DIN 53217 |
Testing of paints, varnishes and similar coating materials; determination of density; lmmersed body method |
2. Pyknometermetoder
2.1 För vätskor
|
ISO 3507 |
Pycnometers |
|
ISO 758 |
Liquid chemical products; determination of density at 20 oC |
|
DIN 12797 |
Gay-Lussac pycnometer (för icke-flyktiga vätskor som inte är för viskösa) |
|
DIN 12798 |
Lipkin pycnometer (för vätskor med kinematisk viskositet under 100 × 10-6 m2s-1 vid 15 oC) |
|
DIN 12800 |
Sprengel pycnometer (för vätskor enligt DIN 12798) |
|
DIN 12801 |
Reischauer pycnometer (för vätskor med kinematisk viskositet under 100 × 10-6 m2s-1 vid 20 oC, särskilt lämplig även för kolväten och vattenlösningar samt för vätskor med högre ångtryck, cirka 1 bar vid 90 oC) |
|
DIN 12806 |
Hubbard pycnometer (för alla typer av viskösa vätskor som inte har för högt ångtryck, särskilt även för färg, lacker och bitumen) |
|
DIN 12807 |
Bingham pycnometer (för vätskor enligt DIN 12801) |
|
DIN 12808 |
Jaulmes pycnometer (särskilt för etanol-vattenblandningar) |
|
DIN 12809 |
Pycnometer with ground-in thermometer and capillary side tubes (för väts-kor som inte är för tjockflytande) |
|
DIN 53217 |
Testing of paints, varnishes and similar products; determination of density by pycnometer |
|
DIN 51757 |
Point 7: Testing of mineral oils and related materials; determination of density |
|
ASTM D 297 |
Section 15: Rubber products – chemical analysis |
|
ASTM D 2111 |
Method C: Halogenated organic compounds |
|
BS 4699 |
Method for determination of specific gravity and density of petroleum products (graduated bicapillary pycnometer method) |
|
BS 5903 |
Method for determination of relative density of petroleum products by the capillary-stoppered pycnometer method |
|
NF T 20-053 |
Chemical products for industrial use – Determination of density of solids in powder and liquids – Pykonometric method |
2.2 För fasta ämnen
|
ISO 1183 |
Metod B i Method for Determining the Density and Relative Density of Plastics excluding Cellular Plastics |
|
NF T 20-053 |
Chemical products for industrial use – Determination of density of solids in powder and liquids – Pycnometric method |
|
DIN 19683 |
Determination of the density of soils |
3. Luftjämförelsepyknometer
|
N |
Part 3: Prüfung von Anstrichstoffen und ähnlichen Beschichtungsstoffen: Pulverlack; Bestimmung der Dichte |
|
DIN 53243 |
Anstrichstoffe; Chlorhaltige Polymere; Prüfung |
A. 4. ÅNGTRYCK
1. METOD
De flesta av de beskrivna metoder som beskrivs är baserade på OECD:s riktlinjer för undersökning av kemikalier (se hänvisning 1). Grundprinciperna anges i hänvisningarna 2 och 3.
1.1 INLEDNING
Vid utförande av detta test är det värdefullt att ha förhandskunskap om ämnets struktur, smältpunkt och kokpunkt.
Det finns inte någon enskild metod som kan användas över hela ångtrycksområdet. Därför rekommenderas flera metoder för mätning av ångtryck från < 10-4 till 105 Pa.
Orenheter inverkar normalt på ångtrycket och graden av denna inverkan beror i hög grad på arten av orenhet.
Om provet innehåller flyktiga orenheter som kan inverka på resultatet kan ämnet renas. Det kan också vara lämpligt att ange ångtrycket för det tekniska materialet.
För vissa av de metoder som beskrivs här används apparater med metalldelar. Detta bör beaktas vid undersökning av frätande ämnen.
1.2 DEFINITIONER OCH ENHETER
Ett ämnes ångtryck definieras som mättnadstrycket över ett fast ämne eller en vätska. Vid termodynamisk jämvikt är ett rent ämnes ångtryck endast en funktion av temperaturen.
Den SI-enhet för tryck som används är pascal (Pa).
Nedan följer några enheter som tidigare har använts, och deras omvandlingsfaktorer:
|
1 Torr (= 1 mm Hg) |
= 1,333 × 102 Pa |
|
1 atmosfär |
= 0,013 × 105 Pa |
|
1 bar |
=105 Pa |
SI-enheten för temperatur är kelvin (K).
Den universella gaskonstanten R är 8,314 J mol-1 K-1.
Temperaturens beroende av ångtrycket beskrivs genom Clausius-Clapeyrons ekvation:

där:
|
p |
= |
ämnets ångtryck i pascal |
|
A Hv |
= |
ämnets forångningsvärme i J mol-1 |
|
R |
= |
den universella gaskonstanten i J mol-1 K-1 |
|
T |
= |
termodynamisk temperatur i K |
1.3 REFERENSÄMNEN
Man behöver inte använda referensämnen varje gång ett nytt ämne ska undersökas. Referensämnen ska först och främst användas för att då och då kontrollera metoden och för jämförelse med resultat som uppnåtts med andra metoder.
1.4 TESTMETODSPRINCIP
Till bestämning av ångtrycket föreslås sju metoder som kan tillämpas i olika ångtrycksområden. För varje metod bestäms ångtrycket vid olika temperaturer. I ett begränsat temperaturområde är logaritmen för en rent ämnes ångtryck en linjär funktion av det inverterade värdet av temperaturen.
1.4.1 Dynamisk metod
Med den dynamiska metoden mäts den kokpunkt som motsvarar ett givet tryck.
Rekommenderat område:
103 till 105 Pa.
Denna metod rekommenderas även för bestämning av normal kokpunkt och kan användas i detta syfte upp till 600 K.
1.4.2 Statisk metod
Med den statiska metoden bestäms ångtrycket i ett slutet system, som är i termodynamisk jämvikt vid en given temperatur. Denna metod lämpar sig för fasta ämnen och vätskor som består av en eller flera komponenter.
Rekommenderat område:
10 upp till 105 Pa.
Denna metod kan också användas i området 1 till 10 Pa, under förutsättning att försiktighet iakttas.
1.4.3 Isoteniskop
Denna standardiserade metod är också en statisk metod men den kan normalt inte användas för flerkomponents-system. Ytterligare information finns i ASTM metod D-2879-86.
Rekommenderat område:
100 till 105 Pa.
1.4.4 Effusionsmetod: Ångtrycksjämvikt
Den mängd ämne som avgår ur en cell per tidsenhet genom en öppning av känd storlek, bestäms under vakuum så att den mängd ämne som återvänder till cellen är försumbar (t.ex. genom mätning av den puls som en ångstråle framkallar på en känslig våg eller genom att mäta viktförlusten).
Rekommenderat område:
1C-3 till 1 Pa.
1.4.5 Effusionsmetod: genom viktförlust eller med en kylfälla
Denna metod ar baserad på bestämning av den mängd testämne som under ultravakuum strömmar ut från en Knudsen-cell (se hänvisning 4) genom en mikroöppning per tidsenhet i gasform. Volymen av den utströmmade ångan kan konstateras antingen genom att bestämma cellens viktförlust eller genom att kondensera ångan vid låg temperatur och bestämma den förångade mängden ämne genom kromatografisk analys. Ångtrycket beräknas med användning av Hertz-Knudsen-relationen.
Rekommenderat område:
10o till 1 Pa.
1.4.6 Gasmättnadsmetod
En ström av inert bärargas leds över ämnet på ett sådant sätt att den mättas med ångan. Den mängd material som en känd bärargas transporterar kan antingen mätas genom insamling i en lämplig fälla eller genom in-line analys. Utifrån detta kan ångtrycket vid en given temperatur beräknas.
Rekommenderat område:
10-4 till 1 Pa.
Denna metod kan också användas i området 1 till 10 Pa, under förutsättning att försiktighet iakttas.
1.4.7 Rotormetod
Med denna metod är det egentliga mätinstrumentet en liten stålkula som svävar i ett magnetfält och roterar med hög hastighet. Gastrycket utleds av den tryckavhängiga bromsningen av stålkulan.
Rekommenderat område:
10-4 till 0,5 Pa.
1.5 KVALITETSKRITERIER
I följande tabell jämförs de olika metoderna för bestämning av ångtrycket med hänsyn till användning, repeterbarhet, reproducerbarhet, mätområde och befintlig standard.
Tabell
Kvalitetskriterier
|
Ebulliometer |
Ämnen |
Uppskattad repeterbarhet (7) |
Uppskattad reproducerbarhet (7) |
Re kommenderat område |
Befintlig standard |
|||
|
Fasta |
Vätskor |
|||||||
|
Låg smältning |
ja |
Upp till 25 % |
Upp till 25 % |
103 Pa till 2 × 103 Pa |
— |
||
|
|
|
|
1 till 5 % |
1 till 5 % |
2 × 103 Pa till 105 Pa |
— |
||
|
ja |
ja |
5 till 10 % |
5 till 10 % |
10 Pa till 105 Pa (8) |
NFT 20-048 (5) |
||
|
ja |
ja |
5 till 10 % |
5 till 10 % |
102 Pa till 105 Pa |
ASTM-D 2879-86 |
||
|
ja |
ja |
5 till 20 % |
Upp till 50 % |
10-3 Pa till 1 Pa |
NFT 20-047 (6) |
||
|
ja |
ja |
10 till 30 % |
— |
10-3 Pa till 1 Pa |
— |
||
|
ja |
ja |
10 till 30 % |
Upp till 50 % |
10-4 Pa till 1 Pa |
— |
||
|
ja |
ja |
10 till 20 % |
— |
10-4 Pa till 0,5 Pa |
— |
||
1.6 METODBESKRIVNING
1.6.1 Dynamisk mätning
1.6.1.1 Apparatur
Mätinstrumentet består typiskt av ett kokkärl med ansluten kylare av glas eller metall (se figur 1), utrustning för mätning av temperaturen och utrustning för reglering och mätning av trycket. En typisk mätutrustning, som den som visas på ritningen, är gjord av värmebeständigt glas och består av fem delar:
Ett stort, delvis dubbelväggigt rör som består av ett slipat förband, en kylare, ett kylkärl och ett inlopp.
En glascylinder med en Cottrell-pump är monterad på rörets koksektion och har en grov yta av krossat glas för att undvika ”stötkokning”.
Temperaturen mäts med en lämplig temperaturgivare (t.ex. motståndstermometer eller termoelement med mantel) som har förts in i instrumentet helt till mätpunkten (se figur 1, nr 5) genom ett lämpligt inlopp (t.ex. slipat hanförband).
De nödvändiga anslutningarna görs till tryckreglerings- och mätutrustningen.
Kolven, som fungerar som en buffertvolym, är ansluten till mätapparaten med ett kapillärrör.
Kokkärlet värms upp med ett värmeelement (t.ex. värmepatron) som sätts in i glasapparaturen nedifrån. Önskad strömstyrka ställs in och mäts med ett termoelement.
Det vakuum på mellan 102 Pa och omkring 105 Pa som krävs ställs in med en vakuumpump.
En passande ventil används för att reglera trycket med luft eller kväve (mätområde ca 102 till 105 Pa) och för utluftning.
Trycket mäts med en manometer.
1.6.1.2 Mätförfarande
Ångtrycket mäts genom bestämning av provets kokpunkt vid olika angivna tryck mellan ungefär 103 och 105 Pa. En jämn temperatur under konstant tryck visar att kokpunkten är nådd. Skummande ämnens ångtryck kan inte mätas med denna metod.
Ämnet placeras i det rena torkade testkärlet. Det kan uppstå problem med fasta ämnen som inte är i pulverform, men detta kan ibland undvikas genom att uppvärmning av kylmanteln. Sedan kärlet fyllts sammanflänsas apparaten och ämnet avgasas. Därefter ställs lägsta önskat tryck in och uppvärmningen sätts på. Samtidigt ansluts temperaturgivaren till en skrivare.
Jämvikt har nåtts när en konstant kokpunkt registreras vid konstant tryck. Man ska särskilt se till att stötkokning inte förekommer. Dessutom måste fullständig kondensation ske i kylaren. Vid bestämning av ångtrycket av fasta ämnen med låg smältpunkt ska man se till att kylaren inte täpps till.
Sedan jämviktspunkten har registrerats ställs ett högre tryck in. Processen fortsätter på samma sätt tills 105 Pa har uppnåtts (sammanlagt ungefär 5–10 mätpunkter). I kontrollsyfte ska jämviktspunkterna upprepas vid avtagande tryck.
1.6.2 Statisk mätning
1.6.2.1 Apparatur
Apparaten består av en provbehållare och ett uppvärmnings- och kylsystem för att reglera och mäta provets temperatur. Apparaten omfattar dessutom utrustning för inställning och mätning av trycket. Grundprinciperna framgår av figur 2a och 2b.
Provkammaren (se figur 2a) är på den ena sidan avgränsad av en lämplig högvakuumventil och på den andra sidan förbunden med ett U-rör som innehåller lämplig manometervätska. Den ena anden av U-roret är ansluten till vakuumpumpen, till kvävecylindern eller utluftningsventilen och en manometer.
En tryckmätare med en separat visare kan användas i stället för U-röret (se figur 2b).
Provets temperatur styrs genom att provkärlet, inklusive ventil och U-rör, placeras i ett bad som har en konstant temperatur av ±0,2 K. Temperaturen mäts på provkärlets utsida eller i själva kärlet.
En vakuumpump med en kylfälla för evakuering av apparaten används.
I metod 2a mäts ämnets ångtryck indirekt med en nollindikator. Därvid beaktas att densiteten av vätskan i U-röret ändrar sig vid stora temperaturförändringar.
Följande vätskor är lämpliga som nollindikatorer i U-röret och väljs med hänsyn till tryckområdet och testämnets kemiska egenskaper: silikonvätskor och ftalater. Testämnet får inte kunna lösas väsentligt i eller reagera med vätskan i U-röret.
I manometern kan man använda kvicksilver i området från normalt lufttryck till 102 Pa, medan silikonvätskor och ftalater är lämpliga från 102 Pa ned till 10 Pa. Värmebeständiga membrankondensatormanometrar kan till och med användas under 10-1 Pa. Det finns även andra tryckmätare som kan användas under 102 Pa.
1.6.2.2 Mätförfarande
Före mätningen ska alla delar av apparaten i figur 2 rengöras och torkas noggrant.
Vid metod 2a fylls U-röret med önskad vätska, som avgasas vid förhöjd temperatur före avläsning.
Testämnet placeras i apparaten som sedan stängs, varvid temperaturen sänks tillräckligt för avgasning. Temperaturen måste vara så pass låg att luften sugs ut, men i fråga om flerkomponentsystem får det inte ske någon ändring av materialets sammansättning. Vid behov kan jämvikt erhållas snabbare genom omrörning.
Provet kan underkylas med t.ex. flytande kväve (varvid försiktighet iakttas för att undvika kondensering av luft eller pumpvätska) eller en blandning av etanol och kolsyresnö. Vid mätning av låga temperaturer används ett temperaturreglerat bad som är anslutet till en ultrakryomat.
Med ventilen över provkammaren i öppet läge pumpas därefter den inneslutna luften ut ur apparaten i flera minuter. Därefter stängs ventilen och provet kyls till lägsta vald temperatur. Vid behov upprepas avgasningsproceduren.
När provet värms upp ökar ångtrycket. Detta ändrar jämvikten hos vätskan i U-röret. För att kompensera detta leds kväve eller luft in i apparaten via en ventil tills tryckindikatorvärskan åter är på noll. Det tryck som krävs för detta avläses på en precisionsmanometer vid rumstemperatur. Detta tryck motsvarar ämnets ångtryck vid den särskilda mättemperatur.
Metod 2b är en liknande metod, men ångtrycket avläses direkt.
Ångtryckets temperaturberoende bestäms med tillräckligt små temperaturintervall (omkring 5 till 10 mätpunkter totalt) upp till önskat maximum. Lågtemperaturmätningar måste upprepas i kontrollsyfte.
En eventuell avvikelse mellan de nya värdena och den kurva som erhållits vid stigande temperatur kan bero på att
|
1. |
provet fortfarande innehåller luft (t.ex. ämnen med hög viskositet) eller lågkokande ämnen, som frigjorts under uppvärmningen och kan avlägsnas genom ytterligare underkylning, |
|
2. |
nedkylningstemperaturen inte är tillräckligt låg, varvid flytande kväve används som kylmedel; om antingen 1 eller 2 är fallet måste mätningarna upprepas; |
|
3. |
det sker en kemisk reaktion i ämnet i det undersökta temperaturområdet (t.ex. dekomposition eller polymerisation). |
1.6.3 Isoteniskop
En fullständig beskrivning av denna metod finns i hänvisning 7. Mätanordningens princip framgår av figur 3. I likhet med den statiska metoden, som beskrivits i 1.6.2, lämpar sig isoteniskopet för undersökning av fasta ämnen eller vätskor.
Vid mätning av vätskor fungerar själva ämnet som hjälpmanometervätska. Den mängd ämne som behövs för att fylla klotet och manometersektionens korta arm placeras i isoteniskopet. Isoteniskopets förbinds med ett vakuumsystem, evakueras och fylls med kväve. Evakueringen och rensningen av systemet upprepas två gånger så att alla kväverester avlägsnas. Det fyllda isoteniskopet placeras horisontellt så att provet fördelas i ett tunt lager i provkolven och manometersektionen (U-delen). Trycket i systemet minskas till 133 Pa och provet värms försiktigt upp tills det nästan kokar (avlägsnande av upplösta fixerade gaser). Isoteniskopet placeras sedan så att provet återgår till klotet och manometerns korta arm och båda är helt fyllda med vätska. Samma tryck som vid avgasning upprätthålls. Provkolvens utdragna topp värms försiktigt upp med en liten låga tills den ånga som bildas utvidgas tillräckligt för att pressa ut en del av provet från klotets översta del och manometerarm till isoteniskopets manometersektion, varvid ett ångfyllt, kvävefritt område skapas.
Isoteniskopet placeras sedan i ett bad med konstant temperatur och kvävetrycket ställs in tills det motsvarar provets tryck. Att tryckjämvikt föreligger framgår av isoteniskopets manometersektion. Vid jämvikt motsvarar kvävets ångtryck ämnets ångtryck.
När det gäller fasta ämnen används, beroende på tryck och temperaturområde, de manometervätskor som anges i 1.6.2. Den avgasade manometervätskan fylls in i utbuktningen på isoteniskopets långa arm. Därefter placeras det fasta testämnet i klotet och avgasas vid förhöjd temperatur. Sedan lutas isoteniskopet så att manometer-vätskan kan flyta in i U-röret. Mätningen av ångtrycket som en funktion av temperaturen utförs enligt 1.6.2.
1.6.4 Effusionsmetod: Ångtrycksvåg
1.6.4.1 Apparatur
I litteraturen (se hänvisning 1) beskrivs flera olika apparaturutformningar. Den apparat som beskrivs här illustrerar de principer som används (se figur 4). Figur 4 visar apparaturens huvudkomponenter: en högvakuumbehållare av rostfritt stål eller glas, utrustning för att producera och mäta vakuum och inbyggda komponenter för mätning av ångtrycket på en våg. Apparaturen innehåller följande inbyggda komponenter:
|
— |
En förångningsugn med fläns och roterande intag. Förångningsugnen är en cylindrisk behållare gjort av t.ex. koppar eller en kemiskt beständig legering med god värmeledningsförmåga. Man kan också använda en glasbehållare med kopparvägg. Ugnen har en diameter av ca 3 till 5 cm och en höjd av 2 till 5 cm. Det finns en till tre öppningar av olika storlek för ångströmmen. Ugnen värms upp antingen av en värmeplatta nedtill eller av en värmeslinga runt utsidan. För att undvika avledning av värme till bottenplattan är värmeaggregatet fastgjort i denna med en metall med låg värmeledningsförmåga (nickel-silver eller kromnickelstål), t.ex. ett nickel-silverrör fastsatt i ett roterade intag, om man använder en ugn med flera öppningar. En sådan uppställning har den fördelen att det möjliggör införande av en kopparstång, vilket tillåter kylning från utsidan med ett vattenbad, |
|
— |
Om ugnslocket, som är gjort av koppar, har tre öppningar med olika tvärsnitt, inbördes förskjutna 90 oC, kan olika ångtrycksområden inom den totala mätområdet täckas (öppningar med diametrar på ca 0,30 till 4,50 mm). Stora öppningar används för lågt ångtryck och vice versa. Genom att vrida ugnen kan önskadugnsöppning eller ett mellanläge i ångströmmen (ugnsöppning – skärm – vågskål) ställas in och molekylstrå-len riktas eller avledas genom ugnsöppningen mot vågskålen. För att kunna mäta ämnets temperatur mon-teras ett termoelement eller en motståndstermometer på lämpligt ställe. |
|
— |
Ovanför skärmen finns en vågskål med en mycket känslig mikrovåg (se nedan). Vågskålen är omkring30 mm i diameter. Guldpläterad aluminium är ett lämpligt material. |
|
— |
Vågskålen omges av en cylindrisk kylbox av mässing eller koppar. Beroende på typ av våg är den förseddmed öppningar för vågbalken och en skärmöppning för molekylströmmen och den ska garantera total kon-densation av ångan på vågskålen. Värmen leds bort till utsidan av t.ex. en kopparstång ansluten till kyl-boxen. Stången leds genom bottenplattan och är värmeisolerad från denna med t.ex. ett rör av kromnick-elstål. Stången sänks ned i ett dewarkärl med flytande kväve under bottenplattan, eller så leds flytande kvävegenom stången. Temperaturen i kylboxen hålls på så sätt vid omkring –120 oC. Vågskålen kyls ned uteslu-tande genom strålning, vilket är lämpligt för det tryckområde som undersöks (nedkylning omkring 1 timmeinnan mätning påbörjas). |
|
— |
Vågen placeras ovanför kylboxen. Lämpliga vågar är t.ex. en högkänslig 2-armad elektronisk mikrovåg (sehänvisning 8) eller ett högkänsligt vridspoleinstrument (se OECD Test Guideline 104, Edition 12.05.81). |
|
— |
På bottenplattan finns också elektriska anslutningar för termoelement (eller motståndstermometrar) och vär-meslingor. |
|
— |
Ett vakuum erhålls i kärlet med hjälp av en partiell vakuumpump eller en högvakuumpump (krävt vakuumomkring 1 till 2 × 10-3 Pa, erhållet efter 2 timmars pumpning). Trycket regleras med en lämplig joniserings-manometer. |
1.6.4.2 Mätförfarande
Kärlet fylls med testämnet och locket stängs. Skärmen och kylboxen skjuts på plats över ugnen. Apparaten stängs och vakuumpumparna sätts på. Sluttrycket innan mätningen påbörjas ska vara ungefär 10-4 Pa. Nedkylning av kylboxen sätts igång vid 10-2 Pa .
När tillräckligt vakuum har uppnåtts inleds kalibreringsserien vid den lägsta temperaturen. Den nödvändiga öppningen öppnas och ångstrålen passerar skärmen, som är direkt monterad över locket, och träffar sedan den nedkylda vägskålen. Vågskålen ska vara så stor att hela den ångsträle som leds genom skärmen träffar den. Ång-strålens impuls överför en kraft på vågskålen och molekylerna kondenseras på dess nedkylda yta.
Impulsen och den samtida kondensationen frambringar en signal på skrivaren. Utvärdering av denna signal ger två upplysningar:
|
1. |
Med den särskilda apparat som beskrivs här bestäms ångtrycket direkt från den impuls som överförs till vågskålen (molekylvikten behöver inte vara känd [se hänvisning 2]). Vid utvärdering av avläsningarna måsteman ta hänsyn till geometriska faktorer, såsom ugnsöppning och molekylstrålens vinkel. |
|
2. |
Samtidigt kan den kondenserade vikten mätas och avdunstningshastigheten beräknas utifrån detta. Ång-trycket kan även beräknas utifrån avdunstningshastigheten och molekylvikten med användning av Hertzekvation (se hänvisning 2): |

där:
|
G |
= |
avdunstningshastig het (kg s-1 m-2) |
|
M |
= |
molekylvikt (g mol-1) |
|
T |
= |
temperatur (K) |
|
R |
= |
universell gaskonstant Jmol-1 K-1) |
|
p |
= |
ångtryck (Pa) |
När tillräckligt vakuum har uppnåtts börjar serien av mätningar vid den lägsta önskade mättemperaturen.
För ytterligare mätningar ökas temperaturen med små intervaller tills den högsta önskade temperaturen är uppnådd. Därefter kyls provet ned igen och en andra ångtryckskurva kan registreras. Om den andra testomgången inte bekräftar resultaten från den första omgången kan detta bero på att ämnet bryts ned i det undersökta temperaturområdet.
1.6.5 Effusionsmetod: Viktförlust
1.6.5.1 Apparatur
En effusionsapparat består av följande baskomponenter:
|
— |
En behållare som kan termostatregleras och evakueras och i vilken effusionscellerna placeras. |
|
— |
En högvakuumpump (t.ex. diffusionspump eller turbomolekylärpump) med vakuummätare. |
|
— |
En kylfålla med flytande kväve eller kolsyresnö. |
En elektriskt uppvärmd vakuumbehållare av aluminium med 4 effusionsceller av rostfritt stål visas i figur 5 som ett exempel. Den rostfria folien som är omkring 0,3 mm tjock har en effusionsöppning med en diameter av 0,2 till 1,0 mm och är ansluten till effusionscellen med ett skruvlock.
1.6.5.2 Mätförfarande
Referens- och testämnet tillsätts i varje effusionscell, metallmembranet med öppningarna sätts fast med skruvlocket, och varje cell vägs till 0,1 mg noggrannhet. Cellen placeras i den termostatreglerade apparaten som sedan evakueras till mindre än en tiondedel av det förväntade trycket. Med i förväg fastställda intervaller på mellan 5 och 30 timmar tillförs luft i apparaten och viktförlusten i effusionscellerna bestäms genom vägning.
För att säkerställa att resultaten inte påverkas av flyktiga orenheter vägs cellerna vid fastställda tidsintervaller för att kontrollera att avdunstningshastigheten är konstant under minst två sådana tidsintervaller.
Ångtrycket p i effusionscellen beräknas genom

där:
|
p |
= |
ångtrycket (Pa) |
|
m |
= |
massan av den mängd ämne som lämnar cellen under tiden t (kg) |
|
t |
= |
tiden (s) |
|
A |
= |
öppningens area (m2) |
|
K |
= |
korrektionsfaktor |
|
R |
= |
universell gaskonstant Jmol-1 Kh-1) |
|
T |
= |
temperaturen (K) |
|
M |
= |
molekylvikten (kg mol-1) |
Korrektionsfaktorn K beror på förhållandet mellan den cylindriska öppningens längd och radie:
|
Förhållande: |
0,1 |
0,2 |
0,6 |
1,0 |
2,0 |
|
K |
0,952 |
0,909 |
0,771 |
0,672 |
0,514 |
Ovanstående ekvation kan också skrivas som

där  är effusionseelkonstanten.
är effusionseelkonstanten.
Denna effusionscellkonstant E kan bestämmas med referensämnen (2,9) med användning av följande ekvation:
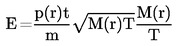
där:
|
p(r) |
= |
referensämnets ångtryck (Pa) |
|
M(r) |
= |
referensämnets molekylvikt (kg mol-1) |
1.6.6 Gasmättnadsmetod
1.6.6.1 Apparatur
En typisk apparat som används till detta test består av de komponenter som visas i figur 6a och beskrivs nedan (se hänvisning 1).
Inert gas:
Bärargasen får inte reagera kemiskt med testämnet. Normalt är det tillräckligt att använda kväve för detta syfte men ibland kan andra gaser krävas (se hänvisning 10). Den använda gasen ska vara torr (se figur 6a, position 4: Givare. Relativ fuktighet).
Flödesreglering:
Ett lämpligt gasregleringssystem krävs för att säkerställa ett konstant och väljbart flöde genom mättnadskolonnen.
Fällor för uppsamling av ånga:
Dessa är beroende av det aktuella provets egenskaper och den valda analysmetoden. Ångan ska infångas kvantitativt och i en form som medger efterföljande analys. För vissa provämnen lämpar sig fällor innehållande vätskor som hexan eller etylenglykol. För andra kan fasta absorptionsmedel användas.
Som alternativ till uppsamling av ånga och efterföljande analys kan in-line analystekniker, t.ex. kromatografi, användas för kvantitativ bestämning av den mängd material som en känd mängd bärargas för med sig. Dessutom kan provets förlust av massa mätas.
Värmeväxlare:
För mätning vid olika temperaturer kan det vara nödvändigt att inkludera en värmeväxlare i utrustningen.
Mättnadskolonn:
Provämnet appliceras i form av losningsbeläggning på ett lämpligt inert bärarmatenal. Det belagda bärarmaterialet packas i mättnadskolonnen, vars dimensioner och flödeshastighet ska vara sådana att en fullständig mättnad av bärargasen säkerställs. Mättnadskolonnen ska vara termostatreglerad. För mätningar över rumstemperatur ska stycket mellan mättnadskolonnen och fällorna uppvärmas för att förhindra kondensation av provämnet.
För att minska masstransport genom diffusion kan ett kapillärrör placeras efter mättnadskolonnen (se figur 6b).
1.6.6.2 Mätförfarande
Förberedelse av mättnadskolonnen:
En upplösning av provämnet i ett mycket flyktigt lösningsmedel tillsätts till en lämplig mängd bärarmaterial. Tillräckligt med provämne ska tillsättas för att upprätthålla mättnaden så länge testet pågår. Lösningsmedlet avdunstar helt i luft eller i en roterande förångare och det noggrant blandade materialet tillsätts till mättnadskolonnen. Efter termostatreglering av provet leds kväve genom apparaten.
Mätning:
Fällorna eller in-line detektoren ansluts till kolonnens utlopp och tiden registreras. Flödeshastigheten kontrolleras i början och med jämna mellanrum under testet med en bubbelmätare (eller kontinuerligt med en massflö-desmätare).
Trycket vid mättnadskolonnens utgångsöppning ska mätas. Detta kan göras
|
a) |
genom att en tryckmätare insätts mellan mättnadskolonnen och fällorna (vilket kan vara otillfredsställandeeftersom det ökar dödvolymen och adsorptionsytan), eller |
|
b) |
genom att bestämma tryckfallet över det använda systemet av fällor som funktion av flödeshastigheten i ettseparat test (eventuellt inte så lyckat för vätskefällor). |
Den tid som krävs till uppsamling av den mängd provämne som behövs för de olika analysmetoderna, bestäms vid preliminära försök eller genom uppskattningar. Som ett alternativ till uppsamling av ämnet för senare analys kan kvantitativa in-line analystekniker användas (t.ex. kromatografi). För beräkning av ångtrycket vid en given temperatur utförs preliminära försök för att bestämma den maximala flödeshastigheten vid vilken bärargasen mättas fullständigt med ämnets ånga. Detta säkerställs om bärargasen leds genom mättnadskolonnen så pass sakta att en lägre hastighet inte ger ett större beräknat ångtryck.
Valet av analysmetod bestäms av arten av det ämne som ska undersökas (t.ex. gaskromatografi eller gravime-tri).
Den mängd ämne som en känd volym bärargas transporterar bestäms.
1.6.6.3 Beräkning av ångtryck
Ångtrycket beräknas utifrån ångans densitet, W/V, genom ekvationen:

där;
|
p |
= |
ångtrycket (Pa) |
|
W |
= |
vikten av adsorberat provämne (g) |
|
V |
= |
volymen mättad gas (m3) |
|
R |
= |
universell gaskonstanten (Jmol-1 K-1) |
|
T |
= |
temperaturen (K) |
|
M |
= |
testämnets molekylvikt (g mol-1). |
De mätta volymerna ska korrigeras för tryck- och temperaturskillnader mellan flödesmätaren och den termostatreglerade mättnadskolonnen. Om flödesmätaren är placerad nedströms från ångfällan kan det vara nödvändigt med korrektioner för att ta hänsyn till eventuella avdunstade beståndsdelar från fällorna (se hänvisning 1).
1.6.7 Rotormetod (se hänvisning 8, 11, 13)
1.6.7.1 Apparatur
Rotormetoden kan utföras med en viskocitetsmätare med en roterande kula enligt figur 8. En schematisk ritning av försöksuppställningen visas i figur 7.
Mätapparaten består typiskt av ett mäthuvud med roterande kula placerad i en termostatreglerad inhägnad (reglerad inom 0,1 oC). Provbehållaren placeras i en termostatreglerad inhägnad (reglerad inom 0,01 oC) och alla andra delar i utrustningen hålls vid en högre temperatur för att förhindra kondensation. En högvakuumpump-anordning ansluts till systemet genom högvakuumventiler.
Mäthuvudet består av en stålkula (4 till 5 mm i diameter) i ett rör. Kulan hålls stabilt svävande av ett magnetfält, normalt genom en kombination av permanentmagneter och styrspolar.
Kulan sätts i rörelse med hjälp av roterande fält som produceras av spolarna. Rotationshastigheten mäts med detektorspolar, som mäter den alltid närvarande svaga tvärmagnetiseringen av kulan.
1.6.7.2 Mätförfarande
När kulan har nått en bestämd rotationshastighet v(o) (normalt omkring 400 varv per sekund), avbryts energitillförseln, och gasfriktionen börjar retardera kulans rotation.
Minskningen av rotationshastighet mäts som en funktion av tiden. När den friktion som orsakats av den magnetiska suspensionen är försumbar i jämförelse med gasfriktionen, bestäms gastrycket p genom:
![]()
där:
|
|
= |
gasmolekylernas medelhastighet |
|
r |
= |
kulans radie |
|
p |
= |
kulans massdensitet |
|
o |
= |
koefficient av tangentiell impulsöverföring (ε = 1 för en ideal sfärisk kulyta) |
|
t |
= |
tid |
|
v(t) |
= |
rotationshastighet efter tid t |
|
v(o) |
= |
initial rotationshastighet |

Denna ekvation kan även skrivas

där tn, tn.-1 är den tid som krävs för ett givet antal N varv. Dessa tidsintervaller tn, tn-1 följer på varandra och tn>tn-1.
Gasmolekylens medelhastighet  bestäms genom
bestäms genom

där:
|
T |
= |
temperaturen |
|
R |
= |
universella gaskonstanten |
|
M |
= |
molekylvikten |
2. DATA
Ångtrycket från någon av föregående metoder ska bestämmas vid minst två temperaturer. För att kontrollera ångtryckskurvans linearitet är det önskvärt med tre eller flera bestämningar i området 0–50 oC.
3. RAPPORTERING
Testrapporten ska, om möjligt, innehålla följande uppgifter:
|
— |
Använd metod. |
|
— |
Exakt specificering av ämnet (beteckning och orenheter) och eventuella inledande reningsåtgärder. |
|
— |
Minst två värden för ångtryck och temperatur, helst i området 0–50 oC. |
|
— |
Alla rådata. |
|
— |
En kurva över log p, som funktion av l/T. |
|
— |
En beräkning av ångtrycket vid 20–25 oC. |
Om en övergång (tillståndsändring, nedbrytning) iakttas, ska följande uppgifter antecknas:
|
— |
Förändringens art. |
|
— |
Temperatur vid vilken förändringen inträffar vid atmosfäriskt tryck. |
|
— |
Ångtryck vid 10 och 20 oC under övergångstemperaturen och 10 och 20 oC över denna temperatur (såvidainte övergången sker från fast ämne till gas) |
Alla uppgifter och upplysningar av betydelse för tolkningen av resultaten ska rapporteras, särskild vad gäller orenheter och ämnets fysiska tillstånd.
4. HÄNVISNINGAR
|
1. |
OECD, Paris, 1981, Test Guideline 104, rådets beslut K(81) 30 slutlig. |
|
2. |
Ambrose, D. i B. Le Neindre, B. Vodar, (Eds.): Experimental Thermodynamics, Butterworths, London,1975, vol. II. |
|
3. |
R. Weissberger ed.: Technique of Organic Chemistry, Physical Methods of Organic Chemistry, 3rd ed.,kapitel IX, Interscience Publ., New York, 1959, vol. I, del 1. |
|
4. |
Knudsen, M. Ann. Phys. Lpz., 1909, vol. 29, 1979; 1911, vol. 34, s. 593. |
|
5. |
NF T 20-048 AFNOR (Sept. 85). Chemical products for industrial use – Determination of vapour pressureof solids and liquids within range from 10-1 tu 105 Pa – Static method. |
|
6. |
NF T 20-047 AFNOR (Sept. 85). Chemical products for industrial use – Determination of vapour pressureof solids and liquids within range from 10-3 to 1 Pa – Vapour pressure balance method. |
|
7. |
ASTM D 2879-86, Standard test method for vapour pressure temperature relationship and initial decomposition temperature of liquids by isoteniscope. |
|
8. |
G. Messer, P. Röhl, G. Grosse, W. Jitschin, J. Vac. Sci. Technol.(A), 1987, vol. 5 (4), s. 2440. |
|
9. |
Ambrose, D.; Lawrenson, I. J.; Sprake, C. H. S, J. Chem. Thermodynamics 1975, vol. 7, s. 1173. |
|
10. |
B. F. Rordorf. Thermochimica Acta, 1985, vol. 85, s. 435. |
|
11. |
G. Comsa, J. K. Fremerey och B. Lindenau, J. Vac. Sci. Technol., 1980, vol. 17 (2), s. 642. |
|
12. |
G. Reich, J. Vac. Sci. Technol., 1982, vol. 20 (4), s. 1148. |
|
13. |
J. K. Fremerey, J. Vac. Sci. Technol. (A), 1985, vol. 3 (3), s. 1715. |
Tillägg 1
Utvärderingsmetod
INLEDNING
Man kan använda beräknade värden av ångtrycket för att
|
— |
bestämma vilken försöksmetod som är lämplig, |
|
— |
göra en bedömning eller fastställa ett gränsvärde om man av tekniska orsaker (inbegripet när ångtrycket är mycket lågt) inte kan använda försöksmetoder, |
|
— |
konstatera i vilket fall det är berättigat att avstå från försöksmätningar till följd av att ångtrycket sannolikt kommer att vara < 10-5 Pa vid omgivningstemperatur. |
BEDÖMNINGSMETOD
Vätskors och fasta ämnens ångtryck kan bedömas genom användning av den modifierade Watson-korrelationen (se hänvisning a). De enda testdata som är nödvändiga är den normala kokpunkten. Metoden kan användas vid ett tryckområde från 105 Pa till 10-5 Pa.
Närmare information om metoden finns i ”Handbook of Chemical Property Estimation Methods” (se hänvisning b).
BERÄKNINGSFÖRFARANDE
I enlighet med hänvisning b beräknas ångtrycket enligt följande:
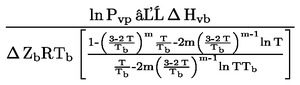
där:
|
T |
= |
Den gällande temperaturen |
|
Tb |
= |
Normal kokpunkt |
|
Pvp |
= |
Ångtryck vid temperatur T |
|
Δ Hvh |
= |
Förångningsvärme |
|
Δ Zb |
= |
Kompressionsfaktor (beräknad till 0,97) |
|
m |
= |
Empirisk faktor beroende av det fysiska tillståndet hos den gällande temperaturen |
Dessutom:

där KF är en empirisk faktor som beaktar ämnets polaritet. I hänvisning b finns det en förteckning med KF-faktorer för flera typer av föreningar.
Det är ganska vanligt med data som anger kokpunkt vid sänkt tryck. I sådana fall kan man i enlighet med hänvisning b beräkna ångtrycket på följande sätt:

där T1 är kokpunkten vid sänkt tryck P1.
RAPPORT
När utvärderingsmetoden används ska rapporten inbegripa en omfattande dokumentation av beräkningen.
HÄNVISNINGAR
|
1. |
K. M. Watson, Ind. Eng. Chem; 1943, vol. 35, s. 398. |
|
2. |
W. J. Lyman, W. F. Reehl, D, H. Rosenblatt. Handbook of Chemical Property Estimation Methods Mc Graw-Hill, 1982. |
Tillagg 2
Figur 1
Apparat för bestämning av ångtryckskurvan enligt den dynamiska metoden

Figur 2a
Apparat för bestämning av ångtryckskurvan enligt den statiska metoden (med U-rörsmanometer)

Figur 2b
Apparat för bestämning av ångtryckskurvan enligt den statiska metoden (med tryckindikator)

Figur 3
Isoteniskop (se hänvisning 7).

Figur 4
Apparat för bestämning av ångtryckskurvan enligt ångtrycksvågsmetoden

Figur 5
Exempel på en apparat för förångning vid lågt tryck genom effusionsmetoden, med en effusionscellvolym av 8 cm

Figur 6a
Exempel på ett flödessystem för bestämning av ångtryck genom gasmättnadsmetoden

Figur 6b
Exempel på ett system för bestämning av ångtryck genom gasmättnadsmetoden med kapillärrör placerat efter gasmättnadskammaren

Figur 7
Exempel på försöksanordning för rotormetoden

Figur 8
Exempel på mathuvud med roterande kula

A.5 YTSPÄNNING
1. METOD
De beskrivna metoderna är baserade på OECD:s riktlinjer för undersökning av kemikalier (se hänvisning 1). Grundprinciperna anges i hänvisning 2.
1.1 INLEDNING
De beskrivna metoderna används för matning av ytspänningen hos vattniga lösningar.
Vid utförande av dessa test är det värdefullt att ha förhandskunskap om ämnets löslighet i vatten, dess struktur, hydrolysegenskaper och kritiska koncentrationer för micellbildning.
Följande metoder kan tillämpas på de flesta kemiska ämnen, oavsett renhetsgrad.
Mätning av ytspänning med ringtensiometermetoden är begränsad till vattniga lösningar med en dynamisk viskositet på mindre än 200 m Pa s.
1.2 DEFINITIONER OCH ENHETER
Den fria entalpinytan per ytareaenhet betecknas som ytspänning.
Ytspänningen anges som:
N/m (SI-enhet) eller
mN/m (SI-underenhet).
1 N/m = 103 dyn/cm
1 mN/m = dyn/cm i det gamla cgs-systemet.
1.3 REFERENSÄMNEN
Man behöver inte använda referensämnen varje gång ett nytt ämne ska undersökas. Referensämnen ska först och främst användas för att då och då kontrollera metoden och för jämförelse med resultat som uppnåtts med andra metoder.
I hänvisningarna 1 och 3 anges referensämnen som täcker ett stort antal ytspänningar.
1.4 TESTMETODSPRINCIPER
Metoderna är baserade på mätning av den största kraft som ska utövas vertikalt på en bygel eller ring i kontakt med vätskeytan av ett prov som ska undersökas i ett mätkärl för att bryta denna kontakt, eller den vertikala kraft som behöver utövas på en platta vars ena kant har kontakt med vätskeytan, för att lösgöra film som har bildats.
Ämnen vars löslighet i vatten är minst 1 mg/liter testas i en vattnig lösning vid en enda koncentration.
1.5 KVALITETSKRITERIER
Dessa metoder kan ge större noggrannhet än vad som kan förväntas krävas vid miljöbetingade undersökningar.
1.6 METODBESKRIVNING
En lösning av ämnet framställs i destillerat vatten. Koncentrationen av denna lösning ska vara 90 % av ämnets mättnadskoncentration i vatten. Om denna koncentration är större än 1 g/1 används en koncentration av 1 g/l för testet. Ämnen med en löslighet i vatten på mindre än 1 mg/l behöver inte testas.
1.6.1 Plattmetoden
Se ISO-304 och NF T 73-060 (Surface active agents – determination of surface tension by drawing up liquid films).
1.6.2 Bygelmetoden
Se ISO 304 och NF T 73-060 (Surface active agents – determination of surface tension by drawing up liquid films).
1.6.3 Ringmetoden
Se ISO 304 och NF T 73-060 (Surface active agents – determination of surface tension by drawing up liquid films).
1.6.4 OECD-harmoniserad ringmetod
1.6.4.1 Apparatur
Denna mätning kan göras med kommersiellt tillgängliga tensiometrar och består av följande delar:
|
— |
flyttbart provbord, |
|
— |
kraftmätningssystem, |
|
— |
mätkropp (ring), |
|
— |
mätkärl. |
1.6.4.1.1
Det flyttbara provbordet används som stöd till det temperaturreglerade mätkärl som innehåller den vätska som ska undersökas. Det är monterat på ett stativ tillsammans med kraftmätningssystemet.
1.6.4.1.2
Kraftmätningssystemet (se figur) är placerat över provbordet. Osäkerheten vid kraftmätningen får inte vara större än ± 10-6 N, motsvarande en felmarginal på ±0,1 mg viktmätning. Oftast är mätskalan på kommersiellt tillgängliga tensiometrar indelad i mN/m så att ytspänningen kan avläsas direkt i mN/m med en noggrannhet på 0,1 mN/m.
1.6.4.1.3
Ringen är vanligtvis gjord av en tråd av platina-indium, ungefär 0,4 mm tjock och med en medelomkrets på 60 mm. Trådringen är upphängd horisontellt på en metallstång och en bärarm av tråd, som utgör anslutningen till kraftmätningssystemet (se figur).
Figur
Mätkropp
(Alla mått är angivna i mm)

1.6.4.1.4
Mätkärlet som innehåller den testlösning som ska mätas ska vara ett temperaturreglerat glaskärl. Det ska vara så utformat att temperaturen på provlösningen och på gasfasen ovanför dess yta förblir konstant under mätningen så att provet inte kan avdunsta. Cylindriska glaskärl med en innerdiameter på minst 45 mm kan användas.
1.6.4.2 Iordningställande av apparaturen
1.6.4.2.1
Glaskärl ska rengöras noggrant. Vid behov ska de diskas med varm kromsvavelsyra och därefter med tjockflytande fosforsyrelösning (83–98 % per vikt H3PO4), sedan noggrant sköljas i kranvatten och slutligen diskas i omdestillerat vatten tills en neutral reaktion erhålls. Slutligen ska de torkas eller sköljas med en del av den provvätska som ska mätas.
Ringen ska först sköljas noggrant i vatten för att avlägsna eventuella vattenlösliga ämnen. Därefter ska den nedsänkas hastigt i kromsvavelsyra, tvättas i omdestillerat vatten tills en neutral reaktion erhålls och slutligen upphettas hastigt över en metanollåga.
Anmärkning:
Förorening med ämnen som inte löses eller förstörs av kromsvavelsyra eller fosforsyra, t.ex. silikonpreparat, ska avlägsnas med hjälp av lämpligt organiskt lösningsmedel.
1.6.4.2.2
Kalibreringen av apparaten består i att nollpunkten kontrolleras och justeras så att instrumentets indikering medger en tillförlitlig avläsning i mN/m.
Montering;
Apparaten ska planriktas, t.ex med användning av ett vattenpass på tensiometerns fot, varefter nivåskruvarna på foten justeras.
Nollpunktsjustering:
När ringen har monterats på apparaten men innan den nedsänks i vätskan, ska tensiometerindikeringen justeras till noll och ringen kontrolleras så att den är parallell med vätskeytan. För detta ändamål kan vätskeytan användas som spegel.
Kalibreringar:
Själva provkalibreringen kan utföras genom en av följande två metoder:
|
a) |
Användning av en vikt: detta är ett förfarande där ryttare av känd vikt mellan 0,1 och 1,0 g placeras på ringen. Kalibreringsfaktorn Φa, med vilken alla instrumentavläsningar ska multipliceras, bestäms enligt följande ekvation (se hänvisning 1):
där:
|

 (mN/m)
(mN/m)|
b) |
Användning av vatten: detta är ett förfarande där det används rent vatten vars ytspänning vid, t.ex. 23 oC, är lika med 72,3 mN/m. Denna metod är snabbare att utföra än viktkalibreringen men risken finns alltid att vattnets ytspänning har ändrats genom förorening av ytaktiva substanser. Kalibreringsfaktorn Φb med vilken alla instrumentavläsningar ska multipliceras, bestäms enligt följande ekvation (2):
där:
|

1.6.4.3 Iordningställande av prov
Vattniga lösningar framställs med passande koncentrationer av de ämnen som ska undersökas. Lösningarna får inte innehålla några olösta ämnen.
Lösningarna ska hållas vid en konstant temperatur (±0,5 oC). Eftersom en lösnings ytspänning i mätkärlet ändras med tiden ska flera mätningar göras vid olika tidpunkter och en kurva ritas över ytspänning som en funktion av tiden. När ytspänningen inte längre ändras har ett jämviktstillstånd uppnåtts.
Damm och gasformig förorening av andra ämnen påverkar mätningen. Arbetet ska därför utföras under en skyddskåpa.
1.6.5 Testbetingelser
Mätningen ska göras vid ungefär 20 oC och hållas inom ±0,5 oC.
1.6.6 Testförfarande
De lösningar som ska mätas fylls i det noggrant rengjorda kärlet varvid skumbildning ska undvikas. Mätkärlet placeras på apparatens provbord. Bordsytan med mätkärlet ska höjas tills ringen är helt nedsänkt i den lösning som ska mätas. Därefter ska bordsytan sänkas gradvis och jämt (med en hastighet av ungefär 0,5 cm/min) för att lösgöra ringen från ytan tills den största kraften har uppnåtts. Det vätskelager som vidhäftar ringen får inte avlägsnas från ringen. När mätningen är gjord ska ringen åter nedsänkas helt i lösningen och mätningen upprepas till dess att ett konstant ytspänningsvärde erhålls. Den tid som går åt från överföring av lösningen till mätkärlet och tills mätningen görs ska antecknas för varje bestämning. Avläsningar ska göras vid den största kraft som behövs för att lösgöra ringen från vätskans yta.
2. DATA
För att beräkna ytspänningen ska det på apparaten i mN/m avlästa värdet först multipliceras med kalibreringsfaktor Φa, eller Φb (beroende på vilken kalibreringsmetod som används). Detta ger endast ett ungefärligt värde som ska korrigeras.
Harkins och Jordan (se hänvisning 4) har empiriskt bestämt korrektionsfaktorer för ytspänningsvärden som är bestämda med ringmetoden. Dessa faktorer är beroende av ringens mått, vätskans densitet och dess ytspänning.
Eftersom det är besvärligt att för varje enskild mätning bestämma korrektionsfaktorn utifrån Harkins och Jordans tabeller, kan man vid beräkning av ytspänningen hos vattenlösningar använda den förenklade metoden att avläsa de korrigerade ytspänningsvärdena direkt i tabellen. (Interpolation ska användas för avläsningar som ligger mellan tabellvärdena.)
Tabell
Korrektion av den mätta ytspänningen
Endast för vattniga lösningar, ρ = 1 g/cm3
|
r |
9,55 mm (genomsnittlig ringradie) |
|
r |
0,185 mm (ringtrådens radie) |
|
Experimentellt värde (mN/m) |
Korrigerat värde (mN/m) |
|
|
Viktkalibrering (se 1.6.4.2.2 a) |
Vattenkalibrering (se 1.6.4.2.2 b) |
|
|
20 |
16,9 |
18,1 |
|
22 |
18,7 |
20,1 |
|
24 |
20,6 |
22,1 |
|
26 |
22,4 |
24,1 |
|
28 |
24,3 |
26,1 |
|
30 |
26,2 |
28,1 |
|
32 |
28,1 |
30,1 |
|
34 |
29,9 |
32,1 |
|
36 |
31,8 |
34,1 |
|
38 |
33,7 |
36,1 |
|
40 |
35,6 |
38,2 |
|
42 |
37,6 |
40,3 |
|
44 |
39,5 |
42,3 |
|
46 |
41,4 |
44,4 |
|
48 |
43,4 |
46,5 |
|
50 |
45,3 |
48,6 |
|
52 |
47,3 |
50,7 |
|
54 |
49,3 |
52,8 |
|
56 |
51,2 |
54,9 |
|
58 |
53,2 |
57,0 |
|
60 |
55,2 |
59,1 |
|
62 |
57,2 |
61,3 |
|
64 |
59,2 |
63,4 |
|
66 |
61,2 |
65,5 |
|
68 |
63,2 |
67,7 |
|
70 |
65,2 |
69,9 |
|
72 |
67,2 |
72,0 |
|
74 |
69,2 |
— |
|
76 |
71,2 |
— |
|
78 |
73,2 |
— |
Denna tabell är utarbetat efter Harkins-Jordans korrektion och liknar den som finns i DIN-standarden (DIN 53914) för vatten och vattniga lösningar (densitet p = 1 g/cm3) och är avsedd för en kommersiellt tillgänglig ring med mitten R = 9,55 mm (genomsnittlig ringradie) och r = 0,185 mm (ringtrådens radie). Tabellen ger korrigerade värden för ytspänning som mätts efter kalibrering med vikt eller vatten.
Alternativt kan ytspänningen utan föregående kalibrering beräknas enligt följande formel:

där:
|
F |
= |
kraften mätt på dynamometern när hinnan bryts |
|
R |
= |
ringens radie |
|
f |
= |
korrektionsfaktorn (se hänvisning 1) |
3. RAPPORTERING
3.1 TESTRAPPORT
Testrapporten ska om möjligt innehålla minst följande uppgifter:
|
— |
Använd metod. |
|
— |
Använd typ av vatten eller lösning. |
|
— |
Exakt specificering av ämnet (beteckning och orenheter). |
|
— |
Mätresultat: ytspänning (avläsning) med uppgift om både enskilda avläsningar och deras aritmetiska medelvärde och det korrigerade medelvärdet (med hänsyn tagen till apparaturfaktorn och korrektionstabellen). |
|
— |
Lösningens koncentration. |
|
— |
Testtemperaturen. |
|
— |
Den använda lösningens ålder, särskilt tidsåtgången mellan framställning och mätning av lösningen. |
|
— |
Beskrivning av ytspänningens tidsberoende efter överföring av lösningen till mätkärlet. |
|
— |
Alla uppgifter och upplysningar som är av betydelse för tolkningen av resultaten ska rapporteras, i synnerhet med avseende på orenheter och ämnets fysiska tillstånd. |
3.2 TOLKNING AV RESULTAT
Med tanke på att destillerat vatten har en ytspänning av 72,75 mN/m vid 20 oC anses ämnen med en ytspänning på mindre än 60 mN/m bestämd genom denna metod vara ytspänningsaktiva ämnen.
4. HÄNVISNINGAR
|
1. |
OECD, Paris, 1981, Test Guideline 115, rådets beslut K(S1) 30 slutligt. |
|
2. |
R. Weissberg ed.: Technique of Organic Chemistry, Physical Methods of Organic Chemistry, 3rd ed., Inter-science Publ., New York, 1959, vol. I, del I, kapitel XIV. |
|
3. |
Pure Appl. Chem., 1976, vol. 48, s. 511. |
|
4. |
Harkins, W. D., Jordan, H. R, J. Amer. Chem. Soc, 1930, vol. 52, s. 1751. |
A.6 VATTENLÖSLIGHET
1. METOD
De beskrivna metoderna är baserade på OECD:s riktlinjer för undersökning av kemikalier (se hänvisning 1).
1.1 INLEDNING
Vid utförande av detta test är det värdefullt att ha förhandskunskap om ämnets strukturformel, ångtryck, dissociationskonstant och hydrolys (som en funktion av pH-värdet).
Det finns inte någon enskild metod som kan användas över hela vattenlöslighetsområdet.
De två testmetoderna som beskrivs nedan täcker hela löslighetsområdet men kan inte tillämpas på flyktiga ämnen:
|
— |
Den ena metoden, som tillämpas på väsentligen rena ämnen med låg löslighet (< 10-2 gram per liter) som ärstabila i vatten, kallas ”kolonnelueringsmetoden”. |
|
— |
Den andra metoden, som tillämpas på väsentligen rena ämnen med högre löslighet (> 10-2 gram per liter)som är stabila i vatten, kallas ”kolvmetoden”. |
Det undersökta ämnets vattenlöslighet kan i hög grad påverkas av närvaron av orenheter.
1.2 DEFINITION OCH ENHETER
Ett ämnes löslighet i vatten anges av mättnadskoncentrationen av ämnet i vatten vid en viss temperatur. Lösligheten i vatten anges i viktenhet per volym lösning. SI-enheten är kg/m3 (gram per liter kan också användas).
1.3 REFERENSÄMNEN
Man behöver inte använda referensämnen varje gång ett nytt ämne ska undersökas. Referensämnen ska först och främst användas för att då och då kontrollera metoden och för jämförelse med resultat som uppnåtts med andra metoder.
1.4 TESTMETODSPRINCIP
Den ungefärliga mängd som behövs av provet och tiden som krävs för att uppnå mättnadskoncentrationen ska bestämmas i ett enkelt förberedande test.
1.4.1 Kolonnelueringsmetoden
Denna metod är baserad på eluering av provämnet med vatten från en mikrokolonn, som innehåller ett inert bärarmaterial, t.ex. glaspärlor eller sand, belagt med överskott av provämnet. Vattenlösligheten bestäms när eluatets viktkoncentration är konstant. Detta framkommer som en koncentrationsplatå som en funktion av tiden.
1.4.2 Kolvmetoden
Med denna metod löses ämnet (fasta ämnen pulvriseras) i vatten vid en temperatur något över testtemperaturen. När mättnad har uppnåtts avkyls blandningen och hålls under omrörning vid testtemperaturen så länge som krävs för att uppnå jämvikt (se hänvisning 2). Alternativt kan mätningen utföras direkt vid testtemperaturen, om man genom lämplig provtagning ser till att mättnadsjämvikt uppnås. Därefter bestäms ämnets viktkoncentration i vattenlösningen, som inte får innehålla olösta partiklar, med lämplig analysmetod.
1.5 KVALITETSKRITERIER
1.5.1 Repeterbarhet
För kolonnelueringsmetoden är det möjligt att uppnå < 30 %, och för kolvmetoden bör den vara < 15 %.
1.5.2 Sensitivitet
Sensitiviteten är beroende av analysmetoden, men det är möjligt att bestämma så låga viktkoncentrationer som 10-6 gram per liter.
1.6 METODBESKRIVNING
1.6.1 Testbetingelser
Testet ska helst utföras vid 20 ±0,5 oC. Om man misstänker att lösligheten är temperaturberoende (> 3 % per oC), ska man även utföra tester vid två andra temperaturer, som ligger minst 10 oC över respektive under den först valda temperaturen. I detta fall ska temperaturen regleras till ±0,1 oC. Den valda temperaturen ska hållas konstant i alla relevanta delar av utrustningen.
1.6.2 Förberedande test
Till cirka 0,1 g av provet (fasta ämnen måste pulvriseras) i ett 10 ml graderat mätglas med glaspropp tillsätts stegvis ökade mängder destillerat vatten vid rumstemperatur enligt tabellen nedan:
|
0,1 g lösligt i ”x” ml vatten |
0,1 |
0,5 |
1 |
2 |
10 |
100 |
> 100 |
|
Ungefärlig löslighet (gram/l) |
> 1 000 |
1 000–200 |
200–100 |
100–50 |
50–10 |
10–1 |
< 1 |
Efter varje tillsättning av angiven mängd vatten skakas blandningen kraftigt i 10 minuter och kontrolleras visuellt så att det inte finns några olösta delar av provet. Om provet eller delar av det efter tillsättning av 10 ml vatten fortfarande inte är löst, ska experimentet upprepas i ett 100 ml mätglas med en större mängd vatten. Vid lägre löslighet kan det ta avsevärt längre tid att lösa upp ett ämne (24 timmar bör medges). Den ungefärliga lösligheten anges i tabellen under den volym av tillsatt vatten i vilken fullständig lösning av provet inträffar. Om ämnet fortfarande inte verkar vara löst ska 24 timmar tillåtas (högst 96 timmar) eller ytterligare utspädning göras för att säkerställa att kolonneluerings- eller kolvmetoden kan användas.
1.6.3 Kolonnelueringsmetoden
1.6.3.1 Bärarmaterial, lösningsmedel och elueringsmedel
Till kolonnelueringsmetoden ska ett inert bärarmaterial användas, t.ex. glaspärlor och sand. Ett lämpligt flyktigt lösningsmedel av analytisk reagenskvalitet ska användas till att överföra provämnet till bärarmaterialet. Som eluerings- eller lösningsmedel används vatten som omdestillerats i glas- eller kvartsapparaten.
Anmärkning:
Vatten direkt från en organisk jonbytare får inte användas.
1.6.3.2 Laddning av bärarmatenal
Ungefär 600 mg bärarmaterial vägs upp och överförs till en 50 ml rundbottnad kolv.
En lämplig, beräknad mängd av provämnet löses i det valda lösningsmedlet. En lämplig mängd av provlösningen tillsätts bärarmaterialet. Lösningsmedlet ska avdunstas fullständigt i t.ex. en rotationsindunstare; i annat fall uppnås inte vattenmättnad av bäraren till följd av fördelningseffekter pä bärarmaterialets yta.
Laddningen av bärarmaterial kan orsaka problem (felaktiga resultat) om provämnet avsätts som en olja eller i en annan kristallfas. Problemet ska undersökas experimentellt och rapporteras.
Det laddade bärarmaterialet ska ligga i blöt i ungefär två timmar i cirka 5 ml vatten, och därefter tillsätts suspensionen till mikrokolonnen. Alternativt kan det torra laddade bärarmaterialet tillsättas mikrokolonnen, som har fyllts med vatten, och därefter får det vila i ungefär två timmar tills jämvikt har uppnåtts.
Testförfarande:
Elueringen av ämnet från bärarmaterialet kan utföras på ett av två olika sätt:
|
— |
Med cirkulationspump (se figur 1). |
|
— |
Med nivåkärl (se figur 4). |
1.6.3.3 Kolonnelueringsmetoden med recirkulationspump
Apparatur
En schematisk uppställning av ett typiskt system visas i figur 1. En lämplig mikrokolonn visas i figur 2, även om valfri storlek kan användas, förutsatt att den uppfyller kriterierna för reproducerbarhet och sensitivitet. Kolonnen ska ha ett tomt utrymme på minst fem bäddvolymer vatten plus minst fem prover. Alternativt kan storleken minskas om mer lösningsmedel tillsätts som ersättning för de första fem bäddvolymerna, som avlägsnas med orenheter.
Kolonnen ska anslutas till en cirkulationspump med kapacitet att styra flöden på cirka 25 ml/timme. Pumpen ansluts med kopplingar av fluoretenplast (PTFE) och/eller glas. När kolonnen och pumpen kopplats ihop ska det vara möjligt att ta prover av eluatet och att hålla det tomma utrymmet i jämvikt vid atmosfäriskt tryck. Kolonnmaterialet stöds med en liten (5 mm) glasullspropp, som även fungerar som filter för partiklar. Recirkulationspumpen kan t.ex. vara en peristaltisk pump (noggrannhet ska iakttas så att slangmaterialet inte utsätts för förorening och/eller adsorption) eller en membranpump.
Mätförfarande
Flödet genom kolonnen startas. Flödeshastigheten bör vara ungefär 25 ml/timme (cirka 10 bäddvolymer/timme för den beskrivna kolonnen). De första fem bäddvolymerna (minimum) kasseras för att avlägsna vattenlösliga orenheter. Därefter körs cirkulationspumpen tills jämvikt har uppnåtts, dvs. när fem på varandra följande prover ger koncentrationer som slumpmässigt inte avviker mer än ± 30 %. Dessa prov ska vara åtskilda från varandra med tidsintervaller som motsvarar genomloppet av minst 10 bäddvolymer elueringsmedel.
1.6.3.4 Kolonnelueringsmetod med nivåkärl
Apparatur (se figur 4 och 3)
Nivåkärl: Anslutningen till nivåkärlet görs med användning av ett slipat glasövergångsstycke som kopplas med en PTFE-slang. Flödeshastigheten bör vara ungefär 25 ml/timme. Successiva fraktioner av eluatet ska samlas och analyseras efter den valda metoden.
Mätförfarande
Fraktioner från det mittersta elueringsområdet, där koncentrationerna är konstanta (± 30 %) vid minst fem på varandra följande fraktioner, används for att bestämma lösligheten i vatten.
I båda fallen (användning av recirkulationspump eller nivåkärl) utförs ännu ett försök med hälften så hög flödeshastighet som vid det första försöket. Om resultaten från de två försöken överensstämmer är testet tillfredsställande. Om lösligheten är större vid testet med lägre flödeshastighet, ska man fortsätta att halvera flödeshastigheten tills två på varandra följande test ger samma löslighet.
I båda fallen (användning av recirkulationspump eller nivåkärl) ska fraktionernas innehåll av kollodialt material kontrolleras med hjälp av Tyndall-effekten (ljusspridning). Förekomst av sådana partiklar innebär att resultaten är ogiltiga och testet måste upprepas efter förbättringar av kolonnens filtreringsfunktion.
Varje provs pH-värde ska registreras. Ytterligare ett försök utförs vid samma temperatur.
1.6.4 Kolvmetoden
1.6.4.1 Apparatur
Till kolvmetoden behövs följande material:
|
— |
vanliga laboratorieglas och -instrument, |
|
— |
lämplig anordning för omskakning av lösningar under reglerade konstanta temperaturer, |
|
— |
en centrifug (helst termostatreglerad), om detta behövs för emulsioner, och |
|
— |
utrustning för analytisk bestämning. |
1.6.4.2 Mätförfarande
Den mängd material som behövs för att mätta den önskade volymen vatten beräknas utifrån det förberedande testet. Den volym vatten som behövs är beroende av analysmetoden och löslighetsområdet. Ungefär fem gånger den materialmängd som bestämts enligt ovan beräknas för var och en av tre glasbehållare med glasproppar (t.ex. centrifugglas, kolvar). Den valda volymen vatten tillsätts i varje kärl, och kärlen tillsluts helt. De tillslutna kärlen omskakas därefter vid 30 oC. (En skaknings- eller omrörningsanordning som kan fungera vid konstant temperatur ska användas, t.ex. magnetomrörning i ett termostatreglerat bad.) Efter en dag tas det ena kärlet bort och får stå i 24 timmar vid testtemperaturen och ska omskakas då och då för att återuppnå jämvikt. Därefter centrifugeras innehållet i kärlet vid rumstemperatur och föreningens koncentration i den klara vattenfasen bestäms enligt lämplig analysmetod. De två andra kolvarna ska, efter att i två respektive tre dagar ha uppnått en första jämvikt vid 30 oC, behandlas likadant. Om koncentrationsresultaten från minst två av kärlen överensstämmer med den erforderliga reproducerbarheten, är testet tillfredsställande. Om resultaten från kärl 1, 2 och 3 visar en tendens att ge stigande värden, ska hela testet upprepas med användning av längre tider for uppnående av jämvikt.
Mätförfarandet kan också utföras utan föregående inkubation vid 30 oC. För att kunna bedöma graden av mättnadsjämvikt tas prover tills omrörningstiden inte längre påverkar testämnets koncentration.
Varje provs pH-värde ska registreras.
1.6.5 Analys
Helst ska en ämnesspecifik analysmetod användas till dessa bestämningar, eftersom små mängder av lösliga orenheter kan orsaka stora fel i den uppmätta lösligheten. Exempel på sådana metoder är: gas- eller vätskekromatografi, titreringsmetoder, fotometriska metoder och voltametriska metoder.
2. DATA
2.1 KOLONNELUERINGSMETODEN
Medelvärdet och standardavvikelsen ska beräknas for minst fem på varandra följande prov, tagna vid mättnadsplatån. Resultaten ska anges i viktenheter per volymenhet lösning.
Genomsnittet från två tester med olika flödeshastigheter jämförs och repeterbarheten ska ligga under 30 %.
2.2 KOLVMETODEN
De enskilda resultaten ska anges för var och en av de tre kolvarna, och för de resultat som bedöms vara konstanta (repeterbarhet mindre än 15 %), ska medelvärdet beräknas och anges i viktenheter per volymenhet lösning. Om lösligheten ar mycket hög (> 100 gram per liter) kan det vara nödvändigt att omvandla viktenhet till volymenhet med hjälp av densiteten.
3. RAPPORTERING
3.1 KOLONNELUERINGSMETODEN
Testrapporten ska om möjligt innehålla följande uppgifter:
|
— |
Resultaten av det förberedande testet. |
|
— |
Exakt specificering av ämnet (beteckning och orenheter). |
|
— |
Koncentration, flödeshastighet och pH-värde för varje prov. |
|
— |
Medelvärde och standardavvikelse för minst fem prov från mättnadsplatån från varje försök. |
|
— |
Genomsnittet av de två successiva, acceptabla försöken. |
|
— |
Vattentemperaturen under mättnadsprocessen. |
|
— |
Använd analysmetod. |
|
— |
Arten av det använda bärarmaterialet. |
|
— |
Laddning av bärarmaterialet. |
|
— |
Använt lösningsmedel. |
|
— |
Tecken på eventuell kemisk instabilitet hos ämnena under testet och den använda metoden. |
|
— |
Alla upplysningar av betydelse för tolkningen av resultaten, särskilt i fråga om orenheter och ämnets fysiska tillstånd. |
3.2 KOLVMETODEN
Rapporten ska om möjligt innehålla följande uppgifter:
|
— |
Resultaten av det förberedande testet. |
|
— |
Exakt specificering av ämnet (beteckning och orenheter). |
|
— |
Enskilda analytiska bestämningar och, om fler än ett värde bestämdes, genomsnittet för varje kolv. |
|
— |
Varje provs pH-värde. |
|
— |
Genomsnittet av värdet för de olika kolvar som överensstämde. |
|
— |
Testtemperaturen. |
|
— |
Använd analysmetod. |
|
— |
Tecken på eventuell kemisk instabilitet hos ämnena under testet och den använda metoden. |
|
— |
Alla uppgifter och upplysningar som är av betydelse för tolkningen av resultaten, särskilt i fråga om orenheter och ämnets fysiska tillstånd. |
4. HÄNVISNINGAR
|
1. |
OECD, Paris, 1981, Test Guideline 105, rådets beslut K(81) 30 slutligt. |
|
2. |
NF T 20-045 AFNOR (Sept., 85). Chemical products for industrial use – Determination of water solubilityof solids and liquids with low solubility – Column elution method. |
|
3. |
NF T 20-045 AFNOR (Sept., 85). Chemical products for industrial use – Determination of water solubilityof solids and liquids with high solubility – Flask method. |
Tillägg
Figur 1
Kolonnelueringsmetod med recirkulationspump

Figur 2
En typisk mikrokolonn
(Alla mått anges i mm)

Figur 3
En typisk mikrokolonn
(Alla mått anges i mm)

Figur 4
Kolonnelueringsmetod med nivåkärl*

A.8 FÖRDELNINGSKOEFFICIENT
1. METOD
Den beskrivna ”skakkolvmetoden” är baserade på OECD:s riktlinjer för undersökning av kemikalier (se hänvisning 1).
1.1 INLEDNING
Vid utförande av detta test är det värdefullt att ha förhandskunskap om ämnets strukturformel, dissociationskonstant, hydrolys, löslighet i n-oktansyra och ytspänning.
Mätningar på joniserbara ämnen utförs på den icke-joniserade formen (fri syra eller fri bas), dvs. genom användning av en passande buffert med ett pH-värde av minst 1 enhet under (fri syra) eller över (fri bas) ämnets pK-värde.
Testmetoden omfattar två särskilda förfaranden: skakkolvmetoden och högpresterande vätskekromatografi (HPLC). Den första metoden är användbar när värden av log Pow (se definitionerna nedan) ligger mellan - 2 och 4, och den andra metoden i området mellan 0 och 6. Innan endera försöksmetoden inleds ska en preliminär uppskattning av fördelningskoefficienten erhållas.
Skakkolvmetoden kan endast användas för väsentligen rena ämnen som är lösliga i vatten och n-oktan. Den kan inte användas för ytaktiva ämnen (för vilka ett beräknat värde eller en uppskattning grundad på lösligheten i vatten respektive n-oktan bör finnas).
HPLC-metoden kan inte användas för starka syror och baser, metallkomplexer, ytaktiva ämnen och ämnen som reagerar med elueringsmedlet. För sådana material används ett beräknat eller uppskattat värde på grundval av lösligheten i n-oktan respektive vatten.
HPLC-metoden är mindre känslig än skakkolvmetoden för närvaron av orenheter i ämnet. Trots detta kan orenheter i vissa tillfällen göra det svårt att tolka resultaten eftersom toppvärden blir osäkra. För blandningar med ett oupplöst band anges en övre och nedre gräns för log P.
1.2 DEFINITIONER OCH ENHETER
Fördelningskoefficienten (P) definieras som förhållandet mellan jämviktskoncentrationerna (ci) för ett ämne som lösts i ett tvåfassystem bestående av två i stort sett icke blandbara lösningsmedel. För n-oktansyra och vatten är

Fördelningskoefficienten (P) är således ett förhållande mellan två koncentrationer och uttrycks vanligtvis i form av dess logaritm för basen 10 (log P).
1.3 REFERENSÄMNEN
Skakkolvmetoden
Man behöver inte använda referensämnen varje gång ett nytt ämne ska undersökas. Referensämnen ska först och främst användas för att då och då kontrollera metoden och för jämförelse med resultat som uppnåtts med andra metoder.
HPLC-metoden
För att kunna korrelera uppmätta HPLC-data för ett ämne med dess P-värde måste en kalibreringskurva för log P mot kromatografiska data fastställas med användning av minst 6 referenspunkter. Det är upp till användaren att välja lämpliga referensämnen. När så är möjligt ska minst ett referensämne ha en Pow-värde som ligger över testämnets, och minst ett med ett Pow-värde under testämnets. För värden av log P under 4 kan kalibreringen baseras på data som erhållits genom skakkolvmetoden. För värden av log P över 4 kan kalibreringen baseras på validerade hänvisningsvärden om dessa motsvarar beräknade värden. För bättre noggrannhet är det att föredra att välja referensämnen som strukturmässigt liknar testämnet.
Det finns omfattande listor av värden av log Pow för många grupper av kemikalier (se hänvisningar 2 och 3). Om det inte finns data om fördelningskoefficienter för strukturmässigt besläktade ämnen kan en mer allmän kalibrering, upprättad med andra referensföreningar, användas.
I tillägg 2 finns en lista med rekommenderade referensämnen och deras Pow-värden.
1.4 TESTMETODSPRINCIPER
1.4.1 Skakkolvmetoden
För att kunna bestämma fördelningskoefficienten måste jämvikt uppnås mellan alla samverkande komponenter i systemet, och koncentrationerna av de ämnen som lösts i de två faserna måste bestämmas. En genomgång av litteraturen i detta ämne visar att man kan använda flera olika tekniker för att lösa detta problem, dvs. noggrann blandning av två faserna för bestämning av det undersökta ämnets jämviktskoncentration efter separation av faserna.
1.4.2 HPLC-metoden
HPLC utförs på analytiska kolonner packade med en kommersiellt tillgänglig fast fas innehållande långa kedjor av kolväte (t.ex. C8, C18) kemiskt bundna till kisel. Om kemikalier insprutas på en sådan kolonn kommer de att vandra genom den med olika hastigheter, eftersom deras fördelning mellan den mobila och den stationära fasen inte är densamma. Kemikalieblandningar elueras i ordningsföljd efter hydrofobicitet, varvid de vattenlösliga kemikalierna elueras först och de oljeupplösliga sist, i proportion till deras kolväte-/vattenfördelningskoefficient. Därefter kan sammanhanget mellan retentionstiden på en sådan kolonn (med omvänd fas) och n-oktan/vattenfördelningskoefficienten fastställas. Fördelningskoefficienten utleds av kapacitetsfaktorn k, som fås genom

där tR är testämnets retentionstid och to den genomsnittliga tid det tar för en lösningsmedelsmolekyl att komma igenom kolonnen (kolonnens dödtid).
Det krävs inga kvantitativa analysmetoder och det är endast nödvändigt att bestämma retentionstiden.
1.5 KVALITETSKRITERIER
1.5.1 Repeterbarhet
Skakkolvmetoden
För att säkerställa en exakt fördelningskoefficient ska dubbla bestämningar göras under tre olika testbetingelser, varvid såväl mängden av det angivna ämnet som förhållandet mellan lösningsmedlens volymer kan varieras. Bestämningen av fördelningskoefficientens värden uttryckta som deras allmänna logaritmer ska ligga inom ±0,3 logaritmenheter.
HPCL-metoden
För att öka tilltron till mätningen görs dubbla bestämningar. Värdena av log P beräknade på grundval av de individuella mätningarna ska ligga inom ett intervall av ±0,1.
1.5.2 Sensitivitet
Skakkolvmetoden
Metodens mätområde bestäms av analysförfarandets detektionsgräns. Denna bör vara tillräcklig för att medge bestämning av värden för log Pow i området - 2 till 4 (under vissa omständigheter kan detta område ökas till log Pow upp till 5) när koncentrationen av det lösta ämnet i endera fasen inte är större än 0,01 mol per liter.
HPLC-metoden
Med HPLC-metoden kan man uppskatta fördelningskoefficienter i log Pow-området från 0 till 6.
Normalt kan en förenings fördelningskoefficient uppskattas med en avvikelse av mindre än ± 1 från det värde som uppnås med skakkolvmetoden. Typiska korrelationer framgår i hänvisningarna (4, 5, 6, 7 och 8). Man upnår normalt större noggrannhet om man använder korrelationskurvor baserade på strukturmässigt besläktade referensämnen {se hänvisning 9).
1.5.3 Specificitet
Skakkolvmetoden
Nernsts fördelningslag gäller endast för utspädda lösningar vid konstant temperatur, tryck och pH-värde. Den gäller endast för ett rent ämne som fördelats mellan två rena lösningsmedel. Om flera olika lösta ämnen förekommer i den ena eller i båda faserna samtidigt, kan detta påverka resultaten.
Dissociation eller association av de lösta molekylerna resulterar i avvikelser från Nernsts fördelningslag. Sådana avvikelser framgår av det faktum att fördelningskoefficienten blir beroende av lösningens koncentration.
På grund av de många inblandade jämvikterna ska denna metod inte användas på joniserbara föreningar utan korrektion. För sådana föreningar kan man överväga att använda buffertlösningar i stället för vatten. I så fall ska buffertlösningens pH-värde ligga minst 1 enhet från ämnets pKa och detta pH-värdes inverkan på miljön ska beaktas.
1.6 METODBESKRIVNING
1.6.1 Preliminär uppskattning av fördelningskoefficienten.
Fördelningskoefficientens värde kan företrädesvis uppskattas genom en enkel beräkning (se tillägg 1) eller, vid behov, på grundval av testämnenas löslighet i de rena lösningsmedlen (se hänvisning 10).
1.6.2 Skakkolvmetoden
1.6.2.1 Förberedelser
n-oktansyra: Fördelningskoefficienten ska bestämmas med användning av reagenser med mycket hög analytisk renhetsgrad.
Vatten: Destillerat eller omdestillerat vatten från glas- eller kvartsapparat ska användas. För joniserbara föreningar används buffertlösmngar i stället för vatten, om detta är motiverat.
Anmärkning:
Vatten som hämtats direkt från en jonbytare får inte användas.
1.6.2.1.1
Innan en fördelningskoefficient bestäms mättas var och en av de faser som ingår i lösningsmedelssystemet ömsesidigt genom omskakning vid testtemperatur. För detta är det praktiskt att använda en mekanisk skakapparat, och låta den i 24 timmar skaka två stora stamkolvar med n-oktansyra med mycket hög analytisk renhetsgrad eller vatten, vardera innehållande tillräcklig mängd av det andra lösningsmedlet, varefter flaskorna får stå tillräckligt länge för att medge separation av faserna och för att uppnå ett mättnadstillstånd.
1.6.2.1.2
Testkärlet ska vara nästan helt fyllt av tvåfassystemet. Detta förhindrar materialförlust till följd av avdunstning. Förhållandet mellan volym och mängder ämne fastställs på grundval av följande:
|
— |
Den preliminära uppskattningen av fördelningskoefficienten (se ovan). |
|
— |
Den minsta mängden testämne som behövs för analysförfarandet. |
|
— |
Begränsningen av en högsta koncentration i endera fasen på 0,01 mol per liter. |
Tre test ska göras. I det första tillsätts det beräknade volymförhållandet av n-oktansyra till vatten; i det andra tillsätts dubbla volymen n-oktansyra; och i det tredje tillsätts halva volymen n-oktansyra (dvs. 1:1, 1:2, 2:1).
1.6.2.1.3
En stamlösning framställs i n-oktansyra förmättad med vatten. Innan stamlösningen används för bestämning av fördelningskoefficienten ska dess faktiska koncentration bestämmas exakt. Denna lösning ska förvaras under stabila betingelser.
1.6.2.2 Testbetingelser
Testtemperaturen ska hållas konstant (± 1 oC) och ligga i området 20–25 oC.
1.6.2.3 Mätförfarande
1.6.2.3.1
För varje testbetingelse används två testkärl som vart och ett innehåller erforderliga, exakt mätta mängder av de två lösningsmedlen och den erforderliga mängden stamlösning.
Delarna n-oktansyra ska mätas per volym. Testkärlen ska antingen placeras i en lämplig skakapparat eller skakas för hand. Nar ett centrifugeringsglas används rekommenderas att snabbt vrida centrifugglaset 180o kring dess tväraxel så att all instängd luft stiger upp genom de två faserna. Erfarenheten visar att 50 sådana vridningar normalt är tillräckligt för att erhålla fördelningsjämvikt. För att vara säker på detta rekommenderas 100 vridningar under fem minuter.
1.6.2.3.2
Vid behov kan blandningen centrifugeras för att separera faserna. Detta görs i en laboratoriecentrifug som hålls vid rumstemperatur, eller, om en centrifug utan temperaturreglering används, ska centrifugglasen minst en timme före analysen bringas i jämvikt med testtemperaturen.
1.6.2.4 Analys
För bestämningen av fördelningskoefficienten är det nödvändigt att bestämma koncentrationerna av testämnet i båda faserna. Detta kan man göra genom att för varje testbetingelse ta ett delprov av varje fas från varje glas och analysera dessa enligt det valda förfarandet. Den totala mängden ämne i båda faserna beräknas och jämförs med den mängd ämne som ursprungligen tillsattes.
Proven från vattenfasen ska tas på så sätt att risken att den innehåller spår av n-oktansyra minimeras: en glasspruta med avtagbar kanyl kan användas till att ta prov från vattenfasen. Sprutan ska först fyllas delvis med luft. Luften ska försiktigt pressas ut medan kanylen förs genom n-oktansyrafasen. En lämplig volym av vattenfasen dras upp i sprutan. Sprutan dras snabbt bort från lösningen och kanylen avlägsnas. Innehållet i sprutan kan sedan användas som vattenprovet. Koncentrationen i de två separerade faserna ska helst bestämmas med en ämnesspecifik metod. Exempel på fysisk-kemiska bestämningar som kan vara lämpliga är
|
— |
fotometriska metoder, |
|
— |
gaskromatografi, |
|
— |
högpresterande vätskekromatografi (HPLC). |
1.6.3 HPLC-metoden
1.6.3.1 Förberedelser
Apparatur
Det krävs en vätskekromatograf med pulsfri pump och passande detektionssystem. Användning av insprutningsventil med insprutningsspole rekommenderas. Närvaro av polära grupper i den stationära fasen kan väsentligt nedsätta HPLC-kolonnernas prestation. Till följd av detta måste de stationära faserna ha lägsta möjliga procentuella innehåll av polära grupper (se hänvisning 11). Ett kommersiellt mikroniserat fyllningsmaterial med omvänd fas eller färdigfyllda kolonner kan användas. Eventuellt kan en förkolonn placeras mellan insprutnings-systemet och den analyskolonnen.
Mobil fas
Metanol och vatten av HPLC-kvalitet används för att framställa elueringsvätskan, vilken avgasas innan användning. Isokratisk eluering används. Den använda metanol/vattenblandningen ska innehålla minst 25 % vatten. För eluering av föreningar med ett log P av 6 på en timme med en flödeshastighet av 1 ml/m räcker det vanligtvis med en blandning av metanol och vatten i förhållandet 3:1 (v/v). För föreningar med ett högt log P-värde kan det vara nödvändigt att minska elueringstiden (även för referensämnena) genom att utnyttja en mindre polär mobil fas eller en kortare kolonn.
Ämnen med mycket låg löslighet i n-oktan har en tendens att ge onormalt låga värden för log Pow med HPLC-metoden. Ibland följer sådana föreningars toppar lösningsmedelsfronten. Detta beror sannolikt på att fördelningsprocessen är för långsam för att jämvikt ska kunna uppnås inom tiden för en HPLC-separation. I detta fall kan en minskning av flödeshastigheten och/eller en minskning av metanol/vattenförhållandet vara effektivt för att erhålla ett pålitligt värde.
Både testämnet och referensämnen ska vara lösliga i den mobila fasen i en så hög koncentration att detta kan påvisas. Endast i undantagsfall får tillsättningsämnen användas till metanol/vattenblandningen, eftersom tillsättningsämnen kommer att ändra kolonnens egenskaper. För kromatografer med tillsättningsämnen ska man alltid använda en separat kolonn av samma slag. Om metanol/vatten inte är lämpligt kan man använda andra blandningar av vatten och organiska lösningsmedel, t.ex. etanol/vatten eller acetonitril/vatten.
För joniserbara ämnen är elueringsvätskans pH-värde kritiskt. Det ska ligga innanför kolonnens driftsområde, vilket normalt är från 2 till 8. Användning av buffert rekommenderas. Det är viktigt att undvika saltutfällning och försämring av kolonnen, vilket kan förekomma med vissa blandningar av organisk fas och buffert. HPLC-mätningar med kiselbaserade faser med pH över 8 rekommenderas inte eftersom alkalisk mobil fas kan leda till att kolonnens prestation hastigt försämras.
Lösningar
Referensämnen ska vara av renast möjliga kvalitet. För testning och kalibrering ska ämnena om möjligt lösas i den mobila fasen.
Försöksbetingelser
Temperaturen under mätningarna bör inte variera med mer än 2 K.
1.6.3.2 Mätningar
Beräkning av dödtiden t0
Dödtiden t0 kan bestämmas antingen med en homolog serie (t.ex. n-alkylmetylketoner) eller med organiska ämnen som inte hålls kvar (t.ex. thiourea eller formamid). För att beräkna dödtiden t0 pä grundval av en homolog serie insprutas minst sju element av en homolog serie, och retentionstiderna bestäms. Bruttoretentionstiderna tr(nc+1) avbildas mot tr(nc), och i regressionsekvationen
tr(nc + 1) = a + b tr(nc)
bestäms avskärningen a och hållningskoefficienten b (nc = antalet kolatomer). Dödtiden ges då genom
to = a/(l – b)
Kalibreringskurva
Nästa steg består i att konstruera en korrelationskurva av log k mot log P för passande referensämnen. I praktiken insprutas det samtidigt mellan fem och tio standardreferensämnen, om log P ligger i det förväntade området, och retentionstiderna bestäms, helst med en integrationsskrivare som är kopplad till detektionssystemet. Motsvarande logaritmer för kapacitetsfaktorerna, log k, beräknas och avsätts mot den log P som är bestämd genom skakkolvmetoden. Kalibreringen företas regelbundet, minst en gång om dagen, för att ta hänsyn till för eventuella ändringar i kolonnens prestation.
Bestämning av testämnets kapacitetsfaktor
Testämnet sprutas in som en så liten mängd mobil fas som möjligt. Retentionstiden bestäms (dubbla bestämningar), varefter kapacitetsfaktorn k kan beräknas. På grundval av korrelationskurvan över referensämnena kan testämnets fördelningskoefficient interpoleras. Vid mycket låga och mycket höga fördelningskoefficienter är det nödvändigt att extrapolera. I sådana fall måste man vara särskilt uppmärksam på regressionslinjens konfidens-gränser.
2. DATA
Skakkolvmetoden
Tillförlitligheten av de bestämda P-värdena kan prövas genom en jämförelse mellan medelvärdena för de dubbla bestämningarna och det totala medelvärdet.
3. RAPPORTERING
Testrapporten ska om möjligt innehålla följande uppgifter:
|
— |
Exakt specificering av ämnet (beteckning och orenheter). |
|
— |
Om metoderna inte kan användas (t.ex. ytaktiva material), en beräknat värde eller en uppskattning på grundval av individuella n-oktan och vattenlöslighet. |
|
— |
Alla uppgifter och upplysningar av betydelse för tolkningen av resultaten, särskild i fråga om orenheter i ämnet och ämnets fysiska tillstånd. |
För skakkolvmetoden:
|
— |
Resultatet från en eventuell preliminär uppskattning. |
|
— |
Temperatur vid bestämningen. |
|
— |
Data om de analysförfaranden som används vid bestämning av koncentrationerna. |
|
— |
Eventuell centrifugeringstid och -hastighet. |
|
— |
De mätta koncentrationerna i båda faserna för varje bestämning. (Detta innebär att totalt 12 koncentrationer ska anges.) |
|
— |
Vikten på testämnet, den använda volymen av varje fas i varje testkärl och den totala beräknade mängden testämne som finns i varje fas efter uppnående av jämvikt. |
|
— |
De beräknade värdena för fördelningskoefficienten (P) och medelvärdet för varje uppsättning testbetingelser, liksom medelvärdet för alla bestämningar. Eventuella tecken på att fördelningskoefficienten är koncentrationsberoende ska anges i rapporten. |
|
— |
De enskilda P-värdenas standardavvikelse från genomsnittet. |
|
— |
Genomsnittsvärdet för P från alla bestämningar anges också som dess logaritm (bas 10). |
|
— |
Det beräknade teoretiska värdet av Pow, när detta värde är bestämt eller när det mätta värdet är > 104. |
|
— |
pH-värdet i vattnet som har använts och i vattenfasen under testet. |
|
— |
Motivering för användning av buffertlösningar i stället för vatten, deras sammansättning, koncentration och pH-värde samt vattenfasens pH-värde före och efter testet. |
För HPLC-metoden:
|
— |
Resultatet av en eventuell preliminär uppskattning. |
|
— |
Test- och referensämnen, och deras renhetsgrad. |
|
— |
Temperaturområde vid bestämningen. |
|
— |
Det pH-värde vid vilket bestämningarna gjorts. |
|
— |
Detaljerade uppgifter om analys- och förkolonn, rörlig fas och detektionssystem. |
|
— |
Retentionsdata och hänvisningsvärden för log P för referensämnen som använts vid kalibrering. |
|
— |
Detaljerade uppgifter om anpassning av retentionslinjen (log k mot log P). |
|
— |
Genomsnittliga retentionsdata och interpolerat log P-värde för testämnet. |
|
— |
Beskrivning av utrustning och driftsbetingelser. |
|
— |
Elueringsprofiler. |
|
— |
Den mängd test- och referensämne som tillförts kolonnen. |
|
— |
Dödtiden och hur den är uppmätt. |
4. HÄNVISNINGAR
|
1. |
OECD, Paris, 1981, Test Guideline 107, rådets beslut K(81) 30 slutligt. |
|
2. |
C. Hansch och A. j. Leo, Substituent Constants for Correlation Analysis in Chemistry and Biology, John Wiley, New York, 1979. |
|
3. |
Log P and Parameter Database, A took for the quantitative prediction of bioactivity (C. Hasch, chairman, A. J. Leo, dir.) – Tillgänglig från Pomona College Medical Chemistry Project 1982, Pornona College, Claremont, California 91711. |
|
4. |
L. Renberg, G. Sundström och K. Sundh-Nygård, Chemosphere, 1980, vol. 80, s. 683. |
|
5. |
H. Ellgehausen, C. D'Hondt och R. Fuerer, Pestic. Sci., 1981, vol, 12, s. 219. |
|
6. |
B. McDuffie, Chemosphere, 1981, vol. 10, s. 73. |
|
7. |
W. E. Hammers et al., J. Chromatogr., 1982, vol. 247, s. 1. |
|
8. |
J. E. Haky och A. M. Youg, J. Liq. Chromat., 1984, vol. 7, s. 675. |
|
9. |
S. Fujisawa och E. Masuhara, J. Biomed. Mat. Res., 1981, vol. 15, s. 787. |
|
10. |
O. Jubermann, Verteilen und Extrahieren, in Methoden der Organischen Chemie (Houben Weyl), Allgemeine Laboratoriumpraxis (editerad av E. Muller), Georg Thieme Verlag, Stuttgart, 1958, band I/l, s. 223–339. |
|
11. |
R. F. Rekker och H. M. de Kort, Euro. J. Med. Chem,. 1979, vol. 14., s. 479. |
|
12. |
A. Leo, C. Hansch och D. Elkins, artition coefficients and their uses. Chem. Rev. 1971, vol. 71, s. 525. |
|
13. |
R. F. Rekker, The Hydrophobic Fragmental Constant, Elsevier, Amsterdam, 1977. |
|
14. |
NF T 20-043 AFNOR (1985). Chemical products for industrial use – Determination of partition coefficient – Flask shake method. |
|
15. |
C. V. Eadsforth och P. Moser, Chemosphere, 1983, vol. 12, s. 1459. |
|
16. |
A. Leo, C. Hansch och D. Elkins, Chem. Rev., 1971, vol. 71, s. 525. |
|
17. |
C. Hansch, A. Leo, S. H. Unger, K. H. Dim, D. Nikaitani och E. J. Lien, J. Med. Chem, 1973, vol. 16, s. 1207. |
|
18. |
W. B. Neely, D. R. Branson och G. E. Blau, Environ. Sci. Technol., 1974, vol. 8, s. 1113. |
|
19. |
D. S. Brown och E. W. Flagg, J. Environ. Qual., 1981, vol. 10, s. 382. |
|
20. |
J. K. Seydel och K. J. Schaper, Chemische Struktur und biologische Aktivität von Wirkstoffen, Verlag Chemiw, Weinheim, New York, 1979. |
|
21. |
R. Franke, Theoretical Drug Design Methods, Elsevier, Amsterdam, 1984. |
|
22. |
Y. C. Martin, Quantitative Drug Design, Marcel Dekker, New York, Basel, 1978. |
|
23. |
N. S. Nirrlees, S. J. Noulton, C. T. Murphy, P. J. Taylor; J. Med. Chem., 1976, vol. 19, s. 615. |
Tillägg 1
Metoder för beräkning/uppskattning
INLEDNING
I Handbook of Chemical Property Estimation Methods finns en allmän introduktion till beräkningsmetoder, data och exempel (se hänvisning a).
Beräknade värden av Pow kan användas
|
— |
för att bestämma vilken testmetod som är lämplig (skakkolvområde: log Pow: 2 till 4, HPLC-område: log Pow: 0 till 6), |
|
— |
för val av lämpliga testbetingelser (t.ex. referensämnen för HPLC-förfaranden, volymratio n-oktanol/vatten för skakkolvmetoden), |
|
— |
som en intern laboratoriekontroll vid eventuella försöksfel, |
|
— |
för att få en uppskattning av Pow i fall där testmetoderna inte kan tillämpas av tekniska skäl. |
UPPSKATTNINGSMETOD
Preliminär uppskattning av fördelningskoefficient.
Fördelningskoefficientens värde kan uppskattas med hjälp av testsubstansens löslighet i rena lösningsmedel. För detta används:

BERÄKNINGSMETODER
Principen för beräkningsmetoderna
Alla beräkningsmetoder är baserade på formell fragmentering av molekylen i lämpliga understrukturer för vilka man har pålitlig kännedom om log Pow-inkrementer. Log Pow för hela molekylen beräknas sedan som summan av dess motsvarande fragmentvärden plus summan av korrektionsuttrycken för intramolekylär växelverkan.
En lista med fragmentkonstanter och korrektionsuttryck finns i hänvisningarna b, c, d och e. Vissa av dem uppdateras regelbundet (hänvisning b).
Kvalitetskriteria
Normalt avtar beräkningsmetodens pålitlighet i takt med komplexiteten hos den testade föreningen. För enkla molekyler med låg molekylvikt och en eller två funktionsgrupper kan en avvikelse av 0,1 till 0,3 förväntas mellan resultatet av olika fragmenteringsmetoder och det mätta värdet för log Pow. Felmarginalen kan vara ännu större för mer komplexa molekyler. Detta beror på pålitligheten och förekomsten av fragmentkonstanter, liksom möjligheten att känna igen intramolekylära växelverkningar (t.ex. väteförbindelser) och om korrektionstermen används korrekt (detta är inte ett lika stort problem med programvaran CLOGP-3). 1 fråga om joniserbara föreningar är det av stor betydelse att laddningen eller joniseringsgraden bedöms korrekt.
Beräkningsmetoder
Hansch Π-metoden
Den ursprungliga konstanten för hydrofoba substituenter, π, som införts av Fujita m.fl. (se hänvisning f) definieras genom
πx = log Pow (PhX) - log Pow (PhH)
I enlighet med dess definition är -metoden lämplig för aromatisk substitution. Det finns tabeller med -värden för ett stort antal substitutioner (se hänvisningar b, c och d). De används för beräkning av log Pow för aromatiska molekyler och understrukturer.
(e.g. πCl = log Pow (C6H5Cl) - log Pow (C6H6) = 2,84 - 2,13 = 0,71).
där Pow (PhX) är fördelningskoefficienten av ett aromatiskt derivat och Pow (PhH) är moderföreningens fördelningskoefficient
Rekkers metod
I enlighet med Rekker (se hänvisning g) beräknas värdet av log Pow på följande sätt:

där fi representerar olika molekylfragmentkonstanter och ai är den frekvens med vilken de förekommer i den molekyl som testas. Korrektionstermerna kan uttryckas som en integral multipel av en enda konstant Cm (den s.k. magiska konstanten). Fragmentkonstanterna fi ch Cm bestäms på grundval av en tabell med 1 054 experimentellt bestämda värden av Pow (825 föreningar) med användning av flerdimensionell regressionsanalys (se hänvisningar c och h). Växelverkningsuttrycken bestäms efter fastställda regler som beskrivs i hänvisningslitteraturen (se hänvisningar e, h och i).
Hansch-Leos metod
I enlighet med Hansch och Leo (se hänvisning c) beräknas värden av log Pow på följande sätt:

där fi representerar de olika molekylfragmentkonstanterna, Fi är korrektionsuttrycken och ai och bj är de motsvarande uppträdandefrekvenserna. På grundval av experimentellt bestämda värden av Pow har man genom att prova sig fram kunnat ställa upp en lista över atom- och gruppfragmentvärden och en lista över korrektionsuttryck Fj (s.k. faktorer). Korrektionsuttrycken har delats in i olika klasser (se hänvisningar a och c). Det är relativt komplicerat och tidsödande att ta hänsyn till alla regler och korrektionsuttryck. Programvarupaket har utvecklats för detta (se hänvisning b).
Kombinerad metod
Beräkningen av log Pow för komplexa molekyler kan förbättras betydligt om molekylen delas upp i större delstrukturer för vilka pålitliga log Pow-värden finns tillgängliga, antingen från tabeller (se hänvisningar b och c) eller från egna mätningar. Sådana fragment (t.ex. heterocykler, anthraquinon, azobensen) kan sedan kombineras med Hanschs Π-värden eller med Rekkers eller Leos fragmentkonstanter.
Anmärkningar
|
i) |
Beräkningsmetoderna kan endast användas på helt eller delvis joniserade föreningar om det är möjligt att beakta de nödvändiga korrektionsfaktorerna. |
|
ii) |
Om intramolekylära väteförbindelser kan antas förekomma måste motsvarande korrektionstermer (ca +0,6 till +1,0 log Pow-enheter) läggas till (se hänvisning a). Indikationer av närvaron av sådana förbindelser kan erhållas från stereomodeller eller spektroskopiska data av molekylen. |
|
iii) |
Om flera tautomeriska former är möjliga används den mest sannolika formen som bas för beräkningen. |
|
iv) |
Revisioner av listor med fragmentkonstanter ska följas noggrant. |
Rapportering
Vid användning av beräknings-/uppskattningsmetoder ska testrapporten om möjligt innehålla följande information:
|
— |
Beskrivning av ämnet (blandning, orenheter etc). |
|
— |
Indikation av eventuella intramolekylära väteförbindelser, dissociation, laddning och andra eventuella särskilda förhållanden (t.ex. tautomeri). |
|
— |
Beskrivning av beräkningsmetoden. |
|
— |
Identifiering eller tillhandahållande av databas. |
|
— |
Särskilda val av fragment. |
|
— |
Fullständig dokumentation av beräkningen. |
HÄNVISNINGAR
|
1. |
W. J. Lyman, W. F. Reehl och D. H. Rosenblatt (ed.), Handbook of Chemical Property Estimation Methods, McGraw-Hill, New York, 1983. |
|
2. |
Pomona College, Medicinal Chemistry Project, Claremont, California 92711, USA, Log P Database och Med. Chem. Software (Program CLOGP - 3). |
|
3. |
C. Hansch och A. J. Leo, Substituent Constants for Correlation Analysis in Chemistry and Biology, John Wiley, New York, 1979. |
|
4. |
A. Leo, C. Hansch, D. Elkins, Chem. Rev, 1971, vol. 71, s. 525. |
|
5. |
R. F. Rekker och H. M. de Kort, Euro. J. Med. Chem,. 1979, vol. 14, s. 479. |
|
6. |
6. T. Fujita, J. Iwasa och C. Hansch, J. Amer. Chem. Soc, 1964, vol. 86, s. 5175. |
|
7. |
R. F. Rekker, The Hydrophobic Fragmental Constant, Pharmacochemistry Library, Elsevier, New York, 1977, vol. 1. |
|
8. |
C. V. Eadsforth, P. Moser, Chemosphere, 1983, vol. 12, s. 1459. |
|
9. |
R. A. Scherrer, ACS, American Chemical Sociery, Washington D. C, 19S4, Symposium Senes 255, s. 225. |
Tillägg 2
Rekommenderade referensämnen för HPLC-metoden
|
Nr |
Referensämne |
Log Pow |
pKa |
|
1 |
2-butanon |
0,3 |
|
|
2 |
4-acetylpyndin |
0,5 |
|
|
3 |
Anilin |
0,9 |
|
|
4 |
Acetanilid |
1,0 |
|
|
5 |
Bensylalkohol |
1,1 |
|
|
6 |
p meloxilenol |
1,3 |
pKa = 10,26 |
|
7 |
Fenoxyättiksyra |
1,4 |
pKa = 3,12 |
|
8 |
Fenol |
1,5 |
pKa = 9,92 |
|
9 |
2,4-dinitrofenol |
1,5 |
pKa = 3,96 |
|
10 |
Bensonitril |
1,6 |
|
|
11 |
Fenylättiknitril |
1,6 |
|
|
12 |
4-metylbensenalkohol |
1,6 |
|
|
13 |
Acetonfenon |
1,7 |
|
|
14 |
2-nitrofenol |
1,8 |
pKa = 7,17 |
|
15 |
3-nitrobcnsoesyra |
1,8 |
pKa = 3,47 |
|
16 |
4-kloranilin |
1,8 |
pKa = 4,15 |
|
17 |
Nitrobensen |
1,9 |
|
|
18 |
Kanelalkohol |
1,9 |
|
|
19 |
Bensoesyra |
1,9 |
pKa = 4,19 |
|
20 |
p-kresol |
1,9 |
pKa = 10,17 |
|
21 |
Kanelsyra |
2,1 |
pKa = 3,89 cis 4,44 träns |
|
22 |
Anisol |
2,1 |
|
|
23 |
Metylbensoat |
2,1 |
|
|
24 |
Bensen |
2,1 |
|
|
25 |
3-metylbensoesyra |
2,4 |
pKa = 4,27 |
|
26 |
4-klorfenol |
2,4 |
pKa = 9,1 |
|
27 |
Trikloretylen |
2,4 |
|
|
28 |
Atrazin |
2,6 |
|
|
29 |
Etylbensoat |
2,6 |
|
|
30 |
2,6-diklorbensonitril |
2,6 |
|
|
31 |
3-klorbensoesyra |
2,7 |
pKa = 3,82 |
|
32 |
Toluen |
2,7 |
|
|
33 |
1-naftol |
2,7 |
pKa = 9,34 |
|
34 |
2,3-dikloranilin |
2,8 |
|
|
35 |
Monoklorbensen |
2,8 |
|
|
36 |
Allyl-fenyleter |
2,9 |
|
|
37 |
Bromobensen |
3,0 |
|
|
38 |
Etylbensen |
3,2 |
|
|
39 |
Bensofenon |
3,2 |
|
|
40 |
4-fenylfenol |
3,2 |
pKa = 9,54 |
|
41 |
Tymol |
3,3 |
|
|
42 |
1,4-diklorbensen |
3,4 |
|
|
43 |
Difenylamin |
3,4 |
pKa = 0,79 |
|
44 |
Naftalen |
3,6 |
|
|
45 |
Fenylbensoat |
3,6 |
|
|
46 |
Isopropylbensen |
3,7 |
|
|
47 |
2,4,6-triklorfenol |
3,7 |
PKa = 6 |
|
48 |
Bifenyl |
4,0 |
|
|
49 |
Bensylbensoat |
4,0 |
|
|
50 |
2,4-dinitro-6 sec. butylfenol |
4,1 |
|
|
51 |
1,2,4-triklorbensen |
4,2 |
|
|
52 |
Dodekansyra |
4,2 |
|
|
53 |
Difenyleter |
4,2 |
|
|
54 |
n-butylbensen |
4,5 |
|
|
55 |
Fenantren |
4,5 |
|
|
56 |
Fluoranten |
4,7 |
|
|
57 |
Dibensyl |
4,8 |
|
|
58 |
2,6-difenylpyridin |
4,9 |
|
|
59 |
Tnfenylamin |
5,7 |
|
|
60 |
DDT |
6,2 |
|
|
Andra referensämnen med låg log Pow |
|||
|
1 |
Nikotinsyra |
-0,07 |
|
A.9 FLAMPUNKT
1. METOD
1.1 INLEDNING
Vid utförande av detta test är det värdefullt att ha förhandskunskap om ämnets brandfarlighet. Testförfarandet kan tillämpas på flytande ämnen vars ångor kan antändas av antändningskällor. De testmetoder som beskrivs i denna text är endast tillförlitliga för de flampunktsområden som anges för de enskilda metoderna.
Vid val av försöksmetod ska man ta hänsyn till en eventuell kemisk reaktion mellan ämnet och provbehållaren.
1.2 DEFINITIONER OCH ENHETER
Flampunkten är den lägsta temperatur, korrigerad vid ett tryck av 101,325 kPa, vid vilken testvätskan i ett slutet testkärl, under de betingelser som definieras för testmetoden, utvecklar ånga i sådan mängd att en brandfarlig ång-luftblandning bildas i testkärlet.
Enheter: oC
t = T – 273,15
(t i oC och T i K)
1.3 REFERENSÄMNEN
Man behöver inte använda referensämnen varje gång ett nytt ämne ska undersökas. Referensämnen ska först och främst användas för att då och då kontrollera metoden och för jämförelse med resultat som uppnåtts med andra metoder.
1.4 TESTMETODSPRINCIPER
Ämnet placeras i ett testkärl och värms upp eller kyls ned till testtemperaturen i enlighet med det beskrivna förfarandet för de individuella testmetoderna. Antändningsförsök utförs för att fastställa om provet antänds vid testtemperaturen.
1.5 KVALITETSKRITERIER
1.5.1 Repeterbarhet
Repeterbarheten varierar med flampunktsområde och använd testmetod: högst ± 2 oC.
1.5.2 Sensitivitet
Sensitiviteten är beroende av använd testmetod.
1.5.3 Specificitet
Vissa testmetoders specificitet är begränsad till vissa flampunktsområden och avhängiga av ämnesrelaterade egenskaper (t.ex. hög viskositet).
1.6 METODBESKRIVNING
1.6.1 Förberedelser
Ett prov av testämnet placeras i en testapparat enligt 1.6.3.1 och/eller 1.6.3.2.
Av säkerhetsskäl rekommenderas att man för energiska eller toxiska ämnen endast använder en metod med en liten provstorlek, ca 2 cm3.
1.6.2 Testbetingelser
Apparaten ska, såvida detta är förenligt med säkerhetskraven, placeras fritt från drag.
1.6.3 Testförfarande
1.6.3.1 Jämviktsmetoden
Se ISO 1516, ISO 3680, ISO 1523, ISO 3679.
1.6.3.2 Icke-jämviktsmetoden
Abel-apparat:
Se BS 2000 del 170, NF M07-011, NF T66-009.
Abel-Pensky-apparat:
Se (EN 57), DIN 51755 del 1 (för temperaturer från 5 till 65 oC), DIN 51755 del 2 (för temperaturer under 5 oC), NF M07-036.
Tag-apparat:
Se ASTM D 56.
Pensky-Mårtens-apparat:
Se ISO 2719, EN 11, DIN 51758, ASTM D 93, BS 2000-34, NF M07-019.
Anmärkningar:
Om flampunkten, bestämd med en icke-jämviktsmetod enligt 1.6.3.2, konstateras vara 0 ± 2 oC, 21 ± 2 oC eller 55 ± 2 oC, ska den bekräftas med en jämviktsmetod med samma apparat.
Endast de metoder som kan utvisa flampunktstemperaturen får användas till en anmälan.
För att bestämma flampunkten för viskösa vätskor (färger, lim och liknande) som innehåller lösningsmedel, får endast apparatur och testmetoder som lämpar sig för bestämning av flampunkten för viskösa vätskor användas.
Se ISO 3679, ISO 3680, ISO 1523, DIN 53213 del 1.
|
2. |
DATA |
3. RAPPORTERING
Testrapporten ska, om möjligt, innehålla följande uppgifter:
|
— |
Exakt specificering av ämnet (beteckning och orenheter). |
|
— |
Den använda metoden ska anges liksom eventuella avvikelser. |
|
— |
Resultaten och kompletterande uppgifter av betydelse för tolkningen av resultaten. |
4. HÄNVISNINGAR
Inga.
A.10 ANTÄNDLIGHET (FASTA ÄMNEN)
1. METOD
1.1 INLEDNING
Vid utförande av detta test är det värdefullt att ha förhandskunskap om ämnets eventuella explosiva egenskaper. Testet utförs endast på pulverformiga, granulära och pastaaktiga ämnen.
För att inte ta med alla ämnen som kan antändas, utan endast de som brinner snabbt eller de som på ett eller annat sätt har särskilt farliga egenskaper vid förbränning, anses endast de ämnen vars förbränningshastighet överstiger ett visst gränsvärde vara lättantändliga.
Det kan vara särskilt farligt om glödningen fortsätter genom metallpulver eftersom en brand är mycket svår att släcka.
Metallpulver anses lättantändliga om glödningen kan breda ut sig i provet inom en viss tid.
1.2 DEFINITIONER OCH ENHETER
Förbränningstiden uttrycks i sekunder.
1.3 REFERENSÄMNEN
Inte angivna.
1.4 TESTMETODSPRINCIP
Ämnet formas till ett 250 mm långt obrutet band och det företas ett preliminärt screeningtest för att bestämma om det, efter antändning med en gaslåga, sker en förbränning som breder ut sig med öppen låga eller genom glödning. Om glödningen breder ut sig mer än 200 mm av bandet inom en bestämd tid ska ett fullständigt testprogram utföras för bestämning av förbränningshastigheten.
1.5 KVALITETSKRITERIER
Inte angivna.
1.6 METODBESKRIVNING
1.6.1 Preliminärt screeningtest
Ämnet formas till ett obrutet band som är omkring 250 mm långt, 20 mm brett och 10 mm högt på en ickebrännbar, icke-porös dåligt värmeledande plats. En gaslåga (minsta diameter 5 mm) hålls vid den ena änden av pulverbandet tills pulvret fattar eld, dock högst 2 minuter (5 minuter för metallpulver och metallegeringar). Man noterar om förbränningen breder ut sig 200 mm över bandet under testperiodens 4 minuter (40 minuter för metallpulver). Om ämnet inte fattar eld eller förbränning med öppen låga eller genom glödning inte breder ut sig 200 mm över bandet under testperiodens 4 minuter (40 minuter) anses ämnet inte vara lättantändligt och ytterligare försök är inte nödvändiga. Om förbränningen i ämnet breder ut sig 200 mm över bandet på mindre än 4 minuter eller mindre än 40 minuter för metallpulver utförs nedanstående test (1.6.2).
1.6.2 Test för förbränningshastighet
1.6.2.1 Förberedelse
Pulverformiga eller granulära ämnen fylls löst packat i en form som har en längd av 250 mm och en triangelformad tvärsnittssektion med en invändig höjd av 10 mm och en invändig bredd av 20 mm. På ömse sidor om formen monteras i längdriktningen en sidoavgränsande metallplatta, som skjuter ut 2 mm ovanför den triangelformade tvärsnittssektionen (se figuren). Därefter låter man formen falla tre gånger från 2 cm höjd på ett fast underlag. Om så behövs fylls formen på igen. Därefter tas de sidoavgränsande metallplattorna bort och överskottsämne skrapas bort. En icke-brännbar, icke-porös dåligt värmeledande platta läggs över formen, apparaten vänds upp och ned och formen avlägsnas.
Pastaaktiga ämnen breds ut på en icke-brännbar, icke-porös dåligt värmeledande platta som en 250 mm lång korv med ett tvärsnitt av ungefär 1 cm2.
1.6.2.2 Testbetingelser
Om ämnet är fuktkänsligt ska testet utföras sä snabbt som möjligt efter det att ämnet har avlägsnats från behållaren.
1.6.2.3 Testförfarande
Anordna testhögen längs draget i ett dragskåp.
Luftdraget ska vara tillräckligt för att förhindra rök från att ta sig in i laboratoriet och ska inte ändras under testet. Ett dragskydd bör ställas upp runt apparaten.
En het flamma från en gasbrännare (minsta diameter 5 mm) används för att antända ena änden av högen.
När 80 mm av högen har bränts ska förbränningshastigheten över de nästa 100 mm mätas. Testet ska utföras sex gånger, varje gång på ett rent kallt underlag, såvida inte ett positivt resultat erhållits tidigare.
2. DATA
Bränntiden från det preliminära screeningtestet (1.6.1) och den kortaste förbränningstiden som bestämts i upp till sex test (1.6.2.3) är nödvändiga för utvärderingen.
3. RAPPORTERING
3.1 TESTRAPPORT
Testrapporten ska om möjligt innehålla minst följande uppgifter:
|
— |
Exakt specificering av ämnet (identifiering och orenheter). |
|
— |
En beskrivning av ämnet som ska undersökas, dess fysiska tillstånd, inklusive fukthalt. |
|
— |
Resultat från det preliminära screeningtestet och från test av brännhastighet om sådant utförts. |
|
— |
Alla ytterligare upplysningar som är av betydelse för tolkningen av resultaten. |
3.2 TOLKNING AV RESULTATEN
Pulverformiga, granulära eller pastaaktiga ämnen ska anses vara mycket brandfarliga om bränntiden vid en av testerna, som utförts enligt det i 1.6 beskrivna testförfarandet, är kortare än 45 sekunder. Metallpulver eller metallegeringar anses mycket lättantändliga om de kan antändas och lågan eller reaktionszonen breder ut sig över hela provet inom 10 minuter.
4. HÄNVISNINGAR
NF T 20-042 (Sept. 85). Chemical products for industrial use. Determination of the flammability of solids.
Tillägg
Figur
Form och tillbehör för anordning av testhögen
(Alla mått i mm)

A.11 ANTÄNDLIGHET (GASER)
1. METOD
1.1 INLEDNING
Denna metod gör det möjligt att bestämma om en gas blandad med luft vid rumstemperatur (cirka 20 oC) och atmosfäriskt tryck kan antändas och i så fall i vilket koncentrationsområde. Blandningar med ökande koncentrationer av testgasen i luft utsätts för en elektrisk gnista varvid iakttas om antändning sker.
1.2 DEFINITIONER OCH ENHETER
Området för antändlighet är koncentrationsområdet mellan den nedre och övre explosionsgränsen. De nedre och övre explosionsgränserna är de koncentrationsgränser för den brandfarliga gasen i en blandning med luft vid vilken en flamma inte breder ut sig.
1.3 REFERENSÄMNEN
Inte angivet.
1.4 TESTMETODSPRINCIP
Koncentrationen av gasen i luft ökas stegvis och för varje steg utsätts blandningen för en elektrisk gnista.
1.5 KVALITETSKRITERIER
Inte angivna.
1.6 METODBESKRIVNING
1.6.1 Apparatur
Testkärlet består av en upprättstående glascylinder med en inre diameter på minst 50 mm och en höjd på minst 300 mm. Antändningselektroderna är placerade 3–5 mm från varandra och 60 mm ovanför cylinderns botten. Cylindern är utrustad med en tryckutlösningsöppning. Apparaten ska avskärmas för att begränsa eventuell explosionsskada.
Som antändningskälla används en stående induktionsgnista med en varaktighet på 0,5 sek. Denna gnista genereras av en högspänningstransformator med en utgångsspänning på 10–15 kV (högsta ingångseffekt 300 W). I hänvisning 2 visas ett exempel på en lämplig apparat.
1.6.2 Testbetingelser
Detta test ska utföras vid rumstemperatur (cirka 20 oC).
1.6.3 Testförfarande
Med användning av doseringspumpar införs en känd koncentration av gas i luft i glascylindern. En gnista förs genom blandningen och därvid iakttas om en flamma lösgör sig från antändningskällan och självständigt breder ut sig. Gaskoncentrationen varieras i steg på 1 % volym tills antändning sker enligt beskrivningen ovan.
Om man utifrån kännedom om gasens kemiska struktur kan förmoda att gasen inte kan antändas och att sammansättningen av den stökiometriska blandningen med luft kan beräknas, behöver man endast testa blandningar från 10 % under till 10 % över det stökiometriska blandningsförhållandet.
2. DATA
Förekomsten av flamutbredning är den enda datauppgiften av betydelse för bestämning av denna egenskap.
3. RAPPORTERING
Testrapporten ska om möjligt innehålla följande uppgifter:
|
— |
Exakt specificering av ämnet (beteckning och orenheter). |
|
— |
En beskrivning av den använda apparaturen med måttangivelser. |
|
— |
Temperaturen vid vilken testet gjordes. |
|
— |
Undersökta koncentrationer och erhållna resultat. |
|
— |
Testresultaten: inte antändlig gas eller lättantändlig gas. |
|
— |
Om slutsatsen blir ”inte antändlig”, ska man ange det koncentrationsområde inom vilket provning med 1 % intervaller har skett. |
|
— |
Alla uppgifter och upplysningar som är av betydelse för tolkningen av resultaten ska rapporteras. |
4. HÄNVISNINGAR
|
1. |
NF T 20-041 (Sept. 85), Chemical products for industrial use. Determination of flammability of gases. |
|
2. |
W. Berthold, D. Conrad, T. Grewser, H. Grosse-Wortmann, T. Redeker och H. Schake. ”Entwicklung einer Standard-Apparatur zur Messung von Explosionsgrenzen”. Chem.-Ing.-Tech., 1984, vol. 56, 2, s. 126–127. |
A.12 ANTÄNDLIGHET (KONTAKT MED VATTEN)
1. METOD
1.1 INLEDNING
Denna testmetod kan användas till att bestämma om ett ämne vid reaktion med vatten eller fuktig luft utvecklar farliga mängder gas som eventuellt är lättantändliga.
Testmetoden kan tillämpas på både fasta och flytande ämnen. Denna metod kan inte tillämpas på ämnen som antänds spontant i kontakt med luft.
1.2 DEFINITIONER OCH ENHETER
Lättantändliga: Ämnen som vid kontakt med vatten eller fuktig luft utvecklar lättantändliga gaser i farliga mängder med en lägsta hastighet på 1 liter/kg per timme.
1.3 TESTMETODSPRINCIP
Ämnet testas enligt det stegförfarande som beskrivs nedan. Testet kan avbrytas vid antändning. Om man vet att ämnet inte reagerar våldsamt med vatten kan man börja med steg 4 (1.3.4).
1.3.1 Steg 1
Testämnet placeras i ett tråg innehållande destillerat vatten vid 20 oC och därvid iakttas om den utvecklade gasen antänds eller inte.
1.3.2 Steg 2
Testämnet placeras på ett filtrerpapper som flyter på ytan av en skål innehållande destillerat vatten vid 20 oC, och man iakttar om den utvecklade gasen antänds eller inte. Filtrerpapperet fyller endast funktionen att hålla ämnet kvar på plats för att öka möjligheten till antändning.
1.3.3 Steg 3
Testämnet formas till en hög, ungefär 2 cm hög och med 3 cm diameter. Ett par droppar vatten tillförs högen och man iakttar om den utvecklade gasen antänds eller inte.
1.3.4 Steg 4
Testämnet blandas med destillerat vatten vid 20 oC och gasutvecklingshastigheten mäts varje timme under sju timmar. Om utvecklingshastigheten efter sju timmar är oregelbunden eller ökande ska mättiden förlängas till högst fem dagar. Testet kan avbrytas om hastigheten vid något tillfälle överstiger 1 liter/kg per timme.
1.4 REFERENSÄMNE
Inte angivet.
1.5 KVALITETSKRITERIER
Inte angivna.
1.6 METODBESKRIVNING
1.6.1 Steg 1
1.6.1.1 Testbetingelser
Testet utförs vid rumstemperatur (ca 20 oC).
1.6.1.2 Testförfarande
En liten mängd (ungefär 2 mm diameter) av testämnet ska placeras i ett tråg innehållande destillerat vatten. Det iakttas om a) gas utvecklas och b) om gasen antänds. Om gasen antänds behöver ämnet inte undersökas ytterligare eftersom ämnet betraktas som farligt.
1.6.2 Steg 2
1.6.2.1 Apparatur
Ett filtrerpapper placeras flytande på ytan av destillerat vatten i lämpligt kärl, t.ex. en avdunstningsskål med 100 mm diameter.
1.6.2.2 Testbetingelser
Testet utförs vid rumstemperatur (ca 20 oC).
1.6.2.3 Testförfarande
En liten mängd av testämnet (ungefär 2 mm diameter) placeras i mitten av ett filtrerpapper. Det iakttas om a) gas utvecklas och b) om gasen antänds. Om gasen antänds behöver ämnet inte undersökas ytterligare eftersom ämnet betraktas som farligt.
1.6.3 Steg 3
1.6.3.1 Testbetingelser
Testet utförs vid rumstemperatur.
1.6.3.2 Testförfarande
Testämnet formas till en hög, ungefär 2 cm hög och med 3 cm diameter, med en liten fördjupning i toppen. Ett par droppar vatten tillsätts i fördjupningen och man iakttar om a) gas utvecklas och b) om gasen antänds. Om gasen antänds behöver ämnet inte undersökas ytterligare eftersom ämnet betraktas som farligt.
1.6.4 Steg 4
1.6.4.1 Apparatur
Apparaturen monteras enligt figuren.
1.6.4.2 Testbetingelser
Undersök om behållaren, i vilken testämnet finns, innehåller pulver < 500 μm (partikelstorlek). Om pulvret utgör mer än 1 % viktprocent av den totala mängden, eller om provet är sprött, ska hela ämnet malas till pulver innan det undersöks för att medge en minskning av partikelstorleken under lagring och hantering. I övrigt ska ämnet användas i det tillstånd det togs emot. Testet ska utföras vid rumstemperatur (20 oC) och atmosfäriskt tryck.
1.6.4.3 Testförfarande
10 till 20 ml vatten tillsätts i apparatens dropptratt, och 10 g av ämnet placeras i den koniska kolven. Den gasvolym som utvecklas kan mätas med lämplig metod. Kranen på dropptratten öppnas för att släppa in vattnet i den koniska kolven och ett stoppur startas. Gasutvecklingen mäts varje timme under en sjutimmarsperiod. Om gasutvecklingen är oregelbunden under denna period, eller om hastigheten för gasutvecklingen ökar vid slutet av denna period, fortsätter mätningarna i upp till fem dagar. Om gasutvecklingen vid någon tidpunkt överstiger 1 liter/timme kan testet avbrytas. Detta test ska utföras tre gånger.
Om gasens kemiska identitet är okänd ska gasen analyseras. Om gasen innehåller mycket brandfarliga komponenter och det är okänt om hela blandningen är mycket farlig, ska en blandning med samma sammansättning framställas och undersökas enligt testmetod A.11.
2. DATA
Ämnet anses farligt om
|
— |
spontan antändning sker under testförfarandet, eller |
|
— |
brandfarlig gas utvecklas i högre mängd än 1 liter/kg substans per timme. |
3. RAPPORTERING
Testrapporten ska om möjligt innehålla följande uppgifter:
|
— |
Exakt specificering av ämnet (beteckning och orenheter). |
|
— |
Uppgifter om eventuell inledande behandling av testämnet. |
|
— |
Testresultaten (steg 1, 2, 3 och 4). |
|
— |
Den utvecklade gasens kemiska sammansättning. |
|
— |
Gasens utvecklingshastighet och steg 4 (1.6.4) utfördes. |
|
— |
Eventuella ytterligare upplysningar som är av betydelse för tolkningen av resultaten. |
4. HÄNVISNINGAR
|
1. |
Recommendations on the transport of dangerous goods, test and criteria, 1990, United Nations, New York. |
|
2. |
NF T 20-040 (Sept. 85). Chemical products for industrial use. Determination of the flammability of gases formed by the hydrolysis of solid and liquid products. |
Tillägg
Figur
Apparatur

A.13 PYROFORISKA EGENSKAPER HOS FASTA OCH FLYTANDE ÄMNEN
1. METOD
1.1 INLEDNING
Testförfarandet kan tillämpas på fasta och flytande ämnen som i små mängder antänds spontant kort tid efter det att de har kommit kontakt med luft vid rumstemperatur (cirka 20 oC).
Ämnen som kräver timmar eller dagar vid rumstemperatur eller förhöjd temperatur innan de självantänds omfattas inte av denna testmetod.
1.2 DEFINITIONER OCH ENHETER
Ämnen anses vara mycket brandfarliga om de antänds eller förkolnar under de betingelser som beskrivs i 1.6.
Det kan även vara nödvändigt att undersöka vätskors självantändlighet med metod A.15: Självantändlighetstemperatur (vätskor och gaser).
1.3 REFERENSÄMNEN
Inte angivna.
1.4 TESTMETODSPRINCIP
Ämnet, fast eller flytande, blandas med en inaktiv bärare och får under fem minuter vara i kontakt med luften vid rumstemperatur. Flytande ämnen som inte fattar eld absorberas sedan de på filterpapper och utsätts under fem minuter för luftens påverkan vid rumstemperatur (cirka 20 oC). Fasta och flytande ämnen som fattar eld och vätskor som fattar eld eller får filterpappret att förkolna anses vara självantändliga.
1.5 KVALITETSKRITERIER
Repeterbarhet: Av säkerhetsskäl räcker det med ett positivt resultat för att ämnet ska bedömas vara självantändligt.
1.6 METODBESKRIVNING
1.6.1 Apparatur
En porslinskopp, diameter ca 10 cm, fylls med kiselgur upp till ungefär 5 mm höjd vid rumstemperatur (ca 20 oC).
Anmärkning:
Kiselgur eller annat jämförbart inert ämne som är allmänt tillgängligt ska anses vara representativt för jord på vilken testämnet kan spillas ut vid en olyckshändelse.
Det behövs ett torrt filterpapper för att testa vätskor som inte fattar eld vid kontakt med luften när de blandas med en inaktiv bärare.
1.6.2 Testförfarande
a) Pulverformiga fasta ämnen
1–2 cm3 av det pulverformiga testämnet hälls från cirka 1 m höjd på en icke brännbar yta, varvid iakttas om ämnet antänds under fallet eller innan det har gått fem minuter efter det att det har lagt sig.
Testet utförs sex gånger såvida inte antändning sker.
b) Vätskor
Cirka 5 cm3 av testvätskan hälls i den förberedda porslinskoppen, varvid iakttas om ämnet antänds inom fem minuter.
Orn inte antändning sker i något av de sex testerna utförs följande test:
Med en injektionsspruta sprutas 0,5 ml av provet på ett böjt filterpapper och det observeras om antändning eller förkolning av filterpappret sker inom fem minuter. Testet utförs tre gånger såvida inte antändning eller förkolning sker.
2. DATA
2.1 TOLKNING AV RESULTAT
Testet kan avbrytas så snart som ett av testen ger positivt resultat.
2.2 UTVÄRDERING
Om ämnet fattar eld inom fem minuter efter det har tillsatts till en inaktiv bärare och exponeras för luft, eller en vätska får en bit filterpapper att förkolna eller fatta eld inom fem minuter efter det att ämnet tillsatts och utsatts för luftens påverkan anses ämnet självantändligt.
3. RAPPORTERING
Testrapporten ska om möjligt innehålla följande uppgifter:
|
— |
Exakt specificering av ämnet (beteckning och orenheter). |
|
— |
Testresultaten. |
|
— |
Eventuella ytterligare upplysningar som är av betydelse för tolkningen av resultaten. |
4. HÄNVISNINGAR
|
1. |
NF T 20-039 (Sept. 85). Chemical products for industrial use. Determination of the spontaneous flammability of solids and liquids. |
|
2. |
Recommendations on the Transport of Dangerous Goods, Test and Criteria, 1990, United Nations, New York. |
A.14 EXPLOSIVA EGENSKAPER
1. METOD
1.1 INLEDNING
Metoden innefattar ett testschema för att bestämma om ett fast, flytande eller pastaaktigt ämne eller preparat utgör en explosionsfara om det utsätts för påverkan av en öppen låga (värmesensitivitet) eller slag- eller friktionspåverkan (sensitivitet för mekanisk påverkan) och om ett flytande ämne utgör en explosionsfara om det utsätts för påverkan av en öppen låga eller slag.
Metoden består av tre delar:
|
a) |
test av värmesensitiviteten (se hänvisning 1), |
|
b) |
test av mekanisk sensitivitet avseende slagpåverkan (se hänvisning 1), |
|
c) |
test av mekanisk sensitivitet avseende friktionspåverkan (se hänvisning 1). |
Metoden ger data för att utvärdera sannolikheten att det vid vissa ovanliga påverkningar framkallas en explosion. Metoden är inte avsedd att säkerställa om ett ämne över huvud taget kan explodera.
Metoden är lämplig för att bestämma om ett ämne innebär en explosionsfara (värme- och mekanisk känslighet) under de särskilda betingelser som anges i direktivet. Metoden grundas på ett antal apparater som är allmänt använda internationellt (se hänvisning 1) och som vanligtvis ger användbara resultat. Det erkänns att metoden inte är definitiv. Alternativ apparatur kan användas förutsatt att den är internationellt erkänd och att de erhållna resultaten kan korreleras med de resultat som erhållits med den angivna apparaturen.
Testen behöver inte utföras om det enligt föreliggande termodynamiska uppgifter (t.ex. bildningsvärme, nedbrytningsvärme) och/eller frånvaro av vissa reaktiva grupper (se hänvisning 2) i strukturformeln är ställt utom rimligt tvivel att ämnet inte kan brytas ned, bilda gaser och frigöra värme mycket snabbt (dvs. materialet utgör inte någon explosionsrisk). Ett test av mekanisk sensitivitet avseende friktion krävs inte för flytande ämnen.
1.2 DEFINITIONER OCH ENHETER
Explosiva:
Ämnen som kan explodera när de utsätts för en öppen låga eller som är känsliga för slag och friktion i den angivna apparaturen (eller är mer mekaniskt känsliga än 1,3-dinitrobensen i alternativ apparatur).
1.3 REFERENSÄMNE
1,3-dinitrobensen, teknisk kristallinprodukt som siktats genom 0,5 mm, för friktions- och slagpåverkanmetoden.
Perhydro-l,3,5-trinitro-l,3,5-triazin (RXD, hexogen, cyclonit – CAS 121-82-4), omkristalliserad från vattnigt cyclohexanon, våtsiktad genom en 250 μm och bibehållen i en 150 μm sil, torkad vid 103 ± 2 oC (i 4 timmar) för den andra serien av friktions- och slagtester.
1.4 TESTMETODSPRINCIP
Förberedande tester är nödvändigt för att fastställa säkra betingelser för utförandet av de tre sensitivitetstestet:
1.4.1 Test för säkerhet vid hantering (se hänvisning 3)
Av säkerhetsskäl ska, innan huvudtestet inleds, mycket små prov (cirka 10 mg) av ämnet utsättas för upphettning, utan att vara inneslutna, med en gaslåga, för slagpåverkan i lämplig typ av apparatur och friktionspåverkan genom användning av en hammare mot ett städ eller någon typ av friktionsmaskin. Syftet är att fastställa om ämnet är så sensitivt och explosivt att de föreskrivna sensitivitetstesterna, särskilt test av värmekänsligheten, ska utföras med särskilda försiktighetsåtgärder för att undvika att den som utför testen skadas.
1.4.2 Värmekänslighet
Metoden innebär att ämnet värms upp i ett stålrör som stängs med en strypfläns med olika håldiametrar. På detta sätt kan man bestämma om ämnet är benäget att explodera under intensiv värmepåverkan vid väl definierad inneslutning.
1.4.3 Mekanisk känslighet (slagpåverkan)
Metoden innebär att ämnet utsätts för slagpåverkan från en bestämd massa från en bestämd höjd.
1.4.4 Mekanisk känslighet (friktion)
Metoden innebär att ämnet eller preparatet utsätts för friktion mellan standardytor under specificerade betingelser för belastning och relativ rörelse.
1.5 KVALITETSKRITERIER
Inte angivna.
1.6 METODBESKRIVNING
1.6.1 Värmekänslighet (effekt av öppen liga)
1.6.1.1 Apparatur
Apparaturen består av ett engångsstålrör med en återanvändbar stängningsanordning (se figur 1) som placeras i en uppvärmnings- och skyddsanordning. Röret är tillverkat av djupdragen stålplåt (se tillägg) och har en innerdiameter av 24 mm, en längd av 75 mm och en väggtjocklek av 0,5 mm. I den öppna änden är röret flänsat så att det kan slutas med strypflänsanordningen (figur 1). Detta består av en tryckmotståndskraftig skyddsfläns med ett hål i mitten, som är stadigt förankrad till ett rör genom ett tvådelat skruvförband (mutter och gängad krage). Muttern och den gängade kragen är tillverkade av krom-manganstål (se tillägg), vilket är gnistfritt upp till 800 oC. Det finns strypflänsar med en rad olika håldiametrar, som är gjorda av 6 mm tjockt värmebeständigt stål (se tillägg).
1.6.1.2 Testbetingelser
Normalt testas ämnet i den form det är mottaget, men i vissa fall, t.ex. om det är pressat, gjutet eller på annat sätt sammanpressat, kan det vara nödvändigt att testa ämnet efter sönderdelning.
För fasta ämnen bestäms den mängd material som ska användas för varje test genom ett förfarande i två steg. I det första steget fylls ett tarterat rör med 9 cm 3av ämnet som sammanpressas med en kraft av 80 N fördelat över hela rörtvärsnittet. Av säkerhetsskäl, eller om ämnet kan ändra fysisk form vid kompression, kan andrapåfyllningsförfaranden användas. Sammanpressning är t.ex. inte lämpligt om ämnet är mycket friktionskänsligt. Om materialet kan komprimeras tillsätts och sammanpressas mera tills röret är fyllt till en höjd av 55 mm från överkanten. Den totala mängd som har använts för att fylla röret till denna höjd bestäms och ytterligare 2 portioner som sammanpressas med en kraft av 80 N tillsätts. Därefter tillsätts material genom sammanpressning eller så avlägsnas material tills röret är fyllt till 15 mm från överkanten. Det andra steget består i att en tredjedel av den samlade mängd som användes i det första steget tillsätts och sammanpressas. Därefter tillsätts ytterligare 2 portioner som sammanpressas med en kraft av 80 N och materialets höjd i röret justeras till 15 mm från överkanten genom tillsättning eller avlägsnande av material. Vid varje försök används den mängd fast ämne som har fastställts i detta andra steg. Påfyllningen sker i tre lika stora portioner som var och en komprimeras till 9 cm3 med den kraft som krävs. (Detta är lättast att göra genom användning av avståndsringar.)
Vätskor och geléaktiga ämnen fylls i röret till en höjd av 60 mm varvid försiktighet iakttas med geléaktiga ämnen för att undvika luftbubblor under påfyllning. Den gängade kragen förs upp på röret underifrån, den valda strypflänsen sätts in och muttern åtstramas efter smörjning med ett molybdensulfidbaserat smörjmedel. Det är viktigt att kontrollera att inget ämne har fastnat mellan flänsen och plattan eller i gängorna.
Uppvärmningen sker med propan, som från en industriell cylinder med tryckreglator (60–70 mbar) leds genom en mätare och fördelas jämnt till fyra brännare genom en anslutning (vilket kontrolleras genom visuell inspektion av brännarnas lågor).
1.6.1.3 Testförfarande
Varje test pågår tills röret antingen sönderdelas eller har varit uppvärmt i 5 minuter. Ett test vid vilket röret sönderdelas i tre eller flera fragment, som eventuellt hänger ihop med smala metallstrimlor enligt figur 2, betraktas som en explosion. Ett test som leder till färre fragment eller ingen fragmentering betraktas inte som en explosion.
Först utförs tre tester med en strypfläns med ett 6 mm hål, och om ingen explosion sker utförs ytterligare tre test med en strypfläns med ett 2 mm hål. Om explosion sker under ett av dessa tester är ytterligare testning inte nödvändig.
1.6.1.4 Utvärdering
Testresultatet anses positivt om en explosion inträffar i en av ovan nämnda testserier.
1.6.2 Mekanisk sensitivitet (slagpåverkan)
1.6.2.1 Apparatur (figur 4)
En typisk fallhammarapparatur består i huvudsak av ett gjutstålsblock med fot, städ, pelare, gejder, fallvikter, utlösningsmekanism och provhållare. Stålstädet, 100 mm (diameter) × 70 mm (höjd) är fastskruvat ovanpå ett stålblock, 230 mm (längd) × 250 mm (bredd) × 200 mm (höjd) med gjuten fot, 450 mm (längd) × 450 mm (bredd) × 60 mm (höjd). En pelare bestående av en heldraget stålrör är fastsatt i en hållare som är fastskruvad bak på stålblocket. Apparaten är förankrad i ett massivt betongblock, 60 x 60 x 60 cm med fyra skruvar, så att gejderna är fullständigt lodräta och fallvikterna kan falla fritt. Det finns fallvikter på 5 och 10 kg framställda i massivt stål. Slagvikternas slagyta är av härdat stål, HRC 60–63, och har en diameter av minst 25 mm.
Provet placeras i en slaganordning som består av två massiva stålcylindrar som är placerade koaxialt ovanför varandra i en ihålig cylindrisk styrring av stål. Stålcylindrarna ska ha en diameter av 10 (–0,003, –0,005) mm och en höjd av 10 mm och ha polerade ytor, rundade kanter (kurvradie 0,5 mm) och en hårdhet av HRC 58–65. Den ihåliga cylindern ska ha en utvändig diameter av 16 mm, en polerad urborrning av 10 (+0,005, +0,010) mm och en höjd av 13 mm. Slaganordningen samlas på ett mellanstäd (diameter 26 mm, höjd 26 mm) av stål och centreras med en ring i vilken det finns perforeringar för att leda bort rökgaser.
1.6.2.2 Testbetingelser
Provet ska ha en volym på 40 mm3 eller en volym som passar till en alternativ apparatur. Fasta ämnen testas i torrt tillstånd och förbereds enligt följande:
|
a) |
Pulverformiga ämnen siktas (siktstorlek 0,5 mm); allt som har passerat sikten används till testet. |
|
b) |
Pressade, gjutna eller på annat sätt sammanpressade ämnen sönderdelas och siktas, siktfraktioner från 0,5–1 mm i diameter används vid testet. |
Ämnen som normalt förekommer i pastaform bör om möjligt testas i torrt tillstånd och under alla omständigheter efter borttagning av så mycket upplösningsmedel som möjligt. Vätskor testas med ett avstånd av 1 mm mellan den översta och den nedersta stålcylindern.
1.6.2.3 Testförfarande
En serie av sex tester utförs i vilka den 10 kg tunga massan får falla från 0,40 m (40 J). Om en explosion erhålls under de sex testerna vid 40 J måste en andra serie bestående av sex tester utföras i vilka en 5 kg tung massa får falla från 0,15 m (7,5 J). I andra apparaturer jämförs provet med det valda referensämnet med användning av ett fastställt förfarande (tvåslagsteknik osv.).
1.6.2.4 Utvärdering
Testresultatet anses positivt om en explosion (eldutveckling och knall är likvärdiga med explosion) inträffar minst en gång i ett av testerna med den beskrivna slagapparaturen eller provet är mer sensitivt än 1,3-dinitrobensen eller RXD i en alternativt slagpåverkantest.
1.6.3 Mekanisk sensitivitet (friktion)
1.6.3.1 Apparatur (figur 5)
Friktionsapparaturen består av en bottenplatta av gjuten stål på vilken själva friktionsanordningen är monterad. Denna består av en fast porslinspinne och rörliga porslinsplåtar. Porslinsplåten är fäst i en glidbar vagga som rör sig mellan två skenor. Vaggan drivs via en drivstång, en excentrisk trissa och en överföringsväxel av en elektrisk motor på sådant sätt att porslinsplåten rörs under porslinspinnen med en bakåt- och framåtgående rörelse på 10 mm. Porslinspinnen ska belastas med t.ex. 120 eller 360 newton.
Porslinsplattorna är tillverkade av vitt tekniskt porslin (uppruggningsdjup 9–32 μm) och har dimensionerna 25 mm (längd) × 25 mm (bredd) × 5 mm (höjd). Även den cylindriska porslinspinnen är tillverkad av vitt tekniskt porslin, är 15 mm lång, har en innerdiameter på 10 mm och uppruggade sfäriska ändytor med en rundningsradie på 10 mm.
1.6.3.2 Testbetingelser
Provet ska ha en volym av 10 mm3 eller en volym som passar till en alternativ apparatur.
Fasta ämnen testas i torrt tillstånd och förbereds enligt följande:
|
a) |
Pulverformiga ämnen siktas (siktstorlek 0,5 mm); allt som har passerat sikten används till testet. |
|
b) |
Pressade, gjutna eller på annat sätt sammanpressade ämnen sönderdelas och siktas, siktfraktioner < 0,5 mm i diameter används vid testet. |
Ämnen som normalt förekommer i pastaform bör om möjligt testas i torrt tillstånd. Om ämnet inte kan testas i torrt tillstånd ska pastan (efter borttagning av så mycket upplösningsmedel som möjligt) testas som en 0,5 mm tjock, 2 mm bred och 10 mm lång film, som framställs med en speciell form.
1.6.3.3 Testförfarande
Porslinspinnen anbringas på testprovet och vikten hängs på. När testet utförs ska slipmärkena på porslinsplattan ligga tvärs emot rörelseriktningen. Kontrollera noggrant att pinnen vilar på provet, att tillräckligt med testämne ligger under pinnen och att plattan rör sig korrekt under pinnen. För pastaaktiga ämnen används en 0,5 mm tjock form med en hål av 2 × 10 för att anbringa ämnet på plattan. Porslinsplattan ska flytta sig 10 mm fram och tillbaka under porslinspinnen under 0,44 sekunder. Varje del av porslinsplattan och porslinspinnen får endast användas en gång. Varje pinnes två ändar räcker till två försök och de två ytorna på en platta räcker vardera för tre försök.
Sex försök med en belastning av 360 N utförs. Om en explosion förekommer i ett av dessa sex försök utförs en ny serie med sex försök med en belastning av 120 N. I andra apparater jämförs proven med det valda referensämnet med hjälp av ett fastställt förfarande (t.ex. up- and down-teknik).
1.6.3.4 Utvärdering
Testresultatet anses positivt om en explosion (krepitation, eldutveckling och knall är likvärdiga med explosion) inträffar minst en gång i ett av testerna med den beskrivna friktionsapparaturen eller de motsvarande kriterierna i ett alternativ friktionstest är uppfyllda.
2. DATA
Ett ämne anses i princip vara explosionsfarligt i direktivets bemärkelse om ett positivt resultat erhålls i testet av värmesensitivitet, slagpåverkantest eller friktionspåverkantest.
3. RAPPORTERING
3.1 TESTRAPPORT
Testrapporten ska om möjligt innehålla minst följande uppgifter:
|
— |
Testämnets beteckning, sammansättning, renhet, fukthalt osv. |
|
— |
Provets fysiska form och om det har krossats, brutits eller siktats. |
|
— |
Iakttagelser under värmesensitivitetstestet (t.ex. provmassa, antal fragment). |
|
— |
Iakttagelser under testet av mekanisk känslighet (t.ex. avsevärd rökutveckling eller fullständig nedbrytning utan knall, lågor, gnistor, sprakning). |
|
— |
Resultat för varje test. |
|
— |
Vetenskapliga grunder för eventuell användning av alternativ apparatur, samt tecken på korrelation mellan resultat som erhållits med beskriven apparatur och de som erhållits med likvärdig apparatur. |
|
— |
Alla relevanta kommentarer, t.ex. hänvisning till test med liknande produkter, som kan vara av betydelse för en korrekt tolkning av resultaten. |
|
— |
Alla ytterligare kommentarer som är relevanta för tolkning av resultaten. |
3.2 TOLKNING OCH UTVÄRDERING AV RESULTAT
I testrapporten ska alla resultat som anses vara falska, avvikande eller icke representativa anges. Om något resultat ska bortses från ska en förklaring ges och resultaten av ett alternativt eller kompletterande test anges. Om ett avvikande resultat inte kan förklaras på detta sätt ska det godtas för vad det är och i enlighet därmed användas vid klassificering av ämnet.
4. HÄNVISNINGAR
|
1. |
Recommendations on the Transport of Dangerous Goods: Test and criteria., 1990, Förenta nationerna, New York. |
|
2. |
Bretherick, L., Handbook of Reactive Chemical Hazards, 4th edition, Butterworths, London, ISBN 0-750- 60103-5, 1990. |
|
3. |
Koenen, H., Ide, K. och Swart, K. H., Explosivstoffe, 1961, vol. 3, s. 6–13 och 30–42. |
|
4. |
NF T 20-038 (Sept. 85), Chemical products for industrial use – Determination of explosion risk. |
Tillägg
Exempel på materialspecifikation för värmesensitivitetstestet (se DIN 1623)
|
1. |
Rör: Materialspecifikation nr 1.0336.505 g. |
|
2. |
Strypfläns: materialspecifikation nr 1.4873. |
|
3. |
Gängad krage och mutter: materialspecifikation nr 1.3817. |
Figur 1
Apparatur för värmesensitivitetstest
(Alla mått i mm)

Figur 2
Värmesensitivitetstest
Exempel på fragmentering

Figur 3
Kalibrering av uppvärmningshastigheten vid test av värmesensitivitet

Kurvan över temperaturförloppet mot tiden vid uppvärmning av dibutylftalat (27 cm3) i ett stängt rör (strypfläns med 1,5 mm hål) med ett propanflöde av 3,2 liter/minut. Temperaturen är mätt med kromel/alumel-termoelement med 1 mm inkapsling i rostfritt stil, placerat centralt 43 mm under rörens överkant. Mellan 135 oC och 285 oC ska uppvärmningshastigheten vara 185–215 K/minut.
Figur 4
Slagpåverkanapparatur
(Alla mått i mm)

Figur 4
Fortsättning

Figur 5
Apparatur för friktionssensitivitet

A.15 SJÄLVANTÄNDNINGSTEMPERATUR (VÄTSKOR OCH GASER)
1. METOD
1.1 INLEDNING
Explosiva ämnen och ämnen som antänds spontant vid kontakt med luft vid omgivningstemperatur, ska inte användas vid detta test. Testförfarandet får tillämpas på gaser, vätskor och ångor som i närvaro av luft kan antändas av en varm yta.
Självantändningstemperaturen kan sänkas avsevärt vid förekomst av katalytiska orenheter, av ytmaterialet eller av en större volym på testkärlet.
1.2 DEFINITIONER OCH ENHETER
Graden av självantändlighet uttrycks som självantändningstemperaturen. Självantändningstemperaturen är den lägsta temperatur vid vilken testämnet antänds när det blandas med luft under de betingelser som definierats för testmetoden.
1.3 REFERENSÄMNEN
Referensämnen återges i standarderna (1.6.3). De ska först och främst användas till att emellanåt kalibrera metoden och till att möjliggöra en jämförelse av resultaten från andra metoder.
1.4 TESTMETODSPRINCIP
Med denna metod bestäms den lägsta temperaturen hos den invändiga ytan i ett slutet rum, vid vilken en gas, ånga eller vätska som sprutas in i rummet, fattar eld.
1.5 KVALITETSKRITERIER
Repeterbarheten varierar beroende på självantändlighetens temperaturområde och den använda testmetoden.
Sensitiviteten och specificiteten beror på den använda testmetoden.
1.6 METODBESKRIVNING
1.6.1 Apparatur
Apparaturen finns beskriven i den metod som avses i 1.6.3.
1.6.2 Testbetingelser
Ett prov av testämnet undersöks enligt den metod som avses i 1.6.3.
1.6.3 Testförfarande
Se IEC 79-4, DIN 51794, ASTM-E 659-78, BS 4056, NF T 20-037.
2. DATA
Testtemperaturen, atmosfäriskt tryck, mängden använt prov och tidsintervall registreras innan antändning sker.
3. RAPPORTERING
Testrapporten ska om möjligt innehålla följande uppgifter:
|
— |
Exakt specifikation av ämnet (beteckning och orenheter). |
|
— |
Mängden använt prov, atmosfäriskt tryck. |
|
— |
Använd apparatur. |
|
— |
Mätresultaten (testtemperaturer, resultat beträffande antändning, motsvarande tidsintervall). |
|
— |
Alla ytterligare upplysningar som är av betydelse för tolkningen av resultaten. |
4. HÄNVISNINGAR
Inga.
A.16 RELATIV SJÄLVANTÄNDNINGSTEMPERATUR (FASTA ÄMNEN)
1. METOD
1.1 INLEDNING
Explosiva ämnen och ämnen som antänds spontant vid kontakt med luft vid omgivningstemperatur ska inte användas vid detta test.
Syftet med detta test är att få preliminära uppgifter om fasta ämnens självantändlighet vid förhöjda temperaturer.
Om den värme som utvecklas, antingen genom ämnets reaktion med syre eller genom exoterm nedbrytning, inte avges tillräckligt snabbt till omgivningen inträffar en självuppvärmning som leder till självantändning. Självantändning inträffar således när värmeproduktionens hastighet överstiger värmeförlustens hastighet.
Tcstförfarandet är värdefullt som ett förberedande screentest för fasta ämnen. Med tanke på det komplicerade förhållandet vid antändning och förbränning av fasta ämnen, ska självantändningstemperaturen som bestäms enligt denna metod endast användas i jämförande syfte.
1.2 DEFINITIONER OCH ENHETER
Den självantändningstemperatur som erhållits med denna metod är den lägsta omgivningstemperatur, uttryckt i oC, vid vilken en viss volym av ett ämne självantänds under definierade betingelser.
1.3 REFERENSÄMNEN
Inga.
1.4 TESTMETODSPRINCIP
En viss volym av testämnet placeras i en ugn vid rumstemperatur; temperatur-/tidskurvan avseende betingelserna i provets mitt registreras medan temperaturen i ugnen ökas till 400 oC, eller till smältningspunkten om denna är lägre, med en hastighet av 0,5 oC per minut. Den ugnstemperatur vid vilken provets temperatur uppnår 400 oC genom självuppvärmning kallas i detta test självantändningstemperaturen.
1.5 KVALITETSKRITERIER
Inga.
1.6 METODBESKRIVNING
1.6.1 Apparatur
1.6.1.1 Ugn
En temperaturprogrammerad laboratorieugn (volym ca 2 liter) utrustad med naturlig luftcirkulation och explosionssäkring. För att undvika eventuell explosionsrisk ska det säkerställas att nedbrytningsgaser inte kan komma i kontakt med de elektriska värmeelementen.
1.6.1.2 Kub av ståltrådsnät
En bit rostfritt ståltrådsnät med 0,045 mm maskor skärs till enligt mallen i figur 1. Nätet viks och fästs ihop med tråd till kuber som är öppna på ovansidan.
1.6.1.3 Termoelement
Passande termoelement.
1.6.1.4 Skrivare
Valfri tvåkanalsskrivare som ar kalibrerad från 0 till 600 oC eller motsvarande spänning.
1.6.2 Testbetingelser
Ämnen testas i sin kommersiella form.
1.6.3 Testförfarande
Kuben fylls med testämnet och slås med lätta slag varefter ytterligare ämne tillsätts tills kuben är helt fylld. Provet hängs därefter upp i mitten av ugnen vid rumstemperatur. Ugnstemperaturen registreras med ett termoelement som är placerat mitt i kuben och ett annat som är placerat mellan kuben och ugnsväggen.
Ugnens och provets temperaturer avläses fortlöpande medan ugnstemperaturen ökas till 400 oC eller till smältpunkten, om denna är lägre, med en hastighet av 0,5 oC per minut.
När ämnet antänds visar termoelementet i provet en mycket kraftig temperaturhöjning över ugnstemperaturen.
2. DATA
Den ugnstemperatur vid vilken provets temperatur uppnår 400 oC genom självuppvärmning är av betydelse för utvärderingen (se figur 2).
3. RAPPORTERING
Testrapporten ska om möjligt innehålla följande uppgifter:
|
— |
En beskrivning av testämnet. |
|
— |
Mätresultaten inklusive temperatur/tidskurvor. |
|
— |
Alla ytterligare upplysningar som är av betydelse för tolkningen av resultaten. |
4. HÄNVISNINGAR
NF T 20-036 (September 1985). Chemical products for industrial use. Determination of the relative tempe-rature of the spontaneous flammability of solids.
Figur 1
Mall till 20 mm testkub

Figur 2
Typisk temperatur/tidskurva

A.17 OXIDERANDE EGENSKAPER (FASTA ÄMNEN)
1. METOD
1.1 INLEDNING
Vid utförande av detta test är det värdefullt att ha förhandskunskap om ämnets potentiellt explosiva egenskaper och toxicitet.
Testet får inte tillämpas på vätskor och gaser, explosiva eller mycket brandfarliga ämnen eller organiska peroxider.
Detta test behöver inte utföras om en undersökning av strukturformeln fastslår utom rimligt tvivel att ämnet inte kan reagera exotermt med ett brännbart material.
För att säkerställa om testet ska utföras med iakttagande av särskilda försiktighetsåtgärder, ska ett förberedande test utföras.
1.2 DEFINITIONER OCH ENHETER
Bränntid: Den reaktionstid, i sekunder, som krävs för att reaktionszonen breder ut sig längs en hög, enligt det förfarande som beskrivs i 1.6.
Brännhastighet: Uttrycks i mm per sekund.
Maximal brännhastighet: det högsta värdet för brännhastigheter som erhållits för blandningar innehållande 10–90 viktprocent av det oxiderande ämnet.
1.3 REFERENSÄMNE
Bariumnitrat (analytisk kvalitet) används som referensämne för testet och det inledande screeningtestet.
Referensblandningen är den blandning av bariumnitrat och cellulosapulver som framställts enligt 1.6 och som har den maximala brännhastigheten (vanligtvis en blandning med 60 viktprocent bariumnitrat).
1.4 TESTMETODSPRINCIP
Av säkerhetsskäl utförs ett inledande screeningtest. Detta test är tillräckligt om det klart utvisar att testämnet eller testpreparatet har oxiderande egenskaper. Om så inte är fallet ska ämnet eller preparatet underkastas ett fullständigt test.
Vid det fullständiga testet ska testämnet och en definierat brännbart ämne blandas i olika proportioner. Varje blandning formas sedan till en hög som antänds i ena änden. Den maximala brännhastigheten bestäms därefter och jämförs med referensblandningens maximala brännhastighet.
1.5 KVALITETSKRITERIER
Om så behövs kan valfri malnings- och blandningsmetod användas, förutsatt att skillnaden mellan den maximala brännhastigheten i de sex separata testen inte avviker mer än 10 % från det aritmetiska medelvärdet.
1.6 METODBESKRIVNING
1.6.1 Förberedelser
1.6.1.1 Testämne
Testprovet reduceras till en partikelstorlek < 0,125 mm enligt följande förfarande: Sikta testämnet; mal återstående fraktion, upprepa förfarandet tills hela testfraktionen har passerat sikten.
Valfri malnings- och siktningsmetod som uppfyller kvalitetskriterierna får användas.
Innan blandningen framställs ska ämnet torkas vid 105 oC tills konstant vikt har uppnåtts. Om testämnets nedbrytningstemperatur är lägre än 105 oC ska ämnet torkas vid en passande lägre temperatur.
1.6.1.2 Brännbart ämne
Cellulosapulver används som brännbart ämne. Cellulosan ska vara av den typ som används vid tunnskiktskromatografi eller kolonnkromatografi. En typ där mer än 85 % av fiberlängderna ligger mellan 0,020 och 0,075 mm har visat sig vara lämplig. Cellulosapulvret passerar genom en sikt med en maskstorlek på 0,125 mm. Samma blandning ska användas i hela testet.
Innan blandningen framställs ska ämnet torkas vid 105 oC tills konstant vikt har uppnåtts.
Om trämjöl används i det förberedande testet ska ett barrvedsmjöl tillredas genom att den del som passerar en sikt med maskstorleken 1,6 mm insamlas, blandas noggrant och sedan torkas vid 105 oC i fyra timmar i ett högst 25 mm tjockt lager. Efter avkylning ska det förvaras i en lufttät behållare, fylld till brädden, tills det ska användas, helst inom 24 timmar efter torkning.
1.6.1.3 Antändningskälla
Som antändningskälla används lågan från en gasbrännare (minsta diameter 5 mm). Om en annan antändningskälla används (t.ex. vid testning i en inert atmosfär) ska en beskrivning och en motivering bifogas.
1.6.2 Testförfarande
Anmärkning:
Blandningar av oxiderande ämnen med cellulosa eller trämjöl ska behandlas som potentiellt explosiva och hanteras med vederbörlig försiktighet.
1.6.2.1 Förberedande test
Det torkade ämnet blandas med det torkade cellulosapulvret eller trämjöl i proportionerna 2 viktdelar av testämnet till 1 viktdel cellulosa eller träm)öl och blandningen formas till en liten konformad hög med måtten 3,5 cm (basens diameter) × 2,5 cm (höjd) genom att en konliknande form (t.ex. ett laboratorieglastratt vars pip är tillsluten) fylls, utan att packas.
Högen placeras på en kall, icke ledande, ogenomtränglig basplatta. Testet ska utföras i ett dragskåp som i 1.6.2.2.
Antändningskällan sätts i kontakt med konen. Kraften och varaktigheten av den påföljande reaktionen iakttas och registreras.
Ämnet anses vara oxiderande om reaktionen är kraftfull.
Om resultaten på något sätt är tvetydiga ska det nedan beskrivna fullständiga testförfarandet utföras.
1.6.2.2 Fullständigt test
En blandning av oxiderande ämne och cellulosa, innehållande 10–90 viktprocent oxiderande ämne, framställs i steg om 10 viktprocent. I gränsfall ska mellanblandningar av oxiderande ämne och cellulosa användas för att få fram en noggrannare maximal brännhastighet.
Högen byggs upp med användning av en form. Formen är tillverkad av metall, har en längd på 250 mm, och en triangelformad tvärsnittssektion med en inre höjd på 10 mm och en inre bredd på 20 mm. På båda sidor om formen monteras i längdriktningen två sidoavgränsande metallplåtar som skjuter upp 2 mm ovanför den triangelformade tvärsnittssektionens övre kant (se figuren). Denna anordning fylls löst och med råge av blandningen. Efter det att formen har fatt falla en gång från 2 cm höjd på en fast yta skrapas återstående överskottsämne bort med en skråställd platta. Därefter avlägsnas sidoavgränsningarna och resterande pulver slätas ut med en kavel. En icke-brännbar plåt placeras sedan ovanpå formen, apparaturen vänds och formen avlägsnas.
Högen placeras i dragskåpet tvärs över luftdragets riktning.
Lufthastigheten ska vara så hög att den förhindrar att rökgaser släpps ut till laboratoriet och ska inte varieras under testet. En dragskärm ska placeras runt apparaturen.
Eftersom cellulosan och testämnena är hygroskopiska ska testet utföras så snabbt som möjligt.
Ena änden av högen tänds på genom beröring av lågan.
När reaktionszonen har förflyttat sig de första 30 mm mäts reaktionstiden över följande 200 mm.
Testet utförs med referensämnet och minst en gång med varje serie av blandningar av testämnet och cellulosa.
Om den maximala brännhastigheten konstateras vara signifikant högre än referensämnets, kan testet avbrytas. I annat fall ska testet upprepas fem gånger med de tre blandningar som gav de högsta brännhastigheterna.
Om man misstänker att resultatet är felaktigt positivt upprepas testet med ett inert ämne med motsvarande partikelstorlek, t.ex. kiselgur, i stället för cellulosa. Alternativt kan den blandning av testämne och cellulosa som har den snabbaste reaktionshastigheten testas i inert atmosfär (< 2 % v/v syreinnehåll).
2. DATA
Av säkerhetsskäl ska den maximala brännhastigheten – inte medelvärdet – anses vara testämnets karakteristiska oxiderande egenskap.
Det högsta värdet för brännhastighet som erhålls vid en testomgång med sex test av en given blandning är relevant for utvärderingen.
Rita en kurva över det högsta värdet för brännhastighet för varje blandning som en funktion av dess innehåll av oxiderande medel. Bestäm den maximala brännhastigheten utifrån kurvan.
De sex matta värdena för brännhastighet, som vid en testomgång erhållits för blandningen med den maximala brännhastigheten, får inte avvika mer än 10 % från det aritmetiska medelvärdet. I annat fall ska malnings- och blandningsmetoderna förbättras.
Jämför den erhållna maximala brännhastigheten med referensblandningens maximala brännhastighet (se 1.3).
Om testen utförs i en inert atmosfär jämförs den maximala brännhastigheten med den från referensblandningen i en inert atmosfär.
3. RAPPORTERING
3.1 TESTRAPPORT
Testrapporten ska om möjligt innehålla följande uppgifter:
|
— |
Testämnets beteckning, sammansättning, renhet, fukthalt osv. |
|
— |
Eventuell behandling av testprovet (t.ex. malning eller torkning). |
|
— |
Använd antändningskälla. |
|
— |
Mätresultaten. |
|
— |
Reaktionssättet (t.ex. brinnande låga vid ytan, genombränning av hela massan och alla uppgifter om för bränningsprodukter). |
|
— |
Alla ytterligare upplysningar som är av betydelse för tolkningen av resultaten, inbegripet en beskrivning av kraften (lågor, gnistor, rök, långsamt pyrande glöd osv.) och ungefärlig varaktighet som iakttagits vid det för beredande säkerhets-screeningtestet för både test- och referensämne. |
|
— |
Resultat av eventuella tester med ett inert ämne. |
|
— |
Resultat av eventuella tester i inert atmosfär. |
3.2 TOLKNING AV RESULTATEN
Ett ämne ska anses vara ett oxiderande ämne om
|
a) |
en kraftig reaktion inträffar under det inledande screeningtestet |
|
b) |
den maximala brännhastighet av de blandningar som undersökts vid fullständig testning är högre än eller lika med den maximala brännhastigheten för referensblandningen av cellulosa och bariumnitrat. |
För att undvika falska positiva resultat ska de resultat som erhålls vid test av ämnet med ett inert material och vid test i inert atmosfär också beaktas vid tolkning av resultaten.
4. HÄNVISNINGAR
NF T 20-035 (september 85), Chemical products for industrial use. Determination of the oxidizing properties of solids.
Tillägg
Figur
Form och tillbehör för iordningställande av högen
(Alla mått i mm)

A.18 MOLEKYLÄR MEDEL VIKT OCH MOLEKYLÄR VIKTFÖRDELNING AV POLYMERER
1. METOD
Denna kromatografiska metod (genomträngning i gelé) är en kopia på OECD TG 118 (1996). Grundläggande principer och annan teknisk information finns under hänvisning 1.
1.1 INLEDNING
Eftersom polymerernas egenskaper är så varierande är det omöjligt att beskriva en enda metod som ger exakta förhållanden för separation och bedömning som täcker alla eventualiteter och särskilda företeelser vid separation av polymerer. Särskilt komplexa polymera system är ofta inte mottagliga för kromatografisk analys av genomträngning i gelé (GPC). När GPC inte är praktiskt genomförbart kan den molekylära vikten fastställas med andra metoder (se bilaga). I sådana fall ska fullständig information om och motivering för den använda metoden anges.
Den beskrivna metoden grundas på DIN-standard 55672 (1). Detaljerad information om hur experimentet ska utföras och hur datan ska bedömas finns i denna DIN-standard. Om experimentförhållandena måste ändras, ska dessa förändringar motiveras. Andra standarder får tillämpas om full hänvisning därtill görs. I den beskrivna metoden används polystyrenprover med känd polydispertion för kalibrering, som kan behöva anpassas till vissa polymerer, t.ex. vattenlösliga och anpassade polymerer med långa molekylkedjor.
1.2 DEFINITIONER OCH ENHETER
Den numerära medelmolekylvikten Mn och den viktmässigt molekylära medelvikten Mw fastställs med följande ekvationer:
|
|
|


där:
Hi är nivån på detektorsignalen från baslinjen för den kvarhållna volymen Vi,
Mi är polymerfraktionens molekylvikt vid den kvarhållna volymen Vi, och
n är antalet datapunkter.
Bredden på den molekylära viktfördelningen, som är ett mått på systemdispertionen, fås med förhållandet Mw/Mn.
1.3 REFERENSÄMNEN
Eftersom GPC är en relativ metod måste kalibrering göras. Normalt används tätt fördelad, linjärt konstruerad standardpolystyren med känd medelmolekylvikt Mn och Mw och en känd molekylviktfördelning för detta ändamål. Kalibreringskurvan kan bara användas för fastställande av molekylvikten för det okända provet om separationsförhållandena för provet och standardämnena valts på exakt samma sätt.
Det fastställda förhållandet mellan molekylvikten och urlakningsvolymen gäller enbart under det aktuella experimentets specifika förhållanden. Dessa förhållanden omfattar framför allt temperatur, lösning eller lösningsblandning, kromatografiska förhållanden och separationskolonn eller kolonnsvstem.
Den molekylvikt som fastställs för provet på detta sätt utgörs av relativa värden och beskrivs som polystyrenets molekylära ekvivalensvikt. Detta innebär att molekylvikterna, beroende på struktur och kemisk skillnad mellan provet och standardämnet, kan avvika från de absoluta värdena i större eller mindre utsträckning. Om andra standardämnen används, t.ex. polyetylenglykol, polyetylenoxid, poly-metylmetakrylat, polyakrylsyra, ska skälet därtill anges.
1.4 PRINCIPEN FÖR PROVNINGSMETODEN
Både provets molekylviktfördelning och medelmolekylvikter (Mn, Mw) kan fastställas med hjälp av GPC. GPC är en särskild typ av vätskekromatografi där provet separeras beroende på de enskilda komponenternas hydrodynamiska volymer (2).
Separationen utförs när provet passerar genom en kolonn fylld med poröst material, vanligtvis en organisk gelé. Små molekyler kan passera igenom porerna medan stora molekyler kvarhålls. De stora molekylernas passage blir därför kortare och dessa tas bort först. Medelstora molekyler passerar genom en del av porerna och/eller elueras senare. De minsta molekylerna, med en hydrodynamisk medelradie mindre än porerna i gelén, kan passera genom samtliga porer. Dessa elueras då sist.
I idealsituationen styrs separeringen helt och hållet av molekylstorleken, men i praktiken är det svårt att förhindra att åtminstone en viss absorbtionseffekt inverkar störande. Ojämn packning i kolonnen och döda volymer kan förvärra situationen (2).
Detektionen utförs med t.ex. reflektionsindex eller UV-absorbtion och ger en enkel fördelningskurva. För att kunna tillskriva kurvan den faktiska molekylvikten måste kolonnen kalibreras, genom att polymerer med känd molekylvikt och helst i stort sett liknande struktur, t.ex. olika standardpolysterener, tillåts passera genom kolonnen. Normalt fås en typisk Gaus-kurva, ibland utdragen i en mindre svans på sidan för den låga molekylvikten, om de vertikala axlarna anger mängden, i förhållande till vikten, för de olika molekylvikter som elueras, och den horisontella axeln står för den logaritmiska molekylvikten.
1.5 KVALITETSKRITERIER
Repeterbarheten (relativ standardavvikelse: RSD) för elutionsvolymen ska vara bättre än 0,3 %. Nödvändig repeterbarhet för analysen måste säkerställas genom korrigering enligt intern standard om ett kromatogram bedöms vara tidsberoende och inte omfattas av ovannämnda kriterier (1). Spridningen beror på standardämnets molekylvikt. Följande värden är typiska för standardpolystyren:
|
Mp < 2 000 |
Mw/Mn < 1,20 |
|
2 000 < Mp < 106 |
Mw/Mn < 1,05 |
|
Mp > 106 |
Mw/Mn < 1,20 |
(Mp är standardämnets molekylvikt vid toppvärdet.)
1.6 BESKRIVNING AV PROVNINGSMETODEN
1.6.1 Beredning av lösningar med standardpolystyren
Standardpolystyrenen löses noggrant genom blandning i vald eluent. Hänsyn måste tas till tillverkarens rekommendationer vid beredning av lösningarna.
Koncentrationen av de standardämnen som väljs beror på olika faktorer, t.ex. injektionsvolym, lösningsviskositet och den analytiska detektorns känslighet. Maximal injektionsvolym måste anpassas till kolonnens längd för undvikande av överbelastning. Typiska injektionsvolymer för analytiska separationer med GPC och en kolonn på 30 cm × 7,8 mm ligger mellan 40 och 100 μl. Större volymer kan användas men bör inte överstiga 250 μl. Det optimala förhållandet mellan injektionsvolym och koncentration måste fastställas innan kolonnen kalibreras.
1.6.2 Beredning av provlösningen
I princip gäller samma krav för beredning av provlösning. Provet löses upp i lämpligt lösningsmedel, t.ex. tetrahydrofuran (THF), genom omsorgsfull skakning. Det får under inga omständigheter lösas upp i ultraljudsbad. Vid behov kan provlösningen renas genom ett membranfilter, med en finhet på mellan 0,2 och 2 μm.
Förekomst av oupplösta partiklar måste registreras i slutrapporten, eftersom dessa kan bero på högviktsmolekyler. Lämplig metod ska användas för fastställande av viktandelen oupplösta partiklar. Lösningen bör användas inom 24 timmar.
1.6.3 Apparater
|
— |
Lösningsbehållare. |
|
— |
Avluftningsapparat (när så krävs). |
|
— |
Pump. |
|
— |
Pulsdämpare (när så krävs). |
|
— |
Injektionssystem. |
|
— |
Kromatografikolonner. |
|
— |
Detektor. |
|
— |
Flödesmätare (när så krävs). |
|
— |
Apparat för registrering/bearbetning av data. |
|
— |
Avfallskärl. |
Det måste säkerställas att GPC-systemet är inert avseende använda lösningsmedel, t.ex. genom användning av stålkapilär för THF-lösningen.
1.6.4 Injektions- och lösningstillförselsystem
En definierad volym av lösningsprovet fylls i kolonnen, antingen automatiskt eller manuellt, i en tydligt definierad zon. Alltför snabb utdragning eller intryckning av kolven i sprutan, om fyllningen sköts manuellt, kan förorsaka förändringar i den observerade molekylviktfördelningen. Systemet för tillförsel av lösning ska så långt möjligt vara pulsfritt, helst med inbyggd pulsdämpning. Flödeshastigheten ska vara ca 1 ml/min.
1.6.5 Kolonn
Polymeren kännetecknas, beroende på provet, antingen genom användning av enkel kolonn eller genom flera kolonner kopplade i serie. Ett antal porösa kolonnmaterial med definierade egenskaper, t.ex. porstorlek, exklutionsgränser osv., finns kommersiellt tillgängliga. Val av separationsgelé och kolonnlängd beror både på provets egenskaper (hydrodynamisk volym, molekylviktfördelning) och specifika förhållandena för separationen, som t.ex. lösning, temperatur och flödeshastighet (1) (2) (3).
1.6.6 Teoretiska plattor
Den kolonn eller kolonnkombination som används för separationen måste karakteriseras av antalet teoretiska nivåer. Detta inkluderar, när THF används som elutionslösning, fyllning av en lösning av etylbensen eller annan lämplig icke-polär lösning i en kolonn med känd längd. Antalet teoretiska nivåer fås med följande ekvation:
|
|
eller |
|


där:
|
N |
= |
är antalet teoretiska nivåer, |
|
Ve |
= |
är elutionsvolymen vid toppvärdet, |
|
W |
= |
är toppvärdets baslinjebredd, och |
|
W1/2 |
= |
är toppbredden vid halva höjden. |
1.6.7 Separationseffektivitet
Förutom antalet teoretiska nivåer som styr bandbredden, spelar också separationseffektiviteten, som bestäms av lutningen på kalibreringskurvan, en viss roll. Separationseffektiviteten för en kolonn fås med följande förhållande:

där:
|
Ve, Mx |
= |
är elutionsvolymen för polystyren med molekylvikten Mx och |
|
Ve,(10Mx) |
= |
är elutionsvolymen för polystyren med en tio gånger större molekylvikt. |
Systemets upplösning definieras normalt som:

där:
|
Ve1 och Ve2 |
= |
är elutionsvolymerna för de två standardpolystyrenerna vid maximalt toppvärde, |
|
W1 och W2 |
= |
är baslinjebredden vid toppvärde och |
|
M1 och M2 |
= |
är molekylvikten vid maximalt toppvärde (bör skilja sig åt med en faktor på 10). |
R-värdet för kolonnsystemet bör vara större än 1,7 (4).
1.6.8 Lösningsmedel
Samtliga lösningsmedel måste ha hög renhet (för THF används en renhet på 99,5 %). Lösningsmedelsbehållaren (om nödvändigt i en inert gasatmosfär) ska vara tillräckligt stor för kalibrering av kolonnen och flera provanalyser. Lösningsmedlet ska avgasas innan det via pumpen transporteras till kolonnen.
1.6.9 Temperaturövervakning
Temperaturen i de kritiska interna komponenterna (injektionsslinga, kolonn, detektor och rörsystem) ska vara jämn och anpassad till valt lösningsmedel.
1.6.10 Detektor
Syftet med detektorn är att kvantitativt registrera koncentrationen i det prov som elueras från kolonnen. För att undvika onödig breddning av toppvärden ska detektorcellens kuvettvolym hållas så liten som möjligt. Den bör inte vara större än 10 μl, utom för lättflyktiga och tunna detektorer Differentialrefraktometri används vanligtvis för detektion. Om provets eller elutionslösningens särskilda egenskaper så kräver kan emellertid andra detektortyper användas t.ex. UV/VIS, IR- eller viskositetsdetektorer.
2. DATA OCH RAPPORTERING
2.1 DATA
Detaljerade utvärderingskriterier och krav avseende insamling och bearbetning av data ska uppfylla kraven i DIN-standarden (1).
Två sinsemellan oberoende experiment ska utföras för varje prov. Experimenten ska analyseras individuellt.
Mn, Mw, Mw/Mn och Mp ska anges för varje mätning. Det är nödvändigt att uttryckligen ange att uppmätta värden är relativa värden, ekvivalenta mot molekylvikten för det standardämne som använts.
Efter fastställande av retentionsvolymerna eller retentionstiderna (eventuellt korrigerade med en intern standard), plottas log Mp-värdena (Mp är maximalt toppvärde för kalibreringsämnet) mot en av nämnda mängder. Minst två kalibreringspunkter krävs per tiotal molekylvikter och minst fem mätpunkter för den totala kurvan, som ska täcka provets uppskattade molekylvikt. Kalibreringskurvans ändpunkt för den låga molekylvikten definieras av n-hexylbenzen eller annan lämplig icke-polär lösning. Medelantal och medelvikt för molekylvikterna fastställs allmänt med elektronisk databearbetning, baserad på formlerna i avsnitt 1.2. Om manuell digitalisering används kan råd hämtas från ASTM D 3536-91 (3).
Fördelningskurvan ska presenteras i tabellform eller som ett diagram (differentialfrekvens eller summa procentandel mot log M). I den grafiska presentationen ska varje tiotal molekylvikter normalt ha en bredd på 4 cm, och maximalt toppvärde ska ligga på ungefär 8 cm höjd. Vid intregralfördelningskurvor ska skillnaderna mellan 0 och 100 % vara ungefär 10 cm.
2.2 PROVNINGSRAPPORT
Provningsrapporten ska innehålla följande information:
2.2.1 Undersökt ämne
|
— |
Tillgänglig information om det undersökta ämnet (identitet, tillsatser, föroreningar). |
|
— |
Beskrivning av hanteringen av provet, anmärkningar, problem. |
2.2.2 Instrument
|
— |
Eluentbehållare, inertgas, avgasning av eluenten, eluentens sammansättning, föroreningar. |
|
— |
Pump, pulsdämpare, injektionssystem. |
|
— |
Separationskolonner (tillverkare, all information om kolonnernas karakteristika, som porstorlek, typ av separationsmaterial osv., antal, längd och inbördes ordning på använda kolonner). |
|
— |
Antal teoretiska nivåer i kolonnen eller kolonnkombinationen, separationseffektivitet (systemupplösning). |
|
— |
Information om topparnas symmetri. |
|
— |
Kolonntemperatur, typ av temperaturövervakning. |
|
— |
Detektor (mätprincip, typ, kuvettvolym). |
|
— |
Flödesmätare om sådan används (tillverkare, mätprincip). |
|
— |
System för registrering och bearbetning av data (hårdvara och mjukvara). |
2.2.3 Systemkalibrering
|
— |
Detaljerad beskrivning av den metod som används för konstruktion av kalibreringskurvan. |
|
— |
Information om kvalitetskriterier för den aktuella metoden (t.ex. koordinationskoefficient, kvadraternas summeringsfel osv.). |
|
— |
Information om all extrapolering, alla antaganden och approximeringar gjorda under experimentproceduren samt om bedömning och bearbetning av data. |
|
— |
Alla mätningar som gjorts för konstruktion av kalibreringskurvan ska dokumenteras i en tabell som inkluderar följande information för varje kalibreringspunkt:
|
2.2.4 Bedömning
|
— |
Bedömning på tidsbasis: metoder som används för att säkerställa önskad reproducerbarhet (korrektionsmetod, intern standard osv.). |
|
— |
Information om huruvida bedömningen gjorts på basis av elutionsvolymen eller retentionstiden. |
|
— |
Information om gränserna för bedömningen, om ett toppvärde inte analyserats fullständigt. |
|
— |
Beskrivning av utjämningsmetoder, om sådana används. |
|
— |
Beredning av och förbehandlingsprocedurer för provet. |
|
— |
Närvaro av oupplösta partiklar, om sådana förekommit. |
|
— |
Injektionsvolym (μl) och injektionskoncentration (mg/ml). |
|
— |
Anmärkningar om effekter som lett till avvikelser från den ideala GPC-profilen. |
|
— |
Detaljerad beskrivning av alla ändringar i provningsförfarandet. |
|
— |
Detaljerade uppgifter om felområden. |
|
— |
All övrig information och anmärkningar relevanta för tolkningen av resultatet. |
3. HÄNVISNINGAR
|
1. |
DIN 55672 (1995), Gelégenomträngningskromatografi (GPC) med tetrahydrofuran (THF) som elutionsmedel, del 1. |
|
2. |
Yau, W.W., Kirkland, J.J., och Bly, D.D. eds. (1979), Modern Size Exclusion Liquid Chromatography, J. Wiley and Sons. (ung. Modern vätskeuteslutningskromatografi för uteslutning) |
|
3. |
ASTM D 3536-91, (1991), Standardmetod för analys av medelmolekylvikter och molekylviktfördelning med vätskeuteslutningskromatografi (gelégenomträngningskromatografi – GPC). American Society for Testing and Materials, Philadelphia, Pennsylvania. |
|
4. |
ASTM D 5296-92, (1992), Standardmetod för analys av medelmolekylvikter och molekylviktfördelning i polystyren med storleksseparationskromatografi. American Society for Testing and Materials, Philadelphia, Pennsylvania. |
Bilaga
Exempel på andra metoder för fastställande av medelantalet molekylvikt (Mn) för polymerer
Gelégenomträngningskromatografi (GPC) är den metod som är att föredra för fastställande av Mn, framför allt när en uppsättning standardämnen finns tillgängliga, vilkas struktur är jämförbar med polymerens struktur. När det föreligger praktiska svårigheter med användning av GPC eller det redan finns en förväntan att ämnet inte kommer att uppfylla ett vanligt Mn-kriterium (och detta kräver bekräftelse) finns alternativa metoder, som t.ex.:
1. Utnyttjande av kolligativa egenskaper
|
1.1 |
Ebullioscopi/kryoscopi inkluderar mätning av kokpunktshöjning (ebullioscopi) eller fryspunktssänkning (cryoscopi) för ett lösningsmedel när polymeren tillsätts. Metoden grundas på det faktum att den upplösta polymerens effekt på vätskans kok- och/eller fryspunkt beror på polymerens molekylvikt (1) (2). Tillämpningsbarhet: Mn < 20 000. |
|
1.2 |
Sänkning av ångtryck inkluderar mätning av ångtrycket för en vald referensvätska före och efter tillsats av kända mängder polymerer (1) (2). Tillämpningsbarhet: Mn < 20 000 (teoretiskt, i praktiken emellertid ett begränsat värde). |
|
1.3 |
Membranosmometri grundas på osmosprincipen, dvs. den naturliga tendensen för molekylerna i en lösning att passera genom ett halvgenomträngligt membran från en lösning till en koncentrerad lösning för att uppnå balans. I detta prov har den uppblandade lösningen nollkoncentration, medan den koncentrerade lösningen innehåller polymeren. Lösningens passage genom membranet förorsakar ett differenstryck som är avhängigt koncentrationen och polymerens molekylvikt (1) (3) (4). Tillämpningsbarhet: Mn < 20 000–200 000. |
|
1.4 |
Ångfasosmometri inkluderar jämförelse av förångningshastigheten för en ren aerosollösning till minst tre aerosoler innehållande polymeren vid olika koncentrationer (1) (2) (4). Tillämpningsbarhet: Mn < 20 000. |
2. Ändgruppanalys
För att kunna använda denna metod krävs kunskap både om polymerens övergripande struktur och om den typ av kedja som avslutar ändgrupperna (som måste vara urskiljningsbar från huvudstrukturen med t.ex. NMR eller titrering/derivatisering) Fastställandet av molekylkoncentrationen i polymerens ändgrupper kan ge ett värde for rnolekylvikten (7) (8) (9).
Tillämpningsbarhet: Mn upp till 50 000 (med minskande tillförlitlighet).
3. Hänvisningar
|
1. |
Billmeyer, F.W. Jr., (1984), Textbook of Polymer Science (ung. Textbok om polymerforskning), tredje utg., John Wiley, New York. |
|
2. |
Glover, C.A., (1975), Absolute Colligative Property Methods (ung. Absoluta metoder för bestämning av kolligativa egenskaper), kapitel 4, In: Polymer Molecular Weights (ung. Polymerers molekylvikt), Part I P.E. Siade, Jr. ed., Marcel Dekker, New York. |
|
3. |
ASTM D 3750-79, (1979), Standard Practice for Determination of Number-Average Molecular Weight of Polymers by Membrane Osmometry (ung. Standardmetod för bestämning av numerisk medelmolekylvikt med membranosmometri i polymerer). American Society for Testing and Materials, Philadelphia, Pennsylvania. |
|
4. |
Coll, H. (1989), Membrane Osmometry (Membranosmometri). In: Determination of Molecular Weight (Bestämning av molekylvikt), A.R. Cooper ed., J. Wiley and Sons, s. 25–52. |
|
5. |
ASTM 3592-77, (1977), Standard Recommended Practice for Determination of Molecular Weight by Vapour Pressure (ung. Rekommenderad standardmetod för bestämning av molekylvikt med hjälp av ångtryck), American Society for Testing and Materials, Philadelphia, Pennsylvania. |
|
6. |
Morris, C.E.M., (1989), Vapour Pressure osmometry (Ångtrycksosmometri). In: Determination of Molecular Weight (Bestämning av molekylvikt), A.R. Cooper ed., John Wiley and Sons. |
|
7. |
Schröder, E., Muller, G., och Arndt, K-F., (1989), Polymer Characterisation (Polymerkarakterisering), Carl Hanser Verlag, München. |
|
8. |
Garmon, R.G., (1975), End-Group Determinations (ung. Bestämning av ändgrupper) kapitel 3 In: Polymer Molecular Weights (Polymerers molekylvikt), Del 1, P.E. Siade, Jr. ed., Marcel Dekker, New York. |
|
9. |
Amiya, S., et al. (1990), Pure and Applied Chemistry (ung. Ren och tillämpad kemi), 62, 2139–2146. |
A.19 INNEHÅLL AV LÅGMOLEKYLVIKTSPOLYMERER
1. METOD
Denna kromatografiska metod (genomträngning i gelé) är en kopia på OECD TG 119 (1996). Grundläggande principer och annan teknisk information finns i hänvisningarna.
1.1 INLEDNING
Eftersom polymerernas egenskaper är så varierande är det omöjligt att beskriva en enda metod som ger exakta förhållanden för separation och bedömning som täcker alla eventualiteter och särskilda företeelser vid separation av polymerer. Särskilt komplexa polymera system är ofta inte mottagliga för kromatografisk analys av genomträngning i gelé (GPC). När GPC inte är praktiskt genomförbart kan den molekylära vikten fastställas med andra metoder (se bilaga). I sådana fall ska fullständig information om och motivering för den använda metoden anges.
Den beskrivna metoden grundas på DIN-standard 55672 (1). Detaljerad information om hur experimentet ska utföras och hur datan ska bedömas finns i denna DIN-standard. Om experimentförhållandena måste ändras, ska dessa förändringar motiveras. Andra standarder får tillämpas om full hänvisning därtill görs. I den beskrivna metoden används polystyrenprover med känd polydispertion för kalibrering, som kan behöva anpassas till vissa polymerer, t.ex. vattenlösliga och anpassade polymerer med långa molekylkedjor.
1.2 DEFINITIONER OCH ENHETER
Låg molekylvikt definieras som en molekylvikt under 1 000 dalton.
Den numerära medelmolekylvikten Mn och den viktmässigt molekylära medelvikten Mw fastställs med följande ekvationer:
|
|
|


där:
|
Hi |
= |
är nivån på detektorsignalen från baslinjen för den kvarhållna volymen Vi, |
|
Mi |
= |
är polymerfraktionens molekylvikt vid den kvarhållna volymen Vi, och n är antalet datapunkter. |
Bredden på den molekylära viktfördelningen, som är ett mått på systemdispertionen, fås med förhållandet Mw/Mn.
1.3 REFERENSÄMNEN
Eftersom GPC är en relativ metod måste kalibrering göras. Normalt används tätt fördelad, linjärt konstruerad standardpolystyren med känd medelmolekylvikt Mn och Mw och en känd molekylviktfördelning för detta ändamål. Kalibreringskurvan kan bara användas för fastställande av molekylvikten för det okända provet om separationsförhållandena för provet och standardämnena valts på exakt samma sätt.
Det fastställda förhållandet mellan molekylvikten och urlakningsvolymen gäller enbart under det aktuella experimentets specifika förhållanden. Dessa förhållanden omfattar framför allt temperatur, lösning eller lösningsblandning, kromatografiska förhållanden och separationskolonn eller kolonnsystem.
Den molekylvikt som fastställs för provet på detta sätt utgörs av relativa värden och beskrivs som polystyrenets molekylära ekvivalensvikt. Detta innebär att molekylvikterna, beroende på struktur och kemisk skillnad mellan provet och standardämnet, kan avvika från absoluta värdena i större eller mindre utsträckning. Om andra standardämnen används, t.ex. polyetylenglykol, polyetylenoxid, poly-metylmetakrylat, polyakrylsyra ska skälet därtill anges.
1.4 PRINCIPEN FÖR PROVNINGSMETODEN
Både provets molekylviktfördelning och medelmolekylvikter (Mn, Mw) kan fastställas med hjälp av GPC. GPC är en särskild typ av vätskekromatografi där provet separeras beroende på de enskilda komponenternas hydrodynamiska volymer (2).
Separationen utförs när provet passerar genom en kolonn fylld med poröst material, vanligtvis en organisk gelé. Små molekyler kan passera igenom porerna medan stora molekyler kvarhålls. De stora molekylernas passage blir därför kortare och dessa tas bort först. Medelstora molekyler passerar genom en del av porerna och/eller elueras senare. De minsta molekylerna, med en hydrodynamisk medelradie mindre än porerna i gelén, kan passera genom samtliga porer. Dessa elueras då sist.
I idealsituationen styrs separeringen helt och hållet av molekylstorleken, men i praktiken är det svårt att förhindra att åtminstone en viss absorbtionseffekt inverkar störande. Ojämn packning i kolonnen och döda volymer kan förvärra situationen (2).
Detektionen utförs med t.ex. reflektionsindex eller UV-absorbtion och ger en enkel fördelningskurva. För att kunna tillskriva kurvan den faktiska molekylvikten måste kolonnen kalibreras genom att polymerer med känd molekylvikt och helst i stort sett liknande struktur, t.ex. olika standardpolysterener, tillåts passera genom kolonnen. Normalt fås en typisk Gaus-kurva, ibland utdragen i en mindre svans på sidan för den låga molekylvikten, om de vertikala axlarna anger mängden, i förhållande till vikten, för de olika molekylvikter som elueras, och den horisontella axeln står för den logaritmiska molekylvikten.
Innehållet med låg molekylvikt härleds från denna kurva. Beräkningen kan bara bli korrekt om innehållet av lågmolekylviktarter är jämnt fördelat i polymermassan.
1.5 KVALITETSKRITERIER
Repeterbarheten (relativ standardavvikelse: RSD) för elutionsvolymen ska vara bättre än 0,3 %. Nödvändig repeterbarhet för analysen måste säkerställas genom korrigering enligt intern standard om ett kromatogram bedöms vara tidsberoende och inte omfattas av ovannämnda kriterier (1). Spridningen beror på standardämnets molekylvikt. Följande värden är typiska för standardpolystyren:
|
Mp < 2 000 |
Mw/Mn < 1,20 |
|
2 000 < Mp < 106 |
Mw/Mn < 1,05 |
|
Mp > 106 |
Mw/Mn < 1,20 |
(Mp är standardämnets molekylvikt vid toppvärdet.)
1.6 BESKRIVNING AV PROVNINGSMETODEN
1.6.1 Beredning av lösningar med standardpolystyren
Standardpolystyrenen löses noggrant genom blandning i vald eluent. Hänsyn måste tas till tillverkarens rekommendationer vid beredning av lösningarna.
Koncentrationen av de standardämnen som väljs beror på olika faktorer, t.ex. injektionsvolym, lösningsviskositet och den analytiska detektorns känslighet. Maximal injektionsvolym måste anpassas till kolonnens längd för undvikande av överbelastning. Typiska injektionsvolymer for analytiska separationer med GPC och en kolonn på 30 cm × 7,8 mm ligger mellan 40 och 100 μl. Större volymer kan användas men bör inte överstiga 250 μl. Det optimala förhållandet mellan injektionsvolvm och koncentration måste fastställas innan kolonnen kalibreras.
1.6.2 Beredning av provlösningen
I princip gäller samma krav för beredning av provlösning. Provet löses upp i lämpligt lösningsmedel, t.ex. tetrahydrofuran (THF), genom omsorgsfull skakning. Det får under inga omständigheter lösas upp i ultraljudsbad. Vid behov kan provlösningen renas genom ett membranfilter, med en finhet på mellan 0,2 och 2 μm.
Förekomst av oupplösta partiklar måste registreras i slutrapporten, eftersom dessa kan bero på högviktsmolekyler. Lämplig metod ska användas för fastställande av viktandelen oupplösta partiklar. Lösningen bör användas inom 24 timmar.
1.6.3 Korrigering för innehåll av föroreningar och tillsatser
Korrigering av artinnehållet av M < 1 000 för bidraget från närvarande icke-po! ymera specifika komponenter (t.ex. föroreningar och/eller tillsatser) krävs normalt, med mindre det uppmätta innehållet redan är mindre än 1 %. Detta fås genom direkt analys av polymerlösningen eller GPC-eluaten.
När eluaten efter passage genom kolonnen ska spädas för ytterligare analys, ska den vara koncentrerad. Det kan vara nödvändigt att evaporera eluaten till torrhet och lösa upp den igen. Eluatkoncentrationen ska göras under förhållanden som garanterar att inga förändringar uppstår i eluaten. Hanteringen av eluaten efter GPC-steget är beroende av den analysmetod som används för mängdbestämningen.
1.6.4 Apparater
GPC-apparater består av följande komponenter:
|
— |
Lösningsbehållare. |
|
— |
Avluftningsapparat (när så krävs). |
|
— |
Pump. |
|
— |
Pulsdämpare (när så krävs). |
|
— |
Injektionssystem. |
|
— |
Kromatografikolonner. |
|
— |
Detektor. |
|
— |
Flödesmätare (när så krävs). |
|
— |
Apparat för registrering/bearbetning av data. |
|
— |
Avfallskärl. |
Det måste säkerställas att GPC-systemet är inert avseende använda lösningsmedel, t.ex. genom användning av stålkapilär för THF-lösningen.
1.6.5 Injektions- och lösningstillförselsystem
En definierad volym av lösningsprovet fylls i kolonnen, antingen automatiskt eller manuellt, i en tydligt definierad zon. Alltför snabb utdragning eller intryckning av kolven i sprutan, om fyllningen sköts manuellt, kan förorsaka förändringar i den observerade molekylviktfördelningen. Systemet för tillförsel av lösning ska så långt möjligt vara pulsfritt, helst med inbyggd pulsdämpning. Flödeshastigheten ska vara ca 1 ml/min.
1.6.6 Kolonn
Polymeren kännetecknas, beroende på provet, antingen genom användning av enkel kolonn eller flera kolonner kopplade i serie. Ett antal porösa kolonnmaterial med definierade egenskaper, t.ex. porstorlek, exklutionsgränser osv. finns kommersiellt tillgängliga. Val av separationsgelé och kolonnlängd beror både på provets egenskaper (hydrodynamisk volym, molekylviktfördelning) och specifika förhållandena för separationen, som t.ex. lösning, temperatur och flödeshastighet (1) (2) (3).
1.6.7 Teoretiska nivåer
Den kolonn eller kolonnkombination som används för separationen måste karakteriseras av antalet teoretiska nivåer. Detta inkluderar, när THF används som elutionslösning, fyllning av en lösning av etylbensen eller annan lämplig icke-polär lösning i en kolonn med känd längd. Antalet teoretiska nivåer fås med följande ekvation:
|
|
där: |
|


eller,
|
N |
= |
är antalet teoretiska nivåer, |
|
Ve |
= |
är elutionsvolymen vid toppvärdet, |
|
W |
= |
är toppvärdets baslinjebredd, och |
|
W1/2 |
= |
är toppbredden vid halva höjden. |
1.6.8 Separationseffektivitet
Förutom antalet teoretiska nivåer, som styr bandbredden, spelar också separationseffektiviteten, som bestäms av lutningen på kalibreringskurvan, en viss roll. Separationseffektiviteten för en kolonn fås med följande förhållande:

där:
|
Ve, Mx |
= |
är elutionsvolymen för polystyren med molekylvikten Mx, och |
|
Ve,(10Mx) |
= |
är elutionsvolymen för polystyren med en tio gånger större molekylvikt. |
Systemets upplösning definieras normalt som:

där:
|
Ve1 och Ve2 |
= |
är elutionsvolymerna för de två standardpolystyrenerna vid maximalt toppvärde, |
|
W1 och W2 |
= |
är baslinjebredden vid toppvärde, och |
|
M1 och M2 |
= |
är molekylvikten vid maximalt toppvärde (bör skilja sig åt med en faktor på 10). |
R-värdet för kolonnsystemet bör vara större än 1,7 (4)
1.6.9 Lösningsmedel
Samtliga lösningsmedel måste ha hög renhet (för THF används en renhet på 99,5 %). Lösningsmedelsbehållaren, om nödvändigt i en inert gasatmosfär ska vara tillräckligt stor för kalibrering av kolonnen och flera provanalyser. Lösningsmedlet ska avgasas innan det via pumpen transporteras till kolonnen.
1.6.10 Temperaturövervakning
Temperaturen i de kritiska, interna komponenterna (injektionsslinga, kolonn, detektor och rörsystem) ska vara jämn och anpassad till valt lösningsmedel.
1.6.11 Detektor
Syftet med detektorn är att kvantitativt registrera koncentrationen i det prov som elueras från kolonnnen. För att undvika onödig breddning av toppvärden ska detektorcellens kuvettvolym hållas sä liten som möjligt. Den bör inte vara större än 10 μl, utom for lattffyktiga och tunna detektorer. Differentialrefraktometri används vanligtvis för detektion. Om provets eller elutionslösningens särskilda egenskaper så kräver kan emellertid andra detektortyper användas, t.ex. UV/VIS, IR- eller viskositetsdetektorer osv.
2. DATA OCH RAPPORTERING
2.1 DATA
Detaljerade utvärderingskriterier och krav avseende insamling och bearbetning av data ska uppfylla kraven i DIN-standarden (1).
Två sinsemellan oberoende experiment ska utföras för varje prov. Experimenten ska analyseras individuellt. Det är alltid viktigt att också ta fram data från blankprover, behandlade på samma sätt som det verkliga provet.
Det är nödvändigt att uttryckligen ange att uppmätta värden är relativa värden, ekvivalenta mot molekylvikten för det standardämne som använts.
Efter fastställande av retentionsvolymerna eller retentionstiderna (eventuellt korrigerade med en intern standard), plottas log Mp-värdena (Mp är maximalt toppvärde för kalibreringsämnet) mot en av nämnda mängder. Minst två kalibreringspunkter krävs per tiotal molekylvikter och minst fem mätpunkter för den totala kurvan, som ska täcka provets uppskattade molekylvikt. Kalibreringskurvans ändpunkt för den låga molekylvikten definieras av n-hexylbenzen eller annan lämplig icke-polär lösning. Den del av kurvan som svarar mot molekylvikter under 1 000 bestäms och korrigeras för föroreningar och tillsatser efter behov. Elutionskurvorna bedöms i allmänhet med hjälp av elektronisk databehandling. Om manuell digitalisering används kan råd hämtas från ASTM D 3536-91 (3).
Om någon olöslig polymer blir kvar i kolonnen är dess molekylvikt troligen högre än den upplösta fraktionen och om hänsyn inte tas därtill kommer den; att ge en för hög bedömning av innehållet lågmolekylvikter. Riktlinjer för korrigering av innehållet av lågmolekylvikten för olöslig polymer ges i bilagan.
Fördelningskurvan ska presenteras i tabellform eller som ett diagram (differentialfrekvens eller summa procentandel mot log M). I den grafiska presentationen ska varje tiotal molekylvikter normalt ha en bredd på 4 cm, och maximalt toppvärde ska ligga på ungefär 8 cm höjd. Vid intregralfördelningskurvor ska skillnaderna mellan 0 och 100 % vara ungefär 10 cm.
2.2 PROVNINGSRAPPORT
Provningsrapporten ska innehålla följande information.
2.2.1 Undersökt ämne
|
— |
Tillgänglig information om det undersökta ämnet (identitet, tillsatser, föroreningar). |
|
— |
Beskrivning av hanteringen av provet, anmärkningar, problem. |
2.2.2 Instrument
|
— |
Eluentbehållare, inertgas, avgasning av eluenten, eluentens sammansättning, föroreningar. |
|
— |
Pump, pulsdämpare, injektionssystem. |
|
— |
Separationskolonner (tillverkare, all information om kolonnernas karakteristika, som porstorlek, typ av separationsmaterial osv., antal, längd och inbördes ordning på använda kolonner). |
|
— |
Antal teoretiska nivåer i kolonnen eller kolonnkombinationen, separationseffektivitet (systemupplösning). |
|
— |
Information om topparnas symmetri. |
|
— |
Kolonntemperatur, typ av temperaturövervakning. |
|
— |
Detektor (mätprincip, typ, kuvettvolym). |
|
— |
—Flödesmätare om sådan används (tillverkare, mätprincip). |
|
— |
System för registrering och bearbetning av data (hårdvara och mjukvara). |
2.2.3 Systemkalibrering
|
— |
Detaljerad beskrivning av den metod som används för konstruktion av kalibreringskurvan. |
|
— |
Information om kvalitetskriterier för den aktuella metoden (t.ex. koordinationskoefficient, kvadraternas summeringsfel osv.). |
|
— |
Information om all extrapolering, alla antaganden och approximeringar gjorda under experimentproceduren samt om bedömning och bearbetning av data. |
|
— |
Alla mätningar som gjorts för konstruktion av kalibrenngskurvan ska dokumenteras i en tabell som inkluderar följande information för varje kalibreringspunkt:
|
2.2.4 Information om innehåll av lågmolekylvikt i polymer
|
— |
Beskrivning av de metoder som används för analys och hur experimentet utfördes. |
|
— |
Information om andelen innehåll lågmolekylviktarter (w/w) för hela provet. |
|
— |
Information om föroreningar, tillsatser och andra icke-polymera arter, som viktandel i förhållande till hela provet. |
2.2.5 Bedömning
|
— |
Bedömning på tidsbasis: metoder som används för att säkerställa önskad reproducerbarhet (korrek tionsmetod, intern standard osv.). |
|
— |
Information om huruvida bedömningen gjorts på basis av elutionsvolymen eller retentionstiden. |
|
— |
Information om gränserna för bedömningen, om ett toppvärde inte analyserats fullständigt. |
|
— |
Beskrivning av utjämningsmetoder, om sådana används. |
|
— |
Beredning av och förbehandlingsprocedurer för provet. |
|
— |
Närvaro av oupplösta partiklar, om sådana förekommit. |
|
— |
Injektionsvolym (μ1) och injektionskoncentration (mg/ml). |
|
— |
Anmärkningar om effekter som lett till avvikelser från den ideala GPC-profilen. |
|
— |
Detaljerad beskrivning av alla ändringar i provningsförfarandet. |
|
— |
Detaljerade uppgifter om felområden. |
|
— |
All övrig information och anmärkningar relevanta för tolkningen av resultatet. |
3. HÄNVISNINGAR
|
1. |
DIN 55672 (1995), Gelégenomträngningskromatografi (GPC) med tetrahydrofuran (THF) som elutionsmedel, del 1. |
|
2. |
Yau, W.W., Kirkland, J.J., och Bly, D.D. eds. (1979), Modern Size Exclusion Liquid Chromato- graphy, J. Wiley och Sons. (ung. Modern vätskeuteslutningskromatografi för uteslutning). |
|
3. |
ASTM D 3536-91, (1991), Standardmetod för analys av medelmolekylvikter och molekylviktfördelning med vätskeuteslutningskromatografi (gelégenomträngningskromatografi – GPC). American Society for Testing and Materials, Philadelphia, Pennsylvania. |
|
4. |
ASTM D 5296-92, (1992), Standardmetod för analys av medelmolekylvikter och molekylviktfördelning i polystyren med storleksseparationskromatografi. American Society for Testing and Materials, Philadelphia, Pennsylvania. |
Bilaga
Riktlinjer för korrigering av lågmolekylärt innehåll beroende på närvaro av olöslig polymer
När olösliga polymerer förekommer i ett prov, leder detta till massaförlust under GPC-analysen. Den olösliga polymeren kvarhålls definitivt i kolonnen eller provfiltret, medan den lösbara delen av provet passerar genom kolonnen. När polymerens refraktiva indexökning (dn/dc) kan uppskattas eller mätas, kan provets massaförlust i kolonnen uppskattas. I detta fall görs en korrigering med hjälp av en extern kalibrering med standardmaterial med känd koncentration och dn/dc för kalibrering av refraktometerns reaktion. I nedanstående exempel används en standardpolymer (merylmetakrylat) (pMMA).
Vid den externa kalibreringen för analys av akrylpolymerer analyseras en standard-pMMA med känd koncentration i terahydrofuran med GPC, och resulterande data används för att få fram refraktometerkonstanten med följande ekvation:
K = R/(C × V × dn/dc)
där:
|
K |
= |
står för refraktometerkonstanten i μvs/ml, |
|
R |
= |
för reaktionen på standard-pMMA i μvs, |
|
C |
= |
för koncentrationen av standard-pMMA i mg/ml, |
|
V |
= |
för insprutningsvolymen i ml, och |
|
dn/dc |
= |
är ökningen av refrakdonsindexet för pMMA i tetrahydrofuran (i ml/mg). |
Följande data är normala för en standard-pMMA:
|
R |
= |
2 937 891 |
|
C |
= |
1,07 mg/ml |
|
V |
= |
0,1 ml |
|
dn/dc |
= |
9 × 10-5 ml/mg |
Resulterande K-värde, 3,05 × 1011, används därefter för att beräkna den teoretiska detektorreaktionen, om 100 % av den insprutade polymeren hade eluerat genom detektorn.
A.20 POLYMERERS BETEENDE I VATTEN VID LÖSNING/EXTRAKTION
1. METOD
Den beskrivna metoden är en kopia av en reviderad version av OECD TG 120 (1997). Ytterligare teknisk information finns i hänvisning 1.
1.1 INLEDNING
För vissa polymerer, som t.ex. emultionspolymerer, kan visst inledande förberedande arbete krävas innan nedanstående metod kan användas. Metoden är inte tillämplig på flytande polymerer och på polymerer som under provningsförhållandena reagerar med vatten.
När metoden inte är praktisk eller möjlig kan beteendet vid lösning/extraktion undersökas med andra metoder. I så fall ska fullständiga detaljer och motivering för den använda metoden anges.
1.2. REFERENSÄMNEN
Inga.
1.3. UNDERSÖKNINGSMETODENS PRINCIP
Polymerernas beteende vid lösning/extraktion i vatten fastställs med flaskmetoden (se A.6 lösnings-barhet i vatten, flaskmetod) med nedan beskrivna ändringar.
1.4. KVALITETSKRITERIER
Inga,
1.5. BESKRIVNING AV UNDERSÖKNINGSMETODEN
1.5.1 Utrustning
Följande utrustning krävs för denna metod:
|
— |
Krossanordning, t.ex. kvarn för produktion av partiklar med känd storlek. |
|
— |
Apparater för skakning med möjlighet till temperaturövervakning. |
|
— |
Membranfiltersystem. |
|
— |
Lämplig analysutrustning. |
|
— |
Standardsil. |
1.5.2 Beredning av prover
Ett representativt prov ska först reduceras till en partikelstorlek på mellan 0,125 och 0,25 mm med lämplig sil. Kylning kan krävas för provets stabilitet eller för malningsprocessen. Material med gummiliknande natur kan krossas vid temperaturen på flytande kvävgas (1).
Om den önskade partikelstorleksfraktionen inte kan skapas ska åtgärd vidtas för att i största utsträckning minska partikelstorleken och resultatet därav registreras. Det ska i rapporten anges på vilket sätt det krossade provet förvarats fram till undersökningen.
1.5.3 Förfarande
Tre prover på 10 g av det undersökta ämnet ska vägas i vart och ett av tre kärl försedda med glaspropp, och därefter ska 1 000 ml vatten tillsättas till varje kärl. Om hantering av 10 g polymer visar sig vara opraktiskt, ska den närmast högre mängd som kan hanteras användas och vattenvolymen justeras i enlighet därmed.
Kärlen ska tillslutas noggrant och därefter upphettas till 20 oC. En skak- eller omrörningsanordning som kan arbeta vid konstant temperatur ska användas. Efter en period på 24 timmar ska innehållet i varje kärl centrifugeras eller filtreras och polymerkoncentrationen i den klara vätskefasen fastställas med lämplig analysmetod. Om ingen lämplig analysmetod för vätskefasen finns kan den totala lösbarheten/extraktionsbarheten uppskattas utifrån filterrestens eller centrifugrestens torra vikt.
Det är vanligtvis nödvändigt att kvantitativt differentiera å ena sidan föroreningarna och tillsatserna och å andra sidan de med låg molekylvikt. Det är också viktigt att vid gravimetriskt fastställande göra ett blankprov utan undersökt ämne, för att kunna ta hänsyn till resterna från experimentet.
Polymerernas beteende vid lösning/extraktion i vatten vid 37 oC och pH 2 samt pH 9 kan fastställas på samma sätt såsom beskrivet för utförandet av experimentet vid 20 oC. pH-värdena kan erhållas genom tillsats av antingen lämplig buffert eller syra eller bas, som t.ex. saltsyra, ättiksyra, analytiskt natrium eller kaliumhydroxid eller NH3.
En eller två undersökningar ska göras, beroende på vilken analysmetod som används. När tillräckligt specifika metoder finns för direktanalys av vätskefasen avseende polymerkomponenten, är det tillräckligt med en sådan undersökning som beskrivits här ovan. När sådana metoder inte finns tillgängliga och fastställande av polymerens beteende vid lösning/extraktion begränsas till indirekt analys, genom fastställande enbart genom det totala innehållet organiskt kol (TOC) i vätskeextraktet, ska ytterligare en undersökning göras. Denna kompletterande undersökning ska också göras på tre prover, med tio gånger mindre polymer och samma mängd vatten som använts vid den första undersökningen.
1.5.4 Analys
1.5.4.1 Undersökningar utförda med en provstorlek
Det kan finnas metoder tillgängliga för direktanalys av polymerkomponenter i vätskefasen.
Alternativt kan eventuellt också indirekt analys av upplösta/extraherade polymerkomponenter, genom fastställande av det totala innehållet upplösningsbara delar och korrigering för ej polymerspecifika komponenter, göras.
Analys av vätskefasen avseende totala polymeriska arter är möjlig antingen genom tillräckligt känslig metod, t.ex.:
|
— |
TOC med hjälp av persulfat- eller dikromatupplösning för att producera CO2, följt av uppskattning med IR-analys eller kemisk analys, |
|
— |
Atomabsorbtionsspektrometri (AAS) eller dess induktivt kopplade plasmautsläpp (ICP) motsvarande den för kisel- eller metallinnehållande polymerer, |
|
— |
UV-absorbtion eller spektrofluorimetri för akrylpolymerer, |
|
— |
LC-MS för lågmolekylviktsprover, |
eller genom vakuumevaporation till torrhet av vätskeextraktet och spektroskopisk analys IR, UV, osv.) eller AAS/ICP-analys av resten.
Om analys av vätskefasen som sådan inte är praktiskt genomförbar, ska vätskeextraktet extraheras med ett vattenoupplösligt organiskt lösningsmedel, t.ex. klorerat kolväte. Lösningsmedlet evaporeras därefter och resten analyseras enligt ovan avseende angivet polymerinnehåll. Komponenter i denna rest som identifieras som föroreningar eller tillsatser ska subtraheras för bestämning av polymerens egen upplösning/extraktion.
När förhållandevis stora mängder sådant material finns närvarande, kan det vara nödvändigt att analysera resterna, t.ex. HPLC- eller GC-analys för att differentiera föroreningarna från monomera och monomerderiverade ämnen, så att det faktiska innehållet av det senare kan fastställas.
I vissa fall kan vanlig evaporation av det organiska lösningsmedlet till torrhet och vägning av den torra resten vara tillräckligt.
1.5.4.2 Provning med två olika provstorlekar
Alla vätskeextrakt analyseras avseende total mängd organiskt kol.
Gravimetrisk bestämning ska göras på den oupplösta/ej extraherade delen av provet. Om polymer-resten, efter centrifugering eller filtrering av innehållet i varje kärl, sitter kvar på kärlväggen, ska kärlet sköljas med filtratet tills kärlet är rent från alla synliga rester. Därefter ska filtratet återigen centrifugeras eller filtreras. Kvarvarande rester på filtret eller i centrifugröret torkas vid 40 oC under vakuum och vägs. Torkningen ska fortsätta tills konstant vikt nås.
2. DATA
2.1 PROVNING MED EN ENDA PROVSTORLEK
De enskilda resultaten för var och en av de tre flaskorna samt medelvärdena ska anges och uttryckas i enheter för massa/volym lösning (normalt mg/l) eller massa/massa av polymerprovet (normalt mg/g). Dessutom ska provets viktförlust anges, beräknad som lösningens vikt dividerad med det ursprungliga provets vikt. Den relativa standardavvikelsen ska beräknas. Enskilda värden ska ges för hela ämnet (polymer plus väsentliga tillsatser osv.) och för polymeren ensam (dvs. efter subtraktion av bidraget från sådana tillsatser).
2.2 PROVNING MED TVÅ OLIKA PROVSTORLEKAR
De enskilda värdena för total mängd organiskt kol i vätskeextrakten för de två trippelexperimenten och medelvärdet för varje experiment ska anges, uttryckt som massa per volym lösning (normalt mg C/l), samt som massa per vikt för det ursprungliga provet (normalt mg C/g).
Om ingen skillnad föreligger mellan resultaten för stort respektive litet förhållande mellan prov och vatten, kan detta innebära att alla extraherbara komponenter faktiskt extraherats. I sådana fall krävs normalt ingen direkt analys.
Resternas individuella vikter ska anges och uttryckas i procent av provernas initialvikt. Medelvärden ska beräknas för varje experiment. Skillnaderna mellan 100 och funnen procentandel representerar andelen lösningsbart och extraherbart material i ursprungsprovet.
3. RAPPORT
3.1 PROVNINGSRAPPORT
Provningsrapporten ska innehålla följande information:
3.1.1 Undersökt ämne
|
— |
Tillgänglig information om ämnet, identitet, tillsatser, föroreningar, innehåll av varianter med låg molekylvikt. |
3.1.2 Experimentella förhållanden
|
— |
Beskrivning av tillämpat förfarande och experimentella förhållanden. |
|
— |
Beskrivning av analys- och detektionsmetoder. |
3.1.3 Resultat
|
— |
Resultatet avseende lösningsbarhet/extraktionsbarhet i mg/l, individuella värden och medelvärden för extraktionsprovet för de olika lösningarna, uppdelat i polymerinnehåll och föroreningar, tillsatser osv. |
|
— |
Resultatet av upplösningen/extraktionen av polymeren i mg/g. |
|
— |
Värden för det totala innehållet organiskt kol i vätskeextrakten, lösningens vikt och beräknade andel, om denna mäts. |
|
— |
pH-värde för varje prov. |
|
— |
Information om blankvärden. |
|
— |
Vid behov hänvisningar till det undersökta ämnets kemiska instabilitet, både under provnings- och analysprocessen. |
|
— |
All information som är viktig för tolkningen av resultaten. |
4. HÄNVISNINGAR
|
1. |
DIN 53733 (1976), Provtagning från plastprodukter för provningsändamål. |
A.21 OXIDERANDE EGENSKAPER (VÄTSKOR)
1. METOD
1.1 INLEDNING
Denna provningsmetod är utformad för att mäta ett flytande ämnes potential att öka förbränningshastigheten eller förbränningsintensiteten hos ett brännbart ämne, eller att bilda en blandning med ett brännbart ämne som självantänds när de två blandas noggrant. Metoden är baserad på FN:s provningsmetod Provning för oxiderande vätskor (1) och är likvärdig med den. Då denna metod A.21 emellertid i första hand är utformad för att uppfylla kraven i förordning (EG) nr 1907/2006, krävs endast jämförelse med ett referensämne. Provning och jämförelse med ytterligare referensämnen kan bli nödvändiga om provningsresultaten förväntas användas för andra syften (9).
Denna provning behöver inte utföras när en undersökning av strukturformeln kan fastställa att ämnet utan rimligt tvivel är oförmöget att reagera exotermiskt med ett brännbart material.
Det är värdefullt att ha förhandsinformation om ämnets alla möjliga explosiva egenskaper innan denna provning utförs.
Denna provning är inte tillämplig på fasta ämnen, gaser, explosiva eller mycket brandfarliga ämnen, eller peroxider.
Denna provning behöver eventuellt inte utföras, om resultat för provsubstansen ifråga från provning enligt FN:s Provning för oxiderande vätskor (1) redan finns tillgängliga.
1.2 DEFINITIONER OCH ENHETER
Med ”medeltid för tryckstegring” avses medelvärdet av de uppmätta tiderna för att en blandning vid provning ska åstadkomma en tryckökning från 690 kPa till 2 070 kPa över det atmosfäriska trycket.
1.3 REFERENSÄMNE
65 % (viktprocent) salpetersyra (analysgrad) ska användas som referensämne. (10)
Om den som utför försöken förutser att resultaten från denna provning eventuellt kommer att användas för andra syften (9), kan även provning med ytterligare referensämnen vara lämpligt (11).
1.4 PRINCIPEN FÖR PROVNINGSMETODEN
Vätskan som ska provas blandas med fibercellulosa, i ett viktförhållande 1 till 1, och införs i ett tryckkärl. Om vid blandning eller fyllning självantändning inträffar krävs ingen ytterligare provning.
Om självantändning inte inträffar ska hela provningen utföras. Blandningen upphettas i ett tryckkärl och medelvärdet fastställs för den tid det tar för trycket att stiga från 690 kPa till 2 070 kPa över det atmosfäriska trycket. Denna medeltid jämförs med medeltiden för trycket att stiga för en 1:1-blandning av referensämnet (-ämnena) och cellulosa.
1.5 KVALITETSKRITERIER
I en serie om fem försök med ett och samma ämne ska inget resultat avvika med mer än 30 % från det aritmetiska medelvärdet. Resultat som avviker med mer än 30 % från medelvärdet ska förkastas, tillvägagångssättet för blandning och fyllning förbättras och provningen upprepas.
1.6 BESKRIVNING AV METODEN
1.6.1 Preparering
1.6.1.1 Brännbart ämne
Torkad fibercellulosa med en fiberlängd mellan 50 och 250 μm och en medeldiameter av 25 μm (12), används som brännbart material. Den torkas tills vikten inte ändras, i ett lager som är högst 25 mm tjockt vid 105 oC under fyra timmar, och förvaras i en exikator med torkmedel tills den svalnat och ska användas. Vattenhalten i den torkade cellulosan ska vara mindre än 0,5 % av torrsubstansen (13). Om det behövs ska torktiden förlängas för att uppnå detta (14). Samma parti cellulosa ska användas under hela provningen.
1.6.1.2 Apparat
1.6.1.2.1
Ett tryckkärl krävs. Kärlet består av ett cylindriskt tryckkärl som är 89 mm långt med en ytterdiameter av 60 mm (se figur 1). Två plana ytor bearbetas fram på motstående sidor (minskar kärlets tvärsnitt till 50 mm) för att underlätta fasthållning när tändstift och urluftningsplugg monteras. Kärlet som har en urborrning med en diameter av 20 mm har falsats på insidan i båda ändar till ett djup av 19 mm och gängats till 1”British Standard Pipe (BSP) eller metrisk motsvarighet. En tryckavlastare, i form av ett sidostycke, är inskruvad i den rundade delen av tryckkärlet 35 mm från ena änden och vinkelrätt mot de plana ytorna. Uttaget för detta borras till ett djup av 12 mm och gängas till 1/2” BSB (eller metrisk motsvarighet) för att passa till gängan på sidostyckets ände. Om så behövs kan en inert tätning anbringas för att se till att anslutningen blir gastät. Sidostycket sticker ut 55 mm från tryckkärlets huvuddel och har ett hål med en diameter av 6 mm. Sidostyckets ände är falsad och gängad för att passa en tryckgivare av membrantyp. Alla tryckmätningsanordningar kan användas under förutsättning att de inte påverkas av de heta gaserna eller upplösningsprodukterna och klarar att svara på tryckhöjningshastigheter motsvarande 690–2 070 kPa på mindre än 5 ms.
Tryckkärlets ände som är längst från sidostycket är förseglad med ett tändstift som är utrustat med två elektroder, den ena isolerad från och den andra ansluten till tändstiftets huvuddel. Den andra änden av tryckkärlet är förseglad med ett sprängbleck (sprängtryck cirka 2 200 kPa) som hålls på plats av en fästplugg med ett hål med en diameter av 20 mm. Om så behövs kan en inert tätning användas tillsammans med tändstiftet för att garantera en gastät anslutning. En stödställning (figur 2) håller enheten i rätt läge vid användning. Denna omfattar vanligen en gjutstålsplatta med måtten 235 mm × 184 mm × 6 mm och ett 185 mm långt fyrkantsrör 70 mm × 70 mm × 4 mm.
En sektion är bortskuren från motstående sidor i ena änden av fyrkantsrörets längd så att en struktur med två plansidiga ben som sticker ut från ett 86 mm långt helt fyrkantrör återstår. Ändarna på dessa plana sidor kapas av med en vinkel av 60o mot horisontalplanet och svetsas fast på bottenplattan. Ett urtag som är 22 mm brett och 46 mm djupt bearbetas fram på den ena sidan av den övre änden av fyrkantsröret, på så sätt att sidostycket passar in i urtaget när tryckkärlsenheten – med tändstiftsänden först – sänks ned i stödställningen. En stålbit som är 30 mm bred och 6 mm tjock svetsas fast på den undre inre sidan av fyrkantsröret för att fungera som en distans. Två 7 mm tumskruvar, gängade i motsatta sidor, håller tryckkärlet stadigt på plats. Två 12 mm breda band av 6 mm tjockt stål, svetsade på de sidobitar som sitter på fyrkantsrörets undre del, stödjer tryckkärlet från undersidan.
1.6.1.2.2
Tändsystemet består av en 25 cm lång Ni/Cr-tråd med en diameter av 0,6 mm och ett motstånd av 3,85 ohm/m. Tråden är lindad, med hjälp av en 5 mm tjock stång, till formen av en spiral och är fastsatt vid tändstiftets elektroder. Spiralen ska ha ett av de utseenden som visas i figur 3. Avståndet mellan tryckkärlets undersida och undersidan av tändspiralen ska vara 20 mm. Om elektroderna inte går att justera ska tändtrådens ändar mellan spiralen och kärlets undersida isoleras med ett keramiskt överdrag. Tråden värms upp av en kraftkälla med konstant spänning som kan ge minst 10 A.
1.6.2 Provningens utförande (15)
Apparaten, helt monterad med tryckgivare och uppvärmningssystem men utan sprängblecket på plats, stöds med tändstiftsänden nedåt. 2,5 g av den vätska som ska provas blandas med 2,5 g torkad cellulosa i dekanteringsglas med hjälp av en omrörningsstav av glas (16). För säkerhets skull ska blandningen utföras med en skyddsskärm mellan operatören och blandningen. Om vid blandning eller fyllning självantändning uppstår krävs ingen ytterligare provning. Blandningen tillsätts tryckkärlet i små portioner under knackning, så att blandningen säkert packas runt tändspiralen och är i god kontakt med den. Det är viktigt att spiralen inte förvrids under packningsprocessen då detta kan leda till oriktiga resultat (17). Sprängblecket sätts på plats och fästpluggen skruvas åt ordentligt. Det fyllda kärlet placeras, med sprängblecket överst, i stödställningen vilken ska stå i ett lämpligt, förstärkt dragskåp eller eldningsutrymme. Kraftförsörjningen ansluts till tändstiftets yttre anslutningar och 10 A läggs på. Tiden mellan det att blandningen påbörjas och inkopplingen av strömmen får inte överstiga 10 minuter.
Signalen som avges av tryckgivaren registreras med ett lämpligt system som möjliggör både utvärdering och alstring av en permanent dokumentering av den tidtrycksprofil som erhålls (t.ex. en transientregistrerare kopplad till en skrivare). Blandningen upphettas tills sprängblecket går sönder eller tills minst 60 s har gått. Om sprängblecket inte går sönder, ska man låta blandningen svalna innan man försiktigt demonterar apparaten, med vidtagande av försiktighetsåtgärder för att hantera eventuellt övertryck. Fem försök utförs med provämnet och referensämnet (-ämnena). Den tid det tar för trycket att stiga från 690 kPa till 2 070 kPa över det atmosfäriska trycket noteras. Medelvärdet för tryckstegringstiden beräknas.
I vissa fall kan ämnen alstra en tryckstegring (alltför snabb eller alltför långsam) som orsakas av kemiska reaktioner och som inte är relaterad till ämnets oxiderande egenskaper. I dessa fall kan det vara nödvändigt att upprepa provningen med ett inert ämne, t.ex. diatomit (kiselgur), i stället för cellulosa för att klargöra reaktionens natur.
2. DATA
Tryckstegringstiderna både för provämnet och referensämnet (-ämnena). Tryckstegringstiderna för provningarna med ett inert ämne, om sådana utförts.
2.1 BEHANDLING AV RESULTAT
Tryckstegringstiderna både för provämnet och referensämnet (-ämnena) beräknas.
Medelvärdet för tryckstegringstiderna för provningarna med ett inert ämne (om sådana utförts) beräknas.
I tabell 1 visas några exempel på resultat.
Tabell 1
Exempel på resultat (21)
|
Ämne (18) |
Medeltryckstegringstid för en 1:1-blandning med cellulosa (ms) |
|
Ammoniumdikromat, mättad vattenlösning |
20 800 |
|
Kalciumnitrat, mättad vattenlösning |
6 700 |
|
Järn(III)nitrat, mättad vattenlösning |
4 133 |
|
Litiumperklorat, mättad vattenlösning |
1 686 |
|
Magnesiumperklorat, mättad vattenlösning |
777 |
|
Nickelnitrat, mättad vattenlösning |
6 250 |
|
Salpetersyra, 65 % |
4 767 (20) |
|
Perklorsyra, 55 % |
121 (20) |
|
Perchloric acid, 55 % |
59 |
|
Kaliumnitrat, 30 % vattenlösning |
26 690 |
|
Silvernitrat, mättad vattenlösning |
|
|
Natriumklorat, 40 % vattenlösning |
2 555 (20) |
|
Natriumnitrat, 45 % vattenlösning |
4 133 |
|
Inert ämne |
|
|
Vatten: cellulosa |
3. RAPPORT
3.1 PROVNINGSRAPPORT
Provningsrapporten ska omfatta följande information:
|
— |
Identiteten, sammansättningen, renheten, etc. för det ämne som provas. |
|
— |
Provämnets koncentration. |
|
— |
Torkprocessen för använd cellulosa. |
|
— |
Vattenhalten för använd cellulosa. |
|
— |
Resultatet av mätningarna. |
|
— |
Resultatet från provningar med ett inert ämne, om sådana utförts. |
|
— |
Det beräknade medelvärdet för tryckstegringstiden. |
|
— |
Alla avvikelser från denna metod och orsaken till dem. |
|
— |
All ytterligare information eller kommentarer som har betydelse för resultatets tolkning. |
3.2 TOLKNING AV RESULTATET (22)
Provningsresultaten har bedömts på grundval av
|
a) |
huruvida blandningen av provämne och cellulosa självantänder, och jämförelse av medelvärdet för den tid det tar för trycket att stiga från 690 kPa till 2 070 kPa |
|
b) |
med respektive referensämne (-ämnen). |
Ett flytande ämne anses vara ett oxidationsmedel om
|
a) |
en 1:1-blandning, i viktdelar, av ämnet och cellulosa självantänder, eller |
|
b) |
en 1:1-blandning, i viktdelar, av ämnet och cellulosa uppvisar ett medelvärde för tryckstegringstiden som är mindre än eller lika med medelvärdet för tryckstegringstiden för en 1:1-blandning, i viktdelar, av 65 % (viktprocent) salpetersyra och cellulosa. |
För att undvika falska positiva resultat ska, om så behövs, även de resultat som erhålls vid provning av ämnet med ett inert material beaktas då resultaten tolkas.
4. HÄNVISNINGAR
|
1. |
FN:s rekommendationer om transport av farligt gods, manual för provningar och kriterier. 3:e reviderade utgåvan. FN:s publikation nr: ST/SG/AC.10/1 l/rev. 3, 1999, sid. 342. Provning O.2: Provning för oxiderande vätskor. |
Figur 1
Tryckkärl

Figur 2
Stödställning

Figur 3
Tändsystem

Anm.: Endera av de här systemen kan användas.
(1) Beroende på typ av instrument och ämnets renhetsgrad.
(2) Beroende på typ av instrument och ämnets renhetsgrad.
(3) Beroende på typ av instrument och ämnets renhetsgrad.
(4) Beroende på typ av instrument och ämnets renhetsgrad.
(5) Denna noggrannhet gäller endast för den enkla anordning som beskrivs i t.ex. ASTM D 1120-72; den kan förbättras med mer avancerade ebulliometeranordningar.
(6) Gäller endast för rena ämnen. Användning under andra omständigheter ska motiveras.
(7) Beroende på renhetsgrad.
(8) Denna metod kan också användas i området 1 till 10 Pa, under förutsättning att försiktighet iakktas.
(9) Såsom, till exempel, inom ramen för FN:s transportföreskrifter.
(10) Såsom, till exempel, inom ramen för FN:s transportföreskrifter.
(11) I hänvisning 1 används t.ex. 50 % (viktprocent) perklorsyra och 40 % (viktprocent) natriumklorat.
(12) T.ex. Whatmans kolonn kromatografiska cellulosapulver CF 11, katalognummer 4021 050.
(13) Bekräftad genom (t.ex.) Karl-Fisher titrering.
(14) Denna vattenhalt kan alternativt åstadkommas genom (t.ex.) uppvärmning vid 105 oC i vakuum under 24 timmar.
(15) Blandningar av oxidationsmedel och cellulosa ska behandlas som potentiellt explosiva och hanteras med försiktighet.
(16) I praktiken kan detta åstadkommas genom att göra i ordning en 1:1-blandning av vätskan, som ska provas, och cellulosa i en större mängd än vad som behövs för försöket och överföra 5 ±0,1 g till tryckkärlet. Blandningen måste vara nygjord för varje försök.
(17) Framför allt måste kontakt mellan spiralens på varandra följande varv undvikas.
(18) Se hänvisning (1) för klassificering enligt FN:s transportsystem
(19) Mättade lösningar ska iordningställas vid 20 oC.
(20) Medelvärde från jämförande försök från flera laboratorier
(21) Det maximala trycket 2 070 kPa uppnåddes ej.
(22) Se referens 1 för tolkning av resultaten enligt FN:s transportföreskrifter med användning av flera referensämnen.
DEL B: METODER FÖR BESTÄMNING AV TOXICITET OCH ANDRA HÄLSOEFFEKTER
INNEHÅLLSFÖRTECKNING
ALLMÄN INLEDNING
|
B.1a |
AKUT TOXICITET (ORALT) – METOD MED FASTA SÄRSKILJANDE DOSER |
|
B.1b |
AKUT TOXICITET ORALT – METOD FÖR BESTÄMNING AV AKUT TOXICITETSKLASS |
|
B.2 |
AKUT TOXICITET (INANDNING) |
|
B.3 |
AKUT TOXICITET (DERMALT) |
|
B.4 |
AKUT TOXICITET: HUDIRRITATION/HUDKORROSION |
|
B.5 |
AKUT TOXICITET: ÖGONIRRITATION/ÖGONKORROSION |
|
B.6 |
HUDSENSIBILISERING |
|
B.7 |
28 DAGARS STUDIE AV TOXICITET MED UPPREPAD ORAL DOSERING |
|
B.8 |
TOXICITET VID UPPREPAD DOSERING (28 DAGAR, INANDNING) |
|
B.9 |
TOXICITET VID UPPREPAD DOSERING (28 DAGAR, DERMALT) |
|
B.10 |
MUTAGENICITET – TEST IN VITRO AV KROMOSOMAVVIKELSER HOS DÄGGDJUR |
|
B.11 |
MUTAGENICITET – TEST IN VIVO AV KROMOSOMAVVIKELSER I BENMÄRG HOS DÄGGDJUR |
|
B.12 |
MUTAGENICITET – TEST AV MIKROKÄRNOR IN VIVO I ERYTROCYTER HOS DÄGGDJUR |
|
B.13/14 |
MUTAGENICITET – TEST AV ÅTERMUTATION HOS BAKTERIER |
|
B.15 |
MUTAGENICITETSTEST OCH SVREENING FÖR CANCERGENICITET |
|
B.16 |
MITOTISK REKOMBINATION – SACCHAROMYCES CEREVISIAE |
|
B.17 |
MUTAGENICITET – TEST AV GENMUTATIONER HOS DÄGGDJURSCELLER IN VITRO |
|
B.18 |
DNA-SKADA OCH DNS-REPARATION – FELAKTIG DNA-SYNTES – DÄGGDJURSCELLER IN VITRO |
|
B.19 |
TEST AVSEENDE SYSTERKROMATIDSBYTEN IN VITRO |
|
B.20 |
KÖNSBUNDEN RECESSIV LETAL TEST PÅ DROSOPHILA MELANOGASTER |
|
B.21 |
CELLTRANSFORMATIONSTEST PÅ DÄGGDJURSCELLER IN VITRO |
|
B.22 |
DOMINANT LETAL TEST PÅ GNAGARE |
|
B.23 |
SPERMATOGENIAL TEST AV KROMOSOMAVVIKELSER HOS DÄGGDJUR |
|
B.24 |
FLÄCKTEST PÅ MUS |
|
B.25 |
TEST AVSEENDE ÄRFTLIG TRANSLOKATION PÅ MUS |
|
B.26 |
TEST AVSEENDE SUBKRONISK ORAL TOXICITET (90-DAGARS UPPREPAT ORALTEST PÅ GNAGARE) |
|
B.27 |
TEST AVSEENDE SUBKRONISK ORAL TOXICITET (90-DAGARS UPPREPAT ORALTEST PÅ ICKE-GNAGARE) |
|
B.28 |
TEST AVSEENDE SUBKRONISK DERMAL TOXICITET (90-DAGARS REPETERAD DERMALTEST PÅ GNAGARE) |
|
B.29 |
TEST AVSEENDE SUBKRONISK INHALATIONSTOXICITET (90-DAGARS REPETERAD INHALATIONSTEST PÅ GNAGARE) |
|
B.30 |
TEST AVSEENDE KRONISK TOXICITET |
|
B.31 |
TEST AVSEENDE PRENATAL UTVECKLINGSTOXICITET |
|
B.32 |
TEST AVSEENDE CANCERGENICITET |
|
B.33 |
KOMBINERAT TEST AVSEENDE KRONISK TOXICITET OCH CANCERGENICITET |
|
B.34 |
REPRODUKTIONSTOXICITETSTEST PÅ EN GENERATION |
|
B.35 |
TEST AVSEENDE REPRODUKTIONSTOXICITET PÅ TVÅ GENERATIONER |
|
B.36 |
TOXIKOKINETIK |
|
B.37 |
FÖRDRÖJD NEUROTOXICITET ORSAKAD AV ORGANISKA FOSFORFÖRENINGAR TILL FÖLJD AV AKUT EXPONERING |
|
B.38 |
FÖRDRÖJD NEUROTOXICITET ORSAKAD AV ORGANISKA FOSFORFÖRENINGAR (28 DAGARS UPPREPAD DOSUNDERSÖKNING) |
|
B.39 |
TEST AV REPARATIONSSYNTES (uds) HOS LEVERCELLER FRÅN DÄGGDJUR IN VIVO |
|
B.40 |
IN VITRO-TEST AV HUDKORROSIVITET: BESTÄMNING AV TRANSKUTANT ELEKTRISKT MOTSTÅND (TER) |
|
B.40a |
IN VITRO-TEST AV HUDKORROSIVITET: TEST MED MODELL AV HUMAN HUD |
|
B.41 |
3T3 NRU-FOTOTOXICITETSTEST IN VITRO |
|
B.42 |
HUDSENSIBILISERING: LLNA-METODEN (LOCAL LYMPH NODE ASSAY) |
|
B.43 |
TEST AVSEENDE NEUROTOXICITET PÅ GNAGARE |
|
B.44 |
HUDABSORPTION: IN VIVO-METOD |
|
B.45 |
HUDABSORPTION: IN VITRO-METOD |
ALLMÄN INLEDNING
A. BESKRIVNING AV TESTSUBSTANSEN
Testsubstansens sammansättning, inbegripet de viktigaste orenheterna, och dess relevanta fysikaliskt-kemiska egenskaper, inbegripet ämnets stabilitet, ska vara kända innan en toxicitetsundersökning inleds.
Testsubstansens fysikaliskt-kemiska egenskaper ger viktig vägledning vid val av tillförselväg, utformning av varje enskild undersökning samt hantering och lagring av testsubstansen.
En analysmetod för kvalitativ och kvantitativ bestämning av testsubstansen (om möjligt inbegripet viktiga orenheter) i doseringsmediet och i biologiska material bör utvecklas innan en undersökning inleds.
Alla uppgifter om identifiering av testsubstansen, dess fysikaliskt-kemiska egenskaper, renhet och beteende bör omfattas av testrapporten.
B. DJURVÅRD
En strikt kontroll av miljöförhållandena samt tillämpliga djurvårdsmetoder är grundläggande i fråga om toxicitetstester.
i) Förvaltningsförhållanden
Miljön i försöksdjurens rum eller burar ska vara avpassade efter djurarten. Lämpliga betingelser för råttor, möss och marsvin är en rumstemperatur på 22 ±3 oC med en relativ luftfuktighet på 30 – 70 %. För kaniner och marsvin bör temperaturen hållas vid 20 ±3 oC med en relativ luftfuktighet på 30 – 70 %.
Vissa testmetoder är särskilt känsliga för temperatureffekter och i dessa fall ingår detaljuppgifter om lämpliga betingelser i metodbeskrivningen. I alla undersökningar av toxiska effekter ska temperaturen och luftfuktigheten kontrolleras, registreras och anges i den slutliga testrapporten.
Artificiell belysning bör användas, med en dygnsrytm på 12 timmar ljus, 12 timmar mörker. Detaljuppgifter om belysningsrytmen ska registreras och anges i den slutliga testrapporten.
Om inte annat anges för metoden får djuren inhysas individuellt eller i små grupper av samma kön; i fråga om grupper bör högst fem djur inhysas i varje bur.
I rapporter om djurförsök är det viktigt att ange vilken typ av bur som används och antalet djur i varje bur, både under exponering för det kemiska ämnet och under en påföljande observationstid.
ii) Utfodring
Fodret ska uppfylla alla näringsmässiga krav som kan ställas för den djurart som används i försöket. Om testsubstansen tillförs djuren via fodret kan näringsvärdet sjunka till följd av en samverkan mellan ämnet och någon beståndsdel i fodret. Möjligheten av en sådan reaktion ska övervägas vid tolkningen av testresultaten. Konventionellt laboratoriefoder får användas tillsammans med obegränsade mängder vatten. Valet av foder kan påverkas av behovet av att säkerställa en lämplig blandning med testsubstansen när denna tillförs enligt denna metod.
I fodret får inte finnas störande koncentrationer av föroreningar som man vet påverkar toxiciteten.
C. ALTERNATIVA TESTER
För Europeiska unionen är det ett åtagande att främja utvecklandet och valideringen av alternativa metoder som kan ge samma informationsnivå som nuvarande djurförsök, men som använder färre djur, orsakar mindre lidande eller undviker användning av djur fullständigt.
När sådana metoder blir tillgängliga ska de om möjligt beaktas för riskbedömning och efterföljande klassificering och märkning av eventuella risker.
D. UTVÄRDERING OCH TOLKNING
När tester utvärderas och tolkas ska begränsningarna för i vilken utsträckning resultaten av djurförsök och in vitro-undersökningar går att tillämpa på människor beaktas och därför får bevis för negativa effekter på människor i förekommande fall användas som bekräftelse av testresultaten.
E. LITTERATURHÄNVISNINGAR
De flesta av dessa metoder har utvecklats inom ramen för OECD:s program för riktlinjer för tester och bör utföras i enlighet med principerna för god laboratoriepraxis, i syfte att så långt möjligt säkerställa ”ömsesidigt godtagande av uppgifter”.
Ytterligare information finns i de hänvisningar som görs i OECD:s riktlinjer och relevant litteratur som publicerats på annat håll.
B.1a AKUT TOXICITET (ORALT) – METOD MED FASTA SÄRSKILJANDE DOSER
1. METOD
Denna testmetod motsvarar OECD TG 420 (2001).
1.1 INLEDNING
Vid traditionella metoder för bedömning av akut toxicitet används djurens död som slutpunkt. 1984 föreslog British Toxicology Society en ny metod för test avseende akut toxicitet. Den nya metoden bygger på att djuren ges en serie doser på definierade nivåer (se hänvisning 1). Då behöver djurens död inte användas som slutpunkt, eftersom metoden bygger på observationer av klara tecken på toxicitet vid någon av de definierade dosnivåerna. Efter brittiska och internationella (se hänvisning 2 respektive 3) valideringsstudier in vivo 1992 detta förfarande som en testmetod. Därefter har metodens statistiska egenskaper utvärderats med hjälp av matematiska modeller i en serie studier (se hänvisning 4, 5, och 6). Med hjälp av dessa in vivo- och modelleringsstudier har man kunnat demonstrera att metoden är reproducerbar, kräver ett mindre antal djur och orsakar mindre lidande än de traditionella metoderna, och att metoden även gör det möjligt att klassificera ämnen på ett liknande sätt som med de övriga testmetoderna för akut toxicitet.
Riktlinjer för valet av den lämpligaste testmetoden för ett visst ändamål finns i vägledningen om testning av akut toxicitet oralt (se hänvisning 7). I vägledningen finns även tilläggsinformation rörande utförandet av testmetod B.1b och tolkningen av resultaten.
Metoden bygger på principen om att endast skäligen toxiska doser används i huvudtestet, och att doser som förväntas vara dödliga ska undvikas. Doser som, på grund av frätande eller allvarligt irriterande verkningar, är kända att orsaka klar smärta och svårt lidande behöver inte användas. Döende djur eller djur som uppvisar klara tecken på smärta eller allvarligt och ihållande lidande ska avlivas skonsamt, och vid tolkningen av testresultaten ska dessa djur beaktas på samma sätt som djur som har dött under testets gång. Kriterierna för beslut rörande avlivning av döende eller svårt lidande djur, liksom även riktlinjer för tecknen på förutsägbar eller annalkande död, behandlas i en separat vägledning (se hänvisning 8).
Metoden ger information om ett ämnes farliga egenskaper och möjliggör klassificering av ämnet enligt det globala harmoniserade systemet (GHS) för klassificering av kemikalier som orsakar akut toxicitet (se hänvisning 9).
Testningslaboratoriet bör analysera all tillgänglig information om testämnet innan själva testningen genomförs. Sådan information inbegriper ämnets identitet och kemiska struktur, fysikalkemiska egenskaper, resultaten från andra eventuella in vitro- eller in vivo-test avseende toxicitet, toxikologiska data om strukturellt närbesläktade ämnen och ämnets tilltänkta användningsändamål. Denna information behövs för att avgöra huruvida testet är relevant för skyddet av människors hälsa, och hjälper vid valet av en optimal startdos.
1.2 DEFINITIONER
Akut toxicitet (oralt): De skadliga verkningar som uppträder efter oral tillförsel av en engångsdos av ett ämne eller upprepade doser som ges inom 24 timmar.
Fördröjd död: Det att ett djur inte dör eller är döende inom 48 timmar, men dör senare under den 14 dagar långa observationsperioden.
Dos: Den mängd testämne som tillförs. Dosen uttrycks som vikten testämne per viktenhet försöksdjur (exempelvis mg/kg).
Uppenbar toxicitet: En allmän term för att beskriva klara tecken på toxicitet efter tillförsel av testämnet (exempel finns i hänvisning 3). Verkningarna är då så allvarliga att närmaste högre dos troligen leder till att största delen av djuren utsätts för svår smärta eller uppvisar ihållande tecken på allvarligt lidande, är döende (kriterierna anges i vägledningen om konsekvenser för människor, se hänvisning 8) eller sannolikt dör.
GHS: Globala harmoniserade systemet för klassificering och märkning av kemikalier och blandningar. Systemet utarbetas gemensamt inom OECD (människors hälsa och miljö), FN:s expertkommitté om transport av farligt gods (fysikalkemiska egenskaper) och ILO (varningar). Samordningen sker genom det gemensamma programmet för sund kemikaliehantering (IOMC).
Annalkande död: När ett djur troligen är döende eller har dött före den nästa planenliga observationstidpunkten. Hos gnagare kan tecknen på annalkande död inbegripa konvulsioner, lateral kroppsställning, halvliggande ställning och tremor (närmare uppgifter finns i vägledning om konsekvenserna för människor, se hänvisning 8).
LD50 (median letal dos): En statistiskt fastställd engångsdos som kan förväntas leda till döden för 50 % av de djur som har tillförts dosen oralt. LD 50 -värdet uttrycks som vikten testämne per viktenhet försöksdjur (mg/kg).
Gränsdos: En dos på den övre gränsnivån (2 000 eller 5 000 mg/kg).
Döende djur: Ett djur som håller på att dö eller som har små chanser att överleva, även om det får behandling (närmare uppgifter finns i vägledningen om konsekvenserna för människors hälsa, se hänvisning 8).
Förutsägbar död: Förekomsten av kliniska tecken som tyder på död vid en känd framtida tidpunkt som infaller före försökets planerade avslutningstidpunkt, exempelvis oförmåga att nå vatten eller föda (närmare uppgifter finns i vägledningen om konsekvenserna för människors hälsa, se hänvisning 8).
man når den dos som orsakar uppenbar toxicitet eller ett dödsfall, eller tills inga verkningar kan iakttas på den högsta dosnivån eller dödsfall inträffar på den lägsta dosnivån.
1.4 BESKRIVNING AV TESTMETODEN
1.4.1 Val av djurart
Rekommenderad gnagare är råtta, även om testet kan utföras med andra gnagare. I regel används hondjur (se hänvisning 7). Detta beror på att litteraturstudier av konventionella LD50-test indikerar att känslighetsskillnaderna mellan könen är små, men att honorna i förekommande fall ofta är en aning mer känsliga (se hänvisning 10). Om däremot kunskaperna om strukturellt närbesläktade ämnens toxikologiska eller toxiko-kinetiska egenskaper tyder på att hanarna sannolikt är känsligare än honorna bör hanar användas. Om testet görs på hanar bör tillbörlig motivering ges.
Djuren bör vara unga och friska vuxna exemplar som härrör från normala laboratoriestammar. Hondjuren får inte ha fött ungar och får inte vara dräktiga. När dostillförseln inleds ska djuret vara mellan 8 och 12 veckor gammalt, och variationerna i vikt får vara högst ± 20 % av medelvikten för alla tidigare djur som har doserats.
1.4.2 Förvarings- och utfodringsbetingelser
Temperaturen i testdjurens utrymmen bör vara 22 oC (± 3 oC). Den relativa fuktigheten bör vara minst 30 % och ska helst inte överstiga 70 %, utom när rummet rengörs. Målvärdet bör dock vara 50–60 %. Belysningen bör vara artificiell med omväxlande 12 timmar ljus och 12 timmar mörker. För utfodringen kan konventionellt laboratoriefoder användas, och djuren bör ha obegränsad tillgång till dricksvatten. Djur som tillförs doser på samma nivå kan inhysas i samma bur, men antalet djur per bur får inte vara högre än att klara observationer kan göras av varje djur.
1.4.3 Förberedelse av djuren
Djuren väljs ut slumpmässigt och förses med märkning så att de kan identifieras individuellt Därefter får djuren acklimatisera sig till laboratoriets betingelser i sina burar i minst 5 dagar före doseringsstart.
1.4.4 Förberedelse av doserna
I regel ska de olika doserna av testämnet tillföras i en konstant volym, vilket innebär att doserna har varierande koncentration. När en slutprodukt i vätskeform eller en blandning testas, kan det med tanke på riskbedömningen vara mer relevant att använda testämnet i outspädd form (konstant koncentration), vilket även förutsätts av vissa tillsynsmyndigheter. Den maximala dosvolymen för tillförsel får aldrig överskridas. Den största mängd vätska som kan tillföras vid ett och samma tillfälle beror på testdjurets storlek. För gnagare bör denna volym i regel inte överskrida 1 ml per 100 g kroppsvikt. I fråga om vattenlösningar får dock 2 ml per 100 g kroppsvikt användas. För tillförsel av dosen rekommenderas alltid i första hand lösning, suspension eller emulsion i vatten, sedan lösning, suspension eller emulsion i olja (exempelvis majsolja) och därefter eventuella andra vehiklar. I fråga om andra vehiklar än vatten måste de toxikologiska egenskaperna vara kända. Doserna måste beredas strax före tillförseln, utom i fall där preparatets stabilitet är känd och har påvisats vara godtagbar över hela användningsperioden.
1.5 FÖRFARANDE
1.5.1 Tillförsel av doser
Testämnet tillförs som en engångsdos genom sondmatning med hjälp av magsond eller lämplig intubationskanyl. I det sällsynta fallet att det inte är möjligt med en engångsdos, kan dosen tillföras i mindre delar över en period som inte överskrider 24 timmar.
Före dostillförseln hålls djuren fastande (utan föda och med tillgång till vatten, för råttor över natten och för möss i 3–4 timmar). Efter fasteperioden vägs djuren och testämnet tillförs. Efter tillförsel av ämnet kan djuren hållas utan föda i ytterligare 3–4 timmar (råttor) respektive 1–2 timmar (möss). Om dosen tillförs i delar över en tidsperiod kan det vara nödvändigt att ge djuren föda och vatten, beroende på periodens längd.
1.5.2 Inledande test
Avsikten med ett inledande test är att få en grund för valet av optimal startdos för huvudtestet. Testämnet tillförs enskilda djur enligt den sekvens som anges i flödesschemat i bilaga 1. Det inledande testet kan avslutas när sekvensen når fram till ett utfall om en startdos för huvudtestet (eller om ett djur dör på den lägsta angivna dosnivån).
Som startdos för det inledande testet väljs en av de angivna dosnivåerna 5, 50, 300 och 2 000 mg/kg. Startdosen väljs så att den troligen ger tecken på uppenbar toxicitet, och den ska i mån av möjlighet grunda sig på resultat från in vivo- och in vitro-test av samma kemikalie och strukturellt närbesläktade kemikalier. Om sådan information inte finns att tillgå, används startdosen 300 mg/kg.
Det bör gå minst 24 timmar från doseringen av att djur till doseringen av följande djur. Alla djur bör observeras i minst 14 dagar.
Undantagsvis, och endast när det kan motiveras med specifika tillsynskrav, får en ytterligare högre dosnivå på 5 000 mg/kg användas (se bilaga 3). Med hänsyn till djurens välbefinnande avråds testning på djur när det gäller GHS-kategori 5 (2 000–5 000 mg/kg). Sådan testning får endast göras när det finns en stark sannolikhet för att resultaten har en direkt relevans för skyddet av djurs eller människors hälsa eller av miljön.
I ett inledande test där ett djur dör vid den lägsta dosnivån (5 mg/kg), bör förfarandet i regel avslutas och ämnet klassificeras som tillhörande GHS-kategori 1 (se bilaga 1). Om det ändå behövs ytterligare bekräftelse av klassificeringen kan ett valfritt tilläggsförfarande genomföras. Detta går ut på att ett andra djur doseras vid nivån 5 mg/kg. Om detta andra djur dör, anses GHS-kategorin 1 vara bekräftad och testet avslutas omedelbart. Om det andra djuret överlever doseras högst tre tilläggsdjur vid nivån 5 mg/kg. Eftersom mortalitetsrisken är hög bör dessa djur av hänsyn till djurens välbefinnande doseras sekventiellt. Efter doseringen av ett djur får följande djur inte doseras förrän det är klart att det tidigare djuret sannolikt överlever. Om något av dessa djur dör ska doseringssekvensen omedelbart avslutas, och inga ytterligare djur ska doseras. Eftersom förekomsten av ett dödsfall till (oavsett hur många djur som har testats vid testets slut) leder till utfall A (två eller flera dödsfall), ska klassificeringen enligt bilaga 2 göras för dosen 5 mg/kg (kategori 1 om det förekommit två eller flera dödsfall eller kategori 2 om det inte förekommit fler än ett dödsfall). Dessutom innehåller bilaga 4 vägledning om klassificeringen enligt EU-systemet, som gäller tills det att det nya GHS-systemet har genomförts.
1.5.3 Huvudtest
1.5.3.1 Antalet djur och dosnivåer
I flödesschemat i bilaga 2 anges de steg som bör följas efter testning vid startdosnivå. Ett av tre steg måste tas – avsluta testet och riskklassificera ämnet på grundval av utfallet, testa vid en högre dosnivå eller testa vid en lägre dosnivå. Med hänsyn till djurens välbefinnande bör dosnivåer som har orsakat dödsfall i det inledande testet inte användas i huvudtestet (se bilaga 2). Erfarenheterna tyder på att det mest sannolika utfallet vid startdosnivån är att ämnet kan klassificeras och att ingen ytterligare testning behövs.
I regel används totalt fem djur av samma kön vid varje dosnivå. Dessa fem djur inbegriper det djur som i det inledande testet doserades vid den valda nivån och därutöver fyra djur (utom, i undantagsfall, om en dosnivå som används i huvudtestet inte har ingått i det inledande testet).
Tidsintervallet mellan doseringen vid varje nivå bestäms av start, varaktighet och allvarlighetsgrad för de toxiska tecknen. Doseringen av djuren på följande dosnivå bör inte inledas förrän det råder säkerhet om de tidigare doserade djurens överlevnad. I förekommande fall rekommenderas ett intervall på 3–4 dagar mellan doseringen vid de olika nivåerna, så att eventuella tecken på fördröjd toxicitet kan observeras. Tidsintervallet kan anpassas på tillbörligt sätt, exempelvis i fall där responsen är tvetydig.
I fall där man överväger att använda en övre dos på 5 000 mg/kg bör förfarandet enligt bilaga 3 följas (se även avsnitt 1.6.2).
1.5.3.2 Gränstest (limit test)
Gränstest används primärt i situationer där laboratoriet har information som tyder på att testämnet sannolikt inte är toxiskt (toxiskt endast ovanför de gränsdoser som anges i bestämmelser). Information om testämnets toxicitet kan härledas från kunskaperna om liknande testade ämnen eller liknande testade blandningar eller produkter, med hänsyn tagen till identiteten och halten av de komponenter som har känd toxikologisk signifikans. Om informationen om ämnets toxicitet är bristfällig eller saknas, eller om ämnet troligen är toxiskt, bör huvudtestet genomföras.
Som gränstest för den metod som beskrivs här kan man använda ett inledande test med en startdos på 2 000 mg/kg (eller undantagsvis 5 000 mg/kg) som åtföljs av dosering av ytterligare fyra djur vid denna nivå.
1.6 OBSERVATIONER
Efter doseringen observeras djuren individuellt minst en gång under de första 30 minuterna, periodiskt under de första 24 timmarna (med särskild uppmärksamhet under de första 4 timmarna) och därefter dagligen under totalt 14 dagar. Detta gäller inte för djur som av hänsyn till djurens välbefinnande måste avlivas skonsamt eller för djur som hittas döda. Observationsperiodens längd bör dock ha en anpassningsmån. Längden bör bestämmas enligt de toxiska reaktionerna, tidpunkten för när de sätter in och återhämtningsperiodens längd. Observationsperioden kan således förlängas om det anses vara nödvändigt. Tidpunkten då tecken på toxicitet uppträder och försvinner är viktig, särskilt om de toxiska tecknen tenderar att vara fördröjda (se hänvisning 11). Alla observationer registreras systematiskt, med individuella registreringar för varje djur.
Tilläggsobservationer behövs om djuren fortsätter att uppvisa tecken på toxicitet. Observationerna bör omfatta ändringar i hud och päls, ögon och slemhinnor samt även andningsorganen, cirkulationsorganen och det autonoma och centrala nervsystemet. Likaså bör den somatomotoriska aktiviteten och beteendemönstret observeras. Uppmärksamhet bör fästas vid observationer av tremor, konvulsioner, salivutsöndring, diarré, letargi, sömn och koma. Hänsyn bör tas till de principer och kriterier som anges i vägledningen om konsekvenserna för människor (se hänvisning 8). Djur som befinns vara döende och djur som visar tecken på svår smärta eller ihållande lidande bör avlivas skonsamt. I fall där djur avlivas av hänsyn till djurens välbefinnande eller hittas döda, bör tidpunkten för dödsfallet registreras så exakt som möjligt.
1.6.1 Kroppsvikt
Djurens individuella vikter bör registreras strax innan testämnet tillförs och därefter minst en gång per vecka. Alla viktändringar bör bestämmas och registreras. Vid testets slut vägs alla överlevande djur och därefter avlivas de skonsamt.
1.6.2 Patologi
Alla försöksdjur (inbegripet de som dött under testet eller som har avlivats av hänsyn till djurens välbefinnande) bör obduceras. Alla betydande patologiska förändringar bör registreras för varje djur. Användbar information kan även fås genom mikroskopundersökning av organ som uppvisar betydande skador och som härrör från djur som överlevt minst 24 timmar efter tillförsel av startdosen.
2. DATA
Data bör rapporteras individuellt per djur. Dessutom bör alla data sammanställas i tabellform. Tabellen ska för varje testgrupp visa antalet djur som använts, antalet djur som uppvisat tecken på toxicitet, antalet djur som under testets gång har dött eller avlivats av hänsyn till djurens välbefinnande, tidpunkterna för alla individuella dödsfall, en beskrivning av och förloppet för de toxiska verkningarna och deras reversibilitet samt obduktionsfynd.
3. RAPPORTERING
3.1 TESTRAPPORT
Testrapporten ska innehålla nedan angivna uppgifter, enligt det som är relevant:
|
|
Testämne:
|
|
|
Vehikel (i förekommande fall).
|
|
|
Försöksdjur:
|
|
|
Testbetingelser:
|
|
|
Resultat:
|
|
|
Diskussion och tolkning av resultaten. |
|
|
Slutsatser. |
4. HÄNVISNINGAR
|
1. |
British Toxicology Society Working Party on Toxicity (1984), ”Special report: a new approach to the classification of substances and preparations on the basis of their acute toxicity”, Human Toxicol., 3, 85–92. |
|
2. |
M. J. Van den Heuvel, A.D. Dayan och R.O. Shillaker (1987), ”Evaluation of the BTS approach to the testing of substances and preparations for their acute toxicity”, Human Toxicol., 6, 279–291. |
|
3. |
M. J. Van den Heuvel, D.G. Clark, R.J. Fielder, P.P. Koundakjian, G.J.A. Oliver, D. Pelling, N.J. Tomlinson och A.P. Walker (1990), ”The international validation of a fixed-dose procedure as an alternative to the classical LD50 test”, Fd. Chem. Toxicol., 28, 469–482. |
|
4. |
A. Whitehead och R.N. Curnow (1992), ”Statistical evaluation of the fixed-dose procedure”, Fd. Chem. Toxicol., 30, 313–324. |
|
5. |
N. Stallard och A. Whitehead (1995), ”Reducing numbers in the fixed-dose procedure”, Human Exptl. Toxicol., 14, 315–323. |
|
6. |
Stallard, N., Whitehead, A. and Ridgeway, P. (2002), Statistical evaluation of the revised fixed dose pro cedure. Hum. Exp. Toxicol., 21, 183–196. |
|
7. |
OECD (2001), ”Guidance Document on Acute Oral Toxicity Testing”, Environmental Health and Safety Monograph Series on Testing and Assessment, 24, Paris. |
|
8. |
OECD (2000), ”Guidance Document on the Recognition”, Assessment and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluation, Environmental Health and Safety Monograph Series on Testing and Assesment, 19. |
|
9. |
OECD (1998), ”Harmonised Integrated Hazard Classification for Human Health and Environmental Effects of Chemical Substances as endorsed by the 28th Joint Meeting of the Chemicals Committee and the Working Party on Chemicals in November 1998, Part 2”, s. 11 (http://webnet1.oecd.org/oecd/ pages/home/displaygeneral/0,3380,EN-documents-521-14-no-24-no-0,FF.html). |
|
10. |
R.L. Lipnick, J.A. Cotruvo, R.N. Hill, R.D. Bruce, K.A. Stitzel, A.P. Walker, I. Chu, M. Goddard, L. Segal, J.A. Springer och R.C. Myers (1995), ”Comparison of the Up-and-Down, Conventional LD50, and Fixed-Dose Acute Toxicity Procedures”, Fd. Chem. Toxicol., 33, 223–231. |
|
11. |
P.K Chan och A.W. Hayes (1994), ”Chapter 16 Acute Toxicity and Eye Irritation, Principles and Methods of Toxicology”, 3rd Edition, red. A.W. Hayes, Raven Press, Ltd. New York, USA. |
BILAGA 1
FLÖDESSCHEMA FÖR INLEDANDE TEST


BILAGA 2
FLÖDESSCHEMA FÖR HUVUDTEST


Bilaga 3
KRITERIER FÖR HUR TESTÄMNEN MED FÖRVÄNTADE LD50-VÄRDEN ÖVER 2 000 MG/KG KAN KLASSIFICERAS UTAN TESTNING
Med hjälp av kriterierna för riskkategori 5 kan man identifiera testämnen som har en relativt låg risk för akut toxicitet men som under vissa omständigheter kan utgöra en risk för sårbara populationer. Dessa ämnen förväntas ha ett LD50-värde (oralt eller via huden) inom området 2 000–5 000 mg/kg eller motsvarande doser i fråga om andra tillförselvägar. Ett testämne kan klassificeras enligt riskkategorin 2 000 mg/kg < LD50 < 5 000 mg/kg (GHS-kategori 5) i följande fall:
|
a) |
Om ämnet hänförs till denna kategori enligt utfallet i något av de testningsscheman som finns i bilaga 2, på grundval av mortalitet. |
|
b) |
Om det redan finns tillförlitliga bevis för att LD50-värdet ligger inom det intervall som motsvarar kategori 5 eller om tidigare djurförsök eller toxiska verkningar hos människor tyder på akut fara för människors hälsa. |
|
c) |
På grundval av extrapolering, uppskattning eller mätning av data, om det inte är motiverat att placera ämnet i en högre riskklass.
|
TESTNING PÅ DOSNIVÅER ÖVER 2 000 MG/KG
Undantagsvis, och endast när det kan motiveras med specifika tillsynskrav, får en ytterligare övre dosnivå på 5 000 mg/kg användas. Med hänsyn till djurens välbefinnande avråds testning på djur vid dosnivån 5 000 mg/kg. Sådan testning bör endast övervägas när det finns en stark sannolikhet för att resultaten har en direkt relevans för skyddet av djurs eller människors hälsa (9).
Inledande test
De beslutsregler som styr det sekventiella förfarandet enligt bilaga 1 utökas till att innefatta en dosnivå på 5 000 mg/kg. När en startdos på 5 000 mg/kg används vid ett inledande test och utfallet blir A (död) måste således ytterligare ett djur testas vid nivån 2 000 mg/kg, medan utfallen B och C (uppenbar toxicitet eller ingen toxicitet) gör det möjligt att använda 5 000 mg/kg som startdos i huvudtestet. På samma sätt gäller att om en startdos annan än 5 000 mg/kg används i ett test där man går vidare till 5 000 mg/kg efter att ha fått utfall B eller C vid nivån 2 000 mg/kg, leder ett utfall A på nivån 5 000 mg/kg till att huvudtestets startnivå blir 2 000 mg/kg medan utfallen B och C leder till 5 000 mg/kg som startnivå.
Huvudtest
De beslutsregler som styr det sekventiella förfarandet enligt bilaga 2 utökas till att innefatta en dosnivå på 5 000 mg/kg. Detta innebär att utfallet A (≥ 2 dödsfall) i ett huvudtest med startdosen 5 000 mg/kg leder till att en ytterligare grupp måste testas vid nivån 2 000 mg/kg, medan utfallet B (uppenbar toxicitet och/eller ≤ 1 dödsfall) eller C (ingen toxicitet) leder till att ämnet inte får någon klassificering enligt GHS. På liknande sätt gäller att om en startdos annan än 5 000 mg/kg används i ett test och man har gått vidare till dosnivån 5 000 mg/kg efter att ha fått utfall C vid dosnivån 2 000 mg/kg, leder utfallet A vid dosnivån 5 000 mg/kg till att ämnet anses tillhöra GHS-kategorin 5 medan utfall B eller C leder till att ämnet inte får någon klassificering.
Bilaga 4
TESTMETOD B.l b
– Vägledning för klassificering enligt EU-Systemet, som gäller under övergängsperioden tills det globala harmoniserade systemet (GHS) för kassificering har genomförts helt (ur hänvisning 8)


B.1b AKUT TOXICITET ORALT – METOD FÖR BESTÄMNING AV AKUT TOXICITETSKLASS
1. METOD
Denna testmetod motsvarar OECD TG 423 (2001).
1.1 INLEDNING
Denna metod för bestämning av akut toxicitetsklass (se hänvisning 1) är ett stegvis förfarande där tre djur av samma kön används per steg. Beroende på mortaliteten och/eller förekomsten av döende djur behövs det i genomsnitt 2–4 steg för bedömningen av testämnets akuta toxicitet. Förfarandet är reproducerbart, kräver mycket få djur och ger en möjlighet att klassificera ämnen på ett liknande sätt som med de övriga metoderna för testning av akut toxicitet. Metoden för bestämning av akut toxicitetsklass baserar sig på biometriska bedömningar (se hänvisningarna 2, 3, 4 och 5) och fast definierade doser på tillräckligt separerade nivåer för att testämnet ska kunna kategoriseras för klassificering och riskbedömning. Metoden antogs 1996 och har validerats omfattande in vivo mot LD50-data som erhållits vid litteraturstudier, både nationellt (se hänvisning 6) och internationellt (se hänvisning 7).
Riktlinjer för valet av den lämpligaste testmetoden för ett bestämt ändamål finns i vägledningen för testning av akut oral toxicitet (se hänvisning 8). I vägledningsdokumentet finns även tilläggsinformation rörande utförandet av testmetod B.1b och tolkningen av resultaten.
Testämnesdoser som, på grund av frätande eller allvarligt irriterande verkningar, är kända att orsaka klar smärta och svårt lidande behöver inte ges. Döende djur eller djur som uppvisar klara tecken på smärta eller allvarligt och ihållande lidande ska avlivas skonsamt, och vid tolkningen av testresultaten ska dessa djur beaktas på samma sätt som djur som har dött under testets gång. Kriterierna för beslut rörande avlivning av döende eller svårt lidande djur, liksom även vägledning om igenkänning av förutsägbar eller annalkande död, behandlas i en separat vägledning (se hänvisning 9).
Metoden bygger på användning av fast definierade doser, och resultatet ger en möjlighet att kategorisera och klassificera ett ämne enligt det globala harmoniserade systemet (GHS) för klassificering och märkning av kemikalier som orsakar akut toxicitet (se hänvisning 10).
Även om metoden i princip inte är avsedd att användas för beräkning av exakta LD50-värden ger den ändå en möjlighet att bestämma exponeringsområden där ämnets verkan sannolikt är dödlig, eftersom den viktigaste ändpunkten för detta test fortfarande är andelen djur som dör. Bestämning av ett LD50-värde med denna metod kan endast göras när minst två doser leder till en mortalitet som är högre än 0 % och lägre än 100 %. Eftersom testningen alltid utförs med fast definierade doser, oavsett vilket ämne som testas, och klassificeringen uttryckligen är bunden till antalet djur som reagerar på ett visst sätt, ökas möjligheterna till enhetliga testrapporter och repeterbarhet mellan olika laboratorier.
Testningslaboratoriet bör analysera all tillgänglig information om testämnet innan själva testningen genomförs. Sådan information inbegriper ämnets identitet och kemiska struktur, fysikalkemiska egenskaper, resultaten från andra eventuella in vivo- eller in vitro-test avseende toxicitet, toxikologiska data om strukturellt närbesläktade ämnen och ämnets förutsedda användningsändamål. Denna information behövs för att avgöra huruvida testet är relevant för skyddet av människors hälsa, och utgör en hjälp vid valet av en optimal startdos.
1.2 DEFINITIONER
Akut toxicitet (oralt): De skadliga verkningar som uppträder efter oral tillförsel av en engångsdos av ett ämne eller upprepade doser som ges inom 24 timmar.
Fördröjd död: Det att ett djur inte dör eller är döende inom 48 timmar, men dör senare under den 14 dagar långa observationsperioden.
Dos: Den mängd testämne som tillförs. Dosen uttrycks som vikten testämne per viktenhet försöksdjur (exempelvis mg/kg).
GHS: Globala harmoniserade systemet för klassificering och märkning av kemikalier och blandningar. Systemet utarbetas gemensamt inom OECD (människors hälsa och miljö), FN:s expertkommitté om transport av farligt gods (fysikalkemiska egenskaper) och ILO (varningar). Samordningen sker genom det gemensamma programmet för sund kemikaliehantering (IOMC).
Annalkande död: När ett djur troligen är döende eller har dött före den nästa planenliga observationstidpunkten. Tecken som tyder på detta tillstånd kan hos gnagare inbegripa konvulsioner, kroppsställning på sidan, bakåtlutad kroppsställning och tremor (närmare uppgifter finns i vägledningen om konsekvenser för människor, se hänvisning 9).
LD50 (median letal dos, oralt): En statistiskt fastställd engångsdos som troligen leder till döden för 50 % av de djur som har tillförts dosen oralt. LD50-värdet uttrycks som vikten testämne per viktenhet försöksdjur (mg/kg).
Gränsdos: En testdos vid den övre gränsnivån (2 000 eller 5 000 mg/kg).
Döende djur: Ett djur som håller på att dö eller som har små chanser att överleva, även om det får behandling (närmare uppgifter finns i vägledningen om konsekvenser för människor, se hänvisning 9).
Förutsägbar död: Förekomsten av kliniska tecken som tyder på död vid en känd framtida tidpunkt som infaller före försökets planerade avslutningstidpunkt, exempelvis oförmåga att nå vatten eller föda (närmare uppgifter finns i vägledningen om konsekvenser för människor, se hänvisning 9).
1.3 PRINCIP FÖR TESTMETODEN
Testmetoden bygger på principen om att man genom ett stegvis förfarande med ett minimalt antal djur per steg kan få tillräcklig information om testämnets akuta toxicitet för att det ska vara möjligt att klassificera ämnet. Ämnet tillförs oralt till en grupp försöksdjur vid en av de definierade dosnivåerna. Ämnet testas genom ett stegvis förfarande med tre djur av samma kön (i regel honor) per steg. Avsaknaden eller förekomsten av ämnesrelaterad mortalitet hos de djur som doserats på ett visst steg avgör vilket av följande som blir nästa steg:
|
— |
Ingen ytterligare testning behövs. |
|
— |
Ytterligare tre djur tillförs en lika stor dos. |
|
— |
Ytterligare tre djur tillförs en dos på nästa högre eller lägre dosnivå. |
Testningsförfarandet beskrivs närmare i bilaga 1. När denna metod används kan testämnet genom de olika utfallen placeras i en av de toxicitetsklasser som definieras genom fasta LD50-gränsvärden.
1.4 BESKRIVNING AV TESTMETODEN
1.4.1 Val av djurart
Rekommenderad gnagare är råtta, även om testet kan utföras med andra gnagare. I regel används hondjur (se hänvisning 9). Skälet till detta är att litteraturundersökningar av konventionella LD50-test tyder på att känslighetsskillnaderna mellan könen är små, men att honorna i förekommande fall ofta är en aning mer känsliga (se hänvisning 11). Om däremot kunskaperna om strukturellt närbesläktade ämnens toxikologiska eller toxikokinetiska egenskaper tyder på att hanarna sannolikt är känsligare än honorna bör hanar användas. Om testet görs på hanar bör tillbörlig motivering ges.
Djuren bör vara unga och friska vuxna exemplar som härrör från normala laboratoriestammar. Hondjuren får inte ha fött ungar och får inte vara dräktiga. När dostillförseln inleds ska djuret vara mellan 8 och 12 veckor gammalt, och variationerna i vikt får vara högst ± 20 % av medelvikten för alla tidigare djur som har doserats.
1.4.2 Förvarings- och utfodringsbetingelser
Temperaturen i testdjurens utrymmen bör vara 22 oC (± 3 oC). Den relativa fuktigheten bör vara minst 30 % och ska helst inte överstiga 70 %, utom när rummet rengörs. Målvärdet bör dock vara 50–60 %. Belysningen bör vara artificiell med omväxlande 12 timmar ljus och 12 timmar mörker. För utfodringen kan konventionellt laboratoriefoder användas, och djuren bör ha obegränsad tillgång till dricksvatten. Djur som tillförs doser på samma nivå kan inhysas i samma bur, men antalet djur per bur får inte vara högre än att klara observationer kan göras av varje djur.
1.4.3 Förberedelse av djuren
Djuren väljs ut slumpmässigt och förses med märkning så att de kan identifieras individuellt Därefter får djuren acklimatisera sig till laboratoriets betingelser i sina burar i minst 5 dagar före doseringsstart.
1.4.4 Förberedelse av doserna
I regel ska de olika doserna av testämnet tillföras i en konstant volym, vilket innebär att doserna har varierande koncentration. När slutprodukter i vätskeform eller blandningar testas, kan det med tanke på riskbedömningen vara mer relevant att använda testämnet i outspädd form (konstant koncentration), vilket även förutsätts av vissa tillsynsmyndigheter. Den maximala dosvolymen för tillförsel får aldrig överskridas. Den största mängd vätska som kan tillföras vid ett och samma tillfälle beror på testdjurets storlek. För gnagare bör denna volym i regel inte överskrida 1 ml per 100 g kroppsvikt. I fråga om vattenlösningar får dock 2 ml per 100 g kroppsvikt användas. För tillförsel av dosen rekommenderas alltid i första hand lösning, suspension eller emulsion i vatten, sedan lösning, suspension eller emulsion i olja (exempelvis majsolja) och därefter eventuella andra vehiklar. I fråga om andra vehiklar än vatten måste de toxikologiska egenskaperna vara kända. Doserna måste beredas strax före tillförseln, om inte preparatets stabilitet är känd och har påvisats vara godtagbar över hela användningsperioden.
1.5 FÖRFARANDE
1.5.1 Tillförsel av doser
Testämnet tillförs som en engångsdos genom sondmatning med hjälp av magsond eller lämplig intubationskanyl. I det sällsynta fall det inte är möjligt med en engångsdos, kan dosen tillföras i mindre delar över en period som inte överskrider 24 timmar.
Före dostillförseln hålls djuren fastande (utan föda och med tillgång till vatten, för råttor över natten och för möss i 3–4 timmar). Efter fasteperioden vägs djuren och testämnet tillförs. Efter tillförsel av ämnet kan djuren hållas utan föda i ytterligare 3–4 timmar (råttor) respektive 1–2 timmar (möss). Om dosen tillförs i delar över en tidsperiod kan det vara nödvändigt att ge djuren föda och vatten, beroende på tidsperiodens längd.
1.5.2 Antalet djur och dosnivåer
Tre djur används per steg. För startdosen väljs en av fyra fast definierade nivåer (5, 50, 300 eller 2 000 mg per kg kroppsvikt). För startdosen väljs den nivå som högst sannolikt leder till att ett antal av de doserade djuren dör. I flödesschemat i bilaga 1 beskrivs de förfaranden som gäller för de olika startdoserna. Dessutom innehåller bilaga 4 vägledning om klassificeringen enligt EU-systemet, som gäller tills det att det nya GHS-systemet har genomförts.
Om den tillgängliga informationen tyder på att inga djur dör vid den högsta startdosnivån (2 000 mg per kg kroppsvikt) bör ett gränstest utföras. Om det inte finns någon information tillgänglig om testämnet, rekommenderas med hänsyn till djurens välbefinnande att startdosen 300 mg per kg kroppsvikt används.
Tidsintervallet mellan dostillförseln till de olika grupperna bestäms av start, varaktighet och allvarlighets-grad för de toxiska tecknen. Doseringen av djuren på nästa dosnivå bör inte inledas förrän det råder säkerhet om de tidigare doserade djurens överlevnad.
Undantagsvis, och endast när det kan motiveras med specifika tillsynskrav, får en ytterligare övre dosnivå på 5 000 mg per kg kroppsvikt användas (se bilaga 2). Med hänsyn till djurens välbefinnande avråds testning på djur när det gäller GHS-kategori 5 (2 000–5 000 mg per kg kroppsvikt). Sådan testning bör endast övervägas när det finns en stark sannolikhet för att resultaten har direkt relevans för skyddet av människors eller djurs hälsa eller av miljön.
1.5.3 Gränstest
Gränstest används primärt i situationer där laboratoriet har information som tyder på att testämnet sannolikt inte är toxiskt (toxiskt endast ovanför de gränsdoser som anges i gällande föreskrifter). Information om testämnets toxicitet kan härledas från kunskaperna om liknande testade ämnen eller liknande testade blandningar eller produkter, med hänsyn tagen till identiteten och halten av de komponenter som har känd toxikologisk signifikans. Om informationen om ämnets toxicitet är bristfällig eller saknas, eller om ämnet troligen är toxiskt, bör huvudtestet genomföras.
Ett gränstest vid en dosnivå 2 000 mg per kg kroppsvikt kan utföras med sex djur (tre djur per steg). Undantagsvis kan ett gränstest på dosnivån 5 000 mg per kg kroppsvikt utföras med tre djur (se bilaga 2). Om testämnet orsakar dödsfall måste eventuellt ytterligare testning utföras på närmaste lägre nivå.
1.6 OBSERVATIONER
Efter doseringen observeras djuren individuellt minst en gång under de första 30 minuterna, periodiskt under de första 24 timmarna (med särskild uppmärksamhet under de första 4 timmarna) och därefter dagligen under totalt 14 dagar. Detta gäller inte för djur som av hänsyn till djurens välbefinnande måste avlivas skonsamt eller för djur som hittas döda. Observationsperiodens längd bör dock ha en anpassningsmån. Längden bör bestämmas enligt de toxiska reaktionerna, tidpunkten för när de sätter in och återhämtningsperiodens längd. Observationsperioden kan således förlängas om det anses vara nödvändigt. Tidpunkten då tecken på toxicitet uppträder och försvinner är viktig, särskilt om de toxiska tecknen tenderar att vara fördröjda (se hänvisning 12). Alla observationer registreras systematiskt, med individuella registreringar för varje djur.
Tilläggsobservationer behövs om djuren fortsätter att uppvisa tecken på toxicitet. Observationerna bör omfatta förändringar i hud och päls, ögon och slemhinnor samt även andningsorganen, cirkulationsorganen och det autonoma och centrala nervsystemet. Likaså bör den somatomotoriska aktiviteten och beteendemönstret observeras. Uppmärksamhet bör fästas vid observationer av tremor, konvulsioner, salivutsöndring, diarré, letargi, sömn och koma. Hänsyn bör tas till de principer och kriterier som anges i vägledningen om konsekvenser för människor (se hänvisning 8). Djur som befinns vara döende och djur som visar tecken på svår smärta eller ihållande lidande bör avlivas skonsamt. I fall där djur avlivas av hänsyn till djurens välbefinnande eller hittas döda, bör tidpunkten för dödsfallet registreras så exakt som möjligt.
1.6.1 Kroppsvikt
Djurens vikt bör registreras individuellt strax före testämnet tillförs och därefter minst en gång per vecka. Alla viktändringar bör bestämmas och registreras. Vid testets slut vägs alla överlevande djur och avlivas därefter skonsamt.
1.6.2 Patologi
Alla försöksdjur (inbegripet de som dött under testet eller som har avlivats av hänsyn till djurens välbefinnande) bör obduceras. Alla betydande patologiska förändringar bör registreras för varje djur. Användbar information kan även fås genom mikroskopundersökning av organ som uppvisar betydande skador och som härrör från djur som överlevt minst 24 timmar efter tillförsel av startdosen.
2. DATA
Data bör rapporteras individuellt per djur. Dessutom bör alla data sammanställas i tabellform. Tabellen ska för varje testgrupp visa antalet djur som använts, antalet djur som uppvisat tecken på toxicitet, antalet djur som under testets gång har dött eller avlivats av hänsyn till djurens välbefinnande, tidpunkterna för alla individuella dödsfall, en beskrivning av och förloppet för de toxiska verkningarna och deras reversibili-tet samt obduktionsfynd.
3. RAPPORTERING
3.1 Testrapport
Testrapporten ska innehålla nedan angivna uppgifter, enligt det som är relevant:
|
|
Testämne:
|
|
|
Vehikel (i förekommande fall).
|
|
|
Försöksdjur:
|
|
|
Testbetingelser:
|
|
|
Resultat:
|
|
|
Diskussion och tolkning av resultaten. |
|
|
Slutsatser. |
4. HÄNVISNINGAR
|
1. |
Roll R., Höfer-Bosse T. and Kayser D. (1986), New Perspectives in Acute Toxicity Testing of Chemicals. Toxicol. Lett., Suppl. 31, 86. |
|
2. |
Roll R., Riebschläger M., Mischke U. and Kayser D. (1989), Neue Wege zur Bestimmung der akuten Toxizität von Chemikalien. Bundesgesundheitsblatt 32, 336–341. |
|
3. |
Diener W., Sichha L., Mischke U., Kayser D. and Schlede E. (1994), The Biometric Evaluation of the Acute-Toxic-Class Method (Oral), Arch. Toxicol. 68, 559–610. |
|
4. |
Diener W., Mischke U., Kayser D. and Schlede E. (1995), The Biometric Evaluation of the OECD Modified Version of the Acute-Toxic-Class Method (Oral). Arch. Toxicol. 69, 729–734. |
|
5. |
Diener W. and Schlede E. (1999), Acute Toxicity Class Methods: Alterations to LD/LC50 Tests. ALTEX 16, 129–134. |
|
6. |
Schlede E., Mischke U., Roll R. and Kayser D. (1992), A National Validation Study of the Acute-Toxic- Class Method – An Alternative to the LD50 Test. Arch. Toxicol. 66, 455–470. |
|
7. |
Schlede E., Mischke U., Diener W. and Kayser D. (1994), The International Validation Study of the Acute-Toxic-Class Method (Oral). Arch. Toxicol. 69, 659–670. |
|
8. |
OECD (2001), Guidance Document on Acute Oral Toxicity Testing. Environmental Health and Safety Monograph Series on Testing and Assessment N. 24. Paris. |
|
9. |
OECD (2000), Guidance Document on the Recognition, Assessment and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluation. Environmental Health and Safety Monograph Series on Testing and Assessment N. 19. |
|
10. |
OECD (1998), Harmonized Integrated Hazard Classification System For Human Health And Environ mental Effects Of Chemical Substances as endorsed by the 28th Joint Meeting of the Chemicals Committee and the Working Party on Chemicals in November 1998, Part 2, p. 11 [http://webnet1. oecd.org/oecd/pages/home/displaygeneral/0,3380, EN-documents-521-14-no-24-no-0,FF.html]. |
|
11. |
Lipnick R.L., Cotruvo J.A., Hill R.N., Bruce R.D., Stitzel K.A., Walker A.P., Chu I., Goddard M., Segal L., Springer J.A. and Myers R.C. (1995), Comparison of the Up-and Down, Conventional LD50, and Fixed Dose Acute Toxicity Procedures. Fd. Chem. Toxicol. 33, 223–231. |
|
12. |
Chan P.K. and Hayes A.W. (1994), Chap. 16. Acute Toxicity and Eye Irritancy. Principles and Methods of Toxicology. Third Edition. A.W. Hayes, Editor. Raven Press, Ltd., New York, USA. |
Bilaga 1
FÖRFARANDEN SOM BÖR FÖLJAS BEROENDE PÅ STARTDOS
ALLMÄNNA ANMÄRKNINGAR
För de olika startdoserna finns det olika testningsscheman som bör följas, enligt beskrivningen i denna bilaga.
|
— |
Bilaga 1 a: Startdosen är 5 mg per kg kroppsvikt. |
|
— |
Bilaga 1 b: Startdosen är 50 mg per kg kroppsvikt. |
|
— |
Bilaga 1 c: Startdosen är 300 mg per kg kroppsvikt. |
|
— |
Bilaga 1 d: Startdosen är 2 000 mg per kg kroppsvikt. |
Testningens förlopp anges av pilarna och beror på antalet avlivade djur eller djur som dött.
Bilaga 1 A
TESTFÖRFARANDE MED STARTDOSEN 5 MG PER KG KROPPSVIKT

Bilaga 1 B
TESTFÖRFARANDE MED STARTDOSEN 50 MG PER KG KROPPSVIKT

Bilaga 1 C
TESTFÖRFARANDE MED STARTDOSEN 300 MG PER KG KROPPSVIKT

Bilaga 1 D
TESTFÖRFARANDE MED STARTDOSEN 2 000 MG PER KG KROPPSVIKT

Bilaga 2
KRITERIER FÖR HUR TESTÄMNEN MED FÖRVÄNTADE LD50-VÄRDEN ÖVER 2 000 MG/KG KAN KLASSIFICERAS UTAN TESTNING
Med hjälp av kriterierna för riskkategori 5 kan man identifiera testämnen som har en relativt låg risk för akut toxicitet men som under vissa omständigheter kan utgöra en risk för sårbara populationer. Dessa ämnen förväntas ha ett LD50-värde (oralt eller via huden) inom området 2 000–5 000 mg/kg eller motsvarande doser i fråga om andra tillförselvägar. Ett testämne kan klassificeras enligt riskkategorin 2 000 mg/kg < LD50 < 5 000 mg/kg (GHS-kategori 5) i följande fall:
|
a) |
Om ämnet hänförs till denna kategori enligt utfallet i något av de testningsscheman som finns i bilaga 1A–1 D, på grundval av mortalitet. |
|
b) |
Om det redan finns tillförlitliga bevis för att LD50-värdet ligger inom det intervall som motsvarar kategori 5 eller om tidigare djurförsök eller toxiska verkningar hos människor tyder på akut fara för människors hälsa. |
|
c) |
På grundval av extrapolering, uppskattning eller mätning av data, om det inte är motiverat att placera ämnet i en högre riskklass.
|
TESTNING PÅ DOSNIVÅER ÖVER 2 000 MG/KG
Med hänsyn till djurens välbefinnande avråds testning på djur när det gäller kategori 5 (5 000 mg/kg). Sådan testning bör endast övervägas när det finns en hög sannolikhet för att resultaten har direkt relevans för skyddet av djurens eller människors hälsa (se hänvisning 10). Ingen ytterligare testning bör utföras vid högre dosnivåer.
I nödvändiga fall används en dos på 5 000 mg/kg i ett enda steg (tre djur). Om det först doserade djuret dör, fortsätts doseringen till 2 000 mg/kg i enlighet med flödesschemat i bilaga 1. Om det första djuret överlever, doseras ytterligare två djur. Om endast ett av de tre djuren dör antas LD50-värdet överskrida 5 000 mg/kg. Om de två djuren dör fortsätts doseringen med 2 000 mg/kg.
Bilaga 3
TESTMETOD B.1b: Vägledning för klassificering enligt EU-systemet, som gäller tills det globala harmoniserade systemet för klassificering och märkning av kemikalier (GHS) har genomförts helt (ur hänvisning 8)




B.2 AKUT TOXICITET (INANDNING)
1. METOD
1.1 INLEDNING
Det är användbart att ha förhandsupplysningar om ämnets partikelstorleksfördelning, ångtryck, smältpunkt, kokpunkt, flampunkt och eventuellt explosiva egenskaper.
Se allmän inledning till del B (A).
1.2 DEFINITIONER
Se allmän inledning till del B (B).
1.3 REFERENSÄMNEN
Inga.
1.4 TESTMETODSPRINCIP
Flera grupper av försöksdjur exponeras under en angiven period för testämnet i graderade koncentrationer, en bestämd koncentration per grupp. Därefter observeras effekter och dödsfall. Djur som dör under försöket ska obduceras och i slutet av försöket obduceras överlevande djur.
Det kan vara nödvändigt att på ett humant sätt avliva djur som uppvisar ihållande tecken på stark smärta och lidande. Undersökningar av ämnen på ett sätt som på grund av deras frätande eller irriterade egenskaper vållar avsevärd smärta och lidande behöver inte utföras.
1.5 KVALITETSKRITERIER
Inga.
1.6 METODBESKRIVNING
1.6.1 Förberedelser
I minst fem dagar före försöket ska djuren hållas under testbetingelser vad beträffar miljö och utfodring. Före försöket fördelas friska könsmogna djur slumpmässigt i erforderligt antal behandlingsgrupper. De behöver inte utsättas för simulerad exponering om detta inte indiceras av den typ av exponeringsapparatur som används.
Det kan vara nödvändigt att mikronisera fasta testämnen för att erhålla lämplig partikelstorlek.
Vid behov kan lämplig vehikel tillsättas till testämnet för att göra det lättare att uppnå en lämplig koncentration av testämnet i atmosfären. I sådana fall ska försöket omfatta en kontrollgrupp som exponeras för vehikeln. Om den vehikel eller annan additiv substans används för att underlätta dosering ska det vara påvisat att dessa inte framkallar toxiska effekter. Här kan tidigare insamlade data användas, om så bedöms vara lämpligt.
1.6.2 Testbetingelser
1.6.2.1 Försöksdjur
Om det inte finns kontraindikationer är råttan förstahandsvalet. De vanligen nyttjade laboratoriestammarna ska användas. För både han- och honkön gäller att vikten hos djur som används i ett försök inte får avvika mer än ± 20 % från en rimlig genomsnittsvikt.
1.6.2.2 Antal och kön
Minst 10 gnagare (fem honor och fem hanar) används på varje koncentrationsnivå. Honorna får inte ha fött ungar och får inte vara dräktiga.
Anmärkning: Vid användning av djur som är högre upp i näringskedjan än gnagare vid tester av akut toxicitet ska man överväga användning av ett mindre antal djur. Doserna ska fastställas med omsorg och man ska anstränga sig för att inte utnyttja högre doser än moderat toxiska doser. Vid sådana undersökningar bör tillförsel av dödliga doser av testämnet undvikas.
1.6.2.3 Exponeringskoncentrationer
Till försöket ska användas ett tillräckligt antal exponeringskoncentrationer, minst tre, som ska vara så valda att man får en gradering av de toxiska effekterna och av dödligheten i behandlingsgrupperna. Datauppgifterna ska vara tillräckliga för framställning av en koncentrations-/dödlighetskurva och, om möjligt, för att medge en rimlig bestämning av LC50.
1.6.2.4 Gränstest
Om en exponering av fem han- och fem hondjur med 20 mg/l av en gas eller 5 mg/l av en aerosol eller ett stoft under fyra timmar (eller om detta inte är möjligt på grund av det testade ämnets fysikaliska eller kemiska, inbegripet explosiva, egenskaper, den högsta koncentration som kan uppnås) inte har produktrelaterad dödlig verkan inom 14 dagar, kan ytterligare försök anses som överflödiga.
1.6.2.5 Exponeringstid
Exponeringstiden är fyra timmar.
1.6.2.6 Utrustning
Djuren ska testas med inhalationsutrustning som utformats så att den upprätthåller en dynamisk luftcirkulation med minst 12 luftväxlingar per timme, för att säkerställa en tillräcklig syrehalt och en jämnt fördelad exponeringsatmosfär. Om exponeringskammare används ska den vara så utformad att trängseln bland försöksdjuren minimeras och exponeringen för testämnet genom inandning maximeras. Som en allmän regel för att säkerställa en stabi] atmosfär i en kammare, ska försöksdjurens totala ’volym’ inte överstiga 5 % av exponeringskammarens volym. Exponeringen kan göras genom kammare för mun-näsa, enbart huvudet eller hela försöksdjurets kropp; vid användning av de två första metoderna kan man minska möjligheten att testämnet upptas via andra vägar.
1.6.2.7 Observationstid
Observationstiden ska vara minst 14 dagar. Observationstiden ska emellertid inte vara strikt fastställd utan ska bestämmas utifrån de toxiska reaktionerna, tidpunkten för deras uppträdande och återhämtningsperiodens längd. Den kan följaktligen förlängas när detta anses nödvändigt. Tidpunkten vid vilken tecken på toxicitet uppträder respektive försvinner och tidpunkten för dödsfall är viktiga, särskilt om det finns en tendens till att sena dödsfall inträffar.
1.6.3 Förfarande
Omedelbart före exponering ska djuren vägas varefter de exponeras för testkoncentrationen i den aktuella apparaturen i minst fyra timmar, efter det att jämvikt har uppnåtts för koncentrationen i kammaren. Jämvikt ska uppnås på kort tid. Temperaturen under försöket ska hållas vid 22 ± 3 oC. Under ideala betingelser ska den relativa luftfuktigheten hållas mellan 30 % och 70 %, men i vissa fall (t.ex. test med aerosoler) kan detta vara praktiskt ogenomförbart. Ett svagt undertryck i kammaren (5 mm vatten) förhindrar att testämnet läcker ut i det omgivande rummet. Under exponeringen ska djuren undanhållas foder och vatten. Ett dynamiskt respirationssystem med ett lämpligt analytiskt kontrollsystem ska användas. Ett försökstest rekommenderas för att fastställa lämpliga exponeringskoncentrationer. Systemet ska vara sådant att det så snabbt som möjligt går att säkerställa stabila exponeringsbetingelser. Lufthastigheten ska justeras för att säkerställa att betingelserna är homogena i hela exponeringskammaren.
Mätning eller kontroll ska göras av:
|
a) |
Lufthastighet (fortlöpande). |
|
b) |
Den faktiska koncentrationen av testämnet mätt i respirationszonen minst tre gånger under exponeringen (vissa atmosfärer, t.ex. aerosoler med höga koncentrationer, kan behöva kontrolleras oftare). Under exponeringsperioden ska koncentrationen inte avvika mer än ± 15 % från den genomsnittliga koncentrationen. En sådan kontrollnivå kan emellertid var omöjlig att uppnå vid test med stoft och vissa aerosoler, och i sådana fall kan större avvikelser accepteras. För aerosoler ska analys av partikelstorleken göras så ofta som möjligt (minst en gång per försöksgrupp). |
|
c) |
Temperatur och luftfuktighet, fortlöpande om möjligt. |
Under och efter exponering ska observationer göras och registreras systematiskt för varje enskilt djur. Den första dagen ska täta observationer göras. En grundlig klinisk undersökning ska göras minst en gång varje arbetsdag, andra observationer ska göras dagligen och lämpliga åtgärder ska vidtas för att minimera förlust av djur som ingår i studien, t.ex. ska djur som hittas döda obduceras eller nedfrysas och svaga eller döende djur ska isoleras eller avlivas.
Observation ska omfatta hud- och pälsförändringar, ögon, slemhinnor, respirations- och cirkulationssystem, autonoma och centrala nervsystemet samt kroppsmotorik och beteendemönster. Särskilt uppmärksamhet ska ägnas åt observation av andningsbeteende, skälvningar, kramper, salivavsöndring, diarré, letargi, sömn och koma. Tidpunkten för dödsfall ska registreras så noggrant som möjligt. De enskilda djurens vikt ska bestämmas varje vecka efter exponeringen och vid dödsfall.
Djur som dör under försöket ska obduceras och i slutet av försöket ska överlevande djur obduceras, särskilt med avseende på eventuella förändringar av övre och nedre andningsvägarna. Alla makroskopiska patologiska förändringar ska registreras. Vid sådana indikationer ska vävnadsprover tas för histopatologisk undersökning.
2. DATA
Data ska ställas upp i tabellform och för varje behandlingsgrupp ska anges antal djur i början av försöket, tidpunkten för enskilda djurs dödsfall, antal djur som visar andra tecken på toxicitet, beskrivning av toxiska effekter och obduktionsfynd. De enskilda försöksdjurens vikt ska bestämmas och registreras om överlevnaden överstiger en dag efter exponeringen. Avlivning av djur på grund av ämnesrelaterat lidande och smärta ska registreras som ämnesrelaterade dödsfall. LC50 ska bestämmas enligt en vedertagen metod. Utvärderingen av data ska omfatta eventuellt samband mellan djurens exponering för testämnet och omfattningen och svårighetsgrad av alla abnormiteter, inklusive beteendemässiga och kliniska abnormiteter, makroskopiska lesioner, ändringar av kroppsvikt, dödlighet och eventuella andra toxikologiska effekter.
3. RAPPORTERING
3.1 TESTRAPPORT
Testrapporten ska om möjligt innehålla följande uppgifter:
|
— |
Djurart, stam, härkomst, miljöbetingelser, foder osv. |
|
— |
Testbetingelser; beskrivning av exponeringsapparaturen inklusive utformning, typ, dimensioner, luftkälla, system för framställning av finfördelade ämnen och aerosoler, metod att konditionera luften och sätt att hålla djuren i en försökskammare när sådan används. Utrustning för mätning av temperatur, luftfuktighet och finfördelade aerosolers koncentration och storlek ska beskrivas. |
Exponeringsdata
Dessa ska ställas upp i tabellform och presenteras med medelvärden och ett spridningsmått (t.ex. standardavvikelse) och ska, om möjligt, omfatta
|
a) |
lufthastighet genom inhalationsutrustningen, |
|
b) |
luftens temperatur och fuktighet, |
|
c) |
nominella koncentrationer (total mängd testämne som tillförts inhalationsutrustningen, dividerat med luft volymen), |
|
d) |
typ av vehikel, om sådan används, |
|
e) |
faktiska koncentrationer i försökets respirationszon, |
|
f) |
massmedianen av den aerodynamiska diametern (MMAD) och den geometriska standardavvikelsen (GSD), |
|
g) |
jämviktsperiod, |
|
h) |
exponeringstid
|
3.2 UTVÄRDERING OCH TOLKNING
Se allmän inledning till del B (D).
4. HÄNVISNINGAR
Se allmän inledning till del B (E).
B.3 AKUT TOXICITET (DERMALT)
1. METOD
1.1 INLEDNING
Se allmän inledning till del B (A).
1.2 DEFINITIONER
Se allmän inledning till del B (B).
1.3 REFERENSÄMNEN
Inga.
1.4 TESTMETODSPRINCIP
Testämnet appliceras i graderade doser på huden till flera grupper av försöksdjur, en bestämd dos per grupp. Därefter observeras effekter och dödsfall. Djur som dör under försöket ska obduceras och i slutet av försöket obduceras överlevande djur.
Det kan vara nödvändigt att på ett humant sätt avliva djur som uppvisar ihållande tecken på stark smärta och lidande. Undersökningar av ämnen på ett sätt som på grund av deras frätande eller irriterade egenskaper vållar avsevärd smärta och lidande behöver inte utföras.
1.5 KVALITETSKRITERIER
Inga.
1.6 METODBESKRIVNING
1.6.1 Förberedelser
I minst fem dagar före försöket ska djuren hållas under testbetingelserna vad beträffar miljö och utfodring. Före försöket fördelas friska könsmogna djur slumpmässigt i behandlingsgrupper. Ungefär 24 timmar före testet ska päls avlägsnas från djurens rygg genom klippning eller rakning. Vid klippning eller rakning ska försiktighet iakttas så att huden inte skadas, eftersom det kan ändra dess genomtränglighet. Minst 10 % av kroppsytan ska göras klar för applicering av testämnet. Vid test av fasta ämnen, som får pulvriseras om det är lämpligt, ska testämnet fuktas tillräckligt med vatten eller, vid behov med lämplig vehikel, för att säkerställa god kontakt med huden. När vehikel används ska hänsyn tas till vehikelns inverkan på testämnets penetrering av huden. Flytande testämnen används vanligen outspädda.
1.6.2 Testbetingelser
1.6.2.1 Försöksdjur
Könsmogna råttor eller kaniner får användas. Andra arter kan användas men i så fall ska detta motiveras. De vanligen nyttjade laboratoriestammarna ska användas. För både han- och honkön gäller att vikten hos djur som används i ett försök inte får avvika mer än ± 20 % från en rimlig genomsnittsvikt.
1.6.2.2 Antal och kön
Minst 5 djur ska användas på varje dosnivå. De ska alla vara av samma kön. Om hondjur används får de inte ha fött ungar och får inte vara dräktiga. Om det finns information som visar att ett kön är väsentligt mer känsligt ska djur av detta kön användas.
Anmärkning: Vid användning av djur som är högre upp i näringskedjan än gnagare vid tester av akut toxicitet ska man överväga användning av ett mindre antal djur. Doserna ska fastställas med omsorg och man ska anstränga sig för att inte utnyttja högre doser än moderat toxiska doser. Vid sådana undersökningar bör tillförsel av dödliga doser av testämnet undvikas.
1.6.2.3 Dosnivåer
Till försöket ska användas ett tillräckligt antal dosnivåer, minst tre, som ska vara så valda att man får en gra-dering av de toxiska verkningarna och av dödligheten i behandlingsgrupperna. När dosnivåerna bestäms ska hänsyn tas till eventuellt irriterande eller trätande effekter. Datauppgifterna ska vara tillräckliga för framställning av en dos-/responskurva och, om möjligt, för att medge en rimlig bestämning av LD50.
1.6.2.4 Gränstest
Ett gränstest med en dosnivå av minst 2 000 mg/kg kroppsvikt kan utföras med en grupp av fem djur av varje kön enligt ovan beskrivna förfarande. Om ämnesrelaterade dödsfall inträffar bör en fullständig undersökning övervägas.
1.6.2.5 Observationsperiod
Observationstiden ska vara minst 14 dagar. Observationstiden ska emellertid inte vara strikt fastställd utan ska bestämmas utifrån de toxiska reaktionerna, tidpunkten för deras uppträdande och återhämtningsperiodens längd. Den kan följaktligen förlängas när detta anses nödvändigt. Tidpunkten vid vilken tecken på toxicitet uppträder respektive försvinner och tidpunkten för dödsfall är viktiga, särskilt om det finns en tendens till att sena dödsfall inträffar.
1.6.3 Förfarande
Djuren ska hållas i var sin bur. Testämnet ska appliceras jämnt över ett område som motsvarar ungefär 10 % av den totala kroppsytan. Vid användning av kraftigt toxiska ämnen kan en mindre yta täckas, men ämnet ska appliceras på ytan i ett så tunt och jämnt lager som möjligt.
Testämnen ska hållas i kontakt med huden med en porös gaskompress och icke hudirriterande tejp under en 24 timmar lång exponeringstid. Testytan ska vidare täckas på lämpligt sätt för att säkerställa att gaskompressen och testämnet hålls på plats så att djuren inte kan svälja testämnet. För att förhindra att djuren intar testämnet kan
rörelsehindrande anordningar användas, men total immobilisering rekommenderas inte.
I slutet av exponeringsperioden ska resterande testämne avlägsnas, i lämpliga fall med vatten eller med annan lämplig metod för rengöring av huden. Observationer ska registreras systematiskt när de görs. Varje enskilt djur ska registreras separat. Den första dagen ska täta observationer göras. En grundlig klinisk undersökning ska göras minst en gång varje arbetsdag, andra observationer ska göras dagligen och lämpliga åtgärder ska vidtas för att minimera förlust av djur som ingår i studien, t.ex. ska djur som hittas döda obduceras eller nedfrysas och svaga eller döende djur ska isoleras eller avlivas.
Observation av djuren i burarna ska omfatta pälsförändringar, behandlad hud, ögon och slemhinnor, respirations- och cirkulationssystem, autonoma och centrala nervsystemet samt kroppsmotorik och beteendemönster. Särskilt uppmärksamhet ska ägnas åt observation av skälvningar, kramper, salivavsöndring, diarré, letargi, sömn och koma. Tidpunkten för dödsfall ska registreras så noggrant som möjligt. Djur som dör under försöket ska obduceras och i slutet av försöket ska överlevande djur obduceras. Alla makroskopiska patologiska förändringar ska registreras. När det är indicerat ska vävnadsprov tas för histopatologisk undersökning.
Bedömning av toxicitet bos det motsatta könet
Sedan en undersökning av det ena könet avslutats ska minst en grupp med fem djur av det motsatta könet testas för att fastställa om djur av detta kön är avsevärt mer känsliga för testämnet. I vissa fall kan användning av färre djur vara motiverat. Om det finns tillräckligt med information om att djur av det kön som har testats är avsevärt mer känsliga, behöver djur av det motsatta könet inte testas.
2. DATA
Data ska ställas upp i tabellform och för varje behandlingsgrupp ska anges antal djur i början av försöket, tidpunkten för enskilda djurs dödsfall, antal djur som visar andra tecken på toxicitet, beskrivning av toxiska effekter och obduktionsfynd. De enskilda försöksdjurens vikt ska bestämmas och registreras omedelbart innan testämnet appliceras, därefter varje vecka och vid dödsfall. Viktförändringar ska beräknas och registreras om överlevnaden överstiger en dag efter applicering av testämnet. Avlivning av djur på ett humant sätt till följd av ämnesrelaterat lidande och smärta ska registreras som ämnesrelaterade dödsfall. LD50 ska bestämmas enligt en erkänd metod.
Utvärderingen av data ska omfatta eventuellt samband mellan djurens exponering för testämnet och omfattningen och svårighetsgraden av alla abnormiteter, inklusive beteendemässiga och kliniska abnormiteter, makro-skopiska lesioner, ändringar av kroppsvikt, dödlighet och eventuella andra toxikologiska effekter.
3. RAPPORTERING
3.1 TESTRAPPORT
Testrapporten ska om möjligt innehålla följande uppgifter:
|
— |
Djurart, stam, härkomst, miljöbetingelser, foder osv. |
|
— |
Testbetingelser (inklusive metod för hudrengöring och slag av bandage: med eller utan ocklusion). |
|
— |
Dosnivåer (med angivande av vehikel, om sådan används, och koncentrationer). |
|
— |
Försöksdjurens kön. |
|
— |
Tabelluppställning av responsdata efter kön och dosnivå (dvs. antal döda djur, antal djur som uppvisar tecken på toxicitet, antal exponerade djur). |
|
— |
Dödstidpunkt efter dosering, motivering och kriterier för human avlivning av djur. |
|
— |
Alla observationer. |
|
— |
LD50-värde för det kön som har varit föremål för en fullständig undersökning bestämt efter 14 dagar med angivande av bestämningsmetod. |
|
— |
95 % tillförlitlighetsintervall för LD50 (där detta är möjligt). |
|
— |
Dos-dödlighetskurva och lutning (om bestämningsmetoden medger detta). |
|
— |
Obduktionsfynd. |
|
— |
Eventuella histopatologiska fynd. |
|
— |
Resultat av försök med motsatt kön. |
|
— |
Diskussion av resultaten (med särskild uppmärksamhet på om human avlivning av djur under försöket kan ha inverkat på det beräknade LD50-värdet). |
|
— |
Tolkning av resultaten. |
3.2 UTVÄRDERING OCH TOLKNING
Se allmän inledning till del B (D).
4. HÄNVISNINGAR
Se allmän inledning till del B (E).
B.4 AKUT TOXICITET: HUDIRRITATION/HUDKORROSION
1. METOD
Denna testmetod motsvarar OECD TG 404 (2002).
1.1 INLEDNING
Vid utarbetandet av denna uppdaterade metod lades särskilt stor vikt vid möjligheterna till bättre testningsförfaranden, där djurskyddsfrågor beaktas så väl som möjligt. Sådana förfaranden innebär att all existerande information om testämnet utvärderas i syfte att undvika onödiga djurförsök. Metoden inbegriper en rekommendation om att laboratoriet, innan det beskrivna in vivo-testet för hudkorrosion/hudirritation utförs, gör en weight-of-the-evidence-analys av existerande relevanta data. Om de data som finns att tillgå inte är tillräckliga, kan behövliga data genereras med hjälp av stegvis testning (1). Den rekommenderade testningsstrategin inbegriper validerade och godkända in vitro-test (se bilagan till denna metod). Dessutom rekommenderas att de tre testkompresserna, där det är lämpligt, appliceras på djuret i följd, i stället för samtidigt, vid det inledande in vivo-testet.
Med hänsyn till sund vetenskaplig praxis och djurens välbefinnande bör in vivo-testning inte genomföras förrän alla tillgängliga data som är relevanta för ämnets förmåga att framkalla hudkorrosion/hudirritation har utvärderats genom en weight-of-the-evidence-analys. Sådana data är belägg från gjorda undersökningar på människor och/eller försöksdjur, belägg på korrosion/irritation i fråga om ett eller flera strukturellt besläktade ämnen eller blandningar av sådana ämnen, data som demonstrerar att ämnet är stark surt eller alkaliskt (2), (3) samt resultat från validerade och godkända in vitro- eller ex vivo-test (4), (5), (5a). Genom analysen bör man kunna minska behovet av in vivo-testning med avseende på hudkorrosion/hudirritation när det gäller ämnen för vilka det redan finns tillräckliga belägg från andra undersökningar av dessa två end points.
En rekommenderad strategi för stegvis testning, som inbegriper validerade och godkända in vitro- eller ex vivo-test med avseende på korrosion/irritation finns som bilaga till denna metod. Strategin utvecklades av och rekommenderades enhälligt av deltagarna i en OECD-arbetsgrupp (6). Strategin har antagits som den rekommenderade testningsstrategin i Globally Harmonised System for the Classification of Chemical Sub-stances (GHS) (7). Strategin för stegvis testning ingår inte i testmetod B.4, men användning av den rekommenderas ändå innan man går in för in vivo-testning. För nya ämnen rekommenderas den som ett stegvis testningsförfarande för generering av vetenskapligt sunda data om ett ämnes förmåga att framkalla korrosion/irritation. För befintliga ämnen med otillräckliga data om hudkorrosion/hudirritation bör strategin användas för att fylla upp luckor i tillgängliga data. Om en annan testningsstrategi eller en annan procedur används, eller om man beslutar att inte använda ett stegvis testningsförfarande, bör detta motiveras.
Om bestämningen av korrosivitet eller irritationsförmåga inte kan göras med hjälp av en weight-of-the-evidence-analys som står i överensstämmelse med strategin för stegvis testning, bör ett in vivo-test övervägas (se bilagan).
1.2 DEFINITIONER
Hudirritation: Uppkomsten av reversibel skada på huden efter att testämnet har applicerats i upp till 4 timmar.
Hudkorrosion (hudfrätning): Uppkomsten av irreversibel skada på huden i form av synlig nekros genom epidermis och in i dermis, efter att ett testämne har applicerats i upp till fyra timmar. Frätningsreaktioner karakteriseras av sår, blödning, blodiga sårskorpor och, vid slutet av observationsperioden på 14 dagar, avfärgning genom blekning av huden, hela områden med alopeci (håravfall) och ärr. Histopatologi bör övervägas för utvärdering av skador som inte är entydiga.
1.3 PRINCIP FÖR TESTMETODEN
Testämnet appliceras som en engångsdos på försöksdjurets hud. Djurets obehandlade hudområden används som referens. Graden av irritation eller korrosion granskas och poängsätts med specifierade intervall, och beskrivs närmare i syfte att få en fullständig utvärdering av verkningarna. Testet bör pågå tillräckligt länge för att ge möjlighet att utvärdera om de observerade verkningarna är reversibla eller irreversibla.
Djur som uppvisar ihållande tecken på allvarligt lidande eller smärta vid något skede av testningen bör avlivas på ett skonsamt sätt, och detta bör beaktas vid utvärderingen av ämnet. Kriterier för att fatta beslut om skonsam avlivning av döende eller svårt lidande djur finns i hänvisning (8).
1.4 BESKRIVNING AV TESTMETODEN
1.4.1 Förberedelser för in vivo-test
1.4.1.1 Val av djurart
Rekommenderat försöksdjur är albinokanin. Friska, unga vuxna exemplar bör användas. Om andra arter används bör detta motiveras.
1.4.1.2 Förberedelse av djuren
Cirka 24 timmar före testet bör djurens päls avlägsnas genom kortklippning av flankens ryggsida. Varsamhet bör iakttas för att inte repa huden, och endast djur med frisk och intakt hud bör användas.
Kaniner från vissa stammar har områden med speciellt tät päls, särskilt under vissa årstider. Sådana områden bör inte användas för testning.
1.4.1.3 Förvarings- och utfodringsbetingelser
Djuren ska inhysas individuellt. Temperaturen i försöksdjurens utrymmen bör vara 20 oC (± 3 oC) för kaniner. Den relativa fuktigheten bör vara minst 30 % och ska helst inte överstiga 70 %, utom när rummet rengörs. Målvärdet bör dock vara 50–60 %. Belysningen bör vara artificiell med omväxlande 12 timmar ljus och 12 timmar mörker. Konventionellt laboratoriefoder kan användas för utfodringen, och djuren bör ha obegränsad tillgång till dricksvatten.
1.4.2 Testförfarande
1.4.2.1 Applicering av testämnet
Testämnet appliceras på en liten yta (cirka 6 cm2) av huden och täcks med en kompress som hålls på plats med ett icke-irriterande häftförband. Om det inte är möjligt att applicera ämnet direkt (exempelvis vätskor eller vissa pastor) bör ämnet först appliceras på kompressen, som därefter placeras på huden. Kompressen ska under exponeringsperioden hållas i lös kontakt med huden med hjälp av ett lämpligt halvocklusivt förband. Om testämnet appliceras på kompressen bör denna fästas vid huden på sådant sätt att det finns god kontakt och en jämn fördelning av ämnet på huden. Man bör se till att djuret inte når kompressen, eller kan äta eller andas in ämnet.
Ämnen i vätskeform används i regel outspädda. Vid testning av fasta ämnen (som vid behov kan pulverise-ras) ska ämnet vätas med minsta möjliga mängd vatten (eller vid behov någon annan lämplig vehikel), som behövs för att säkerställa god hudkontakt. Om annan vehikel än vatten används bör vehikelns potentiella inverkan på den hudirritation som testämnet framkallar vara minimal eller noll.
I slutet av exponeringsperioden (som i regel varar fyra timmar) ska överskottsämne om möjligt avlägsnas med vatten eller ett lämpligt lösningsmedel, utan att störa existerande respons eller skada huden.
1.4.2.2 Dosnivå
En dos på 0,5 ml vätska eller 0,5 g fast ämne eller pasta appliceras på huden.
1.4.2.3 Inledande test (test avseende hudirritation/hudkorrosion in vivo på ett enskilt djur)
Det rekommenderas starkt att ett första in vivo-test görs på ett enskilt djur, särskilt om det föreligger misstanke om att ämnet kan vara korrosivt. Detta är i enlighet med strategin för stegvis testning (se bilaga 1).
Om ett ämne har bedömts vara korrosivt på grundval av en weight-of-the-evidence-analys, behövs ingen ytterligare testning på djur. För de flesta ämnen som misstänks vara korrosiva, behövs i regel ingen ytterligare in vivo-testning. I fall där det på grund av otillräckliga bevis är motiverat att generera ytterligare data, kan begränsade test på djur genomföras enligt följande princip: Upp till tre testkompresser appliceras sekventiellt på djuret. Den första kompressen tas bort efter tre minuter. Om ingen allvarlig hudreaktion kan observeras, appliceras en andra kompress, som i sin tur tas bort efter en timme. Om observationerna i detta skede tyder på att exponeringen kan förlängas till fyra timmar utan lidande för testdjuret, appliceras en tredje kompress, som tas bort efter fyra timmar. Därefter kan responsen graderas.
Om korrositivitet kan observeras efter någon av de tre sekventiella exponeringarna, avslutas testet omedelbart. Om ingen korrosion observeras efter att den sista kompressen har tagits bort, hålls djuret under observation i 14 dagar, utom om korrosion framkallas tidigare.
I fall där testämnet inte förväntas framkalla korrosion men där det kan vara irriterande, appliceras en enstaka kompress på ett djur i fyra timmar.
1.4.2.4 Bekräftande test (in vivo-test avseende hudirritation med ytterligare djur)
Om ingen korrosion observeras vid det inledande testet bör irritationsrespons eller negativ respons bekräftas med hjälp av ytterligare två djur. En kompress appliceras på vardera djuret för en exponeringsperiod på fyra timmar. Om irritation har observerats vid det inledande testet, kan bekräftelsetestet göras enligt det sekventiella förfarandet eller genom samtidig exponering på ytterligare två djur. Om inledande test i undantagsfall inte utförs, kan två eller tre djur behandlas med en enskild kompress som tas bort efter fyra timmar. Om två djur används, och samma respons observeras hos båda, behövs ingen ytterligare testning. I annat fall testas även det tredje djuret. Om den respons som observeras inte är entydig, kan det vara nödvändigt att klargöra responsen med hjälp av ytterligare djur.
1.4.2.5 Observationsperiod
Observationsperioden bör vara tillräckligt lång för att man till fullo ska kunna utvärdera om de observerade verkningarna är reversibla eller inte. Försöket bör dock avslutas omedelbart, oavsett hur länge det pågått, om ett djur uppvisar tecken på svåra smärtor eller lidande. För utvärderingen av om verkningarna är reversibla, bör djuren observeras i upp till 14 dagar efter att kompresserna har tagits bort. Om reversibilitet kan observeras innan 14 dagar har gått kan försöket omedelbart avslutas.
1.4.2.6 Kliniska observationer och gradering av hudreaktionerna
Alla djur bör undersökas för tecken på erytem och ödem, och den respons som erhållits vid 60 minuter och därefter vid 24, 48 och 72 timmar efter borttagning av kompresserna registreras. Vid inledande test på ett enskilt djur undersöks applikationsstället omedelbart efter att kompressen har tagits bort. Hudreaktionerna graderas och registreras enligt klassificeringsskalan i tabellen nedan. Om djuret har fått en hudskada som vid 72 timmar inte kan identifieras som irritation eller korrosion, kan observationer behövas fram till dag 14 för att avgöra om verkningarna är reversibla. Utöver observationerna av irritation, bör alla lokala toxiska effekter, såsom avfettning av huden och alla eventuella systemiska skadliga verkningar (exempelvis kliniska tecken på toxicitet eller verkningar på kroppsvikten) till fullo beskrivas och registreras. I fall av tvetydig respons bör en histopatologisk undersökning övervägas i klargörande syfte.
Graderingen av hudrespons är oundgängligen subjektiv. För att främja en harmonisering i fråga om graderingen av hudrespons och som stöd för testningslaboratorier och alla som är med om att utföra och tolka observationer, bör den personal som gör observationsarbetet få tillbörlig utbildning i det poängsättnings-system som används (se tabellen nedan). En illustrerad vägledning för gradering av hudirritation och andra hudskador kan vara till hjälp (9).
2. DATA
2.1 PRESENTATION AV RESULTATEN
Undersökningens resultat bör sammanställas i tabellform i den slutliga testrapporten. Resultaten bör omfatta alla aspekter som uppräknas i avsnitt 3.1.
2.2 UTVÄRDERING AV RESULTATEN
Graderna av hudirritation bör bedömas med hänsyn till skadornas natur och allvarlighetsgrad samt till om skadorna är reversibla eller irreversibla. Individuella resultat av graderingen representerar ingen absolut standard för ett ämnes förmåga att framkalla irritation, eftersom även andra verkningar av testämnet utvärderas. De enskilda bedömningarna bör däremot ses som referensvärden, som bör utvärderas i kombination med alla andra observationer från undersökningen.
Reversibiliteten hos hudskador bör beaktas när man utvärderar respons med avseende på irriterande egenskaper. Om verkningar av typen alopeci (begränsat område), hyperkeratos, hyperplasi och flagning håller i till slutet av den 14 dagar långa observationsperioden, bör testämnet betraktas som irriterande.
3. RAPPORTERING
3.1 TESTRAPPORT
Testrapporten ska innehålla nedan angivna uppgifter.
|
|
Motivering för in vivo-testning: weight-of-evidence-analys av tidigare existerande data, inbegripet resultat från stegvis testning enligt den rekommenderade strategin, enligt följande:
|
|
|
Testämne:
|
|
|
Vehikel:
|
|
|
Testdjur:
|
|
|
Testbetingelser:
|
|
|
Resultat:
|
|
|
Diskussion om resultaten. |
4. HÄNVISNINGAR
|
1. |
Barratt, M.D., Castell, J.V., Chamberlain, M., Combes, R.D., Dearden, J.C., Fentem, J.H., Gerner, I., Giuliani, A., Gray T.J.B., Livingston, D.J., Provan, W.M., Rutten, F.A.J.J.L., Verhaar, H.J.M., Zbinden, P. (1995), The Integrated Use of Alternative Approaches for Predicting Toxic Hazard. ECVAM Workshop Report 8. ATLA 23, 410–429. |
|
2. |
Young, J.R., How, M.J., Walker, A.P., Worth W.M.H. (1988), Classification as Corrosive or Irritant to Skin of Preparations Containing Acidic or Alkaline Substance Without Testing on Animals. Toxicol. In Vitro, 2, 19–26. |
|
3. |
Worth, A.P., Fentem, J.H., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Esdaile, D.J., Liebsch, M. (1998), Evaluation of the proposed OECD Testing Strategy for skin corrosion. ATLA 26, 709–720. |
|
4. |
ECETOC (1990), Monograph No. 15, Skin Irritation, European Chemical Industry, Ecology and Toxicology Centre, Brussels. |
|
5. |
Fentem, J.H., Archer, G.E.B., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Edsail, D.J., Holzhutter, H.G. and Liebsch, M. (1998), The ECVAM international validation study on in vitro tests for skin corrosivity. 2. Results and evaluation by the Management Team. Toxicology in Vitro 12, pp.483–524. |
|
5a. |
Testing Method B.40 Skin Corrosion. |
|
6. |
OECD (1996), OECD Test Guidelines Programme: Final Report of the OECD Workshop on Harmonization of Validation and Acceptance Criteria for Alternative Toxicological Test Methods. Held in Solna, Sweden, 22–24 January 1996 (http://www.oecd1.org/ehs/test/background.htm). |
|
7. |
OECD (1998), Harmonized Integrated Hazard Classification System for Human Health and Environmental Effects of Chemical Substances, as endorsed by the 28th Joint Meeting of the Chemicals Committee and the Working Party on Chemicals, November 1998 (http://www.oecd1.org/ehs/Class/HCL6.htm). |
|
8. |
OECD (2000), Guidance Document on the Recognition, Assessment and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluation. OECD Environmental Health and Safety Publications. Series on Testing and Assessment No. 19 (http://www.oecd1.org/ehs/test/ monos.htm). |
|
9. |
EPA (1990), Atlas of Dermal Lesions, (20T-2004). United States Environmental Protection Agency, Office of Pesticides and Toxic Substances, Washington, DC, August 1990. (Available from OECD Secretariat upon request.) |
Tabell 1
GRADERING AV HUDREAKTIONER
Rodnad och sårskorpebildning
|
Ingen rodnad … |
0 |
|
Mycket svag rodnad (knappt skönjbart) … |
1 |
|
Väldefinierad rodnad … |
2 |
|
Moderat till svår rodnad … |
3 |
|
Svår rodnad (köttrodnad) till sårskorpebildning som utgör hinder för gradering av rodnaden … |
4 |
Maximum: 4
Ödembildning
|
Inget ödem … |
0 |
|
Mycket svagt ödem (knappt skönjbart) … |
1 |
|
Milt ödem (området är väl avgränsat genom klar svullnad) … |
2 |
|
Moderat ödem (svullnadens höjd cirka 1 mm) … |
3 |
|
Svårt ödem (höjd över 1 mm och svullnaden sträcker sig utöver exponeringsområdet) … |
4 |
Maximum: 4
I fall av tvetydig respons kan en histopatologisk undersökning utföras i klargörande syfte.
Bilaga
Strategi för stegvis testning med avseende på hudirritation och hudkorrosion
ALLMÄNT
Även om denna strategi för stegvis testning inte ingår som en del av testmetod B.4, är den ett uttryck för det tillvägagångssätt som rekommenderas för bestämning av hudirriterande eller hudkorrosiva egenskaper. Detta tillvägagångssätt representerar både bästa praxis och etiska riktlinjer för in vivo-testning med avseende på hudirritation/hudkorrosion. Testmetoden utgör en vägledning för utförandet av in vivo-test och en sammanfattning av de faktorer som bör beaktas innan ett sådant test påbörjas. Strategin utgör ett tillvägagångssätt för utvärderingen av existerande data om olika testämnens egenskaper med avseende på hudirritation/hudkorrosion. Den utgör också ett stegvis förfarande för generering av relevanta data om ämnen för vilka det krävs ytterligare testning eller för ämnen som ännu inte har testats. Enligt strategin rekommenderas även validerade och godkända test in vitro eller ex vivo med avseende på hudkorrosion/hudirritation under specifika betingelser.
Med hänsyn till sund vetenskaplig praxis och djurens välbefinnande är det viktigt att undvika onödig användning av djur, och att minimera all testning som sannolikt framkallas allvarlig respons hos djuren. All information som är relevant för ämnets förmåga att framkalla hudkorrosion/hudirritation bör utvärderas innan man överväger in vivo-testning. Det kan eventuellt redan finnas tillräckliga bevis för att klassificera ett testämne med avseende på dess förmåga att framkalla hudkorrosion eller hudirritation, utan att för den skull behöva utföra testning på laboratoriedjur. När man använder en weight-of-the-evidence-analys och en strategi för stegvis testning minimeras behovet av in vivo-testning, särskilt för ämnen som sannolikt framkallar allvarliga reaktioner.
Det rekommenderas att en weight-of-the-evidence-analys används för att utvärdera existerande information om olika ämnens förmåga att framkalla hudirritation eller hudkorrosion för att avgöra om det behövs ytterligare testningar, andra än hudtestningar in vivo, som hjälp för att karakterisera en sådan egenskap. I fall där ytterligare undersökningar behövs, rekommenderas strategin för stegvis testning när det gäller att generera relevanta försöksdata. För ämnen som inte har testats tidigare bör strategin för stegvis testning användas för att generera de data som behövs för att utvärdera ämnets förmåga att framkalla hudkorrosion eller hudirritation. Den testningsstrategi som beskrivs i denna bilaga har utarbetats inom ramen för en OECD-arbetsgrupp (1). Strategin bekräftades och utvidgades senare i Harmonised Integrated Hazard Classification System for Human Health and Environmental Effects of Chemical Substances, i den form det godkändes vid 28th Joint Meeting ofthe Chemicals Committee and the Working Party on Chemicals, i november 1998 (2).
BESKRIVNING AV UTVÄRDERINGS- OCH TESTNINGSSTRATEGIN
Innan testning utförs som en del av strategin för stegvis testning (se schemat), bör all tillgänglig information utvärderas i syfte att fastställa behovet av in vivo-testning. Trots att signifikant information kan fås genom utvärdering av enskilda parametrar (till exempel extrema pH-värden), bör den tillgängliga informationen utvärderas som en helhet. Alla relevanta data om det berörda ämnets (eller analoga ämnens) verkningar bör utvärderas genom beslut som grundas på en weight-of-the-evidence-analys, och en motivering för beslutet bör anges. Huvudvikten bör läggas vid existerande data om ämnet (både i fråga om människor och djur). Därefter följer resultatet av testning in vitro eller ex vivo. In vivo-testning av korrosiva ämnen bör alltid undvikas när det är möjligt. Till de faktorer som bör beaktas inom ramen för testningsstrategin hör följande:
Utvärdering av existerande data rörande människor och djur (steg 1). Existerande data rörande människor, till exempel kliniska undersökningar eller undersökningar inom arbetslivet och fallstuder och/eller data om testning på djur, såsom data från toxicitetsundersökningar med enstaka eller upprepadhudexponering, bör beaktas först, eftersom de ger tillgång till information som direkt hör samman med verkningarna på huden. Ämnen med kända irriterande eller korrosiva egenskaper, samt ämnen med klara bevis om att de inte har sådana egenskaper, behöver inte testas in vivo.
Analys av strukturaktivitetssamband (SAR) (steg 2). I mån av tillgänglighet bör resultaten från testning av strukturellt närbesläktade ämnen beaktas. Om det finns tillgång till data rörande människa och/eller djur för strukturellt närbesläktade ämnen eller blandningar av sådana ämnen, och dessa data är tillräckliga för att indikera att dessa ämnen har förmåga att framkalla hudkorrosion/hudirritation, kan man anta att det ämne som utvärderas kommer att framkalla samma respons. I sådana fall behöver ämnet eventuellt inte testas. Negativa data från undersökningar med strukturellt närbesläktade ämnen eller blandningar av sådana ämnen utgör inte, inom ramen för strategin för stegvis testning, tillräckliga bevis på att ett ämne inte skulle ha korrosiva eller irriterande egenskaper. Validerade och godkända metoder för bestämning av strukturaktivitetssamband bör användas för att identifiera förmågan att framkalla hudkorrosion och hudirritation.
Fysikalkemiska egenskaper och kemisk reaktivitet (steg 3). Ämnen med extrema pH-värden, till exempel ≤ 2,0 eller ≥ 11,5 kan ha kraftiga lokala verkningar. Om ett extremt pH-värde används som grund för att konstatera att ett ämne är korrosivt eller irriterande för huden, behöver eventuellt också ämnets syra/bas-reserv (buffertkapacitet) beaktas (3), (4). Om buffertkapaciteten tyder på att ämnet eventuellt inte är korrosivt för huden, bör ytterligare testning utföras för att bekräfta detta. Härvid bör man helst använda validerade och godkända in vitro- eller ex vivo-tester (se stegen 5 och 6).
Toxicitet för huden (steg 4). Om ett ämne har konstaterats vara mycket toxiskt när det tillförs via huden, kan en in vivo-undersökning med avseende på hudirritation/hudkorrosion eventuellt inte utföras på grund av att den normalt applicerade mängden testämne skulle överskrida den mycket toxiska dosen, vilket skulle leda till att djuren dör eller utsätts för svårt lidande. Om dessutom undersökningar med avseende på toxicitet för huden har gjorts på albinokanin med doser upp till gränsnivån på 2 000 mg/kg kroppsvikt eller högre, och ingen hudirritation eller hudkorrosion har observerats, behövs sannolikt ingen ytterligare testning för hudirritation eller hudkorrosion. Ett antal faktorer bör beaktas vid utvärderingen av resultaten från tidigare utförda undersökningar med avseende på akut toxicitet för huden. Som exempel kan nämnas att den rapporterade informationen om hudskador kan vara ofullständig. Testningen och observationerna kan ha gjorts på en annan art än kanin, och arterna kan vara mycket olika när det gäller responskänsligheten. Testämnet kan också ha applicerats på djuren i en form som inte är lämplig när det gäller bedömning av hudirritation/hudkorrosion, till exempel utspädda ämnen för testning av toxicitet för huden (5). I fall där välplanerade och välutförda undersökningar med avseende på toxicitet för huden har gjorts på kanin, kan dock negativa fynd anses vara tillräckligt bevis på att ämnet i fråga inte är korrosivt eller irriterande.
Resultat från in vitro- eller ex vivo-test (steg 5 och 6). Ämnen som har uppvisat korrosiva eller starkt irriterande egenskaper i ett validerat och godkänt test in vitro eller ex vivo (6), (7) avsett för utvärdering av dessa specifika verkningar, behöver inte testas på djur. Man kan anta att sådana ämnen framkallar liknande allvarliga verkningar in vivo.
In vivo-test på kanin (steg 7 och 8). Om ett beslut grundat på en weight-of the-evidence-analys fattats för in vivo-testning, bör man börja med ett inledande test där ett enskilt djur används. Om resultatet av ett sådant test tyder på att ämnet framkallar korrosion på huden, bör ingen ytterligare testning utföras. Om ingen korrosion observeras vid det inledande testet bör irritationsrespons eller negativ respons bekräftas med hjälp av ytterligare två djur och en exponeringsperiod på fyra timmar. Om irritation har observerats vid det inledande testet, kan bekräftelsetestet göras enligt den stegvisa principen eller genom samtidig exponering på ytterligare två djur.
HÄNVISNINGAR
|
1. |
OECD (1996), ”Test Guidelines Programme: Final Report on the OECD Workshop on Harmonization of Validation and Acceptance Criteria for Alternative Toxicological Test Methods”, Solna, Sverige, den 22–24 januari 1996 (http://www1.oecd.org/ehs/test/background.htm). |
|
2. |
OECD (1998), Harmonized Integrated Hazard Classification System for Human Health and Environmental Effects of Chemical Substances, as endorsed by the 28th Joint Meeting of the Chemicals Committee and the Working Party on Chemicals, november 1998 (http://www1.oecd.org/ehs/Class/HCL6.htm). |
|
3. |
A.P. Worth, J.H. Fentem, M. Balls, P.A. Botham, R.D. Curren, L.K. Earl, J. Esdaile, M. Liebsch (1998), ”An Evaluation of the Proposed OECD Testing Strategy for Skin Corrosion”, ATLA. 26, 709–720. |
|
4. |
J.R. Young, M.J. How, A.P. Walker, W.M.H.Worth (1988), ”Classification as Corrosive or Irritant to Skin of Preparations Containing Acidic or Alkaline Substances, Without Testing on Animals”, Toxic In Vitro, 2 (1) 19–26. |
|
5. |
S.M. Patil, E. Patrick, H.I. Maibach (1996), ”Animal, Human, and In Vitro Test Methods for Predicting Skin Irritation”, Francis N. Marzulli och Howard I. Maibach (red.), Dermatotoxicology, femte utgåvan ISBN 1-56032-356-6, kapitel 31, 411–436. |
|
6. |
Testmetod B.40. |
|
7. |
J.H. Fentem, G.E.B. Archer, M. Balls, P.A. Botham, R.D. Curren, L.K. Earl, D.J. Esdaile, H.G. Holzhutter och M. Liebsch (1998), ”The ECVAM international validation study on in vitro tests for skin corrosivity. 2. Results and evaluation by the Management Team”, Toxicology in Vitro, 12, 483–524. |
Schema
TESTNINGS- OCH UTVÄRDERINGSSTRATEGI FÖR HUDIRRITATION/HUDKORROSION

B.5 AKUT TOXICITET: ÖGONIRRITATION/ÖGONKORROSION
1. METOD
Denna testmetod motsvarar OECD TG 405 (2002).
1.1 INLEDNING
Vid utarbetandet av denna uppdaterade metod lades särskilt stor vikt vid möjligheterna till bättre testningsförfaranden, där djurskyddsfrågor beaktas så väl som möjligt. Sådana förfaranden innebär att all existerande information om testämnet utvärderas i syfte att undvika onödiga djurförsök. Metoden innehåller en rekommendation om att laboratoriet, innan det beskrivna in vivo-testet för akut ögonkorrosion/ögonirritation inleds, gör en weight-of-the-evidence-analys av existerande relevanta data. Om de data som finns att tillgå inte är tillräckliga, kan behövliga data genereras med hjälp av stegvis testning (2) (3). Den rekommenderade testningsstrategin inbegriper validerade och godkända in vitro-test (se bilagan till denna metod). Likaså rekommenderas, innan in vivo-ögontest övervägs, utförandet av ett in vivo-test med avseende på hudirritation/hudkorrosion i syfte att förutsäga eventuell ögonkorrosion.
Med hänsyn till sund vetenskaplig praxis och djurens välbefinnande bör in vivo-testning inte övervägas förrän alla tillgängliga data som är relevanta för ämnets förmåga att framkalla ögonkorrosion/ögonirritation har utvärderats genom en weight-of-the-evidence-analys. Till sådana data hör bevis från gjorda undersökningar på människor och/eller försöksdjur, bevis på förmåga att framkalla korrosion/irritation som finns hos ett eller flera strukturellt närbesläktade ämnen eller blandningar av sådana ämnen, data om att ämnet är starkt surt eller alkaliskt (4) (5) samt resultat från validerade och godkända in vitro- eller ex vivo-test med avseende på hudkorrosion och hudirritation (6) (6a). Sådana undersökningar kan ha gjorts före, eller motiverade av, weight-of-the-evidence-analys.
För vissa ämnen kan en sådan analys peka på att det behövs in vivo-testning av ämnets förmåga att framkalla hudkorrosion/hudirritation. I alla sådana fall bör man, innan ett in vivo-ögontest övervägs, helst göra ett in vivo-test (testmetod B.4) för utvärdering av ämnets verkningar på huden (7). Med hjälp av weight-of-the-evidence-analys och strategin för stegvis testning bör man kunna minska behovet av in vivo-testning med avseende på ögonkorrosion/ögonirritation när det gäller ämnen för vilka det redan finns tillräckliga bevis från andra undersökningar. Om det inte går att fastställa ett ämnes förmåga att framkalla ögonkorrosion eller ögonirritation i enlighet med strategin för stegvis testning, inte ens efter det att man utfört en in vivo-undersökning med avseende på hudkorrosion och hudirritation, kan ett in vivo-test med avseende på ögonkorrosion och ögonirritation genomföras.
En rekommenderad strategi för stegvis testning, som inbegriper validerade och godkända in vitro- eller ex vivo-test med avseende på korrosion/irritation, finns som bilaga till denna metod. Strategin utvecklades av och rekommenderades enhälligt av deltagarna i en OECD-arbetsgrupp (8). Strategin har antagits som den rekommenderade testningsstrategin i Globally Harmonised System for the Classification of Chemical Substances (GHS) (9). Denna strategi för stegvis testning ingår inte i testmetod B.4, men användning av den rekommenderas ändå innan man går in för in vivo-testning. För nya ämnen rekommenderas den som en stegvis testningsmetod för generering av vetenskapligt sunda data om ett ämnes korrosion/irritation. För befintliga ämnen med otillräckliga data om korrosion/irritation för hud och ögon bör strategin användas för att fylla upp luckor i tillgängliga data. Om en annan testningsstrategi eller ett annat förfarande används, eller om man beslutar att inte använda ett förfarande med stegvis testning, bör detta motiveras.
1.2 DEFINITIONER
Ögonirritation: Ändringar som uppstår i ögat efter applicering av ett testämne på ögats främre yta och som inte är fullt reversibla inom 21 dagar från appliceringen.
Ögonkorrosion (ögonfrätning): Vävnadsskador som uppstår i ögat, eller allvarlig fysisk synnedsättning, efter applicering av ett testämne på ögats främre yta, och som inte är fullt reversibla inom 21 dagar från appliceringen.
1.3 PRINCIP FÖR TESTMETODEN
Testämnet appliceras som en engångsdos i försöksdjurets ena öga. Det obehandlade ögat används som referens. Graden av ögonirritation eller ögonkorrosion utvärderas genom klassificering av skadorna på konjunktiva, hornhinnan och iris med specifika intervall. Likaså bör övriga verkningar i ögat och skadliga systemiska verkningar beskrivas för att en fullständig utvärdering av verkningarna ska fås. Testet bör pågå tillräckligt länge för att ge möjlighet att utvärdera om de observerade verkningarna är reversibla eller irreversibla.
Djur som uppvisar ihållande tecken på allvarligt lidande eller smärta vid något skede av testningen bör avlivas på ett skonsamt sätt, och detta bör beaktas vid utvärderingen av ämnet. Kriterier för att fatta beslut om skonsam avlivning av döende eller allvarligt lidande djur finns i hänvisning (10).
1.4 BESKRIVNING AV TESTMETODEN
1.4.1 Förberedelser för in vivo-test
1.4.1.1 Val av art
Rekommenderat försöksdjur är albinokanin. Friska, unga vuxna exemplar bör användas. Om andra arter eller stammar används bör detta motiveras.
1.4.1.2 Förberedelse av djuren
De djur som preliminärt har valts ut för testning bör undersökas med avseende på båda ögonen inom 24 timmar före testets början. Djur som har irriterade ögon, okulära defekter eller existerande skada på hornhinnan bör inte användas.
1.4.1.3 Förvarings- och utfodringsbetingelser
Djuren ska inhysas individuellt. Temperaturen i försöksdjurens utrymmen bör vara 20 oC (±3 oC) för kaniner. Den relativa fuktigheten bör vara minst 30 % och ska helst inte överstiga 70 %, utom när rummet rengörs. Målvärdet bör dock vara 50–60 %. Belysningen bör vara artificiell med omväxlande 12 timmar ljus och 12 timmar mörker. Konventionellt laboratoriefoder kan användas för utfodringen, och djuren bör ha obegränsad tillgång till dricksvatten.
1.4.2 Testförfarande
1.4.2.1 Applicering av testämnet
Testämnet tillförs i ena ögats konjunktivasäck (alla djur). Före tillförseln bör det undre ögonlocket varsamt dras ut en bit ut från ögongloben. Ögonlocken förs därefter samman för cirka en sekund för att förhindra att ämnet kommer ut. Det andra, obehandlade ögat används som referens.
1.4.2.2 Sköljning
Försöksdjurens ögon bör inte sköljas under minst 24 efter tillförseln av testämnet, utom när det gäller fasta ämnen (se avsnitt 1.4.2.3.2) eller om det uppstår omedelbara korrosiva eller irriterande verkningar. Efter 24 timmar kan ögonen sköljas, om detta bedöms vara lämpligt.
Användningen av en satellitgrupp för att undersöka inverkan av sköljningen rekommenderas inte, utom om det är vetenskapligt motiverat. Om en satellitgrupp används, ska den bestå av två kaniner. Sköljningsbetingelserna bör dokumenteras omsorgsfullt, till exempel sköljningstidpunkten, lösningens sammansättning och temperatur samt sköljningstiden, applicerad volym och sköljningsintensitet (volymström).
1.4.2.3 Dosnivå
1.4.2.3.1 Testning av vätskor
Vid testning av vätskor används en dos på 0,1 ml. Pumpsprej bör inte användas för att tillföra ämnet direkt i ögat. I stället bör vätskan sprejas in i en behållare, som används för att tillföra 0,1 ml i ögat.
1.4.2.3.2 Testning av fasta ämnen
Vid testning av fasta ämnen, pastor och ämnen i partikelform, bör den tillförda mängden ha en volym på 0,1 ml eller en medelvikt på högst 100 mg. Testämnet bör malas ned till fint stoft. Det fasta materialets volym bör mätas efter en varsam kompaktering, som görs till exempel genom att klappa mätbehållaren. Om ett fast testämne inte har tagis bort ur försöksdjurets öga med hjälp av fysiologiska mekanismer vid den första observationstidpunkten (en timme efter behandlingen), kan ögat sköljas med saltslösning eller destillerat vatten.
1.4.2.3.3 Testning av aerosoler
Det rekommenderas att material i form av pumpsprej eller aerosol sprejas i en behållare, som sedan används för tillförsel i ögat. Det enda undantaget är aerosoler i tryckbehållare som inte går att samla in på grund av förångning. I sådana fall hålls djurets öga öppet och testämnet sprutas in i ögat under cirka en sekund, på cirka 10 cm avstånd direkt framför ögat. Avståndet kan variera beroende på behållarens tryck och innehåll. Det är viktigt att se till att ögat inte skadas av sprejtrycket. I tillämpliga fall kan det vara nödvändigt att utvärdera om sprejstyrkan kan framkalla ’mekanisk’ skada i ögat.
En uppskattning av dosen från en aerosol kan göras genom en simulering på följande sätt: Ämnet sprejas på ett vägningspapper genom en öppning som har storleken av ett kaninöga och som hålls strax framför papperet. Papperets viktökning används som grund för att uppskatta den mängd som har sprejats i ögat. För flyktiga ämnen kan dosen uppskattas genom att väga en mottagningsbehållare före och efter borttagning av testämnet.
1.4.2.4 Inledande test (in vivo-test avseende ögonirritation/ögonkorrosion på ett enskilt djur)
I enlighet med det som anges i strategin för stegvis testning (se bilaga 1), rekommenderas starkt att in vivo-testet först görs på ett enskilt djur.
Om resultatet av ett sådant test tyder på att ämnet är korrosivt eller framkallar allvarlig irritation i ögat när man använder det beskrivna förfarandet, bör ingen ytterligare testning utföras.
1.4.2.5 Lokalbedövning
Lokalbedömning kan användas beroende på fall. Om weight-of-the-evidence-analysen tyder på att ämnet kan framkalla smärta, eller om en inledande testning tyder på att en smärtsam reaktion kommer att ske, kan djuret få lokalbedövning innan testämnet tillförs. Typen, koncentrationen och dosen för lokalbedövningsmedlet bör väljas omsorgsfullt för att säkerställa att lokalbedövningen inte påverkar den reaktion som testämnet framkallar. Lokalbedövning bör även ges i det andra ögat (referensögat).
1.4.2.6 Bekräftande test (in vivo-test avseende ögonirritation med ytterligare djur)
Om ingen korrosiv verkan observeras vid det inledande testet, bör responsen (irriterande eller negativ) bekräftas med hjälp av högst två ytterligare djur. Om en allvarlig irritation har observerats vid det inledande testet, vilket tyder på en eventuell stark (irreversibel) verkan vid den bekräftande testningen, rekommenderas bekräftande test som görs stegvis på ett djur i taget, i stället för samtidigt test på båda tilläggsdjuren. Om frätning eller kraftig irritation kan observeras på det tilläggsdjur som först används, bör testningen avbrytas. Om irritationsresponsen är mild till moderat behövs eventuellt även det andra tilläggsdjuret för bekräftelse.
1.4.2.7 Observationsperiod
Observationsperioden bör vara tillräckligt lång för att det till fullo ska gå att utvärdera omfattningen och reversibiliteten hos de observerade verkningarna. Försöket bör dock avslutas omedelbart, oavsett hur länge det pågått, om det berörda djuret uppvisar tecken på svåra smärtor eller lidande (9). För fastställandet av verkningarnas reversibilitet bör djuren i regel observeras i 21 dagar efter tillförsel av testämnet. Om reversibilitet kan observeras innan 21 dagar har gått kan försöket omedelbart avslutas.
1.4.2.7.1 Kliniska observationer och gradering av ögonreaktionerna
Ögonen bör undersökas 1, 24, 48 och 72 timmar efter tillförsel av testämnet. Djuren bör inte utsättas för testning längre än det är nödvändigt för att få definitiv information. Djur som uppvisar allvarliga och ihållande tecken på smärta eller lidande bör omedelbart avlivas på ett skonsamt sätt, och detta bör beaktas vid utvärderingen av ämnet. Djur som fått följande ögonskador efter tillförsel av ämnet bör avlivas på ett skonsamt sätt: perforering eller signifikant ulceration av kornea inklusive stafylom, blod i främre ögonkammaren, korneaopacitet av grad 4 som varar i 48 timmar; ingen ljusreaktion (irisreaktion av grad 2) som varar i 72 timmar, ulceration av konjuktivamembran, nekros i konjunktiva- eller blinkhinna eller bildning av sårskorpa. Skälet till avlivningen är att dessa skador i regel är irreversibla.
Djur som inte utvecklar okulära skador ska inte avlivas förrän det har gått tre dagar från tillförsel av ämnet. Djur med milda till moderata skador bör observeras tills skadorna läks, eller under 21 dagar. Efter det avslutas undersökningen. Observationer bör göras på dag 7, 14 och 21 i syfte att fastställa skadornas status och huruvida de är reversibla eller inte.
Graderna av okulär reaktion (konjunktiva, kornea och iris) bör registreras vid varje undersökning (se tabell 1). Likaså bör alla andra ögonskador (till exempel pannus, missfärgning) eller skadliga systemiska verkningar även rapporteras.
Som hjälpmedel vid undersökningen av reaktionerna kan användas en binokulärlupp, ficklampa, biomikroskop eller annan lämplig utrustning. Efter det att observationerna har registrerats vid 24 timmar efter tillförseln, kan ögonen undersökas ytterligare med hjälp av fluorescein.
Graderingen av okulär respons är oundgängligen subjektiv. För att främja en tillnärmning av graderingen av okulär respons, och som stöd för testningslaboratorier och alla som är med om att utföra och tolka observationer, bör den personal som gör observationsarbetet få tillbörlig utbildning om klassificeringssystemet. Graderingen av okulär respons bör göras i blindo.
2. DATA
2.2 UTVÄRDERING AV RESULTATEN
Klassificeringen av okulär respons bör göras med hänsyn till skadornas natur och allvarlighetsgrad samt deras reversibilitet eller irreversibilitet. Individuella klassificeringsresultat representerar ingen absolut standard för ett ämnes förmåga att framkalla irritation, eftersom även andra verkningar av testämnet utvärderas. Enskilda klassificeringsresultat bör i stället ses som referensvärden, och de är meningsfulla endast när de åtföljs av en fullständig beskrivning och utvärdering av alla observationer.
3. RAPPORTERING
3.1 TESTRAPPORT
Testrapporten ska innehålla nedan angivna uppgifter.
|
|
Motivering för in vivo-testning: weight-of-evidence-analys av tidigare existerande data, inbegripet resultat från ett förfarande enligt strategin för stegvis testning, enligt följande:
|
|
|
Testämne:
|
|
|
Vehikel:
|
|
|
Testdjur:
|
|
|
Resultat:
|
|
|
Diskussion om resultaten. |
3.2 TOLKNING AV RESULTATEN
En extrapolering av resultaten från undersökningar av ögonirritation som gjorts på försöksdjur är valid endast till en begränsad utsträckning. I många fall är albinokaniner mycket känsligare än människor i fråga om ämnen som är irriterande eller korrosiva för ögat.
Särskild noggrannhet bör iakttas vid tolkning av resultaten i syfte att utesluta irritation som beror på en sekundär infektion.
4. HÄNVISNINGAR
|
1. |
Barratt, M.D., Castell, J.V., Chamberlain, M., Combes, R.D., Dearden, J.C., Fentem, J.H., Gerner, I., Giuliani, A., Gray, T.J.B., Livingston, D.J., Provan, W.M., Rutten, F.A.J.J.L., Verhaar, H.J.M., Zbinden, P. (1995), The Integrated Use of Alternative Approaches for Predicting Toxic Hazard. ECVAM Workshop Report 8. ATLA 23, 410–429. |
|
2. |
de Silva, O., Cottin, M., Dami, N., Roguet, R., Catroux, P., Toufic, A., Sicard, C., Dossou, K.G., Gerner, I., Schlede, E., Spielmann, H., Gupta, K.C., Hill, R.N. (1997), Evaluation of Eye Irritation Potential: Statistical Analysis and Tier Testing Strategies. Food Chem. Toxicol 35, 159–164. |
|
3. |
Worth, A.P. och Fentem, J.H. (1999), A general approach for evaluating stepwise testing strategies ATLA 27, 161–177. |
|
4. |
Young, J.R., How, M.J., Walker, A.P., Worth, W.M.H. (1988), Classification as Corrosive or Irritant to Skin of Preparations Containing Acidic or Alkaline Substances Without Testing on Animals. Toxicol. In Vitro, 2, 19–26. |
|
5. |
Neun, D.J. (1993), Effects of Alkalinity on the Eye Irritation Potential of Solutions Prepared at a Single pH. J. Toxicol. Cut. Ocular Toxicol. 12, 227–231. |
|
6. |
Fentem, J.H., Archer, G.E.B., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Esdaile, D.J., Holzhutter, H.G. och Liebsch, M. (1998), The ECVAM international validation study on in vitro tests for skin corrosivity. 2. Results and evaluation by the Management Team. Toxicology in Vitro 12, s. 483–524. |
|
(6a) |
Testmetod B.40 Hudkorrosion. |
|
(7) |
Testmetod B.4. Akut toxicitet: hudirriation/hudkorrosion. |
|
(8) |
OECD (1996), OECD Test Guidelines Programme: Final Report of the OECD Workshop on Harmonization of Validation and Acceptance Criteria for Alternative Toxicological Test Methods, Solna, Sverige, den 22–24 januari 1996 (http://www.oecd.org/ehs/test/background.htm). |
|
(9) |
OECD (1998), Harmonized Integrated Hazard Classification System for Human Health and Environmental Effects of Chemical Substances, as endorsed by the 28th Joint Meeting of the Chemicals Committee and the Working Party on Chemicals, november 1998 (http://www.oecd.org/ehs/Class/HCL6.htm). |
|
(10) |
OECD (2000), Guidance Document on the Recognition, Assessment and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluation, OECD Environmental Health and Safety Publications, Series on Testing and Assessment, No. 19 (http://www.oecd.org/ehs/test/monos.htm). |
Tabell 1
GRADERING AV OKULÄRA SKADOR
Hornhinna (kornea)
Opacitet: graden av densitet (avläsningarna bör göras på området med den högsta densiteten) (1)
|
Ingen ulceration eller opacitet … |
0 |
|
Spridda eller diffusa områden med opacitet (annan än svag avmattning av den normala lystern), iris alla detaljer är klart synliga … |
1 |
|
Enkelt skönjbart translucent område, detaljerna i iris är lätt förmörkade … |
2 |
|
Pärlemorliknande område, inga detaljer i iris synliga, pupillens storlek kan knappt skönjas … |
3 |
|
Opak hornhinna, iris kan inte skönjas genom opaciteten … |
4 |
Maximum: 4
ANMÄRKNINGAR
NOTER
Iris
|
Normal … |
0 |
|
Märkbart fördjupade rugae, blodstockning, svullnad, moderate hyperemi kring kornea eller injektion; iris reagerar för ljus (en långsam reaktion bedöms som en verkning) … |
1 |
|
Blödning, kraftig destruktion eller ingen reaktion för ljus … |
2 |
Maximum: 2
Konjunktiva
Rodnad (hänför sig till palpebral och bulbär konjunktiva, exklusive hornhinnan och iris)
|
Normal … |
0 |
|
Vissa blodkärl är hyperemiska (injekterade) … |
1 |
|
Diffus, blodröd färg; individuella blodkärl kan inte skönjas med lätthet … |
2 |
|
Diffust köttröd … |
3 |
Maximum: 3
Chemos
Svullnad (hänför sig till ögonlocken och/eller blinkhinnor)
|
Normalt … |
0 |
|
En viss svullnad över det normala … |
1 |
|
Klar svullnad, delvis eversion av ögonlocken … |
2 |
|
Svullnad, ögonlocken ungefär halvstängda … |
3 |
|
Svullnad, ögonlocken mer än halvstängda … |
4 |
Maximum: 4
Bilaga
Strategi för stegvis testning med avseende på ögonirritation och ögonkorrosion
ALLMÄNT
För en sund vetenskaplig praxis och för djurens välbefinnande är det viktigt att undvika onödig användning av djur, och minimera all testning som sannolikt framkallar allvarlig respons hos djuren. All information som är relevant för ett ämnes förmåga att framkalla ögonkorrosion/ögonirritation bör utvärderas innan in vivo-testning övervägs. Det finns eventuellt redan tillräckliga bevis för att klassificera ett testämne med avseende på dess förmåga att framkalla ögonkorrossion eller ögonirritation, utan behov av testning på laboratoriedjur. Man kan därför, genom att använda en weight-of-the-evidence-analys och följa strategin för stegvis testning, minimera behovet av in vivo-testning, särskilt i fråga om ämnen som sannolikt framkallar allvarliga reaktioner.
Det rekommenderas att en weight-of-the-evidence-analys används för att utvärdera existerande information om olika ämnens förmåga att framkalla ögonirritation eller ögonkorrosion. Därefter kan man avgöra om det behövs ytterligare undersökningar, andra än in vivo-test, som hjälp för att karakterisera sådan egenskap. I fall där ytterligare undersökningar behövs, rekommenderas ett förfarande enligt strategin för stegvis testning för att få fram relevanta försöksdata. För ämnen som inte har testats tidigare bör strategin för stegvis testning följas för att få fram de data som behövs för att utvärdera ämnets förmåga att framkalla ögonkorrosion eller ögonirritation. Testningsstrategin som beskrivs i denna bilaga har utarbetats inom ramen för en OECD-arbetsgrupp (1). Strategin bekräftades och utvidgades senare i Harmonised Integrated Hazard Clas-sification System for Human Health and Environmental Effects of Chemical Substances, i den form systemet godkändes av vid det 28:e gemensamma mötet för kemikaliekommittén och kemikaliearbetsgruppen (Chemicals Committee och Working Party on Chemicals), i november 1998 (2).
Även om denna strategi för stegvis testning inte ingår som en del av testmetod B.5, är den ett uttryck för de rekommenderade principerna för bestämning av ett ämnes förmåga att framkalla ögonirritation eller ögonkorrosion. Dessa principer representerar både bästa praxis och etiska riktlinjer för in vivo-testning med avseende på ögonirritation/ögonkorrosion. Testmetoden är en vägledning för utförandet av test in vivo, och utgör en sammanfattning av de faktorer som bör beaktas innan ett sådant test övervägs. Inom ramen för strategin för stegvis testning används weight-of-the-evidence-analyser för utvärderingen av existerande data om olika testämnens egenskaper med avseende på ögonirritation/ögonkorrosion. Samtidigt fås tillgång till en stegvis metod för generering av relevanta data om ämnen för vilka det krävs ytterligare testning eller för ämnen som ännu inte har testats. Enligt strategin utför man först validerade och godkända in vitro- eller ex vivo-tester, och därefter görs testning enligt testmetod B.4 (hudirritation/hudkorrosion) under specifika omständigheter (3) (4).
BESKRIVNING AV DEN STEGVISA TESTNINGSSTRATEGIN
Innan testning utförs som en del av förfarandet enligt strategin för stegvis testning (se figuren), bör all tillgänglig information utvärderas i syfte att fastställa behovet av in vivo-test på ögonen. Trots att signifikant information kan fås genom utvärdering av enskilda parametrar (till exempel extrema pH-värden), bör den tillgängliga informationen utvärderas som en helhet. Alla relevanta data om det berörda ämnets verkningar och strukturellt närbesläktade ämnen bör utvärderas när man fattar ett beslut grundat på en weight-of-the-evidence-analys, och en motivering för beslutet bör anges. Huvudvikten bör läggas vid existerande data om ämnet (både i fråga om människor och djur). Därefter följer resultatet av testning in vitro eller ex vivo. Testning in vivo av korrosiva ämnen bör undvikas alltid när det är möjligt. Till de faktorer som bör beaktas inom ramen för testningsstrategin hör följande:
Utvärdering av existerande data rörande människor och djur (steg 1). Existerande data rörande människor, till exempel kliniska undersökningar eller undersökningar inom arbetslivet och fallstudier och/eller data från undersökningar med avseende på ögon bör beaktas först, eftersom de ger tillgång till information som direkt hör samman med verkningarna på ögonen. Därefter bör tillgängliga data från human- och/eller djurstudier rörande hudkorrosion/hudirritation utvärderas. Ämnen som veterligen har förmåga att framkalla korrosion eller allvarlig irritation i ögat bör inte tillföras i djurögon. Samma gäller för ämnen som har korrosiva eller irriterande egenskaper för huden, eftersom sådana ämnen kan anses vara korrosiva och/eller irriterande även för ögonen. Om resultaten från tidigare utförda okulära undersökningar visar att ett ämne inte är korrosivt eller irriterande, bör det inte heller testas på ögonen in vivo.
Analys av strukturaktivitetssamband (SAR) (steg 2). I mån av tillgänglighet bör resultaten från testning av strukturellt närbesläktade ämnen beaktas. Om det finns tillgång till data rörande människa och/eller djur för strukturellt närbesläktade ämnen eller blandningar av sådana ämnen, och dessa data är tillräckliga för att indikera att dessa ämnen har förmåga att framkalla ögonkorrosion/ögonirritation, kan man anta att testämnet kommer att framkalla samma respons. I sådana fall behöver ämnet eventuellt inte testas. Negativa data från undersökningar med strukturellt närbesläktade ämnen eller blandningar av sådana ämnen utgör inte, inom ramen för strategin för stegvis testning, tillräckliga bevis på att ett ämne skulle sakna korrosiva eller irriterande egenskaper. Validerade och godkända metoder som bygger på strukturellt aktivitetssamband bör användas för att identifiera förmågan att framkalla korrosion och irritation på huden och ögonen.
Fysikalkemiska egenskaper och kemisk reaktivitet (steg 3). Ämnen med extrema pH-värden, till exempel ≤2,0 eller ≥11,5 kan ha kraftiga lokala verkningar. Om ett extremt pH-värde används som grund för att konstatera att ett ämne är korrosivt eller irriterande för ögonen, behöver eventuellt också ämnets syra/bas-reserv (buffertkapacitet) beaktas (5) (6). Om buffertkapaciteten tyder på att ämnet eventuellt inte är korrosivt för ögonen, bör ytterligare testning utföras för att bekräfta detta. Härvid bör man helst använda validerade och godkända in vitro- eller ex vivo-tester (se steg 5 och 6).
Beaktande av annan existerande information (steg 4). På detta steg bör man beakta all tillgänglig information om systemisk toxicitet när ett ämne tillförs via huden. Även testämnets akuta toxicitet för huden bör beaktas. Om testämnet har konstaterats vara mycket toxiskt när det tillförs via huden, behöver det eventuellt inte testas på ögonen. Även om det inte nödvändigt finns ett förhållande mellan akut toxicitet för huden och förmåga att framkalla ögonirritation/ögonkorrosion, kan man anta att ett ämne som har hög toxicitet vid tillförsel via huden, även har hög toxicitet när ämnet tillförs i ögonen. Sådana data kan även beaktas mellan steg 2 och steg 3.
Resultat från in vitro- eller ex vivo-test (steg 5 och 6). Ämnen som har visat sig framkalla korrosion eller allvarlig irritation i ett in vitro- eller ex vivo-test (7) (8) som har validerats och godkänts specifikt för utvärdering av korrosion eller irritation på huden eller ögonen, behöver inte testas på djur. Man kan anta att sådana ämnen framkallar liknande allvarliga verkningar in vivo. Om det inte finns tillgång till resultat från validerade och godkända in vitro- eller ex vivo-test, ska man hoppa över stegen 5 och 6 och fortsätta förfarandet direkt från steg 7.
Utvärdering in vivo av ämnets irriterande eller korrosiva egenskaper för huden (steg 7). Om det på grundval av data från de undersökningar som uppräknas ovan inte finns tillräckliga bevis för en avgörande weight-of-the-evidence-analys av ett ämnes förmåga att framkalla ögonirritation/ögonkorrosion, bör förmågan att framkalla hudirritation/hudkorrosion in vivo utvärderas först, med användning av testmetod B.4 (4) och dess bilaga (9). Om ämnet veterligen framkallar korrosion eller allvarlig hudirritation, bör det betraktas som ett korrosivt ögonirriterande ämne, utom om det finns någon annan information som stöd för en alternativ slutsats. Då behöver ögontestning in vivo inte utföras. Om ämnet inte är korrosivt eller allvarligt irriterande för huden, bör ögontest in vivo utföras.
In vivo-test på kanin (steg 8 och 9): In vivo-ögontest ska börja med ett inledande test på ett enskilt djur. Om resultatet av ett sådant test tyder på att ämnet framkallar korrosion eller allvarlig irritation i ögat, bör ingen ytterligare testning utföras. Om testresultatet tyder på att ämnet inte framkallar korrosion eller allvarlig irritation, utförs ett bekräftande test med ytterligare två djur.
HÄNVISNINGAR
|
1. |
OECD (1996), OECD Test Guidelines Programme: Final Report of the OECD Workshop on Harmonization of Validation and Acceptance Criteria for Alternative Toxicological Test Methods, Solna, Sverige, den 22–24 januari 1996 (http://www1.oecd.org/ehs/test/background.htm). |
|
2. |
OECD (1998), Harmonized Integrated Hazard Classification System for Human Health and Environmental Effects of Chemical Substances, as endorsed by the 28th Joint Meeting of the Chemicals Committee and the Working Party on Chemicals, november 1998 (http://www1.oecd.org/ehs/Class/HCL6.htm). |
|
3. |
A.P. Worth och J.H. Fentem (1999), A General Approach for Evaluating Stepwise Testing Strategies, ATLA, 27, 161–177. |
|
4. |
Testmetod B.4. Akut toxicitet: Hudirritation/hudkorrosion. |
|
5. |
Young, J.R., How, M.J., Walker, A.P., Worth W.M.H. (1988), Classification as Corrosive or Irritant to Skin of Preparations Containing Acidic or Alkaline Substances Without Testing on Animals. Toxicol. In Vitro, 2, 19–26. |
|
6. |
Neun, D.J. (1993), Effects of Alkalinity on the Eye Irritation Potential of Solutions Prepared at a Single pH. J. Toxicol. Cut. Ocular Toxicol. 12, 227–231. |
|
7. |
Fentem, J.H., Archer, G.E.B., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Esdaile, D.J., Holzhutter, H.G. och Liebsch, M. (1998), The ECVAM international validation study on in vitro tests for skin corrosivity. 2. Results and evaluation by the Management Team. Toxicology in Vitro 12, s. 483–524. |
|
8. |
Testmetod B.40 Hudkorrosion. |
|
9. |
Bilaga till testmetod B.4: En strategi för stegvis testning med avseende på hudirritation och hudkorrosion. |
Schema
TESTNINGS- OCH UTVÄRDERINGSSTRATEGI FÖR ÖGONIRRITATION/ÖGONKORROSION


B.6 HUDSENSIBILISERING
1. METOD
1.1 INLEDNING
Kommentarer:
Testers noggrannhet och förmåga att spåra sådana ämnen som kan orsaka hudallergi hos människan anses betydelsefulla i ett klassificeringssystem för toxicitet som avser folkhälsan.
Det finns ingen enstaka testmetod som på ett lämpligt sätt kan identifiera samtliga de ämnen som kan vara sensibiliserande och som är relevant för alla ämnen.
Faktorer som ett ämnes fysikaliska egenskaper, inbegripet dess förmåga att penetrera huden, ska beaktas vid valet av test.
Två typer av test på marsvin har utvecklats: Ett test av adjuvanstyp i vilket man åstadkommer ett allergiskt tillstånd genom att upplösa eller suspendera (blanda) testsubstansen i Freunds Complete Adjuvant (FCA), och test av icke-adjuvanstyp.
Adjuvanstester är sannolikt mer precisa vad gäller att förutsäga ämnets eventuella kapacitet att orsaka hudsensibilisering hos människan än metoder där inte Freunds Complete Adjuvant används och de är därför att föredra.
Maximeringstest på marsvin (GPMT) används ofta som adjuvanstest. Trots att flera andra metoder kan användas för att upptäcka ett ämnes potentiella förmåga att orsaka hudsensibilisering anses maximeringsförsöket på marsvin vara den bästa metoden.
Icke-adjuvanstester (av vilka Buehler-testet föredras) med många kemiska klasser anses mindre känsliga.
I vissa fall anses det finnas goda skäl att välja Buehler-testet, som innebär lokal applicering i stället för den intradermala injektion som används i GPMT. Då Buehler-testet används ska detta vara vetenskapligt välmotiverat.
Här beskrivs GPMT och Buehler-testet. Andra metoder får användas, förutsatt att de är väldokumenterade och vetenskapligt motiverade.
Om en erkänd kontrollundersökning ger positiva resultat, kan testämnet betecknas som potentiellt sensibiliserande, och det behöver inte vara nödvändigt att genomföra ytterligare test på marsvin. Om en sådan kontrollundersökning dock ger negativt resultat ska ett test på marsvin genomföras enligt det förfarande som beskrivs i denna testmetod.
Se även Allmän inledning, del B.
1.2 DEFINITIONER
Hudsensibilisering (allergisk kontaktdermatit): En immunologiskt utlöst kutan reaktion på ett ämne. Hos människan kan reaktionerna kännetecknas av klåda, hudrodnad, ödem, blemmor och blåsor eller en kombination av dessa. Andra arter kan reagera annorlunda så att endast hudrodnad och ödem observeras.
Induktionsexponering: En försöksexponering av ett försöksobjekt för en testsubstans i syfte att inducera hypersensibilitet.
Induktionsperiod: En period på minst en vecka som följer efter induktionsexponering då hypersensibilitet kan utvecklas.
Provokationsexponering: En försöksexponering av ett tidigare behandlat objekt för att testa ämnet efter en induktionsperiod i syfte att bestämma om objektet reagerar med hypersensibilitet.
1.3 REFERENSÄMNEN
Den tillämpade försöksmetodens noggrannhet och tillförlitlighet bör utvärderas var sjätte månad genom användning av ämnen som är kända för att ha milda till moderata hudsensibiliseringsegenskaper.
I ett korrekt utfört test bör en reaktion på minst 30 % i adjuvanstest och minst 15 % i icke-adjuvanstest förväntas för milda/moderata sensibiliserande ämnen.
Följande ämnen är att föredra:
|
CAS-nummer |
EINECS-nummer |
EINECS-namn |
Vanliga namn |
|
101-86-0 |
202-983-3 |
α-hexylcinnamaldehyd |
α-hexylcinnamaldehyd |
|
149-30-4 |
205-736-8 |
benzotiasol-2-tiol (merkapto benzotiasol) |
kaptax |
|
94-09-7 |
202-303-5 |
benzokain |
norkain |
Då det är motiverat på grund av vissa omständigheter får andra kontrollämnen som uppfyller ovan angivna kriterier användas.
1.4 PRINCIP FÖR TESTMETODEN
Försöksdjuren exponeras inledningsvis för testsubstansen genom intradermala injektioner och/eller epidermal applicering (induktionsexponering). Efter en paus på 10–14 dagar (induktionsperiod) under vilken en immunreaktion kan utvecklas exponeras djuren för en provokationsdos. Omfattningen och graden av hudens reaktion på provokationsexponeringen hos försöksdjuren jämförs med den som uppträder hos kontrollgruppens djur som under induktionsexponeringen får skenbehandling och därefter utsätts för provokationsexponering.
1.5 BESKRIVNING AV TESTMETODERNA
Om det anses nödvändigt att avlägsna testsubstansen görs det med vatten eller en lämplig lösning utan att befintlig reaktion eller hudens tillstånd ändras.
1.5.1 Maximeringstest på marsvin (GPMT)
1.5.1.1
Friska unga vuxna albinomarsvin acklimatiseras till laboratoriemiljö under minst fem dagar före försöket. Före försöket väljs djuren ut slumpvis och fördelas i behandlingsgrupper. Pälsen avlägsnas genom klippning, rakning eller eventuellt kemiskt, beroende på vilken testmetod som används. Försiktighet ska iakttas för att inte skada huden. Djuren vägs innan försöket påbörjas och då det avslutas.
1.5.1.2
1.5.1.2.1 Försöksdjur
Vanliga stammar av albinomarsvin används.
1.5.1.2.2 Antal och kön
Hannar och/eller honor kan användas. Om honor används ska de inte ha fått ungar och inte vara dräktiga.
Minst 10 djur används i behandlingsgruppen och minst 5 djur i kontrollgruppen. Om färre än 20 testdjur och 10 kontrolldjur har använts och det inte är möjligt att konstatera att testsubstansen är hudsensibiliserande, rekommenderas starkt tester med ytterligare djur upp till totalt 20 testdjur och 10 kontrolldjur.
1.5.1.2.3 Dosnivåer
Den testsubstanskoncentration som används vid varje induktionsexponering ska tolereras väl av hela organismen och vara den högsta nivå som orsakar mild till måttlig hudirritation. Den koncentration som används vid provokationsexponering ska vara den högsta icke-irriterande dosen. Om nödvändigt, kan de lämpliga koncentrationerna fastställas med hjälp av en pilotundersökning med två eller tre djur. För detta ändamål bör användningen av FCA-behandlade djur beaktas.
1.5.1.3
1.5.1.3.1 Induktion
Dag 0-behandlingsgrupp
Tre par intradermala injektioner på 0,1 ml ges i skuldertrakten, där pälsen avlägsnats, så att varje par injektioner görs på var sida om mittlinjen.
Injektion l: en 1:1-blandning (viktprocent) FCA/vatten eller fysiologisk saltlösning.
Injektion 2: Testsubstansen i lämplig vehikel i vald koncentration.
Injektion 3: Testsubstansen i vald koncentration i en 1:1-blandning (viktprocent) FCA/vatten eller fysiologisk saltlösning.
I injektion 3 löses de vattenlösliga ämnena i vattenfasen innan de blandas med FCA. Fettlösliga eller olösliga ämnen suspenderas (blandas) i FCA innan de blandas med vattenfasen. Testsubstansens slutliga koncentration ska vara densamma som den i injektion 2.
Injektionerna 1 och 2 ges med ett kort intervall och närmast huvudet medan injektion 3 ges i testområdets nedersta del.
Dag 0-kontrollgrupp
Tre par intradermala injektioner på 0,1 ml ges i samma område som på djuren i behandlingsgruppen.
Injektion l: en 1:1-blandning (viktprocent) FCA/vatten eller fysiologisk saltlösning.
Injektion 2: Outspädd vehikel.
Injektion 3: En 50 % blandning av vehikeln i en 1:1-blandning (viktprocent) FCA/vatten eller fysiologisk saltlösning.
Dag 5–7-behandlings- och kontrollgrupp
Om ämnet inte är hudirriterande behandlas testområdet, efter klippning eller rakning, ungefär 24 timmar före lokal applicering med 0,5 ml 10 % natriumlaurylsulfat i vaselin i syfte att orsaka lokal irritation.
Dag 6–8-behandlingsgrupp
Pälsen avlägsnas på nytt från testområdet. Testsubstansen i lämplig vehikel fördelas på ett filterpapper (2 × 4 cm), appliceras på testområdet och hålls i kontakt med huden genom täckande förband under 48 timmar. Valet av vehikel ska vara välmotiverat. Fasta ämnen pulveriseras och införlivas i en lämplig vehikel. Vätskor kan om det är lämpligt appliceras outspädda.
Dag 6–8-kontrollgrupp
Pälsen avlägsnas på nytt från testområdet. Vehikeln appliceras på liknande sätt på testområdet och hålls i kontakt med huden genom täckande förband under 48 timmar.
1.5.1.3.2 Provokation
Dag 20–22-behandlings- och kontrollgrupp
Pälsen avlägsnas frän behandlings- och kontrolldjurens flanker. En kompress eller kammare innehållande testsubstansen appliceras på djurens ena flank och, om relevant, en kompress eller kammare innehållande enbart vehikel på den andra flanken. Kompresserna hålls i kontakt med huden genom täckande förband under 24 timmar.
1.5.1.3.3 Observation och gradering: Behandlings- och kontrollgrupp
|
— |
Cirka 21 timmar efter det att kompressen avlägsnats rengörs det provocerade området och klipps eller rakas och vid behov avlägsnas pälsen med hårborttagningsmedel. |
|
— |
Cirka tre timmar senare (ungefär 48 timmar från det att provokationsexponeringen påbörjats) observeras hudreaktionen och noteras i enlighet med graderingen i tillägget. |
|
— |
Cirka 24 timmar efter föregående observation görs en andra observation (72 timmar) och resultaten noteras. |
Blindläsning av test- och kontrolldjur rekommenderas.
Om det är nödvändigt för att klargöra resultaten från den första provokationen bör en andra provokation (dvs. en omprovokation) övervägas en vecka efter den första, i tillämpliga fall med en ny kontrollgrupp. En omprovokation får också göras på den ursprungliga kontrollgruppen.
Alla hudreaktioner och ovanliga resultat, inbegripet systematiska reaktioner, som orsakas av induktions- och provokationsförfarandena bör observeras och noteras i enlighet med graderingsskalan enligt Magnusson/Kligman (se tillägget), övriga förfaranden t.ex. histopatologisk undersökning eller mätning av hudvecks tjocklek, bör utföras för att klargöra tveksamma reaktioner.
1.5.2 Buehler-test
1.5.2.1
Friska unga vuxna albinomarsvins acklimatiseras till laboratoriemiljön under minst fem dagar före försöket. Före försöket väljs djuren ut slumpvis och fördelas i behandlingsgrupper. Pälsen avlägsnas genom klippning, rakning eller eventuellt kemiskt, beroende på den testmetod som används. Försiktighet ska iakttas för att inte skada huden. Djuren vägs innan försöket påbörjas och då det avslutas.
1.5.2.2
1.5.2.2.1 Försöksdjur
Vanliga laboratoriestammar av albinomarsvin används.
1.5.2.2.2 Antal och kön
Hannar och/eller honor kan användas. Om honor används ska de inte ha fått ungar och inte vara dräktiga.
Minst 20 djur används i behandlingsgruppen och minst 10 djur i kontrollgruppen.
1.5.2.2.3 Dosnivåer
Testsubstansens koncentration anpassas till den högsta nivå som djuren har tolererat väl i varje induktionsstadium och ska vara den högsta nivå som orsakar mild till uthärdlig hudirritation. Den koncentration som används vid provokationsexponering ska vara den högsta icke-irriterade dosen. Om nödvändigt, kan de lämpliga koncentrationerna fastställas med hjälp av en pilotundersökning med två eller tre djur.
I fråga om vattenlösliga testmaterial är det lämpligt att använda vatten eller en icke-irriterande lösning av anjon-ytaktiva ämnen som vehikel. I fråga om övriga testmaterial är 80 % etanol/vatten att föredra för induktion och aceton för provokation.
1.5.2.3
1.5.2.3.1 Induktion
Dag 0-behandlingsgrupp
Pälsen avlägsnas från ena flanken (klipps).
Testkompressystemet dränks in med testsubstansen i lämplig vehikel (valet av vehikel ska vara välmotiverat; om lämpligt får testsubstanser i vätskeform appliceras outspädda). Testkompressystemet appliceras på testområdet och hålls i kontakt med huden med ocklusivkompress eller -kammare och lämpligt förband under sex timmar.
Testkompressystemet ska vara ocklusivt. En bomullskompress är lämplig och kan vara fyrkantig eller rund och cirka 4–6 cm2. Fasthållning medelst lämpligt förband är lämpligt för att säkerställa ocklusion. Om omslag används kan ytterligare exponering krävas.
Dag 0-kontrollgrupp
Pälsen avlägsnas från ena flanken (klipps). Endast vehikeln appliceras på liknande sätt som för behandlingsgruppen. Testkompressystemet hålls i kontakt med huden genom ocklusivkompress eller -kammare och lämpligt förband under sex timmar. Om det kan visas att en simulerad kontrollgrupp inte krävs kan en oexponerad kontrollgrupp användas.
Dag 6–8- och 13–15-behandlings- och kontrollgrupp
Samma applicering som dag 0 utförs på samma testområde (vid behov efter avlägsnande av päls) på samma flank dag 6–8 och åter dag 13–15.
1.5.2.3.2 Provokation
Dag 27–29- behandlings- och kontrollgrupp
Pälsen avlägsnas från den obehandlade flanken på behandlings- och kontrolldjuren (klippning). En ocklusivkompress eller -kammare innehållande lämplig mängd testsubstans appliceras vid maximal icke-irriterande koncentration, på den bakre obehandlade flanken på behandlings- och kontrolldjuren.
I relevanta fall appliceras också en ocklusivkompress eller -kammare på den främre obehandlade flanken på både behandlings- och kontrolldjur. Kompresserna eller kamrarna hålls i kontakt med huden genom lämpligt förband under sex timmar.
1.5.2.3.3 Observation och gradering
|
— |
Cirka 21 timmar efter det att kompressen tagits bort avlägsnas pälsen från provokationsområdet. |
|
— |
Cirka tre timmar senare (ungefär 30 timmar efter det att provokationskompressen applicerades) observeras hudreaktionerna och noteras i enlighet med graderingen i tillägget. |
|
— |
Cirka 24 timmar efter 30-timmarsobservationen (ungefär 54 timmar efter det att provokationskompressen applicerades) observeras hudreaktionerna på nytt och noteras. |
Blindläsning av test och kontrolldjur rekommenderas.
Om det är nödvändigt för att klargöra resultaten från den första provokationen bör en andra provokation (dvs. en förnyad provokation) övervägas en vecka efter den första, i tillämpliga fall med en ny kontrollgrupp. En förnyad provokation får också göras på den ursprungliga kontrollgruppen.
Alla hudreaktioner och ovanliga resultat, inbegripet systemiska reaktioner, som följer av induktions- och provokationsförfarandena bör observeras och noteras i enlighet med graderingsskalan enligt Magnusson/Kligman (se tillägget), övriga förfaranden, t.ex. histopatologisk undersökning eller mätning av hudvecks tjocklek, får utföras för att klargöra tveksamma reaktioner.
2. DATA (GPMT och Buehler-test)
Uppgifterna bör sammanfattas i tabellform som i fråga om varje djur visar hudreaktioner vid varje observation.
3. RAPPORTERING (GPMT och Buehler-test)
Om ett screeningsförsök utförs före marsvinsförsöket ska en beskrivning av eller hänvisning till försöket (t.ex. Local Lymph Node Essay (LLNA), Mouse Ear Swelling Test (MEST), inbegripet uppgifter om förfarandet, lämnas tillsammans med de resultat som erhållits genom försöket och referensämnena.
Försöksrapport (GPMT och Buehler-test)
Försöksrapporten ska om möjligt innehålla följande uppgifter:
|
|
Försöksdjur:
|
|
|
Försöksförhållanden:
|
|
|
Resultat:
|
|
|
Diskussion om resultaten. |
|
|
Slutsatser. |
4. HÄNVISNINGAR
Denna metod motsvarar OECD TG 406.
Tillägg
TABELL
Graderingsskala enligt Magnusson/Kligman för utvärdering av reaktioner på provokationskompresstest
0 = ingen synbar förändring
1 = lätt eller måttlig hudrodnad
2 = måttlig till stark hudrodnad
3 = intensiv hudrodnad och svullnad
B.7 28 DAGARS STUDIE AV TOXICITET MED UPPREPAD ORAL DOSERING
1. METOD
1.1 INLEDNING
Se Allmän inledning, del B.
1.2 DEFINITIONER
Se Allmän inledning, del B.
1.3 PRINCIP FÖR TESTMETODEN
Testsubstansen tillförs dagligen oralt i gradvist ökande doser till flera grupper försöksdjur, en dosnivå per grupp, under 28 dagar. Under doseringsperioden observeras djuren noggrant varje dag efter tecken på toxicitet. Djur som dör eller avlivas under försöket obduceras och vid försökets slut avlivas de överlevande djuren och obduceras.
Denna metod lägger större vikt vid de neurologiska effekterna som särskild sluteffekt samt behovet av noggranna kliniska observationer av djuren i syfte att erhålla mesta möjliga information. Metoden ska identifiera kemikalier med eventuell neurotoxisk verkan vilket kan motivera mer ingående undersökningar av den aspekten. Därutöver kan metoden ge indikationer på immunologiska och reproduktionstoxiska effekter.
1.4 BESKRIVNING AV TESTMETODEN
1.4.1 Förberedelser
Friska unga vuxna djur väljs slumpvis ut till kontroll- och dosgrupperna. Burarna bör ordnas så att eventuella effekter på grund av deras placering minimeras. Djuren identifieras individuellt och hålls i sina burar minst fem dagar innan försöket påbörjas för att de ska acklimatisera sig till laboratoriemiljön.
Testsubstansen tillförs via sondmatning, foder eller vatten. Den metod som väljs för oral tillförsel beror på syftet med studien och ämnets fysikaliskt–kemiska egenskaper.
Testsubstansen löses vid behov i lämplig vehikel. Om möjligt bör i första hand vattenlösning eller vattensuspension övervägas, följt av oljelösning eller oljeemulsion (t.ex. majsolja) samt därefter andra vehiklar. I fråga om andra vehiklar än vatten ska vehikelns toxiska egenskaper vara kända. Testsubstansens stabilitet i vehikeln bör fastställas.
1.4.2 Försöksförhållanden
1.4.2.1 Försöksdjur
Helst ska råttor användas även om andra gnagare kan användas. Vanligen använda laboratoriestammar av unga friska vuxna djur bör användas. Honorna ska inte ha fått ungar och inte vara dräktiga. Doseringen bör inledas snarast möjligt efter avvänjningen och under alla förhållanden innan djuren är nio veckor gamla.
Då undersökningen påbörjas bör variationen i vikt mellan de använda djuren vara minimal och inte överstiga ± 20 % av vardera könets genomsnittsvikt.
Om upprepad oralstudie utförs som inledning till en långtidsstudie ska helst djur från samma stam och med samma ursprung användas i båda försöken.
1.4.2.2 Antal och kön
Minst 10 djur (fem honor och fem hannar) bör användas för varje dosnivå. Om avlivningar är inplanerade under försökets gång bör antalet djur ökas med det antal som avlivas innan försöket slutförts.
Därutöver bör en satellitgrupp på 10 djur (fem av vardera könet) behandlas med den höga dosnivån under 28 dagar och observeras angående reversibla eller kvarstående effekter eller fördröjt uppträdande av toxiska effekter under 14 dagar efter det att försöket avslutats. En satellitgrupp på 10 kontrolldjur (fem av vardera könet) ska också användas.
1.4.2.3 Dosnivåer
I allmänhet bör minst tre dosgrupper och en kontrollgrupp användas. Med undantag för behandling med testsubstansen bör djuren i kontrollgruppen hanteras på exakt samma sätt som djuren i dosgrupperna. Om en vehikel används vid tillförsel av testsubstansen bör kontrollgruppen tillföras vehikeln i den största dos som används.
Om det, efter utvärdering av andra uppgifter, inte förväntas några effekter vid en dos på 1 000 mg/kg kroppsvikt får ett ’limit-test’ utföras. Om det inte finns tillgång till relevant underlag får en pilotstudie genomföras till hjälp för att bestämma doser som ska användas.
Dosnivåerna bör väljas med hänsyn till eventuellt befintliga toxicitets- och toxikokinetiska uppgifter som finns tillgängliga för testsubstansen eller närbesläktade ämnen. Den högsta dosnivån bör väljas i syfte att inducera toxiska effekter utan att orsaka dödsfall eller allvarligt lidande. Därefter bör dosnivåerna väljas i fallande skala för att påvisa en dosrelaterad reaktion samt vid den lägsta dosnivån inga påvisbara skadliga effekter (NOAEL. – no-observed-adverse effects). Två- till fyrdubbla dosintervall är ofta optimala för att fastställa de fallande dosnivåerna; tillägg av en fjärde testgrupp är att föredra framför användning av mycket breda intervall (t.ex. över faktor 10) mellan dosnivåerna.
För substanser som tillförs via foder eller dricksvatten är det viktigt att försäkra sig om att de kvantiteter av testsubstansen som används inte stör det normala näringsupptaget eller den normala vattenbalansen. När testsubstansen tillförs via fodret kan man använda antingen en konstant koncentration (ppm) eller en konstant dosnivå i förhållande till djurens kroppsvikt. Det använda alternativet ska anges. För ett ämne som tillförs genom sondmatning ska dosen ges vid samma tid varje dag och vid behov justeras för att hålla en konstant dosnivå i förhållande till djurens kroppsvikt.
Om försök med upprepad oral dosering används som förstudie till en långtidsstudie ska samma foder användas i båda försöken.
1.4.2.4 ”Limit-test”
Om ett försök med en dosnivå om minst 1 000 mg per kg kroppsvikt och dag eller, vid tillförsel i föda eller dricksvatten, en motsvarande andel i födan eller dricksvattnet (baserad på fastställd kroppsvikt) och med användning av det tillvägagångssätt som beskrivits för denna studie, inte ger några observerbara toxiska effekter och om toxicitet inte förväntas på grundval av uppgifter från strukturellt närbesläktade ämnen, kan en fullständig undersökning med tre dosnivåer anses överflödig. Ett ”limit-test” används utom när exponering på människor visar att en högre dosnivå behövs.
1.4.2.5 Observationsperiod
Observationsperioden bör vara 28 dagar. Djur i en satellitgrupp avsedd för fortsatta observationer bör hållas i minst ytterligare 14 dagar utan behandling för att fördröjda eller kvarstående effekter eller återhämtning från toxiska effekter ska kunna upptäckas.
1.4.3 Förfarande
Djuren doseras med testsubstansen dagligen sju dagar per vecka under en period på 28 dagar. Dosering fem dagar per vecka måste motiveras. Om testsubstansen tillförs genom sondmatning ska ämnet ges till djuren i en enda dos via magsond eller lämplig intubationskanyl. Den största mängd vätska som kan tillföras vid ett och samma tillfälle beror på försöksdjurets storlek. Volymen bör inte överstiga 1 ml/100 g kroppsvikt utom i fråga om vattenlösningar då 2 ml/100 g kroppsvikt får användas. Med undantag för irriterande eller frätande ämnen som normalt visar kraftigare effekt med högre koncentration bör variationer i testvolymen minimeras genom justering av koncentrationen för att säkerställa en konstant volym på alla dosnivåer.
1.4.3.1 Generella observationer
Generella kliniska observationer bör göras minst en gång per dag, helst vid samma tidpunkt(er) varje dag och med hänsyn till de maximalt förväntade effekterna efter dosering. Djurens hälsostatus bör noteras. Minst två gånger per dag kontrolleras om djur är döende eller döda. Döende djur och djur som lider allvarligt bör avlägsnas, avlivas skonsamt och obduceras.
Noggranna kliniska observationer görs en gång av samtliga djur före den första exponeringen (för att möjliggöra jämförelser på samma objekt) och därefter minst en gång i veckan. Dessa observationer bör göras utanför buren på en ”standardarena” och helst vid samma tidpunkt varje gång. Observationerna bör noteras noggrant, helst genom ett poängsystem som definieras utförligt av testlaboratoriet. Minimala variationer i försöksbetingelserna bör eftersträvas, och observationerna ska helst utföras av någon som inte känner till behandlingen. De tecken som noteras bör omfatta, men inte begränsas till, förändringar av hud, hårrem, ögon och slemhinnor, uppträdande av sekret och utsöndringar samt autonom aktivitet (tårflöde, upprest hårrem, pupillstorlek, ovanligt andningsmönster). Förändringar i gång, hållning och reaktion på hantering liksom eventuella kramper eller spasmer, stereotypt beteende (t.ex. överdrivet putsande, repetitivt cirklande) eller bisarrt beteende (t.ex. självstympning, baklängesgång) bör också noteras.
Under den fjärde exponeringsveckan bör den sensoriska reaktionen på stimuli av olika slag utföras (t.ex. hörsel-, syn- och djupsensoriska stimuli) liksom bedömning av greppstyrka och motorisk aktivitet. Ytterligare uppgifter om de förfaranden som kan följas finns publicerad (se Allmän inledning, del B).
De funktionsobservationer som utförs under den fjärde exponeringsveckan kan utelämnas om en förstudie genomförs som inledning till ett efterföljande subkroniskt 90-dagarstest. I sådana fall bör funktionsobservationerna inkluderas i den efterföljande undersökningen. Å andra sidan kan tillgång på uppgifter om funktionsobservationer från en förstudie med upprepad dosering öka möjligheterna att välja dosnivåer till en efterföljande subkronisk undersökning.
I undantagsfall får funktionsobservationerna också utelämnas för grupper som annars skulle visa tecken på toxicitet i en sådan utsträckning att det väsentligt skulle påverka utförandet av funktionsförsöket.
1.4.3.2 Kroppsvikt samt foder- och vattenkonsumtion
Alla djuren bör vägas minst en gång i veckan. Mätning av foder- och vattenkonsumtion bör göras minst en gång per vecka. Om testsubstansen tillförs via dricksvattnet bör även vattenkonsumtionen mätas minst en gång i veckan.
1.4.3.3 Hematologi
Följande hematologiska undersökningar bör göras då testperioden avslutas: hematokrit, hemoglobinkoncentration, räkning av röda blodkroppar, total- och differentialräkning av vita blodkroppar, blodplättsräkning samt ett mått på koaguleringstid och koaguleringsförmåga.
Blodprov bör tas från ett angivet ställe just före eller som ett led i avlivningsförfarandet och förvaras under lämpliga förhållanden.
1.4.3.4 Klinisk kemi
Kliniskt kemiska bestämningar för att undersöka större toxiska effekter i vävnader, särskilt effekter på lever och njurar, bör utföras på de blodprov som tas från samtliga djur strax före eller som ett led i avlivningsförfarandet (med undantag för de djur som hittas döende eller avlivas under försökets gång). Djuren bör fasta natten före blodprovstagningen (2). Undersökningar av serum eller plasma ska omfatta natrium, kalium, glukos, totalkolesterol, urinämne, kreatinin, totalprotein och albumin. minst två enzymer som indikerar effekter på leverceller (t.ex. alanin-aminotransferas, aspartat-aminotransferas, alkaliskt fosfatas, gamma-glutamyl-transpeptidas och sorbitol-dehydrogenas). Mätningar av ytterligare enzymer (från lever eller av annat ursprung) och gallsyror kan under vissa förhållanden ge användbar information.
Följande urinanalysbestämningar kan utföras frivilligt under sista veckan av försöket med samling av urin på fasta tider: utseende, volym, osmolalitet eller specifik vikt, pH, protein, glukos och blod eller blodceller.
Dessutom bör tester för att undersöka serummarkörer av allmänna vävnadsskador övervägas. Andra bestämningar som bör utföras, om testsubstansens kända egenskaper kan eller misstänks påverka närbesläktade metaboliska profiler, omfattar kalcium, fosfat, fastevärden av triglycerider, specifika hormoner, methemoglobin och kolinesteras. Dessa behöver identifieras för ämnen inom vissa grupper eller från fall till fall.
Generellt krävs flexibla tillvägagångssätt beroende på djurart och på den observerade eller förväntade effekten av ett givet ämne.
Om historiska basdata är otillräckliga bör hänsyn tas till hematologiska och kliniskt kemiska variabler innan doseringen påbörjas.
1.4.3.5 Obduktion
Samtliga djur i undersökningen bör underkastas en fullständig obduktion som omfattar yttre granskning av kroppen, alla kroppsöppningar liksom kranium, bröst- och bukhåla samt deras innehåll. Lever, njurar, binjurar, testiklar, bitestiklar, bräss, mjälte, hjärna och hjärta från samtliga djur ska putsas från angränsande vävnad och deras våtvikt vägas så snart möjligt efter dissekering för att undvika uttorkning.
Följande vävnader och organ bör bevaras i det mest lämpade mediet för histopatologiska undersökningar: alla stora skador, hjärnan (representativa regioner inbegripet storhjärna, lillhjärna och hjärnbrygga), ryggrad, magsäck, tunntarm och tjocktarm (inklusive Peyer's patches), lever, njurar, binjurar, mjälte, hjärta, bräss, sköldkörtel, luftstrupe och lungor (konserverade genom inflation med fixermedel och därefter immersion), könskörtlar, accessoriska könsorgan (t.ex. livmoder, prostata), urinblåsa, lymfkörtlar (helst en lymfkörtel som omfattar tillförselvägen och en annan långt från tillförselvägen för att täcka systematiska effekter), perifer nerv (ischias- eller tibialnerven), helst nära muskeln, och ett snitt av ryggmärgen (alternativt färskt benmärgsaspirat). Kliniska eller andra fynd indikerar att ytterligare vävnader behöver undersökas. Dessutom bör de organ bevaras som på grund av testsubstansens kända egenskaper förmodas vara målorgan.
1.4.3.6 Histopatologisk undersökning
En fullständig histopatologisk undersökning bör utföras på bevarade organ och vävnader från samtliga djur i kontroll- och högdosgrupperna. Undersökningarna bör utsträckas till djur i alla andra dosgrupper om behandlingsrelaterade förändringar observeras i de högre dosgrupperna.
Alla stora skador ska undersökas.
Om en satellitgrupp används bör histopatologisk undersökning utföras på de vävnader och organ som visat effekter i dosgrupperna.
2. DATA
Individuella data bör tillhandahållas. Därutöver bör alla data sammanfattas i en tabell som anger antalet djur i varje testgrupp vid försökets början, antalet djur som hittats döda under försöket eller som avlivats av humanitära skäl samt tidpunkten för avlivning, antalet djur som visar tecken på toxicitet, en beskrivning av de tecken på toxicitet som observerats, inbegripet tidpunkten då de först visar sig, duration och hur allvarliga de toxiska effekterna är, antalet djur som visar skador, typ av skada och procentandelen djur för varje typ av skada.
Om möjligt bör numeriska resultat utvärderas genom en lämplig och allmänt godtagbar statistisk metod. De statistiska metoderna bör väljas vid utformningen av undersökningen.
3. RAPPORTERING
FÖRSÖKSRAPPORT
Försöksrapporten ska om möjligt innehålla följande uppgifter:
|
|
Försöksdjur:
|
|
|
Försöksförhållanden:
|
|
|
Resultat:
|
|
|
Diskussion om resultaten. |
|
|
Slutsatser. |
4. HÄNVISNINGAR
Denna metod motsvarar OECD TG 407.
B.8 TOXICITET VID UPPREPAD DOSERING (28 DAGAR, INANDNING)
1. METOD
1.1 INLEDNING
Det är fördelaktigt att ha förhandsupplysningar om ämnets partikelstorleksfördelning, ångtryck, smältpunkt, kokpunkt, flampunkt och eventuellt explosiva egenskaper.
Se också allmän inledning till del B (A).
1.2 DEFINITION
Se allmän inledning till del B (B).
1.3 REFERENSÄMNEN
Inga.
1.4 TESTMETODSPRINCIP
Flera grupper av försöksdjur exponeras dagligen i 28 dagar för testämnet i graderade koncentrationer, en bestämd koncentration per grupp. Om vehikel används för att underlätta framtagning av lämplig koncentration av testämnet i luften, ska en kontrollgrupp som utsätts för vehikeln användas. Under administrationsperioden observeras djuren dagligen för tecken på toxicitet. Djur som dör under testet ska obduceras och i slutet av testet avlivas och obduceras överlevande djur.
1.5 KVALITETSKRITERIER
Inga.
1.6 METODBESKRIVNING
1.6.1 Förberedelser
I minst fem dagar före testet ska djuren hållas under testbetingelser med hänsyn till miljö och utfodring. Före testet fördelas friska könsmogna djur slumpmässigt i erforderligt antal grupper. Vid behov ska en lämplig vehikel tillsättas till testämnet för att underlätta framtagning av lämplig koncentration av ämnet i luften. Om vehikel eller annan tillsats används för att underlätta dosering ska det vara fastställt att den inte framkallar några toxiska effekter. I lämpliga fall kan tidigare insamlade data användas.
1.6.2 Testbetingelser
1.6.2.1 Försöksdjur
Om det inte finns kontraindikationer är råttan förstahandsvalet. De vanligen nyttjade laboratoriestammarna med unga friska djur ska användas.
Vid testets början får vikten hos de aktuella djuren inte avvika mer än ± 20 % från en rimlig genomsnittsvikt.
1.6.2.2 Antal och kön
Minst 10 djur (fem honor och fem hanar) ska ingå i varje grupp. Honorna får inte ha fött ungar och får inte vara dräktiga. Vid planenligt avlivande av djur under testet ska det ursprungliga antalet djur ökas med det antal djur som planenligt ska avlivas innan studien slutförs. Dessutom kan en satellitgrupp bestående av 10 djur (fem av vardera könet) tillföras den högsta koncentrationen i 28 dagar och därefter observeras i 14 dagar efter testet för reversibilitet, persistens eller fördröjt uppträdande av toxiska effekter. Även en satellitgrupp bestående av 10 kontrolldjur (fem av vardera könet) ska användas.
1.6.2.3 Exponeringskoncentration
Till testet används minst tre koncentrationsnivåer och en kontrollgrupp eller en kontrollgrupp som tillförs vehikeln (i en koncentration som svarar mot den högsta koncentrationen av vehikeln). Djuren i kontrollgruppen ska behandlas på samma sätt som djuren i behandlingsgruppen, bortsett från att de inte ska tillföras testämnet. Den högsta koncentrationsnivån ska ge toxiska effekter men inga, eller få, dödsfall. Den lägsta koncentrationsnivån ska inte ge några tecken på toxicitet. Om det finns en användbar uppskattning av människans exponering för ämnet ska den lägsta nivån vara högre än denna. Helst ska medelkoncentrationen ge minsta möjliga observerbara toxiska effekter. Om fler än en medelkoncentration används ska koncentrationsnivåerna väljas på ett sådant sätt att en gradering av de toxiska effekterna erhålls. I grupperna som utsätts för låg- och medelkoncentrationer och i kontrollgruppen ska antalet dödsfall hållas på en låg nivå för att utvärderingen av resultaten ska vara meningsfylld.
1.6.2.4 Exponeringstid
Durationen av den dagliga exponeringen ska vara sex timmar men andra perioder kan behövas för att uppfylla särskilda krav.
1.6.2.5 Utrustning
Djuren ska testas i inhalationsapparatur som är så utformad att den upprätthåller en dynamisk luftcirkulation med minst 12 luftväxlingar per timme för att säkerställa en tillräcklig syrehalt och en jämnt fördelad exponeringsluft. Om exponeringskammare används ska den vara så utformad att trängseln bland försöksdjuren minimeras och exponeringen för testämnet genom inandning maximeras. Som en allmän regel för att säkerställa en stabil atmosfär i kammaren, ska försöksdjurens totala ’volym’ inte överstiga 5 % av exponeringskammarens volym. Exponeringen kan göras genom kammare för mun–näsa, enbart huvudet eller hela försöksdjurets kropp; genom användning av de två första metoderna kan man minska möjligheten att testämnet upptas via andra vägar.
1.6.2.6 Observationstid
Under hela testet och återhämtningsperioden ska försöksdjuren observeras dagligen för tecken på toxicitet. Tidpunkten för dödsfall och tidpunkten då tecken på toxicitet uppträder och försvinner ska registreras.
1.6.3 Förfarande
Djuren exponeras för testämnet dagligen, fem à sju dagar i veckan i 28 dagar. Djur i en eventuell satellitgrupp som planerats för uppföljning ska observeras i ytterligare 14 dagar utan någon behandling så att det kan fastställas om de toxiska effekterna försvinner eller om de håller i sig. Testtemperaturen ska hållas vid 22 ± 3 oC.
Under ideala betingelser ska den relativa luftfuktigheten hållas mellan 30 och 70 %, men i vissa fall (t.ex. test av aerosoler) kan detta vara praktiskt ogenomförbart. Ett svagt undertryck i kammaren (≤ 5 mm vatten) förhindrar att testämnet läcker ut i det omgivande rummet. Foder och vatten ska undanhållas djuren under exponeringen.
Till testet används ett dynamiskt inhalationssystem med lämpligt analytisk kontrollsystem för att säkerställa exponeringskoncentrationen. Ett provtest rekommenderas för att fastställa lämpliga exponeringskoncentrationer. Luftcirkulationen ska justeras för att säkerställa homogena betingelser i hela exponeringskammaren. Systemet ska vara av en sådan beskaffenhet att stabila exponeringsbetingelser uppnås så snart som möjligt.
Mätning eller kontroll ska göras av:
|
a) |
Lufthastigheten (fortlöpande). |
|
b) |
Den faktiska koncentrationen av testämnet mätt i respirationszonen. Under den dagliga exponeringsperioden ska koncentrationen inte avvika mer än ± 15 % från den genomsnittliga koncentrationen. En sådan kontrollnivå kan emellertid vara omöjlig att uppnå vid test med vissa aerosoler, och i sådana fall kan större avvikelser accepteras. Under hela testet ska de dagliga koncentrationerna hållas så konstanta som möjligt. För aerosoler analyseras partikelstorleken minst en gång i veckan per försöksgrupp. |
|
c) |
Temperatur och luftfuktighet, helst fortlöpande. |
Under och efter exponering görs systematiska observationer som registreras för varje enskilt djur. Alla djur ska observeras dagligen och tecken på toxicitet registreras tillsammans med tidpunkten då de uppträdde samt graden och varaktigheten av toxiciteten. Observationer ska omfatta hud- och pälsförändringar, ögon och slemhinnor och även respirations- och cirkulationssystem, autonoma och centrala nervsystemet samt kroppsmotorik och beteendemönster. Djurens ska vägas varje vecka och vidare rekommenderas att foderförbrukningen mäts per vecka. Det är nödvändigt att observera djuren regelbundet för att säkerställa att så få djur som möjligt faller bort från studien till följd av kannibalism, autolys av vävnader eller förväxling. I slutet av testet avlivas och obduceras alla överlevande djur utom djuren i satellitgruppen. Döende djur och djur som visar symptom på stark smärta eller lidande avlägsnas så snart de iakttas och avlivas sedan på humant sätt samt obduceras.
I slutet av testet ska alla djur inklusive kontrollgruppen genomgå följande undersökning:
|
i) |
Hematologi omfattande minst hematokrit, hemoglobinkoncentration, erytrocyträkning, total och differential räkning av leukocyter samt mätning av koaguleringsförmågan. |
|
ii) |
Klinisk biokemisk analys av blodet omfattande minst en lever- och njurfunktionsparameter: alanin-aminotransferas (tidigare kallat glutaminsyra-pyrodruvsyra-transaminas, GPT), serum-aspartat-aminotransferas (tidigare kallat glutaminsyra-oxalättiksyra-transaminas, GOT), och urinkväve, äggviteämnen, blod-kreatinin, totalt bilirubin och totalt serumprotein. |
Andra bestämningar som kan vara nödvändiga för en adekvat toxikologisk utvärdering är kalcium, fosfor, klorid, natrium, kalium, glukos efter fasta, lipidanalys, hormoner, syra/basbalansen, methemoglobin och kolinesterasaktivitet.
Ytterligare kliniska biokemiska analyser kan vid behov utföras för att vidare utreda de observerade effekterna.
1.6.3.1 Makroskopisk undersökning
Alla försöksdjur ska genomgå en fullständig makroskopisk undersökning. Åtminstone lever, njurar, binjurar, lungor och testiklar ska vägas våta så snart som möjligt efter dissektion för att undvika uttorkning. Organ och vävnader (andningsvägarna, lever, njure, mjälte, testiklar, binjurar, hjärta och andra organ som företer allvarliga skador eller storleksförändringar) ska förvaras i lämpligt medium för eventuell senare histopatologisk undersökning. Näs–svalgrummet och lungor ska tas ut hela och oskadade, vägas och behandlas med lämpligt fixativ för att säkerställa att lungstrukturen bibehålls.
1.6.3.2 Histopatologisk undersökning
Histologisk undersökning ska göras av bevarade organ och vävnader från högkoncentrationsgruppen och kontrollgruppen/grupperna. Organ och vävnader, hämtade från gruppen som fått den högsta koncentrationen, som uppvisar defekter som kan tillskrivas testämnet, ska undersökas i alla grupper som fått lägre dos. Djur i en eventuell satellitgrupp ska genomgå histologisk undersökning, särskilt de organ och vävnader som har utvisat effekter i de övriga behandlingsgrupperna.
2. DATA
Data ska ställas upp i tabellform och för varje behandlingsgrupp anges antal djur i början av testet och antal djur som företer varje typ av skada.
Alla observerade resultat ska utvärderas med lämplig statistisk metod. Alla vedertagna statistiska metoder kan användas.
3. RAPPORTERING
3.1 TESTRAPPORT
Testrapporten ska om möjligt innehålla följande uppgifter:
|
— |
Djurart, stam, härkomst, miljöbetingelser, foder osv. |
|
— |
Testbetingelser: Beskrivning av exponeringsapparaturen, inklusive utformning, typ, mått, luftkälla, system för att generera partiklar och aerosoler, metod för konditionering av luften, behandling av utpumpad luft och metoden att hålla djuren i en exponeringskammare, om sådan används. Utrustningen för att mäta temperatur, luftfuktighet och i förekommande fall aerosolkoncentrationers eller partikelstorlekars stabilitet, ska beskrivas. Exponeringsdata: Dessa ska ställas upp i tabellform med angivande av medelvärden och ett spridningsmått (t.ex. standardavvikelse) och ska om möjligt omfatta:
|
|
— |
Uppgifter om toxisk reaktion uppdelade efter kön och koncentration. |
|
— |
Tidpunkten för dödsfall under testet eller överlevnad till slutet av testet. |
|
— |
Beskrivning av toxiska och andra effekter. |
|
— |
Tidpunkten för observation av varje abnormt tecken och dess senare utveckling. |
|
— |
Uppgifter om foder och kroppsvikt. |
|
— |
Gjorda hematologiska undersökningar och alla resultat. |
|
— |
Gjorda kliniska biokemiska analyser och alla resultat. |
|
— |
Obduktionsfynd. |
|
— |
Detaljerad beskrivning av alla histopatologiska fynd. |
|
— |
Statistisk behandling av resultaten när detta är möjligt. |
|
— |
Diskussion om resultaten. |
|
— |
Tolkning av resultaten. |
3.2 UTVÄRDERING OCH TOLKNING
Se allmän inledning till del B (D).
4. HÄNVISNINGAR
Se allmän inledning till del B (E).
B.9 TOXICITET VID UPPREPAD DOSERING (28 DAGAR, DERMALT)
1. METOD
1.1 INLEDNING
Se allmän inledning till del B (A).
1.2 DEFINITIONER
Se allmän inledning till del B (B).
1.3 REFERENSÄMNEN
Inga.
1.4 TESTMETODSPRINCIP
Testämnet appliceras dagligen i graderade doser på huden till flera grupper av försöksdjur, varvid varje grupp ges en bestämd dos i 28 dagar. Under appliceringsperioden observeras djuren dagligen för tecken på toxicitet. Djur som dör under testet obduceras, och i slutet av testet avlivas och obduceras de överlevande djuren.
1.5 KVALITETSKRITERIER
Inga.
1.6 METODBESKRIVNING
1.6.1 Förberedelser
I minst fem dagar före testet ska djuren hållas under testbetingelser med hänsyn till miljö och utfodring. Före testet fördelas friska unga djur slumpmässigt i behandlings- och kontrollgrupper. Omedelbart innan testet inleds ska päls klippas bort från försöksdjurens rygg. Om pälsen rakas bort ska detta göras ungefär 24 timmar före testet. Klippning eller rakning behöver vanligtvis upprepas ungefär en gång i veckan. När pälsen klipps eller rakas bort ska försiktighet iakttas så att huden inte skadas. Minst 10 % av kroppsytan ska göras klar för applicering av testämnet. Vid fastställande av vilket och hur stort område som ska förberedas ska hänsyn tas till djurets kroppsvikt. Vid test av fasta ämnen, som får pulvriseras om det är lämpligt, ska testämnet fuktas tillräckligt med vatten eller, vid behov med lämplig vehikel, för att säkerställa god kontakt med huden. Flytande testämnen används vanligen outspädda. Testämnet appliceras dagligen fem till sju dagar i veckan.
1.6.2 Testbetingelser
1.6.2.1 Försöksdjur
Könsmogna råttor, kaniner eller marsvin får användas. Andra arter kan användas men i så fall ska detta motiveras.
Vid testets början får vikten hos de aktuella djuren inte avvika mer än ± 20 % från en rimlig genomsnittsvikt.
1.6.2.2 Antal och kön
Minst 10 djur (fem honor och fem hanar) med frisk hud ska användas på varje dosnivå. Honorna får inte ha fött ungar och får inte vara dräktiga. Vid planenligt avlivande av djur under testet ska det ursprungliga antalet djur ökas med det antal djur som planenligt ska avlivas innan studien slutförs. Dessutom kan en satellitgrupp bestående av 10 djur (fem av vardera könet) tillföras den högsta dosnivån i 28 dagar och därefter observeras i 14 dagar efter testet för reversibilitet, persistens eller fördröjt uppträdande av toxiska effekter. Även en satellitgrupp bestående av 10 kontrolldjur (fem av vardera könet) ska användas.
1.6.2.3 Dosnivåer
Till testet ska användas minst tre dosnivåer samt en kontrollgrupp eller en vehikelkontroll, om vehikel används. Testämnet ska appliceras vid samma tid varje dag och dosnivåerna ska justeras med jämna mellanrum (varje eller varannan vecka) så att dosnivån hålls konstant i förhållande till djurens kroppsvikt. Djuren i kontrollgruppen ska behandlas på samma sätt som djuren i behandlingsgruppen, bortsett från att de inte ska tillföras testämnet. Om vehikel används för att underlätta dosering ska kontrollgruppen tillföras vehikeln på samma sätt som behandlingsgrupperna. Vidare ska vehikeln tillföras kontrollgruppen i samma mängd som till de försöksdjur i gruppen som ges den högsta dosnivån. Den högsta dosnivån ska ge toxiska effekter men inga eller få dödsfall. Den lägsta dosnivån ska inte ge några tecken på toxicitet. Om det finns en användbar uppskattning av människans exponering för ämnet ska den lägsta nivån vara högre än denna. Helst ska medeldosen ge minsta möjliga observerbara toxiska effekter. Om fler än en medeldos används ska dosnivåerna väljas på ett sådant sätt att en gradering av de toxiska effekterna erhålls. I grupperna som utsätts för låg- och medeldoser och i kontrollgruppen ska antalet dödsfall hållas på en låg nivå för att utvärderingen av resultaten ska vara meningsfylld.
Om appliceringen av testämnet ger allvarlig hudirritation ska koncentrationerna sänkas, vilket kan medföra att andra toxiska effekter minskar eller uteblir på högsta dosnivån. Om huden har skadats allvarligt kan det bli nödvändigt att avsluta det pågående testet och påbörja en ny med lägre koncentrationer.
1.6.2.4 Gränstest
Om det vid ett förberedande test fastställs att en dosnivå på 1 000 mg/kg, eller en högre dosnivå om detta motiveras av kunskap om människans möjliga exponering, inte ger några toxiska effekter kan ytterligare test anses vara överflödiga.
1.6.2.5 Observationsperiod
Försöksdjuren ska observeras dagligen för tecken på toxicitet. Tidpunkten för dödsfall och tidpunkten då tecken på toxicitet uppträder ska registreras.
1.6.3 Förfarande
Djuren ska hållas i var sin bur. Den ideala doseringen är att djuren utsätts för testämnet sju dagar i veckan i 28 dagar. Djur i en eventuell satellitgrupp som planerats för uppföljning ska observeras i ytterligare 14 dagar utan tillförsel av testämnet så att det kan fastställas om de toxiska effekterna försvinner eller om de håller i sig. Exponeringstiden ska vara minst sex timmar per dag.
Testämnet ska appliceras jämnt över ett område som motsvarar ungefär 10 % av den totala kroppsytan. Med starkt toxiska ämnen kan testområdet vara mindre men så stor del av området som möjligt ska täckas med ett så tunt och jämnt fördelat lager som möjligt.
Under exponeringen ska testämnet hållas i kontakt med huden med en porös kompress och icke hudirriterande tejp. Testområdet ska vidare täckas på lämpligt sätt så att gaskompressen och testämnet hålls på plats och djuren inte kan inta testämnet. För att förhindra intag av testämnet kan rörelsehindrande anordningar användas men total immobilisering rekommenderas inte. Alternativt kan en ’skyddskrage’ användas.
I slutet av exponeringsperioden ska kvarvarande testämne avlägsnas, om så är lämpligt med användning av vatten eller någon annan passande metod att rengöra huden.
Alla djuren ska observeras dagligen och tecken på toxicitet ska registreras tillsammans med tidpunkten då de uppträdde samt graden och varaktigheten av toxiciteten. Observationer ska omfatta hud- och pälsförändringar, ögon och slemhinnor och även respirations- och cirkulationssystem, autonoma och centrala nervsystemet samt kroppsmotorik och beteendemönster. Djurens ska vägas varje vecka och vidare rekommenderas att foderförbrukningen mäts per vecka. Det är nödvändigt att observera djuren regelbundet för att säkerställa att så få djur som möjligt faller bort från studien till följd av kannibalism, autolys av vävnader eller förväxling. I slutet av testet avlivas och obduceras alla överlevande djur utom djuren i satellitgruppen. Djur som dör under testet obduceras, och i slutet av testet avlivas och obduceras de överlevande djuren.
I slutet av testet ska alla djur inklusive kontrollgruppen genomgå följande undersökning:
|
1. |
Hematologi omfattande minst hematokrit, hemoglobinkoncentration, erytrocyträkning, total och differential räkning av leukocyter samt mätning av koaguleringsförmågan. |
|
2. |
Klinisk biokemisk analys av blodet omfattande minst en lever- och njurfunktionsparameter: alanin-aminotransferas (tidigare kallat glutaminsyra-pyrodruvsyra-transaminas, GPT), serum-aspartat-aminotransferas (tidigare kallat glutaminsyra-oxalättiksyra-transaminas, GOT), och urinäkväve, äggviteämnen, blod-kreatinin, totalt bilirubin och totalt serumprotein. |
Andra bestämningar som kan vara nödvändiga för en adekvat toxikologisk utvärdering är kalcium, fosfor, klorid, natrium, kalium, glukos efter fasta, lipidanalys, hormoner, syra/basbalansen, metahemoglobin och kolinesterasaktivitet.
Ytterligare kliniska biokemiska analyser kan vid behov utföras för att vidare utreda de observerade effekterna.
1.6.4 Makroskopisk undersökning
Alla försöksdjur ska genomgå en fullständig makroskopisk undersökning. Lever, njurar, binjurar och testiklar ska vägas våta så snart som möjligt efter dissektion för att undvika uttorkning. Organ och vävnader, dvs. obehandlad och behandlad hud, lever, njure, mjälte, testiklar, binjurar, hjärta och målorgan (de organ som företer allvarliga skador eller storleksförändringar) ska förvaras i lämpligt medium för eventuell senare histopatologisk undersökning.
1.6.5 Histopatologisk undersökning
Histologisk undersökning ska göras av bevarade organ och vävnader från högdosgruppen och kontrollgruppen. Organ och vävnader, hämtade från gruppen som fått den högsta dosnivån, som uppvisar defekter som kan tillskrivas testämnet, ska undersökas i alla grupper som fått lägre dos. Djur i satellitgruppen ska genomgå histologisk undersökning, särskilt de organ och vävnader som har utvisat effekter i de övriga behandlingsgrupperna.
2. DATA
Data ska ställas upp i tabellform och för varje behandlingsgrupp anges antal djur i början av testet och antal djur som företer varje typ av skada.
Alla observerade resultat ska utvärderas med lämplig statistisk metod. Alla vedertagna statistiska metoder kan användas.
3. RAPPORTERING
3.1 TESTRAPPORT
Testrapporten ska om möjligt innehålla följande uppgifter:
|
— |
Data om djuren (art, stam, härkomst, miljöbetingelser, foder osv.). |
|
— |
Testbetingelser (inklusive slag av bandage: med eller utan ocklusion). |
|
— |
Dosnivåer (inklusive vehikel, om sådan använts) och koncentrationer. |
|
— |
Uppgifter om toxisk reaktion uppdelade efter kön och dos. |
|
— |
Icke toxisk dos, om sådan har fastställts. |
|
— |
Tidpunkten för dödsfall under testet eller överlevnad till slutet av testet. |
|
— |
Toxiska och andra effekter. |
|
— |
Tidpunkten för observation av varje abnormt tecken och dess senare utveckling |
|
— |
Uppgifter om foder och kroppsvikt. |
|
— |
Hematologiska undersökningar och alla resultat. |
|
— |
Kliniska biokemiska analyser och alla resultat. |
|
— |
Obduktionsfynd. |
|
— |
Detaljerad beskrivning av alla histopatologiska fynd. |
|
— |
Statistisk behandling av resultaten i lämpliga fall. |
|
— |
Diskussion om resultaten. |
|
— |
Tolkning av resultaten. |
3.2 UTVÄRDERING OCH TOLKNING
Se allmän inledning till del B (D).
4. HÄNVISNINGAR
Se allmän inledning till del B (E).
B.10 MUTAGENICITET – TEST IN VITRO AV KROMOSOMAVVIKELSER HOS DÄGGDJUR
1. METOD
Denna metod är en kopia av metoden OECD TG 47 3, Test In Vitro av kromosomavvikelser hos däggdjur (1997).
1.1 INLEDNING
Avsikten med testen av kromosomavvikelser in vitro är att identifiera ämnen som orsakar strukturella kromosomavvikelser hos odlade däggdjursceller (1) (2) (3). Strukturella avvikelser kan vara av två typer, kromosomala eller kromatida. Majoriteten av de kemiska mutagenerna inducerar avvikelser av kromatidtyp, men även avvikelser av kromosomtyp förekommer. En ökning av polyploidin kan indikera att ett kemiskt ämne har potential att inducera numeriska avvikelser. Denna metod är dock inte utformad för att mäta numeriska avvikelser och används inte rutinmässigt för det ändamålet. Kromosommutationer och liknande händelser är orsaken till många genetiska sjukdomar och det finns påtagliga bevis för att kromosommutationer och liknande händelser som ger upphov till förändringar i onkogener och tumörsuppressorgener i somatiska celler är inblandade i cancerinduktion hos människa och hos försöksdjur.
Test av kromosomavvikelser in vitro kan göras med kulturer av etablerade cellinjer, cellstammar eller primära cellkulturer. De celler som används väljs ut på grundval av tillväxtförmåga vid odling, karyotypens stabilitet, antalet kromosomer, kromosomernas mångfald och den spontana frekvensen av kromosomavvikelser.
Tester som utförs in vitro kräver i allmänhet att en exogen källa för metabolisk aktivering används. Detta metaboliska system för aktivering kan inte helt och hållet efterlikna de förhållanden som råder i däggdjuret in vitro. Man bör se till att undvika sådana förhållanden som skulle leda till positiva resultat som inte återspeglar den verkliga mutageniciteten och kan ha sin orsak i pH-förändringar, osmolalitet eller hög cytotoxicitet (4) (5).
Detta test används för att söka efter möjliga mutagener och karcinogener hos däggdjur. Många ämnen som är positiva i detta test är karcinogena hos däggdjur, men det finns ingen perfekt korrelation mellan detta test och karcinogenicitet. Korrelationen är beroende på den kemiska klassen och det finns allt starkare bevis för att det finns karcinogener som inte detekteras med detta test, eftersom det ser ut som om de verkar genom andra mekanismer än direkta DNA-skador.
Se även Allmän inledning, del B.
1.2 DEFINITIONER
avvikelser av kromatidtyp: en strukturell kromosomskada, uttryckt i form av brott på enstaka kromatider eller brott och återförening mellan kromatider.
avvikelser av kromosomtyp: en strukturell kromosomskada, uttryckt som brott, eller brott och återförening, hos båda kromatiderna pä samma ställe.
endoreduplikation: en process där kärnan efter den S-fas under DNA-replikationen inte övergår i mitos utan inleder ännu en S-fas. Resultatet är kromosomer med 4, 8, 16, ... kromatider.
lucka: en akromatisk skada som är mindre än bredden hos en kromatid, och med ett minimum av misspassning hos kromatiderna.
mitotiskt index: den andel celler som är i metafas delat med det totala antalet celler som kan observeras i en cellpopulation; en indikation på graden av tillväxt hos den populationen.
numerisk avvikelse: en förändring av antalet kromosomer jämfört med det normala antalet karakteristika hos de celler som används.
polyploidi: en multipel av det haploida antalet kromosomer (n) som skiljer sig från det diploida antalet (dvs. 3n, 4n och så vidare).
strukturell avvikelse: en förändring i kromosomstrukturen som kan detekteras genom en mikroskopisk undersökning av celldelningens metafasstadium, och som uppträder i form av deletioner och fragment, interna eller externa utbyten.
1.3 TESTMETODENS PRINCIP
Cellodlingar exponeras för testsubstansen både med och utan metabolisk aktivering. Vid förutbestämda intervall, efter det att cellodlingen har exponerats för testsubstansen, utförs behandling med ett ämne som stoppar celldelningen i metafas (t.ex. Colcemid® eller colchicin). Därefter skördas cellerna och färgas in, varpå närvaro av kromosomavvikelser i metafascellerna analyseras mikroskopiskt.
1.4 BESKRIVNING AV TESTMETODEN
1.4.1 Förberedelser
1.4.1.1 Celler
Ett antal olika cellinjer, stammar eller primära cellkulturer, inklusive humanceller, kan användas (t.ex. fibroblaster från kinesisk hamster, perifera blodlymfocyter från människor eller andra däggdjur).
1.4.1.2 Typ av medium och kultur
Lämpliga odlingsmedier och inkubationsförhållanden (odlingskärl, CO2-koncentration, temperatur och luftfuktighet) bör väljas för odlingarna. Etablerade cellinjer och stammar bör rutinmässigt kontrolleras med avseende på stabilitet hos det modala kromosomantalet och frånvaro av kontaminerad mykoplasma. Om kontaminering har skett bör de inte användas. Cellernas normala cellcykeltid och de odlingsförhållanden som används bör vara kända.
1.4.1.3 Förberedelse av kulturer
Etablerade cellinjer och stammar: celler dras upp från stamkulturer, inokuleras i ett odlingsmedium som har en sådan densitet att kulturerna inte kommer att växa samman innan det är dags att skörda, och inkuberas vid 37 oC.
Lymfocyter: helblod, behandlat med en antikoagulant (t.ex. heparin} eller separerade lymfocyter som erhållits från friska individer, tillsätts till odlingsmediet, vilket innehåller en mitogen substans (t.ex. fytohemaglutinin), varpå inkubering sker vid 37 oC.
1.4.1.4 Metabolisk aktivering
Celler bör exponeras för testsubstansen både i närvaro och frånvaro av ett lämpligt metaboliskt system för aktivering. Det mest använda systemet är en kofaktorstödd post-mitokondriell fraktion (S9) beredd ur lever från gnagare vilka behandlats med enzyminducerande ämnen såsom Aroclor 1254 (6) (7) (8) (9) eller en blandning av fenobarbiton och β-naftoflavon (10) (11) (12).
Den postmitokondriella fraktionen används vanligen vid koncentrationer i området 1 till 10 % v/v i det slutliga testmediet. Tillståndet hos ett system för metabolisk aktivering kan bero på den klass av kemikalier som testas. I vissa fall kan det vara lämpligt att använda mer än en koncentration av den post-mitokondriella fraktionen.
Ett antal vidareutvecklingar, inklusive skapandet av genetiskt modifierade cellinjer som uttrycker specifika aktiverande enzymer, kan erbjuda en potential för endogen aktivering. Valet av de cellinjer som ska användas bör vara vetenskapligt underbyggt (t.ex. genom relevansen av cytokrom P450 isoenzym för metabolismen hos testsubstansen).
1.4.1.5 Beredning av testsubstansen
Fasta testsubstanser bör lösas upp i eller blandas med lämpligt lösningsmedel eller vehikel och spädas ut om detta är lämpligt före det att cellerna behandlas. Flytande testsubstanser kan tillsättas direkt till testsystemen och/eller spädas ut innan behandlingen startas. Nygjorda beredningar av testsubstansen bör användas om inte stabilitetsdata påvisar att lagring är acceptabelt.
1.4.2 Försöksbetingelser
1.4.2.1 Lösningsmedel/vehikel
Lösningsmedlet/vehikeln bör inte kunna misstänkas för att reagera kemiskt med testsubstansen och bör vara förenlig med överlevnaden för cellerna samt S9-aktiviteten. Om andra lösningsmedel/vehiklar än gängse brukliga används bör det finnas data som visar att de är kompatibla. Det rekommenderas att vattenbaserade lösningsmedel/vehiklar i första hand bör övervägas närhelst det är möjligt. När substanser som är instabila i vatten testas, bör de organiska lösningsmedel som används vara fria från vatten. Vatten kan avlägsnas genom tillsats av en molekylsikt.
1.4.2.2 Exponeringskoncentrationer
Bland de kriterier som ska övervägas när den högsta koncentrationen ska fastställas är cytotoxicitet, löslighet i testsystemet och förändringar i pH eller osmolalitet.
Cytotoxicitet bör fastställas, såväl med som utan metabolisk aktivering i huvudexperiment, med hjälp av en lämplig indikation på cellernas integritet och tillväxt, såsom graden av sammanväxning, viabilitetsräkning av cellerna, eller ett mitotiskt index. Det kan vara lämpligt att fastställa cytotoxicitet och löslighet i ett förberedande experiment.
Minst tre analyserbara koncentrationer bör användas. Där cytotoxicitet uppträder bör dessa koncentrationer täcka in ett område som sträcker sig från maximal toxicitet ner till liten eller ingen toxicitet. Detta innebär normalt att koncentrationerna bör separeras av minst en faktor mellan 2 och 10. Vid tiden för skörd av cellerna bör den högsta koncentrationen visa en signifikant reduktion i graden av konfluens, cellräkning eller mitotiskt index (alla större än 50 %). Det mitotiska indexet är endast ett indirekt mått på cytotoxiska/cytosta-tiska effekter och är beroende av tiden efter behandlingen. Det mitotiska indexet är dock acceptabelt för suspensionskulturer där andra toxicitetsmätningar kan vara besvärliga och opraktiska. Information om kinetiken för cellcykler, såsom medelgenerationstid (AGT), kan användas som extra information. AGT är dock ett övergripande medelvärde som inte alltid avslöjar existensen av försenade subpopulationer, och även små ökningar av medelgenerationstiden kan associeras med en mycket påtaglig senareläggning av tiden för optimalt utbyte av avvikelser.
För substanser som är relativt icke-cytotoxiska bör den maximala testkoncentrationen vara 5 μl/ml, 5 mg/ml eller 0,01 M, beroende på vilket av dessa värden som är det lägsta.
För relativt olösliga substanser, som inte är toxiska vid koncentrationer som är lägre än koncentrationen där olöslighet inträder, bör den högsta använda dosen vara en koncentration som ligger över löslighetsgränsen i det avslutande odlingsmediet mot slutet av behandlingsperioden. I vissa fall (t.ex. när toxicitet endast uppträder vid koncentrationer som är högre än den lägsta olösliga koncentrationen) är det lämpligt att testa vid mer än en koncentration med synlig fällning. Det kan vara lämpligt att fastställa lösligheten vid början och slutet av behandlingen, eftersom lösligheten kan ändras under det att exponeringen i testsystemet pågår, beroende på närvaro av celler, S9, serum, etc. Löslighet kan detekteras med blotta ögat. Fällningen torde inte störa bestämningen.
1.4.2.3 Negativa och positiva kontroller
Samtidiga positiva och negativa kontroller (lösningsmedel eller vehikel), både med och utan metabolisk aktivering, bör inkluderas i varje experiment. När metabolisk aktivering används, bör den positiva kontrollkemikalien vara av en sådan typ att aktivering krävs för att ge ett mutagent svar.
Positiva kontroller bör utnyttja en känd klastogen vid exponeringsnivåer som förväntas ge en reproducerbar och detekterbar ökning jämfört med bakgrunden och som demonstrerar känsligheten hos testsystemet.
Positiva kontrollkoncentrationer bör väljas på ett sådant sätt att effekterna är tydliga men inte omedelbart avslöjar identiteten hos kodade utstryksglas för läsaren. I tabellen nedan ges några exempel på positiva kontrollsubstanser.
|
Betingelser för metabolisk aktivering |
Substans |
CAS-nummer |
EINECS-nummer |
|
Frånvaro av exogen metabolit Aktivering |
Metylmetansulfonat |
66-27-3 |
200-625-0 |
|
Etylmetansulfonat |
62-50-0 |
200-536-7 |
|
|
Etylnitrosourea |
759-73-9 |
212-072-2 |
|
|
Mitomycin C |
50-07-7 |
200-008-6 |
|
|
4-Nitroquinolin-N-oxid |
56-57-5 |
200-281-1 |
|
|
Närvaro av exogen metabolit Aktivering |
Bens[a]pyren |
50-32-8 |
200-028-5 |
|
Cyklofosfamid Cyklofosfamidmonohydrat |
50-18-0 6055-19-2 |
200-015-4 |
Andra lämpliga positiva kontrollsubstanser kan användas. Användning av kemiska klassrelaterade positiva kontrollkemikalier bör övervägas när sådana finns att tillgå.
Negativa kontroller, bestående av lösningsmedel eller enbart en vehikel i behandlingsmediet, och hanterade på samma sätt som behandlingsodlingarna, bör inkluderas var gång som odlingen skördas. Dessutom bör obehandlade kontroller också användas om det inte finns historiska kontrolldata som påvisar att inga skadliga eller mutagena effekter induceras av det valda lösningsmedlet.
1.4.3 Förfarande
1.4.3.1 Behandling med en testsubstans
Prolifererande celler behandlas med testsubstansen i såväl närvaro som frånvaro av ett system för metabolisk aktivering. Behandling av lymfocyter bör inledas när cirka 48 timmar gått efter den mitogena stimuleringen.
|
1.4.3.2 |
Två kulturer bör normalt användas för varje koncentration, och rekommenderas varmt för negativa kontrollkulturer eller kontrollkulturer för lösningsmedel. Där minimala variationer mellan två kulturer kan påvisas (13) (14) ur historiska data, kan det vara acceptabelt att en enda kultur används för varje koncentration. Gasformiga eller flyktiga substanser bör testas med hjälp av lämpliga metoder, t.ex. test i slutna odlingskärl (15) (16). |
1.4.3.3 Tid för skörd av odlingen
I det första experimentet bör cellerna exponeras för testsubstansen, både med och utan metabolisk aktivering, under 3–6 timmar och prov bör tas ut vid en tid som motsvarar cirka 1,5 gånger den normala cellcykelns längd efter det att behandlingen har påbörjats (12). Om detta protokoll ger negativa resultat både med och utan aktivering, bör ytterligare ett experiment utan aktivering utföras, med kontinuerlig behandling fram till dess att provtagning sker vid en tidpunkt som motsvarar cirka 1,5 gånger den normala cellcyklens längd. Vissa kemikalier kan vara enklare att detektera vid behandlings-/provtagningstider som är längre är 1,5 cykellängder. Negativa resultat med metabolisk aktivering måste bekräftas från fall till fall. I de fall där det inte anses nödvändigt att bekräfta negativa resultat bör detta motiveras.
1.4.3.4 Kromosomberedning
Cellkulturer behandlas vanligtvis med Colcemid® eller colchicin under 1–3 timmar innan skördning sker. Varje cellkultur skördas och behandlas separat för preparationen av kromosomer. Kromosomberedning innefattar hypoton behandling av cellerna, fixering och infärgning.
1.4.3.5 Analys
Alla utstryksglas, inklusive de med positiva och negativa kontroller, bör kodas upp oberoende av varandra innan den mikroskopiska analysen utförs. Eftersom fixeringen ofta ger upphov till brott på en del av de celler som är i metafas, med åtföljande förlust av kromosomer, bör de celler som påträffas därför innehålla ett centromerantal som är ekvivalent med det modala antalet ± 2 för alla celltyper. Åtminstone 200 väl spridda metafaser bör förekomma per koncentration och kontroll, med lika fördelning mellan dubbelproven, om det är tillämpligt. Detta antal kan reduceras när ett stort antal avvikelser observeras.
Även om syftet med testet är att detektera strukturella kromosomavvikelser, är det viktigt att notera polyploidi och endoreduplikation när dessa händelser observeras.
2. DATA
2.1 BEHANDLING AV RESULTATEN
Den experimentella enheten utgörs av cellen, och därför bör den procentandel av cellerna som uppvisar strukturella kromosomavvikelser utvärderas. Olika typer av strukturella kromosomavvikelser bör listas med antal och frekvenser för experimentella kulturer samt kontrollkulturer. Luckor i DNA-strängen noteras separat och rapporteras men inkluderas inte generellt i den totala frekvensen för avvikelser.
Samtidiga mätningar av cytotoxicitet för alla behandlade och negativa kontrollkulturer i huvudexperimenten avseende avvikelser bör även samlas in.
Individuella data för kulturen bör tas fram. Dessutom bör alla data sammanställas i form av en tabell.
2.2 UTVÄRDERING OCH TOLKNING AV RESULTATEN
Det finns ett flertal kriterier som används för att bestämma om ett resultat är positivt, t.ex. en koncentrationsrelaterad ökning eller en reproducerbar ökning av antalet celler med kromosomavvikelser. Man bör i första hand överväga resultatets biologiska relevans. Statistiska metoder kan användas som en hjälp vid utvärderingen av testresultaten (3) (13). Statistisk signifikans bör dock inte vara den enda faktor som bestämmer om resultatet är positivt.
En ökning av antalet polyploida celler kan utgöra en indikation på att testsubstansen har potential att inhibera mitotiska processer och att inducera numeriska kromosomavvikelser. En ökning av antalet celler med endoreduplicerade kromosomer kan utgöra en indikation på att testsubstansen har potential att inhibera cellcykelns förlopp (17) (18).
En testsubstans för vilken resultatet inte uppfyller ovanstående kriterier bedöms såsom icke-mutagen i detta system.
Även om de flesta experiment kommer att ge tydligt positiva eller negativa resultat, så kan i sällsynta fall uppsättningen av data göra det omöjligt att utföra en otvetydig bedömning med avseende på testsubstansens aktivitet. Resultatet kan förbli tvetydigt eller tveksamt, oavsett hur många experiment som utförs.
Positiva resultat från testet av kromosomavvikelser in vitro indikerar att testsubstansen inducerar strukturella kromosomavvikelser hos odlade somatiska däggdjursceller. Negativa resultat indikerar att testsubstansen inte inducerar kromosomavvikelser hos odlade somatiska däggdjursceller under de betingelser som rådde under testet.
3. RAPPORTERING
TESTRAPPORT
Testrapporten måste innehålla följande information:
|
|
Lösningsmedel/vehikel:
|
|
|
Celler:
|
|
|
Försöksbetingelser:
|
|
|
Resultat:
|
|
|
Diskussion av resultaten. |
|
|
Slutsatser. |
4. HÄNVISNINGAR
|
1. |
Evans, H. J. (1976), Cytological Methods for Detecting Chemical Mutagens. In: Chemical mutagens, Principles and Melhods for their Detection, Vol. 4, Hollaender, A. (ed) Plenum Press, New York and London, s. 1–29. |
|
2. |
Ishidate, M. Jr. och Sofuni, T. (1985), The In Vitro Chromosomal Aberration Test Using Chinese Hamster Lung (CHL) Fibroblast Cells in Culture. In: Progress in Mutation Research, Vol. 5, Ashby, J. et al., (eds) Elsevier Science Publishers, Amsterdam-New York-Oxford, s. 427–432. |
|
3. |
Galloway, S. M, Armstrong, M. J., Reuben, C, Colman, S., Brown, B., Cannon, C, Bloom, A. D., Nakamura, K, Ahmed, M., Duk, S., Rimpo, J., Margolin, G.H., Resnick, M. A, Anderson, G. och Zeiger, E. (1978), Chromosome aberration and sister chromatic exchanges in Chinese hamster ovary cells: Evaluation of 108 chemicals. Environs. Molec. Mutagen 10 (suppl.10), s. 1–175. |
|
4. |
Scott, D., Galloway, S. M, Marshall, R. R., Ishidate, M. Jr., Brusick, D., Ashby, J. och Myhr, B. C. (1991), Genotoxicity under Extreme Culture Conditions. A report from ICPEMC Task Group 9. Mutation Res., 257, s. 147–204. |
|
5. |
Morita, T., Nagaki, T., Fukuda, I. och Okumura, K., (1992), Clastogenicity of low pH to Various Cultured Mammalian Cells. Mutation Res., 268, s. 297–305. |
|
6. |
Arnes, B. N., McCann, J. och Yamasaki, E. (1975), Methods for Detecting Carcinogens and Mutagens with the Salmonella/Mammalian Microsome Mutagenicity Test. Mutation Res., 31, s. 347–364. |
|
7. |
Maron, D. M. och Arnes, B. N. (1983), Revised Methods for the Salmonella Mutagenicity Test. Mutation Res., 113, s. 173–215. |
|
8. |
Natarajan. A. T., Tates, A. D., van Buul, P. P. W., Meijers, M. och de Vogel, N. (1976), Cytogenetic Effects of Mutagen/Carcinogens after Activation in a Microsomal System In Vitro, I. Induction of Chromosome Aberrations and Sister Chromatid Exchange by Diethylnitrosamine (DEN) and Dimethylnitrosamine (DMN) in CHO Cells in the Presence of Rat-Liver Microsomes. Mutation Res., 37, s. 83–90. |
|
9. |
Matsuoka, A., Hayashi, M. och Ishidate, M. Jr. (1979), Chromosomal Aberration Tests on 29 Chemicals Combined with S9 Mix In Vitro. Mutation Res., 66, s. 277–290. |
|
10. |
Elliot, B. M., Combes, R. D., Elcombe, C. R., Gatehouse, D. G., Gibson, G. G., Mackay, J. M. och Wolf, R. C. (1992), Report of UK Environmental Mutagen Society Working Party. Alternatives to Aroclor 1254-induced S9 in In Vitro Genotoxicity Assays. Mutagenesis, 7, s. 175–177. |
|
11. |
Matsushima, T., Sawamura, M, Hara, K. och Sugimura, T. (1976), A Safe Substitute for Polychlorinated Biphenyls as an Inducer of Metabolic Activation Systems. In: de Serres, F. J., Fouts, J. R., Bend, J. R. och Philpot, R. M. (eds) In Vitro Metabolic Activation in Mutagenesis Testing, Elsevier, North-Holland, s. 85–88. |
|
12. |
Galloway, S. M., Aardema. M. J., Ishidate, M. Jr., Ivett, J. L., Kirkland, D. J., Morita, T., Mosesso, P, Sofuni, T. (1994), Report from Working Group on in In Vitro Tests for Chromosomal Aberrations. Mutation Res., 312, s. 241–261. |
|
13. |
Richardson, C, Williams, D. A., Allen, J. A., Amphlett, G., Chanter, D. O. och Phillips, B. (1989), Analysis of Data from In Vitro Cytogenetic Assays. In Statistical Evaluation of Mutagenicity Test Data. Kirkland, D. J., (ed) Cambridge University Press, Cambridge, s. 141–154. |
|
14. |
Soper, K. A. och Galloway, S. M. (1994), Replicate Flasks are not Necessary for In Vitro Chromosome Aberration Assays in CHO Cells. Mutation Res., 312, s. 139–149. |
|
15. |
Krahn, D. F, Barsky, F. C. och McCooey, K. T. (1982), CHO/HGPRT Mutation Assay: Evaluation of Gases and Volatile Liquids. In: Tice, R. R., Costa, D. L., Schaich, K. M. (eds). Genotoxic Effects oj Airborne Agents. New York, Plenum, s. 91–103. |
|
16. |
Zamora, P. O., Beson, J. M., Li, A. P. och Brooks, A. L. (1983), Evaluation of an Exposure System Using Cells Grown on Collagen Gels of Detecting Highly Volatile Mutagens in the CHO/HGPRT Mutation Assay. Environmental Mutagenesis, 5, s. 795–801. |
|
17. |
Locke-Huhle, C. (1983), Endoreduplication in Chinese hamster cells during alpha-radiation induced G2 arrest. Mutation Res., 119, s. 403–413. |
|
18. |
Huang, Y., Change, C. och Trosko, J. E. (1983), Aphidicolin-induced endoreduplication in Chinese hamster cells. Cancer Res., 43, s. 1362–1364. |
B.11 MUTAGENICITET – TEST IN VIVO AV KROMOSOMAVVIKELSER I BENMÄRG HOS DÄGGDJUR
1. METOD
Denna metod är en kopia av OECD TG 475, Test av kromosomavvikelser i benmärg hos däggdjur (Mammalian Bone Marrow Chromosome Aberration Test (1997)).
1.1 INLEDNING
Testet in vivo avseende kromosomavvikelser hos däggdjur används för att detektera strukturella kromosomavvikelser inducerade av testsubstansen i benmärgsceller hos djur, vanligen hos gnagare (1) (2) (3) (4). Strukturella kromosomavvikelser kan vara av två typer, kromosomala eller kromatida. En ökning av polyploidin kan indikera att en kemikalie har potential att inducera numeriska avvikelser. För huvuddelen av de kemiska mutagenerna utgörs de inducerade avvikelserna av kromatidtypen, men även avvikelser av kromosomtyp kan förekomma. Kromosommutationer och liknande händelser är orsaken till många genetiska sjukdomar hos människa och det finns omfattande bevis för att kromosommutationer och liknande händelser som ger upphov till förändringar i onkogener samt att tumörsuppressorgener är inblandade i cancer hos människa och i experimentella system.
Gnagare används rutinmässigt i detta test. Benmärg är målvävnaden i detta test, eftersom det är en mycket kärlrik vävnad som innehåller en cellpopulation med en snabb cellcykel, och där cellerna enkelt kan isoleras och bearbetas. Andra arter och målvävnader används inte med denna metod.
Detta test av kromosomavvikelser är särskilt relevant för att fastställa mutagena risker, eftersom det medger överväganden av faktorer in vivo för metabolism, farmakokinetik och processer för DNA-reparationer, även om dessa faktorer kan variera mellan olika arter och vävnader. Ett test in vivo är även användbart för fortsatta undersökningar av en mutagen effekt, med detektion genom tester in vitro.
Om det finns bevis för att testsubstansen, eller en reaktiv metabolit, inte kommer att nå målvävnaden, är det inte lämpligt att använda detta test.
Se även Allmän inledning, del B.
1.2 DEFINITIONER
avvikelser av kromatidtyp: en strukturell kromosomskada, uttryckt i form av brott på enstaka kromatider eller brott och återförening mellan kromatider.
avvikelser av kromosomtyp: en strukturell kromosomskada, uttryckt som brott, eller brott och återförening, hos båda kromatiderna på samma ställe.
endoreduplikation: en process där kärnan efter den S-fas under DNA-replikationen inte övergår i mitos utan inleder ännu en S-fas. Resultatet är kromosomer med 4, 8, 16, ... kromatider.
lucka: en akromatisk skada som är mindre än bredden hos en kromatid, och med ett minimum av misspassning hos kromatiderna.
numerisk avvikelse: en förändring av antalet kromosomer jämfört med det normala antalet karakteristika hos de celler som används.
polyploidi: en multipel av det haploida antalet kromosomer (n) som skiljer sig från det diploida antalet (dvs. 3n, 4n.).
strukturell avvikelse: en förändring i kromosomstrukturen som kan detekteras genom en mikroskopisk undersökning av celldelningens metafasstadium, och som uppträder i form av deletioner och fragment, interna eller externa utbyten.
1.3 TESTMETODENS PRINCIP
Djur exponeras för testsubstansen genom en lämplig metod för exponering och de avlivas på lämpliga tider efter behandlingen. Innan de avlivas behandlas djuren med ett ämne som gör att celler stannar upp i metafas (t.ex. colchicin eller Colcemid®). Kromosomberedningar görs sedan från benmärgscellerna och färgas in, varpå celler i metafas analyseras med avseende på kromosomavvikelser.
1.4 BESKRIVNING AV TESTMETODEN
1.4.1 Förberedelser
1.4.1.1 Val av djurarter
Vanligen används råttor, möss och kinesiska hamstrar, även om vilket lämpligt däggdjur som helst kan utnyttjas. Man bör välja de stammar av unga, friska vuxna djur som vanligen används på laboratorier. När undersökningen ska påbörjas, är det lämpligt att viktskillnaden mellan djuren är så liten som möjligt och den bör inte överstiga ± 20 % av medelvikten för varje kön.
1.4.1.2 Förhållanden vid förvaring och utfodring
De generella förhållanden som anges i den Allmänna inledningen till del B gäller. Man bör dock sträva efter en relativ luftfuktighet på 50–60 %.
1.4.1.3 Förberedelse av djuren
Friska, unga vuxna djur väljs ut slumpartat för placering i kontroll- och behandlingsgrupperna. Burar bör arrangeras på ett sådant sätt att möjliga effekter beroende på placeringen av burarna blir så små som möjligt. Djuren ska ha en unik identifiering. De bör vänjas vid förhållandena i laboratoriet under minst fem dagar.
1.4.1.4 Beredning av doser
Fasta testsubstanser bör lösas upp i eller blandas med lämpliga lösningsmedel eller vehiklar och spädas ut, om så är lämpligt, innan dosen administreras till djuren. Testsubstanser i flytande form kan doseras direkt eller spädas ut före dosering. Nygjorda beredningar av testsubstansen bör användas, om inte stabilitetsdata visar att lagring är acceptabel.
1.4.2 Försöksbetingelser
1.4.2.1 Lösningsmedel/vehikel
Lösningsmedlet/vehikeln bör inte ge upphov till toxiska effekter vid de dosnivåer som används och bör inte kunna misstänkas för att reagera kemiskt med testsubstansen. Om andra lösningsmedel/vehiklar än gängse brukliga används, bör användningen av dem ha stöd från data som visar på deras kompatibilitet. Därhelst det är möjligt rekommenderas att man i första hand överväger att använda vattenbaserade lösningsmedel/vehiklar.
1.4.2.2 Kontroller
Samtidiga positiva och negativa kontroller (lösningsmedel/vehikel) bör ingå för varje kön i varje test. Förutom behandling med testsubstansen, bör djuren i kontrollgrupperna hanteras på ett identiskt sätt i förhållande till djuren i de grupper där behandling skett.
Positiva kontroller bör ge strukturella avvikelser in vivo vid exponeringsnivåer som förväntas ge en detekterbar ökning i förhållande till bakgrunden. Positiva kontrolldoser bör väljas på ett sådant sätt att effekterna är tydliga men att identiteten hos kodade utstryksglas inte omedelbart avslöjas för läsaren. Det är acceptabelt att den positiva kontrollen administreras via en annan tillförselväg än testsubstansen och att provtagning endast sker vid ett enstaka tillfälle. Användning av kemiska klassrelaterade positiva kontrollkemikalier kan dessutom övervägas när dessa finns tillgängliga. I tabellen nedan ges några exempel på positiva kontrollsubstanser.
|
Substans |
CAS-nummer |
EINECS-nummer |
|
Etylmetansulfonat |
62-50-0 |
200-536-7 |
|
Etylnitrosourea |
759-73-9 |
212-072-2 |
|
Mitomycin C |
50-07-7 |
200-008-6 |
|
Cyklofosfamid Cyklofosfamidmonohydrat |
50-18-0 6055-19-2 |
200-015-4 |
|
Trietylenmelamin |
51-18-3 |
200-083-5 |
Negativa kontroller, behandlade enbart med lösningsmedel eller vehikel, och i övrigt behandlade på samma sätt som behandlingsgrupperna, bör inkluderas för varje provtagningstillfälle, om inte acceptabel variabilitet och frekvens hos celler med kromosomavvikelser hos djuren kan erhållas från historiska kontrolldata. Om enstaka provtagning används för negativa kontroller, är den lämpligast tiden det första provtagningstillfället. Dessutom bör obehandlade kontroller även användas om det inte finns historiska eller publicerade kontrolldata som visar att inga skadliga eller mutagena effekter induceras av det valda lösningsmedlet/vehikeln.
1.5 FÖRFARANDE
1.5.1 Försöksdjurens antal och kön
Varje behandlad grupp samt kontrollgruppen måste bestå av minst 5 analyserbara djur av varje kön. Det räcker med att testa enbart det ena könet, om det vid tiden för undersökningens genomförande finns data tillgängliga från undersökningar av samma art och där man har använt sig av samma exponeringssätt, så att det går att visa att det inte finns några substantiella skillnader i toxicitet mellan könen. I de fall där exponering för kemikalier hos människa kan vara könsspecifik, vilket t.ex. kan vara fallet för vissa läkemedel, bör testet utföras med försöksdjur av lämpligt kön.
1.5.2 Behandlingsschema
Testsubstanser administreras med fördel som en enstaka behandling. Testsubstanser kan även administreras i form av en delad dos, två behandlingar under samma dag med högst några timmars intervall, för att underlätta administrering av en stor volym. Andra doseringsscheman bör vara vetenskapligt motiverade.
Prover bör tas vid två separata tillfällen efter behandling under en dag. Det första provtagningstillfället för gnagare ligger på 1,5 gånger den normala cellcykelns längd (den senare är normalt 12–18 timmar) efter behandling. Eftersom den tid som går åt för upptag och metabolism av testsubstansen, likväl som dess effekt på cellcykelns kinetik, kan påverka den optimala tiden för detektion av kromosomavvikelser, så rekommenderas att provtagningen senareläggs till 24 timmar efter det första provtagningstillfället. Om doseringsscheman på mer än en dag används, bör man använda en provtagningstid på 1,5 gånger den normala cellcykelns längd efter den slutliga behandlingen.
Innan djuren avlivas bör de injiceras intraperitonealt med en lämplig dos av ett ämne som stoppar celldelningen i metafas (t.ex. Colcemid® eller colchicin). Prov på försöksdjur bör därefter tas vid lämpliga intervall. För möss är detta intervall ca 3–4 timmar; för kinesiska hamstrar ligger intervallet på ca 4–5 timmar. Celler skördas från benmärgen och analyseras med avseende på kromosomavvikelser.
1.5.3 Dosnivåer
Om det inte finns några lämpliga data tillgängliga och en undersökning därför utförs för att fastställa omfattningen, bör denna undersökning utföras i samma laboratorium, och med samma arter, stammar, kön och behandlingsschema som senare ska användas i huvudundersökningen (5). Vid toxicitet används tre dosnivåer för det första provtagningstillfället. Dessa dosnivåer bör täcka in ett område som sträcker sig från maximal till liten eller ingen toxicitet. Vid senare provtagningstillfällen räcker det med att endast den högsta dosen används. Den högsta dosen definieras som den dos som producerar tecken på toxicitet och så att högre dosnivåer baserade på samma doseringsschema skulle förväntas leda till att försöksdjuret dör. Substanser med specifika biologiska verkningar vid låga icke-toxiska doser (såsom hormoner och mitogena substanser) kan utgöra undantag från kriterier som är dosbestämmande och bör utvärderas från fall till fall. Den högsta dosen kan även definieras som en dos vilken ger upphov till någon indikation på toxicitet i benmärgen (t.ex. mer än 50 % reduktion av mitotiskt index).
1.5.4 ”Limit-test”
En fullständig undersökning med tre dosnivåer kan inte anses vara nödvändig om ett test på en dosnivå på minst 2 000 mg/kg kroppsvikt och med en enstaka behandling, eller i form av två behandlingar under samma dag, inte ger upphov till observerbara toxiska effekter, och om ingen genotoxicitet kan förväntas utifrån data om strukturellt relaterade substanser. För undersökningar med en längre varaktighet ligger gränsdosen på 2 000 mg/kg/kroppsvikt/dag vid behandlingar upp till 14 dagar, och 1 000 mg/kg/kroppsvikt/dag för behandlingar som varar längre än 14 dagar. Förväntad exponering av människor kan utgöra en indikation på behovet av att använda en högre dosnivå i ett ’limit-test’.
1.5.5 Administrering av doser
Testsubstansen administreras normalt genom sondmatning med hjälp av en magsond, en lämplig intuberingskanyl eller genom en intraperitoneal injektion. Andra vägar för exponering kan vara acceptabla där de kan motiveras. Den maximala vätskevolymen som kan administreras med hjälp av sondmatning eller injektion vid ett tillfälle beror på försöksdjurets storlek. Volymen bör inte överstiga 2 ml/100 g kroppsvikt. Användning av volymer som är högre än dessa måste motiveras. Förutom för irriterande eller korrosiva substanser, som normalt kommer att visa på stegrade effekter med högre koncentrationer, bör variationer av testvolymen minimeras genom en justering av koncentrationen för att säkerställa att volymen är konstant vid alla dosnivåer.
1.5.6 Kromosomberedning
Omedelbart efter avlivningen tas benmärgen ut, tillsätts hypoton lösning och fixeras. Cellerna sprids sedan ut på utstryksglas och färgas in.
1.5.7 Analys
Det mitotiska indexet bör fastställas om ett mått på cytotoxicitet hos minst 1 000 celler per djur för alla behandlade försöksdjur (inklusive positiva kontroller) och obehandlade försöksdjur för negativ kontroll.
Minst 100 celler bör analyseras för varje försöksdjur. Detta antal kan reduceras när ett stort antal avvikelser observeras. Alla utstryksglas, inklusive de för positiva och negativa kontroller, bör kodas oberoende av varandra innan de analyseras i mikroskop. Eftersom beredningen av utstryksglas ofta resulterar i brott på en del av de celler som är i metafas med förlust av kromosomer som följd, bör de celler som påträffas därför innehålla ett centromererantal som är lika med antalet 2n ± 2.
2. DATA
2.1 BEHANDLING AV RESULTATEN
Individuella data för försöksdjuren bör presenteras i tabellform. Den experimentella enheten utgörs av djuret. För varje försöksdjur bör antalet celler som påträffas, antalet avvikelser per cell och procentandelen celler med strukturella kromosomavvikelser utvärderas. Antalet och frekvensen av olika typer av strukturella kromosomavvikelser bör listas för behandlade grupper och kontrollgrupper. Luckor i DNA-strängen noteras separat och rapporteras men inkluderas inte generellt i den totala frekvensen av avvikelser. Om det inte finns några bevis för en skillnad i resultaten mellan könen, kan data från båda könen kombineras för statistisk analys.
2.2 UTVÄRDERING OCH TOLKNING AV RESULTATEN
Det finns ett flertal kriterier som används för att bestämma om ett resultat är positivt, t.ex. en dosrelaterad ökning i det relativa antalet celler med kromosomavvikelser eller en tydlig ökning av antalet celler som uppvisar avvikelser i en grupp som fått en enstaka dos vid ett enstaka provtagningstillfälle. Man bör i första hand överväga resultatets biologiska relevans. Statistiska metoder kan vara till hjälp vid utvärdering av testresultatet (6). Statistisk signifikans bör dock inte vara den enda faktor som bestämmer om resultatet är positivt. Tvetydiga resultat bör klarläggas genom fortsatta tester, lämpligen vid ändrade experimentella betingelser.
En ökning av polyploidin kan utgöra en indikation på att testsubstansen har potential att inducera numeriska kromosomavvikelser. En ökning av endoreduplikationen kan utgöra en indikation på att testsubstansen har potential att inhibera cellcykelns förlopp (7) (8).
En testsubstans för vilken resultatet inte uppfyller ovanstående kriterier anses vara icke-mutagen i detta test.
Även om de flesta experiment kommer att ge tydligt positiva eller negativa resultat, så kan i sällsynta fall uppsättningen av data göra det omöjligt att utföra en otvetydig bedömning med avseende på testsubstansens aktivitet. Resultatet kan förbli tvetydigt eller tveksamt, oavsett hur många experiment som utförs.
Positiva resultat från testet av kromosomavvikelser in vitro, indikerar att en substans inducerar kromosomavvikelser i benmärgen hos de arter som testades. Negativa resultat indikerar att testsubstansen, under dessa försöksbetingelser, inte inducerar kromosomavvikelser i benmärgen hos de arter som testades.
Sannolikheten för att testsubstansen eller dess metaboliter når ut till det stora kretsloppet eller specifikt till målvävnaden (t.ex. systemisk toxicitet) bör diskuteras.
3. RAPPORTERING
TESTRAPPORT
Testrapporten måste innehålla följande information:
|
|
Lösningsmedel/vehikel:
|
|
|
Försöksdjur:
|
|
|
Försöksbetingelser:
|
|
|
Resultat:
|
|
|
Diskussion av resultaten. |
|
|
Slutsatser. |
4. HÄNVISNINGAR
|
1. |
Adler, I. D. (1984), Cytogenetic tests in Mammals. In: Mutagenicity Testing: a Practical Approach. S. Venitt och J. M. Parry (eds). IRL Press, Oxford, Washington D.C., s. 275–306. |
|
2. |
Preston, R. J., Dean, B. J., Galloway, S., Holden, H., McFee, A. K. och Shelby, M. (1987), Mammalian In Vivo Cytogenetic Assays: Analysis of Chromosome Aberrations in Bone Marrow Cells. Mutation Res., 189. s. 157–165. |
|
3. |
Richold, M., Chandley, A., Ashby, J., Gatehouse, D. G., Bootman, J. och Henderson, L. (1990), In Vivo Cytogenetic Assays. In: D. J. Kirkland (ed.) Basic Mutagenicity Tests, UKEMS Recommended Procedures. UKEMS Sub-Committee on Guidelines for Mutagenicity Testing. Report. Part I revised. Cambridge University Press, Cambridge, New York, Port Chester, Melbourne, Sydney, s. 115–141. |
|
4. |
Tice, R. R., Hayashi, M., MacGregor, J. T., Anderson, D., Blakey, D. H. Holden, H. E., Kirsch-Volders, M., Oleson Jr., F. B., Pacchierotti, F., Preston, R. J., Romagna, F., Simada, H., Sutou, S. och Vannier, B. (1994), Report from the Working Group on the In Vivo Mammalian Bone Marrow Chromosomal Aberration Test. Mutation Res., 312, s. 305–312. |
|
5. |
Fielder, R. J., Allen, J. A., Boobis, A. R., Botham, P. A., Doe, J., Esdaile, D. J., Gatehouse, D. G., Hodson-Walker. G., Morton, D. B., Kirkland, D. J. och Richold, M. (1992), Report of British Toxicology Society/UK Environmental Mutagen Society Working Group: Dose setting in In Vivo Mutagenicity Assays. Mutagenesis, 7, s. 313–319. |
|
6. |
Lovell, D. P., Anderson, D., Albanese, R., Amphlett, G. E., Clare, G., Ferguson, R., Richold, M., Papworth, D. G. och Savage, J. R. K. (1989), Statistical Analysis of In Vivo Cytogenetic Assays. In: UKEMS Sub-Comntittee on Guidelines for Mutagenicity Testing. Report Part. III. Statistical Evaluation of Mutagenicity Test Data. D. J. Kirkland, (ed.) Cambridge University Press, Cambridge, s. 184–232. |
|
7. |
Locke-Huhle, C. (1983), Endoreduplication in Chinese hamster cells during alpha-radiation induced G2 arrest. Mutation Res., 119, s. 403–413. |
|
8. |
Huang, Y., Change, C. and Trosko, J. E. (1983), Aphidicolin-induced endoreduplication in Chinese hamster cells. Cancer Res., 43, s. 1362–1364. |
B.12 MUTAGENICITET – TEST AV MIKROKÄRNOR IN VIVO I ERYTROCYTER HOS DÄGGDJUR
1. METOD
Denna metod är en kopia av testet OECD TG 474, Test av mikrokämor i erytrocyter hos däggdjur (Mammalian Erythrocyte Micronucleus Test [1997]).
1.1 INLEDNING
Testet av mikrokärnor in vivo hos däggdjur används för detektion av sådana skador på kromosomer eller på den mitotiska apparaten hos erytroblaster som induceras av testsubstansen, genom analys av erytrocyter från provtagning på benmärg och/eller perifert blod från djurceller, vanligtvis gnagare.
Avsikten med testet av mikrokärnor är att identifiera substanser som orsakar cytogen skada, vilket resulterar i bildning av mikrokärnor som innehåller isolerade kromosomfragment eller hela kromosomer.
När en erytroblast från benmärg utvecklas till en polykromatisk erytrocyt stöts huvudkärnan ut. Varje mikrokärna som har bildats kan bli kvar i den annars kärnfria cytoplasman. Visualisering av mikrokärnor underlättas i dessa celler eftersom de saknar en huvudkärna. En ökning av frekvensen av polykromatiska erytrocyter med mikrokärnor hos behandlade försöksdjur utgör en indikation på inducerade kromosomskador.
Benmärg från gnagare används rutinmässigt i detta test eftersom polykromatiska erytrocyter produceras i sådan vävnad. Mätningen av omogna (polykromatiska) erytrocyter med mikrokärnor i perifert blod är lika acceptabelt hos alla arter där oförmågan hos mjälten att avlägsna erytrocyter med mikrokärnor har kunnat påvisas, eller som har uppvisat en adekvat sensitivitet för att detektera ämnen som orsakar strukturella eller numeriska kromosomavvikelser. Mikrokärnor kan särskiljas genom ett antal olika kriterier. Dessa innefattar identifiering av närvaron eller frånvaron av en kinetochor eller av centromer-DNA i mikrokärnor. Frekvensen av omogna (polykromatiska) erytrocyter med mikrokärnor utgör den huvudsakliga slutpunkten. Antalet mogna (normokromatiska) erytrocyter i perifert blod som innehåller mikrokärnor bland ett givet antal mogna erytrocyter kan även användas som slutpunkt på undersökningen, när försöksdjur behandlas kontinuerligt under fyra veckor eller längre.
Detta test av mikrokärnor in vivo på däggdjur är särskilt relevant för att fastställa mutagena risker, eftersom det möjliggör överväganden av faktorer för in vivo metabolism, farmakokinetik och processer för DNA-reparationer, även om sådana faktorer kan variera mellan olika arter, vävnader och genetiska slutpunkter. Ett test in vivo är även användbart för fortsatta undersökningar av en mutagen effekt, med detektion i ett in vitro-system.
Om det finns bevis för att testsubstansen eller en reaktiv metabolit inte kommer att nå målvävnaden, är det inte lämpligt att använda detta test.
Se även Allmän inledning, del B.
1.2 DEFINITIONER
centromer (Kinetochor): delar av en kromosom till vilka spindelfibrer är fästa under celldelning, vilket ger upphov till en ordnad rörelse av dotterkromosomerna mot dottercellernas poler.
mikrokärnor: små extra kärnor som är separata från cellernas huvudkärna, och som produceras under mitosens telofas (meios) från isolerade kromosomfragment eller hela kromosomer.
normokromatisk erytrocyt: mogna erytrocyter som saknar ribosomer och som kan särskiljas från omogna polykromatiska erytrocyter genom infärgning som är selektiv för ribosomer.
pofykromalisk erytrocyt: omogen erytrocyt, i ett intermediärt utvecklingsstadium, vilken fortfarande innehåller ribosomer och därför kan särskiljas från mogna, normokromatiska erytrocyter genom infärgning som är selektiv för ribosomer.
1.3 TESTMETODENS PRINCIP
Försöksdjur bör exponeras för testsubstansen på ett lämpligt sätt. Om benmärg används, bör försöksdjuren avlivas vid lämpliga tider efter behandling, varvid benmärg extraheras, preparationer görs och färgas in (1) (2) (3) (4) (5) (6) (7). När perifert blod används, samlas blodet upp vid lämpliga tidpunkter efter behandling och utstrykspreparationer görs, vilka sedan färgas in (4) (8) (9) (10). Vid undersökningar med perifert blod, bör det gå så kort tid som möjligt mellan den sista exponeringen och skörden av cellerna. Preparationerna analyseras med avseende på närvaron av mikrokärnor.
1.4 BESKRIVNING AV TESTMETODEN
1.4.1 Förberedelser
1.4.1.1 Val av djurarter
Råttor, möss och kinesiska hamstrar används vanligen, även om vilket lämpligt däggdjur som helst kan användas. Vid användning av perifert blod rekommenderas möss. Det är emellertid även möjligt att använda andra däggdjursarter, förutsatt att det är en art där mjälten inte avlägsnar erytrocyter med mikrokärnor eller en art som har uppvisat en adekvat känslighet när det gäller att detektera ämnen som kan orsaka strukturella eller numeriska kromosomavvikelser. Vanligt använda laboratoriestammar av unga, friska försöksdjur bör komma i fråga. Vid undersökningens början bör viktskillnaden mellan försöksdjuren vara minimal och inte överskrida + 20 % av medelvikten för varje kön.
1.4.1.2 Förhållanden vid förvaring och utfodring
De generella förhållanden som anges i den allmänna inledningen till del B gäller. Man bör dock sträva efter en relativ luftfuktighet på 50-60 %.
1.4.1.3 Förberedelse av djuren
Friska, unga vuxna djur väljs ut slumpartat för placering i kontroll- och behandlingsgrupperna. Burar bör arrangeras på ett sådant sätt att möjliga effekter beroende på placeringen av burarna blir så små som möjligt. Djuren ska ha en unik identifiering. De bör vänjas vid förhållandena i laboratoriet under minst fem dagar.
1.4.1.4 Beredning av doser
Fasta testsubstanser bör lösas upp i eller blandas med lämpliga lösningsmedel eller vehiklar och spädas ut, om så är lämpligt, innan dosen administreras till djuren. Testsubstanser i flytande form kan doseras direkt eller spädas ut före dosering. Nygjorda beredningar av testsubstansen bör användas om inte stabilitetsdata visar att lagring är acceptabel.
1.4.2 Försöksbetingelser
1.4.2.1 Lösningsmedel/vehikel
Lösningsmedlet/vehikeln bör inte ge upphov till toxiska effekter vid de dosnivåer som används och bör inte kunna misstänkas för att reagera kemiskt med testsubstansen. Om andra lösningsmedel/vehiklar än gängse brukliga används, bör användningen av dem ha stöd från data som visar på deras kompatibilitet. Det rekommenderas att, därhelst det är möjligt, man i första hand överväger vattenbaserade lösningsmedel/vehiklar.
1.4.2.2 Kontroller
Samtidiga positiva och negativa kontroller (lösningsmedel/vehikel) bör ingå för varje test. Förutom behandling med testsubstansen, bör djuren i kontrollgrupperna hanteras på ett identiskt sätt i förhållande till djuren i de grupper där behandling skett.
Positiva kontroller bör ge mikrokärnor in vivo vid de exponeringsnivåer som förväntas ge en detekterbar ökning i förhållande till bakgrunden. Positiva kontrolldoser bör väljas på ett sådant sätt att effekterna är tydliga, men att identiteten hos kodade utstryksglas inte omedelbart avslöjas för läsaren. Det är acceptabelt att den positiva kontrollen administreras via en annan tillförselväg än testsubstansen och att provtagning endast sker vid ett enstaka tillfälle. Användning av kemiska klassrelaterade positiva kontrollkemikalier kan dessutom övervägas när dessa finns tillgängliga. I tabellen nedan ges några exempel på positiva kontrollsubstanser.
|
Substans |
CAS-nummer |
EINECS-nummer |
|
Etylmetansulfonat |
62-50-0 |
200-536-7 |
|
N-etyl-N-nitrosourea |
759-73-9 |
212-072-2 |
|
Mitomycin C |
50-07-7 |
200-008-6 |
|
Cyklofosfoamid Cyklofosfoamidmonohydrat |
50-18-0 6055-19-2 |
200-015-4 |
|
Trietylenmelamin |
51-18-3 |
200-083-5 |
Negativa kontroller, behandlade enbart med lösningsmedel eller vehiklar, och i övrigt behandlade på samma sätt som behandlingsgrupperna, bör inkluderas för varje provtagningstillfälle, om inte acceptabel variabilitet och frekvens hos celler med kromosomavvikelser hos djuren kan erhållas ur historiska kontrolldata. Om enstaka provtagningar används för negativa kontroller, är den lämpligaste tiden det första provtagningstillfället. Dessutom bör obehandlade kontroller även användas om det inte finns historiska eller publicerade kontrolldata som visar att inga skadliga eller mutagena effekter induceras av det valda lösningsmedlet/vehikeln.
Om perifert blod används, kan även ett prov taget innan behandlingen påbörjas vara acceptabelt som en samtidig negativ kontroll, men endast för de korta perifera undersökningarna av blod (t.ex. 1–3 behandling(ar)) när resulterande data ligger inom det förväntade området för den historiska kontrollen.
1.5 FÖRFARANDE
1.5.1 Försöksdjurens antal och kön
Varje behandlad grupp samt kontrollgruppen måste bestå av minst 5 analyserbara djur av varje kön. Det räcker med att testa enbart det ena könet, om det vid tiden för undersökningens genomförande finns data tillgängliga från undersökningar av samma art och där man har använt sig av samma exponeringssätt, så att det går att visa att det inte finns några substantiella skillnader i toxicitet mellan könen. I de fall där exponering för kemikalier hos människa kan vara könsspecifik, vilket t.ex. kan vara fallet för vissa läkemedel, bör testet utföras med försöksdjur av lämpligt kön.
1.5.2 Behandlingsschema
Inget standardiserat behandlingsschema (t.ex en, två eller flera behandlingar med 24 timmars intervall) kan rekommenderas. Proverna från utökade doseringsscheman är acceptabla så länge som en positiv effekt har demonstrerats för denna typ av undersökning eller, för en negativ undersökning, så länge som toxicitet har påvisats eller gränsvärdesdosen har använts, och doseringen fortsatte tills det var dags för provtagning. Testsubstanser kan även administreras i form av en delad dos, dvs. två behandlingar under en och samma dag med högst några timmars intervall, för att på så sätt underlätta administrering av stora volymer med material.
Testet kan utföras på två sätt
|
a) |
Försöksdjur behandlas en gång med testsubstansen. Prover på benmärg tas ut minst två gånger, med början tidigast 24 timmar och senast 48 timmar efter behandling, och med lämpliga intervall mellan proven. Om prover tas tidigare än 24 timmar efter behandling bör detta motiveras. Prover på perifert blod tas ut minst två gånger, med början tidigast 36 timmar efter behandling, och i lämpliga intervall efter det första provet, men inte senare än efter 11 timmar. När ett positivt svar identifieras vid ett provtagningstillfälle, så är ytterligare provtagning inte nödvändig. |
|
b) |
Om två eller flera dagliga behandlingar utförs (t.ex. två eller flera behandlingar med 24 timmars intervall), bör prover tas ut en gång mellan 18 och 24 timmar efter den slutliga behandlingen av benmärgen och en gång mellan 36 och 48 timmar efter den slutliga behandlingen av det perifera blodet (12). |
Prover kan dessutom tas vid andra tidpunkter när detta är lämpligt.
1.5.3 Dosnivåer
Om det inte finns några lämpliga data tillgängliga och en undersökning därför utförs för att fastställa omfattningen, bör denna undersökning utföras i samma laboratorium, och med samma arter, stammar, kön och behandlingsschema som senare ska användas i huvudundersökningen (13). Vid toxicitet används tre dosnivåer för det första provtagningstillfället. Dessa dosnivåer bör täcka in ett område som sträcker sig från maximal till liten eller ingen toxicitet. Vid senare provtagningstillfällen räcker det med att endast den högsta dosen används. Den högsta dosen definieras som den dos som producerar tecken på toxicitet och så att högre dosnivåer baserade på samma doseringsschema skulle förväntas leda till att försöksdjuret dör. Substanser med specifika biologiska verkningar vid låga icke-toxiska doser (såsom hormoner och mitogena substanser) kan utgöra undantag från kriterier som är dosbestämmande och bör utvärderas från fall till fall. Den högsta dosen kan även definieras som en dos vilken ger upphov till någon indikation på toxicitet i benmärgen (t.ex. en reduktion av andelen omogna erytrocyter jämfört med det totala antalet erytrocyter i benmärgen eller i perifert blod).
1.5.4 ”Limit-test”
En fullständig undersökning med tre dosnivåer kan inte anses vara nödvändig om ett test på en dosnivå på minst 2 000 mg/kg kroppsvikt och med en enstaka behandling, eller i form av två behandlingar under samma dag, inte ger upphov till observerbara toxiska effekter, och om ingen genotoxicitet kan förväntas utifrån data om strukturellt relaterade substanser. För undersökningar med en längre varaktighet ligger gränsdosen på 2 000 mg/kg/kroppsvikt/dag vid behandlingar upp till 14 dagar, och på 1 000 mg/kg/kroppsvikt/dag för behandlingar som varar längre än 14 dagar. Förväntad exponering av människor kan utgöra en indikation på behovet av att använda en högre dosnivå i ett ”limit-test”.
1.5.5 Administrering av doser
Testsubstansen administreras normalt genom sondmatning med hjälp av en magsond, en lämplig intuberingskanyl eller genom en intraperitoneal injektion. Andra tillförselvägar för exponering kan vara acceptabla där de kan motiveras. Den maximala vätskevolymen som kan administreras med hjälp av sondmatning eller injektion vid ett tillfälle beror på försöksdjurets storlek. Volymen bör inte överstiga 2 ml/100 g kroppsvikt. Användning av volymer som är högre än dessa måste motiveras. Förutom för irriterande eller korrosiva substanser, som normalt kommer att visa på stegrade effekter med högre koncentrationer, bör variationer av testvolymen minimeras genom en justering av koncentrationen för att säkerställa att volymen är konstant vid alla dosnivåer.
1.5.6 Beredning av benmärg/blod
Benmärgsceller erhålls vanligen från lårbenet eller skenbenet omedelbart efter det att djuret avlivats. Normalt avlägsnas cellerna från lårbenet eller skenbenet, prepareras och färgas in med hjälp av etablerade metoder. Perifert blod erhålls från svansvenen eller någon annan lämplig blodåder. Blodceller färgas omedelbart in supravitalt (8) (9) (10) eller så görs utstrykspreparationer vilka sedan färgas in. Användningen av ett DNA-specifikt färgämne (t.ex. akridinorange (14) eller Hoechst 33258 plus pyronin-Y (15)) kan eliminera några av de artefakter som ar associerade med användning av ett färgämne som inte är DNA-specifikt. Denna fördel utesluter inte användningen av konventionella färgämnen (t.ex. Giemsa). Ytterligare system (t.ex. cellulosakolonner för att avlägsna celler med kärnor (16)) kan även användas, förutsatt att dessa system har visat sig fungera på ett adekvat sätt vid preparation av mikrokärnor i laboratoriet.
1.5.7 Analys
Andelen omogna erytrocyter bland det totala antalet erytrocyter (omogna + mogna) bestäms för varje djur genom att totalt räkna minst 200 erytrocyter för benmärg och 1 000 erytrocyter för perifert blod (17). Alla utstryksglas, inklusive de från positiva och negativa kontroller, bör ges en oberoende kodning innan analys i mikroskop utförs. Minst 2 000 omogna erytrocyter per djur undersöks med avseende på förekomst av omogna erytrocyter med mikrokärnor. Ytterligare information kan erhållas genom att söka efter mogna erytrocyter med mikrokärnor. Vid analys av utstryksglas bör antalet omogna erytrocyter jämfört med det totala antalet erytrocyter inte vara mindre än 20 % av kontrollvärdet. När djur behandlas kontinuerligt under fyra veckor eller mer kan även minst 2 000 mogna erytrocyter per djur undersökas med avseende på förekomst av mikrokärnor. System för automatiserad analys (bildanalys och flödescytometri av cellsuspensioner) är acceptabla alternativ till manuell utvärdering om lämplig verifiering och validering sker.
2. DATA
2.1 BEHANDLING AV RESULTATEN
Individuella data från försöksdjur bör presenteras i tabellform. Den experimentella enheten utgörs av försöksdjuret. Antalet omogna erytrocyter som påträffas, antalet omogna erytrocyter med mikrokärnor, och antalet omogna bland det totala antalet erytrocyter bör listas separat för varje djur som analyseras. När försöksdjur behandlas kontinuerligt under fyra veckor eller mer, bör data över mogna erytrocyter också presenteras, om dessa har samlats in. Andelen omogna erytrocyter bland det totala antalet erytrocyter och, om det anses tilllämpligt, procentandelen av erytrocyter med mikrokärnor, bör anges för varje försöksdjur. Om det inte finns några bevis för en skillnad i resultaten mellan könen, kan data från båda könen kombineras för statistisk analys.
2.2 UTVÄRDERING OCH TOLKNING AV RESULTATEN
Det finns ett flertal kriterier som används för att bestämma om ett resultat är positivt, t.ex. en dosrelaterad ökning av antalet celler med mikrokärnor eller en tydlig ökning av antalet celler med mikrokärnor hos en grupp som fått en enstaka dos vid ett enstaka provtagningstillfälle. Man bör i första hand överväga resultatets biologiska relevans. Statistiska metoder kan användas som en hjälp vid utvärdering av testresultatet (18) (19). Statistisk signifikans bör dock inte vara den enda faktor som bestämmer om resultatet är positivt. Tvetydiga resultat bör klarläggas genom fortsatta tester, lämpligen vid ändrade experimentella betingelser.
En testsubstans för vilken resultatet inte uppfyller ovanstående kriterier anses vara icke-mutagen i detta test.
Även om de flesta experiment kommer att ge tydligt positiva eller negativa resultat, kan i sällsynta fall uppsättningen av data göra det omöjligt att utföra en otvetydig bedömning med avseende på testsubstansens aktivitet. Resultatet kan förbli tvetydigt eller tveksamt, oavsett hur många gånger experimentet upprepas.
Positiva resultat i testet av mikrokärnor tyder på att substansen inducerar mikrokärnor som är ett resultat av en kromosomal skada eller skada på den mitotiska apparaten i erytroblasterna hos den art som testas. Negativa resultat indikerar att testsubstansen, under dessa försöksbetingelser, inte ger upphov till mikrokärnor i de omogna erytrocyterna hos den art som testas.
Sannolikheten för att testsubstansen eller någon av dess metaboliter ska nå ut i det stora kretsloppet eller specifikt till målvävnaden (t.ex. systemisk toxicitet) bör diskuteras.
3. RAPPORTERING
TESTRAPPORT
Testrapporten måste innehålla följande information:
|
|
Lösningsmedel/vehikel:
|
|
|
Försöksdjur:
|
|
|
Försöksbetingelser:
|
|
|
Resultat:
|
|
|
Diskussion av resultaten. |
|
|
Slutsatser. |
4. HÄNVISNINGAR
|
1. |
Heddle, J. A. (1973), A Rapid In Vivo Test for Chromosomal Damage, Mutation Res., 18, s. 187–190. |
|
2. |
Schmid, W. (1975), The micronucleus Test, Mutation Res., 31, s. 9–15. |
|
3. |
Heddle, J. A., Salamone, M. F., Hite, M., Kirkhart, B., Mavournin, K., MacGregor, J. G. and Newell, G. W. (1983), The Induction of Micronuclei as a Measure of Genotoxicity. Mutation Res. 123. s. 61–118. |
|
4. |
Mavourmin, K. H., Blakey, D. H., Cimino, M. C, Salamone, M. F. and Heddle, J. A. (1990), The In Vivo micronucleus Assay in Mammalian Bone Marrow and Peripheral Blood. A report of the U.S. Environmental Protection Agency Gene-Tox Program, Mutation Res., 239, s. 29–80. |
|
5. |
MacGregor, J. T., Schlegel, R. Choy, W. N., and Wehr, C. M. (1983), Micronuclei in Circulating Erythrocytes: A Rapid Screen for Chromosomal Damage During Routine Toxicity Testing in Mice. in: ’Developments in Science and Practice of Toxicology’, ed. A. W. Hayes, R. C. Schnell and T. S. Miya, Elsevier, Amsterdam, s. 555–558. |
|
6. |
MacGregor,). T., Heddle, J. A. Hite, M., Margolin, G. H., Ramel, C, Salamone, M. F., Tice, R. R., and Wild, D. (1987), Guidelines for the Conduct of micronucleus Assays in Mammalian Bone Marrow Erythrocytes. Mutation Res., 189: s. 103–112. |
|
7. |
MacGregor, J. T., Wehr, C. M., Henika, P. R., and Selby, M. E. (1990), the In vivo Erythrocyte Micronuc- leus Test: Measurement at Steady State Increases Assay Efficiency and Permits Integration with Toxicity Studies. Fund. Appl. Toxicol. 14, s. 513–522. |
|
8. |
Hayashi, M., Morita, T., Kodarna, Y., Sofuni, T. and Ishidate, M. Jr. (1990), The Micronucleus Assay with Mouse Peripheral Blood Reticulocytes Using Acridine Orange-Coated Slides. Mutation Res., 245, s. 245–249. |
|
9. |
The Collaborative Study Group for the Micronucleus Test (1992), Micronucleus Test Wirth Morse Perip heral Blood Erythrocytes by Acridine Orange supravital Staining: The Summary Report of the 5th Col laborative Study by CSGMT/JEMMS. MMS. Mutation Res. 278, s. 83–98. |
|
10. |
The Collaborative Study Group for the Micronucleus Test (CSGMT/JEMMS. MMS: The Mammalian Mutagenesis Study Group of the Environmental Mutagen Society of Japan) (1995), Protocol recommended for the short-term mouse peripheral blood micronucleus test. Mutagenesis, 10, s. 153–159. |
|
11. |
Hayashi, M., Tice, R. R., MacGregor, J. T., Anderson, D., Blackey, D. H., Kirsch-Volders, M., Oleson, Jr. F. B., Pacchierotti, F., Romagna, F., Shimada, H., Sutou, S. and Vannier, B. (1994), In vivo Rodent Eryth rocyte Micronucleus Assay. Mutation Res., 312, s. 293–304. |
|
12. |
Higashikuni, N. and Sutou, S. (1995), An optimal, generalized sampling time of 30 ± 6 h after double dosing in the mouse peripheral blood micronucleus test. Mutagenesis, 10, s. 313–319. |
|
13. |
Fielder, R. J., Allen, J. A., Boobis, A. R., Botham, P. A., Doc, J., Esdaile, D.]., Gatehouse, D. G., Hodson- Walker, G., Morton, D. B.. Kirkland. D. J. and Rochold, M. (1992), Report of British Toxicology Society/UK Environmental Mutagen Society Working Group: Dose Setting in In Vivo Mutagenicity Assays. Mutagenesis, 7, s. 313–319. |
|
14. |
Hayashi, M., Sofuni, T. and Ishidate, M. Jr. (1983), An Application of Acridine Orange Fluorescent Stai ning to the micronucleus Test. Mutation Res. 120, s. 241–247. |
|
15. |
MacGregor, J. T., Wehr, C. M. and Langlois, R. G. (1983), A Simple Fluorescent Staining Procedure for Micronuclei and RNA in Erythrocytes Using Hoechst 33258 and Pyronin Y. Mutation Res., 120, s. 269–275. |
|
16. |
Romagna, F. and Staniforth, C. D. (1989), The automated bone marrow micronucleus test. Mutation Res. 213, s. 91–104. |
|
17. |
Gollapudi, B. and McFadden, L. G. (1995), Sample size for the estimation of polychromatic to normochromatic erythrocyte ratio in the bone marrow micronucleus test. Mutation Res., 347, s. 97–99. |
|
18. |
Richold, M., Ashby, J., Bootman, J., Chandley, A., Gatehouse, D. G. and Henderson, L. (1990), In Vivo Cytogenetics Assay. In: D. J. Kirkland (ed) Basic Mutagenicity tests. UKEMS Recommended Procedures. UKEMS Sub-Committee on Guidelines for Mutagenicity testing. Report, Part I, revised. Cambridge University Press, Cambridge, New York, Port Chester, Sydney, s. 115–141. |
|
19. |
Loveli, D. P., Anderson, D., Albanese, R., Amphlett, G. E., Clare, G., Ferguson, R., Richold, M., Papworth, D. G. and Savage, J. R. K. (1989), Statistical Analysis of In Vivo Cytogenetic Assays. In: D. J. Kirkland (cd) Statistical Evaluation oj Mutagenicity Test Dala. UKEMS Sub-Committee on Guidelines for Mutagenicity Testing. Report, Part III. Cambridge University Press, Cambridge, New York, Port Chester, Melbourne, Sydney, s. 184–232. |
B.13/14 MUTAGENICITET – TEST AV ÅTERMUTATION HOS BAKTERIER
1. METOD
Denna metod är en kopia av OECD TG 471, Omvänt bakteriellt mutationstest (Bakterial Reserve Mutation Test (1997)).
1.1 INLEDNING
1 detta test av återmutation hos bakterier används aminosyraberoende stammar av Salmonelle typhimurium och Escherichia coli för att detektera punktmutationer, vilka innefattar substitution, addition eller deletion av ett eller några DNA-baspar (1) (2) (3). Testet bygger på principen att man detekterar mutationer som gör att redan existerande mutationer i teststammarna går tillbaka varvid den funktionella förmågan hos bakterierna att syntetisera en essentiell aminosyra återställs. De återmuterade bakterierna detekteras på grundval av sin förmåga att växa i frånvaro av den aminosyra som krävs hos den ursprungliga teststammen.
Punktmutationer är orsaken till många genetiska sjukdomar hos människor och mycket tyder på att punktmutationer i onkogener och suppressorgener för tumörer i somatiska celler är inblandade i bildning av tumörer hos människor och försöksdjur. Testet av återmutation hos bakterier är snabbt, billigt och relativt enkelt att utföra. Många av testarterna har ett flertal egenskaper som gör dem mera känsliga för detektion av mutationer inklusive responsiva DNA-sekvenser vid reversionssäten, ökad cellerpermeabilitet för stora molekyler och eliminering av reparationssystem för DNA eller förstärkning av felbenägna reparationsprocesser för DNA. Specificiteten hos teststammarna kan erbjuda en del nyttig information om de typer av mutationer som induceras av genotoxiska ämnen. En mycket stor databas med resultat för ett stort antal olika strukturer finns tillgänglig för tester av återmutation hos bakterier och väletablerade metoder har utvecklats för testning av kemikaler med olika fysisk-kemiska egenskaper, inklusive flyktiga ämnen.
Se även Allmän inledning, del B.
1.2 DEFINITIONER
ett test av återmutation hos antingen Salmonella typhimurium eller Escherichia coli detekterar mutation i en aminosyraberoende stam (histidin respektive tryptofan), vilket ger en stam som är oberoende av en extern tillförsel av aminosyror.
mutagener som ger basparsubstitutioner är ämnen som orsakar en basförändring i DNA. I ett test av återmutation kan denna förändring uppträda vid sätet för den ursprungliga mutationen eller på ett andra säte i det bakteriella genomet.
mutagener som ger förskjutningar (frameshift) är ämnen som orsakar addition eller deletion av ett eller flera baspar i DNA, vilket då ger en ändrad avläsningsordning i RNA.
1.3 INLEDANDE ÖVERVÄGANDEN
Testet av återmutation hos bakterier utnyttjar prokaryota celler, vilka skiljer sig från däggdjursceller med avseende på sådana faktorer som upptag, metabolism, kromosomstruktur och processer för DNA-reparation. Tester som utförs in vitro kräver i allmänhet att det finns en yttre källa för metabolisk aktivering. Hos system för metabolisk aktivering in vitro kan inte förhållandena för däggdjur in vivo efterliknas helt och hållet. Testet ger därför ingen direkt information om den mutagena och den karcinogena potensen hos en substans i däggdjur.
Testet av återmutation hos bakterier används vanligen som en inledande undersökning av genotoxisk aktivitet och, i synnerhet, av aktivitet som inducerar punktmutationer. En omfattande databas har demonstrerat att många kemikalier som ger positiva resultat i detta test även uppvisar mutagen aktivitet i andra tester. Det finns exempel på mutagena ämnen som inte kan detekteras med hjälp av detta test. Skälen till dessa tillkortakommanden kan tillskrivas den specifika naturen hos den detekterade slutpunkten, skillnader i metabolisk aktivering, eller skillnader i biotillgänglighet. Å andra sidan kan faktorer som höjer känsligheten hos testet leda till en överskattning av den mutagena aktiviteten.
Testet av återmutation hos bakterier kan visa sig olämpligt för utvärdering av vissa klasser av kemikalier såsom starkt baktericida ämnen (t.ex. vissa antibiotika) och de som anses (eller är kända för att) interferera specifikt med systemet för cellreplikation hos däggdjur (t.ex. vissa inhibitorer av topoisomeras och vissa nukleosidanaloger). 1 sådana fall kan mutationstester på däggdjur vara mera lämpliga.
Även om många ämnen som ger positiva resultat i detta test är karcinogena för däggdjur, är korrelationen inte absolut. Den är beroende på kemisk klass och det finns karcinogener som inte kan detekteras med hjälp av detta test, eftersom de verkar genom andra, icke-genotoxiska mekanismer eller mekanismer som saknas i bakterieceller.
1.4 TESTMETODENS PRINCIP
Suspensioner med bakterieceller exponeras för testsubstansen i närvaro och i frånvaro av ett exogent system för metabolisk aktivering. 1 plattinkorporeringsmetoden blandas dessa suspensioner med en toppskiktsagar varpå plattor omedelbart görs på minimalmedium. I preinkubationsmetoden inkuberas behandlingsblandningen och sedan blandas denna med en toppskiktsagar innan plattor görs på minimalmedium. I båda metoderna räknas återmuterade kolonier efter två eller tre dagars inkubering och jämförs med antalet spontant återmuterade kolonier på kontrollplattor med lösningsmedel.
Ett flertal procedurer för att utföra testet av återmutation hos bakterier har beskrivits. Bland de som vanligtvis används ingår plattinkorporeringsmetoden (1) (2) (3) (4), preinkubationsmetoden (2) (3) (5) (6) (7) (8), fluktue-ringsmetoden (9) (10) och suspensionsmetoden (11). Modifikationer för testning av gaser eller ångor har tidigare beskrivits (12).
De procedurer som beskrivs i metoden hänför sig primärt till plattinkorporerings- och preinkubationsmetoder. Inga av dem är acceptabla för att genomföra experiment både med och utan metabolisk aktivering. Vissa substanser kan detekteras mer effektivt med hjälp av preinkubationsmetoden. Dessa substanser tillhör kemiska klasser som innefattar korta kedjor med alifatiska nitrosaminer, tvåvärda metaller, aldehyder, azofärger och diazoföreningar, pyrolizidinalkoloider, allylföreningar och nitroföreningar (3). Det är även känt att vissa klasser av mutanter inte alltid detekteras med hjälp av standardprocedurer såsom plattinkorporeringsmetoden eller preinkubationsmetoden. Dessa bör uppfattas som ’speciella fall’ och det rekommenderas varmt att alternativa procedurer används för detektionen av dem. De följande ’speciella fallen’ bör identifieras (tillsammans med exempel på procedurer som kan användas för detektionen av dem): azofärger och diazoföreningar (3) (5) (6) (13), gaser och flyktiga kemikaler (12) (14) (15) (16) samt glykosider (17) (18). En avvikelse från standardproceduren måste vara vetenskapligt motiverad.
1.5 BESKRIVNING AV TESTMETODEN
1.5.1 Förberedelser
1.5.1.1 Bakterier
Nya kulturer med bakterier bör få växa till sig fram till den sena exponentiella eller tidiga stationära tillväxtfasen (ungefär 109 celler per ml). Kulturer som befinner sig i sen stationär fas bör inte användas. Det är viktigt att de kulturer som används i experimentet har en hög tider med livsdugliga bakterier. Titern kan fås fram antingen ur tillväxtkurvor från historiska kontrolldata, eller ur varje försök genom att bestämma antalet livsdugliga celler med hjälp av ett utstrykningsexperiment.
Den rekommenderade inkubationstemparaturen är 37 oC.
Minst fem bakteriestammar bör användas. Dessa bör inkludera fyra av S. typhimurium (TA 1535; Ta 1537 eller TA97a eller TA97; TA9S; och TA100) som har visat sig vara tillförlitliga och ge en reproducerbar respons i olika laboratorier. Dessa fyra stammar av S, typhimurium har GC-baspar vid det primära återmutationsstället och det är känt att de inte kan detektera vissa oxiderande mutagener, tvärbindande ämnen och hydraziner. Sådana substanser kan detekteras med hjälp av E. coli WP2-stammar eller S. typhimurium TA 102 (19) som har ett AT-baspar vid det primära återmutationsstället. Den rekommenderade kombinationen av stammar är därför:
|
— |
S. lyphimurium TA 1535, |
|
— |
S. typhimurium TA1537 eller TA97 eller TA97a, |
|
— |
5. typhimuriurn TA98, |
|
— |
S. typhimurium TA100, och |
|
— |
E. coli WP2 uvrA, eller C. coli WP2 uvrA (pKM101), eller S. typhimurium TA102. |
Fär att kunna detektera tvärbindande mutagener kan det vara lämpligt att inkludera TA102 eller att också använda en DNA-reparationsduglig stam av E. coli (t.ex. E.coli WP2 eller E. coli WP2 (pKM101)).
Etablerade procedurer för beredning av baskulturer, markörverifiering och lagring bör användas. Behovet av aminosyror för tillväxten bör påvisas för varje frusen preparation av baskulturer (histidin för stammar av S. typhimurium och tryptofan för stammar av E. coli). Andra fenotypiska karakteristika bör kontrolleras på liknande sätt, nämligen närvaron eller frånvaron av R-faktorplasmider där detta är lämpligt (dvs. ampicillin-resi-stens i stammarna TA98, TA100 och TA97a eller TA97, WP2 uvrA och WP2 uvrA (pKM101) och ampicillin + tetracyklinreststens i stammen TA102); närvaron av karakteristiska mutationer (dvs. rfa-mutation i S. typhimurium genom känslighet för kristallviolett och uvrA-mutation i E. coli eller uvrB-mutation i S. typhimurium genom känslighet för ultraviolett ljus) (2) (3). Stammarna bör även ge spontant återmuterande platträkningar av kolonier inom de frekvensområden som kan förväntas från laboratoriets historiska kontrolldata och helst inom det område som rapporteras i litteraturen.
1.5.1.2 Medium
Använd en lämplig minimalagar (t.ex. innehållande Vogel-Bonner minimalmedium E och glukos) och ett agartoppskikt som innehåller histindin och biotin eller tryptofan, så att några stycken celldelningar ska kunna ske (1) (2) (9).
1.5.1.3 Melabolisk aktivering
Bakterier bör exponeras för testsubstansen både i närvaro och frånvaro av ett lämpligt system för metabolisk aktivering. Det vanligaste systemet som används är en post-mitokondriell fraktion där en kofaktor tillsatts (S9) vilken beretts ur lever från gnagare vilka behandlats med enzyminducerande ämnen såsom Aroclor 1254 (1) (2) eller en kombination av fenobarbiton och β-naftoflavon (18) (20) (21). Den post-mitokondriella fraktionen används vanligen vid koncentrationer i området från 5 till 30 % v/v i S9-blandningen. Valet av och betingelserna hos ett system för metabolisk aktivering kan bero på den klass av kemikalier som testas. I vissa fall kan det vara lämpligt att använda mer än en koncentration av den postmitokondriella fraktionen. För azofärger och diazoämnen, kan användning av ett reduktivt system för metabolisk aktivering vara lämpligare (6) (13).
1.5.1.4 Beredning av testsubstansen
Fasta testsubstanser bör lösas upp i eller blandas med lämpliga lösningsmedel eller vehiklar och spädas ut, om detta är lämpligt, före det att bakterierna behandlas. Flytande testsubstanser kan tillsättas direkt till testsystemen och/eller spädas ut före behandling. Nygjorda beredningar bör användas om inte stabilitetsdata visar att lagring kan accepteras.
Lösningsmedel/vehiklar bör inte kunna misstänkas för att reagera kemiskt med testsubstansen och bör vara förenliga med bakteriernas överlevnad samt S9-aktivitet (22). Om man använder andra lösningsmedel/vehiklar än gängse brukliga, bör användningen av dem ha stöd från data som visar på deras kompatibilitet. Därhelst det är möjligt, rekommenderas att man i första hand överväger vattenbaserade lösningsmedel/vehiklar.
1.5.2 Försöksbetingelser
1.5.2.1 Teststammar (se 1.5.1.1)
1.5.2.2 Exponeringskoncentration
Cytotoxiciteteri och lösligheten i den slutliga behandlingsblandningen är några av de kriterier som man måste ta hänsyn till vid bestämningen av den högsta mängd av testsubstansen som ska användas.
Det kan vara bra att bestämma toxicitet och löslighet i ett förberedande experiment. Cytotoxicitet kan detekteras genom en minskning av antalet återmuterande kolonier, en uttunning eller minskning av bakgrundstillväxten, eller graden av överlevnad hos de behandlade kulturerna. Cytotoxiciteten hos en substans kan ändras i närvaro av system för metabolisk aktivering. Olöslighet bör fastställas som en utfällning i den slutliga blandningen, under de verkliga försöksbetingelserna och ska vara synlig för blotta ögat.
Den maximala rekommenderade testkoncentrationen för lösliga icke-cytotoxiska substanser är 5 mg/platta eller 5 μl/platta. För icke-cytotoxiska substanser som inte är lösliga vid 5 mg/platta eller 5 μl/platta, bör en eller flera av de koncentrationer som testas vara olösliga i den slutliga behandlingsblandningen. Testsubstanser som är cytotoxiska redan under 5 mg/platta eller 5 μl/platta bör testas upp till en cytotoxisk koncentration. Fällningen torde inte störa bestämningen.
Minst fem olika analyserbara koncentrationer av testsubstansen bör användas, med ungefär halva log-intervall (dvs. √10) mellan testpunkterna för ett inledande experiment. Mindre intervall kan vara lämpliga när ett koncentrationssvar håller på att undersökas. Testning över en koncentration på 5 mg/platta eller 5 μl/platta kan övervägas vid utvärdering av substanser som innehåller avsevärda mängder av potentiellt mutagena föroreningar.
1.5.2.3 Negativa och positiva kontroller
Samtidiga stamspecifika positiva och negativa kontroller (lösningsmedel eller vehikel), både med och utan metabolisk aktivering, bör inkluderas i varje försök. Positiva kontrollkoncentrationer som visar på det effektiva utfallet för varje försök bör väljas.
För försök där ett system för metabolisk aktivering används, bör den positiva kontrollreferenssubstansen väljas på basis av de typer av bakteriestammar som används.
De följande substanserna utgör exempel på lämpliga positiva kontroller för försök med metabolisk aktivering:
|
Substans |
CAS-nummer |
EINECS-nummer |
|
9,10-dimetylantracen |
781-43-1 |
212-308-4 |
|
7,12-dimetylbensantracen |
57-97-6 |
200-359-5 |
|
bens[a]pyren |
50-32-8 |
200-028-5 |
|
2-aminoantracen |
613-13-8 |
210-330-9 |
|
Cyklofosfoamid |
50-18-0 |
200-015-4 |
|
Cyklofosfoamidmonohydrat |
6055-19-2 |
|
Höljande substans är en lämplig positiv kontroll for den reduktiva metaboliska aktiveringsmetoden:
|
Substans |
CAS-nummer |
EINECS-nummer |
|
kongorött |
573-58-0 |
209-358-4 |
2-aminoantracen bör inte användas som den enda indikatorn på effektiviteten hos S9-blandningen. Om 2-aminoanracen används, bör varje sats av S9 även karakteriseras med en mutagen som kräver metabolisk aktivering med hjälp av mikrosomala enzymer, t.ex. bens[a]pyren, dimetylbensantracen.
Följande substanser utgör exempel på sortspecifika positiva kontroller för försök som utförs utan exogena system för metabolisk aktivering:
|
Substans |
CAS-nummer |
EINECS-nummer |
Stam |
|
Natriumazid |
26628-22-8 |
247-852-1 |
TA 1535 och TA 100 |
|
2-nitrofluoren |
607-57-8 |
210-138-5 |
TA 98 |
|
9-aminoakridin |
90-45-9 |
201-995-6 |
TA 15 37, TA 97 och TA 97a |
|
ICR 191 |
17070-45-0 |
241-129-4 |
TA 1537, TA 97 och TA 97a |
|
Kumenhydroperoxid |
80-15-9 |
201-254-7 |
TA 102 |
|
Mitomycin C |
50-07-7 |
200-008-6 |
WP2uvrA och TA 102 |
|
N-etyl-N-nitro-N-nitrosoguanidin |
70-25-7 |
200-730-1 |
WP2, WP2uvrA och WP2uvrA (pKM101) |
|
4-nitroquinolin-1 -oxid |
56-57-5 |
200-281-1 |
WP2, WP2uvrA och WP2uvrA (pKM101) |
|
Furylfuramid (AF2) |
3688-53-7 |
|
plasmidinnehållande stammar |
Andra lämpliga positiva kontrollreferenssubstanser kan användas. Användningen av kemiska klassrelaterade positiva kontrollkemikalier bör övervägas när sådana finns att tillgå.
Negativa kontroller, bestående av enbart lösningsmedel eller vehikel, utan testsubstans, och i övrigt hanterade på samma sätt som för de behandlade grupperna, bör inkluderas. Obehandlade kontroller bör dessutom även användas, om det inte finns historiska kontrolldata som visar att inga farliga eller mutagena effekter induceras av det valda lösningsmedlet.
1.5.3 Förfarande
Vid plattinkorporeringsmetoden (1) (2) (3) (4), utan metabolisk aktivering, blandas vanligen 0,05 ml eller 0,1 ml av testlösningarna, 0,1 ml av färsk bakteriekultur (innehållande ungefär 108 livsdugliga celler) och 0,5 ml av steril buffert med 2,0 ml av toppskiktsagar. För ett försök med metabolisk aktivering blandas vanligen 0,5 ml av en metabolisk aktiveringsblandning, vilken innehåller en adekvat mängd postmitokondriell fraktion (i området från 5 till 30 % v/v i den metaboliska aktiveringsblandningen) med toppskiktsagar (2,0 ml), tillsammans med bakterierna och testsubstansen/testlösningen. Innehållet i varje rör blandas och hälls över ytan på en minimalagarplatta. Toppskiktsagarn tillåts att stelna innan inkubering sker.
För förinkubationsmetoden (2) (3) (5) (6), förinkuberas testsubstansen/testlösningen med teststamman (innehållande ungefär 108 livsdugliga celler) och steril buffert eller med system för metabolisk aktivering (0,5 ml), vanligen under 20 minuter eller längre vid 30–37 oC, innan den blandas med toppskiktsagarn och hälls uppå ytan på en minimalagarplatta. Vanligen blandas 0,05 eller 0,1 ml av testsubstansen/testlösningen, 0,1 ml bakterier och 0,5 ml av S9-blandningen eller steril buffert blandat med 2,0 ml av toppskiktsagar. Rören bör luftas under förinkubationen med hjälp av en skakmaskin.
För att möjliggöra en rimlig uppskattning av variationen bör tre plattor användas vid varje dosnivå. Användning av två plattor är acceptabelt när detta är vetenskapligt motiverat. Förlust av en enstaka platta innebär inte nödvändigtvis att ett försök blir ogiltigt.
Gasformiga eller flyktiga substanser bör testas med lämpliga metoder, såsom i slutna kärl (12) (14) (15) (16).
1.5.4 Inkubering
Alla plattor i en given assay bör inkuberas vid 37 oC under 48–72 timmar. Efter inkubationsperioden räknas antalet återmuterande kolonier per plattor.
2. DATA
2.1 BEHANDLING AV RESULTATEN
Data bör presenteras som antalet återmuterande kolonier per platta. Antalet återmuterande kolonier på såväl negativa (lösningsmedelskontroll och obehandlad kontroll, om en sådan används) och positiva kontrollplattor bör även anges. Individuella platträkningar, medelantalet för de återmuterande kolonierna per platta och standardavvikelsen bör uppges för såväl testsubstansen som för positiva och negativa (obehandlade och/eller (lösningsmedels) kontroller).
Der finns inget krav på verifiering av ett tydligt positivt svar. Tvetydiga resultat bör klarläggas genom fortsatta tester, lämpligen vid ändrade experimentella betingelser. Negativa resultat måste bekräftas från fall till fall. I de fall där det inte anses nödvändigt att bekräfta negativa resultat bör detta motiveras. Vid uppföljningsexperiment bör man överväga att ändra parametrarna i undersökningen för att utvidga de bedömda försöksbetingelserna. De försöksparametrar som skulle kunna modifieras innefattar koncentrationsindelningen, metoden för behandling (plattinkorporering eller förinkubation i vätska), och betingelser för metabolisk aktivering.
2.2 UTVÄRDERING OCH TOLKNING AV RESULTATEN
Det finns ett flertal kriterier som används för att bestämma om ett resultat är positivt, t.ex. en koncentrationsrelaterad ökning över hela det testade området och/eller en reproducerbar ökning, vid en eller flera koncentrationer, av antalet återmuterande kolonier per platta för minst en stam, med eller utan ett system för metabolisk aktivering (23). Man bör i första hand överväga resultatets biologiska relevans. Statistiska metoder kan användas som en hjälp vid utvärdering av testresultatet (24). Statistisk signifikans bör dock inte vara den enda faktor som bestämmer om svaret är positivt
En testsubstans för vilken resultatet inte uppfyller ovanstående kriterier anses vara icke-mutagen i detta test.
Även om de flesta experiment kommer att ge tydligt positiva eller negativa resultat, kan uppsättningen av data i sällsynta fall göra det omöjligt att utföra en otvetydig bedömning med avseende på testsubstansens aktivitet. Resultatet kan förbli tvetydigt eller tveksamt, oavsett hur många gånger experimentet upprepas.
Positiva resultat från testet av återmutation hos bakterier indikerar att substansen inducerar punktmutationer genom bassubstitutioner eller förskjutningar i genomet hos antingen Salmonella typhimurium och/eller Escheri-chia coli. Negativa resultat indikerar att testsubstansen inte är mutagen hos den testade sorten under dessa försöksbetingelser.
3. RAPPORTERING
TESTRAPPORT
Följande information måste ingå i testrapporten:
|
|
Lösningsmedel/vehikel:
|
|
|
Stammar:
|
|
|
Försöksbetingelser:
|
|
|
Resultat:
|
|
|
Diskussion av resultaten. |
|
|
Slutsatser. |
4. HÄNVISNINGAR
|
1. |
Ames, B. N., McCann,). and Yamasaki E. (1975), Metods for Detecting Carcinogens and Mutagens with the Salmonella/Mammalian-Microsome Mutagenitic Test. Mutation Res., 31, s. 347–364. |
|
2. |
Maron, D. M. and Ames, B. N. (1983), Revised Methods for the Salmonella Mutagenicity Test. Mutation Res. 113, s. 173–215. |
|
3. |
Gatehouse, D., Haworth, S., Cebula, T., Gocke. E., Kier, L. Matsushima, T. Melcion, C, Nohmi, T., Venitt, S. and Zeiger, E. (1994), Recommendations for the Performance of Bacterial Mutation Assays. Mutation Res. 312, s. 217–23 3. |
|
4. |
Kier, L. D. , Brusick I., Auletta, A. E., Von Halle, E. S., Brown, M. M, Simmon, V. R, dunkel, V., McCann, J., Mortelmans, K., Prival, M., Rao, T.K. and Ray V. (1986), The Salmonella typhimurium/Mam malian Microsomal Assay: A Report of the U.S. Environmental Protection Agency Gene-Tox Program. Mutation Res. 168, s. 69–240. |
|
5. |
Yahagi, T., Degawa, M., Seino, Y. Y. , Matsushima, T., Nagao, M., Sugimura, T. and Hashimoto, Y. (1975), Mutagenicity of Carcinogen Azo Dyes and their Derivatives, Cancer Letters, 1, s. 91–96. |
|
6. |
Matsushima, M., Sugimura, T., Nagao, M., Yahagi, T., Shirai, A. and Sawamura, M. (1980), Factors Modulating Mutagenicity Microbial Tests, in: Short-term Test Systems for Detecting Carcinogens, ed. Norpoth K. H. and Garner, R. C, Springer, Berlin-Heidelberg-New York, s. 273–285. |
|
7. |
Gatehouse, D. G., Rowland, 1. R., Wilcox. P., Callender, R. D. and Foster, R. (1980), Bacterial Mutation Assays, in: Basic Mutagenicily Tests: UKEMS Part I Revised. ed. D.). Kirkland, Cambridge University Press, s. 13–61. |
|
8. |
Aeschacher, H. U., Wolleb. U. and Porchet, L. (1987), Liquid Preincubation Mutagenicity Test for Foods. J. l:ood Safety, 8, s. 167–177. |
|
9. |
Green, M. H. L., Muriel, W. J. and Bridges, B. A. (1976), Use of a simplified fluctuation test to detect low levels of mutagens, Mutation Res., 38, s. 33–42. |
|
10. |
Hubbard, S. A., Green, M. H. L, Gatehouse, D. and Bridges,]. W. (1984), The Fluctuation Test in Bacteria, in: Handbook of Mutagenicity Test Procedures, 2nd Edition, cd. Kilbey, B. J., Legator, M., Nichols, W. and Ramel, C, Elsevier, Amsterdam-New York-Oxford, s. 141–161. |
|
11. |
Thompson, E. D. and Melampy, P. J. (1981), An Examination of the Quantitative Suspension Assay for Mutagenesis with Strains of Salmonella lyphimuriurn, Environmental Mutagenesis. 3, s. 453–465. |
|
12. |
Araki, A., Noguchi, T., Kato, F. and Matsushima, T. (1994), Improved Method for Mutagenicity Testing of Gaseous Compounds by Using a Gas Sampling Bag, Mutation Res., 307, s. 335–344. |
|
13. |
Prival, M. J., Bell, S. J., Mitchell, V. D., Reipert, M. D. and Vaughan, V. L. (1984), Mutagenicity of Benzidine and Benzidine-Congener Dyes and Selected Monoazo Dyes in a Modified Salmonella Assay, Muta tion Res., 136, s. 33–47. |
|
14. |
Zeiger, E., Anderson. B. E., Haworth, S., Lawlor, T. and Mortelmans, K. (1992), Salmonella Mutagenicity Tests. V. Results from the Testing of 311 Chemicals, Environ. Mol. Mutagen., 19, s. 2–141. |
|
15. |
Simmon, V., Kauhanen, K. and Tardiff, R. G. (1977), Mutagenic Activity of Chemicals Identified in Drinking Water, In Progress in Genetic Toxicology, D. Scott, B. Bridges and F., Sobels (eds.) Elsevier, Ams terdam, s. 249–258. |
|
16. |
Hughes, T.)., Simmons, D. M, Monteith, I. G. and Claxton, L. D. (1987), Vaporization Technique to Measure Mutagenic Activity of Volatile Organic Chemicals in the Ames/Salmonella Assay, Environmenlal Mutagenesis, 9, s. 421–441. |
|
17. |
Matsushima, T., Matsumoto, A., Shirai, M., Sawamura, M., and Sugimura, T. (1979), Mutagenicity of the Naturally Occurring Carcinogen Cycasin and Synthetic Methylazoxy Methane Conjugates in Salmo nella typhimurium, Cancer Res., 39, s. 3780–3782. |
|
18. |
Tamura, G., Gold, C, Ferro-Luzzi, A. and Arnes, B. N. (1980), Fecalase: A Model for Activation of Dietary Glycosides to Mutagens by Intestinal Flora, Proc. Natl. Acad. Sci,. USA, 77, s. 4961–4965. |
|
19. |
Wilcox, P., Naidoo, A., Wedd, D.). and Gatehouse, D. G. (1990), Comparison of Salmonella typhimurium TA 102 with Escherichia coli WP2 Tester strains, Mutagenesis, 5, s. 285–291. |
|
20. |
Matsushima, T., Sawamura, M., Hara, K. and Sugimura, T. (1976), A Safe Substitute for Polychlorinated Biphenyls as an Inducer or Metabolic Activation Systems, in: in vitro Metabolic Activation in Mutagenesis Testing, eds. F. (. de Serres et al. Elsevier, North Holland, s. 85–88. |
|
21. |
Elliot, B. M., Combes, R. D., Elcombe, C. R., Gatehouse, D. G., Gibson, G. G., Mackay, J. M. and Wolf, R. C. (1992), Alternatives to Aroclor 1254-induced S9 in in vitro Genotoxicity Assays, Mutagenesis, 7, s. 175–177. |
|
22. |
Maron, D., Katzenellenbogen, J. and Arnes, B. N. (1981), Compatibility of Organic Solvents with the Salmonella/Microsome Test, Mutation Res., 88, s. 343–350. |
|
23. |
Claxton, L. D., Allen, J., Auletta, A., Mortelmans, K., Nestmann, E. and Zeiger, E. (1987), Guide for the Salmonella typhimurium/Mammalian Microsome Tests for Bacterial Mutagenicity, Mutation Res., 189, s. 83–91. |
|
24. |
Mahon, G. A. T., Green, M. H. L, Middleton, B., Mitchel. I., Robinson, W. D. and Tweats, D. J. (1989), Analysis of Data from Microbial Colony Assays, in: UKEMS Sub-Committee on Guidelines for Mutagenicity Testing. Part II. Statistical Evaluation of Mutagenicity Test Dala, ed. Kirkland, D.]., Cambridge University Press, s. 28–65. |
B. 15 MUTAGENICITETSTEST OCH SCREENING FÖR CANCEROGENICITET GENMUTATION – SACCHAROMYCES CEREVISIAE
1. METOD
1.1 INLEDNING
Se allmän inledning till del B
1.2 DEFINITION
Se allmän inledning till del B.
1.3 REFERENSÄMNEN
Inga.
1.4 TESTMETODSPRINCIP
En rad olika haploida och diploida stammar av jästen Saccaromyces cerevisiae kan användas vid undersökning av genmutationer, som induceras av kemiska ämnen med och utan metabolisk aktivering.
Man har använt framåtmutationssystem med haploida stammar vid t.ex. bestämning av mutation från röda, adeninberoende mutanter (ade-1, ade-2) till dubbelt adeinberoende vita mutanter och selektiva system t.ex. induktion av canavanin-resistens och cycloheximid-resistens.
De bäst validerade tillbakamutationssystemen bygger på användning av den haploida stammen XV 185-14C, som är bärare av de ockra nonsensmutationerna ade 2-1, arg 4-17, lys 1-1 och trp 5-48, som tillbakamuterar med basparsubstitutionsmutagener, som inducerar punktmutationer eller ockra suppressormutationer. XV 185-14C är också bärare av bis 1-7 markören som är en missensemutation som huvudsakligen tillbakamuterar vid mutationer i andra loci, och markören bom 3-10 som tillbakamuterar med frameshift-mutagener.
I diploida stammar av Saccaromyces cerevisiae är D7 som är homozygot för ilv 1-92, den enda med omfattande användning.
1.5 KVALITETSKRITERIER
Inga.
1.6 METODBESKRIVNING
Förberedelser
Lösningar av testämne och kontrollämne framställs omedelbart före försöket med hjälp av en passande vehikel. Om organiska kemiska förbindelser som inte går att lösa i vatten används, bör organiska lösningsmedel som etanol, aceton eller dimetylsulfoxid (DMSO) användas i koncentrationer på upp till 2 % v/v. Koncentrationen av vehiklen bör inte orsaka någon ändring av cellers livskraft eller växtegenskaper.
Cellerna exponeras för testämnet både i närvaro och i frånvaro av ett lämpligt exogent metaboliskt aktiveringssystem.
Det mest använda systemet består av en post-mitokondrial fraktion som kompletterats med en co-faktor, framställd från lever av gnagare som förbehandlats med enzyminducerande medel. Användningen av andra arter, vävnader, post-mitokondriala faktorer, eller förfarande kan också vara lämplig för metabolisk aktivering.
Testbetingelser
Den haploida stammen XV 185-14C och den diploida stammen D, är de mest vanligen använda i genmutationstester. Även andra stammar kan vara lämpliga.
Vid bestämning av överlevnadsgrad och antal mutanter används lämpliga kulturmedier.
Positiva kontroller, obehandlade kontroller och kontroller med lösningsmedel ska användas jämsides. Lämpliga positiva kontrollämnen ska användas för varje enskild genetisk endpoint.
Minst fem olika koncentrationer av testämnet med passande intervaller mellan koncentrationerna ska användas. För toxiska ämnen ska den högsta koncentrationen som testas inte minska överlevnadsgraden till mindre än 5–10 %. Ämnen som är relativt oupplösliga i vatten testas på lämpligt sätt upp till gränsen för löslighet med användning av lämliga förfaranden. De högsta koncentrationerna av icke-toxiska testämnen som är fritt lösliga i vatten fastställs från test till test.
Plattorna inkuberas i 4–7 dagar vid 28–30 oC i mörker.
Subkulturer bör användas med spontana mutationsfrekvenser inom allmänt accepterade gränser.
Minst tre parallella plattor används för varje koncentration vid bestämning av prototrofer som framkommit vid genmutation, och av cellernas livskraft. När man testar markörer med, som t.ex. horn 3–10, en låg mutationsfrekvens, kan antalet plattor ökas så att en statistiskt relevant mängd data erhålls.
Testförfarande
Stammar av Saccaromyces cerevisiae exponeras normalt genom ett test i vätska som inbegriper antingen celler i stationär fas eller celler i växtfas. Inledande undersökningar bör utföras med celler i växtfas. 1-5 x 107 celler per ml exponeras för testämnet i upp till 18 timmar vid 28–37 oC under skakning. Om det behövs tillsätts en passande mängd metabolisk aktiveringssystem under exponeringen. Efter behandlingen centrifugeras och tvättas cellerna varefter de utsås på ett lämpligt kulturmedium. Efter inkubationen registreras induktionen av genmutationer och antal överlevande per platta. Om det första testet utfaller negativt bör ett andra utföras med användning av celler i stationär fas. Om det första testet utfaller positivt ska det bekräftas genom ett oberoende test.
2. DATA
Data ska ställas upp i tabellform och ange antal räknade kolonier, antal mutanter, överlevnadsfrekvens och mutationsfrekvens. Alla resultat ska bekräftas genom ett oberoende test. Utvärderingen av resultaten bör ske på grundval av en lämplig statistisk metod.
3. RAPPORTERING
3.1 TESTRAPPORTEN
Testrapporten ska om möjligt innehålla minst följande uppgifter:
|
— |
Använda stammar. |
|
— |
Testbetingelser: stationär fas eller växtceller, sammansättning av media, inkubationstemperatur och inkubationsperiod, metaboliskt aktiveringssystem. |
|
— |
Exponeringsbetingelser, exponeringskoncentrationer, förfarande och varaktighet, exponeringstemperatur positiva och negativa kontroller. |
|
— |
Antal räknade kolonier, antal mutanter, Överlevnadsfrekvens och mutationsfrekvens, förhållandet mellan dos och effekt när detta är möjligt, statistisk utvärdering av data. |
|
— |
Diskussion om resultaten. |
|
— |
Tolkning av resultaten. |
3.2 UTVÄRDERING OCH TOLKNING
Se allmän inledning till del B.
4. HÄNVISNINGAR
Se allmän inledning till del B.
B.16 MITOTISK. REKOMBINATION — SACCHAROMYCES CEREVISIAE
1. METOD
1.1 INLEDNING
Se allmän inledning till del B.
1.2 DEFINITION
Se allmän inledning till del B.
1.3 REFERENSÄMNEN
Inga.
1.4 TESTMETODSPRINCIP
Hos Saccaromyces cerevisiae kan mitotisk rekombination mellan gener (eller mer generellt mellan en gen och dess centromer) och inom samma gen konstateras. Den förstnämnda formen kallas mitotisk överkorsning och framkallar reciproka produkter medan den andra formen för det mesta inte är reciprok och kallas genkonversion. Överkorsning bestäms normalt genom framkallande av recessiva homozygotiska kolonier eller sektorer i en heterozygotisk stam, medan genkonversion bestäms genom framkallande av prototrofa revertanter i en auxotrof heteroallelisk stam, som är bärare till olika, defekta alleler av samma gen. De mest använda stammarna vid undersökning av mitotisk genkonversion är D4 (med heteroallel ade 2 och trp 5), D7 (med heteroallel trp 5), BZ34 (med heteroallel arg 4) och JD1 (med heteroallel bis 4 och trp 5). Mitotisk överkorsning som framkallar röda och ljusröda homozygotiska sektorer kan undersökas i D5 eller i D7 (som även mäter mitotisk genkonversion och omvänd mutation av ilv 1-92), eftersom bägge stammar är heteroalleliska för att komplettera alleler av ade 2.
1.5 KVALITETSKRITERIER
Inga.
1.6 METODBESKRIVNING
Förberedelser
Lösningar av testämne och kontroll- eller referensämne framställs omedelbart före försöket med hjälp av passande vehikel. Om organiska kemiska förbindelser som inte går att lösa i vatten används, bör organiska lösningsmedel som etanol, aceton eller dimetylsulfoxid (DMSO) användas i koncentrationer på upp till 2 % v/v. Koncentrationen av vehiklen bör inte orsaka någon ändring av cellers livskraft eller växtegenskaper.
Cellerna exponeras för testämnet både i närvaro och i frånvaro av ett lämpligt exogent metaboliskt aktiveringssystem. Det vanligen använda systemet består av en post-mitokondrial fraktion som kompletterats med en co-faktor, framställd från lever av gnagare som förbehandlats med enzyminducerande medel. Användningen av andra arter, vävnader, post-mitokondriala faktorer, eller förfaranden kan också vara lämplig för metabolisk aktivering.
Testbetingelser
De mest vanligen använda stammarna är de diploida D4, D5, D7 och JD1. Även andra stammar kan vara lämpliga.
Vid bestämning av överlevnadsgrad och mitotisk rekombination används lämpliga kulturmedier.
Positiva kontroller, obehandlade kontroller och kontroller med lösningsmedel ska användas jämsides. Lämpliga positiva kontrollämnen ska användas för varje enskild genetisk endpoint.
Minst fem olika koncentrationer av testämnet med passande intervaller mellan koncentrationerna ska användas. Här måste man ta hänsyn till faktorer som cytotoxicitet och löslighet. De lägsta koncentrationerna bör inte ha någon verkan på cellernas överlevnadsfrekvens. För toxiska ämnen ska den högsta koncentrationen som testas inte minska överlevnadsgraden till mindre än 5–10 %. Ämnen som är relativt oupplösliga i vatten testas på lämpligt sätt upp till gränsen för löslighet. Den högsta koncentrationen av icke-toxiska testämnen, som är fritt lösliga i vatten, fastställs från test till test.
Celler i stationär fas eller växtfas kan exponeras för testämnet i upp till 18 timmar. Om långa exponeringsperioder används bör dock kulturerna undersökas mikroskopiskt för sporbildning vars närvaro gör testet värdelöst.
Plattorna inkuberas i 4–7 dagar vid 28–30 oC i mörker. Plattor som används för bestämning av röda och ljusröda homozygota sektorer, som framkallats genom mitotisk överkorsning, förvaras i kylskåp (ca 4 oC) i ytterligare 1-2 dagar innan räkning för att möjliggöra utveckling av lämpliga pigmenterade kolonier.
Subkulturer bör användas med spontana mutationsfrekvenser inom allmänt accepterade gränser.
Minst tre parallella plattor används för varje koncentration vid bestämning av prototrofer som framkommit vid genmutation, och av cellernas livskraft. När man testar recessiva homozygoter, som framkallats genom mitotisk överkorsning, ökas antalet plattor så att ett lämpligt antal kolonier erhålls.
Testförfarande
Stammar av Saccaromyces cerevisiae exponeras normalt genom test i vätska som inbegriper antingen celler i stationär fas eller celler i växtfas. Inledande undersökningar bör utföras med celler i växtfas. 1-5 x 107 celler per ml exponeras för testämnet i upp till 18 timmar vid 28–37 oC under skakning. Om det behövs tillsätts en passande mängd metabolisk aktiveringssystem under exponeringen.
Efter behandlingen centrifugeras och tvättas cellerna varefter de utsås på ett lämpligt kulturmedium. I slutet av inkubationen registreras induktionen av genmutationer och antal överlevande per platta.
Om det första testet utfaller negativt bör ett andra utföras med användning av celler i stationär fas. Om det första testet utfaller positivt ska det bekräftas genom ett oberoende test.
2. DATA
Data ska ställas upp i tabellform och ange antal räknade kolonier, antal rekombinanter samt rekombinationsfrekvens.
Alla resultat ska bekräftas genom ett oberoende test.
Utvärderingen av resultaten bör ske på grundval av en lämplig statistisk metod.
3. RAPPORTERING
3.1 TESTRAPPORT
Testrapporten ska om möjligt innehålla minst följande uppgifter:
|
— |
Använda stammar. |
|
— |
Testbetingelser: stationär fas eller växtceller, sammansättning av medier, inkubationstemperatur och inkubationsperiod, metaboliskt aktiveringssystem. |
|
— |
Exponeringsbetingelser, exponeringskoncentrationer, förfarande och varaktighet, exponeringstemperatur positiva och negativa kontroller. |
|
— |
Antal räknade kolonier, antal mutanter, överlevnadsfrekvens och mutationsfrekvens, förhållandet mellan dos och effekt när detta är möjligt, statistisk utvärdering av data. |
|
— |
Diskussion om resultaten. |
|
— |
Tolkning av resultaten. |
3.2 UTVÄRDERING OCH TOLKNING
Se allmän inledning till del B.
4. HÄNVISNINGAR
Se allmän inledning till del B.
B.17 MUTAGENICITET – TEST AV GENMUTATIONER HOS DÄGGDJURSCELLER IN VITRO
1. METOD
Denna metod är en kopia av OECD TG 476, Test av genmutationer hos däggdjursceller in vitro (In Vitro Mammalian Cell Gene Mutation Test (1997)).
1.1 INLEDNING
Genmutationstestet in vitro för däggdjursceller kan användas för att detektera genmutationer som inducerats med hjälp av kemiska substanser. Lämpliga cellinjer innefattar L5178Y muslymfomceller, CHO-, CHO-AS52-och V79-linjerna från kinesiska hamsterceller och TK6 lymfoblastoidceller från människa (1). 1 dessa cellinjer mäter de vanligast använda genetiska slutpunkterna mutation av tymidinkinas (TK) och hypoxanthinguanin fosforibosyltransferas (HPRT), och en transgen av xanthin-guanin fosforibosyltransferas (XPRT). Mutationstesterna TK, HPRT och XPRT detekterar olika spektra av genetiska händelser. Den autosomala lokaliseringen av TK och XPRT kan göra det möjligt att detektera genetiska händelser (t.ex. stora deletioner) som inte detekteras vid HPRT-lokuset på X-kromosomer (2) (3) (4) (5) (6).
I genmutationstestet in vitro för däggdjursceller kan kulturer med etablerade cellinjer eller celltyper användas. De celler som används väljs ut på basis av tillväxtförmåga vid odling och stabilitet med avseende på spontan mutationsfrekvens.
Tester som utförs in vitro kräver i allmänhet att en exogen källa för metabolisk aktivering används. Detta system för metabolisk aktivering kan inte fullt ut efterlikna förhållandena in vivo hos däggdjur. Försiktighet bör iakttagas så att man undviker förhållanden som skulle kunna leda till resultat som inte avspeglar den verkliga mutageniciteten. Positiva resultat som inte avspeglar den verkliga mutageniciteten kan uppkomma från förändringar i pH, osmolalitet eller hög cytotoxicitet (7).
Detta test används för att undersöka möjliga mutagener och karcinogener i däggdjur. Många ämnen som är positiva i detta test är karcinogena i däggdjur. Det föreligger dock ingen perfekt korrelation mellan detta test och karcinogenicitet. Korrelationen är beroende på den kemiska klassen och det finns allt fler bevis för att det finns karcinogener som inte detekteras med hjälp av detta test, eftersom de verkar uppträda genom andra, icke-genotoxiska mekanismer eller mekanismer som är frånvarande i bakterieceller (6).
Se även Allmän inledning, del B.
1.2 DEFINITIONER
framåtmutation: en genmutation från den parentala typen till den mutanta formen, vilket ger upphov till en förändring eller en förlust av den enzymatiska aktiviteten i funktionen hos det protein som bildas.
mulagener som ger basparsubstitutioner: substanser som orsakar substitution av ett eller flera baspar i DNA.
förskjutnings (frameshift)-mutagene: substanser som orsakar addition eller deletion av ett eller flera baspar i DNA-molekylen.
fenotypisk expressionslid: en period under vilken celler som nyligen muterat töms på oförändrade genprodukter. mutantfrekvens: antalet mutanta celler som observerats, delat med antalet livskraftiga celler.
relativ total tillväxt: ökningen av antalet celler över tiden jämfört med en kontrollpopulation av celler, beräknat som produkten av suspensionstillväxten i förhållande till den negativa kontrollen, gånger kloningseffektiviteten i förhållande till den negativa kontrollen.
relativ suspensionstillväxt: ökning av cellantalet delat med expressionsperioden i relation till den negativa kontrollen.
viabilitel: kloningens effektivitet hos de behandlade cellerna vid tiden för utstryk på platta, under selektiva betingelser efter expressionsperioden.
överlevnad: kloningens effektivitet hos de behandlade cellerna när utstryk på platta sker vid behandlingsperiodens slut.
Överlevnaden uttrycks vanligen i förhållande till överlevnaden hos kontrollens cellpopulation.
1.3 TESTMETODENS PRINCIP
Celler som lider brist pä tymidinkinas (TK), beroende på mutationen TK± —> TK+/-, är resistenta mot de cytotoxiska effekterna av pyrimidinanalogen trifluortymidin (TFT). Celler med väl fungerande tymidinkinas är känsliga för TFT, eftersom TFT ger upphov till inhibering av cellmetabolismen och stoppar fortsatt celldelning. Mutanta celler kan således utvecklas i närvaro av TFT, medan däremot normala celler, som innehåller tymidinkinas, inte klarar detta. Celler som lider brist på HPRT eller XPRT kan på liknande sätt väljas ut genom resistens mot 6-tioguanin (TG) eller 8-azaguanin (AG). Egenskaperna hos testsubstansen bör noggrant gås igenom om en basanalog eller ett ämne som är relaterat till det selektiva ämnet testas i något av testerna för genmutationer i däggdjursceller. Exempelvis bör varje misstänkt selektiv toxicitet hos testsubstansen mot mutanta och icke-mutanta celler undersökas. Det sätt som det selekterande systemet/ämnet fungerar på, måste således bekräftas när man testar kemikalier som är strukturellt relaterade med det selektiva ämnet (8).
Celler i suspension eller kulturer i form av ett enkelskikt exponeras för testsubstansen, både med och utan metabolisk aktivering, under en lämplig tidsperiod och subkultiveras för att bestämma cytotoxiciteten och för att tillåta fenotypisk expression innan mutantvalet sker (9) (10) (11) (12) (13). Cytotoxiciteten bestäms i vanliga fall genom att mäta den relativa kloningseffektiviteten (överlevnad) eller den relativa totala tillväxten för kulturerna efter behandlingsperioden. De behandlade kulturerna bibehålls i ett tillväxtmedium under en tillräckligt lång tid, karakteristisk för varje selekterat lokus och celltyp, för att tillåta i det närmaste optimal fenotypisk expression av inducerade mutationer. Mutationsfrekvensen bestäms genom att inockulera ett känt antal celler i ett medium som innehåller det selektiva ämnet för detektion av mutanta celler, och i ett medium utan selektivt ämne för bestämning av kloningseffektiviteten (viabiliteten). Efter en lämplig inkubationstid räknas cellerna. Mutationsfrekvensen erhålls från antalet mutanta kolonier i selektivt medium och antalet kolonier i icke-selektivt medium.
1.4 BESKRIVNING AV TESTMETODEN
1.4.1 Förberedelser
1.4.1.1 Celler
Det finns ett antal olika celltyper som kan användas i detta test, inklusive subkloner från L5178Y, CHO, CHO-AS52, V79 eller TK6-celler. De celltyper som används i detta test bör ha en demonstrerad känslighet mot kemiska mutagener, en hög kloningseffektivitet och en stabil spontan mutationsfrekvens. Cellerna bör kontrolleras med avseende på kontaminering från mykoplasma och bör inte användas om kontaminering skett.
Testet bör utformas på ett sådant sätt att det har en förutbestämd känslighet och styrka. Antalet celler, kulturer och koncentrationer av testsubstansen som används bör reflektera dessa definierade parametrar (14). Det minsta antalet livskraftiga celler som överlever behandlingen och används vid varje stadium av testet bör baseras på den spontana mutationsfrekvensen. En generell regel är att använda ett cellantal som är minst tio gånger det inverterade värdet av den spontana mutationsfrekvensen. Det rekommenderas dock att minst 106 celler används. Adekvata historiska data för det cellsystem som används bör vara tillgängliga för att kontrollera att testet ger reproducerbara resultat.
1.4.1.2 Betingelser för media och kulturer
Lämpliga kulturmedia och inkubationsbetingelser (odlingskärl, temperatur, CO2-koncentration och luftfuktighet) bör användas. Mediet bör väljas ut i enighet med de selektiva system och celltyper som används i testet. Det är särskilt viktigt att betingelserna för kulturerna väljs på ett sådant sätt att man säkrar en optimal celltillväxt under expressionsperioden och en förmåga till bildning av kolonier för både mutanta och icke-mutanta celler.
1.4.1.3 Förberedelse av kulturer
Celler dras upp från stamkulturer, inokuleras i ett odlingsmedium och inkuberas vid 37 oC. Innan de används i detta test kan kulturerna behöva tvättas rena från redan existerande mutanta celler.
1.4.1.4 Metabolisk aktivering
Celler bör exponeras för testsubstansen både i närvaro och frånvaro av ett lämpligt system för metabolisk aktivering. Det system som vanligen används är en postmitokondriell fraktion som tillsatts en kofaktor (S9), beredd ur levern från gnagare vilka behandlats med enzyminducerande ämnen såsom Aroclor 1254 (15) (16) (17) (18) eller en kombination av fenobarbiton och β-naftoflavon (19) (20).
Den postmitokondriella fraktionen används vanligen vid koncentrationer i området 1–10 % v/v i det slutliga testmediet. Valet av och betingelserna för ett system för metabolisk aktivering kan vara beroende av klassen av kemikalier som testas. I vissa fall kan det vara lämpligt att använda mer än en koncentration av den postmitokondriella fraktionen.
Ett antal vidareutvecklingar, inklusive konstruktion av genetiskt modifierade cellinjer som uttrycker specifika aktiverande enzymer, kan ge en potential för endogen aktivering. Valet av de cellinjer som används bör vara vetenskapligt motiverat (t.ex. genom relevansen av cytokrom P450 isoenzym för metabolismen av testsubstansen).
1.4.1.5 Beredning av testsubstansen
Fasta testsubstanser bör lösas upp i eller blandas med lämpliga lösningsmedel eller vehiklar och spädas ut innan cellerna behandlas, om detta är lämpligt. Flytande testsubstanser kan tillsättas direkt till testsystemen och/eller spädas ut innan behandlingen startas. Nygjorda beredningar av testsubstansen bör användas om inte stabilitetsdata tyder på att lagring kan accepteras.
1.4.2 Försöksbetingelser
1.4.2.1 Lösningsmedel/vehikel
Lösningsmedlet/vehikeln bör inte kunna misstänkas för att reagera kemiskt med testsubstansen och bör vara förenlig med överlevnaden för cellerna samt S9-aktiviteten. Om andra lösningsmedel/vehiklar än gängse brukliga används bör det finnas data som visar att de är kompatibla. Det rekommenderas att vattenbaserade lösningsmedel/vehiklar i första hand bör övervägas närhelst det är möjligt. När substanser som är instabila i vatten testas, bör de organiska lösningsmedel som används vara fria från vatten. Vatten kan avlägsnas genom tillsats av en molekylsikt.
1.4.2.2 Exponeringskoncentrationer
Bland de kriterier som ska övervägas när den högsta koncentrationen ska fastställas är cytotoxicitet, löslighet i testsystemet och förändringar i pH eller osmolalitet.
Cytotoxicitet bör fastställas såväl med som utan metabolisk aktivering i huvudexperimentet med hjälp av lämplig indikation på cellernas integritet och tillväxt, såsom relativ kloningseffektivitet (överlevnad) eller relativ total tillväxt. Det kan vara lämpligt att fastställa cytotoxicitet och löslighet i ett förberedande experiment.
Minst fyra analyserbara koncentrationer bör användas. Där cytotoxicitet uppträder bör dessa koncentrationer täcka in ett område som sträcker sig från maximal toxicitet ner till liten eller ingen toxicitet. Detta innebär normalt att koncentrationsnivåerna bör separeras av minst en faktor mellan 2 och \10. Om den maximala koncentrationen är baserad på cytotoxicitet bör detta då resultera i ungefär 10–20 % (men inte mindre än 10 %) relativ överlevnad (relativ kloningseffektivitet) eller relativ total tillväxt. För relativt icke-cytotoxiska substanser, bör den maximala testkoncentrationen vara 5 mg/ml, 5 μl/ml eller 0,01 M, beroende på vilket som är det lägsta.
Relativt olösliga substanser bör testas upp till eller över sin löslighetsgräns under odlingsbetingelserna för kulturen. Bevis för olöslighet bör bestämmas i det slutliga behandlingsmediet för vilket cellerna exponeras. Det kan vara lämpligt att fastställa lösligheten vid behandlingens början och slutpunkt, eftersom lösligheten kan förändras under exponeringsförloppet i testsystemet beroende på närvaron av celler, S9, serum, etc. Olöslighet kan detekteras med hjälp av blotta ögat. Fällningen torde inte störa bestämningen.
1.4.2.3 Kontroller
Samtidiga positiva och negativa kontroller (lösningsmedel eller vehikel), både med och utan metabolisk aktivering, bör inkluderas i varje experiment. När metabolisk aktivering används, bör den positiva kontrollkemikalien vara av sådant slag att den kräver aktivering för att ge ett mutagent svar.
I tabellen nedan ges några exempel på positiva kontrollsubstanser.
|
Betingelser för metabolisk akti-vering |
Lokus |
Substans |
CAS-nummer |
EINECS-nummer |
|
Frånvaro av exogen metabolisk aktivering |
HPRT |
Etylmetansulfonat |
62-50-0 |
200-536-7 |
|
Etylnitrosourea |
759-73-9 |
212-072-2 |
||
|
TK (små och stora kolonier) |
Metylmetansulfonat |
66-27-3 |
200-62 5-0 |
|
|
XPRT |
Etylmetansulfonat |
62-50-0 |
200-536-7 |
|
|
Etylnitrosourea |
759-73-9 |
212-072-2 |
||
|
Närvaro av exogen metabolisk aktivering |
HPRT |
3-Metylkolantren |
56-49-5 |
200-276-4 |
|
N-Nitrosodimetylamin |
62-75-9 |
200-549-8 |
||
|
7,12-Dimetylbensantraccn |
57-97-6 |
200-359-5 |
||
|
TK (små och stora kolonier) |
Cyklofosfoamid |
50-18-0 |
200-015-4 |
|
|
Cyklofosfoamidmonohydrat |
6055-19-2 |
|
||
|
Bcns[a]pyrcn |
50-32-8 |
200-028-5 |
||
|
3-Metylkolantren |
56-49-5 |
200-276-5 |
||
|
XPRT |
N-Nitrosodimetylamin (för höga nivåer av S-9) |
62-75-9 |
200-549-8 |
|
|
Bens[a]pyren |
50-32-8 |
200-028-5 |
Andra lämpliga referenssubstanser för positiv kontroll kan användas, t.ex. om ett laboratorium har en historisk databas med 5-brom-2'-deoxiuridin [CAS-nummer 59-14-3, ElNECS-nummer 200-415-9] skulle även denna referenssubstans kunna användas. Användning av kemiska klassrelaterade positiva kontrollkemikalier bör övervägas när sådana finns att tillgå.
Negativa kontroller, bestående av enbart ett lösningsmedel eller en vehikel i behandlingsmediet och behandlade på samma sätt som behandlingsgrupperna bör inkluderas. Dessutom bör även obehandlade kontroller användas, om det inte finns historiska kontrolldata som visar att inga skadliga eller mutagena effekter induceras av det valda lösningsmedlet.
1.4.3 Förfarande
1.4.3.1 Behandling med testsubstansen
Prolifererande celler bör exponeras för testsubstansen både med och utan metabolisk aktivering. Exponering bör ske under en lämplig tidsperiod (normalt är 3–6 timmar effektivt). Exponeringstiden kan utsträckas över en eller flera cellcykler.
Antingen en eller två behandlade kulturer kan användas vid varje koncentration som testas. När enstaka kulturer används, bör antalet koncentrationer ökas så att ett adekvat antal kulturer säkerställs för analys (t.ex. minst åtta analyserbara koncentrationer). Två negativa (lösningsmedels-) kontrollkulturer bör användas.
Gasformiga eller flyktiga substanser bör testas med hjälp av lämpliga metoder, såsom slutna odlingskärl (21) (22).
1.4.3.2 Mätning av överlevnad, viabilitet och mutantfrekvens
Mot slutet av exponeringsperioden tvättas cellerna och odlas för att bestämma överlevnad och för att medge att den mutanta fenotypen uttrycks. Mätning av cytotoxicitet, genom en bestämning av den relativa kloningseffektiviteten (överlevnad) eller relativa totala tillväxten för kulturerna, initieras normalt efter behandlingsperioden.
Varje lokus har definierade krav på minimitid, så att närmast optimal fenotypisk expression av nyligen inducerade mutanter medges (HPRT och XPRT kräver minst 6–8 dagar och TK minst två dagar). Celler odlas ofta i ett medium med och utan selektiva ämnen för att såväl antalet mutanter som kloningens effektivitet ska kunna bestämmas. Mätningen av viabiliteten (används för att beräkna mutantfrekvensen) initieras vid slutet av expressionstiden genom utstryk på platta i ett icke-selektivt medium.
Om testsubstansen är positiv i testet L5178Y TK+/-, bör storleksmätning av kolonierna utföras på åtminstone en av testkulturerna (den högsta positiva koncentrationen) och på de negativa och positiva kontrollerna. Om testsubstansen är negativ i testet L5178Y TK+/-, bör storleksmätning av kolonierna utföras på de negativa och positiva kontrollerna. I undersökningar där TK6TK+/- används, kan storleksmätning av kolonierna utföras även där.
2. DATA
2.1 BEHANDLING AV RESULTATEN
Data bör inkludera en bestämning av cytotoxicitet och viabilitet, räkning av kolonier och mutantfrekvenser för de behandlade kulturerna och kontrollkulturerna. Om testet L5178Y TK+/- ger ett positivt svar, påvisas kolonier utifrån kriterier för små och stora kolonier för åtminstone en koncentration av testsubstansen (högsta positiva koncentrationen) och för de negativa och positiva kontrollerna. Den molekylära och cytogenetiska naturen hos både stora och små mutanta kolonier har undersökts mycket ingående (23) (24). I testet TK+/-påvisas kolonier utifrån kriterier för normal tillväxt (stora) och långsam tillväxt (små) av kolonier (25). Mutanta celler som har utsatts för den mest omfattande genetiska skadan uppvisar förlängda dubbleringstider och bildar därför små kolonier. Denna skada sträcker sig typiskt i en skala från förlust av hela genen till karyotypiskt synliga kromosomavvikelser. Induktion av mutanter som ger små kolonier har associerats med kemikalier som inducerar omfattande kromosomavvikelser (26). Mutanta celler som är mindre allvarligt påverkade växer med hastigheter liknande de som parentala celler uppvisar och bildar stora kolonier.
Överlevnad (relativa kloningseffektiviteter) eller relativ total tillväxt bör anges. Mutantfrekvensen bör uttryckas som antalet mutanta celler per antalet celler som överlever.
Individuella data för kulturerna bör anges. Alla data bör dessutom sammanfattas i tabellform.
Det finns inget krav på verifiering av ett tydligt positivt svar. Tvetydiga resultat bör klarläggas genom fortsatta tester, lämpligen vid ändrade experimentella betingelser. Negativa resultat måste bekräftas från fall till fall. 1 de fall där det inte anses nödvändigt att bekräfta ett negativt resultat bör detta motiveras. Vid tvetydiga eller negativa resultat bör man vid uppföljningsexperiment överväga att ändra parametrarna i undersökningen för att utvidga de bedömda försöksbetingelserna. Försöksparametrar som kan behöva modifieras inkluderar koncentrationsstegen och villkoren för metabolisk aktivering.
2.2 UTVÄRDERING OCH TOLKNING AV RESULTATET
Det finns ett flertal kriterier som används för att bestämma om ett resultat är positivt, t.ex. en koncentrationsrelaterad ökning eller en reproducerbar ökning i mutationsfrekvensen. Man bör i första hand överväga resultatets biologiska relevans. Statistiska metoder kan vara till hjälp vid utvärdering av testresultatet. Statistisk signifikans bör dock inte vara den enda faktor som bestämmer om resultatet är positivt.
En testsubstans för vilken resultatet inte uppfyller ovanstående kriterier anses vara icke-mutagen i detta test.
Även om de flesta experiment kommer att ge tydligt positiva eller negativa resultat, kan i sällsynta fall uppsättningen av data göra det omöjligt att utföra en otvetydig bedömning med avseende på testsubstansens aktivitet. Resultatet kan förbli tvetydigt eller tveksamt, oavsett hur många gånger som experimentet upprepas.
Positiva resultat från testet av genmutationen in vitro hos däggdjursceller indikerar att testsubstansen inducerar genmutationer i de odlade däggdjursceller som används. Ett positivt koncentrationssvar som är reproducerbart är mycket meningsfullt. Negativa resultat indikerar att testsubstansen, under försöksbetingelserna, inte inducerar genmutationer i de odlade däggdjursceller som används.
3. RAPPORTERING
TESTRAPPORT
Följande information måste ingå i testrapporten:
|
|
Lösningsmedel/vehikel:
|
|
|
Celler:
|
|
|
Försöksbetingelser:
|
|
|
Resultat:
|
|
|
Diskussion av resultaten. |
|
|
Slutsatser. |
4. HÄNVISNINGAR
|
1. |
Moore, M. M., DeMarini, D. M. DeSerres, F. J. and Tindal, K. R. (eds.) (1987), Banbury Report 28; Mam-malian Cell Mutagenesis, Cold Spring Harbor Laboratory, New York. |
|
2. |
Chu, E. H. Y. and Malling H. V. (1968), Mammalian Cell Genetics. II. Chemical Induction of Specific Locus Mutations in Chinese Hamster Cells In Vitro, Proc. Natl. Acad. Sci., USA, 61, s. 1306–1312. |
|
3. |
Liber, H. L. and Thilly, W. G. (1982), Mutation Assay at the Thymidine Kinase Locus in Diploid Human Lymphoblasts, Mutation Res., 94, s. 467–485. |
|
4. |
Moore, M. M.. Harington-Brock, K., Doerr, C. L. and Dearfield, K. L. (1989), Differential Mutant Quanti tation at the Mouse Lymphoma TK and CHO HGPRT Loci. Mutagenesis, s. 394–403. |
|
5. |
Aaron, C. S., and Stankowski, Jr. L. F., (1989), Comparison of the AS52/XPRT and the CHO/HPRT Assays: Evaluation of the Six Drug Candidates, Mutation Res., 223, s. 121–128. |
|
6. |
Aaron, C. S., Bolcsfoldi, G., Glatt, H. R., Moore, M., Nishi, Y., Stankowski, Jr. L. F., Theiss, J. and Thompson, E. (1994), Mammalian Cell Gene Mutation Assays Working Group Report. Report of the International Workshop on Standardisation of Genotoxicity Test Procedures. Mutation Res., 312, s. 235–239. |
|
7. |
Scott, D., Galloway, S. M., Marshall, R. R., Ishidate, M., Brusick, D., Ashby, J. and Myhr, B. C. (1991) Genotoxicity Under Extreme Culture Conditions. A report from ICPEMC Task Group 9, Mutation Res., 25 7. s. 147–204. |
|
8. |
Clive, D., McCuen, R., Spector, J. F. S., Piper, C. and Mavournin, K. H. (1983), Specific Gene Mutations in L5178Y Cells in Culture. A Report of the U.S. Environmental Protection Agency Gene-Tox Program. Mutation Res., 115, s. 225–251. |
|
9. |
Li, A. P., Gupta, R. S., Heflich, R. H. and Wasson, J. S. (1988), A Review and Analysis of the Chinese Hamster Ovary/Hypoxanthine Guanine Phosphoribosyl Transferase System to Determine the Mutagenicity of Chemical Agents: A Report of Phase III of the U.S. Environmental Protections Agency Gene-Tox Program, Mutation Res,, 196, s. 17–36. |
|
10. |
Li, A. P., Carver, J. H., Choy, W. N., Hsie, A. W., Gupta, R. S., Loveday, K. S., O'Neill, J. P., Riddle, J. C, Stankowski, L. F. Jr. and Yang, L. L. (1987), A Guide for the Performance of the Chinese Hamster Ovary Cell/Hypoxanthine-Guanine Phosphoribosyl Transferase Gene Mutation Assay, Mutation Res., 189, s. 135–141. |
|
11. |
Liber, H. L, Yandell, D. W. and Little, J. B. (1989), A Comparison of Mutation Induction at the TK and HPRT Loci in Human Lymphoblastoid Cells: Quantitative Differences are Due to an Additional Class of Mutations at the Autosomal TK Locus, Mutation Res., 216, s. 9–17. |
|
12. |
Stankowski, L. F. Jr., Tindal,, K. R., and Hsie, A. W. (1986), Quantitative and Molecular Analyses of Ethyl Methanosulphonate and ICR 191-Induced Molecular Analyses of Ethyl Methanosulphonate and 1CR 191 Induced Mutation in AS52 Cells, Mutation Res.. 160, s. 133–147. |
|
13. |
Turner, N. T., Balton, A. G. and Clive, D. (1984), Procedure for the L51 78Y/TK+/- – TK+/- Mouse Lymphoma Cell Mutagenicity Assay, in: Kilbey, B.J. et all (eds.) Handbook oj Mutagenicity Test Procedures, Else-vier Science Publishers, New York, s. 239–268. |
|
14. |
Arlett, C. F.. Smith, D. M., Clarke, G. M., Green, M. H. L., Cole, J., McGregor, D. B. and Asquith S. C. (1989), Mammalian Cell Gene Mutation Assays Based upon Colony Formation, in: Statistical Evaluation of Mutagenicity Test Data, Kirkland, D. J., ed., Cambirdge University Press, s. 66–101. |
|
15. |
Abbondandolo, A., Bonatti, S., Corti, G., Fiorio, R., Loprieno, N. and Mazzaccaro, A. (1977), Induction of 6-Thioguanine-Resistant Mutants in V79 Chinese Hamster Cells by Mouse-Liver Microsome- Activated Dimethyllnitrosamine, Mutation Res., 46, s. 365–373. |
|
16. |
Ames, B. N., McCann, J. and Yamasaki, E. (1975), Methods for Detecting Carcinogens and Mutagens with the Salmonella/Mammalian-Microsome Mutagenicity Test, Mutation Res., 31, s. 347–364. |
|
17. |
Clive, D., Johnson, K. O., Spector, J. F. S., Batson A. G. and Brown M. M. M. (1979), Validation and Characterization of the L5178Y/TK+/- – Mouse Lymphoma Mutagen Assay System. Mutat. Res., 59, s. 61–108. |
|
18. |
Maron, D. M. and Arnes, B. N. (1983), Revised Methods for the Salmonella Mutagenicity Test, Mutuation Res.. 113, s. 173–215. |
|
19. |
Elliott, E. M., Combes, R. D., Elcombe, C. R., Gatehouse, D. G., Gibson, G. G., Mackay, J. M. and Wolf, R. C. (1992), Alternatives to Aroclor 1254-Induced S9 in: In Vitro Genotoxicity Assays, Mutagenesis 7, s. 175–177. |
|
20. |
Matsushima, T., Sawamura, M., Hara, K. and Sugimura, T. (1976), A Safe Substitute for Polycholrinated Biphenyls as an Inducer of Metabolic Activation Systems, in:In Vitro Metabolic Activation in Mutagenesis Testing, de Serres, F. J., Fouts, J. R., Bend, J. R. and Philpot, R. M. (eds), Elsevier, North-Holland, s. 85–88. |
|
21. |
Krahn, D. F., Barsky, F. C. and McCooey, K. T. (1982), CHO/HGPRT Mutation Assay: Evaluation of Gases and Volatile Liquids, in: Tice, R. R., Costa, D. L, Schaich, K. M. (eds), Genotoxic Effects of Airborne Agents, New York, Plenum, s. 91–103. |
|
22. |
Zamora, P. O., Benson, J. M., Li, A. P. and Brooks, A. L. (1983), Evaluation of an Exposure System Using Cells Grown on Gollagen Gels tor Detecting Highly Volatile Mutagens in the CHO/HGPRT Mutation Assay, Environmental Mutagenesis, 5, s. 795–801. |
|
23. |
Applegate, M. L., Moore, M. M., Broder, C. B., Burrell, A. and Hozier. J. C. (1990), Molecular Dissection of Mutations at the Heterozygous Thymidine Kinase Locus in Mouse Lymphoma Cells, Proc. Natl. Acad. Sci. USA, 87, s. 51–55. |
|
24. |
Moore, M. M., Clive, D. Hozier, J. C, Howard, B. E., Batson, A. G., Turner, N. T. and Sawyer, J. (1985), Analysis of Trifluoronthymidine, Resistant (TFT) Mutants of L5178Y/TK+/- – Mouse Lymphoma Cells, Mutation Res., 151, s. 161–174. |
|
25. |
Yandell, D. W., Dryja, T. P. and Little, J. B. (1990), Molecular Genetic Analysis of Recessive Mutations at a Heterozygous Autosomal Locus in Human Cells, Mutation Res., 229, s. 89–102. |
|
26. |
Moore, M. M. and Doerr, C. L. (1990), Comparison of Chromosome Aberration Frequency and Small Colony TK-Deficient Mutant Frequency in L5178Y/TK+/- – 3.7.2C Mouse Lymphoma Cells, Mutagenesis, 5, s. 609–614. |
B.18 DNA-SKADA OCH DNA-REPARAT1ON – FELAKTIG DNA-SYNTES –DÄGGDJURSCELLER IN VITRO
1. METOD
1.1 INLEDNING
Se allmän inledning till del B.
1.2 DEFINITION
Se allmän inledning till del B.
1.3 REFERENSÄMNEN
Inga.
1.4 TESTMETODSPRINCIP
Vid test av sporadisk DNA-syntes (UDS) mäts DNA-reparationssyntesen efter bortskärning och avlägsnade av det avsnitt av DNA-strängen som innehåller det skadade område som inducerats av kemiska eller fysiska agenser. Testen bygger på inkorporering av tritiummarkerat thymidin (3H-TdR) i däggdjurscellers DNA när dessa celler inte är i S-fasen av cellcykeln. Upptaget av 3H-TdR kan besämmas genom autoradiografi eller genom vätskescintillationsräking (LSC) av DNA från de exponerade cellerna. Såvida inte primärkulturer av råtthepatocyter används, exponeras odlade däggdjursceller för testämnet med och utan ett exogent metaboliskt aktiveringssystem. UDS-test kan också företas i in vivo-system.
1.5 KVALITETSKRITERIER
Inga.
1.6 METODBESKRIVNING
Förberedelser
Testämne samt kontroll- och referensämnen tillsätts i ett kulturmedium eller upplöses eller suspenderas i lämpliga vehiklar innan cellerna exponeras. Koncentrationen av vehikeln bör inte orsaka någon ändring av cellers livskraft.
Primärkulturer av råtthepatocyter, humana lymfocyter eller etablerade cellinjer (t.ex. humana diploida fibroblaster) kan användas.
Cellerna exponeras för testämnet både i närvaro och i frånvaro av ett lämpligt metaboliskt aktiveringssystem.
Försöksbetingelser
Vid autoradiografisk bestämning av UDS används minst två och vid LSC sex kulturer (eller mindre om det är vetenskapligt berättigat) för varje experimentpunkt.
Positiva och negativa (obehandlade och/eller vehikel) kontroller ska användas jämsides med och utan metabolisk aktivering i varje försök.
Som positiv kontroll vid användning av råtthepatocyter kan t.ex. 7,12-DMBA (7,12-dimetylbenzanthracen) eller 2-AAF (2-acetylaminofluoren) användas. Som positiv kontroll vid användning av etablerade cellinjer kan t.ex. 4-NQO (4-nitrouinloin-N-oxid) användas både i samband med autoradiografisk registering och LSC utan rnetabolisk aktivering. När metaboliska aktiveringssystem används kan t.ex. N-dimetylnitrosamin användas för positiv kontroll.
En rad olika koncentrationer av testämnet med passande intervaller mellan koncentrationerna som täcker alla verkningar ska användas. Den högsta koncentrationen bör ha en viss cytotoxisk verkan. Ämnen som är relativt oupplösliga i vatten testas på lämpligt sätt upp till gränsen för löslighet. Den högsta koncentrationen av icke-toxiska testämnen, som är fritt lösliga i vatten, fastställs från test till test.
Lämpligt växtmedium, CO2-koncentration, temperatur och fuktighet för att bibehålla kulturerna ska användas. Etablerade cellinjer ska kontrolleras regelbundet för mykoplasma-förorening.
I samband med primärkulturer av hepatocyter används inte metaboliska aktiveringssystem. Etablerade cellinjer och lymfocyter exponeras för testämnet både i närvaro och i frånvaro av ett lämpligt metaboliskt aktiveringssystem.
Förfarande
Etablerade cellinjer framställs på grundval av stamkulturer (t.ex. genom trypsinering eller skakning) och utsås med passande täthet i odlingskärl, varefter de inkuberas vid 37 oC.
Primärkulturer av råtthepatocyter etableras när hepatocyter omedelbart efter isolering förs över i ett passande medium och tillåts att häfta sig vid den växande ytan.
Humana lymfocytkulturer odlas genom därtill lämpade metoder.
Primärkulturer av råtthepatocyter
Färskt isolerade råtthepatocyter exponeras för testämnet under lämplig tid i ett medium som innehåller 3H-TdR. Efter exponering avlägsnas cellerna från mediet och tvättas, fixeras och torkas. Objektglasen bör doppas i autoradiografisk emulsion (alternativt kan ”stripping” film användas) och belyses, framkallas, färgas och räknas.
Etablerade cellinjer och lymfocyter
Autoradiografisk teknik: Cellkulturerna exponeras först för testämnet under en passande tid och därefter för 3H-TdR. Exponeringsperioderna beror på ämnena, de metaboliska aktiveringssystemen och celltyperna. Exponering för 3H-TdR bör ske samtidigt eller inom några minuter efter exponering för testämnet så att UDS mäts vid den tid då aktiviteten är störst. Valet mellan dessa två förfaranden beror på risken för interaktion mellan testämnet och 3H-TdR. För att skilja mellan UDS och semi-konservativ DNA-replikation kan den senare inhiberas genom användning av t.ex. argininfattiga medier, ett lågt seruminnehåll eller tillsättning av hydroxyurinämne till kulturmediet.
LSC-mätning av UDS: Före exponering för testämnet blockeras cellernas övergång till S-fasen som beskrivits ovan. Därefter exponeras cellerna på samma sätt som vid den autoradiografiska tekniken. Omedelbart efter inkubationen extraheras cellernas DNA och det totala DNA-innehållet samt mängden av inkorporerat 3H-TdR mäts.
När de nämnda teknikerna används på humana lymfocyter är det inte nödvändigt att hämma den semi-konservativa DNA-replikationen, såvida det inte rör sig om stimulerade kulturer.
Analys
Vid mätning av UDS i odlade celler medräknas inte kärnor i S-fasen. Minst 50 celler per koncentration ska räknas. Objektglasen bör kodmärkas innan räkning. På varje platta bör flera olika slumpmässigt valda områden med stort inbördes avstånd räknas. Mängden av inkorporerat 3H-TdR i cytoplasman bestäms genom räkning i tre områden i kärnstorlek i varje räknad cells cytoplasma.
Vid LSC-mätning av UDS används ett passande antal kulturer per koncentration och per kontrollförsök.
Alla resultat ska bekräftas genom ett oberoende test.
2. DATA
Data ställs upp i tabellform.
2.1 AUTORADIOGRAFISK MÄTNING
Mängden av inkorporerat 3H-TdR i cytoplasman och antalet korn som registreras i cellkärnområdet anges separat.
Fördelning av mängden av inkorporerat 3H-TdR i cytoplasman och antalet korn per kärna kan beskrivas genom användning av medel, median eller modus.
2.2 LSC-MÄTNING
Vid LSC-mätning anges inkorporeringen av 3H-TdR som dpm/μg DNA. Distributionen av inkorporeringen kan beskrivas genom användning av medel dpm/μg DNA med tillhörande standardavvikelse.
Utvärderingen av resultaten ska grundas på en lämplig statistisk metod.
3. RAPPORTERING
3.1 TESTRAPPORT
Testrapporten ska om möjligt innehålla minst följande uppgifter:
|
— |
Använda celler, täthet och antal passager på exponeringstidpunkten, antal cellkulturer. |
|
— |
Metoder för bibehållande av cellkulturer, inbegripet medium, temperatur och CO2– koncentration. |
|
— |
Testämne, vehikel, koncentrationer och motivering för val av använda koncentrationer. |
|
— |
Detaljerad information om de metaboliska aktiveringssystemen. |
|
— |
Exponeringsschema. |
|
— |
Positiva och negativa kontroller. |
|
— |
Använd autoradiografisk teknik. |
|
— |
Förfarande vid blockering av cellernas övergång till S-fasen. |
|
— |
Förfarande vid extraktion av DNA och bestämning av det totala DNA-innehållet vid LSC- mätningen. |
|
— |
Förhållandet mellan dos och reaktion när detta är möjligt. |
|
— |
Statistisk utvärdering. |
|
— |
Diskussion om resultaten. |
|
— |
Tolkning av resultaten. |
3.2 UTVÄRDERING OCH TOLKNING
Se allmän inledning till del B.
4. HÄNVISNINGAR
Se allmän inledning till del B.
B.19 TEST AVSEENDE SYSTERK ROMATIDBYTEN IN VITRO
1. METOD
1.1 INLEDNING
Se allmän inledning till del B.
1.2 DEFINITION
Se allmän inledning till del B.
1.3 REFERENSÄMNEN
Inga.
1.4 TESTMETODSPRINCIP
Test av systerkromaidbyten (SCE) är ett korttidstest för bestämning av reciprok utväxling av DNA mellan två systerkromatider i en kromosom under replikation. Vid SCE sker en utväxling av DNA-replikationsprodukter vid skenbart homolog loci. Man tror att utväxlingsprocessen sannolikt omfattar brytning och återförening av DNA-koder, men det finns nästan ingen kännedom om dess molekylbas. För att kunna upptäcka SCE är det nödvändigt att differentialmärka systerkromatiderna, vilket kan göras genom att inkorporera bromdeoxyuridin (BrdU) i kromosomernas DNA under två cellcykler.
Däggdjursceller exponeras in vitro för testämnet med och utan ett exogent metaboliskt aktiveringssystem från däggdjur, om detta anses relevant, och odlas under två cellcykler i ett medium som innehåller BrdU. Sedan cellerna behandlats med en spindelfiberinhibitor (t.ex. kolchicin) för att cellerna ska ackumuleras i ett metafasliknande stadium av mitosen (c-metafas), skördas de och prepareras för kromosomanalys.
1.5 KVALITETSKRITERIER
Inga.
1.6 METODBESKRIVNING
Förberedelser
|
— |
För detta test kan primärkulturer (t.ex. humana lymfocyter) eller etablerade cellinjer (t.ex. äggstocksceller från kinesisk hamster) användas. Cellinjer bör kontrolleras för mykoplasmaförorening. |
|
— |
Lämpligt kulturmedium och lämpliga inkubationsbetingelser (t.ex. temperatur, odlingskärl, CO2-koncentrationer och fuktighet) ska användas. |
|
— |
Testämnet tillsätts i ett kulturmedium eller upplöses eller suspenderas i lämpliga vehiklar innan cellerna exponeras. Koncentrationen av vehiklen bör inte orsaka någon ändring av cellers livskraft eller växtegenskaper. En eventuell påverkan av SCE-frekvensen kontrolleras genom odling av en kontrollkultur i lösningsmedlet. |
|
— |
Cellerna exponeras för testämnet både i närvaro och i frånvaro av ett lämpligt exogent metaboliskt aktiveringssystem från däggdjursceller. Om celltyper med endogen metabolisk aktiveringanvänds bör det vara bevisat att aktiveringsform och -hastighet är lämpliga för den kemiska klass som testas. |
Testbetingelser
För varje experimentell punkt används minst två parallella kulturer.
Som positiv kontroll används vid varje enskilt försök både en direkt verkande kemisk förbindelse och en förbindelse som kräver metabolisk aktivering. Dessutom företas vehikelkontroll.
Följande kemiska förbindelser kan t.ex. användas till positiv kontroll:
|
— |
direktverkande förbindelser:
|
|
— |
indirekt verkande förbindelser:
|
Vid behov kan en supplerande positiv kontroll göras med användning av en kemisk förbindelse av samma typ som testämnet.
Minst tre koncentrationer av testämnet med passande intervaller mellan koncentrationerna ska användas. Den högsta koncentrationen bör ha en tydlig cytotoxisk verkan men får inte förhindra den nödvändiga celldelningen. Ämnen som är relativt oupplösliga i vatten testas på lämpligt sätt upp till gränsen for löslighet. Den högsta koncentrationen av icke-toxiska testämnen, som är fritt lösliga i vatten, fastställs från test till test.
Testförfarande
Etablerade cellinjer framställs på grundval av stamkulturer (t.ex. genom trypsinering eller skakning) och utsås med passande täthet i odlingskärl, varefter de inkuberas vid 37 oC. För monolagskulturer justeras antalet celler per kulturkärl så att kulturerna vid tidpunkten för skörden högst täcker 50 % av växtarealen. Cellerna kan också odlas i suspension. Humana lymfocytkulturer odlas med användning av därtill lämpliga metoder på grundval av hepariniserat blod och inkuberas vid 37 oC.
Celler i den exponentiella växtfasen exponeras för testämnet under en passande period. I de flesta fall räcker det med 1–2 timmar men i vissa fall kan exponeringsperioden förlängas till att omfatta två hela cellcykler. Celler vars endogena metaboliska aktivering är otillräcklig exponeras både med och utan tillsättning av ett lämpligt metaboliskt aktiveringssystem. Efter exponeringen tvättas cellerna fria från testämne. Därefter odlas de under två replikationscykler i ett medium som innehåller BrdU. Alternativt kan cellerna exponeras för testämnet och BrdU samtidigt under en period som motsvarar två cellcykler.
Kulturer från humana lymfocyter exponeras medan de befinner sig i semisynkront tillstånd.
Cellerna analyseras under deras andra delning efter exponering vilket säkerställer att exponeringsperioden täcker den största delen av de följsamma faserna i cellcykeln. Alla kulturer som tillsatts BrdU bevaras och manipuleras i mörker eller i svagt ljus från glödlampor till dess att cellerna skördas för att minimiera fotolysen av BrdU-haltigt DNA.
Cellkulturerna behandlas med en spindelfiberinhibitor (t.ex. kolchicin) i 1–4 timmar innan de skördas. Varje kultur skördas för sig och behandlas separat för kromosompreparering
Vid kromosompreparation används cytogeniska standardtekniker. Färgning på objektglas för att visa SCE kan ske på flera olika sätt (t.ex. fluorescens plus Giemsametoden).
Antalet av de celler som ska analyseras grundas på den spontana SCE-frekvensen i kontrolltest. Normalt analyseras minst 25 väl utspridda metafaser per kultur för SCE. Objektglas kodas före analys. Vid analys av humana lymfocyter analyseras endast metafaser med 46 centromerer. Vid analys av etablerade cellinjer analyseras endast metafaser med det modala antal centromerer ± 2. Det bör anges huruvida omväxling i märkningen omkring centromeret betraktas som SCE. Resultaten bör bekräftas genom ett oberoende test.
2. DATA
Data ställs upp i tabellform. Antal SCE för varje metafas samt antalet SCE per kromosom för varje metafas bör anges separat för alla exponerade kulturer och kontrollkulturer.
Utvärderingen av data grundas på lämpliga statistiska metoder.
3. RAPPORTERING
3.1 TESTRAPPORT
Testrapporten ska om möjligt innehålla följande uppgifter:
|
— |
Använda celler, metoder för bibehållande av cellkulturer. |
|
— |
Testbetingelser: Sammansättning av mediet, CO2-koncentration, testämnekoncentration, använd vehikel, inkubationstemperatur, exponeringsperiod, använd spindelfiberinhibitor plus upplysningar och dess koncentration och behandlingens varaktighet, använt metaboliskt aktiveringssystem, positiva och negativa kontroller. |
|
— |
Antal cellkulturer per experimentell punkt. |
|
— |
Detaljerade upplysningar om objektprepareringstekniken. |
|
— |
Antal analyserade metafaser (data anges separat för varje kultur). |
|
— |
Genomsnittligt antal SCE per cell och per kromosom (data anges separat för varje kultur). |
|
— |
Kriterier för utvärdering av SCE. |
|
— |
Motivering för val av doser. |
|
— |
Förhållandet mellan dos och reaktion när detta är möjligt. |
|
— |
Statistisk utvärdering. |
|
— |
Diskussion om resultaten. |
|
— |
Tolkning av resultaten. |
3.2 UTVÄRDERING OCH TOLKNING
Se allmän inledning till del B.
4. HÄNVISNINGAR
Se allmän inledning till del B.
B.20 KÖNSBUNDEN RECESSIV LETAL TEST PÅ DROSOPHILA MELANOGASTER
1. METOD
1.1 INLEDNING
Se allmän inledning till del B.
1.2 DEFINITION
Se allmän inledning till del B.
1.3 REFERENSÄMNEN
Inga.
1.4 TESTMETODSPRINCIP
Vid könsbunden recessiv letal test (SLRL) på Drosophila melanogaster undersöks förekomsten av både punktmutationer och mindre deletioner hos insektens avkomma. Det är en framåtmutationstest genom vilken det är möjligt att undersöka ca 800 loci på X-kromosomen i syfte att registrera mutationer. Detta antal utgör ca 80 % av alla X-kromosomens loci. X-kromosomen utgör omkring en femtedel av den samlade haploida kromosommängden.
Mutationer i Drosophila melanogasters X-kromosom kommer till fenotypiskt uttryck hos handjur som är bärare av den muterade genen. När mutationen under hemizygota betingelse är letal konstateras detta genom frånvaron av en av de två typer av handjur som normalt finns i ett meterozy-gotiskt hondjurs avkomma. I SLRL-testet utnyttjas detta förhållande med hjälp av speciellt märkta och arrangerade kromosomer.
1.5 KVALITETSKRITERIER
Inga.
1.6 METODBESKRIVNING
Förberedelser
Handjur av väldefinierade vildtypstammar och hondjur från Muller-5-stammen kan användas. Lämpligt märkta hondjur från andra stammar och multipelt inverterade X-kromosomer kan också användas.
Testämnet upplöses i vatten. Ämnen som är oupplösliga i vatten kan upplösas eller suspenderas i lämpliga vehikler (t.ex. en blandning av etanol och Tween-60 eller - 80) och därefter spädas ut i vatten eller salin före tillförsel. Dimetylsulfoxid (DMSO) bör undvikas som vehikel.
Testet bör utformas så att följsamheten och betydelsen av testet är fastställt på förhand. Den spontana mutationsfrekvens som registreras i kontrollförsök är av stor betydelse för bestämning av det antal exponerade kromosomer som ska analyseras.
Exponeringen kan ske genom oral tillförsel, injektion eller genom exponering för gaser eller ångor. Testämnet kan eventuellt tillföras i en sockerlösning. Vid behov kan den upplösas i en 0,7 % NaCl-lösning och injiceras i bröstkorgen eller buken.
En negativ (vehikel) och en positiv kontroll ska utföras. Om det finns data från tidigare laboratorieförsök är det dock inte nödvändigt att utföra sidlöpande kontroll.
Tre koncentrationsnivåer ska användas. För en preliminär undersökning kan en enkel koncentration användas som antingen kan vara den högsta koncentration som insekterna tål eller en koncentration som orsakar en viss grad av toxisk verkan. Vid icke-toxiska testämnen exponeras insekterna för de största möjliga koncentrationerna.
Vildtyphandjur (3–5 dagar gamla) exponeras för testämnet och paras individuellt med flera jungfruliga hondjur från Muller-5-stammen eller från andra lämpligt märkta stammar (med multipelt inverterade X-kromosomer). Hondjuren byts ut mot nya jungfruliga hondjur varannan eller var tredje dag tills hela könscellcykeln är täckt. Hondjurens avkomma registreras för att konstatera letala verkningar som motsvarar exponeringens verkan på mogen sperma, spermatider i sena stadier eller medelstadier, spermatider i tidiga stadier, spermatocyter och spermatogonia.
Heterozygota F1-hondjur från de ovan nämnda korsningarna paras individuellt (dvs. ett hondjur per flaska) med sina bröder. I F2-generationen registreras varje kultur för bestämning av frånvaron av vildtyphandjur. Om en kultur har uppstått från ett F1-hondjur som bär en letal gen i den parentala X-kromosomen (dvs. det finns inga handjur med den exponerade kromosomen) testas döttrar till detta hondjur med samma genotyp för att konstatera om den letala verkan upprepas i nästa generation.
2. DATA
Data ska ställas upp i tabellform och ange antal testade X-kromosomer, antal ofruktsamma handjur, antal letala kromosomer för varje exponeringskoncentration och för var och en av de exonerade handjurens parningsperioder. Dessutom anges antalet klungor av olika storlek per handjur. Resultaten bör bekräftas genom ett oberoende test.
Utvärderingen av en könsbunden recessiv letal test ska grundas på lämpliga statistiska metoder. Kluster av generationer med recessiva letala gener som stammar från ett enda handjur ska registreras och utvärderas på grundval av en lämpligt statistisk metod.
3. RAPPORTERING
3.1 TESTRAPPORT
Testrapporten ska om möjligt innehålla minst följande uppgifter:
|
— |
Stammar: Använda drosophila-stammar, insekternas ålder, antal exponerade handjur, antal sterila handjur, antal etablerade F2-bestånd, antal F2-bestånd utan avkomma, antal kromosomer med letala gener i varje könscellstadium. |
|
— |
Kriterier för fastställande av storleken av de exponerade grupperna. |
|
— |
Testbetingelser: Detaljerade beskrivning av exponerings- och stickprovsschema, exponeringskoncentrationer, toxicitetsdata, negativ (lösningsmedel) och eventuella positiva kontroller. |
|
— |
Kriterier för registrering av letala mutationer. |
|
— |
Förhållandet mellan dos och reaktion när detta är möjligt. |
|
— |
Statistisk utvärdering. |
|
— |
Diskussion om resultaten. |
|
— |
Tolkning av resultaten. |
3.2 UTVÄRDERING OCH TOLKNING
Se allmän inledning till del B.
4. HÄNVISNINGAR
Se allmän inledning till del B.
B.21 CELLTRANSFORMATIONSTEST PÅ DÄGGDJURSCELLER IN VITRO
1. METOD
1.1 INLEDNING
Se allmän inledning till del B.
1.2 DEFINITION
Se allmän inledning till del B.
1.3 REFERENSÄMNEN
Inga.
1.4 TESTMETODSPRINCIP
Kulturer av däggdjursceller kan användas för upptäckt av fenotypiska förändringar in vitro som inducerats av kemiska förbindelser i samband med maligna transformationer in vivo. De mest använda celltyperna är C3H10TlVi, 3T3, SHE, Fischer-råtta, och testerna bygger på förändringar i cellmorfologin, fokusbildning eller egenskapen att kunna växa i mjuk agar. Det finns också mindre vanligt använda stammar som används för undersökning av andra fysiologiska eller morfologiska förändringar i celler till följd av exponering för cancerogena kemiska förbindelser. Inga direkta verkningsmekanismförbindelser mellan cancer och de genetiska endpoints som används i dessa tester in vitro finns beskrivna. Några av testsystemen kan användas för upptäckt av tumörpromotorer. Den cytotoxiska effekten kan bestämmas genom mätning av testämnets verkan på kolonibildande (kloniseringseffektiviteten) eller kulturernas växtfrekvens. Syftet med mätningen av den cytotoxiska effekten är att konstatera om exponering för testämnet är toxikologiskt relevant, men kan inte användas för beräkning av transformationsfrekvensen i alla tester, eftersom vissa kräver lång inkubationstid och/eller överföring till nya plattor.
1.5 KVALITETSKRITERIER
Inga.
1.6 METODBESKRIVNING
Förberedelser
En rad olika cellinjer eller primärlinjer kan användas beroende på vilken transformationstest som görs. Försöksledaren måste säkerställa att använda celler genomgår relevanta fenotypiska förändringar efter exponering för kända cancerogener, och försöksledarens eget laboratorium måste ha bevisat och dokumenterat det använda testets validitet och trovärdighet.
Medium och försöksbetingelser som är lämpliga för transformationstest ska användas.
Testämnet tillsätts i ett kulturmedium eller upplöses eller suspenderas i lämpliga vehiklar innan cellerna exponeras. Koncentrationen av vehikeln bör inte orsaka någon ändring av cellers livskraft eller växtegenskaper.
Cellerna exponeras för testämnet både i närvaro och i frånvaro av ett lämpligt exogent metaboliskt aktiveringssystem. Om celltyper med endogen metabolisk aktivering används bör det vara bevisat att aktiveringsform och -hastighet är lämpligt för den kemiska klass som testas.
Testbetingelser
Som positiva kontroll används vid varje enskilt försök både en direkt verkande kemisk förbindelse och en förbindelse som kräver metabolisk aktivering. Dessutom företas negativ (vehikel) kontroll.
Följande kemiska förbindelser kan t.ex. användas till positiv kontroll:
|
— |
direktverkande förbindelser:
|
|
— |
förbindelser som kräver metabolisk aktivering:
|
Vid behov kan en supplerande positiv kontroll göras med användning av en kemisk förbindelse av samma typ som testämnet.
Flera koncentrationer av testämnet ska användas. Dessa koncentrationer bör framkalla en koncentrationsrelaterad toxisk verkan. Den högsta koncentrationen bör medföra en låg överlevnadsgrad och överlevnadsgraden vid den lägsta koncentrationen bör vara av samma storlek som vid det negativa kontrollförsöket. Ämnen som är relativt oupplösliga i vatten testas på lämpligt sätt upp till gränsen för löslighet. Den högsta koncentrationen av icke-toxiska testämnen, som är fritt lösliga i vatten, fastställs från test till test.
Testförfarande
Cellerna bör exponeras under för det använda testsystemet passande period. Detta kan inbegripa upprepad dosering åtföljt av en ändring av medium (och vid behov ny metabolisk aktivering) om det är fråga om långvarig exponering. Celler vars endogena metaboliska aktivering är otillräcklig testas både med och utan ett lämpligt metaboliskt aktiveringssystem. Efter exponeringen tvättas cellerna fria från testämne. Därefter odlas de i syfte att låta den transformerade fenotyp som testet bygger på komma till uttryck och i syfte att bestämma transformationsfrekvensen. Alla resultat ska bekräftas genom ett oberoende test.
2. DATA
Data ska ställs upp i tabellform och kan ta sig olika uttryck beroende på använt testsystem. Det kan t.ex. röra sig om räkning av plattor, positiva plattor eller antal transformerade celler. Vid behov anges överlevnadsgraden som en procentdel av överlevnaden i kontrollförsöken, och transformationsfrekvensen uttrycks som antal transformerade celler per överlevande. Utvärderingen av data grundas på lämpliga statistiska metoder.
3. RAPPORTERING
3.1 TESTRAPPORT
Testrapporten ska om möjligt innehålla minst följande uppgifter:
|
— |
Använd celltyp, antal cellkulturer, metoder för bibehållande av cellkulturer. |
|
— |
Testbetingelser: Testämnekoncentration, använd vehikel, inkubationstid, testets varaktighet och frekvens, celltäthet under exponeringen, använt exogent metaboliskt aktiveringssystem, positiva och negativa kontroller, specifikation av den fenotyp som testet bygger på, använt selektivt system (om sådant har använts), motivering för val av doser. |
|
— |
Metoder för registrering av livskraftiga och transformerade celler. |
|
— |
Statistisk utvärdering. |
|
— |
Diskussion om resultaten. |
|
— |
Tolkning av resultaten. |
3.2 UTVÄRDERING OCH TOLKNING
Se allmän inledning till del B.
4. HÄNVISNINGAR
Se allmän inledning till del B.
B.22 DOMINANT LETAL TEST PÅ GNAGARE
1. METOD
1.1 INLEDNING
Se allmän inledning till del B.
1.2 DEFINITIONER
Se allmän inledning till del B.
1.3 REFERENSÄMNEN
Inga.
1.4 TESTMETODSPRINCIP
Dominanta letala verkningar leder till att embryot eller fostret dör. När dominanta letala mutationer framkallas genom exponering för en kemisk ämne är det ett tecken på att försöksdjurartens könscellvävnader har skadats. Det råder bred enighet om att dominanta letala gener uppstår till följd av kromosomskador (strukturella och numeriska anomalier). Om exponerade handjurs avkomma dör på det embryonala stadiet kan även detta skyllas på toxiska verkningar.
Normalt exponeras handjur för testämnet och paras med jungfruliga hondjur. Könscellernas olika stadier kan undersökas separat genom att använda på varandra följande parningsperioder. Ökningen av antalet döda implantater per hondjur i den exponerade gruppen i förhållande till antalet döda implantater per hondjur i kontrollgruppen visar den letala verkan efter implantation. Det letala verkan före implantation kan bedömas på grundlag av räkning av corpora lutea eller genom sammanslagning av det totala antalet implantater per hondjur i de exponerade grupperna och kontrollgruppen. Den totala dominanta letala verkan är summan av de letala verkningar före och efter implantation. Beräkningen av den totala dominanta letala verkan grundas på sammanräkning av antalet levande implantater per hondjur i försöksgruppen och antalet levande implantater per hondjur i kontrollgruppen. En minskning av antalet implantater under bestämda tidsperioder kan skyllas på celldöd (av spermacyter och/eller spermatogoner).
1.5 KVALITETSKRITERIER
Inga.
1.6 METODBESKRIVNING
Förberedelser
Testämnet upplöses eller suspenderas i fysiologisk saltlösning. Kemiska förbindelser som inte är lösliga i saltlösning ska lösas eller suspenderas i lämplig vehikel. Den vehikel som används får inte inverka på testämnet eller framkalla toxiska effekter. Nyligen framställda lösningar av testämnet ska användas.
Försöksbetingelser
Testämnet ska vanligen endast tillföras en gång. På grundval av toxikologisk information kan upprepad behandling användas. Vanligtvis tillförs testämnet genom oral intubering eller intraperitoneal injicering. Även andra tillförselvägar kan användas
Råttor och möss rekommenderas som försöksdjur. Friska könsmogna djur fördelas slumpmässigt i behandlings- och kontrollgrupper.
Ett lämpligt antal handjur bör användas, med hänsyn tagen till den spontana variationen i den biologiska egenskap som utvärderas. Beslutet om hur många djur som ska användas bör grundas på den på förhand fastställda känsligheten för upptäckt och betydelsegrad. Vid ett typiskt försök ska det finnas tillräckligt med handjur för att uppnå 30–50 dräktiga hondjur varje parningsperiod.
Normalt görs sidlöpande positiva och negativa (vehikel) kontroller för varje försök. Om det finns godtagbara positiva kontrollresultat från nyligen genomförda försök i samma laboratorium kan dock dessa användas i stället för sidlöpande positiv kontroll. Positiva kontrollämnen används i tillräcklig låga doser (t.ex. MMS i. p., 10 mg/kg) för att visa testens känslighet.
Normalt används tre olika dosnivåer. Den högsta dosnivån bör orsaka tecken på toxiska verkningar eller minskad fertilitet hos de exponerade djuren. I vissa fall räcker det med en enda hög dosnivå.
Icke-toxiska ämnen testas vid 5 g/kg i en enda dos eller vid upprepad tillförsel av 1 g/kg/dag.
Förfarande
Flera olika doseringsscheman kan användas. Normalt ges hela dosen på en gång men andra behandlingsscheman kan också användas.
Efter behandling paras handjuren med lämpliga intervaller individuellt med 1–2 jungfruliga hondjur. Hondjuren placeras hos handjuren under en period som motsvarar minst en äggcykel, eller tills parning har skett, vilket konstateras genom närvaron av sperma i vaginan eller närvaron av en vaginalpropp.
Antal parningar efter behandlingen fastställs på grundval av doseringsschemat och bör säkerställa att alla könscellsstadier är representerade efter doseringen.
Hondjuren avlivas under den andra hälften av dräktigheten och innehållet i livmodern undersöks för bestämning av antalet levande och döda implantat. Äggstockarna undersöks för bestämning av antalet corpora lutea.
2. DATA
Data ska ställas upp i tabellform och ange antal handjur samt antal dräktiga och icke dräktiga hondjur. Resultaten av varje enskild parning, inbegripet identifiering av varje han- och hondjur, ska anges separat. För varje hondjur anges parningsvecka, handjurets dos och antalet levande och döda implantat.
Beräkningen av den totala dominanta letala verkan grundas på jämförelse av antal levande implantat per hondjur hondjur i försöksgruppen och antalet levande implantat per hondjur i kontrollgruppen. Förhållandet mellan döda och levande implantat i försöksgruppen i jämförelse med förhållandet i kontrollgruppen analyseras för bestämning av den letala verkan efter implantation.
Om data registreras som tidiga eller sena dödsfall ska detta framgå av tabellerna. Om en utvärdering av den letala verkan sker före implantation ska denna anges. Den letala verkan före implantation kan beräknas som skillnaden mellan antalet corpora lutea och antalet implantater eller som en minskning av det genomsnittliga antalet implantat per livmoder i jämförelse med kontrollparningarna.
Utvärderingen av resultat ska grundas på en lämplig statistisk metod.
3. RAPPORTERING
3.1 TESTRAPPORT
Testrapporten ska om möjligt innehålla minst följande uppgifter:
|
— |
Djurart, stam, de använda djurens ålder och vikt, antal djur av varje kön i försöks- och kontrollgrupperna. |
|
— |
Testämne, vehikel, testade dosnivåer och motivering för val av doser, negativa och positiva kontroller. |
|
— |
Tillförselväg och dosens varaktighet. |
|
— |
Parningsschema. |
|
— |
Metod för bestämning av om parning skett. |
|
— |
Tidpunkt för avlivning. |
|
— |
Kriterier för registrering av dominanta letala gener. |
|
— |
Förhållandet mellan dos och reaktion när detta är möjligt. |
|
— |
Statistisk utvärdering. |
|
— |
Diskussion om resultaten. |
|
— |
Tolkning av resultaten. |
3.2 UTVÄRDERING OCH TOLKNING
Se allmän inledning till del B.
4. HÄNVISNINGAR
Se allmän inledning till del B.
B.23 SPERMATOGONIAL TEST AV KROMOSOMAVVIKELSER HOS DÄGGDJUR
1. METOD
Denna metod är en kopia av testet OECD TG 483, Spermatogonial test av kromosomavvikelser hos däggdjur (Mammalian Spermatogonial Chromosomc Aberration Test (1997)).
1.1 INLEDNING
Ändamålet med testet av spermatogoniala avvikelser in vivo hos däggdjur, är att identifiera de substanser som orsakar strukturella kromosomavvikelser i spermatogoniala celler hos däggdjur (1) (2) (3) (4) (5). Strukturella avvikelser kan vara av två typer, kromosomala eller kromatida. Majoriteten av de kemiska mutagenerna inducerar avvikelser av kromatidtyp, men även avvikelser av kromosomtyp förekommer. Denna metod är inte utformad för att mäta numeriska avvikelser och används inte rutinmässigt för detta ändamål. Kromosommutationer och relaterade händelser är orsaken till många genetiska sjukdomar hos människor.
Detta test mäter kromosomhändelser i spermatogoniala könsceller och förväntas därför utgöra en förutsägelse på induktion av ärftliga mutationer i könsceller.
Gnagare används rutinmässigt i detta test. Detta cytogenetiska test in vivo detekterar kromosomavvikelser i spermatogoniala mitoser. Andra målceller omfattas inte av denna metod.
För att detektera avvikelser av kromatidtyp i spermatogoniala celler, bör den första mitotiska celldelningen efter behandlingen undersökas innan dessa skador förloras i de påföljande celldelningarna. Ytterligare information från behandlade spermatogoniala stamceller kan erhållas genom meiotisk kromosomanalys av avvikelser av kromosomtyp vid diakinesmetafas I, när de behandlade cellerna förvandlas till spermatocyter.
Detta in vivo-test är utformat för att undersöka om mutagener för somatiska celler även är aktiva i könsceller. Det spermatogoniala testet är dessutom relevant för att fastställa risken för mutagenicitet, eftersom det medger att faktorer för in vivo metabolism, farmakokinetik och reparationsprocesser för DNA beaktas.
Ett antal generationer av spermatogonier finns närvarande i testikeln med ett spektrum av känslighet för kemisk behandling. De avvikelser som detekteras representerar alltså ett samlat svar på behandlade spermatogoniala cellpopulationer, där de mera talrika differentierade spermatogoniala cellerna överväger. Olika generationer av spermatogonier kan där komma i kontakt med det stora kretsloppet. Om detta sker eller ej avgörs av deras position inne i testikeln, beroende på den fysiska och fysiologiska Sertoli-cellbarriären och på blod/testikelbarriären.
Om det finns bevis för att testsubstansen, eller en reaktiv metabolit, inte kommer att nå fram till målvävnaden, är det inte lämpligt att använda detta test.
Se även Allmän inledning, del B.
1.2 DEFINITIONER
avvikelse av kromatidtyp: strukturell kromosomskada uttryckt som brott på enstaka kromatider eller brott och återförening mellan kromatider.
avvikelse av kromosomtyp: strukturell kromosomskada uttryckt som brott och återförening, på båda kromatiderna på samma ställe.
lucka: en akromatisk skada som är mindre än bredden på en kromatid, och med ett minimum av misspassning hos kromatiderna.
numerisk avvikelse: en förändring av antalet kromosomer från det normala antalet karakteristiskt för det försöksdjur som används.
polyploidi: en multipel av det haploida kromosomantalet (n) som skiljer sig från det diploida (dvs. 3n, 4n osv.).
strukturell avvikelse: en förändring av kromosomstrukturen som är detekterbar med hjälp av en mikroskopisk undersökning av metafasstadiet vid celldelning, observerad i form av deletioner, interna eller externa utbyten.
1.3 TESTMETODENS PRINCIP
Försöksdjur exponeras för testsubstansen genom ett lämpligt exponeringssätt och avlivas sedan vid en lämplig tid efter behandling. Innan avlivningen sker, behandlas försöksdjuren med en substans som gör att celldelningen stannar upp i metafas (t.ex. colchicin eller Colcemid®). Kromosomberedninger görs sedan från könsceller och färgas in, varpå de celler som är i metafas analyseras med avseende på kromosomavvikelser.
1.4 BESKRIVNING AV TESTMETODEN
1.4.1 Förberedelser
1.4.1.1 Val av djurarter
Handjur av kinesiska hamstrar och möss används i stor utsträckning. Det är även möjligt att använda hanar från andra lämpliga däggdjursarter. Man bör välja de stammar av unga, friska vuxna djur som vanligen används på laboratorier. När undersökningen ska påbörjas är det lämpligt att viktskillnaden mellan djuren är så liten som möjligt och den bör inte överstiga ± 20 % av medelvikten.
1.4.1.2 Förhållanden vid förvaring och utfodring
De generella förhållanden som anges i den Allmänna inledningen till del B gäller. Man bör dock sträva efter en relativ luftfuktighet på 50–60 %.
1.4.1.3 Förberedelse av djuren
Friska, unga vuxna djur väljs ut slumpartat för placering i kontroll- och behandlingsgrupperna. Burar bör arrangeras på ett sådant sätt att möjliga effekter beroende på placeringen av burarna blir så små som möjligt. Djuren ska ha en unik identifiering. De bör vänjas vid förhållandena i laboratoriet under minst fem dagar innan undersökningen börjar.
1.4.1.4 Beredning av doser
Festa testsubstanser bör lösas upp i eller blandas med lämpliga lösningsmedel eller vehiklar och spädas ut, om så är lämpligt, innan dosen administreras till djuren. Testsubstanser i flytande form kan doseras direkt eller spädas ut före dosering. Nygjorda beredningar av testsubstansen bör användas, om inte stabilitetsdata visar att lagring är acceptabel.
1.4.2 Försöksbetingelser
1.4.2.1 Lösningsmedel/vehikel
Lösningsmedlet/vehikeln bör inte ge upphov till toxiska effekter vid de dosnivåer som används och bör inte kunna misstänkas för att reagera kemiskt med testsubstansen. Om andra lösningsmedel/vehiklar än gängse brukliga används, bör användningen av dem ha stöd från data som visar på deras kompatibilitet. När det är möjligt, rekommenderas att man i första hand överväger att använda vattenbaserade lösningsmedel/vehiklar.
1.4.2.2 Kontroller
Samtidiga positiva och negativa (lösningsmedel/vehikel) kontroller bör inkluderas i varje test. Utom vid behandling med testsubstansen, bör försöksdjuren i kontrollgrupperna hanteras på ett sätt om är identiskt med sättet för försöksdjuren i de grupper som behandlas.
Positiva kontroller bör de strukturella kromosomavvikelser in vivo i spermatogoniala celler när administrering sker vid exponeringsnivåer som förväntas ge en detekterbar ökning i förhållande till bakgrunden.
Positiva kontrolldoser bör väljas på ett sådant sätt att effekterna är tydliga men inte omedelbart avslöjar identiteten hos kodade utstryksglas för läsaren. Det är acceptabelt att den positiva kontrollen administreras via en annan tillförselväg än testsubstansen och att provtagningen endast sker vid ett tillfälle. Dessutom kan användningen av kemiska klassrelaterade positiva kontrollkemikalier övervägas när sådana finns att tillgå. 1 tabellen nedan ges några exempel på positiva kontrollsubstanser.
|
Substans |
CAS-nummer |
Einecs-nummer |
|
Cyklofosfoamid Cyklofosfoamidmonohydrat |
50-18-0 6055-19-2 |
200-015-4 |
|
Cyclohexylamin |
108-91-8 |
203-629-0 |
|
Mitomycin C |
50-07-7 |
200-008-6 |
|
Monomer akrylamid |
79-06-1 |
201-173-7 |
|
Trietylenmelamin |
51-18-3 |
200-083-5 |
Negativa kontroller, behandlade med enbart ett lösningsmedel eller en vehikel, och i övrigt behandlade på samma sätt som behandlingsgrupperna, bör inkluderas för varje provtagningstillfälle, om inte acceptabel variabilitet och frekvens för celler med kromosomavvikelser mellan försöksdjuren kan påvisas från historiska kontrolldata. Dessutom bör obehandlade kontroller även användas, om det inte finns historiska eller publicerade kontrolldata som visar att inga skadliga eller mutagena effekter induceras av det valda lösningsmedlet/vehikeln.
1.5 FÖRFARANDE
1.5.1 Antal försöksdjur
Varje behandlad grupp och kontrollgrupp måste inkludera minst fem analyserbara hanar.
1.5.2 Behandlingsschema
Testsubstanser bör lämpligen administreras en eller två gånger (dvs. som en enstaka behandling eller som två behandlingar). Testsubstanser kan även administreras i form av en delad dos, dvs. två behandlingar under samma dag med högst några timmars intervall, för att underlätta administrering av stora volymer. Andra doseringsscheman bör vara vetenskapligt motiverade.
I den högsta dosgruppen används två provtagningstider efter behandlingen. Eftersom cellcykelns kinetik kan påverkas av testsubstansen, används en tidig och en sen provtagningstid vid ungefär 24 och 48 timmar efter behandlingen. För andra doser än den högsta dosen bör en provtagningstid vid 24 timmar eller 1,5 cellcyklers längd efter behandlingen användas, om inte andra provtagningstider är kända för att vara lämpligare för detektion av effekter (6).
Dessutom kan andra provtagningstider användas. När det t.ex. gäller fall där kemikalier kan inducera kromosom-”lagging”, eller när de kan ge upphov till S-oberoende effekter, kan tidigare provtagningstider visa sig vara lämpligare (1).
Man måste i varje enskilt fall fastställa om det är lämpligt att använda ett upprepat behandlingsschema. Efter ett upprepat behandlingsschema bör försöksdjuren avlivas 24 timmar (1,5 gånger cellcykelns längd) efter den sista behandlingen. Ytterligare provtagningstider kan användas där så är lämpligt.
Innan avlivningen sker, injiceras försöksdjuren intraperitonealt med en lämplig dos av en substans som stoppar celldelningen i metafas (t.ex. Colcemid® eller colchicin). Provtagning på försöksdjur sker därefter med lämpliga intervall. För möss ligger detta intervall på ungefär 3–5 timmar och för kinesiska hamstrar ligger intervallet pä ungefär 4–5 timmar.
1.5.3 Dosnivåer
Om det inte finns några lämpliga data tillgängliga och en undersökning därför utförs för att fastställa omfattningen, bör denna undersökning utföras i samma laboratorium, och med samma arter, stammar och behandlingsschema som senare ska användas i huvudundersökningen. Vid toxicitet används tre dosnivåer för det första provtagningstillfället. Dessa dosnivåer bör täcka in ett intervall som sträcker sig från maximal till liten eller ingen toxicitet. Vid senare provtagningstillfällen räcker det med att endast den högsta dosen används. Den högsta dosen definieras som den dos som producerar tecken på toxicitet och så att högre dosnivåer baserade på samma doseringsschema skulle förväntas leda till att försöksdjuret dör.
Substanser med specifika biologiska verkningar vid låga icke-toxiska doser (såsom hormoner och mitogena substanser) kan utgöra undantag från kriterier som är dosbestämmande och bör utvärderas ifrån fall till fall. Den högsta dosen kan även definieras som en dos vilken ger upphov till någon indikation på toxicitet i den spermatogoniala cellerna (t.ex. en reduktion i kvoten av spermatogoniala mitoser hos de första och andra meiotiska metafaserna; denna reduktion bör inte överskrida 50 %).
1.5.4 ”Limit-test”
En fullständig undersökning med tre dosnivåer kan inte anses vara nödvändig om ett test på en dosnivå vid minst 2 000 mg/kg kroppsvikt och med en enstaka behandling, eller i form av två behandlingar under samma dag, inte per upphov till observerbara toxiska effekter, och om ingen genotoxicitet kan förväntas utifrån data om strukturellt relaterade substanser. En förväntad exponering av människor kan visa på behovet av att en högre dosnivå används i ’limit-testen’.
1.5.5 Administrering av doser
Testsubstansen administreras normalt genom sondmatning med hjälp av en magsond, en lämplig intuberingskanyl eller genom en intraperitoneal injektion. Andra tillförselvägar för exponering kan vara acceptabla där de kan motiveras. Den maximala vätskevolymen som kan administreras med hjälp av sondmatning eller injektion vid ett tillfälle beror på försöksdjurets storlek. Volymen bör inte överstiga 2 ml/100 g kroppsvikt. Användning av volymer som är större än dessa måste motiveras. Förutom för irriterande eller korrosiva substanser, som normalt kommer att visa på stegrade effekter med högre koncentrationer, bör variationer av testvolymen minimeras genom att en justering av koncentrationen sker för att säkerställa att volymen är konstant vid alla dosnivåer.
1.5.6 Kromosomberedning
Omedelbart efter avlivning, tas cellsuspensioner ut från en eller båda testiklarna, exponeras för en hypoton lösning och fixeras. Cellerna stryks sedan ut på utstryksglas och färgas in.
1.5.7 Analys
För varje försöksdjur bör minst 100 väl spridda metafaser analyseras (dvs. ett minimum på 500 metafaser per grupp). Detta antal skulle kunna reduceras när ett stort antal avvikelser observeras. Alla utstryksglas, inklusive de från positiva och negativa kontroller, bör få en oberoende kodning innan den mikroskopiska analysen utförs. Eftersom fixeringsprocedurerena ofta resulterar i brott på en del av de celler som är i metafas, med förlust av kromosomer som följd, bör de celler som påvisas innehålla ett antal centromerer som motsvarar antalet 2 N ± 2.
2. DATA
2.1 BEHANDLING AV RESULTATEN
Individuella data från försöksdjur bör presenteras i tabellform. Den experimentella enheten utgörs av försöksdjuret. För varje individuellt försöksdjur bör antalet celler med strukturella kromosomavvikelser och antalet kromosomavvikelser per cell utvärderas. Olika typer av strukturella kromosomavvikelser bör listas med antal och frekvenser för behandlade grupper och kontrollgrupper. Luckor i DNA-strängen registreras separat och rapporteras men inkluderas i allmänhet inte i den totala avvikelse frekvensen.
Om såväl mitos som meios observeras, bör förhållandet mellan spermatogoniala mitoser och de första och andra meiotiska metafaserna fastställas som ett mått på cytotoxicitet för alla behandlade och negativa kontrollförsöksdjur vid ett totalt prov pä 100 celler som delar sig per djur för att etablera en möjligt cytotoxisk effekt. Om endast mitos kan observeras, bör mitosindexet bestämmas i minst 1 000 celler för varje försöksdjur.
2.2 UTVÄRDERING OCH TOLKNING AV RESULTATEN
Det finns ett flertal kriterier som används för att bestämma om ett resultat är positivt, t.ex. en dosrelaterad ökning av det relativa antalet celler med kromosomavvikelser eller en tydlig ökning av antalet celler hos en grupp som fått en enstaka dos vid ett enstaka provtagningstillfälle. Man bör i första hand överväga resultatets biologiska relevans. Statistiska metoder kan användas som en hjälp vid utvärdering av testresultatet (8). Tvetydiga resultat bör klarläggas genom fortsatta tester, lämpligen vid ändrade experimentella betingelser.
En testsubstans för vilken resultatet inte uppfyller ovanstående kriterier anses vara icke-mutagen i detta test.
Även om de flesta experiment kommer att ge tydligt positiva eller negativa resultat, så kan i sällsynta fall uppsättningen av data göra det omöjligt att utföra en otvetydig bedömning med avseende på testsubstansens aktivitet. Resultatet kan förbli tvetydigt eller tveksamt, oavsett hur många gånger som experimentet upprepas.
Positiva resultat från in vivo-test för spermatogoniala kromosomavvikelser indikerar att testsubstansen inducerar strukturella kromosomavvikelser i könscellerna hos den art som testas. Negativa resultat indikerar att testsubstansen, under försöksbetingelserna, inte inducerar kromosomavvikelser i könscellerna på den art som testas.
Sannolikheten för att testsubstansen eller dess metaboliter når målvävnaden bör diskuteras.
3. RAPPORTERING
TESTRAPPORT
Testrapporten måste innehålla följande information:
|
|
Lösningsmedel/vehikel:
|
|
|
Försöksdjur:
|
|
|
Försöksbetingelser:
|
|
|
Resultat:
|
|
|
Diskussion av resultaten. |
|
|
Slutsatser. |
4. HÄNVISNINGAR
|
1. |
Adler, 1. D., (1986), Clastogenic Potential in Mouse Spermatogonia of Chemical Mutagens Related to their Cell-Cycle Specifications, in: Genetic Toxicology of Environmental Chemicals, Part B: Genetic Effects and Applied Mutagenesis, Ramel, C, Lambert, B. and Magnusson, J. (eds) Liss, New York, s. 477–484. |
|
2. |
Adler, I. D., (1984), Cytogenetic tests in Mammals, in: Mutagenicity Testing: a Practical Approach, (ed.) S. Venitt and J. M. Parry, 1RL Press, Oxford, Washington DC, s. 275–306. |
|
3. |
Evans, E. P., Breckon, G. and Ford, C. E. (1964), An Air-drying Method for Meiotic Preparations from Mammalian Testes, Cytogenetics and Cell Genetics, 3, s. 289–294. |
|
4. |
Richold, M., Ashby, J., Chandley, A., Gatehouse, D. G. and Henderson, L. (1990), In Vivo Cytogenetic Assays, in: D. J. Kirkland (ed.), Basic Mutagenicity Tests, UKEMS Recommended Procedures. UKEMS Subcommittee on Guidelines for Mutagenicity Testing. Report. Part I revised, Cambridge University Press, Cambridge, New York, Port Chester, Melbourne, Sydney, s. 115–141. |
|
5. |
Yamamoto, K. and Kikuchi, Y. (1978), A New Method for Preparation of Mammalian Spermatogonial Chromosomes, Mutation Res. 52, s. 207–209. |
|
6. |
Adler, 1. D., Shelby M. D., Bootman, J., Favor, J., Generoso, W., Pacchierotti, F.. Shibuya, T. and Tanaka N. (1994), International Workshop on Standardisation of Genotoxicity Test Procedures. Summary Report of the Working Group on Mammalian Germ Cell Tests, Mutation Res. 312, s. 313–318. |
|
7. |
Fielder, R. J., Allen, J. A., Boobis, A. R., Botham, P. A., Doe, J., Esdaile, D. J., Gatehouse, D. G., Hodson- Walker, G.. Morton, D. B., Kirkland, D. J. and Richold, M. (1992), Report of British Toxicology Society/UK Environmental Mutagen Society Working Group: Dose setting in In Vivo Mutagenicity Assays, Mutagenesis, 7, s. 313–319. |
|
8. |
Lovell, D. P., Anderson, D., Albanese, R., Amphlett, G. E., Clarc, G., Ferguson, R., Richold, M., Papworth, D. G. and Savage, J. R. K. (1989), Statistical Analysis of In Vivo Cytogenetic Assays, in: D. J. Kirkland (ed.), Statistical Evaluation of Mutagenicity Test Data. UKEMS Subcommittee on Guidelines for Mutagenicity Testing, report, Part 111. Cambridge University Press, Cambridge, New York, Port Chester, Melbourne, Sydney, s. 184–232. |
B.24 FLÄCKTEST PÅ MUS
1. METOD
1.1 INLEDNING
Se allmän inledning till del B.
1.2 DEFINITIONER
Se allmän inledning till del B.
1.3 REFERENSÄMNEN
Inga.
1.4 TESTMETODSPRINCIP
Fläcktestet är ett in vivo-test på mus där musfoster på det embryonala stadiet exponeras för kemiska förbindelser. Målcellerna i fostren är melanoblasterna, och målgenerna är de gener som bestämmer pälshårens pigmentering. Fostren är heterozygota i ett antal av dessa pälsfärger. Vid mutation eller förlust av det dominanta allele i en melanoblaster genom en rad genetiska händelser kommer den recessiva fenotypen till uttryck i de celler som bildas från denna cellstam, vilket medför en annorlunda färgade fläckar i musens päls. Mängden av avkommor med sådana fläckar, dvs. mutationer, registreras och jämförs med motsvarande mängd blandad avkomma av hondjur som har exponerats för lösningsmedlet. Vid fläcktest på mus bestäms förmodade somatiska mutationer i fosterceller.
1.5 KVALITETSKRITERIER
Inga.
1.6 METODBESKRIVNING
Förberedelser
Testämnet upplöses eller suspenderas i isotonisk saltlösning. Kemiska förbindelser som inte är lösliga i vatten ska lösas eller suspenderas i lämplig vehikel. Vehikel som används för att underlätta dosering får inte inverka på testämnet eller framkalla toxiska effekter. Nyligen framställda lösningar av testämnet ska användas.
Mus av stammen T-stock (nonagouti, a/a; chinchilla, pink eye cchp/cchp; brown, b/b; dilute, short ear, d se/d se; piebald spotting, s/s) paras med antingen HT-stock (pallid, nonagouti, brachypody, pa a bp/pa a bp; leaden fuzzy, In fz/In fz; pearl pe/pe) eller C57 BL (nonagouti, a/a). Andra lämpliga korsningar såsom NMRI (nonagouti, a/a; albino, c/c) och DBA (nonagouti, a/a; brown b/b; dilute, d/d) kan användas under förutsättning att avkomman är nonagouti.
Tillräckligt många dräktiga hondjur för att producera en passande mängd avkomma för var och en av de använda doserna används. Det antal som är lämpligt kan fastställas på grundval av antalet observerade fläckar hos de exponerade mössen och kontrolldatas storleksordning. Ett negativt resultat är acceptabelt först sedan minst 300 ungar av hondjur som exponerats för den största dosen har undersökts.
Sidlöpande kontrolldata från möss som endast exponerats för vehikel (negativa kontroller) bör finnas tillgängliga. Kontrolldata från tidigare försök vid samma laboratorium kan räknas med för att höja testets känslighet, under förutsättning att de är homogena. Positiva kontrolldata bör finnas tillgängliga från försök som nyligen genomförts i samma laboratorium med en kemisk förbindelse som är känd för att visa mutagenicitet vid denna test, om det inte konstateras någon mutagenicitet av testämnet.
Normalt tillförs testämnet genom oral intubering eller intraperitoneal injicering i de dräktiga hondjuren. Inhalation och andra tillförselvägar kan vara lämpliga.
Minst två dosnivåer används, av vilka den ena ska medföra tecken på toxisk verkan eller minskad kullstorlek. Icke-toxiska ämnen testas genom användning av så höga doser som möjligt.
Förfarande
Vanligen ges försöksdjuren hela dosen på en gång under dräktighetsperiodens dag 8, 9 eller 10, varvid dag 1 räknas som den dag vaginalproppen observeras för första gången. Dessa dagar motsvarar dag 7,25, 8,25 och dag 9,25 efter befruktning. Upprepad behandling kan behövas på dessa dagar.
Avkomman blindkodas och eventuella pigmenterade fläckar registreras 3–4 veckor efter födseln. Man skiljer på tre typer av fläckar:
|
a) |
Vita fläckar inom ett avstånd av 5 mm från den ventrala mittlinjen. Dessa fläckar antas vara ett resultat av celldöd (WMVS). |
|
b) |
Gula agouti-aktiga fläckar på eller omkring spene, genitalia, strupe, axillär- och inguinalområden, och mitt i pannan. Dessa fläckar antas vara ett resultat av feldifferentiering (MDS). |
|
c) |
Pigmenterade och vita fläckar slumpmässigt fördelade i pälsen. Dessa fläckar antas vara ett resultat av somatiska mutationer (RS). |
Alla tre typer registreras men endast den sistnämnda, RS, är genetiskt relevant. Om det är svårt att skilja mellan MDS och RS kan fluorecensmikroskopi av hårprover användas.
Synliga morfologiska abnormiteter hos avkomman registreras också.
2. DATA
Det totala antalet undersökta ungar och antal ungar med en eller flera fläckar som antas vara ett resultat av somatisk mutation anges. Data från försöksgrupper och negativ kontrollgrupp jämförs med hjälp av lämpliga statistiska metoder. Data anges även per kull.
3. RAPPORTERING
3.1 TESTRAPPORT
Testrapporten ska om möjligt innehålla minst följande uppgifter:
|
— |
Korsning av använda stammar. |
|
— |
Antal dräktiga hondjur i försöks- och kontrollgrupperna. |
|
— |
Den genomsnittliga kullstorleken i försöks- och kontrollgrupperna vid födseln, och när ungarna är avvanda. |
|
— |
Dosnivåer. |
|
— |
Använt lösningsmedel. |
|
— |
Den dag i dräktighetsperioden då dosering sker. |
|
— |
Tillförselväg. |
|
— |
Det totala antal undersökta ungar och antal ungar med WMVS, MDS och RS i försöks- och kontrollgrupperna. |
|
— |
Makroskopiska morfologiska abnormiteter. |
|
— |
Förhållandet mellan dos och reaktion för RS när detta är möjligt. |
|
— |
Statistisk utvärdering. |
|
— |
Diskussion om resultaten. |
|
— |
Tolkning av resultaten. |
3.2 UTVÄRDERING OCH TOLKNING
Se allmän inledning till del B.
4. HÄNVISNINGAR
Se allmän inledning till del B.
B.25 TEST AVSEENDE ÄRFTLIG TRANSLOKATION PÅ MUS
1. METOD
1.1 INLEDNING
Se allmän inledning till del B.
1.2 DEFINITIONER
Se allmän inledning till del B.
1.3 REFERENSÄMNEN
Inga.
1.4 TESTMETODSPRINCIP
Vid test av ärftlig translokation hos mus konstateras strukturella och numeriska kromosomförändringar i däggdjursceller uttryckt i förstagenerationens avkomma. De kromosomförändringar som mäts är reciproka translokationer och, när avkomman även omfattar hondjur, förlust av en X-kromosom. Bärare av translokationer och XO-hondjur har nedsatt fertilitet, vilket utnyttjas vid valet av F1-avkomma för cytogenetisk analys. Vissa typer av translokationer (X-autosome och c-t-typen) orsakar fullständig sterilitet. Translokationer konstateras cytogenetiskt i meiosens diakines-metafas hos handjur, som antingen kan vara av F1-generationen eller söner till F,-hondjur. XO-hondjur identifieras cytogenetiskt genom att kromosomtalet i benmärgsmitoser endast är 39.
1.5 KVALITETSKRITERIER
Inga.
1.6 METODBESKRIVNING
Förberedelser
Testämnet upplöses i isotonisk saltlösning. Ämnen som inte är lösliga i saltlösning ska lösas eller uppslammas i lämplig vehikel. Nyligen framställda lösningar av testämnet ska användas. Vehikel som används för att underlätta dosering får inte inverka på testämnet eller framkalla toxiska effekter.
Vanligen tillförs testämnet genom oral intubering eller intraperitoneal injicering. Även andra tillförselvägar kan användas.
Som försöksdjur används mus eftersom det är enklast med hänsyn till uppfödning och cytologisk verifikation. Ingen särskild stam krävs. Om man planerar att testa fertiliteten bör den använda stammens genomsnittliga kullstorlek vara över åtta och relativt konstant.
Friska könsmogna djur används.
Det antal djur som behövs beror på den spontana translokationsfrekvensen och den induktionsfrekvens som är nödvändig för att uppnå ett positivt resultat.
Normalt görs testet genom undersökning av F1-handjur. Minst 500 F1-handjur undersöks per dos. Om även kvinnlig F1-avkomma används undersöks 300 av varje kön.
Tilllräckliga kontrolldata från sidlöpande och tidigare, kontrollförsök ska finnas tillgängliga. Om det finns godtagbara positiva kontrollresultat från tidigare försök vid samma laboratorium kan dessa användas istället för en sidlöpande positiv kontroll.
En dos testas, normalt den högsta dosen med minimal toxisk verkan, men utan inflytande på reproduktion eller överlevnad. Om förhållandet mellan dos och reaktion ska bestämmas används ytterligare två lägre doser. Icke-toxiska ämnen testas genom användning av så höga doser som möjligt.
Förfarande
Två olika behandlingsscheman kan användas. I det mest använda schemat ges hela dosen på en gång. Testämnet kan också ges 7 dagar i veckan i 35 dagar. Antalet parningar efter exponeringen beror på exponeringsschemat och ska vara tillräckligt stort för att säkerställa att alla exponerade könscellstadier kan undersökas. Efter parningen placeras hondjuren i var sin bur. Vid födseln registreras datumet, kullstorleken och avkommans kön. Alla handjuren bland avkomman avvänjs och hondjuren avlägsnas såvida de inte inbegrips i försöket
En av två möjliga metoder används:
|
— |
Fertilitetsundersökning av F1-generationen med efterföljande verifikation av eventuella translokationsbärare genom cytologisk analys. |
|
— |
Cytogenetisk analys av alla F1-handjur utan föregående urval på grundval av fertilitetsundersökningar. |
|
a) |
Fertilitetsundersökning Nedsatt fertilitet hos F1-generationens medlemmar kan konstateras vid observation av kullstorleken och/eller analys av de parade hondjurens livmoderinnehåll. Kriterier för bestämning av normal och nedsatt fertilitet måste fastställas innan fläcktestet på mus inleds. Observation av kullstorlek: De F1-handjur som ska undersökas placeras i var sin bur tillsammans med hondjur från samma försök eller från bestånden. Från och med den 18:e dagen efter parning iakttas burarna dagligen. Kullstorleken och kön i F2-generationen registreras vid födseln, varefter kullarna avlägsnas. Om F1-hondjur testas bevaras de små F2-kullarna för ytterligare undersökningar. Konstatering av om hondjur är translokationsbärare sker genom cytogenetisk analys av translokation hos någon av deras manliga avkomma. XO-hondjur identifieras genom ändring av förhållandet mellan han- och hondjur i deras avkomma från 1:1 till 1:2. Vid ett sekvensiellt ordnat förfarande elimineras normalt F1-djur från den vidare undersökningen om storleken av den första F2-kullen är lika med eller större än ett på förhand fastställt normalvärde. I motsatt fall observeras den andra eller tredje F2-kullen. F1-djur som inte kan klassificeras som normala efter observation av upp till tre F2-kullar underkastas antingen ytterligare undersökning genom analys av de parade hondjurens livmoderinnehåll eller direkt cytogenetisk analys. Analys av innehållet i livmodern: Den mindre kullstorleken hos translokationsbärare skylls på att fostren dör på det embryonala stadiet vilket innebär att ett stort antal döda implantat tyder på translokation hos försöksdjuret. De F1-handjur som ska undersökas paras vart och ett separat med upp till tre hondjur. Vid daglig undersökning mellan kl. 10 och kl. 12 på förmiddagen av förekomsten av vaginalpropp, konstateras om befruktning har skett. Hondjuren avlivas 14–16 dagar senare och antalet levande och döda implantat i livmodern registreras. |
|
b) |
Cytogenetisk analys Testpreparat framställs genom lufttorkningsteknik. Translokationsbärarna identifieras på grundval av multivalenta konfigurationer i primärar spermatocyters diakines-metafas I. Om det observeras minst två celler med multivalenta konfigurationer anses det bevisat att försöksdjuret är translokationsbärare. När inget urval görs på grundval av fertilitetsundersökningar undersöks alla F1-handjur cytogene-tiskt. Mikroskopisk undersökning görs av minst 25 diakines-metafaser I per handjur. F1-handjur med små testiklar och defekt meiose innan diakines, samt F1,-hondjur som förmodas vara XO-bärare, underkastas undersökning av mitotiska metafaser i spermatogoner eller benmärg. Om ovanligt långa och/eller korta kromosomer observeras i tio celler anses det bevisat att det är tal om en translokation som gör handjur sterila (c-typen). Vissa X-automsoma translokationer som gör handjur sterila kan endast identifieras genom bandanalys av mitosekromosomer. Om 39 kromosomer observeras i tio mitoser anses det bevisat att det är tal om ett XO-hondjur. |
2. DATA
Data ställs upp i tabellform.
Den genomsnittliga kullstorleken och könsfördelningen i föräldragenerationens avkomma vid födseln och vid avvänjning anges för varje parningsperiod.
För bedömning av F1-djurens fertilitet anges den genomsnittliga kullstorleken efter de normala parningarna och varje enskild kullstorlek efter parning av F1-translokationsbärare. Efter analys av livmoderinnehåll anges det genomsnittliga antalet levande och döda implantat efter normala parningar och antalet levande och döda implantat efter varje enskild parning av F1-translokationsbärare.
Efter cytogenetisk analys av diakines-metafaser I anges antalet multivalenta konfigurationer och det totala antalet undersökta celler för varje enskild translokationsbärare.
Det totala antalet parningar och parningsperiodernas längd anges för sterila F1-djur. Detaljerade upplysningar lämnas om testikelvikt och cytogenetisk analys.
Den genomsnittliga kullstorleken, F2-avkommans könsfördelning och resultaten av den cytogenetiska analysen anges för XO-hondjur.
Om eventuella F1-translokationsbärare väljs ut vid fertilitetsundersökningar ska tabellerna innehålla upplysningar om hur många av de utvalda djuren som senare visade sig vara heterozygota efter translokation.
Data från negativa och positiva kontrollförsök rapporteras på liknande sätt.
3. RAPPORTERING
3.1 TESTRAPPORT
Testrapporten ska om möjligt innehålla minst följande uppgifter:
|
— |
Musstam, djurens ålder, de doserade djurens vikt, antal djur av varje kön i försöks- och kon trollgrupperna i föräldragenerationen. |
|
— |
Antal föräldradjur av varje kön i försöks- och kontrollgrupperna. |
|
— |
Försöksbetingelser, detaljerad beskrivning av behandlingen av djuren, dosnivå, lösningsmedel, parningsschema. |
|
— |
Avkommans antal och kön för varje enskilt hondjur, antal och kön av avkomma som föds upp för translokationsanalys. |
|
— |
Tidpunkt och kriterier för translokationsanalys. |
|
— |
Antal translokationsbärare samt detaljerad beskrivning av dessa, inbegripet avlivningsdata och upplysningar om livmoderinnehåll, om sådana finns. |
|
— |
Cytogenetiska förfaranden och detaljerade upplysningar om den mikroskopiska analysen, helt med bilder. |
|
— |
Statistisk utvärdering. |
|
— |
Diskussion om resultaten. |
|
— |
Tolkning av resultaten. |
3.2 UTVÄRDERING OCH TOLKNING
Se allmän inledning till del B.
4. HÄNVISNINGAR
Se allmän inledning till del B.
B.26 TEST AVSEENDE SUBKRONISK ORAL TOXICITET 90 DAGARS UPPREPAT ORALTEST PÅ GNAGARE
1. METOD
Denna testmetod för subkronisk oral toxicitet motsvarar OECD TG 408 (1998).
1.1 INLEDNING
Vid bedömning och utvärdering av en kemikalies toxiska egenskaper kan bestämningen av subkronisk oral toxicitet med användning av upprepade doser genomföras efter det att man har fått preliminär toxicitetsinformation från test avseende akut toxicitet eller från test med upprepad dosering (28 dagar). Från 90-dagarstestet fås information om eventuella hälsoskador som kan uppstå till följd av upprepad exponering under en lång tidsperiod som täcker mognaden efter avvänjning och uppväxten ända in i vuxenstadiet. Genom testet fås information om de mest betydande toxiska verkningarna, indikation om vilka organ som påverkas och huruvida det finns en risk för ackumulering. Likaså kan man få en uppskattning om en exponeringsnivå vid vilken inga skadliga verkningar observeras. Denna information kan användas för att välja dosnivåer för test avseende kronisk toxicitet och för att fastställa säkerhetskriterier gällande exponering på människor.
Metoden innebär starkare betoning av neurologiska slutpunkter (endpoints) och ger indikationer om immunologiska och reproduktionstoxiska verkningar. Likaså betonas behovet av noggranna kliniska observationer av djuren i syfte att få fram så mycket information som möjligt. Testets syfte är att möjliggöra identifiering av kemikalier som har potential att framkalla sådana neurotoxiska, immunologiska eller reproduktionstoxiska verkningar som kan motivera närmare undersökningar.
Se även Allmän inledning, del B.
1.2 DEFINITIONER
Dos: Den mängd testämne som tillförs. Dosen uttrycks som vikt (g, mg), som vikten testämne per viktenhet försöksdjur (t.ex. mg/kg) eller som en konstant utfodringskoncentration (ppm).
Dosering: Ett allmänt begrepp som inbegriper dos, dess frekvens och varaktighet.
NOAEL: Förkortning för ’no-observed-adverse-effect level’ (nivå vid vilken inga skadliga verkningar observeras). Avser den högsta dosnivån vid vilken inga skadliga behandlingsrelaterade verkningar kan observeras.
1.3 PRINCIP FÖR TESTMETODEN
Testämnet tillförs oralt dagligen under 90 dagar i stegvis ökande doser till flera grupper försöksdjur, en dosnivå per grupp. Under denna period observeras djuren noggrant för att se om de uppvisar några tecken som tyder på att testämnet är toxiskt. Djur som dör eller avlivas under testet obduceras och vid testets slut avlivas de överlevande djuren och obduceras.
1.4 BESKRIVNING AV TESTMETODEN
1.4.1 Förberedelse av djuren
För testet används friska djur som har acklimatiserats till laboratoriemiljön under minst fem dagar och som inte har använts för tidigare test. Försöksdjuren bör karakteriseras enligt art, stam, ursprung, kön, vikt och ålder. Djuren fördelas slumpvis i kontroll- och behandlingsgrupper. Burarna placeras på sådant sätt att eventuella verkningar på grund av placeringen minimeras. Varje djur tilldelas ett unikt identifieringsnummer.
1.4.2 Förberedelse av doserna
Testämnet tillförs via sondmatning, foder eller dricksvatten. Den metod som väljs för oral tillförsel beror på testets syfte och testämnets fysikalisk-kemiska egenskaper.
Vid behov kan testämnet lösas upp eller suspenderas i en lämplig vehikel. Om möjligt bör man i första hand överväga vattenlösning eller vattensuspension, sedan oljelösning eller oljeemulsion (t.ex. majsolja) och därefter eventuella övriga vehiklar. I fråga om andra vehiklar än vatten måste de toxiska egenskaperna vara kända. Testämnets stabilitet vid tillförselförhållandena bör fastställas.
1.4.3 Testförhållanden
1.4.3.1 Försöksdjur
Rekommenderat försöksdjur är råtta, även om testet kan utföras med andra gnagare (t.ex. mus). Djuren bör vara unga och friska vuxna exemplar som härrör från normala laboratoriestammar. Hondjuren får inte ha fött ungar och får inte vara dräktiga. Doseringen bör inledas snarast möjligt efter avvänjningen och under alla förhållanden innan djuren är nio veckor gamla. Vid testets början bör variationen i vikt mellan de använda djuren vara minimal och får inte överstiga ± 20 % av vardera könets medelvikt. Om testet utförs som inledning till ett långtidstest avseende kronisk toxicitet måste djur från samma stam och ursprung användas för båda testen.
1.4.3.2 Antal och kön
Minst 20 djur (tio honor och tio hanar) bör användas för varje dosnivå. Om avlivningar är inplanerade under testets gång bör antalet djur ökas med det antal djur som avlivas innan testet har slutförts. Beroende på tidigare kunskaper om kemikalien eller en nära besläktad kemikalie bör man överväga att ta med ytterligare en satellitgrupp på tio djur (fem per kön) för kontrollgruppen och toppdosgruppen. Efter behandlingsperioden observeras satellitgruppen med avseende på reversibla eller kvarstående toxiska verkningar. Längden för denna period fastställs med beaktande av de observerade verkningarna.
1.4.3.3 Dosnivåer
Minst tre dosgrupper och en parallell kontrollgrupp bör användas, utom om gränstest utförs (se 1.4.3.4). Dosnivåerna kan basera sig på resultaten från test med upprepad dos eller från preliminära test för dosbestämning, och de bör fastställas med beaktande av alla toxikokinetiska data som finns tillgängliga för testämnet eller närbesläktade ämnen. Den högsta dosnivån väljs så att den framkallar toxiska verkningar utan att orsaka dödsfall eller svårt lidande, förutsatt att det inte finns begränsningar som beror på testämnets fysikalisk-kemiska natur eller biologiska verkningar. Därefter väljs dosnivåerna i fallande skala så att en dosrelaterad respons kan påvisas, och samtidigt så att inga påvisbara skadliga verkningar (NOAEL) kan observeras vid den lägsta dosnivån. Två- till fyrfaldiga dosintervall är ofta optimala för de fallande dosnivåerna. Tillägg av en fjärde testgrupp är att föredra framför användning av mycket breda intervall mellan dosnivåerna (t.ex. en faktor som är större än cirka 6–10).
Kontrollgruppen är en obehandlad grupp, eller en vehikelkontrollgrupp om vehikel används för tillförsel av testämnet. Förutom att djuren i kontrollgruppen inte behandlas med testämnet bör de hanteras på exakt samma sätt som djuren i dosgrupperna. Om en vehikel används bör kontrollgruppen tillföras vehikeln i den största volym som används. Om testämnet tillförs via fodret och man märker att foderintaget minskar, kan det vara till fördel med en parallellt utfodrad kontrollgrupp för att kunna avgöra huruvida minskningen beror på att testämnet påverkar fodrets smak eller på att det förekommer toxikologiska förändringar i testmodellen.
Följande egenskaper hos vehikeln och övriga eventuella tillsatsämnen bör beaktas: verkningarna på absorption, distribution, metabolism eller kvarhållande av testämnet; verkningarna på testämnets kemiska egenskaper i den utsträckning dessa verkningar kan påverka testämnets toxiska egenskaper samt verkningarna på försöksdjurens foder- eller vattenintag eller deras näringstillstånd.
1.4.3.4 Gränstest (Limit-test)
Om ett test med en dosnivå om minst 1 000 mg per kg kroppsvikt och dag genomförs på det sätt som beskrivs här och inga toxiska verkningar kan observeras, och om toxicitet inte förväntas på grundval av uppgifter om strukturellt närbesläktade ämnen, kan ett fullständigt test med tre dosnivåer anses vara överflödigt. Gränstest tillämpas utom när exponering på människor indikerar att en högre dosnivå behövs.
1.5 FÖRFARANDE
1.5.1 Tillförsel av doser
Djuren ges dagliga doser av testämnet under 90 dagar. Varje annat doseringsschema, t.ex. dosering fem dagar per vecka, måste motiveras. Om testämnet tillförs genom sondmatning bör hela dosen tillföras på en gång via magsond eller lämplig intubationskanyl. Den största mängd vätska som kan tillföras vid ett och samma tillfälle beror på försöksdjurets storlek. Volymen får inte överstiga 1 ml per 100 g kroppsvikt utom i fråga om vattenlösningar för vilka 2 ml per 100 g kroppsvikt får användas. Med undantag för irriterande eller frätande ämnen som normalt ger kraftigare verkan med högre koncentration bör variationer i testvolym minimeras genom justering av koncentrationen, för att säkerställa en konstant volym på alla dosnivåer.
För ämnen som tillförs via foder eller dricksvatten är det viktigt att försäkra sig om att testämnet ges i kvantiteter som inte stör det normala näringsupptaget eller den normala vattenbalansen. När testämnet tillförs via fodret kan man använda en konstant koncentration (ppm) eller en konstant dosnivå i förhållande till djurens kroppsvikt. Det använda alternativet måste anges. När ett ämne tillförs genom sondmatning bör dosen ges vid samma tid varje dag och vid behov justeras dosen så att dosnivån hålls konstant i förhållande till djurens kroppsvikt. Om ett 90-dagarstest används som inledning till ett långtidstest avseende kronisk toxicitet måste samma foder användas i båda testen.
1.5.2 Observationer
Observationsperioden bör vara minst 90 dagar. Djur i en satellitgrupp avsedd för fortsatta observationer hålls obehandlade under en lämplig period så att man kan upptäcka kvarstående verkningar eller återhämtning från toxiska verkningar.
Generella kliniska observationer bör göras minst en gång per dag, helst vid samma tid(er) varje dag och med beaktande av perioden för när maximala verkningar förväntas efter dosering. Djurens hälsostatus bör registreras. Minst två gånger per dag, i regel i början och slutet av varje dag, undersöks alla djur i syfte att hitta döende eller döda djur.
Noggranna kliniska observationer görs minst en gång av alla djur före den första exponeringen (för att möjliggöra jämförelser på samma objekt) och därefter minst en gång i veckan. Dessa observationer bör göras utanför buren, helst på en ’standardarena’ och vid samma tid på dagen varje gång. Observationerna bör noteras noggrant, helst genom ett poängsystem som testlaboratoriet har definierat exakt. Man bör eftersträva minimala variationer i observationsbetingelserna. Observationerna bör omfatta, men inte begränsas till, förändringar av hud, päls, ögon och slemhinnor, förekomst av sekret och utsöndringar samt autonom aktivitet (tårflöde, upprest päls, pupillstorlek, ovanligt andningsmönster). Förändringar i gång, hållning och reaktion på hantering liksom eventuella kramper eller spasmer, stereotypt beteende (t.ex. överdrivet putsande, eller att djuren springer runt i cirklar) eller bisarrt beteende (t.ex. självstympning, baklängesgång) bör också noteras (1).
Innan testämnet tillförs och efter avslutat test undersöks djuren oftalmologiskt med ett oftalmoskop eller motsvarande utrustning. Denna undersökning bör helst omfatta alla djur men åtminstone djuren i högdos- och kontrollgrupperna. Om ögonförändringar upptäcks bör alla djur undersökas.
Mot slutet av exponeringsperioden, och under alla förhållanden inte tidigare än vecka 11, görs en bedömning av den sensoriska reaktionen på stimuli (1) (t.ex. hörsel-, syn- och djupsensoriska stimuli) (2), (3), (4), samt bedömning av greppstyrka (5) och motorisk aktivitet (6). Närmare uppgifter om de förfaranden som kan följas finns i respektive hänvisning. Alternativt kan även andra förfaranden än de som nämns i hänvisningarna användas.
Mot slutet av testet kan funktionsobservationerna utelämnas om det finns uppgifter om motsvarande observationer tillgängliga från övriga test och man inte har kunnat konstatera några funktionsstörningar vid de dagliga kliniska observationerna.
I undantagsfall kan funktionsobservationerna också utelämnas för grupper vars djur i övrigt uppvisar tecken på toxicitet i en sådan utsträckning att det väsentligt skulle påverka funktionstestet.
1.5.2.1 Kroppsvikt samt foder- och vattenkonsumtion
Alla djur bör vägas minst en gång i veckan. Foder- och vattenkonsumtion bör mätas minst en gång varje vecka. Om testämnet tillförs via dricksvattnet bör även vattenkonsumtionen mätas minst en gång i veckan. Mätning av vattenkonsumtionen kan också övervägas för foder- eller sondmatningstest under vilka vattendrickandet kan förändras.
1.5.2.2 Hematologi och klinisk biokemi
Blodprov bör tas från ett angivet ställe och förvaras under lämpliga förhållanden. I slutet av testperioden tas blodprov strax före eller som ett led i avlivningsförfarandet.
Följande hematologiska undersökningar bör göras då testperioden avslutas och varje gång eventuella blodprover tas under testperiodens gång: hematokrit, hemoglobinkoncentration, räkning av röda blodkroppar, total-och differentialräkning av vita blodkroppar, blodplättsräkning samt mätning av koaguleringstid och koaguleringsförmåga.
Kliniska biokemiska bestämningar för att undersöka större toxiska effekter i vävnader, särskilt verkningar på lever och njurar, bör utföras på de blodprov som tas från alla djur strax före eller som ett led i avlivningsförfarandet (med undantag för de djur som hittas döende eller avlivas under testets gång). På samma sätt som för hematologiska undersökningar kan blodprover för kliniska biokemiska undersökningar tas under testets gång. Det rekommenderas att djuren fastar natten före blodprovstagningen (3). Undersökningarna av plasma eller serum bör omfatta natrium, kalium, glukos, totalkolesterol, urinämne, urinämneskväve, kreatinin, totalprotein och albumin, minst två enzymer som indikerar verkningar på leverceller (t.ex. alaninaminotransferas, aspartataminotransferas, alkaliskt fosfatas, gamma-glutamyltranspeptidas och sorbitoldehydrogenas). Mätningar av ytterligare enzymer (från lever eller av annat ursprung) och gallsyror kan under vissa förhållanden ge användbar information.
Eventuellt kan dessutom följande urinanalyser utföras under sista veckan av testet med uppsamling av urin på fasta tider: utseende, volym, osmolalitet eller specifik vikt, pH, protein, glukos och blod eller blodceller.
Dessutom bör undersökning av serummarkörer av allmänna vävnadsskador övervägas. Ytterligare bestämningar som bör utföras, i det fall att testämnets kända egenskaper kan eller misstänks påverka närbesläktade metaboliska profiler, omfattar kalcium, fosfor, fastevärden av triglycerider, specifika hormoner, metahemoglobin och kolinesteras. Dessa bestämningar behövs för kemikalier inom vissa klasser eller från fall till fall.
Överlag krävs en viss flexibilitet i fråga om tillvägagångssätt, beroende på djurart och på de observerade eller förväntade verkningarna av ett givet ämne.
Om historiska basdata är otillräckliga bör man överväga bestämning av hematologiska och kliniska biokemiska variabler innan doseringen påbörjas. I allmänhet rekommenderas inte att dessa uppgifter genereras före behandlingen (7).
1.5.2.3 Obduktion
Alla försöksdjur bör genomgå en fullständig obduktion som omfattar omsorgsfull yttre granskning av kroppen, alla kroppsöppningar samt kranium, bröst- och bukhåla och deras innehåll. Lever, njurar, binjurar, testiklar, bitestiklar, livmoder, äggstockar, bräss, mjälte, hjärna och hjärta från alla djur (med undantag för de djur som hittas döende eller avlivas under testets gång) putsas från angränsande vävnad och deras våtvikt bestäms så snart möjligt efter dissekering för att undvika uttorkning.
Följande vävnader och organ bör bevaras i det mest lämpade mediet med hänsyn till typen av vävnad och planerade efterföljande histopatologiska undersökningar: alla organ och vävnader med stora skador, hjärnan (representativa regioner inbegripet storhjärna, lillhjärna och hjärnbrygga), ryggrad (på tre nivåer: cervikal, thorakal (mellersta) och lumbal), hypofys, sköldkörtel, bisköldkörtel, bräss, matstrupe, salivkörtlar, magsäck, tunntarm och tjocktarm (inklusive Peyers plack), lever, bukspottkörtel, njurar, binjurar, mjälte, hjärta, luftstrupe och lungor (konserverade genom inflation med fixermedel och därefter immersion), aorta, könskörtlar, livmoder, accessoriska könsorgan, honornas bröstkörtlar, prostata, urinblåsa, gallblåsa (mus), lymfkörtlar (helst en lymfkörtel som omfattar tillförselvägen och en annan långt från tillförselvägen för att täcka systematiska effekter), perifer nerv (ischias- eller tibialnerven), helst nära muskeln, ett snitt av ryggmärgen (alternativt färskt benmärgsaspirat), hud och ögon (om förändringar har observerats under oftalmologiska undersökningar). Kliniska eller övriga fynd kan indikera att ytterligare vävnader behöver undersökas. Dessutom bör man bevara alla organ som på grund av testämnets kända egenskaper förmodas vara målorgan.
1.5.2.4 Histopatologi
En fullständig histopatologisk undersökning bör utföras på bevarade organ och vävnader från alla djur i kontroll- och högdosgrupperna. Om behandlingsrelaterade förändringar observeras i högdosgruppen bör undersökningarna utsträckas till djuren i alla övriga dosgrupper.
Alla stora skador bör undersökas.
Om en satellitgrupp används bör histopatologisk undersökning utföras på de vävnader och organ på vilka verkningar har konstaterats i dosgrupperna.
2. DATA OCH RAPPORTERING
2.1 DATA
Uppgifter bör rapporteras individuellt. Därutöver bör alla uppgifter sammanställas i en tabell över antalet djur i varje testgrupp vid testets början, antalet djur som hittats döda under testet eller som avlivats av humanitära skäl samt tidpunkten för avlivning, antalet djur som uppvisat tecken som tyder på att testämnet är toxiskt, en beskrivning av observerade tecken på toxicitet, inbegripet tidpunkten då sådana första gången upptäcktes, varaktighet och hur allvarliga de toxiska verkningarna är, antalet djur som har skador, typ av skada och procentandelen djur för varje typ av skada.
Om möjligt bör numeriska resultat utvärderas genom en tillämplig och allmänt godtagbar statistisk metod. De statistiska metoderna och data för vilka analys planeras bör väljas när testet utformas.
2.2 TESTRAPPORT
Testrapporten ska innehålla nedan angivna uppgifter.
2.2.1 Testämne
|
— |
Aggregationstillstånd, renhet och fysikalisk-kemiska egenskaper. |
|
— |
Identifieringsdata. |
|
— |
Vehikel (i förekommande fall). Motivering för val av vehikel, om annan än vatten. |
2.2.2 Försöksdjur
|
— |
Art och stam som använts. |
|
— |
Djurens antal, ålder och kön. |
|
— |
Ursprung, förvaringsförhållanden, foder osv. |
|
— |
Djurens individuella vikter vid testets början. |
2.2.3 Testförhållanden
|
— |
Motivering för val av dosnivåer. |
|
— |
Utförliga uppgifter om testämnets formulering och beredning av fodret, uppnådd koncentration, prepara tets stabilitet och homogenitet. |
|
— |
Utförliga uppgifter om hur testämnet har tillförts. |
|
— |
Faktiska doser (mg per kg kroppsvikt och dag) och omräkningsfaktor från testämneskoncentrationen i foder eller vatten (ppm) till faktisk dos, om tillämpligt. |
|
— |
Uppgifter om foder- och vattenkvalitet. |
2.2.4 Resultat
|
— |
Kroppsvikt och förändringar av kroppsvikt. |
|
— |
Foderkonsumtion och i tillämpliga fall vattenkonsumtion. |
|
— |
Uppgifter om toxiska reaktioner per kön och dosnivå, inklusive tecken på toxicitet. |
|
— |
Art, grad och varaktighet för kliniska observationer (oavsett om de är reversibla eller inte). |
|
— |
Resultat av oftalmologisk undersökning. |
|
— |
Bedömning av sensorisk aktivitet, greppstyrka och motorisk aktivitet (i mån av tillgänglighet). |
|
— |
Blodundersökningar med relevanta basvärden. |
|
— |
Kliniska biokemiska undersökningar med relevanta basvärden. |
|
— |
Kroppsvikt vid avlivning, organvikt och förhållandena mellan organ- och kroppsvikt. |
|
— |
Obduktionsfynd. |
|
— |
En detaljerad beskrivning av alla histopatologiska fynd. |
|
— |
Absorptionsdata om de finns tillgängliga. |
|
— |
Statistisk bearbetning av resultaten, i tillämpliga fall. |
Diskussion om resultaten.
Slutsatser.
3. HÄNVISNINGAR
|
1. |
IPCS (1986). Principles and Methods for the Assessment of Neurotoxicity Associated with Exposure to Chemicals. Environmental Health Criteria Document No 60. |
|
2. |
Tupper, D. E., Wallace, R.B. (1980). Utility of the Neurologic Examination in Rats. Acta Neurobiol. Exp., 40, pp. 999–1003. |
|
3. |
Gad, S. C. (1982). A Neuromuscular Screen for Use in Industrial Toxicology. J. Toxicol. Environ. Health, 9, pp. 691–704. |
|
4. |
Moser, V. C, Mc Daniel, K. M., Phillips, P. M. (1991). Rat Strain and Stock Comparisons Using a Functional Observational Battery: Baseline Values and Effects of Amitraz. Toxicol. Appl. Pharmacol., 108, pp. 267–283. |
|
5. |
Meyer O. A., Tilson H. A., Byrd W. C, Riley M. T. (1979). A Method for the Routine Assessment of Fore- and Hindlimb grip Strength of Rats and Mice. Neurobehav. Toxivol., 1, pp. 233–236. |
|
6. |
Crofton K. M., Howard J. L., Moser V. C, Gill M. W., Reiter L. W., Tilson H. A., MacPhail R. C. (1991). Interlaboratory Comparison of Motor Activity Experiments: Implication for Neurotoxicological Assessments. Neurotoxicol. Teratol., 13, pp. 599–609. |
|
7. |
Weingand K., Brown G., Hall R. et al. (1996). ’Harmonisation of Animal Clinic Pathology Testing in Toxicity and Safety Studies’, Fundam. & Appl. Toxicol., 29, pp. 198–201. |
B.27 TEST AVSEENDE SUBKRONISK ORAL TOXICITET 90 DAGARS UPPREPAT ORALTEST PÅ ICKE-GNAGARE
1. METOD
Denna testmetod för subkronisk oral toxicitet motsvarar OECD TG 409 (1998).
1.1 INLEDNING
Vid bedömning och utvärdering av en kemikalies toxiska egenskaper kan bestämningen av subkronisk oral toxicitet med användning av upprepade doser genomföras efter det att man har fått preliminär toxicitetsinformation från test avseende akut toxicitet eller från test med upprepad dosering (28 dagar). Genom 90-dagarsstestet fås information om eventuella hälsoskador som kan uppstå till följd av upprepad exponering under en period av snabb tillväxt och in i ungt vuxenstadium. Genom testet fås information om de mest betydande toxiska verkningarna, indikation om vilka organ som påverkas och huruvida det finns en risk för ackumulering. Likaså kan man få en uppskattning om en exponeringsnivå vid vilken inga skadliga verkningar observeras. Denna information kan användas för att välja dosnivåer för test avseende kronisk toxicitet och för att fastställa säkerhetskriterier gällande exponering på människor.
Testmetoden kan användas för identifiering av skadliga verkningar hos icke-gnagare som utsätts för exponering av en kemikalie och den bör endast användas i följande fall:
|
— |
Verkningar som har observerats i övriga test indikerar att det finns ett behov av förtydligande eller karakterisering med hjälp av en art som inte är gnagare. |
|
— |
Toxikokinetiska test indikerar att en specifik art av icke-gnagare är det mest relevanta alternativet för valet av försöksdjur. |
|
— |
Andra särskilda orsaker motiverar att en art som inte är gnagare används. |
Se även Allmän inledning, del B.
1.2 DEFINITIONER
Dos: Den mängd testämne som tillförs. Dosen uttrycks som vikt (g, mg), som vikten testämne per viktenhet försöksdjur (t.ex. mg/kg) eller som en konstant utfodringskoncentration (ppm).
Dosering: Ett allmänt begrepp som inbegriper dos, dess frekvens och varaktighet.
NOAEL: Förkortning för ”no-observed-adverse-effect level” (nivå vid vilken inga skadliga verkningar observeras). Avser den högsta dosnivån vid vilken inga skadliga behandlingsrelaterade verkningar kan observeras.
1.3 PRINCIP FÖR TESTMETODEN
Testämnet tillförs oralt dagligen under 90 dagar i stegvis ökande doser till flera grupper försöksdjur, en dosnivå per grupp. Under denna period observeras djuren noggrant för att se om de uppvisar några tecken som tyder på att testämnet är toxiskt. Djur som dör eller avlivas under testet obduceras och vid testets slut avlivas de överlevande djuren och obduceras.
1.4 BESKRIVNING AV TESTMETODEN
1.4.1 Val av djurart
En ofta använd icke-gnagare är hund. Hunden bör vara av definierad ras, ofta används beagle. Även andra arter, t.ex. svin eller minigrisar, kan användas. Primater rekommenderas inte och användning av dem måste motiveras. Djuren bör vara unga och friska. När det gäller hund bör doseringen helst inledas vid 4–6 månaders ålder och aldrig senare än vid 9 månaders ålder. Om testet utförs som inledning till ett långtidstest avseende kronisk toxicitet måste djur av samma art och ras användas för båda testen.
1.4.2 Förberedelse av djuren
För testet används unga och friska djur som har acklimatiserats till laboratoriemiljön och som inte har använts för tidigare test. Acklimatiseringsperiodens längd beror på djurarterna och deras ursprung. För hund eller för ändamålet uppfödda svin från en resident koloni rekommenderas minst fem dagar och för djur av externt ursprung rekommenderas minst två veckor. Försöksdjuren bör karakteriseras enligt art, stam, ursprung, kön, vikt och ålder. Djuren fördelas slumpvis i kontroll- och behandlingsgrupper. Burarna placeras på sådant sätt att eventuella verkningar på grund av placeringen minimeras. Varje djur tilldelas ett unikt identifieringsnummer.
1.4.3 Beredning av doserna
Testämnet kan tillföras via foder eller dricksvatten, genom sondmatning eller i kapslar. Den metod som väljs för oral tillförsel beror på testets syfte och testämnets fysikalisk-kemiska egenskaper.
Vid behov kan testämnet lösas upp eller suspenderas i en lämplig vehikel. Om möjligt bör man i första hand överväga vattenlösning eller vattensuspension, sedan oljelösning eller oljeemulsion (t.ex. majsolja) och därefter eventuellt andra vehiklar. I fråga om andra vehiklar än vatten måste de toxiska egenskaperna vara kända. Testämnets stabilitet vid tillförselförhållandena bör fastställas.
1.5 FÖRFARANDE
1.5.1 Djurens antal och kön
Minst åtta djur (fyra honor och fyra hanar) bör användas för varje dosnivå. Om avlivningar är inplanerade under testets gång bör antalet djur ökas med det antal djur som avlivas under testets gång. Antalet djur vid testets slut måste vara tillräckligt med tanke på en meningsfylld utvärdering av de toxiska verkningarna. Beroende på tidigare kunskaper om ämnet eller en nära besläktad kemikalie bör man överväga att ta med ytterligare en satellitgrupp på åtta djur (fyra per kön) för kontrollgruppen och toppdosgruppen. Efter behandlingsperioden observeras satellitgruppen med avseende på reversibla eller kvarstående toxiska verkningar. Längden för denna period fastställs med beaktande av de observerade verkningarna.
1.5.2 Dosering
Minst tre dosgrupper och en parallell kontrollgrupp bör användas, utom om gränstest utförs (se 1.5.3). Dosnivåerna kan basera sig på resultaten från test med upprepad dos eller från preliminära test för dosbestämning och de fastställs med beaktande av alla toxikokinetiska data som finns tillgängliga för testämnet eller närbesläktade ämnen. Den högsta dosnivån väljs så att den framkallar toxiska verkningar utan att orsaka dödsfall eller svårt lidande, förutsatt att det inte finns begränsningar som beror på testämnets fysikalisk-kemiska natur eller biologiska verkningar. Därefter väljs dosnivåerna i fallande skala så att en dosrelaterad reaktion kan påvisas, medan inga påvisbara skadliga verkningar (NOAEL) bör uppstå vid den lägsta dosnivån. Två- till fyrfaldiga dosintervall är ofta optimala för de fallande dosnivåerna. Tillägg av en fjärde testgrupp är att föredra framför användning av mycket breda intervall mellan dosnivåerna (t.ex. över en faktor på cirka 6–10).
Kontrollgruppen är en obehandlad grupp, eller en vehikelkontrollgrupp om vehikel används för tillförsel av testämnet. Förutom att djuren i kontrollgruppen inte behandlas med testämnet bör de hanteras på exakt samma sätt som djuren i dosgrupperna. Om en vehikel används bör kontrollgruppen tillföras vehikeln i den största volym som används. Om testämnet tillförs via fodret och man märker att foderintaget minskar, kan det vara till fördel med en parallellt utfodrad kontrollgrupp for att kunna avgöra huruvida minskningen beror på att testämnet påverkar fodrets smak eller på att det förekommer toxikologiska förändringar i test-modellen.
Följande egenskaper hos vehikeln och övriga eventuella tillsatsämnen bör beaktas: verkningarna på absorption, distribution, metabolism eller kvarhållande av testämnet; verkningarna på testämnets kemiska egenskaper i den utsträckning dessa verkningar kan påverka testämnets toxiska egenskaper samt verkningarna på försöksdjurens foder- eller vattenintag eller deras näringstillstånd.
1.5.3 Gränstest (Limit-test)
Om ett test med en dosnivå om minst 1 000 mg per kg kroppsvikt och dag genomförs på det sätt som beskrivs här och inga toxiska verkningar kan observeras, och om toxicitet inte förväntas på grundval av uppgifter om strukturellt närbesläktade ämnen, kan ett fullständigt test med tre dosnivåer anses vara överflödigt. Gränstest tillämpas utom när exponering på människor indikerar att en högre dosnivå behövs.
1.5.4 Tillförsel av doser
Djuren ges dagliga doser av testämnet under 90 dagar. Varje annat doseringsschema, t.ex. dosering fem dagar per vecka, måste motiveras. Om testämnet tillförs genom sondmatning bör hela dosen tillföras på en gång via magsond eller lämplig intubationskanyl. Den största mängd vätska som kan tillföras vid ett och samma tillfälle beror på försöksdjurets storlek. I regel bör volymen hållas så låg som möjligt. Med undantag för irriterande eller frätande ämnen som normalt ger kraftigare verkan med högre koncentration bör variationer i testvolym minimeras genom justering av koncentrationen, för att säkerställa en konstant volym på alla dosnivåer.
För ämnen som tillförs via foder eller dricksvatten är det viktigt att försäkra sig om att testämnet ges i kvantiteter som inte stör det normala näringsupptaget eller den normala vattenbalansen. När testämnet tillförs via fodret kan man använda en konstant koncentration (ppm) eller en konstant dosnivå i förhållande till djurens kroppsvikt. Det använda alternativet måste anges. För ett ämne som tillförs genom sondmatning eller kapslar bör dosen ges vid samma tid varje dag och vid behov justeras så att dosnivån hålls konstant i förhållande till djurens kroppsvikt. Om ett 90-dagarstest används som inledning till ett långtidstest avseende kronisk toxicitet måste samma foder användas i båda testen.
1.5.5 Observationer
Observationsperioden bör vara minst 90 dagar. Djur i en satellitgrupp avsedd för fortsatta observationer hålls obehandlade under en lamplig period så att man kan upptäcka kvarstående verkningar eller återhämtning från toxiska verkningar.
Generella kliniska observationer bör göras minst en gång per dag, helst vid samma tid(er) varje dag och med beaktande av perioden för när maximala verkningar förväntas efter dosering. Djurens hälsostatus bör registreras. Minst två gånger per dag, i regel i början och slutet av varje dag, undersöks alla djur i syfte att hitta döende eller döda djur.
Noggranna kliniska observationer görs minst en gång av alla djur före den första exponeringen (för att möjliggöra jämförelser på samma objekt) och därefter minst en gång i veckan. Dessa observationer bör göras, när det är genomförbart, utanför buren, helst på en ”standardarena” och vid samma tidpunkt varje gång. Man bör eftersträva minimala variationer i observationsbetingelserna. Alla tecken på toxicitet bör registreras i detalj, inklusive tidpunkt för första upptäckt, svårighetsgrad och varaktighet. Observationerna bör omfatta, men inte begränsas till, förändringar av hud, päls, ögon och slemhinnor, förekomst av sekret och utsöndringar samt autonom aktivitet (tårflöde, upprest päls, pupillstorlek, ovanligt andningsmönster). Förändringar i gång, hållning och reaktion på hantering liksom eventuella kramper eller spasmer, stereotypt beteende (t.ex. överdrivet putsande, eller att djuren springer runt i cirklar) eller eventuellt bisarrt beteende (t.ex. självstympning, baklängesgång) bör också noteras.
Innan testämnet tillförs och efter avslutat test undersöks djuren oftalmologiskt med ett oftalmoskop eller motsvarande utrustning. Denna undersökning bör helst omfatta alla djur men åtminstone djuren i högdos-och kontrollgrupperna. Om behandlingsrelaterade ögonförändringar upptäcks bör alla djur undersökas.
1.5.5.1 Kroppsvikt samt foder- och vattenkonsumtion
Alla djur bör vägas minst en gång i veckan. Foder- och vattenkonsumtion bör mätas minst en gång varje vecka. Om testämnet tillförs via dricksvattnet bör även vattenkonsumtionen mätas minst en gång i veckan. Mätning av vattenkonsumtionen kan också övervägas för test med tillförsel via foder eller sond under vilka vattendrickandet kan förändras.
1.5.5.2 Hematologi och klinisk biokemi
Blodprov bör tas från ett angivet ställe och förvaras under lämpliga förhållanden. I slutet av testperioden tas blodprov strax före eller som ett led i avlivningsförfarandet.
Blodprov, omfattande hematokrit, hemoglobinkoncentration, räkning av röda blodkroppar, total- och differentialräkning av vita blodkroppar, blodplättsräkning samt mätning av koaguleringstid och koaguleringsförmåga, protrombintid eller tromboplastintid tas vid testets början och därefter varje månad eller när halva testperioden gått, och när testet avslutas.
Kliniska biokemiska bestämningar för undersökning av betydande toxiska verkningar i vävnader, särskilt verkningar på njurar och lever, görs på blodprov som tagits från alla djur när testets inleds, varje månad eller när halva testperioden har gått, och när testet avslutas. Undersökning kan även övervägas i fråga om elektrolytbalans, kolhydratmetabolism samt lever- och njurfunktion. De specifika undersökningar som väljs beror på observationerna av hur testämnet agerar. Innan blodprov tas bör djuren fasta under en för arten lämplig period. Till de bestämningar som föreslås hör mätningar av kalcium, fosfor, klorid, natrium, kalium, fasteglukos, alaninaminotransferas, aspartataminotransferas, ornitindekarboxylas, gammaglutamyltranspep-tidas, urinämneskväve, albumin, blodkreatinin, totalbilirubin och totalserumprotein.
Urinanalyser bör göras minst när testet inleds, när halva testperioden gått och när testet avslutas, med uppsamling av urin på fasta tider. Urinanalyserna omfattar utseende, volym, osmolalitet eller specifik vikt, pH, protein, glukos och blod eller blodceller. Ytterligare parametrar kan vid behov användas för att utvidga undersökningen av observerade verkningar.
Dessutom bör undersökning av markörer för allmänna vävnadsskador övervägas. För en tillräcklig toxikologisk bedömning kan även behövas analys av lipider, hormoner, syra-basbalans, metahemoglobin och kolinesterasinhibition. Ytterligare klinisk biokemi kan vid behov användas för att utvidga undersökningen av observerade verkningar. Dessa bestämningar behövs för kemikalier inom vissa klasser eller från fall till fall.
Överlag krävs en viss flexibilitet i fråga om tillvägagångssätt, beroende på djurart och på de observerade eller förväntade verkningarna av ett givet ämne.
1.5.5.3 Obduktion
Alla försöksdjur bör genomgå en fullständig obduktion som omfattar omsorgsfull yttre granskning av kroppen, alla kroppsöppningar samt kranium, bröst- och bukhåla och deras innehåll. Lever med gallblåsa, njurar, binjurar, testiklar, bitestiklar, äggstockar, livmoder, sköldkörtel (med bisköldkörtlar), bräss, mjälte, hjärna och hjärta från alla djur (med undantag för de djur som hittas döende eller avlivas under testets gång) putsas från angränsande vävnad och deras våtvikt bestäms så snart som möjligt efter dissekering för att undvika uttorkning.
Följande vävnader och organ bör bevaras i det mest lämpade mediet med hänsyn till typen av vävnad och planerade efterföljande histopatologiska undersökningar: alla organ och vävnader med stora skador, hjärnan (representativa regioner inbegripet storhjärna, lillhjärna och hjärnbrygga), ryggrad (på tre nivåer: cervikal, thorakal (mellersta) och lumbal), hypofys, ögon, sköldkörtel, bisköldkörtel, bräss, matstrupe, salivkörtlar, magsäck, tunntarm och tjocktarm (inklusive Peyers plack), lever, gallblåsa, bukspottkörtel, njurar, binjurar, mjälte, hjärta, luftstrupe och lungor, aorta, könskörtlar, livmoder, accessoriska könsorgan, honornas bröstkörtlar, prostata, urinblåsa, lymfkörtlar (helst en lymfkörtel som omfattar tillförselvägen och en annan långt från tillförselvägen för att täcka systematiska effekter), perifer nerv (ischias- eller tibialnerven), helst nära muskeln, ett snitt av ryggmärgen (alternativt färskt benmärgsaspirat) och hud. Kliniska eller övriga fynd kan indikera att ytterligare vävnader behöver undersökas. Dessutom bör man bevara alla organ som på grund av testämnets kända egenskaper förmodas vara målorgan.
1.5.5.4 Histopatologi
En fullständig histopatologisk undersökning bör utföras på bevarade organ och vävnader från åtminstone alla djur i kontroll- och högdosgruppen. Undersökningarna bör utsträckas till djur i alla andra dosgrupper om behandlingsrelaterade förändringar observeras i högdosgruppen.
Alla stora skador bör undersökas.
Om en satellitgrupp används bör histopatologisk undersökning utföras på de vävnader och organ på vilka verkningar har konstaterats i dosgrupperna.
2. DATA OCH RAPPORTERING
2.1 DATA
Uppgifter bör rapporteras individuellt. Därutöver bör alla uppgifter sammanställas i en tabell över antalet djur i varje testgrupp vid testets början, antalet djur som hittats döda under testet eller som avlivats av humanitära skäl samt tidpunkten för avlivning, antalet djur som uppvisat tecken som tyder på att testämnet är toxiskt, en beskrivning av observerade tecken på toxicitet, inbegripet tidpunkten då sådana första gången upptäcktes, varaktighet och hur allvarliga de toxiska verkningarna är, antalet djur som har skador, typ av skada och procentandelen djur för varje typ av skada.
Om möjligt bör numeriska resultat utvärderas genom en tillämplig och allmänt godtagbar statistisk metod. De statistiska metoderna och data för vilka analys planeras bör väljas när testet utformas.
2.2 TESTRAPPORT
Testrapporten ska innehålla nedan angivna uppgifter.
2.2.1 Testämne
|
— |
Aggregationstillstånd, renhet och fysikalisk-kemiska egenskaper. |
|
— |
Identifieringsdata. |
|
— |
Vehikel (i förekommande fall). Motivering för val av vehikel, om annan än vatten. |
2.2.2 Försöksdjur
|
— |
Art och stam som använts. |
|
— |
Djurens antal, ålder och kön. |
|
— |
Ursprung, förvaringsförhållanden, foder osv. |
|
— |
Djurens individuella vikter vid testets början. |
2.2.3 Testförhållanden
|
— |
Motivering för val av dosnivåer. |
|
— |
Utförliga uppgifter om testämnets formulering och beredning av fodret, uppnådd koncentration, prepa ratets stabilitet och homogenitet. |
|
— |
Utförliga uppgifter om hur testämnet har tillförts. |
|
— |
Faktiska doser (mg per kg kroppsvikt och dag) och omräkningsfaktor från testämneskoncentrationen i foder eller vatten (ppm) till faktisk dos, om tillämpligt. |
|
— |
Uppgifter om foder- och vattenkvalitet. |
2.2.4 Resultat
|
— |
Kroppsvikt och förändringar av kroppsvikt. |
|
— |
Foderkonsumtion och i tillämpliga fall vattenkonsumtion. |
|
— |
Uppgifter om toxiska reaktioner per kön och dosnivå, inklusive tecken på toxicitet. |
|
— |
Art, grad och varaktighet för kliniska observationer (oavsett om de är reversibla eller inte). |
|
— |
Oftalmologisk undersökning. |
|
— |
Blodundersökningar med relevanta basvärden. |
|
— |
Kliniska biokemiska undersökningar med relevanta basvärden. |
|
— |
Kroppsvikt vid avlivning, organvikt och förhållandena mellan organ- och kroppsvikt. |
|
— |
Obduktionsfynd. |
|
— |
En detaljerad beskrivning av alla histopatologiska fynd. |
|
— |
Absorptionsdata om de finns tillgängliga. |
|
— |
Statistisk bearbetning av resultaten, i tillämpliga fall. |
Diskussion om resultaten.
Slutsatser.
B.28 TEST AVSEENDE SUBKRONISK DERMAL TOXICITET 90 DAGARS REPETERAD DERMALTEST PÅ GNAGARE
1. METOD
1.1 INLEDNING
Se allmän inledning till del B.
1.2 DEFINITIONER
Se allmän inledning till del B.
1.3 REFERENSÄMNEN
Inga.
1.4 TESTMETODSPRINCIP
Testämnet appliceras dagligen på huden i graderade doser till flera grupper av försöksdjur, en bestämd dos per grupp i 90 dagar. Under försöksperioden ska djuren observeras dagligen för tecken på toxicitet. Djur som dör under försöket obduceras och i slutet av försöket avlivas och obduceras överlevande djur,
1.5 KVALITETSKRITERIER
Inga.
1.6 METODBESKRIVNING
Förberedelser
I minst fem dagar före försöket ska djuren hållas under testbetingelserna vad beträffar miljö och utfodring. Före försöket fördelas unga friska djur slumpmässigt i behandlings- och kontrollgrupper. Kort innan försöket klipps ryggpälsen bort på försöksdjuren. Om pälsen rakas bort bör detta ske ca 24 timmar innan försöket inleds. Klippning och rakning måste normalt upprepas varje vecka. Vid klippning eller rakning ska försiktighet iakttas så att huden inte skadas. Minst 10 % av djurets kroppsyta ska göras klar för applicering av testämnet. Djurets vikt måste beaktas vid bestämning av hur stort område som ska klippas. Vid test av fasta ämnen, vilka vid behov kan pulveriseras, ska testämnet fuktas tillräckligt med vatten eller, vid behov, med lämplig vehikel, för att säkerställa god kontakt med huden. Flytande testämnen används vanligen outspädda. Daglig applicering fem till sju dagar i veckan ska ske.
Försöksbetingelser
Vuxna råttor eller kaniner får användas. Andra djurarter kan användas men detta måste motiveras. Djur av vanligen nyttjade laboratoriestammar ska användas. Vid inledning av försöket får vikten hos de djur som används inte avvika mer än ± 20 % från genomsnittsvikten. Om ett test av subkronisk toxicitet med dermal tillförsel utförs som en förundersökning till en långtidsundersökning ska samma djurart och stam användas i båda undersökningarna.
Minst 20 djur (10 hondjur och 10 handjur) med frisk hud ska användas på varje doseringsnivå. Hondjuren får inte ha fött ungar och får inte vara dräktiga. Vid planenligt avlivande av försöksdjur under försöket ska det ursprungliga antalet djur ökas med det antal djur som planenligt ska avlivas innan försöket slutförs. Dessutom kan en satellitgrupp bestående av 20 djur (10 av vardera kön) behandlas med den högsta dosen i 90 dagar och därefter observeras i 28 dagar för reversibilitet, per-sistens eller fördröjt uppträdande av toxiska verkningar.
Minst tre dosnivågrupper och en kontrollgrupp ska användas. Om vehikel används ska, förutom kontrollgruppen, en vehikelkontrollgrupp användas. Exponeringsperioden bör vara minst sex timmar per dag. Applicering av testämnet bör ske vid samma tidpunkt varje dag och den mängd ämne som används ska justeras regelbundet (varje eller varannan vecka) för att säkerställa att den hålls konstant i förhållande till djurets kroppsvikt. Djuren i kontrollgrupen ska behandlas på samma sätt som försöksdjuren, bortsett från applicering av testämnet. Om vehikel används för att underlätta doseringen ska vehikelkontrollgruppen doseras på samma sätt som försöksgrupperna och ges samma mängd ämne som den som appliceras på djuren i den försöksgrupp med den högsta doseringsnivån. Den högsta doseringsnivån bör medföra toxiska verkningar men inte leda till någon, eller endast begränsad, dödlighet. Den lägsta doseringsninvån bör inte ha någon toxisk verkan. Om det finns en användbar uppskattning av människors högsta exponering för ämnet bör den lägsta dosnivån vara högre än denna. Idealt ska medeldosen medföra minsta möjliga observerbara toxiska verkningar. Om flera medeldosnivåer används ska de väljas på ett sätt att en gradering av de toxiska verkningarna, erhålls. I lågdos-, medeldos- och kontrollgrupperna bör antalet dödsfall vara få för att möjliggöra en meningsfull utvärdering av resultaten.
Om testämnet medför allvarlig hudirritation bör koncentrationen sänkas, vilket kan leda till en minskning eller ett fullständigt bortfall av de övriga toxiska verkningarna vid den högsta doseringsnivån. Om huden har skadats allvarligt kan det vara nödvändigt att avbryta försöket och utföra ett nytt försök med en lägre koncentration.
Om en dos av 1 000 mg/kg kroppsvikt och dag eller en högre dosnivå som fastställts på grundval av kännedom om exponering för människor, inte medför någon toxisk verkan kan ytterligare testning anses överflödig.
Samtliga djur ska observeras dagligen och tecken på toxicitet registreras. Tidpunkten för dödsfall och tidpunkten vid vilken tecken på toxicitet uppträder respektive försvinner ska registreras.
Djuren ska hållas i var sin bur. Idealt appliceras testämnet varje dag i 90 dagar.
Djur i en eventuell satellitgrupp som planerats för uppföljning bör hållas under observation i ytterligare 28 dagar utan applicering av testämne för att fastställa om de toxiska verkningarna försvinner eller håller i sig. Exponeringsperioden bör vara sex timmar per dag.
Testämnet ska appliceras jämnt över ett område som motsvarar ca 10 % av den totala kroppsytan. Vid användning av kraftigt toxiska ämnen kan en mindre yta täckas, men ämnet ska appliceras på ytan i ett så tunt och jämnt lager som möjligt.
Testämnet hållas i kontakt med huden med en porös gaskompress och icke hudirriterande tejp. Testytan ska vidare täckas på lämpligt sätt för att säkerställa att gaskompressen och testämnet hålls på plats och att djuren inte kan svälja testämnet. För att förhindra att djuren intar testämnet kan rörelsehindrande anordningar användas, men total immobilisering rekommenderas inte.
I slutet av exponeringsperiodens ska resterande testämne avlägsnas, i lämpliga fall med vatten eller med annan lämplig metod för rengöring av huden.
Samtliga djur ska observeras dagligen och tecken på toxicitet ska registreras tillsammans med tidpunkten då de uppträdde för första gången samt graden och varaktigheten av toxiciteten. Observationer av djuren i burarna ska omfatta hud- och pälsförändringar, ögon, slemhinnor, respirations-och cirkulationssystem, autonoma och centrala nervsystemet, kroppsmotorik samt beteendemönster. Foderintag och djurens vikt ska registreras en gång i veckan. Det är nödvändigt att observera djuren regelbundet för att säkerställa att så få djur som möjligt faller bort från studien till följd av kannibalism, autolys av vävnader eller förväxling. I slutet av testperioden avlivas och obduceras alla överlevande djur, utom djuren i satellitgruppen. Så snart döende djur upptäcks ska dessa avlägsnas, avlivas och obduceras.
Följande undersökningar utförs normalt på alla djur, inbegripet kontrollgruppen:
|
a) |
Innan testämnet appliceras samt sedan försöket avslutats utförs en oftamologisk undersökning med användning av ett oftalmoskop eller liknande lämplig utrustning, helst på alla djur men åtminstone på djuren i högdos- och kontrollgrupperna. Om förändringar i ögonen observeras ska alla djur undersökas. |
|
b) |
Vid slutet av testperioden ska en hematologisk undersökning utföras, inbegripet hematokrit-och hemoglobinkoncentration, erytrocyträkning, total och differentiell leukocyträkning, mätning av koagulationsnivån, t.ex. koagulationstid, protombintid, tromboplastintid eller blodplättsräkning. |
|
c) |
Vid slutet av försöksperioden ska en klinisk-biokemisk blod analys utföras. För denna undersökning är följande analysområden lämpliga: elektrolytbalans, kolhydratmetabolism, lever- och njurfunktion. Valet av specifika analyser kommer att påverkas av observationer av testämnets verkningssätt. Följande bestämningar föreslås: kalcium, fosfor, klorid, natrium, kalium, fasteglukos (fasteperioden beror på djurarten), glutaminsyra-pyrodruvsyra-transaminas (4), glutamin-syra-oxalättiksyra-transaminas (5), ornitin dekarboxylas, gamma-glutamyl-transpeptidas, urinkväve, albumin, blodkreatinin, totalt bilirubin och totalt serumprotein. Andra bestämningar som kan vara nödvändiga för en tillfredsställande toxikologisk bedömning inbegriper analyser av lipider, hormoner, syra/basbalans, methemoglobin och kolinesterasaktivitet. Ytterligare klinisk-biokemiska analyser kan utföras då sådana behövs för en vidare undersökning av observerade verkningar. |
|
d) |
En urinanalys krävs i regel inte, men kan dock vara nödvändig om en toxisk verkan förväntas eller observeras. |
Om tidigare insamlade basdata är otillräckliga bör man överväga att bestämma hematologiska och klinisk-biokemiska parametrar innan man inleder försöket.
Alla djur bör underkastas en fullständig obduktion som inbegriper undersökning av alla externa kroppsytor, alla kroppsöppningar liksom kranie-, bröstkorgs- och bukhålan samt deras innehåll. Lever, njurar, binjurar och testiklar ska vägas våta så snart som möjligt efter dissektion för att undgå uttorkning. Följande vävnader och organ bör bevaras i ett lämpligt medium för en eventuell framtida histopatologisk undersökning: alla vävnader och organ som uppvisar lesioner, hjärnan –inbegripet snitt av medulla/pons, lillhjärnbarken och hjärnbarken, hypofysen, sköldkörteln/bisköldkörteln, thymusvävnad, (luftstrupe) och lungor, hjärta, stora kroppspulsådern, spottkörtlar, lever, mjälte, njurar, binjurar, bukspottskörtel, könskörtlar, livmoder, associerade könsorgan, gallblåsa (om sådan finns), matstrupe, magsäck, tolvfingertarm, tomtarmen, ileum, cekum, colon, ändtarm, urinblåsan, representativa lymfkörtlar, (kvinnliga bröstkörtlar), (lårmuskulatur), nerver från det perifera nervsystemet, (bröstbenet med benmärg), (ögon), (femur, inbegripet ledyta), (ryggmärg på tre nivåer – vid halsen, mitt för bröstkorgen och från landområdet), och (exorbitala tårkörtlar). De vävnader som anges inom parentes behöver endast undersökas om det finns tecken på toxicitet eller i samband med målorgan.
|
a) |
En fullständig histopatologisk undersökning ska utföras på obehandlad och behandlad hud och på organ och vävnader frän alla djur i kontroll- och högdosegrupperna. |
|
b) |
Alla allvarliga lesioner ska undersökas. |
|
c) |
Målorgan i andra dosgrupper ska också undersökas. |
|
d) |
Om råttor används ska lungorna från djur i låg- och medeldosgrupperna undersökas histopatologiskt för tecken på infektion, eftersom detta ger en lämplig uppfattning om djurens hälsotillstånd. Ytterligare histopatologiska undersökningar av djur i dessa grupper krävs inte rutinmässigt, men organ som har uppvisat tecken på lesioner i högdosgruppen ska alltid undersökas. |
|
e) |
Djur i en eventuell satellitgrupp bör underkastas en histopatologisk undersökning av de organ och vävnader som har visat tecken på toxisk verkan hos djur i de behandlade grupperna. |
2. DATA
Data ska ställas upp i tabellform och för varje grupp ska anges antal djur i början av försöket, antal djur med lesioner och den procent djur som uppvisar varje slag av lesion. Resultaten ska utvärderas på grundval av en lämplig statistisk metod. Alla erkända statistiska metoder får användas.
3. RAPPORTERING
3.1. TESTRAPPORT
Testrapporten ska om möjligt innehålla följande:
|
— |
Djurart, stam, härkomst, miljöbetingelser, foder. |
|
— |
Försöksbetingelser. |
|
— |
Dosnivåer (med angivande av vehikel om sådan använts) och koncentrationer. |
|
— |
Upplysningar om toxisk reaktion uppdelade efter kön och dos. |
|
— |
Icke-toxisk dos, om sådan har fastställts. |
|
— |
Tidpunkten för dödsfall under försöket eller överlevnad till slutet av försöket. |
|
— |
Beskrivning av toxiska och andra verkningar. |
|
— |
Tidpunkt för observation av varje abnormalt tecken och dess senare utveckling. |
|
— |
Uppgifter om foder och kroppsvikt. |
|
— |
Oftamologiska rön. |
|
— |
Hematologiska undersökningar och samtliga resultat. |
|
— |
Använda klinisk-biokemiska undersökningar och samtliga resultat (inbegripet resultat av en eventuell urinanalys). |
|
— |
Obduktionsrön. |
|
— |
Detaljerad beskrivning av alla histopatologiska rön. |
|
— |
Eventuell statistisk bearbetning av resultaten. |
|
— |
Diskussion av resultaten. |
|
— |
Tolkning av resultaten. |
3.2. UTVÄRDERING OCH TOLKNING
Se den allmänna inledningen till del B.
4. HÄNVISNINGAR
Se den allmänna inledningen till del B.
B.29 TEST AVSEENDE SUBKRONISK INHALATIONSTOXICITET 90 DAGARS REPETERAD INHALATIONSTEST PÅ GNAGARE
1. METOD
1.1 INLEDNING
Se allmän inledning till del B.
1.2 DEFINITIONER
Se allmän inledning till del B.
1.3 REFERENSÄMNEN
Inga.
1.4 TESTMETODSPRINCIP
Flera grupper av försöksdjur exponeras dagligen under en angiven period för graderade koncentrationer av testämnet, en bestämd koncentration per grupp i 90 dagar. Om vehikel används för att underlätta framtagning av lämplig koncentration av testämnet i luften, ska en kontrollgrupp som utsätts för vehikeln användas. Under försöksperioden observeras djuren dagligen för tecken på toxicitet. Djur som dör under testet ska obduceras och i slutet av testet avlivas och obduceras överlevande djur.
1.5 KVALITETSKRITERIER
Inga.
1.6 METODBESKRIVNING
I minst fem dagar före försöket ska djuren hållas under testbetingelser vad beträffar miljö och utfodring. Före testet fördelas friska unga djur slumpmässigt i behandlings- och kontrollgrupper. Vid behov kan en vehikel tillsättas för att underlätta framtagning av lämplig koncentration av testämnet i luften. Om vehikel eller annan tillsats används för att underlätta dosering ska det vara fastställt att den inte framkallar några toxiska verkningar. I lämpliga fall kan tidigare insamlade data användas.
Om det inte finns kontraindikationer ska i första hand råttor användas. Unga friska djur från vanligen nyttjade laboratoriestammar ska användas. Vid inledning av testet får vikten hos de djur som används inte avvika mer än ± 20 % från en lämplig genomsnittsvikt. Om ett test av subkronisk toxicitet genom inhalation utförs som en förundersökning till en långtidsundersökning ska samma djurart och stam användas i båda undersökningarna.
Minst 20 djur (10 hondjur och 10 handjur) används på varje koncentrationsnivå. Hondjuren får inte ha fött ungar och får inte vara dräktiga. Vid planenligt avlivande av djur under testet ska det ursprungliga antalet djur ökas med det antal djur som planenligt ska avlivas innan studien slutförs. Dessutom kan en satellitgrupp bestående av 20 djur (10 av vardera könet) tillföras den högsta koncentrationen i 90 dagar och därefter observeras i 28 dagar för reversibilitet, persistens eller fördröjt uppträdande av toxiska verkningar.
Minst tre koncentrationsnivågrupper och en kontrollgrupp eller en kontrollgrupp som tillförs vehikeln (i en koncentration som motsvarar den högsta koncentrationen av vehikeln) ska användas. Djuren i kontrollgruppen ska behandlas på samma sätt som djuren i behandlingsgrupperna, bortsett från att de inte ska utsättas för testämnet. Den högsta koncentrationsnivån bör medföra toxiska verkningar men inte leda till någon, eller begränsad, dödlighet. Den lägsta koncentrationsnivån ska inte medföra några tecken på toxicitet. Om det finns en användbar uppskattning av människans exponering för ämnet ska den lägsta nivån vara högre än denna. Idealt ska medelkoncentrationen ge minsta möjliga observerbara toxiska verkningar. Om fler än en medelkoncentration används ska koncentrationsnivåerna väljas på ett sådant sätt att en gradering av de toxiska verkningarna erhålls. I låg-, medelkoncentrations- och kontrollgrupperna ska antalet dödsfall vara få för att möjliggöra en meningsfull utvärderingen av resultaten.
Den dagliga exponeringsprioden är sex timmar och en jämn fördelning av koncentrationen i kamrarna måste säkerställas. Vid särskilda omständigheter kan även andra perioder användas.
Djuren ska testas med inhalationsutrustning som utformats så att den upprätthåller en dynamisk luftcirkulation med minst 12 luftväxlingar per timme, för att säkerställa en tillräcklig syrehalt och en jämnt fördelad exponeringsatmosfär. Om exponeringskammare används ska den vara så utformad att trängseln bland försöksdjuren minimeras och exponeringen för testämnet genom inhalation maximeras. Som en allmän regel för att säkerställa en stabil atmosfär i en kammare, ska försöksdjurens totala ’volym’ inte överstiga 5 % av exponeringskammarens volym. Exponeringen kan göras genom kammare för mun–näsa, enbart huvudet eller hela försöksdjurets kropp; vid användning av de två första metoderna minskar man möjligheten att testämnet upptas via andra vägar.
Alla försöksdjur ska observeras dagligen för tecken på toxicitet under hela försöket och under återhämtningsperioden. Tidpunkten för dödsfall och tidpunkten vid vilken tecken på toxicitet uppträder respektive försvinner ska registreras.
Djuren ska exponeras för testämnet dagligen, fem till sju dagar i veckan, i 90 dagar. En eventuell satellitgrupp avsedd för efterföljande observationer bör hållas under observation i ytterligare 28 dagar, utan tillförsel av testämne, så att man har möjlighet att registrera om de toxiska verkningarna försvinner eller persisterar. Temperaturen under försöket ska hållas vid 22 ± 3 oC. Under ideala betingelser ska den relativa luftfuktigheten hållas mellan 30 % och 70 %, men i vissa fall (t.ex. test med aerosoler) kan detta vara praktiskt ogenomförbart. Under exponeringen ska djuren undanhållas foder och vatten.
Ett dynamiskt respirationssystem med ett lämpligt analytiskt kontrollsystem ska användas. Ett försökstest rekommenderas för att fastställa lämpliga exponeringskoncentrationer. Luftgenomströmningen ska justeras för att säkerställa att betingelserna är homogena i hela exponeringskammaren. Systemet ska vara sådant att det så snabbt som möjligt går att säkerställa stabila exponeringsbetingelser.
Följande ska mätas eller kontrolleras:
|
a) |
Luftgenomströmningen: Luftgenomströmningen i kammaren ska helst kontrolleras fortlöpande. |
|
b) |
Den faktiska koncentrationen av testämnet mätt i respirationszonen. Under den dagliga exponeringsperioden ska koncentrationen inte avvika mer än ± 15 % från den genomsnittliga koncentrationen. En sådan kontrollnivå kan emellertid var omöjlig att uppnå vid test med stoft och vissa aerosoler, och i sådana fall kan större avvikelser accepteras. Under inkörning av utrustningen bör analys göras av partikelstorleken för att bestämma aerosolkoncentrationens stabilitet. Under exponeringen ska analyser göras så ofta som möjligt för att bestämma fördelningen av partikelstorleken. |
|
c) |
Temperatur och luftfuktighet. |
|
d) |
Under och efter exponering ska observationer göras och registreras systematiskt för varje enskilt djur. Alla djur ska observeras dagligen och tecken på toxicitet ska registeras, inbegripet när dessa tecken uppträder för första gången, hur länge de varar, och hur kraftiga de är. Inspektioner av djuren i burarna ska omfatta hud- och pälsförändringar, ögon, slemhinnor, respirations- och cirkulationssystem, autonoma och centrala nervsystemet samt kroppsmotorik och beteendemönster. Foderintaget och djurens vikt ska registreras en gång i veckan. Regelbunden inspektion av djuren är nödvändig för att minimera förlust av djur som ingår i försöket till följd av kannibalism, autolys av vävnader eller förväxling. Vid försökets avslutning ska alla överlevande djur avlivas och obduceras. Så snart döende djur upptäcks ska dessa avlägsnas och obduceras. |
Alla djur inklusive kontrollgruppen ska genomgå följande undersökning:
|
a) |
Före exponering för testämnet samt sedan försöket avslutats ska en oftamologisk undersökning med användning av ett oftalmoskop eller liknande lämplig utrustning utföras, helst på alla djur men åtminstone på högdos- och kontrollgrupperna. Om förändringar i ögonen observeras ska alla djur undersökas. |
|
b) |
Vid slutet av försöket ska en hematologisk undersökning utföras, inbegripet hematokrit, hemoglobinkoncentration, erytrocyträkning, total och differentiell leukocyträkning samt ett mått på koaguleringsförmågan, t.ex. koagulationstid, protombintid, tromboplastintid eller blodplättsräkning. |
|
c) |
Vid slutet av försöksperioden ska en klinisk-biokemisk blodanalys utföras. För denna undersökning är följande analyser lämpliga: elektrolytbalans, kolhydratmetabolism, lever- och njurfunktion. Valet av specifika analyser kommer att påverkas av observationer av testämnets verkningssätt. Följande bestämningar föreslås: kalcium, fosfor, klorid, natrium, kalium, fasteglukos (fasteperioden beror på djurarten), glutaminsyra-pyrodruvsyra-transaminas (4), glutaminsyra-oxalättiksyra-transaminas (5), ornitin dekarboxlas, gamma-glutamyltranspeptidas, urinkväve, albumin, blod-kreatinin, totalt bilirubin och totalt serumprotein. Andra bestämningar som kan vara nödvändiga för en adekvat toxikologisk utvärdering inbegriper analyser av lipider, hormoner, syra/basbalansen, metehemoglobin och kolinesterasaktivitet. Ytterligare klinisk-biokemiska analyser kan vid behov utföras för att vidare utreda de observerade verkningar. |
|
d) |
En urinanalys krävs i regel inte, men kan dock vara nödvändig om en toxisk verkan förväntas eller observeras. |
Om tidigare insamlade grundläggande uppgifter är otillräckliga bör man överväga att bestämma hematologiska och klinisk-biokemiska parametrar innan man inleder försöket.
Alla försöksdjur ska genomgå en fullständig makroskopisk undersökning som inbegriper undersökning av alla externa kroppsytor, alla kroppsöppningar liksom kranie-, bröstkorgs- och bukhålan samt deras innehåll. Lever, njurar, binjurar och testiklar ska vägas våta så snart som möjligt efter dissektion för att undvika uttorkning. Följande vävnader och organ ska förvaras i lämpligt medium för eventuell senare histopatologisk undersökning: alla vävnader och organ som företer allvarliga lesioner, lungor – som ska tas ut hela och oskadade, vägas och behandlas med ett lämpligt fixativ för att säkerställa att lungstrukturen bibehålls (perfusion med ett fixativ anses vara ett effektivt förfarande), vävnader i näs–svalgrummet, hjärnan – inbegripet snitt av medulla/pons, lilllhjärnbarken och hjärnbarken, hypofysen, sköldkörteln/bisköldkörteln, thymusvävnad, luftstrupe och lungor, hjärta, stora kroppspulsådern, spottkörtlar, lever, mjälte, njurar, binjurar, bukspottskörtel, könskörtlar, livmoder, (associerade könsorgan), (hud), gallblåsa (om sådan finns), matstrupe, magsäck, tolvfingertarm, tomtarmen, ileum, cekum, colon, ändtarm, urinblåsan, representativa lymfkörtlar, (kvinnliga bröstkörtlar), (lårmuskulatur), nerver från det perifera nervsystemet, (ögon), bröstbenet med benmärg, (femur, inbegripet ledyta) och (ryggmärg på tre nivåer – vid halsen, mitt för bröstkorgen och från ländområdet). De vävnader som anges inom parenteser behöver endast undersökas om det finns tecken på toxicitet eller i samband med målorgan.
|
a) |
En fullständig histopatologisk undersökning ska utföras på luftvägarna och andra organ och vävnader från alla djur i kontroll- och högdosgrupperna. |
|
b) |
Alla allvarliga lesioner ska undersökas. |
|
c) |
Målorganen från andra dosgrupper ska undersökas. |
|
d) |
Lungor från djur i lågdos- och medeldosgrupperna ska underkastas en histopatologisk undersökning eftersom detta ger en lämplig uppfattning om djurens hälsotillstånd. Ytterligare histopatologiska undersökningar av djur från dessa grupper krävs inte rutinmässigt, men sådana organ som har visat tecken på förändringar i högdosgruppen ska alltid undersökas. |
|
e) |
Om en satellitgrupp har använts bör en histopatologisk undersökning göras på sådana organ och vävnader som har visat tecken på toxisk verkan hos djur i behandlade grupperna. |
2. DATA
Data ska ställas upp i tabellform och för varje grupp ska anges antalet djur i början av försöket, antal djur med lesioner och den procent djur som uppvisar varje slag av lesion. Resultaten ska utvärderas på grundval av en lämplig statistisk metod. Alla erkända statistiska metoder får användas.
3. RAPPORTERING
3.1 TESTRAPPORT
Testrapporten ska om möjligt innehålla följande:
|
— |
Djurart, stam, härkomst, miljöbetingelser, foder. |
|
— |
Testbetingelser: Beskrivning av exponeringsapparaturen: inklusive utformning, typ, dimensioner, luftkälla, system för framställning av finfördelade ämnen och aerosoler, metod att konditionera luften och sätt att hålla djuren i en försökskammare när sådan används. Utrustning för mätning av temperatur, luftfuktighet och finfördelade aerosolers koncentration och storlek ska beskrivas. Exponeringsdata: Dessa ska ställas upp i tabellform och presenteras med medelvärden och ett spridningsmått (t.ex. standardavvikelse) och ska, om möjligt, omfatta
|
|
— |
Upplysningar om toxisk reaktion uppdelade efter kön och koncentration. |
|
— |
Icke toxisk dos, om sådan har fastställts. |
|
— |
Tidpunkten för dödsfall under testet eller överlevnad till slutet av testet. |
|
— |
Beskrivning av toxiska och andra verkningar. |
|
— |
Tidpunkten för observation av varje abnormt tecken och dess senare utveckling. |
|
— |
Uppgifter om foder och kroppsvikt. |
|
— |
Oftalmologiska rön. |
|
— |
Utförda hematologiska undersökningar och alla resultat. |
|
— |
Utförda klinisk-biokemiska undersökningar och alla resultat (inbegripet resultat av en eventuell urinanalys). |
|
— |
Obduktionsrön. |
|
— |
Detaljerad beskrivning av alla histopatologiska rön. |
|
— |
Statistisk behandling av resultaten när detta är möjligt. |
|
— |
Diskussion om resultaten. |
|
— |
Tolkning av resultaten. |
3.2. UTVÄRDERING OCH TOLKNING
Se allmän inledning till del B.
4. HÄNVISNINGAR
Se allmän inledning till del B.
B.30 TEST AVSEENDE KRONISK TOXICITET
1. METOD
1.1 INLEDNING
Se allmän inledning till del B.
1.2 DEFINITIONER
Se allmän inledning till del B.
1.3 REFERENSÄMNEN
Inga.
1.4 TESTMETODSPRINCIP
Testämnet tillförs normalt sju dagar i veckan på lämpligt sätt till flera grupper av försöksdjur, en dos per grupp, under huvuddelen av deras livslängd. Under och efter exponering för testämnet observeras försöksdjuren dagligen för tecken på toxicitet.
1.5 KVALITETSKRITERIER
Inga.
1.6 METODBESKRIVNING
Förberedelser
I minst fem dagar före försöket ska djuren hållas under testbetingelser vad beträffar miljö och utfodring. Före försöket fördelas friska unga djur slumpmässigt i behandlings- och kontrollgrupper.
Försöksbetingelser
I första hand ska råttor användas.
På grundval av resultat från tidigare utförda undersökningar kan andra arter (gnagare eller icke-gnagare) användas. Unga friska djur av vanligen nyttjade laboratoriestammar ska användas och dosering ska inledas så snart som möjligt efter avvänjning.
I början av försöket får vikten hos de djur som används inte avvika mer än ± 20 % från genomsnittsvikten. Om ett test av subkronisk toxicitet utförs som en förundersökning till en långtidsundersökning ska samma djurart/ras och stam användas i båda undersökningarna.
I fråga om gnagare ska minst 40 djur (20 hondjur och 20 handjur) användas för varje dosnivågrupp och för kontrollgruppen. Hondjuren får inte ha fött ungar och får inte vara dräktiga. Vid planenligt avlivande av försöksdjur under försöket ska det ursprungliga antalet djur ökas med det antal djur som planenligt ska avlivas innan försöket slutförs.
För icke-gnagare godkäns ett mindre antal djur, dock minst fyra till sex per grupp.
Minst tre dosnivågrupper och en kontrollgrupp ska användas. Den högsta dosen bör ha en tydlig toxisk verkan utan att förorsaka omfattande dödlighet.
Den lägsta dosen bör inte ha någon toxisk verkan.
Medeldosen (eventuellt flera) bör ligga mitt emellan den högsta och den lägsta dosen.
Vid val av dosnivåer bör hänsyn tas till data från tidigare tester av toxicitet.
Djuren exponeras normalt dagligen för testämnet. Om ämnet tillförs via dricksvattnet eller fodret bor detta finnas tillgänglig kontinuerligt.
En kontrollgrupp som i alla avseenden motsvarar de behandlade grupperna, bortsett från tillförseln av testämnet, ska användas.
Under särskilda omständigheter, t.ex. vid inhalationstester där aerosoler används, eller där en emulgator med okänd biologisk verkan används i tester med oral tillförsel, ska även en negativ motsvarande kontrollgrupp användas. Den negativa kontrollgruppen ska behandlas på samma sätt som försöksgrupperna, bortsett från exponering för testämne eller eventuell vehikel.
Det finns två huvudsakliga tillförselvägar: oralt och genom inhalation. Valet av tillförselväg beror på testämnets fysikaliska och kemiska egenskaper och det sätt på vilket människor sannolikt exponeras för testämnet.
Dermal tillförsel av testämnet är kopplat till en mängd praktiska problem. Om kronisk systemisk toxicitet är ett resultat av perkutan absorption framgår detta normalt genom en ytterligare test med oral tillförsel, kombinerat med kännedom om graden av perkutan absorption på grundval av tidigare perkutana toxicitetstester.
Om testämnet absorberas i mag–tarmkanalen och om människor utsätts för tillförsel genom ingestion, är oral tillförsel att föredra såvida inte kontraindiktioner föreligger. Djuren tillförs testämnet via fodret, upplöst i dricksvattnet, eller i en kapsel. Idealt tillförs djuren testämnet sju dagar i veckan eftersom en begränsning av doseringen till fem dagar i veckan kan leda till återhämtning eller abstinenstoxicitet mellan exponeringsperioderna, vilket kan påverka resultaten och utvärdering av resultaten. Av praktiska skäl godkänns dock dosering fem dagar i veckan.
Eftersom inhalationstester inbegriper mer komplexa tekniska problem än andra tillförselvägar för testämnen behövs en mer detaljerad vägledning av denna tillförselväg. Det bör påpekas att under vissa omständigheter kan intratrakeal installation vara ett alternativ.
Försök med långtidsexponering utförs normalt på grundval av planerad exponering för människor. Djuren exponeras antingen fem dagar i veckan, sex timmar dagligen, sedan jämvikt uppnåtts för koncentrationen i kammaren (intermittent exponering), eller så utgår man från möjlig miljöexponering, varvid djuren exponeras sju dagar i veckan, 22–24 timmar om dagen sju dagar i veckan (kontinuerlig exponering), med omkring en timme om dagen vid ett bestämt klockslag för utfodring av djuren och rengöring av kamrarna.
I båda fallen exponeras djuren normalt för bestämda koncentrationer av testämnet. En viktig skillnad mellan intermittent och kontinuerlig exponering, som man bör vara uppmärksam på, är att intermittent exponering inbegriper en exponeringsfri period av 17–18 timmar per dag, och en ännu längre period under helgerna, då djuren kan återhämta sig från verkningarna av varje daglig exponering.
Valet mellan intermittent och kontinuerlig exponering beror på syftet med försöket och den exponering för människor som simuleras. Man måste dock beakta en rad tekniska problem. Fördelen med kontinuerlig exponering, när det handlar om att simulera miljöbetingelser, kan t.ex. gå förlorad till följd av nödvändigheten att vattna och utfodra djuren under exponeringsperioden och även behovet av mer komplicerad (och pålitlig) teknik för generering och kontroll av aerosoler och luftfuktighet.
Djuren ska testas i en inhalationskammare som utformats så att den upprätthåller en dynamisk luftcirkulation med minst 12 luftväxlingar per timme, för att säkerställa en tillräcklig syrehalt och en jämnt fördelad exponeringsatmosfär. Kontroll- och exponeringskammare ska vara identiska i fråga om konstruktion och utformning för att säkerställa att exponeringsförhållandena är de samma i alla avseenden förutom exponering för testämnet. Ett normalt undertryck ska upprätthållas i kammaren för att förhindra att testämnet sipprar ut i det omgivande rummet. Exponeringskammaren ska vara så utformad att trängseln bland försöksdjuren minimeras. Som en allmän regel for att säkerställa en stabil atmosfär i en kammare, ska försöksdjurens totala ’volym’ inte överstiga 5 % av exponeringskammarens volym.
Följande ska mätas eller kontrolleras:
|
i) |
Luftgenomströmning: luftgenomströmningen i kammaren ska helst kontrolleras fortlöpande: |
|
ii) |
Koncentration: Under den dagliga exponeringsperioden ska koncentrationen av testämnet inte avvika mer än ± 15 % från den genomsnittliga koncentrationen. |
|
iii) |
Temperatur och luftfuktighet: För gnagare hålls temperaturen vid 22 ± 2 oC och fuktigheten i kammaren mellan 30 % och 70 %, förutom när vatten används för suspension av testämnet i kammarens atmosfär. Helst ska båda kontrolleras fortlöpande. |
|
iv) |
Mätning av partikelstorlek: Om atmosfärer med flytande eller fasta aerosoler används i kammare bör man analysera fördelningen av partikelstorleken. Aerosolpartiklarna ska vara möjliga att inhalera för de försöksdjur som används. Provtagning ska ske av kammarens atmosfär i djurens respirationszon. Luftproven ska vara representativa för den partikelfördelning som djurens exponeras för och det ska genom gravimetrisk analys vara möjligt att på grundval av dessa luftprover beskriva hela aerosolen, även om en stor del av den inte är möjlig att inhalera. Under inkörning av utrustningen bör analys göras av partikelstorleken för att bestämma aerosolkoncentrationens stabilitet. Under exponeringen ska analyser göras så ofta som det är nödvändigt för att korrekt bestämma om den fördelning av partikelstorleken som djuren exponeras för är konstant. |
Exponeringsperioden ska vara minst 12 månader.
Förfarande
En grundlig klinisk undersökning ska göras minst en gång om dagen. Andra observationer ska göras dagligen och lämpliga åtgärder ska vidtas för att minimera förlust av djur som ingår i studien, t.ex. ska djur som hittas döda obduceras eller nedfrysas, och svaga eller döende djur ska isoleras eller avlivas. Noggranna observationer är nödvändiga för att säkerställa att så få djur som möjligt faller bort från studien till följd av sjukdom, kannibalism eller autolys av vävnader.
Kliniska tecken, inbegripet neurologiska förändringar, förändringar i ögon och dödlighet ska registreras för alla djur. Tidpunkten då toxiska tecken uppträder för första gången, hur denna toxicitet utvecklas samt misstanke om förekomst av tumörer ska registreras.
Varje djur ska vägas en gång i veckan under försöksperiodens första 13 veckor och därefter minst en gång var fjärde vecka. Foderintaget ska fastställas en gång i veckan under försöksperiodens första 13 veckor och därefter cirka var tredje månad, om inte hälsotillstånd eller förändring i kroppsvikt kräver annat.
En hematologisk undersökning (t.ex. hemoglobinkoncentration, erytrocyträkning, total leukocyträkning, räkning av blodplättar samt mått på koaguleringsförmågan) ska företas efter tre månader, sex månader samt därefter cirka var sjätte månad samt vid försökets avslutning på blodprover som i fråga om icke-gnagare tas på varje enskilt djur, och i fråga om råttor på tio djur av varje kön i alla grupper. Om det är möjligt ska proverna tas från samma råttor varje gång. I fråga om icke-gnagare ska dessutom ett prov tas innan försöket inleds.
Om kliniska observationer tyder på att djurens hälsotillstånd har försämrats under försöket ska en differentialräkning på prover från de ifrågavarande djuren företas.
En differentialräkning ska företas på prover från djur i den grupp som har varit exponerad för den högsta dosen och djur från kontrollgruppen. En differentialräkning på prover från djur i den grupp som har fått den näst högsta dosen ska endast företas om det är stor skillnad mellan den högst doserade gruppen och kontrollgruppen, eller om patologiska rön indikerar detta.
Urinprov för analys ska tas från alla icke-gnagare och från tio råttor per kön i alla grupper, helst från samma råttor varje gång med samma intervaller som vid den hematologiska undersökningen. Följande undersökningar ska utföras på prover antingen från varje enskild djur eller, i fråga om gnagare, på en samlad urinprovs/kön/grupp:
|
— |
Utseende: volym och densitet för enskilda djur. |
|
— |
Protein, glukos, keton, ockult blod (semikvantitativt). |
|
— |
Sedimentmikroskopi (semikvantitativt). |
Omkring var sjätte månad samt vid försökets avslutning ska blodprov för klinisk-kemisk undersökning tas från varje enskild djur när det gäller icke-gnagare och tio råttor per kön i alla grupper, helst från samma råttor varje gång. I fråga om icke-gnagare ska dessutom prov tas innan försöket inleds. Plasma från proverna ska underkastas följande undersökningar:
|
— |
Total proteinkoncentration. |
|
— |
Albuminkoncentration. |
|
— |
Test av leverfunktion t.ex. alkalisk fosfataktivitet, glutaminsyra-pyrodruvsyra-transaminasak-tivitet (4), och glutaminsyra-oxaläniksyra-transaminasaktivitet (5), gamma-glutamyl-transpep-tidas, ornitin dekaroxylas. |
|
— |
Kolhydratmetabolism, t.ex. fasteglukos. |
|
— |
Test av njurfunktion, t.ex. urinämne. |
Alla försöksdjur, även de som dör under försöket eller döende djur som avlivas, ska underkastas en fullständig makroskopisk undersökning. Vid avlivning ska blodprov tas från alla djur för differentialräkning. Alla makroskopiska synliga lesioner, tumörer eller lesioner som misstänks vara tumörer, ska bevaras. Makroskopiska observationer ska, så långt det går, korreleras med mikroskopiska rön.
Alla vävnader och organ bör bevaras för histopatologisk undersökning. Detta gäller normalt följande organ och vävnader: hjärnan (6), (medulla/pons, lillhjärnbarken och hjärnbarken), hypofysen, sköldkörteln (inbegripet bisköldkörteln), thymus, lungor (inbegripet luftstrupe), hjärta, stora kroppspulsådern, spottkörtlar, lever (6), mjälte, njurar (6), binjurar (6), matstrupe, magsäck, tolvfinger-tarm, tomtarmen, ileum, cekum, colon, ändtarm, livmoder, urinblåsan, lymfkörtlar, bukspottskörtel, könskörtlar (6), associerade könsorgan, kvinnliga bröstkörtlar, hud, muskulatur, nerver från det perifera nervsystemet, ryggmärg (på tre nivåer – vid halsen, mitt för bröstkorgen och från landområdet), bröstbenet med benmärg och femur (inbegripet ledyta) och ögon. Det bästa sättet att bevara lungor och urinblåsa är att blåsa upp dem med ett fixermedel. I samband med inhalationstester är det av grundläggande betydelse för de hispatologiska testerna att lungorna blåses upp. Vid särskilda tester såsom inhalationstester ska hela luftsystemet undersökas, inbegripet näsa, svalg och luftrör.
Om andra kliniska undersökningar utförs ska den information som erhålls från dessa finnas tillgänglig innan en mikroskopisk undersökning företas, eftersom detta kan vara av betydande värde för patologen.
Alla synliga förändringar, särskilt tumörer och andra lesioner som inträffar i ett organ ska undersökas mikroskopiskt. Dessutom rekommenderas följande:
|
a) |
Mikroskopisk undersökning av alla bevarade organ och vävnader med en fullständigt beskrivning av alla lesioner som påträffats i
|
|
b) |
Sådana organ eller vävnader som uppvisar abnormitet som orsakats av, eller kan ha orsakats av, testämnet ska även undersökas i lågdosgrupperna. |
|
c) |
Om försöksresultaten visar på betydliga förändringar i djurets normala livslängd eller induktion av verkningar som kan påverka en toxisk reaktion, bör djuren i nästa lägre dosnivå undersökas i enlighet med ovan beskrivna riktlinjer. |
|
d) |
Information om normalt förekommande lesioner hos den använda laboratoriestammen, under samma laboratorievillkor, dvs. tidigare insamlade data, är oumbärligt för en korrekt bedömning av betydelsen av de förändringar som observerats i de behandlade djuren. |
2. DATA
Data ska ställas upp i tabellform och för varje behandlingsgrupp ska anges antal djur i början av försöket, antal djur som uppvisar lesioner och den procentdel djur som uppvisar varje slag av lesion. Utvärderingen av resultaten bör ske på grundval av en lämplig statistisk metod. Alla erkända statistiska metoder får användas.
3. RAPPORTERING
3.1 TESTRAPPORT
Testrapporten ska om möjligt innehålla följande uppgifter:
|
— |
Djurart, stam, härkomst, miljöbetingelser, foder. |
|
— |
Försöksbetingelser: Beskrivning av exponeringsapparaturen: Inklusive utformning, typ, dimensioner, luftkälla, system för framställning av finfördelade ämnen och aerosoler, metod att konditionera luften och sätt att hålla djuren i en försökskammare när sådan används. Utrustning för mätning av temperatur, luftfuktighet och finfördelade aerosolers koncentration och storlek ska beskrivas. Exponeringsdata: Dessa ska ställas upp i tabellform och presenteras med medelvärden och ett spridningsmått (t.ex. standardavvikelse) och ska, om möjligt, omfatta
|
|
— |
Dosnivåer (med angivande av vehikel, om sådan har använts) och koncentrationer. |
|
— |
Uppgifter om toxisk reaktion uppdelade efter kön och dos. |
|
— |
Icke-toxisk dos, om sådan har fastställts. |
|
— |
Tidpunkten för observation av varje abnormt tecken och dess senare utveckling. |
|
— |
Beskrivning av toxiska och andra verkningar. |
|
— |
Tidpunkten för observation av varje abnormt tecken och dess senare utveckling. |
|
— |
Uppgifter om foder och kroppsvikt, |
|
— |
Oftalmologiska rön. |
|
— |
Utförda hematologiska undersökningar och alla resultat. |
|
— |
Utförda klinisk-biokemiska undersökningar och alla resultat (inbegripet resultat av en eventuell urinanalys). |
|
— |
Obduktionsrön. |
|
— |
Detaljerad beskrivning av alla histopatologiska rön. |
|
— |
Statistisk behandling av resultaten i lämpliga fall. |
|
— |
Diskussion av resultaten. |
|
— |
Tolkning av resultaten. |
3.2 UTVÄRDERING OCH TOLKNING
Se allmän inledning till del B.
4. HÄNVISNINGAR
Se allmän inledning till del B.
B.31 TEST AVSEENDE PRENATAL UTVECKLINGSTOXICITET
1. METOD
Denna metod är i stort sett identisk med OECD TG 414 (2001).
1.1 INLEDNING
Denna metod för test av utvecklingstoxicitet är avsedd att ge allmän information om verkningarna av prenatal exponering på dräktiga försöksdjur och på den organism som utvecklas i livmodern. Detta kan inbegripa utvärdering av verkningarna på såväl modern som på fostret (död, strukturella abnormaliteter eller avvikande tillväxt). Funktionsstörningar omfattas inte av denna testmetod, även om de utgör en viktig del av utvecklingen. Funktionsstörningar kan testas separat eller i kombination med detta test genom användning av metoden för test avseende utvecklingsneurotoxicitet. För information om testning av funktionsstörningar och andra postnatala verkningar hänvisas till testmetoden för reproduktionstoxicitet på två generationer och till testmetoden för utvecklingsneurotoxicitet.
Denna testmetod kan i enskilda fall behöva anpassas i enlighet med speciell kännedom om t.ex. de fysikalisk-kemiska eller toxikologiska egenskaperna hos det ämne som testas. Sådan anpassning kan godtas om det föreligger övertygande vetenskapliga bevis om att anpassningen leder till ett mer informativt test. I sådana fall bör testrapporten innehålla en detaljerad redogörelse för de vetenskapliga bevis som åberopas.
1.2 DEFINITIONER
Utvecklingstoxikologi: Undersökning av sådana skadliga verkningar på en organism under utveckling som kan uppstå till följd av exponering före befruktningen, under den prenatala utvecklingen eller postnatalt fram till könsmognaden. Utvecklingstoxiciteten tar sig främst uttryck som 1) organismens död, 2) strukturell abnormalitet, 3) avvikande tillväxt och 4) funktionsstörningar. Utvecklingstoxicitet benämndes tidigare ofta teratogenicitet.
Skadlig verkning: Varje sådan behandlingsrelaterad avvikelse från normaltillståndet som minskar organismens förmåga att överleva, föröka sig eller anpassa sig till omgivningen. Begreppet utvecklingstoxicitet i dess vidaste mening omfattar alla verkningar som stör den normala utvecklingen hos conceptus, både före och efter födseln.
Avvikande tillväxt: En vikt- eller storleksavvikelse hos avkommans organ eller kropp.
Avvikelser (anomalier): Strukturella utvecklingsavvikelser; här inbegrips både missbildningar och variationer (28).
Missbildning/betydande abnormalitet: En strukturell ändring som bedöms vara skadlig för djuret (kan också vara dödlig) och som i regel är sällsynt.
Variation/mindre abnormalitet: En strukturell ändring som bedöms vara endast lite eller inte alls skadlig för djuret; den kan vara övergående och vara relativt vanlig hos kontrollpopulationen.
Conceptus: Summan av derivat från ett befruktat ägg vid varje utvecklingsstadium från befruktning till födsel; här inbegrips omgivande membran samt själva embryot eller fostret.
Implantation (nidation): Det förlopp då blastocyten fäster sig vid livmoderns epitelhinna; här ingår blastocytens penetrering av livmoderepitelet och inbäddning i endometriet.
Embryo: Det tidiga utvecklingsstadiet hos en organism, särskilt den produkt som utvecklas till följd av befruktningen av ett ägg, från det att den långa axeln har framträtt tills alla viktiga strukturer finns närvarande.
Embryotoxicitet: Skadlig för den normala strukturen, utvecklingen, tillväxten eller livsdugligheten hos ett embryo.
Foster: Den ofödda avkomman under den postembryonala perioden.
Fostertoxicitet: Skadlig för den normala strukturen, utvecklingen, tillväxten eller livsdugligheten hos ett foster.
Abort: Prematur utstötning av befruktningsprodukterna (embryot eller ett icke-livsdugligt foster) ur livmodern.
Resorption: Conceptus som har dött efter implantationen i livmodern och som resorberas eller har resorberats.
Tidig resorption: Tecken på implantation utan ett igenkännbart embryo eller foster.
Sen resorption: Dött embryo eller foster med externa degenerativa förändringar.
NOAEL: Förkortning för ’no-observed-adverse-effect level’ (nivå där inga skadliga verkningar observeras), dvs. den högsta dos- eller exponeringsnivå vid vilken inga skadliga behandlingsrelaterade verkningar kan observeras.
1.3 REFERENSÄMNE
Inga.
1.4 PRINCIP FÖR TESTMETODEN
Normalt tillförs testämnet till de dräktiga djuren minst från tidpunkten för implantationen till en dag före den planenliga avlivningsdagen. Den senare bör ligga så nära den normala nedkomstdagen som möjligt utan att det finns risk för att testdata går förlorade på grund av prematur nedkomst. Testmetoden är inte avsedd för undersökning enbart av organogenesperioden (t.ex. dag 5–15 hos gnagare och dag 6–18 hos kanin), utan även av verkningarna från tidpunkten för preimplantation (när det är tillämpligt) genom hela dräktighetsperioden fram till dagen före kejsarsnittet. Kort före kejsarsnittet avlivas hondjuren, livmoderinnehållet undersöks och fostren undersöks med avseende på externt observerbara anomalier och med avseende på mjukvävnads- och skelettavvikelser.
1.5 BESKRIVNING AV TESTMETODEN
1.5.1 Val av djurart
Det rekommenderas att testningen görs på de mest relevanta arterna, och att laboratoriearterna och stammarna är desamma som normalt används för testning av prenatal utvecklingstoxicitet. Rekommenderad gnagare är råtta och rekommenderad icke-gnagare är kanin. Om andra arter används måste detta motiveras.
1.5.2 Förvarings- och utfodringsbetingelser
Temperaturen i försöksdjurens rum bör vara 22 oC (± 3 oC) för gnagare och 18 oC (± 3 oC) för kaniner. Den relativa fuktigheten bör vara minst 30 % och helst inte överstiga 70 %, utom när rummet rengörs. Målvärdet bör dock vara 50–60 %. Belysningen bör vara artificiell med omväxlande 12 timmar ljus och 12 timmar mörker. För utfodringen kan konventionellt laboratoriefoder användas, och djuren bör ha obegränsad tillgång till dricksvatten.
För parningsprocedurerna används burar som är lämpliga för ändamålet. Även om individuell inhysning av parade djur är att rekommendera, godtas även inhysning i små grupper.
1.5.3 Förberedelse av djuren
För testet används friska djur som har acklimatiserats till laboratoriemiljön under minst fem dagar och som inte har använts för tidigare test. Försöksdjuren karakteriseras enligt art, stam, ursprung, kön, vikt och ålder. Djuren i alla testgrupper bör vara så lika som möjligt med avseende på vikt och ålder. För alla dosnivåer används unga vuxna hondjur som inte har fått ungar. Hondjuren bör paras med handjur av samma art och stam. Parning av syskon bör undvikas. För gnagare räknas dräktighetsdag 0 som den dag då en vaginalpropp eller sperma kan observeras. För kaniner räknas dag 0 i regel som dagen för coitus eller artificiell insemination, om sådan används. De parade hondjuren fördelas slumpmässigt i kontroll- och behandlingsgrupper. Burarna placeras i sådan ordning att eventuella verkningar på grund av placeringen minimeras. Varje djur förses med ett unikt identifieringsnummer. De parade hondjuren fördelas slumpmässigt i kontroll- och behandlingsgrupper. Om hondjuren paras i satser bör djuren i varje sats fördelas jämnt över grupperna. Likaså bör hondjur som har befruktats av samma handjur fördelas jämnt över grupperna.
1.6 FÖRFARANDE
1.6.1 Djurens antal och kön
Varje test- och kontrollgrupp bör innehålla ett tillräckligt antal hondjur för att det vid obduktionen ska finnas cirka 20 hondjur som har implantation. Grupper där mindre än 16 djur har implantation kan vara olämpligt. Mortalitet hos moderdjuren leder inte nödvändigtvis till att testet blir ogiltigt, förutsatt att mortaliteten inte överskrider cirka 10 %.
1.6.2 Förberedelse av doserna
Om vehikel eller någon annan tillsats används för att underlätta tillförseln bör följande beaktas: Verkningarna på absorption, distribution, metabolism och kvarhållande eller utsöndring av testämnet; verkningarna på testämnets kemiska egenskaper till den del dessa verkningar kan påverka testämnets toxiska egenskaper samt verkningarna på försöksdjurens foder- eller vattenintag eller deras näringstillstånd. Den vehikel som används får inte vara utvecklingstoxisk eller påverka reproduktionen.
1.6.3 Dosering
Testämnet bör i regel ges dagligen från och med implantationen (t.ex. dag 5 efter parningen) fram till dagen före det planenliga kejsarsnittet. Om preliminära test, i den mån sådana har gjorts, inte indikerar en hög potential för förluster före implantationen kan behandlingen förlängas till att omfatta hela dräktighets-perioden, från parningen till dagen före den planenliga avlivningen. Det är allmänt känt att olämplig hantering eller stress under dräktigheten kan leda till prenatala förluster. För att förebygga prenatala förluster på grund av faktorer som inte är behandlingsrelaterade bör onödig hantering av dräktiga djur undvikas. Likaså bör man undvika att utsätta sådana djur för stress på grund av externa faktorer såsom buller.
Minst tre dosnivåer och en parallell kontrollgrupp bör användas. Friska djur fördelas slumpmässigt i kontroll- och behandlingsgrupper. Dosnivåerna bör väljas så att en gradering av de toxiska effekterna erhålls. Såvida det inte finns några begränsningar till följd av testämnets fysikalisk-kemiska eller biologiska egenskaper bör den högsta dosen väljas så att den förväntas förorsaka en viss utvecklingstoxicitet och/eller toxiska verkningar på moderdjuret (kliniska tecken eller minskning av kroppsvikten), men inte död eller allvarligt lidande. Minst en av de mellanliggande dosnivåerna bör ge minsta möjliga observerbara toxiska verkningar. Den lägsta dosnivån bör inte ge några tecken på toxicitet hos moderdjuret eller utvecklingstoxicitet. Därefter väljs dosnivåerna i fallande skala så att en dosrelaterad reaktion kan påvisas. En nivå bör väljas så att inga påvisbara skadliga verkningar (NOAEL) uppstår. För fallande dosnivåer är två- till fyrfaldiga dosintervall ofta optimala. Tillägg av en fjärde testgrupp är att föredra framför användning av mycket breda intervall mellan dosnivåerna (t.ex. en faktor som är högre än cirka 6–10). Även om fastställandet av en NOAEL-nivå för moderdjuret är ett mål, kan även test där en sådan nivå inte fastställs vara godtagbara
Dosnivåerna bör väljas med hänsyn till alla kända toxicitetsuppgifter och all känd tilläggsinformation om metabolismen och de toxikokinetiska egenskaperna hos testämnet eller närbesläktade material. Denna information är också till hjälp när det gäller att demonstrera lämpligheten hos dossystemet.
En parallell kontrollgrupp bör användas. Kontrollgruppen ska vara en obehandlad grupp eller en vehikelkontrollgrupp, om vehikel används för tillförsel av testämnet. Alla grupper ges samma volym av testämnet eller vehikeln. Djuren i kontrollgruppen (kontrollgrupperna) bör hanteras på samma sätt som testgruppsdjuren. Vehikelkontrollgrupperna bör få vehikel i den största mängd som används (samma mängd som ges till den grupp som får den lägsta dosen).
1.6.4 Gränstest (limit-test)
Om ett test med en dosnivå om minst 1 000 mg oralt per kroppsvikt och dag genomförs på det sätt som beskrivs här, och inga toxiska verkningar kan observeras hos de dräktiga djuren eller deras avkomma, och om verkningar inte förväntas på grundval av kända data (t.ex. data om strukturellt eller metaboliskt närbesläktade ämnen), kan ett fullständigt test med tre dosnivåer bedömas vara överflödigt. Om det är sannolikt att människor kommer att utsättas för exponering måste eventuellt en högre oral dosnivå användas vid gränstestet. För andra former av tillförsel, såsom inandning eller applicering på huden, kan testämnets fysikalisk-kemiska egenskaper ofta indikera och begränsa den maximala exponeringsnivån (t.ex. applicering på huden får inte förorsaka allvarlig lokal toxicitet).
1.6.5 Tillförsel av doser
Testämnet eller vehikeln tillförs i regel oralt via intubation. Om någon annan tillförselväg används bör detta motiveras och det kan vara nödvändigt med tillämpliga modifikationer (2)(3)(4). Testämnet bör tillföras vid ungefär samma tid varje dag.
Dosen till individuella djur bör i regel basera sig på den senaste bestämningen av djurets kroppsvikt. Försiktighet bör dock iakttas när dosen justeras under dräktighetens sista tredjedel. Dosen bör väljas med hänsyn till kända data så att överdrivet kraftig toxicitet på moderdjuret kan undvikas. Om kraftig toxicitet ändå observeras på behandlade moderdjur bör dessa avlivas på ett humant sätt. Om flera dräktiga djur visar tecken på kraftig toxicitet bör man överväga att avsluta den dosgruppen. Om testämnet tillförs genom sondmatning rekommenderas att hela dosen tillförs på en gång via magsond eller lämplig intubationskanyl. Den största mängd vätska som kan tillföras vid ett och samma tillfälle beror på försöksdjurets storlek. Volymen får inte överstiga 1 ml per 100 g kroppsvikt, utom för vattenlösningar där volymen kan uppgå till 2 ml per 100 g kroppsvikt. Om majsolja används som vehikel får volymen inte överskrida 0,4 ml per 100 g kroppsvikt. Variationerna i testvolym bör minimeras genom att man justerar koncentrationerna så att en konstant volym kan säkerställas på alla dosnivåer.
1.6.6 Observationer av moderdjuren
Kliniska observationer bör göras och registreras minst en gång per dag, helst vid samma tid(er) varje dag. Tidpunkterna bör väljas med beaktande av perioden för när maximala verkningar förväntas efter dosering. Vid observationerna registreras djurens tillstånd (mortalitet, döende djur, relevanta beteendeändringar och alla tecken på uppenbar toxicitet).
1.6.7 Kroppsvikt och foderkonsumtion
Djuren bör vägas på dräktighetsdag 0 (eller inte senare än dräktighetsdag 3 om djuren kommer från en utomstående uppfödare hos vilken djuren har parats vid en viss tidpunkt), på den första doseringsdagen, därefter minst var tredje dag under doseringsperioden och till slut på den planenliga avlivningsdagen.
Foderkonsumtionen bör registreras med tre dagars intervall, på samma dagar som djuren vägs.
1.6.8 Undersökning post-mortem
Hondjuren avlivas en dag före den förväntade födslodagen. Hondjur som visar tecken på abort eller prematur födsel före den planenliga avlivningen bör avlivas och bli föremål för en grundlig makroskopisk undersökning.
Vid testets slut eller om djur dör under pågående test ska moderdjuret undersökas makroskopiskt med avseende på strukturella abnormaliteter eller patologiska förändringar. För att få en så hög grad av objektivitet som möjligt bör utvärderingen av moderdjuren under kejsarsnittet och fosteranalyserna helst göras utan kännedom om behandlingsgrupp.
1.6.9 Undersökning av livmoderinnehåll
Omedelbart efter testets slut eller så snart som möjligt efter ett dödsfall avlägsnas livmodern och djurens dräktighetsstatus fastställs. Om en livmoder inte har några tecken på dräktighet bör dess status bekräftas t.ex. genom ammoniumsulfidfärgning (för gnagare) eller Salewski-färgning eller någon annan lämplig metod (för kaniner) (5).
Varje dräktig livmoder vägs inklusive livmoderhals. Livmodern behöver inte vägas för djur som har självdött under testets gång.
För dräktiga djur fastställs antalet corpora lutea.
Livmoderinnehållet undersöks med avseende på eventuella döda embryon eller foster och antalet levande foster. Graden av resorption bör beskrivas i syfte att uppskatta den relativa tidpunkten för när conceptus har dött (se avsnitt 1.2).
1.6.10 Undersökning av foster
För varje foster fastställs kön och kroppsvikt.
Fostren undersöks för externa avvikelser (6).
Fostren undersöks också för avvikelser i skelettet och mjukdelarna (t.ex. variationer och missbildningar eller anomalier) (7)(8)(9)(10)(11)(12)(13)(14)(15)(16)(17)(18)(19)(20)(21)(22)(23)(24). Kategorisering av fosteravvikelserna rekommenderas, men krävs inte. Om kategorisering görs, bör kriterierna för definition av de olika kategorierna klart anges. Särskild uppmärksamhet bör fästas vid reproduktionsorganen, som ska undersökas för tecken på avvikande utveckling.
I fråga om gnagare förbereds cirka hälften av varje kull för undersökningar avseende avvikelser i skelettet. Återstoden förbereds och undersöks för avvikelser i mjukdelarna. För detta används godkända eller lämpliga metoder för seriell sektionering eller tekniker för noggrann dissektion.
I fråga om icke-gnagare, t.ex. kaniner, bör alla foster undersökas för avvikelser i både mjukdelarna och skelettet. Fosterkropparna dissekeras noggrant och utvärderas med avseende på avvikelser i mjukdelarna. Detta kan inbegripa procedurer för ytterligare utvärdering av den interna hjärtstrukturen (25). Från hälften av de foster som har undersökts på detta sätt avlägsnas huvudet och förbereds för utvärdering av avvikelser i mjukvävnad (ögon, hjärna, nasalpassager och tunga) med användning av normala seriella sektioneringsme-toder (26) eller någon annan lika känslig metod. Kropparna hos dessa foster och återstående intakta foster förbereds och undersöks för avvikelser i skelettet, med användning av samma metoder som har beskrivits för gnagare.
2. DATA
2.1 BEHANDLING AV RESULTATEN
Data rapporteras individuellt för moderdjuren och deras avkomma och sammanställs i en tabell. Tabellen ska för varje testgrupp visa antalet djur vid testets början, antalet djur som under testets gång har dött eller avlivats av humana skäl, tidpunkten för alla dödsfall eller humana avlivningar, antalet dräktiga hondjur, antalet djur som visat tecken på toxicitet, en beskrivning av observerade toxicitetstecken (tidpunkten då sådana först upptäcktes, de toxiska verkningarnas varaktighet och allvarlighetsgrad), typerna av observationer om embryon/foster och alla relevanta data om kullarna.
De numeriska resultaten utvärderas med en lämplig statistisk metod, med kullen som enhet för analysen av data. Den statistiska metod som används bör vara allmänt erkänd. Valet av statistiska metoder bör ingå som en del av testets utformning och bör motiveras. Rapporten bör även omfatta data om djur som inte överlever fram till den planenliga avlivningen. Sådana data kan, när det är relevant, inkluderas i gruppens medelvärden. Relevansen hos data från sådana djur bör motiveras och bedömas på individuell grund. Detsamma gäller för avgöranden om huruvida dessa data ska ingå i en grupps medelvärden eller inte.
2.2 UTVÄRDERING AV RESULTATEN
Resultaten från testet avseende prenatal utvecklingstoxicitet ska utvärderas med avseende på de observerade verkningarna. Utvärderingen bör omfatta följande information:
|
— |
Resultaten från test på moderdjur och embryo/foster, inbegripet en utvärdering av förhållandet (eller avsaknaden av ett förhållande) mellan djurens exponering för testämnet och förekomsten av och allvarligheten hos alla fynd. |
|
— |
Kriterier som används för att kategorisera avvikelser på fostret (externt, mjukvävnad och skelett), om kategorisering har gjorts. |
|
— |
I tillämpliga fall historiska kontrolldata som stöd för tolkningen av resultaten. |
|
— |
De antal som har använts vid beräkning av procenttal eller index. |
|
— |
I tillämpliga fall adekvat statistisk analys av testresultaten. En sådan analys bör inbegripa tillräcklig information om analysmetoden så att en oberoende granskare eller statistiker kan göra en ny utvärdering och rekonstruera analysen. |
Om utfallet tyder på frånvaro av toxiska verkningar bör ytterligare undersökningar för att fastställa testämnets absorption och biotillgänglighet övervägas.
2.3 TOLKNING AV RESULTATEN
Ett test avseende prenatal utvecklingstoxicitet ger information om verkningarna av upprepad exponering för ett ämne på moderdjuren under dräktigheten och på avkommans utveckling inne i livmodern. Testresultaten ska tolkas tillsammans med resultaten från test avseende subkronisk toxicitet, reproduktionstoxi-citet, toxikokinetik och andra test. Eftersom betoning läggs både vid endpointen för allmän toxicitet uttryckt som toxicitet för moderdjuret och vid endpointen för utvecklingstoxicitet, ger testresultatet en viss möjlighet till diskriminering mellan verkningar på utvecklingen i frånvaron av allmän toxicitet och verkningar som endast induceras på nivåer som även är toxiska för moderdjuret (27).
3. RAPPORTERING
3.1 TESTRAPPORT
Testrapporten ska innehålla nedan angivna specifika uppgifter.
|
|
Testämne:
|
|
|
Vehikel (i förekommande fall):
|
|
|
Försöksdjur:
|
|
|
Testbetingelser:
|
|
|
Resultat: |
|
|
Data om moderdjurens toxicitetsrespons, inklusive men inte begränsat till följande:
|
|
|
”Endpoints” för utvecklingen per dos för kullar med implantation, inklusive följande:
|
|
|
”Endpoints” för utvecklingen per dos för kullar med levande foster, inklusive följande:
|
|
|
Diskussion om resultaten. |
|
|
Slutsatser. |
4. HÄNVISNINGAR
|
1. |
Kavlock R.J. et al. (1996) A Simulation Study of the Influence of Study Design on the Estimation of Benchmark Doses for Developmental Toxicity. Risk Analysis 16; 399–410. |
|
2. |
Kimmel, C.A. and Francis, E.Z. (1990) Proceedings of the Workshop on the Acceptability and Interpretation of Dermal Developmental Toxicity Studies. Fundamental and Applied Toxicology 14; 386–398. |
|
3. |
Wong, B.A., et al. (1997) Developing Specialized Inhalation Exposure Systems to Address Toxicological Problems. CIIT Activities 17; 1–8. |
|
4. |
US Environmental Protection Agency (1985) Subpart E-Specific Organ/Tissue Toxicity, 40 CFR 798.4350: Inhalation Developmental Toxicity Study. |
|
5. |
Salewski, E. (1964) Faerbermethode zum Makroskopischen Nachweis von Implantations Stellen am Uterusder Ratte. Naunyn-Schmeidebergs Archiv fur Pharmakologie und Experimentelle Pathologie 247:367. |
|
6. |
Edwards, J.A. (1968) The external Development of the Rabbit and Rat Embryo. In Advances in Teratology. D.H.M. Woolam (ed.) Vol. 3. Academic Press, NY. |
|
7. |
Inouye, M. (1976) Differential Staining of Cartilage and Bone in Fetal Mouse Skeleton by Alcian Blue and Alizarin Red S. Congenital Anomalies 16; 171–173. |
|
8. |
Igarashi, E. et al. (1992) Frequency Of Spontaneous Axial Skeletal Variations Detected by the Double Staining Techniquefor Ossified and Cartilaginous Skeleton in Rat Foetuses. Congenital Anomalies 32; 381–391. |
|
9. |
Kimmel, C.A. et al. (1993) Skeletal Development Following Heat Exposure in the Rat. Teratology 47; 229–242. |
|
10. |
Marr, M.C. et al. (1988) Comparison of Single and Double Staining for Evaluation of Skeletal Development: The Effects of Ethylene Glycol (EG) in CD Rats. Teratology 37; 476. |
|
11. |
Barrow, M.V. and Taylor, W.J. (1969) A Rapid Method for Detecting Malformations in Rat Foetuses. Journal of Morphology 127:291–306. |
|
12. |
Fritz, H. (1974) Prenatal Ossification in Rabbits ss Indicative of Foetal Maturity. Teratology 11; 313–320. |
|
13. |
Gibson, J.P. et al. (1966) Use of the Rabbit in Teratogenicity Studies. Toxicology and Applied Pharmacology 9; :398–408. |
|
14. |
Kimmel, C.A. and Wilson, J.G. (1973) Skeletal Deviation in Rats: Malformations or Variations? Teratology 8; 309–316. |
|
15. |
Marr, M.C. et al. (1992) Developmental Stages of the CD (Sprague-Dawley) Rat Skeleton after Maternal Exposure to Ethylene Glycol. Teratology 46; 169–181. |
|
16. |
Monie, I.W. et al. (1965) Dissection Procedures for Rat Foetuses Permitting Alizarin Red Staining of Skeleton and Histological Study of Viscera. Supplement to Teratology Workshop Manual, pp. 163–173. |
|
17. |
Spark, C. and Dawson, A.B. (1928) The Order and Time of appearance of Centers of Ossification in the Fore and Hind Limbs of the Albino Rat, with Special Reference to the Possible Influence of the Sex Factor. American Journal of Anatomy 41; 411–445. |
|
18. |
Staples, R.E. and Schnell, V.L. (1964) Refinements in Rapid Clearing Technique in the KOH-Alizarin Red S Method for Fetal Bone. Stain Technology 39; 61–63. |
|
19. |
Strong, R.M. (1928) The Order Time and Rate of Ossification of the Albino Rat (Mus Norvegicus Albinus) Skeleton. American Journal of Anatomy 36; 313–355. |
|
20. |
Stuckhardt, J.L. and Poppe, S.M. (1984) Fresh Visceral Examination of Rat and Rabbit Foetuses Used in Teratogenicity Testing. Teratogenesis, Carcinogenesis, and Mutagenesis 4; 181–188. |
|
21. |
Walker, D.G. and Wirtschafter, Z.T. (1957) The Genesis of the Rat Skeleton. Thomas, Springfield, IL. |
|
22. |
Wilson, J.G. (1965) Embryological Considerations in Teratology. In Teratology: Principles and Techniques, Wilson J.G. and Warkany J. (eds). University of Chicago, Chicago, IL, pp 251–277. |
|
23. |
Wilson, J.G. and Fraser, F.C. (eds). (1977) Handbook of Teratology, Vol. 4. Plenum, NY. |
|
24. |
Varnagy, L. (1980) Use of Recent Fetal Bone Staining Techniques in the Evaluation of Pesticide Teratogenicity. Acta Vet. Acad. Sci. Hung. 28; 233–239. |
|
25. |
Staples, R.E. (1974) Detection of visceral Alterations in Mammalian Foetuses. Teratology 9; 37–38. |
|
26. |
Van Julsingha, E.B. and C.G. Bennett (1977) A Dissecting Procedure for the Detection of Anomalies in the Rabbit Foetal Head. In: Methods in Prenatal Toxicology Neubert, D., Merker, H.J. and Kwasigroch, T.E. (eds.). University of Chicago, Chicago, IL, pp. 126–144. |
|
27. |
US Environmental Protection Agency (1991) Guidelines for Developmental Toxicity Risk Assessment. Federal Register 56; 63798–63826. |
|
28. |
Wise, D.L. et al. (1997) Terminology of Developmental Abnormalities in Common Laboratory Mammals (Version 1) Teratology 55; 249–292. |
B.32 TEST AVSEENDE CANCEROGENICITET
1. METOD
1.1 INLEDNING
Se allmän inledning till del B.
1.2 DEFINITIONER
Se allmän inledning till del B.
1.3 REFERENSÄMNEN
Inga.
1.4 TESTMETODSPRINCIP
Testämnet tillförs normalt sju dagar i veckan på lämpligt sätt till flera grupper av försöksdjur, en bestämd dos per grupp under huvuddelen av deras livslängd. Under och efter exponering för testämnet observeras försöksdjuren dagligen för tecken på toxicitet, särskilt utveckling av tumörer.
1.5 KVALITETSKRITERIER
Inga.
1.6 METODBESKRIVNING
I minst fem dagar före försöket ska djuren hållas under testbetingelser vad beträffar miljö och utfodring. Före försöket fördelas friska unga djur slumpmässigt i behandlings- och kontrollgrupper.
På grundval av resultaten från tidigare utförda tester kan andra arter (gnagare eller icke-gnagare) användas. Unga friska djur av vanligen nyttjade laboratoriestammar ska användas och dosering ska inledas så snart som möjligt efter avvänjning.
I början av försöket får vikten hos de djur som används inte avvika mer än ± 20 % från genomsnittsvikten. Om ett test av subkronisk toxicitet med oral tillförsel utförs som en förundersökning till en långtidsundersökning ska samma djurart användas i båda undersökningarna.
I fråga om gnagare ska minst 100 djur (50 hondjur och 50 handjur) användas på varje dosnivå och för motsvarande kontrollgrupp. Hondjuren får inte ha fött ungar och får inte vara dräktiga. Vid planenligt avlivande av försöksdjur under försöket ska det ursprungliga antalet djur ökas med det antal djur som planenligt ska avlivas innan försöket slutförs.
Minst tre dosnivågrupper ska användas förutom den motsvarande kontrollgruppen. Den högsta dosen bör framkalla minimala toxicitetstecken, t.ex. en liten minskning av kroppsviktstillväxten (mindre än 10 %), utan att leda till väsentliga ändringar av den normala livslängden, med undantag för fall av tumörer.
Den lägsta dosen bör inte påverka normal tillväxt, utveckling och livslängd eller framkalla några tecken på toxicitet. I allmänhet bör den lägsta dosen inte vara mindre än 10 % av den högsta dosen.
Medeldosen (eventuellt flera) bör ligga mitt emellan den högsta och den lägsta dosen.
Vid val av dosnivåer bör hänsyn tas till data från tidigare tester och undersökningar av toxicitet.
Djuren exponeras normalt dagligen.
Om ämnet administreras via dricksvattnet eller fodret bör detta finnas tillgänglig kontinuerligt.
En motsvarande kontrollgrupp ska användas och behandlas på samma sätt som försöksgrupperna, bortsett från exponering för testämnet.
Under särskilda omständigheter, t.ex. vid inhalationstester där aerosoler används, eller där en emulgator med okänd biologisk verkan används i tester med oral tillförsel, används en ytterligare kontrollgrupp som inte ska exponeras för vehikel eller testämne.
Det finns tre huvudsakliga tillförselvägar: oral tillförsel, dermal tillförsel respektive tillförsel genom inhalation. Valet av tillförselväg beror på testämnets fysikaliska och kemiska egenskaper och det sätt på vilket människor sannolikt exponeras för testämnet.
Oral tillförsel
Om testämnet absorberas i mag-tarmkanalen och om människor exponeras genom munnen, är test med oral tillförsel att föredra såvida inte kontraindikationer föreligger. Djuren tillförs testämnet via fodret, upplöst i dricksvattnet, eller via en kapsel.
Idealt ska djuren tillföras testämnet sju dagar i veckan eftersom en begränsning av doseringen till fem dagar i veckan kan leda till återhämtning eller abstinenstoxicitet mellan exponeringsperioderna, vilket kan påverka resultaten och efterföljande utvärdering. Av praktiska skäl godkänns dock dosering fem dagar i veckan.
Dermal tillförsel
Exponering på huden genom pensling kan väljas som modell för framkallande av hudlesioner i avsikt att simulera en huvudsaklig form av exponering för människor.
Tillförsel genom inhalation
Eftersom inhalationstester inbegriper mer komplexa tekniska problem än andra tillförselvägar för testämnen behövs en mer detaljerad vägledning av detta administrationssätt. Det bör påpekas att intratrakeal installation kan vara ett alternativ under vissa omständigheter.
Försök med långtidsexponering utförs normalt på grundval av möjlig mänsklig exponering varvid djuren exponeras sex timmar om dagen fem dagar i veckan sedan jämvikt uppnåtts för koncentrationen i kammaren (intermittent exponering), eller så utgår man från möjlig miljöexponering varvid djuren exponeras sju dagar i veckan 22–24 timmar om dagen (kontinuerlig exponering) med omkring en timme om dagen vid ett bestämt klockslag för utfodring av djuren och rengöring av kamrarna. I båda fallen exponeras djuren normalt för bestämda exponeringskoncentrationer. En viktig skillnad mellan intermittent och kontinuerlig exponering, som man bör vara uppmärksam på, är att intermittent exponering inbegriper en exponeringsfri period av 17–18 timmar per dag, och en ännu längre period under helgerna, då djuren kan återhämta sig från verkningarna av varje daglig exponering.
Valet mellan intermittent och kontinuerlig exponering beror på syftet med försöket och den mänskliga exponering som simuleras. Man måste dock ta hänsyn till en rad tekniska problem. Fördelen vid kontinuerlig exponering, när det handlar om att simulera miljöbetingelser, kan t.ex. gå förlorad till följd av nödvändigheten att vattna och utfodra djuren under exponeringsperioden och även behovet av bättre utvecklade (och pålitliga) tekniker för generering och kontroll av aerosoler och luftfuktighet.
Djuren ska testas i en inhalationskammare som utformats så att den upprätthåller en dynamisk luftcirkulation med minst 12 luftväxlingar per timme, för att säkerställa en tillräcklig syrehalt och en jämnt fördelad exponeringsatmosfär. Kontroll- och exponeringskammare ska vara identiska i fråga om konstruktion och utformning för att säkerställa att exponeringsförhållandena är desamma i alla avseende förutom exponering för testämnet. Ett normalt undertryck ska upprätthållas i kammaren för att förhindra att testämnet sipprar ut i det omgivande rummet. Exponeringskammaren ska vara så utformade att trängseln bland försöksdjuren minimeras. Som en allmän regel för att säkerställa en stabil atmosfär i en kammare, ska försöksdjurens totala ”volym” inte överstiga 5 % av exponeringskammarens volym.
Följande ska mätas eller kontrolleras:
|
i) |
Luftgenomströmningen: Luftgenomströmningen i kammaren ska helst kontrolleras fortlöpande, |
|
ii) |
Koncentrationen: Under den dagliga exponeringsperioden ska koncentrationen inte avvika mer än ± 15 % från den genomsnittliga koncentrationen. Under hela försöksperioden ska den dagliga koncentrationen hållas så konstant som möjligt. |
|
iii) |
Temperatur och luftfuktighet: För gnagare hålls temperaturen vid 22 ± 2 oC och fuktigheten i kammaren mellan 30 % och 70 %, förutom när vatten används för suspension av testämnet i kammarens atmosfär. Helst ska båda kontrolleras fortlöpande. |
|
iv) |
Mätning av partikelstorlek: Om atmosfärer med flytande eller fasta aerosoler används i kammare bör man analysera fördelningen av partikelstorleken. Aerosolpartiklarna ska vara möjliga att inhalera för de försöksdjur som används. Provtagning ska ske av kammarens atmosfär i djurens respirationszon. Luftproven ska vara representativa för den partikelfördelning som djurens exponeras för och det ska genom gravimetrisk analys vara möjligt att på grundval av dessa luftprover beskriva hela aerosolen, även om en stor del av den inte är möjlig att inhalera. Under inkörning av utrustningen bör analys göras av partikelstorleken för att bestämma aerosolkoncentrationens stabilitet och därefter ska analyser göras under exponeringsperioden så ofta som det är nödvändigt för att korrekt bestämma om den fördelning av partikelstorleken som djuren exponeras för är konstant. |
En test för cancerogenicitet omfattar merparten av försöksdjurens normala livslängd. Om mus eller hamster används bör försöket vara i 18 månader, och om råttor används, i 24 månader. Vid användning av vissa djurstammar med längre livslängd och/eller en låg spontan förekomst av tumörer bör försöket vara i 24 månader med mus och hamster och i 30 månader med råtta. Det accepteras dock att ett sådant förlängt försök avslutas när antalet överlevande djur i den grupp som exponeras för den lägsta dosen, eller kontrollgruppen har nått 25 %. Om det är en uppenbar skillnad i djurens kön bör varje kön betraktas som ett försök för sig, och försökets längd anpassas till detta. Om prematura dödsfall uppträder i den grupp som exponeras för den högsta dosen och om det är uppenbart att detta orsakats av toxiska verkningar, är det inte nödvändigt att avsluta försöket, under förutsättning att de toxiska verkningarna inte skapar problem i andra grupper. För att ett negativt testresultat ska kunna accepteras får högst 10 % av varje grupp falla utanför försöket på grund av autolys av vävnader, kannibalism eller praktiska problem, och överlevandeprocenten i alla grupper får inte vara lägre än 50 % efter 18 månaders försök med användning av mus och hamster, och efter 24 månader med användning av råtta.
Förfarande
Daglig observation av djuren i burarna ska omfatta hud- och pälsförändringar, ögon, slemhinnor, respirations- och cirkulationssystem, autonoma och centrala nervsystemet samt kroppsmotorik och beteendemönster.
Det är nödvändigt att observera djuren regelbundet för att säkerställa att så få djur som möjligt faller bort från studien till följd av kannibalism, autolys av vävnader eller förväxling. Så snart döende djur upptäcks ska dessa avlägsnas, avlivas och obduceras.
Kliniska tecken och dödlighet ska registreras för varje djur. Särskilt uppmärksamhet ska ägnas åt tumörutveckling: den tidpunkt då tecken uppträder för första gången, lokalisering, dimensioner, framträdande och utveckling av varje tydligt synliga eller palpabla tumörer.
Foderintaget (och vattenkonsumtionen om testämnet tillförs via dricksvattnet) ska mätas en gång i veckan under försöksperiodens första 13 veckor och därefter omkring var tredje månad, såvida inte djurens hälsotillstånd eller förändring av kroppsvikt kräver annat.
Varje enskild djur ska vägas en gång i veckan under försöksperiodens första 13 veckor och därefter minst en gång var fjärde vecka.
Kliniska undersökningar
Om kliniska observationer av djuren i burarna tyder på att djurens hälsotillstånd har försämrats under försöket ska en differentialräkning på prover från de ifrågavarande djuren företas.
Efter 12 månader, 18 månader och innan djuren avlivas ska ett blodutstryk tas från varje djur. En differentialräkning ska företas på prover från djur i den högdos- och kontrollgrupperna. Om dessa data, särskilt de som tagits fram strax före avlivning, eller data från den patologiska undersökningen så kräver, ska en differentialräkning företas på djur i den grupp som har fått den näst högsta dosen.
Alla försöksdjur, även de som dör under försöket eller döende djur som avlivas under försöket ska genomgå en fullständig makroskopisk undersökning. Alla makroskopiska synliga lesioner, tumörer eller lesioner som misstänks vara tumörer, ska bevaras.
Följande vävnader och organ bör bevaras i lämplig media för eventuell senare histopatologisk undersökning: hjärnan (inberipet snitt av medulla/pons, lillhjärnbarken och hjärnbarken), hypofysen, sköldkörteln och bisköldkörteln, thymusvävnad, luftstrupe och lungor, hjärta, stora kroppspulsådern, spottkörtlar, lever, mjälte, njurar, binjurar, bukspottskörtel, könskörtlar, livmoder, associerade könskörtlar, hud, matstrupe, magsäck, tolvfingertarm, tomtarmen, ileum, cekum, colon, ändtarm, urinblåsan, representativa lymfkörtlar, kvinnliga bröstkörtlar, lårmuskulatur, nerver från det perifera nervsystemet, bröstbenet med benmärg, femur (inbegripet ledyta), ryggmärg på tre nivåer (vid halsen, mitt för bröstkorgen och från ländområdet) och ögon.
Det bästa sättet att bevara lungor och urinblåsa är att blåsa upp dem med ett fixermedel. I samband med inhalationstester är det av grundläggande betydelse för de hispatologiska testerna att lungorna blåses upp. Vid inhalationstester ska hela luftsystemet undersökas, inbegripet näsa, svalg och luftrör.
|
a) |
En fullständig histopatologisk undersökning ska göras av organ och vävnader hos alla djur som dör eller avlivas under försöket och alla djur i högdos- och kontrollgrupperna. |
|
b) |
En mikroskopisk undersökning av alla klart synliga tumörer, samt lesioner som misstänks för att vara tumörer ska göras i alla grupper. |
|
c) |
Om det föreligger väsentlig skillnad på förekomsten av neoplastiska lesioner i den högst doserade gruppen och kontrollgruppen ska de relevanta organen eller vävnaderna underkastas en histopatologisk undersökning i de övriga grupperna, |
|
d) |
Om antalet överlevande i den högst doserade gruppen är väsentligt lägre än i kontrollgruppen ska den grupp som ar doserad med den näst högsta dosen underkastas en fullständig histopatologisk undersökning. |
|
e) |
Om undersökningen av den högst doserade gruppen ger anledning att misstänka att det är fråga om induktion av toxiska eller andra verkningar som kan ha betydelse för en neoplastisk reaktion, bör den näst högst doserade gruppen underkastas en fullständig histopatologisk undersökning. |
2. DATA
Data ska ställas upp i tabellform och för varje behandlingsgrupp ska anges antal djur i början av försöket, antal djur med tumörer som registrerats under försöket, tidpunkten för registrering samt antal djur med tumörer som upptäckts först efter avlivning. Utvärderingen av resultaten ska ske på grundval av en lämplig statistisk metod. Alla erkända statistiska metoder får användas.
3. RAPPORTERING
3.1. TESTRAPPORT
Testrapporten ska om möjligt innehålla minst följande uppgifter:
|
— |
Djurart, stam, härkomst, miljöbetingelser, foder. |
|
— |
Försöksbetingelser
|
|
— |
Doseringsnivåer (med angivelse av vehikel om en sådan har använts) och koncentrationer. |
|
— |
Upplysningar om förekomsten av tumörer indelat efter kön, dos och slag av tumör. |
|
— |
Tidpunkt för dödsfall under försöket eller om djuren överlevde försöket. |
|
— |
Uppgifter om toxisk reaktion indelat efter kön och dos. |
|
— |
Beskrivning av toxisk eller annan verkan. |
|
— |
Tidpunkt för observation av varje abnormalt tecken och dess senare utveckling. |
|
— |
Uppgifter om foder och kroppsvikt. |
|
— |
Utförda hematologiska analyser och alla resultat. |
|
— |
Obduktionsrön. |
|
— |
Detaljerad beskrivning av alla histopatologiska rön. |
|
— |
Statistisk bearbetning av resultaten och beskrivning av den använda metoden. |
|
— |
Diskussion av resultaten. |
|
— |
Tolkning av resultaten. |
3.2 UTVÄRDERING OCH TOLKNING
Se allmän inledning till del B.
4. HÄNVISNINGAR
Se allmän inledning till del B.
B.33 KOMBINERAT TEST AVSEENDE KRONISK TOXICITET OCH CANCEROGENICITET
1. METOD
1.1 INLEDNING
Se allmän inledning till del B.
1.2 DEFINITIONER
Se allmän inledning till del B.
1.3 REFERENSÄMNEN
Inga.
1.4 TESTMETODSPRINCIP
Syftet med en kombinerad test av kronisk toxicitet/cancerogenicitet är att bestämma ett ämnes kroniska och cancerogena verkningar på däggdjur efter långvarig exponering.
För att göra detta kompletteras testet av cancerogenicitet med minst en satellitgrupp som exponeras för testämnet, och en kontrollsatellitgrupp. De doser som den högst doserade satellitgruppen exponeras för kan vara större än den största dos som används i testet av cancerogenicitet. De djur som används i testet av cancerogenicitet undersöks både för toxisk reaktion och för cancerogenetiska reaktioner. Djuren i den satellitgrupp som exponeras för ämnet undersöks för toxisk reaktion.
Testämnet tillförs normalt sju dagar i veckan på lämpligt sätt till flera grupper av försöksdjur, en dos per grupp under huvuddelen av djurens livslängd. Under och efter exponering för testämnet observeras försöksdjuren dagligen för tecken på toxicitet och utveckling av tumörer.
1.5 KVALITETSKRITERIER
Inga.
1.6 METODBESKRIVNING
I minst fem dagar före försöket ska djuren hållas under testbetingelser vad beträffar miljö och utfodring. Före försöket fördelas friska unga djur slumpmässigt i behandlings- och kontrollgrupper.
I första hand ska råttor användas. På grundval av resultaten från tidigare utförda undersökningar kan andra arter (gnagare eller icke-gnagare) användas. Unga friska djur av vanligen nyttjade laboratoriestammar ska användas och dosering ska inledas så snart som möjligt efter avvänjning.
I början av försöket får vikten hos de djur som används inte avvika mer än ± 20 % från genomsnittsvikten. Om ett test av subkronisk toxicitet med oral tillförsel utförs som en förundersökning till en långtidsundersökning ska samma djurart användas i båda undersökningarna.
I fråga om gnagare ska minst 100 djur (50 hondjur och 50 handjur) används på varje dosnivå och motsvarande kontrollgrupp. Hondjuren får inte ha fött ungar och får inte vara dräktiga. Vid plan-enligt avlivande av försöksdjur under försöket ska det ursprungliga antalet djur ökas med det antal djur som planenligt ska avlivas innan försöket slutförs.
Den eller de behandlade satellitgrupper som används för utvärdering av andra patologiska verkningar än tumörer ska omfatta 20 djur av varje kön, medan kontrollsatellitgruppen ska omfatta tio djur av varje kön.
Vid testning av cancerogenicitet ska minst tre dosnivågrupper användas förutom motsvarande kontrollgrupp. Den högsta dosen bör framkalla minimala toxicitetstecken, t.ex. en liten minskning av kroppsvikten (mindre än 10 %), utan att leda till väsentliga ändringar av den normala livslängden, med undantag för fall av tumörer.
Den lägsta dosen bör inte påverka normal tillväxt, utveckling och livslängd eller framkalla några tecken på toxicitet. I allmänhet bör den inte vara mindre än 10 % av den högsta dosen.
Medeldosen (eventuellt flera) bör ligga mitt emellan den högsta och den lägsta dosen.
Vid val av dosnivåer bör hänsyn tas till data från tidigare tester och undersökningar av toxicitet.
Vid testning av kronisk toxicitet ska ytterligare försöksgrupper och en motsvarande satellitkontrollgrupp användas i försöket. Den högsta dosen som djuren i den eller de behandlade satellitgrupperna exponeras för bör framkalla tydliga tecken på toxicitet.
Djuren doseras normalt dagligen med testämnet.
Om ämnet administreras via dricksvattnet eller via fodret bör detta finnas tillgänglig kontinuerligt.
En kontrollgrupp som är identisk som och behandlas på samma sätt som försöksgrupperna, bortsett från tillförsel av testämnet, ska användas.
Under särskilda omständigheter, t.ex. vid inhalationstester där aerosoler används, eller där en emulgator med okänd biologisk verkan används i tester med oral tillförsel, ska en ytterligare kontrollgrupp som inte exponeras för vehikel användas.
Det finns tre huvudsakliga tillförselvägar: oral tillförsel, dermal tillförsel respektive tillförsel genom inhalation. Valet av tillförselväg beror på testämnets fysikaliska och kemiska egenskaper och det sätt på vilket människor sannolikt exponeras för testämnet.
Oral tillförsel
Om testämnet absorberas i mag-tarmkanalen och om människor utsätts för tillförsel genom munnen, föredras den orala tillförselvägen såvida inte kontraindikationer föreligger.
Djuren tillförs testämnet via fodret, upplöst i dricksvattnet, eller via en kapsel. Idealt tillförs djuren testämnet sju dagar i veckan eftersom en begränsning av doseringen till fem dagar i veckan kan leda till återhämtning eller abstinenstoxicitet mellan exponeringsperioderna, vilket kan påverka resultaten och värderingen av resultaten. Av praktiska skäl godkänns dock dosering fem dagar i veckan.
Påföring på huden
Exponering på huden genom pensling kan väljas som modell för framkallande av hudlesioner i avsikt att simulera en huvudsaklig form av exponering för människor.
Tillförsel genom inhalation
Eftersom inhalationstester inbegriper mer komplexa tekniska problem än andra tillförselvägar för testämnen ges här en mer detaljerad vägledning av detta administrationssätt. Det bör påpekas att intratrakeal installation kan vara ett alternativ under vissa omständigheter.
Försök med långtidsexponering utförs normalt på grundval av möjlig mänsklig exponering varvid djuren exponeras sex timmar om dagen fem dagar i veckan sedan jämvikt uppnåtts för koncentrationen i kammaren (intermittent exponering), eller så utgår man från möjlig miljöexponering varvid djuren exponeras sju dagar i veckan 22–24 timmar om dagen (kontinuerlig exponering) med omkring en timme om dagen vid ett bestämt klockslag för utfodring av djuren och rengöring av kamrarna. I båda fallen exponeras djuren normalt för bestämda exponeringskoncentrationer. En viktig skillnad mellan intermittent och kontinuerlig exponering, som man bör vara uppmärksam på, är att intermittent exponering inbegriper en exponeringsfri period av 17–18 timmar per dag, och en ännu längre period under helgerna, då djuren kan återhämta sig från verkningarna av varje daglig exponering.
Valet mellan intermittent och kontinuerlig exponering beror på syftet med försöket och den mänskliga exponering som simuleras. Man måste dock ta hänsyn till en rad tekniska problem. Fördelen vid kontinuerlig exponering, när det handlar om att simulera miljöbetingelser, kan t.ex. gå förlorad till följd av nödvändigheten att vattna och utfodra djuren under exponeringsperioden och även behovet av bättre utvecklade (och pålitliga) tekniker för generering och kontroll av aerosoler och luftfuktighet.
Djuren ska testas i en inhalationskammare som utformats så att den upprätthåller en dynamisk luftcirkulation med minst 12 luftväxlingar per timme, för att säkerställa en tillräcklig syrehalt och en jämnt fördelad exponeringsatmosfär. Kontroll- och exponeringskammare ska vara identiska i fråga om konstruktion och utformning för att säkerställa att exponeringsförhållandena är de samma i alla avseende förutom exponering för testämnet. Ett lätt undertryck ska normalt upprätthållas i kammaren för att förhindra att testämnet sipprar ut i det omgivande rummet. Exponeringskammaren ska vara så utformade att trängseln bland försöksdjuren minimeras. Som en allmän regel för att säkerställa en stabil atmosfär i en kammare, ska försöksdjurens totala ”volym” inte överstiga 5 % av exponeringskammarens volym.
Följande ska mätas eller kontrolleras:
|
i) |
Luftgenomströmningen: Luftgenomströmningen i kammaren ska helst kontrolleras fortlöpande. |
|
ii) |
Koncentrationen: Under den dagliga exponeringsperioden ska koncentrationen inte avvika mer än ± 15 % från den genomsnittliga koncentrationen. Under hela försöksperioden ska den dagliga koncentrationen hållas så konstant som möjligt. |
|
iii) |
Temperatur och luftfuktighet: För gnagare hålls temperaturen vid 22 ± 2 oC och fuktigheten i kammaren mellan 30 % och 70 %, förutom när vatten används för suspension av testämnet i kammarens atmosfär. Helst ska båda kontrolleras fortlöpande. |
|
iv) |
Mätning av partikelstorlek: Om atmosfärer med flytande eller fasta aerosoler används i kammare bör man analysera fördelningen av partikelstorleken. Aerosolpartiklarna ska vara möjliga att inhalera för de försöksdjur som används. Provtagning ska ske av kammarens atmosfär i djurens respirationszon. Luftproven ska vara representativa för den partikelfördelning som djurens exponeras för och det ska genom gravimetrisk analys vara möjligt att på grundval av dessa luftprover beskriva hela aerosolen, även om en stor del av den inte är möjlig att inhalera. Under inkörning av utrustningen bör analys göras av partikelstorleken för att bestämma aerosolkoncentrationens stabilitet och därefter ska analyser göras under exponeringsperioden så ofta som det är nödvändigt för att korrekt bestämma om den fördelning av partikelstorleken som djuren exponeras för är konstant. |
Den del av testet som handlar om cancerogenicitet ska omfatta merparten av försöksdjurens normala livslängd. Om mus eller hamster används bör försöket vara i 18 månader, och om råttor används, i 24 månader. Vid användning av vissa djurstammar med längre livslängd och/eller en låg spontan förekomst av tumörer bör försöket vara i 24 månader med mus och hamster och i 30 månader med råtta. Det accepteras dock att ett sådant förlängt försök avslutas när antalet överlevande djur i den grupp som exponeras för den lägsta dosen, eller kontrollgruppen har nått 25 %. Om det är en uppenbar skillnad i djurens kön bör varje kön betraktas som ett försök för sig själv, och försökets längd anpassas till detta. Om prematura dödsfall uppträder i den grupp som exponeras för den högsta dosen och om det är uppenbart att detta orsakats av toxiska verkningar, är det inte nödvändigt att avsluta försöket, under förutsättning att de toxiska verkningarna inte skapar problem i andra grupper. För att ett negativt testresultat ska kunna accepteras får högst 10 % av varje grupp falla utanför försöket på grund av autolys av vävnader, kannibalism eller förväxling, och överlevandeprocenten i alla grupper får inte vara lägre än 50 % efter 18 månaders försök med användning av mus och hamster, och efter 24 månader med användning av råtta.
Satellitgrupperna, som består av 20 doserade djur av varje kön och en kontrollgrupp med tio djur per kön och som används för test av kronisk toxicitet, ska hållas under försöksbetingelserna i minst 12 månader. Tidpunkten för avlivning av dessa djur bör fastställas utifrån en bedömning av patologiska rön i förhållande till testämnet, och på ett sådant sätt att eventuella geriatriska förändringar inte har en negativ inverkan.
Förfarande
Daglig observation av djuren i burarna ska omfatta hud- och pälsförändringar, ögon, slemhinnor, respirations- och cirkulationssystem, autonoma och centrala nervsystemet samt kroppsmotorik och beteendemönster.
Djuren i den eller de doserade satellitgrupperna ska undersökas kliniskt med passande mellanrum.
Det är nödvändigt att observera djuren regelbundet för att säkerställa att så få djur som möjligt faller bort från studien till följd av kannibalism, autolys av vävnader eller förväxling. Så snart döende djur upptäcks ska dessa avlägsnas, avlivas och obduceras.
Kliniska tecken, inbegripet neurologiska förändringar och förändringar i ögonen, samt dödlighet ska registreras för varje djur. Särskilt uppmärksamhet ska ägnas åt tumörutveckling: den tidpunkt då tecken uppträder för första gången, lokalisering, dimensioner, framträdande och utveckling av alla tydligt synliga eller palpabla tumörer ska registreras.
Foderintaget (och vattenkonsumtionen om testämnet tillförs via dricksvattnet) ska mätas en gång i veckan under försöksperiodens första 13 veckor och därefter omkring var tredje månad, såvida inte djurens hälsotillstånd eller förändring i kroppsvikten kräver annat.
Varje enskild djur ska vägas en gång i veckan under försöksperiodens första 13 veckor och därefter minst en gång var fjärde vecka.
Kliniska undersökningar
En hematologisk undersökning (t.ex. hemoglobinkoncentration, hematokrit, erytrocyträkning, total leukocyträkning, räkning av blodplättar samt mått på koaguleringsförmågan) ska företas efter tre månader, sex månader samt därefter cirka var sjätte månad samt vid försökets avslutning på blodprover från 10 råttor av varje kön i alla grupper. Om det är möjligt ska proverna tas från samma råttor varje gång.
Om kliniska observationer av djuren i burarna tyder på att djurens hälsotillstånd har försämrats under försöket ska en differentialräkning på prover från de ifrågavarande djuren företas. En differentialräkning ska företas på prover från djur i högdos- och kontrollgrupperna. En differentialräkning på prover från djur i den grupp som har fått den näst högsta dosen ska endast företas om det är stor skillnad mellan den högst doserade gruppen och kontrollgruppen, eller om den patologiska undersökningen tyder på detta.
Urinprov för analys ska tas från tio råttor av varje kön i alla grupper, helst från samma råttor varje gång med samma intervaller som vid den hematologiska undersökningen. Följande undersökningar ska utföras på prover antingen från varje enskild djur eller, i fråga om råttor, på en samlad urinprov/kön/grupp:
|
— |
Utseende: volym och densitet för enskilda djur. |
|
— |
Protein, glukos, keton, ockult blod (semikvantitativt). |
|
— |
Sedimentmikroskopi (semikvantitativt). |
Omkring var sjätte månad samt vid försökets avslutning ska blodprov tas för klinisk-kemisk undersökning från varje enskild djur när det gäller icke-gnagare och från tio råttor av varje kön i alla grupper, helst från samma råttor varje gång. 1 fråga om icke-gnagare ska dessutom prov tas innan försöket inleds. Plasma från proverna ska underkastas följande undersökningar:
|
— |
Total proteinkoncentration. |
|
— |
Albuminkoncentration. |
|
— |
Leverfunktion t.ex. alkalisk fosfataktivitet, glutaminsyra-pyrodruvsyra-transaminasaktivitet (4), och glutaminsyra-oxalättiksyra-transaminasaktivitet (5), gamma-glutamyl-transpeptidas, ornitin dekaroxylas. |
|
— |
Kolhydratmetabolism, t.ex. fasteglukos. |
|
— |
Njurfunktion, t.ex. urinämne. |
Alla försöksdjur, även de som dör under försöket eller döende djur som avlivas under försöket ska underkastas en fullständig makroskopisk undersökning. Vid avlivning ska blodprov tas från alla djur för differentialräkning. Alla makroskopiska synliga lesioner, tumörer eller lesioner som misstänks vara tumörer, ska bevaras. Makroskopiska observationer ska så långt det går korreleras med mikroskopiska rön.
Alla vävnader och organ bör bevaras för histopatologisk undersökning. Detta gäller normalt följande organ och vävnader: hjärnan (7), (medulla/pons, lilllhjärnbarken och hjärnbarken), hypofysen, sköldkörteln (inbegripet bisköldkörteln), thymus, lungor (inbegripet luftstrupe), hjärta, stora kroppspulsådern, spottkörtlar, lever (7), mjälte, njurar (7), binjurar (7), matstrupe, magsäck, tolvfinger-tarm, tomtarmen, ileum, cekum, colon, ändtarm, urinblåsan, lymfkörtlar, bukspottskörtel, könskörtlar (7), associerade könsorgan, kvinnliga bröstkörtlar, hud, muskulatur, nerver från det perifera nervsystemet, ryggmärg (på tre nivåer – vid halsen, mitt för bröstkorgen och från ländområdet), bröstbenet med benmärg och femur (inbegripet ledyta), och ögon.
Det bästa sättet att bevara lungor och urinblåsa är att blåsa upp dem med ett fixeringsmedel. I samband med inhalationstester är det av grundläggande betydelse för de hispatologiska testerna att lungorna blåses upp. Vid inhalationstester ska hela luftsystemet undersökas, inbegripet näsa, svalg och luftrör.
Om andra kliniska undersökningar utförs ska den information som erhålls från dessa finnas tillgänglig innan en mikroskopisk undersökning företas, eftersom detta kan vara av betydande värde för patologen.
För den del av undersökningen som gäller kronisk toxicitet gäller följande:
En detaljerad undersökning ska göras av alla bevarade organ från alla djur i högdossatellit- och kontrollgrupperna Om det finns patologiska tecken som kan sättas i samband med testämnet i högdossatellitgruppen ska målorganen hos djuren i övriga doserade satellitgrupper samt i de doserade grupperna i den del av undersökningen som gäller cancerogenicitet underkastas en fullständig och detaljerad histologisk undersökning vid försökets avslutning.
För den del av undersökning som gäller cancerogenicitet gäller följande:
|
a) |
En fullständig histopatologisk undersökning ska göras av organ och vävnader från de djur som dör eller avlivas under försöket samt djuren i högdos- och kontrollgrupperna. |
|
b) |
En mikroskopisk undersökning av alla klart synliga tumörer, samt lesioner som misstänks för att vara tumörer ska göras i alla grupper. |
|
c) |
Om det föreligger väsentlig skillnad på förekomsten av neoplastiska lesioner i den högst doserade gruppen och kontrollgruppen ska de relevanta organen eller vävnaderna underkastas en histopatologisk undersökning i de övriga grupperna. |
|
d) |
Om antalet överlevande i högdosgruppen är väsentligt lägre än i kontrollgruppen ska den grupp som är doserad med den näst högsta dosen underkastas en fullständig histopatologisk undersökning. |
|
e) |
Om undersökningen av högdosgruppen ger anledning att misstänka att det är fråga om induktion av toxiska eller andra verkningar som kan ha betydelse för en neoplastisk reaktion, bör den näst högst doserad gruppen underkastas en fullständig histopatologisk undersökning. |
2. DATA
Data ska ställas upp i tabellform och för varje behandlingsgrupp anges antal djur i början av försöket, antal djur som uppvisar tumörer eller toxiska verkningar under försöket, tidpunkten för registrering samt antal djur med tumörer som upptäckts efter avlivning. Utvärderingen av resultaten ska ske på grundval av en lämplig statistisk metod, Alla erkända statistiska metoder får användas.
3. RAPPORTERING
3.1 TESTRAPPORT
Testrapporten ska om möjligt innehålla minst följande uppgifter:
|
— |
Djurart, stam, härkomst, miljöbetingelser, foder. |
|
— |
Försöksbetingelser:
|
|
— |
Doseringsnivåer (med angivelse av vehikel om en sådan har använts) och koncentrationer. |
|
— |
Upplysningar om förekomsten av tumörer indelat efter kön, dos och slag av tumör. |
|
— |
Tidpunkt för dödsfall under försöket eller om djuren överlevde försöket. |
|
— |
Uppgifter om toxisk reaktion indelat efter kön och dos. |
|
— |
Beskrivning av toxisk eller annan verkan. |
|
— |
Tidpunkt för observation av varje abnormalt tecken och dess senare utveckling, |
|
— |
Oftamologiska rön. |
|
— |
Uppgifter om foder och kroppsvikt. |
|
— |
Utförda hematologiska analyser och alla resultat. |
|
— |
Utförda klinisk-biokemiska undersökningar och alla resultat (inbegripet eventuell urinanalys). |
|
— |
Obduktionsrön. |
|
— |
Detaljerad beskrivning av alla histopatologiska rön. |
|
— |
Statistisk bearbetning av resultaten och beskrivning av den använda metoden. |
|
— |
Diskussion av resultaten. |
|
— |
Tolkning av resultaten. |
3.2 UTVÄRDERING OCH TOLKNING
Se allmän inledning till del B.
4. HÄNVISNINGAR
Se allmän inledning till del B.
B.34 REPRODUKTIONSTOXICITETSTEST PÅ EN GENERATION
1. METOD
1.1 INLEDNING
Se allmän inledning till del B.
1.2 DEFINITIONER
Se allmän inledning till del B.
1.3. REFERENSÄMNEN
Inga.
1.4 TESTMETODSPRINCIP
Testämnet tillförs i graderade doser till flera grupper av handjur och hondjur. Handjuren bör tillföras ämnet under tillväxtperioden och under minst en fullständig spermiebildande cykel (ca 56 dagar för mus och 70 dagar för råtta) för att framkalla eventuella hälsomässiga oönskade verkningar som testämnet kan ha på spermatogenes.
Hondjur i föräldra(Fo)generationen tillförs testämnet under minst två fullständiga brunstighetscykler för att framkalla eventuella skadliga verkningar som testämnet kan ha på brunsten. Därefter paras djuren. Båda kön tillförs testämnet under parningsperioden, och därefter tillförs endast hondjuren testämnet under dräktighetsperioden och diperioden. Vid tillförsel genom inhalation måste metoden anpassas.
1.5 KVALITETSKRITERIER
Inga.
1.6 METODBESKRIVNING
Förberedelser
Före försöket fördelas friska unga djur slumpmässigt i behandlings- och kontrollgrupper. I minst fem dagar före försöket ska djuren hållas under testbetingelser vad beträffar miljö och utfodring. Det rekommenderas att testämnet tillförs via fodret eller dricksvattnet. Andra tillförselvägar är också godtagbara Samma tillförselmetod ska användas för alla djur under hela behandlingsperioden. Om vehikel eller annan tillsats används för att underlätta doseringen ska det vara fastställt att den inte framkallar några toxiska verkningar. Djuren bör tillföras testämnet sju dagar i veckan.
Val av djurart
I första hand ska råttor och möss användas. Unga friska djur, som inte tidigare har använts i försök, ska användas. Stammar med låg fertilitet bör inte användas. Försöksdjuren bör vara representativa för sin art, stam, kön, vikt och/eller ålder.
För att korrekt kunna bedöma fertiliteten ska både han- och hondjur testas. Alla försöks- och kontrolldjur ska vara avvanda innan dosering inleds.
Antal och kön
Varje behandlings- och kontrollgrupp ska bestå av ett tillräckligt högt antal djur för att resultera i omkring 20 dräktiga hondjur som är nära eller vid födslotidpunkten.
Syftet med detta är att säkerställa att det föds tillräckligt många ungar för att säkerställa en meningsfull utvärdering av hur testämnet kan påverka fertiliteten, dräktigheten och moderdjurens beteende i F0-generationen samt diandet, tillväxten och utvecklingen i F1-generationen från nedkomsten till avvänjning.
Foder och vatten ges ad libitum. När födslotidpunkten närmar sig placeras de dräktiga hondjuren i var sin födslo- eller dräktighetsbur och djuren kan eventuellt förses med material för att bygga bo.
Minst tre olika dosnivågrupper samt en kontrollgrupp ska användas. Om vehikel används för tillförsel av testämnet ska kontrollgruppen tillföras samma mängd vehikel som högdosgruppen. Om ett testämne leder till ett minskat foderintag eller påverkar absorption eller metabolism kan det vara nödvändigt att använda en motsvarande restriktivt utfodrad kontrollgrupp. Såvida det inte finns någora begränsningar till följd av testämnets fysikaliska-kemiska eller biologiska egenskaper bör den högsta dosen idealt förorsaka toxiska verkningar, men inte dödsfall i föräldra(F0)-generationen. Medeldosnivåerna (eventuellt flera) bör framkalla minsta möjliga observerbara toxiska verkningar som orsaktas av testämnet, och den lägsta dosen bör inte ha någon toxisk effekt på föräldragenerationen eller avkomman. Om ämnet tillförs via magsond eller i kapsel bör den dos som varje enskilt djur mottar beräknas på grundval av djurets kroppsvikt och varje vecka justeras med hänsyn till förändringar i kroppsvikt. Doser som tillförs dräktiga hondjur kan beräknas efter kroppsvikt vid dag 0 eller 6 av dräktigheten, om så önskas.
Om ett testämne med låg toxicitet i en dosnivå på 1 000 mg/kg kroppsvikt inte har någon påverkan på reproduktionen kan ytterligare tester anses vara överflödiga. Om en inledande undersökning, i vilken endast den högsta dosen används, klart visar på toxisk verkan på moderdjuren, men inte har någon skadlig verkan på fertiliteten, kan ytterligare tester anses vara överflödiga.
Förfarande
Daglig dosering av föräldra(F0)-generationen inleds när djuren är mellan fem och nio veckor gamla, och efter det att de är avvanda och har acklimatiserat sig i minst fem dagar. Om råttor används fortsätter doseringen i tio veckor före parningsperioden (för möss åtta veckor). Handjuren bör avlivas och undersökas antingen vid slutet av parningsperioden eller så kan man fortsätta att tillföra dem testämnet fram till en eventuell produktion av en andra kull, varefter de avlivas och undersöks någongång före försökets avslutning. Dosering av föräldra(F0)-generationens hondjur inleds efter minst fem dagars acklimatisering och fortsätter under minst två veckor före parning. Daglig dosering av föräldra(F0)-generationens hondjur fortsätter under den tre veckor långa parningsperioden, under dräktighetsperioden och fram till det att F1-generationen är avvand. Man bör överväga eventuella justeringar av försöksschemat om det föreligger uppgifter om testämnet, t.ex. induktion av metabolism eller bioackumulering.
Parning kan utföras i förhållandet 1:1 (ett handjur till ett hondjur) eller 1:2 (ett handjur till två hondjur) i undersökningar av reproduktionstoxicitet.
Om parning i förhållandet 1:1 används förs ett handdjur och ett hondjur samman tills dräktighet inträffar eller tre veckor har förflutit. Varje morgon ska hondjuren undersökas för närvaron av spermier eller vaginalproppar. Som dag 0 i dräktighetsperioden räknas den dag då man observerar en vaginalpropp eller sperma.
De par som inte parar sig bör undersökas för att fastställa orsaken till den påtagliga ofruktsamheten.
Detta kan göras genom att djuren t.ex. får möjlighet att para sig med han- eller hondjur vars fruk-barhet är fastställd, genom mikroskopisk undersökning av reproduktionsorganen och genom undersökning av brunstighetsperioden eller speratogenes.
De djur som doseras i fertilitetsundersökningen bör ha möjlighet att föda normalt och att behålla sin avkomma tills den är avvand utan standardisering av kullar.
Om kullar standardiseras föreslås följande förfarande: Mellan dag 1 och 4 efter födseln standardiseras varje kull genom att extra ungar avlägsnas så att varje kull så långt det är möjligt består av fyra handjur och fyra hondjur.
Om det på grund av antalet han- och hondjur inte är möjligt att standardisera varje kulle att de innehåller fyra handjur och fyra hondjur kan en delvis standardisering godkännas (t.ex. fem handjur och tre hondjur). Ingen standardisering ska ske på kullar som består av mindre än åtta ungar.
Djuren ska inspekteras minst en gång om dagen under hela försöket. Relevanta beteendeförändringar, tecken på svårartad eller förlängd födsel och alla toxicitetstecken, inbegripet dödlighet, ska registreras. Före och under parningstiden registreras foderintaget en gång i veckan. Efter födseln och under diperioden ska foderintaget (och vattenkonsumtionen om testämnet tillförs via dricksvattnet) mätas varje vecka. Han- och hondjur av F0-generationen vägs den dag då doseringen inleds och därefter en gång i veckan. Dessa observationer bör rapporteras individuellt för varje enskilt djur.
Dräktighetsperioden beräknas från dag 0. Varje kull undersöks så snart som möjligt efter födseln för att fastställa ungarnas antal och kön, antal dödfödda och levande ungar och förekomsten av mak-roskopiskt synliga abnormiteter.
Döda ungar och ungar som avlivas dag 4 ska bevaras och undersökas för eventuella defekter. Levande ungar ska räknas och kullen vägas morgonen efter födseln, på dag 4 och dag 7 och därefter en gång i veckan tills försöket avslutats då djuren ska vägas individuellt.
Fysiska eller beteendemässiga abnormiteter som observeras i hondjuret eller avkomman ska registreras.
F0-generationsdjur som avlivas efter eller dör under försöket ska undersökas makroskopiskt för strukturella abnormiteter eller patologiska förändringar, med särskilt uppmärksamhet riktad mot organen i reproduktionssystemet. Döda eller döende ungar ska undersökas för defekter.
Äggstockar, livmoder, livmoderhals, slida, testiklar, bitestiklar, sädesblåsa, prostata, koagulerings-körtlar, hypofys och målorgan (eventuellt flera) från alla djur i F0-generationen ska bevaras för mikroskopisk undersökning. Om dessa organ inte har undersökts i andra försök med multipel dosering ska organen från djuren i högdos- och kontrollgrupperna, samt de djur som dör under försöket, undersökas mikroskopiskt om detta är praktiskt möjligt.
De organ från dessa djur som uppvisar abnormiteter ska även undersökas hos alla andra djur i F0-generationen. I dessa fall ska alla vävnader som uppvisar makroskopiska patologiska förändringar undersökas mikroskopiskt. Liksom det föreslås i parningsavsnittet kan även reproduktionsorganen från djur som antas vara ofruktsamma undersökas mikroskopiskt.
2. DATA
Data ska ställas upp i tabellform och för varje grupp anges antal djur i början av försöket, antal fertila handjur, antal dräktiga hondjur, slag av förändring och den procent djur som upvisar varje slag av förändring.
Utvärderingen av resultaten ska om möjligt grundas på en lämplig statistisk metod. Alla erkända statistiska metoder får användas.
3. RAPPORTERING
3.1 TESTRAPPORT
Testrapporten ska om möjligt innehålla minst följande uppgifter:
|
— |
Djurart, stam. |
|
— |
Uppgifter om toxisk reaktion uppdelade efter kön och dos, inbegripet fertilitet, dräktighet och avkommans livsduglighet. |
|
— |
Tidpunkten för dödsfall under försöket eller överlevnad till slutet av försöket eller tidpunkt för planenlig avlivning. |
|
— |
Tabell med angivelse av vikten för varje enskild kulle, ungarnas genomsnittsvikt och varje enskild unges vikt vid försökets avslutning. |
|
— |
Toxiska och andra verkningar på reproduktion, avkomma, postnatal tillväxt. |
|
— |
Tidpunkten för observation av varje abnormalt tecken och dess senare utveckling. |
|
— |
Uppgifter om foder och kroppsvikt för F0-generationen. |
|
— |
Obduktionsrön. |
|
— |
Detaljerad beskrivning av alla mikroskopiska rön. |
|
— |
Eventuell satistisk behandling av resultaten i lämpliga fall. |
|
— |
Diskussion om resultaten. |
|
— |
Tolkning av resultaten. |
3.2 UTVÄRDERING OCH TOLKNING
Se allmän inledning till del B.
4. HÄNVISNINGAR
Se allmän inledning till del B.
B.35 TEST AVSEENDE REPRODUKTIONSTOXICITET PÅ TVÅ GENERATIONER
1. METOD
Denna metod är i stort sett identisk med OECD TG 416 (2001).
1.1 INLEDNING
Denna metod för test avseende reproduktionstoxicitet på två generationer har som syfte att ge allmän information om ett testämnes verkningar på integriteten och prestanda hos hondjurs och handjurs reproduktionssystem, inklusive körtelfunktion, brunstighetscykel, parningsbeteende, befruktning, dräktighet, födsel, diperiod och avvänjning samt avkommans tillväxt och utveckling. Testet kan också ge information om testämnets verkningar på neonatal morbiditet och mortalitet, preliminära data om prenatal och postnatal utvecklingstoxicitet samt vägledning för senare test. Utöver undersökning av tillväxt och utveckling hos F1-generationen är testmetoden också avsedd för bedömning av integritet och prestanda hos hondjurs och handjurs reproduktionssystem samt F2-generationens tillväxt och utveckling. För att erhålla ytterligare information om utvecklingstoxicitet och funktionsstörningar kan segment med tilläggstest införlivas i detta protokoll, där metoderna för utvecklingstoxicitet eller utvecklingsneurotoxicitet används. Alternativt kan dessa endpoints undersökas i separata test där lämpliga testmetoder används.
1.2 PRINCIP FÖR TESTMETODEN
Testämnet tillförs i graderade doser till flera grupper av handjur och hondjur. Handjur i P-generationen bör tillföras ämnet under tillväxtperioden och under minst en fullständig spermiebildande cykel (cirka 56 dagar för mus och 70 dagar för råtta) för att framkalla eventuella skadliga verkningar på spermatogenesen. Verkningarna på sperman bestäms genom ett antal spermaparametrar (t.ex. spermans morfologi och motilitet), genom undersökning av vävnadspreparat och genom detaljerad histopatologi. Om data om spermatogenesen finns att tillgå från tidigare tillräckligt långa test med upprepad dos, t.ex. ett 90-dagars test, behöver P-generationens handjur inte inkluderas i utvärderingen. Det rekommenderas dock att prover eller digitala registreringar av P-generationens sperma bevaras, så att senare utvärderingar vid behov kan göras. P-gene-rationens hondjur bör ges doser under tillväxten och under flera fullständiga brunstighetscykler för att framkalla eventuella skadliga verkningar av testämnet på brunstighetscykelns normalitet. Testämnet tillförs föräldradjuren (P) under parningsperioden, under dräktighetsperioden och under F1-avkommans hela avvänjningsperiod. Efter avvänjningen tillförs testämnet fortsättningsvis till F1-avkomman under tillväxtperioden och under perioden för parning och produktion av en F2-generation, fram till det att F2-generatio-nen är avvand.
Kliniska observationer och patologiska undersökningar görs på alla djur med avseende på toxicitetstecken, med särskild tonvikt på verkningarna på integriteten och prestanda hos hondjurens och handjurens reproduktionssystem och på avkommans tillväxt och utveckling.
1.3 BESKRIVNING AV TESTMETODEN
1.3.1 Val av djurart
Rekommenderat testdjur är råtta. Om andra arter används bör detta motiveras och testmetoden måste modifieras på tillbörligt sätt. Stammar med låg fertilitet eller välkänd hög incidens av utvecklingsdefekter ska inte användas. Vid testets början bör variationen i vikt mellan djuren vara minimal och får inte överstiga 20 % av vardera könets medelvikt.
1.3.2 Förvarings- och utfodringsbetingelser
Temperaturen i testdjurens utrymmen bör vara 22 oC (± 3o). Den relativa fuktigheten bör vara minst 30 % och ska helst inte överstiga 70 %, utom när rummet rengörs. Målvärdet bör dock vara 50–60 %. Belys-ningen bör vara artificiell med omväxlande 12 timmar ljus och 12 timmar mörker. För utfodringen kan konventionellt laboratoriefoder användas, och djuren bör ha obegränsad tillgång till dricksvatten. Valet av foder kan påverkas av behovet att säkerställa att en lämplig blandning med testämnet fås, om man har valt denna tillförselmetod.
Djuren kan inhysas individuellt eller i burar med små grupper av samma kön. För parningsprocedurerna används burar som är lämpliga för ändamålet. Efter tecken på kopulation flyttas de parade hondjuren till var sin födslo- eller maternitetsbur. Parade råttor kan också hållas i små grupper och separeras en eller två dagar före födseln. När tidpunkten för födsel närmar sig ska de parade djuren ges lämpliga och definierade material för beredning av ett bo.
1.3.3 Förberedelse av djuren
För testet används unga friska djur som har acklimatiserats till laboratoriemiljön under minst fem dagar och som inte har använts för tidigare test. Testdjuren karakteriseras enligt art, stam, ursprung, kön, vikt och ålder. Alla syskonförhållanden mellan djuren bör vara kända så att parning mellan sådana djur kan undvikas. Djuren fördelas slumpmässigt i kontroll- och behandlingsgrupper (stratifiering enligt kroppsvikt rekommenderas). Burarna placeras i sådan ordning att eventuella verkningar på grund av placeringen mini-meras. Varje djur förses med ett unikt identifieringsnummer. För P-generationen bör detta göras innan tillförseln av testämnet inleds. För F1-generationen bör detta göras efter avvänjningen av de djur som har valts för parning. För alla utvalda F1-djur bör uppgifter registreras om vilken kull de härstammar från. Dessutom rekommenderas att ungarna identifieras individuellt så snart som möjligt efter födseln, i fall där individuell vägning av ungar eller funktionstest övervägs.
Föräldradjuren (P) bör vara 5–9 veckor gamla när tillförseln av testämnet inleds. Djuren i alla testgrupper bör vara så lika som möjligt med avseende på vikt och ålder.
1.4 FÖRFARANDE
1.4.1 Djurens antal och kön
Varje behandlings- och kontrollgrupp ska bestå av så många djur att det nära eller vid tidpunkten för födseln helst finns minst 20 dräktiga hondjur. I fråga om ämnen som förorsakar oönskade behandlingsrelaterade verkningar (t.ex. sterilitet, övertoxicitet vid den höga dosen) är detta kanske inte möjligt. Syftet är att få ett tillräckligt antal dräktigheter för att säkerställa en meningsfull utvärdering av testämnets potential att påverka fertiliteten, dräktigheten och moderdjurens beteende samt diandet, tillväxten och utvecklingen hos F1-avkomman från befruktning till mognad och utvecklingen av dennas avkomma (F2) till avvänjning. Om det önskade antalet dräktiga djur (dvs. 20) inte erhålls, behöver testet således inte nödvändigtvis förkastas, utan detta bör bedömas från fall till fall.
1.4.2 Förberedelse av doserna
Det rekommenderas att testämnet tillförs oralt (med fodret, dricksvattnet eller genom sondmatning), utom om någon annan tillförselväg (t.ex. via huden eller genom inandning) bedöms vara lämpligare.
Vid behov kan testämnet upplösas eller suspenderas i en lämplig vehikel. Om möjligt bör man i första hand överväga vattenlösning eller vattensuspension, sedan oljelösning eller oljeemulsion (t.ex. majsolja) och därefter eventuellt andra vehiklar. I fråga om andra vehiklar än vatten måste de toxiska egenskaperna vara kända. Testämnets stabilitet i vehikeln bör fastställas.
1.4.3 Dosering
Minst tre dosnivåer och en parallell kontrollgrupp bör användas. Den högsta dosnivån väljs så att den framkallar toxiska verkningar men inte dödsfall eller allvarligt lidande, förutsatt att det inte finns begränsningar som beror på testämnets fysikalisk-kemiska natur eller biologiska verkningar. I fall av oväntad mortalitet kan testet i regel ändå godkännas, så länge som mortalitetsgraden är lägre än cirka 10 procent hos föräldradjuren (P). Därefter väljs dosnivåerna i fallande skala så att dosrelaterade verkningar kan påvisas. En nivå bör väljas så att inga påvisbara skadliga verkningar (NOAEL) uppstår. Två- till fyrfaldiga dosintervall är ofta optimala för de fallande dosnivåerna. Tillägg av en fjärde testgrupp är att föredra framför användning av mycket breda intervall mellan dosnivåerna (t.ex. en faktor som överstiger cirka 10). För utfodringstest bör dosintervallet inte vara mer en trefaldigt. Dosnivåerna bör väljas med hänsyn till kända toxicitetsdata, särskilt resultat från test med upprepad dos. Likaså bör hänsyn tas till känd information om metabolism och kinetik för testämnet eller närbesläktade material. Denna information är också till hjälp när det gäller att demonstrera lämpligheten hos dossystemet.
Kontrollgruppen ska vara en obehandlad grupp eller en vehikelkontrollgrupp, om vehikel används för tillförsel av testämnet. Med undantag för behandling med testämnet bör djuren i kontrollgruppen hanteras på exakt samma sätt som djuren i dosgrupperna. Om en vehikel används bör kontrollgruppen få vehikeln i den största volym som används. Om testämnet tillförs med fodret och detta leder till minskat foderintag eller minskad foderanvändning, kan det vara nödvändigt med en parallellt utfodrad kontrollgrupp. Alternativt kan man i stället för en sådan kontrollgrupp använda data från kontrollerade test för utvärdering av hur ett minskat foderintag påverkar reproduktionsparametrarna.
Följande egenskaper hos vehikeln och övriga eventuella tillsatsämnen bör i tillämplig mån beaktas: Verkningarna på absorption, distribution, metabolism eller kvarhållande av testämnet, verkningarna på testämnets kemiska egenskaper till den del dessa verkningar kan påverka testämnets toxiska egenskaper samt verkningarna på testdjurens foder- eller vattenintag eller deras näringstillstånd.
1.4.4 Gränstest (Limit-test)
Om ett oralt test med en dosnivå om minst 1 000 mg/kg kroppsvikt per dag (eller, om testämnet tillförs med foder eller dricksvatten, en motsvarande procenthalt i foder eller dricksvatten med användning av de förfaranden som beskrivs för detta test) inte leder till några observerbara toxiska verkningar varken hos för-äldradjuren eller deras avkomma, och om toxicitet inte kan förväntas på grundval av data från strukturellt eller metaboliskt närbesläktade ämnen, kan ett fullständigt test med flera dosnivåer bedömas vara överflödigt. Detta gränstestkriterium är tillämpligt förutom i fall där sannolik exponering på människor indikerar att en högre oral dosnivå behövs. För andra former av tillförsel, såsom inandning eller applicering på huden, kan testämnets fysikalisk-kemiska egenskaper, såsom löslighet, ofta indikera och begränsa den maximala exponeringsnivån.
1.4.5 Tillförsel av doser
Djuren ska ges doser av testämnet alla dagar i veckan. Tillförsel på oral väg (foder, dricksvatten eller sond-matning) rekommenderas. Om någon annan tillförselväg används måste detta motiveras och tillbörliga justeringar kan vara nödvändiga. Samma tillförselmetod ska användas för alla djur under den valda testperioden. Om testämnet tillförs genom sondmatning bör magsond användas. Den volym vätska som tillförs på en och samma gång får inte överskrida 1 ml per l00 g kroppsvikt (0,4 ml per 100 g kroppsvikt om majsolja används), utom för vattenlösningar där volymen kan uppgå till 2 ml per 100 g kroppsvikt. Med undantag för irriterande eller frätande ämnen som normalt ger kraftigare verkan med högre koncentration bör variationer i testvolym minimeras genom justering av koncentrationen, för att säkerställa en konstant volym på alla dosnivåer. Om tillförsel genom sondmatning används får ungarna normalt testämnet endast indirekt genom mjölken, tills direkt tillförsel inleds efter avvänjningen. Om tillförsel via foder eller vatten används kommer ungarna dessutom att få testämnet direkt så snart de börjar äta själva under den sista veckan av diperioden.
För ämnen som tillförs via fodret eller dricksvattnet är det viktigt att försäkra sig om att testämnet ges i mängder som inte stör det normala näringsupptaget eller den normala vattenbalansen. Testämne som tillförs via fodret kan ges som en konstant koncentration (ppm) eller en konstant dosnivå i förhållande till djurens kroppsvikt. Det använda alternativet ska anges. Testämne som tillförs genom sondmatning bör ges vid samma tid varje dag, och justeras minst varje vecka så att dosnivån hålls konstant i förhållande till djurens kroppsvikt. Informationen om placentadistribution bör beaktas när sondmatade doser justeras enligt vikten.
1.4.6 Testschema
Daglig dosering av föräldragenerationen (P) inleds när djuren är mellan fem och nio veckor gamla. Daglig dosering av F1-generationens han- och hondjur inleds efter avvänjningen. Om testämnet tillförs med fodret eller dricksvattnet bör hänsyn tas till att F1-ungarna redan under diperioden kan utsättas för direkt exponering av testämnet. Tillförseln ska fortsätta för båda könen (P och F1) under minst 10 veckor före parningsperioden. Tillförseln fortsätts för båda könen under den två veckor långa parningsperioden. Handjuren avlivas humant och undersöks när de inte längre behövs för bedömningen av reproduktionstoxiska verkningar. Tillförseln till föräldragenerationens (P) hondjur fortsätts under hela dräktigheten och fram till det att F1-avkomman har avvants. Eventuella justeringar av testschemat bör övervägas om känd information om testämnet indikerar detta, t.ex. toxicitetsdata, induktion av metabolism eller bioackumulering. Dosen till varje djur bör i regel basera sig på den senaste bestämningen av djurets kroppsvikt. Försiktighet bör dock iakttas när dosen justeras under dräktighetens sista tredjedel.
Behandlingen av P- och F1-generationens handjur och hondjur fortsätts fram till det att testet avslutas. Alla P- och F1-generationens handjur och hondjur avlivas humant när de inte längre behövs för bedömningen av reproduktionstoxiska verkningar. F1-avkomma som inte väljs ut för parning och all F2-avkomma avlivas humant efter avvänjningen.
1.4.7 Parning
1.4.7.1 Parning i föräldragenerationen (P)
För parningen sammanförs ett hondjur med ett ensamt handjur från samma dosnivå (parning i förhållandet 1:1) tills kopulation inträffar eller 2 veckor har gått. Hondjuren undersöks varje dag för närvaron av sperma eller vaginalproppar. Som dräktighetens dag 0 räknas den dag då en vaginalpropp eller sperma observeras. Om parningen inte lyckas kan man överväga ett nytt parningsförsök där hondjuret sammanförs med ett handjur med fastställd fertilitet från samma grupp. De djur som har parat sig bör identifieras entydigt i testets data. Parning av syskon bör undvikas.
1.4.7.2 F1-parning
För parning av F1-avkomman väljs minst ett handjur och ett hondjur från varje kull efter avvänjningen, vilka korsas med ungar ur en annan kull på samma dosnivå. På detta sätt produceras F2-generationen. Ungarna väljs ut slumpmässigt ur varje kull i fall där inga signifikanta skillnader kan observeras rörande kroppsvikt eller utseende mellan ungarna i en kull. Om skillnader kan observeras bör de djur som bäst representerar kullen väljas ut. I praktiken görs detta bäst på grundval av kroppsvikt, även om det i vissa fall kan vara lämpligare att använda utseendet som grund. F1-avkomman ska inte paras förrän den har nått fullständig sexuell mognad.
Par utan avkomma ska undersökas i syfte att fastställa uppenbara orsaker till infertiliteten. Detta kan göras t.ex. genom att djuren får möjlighet att para sig med han- eller hondjur vars fertilitet är fastställd, genom mikroskopisk undersökning av reproduktionsorganen eller genom undersökning av brunstighetspe-rioden eller spermatogenesen.
1.4.7.3 Andra parningen
I vissa fall, t.ex. vid behandlingsrelaterade förändringar i kullstorlek eller om entydig effekt av den första parningen inte kan observeras, rekommenderas att vuxna P- eller F1-djur får para sig på nytt för att producera en ny kull. För denna andra parning rekommenderas korsning av djur som inte har producerat någon kull, med hondjur och handjur vars fertilitet är fastställd. Om en ny kull bedöms vara nödvändig i någondera generationen, bör den andra parningen ske cirka en vecka efter det att föregående kull har avvants.
1.4.7.4 Kullstorlek
Djuren får föda normalt och hålla sin avkomma tills den är avvand. Standardisering av kullarna är inte obligatorisk. Om standardisering görs, bör den metod som används beskrivas i detalj.
1.5 OBSERVATIONER
1.5.1 Kliniska observationer
En allmän klinisk observation bör göras varje dag. Om testämnet tillförs genom sondmatning bör observationstidpunkten väljas med beaktande av perioden för när maximala verkningar förväntas efter tillförseln. Avvikande beteende, tecken på svår eller förlängd födsel och alla tecken på toxicitet bör registreras. Dessutom bör alla djur inspekteras närmare minst en gång per vecka, lämpligen i samband med vägningen. Två gånger per dag (under veckosluten en gång per dag, om lämpligt) bör alla djur inspekteras för morbiditet och mortalitet.
1.5.2 Föräldradjurens kroppsvikt samt foder- och vattenkonsumtion
Föräldradjuren (P och Fl) ska vägas på den första doseringsdagen och därefter minst varje vecka. Föräldragenerationernas hondjur (P och F1) ska vägas minst på dräktighetsdagarna 0, 7, 14 och 20 eller 21, under diperioden på samma dagar då kullarna vägs och slutligen på den dag då djuren avlivas. Dessa observationer bör rapporteras individuellt för varje vuxet djur. Före parningen och under diperioden ska foderkonsumtionen mätas minst varje vecka. Vattenkonsumtionen ska mäta minst varje vecka om testämnet tillförs med vattnet.
1.5.3 Brunstighetscykel
Brunstighetscykelns längd och normalitet utvärderas hos P- och F1-hondjuren genom vaginala smear-test före parningen och eventuellt under parningsperioden fram till det att tecken på parning kan observeras. Tagningen av vaginala eller cervikala celler bör göras med försiktighet för att undvika störningar på slemhinnan och induktion av skendräktighet (1).
1.5.4 Spermaparametrar
Vid testets slut registreras för alla P- och F1-handjur vikten för testiklar och bitestiklar. Ett exemplar av varje organ bör bevaras för histopatologisk undersökning (se avsnitt 1.5.7, 1.5.8.1). Återstående testiklar och bitestiklar från en undergrupp med minst tio handjur från varje grupp av P- och F1-handjur används för räkning av homogeniseringsresistenta spermatider respektive cauda epididymala spermareserver. Från samma undergrupp av handdjur insamlas sperma från cauda epididymis eller vas deferens för utvärdering av spermans motilitet och morfologi. Om behandlingsrelaterade verkningar observeras eller om resultaten från andra test tyder på verkningar på spermatogenes, ska sperman utvärderas för alla handjur i varje dosgrupp. I annat fall kan räkningen begränsas till P- och F1-handjuren i kontroll- och högdosgruppen.
Totalantalet homogeniseringsresistenta testikulära spermatider och cauda epididymala spermier bör räknas (2)(3). Cauda-spermareserverna kan härledas från koncentrationen och volymen hos sperman i den suspension som används för de kvalitativa bedömningarna och från antalet spermier som räknats efter påföljande malning eller homogenisering av återstående cauda-vävnad. Räkningen bör utföras omedelbart efter avlivningen för den valda undergruppen av handjur från alla dosgrupper. Undantag till detta är om man gör video- eller digitalregistreringar eller om proverna djupfryses och analyseras senare. I sådana fall kan kontroll- och högdosgruppen analyseras först. Om inga behandlingsrelaterade verkningar (t.ex. på spermiernas antal, motilitet eller morfologi) kan observeras, behöver de övriga dosgrupperna inte analyseras. Om behandlingsrelaterade verkningar kan observeras i högdosgruppen bör även grupperna med lägre doser utvärderas.
Motiliteten hos epididymal sperma (eller ductus deferens-sperma) bör bedömas eller videofilmas omedelbart efter avlivningen. Sperman bör skördas på sådant sätt att skador minimeras. Utspädningen av sperman för motilitetsanalysen bör göras med användning av godtagbara metoder (4). Procentandelen progressivt motila spermier bestäms subjektivt eller objektivt. Om datorstödd motilitetsanalys används (5)(6)(7)(8)(9)(10) görs härledningen av den progressiva motiliteten på grundval av användardefinierade trösklar för genomsnittlig banhastighet och linearitetsindex. Om proverna videofilmas (11) eller om bilderna registreras på annat sätt vid obduktionen, kan den efterföljande analysen begränsas till P- och F1-handjuren i kontroll- och högdosgruppen. Undantag till detta är om behandlingsrelaterade verkningar kan observeras, i vilket fall även grupperna med lägre doser bör utvärderas. Om det inte förekommer video-eller digitalbildsregistrering måste alla prover i alla behandlingsgrupper analyseras vid obduktionen.
Ett spermaprov från epididymis (eller vas deferens) utvärderas morfologiskt. Spermierna (minst 200 per prov) bör undersökas i form av fixerade våta preparat (12) och klassificeras som normala eller abnormala. Exempel på morfologiska abnormaliteter hos spermier är fusion, isolerade huvuden och missformade huvuden eller svansar. Utvärderingen bör göras omedelbart efter avlivningen för den valda undergruppen av handjur från alla dosgrupper. Om det finns video- eller digitalregistreringar kan utvärderingen göras senare. Även cellproverna från smear-test kan, om de fixeras, avläsas senare. I sådana fall kan kontroll- och högdosgruppen analyseras först. Om inga behandlingsrelaterade verkningar (t.ex. på spermans morfologi) kan observeras behöver de övriga dosgrupperna inte analyseras. Om behandlingsrelaterade verkningar kan observeras i högdosgruppen bör även grupperna med lägre doser utvärderas.
Om någon av de ovan nämnda spermabedömningsparametrarna har undersökts tidigare som ett led i ett systematiskt toxicitetstest som varat minst 90 dagar, behöver bedömningen inte nödvändigtvis upprepas i tvågenerationstestet. Det rekommenderas dock att prover eller digitala registreringar av P-generationens sperma bevaras, så att en utvärdering vid behov kan göras senare.
1.5.5 Avkomma
Varje kull undersöks så snart som möjligt efter födseln (diperiodens dag 0) för att fastställa antalet ungar och deras kön, antalet dödfödda ungar, antalet levande ungar och förekomsten av betydande anomalier. Ungar som konstateras vara döda på dag 0 bör helst, om de är tillräckligt intakta, undersökas för eventuella defekter och dödsorsak, samt bevaras. De levande ungarna räknas och vägs individuellt efter födseln (diperiodens dag 0) eller på dag 1. Därefter vägs ungarna på de reguljära vägningsdagarna, t.ex. diperiodens dagar 4, 7, 14 och 21. Fysiska eller beteendemässiga abnormaliteter hos moderdjuren eller avkomman bör registreras.
Avkommans fysiska utveckling bör registreras, främst genom ökningen i kroppsvikt. Övriga fysiska parametrar (t.ex. öron- och ögonöppningar, tandsprickning, hårväxt) kan ge tilläggsinformation, men dessa data bör helst utvärderas i samband med data om sexuell mognad (t.ex. ålder och kroppsvikt när vagina öppnas eller när balano-preputial separation sker) (13). Funktionella undersökningar (t.ex. motorisk aktivitet, sensorisk funktion, reflex ontogeni) hos F1-avkomman före och/eller efter avvänjningen, särskilt faktorer som hör samman med den sexuella mognaden, rekommenderas om sådana undersökningar inte ingår i separata test. Åldern då vagina öppnas och preputial separation sker bör bestämmas för de avvanda F1-ungar som väljs ut för parning. Anogenitalt avstånd mäts på dag 0 efter födseln hos F2-ungar, om detta bedöms vara nödvändigt på grund av att F1-generationen har avvikelser rörande kvoten mellan könen eller tidpunkten för sexuell mognad.
De funktionella observationerna kan utelämnas för grupper som på annat sätt uppvisar tecken på skadliga verkningar (t.ex. betydande minskning av viktökningen). Om funktionella undersökningar görs, ska de inte omfatta de ungar som har valts ut för parning.
1.5.6 Obduktion
När testet avslutas eller om djur dör under testets gång, ska alla föräldradjur (P och F1), alla ungar med externa abnormaliteter eller kliniska tecken samt en slumpmässigt vald unge per kön och kull från både F1- och F2-generationen undersökas makroskopiskt för eventuella strukturella abnormaliteter eller patologiska avvikelser. Särskild uppmärksamhet bör fästas vid reproduktionssystemets organ. Döende ungar som avlivas humant eller döda ungar bör, förutsatt att de är tillräckligt intakta, undersökas för eventuella defekter och dödsorsak, samt bevaras.
Livmodern hos alla förstfödande hondjur ska undersökas på ett sätt som inte stör den histopatologiska utvärderingen, för förekomsten av implantationer och deras antal.
1.5.7 Organvikter
Vid testets slut bestäms kroppsvikten och vikten hos följande organ för alla P- och F1-föräldradjur (pariga organ vägs individuellt):
|
— |
Livmoder, äggstockar. |
|
— |
Testiklar, bitestiklar (totalt och cauda). |
|
— |
Prostata. |
|
— |
Sädesblåsa med koaguleringskörtlar och deras vätskor samt prostatan (som en enhet). |
|
— |
Hjärna, lever, njurar, mjälte, hypofys, sköldkörtel och adrenalinkörtel samt kända målorgan. |
Kroppsvikten vid testets slut bestäms för de F1- och F2-ungar som väljs ut för obduktion. Vikten bestäms även för följande organ från den slumpmässigt valda ungen per kön per kull (se avsnitt 1.5.6): hjärna, mjälte och bisköldkörtel.
Resultaten från obduktionen och vägningen av organ bör i tillämplig mån bedömas med hänsyn till observationer från andra test med upprepad dos.
1.5.8 Histopatologi
1.5.8.1 Föräldradjur
Följande organ och vävnader (eller representativa prover) från föräldradjuren (P och F1) ska fixeras och förvaras i ett lämpligt medium för histopatologisk undersökning:
|
— |
Vagina, livmoder med livmoderhals samt äggstockar (bevarade i adekvat fixervätska). |
|
— |
En testikel (bevarad i Bouins fixervätska eller motsvarande), en bitestikel, sädesblåsa, prostata och koaguleringskörtel. |
|
— |
Tidigare identifierade målorgan från alla P- och F1-djur som har valts ut för parning. |
Fullständig histopatologi görs av de ovan uppräknade bevarade organen och vävnaderna för alla sådana P-och F1-djur i högdos- och kontrollgruppen som har valts ut för parning. Undersökning av äggstockarna hos P-djuren är inte obligatorisk. Organ som har behandlingsrelaterade avvikelser bör även undersökas i låg- och medeldosgrupperna som stöd för bestämningen av NOAEL. Dessutom bör histopatologisk undersökning göras på reproduktionsorganen från sådana djur i låg- och medeldosgruppen som misstänks för reducerad fertilitet, t.ex. djur som inte parade sig, inte befruktades, inte avlade, eller djur som inte födde frisk avkomma. Alla betydande skador såsom atrofi eller tumörer bör undersökas.
Detaljerad histopatologisk undersökning görs på testiklarna (t.ex. med användning av Bouins fixervätska, paraffininbäddning och transverssektioner av 4–5 μm tjocklek) för att identifiera behandlingsrelaterade verkningar såsom tillbakabildade spermatider, uteblivna germinalcellslager eller -typer, multinukleära jätteceller eller degraderingen av spermatogeniska celler in i lumen (14). Undersökning av den intakta bitesti-keln ska inbegripa caput, corpus och cauda, vilket kan åtföljas av en utvärdering av en longitudinell sektion. Bitestikeln utvärderas för leukocytinfiltration, förändringar i prevalensen av celltyper, avvikande celltyper och spermafagocytos. PAS och hematoxylinfärgning kan användas för undersökning av handjurens reproduktionsorgan.
Äggstockarna efter diperioden bör innehålla primordiala och växande folliklar samt de stora corpora lutea från diperioden. Den histopatologiska undersökningen bör detektera kvalitativ utarmning av den primordiala follikelpopulationen. En kvantitativ utvärdering av de primordiala folliklarna görs för F1-hondjuren. Antalet djur, valet av äggstockssektion och sektionens storlek bör vara statistiskt lämpliga för det utvärderingsförfarande som används. Utvärderingen bör inbegripa räkning av antalet primordiala folliklar, som kan kombineras med små växande folliklar, för jämförelse av äggstockarna hos behandlinggruppernas och kontrollgruppens djur (15)(16)(17)(18)(19).
1.5.8.2 Avvanda ungar
Grovt abnormala vävnader och målorgan från alla ungar med externa abnormaliteter eller kliniska tecken, samt från en slumpmässigt vald unge per kön och per kull i både F1- och F2-generationen (som inte valts ut för parning), ska fixeras och förvaras i ett lämpligt medium för histopatologisk undersökning. Fullständig histopatologisk karakterisering av bevarade vävnader bör göras, med särskild tonvikt på reproduktionssystemets organ.
2. DATA
2.1 BEHANDLING AV RESULTATEN
Data rapporteras individuellt för moderdjuren och deras avkomma och sammanställs i en tabell. Tabellen ska för varje testgrupp och varje generation visa antalet djur vid testets början, antalet djur som under testets gång har dött eller avlivats av humana skäl, tidpunkten för alla dödsfall eller humana avlivningar, antalet fertila djur, antalet dräktiga hondjur, antalet djur som uppvisar tecken på toxicitet, en beskrivning av de toxicitetstecken som har observerats (tidpunkten då sådana först upptäcktes, de toxiska verkningarnas varaktighet och allvarlighetsgrad), typerna av observationer om föräldradjur och avkomma, typerna av histopatologiska avvikelser och alla relevanta data om kullarna.
De numeriska resultaten ska utvärderas med en lämplig och allmänt erkänd statistisk metod. Valet av statistiska metoder bör ingå som en del av testets utformning och bör motiveras. Statistiska dos-responsmodeller kan vara användbara för analysen av data. Rapporten ska innehålla tillräckligt med information om analysmetoden och det datorprogram som används, så att en oberoende granskare eller statistiker kan göra en ny utvärdering och rekonstruera analysen.
2.2 UTVÄRDERING AV RESULTATEN
Resultaten från detta test för reproduktionstoxicitet på två generationer ska utvärderas med avseende på de observerade verkningarna, inbegripet resultaten från obduktion och mikroskopiska undersökningar. Utvärderingen ska omfatta förhållandet mellan, eller avsaknaden därav, mellan testämnesdosen och närvaron eller frånvaron, incidensen och allvarlighetsgraden för abnormaliteter, inbegripet grova skador, identifierade målorgan, verkningar på fertiliteten, kliniska abnormaliteter, verkningar på reproduktionsförmågan och kullen, avvikelser rörande kroppsvikt, verkningar på mortaliteten samt alla andra toxiska verkningar. Vid utvärdering av testresultaten bör hänsyn tas till testämnets fysikalisk-kemiska egenskaper och de toxikokinetiska data som är kända.
Ett korrekt genomfört test för reproduktionstoxicitet bör inbegripa en tillfredsställande uppskattning av den nivå där inga verkningar observeras och ge en förståelse för de skadliga verkningarna på reproduktion, födsel och digivning, samt på den postnatala utvecklingen inklusive tillväxt och sexuell utveckling.
2.3 TOLKNING AV RESULTATEN
Ett test för reproduktionstoxicitet på två generationer ska ge information om verkningarna av upprepad exponering för ett ämne under alla faser av reproduktionscykeln. Testet ska särskilt ge information om reproduktionsparametrarna samt om avkommans utveckling, tillväxt, mognad och överlevnad. Testresultaten ska tolkas tillsammans med resultaten från test avseende subkronisk toxicitet, prenatal utvecklingstoxicitet och toxikokinetik samt andra kända testresultat. Resultaten från detta test kan användas för att bedöma behovet av ytterligare testning av en kemikalie. Extrapolering av testresultaten till att gälla för exponering på människor kan i viss utsträckning godtas. Resultaten kan bäst tillämpas på information om dosnivåer där inga verkningar kan observeras och information om tillåten exponering på människor (20)(21)(22)(23).
3. RAPPORTERING
3.1 TESTRAPPORT
Testrapporten ska innehålla nedan angivna uppgifter.
|
|
Testämne:
|
|
|
Vehikel (i förekommande fall).
|
|
|
Testdjur:
|
|
|
Testbetingelser:
|
|
|
Resultat:
|
|
|
Diskussion om resultaten. |
|
|
Slutsatser, inbegripet värden för den lägsta nivå där inga verkningar kan observeras (NOAEL) hos moderdjur och avkomma. |
4. HÄNVISNINGAR
|
1. |
Sadleir, R.M.F.S. (1979), ”Cycles and Seasons”, Reproduction in Mammals, I. Germ Cells and Fertilization, C.R. Auston och R.V. Short (red.), Cambridge, New York. |
|
2. |
Gray, L.E. et al. (1989), ”A Dose-Response Analysis of Methoxychlor-Induced Alterations of Reproductive Development and Function in the Rat”, Fundamental and Applied Toxicology, 12:92–108. |
|
3. |
Robb, G.W. et al. (1978), ”Daily Sperm Production and Epididymal Sperm Reserves of Pubertal and Adult Rats”, Journal of Reproduction and Fertility, 54:103–107. |
|
4. |
Klinefelter, G.R. et al. (1991), ”The Method of Sperm Collection Significantly Influences Sperm Motion Parameters Following Ethane Dimethanesulfonate Administration in the Rat”, Reproductive Toxicology, 5:39–44. |
|
5. |
Seed, J. et al. (1996), ”Methods for Assessing Sperm Motility, Morphology, and Counts in the Rat, Rabbit, and Dog: a Consensus Report”, Reproductive Toxicology, 10(3):237–244. |
|
6. |
Chapin, R.E. et al. (1992), ”Methods for Assessing Rat Sperm Motility”, Reproductive Toxicology, 6:267–273. |
|
7. |
Klinefelter, G.R. et al. (1992), ”Direct Effects of Ethane Dimethanesulphonate on Epididymal Function in Adult Rats: an In Vitro Demonstration”, Journal of Andrology 13:409–421. |
|
8. |
Slott, V.L. et al. (1991), ”Rat Sperm Motility Analysis: Methodologic Considerations”, Reproductive Tox icology, 5:449–458. |
|
9. |
Slott, V.L. och Perreault, S.D. (1993), ”Computer-Assisted Sperm Analysis of Rodent Epididymal Sperm Motility Using the Hamilton-Thorn Motility Analyzer”, Methods in Toxicology, Part A., Academic, Orlando, Florida, 319–333. |
|
10. |
Toth, G.P. et al. (1989), ”The Automated Analysis of Rat Sperm Motility Following Subchronic Epichlorhydrin Administration: Methodologic and Statistical Considerations”, Journal of Andrology, 10:401–415. |
|
11. |
Working, P.K. och M. Hurtt (1987), ”Computerized Videomicrographic Analysis of Rat Sperm Motility”, Journal of Andrology, 8:330–337. |
|
12. |
Linder, R.E. et al. (1992), ”Endpoints of Spermatoxicity in the Rat After Short Duration Exposures to Fourteen Reproductive Toxicants”, Reproductive Toxicology, 6:491–505. |
|
13. |
Korenbrot, C.C. et al. (1977), ”Preputial Separation as an External Sign of Pubertal Development in the Male Rat”, Biological Reproduction, 17:298–303. |
|
14. |
Russell, L.D. et al. (1990), ”Histological and Histopathological Evaluation of the Testis”, Cache River Press, Clearwater, Florida. |
|
15. |
Heindel, J.J. och R.E. Chapin, (red.) (1993), ”Part B. Female Reproductive Systems, Methods in Toxico logy”, Academic, Orlando, Florida. |
|
16. |
Heindel, J.J. et al. (1989), ”Histological Assessment of Ovarian Follicle Number in Mice As a Screen of Ovarian Toxicity, Growth Factors and the Ovary”, A.N. Hirshfield (red.), Plenum, New York, 421–426. |
|
17. |
Manson, J.M. och Y.J. Kang, (1989), ”Test Methods for Assessing Female Reproductive and Developmental Toxicology”, Principles and Methods of Toxicology, A.W. Hayes (red.), Raven, New York. |
|
18. |
Smith, B.J. et al. (1991), ”Comparison of Random and Serial Sections in Assessment of Ovarian Toxicity”, Reproductive Toxicology, 5:379–383. |
|
19. |
Heindel, J.J. (1999), ”Oocyte Quantitation and Ovarian Histology. In: An Evaluation and Interpretation of Reproductive Endpoints for Human Health Risk Assessment”, G. Daston och C.A. Kimmel, (red.), ILSI Press, Washington, DC. |
|
20. |
Thomas, J. A. (1991), ”Toxic Responses of the Reproductive System”, Casarett and Doull's Toxicology, M.O. Amdur, J. Doull och C.D. Klaassen (red.), Pergamon, New York. |
|
21. |
Zenick, H. och E.D. Clegg (1989), ”Assessment of Male Reproductive Toxicity: A Risk Assessment Approach”, Principles and Methods of Toxicology, A.W. Hayes (red.), Raven Press, New York. |
|
22. |
Palmer, A.K. (1981), Developmental Toxicology, Kimmel, C.A. och J. Buelke-Sam (red.), Raven Press, New York. |
|
23. |
Palmer, A.K. (1978), Handbook of Teratology, Vol. 4, J.G. Wilson och F.C Fraser (red.), Plenum Press, New York. |
B.36 TOXIKOKINETIK
1. METOD
1.1 INLEDNING
Se allmän inledning till del B.
1.2 DEFINITIONER
Se allmän inledning till del B.
1.3 REFERENSÄMNEN
Inga.
1.4 TESTMETODSPRINCIP
Testämnet tillförs på ett passande sätt. Beroende på syftet med försöket kan testämnet ges i en enkel dos eller i flera doser fördelat över fastställda perioder till en eller flera grupper av försöksdjur. Beroende på vilken typ av undersökning som utförs fastställs ämnet och/eller dess metaboliter i kroppsvätska, vävnader och/eller exkret.
Försöket kan ske med ’radioaktivt markerat’ eller ’omarkerat’ testämne. Om radioaktivt markerat testämne används ska detta ingå i en sådan form i testämnet det ger största möjliga antal upplysningar om ämnets öde.
1.5. KVALITETSKRITERIER
Inga.
1.6. METODBESKRIVNING
Förberedelser
I minst fem dagar före försöket ska unga friska djur acklimatiseras till testbetingelserna. Före försöket fördelas djur slumpmässigt i behandlingsgrupper. I vissa fall kan mycket unga, dräktiga eller förbehandlade djur användas.
Försöksbetingelser
Försöksdjur
Toxikokinetiska försök kan utföras på en eller flera lämpliga djurarter och bör ta hänsyn till vilka djurarter man har använd eller planerar att använda i andra toxikologiska undersökningar av samma testämne. Om gnagare använts får djurens vikt vid inledningen av försöket inte avvika mer än ± 20 % från genomsnittsvikten.
Antal och kön
Vid undersökning av absorption och utsöndring används fyra djur i varje doseringsgrupp. Inga krav ställs på djurens kön men i vissa fall kan det vara nödvändigt att använda djur av båda könen. Om könstbetingade skillnader uppträder ska fyra djur av varje kön användas. I försök med icke-gnagare kan ett mindre antal djur användas.
Vid undersökning av fördelning i vävnader fastställs den storlek som försöksgrupperna ska ha vid försökets inledning, med hänsyn till hur många djur som ska avlivas vid på förhand fastställda tidspunkter och hur många sådana tidpunkter försöket ska omfatta.
Vid undersökning av metabolism fastställs gruppstorleken efter behov.
Vid undersökning med flera doser och flera undersökningstidpunkter fastställs gruppstorleken med hänsyn till planenlig avlivning och antal undersökningstidpunkter. Varje grupp måste dock bestå av minst två djur. Gruppstorleken ska vara tillräcklig för att korrekt kunna få fram data om absorption, koncentrationsnivåer och uttömning (i tillämpliga fall) av testämnet och/eller dess metaboliter.
Dosnivåer
När testämnet ges i en enkel dos används minst två olika doseringsnivåer. Det bör finnas en lågdosnivå vid vilken ingen toxisk verkan observeras, och en högdosnivå där det kan vara tal om tox-lkokinetiska parametrar eller toxiska verkningar.
Om testämnet ges i flera doser räcker det normalt att använda den låga dosen. 1 vissa fall kan det dock vara nödvändigt att även använda en hög dos.
Tillförselväg
Vid toxikokinetiska undersökningar ges testämnet på samma sätt och om möjligt med hjälp av samma vehikel som i redan företagna eller planerade toxikologiska undersökningar av samma testämne. Normalt tillförs testämnet antingen oralt med magsond eller via fodret, genom påföring på huden eller genom inhalation under på förhand fastställda perioder till grupper av försöksdjur. Intravenös tillförsel av testämnet kan vara användbart för att bestämma relativ absorption genom andra tillförselvägar. Dessutom kan nyttig information komma fram om distnbutionsmönstret kort efter intravenös tillförsel av ett ämne.
Man bör ta hänsyn till möjligheten att vehikeln påverkar testämnet. Samtidigt måste man vara uppmärksam på skillnader i absorption mellan tillförsel av testsubsansen via magsond och via fodret och att det därför behövs en exakt bestämning av dosens, särskilt när testämnet tillförs via fodret.
Observationsperiod
Samtliga djur ska observeras dagligen och tecken på toxicitet och andra kliniska symptom ska registreras, inbegripet när de uppträdde för första gången, graden och varaktigheten.
Förfarande
Sedan försöksdjuren vägts tillförs testämnet på lämpligt sätt.. Vid behov kan kan man låta djuren fasta innan testämnet tillförs.
Absorption
Den mängd av den tillförda ämnet som absorberas, och den hastigheten med vilken den absorberas, kan värderas med hjälp av olika metoder med eller utan referensgruppen (8), t.ex. genom
|
— |
bestämning av mängden testämne och/eller dess metaboliter i exkret, t.ex. i urin, galla, avföring, utandningsluft samt i kroppen efter avlivning, |
|
— |
jämförelse av biologisk verkan (t.ex. i undersökningar av akut toxicitet) i försöks-, kontroll- och referensgrupper, |
|
— |
jämförelse av mängden av testämne och/eller dess metaboliter som utsöndras genom njurarna i försöks- och referensgrupper, |
|
— |
bestämning av arean under kurvan för testämnets plasmakoncentration och/eller dess metabo liter som en funktion av tiden och jämförelse med data från en referensgrupp. |
Distribution
Det är i dag möjligt att analysera distributionsmönstret på två olika sätt:
|
— |
Kvalitativ information erhålls genom användning av helkroppsautoradiografisk teknik. |
|
— |
Kvantitativ information erhålls genom avlivning av djur vid olika tidpunkter efter exponering och bestämning av mängd och koncentration av testämne och/eller dess metaboliter i vävnader och organ. |
Utsöndring
Vid undersökning av utsöndring uppsamlas urin, avföring och utandningsluft samt, i vissa fall, galla. Mängden av testämne och/eller dess metaboliter i dessa exkret bör mätas flera gånger efter exponering, tills ca 95 % av den tillförda dosen har utsöndrats, dock högst i sju dagar.
I vissa fall kan det vara nödvändigt att ta hänsyn till utsöndring av testämne i mjölken från digivande försöksdjur.
Metabolism
För att bestämma metabolismens intensitet och mönster analyseras biologiska prover med hjälp av en för detta syfte lämplig metod. Om frågor som uppstått vid tidigare toxikologiska undersökningar måste besvaras klargörs metaboliternas struktur och förslag ges om möjliga metabolismvägar. Undersökningar in vitro kan bidra till att skaffa fram upplysningar om metabolismvägar.
Ytterligare information om sambanden mellan metabolism och toxisk verkan kan fås genom biokemiska undersökningar, t.ex. genom bestämning av verkningar på metaboliserande enzymsystem, reduktion av mängden endogena icke-protein-sulfhydrylförbindelser och testämnet bindning til! makromolekyler.
2. DATA
Beroende på vilken slags undersökning som utförts ska data ställas upp i tabellform och stödjas, när så är möjligt, av grafiska framställningar. Om det är möjligt ska för varje försöksgrupp anges medelvärde och statistisk variation av alla mätningar i förhållande till tid, doser, vävnader och organ. Den absorberade och utsöndrade mängden samt utsöndringens hastighet bestäms genom för detta syfte lämpliga metoder. Vid undersökningar av metabolism klargörs de identifierade metaboliternas struktur och möjliga metabolismvägar läggs fram.
3. RAPPORTERING
3.1 TESTRAPPORT
Testrapporten ska om möjligt innehålla minst följande uppgifter:
|
— |
Djurart, stam, härkomst, miljöbetingelser, foder. |
|
— |
Beskrivning av markerade ämnen, om sådana har använts. |
|
— |
Dosmväer och använda intervaller. |
|
— |
Tillförselväg och eventuella vehikler. |
|
— |
Toxisk och annan verkan. |
|
— |
Metoder för bestämning av testamne och/eller dess metaboliter i biologiska prover, inbegripet inhaltionsluft. |
|
— |
Mätresultat uppställda i tabellform efter kön, doser, andra testparametrar, tid, vävnader och organ. |
|
— |
Presentation av den avsorberade och utsöndrade mängden som en funktion av tiden. |
|
— |
Metoder för att karakterisera och identifiera metaboliter i biologiska prov. |
|
— |
Metoder för biokemiska mätningar i sambnad med metabolism |
|
— |
Förslag av väg för metabolism. |
|
— |
Diskussion om resultaten. |
|
— |
Tolkning av resultaten. |
3.2 UTVÄRDERING OCH TOLKNING
Se allmän inledning till del B.
4. HÄNVISNINGAR
Se allmän inledning till del B.
B.37 FÖRDRÖJD NEUROTOXICITET ORSAKAD AV ORGANISKA FOSFORFÖRENINGAR TILL FÖLJD AV AKUT EXPONERING
1. METOD
1.1 INLEDNING
Vid bedömning och utvärdering av ämnens toxiska effekter är det viktigt att beakta potentialen hos substanser i vissa ämnesgrupper att framkalla specifika typer av neurotoxicitet som kanske inte upptäcks vid andra toxicitetsundersökningar. Det har observerats att vissa organiska fosforföreningar framkallar fördröjd neurotoxicitet och dessa ska beaktas för eventuell utvärdering.
Screening-tester in vitro får tillämpas för att identifiera de ämnen som kan framkalla fördröjd polyneuropati; negativa resultat från in vitro-undersökningar bevisar dock inte att testsubstansen inte skulle vara neurotoxisk.
Se Allmän inledning, del B.
1.2 DEFINITIONER
Organiska fosforföreningar omfattar icke joniserande organiska fosforestar, tioestrar eller anhydrider av organofosfor-, organofosforo- eller organofosforamidsyror eller av besläktade fosfonthio-, fosforthio- eller fosforthioamidsyror eller andra ämnen som kan framkalla sådan fördröjd neurotoxicitet som ibland observeras i denna ämnesgrupp.
Fördröjd neurotoxicitet är ett syndrom som förknippas med förlängd fördröjd debut för ataxi, distala axonopatier i ryggmärgen och perifera nerver samt hämmande och åldrande av neurotoxiskt esteras (NTE) i nervvävnad.
1.3 REFERENSÄMNEN
Ett referensämne ska testas i en positiv kontrollgrupp för att påvisa att den testade artens respons inte ändras väsentligt under försöksförhållandena i laboratoriet.
Ett exempel på en mycket använd neurotoxisk substans är trio-tolylfosfat (CAS 78-30-8, EINECS 201-103-5, CAS-namn: fosforsyra, tris(2-metyl-fenyl)ester), även kallad tris-o-kresylfosfat.
1.4 PRINCIP FÖR TESTMETODEN
Testsubstansen tillförs oralt i en enda dos till tamhöns som i tillämpliga fall skyddats från akuta kolinerga effekter. Djuren observeras under 21 dagar i fråga om beteendestörningar, ataxi och förlamning. Biokemiska mätningar, särskilt inhibition av neurotoxiskt esteras (NTE) utförs på höns som väljs slumpvis ur varje grupp, normalt 24–48 timmar efter doseringen. 21 dagar efter exponeringen avlivas resterande höns och en histopatologisk undersökning av valda nervvävnader genomförs.
1.5 BESKRIVNING AV TESTMETODEN
1.5.1 Förberedelse
Friska unga vuxna höns som inte får medicin och som inte har virussjukdomar eller gångdefekter ska väljas slumpmässigt till behandlings- respektive kontrollgrupper och acklimatiseras till laboratorieförhållandena under minst fem dagar före undersökningens skalljan.
Burar eller inhängnader som är stora nog att ge hönsen fri rörlighet och som underlättar observation av hönsens gång ska användas.
Dosering av testsubstansen ska normalt ske oralt genom sondmatning, gelatinkapslar eller motsvarande metod. Vätska kan ges outspädd eller löst i en lämplig vehikel som majsolja; fasta ämnen ska om möjligt lösas eftersom stora doser fasta ämnen i gelatinkapslar kanske inte kan absorberas effektivt. I fråga om icke-flytande vehiklar ska vehikelns toxiska egenskaper vara kända; i annat fall ska de bestämmas före försöket.
1.5.2 Försöksförhållanden
1.5.2.1 Försöksdjur
Unga vuxna värphöns (Gallus gallus domesticus) i åldern 8–12 månader rekommenderas. Standardstora arter och stammar ska användas och hönsen ska normalt ha fötts upp under sådana förhållanden att de kunnat röra sig fritt.
1.5.2.2 Antal och kön
Utöver behandlingsgruppen ska både en vehikelkontrollgrupp och en positiv kontrollgrupp användas. Vehikelkotrollgruppen ska behandlas på samma sätt som behandlingsgruppen med undantag för att tillförsel av testsubstansen utesluts.
Tillräckligt många höns ska användas i varje grupp för att minst sex fåglar ska kunna avlivas för biokemisk bestämning (tre stycken vid vardera av två tidpunkter) och sex överleva observationsperioden på 21 dagar för patologisk undersökning.
Den positiva kontrollgruppen kan hållas parallellt eller vara en aktuell historisk kontrollgrupp. Den ska omfatta minst sex höns som behandlats med ett välkänt neurotoxiskt ämne som ger fördröjd polyneuropati, tre för biokemisk och tre för patologisk undersökning. Regelbunden uppdatering av historiska data rekommenderas. Nya positiva kontrolldata ska tas fram om någon viktig komponent (t.ex. stam, foder, förvaringsutrymmen) har ändrats av det berörda laboratoriet efter det att det tidigare försöket genomfördes.
1.5.2.3 Dosnivåer
En preliminär undersökning med ett lämpligt antal höns och dosnivågrupper ska utföras för att fastställa vilken nivå som ska användas i huvudundersökningen. En viss dödlighet i den preliminära undersökningen är nödvändig för att fastställa en tillräcklig dos för huvudundersökningen. För att förhindra dödsfall beroende på akuta kolinerga effekter får atropin eller något annat skyddsämne, om vilket det är känt att det inte påverkar fördröjda neurotoxiska responser, användas. En variation av testmetoder får användas för att beräkna den maximala icke-dödliga dosen av testsubstans (se metod B.la). Historiska data om hönsen eller annan toxikologisk information kan också vara till hjälp vid valet av dos.
Testsubstansens dosnivå i huvudundersökningen ska vara den högsta möjliga med beaktande av resultaten från den preliminära undereökningen för dosval och den övre dosgränsen på 2 000 mg/kg kroppsvikt. Eventuella dödsfall ska inte vara fler än att tillräckligt många djur överlever för biokemi (sex stycken) och histologi (sex stycken) efter 21 dagar. Atropin eller andra skyddsämnen, om vilka det är känt att de inte påverkar fördröjda neurotoxiska responser, ska användas för att förhindra dödsfall till följd av akuta kolinerga effekter.
1.5.2.4 ”Limit-test”
Om ett test vid en dosnivå på minst 2 000 mg/kg kroppsvikt och dag, med användning av de förfaranden som beskrivs för denna metod, inte framkallar några observerbara toxiska effekter och om toxicitet inte är att förvänta på grundval av data från strukturellt besläktade ämnen, kan en undersökning med användning av en högre dos anses överflödig. ”Limit-test” är tillämpligt, med undantag för om exponering på människor påvisar ett behov av att använda en högre dosnivå.
1.5.2.5 Observationsperiod
Observationsperioden ska vara 21 dagar.
1.5.3 Förfarande
Efter tillförsel av ett skyddsämne för att förhindra dödsfall till följd av akuta kolinerga effekter tillförs testsubstansen som engångsdos.
1.5.3.1 Allmänna observationer
Observationerna ska påskalljas direkt efter exponeringen. Samtliga höns ska observeras noggrant flera gånger under de första två dagarna och därefter minst en gång dagligen under en period på 21 dagar eller fram till planerad avlivning. Alla tecken på toxicitet ska noteras, inbegripet tid för beteendestörningarnas debut, deras typ, allvar och duration. Ataxi ska mätas enligt en ordningsskala pä minst fyra nivåer, och förlamning ska noteras. Minst två gånger per vecka ska de höns som valts ut för patologiska undersökningar tas ut från burarna och underkastas en period av påtvingad motorisk aktivitet, t.ex. klättring på stege, för att underlätta observation av minimala toxiska effekter. Döende djur och djur som lider av svår smärta ska tas bort när detta upptäcks, avlivas skonsamt och obduceras.
1.5.3.2 Kroppsvikt
Samtliga höns ska vägas just före tillförseln av testsubstansen och därefter minst en gång i veckan.
1.5.3.3 Biokemi
Sex höns väljs slumpvis från var och en av behandlings- respektive vehikelkontrollgrupperna och tre höns från den positiva kontrollgruppen (om denna grupp hålls parallellt.) Hönsen ska avlivas några dagar efter dosering, och hjärnan och lumbala delar av ryggmärgen prepareras och testas i fråga om inhibition av neurotoxiskt esteras. Därutöver kan det också vara lämpligt att preparera och testa ischiasnerwävnad i fråga om inhibition av neurotoxiskt esteras. Normalt ska tre fåglar från kontrollgruppen och varje behandlingsgrupp avlivas efter 24 timmar och tre efter 48 timmar, medan de tre hönsen från de positiva kontrollgrupperna ska avlivas efter 24 timmar. Om eventuella kliniska tecken på förgiftning (detta kan ofta bedömas genom observation vid debuten på kolinerga svar) indikerar att det toxiska ämnet kan tas upp mycket långsamt är det bättre att ta vävnadsprov från tre fåglar vid vardera av två tidpunkter mellan 24 och så sent som 72 timmar efter dosering.
Om det bedöms lämpligt kan även analyser av acetylkolinesteras (AChE) utföras på dessa prover. Spontan reaktivering av AChE kan dock uppträda in vivo och därmed leda till en underskattning av ämnets potential som AChE-hämmare.
1.5.3.4 Obduktion
Obduktion av samtliga djur (avlivade enligt planen och döende djur som avlivats) ska omfatta observation av hjärnans och ryggmärgens utseende.
1.5.3.5 Histopatologisk undersökning
Nervvävnad från djur som överlevt observationsperioden och som inte används för biokemiska undersökningar ska underkastas mikroskopisk undersökning. Vävnaderna ska fixeras in situ med användning av perfusionsteknik. Snitten ska omfatta lillhjärnan (longitudiellt, medialt), förlängda märgen, ryggmärgen och perifera nerver. Ryggradssnitten ska tas från halsområdet, mitten av brösthålan och det lumbosakrala området. Snitt från tibialnervens distala region och dess förgreningar till musculus gastrocnemius och ischiasnerven ska tas. Snitten ska färgas med lämplig myelin- och axonspecifik färg.
2. DATA
Negativa resultat i fråga om de sluteffekter som valts för denna metod (biokemi, histopatologi och beteendeobservationer) skulle normalt inte kräva ytterligare tester av fördröjd neurotoxicitet. Tvetydiga eller oklara resultat i fråga om dessa sluteffekter kan komma att kräva ytterligare utvärdering.
Individuella data ska anges. Därutöver ska alla data sammanfattas i tabellform som för varje testgrupp visar antalet djur vid försökets skalljan, antalet djur som uppvisar skador, beteendeeffekter eller biokemiska effekter, typen och allvaret av dessa skador eller effekter och procentandelen djur som uppvisar respektive typ och grad av allvar av skada eller effekt.
Resultaten av denna undersökningen ska utvärderas i fråga om fördelning, allvar och korrelation mellan beteendeeffekter, biokemiska och histopatologiska effekter och eventuellt andra observerade effekter i behandlings- och kontrollgrupperna.
Numeriska resultat ska utvärderas genom lämpliga och allmänt erkända statistiska metoder. De statistiska metoder som används ska väljas vid utformandet av undersökningen.
3. RAPPORTERING
FÖRSÖKSRAPPORT
Försöksrapporten ska om möjligt innehålla följande uppgifter:
|
|
Försöksdjur
|
|
|
Försöksförbållanden:
|
|
|
Resultat: |
|
|
|
|
|
Diskussion om resultaten. |
|
|
Slutsatser. |
4. HÄNVISNINGAR
Denna metod motsvarar OECD TG 418.
B.38 FÖRDRÖJD NEUROTOXICITET ORSAKAD AV ORGANISKA FOSFORFÖRENINGAR 28 DAGARS UPPREPAD DOSUNDERSÖKNING
1. METOD
1.1 INLEDNING
Vid bedömning och utvärdering av ämnens toxiska effekter är det viktigt att beakta potentialen hos substanser inom vissa ämnesgrupper att framkalla vissa typer av neurotoxicitet som kanske inte upptäcks vid andra toxicitetsundersökningar. Det har observerats att vissa organiska fosforföreningar framkallar fördröjd neurotoxicitet och dessa ska beaktas för eventuell utvärdering.
Screening-tester in vitro får tillämpas för att identifiera de ämnen som kan framkalla fördröjd polyneuropati; negativa resultat från in vitro-undersökningar bevisar dock inte att testsubstansen inte skulle vara neurotoxisk.
Denna 28 dagars undersökning av fördröjd neurotoxicitet ger information om de tänkbara hälsorisker som troligen uppstår till följd av upprepad exponering under en begränsad tidsperiod. Den ger information om dosrespons och kan ge en beräkning av en NOAEL-nivå som kan vara användbar för att fastställa säkerhetsföreskrifter om exponering.
Se även Allmän inledning, del B.
1.2 DEFINITIONER
Organiska fosforföreningar omfattar icke joniserande organiska fosforestrar, tioestrar eller anhydnder av organofosfor-, organofosforo- eller organofosforamidsyror eller av besläktade fosfonthio-, fosforthio- eller fosforthioamidsyror eller andra ämnen som kan framkalla sådan fördröjd neurotoxitet som ibland observeras i denna ämnesgrupp.
Fördröjd neurotoxicitet är ett syndrom som förknippas med förlängd fördröjd debut för ataxi, di-stala axonopatier i ryggmärgen och perifera nerver samt hämmande och åldrande av neurotoxiskt esteras (NTE) i nervvävnad.
1.3 PRINCIP FÖR TESTMETODEN
Testsubstansen tillförs dagligen oralt till tamhöns under 28 dagar. Djuren observeras minst en gång dagligen i fråga om beteendestörningar, ataxi och förlamning till dess det gått 14 dagar efter den sista dosen. Biokemiska mätningar, särskilt inhibition av neurotoxiskt esteras (NTE) utförs på höns som väljs slumpvis ur varje grupp, normalt 24 – 48 timmar efter doseringen. Två veckor efter den sista dosen avlivas resterande höns och en histopatologisk undersökning av valda neuralvävnader genomförs.
1.4 BESKRIVNING AV TESTMETODEN
1.4.1 Förberedelser
Friska unga vuxna höns som inte får medicin och som inte har virussjukdomar eller gångdefekter ska väljas slumpmässigt till behandlings- respektive kontrollgrupper och acklimatiseras till laboratorieförhållandena under minst fem dagar före undersökningens skalljan.
Burar eller inhängnader som är stora nog att ge hönsen fri rörlighet och som underlättar observation av hönsens gång ska användas.
Oral dosering ska ske dagligen sju dagar i veckan, helst genom sondmatning eller tillförsel av gelatinkapslar. Vätska kan ges outspädd eller löst i en lämplig vehikel som majsolja; fasta ämnen ska om möjligt lösas eftersom stora doser fasta ämnen i gelatinkapslar kanske inte kan absorberas effektivt. I fråga om icke-flytande vehiklar ska vehikelns toxiska egenskaper vara kända; i annat fall ska de bestämmas före försöket.
1.4.2 Försöksförhållanden
1.4.2.1 Försöksdjur
Unga vuxna värphöns (Gallus gallus domesticus) i åldern 8 – 12 månader rekommenderas. Standardstora arter och stammar ska användas och hönsen ska normalt ha fötts upp under sådana förhållanden att de kunnat röra sig fritt.
1.4.2.2 Antal och kön
Vanligen bör minst tre behandlingsgrupper och en vehikelkontrollgrupp användas. Vehikelkontrollgruppen ska behandlas på samma sätt som behandlingsgruppen med undantag för att tillförsel av testsubstansen utesluts.
Tillräckligt många höns ska användas i varje grupp för att minst sex fåglar ska kunna avlivas för biokemisk bestämning (tre stycken vid vardera två tidpunkter) och sex överleva en 14 dagar lång observationsperiod efter behandlingen för patologisk undersökning.
1.4.2.3 Dosnivåer
Dosnivåerna ska väljas med beaktande av resultaten från ett akut test av fördröjd neurotoxicitet eller andra befintliga toxicitets- eller kinetiska data som finns tillgängliga i fråga om testsubstansen. Den högsta dosnivån ska väljas i syfte att inducera toxiska effekter, helst fördröjd toxicitet, men inte dödsfall eller svårt lidande. Därefter ska dosnivåer fastställas i en fallande skala i syfte att påvisa eventuella dos-responssamband och NOAEI. vid den lägsta dosnivån.
1.4.2.4 ”Limit-test”
Om ett test vid en dosnivå på minst 1 000 mg/kg kroppsvikt och dag, med användning av de förfaranden som beskrivs för denna undersökning, inte framkallar några observerbara toxiska effekter och om toxicitet inte är att förvänta på grundval av data om strukturellt besläktade ämnen kan en undersökning med en högre dos anses överflödig. ”Limit-test” är tillämpligt, med undantag för om exponering på människor indikerar att det finns behov av att använda en högre dosnivå.
1.4.2.5 Observationsperiod
Samtliga djur ska observeras minst en gång dagligen under exponeringsperioden och därefter under 14 dagar, med undantag för planerad dissektion.
1.4.3 Förfarande
Djuren doseras med testsubstansen sju dagar i veckan under en period på 28 dagar.
1.4.3.1 Allmänna observationer
Observationerna ska påskalljas direkt efter exponeringen. Samtliga höns ska observeras minst en gång dagligen under de 28 behandlingsdagarna och under minst 14 dagar efter dosering eller fram till planerad avlivning. Alla tecken på toxicitet ska noteras, inbegripet tid för beteendestörningarnas debut, deras typ, allvar och duration. Observationerna ska omfatta, men inte begränsas till, beteendeavvikelser. Ataxi ska mätas enligt en ordningsskala på minst fyra nivåer, och förlamning ska noteras. Minst två gånger per vecka ska hönsen tas ut från burarna och underkastas en period av påtvingad motorisk aktivitet, t.ex. klättring på stege, för att underlätta observation av minimala toxiska effekter. Döende djur och djur som lider av svår smärta ska tas bort när detta upptäcks, avlivas skonsamt och obduceras.
1.4.3.2 Kroppsvikt
Samtliga höns ska vägas just före tillförseln av testsubstansen och därefter minst en gång i veckan.
1.4.3.3 Biokemi
Sex höns väljs slumpvis från var och en av behandlings- respektive vehikelkontrollgrupperna och avlivas några dagar efter doseringen, och hjärnan och lumbala delar av ryggmärgen prepareras och testas i fråga om inhibition av neurotoxiskt esteras. Därutöver kan det också vara lämpligt att preparera och testa ischiasnervvävnad i fråga om inhibition av neurotoxiskt esteras. Normalt ska tre fåglar från kontrollgruppen och var och en av behandlingsgrupperna avlivas efter 24 timmar och tre 48 timmar efter den sista doseringen. Om data från den akuta undersökningen eller andra undersökningar (t.ex. toxikokinetik) indikerar att andra tidpunkter för avlivning är att föredra efter slutdosering så ska dessa tidpunkter tillämpas och grunden för detta dokumenteras.
Om det bedöms lämpligt kan även analyser av acetylkolinesteras (AChE) utföras på dessa prover. Spontan reaktivering av AChE kan dock uppträda in vivo och därmed leda till en underskattning av ämnets potential som AChE-hämmare.
1.4.3.4 Obduktion
Obduktion av samtliga djur (avlivade enligt planen och döende djur som avlivats) ska omfatta observation av hjärnans och ryggmärgens utseende.
1.4.3.5 Histopatologisk undersökning
Nervvävnad från djur som överlevt observationsperioden och som inte används för biokemiska undersökningar ska underkastas mikroskopisk undersökning. Vävnaderna ska fixeras in situ med användning av perfusionsteknik. Snitten ska omfatta lillhjärnan (longitudiellt, medialt), förlängda märgen, ryggmärgen och perifera nerver. Ryggradssnitten ska tas från halsområdet, mitten av brösthålan och det lumbosakrala området. Snitt från tibialnervens distala region och dess förgreningar till musculus gastrocnemius och ischiasnerven ska tas. Snitten ska färgas med lämplig myelin- och axonspecifik färg. Inledningsvis ska mikroskopundersökning utföras på de bevarade vävnaderna av samtliga djur i kontroll- respektive högdosgruppen. Om det finns bevis på effekter i högdosgruppen ska mikroskopundersökning även utföras på hönsen från mellan- och lågdos-grupperna.
2. DATA
Negativa resultat i fråga om de sluteffekter som valts för denna metod (biokemi, histopatologi och beteendeobservationer) skulle normalt inte kräva ytterligare tester av fördröjd neurotoxicitet. Tvetydiga eller oklara resultat i fråga om dessa sluteffekter kan komma att kräva ytterligare utvärdering.
Individuella data ska anges. Därutöver ska alla data sammanfattas i tabellform som för varje testgrupp visar antalet djur vid försökets skalljan, antalet djur som uppvisar skador, beteendeeffek-tcr eller biokemiska effekter, typen och allvaret av dessa skador eller effekter och procentandelen djur som uppvisar respektive typ och grad av allvar av skada eller effekt.
Resultaten av denna undersökning ska utvärderas i fråga om fördelning, allvar och korrelation mellan beteendeeffekter, biokemiska och histopatologiska effekter och eventuellt andra observerade effekter i behandlings- och kontrollgrupperna.
Numeriska resultat ska utvärderas genom lämpliga och allmänt erkända statistiska metoder. De statistiska metoder som används ska väljas vid utformandet av undersökningen.
3. RAPPORTERING
FÖRSÖKSRAPPORT
Försöksrapporten skal! om möjligt innehålla följande uppgifter:
|
|
Försöksdjur
|
|
|
Försöksförhållanden:
|
|
|
Resultat:
|
|
|
Diskussion om resultaten. |
|
|
Slutsatser. |
HÄNVISNINGAR
Denna metod motsvarar OECD TG 419.
B.39 TEST AV REPARATIONSSYNTES (UDS) HOS LEVERCELLER FRÅN DÄGGDJUR IN-VIVO
1. METOD
Denna metod är en kopia av testet OECD TG 486, Test av reparationssyntes (UDS) hos leverceller från däggdjur in vivo (Unscheduled DNA Synthesys (UDS) Test with Mammalian Liver Celler in Vivo (1997)).
1.1 INLEDNING
Ändamålet med testet av reparationssyntes (UDS) hos leverceller från däggdjur in vivo är att identifiera testsubstanscr som inducerar DNA-reparation i leverceller från behandlade försöksdjur (se (1) (2) (3) (4).
Detta in vivo-test erbjuder en metod för undersökning av genotoxiska effekter av kemikaler i levern. Slutpunktsmätning är indikativt för DNA-skador och påföljande reparation i leverceller. Levern är vanligen det huvudsakliga sätet för metabolismen av absorberade ämnen. Det är således ett lämpligt säte för att mäta DNA-skador in vivo.
Om det finns bevis för att testsubstansen inte kommer att nå målvävnaden är det inte lämpligt att använda detta test.
Reparationssyntesens slutpunkt mäts genom att bestämma upptaget av märkta nukleosider i celler som inte undergår reguljär DNA syntes (S-fas). Den teknik som är den mest använda är bestämning av upptag av triti-ummärkt tymidin (H-TdR) med hjälp av autoradiografi. Råttlever används med fördel för UDS-tester in vivo. Andra vävnader än lever kan användas, men omfattas inte av denna metod.
Detektionen av ett UDS-svar är beroende av antalet DNA-baser som ”klipps bort” och byts ut vid platsen för skadan. UDS-testet är därför särskilt värdefullt för att detektera substansinducerad ”longpatch”-reparation (20–30 baser). ”Shortpatch”-reparation (1–3 baser) detekteras däremot med mycket lägre känslighet. Vidare kan mutagena händelser inträffa på grund av utebliven reparation, felreparation eller felreplikation av DNA-skador. Omfattningen av UDS-svaret ger ingen indikation på noggrannheten i reparationsprocessen. Dessutom är det möjligt att ett mutagen reagerar med DNA men att DNA-skadan inte repareras via en reparationsprocess där DNA-baser ”klipps bort” och byts ut. Bristen på specifik information rörande mutagen aktivitet, erhållen genom UDS-testet, kompenseras för genom dess potentiella känslighet, eftersom den mäts i hela genomet.
Se även Allmän inledning, del B.
1.2 DEFINITIONER
celler som repareras: ett nettoantal korn i kärnan (net nuclear grain, NNG) som är större än ett förvalt värde, vilket ska verifieras av det laboratorium som utför testet.
nettoantal korn i kärnan (NNG): kvantitativ mätning av UDS-aktivitet hos celler i autoradiografiska UDS-tester, beräknade genom att subtrahera medelantalet av de cytoplasmiska kornen i kärnekvivalenta cytoplasmiska områden (CG) från antalet korn i kärnan (nuclear grains, NG): NNG = NG-CG. NNG-räkningar beräknas för individuella celler och slås sedan samman för celler i en kultur, i parallella kulturer, etc.
reparationssyntes (UDS): DNA-reparationssyntes efter ’bortklippning’ och avlägsnande av en DNA-sträng som innehåller ett avsnitt med skador inducerats av kemiska substanser eller fysiska ämnen. (UDS)
1.3 TESTMETODENS PRINCIP
UDS-testet med leverceller från däggdjur in vivo indikerar DNA-reparationssyntes efter ”bortklippning” och avlägsnande av en sträng med DNA innehållande ett avsnitt med skador, vilka inducerats av kemiska substanser eller fysiska ämnen. Testet baseras i vanliga fall på en inkorporering av 3H-TdR i DNA hos leverceller, vilka har en låg frekvens av celler som befinner sig i cellcykelns S-fas. Upptaget av 3H-TdR bestäms vanligen genom autoradiografi. eftersom denna teknik inte ar sä mottaglig för interferens frän celler i S-fas som, t.ex. vätske-scintillationsräkning
1.4 BESKRIVNING AV METODEN
1.4.1 Förberedelser
1.4.1.1 Val av djurarter
Vanligen används råttor men det går även bra att använda ett annat lämpligt däggdjur. Man bör välja de stammar av unga, friska vuxna försöksdjur som vanligen används på laboratorier. När undersökningen ska påbörjas, är det lämpligt att viktskillnaden mellan djuren är så liten möjligt och den bör inte överstiga ±20 % av medelvikten för varje kön.
1.4.1.2 Förhållanden vid förvaring och utfodring
De generella förhållanden som anges i den Allmänna inledningen till del B gäller. Man bör dock sträva efter en relativ luftfuktighet på 50–60 %.
1.4.1.3 Förberedelse av djuren
Friska, unga och vuxna försöksdjur väljs ut slumpartat för placering i kontroll- och behandlingsgrupperna. Burar bör arrangeras på ett sådant sätt att möjliga effekter beroende på placeringen av burarna blir så små som möjligt. Djuren ska ha en unik identifiering och hållas i sina burar under minst fem dagar innan undersökningen börjar, för att göra det möjligt för dem att acklimatiseras till förhållandena i laboratoriet.
1.4.1.4 Berdninq av testsubstansen
Testsubstanser i fast form bör lösas upp i eller blandas med lämpliga lösningsmedel eller vehiklar och spädas ut, om så är lämpligt, innan dosen administreras till försöksdjuren. Testsubstanser i flytande form kan doseras direkt eller så späds de ut före dosering. Nygjorda beredningar av testsubstansen bör användas om inte stabilitetsdata visar att lagring är acceptabelt.
1.4.2 Försöksbetingelser
1.4.2.1 Lösningsmedel/vehikel
Lölsningsmedlet/vehikeln bör inte ge upphov till toxiska effekter vid de dosnivåer som används och bör inte kunna misstänkas för att reagera kemiskt med testsubstansen. Om andra lösningsmedel/vehiklar än gängse brukliga används, bör användningen av dem ha stöd från data som visar på deras kompatibilitet. När det är möjligt, rekommenderas att man i första hand överväger vattenbaserade lösningsmedel/vehiklar.
1.4.2.2 Kontroller
Samtidiga positiva och negativa kontroller (lösningsmedel/vehikel) bör ingå i varje oberoende utförd del av experimentet. Förutom vid behandling med testsubstansen, bör försöksdjuren i kontrollgrupperna hanteras på ett identiskt sätt i förhållande till djuren i de grupper där behandling skett.
Positiva kontroller bör utgöras av substanser som är kända för att ge UDS när de administreras vid exponeringsnivåer som kan förväntas att ge en detekterbar ökning jämfört med bakgrunden. Positiva kontroller som kräver metabolisk aktivering bör användas vid doser som framkallar ett måttligt svar (4). Doserna kan väljas på ett sådant sätt att effekterna är tydliga men att de inte omedelbart avslöjar identiteten hos kokade utstryksglas för läsaren. I tabellen nedan ges några exempel på positiva kontrollsubstanser.
|
Tider for provtagning |
Substans |
CA S-nummer |
EINECS-nummer |
|
Tidiga provtagningstider (2–4 tim.) |
N-Nitrosodimethylamin |
62-75-9 |
200-249-8 |
|
Sena provtagningstider (12–16 tim.) |
N-2-Fluorenylacetamid (2-AAF) |
53-96-3 |
200-188-6 |
Andra lämpliga positiva kontrollsubstanser kan används. Det är acceptabelt att den positiva kontrollen administreras via en annan tillförselväg än testsubstansen.
1.5 FÖRFARANDE
1.5.1 Försökdjurens antal och kön
Ett lämpligt antal försöksdjur bör användas, för att man ska kunna ta hänsyn till den naturliga biologiska variationen hos testresultaten. Antalet försöksdjur bör utgöras av minst tre analyserbara försöksdjur per grupp. Där det finns en signifikant historiks databas som har ackumulerats, krävs det endast ett eller två försöksdjur för de samtidiga negativa och positiva kontrollgrupperna.
Om det vid tiden för undersökningen visar sig att det finns data som är tillgängliga från undersökningar på samma stam och där samma tillförselväg för exponering har använts, samt att dessa visar att det inte finns några betydande skillnader i toxicitet mellan könen, kan testning på endast ett kön, helst på hanar, vara tillräckligt. I de fall där exponering av människa för kemikaler kan ge könsspecifika resultat, som t.ex. för vissa läkemedel, bör testet utföras med försöksdjur av lämpligt kön.
1.5.2 Behandlingsschema
Testsubstanser administreras i allmänhet i form av en enstaka behandling.
1.5.3 Dosnivåer
Normalt används minst tvä olika dosnivåer. Den högsta dosen definieras som dos vilken producerar tecken på toxicitet på ett sådant sätt att högre dosnivåer, baserade på en likadan doseringsregim, skulle förväntas leda till letalitet. 1 allmänhet bör den lägre dosen vara 50 % till 25 % av den högre dosen.
Substanser med specifika biologiska verkningar vid låga icke-toxiska doser (såsom hormoner och mitogena substanser) kan utgöra undantag med avseende pa de dosbestämmande kriterierna och bör utvärderas från fall till fall. Om en undersökning för att bestämma intervallet utförs på grund av att det inte finns några lämpliga data tillgängliga, bör denna utföras i samma laboratorium, och där man använder samma arter, stammar, kön och behandlingsschema som i huvudundersökningen.
Den högsta dosen kan även definieras som en dos vilken producerar en indikation på toxicitet i levern (t.ex. pyknotiska kärnor).
1.5.4 Limit-test
En fullständig undersökning kan inte anses vara nödvändig om ett test på en dosnivå vid minst 2 000 mg/kg kroppsvikt och med en enstaka behandling, eller i form av två behandlingar under samma dag, inte ger upphov till observerbara toxiska effekter, och om ingen genotoxicitet kan förväntas utifrån data om strukturellt relaterade substanser. En förväntad exponering av människor kan visa på behovet av att en högre dosnivå används i ’limit-testen’.
1.5.5 Administrering av doser
Testsubstansen administreras vanligen via sondmatning genom att använda en magsond eller en lämplig intubationskanyl. Andra tillförselvägar för exponering kan vara acceptabla, där de kan motiveras. Den intraperitoneala vägen rekommenderas dock inte, eftersom den skulle kunna exponera levern direkt för testsubstansen snarare än via det cirkulatoriska systemet. Den maximala vätskevolymen som kan administreras genom sondmatning eller injektion vid ett tillfälle beror av försöksdjurets storlek. Volymen bör inte överskrida 2 ml/100 g kroppsvikt. Användningen av större volymer måste motiveras. Förutom när det gäller irriterande eller korro-.siva substanser, vilka normalt kommer att avslöja förvarade effekter vid högre koncentrationer, bör variationer av testvolymen minimeras genom en justering av koncentrationen, så att konstant volym kan säkerställas vid alla dosnivåer.
1.5.6 Beredning av leverceller
I normala fall bereds leverceller frän behandlade försöksdjur 12–16 timmar efter dosering. Ytterligare en tidig provtagningstidpunkt (normalt 2–4 timmar efter behandlingen) är normalt nödvändig om inte det finns ett tydligt positivt svar efter 12–16 timmar. Alternativa provtagningstidpunkter kan används när det är motiverat utifrån toxikokinetiska data.
Kortvariga kulturer med leverceller från däggdjur startas genom att skölja lever in situ med kollagenas och att låta nyligen dissocierade leverceller fästa sig på en lämplig yta. Leverceller från negativa kontrollförsöksdjur bör ha en viabilitet (5) på minst 50 %.
1.5.7 Bestämning av UDS
Nyligen isolerade leverceller från däggdjur inkuberas normalt med ett medium som innehåller 3H-TdR under en tid som är tillräckligt lång, t.ex. 3–8 timmar. När inkubationsperioden går mot sitt slut, bör mediet avlägsnas från cellerna, vilken sedan kan inkuberas i ett medium som innehåller ett överskott på omärkt tymidin för att försvaga oinkorporerad radioaktivitet (’cold chase’). Cellerna tvättas sedan, fixeras och torkas. Vid längre inkubationstider, kan ’cold chase’ visa sig vara onödigt. Utstryksglas doppas sedan i en autoradiografisk emulsion, exponeras i mörker (t.ex. nedkylda under 7–14 dagar) framkallas och färgas in, varpå exponerade silverkorn sedan räknas. Två till tre utstryksglas bereds från varje försöksdjur.
1.5.8 Analys
Preparationerna av utstryksglas bör innehålla tillräckligt många celler med normal morfologi för att medge en meningsfull bestämning av UDS. Preparationer undersöks mikroskopiskt efter tecken på uppenbar cytotoxicitct (t.ex. pyknos, reducerade nivåer av radioaktiv märkning).
Utstryksglas bör kodas innan kornen räknas. Normalt påvisas 100 celler från varje försöksdjur från minst två utstryksglas. om mindre än 100 celler/försöksdjur påvisas bör detta motiveras. Vid kornräkningar påvisas inte kärnor i S-fas, men proportionen av celler i S-fas kan registreras.
Mängden av 3H-TdR som inkorporerats i kärnan och cytoplasman hos morfologiskt normala celler, som bevisats av upplagringen av silverkorn, bör bestämmas med hjälp av lämpliga metoder.
Kornräkningar bestäms i kärnorna (nuclear grains, NG) och kärnekvivalenta områden i cytoplasman (cytoplasmic grains, CG). CG-räkningar mäts genom att antingen ta det område av cytoplasman som har den mest omfattande märkningen, eller genom att ta ett medeltal av två eller tre slumpmässiga cytoplasmaräkningar nära kärnan. Andra räknemetoder (t.ex. helcellsräkning) kan användas om de kan motiveras (6).
2. DATA
2.1 BEHANDLING AV RESULTATEN
Individuella utstryksglas och försöksdjursdata bör tas fram. Dessutom bör alla data sammanfattas i tabellform. Räkningar av nettoantalet korn i kärnan (NNG) bör tas fram för varje cell, för varje försöksdjur och för varje dos och tid genom att subtrahera CG-räkningar från NG-räkningar. Om ’celler under reparation’ räknas in, bör kriterierna för definition av ’celler under reparation’ vara motiverade och baserade på historiska eller samtidiga negativa kontrolldata. Numeriska resultat kan utvärderas utifrån statistiska metoder. Om statistiska tester används, bör de selekteras och motiveras innan undersökningen utförs.
2.2 UTVÄRDERING OCH TOLKNING AV RESULTATEN
Exempel på kriterier för positiva/negativa svar innefattar:
|
positiva |
i) |
NNG-värden över ett förvalt tröskelvärde som är motiverat på basis av historiska laboratoriedata |
|
eller |
ii) |
NNG-värden som är signifikant större än den samtidiga kontrollen |
|
negativa |
i) |
NNG-värden inom/under det historiska kontrolltröskelvärdet |
|
eller |
ii) |
NNG-värden som inte är signifikant större än den samtidiga kontrollen |
Den biologiska relevansen hos registrerade data bör uppmärksammas: d.v.s. parameter såsom variationer mellan olika försöksdjur, dos/respons-förhållanden och cytotoxicitetet bör också beaktas. Statistiska metoder kan användas såsom en hjälp vid utvärderingen av testresultaten. Statistisk signifikans bör dock inte vara den enda faktor som bestämmer om resultatet är positivt.
Även om de flesta experiment kommer att ge tydligt positiva eller negativa resultat, kan uppsättningen av data i sällsynta fall göra det omöjligt att utföra en otvetydig bedömning med avseende på testsubstansens aktivitet. Resultatet kan förbli tvetydigt eller tveksamt, oavsett hur många gånger som experimentet upprepas.
Ett positivt resultat från UDS-testet på leverceller från däggdjur in vivo indikerar att testsubstansen inducerar DNA-skador i leverceller från däggdjur in vivo, vilka kan repareras av reparartionssyntes in vitro. Ett negativt resultat indikerar att testsubstansen, under försöksbetingelserna, inte inducerar skador på DNA som är detekterbara med hjälp av detta test.
Sannolikheten för att testsubstansen eller dess metaboliter ska nå ut i det stora kretsloppet eller specifikt till målvävnaden (t.ex. systemisk toxicitet) bör diskuteras.
3. RAPPORTERING
TESTRAPPORT
Testrapporten måste innehålla följande information:
|
|
Lösningsmedel/vehikel:
|
|
|
Försöksdjur:
|
|
|
Försöksbetingelser:
|
|
|
Resultat:
|
|
|
Diskussion av resultaten. |
|
|
Slutsatser. |
4. HÄNVISNINGAR
|
1. |
Ashby, J., Lefevre, P. A., Burlison, B. and Penman, M. G. (198 5). An Assessment of the In Vivo Rat Hepatocyte DNA Repair Assay. Mutation Res., 156, s. 1–18. |
|
2. |
Buttervvorth, B. E., Ashby, J., Bermudez, E., Casciano, D., Mirsalis, J., Probst, G. and Williams, G. (1987). A Protocol and Guide for the In Vivo Rat Hepatocyte DNA-Repair Assay. Mutation Res., 189, s. 123–133. |
|
3. |
Kennelly, J. C, Waters, R., Ashby, J., Lefevre, P. A., Burlison, B., Benford, D. J., Dean, S. W. and Mitchell, I. de G. (1993). In Vivo Rat Liver UDS Assay, in Kirkland D. J. and Fox M., (eds) Supplementary Mutagenicity Tests: UKEM Recommended Procedures. UKEMS Subcommitee on Guidelines for Mutagenicity Testing. Report Part II revised. Cambridge University Press, Cambridge, New York, Port Chester, Melbourne, Sydney s. 52–77. |
|
4. |
Madle, S., Dean, S. W., Andrae, U., Brambilla, G., Burlinson, B., Doolittle, D. J., Furihata, C, Herner, T., McQueen, C. A. and Mori, H. (1993). Recommendations for the Performance of UDS Tests In Vitro and In Vivo. Mutation Res., 312, s. 263–285. |
|
5. |
Fautz, R., Hussain, B., Efstathiou, E. and Hechenberger-Freudl, C. (1993). Assesment of the Relation Between the Initial Viability and the Attachment of Freshly Isoladet Rat Hepatocytes Used for the In Vivo/ln Vitro DNA Repair Assay (UDS). Mutation Res., 291, s. 21–27. |
|
6. |
Mirsalis,). C, Tyson, C. K. and Butterworth, B. E. (1982). Detection of Genotoxic Carcinogens in the In Vivo/ln Vitro Hepatocyte DNA Repair Assay. Environ. Mutagen, 4, s. 553–562. |
B.40 IN VITRO-TEST AV HUDKORROSIVITET: BESTÄMNING AV TRANSKUTANT ELEKTRISKT MOTSTÅND (TER)
1. METOD
Denna testmetod motsvarar OECD TG 430 (2004).
1.1 INLEDNING
Hudkorrosivitet innebär att en irreversibel vävnadsskada uppstår i huden som en följd av att ett testmaterial [definierat enligt det internationella systemet för klassificering och märkning av kemikalier (GHS)] (1) appliceras. Denna metod bygger på ett förfarande som innebär att testmaterialets hudkorrosivitet inte testas på levande djur.
Bedömning av hudkorrosiviteten har normalt inneburit användning av försöksdjur (2). På grund av den smärta och det lidande som försöksdjuren utsätts för har översynen av testmetod B.4 gjorts på ett sådant sätt att bedömningen av hudkorrosiviteten går att göra med alternativa metoder in vitro, så att försöksdjur inte längre behöver användas.
Ett första steg mot att definiera alternativa testmetoder som kunde användas för testning av hudkorrosiviteten var att genomföra förvalideringsstudier (3). Efter detta gjordes en formell valideringsstudie (6)(7)(8) av in vitro-metoder för att bedöma hudkorrosiviteten (4)(5). Utfallet av dessa studier sammantaget med annan publicerad litteratur ledde till rekommendationen att följande tester skulle användas för bedömning av hudkorrosiviteten in vivo (9)(10)(1 1): ett test där en modell av human hud används (se testmetod B.40b) och bestämning av transkutant elektriskt motstånd (denna metod).
Från en valideringsstudie och andra publicerade studier rapporteras att prövningen av transkutant elektriskt motstånd (TER) på råtthud (12)(13) kan användas för en tillförlitlig särskiljning mellan ämnen som är kända för att vara hudkorrosiva och kända icke-korrosiva ämnen (5)(9).
Testet enligt denna metod gör det möjligt att identifiera kemikalier och blandningar som är hudkorrosiva. Dessutom gör testet det möjligt att identifiera icke hudkorrosiva ämnen och blandningar med stöd av annan befintlig information (t.ex. pH, förhållandet mellan struktur och aktivitet, data som gäller människor och/eller djur) (1)(2)(11)(14). Testet ger ingen information om hudirritation, inte heller ger det möjlighet till den underklassificering som är möjlig enligt det internationella systemet för klassificering och märkning av kemikalier (GHS) (1).
För en fullständig utvärdering av lokala hudeffekter efter en enda hudexponering rekommenderas den sekventiella teststrategi som finns som tillägg till testmetod B.4 (2) och som anges i det internationella systemet för klassificering och märkning (1). Denna teststrategi inbegriper in vitro-tester av hudkorrosivitet (enligt den häri beskrivna metoden) och hudirritation innan tester på levande djur övervägs.
1.2 DEFINITION
Hudkorrosivitet in vivo: innebär att en irreversibel skada uppstår i huden, det vill säga synlig nekros genom epidermis och ner i dermis efter att ett testämne har applicerats och fått verka i upp till fyra timmar. Korrosiva reaktioner kännetecknas av sår, blödning, blodiga sårskorpor och i slutet av observationsperioden på 14 dagar av missfärgning på grund av blekning av huden, håravfall på hela hudområden och ärrbildning. Histopatologi bör övervägas för att bedöma tveksamma skador.
Transkutant elektriskt motstånd (TER): är ett mått på hudens elektriska impedans, uttryckt i kΩ. Det är en enkel och robust metod för att bedöma hudens barriärfunktion genom att registrera jonflödet genom huden med en Wheatstonebrygga.
1.3 REFERENSÄMNEN
Tabell 1
Referenskemikalier
|
Namn |
EINECS-nr |
CAS-nr |
|
|
1,2-Diaminopropan |
201-155-9 |
78-90-0 |
Starkt korrosivt |
|
Akrylsyra |
201-177-9 |
79-10-7 |
Starkt korrosivt |
|
2-tert-butylfenol |
201-807-2 |
88-18-6 |
Korrosivt |
|
Kaliumhydroxid (10 %) |
215-181-3 |
1310-58-3 |
Korrosivt |
|
Svavelsyra (10 %) |
231-639-5 |
7664-93-9 |
Korrosivt |
|
Oktansyra (kaprylsyra) |
204-677-5 |
124-07-02 |
Korrosivt |
|
4-Amino-1,2,4-triazol |
209-533-5 |
584-13-4 |
Icke-korrosivt |
|
Eugenol |
202-589-1 |
97-53-0 |
Icke-korrosivt |
|
Fenetylbromid |
203-130-8 |
103-63-9 |
Icke-korrosivt |
|
Tetrakloretylen |
204-825-9 |
27-18-4 |
Icke-korrosivt |
|
Isostearinsyra |
250-178-0 |
30399-84-9 |
Icke-korrosivt |
|
4-(Metyltio)-bensaldehyd |
222-365-7 |
3446-89-7 |
Icke-korrosivt |
De flesta av kemikalierna i förteckningen har tagits från den förteckning över kemikalier som valdes för den internationella valideringsstudien ECVAM (4). Urvalet bygger på följande kriterier:
|
i) |
Lika många korrosiva som icke-korrosiva ämnen. |
|
ii) |
Kommersiellt tillgängliga ämnen som täcker in de flesta relevanta kemiskalieklasser. |
|
iii) |
Medtagande av såväl starkt korrosiva som mindre korrosiva ämnen för att göra det möjligt att särskilja ämnen på grundval av deras hudkorrosivitet. |
|
iv) |
Val av kemikalier som kan hanteras i ett laboratorium utan att utgöra någon annan allvarlig risk utöver hudkorrosiviteten. |
1.4 PRINCIPEN FÖR TESTMETODEN
Testämnet appliceras och får verka i upp till 24 timmar på den epidermala ytan av hudbitar i ett testsystem med två kammare där hudbitarna fungerar som skiljemembran mellan kamrarna. Hudbitarna tas från humant avlivade, 28-30 dagar gamla råttor. Korrosivt material identifieras genom sin förmåga att framkalla skador på hudens hornlager och störningar i den normala barriärfunktionen, vilket kan fastställas som en sänkning av det transkutana elektriska motståndet (TER) under ett tröskelvärde (12). För TER för råtthud har ett brytvärde på 5 kΩ valts, baserat på omfattande data för ett brett urval av kemikalier där den absoluta majoriteten av värden antingen låg klart över (ofta > 10 kΩ) eller klart under (ofta < 3 kΩ) detta värde (12). I allmänhet sänker material som inte har någon korrosiv verkan på djur inte TER under detta brytvärde, oavsett om de är hudirriterande eller inte. Vidare kan användning av andra hudberedningar eller annan utrustning ändra brytvärdet, vilket kräver ytterligare validering.
Ett färgämnesbindningssteg ingår i testförfarandet för verifiering av positiva resultat vid TER-värden runt 5 kΩ. Färgämnesbindningssteget visar om ökningen av jongenomsläpplighet beror på fysisk nedbrytning av hornlagret. TER-metoden med råtthud har visats ge förutsägbarhet för korrosiv verkan in vivo på kanin, fastställd med testmetod B.4 (2). Det ska påpekas att in vivo-kanintestet är mycket konservativt med avseende på hudkorrosiv verkan och hudirritation jämfört med lapptest på mänsklig hud (15).
1.5 BESKRIVNING AV TESTMETODEN
1.5.1 Djur
Råtta är den föredragna arten eftersom råttors hudkänslighet för kemikalier i detta test har visats tidigare (10). Åldern (då huden tas till vara) och råttstammen är av särskild betydelse för att säkerställa att hårsäckarna är i vilofas innan den vuxna hårtillväxten börjar.
Rygg- och flankhår från unga, ungefär 22 dagar gamla han- eller honråttor (erhållna från Wistar eller en jämförbar stam) avlägsnas noggrant med en liten sax. Därefter tvättas djuren noga och torkas sedan omsorgsfullt medan det klippta området hålls ner i antibiotikalösning (som innehåller t.ex. streptomycin, penicillin, kloramfenikol och amfotericin, i koncentrationer som hämmar bakterietillväxt). Djuren tvättas på nytt med antibiotika på tredje eller fjärde dagen efter den första tvätten och används inom 3 dagar från den andra tvätten, då hornlagret har återhämtat sig från hårborttagningen.
1.5.2 Beredning av hudbitar
Djuren avlivas med en human metod när de är 28-30 dagar gamla. Åldern är av stor betydelse. Huden från rygg och flank från varje djur avlägsnas sedan och överflödet av underhudsfett skalas försiktigt bort från huden. Hudbitar med en diameter på ungefär 20 mm tas ut. Hudbitarna kan lagras innan de används om det kan visas att positiva och negativa kontrolldata är likvärdiga med dem som erhålls för färsk hud.
Varje hudbit placeras över änden av ett PTFE-rör (polytetrafluoreten) så att överhuden är i kontakt med röret. En o-ring i gummi presspassas över rörets ände för att hålla huden på plats och överflödig vävnad putsas bort. Rörets och o-ringens mått visas i figur 2. O-ringen i gummi förseglas sedan till PTFE-rörets ände med vaselin. Röret fästs med en fjäderklämma i en kammare som innehåller MgSO4-lösning (154 mM) (figur 1). Hudbiten ska vara helt nedsänkt i MgSO4-lösningen. Upp till 10–15 hudbitar kan erhållas från ett enda råttskinn.
Innan testningen börjar mäts de båda hudbitarnas elektriska motstånd som ett kvalitetskontrollförfarande för varje djurhud. Båda bitarna ska ge värden för motståndet som är större än 10 kΩ för att återstoden av hudbitarna ska kunna användas för testet. Om motståndet är mindre än 10 kΩ ska återstående hudbitar från den aktuella huden kastas.
1.5.3 Applicering av test- och kontrollämnen
Parallella positiva och negativa kontroller ska användas för varje studie för att säkerställa att försöksmodellen fungerar adekvat. Hudbitar från ett enda djur ska användas. Förslag på ämnen för positiv och negativ kontroll är 10 M saltsyra respektive destillerat vatten.
Testämnen i vätskeform (150 μl) appliceras på epidermisytan inuti röret. Vid testning av ämnen i fast form appliceras en tillräcklig mängd för att säkerställa att hela epidermisytan täcks. Därefter tillförs avjoniserat vatten (150 μl) på det fasta ämnet och röret omskakas varsamt. Testämnet bör ha maximal kontakt med huden. För vissa fasta ämnen kan detta uppnås genom uppvärmning till 30 oC så att testämnet smälter eller mjuknar. Alternativt kan ämnet finfördelas till granulat eller pulver.
Varje testämne appliceras på tre hudbitar. Testämnet får verka i 24 timmar vid 20-23 oC och avlägsnas därefter genom sköljning med kranvattenstråle (upp till 30 oC). Sköljningen bör pågå till dess att inget ämne längre fås bort.
1.5.4 TER-mätningar
Hudens elektriska impedans uttryckt som transkutant elektriskt motstånd, TER, mäts med hjälp av växelström med låg spänning med en Wheatstonebrygga (13). Allmänna tekniska data för bryggan är 1–3 V driftspänning, en sinusformad eller rektangulär växelström på 50–1 000 Hz och ett mätområde på minst 0,1–30 kΩ. Den brygga som användes i valideringsstudien mätte induktans, kapacitans och motstånd upp till värden på 2 000 H, 2 000 μF respektive 2 Mω vid frekvenser på 100 Hz eller 1 kHz med användning av seriella eller parallella värden. För mätningen av hudkorrosivitet genom TER-bestämning registrerades motståndet vid frekvensen 100 Hz och seriella värden användes. För att sänka hudens ytspänning tillförs etanol (70 %) i tillräcklig mängd för att täcka epidermisytan. Etanolen får verka några sekunder och hälls sedan ur röret. Därefter behandlas huden med 3 ml MgSO4-lösning (154 mM). För motståndsmätningen (kΩ/hudbit) placeras mätutrustningens elektroder på hudbitens motstående båda ytor (figur 1). Elektrodernas mått och elektrodlängden under krokodilklämman framgår av figur 2. Den inre elektrodklämman hålls uppe på PTFE-röret under motståndsmätningen för att säkerställa att elektrodlängden i magnesiumsulfatlösningen hålls konstant. Den yttre elektroden placeras innanför receptorkammaren så att elektroden ligger mot kammarens botten. Avståndet mellan fjäderklämman och PTFE-rörets botten ska hållas konstant (figur 2) eftersom avståndet påverkar det erhållna motståndsvärdet. Följaktligen ska avståndet mellan den inre elektroden och hudbiten vara konstant och minimalt (1–2 mm).
Om det uppmätta motståndsvärdet överstiger 20 kΩ kan detta bero på att hudbitens epidermisyta är täckt av testämne. För att få bort ämnet kan man t.ex. sluta till PTFE-röret med tummen (som bör skyddas med handske) och skaka om röret i cirka 10 sekunder, hälla ut magnesiumsulfatlösningen och upprepa mätningen med ny MgSO4-lösning.
Såväl testanordningens egenskaper och mått som försöksförfarandet kan påverka de TER-värden som erhålls. Tröskelvärdet på 5 kΩ för hudkorrosivitet togs fram utifrån data som erhållits med den specifika anordning och det specifika förfarande som beskrivs för denna metod. Andra tröskelvärden och kontrollvärden kan gälla om testbetingelserna ändras eller om en annan testanordning används. Därför måste metoden och tröskelvärdena för motstånd kalibreras genom test av ett antal referensstandarder som väljs bland de kemikalier som användes i valideringsstudien (4) (5) eller från liknande kemikalieklasser som de kemikalier som undersöks. En uppsättning lämpliga referenskemikalier listas i tabell 1.
1.5.5 Metoder som bygger på färgämnesbindning
Exponering för vissa icke-korrosiva material kan sänka motståndet under tröskelvärdet 5 kΩ så att joner kan flöda genom hornlagret då det elektriska motståndet minskar (5). Exempelvis kan neutrala organiska ämnen och kemikalier som har ytaktiva egenskaper (bland annat detergenter, emulgeringsmedel och andra ytaktiva ämnen) avlägsna hudlipider och därmed försämra barriärfunktionen och göra huden mer genomsläpplig för joner. Om TER-värdena för testämnena är lägre än eller omkring 5 kΩ utan någon synlig skada bör en bestämning av färgämnespenetrationen göras i kontroll och behandlad vävnad för att fastställa om de erhållna TER-värdena är en följd av ökad genomsläpplighet hos huden eller av hudkorrosivitet (3)(5). Om hudkorrosivitet föreligger och hornlagret är skadat kommer färgämnet sulforhodiamin B snabbt att tränga igenom hudytan och färga underliggande vävnad då det appliceras. Detta färgämne är stabilt mot ett brett urval av kemikalier och påverkas inte av det extraktionsförfarande som beskrivs nedan.
1.5.5.1 Applicering och avlägsnande av färgämnet sulforhodamin B
Efter TER-testet hälls magnesiumsulfatet bort från röret och huden granskas noga med avseende på synlig skada. Om ingen tydlig och mer omfattande skada observeras appliceras 150 μL 10-procentig (vikt i volym) lösning av färgämnet sulforhodamine B (Acid Red 52; C.I. 45100; EINECS-nummer 222-529-8; CAS-nummer 3520-42-1) i destillerat vatten på epidermisytan av alla hudbitar. Lösningen får verka i 2 timmar. Därefter avlägsnas överskott (obundet färgämne) genom tvättning av hudbitarna med kranvattenstråle (upp till rumstemperatur) i ungefär 10 sekunder. Hudbitarna tas försiktigt bort från PTFE-rören och varje hudbit placeras i en burk (t.ex. en 20 ml scintillationsburk i glas) som innehåller destillerat vatten (8 ml). Brukarna omskakas varsamt under 5 minuter för att avlägsna ytterligare överskott (obundet färgämne). Därefter upprepas sköljningen. Hudbitarna tas bort, placeras i burkar som innehåller 5 ml 30-procentig (vikt i volym) natriumdodecylsulfat (SDS) i destillerat vatten och inkuberas över natten vid 60 oC.
Efter inkubationen avlägsnas hudbitarna och slängs. Den kvarvarande lösningen centrifugeras under 8 minuter vid 21 oC (relativ centrifugalkraft cirka 175 x g). Ett 1 ml prov av supernatanten späds ut i volymförhållandet 1:5 (t.ex. 1 ml + 4 ml) med 30-procentig (vikt i volym) SDS i destillerat vatten. Lösningens optiska densitet (OD) mäts vid 565 nm.
1.5.5.2 Beräkning av färgämneshalten
Halten av sulforhodamin B per hudbit beräknas utifrån OD-värdena (5) (färgämnets molära extinktionskoefficient vid 565 nm = 8,7 x l04; molekylvikt = 580). Färgämneshalten bestäms för varje hudbit med hjälp av en lämplig kalibreringskurva och därefter beräknas en medelhalt för alla hudbitarna.
2. DATA
Motståndsvärden (kΩ) och medelhalter av färgämne (μg/hudbit) ska där det lämpar sig för testämnet liksom för positiva och negativa kontroller rapporteras i tabellform (enskilda försöksdata och medelvärden ± standarddeviation), inbegripet data för replikat och upprepade försök, medelvärden och enskilda värden.
2.1 TOLKNING AV RESULTAT
TER-medelvärdena godtas om parallella positiva och negativa kontrollvärden faller inom de godtagbara intervallen för den metod som används på testlaboratoriet. De godtagbara motståndsintervallen för metoden och anordningen som beskrivs ovan ges i följande tabell:
|
Kontroll |
Ämne |
Motståndsintervall (kΩ) |
|
Positiv |
10 M saltsyra |
0,5–1,0 |
|
Negativ |
Destillerat vatten |
10–25 |
Medelhalterna för färgämnesbindning godtas på villkor att värdena för de parallella kontrollerna ligger inom de godtagbara intervallen för metoden. Föreslagna intervall för färgämneshalt för kontrollämnena för den metod och den anordning som beskrivs ovan ges nedan:
|
Kontroll |
Ämne |
Intervall för färgämneshalt (μg/hudbit) |
|
Positiv |
10 M saltsyra |
40–100 |
|
Negativ |
Destillerat vatten |
15–35 |
Testämnet anses vara icke-korrosivt
|
i) |
om TER-medelvärdet för testämnet är högre än 5 kΩ eller |
|
ii) |
TER-medelvärdet är lägre än eller lika med 5 kΩ och
|
Testämnet anses vara korrosivt
|
i) |
om TER-medelvärdet är mindre än eller lika med 5 kΩ och hudbiten är uppenbart skadad eller |
|
ii) |
TER-medelvärdet är lägre än eller lika med 5 kΩ och
|
3. RAPPORTERING
3.1 TESTRAPPORT
Testrapporten måste innehålla följande information:
|
|
Test- och kontrollämnen:
|
|
|
Försöksdjur:
|
|
|
Försöksförhållanden:
|
|
|
Resultat:
|
|
|
Diskussion av resultaten. |
|
|
Slutsatser. |
4. HÄNVISNINGAR
|
1. |
OECD (2001), Harmonised Integrated Classification System for Human Health and Environmental Hazards of Chemical Substances and Mixtures. OECD Series on Testing and Assessment Number 33. ENV/JM/MONO(2001)6, Paris. http://www.olis.oecd.org/olis/2001doc.nsf/LinkTo/env-jm-mono(2001)6. |
|
2. |
Testmethod B.4. Akut toxicitet: hudirriation/hudkorrosion. |
|
3. |
Botham, P.A., Chamberlain, M., Barratt, M.D., Curren, R.D., Esdaile, D.J., Gardner, J.R., Gordon, V.C., Hildebrand, B., Lewis, R.W., Liebsch, M., Logemann, P., Osborne, R., Ponec, M., Regnier, J.F., Steiling, W., Walker, A.P., and Balls, M. (1995), A prevalidation study on in vitro skin corrosivity testing. The report and recommendations of ECVAM Workshop 6. ATLA 23, 219–255. |
|
4. |
Barratt, M.D., Brantom, P.G., Fentem, J.H., Gerner, I., Walker, A.P., and Worth, A.P. (1998), The ECVAM international validation study on in vitro tests for skin corrosivity. 1. Selection and distribution of the test chemicals. Toxic. in Vitro 12, 471–482. |
|
5. |
Fentem, J.H., Archer, G.E.B., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Esdaile, D.J., Holzhütter, H.-G., and Liebsch, M. (1998), The ECVAM international validation study on in vitro tests for skin corrosivity. 2. Results and evaluation by the Management Team. Toxic. in Vitro 12, 483–524. |
|
6. |
OECD (1996), Final Report of the OECD Workshop on Harmonization of Validation and Acceptance Criteria for Alternative Toxicological Test Methods, 62pp. |
|
7. |
Balls, M., Blaauboer, B.J., Fentem. J.H., Bruner. L., Combes, R.D., Ekwall, B., Fielder. R.J., Guillouzo, A., Lewis, R.W., Lovell, D.P., Reinhardt, C.A., Repetto, G., Sladowski. D., Spielmann, H., and Zucco, F. (1995), Practical aspects of the validation of toxicity test procedures. The report and recommendations of ECVAM workshops. ATLA 23, s. 129–147. |
|
8. |
ICCVAM (Interagency Coordinating Committee on the Validation of Alternative Methods). (1997), Validation and Regulatory Acceptance of Toxicological Test Methods. NIH Publication No. 97-3981. National Institute of Environmental Health Sciences, Research Triangle Park, NC, USA. http://iccvam.niehs.nih.gov/docs/guidelines/validate.pdf |
|
9. |
ECVAM (1998). ECVAM News & Views. ATLA 26, s. 275–280. |
|
10. |
ICCVAM (Interagency Coordinating Committee on the Validation of Alternative Methods). (2002), ICCVAM evaluation of EpiDermTM, EPISKINTM (EPI-200), and the Rat Skin Transcutaneous Electrical Resistance (TER) assay: In Vitro test methods for assessing dermal corrosivity potential of chemicals. NIH Publication No. 02-4502. National Toxicology Program Interagency Center for the Evaluation of Alternative Toxicological Methods, National Institute of Environmental Health Sciences, Research Triangle Park, NC, USA. http://iccvam.niehs.nih.gov/methods/epiddocs/epis_brd.pdf. |
|
11. |
OECD (2002), Extended Expert Consultation Meeting on The In Vitro Skin Corrosion Test Guideline Proposal, Berlin, 1st - 2nd November 2001, Secretariat's Final Summary Report, 27th March 2002, OECD ENV/EHS, available upon request from the Secretariat |
|
12. |
Oliver, G.J.A., Pemberton, M.A., and Rhodes, C. (1986), An in vitro skin corrosivity test -modificationsand validation. Fd. Chem. Toxicol. 24, s. 507–512. |
|
13. |
Botham, P.A., Hall, T.J., Dennett, R., McCall, J.C., Basketter, D.A., Whittle, E., Cheeseman, M., Esdaile, D.J., and Gardner, J. (1992), The skin corrosivity test in vitro: results of an interlaboratory trial. Toxic. in Vitro 6, s. 191–194. |
|
14. |
Worth AP, Fentem JH, Balls M, Botham PA, Curren RD, Earl LK, Esdaile DJ, Liebsch M (1998), An Evaluation of the Proposed OECD Testing Strategy for Skin Corrosion. ATLA 26, s. 709–720. |
|
15. |
Basketter, D.A., Chamberlain, M., Griffiths, H.A., Rowson, M., Whittle, E., York, M. (1997), The classification of skin irritants by human patch test. Fd. Chem. Toxicol. 35, s. 845–852. |
|
16. |
Oliver G.J.A, Pemberton M.A and Rhodes C. (1988), An In Vitro model for identifying skin-korrosivt chemicals. I. Initial Validation. Toxic. in Vitro. 2, s. 7–17. |
Figur 1
Anordning för TER-analys av råtthud
Figur 2
Mått på polytetrafluoretenrör (PFTE-rör), mottagarrör och elektroder
Kritiska faktorer för den anordning som visas ovan:
|
— |
PTFE-rörets innerdiameter. |
|
— |
Elektrodernas längd jämfört med PTFE-rörets och mottagarrörets. Hudbiten ska inte vara i kontakt med elektroderna och en standardlängd av elektroden ska vara i kontakt med MgSO4-lösningen. |
|
— |
Mängden MgSO4-lösning i mottagarröret ska ge ett visst djup i vätskan jämfört med nivån i PFTE-röret, såsom visas i figur 1. |
|
— |
Hudbiten ska vara ordentligt fäst vid PFTE-röret, så att det elektriska motståndet är ett verkligt mått på hudens egenskaper. |
B.40B IN VITRO-TEST AV HUDKORROSIVITET: TEST MED MODELL AV HUMAN HUD
1. METOD
Denna testmetod motsvarar OECD TG 431 (2004).
1.1 INLEDNING
Hudkorrosivitet innebär att en irreversibel vävnadsskada uppstår i huden som en följd av att ett testmaterial [definierat enligt det internationella systemet för klassificering och märkning av kemikalier (GHS)] (1) appliceras. Denna metod bygger på ett förfarande som innebär att testmaterialets hudkorrosivitet inte testas på levande djur.
Bedömning av hudkorrosiviteten har normalt inneburit användning av försöksdjur (2). På grund av den smärta och det lidande som försöksdjuren utsätts för har översynen av testmetod B.4 gjorts på ett sådant sätt att bedömningen av hudkorrosiviteten går att göra med alternativa metoder in vitro, så att försöksdjur inte längre behöver användas.
Ett första steg mot att definiera alternativa testmetoder som kunde användas för testning av hudkorrosiviteten var att genomföra förvalideringsstudier (3). Efter detta gjordes en formell valideringsstudie (6)(7)(8) av in vitro-metoder för att bedöma hudkorrosiviteten (4)(5). Utfallet av dessa studier sammantaget med annan publicerad litteratur (9) ledde till rekommendationen att följande tester kan användas för bedömning av hudkorrosiviteten in vivo (10)(11)(12)(13): ett test där en modell av human hud används (denna metod) och bestämning av transkutant elektriskt motstånd (se testmetod B.40).
Enligt valideringsstudier gör tester med modeller av human hud (3)(4)(5)(9) det möjligt att på ett tillförlitligt sätt skilja mellan kända hudkorrosiva ämnen och icke-korrosiva ämnen. Testprotokollet kan också ge en indikation för att skilja mellan mycket hudkorrosiva och mindre hudkorrosiva ämnen.
Testet enligt denna metod gör det möjligt att identifiera kemikalier och blandningar som är hudkorrosiva. Dessutom gör testet det möjligt att identifiera icke hudkorrosiva ämnen och blandningar med stöd av annan befintlig information (t.ex. pH, förhållandet mellan struktur och aktivitet, data som gäller människor och/eller djur) (1)(2)(13)(14). Testet ger ingen tillräcklig information om hudirritation, inte heller ger det möjlighet till den underklassificering som är möjlig enligt det internationella systemet för klassificering och märkning av kemikalier (GHS) (1).
För en fullständig utvärdering av lokala hudeffekter efter en enda hudexponering rekommenderas den sekventiella teststrategi som finns som tillägg till testmetod B.4 (2) och som anges i det internationella systemet för klassificering och märkning (1). Denna teststrategi inbegriper in vitro-tester av hudkorrosiviteten (enligt den häri beskrivna metoden) och hudirritation innan tester på levande djur övervägs.
1.2 DEFINITIONER
Hudkorrosion in vivo: innebär att en irreversibel skada uppstår i huden, det vill säga synlig nekros genom epidermis och ner i dermis efter att ett testämne har applicerats och fått verka i upp till fyra timmar. Korrosiva reaktioner kännetecknas av sår, blödning, blodiga sårskorpor och i slutet av observationsperioden på 14 dagar av missfärgning på grund av blekning av huden, håravfall på hela hudområden och ärrbildning. Histopatologi bör övervägas för att bedöma tveksamma skador.
Cellviabilitet: en parameter som är ett mått på en cellpopulations totala aktivitet (t.ex. cellulära mitokondriska dehydrogenasers förmåga att reducera det vitala färgämnet MTT). Denna förmåga är korrelerad till det totala antalet celler och till cellernas viabilitet, beroende på vilken slutpunkt som mäts och på försöksdesignen.
1.3 REFERENSÄMNEN
Tabell 1
Referenskemikalier
|
Namn |
EINECS-nr |
CAS-nr |
|
|
1,2-Diaminopropan |
201-155-9 |
78-90-0 |
Starkt korrosiv |
|
Akrylsyra |
201-177-9 |
79-10-7 |
Starkt korrosiv |
|
2-tert-butylfenol |
201-807-2 |
88-18-6 |
Korrosiv |
|
Kaliumhydroxid (10 %) |
215-181-3 |
1310-58-3 |
Korrosiv |
|
Svavelsyra (10 %) |
231-639-5 |
7664-93-9 |
Korrosiv |
|
Oktansyra (kaprylsyra) |
204-677-5 |
124-07-02 |
Korrosiv |
|
4-Amino-1,2,4-triazol |
209-533-5 |
584-13-4 |
Icke-korrosiv |
|
Eugenol |
202-589-1 |
97-53-0 |
Icke-korrosiv |
|
Fenetylbromid |
203-130-8 |
103-63-9 |
Icke-korrosiv |
|
Tetrakloretylen |
204-825-9 |
27-18-4 |
Icke-korrosiv |
|
Isostearinsyra |
250-178-0 |
30399-84-9 |
Icke-korrosiv |
|
4-(Metyltio)-benzaldehyd |
222-365-7 |
3446-89-7 |
Icke-korrosiv |
De flesta av kemikalierna i förteckningen har tagits från den förteckning över kemikalier som valdes för den internationella valideringsstudien ECVAM (4). Urvalet bygger på följande kriterier:
|
i) |
Lika många korrosiva som icke-korrosiva ämnen. |
|
ii) |
Kommersiellt tillgängliga ämnen som täcker de flesta relevanta kemikalieklasser. |
|
iii) |
Medtagande av såväl starkt korrosiva som mindre korrosiva ämnen för att göra det möjligt att särskilja ämnen på grundval av hudkorrosiviteten. |
|
iv) |
Val av kemikalier som kan hanteras i ett laboratorium utan att utgöra någon annan allvarlig risk utöver hudkorrosiviteten. |
1.4 PRINCIPEN FÖR TESTMETODEN
Testämnet appliceras på ytan av en tredimensionell modell av human hud som består minst av en rekonstruerad epidermis med funktionellt hornlager. Korrosiva ämnen identifieras genom sin förmåga att efter fastställda exponeringsperioder framkalla en sänkning av cellernas viabilitet (vilket kan fastställas t.ex. med MTT-reduktionsbestämning (15)) under definierade tröskelvärden. Principen för bestämningen följer hypotesen om att kemikalier är korrosiva om de kan penetrera hornlagret genom diffusion eller erosion och är tillräckligt cytotoxiska för att orsaka celldöd i underliggande cellskikt.
1.4.1 Förfarande
1.4.1.1 Modeller av human hud
Modeller av human hud kan konstrueras eller erhållas från kommersiella källor (t.ex. EpiDermTM- och EPISKINTM-modellerna) (16)(17)(18)(19) eller utvecklas eller konstrueras på försökslaboratoriet (20)(21). Det är vedertaget att användning av human hud är föremål för nationella och internationella etiska hänsyn och begränsningar. Alla nya modeller ska valideras (minst i den omfattning som beskrivs i 1.4.1.1.2). Modeller av human hud som används för detta test måste uppfylla följande villkor:
1.4.1.1.1
Epitelet ska konstrueras av humana keratinocyter. Flera lager av viabla epitelceller ska finnas under ett fungerande hornlager. Hudmodellen kan också ha ett lager med en stromal komponent. Hornlagret ska bestå av flera skikt och ha den lipidprofil som krävs för att barriärfunktionen ska räcka för att motstå snabb penetrering av cytotoxiska markörer. Modellens inneslutningsegenskaper ska vara sådana att ämnen inte ska kunna passera runt hornlagret till den viabla vävnaden. Om testkemikalier kan passera runt hornlagret blir modelleringen av hudexponering dålig. Hudmodellen får inte vara kontaminerad med bakterier (inbegripet mykoplasma) eller svamp.
1.4.1.1.2
Viabiliteten kvantifieras normalt genom användning av MTT eller andra färgämnen som omvandlas metaboliskt. I dessa fall ska den optiska densiteten (OD) för extraherat (löst) färgämne från den negativa kontrollvävnaden vara minst 20 gånger större än OD för enbart extraktionslösningsmedlet (se (22) för en översikt). Den negativa kontrollvävnaden ska vara stabil i kultur (ge motsvarande resultat vid viabilitetsmätning) under hela exponeringstiden under försöket. Hornlagret ska vara tillräckligt robust för att stå emot snabb penetrering av vissa cytotoxiska markörer (t.ex. 1-procentig Triton X-100). Denna egenskap kan uppskattas på grundval av den exponeringstid som krävs för att minska cellviabiliteten med 50 procent (ET50) (denna tid är t.ex. > 2 timmar för EpiDermTM- och EPISKINTM-modellerna). Vävnaden ska vara reproducerbar över tiden och helst mellan laboratorier. Dessutom ska den kunna användas för att förutsäga den korrosiva potentialen hos referenskemikalier (se tabell 1) då den används enligt det valda försöksprotokollet.
1.4.1.2 Applicering av test- och kontrollämnen
Två vävnadsreplikat används för varje behandling (exponeringstid), även för kontrollerna. Ämnen i vätskeform måste appliceras i tillräcklig mängd för att täcka hudens yta (minst 25 μl/cm2). Ämnen i fast form måste appliceras i tillräcklig mängd för att täcka huden och därefter måste ämnet fuktas med avjoniserat eller destillerat vatten för att säkerställa god kontakt med huden. I vissa fall måste ämnen i fast form pulveriseras före appliceringen. Appliceringsmetoden måste vara lämplig för testämnet (se hänvisning 5). I slutet av exponeringsperioden ska testämnet omsorgsfullt tvättas bort från hudytan med en lämplig buffert eller med 0,9-procentig NaCl.
Parallella positiva och negativa kontroller ska användas i varje studie för att säkerställa att försöksmodellen fungerar adekvat. Föreslagna ämnen för positiv kontroll är isättika eller 8N KOH. Föreslagna ämnen för negativ kontroll är 0,9-procentig NaCl eller vatten.
1.4.1.3 Mätning av cellernas viabilitet
Endast kvantitativa, validerade metoder får användas för att mäta cellernas viabilitet. Vidare måste viabilitetsmåttet vara förenligt med användning av den tredimensionella vävnadskonstruktionen. Ospecifik bindning av färgämne får inte störa viabilitetsmätningen. Proteinbindande färgämnen och sådana som inte genomgår metabolisk omvandling (t.ex. neutralrött) är därför olämpliga. Den oftast använda bestämningsmetoden är MTT-reduktion [3-(4,5-dimetylthiazol-2yl)-2,5-difenyltetrazoliumbromid; EINECS-nummer 206-069-5, CAS-nummer 298-93-1)] , som har visat sig ge exakta och reproducerbara resultat (5), men även andra metoder kan användas (t.ex. 0,3-1 mg/ml) vid en lämplig inkubationstemperatur under 3 timmar. Den utfällda blå formazanprodukten extraheras sedan med lösningsmedel (isopropanol) och formazanhalten mäts genom OD-bestämning vid en våglängd mellan 540 och 595 nm.
Kemisk inverkan av testämnet på färgämnet kan likna inverkan av cellulär metabolism och leda till en falsk bedömning av viabiliteten. Detta har visats inträffa då ett testämne inte avlägsnas helt från huden genom sköljning (9). Om testämnet påverkar färgämnet direkt måste ytterligare kontroller användas för att upptäcka och korrigera för dessa testämnens störning av viabilitetsmätningarna (9)(23).
2. DATA
För varje vävnad ska OD-värden och beräknade procentvärden för cellviabilitet rapporteras i tabellform såväl för testämne som för positiva och negativa kontroller, inbegripet data för replikat och upprepade försök, medelvärden och enskilda värden
2.1 TOLKNING AV RESULTAT
De OD-värden som erhållits för varje testprov kan användas för att beräkna en procentuell viabilitet i förhållande till den negativa kontrollen, som sätts till 100 %. Tröskelvärdet för cellviabilitet som skiljer korrosiva ämnen från icke-korrosiva (eller som skiljer mellan olika korrosiva klasser) eller det statistiska förfarande eller de statistiska förfaranden som används för att utvärdera resultaten och för att identifiera korrosiva ämnen måste definieras klart och dokumenteras. Dessutom ska lämpligheten visas. Generellt fastställs dessa tröskelvärden vid optimeringen av försöket under en förvalideringsfas och bekräftas under en valideringsstudie. Ett exempel är förutsägbarheten för korrosivitet associerad till EpiDermTM-modellen (9).
Testämnet anses vara korrosivt för huden
|
i) |
om viabiliteten efter 3 minuters exponering är mindre än 50 % eller |
|
ii) |
om viabiliteten efter 3 minuters exponering är större än eller lika med 50 % och viabiliteten efter 1 timmes exponering är mindre än 15 %. |
Testämnet anses vara icke-korrosivt för huden
|
i) |
om viabiliteten efter 3 minuters exponering är större än eller lika med 50 % och viabiliteten efter 1 timmes exponering är större än eller lika med 15 %. |
3. RAPPORTERING
3.1 TESTRAPPORT
Testrapporten måste innehålla följande information:
|
|
Test- och kontrollämne:
|
|
|
Motivering av hudmodellen och det använda försöksprotokollet. |
|
|
Försöksförhållanden:
|
|
|
Resultat:
|
|
|
Diskussion av resultaten. |
|
|
Slutsats. |
4. HÄNVISNINGAR
|
1. |
OECD (2001), Harmonised Integrated Classification System for Human Health and Environmental Hazards of Chemical Substances and Mixtures. OECD Series on Testing and Assessment Number 33. ENV/JM/MONO(2001)6, Paris. http://www.olis.oecd.org/olis/2001doc.nsf/LinkTo/env-jm-mono(2001)6. |
|
2. |
Testmetod B.4. Akut toxicitet: hudirriation/hudkorrosion. |
|
3. |
Botham, P.A., Chamberlain, M., Barratt, M.D., Curren, R.D., Esdaile, D.J., Gardner, J.R., Gordon, V.C., Hildebrand, B., Lewis, R.W., Liebsch, M., Logemann, P., Osborne, R., Ponec, M., Regnier, J.F., Steiling, W., Walker, A.P., and Balls, M. (1995), A prevalidation study on in vitro skin corrosivity testing. The report recommendations of ECVAM Workshop 6 ATLA 23, s. 219–255. |
|
4. |
Barratt, M.D., Brantom, P.G., Fentem, J.H., Gerner, I., Walker, A.P., and Worth, A.P. (1998), The ECVAM international validation study on in vitro tests for skin corrosivity. 1. Selection and distribution of the test chemicals. Toxic. In Vitro 12, s. 471–482. |
|
5. |
Fentem, J.H., Archer, G.E.B., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Esdaile, D.J., Holzhutter, H.G. and Liebsch, M. (1998), The ECVAM international validation study on in vitro tests for skin corrosivity. 2. Results and evaluation by the Management Team. Toxic. In Vitro 12, s. 483–524. |
|
6. |
OECD (1996), Final Report of the OECD Workshop on Harmonization of Validation and Acceptance Criteria for Alternative Toxicological Test Methods, 62pp. |
|
7. |
Balls, M., Blaauboer, B.J., Fentem, J.H., Bruner, L., Combes, R.D., Ekwall, B., Fielder, R.J., Guillouzo, A., Lewis, R.W., Lovell, D.P., Reinhardt, C.A., Repetto, G., Sladowski, D., Spielmann, H., and Zucco, F. (1995), Practical aspects of the validation of toxicity test procedures. Test report and recommendations of ECVAM workshops. ATLA 23, s. 129–147. |
|
8. |
ICCVAM (Interagency Coordinating Committee on the Validation of Alternative Methods). (1997), Validation and Regulatory Acceptance of Toxicological Test Methods. NIH Publication No. 97-3981. National Institute of Environmental Health Sciences, Research Triangle Park, NC, USA. http://iccvam.niehs.nih.gov/docs/guidelines/validate.pdf. |
|
9. |
Liebsch, M., Traue, D., Barrabas, C., Spielmann, H., Uphill, P., Wilkins, S., McPherson, J.P., Wiemann, C., Kaufmann, T., Remmele, M. and Holzhutter, H.G. (2000), The ECVAM prevalidation study on the use of EpiDerm for skin Corrosivity testing. ATLA 28, pp., s. 371–401. |
|
10. |
ECVAM (1998), ECVAM News & Views. ATLA 26, s. 275–280. |
|
11. |
ECVAM (2000), ECVAM News & Views. ATLA 28, s. 365–67. |
|
12. |
ICCVAM (Interagency Coordinating Committee on the Validation of Alternative Methods), (2002), ICCVAM evaluation of EpiDermTM, EPISKINTM (EPI-200), and the Rat Skin Transcutaneous Electrical Resistance (TER) assay: In Vitro test methods for assessing dermal corrosivity potential of chemicals. NIH Publication No. 02-4502. National Toxicology Program Interagency Center for the Evaluation of Alternative Toxicological Methods, National Institute of Environmental Health Sciences, Research Triangle Park, NC, USA. http://iccvam.niehs.nih.gov/methods/epiddocs/epis_brd.pdf. |
|
13. |
OECD (2002), Extended Expert Consultation Meeting on The In Vitro Skin Corrosion Test Guideline Proposal, Berlin, 1st — 2nd November 2001, Secretariat's Final Summary Report, 27th March 2002, OECD ENV/EHS, available upon request from the Secretariat |
|
14. |
Worth AP, Fentem JH, Balls M, Botham PA, Curren RD, Earl LK, Esdaile DJ, Liebsch M (1998). An Evaluation of the Proposed OECD Testing Strategy for Skin Corrosion. ATLA 26: 709-720. |
|
15. |
Mosmann, T. (1983), Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity asssays. J.Immunol. Meth. 65, s. 55–63. |
|
16. |
Cannon, C.L., Neal, P.J., Southee, J.A., Kubilus, J., and Klausner, M., (1994), New epidermal model for dermal irritancy testing. Toxic. In Vitro 8, s. 889–891. |
|
17. |
Ponec, M., Boelsma, E., Weerheim, A., Mulder, A., Boutwstra, J., and Mommaas, M., (2000), Lipid and ultrastructural characterization of reconstructed skin models. International Journal of Pharmaceutics. s. 203, 211–225. |
|
18. |
Tinois E, Gaetani Q, Gayraud B, Dupont D, Rougier A, Pouradier DX (1994), The Episkin model: Successful reconstruction of human epidermis in vitro. In In vitro Skin Toxicology. Edited by A Rougier, AM Goldberg and HI Maibach: s. 133–140 |
|
19. |
Tinois E, Tiollier J, Gaucherand M, Dumas H, Tardy M, Thivolet J (1991), In vitro and post -transplantation differentiation of human keratinocytes grown on the human type IV collagen film of a bilayered dermal substitute. Experimental Cell Research 193:310-319 |
|
20. |
Parentau, N.L., Bilbo, P., Molte, C.J., Mason, V.S., and Rosenberg, H. (1992), The organotypic culture of human skin keratinocytes and fibroblasts to achieve form and function. Cytotechnology 9, s. 163–171. |
|
21. |
Wilkins, L.M., Watson, S.R., Prosky, S.J., Meunier, S.F., Parentau, N.L. (1994), Development of a bilayered living skin construct for clinical applications. Biotechnology and Bioengineering 43/8, s. 747–756. |
|
22. |
Marshall, N.J., Goodwin, C.J., Holt, S.J. (1995), A critical assessment of the use of microculture tetrazolium assays to measure cell growth and function. Growth Regulation 5, s. 69–84. |
|
23. |
Fentem, J.H., Briggs, D., Chesne, C., Elliot, G.R., Harbell, J.W., Heylings, J.R., Portes, P., Rouget, R., and van de Sandt, J.J.M., and Botham, P.A. (2001), A prevalidation study on in vitro tests for acute skin irritation: results and evaluation by the Management Team. Toxic. InVitro 15, s. 57–93. |
B.41 3T3 NRU-FOTOTOXICITETSTEST IN VITRO
1. METOD
Denna testmetod motsvarar OECD TG 432 (2004).
1.1 INLEDNING
Fototoxicitet definieras såsom toxisk respons som framkallas när ett ämne appliceras på kroppen och responsen sedan antingen utlöses eller ökar (tydligt vid låga dosnivåer) efter exponering för ljus eller toxisk respons som framkallas genom hudbestrålning efter administrering av en kemikalie till organismen.
3T3 NRU-fototoxicitetstestet in vitro används för att identifiera den fototoxiska potentialen hos ett testämne vilken induceras av den exciterade kemikalien efter exponering för ljus. Testet ger en utvärdering av foto-cytotoxiciteten genom att mäta den relativa minskningen av viabilitet hos celler som exponeras för kemikalien i närvaro respektive i frånvaro av ljus. Ämnen som identifieras med detta test är sannolikt fototoxiska in vivo efter administrering till organismen och fördelning i huden eller efter applicering på hudens yta.
Många typer av kemikalier har rapporterats ge upphov till fototoxiska effekter (1)(2)(3)(4). Deras gemensamma egenskap är förmågan att absorbera ljusenergi inom solljusets våglängdsområde. Enligt fotokemins första lag (Grotthaus-Drapers lag) är tillräcklig absorption av ljuskvanta förutsättningen för en fotoreaktion. Innan man planerar biologisk testning av en kemikalie bör man därför fastställa den berörda kemikaliens absorptionsspektrum avseende UV/synligt ljus enligt OECD:s testriktlinjer 101. Om den molära extinktions- eller absorptionskoefficienten ligger under 10 liter × mol--1 × cm-1 har kemikalien troligen ingen fotoreaktiv potential och behöver sannolikt inte testas med 3T3 NRU-fototoxicitetstest in vitro eller annat biologiskt test av skadliga fotokemiska effekter (1)(5) (bilaga 1).
3T3 NRU-fototoxicitetstest in vitro utvärderades nyligen med avseende på tillförlitlighet och relevans (6)(7)(8)(9). 3T3 NRU-fototoxicitetstestet in vitro visades ha förutsägbarhet vad gäller akuta fototoxiska effekter på djur och människa in vivo. Testet är inte utformat för att förutsäga andra skadliga verkningar som kan uppstå genom kombinerad verkan av en kemikalie och ljus, t.ex. fotogenotoxicitet, fotoallergi eller fotokarcinogenitet. Testet är inte heller avsett att användas för bedömning av fototoxisk potential. Det har inte heller utformats för att förutsäga indirekta mekanismer kopplade till fototoxicitet, effekter av metaboliter av testämnet eller effekten av blandningar.
Metaboliserande system är ett allmänt krav för alla in vitro-test som används för förutsägelse av genotoxisk och karcinogen potential, men när det gäller fototoxicitet finns hittills bara några få exempel när metabolisk omvandling krävs för att ett fototoxin ska verka in vivo eller in vitro. Det är därför varken nödvändigt eller vetenskapligt motiverat att det aktuella testet genomförs med ett system som har metabolisk aktivering.
1.2 DEFINITIONER
strålning: Intensiteten hos ultraviolett (UV) eller synligt ljus som når en yta, uttryckt i W/m2 eller mW/cm2.
ljusdos: Den mängd (= intensitet x tid) ultraviolett (UV) eller synlig strålning som når en yta, uttryckt i Joule (= W x s) per ytenhet, t.ex. J/m2 eller J/cm2.
uv-ljusets våglängdsområde: Den uppdelning som rekommenderas av CIE (Commission Internationale de L'Eclairage) är: UVA (315-400 nm), UVB (280-315 nm) och UVC (100-280 nm). Även andra uppdelningar förekommer – gränsen mellan UVB och UVA sätts ofta vid 320 nm och UVA kan uppdelas i UV-A1 och UV-A2 med en gräns vid cirka 340 nm.
cellviabilitet: En parameter för att mäta en cellpopulations totala aktivitet (t.ex. upptag av det vitala färgämnet neutralrött i cellens lysosomer). Denna aktivitet korrelerar, beroende på den slutpunkt som uppmäts och den testutformning som används, med det totala antalet celler och deras vitalitet.
relativ cellviabilitet: Cellviabiliteten uttryckt i förhållande till negativa kontroller (med lösningsmedel) som har tagits fortlöpande under hela testförfarandet (+ UV eller – UV), utan behandling med testkemikalie.
PIF (fotoirritationsfaktor): En faktor som fås genom jämförelse mellan två cytotoxiska koncentrationer (EC50) av testkemikalien som ger samma effekt i frånvaro (- UV) respektive närvaro (+ UV) av icke-cytotoxisk bestrålning med UVA/synligt ljus.
EC50: Den koncentration av testkemikalien som minskar cellviabiliteten med 50 %.
MPE (Mean Photo Effect): Ett mått som har härletts från matematisk analys av de koncentrations-responskurvor som erhålls i frånvaro (- UV) respektive närvaro (+ UV) av icke-cytotoxisk bestrålning med UVA/synligt ljus.
fototoxicitet: Akut toxisk respons som framkallas när huden först exponeras för vissa kemikalier och därefter för ljus, eller som framkallas på liknande sätt genom hudbestrålning efter administrering av en kemikalie till organismen.
1.3 PRINCIP FÖR TESTMETODEN
3T3 NRU-fototoxicitetstestet in vitro bygger på en jämförelse av en kemikalies toxicitet då den testas i närvaro och i frånvaro av en icke-cytotoxisk dos av simulerat solljus. Cytotoxiciteten i detta test uttrycks som en koncentrationsberoende minskning av upptaget av det vitala färgämnet neutralrött vid mätning 24 timmar efter behandling med testkemikalien och påföljande bestrålning (10). Neutralrött är ett svagt katjoniskt färgämne som lätt tränger igenom cellmembranen genom icke-diffusion och ackumuleras intracellulärt i lysosomer. Förändringar i ytan av det känsliga lysosommembranet gör lysosomerna sköra och ger andra förändringar som gradvis blir irreversibla. Sådana förändringar orsakade av verkan av xenobiotika ger minskat upptag och minskad bindning av neutralrött. Därmed går det att skilja mellan viabla, skadade och döda celler, vilket är grunden för detta test.
Balb/c 3T3-celler hålls i kultur under 24 timmar för monolayerbildning. Två 96-hålsplattor per testkemikalie förinkuberas i en timme med åtta olika koncentrationer av testämnet. Därefter exponeras den ena av de två plattorna för en icke-cytotoxisk ljusdos medan den andra plattan hålls i mörker. Sedan byter man ut båda plattornas behandlingsmedium mot kulturmedium och efter ytterligare 24 timmars inkubering bestäms cellviabiliteten med neutralröttupptag. Cellviabiliteten uttrycks som en procentandel jämfört med obehandlade kontroller med lösningsmedel och beräknas för varje testkoncentration. Därefter uppskattas den fototoxiska potentialen genom jämförelse av den koncentrationsrespons som erhålls i närvaro och i frånvaro av strålning, i regel på nivån EC50, dvs. den koncentration som minskar cellviabiliteten med 50 % jämfört med de obehandlade kontrollproverna.
1.4 BESKRIVNING AV TESTMETODEN
1.4.1 Förberedelser
1.4.1.1 Celler
En etablerad musfibroblastcellinje, Balb/c 3T3, klon 31, antingen från ATCC (American Type Culture Collection, Manassas, VA, USA) eller från ECACC (European Collection of Cell Cultures, Salisbury, Wiltshire, UK) användes för valideringsstudien, och det rekommenderas därför att sådana celler erhålls från en välrenommerad cellbank. Andra celler eller cellinjer kan användas med samma testförfarande, förutsatt att odlingsbetingelserna anpassas till cellernas specifika behov, men motsvarigheten måste demonstreras.
Cellerna ska regelbundet kontrolleras med avseende på mykoplasmaförorening och får endast användas om ingen sådan påvisas (11).
Det är viktigt att Balb/c 3T3-cellernas UV-känslighet kontrolleras regelbundet enligt det kvalitetskontrollförfarande som beskrivs i denna metod. Eftersom cellernas UVA-känslighet kan öka med antalet passager ska Balb/c 3T3-celler med lägsta möjliga passagenummer användas, helst under 100 (se avsnitt 1.4.2.2.2 och bilaga 2).
1.4.1.2 Media och odlingsbetingelser
Lämpliga näringslösningar och inkuberingsbetingelser bör användas för vanliga cellpassager och under testförfarandet. För Balb/c 3T3-celler innebär detta t.ex. DMEM (Dulbecco's Modified Eagle's Medium) kompletterat med 10 % serum från nyfödd kalv, 4 mM glutamin, penicillin (100 IE) och streptomycin (100 μg/ml) samt inkubering i fuktig atmosfär vid 37 oC och 5–7,5 % CO2 beroende på bufferten (se avsnitt 1.4.1.4, andra stycket.). Det är särskilt viktigt att man med cellodlingsbetingelserna kan säkerställa en cellcykeltid inom det normala historiska intervallet för de celler eller den cellinje som används.
1.4.1.3 Beredning av kulturer
Celler från djupfrysta stamkulturer sås ut i näringslösningen i lämplig täthet och subkultiveras minst en gång innan de används i 3T3 NRU-fototoxicitetstestet in vitro.
För fototoxicitetstestet sås testcellerna ut i näringslösningen med sådan täthet att odlingarna inte växer samman före avslutat test, dvs. när cellviabiliteten bestäms 48 timmar efter utsådden. För Balb/c 3T3-celler som odlas på 96-hålsplattor rekommenderas tätheten 1 × 104 celler per hål.
För varje testkemikalie sås celler ut på samma sätt på två separata 96-hålsplattor som därefter parallellt används genom hela testförfarandet under identiska odlingsförhållanden, med undantag av den tidsperiod då den ena plattan bestrålas (+ UVA/synligt ljus) och den andra hålls i mörker (– UVA/synligt ljus).
1.4.1.4 Beredning av testämne
Testämnena måste ha beretts omedelbart före användningen, om man inte med stöd av stabilitetsdata kan visa att ämnena är lagringsbeständiga. Det rekommenderas att alla hantering och inledande behandling av cellerna görs under ljusbetingelser som inte kan ge fotoaktivering eller nedbrytning av testämnet före bestrålning.
Testkemikalierna ska lösas upp i buffrade saltlösningar, t.ex. EBSS (Earle's Balanced Salt Solution) eller fysiologiskt balanserade buffertlösningar som inte får innehålla proteinkomponenter, ljusaborberande komponenter (t.ex. pH-indikatorfärger och vitaminer) som kan störa bestrålningsresultatet. Eftersom cellerna under bestrålningen hålls utanför CO2-inkubatorn under cirka 50 minuter, är det viktigt att undvika alkalisering. Om svaga buffertar som EBSS används kan detta uppnås genom att cellerna inkuberas vid 7,5 % CO2. Om cellerna inkuberas vid endast 5 % CO2 bör en starkare buffert väljas.
Testkemikalier med begränsad vattenlöslighet ska lösas upp i lämpligt lösningsmedel. Om lösningsmedel används måste detta förekomma i en konstant volymkoncentration i alla odlingar, dvs. både i de negativa kontrollerna och i alla koncentrationer av testkemikalien, och lösningsmedlet måste vara icke-cytotoxiskt vid denna koncentration. Testkemikalierna bör väljas så att utfällning och grumling i lösningarna undviks.
Som lösningsmedel rekommenderas dimetylsulfoxid (DMSO) och etanol (ETOH). Andra lösningsmedel med låg cytotoxicitet kan vara lämpliga men måste före användning utvärderas med avseende på specifika egenskaper, t.ex. reaktionsbenägenhet med testkemikalien, dämpning av den fototoxiska verkan eller benägenhet att fånga upp radikaler och/eller kemisk stabilitet i lösningsmedlet.
Vortex-blandning och/eller ultraljudsbehandling och/eller uppvärmning till lämplig temperatur kan användas för att underlätta upplösningen, förutsatt att detta inte påverkar testkemikaliens stabilitet.
1.4.1.5 Bestrålningsbetingelser
1.4.1.5.1
Valet av lämplig ljuskälla i kombination med lämplig filtrering är den viktigaste faktorn vid testning av fototoxicitet. I regel förknippas UVA och de synliga våglängdsområdena med fototoxiska reaktioner in vivo (3)(12), medan UVB har mindre relevans men är mycket cytotoxisk – cytotoxiciteten ökar 1 000-faldigt när våglängden minskar från 313 till 280 nm (13). Kriterierna för val av lämplig ljuskälla måste inbegripa kravet att ljuskällan emitterar våglängder som absorberas av testkemikalien (absorptionsspektrum) och att ljusdosen (den dos som uppnås inom skälig tid) är tillräcklig för detektion av kända fotocytotoxiska kemikalier. Dessutom får de våglängder och doser som används inte vara otillbörligt skadliga för testsystemet, vilket inbegriper värmeemission (infraröda området).
Det optimala är att använda simulerat solljus från en solsimulator. Fördelningen av stråldosen från den filterförsedda solsimulatorn ska vara nära den i utomhusljus som ges i (14). I solsimulatorer används både xenonbåglampor och (dopade) kvicksilver-metallhalidbåglampor (15). Fördelen med de senare är att de emitterar mindre värme och är billigare, men ljusets likhet med solljus är mindre än för xenonbåglampor. Eftersom alla solsimulatorer emitterar betydande mängder UVB bör de förses med lämpliga filter för att minska de mycket cytotoxiska UVB-våglängderna. Alla plastmaterial för cellodling innehåller UV-stabilisatorer. Därför bör spektret mätas genom samma typ av lock till 96-hålsplattor som de som används vid bestämningen. Oavsett vilka åtgärder som vidtas för att dämpa delar av spektret genom filtrering eller genom ofrånkomlig inverkan från utrustningen får det spektrum som registreras under filtren inte avvika från standardiserat utomhusdagsljus (14). Ett exempel på bestrålningsfördelning för den filterförsedda solsimulator som användes i valideringsstudien av 3T3-NRU-fototoxicitetstestet in vitro ges i (8)(16). Se även figur 1 i bilaga 2.
1.4.1.5.2
Ljusets (strålningens) intensitet ska före varje fototoxicitetstest rutinmässigt kontrolleras med hjälp av en lämplig bredbands-UV-mätare. Intensiteten ska mätas genom samma typ av lock som används för de 96-hålsplattor som används för toxicitetsbestämningen. UV-mätaren måste ha kalibrerats mot källan och dess prestanda bör kontrolleras. För detta ändamål rekommenderas användning av en referensmätare, dvs. en annan, identiskt kalibrerad UV-mätare av samma typ. Helst ska man även då och då använda en spektroradiometer för att mäta den spektrala strålningen från den filtrerade ljuskällan och för att kontrollera bredbands-UV-mätarens kalibrering.
En dos på 5 J/cm2 (UVA) fastställdes vara icke-toxisk för Balb/c 3T3-celler men tillräckligt stark för att excitera kemikalier så att de utlöste fototoxiska reaktioner (6)(17). För att erhålla 5 J/cm2 inom 50 minuter ställdes t.ex. strålningen in på 1,7 mW/cm2 (figur 2, bilaga 2). Om en annan cellinje eller en annan ljuskälla används måste strålningsdosen eventuellt justeras så att en dosregim kan väljas som inte är skadlig för cellerna men som räcker för att excitera vanliga fototoxiska ämnen. Tiden för ljusexponeringen beräknas på följande sätt:
|
|
(1J = 1 Ws) |

1.4.2 Försöksförhållanden
1.4.2.1 Koncentrationer av testämne
Koncentrationsområdena för en kemikalie som ska testas i närvaro (+ UVA) och i frånvaro (– UVA) av ljus ska först fastställas på lämpligt sätt i förberedande försök. Det kan vara lämpligt att testa lösligheten initialt och efter 60 minuter (eller den behandlingstid som ska användas) eftersom lösligheten kan förändras med tiden eller under loppet av exponeringen. För att undvika toxicitet på grund av olämpliga kulturbetingelser eller på grund av starkt sura eller basiska kemikalier ska pH för cellkulturerna med tillsatt testkemikalie ligga i området 6,5–7,8.
Den högsta koncentrationen av testämne bör ligga inom fysiologiska testbetingelser, dvs. så att osmotisk stress och pH-stress undviks. Beroende på testkemikalien kan det vara nödvändigt att beakta andra fysikalkemiska egenskaper som faktorer som begränsar den högsta testkoncentrationen. För relativt svårlösliga ämnen som inte är toxiska vid koncentrationer upp till mättnadspunkten ska den högsta möjliga koncentrationen testas. I regel ska utfällning av testkemikalien vid någon testkoncentration undvikas. Den maximala koncentrationen av ett testämne ska inte överstiga 1 000 μg/ml och osmolariteten ska inte överstiga 10 mmolar. En geometrisk spädningsserie med 8 testämneskoncentrationer och konstant utspädningsfaktor ska användas (se avsnitt 2.1, andra stycket).
Om information finns (från ett försök för att fastställa koncentrationsområde) om att testkemikalien inte är cytotoxisk upp till gränskoncentrationen i mörkerförsöket (– UVA) men är starkt cytotoxisk vid bestrålning (+ UVA) kan de koncentrationsområden som ska väljas för ljusförsöket (+ UVA) skilja sig från dem som har valts för mörkerförsöket för att kravet på adekvat datakvalitet ska uppfyllas.
1.4.2.2 Kontroller
1.4.2.2.1
Cellerna bör rutinmässigt (ungefär vid var femte passage) kontrolleras med avseende på känslighet för ljuskällan genom att deras viabilitet följs efter exponering för ökande stråldoser. Flera stråldoser, inbegripet nivåer betydligt över dem som används för 3T3 NRU-fototoxicitetstestet, ska användas för denna bestämning. Doserna kvantifieras lättast genom mätningar av ljuskällans UV-del. Cellerna sås med den täthet som används för 3T3 NRU-fototoxicitetstestet in vitro och bestrålas påföljande dag. Cellviabiliteten bestäms en dag senare med neutralröttupptag. Det ska visas att den resulterande högsta icke-cytotoxiska dosen (t.ex. vid 5 J/cm2 (UVA) vid valideringsstudien) räckte för att klassificera referenskemikalierna (tabell 1) korrekt.
1.4.2.2.2
Testet uppfyller kriterierna om de bestrålade negativkontrollerna med lösningsmedel uppvisar en viabilitet på över 80 % då de jämförs med obestrålade negativkontroller med lösningsmedel.
1.4.2.2.3
Den absoluta optiska densiteten (OD540 NRU) för neutralrött extraherat från lösningsmedelskontrollerna visar om de 1×104 cellerna som såtts per hål har tillvuxit med normal fördubblingstid under de två dagar bestämningen pågått. Ett test uppfyller godtagbarhetskriterierna om medelvärdet på OD540 NRU för de obehandlade kontrollerna är ≥ 0,4 (dvs. cirka tjugo gånger bakgrundsabsorbansen för lösningsmedlet).
1.4.2.2.4
En känd fototoxisk kemikalie ska testas samtidigt med varje 3T3 NRU-fototoxicitetstest in vitro. Klorpromazin (CPZ) rekommenderas. Följande godtagbarhetskriterier definierades för test av CPZ enligt standardprotokollet för 3T3 NRU-fototoxicitetstestet in vitro: bestrålad CPZ (+ UVA): EC50 = 0,1 till 2,0 μg/ml, obestrålad CPZ (– UV): EC50= 7,0 till 90,0 μg/ml. Fotoirritationsfaktorn (PIF) ska vara > 6. Historisk prestanda för positivkontrollen ska följas.
Andra fototoxiska kemikalier som är lämpliga med tanke på de testade kemikaliernas kemikalieklass eller löslighetsegenskaper kan användas som parallella positivkontroller i stället för klorpromazin.
1.4.3 Testförfarande (6)(7)(8)(16)(17)
1.4.3.1 Dag 1
Dosera 100 μl näringslösning i de perifera hålen i en 96-håls mikrotitterplatta för vävnadsodling (= blindprov). Dosera 100 μl cellsuspension med 1 x l05 celler/ml i näringslösning (= 1x104 celler/hål) i de återstående hålen. Två plattor ska beredas för varje serie av enskilda testämneskoncentrationer, för lösningsmedel och för positivkontroller.
Inkubera cellerna i 24 timmar (se avsnitt 1.4.1.2) tills de bildar ett halvkonfluent monolayer. Denna inkuberingsperiod är tillräcklig för cellernas återhämtning och vidhäftning samt exponentiell tillväxt.
1.4.3.2 Dag 2
Dekantera näringslösningen från cellerna efter inkuberingen och tvätta två gånger med 150 μl av den buffertlösning som används för inkuberingen. Tillsätt 100 μl av buffertlösningen med lämplig koncentration av testkemikalie eller lösningsmedel (lösningsmedelskontroll). Använd 8 olika koncentrationer av testkemikalien. Inkubera cellerna med testämnet i mörker i 60 minuter (se avsnitt 1.4.1.2 och 1.4.1.4, andra stycket).
Från de två plattor som beretts för varje koncentrationsserie av testämne och kontroll väljs en, i regel slumpmässigt, för bestämning av cytotoxicitet (– UVA) (dvs. kontrollplattan), och en (behandlingsplattan) för bestämning av fotocytotoxicitet (+ UVA).
Utför bestämningens + UVA-del genom att bestråla cellerna vid rumstemperatur i 50 minuter genom locket på 96-hålsplattan med den högsta icke-cytotoxiska stråldosen (se även bilaga 2). Håll obestrålade plattor (– UVA) i en mörk låda i 50 minuter (= UVA-exponeringstiden).
Dekantera testlösningen och tvätta två gånger med 150 μl av den buffertlösning som används för inkuberingen, men utan testämnet. Ersätt bufferten med näringslösning och inkubera (se avsnitt 1.4.1.2) över natt (18–22 timmar).
1.4.3.3 Dag 3
1.4.3.3.1
Cellerna ska undersökas i faskontrastmikroskop med avseende på tillväxt, morfologi och monolayerintegritet. Förändringar av cellernas morfologi och effekter på celltillväxten ska noteras.
1.4.3.3.2
Tvätta cellerna med 150 μl föruppvärmd buffert. Avlägsna tvättlösningen genom att klappa försiktigt. Tillsätt 100 μl av 50 μg/ml neutralrött (NR) (3-amino-7-dimetylamino-2-metylfenazinhydroklorid, Einecs-nummer 209-035-8; CAS-nummer 553-24-2; C.I. 50040) i medium utan serum (16) och inkubera enligt beskrivningen i avsnitt 1.4.1.2 i tre timmar. Avlägsna NR-mediet efter inkuberingen och tvätta cellerna med 150 μl buffert. Dekantera och avlägsna överskottet av buffert genom absorption eller centrifugering.
Tillsätt exakt 150 μl NR-desorptionslösning (färsk lösning av 49 delar vatten + 50 delar etanol + 1 del ättiksyra).
Skaka mikrotiterplattan varsamt på en skakmaskin i 10 minuter tills NR har extraherats ur cellerna och en homogen lösning har bildats.
Mät NR-extraktets optiska densitet vid 540 nm i en spektrofotometer och använd blindproven som referens. Spara data i lämpligt filformat för efterföljande analys.
2. DATA
2.1 KVALITET OCH KVANTITET AV DATA
Erhållna testdata bör ge möjlighet till meningsfull analys av den koncentrationsberoende respons som konstaterats i närvaro och i frånvaro av bestrålning och om möjligt den koncentration av testkemikalien som minskar cellviabiliteten till 50 % (EC50). Om cytotoxicitet detekteras ska både koncentrationsområdet och intervallet för enskilda koncentrationer anpassas så att försöksdata kan sammanställas till en kurva.
För både klart positiva och klart negativa resultat (se avsnitt 2.3, första stycket), kan det räcka med huvudförsöket, underbyggt med ett eller flera preliminära försök för bestämning av område.
Om man får testresultat som är oklara eller kommer nära förutsägbarhetsmodellens gräns måste resultaten verifieras genom upprepat test (se även avsnitt 2.4, andra stycket). I sådana fall bör man överväga att variera försöksbetingelserna. Försöksbetingelser som kan varieras inbegriper koncentrationsområdet eller koncentrationsintervallet, förinkuberingstiden och bestrålningstiden. En kortare exponeringstid kan vara lämplig för kemikalier som är instabila i vatten.
2.2 UTVÄRDERING AV RESULTATEN
En fotoirritationsfaktor (PIF) eller ett MPE-värde (Mean Photo Effect) kan beräknas för utvärderingen av data.
Beräkning av måtten på fototoxicitet (se nedan) kräver att de diskreta värdena för koncentrationsrespons uppskattas med hjälp av en lämplig fullständig koncentrations-responskurva (försöksmodell). Kurvanpassningen till data görs i regel med icke-linjär regression (18). För att bedöma datavariabilitetens inverkan på den anpassade kurvan rekommenderas bootstrap-metoden.
En fotoirritationsfaktor (PIF) beräknas med hjälp av följande formel:
![]()
Om inget värde på EC50 i närvaro eller frånvaro av ljus kan beräknas, går det inte att beräkna någon PIF för testämnet. MPE (Mean Photo Effect) bygger på en jämförelse mellan de fullständiga koncentrations-responskurvorna (19). MPE definieras som det viktade medelvärdet över en representativ uppsättning fotoeffektvärden:

Fotoeffekten PEc vid koncentrationen C definieras som produkten av responseffekten REc och doseffekten DEc dvs. PEc = REc x DEc. Responseffekten REc är skillnaden mellan observerade respons i frånvaro och i närvaro av ljus, dvs. REc = Rc (– UVA) –Rc (+ UVA). Doseffekten ges av
där C* representerar ekvivalenskoncentrationen, dvs. den koncentration där + UVA-responsen är lika stor som – UVA-responsen vid koncentrationen C. Om C* inte kan bestämmas därför att svarsvärdena på +UVA-kurvan systematiskt är högre eller lägre än RC (– UVA) sätts doseffekten till 1. Viktningsfaktorerna wi ges av det högsta responsvärdet, dvs. wi = MAX {Ri (+ UVA), Ri (– UVA) }. Koncentrationsrastret C väljs så att samma antal punkter faller inom vart och ett av de koncentrationsintervall som definieras av de koncentrationsvärden som användes i försöket. Beräkningen av MPE begränsas till det maximala koncentrationsvärde vid vilket minst en av de två kurvorna ännu uppvisar ett responsvärde på minst 10 %. Om denna maximala koncentration är högre än den högsta koncentration som användes i + UVA-försöket sätts den återstående delen av + UVA-kurvan till responsvärdet ”0”. Beroende på om MPE-värdet är större än ett korrekt valt tröskelvärde (MPEc=0,15) eller inte, klassificeras kemikalien som fototoxisk.
Ett programpaket för beräkning av PIF och MPE finns tillgängligt från (20).
2.3 TOLKNING AV RESULTATEN
Baserat på valideringsstudien (8) förutsäger PIF < 2 eller MPE < 0,1”ingen fototoxicitet”. PIF > 2 och <5 eller MPE > 0,1 och < 0,15 förutsäger ”sannolik fototoxicitet”. PIF > 5 eller MPE > 0,15 förutsäger ”fototoxicitet”.
Ett laboratorium som ska börja använda denna bestämningsmetod ska testa de referensmaterial som listas i tabell 1 innan man börjar testa ämnen för fototoxisk bestämning. PIF- eller MPE-värdena ska ligga nära dem som anges i tabell 1.
Tabell 1
|
Kemiskt namn |
EINECS-nr |
CAS-nr |
PIF |
MPE |
Absorptions-topp |
Lösningsmedel (9) |
|
Amiodaron-HCl |
243-293-2 |
[19774-82-4] |
> 3,25 |
0,27–0,54 |
242 nm 300 nm (axel) |
etanol |
|
Kloropromazin-HCl |
200-701-3 |
[69-09-0] |
> 14,4 |
0,33–0,63 |
309 nm |
etanol |
|
Norfloxacin |
274-614-4 |
[70458-96-7] |
> 71,6 |
0,34–0,90 |
316nm |
acetonitril |
|
Antracen |
204-371-1 |
[120-12-7] |
> 18,5 |
0,19–0,81 |
356 nm |
acetonitril |
|
Protoporfyrin IX, Dinatrium |
256-815-9 |
[50865-01-5] |
>4 5,3 |
0,54–0,74 |
402 nm |
etanol |
|
L-Histidin |
|
[7006-35-1] |
no PIF |
0,05–0,10 |
211nm |
vatten |
|
Hexaklorofen |
200-733-8 |
[70-30-4] |
1,1–1,7 |
0,00–0,05 |
299 nm 317nm (axel) |
etanol |
|
Natriumlaurylsulfat |
205-788-1 |
[151-21-3] |
1,0–1,9 |
0,00–0,05 |
Ingen absorption |
vatten |
2.4 TOLKNING AV DATA
Om fototoxisk verkan observeras endast vid den högsta testkoncentrationen (särskilt för vattenlösliga testkemikalier), kan ytterligare faktorer behöva beaktas för bestämning av skadligheten. Dessa faktorer kan inbegripa data om hudabsorption och ackumulering av kemikalien i huden och/eller data från andra test, t.ex. test av kemikalien på djur- eller människohud in vitro eller på hudmodeller.
Om ingen toxicitet påvisas (+ UVA och – UVA) och om dålig löslighet begränsade de koncentrationer som kunde testas bör testämnets lämplighet för bestämningsmetoden ifrågasättas och verifierande testning bör övervägas, t.ex. en annan modell.
3. RAPPORTERING
TESTRAPPORT
Testrapporten ska innehålla följande uppgifter:
|
|
Testämne:
|
|
|
Lösningsmedel:
|
|
|
Celler:
|
|
|
Försöksförhållanden (1) – inkubering före och efter behandlingen:
|
|
|
Försöksförhållanden (2) – behandling med kemikalien:
|
|
|
Försöksförhållanden (3) – bestrålning:
|
|
|
Försöksförhållanden (4) – viabilitetstest med neutralrött (NR):
|
|
|
Resultat:
|
|
|
Diskussion av resultaten. |
|
|
Slutsatser. |
4. HÄNVISNINGAR
|
1. |
Lovell W.W. (1993). A scheme for in vitro screening of substances for photoallergenic potential. Toxic. In Vitro 7: 95–102. |
|
2. |
Santamaria, L. and Prino, G. (1972). List of the photodynamic substances. In ”Research Progress in Organic, Biological and Medicinal Chemistry” Vol. 3 part 1. North Holland Publishing Co. Amsterdam. p XI–XXXV. |
|
3. |
Spielmann, H., Lovell, W.W., Hölzle, E., Johnson, B.E., Maurer, T., Miranda, M.A., Pape, W.J.W., Sapora, O., and Sladowski, D. (1994). In vitro phototoxicity testing: The report and recommendations of ECVAM Workshop 2. ATLA, 22, 314–348. |
|
4. |
Spikes, J.D. (1989). Photosensitization. In ”The science of Photobiology” Edited by K.C. Smith. Plenum Press, New York. 2nd edition, p 79–110. |
|
5. |
OECD (1997) Environmental Health and Safety Publications, Series on Testing and Assessment No.7 ”Guidance Document on Direct Phototransformation of Chemicals in Water” Environment Directorate, OECD, Paris. |
|
6. |
Spielmann, H., Balls, M., Döring, B., Holzhütter, H.G., Kalweit, S., Klecak, G., L'Eplattenier, H., Liebsch, M., Lovell, W.W., Maurer, T., Moldenhauer. F. Moore. L., Pape, W., Pfannbecker, U., Potthast, J., De Silva, O., Steiling, W., and Willshaw, A. (1994). EEC/COLIPA project on in vitro phototoxicity testing: First results obtained with a Balb/c 3T3 cell phototoxicity assay. Toxic. In Vitro 8, 793–796. |
|
7. |
Anon (1998). Statement on the scientific validity of the 3T3 NRU PT test (an in vitro test for phototoxicity), European Commission, Joint Research Centre: ECVAM and DGXI/E/2, 3 November 1997, ATLA, 26, 7–8. |
|
8. |
Spielmann, H., Balls, M., Dupuis, J., Pape, W.J.W., Pechovitch, G. De Silva, O., Holzhütter, H.G., Clothier, R., Desolle, P., Gerberick, F., Liebsch, M., Lovell, W.W., Maurer, T., Pfannenbecker, U., Potthast, J. M., Csato, M., Sladowski, D., Steiling, W., and Brantom, P. (1998). The international EU/COLIPA In vitro phototoxicity validation study: results of phase II (blind trial), part 1: the 3T3 NRU phototoxicity test. Toxic. In Vitro 12, 305–327. |
|
9. |
OECD (2002) Extended Expert Consultation Meeting on The In Vitro 3T3 NRU Phototoxicity Test Guideline Proposal, Berlin, 30th–31th October 2001, Secretariat's Final Summary Report, 15th March 2002, OECD ENV/EHS, available upon request from the Secretariat. |
|
10. |
Borenfreund, E., and Puerner, J.A. (1985). Toxicity determination in vitro by morphological alterations and neutral red absorption. Toxicology Lett., 24, 119–124. |
|
11. |
Hay, R.J. (1988) The seed stock concept and quality control for cell lines. Analytical Biochemistry 171, 225–237. |
|
12. |
Lambert L.A, Warner W.G., and Kornhauser A. (1996) Animal models for phototoxicity testing. In ”Dermatotoxicology”, edited by F.N. Marzulli and H.I. Maibach. Taylor & Francis, Washington DC. 5th Edition, p 515–530. |
|
13. |
Tyrrell R.M., Pidoux M (1987) Action spectra for human skin cells: estimates of the relative cytotoxicity of the middle ultraviolet, near ultraviolet and violet regions of sunlight on epidermal keratinocytes. Cancer Res., 47, 1825–1829. |
|
14. |
ISO 10977. (1993). Photography – Processed photographic colour films and paper prints – Methods for measuring image stability. |
|
15. |
Sunscreen Testing (UV.B) TECHNICAL REPORT, CIE, International Commission on Illumnation, Publication No. 90, Vienna, 1993, ISBN 3 900 734 275. |
|
16. |
ZEBET/ECVAM/COLIPA – Standard Operating Procedure: In Vitro 3T3 NRU Phototoxicity Test. Final Version, 7 September, 1998. 18 pgs. |
|
17. |
Spielmann, H., Balls, M., Dupuis, J., Pape, W.J.W., De Silva, O., Holzhütter, H.G., Gerberick, F., Liebsch, M., Lovell, W.W., and Pfannenbecker, U. (1998) A study on UV filter chemicals from Annex VII of the European Union Directive 76/768/EEC, in the in vitro 3T3 NRU phototoxicity test. ATLA 26, 679–708. |
|
18. |
Holzhütter, H.G., and Quedenau, J. (1995) Mathematical modeling of cellular responses to external signals. J. Biol. Systems 3, 127–138. |
|
19. |
Holzhütter, H.G. (1997). A general measure of in vitro phototoxicity derived from pairs of dose-response curves and its use for predicting the in vivo phototoxicity of chemicals. ATLA, 25, 445–462. |
|
20. |
http://www.oecd.org/document/55/0,2340,en_2649_34377_2349687_1_1 _1_1,00.html |
Bilaga 1
Rollen för 3T3 NRU-fototoxicitetstest vid ett sekventiellt tillvägagångssätt för fototoxicitetstestning av kemikalier

Bilaga 2
Figur 1
Spektral strålningsdistribution från en filtrerad solsimulator

(se avsnitt 1.4.1.5, andra stycket)
Figur 1 ger ett exempel på en godtagbar strålningsdistribution för en filtrerad solsimulator. Värdena kommer från den dopade metallhalidkälla som användes i valideringsstudien för 3T3 NRU-fototoxicitetstest (6)(8)(17). Effekten av två olika filter och den ytterligare filtreringsverkan från locket på 96-hålsplattan visas. H2-filtret användes endast med testsystem som tål en större mängd UVB (test med hudmodell och fotohemolystest med röda blodkroppar). I 3T3 NRU-fototoxicitetstestet användes H1-filtret. Figuren visar den ytterligare filtreringsverkan som plattlocket ger, huvudsakligen i UVB-området, som dock fortfarande lämnar tillräckligt med UVB i strålningsspektret för att excitera kemikalier som normalt absorberar i UVB-området, t.ex. amiodaron (se tabell 1).
Figur 2
Strålningskänslighet hos Balb/c 3T3-celler (mätt i UVA-området)
Cellviabilitet ( % NR-upptag hos mörkerkontroller)

(se avsnitt 1.4.1.5.2, andra stycket, 1.4.2.2.1, 1.4.2.2.2)
Känslighet hos Balb/c 3T3-celler mot bestrålning med den solsimulator som användes i valideringsstudien av 3T3 NRU-fototoxicitetstestet, mätt i UVA-området. Figuren visar resultat erhållna på 7 olika laboratorier vid förvalideringsstudien (1). De två kurvorna med öppna symboler erhölls med åldrade celler (stort antal passager) som man hade varit tvungen att ersätta med nya celler. Kurvorna med symboler i fetstil visar celler med godtagbar strålningstolerans.
Den högsta icke-cytotoxiska stråldosen på 5 J/cm2 erhölls från dessa data (vertikal streckad linje). Den horisontella streckade linjen visar dessutom den maximala godtagbara strålningseffekten som ges i stycke 1.4.2.2.
B.42 HUDSENSIBILISERING: LLNA-METODEN (LOCAL LYMPH NODE ASSAY)
1. METOD
Denna testmetod motsvarar OECD TG 429 (2002).
1.1 INLEDNING
LLNA-metoden (Local Lymph Node Assay) har validerats och godkänts i den utsträckning som krävs för en ny metod (1), (2), (3). LLNA är den andra metoden för bedömning av kemikaliers förmåga att framkalla hudsensibilisering hos djur. Den tidigare metoden (B.6) innebär test på marsvin, speciellt maximeringstest på marsvin och Buehler-test (4).
LLNA-metoden är en alternativ metod för identifiering av kemikalier som framkallar hudsensibilisering, och för bekräftelse av att en kemikalie inte har någon signifikant förmåga att framkalla hudsensibilisering. Detta betyder dock inte nödvändigtvis att LLNA alltid ska användas i stället för test på marsvin., utan att LLNA-metoden är likvärdig och kan användas som ett alternativ i fall där positiva och negativa resultat i regel inte kräver någon ytterligare bekräftelse.
LLNA-metoden har vissa fördelar både vetenskapligt och djurskyddsmässigt sett. Metoden används för att undersöka hudsensibiliseringens induktionsfas, vilket ger tillgång till kvantitativa data som är lämpliga för dos-responsbedömning. Närmare uppgifter om valideringen av LLNA och en översikt av det associerade arbetet finns i ett antal publikationer (5), (6), (7), (8). Det bör dessutom noteras att de ämnen som har milda till moderata sensibiliseringsegenskaper och som anses vara lämpliga som positivt kontrollämne för testmetoder med marsvin, även är lämpliga i fråga om LLNA-metoden (6), (8), (9).
LLNA är en in vivo-metod, och därför går det inte att undvika användningen av djur för bedömning av sensibilisering genom kontakt. Metoden ger dock möjligheter att minska antalet djur som behövs. Dessutom ger LLNA möjligheter till att använda djuren på ett betydligt bättre sätt vid test avseende sensibilisering. LLNA baserar sig på observationer av immunologiska händelser som stimuleras av kemikalier under sensi-biliseringens induktionsfas. Till skillnad från test på marsvin behöver man med LLNA inte framkalla provo-kationsinducerade dermala hypersensitivitetsreaktioner. Dessutom behöver adjuvant inte användas, vilket är fallet vid maximeringstest på marsvin. LLNA är därför en mindre plågsam metod för djuren. Trots fördelarna med LLNA jämfört med traditionella test på marsvin, bör man vara medveten om att LLNA har vissa begränsningar. Det kan därför i vissa fall vara nödvändigt att använda traditionella test på marsvin (till exempel falska negativa fynd med LLNA för vissa metaller eller falska positiva fynd för vissa hudirriterande ämnen (10)).
Se även Inledning, del B.
1.2 PRINCIP FÖR TESTMETODEN
LLNA bygger på principen om att sensibiliserande ämnen inducerar en primär proliferation av lymfocyter i den lymfnod som dränerar det område där kemikalien appliceras. Denna proliferation står i proportion till dosen (och det allergiframkallande ämnets styrka), och är ett enkelt sätt för att få en objektiv och kvantitativ mätning av sensibiliseringen. Med hjälp av LLNA kan man utvärdera denna proliferation som ett dosresponsförhållande där proliferationen i testgruppen jämförs med proliferationen i vehikelbehandlade kontrollgrupper. Därefter bestäms förhållandet mellan proliferationen i behandlings- och kontrollgrupperna, det så kallade stimulationsindexet (Stimulation Index, SI). Indexet måste minst ha värdet tre för att det ska vara motiverat med ytterligare undersökning av ett testämne med avseende på ämnets förmåga att framkalla hudsensibilisering. Den metod som beskrivs här baserar sig på radioaktiv märkning för mätningen av cellproliferationen. Även andra end points kan användas för bedömning av proliferationen, förutsatt att dessa kan motiveras och har tillräcklig vetenskaplig grund, inbegripet fullständiga hänvisningar och beskrivning av metoden.
1.3 BESKRIVNING AV TESTMETODEN
1.3.1 Beredning
1.3.1.1 Förvarings- och utfodringsbetingelser
Djuren ska inhysas individuellt. Temperaturen i testdjurens utrymmen bör vara 22 oC (± 3 oC). Den relativa fuktigheten bör vara minst 30 % och ska helst inte överstiga 70 %, utom när rummet rengörs. Målvärdet bör dock vara 50–60 %. Belysningen bör vara artificiell med omväxlande 12 timmar ljus och 12 timmar mörker. För utfodringen kan konventionellt laboratoriefoder användas, och djuren bör ha obegränsad tillgång till dricksvatten.
1.3.1.2 Förberedelse av djuren
Djuren väljs ut slumpmässigt och förses med märkning så att de kan identifieras individuellt (öronmärkning får dock inte användas). Därefter får djuren acklimatisera sig till laboratoriets betingelser i sina burar i minst 5 dagar före doseringsstart. Innan behandlingen inleds undersöks alla djur för att säkerställa att de inte har några synliga hudskador.
1.3.2 Testbetingelser
1.3.2.1 Testdjur
För detta test används mus. Testdjuren ska vara unga vuxna mushonor av stammen CBA/Ca eller CBA/J. De bör inte ha fått ungar och får inte vara dräktiga. När försöket inleds bör djuren vara 8–12 veckor gamla. Djurens viktvariation bör vara minimal och får inte överskrida 20 % av medelvikten. Övriga stammar eller hanar kan användas förutsatt att det finns tillräckliga data som stöd för att det inte förekommer några betydande stam- eller könsberoende skillnader i LLNA-respons.
1.3.2.2 Tillförlitlighetskontroll
Positiva kontrollämnen används för att demonstrera metodens prestanda och laboratoriets kompetens att genomföra testet på ett framgångsrikt sätt. Ett positivt kontrollämne bör ge en positiv LLNA-respons på den exponeringsnivå som förväntas ge ett stimuleringsindex (SI) > 3 i förhållande till negativa kontrollgruppen. Dosen för det positiva kontrollämnet bör väljas så att induktionen är tydlig men inte för kraftig. Rekommenderade ämnen är hexylkanelaldehyd (CAS-nr 101-86-0, Einecs-nr 202-983-3) och merkaptobensotiazol (CAS-nr 149-30-4, EINECS-nr 205-736-8). I vissa fall kan även andra kontrollämnen som uppfyller de ovan angivna kriterierna användas, förutsatt att tillbörlig motivering kan ges. Trots att det i regel krävs en positiv kontrollgrupp för varje försök, kan det förekomma situationer där testlaboratoriet har tillgång till historiska positiva kontrolldata som påvisar enhetlig och nöjaktig respons över en sexmånadersperiod eller längre. I sådana situationer kan det vara lämpligt att testningen med positiva kontrollämnen görs med längre intervall, dock inte längre än 6 månader. Även om det positiva kontrollämnet bör testas i en vehikel som veterligen framkallar en enhetlig respons (till exempel aceton: olivolja), kan det förekomma vissa föreskriftsreglerade situationer där det även behövs testning i en icke-standardmässig vehikel (kliniskt och kemiskt relevant formel). I en sådan situation bör man utföra test med avseende på en eventuell växelverkan mellan det positiva kontrollämnet och den icke-standardmässiga vehikeln.
1.3.2.3 Antalet djur, dosnivåer och valet av vehikel
Minst fyra djur per dosgrupp och minst tre koncentrationer av testämnet bör användas. Dessutom används en negativ kontrollgrupp som endast behandlas med den använda vehikeln samt, där det är lämpligt, en positiv kontroll. I fall där man samlar in data om individuella djur, bör varje dosgrupp omfatta minst fem djur. Med undantag för behandling med testämnet bör djuren i kontrollgrupperna hanteras på exakt samma sätt som djuren i dosgrupperna.
Doserna och vehikeln bör väljas på grundval av rekommendationerna i hänvisning (1). Doserna väljs ur koncentrationsserien 100 %, 50 %, 25 %, 10 %, 5 %, 2,5 %, 1 %, 0,5 % osv. Tillgängliga data om toxicitet och hudirritation bör beaktas vid valet av de tre koncentrationerna, så att den högsta koncentrationen ger maximal exponering samtidigt som systemisk toxicitet och överdriven lokal hudirritation undviks (2), (11).
Valet av vehikel bör basera sig på en maximering av testkoncentrationerna och lösligheten vid beredning av en lämplig lösning eller suspension för applicering av testämnet. Som vehikel rekommenderas, i preferensordning, aceton/olivolja (4:1 v/v), dimetylformamid, metyletylketon, propylenglykol och dimetylsulpoxid (2), (10). Även andra vehiklar kan användas, förutsatt att tillräcklig vetenskaplig motivering kan ges. I vissa situationer kan det vara nödvändigt att som extra kontroll använda ett kliniskt relevant lösningsmedel eller en kommersiellt tillgänglig blandning där testämnet ingår. Särskild vikt bör fästas vid att säkerställa att hydrofila material inkorporeras i ett vehikelsystem som väter huden och som inte omedelbart rinner bort. Det betyder att helt vattenbaserade vehiklar bör undvikas.
1.3.3 Testförfarande
1.3.3.1 Försöksschema
För testet används följande försöksschema:
|
|
Dag 1 Djur identifieras individuellt och vikten registreras. 25 μl av en lämplig koncentration av testämnet, enbart vehikel eller positivt kontrollämne (enligt det som är lämpligt) appliceras öppet på båda öronens baksida. |
|
|
Dagarna 2 och 3 Upprepa appliceringen enligt dag 1. |
|
|
Dagarna 4 och 5 Ingen behandling. |
|
|
Dag 6 Registrera vikten för varje djur. Injicera 250 μl fosfatbuffrad saltlösning (PBS) som innehåller 20 μCi (7,4e + 8 Bq) 3H-metyltymidin i alla test- och kontrolldjur via svansvenen. Alternativt kan 250 μL PBS som innehåller 2 μCi (7,4e + 7 Bq) 125I-jododeoxyuridin och 10-5 M fluorodeoxyuridin injiceras i alla testdjur via svansvenen. Efter fem timmar avlivas djuren. De aurikulära dränerande lymfnoderna från varje öra skärs bort och poolas i PBS för varje försöksgrupp (metoden med poolad behandlingsgrupp). Alternativt kan lymfnodspar från individuella djur skäras bort och poolas i PBS för varje djur (metoden med individuellt djur). Närmare uppgifter samt scheman för nodidentifiering och dissekering finns i bilaga 1 i hänvisning 10. |
1.3.3.2 Beredning av cellsuspensioner
En enskild cellsuspension bestående av lymfnodsceller (LNC) från poolade behandlingsgrupper eller bilateralt från individuella djur bereds genom försiktig mekanisk sönderdelning med hjälp av ett nät av rostfritt stål (200 mesh). Lymfnodscellerna tvättas två gånger med ett överskott PBS och fälls ut med 5 % triklorättiksyra (TCA) vid 4 oC under 18 timmar (1). Fällningen återupplöses i 1 ml TCA och överförs till skintillationskärl med 10 ml skintillationsvätska för 3H-räkning eller överförs direkt till gammaräkningsrör för 125I-räkning.
1.3.3.3 Bestämning av cellproliferationen (radioaktivt innehåll)
Innehållet av 3H-metyltymidin bestäms genom β-skintillationsräkning såsom sönderfallet per minut (DPM). Innehållet av 125I-jododeoxyuridin bestäms genom 125I-räkning och uttrycks även som DPM. Beroende på vilken metod som har valts uttrycks innehållet som DPM/behandlingsgrupp (metod med pooling) eller DPM/djur (metod med individuella djur).
1.3.3.4 Observationer
1.3.3.4.1
Djuren bör observeras dagligen med tanke på eventuella kliniska tecken – lokal irritation vid appliceringsstället eller tecken på systemisk toxicitet. Alla observationer registreras systematiskt, med individuella registreringar för varje djur.
1.3.3.4.2
Såsom konstateras i avsnitt 1.3.3.1 ska de individuella djurens vikt registreras vid testets början och strax före den schemalagda avlivningen.
1.3.4 Beräkning av resultaten
Resultaten uttrycks i form av ett stimulationsindex (SI). När pooling används erhålls SI genom att dividera det poolade radioaktiva innehållet för varje behandlingsgrupp med innehållet i den poolade vehikelkontrollgruppen, vilket ger ett genomsnittligt SI. När individuella djur används erhålls SI genom att dividera det genomsnittliga DPM/djur inom varje ämnes testgrupp och den positiva kontrollgruppen med det genomsnittliga DPM/djur för lösningsämnet/vehikelkontrollgruppen. Det genomsnittliga SI-värdet för den vehikelbehandlade kontrollgruppen är då 1.
När beräkningen av SI görs för individuella djur kan man göra en statistisk analys av försöksdata. Vid valet av en lämplig metod för den statistiska analysen bör man vara medveten om eventuella olikheter hos varianserna samt andra relaterade problem, som kan göra det nödvändigt att utföra en datatransformation eller en icke-parametrisk statistisk analys. Tolkningen av data kan lämpligen göras genom utvärdering av alla individuella data för behandlade grupper och vehikelkontrollgrupper. Med utvärderingen som grund uppritas sedan en anpassad dosresponskurva med beaktande av konfidensgränserna (8), (12), (13). Man bör dock vara uppmärksam på eventuell ”utanförliggande” respons hos individuella djur inom en grupp. Sådan respons kan göra det nödvändigt att använda alternativa metoder för responsmätningen (till exempel median i stället för medeltal) eller att eliminera respons som ligger utanför området.
För avgöranden rörande positiv respons förutsätts ett stimulationsindex ≥ 3, beaktande av dosrespons och, där det är lämpligt, beaktade av statistisk signifikans (3), (6), (8), (12), (14).
Vid behov kan de erhållna resultaten klargöras ytterligare genom en granskning av testämnets olika egenskaper, inbegripet eventuell strukturell släktskap med kända hudsensibiliserande ämnen, huruvida ämnet orsakar särskilt kraftig hudirritation samt naturen hos den dosrespons som har iakttagits. Dessa och andra aspekter diskuteras närmare i andra källor (7).
2. DATA
Data bör sammanställas i tabellform med genomsnittliga och individuella DPM-värden och stimulationsindex för varje dosgrupp (inbegripet vehikelkontrollgruppen).
3. RAPPORTERING
3.1 TESTRAPPORT
Testrapporten ska innehålla nedan angivna uppgifter.
|
|
Testämne:
|
|
|
Vehikel:
|
|
|
Testdjur:
|
|
|
Testbetingelser:
|
|
|
Tillförlitlighetskontroll:
|
|
|
Resultat:
|
|
|
Diskussion om resultaten:
|
4. HÄNVISNINGAR
|
1. |
Kimber, I. and Basketter, D.A. (1992). The murine local lymph node assay; collaborative studies and new directions: A commentary. Food and Chemical Toxicology 30, 165–169. |
|
2. |
Kimber, I, Derman, R.J., Scholes E.W, and Basketter, D.A. (1994). The local lymph node assay: developments and applications. Toxicology, 93, 13–31. |
|
3. |
Kimber, I., Hilton, J., Dearman, R.J., Gerberick, G.F., Ryan, C.A., Basketter, D.A., Lea, L., House, R.V., Ladies, G.S., Loveless, S.E., Hastings, K.L. (1998). Assessment of the skin sensitisation potential of topical medicaments using the local lymph node assay: An interlaboratory exercise. Journal of Toxicology and Environmental Health, 53, 563–79. |
|
4. |
Testmetod B.6. |
|
5. |
Chamberlain, M. and Basketter, D.A. (1996). The local lymph node assay: status of validation. Food and Chemical Toxicology, 34, 999–1002. |
|
6. |
Basketter, D.A., Gerberick, G.F., Kimber, I. and Loveless, S.E (1996). The local lymph node assay- A viable alternative to currently accepted skin sensitisation tests. Food and Chemical Toxicology, 34, 985–997. |
|
7. |
Basketter, D.A., Gerberick, G.F. and Kimber, I. (1998). Strategies for identifying false positive responses in predictive sensitisation tests. Food and Chemical Toxicology. 36, 327–33. |
|
8. |
Van Och, F.M.M, Slob, W., De Jong, W.H., Vandebriel, R.J., Van Loveren, H. (2000). A quantitative method for assessing the sensitising potency of low molecular weight chemicals using a local lymph node assay: employement of a regression method that includes determination of uncertainty margins. Toxicology, 146, 49–59. |
|
9. |
Dearman, R.J., Hilton, J., Evans, P., Harvey, P., Basketter, D.A. and Kimber, I. (1998). Temporal stability of local lymph node assay responses to hexyl cinnamic aldehyde. Journal of Applied Toxicology, 18, 281-4. |
|
10. |
National Institute of Environmental Health Sciences (1999). The Murine Local Lymph Node Assay: A Test Method for Assessing the Allergic Contact Dermatitis Potential of Chemicals/Compounds: The Results of an Independent Peer Review Evaluation Coordinated by the Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM) and the National Toxicology Program Center for the Evaluation of Alternative Toxicological Methods (NICETAM). NIH Publication No: 99-4494, Research Triangle Park, N.C. (http://iccvam.niehs.nih.gov). |
|
11. |
Testmetod B.4. |
|
12. |
Basketter, D.A., Selbie, E., Scholes, E.W. Lees, D. Kimber, I. and Botham, P.A. (1993) Results with OECD recommended positive control sensitisers in the maximisation, Buehler and local lymph node assays. Food and Chemical Toxicology, 31, 63–67. |
|
13. |
Basketter D.A., Lea L.J., Dickens A., Briggs D., Pate I., Dearman R.J., Kimber I. (1999). A comparison of statistical approaches to the derivation of EC3 values from local lymph node assay dose responses. J. Appl. Toxicology, 19, 261–266. |
|
14. |
Basketter DA, Blaikie L, Derman RJ, Kimber I, Ryan CA, Gerberick GF, Harvey P, Evans P, White IR and Rycroft RTG (2000). Use of local lymph node assay for the estimation of relative contact allergenic potency. Contact Dermatitis 42, 344–48. |
B.43 TEST AVSEENDE NEUROTOXICITET PÅ GNAGARE
1. METOD
Denna metod motsvarar OECD TG 424 (1997).
Med testmetoden fås information som bekräftar eller ytterligare karakteriserar en kemikalies förmåga att framkalla neurotoxicitet hos vuxna djur. Testmetoden kan kombineras med tidigare metoder för toxicitetstest med upprepad dos eller den kan användas separat. Det rekommenderas att OECD:s vägledning om strategier och metoder rörande testning av neurotoxicitet (se hänvisning 1) används som hjälp när man utformar försök som baserar sig på denna testmetod. Detta är särskilt viktigt när man överväger modifikationer av testmetodens normala observationer och förfaranden. OECD:s vägledning är avsedd som hjälp vid valet av alternativa testförfaranden som kan behövas i specifika omständigheter.
Metoden omfattar inte bedömningen av utvecklingsneurotoxicitet.
1.1 INLEDNING
Vid bedömning och utvärdering av en kemikalies toxiska egenskaper är det viktigt att ta hänsyn till kemikaliens förmåga att framkalla neurotoxiska verkningar. Redan i metoden för test avseende systemisk toxicitet med upprepad dos ingår observationer avsedda att påvisa förmågan att framkalla neurotoxicitet. Med hjälp av föreliggande testmetod kan man få ytterligare information om eller bekräfta de neurotoxiska verkningar som har observerats vid undersökning av systemisk toxicitet med upprepad dos. För vissa klasser av kemikalier kan en analys av förmågan att framkalla neurotoxicitet tyda på att det är lämpligare att använda föreliggande metod direkt, och inte bygga på indikationer på neurotoxicitet från undersökningar av systemisk toxicitet med upprepad dos. En sådan analys kan omfatta exempelvis följande:
|
— |
des signes de neurotoxicité ou des lésions neuropathologiques sont observés dans des études de toxicité autres que des études de toxicité systémique à dose répétée, ou |
|
— |
Strukturellt släktskap eller annan information som länkar kemikalien i fråga till ämnen med känd neurotoxicitet. |
Det kan även finnas andra tillfällen då det är lämpligt att använda föreliggande testmetod (se hänvisning 1).
Metoden har utformats så att den kan anpassas till särskilda behov att bekräfta en kemikalies specifika histopatologiska och beteenderelaterade neurotoxicitet, eller ge en möjlighet att karakterisera och kvantifiera neurotoxisk respons.
Tidigare likställdes neurotoxicitet med neuropati som inbegriper neuropatologiska skador eller neurologiska funktionsstörningar såsom förlamning, paralys eller tremor. Även om neuropati är en betydande manifestation av neurotoxicitet, vet man numera att det finns många andra tecken på nervsystemtoxicitet (förlust av motorisk koordination, sensoriska brister, inlärnings- och minnesstörningar), vilka inte alltid kommer fram vid undersökning av neuropati eller andra slags studier.
Syftet med föreliggande testmetod är att få fram betydande neurobeteenderelaterade och neuropatologiska verkningar hos vuxna gnagare. Trots att beteenderubbningar, även i frånvaron av morfologiska ändringar, kan tyda på skadliga verkningar på organismen, är dock inte alla beteenderubbningar specifika för nervsystemet. Alla observerade ändringar bör därför utvärderas tillsammans med korrelativa histopatologiska, hematologiska eller biokemiska data samt data om andra typer av systemisk toxicitet. Den testmetod som beskrivs här och vars syfte är att karakterisera och kvantifiera neurotoxisk respons inbegriper specifika histopatologiska och beteenderelaterade förfaranden som ytterligare kan kompletteras med elektrofysiologiska och/eller biokemiska undersökningar (se hänvisning 1, 2, 3 och 4).
Neurotoxiska ämnen kan verka på flera olika mål inom nervsystemet och genom flera olika mekanismer. Så länge som det inte finns ett enda system med vars hjälp alla ämnen kunde testas på djupet med avseende på neurotoxicitet, kan det vara nödvändigt att använda andra in vivo- eller in vitro-test som är specifika för den typ av neurotoxicitet som observeras eller förväntas.
Föreliggande testmetod kan även, i kombination med OECD:s vägledning om strategier och metoder rörande testning av neurotoxicitet (se hänvisning 1), användas som grund för test där man vill ytterligare karakterisera eller öka känsligheten hos dosresponskvantifieringen, och på så sätt få en bättre uppskattning av nivån för inga påvisbara skadliga verkningar eller bekräfta en kemikalies kända eller misstänkta skadliga verkningar. Man kan exempelvis utforma test för identifiering och utvärdering av de neurotoxiska mekanismerna eller test vars resultat kompletterar tillgängliga data från tidigare grundläggande observationer rörande neurobeteende och neuropatologi. Sådana test behöver inte ge samma resultat som kan fås med de standardförfaranden som rekommenderas i föreliggande metod, till den del dessa data redan finns och inte kan anses vara nödvändiga för tolkning av resultaten.
Detta neurotoxicitetstest ger, fristående eller som del av en kombination, följande möjligheter:
|
— |
Identifiera om nervsystemet påverkas permanent eller tillfälligt av den kemikalie som testas. |
|
— |
Bidra till karakteriseringen av de nervsystemrubbningar som hör samman med exponering för kemikalien, och skapa en förståelse för de underliggande mekanismerna. |
|
— |
Bestämma dos- och tid-responsförhållanden i syfte att uppskatta en nivå där inga verkningar kan påvisas (och som kan användas för att fastställa säkerhetskriterier för kemikalien). |
Vid test enligt denna metod används oral tillförsel av testämnet. Det kan vara lämpligare med andra tillförselvägar (exempelvis via huden eller inandning). I förekommande fall måste då eventuellt de rekommenderade förfarandena modifieras. Överväganden om valet av tillförselväg ska basera sig på profilen för exponering på människor och tillgänglig toxikologisk eller kinetisk information.
1.2 DEFINITIONER
Skadlig verkning: Varje sådan behandlingsrelaterad avvikelse från normaltillståndet som minskar organismens förmåga att överleva, föröka sig eller anpassa sig till omgivningen.
Dos: Den mängd testämne som tillförs. Dosen uttrycks som vikt (g, mg), vikten testämne per viktenhet försöksdjur (exempelvis mg/kg) eller en konstant utfodringskoncentration (ppm).
Dosering: Ett allmänt begrepp som inbegriper dos, dess frekvens och varaktighet.
Neurotoxicitet: En skadlig förändring i nervsystemets struktur eller funktion till följd av exponering för ett kemiskt, biologiskt eller fysikaliskt ämne.
Neurotoxiskt ämne: Varje kemiskt, biologiskt eller fysikaliskt ämne som har förmågan att framkalla neurotoxicitet.
NOAEL: Förkortning för ”no-observed-adverse-effect level” (nivå vid vilken inga skadliga verkningar observeras). Detta är den högsta dosnivån vid vilken inga skadliga behandlingsrelaterade verkningar kan observeras.
1.3 PRINCIP FÖR TESTMETODEN
Testämnet tillförs oralt i ett urval doser till flera grupper med laboratoriegnagare. I regel behövs upprepade doser. Doseringsschemat kan gälla subakut toxicitet (28 dagar), subkronisk toxicitet (90 dagar) eller kronisk toxicitet (1 år eller längre). Förfarandet som beskrivs här kan även användas för undersökning av akut neurotoxicitet. Djuren testas i syfte att få fram eller karakterisera beteenderelaterade och/eller neurologiska rubbningar. Under observationsperioderna görs bedömning av ett antal beteenden som kan påverkas av neurotoxiska ämnen. Efter avslutat test tas en undergrupp per kön från varje grupp för perfusion in situ, och snitt av hjärna, ryggrad och perifera nerver prepareras och undersöks.
Om testet görs fristående i syfte att påvisa neurotoxicitet eller karakterisera neurotoxiska verkningar, kan de djur som inte har använts för perfusion och efterföljande histopatologi (se tabell 1) användas för specifika undersökningar av neurobeteende, neuropatologi, neurokemi eller elektrofysiologi. Resultaten från sådana undersökningar kan komplettera data som fås med den föreliggande metodens standardundersökningar (se hänvisning 1). Tilläggsundersökningar kan vara till särskilt stor nytta i fall där empiriska observationer eller de förväntade verkningarna tyder på att en kemikalies neurotoxicitet är av en specifik typ eller har ett specifikt mål. Alternativt kan de återstående djuren användas för bedömningar som bygger på metoderna för toxicitetstest med upprepad dos på gnagare.
När test enligt denna metod kombineras med test enligt andra metoder måste djurens antal vara tillräckligt för att observationskraven ska uppfyllas för båda testerna.
1.4 BESKRIVNING AV TESTMETODEN
1.4.1 Val av djurart
Rekommenderad gnagare är råtta. Om testet utförs med andra gnagare bör detta motiveras. Djuren bör vara unga och friska vuxna exemplar som härrör från normala laboratoriestammar. Hondjuren får inte ha fött ungar och får inte vara dräktiga. Doseringen bör i regel inledas så snart som möjligt efter avvänjningen, helst inte senare än vid sex veckors ålder och under alla omständigheter innan djuren fyllt nio veckor. När testet kombineras med andra test måste denna faktor eventuellt anpassas. Vid testets början får variationen i vikt mellan de använda djuren inte överstiga ±20 % av vardera könets medelvikt. Om ett kortvarigt test med upprepad dos utförs som inledning till ett långtidstest måste djur från samma stam och ursprung användas vid båda testerna.
1.4.2 Förvarings- och utfodringsbetingelser
Temperaturen i försöksdjurens utrymmen bör vara 22 oC (± 3 oC). Den relativa fuktigheten bör vara minst 30 % och ska helst inte överstiga 70 %, utom när rummet rengörs. Målvärdet bör dock vara 50–60 %. Belysningen bör vara artificiell med omväxlande 12 timmar ljus och 12 timmar mörker. Periodiska höga ljud bör minimeras. För utfodringen kan konventionellt laboratoriefoder användas, och djuren bör ha obegränsad tillgång till dricksvatten. Om tillförseln görs genom fodret, kan valet av foder kan påverkas av behovet att säkerställa en lämplig blandning med testämnet. Djuren kan inhysas individuellt eller i burar med små grupper av samma kön.
1.4.3 Förberedelse av djuren
Friska unga djur fördelas slumpvis i behandlings- och kontrollgrupper. Burarna placeras i sådan ordning att eventuella verkningar på grund av placeringen minimeras. Djuren förses med individuell märkning och hålls i sina burar under minst 5 dagar före försökets start så att de får acklimatisera sig till laboratoriets betingelser.
1.4.4 Tillförselväg och beredning av doserna
Denna testmetod är särskilt avsedd för oral tillförsel av testämnet. Den orala tillförseln kan göras genom sond, foder, dricksvatten eller kapsyler. Om andra tillförselvägar används (exempelvis via huden eller inandning) måste eventuellt de förfaranden som rekommenderas enligt metoden modifieras. Överväganden om valet av tillförselväg ska basera sig på profilen för exponering på människor och tillgänglig toxikologisk eller kinetisk information. Motivering bör ges för valet av tillförselväg liksom även en redogörelse för eventuella modifikationer av testmetodens förfaranden.
Vid behov kan testämnet upplösas eller suspenderas i en lämplig vehikel. Om möjligt bör man i första hand överväga vattenlösning eller vattensuspension, sedan oljelösning eller oljesuspension (exempelvis majsolja) och därefter eventuell lösning eller suspension i andra vehiklar. Vehikelns toxiska egenskaper måste vara kända. Dessutom bör hänsyn tas till följande egenskaper hos vehikeln: Verkningarna på absorption, distribution, metabolism eller kvarhållande av testämnet till den del dessa verkningar kan påverka testämnets toxiska egenskaper samt verkningarna på testdjurens foder- eller vattenintag eller deras näringstillstånd.
1.5 FÖRFARANDEN
1.5.1 Djurens antal och kön
När testet utförs fristående bör minst 20 djur (10 honor och 10 hanar) användas i varje dos- och kontrollgrupp med tanke på utvärderingen av detaljerade kliniska och funktionella observationer. Minst fem hanar och fem honor som har valts ut bland dessa tio hanar och tio honor bör vid testets slut perfuseras in situ och användas för detaljerad neurohistopatologi. Om endast ett begränsat antal djur i en given dosgrupp observeras med avseende på tecken på neurotoxiska verkningar bör man överväga att införliva dessa djur i det urval som perfuseras. Om testet utförs i kombination med toxicitetstest med upprepad dos, bör djurens antal vara tillräckligt för att båda testernas syfte ska uppnås. Minimiantalet djur per grupp för olika testkombinationer anges i bilaga 1. Om man planerar avlivningar under testets gång eller återhämtningsgrupper för observation av reversibilitet, varaktighet eller fördröjd start för de toxiska verkningarna efter doseringen, eller om man planerar tilläggsobservationer, bör antalet djur ökas i syfte att säkerställa tillgången till nödvändigt antal djur för observation och histopatologi.
1.5.2 Doserings- och kontrollgrupp
I regel bör minst tre dosgrupper och en parallell kontrollgrupp användas. Om analys av tillgängliga data tyder på att inga verkningar förväntas vid en upprepad dos på 1 000 mg per kg kroppsvikt och dag, kan ett gränstest utföras. Om det inte finns lämpliga data att tillgå kan ett inledande test utföras som hjälp för bestämning av doserna. Med undantag för behandling med testämnet bör djuren i kontrollgruppen hanteras på exakt samma sätt som djuren i dosgrupperna. Om en vehikel används för tillförsel av testämnet bör kontrollgruppen få vehikeln i den största volym som används.
1.5.3 Tillförlitlighetskontroll
Testningslaboratoriet bör redogöra för laboratoriets kompetens att utföra försöket samt för känsligheten hos de förfaranden som används. Redogörelsen bör ge belägg för förmågan att detektera och kvantifiera förändringar i de olika ändpunkter som rekommenderas för observation (autonoma tecken, sensorisk reaktivitet, greppstyrka och motorisk aktivitet). Information om kemikalier som orsakar olika typer av neurotoxisk respons och som kan användas som positivkontrollämnen finns i hänvisning 2–9. Historiska data kan användas förutsatt att testförfarandena till väsentliga delar förblir desamma. Periodisk uppdatering av historiska data rekommenderas. Om det testande laboratoriet gör ändringar av testförloppet eller testets förfaranden bör laboratoriet lägga fram nya data som ger belägg på förfarandenas fortsatta känslighet.
1.5.4 Val av dos
Dosnivåerna bör väljas med hänsyn tagen till alla tillgängliga tidigare data om observerad toxicitet och kinetik för testämnet och närbesläktade material. Den högsta dosnivån bör väljas så att den framkallar neurotoxiska verkningar eller tydliga systemiska toxiska verkningar. Därefter väljs dosnivåerna i fallande skala så att en dosrelaterad respons kan påvisas, och samtidigt så att inga påvisbara skadliga verkningar (NOAEL) kan observeras på den lägsta dosnivån. Dosnivåerna bör väljas så att primära toxiska verkningar på nervsystemet kan särskiljas från verkningar som härrör från systemisk toxicitet. Det är ofta optimalt med 2–3 intervall, och tillägg av en fjärde testgrupp är ofta att föredra framför användning av mycket breda intervall (exempelvis mer än faktorn 10) mellan dosnivåerna. Hänsyn bör tas till en eventuellt existerande skälig uppskattning av exponering på människor.
1.5.5 Gränstest (limit test)
Om ett test med en dosnivå på minst 1 000 mg per kroppsvikt och dag genomförs på det sätt som beskrivs här och inga neurotoxiska verkningar kan observeras, och om toxicitet inte förväntas på grundval av uppgifter om strukturellt närbesläktade ämnen, kan ett fullständigt test med tre dosnivåer anses vara överflödigt. Om det är sannolikt att människor kommer att utsättas för exponering måste eventuellt en högre dosnivå (oralt) användas vid gränstestet. För andra former av tillförsel, såsom inandning eller applicering på huden, kan testämnets fysikalkemiska egenskaper ofta utnyttjas för att indikera och begränsa den maximala exponeringsnivån. För ett test avseende akut toxicitet oralt bör gränstestets dos vara minst 2 000 mg/kg.
1.5.6 Tillförsel av doser
Djuren doseras dagligen med testämnet, 7 dagar i veckan under minst 28 dagar. Om doseringen görs 5 dagar i veckan eller under en kortare period bör detta motiveras. Om testämnet tillförs genom sondmatning bör hela dosen tillföras på en gång via magsond eller lämplig intubationskanyl. Den största mängd vätska som kan tillföras vid ett och samma tillfälle beror på testdjurets storlek. Volymen bör inte överskrida 1 ml per 100 g kroppsvikt. I fråga om vattenlösningar får dock volymer på upp till 2 ml per 100 g kroppsvikt användas. Med undantag för irriterande eller frätande ämnen som normalt ger kraftigare verkningar vid högre koncentrationer bör variationer i testvolym minimeras genom justering av koncentrationen, för att säkerställa en konstant volym på alla dosnivåer.
För ämnen som tillförs via fodret eller dricksvattnet är det viktigt att försäkra sig om att testämnet ges i mängder som inte stör det normala näringsupptaget eller den normala vattenbalansen. När testämnet tillförs via fodret kan man använda en konstant koncentration (ppm) eller en konstant dosnivå i förhållande till djurens kroppsvikt. Det använda alternativet måste anges. När ett ämne tillförs genom sondmatning bör dosen ges vid samma tid varje dag, och vid behov justeras dosen så att dosnivån hålls konstant i förhållande till djurens kroppsvikt. Om ett test med upprepad dos används som inledning till ett långtidstest måste samma foder användas vid båda testningarna. Om det inte är möjligt med en engångsdos vid test avseende akut toxicitet, kan dosen tillföras i mindre delar över en period som inte överskrider 24 timmar.
1.6 OBSERVATIONER
1.6.1 Frekvens för observationer och tester
Vid test med upprepad dos bör observationsperioden vara lika lång som doseringsperioden. Vid test avseende akut toxicitet bör djuren observeras i ytterligare 14 dagar efter doseringsperiodens slut. För djur i satellitgrupper som inte exponeras under en period efter behandlingen, bör observationerna även täcka denna period.
Observationerna bör göras tillräckligt ofta för att maximera sannolikheten för upptäckt av beteenderelaterade och/eller neurologiska avvikelser. Observationerna bör helst göras vid samma tid varje dag, med hänsyn tagen till när man förväntar sig de starkaste verkningarna efter doseringen. En översikt av frekvensen för kliniska och funktionella observationer finns i tabell 2. Om kinetiska eller andra data från tidigare studier tyder på avvikande tidpunkter för observationer, prov eller förlängda observationsperioder, bör schemat anpassas så att maximal information erhålls. Motivering bör ges för alla ändringar av schemat.
1.6.1.1 Observationer av allmänt hälsotillstånd och mortalitet/morbiditet
Alla djur bör observeras omsorgsfullt minst en gång per dag med avseende på hälsotillstånd och minst två gånger per dag med avseende på morbiditet och mortalitet.
1.6.1.2 Detaljerade kliniska observationer
Detaljerade kliniska observationer bör göras på alla djur som har valts ut för detta ändamål (se tabell 1), en gång före den första exponeringen (för att få en jämförelsegrund per djur) och därefter med olika tidsintervall beroende på testets varaktighet (se tabell 2). Detaljerade kliniska observationer av återhämtningen i satellitgrupper bör göras i slutet återhämtningsperioden. De kliniska observationerna bör göras utanför buren på en standardarena. Observationerna bör registreras omsorgsfullt med hjälp av ett poängsystem som inbegriper kriterier eller poängskalor för varje mätning. Testningslaboratoriet bör tydligt ange vilka kriterier eller skalor som har använts. Det är viktigt att säkerställa att variationerna i testbetingelserna är minimala (inte systematiskt relaterade till behandlingen) och att observationerna görs av en kompetent person som inte känner till den aktuella behandlingen.
Det rekommenderas att observationerna genomförs på ett strukturerat sätt där väldefinierade kriterier (inbegripet definition av området för normal) systematiskt tillämpas på varje djur vid varje observationstidpunkt. Området för normal bör dokumenteras på tillbörligt sätt. Alla observerade tecken bör registreras. I tillämplig mån bör även omfattningen av de observerade tecknen registreras. De kliniska observationerna bör omfatta, men inte begränsas till, förändringar av hud, päls, ögon och slemhinnor, förekomst av sekret och utsöndringar samt autonom aktivitet (tårflöde, upprest päls, pupillstorlek, ovanligt andningsmönster och/eller andning genom munnen, alla ovanliga tecken rörande urinering eller defekation samt missfärgning av urinen).
Registreringarna bör även omfatta all ovanlig respons rörande kroppsställning, aktivitetsnivå (exempelvis minskad eller ökad utforskning av standardarenan) och rörelsekoordinationen. Förändringar i gång (exempelvis vaggande gång, ataxi), kroppsställning (exempelvis kutad rygg) och respons på hantering, placering eller andra omgivningsstimuli, liksom eventuella kloniska eller toniska rörelser, konvulsioner eller skälvningar, stereotypier, (exempelvis överdrivet putsande, ovanliga huvudrörelser, upprepat cirklande) eller bisarrt beteende (exempelvis bitning eller överdrivet slickande, självstympning, bakåtgång, utstötning av ljud) eller aggressioner bör registreras.
1.6.1.3 Funktionella observationer
Liksom i fråga om de kliniska observationerna bör funktionella observationer göras en gång före exponeringen och därefter med tillbörlig frekvens på alla djur som har valts ut för detta ändamål (se tabell 1). Frekvensen för de funktionella observationerna beror även på testets varaktighet (se tabell 2). Utöver de observationsperioder som anges i tabell 2 bör satellitgrupper för återhämtning bli föremål för funktionella observationer så nära avlivningen som möjligt. De funktionella observationerna bör omfatta sensorisk reaktion på olika slags stimuli, exempelvis hörsel-, syn- och djupsensoriska stimuli (se hänvisning 5, 6 och 7), bedömning av greppstyrka (se hänvisning 8) och bedömning av motorisk aktivitet (se hänvisning 9). Den motoriska aktiviteten bör mätas med automatiserad utrustning som kan detektera både minskningar och ökningar av aktiviteten. Om något annat definierat system används bör detta vara kvantitativt och dess känslighet och tillförlitlighet bör påvisas. Alla apparater bör testas med avseende på tillförlitlighet över tiden och enhetliga resultat från olika apparater. Närmare uppgifter om lämpliga förfaranden finns i respektive hänvisning. Om det inte finns några data (exempelvis struktur-aktivitet, epidemiologiska data, tidigare toxikologiska test) som tyder på förmågan att framkalla neurotoxiska verkningar, bör man överväga att införliva mer specialiserade observationer av sensorisk och motorisk funktion eller inlärning och minne, för att undersöka dessa eventuella verkningar närmare. Mer information om specialiserade observationer och hur de görs finns i hänvisning 1.
Undantagsvis kan djur som uppvisar så svåra tecken på toxicitet att de i betydande mån skulle störa de funktionella observationerna utelämnas från dessa. Om djur utelämnas från de funktionella observationerna bör detta motiveras.
1.6.2 Kroppsvikt samt foder- och vattenkonsumtion
Vid test som varar upp till 90 dagar bör alla djur vägas minst en gång per vecka och foderkonsumtionen bör mätas (vattenkonsumtionen i de fall då testämnet tillförs i vattnet) minst varje vecka. Vid långtidstest bör alla djur vägas minst en gång per vecka under de första 13 veckorna och därefter minst var fjärde vecka. Foderkonsumtionen (vattenkonsumtionen i de fall då testämnet tillförs i vattnet) bör mätas minst varje vecka under de första 13 veckorna och därefter ungefär var tredje månad, utom om förändringar i hälsotillstånd eller kroppsvikt motiverar ett annat schema.
1.6.3 Oftalmologi
Vi test som varar längre än 28 dagar bör djuren, innan testämnet tillförs och vid testets slut, undersökas oftalmologiskt med ett oftalmoskop eller motsvarande utrustning. Denna undersökning bör helst omfatta alla djur men minst djuren i högdos- och kontrollgrupperna. Om ögonförändringar upptäcks, eller om kliniska tecken motiverar det, bör alla djur undersökas. Vid långtidstest bör oftalmologisk undersökning göras även när testet varat 13 veckor. Oftalmologisk undersökning behöver inte göras om det redan finns tillgång till data från tidigare gjorda test av liknande varaktighet och vid liknande dosnivåer.
1.6.4 Hematologi och klinisk biokemi
När testet avseende neurotoxicitet utförs i kombination med test avseende systemisk toxicitet med upprepad dos, bör hematologiska undersökningar och klinisk-biokemiska bestämningar göras enligt det som gäller för testet avseende systemisk toxicitet. Provtagningen bör göras på sådant sätt att alla potentiella verkningar på det neurologiska beteendet minimeras.
1.6.5 Histopatologi
Den neuropatologiska undersökningen bör utformas så att den kompletterar och utökar de observationer som har gjorts under testets in vivo-fas. Vävnad från minst fem djur per kön och grupp (se tabell 1 och nästa avsnitt) bör fixeras in situ med hjälp av allmänt erkänd perfusions- och fixeringsteknik (se kapitel 5 i hänvisning 3 och kapitel 50 i hänvisning 4). Alla observerade betydande förändringar bör registreras. Om testet utförs fristående i syfte att påvisa neurotoxicitet eller karakterisera neurotoxiska verkningar, kan återstoden av djuren användas för specifika undersökningar av neurobeteende (se hänvisning 10 och 11), neuropatologi (se hänvisning 10, 11, 12 och 13), neurokemi (se hänvisning 10, 11, 14 och 15) eller elektrofysiologi (se hänvisning 10, 11, 16 och 17) i den mån sådana undersökningar kan komplettera de förfaranden och undersökningar som beskrivs i denna metod. Återstoden kan även utnyttjas för att få fler djur för de histopatologiska undersökningarna. Dessa tilläggsförfaranden kan vara särskilt motiverade i fall där empiriska observationer eller förväntade verkningar tyder på att en kemikalies neurotoxicitet är av en specifik typ eller har ett specifikt mål (se hänvisning 2 och 3). Alternativt kan återstoden av djuren användas för rutinmässiga patologiska bedömningar enligt det som beskrivs i metoden för test med upprepad dos.
All vävnad som inbäddats i paraffin bör färgas med ett standardförfarande (såsom hematoxylin och eosin), och därefter undersökas i mikroskop. Om tecken på perifer neuropati observeras eller misstänks, bör plastinbäddade prover av perifer nervvävnad undersökas. De kliniska tecknen kan också ge indikationer på ytterligare objekt som bör undersökas eller på behovet att använda speciella färgningsförfaranden. Vägledning om ytterligare undersökningsobjekt finns i hänvisning 3 och 4. Det kan också vara motiverat med vissa specialfärgmedel i syfte att påvisa specifika typer av patologiska förändringar (se hänvisning 18).
Representativa snitt av centrala och perifera nervsystemet bör undersökas histologiskt (se kapitel 5 i hänvisning 3 och kapitel 50 i hänvisning 4). De områden som i regel ska undersökas inbegriper framhjärnan, centrum av storhjärnan, inbegripet ett snitt genom hippocampus, mitthjärnan, lillhjärnan, hjärnbryggan, förlängda märgen, ögat med synnerv och näthinna, ryggraden vid nacken och ländryggen, dorsala rotganglier, dorsala och ventrala rotfibrer, proximala ischiasnerven, proximala tibialnerven (vid knät) och tibialnervens vadmuskelförgrening. De snitt som görs av ryggraden och perifera nerver bör inbegripa diagonala, transversala och längsgående snitt. Uppmärksamhet bör fästas vid nervsystemets vaskulatur. Likaså bör man undersöka ett skelettmuskelprov, särskilt vadmuskeln. Särskild uppmärksamhet bör fästas vid sådana punkter i centrala och perifera nervsystemet som har cellulär och fibrös struktur och mönster och som man vet att särskilt påverkas av neurotoxiska ämnen.
Vägledning om neuropatologiska förändringar som typiskt uppstår efter exponering av toxiska ämnen finns i hänvisning 3 och 4. För vävnadsproverna rekommenderas en stegvis undersökning där snitt från djuren i högdosgruppen först jämförs med snitt från djuren i kontrollgruppen. Om inga neuropatologiska förändringar kan iakttas i proverna från dessa grupper behövs inga ytterligare analyser. Om neuropatologiska förändringar kan iakttas i högdosgruppen bör prover av alla potentiellt påverkade vävnader från mellandos- och lågdosgruppen kodas och undersökas.
Om tecken på neuropatologiska förändringar kan iakttas vid den kvalitativa undersökningen bör en tillläggsundersökning göras på nervsystemets alla regioner som uppvisar sådana förändringar. Snitt från djuren i alla dosgrupper, från alla potentiellt påverkade regioner bör kodas och undersökas randomiserat av personer som inte känner till koden. Frekvens och allvarlighetsgrad bör registreras för alla skador. Efter bedömning av alla regioner från alla dosgruppers djur avtäcks koderna, och en statistisk analys genomförs för utvärdering av förhållandet mellan dos och respons. För varje skada bör man beskriva exempel på olika allvarlighetsgrader.
De neuropatologiska fynden bör utvärderas med hänsyn tagen till resultaten från observationer och mätningar av beteende, liksom även till övriga data från tidigare och parallella tester av det berörda ämnets systemiska toxicitet.
2. DATA
2.1 BEHANDLING AV RESULTATEN
Data bör rapporteras individuellt. Därutöver bör alla data sammanfattas i en tabell över antalet djur i varje testgrupp eller kontrollgrupp vid testets början, antalet djur som hittats döda under testets gång eller som avlivats av hänsyn till djurens välbefinnande inbegripet avlivningstidpunkterna, antalet djur som visat tecken på toxicitet, en beskrivning av observerade tecken på toxicitet, inbegripet tidpunkten då sådana första gången upptäcktes, varaktighet och hur allvarliga de toxiska verkningarna är, antalet djur som har skador samt skadornas typ och allvarlighetsgrad.
2.2 UTVÄRDERING OCH TOLKNING AV RESULTATEN
Alla fynd bör utvärderas med avseende på förekomst, allvarlighetsgrad och korrelationen mellan neurobeteenderelaterade och neuropatologiska verkningar (även neurokemiska eller elektrofysiologiska verkningar, om tilläggsundersökningar ingår) samt alla andra skadliga verkningar som har observerats. Om möjligt bör de numeriska resultaten utvärderas genom en tillämplig och allmänt godtagbar statistisk metod. De statistiska metoderna bör fastställas vid utformningen av testet.
3. RAPPORTERING
3.1 TESTRAPPORT
Testrapporten ska innehålla nedan angivna uppgifter.
|
|
Testämne:
|
|
|
Vehikel (i förekommande fall):
|
|
|
Försöksdjur:
|
|
|
Testbetingelser:
|
|
|
Observation och testförfaranden:
|
|
|
Resultat:
|
|
|
Diskussion om resultaten:
|
|
|
Slutsatser:
|
4. HÄNVISNINGAR
|
1. |
”OECD Giudance Document on Neurotoxicity Testing Strategies and Test Methods”, OECD, Paris (under beredning). |
|
2. |
”Test Guidline for a Developmental Neurotoxicitu Study, OECD Guidelines for the Testing of Chemicals” (under beredning). |
|
3. |
World Health Organization (WHO) (1986), ”Environmental Health Criteria document 60: Principles and Methods for the Assessment of Neurotoxicity associated with Exposure to Chemicals”. |
|
4. |
P.S. Spencer och H.H. Schaumburg (1980), ”Experimental and Clinical Neurotoxicology”, red. P.S. Spencer och H.H. Schaumburg, Williams and Wilkins, Baltimore/London. |
|
5. |
D.E. och R.B. Wallace (1980), ”Utility of the Neurological Examination in Rats”, Acta Neurobiol. Exp., 40, 999–1003. |
|
6. |
S.C. Gad (1982), ”A Neuromuscular Screen for Use in Industrial Toxicology”, J. Toxicol. Environ. Health, 9, 691–704. |
|
7. |
V.C. Moser, K.M. McDaniel och P.M. Phillips (1991), ”Rat Strain and Stock Comparisons Using a Functional Observational Battery: Baseline Values and Effects of amitraz”, Toxic. Appl. Pharmacol., 108, 267–283. |
|
8. |
O.A. Meyer, H.A. Tilson, W.C. Byrd och M.T. Riley (1979), ”A Method for the Routine Assessment of Fore- and Hind-limb Grip Strength of Rats and Mice”, Neurobehav. Toxicol., 1, 233–236. |
|
9. |
K.M. Crofton, J.L. Haward, V.C. Moser, M.W. Gill, L.W. Reirer, H.A. Tilson och R.C. MacPhail (1991), ”Interlaboratory Comparison of Motor Activity Experiments: Implication for Neurotoxicological Assessments”, Neurotoxicol. Teratol., 13, 599–609. |
|
10. |
H.A. Tilson och C.L. Mitchell (red.) (1992), ”Neurotoxicology Target Organ Toxicology Series”, Raven Press, New York. |
|
11. |
L.W. Chang (red.) (1995), ”Principles of Neurotoxicology”, Marcel Dekker, New York. |
|
12. |
B. Broxup (1991), ”Neuropathology as a screen for Neurotoxicity Assessment”, J. Amer. Coll. Toxicol., 10, 689–695 |
|
13. |
V.C. Moser, D.C. Anthony, W.F. Sette och R.C. MacPhail (1992), ”Comparison of Subchronic Neurotoxicity of 2-Hydroxyethyl Acrylate and Acrylamide in Rats”, Fund. Appl.Toxicol., 18, 343–352. |
|
14. |
J.P. O'Callaghan (1988), ”Neurotypic and Gliotypic Proteins as Biochemical Markers of Neurotoxicity”, Eurotoxicol. Teratol., 10, 445–452. |
|
15. |
J.P. O'Callaghan och D.B. (1988), ”Acute Exposure of the Neonatal Rat to Triethyltin Results in Persistent Changes in Neurotypic and Gliotypic Proteins”,J. Pharmacol. Exp. Ther., 244, 368–378. |
|
16. |
D.A. Fox, H.E. Lowndes och G.G. Birkamper (1982), ”Electrophysiological Techniques in Neurotoxicology”, Nervous System Toxicology, red. C. L. Mitchell, Raven Press, New York, 299–335. |
|
17. |
B.L. Johnson (1980), ”Electrophysiological Methods in neurotoxicity Testing”, Experimental and Clinical Neurotoxicology. red. P.S. Spencer och H. H. Schaumburg, Williams and Wilkins Co., Baltimore/London, 726–742. |
|
18. |
J.D. Bancroft och A. Steven (1990), ”Theory and Pratice of Histological Techniques, Chapter 17”, Neuropathological Techniques, red. Lowe, James och Cox, Gordon, Churchill Livingstone. |
Tabell 1
Minimiantalet djur per grupp när test avseende neurotoxicitet utförs dels fristående, dels i kombination med andra test
|
|
OLIKA UTFÖRANDEN AV TEST AVSEENDE NEUROTOXICITET |
|||
|
Separat test |
I kombination med 28-dagarstest |
I kombination med 90-dagarstest |
I kombination med test avseende kronisk toxicitet |
|
|
Sammanlagt antal djur per grupp |
10 hanar och 10 honor |
10 hanar och 10 honor |
15 hanar och 15 honor |
25 hanar och 25 honor |
|
Antalet djur som valts ut för funktionella observationer inbegripet detaljerade kliniska observationer |
10 hanar och 10 honor |
10 hanar och 10 honor |
10 hanar och 10 honor |
10 hanar och 10 honor |
|
Antalet djur som valts ut för perfusion in situ och neurohistopatologi |
5 hanar och 5 honor |
5 hanar och 5 honor |
5 hanar och 5 honor |
5 hanar och 5 honor |
|
Antalet djur som valts ut för observationer vid toxicitetstest med upprepad dos, vid test av subkronisk eller kronisk toxicitet, för hematologisk undersökning, klinisk-biokemisk undersökning, histopatologi och dylikt, enligt respektive vägledning |
|
5 hanar och 5 honor |
||
|
Tilläggsobservationer, i förekommande fall |
5 hanar och 5 honor |
|
|
|
Tabell 2
Frekvens för kliniska och funktionella observationer
|
Typ av observationer |
Testets varaktighet |
||||||||||||||||||||||||||
|
Akut |
28-dagars |
90-dagars |
Kronisk |
||||||||||||||||||||||||
|
För alla djur |
Allmänt hälsotillstånd |
Dagligen |
Dagligen |
Dagligen |
Dagligen |
||||||||||||||||||||||
|
Mortalitet/morbiditet |
Två gånger per dag |
Två gånger per dag |
Två gånger per dag |
Två gånger per dag |
|||||||||||||||||||||||
|
I djur som valts ut för funktionella observationer |
Detaljerade kliniska observationer |
|
|
|
|
||||||||||||||||||||||
|
Funktionella observationer |
|
|
|
|
|||||||||||||||||||||||
B.44 HUDABSORPTION: IN VIVO-METOD
1. METOD
Denna testmetod motsvarar OECD TG 427 (2004).
1.1 INLEDNING
Exponering för många kemikalier sker i huvudsak genom huden, medan de flesta toxikologiska studier som görs med försöksdjur gäller oral administrering. Den in vivo-studie av hudabsorption som beskrivs i dessa riktlinjer ger den koppling som behövs för att kunna extrapolera från orala studier vid säkerhetsbedömning efter dermal exponering.
Ett ämne måste passera ett stort antal cellager i huden innan det når blodcirkulationen. För de flesta ämnen är stratum corneum, hornlagret som består av döda celler, det hastighetsbestämmande lagret. Permeabiliteten genom huden beror både på hur lipofil kemikalien är och hur tjockt epidermis yttersta lager är samt på faktorer som ämnets molekylvikt och koncentration. I regel har rått- och kaninhud högre permeabilitet än human hud, medan hudpermeabiliteten hos marsvin och apor ligger närmare människans.
Metoderna för att mäta perkutan absorption kan indelas i två kategorier, in vivo och in vitro. In vivo-metoden kan ge god information om hudabsorption hos olika arter av försöksdjur. Under senare tid har in vitro-metoder utvecklats. I dessa används transport över hela eller delar av tvärsnitt av djurhud eller human hud till en vätskereservoar. In vitro-metoden beskrivs i en separat testmetod (1). Man bör konsultera OECD:s riktlinjer för genomförande av hudabsorptionsstudier (2) som ett stöd för valet av lämpligaste metod under givna betingelser, eftersom de innehåller detaljerade uppgifter om lämpligheten hos både in vivo- och in vitro-metoder.
Den in vivo-metod som beskrivs i denna metod gör det möjligt att bestämma testämnets penetration genom huden in till organismen. Tekniken har haft bred användning under många år (3)(4)(5)(6)(7). I många fall kan hudabsorptionsstudier in vitro vara lämpliga, men det kan finnas lägen där endast en in vivo-studie kan ge de data som krävs.
Fördelar med in vivo-metoden är att den utnyttjar fysiologiskt och metaboliskt intakta system och en art som är vanlig i många toxicitetsstudier samt att den kan modifieras för användning med andra arter. Nackdelarna är att levande djur används, behovet av att använda radioaktivmärkt material för att göra det lättare att få tillförlitliga resultat, svårigheter att fastställa den tidiga absorptionsfasen och skillnader i permeabilitet mellan den föredragna artens (råtta) hud och mänsklig hud. Djurhud är i regel mer permeabel och kan därför ge en överskattning av den humana hudabsorptionen (6)(8)(9). Frätande/korrosiva ämnen bör inte testas på levande djur.
1.2 DEFINITIONER
Oabsorberad dos: representerar det som tvättas bort från hudytan efter exponering och eventuellt finns kvar på det icke-tättslutande täckmaterialet, inbegripet eventuell dos som kan visas avdunsta från huden under exponeringen.
Absorberad dos (in vivo): innefattar det som finns i urinen, i tvättvatten från buren, i avföring, i utandningsluften (om detta mäts), i blod, i vävnader (om sådana tas till vara) och i det återstående kadavret efter avlägsnande av huden på appliceringsstället.
Absorberbar dos: representerar det som finns på eller i huden efter tvätt.
1.3 PRINCIPEN FÖR TESTMETODEN
Testämnet, som helst ska vara radioaktivmärkt, appliceras på djurets rakade hud i en eller flera lämpliga doser i form av en representativ beredning som är i bruk. Testberedningen får vara i kontakt med huden under en bestämd tidsperiod under ett lämpligt täckmaterial (icke-tättslutande, halvtättslutande eller tättslutande) för att förhindra att djuret äter av testberedningen. I slutet av exponeringstiden tas täckmaterialet bort och huden rengörs med lämpligt rengöringsmedel. Täckmaterial och tvättmaterial behålls för analys och ett nytt täckmaterial appliceras. Före, under och efter exponeringsperioden hålls djuren i individuella metabolismburar och exkrementer och utandningsluft från dessa perioder samlas upp för analys. Insamlingen av utandningsluft kan uteslutas om det finns tillräckliga data som visar att inga eller få flyktiga radioaktiva metaboliter bildas. Varje studie omfattar i regel flera grupper av djur som exponeras för testberedningen. En grupp avlivas i slutet av exponeringsperioden. Andra grupper avlivas efter fastställda tidsintervall därefter (2). I slutet av provtagningstiden avlivas de återstående djuren, blod samlas in för analys, huden på applikationsstället avlägsnas för analys och kadavret analyseras med avseende på outsöndrat material. Proverna analyseras med lämpliga medel och graden av hudabsorption uppskattas (6)(8)(9).
1.4 BESKRIVNING AV METODEN
1.4.1 Val av djurart
Råtta är den art som oftast används, men även hårlösa stammar och arter som har hudabsorptionhastigheter som mer liknar människans kan användas (3)(6)(7)(8)(9). Unga vuxna, friska djur av samma kön (hankön är standard) av rutinmässigt använda laboratoriestammar ska användas. I början av studien bör djurens viktvariation inte överstiga ± 20 % av medelvikten. Exempelvis är hanråttor på 200 g – 250 g lämpliga, särskilt i övre delen av viktintervallet.
1.4.2 Djurens antal och kön
En grupp på minst fyra djur av ett kön ska användas för varje testberedning och varje schemalagd avslutningstid. Varje djurgrupp avlivas efter olika tidsintervall, t.ex. i slutet av exponeringstiden (i regel efter 6 eller 24 timmar) och vid de efterföljande tidpunkterna (t.ex. 48 och 72 timmar). Om data finns tillgängliga som visar betydande skillnader i dermal toxicitet mellan hanar och honor ska det känsligare könet väljas. Om inga sådana data finns kan någondera könet användas.
1.4.3 Inhysning och diet
Temperaturen i försöksdjursrummet ska vara 22 oC (± 3 oC). Den relativa luftfuktigheten ska vara minst 30 % och ska helst inte överstiga 70 % annat än vid rengöring, men målet ska vara att hålla 50–60 %. Artificiell belysning ska användas, med en sekvens på 12 timmar ljus och 12 timmar mörker. Konventionella laboratoriedieter kan användas och föda ska finnas fritt tillgänglig tillsammans med obegränsat med dricksvatten. Under studien och helst även under acklimatiseringen ska djuren inhysas enskilt i metabolismburar. Eftersom spill av föda och vatten skulle äventyra resultaten ska risken för spill minimeras.
1.4.4 Förberedelse av djuren
Djuren märks för att möjliggöra identifiering av individerna och hålls i sina burar i minst fem dagar innan studien inleds för att de ska acklimatisera sig till laboratoriebetingelserna.
Efter acklimatiseringsperioden, och ungefär 24 timmar före dosering, rakas ett hudområde på djuren runt hals och rygg. Eftersom permeabilitetsegenskaperna för skadad hud skiljer sig från dem hos intakt hud bör man undvika att skada huden. Efter rakning och ungefär 24 timmar innan testämnet appliceras på huden (se avsnitt 1.4.7) ska hudytan torkas av med aceton för att avlägsna sebum. En kompletterande tvätt med tvål och vatten rekommenderas inte, eftersom eventuella tvålrester kan främja absorption av testämne. Området måste vara tillräckligt stort för att det ska gå att göra en tillförlitlig beräkning av den absorberade mängden testkemikalie per cm2 hud, helst minst 10 cm2. Detta område är praktiskt möjligt att ha på råttor med kroppsvikten 200–250 g. Efter förberedelsen återförs djuren till metabolismburarna.
1.4.5 Testämne
Testämnet är den entitet vars penetrationsegenskaper ska studeras. Helst ska testämnet vara radioaktivmärkt.
1.4.6 Testberedningen
Beredningen av testämne (t.ex. rent, utspätt eller formulerat material innehållande testkemikalien som appliceras på huden) ska vara detsamma (eller en rimlig ersättning) som det som människor eller andra målarter kan exponeras för. Alla avvikelser från den beredning som ”är i bruk” måste motiveras. Vid behov löses eller suspenderas testämnet i en lämplig vehikel. För andra vehikler än vatten ska absorptionsegenskaper och potentiella interaktioner vara kända.
1.4.7 Applicering på huden
Ett appliceringsställe med en viss yta definieras på hudytan. En känd mängd av testberedningen appliceras sedan jämnt på detta ställe. Mängden ska normalt efterlikna den potentiella exponering människor utsätts för, i regel 1–5 mg/cm2 för ett fast ämne och upp till 10 μl/cm2 för vätskor. Alla andra mängder ska motiveras av de förväntade användningsbetingelserna, studiens syfte eller testberedningens fysikaliska egenskaper. Efter applicering ska det behandlade stället skyddas från putsning. Ett exempel på en typisk anordning visas i figur 1. Normalt skyddas appliceringsstället med ett täckmaterial som inte är tättslutande (t.ex. ett permeabelt skydd av nylonväv). För mycket små mängder bör dock appliceringsstället täckas tätt. Om avdunstning av halvflyktiga testämnen minskar upptagshastigheten av testämnet i oacceptabel omfattning (se även avsnitt 1.4.10, första stycket) måste det avdunstade ämnet fångas i ett träkolsfilter som täcker appliceringsanordningen (se figur 1). Det är viktigt att anordningen inte skadar huden och inte heller absorberar eller reagerar med testberedningen. Djuren återförs till individuella metabolismburar för uppsamling av exkrementer.
1.4.8 Varaktighet för exponering och provtagning
Exponeringens varaktighet är tidsintervallet mellan applicering och avlägsnande av testberedningen genom tvätt av huden. Exponeringsperioden ska vara relevant, i regel 6 eller 24 timmar, i förhållande till den förväntade humana exponeringstiden. Efter exponeringsperioden hålls djuren i metabolismburarna fram till den schemalagda avslutningen. Djuren ska observeras med regelbundna intervall med avseende på tecken på toxicitet/onormala reaktioner under hela studieperioden. I slutet av exponeringsperioden ska den behandlade huden granskas så att man kan hitta eventuella synliga tecken på irritation.
Metabolismburarna ska tillåta separat uppsamling av urin och avföring under hela studien. De ska också tillåta uppsamling av 14C-koldioxid och flyktiga 14C-kolföreningar för analys vid produktion som överstiger 5 %. Urin, avföring och infångningsvätskor (t.ex. med 14C-koldioxid och flyktiga 14C-föreningar) ska samlas upp enskilt från varje grupp vid varje provtagningstillfälle. Om det finns tillräckliga data för att man ska kunna utgå ifrån att inga eller mycket lite flyktiga radioaktiva metaboliter bildas går det att använda öppna burar.
Exkrementer samlas upp under exponeringsperioden, upp till 24 timmar efter den initiala hudkontakten och sedan dagligen till och med försökets sista dag. Tre extra uppsamlingsintervall räcker i regel, men syftet med testberedningen eller befintliga kinetiska data kan ge vid handen att andra eller ytterligare tidpunkter är lämpliga för studien.
I slutet av exponeringsperioden tas skyddsanordningen bort från varje djur och sparas separat för analys. Den behandlade huden på alla djur ska tvättas minst 3 gånger med rengöringsmedel med lämpliga svabbar. Det är viktigt att undvika att kontaminera andra delar av kroppen. Rengöringsmedlet ska vara representativt för normal hygien, t.ex. en tvållösning i vatten. Slutligen ska huden torkas. Alla svabbar och allt tvättvatten ska sparas för analys. Ett nytt täckmaterial appliceras för att skydda det behandlade stället på djur som ingår i grupper med senare provtagningstidpunkter innan de återförs till sina enskilda burar.
1.4.9 Avslutningsförfaranden
För varje grupp ska de enskilda djuren avlivas på den schemalagda tidpunkten och blodprov tas för analys. Skyddsanordningen eller täckmaterialet avlägsnas för analys. Huden från appliceringsstället och en motsvarande yta av odoserad, rakad hud tas från varje djur för separat analys. Appliceringsstället kan delas upp för att skilja hornlagret från underliggande epidermis så att mer information erhålls om testkemikaliens fördelning. Bestämningen av fördelningen under en viss tid efter exponeringsperioden bör ge en indikation om vad som händer med eventuell testkemikalie i hornlagret. För att underlätta uppdelningen av huden (efter att djuret har genomgått sluttvätt och avlivats) tas alla skyddsanordningar bort. Huden på appliceringsstället, med en omgivande ring av hud, skärs ut från råttan och nålas upp på en platta. En remsa häftande tejp appliceras på hudytan med ett varsamt tryck och tejpen tas bort tillsammans med en del av hornlagret. Tejpremsor appliceras successivt tills tejpen inte längre fäster vid hudytan, då hela hornlagret har avlägsnats. För varje djur kan remsorna slås samman i en enda behållare. Till denna behållare sätts en vävnadsnedbrytare för att lösa upp hornlagret. Eventuell potentiell målvävnad kan avlägsnas för separat mätning innan det återstående kadavret analyseras med avseende på absorberad kadaverdos. De enskilda kadavren ska sparas för analys. Vanligen räcker en analys av totalinnehållet. Målorgan kan avlägsnas för separat analys (om andra studier tyder på att detta bör göras). Urin i blåsan vid den schemalagda tidpunkten för avlivning ska läggas till de tidigare uppsamlade urinproven. Efter uppsamling av exkrementer från metabolismburarna vid den schemalagda avlivningstiden ska burarna och tillhörande infångningsanordningar tvättas med lämpligt lösningsmedel. Även annan potentiellt kontaminerad utrustning ska analyseras.
1.4.10 Analys
Adekvat återvinning av radioaktiviteten (dvs. ett medelvärde på 100 ± 10 % av all radioaktivitet) ska uppnås i alla studier. Återvinning utöver detta intervall måste motiveras. Mängden av den administrerade dosen i varje prov analyseras med lämpliga vedertagna förfaranden.
De statistiska beräkningarna ska inbegripa ett mått på variansen för replikaten för varje applicering.
2. DATA
Följande mätningar ska göras för varje djur, vid varje provtagningstillfälle för testkemikalien och/eller metaboliter. Utöver individdata ska data grupperade efter provtagningstidpunkter rapporteras som medelvärden:
|
— |
Kvantitet i skyddsanordningen. |
|
— |
Kvantitet som kan avlägsnas från huden. |
|
— |
Kvantitet i/på huden som inte kan tvättas bort från denna. |
|
— |
Kvantitet i blodprover. |
|
— |
Kvantitet i exkrementer och i utandningsluft (om sådana data finns). |
|
— |
Återstående kvantitet i kadavret och i eventuella organ som har avlägsnats för separat analys. |
Kvantiteten av testämnet och/eller metaboliter i exkrementer, utandningsluft, blod och kadaver gör det möjligt att bestämma den totala mängd som har absorberats vid varje tidpunkt. Även en beräkning av mängden testkemikalie som absorberats för varje cm2 hud som exponerats för testämnet kan göras.
3. RAPPORTERING
3.1 FÖRSÖKSRAPPORT
Testrapporten måste avspegla de krav som ställs enligt försöksprotokollet, inbegripet en motivering för det använda testsystemet. Rapporten ska innehålla följande information:
|
|
Testämne:
|
|
|
Testberedning:
|
|
|
Försöksdjur:
|
|
|
Försöksförhållanden:
|
|
|
Resultat:
|
|
|
Diskussion av resultaten. |
|
|
Slutsatser. |
4. HÄNVISNINGAR
|
1. |
Testing Method B.45. Skin Absorption: In vitro Method. |
|
2. |
OECD (2002). Guidance Document for the Conduct of Skin Absorption Studies. OECD, Paris. |
|
3. |
ECETOC (1993) Percutaneous Absorption. European Centre for Ecotoxicology and Toxicology of Chemicals, Monograph No. 20. |
|
4. |
Zendzian RP (1989) Skin Penetration Method suggested for Environmental Protection Agency Requirements. J. Am. Coll. Toxicol. 8(5), 829–835. |
|
5. |
Kemppainen BW, Reifenrath WG (1990) Methods for skin absorption. CRC Press Boca Raton, FL, USA. |
|
6. |
EPA (1992) Dermal Exposure Assessment: Principles and Applications. Exposure Assessment Group, Office of Health and Environmental Assessment. |
|
7. |
EPA (1998) Health Effects Test Guidelines, OPPTS 870-7600, Dermal Penetration. Office of Prevention, Pedsticides and Toxic Substances. |
|
8. |
Bronaugh RL, Wester RC, Bucks D, Maibach HI and Sarason R (1990) In vivo percutaneous absorption of fragrance ingredients in reshus monkeys and humans. Fd. Chem. Toxic. 28, 369–373. |
|
9. |
Feldman RJ and Maibach HI (1970) Absorption of some organic compounds through the skin in man. J. Invest Dermatol. 54, 399–404. |
Figur 1
Ett exempel på en typisk anordning som används för att definiera och skydda appliceringsstället på huden vid hudabsorptionsstudier in vivo.

B.45. HUDABSORPTION: in vitro-METOD
1. METOD
Denna testmetod motsvarar OECD TG 428 (2004).
1.1 INLEDNING
Denna metod har utformats för att ge information om absorption av ett testämne som appliceras på utskuren hud. Den kan antingen kombineras med metoden för hudabsorption in vivo (1) eller genomföras separat. Det rekommenderas att OECD:s riktlinjedokument för genomförande av hudabsorptionsstudier (2) konsulteras som ett stöd för utformningen av studier som bygger på denna metod. Riktlinjedokumentet har tagits fram för att underlätta urvalet av in vitro-förfaranden för användning under särskilda betingelser, för att garantera att de resultat som erhålls med denna metod blir tillförlitliga.
Metoderna för att mäta hudabsorption och dermal leverans kan indelas i två kategorier: in vivo och in vitro. In vivo-metoder för hudabsorption är väl etablerade och ger farmakokinetisk information i ett urval av djurarter. En in vivo-metod beskrivs separat i en annan testmetod (1). In vitro-metoder har också använts under många år för att testa hudabsorption. Formella valideringsstudier av in vitro-metoder som omfattas av denna testmetod har inte gjorts, men OECD-experter enades 1999 om att tillräckligt med data hade utvärderats för att stödja in vitro-metoden (3). Detaljerade uppgifter som ytterligare stödjer detta, inbegripet ett betydande antal direktjämförelser av in vitro- och in vivo-metoder, finns i riktlinjedokumentet (2). Det finns ett antal monografier som innehåller översikter över detta ämne och ger en detaljerad bakgrund till användningen av en in vitro-metod (4)(5)(6)(7)(8)(9)(10)(11)(12). In vitro-metoder mäter diffusionen av kemikalier in i och genom huden till en vätskereservoar, och det går använda icke-viabel hud för att mäta endast diffusion eller färsk, metaboliskt aktiv hud för att samtidigt mäta diffusion och metabolism i huden. Sådana metoder har särskilt funnit användning för att jämföra leveransen av kemikalier in i och genom huden från olika formuleringar och kan också ge användbara modeller för hudabsorption hos människa.
In vitro-metoden är inte alltid tillämplig för alla situationer och för alla kemikalieklasser. Den kan gå att använda för en initial kvalitativ utvärdering av hudpenetrationen. I vissa fall kan det vara nödvändigt att följa upp denna utvärdering med in vivo-data. Riktlinjedokumentet (2) bör konsulteras för ytterligare information om situationer där in vitro-metoden skulle vara lämplig. Mer detaljerade uppgifter som stöd för beslutet finns i (3).
Denna metod presenterar allmänna principer för att mäta hudabsorption och leverans av ett testämne med hjälp av utskuren hud. Hud från många däggdjursarter, inbegripet människa, kan användas. Hudens permeabilitetsegenskaper bibehålls efter utskärning, eftersom den huvudsakliga diffusionsbarriären är det icke-viabla hornlagret, stratum corneum; aktiv transport genom huden har inte påvisats. Huden har visats kunna metabolisera vissa kemikalier under hudabsorption (6), men denna process är inte hastighetsbegränsande med avseende på faktisk absorberad dos, även om den kan påverka naturen hos det material som kommer in i blodbanan.
1.2 DEFINITIONER
Oabsorberad dos: representerar det som tvättas bort från hudytan efter exponering och eventuellt finns kvar på det icke-tättslutande täckmaterialet, inbegripet eventuell dos som kan visas avdunsta från huden under exponeringen.
Absorberad dos (in vitro) : massan av testämne som når mottagarvätskan eller den systemiska cirkulationen inom en given tidsperiod.
Absorberbar dos (in vitro) : representerar det som finns kvar på eller i huden efter tvättning.
1.3 PRINCIPEN FÖR TESTMETODEN
Testämnet, som kan vara radioaktivmärkt, appliceras på ytan av ett hudprov som skiljer två kammare åt i en diffusionscell. Kemikalien finns kvar på huden under en fastställd tid och under fastställda betingelser innan den avlägsnas med ett lämpligt rengöringsförfarande. Mottagarvätskan provtas vid tidpunkter under hela försöket och analyseras med avseende på testkemikalie och/eller metaboliter.
Om metaboliskt aktiva system används kan metaboliter av testkemikalien analyseras med lämpliga metoder. I slutet av försöket kvantifieras fördelningen av testkemikalien och dess metaboliter, om sådana förekommer.
Under lämpliga betingelser, såsom beskrivs i denna metod och i riktlinjedokumentet (2), mäts absorptionen av en testkemikalie under en given tidsperiod genom analys av en mottagarvätska och av den behandlade huden. Det testämne som återstår i huden ska betraktas som absorberat om det inte kan visas att absorptionen kan bestämmas utifrån enbart värden för receptorvätskan. Analys av övriga komponenter (material som tvättas bort från huden och som finns kvar i hudlagren) gör ytterligare utvärdering av data möjlig, inbegripet total fördelning av testämne och procentandel återvunnet testämne.
Resultat för relevanta referenskemikalier ska finnas tillgängliga och överensstämma med publicerad litteratur om den använda metoden för att visa testsystemets prestanda och tillförlitlighet på det laboratorium som genomför försöket. Detta krav kan uppfyllas genom att en lämplig referenskemikalie (helst ett ämne vars lipofilicitet ligger nära testämnets) testas samtidigt med testämnet eller genom att adekvata historiska data tillhandahålls för ett antal referensämnen med olika lipofilicitet (t.ex. koffein, bensoesyra och testosteron).
1.4 BESKRIVNING AV TESTMETODEN
1.4.1 Diffusionscell
En diffusionscell består av en givarkammare och en mottagarkammare mellan vilka huden placeras (ett exempel på en typisk utformning finns i figur 1). Cellen ska ge en god förslutning runt huden, möjliggöra enkel provtagning och en god blandning av den mottagarlösning som är i kontakt med hudens undersida samt god temperaturkontroll av cellen och dess innehåll. Diffusionsceller av både statisk typ och genomflödestyp är godtagbara. Normalt lämnas givarkammaren öppen under exponering för en begränsad dos av en testberedning. Vid applicering av en obegränsad dos och i vissa fall även av begränsade doser kan givarkammaren inneslutas.
1.4.2 Mottagarvätska
Föredraget används en fysiologiskt anpassad mottagarvätska, även om andra vätskor kan användas om detta är motiverat. Mottagarvätskans exakta sammansättning ska anges. Det ska visas att testkemikaliens löslighet i mottagarvätskan är rimlig så att vätskan inte fungerar som en barriär för absorption. Mottagarvätskan får inte heller skada hudberedningen. I ett genomflödessystem får flödeshastigheten inte förhindra diffusion av testämnet in i mottagarvätskan. I ett statiskt system ska vätskan vara under ständig omrörning och provtas regelbundet. Om metabolism studeras måste receptorvätskan kunna hålla huden viabel under hela försöket.
1.4.3 Hudberedningar
Människo- eller djurhud kan användas. Användning av human hud begränsas är föremål för nationella och internationella etiska hänsynstaganden och begränsningar. Viabel hud är att föredra, men icke-viabel hud kan användas om det kan visas att den inte är skadad. Både epidermala membran (separerade med hjälp av enzymer, värme eller kemikalier) eller hud med anpassad tjocklek (i regel 200–400 μm tjock) preparerad med dermatom är godtagbara. Hud med full tjocklek kan användas, men alltför tjock hud (cirka > 1 mm) bör undvikas, om inte detta särskilt krävs för bestämning av testkemikalien i hudlagren. Valet av art, anatomisk placering och preparationsteknik måste motiveras. Godtagbara data för minst fyra replikat per testberedning krävs.
1.4.4 Hudberedningens integritet
Huden måste prepareras korrekt. Olämplig hantering kan leda till att hornlagret skadas. Därför måste man kontrollera att den preparerade huden inte är skadad. När hudmetabolism undersöks ska den nyutskurna huden användas så snart som möjligt och under betingelser som är kända för att främja metabolisk aktivitet. Som en allmän vägledning kan sägas att nyutskuren hud ska användas inom 24 timmar, men den godtagbara lagringsperioden kan variera beroende på vilket enzymsystem som är involverat i metaboliseringen och på lagringstemperaturen (13). När hudberedningar har lagrats före användning ska det finnas belägg för att barriärfunktionen är bibehållen.
1.4.5 Testämne
Testämnet är den entitet var penetrationsegenskaper studeras. Helst ska testämnet vara radioaktivmärkt.
1.4.6 Testberedning
Beredningen av testämne (t.ex. rent, utspätt eller formulerat material innehållande testkemikalien som appliceras på huden) ska vara detsamma (eller en rimlig ersättning) som det som människor eller andra målarter kan exponeras för. Alla avvikelser från den beredning som ”är i bruk” måste motiveras.
1.4.7 Testämneskoncentration och testämnesformuleringar
Normalt används mer än en koncentration av testämnet så att den övre gränsen för potentiell human exponering ligger inom koncentrationsintervallet. Likaledes bör man överväga att testa ett urval av typiska formuleringar.
1.4.8 Applicering på huden
Under normala betingelser för human exponering för kemikalier är doserna vanligen begränsade. Därför ska en applicering som efterliknar human exponering användas, normalt 1–5 mg/cm2 hud för ett fast ämne och upp till 10 μl/cm2 för en vätska. Mängden ska motiveras av de förväntade användningsbetingelserna, studiens syften och testberedningens fysikaliska egenskaper. Exempelvis kan appliceringen på hudytan vara obegränsad om stora volymer per enhetsyta appliceras.
1.4.9 Temperatur
Passiv diffusion av kemikalier (och därmed deras hudabsorption) påverkas av temperaturen. Diffusionskammaren och huden ska hållas vid en konstant temperatur som ligger nära den normala hudtemperaturen på 32 oC ± 1 oC. Olika cellutföranden kräver olika temperatur på vattenbad eller uppvärmningsblock för att säkerställa att mottagarvätskan/huden hålls vid sin fysiologiska norm. Fuktigheten ska helst ligga mellan 30 och 70 %.
1.4.10 Varaktighet för exponering och provtagning
Huden kan exponeras för testberedningen under hela försökets gång eller under kortare tider (t.ex. för att efterlikna en särskild typ av human exponering). Överflödig testberedning ska tvättas bort från huden med lämpligt rengöringsmedel och tvättvattnet ska samlas upp för analys. Förfarandet för att avlägsna testberedningen beror på de förväntade användningsbetingelserna och ska motiveras. En provtagningsperiod på 24 timmar krävs normalt för att tillåta adekvat karakterisering av absorptionsprofilen. Eftersom hudens integritet kan börja försämras efter 24 timmar bör provtagningstidpunkterna normalt inte ligga efter 24 timmar. För testämnen som tränger igenom huden snabbt kan detta vara onödigt, men för testämnen som tränger igenom huden långsamt kan längre tider krävas. Provtagningsfrekvensen för mottagarvätska ska vara sådan att det går att återge testämnets absorptionsprofil grafiskt.
1.4.11 Avslutningsförfaranden
Alla testsystemets komponenter ska analysers och återvinningen bestämmas. Detta inbegriper givarkammaren, tvättvattnet från hudberedningen, hudberedningen samt mottagarvätska och mottagarkammare. I vissa fall kan huden delas upp i den exponerade hudytan och hudytan under cellens fläns samt i hornlager-, epidermis- och dermisfraktioner för separat analys.
1.4.12 Analys
Adekvat återvinning av radioaktiviteten ska uppnås i alla studier (målet bör vara ett medelvärde på 100 ± 10 % av radioaktiviteten, och alla avvikelser ska motiveras). Mängden testämne i mottagarvätskan och hudberedningen samt i tvättvätskan från hudyta och apparatur ska analyseras med lämplig teknik.
2. DATA
Analysen av mottagarvätskan, fördelningen av testkemikalien i testsystemet och absorptionsprofilen med tid ska presenteras. Då begränsade doser används ska den mängd som tvättas bort från huden, den mängd som finns i huden (och i de olika hudlagren om detta har analyserats) samt den mängd som finns i mottagarvätskan (hastighet samt mängd eller procentandel av applicerad dos) beräknas. Hudabsorption uttrycks ibland endast som data för mottagarvätskan. Om testämnet finns kvar i huden i slutet av försöket kan dock den mängden behöva inkluderas i den totala absorberade mängden (se stycke 66 i hänvisning 3). Om en obegränsad dos har använts vid exponeringen kan det gå att beräkna en permeabilitetskonstant (Kp) med hjälp av erhållna data. Under de senare betingelserna är procentandelen absorberat testämne inte av betydelse.
3. RAPPORTERING
3.1 FÖRSÖKSRAPPORT
Försöksrapporten måste avspegla de krav som ställs enligt försöksprotokollet, inbegripet en motivering för det använda testsystemet. Rapporten måste innehålla följande information:
|
|
Testämne:
|
|
|
Testberedning:
|
|
|
Försöksförhållanden:
|
|
|
Resultat:
|
|
|
Diskussion av resultaten. |
|
|
Slutsatser. |
4. HÄNVISNINGAR
|
1. |
Testmetod B.44. Hudabsorption: in vivo-metod. |
|
2. |
OECD (2002). Guidance Document for the Conduct of Skin Absorption Studies. OECD, Paris. |
|
3. |
OECD (2000). Report of the Meeting of the OECD Extended Steering Committee for Percutaneous Absorption Testing, Annex 1 to ENV/JM/TG(2000)5. OECD, Paris. |
|
4. |
Kemppainen BW and Reifenrath WG. (1990). Methods for skin absorption. CRC Press, Boca Raton. |
|
5. |
Bronaugh RL and Collier, SW. (1991). Protocol for In vitro Percutaneous Absorption Studies, in In vitro Percutaneous Absorption: Principles, Fundamentals and Applications, RL Bronaugh and HI Maibach, Eds., CRC Press, Boca Raton, pp. 237–241. |
|
6. |
Bronaugh RL and Maibach HI. (1991). In vitro Percutaneous Absorption: Principles, Fundamentals and Applications. CRC Press, Boca Raton. |
|
7. |
European Centre for Ecotoxicology and Toxicology of Chemicals (1993). Monograph No. 20, Percutaneous Absorption, ECETOC, Brussels. |
|
8. |
Diembeck W, Beck H, Benech-Kieffer F, Courtellemont P, Dupuis J, Lovell W, Paye M, Spengler J, Steiling W (1999). Test Guidelines for In Vitro Assessment of Dermal Absorption and Percutaneous Penetration of Cosmetic Ingredients, Fd Chem Tox, 37, 191–205. |
|
9. |
Recommended Protocol for In vitro Percutaneous Absorption Rate Studies (1996). US Federal Register, Vol. 61, No. 65. |
|
10. |
Howes D, Guy R, Hadgraft J, Heylings JR et al. (1996). Methods for assessing Percutaneous absorption. ECVAM Workshop Report ATLA 24, 81 R10. |
|
11. |
Schaefer H and Redelmeier TE. (1996). Skin barrier: principles of percutaneous absorption. Karger, Basel. |
|
12. |
Roberts MS and Walters KA. (1998). Dermal absorption and toxicity assessment. Marcel Dekker, New York. |
|
13. |
Jewell, C., Heylings, JR., Clowes, HM. And Williams, FM. (2000). Percutaneous absorption and metabolism of dinitrochlorobenzene in vitro. Arch Toxicol 74: 356–365. |
Figur 1
Ett exempel på en typisk utformning av en statisk diffusionscell för perkutana absorptionsstudier in vitro.

(1) Området för hornhinneopacitet bör noteras
(2) I fråga om vissa mätningar av serum och plasma, särskilt i fråga om glukos, är fasta under natten att föredra. Huvudskälet är att den ökade variation som skulle följa av icke-fastande skulle tendera att dölja mer subtila effekter och försvåra tolkningen. Å andra sidan kan fasta under natten påverka djurens generella metabolism och särskilt i utfodringsundersökningar störa den dagliga exponeringen för testsubstansen. Om djuren får fasta under natten bör de kliniskt kemiska analyserna göras efter det att funktionsobservationer utförts under försökets fjärde vecka.
(3) För vissa matningar som görs på serum och plasma, särskilt i fråga om glukos, är fasta över natten att föredra. Den viktigaste orsaken till detta är att resultaten annars oundvikligen uppvisar större variationer, vilket tenderar att maskera svaga verkningar och göra det svårare att tolka resultaten. Å andra sidan kan fasta över natten leda till störningar i djurens allmänna metabolism och särskilt i fråga om exponeringstest störa den dagliga exponeringen för testämnet. Om fasta över natten tillämpas bör de kliniska biokemiska bestämningarna göras efter funktionsobservationerna.
(4) Numera känt som serum alanine aminotransferase.
(5) Numera kant som serum aspartate aminotransferase.
(6) Dessa organ, från tio djur per kön per grupp för gnagare och samtliga icke-gnagare, plus thyroid (med parathyroider) för samtliga icke-gnagare, ska vägas.
(7) Dessa organ, från tio djur per kön per grupp för gnagare, ska vägas.
(8) I den metod som beskrivs här avses med referensgrupp en grupp som tillförs testämnet genom en annan tillförselväg som säkerställer att hela dosen tillförs.
(9) Lösningsmedel som används vid absorptionsmätning.
|
† |
Inbegriper 5 djur som valts ut för funktionella observationer och detaljerade kliniska observationer som en del av testet avseende neurotoxicitet. |
DEL C: METODER FÖR BESTÄMNING AV EKOTOXICITET
INNEHÅLLSFÖRTECKNING
|
C.1 |
AKUT TOXICITET FÖR FISK |
|
C.2 |
TEST AV AKUT IMMOBILISERING AV DAPHNIA-ARTER |
|
C.3 |
TEST AV TILLVÄXTHÄMNING PÅ ALGER |
|
C.4 |
BESTÄMNING AV ”LÄTT” BIONEDBRYTBARHET |
|
DEL 1 |
ALLMÄNNA PRINCIPER |
|
DEL 2 |
DOC-ELIMINERING (DOC DIE-AWAY) (metod C.4-A) |
|
DEL 3 |
MODIFIERAT OECD-SCREENINGTEST (metod C.4-B) |
|
DEL 4 |
CO2-UTVECKLING (metod C.4-C) |
|
DEL 5 |
MANOMETRISK RESPIROMETRITEST (metod C.4-D) |
|
DEL 6 |
CLOSED BOTTLE-TEST (metod C.4-E) |
|
DEL 7 |
MITI TEST (metod C.4-F) |
|
C.5 |
NEDBRYTNING – BIOKEMISK SYREFÖRBRUKNING |
|
C.6 |
NEDBRYTNING – KEMISK SYREFÖRBRUKNING |
|
C.7 |
NEDBRYTNING – ABIOTISK NEDBRYTNING: HYDROLYS SOM FUNKTION AV PH |
|
C.8 |
TOXICITET HOS DAGGMASKAR |
|
C.9 |
BIOLOGISK NEDBRYTBARHET: ZAHN-WELLENS TEST |
|
C.10 |
BIOLOGISK NEDBRYTBARHET: AKTIVERAT SLAM – SIMULATIONSTEST |
|
C.11 |
BIOLOGISK NEDBRYTBARHET: AKTIVERAT SLAM – RESPIRATIONSHÄMNINGSTEST |
|
C.12 |
BIOLOGISK NEDBRYTBARHET: MODIFIERAT SCAS-TEST |
|
C.13 |
BIOKONCENTRATION: GENOMFLÖDESPROVNING MED FISK |
|
C.14 |
TILLVÄXTTEST PÅ UNGA EXEMPLAR AV FISK |
|
C.15 |
FISK, KORTTIDSTEST AVSEENDE TOXICITET PÅ EMBRYO-OCH SÄCKYNGELSTADIERNA |
|
C.16 |
HONUNGSBIN – TEST AVSEENDE AKUT TOXICITET (ORALT) |
|
C.17 |
HONUNGSBIN – TEST AVSEENDE AKUT TOXICITET (KONTAKT) |
|
C.18 |
BESTÄMNING AV ADSORPTION OCH DESORPTION I JORD VID JÄMVIKT I UPPSLAMMAT JORDPROV |
|
C.19 |
UPPSKATTNING AV ADSORPTIONSKOEFFICIENT (KOC ) I JORD OCH AVLOPPSSLAM MED ANVÄNDNING AV HÖGTRYCKSVÄTSKEKROMATOGRAFI (HPLC) |
|
C.20 |
REPRODUKTIONSTEST PÅ DAPHNIA MAGNA |
|
C.21 |
JORDLEVANDE MIKROORGANISMER: TEST RÖRANDE OMVANDLING AV KVÄVE |
|
C.22 |
JORDLEVANDE MIKROORGANISMER: TEST RÖRANDE OMVANDLING AV KOL |
|
C.23 |
AEROB OCH ANAEROB OMVANDLING I JORD |
|
C.24 |
AEROB OCH ANAEROB OMVANDLING I AKVATISKA SEDIMENTSYSTEM |
C.1 AKUT TOXICITET FÖR FISK
1. METOD
1.1 INLEDNING
Syftet med detta test är att bestämma ett ämnets akuta dödliga toxicitet för sötvattenfisk. Det är värdefullt att känna till ämnets löslighet i vatten, ångtryck, kemiska stabilitet, dissociationskonstanter och bionedbrytbarhet när man ska välja den mest lämpliga testmetoden (statisk, semistatisk eller genomströmning) för att säkerställa tillfredsställande konstanta koncentrationer av testämnet under hela testperioden.
Vid planeringen av testet och tolkningen av testresultaten bör man ta hänsyn till andra uppgifter (t.ex. strukturformel, renhetsgrad, art och procentdel av signifikanta orenheter, förekomst och mängd av tillsatser, samt n-oktanol/vattendelningskoefficienten).
1.2 DEFINITIONER OCH ENHETER
Akut toxicitet är de märkbara skadliga verkningar som på kort tid (dagar) framkallas hos en organism som exponeras för ett ämne. I detta test uttrycks akut toxicitet som median letal koncentration (LC50), dvs. den koncentration i vatten som medför att 50 % av fiskarna i en försöksgrupp dör under kontinuerlig exponering i en viss tid. Exponeringstiden ska anges.
Alla koncentrationer av testämnet anges i vikt per volymenhet (milligram per liter). De kan också uttryckas som vikt per vikt (mg/kg-1).
1.3 REFERENSÄMNEN
Ett referensämne kan användas för att visa att försöksdjurens respons inte har ändrats signifikant under testbetingelserna i laboratoriet.
Inga referensämnen finns angivna för detta test.
1.4 TESTMETODSPRINCIP
Ett gränstest kan utföras med 100 mg per liter för att konstatera om LC50 är större än denna koncentration.
Fisken exponeras för testämnet som i en rad koncentrationer tillsätts vattnet under 96 timmar. Dödligheten registreras med högst 24 timmars mellanrum och om så är möjligt beräknas de koncentrationer som vid varje observationstillfälle har medfört att 50 % av fisken (LC50) har dött.
1.5 KVALITETSKRITERIER
Kvalitetskriterierna gäller för såväl gränstestet som den fullständiga testmetoden.
Dödligheten i kontrollgruppen får inte överstiga 10 % (eller 1 fisk om färre än 10 fiskar används) i slutet av testet.
Under hela testet ska koncentrationen av upplöst syre vara högre än 60 % av luftmättnadsvärdet.
Koncentrationer av testämnet ska ligga över 80 % av startkoncentrationen under hela testperioden.
För ämnen som är lättlösliga i testmediet och ger stabila lösningar, dvs. ämnen som inte i hög grad förångas, bryts ned, hydrolyseras eller adsorberas, kan startkoncentrationen antas vara lika med den nominella koncentrationen. Man ska kunna bevisa att koncentrationen har bibehållits under hela testet och att kvalitetskriterierna är uppfyllda.
För ämnen som är
|
i) |
svårlösliga i testmediet, eller |
|
ii) |
kan skapa stabila emulsioner eller dispersioner, eller |
|
iii) |
är ostabila i vattniga lösningar |
ska man som värde för startkoncentrationen använda den koncentration som är uppmätt i lösningen (eller om detta inte är tekniskt möjligt, mätt i vattenkolonnen) vid testets inledning. Koncentrationen bestäms sedan jämvikt uppnåtts, men innan fisken släpps i.
I alla dessa fall ska ytterligare mätningar göras under testets gång för att säkerställa att koncentrationen har bibehållits och att kvalitetskriterierna har varit uppfyllda.
pH-värdet får inte variera med mer än 1 enhet.
1.6. METODBESKRIVNING
Tre typer av förfaranden kan användas:
Statiskt test:
Toxicitetstest utan något genomflöde av testlösningen. (Lösningarna förblir oförändrade under hela testet.)
Semi-statiskt test:
Test utan genomflöde av testlösningen men där hela testlösningar regelbundet byts ut helt efter längre perioder (t.ex. 24 timmar).
Genomströmningstest:
Toxicitetstest där vattnet i testkamrarna förnyas hela tiden och där testämnet transporteras med det vatten som används till att förnya testmediet.
1.6.1 Reagenser
1.6.1.1 Lösningar av testämnen
Stamlösningar av erforderlig styrka framställs genom att testämnet upplöses i avjoniserat vatten eller vatten enligt 1.6.1.2.
De valda testkoncentrationerna framställs genom utspädning av stamlösningen. Om höga koncentrationer testas kan ämnet lösas direkt i utspädningsvattnet.
Ämnen testas normalt endast upp till gränsen för löslighet. För vissa ämnen (t.ex. ämnen som är svårlösliga i vatten, som har ett högt Pow-värde, eller som bildar en stabil dipersion och inte en riktig lösning i vatten) accepteras att man har använt en testkoncentration som är större än lösligheten för att säkerställa att den högsta upplösta/stabila koncentration har nåtts. Det är dock viktigt att denna koncentration inte på annat sätt förstör testmetoden (t.ex. bildar en hinna av ämnet på vattenytan som förhindrar syrsättning av vattnet, etc.).
Ultraljud, organiska lösningsmedel, emulgeringsmedel eller dispergeringsmedel kan användas för att framställa stamlösningar av ämnen som är svårlösliga i vatten och för att dispergera sådan ämnen i testmediet. Om tillsatsämnen används ska alla testkoncentrationer innehålla samma mängd av tillsatsämnet och en extra kontrollgrupp ska användas som utsätts för samma koncentration av tillsatsämnet som den som används i testserien. Sådana tillsatsämnen används i lägsta möjliga koncentration och får aldrig vara större än 100 mg per liter i testmediet.
Testet utförs utan justering av pH-värdet. Om det finns tecken på en markant förändring av pH-värdet rekommenderas att testet upprepas med pH-justering och att dessa resultat rapporteras. I detta fall ska pH-värdet i stamlösningen justeras efter pH-värdet i utspädningsvattnet, om det inte finns särskilda skäl som talar mot detta. HC1 och NaOH är förstahandsvalen för justering av pH-värdet. Denna pH-justering ska göras på ett sådant sätt att testämnets koncentration i stamlösningen inte ändras i någon signifikant grad. Om justeringen medför någon kemisk reaktion eller fysisk utfällning av testämnet ska detta rapporteras.
1.6.1.2 Vatten till förvaring och utspädning
Dricksvatten (som inte är förorenat av skadliga koncentrationer av klor, tungmetaller eller andra ämnen), råvatten av god kvalitet eller rekonstituerat vatten (se tillägg 1) kan användas. Vatten med en total hårdhet på mellan 10 och 250 mg per liter (som CaCO3) och med ett pH-värde av 6,0–8,5 är att föredra.
1.6.2 Apparatur
All apparatur ska vara tillverkad av kemiskt inert material:
|
— |
Automatiskt utspädningssystem (till genomströmningstestet). |
|
— |
Syremätare. |
|
— |
Utrustning för bestämning av vattnets hårdhet. |
|
— |
Lämplig apparatur för temperaturkontroll. |
|
— |
pH-mätare. |
1.6.3 Testfisk
Fiskarna ska vara friska och utan synliga missbildningar.
De arter som ska användas bör väljas på grundval av praktiska kriterier, t.ex. att de är lätt tillgängliga under hela året, enkla att hålla, lämpliga för testet, har en relativ känslighet, samt på grundval av ekonomiska, biologiska och ekologiska faktorer av betydelse. Nödvändigheten att kunna jämföra de data som erhålls och redan genomförd internationell harmonisering (hänvisning 1) bör också beaktas vid val av fiskart.
I tillägg 2 förtecknas de fiskarter som rekommenderas för detta test. Sebrafisk och regnbåge är att föredra.
1.6.3.1 Förvaring
Testfisk ska helst komma från ett enda bestånd och vara av ungefär samma längd och ålder. Fisken ska förvaras i minst 12 dagar under följande betingelser:
Belastning:
Anpassad efter systemet (cirkulation eller genomströmning) och fiskarterna.
Vatten:
Se punkt 1.6.1.2.
Ljus:
12–16 timmars ljus per dag.
Koncentration av upplöst syre:
Minst 80 % av luftmättnadsvärdet.
Utfodring:
Dagligen eller tre gånger per vecka, sista gången 24 timmar innan testet inleds.
1.6.3.2 Dödlighet
Efter 48 timmars acklimatisering registreras dödligheten och följande kriterier tillämpas:
|
— |
Högre än 10 % av populationen på 7 dagar: hela partiet kasseras. |
|
— |
Mellan 5 och 10 % av populationen: observationsperioden förlängs med ytterligare 7 dagar. Om inga fler dödsfall inträffar kan partiet accepteras, i annat fall kasseras det. |
|
— |
Lägre än 5 % av populationen: partiet kan accepteras. |
1.6.4 Tillvänjning
All fisk ska i minst 7 dagar före testet exponeras för vatten av den kvalitet och temperatur som ska användas i testet.
1.6.5 Testförfarande
Före det slutliga testet kan ett inledande test göras för att fastställa vilka koncentrationer som ska användas i huvudtestet.
Förutom testserien inbegrips en kontrollgrupp som inte tillförs testämnet och, i vissa fall, en kontrollgrupp som tillförs tillsatsämnet.
Beroende på testämnets fysiska och kemiska egenskaper ska ett statiskt test, ett semi-statiskt test eller ett genomströmningstest väljas så att kvalitetskriteriet uppfylls.
Fisken exponeras för testämnet enligt följande:
|
— |
Varaktighet: 96 timmar. |
|
— |
Antal djur: Minst 7 per koncentration. |
|
— |
Kar: Av passande storlek i förhållande till den rekommenderade belastningen. |
|
— |
Belastning: För statisk och semistatiska test rekommenderas 1,0 g per liter som maximal belastning. För genomströmningssystem kan större belastning accepteras. |
|
— |
Testkoncentrationer: Minst 5 koncentrationer med ett konstant inbördes förhållande av högst 2,2 som så långt det för täcker intervallet 0–100 % dödlighet. |
|
— |
Vatten: Se 1.6.1.2. |
|
— |
Ljus: 12–16 timmar ljus per dag. |
|
— |
Temperatur: Lämplig för arten (tillägg 2) men inom ±1 oC i ett enskilt test. |
|
— |
Koncentration av upplöst syre: Inte mindre än 60 % av luftmättnadsvärdet vid den valda temperaturen. |
|
— |
Utfodring: Ingen. |
Fisken besiktas efter de första 2–4 timmarna och minst var 24:e timme. Fisk anses vara död om beröring av stjärtfenan inte framkallar någon reaktion och inga andningsrörelser kan iakttas. Död fisk avlägsnas när den upptäcks och dödsfall registreras. Register förs över synliga abnormiteter (t.ex. förlust av jämvikten, ändrat simbeteende, andningsfunktion, pigmentering osv.).
Mätningar av pH, upplöst syre och temperatur ska göras dagligen.
Gränstest
Genom användning av förfarandena som beskrivs i denna testmetod kan ett gränstest utföras med 100 mg per liter för att konstatera om LC50 är större än denna koncentration.
Om ämnet är av sådan art att man inte kan erhålla en koncentration av 100 ml per liter i testvattnet utförs gränstestet vid ämnets mättnadskoncentration (eller den högsta koncentrationen vid vilken det bildas en stabil disper-sion) i det använda mediet (se även 1.6.1.1).
Gränstestet utförs med 7–10 fiskar och en eller två kontrollgrupper av samma storlek. (Enligt binomialteorin är det vid användning av 10 fiskar 99,9 % säkerhet för att LC50 är större än den använda koncentrationen, om dödligheten är noll. Med 7, 8 eller 9 fiskar ger en dödlighet av noll 99 % säkerhet för att LC50 är större än den använda koncentrationen.)
Om dödsfall sker måste en fullständig undersökning utföras. Eventuella subletala verkningar registreras.
2. DATA OCH UTVÄRDERING
För var och en av de tidsperiod då observationer företas (efter 24, 48, 72 och 96 timmar) avbildas den procentuella dödligheten mot koncentrationen på logaritmisk sannolikhetspapper.
När så är möjligt och för varje observationstid ska LC50 och dess konfidensgränser (p = 0,05) uppskattas genom standardförfaranden. Dessa värden ska avrundas till en (eller högst två) signifikanta siffror (exempel på avrundning till två siffror: 170 för 173,5, 0,13 för 0,127, 1,2 för 1,21).
I de fall där koncentrations-procentresponskurvans lutning är för brant för att medge en beräkning av LC50 räcker det med en grafisk uppskattning av detta värde.
Om två på varandra följande koncentrationer med ett förhållande på 2,2 endast ger 0 och 100 % dödlighet, räcker dessa två värden för att ange inom vilket område LC50 ligger.
Om det framgår att det inte går att hålla testämnet stabilt eller homogent ska detta rapporteras och försiktighet ska iakttas vid tolkningen av resultaten.
3. RAPPORTERING
Testrapporten ska om möjligt innehålla:
|
— |
Uppgifter om testfisken (vetenskapligt namn, stam, leverantör, eventuell förbehandling, storlek och antal som använts for varje koncentration). |
|
— |
Källan till utspädningsvattnet och viktiga kemiska egenskaper (Ph, hårdhet, temperatur). |
|
— |
För ämnen med låg vattenlöslighet anges metod att framställa stam- och testlösning. |
|
— |
Koncentrationen av eventuella tillsatsämnen. |
|
— |
En lista över de koncentrationer som använts och alla tillgängliga uppgifter om testämnets stabilitet vid kon centrationerna i testlösningen. |
|
— |
Om kemiska analyser görs ska metoder och resultat rapporteras. |
|
— |
Resultat från gränstest om sådant utförts. |
|
— |
Skälen till valet av och detaljer om använt testförfarande (t.ex. statisk, semi-statisk, doseringstakt, genomströmningstakt, eventuell syrsättning, antal fiskar osv.). |
|
— |
Beskrivning av testutrustningen. |
|
— |
Ljusbetingelser. |
|
— |
Testlösningens koncentration av upplöst syre, pH-värden och temperatur var 24:e timme. |
|
— |
Bevis på att kvalitetskriterierna har uppfyllts. |
|
— |
Kumulativ dödlighet för varje koncentration och kontrollprov (eller kontroll med tillsatsämne om sådant behövs) vid de rekommenderade observationstiderna. |
|
— |
Grafisk framställning av denna responskurva över koncentration/procent i slutet av testet. |
|
— |
Om möjligt, LC50-värden för varje rekommenderad observationstid (med 95 % konfidensgränser). |
|
— |
Statistiska förfaranden som använts vid bestämning av LC50-värdena. |
|
— |
Om referensämne har använts ska erhållna resultat anges. |
|
— |
Högsta testade koncentration som inte medför dödsfall inom testperioden. |
|
— |
Lägsta testade koncentrationen som medför 100 % dödlighet inom testperioden. |
4. HÄNVISNINGAR
|
1. |
OECD, Paris, 1981, Test Guideline 203, rådets beslut K(81) 30 slutligt och uppdaterat. |
|
2. |
AFNOR – Determination of the acute toxicity of a substance to Brachydanio rerio – Static and Flow Through methods – NFT 90-303 juni 1985. |
|
3. |
AFNOR – Determination of the acute toxicity of a substance to Salmo gairdneri – Static and Flow Through methods – NFT 90-305 juni 1985. |
|
4. |
ISO 7346/1,/2 och/3 – Water Quality – Determination of the acute lethal toxicity of a substance to a fresh water fish (Brachydanio rerio Hamilton-Buchanan – Teleostei, Cyprinidae). Del 1: Static method. Del 2: Semi-static method. Del 3: Flow-through method. |
|
5. |
Eidgenössiches Departament des Innern, Schweiz: Richtlinien fur Probenahme und Normung von Wasseruntersuchungsmetoden – Del II 1974. |
|
6. |
DIN Testverfahren mit Wasserorganismen, 38 412 (Ll) och L (15). |
|
7. |
JIS K 0102, Acute toxicity test for fish. |
|
8. |
NEN 6506 – Water – Bepailing van de akute toxiciteit met behulp van Poecilia reticulata, 1980. |
|
9. |
Environmental Protection Agency, Methods for the acute toxicity tests with fish, macroinvertebrates and amphibians, The committee on Methods for Toxicity Tests with Aquatic Organisms, Ecological Research Series EPA-660-75-009, 1975. |
|
10. |
Environmental Protection Agency, Environmental monitoring and support laboratory, Office of Research and Development, EPA-600/4-78-012, January 1978. |
|
11. |
Environmental Protection Agency, Toxic Substance Control, del IV, 16 mars 1979. |
|
12. |
Standard methods for the examination of water and wastewater, 14th edition, APHA-AWWA-WPCF, 1975. |
|
13. |
Commission of the European Communities, Inter-laboratory test programme concerning the study of the ecotoxicity of a chemical substance with respect to the fish, EEC Study D.8368, 22 March 1979. |
|
14. |
Verfahrensvorschlag des Umweltsbundesamt zum akuten Fisch-Test. Rudolph, P. och Boje, R. Okotoxikologie, Grundlagen fur die okotoxikologische Bewertung von Umweltchemikalien nach dem Chemikalienge-setz, ecomed 1986. |
|
15. |
Litchfield, J. T. and Wilcoxon, E, A simplified method for evaluating dose effects experiments, J. Pharm, Exp. Therap., vol. 96, 1949, s. 99. |
|
16. |
Finney, D. J. Statistical Methods in biological Assay. Griffin, Weycombe, U. K., 1978. |
|
17. |
Sprague, J. B. Measurement of pollutant toxicity to fish. II Bioassay methods for acute toxicity. Water Res. 1969, vol. 3, s. 793–821. |
|
18. |
Sprague, J. B. Measurement of pollutant toxicity to fish. II Utilising and applying bioassay results. Water Res. 1970, vol. 4, s. 3–32. |
|
19. |
Stephan, C. E. Methods for calculating an LC50. In Aquatic Toxicology and Hazard Evaluation (edited by F. I. Mayer och J. L. Hamelink). American Society for Testing and Materials, ASTM STP 634, 1977, s. 65–84. |
|
20. |
Stephan, C. E., Busch, K. A., Smith, R., Burke, J. och Andrews, R. W. A computer program for calculating an LC50. US EPA. |
Tillägg 1
Rekonstituterat vatten
Exempel på lämpligt utspädningsvatten
Alla kemikalier ska vara av analytisk renhetsgrad.
Vattnet ska vara destillerat vatten av god kvalitet eller avjoniserat vatten med en ledningsförmåga på mindre än 5μScm-1.
Apparatur för destillering av vatten får inte innehålla några delar gjorda av koppar.
Stamlösningar
|
CaCl2 · 2H2O (kalciumklorid dihydrat) Upplöses i vatten så att 1 liter erhålls. |
11,76 g |
|
MgSO4.7H2O (magnesiumsulfat heptahydrat): Upplöses i vatten så att 1 liter erhålls. |
4,93 g |
|
NaHCO3 (natriumvätekarbonat): Upplöses i vatten så att 1 liter erhålls. |
2,59 g |
|
KCl (kaliumklond): Upplöses i vatten så att 1 liter erhålls. |
0,23 g |
Rekonstituerat utspädningsvatten
Blanda 25 ml av de fyra stamlösningarna med vatten så att 1 liter erhålls.
Syrsätt tills koncentrationen av upplöst syre är lika med luftmättnadsvärdet.
pH ska vara 7,8 ±0,2.
Justera vid behov pH-värdet med NaOH (natriumhydroxid) eller HCl (saltsyra).
Det således framställda utspädningsvattnet sätts åt sidan i 12 timmar och ska inte syrsättas ytterligare.
Summan av Ca- och Mg-jonerna i denna lösning är 2,5 mmol per liter. Förhållandet mellan Ca- och Mg-jonerna är 4:1 och mellan Na- och K-jonerna 10:1. Den totala alkaliniteten hos denna lösning är 0,8 mmol per liter.
Avvikelser vid framställningen av utspädningsvattnet får inte ändra vattnets sammansättning eller egenskaper.
Tillägg 2
Fiskarter som rekommenderas till test
|
Rekommenderade arter |
Rekommenderad temperaturintervall ( oC) |
Rekommenderad total längd på testdjur (cm) |
|
Bracbydanio rerio (Teleostei, Cyprinidae) (Hamilton-Buchanan) Sebrafisk |
20–24 |
3,0 ± 0,5 |
|
Pimephales promelas (Teleostei, Cyprinidae) (Rafinesque) Fathead minnow |
20–24 |
5,0 ± 2,0 |
|
Cypnnus carpio (Teleostei, Cyprinidae) (Linné 1758) Karp |
20–24 |
6,0 ± 2,0 |
|
Oryzias latipes (Teleostei, Poeciliidae) Cyprinodontidae (Tomminck och Schlegel 1850) Red killifish |
20–24 |
3,0 ± 1,0 |
|
Poecilia reticulata (Teleostei, Poeciliidae) (Peters 1859) Guppy |
20–24 |
3,0 ± 1,0 |
|
Lepomis macrochirus (Teleostei, Centrarchidae) (Rafinesque Linneaus 1758) Bluegill |
20–24 |
5,0 ± 2,0 |
|
Onchorhynchus mykiss (Teleostei, Salmonidae) (Walbaum 1988) Regnbågsforell |
12–17 |
6,0 ± 2,0 |
|
Leuciscus idus (Teleostei, Cyprinidae) (Linné 1758) Guldfisk |
20–24 |
6,0 ± 2,0 |
Odlingsbetingelser
Ovan angivna fiskar är lätta att odla och/eller är lätt tillgängliga året runt. De kan förädlas och odlas i fiskdammar eller i laboratorier under sjukdoms- och parasitkontrollerade betingelser så att testdjuren är friska och av känd härstamning. Dessa fiskar finns i många delar av världen.
Tillägg 3
Exempel på kurva över koncentration mot dödlighet
Exempel på bestämning av LC50 på log-probit-papper

C.2 TEST AV AKUT IMMOBILISERING AV DAPHNIA-ARTER
1. METOD
Denna testmetod för akut immobilisering motsvarar OECD TG 202 (2004).
1.1 INLEDNING
Denna metod beskriver ett akut toxicitetstest för att bedöma effekten av kemikalier på hinnkräftor (Daphnia). Såvitt möjligt har befintliga testmetoder använts (1)(2)(3).
1.2 DEFINITIONER
För denna metods ändamål gäller följande definitioner:
|
|
EC 50 : Den koncentration som beräknas immobilisera 50 % av hinnkräftorna inom en given exponeringstid. Om någon annan definition används måste denna rapporteras tillsammans med en hänvisning. |
|
|
Immobilisering: De djur som inte längre kan simma inom 15 sekunder efter varsam skakning av testkärlet anses vara immobiliserade (även om de fortfarande kan röra på antennerna). |
1.3 PRINCIP FÖR TESTMETODEN
Unga havskräftor, yngre än 24 timmar då testet inleds, exponeras för testämnet i en serie koncentrationer under en period av 48 timmar. Immobilisering registreras vid 24 timmar och 48 timmar och jämförs med kontrollvärden. Resultaten analyseras för beräkning av EC50 vid 48 timmar (se avsnitt 1.2 för definitioner). Bestämning av EC50 vid 24 timmar är valfri.
1.4 INFORMATION OM TESTÄMNET
Testämnets vattenlöslighet och ångtryck ska vara kända och en tillförlitlig analysmetod ska finnas tillgänglig för kvantifiering av ämnet i testlösningen, med rapporterad återvinningseffektivitet samt analysgräns. Användbar information inbegriper strukturformel, ämnets renhet, stabiliteten i vatten och i ljus, Pow och resultat från ett test för biologisk nedbrytbarhet (se metod C.4).
Anmärkning: Vägledning för testämnen med fysikalkemiska egenskaper som gör dem svåra att testa ges i (4).
1.5 REFERENSÄMNEN
Ett referensämne kan testas med avseende på EC50 som ett sätt att garantera att försöksbetingelserna är tillförlitliga. Giftämnen som används vid internationell ringtestning (1)(5) rekommenderas för detta ändamål (1). Försök med ett referensämne ska helst göras varje månad och minst två gånger per år.
1.6 KVALITETSKRITERIER
För att ett försök ska vara giltigt gäller följande kriterier för prestanda:
|
— |
I kontrollerna, inbegripet den kontroll som innehåller solubiliseringsagenset, får inte mer än 10 % av hinnkräftorna immobiliseras. |
|
— |
Koncentrationen av löst syre i slutet av försöket ska vara > 3 mg/l i kontroll- och testkärl. |
Anmärkning: För det första kriteriet ska inte mer än 10 % av kontrollhinnkräftorna uppvisa immobilisering eller andra tecken på sjukdom eller stress, t.ex. missfärgning eller ovanligt beteende såsom infångning vid vattenytan.
1.7 BESKRIVNING AV TESTMETODEN
1.7.1 Utrustning
Försökskärl och annan apparatur som kommer i kontakt med testlösningarna bör vara tillverkade helt i glas eller annat kemiskt inert material. I regel används glasprovrör eller glasbägare. Dessa bör rengöras före varje rutinmässigt laboratorieförfarande. Försökskärlen ska vara löst täckta för att minska vattenförlusten på grund av avdunstning och för att förhindra att damm kommer ner i lösningarna. Flyktiga ämnen ska testas i helt fyllda, slutna kärl som är tillräckligt stora för att förhindra att syret blir begränsande eller att det blir syrebrist (se avsnitt 1.6 och första stycket i avsnitt 1.8.3).
Dessutom behövs åtminstone en del av följande utrustning: syrgasmätare (med mikroelektrod eller annan lämplig anordning för att mäta löst syre i lågvolymsprover), pH-meter, adekvat anordning för temperaturkontroll, utrustning för bestämning av totalt organiskt kol (TOC), utrustning för bestämning av kemisk syreförbrukning (COD), utrustning för bestämning av vattnets hårdhetsgrad.
1.7.2 Försöksorganism
Daphnia magna Straus är den föredragna testarten, även om andra lämpliga arter av hinnkräfta kan användas i detta test (t.ex. Daphnia pulex). Vid testets början måste djuren vara yngre än 24 timmar och för att minska variabiliteten är en stark rekommendation att de inte tillhör den första satsen avkomma. De ska komma från en frisk stam (dvs. inte visa tecken på stress, t.ex. hög dödlighet, förekomst av hanar och ephippia, fördröjd produktion av första satsen avkomma, missfärgade djur osv.). Alla organismer som används för ett visst försök ska ha sitt ursprung i odlingar som etablerats från samma stam av hinnkräftor. Stamdjuren måste hållas i samma odlingsbetingelser (ljus, temperatur, medium) som används vid testet. Om annat än normalt odlingsmedium för hinnkräftor används vid testet är det tillrådligt med en acklimatiseringsperiod före testet. Under denna period ska Dapnia-avkomman hållas i utspädningsvatten vid testtemperaturen under minst 48 timmar innan testet inleds.
1.7.3 Förvarings- och utspädningsvatten
Naturligt vatten (yt- eller grundvatten), rekonstituerat vatten eller avklorerat kranvatten är godtagbart som förvarings- och utspädningsvatten om hinnkräftorna överlever i det under odling, acklimatisering och testning utan att uppvisa tecken på stress. Vatten som uppfyller de kemiska egenskaper som listas i bilaga 1 är lämpliga som testvatten. Vattnet ska hålla konstant kvalitet under testperioden. Rekonstituerat vatten kan framställas genom tillsats av specifika mängder reagens av vedertagen analysrenhetsgrad till avjonat eller destillerat vatten. Exempel på rekonstituerat vatten ges i (1) (6) och i bilaga 2. Observera att medier som innehåller kända kelateringsmedel, t.ex. M4- och M7-medium i bilaga 2, ska undvikas för testämnen som innehåller metaller. pH ska ligga i området 6 till 9. En hårdhetsgrad på mellan 140 och 250 mg/l (i form av CaCO3) rekommenderas för Daphnia Magna, medan även lägre hårdhetsgrader kan vara lämpliga för andra Daphnia-arter. Utspädningsvattnet kan luftas före testet så att koncentrationen av löst syre når mättnad.
Om naturligt vatten används ska kvalitetsparametrarna mätas minst två gånger per år eller närhelst man kan misstänka att dessa egenskaper kan ha ändrats väsentligt (se stycket ovan och bilaga 1). Mätningar av tungmetaller (t.ex. Cu, Pb, Zn, Hg, Cd, Ni) ska också göras. Om avklorerat kranvatten används är en daglig kloranalys önskvärd. Om utspädningsvattnet kommer från en yt- eller grundvattenkälla ska konduktivitet samt totalt organiskt kol (TOC) eller kemisk syreförbrukning (COD) mätas.
1.7.4 Testlösningar
Testlösningar av de valda koncentrationerna bereds i regel genom utspädning av en stamlösning. Stamlösningarna bör helst beredas genom upplösning av testämnet i utspädningsvattnet. Så långt det är möjligt bör bruk av lösningsmedel, emulgeringsmedel eller dispergeringsmedel undvikas. Sådana föreningar kan dock krävas i vissa fall för att få en stamlösning av lämplig koncentration. Vägledning när det gäller lämpliga lösningsmedel, emulgeringsmedel och dispergeringsmedel ges i (4). Testämnets halt i testlösningarna bör under inga omständigheter överskrida lösligheten i utspädningsvattnet.
Testet ska göras utan pH-justering. Om pH inte ligger kvar i området 6–9 görs ett andra test, med justering av stamlösningens pH till utspädningsvattnets före tillsats av testämne. pH-justeringen ska göras på ett sådant sätt att stamlösningens koncentration inte förändras väsentligt och så att ingen kemisk reaktion eller utfällning av testämnet orsakas. HC1 och NaOH är föredragna.
1.8 FÖRFARANDE
1.8.1 Exponeringsförhållanden
1.8.1.1 Testgrupper och kontroller
Testkärlen fylls med lämpliga volymer utspädningsvatten och lösningar av testämne. Kvoten luftvolym/vattenvolym i kärlet ska vara identisk för test- och kontrollgruppen. Hinnkräftorna placeras sedan i testkärlen. Minst 20 djur, helst indelade i fyra grupper om fem vardera, ska användas för varje testkoncentration och för kontrollerna. Minst 2 ml testlösning ska tillhandahållas för varje djur (dvs. en volym på l0 ml för fem hinnkräftor per testkärl). Testet kan utföras med halvstatisk förnyelse eller med genomflödessystem där koncentrationen av testämne inte är stabil.
En kontrollserie med utspädningsvatten och också, om detta är relevant, en kontrollserie som innehåller solubiliseringsagenset måste köras utöver behandlingsserierna.
1.8.1.2 Testkoncentrationer
Ett test för att söka intervall kan genomföras för att bestämma koncentrationsområdet för det egentliga testet om ingen information om testämnets toxicitet finns att tillgå. För detta ändamål exponeras hinnkräftorna för en serie koncentrationer av testämnet som ligger långt ifrån varandra. Fem hinnkräftor ska utsättas för varje testkoncentration i 48 timmar eller mindre, och inga replikat krävs. Exponeringstiden kan förkortas (t.ex. 24 timmar eller mindre) om data lämpliga för intervalltestets ändamål kan erhållas på kortare tid.
Minst fem testkoncentrationer ska användas. Dessa arrangeras i geometriska serier med en separationsfaktor som helst inte överskrider 2,2. Om färre än fem koncentrationer används ska detta motiveras. Den högsta testade koncentrationen ska helst ge l00-procentig immobilisering, och den lägsta koncentrationen som testas ska helst inte ge någon observerbar effekt alls.
1.8.1.3 Inkuberingsförhållanden
Temperaturen ska ligga i området 18 oC till 22 oC, och för varje enskilt test ska den vara konstant inom ± 1 oC. En dygnscykel med 16 timmar ljus och 8 timmar mörker rekommenderas. Fullständigt mörker är också godtagbart, särskilt om testämnet är instabilt i ljus.
Testkärlen måste luftas under testet. Testet genomförs utan pH-justering. Hinnkräftorna ska inte utfodras under testet.
1.8.1.4 Varaktighet
Testets varaktighet är 48 timmar.
1.8.2 Observationer
Varje testkärl ska kontrolleras med avseende på immobiliserade hinnkräftor 24 och 48 timmar efter det att testet har påbörjats (se avsnitt 1.2 för definitioner). Utöver immobilisering ska alla onormala förändringar av beteende och utseende rapporteras.
1.8.3 Analyser
Löst syre och pH mäts i början och i slutet av testet i kontrollen eller i kontrollerna och i den högsta testämneskoncentrationen. Koncentrationen av löst syre i kontrollerna ska överensstämma med giltighetskriteriet (se avsnitt 1.6). pH-värdet ska normalt inte variera med mer än 1,5 enheter i något enskilt test. Temperaturen mäts i regel i kontrollkärlen eller som omgivningstemperatur och ska helst registreras kontinuerligt under testet eller åtminstone i början och i slutet av testet.
Koncentrationen av testämne ska mätas minst vid den högsta och den lägsta testkoncentrationen i början och i slutet av testet (4). Det rekommenderas att resultaten baseras på uppmätta koncentrationer. Om det finns belägg för att koncentrationen av testämnet har hållit sig inom ± 20 % av den nominella eller uppmätta initiala koncentrationen genom hela testet kan resultaten dock baseras på nominella eller uppmätta initialvärden.
1.9 LIMIT-TEST
Genom användning av de förfaranden som beskrivs i denna metod kan ett så kallat limit-test göras upp vid 100 mg/l testämne eller upp till ämnets löslighetsgräns i testmediet (den av dessa koncentration som är lägst) för att visa att EC50 är högre än denna koncentration. Limit-testet ska göras med 20 hinnkräftor (helst indelade i fyra grupper om fem), med samma antal i kontrollen eller kontrollerna. Om någon immobilisering sker ska en fullständig studie genomföras. Allt onormalt beteende som observeras ska registreras.
2. DATA
Data ska sammanfattas i tabellform som för varje behandlingsgrupp och kontroll visar antalet hinnkräftor som använts och immobilisering vid varje observation. Procentandelen immobiliserade hinnkräftor vid 24 timmar och 48 timmar plottas mot testkoncentrationerna. Data analyseras med lämpliga statistiska metoder (t.ex. probitanalys osv.) för att beräkna kurvornas lutningar och EC50 med 95 % konfidensintervall (p = 0,05) (7) (8).
Om standardmetoderna för att beräkna EC50 inte går att tillämpa på erhållna data ska den högsta koncentration som inte ger någon immobilisering och den lägsta koncentration som ger 100 % immobilisering användas som approximering för EC50 (eftersom detta värde betraktas som det geometriska medelvärdet för dessa båda koncentrationer).
3. RAPPORTERING
3.1 FÖRSÖKSRAPPORT
Försöksrapporten måste innehålla följande information:
Testämne:
|
— |
Fysikaliskt tillstånd och relevanta fysikalkemiska egenskaper. |
|
— |
Kemiska identifieringsdata, inbegripet renhet. |
Försöksart:
|
— |
Källa och art av Daphnia, leverantör av källan (om denna är känd) och odlingsbetingelser (inbegripet fodrets källa, typ och mängd samt utfodringsfrekvens). |
Försöksförhållanden:
|
— |
Beskrivning av försökskärlen: typ av kärl, lösningsvolym, antal hinnkräftor per försökskärl (replikat) per koncentration. |
|
— |
Beredningsmetoder för stam- och testlösningar, inbegripet användning av lösningsmedel eller dispergeringsmedel, använda koncentrationer. |
|
— |
Detaljerade uppgifter om utspädningsvattnet: källa och detaljerade uppgifter om vattenkvalitet (pH, hårdhetsgrad, Ca/Mg-kvot, Na/K-kvot, alkalinitet, konduktivitet osv.) samt sammansättning om rekonstituerat vatten används. |
|
— |
Inkuberingsförhållanden: temperatur, ljusintensitet och periodicitet, löst syre, pH osv. |
Resultat:
|
— |
Antal och procentandel hinnkräftor som immobiliserats eller visat tecken på negativ inverkan (inbegripet onormalt beteende) i kontrollerna och i varje behandlingsgrupp vid varje observationstidpunkt samt en beskrivning av de effekter som observerats. |
|
— |
Resultat och datum för tester som utförts med referensämnen, om detta finns att tillgå. |
|
— |
De nominella testkoncentrationerna och resultaten av alla analyser för bestämning av koncentrationen av testämne i försökskärlen. Även metodens återvinningseffektivitet och analysgräns ska rapporteras. |
|
— |
Alla fysikalkemiska mätningar av temperatur, pH och löst syre som gjorts under testet. |
|
— |
EC50 vid 48 timmar för immobilisering med konfidensintervall och diagram över den anpassade modell som använts för att beräkna dessa värden, lutningarna för dos-responskurvorna och deras standardfel; statistiska förfaranden som använts för bestämning av EC50; (dessa data för immobiliseringen vid 24 timmar ska också rapporteras om de mäts). |
|
— |
Förklaring till eventuell avvikelse från testmetoden och uppgift om huruvida avvikelsen påverkade resultaten. |
4. HÄNVISNINGAR
|
1. |
ISO 6341. (1996). Water quality – Determination of the inhibition of the mobility of Daphnia magna Straus (Cladocera, Crustacea) – Acute toxicity test. Third edition, 1996. |
|
2. |
EPA OPPTS 850.1010. (1996). Ecological Effects Test Guidelines – Aquatic Invertebrate Acute Toxicity Test, Freshwater Daphnids. |
|
3. |
Environment Canada. (1996) Biological test method. Acute Lethality Test Using Daphnia spp. EPS 1/RM/11, Environment Canada, Ottawa, Ontario, Kanada. |
|
4. |
Guidance Document on Aquatic Toxicity Testing of Difficult Substances and Mixtures. OECD Environmental Health and Safety Publication. Series on Testing and Assessment. No. 23. Paris, 2000. |
|
5. |
Commission of the European Communities. Study D8369. (1979). Inter-laboratory Test Programme concerning the study of the ecotoxicity of a chemical substance with respect to Daphnia. |
|
6. |
OECD Guidelines for the Testing of Chemicals. Guideline 211: Daphnia magna Reproduction Test, adopted September 1998. |
|
7. |
Stephan C.E. (1977). Methods for calculating an LC50. In Aquatic Toxicology and Hazard Evaluation (edited by F.I. Mayer and J.L. Hamelink). ASTM STP 634 – American Society for Testing and Materials. Pp65–84. |
|
8. |
Finney D.J. (1978). Statistical Methods in Biological Assay. 3rd ed. London. Griffin, Weycombe, UK. |
Bilaga 1
VISSA KEMISKA EGENSKAPER FÖR ETT GODTAGBART SPÄDNINGSVATTEN
|
Ämne |
Koncentration |
|
Partiklar |
< 20 mg/l |
|
Totalt organiskt kol |
< 2 mg/l |
|
Icke-joniserat ammonium |
< 1 μg/l |
|
Restklor |
< 10 μg/l |
|
Total mängd pesticider med organiskt fosfat |
< 50 ng/l |
|
Total mängd pesticider med organiskt klor plus polyklorerade bifenyler |
< 50 ng/1 |
|
Totalt organiskt klor |
< 25 ng/1 |
Bilaga 2
EXEMPEL PÅ LÄMPLIGT REKONSTITUERAT TESTVATTEN
ISO för testvatten (1)
|
Stamlösningar (enstaka ämnen) |
För framställning av rekonstituerat vatten, sätt följande volymer av stamlösningarna till 1 liter vatten (2) |
|
|
Ämne |
Mängd tillsatt 1 liter vatten (2) |
|
|
Kalciumklorid CaCl2, 2H2O |
11,76 g |
25 ml |
|
Magnesiumsulfat MgSO4, 7H2O |
4,93 g |
25 ml |
|
Natriumbikarbonat NaHCO3 |
2,59 g |
25 ml |
|
Kaliumklorid K Cl |
0,23 g |
25 ml |
Elendt M7- och M4-medium
Acklimatisering till Elendt M4- och M7-medium
Vissa laboratorier för över hinnkräftorna direkt till M4- och M7-medier. En viss framgång har uppnåtts med gradvis acklimatisering, dvs. överflyttning från eget medium till 30 % Elendt, sedan till 60 % Elendt och slutligen till 100 % Elendt. Acklimatiseringsperioderna kan behöva vara upp till en månad långa.
Beredning
Spårelement
Separata stamlösningar (I) av enskilda spårelement bereds först i vatten av lämplig renhet, t.ex. avjoniserat, destillerat eller renat med omvänd osmos. Från dessa olika stamlösningar (I) bereds en andra stamlösning (II) som innehåller alla spårelement (kombinerad lösning), dvs.:
|
Stamlösning(ar) I (enskilt ämne) |
Mängd satt till 1 1 vatten (mg/l) |
Koncentration (relaterad till medium M4) |
För beredning av den kombinerade stamlösningen II tillsätts följande mängd av stamlösning I till 1 1 vatten (ml/l) |
|
|
M4 |
M7 |
|||
|
H3 BO3 |
57 190 |
20 000-faldig |
1,0 |
0,25 |
|
MnCl2.4H2O |
7 210 |
20 000-faldig |
1,0 |
0,25 |
|
LiCl |
6 120 |
20 000-faldig |
1,0 |
0,25 |
|
RbCl |
1 420 |
20 000-faldig |
1,0 |
0,25 |
|
SrCl2.6H2O |
3 040 |
20 000-faldig |
1,0 |
0,25 |
|
NaBr |
320 |
20 000-faldig |
1,0 |
0,25 |
|
Na2MoO4.2H2O |
1 230 |
20 000-faldig |
1,0 |
0,25 |
|
CuCl2.2H2O |
335 |
20 000-faldig |
1,0 |
0,25 |
|
ZnCl2 |
260 |
20 000-faldig |
1,0 |
1,0 |
|
CoCl2.6H20 |
200 |
20 000-faldig |
1,0 |
1,0 |
|
KI |
65 |
20 000-faldig |
1,0 |
1,0 |
|
Na2 SeO3 |
43,8 |
20 000-faldig |
1,0 |
1,0 |
|
NH4 VO3 |
11,5 |
20 000-faldig |
1,0 |
1,0 |
|
Na2 EDTA.2H2O |
5 000 |
2 000-faldig |
— |
— |
|
FeSO4.7H2O |
1 991 |
2 000-faldig |
— |
— |
|
Både Na2EDTA- och FeSO4-lösningarna förbereds var för sig, hälls samman och autoklaveras omedelbart. |
||||
|
Detta ger: |
||||
|
21 Fe-EDTA-lösning |
|
1 000-faldig |
20,0 |
5,0 |
M4- och M7-medier
M4- och M7-medier bereds med stamlösning II, makro-näringsämnen och vitaminer enligt följande:
|
|
Mängd tillsatt vatten (mg/l) |
Koncentration (relaterad till medium M4) |
Mängd stamlösning II som tillsätts för beredning av medium (ml/l) |
|
|
M4 |
M7 |
|||
|
Stamlösning II (kombinerad med spårelement) |
|
20-faldig |
50 |
50 |
|
Stamlösningar för makronäringsämnen (enskilt ämne) |
|
|
|
|
|
CaCl2 . 2H20 |
293 800 |
1 000-faldig |
1,0 |
1,0 |
|
MgSO4 . 7H2O |
246 600 |
2 000-faldig |
0,5 |
0,5 |
|
KC1 |
58 000 |
10 000-faldig |
0,1 |
0,1 |
|
NaHCO3 |
64 800 |
1 000-faldig |
1,0 |
1,0 |
|
Na2 SiO3 . 9H2O |
50 000 |
5 000-faldig |
0,2 |
0,2 |
|
NaNO3 |
2 740 |
10 000-faldig |
0,1 |
0,1 |
|
KH2 PO4 |
1 430 |
10 000-faldig |
0,1 |
0,1 |
|
K2HPO4 |
1 840 |
10 000-faldig |
0,1 |
0,1 |
|
Kombinerad vitaminstamlösning |
— |
10 000-faldig |
0,1 |
0,1 |
|
Den kombinerade vitaminstamlösningen bereds genom tillsats av de tre vitaminerna till 1 1 vatten, såsom visas nedan: |
||||
|
Tiaminhydroklorid |
750 |
10 000-faldig |
|
|
|
Cyanokobalamin (B12) |
10 |
10 000-faldig |
|
|
|
Biotin |
7,5 |
10 000-faldig |
|
|
Den kombinerade vitaminstamlösningen lagras fryst i små alikvoter. Vitaminer sätts till medierna strax före användning.
|
Märk |
: |
För att undvika utfällning av salter vid beredning av kompletta medier ska alikvoter av stamlösningar sättas till cirka 500–800 ml avjonat vatten som sedan späds upp till 1 liter. |
|
Märk |
: |
Den första publiceringen av M4-mediet finns i Elendt, B. P. (1990). Selenium deficiency in crustacea; an ultrastructual approach to antennal damage in Daphnia magna Straus. Protoplasma, 154, 25–33. |
C.3 TEST AV TILLVÄXTHÄMNING PÅ ALGER
1. METOD
1.1 INLEDNING
Syftet med denna test är att bestämma ett ämnes verkningar på encelliga grönalgarters tillväxt. Vid relativt kortvariga (72 timmar) tester kan verkningarna över flera generationer utvärderas. Denna metod kan justeras så att den kan användas i samband med flera encelliga algarter varvid en beskrivning av den använda metoden måste anges tillsammans med testrapporten.
Denna metod är enklast att använda för vattenlösliga ämnen som under testbetingelserna sannolikt kommer att förbli i vattnet.
Metoden kan användas för ämnen som inte har direkt inverkan på mätningen av algtillväxt.
Det är önskvärt att innan testet inleds ha så mycket information som möjligt om ämnets löslighet i vatten, ångtryck, kemiska stabilitet, dissociationskonstanter och nedbrytbarhet.
Övrig information (t.ex. strukturformel, renhetsgrad, art och procent av betydande orenheter, närvaro och mängd av tillsatsämnen samt n-oktanol/vatten-fördelningskoefficient) bör beaktas både vid planeringen av testet och vid tolkning av resultaten.
1.2 DEFINITIONER OCH ENHETER
Cellkoncentration: antalet celler per milliliter.
Tillväxt: förökningen av cellkoncentrationen under testperioden.
Tillväxttakt: förökningen av cellkoncentrationen per tidsenhet.
I denna metod avses med EC50 den testämneskoncentration som medför en 50-procentig reduktion av antingen tillväxten (EbC50) eller tillväxttakten (ErC50) i förhållande till kontrollproven.
I denna metod avses med NOEC (no observed effect concentration) den högsta testkoncentration där de uppmätta koncentrationerna inte visar någon betydande tillväxthämning i förhållande till kontrollvärdet.
Alla koncentrationer av testämnet anges i vikt per volym (milligram per liter). De kan också uttryckas som vikt per vikt (mg. kg-1).
1.3 REFERENSÄMNEN
En referensämne kan användas för att påvisa att testartens känslighet inte har ändrats väsentligt under försöksbetingelserna i laboratoriet.
Om ett referensämne används bör resultaten anges i testrapporten. Kaliumdikromat kan användas som referensämne, men dess färg kan påverka den ljuskvalitet och intensitet som är tillgängligt för cellerna och även spektrofotomeriska bestämningar om sådana används. Kaliumdikromat har använts i en internationell interlaboratorietest (se hänvisning 3 och tillägg 2).
1.4 TESTMETODSPRINCIP
Ett gränstest kan utföras med 100 mg per liter för att konstatera att EC50 är större än denna koncentration.
Kulturer av utvalda grönalger med expotentiell tillväxt exponeras för olika koncentrationer av testämnet över flera generationer under väl definierade testbetingelser.
Testlösningarna inkuberas i 72 timmar och under denna period mäts celltätheten minst var 24:e timme. Tillväxthämningen i förhållande till en kontrollkultur bestäms.
1.5 KVALITETSKRITERIER
Kvalitetskriterierna gäller för såväl gränstestet som den fullständiga testmetoden.
Cellkoncentrationen i kontrollkulturerna ska ha ökats med en faktor på minst 16 inom tre dagar.
Koncentrationer av testämnet ska ligga över 80 % av startkoncentrationen under hela testperioden.
För ämnen som är lättlösliga i testmediet och ger stabila lösningar, dvs. ämnen som inte i hög grad förångas, bryts ned, hydrolyseras eller adsorberas, kan startkoncentrationen antas vara lika med den nominella koncentrationen. Det ska bevisas att koncentrationen bibehållits under hela testet och att kvalitetskriterierna är uppfyllda.
För ämnen som är
|
i) |
svårlösliga i testmediet eller |
|
ii) |
kan skapa stabila emulsioner eller dispersioner eller |
|
iii) |
är ostabila i vattniga lösningar |
ska det som värde för startkoncentrationen användas den koncentration som är uppmätt vid testets inledning. Koncentrationen bestäms sedan jämvikt uppnåtts.
I alla dessa fall ska ytterligare mätningar göras under testets gång för att säkerställa att koncentrationen har bibehållits och att kvalitetskriterierna har varit uppfyllda.
Det erkänns att en väsentlig mängd testämne kommer att finnas i algbiomassan vid testets avslutning. För att bevisa överensstämmelse med ovan nämnda kvalitetskriterier bör både den del av testämnet som finns i biomassan, och den del som finns i lösningen (eller om det inte är tekniskt möjligt, mätt i vattenkolonnen) beaktas. Eftersom det kan vålla betydliga tekniska problem att bestämma testämneskoncentrationen i algbiomassan kan överensstämmelse med kvalitetskriterierna uppfyllas genom att använda en testbehållare med den högsta möjliga ämneskoncentrationen men utan alger, i vilken koncentrationen i lösningen (eller om det inte är tekniskt möjligt, i vattenkolonnen) mäts vid testperiodens början och slut.
1.6 METODBESKRIVNING
1.6.1 Reagenser
1.6.1.1 Lösningar av testämnen
Stamlösningar av erforderlig styrka framställs genom att testämnet upplöses i avjoniserat vatten eller vatten enligt 1.6.1.2.
De valda testkoncentrationerna framställs genom tillsättning av en passande mängd testämne till algförkulturer (se tillägg 1).
Ämnen testas normalt endast upp till gränsen för löslighet. För vissa ämnen (t.ex. ämnen som är svårlösliga i vatten, som har ett högt Pow-värde, eller som bildar en stabil dispersion och inte en riktig upplösning) accepteras att det används en testkoncentration som är större än lösligheten för att säkerställa att den högsta upplösta/stabila koncentration har nåtts. Det är dock viktigt att denna koncentration inte på annat sätt förstör testmetoden (t.ex. bildandet av en hinna av ämnet på vattenytan som förhindrar syrsättning av vattnet, etc.).
Ultraljud, organiska lösningsmedel, emulgeringsmedel eller dispergeringsmedel kan användas för att framställa stamlösningar av ämnen som är svårlösliga i vatten och för att dispergera sådana ämnen i testmediet. Om tillsatsämnen används ska alla testkoncentrationer innehålla samma mängd av tillsatsämnet och en extra kontrollgrupp ska användas som utsätts för samma koncentration av tillsatsämnet som används i testserien. Sådana tillsatsämnen används i lägsta möjliga koncentration och får aldrig vara större än 100 mg per liter i testmediet.
Testet ska göras utan justering av pH-värdet. Om det finns tecken på en markant förändring av pH-värdet rekommenderas att testet upprepas med pH-justering och att resultaten rapporteras. 1 detta fall ska pH-värdet i stamlösningen justeras efter pH-värdet i utspädningsvattnet, om det inte finns särskilda skäl som talar mot detta. HC1 och NaOH är förstahandsvalen för detta ändamål. Denna pH-justering ska göras på ett sådant sätt att testämnets koncentration i stamlösningen inte ändras i någon signifikant grad. Om justeringen medför någon kemisk reaktion eller fysisk utfällning av testämnet ska detta rapporteras.
1.6.1.2 Testmedium
Vattnet ska vara destillerat vatten av god kvalitet och avjoniserat vatten med en ledningsförmåga på mindre än 5 μScm-1. Vattendestillationsapparatur får inte innehålla delar av koppar.
Följande medium rekommenderas:
Fyra stamlösningar framställs i enlighet med nedanstående tabell. Stamlösningarna steriliseras genom autoklave-ring eller membranfiltrering, varefter de förvaras vid 4 oC skyddade mot ljus. Stamlösning nummer 4 ska steriliseras genom membranfiltrering. Testlösningarnas slutliga näringsämneskoncentration uppnås genom förtun-mng av stamlösningarna.
|
Näringsämne |
Koncentration stamlösningen |
Slutkoncentration stamlösningen |
|
Stamlösning 1: makronäringsämnen |
||
|
NH4Cl |
1,5 g/l |
15 mg/l |
|
MgCl2.6H2O |
1,2 g/l |
12 mg/l |
|
CaCl2.2H2O |
1,8 g/l |
18 mg/l |
|
MgSO4.7H2O |
1,5 g/l |
15 mg/l |
|
KH2PO4 |
0,16 g/l |
1,6 mg/l |
|
Stamlösning 2: Fe-EDTA |
||
|
FeCl3.6H2O |
80 mg/l |
0,08 mg/l |
|
Na2EDTA.2H2O |
100 mg/l |
0,1 mg/l |
|
Stamlösning 3: spårämnen |
||
|
H3BO3 |
185 mg/l |
0,185 mg/l |
|
MnCl2.4H2O |
415 mg/l |
0,415 mg/l |
|
ZnCl2 |
3 mg/l |
3 × 10-3 mg/l |
|
CoCl2.6H2O |
1,5 mg/l |
1,5 × 10-3 mg/l |
|
CuCl2.2H2O |
0,01 mg/l |
10 5 mg/l |
|
Na2MoO4.2H2O |
7 mg/l |
7 × 10-3 mg/l |
|
Stamlösning 4: NaHCO3 |
||
|
NaHCO3 |
50 g/l |
50 mg/l |
Efter ekvilibrering med luft är detta mediums pH ca 8.
1.6.2 Apparatur
|
— |
Normal laboratorieutrustning. |
|
— |
Testkolvar av passande volym (250 ml koniska flaskor är t.ex. lämpligt när testämnets volym är 100 ml). Alla testkolvar ska vara identiska, både vad beträffar material och dimensioner. |
|
— |
Odlingsapparatur: ett skåp eller en kammare med en temperatur av 21–25 oC med en tolerans av ± 2 oC och med oavbruten enhetlig belysning i spektralområdet 400–700 nm. Växtbetingelserna, inbegripet ljusstyrkan, anses tillfredsställande om kontrollkulturer uppnår den rekommenderade tillväxttakten. En ljusintensitet i området 60–120 μE/m-2·s-1 (35 till 70 × 1018 fotonerm-2. s-1) rekommenderas när den mäts i området 400-700 nm med en lämplig mottagare. För ljusmätningsinstrument kalibrerade i lux är den motsvarande lämpliga nivån 6 000–10 000 lux. Denna ljusintensitet kan uppnås med hjälp av 4–7 universella fluorescerande vita lampor (ljustemperatur på ca 4 300 K) placerade på ett avstånd av 0,35 m från algkulturen. |
|
— |
Mätningar av cellkoncentrationen ska göras genom direkt räkning av levande celler, t.ex. med hjälp av ett mikroskop med räkningskammare. Andra metoder (fotometri, turbidimetri, m.fl.) kan dock användas om de är tillräckligt känsliga och visar sig väl korrelerade med cellkoncentrationen. |
1.6.3 Testorganismer
Det föreslås att de grönalger som används är av en snabbväxande art som är lämplig för odling och test. Följande arter anses lämpliga:
|
— |
Selenastrum capricornutum, t.ex. ATCC 22662 eller CCAP 278/4, |
|
— |
Scenedesmus subspicatus, t.ex. 86.81 SAG. |
Anmärkning:
|
ATCC |
= |
American Type Culture Collection (USA) |
|
CCAP |
= |
Culture Centre of Algae and Protozoa (UK) |
|
SAG |
= |
Collection of algal culture (Göttingen, Tyskland) |
Om andra arter används måste stammen anges.
1.6.4 Testets utformning
Genom inledande tester bestäms det koncentrationsområde där verkningar kan förväntas inträda.
De två mätningarna av växten (biomassa och tillväxttakt) kan leda till vitt skilda resultat för tillväxthämning. Båda ska användas i de inledande testerna så att det säkerställs att både EbC50 och ErC50 kan värderas utifrån den geometriska serie som väljs för koncentrationerna.
Cellkoncentrationen vid försökets inledning
Det rekommenderas att den initiala cellkoncentrationen i testkulturerna är ca 104 celler/ml för Selenastrum capricornutum och Scenedesmus subspicatus. Om andra arter används ska biomassan vara jämförbar.
Testämnekoncentrationer
För testet används minst fem koncentrationer som bildar en geometrisk serie med en koncentration av högst 2,2 väljas ut. Den lägsta koncentrationen bör inte ha några synbara verkningar på algens tillväxt. Den högsta koncentration som testas bör hämma tillväxten med minst 50 % i förhållande till kontrollkulturen och helst stoppa tillväxten helt.
Replikater och kontrollkoncentrationer
Testets utformning bör inbegripa tre replikater vid varje testkoncentrationsnivå. Tre kontrollkulturer utan testämne ska användas och vid behov, ska även tre kontrollkulturer som innehåller tillsatsämnet användas. Om det finns rimliga skäl kan testets utformning ändras så att antalet koncentrationer ökas och antalet replikater per koncentration minskas.
Testförfarande
Testkulturer framställs med den önskade testämneskoncentrationen och den önskade mängd alginokulat genom tillsättning av stamlösning av testämnet till en passande mängd algförkultur (se tillägg 1).
Testkolvarna skakas och placeras i odlingsapparaturen. Algcellerna hålls i suspension genom skakning, omrör-ning eller genombubbling med luft så att det blir bättre luftväxling och mindre pH-variationer i testlösningarna. Kulturerna förvaras i en temperatur mellan 21 och 25 oC som hålls konstant inom ± 2 oC.
Cellkoncentrationen i varje kolv bestäms minst tre gånger, nämligen 24, 48 och 72 timmar efter inledning av försöket. När cellkoncentrationen mäts på annat sätt än genom direkt räkning används filtrerat algmedium med den ifrågavarande testämneskoncentrationen för bestämning av bakgrundsvärden.
pH mäts vid försökets inledning och efter 72 timmar.
Kontrollkulturernas pH får normalt inte avvika med mer än 1,5 under försöket.
Test av flyktiga ämnen
Det finns ännu ingen allmänt erkänd metod för test av flyktiga ämnen. Om man vet att ett ämne har en tendens att förångas kan man använda slutna testkolvar med ökat luftutrymme ovanför vätskan. Möjligheten av brist på CO2 bör beaktas vid beräkning av detta extra luftutrymme. Förslag på variationer av denna metod har lagts fram (se hänvisning 4).
Man bör försöka att bestämma den mängd ämne som återstår i lösningar, varvid yttersta försiktighet bör iakttas vid tolkning av resultaten av tester med flyktiga kemikalier i ett slutet system.
Gränstest
Genom användning av förfarandena som beskrivs i denna testmetod kan ett gränstest utföras med 100 mg per liter för att konstatera om LC50 är större än denna koncentration.
Om ämnet är av sådan art att man inte kan erhålla en koncentration av 100 ml per liter i testvattnet utförs gränstestet vid ämnets mättnadskoncentration (eller den högsta koncentrationen vid vilken det bildas en stabil dispersion) i det använda mediet (se även 1.6.1.1).
Gränstestet utförs minst som en tredubbelbestämning och med kontrollkulturer av samma storlek. Både mätning av växten (biomassa och tillväxttakt) används i gränstestet.
Om biomassan eller tillväxttakten är minst 25 % lägre i gränstestet än i kontrollkulturen ska en fullständig undersökning utföras.
2. DATA OCH UTVÄRDERING
De uppmätta cellkoncentrationerna i testkulturerna och kontrollkulturerna ska ställas upp i tabellform tillsammans med koncentrationerna av testämnena och tidpunkterna för mätningarna. Medelvärdet av cellkoncentrationen för varje testämneskoncentration och för kontrollkulturerna ritas in mot tiden (0–72 timmar) så att växt-kurvor erhålls.
För bestämning av förhållandet mellan koncentration och verkan bör följande två metoder användas. Vissa ämnen kan stimulera tillväxt vid låga koncentrationer. Endast datapunkter som anger att hämningen är mellan 0 och 100 % ska beaktas.
2.1 JÄMFÖRELSE AV AREALER UNDER VÄXTKURVORNA
Arealen mellan växtkurvorna och den horisontella linjen N = No kan beräknas efter denna formel:

där:
|
A |
= |
areal |
|
No |
= |
nominellt antal celler per ml vid tidpunkten t0 (inledningen av testet) |
|
N1 |
= |
uppmätt antal celler vid tidpunkten t1 |
|
Nn |
= |
uppmätt antal celler vid tidpunkten tn |
|
t1 |
= |
tidpunkt för första mätning efter testets inledning |
|
tn |
= |
tidpunkt för nde mätning efter testets inledning |
|
n |
= |
antal mätningar gjorda efter inledning av testet |
Den procentuella hämning av celltillväxten för varje testkoncentration (IA) beräknas efter följande formel:

där:
|
Ac |
= |
arealen mellan växtkurvan för kontrollkulturen och den horisontella linjen N = No |
|
At |
= |
arealen mellan växtkurvan vid koncentrationen t och den horisontella linjen N = No. |
|
IA |
= |
värdena avbildas på semilogaritmiskt papper eller semilogaritmiskt probitpapper mot motsvarande koncentrationer. Om probitpapper används dras den bästa räta linjen, antingen med ögonmått eller genom regressionsberäkning. |
EC50-värde beräknas från regressionslinjen genom att läsa av koncentrationen som motsvarar 50 % hämning (IA = 50 %). Som en entydig beteckning för detta värde föreslås att man i samband med denna beräkningsmetod använder symbolen EbC50. Det är viktigt att EbC50 anges med lämplig exponeringsperiod, t.ex. EbC50 (0–72 h).
2.2 JÄMFÖRELSE AV TILLVÄXTTAKT
Den genomsnittliga specifika tillväxttakten (μ) för kulturer med exponentiell tillväxt kan beräknas som:

där t0 är tiden vid testets inledning.
Alternativt kan den genomsnittliga specifika tillväxttakten för varje tillväxttakt härledas av regressionslinjens lutning i en avbildning av ln N mot tiden.
Den procentuella reduktionen av den genomsnittliga tillväxttakten för varje testämnekoncentration (Iμt) beräknas efter följande formel:

där:
|
μc |
= |
genomsnittliga tillväxttakten för kontrollkulturen |
|
μt |
= |
specifika genomsnittliga tillväxttakten för testkoncentration |
Den procentuella reduktionen av den genomsnittliga tillväxttakten för varje testämnekoncentration jämfört med kontrollvärdena avbildas mot logaritmen till koncentrationen. Deras EC50 kan avläsas av den därvid framkomna grafen. Som en entydig beteckning för det EC50-värde som beräknas med denna metod föreslås att man använder symbolen ErC50. Tidpunkterna för mätningarna ska anges. Om t.ex. värdet gäller observationstidpunkterna 0 och 72 timmar blir symbolen ErC50 (0–72 h).
Obs.: Tillväxttakten är en logaritmisk term, och små ändringar i tillväxttakten kan medföra stora ändringar i biomassan. EbC och ErC är därför inte numeriskt jämförbara värden.
2.3 BERÄKNING AV NOEC-VÄRDEN
Den koncentration där man inte kan iaktta någon verkan (NOEC-värden) bestäms genom en lämplig statistisk metod för jämförelse av flera prover (t.ex. variationsanalys eller Dunnetts test), som används på de enskilda värdena av arealerna under växtkurvorna A (se 2.1) eller av de specifika tillväxttakterna μ (se 2.2).
3. RAPPORTERING
Testrapporten ska om möjligt innehålla följande uppgifter:
|
— |
Testämne: Data för kemisk identifiering. |
|
— |
Testorganismer: Härkomst, laboratoriekultur, stamnummer, odlingsmetod. |
|
— |
Testbetingelser:
|
|
— |
Resultat:
|
4. HÄNVISNINGAR
|
1. |
OECD, Paris, 1981, Test Guidelines 201, rådets beslut K(81) 30 slutlig. |
|
2. |
Umweltsbundesamt, Berling, 1984, Verfahrensvorschalg ”Hemmung der Zellvermehrung bei der Grünalge Scenedesmus subspicatus”, i: Rudolph/Boje: Ökotoxikologie, ecomed. Landsberg, 1986. |
|
3. |
ISO 8692 – Water quality – Fresh water algal growth inhibition test with Scenedesmus subspicatus and Selenastrum capricornutum. |
|
4. |
S. Galassi and M. Vighi-Chemosphere, 1981, vol. 10, 1123–1126. |
Tillägg 1
Exempel på en metod för odling av alger
Allmänna kommentarer
Syftet med odling på grundval av nedanstående metoder är att framställa algkulturer för toxicitetstest.
Lämpliga metoder används för att säkerställa att algkulturerna inte infekteras med bakterier (ISO 4833). Axe-niska kulturer kan under vissa omständigheter vara önskvärda, men det är väsentligt att kulturerna uteslutande består av alger.
Alla operationer ska företas under sterila förhållanden för att undvika förorening med bakterier eller med andra alger. Kontaminerade kulturer kasseras.
Metoder för framställning av algkulturer
Framställning av näringslösningar (medier)
Substratet kan framställas genom förtunning av koncentrerade näringsämneslösningar. För fasta substrater tillsätts 0,8 % agar. Endast sterila substrat används. Autoklavering kan leda till förlust av NH3.
Stamkultur
Stamkulturerna är små algkulturer som regelbundet överförs till ett friskt medium och som används som utgångstestmaterial. Om kulturerna inte används regelbundet stryks de ut på böjda agarrör. De överförs till ett färskt medium minst varannan månad.
Stamkulturerna odlas i koniska kolvar med det lämpliga mediet (volymen ca 100 ml). När algerna inkuberas vid 20 oC i konstant ljus krävs veckovis överföring.
Under överföringen avsätts en viss mängd ”gammal” kultur med sterila pipetter i en kolv med färskt medium så att initialkoncentrationen i fråga om de snabbväxande arterna är ca 100 gånger mindre än i den gamla kulturen.
En arts tillväxttakt kan bestämmas utifrån tillväxtkurvan. Om denna är känd är det möjligt att se vid vilken täthet kulturen ska överföras till det nya mediet. Detta måste göras innan kulturen når dödfasen.
Förkultur
Avsikten med förkulturen är att få en mängd alger som är lämpliga för inokulering av testkulturer. Förkulturen inkuberas under testbetingelser och används medan den fortfarande växer exponentiellt, normalt efter en inkubationsperiod på omkring tre dagar. Om algkulturerna innehåller missbildade eller abnorma celler ska de slängas.
Tillägg 2
ISO 8692 – Water quality – Fresh water algal growth inhibition test with Scenedesmus subspicatus and Selenast-rum capricornutum rapporterar följande resultat för ett test av kaliumdikromat med 16 deltagande laboratorier:
|
|
Medel (mg/l) |
Område (mg/I) |
|
ErC50 (0–72 h) |
0,84 |
0,60 till 1,03 |
|
EbC50 (0–72 h) |
0,53 |
0,20 till 0,75 |
C.4 BESTÄMNING AV ”LÄTT” BIONEDBRYTBARHET
DEL I. ALLMÄNNA PRINCIPER
I.1 INLEDNING
Sex metoder för screening av kemikalier för lätt bionedbrytning i ett aerobt vattenmedium beskrivs:
|
a) |
Eliminering av upplöst organiskt kol (DOC) (Metod C.4-A). |
|
b) |
Modifierat OECD-screeningtest – eliminering av DOC (Metod C.4-B). |
|
c) |
Koldioxid (CO2) utvärdering (Modifierat Sturmtest) (Metod C.4-C). |
|
d) |
Manometrisk respiration (Metod C.4-D). |
|
e) |
Closed Bottle Test (Metod C.4-E). |
|
f) |
MITI (Ministry of International Trade and Industry – Japan) (Metod C.4-F). |
Del 1 innehåller allmänna och generella kommentarer till alla sex tester. I del II–VI1 finns det som är specifikt för de enskilda testerna. Tilläggen innehåller definitioner, formler och vägledande upplysningar.
1988 genomförde OECD ett ringtest som visade att metoderna ger inbördes överensstämmande resultat. En bestämd metod kan dock föredras allt efter det undersökta ämnets fysiska egenskaper.
I.2 VAL AV METOD
För att kunna välja den lämpligaste metoden är det viktigt att känna till kemikaliens löslighet, ångtryck och adsorptionsegenskaper. Den kemiska strukturen eller formeln bör vara känd för att kunna beräkna teoretiska värden och kontrollera uppmätta värden av parametrar, t.ex. ThOD, ThCO2, DOC, TOC och COD (se bilagorna 1 och 2).
Testkemikalier som är lösliga i vatten med 100 mg/liter kan utvärderas genom samtliga metoder, under förutsättning att de är icke-flyktiga och icke adsorberande. Av tabell 1 framgår vilka metoder som är lämpliga för kemikalier som är svårlösliga i vatten, flyktiga eller adsorberande. I bilaga I beskrivs hur man ska göra med kemikalier som är svårupplösliga i vatten och flyktiga kemikalier. Moderat flyktiga kemiska ämnen kan testas genom DOC elimineringsmetoden om det finns tillräckligt med luftrum i testkärlet (som ska stängas på lämpligt sätt). 1 detta fall måste en abiotisk kontroll användas som tillåter eventuell fysisk förlust.
Tabell 1
Tillämpning av testmetoder
|
Test |
Analytisk metod |
Lämplighet för ämnen som är |
||
|
svårlösliga |
flyktiga |
adsorberande |
||
|
DOC-eliminering |
Upplöst organiskt kol |
— |
— |
+/- |
|
Mod. OECD DOC-elim. |
Upplöst organiskt kol |
— |
— |
+/- |
|
CO2 utvärdering |
Respirometri: CO2 bedömning |
+ |
— |
+ |
|
Manometrisk respirometri |
Manometrisk respirometri: syreförbrukning |
+ |
+/- |
+ |
|
Closed bottle |
Respirometri: upplöst syre |
+/- |
+ |
+ |
|
MITI |
Respirometri: syreförbrukning |
+ |
+/- |
+ |
Uppgifter om de relativa proportionerna av testmaterialets huvudbeståndsdelar är till nytta vid tolkningen av de erhållna resultaten, särskilt om resultaten är låga eller marginella.
Uppgifter om testkemikaliens toxicitet för bakterier (bilaga IV) kan vara till nytta vid valet av lämpliga testkoncentrationer och vara avgörande vid tolkningen av låga bionedbrytbarhetsvärden.
I.3 REFERENSÄMNEN
För att kontrollera förfarandet används referensämnen som uppfyller kriterierna för lätt bionedbrytbarhet och som testas i en separat kolv parallellt med de övriga proverna.
Anilin (färskt destillerat), natriumacetat och natriumbensoat är lämpliga kemikalier. Alla dessa referenskemikalier bryts ned vid dessa metoder även när inget inokulat tillsätts.
Det har föreslagits att man borde söka efter en referenskemikalie som är lätt nedbrytbar men som kräver tillsättning av inokulat. Kaliumhydrogenftalat har föreslagits men det behövs mer bevis innan detta ämne kan godtas som ett referensämne.
I metoder som baseras på respirometri kan kvävehaltiga föreningar påverka syreupptagningen på grund av nitri-fikation (se bilagor II och V).
I.4 TESTMETODSPRINCIP
Testämnet upplöses eller uppslammas i ett oorganiskt testmedium, inokuleras och inkuberas under aeroba förhållanden i mörker eller diffust ljus. Den del av DOC i lösningen som stammar från inokulat ska vara så lågt som möjligt i jämförelse med den del som stammar från testämnet. Man ska ta hänsyn till inokulatets endogena aktiviteter genom att köra parallella blindprov med inokulat men utan testämne i lösningen, även om cellernas endogena aktivitet under närvaro av ämnet inte exakt motsvarar denna i den endogena kontrollen. Ett referensämne används parallellt för att kontrollera hur förfarandena fungerar.
I allmänhet följs nedbrytning genom bestämning av parametrar, såsom DOC, CO2-produktion och syreupptagning, och mätningar görs tillräckligt ofta för att möjliggöra identifiering av början och slutet av bionedbrytningen. Mätningarna görs kontinuerligt med automatiska respirometrer. Under tiden mäts både DOC och en annan parameter, men detta sker normalt endast i början och slutet av testet. Specifika kemiska analyser kan också användas för att bedöma primär nedbrytning av testämnet, och för bestämning av koncentrationen av eventuella mellanprodukter (obligatoriskt i MITI-testet).
Testet vara normalt i 28 dagar. När bionedbrytningskurvan har nått en platå vid minst 3 mätningar kan dock testet avbrytas. Om kurvan visar att bionedbrytning har börjat men ännu inte nått en platå inom 28 dagar kan testet förlängas utöver denna period.
I.5 KVALITETSKRITERIER
I.5.1 Repeterbarhet
Till följd av bionedbrytningens natur och den blandade bakteriepopulationen i inokulat, bör minst dubbla bestämningar göras.
Det är en allmän erfarenhet att variationen mellan replikat blir mindre ju större startkoncentrationer av mikroorganismer i testmediet är. Ringtester har dessutom visat att det kan finnas stora variationer mellan resultat som erhållits av olika laboratorier, men generellt uppnås god överensstämmelse med lätt bionedbrytbara föreningar.
I.5.2 Testets giltighet
Ett test anses giltigt om skillnaden mellan de två yttersta värdena för testämnets försvinnande vid en multibestämning i platåfasen, vid avslutning av testet eller vid avslutningen av 10-dagarsfönstret är mindre än 20 % och om den procentuella nedbrytbarheten av referensämnet har nått nivån för lätt bionedbrytbarhet inom 14 dagar. Om inte båda villkoren uppfylls upprepas testet. På grund av stringensen i detta test behöver ett lågt resultat inte nödvändigtvis betyda att testämnet inte är bionedbrytbart under miljöbetingelser utan det pekar på att det krävs mer arbete för att fastställa detta.
Om nedbrytningen i ett toxicitetstest med både testämne och referenskemikalie är mindre än 35 % (baserad på DOC) eller 25 % (baserad på ThOD eller ThCO2) inom 14 dagar måste man anta att testämnet är hämmande (se även bilaga 4). Testserien upprepas, om möjligt med lägre testämneskoncentrationer och högre inokulatkoncentration, dock högst 30 mg/fast ämne/liter.
I.6 ALLMÄNNA FÖRFARANDEN OCH FÖRBEREDELSER
Allmänna villkor som gäller för testerna sammanfattas i tabell 2. Apparatur och andra försöksbetingelser som gäller specifikt för ett individuellt test beskrivs senare under rubriken för det testet.
Tabell 2
Testbetingelser
|
Test |
DOC- eliminering |
CO2-utveckling |
Manometrisk respirometri |
Modifierad OECD screening |
Closed bottle |
MITI(I) |
|||||||||
|
Koncentration av testämnet |
|
|
|
|
|
|
|||||||||
|
som mg/l |
|
|
100 |
|
2–10 |
100 |
|||||||||
|
mg/DOC/l |
10–40 |
10–20 |
|
10–40 |
|
|
|||||||||
|
mg/TbOD/l |
|
|
50–100 |
|
5–10 |
|
|||||||||
|
Koncentration av inokulat (i celler/l, cirka) |
≤ 30 mg/l SS eller ≤ 100 ml utflöde/l (107–108) |
0,5 ml sekundärt utflöde/l (I05) |
≤ 5 ml av utflöde/l (104–106) |
30 mg/l SS (107–108) |
|||||||||||
|
Koncentration av element i mineralmedium (i mg/l) |
|
|
|
|
|
|
|||||||||
|
P |
116 |
11,6 |
29 |
||||||||||||
|
N |
1,3 |
0,13 |
1,3 |
||||||||||||
|
Na |
86 |
8,6 |
17,2 |
||||||||||||
|
K |
122 |
12,2 |
36,5 |
||||||||||||
|
Mg |
2,2 |
2,2 |
6,6 |
||||||||||||
|
Ca |
9,9 |
9,9 |
29,7 |
||||||||||||
|
Fe |
0,05–0,1 |
0,05–0,1 |
0,15 |
||||||||||||
|
pH-värde |
7,4±0,2 |
helst 7,0 |
|||||||||||||
|
Temperatur |
22 ± 2 oC |
25 ±1 oC |
|||||||||||||
|
|
|
|||||||||||||
I.6.1 Utspädningsvatten
Avjoniserat eller destillerat vatten utan innehåll av toxiska substanser (t.ex. Cu++ joner) används. Det får inte innehålla mer än 10 % av det organiska kol som tillförs med testämnet. Denna höga renhet är nödvändig för att undvika höga blindvärden. Föroreningar kan stamma från naturliga orenheter och från jonbyteshartser och material från bakterier och alger. Till en testserie får endast vatten från samma behållare användas och detta vatten ska kontrolleras i förväg genom DOC-analys. En sådan kontroll är inte nödvändig för Closed Bottle-test, men syreförbrukningen från vattnet ska vara låg.
I.6.2 Stamlösningar av oorganiska medier
Stamlösningar framställs av oorganiska medier med passande koncentrationer av salt för användning vid framställning av testlösningarna. Följande stamlösningar kan (med olika förtunningsförhållanden) användas för DOC-eliminering, modifierad OECD-screening, CO2-utveckling, manometrisk respirometri och Closed Bottle-test.
Stamlösningar:
|
a) |
Monokaliumdiväteortofosfat, KH2PO4 |
8,50 g |
|
Dikaliumväteortofosfat, K2HPO4 |
21,75 g |
|
|
Dinatriumväteortofosfat-dihydrat, Na2HPO4 • 2H2O |
33,40 g |
|
|
Ammoniumklorid, NH4Cl |
0,50 g |
|
|
Upplöses i vatten så att 1 000 ml erhålls. pH-värdet ska vara 7,4. |
|
|
|
b) |
Kalciumklorid CaCl2 |
27,50 g |
|
eller kalciumklorid, dihydrat, CaCl2 · 2H2O |
36,40 g |
|
|
Upplöses i vatten så att 1 000 ml erhålls |
|
|
|
c) |
Magnesiumsulfat-heptahydrat, MgSO4 · 7H2O |
22,50 g |
|
Upplöses i vatten så att 1 000 ml erhålls |
|
|
|
d) |
Järn(III)klorid-hexahydrat FeCl3 · 6H2O |
0,25 g |
|
Upplöses i vatten så att 1 000 ml erhålls (1.6.1.1). |
|
Kommentar: För att undvika att behöva framställa denna lösning omedelbart före användning tillsätts 1 droppe koncentrerat HC1 eller 0,40 g etylendiaminotetraättiksyra (EDTA) per liter.
I.6.3 Stamlösningar av kemikalier
Om lösligheten är större än 1 g/l upplöses t.ex. 1–10 g av test- eller referenskemikalien i avjoniserat vatten, och det förtunnas till 1 liter. I motsatt fall framställs stamlösningen i det oorganiska mediet, eller kemikalier tillsätts direkt till det oorganiska mediet. Jämför tillägg III för hantering av mindre lösliga kemikalier. I MITI-testet (metod C.4-F) får dock varken lösningsmedel eller emulgatorer användas.
I.6.4 Inokulat
Inokulat kan stamma från en rad olika källor, t.ex. aktivt slam, spillvattensavlopp (ej klorerat), ytvvatten och ytjord eller en blandning av dessa. Aktivt slam för DOC-eliminering, CO2-utveckling och manometrisk respirometri ska tas från ett reningsverk eller ett laboratorium, som huvudsakligen behandlar avloppsvatten från hushåll. Inokulat från andra källor har visat sig leda till större spridning av resultaten. För modifierad OECD-screening och Closed Bottle-testet krävs ett mer utspätt inokulat utan slampartiklar, och som källa föredras sekundärt avloppsvatten från ett reningsverk som behandlar avloppsvatten från hushåll eller en anläggning i laboratorieskala. För MITI-testet framställs inokulat från en blandning av källor, vilket beskrivs under detta specifika test.
I.6.4.1 Inokulat från aktiverat slam
Ett färskt prov av aktiverat slam tas från en luftningsbassäng vid ett reningsverk eller en laboratorieanläggning som huvudsakligen behandlar avloppsvatten från hushåll. Vid behov avlägsnas grova partiklar genom filtrering genom en finmaskigt sikt och slammet hålls därefter aerobt.
Alternativt utfälls eller centrifugeras slammet (t.ex. vid 1 100 g i 10 minuter) sedan grova partiklar tagits bort. Supernatanten avlägsnas. Slammet kan tvättas i oorganiskt medium. Det koncentrerade slammet uppslammas till en koncentration av 3–5 g uppslammat torrämne per liter, varefter det luftas fram till användning.
Slammet ska tas från en ordentligt fungerande normal anläggning. Om det är nödvändigt att ta slammet från ett starkt belastat reningsverk, eller om man anser att det innehåller hämmande ämnen, måste det tvättas. Utfäll eller centrifugera det omsuspenderade slammet efter noggrann blandning, avlägsna supernatanten och slamma återigen upp det tvättade slammet i en ytterligare mängd oorganiskt medium.
Strax före användning tas ett prov av det fullständigt uppslammade slammet eller av det obehandlade slammet, för bestämning av mängden uppslammat torrämne.
Ett annat alternativ är att homogenisera aktiverat slam (3–5 g uppslammat torrämne per liter). Slammet behandlas i en mekanisk blandare i 2 minuter vid medelhastighet. Det homogeniserade slammet får utfälla i 30 minuter eller vid behov längre, och vätskan dekanteras för användning som inokulat i en mängd av 10 ml per liter oorganiskt medium.
I.6.4.2 Andra inokulatkällor
Det kan framställas av sekundärt avloppsvatten från ett reningsverk eller en laboratorieanläggning som huvudsakligen behandlar avloppsvatten från hushåll. Ett färskt prov tas och förvaras under aerobiska förhållanden under transporten. Detta får utfälla i 1 timma eller filtreras genom ett grovt filterpapper och denna vätska förvaras under aeroba förhållanden fram till användning. Upp till 100 ml av detta slags inokulat kan användas per liter medium.
Ytvatten är en ytterligare inokulatkälla. I detta fall tas ett prov av lämpligt ytvatten, t.ex. flod- eller sötvatten, som förvaras under aeroba förhållanden fram till användning. Vid behov kan inokulat koncentreras genom filtrering eller centrifugering.
I.6.5 Förbehandling av inokulat
Det framställda inokulatet kan förbehandlas till försöksbetingelserna, men får inte tillvänjas till testkemikalien. Förbehandlingen består i luftning av aktivt slam i oorganiskt medium eller av sekundärt avloppsvatten i 5–7 dagar vid rumstemperatur. Förbehandling för MITI-test anses onödigt.
I.6.6 Abiotisk kontroll
Vid behov kontrolleras eventuell abiotisk nedbrytning av testämnet genom bestämning av DOC-förlust, syreupptagning och koldioxidutveckling i sterila kontrollprover som inte innehåller okulat. Sterilisering sker genom membranfiltrering (0,2–0,45 um) eller genom tillsättning av ett passande toxiskt ämne i passande koncentration. Om membranfiltrering används ska proverna tas aseptiskt för att upprätthålla steriliteten. Såvida inte adsorption av testämnet har uteslutits på förhand ska test som mäter bionedbrytning som DOC-förlust inkludera en abiotisk kontroll som inokuleras och förgiftas. Detta gäller särskilt för inokulat med aktiverat slam.
I.6.7 Antal kolvar
För varje test beskrivs antalet kolvar i en typisk försöksserie under rubriken för varje test.
Följande kolvtyper kan användas:
|
— |
Testuppslamning: innehåller testämne och inokulat. |
|
— |
Inokulatblind: innehåller endast inokulat. |
|
— |
Förfarandekontroll: innehåller referensämne och inokulat. |
|
— |
Abiotisk steril kontroll: sterilt, innehåller testämne (1.6.6). |
|
— |
Adsorptionskontroll: innehåller testämne, inokulat och steriliserat ämne. |
|
— |
Toxicitetskontroll: innehåller testämne, referensämne och inokulat. |
Bestämningar i testuppslamningen och inokulatblind ska obligatoriskt göras parallellt. Dessutom rekommenderas parallella bestämningar i den andra kolven.
Detta är dock inte alltid möjligt. Man måste se till att tillräckligt många prover tas eller att tillräckligt många avläsningar görs för att kunna värdera den procentuella elimineringen i 10-dagarsfönstret.
1.7 DATA OCH UTVÄRDERING
Vid beräkning av den procentuella nedbrytningen Dt används genomsnittsvärdena av de dubbla bestämningarna av parametern i testuppslamningen och inokulatblindproven. Formlerna ges i avsnitten om de enskilda testmetoderna. Nedbrytningens förlopp visas grafiskt med angivelse av 10-dagarsfönstret. Den procentuella elimineringen vid avslutningen av 10-dagarsfönstret och vid platån eller testets avslutning beräknas och rapporteras.
I metoder som baseras på respirometri kan kvävehaltiga föreningar påverka syreupptagningen på grund av nitrifikation (se bilagor II och V).
I.7.1 Nedbrytning mätt genom DOC-bestämning
Den procentuella nedbrytningen (Dt) vid varje provtagningstillfälle ska beräknas särskilt för kolvarna som innehåller testämne varvid genomsnittsvärdena av de dubbla bestämningarna av DOC används för att kunna bedöma testets pålitlighet (se 1.5.2). Den beräknas genom användning av följande formel:

där:
|
Dt |
= |
% nedbrytning vid tidpunkten t |
|
Co |
= |
genomsnittlig startkoncentration av DOC i kulturmediet med testämne och inokulat (mg DOC/l) |
|
Ct |
= |
genomsnittskoncentrationen av DOC i kulturmediet med oorganiskt medium och inokulat mg DOC/l) |
|
Cbo |
= |
genomsnittlig startkoncentration av DOC i blindprov med oorganiskt medium och inokulat (mg DOC/1) |
|
Cbt |
= |
genomsnittskoncentration av DOC i blindprov med oorganiskt medium och inokulat vid tidpunkten t (mg DOC/1) |
Alla koncentrationer bestäms experimentellt.
I.7.2 Nedbrytning mätt genom specifik analys
Om specifika analysdata föreligger beräknas den primära bionedbrytningen genom formeln

där:
|
Dt |
= |
% nedbrytning vid tidpunkten t, normalt 28 dagar |
|
Sa |
= |
restmängd testämne i medium med inokulat vid testets avslutning (mg) |
|
Sb |
= |
restmängd testämne i blindprov endast med vatten/medium och testämne (mg) |
I.7.3 Abiotisk nedbrytning
När abiotisk nedbrytning används beräknas den procentuella abiotiska nedbrytningen med användning av
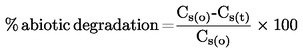
där:
|
Cs(o) |
= |
DOC-koncentration, sterilkontrollen på dag 0 |
|
Cs(t) |
= |
DOC-koncentration, sterilkontrollen på dag t. |
I.8 RAPPORTERING
Testrapporten ska om möjligt innehålla följande:
|
— |
Test- och referenskemikalie och deras renhet. |
|
— |
Testbetingelser. |
|
— |
Inokulat: art och provtagningsställe, koncentration och eventuell förbehandling. |
|
— |
Art och andel av industriavloppsvatten i avloppsvattnet, om känt. |
|
— |
Testets varaktighet och temperatur. |
|
— |
I fråga om svårlösliga testkemikalier: genomförd behandling. |
|
— |
Använd testmetod; ändringar av förfarandet ska vara vetenskapligt motiverat. |
|
— |
Försöksresultaten uppställda i tabellform. |
|
— |
Eventuellt observerade hämningsfenomen. |
|
— |
Eventuellt observerad abiotisk nedbrytning. |
|
— |
Eventuella data från specifika kemiska analyser. |
|
— |
Om möjligt analytiska data om nedbrytningsprodukter. |
|
— |
Avbildning av den procentuella nedbrytningen mot tiden för både test- och referensämnen med tydlig angivelse av ”lag”-fas, nedbrytningstid, 10-dagarsfönster och lutning (se bilaga I). Om man i testet använder pålitlighetskriterier kan genomsnittet av nedbrytningsprocenten i kolvarna innehållande testämnet användas för framställning av grafen. |
|
— |
Procentuell eliminering efter 10-dagarsfönstret och vid platån eller testets avslutning. |
DEL II. DOC-ELIMINERING (DOC DIE-AWAY) (Metod C.4-A)
II.1 TESTMETODSPRINCIP
En uppmätt mängd oorganiskt medium innehållande inokulat och, som nominella enda organiska kolkälla, testämne av känd koncentration (10–40 mg/l), luftas i mörker eller diffust ljus vid 22 ± 2 oC.
Nedbrytningen följs genom regelbunden DOC-analys i 28 dagar. Bionedbrytningen beräknas genom att uttrycka koncentrationen av avlägsnat DOC (korrigerat för koncentration i inokulatblindprovet) i procent av startkoncentrationen av DOC. Den primära bionedbrytningen kan också beräknas utifrån kompletterande kemisk analys vid inkubationsperiodens början och slut.
II.2 METODBESKRIVNING
II.2.1 Apparatur
|
a) |
Koniska kolvar, t.ex. från 250 ml till 2 liter, beroende på den volym som krävs för DOC-analysen. |
|
b) |
Skakapparatur för de koniska kolvarna, antingen med automatisk temperaturkontroll eller placerad i en lokal med konstant temperatur, och tillräckligt kraftig för att hålla förhållandena aeroba i alla kolvarna. |
|
c) |
Filtreringsapparatur med passande membran. |
|
d) |
DOC-analysator. |
|
e) |
Apparatur för bestämning av upplöst syre. |
|
f) |
Centrifug. |
II.2.2 Framställning av oorganiskt medium
Se 1.6.2 om framställning av stamlösning.
10 ml av lösningen (a) blanda med 800 ml utspädningsvatten, och av lösning (b), (c) och (d) tillsätts 1 ml av varje, varefter det fylls upp till 1 liter med förtunningsvatten.
II.2.3 Framställning och förbehandling av inokulat
Inokulat kan stamma från en rad olika källor: aktiverat slam, avloppsvatten, ytvatten och ytjord eller en blandning av dessa.
Se 1.6.4, 1.6.4.1, 1.6.4.2 och 1.6.5.
II.2.4 Förberedelse av kolvarna
Som ett exempel kan 800 ml portioner av oorganiskt medium överföras till koniska 2-literskolvar, varefter det tillsätts så mycket av stamlösningarna av test- och referensämne till de olika kolvarna att kemikaliekoncentrationen blir 10–40 mg DOC per liter. Kontrollera pH-värden och justera om nödvändigt till 7,4. Inokulat från aktiverat slam eller en annan källa tillsätts (se I.6.4) till en slutkoncentration av högst 30 mg uppslammat torrämne per liter. Inokulatprover förbereds också i oorganiskt medium utan test- och referenskemikalier.
Vid behov inkluderas även en kolv för kontroll av om testkemikalien har en hämmande verkan, genom tillsättning av jämförbart lika stora mängder test- och referenskemikalie till oorganiskt medium innehållande inokulat.
Man kan också vid behov inkludera en steril kolv, dvs. en kolv utan inokulat för att undersöka om testkemikalien bryts ned abiotiskt (se 1.6.6).
Om man misstanker att testkemikalien i väsentlig grad kan adsorberas till gas, slampartiklar eller liknande, företas dessutom en preliminär värdering av denna adsorption och därmed av testens lämplighet för den berörda testkemikalien (se tabell 1). Använd en kolv innehållande testämnet, inokulat och det steriliserande ämnet.
Alla kolvar fylls upp till 1 liter med oorganiskt medium, och efter blandning uttas ett prov från varje kolv för bestämning av startkoncentrationen av DOC (se bilaga II, punkt 4). Kolvarnas öppning sluts, t.ex. med aluminiumfolie, på ett sådant sätt att luften fritt kan passera in och ut ur kolvarna. Därefter inleds testet genom att kolvarna placeras i skakapparaturen.
II.2.5 Antal kolvar i en typisk testserie
Kolv 1 och 2: testuppslamning
Kolv 3 och 4: inokulatblind
Kolv 5: förfarandekontroll
Rekommenderas vid behov:
Kolv 6: abiotisk steril kontroll
Kolv 7: adsorptionskontroll
Kolv 8: toxicitetskontroll
Se I.6.7.
II.2.6 Utförande av testet
Under testet görs dubbla bestämningar av DOC-koncentrationerna i alla kolvarna med bestämda tidsintervaller och så ofta att både 10-dagarsfönstret och den procentuella förlusten vid 10-dagarsfönstrets avslutning kan konstateras. Endast så mycket av testuppslamningen tas som är nödvändigt för analysen.
Före provtagningen kompenseras för eventuell förångningsförlust från kolvarna genom tillsättning av vatten (1.6.1). Innan proven tas blandas kulturmediet noggrant och det säkerställs att materialet på kolvens sidor löses upp eller uppslammas före provtagning. Omedelbart efter provtagningen membran-filtreras eller centrifugeras provet (se tillägg 4). Det filtrerade eller centrifugerade provet analyseras samma dag. Det kan förvaras i högst 48 timmar vid 2–4 oC eller längre vid högst – 18 oC.
II.3 DATA OCH RAPPORTERING
II.3.1 Behandling av resultaten
Den procentuella nedbrytningen vid tidpunkt t beräknas enligt 1.7.1 (DOC-bestämning) och eventuellt enligt 1.7.2 (specifik analys).
Alla resultat antecknas på tillhörande scheman.
II.3.2 Resultatens giltighet
Se I.5.2.
II.3.3 Rapportering
Se I.8.
II.4 FORMULÄR
Nedanför visas ett exempel på ett resultatschema.
DOC-ELIMINERING
|
1. |
LABORATORIUM |
|
2. |
DATUM VID INLEDNING AV TEST |
3. TESTÄMNE
Namn:
Stamlösningens koncentration: .... mg/l ämne
Startkoncentration i mediet: t0: .... mg/l ämne
4. INOKULAT
Källa:
Behandling:
Eventuell förbehandling:
Koncentration av uppslammat torrämne i reaktionsblandningen: ... mg/l:
5. KOLBESTÄMNINGAR
Kolanalyser:
|
|
Kolv nr |
|
DOC efter n dagar (mg/l) |
||||
|
0 |
n1 |
n2 |
n3 |
nx |
|||
|
Testkemikalien med inokulat |
1 |
a1 |
|
|
|
|
|
|
a2 |
|
|
|
|
|
||
|
a, medel Ca(t) |
|
|
|
|
|
||
|
2 |
b1 |
|
|
|
|
|
|
|
b2 |
|
|
|
|
|
||
|
b, medel Cb(t) |
|
|
|
|
|
||
|
Inokulatblindprovet utan testkemikalie |
3 |
c1 |
|
|
|
|
|
|
c2 |
|
|
|
|
|
||
|
c, medel Cc(t) |
|
|
|
|
|
||
|
4 |
d1 |
|
|
|
|
|
|
|
d2 |
|
|
|
|
|
||
|
d, medel Cd(t) |
|
|
|
|
|
||
|
|
|
|
|
|
|
||
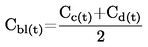
6. UTVÄRDERING AV RÅDATA
|
Kolv nr. |
|
% nedbrytning efter n dagar |
||||
|
Q |
n1 |
n2 |
n3 |
nx |
||
|
1 |
|
0 |
|
|
|
|
|
2 |
|
0 |
|
|
|
|
|
medel (3) |
|
0 |
|
|
|
|

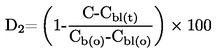
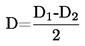
Anmärkning: Motsvarande formler kan användas för referenskemikalien och toxicitetskontrollproverna.
7. ABIOTISK KONTROLL (valfritt)
|
|
Tid (dagar) |
|
|
0 |
t |
|
|
DOC-koncentration (mg/l) i sterilkontroll |
Cs(0) |
Cs(t) |

8. SPECIFIK KEMISK ANALYS (valfritt)
|
|
Restmängd av testämne vid avslutning av test (mg/l) |
% primär nedbrytning |
|
Sterilkontroll |
sb |
|
|
Inokulerat testmedium |
sa |
|
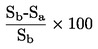
DEL III. MODIFIERAT OECD-SCREEENINGTEST (metod C.4-B)
III.1 TESTMETODSPRINCIP
En uppmätt mängd oorganiskt medium innehållande inokulat och, som nominella enda organiska kolkälla, testämne av känd koncentration (10–40 mg/l), luftas i mörker eller diffust ljus vid 22 ± 2 oC.
Nedbrytningen följs genom regelbunden DOC-analys i 28 dagar. Bionedbrytningen beräknas genom att uttrycka koncentrationen av avlägsnat DOC (korrigerat för koncentration i inokulatblindprovet) i procent av startkoncentrationen av DOC. Den primära bionedbrytningen kan också beräknas utifrån kompletterande kemisk analys vid inkubationsperiodens början och slut.
III.2 METODBESKRIVNING
III.2.1 Apparatur
|
a) |
Koniska kolvar, t.ex. från 250 ml till 2 liter, beroende på den volym som krävs för DOC-analysen. |
|
b) |
Skakapparatur för de koniska kolvarna, antingen med automatisk temperaturkontroll eller placerad i en lokal med konstant temperatur, och tillräckligt kraftig för att hålla förhållandena aeroba i alla kolvarna. |
|
c) |
Filtreringsapparatur med passande membran. |
|
d) |
DOC-analysator. |
|
e) |
Apparatur för bestämning av upplöst syre. |
|
f) |
Centrifug. |
III.2.2 Framställning av oorganiskt medium
Se I.6.2 om framställning av stamlösning.
10 ml av lösningen (a) blandas med 80 ml utspädningsvatten, och av lösning (b), (c) och (d) tillsätts 1 ml av varje, varefter det fylls upp till 1 liter med förtunningsvatten.
I denna metod används endast 0,5 liter spillvatten per liter som inokulat, och därför kan det vara nödvändigt att tillsätta ytterligare spårämnen och växtfaktorer till mediet. Detta görs genom tillsättning av 1 ml av var och en av följande lösningar per liter medium (slutvolymen):
Lösningar av spårämnen:
|
Mangansulfat tetrahydrat, MnSO4 . 4H2O |
39,9 mg |
|
Borsyra, H3BO3 |
57,2 mg |
|
Zinksulfat heptahydrat, ZnSO4 .7H2O |
42,8 mg |
|
Ammoniumheptamolybdat (NH4)6M07O24 |
34,7 mg |
|
Järn-chelat (FeCl3 etylendiamintetraättiksyra) |
100,0 mg |
|
löses i vatten och förtunnas till 1 000 ml |
|
|
Vitaminlösning: |
|
|
Jästextrakt |
15,0 mg |
löses i 100 ml vatten. Det steriliseras genom filtrering genom 0,2 μm membranfilter om lösningen framställs färsk varje gång.
III.2.3 Framställning och förbehandling av inokulat
Inokulat kan stamma från en rad olika källor: aktiverat slam, avloppsvatten, ytvatten och ytjord eller en blandning av dessa. Se 1.6.4.2 och 1.6.5.
0,5 ml per liter oorganisk lösning används.
III.2.4 Förberedelse av kolvarna
Som ett exempel kan 800 ml portioner av oorganiskt medium överföras till koniska 2-literskolvar, varefter det tillsätts så mycket av stamlösningarna av test- och referensämne till de olika kolvarna att kemikaliekoncentrationen blir 10–40 mg DOC per liter. Kontrollera pH-värden och justera till 7,4 om nödvändigt. 0,5 ml spillvatten tillsätts per liter (se I.6.4.2) till kolvarna som inokulat. Inokulatprover förbereds också i oorganiskt medium utan test- och referenskemikalier.
Vid behov inkluderas även en kolv för kontroll av om testkemikalien har en hämmande verkan, genom tillsättning av jämförbart lika stora mängder test- och referenskemikalie till oorganiskt medium innehållande inokulat.
Man kan också vid behov inkludera en steril kolv, dvs. en kolv utan inokulat för att undersöka om testkemika-lien bryts ned abiotiskt (se 1.6.6).
Om man misstänker att testkemikalien i väsentlig grad kan adsorberas till gas, slampartiklar eller liknande, företas dessutom en preliminär värdering av denna adsorption och därmed av testens lämplighet för den berörda testkemikalien (se tabell 1). Använd en kolv innehållande testämnet, inokulat och det steriliserande ämnet.
Alla kolvar fylls upp till 1 liter med oorganiskt medium, och efter blandning uttas ett prov från varje kolv för bestämning av startkoncentrationen av DOC (se bilaga II, punkt 4). Kolvarnas öppning sluts, t.ex. med aluminiumfolie, på ett sådant sätt att luften fritt kan passera in och ut ur kolvarna. Därefter inleds testet genom att kolvarna placeras i skakapparaturen.
III.2.5 Antal kolvar i en typisk testserie
Kolv 1 och 2: testuppslamning
Kolv 3 och 4: inokulatblind
Kolv 5: förfarandekontroll
Rekommenderas vid behov:
Kolv 6: abiotisk steril kontroll
Kolv 7: adsorptionskontroll
Kolv 8: toxicitetskontroll
Se I.6.7.
III.2.6 Utförande av testet
Under testet görs dubbla bestämningar av DOC-koncentrationerna i alla kolvarna med bestämda tidsintervaller och så ofta att både 10-dagarsfönstret och den procentuella förlusten vid 10-dagarsfönstrets avslutning kan konstateras. Endast så mycket av testuppslamningen tas som är nödvändigt för analysen.
Före provtagningen kompenseras för eventuell förångningsförlust från kolvarna genom tillsättning av förtunningsvatten (I.6.1). Innan proven tas blandas kulturmediet noggrant och det säkerställs att materialet på kolvens sidor löses upp eller uppslammas innan provtagning. Omedelbart efter provtagningen membranfiltreras eller centrifugeras provet (se tillägg 4). Det filtrerade eller centrifugerade provet analyseras samma dag. Det kan förvaras i högst 48 timmar vid 2–4 oC eller längre vid högst – 18 oC.
III.3 DATA OCH RAPPORTERING
III.3.1 Behandling av resultaten
Den procentuella nedbrytningen vid tidpunkt t beräknas enligt 1.7.1 (DOC-bestämning) och eventuellt enligt 1.7.2 (specifik analys).
Alla resultat antecknas på tillhörande scheman.
III.3.2 Resultatens giltighet
Se. I.5.2.
III.3.3 Rapportering
Se I.8.
III.4 FORMULÄR
Nedan visas ett exempel på ett formulär.
MODIFIERAT OECD-SCREENINGTEST
|
1. |
LABORATORIUM |
|
2. |
DATUM VID INLEDNING AV TEST |
3. TESTÄMNE
Namn:
Stamlösningens koncentration: .... mg/l ämne
Startkoncentration i mediet, till: .... mg/l ämne
4. INOKULAT
Källa:
Behandling:
Eventuell förbehandling:
Koncentration av uppslammat torrämnen i reaktionsblandningen: ... mg/l
5. KOLBESTÄMNINGAR
Kolanalyser:
|
|
Kolv nr |
|
DOC efter n dagar (mg/l) |
||||
|
0 |
N1 |
n2 |
n3 |
nx |
|||
|
Testkemikalie med inokulat |
1 |
a1 |
|
|
|
|
|
|
a2 |
|
|
|
|
|
||
|
a, medel Ca(t) |
|
|
|
|
|
||
|
2 |
b1 |
|
|
|
|
|
|
|
b2 |
|
|
|
|
|
||
|
b, medel Cb(t) |
|
|
|
|
|
||
|
Inokulatblindprov utan testkemikalie |
3 |
c1 |
|
|
|
|
|
|
c2 |
|
|
|
|
|
||
|
c, medel Cc(t) |
|
|
|
|
|
||
|
4 |
d1 |
|
|
|
|
|
|
|
d2 |
|
|
|
|
|
||
|
d, medel Cd(t) |
|
|
|
|
|
||
|
|
|
|
|
|
|
||
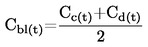
6. UTVÄRDERING AV RÅDATA
|
Kolv nr |
|
% nedbrytning efter n dagar |
||||
|
0 |
n1 |
n2 |
n3 |
nx |
||
|
1 |
|
0 |
|
|
|
|
|
2 |
|
0 |
|
|
|
|
|
medel (4) |
|
0 |
|
|
|
|



Anmärkning: Motsvarande formler kan användas för referenskemikalie och toxicitetskontrollproverna.
7. ABIOTISK KONTROLL (valfritt)
|
|
Tid (dagar) |
|
|
0 |
t |
|
|
DOC-koncentration (mg/l) i sterilkontroll |
Cs(0) |
Cs(t) |

8. SPECIFIK KEMISK ANALYS (valfritt)
|
|
Restmängd av testämne vid avslutning av test (mg/I) |
% primär nedbrytning |
|
Sterilkontroll |
sb |
|
|
Inokulerat testmedium |
sa |
|

DEL IV. CO2-UTVECKLING (metod C.4-C)
IV.1 TESTMETODSPRINCIP
En uppmätt mängd oorganiskt medium innehållande inokulat och, som nominella enda organiska kolkälla, testämne av känd koncentration (10–40 mg DOC eller TOC/l), luftas genom genomledning av koldioxidfri luft med kontrollerad hastighet i mörker eller diffust ljus. Nedbrytningen följs i 28 dagar genom bestämning av den bildade mängden koldioxid i det att det samlas upp i barium- eller natriumhydroxid och mäts antingen genom titrering av resterade hydroxid eller som oorganiskt kol. Den mängd koldioxid som utvecklats av testkemikalien (korrigerat för inokulatblindvärden) uttrycks i procent av startkoncentrationen av ThCO2. Graden av bionedbrytning kan också beräknas utifrån kompletterande DOC-analyser vid inkubationsperiodens början och slut.
IV.2 METODBESKRIVNING
IV.2.1 Apparatur
|
a) |
Kolvar 2–5 liter, alla försedda med luftningsrör, som nästan når ned till botten av behållaren, och avgångsöppning. |
|
b) |
Magnetomrörare, när svårlösliga kemikalier testas. |
|
c) |
Gasabsorptionskolvar. |
|
d) |
Utrustning för kontroll och mätning av luftgenomströmningen. |
|
e) |
Apparatur för tvättning av koldioxid, för framställning av koldioxidfri luft. Alternativt kan CO2-fri syre och CO2-fritt kväve användas i stålkolvar i det riktiga blandningsförhållandet (20 % O2 och 80 % N2). |
|
f) |
Utrustning för bestämning av koldioxid, antingen trimetriskt eller genom en annan form av analys för oorganiskt kol. |
|
g) |
Membranfiltreringsapparatur (valfritt). |
|
h) |
DOC-analysator (valfritt). |
IV.2.2 Framställning av oorganiskt medium
Se 1.6.2 om framställning av stamlösning.
10 ml av lösningen (a) blandas med 800 ml utspädningsvatten, och av lösning (b), (c) och (d) tillsätts 1 ml av varje, varefter det fylls upp till 1 liter med förtunningsvatten.
IV.2.3 Framställning och förbehandling av inokulat
Inokulat kan stamma från en rad olika källor: aktiverat slam, avloppsvatten, ytvatten och ytjord eller en blandning av dessa.
Se I.6.4, I.6.4.1, I.6.4.2 och I.6.5.
IV.2.4 Förberedelse av kolvarna
Följande volym- och viktangivelser gäller som exempel för 5-literskolvar med 3 1 uppslamning. Om mindre volymer används anpassas värdena, dock ska den bildade mängden koldioxid kunna bestämmas korrekt.
2 400 ml oorganiskt medium överförs till varje 5-literskolv. Man tillsätter så mycket förberett aktiverat slam (se I.6.4.1 och I.6.5) att koncentrationen av uppslammat torrämne blir högst 30 mg/l i 3 liter slutblandning. Alternativt kan det förberedda slammet först förtunnas med oorganiskt medium till en uppslamning innehållande 500–1 000 mg/l, varefter en det av detta tillsätts till 5-literkolvens innehåll till en koncentration av 30 mg/l. Detta ger större precision. Andra inokulatkällor kan användas (se I.6.4.2).
Dessa blandningar luftas över natten med CO2-fri luft så all koldioxid drivs ut ur systemet.
Test- och referensämne tillsätts var för sig som kända mängder av stamlösningar till replikatkolvarna så att man uppnår en koncentration av 10–20 mg DOC eller TOC per liter. Till vissa av kolvarna tillsätts inga kemikalier och dessa fungerar som inokulatkontrollprover. Svårlösliga testämnen tillsätts direkt till kolvarna på vikt- eller volyrnbasis, eller hanteras i enlighet med bilaga III.
Om det är nödvändigt används en kolv för att kontrollera om testkemikalien verkar hämmande. Detta görs genom att tillsätta både test- och referenskemikalie av samma koncentration som i de andra kolvarna.
Det kan vara nödvändigt att inkludera en steril kolv, dvs. en kolv utan inokulat för att undersöka om testkemikalien bryts ned abiotiskt (se 1.6.6). Sterilisera genom tillsättning av ett giftigt ämne i passande koncentration.
Alla kolvarna fylls upp till en uppslamningsvolym av 3 liter genom tillsättning av oorganiskt medium, som i förväg har genomluftats med CO2-fri luft. Eventuellt kan man ta prover för DOC-analys (se bilaga II.4) och specifik analys. Absorptionskolvarna förbinds till kolvarnas avgångsöppning.
Om banumhydroxid används ansluts de 3 absorptionskolvarna, var och en innehållande 100 ml 0,0125 M bariumhydroxidlösning, i en serie till varje 5-literskolv. Lösningen ska vara fri från fällt sulfat och karbonat och dess styrka ska bestämmas omedelbart före användning. Om natriumhydroxid används ansluts 2 kolvar varvid den sista fungerar som kontroll av att koldioxiden är absorberad i den första. Absorptionskolvar med serum-kolvstängning är lämpliga. 200 ml 0,05 natriumhydroxid placeras i varje kolv, vilket är tillräckligt för att absorbera hela den mängd koldioxid som utvecklas när testkemikalien bryts ned fullständigt. Natriumhydroxidlösning kommer, även om färskt framställd, dock att innehålla spår av karbonat. Detta korrigeras genom subtraktion av blindprovens karbonatinnehåll.
IV.2.5 Antal kolvar i en typisk testserie
Kolv 1 och 2: testuppslamning
Kolv 3 och 4: inokulatblind
Kolv 5: förfarandekontroll
Rekommenderas vid behov:
Kolv 6: abiotisk steril kontroll
Kolv 7: adsorptionskontroll
Kolv 8: toxicitetskontroll
Se I.6.7.
IV.2.6 Utförande av testet
Testet inleds genom genombubbling av uppslamningarna med 30–100 ml CO2-fri luft per minut. Prover tas regelbundet av kodioxidabsorbenten för analys av CO2-innehåll. Det rekommenderas att man under de första 10 dagarna gör analyser varannan eller var tredje dag och därefter var femte dag fram till dag 28, så att 10-dagarsfönstret kan identifieras.
Den 28:e dagen tas (eventuellt) prover för DOC-analys och specifik analys, uppslamningarnas pH mäts och 1 ml koncentrerad saltsyra tillsätts till varje kolv. Kolvarna luftas över natten så att all koldioxid drivs ut av testuppslamningarna. Dag 29 företas den sista analysen av utvecklad koldioxid.
De dagar då CO2 bestäms avlägsnas den bariumhydroxidabsorbtionskolv som är närmast kolven och innehåller titreras med 0,05 M HCl med fenoftalein som indikator. De övriga absorbtionskolvarna flyttas fram en plats närmare kolven och en ny kolv innehållande 100 ml färskt framställt 0,0125 M bariumhydroxid placeras längst bak i serien. Titreringarna görs enligt behov, t.ex. när betydande fällning syns i den första kolven men inget syns i den andra, dock minst en gång i månaden. Om NaOH används om absorbtionsmedel tas med en injektionsspruta ett litet prov (beroende på den använda kolämnesanaiysatorn) av natriumhydroxidlösningen i den kolv som är närmast kolven. Provet sprutas in i IC-delen av kolämnesanalysatorn för direkt analys av den utvecklade koldioxiden.
Innehållet i den andra absorbtionskolven analyseras först vid testets avslutning så att man kan korrigera för eventuell överföring av koldioxid
IV.3 DATA OCH RAPPORTERING
IV.3.1 Behandling av resultaten
Den absorberade CO2-mängden är efter titrering given genom
mgCO2 = (100 × CB-0,5 × V × CA) × 44
där:
|
V |
= |
den mängd HCl som förbrukas vid titrering av de 100 ml absorptionsmedel (ml) |
|
CB |
= |
bariumhydroxidlösningens koncentration (M) |
|
CA |
= |
saltsyreupplösningens koncentration (M) |
Om CB = 0,0125 M och CA = 0,05 M ska man använda 50 ml för titrering av 100 ml bariumhydroxid, och CO2-mängden är given genom:

I detta fall ska man alltså använda en faktor på 1,1 vid omräkning av den volym HCl som går åt vid titrering, till det antal mg Cl2 som utvecklats.
Den viktmängd CO2 som bildas av bara inokulat och av inokulat med testkemikalie, beräknas utifrån de gällande titreringsvärdena, och skillnaden är den viktmängd CO2 som endast testkemikalien utvecklar.
Om t.ex. enbart inokulat kräver 48 ml för titrering och inokulat plus testkemikalie kräver 45 ml fås
CO2 från inokulat = 1,1 × (50.48) = 2,2 mg
CO2 från inokulat plus testkemikalie = 1,1 × (50-45) = 5,5 mg,
och den viktmängd CO2 som testkemikalien har utvecklat blir alltså 3,3 mg.
Den procentuella bionedbrytningen beräknas genom
![]()
eller
![]()
varvid 3,67 är omräkningsfaktorn (44/12) från kol till koldioxid.
Den procentuella nedbrytningen efter en viss tid beräknas genom att lägga ihop de enskilda procentdelarna av ThCO2 som är uppmätta fram till den gällande tidpunkten.
Om natriumhydroxid används som absorptionsmedel beräknas den utvecklade CO2-mängden, uttryckt som IC (mg), genom att multiplicera absorbtionsmedlets IC-koncentration med dess volym.
Den procentuella nedbrytningen beräknas genom
![]()
IV.3.2 Resultatens giltighet
IC-innehållet i testkemikalieuppslamningen i oorganiskt medium vid testets inledning ska vara mindre än 5 % av TC, och den utvecklade CO2-mängden i inokulatblindprovet får vid testets avslutning inte överstiga 40 mg per liter medium. Om det finns värden på över 70 mg CO2 per liter granskas data och försökstekniker noggrant.
Se också I.5.2.
IV.3.3 Rapportering
Se I.8.
IV.4 FORMULÄR
Nedan visas ett exempel på ett formulär.
KOLDIOXIDUTVECKLING
|
1. |
LABORATORIUM |
|
2. |
DATUM VID INLEDNING AV TEST |
3. TESTÄMNE
Namn:
Stamlösningens koncentration: .... mg/l ämne
Startkoncentration i mediet: .... mg/l ämne
Total C tillsatt till kolven: .... mg C
ThCO2: ... mg CO2
4. INOKULAT
Källa:
Behandling:
Eventuell förbehandling:
Koncentration av uppslammat torrämne i reaktionsblandningen: ... mg/l:
5. KOLDIOXIDUTVECKLING OCH NEDBRYTBARHET
Metod: Ba(OH)2/NaOH/annat
|
Tid (dagar) |
Utvecklat CO2 test (mg) |
Utvecklat CO2 blind (mg) |
utvecklat CO2 kumulerat (mg) (test minus blindprov) |
% ThCO2
|
|||||
|
1 2 |
medel |
3 4 |
medel |
1 |
2 |
1 |
2 |
medel |
|
|
0 |
|
|
|
|
|
|
|
|
|
|
n1 |
|
|
|
|
|
|
|
|
|
|
n2 |
|
|
|
|
|
|
|
|
|
|
n3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
28 |
|
|
|
|
|
|
|
|
|
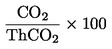
Anmärkning: Motsvarande uppställning kan användas för referenskemikalie och toxicitetskontrollproverna.
6. KOLANALYS (valfritt)
Kolanalysator:
|
Tid (dagar) |
Blindprov mg/l |
Testkemikalie mg/l |
|
0 |
Cb(o) |
Co |
|
28 (5) |
Cb(t) |
ct |

7. ABIOTISK NEDBRYTNING (valfritt)

DEL V. MANOMETRISK RESPIROMETRITEST (metod C.4-D)
V.l TESTMETODSPRINCIP
En uppmätt mängd oorganiskt medium innehållande inokulat och, som nominella enda organiska kolkälla, testämne av känd koncentration (100 mg testämne per liter motsvarande minst 50–100 mg ThOD per liter) rörs om i en stängd kolv vid konstant temperatur (± 1 oC eller mindre) i upp till 20 dagar. Syreförbrukningen bestäms antingen genom mätning av den mängd syre (elektrolytiskt framställt) som krävs för att upprätthålla konstant gasvolym i respirometerkolven, eller utifrån volym- eller tryckändringen (eller en kombination av dessa) i apparaten. Utvecklat koldioxid absorberas i en kaliumhydroxidlösning eller annat lämpligt absorptionsmedel. Den syremängd som testkemikalien har tagit upp (korrigerat mot den parallella inokulatblindprovets upptagning) uttrycks i procent av ThOD eller COD. Frivilligt kan primär bionedbrytning också beräknas genom kompletterande specifik analys vid inkubationsperiodens början och slut, och fullständig bionedbrytning genom DOC-analys.
V.2 TESTMETODSBESKRIVNING
V.2.1 Apparatur
|
a) |
Lämplig respirator. |
|
b) |
Temperaturreglering inom ± 1 oC eller mindre. |
|
c) |
Membranfiltreringsapparatur (valfritt). |
|
d) |
Kolanalysator (valfritt). |
V.2.2 Framställning av oorganiskt medium
Se I.6.2 om framställning av stamlösning.
10 ml av lösningen a) blandas med 800 ml utspädningsvatten, och av lösning b), c) och d) tillsätts 1 ml av varje, varefter det fylls upp till 1 liter med utspädningsvatten.
V.2.3 Framställning och förbehandling av inokulat
Inokulat kan stamma från en rad olika källor: aktiverat slam, avloppsvatten, ytvatten och ytjord eller en blandning av dessa.
Se I.6.4, I.6.4.1, I.6.4.2 och I.6.5.
V.2.4 Förberedelse av kolvarna
Utifrån stamlösningarna framställs det särskilda lösningar av test- och referensämne i oorganiskt medium med en kemikaliekoncentration av normalt 100 mg/l (motsvarande minst 50–100 mg ThOD per liter).
ThOD beräknas på grundval av ammoniumsalt, såvida inte nitrifikation förväntas, i vilket fall beräkningen baseras på nitratbildning (se bilaga II.2).
pH justeras vid behov till 7,4±0,2.
Svårlösliga ämnen tillsätts vid en senare tidpunkt.
Om testämnets toxicitet ska bestämmas framställs ytterligare en lösning i oorganiskt medium, som innehåller både test- och referensämne i samma koncentration som i de individuella lösningarna.
Om det är nödvändigt att bestämma den fysiska-kemiska syreupptagningen framställs en lösning av testämne i en koncentration av normalt 100 mg ThOD per liter som steriliseras vid tillsättningen av ett passande giftigt ämne (se I.6.6).
Den nödvändiga mängden av test- respektive referensämneslösningarna överförs till minst två kolvar (dubbel bestämning). Ytterligare kolvar med enbart oorganiskt medium förbereds (inokulatblindprover) och om det är nödvändigt med den blandade test- och referensämneslösningen och med den sterila lösningen.
Om testämnet är svårlösligt tillsätts det direkt vid denna tidpunkt, antingen på grundval av vikt eller volym eller genom hanteringen enligt bilaga III. Dessutom tillsätts kaliumhydroxid, natriumhydroxidparlor eller ett annat absorptionsmedel till CO2-kamrarna.
V.2.5 Antal kolvar i en typisk testserie
Kolv 1 och 2: testuppslammning
Kolv 3 och 4: inokulatblind
Kolv 5: förfarandekontroll
Rekommenderas, och vid behov:
Kolv 6: abiotisk steril kontroll
Kolv 7: adsorptionskontroll
Kolv 8: toxicitetskontroll
Se I.6.7.
V.2.6 Utförande av testet
Låt behållarna stå tills de har nått den önskade temperaturen och tillsätt förberett aktiverat slam eller ett annat inokulat till de ifrågavarande behållarna så att en koncentration av uppslammat torrämne på högst 30 mg/l erhålls. Montera apparaturen, starta omrörningen, kontrollera lufttätheten och börja mäta syreupptagningen. Normalt krävs det ingen annan passning än de nödvändiga avlåsningarna och daglig kontroll av att temperaturen är korrekt och omrörningen tillräcklig.
Syreupptagningen beräknas på grundval av ofta förekommande regelbundna avläsningar som görs enligt apparaturtillverkarens anvisningar. Vid inkubationstidens avslutning, normalt 28 dagar, mäts pH-värdet i kolvarna, särskilt om syreupptagningen har varit mycket låg eller större än ThODNH4 (för kvävehaltiga föreningar).
Om det är nödvändigt tas prover från respirometerkolvarna före och efter inkuberingen för DOC-analys eller specifik kemisk analys (se bilaga II.4). Man ska se till att volymen av den testuppslamning som kvarstår i kolven efter den första provtagningen är känd. Om testämnet är N-haltigt bestäms koncentrationsökningen av nitrit och nitrat under 28 dagar, och korrektionen för syreförbrukningen genom nitrifikation beräknas (bilaga V).
V.3 DATA OCH RAPPORTERING
V.3.1 Behandling av resultaten
Testkemikaliens syreupptagning (mg) efter en viss tid (korrigerad för inokulatblindprovens syreupptagning under samma tid) divideras med vikten av använd mängd testämne. Av detta framkommer BOD uttryckt i mg syre per mg testämne, dvs.

= mg O2 per mg testämne
Den procentuella bionedbrytningen beräknas antingen genom

eller
![]()
Det bör noteras att de två metoderna inte nödvändigtvis ger samma resultat. Den förstnämnda metoden föredras.
För testämnen som innehåller kväve används passande ThOD (NH4 eller NO3) i enlighet med vad som är känt eller förväntas om förekomst av nitrifikation (bilaga II.2). Om ofullständig nitrifikation sker beräknas korrektionen för syreförbrukning till nitrifikation på grundval av ändringarna i nitrit- och nitratkoncentrationen (bilaga V).
Om man beslutar sig för att göra bestämningar av organiskt kol och en specifik kemikalie beräknas den procentuella nedbrytningen i enlighet med punkt I.7.
Alla resultat noteras på tillhörande formulär.
V.3.2 Resultatens giltighet
Inokulatblindprovens syreupptagning är normalt 20–30 mg O2/l på 28 dagar och bör inte vara större än 60 mg/l. Om det finns värden över 60 mg/l måste data och försökstekniker granskas noggrant. Om pH ligger utanför intervallet 6–8,5 och testämnets syreupptagning är mindre än 60 % upprepas testet med lägre testämneskoncentrationer.
Se också I.5.2.
V.3.3 Rapportering
Se I.8.
V.4 FORMULÄR
Nedan visas ett exempel på ett formulär.
MANOMETRISK RESPIROMETRI
|
1. |
LABORATORIUM |
|
2. |
DATUM VID INLEDNING AV TEST |
3. TESTÄMNE
Namn:
Stamlösningens koncentration: .... mg/l ämne
Startkoncentration i mediet: Co: .... mg/l ämne
Volymen i testkolv (V): .... ml
ThOD eller COD: ... mg O2/mg testämne (NH4, NO3).
4. INOKULAT
Källa:
Behandling:
Eventuell förbehandling:
Koncentration av uppslammat torrämne i reaktionsblandningen: ... mg/l:
5. SYREUPPTAGNING: BIONEDBRYTNING
|
|
Tid (dagar) |
||||||||||||
|
0 |
|
7 |
|
14 |
|
|
21 |
|
|
28 |
|
||
|
O2 uppt. (mg) tescämne |
1 |
|
|
|
|
|
|
|
|
|
|
|
|
|
2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
a, medel |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
O2 uppt. (mg) blindprov |
3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
4 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
b, medel |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Korrigerat BOD (mg) |
(a1 – bm) |
|
|
|
|
|
|
|
|
|
|
|
|
|
(a2 – bm) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
BOD per mg testämne |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
% nedbrytning
|
D1 (a1) |
|
|
|
|
|
|
|
|
|
|
|
|
|
D2 (a2) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
medel (6) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
V = volymen av medium i testkolven. |
|||||||||||||


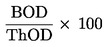
Anmärkning: En motsvarande uppställning kan användas för referenskemikalien och toxicitetskontrollproven.
6. KORREKTION FÖR NITRIFIKATION (se bilaga V)
|
Dag |
0 |
28 |
skillnad |
||
|
|
|
(N) |
||
|
— |
— |
|
||
|
|
|
(N) |
||
|
— |
— |
|
||
|
— |
— |
|
7. KOLBESTÄMNING (valfritt)
Kolanalysator:
|
Tid (dagar) |
Blindprov mg/1 |
Testkemikalie mg/l |
|
0 |
(Cblo) |
(C0) |
|
28 (7) |
(Cblt) |
(Ct) |

8. SPECIFIK KEMIKALIEBESTÄMNING (valfritt)
|
Sb |
= |
koncentrationen av testkemikalien i steril kontrollkolv efter 28 dagar |
|
Sa |
= |
koncentrationen av testkemikalien i kolv med inokulat efter 28 dagar |

9. ABIOTISK NEDBRYTNING (valfritt)
|
a |
= |
syreförbrukningen i sterila kolvar efter 28 dagar (mg) |

(se avsnitt 1 och 3)

DEL VI. CLOSED BOTTLE-TEST (metod C.4-E)
VI.1 TESTMETODSPRINCIP
Till en normal 2–5 mg/l lösning av testämnet i oorganiskt medium tillsätts inokulat bestående av ett förhållandevis litet antal mikroorganismer från en blandad population, och lösningen förvaras i mörker vid konstant temperatur i helt fyllda slutna kolvar. Nedbrytningen följs under 28 dagar genom analys av upplöst syre. Den syremängd som testämnet har förbrukat korrigeras för den syremängd som har förbrukats i det parallella inokulatblindprovet, och uttrycks i procent av ThOD eller COD.
VI.2 TESTMETODSBESKRIVNING
VI.2.1 Apparatur
|
a) |
BOD-kolvar med glaspropp, t.ex. 250–300 ml. |
|
b) |
Vattenbad eller inkuberingsapparat, som kan hålla kolvarna vid konstant temperatur (± 1 oC eller mindre) i mörker. |
|
c) |
Stora glasflaskor (2–5 1) för framställning av medium och uppfyllning av BOD-kolvarna. |
|
d) |
Syreelektrod och syremätare eller utrustning och reagenser för Winkler-titrering. |
VI.2.2 Framställning av oorganiskt medium
Se I.6.2 om framställning av stamlösning.
1 ml lösning a), b), c) och d) förtunnas till 1 liter med vatten.
VI.2.3 Framställning av inokulat
Inokulat tas normalt frän sekundärt avloppsvatten från ett reningsverk eller en laboratorieanläggning som huvudsakligen behandlar avloppsvatten frän hushåll. En alternativ inokulatkälla är ytvatten. Normalt används från en droppe (0,5 ml) till 5 ml filtrat per liter medium. Av-provningar kan vara nödvändiga för att klarlägga den optimala volymen för ett visst avloppsvatten (se I.6.4.2 och I.6.5).
VI.2.4 Förberedelse av kolvar
Det oorganiska mediet luftas kraftigt i minst 20 minuter. En given försöksserie utförs med oorganiskt medium från samma sats. Normalt är mediet färdigt för användning efter att ha stått i 20 minuter vid testtemperaturen. Som kontroll bestäms koncentrationen av upplöst syre. Värdena ska ligga omkring 9 mg/l vid 20 oC. All överföring och uppfyllning med det luftmättade mediet utförs utan att det bildas bubblor, t.ex. genom användning av hävert.
Parallella grupper av BOD-kolvar förbereds för samtidiga försök med test- och referensämne. Minst så många BOD-kolvar, inbegripet inokulatblindprover, samlas så att minst dubbla bestämningar kan företas av syreförbrukningen med önskade regelbundenhet, t.ex. efter 0, 7, 14, 21 och 28 dagar. Det kan vara nödvändigt att använda flera kolvar om man säkert ska kunna identifiera 10-dagarsfönstret.
De stora kolvarna fylls upp till en tredjedel med fullt luftat oorganiskt medium. Därefter tillsätts så mycket stamlösning av test- och referensämne till olika kolvar att deras slutkoncentration normalt inte blir större än 10 ml/l. Till en särskild kolv med kontrollmedium tillsätts inga kemikalier.
För att vara säker på att inokulataktiviteten inte begränsas får koncentrationen av upplöst syre inte understiga 0,5 mg/l i BOD-kolvarna, vilket begränsar testkemikaliens koncentration till ca 2 mg/l. Av svårt nedbrytbara ämnen och ämnen med lågt ThOD kan det dock användas 5–10 mg/l. I vissa fall rekommenderas parallella serier med testämne i två olika koncentrationer, t.ex. 2 och 5 mg/l. Normalt beräknas ThOD på grundval av bildandet av ammoniumsalt, men om nitrifikation förväntas beräknas värdena på grundval av nitratbildning (ThODNO3; se bilaga II.2). Om ofullständig nitrifikation sker korrigeras det för ändringar i nitrit- och nitratkoncentrationen, som är bestämd genom analys (se bilaga V).
Ytterligare en serie kolvar krävs om testämnets toxicitet ska undersökas (t.ex. om ett lågt bionedbrytbarhetsvärde tidigare påvisats).
För detta förbereds ytterligare en stor kolv med luftat oorganiskt medium (ca en tredjedel full) plus testämne och referensämne normalt i samma slutkoncentration som i de andra kolvarna.
Inokulat tillsätts till kolvarna i form av sekundärt avloppsvatten (från 1 droppe [0,5 ml] till 5 ml per liter) eller t.ex. flodvatten (se I.6.4.2). Slutligen fylls det upp med luftat oorganiskt medium med användning av en slang som når ända ner till botten, så att tillräcklig omrörning säkerställs.
VI.2.5 Antal kolvar i en typisk testserie
I en typisk testserie används följande antal kolvar:
|
— |
minst 10 med testämne och inokulat (testuppslamning), |
|
— |
minst 10 med inokulat (inokulatblindprov), |
|
— |
minst 10 med referensämne och inokulat (procedurkontroll), |
|
— |
och i vissa fall 6 kolvar med testämne, referensämne och inokulat (toxicitetskontroll). För att säkert kunna identifiera 10-dagarsfönstet krävs dock dubbelt antal kolvar. |
VI.2.6 Genomförande av testet
De framställda lösningarna fördelas omedelbart i de berörda grupperna av BOD-kolvar, i det att de sugs upp från den nedersta fjärdedelen (inte botten) av den passande stora kolven med en slang, och således att alla BOD-kolvarna fylls helt. Eventuella luftbubblor avlägsnas genom lätta slag på kolvens sida. Starttidskolvarna analyseras omedelbart för upplöst syre antingen genom Winkler-metoden eller med elektrod. Innehållet i kolvarna kan förvaras för senare analys med Winkler-metoden om man omedelbart tillsätter mangan(II)sulfat och natriumhydroxid (den första Winkler-reagensen). De väl tillproppade kolvarna, där syret är fixerat som brunt hydratiserat mangan(III)oxid kan förvaras i mörker vid 10–20 oC i högst 24 timmar innan Winkler-testet avslutas. Resterande kolvar tillproppas och man ser till att inga luftbubblor innesluts, och de inkuberas vid 20 oC i mörker. Parallellt med varje serie ska det göras en fullständig serie för bestämning av inokulatblindvärden. Under de 28 dagarnas inkubering uttas med bestämda intervaller (minst en gång i veckan) minst 2 kolvar av varje serie för analys av upplöst syre.
Med veckovis provtagning skulle det procentuella avlägsnandet kunna bedömas i ett 14-dagarfönster, medan det vid provtagning var 3–4 dag, vilket skulle kräva ca dubbelt så många kolvar, blir möjligt att identifiera 10-dagarfönstret.
För N-haltiga ämnen måste det korrigeras för syreförbrukning vid eventuell nitrifikation. För detta används en O2-elektrod för bestämning av koncentrationen av upplöst syre, varefter det tas ett prov från BOD-kolven för analys av nitrit och nitrat. Den förbrukade syremängden beräknas på grundval av ökningen av nitrit- och nitrat-koncentrationen (se bilaga V).
VI.3 DATA OCH RAPPORTERING
VI.3.1 Behandling av resultat
Först beräknas BOD efter varje tidsintervall genom att dra ifrån syreförlusten (mg O2 per liter) i inokulatblindproven från syreförlusten i kolven med testämne. Denna korrigerade syreförlust divideras med testämnets koncentration (mg/l), varefter den specifika BOD framkommer i mg syre per mg testämne. Den procentuella bionedbrytbarheten beräknas genom att dividera den specifika BOD med den specifika ThOD (beräknad i enlighet med bilaga II.2) eller COD (bestämt genom analys, se bilaga II.3), dvs.

= mg O2 per mg testämne
![]()
eller
![]()
Observera att dessa två metoder inte nödvändigtvis ger samma resultat. Den först nämnda metoden rekommenderas.
Den ThOD (NH4 eller NO3) som motsvarar det föreliggande kännedomen om nitrifikation (bilaga II.2) används. Om ofullständig nitrifikation sker beräknas korrektionen för syreförbrukningen till nitrifikationen ut i från ändringarna i nitrit- och nitratkoncentrationen (bilaga V).
VI.3.2 Resultatens giltighet
Inokulatblindprovens syreförlust bör inte vara större än 1,5 mg upplöst syre per liter efter 28 dagar. Om det finns högre värden måste försöksteknikerna granskas kritiskt. Restkoncentrationen av syre i testkolvarna får inte understiga 0,5 mg/l vid något tillfälle. Mätningar av så lågt syreinnehåll är endast giltiga om den använda mätmetoden för upplöst syre tillåter noggrann mätning av så lågt innehåll.
Se också I.5.2.
VI.3.3 Rapportering
Se 1.8.
VI.4 FORMULÄR
Nedan visas ett exempel på ett formulär.
CLOSED BOTTLE TEST
|
1. |
LABORATORIUM |
|
2. |
DATUM VID INLEDNING AV TEST |
3. TESTÄMNE
Namn:
Stamlösningens koncentration: .... mg/l
Startkoncentration i kolven: .... mg/l
ThOD eller COD: ... mg O2/mg testämne
4. INOKULAT
Källa:
Behandling:
Eventuell förbehandling:
Koncentration i reaktionsblandningen: ... mg/l:
5. DOC-BESTÄMNING
Metod: Winkler/elektrod
Kolvanalys
|
Inkuberingstid (dagar) |
DO (mg/l) |
|||||
|
0 |
n1 |
n2 |
|
|||
|
Blindprov (utan testämne) |
1 |
C1 |
|
|
|
|
|
2 |
C2 |
|
|
|
|
|
|
Genomsnitt |
|
|
|
|
|
|
|
Testämne |
1 |
a1 |
|
|
|
|
|
|
2 |
a2 |
|
|
|
|
|
Genomsnitt |
|
|
|
|
|
|


Anmärkning: En motsvarande uppställning kan användas för referenskemikalien och toxicitetskontrollproverna.
6. KORREKTION FÖR NITRIFIKATION (se bilaga V)
|
Inkuberingstid (dagar) |
0 |
n1 |
n2 |
n3 |
||
|
|
|
|
|
||
|
— |
|
|
|
||
|
— |
|
|
|
||
|
|
|
|
|
||
|
— |
|
|
|
||
|
— |
|
|
|
||
|
— |
|
|
|
7. FÖRLUST AV DO: % NEDBRYTNING
|
|
Förlust efter n dagar (mg/l) |
|||
|
n1 |
n2 |
n3 |
|
|
|
KOLV 1: (mto – mtx) – (mbo – mbx) |
|
|
|
|
|
KOLV 2: (mto – mtx) – (mbo – mbx) |
|
|
|
|
|
KOLV 1:
|
|
|
|
|
|
KOLV 2:
|
|
|
|
|
| % D genomsn. (8) =
|
|
|
|
|



|
mto |
= |
värde i testkolven vid tid 0 |
|
mtx |
= |
värde i testkolven vid tid x |
|
mbo |
= |
genomsnittsblindvärde vid tid 0 |
|
mbx |
= |
genomsnittsblindvärde vid tid x |
Dessutom företas korrektion för nitrifikation i enlighet med punkt 6.
8. BLINDPROVER FÖR FÖRLUST AV DO
Syreförbrukningen i blindprov: (mbo – mb28) mg/l. Denna förbrukning är av betydelse för testets giltighet och får högst vara 1,5 mg/l.
DEL VII. MITI TEST (metod C.4-F)
VII.1 TESTMETODSPRINCIP
Syreupptagningen i en omrörd lösning eller uppslamning av testämnet i oorganiskt medium, som är tillsatt inokulat i form av särskild odlade ej tillvanda mikroorganismer, mäts automatiskt under 28 dagar i en mörklagd sluten respirometer vid 25 ± 1 oC. Den utvecklade koldioxiden absorberas i natriumhydroxid. Bionedbrytbarheten uttrycks som den upptagna syremängden (korrigerat för blindprovens upptagning) i procent av den teoretiska syreupptagningen (ThOD). Även den procentuella primära bionedbrytbarheten beräknas, på grundval av kompletterande specifika kemiska analyser vid inkuberingens början och slut och eventuellt genom DOC-analys.
VII.2 TESTMETODSBESKRIVNING
VII.2.1 Apparatur
|
a) |
Automatisk elektrolytisk BOD-mätare eller respirometer, normalt med 6 kolvar på vardera 300 ml och med skålar för CO2-absorbtionsmedel. |
|
b) |
Lokal med konstant temperatur och vattenbad på 25 ± 1 oC eller mindre. |
|
c) |
Membranfiltreringsutrustning (valfritt). |
|
d) |
Kolanalysator (valfritt). |
VII.2.2 Framställning av oorganiskt medium
Följande stamlösningar framställs av reagenser av analyskvalitet och vatten (I.6.1):
|
a) |
monokaliumdihydrogenorthofosfat, KH2PO4 |
8,50 g |
|
dikaliummonohydrogenorthofosfat, K2HPO4 |
21,75 g |
|
|
dinatriummonohydrogenorthofosfat dodecahydrat, Na2HPO4 12 H2O |
44,60 g |
|
|
ammoniumklorid, NH4CI |
1,70 g |
|
|
Upplöses i vatten och förtunnas till 1 liter. |
|
|
|
Upplösningens pH ska vara 7,2. |
|
|
|
b) |
magnesiumsulfat heptahydrat, MgSO4 7 H2O |
22,50 g |
|
Upplöses i vatten och förtunnas till 1 liter. |
|
|
|
c) |
Kalciumklorid, vattenfritt, CaCl2 |
27,50 g |
|
Upplöses i vatten och förrunnas till 1 liter. |
|
|
|
d) |
Järn(III)klorid hexahydrat, FeCl3 6 H2O |
0,25 g |
|
Upplöses i vatten och förtunnas till 1 liter. |
|
3 ml tas av lösning a, b, c och d och förtunnas till 1 liter.
VII.2.3 Framställning av inokulat
Friska prover samlas in från minst 10 platser, huvudsakligen områden där det används och utleds olika kemikalier. Från sådana platser som reningsverk, floder, sjöar och havsområden insamlas 1-liters prover av slam, ytjord, vatten, etc. och allt blandas ordentligt. Material som flyter ovanpå avlägsnas och efter ett tag anpassas supernatanten till pH 7 ± 1 med natriumhydroxid och fosforsyra.
En passande mängd filtrat används för att fylla en ”fill-and-draw” aktiv slambehållare, och vätskan luftas i ca 23 timmar 30 minuter. 30 minuter efter det att luftningen avslutats hälls omkring en tredjedel av den samlade supernatant-volymen bort och det tillsätts en lika stor mängd av en lösning (med pH 7) som innehåller 0,1 % glukos, 0,1 % pepton och 0,1 % monokaliumorthoforfat, och luftningen påbörjas. Detta sker varje dag. Slamenheten drivs efter god praxis: avloppsvattnet ska vara klart, temperaturen ska hållas vid 25 ± 2 oC, pH ska vara 7 ± 1, slam ska bottenfällas bra, tillräcklig luftning ska upprätthållas för att hålla blandningen konstant aerob, protozoer ska finns till hands och slamaktiviteten ska testas mot referensämnet minst var tredje månad. Slammet får tidigast användas som inokulat efter en månads drift och inte senare än efter fyra månaders drift. Därefter uttas regelbundet prover från minst 10 platser var tredje månad.
För att hålla det friska och det gamla slammet på samma aktivitet blandas den filtrerade supernatanten från aktivt slam genom användning av lika mycket filtrerat supernatant från en färskt insamlat blandning från 10 källor, och denna vätska odlas i enlighet med ovan. 18–24 timmar efter det att enheten fyllts upp kan det tas slam för användning som inokulat.
VII.2.4 Förberedelse av kolvar
Sex kolvar förbereds enligt följande:
Nr 1: testämne i förtunningsvatten, 100 mg/l
Nr 2, 3 och 4: testämne förtunnat med oorganiskt medium till 100 mg/l
Nr 5: referensämne (t.ex. anilin) förtunnat med oorganiskt medium till 100 mg/l
Nr 6: rent oorganiskt medium
Svårlösliga testämnen tillsätts direkt på vikt- eller volymbasis eller hanteras i enlighet med bilaga III, bortsett från att varken lösningsmedel eller emulgatorer får användas. CO2-absorptionsmedel placeras i alla kolvar i den särskilda skålen. pH i kolv nr 2, 3 och 4 justeras till 7,0.
VII.2.5 Genomförandet av testet
En liten mängd inokulat tillsätts till kolv nr 2, 3 och 4 (testuppslamningar), nr 5 (aktivitetskontroll) och nr 6 (inokulatblinprov) så att koncentrationen av uppslammat torrämne blir 30 mg/l. Inokulat tillsätts inte till kolv nr 1 som tjänar som abiotisk kontroll. Utrustningen samlas ihop, lufttätheten kontrolleras, omrörarna startas och syreupptagningsmätningen startas i mörker. Temperaturen, omrörningen och den coulometeriska syreupptagningsskrivaren kontrolleras dagligen och eventuella färgändringar i kolvarna noteras. Syreupptagningen i de 6 kolvarna avläses direkt med hjälp av en passande metod, t.ex. på sexkanalsskrivaren, som ger en BOD-kurva. Vid inkuberingens slut, normalt efter 28 dagar, mäts pH i kolvarna, och koncentrationen av resterande testämne och av eventuella biprodukter och, när det gäller vattenlösliga ämnen, eventuellt DOC-koncentrationen bestäms (bilaga II.4). Särskild försiktighet måste iakttas om det är tal om flyktiga kemikalier. Om nitrifikation förväntas bestäms om möjligt nitrat- och nitritkoncentrationen.
VII.3 DATA OCH RAPPORTERING
VII.3.1 Behandling av resultat
Testämnets syreförbrukning (mg) efter en viss tid, korrigerat för syreupptagningen i inokulatblinproven efter samma tid, divideras med den använda viktmängden testämne. Av detta framkommer BOD uttryck som mg syre per mg testämne, dvs.

= mg O2 per mg testämne
Den procentuella bionedbrytningen framkommer av:

För blandningar beräknas ThOD på samma sätt som för enskilda ämnen, nämligen pä grundval av grundämnesanalysen. Det ThOD som används (ThODNH4 eller ThODNO3) beror på om nitrifikationen är frånvarande eller fullständig (bilaga II.2). Om ofullständig nitrifikation sker beräknas korrektionen för syreförbrukningen till nitrifikationen utifrån ändringarna i nitrit- och nitratkoncentrationen (bilaga V).
Den procentuella primära bionedbrytningen beräknas på grundval av den mängd av den specifika (ursprungliga) kemikalie som försvinner (se I.7.2).

Om förlust av testämne har skett i kolv nr 1, som mäter fysisk och kemisk nedbrytning, antecknas detta och testämnets koncentration (Sb) i denna kolv efter 28 dagar används för att beräkna den procentuella bionedbrytningen.
Om (valfria) bestämningar görs av DOC beräknas den procentuella fullständiga bionedbrytningen genom:

i enlighet med punkt I.7.1. Om testämne (DOC) har försvunnit från kolv nr 1, som mäter fysisk och kemisk nedbrytning, används DOC-koncentrationen i denna kolv för att beräkna den procentuella bionedbrytningen.
Alla resultat noteras på de tillhörande formulären.
VII.3.2 Resultatens giltighet
Inokulatblinprovens syreförbrukning är normalt 20–30 mg O2/l efter 28 dagar och bör inte vara större än 60 mg/l. Om det finns värden över 60 mg/l måste data och försöksteknikerna granskas kritiskt. Om pH ligger utanför intervallet 6–8,5 och testämnets syreförbrukning är mindre än 60 % upprepas testet med lägre testämneskoncentrationer.
Se även I.5.2.
Om den procentuella nedbrytningen av anilin, beräknat utifrån syreförbrukningen, inte är större än 40 % efter 7 dagar och 65 % efter 14 dagar anses testet vara ogiltigt.
VII.3.3 Rapportering
Se I.8.
VII.4 RESULTATFORMULÄR
Nedan visas ett exempel på ett formulär.
MITI (I) TEST
|
1. |
LABORATORIUM |
|
2. |
DATUM VID INLEDNING AV TEST |
3. TESTÄMNE
Namn:
Stamlösningens koncentration: .... mg/l ämne
Startkoncentration i kolven, C0: .... mg/l ämne
Reaktionsblandningens volym, V: .... ml
ThOD: ... mg O2/l
4. INOKULAT
Platser för provtagningen:
|
|
||||
|
|
||||
|
|
||||
|
|
||||
|
|
Koncentrationen av uppslammat torrämne i det aktiva slammet efter acklimatisering med syntetiskt avloppsvatten: .... mg/l.
Volym av aktivt slam per liter slutmedium: ... ml.
Slamkoncentration i slutmedium: … mg/l
5. SYREUPPTAGNING: BIONEDBRYTBARHET
Respirometertyp:
|
|
Tid (dagar) |
||||||
|
0 |
7 |
14 |
21 |
28 |
|||
|
O2 upptag. (mg) testämne |
a1 |
|
|
|
|
|
|
|
a2 |
|
|
|
|
|
||
|
a3 |
|
|
|
|
|
||
|
O2 upptag. (mg) blindprov |
b |
|
|
|
|
|
|
|
Korrigerat O2 upptag. (mg) |
(a1 – b1) (a1 – b1) (a1 – b1) |
|
|
|
|
|
|
|
BOD per mg testämne |
|
Kolv 1 |
|
|
|
|
|
|
Kolv 2 |
|
|
|
|
|
||
|
Kolv 3 |
|
|
|
|
|
||
|
% nedbrytning
|
|
1 |
|
|
|
|
|
|
2 |
|
|
|
|
|
||
|
3 |
|
|
|
|
|
||
|
genomsn. (9) |
|
|
|
|
|
||


Anmärkning: En motsvarande uppställning kan användas för referenskemikalien och toxicitetskontrollproven.
6. KOLANALYS (valfritt)
Kolanalysator:
|
Kolv |
DOC |
% DOC avlägsnat |
Genomsnitt |
|||
|
Mätt |
Korrigerat |
|||||
|
Vatten + testämne |
a |
|
|
|
— |
— |
|
Slam + testämne |
b1 |
|
b1 – c |
|
|
|
|
Slam + testämne |
b2 |
|
b2 – c |
|
|
|
|
Slam + testämne |
b3 |
|
b3 – c |
|
|
|
|
Kontrollblind |
c |
|
— |
|
— |
— |

7. DATA FRÄN SPECIFIK KEMISK ANALYS
|
|
Restmängd av testämne vid testets avslutning |
% nedbrytning |
|
Blindprov med vatten |
Sb |
|
|
Medium med inokulat |
Sa1 |
|
|
Sa2 |
|
|
|
Sa3 |
|

% nedbrytning beräknas för kolv a1, a2 och a3.
8. ANMÄRKNINGAR
En kurva över BOD mot tiden ska bifogas, om sådan föreligger.
Bilaga I
FÖRKORTNINGAR OCH DEFINITIONER
|
DO |
: |
Upplöst syre (mg/l) definieras som koncentrationen av upplöst syre i ett vattnigt prov. |
|
BOD |
: |
Biokemisk syreförbrukning (g) definieras som den mängd syre som mikroorganismer förbrukar vid metabolism av testämnet. Resultatet uttrycks också som gram upptaget syre per gram testat ämne (se metod C.5). |
|
COD |
: |
Kemisk syreförbrukning (g) definieras som den mängd syre som förbrukas vid oxidering av testämnet med varm dikromat i sur vätska. Det är ett mått på den motsvarande mängden oxiderbart ämne; uttrycks också i g förbrukat syre per g testämne (se metod C.6). |
|
DOC |
: |
Upplöst organiskt kol definieras som den mängd organiskt kol som finns i lösningen, passerar genom ett 0,45 μm filter eller förblir i centrifugatet efter centrifugering vid 40 000 m • s-2 (±4 000 g) i 15 minuter. |
|
ThOD |
: |
Teoretisk syreförbrukning (mg) definieras som den samlade mängden syre som krävs för fullständig oxidering av ett kemiskt ämne. Det beräknas på grundval av bruttoformeln (se bilaga II, punkt 2) och uttrycks också i mg syre per mg testämne. |
|
ThCO2 |
: |
Teoretiskt koldioxid (mg) definieras som den mängd koldioxid som enligt beräkningar på grundval av testämnets kända eller mätta kolinnehåll skulle bildas vid fullständig nedbrytning av testämnet. Det uttrycks också i mg utvecklad koldioxid per mg testämne. |
|
TOC |
: |
Ett provs totala innehåll av organiskt kol definieras som summan av organiskt kol i lösning och uppslamning. |
|
IC |
: |
Oorganiskt kol. |
|
TC |
: |
Ett provs totala kolinnehåll definieras som summan av organiskt och oorganiskt kol. |
Primär bionedbrytning
definieras som en ändring av ett ämnes kemiska struktur, framkallad på biologisk väg, som resulterar i att ämnet mister sina specifika egenskaper.
Fullständig bionedbrytning (aerob)
definieras som den nedbrytningsgrad som har nåtts när mikroorganismer har förbrukat ett testämne fullständigt samtidigt som koldioxid, vatten, oorganiska salter och nya celldelar bildas (biomassa).
Lätt bionedbrytning
kännetecknar en godtycklig klass kemikalier som har klarat vissa specifika screeningtester för fullständig bionedbrytning. Dessa tester är så krävande att sådana ämnen snabbt kommer att fullständigt brytas ned i vattnig miljö under aeroba förhållanden.
Inherent (potentiell) bionedbrytbarhet
karakteriserar en klass kemikalier för vilka det finns klara bevis på bionedbrytning (primär eller fullständig) vid varje erkänt test av bionedbrytbarhet.
Behandlingsmöjlighet
definieras som möjligheten av avlägsna ett ämne genom biologisk avloppsvattenrening utan att avloppsvattenreningsprocessernas normala förlopp påverkas. Generellt är alla lätt bionedbrytbara ämnen behandlingsbara, men inte alla inherent bionedbrytbara ämnen. Abiotiska processer kan också inverka på detta.
Lag-period
definieras som den tid som går vid ett elimineringstest från inokuleringen tills dess att nedbrytningsprocenten är minst 10 %. Lag-perioden varierar ofta stort och är svår att reproducera.
Nedbrytningstid
definieras som tiden mellan lagperiodens slut och den tid då nedbrytningen har nått 90 % av sitt maximala värde.
10-dagarsfönstret
definieras som den period på 10 dagar som börjar när nedbrytningen har nått upp till 10 %.
Bilaga II
BERÄKNING OCH BESTÄMNING AV LÄMPLIGA HUVUDPARAMETRAR
Beroende på den metod som väljs kan olika huvudparametrar behövas. I detta avsnitt beskrivs hur dessa värden utleds. Parametrarnas användning beskrivs i de individuella metoderna.
1. Kolhalt
Kolhalten kan antingen beräknas på grundval av testämnets grundämnessammansättning eller bestämmas genom elementäranalys.
2. Teoretisk syreförbrukning (ThOD)
Den teoretiska syreförbrukningen (ThOD) kan beräknas, om grundämnessammansättningen är känd, och annars bestämmas genom elementäranalys. För föreningen
CcHhClclNnNanaOoPpSs
är det utan nitrifikation
 mg/mg
mg/mg
och med nitrifikation
 mg/mg
mg/mg
3. Kemisk syreförbrukning (COD)
Den kemiska syreförbrukningen (COD) bestäms enligt metod C.6.
4. Upplöst organiskt kol (DOC)
Upplöst organiskt kol (DOC) definieras som den organiska mängd kol i en kemikalie eller en kemikalieblandning som i vattnig lösning kan passera genom ett 0,45 μm filter.
Prover tas från testbehållarna och filtreras omedelbart genom filtreringsutrustningen som är försedd med ett passande membranfilter. De första 20 ml av filtratet kasseras (vid små filter kan denna mängd minskas). 10–20 ml samlas upp för kolanalys (vid insprutning mindre, beroende på den använda kolanalysatorn). DOC-koncentrationen bestäms med hjälp av en organisk kolämnesanalysator som gör det möjligt att noggrant bestämma en kolkoncentration på mindre än 10 % av startkoncentrationen av DOC i testet.
Om filtrerade prover inte kan analyseras samma arbetsdag kan de förvaras i högst 48 timmar i kylskåp vid 2–4 oC eller längre tid vid – 18 oC.
Anmärkningar:
Membranfilter är ofta behandlade med ytaktiva ämnen för att göra dem mer hydrofila. De kan därför innehålla upp till flera mg upplösligt organiskt kol som kommer att påverka bionedbrytbarhetsbestämningen. De ytaktiva ämnena och andra upplösliga organiska ämnen avlägsnas från filtren genom kokning i avjoniserat vatten i tre gånger 1 timme. Om engångsfilter används måste varje parti kontrolleras för att bekräfta att de inte avger upplösligt organiskt kol.
På vissa typer av membranfilter kan absorption av testämnet inträffa. Kontroll av att testämnet inte hålls kvar på filtret rekommenderas därför.
Centrifugering vid 40 000 ms-2 (4 000 g) i 15 minuter kan användas i stället för filtrering när man ska skilja mellan TOC och DOC. Metoden är inte pålitlig vid en startkoncentration av < 10 mg DOC per liter, då antingen inte alla bakterier avlägsnas eller något av kolet i bakteriernas cytoplasma återupplöses.
HÄNVISNINGAR
|
— |
Standard Methods for the Examination of Water and Wastewater, 12th ed., Am. Pub. Hlth. Ass., Am. Wat. Poll. Control Fed., Oxygen Demand, 1965, s. 65. |
|
— |
Wagner, R., Von Wasser, 1976, Vol. 46, s. 139. |
|
— |
DIN-Entwurf 38 409 Teil 412 – Deutsche Einheitsverfahren zur Wasser-, Abwasser- und Schlammuntersuchung, Summarisches Wirkungs- und Stoffkenngröβen (Gruppe H). Bestimmung des Chemischen Sauer-stoffbedarfs (CSB) (H 41), Normenausschüβ Wasserwesen (NAW) in DINN Deutsche Institut für Normung e.V. |
|
— |
Gerike, P., The biodegradability testing of poorly water soluble compounds. Chemosphere, 1948, Vol. 13 (1), s. 169. |
Bilaga III
BEDÖMNING AV SVÅRLÖSLIGA ÄMNENS BIONEDBRYTBARHET
Vid test av svårlösliga ämnens bionedbrytbarhet bör särskild uppmärksamhet ges åt nedanstående.
Trots att homogena vätskor sällan kommer att vålla problem vid provtagningen rekommenderas att homogenisera fasta material på lämpligt sätt, så att fel till följd av inhomogenitet undviks. Särskild omsorg ska ske vid uttagning av ett representativt prov på några få mg av blandningar av kemikalier och av ämnen som innehåller stora mängder orenheter.
Under testet kan man använda olika metoder för omrörning. Omrörningen ska dock vara så kraftig att kemikalien hålls dispergerad, och att överhettning, stark skumning och våldsam förskjutning undviks.
En emulgator kan användas för att framställa en stabil dispersion av kemikalien. Emulgatoren får inte vara toxisk för bakterier och får varken kunna brytas ned biologiskt eller vara skumbildande under testförhållandena.
Samma kriterier som för emulgatorer gäller för lösningsmedel.
Användning av fasta bärarmaterial till fasta testämnen rekommenderas inte, däremot kan de vara lämpliga till oljeaktiga ämnen.
När tillsatsämnen såsom emulgatorer, lösningsmedel och bärarmaterial används utförs ett blindprov med tillsatsämnen.
Alla tre respirometertesterna, CO2, BOD och MITI kan användas för att undersöka svårlösliga ämnens bionedbrytbarhet.
HÄNVISNINGAR
|
— |
de Morsier, A. et al., Biodegradation tests for poorly-soluble compounds. Chemosphere, 1987, Vol. 16, s. 833. |
|
— |
Gerike, P., The Biodegradation of poorly water soluble compounds. Chemosphere, 1984, Vol. 13, s. 169. |
Bilaga IV
BEDÖMNING AV BIONEDBRYTBARHETEN HOS KEMISKA ÄMNEN SOM MISSTÄNKS VARA TOXISKA FÖR INOKULAT
Om ett kemiskt ämne som underkastas ett test för lätt bionedbrytbarhet inte verkar vara bionedbrytbart rekommenderas följande förfarande om man vill skilja mellan hämning av bristande bionedbrytbarhet (Reynolds et al., 1987).
Samma eller motsvarande inokulat används för toxicitetstest som för bionedbrytbarhetstest.
För att kunna bedöma toxiciteten hos kemikalier som undersöks i test av lätt bionedbrytbarhet kan man använda en eller en kombination av följande metoder: Hämning av slammets respirationshastighet (aktiverat slam – respirationshämningstest – direktiv 88/302/EEG); BOD och tillväxthämningstest.
Om hämning till följd av toxicitet ska undvikas rekommenderas användning av testämneskoncentrationer i test av lätt bionedbrytbarhet, som högst är 1/10 av de EC50-värden (eller högst de EC20-värden) som erhållits vid toxicitetstest. Ämnen vars EC50 är större än 300 mg/l kommer troligtvis inte att ha toxiska verkningar i test för lätt bionedbrytbarhet.
Ett EC50 på mindre än 20 gl/l kommer troligtvis att skapa betydande svårigheter vid den efterföljande testningen. Testkoncentrationerna ska vara låga, vilket kräver användning av det strikta och känsliga Closed Bottle-testet eller av 14C-markerat material. Alternativt kan det vid tillvänjning av inokulat bli möjligt att använda högre testämneskoncentrationer. I det sistnämnda fallet mister testet för lätt bionedbrytbarhet dock sin specifika karaktär.
HÄNVISNINGAR
Reynolds, L. et al., Evaluation of the toxicity of substances to be assessed for biodegradability. Chemosphere, 1987, Vol. 16, s. 2259.
Bilaga V
KORREKTION AV NITRIFIKATIONENS INTERFERENS PÅ SYREUPPTAGNINGEN
Fel till följd av att inte ta hänsyn til nitrifikation vid bedömning av bionedbrytbarheten hos testämnen, som inte innehåller N, är minimal (högst 5 %) även om oxidationen av mediets innehåll av ammonium-N sker sporadiskt från testprovet till blindprovet. För testämnen som innehåller N kan dock väsentliga fel uppstå.
Om ofullständig nitrifikation sker kan reaktionsblandningens iakttagna syreupptagning korrigeras för den syremängd som har förbrukats för oxidation av ammonium till nitrit och nitrat om ändringarna i nitrit- och nitrat-koncentrationen under inkuberingen bestäms med hänsyn tagen till följande reaktionsutjämningar:
|
2NH4C1 + 3 O2 = 2 HNO2 + 2 HC1 + 2 H2O |
(1) |
|
2 HNO2 + O2 = 2 HNO3 |
(2) |
|
brutto: |
|
|
2 NH4Cl + 4 O2 = 2 HNO3 + 2 HCl + 2 H2O |
(3) |
Från ekvation (1) blir syreupptagningen vid oxidation av 28 g kväve i form av ammoniumklorid (NH4C1) till nitrit 96 g, dvs. en faktor av 3,43 (96/28). På motsvarande sätt blir syreupptagningen vid oxidation av 28 g kväve till nitrat (3) 128 g, dvs. en faktor av 4,57 (128/28).
Eftersom reaktionerna är sekvensiella, då de sker på grund av deras individuella bakteriearter, kan nitratkoncentrationen öka eller minska. I det sistnämnda fallet kommer en ekvivalent mängd nitrat att bildas. Till följd av detta blir syreförbrukningen vid nitratbildning 4,57 gånger ökningen i nitratkoncentrationen, men den syremängd som är involverad i nitritbildningen blir dock 3,43 gånger ökningen av nitritkoncentrationen, eller vid en minskning av nitritkoncentrationen, en syreförlust av -3,43 gånger koncentrationsminskningen.
Dvs.
|
O2-förbrukningen vid nitratbildning = 4,57 × N-nitratkoncentrationsökningen |
(4) |
|
och |
|
|
O2-förbrukningen vid nitritbildning = 3,43 × N-nitritkoncentrationsökningen |
(5) |
|
och |
|
|
O2-förlusten vid nitritförsvinnande = -3,43 × N-nitritkoncentrationsminskningen |
(6) |
|
och därför blir |
|
|
O2-förbrukningen till nitrifikation = ±3,43 × N-nitntkoncentrationsändringen + 4,57 × N-nitratkoncentrationsökningen |
(7) |
|
och därmed |
|
|
O2-förbrukningen till C-oxidation = total iakttagen förbrukning – förbrukning till nitrifikation |
(8) |
Alternativt kan syreupptagningen till följd av nitrifikation som en första uppskattning sättas till 4,57 × tillväxten av oxiderat N, om endast totalt oxiderat N bestäms.
Det korrigerade värdet för syreförbrukningen till C-oxidation jämförs sedan med ThOD NH3, beräknat enligt bilaga II.
C.5 NEDBRYTNING – BIOKEMISK SYREFÖRBRUKNING
1. METOD
1.1 INLEDNING
Syftet med denna metod är att mäta den biokemiska syreförbrukningen (BOD) för fasta eller flytande organiska ämnen.
Data som erhålls vid detta test avser vattenlösliga föreningar. Dock kan, åtminstone principiellt, även flyktiga föreningar undersökas.
Metoden kan endast tillämpas pä organiska testämnen som vid de koncentrationer som används i testet inte inverkar hämmande på bakterier. Om testämnet inte är lösligt vid testkoncentrationen kan det vara nödvändigt att vidta särskilda åtgärder, t.ex. att använda ultraljudsdispersion, för att uppnå en god dispersion av testämnet.
Uppgifter om testämnets toxicitet för mikroorganismer kan vara till nytta vid tolkningen av låga resultat och vid valet av lämpliga testkoncentrationer.
1.2 DEFINITIONER OCH ENHETER
BOD definieras som den mängd upplöst syre som krävs för den biokemiska oxideringsprocessen för en angiven mängd lösning av testämnet under testbetingelser.
Resultaten uttrycks som gram BOD per gram testat ämne.
1.3 REFERENSÄMNEN
Det är önskvärt att lämpligt kontrollämne används för att kontrollera inokulatets aktivitet.
1.4 TESTMETODSPRINCIP
En förbestämd mängd av ämnet, upplöst eller dispergerat i ett väl syrsatt medium, inokuleras med mikroorganismer och inkuberas i mörker vid en konstant, definierad passande temperatur.
BOD bestäms genom skillnaden mellan mängden upplöst syre i början och i slutet av testet. Testet ska pågå i minst fem dagar och högst 28 dagar.
Parallellt med testet ska utföras ett blindprov utan innehåll av testämnet.
1.5 KVALITETSKRITERIER
BOD-bestämningen kan inte anses vara en giltig bestämning av ett ämnes bionedbrytbarhet. Detta test kan endast betraktas som ett screenmg-test.
1.6 METODBESKRIVNING
En preliminär lösning eller dispersion av ämnet förbereds för att uppnå en BOD-koncentration som är förenlig med den använda metoden. BOD bestäms därefter enligt lämplig nationell eller internationell standardmetod.
2. DATA OCH UTVÄRDERING
BOD-innehållet i lösningen beräknas enligt den valda standardmetoden och omvandlas till gram BOD per gram testat ämne.
3. RAPPORTERING
Den använda metoden ska anges.
Den biokemiska syreförbrukningen ska vara ett medeltal av minst tre giltiga mätningar.
Alla uppgifter och upplysningar som är av betydelse för tolkningen av resultaten ska rapporteras, särskilt avseende ämnets sammansättning, orenheter, fysiskt tillstånd och toxiska effekter som kan påverka resultaten.
Eventuella tillsatser som använts för att hämma biologisk nitrifikation ska rapporteras.
4. HÄNVISNINGAR
Lista över standardiserade metoder, t.ex.:
|
|
NF T 90-103: Determination of the biochemical oxygen demand. |
|
|
NBN 407: Biochemical oxygen demand. |
|
|
NEN 3235 5.4: Bepaling van het biochemish zuurstofverbruik (BZV). |
|
|
The determination of biochemical oxygen demand, Methods for the examination of water and associated materials, HMSO, London. |
|
|
ISO 5815: Determination of biochemical oxygen demand after n days. |
C.6 NEDBRYTNING – KEMISK SYREFÖRBRUKNING
1. METOD
1.1 INLEDNING
Syftet med denna metod är att mäta den kemiska syreförbrukningen (COD) för fasta eller flytande organiska ämnen på ett godtyckligt standardsätt under givna laboratoriebetingelser.
Uppgifter om ämnets formel är till nytta vid utförandet av testet och tolkningen av resultaten (t.ex. halogensalter, ferrosalter av organiska föreningar, organiska klorföreningar).
1.2 DEFINITIONER OCH ENHETER
Den kemiska syreförbrukningen är ett mått på ett ämnes förmåga att oxideras, uttryckt som den ekvivalenta mängden syre i en oxiderande reagens som förbrukas av ämnet under givna laboratoriebetingelser.
Resultatet uttrycks i gram COD per gram testat ämne.
1.3 REFERENSÄMNEN
Referensämnen behöver inte användas i samtliga fall när ett nytt ämne ska undersökas. Referensämnen ska först och främst användas till att emellanåt kalibrera metoden och till att möjliggöra en jämförelse av resultaten när en annan metod tillämpas.
1.4 TESTMETODSPRINCIP
En förbestämd mängd av ämnet, upplöst eller dispergerat i vatten, oxideras av kaliumbikromat i ett starkt svavelsyremedium med silversulfat som katalysator under återflöde i två timmar. Restmängden bikromat bestäms genom titrering med standard järnammoniumsulfat.
Vid klorhaltiga ämnen tillsätts kvicksilversulfat (10) för att reducera inverkan av klor.
1.5 KVALITETSKRITERIER
Till följd av det godtyckliga sättet att bestämma COD ska resultatet mera betraktas som en ”indikator på oxideringsförmågan” än som ett mått på mängden organiskt ämne.
Klorid kan inverka på detta test. Oorganiska reducerande eller oxiderande ämnen kan också inverka på BOD-bestämningen.
Vissa cykliska föreningar oxideras inte till fullo vid detta test.
1.6 METODBESKRIVNING
En inledande lösning eller dispersion av ämnet bereds så att ett COD mellan 250 och 600 mg per liter erhålls.
Anmärkningar:
Vid svårlösliga ämnen och ämnen som inte dispergerar kan finpulvriserat eller flytande ämne motsvarande ungefär 5 mg COD överföras till försöksapparaten tillsammans med vatten.
Den kemiska syreförbrukningen (COD) bestäms ofta, och särskilt för svårlösliga ämnen, i förväg med en variant av metoden, nämligen ett slutet system med tryckutjämning (H. Kelkenberg, 1975). Med denna modifikation kan det ofta med framgång företas kvantitativ bestämning av föreningar som endast med svårighet kan bestämmas genom konventionella metoder, t.ex. ättiksyra. Metoden duger dock inte för pyridin. Ökning av kaliumdikromatkoncentrationen till 0,25 N (0,0416 M) i enlighet med hänvisning 1 kan underlätta den direkta invägningen av 5–10 mg ämne, vilket är ett villkor för COD-bestämning på ämnen som är svårlösliga i vatten (hänvisning 2).
För övrig bestäms COD därefter med hjälp av en lämplig nationell eller internationell standardmetod.
2. DATA OCH UTVÄRDERING
COD-innehållet i testkolven beräknas enligt den valda standardmetoden och omvandlas till gram COD per gram testat ämne.
3. RAPPORTERING
Den använda referensmetoden ska anges.
Den kemiska syreförbrukningen ska vara ett medeltal av minst tre mätningar. Alla uppgifter och upplysningar som är av betydelse för tolkningen av resultaten ska rapporteras, särskilt avseende orenheter, ämnets fysiska tillstånd och egenskaper (om de är kända) som kan påverka resultaten.
Om kvicksilversulfat har använts för att minimera inverkan av klor ska detta rapporteras.
4. HÄNVISNINGAR
|
1. |
Kelkenberg, H. Z., von Wasser und Abwasserforschung, 1975, vol. 8., s. 146. |
|
2. |
Gerike, P. The biodegradability testing of poorly water soluble compounds. Chemosphere, 1984, vol. 13, s. 169. Lista över standardiserade metoder, t.ex.:
|
C.7 Nedbrytning – ABIOTISK NEDBRYTNING: Hydrolys som funktion av pH
1. METOD
Denna testmetod motsvarar OECD TG 111 (2004).
1.1 INLEDNING
Kemikalier kan nå ytvatten genom direktutsläpp, vindavdrift, avrinning, dränering, avfallsdeponering, avloppsvatten från industrier, hushåll eller jordbruk och genom atmosfärisk deposition, och de kan där omvandlas genom kemiska (t.ex. hydrolys, oxidation), fotokemiska och mikrobiella processer. I denna riktlinje beskrivs en testmetod för bedömning av abiotisk hydrolytisk omvandling av kemikalier i akvatiska system vid pH-värden som normalt förekommer i miljön (pH 4–9). Riktlinjen grundar sig på nuvarande riktlinjer (1) (2) (3) (4) (5) (6) (7).
Försöken utförs för att fastställa dels i) testsubstansens hydrolyshastighet som funktion av pH, dels ii) vilken typ av hydrolysprodukter som organismer kan exponeras för samt dessas bildnings- och nedbrytningshastighet. Sådana studier kan vara nödvändiga för kemikalier som släpps ut direkt i vatten eller som kan komma att nå miljön genom de andra vägar som nämns ovan.
1.2 DEFINITIONER OCH ENHETER
Se bilaga 2.
1.3 METODENS ANVÄNDBARHET
Metoden kan generellt användas för kemiska ämnen (omärkta eller märkta) för vilka det finns en tillräckligt noggrann och känslig analysmetod. Den kan användas för halvflyktiga och ickeflyktiga ämnen som är tillräckligt vattenlösliga. Testet bör inte användas för kemikalier som är lättflyktiga från vatten (t.ex. gasningsmedel, organiska lösningsmedel) och som därför inte kan hållas i lösning under de förhållanden som råder under testet. Testet kan vara svårt att utföra med ämnen som har mycket låg löslighet i vatten (8).
1.4 PRINCIP FÖR TESTMETODEN
Sterila vattenbaserade buffertlösningar med olika pH-värde (pH 4, 7 och 9) behandlas med testsubstansen och inkuberas i mörker under kontrollerade försöksbetingelser (vid konstant temperatur). Med lämpliga tidsintervall bestäms halterna av testsubstansen och hydrolysprodukter i buffertlösningarna. Om testsubstansen är märkt (t.ex. med 14C) så blir det lättare att upprätta en massbalans.
I testet används en stegvis metod som visas och förklaras i bilaga 1. Varje teststeg utlöses av resultaten från föregående steg.
1.5 INFORMATION OM TESTSUBSTANSEN
Omärkta eller märkta testsubstanser kan användas vid mätning av hydrolyshastigheten. Märkt material är i regel att föredra vid studier av reaktionsvägar och för att upprätta massbalans. I vissa speciella fall är dock märkning inte helt nödvändig. 14C-märkning rekommenderas, men andra isotoper, t.ex. 13C, 15N, 3H, kan också vara användbara. Om möjligt bör märkningen placeras i molekylens mest stabila del(ar). Om det undersökta ämnet exempelvis innehåller en ring så ska märkningen sitta på denna ring. Om ämnet innehåller två eller flera ringar kan det krävas särskilda studier för att utvärdera vad som händer med varje enskild märkt ring och för att få mer information om bildningen av hydrolysprodukter. Testsubstansens renhet bör vara minst 95 %.
Innan ett hydrolystest utförs ska följande uppgifter om testsubstansen finnas tillgängliga:
|
a) |
Löslighet i vatten (testmetod A.6) |
|
b) |
Löslighet i organiska lösningsmedel |
|
c) |
Ångtryck (testmetod A.4) och/eller Henrys konstant |
|
d) |
Fördelningskoefficient n-oktanol/vatten (testmetod A.8) |
|
e) |
Dissociationskonstant (pKa) (OECD:s riktlinje 112) (9) |
|
f) |
Nedbrytningshastighet vid direkt och indirekt fotolys i vatten, i tillämpliga fall |
Analysmetoder för kvantifiering av testsubstansen och, om så behövs, för identifiering och kvantifiering av hydrolysprodukter i vattenlösningar ska finnas tillgängliga (se även avsnitt 1.7.2).
1.6 REFERENSSUBSTANSER
Om möjligt bör referenskemikalier användas för att identifiera och kvantifiera hydrolysprodukter med hjälp av spektroskopiska och kromatografiska metoder eller andra metoder med lämplig känslighet.
1.7 KVALITETSKRITERIER
1.7.1 Utbyte
Analys av minst två identiska prov med buffertlösning eller extrakt av dessa omedelbart efter tillsättning av testsubstansen ger en första indikation på analysmetodens repeterbarhet och på hur enhetligt sättet att tillföra testsubstansen är. Under försökens senare steg erhålls uppgifter om utbyte med hjälp av respektive massbalanser (om märkta material används). Utbytet bör ligga mellan 90 och 110 % för märkta och omärkta kemikalier (7). Om det är tekniskt svårt att få så högt utbyte så kan ett utbyte på 70 % godtas för omärkta kemikalier, men i så fall bör motivering ges.
1.7.2 Analysmetodens repeterbarhet och känslighet
Repeterbarheten hos de analysmetoder som används för att kvantifiera testsubstansen och hydrolysprodukterna i senare skeden kan bestämmas genom analys av dubbelprover av samma buffertlösningar (eller extrakt av dessa) efter det att hydrolysprodukter har bildats i tillräckliga mängder för kvantifiering.
Analysmetoden bör vara så känslig att det är möjligt att kvantifiera testsubstansen i koncentrationer som är högst 10 % av den ursprungliga koncentrationen. I tillämpliga fall bör analysmetoderna också vara så känsliga att det är möjligt att kvantifiera hydrolysprodukter som motsvarar minst 10 % av tillsatsen (i varje skede av undersökningen) och ned till högst 25 % av den högsta koncentrationen.
1.7.3 Konfidensintervall för kinetiska data för hydrolysen
Konfidensintervall ska beräknas och redovisas för alla regressionskoefficienter, hastighetskonstanter, halveringstider och andra kinetiska parametrar (t.ex. DT50).
1.8 BESKRIVNING AV TESTMETODEN
1.8.1 Utrustning
Försöken bör göras i glaskärl (t.ex. provrör eller små kolvar) under mörka och sterila förhållanden om så krävs. Undantag görs om förhandsuppgifter (exempelvis fördelningskoefficienten n-oktanol/vatten) indikerar att testsubstansen kan fastna på glas. I sådana fall bör ett alternativt material (till exempel teflon) övervägas. Problemet med vidhäftning till glas kan eventuellt avhjälpas med en eller flera av följande metoder:
|
— |
Bestämning av den massa av testsubstans och hydrolysprodukter som fastnar på provkärlet. |
|
— |
Användning av ultraljudsbad. |
|
— |
Alla glaskärl tvättas med lösningsmedel vid varje provtagningsintervall. |
|
— |
Användning av bruksfärdiga produkter. |
|
— |
Användning av en större mängd hjälplösningsmedel för tillförsel av testsubstansen till systemet – hjälplösningsmedlet får dock inte leda till hydrolys av testsubstansen. |
Temperaturreglerade skakvattenbad eller termostatreglerade inkubatorer för inkubation av de olika testlösningarna krävs i normala fall.
Normal laboratorieutrustning krävs, och särskilt följande:
|
— |
pH-mätare. |
|
— |
Analysinstrument såsom gaskromatograf, vätskekromatograf och utrustning för tunnskiktskromatografi samt lämpliga detektionssystem för analys av radioaktivt märkta och omärkta ämnen eller metod för isotoputspädning. |
|
— |
Instrument för identifiering, t.ex. masspektrometer, gaskromatograf-masspektrometer, vätskekromatograf-masspektrometer, NMR-spektrometer. |
|
— |
Vätskescintillationsräknare. |
|
— |
Separertrattar för vätske-vätskeextraktion. |
|
— |
Instrument för att koncentrera lösningar och extrakt (t.ex. rotationsevaporator). |
|
— |
Utrustning för temperaturreglering (t.ex. vattenbad). |
Kemiska reagens, exempelvis följande:
|
— |
Organiska lösningsmedel av analytisk renhetsgrad (p.a.), t.ex. hexan, diklormetan. |
|
— |
Scintillationsvätska. |
|
— |
Buffertlösningar (för närmare uppgifter se avsnitt 1.8.3). |
Alla glaskärl, vatten av analytisk renhetsgrad (p.a.) och buffertlösningar som används i hydrolystestet bör vara sterila.
1.8.2 Tillförsel av testsubstans
Testsubstansen tillförs de olika buffertlösningarna i form av vattenlösning (se bilaga 3). För att underlätta upplösningen kan om så krävs små mängder lösningsmedel som är blandbara med vatten (såsom acetonitril, aceton eller etanol) tillåtas för tillförsel och fördelning av testsubstansen. Koncentrationen bör dock i normala fall inte överskrida en volymprocent. Högre koncentrationer av lösningsmedel (t.ex. när det gäller svårlösliga testsubstanser) kan tillåtas endast om det kan visas att lösningsmedlet inte påverkar hydrolysen av testsubstanserna.
Bruksfärdiga produkter bör inte användas rutinmässigt eftersom det inte kan uteslutas att de ingående beståndsdelarna kan påverka hydrolysprocessen. För testsubstanser som är svårlösliga i vatten eller binder till glas kan dock bruksfärdiga lösningar vara ett lämpligt alternativ.
En och samma koncentration av testsubstansen ska användas, och den bör inte överskrida 0,01 M eller halva mättnadskoncentrationen (se bilaga 1).
1.8.3 Buffertlösningar
Hydrolystestet utförs vid pH-värdena 4, 7 och 9. Buffertlösningar för testet bereds av kemikalier och vatten av analytisk renhetsgrad (p.a.). Några användbara buffertsystem beskrivs i bilaga 3. Det bör noteras att hydrolyshastigheten kan påverkas av det buffertsystem som används. Om sådan påverkan observeras bör ett alternativt buffertsystem användas (11).
I varje buffertlösning bör pH kontrolleras med en kalibrerad pH-mätare med en noggrannhet på ±0,1 eller bättre vid relevant temperatur.
1.8.4 Testförhållanden
1.8.4.1 Testtemperatur
Hydrolysförsöken ska utföras vid konstanta temperaturer. För extrapolering är det viktigt att temperaturen inte varierar mer än ±0,5 oC.
Ett inledande test (steg 1) utförs vid 50 oC om testsubstansens hydrolysegenskaper inte är kända. Kinetiska test i senare steg utförs vid minst tre temperaturer (inbegripet testet vid 50 oC) om inte testet i steg 1 visar att testsubstansen är stabil mot hydrolys. Förslagsvis görs testen i temperaturintervallet 10–70 oC (helst minst en temperatur under 25 oC), vilket omfattar rapporteringstemperaturen 25 oC och de flesta temperaturer som förekommer i miljön.
1.8.4.2 Ljus och syre
Alla hydrolystest bör utföras med en lämplig metod för att undvika fotolys. Alla lämpliga åtgärder bör vidtas för att avlägsna syrgas (t.ex. att bubbla med helium, kvävgas eller argon i fem minuter innan lösningen bereds.)
1.8.4.3 Testets varaktighet
Det inledande testet bör pågå i fem dagar, medan testen i senare steg bör pågå tills 90 % av testsubstansen är hydrolyserad, dock inte längre än 30 dagar.
1.8.5 Testets utförande
1.8.5.1 Inledande test (steg 1)
Det inledande testet utförs vid 50 ±0,5 oC och pH 4,0, 7,0 och 9,0. Om mindre än 10 % av hydrolysen har skett efter fem dagar (t0,5 25 oC > 1 år) så bedöms testsubstansen vara hydrolytiskt stabil och i normala fall krävs inga fortsatta tester. Om det är känt att ämnet är instabilt vid normala omgivningstemperaturer (12) så krävs inget inledande test. Analysmetoden måste ha tillräcklig precision och känslighet för att kunna påvisa en tioprocentig minskning av ursprungskoncentrationen.
1.8.5.2 Hydrolys av instabila ämnen (steg 2)
Testet i steg 2 utförs vid de pH-värden där testsubstansen var instabil enligt det inledande testet. Buffertlösningar av testsubstansen hålls vid de valda temperaturerna med hjälp av termostat. Vid test av reaktioner av första ordningen analyseras varje testlösning med tidsintervall som ger minst sex datapunkter som i normala fall är jämnt fördelade mellan 10 och 90 % hydrolys av testsubstansen. Individuella dubbelprov (minst två replikat, i separata reaktionskärl) tas ut och innehållet analyseras vid minst sex provtagningstillfällen (för att få minst tolv replikat av datapunkter). Det bedöms vara olämpligt att använda ett enda bulkprov från vilket enskilda alikvoter av testlösningen tas ut vid varje provtagningsintervall, eftersom detta förfarande inte gör det möjligt att analysera variationen i data och det dessutom kan medföra problem med kontaminering av testlösningen. Test för att bekräfta sterilitet bör utföras i slutet av steg 2 (dvs. vid 90 % hydrolys eller 30 dagar). Om ingen nedbrytning (dvs. omvandling) observeras behöver dock inga sterilitetstest göras.
1.8.5.3 Identifiering av hydrolysprodukter (steg 3)
Alla viktiga hydrolysprodukter, åtminstone de som motsvarar > 10 % av den tillförda dosen, bör identifieras med lämpliga analysmetoder.
1.8.5.4 Valfria test
Ytterligare test vid andra pH-värden än 4, 7 och 9 kan vara nödvändiga för hydrolytiskt instabila testsubstanser. Exempelvis kan det, för fysiologiska ändamål, vara nödvändigt att göra ett test vid surare förhållanden (t.ex. pH 1,2) och vid en enda fysiologiskt relevant temperatur (37 oC).
2. DATA
Mängderna av testsubstans och eventuella hydrolysprodukter bör anges i procent av den tillförda ursprungskoncentrationen eller om så är lämpligt i mg/l för varje provtagningsintervall och för varje pH-värde och testtemperatur. Dessutom bör en massbalans anges i procent av den tillförda ursprungskoncentrationen om en märkt testsubstans har använts.
Testsubstansens koncentration som funktion av tiden bör redovisas grafiskt med logaritmerade värden. Alla viktiga hydrolysprodukter, åtminstone de som motsvarar ≥ 10 % av den tillförda dosen, bör identifieras, och logaritmerade värden av deras koncentrationer bör redovisas grafiskt på samma sätt som för modersubstansen för att visa bildnings- och nedbrytningshastighet.
2.1 BEHANDLING AV RESULTATEN
Mer exakta bestämningar av halveringstiderna eller DT50 bör göras med hjälp av lämpliga kinetiska modeller. Halveringstid och DT50-värden (inklusive konfidensintervall) rapporteras för varje pH-värde och temperatur tillsammans med en beskrivning av den modell som använts, kinetisk ordningsgrad och bestämningskoefficient (r2). Om så är lämpligt bör motsvarande beräkningar göras för hydrolysprodukterna.
Om hastigheterna undersöks vid olika temperaturer bör hastighetskonstanterna (kobs) för hydrolys av pseudoförsta ordningen beskrivas som funktion av temperaturen. Beräkningen bör grundas både på en uppdelning av kobs i hastighetskonstanter för syrakatalyserad, neutral och baskatalyserad hydrolys (kH, kneutral, respektive kOH) och på Arrhenius ekvation:

där Ai och Bi är regressionskonstanter från skärningspunkten respektive lutningen för en anpassad linje som har erhållits genom linjär regression av ln ki mot det reciproka värdet av den absoluta temperaturen i Kelvin (T). Genom att använda Arrheniusrelationerna för syrakatalyserad, neutral och baskatalyserad hydrolys kan hastighetskonstanter av pseudo-första ordningen och därmed halveringstider beräknas för andra temperaturer för vilka en direkt experimentell bestämning av hastighetskonstanten inte är praktiskt genomförbar (10).
2.2 UTVÄRDERING OCH TOLKNING AV RESULTATEN
I de flesta hydrolysreaktioner är reaktionshastigheterna uppenbart av första ordningen och halveringstiderna är därför oberoende av koncentrationen (se ekvation 4 i bilaga 2). Detta innebär att laboratorieresultat som bestämts vid 10-2 till 10-3 M i allmänhet kan tillämpas på de förhållanden som råder i miljön (≤ 10-6 M) (10). Mabey och Mill (11) har rapporterat flera exempel på god överensstämmelse mellan hydrolyshastigheter uppmätta i både rena och naturliga vatten för ett stort antal kemikalier, förutsatt att både pH och temperatur hade mätts.
3. RAPPORTERING
3.1 TESTRAPPORT
Testrapporten ska innehålla åtminstone följande information:
Testsubstans:
|
— |
Trivialnamn, kemiskt namn, CAS-nummer, strukturformel (med angivelse av märkningsposition om radioaktivt märkt material används) och relevanta fysikaliskkemiska egenskaper (se avsnitt 1.5). |
|
— |
Testsubstansens renhet och eventuella föroreningar. |
|
— |
Renhet och aktivitet per mol hos den isotop som används för märkning (i tillämpliga fall). |
|
— |
Buffertlösningar. |
|
— |
Beredningsdag och andra uppgifter om beredningen. |
|
— |
Typ av buffertar och vatten. |
|
— |
Buffertlösningarnas molaritet och pH. |
Testförhållanden:
|
— |
Försöksdag. |
|
— |
Mängd tillförd testsubstans. |
|
— |
Metod och lösningsmedel (typ och mängd) för tillförsel av testsubstansen. |
|
— |
Inkuberad volym av buffertlösning med testsubstans. |
|
— |
Beskrivning av inkubationssystemet. |
|
— |
pH och temperatur under försöket. |
|
— |
Provtagningstidpunkter. |
|
— |
Extraktionsmetod(er). |
|
— |
Metoder för kvantifiering och identifiering av testsubstansen och hydrolysprodukter i buffertlösningarna. |
|
— |
Antal replikat. |
Resultat:
|
— |
Analysmetodernas repeterbarhet och känslighet. |
|
— |
Utbyten (procentvärden för en godtagbar undersökning anges i avsnitt 1.7.1). |
|
— |
Tabeller över data och medelvärden för replikat. |
|
— |
Massbalans under och i slutet av försöken (om märkta testsubstanser används). |
|
— |
Resultat av inledande test. |
|
— |
Diskussion och tolkning av resultaten. |
|
— |
Alla originaldata och siffror. |
Följande uppgifter krävs endast om hydrolyshastigheten har bestämts:
|
— |
Diagram över koncentration som funktion av tiden för testsubstanserna och i tillämpliga fall för hydrolysprodukterna vid varje pH-värde och temperatur. |
|
— |
Tabeller över resultaten av Arrhenius ekvation för temperaturen 20 oC/25 oC, med pH, hastighetskonstant [h-1 eller dag-1], halveringstid eller DT50, temperaturer ( oC) inklusive konfidensintervall samt korrelationskoefficienter (r2) eller motsvarande information. |
|
— |
Föreslagna reaktionsvägar för hydrolysen. |
4. HÄNVISNINGAR
|
1. |
OECD (1981), Hydrolysis as a Function of pH. OECD Guideline for Testing of Chemicals no 111, adopted 12 May 1981. |
|
2. |
US-Environmental Protection Agency (1982), 40 CFR 796.3500, Hydrolysis as a Function of pH at 25 oC. Pesticide Assessment Guidelines, Subdivision N. Chemistry: Environmental Fate. |
|
3. |
Agriculture Canada (1987), Environmental Chemistry and Fate Guidelines for registration of pesticides in Canada. |
|
4. |
Europeiska unionen (EU) (1995), kommissionens direktiv 95/36/EG om ändring av rådets direktiv 91/414/EEG om utsläppande av växtskyddsmedel på marknaden. Bilaga V: Omvandling, spridning och fördelning i miljön. |
|
5. |
Dutch Commission for Registration of Pesticides (1991), Application for registration of a pesticide. Section G: Behaviour of the product and its metabolites in soil, water and air. |
|
6. |
BBA (1980), Merkblatt Nr. 55, Teil I und II: Prüfung des Verhaltens von Pflanzenbehandlungsmitteln im Wasser (oktober 1980). |
|
7. |
SETAC (1995), Procedures for Assessing the Environmental Fate and Ecotoxicity of Pesticides, Mark R. Lynch, Ed. |
|
8. |
OECD (2000), Guidance document on aquatic toxicity testing of difficult substances and mixtures, OECD Environmental Health and Safety Publications Series on Testing and Assessment no 23. |
|
9. |
OECD (1993), Guidelines for the Testing of Chemicals. Paris. OECD (1994–2000): Addenda 6–11 to Guidelines for the Testing of Chemicals. |
|
10. |
Nelson, H, Laskowski D, Thermes S., and Hendley P. (1997), Recommended changes in pesticide fate study guidelines for improving input to computer models. (Text version of oral presentation at the 14th Annual Meeting of the Society of Environmental Toxicology and Chemistry, Dallas TX, november 1993). |
|
11. |
Mabey, W. and Mill, T. (1978), Critical review of hydrolysis of organic compounds in water under environmental conditions. J. Phys. Chem. Ref. Data 7, 383–415. |
Bilaga 1
Översikt över hydrolystestets olika steg

Bilaga 2
Definitioner och enheter
SI-enheter ska alltid användas.
testsubstans: alla ämnen, oavsett om det gäller den ursprungliga föreningen eller relevanta omvandlingsprodukter.
omvandlingsprodukter: omvandlingsprodukter: alla ämnen som uppstår till följd av biotiska eller abiotiska omvandlingsreaktioner av testsubstansen.
hydrolysprodukter: alla ämnen som uppstår till följd av hydrolytiska omvandlingsreaktioner av testsubstansen.
hydrolys: en testsubstans RX reagerar med vatten varvid slutresultatet blir att gruppen X byts ut mot OH.
|
RX + HOH → ROH +HX |
[1] |
I denna förenklade process minskar koncentrationen av RX med en hastighet enligt formeln
|
hastighet = k [H2O] [RX] |
reaktion av andra ordningen |
|
eller |
|
|
hastighet = k [RX] |
reaktion av första ordningen |
beroende på det hastighetsbestämmande steget. Eftersom vatten finns i stort överskott jämfört med testsubstansen brukar denna typ av reaktion beskrivas som en reaktion av första ordningen där den observerade hastighetskonstanten erhålls genom förhållandet
|
kobs = k [H2O] |
[2] |
och kan bestämmas med hjälp av uttrycket (13)
|
|
[3] |


där
t= tiden
och C0, Ct= koncentrationerna av RX vid tiderna 0 och t.
Enheten för denna konstant har dimensionen (tid)-1 och reaktionens halveringstid (den tid det tar för 50 % av RX att reagera) erhålls genom
|
|
[4] |

halveringstid: betecknas med (t0,5) – den tid det tar för en testsubstans att hydrolyseras till 50 % när reaktionen kan beskrivas med kinetik av första ordningen; halveringstiden är oberoende av koncentrationen.
DT50 (Disappearance Time 50): Den tid inom vilken testsubstansens koncentration minskar med 50 %; den skiljer sig från halveringstiden t0,5 om reaktionen inte följer en kinetik av första ordningen.
Uppskattning av k vid olika temperaturer
Om hastighetskonstanterna är kända för två temperaturer så kan hastighetskonstanterna vid andra temperaturer beräknas med hjälp av Arrhenius ekvation:
![]() eller
eller 
Ett diagram över ln k som funktion av 1/T ger en rät linje med lutningen -E/R
där
|
k |
= |
hastighetskonstanten mätt vid olika temperaturer |
|
E |
= |
aktiveringsenergin [kJ/mol] |
|
T |
= |
absolut temperatur [K] |
|
R |
= |
gaskonstanten [8,314 J/mol· K] |
Aktiveringsenergin beräknas genom regressionsanalys eller följande ekvation:

där T2 > T1.
Bilaga 3
Buffertsystem
A. CLARK OCH LUBS:
Buffertblandningar enligt CLARK och LUBS (14)
|
Sammansättning |
pH |
|
0,2 N HCl OCH 0,2 N KCl VID 20 oC |
|
|
47,5 ml HCl + 25 ml KCl utspätt till 100 ml |
1,0 |
|
32,25 ml HCl + 25 ml KCl utspätt till 100 ml |
1,2 |
|
20,75 ml HCl + 25 ml KCl utspätt till 100 ml |
1,4 |
|
13,15 ml HCl + 25 ml KCl utspätt till 100 ml |
1,6 |
|
8,3 ml HCl + 25 ml KCl utspätt till 100 ml |
1,8 |
|
5,3 ml HCl + 25 ml KCl utspätt till 100 ml |
2,0 |
|
3,35 ml HCl + 25 ml KCl utspätt till 100 ml |
2,2 |
|
0,1 M kaliumbiftalat +0,1 N HCl vid 20 oC |
|
|
46,70 ml 0,1 N HCl + 50 ml biftalat utspätt till 100 ml |
2,2 |
|
39,60 ml 0,1 N HCl + 50 ml biftalat utspätt till 100 ml |
2,4 |
|
32,95 ml 0,1 N HCl + 50 ml biftalat utspätt till 100 ml |
2,6 |
|
26,42 ml 0,1 N HCl + 50 ml biftalat utspätt till 100 ml |
2,8 |
|
20,32 ml 0,1 N HCl + 50 ml biftalat utspätt till 100 ml |
3,0 |
|
14,70 ml 0,1 N HCl + 50 ml biftalat utspätt till 100 ml |
3,2 |
|
9,90 ml 0,1 N HCl + 50 ml biftalat utspätt till 100 ml |
3,4 |
|
5,97 ml 0,1 N HCl + 50 ml biftalat utspätt till 100 ml |
3,6 |
|
2,63 ml 0,1 N HCl + 50 ml biftalat utspätt till 100 ml |
3,8 |
|
0,1 M kaliumbiftalat +0,1 N NaOH vid 20 oC |
|
|
0,40 ml 0,1 N NaOH + 50 ml biftalat utspätt till 100 ml |
4,0 |
|
3,70 ml 0,1 N NaOH + 50 ml biftalat utspätt till 100 ml |
4,2 |
|
7,50 ml 0,1 N NaOH + 50 ml biftalat utspätt till 100 ml |
4,4 |
|
12,15 ml 0,1 N NaOH + 50 ml biftalat utspätt till 100 ml |
4,6 |
|
17,70 ml 0,1 N NaOH + 50 ml biftalat utspätt till 100 ml |
4,8 |
|
23,85 ml 0,1 N NaOH + 50 ml biftalat utspätt till 100 ml |
5,0 |
|
29,95 ml 0,1 N NaOH + 50 ml biftalat utspätt till 100 ml |
5,2 |
|
35,45 ml 0,1 N NaOH + 50 ml biftalat utspätt till 100 ml |
5,4 |
|
39,85 ml 0,1 N NaOH + 50 ml biftalat utspätt till 100 ml |
5,6 |
|
43,00 ml 0,1 N NaOH + 50 ml biftalat utspätt till 100 ml |
5,8 |
|
45,45 ml 0,1 N NaOH + 50 ml biftalat utspätt till 100 ml |
6,0 |
Buffertblandningar enligt CLARK och LUBS (fortsättning)
|
0,1 M monokaliumfosfat +0,1 N NaOH vid 20 oC |
|
|
5,70 ml 0,1 N NaOH + 50 ml fosfat utspätt till 100 ml |
6,0 |
|
8,60 ml 0,1 N NaOH + 50 ml fosfat utspätt till 100 ml |
6,2 |
|
12,60 ml 0,1 N NaOH + 50 ml fosfat utspätt till 100 ml |
6,4 |
|
17,80 ml 0,1 N NaOH + 50 ml fosfat utspätt till 100 ml |
6,6 |
|
23,45 ml 0,1 N NaOH + 50 ml fosfat utspätt till 100 ml |
6,8 |
|
29,63 ml 0,1 N NaOH + 50 ml fosfat utspätt till 100 ml |
7,0 |
|
35,00 ml 0,1 N NaOH + 50 ml fosfat utspätt till 100 ml |
7,2 |
|
39,50 ml 0,1 N NaOH + 50 ml fosfat utspätt till 100 ml |
7,4 |
|
42,80 ml 0,1 N NaOH + 50 ml fosfat utspätt till 100 ml |
7,6 |
|
45,20 ml 0,1 N NaOH + 50 ml fosfat utspätt till 100 ml |
7,8 |
|
46,80 ml 0,1 N NaOH + 50 ml fosfat utspätt till 100 ml |
8,0 |
|
0,1 M H3BO3 i 0,1 M KCl +0,1 N NaOH vid 20 oC |
|
|
2,61 ml 0,1 N NaOH + 50 ml borsyra utspätt till 100 ml |
7,8 |
|
3,97 ml 0,1 N NaOH + 50 ml borsyra utspätt till 100 ml |
8,0 |
|
5,90 ml 0,1 N NaOH + 50 ml borsyra utspätt till 100 ml |
8,2 |
|
8,50 ml 0,1 N NaOH + 50 ml borsyra utspätt till 100 ml |
8,4 |
|
12,00 ml 0,1 N NaOH + 50 ml borsyra utspätt till 100 ml |
8,6 |
|
16,30 ml 0,1 N NaOH + 50 ml borsyra utspätt till 100 ml |
8,8 |
|
21,30 ml 0,1 N NaOH + 50 ml borsyra utspätt till 100 ml |
9,0 |
|
26,70 ml 0,1 N NaOH + 50 ml borsyra utspätt till 100 ml |
9,2 |
|
32,00 ml 0,1 N NaOH + 50 ml borsyra utspätt till 100 ml |
9,4 |
|
36,85 ml 0,1 N NaOH + 50 ml borsyra utspätt till 100 ml |
9,6 |
|
40,80 ml 0,1 N NaOH + 50 ml borsyra utspätt till 100 ml |
9,8 |
|
43,90 ml 0,1 N NaOH + 50 ml borsyra utspätt till 100 ml |
10,0 |
B. KOLTHOFF OCH VLEESCHHOUWER:
Citratbuffertar enligt KOLTHOFF och VLEESCHHOUWER
|
Sammansättning |
pH |
|
0,1 M monokaliumfosfat och 0,1 N HCl vid 18 oC (15) |
|
|
49,7 ml 0,1 N HCl + 50 ml citrat utspätt till 100 ml |
2,2 |
|
43,4 ml 0,1 N HCl + 50 ml citrat utspätt till 100 ml |
2,4 |
|
36,8 ml 0,1 N HCl + 50 ml citrat utspätt till 100 ml |
2,6 |
|
30,2 ml 0,1 N HCl + 50 ml citrat utspätt till 100 ml |
2,8 |
|
23,6 ml 0,1 N HCl + 50 ml citrat utspätt till 100 ml |
3,0 |
|
17,2 ml 0,1 N HCl + 50 ml citrat utspätt till 100 ml |
3,2 |
|
10,7 ml 0,1 N HCl + 50 ml citrat utspätt till 100 ml |
3,4 |
|
4,2 ml 0,1 N HCl + 50 ml citrat utspätt till 100 ml |
3,6 |
|
0,1 M monokaliumfostat och 0,1 N NaOH vid 18 oC (15) |
|
|
2,0 ml 0,1 N NaOH + 50 ml citrat utspätt till 100 ml |
3,8 |
|
9,0 ml 0,1 N NaOH + 50 ml citrat utspätt till 100 ml |
4,0 |
|
16,3 ml 0,1 N NaOH + 50 ml citrat utspätt till 100 ml |
4,2 |
|
23,7 ml 0,1 N NaOH + 50 ml citrat utspätt till 100 ml |
4,4 |
|
31,5 ml 0,1 N NaOH + 50 ml citrat utspätt till 100 ml |
4,6 |
|
39,2 ml 0,1 N NaOH + 50 ml citrat utspätt till 100 ml |
4,8 |
|
46,7 ml 0,1 N NaOH + 50 ml citrat utspätt till 100 ml |
5,0 |
|
54,2 ml 0,1 N NaOH + 50 ml citrat utspätt till 100 ml |
5,2 |
|
61,0 ml 0,1 N NaOH + 50 ml citrat utspätt till 100 ml |
5,4 |
|
68,0 ml 0,1 N NaOH + 50 ml citrat utspätt till 100 ml |
5,6 |
|
74,4 ml 0,1 N NaOH + 50 ml citrat utspätt till 100 ml |
5,8 |
|
81,2 ml 0,1 N NaOH + 50 ml citrat utspätt till 100 ml |
6,0 |
C. SÖRENSEN:
Boratblandningar enligt SÖRENSEN
|
Sammansättning |
Sörensen 18 oC |
Walbum, pH vid |
|||
|
ml borax |
ml HCl/NaO H |
10 oC |
40 oC |
70 oC |
|
|
0,05 M borax +0,1 N HCl |
|||||
|
5,25 |
4,75 |
7,62 |
7,64 |
7,55 |
7,47 |
|
5,50 |
4,50 |
7,94 |
7,98 |
7,86 |
7,76 |
|
5,75 |
4,25 |
8,14 |
8,17 |
8,06 |
7,95 |
|
6,00 |
4,00 |
8,29 |
8,32 |
8,19 |
8,08 |
|
6,50 |
3,50 |
8,51 |
8,54 |
8,40 |
8,28 |
|
7,00 |
3,00 |
8,08 |
8,72 |
8,56 |
8,40 |
|
7,50 |
2,50 |
8,80 |
8,84 |
8,67 |
8,50 |
|
8,00 |
2,00 |
8,91 |
8,96 |
8,77 |
8,59 |
|
8,50 |
1,50 |
9,01 |
9,06 |
8,86 |
8,67 |
|
9,00 |
1,00 |
9,09 |
9,14 |
8,94 |
8,74 |
|
9,50 |
0,50 |
9,17 |
9,22 |
9,01 |
8,80 |
|
10,00 |
0,00 |
9,24 |
9,30 |
9,08 |
8,86 |
|
0,05 M borax +0,1 N NaOH |
|||||
|
10,0 |
0,0 |
9,24 |
9,30 |
9,08 |
8,86 |
|
9,0 |
1,0 |
9,36 |
9,42 |
9,18 |
8,94 |
|
8,0 |
2,0 |
9,50 |
9,57 |
9,30 |
9,02 |
|
7,0 |
3,0 |
9,68 |
9,76 |
9,44 |
9,12 |
|
6,0 |
4,0 |
9,97 |
10,06 |
9,67 |
9,28 |
Fosfatblandningar enligt SÖRENSEN
|
Sammansättning |
pH |
|
0,0667 M monokaliumfosfat + 0,0667 M dinatriumfosfat vid 20 oC |
|
|
99,2 ml KH2PO4 + 0,8 ml Na2HPO4 |
5,0 |
|
98,4 ml KH2PO4 + 1,6 ml Na2HPO4 |
5,2 |
|
97,3 ml KH2PO4 + 2,7 ml Na2HPO4 |
5,4 |
|
95,5 ml KH2PO4 + 4,5 ml Na2HPO4 |
5,6 |
|
92,8 ml KH2PO4 + 7,2 ml Na2HPO4 |
5,8 |
|
88,9 ml KH2PO4 + 11,1 ml Na2HPO4 |
6,0 |
|
83,0 ml KH2PO4 + 17,0 ml Na2HPO4 |
6,2 |
|
75,4 ml KH2PO4 + 24,6 ml Na2HPO4 |
6,4 |
|
65,3 ml KH2PO4 + 34,7 ml Na2HPO4 |
6,6 |
|
53,4 ml KH2PO4 + 46,6 ml Na2HPO4 |
6,8 |
|
41,3 ml KH2PO4 + 58,7 ml Na2HPO4 |
7,0 |
|
29,6 ml KH2PO4 + 70,4 ml Na2HPO4 |
7,2 |
|
19,7 ml KH2PO4 + 80,3 ml Na2HPO4 |
7,4 |
|
12,8 ml KH2PO4 + 87,2 ml Na2HPO4 |
7,6 |
|
7,4 ml KH2PO4 + 92,6 ml Na2HPO4 |
7,8 |
|
3,7 ml KH2PO4 + 96,3 ml Na2HPO4 |
8,0 |
C.8 TOXICITET HOS DAGGMASKAR
TEST I SYNTETISK JORD
1. METOD
1.1 INLEDNING
Vid detta laboratorietest tillsätts testämnet till syntetisk jord i vilken daggmaskar placeras i 14 dagar. Efter denna period (alternativt efter sju dagar) undersöks ämnetd dödliga verkan på daggmaskarna. Detta test är en metod för att under en relativt kort tid undersöka kemikaliers verkan på daggmaskar vid upptagning genom huden eller via fodret.
1.2 DEFINITIONER OCH ENHETER
LC50: Koncentrationen av ett ämne som kan förväntas orsaka att 50 % av försöksdjuren dör under testperioden.
1.3 REFERENSÄMNE
En referensämne används regelbundet för att visa att testsystemets känslighet inte har ändrats signifikant.
Som referensämne rekommenderas analytiskt rent kloracetamid.
1.4 TESTMETODSPRINCIP
Eftersom jord är ett variabelt medium används en noggrant definierad syntetisk lerjord för detta test. Vuxna daggmaskar av arten Eisenia foetida (se anmärkningen i tillägget) hålls i en definierad syntetisk jord som behandlats med olika koncentrationer av testämnet. Behållarnas innehåll sprids ut på en bricka 14 dagar (alternativt sju dagar) efter inledningen av testet och det antal daggmaskar som överlever vid varje koncentration räknas.
1.5 KVALITETSKRITERIER
Testet är beräknat att vara så reproducerbart som möjligt med hänsyn till testsubstrat och organism. Om dödligheten hos kontrolldjuren överstiger 10 % vid testets avslutning är testet ogiltigt.
1.6 METODBESKRIVNING
1.6.1 Material
1.6.1.1
Väldefinierad syntetisk jord används som bassubstrat.
|
a) |
Bassubstrat (viktprocent torrämne)
|
|
b) |
Testsubstrat Testsubstratet består av bassubstrat, testämne och avjoniserat vatten. Vatteninnehållet motsvarar ca 25–42 viktprocent av bassubstratets torrvikt. Substratets vatteninnehåll bestäms genom torkning av ett prov till konstant vikt vid 105 oC. Ett viktigt kriterium är att den syntetiska jorden har fuktats så mycket det går, men det får inte finnas något stående vatten. Blandningen sker omedelbart så att det blir en jämn fördelning av testämnet och substratet. Den metod som används för att tillsätta testämnet till substratet ska rapporteras. |
|
c) |
Kontrollsubstrat Kontrollsubstratet består av bassubstrat, testämne och avjoniserat vatten. Om en tillsättningsämne används ska ett extra kontrollsubstrat innehålla samma mängd tillsättningsämne. |
1.6.1.2
Glasbehållare med ett omfång av ca 1 liter (korrekt täckta med plastlock, plattor eller plastfolie med ventilationshål) fyllda med en mängd våt test- eller kontrollsubstrat motsvarande 500 g av substratets torrvikt.
1.6.2 Testbetingelser
Behållarna förvaras i klimatkammare vid en temperatur av 20 oC med en tolerans av ± 2 oC och i konstant ljus. Ljusintensiteten bör vara 400–800 lux.
Testperioden varar i 14 dagar, men dödligheten kan eventuellt bedömas sju dagar efter testets inledning.
1.6.3 Förfarande
Testämnets koncentrationer uttrycks som ämnets vikt i förhållande till grundsubstratets torrsvikt (mg/kg).
Det koncentrationsområde som orsakar 0–100 % dödlighet kan bestämmas genom ett förtest för att få information om vidden av koncentrationer som ska användas vid det definitiva testet.
Ämnet bör testas i följande koncentrationer: 1 000, 100, 10, 1 och 0,1 mg/ämne/kg testsubstrat (torrvikt).
Om en fullständig definitiv test ska göras räcker det att för förtestet använda en uppsättning per koncentration och en för den obehandlade kontrollen med vardera tio daggmaskar.
Resultaten av förtestet används för att välja minst fem koncentrationer i en geometrisk serie som täcker 0–100 % dödlighet och där den konstanta faktorn inte får överstiga 1,8.
Vid test med dessa koncentrationer bör det vara möjligt att med största möjhga exakthet bedöma LC50-värdet och dess konfidensgränser.
Vid det definitiva testet används minst fyra testgrupper per koncentration och fyra obehandlade kontrollgrupper med vardera tio daggmaskar. Resultaten av dessa parallellgrupper anges som ett medelvärde och en standardavvikelse.
Om två på varandra följande koncentrationer, med ett förhållande av 1,8, endast ger 0 och 100 % dödlighet är dessa två värden tillräckliga för att ange LC50-interval! et.
Testämnet bereds, när så är möjligt, utan tillsättning av annat än vatten. Omdelbart före inledningen av testet blandas en emulsion eller dispersion av tesämnet i avjoniserat vatten eller annat lösningsmedel med bassubstratet eller sprayas ut jämnt över detta med en fin kromatografispruta eller liknande.
Om testämnet inte går att lösa i vatten kan det lösas i minsta möjliga mängd av ett passande organiskt lösningsmedel (t.ex. hexan, aceton eller kloroform).
Endast medel som inte går att lösa i vatten för användas för att upplösa, dispergera eller emulgera testämnet. Testsubstratet luftas innan användning. Den förångade vattenmängden ersätts. Kontrollsubstratet får innehålla eventuella tillsättningsämnen i samma mängd.
Om det inte går att lösa, dispergera eller emulgera testämnet i organiska lösningsmedel blandas en blandning av finsand av malen kvarts och den kvantitet av testämnet som är nödvändig för att behandla 500 g torrvikt av syntetisk jord med 490 g torrvikt av testämnet.
För varje testgrupp placeras en mängd våt testsubstrat motsvarande 500 g torrvikt i varje glasbehållare och tio daggmaskar, som under 24 timmar har invänjts i ett liknande vått bassubstrat och därefter snabbt sköljts av och avdroppats på filterpapper innan användning, placeras på testsubstratets yta.
Behållarna täcks med perforerade plastlock, plattor eller plastfolie för att förhindra att substratet torkar, och de förvaras under testbetingelse i 14 dagar.
Utvärderingen sker 14 dagar (alternativt sju dagar) efter testets inledning. Substratet sprids ut på en platta av glas eller rostfritt stål. Daggmaskarna undersöks och antalet överlevande daggmaskar fastställs. Daggmaskar anses vara döda om de inte reagerar på en lätt mekanisk påverkan av framänden.
När undersökningen sker efter sju dagar hälls substratet tillbaka i behållaren och överlevande daggmaskar läggs tillbaka pä testsubstratets yta.
1.6.4 Testorganismer
Testorganismerna bör vara daggmaskar av arten Eisenia foetida (se anmärkningen i tillägget) (minst två månader gamla med clitellum) och väga (vått tillstånd) 300–600 mg. (För uppfödning se tillägget).
2. DATA
2.1 BEHANDLING OCH UTVÄRDERING AV DATA
De använda koncentrationerna av testämnet rapporteras med angivelse av den procentuella dödligheten för varje koncentration.
När erhållna data inte är tillräckliga kan LC50 och dess konfidensgränser (p = 0,05) bestämmas genom användning av standardmetoder (Lichtfield and Wilcoxon, 1949, eller motsvarande metod). LC50 anges som mg testämne per kg testsubstrat (torrvikt)
Om koncentrationskurvans böjning vid ett förhållande av 1,8 endast ger 0 och 100 % dödlighet är dessa två värden tillräckliga för att ange LC50-intervallet.
3. RAPPORTERING
3.1 TESTRAPPORT
Testrapporten ska om möjligt innehålla:
|
— |
Angivelse av att testet utförts i överensstämmelse med ovanstående kvalitetskriterier. |
|
— |
Angivelse av det genomförda testet (förtest och/eller definitivt test). |
|
— |
Noggrann beskrivning av testbetingelserna eller angivelse av att testet utförts i överensstämmelse med metoden. Eventuella avvikelser ska rapporteras. |
|
— |
Noggrann beskrivning av hur testämnet blandats i bassubstratet. |
|
— |
Uppgifter om testorganismer (art, ålder, medelvikt samt lägsta och högsta vikt, betingelserna för uppfödning och hållning samt leverantör). |
|
— |
Använd metod för bestämning av LC50. |
|
— |
Testresultaten, inbegripet alla använda data. |
|
— |
Beskrivning av observerade symptom eller beteendeförändringar hos testorganismerna. |
|
— |
Dödlighet i kontrollgrupperna. |
|
— |
LC50 eller högsta testkoncentration utan dödlighet och lägsta testkoncentration med en dödlig het av 100 % 14 dagar (alternativt sju dagar) efter testets inledning. |
|
— |
Ritning av koncentrations/reaktionskurvan. |
|
— |
Resultat som erhållits med referensämnet antingen i samband med det ifrågavarande testet eller vid tidigare kvalitetskontrollundersökningar. |
4. HÄNVISNINGAR
|
1. |
OECD (1981), Test Guideline 207, rådets beslut K(81) 30 slutlig. |
|
2. |
Edwards, C. A. och Lofty, (1977), Biology of Earthworms. London: Chapman and Hall, s. 331. |
|
3. |
Bouche, M. B. (1972), Lombriciens de France, Écologie et Systématique. Publ. Institut National de la Recherche Agronomique, s. 671. |
|
4. |
Litchfield, J. T. och Wilcoxon J.(1949), A simplified method of evaluating dose-effect experi ments, J. Pharm. Esp. Therap., vol. 96, s. 99–113. |
|
5. |
Europeiska gemenskapernas kommission, Development of a standard laboratory method for assaying the toxicity of chemical substances to earthworms, Rapport EUR 8714 EN. |
|
6. |
Umweltsbundesamt/Biologische Bundesanstalt für Land- und Forstwirtschaft, Berlin (1984), Verfahrensvorschlag Toxizitätstest am Regenwurm Eisenia foetida in künstlichem Boden, i: Rudolph/Boje: Ökotoxikologie, ecomed, Landsberg, 1986. |
Tillägg
Uppfödning och hållning av daggmaskar före test
Vid uppfödning av djuren placeras 30–50 vuxna daggmaskar i en avelsbur med färsk substrat och avlägsnas efter 14 dagar. Dessa djur kan användas för uppfödning av senare grupper. De daggmaskar som kläcks ur kokongerna används för test när de är könsmogna (under föreskrivna villkor efter 2–3 månader).
Förhållanden för hållning och uppfödning
|
Klimatkammare |
: |
Temperatur 20 oC med en tolerans av ± 2 oC, helst med konstant belysning (ljusintensitet 400–800 lux). |
|
Avelsburar |
: |
Lämpliga låga burar med en volym av 10–20 1. |
|
Substrat |
: |
Eisennia foetida kan födas upp i olika djurexkrement. Som uppfödningsmedium rekommenderas en blandning av lika delar av torv och ko- eller hästgödsel. Substratet bör ha ett pH-värde av 6–7 (reglerat med kalciumkarbonat) och låg jonledningsförmåga (under 6 mmhos eller 0,5 % saltkoncentration). Substratet bör vara fuktigt men inte för vått. Andra lämpliga metoder utöver ovan nämnda kan användas. |
Anmärkning: Det finns två raser av Eisenia foetida som vissa systematiker har klassificerat som arter (Bouche, 1972). De är morfologiskt liknande men den ena, Eisenia foetida foetida har typiskt tvärgående ränder på segmenten medan den andra, Eisenia foetida andrei, saknar dessa, och har en rödbrokig färg. Så långt det går bör Eisenia foetida andrei användas. Andra arter kan användas om den nödvändiga metodiken finns tillgänglig.
C.9 BIOLOGISK NEDBRYTBARHET
ZAHN-WELLENS TEST
1. METOD
1.1 INLEDNING
Syftet med denna metod är att mäta bionedbrytbarheten hos vattenlösliga, icke flyktiga organiska föreningar när de utsätts relativt höga koncentrationer av mikroorganismer under ett statiskt test.
Fysikaliska-kemiska reaktioner kan förekomma på de uppslammade artiklarna vilket man måste ta hänsyn till vid tolkning av resultaten (se 3.2).
De ämnen som ska undersökas används i koncentrationer motsvarande DOC-värden av 50–400 mg/l (DOC = dissolved organic carbon, dvs. upplöst organiskt kol), eller COD-värden motsvarande 100–1 000 mg/l (COD = chemical oxygen demand, dvs. kemiskt syrebehov). Fördelen med dessa relativt höga koncentrationer är att de är pålitliga ur analyshänseende.
Vid denna metod mäts koncentrationer av upplöst organiskt kol eller det kemiska syrebehovet i syfte att bedöma testämnets fullständiga bionedbrytbarhet.
Samtidig användning av en specifik analysmetod kan möjliggöra värdering av ämnets primära bionedbrytbarhet (dvs. försvinnandet av den ursprungliga kemiska förbindelsen).
Metoden kan endast tillämpas på organiska testämnen som vid den använda testkoncentrationen
|
— |
är vattenlösliga inom ramen för testbetingelserna, |
|
— |
har försumbart ångtryck inom ramen för testbetingelserna, |
|
— |
inte hämmar bakterier, |
|
— |
endast absorberas i testsystemet i begränsad omfattning, |
|
— |
inte försvinner från testlösningen till följd av skumning. |
Uppgifter om de relativa proportionerna av testämnets huvudbeståndsdelar är till nytta vid tolkningen av de erhållna resultaten, särskilt om resultaten är låga eller marginella.
Uppgifter om testämnets toxicitet för mikroorganismer kan vara till nytta vid tolkningen av låga resultat och vid valet av lämpliga testkoncentrationer.
1.2 DEFINITIONER OCH ENHETER
Nedbrytningen vid testets avslutning definieras som ”bionedbrytbarhet i Zahn-Wellens test”:

där:
|
DT |
= |
Bionedbrytning i procent DOC som avgått vid tid T. |
|
CA |
= |
DOC (eller COD)-värdena i försöksblandningen, mätt tre timmar efter inledning av testet (mg/l) (DOC = dissolved organic carbon/upplöst organiskt kol, COD = chemical oxygen demand/kemiskt syrebehov). |
|
CT |
= |
DOC- eller COD-värdena i försöksblandningen vid tidpunkten för provtagningen (mg/l). |
|
CB |
= |
DOC- eller COD-värdena i blindprovet vid tidpunkten för provtagningen (mg/l). |
|
C BA |
= |
DOC- eller COD-värdena i blindprovet, mätt tre timmar efter inledning av testet (mg/l). |
Nedbrytningsgraden avrundas till närmaste hela procenttal.
Den procentuella nedbrytningen anges som den procent DOC (eller COD) som försvunnit från testämnet.
Skillnaden mellan det värde som mätts efter tre timmmar och det beräknade, helst uppmätta, initiala värdet kan ge nyttiga information om ämnets eliminiering (se 3.2 ”Tolkning av resultat”).
1.3 REFERENSÄMNEN
När ett nytt ämne ska undersökas kan referensämnen i vissa fall vara användbara. Det går dock ännu inte att rekommendera bestämda referensämnen.
1.4 TESTMETODSPRINCIP
Aktiverat slam, oorganiska näringsämnen och testämnet, som är den enda kolämnekällan i vattenlösning, placeras i en glasbehållare med en volym av 1–4 liter, som är försedd med omrörning och luftning. Blandningen rörs och luftas vid 20–25 oC i indirekt ljus eller i mörker i upp till 28 dagar. Nedbrytningen följs genom mätning av DOC-(eller COD-)värden i den filtrerade lösningen en gång om dagen eller regelbundet med en annan lämplig tidsintervall. Förhållandet mellan eliminerat DOC (eller COD) efter varje tidsintervall och värden tre timmar efter testets inledning uttrycks som procentuell bionedbrytning och är ett värde för nedbrytningsgraden vid denna tidpunkt.
Om en specifik analysmetod används kan ändringar av det ursprungliga ämnets koncentration till följd av bionedbrytning mätas (primär bionedbrytning).
1.5 KVALITETSKRITERIER
Metodens reproducerbarhet har fastställts genom ett ringtest.
Metodens känslighet beror till största delen på variationer i blindprovet och till mindre del på noggrannheten vid bestämning av upplöst organiskt kol och testämnekoncentration i vätskan.
1.6 METODBESKRIVNING
1.6.1 Förberedelser
1.6.1.1
Testvatten: Dricksvatten innehållande mindre än 5 mg organiskt kol per liter. Koncentrationen av kalcium- och magnesiumjoner får tillsammans inte överstiga 2,7 mmol per liter. I motsatt fall måste den förtunnas på passande sätt med avjoniserat eller destillerat vatten.
|
Svavelsyra, analytisk reagens (AR.): |
50 g/l. |
|
Natriumhydroxidlösning, AR.: |
40 g/l. |
|
Lösning av oorganiska näringsämnen: i 1 liter avjoniserat vatten löses |
|
|
ammoniumklorid, NH4C1, AR: |
38,5 g. |
|
natriumdihydrogenfosfat, NaH2PO4. 2H2O, AR: |
33,4 g. |
|
kaliumdihydrogenfosfat, KH2PO4, AR: |
8,5 g. |
|
dikaliumhydrogenfosfat, K2HPO4, AR: |
21,75 g. |
1.6.1.2
Glasbehållare med en volym av 1–4 liter (t.ex. cylindrisk behållare).
Omrörare med rörhuvud av glas eller metall på en passande axel (omröraren bör rotera 5–10 cm över behållarens botten). Som ett alternativ kan en magnetomrörare med en 7–10 cm lång mangnetstav användas.
Glasrör med en invändig diameter av 2–4 mm för luftning. Rörets öppning bör ligga ca 1 cm ovanför behållarens botten.
Centrifug (ca 3 550 g).
pH-mätare.
Apparat för mätning av upplöst syre.
Pappersfilter.
Membranfiltreringsapparat.
Membranfilter med en porstorlek av 0,45 μm. Membranfilter är endast lämpligt om det har säkerställts att de varken avger kolämne eller absorberar ämnet under filtreringen.
Analysapparatur för bestämning av innehållet av organiskt kol och den kemiska syrebehovet.
1.6.1.3
Aktiverat slam från ett biologiskt reningsverk tvättas (flera gånger) med testvatten (se ovan) genom centrifugering eller dekantering.
Det aktiverade slammet ska vara i ett lämpligt tillstånd. Sådant slam kan fås från ett väl fungerande reningsverk som huvudsakligen behandlar spillvatten. För att få så många olika bakteriestammar som möjligt kan det vara bättre att blanda inokulat från olika källor (t.ex. från olika reningsverk, från jord, från flodvatten, etc.).
De aktiverade slammets livskraft kontrolleras, se. ”lämplighetskontroll”.
1.6.1.4
1 testbehållarna placeras 500 ml vatten, 2,5 ml lösning av oorganiska näringsämnen per liter samt en mängd aktiverat slam som motsvarar 0,2–1,0 g torrämne per liter färdig blandning. Tillsätt tillräckligt med stamlösning av testämnet så att den färdiga blandningen får en DOC-koncentration av 50–400 mg/l. Motsvarande COD-intervall är 100–1 000 mg/l. Fyll upp med testvatten till en total volym av 1–4 liter. Den totala volym som väljs beror på det antal prover som ska tas för DOC-eller COD-bestämning, och den nödvändiga analysvolymen.
Normalt kan en volym av 2 1 anses tillräcklig. Minst en kontrollbehållare (blindprov) ska göras i ordning samtidigt med varje testsene. Den innehåller endast aktiverat slam och lösning av oorganiska näringsämnen utspätt med vatten till samma totalvolym som i testbehållarna.
1.6.2 Testförfarande
Testbehållarna omrörs med magnetomrörare eller propelleromrörare i indirekt belysning eller i mörker vid 20–25 oC. Luftning sker med komprimerad luft som om nödvändigt rensas med ett vadd-filter och en tvättflaska. Man måste se till att slammet inte sätter sig och att syrekoncentrationen inte sjunker under 2 mg/l.
pH-värdet måste kontrolleras regelbundet (t.ex. dagligen) och vid behov justeras till pH 7–8.
Strax före provtagning kompenseras vattenförlust på grund av förångning genom tillsättning av avjoniserat eller destillerat vatten. Man kan t.ex. märka vätskehöjden i behållaren innan testet inleds. Nya markeringar ritas dit efter varje provtagning (utan luftning och omrörning). De första proverna tas alltid tre timmar efter testets inledning så att man kan konstatera om testmaterialet absorberas på det aktiverade slammet.
Elimineringen av testämnet följs genom DOC- eller COD-bestämningar antingen dagligen eller regelbundet med andra intervaller. Proverna från testbehållaren och blindprovet filtreras genom ett noggrant tvättat pappersfilter. De första 5 ml av filtratet kasseras. Slam som är svårt att filtrera kan avlägsnas först genom centrifugering i tio minuter. Minst dubbla bestämningar av DOC och COD ska göras. Testet varar i upp till 28 dagar.
Obs! Prover som stadigt är grumliga filtreras genom ett membranfilter. Membranfiltren får varken frigöra eller absorbera organiska material.
I varje testserie bör det ingå en behållare som innehåller ett känt ämne så att man kan kontrollera funktionen av det aktiverade slammet. 1 detta syfte är dietynglykol lämpligt.
Om analyser utförs med relativt korta mellanrum (t.ex. dagligen) kommer adaption att framgå tydligt av nedbrytningskurvan (se figur 2). Testet bör därför inte inledas omedelbart före helgen.
Om adaption förekommer i slutet av testet kan testet förlängas tills nedbrytningen är avslutad.
Obs! Om det finns behov av att veta mer om det adapterade slammets beteende utsätts samma aktiverade slam ytterligare en gång för samma testmaterial enligt följande förfarande:
Stäng av omröraren och luftaren och låt det aktiverade slammet sätta sig. Avlägsna överstående vätska, fyll på upp till 2 liter med testvatten och rör om i 15 minuter och låt sedan blandningen sätta sig igen. Sedan överstående vätska avlägsnats används det överblivande slammet för att upprepa testet i enlighet med 1.6.1.4 och 1.6.2 ovan. Det aktiverade slammet kan även isoleras genom centrifugering istället för utfällning.
Det adapterade slammet kan blandas med färskt slam till en koncentration av 0,2–1,0 g torrvikt/l.
Normalt filtreras prover genom ett noggrant tvättat pappersfilter (för tvättning används avjoniserat vatten).
Prover som stadigt är oklara filtreras genom ett membranfilter (0,45 μm).
En dubbelbestämning av DOC-koncentrationen i provfiltraten görs (de första 5 ml kasseras) med TOC-instrument. Om filtratet inte kan analyseras samma dag ska det förvaras i kylskåp till nästa dag. Längre tids förvaring rekommenderas inte.
COD-koncentrationen bestäms i provfiltraten med COD-koncentrationsutrustning i enlighet med det förfarande som beskrivs i hänvisning 2 nedan.
2. DATA OCH UTVÄRDERING
Minst dubbelbestämning av DOC- och COD-koncentrationerna i proverna görs i enlighet med 1.6.2. Nedbrytbarheten vid tidpunkten T beräknas med hjälp av den formel (med definitioner) som anges i 1.2.
Nedbrytningsgraden beräknas till närmaste hela procenttal. De nedbrytning som erhålls vid slutet av testet anges som ”bionedbrytbarheten vid Zahn-Wellens test”.
Obs! Om fullständig nedbrytning inte uppnås innan testperioden är slut och detta resultat bekräftas genom en ytterligare analys följande dag kan testet avslutas.
3. RAPPORTERING
3.1 TESTRAPPORT
Testrapporten ska om möjligt innehålla minst följande:
|
— |
Testämnets initiala koncentration. |
|
— |
Alla andra upplysningar och försöksresultat i fråga om testämnet, en eventuell referensämne och blindförsöket. |
|
— |
Koncentrationen efter tre timmar. |
|
— |
Kurva över bionedbrytningen med förklaring. |
|
— |
När och var provorganismerna är tagna, adaptionsgrad, använd koncentration, etc. |
|
— |
Vetenskapliga motiveringar för eventuella ändringar av testförfarandet. |
3.2 TOLKNING AV RESULTAT
Den förlust av DOC (COD) som sker gradvis över dagar eller veckor är ett tecken på bionedbrytbarhet hos testämnet.
Fysikalisk-kemisk absorption kan dock i vissa fall spela en viss roll och ett tecken på detta är att före de första tre timmarna sker fullständig eller delvis förlust och att skillnaden mellan överstående vätska från kontroll- och testproverna förblir på en oväntat läg nivå.
Det är nödvändigt att göra ytterligare tester om man ska kunna skilja på bionedbrytning (eller delvis bionedbrytning) och absorption.
Detta kan göras på flera olika sätt, men det mest övertygande är att använda överstående vätska som inokulum i en bassettest (helst ett respirometriskt test).
Testämnen som visar en hög förlust av DOC (COD), som inte beror på absorption, vid denna test bör betraktas som potentiellt bionedbrytbara. Delvis förlust som inte beror på absorption, betyder att kemikalien i varje fall i en viss omfattning bryts ned biologiskt.
Låg eller ingen förlust av DOC (COD) kan bero på att testämnet hämmar mikroorganismerna. Detta kan med tiden också visa sig genom att slammet upplöses och försvinner, varvid överstående vätska blir grumlig. Testet bör upprepas med en lägre koncentration testämne. Användning av ett ämnesspecifik analysmetod eller 14C-markerat testämne kan ge större känslighet. Om 14C-markerat testämne används kommer återhämtning av 14CO2 bekräfta att det ror sig om bionedbrytbarhet.
Om resultaten anges som primär bionedbrytbarhet bör det om möjligt anges en förklaring på den kemiska strukturförändringen som leder till avsaknaden av analysresultat för det ursprungliga testämnet.
Valet av analysmetoder ska motiveras och analysresultaten för blindproven ska anges.
4. HÄNVISNINGAR
|
1. |
OECD, Paris (1981), Test Guideline 302 B, rådets beslut K(81) 30 slutlig. |
|
2. |
Bilaga V C.9 Nedbrytning – kemiskt syrebehov, kommissionens direktiv 84/449/EEG, Europeiska gemenskapernas officiella tidning L 251, 19.9.1984. |
Tillägg
EXEMPEL PÅ UTVÄRDERING
|
Organisk förbindelse: |
4-etoxybenzoesyra |
|
Teoretis testkoncentration: |
600 mg/l |
|
Teoretisk DOC: |
390 mg/l |
|
Inoculat: |
Reningsverk |
|
Koncentration: |
1 g torrämne per liter |
|
Enventuell adaption: |
inte adapterat |
|
Analys: |
DOC-bestämning |
|
Testmängd: |
3 ml |
|
Kontrollämne: |
Dietylenglykol |
|
Ämnets toxicitet: |
Ingen toxicitet under 1 000 Använt test: fermenteringsglastestmg/l |
|
Tidpunkt |
Kontrollämne |
Testämne |
|||||
|
Blindprov DOC (16) (mg/l) |
DOC (16) (mg/l) |
DOC netto (mg/l) |
Nedbrytning ( %) |
DOC (16) (mg/l) |
DOC netto (mg/l) |
Nedbrytning ( %) |
|
|
0 |
— |
— |
300,0 |
— |
— |
390,0 |
— |
|
3 timmar |
4,0 |
298,0 |
294,0 |
2 |
371,6 |
367,6 |
6 |
|
1 dag |
6,1 |
288,3 |
282,2 |
6 |
373,3 |
367,2 |
6 |
|
2 dagar |
5,0 |
281,2 |
276,2 |
8 |
360,0 |
355,0 |
9 |
|
5 dagar |
6,3 |
270,5 |
264,2 |
12 |
193,8 |
187,5 |
52 |
|
6 dagar |
7,4 |
253,3 |
245,9 |
18 |
143,9 |
136,5 |
65 |
|
7 dagar |
11,3 |
212,5 |
201,2 |
33 |
104,5 |
93,2 |
76 |
|
8 dagar |
7,8 |
142,5 |
134,7 |
55 |
58,9 |
51,1 |
87 |
|
9 dagar |
7,0 |
35,0 |
28,0 |
91 |
18,1 |
11,1 |
97 |
|
10 dagar |
18,0 |
37,0 |
19,0 |
94 |
20,0 |
2,0 |
99 |
Figur 1
Exempel på kurvor över bionedbrytning

Figur 2
Exempel på slamadaption

C.10 BIOLOGISK NEDBRYTBARHET
AKTIVERAT SLAM – SIMULATIONSTEST
1. METOD
1.1 INLEDNING
1.1.1 Allmänna kommentarer
Metoden kan endast tillämpas på organiska testämnen som vid den använda testkoncentrationen
|
— |
är vattenlösliga i den omfattning som krävs för att tillreda testlösningarna, |
|
— |
har försumbart ångtryck inom ramen för testbetingelserna, |
|
— |
inte hämmar bakterier. |
Uppgifter om de relativa proportionerna av testämnets huvudbeståndsdelar är till nytta vid tolkningen av de erhållna resultaten, särskilt om resultaten är låga eller marginella.
Uppgifter om testämnets toxicitet för mikroorganismer kan vara till nytta vid tolkningen av låga resultat och vid valet av lämpliga testkoncentrationer.
1.1.2 Bestämning av fullständig bionedbrytbarhet (DOC/COD-analys)
Syftet med metoden är att bestämma den fullständiga bionedbrytbarheten vid mätning av avlägsnande av ämnet och eventuella metaboliter i en aktivslamanläggningsmodel vid en koncentration motsvarande 12 mg/l (eller ca 40 mg COD/l). 20 mg DOC/l verkar vara optimalt. (DOC = dissol-ved organic carbon, dvs. upplöst organiskt kol, COD = chemical oxygen demand, dvs. kemiskt syrebehov.)
Testämnets innehåll av organiskt kol (eller dess kemiska syrebehov) ska fastställas.
1.1.3 Bestämning av primär bionedbrytbarhet (specifik analys)
Syftet med metoden är att bestämma den primära bionedbrytbarheten vid mätning av avlägsnandet av ämnet vid en koncentration av ca 20 mg/l genom användning av en specifik analysmetod (lägre eller högre koncentration kan användas om analysmetoden och hänsyn till toxicitet tillåter detta). Detta möjliggör värdering av subtansens primära bionedbrytbarhet (dvs. försvinnandet av den ursprungliga kemiska förbindelsern).
Syftet med denna metod är inte att bestämma testämnets mineralisering.
En passande analysmetod för bestämning av testämnet måste föreligga.
1.2 DEFINITIONER OCH ENHETER
1.2.1 DOC/COD-analys
Nedbrytningen definieras som den de! av testämnet som avlägsnas och beräknas enligt följande formel:
|
|
[1(a)] |

där:
|
DR |
= |
procent DOC (eller COD) som avlägsnats inom den givna medeluppehållstiden med hänsyn till testämnet. |
|
T |
= |
testämnekoncentrationen i inflödet i mg DOC/l (eller mg COD/l). |
|
E |
= |
DOC- (eller COD-)koncentrationen i testenhetens utflöde i mg DOC/l/eller mg COD/1). |
|
Eo |
= |
DOC-(eller COD-)koncentrationen i blindprovenhetens utflöde i mg DOC/1/eller mg (COD/1). |
Nedbrytningen anges som den procent DOC (eller COD) som försvunnit under en given reten-tionstid i fråga om testämnet.
1.2.2 Specifik analys
Den procentdel testämne som elimineras från vattenfasen (RW) inom den givna medeluppehållstiden, beräknas enligt följande formel:
|
|
[1(b)] |

där:
|
C1 |
= |
Ämnekoncentrationen i testenhetens inflöde (mg ämne/l, bestämd genom specifik analys). |
|
C0 |
= |
Ämnekoncentrationen i testenhetens utflöde (mg ämne/1, bestämd genom specifik analys). |
1.3 REFERENSÄMNEN
När ett nytt ämne ska undersökas kan referensämnen i vissa fall vara användbara. Det gir dock ännu inte att rekommendera bestämda referensämnen.
1.4 TESTMETODSPRINCIP
Vid bestämning av den slutliga bionedbrytbarheten används parallellt med med aktiverat slam pilotenheter (OECD confirmatory testenheter eller porous potenheter). Testämnet tillförs till den ena enhetens inflöde (syntetiskt spillvatten eller spillvatten från hushåll) medan den andra enheten endas tillförs spillvatten. Vid bestämning av primär bionedbrytning med specifik analys i inflödet och utflödet används endast en enhet.
DOC-koncentrationer (eller COD-koncentrationer) mäts i utflödena eller så bestäms ämneskoncentrationen genom specifik analys.
DOC som beror på testämnet mäts inte utan anges enbart.
Vid DOC-mätningar (eller COD-mätningar) antas skillnaden mellan test- och kontrollutflödenas genomsnittskoncentrationer bero på onedbrutet testämne.
Om en specifik analysmetod används kan ändringen av stamförbindelsens koncentration mätas (primär bionedbrytning).
Enheterna kan användas enligt ”kopplingsmetoden” genom transinokulering.
1.5 KVALITETSKRITERIER
Den initiala koncentrationen är avhängig den typ av analys som företas och dess begränsningar.
1.6 METODBESKRIVNING
1.6.1 Förberedelser
1.6.1.1
Förutom när det rör sig om specifika analyser krävs det två modelltyper av samma slag. Två slags modelltyper kan användas:
OECD confirmatory test
Utrustningen (tillägg 1) består av ett lagringskärl (A) för syntetiskt spillvatten, doseringspump (B), luftningskärl (C), sedimenteringskärl (D), luftpump (E) för återföring av det aktiva slammet samt kärl (F) för uppsamling av det behandlade spillvattnet.
Kärlen (A) och (F) måste vara av glas eller lämplig plast och rymma minst 24 liter. Pump (B) måste ge ett konstant flöde syntetiskt spillvatten till luftningskärlet. Varje lämpligt system kan användas, under förutsättning att inflödet och koncentration är säkerställt. Normalt är sedimenteringskärlets
höjd (D) så inställd att luftningskärlet innehåller 3 liter blandad vätska. En sintrad luftningskub (G) är upphängd i kärl (C) vid den komiska delens spets. Den luftmängd som blåses genom luftaren ska kontrolleras med en flödesmätare (H).
”Porous pot”:
Den porösa krukan är gjord av plattor av poröst polyetylen (2 mm tjocka, maximal porstorlek 95 μm) som formas till en cylinder med en diameter av 14 cm och en konisk botten som bildar en vinkel på 45o med sidorna (figurerna 1 och 2 i tillägg 2). Den porösa krukan sätts ned i en ogenomtränglig behållare av lämpligt plastmaterial med en diameter av 15 cm som på sin cylindriska del har ett utlopp på 17,2 cm höjd vid en volym av 3 1. På insidan av den inre behållarens översta kant finns det en stark stödjande ring av lämpligt plastmaterial så att det finns 0,5 cm plats till utloppsvatten mellan den inre och den yttre behållaren.
De porösa krukorna kan ställas på botten av ett termostatstyrt vattenbad. Vid ett luftintag till botten av den inre behållaren placeras lämpliga luftningsrör.
Behållare (A) och (F) ska vara av glas eller lämpligt plastmaterial och minst kunna rymma 24 1. Pumpen (B) ska förse luftningstanken med ett konstant flöde av syntetiskt spillvatten. Varje passande system kan användas förutsatt att ett bestämt inflöde och en bestämd koncentration säkerställs.
Det krävs extra inre porösa krukor för att ersätta krukor som blivit igentäppta under användning. Igentäppta krukor läggs i en hypokloritlösning i 24 timmar och tvättas sedan av grundligt med ledningsvatten.
1.6.1.2
Membranfiltreringsapparatur och membranfilter med en porstorlek av 0,45 μm krävs. Membranfilter är lämpliga om det är garanterat att de varken frigör kol eller absorberar ämnet i filtreringssteget.
1.6.1.3
Antingen passande syntetiskt spillvatten eller spillvatten från hushåll kan användas.
Exempel på syntetiskt spillvatten:
Lös upp följande ämnen per liter vattenledningsvatten:
|
Pepton: |
160 mg |
|
Köttextrakt: |
110 mg |
|
Urea: |
30 mg |
|
NaCl: |
7 mg |
|
CaCl2.2H2O: |
4 mg |
|
MgSO4.7H2O: |
2 mg |
|
K2HPO4: |
28 mg |
Spillvatten från hushåll:
Spillvatten från hushåll uppsamlas färskt varje dag från överflödet från primärsedimenteringstanken i en reningsanläggning.
1.6.1.4
En lösning av testämnet, t.ex. 1 %, ska förberedas för tillsättning till testenheten. Ämnets koncentration måste bestämmas, så att rätt mängd kan tillsättas spillvattnet eller direkt i enheten via en andra pump för att ge den testkoncentration som krävs.
1.6.1.5
Obs: Om spillvatten frän hushåll används är det ingen idé att använda ett inokulat med låg bakteriekoncentration, men aktiverat slam kan användas.
Olika slags inokulat kan användas.
Här följer tre exempel på lämpliga inokulat:
|
a) |
Inokulat från ett sekundärt utflöde: Inokulat erhålls från ett sekundärt utflöde av god kvalitet vid ett reningsverk som huvudsakligen behandlar spillvatten från hushåll. Provet ska hållas under aeroba betingelser tills det ska användas. För att förbereda inokulatet filtreras provet genom ett grovt filter och de första 200 ml kasseras. Filtratet hålls under aeroba betingelser tills det ska användas. Inokulatet ska användas samma dag som provet tas. Minst 3 ml ska användas för inokulation. |
|
b) |
Blandat inokulat Inokulat från ett sekundärt utflöde: Se beskrivningen ovan. Inokulat från jord: 100 g trädgårdsjord (bördig, inte steril) uppslammas i 1 000 ml klorfritt dricksvatten (jordar med extremt hög halt av lera, sand eller humus är inte lämpliga). Efter omröring får utfällning av suspensionen ske i 30 minuter. Supernatanten filtreras genom ett grovt filterpapper och de första 200 ml kasseras. Filtratet syresätts omedelbart och syresättningen pågår tills filtratet ska användas. Inokulatet ska användas samma dag som provet tas. Inokulat från ytvatten: En ytterligare del av inokulat tas från ett mesosaprobt ytvatten. Provet filtreras genom ett grovt fil-trerpapper och de första 200 ml kasseras. Filtratet hålls under aeroba betingelser tills det ska användas. Inokulatet ska användas samma dag som provet tas. Lika volymer av de tre inokulatproven blandas väl och det slutliga inokulatet tas från denna blandning. Minst 3 ml ska användas till inokulering. |
|
c) |
Inokulat av aktiverat slam: En mängd (högst 3 1) aktiverat slam {med ett innehåll av suspenderade fasta beståndsdelar på upp till 2,5 g/l) som tagits från luftningstanken i ett reningsverk som huvudsakligen behandlar spillvatten från hushåll kan användas som inokulat. |
1.6.2 Förfarande
Testet testet utförs i rumstemperatur, som ska hållas konstant mellan 18 och 25 oC.
Om det är ändamålsenligt kan testet utföras vid en lägre temperatur (ned till 10 oC). Om testämnet bryts ned krävs det normalt inte någon ytterligare test. Om ämnet emellertid inte bryts ned kan testen göras om vid en konstant temperatur på 18–25 oC.
1.6.2.1
Slambildnings-/stabiliseringsperioden är den period under vilken de suspenderade fasta beståndsdelarna av det aktiverade slammet och enheternas prestationsförmåga blir stationär under de använda driftförhållandena.
Inkörningsperioden är den period som löper från den tidpunkt då testkoncentrationen tillsätts för första gången till den tidpunkt dä dess avlägsnande har nått en nivå (relativt konstant värde). Denna period får högst vara sex veckor.
Utvärderingsperioden är en treveckorsperiod, tre veckor från den tidpunkt då testämnets avlägsnande når ett relativt konstant och vanligtvis högt värde. För ämnen som visar låg eller ingen nedbrytning under de första sex veckorna beräknas utvärderingsperioden för de följande tre veckorna.
Man börjar med att fylla den eller de enheter som är nödvändiga för ett test med inokulat blandat med influent.
Luftningen (och luftpumpen (E), om det rör sig om IECD Confirmatory testenheter) och doseringspumpen (B) sätts därefter igång.
Influens utan testämne ska passera genom luftningskärlet (C) med en hastighet av antingen 1 l i timmen eller 0,5 l i timmen. Detta ger en medelretentionstid på antingen tre eller sex timmar.
Luftningshastigheten ska regleras så att innehållet i kärlet (C) ständigt hålls suspenderat och halten löst syre är minst 2 mg/l.
Skumbildning måste förhindras på lämpligt sätt. Skumdämpande medel som hämmar det aktiva slammet får inte användas.
Slam som samlats kring toppen av luftningskärlet (C) (och för OECD confirmatory testenheters vidkommande i botten av sedimenteringskärlet (D) eller i rörsystemet) ska återföras till cirkulationen minst en gång om dagen genom avborstning eller pä annat sätt.
Om slammet inte sedimenterar kan sedimenteringen förbättras genom tillsättning av en 5-procentig lösning av järnklorid, 2 ml i taget, så många gånger som behövs.
Utflöde samlas upp i kärlet (E) i 20–24 timmar och en provtagning görs efter grundlig blandning. Kärlet (E eller F) måste omedelbart regöras.
För att kunna överväga och kontrollera processens effektivitet mäter man minst två gånger i veckan det kemiska syrebehovet (COD) eller upplöst organiskt kol (DOC) både i filtratet av den ackumulerade utflödet och i det filtrerade inflödet (ett membran med en porstorlek av 0,45 μm används, varvid de första (ca) 20 ml av filtratet kasseras).
Reduceringen av COD eller DOC bör plana ut när nedbrytningen är ungefär densamma varje dag.
Halten torrämnen i det aktiva slammet i luftningskärlet bör fastställas i g/l två gånger i veckan. Man kan använda enheterna på två olika sätt: aningen bestäms det aktiverade slammets torrämneinnehåll två gånger i veckan och om halten är högre än 2,5 g/l ska överskottet av det aktiva slammet kasseras, eller avlägsnas dagligen 500 ml blandad vätska från varje kärl så att slammet får en medeluppehållstid av sex dagar.
När de mätta och uppskattade parametrarna (processens effektivitet (med hänsyn till COD- eller DOC-avlägsnande), slamkoncentrationen, slamsedimenteringsegenskaper, utloppens oklarhet, osv.) för de två enheterna är tillräckligt stabil, kan testämnet tillsättas inloppet till den ena av enheterna (i enlighet med 1.6.2.2).
Det är också möjligt att tillsätta testämnet i början av slambildningsperioden (1.6.2.1), särskilt om slammet tillsätts som inokulat.
1.6.2.2
Arbetsvillkoren är desamma som för inkörningsperioden, och det tillsätts så mycket stamlösning (ca 1 %) av testämnet till testenhetens inflöde att den önskade testämnekoncentrationen (ca 10–20 mg DOC/l eller 40 mg COD/l) i spillvattnet uppnås. Detta kan ske genom att stamlösningen blandas i spillvattnet dagligen eller med hjälp av ett särskilt pumpsystem. Denna koncentration kan uppnås gradvis. Om testämnet inte har någon toxisk verkan på det aktiverade slammet kan även högre koncentrationer testas.
Blindprovsenheten tillförs endast via inflödet utan tillförda ämnen. Lämpliga volymer av utflöden analyseras och filtreras genom membranfilter (0,45 μm) varvid de första (ca) 20 ml filtrat kasseras.
De filtrerade proven måste analyseras samma dag annars måste de förvaras med en passande metod, t.ex. genom tillsättning av 0,05 ml av en 1-procentig lösning av kvicksilverklorid (HgCl2) till varje 10 ml filtrat eller genom förvaring vid 2–4 oC i upp till 24 timmar, eller under –18 oC för längre perioder.
Inkörningsperioden, med tillsättning av testämne, bör inte vara längre än sex veckor, och bedömningsperioden bör inte vara kortare än tre veckor, dvs. omkring 14–20 bestämningar bör finnas tillgängliga för beräkning av slutresultatet.
Kopplingsmetoden:
Enheterna kopplas genom att byta 1,5 blandad vätska (inbegripet slam) mellan de två enheternas luftningskärl en gång om dagen. Om det är fråga om starkt absorberande testämnen tas endast 1,5 1 överstående vätska från sedimenteringskärlen, som hälls i den andra enhetens aktivslamkärl.
1.6.2.3
Två slags analysmetoder kan användas för att följa ämnets beteende:
DOC och COD
Dubbla bestämningar av DOC-koncentrationer görs med kolanalysator och/eller COD-värden enligt hänvisningarna (2).
Specifik analys
Testämnekoncentrationerna bestäms genom en passande analysmetod. När det är möjligt ska specifik bestämning av det ämne som är absorberat på slammet genomföras.
2. DATA OCH UTVÄRDERING
2.1 KOPPLINGSMETODEN
När kopplingsmetoden används beräknas det dagliga avlägsnandet, DR, i enlighet med 1.2.1.
Detta dagligen beräknade värde för avlägsnandet korrigeras genom DRc, för att ta hänsyn till den materialöverföring som orsakas av transinokulering. Detta sker genom utjämning (2) för en genomsnittlig retentionstid av tre timmar och efter utjämning (3) för en genomsnittligt retentionstid av sex timmar.
|
|
[2] |
|
|
(3) |


Genomsnittet av DRc-värdena beräknas, samt standardavvikelse™ erhålls efter utjämning (4).
|
|
(4) |

där:
|
SDRc |
= |
DRc-värdenas standardavvikelse. |
|
|
= |
Genomsnitt av DRc-värdena. |
|
n |
= |
Antal bestämningar. |

Outliers i DRc-värdena elimineras genom ett lämpligt statiskt förfarande, t.ex. Nalimov (hänvisning 6), på 95 % sannolikhetsnivå, och genomsnitt och standardavvikelse för DRc-data utan outliers beräknas igen.
Slutresultatet beräknas sedan med utjämning (5) som
|
|
(5) |

där:
|
tn_1a;α |
= |
Tabellvärden av t för n värdepar av E och Eo och med en statistisk konfidens på P (P = 1 – a), där P sätts till 95 % (hänvisning 1). |
Resultaten anges som genomsnittet med toleranser av 95 % sannolikhetsnivå, den motsvarande standardavvikelsen och antalet data i DRc-data utan ouliers samta antalet outliers, t.ex.:
|
DRc |
= |
98,6 % ±2,3 % DOC-avlägsnande. |
|
s |
= |
4,65 % DOC-avlägsnande. |
|
n |
= |
18. |
|
x |
= |
Antal outliers. |
2.2 METOD UTAN KOPPLING
Enheternas prestationsförmåga kan kontrolleras på följande sätt:

Det dagliga avlägsnandet kan avbildas grafiskt så att det framgår om det finns några tendenser til t. ex. acklimatisering.
2.2.1 Tillämpning av COD/DOC-bestämningar
Det dagliga procentuella avlägsnandet, % DR, beräknas enligt 1.2.1.
Genomsnittet av DR-värdena beräknas samt standardavvikelsen erhålls genom följande formel:
|
|
(6) |

där
|
S DR |
= |
DRi-värdenas standardavvikelse. |
|
|
= |
Genomsnitt i DRi-värdena |
|
n |
= |
Antal bestämningar. |

Outliers i DR-värdena elimineras genom ett lämpligt statiskt förfarande, t.ex. Nalimov (hänvisning 6), på 95 % sannolikhetsnivå, och genomsnitt och standardavvikelse för DRc-data utan outliers beräknas igen.
Slutresultatet beräknas sedan med utjämning (7) som
|
|
(7) |

där:
|
tn-1;α |
= |
Tabellvärden av t för n värdepar av E och Eo och med en statistisk konfidens på P (P = 1 – a), där P sätts till 95 % (hänvisning 1). |
Resultaten anges som genomsnittet med toleranser av 95 % sannolikhetsnivå, den motsvarande standardavvikelsen och antalet data i DR-data utan ouliers samt antalet outliers, t.ex.:
|
DR |
= |
(98,6 % ±2,3 %) DOC-avlägsnande. |
|
s |
= |
4,65 % DOC-avlägsnande. |
|
n |
= |
18. |
|
x |
= |
Antal outliers. |
2.2.2 Tillämpning av specifik analys
Den procentdel testämne som elimineras från vattenfasen (Rw) beräknas enligt 1.2.2.
3. RAPPORTERING
3.1 TESTRAPPORT
Testrapporten ska om möjligt innehålla följande:
|
— |
Det schema som återges i tillägg 3, som visar arbetsvillkoren för testet. |
|
— |
Vald apparatur (OECD confirmatory test eller porous pot). |
|
— |
Vald arbetsmetod. Kopplingsmetod eller ej. |
|
— |
Typ av spillvatten: syntetiskt spillvatten eller spillvatten från hushåll. Om det rör sig om spillvatten från hushåll anges datum och plats för provtagning. |
|
— |
Använt inokulat med angivelse om datum och plats för provtagning. |
|
— |
Angivelse av analysmetoden, om specifika analyser gjorts, och en beskrivning av den. |
|
— |
Avbildning av COD- eller DOC-avlägsnande mot tiden, inbegripet inkörnings- och bedömningsperioder. |
|
— |
Analytisk återhämtning av testämnet som COD eller DOC i stamlösningen. |
|
— |
Avbildning av det procentuella avlägsnandet av testämne från det vattniga fasen mot tiden (inkörningsperiod och värderingsperiod), om specifika analyser gjorts. |
|
— |
Det genomsnittliga avlägsnandet av DOC, COD eller testämne samt standardavvikelse beräknat från resultaten från bedömningsperioden, dvs. när vid ett stabilt avlägsnande av testämne. |
|
— |
Avbildning av aktivslamkoncentration mot tiden. |
|
— |
Eventuella kommentarer om det aktiverade slammet (kasserat överstående slam, förekomst av klumbildningarn, FeCl3, etc.). |
|
— |
Testämnekoncentration som använts vid testet. |
|
— |
Eventuella resultat av analyser av slammet. |
|
— |
Alla upplysningar och testresultat i fråga om testämnet och eventuella referensämnen. |
|
— |
Vetenskapliga motiveringar för eventuella ändringar av testförfarandet. |
3.2 TOLKNING AV RESULTAT
Om den procent testämne som avlägsnas från vattenfasen är låg kan detta bero på att testämnet hämmar mikroorganismerna. Detta kan även avslöjas genom lysis och förlust av slam, som ger grumlig överstående vätska, samt nedsatt effektivitet i fråga av avlägsnandet av COD (eller DOC).
Fysikalisk-kemisk adsorbtion kan dock i vissa fall spela en viss en roll. Skillnaden mellan biologisk verkan på molekylen och fysikalisk-kemisk adsorbtion kan avslöjas genom analys av slammet enligt passande desorption.
Det är nödvändigt att göra ytterligare tester om man ska kunna skilja på bionedbrytning (eller delvis bionedbrytning) och adsorption.
Detta kan göras på flera olika sätt, men det mest övertygande är att använda överstående vätska som inokulum i en bassettest (helst ett respirometriskt test).
Observation av en hög procentuell förlust av DOC (COD) orsakas av bionedbrytning, varvid det däremot vid låg förlust inte kan skiljas mellan bionedbrytning och eliminering. Som ett exempel kan nämnas att om ett oupplösligt ämne påvisar en hög adsorbtionskonstant av 98 %, och 10 % över-stående slam kasseras dagligen, är en eliminering på upp till 40 % möjlig. Om det kasseras 30 % överslående slam kan eliminering på grund av adsorbtion på och förlust tillsammans med överstående slam komma upp till 65 % (hänvisning 4).
Vid användning av en specifik analysmetod måste man vara uppmärksam på förhållandet mellan ämnets struktur och den använda specifika analysmetoden. I detta fall kan det observerade fenomenet inte tolkas som en mineralisering av ämnet.
4. HÄNVISNINGAR
|
1. |
OECD (1981), Test Guideline 303 A, rådets beslut K(81) 30 slutlig. |
|
2. |
Bilaga V C.9 Nedbrytning – kemiskt syrebehov, kommissionens direktiv 84/449/EEG, Europeiska gemenskapernas officiella tidning L 251, 19.9.1984. |
|
3. |
Painter, H. A. och King, E. F. (1978), WRC Porous-Pot method for assessing biodegradability. Technical Report TR70, Water Research Center, UK. |
|
4. |
Wierich, P. och Gerike, P. (1981), ”The fate of soluble, recalcitrant, and adsorbing compounds in activated Sludge plants” – Ecotoxicology and Environmental Safety, Vol. 5, nr 2, s. 161–171. |
|
5. |
Rådets direktiv 82/242/EEG och 82/243/EEG, Europeiska gemenskapernas officiella tidning L 109, 22.4.1982, om ändring av rådets direktiv 73/404/EEG och 73/405/EEG om bio- nedbrytbarhet för tvätt- och rengöringsmedel, Europeiska gemenskapernas officiella tidning L 347, 17.12.1973. |
|
6. |
Streuli, H. (1980), Fehlerhafte Interpretation und Anwendung von Ausreiβertests, inbesondere bei Ringversuchen zur Überprüfung analytisch-chemischer Untersuchungsmethoden, Fresenius-Zeitschrift fur Analytische Chemie, 303, s. 406–409. |
Tillägg 1
Figur 1

Figur 2

Tillägg 2
Figur 1
Utrustning för bestämning av bionedbrytbarhet

Figur 2
3-liters poröst luftningskärl

Tillägg 3
Arbetsvillkor för simuleringstest för aktiverat slam
Sätt ett kryss för varje grupp
Apparatur
|
OECD Confirmatory |
|
|
Porous Pot |
|
Arbetsmetod
|
Enkel enhet |
|
|
Kopplade enheter |
|
|
Icke-kopplade enheter |
|
Transinokulering
|
Ingen |
|
|
Aktiverat slam |
|
|
Överstående vätska |
|
Medeluppehållstid
|
Tre timmar |
|
|
Sex timmar |
|
Basnäringsämne
|
Spillvatten från hushåll |
|
|
Syntetiskt spillvatten |
|
Unokulat
|
Sekundärt spillvatten |
|
|
Blandat inokulat |
|
|
Aktiverat slam |
|
Tillsättning av testämne
|
Initialt |
|
|
Gradvis ökning |
|
|
Efter slambildning |
|
Analys
|
Specific |
|
|
COD |
|
|
DOC |
|
C.11 BIOLOGISK NEDBRYTBARHET
AKTIVERAT SLAM – RESPIRATIONSHÄMNINGSTEST
1. METOD
1.1 INLEDNING
Med denna metod bedöms ett testämnes verkan på mikroorganismer genom mätning av respirationshastigheten enligt bestämda villkor vid olika koncentrationer av testämnet.
Syftet med metoden är att ge en snabb screening-metod för identifiering av ämnen som kan påverka ett aerobt mikrobiellt reningsverk, och att indikera lämpliga icke-hämmande koncentrationer av testämnet som kan användas vid bionedbrytbarhetstest.
Ett förtest kan utföras före det slutliga testet. Detta test ger upplysningar om den koncentrationsintervall som ska användas vid huvudtestet.
1 testet ingår två kontrolltester, en vid början och en i slutet av testserierna. Dessutom bör varje parti aktiverat slam också kontrolleras genom användning av en referensämne.
Metoden är enklast att använda för ämnen som på grand av sin oupplöslighet i vatten och ringa flyktighet sannolikt kommer att förbli i vatten.
För ämnen med begränsad löslighet i testämnet kan det visa sig omöjligt att bestämma EC50.
Resultat som grundas på syreupptagning kan leda till felaktiga slutsatser om ämnet har en tendens att avkoppla oxidativ fosforylering.
Det är användbart att känna till följande när man utför testet:
|
— |
Vattenlöslighet, |
|
— |
Ångtryck. |
|
— |
Strukturformel. |
|
— |
Testämnets renhet. |
Rekommendation
Aktiverat slam som innehåller potentiellt patogena organismer bör hanteras med försiktighet.
1.2 DEFINITIONER OCH ENHETER
Respirationshastigheten är syreförbrukningen hos mikroorganismer i aerobt spillvattenslam och uttrycks normalt i mg O2 per mg slam per timme.
För att beräkna ett testämnes hämmande verkan i en bestämd koncentration uttrycks respirationshastigheten som en procentdel av genomsnittet av de två kontrollrespirationshastigheterna.

där:
|
Rs |
= |
Syreförbrukning vid testade koncentrationer av testämnet. |
|
RC1 |
= |
Syreförbrukningen, kontroll 1. |
|
RC2 |
= |
Syreförbrukningen, kontroll 2. |
I samband med denna metod är EC50 den koncentration av testämne där respirationen är 50 % av den som kontrolltestet visar under de förhållanden som beskrivs i denna metod.
1.3 REFERENSÄMNEN
Det rekommenderas att använda 3,5-diklorfenol, som man vet hämmar respirationen, som referensämne och att testa det för EC50 för varje parti aktiverat slam för att kontrollera att slammets känslighet inte är onormal.
1.4 TESTMETODSPRINCIP
Respirationshastigheten i aktiverat slam tillfört en standardmängd syntetiskt spillvatten mäts efter en kontrolltid av 30 minuter och/eller tre timmar. Även respirationshastigheten i samma aktiverade slam vid olika koncentrationer av testämnet under i övrigt identiska förhållanden mäts. Testämnets hämmande verkan vid en bestämd koncentration uttrycks som en procentdel av medelrespirationshastigheten i två kontrolltester. Ett EC50-värde beräknas ut i från bestämningar vid olika koncentrationer.
1.5 KVALITETSKRITERIER
Testresultaten är giltiga om
|
— |
respirationshastigheten i de två kontrolltesterna ligger inom 15 % av varandra, |
|
— |
EC50 (30 minuter och/eller tre timmar) för 3,5-diklorfenol ligger inom det accepterade området av 5–30 mg/l. |
1.6 METODBESKRIVNING
1.6.1 Reagenser
1.6.1.1
Testämnekoncentrationerna nyframställs vid testets inledning genom användning av en stamlösning. En stamlösningskoncentration av 0,5 g/l är lämpligt om nedanstående förfarande används.
1.6.1.2
En lösning av 3,5-diklorfenollösning kan t.ex. framställas genom att lösa 0,5 g 3,5-diklorfenol i 10 ml 1 M NaOH vilket späds med destillerat vatten till ca 30 ml. Därefter tillsätts under omrörning 0,5 M H2SO4 upp till den punkt då utfällning börjar – det krävs ca 8 ml 0,5 M H2SO4 – och slutligen späds blandningen med destillerat vatten till 1 I. pH-värdet ska därefter vara omkring 7–8.
1.6.1.3
Syntetiskt spillvatten framställs genom att följande ämnen löses upp per liter vatten:
|
— |
16 g pepton, |
|
— |
1 1 g köttextrakt, |
|
— |
3 g urea, |
|
— |
0,7 g NaCl, |
|
— |
0,4 g CaCl2.2H2O, |
|
— |
0,2 g MgSO4.7H2O, |
|
— |
2,8 g K2HPO4. |
Not 1: Detta syntetiska spillvatten är 100 gånger så koncentrerat som det som beskrivs i OECD Technical Report ”Proposed method for the determination of the biodegradability of surfactants used in synthetic detergents” av den 11 juni 1976, med tillsättning av dikaliumvätefosfat.
Not 2: Om det framställda mediet inte används omedelbart ska det förvaras i mörker vid 0 — 4 oC, dock högst en vecka och under förhållanden som inte ger anledningen till ändring i sammansättningen. Mediet kan också steriliseras innan det förvaras, eller så kan man tillsätta pepton och köttextrakt strax innan testet utförs. Omedelbart före användningen blandar man noggrant och pH-värdet justeras.
1.6.2 Apparatur
Mätapparatur: Den exakta utformingen är inte avgörande. Det får dock inte finnas luftmellanrum mellan vätska och propp, och sonden ska sluta tätt till kolvens hals.
Normal laboratorieutrustning krävs, särskilt följande:
|
— |
Mätapparatur, |
|
— |
Luftningsapparatur, |
|
— |
pH-elektrod och pH-mätutrustning, |
|
— |
O2-elektrod. |
1.6.3 Förberedelse av inokulat
Som mikrobiellt inokulat till testet används aktiverat slam från ett reningsverk som huvudsakligen behandlar spillvatten från hushåll.
1 laboratoriet kan grövre partiklar om nödvändigt avlägsnas genom utfällning under en kort period, t.ex. 15 minuter, följt av dekantering av det översta laget med finare slampartiklar. Alternativt kan slammet användas efter några få sekunders blandning i en blandare.
Dessutom kan, om man misstänker närvaron av hämmande material, slammet tvättas med kranvatten eller en isotonisk lösning. Efter centrifugering dekanteras överstående lösning (detta förfarande upprepas tre gånger).
En mindre mängd slam vägs och torkas. Utifrån detta kan man beräkna den mängd vått slam som måste suspenderas i vatten för att erhålla ett aktivt slam med ett innehåll av fasta beståndsdelar suspenderad i en blandad vätska på 2–4 g/l. Detta motsvarar en koncentration av 0,8–1,6 g/l i testmediet om nedan beskrivna förfarande följs.
Om slammet inte kan användas samma dag som det insamlas tillsätts 50 ml syntetiskt spillvatten till varje liter aktiverat slam som framställts enligt beskrivningen ovan. Slammet luftas därefter under natten vid 20 ± 2 oC. Därefter hålls det luftat tills det används nästa dag. Före användningen kontrolleras pH-värdet som vid behov justeras till pH 6–8. De fasta beståndsdelarna som är suspenderade i den blandade vätskan bestäms i enlighet med beskrivningen i föregående avsnitt.
Om man måste använda samma parti slam under de följande dagarna (högst fyra dagar) tillsätts ytterligare 50 ml syntetiskt spillvatten vid varje arbetsdags slut.
1.6.4 Testförfarande
|
Varaktighet/kontakttid: |
30 minuter och/eller tre timmar under luftning |
|
Vatten: |
Dricksvatten (om nödvändigt befriat från klor) |
|
Lufttillförsel: |
Ren, oljefri luft. Luftgenomströmning 0,5–1,0 l/min |
|
Mätapparatur: |
Flatbottnad kolv som t.ex. en BOD-kolv (se figur 1) |
|
Syremätning: |
Syreelektrod med tillhörande skrivare |
|
Näringslösning: |
Syntetiskt spillvatten (se ovan) |
|
Testämne: |
Testlösningen framställs kort innan testets inledning |
|
Referensämne: |
T.ex. 3,5-diklorfenol (minst tre koncentrationer) |
|
Kontrolltest: |
Inokulerat prov utan testämne |
|
Temperatur: |
20 ± 2 oC |
Nedan lämnas förslag till ett experimentförfarande som kan följas för både test- och referensämnet i tretimmars kontaktperioder.
Flera kärl används (t.ex. 1-liters glasbägare).
Minst fem koncentrationer med ett fast koncentrationsförhållande som helst inte överstiger 3,2 används.
Vid tidpunkten ”0” späds 16 ml av det syntetiska spillvattnet med vatten till 300 ml. 200 ml av det mikrobiella inokulatet tillsätts och hela blandningen (500 ml) hälls i ett första kärl (första kontroll C1).
Testkärlen ska genomluftas kontinuerligt för att säkerställa att det upplösta O2 inte faller under 2,5 mg/l, och att O2-koncentrationen är i storleksordningen 6,5 mg/l omedelbart innan mätningen av respirationshastigheten.
Vid tidpunkten ”15 minuter” (15 minuter är ett godtyckligt men bekvämt intervall) utförs ovanstående förfarande, bortsett från att det tillsätts 100 ml testämnestamlösning till de 16 ml syntetiskt spillvatten, före det tillsätts vatten till 300 ml och mikrobiellt inokulat till totalt 500 ml. Denna blandning hälls sedan i en annan glasbägare och luftas enligt beskrivningen ovan. Detta förfarande repeteras med 15 minuters intervaller med olika mängder testämnestamlösning, så att man erhåller en rad kärl med olika koncentrationer av testämnet. Till slut framställs en andra kontroll (C2).
Efter tre timmar registreras pH-värdet och efter en noggrann blandning hälls en del av innehållet i det första kärlet över i mätapparaten, och respirationshastigheten mäts under en period av upp till tio minuter.
Denna bestämning upprepas på innehållet i varje kärl vid 15-minutersintervaller, på ett sådant sätt att kontakttiden för varje behållare blir tre timmar.
Referensämnet testas med varje parti mikrobiellt inokulat på samma sätt.
Ett annat system (t.ex. mer än en syremätare) kan vara nödvändigt när man ska utföra mätningar efter 30 minuters kontakt.
Om det krävs mätning av den kemiska syreförbrukningen framställs ytterligare kärl som innehåller testämne, syntetiskt spillvatten och vatten, men inte aktiverat slam. Syreförbrukningen mäts och registreras efter luftning i 30 minuter och/eller tre timmar (kontakttid).
2. DATA OCH UTVÄRDERING
Respirationshastigheten beräknas utifrån skrivkurvan som O2/l x h mellan ca 6,5 mg O2/l och 2,5 mg O2/l, eller under en tiominutersperiod, om respirationshastigheten är låg. Den del av respirationskurvan över vilken man mäter respirationshastigheten ska vara linjär.
Om respirationshastigheterna i de två kontrolltesterna inte ligger inom 15 % av varandra, eller om referensämnets EC50 (30 minuter och/eller tre timmar) inte ligger inom det accepterade området (5–30 mg/l för 3,5-diklorfenol), är testet ogiltigt och måste göras om.
Hämningsprocenten beräknas vid varje testkoncentration (se 1.2). Hämningsprocenten avtecknas mot koncentrationen på semilogaritmiskt papper (eller probit), och ett EC50-värde utläses.
95 %-konfidensgränserna for EC50-värdena kan bestämmas genom standardförfaranden.
3. RAPPORTERING
3.1 TESTRAPPORT
Testrapporten ska om möjligt innehålla följande:
|
— |
Testämne: data för kemisk identifiering. |
|
— |
Testsystem: källa, koncentration och eventuell förbehandling av det aktiverade slammet. |
|
— |
Testbetingelser:
|
|
— |
Resultat:
|
3.2 TOLKNING AV RESULTAT
EC50-värdet bör endast betraktas som vägledande för testämnets sannolika toxicitet för antingen behandling av spillvatten med aktiverat slam eller mikroorganismer i spillvatten, eftersom de komplexa växelverkningar som förekommer i miljön inte kan simuleras korrekt i ett laboratorietest. Dessutom kan testämnen som har en hämmande effekt på ammoniakoxidering även producera atypiska hämningskurvor. Sådana kurvor måste därför tolkas med försiktighet.
4. HÄNVISNINGAR
|
1. |
International Standard ISO 8192 – 1986. |
|
2. |
Broecker, B. och Zahn, R. (1977), Water Research 11, s. 165. |
|
3. |
Brown, D., Hitz, H. R. och Schaefer, L. (1981), Chemosphere 10, s. 245. |
|
4. |
ETAD (Ecological and Toxicological Association and Dyestuffs Manufacturing Industries), Recommended Method No. 103, även beskriven av: |
|
5. |
B. Robra (1976), Wasser/Abwasser 117, s. 80. |
|
6. |
W. Schaefer (1977), Textilveredlung 6, s. 247. |
|
7. |
OECD (1981), Test Guideline 209, rådets beslut K(81) 30 slutlig. |
C.12 BIOLOGISK NEDBRYTBARHET
MODIFIERAT SCAS-TEST
1. METOD
1.1 INLEDNING
Syftet med denna metod är att bedöma den potentiella fullständiga bionedbrytbarheten av vattenlösliga, icke-flyktiga organiska ämnen när dessa utsätts för relativt höga koncentrationer av mikroorganismer under en längre perioder. Under denna period hålls mikroorganismerna vid liv genom daglig tillförsel av näring bestående av bottensatsen av spillvatten. (I samband med helgen kan spillvattnet förvaras vid 4 oC. Alternativt kan det syntetiska spillvattnet till OECD Confirmatory test användas.)
Fysikalisk-kemisk adsorbtion kan inträffa på de suspenderade fasta ämnena, vilket man måste ta hänsyn till vid tolkningen av resultaten (se 3.2).
Till följd av den flytande fasens långa uppehållstid (36 timmar), och den med mellanrum tillförda näringen, simulerar testet inte de normala villkoren i en spillvattenanläggning. De resultat som uppnås med olika testämnen visar att denna metod har en hög bionedbrytningspotential.
De förhållanden som skapas i denna metod underlättar i hög grad val och/eller adaption av mikroorganismer som är i stånd till att bryta ned testämnet. (Detta förfarande kan dessutom användas för att framställa acklimatiserade inokulat för användning i andra test.)
I detta test bedöms testämnenas fullständiga bionedbrytbarhet genom mätning av koncentrationen av upplöst organiskt kol (DOC). Koncentrationen av DOC bör hellre bestämmas efter upprensning av den sura lösningen än som skillnaden mellan total C och oorganiskt C.
Vid samtidig användning av en specifik analysmetod kan testämnet primära nedbrytbarhet (nedbrytning av en ursprunglig kemisk förbindelse) bedömas.
Denna metod kan endast användas på organiska testämnen som vid den i analysen använda koncentrationen
|
— |
är lösliga i vatten, |
|
— |
har ett obetydligt ångtryck, |
|
— |
inte hämmar bakterierna, |
|
— |
inte absorberas väsentligt i testsystemet, |
|
— |
inte förloras genom att testämnet skummar. |
Testämnet innehåll av organiskt kol ska vara känt.
Upplysningar om de relativa mängder av testämnets huvudbeståndsdelar kan vara till h)älp vid tolkningen av de uppnådda resultaten, särskilt när resultaten är låga eller marginella.
Upplysningar om ämnets toxicitet för mikroorganismer kan vara användbar vid tolkningen av låga resultat och vid valet av en lämplig testkoncentration.
1.2 DEFINITIONER OCH ENHETER
|
CT |
= |
Koncentration av organiskt kol i testämnet, i närvaro eller frånvaro av bottensats av spillvatten i början av luftningsperioden (mg/l). |
|
Ct |
= |
Koncentrationen av upplöst organiskt kol i den överstående vätskan vid avslutningen av luftningsperioden (mg/l). |
|
Cc |
= |
Koncentrationen av upplöst organiskt kol i kontrollens överstående vätska vid avslutning av luftningsperioden (mg/l). |
Bionedbrytbarheten definieras i detta test som förlusten av organiskt kol. Bionedbrytningen kan uttryckas som:
|
1. |
Den procentuella förlusten av Dda av den mängd ämne som tillförs dagligen:
där:
|

|
2. |
Den procentuella förlusten av Dssd av den mängd somm finns närvarande vid varje dags början:
där:
Indexen i och (i + 1) hänvisar till testdagen. Utjämning 2(a) rekommenderas om spillvattnets innehåll av upplöst organiskt kol varierar från dag till dag medan utjämning 2(b) kan användas när mängden av upplöst organiskt kol är förhållandevis konstant från dag till dag. |
1.3 REFERENSÄMNEN
I vissa fall kan referensämnen vara användbara vid undersökning av ett nytt ämne. Ett bestämt referensämne kan dock inte rekommenderas.
Upplysningar om flera olika ämnen som är utvärderade i ringtester tillhandahålls (se tillägg 1) först och främst för att testmoden av och till kan kalibreras, men också för att resultaten kan jämföras om en annan metod används.
1.4 TESTMETODSPRINCIP
Aktiverat slam från ett biologiskt reningsverk placeras i en semi-kontinuerlig slamenhet (SCAS = semi-continous activated sludge). Testämnet och bottensatsen av spillvatten från hushåll tillförs och blandningen luftas i 23 timmar. Luftningen stoppas sedan och slammet ges tid att sätta sig och överstående vätska kasseras.
I det slam som finns kvar i luftningskammaren blandas sedan en ytterligare passande mängd av testämnet, varefter cykeln upprepas.
Bionedbrytning konstateras genom bestämning av innehållet av upplöst organiskt kol i överstående vätska. Detta resultat jämförs med resultaten från analysen av vätskan från en kontrollenhet, som endast tillförts bottensats av spillvatten.
Om en specifik analysmetod används kan änderingar av koncentrationen av den ursprungliga ämnet till följd av bionedbrytning mätas (primär bionedbrytbarhet).
1.5 KVALITETSKRITERIER
Denna metods reproducerbarhet, grundad på förlust av upplöst organiskt kol, är ännu inte fastställd. (I fråga om primär bionedbrytning har mycket tillfredsställande data uppnåtts för lätt ned-brytliga ämnen.)
Metodens känslighet är avhängig i vilken utsträckning variabiliteten i blindproven och i mindre grad av den korrekthet med vilken innehållet av upplöst organiskt kol kan bestämmas, eller av vätskans innehåll av testämne vid inledningen av varje enskild cykel.
1.6 METODBESKRIVNING
1.6.1 Förberedelse
Ett tillräckligt antal rena luftningsenheter, alternativt kan den ursprungliga 1,5 liters SCA-testenheten användas, och lufttillförselslangar (figur 1) för varje enskild testämne och till kontrollen sätts ihop. Den komprimerade luft som tillförs testenheterna rensas med ett vaddfilter och bör vara fri från organiskt kol och vara mättat med vatten för att minska ångförlust.
Ett prov av blandad vätska, innehållande 1–4 g suspenderat fast ämne/l, tas från en aktiverad slamanläggning. Cirka 150 ml av den blandade vätskan krävs för varje luftenhet.
Stamlösningar av testämnet framställs i destillerat vatten. Normalt krävs en koncentration av 400 mg organiskt kol per liter, vilket ger en koncentration av testämnet av 20 mg/l vid inledningen av varje luftningscykel, såvida inte bionedbrytning inträffar.
Högre koncentrationer kan användas om toxiciteten för mikroorganismer tillåter detta.
Slamlösningarnas innehåll av organiskt kol mäts.
1.6.2 Testbetingelser
Testet bör utföras vid 20–25 oC.
En hög koncentration av aeroba mikroorganismer (1–4 g suspenderad fast ämne per liter) används, och den effektiva uppehållstiden är 36 timmar. Det kolämneinnehållande materialet i det tillförda spillvattnet oxideras grundligt, normalt inom åtta timmar efter inledning av varje enskild luftningscykel. Därefter respirerar slammet endogent under resten av luftningsperioden, under vilken det enda tillgängliga substratet är testämnet, om inte också detta metaboliseras snabbt. Dessa förhållanden, kombinerade med daglig återinokulation av proven när spillvatten från hushåll används som medium, ger mycket goda förhållanden för såväl acklimatisering som en hög grad av bionedbrytning.
1.6.3 Genomförande av testet
Man framställer ett prov av blandad vätska från en laboratorieenhet som övervägande behandlar spillvatten från hushåll eller från en lämplig aktiverad slamanläggning, varvid proven förvaras under aeroba förhållanden tills de används i laboratoriet. Varje luftningsenhet samt kontrollenheten fylls på med 150 ml (om det ursprungliga SCAS-testet används multipliceras de angivna volymerna med 10) blandad vätska, och luftning inleds. Efter 23 timmar stoppas luftningen och slammet får sätta sig i 45 minuter. Hanen på varje kolv öppnas successivt och 100 ml överstående vätska uttas. Ett prov av bottensatsen av spillvatten från hushåll tas omedelbart före användning och 100 ml av detta tillsätts det resterande slammet i varje luftningsenhet. Luftningen inleds på nytt. På detta stadium tillsätts inget testämne. Enheterna tillsätts dagligen spillvatten från hushåll men endast upp till den tidpunkt utfällningen resulterar i en klar överstående vätska. Detta tar normalt upp till två veckor, varefter det upplösta organiska kolet i den överstående vätskan vid avslutningen av varje luftningscykel närmar sig ett konstant värde.
Vid avslutningen av denna tidsperiod blandas de olika bottensatserna av slamproverna och 50 ml av detta blandade slamprov tillsätts till varje enhet.
95 ml bottensats av spillvatten och 5 ml vatten tillsätts till kontrollenheterna, och 95 ml av bottensatsen av spillvattnet plus 5 ml av den ifrågavarande stamlösningen av testämnet (400 mg/l) till testenheterna. Luftningen påbörjas på nytt och fortsätter i 23 timmar. Slammet sätter sig i 45 minuter och överstående vätska avlägsnas och analyseras för upplöst organiskt kol.
Ovanstående påfyllnings- och avtappningsförfarande upprepas dagligen under hela testet.
Innan utfällning kan det vara nödvändigt att tvätta enheternas väggar för att förhindra ackumulering av fasta ämnen över vätskenivån. Lör att undvika korskontaminering används en separat skrapa eller borste för varje enskild enhet.
Idealt bör innehållet av upplöst organiskt kol i överstående vätskor bestämmas dagligen, även om mindre regellbundna analyser kan tillåtas. Före analysen filtreras vätskorna genom tvättade membranfilter med en porstorlek av 0,45 μm, eller så centritugeras vätskorna. Membranfilter är lämpliga om det är fastställt att de varken avger kolämne eller adsorberar ämnet under filtrering. Provens temperatur får inte översiga 40 oC så länge det är i centrifugen.
Testens varaktighet är obestämd för ämnen som uppvisar obetydlig eller ingen bionedbrytning. Erfarenheten visar att detta bor vara minst tolv veckor, men inte längre än 26 veckor.
2. DATA OCH UTVÄRDERING
Värdena för innehållet av upplöst organiskt kol i de överstående vätskorna i kontrollenheten avbildas mot tiden i ett koordinatsystem.
Efterhand som bionedbrytning sker närmare sig nivån i testenheten nivån i kontrollenheten. När skillnaden mellan de två nivåerna är konstant vid tre på varandra följande mätningar genomförs ett tillräckligt stort antal ytterligare mätningar för en statistisk behandling av data, och den procentuella bionedbrytningen av testämnet beräknas (Dda eller Dssd, se 1.2).
3. RAPPORTERING
3.1 TESTRAPPORT
Testrapporten ska om möjligt innehålla följande:
|
— |
Fullständiga uppgifter om slag av spillvatten, om vilken typ av enhet som har använts samt om försöksresultaten rörande testämnet, ett eventuellt referensämne, och blindproven. |
|
— |
Temperatur. |
|
— |
Kurva för förlusten av organiskt kol, med beskrivning av beräkningsmetoden (se 1.2). |
|
— |
Datum och plats för provtagning av aktiverat slam och spillvatten, status angående adaption, koncentration, osv. |
|
— |
Vetenskapliga motiveringar för eventuella ändringar av testförfarandet. |
|
— |
Underskrift och datum. |
3.2 TOLKNING AV RESULTAT
Eftersom det ämne som kan testas med denna metod inte kommer att vara lätt bionedbrytligt kommer förlusten av organiskt kol, som uteslutande beror på bionedbrytning, normalt att ske gradvis över dagar eller veckor, undantaget sådana fall där acklimatiseringen är plötslig, vilket framgår av en abrupt förslut efter några veckor.
Fysikalisk-kemisk adsorbtion kan emellertid i vissa fall spela en betydande roll. Detta är fallet när det vid inledningen inträder ett fullständigt eller ett delvist försvinnande av det tillsatta upplösta organiska kolet. Vad som därefter inträffar beror på olika faktorer, såsom adsorptionsgraden och koncentrationen av suspenderat fast ämne i det kasserade spillvattnet. Skillnaden mellan koncentrationen av upplöst organiskt kol i kontroll- och testenheternas överstående vätska ökar normalt gradvis från det först inträffade värdet, och denna skillnad förblir sedan vid det nya värdet under resten av testet, såvida inte acklimatisering sker.
När man ska skilja mellan bionedbrytning (eller delvis bionedbrytning) och adsorption krävs ytterligare analyser. Detta kan göras på flera sätt, men den mest pålitliga metoden är att använda överstående vätska, eller slam, som inokulat i en basistest (helst ett respirometiskt test).
Testämnen som resulterar i ett omfattande, icke-adsorptivt försvinnande av upplöst organiskt kol bör i denna test betraktas som potentiellt bionedbrytbara. Delvist icke-adsorptivt försvinnande tyder på att ämnet i varje fall är bionedbrytbart till viss grad.
Obetydligt eller inget försvinnande av upplöst organiskt kol kan bero på testämnets hämning av mikroorganismer. Detta kan också avslöja lysis och förlust av slam vilket ger oklara överstående vätskor. Testet bör i sådana fall göras om med användning av en lägre koncentration av testämnet.
Användning av en specifik analysmetod eller C-14-markerat testämne kan ge större känslighet. Om ett C-14-testämne används kommer återhämtning av 14CO2 att bekräfta att bionedbrytning inträffat.
Om det dessutom anges resultat för primär bionedbrytning bör det om möjligt ges en förklaring på den förändring av den kemiska strukturern som medför att det ursprungliga testämnet inte kan påvisas.
Valideringen av analysmetoden ska anges tillsammans med värdena för blindprovmediet.
4. HÄNVISNINGAR
|
1. |
OECD (1981), Test Guideline 302 A, rådets beslut K(81) 30 slutlig. |
Tillägg 1
SCAS-test: Exempel på resultat
|
Ämne |
cT (mg/l) |
Ct- Cc (mg/l) |
Procent bionedbrytning |
Testets längd (dagar) |
|
4-acetylaminobensulfonat |
17,2 |
2,0 |
85,0 |
40 |
|
Tetrapropylenbenzensulfonat |
17,3 |
8,4 |
51,4 |
40 |
|
4-nitrofenol |
16,9 |
0,8 |
95,3 |
40 |
|
Dietylenglykol |
16,5 |
0,2 |
98,8 |
40 |
|
Anilin |
16,9 |
1,7 |
95,9 |
40 |
|
Cyklopentantetrakavoxylat |
17,9 |
3,2 |
81,1 |
120 |
Tillägg 2
Exempel på apparatur

C.13 BIOKONCENTRATION: GENOMFLÖDESPROVNING MED FISK
1. METOD
Denna biokoncentrationsmetod är en kopia av OECD TG 305 (1996).
1.1 INLEDNING
Denna metod beskriver en procedur för karakterisering av potentialen för biokoncentration av ämnen i fisk vid genomflöde. Även om genomflödesprov är att föredra, kan halvstatiska metoder tillåtas, förutsatt att giltighetskriterierna uppfylls.
Metoden ger tillräckliga upplysningar för genomförandet av provet, samtidigt som den ger lämplig frihet för experimentell utformning av förhållandena i vissa laboratorier och varierande karakteristik för de undersökta ämnena. Metoden gäller framför allt för stabila organiska kemikalier med värden på log P OW mellan 1,5 och 6,0 (1) men kan även gälla för superlipofila ämnen (med log Pow större än 6,0). Förbedömningen av biokoncentrationsfaktorn (BCF) ibland benämnd KB för sådana superlipofila ämnen kommer sannolikt att vara högre än den biokoncentrationsfaktor vid stabilt tillstånd (BCFSS) som kan förväntas fås vid laboratorieexperiment. Uppskattningar av biokoncentrationsfaktorn för organiska kemikalier med värden för log Pow upp till ungefär 9,0 kan fås med Bintein-ekvationen vid al (2). De parametrar som karakteriserar biokoncentrationspotentialen inkluderar upptagningskonstanten (k1), utsöndringskonstanten (k2) och BCFSS.
Radioaktiv märkning av de ämnen som ska undersökas kan underlätta analysen av vatten- och fiskprover och kan användas för att fastställa huruvida degraderingsidentifiering och kvantifiering ska göras. Om den totala radioaktiva resten mäts (t.ex. genom förbränning eller vävnadsupplösning), grundas BCF på huvudblandningen, eventuella kvarvarande metaboliter och assimilerat kol. BCF-värden grundade på de totala radioaktiva resterna kan därför inte jämföras direkt med ett BCF från specifika analyser av enbart huvudblandningen.
Rengöringsprocedurer kan användas i undersökningar där radioaktiv märkning förekommer, för fastställande av BCF grundat på huvudblandningen, och de större metaboliterna kan vid behov karakteriseras. Det är också möjligt att kombinera en fiskmetabolismstudie med en biokoncentrationsstudie genom analys och identifiering av vävnadsrester.
1.2 DEFINITIONER OCH ENHETER
Biokoncenrationen/bioackumulationen är ökningen av koncentrationen av det undersökta ämnet i eller på en organism (specificerad vävnad därav) i förhållande till koncentrationen av det undersökta ämnet i omgivande medium.
Biokoncentrationsfaktorn (BCF eller KB) vid varje tidpunkt under upptagningsfasen av ackumulationsprovet är det undersökta ämnets koncentration i/på fisken eller den specificerade vävnaden därav (C, i μg/g (ppm)), dividerat med det kemiska ämnets koncentration i omgivande medium (Ca som μg/ml (ppm)).
Den stabila biokoncentrationsfaktorn (BCFSS eller KB) genomgår ingen väsentlig förändring under en längre tidsperiod, om koncentrationen av det undersökta ämnet i omgivande medium är konstant under denna tidsperiod.
Plattån eller stabiliteten nås när mängden undersökt ämne i fisk (Cf) i den tidpunkt där kurvan blir parallell med tidsaxeln och tre på varandra följande Cf-analyser, på prover tagna med intervaller på minst två dagar, ligger inom ± 20 % på de tre proverna och det inte är några betydande skillnader mellan de tre provningsperioderna. När kombinerade prover analyseras krävs minst fyra på varandra följande analyser. När de undersökta ämnena tas upp långsamt bör provtagningsintervallerna vara sju dagar.
Biokoncentrationsfaktor som beräknas direkt från förhållandet mellan kinetiska konstanter (k1/k2) kallas kin etisk koncentrationsfaktor, BCFk.
Fördelningskoeficienten oktanol/vatten (P ow) är förhållandet då det kemiska ämnet lösbarhet i n-oktanol och vatten nar balans (metod A.8), också uttryckt som Kow. Logaritmen för Pow används som en indikering på det kemiska ämnets potential för biokoncentration i vattenorganismer.
Exponerings- eller upptagningsfasen är den tid under vilken fisken exponeras för det undersökta ämnet.
Konstanten för upptagningsförhållandet (K1) är det numeriska värde som definierar ökningshastigheten av koncentrationen av det undersökta ämnet i/på den undersökta fisken eller specificerad vävnad därav, när fisken exponeras för nämnda kemiska ämne (k1 uttrycks som dag-1).
Efterexponerings- eller utsöndringsfasen (förlustfasen) är den period, efter det att den undersökta fisken flyttats från ett medium innehållande det undersökta ämnet till ett medium fritt från detta ämne, under vilken utsöndringen (eller nettoförlusten) av det aktuella ämnet från den undersökta fisken eller specificerad vävnad därav studeras.
Utsöndrings-konstanten (förlustkonstanten) (k2) är det numeriska värdet för minskningshastigheten av koncentrationen av det undersökta ämnet i den undersökta fisken eller i specificerad vävnad därav, efter överföring av den undersökta fisken från ett medium innehållande det undersökta ämnet till ett medium fritt från detta ämne (k2 uttrycks som dag-1).
1.3 PROVNINGSMETODEN
Provet består av två faser: exponerings-(upptagnings-)fasen och efterexponerings-(utsöndrings-)fasen. Under upptagningsfasen exponeras separata grupper av en fiskart för minst två koncentrationer av det undersökta ämnet. Fiskarna överförs därefter till ett medium fritt från det undersökta ämnet för utsöndringsfasen. Utsöndringsfas krävs alltid med mindre ämnesupptagningen under upptagningsfasen varit obetydlig, t.ex. om BCF är mindre än 10. Koncentrationen av det undersökta ämnet i/på fisken eller specificerad vävnad därav ska följas under de bägge provningsfaserna. Utöver de två provningskoncentrationerna ska en kontrollgrupp fiskar hållas under identiska förhållanden, förutom frånvaron av det undersökta ämnet, för att kunna jämföra eventuella negativa effekter i biokoncentrationsprovet mot en jämförbar kontrollgrupp och för att få bakgrundskoncentrationen av det undersökta ämnet.
Upptagningsfasen sträcker sig över 28 dagar, med mindre det kan visas att balans nås tidigare. Beräkning av upptagningsfasens längd fram till stabilitet kan göras med den ekvation som visas i bilaga 3. Utsöndringsperioden inleds därmed genom överföring av fisken till samma medium, men utan det undersökta ämnet, i ett annat rent kärl. När så är möjligt ska biokoncentrationsfaktorn beräknas helst både som koncentrationsförhållandet (BCFSS) i fisken (Cf) och i vattnet (Cw) vid uppenbar stabilitet och som en kinetisk biokoncentrationsfaktor, BCFK, som förhållandet mellan konstanterna för upptagningshastigheten (k1) och utsöndringen (k2), varvid första ordningens kinetik ska förutsättas. Om första ordningens kinetik uppenbarligen inte följs, ska mer komplexa modeller tillämpas (se bilaga 5).
Om stabilitet inte uppnås inom 28 dagar ska upptagningsfasen förlängas tills stabilitet nås eller till 60 dagar, beroende på vilket som inträffar först, varefter utsöndringsfasen påbörjas.
Konstanten för upptagningshastigheten, konstanten för utsöndringshastigheten (eller konstanterna om mer komplexa modeller används), biokoncentrationsfaktorn och när så är möjligt tillförlitlighetsgränserna för var och en av dessa parametrar beräknas med den modell som bäst beskriver den uppmätta koncentrationen av det undersökta ämnet i fisken och vattnet.
BCF uttrycks som en funktion av fiskens totala, våta vikt. I specialfall kan emellertid specificerade vävnader eller organ, som t.ex. muskler, lever osv. användas, om fisken är tillräckligt stor eller den kan delas upp i ätbara (filé) och icke ätbara (inälvor) delar. Eftersom det för många organiska ämnen föreligger ett klart samband mellan potentialen för biokoncentrationen och fettupptagningsförmågan, finns det också ett motsvarande förhållande för fettinnehållet i den undersökta fisken och observerade biokoncentrationen av sådana ämnen. För att minska denna källa till variationer i provningsresultaten för ämnen med hög fettupptagningsförmåga, dvs. med log Pow > 3, bör därför biokoncentrationen uttryckas i förhållande till fettinnehållet förutom i förhållande till hela kroppsvikten.
Fettinnehållet ska fastställas för samma biologiska materia som används för fastställande av koncentrationen av det undersökta ämnet om så är genomförbart.
1.4 INFORMATION OM DET UNDERSÖKTA ÄMNET
Innan biokoncentrationsprovet utförs ska följande information om det undersökta ämnet finnas tillgänglig:
|
a) |
Lösningsbarhet i vatten. |
|
b) |
Uppdelningskoefficienten Pow för oktanol/vatten (också beteckand Kow fastställd med HPLC-metod i A.8). |
|
c) |
Hydrolys. |
|
d) |
Ljusöverföring i vatten fastställd vid solbestrålning eller simulerad solstrålning och under de strålningsförhållanden som gäller för undersökningen av biokoncentrationen (3). |
|
e) |
Ytspänning, dvs. för ämnen där log Pow inte kan fastställas. |
|
f) |
Ångtryck. |
|
g) |
Biologisk nedbrytbarhet (när så är tillämpligt). |
Annan information som krävs är giftigheten för de fiskarter som ska användas under provet, företrädesvis det asymptotiska LC50, dvs. tidsoberoende. Det måste finnas lämplig analysmetod med känd noggrannhet, precision och känslighet för fastställande av mängden undersökt ämne i provlösningar och biologisk materia, tillsammans med uppgifter om beredning och förvaring av proverna. Den analytiska detektionsgränsen för det undersökta ämnet ska också vara känd för både vatten och fiskvävnad. När 14C-märkta ämnen används ska procentandelen radioaktivitet kopplad till föroreningar vara känd.
1.5 PROVETS GILTIGHET
Följande omständigheter ska vara uppfyllda för att provet ska vara giltigt:
|
— |
Temperaturvariationen ska vara mindre än ± 2 oC. |
|
— |
Koncentrationen av löst syre får inte falla under en mättnad på 60 %. |
|
— |
Koncentrationen av det undersökta ämnet i kärlen ska hållas inom ± 20 % av det medelvärde som mäts under upptagningsfasen. |
|
— |
Dödligheten eller andra negativa effekter/sjukdomar i både kontroll- och behandlingsgrupp ska vara mindre än 10 % vid slutet av försöket. Om försöket förlängs över flera veckor eller månader ska dödligheten eller andra negativa effekter i bägge fiskgrupperna vara mindre än 5 % per månad och får inte överstiga 30 % totalt. |
1.6 REFERENSBLANDNINGAR
Användning av referensblandningar med känd biokoncentrationspotential kan vara lämpligt vid kontroll av experimentförfarandet när så krävs. Specifika ämnen kan emellertid ännu inte rekommenderas.
1.7 BESKRIVNING AV PROVNINGSMETODEN
1.7.1 Apparater
Material som kan lösas upp, absorberas eller läcka och som har negativ effekt på fisken ska nogsamt undvikas i alla delar av utrustningen. Vanliga rektangulära eller cylindriska tankar, tillverkade av kemiskt beständigt material och med lämplig kapacitet vad gäller fyllningshastigheten ska användas. Användning av mjuka plaströr bör minimeras. Helst bör rör av teflon (R), rostfritt stål och/eller glas användas. Erfarenheter visar att kiselglas kan krävas för ämnen med hög absorptionskoefficient, som t.ex. syntetiska pyretroider. I dessa fall ska utrustningen slängas efter användningen.
1.7.2 Vatten
Naturligt vatten används vanligtvis vid undersökningen och bör tas från en oförorenad källa med jämn kvalitet. Lösningsvattnet måste hålla en kvalitet som möjliggör överlevnad för valda fiskarter under acklimatiserings- och provningsperioderna, utan att fiskarna uppvisar onormala förändringar eller beteende. I idealfallet ska det kunna visas att de arter som används kan överleva, växa och reproducera sig i vattenlösningen, t.ex. vid laboratorieodling eller en giftundersökning under livscykeln. Vattnet ska bestämmas avseende pH, hårdhet, partikelförekomst, total mängd organiskt kol och helst även ammonium, nitrit och alkalitet, samt för havslevande arter även salthalt. De parametrar som är viktiga för fiskens optimala välstånd är fullt ut kända, men i bilaga 1 anges rekommenderade maximala koncentrationer för ett antal parametrar för söt- och havsvattenprover.
Vattnet ska hälla jämn kvalitet under hela undersökningen. pH-värdet ska ligga mellan 6,0 och 8,5, men under pågående provning ska det ligga inom ±0,5 enheter. För att säkerställa att vattnet inte påverkar undersökningsresultatet på oönskat sätt, t.ex. genom förändring av det undersökta ämnet, eller negativt påverkar fiskbeståndets förutsättningar, ska prover tas med jämna mellanrum för analys. Tungmetaller, t.ex. Cu, Pb, Zn, Hg, Cd, Ni, viktigare an- och katjoner, t.ex. Ca, Mg, Na, K, Cl, SO4, bekämpningsmedel, t.ex. medel som helt består av organisk fosfor eller organiska kloriner, totala mängden organiskt kol och utfällda fasta partiklar ska göras t.ex. var tredje månad när vattenlösningen är känd för att hålla en relativt jämn kvalitet. Om vattenkvaliteten har visat sig vara jämn under minst ett år kan dessa kontroller göras mer sällan och intervallerna utökas, till t.ex. var sjätte månad.
Den naturliga partikelinnehållet samt den totala mängden organiskt kol (TOC) i vattenlösningen ska vara så låg som möjligt för undvikande av absorbtion av det undersökta ämnet i organisk materia, vilket skulle kunna minska dess biotillgänglighet (4). Det maximalt acceptabla värdet är 5 mg/l för partiklar (torr materia som inte passerar ett filter på 0,45 μm) och 2 mg/l för helorganiskt kol (se bilaga 1). Vattnet ska vid behov filtreras före användning. Bidraget till innehållet av organiskt kol i vattnet (exkrementer) från fisk och foderrester ska vara så litet som möjligt. Koncentrationen av organiskt kol i vattnet (exkrementer) från fisk och foderrester ska vara så litet som möjligt. Koncentrationen av organiskt kol i provningskärlet ska under undersökningen inte överstiga den koncentration av organiskt kol som härrör från det undersökta ämnet och om sådan används, från lösningsagenten med mer än 10 mg/l (± 20 %).
1.7.3 Provlösningar
Det undersökta ämnet ska lösas upp vid lämplig koncentration för förvaring. Förrådslösningen ska helst beredas genom att det undersökta ämnet helt enkelt blandas eller rörs i vattenlösningen. Användningen av lösningsmedel eller dispersanter (lösningsagenter) rekommenderas inte. Det kan emellertid i vissa fall vara nödvändigt för att få en lämpligt koncentrerad förrådslösning. Lösningsmedel som kan användas är etanol, metanol, etylenglykolmonometyleter, etylenglykoldimetyleter, dimetylformamid och trietylenglykol. Dispersanter som kan användas är kremofor RH40, Tween 80, metylcellulosa på 0,01 % och HCO-40. Försiktighet ska iakttas vid användning av lätt biologiskt nedbrytbara agenter, eftersom dessa kan förorsaka problem med bakterietillväxt vid genomflödesprovning. Det undersökta ämnet kan vara radioaktivt märkt och ska hålla högsta renhet, företrädesvis > 98 %.
Vid genomflödesprovet krävs ett system som kontinuerligt släpper ut och löser upp förrådslösningen av det undersökta ämnet, t.ex. med mätpump, proportionalitetslösningssystem, för att leverera provningskoncentrationerna till provningskärlen. Helst ska minst fem volymsombyten per dag göras i provningskärlen. Genomflödessystemet är att föredra, men när detta inte är genomförbart, t.ex. om den undersökta organismen påverkas negativt därav, kan en halvstatisk teknik användas, förutsatt att giltighetskriterierna uppfylls. Förrådslösningens och lösningsvattnets flöden ska kontrolleras både 48 timmar före och minst en gång dagligen under pågående provning, Denna kontroll ska omfatta fastställande av flödeshastigheten genom varje provningskammare, samt ska den garantera att flödeshastigheten inte varierar med mer än 20 % inom eller mellan kärlen.
1.7.4 Val av arter
Viktiga kriterier vid val av arter är att de ska finnas enkelt tillgängliga, kan skaffas i lämpliga storlekar och kan hållas på ett tillfredsställande sätt i laboratoriemiljö. Andra kriterier vid val av fiskart är betydelsen för rekreation, handel och ekologi samt jämförbar känslighet, tidigare framgångsrik användning osv.
Rekommenderade provarter anges i bilaga 2. Andra arter kan användas, men provningsförfarandet kan då behöva anpassas för att ge lämpliga provningsförhållanden. Underlaget för val av art och experimentmetod ska i detta fall rapporteras.
1.7.5 Hållande av fisk
Fiskbeståndet ska acklimatiseras under minst två veckor i vatten med provningstemperaturen och hela tiden matas med tillräcklig diet av samma typ som ska användas under provet.
Efter en anpassningsperiod på 48 timmar ska dödligheten registreras och följande kriterier tillämpas:
|
— |
Större dödlighet än 10 % av beståndet under sju dagar – fiskbeståndet ska inte användas. |
|
— |
Dödlighet på mellan 5 och 10 % av beståndet på sju dagar – beståndet ska acklimatiseras under ytterligare sju dagar. |
|
— |
Dödlighet på mindre än 5 % av beståndet under sju dagar – acceptera beståndet, om mer än 5 % av beståndet dött under den andra sjudagarsperioden ska hela beståndet avlägsnas. |
Kontrollera att fisk som använts vid undersökningen inte har några synliga sjukdomar eller abnormaliteter. Bortskaffa eventuell sjuk fisk. Fisken ska inte behandlas mot sjukdomen under de två närmast på undersökningen följande veckorna.
1.8 FÖRFARANDE VID UNDERSÖKNINGEN
1.8.1 Preliminärt prov
Det kan vara lämpligt att göra ett preliminärt experiment för att optimera provningsförhållandena för den slutgiltiga provningen, t.ex. val av koncentration för det undersökta ämnet och längd på upptagnings- och utsöndringsfaserna.
1.8.2 Exponeringsförhållanden
1.8.2.1
Upptagningsfasens längd kan bedömas utifrån praktiska erfarenheter, t.ex. från tidigare undersökning eller en ackumulationsrelaterad kemikalie, eller från vissa empiriska förhållanden tillsammans med kunskap om antingen det undersökta ämnets vattenlöslighet eller partitionskoeffecient i oktanol/vatten (se bilaga 3).
Upptagningsfasen ska vara 28 dagar, med mindre det kan visas att balans nås tidigare. Om stabilitet inte nåtts inom 28 dagar, ska upptagningsfasen förlängas med ytterligare mätningar, tills stabilitet nås eller 60 dagar, om detta inträffar först.
1.8.2.2
En period motsvarande halva upptagningsfasen är normalt tillräcklig för att få tillräcklig minskning, t.ex. 95 %, av det undersökta ämnet i fiskkroppen (se bilaga 3 för förklaring av bedömningen). Om den tid som krävs för att få en 95-procentig minskning blir orimligt lång, dvs. att den överskrider dubbla normala varaktigheten för upptagningsfasen, dvs. mer än 56 dagar, kan en kortare period användas, dvs. tills koncentrationen av det undersökta ämnet är mindre an 10 % av koncentrationen vid stabilitet. För ämnen med ett mer komplext mönster för upptagning och utsöndring än det som representeras av en enstaka fiskmodell med första ordningens kinetik, krävs emellertid utsöndringsfas för fastställande av utsöndringshastigheten. Periodens längd bör emellertid styras utifrån längden på den period som koncentrationen av det undersökta ämnet i fisken ligger kvar över analytiskt mätbar gräns.
1.8.2.3
Välj antalet fiskar per undersökt koncentration på ett sådant satt att minst fyra fiskar per prov finns tillgängliga för varje provtagning. Om större statistisk noggrannhet krävs, behövs fler fiskar per prov.
Om vuxna fiskar användas ska det i rapporten anges huruvida han- eller honfiskar eller bägge sorter använts vid experimentet. Om bägge könen används ska skillnader avseende fettinnehåll mellan könen visas vara utan betydelse innan exponeringen påbörjas. Det kan vara nödvändigt att hålla han-och honfiskar var för sig.
Vid denna typ av undersökningar ska fiskar med jämn vikt alltid väljas på ett sådant sätt att den minsta fisken inte väger mindre än 2/3 av vad den största väger. Samtliga fiskar ska vara från samma årskull och från samma källa. Eftersom fiskens vikt och ålder ibland förefaller ha stor betydelse för BCF-värdena (1), ska dessa uppgifter registreras korrekt. Det rekommenderas att delar av fiskbeståndet vägs före undersökningen, för fastställande av medelvikten.
1.8.2.4
En hög fisktäthet används för att minimera minskningen av Cw provningskärlet ska under undersökningen inte överstiga den koncentration av organiskt kol p.g.a. tillförsel av fiskar vid undersökningens början samt för att undvika en ökning av koncentrationen upplöst syre. Det är viktigt att fisktätheten är anpassad för den art som används. En täthet på 0,1–1,0 g fisk (våt vikt) per liter vatten och dag rekommenderas normalt. Hög täthet kan användas om det visar sig att önskad koncentration av det undersökta ämnet kan upprätthållas inom ± 20 % och att koncentrationen av upplöst syre inte faller under 60 % mättnad.
Vid val av lämpliga system ska hänsyn tas till fiskartens normala beteende. Bottenlevande fisk kan således kräva en större bottenyta i ett akvarium för en viss vattenvolym i förhållande till ytlevande fiskarter.
1.8.2.5
Under acklimatiserings- och provningsperioderna ska fisken utfodras med lämplig diet med känd fetthalt och totalt proteininnehåll, i sådan utsträckning att de hålls friska och behåller kroppsvikten, Fisken ska under acklimatiserins- och provningsperioderna utfodras med ungefär 1 — 2 % av kroppsvikten per dag. Detta håller fetthalten relativt konstant i de flesta fiskarter under provet. Fodermängden ska räknas om, t.ex. en gång per vecka, för att upprätthålla jämn kroppsvikt och fetthalt. Vid denna beräkning kan fiskens vikt i respektive provningskammare bedömas utifrån vikten på den fisk som senast tagits som prov från respektive kammare. Väg inte den fisk som lämnas kvar i kammaren.
Kvarvarande foder och exkrementer ska sugas ut från provningskammaren kort efter utfodringen (30 minuter till 1 timma). Kärlen ska hållas så rena som möjligt under provningen, så att koncentrationen av organisk materia hålls så låg som möjligt, eftersom förekomst av organisk kol kan begränsa det undersökta ämnets biotillgänglighet (1).
Eftersom många foder tillverkas av fiskmjöl, ska fodret analyseras avseendes det undersökta ämnet. Det är också önskvärt att fodret analyseras avseede bekämpningsmedel och tungmetaller.
1.8.2.6
Ljusperioden är vanligtvis 12 till 16 timmar, och temperaturen (± 2 oC) ska vara anpassad för den undesökta arten (ser bilaga 2). Belysningens typ och egenskaper ska vara kända. Hänsyn ska tas till det undersökta ämnets eventuella fototransformation under strålningsförhållanden vid försöket. Lämplig belysning ska användas för undvikande av exponering av fisken för onaturliga ljusprodukter. Det kan i vissa fall vara lämpligt att använda ett filter för bortfiltrering av UV-strålning under 290 nm.
1.8.2.7
Fisken exponeras under genomflödesförhållandena för minst två koncentrationer av det undersökta ämnet i vatten. Normalt väljs den högre (eller högsta) koncentrationen av det undersökta ämnet till ungefär 1 % av dess akuta asymptotiska LC50 och så att den är minst tio gånger högre än den gräns som ska överskridas för detektion i vatten med den använda analysmetoden.
Den högsta provningskoncentrationen kan också fastställas genom division av den akuta LC50 under 96 timmar med lämpligt akut/kroniskt förhållande (lämpliga förhållanden för vissa kemikalier kan vara från ungefär 3 upp till 100). Om möjligt bör andra koncentrationer väljas på ett sådant sätt att de avviker från ovannämnda med en fartor på 10. Om detta inte är möjligt p.g.a. kravet på 1 % av LC5O och det analytiska gränsvärder, kan en lägre faktor än 10 tillämpas eller 14C-märkning av det undersökta ämnet beaktas. Ingen av använda koncentrationer får ligga över det undersökta ämnets lösningsbarhet.
När lösningsagent avvänds ska dess koncentration inte vara större än 0,1 ml/l och vara lika stor i samtliga kärl. Dess bidrag tillsammans med det undersökta ämnet till det totala innehållet av organsikt kol i provningsvattnet ska vara känt. Denna typ av ämne bör emellertid undvikas i största möjliga utsträckning.
1.8.2.8
Kontroll av lösningsvattnet eller, om så är tillämpligt, kontroll av lösningsagenten, ska göras utöver provningsserierna, förutsatt att det kunnat fastställas att agenten inte har någon effekt på fisken. Om så inte är fallet ska bägge kontrollerna göras.
1.8.3 Frekvens för mätning av vattenkvalitet
Upplöst syre, TOC, pH och temperatur ska mätas i samtliga kärl under provningen. Den totala hårdheten och eventuellt salthalten ska mätas vid kontrollerna i ett kärl med högre (eller högst) koncentration. Det upplösta syret och eventuellt salthalten ska mätas minst tre gånger – i början, ungefär i mitten och mot slutet av upptagningsperioden – och en gång i veckan under utsöndringsperioden. TOC ska mätas vid början av provet (24 och 48 timmar innan upptagningsfasen initieras) före det att fisken tillsätts och minst en gång i veckan under både upptagnings- och utsöndringsfaserna. Temperaturen ska mätas dagligen, pH i början och slutet av varje period och hårdheten en gång under varje provning. Temperaturen ska helst avläsas kontinuerligt i åtminstone ett kärl.
1.8.4 Provtagning och analys ar fisk och vatten
1.8.4.1
Vatten frän provningskärlen ska tas både före det att fisken tillsätts samt under upptagnings- och utsöndringsfaserna, för fastställande av koncentrationen av det undersökta ämnet. Prover ska tas av vattnet åtminstone samtidigt som av fisken samt före utfodring. Under upptagningsfasen fastställs koncentrationen av det undersökta ämnet för kontroll av överensstämmelse med giltighetskriterierna.
Prover ska tas på fisken minst fem gånger under upptagningsfasen och vid minst fyra tillfällen under utsöndringsfasen. Eftersom det i bland är svårt att göra en exakt uppskattning av BCF-värdet grundat på detta antal prover, framför allt när annan utsöndringskinetik än första ordningens kan påvisas, kan det vara tillrådligt att ta prover med tätare mellanrum under bägge perioderna (se bilaga 4). Extraproverna ska lagras och bara analyseras om resultaten från första omgångens analyser visar sig vara otillräckliga för beräkning av BCF med önskad noggrannhet.
I bilaga 4 ges ett exempel på ett acceptabelt provtagningsschema. Andra scheman kan lätt räknas ut med andra antagna värden för Pow, för beräkning av exponeringstiden för att få 95-procentig upptagning.
Provtagningen ska försätta under upptagningsfasen tills stabil nivå uppnås, dock minst 28 dagar. Om stabil nivå inte nåtts inom 28 dagar, ska provtagningen fortsätta tills stabil nivå nås, dock minst 60 dagar, om detta inträffar först. Innan utsöndringsfasen inleds ska fisken överföras till rena tankar.
1.8.4.2
Vattenprover för analys tas genom en tät röranslutning i provningskammarens mittpunkt. Eftersom varken filtrering eller centrifugering alltid separerar den icke-biologiska fraktionen från det undersökta ämnet från den biotillgängliga delen (särskilt för superlipofila kemikalier, dvs. kemikalier med log Pow > 5) (1) (5), ska proverna inte utsättas för sådan behandling.
Åtgärder bör istället vidtas för att hålla tankarna så rena som möjligt och innehållet fullorganiskt kol ska övervakas under både upptagnings- och utsöndringsfasen.
Ett lämpligt antal fiskar, i normalfallet minst fyra, ska tas från provningskammaren vid varje provtagning. Provtagningsfisken ska sköljas snabbt med vatten, torkas torr, dödas omedelbart på lämpligaste och humanaste sätt och därefter vägas.
Det är lämpligt att under den tid som provet pågår analysera fisken och vattnet omedelbart efter provtagning, för att förhindra försämring eller andra förluster samt för beräkning av ungefärlig upptagnings- och utsöndringshastighet. Mellananalyser förhindrar också förseningar vid fastställande av när en nivå har nåtts.
Om mellananalyser inte görs ska proverna lagras pa lämpligt sätt. Innan undersökningen påbörjas ska information om lämplig förvaringsmetod för det aktuella ämnet, t.ex. djupfrysning, nedkylning vid 4 oC, förvaringstid, extraktion osv., skaffas.
1.8.4.3
Eftersom hela proceduren i huvudsak styrs av noggrannhet, precision och känslighet i den analysmetod som används för det undersökta ämnet, ska precision och reproduktionsbarhet för den kemiska analysen kontrolleras experimentellt, vilket även gäller för återhämtning av det undersökta ämnet från både vatten och fisk, vilket ska vara tillfresställande med den aktuella metoden. Kontrollera också att det undersökta ämnet inte kan upptäckas i det lösningsvatten som används.
Om så krävs ska Cw- och Cf-värden som fås från undersökningen korrigeras för återhämtnings- och bakgrundsvärden från kontrollerna. Fisk- och vattenprover ska alltigenom hanteras på ett sådant sätt att förorening och förlust minimeras, t.ex. på grund av absorbtion från provtagningsanordningarna.
1.8.4.4
Om radioaktivt märkt material används vid undersökningen är det möjligt att analysera den totala radioaktiva märkningen, dvs. moderblandning och metaboliter, eller kan proverna rengöras så att moderblandningen kan analyseras separat. De viktigare metabohterna kan karakteriseras vid stabil nivå, dock senast vid upptagningsfasens slut. Om BCF som total radioaktivt märkt rest är ≥ 1 000 %, kan det vara lämpligt och för vissa kemikaliekategorier, som t.ex. bekämpningsmedel, starkt rekommendabelt att identifiera och mängdbestämma de rester dom uppgår till ≥ 10 % av den totala resten i fiskvävnaden vid stabil nivå. Om resterna representerar ≥ 10 % av den totala radioaktivt märkta resten if fiskvävnaden identifieras och kvantifieras, är det också rekommendabelt att identifiera och mängdbestämma resterna i provningsvattnet.
Koncentrationen av det undersökta ämnet ska normalt fastsfällas för varje vägd fiskindivid. Om detta inte är möjligt kan sammanslagning av proverna vid varje provtagningstillfälle göras, men detta begränsar det statistiska förfarandet för erhållna data. Om ett visst statistiskt förfarande och dess funktion är viktiga, skal! ett tillräckligt antal fiskar för att tillfredsställa den önskade sammanslagningsproceduren och effekten införas i undersökningen (6) (7).
BCF ska uttryckas både som en funktion av total våt vikt och, för kraftigt lipofila ämnen, som en funktion av fettinnehållet. Fettinnehållet i fisken fastställs om möjligt vid varje provtagningstillfälle. Lämpliga metoder ska användas för fastställandet av fettinnehållet (punkterna 8 och 2 i bilaga 3). Extraktionstekniker med kloroform/metanol kan vara att rekommendera som standardmetod (9). De olika metoderna ger inte identiska värden (10), varför det är viktigt att ange vilken metod som använts. När så är möjligt ska fettanalysen göras på samma extrakt som det som tagits fram för analys av det undesökta ämnet, eftersom fettet ofta måste tas bort frän extraktet innan kromatografisk analys kan göras. Fettinnehållet i fisken (som mg fett per kg våt vikt) får vid slutet av experimentet inte skilja sig frän innehållet vid experimentets början med mer än ± 25 %. Andelen fasta partiklar i vävnaden ska också registreras för att möjliggöra omvandling av fettkoncentrationen från vått till torrt värde.
2. DATA
2.1 RESULTATBEARBETNING
Upptagningskurvan för det undersökta ämnet fås genom plottning av dess koncentration i/på fisk (eller specificerad vävnad därav) under upptagningsfasen i förhållande till tiden på aritmetisk skala. Om kurvan när en platå, dvs. blir ungefär asymptotisk med tidsaxeln, beräknas BCFSS med följande formel:
![]()
När stabilitet inte nås, kan BCFSS beräknas med tillräcklig noggrannhet för riskbedömning från en stabilitet vid 80 % (l,6/k2) eller 95 % (3,0/k2) balans.
Koncentrationsfaktorn (BCFk) fastställs också som förhållandet k1/k2 de två kinetiska konstanterna av första ordningen. Konstanten för utsöndringshastighet, k, fastsälls normalt ur utsöndringskurvan, dvs. en plottning av ökningen av det undersökta ämnets koncentration i fisken i tiden. Konstanten för upptagningshastigheten, k1, beräknas utifrån konstanten k2 och värdet på Cf som fås från upptagningskurvan (se också bilaga 5). Den bästa metoden för erhållande av BCFk och hastighetskonstanterna k1 och k2 är att göra en icke-linjär parameterbedömning i en dator (11). I annat fall kan grafiska metoder användas för beräkning av k1 och k2. Om det är uppenbart att utsöndringskurvan inte är en kurva av första ordningen, ska mer komplexa modeller tillämpas (se referenser i bilaga 3) och råd inhämtas från biostatistiker.
2.2 TOLKNING AV RESULTAT
Resultaten ska tolkas med försiktighet när uppmätta koncentrationer av den undersökta lösningen påträffas på nivåer nära den analytiska metodens gränsvärdet för detektion.
Tydligt definierade upptagnings- och förlustkurvor är en indikation på att biokoncentrationsdatan är av god kvalitet. Skillnaden i upptagnings/utsöndringskonstanterna mellan de två provningskoncentrationerna ska vara mindre än 20 %. Observera att väsentliga skillnader i upptagnings-/utsöndringshas-tigheterna mellan de två tillämpade provningskoncentrationerna ska registreras och om möjligt förklaras. Tillförlitlighetsgränsen för BCF kan för väl utformade studier sägs ligga runt ± 20 %.
3. RAPPORTERING
Undersökningsrapporten måste innehålla följande information:
3.1 UNDERSÖKT ÄMNE
|
— |
Fysisk form och, när så är tillämpligt, fysiokemikaliska egenskaper. |
|
— |
Kemisk identifiering (inklusive eventuellt innehåll av organiskt kol). |
|
— |
Om ämnet är radioaktivt märkt ska den märkta atomens exakta position och andelen radioaktivitet kopplad till föroreningar anges. |
3.2 UNDERSÖKTA ARTER
|
— |
Vetenskapligt namn, variant, källa, eventuell förbehandling, acklimatisering, ålder, storleksområde osv. |
3.3 PROVNINGSFÖRHÅLLANDEN
|
— |
Tillämpat provningsförfarande, t.ex. genomflöde eller halvstatiskt. |
|
— |
Typ och karakteristik för använd belysning och ljusperiod. |
|
— |
Provets utformning, t.ex. antal och storlek på provningskammare, vattenväxlingshastighet, antal kopior, antal fiskar per kopia, antal provningskoncentrationer, upptagnings- och utsöndringsfasernas längd, provtagningsfrekvens för fisk och vatten. |
|
— |
Metod för beredning av förrådslösningar och förnyelsefrekvensen (lösningsagent, dess koncentration och bidrag till innehåll av orgnaiskt kol i provningsvattnet måste anges när sådan används). |
|
— |
Nominella provningsförhållanden, medelvärden för uppmätta värden samt deras standardavvikelser i provningskärlen och metod som använts för beräkning av dessa värden. |
|
— |
Vattenkälla, beskrivning av eventuell förbehandling, resultat från eventuell undersökning av fiskens förmåga att överleva i vattnet, samt vattnets karakteristik: pH, hårdhet, temperatur, syrehalt, klorrestvärde om detta mäts, total mängd organiskt kol, utfällda fasta partiklar, salthalt i det undersökta mediet, om tillämpligt, och eventuella andra gjorda mätningar. |
|
— |
Vattenkvalitet i provningskärlen, pH, hårdhet, TOC, temperatur och syrehalt. |
|
— |
Detaljerad information om utfodringen, t.ex. fodertyp, källa, sammansättning (åtminstone fett- och proteininnehåll) mängd och frekvens. |
|
— |
Information om behandlingen av fisk- och vattenprover, inklusive uppgifter om beredning, förvaring, extraktion och analytiska förfaranden, samt deras noggrannhet, för det undersökta ämnet och fettinnehället (om detta mäts). |
3.4 RESULTAT
|
— |
Resultat från eventuell preliminär studie. |
|
— |
Dödligheten för kontrollfisken och fisken i varje exponerat kärl, samt eventuellt observerat onormalt beteende. |
|
— |
Fettinnehållet i fisken (om detta fastställs vid provningen). |
|
— |
Kurvor, inklusive samtliga mätdata, över upptagningen och utsöndringen av den undersökta kemikalien i fisken, tiden till stabilitet. |
|
— |
Cf en Cw (med standardavvikelse och område, om tillämpligt) för samtliga provtagningstillfällen (C, uttryckt som μg/g våt vikt (ppm) i hela kroppen eller den specificerade vävnaden därav, t.ex. fett, och Cw som μg/ml (ppm)), Cw-värden för kontrollserierna (bakgrundsvärden ska också rapporteras). |
|
— |
Biokoncentrationsfaktorn vid stabilitet (BCFss) och/eller den kinetiska koncentrationsfaktorn (BCFk) och om tillämpligt den 95-procentiga tillförlitlighetsgränsen för upptagnings- och utsöndringshastighetskonstanterna (samtliga uttryckta i förhållande till hela kroppen och det totala fettinnehållet, om detta mäts, i djuret eller den specificerade vävnaden därav), tillförlitlighetsgränser och standardavvikelser (om tillgängliga) samt metoder för beräkning/dataanalys av samtliga koncentrationer av det undersökta ämnet. |
|
— |
Om strålningsmarkerade ämnen används och om så krävs, kan ackumulationen av eventuella upptäckta metaboliter visas. |
|
— |
Eventuella anmärkningar avseende undersökning, eventuell avvikelse från förfarandet och annan relevant information. |
Undvik resultat som t.ex. ”ej upptäckt vid detektionsgränsen” vid framtagningen av undersökningsmetoden och dess experimentella utformning, eftersom sådana resultat inte kan användas för beräkning av hastighetskonstanter.
4. HÄNVISNINGAR
|
1. |
Connell D.W. (1988), Bioaccumulation behaviour of persistent chemicals with aquatic organisms (ung. Bestående kemikaliers bioackumulationsheteende i vattenorganismer), Rev. Environ. Contam. Toxicol. 102, s. 117–156. |
|
2. |
Bintein S., Devillers, J. and Karcher W. (1993), Nonlinear dependence of fish bioconcentration on n-octanol/water partition coefficient. (Fiskbiokoncentrationens icke-linjära beroende av parti tionskoefficienten för non-oktanol/vatten). SAR and QSAR in Environmental Research, s. 29–390. |
|
3. |
OECD (1996), Direct Phototransformation of chemicals in water. Environmental Health and Safety Guidance Document Series on Testing and Assessment of Chemicals (ung. Direkt ljustransformering av kemikalier i vatten. Riktlinjer för miljöskydd – provning och bedömning av kemikalier), nr 3. |
|
4. |
Kristensen P. (1991), Bioconcentration in fish: Comparison of bioconcentration factors derived from OECD and ASTM testing methods; influence of particulate organic matter to the bioavaila- bility of chemicals (ung. Biokoncentration i fisk: Jämförelse av biokoncentrationsfaktorer från OECD:s och ASTM:s provningsmetoder, partikelformigt organiskt materials påverkan på kemikaliers biotillänglighet), Water Quality Institute, Danmark. |
|
5. |
US EPA 822-R-94-002 (1994), Great Lake Water Quality Initiative Technical Support Doc. for the Procedure to Determine Bioaccumulation Factors (ung. Tekniskt underlag för kvalitetsinitiativet för vattnet i Stora sjöarna, för fastställande av bioackumulationsfaktorer), juli 1994. |
|
6. |
US FDA (Myndigheten för livsmedel och droger) Revision. Handbok för analys av bekämpningsmedel, 1, 5600 Fisher's Lane, Rockville, MD 20852, juli 1975. |
|
7. |
US EPA (1974), Avsnitt 5, A(l) Analysis of Human or Animal Adipose Tissue, in Analysis of Pesticide Residues in Human and Environmental Samples (ung. Analys av fettvävnad från människa eller djur vid analys av bekämpningsmedelsrester i prov från människa eller djur), Thompson J.F. (ed.) Research Triangle Park, N.C. 27711. |
|
8. |
Compaan H. (1980), in ”The determination of the possible effects of chemicals and wastes on the aquatic environment: degradation, toxicity, bioaccumulation” (ung. Fastställande av möjliga effekter från kemikalier och avfall på vattenmiljön: degradering, giftighet, bioackumulation), kap. 2.3, del 2. Government Publisching Office, The Hague, The Netherlands. |
|
9. |
Gardner et al. (1995), Limn. & Oceanogr. 30, 1099–1105. |
|
10. |
Randall R.C., Lee H., Ozretich R.J, Lake J.L. and Pruell R.J. (1991), Evaluation of selected lipid methods for normalising pollutant bioaccumulation. Envir. Toxicol. Chem (ung. Utvärdering av vissa fettmetoder för oskadliggörande av förorenande bioackumulation) 10, s. 1431–1436. |
|
11. |
CEC. Bioconcentration of chemical substances in fish: the flow-through method-Ring Test Programme, 1984–1985 (ung. Biokoncentration av kemikalier i fisk: genomflödesmetoden – Ringprovningsprogrammet 1984–1985), slutrapport 1987, författare: P. Kristensen and N. Nyholm. |
|
12. |
ASTM E-l022-84 (Återantagen 1988), Standard Practice for conducting Bioconcentration Tests with Fisches and Saltwater Bivalve Molluscs (ung. Standardförfarande för biokoncentrationsundersökning med fisk och saltvattenblötdjur med två kanaler). |
Bilaga 1
Kemisk karakteristik för acceptabelt lösningsvatten
|
|
Ämne |
Koncentrations-gränsvärde |
|
1 |
Partiklar |
5 mg/l |
|
2 |
Total mängd organiskt kol |
2 mg/l |
|
3 |
Ojoniserad ammonium |
l μg/l |
|
4 |
Klorrester |
10 μg/l |
|
5 |
Total mängd fosfororganiska bekämpningsmedel |
50 ng/l |
|
6 |
Total mängd klororganiska bekämpningsmedel plus flerklorerade bifenyler |
50 ng/1 |
|
7 |
Total mängd organiskt klor |
25 ng/1 |
|
8 |
Aluminium |
1 μg/l |
|
9 |
Arsenik |
1 μg/l |
|
10 |
Krom |
1 μg/l |
|
11 |
Kobolt |
1 μg/l |
|
12 |
Koppar |
1 μg/l |
|
13 |
Järn |
1 μg/l |
|
14 |
Bly |
1 μg/l |
|
15 |
Nickel |
1 μg/l |
|
16 |
Zink |
1 μg/1 |
|
17 |
Kadmium |
100 ng/1 |
|
18 |
Kvicksilver |
100 ng/1 |
|
19 |
Silver |
100 ng/1 |
Bilaga 2
Fiskarter som rekommenderas för undersökning
|
|
Rekommenderade arter |
Rekommenderat temperaturområde för undersökningen ( oC) |
Rekommenderad total längd på det undersökta djuret (cm) |
|
1 |
Danio rerio (17) (Teleostei, Cyprinidae) (Hamilton-Buchanan) Zebrafisk |
20–25 |
3,0±0,5 |
|
2 |
Pimephales promelas (Teleostei, Cyprinidae) (Rafinesque) Fathead minnow (art av kvidd) |
20–25 |
5,0±2,0 |
|
3 |
Cyprinus carpio (Teleostei, Cyprinidae) (Linnaeus) Vanlig karp |
20–25 |
5,0±3,0 |
|
4 |
Oryzias latipes (Teleostei, Poeciliidae) (Temminck and Schlegel) Ricefish (risfiskart) |
20–25 |
4,0±1,0 |
|
5 |
Poecilia reticulata (Teleostei, Poeciliidae) (Peters) Guppy |
20–25 |
3,0±1,0 |
|
6 |
Lepomis macrochirus (Teleostei, Centrarchidae) (Rafinesque) Bluegill (art av solabborre) |
20–25 |
5,0±2,0 |
|
7 |
Oncorhynchus mykiss (Teleostei, Salmonidae) (Walbaum) Regnbågslax |
13–17 |
8,0±4,0 |
|
8 |
Gasterosteus aculeatus (Teleostei, Gasterosteidae) (Linnaeus) Stenspigg |
18–20 |
3,0±1,0 |
Flera söt- och saltvattenarter har använts i olika länder, t.ex:
|
Spot |
Leiostomus xantburus |
|
Sheepshead minnow (art av tandkarp) |
Cyprinodon variegatus |
|
Silverside |
Menidia beryllina |
|
Shiner perch |
Cymatogaster aggregata |
|
English sole |
Parophrys vertulus |
|
Staghorn sculpin |
Leptocottus armatus |
|
Stenspigg |
Gasteroteus aculeatus |
|
Havsaborre |
Dicentracus labrax |
|
Benlöja |
Alburnus alburnus |
Insamling
Den sötvattenfisk som anges i ovanstående tabell är lätt att skaffa och/eller finns lätt tillgänglig under hela aret, medan tillgängligheten for havs- och bräckvattenarterna delvis begränsas till respektive länder. De kan födas upp och odlas på fiskodlingar eller laboratorier under sjukdoms- och parasitkontrollerade förhållanden, så att det använda djuret är friskt och har känd stamtavla. Dessa fiskar finns tillgängliga i stora delar av världen.
Bilaga 3
Förbestämning av längden på upptagnings- och utsöndringsfaserna
1. Förbestämning av upptagningsfasens längd
Innan provet påbörjas ska en uppskattning av k2 göras, och hänsyn till hur stor del av tiden som krävs för att nå stabilitet kan inhämtas från empiriska förhållanden mellan k2 och n- oktanol/vattenfördelningskoeffecienten (Pow) eller k2, och vattenlösligheten (s).
En uppskattning av k2 (dag-1) kan erhållas med t.ex. följande empiriska förhållande (1):
|
log10k2 = 0,414 log10(Pow) + 1,47 (r2 = 0,95) |
(ekvation 1) |
För andra förhållanden se hänvisning 2.
Om partitionskoeffecienten (Pow) inte är känd, kan en bedömning göras (3) utifrån kunskapen om ämnets vatten löslighet (s) med följande formel:
|
log10(Pow) = 0,862 log10(s) + 0,710 (r2 = 0,994) |
(ekvation 2) |
där:
s står för lösligheten i mol/l: (n = 36).
Dessa förhållanden gäller enbart för kemikalier med log Pow-värden mellan 2 och 6,5 (4).
Tiden för att nå en viss del av den stabila nivån kan fås genom tillämpning av så kallad k2-bedömning, med den allmänna kinetiska ekvationen för upptagning och utsöndring (första ordningens kinetik):

eller om Cw är konstant:
|
|
(ekvation 3) |

När stabiliteten börjar närma sig (t → ∞), kan ekvation 3 reduceras (5) (6) till:
 eller Cf/Cw = k1/k2
= BCF
eller Cf/Cw = k1/k2
= BCF
(k1/k2) Cw närmar sig då koncentrationen i fisken vid stabilitet (Cf, s).
Ekvation 3 kan skrivas om på följande sätt:
|
|
(ekvation 4) |


Vid användning av ekvation 4 kan tiden för att nå viss stabilitet förutsägas när k2 förutbestäms med ekvation 1 eller 2.
Som riktlinje kan sägas att den statistiskt optimala längden på upptagningsfasen, för produktion av statistiskt acceptabla data (BCFk) är den period som krävs för att kurvan över koncentrationslogaritmen för det undersökta ämnet i fisken, plottad mot linjär tidsaxel, till medelpunkten är 1,6/k2 eller 80 % av den stabila nivån, men inte mer än 3,0/k2 eller 9.5 -procentig stabilitet (7).
Tiden för att nå 80-procentig stabilitet är (ekvation 4)
|
|
(ekvation 5) |

95-procentig stabilitet fås på motsvarande sätt med ekvationen:
|
|
(ekvation 6) |

Upptagningsfasens (upp) längd för ett undersökt ämne med log Pow = 4 blir då (med ekvationerna 1,5 och 6):
|
log10k2 = -0,414.(4) + 1,47 |
k2 = 0,652 dag-1 |
upp (80 %) = 1,6/0,652, t.ex. 2,45 dagar (59 timmar)
eller upp (95 %) = 3,0/0,652, t.ex. 4,60 dagar (110 timmar)
För ett undersökt ämne med s = 10-5 mol/l (log(s) = -5,0), är upptagningsfasen (med ekvationerna 1,2,5 och 6):
log10 (Pow) = -0,862 . (-5,0) + 0,710 = 5,02
log10 k2 = -0,414 . (5,02) + 1,47
k2 = 0,246 dagar-1
upp (80 %) = 1,6/0,246, t.ex. 6,5 dagar (156 timmar)
eller upp (95 %) = 3,0/0,246, t.ex. 12,2 dagar (293 timmar).
Alternativt kan följande uttryck användas:
teq = 6,54 × 103 Pow + 55,31 (timmar)
Detta uttryck används för beräkning av tiden till det att faktiskt stabilitet nås (4).
2. Förutbestämning av utsöndringsfasens varaktighet
Förutbestämning av den tid som krävs för att minska mängden ämne i kroppen, till en viss del av den ursprungliga koncentrationen, kan också göras med den allmänna ekvationen för upptagning och utsöndring (första ordningens kinetik) (1) (8).
Cw antas vara noll för utsöndringsfasen. Ekvationen kan reduceras till:
 eller
eller 
där Cf,0 är koncentrationen vid början av utsöndringsperioden. 50-procentig utsöndring nås då vid tiden (t50):
 eller
eller 
95-procentig utsöndring fås på motsvarande sätt med ekvationen:

Om en upptagning på 80 % används för den första perioden (1,6/k2) och en 95-procentig förlust i utsöndringsfasen (3,0/k2), är utsöndringsfasen ungefär dubbelt så lång som utsöndringen i upptagningsfasen.
Det är emellertid viktigt att observera att dessa beräkningar är grundade på förutsättningen att upptagnings-och utsöndringsmönstren följer första ordningens kinetik. Om det är uppenbart att första ordningens kinetik inte följs, ska mer komplexa beräkningsmodeller användas (se hänvisning 1).
Hänvisningar (för bilaga 3)
|
l. |
Spacie A. and Hamelink J.L. (1982), Alternative models for describing the bioconcentration of organics in fish. Environ. Toxicol. and Chem. (ung. Alternativa modeller för beskrivning av biokoncentrationen av organiska ämnen i fisk; miljö; gift; kemikalie) 1, s. 309–320. |
|
2. |
Kristensen P. (1991), Bioconcentration in fish: comparison of BCF's derived from OECD and ASTM testing methods; influence of particulate matter to the bioavailability of chemicals (ung. Biokoncen tration i fisk: jämförelse av biokoncentrationsfaktorerna från OECD:s och ASTM:s provningsmetoder, partikelformiga ämnens påverkan på kemikaliernas biotillgänglighet). Danska vattenkvalitetsinstitutet. |
|
3. |
Chiou C.T. and Schmedding D.W. (1982), Partitioning of organic compounds in octanol-water systems. Environ. Sci. Technol (ung. Fördelning av organiska blandningar i oktanol-vattensystem; miljö; forsk ning; teknik). 16 (1), s. 4–10. |
|
4. |
Hawker D.W. and Connell D.W. (1988), Influence of partition coefficient of lipophilic compounds on bioconcentration kinetics with fish (ung. Lipofila blandningars partitionskoefficients påverkan på biokoncentrationskinetiken med fisk). Wat. Res. 22 (6), s. 701–707. |
|
5. |
Branson D.R., Blau G.E., Alexander H.C. and Neely W.B. (1975), Transactions of the American Fisheries Society (ung. Amerikanska fiskeförbundets transaktioner), 104 (4), s. 785–792. |
|
6. |
Ernst W. (1985), Accumulation in aquatic organisms. In: Appraisal of tests to predict the environ mental behaviour of chemicals (ung. Ackumulering i vattenorganismer. In: Bedömning av provningar för att förutsäga kemikaliers beteende i miljön). Författad av Sheehman P., Korte F., Klein W. och Bourdeau P.H., del 4.4 sid 243–255. SCOPE, John Wiley & Sons Ltd. N.Y. |
|
7. |
Reilly P.M., Bajramovic R., Blau G.E., Branson D.R. and Sauerhoff M.W. (1977), Guidelines for the optimal design of experiments to estimate parameters in first order kenetic models (ung. Riktlinjer för optimal utformning av experiment för bedömning av parametrar i modeller för första ordningens kinetik), Can. J. Chem. Eng. SS, s. 614–622. |
|
8. |
Könemann H. and Van Leeuwen K. (1980), Toxicokinetics in fish: Accumulation and elimination of six chlorobenzenes by guppies. Chemosphere (ung. Toxicokinetik i fisk: Ackumulering och eliminerig av sex klorbenzener i guppy), 9, s. 3–19. |
Bilaga 4
Teoretiskt exempel på provtagningsschema för biokoncentrationsundersökningar för ämnen med log Pow = 4
|
Provtagning av fisk |
Tidsschema för provtagning |
Antal vattenprover |
Antal fiskar per prov |
||||
|
Längsta tänkbara provtagningsfrekvens (dagar) |
Tilläggsprover |
||||||
|
Upptagningsfas |
- 1 0 |
|
2 (18) 2 |
tillsätt 45–80 fiskar |
|||
|
Första |
0,3 |
0,4 |
2 (2) |
4 (4) |
|||
|
Andra |
0,6 |
0,9 |
2 (2) |
4 (4) |
|||
|
Tredje |
1,2 |
1,7 |
2 (2) |
4 (4) |
|||
|
Fjärde |
2,4 |
3,3 |
2 (2) |
4 (4) |
|||
|
Femte |
4,7 |
|
2 |
6 |
|||
|
Utsöndringsfas |
|
|
|
Flytta över fisken till vatten fritt från den undersökta kemikalien |
|||
|
Sjätte |
5,0 |
5,3 |
|
4 (4) |
|||
|
Sjunde |
5,9 |
7,0 |
|
4 (4) |
|||
|
Ättonde |
9,3 |
11,2 |
|
4 (4) |
|||
|
Nionde |
14,0 |
17,5 |
|
6 (4) |
|||
|
Värden inom parentes står för antal prover (vatten, fisk} som ska tas om tilläggsprovtagning görs.
|
|||||||
Bilaga 5
Modellbeskrivning
De flesta biokoncentrationsdata har förutsatts bli skäligen väl beskrivna med en enkel tvådelad modell eller tvåparametersmodell, på det sätt som visas med den räta kurva som närmar sig punkterna för koncentrationen i fisk under utsöndringsfasen, när dessa linjer plottas på halvlogaritmiskt papper. Om dessa punkter inte kan beskrivas av en rät kurva ska mer komplexa modeller användas, se t.ex Spacie och Hamelink, hänvisning 1 i bilaga 3.
Grafisk metod för bestämmning av konstanten k2 för utsöndringshastigheten
Plotta en koncentration av det undersökta ämne som återfinns i varje fiskprov mot provtagningstiden på halvlogaritmiskt papper. Linjens lutning är k2.


Observera att avvikelser från rät linje kan vara ett tecken på ett mer komplext utsöndringsmönster än första ordningens kinetik. En grafisk metod kan användas för att särskilja utsöndringstyper som avviker från första ordningens kinetik.
Grafisk metod för fastställande av konstanten k1 för upptagningshastigheten
När k2 tagits fram kan konstanten k1 beräknas med följande formel:
|
|
[ekvation 1] |

Värdet på Cf, avläses i mittpunkten på den jämna upptagningskurva som fås av datan när den logaritmiska koncentrationen plottas i förhållande till tiden på en aritmetrisk skala.
Metod för beräkning av konstanterna för upptagnings- och utsöndringshastigheten
De medel som föredras för erhållande av biokoncentrationsfaktorn och hastighetskonstanterna k1 och k2 är att använda en icke-linjär metod för parameteruppskattning på dator. Dessa program hittar värdena på k1 och k2 utifrån en uppsättning koncentrationsdata sekventiella i tiden och med följande ekvationer:
|
|
[ekvation 2] |
|
|
[ekvation 3] |


där tc står för tiden vid slutet av upptagningsfasen.
Med denna metod kan vanliga avvikelsebedömningar göras för k1 och k2.
Eftersom k2 i de flesta fall kan uppskattas i utsöndringskurvan med förhållandevis hög precision och eftersom ett starkt samband finns mellan de två parametrarna k1 och k2 om de beräknas samtidigt, kan det vara tillrådligt att först beräkna k2 från enbart utsöndringsdata och därefter beräkna k1 från upptagningsdata med icke-linjär regression.
C.14 TILLVÄXTTEST PÅ UNGA EXEMPLAR AV FISK
1. METOD
Denna metod för tillväxttest avseende toxicitet motsvarar OECD TG 215 (2000).
1.1 INLEDNING
Testet är avsett för bestämning av vilka verkningar långvarig exponering för kemikalier har på tillväxten hos unga exemplar av fisk. Testet baserar sig på en metod för bestämning av verkningarna av kemikalier på tillväxten hos unga exemplar av regnbågslax (Oncorynchus mykiss) under genomflödesförhållanden, en metod som har utvecklats och ringtestats (1) (2) inom Europeiska unionen. Även andra väldokumenterade arter kan användas. Man har t.ex. erfarenheter från tillväxttest med sebrafisk (Danio rerio) (19) (3) (4) och ricefish medaka Oryzias latipes) (5) (6) (7).
Se också den allmänna inledningen, del C.
1.2 DEFINITIONER
Lägsta koncentration vid vilken verkningar observeras (Lowest observed effect concentration, LOEC):
den lägsta testade koncentrationen av ett testämne vid vilken man kan observera signifikanta verkningar (vid p < 0,05) jämfört med kontrollgruppen. Alla testkoncentrationer ovanför LOEC måste framkalla skadliga verkningar som är lika stora eller större än de verkningar som kan observeras vid LOEC.
Koncentration vid vilken inga verkningar observeras (No observed effect concentration, NOEC): test-koncentrationen omedelbart under LOEC.
ECx: den koncentration av testämnet som orsakar en tillväxtvariation på x % hos testexemplaren jämfört med kontrollgruppen.
Fisktäthet: våtvikten fisk per vattenvolym. Antalstäthet: antalet exemplar per vattenvolym.
Individuell specifik tillväxthastighet: tillväxthastigheten hos en individ, uppmätt med startvikten som utgångspunkt.
Kärlmedelvärdet för specifik tillväxthastighet: medeltillväxthastigheten hos en kärlpopulation vid en viss koncentration.
Pseudospecifik tillväxthastighet: den individuella tillväxthastigheten jämförd med kärlpopulationens medel-begynnelsevikt.
1.3 PRINICP FÖR TESTMETODEN
Unga fiskexemplar i exponentiell tillväxtfas vägs och placeras i testkärl där de exponeras för en serie subletala koncentrationer av testämnet upplöst i vatten. Exponeringen ska helst ske under genomflödesförhållanden eller, om detta inte är möjligt, under lämpliga halvstatiska förhållanden (statisk förnyelse). Testperiodens längd är 28 dagar. Fiskarna utfodras dagligen. Foderportionen baserar sig på fiskarnas begynnelsevikter och kan räknas om efter 14 dagar. Vid slutet av testet vägs fiskarna på nytt. Verkningarna på tillväxthastigheterna analyseras med en regressionsmodell för att bestämma den koncentration som leder till en variation på x % av tillväxthastigheten, dvs. ECx (t.ex. EC10, EC20 eller EC30). Alternativt kan testdata jämföras med kontrollgruppens värden för att bestämma den lägsta koncentration vid vilken verkningar observeras (LOEC) och den koncentration vid vilken inga verkningar observeras (NOEC).
1.4 INFORMATION OM TESTÄMNET
Det bör finnas tillgäng till resultaten frän ett test avseende akut toxicitet (se metod Cl), som helst ska ha utförts med samma art som används för detta test. Testämnets vattenlöslighet och ångtryck måste vara kända. Det bör finnas tillgång till en tillförlitlig analysmetod for kvantifiering av ämnet i testlösningen och denna metod blir ha känd och rapporterad noggrannhet och detektionsgräns.
Till information som bör anges hör bl.a. testämnets strukturformel, ämnets renhet, stabilitet i vatten och ljus, pKa, Pow och resultaten från test avseende biologisk lättnedbrytbarhet (se metod C.4).
1.5 TESTETS GILTIGHET
För att testet ska vara giltigt måste följande villkor uppfyllas:
|
— |
Mortaliteten i kontrollgruppen (kontrollgrupperna) får inte överskrida 10 % vid testets slut. |
|
— |
Medelvikten hos fiskarna i kontrollgruppen (kontrollgrupperna) måste ha ökat tillräckligt för att ge möjlighet att detektera den minsta variation som anses vara signifikant. Resultaten från ett ringtest (3) har visat att medelvikten för exemplar av regnbågslax i kontrollgrupperna under de 28 testdagarna måste ha ökat med minst hälften (50 %) av begynnelsevikten. Om t.ex. begynnelsevikten är 1 g per fisk (= 100 %) bör slutvikten efter 28 dagar vara ≥ 1,5 g per fisk (≥ 150 %). |
|
— |
Koncentrationen upplöst syrgas måste under hela testet ha varit minst 60 % av luftmättnadsvärdet (ASV). |
|
— |
Under testet får vattentemperaturen inte vid någon tidpunkt variera med mer än ± 1 oC mellan olika testkärl och den får variera med högst 2 oC från de temperaturgränser som specificerats för den art som används (se tillägg 1). |
1.6 BESKRIVNING AV TESTMETODEN
1.6.1 Utrustning
Normal laboratorieutrustning och särskilt
|
a) |
syrgas- och pH-mätare, |
|
b) |
utrustning för bestämning av hårdhet och alkalinitet hos vatten, |
|
c) |
lämplig utrustning för temperaturreglering, helst med kontinuerlig övervakning, |
|
d) |
kärl som är tillverkade av kemiskt inert material och tillräckligt stora med tanke på den rekommenderade fisktätheten och antalstätheten (se avsnitt 1.8.5 och tillägg 1), |
|
e) |
lämplig precisionsvåg (t.ex. noggrannhet ±0,5 %). |
1.6.2 Vatten
Som testvatten kan användas allt vatten i vilket den berörda arten uppvisar lämplig överlevnad och tillväxt under en längre period. Testvattnet bör ha konstant kvalitet under hela testperioden. Vattnets pH bör ligga inom intervallet 6,5–8,5 men får dock under ett givet test variera med högst ±0,5 pH-enheter. Hårdhet over 140 mg/l (räknat som CaCO3) rekommenderas. För att säkerställa att spädvattnet inte påverkar testresultaten på oönskat sätt (t.ex. genom komplexbildning med testämnet), bör vattenprover regelbundet tas för analys. Mätningar av tungmetaller (t.ex. Cu, Pb, Zn, Hg, Cd och Ni), viktiga anjoner och katjoner (t.ex. Ca, Mg, Na, K, Cl och SO4), bekämpningsmedel (t.ex. total mängd organiska fosforföreningar eller total mängd organiska klorföreningar), totalt organiskt kol och uppslammade partiklar bör göras t.ex. var tredje månad i fall där man vet att spädvattnet håller en relativt jämn kvalitet. Om vattenkvaliteten har visat sig vara jämn under minst ett år kan dessa kontroller göras med längre intervall (t.ex. var sjätte månad). I tillägg 2 anges ett antal kemiska egenskaper för godtagbart spädvatten.
1.6.3 Testlösningar
Testlösningar med de valda koncentrationerna bereds genom utspädning av en stamlösning.
Stamlösningen bör helst beredas genom enkel blandning eller omskakning av testämnet i spädvattnet på mekanisk väg (t.ex. omrörning och ultraljud). Mättnadskolonner (löslighetskolonner) kan användas för att få stamlösning av lämplig koncentration.
I vissa fall kan lösnings- eller dispergeringsmedel (lösningsagenter) behövas för att få en stamlösning av lämplig koncentration. Lämpliga lösningsmedel är t.ex. aceton, etanol, metanol, dimetylsulfoxid, dimetylformamid och trietylenglykol. Lämpliga dispergeringsmedel är t.ex. Cremophor RH40, Tween 80, metylcellulosa (0,01 %) och HCO-40. Om man använder agenter som är biologiskt lättnedbrytbara (t.ex. aceton) eller agenter med hög flyktighet bör man iaktta särskild försiktighet eftersom sådana agenter kan orsaka problem med bakterieansamling vid genomflödestest. Om en lösningsagent används får den inte ha signifikant verkan på fisktillväxten eller framkalla synliga skadliga verkningar på de unga exemplaren (verkningar som framkommit vid test med rent lösningsmedel).
För genomflödestest behövs ett system som kontinuerligt portionerar och späder ut stamlösning av testämnet så att man får en tillförsel av en serie koncentrationer till testkärlen. Stamlösningens och spädvattnets flödeshastighet bör kontrolleras regelbundet, helst dagligen, och får inte variera med mer än 10 % under hela testet. Resultaten från ett ringtest (3) har visat att en vattenavlägsning på 6 liter per gram fisk och dag är godtagbar vid test på regnbågslax (se avsnitt 1.8.2.2.).
För halvstatiskt test (statisk förnyelse) beror mediets förnyelsefrekvens på testämnets stabilitet, men daglig förnyelse rekommenderas. Om resultaten från preliminära stabilitetstest (se avsnitt 1.4) visar att testämnet inte är stabilt (dvs. utanför området 80 — 120 % av nominell koncentration eller under 80 % av uppmätt startkoncentration) under ett helt förnyelseintervall måste genomflödestest övervägas.
1.6.4 Val av fiskart
De flesta erfarenheterna från ringtest (1) (3) gäller regnbågslax (Oncorhynchus mykiss) och därför rekommenderas denna art för testet. Även andra väldokumenterade arter kan användas, men då måste testförfarandet eventuellt anpassas så att lämpliga testförhållanden erhålls. Det finns t.ex. erfarenheter från test med sebrafisk (Da-nio rerio) (4) (5) och ricefish (medaka, Oryzias latipes) (6) (7) (8). 1 ett sådant fall måste motiveringen för valet av art och testmetod rapporteras.
1.6.5 Förvaring av fisk
Testfiskarna ska tas från en population från en enda stam, helst från samma romläggning. Fiskarna bör, under minst två veckor före testets början, hållas i vatten med samma kvalitet och ljusförhållanden som det som används under testet. Fiskarna bör utfodras med en minimiportion på 2 % kroppsvikt per dag, helst 4 % kroppsvikt per dag, under hela förvaringsperioden och under testet.
Efter en inledande period på 48 timmar registreras mortaliteten och följande kriterier tillämpas:
|
— |
Om mortaliteten under sju dagar är högre än 10 % av populationen bör hela satsen förkastas. |
|
— |
Om mortaliteten ligger mellan 5 och 10 % av populationen fortsätts acklimatiseringen under ytterligare sju dagar. Om mortaliteten under den andra sjudagarsperioden är högre än 5 % bör hela satsen förkastas. |
|
— |
Om mortaliteten under sju dagar är lägre än 5 % av populationen kan satsen godtas. |
Fiskarna bör inte ges behandling mot sjukdomar under de två veckorna före testet eller under testets gång.
1.7 TESTUTFORMNING
Begreppet ”testutformning” hänför sig till valet av antalet testkoncentrationer och deras inbördes nivåskillnad, antalet kärl vid varje koncentration och antalet fiskar per kärl. Vid testutformningen bör följande beaktas:
|
a) |
Undersökningens syfte. |
|
b) |
Den statistiska analysmetod som kommer att användas. |
|
c) |
Tillgången till försöksresurser och deras kostnader. |
När det gäller undersökningens syfte bör man, om möjligt, specificera de statistiska krav som gäller för detekteringen av en given skillnad (t.ex. avseende tillväxthastighet) eller, alternativt, den noggrannhet med vilken ECx (t.ex. x = 10, 20 eller 30 och helst inte under 10) måste bestämmas. Utan detta kan en hållbar definition av undersökningens storlek inte ges.
Det är viktigt att vara medveten om att en utformning som är optimal (bästa utnyttjande av resurser) för en viss metod för statistisk analys inte nödvändigtvis är optimal för en annan. Den utformning som rekommenderas för uppskattning av LOEC/NOEC är därför inte densamma som den utformning som rekommenderas för regressionsanalys.
1 de flesta fall är regressionsanalys att rekommendera framför variansanalys. Motiveringen till detta diskuteras av Stephan och Rogers (9). Om ingen lämplig regressionsmodell finns att tillgå (r2 < 0,9) bör NOEC/LOEC användas.
1.7.1 Utformning för regressionsanalys
Följande faktorer är viktiga vid utformningen av ett test som ska analyseras med regression:
|
a) |
Den koncentration vid vilken verkningar observeras (t.ex. EC10,20,30) och det koncentrationsområde som är av intresse med tanke på testämnets verkningar måste omfattas av de koncentrationer som används vid testet. Den noggrannhet med vilken man kan bestämma den koncentration vid vilken verkningar observeras är bäst när den berörda koncentrationen ligger i mitten av det testade koncentrationsområdet. Ett preliminärt orienterande försök kan vara till hjälp vid valet av lämpliga testkoncentrationer. |
|
b) |
För att göra det möjligt att uppnå en tillfredsställande statistisk modell bör testet inbegripa minst ett kontrollkärl och fem extra kärl vid olika koncentrationer. När en lösningsagent används och när det är lämpligt bör ett kontrollkärl som innehåller lösningsagenten vid den högsta testade koncentrationen köras utöver testserien (se avsnitten 1.8.3 och 1.8.4). |
|
c) |
En lämplig geometrisk eller logaritmisk serie (10) (se tillägg 3) kan användas. Fördelningen av testkoncentrationerna ska helst vara logaritmisk. |
|
d) |
Om man har tillgång till fler än sex kärl bör de extra kärlen användas för replikat eller fördelas över koncentrationsområdet i syfte att minska skillnaderna mellan de olika koncentrationsnivåerna. Dessa båda alternativ är likvärdiga. |
1.7.2 Utformning för bestämning av NOEC/LOEC med variansanalys (ANOVA)
Det ska helst finnas replikatkärl vid varje koncentration och den statistiska analysen bör göras på kärlnivå (10). Utan replikatkärl kan man inte beakta variabiliteten mellan kärl utöver den som beror på individuella skillnader mellan fiskar. Erfarenheterna har dock visat (11) att variabiliteten mellan kärl i de undersökta fallen har varit mycket liten jämförd med variabiliteten inom kärlet (dvs. mellan olika fiskar). Det är därför ett relativt godtagbart alternativ att göra den statistiska analysen pä nivån för individuella exemplar.
I regel används minst fem testkoncentrationer i en geometrisk serie med en faktor som helst inte bör överskrida 3,2.
När ett test utförs med replikatkärl bör antalet replikatkontrollkärl och därmed antalet fiskar i regel vara det dubbla jämfört med antalet vid testkoncentrationerna, och vid testkoncentrationerna bör antalet vara lika stort (13) (14) (15). Om däremot replikatkärl inte används bör antalet fiskar i kontrollgruppen vara lika stort som antalet vid varje testkoncentration.
Om man utgår från att ANOVA ska grunda sig på kärl i stället för individuella fiskar (det senare fallet skulle innebära antingen individuell märkning av fiskarna eller användning av pseudospecifika tillväxthastigheter [se avsnitt 2.1.2]), måste man ha ett tillräckligt antal replikatkärl för att möjliggöra bestämning av standardavvikelsen för koncentrationerna inom kärlen. Det betyder att frihetsgraderna för fel i variansanalysen bör vara minst 5 (10). Om endast kontrollgrupperna har replikat föreligger det fara för att felvariabiliteten ökar med ökande medelvärde för den berörda tillväxthastigheten. Eftersom tillväxthastigheten sannolikt minskar med ökande koncentration tenderar detta att leda till en överskattning av variabiliteten.
1.8 FÖRFARANDE
1.8.1 Val och vägning av testfisk
Det är viktigt att minimera fiskarnas viktvariationer vid testets början. 1 tillägg 1 anges lämpliga storleksområden för de olika arter som rekommenderas för detta test. De individuella vikterna för hela den fisksats som används för testet bor helst ligga inom ± 10 % av den aritmetiska medelvikten, och får under inga omständig- heter överskrida 25 %. Det rekommenderas att ett delprov av fiskar vägs före testet i syfte att uppskatta medelvikten.
Stampopulationen bör hållas utan foder under 24 timmar före testet. Därefter väljs fiskarna ut slumpmässigt. Fiskarna ges allmän bedövning (t.ex. med en vattenlösning av 100 mg/l trikainmetansulfonat (MS 222) neutraliserad genom tillsats av två delar natriumbikarbonat per del MS 222) och deras våtvikt (avtorkad fisk) bestäms individuellt med den noggrannhet som anges i tillägg 1. Fiskar med vikter inom det planerade området väljs ut och fördelas sedan slumpmässigt mellan testkärlen. Den totala vårvikten för fiskarna i vart och ett testkärl bör registreras. Bedövningen och hanteringen av fiskarna (inbegripet avtorkning och vägning) kan orsaka stress och skador pä de unga exemplaren, särskilt när det gäller små arter. Därför bör unga exemplar hanteras ytterst försiktigt så att stress och skador på testdjuren undviks.
Fiskarna vägs på nytt på testdag 28 (se avsnitt 1.8.6). Om det bedöms vara nödvändigt att räkna om foderportionen, kan fiskarna dock vägas på nytt på testdag 14 (se avsnitt 1.8.2.3). Det finns också andra metoder, t.ex. fotografiska metoder, för att bestämma om fiskarna har vuxit så mycket att portionerna behöver justeras.
1.8.2 Exponeringsförhållanden
1.8.2.1 Varaktighet
Testets varaktighet är ≥ 28 dagar.
1.8.2.2 Fïsktäthet och antalstäthet
Det är viktigt att fisktätheten och antalstätheten är anpassade för de arter som testas (se tillägg 1). Om fisktätheten är för hög uppstår trängselstress som leder till minskade tillväxthastigheter och eventuellt också till sjukdomar. Om fisktätheten är for låg kan det uppstå ett territorialt beteende som också kan påverka tillväxten. Fisktätheten ska under alla omständigheter vara så låg att koncentrationen upplöst syrgas kan hållas på minst 60 % ASV utan luftning. Resultaten från ringtest (3) har visat att det för regnbågslax är lämpligt med en fisktäthet på 16 exemplar à 3 — 5 g i en 40-litersvolym. Rekommenderad vattenavlägsning under testet är 6 liter per fisk och dag.
1.8.2.3 Utfodring
Fiskarna ska ges lämpligt foder (tillägg 1) i tillräcklig mängd för att möjliggöra en godtagbar tillväxthastighet. Det är viktigt att se till att det inte uppstår mikrobtillväxt och vattenturbiditet. För regnbågslax kan man utgå från att villkoren uppfylls om fiskarna ges en portion på 4 % av kroppsvikten per dag (3) (16) (17) (18). Den dagliga portionen kan delas upp i två lika delar och ges vid två utfodringstillfällen per dag med minst 5 timmars mellanrum. Portionen baserar sig på fiskarnas totala begynnelsevikt i vart och ett av testkärlen. Om fiskarna vägs på nytt på dag 14 kan portionen därefter räknas om. Fiskarna ska inte ges foder under de 24 timmar som föregår vägningen.
Okonsumerat foder och fekalt material ska dagligen avlägsnas från testkärlen genom omsorgsfull rengöring av kärlets botten genom sugning.
1.8.2.4 Ljus och temperatur
Ljusperioden och vattentemperaturen bör vara anpassade för den art som testas (se tillägg 1).
1.8.3 Testkoncentrationer
I regel krävs fem koncentrationer av testämnet, oavsett vilken testutformning som används (se avsnitt 1.7.2). Tidigare kunskaper om testämnets toxicitet (t.ex. från test av akut toxicitet eller preliminära orienterande undersökningar) ar till hjälp vid valet av lämpliga testkoncentrationer. Om färre än fem testkoncentrationer används måste detta motiveras. Den högsta testkoncentrationen får inte överskrida testämnets löslighetsgräns i vatten.
Om en lösningsagent används för att underlätta beredningen av testlösningar får agentens slutkoncentration i testkärlet inte överskrida 0,1 ml/l och samma slutkoncentration bör helst användas i alla testkärl (se avsnitt 1.6.3). Man bör dock sträva efter att undvika användning av lösningsagenter.
1.8.4 Kontrollsatser
Antalet spädvattenskontrollsatser beror på testutformningen (se avsnitten 1.7–1.7.2). Om en lösningsagent används bör testet även omfatta lika stort antal kontrollsatser för lösningsagenten som för spädvattnet.
1.8.5 Frekvensen för analytiska bestämningar och mätningar
Under testets gång bör testämnets koncentrationer bestämmas med regelbundna intervall (se nedan).
Vid genomflödestest bör flödeshastigheterna för spädlösning och testämnets stamlösning kontrolleras regelbundet, helst dagligen. Under testets förlopp får flödeshastigheterna inte variera med mer än 10 %. Om det förväntas att testämneskoncentrationerna kommer att hållas inom ± 20 % av de nominella värdena (dvs. inom området 80–120 %, se avsnitten 1.6.2 och 1.6.3) är det tillrådligt att analysera de högsta och lägsta testkoncentrationerna åtminstone vid testets början och därefter varje vecka. Vid test där det inte förväntas att testämnes-koncentrationen kommer att hållas inom ± 20 % av de nominella värdena (med hänvisning till testämnets stabilitetsdata) är det nödvändigt att analysera alla testkoncentrationer, men enligt samma schema.
Vid halvstatiska test (förnyelsetest) där det förväntas att testämneskoncentrationen kommer att hållas inom ± 20 % av de nominella värdena är det tillrådligt att analysera de högsta och lägsta testkoncentrationerna åtminstone när de är nyberedda och strax före förnyelsen. Detta görs vid testets början och därefter varje vecka. Vid test där det inte förväntas att testämneskoncentrationen kommer att hållas inom dessa gränser måste alla testkoncentrationer analyseras enligt samma schema som används för stabilare ämnen.
Det rekommenderas att resultaten grundar sig på de uppmätta koncentrationerna. Om det däremot finns bevis för att testämnets koncentration i lösningen på ett tillfredsställande sätt har hållits inom ± 20 % av den nominella eller uppmätta startkoncentrationen under hela testperioden, kan resultaten grunda sig på nominella eller uppmätta värden.
Proverna kan behöva filtreras (t.ex. med porstorlek 0,45 μm) eller centrifugeras. Centrifugering rekommenderas. Om testmaterialet inte adsorberas på filter kan dock även filtrering godtas.
Under testets gång mäts upplöst syrgas, pH och temperatur i alla testkärl. Totalhårdhet och salthalt (om relevant) mäts i kontrollsatserna och i ett kärl med den högsta koncentrationen. Upplöst syrgas och salthalt (om relevant) mäts minst tre gånger (vid testets början, mitt och slut). Vid halvstatiskt test rekommenderas att upplöst syrgas mäts oftare, helst före och efter varje vattenförnyelse eller minst en gång i veckan. Vid halvstatiskt test mäts pH i början och i slutet av varje vattenförnyelse och vid genomflödestest minst varje vecka. Hårdheten och alkaliniteten mäts en gång per test. Temperaturen bör mätas dagligen och helst övervakas kontinuerligt i minst ett testkärl.
1.8.6 Observationer
Vikt: Vid testets slut vägs alla fiskar (våtvikt, dvs. avtorkade exemplar) antingen i grupper per testkärl eller individuellt. Vägning per testkärl är att föredra eftersom individuell vägning förutsätter att fiskarna har märkts individuellt. Om man gör individuella vägningar för bestämning av individuell specifik tillväxthastighet bör den märkningsteknik som väljs stressa djuren minimalt (ett lämpligt alternativ till frysmärkning är t.ex. användning av färgad tunn fisklina).
Under testperioden bör fiskarna inspekteras dagligen och alla yttre abnormaliteter (t.ex. blödningar eller avfärgning) och onormalt beteende bör registreras. Varje fall av fiskdöd bör registreras, och döda fiskar bör avlägsnas så snart som möjligt. Döda fiskar ska inte ersättas eftersom fisktätheten och antalstätheten är tillräcklig för att tillväxten inte ska påverkas av ändringar i antalet fiskar per kärl. Mängden foder som ges måste dock justeras.
2. DATA OCH RAPPORTERING
2.1 BEHANDLING AV RESULTATEN
Det rekommenderas att en statistiker deltar i både utformning och analys av testet, eftersom metoden medger betydande variationer i fråga om testutformning, t.ex. antalet testkärl, antalet testkoncentrationer, antalet fiskar osv. Med hänvisning till de många alternativen för testutformning ges inga specifika anvisningar om statistiska procedurer.
Tillväxthastigheterna bör inte beräknas för testkärl där mortaliteten överskrider 10 %. Mortaliteten bör dock anges för alla testkoncentrationer.
Oavsett vilken metod som väljs för analys av data består det centrala konceptet av den specifika tillväxthastigheten mellan tidpunkten t, och tidpunkten t,. Denna kan definieras på flera olika sätt, beroende på om fiskarna är individuellt märkta eller inte och om det krävs ett kärlmedelvärde.



där:
|
r1 |
= |
individuell fiskspecifik tillväxthastighet |
|
r2 |
= |
specifik medeltillväxthastighet i kärlet |
|
r3 |
= |
pseudospecifik tillväxthastighet |
|
w1, w2 |
= |
vikten hos en viss fisk vid tidpunkten t1 respektive t2 |
|
loge w1 |
= |
logaritmen av vikten hos en individuell fisk vid testperiodens början |
|
loge w2 |
= |
logaritmen av vikten för en individuell fisk vid testperiodens slut |
|
loge W1 |
= |
medelvärdet för logaritmerna av värdena w1 för fiskarna i kärlet vid testperiodens början |
|
loge W2 |
= |
medelvärdet för logaritmerna av värdena w2 för fiskarna i kärlet vid testperiodens slut |
|
t1, t2 |
= |
tidpunkten (dagar) vid testperiodens början och slut |
r1, r2, r3 kan beräknas för perioden dag 0–28 och i tillämpliga fall (t.ex. om mätning har gjorts dag 14) för perioderna dag 0–14 och dag 14–28.
2.1.1 Analys av resultaten genom regression (koncentration-respons-modellering)
Denna analysmetod innebär att en lämplig matematisk modell anpassas till förhållandet mellan den specifika tillväxthastigheten och koncentrationen. Genom modellen kan man uppskatta ECX. dvs. önskat EC-värde. När denna metod används behöver r inte beräknas för individuella fiskar (r1), utan analysen kan i stället basera sig på kärlets medelvärde for r (r1). Den senare metoden är att föredra. Den är också lämpligare när man använder de minsta arterna.
Karlmedelvärdena för de specifika tillväxthastigheterna (r2) plottas grafiskt mot koncentrationen, vilket ger möjlighet att granska förhållandet mellan koncentration och respons.
En lämplig modell bör väljas för att uttrycka förhållandet mellan r2 och koncentrationen. Valet måste motiveras på tillbörligt sätt.
Om antalet fiskar som överlever inte är lika stort i varje kärl bör modellanpassningen, antingen den är enkel eller icke-linjär. viktas för att kompensera för de olika stora grupperna.
Den metod som används för anpassning av modellen måste ge möjlighet att uppskatta t.ex. EC20 och få fram spridningen kring värdet (standardfel eller konfidensintervall). Kurvan för den anpassade modellen bör visualiseras i förhållande till data så att det framgår i hur hög grad modellen stämmer (9) (19) (20) (21).
2.1.2 Analys av resultaten för uppskattning av LOEC
Om testet har omfattat replikering av kärl vid alla koncentrationsnivåer kan uppskattningen av LOEC basera sig på en variansanalys (ANOVA) av kärlmedelvärdet för specifik tillväxthastighet (se avsnitt 2.1) åtföljd av en lämplig metod (t.ex. Dunnetts eller Williams test (13) (14) (15 (22)) för jämförelse av medelvärdet r för varje koncentration med medelvärdet r för kontrollsatserna, i syfte att hitta den lägsta koncentration för vilken denna skillnad är signifikant vid en sannolikhetsnivå på 0,05. Om de antaganden som krävs för parametriska metoder inte uppfylls – onormal fördelning (t.ex. Shapiro-Wilks test) eller variansen är heterogen (Bartletts test) – bör man överväga att homogenisera varianserna genom transformering av data före ANOVA. Alternativt kan viktad ANOVA övervägas.
Om testet inte har omfattat replikering av kärl vid varje koncentration är kärlbaserad ANOVA okänslig eller omöjlig. I en sådan situation är det en godtagbar kompromiss att basera ANOVA på den pseudospecifika tillväxthastigheten r3 för individuella fiskar.
Medelvärdet r3 för varje testkoncentration kan sedan jämföras med medelvärdet r3 för kontrollsatserna. LOEC kan därefter fås fram på ovan beskrivet sätt. Det bör konstateras att denna metod inte beaktar eller ger skydd mot variabiliteten mellan kärl, utöver den variabilitet som beaktas genom variabiliteten mellan individuella fiskar. Erfarenheter har dock visat (9) att variabiliteten mellan kärl är mycket liten jämfört med variabiliteten inom ett kärl (dvs. mellan fiskar). Om individuella fiskar inte inkluderas i analysen bör metoden för identifiering av utanförliggande värden anges tillsammans med en motivering.
2. TOLKNING AV RESULTATEN
Resultaten bör tolkas med försiktighet när de uppmätta koncentrationerna av det toxiska ämnet i testlösningen ligger på nivåer nära analysmetodens detektionsgräns eller – vid halvstatiska test – när koncentrationen av testämnet minskar mellan nyss beredd lösning och lösningen före förnyelsen.
2.3 TESTRAPPORT
Testrapporten ska innehålla nedan angivna uppgifter.
2.3.1 Testämne:
|
— |
Aggregationstillstånd och relevanta fysikalisk-kemiska egenskaper. |
|
— |
Kemiska identifieringsdata, inklusive renhet och i tillämpliga fall analysmetod för identifiering av testäm net. |
2.3.2 Testdjur:
|
— |
Vetenskapligt namn. |
|
— |
Tillgängliga uppgifter om stam, storlek, leverantör, eventuella förbehandlingar osv. |
2.3.3 Testförhållanden:
|
— |
Testförfarande som har använts (t.ex. halvstatiskt test eller genomflödestest, fisktäthet, antalstäthet, osv.). |
|
— |
Testutformning (t.ex. antalet testkarl, testkoncentrationer och replikat, antalet fiskar per karl). |
|
— |
Metoden för beredning av stamlösningar och förnyelsefrekvensen (i förekommande fall bör även lösningsagenten och dess koncentration anges). |
|
— |
Nominella testkoncentrationer, de uppmätta värdenas medelvärden och standardavvikelser i testkärlen och den metod med vilken dessa erhölls samt bevis för att mätningarna hänför sig till testämnets koncentratio ner i lösningen. |
|
— |
Spädvattnets egenskaper: pH, hårdhet, alkalinitet, temperatur, koncentrationen upplöst syrgas, restklornivåer (om de har uppmätts), totalt organiskt kol, partikelinnehåll, testmediets salthalt (om den har uppmätts) och resultatet av alla övriga mätningar. |
|
— |
Vattenkvaliteten i testkärlen: pH, hårdhet, temperatur och halten upplöst syrgas. |
|
— |
Detaljerade uppgifter om utfodringen (t.ex. typ av foder, källa, hur mycket fiskarna har fått och hur ofta). |
2.3.4 Resultat:
|
— |
Bevis för att kontrollgrupperna uppfyller validitetskriterierna för överlevnad. Data om varje förekomst av fiskdöd. |
|
— |
Statistiska analystekniker som har använts, statistik baserad på replikat eller fisk, behandlingen av data och motivering för de tekniker som har använts. |
|
— |
Data i tabellform över individuella fiskvikter och medeltal dag 0, dag 14 (om uppmätt) och dag 28, kärl medelvärde eller pseudospecifika tillväxthastigheter (enligt det som är lämpligt) för periodernadag 0–28, eller eventuellt dag 0–14 och dag 14–28. |
|
— |
Resultat av den statistiska analysen (regressionsanalys eller ANOVA), helst i tabellform och grafisk form samt LOEC (p=0,05) och NOEC eller ECX, om möjligt med standardfel (enligt det som är lämpligt). |
|
— |
Förekomsten av eventuella ovanliga reaktioner hos fiskarna och eventuella synliga verkningar som har orsakats av testämnet. |
3. HÄNVISNINGAR
|
1. |
Solbe J. K de LG (1987), Environmental Effects of Chemicals (CFM 9350 SLD). Report on a UK Ring Test of a Method for Studying the Effects of Chemicals on the Growth Rate of Fish. WRc Report No PRD 1388-M/2. |
|
2. |
Ashley S., Mallett M. J. and Grandy N.). (1990), EEC Ring Test of a Method for Determining the Effects of Chemicals on the Growth Rate of Fish. Final Report to the Commission of the European Communities. WRc Report No EEC 2600-M. |
|
3. |
Grossland N. O. (1985), A method to evaluate effects of toxic chemicals on fish growth. Chemosphere, 14, s. 1855–1870. |
|
4. |
Nagel R., Bresh H., Caspers N., Hansen P. D., Market M, Munk R., Scholz N. and Höfte B. B. (1991), Effect of 3,4-dichloroanilinc on the early life stages of the Zebrafish (Brachydanio rerio): results of a comparative laboratory study. Ecotox. Environ. Safety, 21, s. 157–164. |
|
5. |
Yamamoto, Tokio. (1975), Series of stock cultures in biological field. Medaka (killifish) biology and strains. Keigaku Publish. Tokio, Japan. |
|
6. |
Holcombe, G./V.. Benoit D. A., Hammermeister, D. E., Leonard, E. N. and Johnson, R. D. (1995), Acute and long-term effects of nine chemicals on the Japanese medaka (Oryzias latipes). Arch. Environ. Conta. Toxicol. 28, s. 287–297. |
|
7. |
Benoit, D. A., Holcombe, G. \V. and Spehar, R. L. (1991), Guidelines for conducting early life toxicity tests with Japanese medaka (Oryzias latipes). Ecological Research Series EPA-600/3-91-063. US Environmental Protection Agency, Duluth, Minnesota. |
|
8. |
Stephan C. E. and Rogers J. W. (1985), Advantages of using regression analysis to calculate results of chronic toxicity tests. Aquatic Toxicology and Hazard Assessment: Eighth Symposium, ASTM STP 891, R. C. Bahner and D. J. Hansen, eds., American Society for Testing and Materials, Philadelphia, s. 328–338. |
|
9. |
Environment Canada (1992), Biological test method: toxicity tests using early life stages of salmonid fish (rainbow trout, coho salmon, or atlantic salmon). Conservation and Protection, Ontario, Report EPS l/RM/28, 81 pp. |
|
10. |
Cox D. R. (1958), Planning of experiments. Wiley Edt. |
|
11. |
Pack S. (1991), Statistical issues concerning the design of tests for determining the effects of chemicals on the growth rate of fish. Room Document 4, OECD Ad Hoc Meeting of Experts on Aquatic Toxicology, WRc Medmenham, UK, 10–12 December 1991. |
|
12. |
Dunnett C. W. (1955), A Multiple Comparisons Procedure for Comparing Several Treatments with a Control,]. Amer. Statist. Assoc, 50, s. 1096–1121. |
|
13. |
Dunnett C. W. (1964), New tables for multiple comparisons with a control. Biometrics, 20, s. 482–491. |
|
14. |
Williams D. A. (1971), A test for differences between treatment means when several dose levels are compared with a zero dose control. Biometrics 27, s. 103–117. |
|
15. |
Johnston, W. L., Atkinson, J. L., Glanville N. T. (1994), A technique using sequential feedings of different coloured food to determine food intake by individual rainbow trout, Oncorhynchus mykiss: effect of feeding level. Aquaculture 120, s. 123–133. |
|
16. |
Quinton, J. C. and Blake, R. W. (1990), The effect of feed cycling and ration level on the compensatory growth response in rainbow trout, Oncorhynchus mykiss. Journal of Fish Biology, 37, s. 33–41. |
|
17. |
Post, G. (1987), Nutrition and Nutritional Diseases of Fish. Chapter IX in Testbook of Fish Health. T.F.H. Publications, Inc. Neptune City, New Jersey, USA. 288 pp. |
|
18. |
Bruce, R. D. and Versteeg D. J. (1992), A statistical procedure for modelling continuous toxicity data . Environ. Toxicol. Chem. 11, s. 1485–1494. |
|
19. |
DeGraeve, G. M., Cooncy, |. M., Pollock, T. L, Reichenbach, J. H., Dean, Marcus, M. D. and Mclntyre, D. O. (1989), Precision of EPA seven-day fathead minnow larval survival and growth test; intra and interlaboratory study. Report EA-6189 (American Petroleum Institute Publication, No 4468). Electric Power Research Institute, Palo Alto, CA. |
|
20. |
Norbert-King T. J. (1988), An interpolation estimate for chronic toxicity: the ICp approach. US Environmental Protection Agency. Environmental Research Lab., Duluth, Minnesota. Tech. Rep. No 05–88 of National Effluent Toxicity Assesment Center. Sept. 1988. 12 pp. |
|
21. |
Williams D. A. (1972), The comparison of several dose levels with a zero dose control. Biometrics 28, s. 510–531. |
Tillägg 1
LÄMPLIGA TESTBETINGELSER OCH FISKARTER SOM REKOMMENDERAS FÖR TESTNING
|
Art |
Rekommenderat temperaturområde oC) |
l.jusperiod (timmar) |
Rekommenderat intervall för fiskarnas begynnelsevikt (fe) |
Mätningsnoggrannhet |
Fisktäthet (g/l) |
Antalstäthet (antal per liter) |
Foder |
Testets varaktighet (dagar) |
|
Rekommenderad art |
|
|
|
|
|
|
|
|
|
Oncorhynchus mykiss Regnbågslax |
12,5–16,0 |
12–16 |
1–5 |
Närmaste 100 mg |
1,2–2,0 |
4 |
Torrfoder som är särskilt ägnat för laxyngel |
≥ 28 |
|
Andra väldokumenterade arter |
|
|
|
|
|
|
|
|
|
Danio rerio Sebrafisk |
21–25 |
12–16 |
0,050–0,100 |
Närmaste 1 mg |
0,2–1,0 |
5–10 |
Levande föda (Brachionus Artemia) |
> 28 |
|
Oryzias latipes ”Ricefish” (Medaka) |
21–25 |
12–16 |
0,050–0,100 |
Närmaste 1 mg |
0,2–1,0 |
5–20 |
Levande föda (Brachionus Artemia) |
> 28 |
Tillägg 2
NÅGRA KEMISKA EGENSKAPER FÖR GODTAGBART SPÄDVATTEN
|
Ämne |
Koncentrationer |
|
Partikelinnehåll |
< 20 mg/l |
|
Totalt organiskt kol |
< 2 mg/l |
|
Ojoniserad ammoniak |
< 1 μg/l |
|
Restklor |
< 10 μg/l |
|
Total mängd bekämpningsmedel typ organiska fosforföreningar |
< 50 ng/1 |
|
Total mängd bekämpningsmedel typ organiska klorföreningar samt polyklorerade bifeny-er |
< 50 ng/1 |
|
Total mängd organiskt bundet klor |
< 25 ng/1 |
Tillägg 3
i logaritmisk serie av koncentrationer lämpliga för toxicitetstest (9)
|
Kolumn (antalet koncentrationer mellan 100 och 10 eller mellan 10 och 1) (20) |
||||||
|
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
100 |
100 |
100 |
100 |
100 |
100 |
100 |
|
32 |
46 |
56 |
63 |
68 |
72 |
75 |
|
10 |
22 |
32 |
40 |
46 |
52 |
56 |
|
3,2 |
10 |
18 |
25 |
32 |
37 |
42 |
|
1,0 |
4,6 |
10 |
16 |
22 |
27 |
32 |
|
|
2,2 |
5,6 |
10 |
15 |
19 |
24 |
|
|
1,0 |
3,2 |
6,3 |
10 |
14 |
18 |
|
|
|
1,8 |
4,0 |
6,8 |
10 |
13 |
|
|
|
1,0 |
2,5 |
4,6 |
7,2 |
10 |
|
|
|
|
1,6 |
3,2 |
5,2 |
7,5 |
|
|
|
|
1,0 |
2,2 |
3,7 |
5,6 |
|
|
|
|
|
1,5 |
2,7 |
4,2 |
|
|
|
|
|
1,0 |
1,9 |
3,2 |
|
|
|
|
|
|
1,4 |
2,4 |
|
|
|
|
|
|
1,0 |
1,8 |
|
|
|
|
|
|
|
1,3 |
|
|
|
|
|
|
|
1,0 |
C.15 FISK, KORTTIDSTEST AVSEENDE TOXICITET PA EMBRYO- OCH SÄCKYNGELSTADIERNA
1. Metod
Denna metod för korttidstest avseende toxicitet motsvarar OECD TG 212 (1998).
1.1 INLEDNING
Vid detta korttidstest avseende toxicitet på fiskembryon och säckyngel exponeras levnadsstadierna från nyligen befruktade ägg till slutet av säckyngelstadiet. Inget foder ges vid detta test, och testet avslutas medan säckynglen fortfarande får sin näring från gulesäcken.
Testet är avsett för bestämning av de letala och, i begränsad utsträckning, subletala verkningarna av kemikalier på de specifika stadier och specifika arter som testas. Testet kan ge användbar information i flera avseenden: dels bildar det en brygga mellan test avseende letala verkningar och test avseende subletala verkningar, dels kan det användas som ett screeningtest inför ett komplett test av de tidiga levnadsstadierna eller för test avseende kronisk toxicitet. Dessutom kan det användas för test av arter för vilka uppfödningsteknikerna inte är tillräckligt utvecklade för att täcka perioden för övergång från endogen till exogen utfodring.
Man bör beakta att endast tester som täcker alla stadier av fiskens livscykel i regel kan ge en tillförlitlig uppskattning om en kemikalies kroniska toxicitet för fisk, och att minskad exponering av levnadsfaser kan medföra lägre känslighet och underskattning av den kroniska toxiciteten. Man kan därför anta att ett test på embryon och säckyngel är mindre känsligt än ett komplett test av de tidiga levnadsstadierna, särskilt när det gäller kemikalier som är starkt lipofila (log Pow > 4) och kemikalier med ett specifikt toxiskt verkningssätt. Man kan dock förvänta sig mindre skillnader i känslighet mellan de två testerna när det gäller kemikalier som har ett icke-specifikt, narkotiskt verkningssätt (1).
Fram till offentliggörandet av detta test har de flesta erfarenheterna från test på embryon och säckyngel erhållits med sötvattenfisken Danio rerio Hamilton-Buchanan (Teleostei, Cyprinidae – sebrafisk). Närmare anvisningar om testförfarandet för denna art finns i tillägg 1. Det finns dock inga hinder för att använda andra arter för vilka det finns erfarenheter att tillgå (tabell 1).
1.2 DEFINITIONER
Lägsta koncentration vid vilken verkningar observeras (Lowest observed effect concentration, LOEC): den lägsta testade koncentrationen av ett testämne vid vilken man kan observera signifikanta verkningar (vid p < 0,05) jämfört med kontrollgruppen. Alla testkoncentrationer ovanför LOEC måste framkalla skadliga verkningar som är lika stora eller större än de verkningar som kan observeras vid LOEC.
Koncentration vid vilken inga verkningar observeras (No observed effect concentration, NOEC): testkoncentrationen omedelbart under LOEC.
1.3 PRINCIP FÖR TESTMETODEN
Fiskens embryo- och säckyngelstadier exponeras för en serie koncentrationer av testämnet upplöst i vatten. Testprotokollet innehåller en möjlighet att välja mellan halvstatiskt test och genomflödestest. Det valda alternativet beror på testämnets egenskaper. Testet inleds med att befruktade ägg placeras i testkärl. Testet avslutas strax innan någon av gulesäckarna i något av testkärlen har absorberats helt eller innan ynglen i kontrollgrupperna börjar dö på grund av svält. De letala och subletala verkningarna bedöms och jämförs med kontrollgruppernas värden i syfte att bestämma den lägsta koncentrationen vid vilken verkningar observeras och den koncentration vid vilken inga verkningar observeras. Alternativt kan verkningarna analyseras med hjälp av en regressionsmodell i syfte att uppskatta den koncentration som framkallar en given procentuell verkan (t.ex. LC/ECx, där x är en definierad procentuell verkan).
1.4 INFORMATION OM TESTÄMNET
Man bör ha tillgång till resultaten från ett test avseende akut toxicitet (se metod C.1), som helst ska ha utförts med samma art som används för detta test. Sådana resultat kan vara till hjälp vid valet av lämpliga testkoncentrationer för testet på tidiga levnadsstadier. Testämnets vattenlöslighet (inbegripet lösligheten i testvattnet) och ångtryck måste vara kända. Man bör ha tillgång till en tillförlitlig analysmetod för kvantifiering av ämnet i testlösningen. Metoden bör ha känd och rapporterad noggrannhet och detektionsgräns.
För definition av testförhållandena bör man ange bl.a. testämnets strukturformel, ämnets renhet, stabilitet i ljus, stabilitet i testförhållandena. pKa, Pow och resultaten från test avseende biologisk lättnedbrytbarhet (se metod C. 4).
1.5 TESTETS GILTIGHET
För att testet ska vara giltigt måste följande villkor uppfyllas:
|
— |
Den totala överlevnaden hos de befruktade äggen i kontrollgrupperna, och i tillämpliga fall i kärlen med rent lösningsmedel, måste vara högre eller lika med de gränsnivåer som definieras i tilläggen 2 och 3. |
|
— |
Koncentrationen löst syrgas måste under hela testet ligga mellan 60 och 100 % av luftmättnadsvärdet (ASV). |
|
— |
Under testet får vattentemperaturen inte variera med mer än ±1,5 oC mellan olika testkärl eller mellan påföljande dagar. Vattentemperaturen bör hållas inom de temperaturgränser som specificerats för den art som används (se bilagorna 2 och 3). |
1.6 BESKRIVNING AV TESTMETODEN
1.6.1 Testkärl
Kärl av glas eller annat kemiskt inert material kan användas. Kärlen måste vara tillräckligt stora med tanke på lämplig fisktäthet (se avsnitt 1.7.1.2). Det rekommenderas att kärlen placeras slumpvis i testutrymmet. En randomiserad blockutformning med en behandling per block är att föredra framför en fullständigt randomiserad utformning, i de fall där det i laboratoriet förekommer sådana systematiska effekter som kan kontrolleras med hjälp av blockuppdelning. Eventuell blockuppdelning måste beaktas i de efterföljande dataanalyserna. Testkärlen bör avskärmas från oönskade störningar.
1.6.2 Val av fiskart
I tabell 1A anges rekommenderade fiskarter. Det hindrar inte att andra arter används (se exemplen i tabell 1B), men då måste testförfarandet eventuellt justeras så att lämpliga testförhållanden kan säkerställas. I ett sådant fall måste testrapporten innehålla en motivering för valet av art och testmetod.
1.6.3 Förvaring av fiskyngel
Närmare uppgifter om lämplig förvaring av fiskyngelstammen finns i OECD TG 210 (21) och i hänvisningarna (2), (3), (4), (5), (6).
1.6.4 Hantering av embryon och yngel
Embryon och yngel kan exponeras i mindre kärl, placerade i huvudkärlet, med sidor eller ändar av nät som möjliggör ett flöde av testlösning genom kärlet. För att få ett icke-turbulent flöde genom dessa små kärl kan de hållas hängande från en arm som sänker och höjer kärlen (de får dock inte höjas över ytan). Ett sifonspolat system kan också användas. Befruktade ägg av laxfiskar kan hållas på ställningar eller galler med så stora öppningar att ynglen kan falla igenom efter kläckningen. Om halvstatiskt testförfarande med fullständig daglig förnyelse används rekommenderas pasteur-pipetter för avlägsnandet av embryon och yngel (se avsnitt 1.6.6).
Om äggbehållare, galler eller nät har använts för att hålla äggen inom huvudkärlet tas dessa tillbehör bort efter att ynglen har kläckts (21), med undantag av nät som behövs för att hålla fiskarna på plats. Om yngel måste flyttas får de inte utsättas för luft, och nät får inte användas för att släppa ut fisk från äggbehållare (dessa säkerhetsåtgärder ar eventuellt inte nödvändiga om arten inte är så känslig, t.ex. när man använder karp). Överföringstidpunkten varierar beroende på art och det är inte alltid nödvändigt med en överföring. Vid halvstatiskt test kan man använda bägare eller grunda behållare, vid behov försedda med ett nät som finns en bit ovanför bägarens botten. Om bägarnas volym är tillräcklig med tanke på fisktätheten (se 1.7.1.2) är det eventuellt inte nödvändigt att överföra embryon eller yngel.
1.6.5 Vatten
Som testvatten kan användas allt vatten som uppfyller de kemiska kraven för godtagbart spädvatten enligt tillägg 4 och i vilket testarternas kontrollgrupper har minst den överlevnadsgrad som anges i tilläggen 2 och 3. Testvattnet bör ha konstant kvalitet under hela testperioden. Vattnets pH-värde får variera med högst ±0,5 pH-enheter. För att säkerställa att spädvattnet inte påverkar testresultaten på oönskat sätt (t.ex. genom komplexbildning med testämnet) eller har skadliga verkningar på yngelstammens prestanda, tas vattenprover regelbundet för analys. Mätningar av tungmetaller (t.ex. Cu, Pb, Zn, Hg, Cd och Ni), viktiga anjoner och katjoner (t.ex. Ca, Mg, Na, K, Cl och SO4), bekämpningsmedel (t.ex. total mängd organiska fosforföreningar eller total mängd organiska klorföreningar), totalt organiskt kol och uppslammade partiklar bör göras t.ex. var tredje månad i fall där man vet att spädvattnet håller en relativt jämn kvalitet. Om vattenkvaliteten har visat sig vara jämn under minst ett år kan dessa kontroller göras med längre intervall (t.ex. var sjätte månad).
1.6.6 Testlösningar
Testlösningar med de valda koncentrationerna bereds genom utspädning av en stamlösning.
Stamlösningen bör helst beredas genom enkel blandning eller omskakning av testämnet i spädvattnet på mekanisk väg (t.ex. omrörning och ultraljud). Mättnadskolonner (löslighetskolonner) kan användas för att få stamlösning av lämplig koncentration. Man bör så långt som möjligt undvika att använda lösnings- eller dispergeringsmedel (lösningsagenter). Sådana medel kan dock i vissa fall behövas för att få en stamlösning av lämplig koncentration. Lämpliga lösningsmedel är t.ex. aceton, etanol, metanol, dimetylformamid och trietylenglykol. Lämpliga dispergeringsmedel är t.ex. Cremophor RH40, Tween 80, metylcellulosa (0,01 %) och HCO-40. Om man använder agenter som är biologiskt lättnedbrytbara (t.ex. aceton) eller agenter med hög flyktighet bör man iaktta särskild försiktighet eftersom sådana agenter kan orsaka problem med bakterieansamling vid genomflödestest. Om en lösningsagent används får den inte ha signifikant verkan på överlevnaden eller framkalla synliga skadliga verkningar på tidiga levnadsstadier (verkningar som framkommit vid test med rent lösningsmedel). Användningen av sådana ämnen bör undvikas med alla medel.
Vid halvstatiskt test kan man välja mellan två olika förnyelseförfaranden. Det ena innebär att nya testlösningar bereds i rena kärl och överlevande ägg och yngel överförs försiktigt till de nya kärlen i en liten volym gammal lösning, samtidigt som man undviker exponering för luft. Det andra förfarandet innebär att organismerna hålls kvar i kärlen medan en andel (minst tre fjärdedelar) av testvattnet byts ut. Hur ofta mediet måste förnyas beror på testämnets stabilitet, men daglig förnyelse rekommenderas. Om resultaten från preliminära stabilitetstest (se avsnitt 1.4) visar att testämnet inte är stabilt (dvs. utanför området 80–120 % av nominell koncentration eller under 80 % av uppmätt startkoncentration) under ett helt förnyelseintervall, måste genomflödestest övervägas. Under alla omständigheter bör man undvika att stressa ynglen när man förnyar vattnet.
För genomflödestest behövs ett system som kontinuerligt portionerar och späder ut stamlösning av testämnet, så att man får en tillförsel av en serie koncentrationer till testkärlen. Stamlösningens och spädvattnets flödeshastighet bör kontrolleras regelbundet, helst dagligen, och får inte variera med mer än 10 % under hela testet. En flödeshastighet som motsvarar minst fem testkärlsvolymer per 24 timmar har konstaterats vara lämplig (2).
1.7 FÖRFARANDE
I litteraturen finns information om kapaciteten hos toxicitetstest på fiskembryon och säckyngel. Ett antal exempel på litteratur finns i hänvisningarna (7), (8), (9).
1.7.1 Exponeringsförhållanden
1.7.1.1 Varaktighet
Testet bör helst inledas inom 30 minuter efter befruktning av äggen. Embryona läggs i testlösningen före eller så snart som möjligt efter att blastocysten påbörjar klyvning, och under alla omständigheter innan gastrulastadiet inleds. Om äggen kommer från en kommersiell leverantör kan det hända att det inte är möjligt att starta testet strax efter befruktningen. Testets känslighet kan allvarligt påverkas av en försenad teststart. Testet måste inledas inom åtta timmar efter befruktningen. Eftersom ynglen inte utfodras under exponeringsperioden, bör testet avslutas strax innan någon av gulesäckarna i något av testkarlen har absorberats helt eller innan ynglen i kontrollgrupperna börjar dö på grund av svält. Testperiodens längd beror på den art som används. Vissa rekommendationer gällande testperiodens längd finns i tilläggen 2 och 3.
1.7.1.2 Fisktäthet
Antalet befruktade ägg vid testperiodens början bör vara tillräckligt med tanke på statistiska krav. Äggen fördelas slumpmässigt mellan behandlingsgrupperna. För varje koncentration används minst 30 befruktade ägg, jämnt fördelade (eller så jämnt det är möjligt – med vissa arter är det svårt att få likadana satser) mellan minst tre likadana testkärl. Fisktätheten (biomassa per volym testlösning) får inte vara högre än att man kan hålla koncentrationen löst syrgas på minst 60 % ASV utan luftning. För genomflödestest rekommenderas en fisktäthet som inte överskrider 0,5 g/l per 24 timmar och som under inga omständigheter överskrider 5 g/l lösning (2).
1.7.1.3 Ljus och temperatur
Ljusperioden och vattentemperaturen bör anpassas till den art som testas (tilläggen 2 och 3). Temperaturövervakningen kan gärna ordnas med hjälp av ett extra testkärl.
1.7.2 Testkoncentrationer
I regel krävs fem koncentrationer av testämnet. Koncentrationerna bör ligga i en serie med en konstant faktor som inte överskrider 3,2. När man väljer testkoncentrationsområde är det viktigt att beakta kurvan som visar sambandet mellan LC50 och exponeringsperioden vid test avseende akut toxicitet. I vissa fall kan det vara motiverat med mindre än fem koncentrationer, t.ex. vid gränstest, och ett snävare koncentrationsintervall. Om mindre än fem koncentrationer används måste detta motiveras. Ämneskoncentrationer som är högre än LC50 under 96 timmar eller 100 mg/l, beroende på vilken som är lägre, behöver inte testas. Ämnen får inte testas ovanför sin löslighetsgräns i testvattnet.
Om en lösningsagent används för att underlätta beredningen av testlösningar (se avsnitt 1.6.6) får agentens slutkoncentration i testkärlet inte överskrida 0,1 ml/l och samma slutkoncentration bör användas i alla testkärl.
1.7.3 Kontrollsatser
En kontrollsats med spädvatten (med tillämpliga replikat) och, om det är relevant, en kontrollsats med lösningsagenten (med tillämpliga replikat) bör köras utöver testserierna.
1.7.4 Frekvensen för analytiska bestämningar och mätningar
Under testets gång bör testämnets koncentrationer bestämmas med regelbundna intervall.
Vid halvstatiskt test, där testämnets koncentration bör hållas inom ± 20 % av den nominella koncentrationen (dvs. inom området 80–120 %, se avsnitten 1.4 och 1.6.6), rekommenderas att de högsta och lägsta testkoncentrationerna åtminstone analyseras strax efter beredningen och strax före förnyandet vid minst tre tillfällen som är jämnt fördelade över testperioden (analyserna görs på ett prov av samma lösning – när den är nyberedd och när den förnyas).
Om det är förväntat att testämnets koncentration inte kommer att hållas inom ± 20 % av den nominella koncentrationen (på grundval av stabilitetsdata om ämnet) måste alla testkoncentrationer analyseras, när de är nyberedda och när de förnyas, men enligt samma schema (dvs. vid minst tre tillfällen som är jämt fördelade över hela testperioden). Bestämning av testämnets koncentration före förnyandet behöver bara göras på ett replikatkärl per testämneskoncentration. Intervallet mellan bestämningarna får inte vara längre än sju dagar. Det rekommenderas att resultaten grundar sig på de uppmätta koncentrationerna. Om det däremot finns bevis för att testämnets koncentration i lösningen på ett tillfredsställande sätt har hållits inom ± 20 % av den nominella eller uppmätta startkoncentrationen under hela testperioden, kan resultaten grunda sig på nominella eller uppmätta startvärden.
Vid genomflödestest kan man lämpligen använda samma provtagningsschema som för halvstatiskt test (även om mätningen av ”gammal” lösning då inte är tillämplig). Om testperioden sträcker sig över mer än sju dagar kan det vara tillrådligt att utöka antalet provtagningstillfällen under den första veckan (t.ex. tre mätningsomgångar) för att kontrollera att testkoncentrationerna hålls stabila.
Proverna kan behöva centrifugeras eller filtreras (t.ex. med porstorlek 0,45 μm). Eftersom det inte alltid är möjligt att separera den icke-biotillgängliga fraktionen av testämnet från den biotillgängliga fraktionen genom centrifugering eller filtrering bör emellertid dessa behandlingar undvikas.
Under testets förlopp mäts upplöst syrgas, pH och temperatur i alla testkärl. Totalhårdhet och salthalt (om relevant) mäts i kontrollsatserna och i ett kärl med den högsta koncentrationen. Upplöst syrgas och salthalt (om relevant) mäts minst tre gånger (vid testets början, mitt och slut). Vid halvstatiskt test rekommenderas att upplöst syrgas mäts oftare, helst före och efter varje vattenförnyelse eller minst en gång i veckan. Vid halvstatiskt test mäts pH i början och i slutet av varje vattenförnyelse och vid genomflödestest minst varje vecka. Hårdheten mäts en gång per test. Temperaturen bör mätas dagligen och helst övervakas kontinuerligt i minst ett testkärl.
1.7.5 Observationer
1.7.5.1 Stadiet för embryonal utveckling
Det embryonala stadium (dvs. gastrulastadium) som råder när exponeringen för testämnet inleds bör bekräftas så exakt som möjligt. Detta kan göras på ett representativt prov av ägg som har förvarats på lämpligt sätt. Även litteratur kan konsulteras i fråga om beskrivning och illustration av de embryonala stadierna (2), (5), (10) och (11).
1.7.5.2 Kläckning och överlevnad
Observationer av kläckning och överlevnad bör göras minst en gång per dag. Alla antal bör registreras. Det kan vara önskvärt med tätare observationer i början av testperioden (t.ex. med 30 minuters intervall under de tre första timmarna) eftersom överlevnadstiderna i vissa fall kan ha större relevans än blotta antalet dödsfall (t.ex. när det förekommer akuta toxiska verkningar). Döda embryon och yngel bör avlägsnas så snart de observeras, eftersom förruttnelse kan ske snabbt. Yttersta försiktighet bör iakttas när döda individer avlägsnas så att man inte slår till eller orsakar fysiska skador på närliggande ägg eller yngel eftersom de är extremt sköra och känsliga. Kriterierna för död varierar beroende på levnadsstadium:
|
— |
Ägg: särskilt i tidiga stadier en tydligt förlust av genomskinlighet och färgförändringar orsakade av koagulering eller utfällning av protein, vilket leder till ett vitt ogenomskinligt utseende. |
|
— |
Embryon: avsaknad av kroppsrörelse, avsaknad av hjärtslag eller ogenomskinlig missfärgning hos arter vars embryon normalt är genomskinliga. |
|
— |
Yngel: orörlighet, avsaknad av andningsrörelse, avsaknad av hjärtslag, vit ogenomskinlig färgning av centrala nervsystemet eller avsaknad av reaktion på mekanisk stimulering. |
1.7.5.3 Onormalt utseende
Gulesäckens absorptionsstadium och antalet yngel som har onormal kroppsform eller pigmentering bör registreras med intervall vars längd bestäms enligt testperiodens längd och karaktären hos de abnormaliteter det gäller. Man bör beakta att onormala embryon och yngel även förekommer av naturliga skäl, och kan för vissa arter uppgå till flera procent i kontrollgruppen (kontrollgrupperna). Onormala djur bör inte avlägsnas från testkärlet förrän de har dött.
1.7.5.4 Onormalt beteende
Abnormaliteter, t.ex. hyperventilation, okoordinerat simmande och atypisk passivitet bör registreras med intervall vars längd bestäms enligt testperiodens längd. Sådana verkningar kan vara svåra att kvantifiera men kan, när de observeras, vara till hjälp vid tolkningen av dödlighetsdata, dvs. ge upplysningar om ämnets toxiska verkningssätt.
1.7.5.5 Längd
Vid testets slut rekommenderas matning av individuella längder. Om det förekommer röta pä stjärtfenan eller frätning av fenorna bör standardlängder användas. 1 ett väl utfört test bör variationskoefficienten med avseende på längden hos replikaten i kontrollgrupperna i regel vara ≤ 20 %.
1.7.5.6 Vikt
Vid testets slut kan individuella vikter bestämmas. Härad är torrvikt (24 timmar vid 60 oC) att föredra framför våtvikt (avtorkning). I ett väl utfört test bör variationskoefficienten med avseende på vikt för replikaten i kontrollgrupperna i regel vara ≤ 20 %.
Från dessa observationer fås följande data, till alla delar eller delvis, för statistisk analys:
|
— |
Kumulativ mortalitet. |
|
— |
Antalet friska yngel vid försökets slut. |
|
— |
Tidpunkten för kläckningens början och slut (dvs. kläckning till 90 % i varje replikat). |
|
— |
Antalet yngel som kläcks per dag. |
|
— |
Längd (och vikt) för överlevande djur vid testets slut. |
|
— |
Antalet yngel som är missbildade eller har onormalt utseende. |
|
— |
Antalet yngel som beter sig onormalt. |
2. DATA OCH RAPPORTERING
2.1 BEHANDLING AV RESULTATEN
Det rekommenderas att en statistiker deltar i både utformning och analys av testet, eftersom metoden medger betydande variationer i fråga om testutformning, t.ex. antalet testkärl, antalet testkoncentrationer, ursprungligt antal befruktade ägg och vilka parametrar som mäts. Med hänvisning till de många alternativen för testutformning ges inga specifika anvisningar om statistiska procedurer.
Om man avser att uppskatta LOEC- eller NOEC-värden är det nödvändigt att analysera variationerna inom varje replikatgrupp med hjälp av variansanalys (ANOVA) eller kontingenstabellprocedurer. För multipla jämförelser mellan resultaten vid enskilda koncentrationer och resultaten för kontrollgrupperna kan Dunnetts metod rekommenderas (12), (13). Det finns också exempel på andra användbara metoder (14), (15). Storleken på den verkan som kan detekteras med hjälp av ANOVA eller andra procedurer (dvs. testets kapacitet) beräknas och rapporteras. Anmärkningsvis kan konstateras att analys med ANOVA inte lämpar sig för alla observationer som uppräknas i avsnitt 1.7.5.6. Kumulativ mortalitet och antalet friska yngel i slutet av testet kan t.ex. analyseras med probitmetoder.
Om man avser att uppskatta värden på LC eller ECx bör en eller flera kurvor, t.ex. logistisk kurva, anpassas till relevanta data med hjälp av en statistisk metod, t.ex. minsta kvadrat-metoden eller icke-linjär minsta kvadratmetod. Kurvan (kurvorna) parametriseras så att relevanta värden på LC eller ECx och motsvarande standardfel kan uppskattas direkt. Därigenom underlättas beräkningen av konfidensintervallet kring LC eller ECx betydligt. Tvåsidig konfidens på 95 % bör tillämpas, utom när det finns goda skäl för att föredra avvikande konfidensnivåer. Anpassningsproceduren bör helst ge tillgång till ett medel för bedömning av signifikansen eller bristen på signifikans. Grafiska metoder för kurvanpassning kan användas. Regressionsanalys är lämplig för alla observationer som uppräknas i avsnitt 1.7.5.6.
2.2 TOLKNING AV RESULTATEN
Resultaten bör tolkas med försiktighet när de uppmätta koncentrationerna av det toxiska ämnet i testlösningen ligger på nivåer nära analysmetodens detektionsgräns. Likaså bör man vara försiktig vid tolkning av resultaten när det galler koncentrationer ovanför ämnets vattenlöslighetsgräns.
2.3 TESTRAPPORT
Testrapporten ska innehålla nedan angivna uppgifter.
2.3.1 Testämne:
|
— |
Aggregationstillstånd och relevanta fysikalisk-kemiska egenskaper. |
|
— |
Kemiska identifieringsdata, inklusive renhet och i tillämpliga fall analysmetod för kvantificering av testämnet. |
2.3.2 Testdjur:
|
— |
Vetenskapligt namn, stam, antalet föräldrafiskar (dvs. hur många honor som behövdes för produktion av det antal ägg som användes i testet), ursprung, metoden för insamling av befruktade ägg och efterföljande behandling av äggen. |
2.3.3 Testförhållanden:
|
— |
Testförfarande som har använts (t.ex. halvstatiskt test eller genomflödestest, tiden mellan befruktningen och testets början, fisktäthet osv.). |
|
— |
Ljusperiod(er). |
|
— |
Testets utformning (t.ex. antalet testkärl och replikat, antalet embryon per replikat). |
|
— |
Metoden för beredning av stamlösningar och förnyelsefrekvensen (i förekommande fall bör även lösningsagenten och dess koncentration anges). |
|
— |
Nominella testkoncentrationer, uppmätta värden, deras medelvärden och standardavvikelser i testkärlen och den metod med vilken dessa erhölls. Om testämnet är vattenlösligt vid koncentrationer under de testade, måste man bevisa att mätningarna hänför sig till testämnets koncentrationer i lösningen. |
|
— |
Spädvattnets egenskaper: pH, hårdhet, koncentrationen upplöst syrgas, restklornivåer (om de har uppmätts), totalt organiskt kol, partikelinnehåll, testmediets salthalt (om den har uppmätts) och resultatet av alla övriga mätningar. |
|
— |
Vattenkvaliteten i testkärlen: pH, hårdhet, temperatur och halten upplöst syrgas. |
2.3.4 Resultat:
|
— |
Resultaten frän eventuella preliminära test gällande testämnets stabilitet. |
|
— |
Bevis för att kontrollgrupperna uppfyller normerna för total överlevnad för testdjur (tilläggen 2 och 3). |
|
— |
Data om mortalitet och överlevnad på embryo- och yngelstadiet och total mortalitet och överlevnad. |
|
— |
Antalet dagar fram till kläckning och antalet kläckta ägg. |
|
— |
Data om längd (och vikt). |
|
— |
Förekomst och beskrivning av morfologiska abnormaliteter (i förekommande fall). |
|
— |
Förekomst och beskrivning av verkningar pä beteendet (i förekommande fall). |
|
— |
Statistisk analys och behandling av data. |
|
— |
För test som analyseras med hjälp av ANOVA bör man rapportera den lägsta koncentration vid vilken verkningar observeras (LOEC) vid p = 0,05 och den koncentration vid vilken inga verkningar observeras (NOEC) för varje respons som har varit föremål för bedömning, tillsammans med en beskrivning av de statistiska procedurer som har använts och angivelse av hur stark verkan som kunde detekteras. |
|
— |
För test som har analyserats med hjälp av regressionstekniker bör man ange LC eller ECx, konfidensintervall och en kurva med den anpassade modell som har använts för beräkningen. |
|
— |
Motiveringar för varje avvikelse från denna testmetod. |
3. HÄNVISNINGAR
|
(1) |
Kristensen P. (1990), Evaluation of the Sensitivity of Short Term Fish Early Life Stage Tests in Relation to other EELS Test Methods. Final report to the Commission of the European Communities, 60 sidor, juni 1990. |
|
(2) |
ASTM (1988), Standard Guide for Conducting Early Life-Stage Toxicity Tests with Fishes. American Society for Testing and Materials. E 1241-88. 26 sidor. |
|
(3) |
Brauhn J. L. and Schoettger R. A. (1975), Acquisition and Culture of Research Fish: Rainbow trout, Fathead minnows, Channel Catfish and Bluegills. s. 54, Ecological Research Series, EPA-660/3-75-011, Duluth, Minnesota. |
|
(4) |
Brungs W. A. and Jones B. R. (1977), Temperature Criteria for Freshwater Fish: Protocol and Procedures, p. 128, Ecological Research Series EPA-600/3-77-061, Duluth, Minnesota. |
|
(5) |
Laale H. W. (1977), The Biology and Use of the Zebrafish (Brachydanio rerio) in Fisheries Research. A Literature Review. J. Biol. 10, s. 121–173. |
|
(6) |
Legault R. (1958), A Technique for Controlling the Time of Daily Spawning and Collecting Eggs of the Zebrafish, Brachydanio rerio (Hamilton-Buchanan) Copeia, 4, s. 328–330. |
|
(7) |
Dave G., Damgaard B., Grande M., Martelin J. E., Rosander B. and Viktor T. (1987), Ring Test of an Embryo-larval Toxicity Test with Zebrafish (Brachydanio rerio) Using Chromium and Zinc as Toxicants. Environmental Toxicology and Chemistry, 6, s. 61–71. |
|
(8) |
Birge J. W., Black J. A. and Westerman A. G. (1985), Short-term Fish and Amphibian Embryo-larval Tests for Determining the Effects of Toxicant Stress on Early Life Stages and Estimating Chronic Values for Single Compounds and Complex Effluents. Environmental Toxicology and Chemistry 4, s. 807–821. |
|
(9) |
Van Leeuwen C. J., Espeldoorn A. and Mol F. (1986), Aquatic Toxicological Aspects of Dithiocarbamates and Related Compounds. III. Embryolarval Studies with Rainbow Trout (Salmo gairdneri). J. Aquatic Toxicology, 9, s. 129–145. |
|
(10) |
Kirchen R. V. and W. R. West (1969), Teleostean Development. Carolina Tips 32(4): 1–4. Carolina Biological Supply Company. |
|
(11) |
Kirchen R. V. and W. R. West (1976), The Japanese Medaka. Its care and Development. Carolina Biological Supply Company, North Carolina, 36 sidor. |
|
(12) |
Dunnett C. W. (1955), A Multiple Comparisons Procedure for Comparing Several Treatments with Control. J. Amer. Statist. Assoc, 50, s. 1096–1121. |
|
(13) |
Dunnett C. W. (1964), New Tables for Multiple Comparisons with a Control. Biometrics, 20, s. 482–491. |
|
(14) |
Mc Clave J. T., Sullivan J. H. and Pearson J.G. (1980), Statistical Analysis of Fish Chronic Toxicity Test Data. Proceedings of 4th Aquatic Toxicology Symposium, ASTM, Philadelphia. |
|
(15) |
Van Leeuwen C. J., Adema D. M. M. and Hermes J. (1990), Quantitative Structure-Activity Relationships for Fish Early Life Stage Toxicity. Aquatic Toxicology, 16, s. 321–334. |
|
(16) |
Environment Canada. (1992), Toxicity Tests Using Early Life Stages of Salmonid Fish (Rainbow Trout, Coho Salmon or Atlantic Salmon). Biological Test Method Series. Report EPS I/RM/28, December 1992, 81 sidor. |
|
(17) |
Dave G. and Xiu R. (1991), Toxicity of Mercury, Nickel, Lead and Cobalt to Embryos and Larvae of Zebrafish. Brachydanio rerio. Arch. of Environmental Contamination and Toxicology, 21, s. 126–134. |
|
(18) |
Meyer A.. Bierman C. H. and Orti G. (1993), The phylogenetic position of the Zebrafish (Danio rerio), a model system in developmental biology – an invitation to the comperative methods. Proc. Royal Society of London, Series B, 252, s. 231–236. |
|
(19) |
Ghillebaert F., Chaillou C, Deschamps F. and Roubaud P. (1995), Toxic Effects, at Three pH Levels, of Two Reference Molecules on Common Carp Embryo. Ecotoxicology and Environmental Safety 32, s. 19–28. |
|
20. |
US EPA (1991), Guidelines for Culturing the Japanese Medaka, Oryzias latipes. EPA report EPA/600/3-91/064, Dec. 1991, EPA, Duluth. |
|
21. |
US EPA (1991), Guidelines for Conducting Early Life Stage Toxicity Tests with Japanese Medaka, (Oryzias latipes). EPA report EPA/600/3-91/063, Dec. 1991, EPA, Duluth. |
|
22. |
De Graeve G. M., Cooney J. D., McIntyre D. O., Poccocic T. L, Reichenbach N. G., Dean J. H. and Marcus M. D. (1991), Validity in the performance of the seven-day Fathead minnow (Pimephales promelas) larval survival and growth test: an intra- and interlaboratory study. Environ. Tox. Chem. 10, s. 1189–1203. |
|
23. |
Calow P. (1993), Handbook of Ecotoxicology, Blackwells, Oxford. Vol. 1, Chapter 10: Methods for spawning, culturing and conducting toxicity tests with Early Life stages of Estuarine and Marine fish. |
|
24. |
Balon E. K. (1985), Early life history of fishes: New developmental, ecological and evolutionary perspectives, Junk Publ., Dordrecht, 280 sidor. |
|
25. |
Blaxter J. H. S. (1988), Pattern and variety in development, in: W. S. Hoar and D. J. Randall eds., Fish Physiology, Vol. XIA, Academic press, s. 1–58. |
Tabell 1A
Fiskarter som rekommenderas för testning
|
Sötvatten |
|
Oncorhynchus mykis Regnbågslax (9), (16) |
|
Danio rerio Sebrafisk (7), (17), (18) |
|
Cyprinus caprio Karp (8), (19) |
|
Oryzias latipes Japanese ricefish/Medaka (20), (21) |
|
Pimephales promelas Fathead minnow (8), (22) |
Tabell 1B
Exempel på övriga väldokumenterade arter som också har använts
|
Sötvatten |
Saltvatten |
|
Carassius auratus Guldfisk (8) |
Menidia peninsulae Tidewater silverside (23), (24), (25) |
|
Lepomis macrochirus Solaborre (”Bluegill”) (8) |
Clupea harengus Sill (24), (25) |
|
Gadus rnorhua Torsk (24), (25) |
|
|
Cyprinodon variegatus Sheepshead minnow (23), (24), (25) |
Bilaga 1
ANVISNINGAR FÖR TOXICITETSTEST PÅ EMBRYON OCH SÄCKYNGEL AV SEBRAFISK (BRACHYDAMO RERIO)
INLEDNING
Sebrafisken härstammar från indiska Coromandel-kusten där den lever i snabbströmmande vatten. Den är en vanlig akvariefisk som tillhör karpfamiljen. Information om vård och odling finns i allmänt tillgängliga referensböcker om tropiska fiskar. En översikt över sebrafiskens biologi och användning för fiskforskning har sammanställts av Laale (1).
Fisken blir sällan längre än 45 mm. Den har en cylinderformad kropp med 7–9 mörkblå silverskiftande strimmor. Strimmorna löper in i stjärt- och analfenorna. Ryggen är olivgrön. Hanarna är slankare än honorna. Honorna är mer silverskiftande och har en svällande buk, särskilt före romläggningen.
Vuxna exemplar klarar av stora variationer i temperatur, pH och vattenhårdhet. För att få friska exemplar som producerar ägg av god kvalitet bör man dock se till att betingelserna är optimala.
Vid romläggningen följer hanen efter honan och trycker sig mot henne, och äggen befruktas efterhand som de kommer ut. Äggen, som är genomskinliga och icke-vidhäftande, faller till botten där de i vissa fall kan bli uppätna av föräldrarna. Romläggningen påverkas av ljus. Om morgonljuset är tillräckligt lägger fisken i regel sin rom under de första timmarna efter soluppgången.
En hona kan producera satser om hundratals ägg med en veckas intervall.
BETINGELSER FÖR FÖRÄLDRAGENERATIONEN, REPRODUKTION OCH TIDIGA LEVNADSSTADIER
Ett antal friska exemplar väljs ut och hålls i lämpligt vatten (se t.ex. tillägg 4) under minst två veckor före planerad romläggning. Fiskgruppen bör ha producerat minst en sats avkomma innan den producerar de ägg som används för testet. Fisktätheten under denna period får inte överskrida 1 gram fisk per liter. Om vattnet byts regelbundet eller om det finns ett reningssystem kan högre täthet tillåtas. Temperaturen i förvaringskärlen bör hållas på 25 ± 2 oC. Fisken bör få varierande foder, såväl vanligt torrfoder som levande foder, t.ex. nyckläckta artemia, mygglarver, daphnia och vita maskar (enkyträer).
Nedan beskrivs två förfaranden som i praktiken har gett tillräcklig mängd friska och befruktade ägg för testningsändamål:
|
i) |
8 hanar och 16 honor placeras i en behållare som innehåller 50 liter spädvatten. Behållaren bör vara avskärmad från direkt ljus och lämnas så ostörd som möjligt i minst 48 timmar. På eftermiddagen till den dag som föregår testets böljan placeras en romläggningsbricka på akvariets botten. Romläggningsbrickan består av en ram (plexiglas eller annat lämpligt material) som är 5–7 cm hög med ett grovt nät (2–5 mm) fäst upptill och ett fint nät (10–30 μm) nedtill. Ett antal ”romläggningsträd” av otvinnat nylonrep fästes vid det grova nätet. Efter att fiskarna har hållits i mörker i 12 timmar utsätts de för ett svagt ljus som stimulerar till romläggning. Två till fyra timmar efter romläggningen tas romläggningsbrickan bort och äggen samlas in. Romläggningsbrickan hindrar fiskarna från att äta upp äggen och underlättar samtidigt insamling av äggen. Fiskgruppen bör ha lagt rom minst en gång innan den lägger den rom som används för testet. |
|
ii) |
5–10 hanar och honor förvaras separat i minst två veckor före den planerade romläggningen. Efter 5–10 dagar har honorna uppsvälld buk och deras genitalpapiller är synliga. Hanfiskarna har inga papiller. Romläggningen utförs i romläggningsbehållare med natförsedd botten (se ovan). Behållaren fylls med spädvatten så att vattendjupet ovanför nätet är 5–10 cm. En hona och två hanar placeras i behållaren två dagar före den planerade romläggningen. Vattentemperaturen ökas stegvis till ett värde som är en grad högre än acklimatiseringstemperaturen. Ljuset släcks och behållarna lämnas så ostörda som möjligt. På morgonen tänds ett svagt ljus för att stimulera romläggningen. Efter 2–4 timmar tas fiskarna bort och äggen samlas in. Om det behövs större äggmängder än en hona kan producera kan man använda tillräckligt antal parallella romläggningsbehållare. De enskilda honornas reproduktionskapacitet registreras före testet (satsens storlek och kvalitet) för att ge möjlighet att välja ut honorna med de bästa reproduktionsegenskaperna för romläggningen. |
Äggen överförs till testkärlen med hjälp av glasrör (innerdiameter minst 4 mm) som är försedda med en mjuk sugboll. Vid överföring av äggen bör den medföljande vattenmängden vara så liten som möjligt. Äggen är tyngre än vatten och sjunker ut ur röret. Det är viktigt att se till att ägg (och yngel) inte kommer i kontakt med luft. Ett eller flera prover av satsen eller satserna undersöks i mikroskop för att kontrollera att det inte förekommer irregulariteter i de första utvecklingsstadierna. Det är inte tillåtet att desinfektera äggen.
Mortaliteten för äggen är högst under de första 24 timmarna efter befruktningen. Under denna period är det vanligt med en mortalitet på 5–40 %. Äggen kan degenerera till följd av misslyckad befruktning eller utvecklingsstörningar. Äggens kvalitet verkar bero på fiskhonan, eftersom vissa honor genomgående producerar ägg av god kvalitet medan andra aldrig gör det. Även utvecklingshastigheten och kläckningshastigheten varierar mellan olika satser. Framgångsrikt befruktade ägg och säckyngel överlever till stor del, i regel över 90 %. Vid 25 oC kläcks äggen 3–5 dagar efter befruktningen och gulesäcken har absorberats cirka 13 dagar efter befruktningen.
Den embryonala utvecklingen har definierats väl av Hisaoka och Battle (2). Äggen och nykläckta yngel är genomskinliga och därför kan man följa med fiskens utveckling och observera eventuella missbildningar. Cirka fyra timmar efter romläggningen kan man se skillnaden mellan obefruktade ägg och befruktade ägg (3). För denna undersökning placeras ägg och yngel i små testkärl och granskas under mikroskop.
De testbetingelser som bör användas för de tidiga levnadsstaderna uppräknas i tillägg 2. Optimala värden för pH och spädvattnets hårdhet är 7.8 respektive 250 mg CaCO3/l.
BERÄKNINGAR OCH STATISTIK
En tvåstegsmetod rekommenderas. Först görs en statistisk analys av data gällande mortalitet, onormal utveckling och kläckningstid. Därefter görs en statistisk utvärdering av kroppslängden hos de exemplar som behandlats i koncentrationer vid vilka inga skadliga verkningar på någon av de nämnda parametrarna har detekterats. Denna metod rekommenderas eftersom ett toxiskt ämne selektivt kan döda mindre fisk, förlänga kläckningstiden och framkalla grova missbildningar, vilket leder till missvisande längdmätningsresultat. Dessutom mäts ungefär samma antal fiskar per behandling, vilket säkerställer de statistiska resultatens giltighet.
BESTÄMNING AV LC50 OCH EC50
Procentandelen överlevande ägg och yngel beräknas och korrigeras för mortaliteten i kontrollgrupperna, enligt Abbotts formel (4):

där:
|
P |
= |
korrigerad procentandel överlevande, |
|
P' |
= |
procentandel överlevande som observerats i testkoncentrationen, |
|
C |
= |
procentandelen överlevande i kontrollgruppen. |
I mån av möjlighet bestäms LC50 vid testets slut med en lämplig metod.
Anvisningar för hur man kan få in morfologiska abnormaliteter i EC50-statistiken har publicerats av Stephan (5).
UPPSKATTNING AV LOEC OCH NOEC
Ett av syftena med testet på ägg och säckyngel är att jämföra koncentrationer över noll med kontrollgruppens koncentrationer, dvs. att bestämma värdet på LOEC. Därför bör förfaranden med multipeljämförelse användas (6), (7), (8), (9). (10).
HÄNVISNINGAR
|
1. |
Laale FE W. (1977), The Biology and Use of the Zebrafish (Brachydanio rerio) in Fisheries Research. A Literature Review. J. Fish Biol. 10, s. 121–173. |
|
2. |
Hisaoka K. K. and Battle H. I. (1958), The Normal Development Stages of the Zebrafish Brachydanio rerio (Hamilton-Buchanan) J. Morph., 102, 311 sidor. |
|
3. |
Nagel R. (1986), Untersuchungen zur Elproduktion beim Zebrabärbling (Brachydanio rerio Hamilton-Buchanan). Journal of Applied Ichthyology, 2, s. 173–181. |
|
4. |
Finney D. J. (1971), Probit Analysis, 3rd cd., Cambridge University Press, Great Britain, s. 1–333. |
|
5. |
Stephan C. E. (1982), Increasing the Usefulness of Acute Toxicity Tests. Aquatic Toxicology and Hazard Assessment: Fifth Conference, ASTM STP 766, J. G. Pearson, R. B, Foster and W. E. Bishop, Eds., American Society for Testing and Materials, s. 69–81. |
|
6. |
Dunnett C. W. (1955), A Multiple Comparisons Procedure for Comparing Several Treatments with a Control.). Amer. Statist. Assoc, 50, s. 1096–1121. |
|
7. |
Dunnett C. W. (1964), New Tables for Multiple Comparisons with a Control. Biometrics, 20, s. 482–491. |
|
8. |
Williams D. A. (1971), A Test for Differences Between Treatment Means when Several Dose Levels are Compared with a Zero Dose Control. Biometrics, 27, s. 103–117. |
|
9. |
Williams D. A. (1972), The Comparison of Several Dose Levels with a Zero Dose Control. Biometrics 28, s. 519–531. |
|
10. |
Sokal R. R. and Rohlf F. J. (1981), Biometry, the Principles and Practice of Statistics in Biological Research, W. H. Freeman and Co., San Francisco. |
Bilaga 2
TESTBETINGELSER, TESTPERIOD OCH ÖVERLEVNADSKRITERIER FÖR REKOMMENDERADE ARTER
|
Arter |
Temperatur ( oC) |
Salthalt (0/00) |
Ljusperiod (timmar) |
Stadierna s längd (dagar) |
Typisk testperiod |
Överlevnad i kontrollgrupper, (minimum %) |
||
|
Embryo |
Säckyngel |
Kläcknings-framgång |
Efter kläckning |
|||||
|
SÖTVATTEN |
||||||||
|
Brachydanio rerio Sebrafisk |
25 ± 1 |
— |
12–16 |
3–5 |
8–10 |
Så snart som möjligt efter befruktning (tidigt gastrula-stadium) till fem dagar efter kläckning (8–10 dagar) |
80 |
90 |
|
Oncorhvnchus mykiss Regnbågslax |
10 ± 1 (22) 12 ± 1 (23) |
— |
0 (24) |
30–35 |
25–30 |
Så snart som möjligt efter befruktning (tidigt gastrula-stadium) till 20 dagar efter kläckning (50–55 dagar) |
66 |
70 |
|
Cyprinus carpio Karp |
21–25 |
— |
12–16 |
5 |
> 4 |
Så snart som möjligt efter befruktning (tidigt gastrula-stadium) till 4 dagar efter kläckning (8–9 dagar) |
80 |
75 |
|
Oryzias latipes Japanese ricefish/Medaka |
24 ± 1 (22) 23 ± 1 (23) |
— |
12–16 |
8–11 |
4–8 |
Så snart som möjligt efter befruktning (tidigt gastrula-stadium) till 5 dagar efter kläckning (13–16 dagar) |
80 |
80 |
|
Pimephales promelas Fathead minnow |
25 ± 2 |
— |
16 |
4–5 |
5 |
Så snart som möjligt efter befruktning (tidigt gastrula-stadium) till 4 dagar efter kläckning (8–9 dagar) |
60 |
70 |
Bilaga 3
Testsbetingelser, testperiod och överlevnadskriterier för andra väldokumenterade arter
|
Arter |
Temperatur ( oC) |
Salthalt (0/00) |
Ljusperiod (timmar) |
Stadiernas längd (dagar) |
Typisk period för test på embryo och säckyngel |
Överlevnad i kontrollgrupper (minimum %) |
||
|
|
|
|
|
Embryo |
Test på säckyngel |
Kläcknings-framgång |
Efter kläckning |
|
|
SÖTVATTEN |
||||||||
|
Carassius auratus Guldfisk |
24 ± 1 |
— |
— |
3–4 |
> 4 |
Så snart som möjligt efter befruktning (tidigt gastrula-stadium) till 4 dagar efter kläckning (7 dagar) |
— |
80 |
|
Leopomis macrochirus Blugill sunfish |
21 ± 1 |
— |
16 |
3 |
> 4 |
Så snart som möjligt efter befruktning (tidigt gastrula-stadium) till 4 dagar efter kläckning (7 dagar) |
— |
75 |
|
SALTVATTEN |
||||||||
|
Menidia peninsulae Tidewater silverside |
22–25 |
15–22 |
12 |
1,5 |
10 |
Så snart som möjligt efter befruktning (tidigt gastrula-stadium) till 5 dagar efter kläckning (6–7 dagar) |
80 |
60 |
|
Clupea harengus Sill |
10 ± 1 |
8–15 |
12 |
20–25 |
3–5 |
Så snart som möjligt efter befruktning (tidigt gastrula-stadium) till 3 dagar efter kläckning (23–27 dagar) |
60 |
80 |
|
Gadus morhua Torsk |
5 ± 1 |
5–30 |
12 |
14–16 |
3–5 |
Så snart som möjligt efter befruktning (tidigt gastrula-stadium) till 3 dagar efter kläckning (18 dagar) |
60 |
80 |
|
Cyprinodon variegatus Sheepshead minnow |
25 ± 1 |
15–30 |
12 |
— |
— |
Så snart som möjligt efter befruktning (tidigt gastrula-staditim) till 4 eller 7 dagar efter kläckning (28 dagar) |
> 75 |
80 |
BILAGA 4
NÅGRA KEMISKA EGENSKAPER FÖR GODTAGBART SPÄDVATTEN
|
Ämne |
Koncentrationer |
|
Partikelinnehåll |
< 20 mg/l |
|
Totalt organiskt kol |
< 2 mg/l |
|
Ojoniserad ammoniak |
< 1 μg/l |
|
Restklor |
< 10 μg/l |
|
Total mängd bekämpningsmedel typ organiska fosforföreningar |
< 50 ng/l |
|
Total mängd bekämpningsmedel typ organiska klorföreningar samt poly-klorerade bifenyler |
< 50 ng/l |
|
Total mängd organiskt bundet klor |
< 25 ng/l |
C.16 HONUNGSBIN – TEST AVSEENDE AKUT TOXICITET (ORALT)
1. METOD
Denna testmetod för akut toxicitet motsvarar OECD TG 213 (1998).
1.1 INLEDNING
Detta test är en laboratoriemetod for bedömning av växtskyddsprodukters och andra kemikaliers akuta toxicitet (oralt) på vuxna arbetsbin.
Vid bedömning och utvärdering av olika ämnens toxiska egenskaper kan det vara nödvändigt att bestämma den akuta toxiciteten (oralt) på honungsbin, t.ex. när det är sannolikt att bin kommer att exponeras för en viss kemikalie. Testet för akut toxicitet (oralt) används för att bestämma bekämpningsmedels och kemikaliers toxicitet för bin. Resultatet av detta test används för att avgöra om det behövs ytterligare utvärderingar. Metoden kan särskilt användas i stegvisa program för bedömning av de risker som bekämpningsmedel kan medföra för bin. Dessa program bygger på en sekventiell arbetsgång från laboratorietoxicitetstest till semifält- och fältförsök (1). Bekämpningsmedel kan testas som verksamma ämnen eller som formulerade produkter.
En giftekvivalent bör användas för att fastställa binas känslighet och testförfarandets noggrannhet.
1.2 DEFINITIONER
Akut toxicitet (oralt): de skadliga verkningar som uppträder inom en period pä högst 96 timmar efter oral tillförsel av en engångsdos av testämnet.
Dos: den mängd testämne som tillförs. Dosen uttrycks som vikten (μg) testämne per försöksdjur (μg/bi). Den faktiska dosen per enskilt bi kan inte beräknas eftersom bina matas kollektivt, men det går att uppskatta en genomsnittsdos (total konsumtion av testämnet dividerat med antalet testbin i en bur).
LD50 (median letal dose) oralt: en statistiskt fastställd engångsdos av ett ämne som kan förväntas leda till döden för 50 % av de djur som fått dosen på oral väg. LD50-värdet uttrycks som μg testämne per bi. När det gäller bekämpningsmedel kan testämnet vara ett verksamt ämne eller en formulerad produkt som innehåller ett eller flera verksamma ämnen.
Mortalitet: ett djur registreras som dött när det är helt orörligt.
1.3 PRINCIP FÖR TESTMETODEN
Vuxna arbetsbin honungsbi (Apis mellifera) exponeras för en serie doser av testämnet som dispergerats i sackaroslösning. Därefter utfodras bina med samma foder, men utan tillsats av testämne. Mortaliteten registreras dagligen under minst 48 timmar och jämförs med kontrollgruppens värden. Om mortaliteten ökar under perioden 24–48 timmar efter tillförseln, medan kontrollgruppens mortalitet hålls på en godtagbar nivå, dvs. < 10 %, bör testet förlängas till högst 96 timmar. Resultaten analyseras för beräkning av LD50 vid 24 och 48 timmar och, om testet har förlängts, även vid 72 och 96 timmar.
1.4 TESTETS GILTIGHET
För att testet ska vara giltigt måste följande villkor uppfyllas:
|
— |
Genomsnittsmortaliteten för det totala antalet kontrolldjur får inte överskrida 10 % vid testets slut. |
|
— |
De värden som erhålls for LD50 för giftekvivalenterna ska ligga inom det specificerade området. |
1.5 BESKRIVNING AV TESTMETODEN
1.5.1 Insamling av bin
För testet används unga vuxna arbetsbin av samma ras, ålder, näringstillstånd osv. Bina tas från adekvat utfodrade, friska, så långt som möjligt sjukdomsfria samhällen med renrasiga drottningar med känd historia och fysiologisk status. Bina bör samlas in på testdagens morgon eller kvällen före testet och hållas i testförhållandena fram till följande dag. Bin som insamlats från yngelfria ramar rekommenderas. Insamling tidigt på våren eller sent på hösten bör undvikas eftersom bina då har genomgått fysiologiska förändringar. Om ett test måste genomföras tidigt pä våren eller sent på hösten kan bina kläckas i en inkubator och födas upp med ”bibröd” (pollen som insamlats frän vaxkakan) och sackaroslösning under en vecka. Bin som har behandlats med kemiska ämnen såsom antibiotika, varroabekämpningsmedel osv. får inte användas för toxicitetstest förrän det har gått fyra veckor från att den sista behandlingen avslutades.
1.5.2 Förvarings- och utfodringsförhållanden
Burarna bör vara enkla att rengöra och välventilerade. De kan vara tillverkade i t.ex. rostfritt stål, nät eller plast. Även träburar för engångsbruk kan användas. Grupper om tio bin per bur rekommenderas. Burarnas storlek bör anpassas efter antalet bin och vara tillräckligt rymliga.
Bina hålls i mörker i ett testrum med en temperatur på 25 ± 2 oC. Den relativa fuktigheten, i regel kring 50–70 %, registreras under hela testförloppet. Hanteringen, såsom behandling och observationer, kan däremot genomföras i (dags)ljus. Som foder ges sackaroslösning i vatten med en slutkoncentration på 500 g/l (50 % vikt per volym). Efter tillförsel av testdoserna ges bina fri tillgång till foder. Utfodringssystemet bör vara utformat på sådant sätt att foderkonsumtionen per bur kan registreras (se avsnitt 1.6.3.1). För utfodringen kan ett cirka 50 mm långt och 10 mm brett glasrör användas, vars öppna ända smalnar av till en diameter på cirka 2 mm.
1.5.3 Förberedelse av bina
De insamlade bina fördelas slumpvis i testburarna som placeras ut slumpvis i testrummet.
Bina kan hållas utan foder i upp till 2 timmar före testets början. Det rekommenderas att bina inte ges foder före behandlingen för att alla bin ska ha jämförbart tarminnehåll vid testets början. Döende bin avlägsnas och ersätts med friska bin före testets början.
1.5.4 Förberedelse av doserna
Om testämnet är vattenlösligt kan det dispergeras direkt i en 50 % sackaroslösning. För tekniska produkter och ämnen med låg vattenlöslighet kan ett organiskt lösningsmedel, emulgeringsämnen eller dispergeringsämnen med låg toxicitet användas som vehikel (t.ex. aceton, dimetylformamid eller dimetylsulfoxid). Vehikelkoncentrationen beror på testämnets löslighet och bör vara densamma för alla testkoncentrationer. I regel är en vehikelkoncentration på 1 % lämplig och denna koncentration bör inte överskridas.
Det behövs också lämpliga kontrollösningar, dvs. om lösningsmedel eller dispergeringsmedel används för att lösa upp testämnet måste två separata kontrollgrupper användas: en som får vattenlösning och en som får sackaroslösning med lösningsmedlet eller vehikeln i samma koncentration som används för doseringslösningarna.
1.6 FÖRFARANDE
1.6.1 Test- och kontrollgrupper
Antalet doser och replikat som testas bör väljas så att de uppfyller de statistiska kraven för bestämning av LD50 med 95 % konfidensintervall. I regel används fem doser i en geometrisk serie med en faktor på högst 2,2. Doserna bör täcka området för LD50. Utspädningsfaktorn och antalet doskoncentrationer fastställs med beaktande av toxicitetskurvans lutning (dos i förhållande till mortalitet) och den statistiska metod som används för analys av resultaten. Ett preliminärt test kan användas som stöd för valet av lämpliga doskoncentrationer.
Varje testkoncentration bör ges till minst tre likadana testgrupper med tio bin per grupp. Utöver själva testserierna bör minst tre kontrollgrupper med tio bin per grupp också behandlas. Kontrollgrupper bör också användas för eventuella lösningsmedel eller vehiklar som används (se avsnitt 1.5.4).
1.6.2 Giftekvivalent
Testserien bör även omfatta en giftekvivalent. Minst tre doser bör väljas på sådant sätt att de täcker det förväntade LD50-värdet. Minst tre likadana burar med tio bin per bur bör användas för varje testdos. Som giftekvivalent rekommenderas dimetoat, vars LD50-värde (oralt, 24 timmar) enligt rapporterade uppgifter ligger inom området 0,10–0,35 μg verksamt ämne per bi (2). Även andra giftekvivalenter kan godtas förutsatt att man har tillgäng till tillräckliga data om förväntad dosrespons (t.ex. paration).
1.6.3 Exponering
1.6.3.1 Tillförsel av doser
Varje testgrupp med bin ges 100–200 μl 50 % sackaroslösning i vatten som innehåller den valda koncentrationen av testämnet. För ämnen som har låg löslighet, låg toxicitet eller låg koncentration i formuleringen krävs en större volym, eftersom större proportioner måste användas i sackaroslösningen. Mängden behandlat foder som konsumeras per grupp följs upp. När utfodringsbehållaren är tom (i regel inom 3–4 timmar) tas den ut ur buren och ersätts med en behållare som innehåller ren sackaroslösning. Bina får därefter fri tillgång till sackaroslösning. För vissa ämnen kan högre koncentrationer i testdosen leda till att bina avvisar den och endast konsumerar lite eller inget alls. Efter en period på högst 6 timmar bör okonsumerat behandlat foder ersättas med ren sackaroslösning. Mängden konsumerat behandlat foder uppskattas (t.ex. genom mätning av återstodens volym eller vikt).
1.6.3.2 Testperiodens längd
Testet fortsätts i regel i 48 timmar efter det att testlösningen har ersatts med ren sackaroslösning. Om mortaliteten fortsätter att stiga med mer än 10 % efter de första 24 timmarna bör testperioden förlängas (till högst 96 timmar), förutsatt att kontrollgruppens mortalitet inte överskrider 10 %.
1.6.4 Observationer
Mortaliteten registreras när det har gått 4 timmar från testets början och därefter vid 24 timmar och 48 timmar efter tillförsel av dosen. Om det behövs en förlängd testperiod görs efterföljande bedömningar med 24 timmars intervall (upp till högst 96 timmar), förutsatt att kontrollgruppens mortalitet inte överskrider 10 %.
En uppskattning görs av mängden behandlat foder som konsumeras per grupp. Genom att jämföra hur stor mängd behandlat och obehandlat foder som har konsumerats inom de angivna 6 timmarna kan man få upplysningar om i vilken grad bina accepterar det behandlade fodret.
Alla avvikelser i beteende som observeras under testperioden bör registreras.
1.6.5 Gränstest (Limit-test)
1 vissa fall (t.ex. om ett testämne förväntas ha låg toxicitet) kan gränstest utföras med 100 μg verksamt ämne per bi, i syfte att demonstrera att LD50 är högre än detta värde. Förfarandet bör vara detsamma, dvs. tre likadana testgrupper per testdos, relevanta kontrollgrupper, uppskattning av mängden konsumerat behandlat foder och användning av giftekvivalent. Om det förekommer mortalitet bör ett fullständigt test genomföras. Alla eventuella observerade subletala verkningar bör registreras (se avsnitt 1.6.4).
2. DATA OCH RAPPORTERING
2.1 DATA
Alla uppgifter bör sammanställas i en tabell. Av tabellen ska framgå, för varje testgrupp samt för varje kontrollgrupp och varje giftekvivalentgrupp, antalet bin som använts, mortaliteten vid varje observationstidpunkt och antalet bin med ett beteende som tyder på skadliga verkningar. Mortalitetsdata bör analyseras med lämpliga statistiska metoder (t.ex. probit-analys, rörligt medelvärde eller binominal sannolikhet} (3), (4). Dosresponskurvor uppritas för varje rekommenderad observationstidpunkt. Kurvornas lutning och median letal dos (LD50) beräknas med 95 % konfidensintervall. Korrigeringar för kontrollgruppens mortalitet kan göras med hjälp av Abbott-korrigering (4), (5). Om det behandlade fodret inte har konsumerats helt bör man göra en uppskattning av hur stor dos av testämnet som har konsumerats per grupp. LD50-värdet uttrycks som μg testämne per bi.
2.2 TESTRAPPORT
Testrapporten ska innehålla nedan angivna uppgifter.
2.2.1 Testämne
|
— |
Aggregationstillstånd och relevanta fysikalisk-kemiska egenskaper (t.ex. stabilitet i vatten, ångtryck). |
|
— |
Kemiska identifieringsdata, inklusive strukturformel, renhet (för bekämpningsmedel t.ex. identiteten och halten av verksamt ämne eller verksamma ämnen). |
2.2.2 Försöksdjur
|
— |
Vetenskapligt namn. ras, ungefärlig ålder (i veckor), insamlingsmetod, insamlingsdatum. |
|
— |
Information om de samhällen som används för insamling av testbin, inklusive hälsa, eventuella vuxensjukdomar, eventuella förbehandlingar osv. |
2.2.3 Testförhållanden
|
— |
Temperatur och relativ fuktighet i testrummet. |
|
— |
Förvaringsförhållanden, inklusive burarnas typ, storlek och material. |
|
— |
Metoder för beredning av stam- och testlösningar (i förekommande fall anges även lösningsmedlet och dess koncentrationer). |
|
— |
Testets utformning, t.ex. antal testkoncentrationer och deras värden, antalet kontrollgrupper, samt för varje testkoncentration och kontrollgrupp även antalet replikatburar och antalet bin per bur. |
|
— |
Testdatum. |
2.2.4 Resultat
|
— |
Resultaten från preliminära test (i förekommande fall). |
|
— |
Rådata: mortaliteten för varje testad dos vid varje observationstidpunkt. |
|
— |
Dos-responskurvorna vid testets slut. |
|
— |
LD50-värden med 95 % konfidensintervall, vid varje rekommenderad observationstidpunkt, för testämnet och för giftekvivalenten. |
|
— |
Statistiska metoder som har använts för att bestämma LD50. |
|
— |
Mortaliteten i kontrollgrupperna. |
|
— |
Övriga biologiska verkningar som har observerats eller uppmätts, t.ex. om bina har betett sig onormalt (inklusive om de avvisat testdosen), konsumtionen av foder i behandlade och obehandlade grupper. |
|
— |
Varje avvikelse från de här beskrivna testförfarandena samt all annan relevant information. |
3. HÄNVISNINGAR
|
1. |
EPPO/Council of Europe (1993), Decision-Making Scheme for the Environmental Risk Assessment of Plant Protection Products – Honeybees. EPPO Bulletin, Vol. 23, N.l. s. 151–165, March 1993. |
|
2. |
Gough, H. j., McIndoe, EX., Lewis, G.B. (1994), The use of dimethoate as a reference compound in laboratory acute toxicity tests on honeybees (Apis mellifera L.) 1981–1992, Journal of Apicultural Research, 22, s. 119–125. |
|
3. |
Litchfield, J.T. and Wilcoxon, F. (1949), A simplified method of evaluating dose-effect experiments. Jour. Pharmacol. and Exper. Ther., 96, s. 99–113. |
|
4. |
Finney, D. J. (1971), Probit Analysis. 3rd ed., Cambridge, London and New York. |
|
5. |
Abbott, W. S. (1925), A method for computing the effectiveness of an insecticide, Jour. Econ. Entomol, 18, s. 265–267. |
C.17 HONUNGSBIN – TEST AVSEENDE AKUT TOXICITET (KONTAKT)
1. METOD
Denna testmetod för akut toxicitet motsvarar OECD TG 214 (1998).
1.1 INLEDNING
Detta test är en laboratoriemetod för bedömning av växtskyddsprodukters och andra kemikaliers akuta toxicitet (kontakt) på vuxna arbetsbin.
Vid bedömning och utvärdering av de toxiska egenskaperna hos ämnen kan det vara nödvändigt att bestämma den akuta toxiciteten (kontakt) på honungsbin, t.ex. när det är sannolikt att bin kommer att exponeras för en viss kemikalie. Testet för akut toxicitet (kontakt) används för att bestämma bekämpningsmedels och kemikaliers toxicitet för bin. Resultatet av detta test används för att avgöra om det behövs ytterligare utvärderingar. Metoden kan särskilt användas i stegvisa program för bedömning av de risker som bekämpningsmedel kan medföra för bin. Dessa program bygger på en sekventiell arbetsgång från laboratorietoxicitetstest till semifält-och fältförsök (1). Bekämpningsmedel kan testas som verksamma ämnen eller som formulerade produkter.
En giftekvivalent bör användas för att fastställa binas känslighet och testförfarandets noggrannhet.
1.2 DEFINITIONER
Akut toxicitet (kontakt): de skadliga verkningar som uppträder inom en period på högst 96 timmar efter lokal applicering av en engångsdos av testämnet.
Dos: den mängd testämne som appliceras. Dosen uttrycks som vikten (μg) testämne per försöksdjur (μg/bi).
LD50 (median letal dose) kontakt: en statistiskt fastställd engångsdos av ett ämne som kan förväntas leda till döden för 50 % av de djur på vilka dosen har applicerats. LD50-värdet uttrycks som μg testämne per bi. När det gäller bekämpningsmedel kan testämnet vara ett verksamt ämne eller en formulerad produkt som innehåller ett eller flera verksamma ämnen.
Mortalitet: ett djur registreras som dött när der är helt orörligt.
1.3 PRINCIP FÖR TESTMETODEN
Vuxna arbetsbin (honungsbin – Apis mellifera) exponeras för en serie doser av testämnet upplöst i lämplig vehikel. Exponeringen sker genom direkt applicering på bröstkorgen (droppar). Testperiodens längd är 48 timmar. Om mortaliteten ökar under perioden 24–48 timmar efter tillförseln, medan kontrollgruppens mortalitet hålls på en godtagbar nivå, dvs. < 10 %, bör testet förlängas till högst 96 timmar. Mortaliteten registreras dagligen och jämförs med kontrollgruppens värden. Resultaten analyseras för beräkning av LD50 vid 24 och 48 timmar, och om testet förlängs, vid 72 och 96 timmar.
1.4 TESTETS GILTIGHET
För att testet ska vara giltigt måste följande villkor uppfyllas:
|
— |
Genomsnittsmortaliteten för det totala antalet kontrolldjur får inte överskrida 10 % vid testets slut. |
|
— |
De värden som erhålls för LD50 för giftekvivalenten ska ligga inom det specificerade området. |
1.5 BESKRIVNING AV TESTMETODEN
1.5.1 Insamling av bin
För testet används unga vuxna arbetsbin med samma ålder, näringstillstånd, ras osv. Bina tas från adekvat utfodrade, friska, så långt som möjligt sjukdomsfria samhällen med renrasiga drottningar med känd historia och fysiologisk status. Bina bor samlas in pä testdagens morgon eller kvällen före testet och hällas i testförhållandena fram till följande dag. Bin som insamlats från yngelfria ramar rekommenderas. Insamling tidigt på våren eller sent på hösten bör undvikas eftersom bina då har genomgått fysiologiska förändringar. Om test måste genomföras tidigt på våren eller sent på hösten kan bina kläckas i en inkubator och födas upp med ”bibröd” (pollen som insamlats från vaxkakan) och sackaroslösning under en vecka. Bin som har behandlats med kemiska ämnen såsom antibiotika, varroabekämpningsmedel osv. får inte användas för toxicitetstest förrän det har gått fyra veckor från det att den sista behandlingen avslutades.
1.5.2 Förvarings- och utfodringsförhållanden
Burarna bör vara enkla att rengöra och välventilerade. Burarna kan vara tillverkade av t.ex. rostfritt stål, nät eller plast. Även träburar för engångsbruk kan användas. Burarnas storlek bör anpassas efter antalet bin och vara tillräckligt rymliga. Grupper om tio bin per bur rekommenderas.
Bina hålls i mörker i ett testrum med en temperatur på 25 ± 2 oC. Den relativa fuktigheten, i regel kring 50–70 %, bör registreras under hela testförloppet. Hanteringen, såsom behandling och observationer, kan däremot genomföras i (dags)ljus. Sackaroslösning i vatten med en slutkoncentration på 500 g/l (50 % vikt per volym) används som foder och tillhandahålls fritt för bina med hjälp av en utfodringsanordning. För utfodringen kan ett cirka 50 mm långt och 10 mm brett glasrör användas, vars öppna ända smalnar av till en diameter på cirka 2 mm.
1.5.3 Förberedelse av bina
De insamlade bina kan bedövas med koldioxid eller kväve före appliceringen av testämnet. Mängden bedövningsmedel och exponeringstiden för medlet bör minimeras. Döende bin avlägsnas och ersätts med friska bin före testets början.
1.5.4 Förberedelse av doserna
Testämnet appliceras upplöst i en vehikel, t.ex. ett organiskt lösningsmedel eller vattenlösning med ett vätmedel. Som organiskt lösningsmedel rekommenderas aceton men även andra organiska lösningsmedel med låg toxicitet kan användas (t.ex. dimetylformamid, dimetylsulfoxid). I fråga om formulerade produkter som är dispergerade i vatten och mycket polära organiska ämnen som inte är lösliga i organiska vehikellösningsmedel kan lösningarna vara enklare att applicera om de bereds i en svag lösning av ett vanligt vätmedel (t.ex. Agral, Cittowett, Lubrol, Triton eller Tween).
Lämpliga kontrollösningar bör beredas. I fall där ett lösningsmedel eller ett dispergeringsmedel används för att lösa upp testämnet bör två separata kontrollgrupper användas, en som behandlas med vatten och en som behandlas med lösningsmedlet eller dispergeringsmedlet.
1.6 FÖRFARANDE
1.6.1 Test- och kontrollgrupper
Antalet doser och replikat som testas bör väljas så att de uppfyller de statistiska kraven för bestämning av LD50 med ett konfidensintervall på 95 %. För testet används i regel fem doser i en geometrisk serie vars faktor inte överskrider 2,2. Doserna bör täcka området för LD50. Antalet doser fastställs med beaktande av toxicitetskurvans lutning (dos i förhållande till mortalitet) och den statistiska metod som används för analys av resultaten. Ett preliminärt test kan användas som stöd för valet av lämpliga doser.
Varje testkoncentration bör ges till minst tre likadana testgrupper med tio bin per grupp.
Utöver själva testserierna bör minst tre kontrollgrupper med tio bin per grupp också behandlas. Om ett organiskt lösningsmedel eller ett vätmedel används måste tre extra kontrollgrupper med tio bin per grupp användas för lösningsmedlet eller vätmedlet.
1.6.2 Giftekvivalent
En giftekvivalent måste tas med i testserien. Minst tre doser bör väljas på sådant sätt att de täcker det förväntade LD50-värdet. Minst tre likadana burar med tio bin per bur bör användas för varje testdos. Som giftekvivalent rekommenderas dimetoat. vars LD50-värde (kontakt) enligt rapporterade uppgifter ligger inom området 0,10–0,30 μg verksamt ämne per bi (2). Även andra giftekvivalenter kan godtas förutsatt att man har tillgäng till tillräckliga data om förväntad dosrespons (t.ex. paration).
1.6.3 Exponering
1.6.3.1 Tillförsel av doser
De bedövade bina behandlas individuellt genom lokal applicering. Bina väljs slumpmässigt ut för de olika testdoserna och kontrollgrupperna. En volym pä 1 μl lösning innehållande testämnet i lämplig koncentration appliceras med mikroapplikator på flanksidan på bröstkorgen av varje bi. Om det är motiverat kan andra volymer användas. Efter appliceringen fördelas bina i testburarna där de bör ha tillgång till sackaroslösning.
1.6.3.2 Testperiodens längd
Rekommenderad testperiod är 48 timmar. Om mortaliteten ökar med mer än 10 % när det har gått 24–48 timmar från appliceringen bör testperioden förlängas (till högst 96 timmar), förutsatt att kontrollgruppens mortalitet inte överskrider 10 %.
1.6.4 Observationer
Mortaliteten registreras vid 4 timmar efter doseringen och därefter vid 24 och 48 timmar. Om det behövs en längre testperiod görs efterföljande bedömningar med 24 timmars intervall (upp till högst 96 timmar), förutsatt att kontrollgruppens mortalitet inte överskrider 10 %.
Allt avvikande uppförande som observeras under testperioden bör registreras.
1.6.5 Gränstest
I vissa fall (t.ex. om ett testämne förväntas ha låg toxicitet) kan gränstest utföras med 100 μg verksamt ämne per bi, i syfte att demonstrera att LD50 är högre än detta värde. Förfarandet bör vara detsamma, inklusive tre likadana testgrupper för testdosen, relevanta kontrollgrupper och användning av giftekvivalent. Om det förekommer mortalitet bör ett fullständigt test genomföras. Alla eventuella observerade subletala verkningar bör registreras (se avsnitt 1.6.4).
2. DATA OCH RAPPORTERING
2.1 DATA
Alla uppgifter bör sammanställas i en tabell. Av tabellen ska framgå, för varje testgrupp samt för varje kontrollgrupp och varje giftekvivalentgrupp, antalet bin som använts, mortaliteten vid varje observationstidpunkt och antalet bin med ett beteende som tyder på skadliga verkningar. Mortalitetsdata bör analyseras med lämpliga statistiska metoder (t.ex. probit-analys, rörligt medelvärde eller binominal sannolikhet) (3), (4). Dos-responskurvor uppritas för varje rekommenderad observationstidpunkt (24, 48 och i förekommande fall 72 och 96 timmar) och kurvornas lutning och median letal dos (LD50) beräknas med 95 % konfidensintervall. Korrigeringar för kontrollgruppens mortalitet kan göras med hjälp av Abbott-korrigering (4), (5). LD50-värdet uttrycks som μg testämne per bi.
2.2 TESTRAPPORT
Testrapporten ska innehålla nedan angivna uppgifter.
2.2.1 Testämne
|
— |
Aggregationstillstånd och fysikalisk-kemiska egenskaper (t.ex. stabilitet i vatten, ångtryck). |
|
— |
Kemiska identifieringsdata, inklusive strukturformel, renhet (för bekämpningsmedel t.ex. identiteten och halten av verksamt ämne eller verksamma ämnen). |
2.2.2 Försöksdjur
|
— |
Vetenskapligt namn, ras, ungefärlig ålder (i veckor), insamlingsmetod, insamlingsdatum. |
|
— |
Information om de samhällen som används for insamling av testbin, inklusive hälsa, eventuella vuxensjuk domar, eventuella förbehandlingar osv. |
2.2.3 Testförhållanden
|
— |
Temperatur och relativ fuktighet i testrummet. |
|
— |
Förvaringsförhållanden, inklusive burarnas typ, storlek och material. |
|
— |
Metod för tillförsel av testamnet, t.ex. använt vehikellösningsmedel, tillförd volym testlösning och använd bedövning. |
|
— |
Testets utformning, t.ex. antal doser och deras storlek, antalet kontrollgrupper, för varje testdos och kon trollgrupp även antalet likadana burar och antalet bin per bur. |
|
— |
Testdatum. |
2.2.4 Resultat
|
— |
Resultaten från preliminära test (i förekommande fall). |
|
— |
Rådata: mortaliteten för varje koncentration som testats vid varje observationstidpunkt. |
|
— |
Dos-responskurvorna vid testets slut. |
|
— |
LD50-värden med 95 % konfidensintervall, vid varje rekommenderad observationstidpunkt, för testämnet och för giftekvivalenten. |
|
— |
Statistiska metoder som använts för att bestämma LD50. |
|
— |
Mortaliteten i kontrollgrupperna. |
|
— |
Övriga biologiska verkningar som observerats eller uppmätts och all onormal respons från bina. |
|
— |
Varje avvikelse från de testmetodförfaranden som beskrivs här samt all annan relevant information. |
3. HÄNVISNINGAR
|
1. |
EPPO/Council of Europe (1993), Decision-Making Scheme for the Environmental Risk Assessment of Plant Protection Products – Honeybees, EPPO bulletin, Vol. 23, N.l, s. 151–165. March 1993. |
|
2. |
Gough, H. J., McIndoe, E. C, Lewis, G. B. (1994), The use of dimethoate as a reference compound in laboratory acute toxicity tests on honeybees (Apis mellifera L.), 1981–1992, Journal of Apicultural Research 22, s. 119–125. |
|
3. |
Litchfield, J. T. and Wilcoxon, F. (1949), A simplified method of evaluating dose-effect experiments, Jour. Pharmacol. and Exper. Ther., 96, s. 99–113. |
|
4. |
Finney, D. J. (1971), Probit Analysis. 3rd ed.. Cambridge, London and New York. |
|
5. |
Abbott, W. S. (1925), A method for computing the effectiveness of an insecticide. Jour. Econ. Entomol. 18, s. 265–267. |
C.18 BESTÄMNING AV ADSORPTION OCH DESORPTION I JORD VID JÄMVIKT I UPPSLAMMAT JORDPROV
1. METOD
Denna metod är i stort sett identisk med OECD TG 106 – Determination of Soil Adsorption/Desorption, using a Batch Equilibrium Method (2000).
1.1 INLEDNING
Metoden bygger bland annat på ett ringprov och en workshop för val av jordar för adsorptionstest (1) (2) (3) (4) samt existerande riktlinjer på nationell nivå (5) (6) (7) (8) (9) (10) (11).
Adsorptions- och desorptionstest används för att erhålla grundläggande uppgifter om kemiska ämnens rörlighet och deras fördelning i biosfärens jord, vatten och luft (12) (13) (14) (15) (16) (17) (18) (19) (20) (21). Uppgifterna kan utnyttjas för att förutspå eller uppskatta t.ex. ett visst kemiskt ämnes tillgänglighet vid organismernas nedbrytningsprocesser (22) (23) samt, omvandlings- och upptagningsprocesser (24), ämnets urlakning genom jordprofilen (16) (18) (19) (21) (25) (26) (27) (28), ämnets flyktighet från jorden (21) (29) (30) eller ämnets avrinning från land och ytor in i naturliga vatten (18) (31) (32). Adsorptionsdata kan användas för jämförelser och för att skapa modeller (19) (33) (34) (35).
Ett kemiskt ämnes fördelning mellan jord- och vattenfas är en komplex process som beror på flera olika faktorer: ämnets kemiska natur (12) (36) (37) (38) (39) (40), jordfasens egenskaper (4) (12) (13) (14) (41) (42) (43) (44) (45) (46) (47) (48) (49) och klimatbetingade faktorer såsom regn, temperatur, sol och vind. Den mångfald fenomen och mekanismer som deltar i ämnets adsorption i jord kan därför inte beskrivas fullständigt med hjälp av den aktuella testmetoden, som utgör en förenklad laboratoriemodell. Även om detta test inte kan täcka alla tänkbara miljösituationer ger det ändå tillräckliga uppgifter om den betydelse för miljön som adsorptionen av det aktuella ämnet har.
Se även den allmänna inledningen.
1.2 TILLÄMPNINGSOMRÅDE
Syftet med metoden är att kunna uppskatta ett ämnes adsorptions- och desorptionsbeteende i jordar. Målet är att få ett sorptionsvärde som kan användas för att uppskatta fördelningen när miljöbetingelserna varierar. För detta ändamål bestäms ämnets adsorptionskoefficient i jämvikt i olika jordar som funktion av jordens egenskaper (t.ex. halten av organiskt kol och lera samt jordens textur och pH). Olika jordtyper måste användas för att testet i så hög grad som möjligt ska täcka växelverkan mellan ett ämne och naturligt förekommande jordarter.
Inom ramen för denna metod avser man med begreppet adsorption den process när ett ämne binds till en jordyta, utan att i detta sammanhang skilja mellan olika adsorptionsprocesser (fysikalisk eller kemisk adsorption) eller processer såsom ytkatalyserad nedbrytning, bulkadsorption eller kemisk reaktion. Adsorption, som sker på kolloidala partiklar (diameter < 0,2 um) som har bildats av jordarterna, räknas inte.
De viktigaste jordparametrarna med tanke på adsorptionen anses vara följande: halten av organiskt kol (3) (4) (12) (13) (14) (41) (43) (44) (45) (46) (47) (48), lerbaken och jordens textur (3) (4) (41) (42) (43) (44) (45) (46) (47) (48) samt joniserbara föreningars pH (3) (4) (42). Andra jordparametrar som kan ha inverkar på adsorption/desorption för ett särskilt ämne är den effektiva katjonbyteskapaciteten (ECEC), halten av amorft järn och aluminiumoxider, särskilt i fråga om vulkaniska och tropiska jordar (4), samt den specifika ytan (49).
Syftet med testet är att utvärdera ett ämnes adsorption i olika jordtyper för vilka halten av organiskt kol, 1er-halten och jordens textur och pH varierar. Testet omfattar tre skeden:
|
Skede 1: |
Förberedande undersökningar för att bestämma följande:
|
|
Skede 2: |
Screeningtest: undersökning av adsorptionen i fem olika jordtyper genom adsorptionskinetik vid en vald koncentration och bestämning av fordelningskoefficienterna Kd och Koc. |
|
Skede 3: |
Bestämning av Freundlich-adsorptionsisotermer för att fastställa hur koncentrationen inverkar på adsorptionens omfattning i jordar. Undersökning av desorption genom desorptionskinetik och Freundlich-desorptionsisotermer (bilaga 1). |
1.3 MATSTORHETER – DEFINITIONER OCH MATTENHETER
|
Beteckning |
Definition |
Måttenhet |
|
|
procentuell adsorption vid tidpunkten ti |
% |
|
Aeq |
procentuell adsorption vid adsorptionsjämvikt |
% |
|
|
massan av testämne som adsorberats i jorden vid tidpunkten ti |
μg |
|
|
massan av testämne som adsorberats i jorden under tidsintervallet Δti |
μg |
|
|
massan av testämne som adsorberats i jorden vid adsorptionjämvikt |
μg |
|
m0 |
massan av testämne i provröret vid adsorptionstestest början |
μg |
|
|
massan av testämne uppmät i ett delprov (
|
μg |
|
|
massan av testämne i lösningen vid adsorptionsjämvikt |
μg |
|
mjord |
mängd av jordfas, uttryckt som jordens torrmassa |
g |
|
Cst |
masskoncentrationen i ämnets stamlösning |
μg cm-3 |
|
C0 |
ursprunglig masskoncentration i provlösningen som är i kontakt med jord |
μg cm-3 |
|
|
ämnets masskoncentration i vattenfasen vid tidpunkten ti då analysen utförs |
μg cm-3 |
|
|
halten av ämne som adsorberats i jord vid adsorptionsjämvikt |
μg g-1 |
|
|
ämnets masskoncentration i vattenfasen vid adsorptionjämvikt |
μg cm-3 |
|
V0 |
ursprunglig volym av vattenfas i kontakt med jord under adsorptionstestet |
cm3 |
|
|
volymen för det delprov som används för att mätta testämnet |
cm3 |
|
Kd |
adsorptionens fördelningskoefficient |
cm3 g-1 |
|
Koc |
normaliserad adsorptionskoefficient |
cm3 g-1 |
|
Kom |
normaliserad fördelningskoefficient |
cm3 g-1 |
|
|
Freundlich-adsorptionskoefficient |
μg 1-1/n (cm3) 1/n g-1 |
|
1/n |
Freundlich-exponent |
|
|
|
procentuell desorption vid tidpunkten ti |
% |
|
|
procentuell desorption under intervallet Δti |
% |
|
Kdes |
skenbar desorptionskoefficient |
cm3 g-1 |
|
|
Freundlich-desorptionskoefficient |
μg 1-1/n (cm3) 1/n g-1 |
|
|
massan av testämne som desorberats från jord vid tidpunkten ti |
μg |
|
|
massan av testämne som desorberats från jord under tidsintervallet Δti |
μg |
|
|
analytiskt bestämd ämnesmassa i vattenfasen vid desorptionsjämvikt |
μg |
|
|
totala massan av testämne som desorberats vid desorptionsjämvikt |
μg |
|
|
massan av ämne som förblir adsorberat i jorden tidsintervallet Δti |
μg |
|
|
massan av ämne som blivit över från adsorptionsjämvikten på grund av ofullständig volymersättning |
μg |
|
|
halten av testämne som förblir adsorberat i jorden vid desorptionsjämvikt |
μg g-1 |
|
|
testämnets masskoncentration i vattenfasen vid desorptionsjämvikt |
μg cm-3 |
|
VT |
totala volymen vattenfas i kontakt med jord under desorptionskinetiktest enligt seriemetoden |
cm3 |
|
VR |
volymen supernatant som har avlägsnats ur röret efter uppnådd adsorptionsjämvikt och som har ersatts med samma volym av 0,01 M CaCl2-lösning |
cm3 |
|
|
volymen delprov som tagits för analytiskt syfte under ett desorptionskinetiktest enligt seriemetoden |
cm3 |
|
|
volymen lösning som tagits ur röret (i) för mättning av testämnet under desorptionskinetiktest (parallellmetod) |
cm3 |
|
|
volymen lösning som vid jämvikt tagits ur röret för mättning av testämnet |
cm3 |
|
MB |
massbalans |
% |
|
mE |
totala massan av testämne som extraherats ur jorden och provkärlets väggar i två steg |
μg |
|
Vrec |
volymen supernatant som insamlats efter uppnådd adsorptionsjämvikt |
cm3 |
|
Pow |
fördelningskoefficient för oktanol-vatten |
|
|
pKa |
dissociationkoefficient |
|
|
Sw |
vattenlöslighet |
g l-1 |























1.4 PRINCIPEN FOR TESTMETODEN
Kända volymer av testämneslösning, omärkta eller radioaktivt märkta och med kända koncentrationer i 0,01 M CaCl2 blandas med jordprover av känd torrvikt som har bringats till jämvikt i 0,01 M CaCl2. Blandningen rörs om tillräckligt länge. Jordsuspensionerna separeras därefter genom centrifugering, och om så önskas kan filtratet och vattenfasen analyseras. Mängden av testämne som adsorberats i jordprovet bestäms som skillnaden mellan den ursprungliga mängden av testämne i lösningen och mängden av kvarvarande testämne i slutet av testet (indirekt metod).
Alternativt kan mängden av adsorberat testämne bestämmas direkt genom jordanalys (direkt metod). Denna procedur, som inbegriper stegvis jordextraktion med lämpligt lösningsmedel, rekommenderas i fall dar skillnaden i lösningens halt av testämne inte kan bestämmas med tillräcklig noggrannhet. Detta kan förekomma om testämnet adsorberas pä provkärlens yta, om testämnet är instabilt inom den tidsskala testet sträcker sig över, om adsorptionen är så svag att halten bara ändras lite eller om adsorptionen är så stark att den resulterande halten ar for låg för att kunna bestämmas med tillräcklig noggrannhet. Om radioaktivt märkt ämne används kan man undvika jordextraktion genom att analysera jordfasen med förbränning och vätskescintilllationsräknare (LSC). LSC är dock en icke-specifik teknik som inte kan skilja mellan ursprungliga ämnen och omvandlingsprodukter och som därför bara kan användas om testämnet är stabilt under hela testet.
1.5 UPPGIFTER OM TESTÄMNET
De kemiska reagenser som används ska ha hög analytisk renhetsgrad. Rekommenderade testämnen är omärkta ämnen med känd sammansättning och en renhetsgrad på helst minst 95 % eller radioaktivt märkta ämnen med känd sammansättning och radioaktiv renhet. Om spårämnen med kort halveringstid används ska sönderfallskorrigering tillämpas.
Innan adsorptions- och desorptionstestet utförs ska följande uppgifter finnas om testämnet:
|
a) |
vattenlöslighet (A.6) |
|
b) |
ångtryck (A.4) eller Henrys lag-koefficient |
|
c) |
abiotisk nedbrytning: hydrolys som funktion av pH (C.7) |
|
d) |
fördelningskoefficient (A.8) |
|
e) |
”lätt” biologisk nedbrytbarhet (C.4) eller aerobisk och anaerobisk omvandling i jord |
|
f) |
pKa för joniserbara ämnen |
|
g) |
direkt fotolys i vatten (t.ex. UV-Vis-absorptionsspektrum i vatten och kvantutbyte) och fotonedbrytning i jord. |
1.6 TESTETS TILLÄMPBARHET
Testet kan tillämpas på kemiska ämnen som kan analyseras med tillräcklig noggrannhet. En viktig parameter som kan påverka tillförlitligheten hos resultaten, särskilt när den indirekta metoden används, är testämnets stabilitet inom testets tidsskala. Ämnets stabilitet måste därför kontrolleras med en förhandsundersökning, och om den visar att det förekommer omvandling inom testets tidsskala är det tillrådligt att huvudundersökningen utförs med analys av både jord- och vattenfasen.
Det kan vara svårt att genomföra testet med ämnen som har låg vattenlöslighet (Sw, < 10-4 g l-1) eller med ämnen som har hög elektrisk laddning, eftersom koncentrationen i vattenfasen då inte kan analyseras med tillräcklig noggrannhet. För sådana ämnen krävs vissa extra åtgärder. Närmare anvisningar finns i berörda avsnitt.
Vid testning av flyktiga ämnen är det viktigt att se till att det inte uppstår hanteringsförluster.
1.7 METODBESKRIVNING
1.7.1 Apparatur och kemiska reagenser
Normal laboratorieutrustning, särskilt följande:
|
a) |
Provrör eller andra kärl för testet. Det är viktigt att rören eller kärlen
|
|
b) |
Skakapparat: rotationsskak eller motsvarande, i ett utförande där jorden hålls i suspension under omrö ringen. |
|
c) |
Centrifug: helst i höghastighetsutförande, t.ex. med centrifugalkraft > 3 000 g, temperaturkontrollerad och med kapacitet att från vattenlösning separera partiklar med diameter större än 0,2 μm. Under omröring och centrifugering ska behållarna vara täckta med lock så att flyktighets- och vattenförluster kan undvi kas. För att minimera adsorption på locken ska dessa vara deaktiverade, t.ex. teflonbelagda skruvkorkar. |
|
d) |
Gärna även: filtreringsapparat som har sterila engångsfilter med porositeten 0,2 μm. Särskild vikt ska fäs tas vid valet av filtermaterial så att förluster av testämne på filtret kan undvikas. För svårlösliga testämnen avråds användning av organiska filtermaterial. |
|
e) |
Analysinstrument lämpliga för mätning av testämnets koncentration. |
|
f) |
Laboratorieugn som kan hålla en temperatur mellan 103 oC och 110 oC. |
1.7.2 Karakterisering av jordar och urval av jordar för test
Jordarna karakteriseras med hjälp av tre parametrar som anses vara i hög grad avgörande för adsorptionskapaciteten: organiskt kol, lerbak och jordens textur samt pH. Såsom tidigare nämnts (se avsnittet Tillämpningsområde) kan även andra fysikalisk-kemiska egenskaper hos jorden påverka adsorption och desorption av ett visst ämne, och de ska i så fall beaktas.
Valet av metoder för jordkarakteriseringen är av stor vikt och kan ha betydande inverkan på resultaten. Det rekommenderas därför att jordens pH mäts i en lösning av 0,01 M CaCl2 (dvs. den lösning som används vid adsorptions- och desorptionstestet) enligt gällande ISO-metod (ISO-10390-1). Det rekommenderas också att övriga jordegenskaper av betydelse bestäms enligt standardmetoder (ISO Handbook of soil analysis) så att sorptionsdata kan analyseras på basis av globalt standardiserade jordparametrar. Arbetena (50–52) innehåller viss vägledning om existerande standardmetoder för jordanalys och jordkarakterisering. Det är tillrådligt att använda referensjordar för kalibrering av jordtestmetoderna.
Tabell 1 innehåller anvisningar för valet av jordar för adsorptions- och desorptionstest. De sju utvalda jordarna spänner över jordtyper som finns i tempererade geografiska zoner. För joniserbara testämnen ska de utvalda jordarna sträcka sig över ett stort pH-område så att ämnets adsorption kan bestämmas i joniserad och ojoniserad form. Anvisningar för hur många olika jordar som ska användas i de olika teststegen finns i avsnitt 1.9 (Mätförfarande).
Om andra jordtyper väljs ska de karakteriseras med användning av samma parametrar och ska ha liknande variationer i fråga om de egenskaper som beskrivs i tabell 1, även om de inte behöver motsvara kriterierna fullständigt exakt.
Tabell 1
Anvisningar för val av jordprover för adsorption och desorption
|
Jordtyp |
pH-område (i 0,01 M CaCl2) |
Halt av organiskt kol ( %) |
Lerhalt ( %) |
Jordtextur (25) |
|
1 |
4,5–5,5 |
1,0–2,0 |
65–80 |
Clay |
|
2 |
> 7,5 |
3,5–5,0 |
20–40 |
clay loam |
|
3 |
5,5–7,0 |
1,5-3,0 |
15–25 |
silt loam |
|
4 |
4,0–5,5 |
3,0–4,0 |
15–30 |
loam |
|
5 |
< 4,0–6,0 (26) |
< 10–15 (26) |
loamy sand |
|
|
6 |
> 7,0 |
40–65 |
clay loam/clay |
|
|
7 |
< 4,5 |
> 10 |
< 10 |
sand/loamy sand |
1.7.3 Provtagning och förvaring av jordprover
1.7.3.1 Provtagning
Det finns inga särskilda rekommendationer om specifika metoder eller verktyg för provtagning. Provtagningsmetoden beror på undersökningens syfte (53) (54) (55) (56) (57) (58).
Följande aspekter ska beaktas:
|
a) |
Platsen ska beskrivas i detalj, bl.a. genom läge, vegetationstäcke, behandlingar med bekämpnings- och gödningsmedel, biologiska tillsatser eller oavsiktlig förorening. Beskrivningen av provtagningsplatsen ska följa rekommendationerna i ISO-standarden för tagning av jordprover (ISO 10381-6). |
|
b) |
Provtagningsplatsen ska anges med UTM-koordinater (Universal Transversal Mercator-Projection/European Horizontal Datum) eller geografiska koordinater. På så sätt kan man längre fram ta nya prover av viss jord. Det kan också underlätta bestämningen av jorden enligt de olika ländernas klassificeringssystem. Vidare ska endast A-horisont tas, ned till ett maximidjup på 20 cm. För jord 7 gäller särskilt att om det finns Oh-horisont närvarande i jorden ska denna horisont tas med i provet. |
]ordproverna ska transporteras i behållare, och temperaturbetingelserna ska vara sådana att jordprovernas ursprungliga egenskaper inte förändras i någon signifikant grad.
1.7.3.2 Förvaring
Jordproverna ska helst användas färska. Om detta inte är möjligt ska jorden förvaras lufttorkad i omgivningstemperatur. Det finns inga rekommenderade gränser för förvaringstiden, men om ett jordprov är äldre än tre år måste det analyseras pä nytt avseende halten av organiskt kol, pH och CEC.
1.7.3.3 Hantering och beredning av jordprover för test
Jordproverna lufttorkas vid omgivningstemperatur (helst 20–25 oC). Finfördelningen ska göras så varsamt som möjligt för att minimera ändringarna i jordens ursprungliga textur. Jordarna sållas till en partikelstorlek < 2 mm med beaktande av rekommendationerna i ISO-standarden om tagning av jordprover (ISO 10381-6). Omsorgsfull homogenisering rekommenderas eftersom resultatens reproducerbarhet då förbättras. Fukthalten i varje jordprov bestäms i tre delprover genom torkning vid 105 oC tills ingen signifikant viktändring längre kan registreras (cirka 12 timmar). Vid alla beräkningar som omfattar jordmassa avses ugnstorkad massa, dvs. jordens vikt korrigerad med en fukthaltsfaktor.
1.7.4 Beredning av testämnet före kontakten med jord
Testämnet upplöses i en lösning av 0,01 M CaCl2 i destillerat eller avjoniserat vatten. CaCl2-lösningen används som vattenlösningsfas för att förbättra centrifugeringen och minimera katjonbytet. Stamlösningens koncentration ska helst vara tre tiopotenser högre an analysmetodens detektionsgräns. Genom denna tröskel säkerställs tillräcklig mätnoggrannhet med den använda metoden. Dessutom ska stamlösningens koncentration vara lägre än testämnets vattenlöslighet.
Stamlösningen ska helst beredas strax före kontakten med jordproverna och förvaras i stängd behållare vid 4 oC. Förvaringstiden beror på testämnets stabilitet och dess koncentration i lösningen.
Hjälplösningsmedel för upplösning av testämnet ska bara användas om ämnet är svårlösligt (Sw < 10-4 g t-1). Hjälplösningsmedlet ska a) vara blandbart med vatten, t.ex. metanol eller acetonitril, ska b) ha en koncentration som inte överskrider 1 % av stamlösningens totalvolym och en koncentration som är under 1 % i den testämneslösning som kommer i kontakt med jorden (helst under 0,1 %) och får c) inte vara ett ytaktivt ämne eller genomgå solvolytiska reaktioner med testamnet. Användningen av ett hjälplösningsmedel måste vara föreskriven och ska motiveras i rapporten.
Ett annat alternativ för att tillföra ett svårlösligt testämne i testsystemet är att använda tillsats (spiking). Det innebär att testämnet upplöses i ett organiskt lösningsmedel, och ett delprov av denna lösning tillsatts systemet bestående av jord och 0,01 M CaCl2-lösning i destillerat eller avjoniserat vatten. Halten av organiskt lösningsmedel i vattenfasen ska hällas så låg som möjligt, normalt ska den inte överskrida 0,1 %. Tillsats från en organisk lösning kan innebära att reproducerbarheten går förlorad i fråga om volymen, dvs. ett nytt fel kan komma in eftersom testämnets och hjälplösningsmedlets koncentration inte är samma i alla test.
1.8 FÖRUTSÄTTNINGAR FÖR UTFÖRANDE AV ADSORPTIONS- OCH DESORPT1ONSTEST
1.8.1 Analysmetod
Följande nyckelparametrar kan påverka sorptionsmätningarnas noggrannhet: noggrannheten hos den metod som används för att analysera lösningen och de adsorberade faserna, testämnets stabilitet och renhet, uppnåendet av sorptionsjämvikt, storleksordningen på lösningens koncentrationsändring, förhållandet mellan jord och lösning samt ändringar i jordens struktur under jämviktsprocessen (35) (59–62). Noggrannheten tas upp i några av exemplen i tillägg 2.
Analysmetodens tillförlitlighet måste kontrolleras inom testets sannolika koncentrationsområde. Det står laboratoriet fritt att utveckla en lämplig metod med lämpliga värden i fråga om noggrannhet, reproducerbarhet, detektionsgränser och utbyte. Anvisningar för hur ett sådant test kan utföras ges nedan.
En lämplig volym av 0,01 M CaCl2 (t.ex. 100 cm2 rörs om under 4 timmar med en viktmängd (t.ex. 20 g) jord som har hög adsorptionsförmåga, dvs. hög halt av organiskt kol och lera. Dessa vikter och volymer kan variera beroende på analysbehoven, men ett jord-lösningsförhållande på 1:5 är en lämplig utgångspunkt. Blandningen centrifugeras och vattenfasen kan filtreras. En bestämd volym av testämnets stamlösning tillsätts till vattenfasen så att den nominella koncentrationen kommer inom testets sannolika koncentrationsområde. Volymen ska vara högst 10 % av vattenfasens slutliga volym, så att karaktären hos den jämviktade jordlösningen ändras så lite som möjligt. Lösningen analyseras.
Ett blindprov bestående av systemet jord + CaCl2-lösning (utan testämne) måste ingå, för kontroll av falska indikationer i analysmetoden och matriseffekter orsakade av jorden.
Bland de analysmetoder som kan användas för sorptionsmätningar ingår gas-vätskekromatografi (GLC), högtrycksvätskekromatografi (HPLC), spektrometri (t.ex. GC-masspektrometri och HPLC-masspektrometri) och vätskescintillationsräknare (för radioaktivt märkta ämnen). Oavsett vilken metod som används gäller kriteriet att metoden anses lämplig om utbytet är mellan 90 % och 110 % av det nominella värdet. För att möjliggöra detektion och utvärdering efter det att ämnet fördelats måste analysmetodens detektionsgränser vara minst två tiopotenser under den nominella koncentrationen.
Egenskaper och detektionsgränser hos den analysmetod som används vid adsorptionstest spelar en viktig roll när det gäller att definiera testbetingelserna och testets totala provningsprestanda. Den föreliggande metoden följer ett allmänt testförlopp och innehåller rekommendationer och anvisningar för alternativa tillvägagångssätt i fall där analysmetoden och laboratoriets betingelser kan medföra begränsningar.
1.8.2 Val av optimalt förhållande mellan jord och lösning
Valet av lämpligt jord-lösningsförhållande för sorptionsundersökningar är beroende av fördelningskoefficienten Kd och den önskade relativa adsorptionsgraden. Ämnets koncentrationsändring i lösningen avgör mätningens statistiska noggrannhet – grundat på adsorptionsekvationens form och analysmetodens gränser – vid detektion av ämnets koncentration i lösningen. För vanligt bruk är det därför praktiskt att bestämma sig för en uppsättning konstanta standardförhållanden för vilka adsorptionen är över 20 %, och helst över 50 % (62), samtidigt som man ska se till att testämnets koncentration i vattenfasen hålls så pass hög så att den kan mätas med tillräcklig noggrannhet. Detta är särskilt viktigt när adsorptionsprocenterna är höga.
Ett lämpligt tillvägagångssätt för att välja lämpliga jord-lösningsförhållanden grundar sig på estimering av Kd-värdet, antingen genom förberedande undersökningar eller genom vedertagna estimeringsmetoder (tillägg 3). Valet av lämpligt förhållande kan sedan göras med hjälp av ett diagram där jord-lösningsförhållandet har avsatts mot Kd för konstanta adsorptionsprocenter (figur 1). Härvid antas att adsorptionsekvationen är linjär (28). Det sökta sambandet erhålls genom att bilda om ekvation 4 för Kd så att den får formen enligt ekvation 1:
|
|
(1) |

eller logaritmisk form med antagande av att R = msoil/V0 and Aeq %/100 =  :
:
|
|
(2) |


I figur 1 visas jord-lösningsförhållandet som funktion av Kd för olika adsorptionsnivåer. Exempel: För jord-lösningsförhållandet 1:5 och Kd = 20 blir adsorptionen cirka 80 %. För att få en adsorption på 50 % med samma Kd måste förhållandet 1:25 användas. Detta sätt att välja jord-lösningsförhållande ger möjlighet att anpassa till olika testningsbehov.
Däremot är det svårare att hantera områden där ämnet adsorberas väldigt mycket eller väldigt litet. Vid låg adsorption rekommenderas jord-lösningsförhållandet 1:1, men för vissa mycket organiska jordtyper kan ännu lägre förhållanden behövas för att erhålla slurry. Analysmetoden måste ägnas stor uppmärksamhet när det gäller matning av små ändringar i lösningens koncentration, annars blir adsorptionsmätningen inexakt. Å andra sidan kan man vid mycket höga värden på fördelningskoefficienten Kd gå upp till ett jord-lösningsförhållande på 1:100 för att lämna en signifikant mängd av ämnet i lösningen. Då är det viktigt med god omröring och att systemet får tillräcklig tid på sig för att uppnå jämvikt. Ett alternativt sätt är att uppskatta Kd-värdet med hjälp av estimeringsmetoder som t.ex. grundar sig på Pow-värden (tillägg 3). Detta kan vara särskilt användbart för lågadsorberande och polära ämnen med Pow < 20 och för lipofila och starkt sorptiva ämnen med Pow > 104.
1.9 MÄTFÖRFARANDE
1.9.1 Testbetingelser
Alla test ska göras i omgivningstemperatur och om möjligt vid en konstant temperatur mellan 20 oC och 25 oC.
Centrifugeringsbetingelserna ska vara sådana att partiklar större än 0,2 μm kan avlägsnas från lösningen. Detta värde anger den minsta partikelstorleken för fasta partiklar och bildar gränsen mellan fasta och kolloidala partiklar. Anvisningar om lämpliga centrifugeringsbetingelser finns i bilaga 4.
Om det inte går att säkerställa att partiklar större an 0,2 um avlägsnas vid centrifugeringen, kan en kombination av centritugering och filtrering med 0,2 μm filter användas. Filtermaterialet ska vara inert så att det inte uppstår förluster av testamnet pä filtret. Under alla omständigheter måste det dokumenteras så att det inte uppstår förluster av testämnet under filtreringen.
1.9.2 Skede 1 – Förberedande undersökning
Syftet med den förberedande undersökningen beskrivs i avsnittet Tillämpningsområde. Anvisningar för hur en sådan förberedande undersökning kan läggas upp ges nedan.
1.9.2.1 Val av optimala jord-lösningsförhållanden
Två jordtyper och tre jord-lösningsförhållanden används (för sex tester). En av jordtyperna har hög halt av organiskt kol och låg lerhalt, och den andra typen har låg halt avorganiskt kol och hög lerhalt. Följande förhållanden föreslås:
|
— |
50 g jord och 50 cm3 vattenlösning av testämnet (förhållande 1:1), |
|
— |
10 g jord och 50 cm3 vattenlösning av testämnet (förhållande 1:5), |
|
— |
2 g jord och 50 cm3 vattenlösning av testämnet (förhållande 1:25). |
Den minsta mängd jord som behövs för testet beror på laboratoriets utrustning och analysmetodernas prestanda. För tillförlitliga testresultat rekommenderas dock minst 1 g, helst 2 g.
Ett kontrollprov med enbart testämnet i 0,01 M CaCl2-lösning (ingen jord) testas enligt exakt samma procedur som används för testsystemet, för att kontrollera testämnets stabilitet i CaCl2-lösningen och dess eventuella adsorption på testkärlets ytor.
Ett blindprov per jord, med samma mängd jord och en totalvolym på 50 cm30,01 M CaCl2-lösning (utan testämne) testas på samma sätt. Detta blindtest fungerar som bakgrundskontroll under analysen så att man kan detektera störande ämnen eller förorenade jordar.
Alla test, även kontroll- och blindtest, ska utföras minst två gånger. Det totala antalet prover som ska beredas för undersökningen bestäms utifrån den metod som används.
Metoderna för den förberedande undersökningen och huvudundersökningen är i regel desamma. I förekommande fall nämns skillnader som är av betydelse.
De lufttorkade jordproverna bringas till jämvikt genom omröring med en minimivolym på 45 cm30,01 M CaCl2 över natten (12 timmar) dagen före testet. Därefter tillsätts en bestämd volym av testämnets stamlösning för att justera den slutliga volymen till 50 cm3. Den tillsatta mängden av stamlösning ska a) vara högst 10 % av vattenfasens slutliga volym på 50 cm3 för att minimera ändringen av karaktären hos den jämviktade jordlösningen och b) helst ge en ursprunglig koncentration av testämnet som är i kontakt med jorden (C0) som är minst två tiopotenser högre än analysmetodens detektionsgräns. Med denna tröskel säkerställs möjligheten att utföra tillräckligt noggranna mätningar även när stark adsorption inträffar (> 90 %) och möjligheten att längre fram bestämma adsorptionsisotermerna. Helst ska också, om det är möjligt, ämnets ursprungliga koncentration (C0) inte överskrida hälften av dess löslighetsgräns.
Nedan ges ett exempel på beräkning av stamlösningens koncentration (CM). För beräkningen gäller antagandet att detektionsgränsen är 0,01 μg cm-3 och adsorptionen 90 %. Sålunda ska den ursprungliga koncentrationen av testämne i kontakt med jorden helst vara lug cm3 (två tiopotenser högre än detektionsgränsen). Under antagandet att den maximala rekommenderade volymen stamlösning tillsätts, dvs. 5 cm3 till 45 cm30,01 M CaCl2-jämviktslösning (= 10 % stamlösning till 50 cm3 total volym av vattenfas), bör stamlösningens koncentration vara 10 μg cm3, dvs. tre tiopotenser högre än analysmetodens detektionsgräns.
Vattenfasens pH ska mätas före och efter kontakt med jorden eftersom pH-värdet spelar en viktig roll för hela adsorptionsprocessen, särskilt när det gäller joniserbara ämnen.
Lösningen rörs om så länge att adsorptionsjämvikt uppnås. Tiden för att nå jämvikt varierar kraftigt beroende pä ämnet och jorden. I regel räcker 24 timmar (77). Vid den förberedande undersökningen kan sekventiella prover tas under en omröringstid på 48 timmar, t.ex. vid 4, 8, 24 och 48 timmar, men analystidpunkterna kan också anpassas till arbetsschemat på laboratoriet.
Det finns två alternativ för analys av testämnet i vattenlösningen: a) parallellmetoden och b) seriemetoden. Det måste betonas att parallellmetoden, även om den tar längre tid att utföra, innebär enklare matematisk behandling av resultaten (tillägg 5). Valet av metod måste avgöras utifrån laboratoriets tillgängliga utrustning och resurser.
|
a) |
Parallellmetoden: Prover med samma jord-lösningsförhållande bereds. Antalet prover bestäms av antalet tidsintervall som används för undersökning av adsorptionskinetiken. Efter centrifugering och i förekommande fall filtrering tas det första rörets vattenfas till vara så fullständigt som möjligt och mäts efter t.ex. 4 timmar, det andra rörets vattenfas mäts efter 8 timmar, det tredje rörets efter 24 timmar osv. |
|
b) |
Seriemetoden: Endast två likadana prover bereds för varje jord-lösningsförhållande. Faserna separeras genom centrifugering med bestämda tidsintervall. Ett litet delprov av vattenfasen analyseras omedelbart med avseende på testämnet, och därefter fortsätts testet med den ursprungliga blandningen. Om filtrering utförs efter centrifugeringen bör laboratoriet ha lämplig utrustning för filtrering av små delprover av vattenlösning. Det rekommenderas att den totala volymen delprover som tas inte överskrider 1 % av lösningens totalvolym, så att det inte uppstår signifikanta ändringar i jord-lösningsförhållandet och minskningar i den mängd av löst ämne som är tillgänglig för adsorption under testet. |
Den procentuella adsorptionen  beräknas för varje tidpunkt (ti) på grundval av den nominella ursprungskoncentrationen och den uppmätta koncentrationen vid tidpunkten för provtagningen (
beräknas för varje tidpunkt (ti) på grundval av den nominella ursprungskoncentrationen och den uppmätta koncentrationen vid tidpunkten för provtagningen ( ), med korrigering för blindprovets värde. En kurva där avsätts mot tiden (figur 1 i tillägg 5) ritas upp för uppskattning av jämviktsplatån (29). Likaså beräknas Kd-värdet vid jämvikt. Utifrån detta Kd-värde väljs lämpliga jord-lösningsförhållanden i figur 1 så att den procentuella adsorptionen hamnar över 20 %, och helst över 50 % (61). De tillämpliga ekvationerna och principerna för uppritningen av kurvan ges i avsnittet ”Data och rapportering” och i bilaga 5.
), med korrigering för blindprovets värde. En kurva där avsätts mot tiden (figur 1 i tillägg 5) ritas upp för uppskattning av jämviktsplatån (29). Likaså beräknas Kd-värdet vid jämvikt. Utifrån detta Kd-värde väljs lämpliga jord-lösningsförhållanden i figur 1 så att den procentuella adsorptionen hamnar över 20 %, och helst över 50 % (61). De tillämpliga ekvationerna och principerna för uppritningen av kurvan ges i avsnittet ”Data och rapportering” och i bilaga 5.
1.9.2.2 Bestämning av adsorptionens jämviktstid och mängden av testämne som adsorberats vid jämvikt
Kurvor där  ellerhar
ellerhar  avsatts mot tiden används, såsom tidigare nämnts, för att uppskatta när adsorptionsjämvikt inträtt och vilken mängd av testämne som adsorbcrats vid jämvikt. 1 figur 1 och 2 i tillägg 5 finns exempel på sådana kurvor. Jämviktstiden är den tid som systemet behöver för att nå en platå.
avsatts mot tiden används, såsom tidigare nämnts, för att uppskatta när adsorptionsjämvikt inträtt och vilken mängd av testämne som adsorbcrats vid jämvikt. 1 figur 1 och 2 i tillägg 5 finns exempel på sådana kurvor. Jämviktstiden är den tid som systemet behöver för att nå en platå.
Om en viss jord inte ger upphov till någon platå utan en oavbruten ökning, kan detta bero på komplicerande faktorer såsom biologisk nedbrytning eller långsam diffusion. Biologisk nedbrytning kan konstateras genom att man upprepar testet med ett steriliserat prov av jorden. Om man fortfarande inte uppnår någon platå är det skäl att forska efter andra tänkbara faktorer. Detta kan göras med lämpliga ändringar av testbetingelserna (temperatur, omröringstider, jord-lösningsförhållande etc.). Testledaren får avgöra om det är värt att fortsätta testet även om jämvikt inte kan uppnås.
1.9.2.3 Adsorption på testkärlets yta och testämnets stabilitet
Vissa indikationer rörande testämnets adsorption på testkärlets yta och rörande testämnets stabilitet kan fås fram genom analys av kontrollproverna. Om en minskning som är större än analysmetodens standardfel konstateras kan detta bero på abiotiskt sönderfall och/eller adsorption på testkärlets yta. För att avgöra vilket fenomen som är aktuellt tvättas testkärlets grundligt med en känd volym av lämpligt lösningsmedel, varefter tvättlösningen analyseras med avseende på testämnet. Om analysen visar att ingen adsorption på testkärlets yta har skett, beror minskningen på att testämnet är abiotiskt instabilt. Om adsorption kan konstateras, är det nödvändigt att byta till testkärl av annat material. Dessa resultat gällande adsorption på testkärlets yta kan dock inte direkt extrapoleras till testet med jord och lösning, eftersom adsorptionen i det senare fallet påverkas av att jord ar närvarande.
Ytterligare uppgifter om testämnets stabilitet kan fas fram genom att man gör upp en massbalans som funktion av tiden. Detta görs genom att vattenfasen, extrakt av jord samt testkärlets väggar analyseras med avseende pä testamnet. Skillnaden mellan massan av tillsatt ämne och summan av testämnesmassorna i vattenfasen, i jordextrakten och på testkärlets väggar är lika med den massa som brutits ned eller förflyktigats eller som inte har extraherats. En förutsättning för att massbalansen ska kunna bestämmas är att adsorptionsjämvikt uppnås inom den tid som går åt för testet.
Massbalansen bestäms för båda jordarna och för ett jord-lösningsförhällande för respektive jord som vid jämvikt ger en adsorption på över 20 %, och helst över 50 %. När proceduren för att få fram lämpliga jord-lösningsförhållanden har avslutats med analysen av det sista provet av vattenfas efter 48 timmar, separeras faserna genom centrifugering och, om så önskas, filtrering. Så mycket vattenfas som möjligt tas tillvara, och ett lämpligt extraktionslösningsmedel (med extraktionskoefficient på minst 95 %) tillsätts jorden för att testämnet ska extraheras. Minst två extraktioner i följd rekommenderas. Mängden av testämne i extrakten från jord och testkärl bestäms, och massbalansen beräknas (se ekvation 10, Data och rapportering). Om massbalansen är under 90 % anses testämnet vara instabilt inom testets tidsskala. Undersökningarna kan emellertid fortsättas om man tar testämnets instabilitet med i beräkningen, och i sådana fall är det tillrådligt att vid huvudundersökningen analysera båda faserna.
1.9.3 Skede 2 – Adsorptionskinetik vid en given koncentration av testämnet
Fem jordar väljs enligt tabell 1. Det är till fördel om dessa fem jordar inbegriper en del eller alla av de jordar som använts för den förberedande undersökningen. Då behöver skede 2 inte upprepas för den förberedande undersökningens jordar.
Jämviktstiden, jord-lösningsförhållandet, jordprovets vikt, volymen av vattenfasen i kontakt med jord samt testämnets koncentration i lösningen väljs på grundval av resultaten från den förberedande undersökningen. Analys ska helst utföras efter en kontakttid på 2, 4, 6, 8 (eventuellt även 10) och 24 timmar. Omröringstiden kan vid behov förlängas till högst 48 timmar om ämnet kräver längre jämviktstid. Vid behov kan analystidpunkterna anpassas.
Varje test (en jord och en lösning) görs minst två gånger för att man ska få en indikation på resultatens varians. För varje test görs också ett blindtest. Det görs på jord och 0,01 M CaCl2-lösning utan testämne och med samma vikt och volym som vid det riktiga testet. Som kontroll görs samma testprocedur på ett kontrollprov med enbart testämnet i 0,01 M CaCl2-lösning (utan jord) som ett sätt att skydda sig mot oförutsedda faktorer.
Den procentuella adsorptionen beräknas för varje tidpunkt  eller tidsintervall
eller tidsintervall  (efter behov) och avsätts mot tiden i ett diagram. Likaså beräknas fördelningskoefficienten Kd vid jämvikt samt den normaliserade adsorptionskoefficienten Koc (för opolära organiska ämnen).
(efter behov) och avsätts mot tiden i ett diagram. Likaså beräknas fördelningskoefficienten Kd vid jämvikt samt den normaliserade adsorptionskoefficienten Koc (för opolära organiska ämnen).
Resultat av adsorptionskinetiktest
Det linjära Kd-värdet duger i allmänhet till att med tillräcklig noggrannhet beskriva sorptionsbeteendet i jord (35) (78), och det representerar ett uttryck för ämnenas inherenta rörlighet i jord. Som exempel kan nämnas att allmänt förekommande kemiska ämnen med Kd ≤ 1 cm3 g-1 i regel anses vara kvalitativt rörliga. Ett system för rörlighetsklassificering enligt Koc-värden har konstruerats av MacCall et al. (16). Det finns också system för urlakningsklassificering som bygger på ett samband mellan Koc och DT-50 (30) (32) (79).
Enligt felanalyser (61) kan Kd-värden under 0,3 cm3 g-1 inte uppskattas med tillräcklig noggrannhet på grundval av en koncentrationsminskning i vattenfasen, inte ens när det (ur noggrannhetssynpunkt) gynnsammaste jordlösningsförhållandet används, dvs. 1:1. 1 detta fall rekommenderas analys av både jord- och lösningsfasen.
Med hänvisning till ovannämnda anmärkningar är det tillrådligt att undersökningen av ämnets adsorptionsbeteende i jord och dess potentiella rörlighet fortsätts genom bestämning av Freundlich-adsorptionsisotermer för sådana system där en tillräckligt noggrann bestämning av Kd är möjlig enligt schemat för den föreliggande testmetoden. En noggrann bestämning är möjlig om det värde som fås fram genom att multiplicera Kd med jordlösningsförhållandet är > 0,3 när mätningarna grundar sig på en koncentrationsminskning i vattenfasen (indirekt metod), eller > 0,1 när båda faserna analyseras (direkt metod) (61).
1.9.4 Skede 3 – Adsorptionsisotermer, desorptionskinetik och desorptionsisotermer
1.9.4.1
Fem testämneskoncentrationer används, helst så att de sträcker sig över två tiopotenser. Vid valet av koncentrationer ska vattenlösligheten och de resulterande jämviktskoncentrationerna i vattenlösning tas med i beräkningen, jordarnas respektive jord-lösningsförhållanden ska hällas konstanta genom hela undersökningen. Adsorptionstestet utförs enligt beskrivningen ovan, med enda skillnaden att vattenfasen bara analyseras en gång. efter den tid som enligt skede 2 går åt för att uppnå jämvikt. Jämviktskoncentrationerna i lösningen bestäms, och den adsorberade mängden beräknas på grundval av minskningen av testämne i lösningen, eller med hjälp av den direkta metoden. Den adsorberade massan per enhet jordmassa avsätts mot testämnets jämviktskoncentration i ett diagram (se Data och rapportering).
Resultat av adsorptionsisotermtest
Av alla hittills framlagda matematiska adsorptionsmodeller är Freundlich-isotermerna en av de mest använda när det gäller att beskriva adsorptionsprocesser. För närmare uppgifter om hur man tolkar adsorptionsmodeller och om deras betydelse hänvisas till arbetena (41) (45) (80) (81) (82).
Anmärkning: Det bör nämnas att jämförelse av K^-värden (Freundlich-adsorptionskocfficienter) för olika ämnen bara är möjlig om K, -värdena är uttryckta i samma måttenhet (83).
1.9.4.2
Syftet med detta test är att undersöka om ett ämne adsorberas reversibelt eller irreversibelt i en jord. Denna upplysning är betydelsefull eftersom desorptionsprocessen också spelar en viktig roll för hur ett ämne beter sig i fältjord. Dessutom är desorptionsdata värdefulla som indata när man vill skapa datormodeller för simulering av urlakning och avrinning av lösta ämnen. Om även desorptionen ska undersökas är det tillrådligt att den nedan beskrivna undersökningen utförs på varje system för vilket man kunnat göra en noggrann bestämning av Kd i det föregående adsorptionskinetiktestet.
Desorptionskinetiken kan, på samma sätt som adsorptionskinetiken, undersökas på två alternativa sätt: a) parallellmetoden och b) seriemetoden. Vilken metod som används får avgöras utifrån laboratoriets utrustning och resurser.
|
a) |
Parallellmetoden: För varje jord som har valts för desorptionsundersökning bereds ett antal prover med samma jord-lösningsförhållande. Antalet prover är samma som antalet tidpunkter vid vilka man önskar undersöka adsorptionskinetiken. Helst ska man använda samma tidsintervall som vid adsorptionskinetiktestet, även om totaltiden kan förlängas efter behov så att desorptionsjämvikt uppnås. För varje test (en jord och en lösning) körs ett blindprov. Detta består av jorden och 0,1 M CaCl2-lösning, utan testämne, med samma vikt respektive volym som vid det riktiga testet. Som kontroll utförs samma testprocedur på enbart testämnet i 0,01 M CaCl2-lösning (utan jord). Alla blandningar av jord med lösning rörs om tills adsorptionsjämvikt uppnås (enligt resultaten från skede 2). Därefter separeras faserna med centrifugering, och så mycket som möjligt av vattenfasen avlägsnas. Volymen avlägsnad lösning ersätts med en lika stor volym av 0,01 M CaCl2 utan testämne, och den nya blandningen rörs om igen. Vattenfasen från det första röret tas till vara så fullständigt som möjligt och mäts efter t.ex. 2 timmar, vattenfasen från det andra röret efter 4 timmar, från det tredje efter 6 timmar osv., tills desorptionsjämvikt uppnås. |
|
b) |
Seriemetoden: Efter adsorptionskinetiktestet centrifugeras blandningen, och så mycket som möjligt av vattenfasen avlägsnas. Den borttagna lösningens volym ersätts med lika stor volym av 0,01 M CaCl2 utan testämne. Den nya blandningen rörs om tills desorptionsjämvikt uppnås. Under denna omröringstid centrifugeras blandningen med bestämda tidsintervall så att faserna separeras. Ett litet delprov av vattenfasen analyseras omedelbart med avseende på testämnet, och därefter fortsätts testet med den ursprungliga blandningen. Volymen för varje enskilt delprov ska vara mindre än 1 % av totalvolymen. Samma mängd av färsk 0,01 M CaCl2-lösning tillsätts blandningen för att bibehålla förhållandet mellan jord och lösning, och omröringen fortsätts under nästa tidsintervall. |
Den procentuella desorptionen beräknas för varje tidpunkt ( ) eller tidsintervall (
) eller tidsintervall ( ) (beroende pä vad som krävs för testet) och avsätts mot tiden i ett diagram. Likaså beräknas desorptionskoefficienten Kdes vid jämvikt. De aktuella ekvationerna finns i avsnittet Data och rapportering och i tillägg 5.
) (beroende pä vad som krävs för testet) och avsätts mot tiden i ett diagram. Likaså beräknas desorptionskoefficienten Kdes vid jämvikt. De aktuella ekvationerna finns i avsnittet Data och rapportering och i tillägg 5.
Resultat från desorptionskinetiktest
Adsorptionsprocessens reversibilitet kan uppskattas med hjälp av ett diagram där den procentuella desorptionen  och adsorptionen
och adsorptionen  har avsatts mot tiden (i samma diagram). Om desorptionsjämvikt uppnås inom två gånger tiden för adsorptionsjämvikt, och den totala desorptionen ar mer än 75 % av den adsorberade mängden, anses adsorptionen vara reversibel.
har avsatts mot tiden (i samma diagram). Om desorptionsjämvikt uppnås inom två gånger tiden för adsorptionsjämvikt, och den totala desorptionen ar mer än 75 % av den adsorberade mängden, anses adsorptionen vara reversibel.
1.9.4.3
Freundlich-desorptionsisotermerna bestäms på de jordar som används i adsorptionsisotermtestet. Desorptionstestet utförs enligt beskrivningen i avsnittet Desorptionskinetik, med undantag av att vattenfasen bara analyseras en gång, vid desorptionsjämvikt. Mängden av desorberat testämne beräknas. Halten av kvarvarande adsorberat testämne i jorden vid desorptionsjämvikt avsätts i ett diagram mot testämnets jämviktskoncentration i lösningen (se Data och rapportering samt tillägg 5).
2. DATA OCH RAPPORTERING
Data från analysen ska presenteras i tabellform (se bilaga 6). De enskilda mätningarna och beräknade medeltal ska anges. Adsorptionsisotermerna ska presenteras i diagramform. Beräkningarna ska göras på nedan beskrivet sätt.
Vid beräkningarna antas att vikten av 1 cm3 vattenlösning är 1 g. Därför kan jord-lösningsförhållandet uttryckas med samma värde oberoende av om vikt/vikt eller vikt/volym valts som måttenhet.
2.1 ADSORPTION
Adsorptionen ( ) uttrycks i procent och definieras såsom förhållandet mellan den mängd av ämne som under testbetingelserna har adsorberats i jorden och den mängd som fanns närvarande vid testets början. Om testämnet är stabilt och inte i någon signifikant grad adsorberas på kärlets väggar beräknas
) uttrycks i procent och definieras såsom förhållandet mellan den mängd av ämne som under testbetingelserna har adsorberats i jorden och den mängd som fanns närvarande vid testets början. Om testämnet är stabilt och inte i någon signifikant grad adsorberas på kärlets väggar beräknas  vid varje tidpunkt ti enligt följande ekvation:
vid varje tidpunkt ti enligt följande ekvation:
|
|
(3) |

där:
|
|
= |
den procentuella adsorptionen vid tidpunkten ti ( %), |
|
|
= |
massan av testämne som adsorberats i jorden vid tidpunkten ti (μg), |
|
m0 |
= |
massan av testämne i provröret, vid testets början (μg). |


Närmare anvisningar för hur den procentuella adsorptionen  beräknas för parallell- respektive seriemetoden finns i tillägg 5.
beräknas för parallell- respektive seriemetoden finns i tillägg 5.
Fördelningskoefficienten Kd är förhållandet mellan ämnets halt i jordfasen och ämnets masskoncentration i vattenlösningen när adsorptionsjämvikt har inträtt, under testbetingelserna.
|
|
(4) |

där:
|
|
= |
halten av adsorberat ämne i jord vid adsorptionsjämvikt (μg g-1), |
|
|
= |
ämnets masskoncentration i vattenfasen vid adsorptionsjämvikt (μg cm-3). Koncentrationen bestäms på analytisk väg med beaktande av värdet från blindprovet. |
|
|
= |
massan av ämne som adsorberats i jord vid adsorptionsjämvikt (μg), |
|
|
= |
ämnets massa i lösningen vid adsorptionsjämvikt (μg), |
|
mjord |
= |
mängd av jordfas, uttryckt som torr jordmassa (g), |
|
v0 |
= |
ursprunglig volym av vattenfas i kontakt med jord (cm3). |




Sambandet mellan Aeq och Kd ges av:
|
|
(5) |

där:
|
Aeq |
= |
procentuell adsorption vid adsorptionsjämvikt, %. |
Den normaliserade adsorptionskoefficienten för organiskt kol Koc bestämmer sambandet mellan fördelningskoefficienten Kd och halten av organiskt kol i jordprovet:
|
|
(6) |

där:
|
% oc |
= |
procentuell andel organiskt kol i jordprovet (g g-1). |
Koefficienten Koc representerar ett värde som beskriver hur huvudsakligen opolära organiska ämnen fördelas mellan a ena sidan jordens eller sedimentets organiska kol och å andra sidan vattnet. Sådana ämnens adsorption är beroende av den sorberande fasta fasens organiska innehåll (7), och därför beror Koc-värdet på de humösa fraktionernas specifika egenskaper, eftersom sorptionskapaciteten hos dessa fraktioner kan variera kraftigt beroende på deras olika ursprung, uppkomst osv.
2.1.1 Adsorptionsisotermer
Ekvationen för Freundlich-adsorptionsisotermer ger sambandet mellan mängden av adsorberat testämne och testämnets koncentration i lösningen vid jämvikt (ekvation 8).
Data behandlas på samma sätt som i punkten ”Adsorption” ovan, och halten av adsorberat testämne i jorden efter adsorptionstestet  , på andra ställen betecknat som x/m) beräknas för varje provrör, under antagandet att jämvikt har inträtt och att
, på andra ställen betecknat som x/m) beräknas för varje provrör, under antagandet att jämvikt har inträtt och att representerar jämviktsvärdet:
representerar jämviktsvärdet:
|
|
(7) |

Freundlich-adsorptionsekvationen (8) ser ut så här:
|
|
(8) |

eller i linjär form:
|
|
(9) |

där:
|
|
= |
Freundlich-adsorptionskoefficienten, som anges i dimensionen cm3 g-1 bara om exponenten l/n = 1. 1 alla andra fall införs l/n i dimensionen för |
|
n |
= |
regressionskoefficienten. l/n ligger i regel mellan 0,7 och 1,0 som indikation på att sorptionsdata ofta är något olinjära. |

 (μg
(μgEkvationerna (8) och (9) ritas upp i diagram, och värdena på  och l/n beräknas med regressionsanalys med hjälp av ekvation 9. Likaså beräknas den logaritmiska ekvationens korrelationskoefficient r. Exempel på kurvor av detta slag finns i figur 2.
och l/n beräknas med regressionsanalys med hjälp av ekvation 9. Likaså beräknas den logaritmiska ekvationens korrelationskoefficient r. Exempel på kurvor av detta slag finns i figur 2.

2.1.2 Massbalans
Massbalansen (MB) anges i procent och definieras som förhållandet mellan den mängd av ämnet som analytiskt kan tas till vara efter adsorptionstestet och den nominella mängden av ämnet vid testets början.
Behandlingen av resultaten blir annorlunda om lösningsmedlet är fullständigt blandbart med vatten. Då kan man behandla resultaten på samma sätt som i punkten ”Desorption” för att bestämma den mängd av ämnet som tagits till vara genom extraktion av lösningsmedel. Om lösningsmedlet är mindre blandbart med vatten krävs en bestämning av den tillvaratagna mängden.
Massbalansen MB för adsorptionen beräknas enligt följande ekvation, under antagandet att termen (mE) motsvarar summan av de testämnesmassor som extraherats ur jorden och från testkärlets yta med hjälp av ett organiskt lösningsmedel:
|
|
(10) |

där:
|
MB |
= |
massbalans ( %) |
|
mE |
= |
total massa av testämne som extraherats ur jorden och från testkärlets väggar i två steg (μg), |
|
C0 |
= |
ursprunglig masskoncentration i testlösningen som är i kontakt med jord (μg cm3), |
|
Vrec |
= |
volymen supernatant som tagits till vara efter adsorptionsjämvikt (cm-3). |
2.2 DESORPTION
Desorptionen (D) uttrycks i procent och definieras som förhållandet mellan den desorberade mängden av testämnet och den tidigare adsorberade mängden av ämnet, under testbetingelserna:
|
|
(11) |
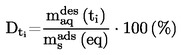
där:
|
|
= |
procentuell desorption vid tidpunkten ti ( %), |
|
|
= |
massan av testämne som desorberats ur jorden vid tidpunkten ti (μg), |
|
|
= |
massan av testämne som adsorberats i jorden vid adsorptionsjämvikt (μg). |


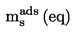
Närmare anvisningar för sättet att beräkna den procentuella desorptionen 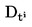 för parallell- och seriemetoden finns i bilaga 5.
för parallell- och seriemetoden finns i bilaga 5.
Den skenbara desorptionskoefficienten (Kdes) är, under de aktuella testbetingelserna, förhållandet mellan massan av ämne som finns kvar i jordfasen och massan av desorberat ämne i vattenlösningen, när desorptionsjämvikt har inträtt:
|
|
(12) |

där:
|
Kdes |
= |
desorptionskoefficient (cm3 g-1): |
|
|
= |
totala massan av testämne som desorberats ur jord vid desorptionsjämvikt (μg), |
|
Vr |
= |
totala volymen av vattenfas i kontakt med jord under desorptionskinetiktestet (cm3). |
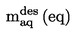
Anvisningar för beräkning av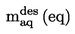 finns i tillagg 5 under rubriken ”Desorption”.
finns i tillagg 5 under rubriken ”Desorption”.
Anmärkning:
Om adsorptionstestet som föregår desorptionstestet utförts med parallellmetoden, kan volymen VT i ekvation (12) sättas lika med Vo.
2.2.1 Desorptionsisotermer
Ekvationen för Frcundlich-desorptionsisotermer beskriver sambandet mellan mängden av testämne som förblir adsorberat i jorden och koncentrationen av testämne i lösningen vid desorptionsjämvikt (ekvation 16).
Halten av ämne som förblir adsorberat i jorden vid desorptionsjämvikt beräknas för varje provrör enligt följande:
|
|
(13) |
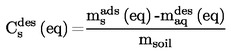
 definieras som:
definieras som:
|
|
(14) |

där:
|
|
= |
balten av testämne som förblir adsorberat i jorden vid desorptionsjämvikt (μg g-1), |
|
|
= |
massan av ämne som bestämts analytiskt i vattenfasen vid desorptionsjämvikt (μg), |
|
|
= |
massan av testämne som blivit över (efter att adsorptionsjämvikt inträtt) på grund av ofullständig volymersättning (μg), |
|
|
= |
massan av ämne i lösningen vid adsorptionsjämvikt (μg), |


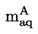

|
|
(15) |

|
|
= |
volymen av lösning som tagits ur röret för uppmätning av testämnet vid desorptionsjämvikt (cm3), |
|
VR |
= |
volymen av supernatant som avlägsnats ur röret efter att adsorptionsjämvikt inträtt och som ersatts med samma volym av 0,01 M CaCl2-lösning (cm3). |

Freundlich-desorptionsekvationen (16) ser ut så här:
|
|
(16) |

eller i linjär form:
|
|
(17) |

där:
|
|
= |
Freundlich-desorptionskoefficienten. |
|
n |
= |
regressionskoefficienren, |
|
|
= |
ämnets masskoncentration i vattenfasen vid desorptionsjämvikt (μg cm-3). |
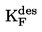

Ekvationerna 16 och 17 kan ritas upp i diagram, och värdet på  och l/n beräknas med regressionsanalys med hjälp av ekvation 17.
och l/n beräknas med regressionsanalys med hjälp av ekvation 17.
Anmärkning:
Om Freundlichs adsorptions- eller desorptionsexponent l/n är lika med 1, blir Freundlichs adsorptions- eller desorptionskoefficient ( respektive
respektive  ) lika med adsorptionens respektive desorptionens jämviktskoefficient (Kd och Kdes), och kurvorna över Cs som funktion av Caq blir linjära. Om exponenten är skild från 1, blir kurvorna över Cs som funktion av Caq olinjära, och adsorptions- och desorptionskoefficienterna kommer att variera längs isotermerna.
) lika med adsorptionens respektive desorptionens jämviktskoefficient (Kd och Kdes), och kurvorna över Cs som funktion av Caq blir linjära. Om exponenten är skild från 1, blir kurvorna över Cs som funktion av Caq olinjära, och adsorptions- och desorptionskoefficienterna kommer att variera längs isotermerna.
2.2.2 Rapport
Testrapporten ska innehålla följande uppgifter:
|
— |
Fullständiga uppgifter om jordproverna, inbegripet följande: |
|
— |
områdets geografiska läge (latitud och longitud), |
|
— |
provtagningsdatum, |
|
— |
bruksändamål (t.ex. jordbruk, skog osv.), |
|
— |
provtagningsdjup, |
|
— |
halt av sand, silt och clay, |
|
— |
pH-värden (i 0,01 M CaCl2), |
|
— |
halt av organiskt kol, |
|
— |
halt av organiskt material, |
|
— |
kvävehalt, |
|
— |
kol-kväveförhållande, |
|
— |
katjonbyteskapacitet (mmol/kg), |
|
— |
alla uppgifter av betydelse som ror provtagning och förvaring av jordproverna, |
|
— |
i förekommande fall, alla uppgifter av betydelse för tolkning av testämnets adsorption och desorption, |
|
— |
uppgift om vilka metoder som använts för bestämning av enskilda parametrar. |
|
— |
Relevanta uppgifter om testämnet |
|
— |
Testtemperaturen |
|
— |
Centrifugeringsbetingelser |
|
— |
Analysprocedurer som använts för analys av testämnet |
|
— |
I förekommande fall motivering för användning av hjälplösningsmedel för beredning av testämnets stam lösning |
|
— |
1 förekommande fall förklaringar av korrigeringar som gjorts i beräkningarna |
|
— |
Data enligt blanketten (bilaga 6) och diagram |
|
— |
Alla uppgifter och observationer som kan vara till hjälp för att tolka testresultaten. |
3. HÄNVISNINGAR
|
1. |
Kukowski H. and Brummer C. (1987), Investigations on the Adsorption and Desorption of Selected Chemicals in Soils. UBA Report 106 02 04 5, Part II. |
|
2. |
Fränzle O., Kuhnt G. and Vetter L. (1987), Selection of Representative Soils in the LC-Territory. UBA Report 106 02045, Part I. |
|
3. |
Kuhnt G. and Muntau H. (Eds.), EURO-Soils: Identification, Collection, Treatment, Characterisation. Special Publication No 1.94.60, Joint Research Centre. European Commission, ISPRA, December 1994. |
|
4. |
OECD Test Guidelines Programme, Final Report of the OECD Workshop on Selection of Soils/Sediments, Belgirate, Italy, 18–20 January 1995 (June 1995). |
|
5. |
US Environment Protection Agency: Pesticide Assessment Guidelines, Subdivision N, Chemistry: Environmental Fate. Series 163-1, Leaching and Adsorption/Desorption Studies, Addendum 6 on Data Reporting. 540/09-88-096, Date: 1/1988. |
|
6. |
US Environment Protection Agency: Prevention, Pesticides and Toxic Substances, OPPTS Harmonized Test Guidelines, Series 835-Fate, Transport and Transformation Test Guidelines, OPPTS No: 835.1220 Sediment and Soil Adsorption/Desorption Isotherm. EPA No: 71 2-C-96-048, April 1996. |
|
7. |
ASTM Standards, E 1195-85, Standard Test Method for Determining a Sorption Constant (Koc) for an Organic Chemical in Soil and Sediments. |
|
8. |
Agriculture Canada: Environmental Chemistry and Fate. Guidelines for registration of pesticides in Canada, 15 July 1987. |
|
9. |
Netherlands Commission Registration Pesticides (1995), Application for registration of a pesticide. Section G. Behaviour of the product and its metabolites in soil, water and air. |
|
10. |
Danish National Agency of Environmental Protection (October 1988), Criteria for registration of pesticides as especially dangerous to health or especially harmful to the environment. |
|
11. |
BBA (1990), Guidelines for the Official Testing of Plant Protection Products, Biological Research Centre for Agriculture and Forestry, Braunschweig, Germany. |
|
12. |
Calvet R. (1989), ”Evaluation of adsorption coefficients and the prediction of the mobilities of pesticides in soils”, in Methodological Aspects of the Study of Pesticide Behaviour in Soil (ed. P. Jamet), INRA, Paris, (Review). |
|
13. |
Calvet R. (1980), ”Adsorption-Desorption Phenomena” in Interactions between herbicides and the soil. (R. J. Hance ed.), Academic Press, London, s. 83–122. |
|
14. |
Hasset J. J. and Banwart W.L. (1989), ”The sorption of nonpolar organics by soils and sediments” in Reactions and Movement of Organic Chemicals in Soils. Soil Science Society of America (S.S.S.A), Special Publication no. 22, s. 31–44. |
|
15. |
van Genuchten M. Th., Davidson J. M. and Wierenga P.J. (1974), ”An evaluation of kinetic and equilibrium equations for the prediction of pesticide movement through porous media”. Soil Sci. Soc. Am. Proc, Vol. 38(1), s. 29–35. |
|
16. |
McCall P.]., Laskowski D. A., Swann R. L. and Dishburger H. J. (1981), ”Measurement of sorption coefficients of organic chemicals and their use, in environmental fate analysis”, in Test Protocols for Environmental Fate and Movement of Toxicants. Proceedings of AOAC Symposium, AOAC, Washington DC. |
|
17. |
Lambert S. M., Porter P. E. and Schieferrstein R. H. (1965), ”Movement and sorption of chemicals applied to the soil”, Weeds, 13, s. 185–190. |
|
18. |
Rhodes R. C, Belasco I. J. and Pease H. L. (1970) ”Determination of mobility and adsorption of agrochemicals in soils”. J.Agric.Food Chem., 18, s. 524–528. |
|
19. |
Russell M. H. (1995), ”Recommended approaches to assess pesticide mobility in soil” in Environmental Behavior of Agrochemicals (ed. T. R. Roberts and P. C. Kearney). John Wiley & Sons Ltd. |
|
20. |
Esser H. O., Hemingway R. J., Klein W., Sharp D. B., Vonk J. W. and Holland P. T. (1988), ”Recommended approach to the evaluation of the environmental behavior of pesticides”, IUPAC Reports on Pesticides (24). Pure Appl. Chem., 60, s. 901–932. |
|
21. |
Guth J. A., Burkhard N. and D. O. Eberle (1976), ”Experimental models for studying the persistence of pesticides in soils”. Proc. BCPC Symposium: Persistence of Insecticides and Herbicides, s. 137–157, BCPC, Surrey, UK. |
|
22. |
Furminge C. G. L. and Osgerby J. M. (1967), ”Persistence of herbicides in soil”. J. Sci. Fd Agric, 18, s. 269–273. |
|
23. |
Burkhard N. and Guth J. A. (1981), ”Chemical hydrolysis of 2-Chloro-4,6-bis(alkylamino)-l,3,5-triazine herbicides and their breakdown in soil under the influence of adsorption”. Pestic. Sci. 12, s. 45–52. |
|
24. |
Guth J. A., Gerber H. R. and Schlaepfer T. (1977), ”Effect of adsorption, movement and persistence on the biological availability of soil-applied pesticides”. Proc. Br. Crop Prot. Conf., 3, s. 961–971. |
|
25. |
Osgerby J. M. (1973), ”Process affecting herbicide action in soil”. Pestic. Sci., 4, s. 247–258. |
|
26. |
Guth J. A. (1972), ”Adsorptions- und Einwascheverhalten von Pflanzenschutzmitteln in Böden”. Schr. Reihe Ver. Wass.-Boden-Lufthyg. Berlin-Dahlem, Heft 37, s. 143–154. |
|
27. |
Hamaker J. W. (1975), ”The interpretation of soil leaching experiments”, in Environmental Dynamics of Pesticides (eds R. Haque and V.H. freed), s. 135–172, Plenum Press, NY. |
|
28. |
Helling C. S. (1971). ”Pesticide mobility in soils”. Soil Sci. Soc. Amer. Proc., 35, s. 732–210. |
|
29. |
Hamaker J. W. (1972), ”Diffusion and volatilization” in Organic chemicals in the soil environment (C.A.I. Goring and J. W. Hamaker eds), Vol. I, s. 49–143. |
|
30. |
Burkhard N. and Guth J. A. (1981), ”Rate of volatilisation of pesticides from soil surfaces; Comparison of calculated results with those determined in a laboratory model system”. Pestic. Sci. 12, s. 37–44. |
|
31. |
Cohen S. Z., Creeger S. M., Carsel R.F. and Enfield CG. (1984), ”Potential pesticide contamination of groundwater from agricultural uses”, in Treatment and Disposal of Pesticide Wastes, s. 297–325, Acs Symp. Ser. 2 59, American Chemical Society, Washington, DC. |
|
32. |
Gustafson D. I. (1989), ”Groundwater ubiquity score: a simple method for assessing pesticide leachability”. J. Environ. Toxic. Chem., 8(4), s. 339–357. |
|
33. |
Leistra M. and Dekkers W. A. (1976). ”Computed effects of adsorption kinetics on pesticide movement in soils”. J. of Soil Sci., 28, s. 340–350. |
|
34. |
Bromilov R. H. and Leistra M. (1980), ”Measured and simulated behavior of aldicarb and its oxydation products in fallow soils”. Pest. Sei., 11, s. 389–395. |
|
35. |
Green R. E. and Karickoff S. W. (1990), ”Sorption estimates for modeling”, in Pesticides in the Soil Environment: Process, Impacts and Modeling (ed. H.H. Cheng). Soil Sci. Soc. Am., Book Series no. 2, s. 80–101, |
|
36. |
Lambert S. M. (1967), ”Functional relationship between sorption in soil and chemical structure”. J. Agri. Food Chem., 15, s. 572–576. |
|
37. |
Hance R. J.(1969), ”An empirical relationship between chemical structure and the sorption of some herbicides by soils”, Agri. Food Chem., 17, s. 667–668. |
|
38. |
Briggs G. G. (1969), ”Molecular structure of herbicides and their sorption by soils”, Nature, 223, 1288. |
|
39. |
Briggs G. G. (1981), ”Theoretical and experimental relationships between soil adsorption, octanol-water partition coefficients, water solubilities, bioconcentration factors, and the parachor”, J. Agric. Food Chem., 29, s. 1050–1059. |
|
40. |
Sabljic A. (1984), ”Predictions of the nature and strength of soil sorption of organic polutance by molecular topology”. J. Agric. Food Chem., 32, s. 243–246. |
|
41. |
Bailey G. W. and White J. L. (1970), ”Factors influencing the adsorption, desorption, and movement of pesticides in soil”. Residue Rev., 32, s. 29–92. |
|
42. |
Bailey G. W., J. L. White and Y. Rothberg (1968), ”Adsorption of organic herbicides by montomorillonite: Role of pH and chemical character of adsorbate”, Soil Sci. Soc. Amer. Proc. 32: s. 222–234. |
|
43. |
Karickhoff S. W. (1981), ”Semi-empirical estimation of sorption of hydrophobic pollutants on natural sediments and soils”, Chemosphere 10, s. 833–846. |
|
44. |
Paya-Perez A., Riaz M. and I.arsen B. (1989), ”Soil Sorption of 6 Chlorobenzenes and 20 PCB Congeners”. Environ. Toxicol. Safety 21, s. 1–17. |
|
45. |
Hamaker). W. and Thompson J. M. (1972), ”Adsorption in organic chemicals' in Organic Chemicals in the Soil Environment” Goring C.A.I, and Hamaker J.W., eds., Vol I och II, Marcel Dekker, Inc., New York, NY, 1972, s. 49–143. |
|
46. |
Deli)., and Warren G. F. (1971), ”Adsorption, desorption and leaching of diphenamid in soils”. Weed Sci. 19: s. 67–69. |
|
47. |
Chu-Huang Wu, Buchring N., Davinson J. M. and Santelmann (1975), ”Napropamide Adsorption, desorption and Movement m soils”. Weed Science, Vol. 23, s. 454–457. |
|
48. |
Haues M. H. B., Stacey M. and Thompson J. M.. (1968), ”Adsorption of s-triazine herbicides by soil organic preparations” in Isotopes and Radiation in Soil Organic Studies, s. 75, International. Atomic Energy Agency, Wien. |
|
49. |
Pionke H. B. and Deangelis R. J. (1980), ”Methods for distributing pesticide loss in field run-off between the solution and adsorbed phase”, CREAMS, in A Field Scale Model for Chemicals, Run-off and Erosion from Agricultural Management Systems, Chapter 19, Vol. III: Supporting Documentation, USDA Conservation Research report. |
|
50. |
ISO Standard Compendium Environment: Soil Quality – General aspects: chemical and physical methods of analysis: biological methods of analysis. First Edition (1994). |
|
51. |
Scheffer F. and Schachtschabel. Lehrbuch der Bodenkunde, F, Enke Verlag, Stuttgart (1982), 11th edition. |
|
52. |
Black, Evans D. D., White J. L., Ensminger L.E. and Clark F. E., eds. ”Methods of Soil Analysis”, Vol 1 and 2, American Society of Agronomy, Madison, \VI, 1982. |
|
53. |
ISO/DIS 10381-1 Soil Quality – Sampling – Part 1: Guidance on the design of sampling programmes. |
|
54. |
ISO/DIS 10381-2 Soil Quality – Sampling – Part 2: Guidance on sampling techniques. |
|
55. |
ISO/DIS 10381-3 Soil Quality – Sampling – Part 3: Guidance on safety of sampling. |
|
56. |
ISO/DIS 10381-4 Soil Quality – Sampling – Part 4: Guidance on the investigation of natural and cultivated soils. |
|
57. |
ISO/DIS 10381-5 Soil Quality – Sampling – Part 5: Guidance on the investigation of soil contamination of urban and industrial sites. |
|
58. |
ISO 10381-6, 1993: Soil Quality – Sampling – Part 6: Guidance on the collection, handling and storage of soil for the assessment of aerobic microbial processes in the laboratory. |
|
59. |
Green R. E. and Yamane V. K. (1970), ”Precision in pesticide adsorption measurements”. Soil Sci. Am. Proc. 34, s. 353–354. |
|
60. |
Grover R. and Hance R. J. (1970), ”Effect of ratio of soil to water on adsorption of linuron and atrazine”, Soil Sci., s. 109–138. |
|
61. |
Boesten, J. J. T. I, ”Influence of soil/liquid ratio on the experimental error of sorption coefficients in pesticide/soil system”, Pest. Sci. 1990, 30, s. 31–41. |
|
62. |
Boesten, J. J. T. I. ”Influence of soil/liquid ratio on the experimental error of sorption coefficients in relation to OECD guideline 106”, Proceedings of 5th international workshop on environmental behaviour of pesticides and regulatory aspects, Brussels, 26–29 April 1994. |
|
63. |
Bastide J., Cantier J. M., et Coste C (1980), ”Comportement de substances herbicides dans le sol en fonction de leur structure chimique”, Weed Res. 21, s. 227–231. |
|
64. |
Brown D. S. and Flagg E. W. (1981), ”Empirical prediction of organic pollutants sorption in natural sediments”, J. Environ.Qual., 10(3), s. 382–386. |
|
65. |
Chiou C. T., Porter P. E., and Schmedding D. W. (1983), ”Partition equilibria of non-ionic organic compounds between soil organic matter and water”. Environ. Sci. Technol., 17(4), s. 227–231. |
|
66. |
Gerstl Z. and Mingelgrin U. (1984), ”Sorption of organic substances by soils and sediments”, J. Environm. Sci. Health, B19 (3), s. 297–312. |
|
67. |
Vowles P. D. and Mantoura R. F. C (1987), ”Sediment-water partition coefficient and HPLC retention factors of aromatic hydrocarbons”, Chemosphere, 16(1), s. 109–116. |
|
68. |
Lyman W. J., Reehl W. F.and Rosenblatt D. H. (1990), Handbook of Chemical Property Estimation Methods. Environmental Behaviour of Organic Compounds. American Chemical Society, Washington DC. |
|
69. |
Keniga E. E., and Goring, C. A. I. (1980), ”Relationship between water solubility, soil sorption, octanol-water partitioning and concentration of chemicals in the biota” in Aquatic Toxicology (eds J.G. Eaton, et al.), s. 78–115, ASTM STP 707, Philadelphia. |
|
70. |
Chiou C. T., Peters L. J. and Freed V. H. (1979), ”A physical concept of soil-water equilibria for non-ionic organic compounds”, Science, Vol. 206, s. 831–832. |
|
71. |
Hassett). J., Banwart W. I., Wood S. G. and Means J. C. (1981), ”Sorption of/-Naphtol: implications concerning the limits of hydrophobic sorption”, Soil Sci. Soc. Am. J. 45, s. 38–42. |
|
72. |
Karickhoff S. W. (1981), ”Semi-empirical estimation of sorption of hydrophobic pollutants on natural sediments and soils”, Chemosphere, Vol. 10(8), s. 833–846. |
|
73. |
Moreale A., van Bladel R. (1981), ”Adsorption de 13 herbicides et insecticides par le sol. Relation solubilité-reactivité”. Revue de l'Agric, 34 (4), s. 319–322. |
|
74. |
Müller M., Kordel W. (1996), ”Comparison of screening methods for the determination/estimation of adsorption coefficients on soil”. Chemosphere, 32(12), s. 2493–2504. |
|
75. |
Kordel W., Kotthoff G. Muller M. (1995), ”HPLC – screening method for the determination of the adsorption coefficient on soil – results of a ring test”. Chemosphere 30 (7), s. 1373–1384. |
|
76. |
Kördel W., Stutte J., Kotthoff C. (1993), ”HPLC – screening method for the determination of the adsorption coefficient on soil – comparison of different stationary phases”. Chemosphere 27 (12), s. 2341–2352. |
|
77. |
Hance. R. J. (1967), ”The Speed of Attainment of Sorption Equilibria in Some Systems Involving Herbicides”, Weed Research, Vol. 7, s. 29–36. |
|
78. |
Koskinen W. C and Harper S. S. (1990), ”The retention processes: mechanisms” in Pesticides in the Soil Environment: Processes, Impacts and Modelling (ed. H.H. Cheng). Soil Sci. Soc. Am. Book Series, No. 2, Madison, Wisconsin. |
|
79. |
Cohen S. Z., Creeger S. M., Carsel R. F. and Enfield C. G. (1984), ”Potential pesticide contamination of groundwater from agricultural uses”, in Treatment and Disposal of Pesticide Wastes, s. 297–325, ACS Symp. Ser. 2 59, American Chemical Society, Washington, DC. |
|
80. |
Giles C. H. (1970), ”Interpretation and use of sorption isotherms” in Sorption and Transport Processes in Soils, S.C.I. Monograph No. 37, s. 14–32. |
|
81. |
Giles, C. H., McEwan J. H., Nakhwa, S.N. and Smith D. (1960), ”Studies in adsorption: XI. A system of classification of solution adsorption isotherms and its use in the diagnosis of adsorption mechanisms and in measurements of pesticides surface areas of soils”. J. Chem. Soo, s. 3973–3993. |
|
82. |
Calvet R., Tercé M. and Arvien J. C (1980), ”Adsorption des pesticides par les sols et leurs constituants: 3. Caractéristiques générales de l'adsorption”, Ann. Agron. 31, s. 239–251. |
|
83. |
Bedbur E. (1996), ”Anomalies in the Freundlich equation”, Proc. COST 66 Workshop, Pesticides in soil and the environment, 13–15 May 1996, Stratford-upon-Avon, UK. |
|
84. |
Guth, J. A. (1985), ”Adsorption/desorption”, in Joint International Symposium, Physicochemical Properties and their Role in Environmental Hazard Assessment, July 1–3, Canterbury, UK. |
|
85. |
Soil Texture Classification (US and FAO systems): Weed Science, 3 3, Suppl. 1 (1985) and Soil Sci. Soc. Amer. Proc. 26:305 (1962). |
Bilaga 1
Testschema
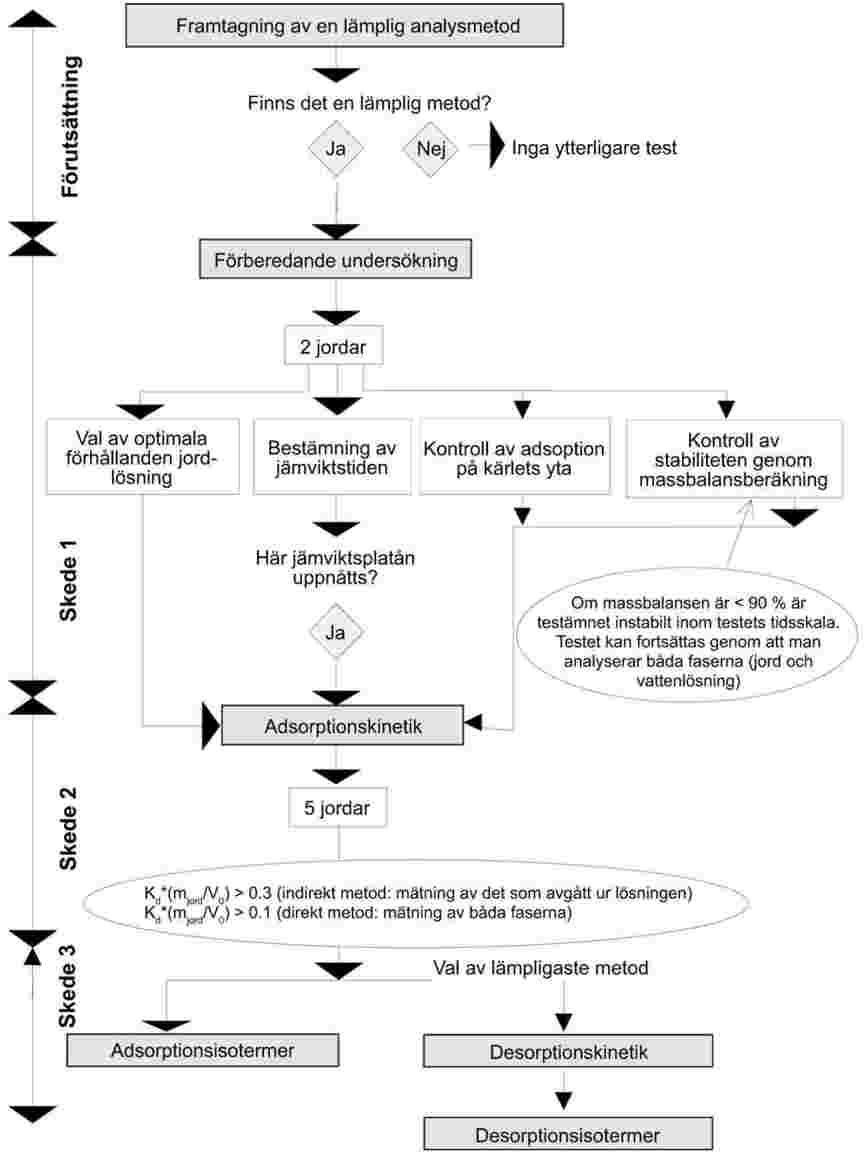
Bilaga 2
ADSORPTIONSRESULTATENS NOGGRANNHET SOM FUNKTION AV ANALYSMETODENS NOGGRANNHET OCH KONCENTRATIONSÄNDRINGEN
Av tabellen nedan (84) framgår klart att när skillnaden mellan massan vid testets början (m0 = 110 μg) och testämnets massa 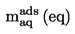 i lösningen vid jämvikt (=100 μg)är mycket liten, så leder ett fel på 5 % vid mätning av jämviktskoncentrationen till ett fel på 50 % i beräkningen av massan av ämne som adsorberats i jorden (
i lösningen vid jämvikt (=100 μg)är mycket liten, så leder ett fel på 5 % vid mätning av jämviktskoncentrationen till ett fel på 50 % i beräkningen av massan av ämne som adsorberats i jorden ( ) och ett fel på 52,4 % i beräkningen av Kd.
) och ett fel på 52,4 % i beräkningen av Kd.
|
Mängd jord |
mjord |
10 g |
|
Lösningens volym |
V0 |
100 cm3 |
|
|
FOR-L_2008142SV.01044401.notes.0187.xml.jpg (μg) |
FOR-L_2008142SV.01044401.notes.0188.xml.jpg (μg cm-3) |
R |
FOR-L_2008142SV.01044401.notes.0189.xml.jpg (μg) |
FOR-L_2008142SV.01044401.notes.0190.xml.jpg (μg g-1) |
R‡ |
Kd* |
R‡ |
|
|
FÖR A = 9 % |
|||||||
|
m0 = 110 μg eller C0=1,100 μg/cm3 |
100 |
1,000 |
verkligt värde |
10 |
1,00 |
verkligt värde |
1 |
|
|
101 |
1,010 |
1 % |
9 |
0,90 |
10 % |
0,891 |
10,9 % |
|
|
105 |
1,050 |
5 % |
5 |
0,50 |
50 % |
0,476 |
52,4 % |
|
|
109 |
1,090 |
9 % |
1 |
0,10 |
90 % |
0,092 |
90,8 % |
|
|
|
FÖR A = 55 % |
|||||||
|
m0= 110 μg eller C0=1,100μg/cm3 |
50,0 |
0,500 |
verkligt värde |
60,0 |
6,00 |
verkligt värde |
12,00 |
|
|
50,5 |
0,505 |
1 % |
59,5 |
5,95 |
0,8 % |
11,78 |
1,8 % |
|
|
52,5 |
0,525 |
5 % |
57,5 |
5,75 |
4,0 % |
10,95 |
8,8 % |
|
|
55,0 |
0,550 |
10 % |
55,0 |
5,50 |
8,3 % |
10,00 |
16,7 % |
|
|
|
FÖR A = 99 % |
|||||||
|
m0 = 110 μg eller C0=1,100 μg/cm3 |
1,100 |
0,011 |
verkligt värde |
108,9 |
10,89 |
verkligt värde |
990 |
|
|
1,111 |
0,01111 |
1 % |
108,889 |
10,8889 |
0,01 % |
980 |
1,0 % |
|
|
1,155 |
0,01155 |
5 % |
108,845 |
10,8845 |
0,05 % |
942 |
4,8 % |
|
|
1,21 |
0,0121 |
10 % |
108,790 |
10,8790 |
0,10 % |
899 |
9,2 % |
|
|
|
= |
|
|
|
= |
testämnets massa i jordfasen vid jämvikt, µg; |
|
|
= |
testämnets massa i vattenfasen vid jämvikt, µg; |
|
|
= |
testämnets halt i jordfasen vid jämvikt, μg g-1; |
|
|
= |
testämnets masskoncentration i vattenfasen vid jämvikt, μg cm-3; |
|
R |
= |
analysfel vid bestämning av |
|
R‡ |
= |
beräkningsfel på grund av analysfelet R. |
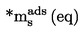

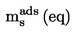
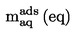

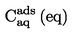
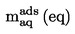 ;
;Bilaga 3
ESTIMERINGSMETODER FÖR Kd
|
1. |
Genom estimeringsmetoder kan Kd uppskattas på grundval av samband med t.ex. Pow-värden (12) (39) (63–68), vattenlöslighetsdata (12) (19) (21) (39) (68–73) eller polaritetsdata som fåtts fram från HPLC-RP (omvänd fas) (74–76). Koc och Kom beräknas med hjälp av ekvationerna i tabell 1 och 2, och sedan räknas Kd fram indirekt ur följande ekvationer:
|
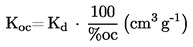

|
2. |
Principen för dessa samband grundar sig på två antaganden: 1) det är främst jordens organiska material som påverkar hur ett ämne adsorberas, och 2) de växelverkansprocesser som förekommer är i huvudsak opolära. Det innebär att dessa samband 1) inte, eller bara delvis, kan tillämpas på polära ämnen, och 2) inte kan tillämpas i fall där jordens halt av organiskt material är mycket låg (12). Vidare gälier att samma sak inte kan konstateras angående sambandet mellan vattenlöslighet och adsorptionens omfattning (19) (21), även om man har funnit tillfredsställande korrelation mellan Pow och adsorption (19). Hittills gjorda undersökningar har gett motsägelsefulla resultat. |
|
3. |
1 tabell 1 ges några exempel på samband mellan adsorptionens fördelningskoefficient och fördelningskoefficienten oktanol-vatten. I tabell 2 ges några exempel på samband mellan adsorptionens fördelningskoefficient och vattenlösligheten. Tabell 1 Exempel på samband mellan adsorptionens fördelningsskoefficient och fördelningskoefficienten oktanol-vatten. Ytterligare exempel finns i (12) (68).
Tabell 2 Exempel på samband mellan adsorptionens fördelningskoefficient och vattenlösligheten. Ytterligare exempel finns i (68) (69).
|
Bilaga 4
BERÄKNINGAR FÖR BESTÄMNING AV CENTRIFUGERINGSBETINGELSER
|
1. |
Centrifugeringstiden fås fram genom följande formel, under antagande att partiklama är sfäriska:
Av praktiska skäl ges parametrarna i måttenheterna g, cm etc. i stället för de normala SI-enheterna. där:
För att säkerställa fullständig separation får centrifugen som regel gå två gånger den framräknade tiden. |

|
2. |
Ekvation 1 kan förenklas ytterligare om lösningens viskositet (η) och densitet (paq) kan anses vara samma som viskositeten och densiteten för vatten vid 25 oC, dvs. η = 8,95 × 10-3 g s-1 cm-1 och paq = 1,0 g, cm-3. Då fås centrifugeringstiden genom ekvation 2:
|

|
3. |
Av ekvation 2 framgår tydligt att det är två faktorer som har stor betydelse när det gäller att bestämma de centrifugeringsbetingelser, dvs. tid (t) och varvtal (rpm), som behövs för att åstadkomma separation av partiklar av en viss storlek (i detta fall med radien 0,1 μm): 1) jordens densitet och 2) blandningens höjd i centrifugeringsröret (Rb-Rt), dvs. det avstånd som en jordpartikel färdas från lösningens övre yta till rörets botten. För en given volym bestäms blandningens höjd självfallet av rörets radie. |
|
4. |
I figur 1 visas centrifugeringstiden (t) som funktion av varvtalet (rpm) för olika jorddensiteter (ps) (figur 1 a) och för olika blandningshöjder i centrifugeringsröret (figur 2 a). Av figur 1 a framgår inverkan av jordens densitet tydligt – vid klassisk centrifugering med 3 000 rpm är centrifugeringstiden cirka 240 minuter när jordens densitet är 1,2 g cm3, men bara 50 minuter när densiteten är 2,0 g cm3. Likaså visar figur 1 b att centrifugeringstiden vid klassisk centrifugering med 3 000 rpm är cirka 50 minuter om blandningens höjd är 10 cm men bara 7 minuter om höjden är 1 cm. Det är dock viktigt att finna en optimal avvägning mellan å ena sidan så liten blandningshöjd som möjligt för centrifugeringen och å andra sidan enkel hantering när det gäller att separera faserna efter centrifugering. |
|
5. |
När testbetingelserna för separering av jord- och lösningsfaserna ska bestämmas är det också viktigt att beakta eventuell förekomst av en tredje ”pseudofas”, dvs. kolloider. Dessa partiklar är mindre än 0,2 μm och kan ha viktig inverkan på hela adsorptionsmekanismen för ett ämne i en jordsuspension. När centrifugeringen utförs på ovan beskrivet sätt blir kolloiderna kvar i vattenfasen och analyseras då tillsammans med den. Då går informationen om deras inverkan förlorad. Om laboratoriet har möjligheter till ultracentrifugering eller ultrafiltrering kan adsorptionen och desorptionen av ett ämne i jord studeras mer ingående, även beträffande ämnets adsorption på kolloiderna. Då ska ultracentrifugering med 60 000 rpm/min eller ultrafiltrering med filterporositeten 100 000 dalton användas för att separera de tre faserna jord, kolloider och lösning. Då ska också testschemat ändras så att alla tre faserna tas med i ämnesanalysen. |
Figur la.

Figur 1b.

Bilaga 5
BERÄKNING AV ADSORPTION A ( %) OCH DESORPTION D ( %)
Procedurens tidsschema är följande:

För samtliga beräkningar gäller antagandet att testämnet är stabilt och att det inte i någon signifikant grad adsorberas på kärlets väggar.
ADSORPTION A (A %)
a) Parallellmetoden
Den procentuella adsorptionen beräknas för varje provrör (i) vid varje tidpunkt (ti), enligt följande ekvation:
|
|
(1) |
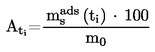
Ekvationens termer beräknas enligt följande:
|
m0 = C0 · V0 (μg) |
(2) |
|
|
(3) |

där:
|
|
den procentuella adsorptionen ( %) vid tidpunkten t1 , |
|
|
testämnets massa i jorden vid tidpunkten ti då analysen utförs (μg), |
|
m0 |
testämnets massa i provröret vid testets början (μg), |
|
C0 |
ursprunglig masskoncentration i testlösningen som är i kontakt med jorden (μg cm-3). |
|
|
ämnets masskoncentration i vattenfasen vid tidpunkten ti då analysen utförs (μg cm-3), denna koncentration bestäms analytiskt med beaktande av värdena från blindproven. |
|
V0 |
ursprunglig volym för den testlösning som är i kontakt med jorden (cm3). |



Värdet på den procentuella adsorptionen  eller
eller 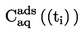 avsätts mot tiden i ett diagram, och kurvan används för att bestämma tiden för när sorptionsjämvikt inträder. Exempel på sådana kurvor finns i figur 1 och figur 2.
avsätts mot tiden i ett diagram, och kurvan används för att bestämma tiden för när sorptionsjämvikt inträder. Exempel på sådana kurvor finns i figur 1 och figur 2.

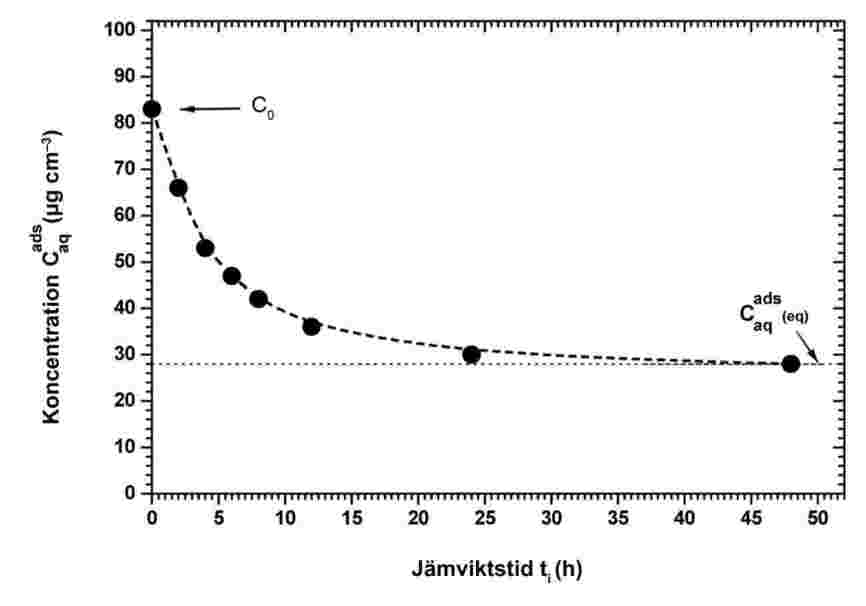
b) Seriemetoden
I ekvationerna som följer är utgångspunkten att adsorptionen undersöks genom mätning av testämnet i små delprover av vattenfasen med bestämda tidsintervall.
|
— |
Den mängd ämne som adsorberats i jorden under varje tidsintervall beräknas på följande sätt:
|
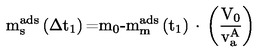



|
— |
Den procentuella adsorptionen under varje tidsintervall
och den procentuella adsorptionen vid tidpunkten ti (
Adsorptionsvärdena |
 beräknas genom ekvationen
beräknas genom ekvationen
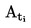 ) fås genom ekvationen
) fås genom ekvationen
 eller
eller  (beroende på vad som krävs i undersökningen) ritas upp som funktion av tiden i ett diagram, och kurvan används för att bestämma tidpunkten för när sorptionsjämvikt inträder.
(beroende på vad som krävs i undersökningen) ritas upp som funktion av tiden i ett diagram, och kurvan används för att bestämma tidpunkten för när sorptionsjämvikt inträder.|
— |
Vid jämviktstiden teq gäller följande:
|



De ovan använda parametrarna definieras enligt följande:
|
|
massan av ämne som adsorberats i jorden under tidsintervallen Δt1, Δt2,…,Δtn (μg), |
|
|
massan av ämne som uppmätts i ett delprov |
|
|
massan av ämne som adsorberats i jorden vid adsorptionsjämvikt (μg), |
|
|
massan av ämne i lösningen vid adsorptionsjämvikt (μg), |
|
|
volymen av det delprov i vilket testämnet uppmäts (cm3), |
|
|
procentuell adsorption under tidsintervallet Δti ( %), |
|
|
procentuell adsorption vid adsorptionsjämvikt ( %). |






DESORPTION D (A %)
Som starttid t0 för desorptionskinetiktestet räknas den tidpunkt då den maximala tillvaratagna volymen av testämneslösning (efter att adsorptionsjämvikt har inträtt) ersätts med en lika stor volym 0,01 M CaCl2-lösning.
a) Parallellmetoden
Vid tidpunkten ti uppmäts massan av testämnet i volymen av den vattenfas som tagits ur rör i ( ), och den desorberade massan beräknas enligt följande ekvation:
), och den desorberade massan beräknas enligt följande ekvation:
|
|
(13) |

Vid desorptionsjämvikt är ti = teq och därför är  =
= 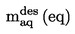 .
.
Massan av testämne som har desorberats under ett tidsintervall (Δti) ges av ekvationen:
|
|
(14) |

Den procentuella desorptionen
beräknas för tidpunkten ti enligt ekvationen
|
|
(15) |

och för tidsintervallet (Δti) enligt ekvationen
|
|
(16) |

där:
|
|
= |
procentuell desorption vid tidpunkten ti ( %), |
|
|
= |
procentuell desorption under tidsintervallet Δti ( %), |
|
|
= |
massan av testämne som desorberats vid tidpunkten ti (μg), |
|
|
= |
massan av testämne som desorberats under tidsintervallet Δti (μg), |
|
|
= |
massan av testämne som uppmätts analytiskt vid tidpunkten ti i den lösningsvolym |
|
|
= |
massan av testämne som blivit över från adsorptionsjämvikten på grund av ofullständig volymersättning (μg) |




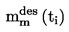
 som har tagits för analysen (μg),
som har tagits för analysen (μg),
|
|
(17) |
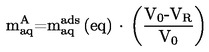
|
|
= |
massan av testämne i lösningen vid adsorptionsjämvikt (μg), |
|
VR |
= |
volymen av supernatant som tagits ur röret efter uppnådd adsorptionsjämvikt och som ersatts med samma volym 0,01 M CaCl2-lösning (cm3), |
|
|
= |
volymen av lösning som tagits ur rör nummer i för uppmätning av testämnet i desorptionskinetiktestet (cm3). |
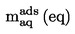

Desorptionsvärdena 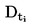 eller
eller  (beroende på vad som krävs i undersökningen) ritas upp som funktion av tiden i ett diagram, och kurvan används för att bestämma när desorptionsjämvikt inträder.
(beroende på vad som krävs i undersökningen) ritas upp som funktion av tiden i ett diagram, och kurvan används för att bestämma när desorptionsjämvikt inträder.
b) Seriemetoden
I ekvationerna som följer är utgångspunkten att den föregående adsorptionsundersökningen utförts genom att mäta testämnet i små delprovsvolymer  av vattenfasen (seriemetoden, se avsnitt 1.9 ”Mätförfarande”). Härvid antas a) att volymen av den supernatant som avlägsnats ur röret efter adsorptionskinetiktestet har ersatts med en lika stor volym av 0,01 M CaCl2-lösning (VR) och b) att den totala volymen av vattenfasen som är i kontakt med jord (VT) under desorptionskinetiktestet förblir konstant och ges av följande ekvation:
av vattenfasen (seriemetoden, se avsnitt 1.9 ”Mätförfarande”). Härvid antas a) att volymen av den supernatant som avlägsnats ur röret efter adsorptionskinetiktestet har ersatts med en lika stor volym av 0,01 M CaCl2-lösning (VR) och b) att den totala volymen av vattenfasen som är i kontakt med jord (VT) under desorptionskinetiktestet förblir konstant och ges av följande ekvation:
|
|
(18) |

Vid tidpunkten ti, gäller följande:
|
— |
Massan av testämne mäts i ett litet delprov (
|
 ) och den desorberade massan beräknas enligt följande ekvation:
) och den desorberade massan beräknas enligt följande ekvation:
|
— |
Vid desorptionsjämvikt är ti = teq, och därför är |
 =
= 
|
— |
Den procentuella desorptionen
|
 beräknas enligt följande ekvation:
beräknas enligt följande ekvation: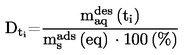
För tidsintervallet (Δti) gäller följande:
Mängden desorberat ämne beräknas för varje tidsintervall, enligt följande:
|
— |
för det första tidsintervallet Δt1 = t1-t0
|

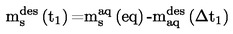
|
— |
för det andra Δt2 = t2-t1
|
 och
och
|
— |
för det n:te tidsintervallet Δtn = tn-tn-1
|
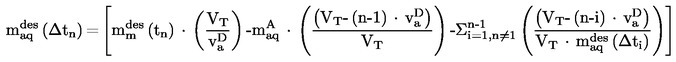 och
och
Till slut beräknas den procentuella desorptionen under varje tidsintervall  med hjälp av ekvationen
med hjälp av ekvationen
|
|
(24) |

och den procentuella desorptionen 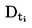 vid tidpunkten ti ges av ekvationen
vid tidpunkten ti ges av ekvationen
|
|
(25) |

där de ovan använda parametrarna definieras på följande sätt:
|
|
= |
massan av testämne som desorberas under tidsintervallen Δt1, Δ t2,… Δtn (μg), |
||
|
|
= |
massan av testämne som desorberas under tidsintervallen Δt1, Δt2,..., Δtn (μg); |
||
|
|
= |
= massan av ämne som uppmätts i ett delprov |
||
|
VT |
= |
totala volymen av vattenfas som är i kontakt med jorden under desorptionskinetiktestet enligt seriemetoden (cm3), |
||
|
|
= |
massan av testämne som blivit över efter uppnådd adsorptionsjämvikt på grund av ofullständig volymersättning (μg),
|
||
|
VR |
= |
volymen av supernatant som avlägsnats ur röret efter att adsorptionsjämvikt inträtt och som ersatts med samma volym av 0,01 M CaCl2-lösning (cm3) |
||
|
|
= |
volymen av det delprov som tagits för analys ur rör nummer i under desorptionskinetiktestet enligt seriemetoden (cm3).
|

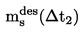
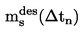






 vid tidpunkterna t
vid tidpunkterna t


Bilaga 6
TEST AV ADSORPTION OCH DESORPTION I JORDAR – RAPPORTBLANKETTER
Testämne:
Jord:
Jordens torrhalt (105 oC, 12 h): … %
Temperatur: … oC
Analysmetodens lämplighet
|
Uppvägt jordprov |
g |
|
|
Jordens torrmassa |
g |
|
|
CaCl2-lösningens volym |
cm3 |
|
|
Slutlösningens nominella koncentration |
μg cm-3 |
|
|
Slutlösningens analyserade koncentration |
μg cm-3 |
|
Princip för den använda analysmetoden:
Kalibrering av analysmetoden:
Testämne:
Jord:
Jordens torrhalt (105 oC, 12 h): … %
Temperatur: … oC
|
Analysmetod: |
Indirekt metod |
|
Parallellmetod |
|
Seriemetod |
|
|
|
Direkt metod |
|
|
|
|
|
Adsorptionstest: testprover
|
|
Beteckning |
Måttenhet |
Jämviktstid |
Jämviktstid |
Jämviktstid |
Jämviktstid |
||||
|
Rör nr |
|
|
|
|
|
|
|
|
|
|
|
Uppvägt jordprov |
— |
g |
|
|
|
|
|
|
|
|
|
Jordens torrmassa |
mjord |
g |
|
|
|
|
|
|
|
|
|
Uppvägda jordprovets vattenvolym (beräknat värde) |
VWS |
cm3 |
|
|
|
|
|
|
|
|
|
Volym av 0,01 M CaCl2-lösning för att få jorden i jämvikt |
|
cm3 |
|
|
|
|
|
|
|
|
|
Stamlösningens volym |
|
cm3 |
|
|
|
|
|
|
|
|
|
Totalvolym av vattenfas i kontakt med jord |
V0 |
cm-3 |
|
|
|
|
|
|
|
|
|
Testlösningens ursprungliga koncentration |
C0 |
μg cm-3 |
|
|
|
|
|
|
|
|
|
Testämnets massa vid testets början |
m0 |
μg |
|
|
|
|
|
|
|
|
|
Efter omröring och centrifugering |
||||||||||
|
INDIREKT METOD |
||||||||||
|
Parallellmetod |
||||||||||
|
Testämnets koncentration i vattenfasen (med blindprovskorrigering) |
|
μg cm-3 |
|
|
|
|
|
|
|
|
|
Seriemetod |
||||||||||
|
Massan av testämne som uppmätts i delprovet VA a |
|
μg |
|
|
|
|
|
|
|
|
|
DIREKT METOD |
||||||||||
|
Massan av testämne som adsorberats i jord |
|
μg |
|
|
|
|
|
|
|
|
|
Beräkning av adsorptionen |
||||||||||
|
Adsorption |
|
% |
|
|
|
|
|
|
|
|
|
|
|
% |
|
|
|
|
|
|
|
|
|
Medelvärde |
|
|
|
|
|
|
||||
|
Adsorptionskoefficient |
Kd |
cm3g-1 |
|
|
|
|
|
|
|
|
|
Medelvärde |
|
|
|
|
|
|
||||
|
Adsorptionskoefficient |
KOC |
cm3 g-1 |
|
|
|
|
|
|
|
|
|
Medelvärde |
|
|
|
|
|
|
||||
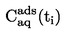




Testämne:
Jord:
Jordens torrhalt (105 oC, 12 h): … %
Temperatur: … oC
Adsorptionstest: blind- och kontrollprov
|
|
Beteckning |
Måttenhet |
Blindprov |
Blindprov |
Kontrollprov |
|||
|
Rör nr |
|
|
|
|
|
|
|
|
|
Uppvägt jordprov |
|
g |
|
|
|
|
0 |
0 |
|
Uppvägda jordprovets vattenvolym (beräknat värde) |
|
cm3 |
|
|
|
|
— |
— |
|
Tillsatt volym av 0,01 M CaCl2-lösning |
|
cm3 |
|
|
|
|
|
|
|
Tillsatt volym av testämnets stamlösning |
|
cm3 |
0 |
0 |
|
|
|
|
|
Vattenfasens totalvolym (beräknat värde) |
|
cm3 |
|
|
|
|
— |
— |
|
Testämnets ursprungliga koncentration i vattenfasen |
|
μg cm-3 |
|
|
|
|
|
|
|
Efter omröring och centrifugering |
||||||||
|
Koncentration i vattenfasen |
|
μg cm-3 |
|
|
|
|
|
|
Anm.: Lägg till kolumner efter behov.
Testämne:
Jord:
Jordens torrhalt (105 oC, 12 h): … %
Temperatur: … oC
Massbalans
|
|
Beteckning |
Måttenhet |
|
|
|
|
|
Rör nr |
|
|
|
|
|
|
|
Uppvägt jordprov |
— |
g |
|
|
|
|
|
Jordens torrmassa |
msoil |
g |
|
|
|
|
|
Uppvägda jordprovets vattenvolym (beräknat värde) |
VWS |
ml |
|
|
|
|
|
Volym av 0,01 M CaCl2-lösning för att få jorden i jämvikt |
|
ml |
|
|
|
|
|
Stamlösningens volym |
|
cm3 |
|
|
|
|
|
Total volym av vattenfas i kontakt med jord |
V0 |
cm3 |
|
|
|
|
|
Testlösningens ursprungliga koncentration |
C0 |
μg cm-3 |
|
|
|
|
|
Jämviktstid |
— |
h |
|
|
|
|
|
Efter omröring och centrifugering |
||||||
|
Testämnets koncentration i vattenfasen (med blindprovskorrigering) |
|
μg cm-3 |
|
|
|
|
|
Jämviktstid |
teq |
h |
|
|
|
|
|
l:a utspädning med lösningsmedel |
||||||
|
Avlägsnad volym av vattenfas |
Vrec |
cm3 |
|
|
|
|
|
Tillsatt volym av lösningsmedel |
ΔV |
cm3 |
|
|
|
|
|
l:a extraktion med lösningsmedel |
||||||
|
Signalutslag vid analys av lösningsmedlet |
SE1 |
varierande |
|
|
|
|
|
Testämnets koncentration i lösningsmedlet |
C E1 |
μg cm-3 |
|
|
|
|
|
Massan av ämne som extraherats från jord och kärlets väggar |
m E1 |
μg |
|
|
|
|
|
2:a utspädning med lösningsmedel |
||||||
|
Avlägsnad volym av lösningsmedel |
ΔVs |
cm3 |
|
|
|
|
|
Tillsatt volym av lösningsmedel |
AV |
cm3 |
|
|
|
|
|
2:a extraktion med lösningsmedel |
||||||
|
Signalutslag vid analys av lösningsmedlet |
SE2 |
varierande |
|
|
|
|
|
Testämnets koncentration i lösningsmedlet |
CE2 |
μg cm-3 |
|
|
|
|
|
Massan av testämne som extraherats frän jord och kärlets väggar |
mE2 |
μg |
|
|
|
|
|
Total massa av testämne som extraherats i två steg |
ME |
Mg |
|
|
|
|
|
Massbalans |
MB |
% |
|
|
|
|

Testämne:
Jord:
Jordens torrhalt (105 oC, 12 h): … %
Temperatur: … oC
Adsorptionsisotermer
|
|
Beteckning |
Måttenhet |
|
|
|
|
|
|
|
|
|
Rör nr |
|
|
|
|
|
|
|
|
|
|
|
Uppvägt jordprov |
— |
g |
|
|
|
|
|
|
|
|
|
Jordens torrmassa |
E |
g |
|
|
|
|
|
|
|
|
|
Uppvägda jordprovets vattenvolym (beräknat värde) |
VWS |
cm3 |
|
|
|
|
|
|
|
|
|
Volym av 0,01 M CaCl2-lösning för att få jorden i jämvikt |
|
cm3 |
|
|
|
|
|
|
|
|
|
Volym av tillsatt stamlösning |
|
cm3 |
|
|
|
|
|
|
|
|
|
Total volym av vattenfas i kontakt med jord (beräknat värde |
V0 |
cm3 |
|
|
|
|
|
|
|
|
|
Lösningens koncentration |
C0 |
μg cm-3 |
|
|
|
|
|
|
|
|
|
Jämviktstid |
— |
h |
|
|
|
|
|
|
|
|
|
Efter omröring och centrifiigering |
||||||||||
|
Testämnets koncentration i vattenfasen (med blindprovskorrigering) |
|
μg cm-3 |
|
|
|
|
|
|
|
|
|
Temperatur |
|
oC |
|
|
|
|
|
|
|
|
|
Adsorberad massa per enhet ord |
|
μg g-1 |
|
|
|
|
|
|
|
|


Regressionsanalys:
värdet på KF ads:
värdet på l/n:
regressionskoefficient r2:
Testämne:
Jord:
Jordens torrhalt (105 oC, 12 h): … %
Temperatur: … oC
|
Analysmetod: |
Indirekt metod |
|
Parallellmetod |
|
Seriemetod |
|
Desorptionstest
|
|
Beteckning |
Måttenhet |
Tidsintervall |
Tidsintervall |
Tidsintervall |
Tidsintervall |
|
|
Numret på röret som kommer från adsorptionssteget |
|
|
|
|
|
|
|
|
Massan av testämne som adsorberats i jord vid adsorptionsjämvikt |
|
μg |
|
|
|
|
|
|
Avlägsnad volym av vattenfas, ersatt med 0,01 M CaCl2 |
VR |
cm3 |
|
|
|
|
|
|
Total volym av vattenfas i kontakt med jord |
PM |
V0 |
cm3 |
|
|
|
|
|
SM |
VT |
cm3 |
|
|
|
|
|
|
Massan av testämne som blivit över efter uppnådd adsorptionsjämvikt på grund av ofullständig volymersättning |
|
μg |
|
|
|
|
|
|
Desorptionskinetik |
|||||||
|
Uppmätt massa av testämne som desorberats ur jorden vid tidpunkten ti |
|
μg |
|
|
|
|
|
|
Volym av lösning som tagits ur rör nummer i för uppmätning av testämnet |
PM |
Vf i |
cm3 |
|
|
|
|
|
SM |
va D |
cm3 |
|
|
|
|
|
|
Massan av testämne som desorberats ur jorden vid tiden ti (beräknat värde) |
|
μg |
|
|
|
|
|
|
Massan av testämne som desorberats ur jorden under tidsintervallet Δti (beräknat värde) |
|
μg |
|
|
|
|
|
|
Procentuell desorption |
|||||||
|
Dcsorption vid tidpunkten ti |
Dti |
% |
|
|
|
|
|
|
Desorption under tidsintervallet Δti |
|
% |
|
|
|
|
|
|
Skenbar desorptionskoefficient |
Kdes |
|
|
|
|
|
|


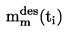

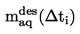
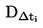
PM: Parallellmetod
SM: Seriemetod
C.19 UPPSKATTNING AV ADSORPTIONSKOEFFICIENTEN (KOC) I JORD OCH AVLOPPSSLAM MED ANVÄNDNING AV HÖGTRYCKSVÄTSKEKROMATOGRAFI (HPLC)
1. METOD
Denna metod är i stort sett identisk med OECD TG121 (2001).
1.1 INLEDNING
Sorptionsbeteendet hos ämnen i jord eller avloppsslam kan beskrivas med hjälp av parametrar som bestämts på experimentell väg med testmetod C.18. En viktig parameter är adsorptionskoefficienten. Den definieras som förhållandet mellan ämnets koncentration i jorden/slammet och ämnets koncentration i vattenfasen vid adsorptionsjämvikt. En adsorptionskoefficient som är normaliserad till jordens halt av organiskt bundet kol (KOC) är en värdefull indikator på en kemikalies bindningsförmåga till det organiska materialet i jord och avloppsslam och ger möjlighet att jämföra olika kemikalier. Denna parameter kan uppskattas genom korrelationer med vattenlösligheten och fördelningskoefficienten n-oktanol/vatten (1) (2) (3) (4) (5) (6) (7).
Vid den försöksmetod som beskrivs här används HPLC för uppskattning av adsorptionskoefficienten KOC i jord och avloppsslam (8). Dessa uppskattningar är tillförlitligare än resultaten från QSAR-beräkningar (9). En uppskattningsmetod kan aldrig helt ersätta mätningar vid jämvikt i uppslammat prov (testmetod C.18). Det uppskattade värdet på KOC kan dock användas som stöd vid valet av lämpliga testparametrar för adsorptions-/desorptionsundersökningar enligt testmetod C.l8, genom beräkning av Kd (distributionskoefficient) eller Kf (Freundlich-adsorptionskoefficient) enligt ekvation 3 (se avsnitt 1.2).
1.2 DEFINITIONER
Kd : Distributionskoefficienten definieras som förhållandet mellan jämviktkoncentrationerna C för ett upplöst testämne i ett tvåfassystem som består av sorbent (jord eller avloppsslam) och en vattenfas. Förhållandet har ingen måttenhet när koncentrationerna i båda faserna uttrycks som vikt per vikt. Om koncentrationen i vattenfasen ges som vikt per volym blir måttenheten ml-g-1. Kd kan variera beroende på sorbentens egenskaper och kan vara koncentrationsberoende.
|
|
(1) |

där:
|
Cjord |
= |
testämnets koncentration i jord vid jämvikt (μg· g-1) |
|
Cslam |
= |
testämnets koncentration i slam vid jämvikt (μg· g-1) |
|
Cvatten |
= |
testämnets koncentration i vattenfasen vid jämvikt (ug· g-1, μg· ml-1) |
Kf: Freundlich-adsorptionskoefficienten definieras som testämnets koncentration i jord eller avloppsslam (x/m) nar jämviktskoncentrationen Cvatten i vattenfasen har värdet ett. Måttenheten är μg· g-1 sorbent. Värdet kan variera beroende på sorbentens egenskaper.
|
|
(2) |
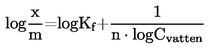
där:
|
x/m |
= |
mängden testämne x (μg) som adsorberats på mängden sorbent m (g) vid jämvikt |
|
l/n |
= |
lutningen hos Freundlich-adsorptionsisotermen |
|
Cvatten |
= |
testämnets koncentration i vattenfasen vid jämvikt (μg· ml-1) |
At
Koc: Distributionskoefficienten (Kd) eller Freundlich-adsorptionskoefficienten (Kt) normaliserad till sorbentens halt av organiskt kol (foc). Särskilt för icke-joniserade kemikalier utgör den en ungefärlig indikator på omfattningen av adsorption mellan ett ämne och sorbenten och ger möjlighet att göra jämförelser mellan olika kemikalier. Beroende på måttenheterna för Kd och Kf kan Koc sakna måttenhet eller ha måttenheten ml g-1 eller μg· g-1 organiskt material.
|
|
(3) |

Förhållandet mellan KOC och Kd är inte alltid linjärt och därför kan Koc-värdena variera mellan olika typer av jord men värdenas variabilitet är betydligt mindre jämfört med Kd- eller Kf-värden.
Adsorptionskoefficienten (Koc) härleds från kapacitetsfaktorn (k') med användning av en kalibreringskurva log k' mot log Koc för valda referensföreningar.
|
|
(4) |

där:
|
tR |
= |
HPLC retentionstiden för test- och referensämne (minuter) |
|
t0 |
= |
HPLC dödtid (minuter) (se avsnitt 1.8.2) |
Pow : Fördelningskoefficienten oktanol-vatten definieras som förhållandet mellan koncentrationerna för upplösta ämnen i n-oktanol och vatten. Värdet har ingen måttenhet.
|
|
(5) |
1.3 REFERENSÄMNEN
Strukturformeln, renheten och dissociationskonstanten (i tillämpliga fall) bör vara kända innan metoden används. Helst ska man också ha tillgång till information om lösligheten i vatten och organiska lösningsmedel, fördelningskoefficienten oktanol-vatten och hydrolysegenskaperna.
För att korrelera uppmätta HPLC-retentionsdata för ett testämne med ämnets adsorptionskoefficient Koc behövs en kalibreringskurva för log Koc. mot log k'. För kurvan krävs minst sex referenspunkter, av vilka minst en ligger ovanför och en under det förväntade värdet för testämnet. Metodens resultat blir betydligt mer exakt om man använder referensämnen som är strukturellt besläktade med testämnet. Om sådana data inte finns tillgängliga ligger valet av lämpliga kalibreringsämnen hos användaren. Då bör en mer allmän uppsättning av strukturellt heterogena ämnen väljas. I tabell 1 i bilagan görs en uppräkning av ämnen och Koc-värden som kan användas för avloppsslam och i tabell 3 ges motsvarande uppgifter för jord. Om andra kalibreringsämnen används måste detta motiveras.
1.4 PRINCIP FOR TESTMETODEN
HPLC utförs med analytiska kolonner som är packade med kommersiellt tillgänglig stationär fas av cyanopropyl med lipofila och polära grupper. Den stationära fasen är måttligt polär och baserad på en silikamatris, enligt följande:
|
–O – Si |
–CH2 – CH2 – CH2 |
–CN |
|
silika |
icke-polär bulk |
polär grupp |
Principen för testmetoden är densamma som för testmetod A.8 (Fördelningskoefficient, HPLC-metod). När testämnet passerar genom kolonnen tillsammans med den rörliga fasen växelverkar testämnet med den stationära fasen. Testämnet fördelas mellan den rörliga och stationära fasen och bromsas därför upp. I och med den stationära fasens sammansättning med både polära och icke-polära bindningsställen kan en molekyls polära och icke-polära grupper växelverka på liknande sätt som när det gäller organiskt material i jord eller i avloppsslam. Det betyder att man kan bestämma förhållandet mellan retentionstiden i kolonnen och adsorptionskoefficienten i organiskt material.
pH-värdet har stor inverkan på sorptionsbeteendet, särskilt när det gäller polära ämnen. I jordbruksjord eller tankar i avloppsreningsverk varierar pH-värdet i regel mellan 5,5 och 7,5. För joniserbara ämnen bör man utföra två test i lämpliga buffertlösningar där det ena testet utförs med joniserad form och det andra med ickejoniserad form, men endast i fall där minst 10 % av testämnet dissocieras inom pH-intervallet 5,5–7,5.
Eftersom endast förhållandet mellan retentionen i HPLC-kolonnen och adsorptionskoefficient används för utvärderingen behövs ingen kvantitativ analytisk metod och endast retentionstiden behöver bestämmas. Om en lämplig uppsättning referensämnen och standardbetingelser kan användas för testet ger denna metod tillgång till ett snabbt och effektivt sätt för bestämning av adsorptionskoefficienten Koc.
1.5 TESTETS TILLÄMPBARHET
HPLC-metoden kan användas på kemiska ämnen (märkta eller omärkta) för vilka det finns tillgång till ett lämpligt detektionssystem (t.ex. spektrofotometer, radioaktivitetsdetektor) och som är tillräckligt stabila under den tid testet varar. Testet kan vara särskilt användbart för kemikalier som är svåra att undersöka i andra försökssystem (t.ex. flyktiga ämnen, ämnen som inte är lösliga i vatten vid koncentrationer som kan mätas analytiskt, ämnen med en hög affinitet till ytan i inkubationssystem). Metoden kan användas för blandningar som ger olösta elutionsband. I sådana fall bör man ange log Koc-värdenas övre och undre gränser för testblandningens föreningar.
Orenheter kan i bland leda till problem vid tolkningen av HPLC-resultaten, men orenheterna har inte så stor betydelse så länge som testämnet klart kan identifieras analytiskt och separeras från orenheterna.
Metoden är validerad för de ämnen som räknas upp i tabell 1 i bilagan och metoden har också tillämpats på ett antal andra kemikalier i följande kemikalieklasser:
|
— |
Aromatiska aminer (t.ex. trifluralin, 4-kloranilin, 3,5-dinitroanilin, 4-metylanilin, N-metylanilin, 1-naftylamin). |
|
— |
Estrar av aromatiska karboxylsyror (t.ex. bensoesyrametylester, 3,5-dinitrobensoesyraetylester). |
|
— |
Aromatiska kolväten (t.ex. toluen, xylen, etylbensen, nitrobensen). |
|
— |
Aryloxifenoxipropionsyraestrar (t.ex. diklofop-metyl, fenoxaprop-etyl, fenoxaprop-P-etyl). |
|
— |
Bensimidazol- och imidazol-fungicider (t.ex. karbendazim, fuberidazol, triazoxid). |
|
— |
Karboxylsyraamider (t.ex. 2-klorbensamid, N,N-dimetylbcnsamid, 3,5-dinitrobensamid, N-metylbensamid, 2-nitrobensamid, 3-nitrobensamid). |
|
— |
Klorerade kolväten (t.ex. endosulfan, DDT, hexaklorbensen, quintozen, 1,2,3-triklorbensen). |
|
— |
Bekämpningsmedel typ organiska fosforföreningar (t.ex. azinfosmetyl, disulfoton, fenamifos, isofenfos, pyrazofos, sulprofos, triazofos). |
|
— |
Fenoler (t.ex. fenol, 2-nitrofenol, 4-nitrofenol, pentaklorofenol, 2,4,6-triklorfenol, 1-naftol). |
|
— |
Fenylurcaderivat (t.e.x. isoproturon, monolmuron, pencycuron). |
|
— |
Pigmentfärgämnen (t.ex. Acid Yellow 219, Basic Blue 41, Direct Red 81). |
|
— |
Polyaromatiska kolväten (t.ex. acenaften, naftalen). |
|
— |
1,3,5-triazin-herbicider (t.ex. prometryn, propazin, simazin, terbutryn). |
|
— |
Triazolderivat (t.ex. tebukonazol, triadimefon, tradimenol, triapentenol). |
Metoden kan inte tillämpas på ämnen som reagerar med eluenten eller den stationära fasen. Metoden är heller inte tillämplig på ämnen som har specifik växelverkan med oorganiska komponenter (t.ex. genom bildning av klusterkomplex med lermineraler). Metoden fungerar eventuellt inte för ytaktiva ämnen, oorganiska föreningar och måttliga eller starka organiska syror och baser. Log Koc-värden från 1,5 till 5,0 kan bestämmas. Joniserbara ämnen måste mätas med hjälp av en buffrad rörlig fas, men stor vikt måste fästas vid att undvika utfällning av buffertkomponenter eller testämne.
1.6 KVALITETSKRITERIER
1.6.1 Noggrannhet
I regel kan adsorptionskoefficient för ett testämne bestämmas inom ±0,5 logaritmiska enheter av det värde som kan bestämmas med mätningar vid jämvikt i uppslammat prov (se tabell 1 i bilagan). Noggrannheten förbättras om det finns tillgång till referensämnen som är strukturellt besläktade med testämnet.
1.6.2 Repeterbarhet
Bestämningarna bör köras minst två gånger. De log Koc-värden som härletts från individuella mätningar bör ligga inom ett område på 0,25 logaritmiska enheter.
1.6.3 Reproducerbarhet
De erfarenheter man hittills har från metoden ger belägg för metodens validitet. En undersökning av HPLC-metoden med 48 ämnen (i huvudsak bekämpningsämnen) för vilka det fanns tillgång till tillförlitliga data om Koc i jord gav en korrelationskoefficient på R = 0,95 (10) (11).
Ett jämförelsetest utfördes av elva deltagande laboratorier för att förbättra och validera metoden (12). Resultaten anges i tabell 2 i bilagan.
1.7 BESKRIVNING AV TESTMETODEN
1.7.1 Preliminär uppskattning av adsorptionskoefficienten
Fördelningskoefficienten oktanol-vatten Pow (= Kow) och, i viss utsträckning, vattenlösligheten kan användas som indikatorer för adsorptionens omfattning, särskilt för icke-joniserande ämnen. Det betyder att dessa indikatorer kan användas för preliminära orienterande test. Korrelationer för flera grupper av kemikalier finns angivna i ett antal publikationer (1) (2) (3) (4) (5) (6) (7).
1.7.2 Apparatur
För testet krävs en vätskekromatograf försedd med en pulsfri pump och lämplig detekteringsutrustning. Det är tillrådligt att använda en injektionsventil med en injektionsslinga. Kolonnen ska vara packad med kommersiellt tillgängligt kemiskt bundet cyanopropylharts på silikabas (t.ex. Hypersil och Zorbax CN). En guardkolonn av samma material kan placeras mellan injektionssystemet och analyskolonnen. Kolonner från olika tillverkare kan vara mycket olika när det gäller separationseffektivitet. Som riktvärden för den kapacitetsfaktor k' som bör uppnås gäller följande: log k' > 0,0 för log Koc = 3,0 och log k' > 0,4 för log Koc = 2,0 när man använder metanol/vatten 55/45 % som rörlig fas.
1.7.3 Rörlig fas
Flera olika rörliga faser har testats och följande två rekommenderas:
|
— |
metanol/vatten (55/45 % v/v) |
|
— |
methanol/0,01 M citratbuffert pH 6,0 (55/45 % v/v). |
För beredning av det eluerande lösningsmedlet används HPLC-klassificerad metanol och destillerat vatten eller citratbuffert. Blandningen avgasas före användningen. Isokratisk eluering bör användas. Om blandningar av metanol och vatten inte är lämpliga kan man försöka med andra blandningar av organiskt lösningsmedel och vatten, t.ex. etanol och vatten eller acetonitril och vatten. För joniserbara blandningar rekommenderas användning av buffertlösningar för stabilisering av pH. Stor vikt måste fästas vid att undvika saltutfällning och kolonnförsämring, vilket kan inträffa med vissa blandningar av organisk fas/buffert.
Inga tillsatser av typen jonparsreagenser bör användas, eftersom de kan påverka den stationära fasens sorptionsegenskaper. Sådana ändringar i den stationära fasen kan i vissa fall vara irreversibla. Därför måste alla försök där tillsatser används utföras med separata kolonner.
1.7.4 Upplösta ämnen
Test- och referensämnen upplöses i den rörliga fasen.
1.8 TESTETS UTFÖRANDE
1.8.1 Testbetingelser
Temperaturen under matningarna bör registreras. Det är högst tillrådligt att hålla kolonnen i temperaturkontrollerad miljö för att garantera konstanta betingelser under kalibrerings- och bestämningskörningarna och vid mätning av testämnet.
1.8.2 Bestämning av dödtiden t0
För bestämning av dödtiden t0 kan två olika metoder användas (se även avsnitt 1.2).
1.8.2.1 Bestämning av dödtiden t0 med användning av en homolog serie
Detta förfarande har visat sig ge tillförlitliga och standardiserade värden på t0. Närmare uppgifter finns i testmetod A.8 (Fördelningskoefficient (n-oktanol/vatten), HPLC-metoden).
1.8.2.2 Bestämning av dödtiden t0 med användning av inerta ämnen som inte bromsas upp i kolonnen
Denna teknik baserar sig på injicering av lösningar av formamid, urea eller natriumnitrat. Mätningarna bör utföras minst två gånger.
1.8.3 Bestämning av retentionstiderna tR
Referensämnena bör väljas enligt beskrivningen i avsnitt 1.3. Vid bestämning av retentionstiderna kan de injiceras som en blandad standard, förutsatt att det finns belägg för att retentionstiden för varje referensstandard inte påverkas av närvaron av de andra referensstandarderna. Kalibreringen bör utföras med regelbundna intervall minst två gånger dagligen så att man kan beakta inverkan av oväntade förändringar i kolonnens prestanda. Det bästa förfarandet är att göra injektionerna före och efter injektionerna av testämnet, för att bekräfta att retentionstiderna inte har förskjutits. Testämnena injiceras i så små mängder som möjligt (för att undvika överbelastning på kolonnen) och deras retentionstider bestäms.
Bestämningarna bör göras minst dubbelt för att öka mätningarnas tillförlitlighet. De värden på log Koc som härletts från individuella mätningar bör falla inom ett område på 0,25 logaritmiska enheter.
1.8.4 Utvärdering
Kapacitetsfaktorerna k' beräknas utifrån dödtiden t0 och retentionstiderna tR hos de valda referensämnena enligt ekvation 4 (se avsnitt 1.2). Referensämnenas värden på log k' plottas sedan mot deras värden på log Koc från mätningar vid jämvikt i uppslammat prov (se tabellerna 1 och 3 i bilagan). Med hjälp av den erhållna kurvan kan log k'-värdet för ett testämne därefter användas för att bestämma ämnets log Koc-värde. Om de aktuella resultaten visar att log Koc för testämnet ligger utanför kalibreringsområdet måste testet upprepas med användning av andra, lämpligare referensämnen.
2. DATA OCH RAPPORTERING
Rapporten måste innehålla följande uppgifter:
|
— |
Identifiering av test- och referensämnen och deras renhet samt, om det är relevant, pKa-värden. |
|
— |
Beskrivning av utrustningen och testbetingelserna, t.ex. analyskolonnens (och guardkolonnens) typ och mått, detekteringsmetoden, den rörliga fasen (komponenternas förhållande, pH), temperaturområdet under matningarna. |
|
— |
Dödtid och den metod som har använts för att bestämma den. |
|
— |
Mängderna test- och referensämnen som har tillförts kolonnen. |
|
— |
Retentionstiderna för de referensämnen som har använts för kalibrering. |
|
— |
Närmare uppgifter om den anpassade regressionslinjen (log k' mot log Koc) och en grafisk framställning av regressionslinjen. |
|
— |
Medelvärden för retentionsdata och uppskattat log Koc-värde för testämnet. |
|
— |
Kromatogram. |
3. HÄNVISNINGAR
|
1. |
W. J. Lyman, W. F. Reehl, D. H. Rosenblatt (ed). (1990), Handbook of chemical property estimation methods, Chap. 4, McGraw-Hill, New York. |
|
2. |
J. Hodson, N. A. Williams (1988), The estimation of the adsorption coefficient (Koc) for soils by HPLC. Chemosphere, 17, 167. |
|
3. |
G. G. Briggs (1981), Theoretical and experimental relationships between soil adsorption, octanol-water partition coefficients, water solubilities, bioconcentration factors, and the parachor. J. Agric. Food Chem., 29, s. 1050–1059. |
|
4. |
C. T. Chiou, P. E. Porter, D.W. Schmedding (1983), Partition equilibria of nonionic organic compounds between soil organic matter and water. Environ. Sci. Technol., 17, s. 227–231. |
|
5. |
Z. Gerstl, U. Mingelgrin (1984), Sorption of organic substances by soils and sediment. J. Environm. Sci. Health, B19, s. 297–312. |
|
6. |
C. T. Chiou, L. J. Peters, V. H. Freed (1979), A physical concept of soil water equilibria for nonionic organic compounds, Science, 106, s. 831–832. |
|
7. |
S. W. Karickhoff (1981), Semi-empirical estimation of sorption of hydrophobic pollutants on natural sediments and soils. Chemosphere, 10, s. 833–846. |
|
8. |
W. Kördel, D. Hennecke, M. Herrmann (1997), Application of the HPLC-screening method for the determination of the adsorption coefficient on sewage sludges. Chemosphere, 35(1/2), s. 121–128. |
|
9. |
M. Mueller, W. Kördel (1996), Comparison of screening methods for the estimation of adsorption coefficients on soil. Chemosphere, 32(12), s. 2493–2504. |
|
10. |
W. Kördel, J. Stutte, G. Kotthoff (1993), HPLC-screening method for the determination of the adsorption coefficient in soil-comparison of different stationary phases, Chemosphere, 27(12), s. 2341–2352. |
|
11. |
B. von Oepen, W. Kördel, W. Klein (1991), Sorption of nonpolar and polar compounds to soils: Processes, measurements and experience with the applicability of the modified OECD Guideline 106, Chemosphere, 22, s. 285–304. |
|
12. |
W. Kördel, G. Kotthoff, J. Müller (1995), HPLC-screening method for the determination of the adsorption coefficient on soil-results of a ring test. Chemosphere, 30(7), s. 1373–1384. |
Bilaga
Tabell 1
Jämförelse av Koc-värden för jord och avloppsslam och beräknade värden från HPLC-screeningmetoden (31), (32)
|
Ämne |
CAS-nr |
log Koc avloppsslam |
log Koc HPLC |
A |
log Koc jord |
HPLC |
A |
|
Atrazin |
1912-24-9 |
1,66 |
2,14 |
0,48 |
1,81 |
2,20 |
0,39 |
|
Linuron |
330-55-2 |
2,43 |
2,96 |
0,53 |
2,59 |
2,89 |
0,30 |
|
Fention |
55-38-9 |
3,75 |
3,58 |
0,17 |
3,31 |
3,40 |
0,09 |
|
Monuron |
150-68-5 |
1,46 |
2,21 |
0,75 |
1,99 |
2,26 |
0,27 |
|
Fenantren |
85-01-8 |
4,35 |
3,72 |
0,63 |
4,09 |
3,52 |
0,57 |
|
Bensoesyrafenylester |
93-99-2 |
3,26 |
3,03 |
0,23 |
2,87 |
2,94 |
0,07 |
|
Bensamid |
55-21-0 |
1,60 |
1,00 |
0,60 |
1,26 |
1,25 |
0,01 |
|
4-nitrobensamid |
619-80-7 |
1,52 |
1,49 |
0,03 |
1,93 |
1,66 |
0,27 |
|
Acetanilid |
103-84-4 |
1,52 |
1,53 |
0,01 |
1,26 |
1,69 |
0,08 |
|
Anilin |
62-53-3 |
1,74 |
1,47 |
0,27 |
2,07 |
1,64 |
0,43 |
|
2,5-dikloranilin |
95-82-9 |
2,45 |
2,59 |
0,14 |
2,55 |
2,58 |
0,03 |
Tabell 2
Resultat av jämförelsetest mellan laboratorier (11 deltagande laboratorier) som har utförts för att förbättra och validera HPLC-metoden (33)
|
Ämne |
CAS-nr |
log Koc |
Koc |
Log Koc |
|
(OECD 106) |
(HPLC-metodcn) |
|||
|
Atrazin |
1912-24-9 |
1,81 |
78 ± 16 |
1,89 |
|
Monuron |
150-68-5 |
1,99 |
100 ± 8 |
2,00 |
|
Triapentenol |
77608-88-3 |
2,37 |
292 ± 58 |
2,47 |
|
Linuron |
330-55-2 |
2,59 |
465 ± 62 |
2,67 |
|
Fention |
55-38-9 |
3,31 |
2062 ± 648 |
3,31 |
Tabell 3
Rekommenderade referensämnen för HPLC-screeningmetoden på grundval av jordadsorptionsdata
|
Referensamne |
CAS-nr |
Log Koc-medelvär-den från matningar vid jamvikt i uppslammat prov |
antal Koc-data |
Log S.D. |
Källa |
|
Acetanilid |
103-84-4 |
1,25 |
4 |
0,48 |
|
|
Fenol |
108-95-2 |
1,32 |
4 |
0,70 |
|
|
2-nitrobensamid |
610-15-1 |
1,45 |
3 |
0,90 |
|
|
N, N-dimetylbensamid |
611-74-5 |
1,52 |
2 |
0,45 |
|
|
4-metylbensamid |
619-55-6 |
1,78 |
3 |
1,76 |
|
|
Metylbensoat |
93-58-3 |
1,80 |
4 |
1,08 |
|
|
Atrazin |
1912-24-9 |
1,81 |
3 |
1,08 |
|
|
Isoproturon |
34123-59-6 |
1,86 |
5 |
1,53 |
|
|
3-nitrobensamid |
645-09-0 |
1,95 |
3 |
1,31 |
|
|
Anilin |
62-53-3 |
2,07 |
4 |
1,73 |
|
|
3,5-dinitrobensamid |
121-81-3 |
2,31 |
3 |
1,27 |
|
|
Karbendazim |
10605-21-7 |
2,35 |
3 |
1,37 |
|
|
Triadimenol |
55219-65-3 |
2,40 |
3 |
1,85 |
|
|
Triazoxid |
72459-58-6 |
2,44 |
3 |
1,66 |
|
|
Triazofos |
24017-47-8 |
2,55 |
3 |
1,78 |
|
|
Linuron |
330-55-2 |
2,59 |
3 |
1,97 |
|
|
Naftalen |
91-20-3 |
2,75 |
4 |
2,20 |
|
|
Endosulfandiol |
2157-19-9 |
3,02 |
5 |
2,29 |
|
|
Metiokarb |
2032-65-7 |
3,10 |
4 |
2,39 |
|
|
Acid Yellow 219 |
63405-85-6 |
3,16 |
4 |
2,83 |
|
|
1,2,3-triklorbensen |
87-61-6 |
3,16 |
4 |
1,40 |
|
|
γ-HCH |
58-89-9 |
3,23 |
5 |
2,94 |
|
|
Fention |
55-38-9 |
3,31 |
3 |
2,49 |
|
|
Direct Red 81 |
2610-11-9 |
3,43 |
4 |
2,68 |
|
|
Pyrazofos |
13457-18-6 |
3,65 |
3 |
2,70 |
|
|
a-endosulfan |
959-98-8 |
4,09 |
5 |
3,74 |
|
|
Diklofop-metyl |
51338-27-3 |
4,20 |
3 |
3,77 |
|
|
benantren |
85-01-8 |
4,09 |
4 |
3,83 |
|
|
Basic Blue 41 (blandning) |
26850-47-5 12270-13-2 |
4,89 |
4 |
4,46 |
|
|
DDT |
50-29-3 |
5,63 |
|
— |
C.20 REPRODUKTIONSTEST PA DAPHNIA MAGNA
1. METOD
Denna testmetod för reproduktionstoxicitet motsvarar OECD TG 211 (1998).
1.1 INLEDNING
Det primära syftet med detta test är att bedöma kemikaliers verkningar på reproduktionskapaciteten hos Daphnia magna.
1.2 DEFINITIONER OCH ENHETER
Föräldrageneration: de Daphnia-honor som finns närvarande vid testets början och vars reproduktionskapacitet testas.
Avkomma: de unga Daphnia som produceras av föräldragenerationen under testets gång.
Lägsta koncentration vid vilken verkningar observeras (Lowest observed effect concentration, LOEC): den lägsta testkoncentrationen vid vilken ämnet inom en fastställd exponeringsperiod framkallar statistiskt signifikanta verkningar på reproduktion och på föräldragenerationens mortalitet (vid p < 0,05) jämfört med kontrollgruppen. Alla testkoncentrationer ovanför LOEC måste framkalla skadliga verkningar som är lika stora eller större än de verkningar som observeras vid LOEC. Om dessa två villkor inte uppfylls måste en fullständig förklaring ges till hur man har valt LOEC (och NOEC).
Koncentration vid vilken inga verkningar observeras (No observed effect concentration, NOEC): den testkoncentration omedelbart under LOEC som inom en fastställd exponeringsperiod inte framkallar några statistiskt signifikanta verkningar (p < 0,05) jämfört med kontrollgruppen.
ECX: den koncentration av testämnet upplöst i vatten som inom en fastställd exponeringsperiod leder till en minskning på x % av reproduktionskapaciteten hos Daphnia magna.
Inneboende tillväxthastighet: ett mått på populationens tillväxt vilket omfattar reproduktionskapacitet och åldersspecifik mortalitet (20) (21) (22). I steady state-populationer är värdet på detta mått noll. För växande populationer är värdet positivt och för minskande populationer är värdet negativt. Det senare är inte hållbart och leder i slutändan till utrotning.
Detektionsgräns: den lägsta koncentration som kan detekteras men inte kvantifieras.
Bestämningsgräns: den lägsta koncentration som kan mätas kvantitativt.
Mortalitet: ett djur registreras som dött när det är orörligt, dvs. när det inte kan simma eller om man inte kan observera rörelser hos bihang eller bakkropp inom 15 sekunder efter en försiktig omrörning i testkärlet. (Eventuell annan definition som används måste rapporteras tillsammans med motsvarande hänvisning.)
1.3 PRINCIP FÖR TESTMETODEN
Unga honor av Daphnia (föräldragenerationen) som är yngre än 24 timmar vid testets början exponeras för testämnet som tillsätts vatten i en serie koncentrationer. Testperiodens längd är 21 dagar. Vid testets slut görs en bedömning av mängden levande avkomma per föräldrageneration. Sådan avkomma som producerats under testet men som dör under testets gång räknas inte med. Föräldragenerationens reproduktionskapacitet kan uttryckas på andra sätt (t.ex. mängden avkomma som producerats per djur per dag från och med första dagen då avkomma observerades). Sådana andra sätt måste i förekommande fall rapporteras separat, utöver varje föräldragenerations totala mängd levande avkomma vid testets slut. Reproduktionskapaciteten hos djur som exponeras för testämnet jämförs med kontrollgruppens (kontrollgruppernas) reproduktionskapacitet i syfte att fastställa den lägsta koncentration vid vilken verkningar observeras (LOEC) och den koncentration vid vilken inga verkningar observeras (NOEC). Dessutom bör data i så stor utsträckning det är möjligt analyseras med en regressionsmodell i syfte att uppskatta den koncentration som skulle leda till en minskning på x % av reproduktionskapaciteten (t.ex. EC50, EC20 eller EC10).
Föräldragenerationens överlevnad och tiden fram till produktion av den första satsen avkomma måste också rapporteras. Övriga testämnesrelaterade verkningar på parametrar såsom tillväxt (t.ex. längd) och eventuellt inneboende tillväxthastighet kan också undersökas.
1.4 INFORMATION OM TESTÄMNET
Resultat frän test avseende akut toxicitet (se metod C.2, del I) som utförts på Daphnia magna bör finnas tillgängliga. Resultaten kan vara användbara vid valet av lämpliga testkoncentrationer för reproduktionstest. Data om testämnets vattenlöslighet och ångtryck bör vara kända och man bör ha tillgång till en tillförlitlig analysmetod för kvantifiering av ämnet i testlösningarna med rapporterad återhämtningsgrad och bestämningsgräns.
För definition av testförhållandena bör man ange bl.a. testämnets strukturformel, renhet, stabilitet i ljus, stabilitet vid testförhållandena, pKa, Pow och resultaten frän test för biologisk lättnedbrytbarhet (se metod C.4).
1.5 TESTETS GILTIGHET
För att ett test ska vara giltigt måste följande kriterier uppfyllas för kontrollgruppen (kontrollgrupperna):
|
— |
Föräldragenerationens mortalitet (honor av Daphnia) får inte överskrida 20 % vid testets slut. |
|
— |
Det genomsnittliga antalet avkommor som har producerats per föräldradjur och som lever vid testets slut måste vara > 60. |
1.6 BESKRIVNING AV TESTMETODEN
1.6.1 Utrustning
Testkärl och annan apparatur som kommer i kontakt med testämnet bör vara tillverkade av glas eller annat kemiskt inert material. I regel används glasbägare.
Dessutom behövs åtminstone en del av följande utrustning:
|
— |
Syrgasmätare (med mikroelektrod eller annan lämplig anordning för mätning av löst syrgas i lågvolymsprover). |
|
— |
Utrustning för temperaturreglering. |
|
— |
pH-meter. |
|
— |
Utrustning för bestämning av vattnets hårdhetsgrad. |
|
— |
Utrustning för bestämning av totalt organiskt kol (TOC) i vattnet eller utrustning för bestämning av kemisk syreförbrukning (COD). |
|
— |
Apparatur för reglering av belysningen och mätning av ljusets intensitet. |
1.6.2 Testorganism
Testet utförs på Daphnia magna Straus. Andra Daphnia-arter kan användas förutsatt att de uppfyller tillämpliga giltighetskriterier (ett relevant giltighetskriterium är reproduktionskapaciteten i kontrollgrupperna). Om andra arter av Daphnia används måste de identifieras ordentligt och det ska motiveras varför de används.
Klonen ska helst identifieras med hjälp av genotypen. Forskningsresultat (1) har visat att reproduktionskapaciteten hos klon A (som härrör från IRCHA i Frankrike) (3) genomgående uppfyller giltighetskriteriet som gäller ett genomsnitt på > 60 överlevande avkommor per föräldradjur vid odling i de betingelser som beskrivs i denna metod. Andra kloner kan godtas förutsatt att Daphnia-odlingen kan påvisas uppfylla giltighetskriterierna för ett test.
Vid testets början måste djuren vara yngre än 24 timmar och får inte tillhöra den första satsen avkomma. Djuren måste komma från en frisk stam (dvs. de får inte uppvisa tecken på stress, t.ex. hög mortalitet, förekomst av hanar och ephippia. fördröjd produktion av första satsen avkomma, missfärgade djur osv.). Stamdjuren måste hållas vid samma odlingsbetingelser (ljus, temperatur, medium, utfodring och djur per volymenhet) som används vid testet. Om annat än normalt odlingsmedium för Daphnia används vid testet är det tillrådligt med en acklimatiseringsperiod på i regel cirka tre veckor (dvs. en generation) innan testets inleds, för att undvika stress på föräldragenerationen.
1.6.3 Testmedium
För testet rekommenderas ett fullständigt definierat medium. Avsaknaden av tillsatser (t.ex. alger, jordextrakt osv.) som är svåra att karakterisera möjliggör bättre standardisering mellan laboratorier. Elendt M4- (4) och M7-medier (se bilaga 1) har konstaterats vara lämpliga för ändamålet. Andra medier (se t.ex. (5) och (6)) kan godtas förutsatt att det kan visas att Daphnia-odlingen uppfyller giltighetskriterierna för testet.
Vid användning av medier som innehåller icke definierade tillsatser ska dessa tillsatser klart specificeras och uppgifter om deras sammansättning lämnas i testrapporten. Det är särskilt viktigt att ange kolhalten, eftersom kol kan utgöra ett fodertillskott. Det rekommenderas att totalt organiskt kol (TOC) och/eller kemisk syreförbrukning (COD) bestäms i en stamberedning av den organiska tillsatsen och att en uppskattning görs av hur denna påverkar TOC/COD i testmediet. TOC-nivån i mediet (dvs. före tillsats av algerna) bör vara lägre än 2 mg/l (7).
Vid testning av ämnen som innehåller metaller är det viktigt att beakta det faktum att testmediets egenskaper (t.ex. hårdhet och kelateringsförmåga) kan ha inverkan på testämnets toxicitet. Därför rekommenderas ett fullständigt definierat medium. För närvarande är Elendt M4 och M7 de enda medier som veterligen är lämpliga för långtidsodling av Daphnia magna. Båda medierna innehåller kelatbildaren EDTA. Det finns resultat (2) som tyder på att toxiciteten hos kadmium i regel förefaller vara lägre när reproduktionstestet genomförs i M4- och M7-medier än i när det genomförs i medier som inte innehåller EDTA. M4 och M7 rekommenderas därför inte för testning av ämnen som innehåller metaller. Likaså bör andra medier som innehåller kända kelatbildare undvikas. I fråga om metallhaltiga ämnen kan det vara tillrådligt att använda ett alternativt medium såsom ASTM-rekonstruerat hårt sötvatten (7) som inte innehåller EDTA, med tillsats av algextrakt (8). Kombinationen av ASTM-rekonstruerat hårt sötvatten och algextrakt är också lämplig för långtidsodling och testning av Daphnia magna (2), även om det fortfarande förekommer en svag keleringseffekt på grund av den organiska komponenten i det tillförda algextraktet.
Vid testets början och under testets gång bör halten upplöst syrgas ligga över 3 mg/l. pH-värdet bör ligga inom området 6–9 och bör i regel inte variera med mer än 1,5 enheter inom ett test. En vattenhårdhet över 140 mg/l (som CaCO3) rekommenderas. Vid test med denna nivå och över har man kunnat observera en reproduktionskapacitet som uppfyller giltighetskriterierna (9) (10).
1.6.4 Testlösningar
Testlösningar av de valda koncentrationerna bereds i regel genom utspädning av en stamlösning. Stamlösningarna bör helst beredas genom upplösning av ämnet i testmediet.
I vissa fall behövs organiska lösningsmedel eller dispergeringsmedel för att få en stamlösning av lämplig koncentration, men användningen av sådana hjälpämnen bör undvikas med alla medel. Lämpliga lösningsmedel är t.ex. aceton, etanol, metanol, dimetylformamid och trietylenglykol. Exempel på lämpliga dispergeringsmedel är Cremophor RH40, metylcellulosa (0,01 %) och HCO-40. Testämnets halt i testlösningarna bör under inga omständigheter överskrida lösligheten i testmediet.
Lösningsmedel används för att producera en stamlösning som kan doseras exakt i vatten. Vid den rekommenderade lösningsmedelskoncentrationen i det slutliga testmediet (< 0,1 ml/l) är de ovan uppräknade lösningsmedlen inte toxiska och ökar inte ämnets vattenlöslighet.
Dispergeringsmedel kan underlätta dispergering och exakt dosering. Vid den rekommenderade koncentrationen i det slutliga testmediet (≤ 0,1 ml/l) är de ovan uppräknade dispergeringsmedlen inte toxiska och ökar inte ämnets vattenlöslighet.
1.7 TESTETS UTFORMNING
Fördelningen av behandlingar och den efterföljande hanteringen av testkärlen bör vara slumpmässig. I annat fall kan man få missvisande resultat som kan tolkas som en koncentrationsrelaterad verkan. Särskilt om testenheterna hanteras i koncentrationsordning kan vissa tidsrelaterade faktorer, såsom trötthet hos personalen eller andra felfaktorer, leda till större verkningar vid de högre koncentrationerna. Om testresultaten sannolikt kommer att påverkas av omständigheter i början av testet eller av miljöbetingelser, såsom placering i laboratoriet, bor man överväga blockuppdelning.
1.8 FÖRFARANDE
1.8.1 Exponeringsförhållanden
1.8.1.1 Testperiodens längd
Testperiodens längd är 21 dagar.
1.8.1.2 Djurtäthet
Föräldradjuren hålls individuellt, ett djur per testkärl, med 50–100 ml medium per kärl.
I vissa fall behövs större volymer med tanke på de krav som gäller för den analytiska procedur som används för bestämning av testämnets koncentration, även om det för den kemiska analysen är tillåtet med pooling av replikat. Om volymer över 100 ml används måste eventuellt den dos som ges till Daphnia ökas för att garantera tillräcklig tillgång på foder och förenlighet med giltighetskriterierna. Vid genomflödestest kan alternativ testutformning övervägas av tekniska skäl (t.ex. fyra grupper med 10 djur per grupp i en större volym), men varje ändring av testutformningen måste rapporteras.
1.8.1.3 Antalet djur
För halvstatiskt test behövs minst 10 djur per testkoncentration och minst 10 individuellt hållna djur i kontrollserierna.
För genomflödestest har en uppdelning med 40 djur i fyra grupper om 10 djur per testkoncentration visat sig vara lämplig (1). Ett mindre antal testorganismer kan användas och man rekommenderar ett minimum på 20 djur per koncentration uppdelade i två eller flera replikat med samma antal djur (t.ex. fyra replikat med fem Daphnia per replikat). I fråga om test där djuren hålls i grupper bör man dock beakta att reproduktionskapaciteten inte kan uttryckas som totalantalet överlevande avkommor per föräldradjur om föräldradjur dör. I sådana fall måste reproduktionskapaciteten uttryckas som ”totalantalet överlevande avkommor som har producerats per föräldradjur som fanns närvarande vid testets början”.
1.8.1.4 Utfodring
Vid halvstatiskt test ges foder helst dagligen men minst tre gånger per vecka (vid byte av medium). Avvikelser från detta (t.ex. vid genomflödestest) måste rapporteras.
Under testet bör föräldradjuren helst utfodras med levande algceller. En eller flera av följande arter kan användas: Chlorella sp. Selenaslrum capricornutum (numera Pseudokirchneriella subcapitata (11)) och Scenedesmus subspicatus. Utfodringen bör grunda sig på mängden organiskt kol (C) som ges till varje föräldradjur. Forskningsresultat (12) för Daphnia magna har visat att det är tillräckligt med dosnivåer mellan 0,1 och 0,2 mg organiskt kol per Daphnia och dag för att nå upp till den mängd avkomma som behövs med tanke på giltighetskriterierna för testet. Dosen kan ges med konstant dosering under hela testet eller, om så önskas, med lägre dosering i början och därefter ökande dosering i takt med föräldradjurens tillväxt. I det senare fallet bör dosen ändå alltid hållas inom det rekommenderade området 0,1–0,2 mg organiskt kol per Daphnia och dag.
Om andra matningssätt (t.ex. antalet algceller eller ljusabsorbans) används för att kontrollera utfodringens dosnivå (av bekvämlighetsskäl, eftersom mätning av kolhalten är tidskrävande) måste varje laboratorium upprätta egna nomogram för mätningarna av algodlingens kolhalt (i bilaga 2 finns anvisningar för hur man upprättar nomogram). Nomogrammen måste ses över minst årligen, och oftare om algodlingsbetingelserna har förändrats. Ljusabsorbans har konstaterats vara en bättre metod än cellräkning när det gäller bestämning av kolhalten (13).
Algsuspensionen som används för utfodring av Daphnia bör vara koncentrerad, så att minimal volym algodlingsmedium överförs till testkärlen. För att höja algkoncentrationen kan man använda centrifugering med påföljande återsuspension i destillerat vatten, avjoniserat vatten eller Daphnia-odlingsmedium.
1.8.1.5 Ljus
Ljuset hälls på i 16 timmar med en intensitet som inte överskrider 15–20 μE· m-2 · s-1.
1.8.1.6 Temperatur
Temperaturen i testmediet bör hållas inom området 18–22 oC. Inom ett och samma försök får temperaturen i mån av möjlighet inte variera med mer än 2 oC inom de angivna gränserna (t.ex. 18–20, 19–21 eller 20–22 oC). Temperaturövervakningen kan gärna ordnas med hjälp av ett extra testkärl.
1.8.1.7 Luftning
Testkärlen får inte luftas under testet.
1.8.2 Testkoncentrationer
I regel används minst fem testkoncentrationer. Koncentrationerna bör ligga i en geometrisk serie med en separationsfaktor som helst inte bör överskrida 3,2. Dessutom bör ett lämpligt antal replikat användas för varje testkoncentration (se avsnitt 1.8.1.3). Om mindre än fem koncentrationer används måste detta motiveras. Ämnen får inte testas i koncentrationer som överskrider löslighetsgränsen i testmediet.
Vid valet av koncentrationsområde bör man beakta följande:
|
i) |
Om syftet är att få värden på LOEC eller NOEC måste den lägsta testkoncentrationen vara tillräckligt låg för att fertiliteten vid den koncentrationen inte ska vara signifikant lägre än i kontrollgruppen. I annat fall måste testet upprepas med en lägre lägsta koncentration. |
|
ii) |
Om syftet är att få värden på LOEC eller NOEC måste den högsta testkoncentrationen vara tillräckligt hög för att fertiliteten vid den koncentrationen ska vara signifikant lägre än i kontrollgruppen. I annat fall måste testet upprepas med en högre högsta koncentration. |
|
iii) |
Om man bestämmer ECX för verkningar på reproduktionskapaciteten är det tillrådligt att använda tillräckligt antal koncentrationer för att möjliggöra bestämning av ECx med tillräcklig konfidensnivå. Om man bestämmer EC5() för verkningar på reproduktionskapaciteten är det tillrådligt att den högsta testkoncentrationen är högre än detta värde på EC50. I annat fall blir konfidensintervallet för EC50 mycket brett, även om det fortfarande är möjligt att bestämma värdet på EC50, och då blir det kanske inte möjligt att få en tillfredsställande bedömning av den anpassade modellens lämplighet. |
|
iv) |
Testkoncentrationsområdet bör helst inte innehålla koncentrationer som framkallar en statistiskt signifikant verkan på vuxna exemplars överlevnad. I ett sådant fall övergår nämligen testets natur från ett enkelt reproduktionstest till ett kombinerat reproduktions- och mortalitetstest, för vilket det krävs en mycket mer komplex statistisk analys. |
Tidigare kunskap om testämnets toxicitet (t.ex. från test avseende akut toxicitet eller preliminära test) är till hjälp vid valet av lämpliga testkoncentrationer.
Om ett lösnings- eller dispergeringsmedel används för att underlätta beredningen av testlösningar (se avsnitt 1.6.4) får slutkoncentrationen i testkärlet inte överskrida 0,1 ml/l och samma slutkoncentration bör användas i alla testkärl.
1.8.3 Kontrollserier
En kontrollserie för testmediet och i förekommande fall en kontrollserie med lösnings- eller dispergeringsmedlet bör köras utöver testserierna. Kontrollseriernas lösningsmedel eller dispergeringsmedel bör ha samma koncentration som i testserierna. Lämpligt antal replikat bör användas (se avsnitt 1.8.1.3).
I ett väl utfört test bör variationskoefficienten kring det genomsnittliga antalet levande avkommor som i kontrollgrupperna produceras per föräldradjur i regel vara 25 %. Värdet måste rapporteras i testutformningar där djuren hålls individuellt.
1.8.4 Förnyelse av testmediet
Frekvensen för förnyelse av testmediet beror på testämnets stabilitet, men får inte underskrida tre gånger i veckan. Om resultaten från preliminära stabilitetstest (se avsnitt 1.4) visar att testämnet inte hålls stabilt (dvs. utanför området 80–120 % av nominell koncentration eller under 80 % av uppmätt startkoncentration) under det maximala förnyelseintervallet (dvs. tre dagar) bör man överväga förnyelse med kortare intervall eller användning av genomflödestest.
Vid halvstatiskt test görs förnyelse av mediet genom att en ny serie testkärl ställs i ordning och föräldradjuren överförs till de nya kärlen t.ex. med en glaspipett av lämplig diameter. Volymen medium som överförs med Daphnia bör vara så liten som möjligt.
1.8.5 Observationer
Resultaten av de observationer som görs under testet registreras på blanketter (se exempel i bilagorna 3 och 4). Om andra mätningar krävs (se 1.3 och 1.8.8) behövs eventuellt tilläggsobservationer.
1.8.6 Avkomma
Det rekommenderas att avkommorna från varje föräldradjur tas bort och räknas dagligen efter det att produktionen av avkomma har satt i gäng, för att hindra avkommorna från att konsumera foder som är avsett för vuxna exemplar. I test enligt denna metod behöver bara levande avkommor räknas, men förekomsten av aborterade ägg eller döda avkommor bör registreras.
1.8.7 Mortalitet
Mortaliteten bland föräldradjuren bör registreras, helst dagligen och minst varje gång avkommorna räknas.
1.8.8 Övriga parametrar
Även om denna metod främst är avsedd för bedömning av verkningarna på reproduktionskapaciteten är det möjligt att övriga verkningar kan kvantifieras i tillräcklig grad med tanke på statistisk analys. Tillväxtmätningar är mycket önskvärda eftersom de ger information om eventuella subletala verkningar. Sådan information kan vara mer användbar än informationen från rena reproduktionsmätningar. Mätning av föräldradjurens längd (t.ex. kroppslängd exklusive analfenan) vid testets slut rekommenderas. Övriga parametrar som kan mätas eller beräknas är tiden fram till att den första satsen avkomma (och efterföljande satser) produceras, satsernas antal och storlek per djur, antalet aborterade satser, förekomsten av hanar eller ephippia och den inneboende tillväxthastigheten.
1.8.9 Frekvensen för analytiska bestämningar och mätningar
Syrgashalten, temperaturen, hårdheten och pH bör mätas minst en gång i veckan i färska och gamla medier, i kontrollsatserna och i den högsta testämneskoncentrationen.
Under testets gång bor testämnets koncentrationer bestämmas med regelbundna intervall.
Vid halvstatiskt test, där testämnets koncentration bör hållas inom 20 % av den nominella koncentrationen (dvs. inom området 80–120 %, se avsnitten 1.4 och 1.8.4), rekommenderas att de högsta och lägsta testkoncentrationerna analyseras minst en gång under testets första vecka, strax efter beredningen och vid förnyelse (analyserna görs alltså på ett prov av samma lösning – när den är nyberedd och när den förnyas). Dessa bestämningar upprepas därefter minst varje vecka.
Vid test där testämnets koncentration inte förväntas hållas inom ± 20 % av den nominella koncentrationen är det nödvändigt att analysera alla testkoncentrationer, när de är nyberedda och när de förnyas. Vid test där testämnets uppmätta startkoncentration inte ligger inom ± 20 % av den nominella koncentrationen men det finns tillräckliga bevis på att startkoncentrationerna är repeterbara och stabila (dvs. inom området 80–120 % av startkoncentrationerna), kan de kemiska bestämningarna under testets vecka 2 och 3 begränsas till de högsta och lägsta testkoncentrationerna. Under alla omständigheter behöver bestämning av testämnets koncentration före förnyandet bara göras för ett enda replikatkärl per testämneskoncentration.
Vid genomflödestest kan man lämpligen använda samma provtagningsschema som vid halvstatiska test (även om mätningen av ”gammal” lösning då inte är tillämplig). Det kan dock vara tillrådligt att utöka antalet provtagningstillfallen under den första veckan (t.ex. tre mätningar) som kontroll på att testkoncentrationerna hålls stabila. Vid dessa typer av försök bör strömningshastigheten för spädningsmedel och testämne kontrolleras dagligen.
Om det däremot finns bevis på att testämnets koncentration i lösningen har hållits tillfredsställande inom ± 20 % av den nominella eller uppmätta startkoncentrationen under hela testperioden, kan nominella eller uppmätta startvärden användas som grund för resultaten. Om avvikelsen från nominell eller uppmätt startkoncentration är större än 20 % bör resultaten uttryckas i form av tidsvägda medelvärden (se bilaga 5).
2. DATA OCH RAPPORTERING
2.1 BEHANDLING AV RESULTATEN
Syftet med testet är att bestämma hur testämnet påverkar antalet levande avkommor som produceras av varje föräldradjur som har överlevt testet. Totalantalet avkommor per föräldradjur beräknas för varje testkärl (varje replikat). Om föräldradjuret i något av replikaten dör under testet eller visar sig vara en hane, tas replikatet i fråga inte med i analysen. I ett sådant fall baserar sig analysen på ett reducerat antal replikat.
För uppskattning av LOEC och NOEC med avseende på kemikaliens verkan på reproduktionskapaciteten är det nödvändigt att beräkna den genomsnittliga reproduktionskapaciteten för replikaten per koncentration och den resterande standardavvikelsen. Detta kan göras med variansanalys (ANOVA). Medelvärdet för varje koncentration bör därefter jämföras med kontrollsatsen med hjälp av en lämplig metod för multipeljämförelse. Dunnetts eller Williams test kan användas för ändamålet (14) (15) (16) (17). Det är nödvändigt att kontrollera hållbarheten hos ANOVA:s antagande om variansens homogenitet. Det är tillrådligt att detta görs grafiskt, hellre än genom ett test avseende formell signifikans (18). Ett lämpligt alternativ är att köra ett Bartlett-test. Om antagandet inte är hållbart måste man överväga att transformera data för att homogenisera varianserna innan ANOVA utförs, eller att utföra viktad ANOVA. Storleken på den verkan som kan detekteras med ANOVA (dvs. den minst signifikanta skillnaden) beräknas och rapporteras.
För uppskattningen av den koncentration som skulle orsaka en minskning på 50 % av reproduktionskapaciteten (dvs. EC50) bör en lämplig kurva, t.ex. den logistiska kurvan, anpassas till data med hjälp av en statistisk metod. t.ex. minsta kvadrat-metoden. Kurvan bör parametriseras så att EC50 och standardfelet kan uppskattas direkt. Därigenom underlättas beräkningen av konfidensintervallen kring EC50. Tvåsidiga konfidensintervall på 95 % bör tillämpas, utom om det finns goda skäl för att välja andra konfidensnivåer. Anpassningsproceduren bör helst ge tillgång till ett medel för bedömning av signifikansen eller bristen på signifikans. Detta kan göras grafiskt eller genom att dela kvadraternas restsumma i ”brist på passning” och ”rena felkomponenter” och utföra ett signifikanstest avseende brist på passning. När man jämför behandlingar som ger låg fertilitet med behandlingar som ger hög fertilitet är det mer sannolikt att de senare leder till en högre varians avseende antalet avkommor som produceras. Därför bör man överväga viktning av de observerade värdena så att de återspeglar de olika varianserna i de olika behandlingsgrupperna (bakgrundsinformation finns i hänvisning 18).
Vid analysen av data från det slutliga ringtestet (2) anpassades en logistisk kurva med hjälp av följande modell (även andra lämpliga modeller kan användas):

där:
|
Y |
= |
det totala antalet avkommor per föräldradjur som är vid liv vid testets slut (beräknat för varje testkärl). |
|
x |
= |
ämnets koncentration. |
|
c |
= |
det förväntade antalet avkommor när x = 0. |
|
x0 |
= |
EC50 i populationen. |
|
b |
= |
lutningsparametern. |
Denna modell kan tillämpas i många situationer, även om den inte är lämplig för alla test. En kontroll av modellens giltighet bör göras på det sätt som föreslås ovan. I vissa fall kan det vara lämpligt med en hormesis-modell där låga koncentrationer ger ökade verkningar (19).
Man kan även uppskatta koncentrationerna för övriga verkningar, t.ex. EC10 eller EC20. Det kan dock vara bättre att använda en annan parametrisering än den som används för uppskattning av EC50.
2.2 TESTRAPPORT
Testrapporten ska innehålla nedan angivna uppgifter.
2.2.1 Testämne:
|
— |
Aggregationstillstånd och relevanta fysikalisk-kemiska egenskaper. |
|
— |
Kemiska identifieringsdata, inbegripet renhet. |
2.2.2 Försöksdjur:
|
— |
Klonen (om den har identifierats med genotypen eller inte), leverantör eller ursprung (om uppgiften är känd) och de odlingsbetingelser som har använts. Om annan art än Daphnia magna har använts måste detta rapporteras och motiveras. |
2.2.3 Testförhållanden:
|
— |
Använt testförfarande (t.ex. halvstatiskt test eller genomflödestest, volym, antal Daphnia per liter). |
|
— |
Ljusperiod och ljusintensitet. |
|
— |
Testets utformning (t.ex. antalet replikat, antalet föräldradjur per replikat). |
|
— |
Detaljuppgifter om det odlingsmedium som har använts. |
|
— |
I förekommande fall uppgifter om tillsatser av organiskt material och information om sammansättning, ursprung, beredningsmetod, TOC och COD för stamberedningar, uppskattning av resulterande TOC och COD i testmediet. |
|
— |
Detaljerad information om utfodringen, inbegripet mängd (uttryckt som mg kol per Daphnia per dag) och schema (t.ex. typen av foder, och i fråga om alger specifikt namn (arter) och i den mån uppgifter finns, stam och odlingsbetingelser). |
|
— |
Metoden för beredning av stamlösningar och förnyelsefrekvensen (i förekommande fall måste även lösnings- eller dispergeringsmedlet och dess koncentration anges). |
2.2.4 Resultat:
|
— |
Resultaten från eventuella preliminära test gällande testämnets stabilitet. |
|
— |
De nominella testkoncentrationerna och resultaten av alla analyser för att fastställa koncentrationen av testämnet i testkärlen (exempel på blanketter finns i bilaga 4). Metodens återhämtningsgrad och bestämningsgränsen måste även rapporteras. |
|
— |
Vattenkvaliteten i testkärlen (pH, temperatur och halten upplöst syrgas, i förekommande fall TOC eller COD och hårdhet) (exempel på blanketter finns i bilaga 3). |
|
— |
Fullständig registrering av levande avkommor från varje föräldradjur (exempel på blankett finns i bilaga 3). |
|
— |
Antalet föräldradjur som dött och den dag de dött (exempel på blankett finns i bilaga 3). |
|
— |
Variationskoefficienten för kontrollgruppens fertilitet (baserad på det totala antalet levande avkommor per föräldradjur som är vid liv vid testets slut). |
|
— |
Kurva som visar det totala antalet levande avkommor per föräldradjur (för varje replikat) som är vid liv vid testets slut som funktion av testämneskoncentrationen. |
|
— |
Den lägsta koncentrationen vid vilken verkan observeras på reproduktionskapaciteten (LOEC), inklusive en beskrivning av de statistiska metoder som har använts, en indikation om de observerade verkningarnas styrka och koncentrationen vid vilken ingen verkan på reproduktionen observeras (NOEC). I tillämpliga fall bör även LOEC och NOEC för föräldradjurens mortalitet rapporteras. |
|
— |
I tillämpliga fall ECX för reproduktion, konfidensintervall och en kurva för den anpassade modell som har använts för beräkningen, lutningen hos dos-responskurvan och dess standardavvikelse. |
|
— |
Övriga observerade biologiska verkningar eller mätningar: Varje observerad eller uppmätt biologisk verkan (t.ex. föräldradjurens tillväxt) tillsammans med alla tillbörliga motiveringar. |
|
— |
Motiveringar for varje avvikelse från testmetoden. |
3. HÄNVISNINGAR
|
1. |
OECD Test Guideline Programme, Report of the Workshop on the Daphnia magna Pilot Ring Test, Sheffield University, UK, 20 March 1993–21 March 1993. |
|
2. |
OECD Environmental Health and Safety Publications. Series on Testing and Assessment No. 6. Report of the Final Ring Test of the Daphnia magna Reproduction Test Paris. 1997. |
|
3. |
Baird D. J., Barber J., Bradley M. C, Soares A.M.V.M. och Calow P. (1991), A comparative study of genotype sensitivity to acute toxic stress using clones of Daphnia magna Strauss. Ecotoxicology and Environmental Safety, 21, s. 257–265. |
|
4. |
Elendt B. P., (1990), Selenium deficiency in Crustacea; An ultrastructural approach to antennal damage in Daphnia magna Straus. Protoplasma, 154, s. 25–33. |
|
5. |
EPA (1993), Methods for Measuring the Acute Toxicity of Effluents and Receiving Waters to Freshwater and Marine Organisms. (Fourth ed.). EPA/600/4-90/027F. C. I. Weber (ed), USEPA, Cincinnati, Ohio. |
|
6. |
Vigano L., (1991), Suitability of commercially available spring waters as standard medium for culturing Daphnia magna. Bull. Environ. Contam. Toxicol., 47, s. 775–782. |
|
7. |
ASTM (1988), Standard Guide for Conducting Acute Toxicity Tests with Fishes, Macroinvertebrates and Amphibians. E729-88a. American Society for Testing and Materials, Philadelphia P.A. 20 pp. |
|
8. |
Baird D. J., Soares A.M.V.M., Girling A., Barber J., Bradley M. C. och Calow P. (1989), The long term maintenance of Daphnia magna Straus for use in ecotoxicological tests; problems and prospects. In: Proceedings of the lst European Conference on Ecotoxicology. Copenhagen 1988 (H. Løkke, H. Tyle & F. Bro-Rasmussen. Eds.), s. 144–148. |
|
9. |
Parkhurst B. R., Forte J. L. och Wright G. P. (1981), Reproducibility of a life-cycle toxicity test with Daphnia magna. Bull. Environ. Contam. and Toxicol., 26, s. 1–8. |
|
10. |
Cowgill U. M. och Milazzo D. P. (1990), The sensitivity of two cladocerans to water quality variables: salinity and hardness. Arch. Hydrobiol., 120(2). s. 185–196. |
|
11. |
Korshikov (1990), Pseudokirchneriella subcapilala Hindak, F-1990. Biologice Prace, 36, 209. |
|
12. |
Sims I. R., Watson S. och Holmes D. (1993), Toward a standard Daphnia juvenile production test. Environmental Toxicology and Chemistry, 12, sp. 2053–2058. |
|
13. |
Sims 1. (1993), Measuring the growth of phytoplankton: the relationship between total organic carbon with three commonly used parameters of algal growth. Arch. Hydrobiol., 128, s. 459–466. |
|
14. |
Dunnett C. W., (1955), A multiple comparisons procedure for comparing several treatments with a control. J. Amer. Statist. Assoc, 50, s. 1096–1121. |
|
15. |
Dunnett C. W., (1964), New tables for multiple comparisons with a control. Biometrics, 20, s. 482–491. |
|
16. |
Williams D. A. (1971), A test for differences between treatment means when several dose levels are compared with a zero dose control. Biometrics 27, s. 103–117. |
|
17. |
Williams D. A. (1972), The comparison of several dose levels with a zero dose control. Biometrics, 28, s. 510–531. |
|
18. |
Draper N. R. och Smith H. (1981), Applied Regression Analysis, second edition, Wiley, N.Y. |
|
19. |
Brain P. och Cousens R. (1989), An equation to describe dose responses where there is stimulation of growth at low doses. Weed Research, 29, s. 93–96. |
|
20. |
Wilson E. O. och Bossert, W. H. (1971), A Primer of Population Biology. Sinauer Associates Inc. Publishers. |
|
21. |
Poole R. W. (1974), An Introduction to quantitative Ecology. Mc Graw-Hill Series in Population Biology, New York, s. 532. |
|
22. |
Meyer J. S., Ingersoll C. G., McDonald L. L. och Boyce M. S. (1986), Estimating uncertainty in population growth rates: Jackknife vs bootstrap techniques. Ecology, 67, s. 1156–1166. |
Bilaga 1
BEREDNING AV FULLSTÄNDIGT DEFINIERADE ELENDT M7- OCH M4-MEDIER
Acklimatisering till Elendt M7- och M4-medier
I vissa laboratorier har man stött på svårigheter vid direkt överföring av Daphnia till M4- (1) och M7-medier. Man har haft vissa framgångar med stegvis acklimatisering, dvs. först en överflyttning till 30 % Elendt, sedan till 60 % Elendt och därefter till 100 % Elendt. I vissa fall måste acklimatiseringsperioden utsträckas till en månad.
BEREDNING
Spårelement
Separata stamlösningar (1) av enskilda spårelement bereds först i vatten av lämplig renhet (renat t.ex. genom avjonisering, destillering eller omvänd osmos). Utifrån dessa stamlösningar (I) bereds en andra stamlösning (II) som innehåller alla spårelement (kombinerad lösning), enligt tabellen nedan.
|
Stamlösningar I (enskilt ämne) |
Mängd som sätts till vallen (mg/l) |
Koncentration (i förhållande till medium M4) (faktor) |
För beredning av kombinerad stamlösning II tillsätts följande mängd stamlösning I till vatten (ml/1) |
|
|
M 4 |
M 7 |
|||
|
H3BO3 |
57 190 |
20 000 |
1,0 |
0,25 |
|
MnCl2 * 4 H2O |
7 210 |
20 000 |
1,0 |
0,25 |
|
LiCl |
6 120 |
20 000 |
1,0 |
0,25 |
|
RbCl |
1 420 |
20 000 |
1,0 |
0,25 |
|
SrCl2 * 6 H20 |
3 040 |
20 000 |
1,0 |
0,25 |
|
NaBr |
320 |
20 000 |
1,0 |
0,25 |
|
Na2MoO4 * 2 H2O |
1 260 |
20 000 |
1,0 |
0,25 |
|
CuCl2 * 2 H2O |
335 |
20 000 |
1,0 |
0,25 |
|
ZnCl2 |
260 |
20 000 |
1,0 |
1,0 |
|
CoCl2 * 6 H2O |
200 |
20 000 |
1,0 |
1,0 |
|
Kl |
65 |
20 000 |
1,0 |
1,0 |
|
Na2SeO3 |
43,8 |
20 000 |
1,0 |
1,0 |
|
NH4VO3 |
11,5 |
20 000 |
1,0 |
1,0 |
|
Na2EDTA * 2 H20 |
5 000 |
2 000 |
— |
— |
|
FeSO4 * 7 H2O |
1 991 |
2 000 |
— |
— |
|
Na2EDTA- och FeSO4-lösningarna bereds separat, hälls samman och sätts omedelbart i autoklav. Det ger följande: |
||||
|
21 Fe-EDTA-lösning |
|
1 000 |
20,0 |
5,0 |
M4- och M7-medier
M4- och M7-medier bereds med användning av stamlösning II, makronäringsämnen och vitaminer, enligt tabellen nedan.
|
|
Mängd som sätts till vatten mg/l |
Koncentration (i förhållande till medium M4) (faktor) |
Mängd stamlösning som tillsätts för beredning av medium (m1/1) |
|||||
|
M 4 |
M 7 |
|||||||
|
Stamlösning II med kombination av spårelement |
|
20 |
50 |
50 |
||||
|
Stamlösningar med makronäringsämnen |
||||||||
|
CaCl2 * 2 H20 |
293 800 |
1 000 |
1,0 |
1,0 |
||||
|
MgSO4 * 7 H2O |
246 600 |
2 000 |
0,5 |
0,5 |
||||
|
KC1 |
58 000 |
10 000 |
0,1 |
0,1 |
||||
|
NaHCO3 |
64 800 |
1 000 |
1,0 |
1,0 |
||||
|
Na2SiO3 * 9 H20 |
50 000 |
5 000 |
0,2 |
0,2 |
||||
|
NaNO3 |
2 740 |
10 000 |
0,1 |
0,1 |
||||
|
KH2PO4 |
1 430 |
10 000 |
0,1 |
0,1 |
||||
|
K2HPO4 |
1 840 |
10 000 |
0,1 |
0,1 |
||||
|
Kombinerad vitaminstamlösning |
— |
10 000 |
0,1 |
0,1 |
||||
|
Den kombinerade vitaminstamlösningen bereds genom att sätta till de tre vitaminerna till en liter vatten enligt nedan. |
||||||||
|
Tiaminhydroklorid |
750 |
10 000 |
|
|
||||
|
Cyanocobalamin (B12) |
10 |
10 000 |
|
|
||||
|
Biotin |
7,5 |
10 000 |
|
|
||||
|
Den kombinerade vitaminstamlösningen lagras nedfryst i små alikvoter. Vitaminerna sätts till medierna strax före användningen.
|
||||||||
Bilaga 2
ANALYS AV TOTALT ORGANISKT KOL (TOC) OCH UPPRÄTTANDE AV NOMOGRAM FÖR TOC I ALGFODER
Det är allmänt känt att kolhalten i algfodret i regel inte mäts direkt utan fås genom korrelationer (nomogram) med surrogatmätningar av t.ex. antalet algceller eller ljusabsorbans.
För mätning av TOC är högtemperaturoxidation att föredra framför UV- eller persulfatmetoder. (Se The Instrumental Determination of Total Organic Carbon. Total Oxygen Demand and Related Determinands, 1979, HMSO 1980; 49 High Holborn, London WC1V 6HB).
Före mätningarna för nomogrammet separeras algerna från tillväxtmediet genom centrifugering med påföljande återsuspension i destillerat vatten. För varje prov mäts surrogatparametern och TOC tre gånger. Blindprov med destillerat vatten bör analyseras och TOC-värdet härledas från det TOC-värde som erhållits för algprovet.
Nomogrammet bör vara linjärt över det avsedda kolhaltområdet. Nedan visas några exempel.
Anm.: Dessa exempel bör inte användas för konversioner. Det är väsentligt att laboratorierna upprättar egna nomogram.



Bilaga 3
EXEMPEL PÅ BLANKETT FÖR REGISTRERING AV MEDIUMFÖRNYELSE, FYSIKALISK-KEMISKA UPPFÖLJNINGSDATA, UTFODRING, DAPHNIA-REPRODUKTION OCH VUXENMORTALITET
|
Försök nr: |
Data fr.o.m.: |
Klon: |
Medium: |
Typ av foder: |
Testämne: |
Nominell konc.: |
||||||||||||||||||
|
Dag |
0 |
] |
2 |
! |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
11 |
12 |
13 |
14 |
15 |
16 |
17 |
18 |
19 |
20 |
21 |
|
|
|
Mediumförnyelse bocka för) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
PH (37) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
färsk |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
gammal |
|
|
|
02 mg/l (37) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
färsk |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
gammal |
|
|
|
Temperatur ( oC) (37) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
färsk |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
gammal |
|
|
|
Utfordring (bocka för) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Antal levande (38) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total |
|
Kärl 1 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
5 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
6 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
7 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
8 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
9 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total |
|
|
Kumulativ vuxenmortalitet (39) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Bilaga 4
EXEMPEL PÅ BLANKETT FÖR REGISTRERING AV RESULTAT FRÅN KEMISK ANALYS
(a) Uppmätta koncentrationer
|
Nominell konc. |
Prov från vecka 1 |
Prov från vecka 2 |
Prov från vecka 3 |
|||
|
Färsk |
Gammal |
Färsk |
Gammal |
Färsk |
Gammal |
|
|
|
|
|
|
|
|
|
(b) Uppmätta koncentrationer uttryckta som procent av nominella koncentrationer
|
Nominell konc. |
Prov från vecka 1 |
Prov från vecka 2 |
Prov från vecka 3 |
|||
|
Färsk |
Gammal |
Färsk |
Gammal |
Färsk |
Gammal |
|
|
|
|
|
|
|
|
|
Bilaga 5
BERÄKNING AV TIDSVÄGT MEDEL VÄRDE
Tidsvägt medelvärde
Utifrån det faktum att testämnets koncentration kan sjunka under perioderna mellan mediumförnyelse är det nödvändigt att överväga vilken koncentration man ska anse utgöra representativ koncentration för det koncentrationsområde som föräldragenerationen av Daphnia exponeras för. Valet ska basera sig på både biologiska och statistiska faktorer. Om man t.ex. tror att fortplantningen mest påverkas av den högsta koncentrationen som förekommer ska den koncentrationen användas som representativ koncentration. Om däremot ackumulerade verkningar eller långtidsverkningar av det toxiska ämnet anses vara viktigare ska medelkoncentrationen användas. I det fallet är det lämpligt att använda tidsvägd medelkoncentration eftersom den beräknas med beaktande av de variationer som den momentana koncentrationen genomgår över tiden.
Figur 1:
Exempel på tidsvägt medelvärde

I figur 1 visas ett exempel på ett (förenklat) test som omfattar 7 dagar och där mediumförnyelse görs dag 0, 2 och 4.
|
|
Den tunna linjen som går upp och ned representerar momentana koncentrationer. Koncentrationssänkningen antas följa en exponentiell sönderfallsprocess. |
|
|
De sex punkterna representerar observerade koncentrationer som har uppmätts vid början och slutet av varje förnyelseperiod. |
|
|
Den tjocka horisontella linjen indikerar nivån för det tidsvägda medelvärdet. |
Det tidsvägda medelvärdet beräknas så, att ytan under det tidsvägda medelvärdet är lika stor som ytan under koncentrationskurvan. Beräkningen för exemplet i figuren ovan illustreras i tabell 1.
Tabell 1:
Beräkning av tidsvägt medelvärde
|
Förnyelse nr |
Dagar |
Konc0 |
Konc1 |
Ln(KoncO) |
Ln(Konc1) |
Yta |
|
1 |
2 |
10,000 |
4,493 |
2,303 |
1,503 |
13,767 |
|
2 |
2 |
11,000 |
6,037 |
2,398 |
1,798 |
16,544 |
|
3 |
3 |
10,000 |
4,066 |
2,303 |
1,403 |
19,781 |
|
Jagar totalt: 7 |
Totalyta |
50,091 |
||||
|
Tidsvägt medeltal |
7,156 |
|||||
|
Dagar avser antalet dagar i förnyelseperioden. Konc0 är den uppmätta koncentrationen vid varje förnyelseperiods början. Konc1 är den uppmätta koncentrationen vid varje förnyelseperiods slut. Ln(konc0) är den naturliga logaritmen av Konc0 Ln(Konc1) ar den naturliga logaritmen av Konc1 Yta år ytan under den exponentiella kurvan för varje förnyelseperiod. Den beräknas enligt följande:
|
||||||
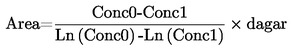
Det tidsvägda medelvärdet är värdet för Totalyta dividerat med värdet för Dagar totalt.
För Daphnia-reproduktionstest måste tabellen självfallet utökas så att den sträcker sig över 21 dagar.
När observationer görs endast i början och slutet av varje förnyelseperiod är det uppenbart att det inte ar möjligt att bekräfta att sönderfallsprocessen verkligen är exponentiell. En annorlunda kurva skulle resultera i en annorlunda beräkning av värdet för Yta. Det är dock inte osannolikt att sönderfallsprocessen är exponentiell och därför är en sådan kurva troligen det bästa alternativet, i brist på annan information.
Däremot finns det anledning att se upp om man genom den kemiska analysen vid förnyelseperiodens slut inte lyckas få fram någon halt av ämnet. Om det inte är möjligt att uppskatta hur snabbt ämnet har försvunnit ur lösningen är det inte möjligt att få en realistisk yta under kurvan, och följaktligen inte heller möjligt att få ett meningsfullt tidsvägt medelvärde.
C.21 JORDLEVANDE MIKROORGANISMER: TEST RÖRANDE OMVANDLING AV KVÄVE
1. METOD
Denna testmetod är i stort sett identisk med OECD TG 216 (2000).
1.1 INLEDNING
Den testmetod som beskrivs här är en laboratoriemetod för undersökning av vilka långtidseffekter en engångsexponering av en kemikalie kan ha på jordlevande mikroorganismers omvandling av kväve. Testmetoden grundar sig i huvudsak på rekommendationerna från Växtskyddsorganisationen för Europa och Medelhavsområdet (EPPO) (1). Även övriga riktlinjer har beaktats, till exempel riktlinjerna från Biologische Bundesanstalt i Tyskland (2), Environmental Protection Agency i USA (3) SETAC (4) och Internationella standardiseringsorganisationen (5). Vid en OECD-workshop om valet av jord och sediment i Belgirate, Italien, 1995 (6), kom man överens om hur många och vilka jordtyper som ska användas i detta test. Rekommendationerna för insamling, hantering och lagring av jordprover grundar sig på ISO-riktlinjer (7) och rekommendationer från workshopen i Belgirate. Vid bedömning och utvärdering av olika ämnens toxiska egenskaper kan det vara nödvändigt att bestämma effekterna på den mikrobiella aktiviteten i jorden, till exempel när det behövs upplysningar om potentiella bieffekter som växtskyddsprodukter kan ha på mikrofloran i jorden, eller när det kan förväntas att de jordlevande mikroorganismerna exponeras för andra kemikalier än växtskyddsprodukter. Testet rörande omvandling av kväve görs för att bestämma sådana kemikaliers effekter på mikrofloran i jorden. Vid testning av jordbrukskemikalier (till exempel växtskyddsprodukter, gödselmedel, skogskemikalier) bör testet omfatta både omvandling av kväve och omvandling av kol. För övriga kemikalier är det tillräckligt med test rörande omvandling av kväve. När sådana kemikalier testas rörande omvandling av kväve och man får EC50-värden som faller inom samma område som gäller för kommersiellt tillgängliga nitrifikationshämmande medel (till exempel nitrapyrin), kan man även göra test rörande omvandling av kol i syfte att få ytterligare upplysningar.
Jord består av levande och icke-levande komponenter som förekommer i komplexa och heterogena sammansättningar. Mikroorganismerna spelar en viktig roll för nedbrytning och omvandling av organiskt material i fertila jordar. Det finns många olika arter av mikroorganismer som inverkar på olika aspekter av jordens fertilitet. Långvariga störningar av dessa biokemiska processer kan även störa kretsloppet av näringsämnen, vilket i sin tur kan leda till förändringar av jordens fertilitet. Omvandling av kol och kväve sker i alla fertila jordar. Även om de mikrobsamhällen som utför dessa processer varierar från jord till jord, följer omvandlingen i huvudsak samma mönster.
Syftet med den testmetod som beskrivs här är att upptäcka de skadliga långtidseffekter som ett ämne kan ha på omvandlingen av kväve i aerob ytjord. Testmetoden kan också användas för att uppskatta olika ämnens effekter på den omvandling av kol som utförs av mikrofloran i jorden. Nitratbildning sker efter att kol-kvävebindningarna har brutits. Om nitratproduktionen sker med samma hastighet i behandlad jord och kontrolljord, är det därför mycket sannolikt att de viktigaste kolnedbrytningsfunktionerna är intakta och välfungerande. Det substrat som har valts för testet (lusernmjöl) har en gynnsam kol/kväve-kvot (i regel mellan 12/1 och 16/1). Därigenom reduceras kolbristen under testets gång, och om ett mikrobsamhälle skadas av en kemikalie är det möjligt att det återhämtar sig inom 100 dagar.
De test som har använts som grund för denna testmetod är primärt utformade för situationer där den mängd ämne som når jorden kan förutses. Så är fallet till exempel för växtskyddsprodukter där man känner till den mängd som har applicerats på en åker. För jordbrukskemikalier räcker det med att testa två sådana doser som är relevanta med tanke på den förväntade dosen. Jordbrukskemikalier kan testas med avseende på aktiva ämnen eller produktsammansättningar. Testet är dock inte begränsat till jordbrukskemikalier. Genom att ändra både mängderna testämne som tillförs jorden och metoderna för utvärdering av data, kan testet även användas för situationer där mängden kemikalie som förväntas nå jorden inte är känd. På så sätt kan man i fråga om andra kemikalier än jordbrukskemikalier bestämma effekterna av en serie koncentrationer på omvandlingen av kväve. Erhållna testdata används för att rita upp en dos-responskurva och beräkna ECx-värden, där x är den procentuella effekten.
1.2 DEFINITIONER
Omvandling av kväve: Den slutliga nedbrytningen av kvävehaltigt material genom inverkan av mikroorganismer. Nedbrytningen sker via ammonifiering och nitrifiering, och utmynnar i den oorganiska slutprodukten nitrat.
ECx (effektiv koncentration): Den koncentration av testämnet i jorden som leder till att omvandlingen av kväve till nitrat hämmas till x procent.
EC50 (median effektiv koncentration): Den koncentration av testämnet i jorden som leder till att omvandlingen av kväve till nitrat hämmas till 50 procent.
1.3 REFERENSÄMNEN
Inga.
1.4 PRINCIP FÖR TESTMETODEN
Siktad jord förbättras med växtmjöl och behandlas med testämnet eller lämnas obehandlad (kontrollprov). Vid testning av jordbrukskemikalier rekommenderas minst två testkoncentrationer, och dessa bör väljas med beaktande av den högsta koncentration som kan förväntas nå jorden. Efter 0, 7, 14 och 28 dagars inkubation behandlas jordproverna (behandlad jord och kontrolljord) med ett lämpligt lösningsmedel och extrakten analyseras med avseende på nitratmängd. Nitratbildningshastigheten i behandlade prover jämförs med den i kontrollproverna, och den procentuella avvikelsen mellan behandlade prover och kontrollprover beräknas. Alla test bör omfatta minst 28 dagar. Om skillnaderna mellan behandlade och obehandlade prover är minst 25 % dag 28, görs fortsatta bestämningar under en period som maximalt sträcker sig till dag 100. Vid testning av andra kemikalier än jordbrukskemikalier tillförs en serie koncentrationer av testämnet till jordproverna och mängderna bildat nitrat i behandlade och obehandlade prover bestäms efter inkubation i 28 dagar. Resultaten från test med flera koncentrationer analyseras med hjälp av en regressionsmodell och relevanta ECX-värden beräknas (till exempel EC50, EC25 och/eller EC10). Se definitionerna.
1.5 TESTETS GILTIGHET
Utvärderingen av testresultat rörande jordbrukskemikalier baserar sig på relativt små skillnader (till exempel medelvärde ±25 %) mellan nitratkoncentrationerna i obehandlade och behandlade jordprover. Därför kan stora variationer i de obehandlade proverna leda till falska resultat. Variationen mellan obehandlade replikatprover bör därför vara mindre än ±15 %.
1.6 BESKRIVNING AV TESTMETODEN
1.6.1 Utrustning
Provbehållarna bör vara tillverkade av kemiskt inert material. Deras volym bör anpassas till det förfarande som används för inkubation av jorden, till exempel inkubation som bulk eller som en serie individuella jordprover (se avsnitt 1.7.1.2). Det är viktigt att minimera vattenförlusterna och sörja för gasväxling under testets förlopp (provbehållarna kan till exempel täckas med perforerad polyetylenfilm). Vid testning av flyktiga ämnen bör förseglingsbara och gastäta behållare användas. Behållarnas storlek bör anpassas så att jordprovet upptar cirka en fjärdedel av behållarens volym.
Därutöver behövs normal laboratorieutrustning, enligt följande:
|
— |
Omrörare: en mekanisk skakapparat eller motsvarande. |
|
— |
Centrifug (3 000 g) eller filtreringsutrustning (med nitratfritt filterpapper). |
|
— |
Instrument för nitratanalys, med adekvat känslighet och reproducerbarhet. |
1.6.2 Valet av jord och antal jordtyper
Endast en typ av jord används. Följande rekommendationer gäller för jorden:
|
— |
Sandhalt: minst 50 % och högst 75 %. |
|
— |
pH: 5,5–7,5. |
|
— |
Halten organiskt kol: 0,5–1,5 %. |
|
— |
Den mikrobiella biomassan bör bestämmas (8), (9) och dess kolhalt bör vara minst 1 % av jordens totala organiska kol. |
I de flesta fall är jord med dessa egenskaper ett exempel på sämsta möjliga situation – adsorptionen av testämnet är minimal, medan mikrofloran har maximal tillgång till testämnet. Det betyder att det i regel inte behövs test med andra typer av jord. I vissa fall, till exempel där testämnet mestadels förväntas hamna i speciella jordar såsom sura skogsjordar eller om testämnet består av elektrostatiskt laddade kemikalier, kan det vara nödvändigt att använda en extra jordtyp.
1.6.3 Insamling och lagring av jordprover
1.6.3.1 Insamling
Man bör ha tillgång till detaljerade upplysningar om historian för den plats där testjorden tas. Upplysningarna bör omfatta exakt läge, växtlighetstäcke, datum för behandlingar med växtskyddsprodukter, behandlingar med organiska och oorganiska gödselmedel, tillförsel av biologiska material eller oavsiktlig förorening. Den plats som väljs för provtagningen bör vara lämpad för långtidsanvändning. Lämpliga platser är flerårig ängsmark, åkrar med ettåriga spannmålsgrödor (utom majs) och tättsådda grönbeten. Vid provtagningstidpunkten måste det ha gått minst ett år från en eventuell behandling av provtagningsplatsen med växtskyddsprodukter. Det bör även ha gått minst sex månader från tillförsel av organiska gödselmedel. Användning av mineralgödselmedel är godtagbar endast när detta sker med utgångspunkt från grödans behov. Jordprover får inte tas förrän det har gått minst tre månader från gödslingen. Jord som har behandlats med gödselmedel med kända biocideffekter (till exempel kalciumcyanid) bör undvikas.
Provtagning bör undvikas under eller strax efter långa perioder (längre än 30 dagar) av torka eller vattenmättnad. På plöjd mark ska jordproverna tas på ett djup mellan 0 och 20 cm. På gräsmark (ängar) eller annan mark som plöjs sällan (minst en växtsäsong mellan varje plöjning) kan maximidjupet för provtagningen vara lite mer än 20 cm (till exempel 25 cm).
Jordproverna ska transporteras i behållare, och temperaturbetingelserna bör vara sådana att det inte finns risk för signifikanta förändringar av jordens ursprungliga egenskaper.
1.6.3.2 Lagring
De jordprover som används ska helst vara nytagna. Om lagring i laboratoriet inte kan undvikas kan jordproverna hållas i mörker vid 4 ±2 oC under högst tre månader. Det är viktigt att lagringsbetingelserna är aeroba. Om jordprover tas från områden där marken är frusen under minst tre månader per år kan proverna lagras vid - 18 oC till - 22 oC under högst sex månader. Den mikrobiella biomassan hos lagrade jordprover bestäms före varje försök. Biomassans kolhalt bör vara minst 1 % av jordens totala kolhalt (se avsnitt 1.6.2).
1.6.4 Hantering och beredning av jordprover som testas
1.6.4.1 Förinkubation
För jordprover som har lagrats (se avsnitt 1.6.3.2) rekommenderas en förinkubation under 2–28 dagar. Under förinkubationen bör jordprovet ha samma temperatur och fukthalt som under själva testet (se avsnitten 1.6.4.2 och 1.7.1.3).
1.6.4.2 Fysikalisk-kemiska egenskaper
Stora föremål (till exempel stenar och växtdelar) avlägsnas manuellt ur jorden, som därefter siktas fuktig, utan att torkas alltför mycket, till en partikelstorlek som är högst 2 mm. Jordprovets fukthalt justeras med destillerat eller avjoniserat vatten till ett värde mellan 40 % och 60 % av den maximala vattenkapaciteten.
1.6.4.3 Förbättring med organiskt substrat
Jorden ska förbättras med ett lämpligt organiskt substrat, till exempel lusern- och gräsmjöl (huvudbeståndsdel: Medicago sativa (blålusern) som har en kol/kväve-kvot mellan 12/1 och 16/1. Den rekommenderade kvoten mellan lusern och jord är 5 g lusern per kilogram jord (torrvikt).
1.6.5 Beredning av testämnet för applicering i jorden
Testämnet appliceras i regel med hjälp av en vehikel. Som vehikel kan vatten användas (för vattenlösliga ämnen) eller ett inert fast material såsom fin kvartssand (partikelstorlek: 0,1–0,5 mm). Vätskeformiga vehiklar andra än vatten (till exempel organiska lösningsmedel såsom aceton och kloroform) bör undvikas eftersom de kan skada mikrofloran. Om sand används som vehikel kan den beläggas med testämnet upplöst eller suspenderat i ett lämpligt lösningsmedel. Lösningsmedlet avlägsnas därefter genom avdunstning innan den belagda sanden blandas i jorden. För en optimal fördelning av testämnet i jorden rekommenderas 10 g sand per kilogram jord (torrvikt). Kontrollproverna behandlas med lika stor mängd rent vatten eller ren kvartssand.
Vid testning av flyktiga kemikalier bör förluster under behandlingen minimeras. Likaså bör en homogen fördelning i jorden säkerställas (testämnet bör exempelvis injiceras på flera ställen i jorden).
1.6.6 Testkoncentrationer
Vid testning av jordbrukskemikalier bör minst två koncentrationer användas. Den lägre koncentrationen bör minst motsvara den maximimängd som i praktiken förväntas nå jorden, och den högre koncentrationen bör vara en multipel av den lägre koncentrationen. Vid beräkning av koncentrationerna för det testämne som tillförs jorden antas att upptaget är enhetligt ned till jorddjupet 5 cm och att bulkdensiteten är 1,5. Vid testning av jordbrukskemikalier som appliceras direkt i jorden eller kemikalier där den mängd som når jorden kan förutses, rekommenderas dels den maximala PEC-koncentrationen (Predicted Environmental Concentration), dels en femfaldig koncentration av denna. Ämnen som förväntas bli applicerade i jorden flera gånger under en säsong bör testas vid koncentrationer som fås genom att multiplicera PEC-koncentrationen med det maximala antalet förväntade appliceringar. Den högsta testkoncentrationen bör dock inte överskrida mer än tio gånger den maximala koncentrationen vid en enskild applicering. Vid testning av andra kemikalier än jordbrukskemikalier används en geometrisk serie med minst fem koncentrationer. Testkoncentrationerna bör täcka det område som behövs för att bestämma ECx-värden.
1.7 TESTETS UTFÖRANDE
1.7.1 Exponeringsbetingelser
1.7.1.1 Behandlade prover och kontrollprover
Vid testning av jordbrukskemikalier delas jorden i tre delar med lika vikt. Två delar blandas med vehikel plus produkt och den tredje blandas med ren vehikel (kontrollprov). Minst tre replikat rekommenderas för behandlade respektive obehandlade jordprover. Vid testning av andra kemikalier än jordbrukskemikalier delas jorden i sex delar med lika vikt. Fem prover blandas med vehikel plus testämne och det sjätte provet blandas med ren vehikel. Tre replikat rekommenderas för behandlade prover respektive kontrollprov. Det är viktigt att säkerställa en homogen fördelning av testämnet i de behandlade jordproverna. Vid inblandningen bör kompaktering eller klumpbildning av jorden undvikas.
1.7.1.2 Inkubation av jordprover
Inkubationen av jordprover kan utföras på två olika sätt. Den kan göras med bulkprover av varje behandlat och obehandlat jordprov eller som en serie individuella och lika stora delprover av varje behandlat och obehandlat jordprov. Vid testning av flyktiga ämnen bör man dock alltid använda en serie individuella delprover. Vid inkubation i form av bulk bereds stora mängder av behandlade och obehandlade jordprover. Under testets gång tas behövligt antal delprover för analys. De mängder som bereds för behandlade och obehandlade jordprover beror på storleken hos de delprover som ingår, antalet replikat som används för analys och det förväntade antalet provtagningsgånger. Jordprover som inkuberas i form av bulk bör blandas grundligt innan delprover tas. När jordprover inkuberas i form av en serie individuella prover, delas varje behandlat och obehandlat prov upp i det antal delprover som behövs och används därefter på planerat sätt. Om det är sannolikt med fler än två provtagningstillfällen bör tillräckligt antal delprover beredas med tanke på alla replikat och alla provtagningstillfällen. Minst tre replikatprover av testjorden bör inkuberas under aeroba betingelser (se avsnitt 1.7.1.1). Alla testbehållare som används bör ha ett tillräckligt utrymme ovanför vätskeytan för att anaeroba betingelser inte ska uppstå. Vid testning av flyktiga ämnen bör testet alltid göras med en serie individuella delprover.
1.7.1.3 Testbetingelser och testperiodens längd
Testet genomförs i mörker vid en rumstemperatur på 20 ±2 oC. Under testets gång bör jordprovernas fukthalt hållas vid 40–60 % av jordens maximala vattenkapacitet (se avsnitt 1.6.4.2). Tillåten variation är ±5 %. Destillerat eller avjoniserat vatten kan tillföras vid behov.
Testet bör omfatta en period på minst 28 dagar. Vid testning av jordbrukskemikalier jämförs nitratbildningshastigheten i behandlade prover och kontrollprover. Om avvikelsen är högre än 25 % dag 28, förlängs testet tills skillnaden är högst 25 % eller tills testet har varat 100 dagar, beroende på vilken gräns som nås först. För andra kemikalier än jordbrukskemikalier avslutas testet efter 28 dagar. På dag 28 bestäms nitratmängderna i behandlade jordprover och kontrollprover, och ECx-värden beräknas.
1.7.2 Provtagning och analys av jordprover
1.7.2.1 Provtagningsschema
Vid testning av jordbrukskemikalier analyseras jordproverna med avseende på nitrat, dag 0, 7, 14 och 28. Om testperioden måste förlängas görs tilläggsbestämningarna med 14 dagars intervall efter dag 28.
Vid testning av andra kemikalier än jordbrukskemikalier används minst fem testkoncentrationer, och nitratanalys görs på jordproverna i början (dag 0) och i slutet av exponeringsperioden (dag 28). En delbestämning kan göras till exempel dag 7 om det bedöms vara nödvändigt. De resultat som erhålls dag 28 används för att bestämma ECx-värdet för kemikalien. Om så önskas kan kontrollprovernas data från dag 0 användas för att rapportera jordens ursprungliga nitrathalt.
1.7.2.2 Analys av jordprover
Mängden nitrat som har bildats i vart och ett av de behandlade proverna och kontrollreplikat bestäms för varje provtagningstidpunkt. Nitratet extraheras ur jorden genom att proverna skakas med ett lämpligt lösningsmedel, till exempel 0,1 M kaliumkloridlösning. 5 ml KCl-lösning per gram jord (torrvikt) rekommenderas. För optimal extraktion bör behållarna med jord och extraktionslösning inte vara fyllda mer än till hälften. Blandningarna skakas vid 150 rpm under 60 minuter. Blandningarna centrifugeras eller filtreras och vätskefasens analyseras med avseende på nitrat. Partikelfria vätskeextrakt kan lagras före analys vid - 20 ±5 oC i högst sex månader.
2. DATA
2.1 BEHANDLING AV RESULTATEN
Vid testning av jordbrukskemikalier registreras mängden nitrat som har bildats i varje replikatprov, och medelvärdena för alla replikat ges i tabellform. Kväveomvandlingshastigheterna utvärderas med lämpliga och allmänt accepterade statistiska metoder (till exempel F-test, 5 % signifikansnivå). Mängderna bildat nitrat uttrycks som mg nitrat/kg torrvikt jord/dag. Nitratbildningshastigheten i varje behandlat prov jämförs med hastigheten i kontrollprovet, och den procentuella avvikelsen från kontrollprovet beräknas.
Vid testning av andra kemikalier än jordbrukskemikalier bestäms mängden nitrat som har bildats i varje replikat, och en dos-responskurva ritas upp för uppskattningen av ECx-värden. Mängderna nitrat i de behandlade proverna (mg nitrat/kg torrvikt jord) dag 28 jämförs med mängderna i kontrollproverna. Resultatet används för beräkning av den procentuella hämningen för varje testkoncentration. Dessa procentvärden ritas upp mot koncentrationen, och ECx-värden beräknas med hjälp av statistiska metoder. Konfidensgränserna (p = 0,95) för de beräknade ECx-värdena bestäms med standardmetoder (10) (11) (12).
Om kvävehalterna i de ämnen som testas är höga kan detta påverka mängden nitrat som bildas under testet. Om sådana ämnen testas vid en hög koncentration (till exempel kemikalier som sannolikt kommer att appliceras vid upprepade tillfällen) måste testet omfatta tillbörliga kontrollprover (jord med testämne men utan växtmjöl). De värden som fås för dessa kontrollprover måste beaktas vid beräkning av ECx.
2.2 TOLKNING AV RESULTATEN
Om resultaten från test rörande jordbrukskemikalier tyder på att nitratbildningshastigheten vid den lägre behandlingsnivån (den maximala förväntade koncentrationen) avviker från hastigheten vid kontrollnivån med högst 25 % vid alla provtagningstidpunkter efter dag 28, kan man anse att produkten inte har någon långtidseffekt på kväveomvandling i jord. Vid utvärdering av test rörande andra ämnen än jordbrukskemikalier används värden för EC50, EC25 och/eller EC10.
3. RAPPORTERING
Testrapporten ska innehålla nedan angivna uppgifter.
|
|
Fullständig identifiering av jorden, inbegripet
|
|
|
Testämne:
|
|
|
Substrat:
|
|
|
Testbetingelser:
|
|
|
Resultat:
|
4. HÄNVISNINGAR
|
1. |
EPPO (1994), Decision-Making Scheme for the Environmental Risk Assessment of Plant Protection Chemicals. Chapter 7: Soil Microflora, EPPO Bulletin, 24: 1–16, 1994. |
|
2. |
BBA (1990), Effects on the Activity of the Soil Microflora, BBA Guidelines for the Official Testing of Plant Protection Products, VI, 1-1 (andra utgåvan, 1990). |
|
3. |
EPA (1987), Soil Microbial Community Toxicity Test. EPA 40 CFR Part 797.3700. Toxic Substances Control Act Test Guidelines; Proposed rule, den 28 september 1987. |
|
4. |
SETAC-Europe (1995), Procedures for assessing the environmental fate and ecotoxicity of pesticides, red. M. R. Lynch, Pub. SETAC-Europe, Bryssel. |
|
5. |
ISO/DIS 14238 (1995), Markundersökningar – Bestämning av kvävemineralisering och nitrifiering i jord och kemikaliers inverkan på dessa processer, Technical Committee ISO/TC 190/SC 4: ”Soil Quality – Biological Methods”. |
|
6. |
OECD (1995), Final Report of the OECD Workshop on Selection of Soils/Sediments, Belgirate, Italien, den 18–20 januari 1995. |
|
7. |
ISO 10381-6 (1993), Soil quality – Sampling. Guidance on the collection, handling and storage of soil for the assessment of aerobic microbial processes in the laboratory. |
|
8. |
ISO 14240-1 (1997), ”Markundersökningar – Bestämning av markens mikrobiella biomassa – Del 1: Sustratinducerad respirationsmetod”. |
|
9. |
ISO 14240-2 (1997), Markundersökningar – Bestämning av markens mikrobiella biomassa – Del 2: Fumigering – extraheringsmetod. |
|
10. |
J. T. Litchfield, och F. Wilcoxon (1949), A simplified method of evaluating dose-effect experiments, Jour. Pharmacol. and Exper. Ther., 96, 99–113. |
|
11. |
D. J. Finney, (1971), Probit Analysis, tredje utgåvan, Cambridge, London och New York. |
|
12. |
D. J. Finney, (1978), Statistical Methods in biological Assay. Griffin, Weycombe, Förenade kungariket. |
C.22 JORDLEVANDE MIKROORGANISMER: TEST RÖRANDE OMVANDLING AV KOL
1. METOD
Denna metod är i stort sett identisk med OECD TG 217 (2000).
1.1 INLEDNING
Den testmetod som beskrivs här är en laboratoriemetod för undersökning av de potentiella långtidseffekter som en engångsexponering av jordbruksprodukter, och eventuellt även andra kemikalier, kan ha på jordlevande mikroorganismers omvandling av kol. Testmetoden grundar sig i huvudsak på rekommendationerna från Växtskyddsorganisationen för Europa och Medelhavsområdet (EPPO) (1). Även övriga riktlinjer har beaktats, till exempel riktlinjerna från Biologische Bundesanstalt i Tyskland (2), Environmental Protection Agency i USA (3) och SETAC (4). Vid en OECD-workshop om valet av jord och sediment i Belgirate, Italien, 1995 (5), kom man överens om hur många och vilka typer av jord som ska användas i detta test. Rekommendationerna för insamling, hantering och lagring av jordprover grundar sig på ISO-riktlinjer (6) och rekommendationer från workshopen i Belgirate.
Vid bedömning och utvärdering av olika ämnens toxiska egenskaper kan det vara nödvändigt att bestämma effekterna på den mikrobiella aktiviteten i jorden, till exempel när det behövs upplysningar om de potentiella bieffekter som växtskyddsprodukter kan ha på mikrofloran i jorden, eller när det är sannolikt att de jordlevande mikroorganismerna exponeras för andra kemikalier än växtskyddsprodukter. Testet rörande omvandling av kol görs för att bestämma sådana kemikaliers effekter på mikrofloran i jorden. Vid testning av jordbrukskemikalier (till exempel växtskyddsprodukter, gödselmedel och skogskemikalier) bör testet omfatta både omvandling av kol och omvandling av kväve. I fråga om övriga kemikalier räcker det att testa omvandling av kväve. När sådana kemikalier testas rörande omvandling av kväve och man får EC50-värden som faller inom samma område som gäller för kommersiellt tillgängliga nitrifikationshämmande medel (till exempel nitrapyrin), kan man även göra test rörande omvandling av kol i syfte att få ytterligare upplysningar.
Jord består av levande och icke-levande komponenter som förekommer i komplexa och heterogena sammansättningar. Mikroorganismerna spelar en viktig roll för nedbrytning och omvandling av organiskt material i fertila jordar. Det finns många olika arter och dessa bidrar till de olika aspekter som gäller för jordens fertilitet. Långvariga störningar av dessa biokemikaliska processer kan även störa kretsloppet av näringsämnen, vilket i sin tur kan leda till förändringar av jordens fertilitet. Omvandling av kol och kväve sker i alla fertila jordar. Även om de mikrobsamhällen som utför dessa processer varierar från jord till jord, följer omvandlingen i huvudsak samma mönster.
Syftet med den testmetod som beskrivs här är att upptäcka skadliga långtidseffekter som ett ämne kan ha på omvandlingen av kol i aerob ytjord. Testet är känsligt för förändringar i storlek och aktivitet hos de mikrobsamhällen som svarar för omvandlingen av kol, eftersom de genom testet utsätts för både kemisk stress och kolsvält. För testet används sandhaltig jord med låg halt organiskt material. Jorden behandlas med testämnet och inkuberas under betingelser som möjliggör snabb mikrobiell metabolism. Under sådana betingelser sker en snabb utarmning av jordens innehåll av lättillgängligt kol. Detta leder till kolbrist som dödar mikrobcellerna och inducerar en viloperiod och/eller sporbildning. Om testet körs under en längre tid än 28 dagar kan summan av dessa reaktioner mätas i de obehandlade jordprovema som en progressiv minskning av metaboliskt aktiv mikrobiell biomassa (7). Om biomassan i en kolstressad jord påverkas av en kemikalie under de betingelser som råder under testet, återställs den eventuellt inte till samma nivå som i ett obehandlat jordprov. Störningar som testämnet har orsakat vid någon försökstidpunkt varar då ofta till slutet av testet.
De test som har använts som grund för denna testmetod är primärt utformade för situationer där den mängd ämne som når jorden kan förutses. Så är fallet till exempel för växtskyddsprodukter där man känner till den mängd som har applicerats på en åker. För jordbrukskemikalier räcker det med att testa två sådana doser som är relevanta med tanke på den förväntade applicerade mängden. Jordbrukskemikalier kan testas med avseende på aktiva ämnen eller produktsammansättningar. Testet är dock inte begränsat till kemikalier med förutsägbara halter i miljön. Genom att ändra både mängderna testämne som tillförs jorden och metoderna för utvärdering av data, kan testet även användas för situationer där mängden kemikalie som förväntas nå jorden inte är känd. På så sätt kan man i fråga om andra kemikalier än jordbrukskemikalier bestämma effekterna av en serie koncentrationer på omvandlingen av kol. Erhållna testdata används för att rita upp en dos-responskurva och beräkna ECx-värden, där x är den procentuella effekten.
1.2 DEFINITIONER
Omvandling av kol: Nedbrytning av organiskt material genom inverkan av mikroorganismer under bildning av slutprodukten koldioxid.
EC X (effektiv koncentration): Den koncentration av testämnet i jorden som leder till att omvandlingen av kol till koldioxid hämmas till x procent.
EC 50 (median effektiv koncentration): Den koncentration av testämnet i jorden som leder till att omvandlingen av kol till koldioxid hämmas till 50 procent.
1.3 REFERENSÄMNEN
Inga.
1.4 PRINCIP FÖR TESTMETODEN
Siktad jord behandlas med testämnet eller lämnas obehandlad (kontrollprov). Vid testning av jordbrukskemikalier rekommenderas minst två testkoncentrationer, och dessa bör väljas med beaktande av den högsta koncentration som kan förväntas nå jorden. Efter inkubation under 0, 7, 14 och 28 dagar blandas de behandlade och obehandlade proverna med glukos, och den glukosinducerade respirationen i varje prov mäts under 12 på varandra följande timmar. Respirationen uttrycks som mängden frigjord koldioxid (mg koldioxid/kg torrvikt jord/h) eller mängden förbrukat syre (mg syre/kg jord/h). Den genomsnittliga respirationen i de behandlade jordproverna jämförs med situationen i kontrollproverna, och den procentuella avvikelsen från kontrollprovet beräknas. Alla test bör omfatta minst 28 dagar. Om skillnaderna mellan behandlade och obehandlade prover är minst 25 % dag 28 görs fortsatta bestämningar med 14 dagars intervall under en period som maximalt sträcker sig till dag 100. Vid testning av andra kemikalier än jordbrukskemikalier appliceras en serie koncentrationer av testämnet i jordprover, och efter 28 dagar bestäms den glukosinducerade respirationen (de genomsnittliga mängderna bildad koldioxid eller förbrukat syre). Resultaten från test med en serie koncentrationer analyseras med hjälp av en regressionsmodell och relevanta ECx-värden beräknas (till exempel EC50, EC25 och/eller EC10). Se definitionerna.
1.5 TESTETS GILTIGHET
Utvärderingen av testresultat rörande jordbrukskemikalier baserar sig på relativt små skillnader (till exempel medelvärde ±25 %) mellan frigjord koldioxid eller förbrukat syre i (eller av) obehandlade och behandlade jordprover. Därför kan stora variationer i de obehandlade proverna leda till falska resultat. Variationen mellan obehandlade replikatprover bör därför vara mindre än ± 15 %.
1.6 BESKRIVNING AV TESTMETODEN
1.6.1 Utrustning
Provbehållarna bör vara tillverkade av kemiskt inert material. Deras volym bör anpassas till det förfarande som används för inkubation av jorden, till exempel inkubation som bulk eller som en serie individuella jordprover (se avsnitt 1.7.1.2). Det är viktigt att minimera vattenförlusterna och sörja för gasväxling under testets förlopp (provbehållarna kan till exempel täckas med perforerad polyetylenfilm). Vid testning av flyktiga ämnen bör förseglingsbara och gastäta behållare användas. Behållarnas storlek bör anpassas så att jordprovet upptar cirka en fjärdedel av behållarens volym.
För bestämning av glukosinducerad respiration krävs inkubationssystem och instrument för mätning av producerad koldioxid eller förbrukat syre. Exempel på sådana system och instruments finns i litteraturen (8) (9) (10) (11).
1.6.2 Valet av jord och antal jordtyper
Endast en typ av jord används. Följande rekommendationer gäller för jorden:
|
— |
Sandhalt: minst 50 % och högst 75 %. |
|
— |
pH: 5,5–7,5. |
|
— |
Halten organiskt kol: 0,5–1,5 %. |
|
— |
Den mikrobiella biomassan bör bestämmas (12) (13) och dess kolhalt bör vara minst 1 % av jordens totala organiska kol. |
I de flesta fall utgör jord med dessa egenskaper ett exempel på sämsta möjliga situation – adsorptionen av testämnet är minimal, medan mikrofloran har maximal tillgång till testämnet. Det betyder att det i regel inte behövs test med andra typer av jord. I vissa fall, till exempel där testämnet mestadels förväntas hamna i särskilda jordar såsom sura skogsjordar eller om testämnet består av elektrostatiskt laddade kemikalier, kan det vara nödvändigt att använda en extra jordtyp.
1.6.3 Insamling och lagring av jordprover
1.6.3.1 Insamling
Man bör ha tillgång till detaljerade upplysningar om historian för den plats där testjorden tas. Upplysningarna bör omfatta exakt läge, växtlighetstäcke, datum för behandlingar med växtskyddsprodukter, behandlingar med organiska och oorganiska gödselmedel, tillförsel av biologiska material eller oavsiktlig förorening. Den plats som väljs för provtagningen bör vara lämpad för långtidsanvändning. Lämpliga platser är flerårig ängsmark, åkrar med ettåriga spannmålsgrödor (utom majs) och tättsådda grönbeten. Vid provtagningstidpunkten måste det ha gått minst ett år från en eventuell behandling av provtagningsplatsen med växtskyddsprodukter. Det bör även ha gått minst sex månader från tillförsel av organiska gödselmedel. Användning av mineralgödselmedel är godtagbar endast när detta sker med utgångspunkt från grödans behov. Jordprover får inte tas förrän det har gått minst tre månader från gödslingen. Jord som har behandlats med gödselmedel med kända biocideffekter (till exempel kalciumcyanid) bör undvikas.
Provtagning bör undvikas under eller strax efter långa perioder (längre än 30 dagar) av torka eller vattenmättnad. På plöjd mark ska jordproverna tas på ett djup mellan 0 och 20 cm. På gräsmark (ängar) eller annan mark som plöjs sällan (minst en växtsäsong mellan varje plöjning) kan maximidjupet för provtagningen vara lite mer än 20 cm (till exempel 25 cm). Jordproverna ska transporteras i behållare, och temperaturbetingelser bör vara sådana att det inte finns risk för signifikanta förändringar av jordens ursprungliga egenskaper.
1.6.3.2 Lagring
De jordprover som används ska helst vara nytagna. Om lagring i laboratoriet inte kan undvikas kan jordproverna hållas i mörker vid 4 ± 2 oC under högst tre månader. Det är viktigt att lagringsbetingelserna är aeroba. Om jordprover tas från områden där marken är frusen under minst tre månader per år kan proverna lagras under högst sex månader vid –18 oC. Den mikrobiella biomassan hos lagrade jordprover bestäms före varje försök. Biomassans kolhalt bör vara minst 1 % av jordens totala kolhalt (se avsnitt 1.6.2).
1.6.4 Hantering och beredning av jordprover som testas
1.6.4.1 Förinkubation
För jordprover som har lagrats (se avsnitten 1.6.4.2 och 1.7.1.3) rekommenderas en förinkubation under 2–28 dagar. Under förinkubationen bör jordprovet ha samma temperatur och fukthalt som under själva testet (se avsnitten 1.6.4.2 och 1.7.1.3).
1.6.4.2 Fysikalisk-kemiska egenskaper
Stora föremål (till exempel stenar och växtdelar) avlägsnas manuellt ur jorden, som därefter siktas fuktig, utan att torkas alltför mycket, till en partikelstorlek som är högst 2 mm. Jordprovets fukthalt justeras med destillerat eller avjoniserat vatten till ett värde mellan 40 % och 60 % av den maximala vattenkapaciteten.
1.6.5 Beredning av testämnet för applicering i jorden
Testämnet appliceras i regel med hjälp av en vehikel. Som vehikel kan vatten användas (för vattenlösliga ämnen) eller ett inert fast material såsom fin kvartssand (partikelstorlek: 0,1–0,5 mm). Vätskeformiga vehiklar andra än vatten (till exempel organiska lösningsmedel såsom aceton och kloroform) bör undvikas eftersom de kan skada mikrofloran. Om sand används som vehikel kan den beläggas med testämnet upplöst eller suspenderat i ett lämpligt lösningsmedel. Lösningsmedlet avlägsnas därefter genom avdunstning innan den belagda sanden blandas i jorden. För en optimal fördelning av testämnet i jorden rekommenderas 10 g sand per kilogram jord (torrvikt). Kontrollproverna behandlas med lika stor mängd rent vatten eller ren kvartssand.
Vid testning av flyktiga kemikalier bör förluster under behandlingen minimeras. Likaså bör en homogen fördelning i jorden säkerställas (testämnet bör exempelvis injiceras på flera ställen i jorden).
1.6.6 Testkoncentrationer
Om testet gäller växtskyddsprodukter eller andra kemikalier med förutsägbara koncentrationer i miljön, bör minst två koncentrationer användas. Den lägre koncentrationen bör minst motsvara den maximimängd som i praktiken förväntas nå jorden, och den högre koncentrationen bör vara en multipel av den lägre koncentrationen. Vid beräkning av koncentrationerna för det testämne som tillförs jorden antas att upptaget är enhetligt ned till jorddjupet 5 cm och att bulkdensiteten är 1,5. Vid testning av jordbrukskemikalier som appliceras direkt på jorden eller kemikalier där den mängd som når jorden kan förutses, rekommenderas PEC-koncentrationen (Predicted Environmental Concentration) och en femfaldig koncentration av denna. Ämnen som förväntas bli applicerade i jorden flera gånger under en säsong bör testas vid koncentrationer som fås genom att multiplicera PEC-koncentrationen med det maximala antalet förväntade appliceringar. Den högsta testkoncentrationen bör dock inte överskrida mer än tiofaldigt den maximala koncentrationen vid en enskild applicering.
Vid testning av andra kemikalier än jordbrukskemikalier används en geometrisk serie med minst fem koncentrationer. Testkoncentrationerna bör täcka det område som behövs för att bestämma ECx-värden.
1.7 TESTETS UTFÖRANDE
1.7.1 Exponeringsbetingelser
1.7.1.1 Behandlade prover och kontrollprover
Vid testning av jordbrukskemikalier delas jorden i tre delar med lika vikt. Två delar blandas med vehikel plus produkt och den tredje blandas med ren vehikel (kontrollprov). Minst tre replikat rekommenderas för behandlade respektive obehandlade jordprover. Vid testning av andra kemikalier än jordbrukskemikalier delas jorden i sex delar med lika vikt. Fem prover blandas med vehikel plus testämne och det sjätte provet blandas med ren vehikel. Tre replikat rekommenderas för behandlade prover respektive kontrollprov. Det är viktigt att säkerställa en homogen fördelning av testämnet i de behandlade jordproverna. Vid inblandningen bör kompaktering eller klumpbildning av jorden undvikas.
1.7.1.2 Inkubation av jordprover
Inkubationen av jordprover kan utföras på två olika sätt. Den kan göras med bulkprover av varje behandlat och obehandlat jordprov eller som en serie individuella och lika stora delprover av varje behandlat och obehandlat jordprov. Vid testning av flyktiga ämnen bör man dock alltid använda en serie individuella delprover. Vid inkubation i form av bulk bereds stora mängder av behandlade och obehandlade jordprover. Under testets gång tas behövligt antal delprover för analys. De mängder som bereds för behandlade och obehandlade jordprover beror på storleken hos de delprover som ingår, antalet replikat som används för analys och det förväntade antalet provtagningstillfällen. Jordprover som inkuberas i form av bulk bör blandas grundligt innan delprover tas. När jordprover inkuberas i form av en serie individuella prover, delas varje behandlat och obehandlat prov upp i behövligt antal delprover som därefter används på planerat sätt. Om det är sannolikt med fler än två provtagningstillfällen bör tillräckligt antal delprover beredas med tanke på alla replikat och alla provtagningsgånger. Minst tre replikatprover av testjorden bör inkuberas under aeroba betingelser (se avsnitt 1.7.1.1). Alla testbehållare som används bör ha ett tillräckligt utrymme ovanför vätskeytan för att det inte ska uppstå anaeroba betingelser. Vid testning av flyktiga ämnen bör testet alltid göras med en serie individuella delprover.
1.7.1.3 Testbetingelser och testperiodens längd
Testet genomförs i mörker vid en rumstemperatur på 20 ± 2 oC. Under testets gång bör jordprovernas fukthalt upprätthållas vid 40–60 % av jordens maximala vattenkapacitet (se avsnitt 1.6.4.2). Tillåten variation är ± 5 %. Destillerat eller avjoniserat vatten kan tillföras vid behov.
Testet bör omfatta en period på minst 28 dagar. Vid testning av jordbrukskemikalier jämförs mängderna koldioxid som frigörs eller syre som förbrukas i behandlade och obehandlade prover. Om avvikelsen är högre än 25 % dag 28, förlängs testet tills skillnaden är högst 25 % eller tills testet har varat 100 dagar, beroende på vilken gräns som nås först. För andra kemikalier än jordbrukskemikalier avslutas testet efter 28 dagar. Dag 28 bestäms mängderna frigjord koldioxid eller förbrukat syre i behandlade jordprover och kontrollprover och ECX-värden beräknas.
1.7.2 Provtagning och analys av jordprover
1.7.2.1 Provtagningsschema
Vid testning av jordbrukskemikalier analyseras jordproverna med avseende på glukosinducerad respiration dag 0, 7, 14 och 28. Om testperioden måste förlängas görs tilläggsbestämningarna med 14 dagars intervall efter dag 28.
Vid testning av andra kemikalier än jordbrukskemikalier används minst fem testkoncentrationer, och jordproverna analyseras med avseende på glukosinducerad respiration i början (dag 0) och i slutet av exponeringsperioden (dag 28). En delbestämning kan göras till exempel dag 7 om det bedöms vara nödvändigt. De resultat som erhålls dag 28 används för att bestämma ECx-värdet för kemikalien. Enligt val kan data rörande kontrollproverna från dag 0 användas för att uppskatta de ursprungliga mängderna metaboliskt aktiv mikrobiell massa i jorden (12).
1.7.2.2 Bestämning av glukosinducerad respiration
Den glukosinducerade respirationen bestäms för varje behandlat och obehandlat replikat och för varje provtagningstidpunkt. Jordproverna blandas med en glukosmängd som är tillräcklig för att framkalla en omedelbar och maximal respirationsrespons. Den mängd glukos som behövs för att framkalla en maximal respirationsrepons i en given jordtyp kan bestämmas genom ett preliminärt test som utförs med en serie glukoskoncentrationer (14). För sandhaltig jord med 0,5–1,5 % organiskt kol är det i regel tillräckligt med 2–4 g glukos per kg jord (torrvikt). Glukosen kan malas till pulver med ren kvartssand (10 g sand/kg torrvikt jord) och blandas sedan homogent in i jorden.
Jordproverna med glukos inkuberas vid 20 ± 2 oC i ett system som är lämpligt för bestämning av respirationen. Bestämningen kan göras kontinuerligt, varje timme eller var annan timme (se avsnitt 1.6.1). Den koldioxid som frigörs eller det syre som förbrukas mäts under 12 på varandra följande timmar och mätningarna bör inledas så snart som möjligt (1–2 timmar) efter glukostillförseln. De totala mängderna frigjord koldioxid eller förbrukat syre under de 12 timmarna uppmäts och den genomsnittliga respirationen bestäms.
2 DATA
2.1 BEHANDLING AV RESULTATEN
Vid testning av jordbrukskemikalier registreras mängden frigjord koldioxid eller förbrukat syre i varje replikatprov, och medelvärdena för alla replikat ges i tabellform. De värden som erhålls utvärderas med lämpliga och allmänt godtagna statistiska metoder (till exempel F-test, 5 % signifikansnivå). De värden som erhålls för den glukosinducerade respirationen uttrycks som mg koldioxid/kg torrvikt jord/h eller mg syre/torrvikt jord/h. Den genomsnittliga koldioxidbildningen eller genomsnittliga syreförbrukningen jämförs med situationen i kontrollprovet och den procentuella avvikelsen från kontrollprovet beräknas.
Vid testning av andra kemikalier än jordbrukskemikalier bestäms mängderna frigjord koldioxid eller förbrukat syre i varje replikat, och en dos-responskurva ritas upp för uppskattningen av ECx-värden. De värden som efter 28 dagar registreras för den glukosinducerade respirationen (mg koldioxid/kg torrvikt jord/h eller mg syre/torrvikt jord/h) i de behandlade proverna jämförs med motsvarande värden för kontrollprovet. Resultatet används för beräkning av den procentuella hämningen för varje testkoncentration. Dessa procentvärden ritas upp mot koncentrationen, och ECx-värden beräknas därefter med hjälp av statistiska metoder. Konfidensgränserna (p = 0,95) för de beräknade ECX-värdena bestäms med standardmetoder (15), (16), (17).
2.2 TOLKNING AV RESULTATEN
Om utvärderingen av testresultaten rörande jordbrukskemikalier visar att respirationen vid den lägre behandlingsnivån (den maximala förväntade koncentrationen) avviker från värdet vid kontrollnivån med högst 25 % vid alla provtagningstidpunkter efter dag 28, kan man anse att produkten inte har någon långtidseffekt på omvandlingen av kol i jord. Vid utvärdering av test rörande andra ämnen än jordbrukskemikalier används värden för EC50, EC25 och/eller EC10.
3. RAPPORTERING
TESTRAPPORT
Testrapporten ska innehålla nedan angivna uppgifter.
|
|
Fullständig identifiering av jorden, inbegripet
|
|
|
Testämne:
|
|
|
Testbetingelser:
|
|
|
Resultat:
|
4. HÄNVISNINGAR
|
1. |
EPPO (1994), ”Decision-Making Scheme for the Environmental Risk Assessment of Plant Protection Chemicals. Chapter 7: Soil Microflora”, EPPO Bulletin, 24, 1–16, 1994. |
|
2. |
BBA (1990), ”Effects on the Activity of the Soil Microflora”, BBA Guidelines for the Official Testing of Plant Protection Products, VI, 1-1 (andra utgåvan, 1990). |
|
3. |
EPA (1987), ”Soil Microbial Community Toxicity Test. EPA 40 CFR Part 797.3700. Toxic Substances Control Act Test Guidelines; Proposed rule”, den 28 september 1987. |
|
4. |
SETAC-Europe (1995), ”Procedures for assessing the environmental fate and ecotoxicity of pesticides”, red. M. R. Lynch, Pub. SETAC-Europe, Bryssel. |
|
5. |
OECD (1995), ”Final Report of the OECD Workshop on Selection of Soils/Sediments”, Belgirate, Italien, den 18–20 januari 1995. |
|
6. |
ISO 10381-6 (1993), ”Soil quality – Sampling. Guidance on the collection, handling and storage of soil for the assessment of aerobic microbial processes in the laboratory”. |
|
7. |
Anderson, J.P.E. (1987), ”Handling and Storage of Soils for Pesticide Experiments”, i ”Pesticide Effects on Soil Microflora”, red. L. Somerville och M.P. Greaves, kap 3, 45–60. |
|
8. |
Anderson, J.P.E. (1982), ”Soil Respiration”, i ”Methods of Soil Analysis – Part 2: Chemical and Microbiological Properties”, Agronomy Monograph, 9, red. A. L. Page, R. H. Miller och D. R. Keeney. 41, 831–871. |
|
9. |
ISO 11266-1 (1993), ”Soil Quality – Guidance on Laboratory Tests for Biodegradation in Soil: Part 1. Aerobic Conditions”. |
|
10. |
ISO 14239 (1997E), ”Markundersökningar – Inkubationssystem för bestämning av mineraliseringen av organiska kemikalier i jord under aeroba förhållanden”. |
|
11. |
O. Heinemeyer, H. Insam, E.A. Kaiser, och G. Walenzik (1989), ”Soil microbial biomass and respiration measurements; an automated technique based on infrared gas analyses”, Plant and Soil, 116, 77–81. |
|
12. |
ISO 14240-1 (1997), ”Markundersökningar – Bestämning av markens mikrobiella biomassa – Del 1: Sustratinducerad respirationsmetod”. |
|
13. |
ISO 14240-2 (1997), ”Markundersökningar – Bestämning av markens mikrobiella biomassa – Del 2: Fumigering – extraheringsmetod”. |
|
14. |
H. P. Malkomes, (1986), ”Einfluβ von Glukosemenge auf die Reaktion der Kurzzeit-Atmung im Boden Gegenüber Pflanzenschutzmitteln, Dargestellt am Beispiel eines Herbizide”, Nachrichtenbl. Deut. Pflanzenschutzd., Braunschweig, 38, 113–120. |
|
15. |
J.T. Litchfield, och F. Wilcoxon (1949), ”A simplified method of evaluating dose-effect – experiments”, Jour. Pharmacol. and Exper. Ther., 96, 99–113. |
|
(16) |
D.J. Finney, (1971), ”Probit Analysis”, tredje utgåvan, Cambridge, London och New York. |
|
(17) |
D.J. Finney (1978), ”Statistical Methods in biological Assay”, Griffin, Weycombe, Förenade kungariket. |
C.23 AEROB OCH ANAEROB OMVANDLING I JORD
1. METOD
Denna testmetod är i stort sett identisk med OECD TG 307 (2002).
1.1 INLEDNING
Testmetoden baserar sig på befintliga riktlinjer (1), (2), (3), (4), (5), (6), (7), (8), (9). Den testmetod som beskrivs här är avsedd för utvärdering av aerob och anaerob omvandling av kemikalier i jord. Bestämningar görs av i) testämnets omvandlingshastighet och ii) karaktären och hastigheterna för bildning och minskning av de omvandlingsprodukter som växter och jordlevande organismer kan exponeras för. Sådana undersökningar behövs för kemikalier som appliceras direkt i jorden eller sannolikt kommer att nå jorden. Resultaten kan också användas för att utarbeta provtagnings- och analysprotokoll för relaterade fältstudier.
För bedömning av omvandlingsmönstren är det i regel tillräckligt med aeroba och anaeroba undersökningar av endast en jordtyp (8), (10), (11). Omvandlingshastigheterna bör bestämmas i minst tre ytterligare jordtyper (8), (10).
Vid en OECD-workshop om valet av jord och sediment som hölls i Belgirate, Italien, 1995 (10), kom man särskilt överens om hur många och vilka typer av jord som ska användas i detta test. De jordtyper som testas bör vara representativa för de miljöbetingelser där användning eller utsläpp förväntas ske. Exempelvis kemikalier som sannolikt kommer att släppas ut i subtropiska till tropiska klimat bör testas med jordmåner i klasserna Ferrasoler eller Nitosoler (FAO-systemet). Workshopen i Belgirate gav också rekommendationer rörande insamling, hantering och lagring av jordprover, baserade på ISO-riktlinjer (15). Även användningen av jord för risodling behandlas i denna metod.
1.2 DEFINITIONER
Testämne: Alla ämnen, oavsett det gäller den ursprungliga föroreningen eller relevanta omvandlingsprodukter.
Omvandlingsprodukter: Alla ämnen som uppstår till följd av biotiska eller abiotiska omvandlingsreaktioner av testämnet, inbegripet CO2 och produkter i bundna rester.
Bundna rester: Dessa är sådana föreningar i jorden, i växter eller i djur som blir kvar i matrisen i form av ursprungligt ämne eller dess metaboliter eller omvandlingsprodukter efter extraktion. Extraktionsmetoden får inte i avsevärd grad leda till ändringar i själva föreningarna eller matrisens struktur. Bindningens karaktär kan delvis klarläggas genom matrisstörande extraktionsmetoder och sofistikerade analysmetoder. Hittills har man på detta sätt kunnat identifiera till exempel kovalenta bindningar, jonbindningar och sorptiva bindningar samt inneslutning. Generellt sett innebär bildningen av bundna rester att biotillgängligheten blir signifikant mindre (12) (modifierat ur IUPAC 1984 (13)).
Aerob omvandling: Reaktioner som sker i närvaro av molekylärt syre (14).
Anaerob omvandling: Reaktioner som sker utan närvaro av molekylärt syre (14).
Jord: En blandning av mineraliska och organisk-kemiska beståndsdelar. De senare består av föreningar med hög kol- och kvävehalt samt hög molekylvikt. Dessa föreningar påverkas av små organismer (nästintill mikroorganismer). Jord kan hanteras i två olika tillstånd:
|
a) |
Ostörd, såsom den har uppstått under tidens gång i karakteristiska skikt av ett flertal olika jordtyper. |
|
b) |
Störd, vilket normalt gäller för odlad mark eller fall där jordprover tas genom grävning och därefter undersöks med denna testmetod (14). |
Mineralisering: Det fullständiga sönderfallet av organiska föreningar till CO2 och H2O under aeroba betingelser samt till CH4, CO2 och H2O under anaeroba betingelser. Med mineralisering avses här ett omfattande sönderfall som sker när en 14C-märkt förening används och en märkt kolatom oxideras under frigörande av motsvarande mängd 14CO2 (14).
Halveringstid: Betecknas med t0,5 – den tid det tar för ett testämne att omvandlas till 50 % när omvandlingen kan beskrivas med kinetik av första ordningen. Denna tid är oberoende av koncentrationen.
DT50 (tid för försvinnande 50): Den tid inom vilken testämnets koncentration går ned med 50 %. Denna tid är inte samma som halveringstiden t0,5 i fall där omvandlingen inte följer kinetik av första ordningen.
DT75 (tid för försvinnande 75): Den tid inom vilken testämnets koncentration går ned med 75 %.
DT90 (tid för försvinnande 90): Den tid inom vilken testämnets koncentration går ned med 90 %.
1.3 REFERENSÄMNEN
Referensämnen bör användas för karakterisering och/eller identifiering av omvandlingsprodukterna genom spektroskopiska och kromatografiska metoder.
1.4 TESTETS TILLÄMPBARHET
Metoden kan tillämpas på alla kemiska ämnen (omärkta eller radioaktivt märkta) för vilka det finns tillgång till en analysmetod som är tillräckligt exakt och känslig. Metoden kan tillämpas på föreningar som är i viss mån flyktiga, ickeflyktiga, vattenlösliga eller vattenolösliga. Testet bör inte tillämpas på kemikalier som är mycket flyktiga från jord (till exempel desinfektionsmedel, organiska lösningsmedel) och som därför inte hålls kvar i jorden under de försöksbetingelser som gäller för detta test.
1.5 UPPLYSNINGAR OM TESTÄMNET
Omärkta eller märkta testämnen kan användas för bestämning av omvandlingshastigheten. Märkt material behövs för undersökning av omvandlingsmönstret och för bestämning av en massbalans. C-märkning rekommenderas, även om andra isotoper, såsom 13C, 15N, 3H och 32P kan användas. Märkningen bör om möjligt finnas i molekylens mest stabila del(ar) (40). Testämnets renhetsgrad bör vara minst 95 %.
Innan test rörande aerob och anaerob omvandling i jord utförs bör följande upplysningar om testämnet finnas tillgängliga:
|
a) |
löslighet i vatten (metod A.6), |
|
b) |
löslighet i organiska lösningsmedel, |
|
c) |
ångtryck (metod A.4) och Henrys lag-konstant, |
|
d) |
fördelningskoefficient n-oktanol/vatten (metod A.8), |
|
e) |
kemisk stabilitet i mörker (hydrolys) (metod C.7), |
|
f) |
pKa om en molekyl är benägen till protonering eller deprotonering (OECD-riktlinje 112) (16). |
Det kan även vara till nytta att känna till exempelvis testämnets toxicitet för jordlevande mikroorganismer (testmetoderna C.21 och C.22)(16).
Det bör finnas tillgång till analysmetoder (inbegripet extraktions- och reningsmetoder) för kvantifiering och identifiering av testämnet och dess omvandlingsprodukter.
1.6 PRINCIP FÖR TESTMETODEN
Jordproverna behandlas med testämnet och inkuberas i mörker i flaskor av biometertyp eller i genomflödessystem under kontrollerade laboratoriebetingelser (konstant temperatur och jordfuktighet). Efter lämpliga tidsintervall extraheras jordproverna och analyseras med avseende på det ursprungliga ämnet och dess omvandlingsprodukter. Flyktiga produkter samlas upp med hjälp av lämplig absorptionsutrustning. Med hjälp av C-märkt material kan de olika mineraliseringshastigheterna av testämnet bestämmas genom att den 14CO2 som bildas fångas upp. Därefter kan en massbalans som inbegriper bildningen av jordbundna rester upprättas.
1.7 KVALITETSKRITERIER
1.7.1 Utbyte
Extraktion och analys av minst dubbla jordprover omedelbart efter tillförsel av testämnet ger en första indikation på analysmetodens repeterbarhet och enhetligheten hos det förfarande som har använts för tillförsel av testämnet. Utbyten för senare stadier av försöken erhålls genom massbalanserna. Utbytena bör vara 90–110 % för radioaktivt märkta kemikalier (8) och 70–110 för omärkta kemikalier (3).
1.7.2 Analysmetodens repeterbarhet och känslighet
Analysmetodens repeterbarhet (med undantag av den initiala extraktionseffektiviteten) när det gäller att kvantifiera testämnet och omvandlingsprodukterna kan kontrolleras genom en duplikatanalys av samma extrakt ur jorden, som bör ha inkuberats tillräckligt länge för att omvandlingsprodukter ska ha bildats.
Analysmetodens detektionsgräns (LOD) för testämnet och för omvandlingsprodukterna bör vara minst 0,01 mg· kg-1 jord (som testämne) eller 1 % av den tillförda dosen, beroende på vilket värde som är lägre. Likaså bör kvantifieringsgränsen (LOQ) anges.
1.7.3 Noggrannheten hos omvandlingsdata
Genom regressionsanalys av testämneskoncentrationerna som en funktion av tiden fås ungefärliga upplysningar om omvandlingskurvans tillförlitlighet och en möjlighet att beräkna konfidensgränserna för halveringstiderna (när det gäller kinetik av pseudoförsta ordningen) eller DT50-värden och, om tillämpligt, värden för DT75 och DT90.
1.8 BESKRIVNING AV TESTMETODEN
1.8.1 Utrustning och kemiska reagenser
Inkubationssystemet kan vara ett statiskt slutet system eller ett lämpligt genomflödessystem (7), (17). Exempel på ett lämplig genomflödessystem för inkubation av jord och på flaskor av biometertyp finns i figur 1 respektive figur 2. Båda typerna av inkubationssystem har sina fördelar och begränsningar (7), (17).
Dessutom behövs normal laboratorieutrustning, särskilt följande:
|
— |
Instrument för analysmetoder såsom gas-vätskekromatografi (GLC), högtrycksvätskekromatografi (HPLC), tunnskiktskromatografi (TLC), inbegripet tillbörliga detektionssystem för analys av radioaktivt märkta eller omärkta ämnen eller metod med invers isotoputspädning. |
|
— |
Instrument för identifiering (till exempel genom masspektrometri, GC-masspektrometri, HPLC-masspektrometri, kärnmagnetisk resonans (NMR)). |
|
— |
Vätskeskintillationsräknare. |
|
— |
Oxidationsmedel för förbränning av radioaktivt material. |
|
— |
Centrifug. |
|
— |
Extraktionsapparatur (till exempel centrifugrör för kallextraktion och Soxhlet-apparatur för kontinuerlig extration med återflöde). |
|
— |
Utrustning för koncentrering av lösningar och extrakt (till exempel roterande evaporator). |
|
— |
Vattenbad. |
|
— |
Mekanisk blandare (till exempel knådningsmaskin eller roterande mixer). |
Kemiska reagenser, till exempel följande:
|
— |
NaOH, p.a. kvalitet, 2 mol dm-3 eller annan lämplig bas (till exempel KOH, etanolamin). |
|
— |
H2SO4, p.a kvalitet, 0,05 mol dm-3. |
|
— |
Etylenglykol, p.a. kvalitet. |
|
— |
Fasta absorptionsmaterial såsom kalk- och polyuretanproppar. |
|
— |
Organiska lösningsmedel (p.a. kvalitet), exempelvis aceton eller metanol. |
|
— |
Skintillationsvätska. |
1.8.2 Tillförsel av testämnet
Innan testämnet tillförs och fördelas i jorden kan det lösas upp i vatten (avjoniserat eller destillerat) eller vid behov i minsta möjliga mängd aceton eller något annat organiskt lösningsmedel (6) i vilket testämnet är tillräckligt lösligt och stabilt. Den mängd lösningsmedel som används får dock inte ha betydande inverkan på jordens mikrobiella aktivitet (se avsnitten 1.5 samt 1.9.2 och 1.9.3). Lösningsmedel som hämmar den mikrobiella aktiviteten, såsom kloroform, diklormetan och andra halogenerade lösningsmedel, bör undvikas.
Testämnet kan också tillföras i fast form, till exempel inblandat i kvartssand (6) eller i ett litet delprov av testjorden som har lufttorkats och steriliserats. Om testämnet tillförs med hjälp av lösningsmedel måste detta få avdunsta innan det spikade delprovet tillförs det ursprungliga icke-sterila jordprovet.
För allmänna kemikalier, vars som huvudsakligen når jorden genom applicering av avloppsslam eller jordbruksprodukter, bör testämnet först tillföras slammet som därefter blandas i jordprovet (se avsnitten 1.9.2 och 1.9.3).
Användningen av formulerade produkter rekommenderas i regel inte. För svårlösliga testämnen kan dock användningen av formulerat material vara ett lämpligt alternativ.
1.8.3 Jord
1.8.3.1 Valet av jord
För bestämning av omvandlingsmönstret används en representativ jordtyp – sandy loam, silty loam, loam eller loamy sand (enligt FAO- och USDA-klassificeringen (18)), pH 5,5–8,0, halten organiskt kol 0,5–2,5 % och en mikrobiell biomassa som utgör minst 1 % av totala mängden organiskt kol (10).
Vid undersökning av omvandlingshastigheter behövs minst tre ytterligare jordtyper som representerar ett urval relevanta jordtyper. Jordtyperna bör vara sinsemellan olika i fråga om halten organiskt kol, pH, lerhalt och mikrobiell biomassa (10).
All jord bör karakteriseras minst med avseende på textur ( % sand, % silt, % clay) (enligt FAO- och USDA-klassificering (18)), pH, katjonbyteskapacitet, organiskt kol, bulktäthet, vattenretentionsegenskaper (41) och mikrobiell biomassa (endast vid aeroba undersökningar). Tilläggsupplysningar om jordens egenskaper kan vara till nytta vid tolkning av resultaten. Jordens egenskaper kan bestämmas med de metoder som rekommenderas i hänvisningarna (19), (20), (21), (22), (23). Den mikrobiella biomassan bör bestämmas med SIR-metoden (substratinducerad respiration) (25), (26) eller alternativa metoder (20).
1.8.3.2 Insamling, hantering och lagring ax jord
Det bör finnas tillgång till detaljerade upplysningar om historian för den plats där testjorden tas. Upplysningarna bör omfatta exakt läge, växtlighetstäcke, behandlingar med kemikalier, behandlingar med organiska och oorganiska gödselmedel, tillförsel av biologiska material eller förorening. Om jorden har behandlats med testämnet eller ett strukturellt analogt ämne inom de fyra senaste åren bör den inte användas för undersökningar rörande omvandling (10), (15).
Jordproverna bör vara nytagna (från A-horisonten eller det översta 20 cm-skiktet) och ha en lämplig vattenhalt med tanke på siktning. För all annan jord än den från risfält bör provtagning undvikas under eller omedelbart efter långa perioder (mer än 30 dagar) av torka, frost eller översvämning (14). Proverna bör transporteras på sådant sätt att förändringar i vattenhalt minimeras. Under transporten bör proverna hållas i mörker med så fri tillgång till luft som det är möjligt. För detta ändamål är det i regel tillräckligt med en löst sluten polyetylenpåse.
Jordproverna bör bearbetas så snart som möjligt efter provtagningen. Först avlägsnas vegetation, större fauna och stenar och därefter får jorden passera ett 2 mm-sikt som fångar upp små stenar, fauna och växtrester. Omfattande torkning eller krossning av jorden före siktningen bör undvikas (15).
Om provtagningen försvåras under vintern (jorden är frusen eller snötäckt) kan proverna tas från en sats jord som har lagrats i växthus under ett växttäcke (till exempel gräs eller gräs–klöverblandning). Alla prover som undersöks bör helst vara nytagna. Om lagring av bearbetade jordprover inte kan undvikas före undersökningen måste betingelserna vara lämpliga och lagringstiden begränsad (4 ± 2 oC och högst tre månader) för att den mikrobiella aktiviteten ska upprätthållas (42). Närmare anvisningar om insamling, hantering och lagring av jordprover avsedda för undersökningar av bioomvandling finns i hänvisningarna (8), (10), (15), (26), (27).
Innan ett bearbetat jordprov används för detta test bör provet förinkuberas för att framkalla groning och avlägsnande av frön, och för att återställa jämvikten för den mikrobiella metabolismen efter den ändring av betingelser som sker mellan provtagning eller lagring och inkubation. Det är i regel tillräckligt med en förinkubationsperiod vars längd är 2–28 dagar och vars temperatur- och fuktbetingelser är ungefär desamma som under det egentliga försöket (15). Lagrings- och förinkubationsperioden bör tillsammans inte vara längre än tre månader.
1.9 TESTETS UTFÖRANDE
1.9.1 Testbetingelser
1.9.1.1 Testtemperatur
Jordproverna bör under hela testperioden inkuberas i mörker vid en konstant temperatur som är representativ för de klimatförhållanden som råder på den plats där testämnet används eller släpps ut. Temperaturen 20 ± 2 oC rekommenderas för testämnen som kan nå jorden i tempererade klimat. Temperaturen bör övervakas.
För kemikalier som appliceras eller släpps ut i kallare klimat (till exempel i de nordiska länderna under hösten och vintern) bör extra jordprover även inkuberas vid en lägre temperatur (till exempel 10 ± 2 oC).
1.9.1.2 Fukthalt
För omvandlingstest som görs under aeroba betingelser bör jordens fukthalt (43) justeras till ett pF-värde mellan 2,0 och 2,5 och upprätthållas på denna nivå (3). Jordens fukthalt uttrycks som massan vatten per massan torr jord. Fukthalten bör regelbundet kontrolleras (till exempel med två veckors intervall) genom vägning av inkubationsflaskorna. Vattenförluster kompenseras genom att tillföra vatten (helst sterilfiltrerat kranvatten). När vatten tillförs är det viktigt att förebygga eller minimera förluster av testämnet eller omvandlingsprodukter som kan ske genom avdunstning eller fotonedbrytning (i förekommande fall).
Vid omvandlingstest som görs under anaeroba betingelser och risfältsbetingelser mättas jorden med vatten genom översvämning.
1.9.1.3 Aeroba inkubationsbetingelser
I genomflödessystem upprätthålls de aeroba betingelserna genom periodisk spolning eller kontinuerlig ventilering med fuktad luft. I biometerflaskorna upprätthålls luftväxlingen genom diffusion.
1.9.1.4 Sterila aeroba betingelser
För att få upplysningar om relevansen hos ett testämnes abiotiska omvandling kan jordproverna steriliseras (steriliseringsmetoder beskrivs i hänvisningarna 16 och 29), behandlas med sterilt testämne (till exempel så att lösningen tillförs genom ett sterilt filter) och luftas med fuktad steril luft (se avsnitt 1.9.1.3. När det gäller jord för risodling bör jord och vatten steriliseras, och inkubationen görs därefter enligt beskrivningen i avsnitt 1.9.1.6.
1.9.1.5 Anaeroba inkubationsbetingelser
För att etablera och upprätthålla anaeroba betingelser inkuberas jorden efter behandlingen med testämnet i aeroba betingelser under en period vars längd är 30 dagar, en halveringstid eller DT50 (beroende på vilken tid som är kortast). Därefter mättas jorden med vatten (1–3 cm vattenskikt) och inkubationssystemet spolas med en inert gas (till exempel kväve eller argon (44). Testsystemet måste vara utformat så att pH, syrehalt och redoxpotential kan bestämmas, och det måste innehålla uppfångare för flyktiga produkter. System av biometertyp måste vara slutna för att luftinträngning genom diffusion ska undvikas.
1.9.1.6 Inkubationsbetingelser för ris
Vid undersökning av omvandlingen i jord för risodling översvämmas jorden med ett vattenskikt på 1–5 cm och testämnet tillförs vattenfasen (9). Jorddjupet bör vara minst 5 cm. Systemet ventileras med luft på samma sätt som vid försök i aeroba betingelser. Vattenskiktets pH, syrehalt och redoxpotential bör övervakas och rapporteras. Före det egentliga försöket behövs en förinkubationsperiod på minst två veckor (se avsnitt 1.8.3.2).
1.9.1.7 Testets varaktighet
Försöken rörande hastighet och mönster bör normalt inte överskrida 120 dagar (45) (3), (6), (8). Det beror på att jordens mikrobiella aktivitet efter denna period sannolikt börjar minska med tiden, eftersom det gäller ett konstgjort laboratoriesystem som är isolerat och inte får naturlig påfyllning. Om det finns ett behov för att karakterisera minskningen av testämnet samt bildningen och minskningen av viktiga omvandlingsprodukter, kan försöksperioden förlängas (till exempel till 6 eller 12 månader) (8). Förlängda inkubationsperioder bör motiveras i testrapporten, som då även bör innehålla resultat från bestämningar av biomassa under och vid slutet av dessa perioder.
1.9.2 Testets utförande
Omkring 50–200 g jord (torrvikt) placeras i varje inkubationsflaska (se figurerna 1 och 2 i bilaga 3), och jorden behandlas med testämnet genom en av de metoder som beskrivs i avsnitt 1.8.2. Om organiska lösningsmedel används för tillförsel av testämnet bör lösningsmedlet avlägsnas från jorden genom avdunstning. Jorden blandas grundligt med en spatel och/eller genom att skaka flaskan. Om försöket görs under risfältsbetingelser, bör jord och vatten blandas grundligt efter tillförseln av testämnet. Små delprover (till exempel 1 g) av den behandlade jorden bör analyseras med avseende på testämnet för att kontrollera att fördelningen är jämn. En alternativ metod beskrivs nedan.
Tillförseln av testämne bör motsvara den högsta dosen som anges i bruksanvisningen för en växtskyddsprodukt, och en jämn tillförsel till ett lämpligt djup i åkern (till exempel 10 cm av det översta jordskiktet (46). Till exempel för kemikalier som appliceras på bladen eller på jorden, utan att blandas ned i jorden, är 2,5 cm ett lämpligt djup för beräkning av hur mycket kemikalie som bör tillföras varje flaska. För kemikalier som tillförs in i jorden används det djup som anges i produktens bruksanvisning. För allmänna kemikalier bör dosen uppskattas enligt den mest relevanta tillförselvägen. Om till exempel den viktigaste tillförselvägen till jorden är via avloppsslam, bör kemikalien doseras i slammet i sådan mängd att koncentrationen motsvarar den förväntade koncentrationen i det slam som används på åkern, och den mängd slam som tillförs testjorden ska motsvara det som normalt tillförs jordbruksjordar. Om denna koncentration inte är tillräckligt hög för att det ska vara möjligt att identifiera de viktigaste omvandlingsprodukterna kan man försöka med att inkubera separata jordprover som innehåller högre halter. Mycket höga halter som påverkar jordens mikrobiella funktioner bör dock undvikas (se avsnitten 1.5 och 1.8.2).
Alternativt kan en större jordsats (till exempel 1–2 kg) behandlas med testämnet. Satsen blandas därefter omsorgsfullt i en lämplig blandare och överförs i mindre portioner (50–200 g) till inkubationsflaskorna (till exempel med hjälp av provfördelare). Små delprover (till exempel 1 g) av den behandlade jordsatsen bör analyseras med avseende på testämnet för att kontrollera att fördelningen är jämn. Detta förfarande rekommenderas eftersom det ger möjlighet till en jämnare fördelning av testämnet i jorden.
Även obehandlade jordprover inkuberas under samma betingelser (aeroba) som de prover som har behandlats med testämnet. Proverna används för bestämningar av biomassa under och vid slutet av försöken.
När testämnet tillförs jorden upplöst i organiskt lösningsmedel bör jordprover som har behandlats med samma mängd lösningsmedel inkuberas under samma betingelser (aeroba) som gäller för de prover som har behandlats med testämnet. Dessa prover används för bestämning av biomassan i början, under och i slutet av undersökningen, i syfte att kontrollera lösningsmedlets inverkan på den mikrobiella massan.
Behållarna som innehåller behandlat jordprov monteras i ett genomflödessystem av det slag som beskrivs i figur 1 eller försluts med en absorptionskolonn av det slag som visas i figur 2 (se bilaga 3).
1.9.3 Provtagning och mätning
Dubbla inkubationsflaskor tas ur systemet med lämpliga intervall, jordproverna extraheras med lämpliga lösningsmedel med olika polaritet och analyseras därefter med avseende på testämnet och/eller omvandlingsprodukter. Ett välplanerat test ska omfatta så många flaskor att två flaskor kan tas ur systemet vid varje provtagningstillfälle. Likaså avlägsnas absorptionslösningar eller fasta absorptionsmaterial med olika intervall (7 dagars intervall under den första månaden och därefter 17 dagars intervall) under och i slutet av inkubationsperioden för varje jordprov, och analyseras med avseende på flyktiga produkter. Utöver ett jordprov som tas direkt efter tillförseln (dag 0) bör testet omfatta minst 5 ytterligare provtagningstillfällen. Tidsintervallerna bör väljas på sådant sätt att mönstret för minskningen av testämnet och mönstren för bildning och minskning av omvandlingsprodukter kan bestämmas (till exempel 0, 1, 3, 7 dagar, 2, 3 veckor, 1, 2, 3 månader).
Vid användning av 14C-märkt testämne kvantifieras icke-extraherbar radioaktivitet genom förbränning och en massbalans beräknas för varje provtagningsintervall.
När det gäller anaerob inkubation och risfältsinkubation analyseras jord- och vattenfasema tillsammans med avseende på testämnet och omvandlingsprodukter, eller separeras genom filtrering eller centrifugering före extraktion och analys.
1.9.4 Valfria tilläggstest
Aeroba, icke-sterila undersökningar vid ytterligare temperaturer och jordfukthalter kan vara till hjälp för uppskattningen av temperaturens och fukthaltens inverkan på omvandlingshastigheten i jord för ett testämne och/eller dess omvandlingsprodukter.
Man kan även försöka få en närmare karakterisering av den icke-extraherbara radioaktiviteten till exempel med hjälp av superkritisk vätskeextraktion.
2. DATA
2.1 BEHANDLING AV RESULTATEN
Mängden testämne, omvandlingsprodukter, flyktiga ämnen (endast i %) och icke-extraherbara ämnen bör anges som procent av initialkoncentrationen för det tillförda ämnet och, där det är lämpligt, som mg· kg-1 jord (torrvikt) för varje provtagningsintervall. En massbalans bör anges som procent av initialkoncentrationen för det tillförda ämnet för varje provtagningsintervall. Med hjälp av en grafisk presentation av testämnets koncentration mot tiden kan halveringstiden för testämnets omvandling eller DT50 uppskattas. De viktigaste omvandlingsprodukterna bör identifieras och deras koncentrationer bör plottas mot tiden så att bildnings- och minskningshastigheterna kan bestämmas. En viktig omvandlingsprodukt är varje produkt som utgör > 10 % av den tillförda dosen vid varje tidpunkt medan undersökningen pågår.
De uppfångade flyktiga produkterna ger en viss indikation om testämnets och dess omvandlingsprodukters potentiella flyktighet ur jord.
Mer exakta bestämningar av halveringstiderna eller DT50 och, vid behov, DT75 och DT90 bör göras med hjälp av lämpliga beräkningar med kinetiska modeller. Värdena för halveringstid och DT50 bör rapporteras tillsammans med en beskrivning av vilken modell som har använts, vilken ordning av kinetik som har tillämpats och bestämningskoefficient (r2. Första ordningens rekommenderas, utom om r2 < 0,7. Vid behov bör beräkningar även göras för viktiga omvandlingsprodukter. Exempel på lämpliga modeller beskrivs i hänvisningarna 31–35.
Om hastigheterna undersöks vid flera temperaturer bör omvandlingshastigheterna beskrivas som en funktion av temperaturen inom försökstemperaturintervallet. För detta används Arrhenius-förhållandet enligt följande formel.
 eller
eller 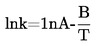 ,
,
där ln A och B är regressionskonstanter från skärningspunkten respektive lutningen för en anpassad linje som har erhållits genom linjär regression av ln k mot 1/T, där k är hastighetskonstanten vid temperaturen T och T är temperaturen uttryckt som grader Kelvin. Det är viktigt att notera att Arrhenius-förhållandet endast är giltigt inom ett begränsat temperaturområde i situationer där omvandlingen sker genom mikrobiell aktivitet.
2.2 UTVÄRDERING OCH TOLKNING AV RESULTATEN
Trots att undersökningarna görs i ett konstgjort system i laboratoriebetingelser, ger resultatet ändå en möjlighet att uppskatta testämnets omvandlingshastighet och omvandlingsprodukternas bildnings- och minskningshastighet i fältbetingelser (36), (37).
En undersökning av mönstret för omvandlingen av ett testämne ger upplysningar om hur ett ämne som tillförs jorden förändras strukturellt genom kemiska och mikrobiella reaktioner.
3. RAPPORTERING
TESTRAPPORT
Testrapporten ska innehålla följande:
|
|
Testämne:
|
|
|
Referenstämnen:
|
|
|
Testjord:
|
|
|
Testbetingelser:
|
|
|
Resultat:
|
4. HÄNVISNINGAR
|
1. |
US Environmental Protection Agency (1982), ”Pesticide Assessment Guidelines, Subdivision N. Chemistry: Environmental Fate”. |
|
2. |
Agriculture Canada (1987), ”Environmental Chemistry and Fate. Guidelines for registration of pesticides in Canada”. |
|
3. |
Europeiska unionen (EU) (1995), ”Kommissionens direktiv 95/36/EG av den 14 juli 1995 om ändring av rådets direktiv 91/414/EEG om utsläppande av växtskyddsprodukter på marknaden. Bilaga II, del A och bilaga III, del A: Omvandling, spridning och fördelning i miljön”. |
|
4. |
Dutch Commission for Registration of Pesticides (1995), ”Application for registration of a pesticide. Section G: Behaviour of the product and its metabolites in soil, water and air”. |
|
5. |
BBA (1986), ”Richtlinie fur die amtliche Prüfung von Pflanzenschutzmitteln, Teil IV, 4-1. Verbleib von Pflanzenschutzmitteln im Boden – Abbau, Umwandlung und Metabolismus”. |
|
6. |
ISO/DIS 11266-1 (1994), ”Soil Quality – Guidance on laboratory tests for biodegradation of organic Chemicals in soil – Part 1: Aerobic conditions”. |
|
7. |
ISO 14239 (1997), ”Markundersökningar – Inkubationssystem för bestämning av mineraliseringen av organiska kemikalier i jord under aeroba förhållanden”. |
|
8. |
SETAC (1995), ”Procedures for Assessing the Environmental Fate and Ecotoxicity of Pesticides”, Mark R. Lynch, red. |
|
9. |
MAFF – Japan 2000, ”Draft Guidelines for transformation studies of pesticides in soil – Aerobic metabolism study in soil under paddy field conditions (flooded)”. |
|
10. |
OECD (1995), ”Final Report of the OECD Workshop on Selection of Soils/Sediments”, Belgirate, Italien, den 18–20 januari 1995. |
|
11. |
J. A. Guth (1980), ”The study of transtbrmations. In Interactions between Herbicides and the Soil (R.J.Hance, red)”, Academic Press, 123–157. |
|
12. |
DFG, ”Pesticide Bound Residues in Soil”, Wiley – VCH, (1998). |
|
13. |
T.R. Roberts, ”Non-extractable pesticide residue in soils and plants”, Pure Appl. Chem, 56, 945–956 (IUPAC 1984). |
|
14. |
OECD Test Guideline 304 A, ”Inherent Biodegradability in Soil” (antagen den 12 maj 1981). |
|
15. |
ISO 10381-6 (1993), ”Soil Quality – Sampling – Part 6: Guidance on the collection, handling and storage of soil for the assessment of aerobic microbial processes in the laboratory”. |
|
16. |
Bilaga V till direktiv 67/548/EEG. |
|
17. |
J. A. Guth (1981), ”Experimental approaches to studying the fate of pesticides in soil”, Progress in Pesticide Biochemislry, (red. D. H. Hutson, T. R. Roberts), J. Wiley & Sons, Vol I, 85–114. |
|
18. |
Soil Texture Classification (US and FAO systems), Weed Science, 33, Suppl. 1 (1985) och Soil Sci. Soc. Amer. Proc, 26, 305(1962). |
|
19. |
Methods of Soil Analysis (1986), ”Part 1, Physical and Mineralogical Methods” (red. A. Klute), Agronomy Series, 9, andra utgåvan. |
|
20. |
Methods of Soil Analysis (1982), ”Part 2, Chemical and Microbiological Properties”, (red. A. L. Page, R. H. Miller och D.R. Kelney), Agronomy Series, 9, andra utgåvan. |
|
21. |
ISO Standard Compendium Environment (1994), ”Soil Quality – General aspects; chemical and physical methods of analysis; biological methods of analysis”, första utgåvan. |
|
22. |
E. Muckenhausen (1975), ”Die Bodenkunde und ihre geologischen, geomorphologischen, mineralogischen und petrologischen Grundlagen”, DLG-Verlag, Frankfurt am Main. |
|
23. |
F. Scheffer, P. Schachtschabel (1975), ”Lehrbuch der Bodenkunde”, F. Enke Verlag, Stuttgart. |
|
24. |
J.P.E. Anderson, K. H. Domsch (1978), ”A physiological method for the quantitative measurement of microbial biomass in soils”, SoilBiol. Biochem., 10, 215–221. |
|
25. |
ISO 14240-1 och 2 (1997). Markundersökningar – Bestämning av markens mikrobiella biomassa – Del 1: Sustratinducerad respirationsmetod. Del 2: Fumigering – extraheringsmetod. |
|
26. |
J.P.E. Anderson (1987), ”Handling and storage of soils for pesticide experiments”, Pesticide Effects on Soil Microflora, (red. M. P. Somerville och Greaves), Taylor & Francis, 45–60. |
|
27. |
Kato, Yasuhiro (1998), ”Mechanism of pesticide transformation in the environment: Aerobic and bio-transformation of pesticides in aqueous environment”. Proceedings of the 16th Symposium on Environmental Science of Pesticide, 105–120. |
|
28. |
O. Keuken, J.P.E. Anderson (1996), ”Influence of storage on biochemical processes in soil”, Pesticides, Soil Microbiology and Soil Quality, 59–63 (SETAC-Europa). |
|
29. |
B. Stenberg, M. Johansson, M. Pel, K. Sjödahl-Svensson, J. Stenström, L. Torstensson (1996), ”Effect of freeze and cold storage of soil on microbial activities and biomass”, Pesticides, Soil Microbiology and Soil Quality, 68–9 (SETAC-Europa). |
|
30. |
. Gennari, M. Negre, R. Ambrosoli (1987), ”Effects of ethylene oxide on soil microbial content and some chemical characteristics”, Plant and Soil, 102, 197–200. |
|
31. |
J. P.E. Anderson (1975), ”Einfluss von Temperatur und Feuchte auf Verdampfung, Abbau und Festlegung von Diallat im Boden”, Z. PflKrankh Pflschutz, Sonderheft VII, 141–146. |
|
32. |
J.W. Hamaker (1976), ”The application of mathematical modelling to the soil persistence and accumulation of pesticides”, Proc. BCPC Symposium: Persistence of Insecticides and Herbicides, 181–199. |
|
33. |
C.A.I. Goring, D.A. Laskowski, J.W. Hamaker, R.W. Meikle (1975), ”Principles of pesticide degradation in soil”, Environmental Dynamics of Pesticides, (red. R. Haque och V.H. Freed), 135–172. |
|
34. |
G. Timme, H. Frehse, V. Laska (1986), ”Statistical interpretation and graphic representation of the degradational behaviour of pesticide residues”II. Pflanzenschutz – Nachrichten Bayer, 39, 188–204. |
|
35. |
G. Timme, H. Frehse (1980), ”Statistical interpretation and graphic representation of the degradational behaviour of pesticide residues”, I. Pflanzenschutz – Nachrichten Bayer, 33, 47–60. |
|
36. |
D.I. Gustafson, L. R. Holden (1990), ”Non-linear pesticide dissipation in soil; a new model based on spatial variability”, Environm. Sci. Technol., 24, 1032–1041. |
|
37. |
K. Hurle, A. Walker (1980), ”Persistence and its prediction”, Interactions between Herbicides and the Soil, (red. R. J. Hance), Academic Press, 83–122. |
Bilaga 1
TENSION, FÄLTKAPACITET (FC) OCH VATTENKAPACITET (WHC) (47)
|
Vattenpelarens höjd (cm) |
pF (48) |
bar (49) |
Anmärkningar |
|
107 |
7 |
104 |
Torr jord |
|
1,6 • 104 |
4,2 |
16 |
Permanent vissningsgräns |
|
104 |
4 |
10 |
|
|
103 |
3 |
1 |
|
|
6 • 102 |
2,8 |
0,6 |
|
|
3,3 • 102 |
2,5 |
0,33 (50) |
Område för fältkapacitet (51) |
|
102 |
2 |
0,1 |
|
|
60 |
1,8 |
0,06 |
|
|
33 |
1,5 |
0,033 |
|
|
10 |
1 |
0,01 |
WHC (ungefärligt värde) |
|
1 |
0 |
0,001 |
Vattenmättad jord |
Tensionen mäts i cm vattenpelare eller i bar. På grund av det stora området för tensionen uttrycks den i praktiken som ett pF-värde som är ekvivalent med logaritmen av cm vattenpelare.
Fältkapaciteten definieras som den mängd vatten som en naturjord kan hålla mot gravitationen i två dagar efter en längre regnperiod eller efter tillräcklig bevattning. Den bestäms på platsen i ostörd jord. Denna bestämning kan således inte göras på störda jordprover i laboratorium. FC-värden som bestäms med störd jord kan uppvisa stora systematiska variationer
Vattenkapaciteten (WHC) bestäms i laboratoriet med ostörd och störd jord genom att mätta en jordkolonn med vatten genom transport genom kapillärkraft. WHC är särskilt användbar för störda jord och kan vara upp till 30 % högre än fältkapaciteten (1). Det är också försöksmässigt enklare att bestämma WHC än att få tillförlitliga FC-värden.
Noter
Bilaga 2
JORDENS FUKTHALT (g vatten per 100 g torr jord) I OLIKA JORDTYPER FRÅN OLIKA LÄNDER
|
|
|
Jordens fukthalt vid |
||
|
Jordtyp |
Land |
|||
|
|
|
WHC (52) |
pF= 1,8 |
pF = 2,5 |
|
Sand |
Tyskland |
28,7 |
8,8 |
3,9 |
|
Loamy sand |
Tyskland |
50,4 |
17,9 |
12,1 |
|
Loamy sand |
Schweiz |
44,0 |
35,3 |
9,2 |
|
Silt loam |
Schweiz |
72,8 |
56,6 |
28,4 |
|
Clay loam |
Brasilien |
69,7 |
38,4 |
27,3 |
|
Clay loam |
Japan |
74,4 |
57,8 |
31,4 |
|
Sandy loam |
Japan |
82,4 |
59,2 |
36,0 |
|
Silt loam |
USA |
47,2 |
33,2 |
18,8 |
|
Sandy loam |
USA |
40,4 |
25,2 |
13,3 |
Bilaga 3
Figur I
Exempel på genomflödesapparatur för undersökning av kemikaliers omvandling i jord (53), (54)
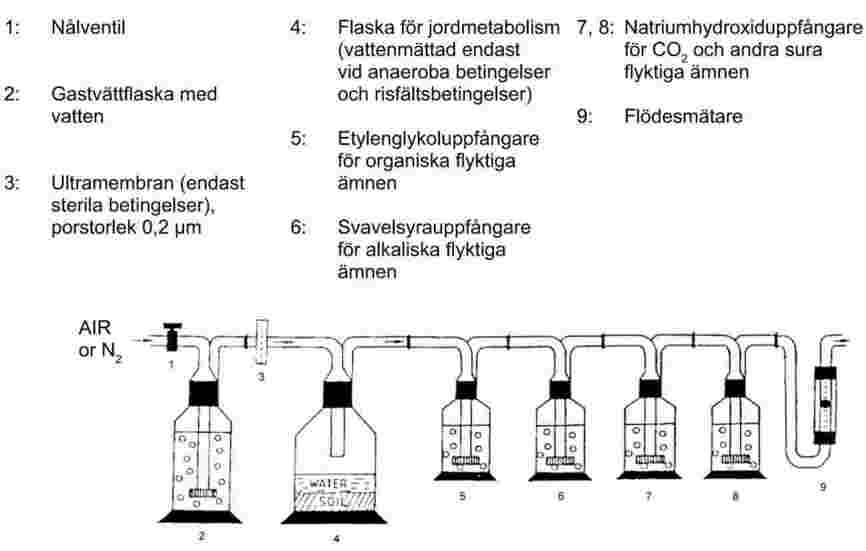
Figur 2
Exempel på en flaska av biometertyp för undersökning av kemikaliers omvandling i jord (55)

C.24 AEROB OCH ANAEROB OMVANDLING I AKVATISKA SEDIMENTSYSTEM
1. METOD
Denna metod är i stort sett identisk med OECD TG 308 (2002).
1.1 INLEDNING
Kemikalier kan nå grunda eller djupa ytvatten exempelvis via direkt applicering, spridning med vinden, avrinning, dränering, bortskaffande av avfall eller utsläpp från industrin, hushåll, jordbruk och från atmosfären. Den testmetod som beskrivs här är en laboratoriemetod för bedömning av aerob och anaerob omvandling av organiska kemikalier i akvatiska sedimentsystem. Metoden grundar sig på existerande riktlinjer (1), (2), (3), (4), (5), (6). Vid en OECD-workshop om valet av jord och sediment som hölls i Belgirate, Italien, 1995 (7), kom man särskilt överens om hur många och vilka typer av sediment som ska användas i detta test. Workshopen i Belgirate gav också rekommendationer rörande insamling, hantering och lagring av sedimentprover, baserade på ISO-riktlinjer (8). Sådana undersökningar behövs för kemikalier som tillsätts direkt i vatten eller som sannolikt når vattenmiljön på det sätt som beskrivs ovan.
Betingelserna i naturliga akvatiska sedimentsystem är ofta aeroba i den övre vattenfasen. Sedimentets ytskikt kan vara aerobt eller anaerobt, medan djupare liggande sediment i regel är anaeroba. Med tanke på detta beskrivs både aerobt och anaerobt test i detta dokument. I det aeroba testet simuleras en aerob vattenpelare över ett aerobt sedimentskikt under vilket det finns en anaerob gradient. I det anaeroba testet simuleras ett fullständigt anaerobt vatten/sediment-system. I situationer där det krävs betydande avvikelser från dessa rekommendationer, till exempel i fråga om intakta sedimentkärnor eller sediment som kan ha exponerats för testämnet, kan andra metoder användas (9).
1.2 DEFINITIONER
Under alla omständigheter bör SI-enheter användas.
Testämne: Alla ämnen, oavsett det gäller en ursprunglig förorening eller relevanta omvandlingsprodukter.
Omvandlingsprodukter: Alla ämnen som uppstår till följd av biotiska eller abiotiska omvandlingsreaktioner av testämnet, inbegripet CO2 och bundna rester.
Bundna rester: Sådana föreningar i jorden, i växter eller i djur som blir kvar i matrisen i form av ursprungligt ämne, dess metaboliter eller dess omvandlingsprodukter efter extraktion. Extraktionsmetoden får inte i avsevärd grad leda till förändringar i själva föreningarna eller matrisens struktur. Bindningens karaktär kan delvis klarläggas genom matrisstörande extraktionsmetoder och sofistikerade analysmetoder. Hittills har man på detta sätt kunnat identifiera till exempel kovalenta bindningar, jonbindningar och sorptiva bindningar samt inneslutning. Generellt sett innebär bildningen av bundna rester en betydande minskning av biotillgängligheten (10) (modifierat ur IUPAC 1984 (11)).
Aerob omvandling (oxidation): Reaktioner som sker i närvaro av molekylärt syre (12).
Anaerob omvandling (reduktion): Reaktioner som sker utan närvaro av molekylärt syre (12).
Naturliga vatten: Ytvatten som fås från dammar, floder, strömmar etc.
Sediment: En blandning av mineraliska och organisk-kemiska beståndsdelar. De senare består av föreningar med hög kol- och kvävehalt samt hög molekylvikt. Sediment deponeras genom inverkan av naturligt vatten och bildar ett gränssnitt med detta vatten.
Mineralisering: Det fullständiga sönderfallet av organiska föreningar till CO2 och H2O under aeroba betingelser, och till CH4, CO2 och H2O under anaeroba betingelser. Med mineralisering avses här ett omfattande sönderfall som sker när en radioaktivt märkt förening används och en märkt kolatom oxideras eller reduceras kvantitativt under frigörande av motsvarande mängd 14CO2 eller 14CH4.
Halveringstid, t0,5 – den tid det tar för ett testämne att omvandlas till 50 % när förloppet kan beskrivas med kinetik av första ordningen. Halveringstiden är oberoende av koncentrationen.
DT 50 (tid för försvinnande 50): Den tid inom vilken testämnets koncentration minskar med 50 %.
DT 75 (tid för försvinnande 75): Den tid inom vilken testämnets koncentration minskar med 75 %.
DT 90 (tid för försvinnande 90): Den tid inom vilken testämnets koncentration minskar med 90 %.
1.3 REFERENSÄMNEN
Referensämnen bör användas för identifiering och kvantifiering av omvandlingsprodukterna genom spektroskopiska och kromatografiska metoder.
1.4 UPPLYSNINGAR OM TESTÄMNET
Omärkta eller isotopmärkta testämnen kan användas för bestämning av omvandlingshastigheten. Helst bör dock märkta ämnen användas. Märkt material behövs för undersökning av omvandlingsmönstret och för bestämning av en massbalans. 14C-märkning rekommenderas, även om andra isotoper, såsom 13C, 15N, 3H och 32P kan användas. Märkningen bör om möjligt finnas i molekylens mest stabila del(ar) (56). Kemikalien eller den radioaktiva kemikalien bör ha en renhetsgrad som är minst 95 %.
Innan ett test utförs bör följande upplysningar om testämnet finnas tillgängliga:
|
a) |
Vattenlöslighet (metod A.6). |
|
b) |
Löslighet i organiska lösningsmedel. |
|
c) |
Ångtryck (metod A.4) och Henrys lag-koefficient. |
|
d) |
fördelningskoefficient n-oktanol/vatten (metod A.8). |
|
e) |
Adsorptionskoefficient (Kd, Kf eller Koc, om lämpligt) (metod C.18). |
|
f) |
Hydrolys (metod C.7). |
|
g) |
Dissociationskonstant (pKa (OECD-riktlinje 112) (13). |
|
h) |
Testämnets struktur och isotopens (isotopernas) placering, om tillämpligt. |
Anmärkning: Den temperatur vid vilken mätningarna görs bör rapporteras.
Övriga användbar information är till exempel upplysningar om testämnets toxicitet för mikroorganismer, upplysningar om lätt eller inneboende bionedbrytbarhet och upplysningar om aerob och anaerob omvandling i jord.
Det bör finnas tillgång till analysmetoder (inbegripet extraktions- och reningsmetoder) för identifiering och kvantifiering av testämnet och dess omvandlingsprodukter i vatten och i sediment (se avsnitt 1.7.2).
1.5 PRINCIP FÖR TESTMETODEN
I den testmetod som beskrivs här används ett aerobt och ett anaerobt akvatiskt sedimentsystem (se bilaga 1). Det ger möjlighet till följande:
|
i) |
bestämning av testämnets omvandlingshastighet i ett vatten/sediment-system, |
|
ii) |
bestämning av testämnets omvandlingshastighet i sedimentet, |
|
iii) |
bestämning av testämnets och/eller omvandlingsprodukternas mineraliseringshastighet (när 14C-märkta testämnen används), |
|
iv) |
identifiering och kvantifiering av omvandlingsprodukterna i vatten- och sedimentfaserna samt upprättandet av en massbalans (när radiomärkt testämne används), |
|
v) |
bestämning av testämnets och omvandlingsprodukternas fördelning mellan de två faserna under en inkubationsperiod i mörker (för att undvika till exempel algblomning) vid konstant temperatur. Halveringstiderna samt värden på DT50, DT75 och DT90 kan bestämmas i de fall där tillräckliga data erhålls, men bör inte extrapoleras till tidpunkter som ligger långt efter försöksperioden (se avsnitt 1.2). |
Minst två sedimenttyper och motsvarande vatten behövs för både den aeroba och anaeroba undersökningen (7). Det kan dock förekomma fall där fler än två akvatiska sediment bör används, till exempel om den kemikalie som testas kan förekomma i sötvattenmiljö och marin miljö.
1.6 TESTETS T1LLÄMPBARHET
Metoden kan allmänt tillämpas på alla kemiska ämnen (omärkta eller radioaktivt märkta) för vilka det finns tillgång till en analysmetod som är tillräckligt exakt och känslig. Metoden kan tillämpas på föreningar som är i viss mån flyktiga, icke-flyktiga, lösliga i vatten eller svårlösliga i vatten. Testet bör inte tillämpas på kemikalier som är mycket flyktiga från vatten (till exempel desinfektionsmedel och organiska lösningsmedel) och som därför inte hålls kvar i vatten eller sediment under de försöksbetingelser som gäller för detta test.
Metoden har hittills använts för att undersöka kemikaliers omvandling i sötvatten och sediment, men kan i princip även tillämpas på flodmynning/havsystem. Metoden är inte lämplig för simulering av betingelserna i strömmande vatten (till exempel floder) eller i öppet hav.
1.7 KVALITETSKRITERIER
1.7.1 Utbyte
Extraktion och analys som görs av minst dubbla vatten- och sedimentprover omedelbart efter tillförsel av testämnet ger en första indikation på analysmetodens repeterbarhet och enhetligheten hos det förfarande som använts för tillförsel av testämnet. Utbyten för senare stadier av försöken erhålls genom massbalanserna (när märkt material används). Utbytena bör vara 90–110 % för märkta kemikalier (6) och 70–110 % för omärkta kemikalier.
1.7.2 Analysmetodens repeterbarhet och känslighet
Analysmetodens repeterbarhet (med undantag av den initiala extraktionseffektiviteten) när det gäller att kvantifiera testämnet och omvandlingsprodukterna, kan kontrolleras genom en duplikatanalys av samma extrakt ur vatten- eller sedimentprover som har inkuberats tillräckligt länge för att omvandlingsprodukter ska ha bildats.
Analysmetodens detektionsgräns (LOD) för testämnet och för omvandlingsprodukterna bör vara minst 0,01 mg· kg-1 i vatten eller sediment (som testämne) eller 1 % av den dos som ursprungligen tillfördes testsystemet, beroende på vilket värde som är lägre. Likaså bör kvantifieringsgränsen (LOQ) anges.
1.7.3 Noggrannheten hos omvandlingsdata
Genom regressionsanalys av testämnets koncentrationer som en funktion av tiden fås ungefärliga upplysningar om omvandlingskurvans exakthet och en möjlighet att beräkna halveringstidernas konfidensgränser (när kinetik av pseudo-första ordningen tillämpas) eller DT50-värden och, om tillämpligt, DT75- och DT90-värden.
1.8 BESKRIVNING AV METODEN
1.8.1 Testsystem och utrustning
Försöken bör utföras i glaskärl (till exempel flaskor eller centrifugrör), utom om förhandsupplysningarna (såsom fördelningskoefficient n-oktanol-vatten eller sorptionsdata) indikerar att testämnet kan bindas till glas, i vilket fall ett alternativt material (till exempel teflon) bör övervägas. Om det är känt att testämnet binds till glas kan problemet eventuellt lindras med en eller flera av följande metoder:
|
— |
bestämning av den massa testämne och omvandlingsprodukter som binds till glas, |
|
— |
säkerställande av att alla glaskärl tvättas med lösningsmedel i slutet av testet, |
|
— |
användning av formulerade produkter (se även avsnitt 1.9.2), |
|
— |
användning av en större mängd hjälplösningsmedel för tillförsel av testämnet till systemet – hjälplösningsmedlet får dock inte leda till så kallad solvolys av testämnet. |
Exempel på typisk testutrustning, till exempel genomflödessystem och system av biometertyp, visas i bilagorna 2 och 3 respektive (14). I hänvisning 15 beskrivs andra lämpliga inkubationssystem. Försöksutrustningen bör vara utformad på sådant sätt att luft- eller kväveväxling kan ske och flyktiga produkter kan uppfångas. Utrustningen bör ha sådana mått att testvillkoren uppfylls (se avsnitt 1.9.1). Ventilation kan ordnas genom försiktig bubbling eller genom att låta luft eller kväve passera över vattenytan. I det senare fallet kan det vara rekommendabelt med en försiktig omblandning av vattnet för bättre fördelning av syre eller kväve i vattnet. CO2-fri luft bör inte användas eftersom detta kan leda till att vattnets pH-värde stiger. Det är under alla omständigheter önskvärt att om möjligt undvika störningar av sedimentet. Kemikalier som är i viss mån flyktiga bör testas i ett system av biometertyp med försiktig omblandning av vattenytan. Man kan även använda stängda kärl där utrymmet ovanför vätskeytan innehåller atmosfärsluft eller kväve och interna kärl för uppfångning av flyktiga produkter (16). Vid aeroba test behövs regelbunden växling av gasen i utrymmet ovanför vätskeytan för att kompensera för biomassans syreförbrukning.
Lämpliga uppfångare för flyktiga omvandlingsprodukter är till exempel kalium- eller natriumhydroxidlösning (1 M) för koldioxid (57), och etylenglykol, etanolamin eller tvåprocentig paraffin i xylen för organiska föreningar. Flyktiga ämnen som bildas under anaeroba betingelser, såsom metan, kan uppfångas till exempel med molekylsiktar. Sådana flyktiga ämnen kan förbrännas exempelvis till CO2 genom att leda gasen genom ett kvartsrör fyllt med CuO vid 900 oC och fånga upp den bildade CO2-gasen i en absorberare med alkali (17).
Det behövs också laboratorieinstrument för kemisk analys av testämnet och dess omvandlingsprodukter (till exempel gas-vätskekromatografi (GLC), högtrycksvätskekromatografi (HPLC), tunnskiktskromatografi (TLC), masspektrometri (MS), gaskromatografi-masspektrometri (GC-MS), vätskekromatografi-masspektrometri (LC-MS), kärnmagnetisk resonans (NMR)), inbegripet detektionssystem för radioaktivt märkta eller omärkta kemikalier enligt behov. När radioaktivt märkt material används krävs också en vätskeskintillationsräknare och ett oxidationsmedel (för förbränning av sedimentprover före analys med avseende på radioaktivitet).
Likaså behövs övrig normal laboratorieutrustning för fysikalisk-kemiska och biologiska bestämningar (se tabell 1, avsnitt 1.8.2.2), glasvaror, kemikalier och reagenser, efter behov.
1.8.2 Valet av akvatiska sediment, antal sediment
Provtagningsplatserna bör alltid väljas i enlighet med testets ändamål. Vid valet av provtagningsplats måste hänsyn tas till eventuella utsläpp från jordbruk, industri eller hushåll till tillrinningsområden och till vatten som ligger uppströms. Sediment som inom de senaste fyra åren har förorenats med testämnet eller strukturellt analoga föreningar får inte användas.
1.8.2.1 Valet av sediment
För de aeroba försöken används i regel två sedimenttyper (7). De två sedimenttyper som väljs ut bör ha olika egenskaper med avseende på halten organiskt kol och textur. Ett av sedimenten bör ha en hög halt organiskt kol (2,5–7,5 %) och en fin textur, medan det andra sedimentet bör ha en låg halt organiskt kol (0,5–2,5 %) och en grov textur. Skillnaden mellan halterna organiskt kol bör i regel vara minst 2 %. Ett sediment har en fin textur när halten clay + silt (58) överstiger 50 % och en grov textur när halten clay + silt understiger 50 %. Skillnaden mellan de två sedimenttyperna med avseende på halterna clay + silt bör i regel vara minst 20 %. I fall där en kemikalie också kan nå havsvatten måste minst det ena vatten/sediment-systemet vara av marint ursprung.
För strikt anaeroba försök bör två sedimenttyper (med motsvarande vatten) tas från de anaeroba zonerna i ytvattenförekomster (7). Både sediment- och vattenfaserna bör hanteras och transporteras varsamt i syrefria betingelser.
Det kan även finnas andra parametrar som är viktiga vid valet av sediment, och dessa bör beaktas från fall till fall. Exempelvis sedimentens pH-område är en viktig faktor vid testning av kemikalier vars omvandling eller sorption kan vara pH-beroende. Ett sådant beroende kan framgå av testämnets pKa-värde.
1.8.2.2 Karakterisering av vatten/sediment-prover
I tabellen som följer finns en översikt av de nyckelparametrar som måste bestämmas och rapporteras (med angivelse av den metod som använts) för både vatten och sediment. Likaså anges i vilket skede av testet dessa parametrar bör bestämmas. Metoder för bestämning av dessa parametrar beskrivs i hänvisningarna (18), (19), (20), (21).
Därutöver kan det, beroende på fall, vara nödvändigt att bestämma och rapportera andra parametrar (till exempel för sötvatten: partiklar, alkalinitet, hårdhet, konduktivitet, NO3/PO4 (förhållande och individuella värden), för sediment: katjonbyteskapacitet, vattenkapacitet, karbonat, totalkväve och totalfosfor samt för marina system: salinitet). Sediment och vatten behöver kanske också analyseras med avseende på nitrat, sulfat, biotillgängligt järn och eventuellt andra elektronmottagare, i syfte att bedöma redoxbetingelserna, särskilt med tanke på den anaeroba omvandlingen.
Bestämning av parametrar för karakterisering av vatten/sediment-prover (7), (22), (23)
|
Parameter |
Skede av testförfarandet |
|||||
|
Fältprovtagning |
Efter-behandl. |
Start av acklimatis. |
Start av test |
Under test |
Slut av test |
|
|
Vatten |
||||||
|
Ursprung/källa |
X |
|
|
|
|
|
|
Temperatur |
X |
|
|
|
|
|
|
pH |
X |
|
X |
X |
X |
X |
|
TOC |
|
|
X |
X |
|
X |
|
Syrehalt2 |
X |
|
X |
X |
X |
X |
|
Redoxpntential* |
|
|
X |
X |
X |
X |
|
Sediment |
||||||
|
Ursprung/källa |
X |
|
|
|
|
|
|
Skiktets djup |
X |
|
|
|
|
|
|
pH |
|
X |
X |
X |
X |
X |
|
Partikelstorleksfördelning |
|
X |
|
|
|
|
|
TOC |
|
X |
X |
X |
|
X |
|
Mikrobiell biomassa (59) |
|
X |
|
X |
|
X |
|
Redoxpotential (60) |
Observationer (färg/lukt) |
|
X |
X |
X |
X |
1.8.3 Insamling, hantering och lagring
1.8.3.1 Insamling
Anvisningarna i utkastet till ISO-riktlinjer om provtagning av bottensediment (8) bör följas vid provtagning av sediment. Sedimentproverna bör tas från sedimentets hela övre skikt (5–10 cm). Tillhörande vatten bör tas från samma plats och vid samma tidpunkt som sedimentprovet tas. För det anaeroba försöket bör sediment- och tillhörande vattenprov tas och transporteras under syrefria betingelser (28) (se avsnitt 1.8.2.1). I litteraturen finns beskrivningar av ett antal olika provtagningsutrustningar (8), (23).
1.8.3.2 Hantering
Sedimentet avskiljs från vatten genom filtrering och våtsiktas (2 mm) med användning av vattenöverskott (vatten från provtagningsplatsen). Detta vatten slängs efter siktningen. Därefter blandas kända mängder sediment och vatten i önskat förhållande (se avsnitt 1.9.1) i inkubationsflaskor och bereds för acklimatiseringsperioden (se avsnitt 1.8.4). Alla hanteringssteg som hör till det anaeroba försöket bör göras under syrefria betingelser (29), (30), (31), (32), (33).
1.8.3.3 Lagring
De sediment- och vattenprover som används ska allra helst vara nytagna. Om proverna ändå måste lagras bör sediment och vatten siktas enligt beskrivningen ovan och lagras tillsammans, med vattenmättnad (6–10 cm vattenskikt) i mörker och vid 4 ± 2 oC4 under högst fyra veckor (7), (8), (23). Proverna för aeroba försök bör lagras med fri tillgång till luft (till exempel i en öppen behållare), medan proverna för anaeroba försök bör lagras i syrefria betingelser. Under transport och lagring får sediment eller vatten inte frysa, och sedimentet får inte torka ut.
1.8.4 Beredning av sediment/vattenproverna för testet
Innan testämnet tillförs bör proverna genomgå en acklimatiseringsperiod. För detta placeras varje sediment/vatten-prov i det inkubationskärl som kommer att användas under själva testet, och acklimatiseringen genomförs under exakt samma betingelser som gäller vid inkubationen under testet (se avsnitt 1.9.1). Acklimatiseringsperioden behövs för att systemet ska nå en tillräcklig stabilitet vad gäller pH, syrehalt i vatten, redoxpotential hos sediment och vatten samt makroskopisk separation av faserna. Acklimatiseringsperioden bör i regel omfatta en till två veckor, och bör inte överskrida fyra veckor. Resultaten från de bestämningar som görs under denna period bör rapporteras.
1.9 TESTETS UTFÖRANDE
1.9.1 Testbetingelser
Testet bör utföras i inkubationsutrustning (se avsnitt 1.8.1) med ett volymförhållande vatten/sediment mellan 3:1 och 4:1, och ett sedimentskikt med en tjocklek av 2,5 cm (±0,5 cm) (61). En minimimängd av 50 g sediment (baserat på torrvikt) per inkubationskärl är rekommenderad.
Testet bör genomföras i mörker vid en konstant temperatur inom intervallet 10–30 oC. Lämplig temperatur är (20 ± 2) oC. Vid behov kan ytterligare en lägre temperatur (till exempel 10 oC) övervägas från fall till fall, beroende på vilka upplysningar man vill få genom testet. Inkubationstemperaturen bör övervakas och rapporteras.
1.9.2 Behandling och tillförsel av testämnet
En enda testkoncentration av kemikalien används (62). För växtskyddskemikalier som appliceras direkt i vattendrag används den maximidos som anges på etiketten, beräknad på grundval av vattenytan i testkärlet. I alla andra fall måste den koncentration som används basera sig på prognoser från utsläpp i miljön. Det är viktigt att säkerställa att den koncentration av testämnet som används är lämplig med tanke på karakterisering av omvandlingvägarna samt bildning och minskning av omvandlingsprodukter. Det kan vara nödvändigt att använda högre doser (till exempel 10-faldiga) i situationer där koncentrationerna av testämnet i början av försöket ligger nära detektionsgränserna, eller om viktiga omvandlingsprodukter inte utan svårighet kan detekteras när halterna är 10 % av testämnesdosen. Om högre testkoncentrationer används får de dock inte ha betydande negativa effekter på den mikrobiella aktiviteten i vatten/sediment-systemet. För att få en konstant koncentration av testämnet i kärl av varierande storlek kan det vara lämpligt med en justering enligt den mängd material som har tillförts. Justeringen görs på grundval av vattenpelarens djup i kärlet i förhållande till vattendjupet på provtagningsplatsen (vilket antas vara 100 cm, även om andra djup kan användas). I bilaga 4 finns ett exempel på en beräkning.
Testämnet ska helst tillföras i form av vattenlösning i testsystemets vattenfas. 1 tvingande fall kan användningen av små mängder vattenlösliga lösningsmedel (såsom aceton eller etanol) tillåtas för tillförsel och fördelning av testämnet. Lösningsmedlets halt får dock inte överskrida 1 % (v/v) och det får inte ha negativa verkningar på den mikrobiella aktiviteten i testsystemet. Vattenlösningen av testämnet bör beredas med omsorg – det kan behövas genereringskolumner och förblandare för att säkerställa fullständig homogenitet. Efter tillsats av vattenlösningen till testsystemet rekommenderas varsam omblandning av vattenfasen, med så liten störning av sedimentet som möjligt.
Användningen av formulerade produkter rekommenderas i regel inte, eftersom de ingående beståndsdelarna kan påverka fördelningen av testämnet eller omvandlingsprodukter mellan vatten- och sedimentfaserna. För testämnen som är svårlösliga i vatten kan dock användningen av formulerat material vara ett lämpligt alternativ.
Antalet inkubationskärl beror på antalet provtagningstillfällen (se avsnitt 1.9.3). Testet bör omfatta tillräckligt många system så att två system kan tas ut vid varje provtagningstillfälle. Om testet omfattar kontrollenheter för varje akvatiskt sedimentsystem bör dessa enheter inte behandlas med testämnet. Kontrollenheterna kan användas för att bestämma den mikrobiella biomassan i sedimentet och totalt organiskt kol i vatten och sediment vid testets försökets slut. Två kontrollenheter (en kontrollenhet för varje akvatiskt sediment) kan användas för att övervaka de behövliga parametrarna i sediment och vatten under acklimatiseringsperioden (se tabellen i avsnitt 1.8.2.2). Två extra kontrollenheter måste tas in i fall där testämnet tillförs med hjälp av ett lösningsmedel, för bedömning av de negativa verkningarna på den mikrobiella aktiviteten i testsystemet.
1.9.3 Testets varaktighet, provtagning
Försöksperioden ska i regel inte sträcka sig över 100 dagar (6), och den bör fortsätta tills nedbrytningvägen och fördelningsmönstret för vatten och sediment har fastställts eller när 90 % av testämnet har omvandlats eller förflyktigats. Antalet provtagningstillfällen bör vara minst sex (inbegripet tidpunkt noll), med en valfri förhandsundersökning (se avsnitt 1.9.4) som görs i syfte att fastställa ett lämpligt provtagningsschema och testets varaktighet, i fall där det inte finns tillgång till tillräckliga data om testämnet från tidigare undersökningar. För hydrofoba testämnen kan det under försökets inledningsperiod behövas ytterligare provtagningstillfällen i syfte att bestämma hur fördelningen sker mellan vatten- och sedimentfaserna.
Vid provtagningstidpunkterna avlägsnas inkubationskärlen i sin helhet (med replikat) för analys. Sediment och ovanliggande vatten analyseras separat (63). Ytvattnet bör avlägsnas omsorgsfullt med minsta möjliga störning av sedimentet. Extraktionen och karakteriseringen av testämnet och omvandlingsprodukterna bör följa tillbörliga analysförfaranden. Det är viktigt att avlägsna material som kan ha adsorberats till inkubationskärlet eller mellanliggande slangar som används för att fånga upp flyktiga ämnen.
1.9.4 Valfritt förförsök
Om testets varaktighet och provtagningsschemat inte kan fastställas med stöd av andra relevanta undersökningar rörande testämnet kan det vara nödvändigt med ett valfritt förförsök. Detta bör utföras under samma betingelser som gäller för det slutliga försöket. 1 förekommande fall bör testrapporten innehålla en översiktlig redogörelse för relevanta försöksbetingelser och resultat från förförsöket.
1.9.5 Mätningar och analys
Vid varje provtagningstillfälle bör testämnets och omvandlingsprodukternas koncentration bestämmas i vatten och sediment, samt rapporteras (som en koncentration och som procentandel av den tillförda koncentrationen). Som allmän regel gäller att identifiering bör göras av alla omvandlingsprodukter som vid något provtagningstillfälle har detekterats med en andel som är ≥ 10 % av den radioaktivitet som tillförts det totala vatten/sediment-systemet, utom om det finns goda skäl för att låta bli. Likaså bör identifiering av omvandlingsprodukter vars koncentrationer kontinuerligt ökar under försökets gång övervägas (även i fall där dessa koncentrationer inte går över de gränser som anges ovan), eftersom ökningen kan tyda på beständighet. Denna fråga bör övervägas från fall till fall, och motiveringarna bör anges i rapporten.
Resultaten från system för uppfångning av gaser och flyktiga ämnen (CO2 och övriga, till exempel flyktiga organiska ämnen) bör rapporteras för varje provtagningstillfälle. Mineraliseringshastigheterna bör rapporteras. Icke-extraherbara (bundna) rester i sediment bör rapporteras för varje provtagningstillfälle.
2 DATA
2.1 BEHANDLING AV RESULTATEN
Den totala massbalansen eller utbytet (se avsnitt 1.7.1) av tillförd radioaktivitet bör beräknas för varje provtagningstillfälle. Resultatet bör rapporteras som en procentandel av den tillförda radioaktiviteten. Fördelningen av radioaktivitet mellan vatten och sediment bör rapporteras som koncentrationer och procentandelar, för varje provtagningstillfälle.
Halveringstiden, DT50 och, om tillämpligt, DT75 och DT90 för testämnet bör beräknas inbegripet konfidensgränser (se avsnitt 1.7.3). Upplysningar om den hastighet med vilken testämnet försvinner i vatten och sediment kan erhållas med hjälp av lämpliga utvärderingsmetoder. Dessa kan omfatta tillämpning av kinetik av pseudoförsta ordningen, empiriska kurvanpassningstekniker där grafiska eller numeriska lösningar används och mer komplexa bedömningar där till exempel modeller med en eller flera behållare används. Närmare upplysningar finns i relevant litteratur (35), (36), (37).
Alla metoder har sina för- och nackdelar samt stora variationer med avseende på komplexitet. Ett antagande om första ordningens kinetik kan innebära en för stark förenkling av nedbrytnings- och fördelningsprocesserna, men ger – i fall där detta antagande kan tillämpas – en term (hastighetskonstanten eller halveringstiden) som är lättbegriplig och av värde vid simuleringsmodellering och beräkningar av förväntade koncentrationer i miljön. Empiriska metoder eller linjära transformationer kan ge bättre anpassning av kurvorna till data, och därmed en bättre uppskattning av halveringstider, DT50 och, om tillämpligt, DT75 och DT90. Användningen av härledda konstanter är dock begränsad. Med modellen med behållare kan man få ett antal användbara konstanter av värde vid riskbedömning. Dessa beskriver nedbrytningshastigheten i olika behållare och fördelningen av kemikalien. De bör också användas för uppskattning av hastighetskonstanter för bildning och nedbrytning av viktiga omvandlingsprodukter. Under alla omständigheter måste den metod som används motiveras, och anpassningsgraden bör demonstreras grafiskt eller statistiskt.
3 RAPPORTERING
3.1 TESTRAPPORT
Testrapporten ska innehålla nedan angivna uppgifter.
|
|
Testämne:
|
|
|
Referensämnen:
|
|
|
Testsediment och -vatten:
|
|
|
Testbetingelser:
|
|
|
Resultat:
|
4. HÄNVISNINGAR
|
1. |
BBA-Guidelines for the examination of plant protectors in the registration process. (1990), Part IV, Section 5-1: Degradability and fate of plant protectors in the water/sediment system, Germany. |
|
2. |
Commission for registration of pesticides: Application for registration of a pesticide. (1991), Part G. Behaviour of the product and its metabolites in soil, water and air, Section G.2.1 (a), The Netherlands. |
|
3. |
MAFF Pesticides Safety Directorate. (1992), Preliminary guideline for the conduct of biodegradability tests on pesticides in natural sediment/water systems. Ref No SC 9046, United-Kingdom. |
|
4. |
Agriculture Canada: Environmental Chemistry and fate (1987), Guidelines for registration of pesticides in Canada. Aquatic (Laboratory) – Anaerobic and aerobic, Canada, s. 35–37. |
|
5. |
US-EPA: Pesticide assessment guidelines, Subdivision N. Chemistry: Environmental fate (1982), Section 162-3, Anaerobic aquatic metabolism. |
|
6. |
SETAC-Europe publication. (1995), Procedures for assessing the environmental fate and ecotoxicity of pesticides. Ed. Dr Mark R. Lynch. SETAC-Europe, Bryssel. |
|
7. |
OECD Test Guidelines Programme. (1995), Final Report of the OECD Workshop on Selection of Soils/sediments, Belgirate, Italy, 18–20 januari 1995. |
|
8. |
ISO/DIS 5667-12 (1994), Water quality – Sampling – Part 12: Guidance on sampling of bottom sediments. |
|
9. |
US-EPA (1998a), Sediment/water microcosm biodegradation test. Harmonised Test Guidelines (OPPTS 835.3180), EPA 712-C-98-080. |
|
10. |
DFG: Pesticide Bound Residues in Soil. Wiley-VCH (1998). |
|
11. |
T.R. Roberts: Non-extractable pesticide residues in soils and plants. Pure Appl. Chem. 56, s. 945–956 (IUPAC 1984). |
|
12. |
OECD Test Guideline 304A: Inherent Biodegradability in Soil (adopted 12 May 1981). |
|
13. |
OECD (1993), Guidelines for Testing of Chemicals, Paris, OECD (1994–2000): Addenda 6-11 to Guidelines for the Testing of Chemicals. |
|
14. |
Scholz, K., Fritz R., Anderson C. and Spiteller M. (1988), Degradation of pesticides in an aquatic model ecosystem. BCPC – Pests and Diseases, 3B-4, 149–158. |
|
15. |
Guth, J.A. (1981), Experimental approaches to studying the fate of pesticides in soil. In Progress in Pesticide Biochemistry (D.H. Hutson, T.R. Roberts, Eds.), Vol. 1, s. 85–114. J. Wiley & Sons. |
|
16. |
Madsen, T., Kristensen, P. (1997), Effects of bacterial inoculation and non-ionic surfactants on degradation of polycyclic aromatic hydrocarbons in soil. Environ. Toxicol. Chem. 16, s. 631–637. |
|
17. |
Steber, J., Wierich, P. (1987), The anaerobic degradation of detergent range fatty alcohol ethoxylates. Studies with 14C-labelled model surfactants. Water Research 21, s. 661–667. |
|
18. |
Black, CA. (1965), Methods of Soil Analysis. Agronomy Monograph No. 9. American Society of Agronomy, Madison. |
|
19. |
APHA (1989), Standard Methods for Examination of Water and Wastewater (17th edition). American Public Health Association, American Water Works Association and Water Pollution Control Federation, Washington D.C. |
|
20. |
Rowell, D.L. (1994), Soil Science Methods and Applications. Longman. |
|
21. |
Light, T.S. (1972), Standard solution for redox potential measurements. Anal. Chemistry 44, s. 1038–1039. |
|
22. |
SETAC-Europe publication (1991), Guidance document on testing Procedures for pesticides in freshwater mesocosms. From the Workshop ”A Meeting of Experts on Guidelines for Static Field Mesocosms Tests”, den 3–4 juli 1991. |
|
23. |
SETAC-Europe publication. (1993), Guidance document on sediment toxicity tests and bioassays for freshwater and marine environments. From the Workshop On Sediment Toxicity Assessment (WOSTA), 8–10 november 1993. Eds.: I.R. Hill, P. Matthiessen and F. Heimbach. |
|
24. |
Vink, J.P.M., van der Zee, S.E.A.T.M. (1997), Pesticide biotransformation in surface waters: multivariate analyses of environmental factors at field sites. Water Research 31, s. 2858–2868. |
|
25. |
Vink, J.P.M., Schraa, G., van der Zee, S.E.A.T.M. (1999), Nutrient effects on microbial transformation of pesticides in nitrifying waters. Environ. Toxicol, s. 329–338. |
|
26. |
Anderson, T.H., Domsch, K.H. (1985), Maintenance carbon requirements of actively-metabolising microbial populations under in-situ conditions. Soil Biol. Biochem. 17, s. 197–203. |
|
27. |
ISO-14240-2. (1997), Soil quality – Determination of soil microbial biomass – Part 2: Fumigation- extraction method. |
|
28. |
Beelen, P. Van and F. Van Keulen. (1990), The Kinetics of the Degradation of Chloroform and Benzene in Anaerobic Sediment from the River Rhine. Hydrobiol, Bull. 24 (1), s. 13–21. |
|
29. |
Shelton, D.R. and Tiedje, J.M. (1984), General method for determining anaerobic biodegradation potential. App. Environ. Microbiol. 47, s. 850–857. |
|
30. |
Birch, R.R., Biver, C, Campagna, R., Gledhill, W.E., Pagga, U., Steber, J., Reust, H. and Bontinck, W.J. (1989), Screening of Chemicals for anaerobic biodegradation. Chemosphere 19, s. 1527–1550. |
|
31. |
Pagga, U. and Beimborn, D.B. (1993), Anaerobic biodegradation tests for organic compounds. Chemoshpere 27, s. 1499–1509. |
|
32. |
Nuck, B.A. and Federle, T.W. (1986), A batch test for assessing the mineralisation of 14C-radiolabelled compounds under realistic anaerobic conditions. Environ. Sci. Technol. 30, s. 3597–3603. |
|
33. |
US-EPA (1998b), Anaerobic biodegradability of organic Chemicals. Harmonised Test Guidelines (OPPTS 835.3400). EPA 712-C-98-090. |
|
34. |
Sijm, Haller and Schrap (1997), Influence of storage on sediment characteristics and drying sediment on sorption coefficients of organic contaminants. Bulletin Environ. Contam. Toxicol. 58, s. 961–968. |
|
35. |
Timme, G., Frehse H. and Laska V. (1986), Statistical interpretation and graphic representation of the degradational behaviour of pesticide residues 11. Pflanzenschutz – Nachrichten Bayer, 39, s. 187–203. |
|
36. |
Timme, G., Frehse, H. (1980), Statistical interpretation and graphic representation of the degradational behaviour of pesticide residues I. Pflanzenschutz – Nachrichten Bayer, 33, s. 47–60. |
|
37. |
Carlton, R.R. and Allen, R. (1994), The use of a compartment model for evaluating the fate of pesticides in sediment/water systems. Brighton Crop Protection Conference – Pest and Diseases, s. 1349–1354. |
Bilaga 1
VÄGLEDNING RÖRANDE AEROBA OCH ANAEROBA TESTSYSTEM
Aerobt testsystem
Det aeroba testsystem som beskrivs i denna testmetod består av ett aerobt vattenskikt (typisk syrehalt är 7 – 10 mg· l-1) och ett sedimentskikt som är aerobt vid ytan och anaerobt under ytan (den typiska genomsnittliga redoxpotentialen (Eh) i sedimentets anaeroba zon ligger mellan – 80 och – 190 mV). Fuktad luft leds över vattenytan i varje inkubationsenhet för att upprätthålla tillräcklig syrehalt i utrymmet ovanför vätskeytan.
Anaerobt testsystem
Testförfarandet är till väsentliga delar samma i ett aerobt som i ett anaerobt testsystem, med undantag av att fuktad kvävgas leds över vattenytan i varje inkubationsenhet för att upprätthålla en kvävgasatmosfär. Sediment och vatten anses vara anaeroba när redoxpotentialen (Eh) är lägre än – 100 mV.
I anaeroba test inbegriper bedömningen av mineraliseringen även bedömning av frigjord koldioxid och metan.
Bilaga 2
EXEMPEL PÅ EN GENOMFLÖDESUTRUSTNING

Bilaga 3
EXEMPEL PÅ EN HIOMETERUTRUSTNING

Bilaga 4
EXAMPELBERÄKNINGAR FÖR DEN DOS SOM TILLFÖRS TESTKÄRLEN
|
Cylinderns innerdiameter: |
= 8 cm |
|
Vattenpelarens djup exklusive sediment: |
= 12 cm |
|
Yta: 3,142 × 42 |
= 50,3 cm2 |
|
Dosering: 500 g testämne/ha motsvarar 5 μg/cm2 |
|
|
Totalt i μg: 5 × 50,3 |
= 251,5 μg |
|
Justering av mängden till att motsvara djupet 100 cm: 12 × 251,5 ÷ 100 |
= 30,18 μg |
|
Vattenpelarens volym: 50,3 × 12 |
= 603 ml |
|
Koncentration i vatten: 30,18 ÷ 603 |
= 0,050 μg/ml eller 50 μg/l |
(1) Resultaten av dessa laboratorieförsök och en teknisk rättelse till ISO 6341 ger ett EC50 – 24 timmar för kaliumdikromat (K2Cr2O7) i intervallet 0,6 mg/l till 1,7 mg/l.
(2) Vatten av lämplig renhet, t.ex. avjonat, destillerat eller renat med omvänd osmos med en konduktivitet som inte överskrider l0 μS.cm-1.
(3) Genomsnittet av D, och D2 ska inte tas om det är en betydande skillnad.
(4) Genomsnittet av D1 och D2 ska inte tas om det är en betydande skillnad.
(5) Eller vid inkuberingens avslutning.
(6) Genomsnittet av D1 och D2 ska inte tas om det föreligger en väsentlig skillnad.
(7) Eller vid inkuberingens slut.
(8) Ta inte genomsnittet om det föreligger väsentlig skillnad med replikaterna.
(9) Ta inte genomsnittet om det föreligger en väsentlig skillnad mellan replikaterna.
(10) Efter användning ska lösningar som innehåller kvicksilversalt behandlas för att undvika spridning av kvicksilver i miljön.
(11) Mabey och Mill rekommenderar borat- eller acetatbuffert i stället för fosfatbuffert (11).
(12) Sådan information kan komma från andra källor, såsom litteraturuppgifter om hydrolys av strukturellt likartade föreningar, eller från andra preliminära, semikvantitativa hydrolystest med testsubstansen på ett tidigare utvecklingsstadium.
(13) Om diagrammet över logaritmerade data som funktion av tiden inte visar att funktionen är linjär (ekvivalent med en reaktionshastighet av första ordningen) så är ekvation 3 olämplig för att bestämma hastighetskonstanten för hydrolys av testsubstansen.
(14) De pH-värden som anges i dessa tabeller har beräknats från de potentiella mätningarna med hjälp av Sörensens standardekvationer (1909). Motsvarande pH-värden är 0,04 enheter högre än tabellvärdena.
(15) Tillsätt en liten kristall tymol eller liknande ämne för att förhindra svampangrepp.
(16) Medelvärde av tre bestämningar.
(17) Meyer A., Orti G. (1993), Proc. Royal Society of London, Series B., Vol. 2.52, s. 231.
(18) Ta prover på vattnet när minst tre kärlvolymer har levererats.
Värden inom parentes står för antal prover (vatten, fisk} som ska tas om tilläggsprovtagning görs.
|
Anmärkning |
: |
Föregående uppskattning för k, för log Pow på 4,0 är 0,652 dagar . Experimentets totala varaktighet är fastställd till: 3 x upp = 3 x 4,6 dagar, dvs. 14 dagar. För uppskattning av ”upp”, se bilaga 3. |
(19) Meyer A.. Bierman C. H. och Orti G. (1993), The phylogenetic position of the Zebrafish (Danio rerio), a model system in developmental biology – an invitation to the comperative methods. Proc. Royal Society of London, Series B, 252, s. 231-236.
(20) En serie av fem (eller flera) på varandra följande koncentrationer kan väljas ur en kolumn, Mittpunkterna mellan koncentrationerna i kolumn (x) finns i kolumn (2x + 1). De listade värdena kan representera koncentrationer uttryckta som procenthalt per volym eller vikt (mg/l eller μg/l). Värdena kan om nödvändigt multipliceras eller divideras med en eller flera tiopotenser. Kolumn 1 kan användas om det råder betydande osäkerhet om toxicitetsnivån.
(21) OECD. Paris. 1992,. Test Guideline 210. ”Fish. Early-life Stage Toxicity Test”.
(22) För embryon.
(23) För yngel.
(24) Mörker for embryon och yngel tills att det gått en vecka efter kläckning, utom när de inspekteras. Därefter dämpad belysning till testets slut.
(25) Enligt FAO och det amerikanska systemet (85).
(26) Variablerna ska helst ha värden inom de angivna intervallen. Om det är svårt att hitta lämpligt jordmaterial kan värden under det angivna minimivärdet godtas.
(27) Jordar med mindre an 0,3 % organiskt kol kan störa korrelationen mellan halten av organiskt material och adsorption. Det är därför tillrådligt att använda jordar med en minimihalt av 0,3 % organiskt kol.

(29) Jämviktsplatån kan också uppskattas genom att i ett diagram avsätta testämnets koncentration i vattenfasen ( )mot tiden (se figur 2 i bilaga 5).
)mot tiden (se figur 2 i bilaga 5).
(30) DT-50: nedbrytningstiden för 50 % av testämnet.
(31) W. Kördel, D. Hennecke, M. Herrmann (1997). Application of the HPLC-screening method for the determination of the adsorption coefficient on sewage sludges. Chemosphere, 35(1/2), 121 — 128.
(32) W. Kördel, D. Hennecke, C. Franke (1997). Determination of the adsorption-coefficients of organic substances on sewage sludges. Chemosphere, 35 (1/2), 107 — 119.
(33) W. Kördel, G. Kotthoff, J. Müller (1995). HPLC-screening method for the determination of the adsorption coefficient on soil-results of a ring test. Chemosphere, 30(7), 1373-1384.
(34) W. Kördel, J. Müller (1994), Bestimmung des Adsorplionskoeffizienten organischer Chemikalien mit der HPLC, UBA R & D Report No. 106 01044 (1994).
(35) B.V. Oepen, \V. Kördel, W. Klein. (1991). Chemosphere, 22, s. 285–304.
(36) (C) Data från industrin.
(37) Ange vilket kärl som användes för försöket.
(38) Markera varje dött provexemplar med ”M” i relevant ruta.
(39) Markera aborterade satser med ”AB” i relevant ruta.
(40) Om testämnet till exempel innehåller en ring måste märkningen finnas på denna ring. Om testämnet innehåller två eller flera ringar kan det vara nödvändigt med separata undersökningar för utvärdering av vad som händer med de enskilda ringarna och för att få lämpliga upplysningar om bildningen av omvandlingsprodukter.
(41) Vattenretentionsegenskaperna hos jord kan mätas som fältkapacitet, vattenkapacitet eller tension (pF). Begreppsförklaringar finns i bilaga 1. I testrapporten bör anges om vattenretentionsegenskaperna och bulktätheten har bestämts i ostörda fältprover eller i störda (bearbetade) prover).
(42) Färska forskningsresultat tyder på att jordprover från tempererade zoner även kan lagras vid -20 C under mer än tre månader (28), (29) utan betydande minskningar av den mikrobiella aktiviteten.
(43) Jorden får inte vara för våt eller för torr med tanke på upprätthållande av tillbörlig luftning och näring för mikroflarn i jorden. De rekommenderade fukthalterna för optimal mikrobiell tillväxt är 40–60 % vattenkapacitet (WHC) och 0,1–0,33 bar (6). Det senare motsvarar pF-området 2,0–2,5. Typiska fukthalter för olika jordtyper anges i bilaga 2.
(44) Aeroba betingelser är dominerande i ytjord och även i jord under ytan, såsom visas i ett forskningsprojekt som fått stöd från EU (K. Takagi et al. (1992), ”Microbial diversity and activity in subsoils: Methods, field site, seasonal variation in subsoil temperatures and oxygen contents”, Proc. Internat. Symp. Enxironm. Aspects Pesticides Microbiol., 270–277, den 17–21 augusti 1992, Sigtuna, Sverige). Anaeroba betingelser förekommer endast sporadiskt vid översvämningar efter skyfall eller när risfältsbetingelser etableras i risfält.
(45) Aeroba försök kan avslutas mycket tidigare, förutsatt att omvandlingsmönstret och mineraliseringen med säkerhet har nått sin absoluta slutpunkt vid försökets slut. Det är möjligt att avsluta testet efter 120 dagar eller när minst 90 % av testämnet har omvandlats, dock endast om minst 5 % CO2 har bildats.
(46) Den ytbaserade initialkoncentrationen beräknas med följande ekvation:
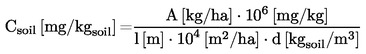
|
Csoil |
= |
Initialkoncentration i jorden (mg· kg-1) |
|
A |
= |
Dosering (kg· ha-1), 1 = jordskiktets tjocklek (m), d = bulktäthet för torr jord (kg· m-3). |
Som en tumregel gäller att en dosering som är 1 kg· ha-1 ger en koncentration i jorden som ligger kring 1 mg· kg-1 i ett 10 cm skikt (när bulktätheten är 1 g cm-3).
(47) E. Muckenhausen (1975), ”Die Bodenkunde und ihre geologischen, geomorphologischen, mineralogischen und petrologischen Grundlagen”, DLG-Verlag, Frankfurt am Main.
(48) pF = logaritmen av cm vattenpelare.
(49) 1 bar= 105 Pa.
(50) Motsvarar en ungefärlig vattenhalt på 10 % i sand, 35 % i loam och 45 % i clay.
(51) Fältkapaciteten är inte konstant utan varierar enligt jordtyp mellan pF 1,5 och 2,5.
(52) Vattenkapacitet
(53) J.A. Guth, (1980), ”The study of transformations”, Interactions between Herbicides and the Soil (red. R. J. Hance.), Academic Press, s. 123–157.
(54) J .A. Guth, (1981), ”Experimental approaches to studying the fate of pesticides in soil”, Progress in Pesticide Biochemistry, (red. D.H. Hutson, T. R. Roberts),./. Wiley & Sons, Vol. 1, s. 85–114.
(55) J.P.E. Anderson, (1975), ”Einfluss von Temperatur und Feuchte auf Verdampfung, Abbau und Festlegung von Diallat im Boden”, Z. PflKrankh Pflschutz, Sonderheft VII, s. 141–146.
(56) Om testämnet till exempel innehåller en ring måste märkningen finnas på denna ring. Om testämnet innehåller två eller flera ringar kan det vara nödvändigt med separata försök för utvärdering av vad som händer med de enskilda ringarna och för att få lämpliga upplysningar om bildningen av omvandlingsprodukter.
(57) Eftersom dessa alkaliska absorptionslösningar även absorberar ventilationsluftens koldioxid och den koldioxid som bildas genom respiration vid aeroba försök, måste lösningarna regelbundet bytas ut för att de inte ska mättas och förlora sin absorptionskapacitet.
(58) Clay + silt är den mineralfraktion vars partikelstorlek är mindre än 50 μm.
(59) Färska forskningsresultat tyder på att bestämningar av vattnets syrehalt och redoxpotential inte har något mekanistiskt eller prognostiskt värde när det gäller tillväxt och utveckling hos de mikrobiella populationerna i ytvatten (24), (25). Bestämning av den biokemiska syreförbrukningen (BOD, vid provtagning, start och slut av testet) och koncentrationerna för mikro- och makronäringsämnena Ca, Mg och Mn (vid start och slut av testet) i vatten, och bestämning av totalt N och totalt P i sediment (vid provtagning och vid slut av testet) kan vara bättre metoder när det gäller att tolka och utvärdera hastigheter och rutter för den aeroba bioomvandlingen.
(60) Metod för mikrobiell respiration (26), desinfektionsmetod (27) eller plate count-mätning (till exempel bakterier, aktinomyceter, svamp och kolonier totalt) för aeroba försök; metanogenes för anaeroba försök.
(61) Färska undersökningar tyder på att lagring vid 4 oC kan leda till en minskning av sedimentets halt av organiskt kol, vilket eventuellt kan leda till en minskning av den mikrobiella aktiviteten (34).
(62) Test med en extra koncentration kan vara till nytta i fråga om kemikalier som når ytvatten via olika rutter (vilket leder till avsevärt varierande koncentrationer) förutsatt att den lägre koncentrationen kan analyseras med tillräcklig exakthet.
(63) I fall där det förekommer en benägenhet till återoxidation av anaeroba omvandlingsprodukter bör de anaeroba betingelserna upprätthållas under provtagning och analys.






