This document is an excerpt from the EUR-Lex website
Document 32017R0735
Commission Regulation (EU) 2017/735 of 14 February 2017 amending, for the purpose of its adaptation to technical progress, the Annex to Regulation (EC) No 440/2008 laying down test methods pursuant to Regulation (EC) No 1907/2006 of the European Parliament and of the Council on the Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH) (Text with EEA relevance. )
Reglamento (UE) 2017/735 de la Comisión, de 14 de febrero de 2017, que modifica, con vistas a su adaptación al progreso técnico, el anexo del Reglamento (CE) n.° 440/2008, por el que se establecen métodos de ensayo de acuerdo con el Reglamento (CE) n.° 1907/2006 del Parlamento Europeo y del Consejo, relativo al registro, la evaluación, la autorización y la restricción de las sustancias y mezclas químicas (REACH) (Texto pertinente a efectos del EEE. )
Reglamento (UE) 2017/735 de la Comisión, de 14 de febrero de 2017, que modifica, con vistas a su adaptación al progreso técnico, el anexo del Reglamento (CE) n.° 440/2008, por el que se establecen métodos de ensayo de acuerdo con el Reglamento (CE) n.° 1907/2006 del Parlamento Europeo y del Consejo, relativo al registro, la evaluación, la autorización y la restricción de las sustancias y mezclas químicas (REACH) (Texto pertinente a efectos del EEE. )
C/2017/0773
DO L 112 de 28.4.2017, p. 1–402
(BG, ES, CS, DA, DE, ET, EL, EN, FR, HR, IT, LV, LT, HU, MT, NL, PL, PT, RO, SK, SL, FI, SV)
 In force
In force
|
28.4.2017 |
ES |
Diario Oficial de la Unión Europea |
L 112/1 |
REGLAMENTO (UE) 2017/735 DE LA COMISIÓN
de 14 de febrero de 2017
que modifica, con vistas a su adaptación al progreso técnico, el anexo del Reglamento (CE) n.o 440/2008, por el que se establecen métodos de ensayo de acuerdo con el Reglamento (CE) n.o 1907/2006 del Parlamento Europeo y del Consejo, relativo al registro, la evaluación, la autorización y la restricción de las sustancias y mezclas químicas (REACH)
(Texto pertinente a efectos del EEE)
LA COMISIÓN EUROPEA,
Visto el Tratado de Funcionamiento de la Unión Europea,
Visto el Reglamento (CE) n.o 1907/2006 del Parlamento Europeo y del Consejo, de 18 de diciembre de 2006, relativo al registro, la evaluación, la autorización y la restricción de las sustancias y mezclas químicas (REACH), por el que se crea la Agencia Europea de Sustancias y Mezclas Químicas, se modifica la Directiva 1999/45/CE y se derogan el Reglamento (CEE) n.o 793/93 del Consejo y el Reglamento (CE) n.o 1488/94 de la Comisión, así como la Directiva 76/769/CEE del Consejo y las Directivas 91/155/CEE, 93/67/CEE, 93/105/CE y 2000/21/CE de la Comisión (1), y, en particular, su artículo 13, apartado 2,
Considerando lo siguiente:
|
(1) |
El Reglamento (CE) n.o 440/2008 de la Comisión (2) incluye los métodos de ensayo para la determinación de las propiedades fisicoquímicas, de la toxicidad y de la ecotoxicidad de las sustancias y mezclas químicas, que deben aplicarse a efectos del Reglamento (CE) n.o 1907/2006. |
|
(2) |
Es necesario actualizar el Reglamento (CE) n.o 440/2008 para incluir los métodos de ensayo nuevos y actualizados que ha adoptado recientemente la Organización de Cooperación y Desarrollo Económicos (OCDE) a fin de tener en cuenta el progreso técnico y de garantizar la reducción del número de animales utilizados en los experimentos, de acuerdo con la Directiva 2010/63/UE del Parlamento Europeo y del Consejo (3). Se ha consultado a los interesados sobre el presente proyecto. |
|
(3) |
La adaptación al progreso técnico contiene veinte métodos de ensayo: un nuevo método para la determinación de una propiedad fisicoquímica, cinco métodos de ensayo nuevos y uno actualizado para la evaluación de la ecotoxicidad, dos métodos de ensayo actualizados para la evaluación del destino y el comportamiento en el medio ambiente, y cuatro métodos de ensayo nuevos y siete actualizados para la determinación de los efectos sobre la salud humana. |
|
(4) |
La OCDE revisa periódicamente sus directrices de ensayo para detectar las que estén científicamente obsoletas. La presente adaptación al progreso técnico suprime seis métodos de ensayo cuyas correspondientes directrices de ensayo de la OCDE han sido canceladas. |
|
(5) |
Procede, por tanto, modificar el Reglamento (CE) n.o 440/2008 en consecuencia. |
|
(6) |
Las medidas previstas en el presente Reglamento se ajustan al dictamen del Comité establecido en virtud del artículo 133 del Reglamento (CE) n.o 1907/2006. |
HA ADOPTADO EL PRESENTE REGLAMENTO:
Artículo 1
El anexo del Reglamento (CE) n.o 440/2008 queda modificado con arreglo a lo dispuesto en el anexo del presente Reglamento.
Artículo 2
El presente Reglamento entrará en vigor a los veinte días de su publicación en el Diario Oficial de la Unión Europea.
El presente Reglamento será obligatorio en todos sus elementos y directamente aplicable en cada Estado miembro.
Hecho en Bruselas, el 14 de febrero de 2017.
Por la Comisión
El Presidente
Jean-Claude JUNCKER
(1) DO L 396 de 30.12.2006, p. 1.
(2) Reglamento (CE) n.o 440/2008 de la Comisión, de 30 de mayo de 2008, por el que se establecen métodos de ensayo de acuerdo con el Reglamento (CE) n.o 1907/2006 del Parlamento Europeo y del Consejo relativo al registro, la evaluación, la autorización y la restricción de las sustancias y mezclas químicas (REACH) (DO L 142 de 31.5.2008, p. 1).
(3) Directiva 2010/63/UE del Parlamento Europeo y del Consejo, de 22 de septiembre de 2010, relativa a la protección de los animales utilizados para fines científicos (DO L 276 de 20.10.2010, p. 33).
ANEXO
El anexo del Reglamento (CE) n.o 440/2008 queda modificado como sigue:
|
1) |
En la parte A se añade el capítulo siguiente: «A.25. CONSTANTES DE DISOCIACIÓN EN AGUA (MÉTODO DE VALORACIÓN VOLUMÉTRICA — MÉTODO DE ESPECTROFOTOMETRÍA — MÉTODO DE CONDUCTIMETRÍA) INTRODUCCIÓN El presente método de ensayo es equivalente a las directrices de ensayo de la OCDE 112 (1981). Condiciones previas
Información orientativa
Indicación general sobre el método de ensayo
Documentos de referencia El presente método de ensayo se basa en métodos que figuran en las referencias de la sección de «Bibliografía» y en el documento Preliminary Draft Guidance for Premanufacture Notification de la EPA, de 18 de agosto de 1978. MÉTODO — INTRODUCCIÓN, OBJETIVO, ÁMBITO DE APLICACIÓN, PERTINENCIA, APLICACIÓN Y LÍMITES DEL ENSAYO La disociación de una sustancia en el agua es importante para evaluar su impacto sobre el medio ambiente. De ella depende la forma de la sustancia, que a su vez determina su comportamiento y transporte. Puede afectar a la adsorción del producto en el suelo y los sedimentos y a su absorción en células biológicas. Definiciones y unidades La disociación es la descomposición reversible en dos o más especies químicas que pueden ser iónicas. El proceso se representa generalmente mediante la ecuación: RX ⇌ R ++ X – y la constante de equilibrio de concentraciones que gobierna la reacción es
Por ejemplo, en el caso particular en que R es el hidrógeno (la sustancia es un ácido), la constante es
o bien
Sustancias de referencia No es preciso emplear las sustancias de referencia siguientes cada vez que se estudia una nueva sustancia. Se facilitan principalmente para poder efectuar de vez en cuando el calibrado del método y ofrecer la posibilidad de comparar los resultados cuando se aplique un método distinto.
Sería útil disponer de una sustancia con varios pK como se indica en la sección Principio del método, más abajo. Esta sustancia podría ser la siguiente:
Principio del método El proceso químico descrito suele depender solo ligeramente de la temperatura, en el intervalo de temperaturas pertinentes para el medio ambiente. La determinación de la constante de disociación requiere una medición de las concentraciones de las formas disociadas y sin disociar de la sustancia. La constante adecuada puede determinarse a partir de la estequiometría de la reacción de disociación indicada en la sección Definiciones y unidades, más arriba. En el caso concreto descrito en el presente método de ensayo, la sustancia se comporta como ácido o base, y la determinación más conveniente se hace midiendo las concentraciones relativas de las formas ionizadas y sin ionizar de la sustancia y el pH de la solución. La relación entre estos términos se da en la ecuación del pKa en la sección Definiciones y unidades, más arriba. Algunas sustancias presentan más de una constante de disociación y pueden obtenerse ecuaciones similares. Algunos de los métodos descritos aquí son también adecuados para disociaciones distintas del tipo ácido/base. Criterios de calidad Repetibilidad La constante de disociación debe repetirse (un mínimo de tres determinaciones) con una variación no superior a ± 0,1 unidades logarítmicas. DESCRIPCIÓN DE LOS PROCEDIMIENTOS DE ENSAYO Hay dos planteamientos básicos para la determinación del pKa. Uno supone la valoración volumétrica de una cantidad conocida de sustancia con ácido o base patrón, según proceda; el otro consiste en determinar la concentración relativa de las formas ionizadas y sin ionizar y su dependencia del pH. Preparación Los métodos basados en estos principios pueden clasificarse como procedimientos de valoración volumétrica, de espectrofotometría y de conductimetría. Soluciones de ensayo Para los métodos de valoración volumétrica y de conductimetría, el producto debe disolverse en agua destilada. Para los métodos de espectrofotometría y otros tipos se utilizan soluciones amortiguadoras. La concentración de la sustancia problema no debe ser superior a 0,01 M o a la mitad de la concentración de saturación (si este valor es menor), y para preparar las soluciones debe emplearse la forma más pura posible de la sustancia. Si la sustancia es poco soluble, podrá disolverse en una pequeña cantidad de un solvente miscible con agua, antes de añadirla hasta conseguir las concentraciones indicadas anteriormente. Las soluciones deben verificarse utilizando el efecto Tyndall con objeto de detectar la presencia de emulsiones, especialmente si se ha utilizado un cosolvente para aumentar la solubilidad. Si se utilizan soluciones amortiguadoras, la concentración de esta no debe ser superior a 0,05 M. Condiciones del ensayo Temperatura La temperatura debe controlarse con una precisión mínima de ± 1 °C. La determinación debe efectuarse preferentemente a 20 °C. Si se sospecha que el resultado depende significativamente de la temperatura, la determinación debe realizarse al menos a otras dos temperaturas. Los intervalos de temperatura deben ser en este caso de 10 °C y el control de la temperatura de ± 0,1 °C. Análisis El método se determinará en función de la naturaleza de la sustancia problema. Debe ser lo suficientemente sensible para permitir la determinación de las distintas especies a la concentración de cada solución de ensayo. Realización del ensayo Método de valoración volumétrica La solución de ensayo se determina por valoración volumétrica con la solución patrón de ácido o de base, según proceda, y midiendo el pH después de cada adición de valorante. Deben efectuarse al menos diez adiciones graduales antes de llegar al punto de equivalencia. Si se alcanza el equilibrio con suficiente rapidez, puede utilizarse un potenciómetro gráfico. A efectos del presente método, es preciso conocer con exactitud tanto la cantidad total de sustancia como su concentración. Deben tomarse las precauciones necesarias para excluir la presencia de dióxido de carbono. En los ensayos normalizados se encuentran pormenores del procedimiento, precauciones y cálculos, por ejemplo en las referencias (1), (2), (3), (4). Método de espectrofotometría Se encuentra una longitud de onda a la que las formas ionizada y sin ionizar de la sustancia tienen coeficientes de extinción sensiblemente diferentes. Se obtiene el espectro de absorción UV/VIS de soluciones de concentración constante a un pH al que la sustancia está esencialmente sin ionizar, a un pH al que está totalmente ionizada, y a varios pH intermedios. Esto puede llevarse a cabo bien añadiendo incrementos de ácido concentrado (o base) a un volumen relativamente grande de una solución de la sustancia de carácter amortiguador y de varios componentes, al principio a pH elevado (o bajo) (ref. 5), o bien añadiendo volúmenes iguales de una solución madre de la sustancia en, por ejemplo, agua o metanol, a volúmenes constantes de diversas soluciones amortiguadoras que abarquen el intervalo deseado de pH. Los valores del pH y de absorbancia a la longitud de onda elegida permiten calcular un número suficiente de valores de pKa, utilizando datos de al menos 5 valores de pH a los que la sustancia esté ionizada como mínimo en un 10 % y sin llegar al 90 %. En la referencia (1) se dan más detalles experimentales y el método de cálculo. Método de conductimetría En una celda de constante pequeña y conocida, se mide la conductividad de una solución aproximadamente 0,1 M de la sustancia en agua de conductividad. Se mide también la conductividad de una serie de diluciones de esta solución efectuadas con exactitud. La concentración se reduce a la mitad cada vez, y la serie debe abarcar al menos un orden de magnitud de concentración. La conductividad limitante a dilución infinita se encuentra realizando un experimento similar con la sal sódica y extrapolando. El grado de disociación puede calcularse entonces a partir de la conductividad de cada solución mediante la ecuación de Onsager, y a continuación, haciendo uso de la ley de dilución de Ostwald, puede calcularse la constante de disociación como K = α2C/(1 – α), donde C es la concentración en moles por litro y α es la fracción disociada. Deben tomarse las precauciones necesarias para excluir la presencia de CO2. En los textos de las normas y en las referencias (1), (6) y (7) se dan más detalles experimentales y el método de cálculo. DATOS E INFORME Tratamiento de los resultados Método de valoración volumétrica Se calcula el valor del pKa en 10 puntos de medición de la curva de valoración. Se calculan la media y la desviación típica de estos valores de pKa. Debe incluirse una representación gráfica del pH frente al volumen de base o ácido patrón, junto con una presentación en forma de cuadro. Método de espectrofotometría Se presentan en forma de cuadro la absorbancia y el pH de cada espectro. Se calculan al menos cinco valores de pKa a partir de los puntos de datos de espectros intermedios, y también se calculan la media y la desviación típica de estos resultados. Método de conductimetría Se calcula la conductividad equivalente Λ para cada concentración de ácido y para cada concentración de una mezcla de un equivalente de ácido más 0,98 equivalentes de hidróxido de sodio exento de carbonato. El ácido se encuentra en exceso para evitar un exceso de OH– debido a la hidrólisis. Se representa 1/Λ frente a Ö_C y puede determinarse la Λo de la sal por extrapolación a concentración cero. La Λo del ácido puede calcularse utilizando los valores de la bibliografía para H+ y Na+. El pKa puede calcularse de α = Λi /Λo y Ka = α2C/(1 – α) para cada concentración. Es posible obtener mejores valores de Ka aplicando correcciones para tener en cuenta la movilidad y la actividad. Deben calcularse la media y la desviación típica de los valores de pKa. Informe del ensayo Deben presentarse todos los datos en bruto y los valores calculados de pKa, junto con el método de cálculo (de preferencia en forma de cuadro, tal como se sugiere en la referencia 1), así como los parámetros estadísticos descritos anteriormente. En el caso de los métodos de valoración volumétrica, deben darse datos de la normalización de los valorantes. En el caso del método de espectrofotometría, deben presentarse todos los espectros. En el caso del método de conductimetría, deben darse datos de la determinación de la constante de celda. Debe aportarse información sobre la técnica utilizada, los métodos analíticos y la naturaleza de los eventuales amortiguadores utilizados. Debe indicarse la temperatura o temperaturas de ensayo. BIBLIOGRAFÍA
|
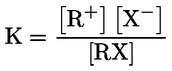
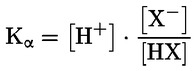

|
2) |
En la parte B, el capítulo B.5 se sustituye por el texto siguiente: «B.5. IRRITACIÓN/CORROSIÓN OCULAR AGUDA INTRODUCCIÓN El presente método de ensayo reproduce las directrices de ensayo (TG) 405 de la OCDE (2012). Las directrices de ensayo de la OCDE para los ensayos de productos se revisan periódicamente a fin de garantizar que reflejan los mejores conocimientos científicos disponibles. En anteriores revisiones de estas directrices de ensayo se prestaba atención especial a las posibles mejoras mediante la evaluación de toda la información existente sobre el producto problema, para no someter a los animales de laboratorio a pruebas innecesarias y responder así a la preocupación por el bienestar de los animales. Las TG 405 (adoptadas en 1981 y actualizadas en 1987, 2002, y 2012) incluyen la recomendación de que, antes de llevar a cabo el ensayo in vivo que se describe para la irritación/corrosión ocular aguda, hay que efectuar una ponderación de las pruebas (1) con los datos relevantes existentes. Si los datos disponibles son insuficientes, se recomienda obtenerlos mediante la aplicación de secuencias de ensayos (2) (3). La estrategia de evaluación recomendada incluye la realización de ensayos in vitro validados y aceptados, y se recoge como suplemento del presente método de ensayo. A efectos del Reglamento (CE) n.o 1907/2006, relativo al registro, la evaluación, la autorización y la restricción de las sustancias y mezclas químicas (REACH) (2), en el correspondiente documento de orientación de la ECHA (21) se incluye también una estrategia de ensayos integrada. Los ensayos con animales solo deben llevarse a cabo si se estima que resultan necesarios tras el examen de los métodos alternativos disponibles, y si se considera que su uso es adecuado. En el momento de la redacción del presente método de ensayo actualizado, hay casos en que la utilización de este método de ensayo sigue siendo necesaria u obligatoria en virtud de algunos marcos normativos. La última actualización se centró principalmente en el uso de analgésicos y anestésicos, sin afectar al concepto de base ni a la estructura de las directrices de ensayo. El ICCVAM (3) y un grupo internacional e independiente de revisión científica por pares revisó la utilidad y las limitaciones de la utilización sistemática de anestésicos tópicos, de analgésicos sistémicos y de parámetros compasivos en los ensayos de irritación ocular in vivo (12). La revisión concluyó que el uso de anestésicos tópicos y de analgésicos sistémicos puede evitar la mayoría o la totalidad del dolor y del sufrimiento, sin afectar a los resultados del ensayo, y recomendó que estas sustancias se utilizaran siempre. El presente método de ensayo tiene en cuenta esta revisión. En los ensayos in vivo de irritación y corrosión ocular aguda deben utilizarse sistemáticamente anestésicos tópicos, analgésicos sistémicos y parámetros compasivos. Las eventuales excepciones habrán de justificarse. Los refinamientos que se describen en el presente método reducirán sustancialmente o evitarán el dolor y el sufrimiento de los animales en la mayoría de las situaciones en las que siga siendo necesario efectuar ensayos de seguridad ocular in vivo. Un tratamiento preventivo y equilibrado del dolor debe incluir: i) un tratamiento previo sistemático con un anestésico tópico (por ejemplo, proparacaína o tetracaína) y un analgésico sistémico (por ejemplo, buprenorfina), ii) un tratamiento posterior sistemático con analgésicos sistémicos (por ejemplo, buprenorfina y meloxicam), iii) un programa de observación, seguimiento y registro de los animales en cuanto a los signos clínicos de dolor o sufrimiento, y iv) un programa de observación, seguimiento y registro de la naturaleza, gravedad y evolución de todas las lesiones oculares. Se encuentran más detalles en los procedimientos actualizados que se describen a continuación. Tras la administración del producto problema, no deben aplicarse más anestésicos ni analgésicos tópicos a fin de evitar interferencias con el estudio. No deben aplicarse de forma tópica analgésicos con actividad antiinflamatoria (p. ej., meloxicam), y las dosis utilizadas sistémicamente no deben interferir con los efectos oculares. Las definiciones se recogen en el apéndice del presente método de ensayo. CONSIDERACIONES INICIALES En aras de una ciencia válida y del bienestar de los animales no debe considerarse la realización de ensayos in vivo hasta que se hayan evaluado todos los datos disponibles relativos al potencial de corrosión/irritación ocular del producto mediante la ponderación de las pruebas. Dichos datos incluyen las pruebas obtenidas en estudios previos realizados con seres humanos o con animales de laboratorio, las pruebas de corrosión/irritación ocular por una o más de sustancias relacionadas estructuralmente o por una mezcla de las mismas, los datos que demuestren una elevada acidez o alcalinidad del producto (4) (5), y los resultados obtenidos en ensayos aceptados y validados de corrosión cutánea y de corrosión/irritación ocular in vitro o ex vivo (6) (13) (14) (15) (16) (17). Los estudios pueden haber sido realizados antes de la ponderación de las pruebas, o a consecuencia de la misma. Para ciertos productos, dicho análisis puede indicar la necesidad de realizar estudios in vivo del potencial de corrosión/irritación ocular del producto. En todos esos casos, antes de considerar el uso de un ensayo ocular in vivo, resulta preferible llevar a cabo en primer lugar un estudio in vitro o in vivo de los efectos de corrosión cutánea del producto, y evaluar este estudio de acuerdo con la estrategia de evaluación secuencial del método de ensayo B.4 (7) o la estrategia de ensayos integrada descrita en el documento de orientación de la ECHA (21). Se adjunta como suplemento del presente método de ensayo, y, a efectos de REACH, en el documento de orientación de la ECHA (21), una estrategia de evaluación secuencial, que incluye la ejecución de ensayos validados de irritación/corrosión ocular in vitro o ex vivo. Se recomienda seguir dicha estrategia de evaluación antes de llevar a cabo ensayos in vivo. Con los productos nuevos se recomienda un enfoque de evaluación por fases para obtener datos científicamente válidos sobre la corrosión/irritación provocada por el producto. Con los productos existentes que cuentan con datos insuficientes sobre su corrosión/irritación cutánea y ocular, puede utilizarse también esta estrategia para obtener los datos que falten. Deben justificarse los casos en que se emplee una estrategia o procedimiento de evaluación diferente, o en que se decida no utilizar un enfoque de evaluación por fases. PRINCIPIO DEL ENSAYO IN VIVO Después de un tratamiento previo con analgésicos sistémicos y la inducción de una anestesia tópica adecuada, el producto problema se aplica en una sola dosis a uno de los ojos del animal de experimentación; el ojo sin tratar sirve de testigo. Se evalúa el grado de irritación/corrosión ocular a intervalos determinados, asignando una puntuación a las lesiones de la conjuntiva, la córnea y el iris. También se describen otros efectos en el ojo y los efectos sistémicos adversos, para proporcionar una evaluación completa de los efectos. La duración del estudio ha de ser suficiente para evaluar la reversibilidad o irreversibilidad de los efectos observados. Los animales que presenten signos de sufrimiento o dolor graves en cualquier fase del ensayo o lesiones compatibles con los parámetros compasivos descritos en el presente método de ensayo (véase el punto 26) deben ser sacrificados de forma compasiva, y el producto evaluado en consecuencia. Los criterios para tomar la decisión de sacrificar de forma compasiva los animales moribundos y que sufren intensamente son objeto de un documento de orientación de la OCDE (8). PREPARACIÓN DEL ENSAYO IN VIVO Selección de las especies El conejo albino es el animal de laboratorio preferido; se emplean individuos adultos jóvenes y sanos. Si se utilizan otras especies será necesario justificarlo. Preparación de los animales En las 24 horas previas al inicio del ensayo se examinarán los dos ojos de los animales de experimentación provisionalmente seleccionados para el ensayo. No se utilizarán animales que muestren irritación ocular, defectos oculares o lesión corneal preexistente. Alojamiento y alimentación Los animales deben ser alojados de manera individual. La temperatura de los animalarios debe ser de 20 °C (± 3 °C) en el caso de los conejos. Aunque la humedad relativa debe ser del 30 % como mínimo y preferiblemente no superar el 70 %, excepto durante la limpieza del animalario, el objetivo debe ser el 50-60 %. La iluminación debe ser artificial, con 12 horas de luz y 12 horas de oscuridad. Debe evitarse una excesiva intensidad de la luz. Para la alimentación se podrán utilizar dietas de laboratorio convencionales, con suministro ilimitado de agua para beber. PROCEDIMIENTO DE ENSAYO Uso de anestésicos tópicos y analgésicos sistémicos Se recomiendan los siguientes procedimientos para evitar o reducir al mínimo el dolor y el sufrimiento en los ensayos de seguridad ocular. Pueden utilizarse otros procedimientos de los que se haya establecido que evitan o alivian de forma igual o mejor el dolor y el sufrimiento.
Aplicación del producto problema El producto problema debe aplicarse a la conjuntiva de un ojo de cada animal, tras separar suavemente el párpado inferior del globo ocular. A continuación se juntan con suavidad los párpados durante un segundo aproximadamente, para que no se pierda el material. El otro ojo no se trata y sirve de testigo. Lavado No se lavarán los ojos de los animales tratados hasta al menos 24 horas después de la instilación del producto problema, excepto en el caso de sólidos (véase el punto 18) y si se producen efectos corrosivos o irritantes inmediatos. Transcurridas 24 horas se podrán lavar los ojos si se considera necesario. No se recomienda utilizar un grupo satélite de animales para investigar la influencia del lavado, a menos que esté justificado desde el punto de vista científico. Si se necesita un grupo satélite, estará formado por dos conejos. Las condiciones del lavado deben quedar minuciosamente documentadas como, por ejemplo, hora del lavado, composición y temperatura de la solución de lavado, duración, volumen y velocidad de la aplicación. Posología 1) Ensayo de líquidos Para el ensayo de líquidos se emplea una dosis de 0,1 ml. No se deben utilizar bombas de aerosol para instilar el producto directamente en el ojo. El aerosol líquido se expulsa de la bomba y se recoge en un recipiente, antes de instilar 0,1 ml en el ojo. 2) Ensayo de sólidos Para el ensayo de sólidos, pastas y productos con partículas, la cantidad utilizada debe tener un volumen de 0,1 ml o no pesar más de 100 mg. El material problema debe estar reducido a polvo fino. El volumen de material sólido se medirá tras compactarlo con suavidad, por ejemplo golpeando suavemente con los dedos el envase medidor. Si en el primer punto temporal de observación (1 hora después de la aplicación) el producto problema sólido no ha sido eliminado del ojo del animal de ensayo por mecanismos fisiológicos, se puede lavar el ojo con suero salino o con agua destilada. 3) Evaluación de aerosoles Se recomienda recoger en un recipiente el contenido de todas las bombas de aerosol antes de instilarlo en el ojo. La única excepción son los productos que van en envases de aerosol presurizados y que no se pueden recoger debido a la vaporización. En esos casos hay que sujetar el ojo bien abierto, administrando el producto problema en el ojo con una sola pulverización de alrededor de un segundo, a una distancia de 10 cm directamente delante del ojo. Esta distancia puede variar dependiendo de la presión del aerosol y de su contenido. Hay que tener cuidado para no lesionar el ojo con la presión del aerosol. En determinados casos puede ser necesario evaluar el potencial de causar una lesión «mecánica» del ojo por la fuerza del aerosol. Es posible calcular la dosis de un aerosol, simulando el ensayo como se indica a continuación: se pulveriza el producto sobre papel de pesar, a través de una abertura del tamaño del ojo de un conejo, colocada directamente delante del papel. El aumento de peso del papel sirve para calcular aproximadamente la cantidad administrada al ojo. En el caso de los productos volátiles se puede calcular la dosis pesando un envase receptor antes y después de retirar el producto problema. Ensayo inicial (ensayo de irritación/corrosión ocular in vivo con un solo animal) Se recomienda encarecidamente realizar el ensayo in vivo inicialmente con un solo animal (véase el suplemento del presente método de ensayo: Estrategia de evaluación secuencial de la irritación y la corrosión oculares). Las observaciones deben permitir determinar la gravedad y la reversibilidad antes de proceder a un ensayo de confirmación con un segundo animal. Si los resultados de este ensayo indican que el producto es irritante intenso o corrosivo en el ojo siguiendo el procedimiento descrito, no se harán más ensayos de irritación ocular. Ensayo de confirmación (ensayo de irritación ocular in vivo con animales adicionales) Si en el ensayo inicial no se observa efecto corrosivo o irritante intenso, la respuesta irritante o negativa debe confirmarse con un máximo de dos animales más. Si se observa efecto irritante en el ensayo inicial, se recomienda efectuar el ensayo de confirmación de forma secuencial exponiendo a los animales de uno en uno, en lugar de exponer a los dos animales adicionales a la vez. Si el segundo animal presenta efectos corrosivos o irritantes intensos se suspenderá el ensayo. Si los resultados del segundo animal son suficientes para permitir una clasificación de peligro, no deben hacerse más ensayos. Período de observación La duración del período de observación debe ser suficiente para evaluar por completo la magnitud y reversibilidad de los efectos observados. No obstante, se dará por finalizado el experimento si en cualquier momento el animal presenta signos de dolor o sufrimiento intensos (8). Para determinar la reversibilidad de los efectos, lo normal es someter a los animales a 21 días de observación a partir de la administración del producto problema. Si se observa la irreversibilidad antes de 21 días, el experimento debe finalizar en ese momento. Observaciones clínicas y graduación de las reacciones oculares Los ojos deben ser objeto de una evaluación exhaustiva en cuanto a la presencia o ausencia de lesiones oculares cuando haya pasado una hora de la APP, seguida de evaluaciones al menos diarias. Los animales deben ser evaluados varias veces al día durante los primeros 3 días para asegurarse de que las decisiones de finalización se adoptan en el momento oportuno. Los animales de ensayo deben ser objeto de evaluación sistemática a lo largo de toda la duración del estudio en cuanto a la presencia de signos clínicos de dolor o sufrimiento (por ejemplo, frotamiento repetido del ojo, parpadeo o lagrimeo excesivo) (9) (10) (11), al menos dos veces al día, con un mínimo de 6 horas entre las observaciones, o con mayor frecuencia si es necesario. Esto es necesario para: i) evaluar adecuadamente los animales en cuanto a la presencia de signos de dolor y sufrimiento a fin de tomar decisiones con conocimiento de causa sobre la necesidad de aumentar la dosis de analgésicos, y ii) evaluar los animales en cuanto a la presencia de parámetros compasivos establecidos a fin de tomar decisiones con conocimiento de causa sobre si procede realizar el sacrificio de los animales de forma compasiva, y garantizar que tales decisiones se tomen de manera oportuna. Se debe utilizar la tinción de fluoresceína sistemáticamente y un biomicroscopio de lámpara de hendidura cuando se considere oportuno (por ejemplo, para evaluar la profundidad de la lesión en caso de úlcera de la córnea) como ayuda en la detección y medición de daños oculares, y para evaluar si se han cumplido los criterios de parámetros establecidos para el sacrificio compasivo. Pueden tomarse fotografías digitales de las lesiones observadas para que sirvan de referencia y proporcionen un registro permanente de la importancia de los daños oculares. Los animales no permanecerán en el ensayo más tiempo del necesario, una vez obtenida la información definitiva. Los animales que muestren dolor o sufrimiento intenso deben ser sacrificados inmediatamente de forma compasiva, y el producto evaluado en consecuencia. Los animales que presenten las siguientes lesiones oculares después de la instilación deben ser sacrificados de forma compasiva (véase en el cuadro 1 una descripción de los grados de las lesiones): perforación de la córnea o ulceración importante de la córnea, incluido el estafiloma; presencia de sangre en la cámara anterior del ojo; opacidad corneal de grado 4; ausencia de reflejo fotomotor (respuesta del iris de grado 2) que persista durante 72 horas; ulceración de la conjuntiva; necrosis de la conjuntiva o de la membrana nictitante, o pérdida de epitelio. Esto se debe a que tales lesiones no suelen ser reversibles. Además, se recomienda que las siguientes lesiones oculares se utilicen como parámetros compasivos para finalizar los estudios antes de que termine el período de observación de 21 días. Se considera que estas lesiones predicen la aparición de lesiones irritantes intensas o corrosivas y de lesiones de las que no se espera que reviertan plenamente al final del período de observación de 21 días: lesión de profundidad importante (por ejemplo, úlcera de la córnea que se extiende más allá de las capas superficiales del estroma), destrucción del limbo > 50 % (puesta de manifiesto por la palidez conjuntival), e infección ocular grave (supuración). Una combinación de vascularización de la superficie de la córnea (es decir, queratitis vascular), mantenimiento de la superficie teñida con fluoresceína a lo largo del tiempo según una evaluación diaria y/o ausencia de nueva epitelización 5 días después de la aplicación del producto problema también podría considerarse como un criterio potencialmente útil para influir en la decisión clínica sobre la finalización precoz del estudio. No obstante, estas observaciones aisladas son insuficientes para justificar la finalización precoz del estudio. Una vez que se han detectado efectos oculares graves, debe pedirse a un veterinario especialista o experto en animales de laboratorio o a una persona con una formación adecuada para identificar lesiones clínicas que realice un examen clínico para determinar si la combinación de tales efectos justifica la finalización precoz del estudio. Deben obtenerse y registrarse los grados de reacción ocular (conjuntiva, córnea e iris) cuando han pasado 1, 24, 48 y 72 horas desde la aplicación del producto problema (cuadro 1). Los animales que no presenten lesiones oculares deben permanecer en observación al menos 3 días después de la instilación. Los animales con lesiones que no sean graves deben permanecer en observación hasta que desaparezcan las lesiones o hasta que transcurran 21 días, momento en que finaliza el estudio. Deben efectuarse y registrarse observaciones al menos cuando hayan pasado 1 hora, 24 horas, 48 horas, 72 horas, 7 días, 14 días y 21 días, para determinar el estado de las lesiones y si son o no reversibles. Deben efectuarse observaciones más frecuentes cuando sea necesario para determinar si el animal de experimentación ha de sacrificarse por motivos compasivos o eliminarse del estudio debido a los resultados negativos. Deben registrarse en cada examen los grados de las lesiones oculares (cuadro 1). También deben consignarse las demás lesiones oculares (por ejemplo, queratitis vascular, manchas, cambios en la cámara anterior) o efectos sistémicos adversos eventuales. Se puede facilitar el examen de las reacciones mediante el uso de una lupa binocular, de una lámpara de hendidura manual, de un biomicroscopio o de otro dispositivo adecuado. Tras registrar las observaciones a las 24 horas, las ulteriores exploraciones del ojo pueden hacerse con la ayuda de fluoresceína. La graduación de las respuestas oculares es subjetiva necesariamente. Para armonizar tal graduación y ayudar a los laboratorios y a quienes efectúan e interpretan las observaciones, el personal encargado recibirá formación adecuada sobre el sistema de puntuación utilizado. DATOS E INFORME Evaluación de los resultados Deben evaluarse las puntuaciones de la irritación ocular, junto con la naturaleza y la gravedad de las lesiones, así como su reversibilidad o irreversibilidad. Las puntuaciones individuales no dan el nivel absoluto de las propiedades irritantes de un producto, ya que también se evalúan otros efectos del producto problema; más bien deben considerarse como valores de referencia, que solo serán significativos cuando sean respaldados por una descripción completa y la evaluación de todas las demás observaciones. Informe del ensayo El informe del ensayo debe incluir la información siguiente:
Interpretación de los resultados La extrapolación a seres humanos de los resultados de los estudios de irritación ocular realizados con animales de laboratorio solo es válida en cierto grado. En muchos casos, los conejos albinos son más sensibles a los irritantes o corrosivos oculares que los seres humanos. Hay que interpretar con cuidado los datos para excluir la irritación resultante de una infección secundaria. BIBLIOGRAFÍA
Cuadro 1: Graduación de las lesiones oculares
Apéndice DEFINICIONES Reserva ácida/alcalina : En el caso de las preparaciones ácidas, es la cantidad (g) de hidróxido de sodio /100 g de preparación que se necesita para obtener un pH determinado. En el caso de las preparaciones alcalinas, es la cantidad (g) de hidróxido de sodio equivalente a la cantidad (g) de ácido sulfúrico /100 g de preparación que se necesita para obtener un pH determinado (Young et al. 1988). 1988. Producto : Sustancia o mezcla. Productos no irritantes : Sustancias que no están clasificadas como irritantes oculares de la categoría I, II o III de la EPA; ni como irritantes oculares de la categoría 1, 2, 2A o 2B del SGA; ni de la categoría 1 o 2 de la UE (17) (18) (19). Productos corrosivos oculares : a) Productos que provocan una lesión irreversible de los tejidos oculares; b) productos clasificados como irritantes oculares de la categoría 1 del SGA, o de la categoría I de la EPA, o de la categoría 1 de la UE (17) (18) (19). Agentes irritantes para los ojos : a) Productos que producen un cambio reversible en el ojo; b) productos que están clasificados como irritantes oculares de la categoría II o III de la EPA; o como irritantes oculares de la categoría 2, 2A o 2B del SGA; o de la categoría 2 de la UE (17) (18) (19). Productos irritantes intensos oculares : a) Productos que provocan en los ojos lesiones tisulares que no se resuelven en los 21 días siguientes a la aplicación, o que provocan una degradación física severa de la vista; b) productos clasificados como irritantes oculares de la categoría 1 del SGA, o de la categoría I de la EPA, o de la categoría 1 de la UE (17) (18) (19). Producto problema : Sustancia o mezcla estudiada con este método de ensayo. Procedimiento escalonado : Estrategia de ensayo por fases, en la que se revisa toda la información existente sobre un producto problema, siguiendo un orden especificado, en un proceso de ponderación de las pruebas en cada escalón, a fin de determinar si se dispone de información suficiente para tomar una decisión sobre la clasificación de un peligro, antes de pasar al escalón siguiente. Si puede establecerse la capacidad de irritación de un producto problema con la información disponible, no hace falta efectuar más ensayos. Si no puede establecerse la capacidad de irritación de un producto problema con la información disponible, se aplica un procedimiento secuencial de ensayos con animales por fases hasta que pueda efectuarse una clasificación inequívoca. Ponderación de las pruebas (proceso) : Los puntos fuertes y débiles de la información recopilada se utilizan como base para alcanzar una conclusión que puede no deducirse directamente de los datos individuales. SUPLEMENTO DEL MÉTODO DE ENSAYO B.5 (4) ESTRATEGIA DE EVALUACIÓN SECUENCIAL DE LA IRRITACIÓN Y LA CORROSIÓN OCULARES Consideraciones generales Es importante evitar el uso innecesario de animales y reducir al mínimo los ensayos que con toda probabilidad producen respuestas graves en los animales, por su bienestar y por motivos científicos. Antes de considerar la realización de ensayos in vivo hay que evaluar toda la información sobre un producto en lo que respeta a su posible poder corrosivo/irritante ocular. Puede que existan pruebas suficientes para clasificar el potencial corrosivo o irritante ocular de un producto problema, sin necesidad de realizar ensayos con animales de laboratorio. Por eso el uso de la ponderación de las pruebas y de una estrategia de evaluación secuencial reducirá al mínimo la necesidad de realizar ensayos in vivo, especialmente si es probable que el producto produzca reacciones graves. Se recomienda utilizar la ponderación de las pruebas para evaluar la información existente sobre el potencial de irritación y corrosión oculares de los productos, para determinar si la realización de estudios adicionales, aparte de los oculares in vivo, ayudaría a caracterizar dicho potencial. Cuando sean necesarios otros estudios, se recomienda utilizar la estrategia de evaluación secuencial para obtener los datos experimentales relevantes. Para sustancias no evaluadas anteriormente, se utilizará la estrategia de evaluación secuencial para obtener los datos necesarios a fin de evaluar su potencial corrosivo/irritante ocular. La estrategia de evaluación inicial que se describe en el prresente suplemento fue redactada en un taller de la OCDE (1). Posteriormente fue confirmada y ampliada por el sistema armonizado integrado de clasificación de peligros para la salud humana y efectos ambientales de los productos químicas (Harmonised Integrated Hazard Classification System for Human Health and Environmental Effects of Chemical Substances), y fue aprobada en la XXVIII reunión del Comité Conjunto para productos químicos (Chemicals Committee and the Working Party on Chemicals) en noviembre de 1998 (2) y actualizada por un grupo de expertos de la OCDE en 2011. Aunque la presente estrategia de evaluación no forma parte integrante del método de evaluación B.5, sí expresa el enfoque recomendado para la determinación de las características de irritación/corrosión oculares. Este enfoque representa la práctica óptima y una referencia desde el punto de vista ético para el análisis in vivo de la irritación/corrosión oculares. El método de ensayo proporciona instrucciones para la realización del ensayo in vivo, y resume los factores que se han de abordar antes de ponerlo en marcha. La estrategia proporciona un enfoque basado en la ponderación de las pruebas para la evaluación de los datos existentes sobre las propiedades de irritación/corrosión oculares de los productos problema, y un enfoque escalonado para generar datos relevantes sobre productos que necesitan estudios adicionales o que no han sido estudiadas. La estrategia incluye la realización en primer lugar de ensayos in vitro o ex vivo validados y aceptados, y luego de estudios según el método de ensayo B.4 en condiciones concretas (3) (4). Descripción de la estrategia de evaluación por fases Antes de llevar a cabo los ensayos que forman parte de la estrategia de evaluación secuencial (véase la figura), se evaluará toda la información disponible, para determinar la necesidad de practicar estudios oculares in vivo. Aunque se puede obtener información significativa a partir de la evaluación de parámetros aislados (por ejemplo, un pH extremo), hay que considerar la totalidad de la información existente. Se evaluarán todos los datos relevantes sobre los efectos del producto en cuestión y de sus análogos estructurales, para tomar una decisión basada en la ponderación de las pruebas, y se presentará la justificación de dicha decisión. Se hará especial hincapié en los datos existentes sobre el producto obtenidos con seres humanos y animales, seguido por el resultado de los ensayos in vitro o ex vivo. Siempre que sea posible se evitará realizar estudios in vivo con productos corrosivos. Los factores que se tienen en cuenta en la estrategia de evaluación son:
ESTRATEGIA DE EVALUACIÓN DE LA IRRITACIÓN/CORROSIÓN OCULAR
BIBLIOGRAFÍA
|
|
3) |
En la parte B, el capítulo B.10 se sustituye por el texto siguiente: «B.10. Ensayo de aberraciones cromosómicas en mamíferos in vitro INTRODUCCIÓN El presente método de ensayo es equivalente a las directrices de ensayo de la OCDE 473 (2016). Forma parte de una serie de métodos de ensayo sobre toxicología genética. Se ha elaborado un documento de la OCDE que aporta información sucinta sobre los ensayos de toxicología genética y una síntesis de los recientes cambios aportados a dichas directrices de ensayo (1). El ensayo de aberraciones cromosómicas in vitro tiene por objeto detectar productos que provocan aberraciones cromosómicas estructurales en células de mamífero en cultivo (2) (3) (4). Las aberraciones estructurales pueden ser cromosómicas o cromatídicas. Puede surgir poliploidía (incluida la endorreduplicación) en los ensayos de aberraciones cromosómicas in vitro. Si bien los anéugenos pueden inducir la aparición de poliploidía, esta por sí sola no indica potencial aneugénico y puede indicar simplemente perturbación del ciclo celular o citotoxicidad (5). Este ensayo no está diseñado para medir la aneuploidía. Para la detección de la aneuploidía se recomendaría un ensayo de micronúcleos in vitro (6). El ensayo de aberraciones cromosómicas in vitro puede emplear cultivos de líneas celulares establecidas o cultivos de células primarias humanas o de roedores. Las células deben seleccionarse sobre la base de su capacidad de crecimiento en cultivo, su estabilidad cariotípica (incluido el número de cromosomas) y su frecuencia de aberraciones cromosómicas espontáneas (7). Por el momento, los datos de que se dispone no permiten formular recomendaciones firmes pero sugieren que es importante, a la hora de evaluar los peligros químicos, examinar la situación de la proteína p53, la estabilidad genética (cariotipo), la capacidad de reparación del ADN y el origen (roedores frente a hombre) de las células elegidas para el ensayo. Se recomienda, pues, a los usuarios de este método de ensayo que consideren la influencia de estas y otras características de las células sobre el comportamiento de una línea celular en la detección de la inducción de aberraciones cromosómicas, ya que la situación de los conocimientos en este ámbito está evolucionando. En el apéndice 1 se dan las definiciones utilizadas. CONSIDERACIONES INICIALES Y LIMITACIONES Los ensayos realizados in vitro suelen exigir la utilización de una fuente exógena de activación metabólica, salvo que las células sean competentes metabólicamente respecto a los productos problema. El sistema exógeno de activación metabólica no reproduce completamente las condiciones in vivo. Deben evitarse las condiciones que puedan conducir a resultados positivos falsos, es decir, lesiones cromosómicas no causadas por la interacción directa entre los productos problema y los cromosomas; tales condiciones incluyen cambios en el pH o en la osmolalidad (8) (9) (10), la interacción con los componentes del medio (11) (12) o unos niveles excesivos de citotoxicidad (13) (14) (15) (16). Este ensayo se emplea para detectar aberraciones cromosómicas que puedan producirse como consecuencia de sucesos clastogénicos. El análisis de la inducción de aberraciones cromosómicas debe efectuarse utilizando células en metafase. Por tanto, es esencial que las células estén en mitosis en los cultivos tanto tratados como sin tratar. En caso de nanomateriales fabricados, puede ser necesario recurrir a adaptaciones específicas del presente método de ensayo, pero estas no se describen aquí. Antes de la utilización del método de ensayo con una mezcla para obtener datos con fines reglamentarios, debe considerarse si podría proporcionar resultados adecuados a tal fin y, en caso afirmativo, por qué. Tales consideraciones no son necesarias si la reglamentación impone el ensayo de la mezcla. PRINCIPIO DEL ENSAYO Los cultivos celulares de origen humano o de otros mamíferos se exponen al producto problema tanto con fuente exógena de activación metabólica como sin ella, salvo que se utilicen células con una capacidad adecuada de metabolización (véase el punto 13). A intervalos predeterminados apropiados tras el inicio de la exposición de los cultivos celulares al producto problema, se tratan estos con un producto que detenga la división celular en la metafase (p. ej., colcemid o colchicina), se recolectan, se tiñen y se analizan al microscopio las células en metafase para evaluar la presencia de aberraciones cromatídicas y cromosómicas. DESCRIPCIÓN DEL MÉTODO Preparación Células Pueden utilizarse cultivos de diversas líneas celulares (por ejemplo, células de ovario de hámster chino (CHO), células de pulmón de hámster chino V79, células de pulmón de hámster chino (CHL)/IU, TK6) o de células primarias, incluidos los linfocitos de sangre periférica de hombre o de otros mamíferos (7). La elección de las líneas celulares debe justificarse científicamente. Cuando se utilizan células primarias, por razones de bienestar animal, debe considerarse la utilización de células primarias de origen humano, cuando sea viable y se hayan tomado de conformidad con los principios éticos humanos y la reglamentación. Los linfocitos de sangre periférica humana deben obtenerse de individuos jóvenes (aproximadamente de 18 a 35 años de edad), no fumadores, sin enfermedades conocidas ni exposiciones recientes a agentes genotóxicos (por ejemplo, productos, radiaciones ionizantes) a niveles que puedan aumentar la incidencia de fondo de las aberraciones cromosómicas. Esto garantizaría una incidencia de fondo de las aberraciones cromosómicas baja y constante. La incidencia de base de las aberraciones cromosómicas aumenta con la edad y esta tendencia es más pronunciada en las mujeres que en los varones (17) (18). Si se combinan células procedentes de más de un donante, debe especificarse el número de donantes. Es necesario demostrar que las células se han dividido entre el inicio de la administración del producto problema y la recolección de las células. Los cultivos celulares se mantienen en fase exponencial de crecimiento celular (líneas celulares) o se estimulan para que se dividan (cultivos primarios de linfocitos), a fin de exponer las células en diferentes fases del ciclo celular, ya que es posible que no se conozca la sensibilidad de las distintas fases celulares a los productos problema. Las células primarias que es preciso estimular con agentes mitogénicos para que se dividan dejan generalmente de estar sincronizadas durante la exposición al producto problema (por ejemplo, los linfocitos humanos tras una estimulación mitogénica de 48 horas). La utilización de células sincronizada durante el tratamiento no es recomendable, pero puede aceptarse si está justificada. Medios y condiciones de cultivo Para el mantenimiento de los cultivos deben utilizarse medios de cultivo y condiciones de incubación adecuados (recipientes de cultivo, atmósfera humidificada con 5 % de CO2 en su caso, temperatura de incubación de 37 °C). Las líneas celulares deben examinarse sistemáticamente para comprobar la estabilidad del número modal de cromosomas y la ausencia de contaminación por Mycoplasma (7) (19), y no deben utilizarse las células que se contaminen o que presenten un cambio en el número modal de cromosomas. Debe determinarse la duración del ciclo celular normal de las líneas celulares o cultivos primarios utilizados en el laboratorio de ensayo, y ser coherente con las características celulares publicadas (20). Preparación de los cultivos Líneas celulares: las células se propagan a partir de cultivos madre, se siembran en el medio de cultivo a una densidad tal que las células en suspensión o en monocapas sigan creciendo exponencialmente hasta el momento de la recolección (por ejemplo, debe evitarse la confluencia de las células cultivadas en monocapas). Linfocitos: se cultiva sangre completa tratada con anticoagulante (p. ej., heparina) o bien linfocitos separados (p.ej., durante 48 horas en el caso de los linfocitos humanos) en presencia de un mitógeno [p. ej., fitohemaglutinina (PHA) en el caso de los linfocitos humanos] a fin de inducir la división celular antes de la exposición al producto problema. Activación metabólica Se debe recurrir a sistemas exógenos de metabolización cuando se utilicen células con una capacidad metabólica endógena inadecuada. El sistema utilizado con más frecuencia que se recomienda por defecto, salvo en casos justificados, es una fracción postmitocondrial (S9) a la que se añaden cofactores y que se obtiene a partir del hígado de roedores (generalmente ratas) tratados con inductores enzimáticos como el aroclor 1254 (21) (22) (23) o una combinación de fenobarbital y β-naftoflavona (24) (25) (26) (27) (28) (29). Esta última combinación no infringe el Convenio de Estocolmo sobre contaminantes orgánicos persistentes (30), y se ha visto que es tan efectiva como el aroclor 1254 para inducir oxidasas de función mixta (24) (25) (26) (28). La fracción S9 se utiliza normalmente a concentraciones que varían entre el 1 y el 2 % (v/v), pero pueden aumentarse hasta el 10 % (v/v) en el medio de ensayo final. Debe evitarse durante el tratamiento el uso de productos que reduzcan el índice mitótico, especialmente productos que acomplejen el calcio (31). La elección del tipo y de la concentración del sistema exógeno de activación metabólica o del inductor metabólico utilizado puede estar influida por la clase de los productos que se someten a ensayo. Preparación del producto problema Los productos problema sólidos deben prepararse en disolventes adecuados y, si es conveniente, diluirse antes de tratar las células (véase el punto 23). Los productos problema líquidos pueden añadirse directamente al sistema de ensayo o diluirse antes del tratamiento del sistema de ensayo. Los productos problema gaseosos o volátiles deben someterse a ensayo aplicando modificaciones adecuadas a los protocolos normales, tales como el tratamiento en recipientes de cultivo sellados (32) (33) (34). La preparación del producto problema debe hacerse justo antes del tratamiento, salvo que se cuente con datos de estabilidad que avalen la posibilidad de su conservación. Condiciones del ensayo Disolventes El disolvente debe elegirse para optimizar la solubilidad de los productos problema sin tener un impacto negativo en la realización del ensayo, por ejemplo por cambiar el crecimiento celular, afectar a la integridad del producto problema, reaccionar con los recipientes de cultivo, o interferir con el sistema de activación metabólica. Siempre que sea posible, se recomienda considerar en primer lugar la utilización de un disolvente (o medio de cultivo) acuoso. Son ejemplos de disolventes bien establecidos el agua o el dimetilsulfóxido. En general, los disolventes orgánicos no deben exceder del 1 % (v/v) y los disolventes acuosos (solución salina o agua) no deben superar el 10 % (v/v) en el medio de tratamiento final. Si se utilizan disolventes que no están bien establecidos (por ejemplo, etanol o acetona), debe disponerse de datos justificativos que indiquen su compatibilidad con el sistema de ensayo y los productos problema, y su ausencia de toxicidad genética a la concentración utilizada. En ausencia de estos datos justificativos, es importante incluir testigos sin tratar (véase el apéndice 1) para demostrar que el disolvente elegido no es nocivo ni tiene efectos clastogénicos. Medición de la proliferación celular y de la citotoxicidad, y selección de las concentraciones de tratamiento Al determinar la concentración máxima de producto problema, debe evitarse llegar a concentraciones que puedan producir respuestas positivas falsas, tales como las que provocan una citotoxicidad excesiva (véase el punto 22), precipitación en el medio de cultivo (véase el punto 23), o cambios marcados del pH o de la osmolalidad (véase el punto 5). Si el producto problema provoca un cambio marcado en el pH del medio en el momento de su adición, el pH puede ajustarse amortiguando el medio de tratamiento final para evitar resultados positivos falsos y mantener unas condiciones de cultivo adecuadas. Se mide la proliferación celular para asegurarse de que las células tratadas han alcanzado la mitosis durante el ensayo en número suficiente y de que los tratamientos se efectúan a los niveles adecuados de citotoxicidad (véanse los puntos 18 y 22). La citotoxicidad se debe determinar con y sin activación metabólica en el experimento principal mediante un indicador adecuado de la muerte y el crecimiento de las células. Si bien la evaluación de la citotoxicidad en un ensayo inicial puede ser útil para definir mejor las concentraciones que deben utilizarse en el experimento principal, no es obligatorio efectuar un ensayo inicial. Si se lleva a cabo, no debe sustituir a la medición de la citotoxicidad en el experimento principal. Para evaluar la citotoxicidad en los ensayos citogenéticos, puede recurrirse a la medición de la duplicación relativa de la población (DRP) o del aumento relativo del recuento celular (ARRC) (13) (15) (35) (36) (55) (véanse las fórmulas en el apéndice 2). En caso de tratamiento a largo plazo y de tiempos de recolección tras el inicio del tratamiento superiores a 1,5 veces la duración del ciclo celular normal (es decir, más de 3 ciclos celulares en total), la DRP puede subestimar la citotoxicidad (37). En tales circunstancias, puede ser preferible medir el ARRC, o puede ser útil evaluar la citotoxicidad tras un tiempo igual a 1,5 veces la duración del ciclo celular normal utilizando la DRP. En el caso de los linfocitos en cultivos primarios, si bien el índice mitótico (IM) es una medida de los efectos citotóxicos o citostáticos, se ve influido por el tiempo al que se mide después del tratamiento, por el mitógeno empleado y por la posible alteración del ciclo celular. No obstante, el IM es aceptable porque es posible que otras mediciones de la citotoxicidad resulten engorrosas y poco prácticas y no puedan aplicarse a la población objetivo de linfocitos que crecen en respuesta a la estimulación con PHA. Si bien el ARRC y la DRP en el caso de las líneas celulares y el IM en el de los cultivos primarios de linfocitos son los parámetros de citotoxicidad recomendados, el uso de otros indicadores (por ejemplo, integridad de las células, apoptosis, necrosis, ciclo celular) podría proporcionar información adicional útil. Deben evaluarse al menos tres concentraciones de ensayo (sin incluir los testigos positivos y de disolvente) que cumplan los criterios de aceptabilidad (citotoxicidad apropiada, número de células, etc.). Cualquiera que sea el tipo de células (líneas celulares o cultivos primarios de linfocitos), pueden utilizarse a cada concentración de ensayo cultivos tratados replicados o sin replicar. Si bien se recomienda el uso de cultivos duplicados, también son aceptables los cultivos sin replicar, siempre que se examine el mismo número total de células, sea en cultivos duplicados o sin replicar. La utilización de cultivos sin replicar es especialmente pertinente cuando se evalúan más de 3 concentraciones (véase el punto 31). Los resultados obtenidos en los cultivos replicados independientes a una concentración determinada se pueden poner en común para el análisis de los datos (38). En el caso de productos problema que muestren escasa o nula citotoxicidad, normalmente serán adecuados los intervalos de concentración de aproximadamente el doble o el triple. Cuando se produce citotoxicidad, las concentraciones de ensayo seleccionadas deben cubrir una gama a partir de la que provoca citotoxicidad según se describe en el punto 22, con inclusión en particular de las concentraciones a las que existe citotoxicidad moderada y débil o nula. Muchos productos problema presentan curvas de respuesta a la concentración de elevada pendiente y, con el fin de obtener datos a niveles bajos y moderados de citotoxicidad o de estudiar en detalle la relación entre dosis y respuesta, entonces es necesario recurrir a concentraciones más próximas entre sí o a más de tres concentraciones (cultivos replicados o sin replicar), en particular en situaciones en que se requiere una repetición del experimento (véase el punto 47). Si la concentración máxima se basa en la citotoxicidad, la concentración más elevada debe aspirar a provocar un 55 ± 5 % de citotoxicidad utilizando los parámetros recomendados de citotoxicidad (es decir, reducción del ARRC y de la DRP con líneas celulares y reducción del IM con cultivos primarios de linfocitos al 45 ± 5 % del testigo negativo en paralelo). Ha de tenerse cuidado al interpretar resultados positivos que solo se encuentren en el extremo superior de este intervalo de citotoxicidad del 55 ± 5 % (13). Para los productos problema poco solubles que no son citotóxicos a concentraciones inferiores a la concentración mínima insoluble, la mayor concentración analizada debe producir turbidez o precipitado visibles a simple vista o con ayuda de un microscopio invertido al final del tratamiento con el producto problema. Incluso si se produce citotoxicidad por encima del límite de solubilidad, es aconsejable hacer el ensayo a una única concentración que produzca turbidez o precipitado visible porque este puede provocar efectos falsos. A la concentración que produce precipitado, se debe evitar que este interfiera con la realización del ensayo (por ejemplo, con la tinción o el examen celular). Puede ser útil determinar la solubilidad en el medio de cultivo antes de que efectuar el experimento. Si no se observa precipitado ni citotoxicidad limitante, la concentración de ensayo más elevada debe corresponder a la más baja de las siguientes: 10 mM, 2 mg/ml o 2 μl/ml (39) (40) (41). Si el producto problema no tiene una composición definida y se trata, por ejemplo, de sustancias de composición desconocida o variable, productos complejos de reacción o materiales biológicos (UVCB) (42), extractos medioambientales, etc., es posible que la concentración superior tenga que ser mayor (por ejemplo, 5 mg/ml), en ausencia de citotoxicidad suficiente, para aumentar la concentración de cada uno de los componentes. Conviene señalar, no obstante, que estos requisitos pueden variar en caso de medicamentos de uso humano (43). Testigos Se incluirán, para cada período de recolección, testigos negativos en paralelo (véase el punto 15), consistentes en el disolvente solo en el medio de tratamiento y sometidos al mismo proceso que los cultivos del ensayo. Es necesario disponer testigos positivos en paralelo a fin de demostrar la capacidad del laboratorio para detectar clastógenos en las condiciones establecidas en el protocolo de ensayo utilizado y la efectividad del sistema exógeno de activación metabólica, si procede. En el cuadro 1 a continuación se encuentran ejemplos de testigos positivos. Es posible utilizar como testigos positivos otros productos, si se justifica. Dado que los ensayos de toxicidad genética con células de mamífero in vitro están suficientemente normalizados, el uso de testigos positivos puede limitarse a un clastógeno que requiera activación metabólica. Siempre que se utilice de forma paralela al ensayo sin activación con la misma duración del tratamiento, la respuesta de este único testigo positivo demostrará tanto la actividad del sistema de activación metabólica como la capacidad de respuesta del sistema de ensayo. No obstante, los tratamiento a largo plazo (sin fracción S9) deben tener su propio testigo positivo, ya que la duración del tratamiento será diferente de la del ensayo con activación metabólica. Cada testigo positivo debe utilizarse a una o varias concentraciones de las que quepa esperar que produzcan incrementos reproducibles y detectables respecto a los valores de fondo para demostrar la sensibilidad del sistema de ensayo (es decir, que los efectos sean claros, pero sin revelar inmediatamente al lector la identidad de los portaobjetos codificados), y la respuesta no debe verse comprometida por una citotoxicidad que exceda de los límites especificados en el método de ensayo. Cuadro 1. Productos de referencia recomendados para evaluar la competencia del laboratorio, y para la selección de los testigos positivos.
PROCEDIMIENTO Tratamiento con el producto problema Se tratan células en crecimiento con el producto problema en presencia y en ausencia de un sistema de activación metabólica. Momento de recolección del cultivo Para una evaluación a fondo, que sería necesaria para deducir un resultado negativo, deben aplicarse las tres condiciones experimentales siguientes, utilizando un tratamiento a corto plazo con y sin activación metabólica y un tratamiento a largo plazo sin activación metabólica (véanse los puntos 43, 44 y 45):
En caso de que alguna de las anteriores condiciones experimentales conduzca a una respuesta positiva, puede no ser necesario investigar los otros regímenes de tratamiento. Preparación de los cromosomas Se tratan los cultivos celulares con colcemid o colchicina, por lo general durante un período de 1 a 3 horas antes de la recolección. Los cultivos celulares se recolectan y procesan por separado para preparar los cromosomas. Esta preparación incluye el tratamiento hipotónico de las células, y su fijación y tinción. En las monocapas, es posible que haya células mitóticas (identificables por su forma redondeada y por despegarse de la superficie) al final del tratamiento de 3-6 horas. Como estas células mitóticas se despegan con facilidad, pueden perderse cuando se retira el medio que contiene el producto problema. Si hay pruebas de un aumento sustancial del número de células mitóticas en comparación con los testigos, lo que indica una probable detención mitótica, las células deben recogerse por centrifugación y volver a añadirse a los cultivos, para evitar la pérdida de células que estén en mitosis, y que podrían presentar aberraciones cromosómicas, en el momento de la recolección. Análisis Todos los portaobjetos, incluidos los de los testigos positivos y negativos, deben codificarse de forma independiente antes de su análisis microscópico para detectar aberraciones cromosómicas. Dado que los métodos de fijación provocan con frecuencia que una parte de las células en metafase pierdan cromosomas, las células examinadas deben contener un número de centrómeros igual al número modal ± 2. Deben examinarse al menos 300 metafases bien extendidas de cada concentración y testigo para concluir que un producto problema es claramente negativo (véase el punto 45). Las 300 células deben repartirse a partes iguales entre las réplicas, cuando se utilizan cultivos replicados. Cuando se utiliza un solo cultivo sin replicar por concentración (véase el punto 21), deben examinarse al menos 300 metafases bien extendidas en este cultivo único. El examen de 300 células tiene la ventaja de aumentar la potencia estadística del ensayo y, además, será poco probable que se observen valores nulos (se espera que sean solo el 5 %) (44). El número de metafases examinadas puede reducirse cuando se observe un elevado número de células con aberraciones cromosómicas y el producto problema se considere claramente positivo. Deben examinarse células con aberraciones cromosómicas estructurales, contando tanto con las separaciones (gaps) como sin ellas. Las roturas y separaciones se definen en el apéndice 1 de conformidad con (45) (46). Las aberraciones cromosómicas y las cromatídicas deben registrarse por separado y clasificarse en subtipos (roturas, intercambios). Los procedimientos seguidos en el laboratorio deben garantizar que el análisis de las aberraciones cromosómicas es realizado por examinadores bien formados y es objeto de una revisión por pares, si procede. Pese a que el objeto del ensayo consiste en detectar aberraciones cromosómicas estructurales, es importante registrar las frecuencias de la poliploidía y de la endorreduplicación cuando se dan estos fenómenos (véase el punto 2). Competencia del laboratorio Con el fin de conseguir la suficiente experiencia con el ensayo antes de utilizarlo en ensayos sistemáticos, el laboratorio debe haber efectuado una serie de experimentos con productos positivos de referencia que actúen a través de diferentes mecanismos y con diversos testigos negativos (con diversos disolventes o vehículos). Las respuestas obtenidas con estos testigos positivos y negativos deben ser coherentes con la bibliografía. Esto no es aplicable a los laboratorios que tienen experiencia, esto es, que disponen de una base de datos históricos, según se define en el punto 37. Debe investigarse una selección de productos testigo positivos (véase el cuadro 1 en el punto 26) con tratamientos cortos y largos en ausencia de activación metabólica, y también con tratamientos cortos en presencia de activación metabólica, para demostrar su capacidad de detectar productos clastogénicos y determinar la efectividad del sistema de activación metabólica. Debe elegirse una gama de concentraciones de los productos seleccionados de forma que produzcan aumentos sobre el nivel de fondo relacionados con la concentración y reproducibles para demostrar la sensibilidad y el intervalo dinámico del sistema de ensayo. Datos sobre testigos históricos El laboratorio debe determinar:
Cuando se obtengan datos por primera vez en relación con la distribución de testigos negativos históricos, los testigos negativos en paralelo deben ser coherentes con los datos publicados de los testigos, si existen. Según se añadan más datos experimentales sobre la distribución de los testigos, los testigos negativos en paralelo deben situarse idealmente dentro de los límites de control del 95 % de dicha distribución (44) (47). La base de datos de testigos negativos históricos del laboratorio debe constituirse en un principio con un mínimo de 10 experimentos, pero preferiblemente con al menos 20 experimentos realizados en condiciones experimentales comparables. Los laboratorios deben utilizar métodos de control de calidad, como gráficos de control [por ejemplo, gráficos C o gráficos de medias (48)], con el fin de determinar la variabilidad de sus datos sobre los testigos positivos y negativos, y de demostrar que la metodología está «controlada» en su laboratorio (44). En la bibliografía pueden encontrarse más recomendaciones sobre cómo conseguir y utilizar los datos históricos (es decir, criterios de inclusión y exclusión de datos en los datos históricos y criterios de aceptabilidad para un determinado experimento) (47). Cualquier cambio en el protocolo experimental debe considerarse en función de su coherencia con las bases de datos de testigos históricos existentes del laboratorio. Cualquier incoherencia importante debería dar lugar a la creación de una nueva base de datos de testigos históricos. Los datos sobre los testigos negativos deben consistir en la incidencia de células con aberraciones cromosómicas procedentes de un solo cultivo o de la suma de cultivos replicados como se describe en el punto 21. Lo ideal sería que los testigos negativos en paralelo estuvieran dentro de los límites de control del 95 % de la distribución de la base de datos de testigos negativos históricos del laboratorio (44) (47). Cuando hay datos de los testigos negativos en paralelo que quedan fuera de los límites de control del 95 %, su inclusión en la distribución de testigos históricos puede ser aceptable en la medida en que dichos datos no sean valores extremos y haya pruebas de que el sistema de ensayo está «controlado» (véase el punto 37) y pruebas de la ausencia de fallos técnicos o humanos. DATOS E INFORME Presentación de los resultados Debe evaluarse el porcentaje de células que presentan aberraciones cromosómicas estructurales. Las aberraciones cromosómicas y cromatídicas, clasificadas por subtipos (roturas, intercambios), deben enumerarse por separado con su número y frecuencia en relación con los cultivos experimentales y testigos. Las separaciones se registran y se recogen en el informe aparte, pero por lo general no se incluyen en la frecuencia total de aberraciones. Se recoge en el informe el porcentaje de poliploidía o de células endorreduplicadas, cuando se observan. Asimismo, deben registrarse las determinaciones de citotoxicidad realizadas en paralelo en todos los cultivos tratados y en los testigos positivos y negativos de los experimentos principales sobre aberraciones. Deben proporcionarse datos de cada cultivo. Además, se resumirán todos los datos en forma de cuadro. Criterios de aceptabilidad La aceptabilidad de un ensayo se basa en los criterios siguientes:
Evaluación e interpretación de los resultados Siempre que se cumplan todos los criterios de aceptabilidad, se considera que el producto problema es claramente positivo si, en alguna de las condiciones experimentales examinadas (véase el punto 28):
Cuando se cumplen todos estos criterios, el producto problema se considera capaz de inducir aberraciones cromosómicas en las células de mamífero cultivadas en este sistema de ensayo. En la bibliografía se encuentran recomendaciones sobre los métodos estadísticos más adecuados (49) (50) (51). Siempre que se cumplan todos los criterios de aceptabilidad, se considera que el producto problema es claramente negativo si, en todas las condiciones experimentales examinadas (véase el punto 28):
el producto problema se considera entonces incapaz de inducir aberraciones cromosómicas en las células de mamífero cultivadas en este sistema de ensayo. No se requiere ninguna verificación de una respuesta claramente positiva o negativa. En caso de que la respuesta no sea ni claramente positiva ni claramente negativa como se describe más arriba, o a fin de ayudar a determinar la relevancia biológica de un resultado, los datos deben ser evaluados por expertos o mediante más investigaciones. Puede ser útil examinar células adicionales (en su caso) o realizar una repetición del experimento modificando las condiciones experimentales, como, por ejemplo, la separación entre concentraciones u otras condiciones de activación metabólica (es decir, concentración u origen de la fracción S9). En casos raros, incluso después de hacer más investigaciones, el conjunto de datos puede no permitir que se extraiga una conclusión de resultado positivo o negativo, por lo que la respuesta del producto problema se considerará dudosa. El aumento del número de células poliploides puede indicar que los productos problema son capaces de inhibir procesos mitóticos y de producir aberraciones cromosómicas numéricas (52). El aumento del número de células con cromosomas endorreduplicados puede indicar que los productos problema tienen la capacidad de inhibir el progreso del ciclo celular (53) (54) (véase el punto 2). Por consiguiente, deben consignarse por separado la incidencia de células poliploides y la de células con cromosomas endorreduplicados. Informe del ensayo El informe del ensayo debe incluir la información siguiente:
BIBLIOGRAFÍA
Apéndice 1 DEFINICIONES Aneuploidía : Desviación respecto al número diploide (o haploide) normal de cromosomas que afecta a uno o varios cromosomas, pero no a dotaciones completas de cromosomas (poliploidía). Apoptosis : Muerte celular programada, caracterizada por una serie de fases que llevan a la desintegración de las células para formar partículas limitadas por membranas que se eliminan a continuación por fagocitosis o por liberación. Proliferación celular : Aumento del número de células como resultado de divisiones celulares mitóticas. Producto : Sustancia o mezcla. Rotura cromatídica : Discontinuidad de una sola cromátida en la que existe una alteración clara de la alineación de una de las cromátidas. Separación (gap) cromatídica : Región sin teñir (lesión acromática) de una sola cromátida en la que existe una alteración mínima de la alineación de la cromátida. Aberración de tipo cromátida : Lesión cromosómica estructural que consiste en la rotura de cromátidas solas o en la rotura y reunión entre cromátidas. Aberración de tipo cromosoma : Lesión cromosómica estructural que consiste en la rotura, o en la rotura y reunión, de ambas cromátidas en el mismo sitio. Clastógeno : Producto que provoca aberraciones cromosómicas estructurales en poblaciones de células u organismos eucarióticos. Concentraciones : Se refiere a las concentraciones del producto problema en el medio de cultivo. Citotoxicidad : Para los ensayos contemplados en este método de ensayo que emplean líneas celulares, la citotoxicidad se identifica como una reducción de la duplicación relativa de la población (DRP) o del aumento relativo del recuento de células (ARRC) de las células tratadas en comparación con el testigo negativo (véanse el punto 17 y el apéndice 2). Para los ensayos contemplados en este método de ensayo que emplean cultivos primarios de linfocitos, la citotoxicidad se identifica como una reducción del índice mitótico de las células tratadas en comparación con el testigo negativo (véanse el punto 18 y el apéndice 2). Endorreduplicación : Proceso en el que, tras una fase S de replicación del ADN, el núcleo no pasa a la mitosis sino que inicia otra fase S. El resultado son cromosomas con 4, 8, 16, etc. cromátidas. Genotoxicidad : Término general que engloba todos los tipos de lesión del ADN o de los cromosomas, con inclusión de roturas, deleciones, adiciones, modificaciones y uniones de nucleótidos, reorganizaciones, mutaciones génicas, aberraciones cromosómicas y aneuploidía. No todos los tipos de efectos genotóxicos resultan en mutaciones o en lesiones cromosómicas estables. Índice mitótico (IM) : Relación entre el número de células en metafase y el número total de células observadas en una población celular; indica el grado de proliferación de dicha población. Mitosis : División del núcleo celular, generalmente compuesta por profase, prometafase, metafase, anafase y telofase. Mutágeno : Agente que provoca un cambio hereditario en una o varias secuencias de pares de bases del ADN en los genes o en la estructura de los cromosomas (aberraciones cromosómicas). Aberración numérica : Cambio del número de cromosomas respecto al número normal característico de las células empleadas. Poliploidía : Aberraciones cromosómicas numéricas en células u organismos que afectan a una o varias dotaciones completas de cromosomas, frente a las aberraciones que afectan solo a cromosomas sueltos (aneuploidía). Situación de la proteína p53 : La proteína p53 participa en la regulación del ciclo celular, en la apoptosis y en la reparación del ADN. Las células deficientes en proteína p53 funcional, incapaces de detener el ciclo celular o de eliminar las células dañadas por apoptosis u otros mecanismos (por ejemplo, inducción de la reparación del ADN) en relación con funciones de la proteína p53 en respuesta a las lesiones del ADN, deben estar, en teoría, más expuestas a las mutaciones génicas o a las aberraciones cromosómicas. Aumento relativo del recuento celular (ARRC) : Aumento del número de células en los cultivos expuestos a los productos frente al aumento en cultivos no tratados, coeficiente expresado en porcentaje. Duplicación relativa de la población (DRP) : Aumento del número de duplicaciones de la población en los cultivos expuestos a los productos frente al aumento en cultivos no tratados, coeficiente expresado en porcentaje. Fracción hepática S9 : Sobrenadante de homogeneizado de hígado después de centrifugación a 9 000 g, es decir, extracto de hígado crudo. Mezcla S9 : Mezcla de la fracción hepática S9 y de cofactores necesarios para la actividad metabólica de las enzimas. Testigo del disolvente : Término general para definir los cultivos testigo que reciben solo el disolvente utilizado para disolver el producto problema. Aberración estructural : Cambio de la estructura cromosómica detectable mediante examen microscópico de la metafase de la división celular, observado como deleciones y fragmentaciones, intracambios o intercambios. Producto problema : Sustancia o mezcla estudiada con este método de ensayo. Testigos sin tratar : Cultivos que no reciben tratamiento (es decir, ni producto problema ni disolvente), pero que se someten al mismo proceso en paralelo que los cultivos que reciben el producto problema. Apéndice 2 FÓRMULAS PARA LA EVALUACIÓN DE LA CITOTOXICIDAD Índice mitótico (IM):
Aumento relativo del recuento celular (ARRC) o duplicación relativa de la población (DRP) (se recomiendan ambos parámetros, ya que tienen en cuenta la proporción de la población celular que se ha dividido):
donde: N.o de duplicaciones de la población = [log (número de células tras el tratamiento ÷ número de células al inicio)] ÷ log 2 Por ejemplo, un ARRC o una DRP del 53 % indica un 47 % de citotoxicidad o citostasis y un 55 % de citotoxicidad o citostasis medido por el IM significa que el IM real es el 45 % del testigo. En cualquier caso, debe medirse el número de células antes de tratamiento, de la misma forma que con los cultivos testigos tratados y negativos. Si bien el recuento celular relativo (RCR, número de células en los cultivos tratados dividido por el número de células en los cultivos testigo) se había utilizado como parámetro de citotoxicidad en el pasado, ya no se recomienda porque puede subestimar la citotoxicidad. En los cultivos testigos negativos, la duplicación de la población debe ser compatible con el requisito de recolectar las células después del tratamiento al cabo de un período igual a aproximadamente 1,5 veces la duración del ciclo celular normal y el índice mitótico debe ser lo bastante elevado para obtener un número suficiente de células en mitosis y poder calcular de forma fiable una reducción del 50 %. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
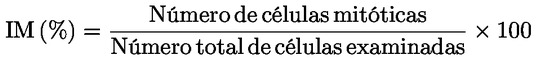

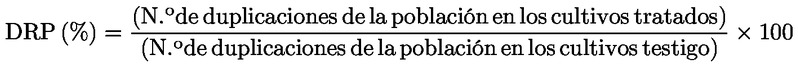
|
4) |
En la parte B, el capítulo B.11 se sustituye por el texto siguiente: «B.11. Ensayo de aberraciones cromosómicas en médula ósea de mamífero INTRODUCCIÓN El presente método de ensayo es equivalente a las directrices de ensayo de la OCDE 475 (2016). Forma parte de una serie de métodos de ensayo sobre toxicología genética. Se ha elaborado un documento de la OCDE que aporta información sucinta sobre los ensayos de toxicología genética y una síntesis de los recientes cambios aportados a dichas directrices de ensayo (1). El ensayo de aberraciones cromosómicas en médula ósea de mamíferos in vivo es especialmente relevante para la genotoxicidad porque, aunque pueden variar según la especie, los factores de los procesos del metabolismo in vivo, de la farmacocinética y de la reparación del ADN están activos y contribuyen a las respuestas. Los ensayos in vivo también resultan útiles para ahondar en el estudio de la genotoxicidad detectada en un sistema in vitro. El ensayo de aberraciones cromosómicas en mamíferos in vivo se realiza para detectar aberraciones cromosómicas estructurales inducidas por los productos problema en células de médula ósea de animales, por lo general roedores (2) (3) (4) (5). Las aberraciones cromosómicas estructurales pueden ser de tipo cromosoma o de tipo cromátida. Si bien la mayoría de las aberraciones provocadas por productos genotóxicos son de tipo cromátida, también pueden producirse aberraciones de tipo cromosoma. Las lesiones cromosómicas y los efectos relacionados ocasionan numerosas enfermedades genéticas humanas y hay pruebas de peso de que, cuando estas lesiones y efectos relacionados provocan alteraciones en los oncogenes y en los genes supresores de tumores, tienen relación con el cáncer en los seres humanos y en sistemas experimentales. Puede surgir poliploidía (incluida la endorreduplicación) en los ensayos de aberraciones cromosómicas in vivo. No obstante, un aumento en la poliploidía no indica por sí solo que haya potencial aneugénico y puede indicar simplemente perturbación del ciclo celular o citotoxicidad. Este ensayo no está diseñado para medir la aneuploidía. Para la detección de la aneuploidía se recomiendan como ensayos in vivo e in vitro, respectivamente, el ensayo de micronúcleos en eritrocitos de mamífero in vivo (capítulo B.12 del presente anexo) o el ensayo de micronúcleos en células de mamífero in vitro (método B.49 del presente anexo). En el apéndice 1 se dan las definiciones de la terminología utilizada. CONSIDERACIONES INICIALES En este ensayo se utilizan habitualmente roedores, pero en algunos casos pueden ser adecuadas otras especies, si está justificado científicamente. El tejido diana es la médula ósea por estar muy vascularizada y por contener una población de células de ciclo corto que pueden aislarse y tratarse con facilidad. La justificación científica para la utilización de especies distintas de las ratas y los ratones debe facilitarse en el informe. Si se utilizan especies que no sean de roedores, se recomienda que la medición de aberraciones cromosómicas en la médula ósea se integre en otro ensayo adecuado de toxicidad. Si hay pruebas de que el producto o productos problema, o alguno o algunos de sus metabolitos, no llegan al tejido diana, puede no proceder la realización del presente ensayo. Antes de la utilización del método de ensayo con una mezcla para obtener datos con fines reglamentarios, debe considerarse si podría proporcionar resultados adecuados a tal fin y, en caso afirmativo, por qué. Tales consideraciones no son necesarias si la reglamentación impone el ensayo de la mezcla. PRINCIPIO DEL MÉTODO DE ENSAYO Se exponen los animales al producto problema por una vía adecuada y se sacrifican de forma compasiva, en un momento adecuado después del tratamiento. Antes de sacrificarlos, se tratan con un agente que detenga la división celular en la metafase (por ejemplo, colchicina o colcemid). A continuación, se realizan preparaciones de cromosomas de células de la médula ósea y se tiñen, tras lo cual se analizan las células en metafase para detectar aberraciones cromosómicas. VERIFICACIÓN DE LA COMPETENCIA DEL LABORATORIO Investigaciones de competencia A fin de establecer que posee la suficiente experiencia con la realización del ensayo antes de utilizarlo para hacer ensayos sistemáticos, el laboratorio debe haber demostrado su capacidad de reproducir los resultados previstos a partir de los datos publicados (por ejemplo, (6)) en cuanto a frecuencias de las aberraciones cromosómicas con un mínimo de dos productos como testigo positivo (incluidas las respuestas débiles inducidas por dosis bajas de los testigos positivos), como las que figuran en el cuadro 1 y con testigos de disolvente/vehículo compatibles (véase el punto 22). Estos experimentos deben utilizar dosis que den aumentos relacionados con la dosis y reproducibles, y demostrar la sensibilidad y el intervalo dinámico del sistema de ensayo en el tejido de interés (médula ósea), empleando el método de examen utilizado en el laboratorio. Este requisito no es aplicable a los laboratorios que tienen experiencia, esto es, que disponen de una base de datos históricos, según se define en los puntos 10-14. Datos sobre testigos históricos En el transcurso de las investigaciones de competencia, el laboratorio debe demostrar:
Cuando se obtengan datos por primera vez en relación con la distribución de testigos negativos históricos, los testigos negativos en paralelo deben ser coherentes con los datos publicados de los testigos, si existen. Según se añadan más datos experimentales sobre la distribución de los testigos históricos, los testigos negativos en paralelo deben situarse idealmente dentro de los límites de control del 95 % de dicha distribución. La base de datos históricos del laboratorio de testigos negativos debe ser estadísticamente sólida para garantizar la capacidad del laboratorio de evaluar la distribución de sus datos sobre los testigos negativos. La bibliografía sugiere que puede ser necesario un mínimo de 10 experimentos, pero que sería preferible contar con al menos 20 experimentos realizados en condiciones experimentales comparables. Los laboratorios deben utilizar métodos de control de calidad, como gráficos de control (por ejemplo, gráficos C o gráficos de medias (7)), con el fin de determinar la variabilidad de sus datos y de demostrar que la metodología está «controlada» en su laboratorio. En la bibliografía pueden encontrarse más recomendaciones sobre cómo conseguir y utilizar los datos históricos (es decir, criterios de inclusión y exclusión de datos en los datos históricos y criterios de aceptabilidad para un determinado experimento) (8). Si el laboratorio no completa un número suficiente de experimentos para establecer una distribución de los testigos negativos estadísticamente sólida (véase el punto 11) durante las investigaciones de competencia (descritas en el punto 9), es admisible que la distribución se establezca durante los primeros exámenes sistemáticos. Este enfoque debe seguir las recomendaciones recogidas en la bibliografía (8) y los resultados de los testigos negativos obtenidos en estos experimentos deben seguir siendo coherentes con los datos publicados de los testigos negativos. Cualquier cambio en el protocolo experimental debe considerarse en función de su impacto en el mantenimiento de la coherencia entre los datos resultantes y la base de datos históricos de testigos existente del laboratorio. Solo si hubiera incoherencias importantes se justificaría el establecimiento de una nueva base de datos históricos de testigos, en caso de que la evaluación por expertos determine que es diferente de la distribución anterior (véase el punto 11). Durante el nuevo establecimiento, puede que no sea necesaria una base de datos completa sobre testigos negativos para poder realizar una prueba real, siempre que el laboratorio pueda demostrar que sus valores de los testigos negativos en paralelo siguen estando en consonancia con su anterior base de datos o con los correspondientes datos publicados. Los datos sobre testigos negativos deben consistir en la incidencia de aberraciones cromosómicas estructurales, excluidas las separaciones (gaps), en cada animal. Lo ideal sería que los testigos negativos en paralelo estuvieran dentro de los límites de control del 95 % de la distribución de la base de datos de testigos negativos históricos del laboratorio. Cuando haya datos de los testigos negativos en paralelo que queden fuera de los límites de control del 95 %, su inclusión en la distribución de testigos históricos puede ser aceptable en la medida en que dichos datos no sean valores discrepantes extremos y haya pruebas de que el sistema de ensayo está «controlado» (véase el punto 11) y no haya pruebas de fallos técnicos o humanos. DESCRIPCIÓN DEL MÉTODO Preparación Selección de la especie animal Deben usarse animales adultos jóvenes y sanos de cepas utilizadas habitualmente en laboratorio. Si bien suelen emplearse ratas, también puede ser adecuado el uso de ratones. Puede utilizarse cualquier otra especie apropiada de mamíferos, si se facilita la justificación científica en el informe. Condiciones de alojamiento y alimentación de los animales Con roedores, la temperatura en el animalario debe ser de 22 °C (± 3 °C). Aunque la humedad relativa debe ser idealmente del 50 al 60 %, debe ser como mínimo del 40 % y preferentemente no superar el 70 %, salvo durante la limpieza del local. La iluminación debe ser artificial, con 12 horas de luz y 12 horas de oscuridad. Para la alimentación se pueden utilizar dietas de laboratorio convencionales, con suministro ilimitado de agua para beber. La elección de la dieta puede verse influida por la necesidad de garantizar una mezcla conveniente del producto problema si se administra por esta vía. Los roedores deben alojarse en grupos pequeños (no más de cinco animales por jaula) del mismo sexo y grupo de tratamiento si no se espera conducta agresiva, preferentemente en jaulas de suelo continuo con un enriquecimiento ambiental adecuado. Los animales podrán alojarse por separado solo si está justificado científicamente. Preparación de los animales Se utilizan normalmente animales adultos jóvenes y sanos (en el caso de los roedores, idealmente de 6-10 semanas de edad al inicio del tratamiento, aunque también pueden aceptarse animales ligeramente más viejos), y se reparten al azar entre los lotes tratados y los testigos. Los animales se identifican individualmente utilizando un método compasivo y mínimamente invasivo (por ejemplo, mediante anillamiento, marcado, implantación de un microchip o identificación biométrica, pero no grapado a las orejas o los dedos), y se deja que se acostumbren a las condiciones del laboratorio durante al menos cinco días. Las jaulas se disponen de forma que se reduzcan al mínimo los posibles efectos debidos al enjaulamiento. Debe evitarse la contaminación cruzada por el testigo positivo y el producto problema. La variación del peso de los animales al empezar el estudio ha de ser mínima y no debe exceder del ± 20 % del peso medio de cada sexo. Preparación de las dosis Los productos problema sólidos deben disolverse o suspenderse en disolventes o vehículos adecuados, o mezclarse con los alimentos o con el agua de bebida, antes de su administración a los animales. Los productos problema líquidos pueden administrarse directamente o diluirse antes de la administración. En lo que respecta a la exposición por inhalación, los productos problema pueden administrarse en forma de gas, de vapor o de aerosol sólido o líquido, dependiendo de sus propiedades fisicoquímicas. Deben utilizarse preparaciones recientes del producto problema, salvo que se cuente con datos de estabilidad que avalen la posibilidad de su conservación y definan las condiciones adecuadas de esta. Disolvente o vehículo El disolvente o vehículo no debe producir efectos tóxicos a las dosis empleadas, y no debe sospecharse que pueda reaccionar químicamente con el producto problema. Si se emplean disolventes o vehículos poco conocidos, debe disponerse de información de referencia que avale su compatibilidad. Siempre que sea posible, se recomienda considerar en primer lugar la utilización de un disolvente o vehículo acuoso. Pueden citarse como ejemplos de disolventes o vehículos compatibles comúnmente utilizados el agua, el suero fisiológico, la solución de metilcelulosa, la solución de sal sódica de carboximetilcelulosa, el aceite de oliva y el aceite de maíz. A falta de información histórica o publicada sobre testigos que demuestre que un disolvente o vehículo elegido atípico no induce ninguna aberración estructural ni otro tipo de efectos nocivos, debe realizarse un estudio inicial a fin de establecer la aceptabilidad del testigo de disolvente/vehículo. Testigos Testigos positivos En principio debe incluirse en cada ensayo un grupo de animales tratados con un producto testigo positivo. Esto puede no aplicarse cuando el laboratorio de ensayo ha demostrado su competencia en la realización del ensayo y ha establecido un intervalo histórico de testigos positivos. Si no se incluye un grupo testigo positivo en paralelo, debe contarse en cada experimento con testigos de examen (portaobjetos fijados y sin teñir). Esto puede conseguirse incluyendo en el examen del estudio muestras de referencias adecuadas que se hayan obtenido y conservado a partir de un experimento con testigo positivo aparte realizado periódicamente (por ejemplo, cada 6 o 18 meses) en el laboratorio donde se realiza el ensayo; por ejemplo, durante las pruebas de competencia y, a continuación, con carácter periódico, en caso necesario. Los productos utilizados como testigo positivo deben provocar con fiabilidad un aumento detectable en la frecuencia de células con aberraciones cromosómicas estructurales respecto al nivel espontáneo. Las dosis del testigo positivo deben elegirse de tal forma que los efectos sean claros, pero no revelen inmediatamente al examinador la identidad de las muestras codificadas. El producto empleado como testigo positivo podrá administrarse por una vía distinta de la del producto problema, con otro programa de tratamiento, y el muestreo podrá efectuarse solo en un único momento. Además, podrá considerarse la utilización como testigo positivo de productos de clases químicas afines, cuando sea posible. En el cuadro 1 se incluyen ejemplos de productos válidos como testigo positivo. Cuadro 1. Ejemplos de productos válidos como testigo positivo
Testigos negativos Deben incluirse animales de un grupo testigo negativo en cada tiempo de muestreo y someterse al mismo proceso que los grupos de tratamiento, salvo que no reciben el tratamiento con el producto problema. Si se utiliza un disolvente o vehículo para la administración del producto problema, el grupo testigo recibirá este disolvente o vehículo. No obstante, si mediante los datos históricos de testigos negativos se demuestra que la variabilidad entre animales y la frecuencia de las células con aberraciones estructurales son coherentes en cada momento de muestreo para el laboratorio de ensayo, puede que solo sea necesario un único muestreo del testigo negativo. En los casos en que se realice un único muestreo de los testigos negativos, debe corresponder al primer momento de muestreo utilizado en el estudio. PROCEDIMIENTO Número y sexo de los animales En general, la respuesta de micronúcleos es similar entre animales machos y hembras (9) y se espera que esto sea así también respecto a las aberraciones cromosómicas estructurales; por lo tanto, la mayor parte de los estudios podría realizarse con cualquier sexo. Unos datos que demuestren la existencia de diferencias pertinentes entre machos y hembras (como diferencias de toxicidad sistémica, metabolismo, biodisponibilidad, toxicidad para la médula ósea, etc., con inclusión por ejemplo de un estudio de determinación del intervalo) favorecerían la utilización de ambos sexos. En este caso puede ser conveniente realizar un estudio con ambos sexos, por ejemplo como parte de un estudio de toxicidad por administración continuada. Podría ser adecuado utilizar el diseño factorial en caso de que se utilizaran animales de ambos sexos. En el apéndice 2 se recogen detalles sobre cómo analizar los datos utilizando dicho diseño. El tamaño de los grupos al principio del estudio debe establecerse de manera que comprendan un mínimo de 5 animales analizables de un mismo sexo, o de cada sexo si se utilizan ambos, por grupo. En caso de que la exposición humana a los productos pueda ser específica de un sexo, como sucede con algunos productos farmacéuticos, el ensayo debe realizarse con animales del sexo correspondiente. Como orientación sobre las necesidades máximas típicas de animales, un estudio de la médula ósea con dos momentos de muestreo con tres grupos tratados y un grupo testigo negativo en paralelo, más un grupo testigo positivo (cada grupo compuesto por cinco animales de un solo sexo), requeriría 45 animales. Dosis Si se lleva a cabo un estudio previo de determinación del intervalo porque no se dispone aún de datos adecuados para ayudar en la selección de las dosis, ha de hacerse en el mismo laboratorio, con la misma especie, cepa, sexo y protocolo de tratamiento que se vayan a utilizar en el estudio principal (10). El estudio debe tratar de determinar la dosis máxima tolerada (DMT), es decir, la dosis más alta que se tolera sin signos de toxicidad que limite el estudio, en relación con la duración del período de estudio (por ejemplo, reducción del peso corporal o citotoxicidad para el sistema hematopoyético) pero sin llegar a la muerte ni presentar signos de dolor, sufrimiento o angustia que requieran el sacrificio compasivo (11). La dosis más alta también puede definirse como la dosis que produce algún signo de toxicidad para la médula ósea. Los productos que presentan saturación de las propiedades toxicocinéticas, o inducen procesos de destoxificación que puedan dar lugar a una disminución de la exposición después de un tratamiento a largo plazo pueden constituir excepciones en cuanto a los criterios de establecimiento de la dosis y han de evaluarse caso por caso. A fin de obtener información sobre la relación dosis-respuesta, los estudios completos deben incluir un grupo testigo negativo y un mínimo de tres dosis separadas por un factor que será en general de 2 y nunca superior a 4. Si el producto problema no produce toxicidad en un estudio de determinación del intervalo o según los datos disponibles, la dosis más alta para una administración única debe ser de 2 000 mg/kg de peso corporal. No obstante, si el producto problema provoca toxicidad, la DMT debe ser la mayor dosis administrada y las dosis deben abarcar preferentemente el intervalo desde la dosis máxima hasta una dosis que provoque toxicidad escasa o nula. Si se observa toxicidad para el tejido diana (médula ósea) a todas las dosis utilizadas en el ensayo, se recomienda hacer otro estudio a dosis no tóxicas. Unos estudios destinados a caracterizar de forma más completa la relación cuantitativa dosis-respuesta pueden necesitar más grupos de dosis. Estos límites pueden variar con determinados tipos de productos problema (por ejemplo, productos farmacéuticos de uso humano) a los que se apliquen requisitos específicos. Ensayo límite Si los experimentos de determinación del intervalo, o los datos disponibles sobre cepas animales afines, indican que un régimen de tratamiento de, como mínimo, la dosis límite (véase más adelante) no produce efectos tóxicos observables (entre los que se incluirían la depresión de la proliferación de la médula ósea u otros signos de citotoxicidad para el tejido diana) y si, basándose en estudios de genotoxicidad in vitro o en datos de productos estructuralmente afines, no cabe esperar genotoxicidad, entonces puede no considerarse necesario realizar un estudio completo con tres dosis, siempre que se haya demostrado que el producto o productos problema alcanzan el tejido diana (médula ósea). En tales casos, podrá ser suficiente una sola dosis, al nivel de la dosis límite. Para un período de administración superior a 14 días, la dosis límite será de 1 000 mg/kg de peso corporal/día. Para un período de administración inferior o igual a 14 días, la dosis límite será de 2 000 mg/kg de peso corporal/día. Administración de las dosis A la hora de diseñar el ensayo, debe tenerse en cuenta la vía prevista de exposición humana. Por lo tanto, cuando esté justificado, podrán elegirse vías de exposición como los alimentos, el agua de bebida, la tópica, la subcutánea, la intravenosa, la oral forzada (por sonda), la inhalación, la intratraqueal o la implantación. En cualquier caso, la vía debe elegirse de forma que se garantice una exposición adecuada del tejido o tejidos diana. En general no se recomienda la inyección intraperitoneal, ya que no es una vía de exposición humana prevista, y debe utilizarse solo en casos que tengan justificación científica específica. Si el producto problema se mezcla con los alimentos o el agua de bebida, en particular en caso de administración única, debe procurarse que el plazo entre el consumo de los alimentos o el agua y el muestreo sea suficiente para permitir la detección de los efectos (véanse los puntos 33-34). El volumen máximo de líquido que puede administrarse de una sola vez por sonda o por inyección depende del tamaño del animal utilizado. Dicho volumen no debe en principio superar 1 ml/100 g de peso corporal, salvo en el caso de las soluciones acuosas, en que puede llegarse a un máximo de 2 ml/100 g. El uso de volúmenes superiores deberá justificarse. Excepto en el caso de productos problema irritantes o corrosivos, que por lo general producen efectos exacerbados a concentraciones superiores, la variabilidad del volumen de ensayo debe reducirse al mínimo ajustando la concentración para conseguir la administración de un volumen constante en relación con el peso corporal a todas las dosis. Pauta de tratamiento Los productos problema se administran normalmente de una vez, pero también puede dividirse la dosis (es decir, realizar dos o más tratamientos el mismo día separados por no más de 2 o 3 horas) con el fin de facilitar la administración de grandes volúmenes. En estas circunstancias, o cuando se administra el producto problema por inhalación, el tiempo de muestreo debe programarse basándose en el momento de la última administración o el final de la exposición. Se dispone de pocos datos sobre la idoneidad de un protocolo de administración continuada para este ensayo. Sin embargo, en circunstancias en las que sea deseable integrar este ensayo con un ensayo de toxicidad por administración continuada, deberá tenerse cuidado para evitar la pérdida de células mitóticas con lesiones cromosómicas, lo que puede suceder a dosis tóxicas. Esta integración es aceptable siempre y cuando la dosis más alta sea superior o igual a la dosis límite (véase el punto 29) y se le administre a un grupo la dosis límite mientras dure el período de tratamiento. El ensayo de micronúcleos (método B.12) debe considerarse el ensayo in vivo de elección para detectar las aberraciones cromosómicas cuando se desee la integración con otros estudios. Deben tomarse muestras de médula ósea en dos momentos distintos tras el tratamiento único. En el caso de los roedores, el primer intervalo de muestreo debe ser el tiempo equivalente a 1,5 veces la duración del ciclo celular normal (suele ser de 12 a 18 horas después del período de tratamiento). Dado que el tiempo necesario para que el producto o productos problema se absorban y se metabolicen, así como su efecto sobre la cinética del ciclo celular, pueden modificar el momento óptimo para detectar aberraciones cromosómicas, se recomienda tomar otra muestra 24 horas después de la primera. En el primer momento de muestreo, se tratan todos los grupos de dosis y se toman muestras de todos ellos para su análisis; no obstante, en los momentos de muestreo posteriores, solo es necesario administrar la dosis más alta. Si se siguen pautas de tratamiento de más de un día con justificación científica, debe hacerse un solo muestreo en general a un máximo de aproximadamente 1,5 veces la duración del ciclo celular normal desde el último tratamiento. Tras el tratamiento y antes de la recogida de la muestra, se les inyecta a los animales por vía intraperitoneal una dosis adecuada de un agente que detenga la división celular en la metafase (por ejemplo, colcemid o colchicina), y las muestras se toman a un intervalo adecuado a partir de ese momento. En el caso de los ratones, este intervalo hasta el muestreo es aproximadamente de 3-5 horas y en el de las ratas es de 2-5 horas. Se toman las células de la médula ósea, se hinchan, fijan y tiñen, y se analizan en busca de aberraciones cromosómicas (12). Observaciones Deben hacerse observaciones clínicas generales de los animales de ensayo y registrarse los signos clínicos al menos una vez al día, preferentemente a la misma hora u horas cada día y teniendo en cuenta el período de mayor intensidad de los efectos previstos tras la administración. Al menos dos veces al día durante el periodo de administración, se debe observar la posible morbilidad y mortalidad de todos los animales. Deben pesarse todos los animales al principio del estudio, al menos una vez por semana durante los estudios de administración continuada, y en el momento del sacrificio. En los estudios de al menos una semana de duración, el consumo de alimentos debe medirse al menos semanalmente. Si el producto problema se administra con el agua de bebida, debe medirse el consumo de agua cada vez que se cambie el agua y al menos una vez por semana. Los animales que presenten signos de toxicidad excesiva pero no letal deben sacrificarse de forma compasiva antes de que termine el período del ensayo (11). Exposición del tejido diana Debe tomarse una muestra de sangre en el momento o momentos adecuados para permitir la investigación de los niveles plasmáticos de los productos problema, a fin de demostrar que ha habido exposición de la médula ósea, cuando esté justificado y cuando no se disponga de otros datos de exposición (véase el punto 44). Preparación de la médula ósea y de los cromosomas Inmediatamente después del sacrificio compasivo, se extraen células de la médula ósea del fémur o la tibia de los animales, se exponen a una solución hipotónica y se fijan. A continuación, las células en metafase se extienden en portaobjetos y se tiñen según métodos establecidos ((3) (12)). Análisis Todos los portaobjetos, incluidos los de los testigos positivos y negativos, deben codificarse independientemente antes del análisis y deben aleatorizarse de forma que el examinador no tenga conocimiento de cómo se han tratado. Se evalúa la citotoxicidad determinando el índice mitótico en un mínimo de 1 000 células por animal, en todos los animales tratados (incluidos los testigos positivos) y en los animales usados como testigos negativos sin tratar o solo con disolvente/vehículo. Deben analizarse al menos 200 metafases en cada animal para detectar aberraciones cromosómicas estructurales, contando tanto con las separaciones (gaps) como sin ellas. No obstante, en caso de que la base de datos históricos de testigos negativos indique que la frecuencia media de aberraciones cromosómicas estructurales de fondo es < 1 % en el laboratorio de ensayo, debe considerarse el examen de células adicionales. Las aberraciones de tipo cromosoma y las de tipo cromátida deben registrarse por separado y clasificarse en subtipos (roturas, intercambios). Los procedimientos seguidos en el laboratorio deben garantizar que el análisis de las aberraciones cromosómicas es realizado por examinadores bien formados y es objeto de una revisión por pares, si procede. Reconociendo que los métodos de preparación de los portaobjetos provocan con frecuencia la rotura de células en metafase y la consiguiente pérdida de cromosomas, las células examinadas deben, por tanto, contener un número de centrómeros no inferior a 2n ± 2, donde n es el número de cromosomas haploides de la especie. DATOS E INFORME Tratamiento de los resultados Los datos relativos a cada animal se presentarán en forma de cuadro. Deben evaluarse respecto a cada animal el índice mitótico, el número de células en metafase examinadas, el número de aberraciones por célula en metafase y el porcentaje de células con aberraciones cromosómicas estructurales. Se debe hacer una relación de los distintos tipos de dichas aberraciones, con su número y frecuencia, respecto a los grupos tratados y los testigos. Las separaciones, así como las células poliploides y las células con cromosomas endorreduplicados, se registran por separado. La frecuencia de las separaciones se recoge en el informe, pero por lo general no se incluye en el análisis de la frecuencia total de aberraciones estructurales. Si no se observan diferencias en la respuesta de un sexo y otro, los datos pueden combinarse para el análisis estadístico. Deben también consignarse los datos sobre toxicidad y signos clínicos de los animales. Criterios de aceptabilidad Los siguientes criterios determinan la aceptabilidad del ensayo:
Evaluación e interpretación de los resultados Siempre que se cumplan todos los criterios de aceptabilidad, se considera que el producto problema es claramente positivo si:
Si se examina únicamente la dosis más alta en un momento particular de muestreo, el producto problema se considera claramente positivo si se ve un aumento estadísticamente significativo en comparación con el testigo negativo en paralelo y los resultados están fuera de la distribución de los datos históricos de los testigos negativos (por ejemplo, límites de control del 95 % según la distribución de Poisson). En la bibliografía se encuentran recomendaciones sobre los métodos estadísticos más adecuados (13). Al realizar un análisis de la relación dosis-respuesta, deben someterse al mismo por lo menos tres grupos tratados con el producto problema. Las pruebas estadísticas deben utilizar como unidad experimental el animal. Un resultado positivo en el ensayo de aberraciones cromosómicas indica que el producto problema provoca aberraciones cromosómicas estructurales en la médula ósea de la especie estudiada. Siempre que se cumplan todos los criterios de aceptabilidad, se considera que el producto problema es claramente negativo si, en todas las condiciones experimentales examinadas:
En la bibliografía se encuentran recomendaciones sobre los métodos estadísticos más adecuados (13). Como pruebas de exposición de la médula ósea a un producto problema pueden citarse la reducción del índice mitótico o la medición de los niveles hemáticos o plasmáticos del producto o productos problema. En caso de administración por vía intravenosa, no hacen falta pruebas de la exposición. Alternativamente, para demostrar la exposición de la médula ósea es posible utilizar los datos de ADME obtenidos en un estudio independiente utilizando la misma vía y la misma especie. Un resultado negativo indica que, en las condiciones del ensayo, el producto problema no provoca aberraciones cromosómicas estructurales en la médula ósea de la especie estudiada. No se requiere ninguna verificación de una respuesta positiva clara o negativa clara. En los casos en que la respuesta no sea claramente negativa ni positiva y con el fin de ayudar a determinar la importancia biológica de un resultado (por ejemplo, un incremento límite o débil), los datos deben ser evaluados por expertos o hay que completar otras investigaciones de los experimentos existentes. En algunos casos, puede ser de utilidad el análisis de más células o la repetición de un experimento en condiciones experimentales modificadas. En casos excepcionales, incluso después de otras investigaciones, los datos no permiten concluir si el producto problema produce resultados positivos o negativos, y el estudio deberá considerarse, por tanto, como de resultado dudoso. Deben consignarse por separado las frecuencias de las metafases poliploides y endorreduplicadas entre las metafases totales. El aumento del número de células poliploides o endorreduplicadas puede indicar que el producto problema es capaz de inhibir procesos mitóticos o la progresión del ciclo celular (véase el punto 3). Informe del ensayo El informe del ensayo debe incluir la información siguiente: Resumen
BIBLIOGRAFÍA
Apéndice 1 DEFINICIONES Aneuploidía : Desviación respecto al número diploide (o haploide) normal de cromosomas en uno o varios cromosomas, pero sin multiplicar dotaciones completas de cromosomas (véase “poliploidía”). Centrómero : Región o regiones del cromosoma con las que se asocian las fibras del huso acromático durante la división celular para permitir el movimiento ordenado de los cromosomas hijos hacia los polos de las células hijas. Producto : Sustancia o mezcla. Aberración de tipo cromátida : Lesión cromosómica estructural que consiste en la rotura de cromátidas solas o en la rotura y reunión entre cromátidas. Aberración de tipo cromosoma : Lesión cromosómica estructural que consiste en la rotura, o en la rotura y reunión, de ambas cromátidas en el mismo sitio. Endorreduplicación : Proceso en el que, tras una fase S de replicación del ADN, el núcleo no pasa a la mitosis sino que inicia otra fase S. El resultado son cromosomas con 4, 8, 16, etc. cromátidas. Separación (gap) : Lesión acromática más estrecha que la anchura de una cromátida, y con alteración mínima de la alineación de las cromátidas. Índice mitótico : Relación entre el número de células en mitosis y el número total de células de una población, que es una medida de la proliferación de dicha población celular. Aberración numérica : Cambio del número de cromosomas respecto al número normal característico de los animales empleados (aneuploidía). Poliploidía : Aberración cromosómica numérica que implica un cambio en el número de toda la dotación cromosómica, en contraste con un cambio numérico en una parte de la dotación cromosómica (aneuploidía). Aberración cromosómica estructural : Cambio de la estructura cromosómica detectable mediante examen microscópico de la metafase de la división celular, observado como deleciones y fragmentaciones, intracambios o intercambios. Producto problema : Sustancia o mezcla estudiada con este método de ensayo. Apéndice 2 DISEÑO FACTORIAL PARA IDENTIFICAR LAS DIFERENCIAS DE SEXO EN EL ENSAYO DE ABERRACIONES CROMOSÓMICAS IN VIVO Diseño factorial y su análisis En este diseño, un mínimo de 5 machos y 5 hembras se someten a ensayo a cada concentración, lo que resulta en la utilización de un mínimo de 40 animales (20 machos y 20 hembras, más los testigos positivos pertinentes). Este diseño, que es uno de los diseños factoriales más simples, es equivalente a un análisis de varianza de dos factores, con el sexo y el nivel de concentración como efectos principales. Los datos pueden analizarse con numerosos paquetes de software estadístico estándar tales como SPSS, SAS, STATA, Genstat, así como utilizando R. El análisis divide la variabilidad de la serie de datos en la variabilidad entre sexos, la variabilidad entre las concentraciones y la relativa a la interacción entre los sexos y las concentraciones. Cada uno de los términos se somete a prueba frente a una estimación de la variabilidad entre los animales replicados dentro de los grupos de animales del mismo sexo que han recibido la misma concentración. Todos los detalles de la metodología subyacente se encuentran en muchos manuales estadísticos conocidos (véanse las referencias) y en las funciones de «ayuda» de los paquetes estadísticos. El análisis se lleva a cabo inspeccionando el término de la interacción sexo x concentración en el cuadro de ANOVA (5). A falta de un término de interacción significativo, los valores combinados de distintos sexos o niveles de concentración ofrecen pruebas estadísticas válidas entre los niveles, sobre la base del término de ANOVA de variabilidad intragrupo puesta en común. El análisis continúa dividiendo la estimación de la variabilidad entre concentraciones en contrastes que permiten una prueba de contrastes lineales y cuadráticos de las respuestas a través de los niveles de concentración. Cuando existe un término de interacción significativa sexo x concentración, este término también puede dividirse en contrastes de interacción lineal x sexo y cuadrático x sexo. Estos términos permiten pruebas de si las respuestas a la concentración son paralelas en los dos sexos, o si existe una diferencia en la respuesta entre estos. La estimación de la variabilidad intragrupo puesta en común se puede utilizar para proporcionar pruebas pareadas de la diferencia entre medias. Estas comparaciones podrían realizarse entre las medias para los dos sexos y entre las medias para los diferentes niveles de concentración, así como para las comparaciones con los niveles de los testigos negativos. En los casos en los que existe una interacción significativa pueden hacerse comparaciones entre las medias de concentraciones diferentes dentro de un sexo o entre las medias de los sexos a la misma concentración. Bibliografía Hay muchos manuales estadísticos que debaten la teoría, el diseño, la metodología, el análisis y la interpretación de los diseños factoriales que van desde los análisis bifactoriales más sencillos hasta las formas más complejas utilizadas en la metodología de diseño de experimentos. La lista que sigue no es exhaustiva. Algunos libros proporcionan ejemplos resueltos de diseños comparables, en algunos casos con el código para aplicar los análisis utilizando diversos paquetes de software.
|
|
5) |
En la parte B, el capítulo B.12 se sustituye por el texto siguiente: «B.12. Ensayo de micronúcleos en eritrocitos de mamífero INTRODUCCIÓN El presente método de ensayo es equivalente a las directrices de ensayo de la OCDE 474 (2016). Forma parte de una serie de métodos de ensayo sobre toxicología genética. Se ha elaborado un documento de la OCDE que aporta información sucinta sobre los ensayos de toxicología genética y una síntesis de los recientes cambios aportados a dichas directrices de ensayo (1). El ensayo de micronúcleos de mamífero in vivo es especialmente relevante para evaluar la genotoxicidad porque, aunque pueden variar según la especie, los factores de los procesos del metabolismo in vivo, de la farmacocinética y de la reparación del ADN están activos y contribuyen a las respuestas. Los ensayos in vivo también resultan útiles para ahondar en el estudio de la genotoxicidad detectada en un sistema in vitro. El ensayo de micronúcleos de mamífero in vivo se emplea para detectar lesiones provocadas por el producto problema en los cromosomas o el aparato mitótico de los eritroblastos. El ensayo sirve para evaluar la formación de micronúcleos en los eritrocitos tomados bien de la médula ósea o bien de la sangre periférica de los animales (por lo general, roedores). El ensayo de micronúcleos tiene por objeto determinar los productos que ocasionan lesiones citogenéticas que dan lugar a la formación de micronúcleos con fragmentos de cromosomas o cromosomas enteros retardados. Cuando los eritroblastos de la médula ósea se transforman en eritrocitos inmaduros (a veces denominados también reticulocitos o eritrocitos policromáticos), el núcleo principal es eliminado, pero los micronúcleos que se hayan formado pueden permanecer en el citoplasma. Resulta más fácil visualizar o detectar los micronúcleos en estas células, pues carecen de núcleo principal. El aumento de la frecuencia de micronúcleos en los eritrocitos inmaduros de animales tratados es indicativo de la inducción de aberraciones cromosómicas numéricas o estructurales. Los eritrocitos micronucleados recién formados se identifican y se cuantifican bien mediante tinción seguida de examen visual con microscópico, o bien mediante análisis automatizado. El recuento de suficientes eritrocitos inmaduros en la médula ósea o la sangre periférica de animales adultos se facilita en gran medida mediante la utilización de un programa automatizado de examen. Estos programas son alternativas aceptables a la evaluación manual (2). En estudios comparativos se ha puesto de manifiesto que tales métodos, utilizando los pertinentes patrones de calibración, pueden proporcionar una mayor sensibilidad y reproducibilidad intralaboratorios e interlaboratorios que el examen microscópico manual (3) (4). Entre los sistemas automatizados que permiten medir la frecuencia de eritrocitos micronucleados se incluyen, por ejemplo, los citómetros de flujo (5), los programas de análisis de imágenes (6) (7), y los citómetros de barrido por láser (8). Aunque no forme parte normalmente del ensayo, es posible distinguir los fragmentos de cromosomas de los cromosomas enteros aplicando diversos criterios. Entre estos se encuentra la presencia o la ausencia de cinetocoro o de ADN centromérico, que son tanto el uno como el otro característicos de los cromosomas intactos. La ausencia de cinetocoro o de ADN centromérico indica que el micronúcleo solo contiene fragmentos de cromosomas, mientras que su presencia es indicativa de la pérdida de cromosomas. En el apéndice 1 se dan las definiciones de la terminología utilizada. CONSIDERACIONES INICIALES La médula ósea de roedores adultos jóvenes es el tejido diana de las lesiones genéticas en este ensayo, ya que los eritrocitos se producen en este tejido. La medición de los micronúcleos en eritrocitos inmaduros en la sangre periférica es aceptable en otras especies de mamíferos respecto a las cuales se haya demostrado que poseen la sensibilidad adecuada para detectar productos que provocan aberraciones cromosómicas numéricas o estructurales (por inducción de la formación de micronúcleos en eritrocitos inmaduros) y se aporte una justificación científica. La frecuencia de la presencia de eritrocitos inmaduros micronucleados es el principal parámetro. La frecuencia de la presencia de eritrocitos maduros que contienen micronúcleos en la sangre periférica también puede utilizarse como parámetro en especies sin una fuerte selección esplénica contra las células micronucleadas y si los animales han sido tratados de forma continua durante un período superior a la vida de los eritrocitos en la especie utilizada (por ejemplo, 4 semanas o más en el caso del ratón). Si hay pruebas de que el producto o productos problema, o sus metabolitos, no llegan al tejido diana, puede no proceder la realización del presente ensayo. Antes de la utilización del método de ensayo con una mezcla para obtener datos con fines reglamentarios, debe considerarse si podría proporcionar resultados adecuados a tal fin y, en caso afirmativo, por qué. Tales consideraciones no son necesarias si la reglamentación impone el ensayo de la mezcla. PRINCIPIO DEL MÉTODO DE ENSAYO Se exponen los animales al producto problema por una vía adecuada. Si se utiliza la médula ósea, los animales se sacrifican de forma compasiva, en uno o varios momentos adecuados tras el tratamiento, se extrae la médula ósea, se hacen los frotis y se tiñen (9) (10) (11) (12) (13) (14) (15). Si se emplea sangre periférica, esta se extrae en uno o varios momentos adecuados tras el tratamiento, se hacen los frotis y se tiñen (12) (16) (17) (18). Cuando el tratamiento se administra de forma aguda, es importante seleccionar los momentos en que se toman las muestras de médula ósea o de sangre para que en ellos se pueda detectar la inducción de la formación de eritrocitos inmaduros micronucleados relacionada con el tratamiento. En el caso de la toma de sangre periférica, también deberá haber transcurrido tiempo suficiente para que estos eritrocitos aparezcan en la sangre circulante. Se analizan los frotis para detectar la presencia de micronúcleos, por visualización utilizando un microscopio, análisis de imágenes, citometría de flujo, o citometría de barrido por láser. VERIFICACIÓN DE LA COMPETENCIA DEL LABORATORIO Investigaciones de competencia A fin de establecer que posee la suficiente experiencia con la realización del ensayo antes de utilizarlo para hacer ensayos sistemáticos, el laboratorio debe haber demostrado su capacidad de reproducir los resultados previstos a partir de los datos publicados (17) (19) (20) (21) (22) en cuanto a frecuencias de los micronúcleos con un mínimo de dos productos como testigo positivo (incluidas las respuestas débiles inducidas por dosis bajas de los testigos positivos), como las que figuran en el cuadro 1, y con testigos de disolvente/vehículo compatibles (véase el punto 26). Estos experimentos deben utilizar dosis que den aumentos relacionados con la dosis y reproducibles, y demostrar la sensibilidad y el intervalo dinámico del sistema de ensayo en el tejido de interés (médula ósea o sangre periférica), empleando el método de examen utilizado en el laboratorio. Este requisito no es aplicable a los laboratorios que tienen experiencia, esto es, que disponen de una base de datos históricos, según se define en los puntos 14-18. Datos sobre testigos históricos En el transcurso de las investigaciones de competencia, el laboratorio debe demostrar:
Cuando se obtengan datos por primera vez en relación con la distribución de testigos negativos históricos, los testigos negativos en paralelo deben ser coherentes con los datos publicados de los testigos, si existen. Según se añadan más datos experimentales sobre la distribución de los testigos históricos, los testigos negativos en paralelo deben situarse idealmente dentro de los límites de control del 95 % de dicha distribución. La base de datos históricos del laboratorio de testigos negativos debe ser estadísticamente sólida para garantizar la capacidad del laboratorio de evaluar la distribución de sus datos sobre los testigos negativos. La bibliografía sugiere que puede ser necesario un mínimo de 10 experimentos, pero que sería preferible contar con al menos 20 experimentos realizados en condiciones experimentales comparables. Los laboratorios deben utilizar métodos de control de calidad, como gráficos de control (por ejemplo, gráficos C o gráficos de medias (23)), con el fin de determinar la variabilidad de sus datos y de demostrar que la metodología está «controlada» en su laboratorio. En la bibliografía pueden encontrarse más recomendaciones sobre cómo conseguir y utilizar los datos históricos (es decir, criterios de inclusión y exclusión de datos en los datos históricos y criterios de aceptabilidad para un determinado experimento) (24). Si el laboratorio no completa un número suficiente de experimentos para establecer una distribución de los testigos negativos estadísticamente sólida (véase el punto 15) durante las investigaciones de competencia (descritas en el punto 13), es admisible que la distribución se establezca durante los primeros exámenes sistemáticos. Este enfoque debe seguir las recomendaciones recogidas en la bibliografía (24) y los resultados de los testigos negativos obtenidos en estos experimentos deben seguir siendo coherentes con los datos publicados de los testigos negativos. Cualquier cambio en el protocolo experimental debe considerarse en función de su impacto en el mantenimiento de la coherencia entre los datos resultantes y la base de datos históricos de testigos existente del laboratorio. Solo si hubiera incoherencias importantes se justificaría el establecimiento de una nueva base de datos históricos de testigos, en caso de que la evaluación por expertos determine que es diferente de la distribución anterior (véase el punto 15). Durante el nuevo establecimiento, puede que no sea necesaria una base de datos completa sobre testigos negativos para poder realizar una prueba real, siempre que el laboratorio pueda demostrar que sus valores de los testigos negativos en paralelo siguen estando en consonancia con su anterior base de datos o con los correspondientes datos publicados. Los datos sobre testigos negativos deben consistir en la incidencia de eritrocitos inmaduros con micronúcleos en cada animal. Lo ideal sería que los testigos negativos en paralelo estuvieran dentro de los límites de control del 95 % de la distribución de la base de datos de testigos negativos históricos del laboratorio. Cuando haya datos de los testigos negativos en paralelo que queden fuera de los límites de control del 95 %, su inclusión en la distribución de testigos históricos puede ser aceptable en la medida en que dichos datos no sean valores discrepantes extremos y haya pruebas de que el sistema de ensayo está «controlado» (véase el punto 15) y no haya pruebas de fallos técnicos o humanos. DESCRIPCIÓN DEL MÉTODO Preparación Selección de la especie animal Deben usarse animales adultos jóvenes y sanos de cepas utilizadas habitualmente en laboratorio. Pueden utilizarse ratones, ratas, u otras especies adecuadas de mamíferos. Si se emplea sangre periférica, se debe comprobar que la eliminación esplénica de células con micronúcleos de la circulación no compromete la detección de la formación de micronúcleos inducida en la especie seleccionada. Esto se ha comprobado claramente en el caso de la sangre periférica de ratón y rata (2). La justificación científica para la utilización de especies distintas de las ratas y los ratones debe facilitarse en el informe. Si se utilizan especies que no sean de roedores, se recomienda que la medición de los micronúcleos inducidos se integre en otro ensayo adecuado de toxicidad. Condiciones de alojamiento y alimentación de los animales Con roedores, la temperatura en el animalario debe ser de 22 °C (± 3 °C). Aunque la humedad relativa debe ser idealmente del 50 al 60 %, debe ser como mínimo del 40 % y preferentemente no superar el 70 %, salvo durante la limpieza del local. La iluminación debe ser artificial, con 12 horas de luz y 12 horas de oscuridad. Para la alimentación se pueden utilizar dietas de laboratorio convencionales, con suministro ilimitado de agua para beber. La elección de la dieta puede verse influida por la necesidad de garantizar una mezcla conveniente del producto problema si se administra por esta vía. Los roedores deben alojarse en grupos pequeños (no más de cinco animales por jaula) del mismo sexo y grupo de tratamiento si no se espera conducta agresiva, preferentemente en jaulas de suelo continuo con un enriquecimiento ambiental adecuado. Los animales podrán alojarse por separado solo si está justificado científicamente. Preparación de los animales Se utilizan normalmente animales adultos jóvenes y sanos (en el caso de los roedores, idealmente de 6-10 semanas de edad al inicio del tratamiento, aunque también pueden aceptarse animales ligeramente más viejos), y se reparten al azar entre los lotes tratados y los testigos. Los animales se identifican individualmente utilizando un método compasivo y mínimamente invasivo (por ejemplo, mediante anillamiento, marcado, implantación de un microchip o identificación biométrica, pero no grapado a las orejas o los dedos), y se deja que se acostumbren a las condiciones del laboratorio durante al menos cinco días. Las jaulas se disponen de forma que se reduzcan al mínimo los posibles efectos debidos al enjaulamiento. Debe evitarse la contaminación cruzada por el testigo positivo y el producto problema. La variación del peso de los animales al empezar el estudio ha de ser mínima y no debe exceder del ± 20 % del peso medio de cada sexo. Preparación de las dosis Los productos problema sólidos deben disolverse o suspenderse en disolventes o vehículos adecuados, o mezclarse con los alimentos o con el agua de bebida, antes de su administración a los animales. Los productos problema líquidos pueden administrarse directamente o diluirse antes de la administración. En lo que respecta a la exposición por inhalación, los productos problema pueden administrarse en forma de gas, de vapor o de aerosol sólido o líquido, dependiendo de sus propiedades fisicoquímicas. Deben utilizarse preparaciones recientes del producto problema, salvo que se cuente con datos de estabilidad que avalen la posibilidad de su conservación y definan las condiciones adecuadas de esta. Condiciones del ensayo Disolvente o vehículo El disolvente o vehículo no debe producir efectos tóxicos a las dosis empleadas, y no debe tener la posibilidad de reaccionar químicamente con los productos problema. Si se emplean disolventes o vehículos poco conocidos, debe disponerse de información de referencia que avale su compatibilidad. Siempre que sea posible, se recomienda considerar en primer lugar la utilización de un disolvente o vehículo acuoso. Pueden citarse como ejemplos de disolventes o vehículos compatibles comúnmente utilizados el agua, el suero fisiológico, la solución de metilcelulosa, la solución de sal sódica de carboximetilcelulosa, el aceite de oliva y el aceite de maíz. A falta de información histórica o publicada sobre testigos que demuestre que un disolvente o vehículo elegido atípico no induce la formación de micronúcleos ni ningún otro tipo de efectos nocivos, debe realizarse un estudio inicial a fin de establecer la aceptabilidad del testigo de disolvente o vehículo. Testigos Testigos positivos En principio debe incluirse en cada ensayo un grupo de animales tratados con un producto testigo positivo. Esto puede no aplicarse cuando el laboratorio de ensayo ha demostrado su competencia en la realización del ensayo y ha establecido un intervalo histórico de testigos positivos. Si no se incluye un grupo testigo positivo en paralelo, debe contarse en cada experimento con testigos de examen (portaobjetos fijados y sin teñir o muestras de suspensiones celulares, según el método de examen). Esto puede conseguirse incluyendo en el examen del estudio muestras de referencias adecuadas que se hayan obtenido y conservado a partir de un experimento con testigo positivo aparte realizado periódicamente (por ejemplo, cada 6 o 18 meses); por ejemplo, durante las pruebas de competencia y, a continuación, con carácter periódico, en caso necesario. Los productos utilizados como testigo positivo deben provocar con fiabilidad un aumento detectable en la frecuencia de los micronúcleos respecto al nivel espontáneo. Cuando se emplee un examen manual por microscopia, las dosis del testigo positivo deben elegirse de tal forma que los efectos sean claros, pero no revelen inmediatamente al examinador la identidad de las muestras codificadas. El producto empleado como testigo positivo podrá administrarse por una vía distinta de la del producto problema, con otro programa de tratamiento, y el muestreo podrá efectuarse solo en un único momento. Además, podrá considerarse la utilización como testigo positivo de productos de clases químicas afines, cuando sea posible. En el cuadro 1 se incluyen ejemplos de productos válidos como testigo positivo. Cuadro 1. Ejemplos de productos válidos como testigo positivo Productos y CASRN Metanosulfonato de etilo [CASRN 62-50-0] Metanosulfonato de metilo [CASRN 66-27-3] Etil-nitrosourea [CASRN 759-73-9] Mitomicina C [CASRN 50-07-7] Ciclofosfamida (monohidrato) [CASRN 50-18-0 (CASRN 6055-19-2)] Trietilenomelamina [CASRN 51-18-3] Colchicina [CASRN 64-86-8] o vimblastina [CASRN 865-21-4] — como anéugenos Testigos negativos Deben incluirse animales de un grupo testigo negativo en cada tiempo de muestreo y someterse al mismo proceso que los grupos de tratamiento, salvo que no reciben el tratamiento con el producto problema. Si se utiliza un disolvente o vehículo para la administración del producto problema, el grupo testigo recibirá este disolvente o vehículo. No obstante, si mediante los datos históricos de testigos negativos se demuestra que la variabilidad entre animales y la frecuencia de las células con micronúcleos son coherentes en cada momento de muestreo en el laboratorio de ensayo, puede que solo sea necesario un único muestreo del testigo negativo. En los casos en que se realice un único muestreo de los testigos negativos, debe corresponder al primer momento de muestreo utilizado en el estudio. Si se emplea sangre periférica, puede aceptarse una muestra previa al tratamiento en lugar de un testigo negativo en paralelo en los estudios a corto plazo si los resultados son coherentes con la base de datos históricos de testigos del laboratorio de ensayo. Se ha demostrado que en las ratas la toma de pequeñas muestras previas al tratamiento (por ejemplo, de menos de 100 μl/día) tiene un impacto mínimo sobre la frecuencia espontánea de micronúcleos (25). PROCEDIMIENTO Número y sexo de los animales En general, la respuesta de micronúcleos es similar entre machos y hembras y, por lo tanto, la mayor parte de los estudios podría realizarse con cualquier sexo (26). Unos datos que demostraran la existencia de diferencias pertinentes entre machos y hembras (como diferencias de toxicidad sistémica, metabolismo, biodisponibilidad, toxicidad para la médula ósea, etc., por ejemplo en un estudio de determinación del intervalo) favorecerían la utilización de ambos sexos. En este caso puede ser conveniente realizar un estudio con ambos sexos, por ejemplo como parte de un estudio de toxicidad por administración continuada. Podría ser adecuado utilizar el diseño factorial en caso de que se utilizaran animales de ambos sexos. En el apéndice 2 se recogen detalles sobre cómo analizar los datos utilizando dicho diseño. El tamaño de los grupos al principio del estudio debe establecerse de manera que comprendan un mínimo de 5 animales analizables de un mismo sexo, o de cada sexo si se utilizan ambos, por grupo. En caso de que la exposición humana a los productos pueda ser específica de un sexo, como sucede con algunos productos farmacéuticos, el ensayo debe realizarse con animales del sexo correspondiente. Como orientación sobre las necesidades máximas típicas de animales, un estudio de la médula ósea realizado de acuerdo con los parámetros establecidos en el punto 37 con tres grupos tratados y testigos negativo y positivo en paralelo (cada grupo compuesto por cinco animales de un solo sexo), requeriría entre 25 y 35 animales. Dosis Si se lleva a cabo un estudio previo de determinación del intervalo porque no se dispone aún de datos adecuados para ayudar en la selección de las dosis, ha de hacerse en el mismo laboratorio, con la misma especie, cepa, sexo y protocolo de tratamiento que se vayan a utilizar en el estudio principal (27). El estudio debe tratar de determinar la dosis máxima tolerada (DMT), es decir, la dosis más alta que se tolera sin signos de toxicidad que limite el estudio, en relación con la duración del período de estudio (por ejemplo, reducción del peso corporal o citotoxicidad para el sistema hematopoyético pero sin llegar a la muerte ni presentar signos de dolor, sufrimiento o angustia que requieran el sacrificio compasivo (28)). La dosis más alta también puede definirse como la dosis que produce toxicidad en la médula ósea (por ejemplo, una reducción de la proporción de eritrocitos inmaduros respecto al total de eritrocitos en la médula ósea o en la sangre periférica superior al 50 %, pero hasta no menos del 20 % del valor del testigo). No obstante, al analizar las células positivas para el CD71 en la circulación sanguínea periférica (es decir, mediante citometría de flujo), esta fracción muy joven de eritrocitos inmaduros responde a los estímulos tóxicos más rápidamente que la cohorte positiva para el ARN de eritrocitos inmaduros, que es más grande. Por lo tanto, puede ponerse de manifiesto una mayor toxicidad aparente con diseños de exposición aguda que examinen la fracción de eritrocitos inmaduros positivos para el CD71 respecto a los que identifican eritrocitos inmaduros sobre la base del contenido de ARN. Por esta razón, cuando en los experimentos se utilizan cinco o menos días de tratamiento, la dosis más alta de los productos problema que causa toxicidad puede definirse como la dosis que produce una reducción estadísticamente significativa de la fracción de eritrocitos inmaduros positivos para el CD71 respecto al total de eritrocitos, pero hasta no menos del 5 % del valor del testigo (29). Los productos que presentan saturación de las propiedades toxicocinéticas, o inducen procesos de destoxificación que puedan dar lugar a una disminución de la exposición después de una administración a largo plazo pueden constituir excepciones en cuanto a los criterios de establecimiento de la dosis y han de evaluarse caso por caso. A fin de obtener información sobre la relación dosis-respuesta, los estudios completos deben incluir un grupo testigo negativo y un mínimo de tres dosis separadas por un factor que será en general de 2 y nunca superior a 4. Si el producto problema no produce toxicidad en el estudio de determinación del intervalo o según los datos existentes, la dosis más alta con un período de administración de 14 días o más debe ser de 1 000 mg/kg de peso corporal/día o, en caso de administración durante un período inferior a 14 días, de 2 000 mg/kg de peso corporal/día. No obstante, si el producto problema provoca toxicidad, la DMT debe ser la mayor dosis administrada y las dosis deben abarcar preferentemente el intervalo desde la dosis máxima hasta una dosis que provoque toxicidad escasa o nula. Si se observa toxicidad para el tejido diana (médula ósea) a todas las dosis utilizadas en el ensayo, se recomienda hacer otro estudio a dosis no tóxicas. Unos estudios destinados a caracterizar de forma más completa la relación cuantitativa dosis-respuesta pueden necesitar más grupos de dosis. Estos límites pueden variar con determinados tipos de productos problema (por ejemplo, productos farmacéuticos de uso humano) a los que se apliquen requisitos específicos. Ensayo límite Si los experimentos de determinación del intervalo, o los datos disponibles sobre cepas animales afines, indican que un régimen de tratamiento de, como mínimo, la dosis límite (véase más adelante) no produce efectos tóxicos observables (entre los que se incluirían la depresión de la proliferación de la médula ósea u otros signos de citotoxicidad para el tejido diana) y si, basándose en estudios de genotoxicidad in vitro o en datos de productos estructuralmente afines, no cabe esperar genotoxicidad, entonces puede no considerarse necesario realizar un estudio completo con tres dosis, siempre que se haya demostrado que el producto o productos problema alcanzan el tejido diana (médula ósea). En tales casos, podrá ser suficiente una sola dosis, al nivel de la dosis límite. Cuando la administración tiene lugar durante un período de 14 días o más, la dosis límite será de 1 000 mg/kg de peso corporal/día. Si el período de administración es inferior a 14 días, la dosis límite será de 2 000 mg/kg de peso corporal/día. Administración de las dosis A la hora de diseñar el ensayo, debe tenerse en cuenta la vía prevista de exposición humana. Por lo tanto, en casos justificados podrán elegirse vías de exposición como los alimentos, el agua de bebida, la tópica, la subcutánea, la intravenosa, la oral forzada (por sonda), la inhalación, la intratraqueal o la implantación. En cualquier caso, la vía debe elegirse de forma que se garantice una exposición adecuada del tejido o tejidos diana. En general no se recomienda la inyección intraperitoneal, ya que no es una vía de exposición humana prevista, y debe utilizarse solo en casos que tengan justificación científica específica. Si el producto problema se mezcla con los alimentos o el agua de bebida, en particular en caso de administración única, debe procurarse que el plazo entre el consumo de los alimentos o el agua y el muestreo sea suficiente para permitir la detección de los efectos (véase el punto 37). El volumen máximo de líquido que puede administrarse de una sola vez por sonda o por inyección depende del tamaño del animal utilizado. Dicho volumen no debe en principio superar 1 ml/100 g de peso corporal, salvo en el caso de las soluciones acuosas, en que puede llegarse a un máximo de 2 ml/100 g. El uso de volúmenes superiores deberá justificarse. Excepto en el caso de productos problema irritantes o corrosivos, que por lo general producen efectos exacerbados a concentraciones superiores, la variabilidad del volumen de ensayo debe reducirse al mínimo ajustando la concentración para conseguir la administración de un volumen constante en relación con el peso corporal a todas las dosis. Pauta de tratamiento De preferencia, se efectúan 2 o más tratamientos a intervalos de 24 horas, especialmente cuando se integra este ensayo en otros estudios de toxicidad. También es posible administrar tratamientos únicos cuando esté científicamente justificado (por ejemplo, productos problema conocidos por bloquear el ciclo celular). Los productos problema también pueden administrarse en dosis divididas (es decir, en dos o más tratamientos el mismo día separados por no más de 2 o 3 horas) con el fin de facilitar la administración de grandes volúmenes. En estas circunstancias, o cuando se administra el producto problema por inhalación, el tiempo de muestreo debe programarse basándose en el momento de la última administración o el final de la exposición. El ensayo puede realizarse con ratas o ratones de tres maneras:
Pueden aplicarse otros regímenes de muestreo o de tratamiento cuando sea pertinente y esté científicamente justificado, y para facilitar la integración con otros ensayos de toxicidad. Observaciones Deben hacerse observaciones clínicas generales de los animales de ensayo y registrarse los signos clínicos al menos una vez al día, preferentemente a la misma hora u horas cada día y teniendo en cuenta el período de mayor intensidad de los efectos previstos tras la administración. Al menos dos veces al día durante el periodo de administración, se debe observar la posible morbilidad y mortalidad de todos los animales. Deben pesarse todos los animales al principio del estudio, al menos una vez por semana durante los estudios de administración continuada, y en el momento del sacrificio. En los estudios de al menos una semana de duración, el consumo de alimentos debe medirse al menos semanalmente. Si el producto problema se administra con el agua de bebida, debe medirse el consumo de agua cada vez que se cambie el agua y al menos una vez por semana. Los animales que presenten signos de toxicidad excesiva pero no letal deben sacrificarse de forma compasiva antes de que termine el período del ensayo (28). En determinadas circunstancias, podría comprobarse la temperatura corporal de los animales, dado que la hipotermia y la hipertermia inducidas por el tratamiento se han relacionado con la producción de falsos resultados (32) (33) (34). Exposición del tejido diana Debe tomarse una muestra de sangre en el momento o momentos adecuados para permitir la investigación de los niveles plasmáticos de los productos problema, a fin de demostrar que ha habido exposición de la médula ósea, cuando esté justificado y cuando no se disponga de otros datos de exposición (véase el punto 48). Preparación de la médula ósea o de la sangre periférica Por lo general, las células de la médula ósea se extraen del fémur o de la tibia de los animales inmediatamente después de su sacrificio compasivo. Las células suelen tomarse, prepararse y teñirse según métodos establecidos. Pueden obtenerse pequeñas cantidades de sangre periférica, conforme a las normas de bienestar animal adecuadas, bien mediante un método que permita la supervivencia del animal, como el sangrado de la vena caudal o de otro vaso sanguíneo adecuado, o bien por punción cardiaca o extracción de un gran vaso en la eutanasia del animal. Tanto en el caso de eritrocitos tomados de la médula ósea como en el de los procedentes de la sangre periférica, dependiendo del método de análisis, las células pueden someterse inmediatamente a una tinción supravital (16) (17) (18), a la preparación de frotis y luego a tinción para microscopia, o a la fijación y tinción de manera adecuada para análisis por citometría de flujo. Si se emplea un colorante específico del ADN [p. ej., naranja de acridina (35) o Hoechst 33258 más pironina-Y (36)] pueden evitarse algunos de los artefactos que aparecen cuando se utiliza un colorante que no es específico del ADN. Esta ventaja no excluye el uso de tinciones convencionales (por ejemplo, Giemsa para el análisis microscópico). También pueden utilizarse otros sistemas [por ejemplo, columnas de celulosa para eliminar las células nucleadas (37) (38)], siempre que se tenga constancia de que son compatibles con la preparación de las muestras en el laboratorio. Si estos métodos son aplicables, pueden utilizarse anticuerpos anti-cinetocoros (39), la técnica FISH con sondas pancentroméricas de ADN (40), o el etiquetado in situ cebado con cebadores pancentroméricos específicos, junto con una tinción de contraste adecuada del ADN (41), para determinar la naturaleza de los micronúcleos (cromosoma/fragmento cromosómico) a fin de determinar si el mecanismo de inducción de la formación de micronúcleos se debe a actividad clastogénica o aneugénica. Pueden utilizarse otros métodos para diferenciar entre clastógenos y anéugenos, siempre que se haya comprobado que son efectivos. Análisis (manual y automatizado) Todos los portaobjetos o muestras para el análisis, incluidos los de los testigos positivos y negativos, deben codificarse independientemente antes de cualquier tipo de análisis y aleatorizarse de forma que el examinador manual no tenga conocimiento de cómo se han tratado; esta codificación no es necesaria cuando se utilizan sistemas de examen automatizados que no dependen de la inspección visual y no pueden verse afectados por el sesgo del operador. Para cada animal, se determina la relación entre los eritrocitos inmaduros y el total de eritrocitos (inmaduros + maduros) en un recuento de al menos 500 eritrocitos si se trata de médula ósea y de 2 000 si se trata de sangre periférica (42). Se debe determinar la incidencia de eritrocitos inmaduros micronucleados en un mínimo de 4 000 eritrocitos inmaduros por animal (43). En caso de que la base de datos históricos de testigos negativos indique que la frecuencia media de eritrocitos inmaduros con micronúcleos de fondo es < 0,1 % en el laboratorio de ensayo, debe considerarse el examen de células adicionales. A la hora de analizar las muestras, la proporción de eritrocitos inmaduros respecto al total de eritrocitos en los animales tratados no debe ser inferior al 20 % de la proporción del testigo del vehículo/disolvente cuando se examinan por microscopia, ni al 5 % aproximadamente de la proporción del testigo del vehículo/disolvente cuando se examinan eritrocitos inmaduros positivos para el CD71 por métodos de citometría (véase el punto 31) (29). Por ejemplo, para un ensayo de médula ósea con examen por microscopia, si la proporción de eritrocitos inmaduros en la médula ósea respecto al testigo es del 50 %, el límite superior de toxicidad sería un 10 % de eritrocitos inmaduros. Debido a que el bazo de la rata secuestra y destruye los eritrocitos micronucleados, para mantener una elevada sensibilidad del ensayo cuando se analiza sangre periférica de rata, es preferible limitar el análisis de eritrocitos inmaduros micronucleados a la fracción más joven. Cuando se utilicen métodos de análisis automatizados, estos eritrocitos más inmaduros podrán identificarse sobre la base de su alto contenido de ARN, o del alto nivel de receptores de transferrina (positivos para el CD71) expresados en su superficie (31). No obstante, la comparación directa de diferentes métodos de tinción ha demostrado que se pueden obtener resultados satisfactorios con diversos métodos, incluyendo la tinción convencional con naranja de acridina (3) (4). DATOS E INFORME Tratamiento de los resultados Los datos relativos a cada animal se presentarán en forma de cuadro. Con cada animal analizado por separado se relacionará el número de eritrocitos inmaduros examinados, el de eritrocitos inmaduros micronucleados y la proporción de eritrocitos inmaduros respecto al total de eritrocitos. Cuando se tratan ratones de forma continua durante 4 semanas o más, también deben darse los datos sobre el número y la proporción de eritrocitos maduros micronucleados, si se han tomado. Deben también consignarse los datos sobre toxicidad y signos clínicos de los animales. Criterios de aceptabilidad Los siguientes criterios determinan la aceptabilidad del ensayo:
Evaluación e interpretación de los resultados Siempre que se cumplan todos los criterios de aceptabilidad, se considera que el producto problema es claramente positivo si también se cumple lo siguiente:
Si se examina únicamente la dosis más alta en un momento particular de muestreo, el producto problema se considera claramente positivo si se ve un aumento estadísticamente significativo en comparación con el testigo negativo en paralelo y los resultados están fuera de la distribución de los datos históricos de los testigos negativos (por ejemplo, límites de control del 95 % según la distribución de Poisson). En la bibliografía se encuentran recomendaciones sobre los métodos estadísticos más adecuados (44) (45) (46) (47). Al realizar un análisis de la relación dosis-respuesta, deben someterse al mismo por lo menos tres grupos tratados con dosis diferentes del producto problema. Las pruebas estadísticas deben utilizar como unidad experimental el animal. Un resultado positivo en el ensayo de micronúcleos indica que el producto problema induce la formación de micronúcleos, que son consecuencia de una lesión de los cromosomas o del aparato mitótico de los eritroblastos de la especie estudiada. En caso de que se haya realizado un ensayo para detectar la presencia de centrómeros en los micronúcleos, si el producto produce micronúcleos con centrómeros (ADN centromérico o cinetocoro, que indica la pérdida de cromosomas enteros), queda demostrado que este producto problema es un anéugeno. Siempre que se cumplan todos los criterios de aceptabilidad, se considera que el producto problema es claramente negativo si, en todas las condiciones experimentales examinadas, también se cumple lo siguiente:
En la bibliografía se encuentran recomendaciones sobre los métodos estadísticos más adecuados (44) (45) (46) (47). Como pruebas de la exposición de la médula ósea a un producto problema pueden citarse la reducción de la proporción de eritrocitos inmaduros y maduros o la medición de los niveles hemáticos o plasmáticos del producto problema. En caso de administración por vía intravenosa, no hacen falta pruebas de la exposición. Alternativamente, para demostrar la exposición de la médula ósea es posible utilizar los datos de ADME obtenidos en un estudio independiente utilizando la misma vía y la misma especie. Un resultado negativo indica que, en las condiciones del ensayo, el producto problema no induce la formación de micronúdeos en los eritrocitos inmaduros de la especie estudiada. No se requiere ninguna verificación de una respuesta positiva clara o negativa clara. En los casos en que la respuesta no sea claramente negativa ni positiva y con el fin de ayudar a determinar la importancia biológica de un resultado (por ejemplo, un incremento límite o débil), los datos deben ser evaluados por expertos o hay que completar otras investigaciones de los experimentos existentes. En algunos casos, puede ser de utilidad el análisis de más células o la repetición de un experimento en condiciones experimentales modificadas. En casos excepcionales, incluso después de otras investigaciones, los datos no permiten concluir si el producto problema produce resultados positivos o negativos, y el estudio deberá considerarse, por tanto, como de resultado dudoso. Informe del ensayo El informe del ensayo debe incluir la información siguiente:
BIBLIOGRAFÍA
Apéndice 1 DEFINICIONES Centrómero : Región o regiones del cromosoma con las que se asocian las fibras del huso acromático durante la división celular para permitir el movimiento ordenado de los cromosomas hijos hacia los polos de las células hijas. Producto : Sustancia o mezcla. Eritroblasto : Fase temprana de desarrollo de los eritrocitos, que precede inmediatamente a la de eritrocitos inmaduros, cuando la célula aún tiene núcleo. Cinetocoro : Estructura proteínica que se forma en el centrómero de las células eucarióticas, que vincula el cromosoma a los polímeros del microtúbulo del huso mitótico durante la mitosis y la meiosis y sirve para separar las cromátidas hermanas durante la división celular. Micronúcleos : Núcleos pequeños, adicionales a los núcleos celulares principales y separados de ellos, y producidos durante la telofase de la mitosis (o la meiosis) por fragmentos cromosómicos o cromosomas enteros retardados. Eritrocito normocromático o maduro : Eritrocito completamente madurado que ha perdido el ARN residual que queda tras la desnucleación o ha perdido otros marcadores celulares de vida corta que desaparecen típicamente tras la desnucleación que tiene lugar después de la división final del eritroblasto. Eritrocito policromático o inmaduro : Eritrocito recién formado en una fase intermedia de desarrollo, que se tiñe con los componentes tanto de color azul como de color rojo de las tinciones clásicas de sangre, como la de Wright-Giemsa, debido a la presencia de ARN residual en las células recién formadas. Estas células recién formadas son aproximadamente lo mismo que los reticulocitos, los cuales se visualizan utilizando una tinción vital que hace que el ARN residual se aglutine en un retículo. Para identificar los glóbulos rojos recién formados se suelen utilizar ahora otros métodos, como la tinción monocromática de ARN con colorantes fluorescentes o el etiquetado de marcadores de superficie de vida corta como el CD71 con anticuerpos fluorescentes. Los eritrocitos policromáticos, los reticulocitos y los eritrocitos positivos para el CD71 son todos eritrocitos inmaduros, aunque cada uno de ellos tiene una distribución por edades algo diferente. Reticulocito : Eritrocito recién formado teñido con una tinción vital que hace que el ARN celular residual se aglutine en un retículo característico. Los reticulocitos y los eritrocitos policromáticos tienen una distribución similar por edades celulares. Producto problema : Sustancia o mezcla estudiada con este método de ensayo. Apéndice 2 DISEÑO FACTORIAL PARA IDENTIFICAR LAS DIFERENCIAS DE SEXO EN EL ENSAYO DE MICRONÚCLEOS IN VIVO Diseño factorial y su análisis En este diseño, un mínimo de 5 machos y 5 hembras se someten a ensayo a cada concentración, lo que resulta en la utilización de un mínimo de 40 animales (20 machos y 20 hembras, más los testigos positivos pertinentes). Este diseño, que es uno de los diseños factoriales más simples, es equivalente a un análisis de varianza de dos factores, con el sexo y el nivel de concentración como efectos principales. Los datos pueden analizarse con numerosos paquetes de software estadístico estándar tales como SPSS, SAS, STATA, Genstat, así como utilizando R. El análisis divide la variabilidad de la serie de datos en la variabilidad entre sexos, la variabilidad entre las concentraciones y la relativa a la interacción entre los sexos y las concentraciones. Cada uno de los términos se somete a prueba frente a una estimación de la variabilidad entre los animales replicados dentro de los grupos de animales del mismo sexo que han recibido la misma concentración. Todos los detalles de la metodología subyacente se encuentran en muchos manuales estadísticos conocidos (véanse las referencias) y en las funciones de “ayuda” de los paquetes estadísticos. El análisis se lleva a cabo inspeccionando el término de la interacción sexo x concentración en el cuadro de ANOVA (6). A falta de un término de interacción significativo, los valores combinados de distintos sexos o niveles de concentración ofrecen pruebas estadísticas válidas entre los niveles, sobre la base del término de ANOVA de variabilidad intragrupo puesta en común. El análisis continúa dividiendo la estimación de la variabilidad entre concentraciones en contrastes que permiten una prueba de contrastes lineales y cuadráticos de las respuestas a través de los niveles de concentración. Cuando existe un término de interacción significativa sexo x concentración, este término también puede dividirse en contrastes de interacción lineal x sexo y cuadrático x sexo. Estos términos permiten pruebas de si las respuestas a la concentración son paralelas en los dos sexos, o si existe una diferencia en la respuesta entre estos. La estimación de la variabilidad intragrupo puesta en común se puede utilizar para proporcionar pruebas pareadas de la diferencia entre medias. Estas comparaciones podrían realizarse entre las medias para los dos sexos y entre las medias para los diferentes niveles de concentración, así como para las comparaciones con los niveles de los testigos negativos. En los casos en los que existe una interacción significativa pueden hacerse comparaciones entre las medias de concentraciones diferentes dentro de un sexo o entre las medias de los sexos a la misma concentración. Bibliografía Hay muchos manuales estadísticos que debaten la teoría, el diseño, la metodología, el análisis y la interpretación de los diseños factoriales que van desde los análisis bifactoriales más sencillos hasta las formas más complejas utilizadas en la metodología de “diseño de experimentos”. La lista que sigue no es exhaustiva. Algunos libros proporcionan ejemplos resueltos de diseños comparables, en algunos casos con el código para aplicar los análisis utilizando diversos paquetes de software.
|
|
6) |
En la parte B, se suprime el capítulo B.15. |
|
7) |
En la parte B, se suprime el capítulo B.16. |
|
8) |
En la parte B, se suprime el capítulo B.18. |
|
9) |
En la parte B, se suprime el capítulo B.19. |
|
10) |
En la parte B, se suprime el capítulo B.20. |
|
11) |
En la parte B, se suprime el capítulo B.24. |
|
12) |
En la parte B, el capítulo B.47 se sustituye por el texto siguiente: «B.47. Método de ensayo de la opacidad y permeabilidad de la córnea de bovino para identificar: i) productos que provocan lesiones oculares graves y ii) productos que no requieren clasificación por irritación ocular ni lesiones oculares graves INTRODUCCIÓN El presente método de ensayo reproduce las directrices de ensayo (TG) 437 de la OCDE (2013). El método de ensayo de la opacidad y permeabilidad de la córnea de bovino (conocido por la siglas de su nombre en inglés, BCOP), fue evaluado por el Comité de Coordinación Interagencias sobre la Validación de Métodos Alternativos estadounidense (Interagency Coordinating Committee on the Validation of Alternative Methods, ICCVAM), junto con el Centro Europeo para la Validación de Métodos Alternativos (European Centre for the Validation of Alternative Methods, ECVAM) y el Centro Japonés para la Validación de Métodos Alternativos (Japanese Center for the Validation of Alternative Methods, JaCVAM), en 2006 y 2010 (1)(2). En la primera evaluación, el método de ensayo BCOP se evaluó en cuanto a su utilidad para identificar productos (sustancias y mezclas) que provocan lesiones oculares graves (1). En la segunda evaluación, el método de ensayo BCOP se evaluó en cuanto a su utilidad para identificar productos (sustancias y mezclas) que no están clasificados por irritación ocular ni por lesiones oculares graves (2). La base de datos de validación del método BCOP incluía 113 sustancias y 100 mezclas en total (2)(3). De estas evaluaciones y de su revisión por pares se dedujo que el método de ensayo permite identificar correctamente los productos (tanto sustancias como mezclas) que provocan lesiones oculares graves (categoría 1), así como los que no requieren clasificación por irritación ocular ni lesiones oculares graves, según se definen en el Sistema Globalmente Armonizado de clasificación y etiquetado de productos químicos (SGA) de las Naciones Unidas (4) y en el Reglamento (CE) n.o 1272/2008 del Parlamento Europeo y del Consejo, sobre clasificación, etiquetado y envasado de sustancias y mezclas (CLP) (7), por lo que el método se considera científicamente válido para ambos objetivos. Una lesión ocular grave es un daño tisular en el ojo o un deterioro físico importante de la visión, como consecuencia de la aplicación de un producto problema en la superficie anterior del ojo, que no es completamente reversible en los 21 días siguientes a la aplicación. Los productos problema que provocan lesiones oculares graves se clasifican en la categoría 1 del SGA de las Naciones Unidas. Los productos no clasificados por irritación ocular o lesiones oculares graves se definen como aquellos que no cumplen los requisitos para la clasificación en las categorías 1 o 2 (2A o 2B), es decir, se mencionan como «sin categoría» del SGA de las Naciones Unidas. El presente método de ensayo incluye el uso recomendado y las limitaciones del método de ensayo BCOP a partir de sus evaluaciones. Las principales diferencias entre la versión inicial de 2009 y la versión actualizada de 2013 de las líneas directrices de la OCDE se refieren, entre otros, a los siguientes elementos: el uso del método de ensayo BCOP para identificar productos que no requieren clasificación según el SGA de las Naciones Unidas (puntos 2 y 7); aclaraciones sobre la aplicabilidad del método de ensayo BCOP a los ensayos de alcoholes, cetonas y sólidos (puntos 6 y 7) y de sustancias y mezclas (punto 8); aclaraciones sobre cómo deben someterse a ensayo las sustancias tensioactivas y las mezclas que contienen tensioactivos (punto 28); actualizaciones y aclaraciones sobre los testigos positivos (puntos 39 y 40); una actualización de los criterios de decisión del método de ensayo BCOP (punto 48); una actualización de los criterios de aceptación del estudio (punto 49); una actualización de los elementos del informe del ensayo (punto 50); una actualización del apéndice 1, sobre las definiciones; la adición del apéndice 2 sobre la capacidad de predicción del método de ensayo BCOP en diferentes sistemas de clasificación; una actualización del apéndice 3 sobre la lista de productos utilizados para demostrar la aptitud; y una actualización del apéndice 4 sobre el soporte de córnea para el método BCOP (punto 1) y sobre el opacímetro (puntos 2 y 3). Actualmente se reconoce en general que, en un futuro próximo, ningún ensayo de irritación ocular in vitro podrá sustituir por sí solo al ensayo ocular de Draize in vivo para predecir toda la gama de irritación causada por las distintas categorías de productos. Sin embargo, unas combinaciones estratégicas de varios métodos de ensayo alternativos dentro de una estrategia de ensayos (escalonados) sí podrán sustituir al ensayo ocular de Draize (5). El enfoque descendente (5) está diseñado para utilizarse cuando, sobre la base de la información existente, se espera de un producto que tenga una alta capacidad de irritación, mientras que el enfoque ascendente (5) está diseñado para utilizarse cuando, sobre la base de la información existente, se espera de un producto que no cause irritación ocular suficiente para exigir una clasificación. El método de ensayo BCOP es un procedimiento in vitro que puede utilizarse, en determinadas circunstancias y con limitaciones específicas con fines de clasificación de peligros para los ojos y de etiquetado de productos. Si bien no se considera válido para sustituir por sí solo al ensayo in vivo con ojo de conejo, el método de ensayo BCOP se recomienda como fase inicial en una estrategia de ensayo tal como el enfoque descendente sugerido por Scott et al. (5) para identificar los productos que provocan lesiones oculares graves, es decir, los productos que han de clasificarse en la categoría 1 del SGA de las Naciones Unidas, sin necesidad de hacer más pruebas (4). El método de ensayo BCOP se recomienda también para identificar productos que no requieren clasificación por irritación ocular ni lesiones oculares graves, según se definen en el SGA de las Naciones Unidas (sin categoría del SGA de las Naciones Unidas) (4) dentro de una estrategia de ensayos como el enfoque ascendente (5). No obstante, un producto del que no se espere que cause lesiones oculares graves o que no esté clasificado por irritación ocular o lesiones oculares graves según el método de ensayo BCOP tendría que someterse a ensayos adicionales (in vitro o in vivo) para establecer una clasificación definitiva. El objetivo del presente método de ensayo es describir los procedimientos utilizados para evaluar el posible peligro para los ojos de un producto problema en función de su capacidad para inducir opacidad y aumento de la permeabilidad en una córnea aislada de bovino. Los efectos tóxicos para la córnea se miden mediante: i) el descenso de la transmisión luminosa (opacidad), y ii) el aumento del paso del pigmento fluoresceína sódica (permeabilidad). Los resultados de la evaluación de la opacidad y de la permeabilidad de la córnea tras su exposición al producto problema se combinan para obtener una puntuación de la capacidad de irritación in vitro (conocida por sus siglas en inglés, IVIS), que se utiliza para clasificar el nivel de capacidad de irritación del producto problema. En el apéndice 1 se dan las definiciones pertinentes. CONSIDERACIONES INICIALES Y LIMITACIONES El presente método de ensayo se basa en el protocolo del método de ensayo BCOP del ICCVAM (6)(7), que fue elaborado originariamente a partir del protocolo del Institute for in vitro Sciences (IIVS) y del protocolo 124 de INVITTOX (8). Este último representa el protocolo utilizado en el estudio de prevalidación patrocinado por la Comunidad Europea y realizado en 1997-1998. Estos dos protocolos se basaban en el método de ensayo BCOP comunicado por primera vez por Gautheron et al. (9). El método de ensayo BCOP puede utilizarse para identificar los productos que provocan lesiones oculares graves, tal como se definen en el SGA de las Naciones Unidas, es decir, que deben clasificarse como productos de la categoría 1 del SGA de las Naciones Unidas (4). Cuando se utiliza para este fin, el método de ensayo BCOP tiene una exactitud general del 79 % (150/191), una tasa de falsos positivos del 25 % (32/126), y una tasa de falsos negativos del 14 % (9/65), cuando se compara con los datos del método de ensayo in vivo con ojo de conejo, clasificados según el sistema de clasificación del SGA de las Naciones Unidas (3) (véase el apéndice 2, cuadro 1). Cuando los productos problema de ciertas clases químicas (en concreto, alcoholes y cetonas) o físicas (en concreto, sólidos), se excluyen de la base de datos, el método de ensayo BCOP tiene una exactitud general del 85 % (111/131), una tasa de falsos positivos del 20 % (16/81), y una tasa de falsos negativos del 8 % (4/50) para el sistema de clasificación del SGA de las Naciones Unidas (3). Las posibles deficiencias del método de ensayo BCOP para identificar los productos que provocan lesiones oculares graves (categoría 1 del SGA de las Naciones Unidas) están relacionadas con la elevada tasa de falsos positivos de alcoholes y cetonas y la elevada tasa de falsos negativos de sólidos, observadas en la base de datos de validación (1)(2)(3). Sin embargo, dado que no todos los alcoholes y cetonas dan resultados positivos excesivos (sobrepredicción) por el método de ensayo BCOP y algunos de ellos se asignan correctamente a la categoría 1 del SGA de las Naciones Unidas, estos dos grupos funcionales orgánicos no se consideran fuera del ámbito de aplicación del método de ensayo. Corresponde al usuario del método de ensayo decidir si puede aceptarse una posible sobrepredicción de un alcohol o cetona, o si deben efectuarse más ensayos con un planteamiento basado en la ponderación de las pruebas. En relación con las tasas de falsos negativos de sólidos, cabe señalar que los sólidos pueden dar lugar a condiciones de exposición variables y extremas en el ensayo in vivo de irritación de Draize, lo cual puede derivar en resultados poco pertinentes sobre el verdadero potencial de irritación (10). También debe señalarse que ninguno de los falsos negativos detectados en la base de datos de validación del ICCVAM (2)(3), en el contexto de la identificación de los productos que inducen lesiones oculares graves (categoría 1 del SGA de las Naciones Unidas), dio lugar a una IVIS ≤ 3, que es el criterio utilizado para identificar un producto problema como sin categoría del SGA de las Naciones Unidas. Además, los falsos negativos del BCOP no son fundamentales en este contexto, ya que todos los productos problema que den 3 < IVIS ≤ 55 se someterían posteriormente a otros ensayos in vitro validados adecuadamente, o como última opción con conejos, en función de las exigencias normativas, utilizando una estrategia de ensayo secuencial en un enfoque de ponderación de las pruebas. Teniendo en cuenta que algunos productos sólidos son clasificados adecuadamente por el método de ensayo BCOP como de la categoría 1 del SGA de las Naciones Unidas, tampoco se considera que este estado físico quede fuera del ámbito de aplicación del método de ensayo. Los investigadores pueden considerar la posibilidad de utilizar este método de ensayo con todos los tipos de productos, aceptándose una IVIS > 55 como indicativa de una respuesta que provoca lesiones oculares graves, con lo que el producto se clasificaría en la categoría 1 del SGA de las Naciones Unidas sin ensayos adicionales. Sin embargo, como se ha mencionado antes, los resultados positivos obtenidos con alcoholes o cetonas deben interpretarse con precaución, debido al riesgo de sobrepredicción. El método de ensayo BCOP puede utilizarse también para identificar productos que no requieren clasificación por irritación ocular o lesiones oculares graves con arreglo al sistema de clasificación del SGA de las Naciones Unidas (4). Cuando se utiliza para este fin, el método de ensayo BCOP tiene una exactitud general del 69 % (135/196), una tasa de falsos positivos del 69 % (61/89), y una tasa de falsos negativos del 0 % (0/107), cuando se compara con los datos del método de ensayo in vivo con ojo de conejo, clasificados según el sistema de clasificación del SGA de las Naciones Unidas (3) (véase el apéndice 2, cuadro 2). La tasa de falsos positivos obtenida (productos sin categoría del SGA de las Naciones Unidas in vivo que presentan una IVIS > 3, véase el punto 48) es considerablemente elevada, pero no es crítica en este contexto, ya que todos los productos problema que den 3 < IVIS ≤ 55 se someterían posteriormente a otros ensayos in vitro validados adecuadamente, o como última opción con conejos, en función de las exigencias normativas, utilizando una estrategia de ensayo secuencial en un enfoque de ponderación de las pruebas. El método de ensayo BCOP no presenta deficiencias específicas para el ensayo de alcoholes, cetonas y sólidos cuando el objetivo es identificar productos que no requieren clasificación por irritación ocular ni lesiones oculares graves (sin categoría del SGA de las Naciones Unidas) (3). Los investigadores pueden considerar la posibilidad de utilizar este método de ensayo con todos los tipos de productos, aceptándose un resultado negativo (IVIS ≤ 3) como indicativo de que no se requiere clasificación (sin categoría del SGA de las Naciones Unidas). Dado que el método de ensayo BCOP solo puede identificar correctamente el 31 % de los productos que no requieren clasificación por irritación ocular o lesiones oculares graves, este método de ensayo no debe ser la primera opción para iniciar un enfoque ascendente (5), si se dispone de otros métodos in vitro validados y aceptados, con gran sensibilidad similar pero mayor especificidad. La base de datos de validación del método BCOP incluía 113 sustancias y 100 mezclas en total (2)(3). El método de ensayo BCOP se considera, por tanto, aplicable a los ensayos de sustancias y de mezclas. El método de ensayo BCOP no se recomienda para la identificación de productos problema que deben clasificarse como irritantes oculares (categoría 2 o categoría 2A del SGA de las Naciones Unidas) o de productos problema que deben clasificarse como moderadamente irritantes para los ojos (categoría 2B del SGA de las Naciones Unidas) debido al considerable número de productos de la categoría 1 del SGA de las Naciones Unidas clasificados de forma insuficiente en las categorías 2, 2A o 2B del SGA de las Naciones Unidas y productos sin categoría del SGA de las Naciones Unidas clasificados de forma excesiva en las categorías 2, 2A o 2B del SGA de las Naciones Unidas (2) (3). A tal fin, pueden resultar necesarias pruebas adicionales con otro método adecuado. Todos los procedimientos con ojos de bovino y córneas de bovino deben ajustarse a las normas y procedimientos aplicables de la instalación de ensayo en relación con la manipulación de materiales derivados de animales, entre los que figuran los tejidos o los líquidos tisulares. Se recomienda observar las precauciones universales de laboratorio (11). Si bien el método de ensayo BCOP no tiene en cuenta las lesiones de la conjuntiva y del iris, sí atiende a los efectos en la córnea, que son un factor de primer orden de la clasificación in vivo cuando se considera la clasificación del SGA de las Naciones Unidas. La reversibilidad de las lesiones de la córnea no puede evaluarse per se en el método de ensayo BCOP. Se ha propuesto, según estudios realizados con ojo de conejo, que se utilice una evaluación de la profundidad inicial de la lesión de la córnea para identificar algunos tipos de efectos irreversibles (12). Sin embargo, se necesitan más conocimientos científicos para comprender cómo se dan efectos irreversibles no relacionados con lesiones iniciales de alto nivel. Finalmente, el método de ensayo BCOP no permite la evaluación del potencial de toxicidad sistémica asociado con la exposición ocular. El presente método de ensayo se actualizará periódicamente a la luz de la nueva información y los datos que se vayan considerando. Por ejemplo, la histopatología puede ser útil si se necesita una caracterización más completa de las lesiones de la córnea. Como se indica en el documento de orientación de la OCDE n.o 160 (13), se insta a los usuarios a conservar las córneas y a preparar muestras de histopatología que puedan utilizarse para compilar una base de datos y criterios de decisión con los que se pueda mejorar más tarde la exactitud de este método. Cuando algún laboratorio establezca inicialmente este método de ensayo, deben utilizarse los productos para demostrar la aptitud que se indican en el apéndice 3. Los laboratorios pueden utilizar estos productos para demostrar su competencia técnica en la realización del método de ensayo BCOP antes de presentar, con fines de clasificación normativa de peligro, los datos obtenidos con este ensayo. PRINCIPIO DEL ENSAYO El método de ensayo BCOP es un modelo organotípico con mantenimiento a corto plazo del funcionamiento fisiológico y bioquímico normal de la córnea de bovino in vitro. En este método de ensayo, la lesión provocada por el producto problema se evalúa mediante la medición cuantitativa de los cambios producidos en la opacidad y en la permeabilidad de la córnea, utilizando un opacímetro y un espectrofotómetro de luz visible, respectivamente. Las dos mediciones se combinan para calcular la IVIS, que se utiliza para asignar una categoría de clasificación de peligro por irritación in vitro a fin de predecir el potencial de irritación ocular in vivo de un producto problema (véanse los criterios de decisión en el punto 48). El método de ensayo BCOP utiliza córneas aisladas de ojos de ganado bovino recién sacrificado. La opacidad de la córnea se mide cuantitativamente como cantidad de luz transmitida a través de la córnea. La permeabilidad se mide cuantitativamente como cantidad del pigmento fluoresceína sódica que atraviesa todo el espesor de la córnea y se detecta así en el medio que se encuentra en la cámara posterior. Los productos problema se aplican a la superficie epitelial de la córnea poniéndolos en la cámara anterior del soporte de córnea. En el apéndice 4 se ofrece una descripción y un diagrama de un soporte de córnea utilizado en el método de ensayo BCOP. Los soportes de córnea pueden obtenerse de diferentes fuentes comerciales o bien construirse en el propio laboratorio. Fuente y edad de los ojos de bovino y selección de la especie animal El ganado bovino enviado a los mataderos se sacrifica normalmente para el consumo humano o con otros fines comerciales. Como fuente de córneas para el método de ensayo BCOP se utilizan solo animales sanos que se consideren adecuados para entrar en la cadena alimentaria humana. Debido a que el ganado bovino presenta una amplia variedad de peso, en función de la raza, edad y sexo, no se recomienda ningún peso concreto del animal en el momento del sacrificio. Puede variar el tamaño de la córnea cuando se utilizan ojos de animales de distintas edades. Las córneas con un diámetro horizontal > 30,5 mm y un espesor central de la córnea (ECC) ≥ 1 100 μm suelen obtenerse de animales de más de ocho años, mientras que las córneas con un diámetro horizontal < 28,5 mm y un ECC < 900 μm suelen proceder de animales de menos de cinco años (14). Por este motivo, no suelen utilizarse ojos de animales de más de sesenta meses de edad. Tradicionalmente no se utilizan tampoco ojos de animales de menos de doce meses, ya que estos ojos están aún en fase de desarrollo y el espesor y el diámetro de su córnea son considerablemente más pequeños que los observados en ojos de bovinos adultos. Sin embargo, es admisible el uso de córneas de animales jóvenes (es decir, de entre seis y doce meses de edad) porque presentan ciertas ventajas, como una mayor disponibilidad, una variabilidad pequeña en cuanto a la edad, y un peligro reducido en cuanto a la posible exposición de los trabajadores a la encefalopatía espongiforme bovina (15). Como puede ser útil una evaluación más completa del efecto del tamaño o del espesor de la córnea sobre la sensibilidad ante productos corrosivos e irritantes, se anima a los usuarios a informar sobre la edad o el peso estimados de los animales de los que proceden las córneas utilizadas en un estudio. Recogida y transporte de los ojos al laboratorio Los ojos son recogidos por los empleados del matadero. Para minimizar las lesiones de los ojos, de tipo mecánico u otro, hay que enuclear los ojos tan pronto como sea posible tras la muerte y enfriarlos inmediatamente después de la enucleación y durante el transporte. A fin de evitar la exposición de los ojos a productos potencialmente irritantes, los empleados del matadero no deben utilizar detergentes cuando laven la cabeza del animal. Los ojos deben sumergirse completamente en solución salina equilibrada de Hank (conocida por sus siglas en inglés, HBSS) refrigerada en un recipiente de tamaño adecuado, y transportarse al laboratorio de manera que se reduzcan al mínimo su deterioro y su contaminación bacteriana. Como los ojos se recogen durante el proceso de sacrificio, pueden estar expuestos a la sangre y a otros materiales biológicos, además de bacterias y otros microorganismos. Por tanto, es importante velar por que se reduzca al mínimo el riesgo de contaminación (por ejemplo, manteniendo en hielo el recipiente con los ojos durante las operaciones de recogida y transporte, y añadiendo antibióticos a la HBSS utilizada para conservar los ojos durante el transporte [por ejemplo, 100 UI/ml de penicilina y 100 μg/ml de estreptomicina]). Debe reducirse al mínimo el tiempo transcurrido entre la recogida de los ojos y la utilización de las córneas en el método de ensayo BCOP (lo normal es efectuar todo el proceso en un solo día), y hay que demostrar que ese tiempo no altera los resultados del ensayo. Estos resultados se basan en los criterios de selección de los ojos, así como en las respuestas obtenidas con testigos positivos y negativos. Todos los ojos utilizados en el ensayo deben proceder del mismo grupo de ojos recogidos un día concreto. Criterios de selección de los ojos utilizados en el método de ensayo BCOP Tras su llegada al laboratorio, los ojos se someten a un atento examen para detectar posibles defectos, como un aumento de la opacidad, arañazos o neovascularización. Solo deben utilizarse las córneas de ojos que no presenten estos defectos. La calidad de cada córnea se evalúa también en otras fases posteriores del ensayo. Deben desecharse las córneas que tengan una opacidad mayor de siete unidades de opacidad o equivalente según el opacímetro y los soportes de córneas que se hayan usado con ellas después del período inicial de equilibrado de una hora (NOTA: El opacímetro debe calibrarse con los patrones de opacidad utilizados para establecer las unidades de opacidad, véase el apéndice 4). Cada grupo de tratamiento (producto problema, testigos negativos y positivos en paralelo) consiste en un mínimo de tres ojos. Para el testigo negativo en el método de ensayo BCPO deben utilizarse tres córneas. Puesto que todas las córneas se extraen del globo entero y se montan en las cámaras de córnea, existe la posibilidad de que haya artefactos procedentes de la manipulación que influyan en los distintos valores de opacidad y permeabilidad de la córnea (incluido el testigo negativo). Por otra parte, los valores de opacidad y permeabilidad de las córneas del testigo negativo se utilizan para corregir los valores de opacidad y permeabilidad de las córneas tratadas con la muestra problema y con los testigos positivos a efectos del cálculo de la IVIS. PROCEDIMIENTO Preparación de los ojos Se disecan córneas libres de defectos, dejándoles un borde de 2 o 3 mm de esclerótica para facilitar la manipulación posterior, y atendiendo a no lesionar el epitelio ni el endotelio de la córnea. Las córneas aisladas se montan en soportes de córnea, diseñados especialmente, que consisten en un compartimento anterior y otro posterior, en contacto respectivamente con el lado epitelial y el lado endotelial de la córnea. Ambas cámaras se llenan hasta rebosar con medio esencial mínimo de Eagle (EMEM) sin rojo de fenol y precalentado, empezando por la cámara posterior y evitando la formación de burbujas. El dispositivo se estabiliza después a 32 ± 1 °C durante al menos una hora para que las córneas alcancen un estado de equilibrio con el medio y consigan una actividad metabólica normal, dentro de lo posible (la temperatura aproximada de la superficie de la córnea in vivo es de 32 °C). Tras el periodo de equilibrado, se añade a ambas cámaras nuevo EMEM sin rojo de fenol y precalentado, y se toman lecturas de la opacidad de base de cada córnea. Deben desecharse las córneas que muestren lesiones tisulares macroscópicas (p. ej., arañazos, pigmentación, neovascularización) o una opacidad mayor de siete unidades de opacidad o equivalente según el opacímetro y los soportes de córneas que se hayan usado. Se selecciona para el testigo negativo (o de disolvente) un mínimo de tres córneas. El resto de las córneas se distribuye después en grupos de tratamiento y de testigo positivo. Como la capacidad térmica del agua es superior a la del aire, el agua ofrece condiciones de temperatura más estables para la incubación. Por tanto, se recomienda utilizar un baño de agua para mantener los soportes de córnea y su contenido a 32 ± 1 °C. Sin embargo, también pueden utilizarse incubadoras de aire, si se toman precauciones para mantener la estabilidad de la temperatura (por ejemplo, precalentando los soportes y los medios). Aplicación del producto problema Se utilizan dos protocolos de tratamiento diferentes, uno para líquidos y agentes tensioactivos (sólidos o líquidos) y otro para sólidos que no son agentes tensioactivos. Los líquidos se someten al ensayo sin diluir. Los semisólidos, cremas y ceras se someten a ensayo en principio como líquidos. Las sustancias tensioactivas sin diluir se someten a ensayo a una concentración del 10 % (p/v) en solución de cloruro sódico al 0,9 %, agua destilada u otro disolvente del que se haya demostrado que no tiene efectos negativos sobre el sistema de ensayo. Debe justificarse adecuadamente si se utilizan otras concentraciones de dilución. Las mezclas que contengan tensioactivos pueden someterse a ensayo sin diluir o diluidas a una concentración adecuada en función del escenario de exposición in vivo pertinente. Debe justificarse adecuadamente la concentración utilizada en el ensayo. Las córneas se exponen a los líquidos y agentes tensioactivos durante 10 minutos. La utilización de otros tiempos de exposición exige una explicación científica adecuada. Véase en el apéndice 1 una definición de tensioactivo y de mezcla que contiene tensioactivos. Los sólidos que no son agentes tensioactivos se someten a ensayo en principio como soluciones o suspensiones a una concentración del 20 % (p/v) en solución de cloruro sódico al 0,9 %, agua destilada u otro disolvente del que se haya demostrado que no tiene efectos negativos sobre el sistema de ensayo. En ciertas circunstancias y con una justificación científica adecuada, es posible someter a ensayo los sólidos sin diluir mediante aplicación directa sobre la superficie de la córnea, utilizando el método de cámara abierta (véase el punto 32). Las córneas se exponen a los sólidos durante cuatro horas, pero, como sucede con los líquidos y agentes tensioactivos, es posible utilizar otros tiempos de exposición si se cuenta con una explicación científica adecuada. Pueden utilizarse distintos métodos de tratamiento, en función de la naturaleza física y de las características químicas del producto problema (por ejemplo, sólidos, líquidos, líquidos viscosos frente a líquidos no viscosos). El factor crítico consiste en velar por que el producto problema cubra adecuadamente la superficie epitelial y se elimine adecuadamente durante las fases de lavado. Suele utilizarse un método de cámara cerrada cuando se estudian productos problema líquidos desde no viscosos hasta ligeramente viscosos, mientras que se usa en principio un método de cámara abierta con productos problema líquidos viscosos y semiviscosos, así como con sólidos sin diluir. En el método de cámara cerrada, se introduce en la cámara anterior (a través de los orificios de administración que se encuentran en la superficie superior de la cámara) una cantidad suficiente de producto problema (750 μl) para cubrir la cara epitelial de la córnea, y los agujeros se tapan a continuación con los tapones de la cámara durante la exposición. Es importante que cada córnea quede expuesta al producto problema durante el periodo adecuado. En el método de cámara abierta, antes del tratamiento se retiran el anillo de cierre de la ventana y la ventana de cristal de la cámara anterior. El producto problema o testigo (750 μl, o una cantidad suficiente de producto problema para cubrir completamente la córnea) se aplica directamente sobre la superficie epitelial de la córnea utilizando una micropipeta. Si es difícil pipetear el producto problema, este puede cargarse a presión en una pipeta de desplazamiento positivo para facilitar la dosificación. La punta de pipeta de la pipeta de desplazamiento positivo se inserta en la punta de dispensación de la jeringa, de forma que el producto problema pueda cargarse en la punta de desplazamiento bajo presión. Simultáneamente, se oprime el émbolo de la jeringa mientras se arrastra hacia arriba el pistón de la pipeta. Si aparece alguna burbuja de aire en la punta de la pipeta, se retira (se expulsa) el producto problema y se repite el proceso hasta que la punta se quede llena sin ninguna burbuja de aire. En caso necesario, puede utilizarse una jeringa normal (sin aguja), ya que permite medir un volumen exacto de producto problema y facilita su aplicación sobre la superficie epitelial de la córnea. Tras la aplicación del producto, se vuelve a colocar la ventana de cristal de la cámara anterior para crear de nuevo un sistema cerrado. Incubación tras la exposición Tras el periodo de exposición, el producto problema, el testigo negativo o el producto de testigo positivo se retiran de la cámara anterior y se lava el epitelio al menos tres veces (o hasta que no quede ningún rastro visible del producto problema) utilizando EMEM con rojo de fenol. Se utiliza para el lavado el medio con rojo de fenol porque la observación del cambio de color de esta sustancia permite determinar la efectividad del lavado de productos problema ácidos o alcalinos. Las córneas se lavan más de tres veces si el rojo de fenol sigue sin colorearse (amarillo o púrpura) o sigue viéndose el producto problema. Una vez el medio ha quedado libre de producto problema, se lavan las córneas una última vez con EMEM sin rojo de fenol. Se utiliza EMEM sin rojo de fenol en el lavado final para eliminar el rojo de fenol de la cámara anterior antes de medir la opacidad. La cámara anterior vuelve a llenarse a continuación con EMEM nuevo sin rojo de fenol. En el caso de líquidos o agentes tensioactivos, las córneas, una vez lavadas, se incuban otras dos horas a 32 ± 1 °C. En ciertas circunstancias puede ser útil prolongar este tiempo tras la exposición, extremo que debe estudiarse en cada caso. Las córneas tratadas con sólidos se lavan a fondo al final del periodo de exposición de cuatro horas, pero no es necesario prolongar su incubación. Al final del periodo de incubación tras la exposición en el caso de líquidos y agentes tensioactivos y al final del periodo de exposición de cuatro horas en el caso de sólidos que no son agentes tensioactivos, se registran la opacidad y la permeabilidad de cada córnea. Asimismo, se observa visualmente cada córnea y se registran las observaciones pertinentes (por ejemplo, abrasión tisular, producto problema residual, opacidad no uniforme). Estas observaciones pueden ser importantes ya que pueden reflejarse en variaciones de las lecturas del opacímetro. Productos testigo En cada experimento se incluyen testigos simultáneos negativos (o del disolvente/vehículo) y positivos. Cuando se somete a ensayo una sustancia líquida al 100 %, se incluye en el método de ensayo BCOP un testigo negativo en paralelo (por ejemplo, solución de cloruro sódico al 0,9 % o agua destilada), de forma que puedan detectarse los eventuales cambios inespecíficos del sistema de ensayo y se disponga de una base de referencia para los parámetros del ensayo. También permite asegurarse de que las condiciones del ensayo no provocan de forma inadecuada una respuesta de irritación. Cuando se somete a ensayo una sustancia líquida diluida, un agente tensioactivo o un sólido, se incluye en el método de ensayo BCOP un grupo de testigo en paralelo del disolvente/vehículo, de forma que puedan detectarse los eventuales cambios inespecíficos del sistema de ensayo y se disponga de una base de referencia para los parámetros del ensayo. Solo pueden utilizarse disolventes/vehículos de los que se haya demostrado que carecen de efectos adversos sobre el sistema de ensayo. Un producto del que se sabe que induce una respuesta positiva se incluye como testigo positivo en paralelo en cada experimento para verificar la integridad del sistema de ensayo y su correcta realización. Sin embargo, para asegurar la posibilidad de evaluar la variabilidad de las respuestas del testigo positivo a lo largo del tiempo, no debe ser excesiva la magnitud de la respuesta de irritación. Como ejemplos de testigos positivos para productos problema líquidos se pueden citar el etanol al 100 % y la dimetilformamida al 100 %. Un ejemplo de testigo positivo para productos problema sólidos es el imidazol al 20 % (p/v) en solución de cloruro sódico al 0,9 %. Es útil disponer de productos de referencia para evaluar el potencial de irritación ocular de productos desconocidos dentro de una clase específica de productos, o para evaluar la capacidad de irritación relativa de un irritante ocular dentro de una gama específica de respuestas de irritación. Parámetros medidos La opacidad se determina mediante la cantidad de luz transmitida a través de la córnea. La opacidad de la córnea se mide cuantitativamente con ayuda de un opacímetro, que permite medir los valores de opacidad en una escala continua. La permeabilidad se determina por la cantidad del pigmento fluoresceína sódica que penetra en todas las capas de células de la córnea (es decir, desde el epitelio de la superficie exterior de la córnea hasta el endotelio de su superficie interior). Se añade a la cámara anterior del soporte de córnea 1 ml de solución de fluoresceína sódica (4 o 5 mg/ml cuando se estudian líquidos y agentes tensioactivos o sólidos no tensioactivos, respectivamente), en contacto con el lado epitelial de la córnea, mientras que la cámara posterior, en contacto con el lado endotelial de la córnea, se llena con EMEM nuevo. El soporte se incuba a continuación en posición horizontal durante 90 ± 5 min a 32 ± 1 °C. La cantidad de fluoresceína sódica que atraviesa la córnea hasta la cámara posterior se mide cuantitativamente mediante espectrofotometría UV/VIS. Las mediciones espectrofotométricas evaluadas a 490 nm se registran como valores de absorbancia o densidad óptica (DO490), medidos en una escala continua. Los valores de permeabilidad a la fluoresceína se determinan utilizando los valores de DO490 conseguidos mediante un espectrofotómetro de luz visible con un camino óptico normal de 1 cm. También puede utilizarse un lector de placa de microvaloración de 96 pocillos siempre que: i) pueda establecerse la banda de linealidad del lector de placas para determinar los valores de DO490 de fluoresceína; y ii) se utilice el volumen correcto de muestras de fluoresceína en la placa de 96 pocillos para obtener unos valores de DO490 equivalentes a los conseguidos con un camino óptico normal de 1 cm [esto puede exigir que los pocillos estén completamente llenos (generalmente, 360 l)]. DATOS E INFORME Evaluación de los datos Una vez corregidos los valores de opacidad y de permeabilidad media (DO490) para tener en cuenta los valores de opacidad de fondo y de permeabilidad (DO490) del testigo negativo, los valores medios de opacidad y permeabilidad (DO490) se combinan en una fórmula empírica para calcular la puntuación de la capacidad de irritación in vitro (IVIS) de cada grupo de tratamiento, de la manera siguiente: IVIS = valor medio de opacidad + [15 × valor medio de permeabilidad (DO490)] Sina et al. (16) comunican que esta fórmula se ha obtenido mediante estudios internos e interlaboratorios. Los datos obtenidos con una serie de 36 compuestos en un estudio interlaboratorios se sometieron a análisis con múltiples variables para determinar la ecuación que se ajustara mejor a los datos in vivo e in vitro. Los científicos de dos empresas distintas realizaron este análisis y llegaron a ecuaciones casi idénticas. Los valores de opacidad y permeabilidad deben evaluarse también independientemente para determinar si un producto problema ha inducido corrosividad o irritación intensa mediante solo uno de los dos parámetros (véase la parte siguiente, sobre criterios de decisión). Criterios de decisión Los valores de corte de la IVIS para la identificación de productos problema que provocan lesiones oculares graves (categoría 1 del SGA de las Naciones Unidas) y productos problema que no requieren clasificación por irritación ocular o lesiones oculares graves (sin categoría del SGA de las Naciones Unidas) figuran a continuación:
Criterios de aceptación del estudio Se considera que un ensayo es aceptable si el testigo positivo da una IVIS comprendida en el intervalo de dos desviaciones típicas respecto a la media histórica del momento, que debe actualizarse al menos cada tres meses, o cada vez que se efectúe un ensayo aceptable en caso de laboratorios en los que no sea frecuente efectuar estos ensayos (es decir, con una frecuencia que no llegue a mensual). Las respuestas de los testigos negativos o del disolvente/vehículo deben llevar a unos valores de opacidad y permeabilidad inferiores a los límites superiores establecidos para los valores de opacidad y permeabilidad de fondo de las córneas de bovino tratadas con el respectivo testigo negativo o del disolvente/vehículo. Una sola tanda de ensayos, formada al menos por tres córneas, debería ser suficiente para un producto problema, si la clasificación resultante es inequívoca. Sin embargo, en casos de resultados dudosos en la primera tanda de ensayos, debe considerarse la realización de una segunda tanda de ensayos (aunque no sea necesariamente obligatoria), así como la de una tercera en caso de resultados medios de IVIS discordantes entre las dos primeras series de pruebas. En este contexto, un resultado de la primera tanda de ensayos se considera dudoso si las predicciones obtenidas de las tres córneas no concuerdan, de forma que:
Informe del ensayo El informe del ensayo debe incluir la siguiente información, en caso de que corresponda a la realización del estudio:
BIBLIOGRAFÍA
Apéndice 1 DEFINICIONES Exactitud : Grado de coincidencia entre los resultados obtenidos con el método de ensayo y los valores de referencia aceptados. Se trata de una medida del comportamiento del método de ensayo y es un aspecto de su “pertinencia”. Este término y el de “concordancia” se suelen usar indistintamente para indicar la proporción de resultados correctos de un método de ensayo. Producto de referencia : Producto utilizado como patrón para comparar con un producto problema. Los productos de referencia deben presentar las siguientes propiedades: i) un origen coherente y fiable; ii) similitud estructural y funcional con la clase de productos problema; iii) características físicas y químicas conocidas; iv) datos de apoyo sobre los efectos conocidos, y v) potencia conocida en la gama de respuestas deseada. Planteamiento ascendente : Enfoque gradual utilizado para un producto del que se sospecha que no requiere clasificación por irritación ocular o lesiones oculares graves, que comienza con la determinación de los productos que no requieren clasificación (resultado negativo) frente a otros productos (resultado positivo). Producto : Sustancia o mezcla. Córnea : Parte transparente delantera del globo ocular que cubre el iris y la pupila y permite el paso de la luz al interior. Opacidad de la córnea : Medida del grado de opacidad de la córnea tras su exposición a un producto problema. Un aumento de la opacidad de la córnea indica que esta ha sufrido una lesión. La opacidad puede evaluarse subjetivamente, como se hace en el ensayo con ojo de conejo de Draize, u objetivamente con un instrumento como un “opacímetro”. Permeabilidad de la córnea : Medida cuantitativa de la lesión del epitelio corneal mediante determinación de la cantidad del pigmento fluoresceína sódica que pasa a través de todas las capas de células corneales. Irritación ocular : Producción de cambios en el ojo, como consecuencia de la aplicación de un producto problema en la superficie anterior del ojo, que son totalmente reversibles en los 21 días siguientes a la aplicación. Intercambiable con “efectos reversibles en el ojo” y con “categoría 2 del SGA de las Naciones Unidas” (4). Tasa de falsos negativos : Proporción de todos los productos positivos identificados erróneamente como negativos por un método de ensayo. Es uno de los indicadores del comportamiento del método de ensayo. Tasa de falsos positivos : Proporción de todos los productos negativos identificados erróneamente como positivos por un método de ensayo. Es uno de los indicadores del comportamiento del método de ensayo. Peligro : Propiedad inherente de un agente o situación que tiene capacidad para provocar efectos adversos cuando un organismo, sistema o (sub)población se expone a dicho agente. Puntuación de la capacidad de irritación in vitro (In Vitro Irritancy Score, IVIS) : Fórmula empírica utilizada en el método de ensayo BCOP mediante la cual se combinan en una sola puntuación in vitro por cada grupo de tratamiento los valores medios de opacidad y de permeabilidad de cada grupo de tratamiento. IVIS = valor medio de opacidad + (15 × valor medio de permeabilidad). Efectos irreversibles en el ojo : Véase la definición de “Lesiones oculares graves”. Mezcla : Mezcla o solución compuesta por dos o más sustancias que no reaccionan (4). Testigo negativo : Muestra replicada no tratada que contiene todos los componentes de un sistema de ensayo. Esta muestra se somete al mismo proceso que las muestras tratadas con producto problema y otras muestras testigo para determinar si el disolvente interactúa con el sistema de ensayo. Sin clasificar : Producto que no está clasificado por irritación ocular (categoría 2, 2A o 2B del SGA de las Naciones Unidas) ni por lesión ocular grave (categoría 1 del SGA de las Naciones Unidas). Intercambiable con “sin categoría del SGA de las Naciones Unidas”. Opacímetro : Instrumento utilizado para medir la opacidad de la córnea evaluando cuantitativamente la transmisión de la luz a través de esta. El instrumento clásico tiene dos compartimentos, cada uno provisto de su propia fuente de luz y una célula fotoeléctrica. Un compartimento se utiliza para la córnea tratada, y el otro para calibrar y poner a cero el instrumento. Se envía a una célula fotoeléctrica la luz de una lámpara halógena a través de un compartimento de control (cámara vacía sin ventanas ni líquido) y se compara con la luz enviada a una célula fotoeléctrica a través del compartimento experimental, que alberga la cámara que contiene la córnea. Se compara la diferencia en la transmisión luminosa a partir de las células fotoeléctricas y se presenta un valor numérico de opacidad en un indicador digital. Testigo positivo : Muestra replicada que contiene todos los componentes de un sistema de ensayo y que se trata con un producto del que se sabe que induce una respuesta positiva. Para asegurar la posibilidad de evaluar la variabilidad de las respuestas del testigo positivo a lo largo del tiempo, no debe ser excesiva la magnitud de la respuesta positiva. Efectos reversibles en el ojo : Véase “Irritación ocular”. Fiabilidad : Medida del grado en que un método de ensayo puede aplicarse de forma reproducible a lo largo del tiempo, en un mismo laboratorio y en distintos laboratorios, utilizando el mismo protocolo. Se evalúa calculando la reproducibilidad intra e interlaboratorios y la repetibilidad intralaboratorios. Lesiones oculares graves : Producción de una lesión tisular en el ojo o una degradación física severa de la vista, como consecuencia de la aplicación de un producto problema en la superficie anterior del ojo, y que no es totalmente reversible en los 21 días siguientes a la aplicación. Intercambiable con “efectos irreversibles en el ojo” y con “categoría 1 del SGA de las Naciones Unidas” (4). Testigo del disolvente/vehículo : Muestra no tratada que contiene todos los componentes de un sistema de ensayo, incluido el disolvente o vehículo, y que se somete al mismo proceso que las muestras tratadas con producto problema y otras muestras testigo a fin de determinar la respuesta de base correspondiente a las muestras tratadas con el producto problema disuelto en el mismo disolvente o vehículo. Cuando se somete a ensayo con un testigo negativo en paralelo, esta muestra pone de manifiesto también si el disolvente o vehículo interactúa con el sistema de ensayo. Sustancia : Elementos químicos y sus compuestos en estado natural u obtenidos mediante cualquier proceso de producción, incluidos los aditivos necesarios para mantener la estabilidad del producto y las eventuales impurezas derivadas del proceso empleado, pero excluidos los disolventes que se puedan separar sin influir en la estabilidad de la sustancia ni cambiar su composición (4). Tensioactivo : También denominado agente tensioactivo, es una sustancia, como un detergente, que puede reducir la tensión superficial de un líquido y, de este modo, permitir que forme espuma o penetre en los sólidos; se conoce también como agente humectante. Mezcla que contiene tensioactivos : En el contexto del presente método de ensayo, es una mezcla que contiene uno o más agentes tensioactivos a una concentración final > 5 %. Enfoque descendente : Enfoque gradual utilizado para un producto del que se sospecha que causa lesiones oculares graves, que comienza con la determinación de los productos que inducen lesiones oculares graves (resultado positivo) frente a otros productos (resultado negativo). Producto problema : Sustancia o mezcla estudiada con este método de ensayo. Estrategia de ensayos escalonados : Estrategia de ensayo por fases, en la que se revisa toda la información existente sobre un producto problema, siguiendo un orden especificado, en un proceso de ponderación de las pruebas en cada escalón, a fin de determinar si se dispone de información suficiente para tomar una decisión sobre la clasificación de un peligro, antes de pasar al escalón siguiente. Si puede establecerse la capacidad de irritación de un producto problema con la información disponible, no hace falta efectuar más ensayos. Si no puede establecerse la capacidad de irritación de un producto problema con la información disponible, se aplica un procedimiento secuencial de ensayos con animales por fases hasta que pueda efectuarse una clasificación inequívoca. Sistema Globalmente Armonizado de clasificación y etiquetado de productos químicos de las Naciones Unidas (SGA de la ONU) : Sistema que propone la clasificación de productos químicos (sustancias y mezclas) según tipos y niveles normalizados de peligros físicos, sanitarios y ambientales, y que hace referencia a los elementos correspondientes de comunicación, como pictogramas, palabras de advertencia, indicaciones de peligro, consejos de prudencia, y fichas de datos de seguridad, a efectos de proporcionar información sobre sus efectos adversos con el fin de proteger a la población (incluidos empresarios, trabajadores, transportistas, consumidores y personal de protección civil) y al medio ambiente (4). Categoría 1 del SGA de las Naciones Unidas : Véase la definición de “Lesiones oculares graves”. Categoría 2 del SGA de las Naciones Unidas : Véase “Irritación ocular”. Sin categoría del SGA de las Naciones Unidas : Productos que no cumplen los requisitos para clasificarse en la categoría 1 o 2 (2A o 2B) del SGA de las Naciones Unidas. Intercambiable con “sin clasificar”. Método de ensayo validado : Método de ensayo sobre el cual se han completado estudios de validación para determinar su pertinencia (incluida su exactitud) y su fiabilidad con un fin específico. Es importante señalar que un método de ensayo validado puede tener un comportamiento insuficiente en términos de exactitud y fiabilidad como para considerarse aceptable a efectos del fin propuesto. Ponderación de las pruebas : Proceso de consideración de los aspectos favorables y desfavorables de los distintos elementos de información a efectos de alcanzar y confirmar una conclusión en cuanto al peligro potencial de un producto problema. Apéndice 2 CAPACIDAD DE PREDICCIÓN DEL MÉTODO DE ENSAYO BCOP Cuadro 1 Capacidad de predicción del método BCOP para identificar los productos que inducen lesiones oculares graves (categoría 1 del SGA de la ONU o del CLP de la UE frente a no categoría 1 (categoría 2 + sin categoría); categoría I de la EPA de EE.UU. frente a no de categoría I (categoría II + categoría III + categoría IV)]
Cuadro 2: Capacidad de predicción del BCOP para identificar los productos que no requieren clasificación por irritación ocular ni por lesiones oculares graves («no irritantes») (sin categoría 1 del SGA de las Naciones Unidas o del CLP de la UE frente a no sin categoría (categoría 1 + categoría 2); categoría IV de la EPA de EE.UU. frente a no de categoría IV (categoría I + categoría II + categoría III)]
Apéndice 3 PRODUCTOS UTILIZADOS PARA DEMOSTRAR LA APTITUD DEL MÉTODO DE ENSAYO BCOP Antes de proceder al uso sistemático de este método de ensayo, los laboratorios deben demostrar su aptitud técnica, identificando correctamente la clasificación de los trece productos recomendados del cuadro 1 en cuanto al peligro para los ojos. Estos productos se seleccionaron para representar la gama de respuestas en cuanto a los peligros para los ojos sobre la base de los resultados del ensayo in vivo con ojo de conejo (TG 405) (17) y el sistema de clasificación SGA de las Naciones Unidas (es decir, categorías 1, 2A, 2B, o sin clasificar) (4). Otros criterios de selección fueron el que los productos estuvieran disponibles en el mercado, que tuvieran datos de referencia in vivo de alta calidad y también datos in vitro de alta calidad obtenidos con el método de ensayo BCOP. Se encuentran datos de referencia en el documento de resumen simplificado (3) y en el documento de revisión de fondo del ICCVAM sobre el método de ensayo BCOP (2)(18). Cuadro 1: Productos recomendados para demostrar la aptitud técnica con el método de ensayo BCOP
Apéndice 4 SOPORTE DE CÓRNEA PARA EL MÉTODO BCOP Los soportes de córnea para el método BCOP se hacen de material inerte, como polipropileno. Los soportes están formados por dos mitades (una cámara anterior y una cámara posterior), y tienen dos cámaras internas cilíndricas similares. Cada cámara está diseñada para albergar un volumen de unos 5 ml y termina en una ventana de cristal, a través de la cual se hacen las mediciones de la opacidad. Cada cámara interna tiene unas dimensiones de 1,7 cm de diámetro y 2,2 cm de profundidad (11). Para evitar fugas se utiliza una junta tórica situada en la cámara posterior. Las córneas se colocan, con el lado endotelial hacia abajo, sobre la junta tórica de las cámaras posteriores, y las cámaras anteriores se colocan sobre el lado epitelial de las córneas. Las cámaras se mantienen en su posición mediante tres tornillos de acero inoxidable situados en los bordes externos de la cámara. El extremo de cada cámara tiene una ventana de cristal, que puede retirarse para facilitar el acceso a la córnea. Para evitar fugas, se pone también una junta tórica entre la ventana de cristal y la cámara. En la parte superior de cada cámara hay dos agujeros que permiten introducir y retirar el medio y los productos problema. Se cierran con tapones de goma durante los periodos de tratamiento y de incubación. La transmisión de la luz a través de los soportes de córnea puede cambiar, ya que los efectos del desgaste o de la acumulación de residuos químicos específicos sobre las perforaciones de la cámara interior o en las ventanas de vidrio pueden afectar a la dispersión o a la reflectancia de la luz. La consecuencia puede ser que aumente o disminuya la transmisión de la luz de base (y a la inversa las lecturas de la opacidad de base) a través de los soportes de las córneas, y puede manifestarse como cambios notables en las mediciones previstas de la opacidad inicial de base de la córnea en distintas cámaras (es decir, los valores iniciales de opacidad de la córnea en distintos soportes de córnea específicos pueden diferir normalmente en más de 2 o 3 unidades de opacidad respecto a los valores de base). Cada laboratorio debe considerar el establecimiento de un programa de evaluación de los cambios en la transmisión de la luz a través de los soportes de córnea, dependiendo de la naturaleza de los productos problema y de la frecuencia de uso de las cámaras. Para fijar los valores de base, los soportes de córnea pueden comprobarse antes de su uso rutinario procediendo a la medición de los valores de opacidad de base (o de transmisión luminosa) de cámaras llenas con medio completo, sin córneas. Los soportes de córnea se comprueban periódicamente después en cuanto a los cambios en la transmisión de la luz en los períodos de utilización. Cada laboratorio puede determinar la frecuencia con la que se deberán comprobar los soportes de córnea, en función de los productos problema, la frecuencia de uso, y las observaciones de cambios en los valores de base de opacidad de la córnea. Si se observan cambios notables en la transmisión de la luz a través de los soportes de córnea, ha de considerarse la posibilidad de proceder al pulido o limpieza adecuados de la superficie interior de los soportes de córnea o a su sustitución. Soporte de córnea: Diagrama despiezado 
Apéndice 5 OPACÍMETRO El opacímetro es un dispositivo que mide la transmisión luminosa. Por ejemplo, en el dispositivo OP-KIT de Electro Design (Riom, Francia) utilizado en la validación del método de ensayo BCOP, se envía a una célula fotoeléctrica la luz de una lámpara halógena a través de un compartimento testigo (cámara vacía sin ventanas ni líquido) y se compara con la luz enviada a una célula fotoeléctrica a través del compartimento experimental, que alberga la cámara que contiene la córnea. Se compara la diferencia en la transmisión luminosa a partir de las células fotoeléctricas y se presenta un valor numérico de opacidad en un indicador digital. Se determina el número de unidades de opacidad. Es posible utilizar otros tipos de opacímetros con una configuración diferente (por ejemplo, que no requieran mediciones paralelas de los compartimentos testigo y experimental) cuando se haya demostrado que dan resultados similares a los del dispositivo validado. Las respuestas del opacímetro deben ser lineales a lo largo de una gama de lecturas de opacidad que incluya los valores de corte utilizados para las diferentes clasificaciones descritas por el modelo de predicción (es decir, hasta el valor de corte que determina la corrosividad / capacidad de irritación intensa). Para garantizar que las lecturas son lineales y exactas hasta la zona de 75-80 unidades de opacidad, es necesario calibrar el opacímetro utilizando una serie de calibradores. Los calibradores se introducen en la cámara de calibración (cámara para córneas diseñada para contener los calibradores) y se toman las lecturas en el opacímetro. La cámara de calibración está designada para mantener los calibradores a la misma distancia aproximadamente entre la luz y la célula fotoeléctrica a la que se colocarían las córneas durante las mediciones de la opacidad. Los valores de referencia y el punto de ajuste inicial dependen del tipo de dispositivo utilizado. La linealidad de las mediciones de la opacidad debe garantizarse mediante procedimientos adecuados (específicos de cada instrumento). Por ejemplo, para los dispositivos OP-KIT de Electro Design (Riom, Francia), el opacímetro se calibra en primer lugar a 0 unidades de opacidad utilizando la cámara de calibración sin ningún calibrador. A continuación se ponen en esta cámara tres calibradores diferentes, uno a uno, y se miden las opacidades correspondientes. A los calibradores 1, 2 y 3 deben corresponderles unas lecturas de opacidad iguales a sus valores establecidos de 75, 150, y 225 unidades de opacidad, respectivamente, ± 5 %. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
13) |
En la parte B, el capítulo B.48 se sustituye por el texto siguiente: «B.48. Método de ensayo de ojo de pollo aislado para identificar: i) productos que provocan lesiones oculares graves y ii) productos que no requieren clasificación por irritación ocular ni lesiones oculares graves INTRODUCCIÓN El presente método de ensayo reproduce las directrices de ensayo (TG) 438 de la OCDE (2013). El método de ensayo de ojo de pollo aislado (conocido por la siglas de su nombre en inglés, ICE), fue evaluado por el Comité de Coordinación Interagencias sobre la Validación de Métodos Alternativos estadounidense (Interagency Coordinating Committee on the Validation of Alternative Methods, ICCVAM), junto con el Centro Europeo para la Validación de Métodos Alternativos (European Centre for the Validation of Alternative Methods, ECVAM) y el Centro Japonés para la Validación de Métodos Alternativos (Japanese Center for the Validation of Alternative Methods, JaCVAM), en 2006 y 2010 (1) (2) (3). En la primera evaluación, se consideró que el ICE es un método de ensayo cientifícamente válido para su uso como ensayo de cribado para identificar productos (sustancias y mezclas) que provocan lesiones oculares graves (categoría 1), según se definen en el Sistema Globalmente Armonizado de clasificación y etiquetado de productos químicos (SGA) de las Naciones Unidas (1) (2) (4) y en el Reglamento (CE) n.o 1272/2008 del Parlamento Europeo y del Consejo, sobre clasificación, etiquetado y envasado de sustancias y mezclas (CLP) (12). En la segunda evaluación, el método de ensayo ICE se evaluó en cuanto a su utilidad como ensayo de cribado para identificar productos que no están clasificados por irritación ocular ni por lesiones oculares graves según se definen en el SGA de las Naciones Unidas (3) (4). Los resultados del estudio de validación y las recomendaciones del grupo de revisión por pares mantuvieron la recomendación original de utilizar el ICE para la clasificación de los productos que inducen lesiones oculares graves (categoría 1 del SGA de las Naciones Unidas), ya que la base de datos disponible se ha mantenido sin cambios desde la validación original por el ICCVAM. En esa fase, no se sugirieron nuevas recomendaciones de ampliación del ámbito de aplicación del ICE para incluir también otras categorías. Se llevó a cabo una reevaluación del conjunto de datos in vitro e in vivo utilizados en el estudio de validación, con el propósito de evaluar la utilidad del ICE para identificar productos que no requieren clasificación por irritación ocular o lesiones oculares graves (5). Esta reevaluación llegó a la conclusión de que el método de ensayo ICE puede utilizarse también para identificar productos que no requieren clasificación por irritación ocular o lesiones oculares graves según se definen en el SGA de las Naciones Unidas (4) (5). El presente método de ensayo incluye los usos recomendados y las limitaciones del método de ensayo ICE sobre la base de estas evaluaciones. Las principales diferencias entre la versión inicial de 2009 y la versión actualizada de 2013 de las directrices de ensayo de la OCDE incluyen, entre otros elementos, el uso del método de ensayo ICE para identificar productos que no requieren clasificación según el sistema de clasificación del SGA de las Naciones Unidas, una actualización de varios aspectos del informe de ensayo, una actualización del apéndice 1 sobre definiciones, y una actualización del apéndice 2 sobre los productos de la prueba de aptitud. Actualmente se reconoce en general que, en un futuro próximo, ningún ensayo de irritación ocular in vitro podrá sustituir por sí solo al ensayo ocular de Draize in vivo para predecir toda la gama de irritación causada por las distintas categorías de productos. Sin embargo, unas combinaciones estratégicas de varios métodos de ensayo alternativos dentro de una estrategia de ensayos (escalonados) sí podrán sustituir al ensayo ocular de Draize (6). El enfoque descendente (7) está diseñado para utilizarse cuando, sobre la base de la información existente, se espera de un producto que tenga una alta capacidad de irritación, mientras que el enfoque ascendente (7) está diseñado para utilizarse cuando, sobre la base de la información existente, se espera de un producto que no cause irritación ocular suficiente para exigir una clasificación. El método de ensayo ICE es un procedimiento in vitro que puede utilizarse, en determinadas circunstancias y con limitaciones específicas como se contempla en los puntos 8 a 10, con fines de clasificación de peligros para los ojos y de etiquetado de productos. Si bien no se considera válido para sustituir por sí solo al ensayo in vivo con ojo de conejo, el método de ensayo ICE se recomienda como fase inicial en una estrategia de ensayo tal como el enfoque descendente sugerido por Scott et al. (7) para identificar los productos que provocan lesiones oculares graves, es decir, los productos que han de clasificarse en la categoría 1 del SGA de las Naciones Unidas, sin necesidad de hacer más pruebas (4). El método de ensayo ICE se recomienda también para identificar productos que no requieren clasificación por irritación ocular ni lesiones oculares graves, según se definen en el SGA de las Naciones Unidas (sin categoría) (4), por lo que puede utilizarse como fase inicial dentro de una estrategia de ensayos de enfoque ascendente (7). No obstante, un producto del que no se espere que cause lesiones oculares graves o que no esté clasificado por irritación ocular o lesiones oculares graves según el método de ensayo ICE tendría que someterse a ensayos adicionales (in vitro o in vivo) para establecer una clasificación definitiva. Además, las autoridades reguladoras competentes deben ser consultadas antes de utilizar el ICE en un enfoque ascendente en relación con otros regímenes de clasificación distintos del SGA de las Naciones Unidas. El objetivo del presente método de ensayo es describir los procedimientos utilizados para evaluar el posible peligro para los ojos de un producto problema en función de su capacidad de inducir o no toxicidad para un ojo enucleado de pollo. Los efectos tóxicos sobre la córnea se miden mediante: i) una evaluación cualitativa de la opacidad, ii) una evaluación cualitativa de las lesiones provocadas al epitelio sobre la base de la aplicación de fluoresceína en el ojo (retención de fluoresceína), iii) una medición cuantitativa del aumento del espesor (inflamación), y iv) una evaluación cualitativa de las lesiones morfológicas macroscópicas de la superficie. Las evaluaciones de la opacidad, la inflamación y las lesiones de la córnea tras la exposición de esta a un producto problema se efectúan por separado y después se combinan para llegar a una clasificación de la capacidad de irritación ocular. En el apéndice 1 se dan las definiciones pertinentes. CONSIDERACIONES INICIALES Y LIMITACIONES El presente método de ensayo se basa en el protocolo sugerido en el documento de orientación de la OCDE 160 (8), elaborado siguiendo el estudio internacional de validación del ICCVAM (1) (3) (9), con aportaciones del Centro Europeo para la Validación de Métodos Alternativos, del Centro Japonés para la Validación de Métodos Alternativos, y del Departamento de Toxicología y Farmacología Aplicada de Calidad de la Vida de TNO de los Países Bajos (TNO Quality of Life Department of Toxicology and Applied Pharmacology). El protocolo se basa en la información obtenida a partir de protocolos publicados, así como del protocolo actualmente utilizado por TNO (10) (11) (12) (13) (14). Se ha estudiado una amplia gama de productos en la validación subyacente al presente método de ensayo, y la base de datos empíricos del estudio de validación contaba con 152 productos, de los que 72 eran sustancias y 80, mezclas (5). El método de ensayo es aplicable a sólidos, líquidos, emulsiones y geles. Los líquidos pueden ser acuosos o no acuosos; los sólidos pueden ser solubles o insolubles en agua. Aún no se ha efectuado ningún estudio de validación con gases y aerosoles. El método de ensayo ICE puede utilizarse para identificar los productos que provocan lesiones oculares graves, es decir, que deben clasificarse como productos de la categoría 1 del SGA de las Naciones Unidas (4). Cuando se utiliza para este fin, las limitaciones que se han detectado en el método de ensayo ICE están relacionadas con las elevadas tasas de falsos resultados positivos de alcoholes y con las elevadas tasas de falsos resultados negativos de sólidos y tensioactivos (1) (3) (9). Sin embargo, las tasas de falsos negativos en este contexto (productos de categoría 1 del SGA de las Naciones Unidas identificados como que no pertenecen a esta categoría) no son críticas, ya que todos los productos problema que dan resultado negativo se someten posteriormente a otro u otros ensayos in vitro validados adecuadamente, o como última opción con conejos, en función de las exigencias normativas, utilizando una estrategia de ensayo secuencial en un enfoque de ponderación de las pruebas. Cabe señalar que los sólidos pueden dar lugar a condiciones de exposición variables y extremas en el ensayo in vivo de irritación ocular de Draize, lo cual puede derivar en resultados poco pertinentes sobre el verdadero potencial de irritación (15). Los investigadores pueden considerar la utilización de este método de ensayo con todos los tipos de productos, aceptándose un resultado positivo como indicativo de lesiones oculares graves, es decir, de clasificación en la categoría 1 del SGA de las Naciones Unidas sin ensayos adicionales. Sin embargo, los resultados positivos obtenidos con alcoholes deben interpretarse con precaución, debido al riesgo de sobrepredicción. Cuando se utiliza para identificar productos que inducen lesiones oculares graves (categoría 1 del SGA de las Naciones Unidas), el método de ensayo ICE tiene una exactitud general del 86 % (120/140), una tasa de falsos positivos del 6 % (7/113), y una tasa de falsos negativos del 48 % (13/27), cuando se compara con los datos del método de ensayo in vivo con ojo de conejo, clasificados según el sistema de clasificación del SGA de las Naciones Unidas (4) (5). El método de ensayo ICE puede utilizarse también para identificar productos que no requieren clasificación por irritación ocular o lesiones oculares graves con arreglo al sistema de clasificación del SGA de las Naciones Unidas (4). Las autoridades reguladoras competentes deben ser consultadas antes de utilizar el ICE en un enfoque ascendente en relación con otros regímenes de clasificación. Este método de ensayo puede utilizarse con todos los tipos de productos, aceptándose un resultado negativo para no clasificar el producto por irritación ocular o lesiones oculares graves. Sin embargo, sobre la base de un solo resultado a partir de la base de datos de la validación, puede haber riesgo de infrapredicción de las pinturas antiincrustantes que contienen disolventes orgánicos (5). Cuando se utiliza para identificar productos que no requieren clasificación por irritación ocular ni lesiones oculares graves, el método de ensayo ICE tiene una exactitud general del 82 % (125/152), una tasa de falsos positivos del 33 % (26/79), y una tasa de falsos negativos del 1 % (1/73), cuando se compara con los datos del método de ensayo in vivo con ojo de conejo, clasificados según el SGA de las Naciones Unidas (4) (5). Cuando los productos problema de ciertas clases (en concreto, pinturas antiincrustantes que contienen disolventes orgánicos) se excluyen de la base de datos, el método de ensayo ICE tiene una exactitud del 83 % (123/149), una tasa de falsos positivos del 33 % (26/78), y una tasa de falsos negativos del 0 % (0/71) para el sistema de clasificación del SGA de las Naciones Unidas (4) (5). El método de ensayo ICE no se recomienda para la identificación de productos problema que deben clasificarse como irritantes oculares (es decir, de categoría 2 o categoría 2A del SGA de las Naciones Unidas) o de productos problema que deben clasificarse como moderadamente irritantes para los ojos (categoría 2B del SGA de las Naciones Unidas), dado el considerable número de productos de la categoría 1 del SGA de las Naciones Unidas clasificados de forma insuficiente en las categorías 2, 2A o 2B del SGA de las Naciones Unidas y de productos sin categoría del SGA de las Naciones Unidas clasificados de forma excesiva en las categorías 2, 2A o 2B del SGA de las Naciones Unidas. A tal fin, pueden resultar necesarias pruebas adicionales con otro método adecuado. Todos los procedimientos con ojos de pollo deben ajustarse a las normas y procedimientos aplicables de la instalación de ensayo en relación con la manipulación de materiales derivados del hombre o de animales, entre los que figuran los tejidos o los líquidos tisulares. Se recomienda observar las precauciones universales de laboratorio (16). Si bien el método de ensayo ICE no tiene en cuenta las lesiones de la conjuntiva y del iris que se evalúan en el método de ensayo de irritación ocular del conejo, sí atiende a los efectos en la córnea, que son un factor de primer orden de la clasificación in vivo cuando se considera la clasificación del SGA de las Naciones Unidas. Asimismo, aunque la reversibilidad de las lesiones de la córnea no puede evaluarse per se con el método de ensayo ICE, se ha propuesto, sobre la base de los estudios en ojo de conejo, que la evaluación de la profundidad inicial de la lesión de la córnea puede utilizarse para identificar ciertos tipos de efectos irreversibles (17). En particular, se necesitan más conocimientos científicos para comprender cómo se dan efectos irreversibles no relacionados con lesiones iniciales de alto nivel. Finalmente, el método de ensayo ICE no permite la evaluación del potencial de toxicidad sistémica asociado con la exposición ocular. El presente método de ensayo se actualizará periódicamente a la luz de la nueva información y los datos que se vayan considerando. Por ejemplo, la histopatología puede ser útil si se necesita una caracterización más completa de las lesiones de la córnea. Para evaluar esta posibilidad, se insta a los usuarios a conservar los ojos y a preparar muestras de histopatología que puedan utilizarse para compilar una base de datos y criterios de decisión con los que se pueda seguir mejorando la exactitud de este método. La OCDE ha elaborado un documento de orientación sobre el uso de métodos de ensayo de la toxicidad ocular in vitro, que incluye procedimientos detallados sobre la recogida de muestras de histopatología, así como información sobre dónde presentar las muestras o los datos de histopatología (8). Cuando algún laboratorio establezca inicialmente este ensayo, deben utilizarse los productos para demostrar la aptitud que se indican en el apéndice 2. Los laboratorios pueden utilizar estos productos para demostrar su competencia técnica en la aplicación del método de ensayo ICE antes de presentar, con fines de clasificación normativa de peligros, los datos obtenidos con este ensayo. PRINCIPIO DEL ENSAYO El método de ensayo ICE es un modelo organotípico con mantenimiento a corto plazo del ojo de pollo in vitro. En este método de ensayo, las lesiones provocadas por el producto problema se evalúan determinando la inflamación, la opacidad y la retención de fluoresceína en la córnea. Mientras que los dos últimos parámetros implican una evaluación cualitativa, el análisis de la inflamación de la córnea permite una evaluación cuantitativa. Cada medición se convierte en una puntuación cuantitativa utilizada para calcular un índice general de irritación, o bien se somete a una categorización cualitativa para asignarle una clasificación de peligro ocular in vitro, bien en la categoría 1 del SGA de las Naciones Unidas o bien sin clasificar en el SGA de las Naciones Unidas. Cualquiera de estos resultados puede utilizarse a continuación para predecir el potencial de causar lesiones oculares graves in vivo o la ausencia de requisito de clasificación en cuanto al peligro para los ojos de un producto problema (véanse los criterios de decisión). No obstante, no se puede dar ninguna clasificación a los productos de los que no se predice que causan lesiones oculares graves o que no se clasifican con el método de ensayo ICE (véase el punto 11). Fuente y edad de los ojos de pollo Tradicionalmente se han venido utilizando para este ensayo los ojos tomados de pollos obtenidos de mataderos donde se sacrifican para el consumo humano, con lo que se suprime la necesidad de disponer de animales de laboratorio. Solo se utilizan los ojos de animales sanos considerados adecuados para entrar en la cadena alimentaria humana. Aunque aún no se ha efectuado ningún estudio comparativo para evaluar la edad óptima de los pollos, la edad y el peso de los pollos usados tradicionalmente en este método de ensayo corresponden a los pollos tiernos que se suelen tratar en los mataderos de aves de corral (es decir, de unas siete semanas de edad y entre 1, 5 y 2,5 kg). Recogida y transporte de los ojos al laboratorio Las cabezas deben retirarse inmediatamente después de la sedación de los pollos, generalmente por choque eléctrico, y de la incisión del cuello para su sangrado. Las fuentes locales de pollos deben estar situadas cerca del laboratorio de forma que las cabezas puedan transferirse desde el matadero al laboratorio en poco tiempo, a fin de reducir al mínimo el deterioro y la contaminación bacteriana. Debe reducirse al mínimo el tiempo transcurrido entre la recogida de las cabezas de pollo y la colocación de los ojos en la cámara de superfusión tras su enucleación (lo normal es que este plazo no exceda de dos horas) a fin de garantizar que se cumplen los criterios de aceptación del ensayo. Todos los ojos utilizados en el ensayo deben proceder del mismo grupo de ojos recogidos un día concreto. Como los ojos se disecan en el laboratorio, las cabezas intactas se transportan desde el matadero a temperatura ambiente (normalmente entre 18 °C y 25 °C) en cajas de plástico humidificadas mediante telas mojadas con solución salina isotónica. Criterios de selección y número de los ojos utilizados en el ICE Se desechan los ojos que tienen una elevada tinción de fluoresceína de base (es decir, > 0,5) o una elevada puntuación de opacidad de la córnea (es decir, > 0,5) una vez han sido enucleados. Cada grupo de tratamiento y de testigo positivo en paralelo consiste en al menos tres ojos. El grupo de testigo negativo o el testigo del disolvente (si se utiliza un disolvente distinto de la solución salina) consiste en al menos un ojo. En el caso de materiales sólidos que dan un resultado de sin clasificar según el SGA, se recomienda realizar el ensayo con una segunda tanda de tres ojos para confirmar o descartar el resultado negativo. PROCEDIMIENTO Preparación de los ojos Se cortan los párpados cuidadosamente, procurando no dañar la córnea. Se evalúa rápidamente la integridad de la córnea mediante la aplicación de una gota de fluoresceína sódica al 2 % (p/v) a la superficie de la córnea durante unos segundos, seguida de lavado con solución salina isotónica. Los ojos tratados con fluoresceína se examinan a continuación con un microscopio de lámpara de hendidura para asegurarse de que la córnea está ilesa (es decir, tiene puntuaciones de retención de fluoresceína y de opacidad de la córnea ≤ 0,5). Los ojos que no presenten lesiones se extraen del cráneo, teniendo cuidado de no lesionar la córnea. El globo ocular se extrae de la órbita sujetando firmemente con pinzas quirúrgicas la membrana nictitante, y los músculos del ojo se cortan con unas tijeras curvas de punta roma. Es importante no causar lesiones a la córnea por una presión excesiva (es decir, artefactos de compresión). Cuando se retira el ojo de la órbita, debe quedar con él una porción visible del nervio óptico. Una vez retirado de la órbita, el ojo se pone en una gasa hidrófila y se retiran, cortándolos, la membrana nictitante y demás tejidos conjuntivos. El ojo enucleado se monta en un soporte de acero inoxidable, con la córnea situada verticalmente. El soporte se transfiere a continuación a la cámara de un aparato de superfusión (18), donde se posiciona de manera que toda la córnea reciba el goteo de solución salina isotónica (3-4 gotas por minuto o 0.1-0,15 ml/min). Las cámaras de los aparatos de superfusión deben estar termostatizadas a 32 ± 1,5 °C. El apéndice 3 incluye un diagrama de un aparato de superfusión normal y del soporte para los ojos, que pueden obtenerse comercialmente o bien construirse en el propio laboratorio. El aparato puede modificarse para cubrir las necesidades de un laboratorio concreto (por ejemplo, para albergar un número diferente de ojos). Una vez colocados en el aparato de superfusión, los ojos se vuelven a examinar con un microscopio de lámpara de hendidura para asegurarse de que no han sufrido ninguna lesión durante el proceso de disección. También debe medirse en este momento el espesor de la córnea en el vértice corneal, utilizando el medidor de profundidad con el microscopio de lámpara de hendidura. Deben sustituirse los ojos que presenten: i) una puntuación de retención de fluoresceína > 0,5; ii) una opacidad corneal > 0,5; o iii) cualquier otro signo de lesión. En cuanto a los ojos que no se hayan rechazado por ninguno de estos criterios, deberán rechazarse también los ojos con un espesor de la córnea que se desvíe en más de un 10 % del valor medio de todos los ojos. Los usuarios deben ser conscientes de que con los microscopios de lámpara de hendidura pueden obtenerse diferentes mediciones del espesor de la córnea con diferentes ajustes de la anchura de la hendidura. Esta debe fijarse en 0,095 mm. Una vez examinados y aprobados todos los ojos, se incuban durante unos 45 o 60 minutos para estabilizarlos con el sistema de ensayo antes de administrar el producto. Tras el periodo de estabilización, se registra una medición de referencia inicial del espesor y de la opacidad de la córnea que sirva como referencia de base (es decir, a tiempo = 0). La puntuación de fluoresceína determinada en el momento de la disección se utiliza como medición de base respecto a este parámetro. Aplicación del producto problema Inmediatamente después de efectuar las mediciones de referencia iniciales, se retira el ojo (en su soporte) del aparato de superfusión, se pone en posición horizontal y se aplica a la córnea el producto problema. Normalmente, los productos problema líquidos se estudian sin diluir, pero, en caso necesario, pueden someterse a dilución (por ejemplo, como parte del diseño del estudio). El disolvente preferido para disolver los productos problema es la solución salina fisiológica. Sin embargo, también pueden utilizarse otros disolventes en condiciones controladas, pero es necesario demostrar la idoneidad de los disolventes que sean distintos de la solución salina fisiológica. Los productos problema líquidos se aplican a la córnea de forma que quede cubierta con el producto problema toda la superficie de la córnea de forma homogénea; el volumen normal es de 0,03 ml. Cuando sea posible, los productos problema sólidos deben triturarse de la manera más fina posible con un mortero o con otro instrumento de trituración comparable. El polvo resultante se aplica a la córnea de forma que su superficie quede cubierta uniformemente con el producto problema; la cantidad normal es de 0,03 g. El producto problema (líquido o sólido) se aplica durante 10 segundos y después se retira del ojo lavando con solución salina isotónica (aproximadamente 20 ml) a temperatura ambiente. El ojo (en su soporte) se vuelve a poner a continuación en el aparato de superfusión, en la posición vertical inicial. En caso necesario, podrá efectuarse un lavado adicional tras la aplicación de 10 segundos y en momentos posteriores (por ejemplo, tras el descubrimiento de residuos del producto problema en la córnea). En general, la cantidad de solución salina utilizada de forma adicional para el lavado no es determinante, pero sí es importante la observación de la adherencia del producto problema a la córnea. Productos testigo En cada experimento se deben incluir testigos en paralelo negativos (o del disolvente/vehículo) y positivos. Cuando se estudian líquidos al 100 % o sólidos, se utiliza solución salina fisiológica como testigo negativo en paralelo en el método de ensayo ICE a fin de detectar cambios inespecíficos del sistema de ensayo y garantizar que las condiciones del ensayo no provocan inadecuadamente una respuesta de irritación. Cuando se estudian líquidos diluidos, se incluye en el método de ensayo un grupo de testigo en paralelo del disolvente/vehículo a fin de detectar cambios inespecíficos del sistema de ensayo y garantizar que las condiciones del ensayo no provocan inadecuadamente una respuesta de irritación. Como se indica en el punto 31, solo pueden utilizarse los disolventes/vehículos de los que se haya demostrado que carecen de efectos adversos sobre el sistema de ensayo. Se incluye como testigo positivo en paralelo en cada experimento un agente irritante ocular conocido, a fin de verificar que se provoca una respuesta adecuada. Como en el presente método se utiliza el ensayo ICE para identificar agentes corrosivos o irritantes intensos, el testigo positivo debe ser un producto de referencia que induzca una respuesta intensa con este método de ensayo. No obstante, para asegurar la posibilidad de evaluar la variabilidad de las respuestas del testigo positivo a lo largo del tiempo, no debe ser excesiva la intensidad de la respuesta. Debe generarse una cantidad suficiente de datos in vitro del testigo positivo, de forma que se pueda calcular una banda aceptable, definida estadísticamente, para el testigo positivo. Si no se dispone de datos anteriores adecuados con el método de ensayo ICE respecto a un testigo positivo determinado, es posible que deban efectuarse estudios para conseguir esta información. Pueden citarse como ejemplo de testigos positivos para productos problema líquidos el ácido acético al 10 % o el cloruro de benzalconio al 5 %, mientras que son ejemplos de testigos positivos para productos problema sólidos el hidróxido sódico o el imidazol. Es útil disponer de productos de referencia para evaluar el potencial de irritación ocular de productos desconocidos dentro de una clase específica de productos, o para evaluar la capacidad de irritación relativa de un irritante ocular dentro de una gama específica de respuestas de irritación. Parámetros medidos Las córneas tratadas se evalúan antes de su tratamiento y a los 30, 75, 120, 180 y 240 minutos (± 5 minutos) después del lavado tras el tratamiento. Esta cadencia permite conseguir un número adecuado de mediciones a lo largo del periodo de tratamiento de cuatro horas, dejando tiempo suficiente entre mediciones para que puedan efectuarse las observaciones requeridas con todos los ojos. Los parámetros evaluados son la opacidad, la inflamación, la retención de fluoresceína y los efectos morfológicos (por ejemplo, lesiones punteadas o aflojamiento del epitelio) de la córnea. A cada uno de los tiempos antes citados se determinan todos los parámetros, salvo la retención de fluoresceína (que se determina solo antes del tratamiento y a los 30 minutos tras la exposición al producto problema). Se recomienda tomar fotografías para documentar la opacidad, la retención de fluoresceína, los efectos morfológicos y, en su caso, la histopatología de la córnea. Tras el examen final a las cuatro horas, se insta a los usuarios a conservar los ojos en un fijador adecuado (por ejemplo, formol neutro amortiguado) para su eventual examen histopatológico; pueden verse más detalles en el punto 14 y la referencia (8). La inflamación de la córnea se determina mediante mediciones del espesor de la córnea efectuadas con un paquímetro óptico en un microscopio de lámpara de hendidura. Se expresa en porcentaje y se calcula con la fórmula siguiente a partir de las mediciones del espesor de la córnea:
Se calcula el porcentaje medio de inflamación de la córnea de todos los ojos del ensayo a todos los tiempos de observación. A partir de la mayor puntuación media de inflamación de la córnea, observada a cualquiera de los tiempos seleccionados, se adjudica a cada producto problema una puntuación global de categoría (véase el punto 51). La opacidad de la córnea se evalúa utilizando el área de esta que se encuentra opacificada más densamente para establecer la puntuación tal como se muestra en el cuadro 1. Se calcula el valor medio de opacidad de la córnea de todos los ojos del ensayo a todos los tiempos de observación. A partir de la mayor puntuación media de opacidad de la córnea, observada a cualquiera de los tiempos seleccionados, se adjudica a cada producto problema una puntuación global de categoría (véase el punto 51). Cuadro 1. Puntuaciones de opacidad de la córnea.
La retención de fluoresceína se evalúa al tiempo de observación de 30 minutos únicamente, como se muestra en el cuadro 2. El valor medio de retención de fluoresceína de todos los ojos del ensayo se calcula entonces al tiempo de observación de 30 minutos, y se utiliza para obtener la puntuación global de categoría asignada a cada producto problema (véase el punto 51). Cuadro 2. Puntuaciones de retención de fluoresceína.
Entre los efectos morfológicos figuran el «punteamiento» de las células del epitelio corneal, el «aflojamiento» del epitelio, la «rugosidad» de la superficie corneal y el «pegado» del producto problema a la córnea. Estas observaciones pueden presentar una intensidad variable y darse simultáneamente. La clasificación de estas observaciones es subjetiva, según la interpretación del investigador. DATOS E INFORME Evaluación de los datos Los resultados de la opacidad de la córnea, de la inflamación y de la retención de fluoresceína deben evaluarse por separado a fin de llegar a una clasificación ICE para cada parámetro. Las clases ICE de cada parámetro se combinan a continuación para generar una clasificación según la capacidad de irritación de cada producto problema. Criterios de decisión Una vez evaluado cada uno de los parámetros, pueden asignarse las clases ICE en función de unas bandas predeterminadas. La interpretación de la inflamación (cuadro 3), la opacidad (cuadro 4), y la retención de fluoresceína (cuadro 5) de la córnea utilizando cuatro clases ICE se efectúa según las siguientes escalas. Es importante señalar que las puntuaciones de inflamación de la córnea que figuran en el cuadro 3 se pueden aplicar solo si el espesor se mide con un microscopio de lámpara de hendidura (por ejemplo, Haag-Streit BP900) con dispositivo medidor de profundidad no 1 y ajuste de la anchura de la hendidura a 9 Cuadro 3. Criterios de clasificación ICE según la inflamación de la córnea.
Cuadro 4. Criterios de clasificación ICE según la opacidad.
Cuadro 5. Criterios de clasificación ICE según la retención media de fluoresceína.
La clasificación in vitro de un producto problema se evalúa leyendo la clasificación del SGA correspondiente a la combinación de categorías obtenidas con la inflamación de la córnea, la opacidad de la córnea y la retención de fluoresceína, tal como se describe en el cuadro 6. Cuadro 6. Clasificaciones in vitro globales.
Criterios de aceptación del estudio Se considera que un ensayo es aceptable si los testigos negativos o del vehículo/disolvente en paralelo y los testigos positivos en paralelo se identifican como sin clasificar en el SGA y en la categoría 1 del SGA, respectivamente. Informe del ensayo El informe del ensayo debe incluir la siguiente información, en caso de que corresponda a la realización del estudio:
BIBLIOGRAFÍA
Apéndice 1 DEFINICIONES Exactitud : Grado de coincidencia entre los resultados obtenidos con el método de ensayo y los valores de referencia aceptados. Se trata de una medida del comportamiento del método de ensayo y es un aspecto de su “pertinencia”. Este término y el de “concordancia” se suelen usar indistintamente para indicar la proporción de resultados correctos de un método de ensayo. Producto de referencia : Producto utilizado como patrón para comparar con un producto problema. Los productos de referencia deben presentar las siguientes propiedades: i) un origen coherente y fiable; ii) similitud estructural y funcional con la clase de productos problema; iii) características físicas y químicas conocidas; iv) datos de apoyo sobre los efectos conocidos; y v) potencia conocida en la banda de la respuesta deseada. Planteamiento ascendente : Enfoque gradual utilizado para un producto del que se sospecha que no requiere clasificación por irritación ocular o lesiones oculares graves, que comienza con la determinación de los productos que no requieren clasificación (resultado negativo) frente a otros productos (resultado positivo). Producto : Sustancia o mezcla. Córnea : Parte transparente delantera del globo ocular que cubre el iris y la pupila y permite el paso de la luz al interior. Opacidad de la córnea : Medida del grado de opacidad de la córnea tras su exposición a un producto problema. Un aumento de la opacidad de la córnea indica que esta ha sufrido una lesión. Inflamación de la córnea : Medición objetiva en el ensayo ICE de la magnitud de la dilatación de la córnea tras su exposición a un producto problema. Se expresa en porcentaje y se calcula a partir de las mediciones del espesor de la córnea de base (antes de la administración del producto) y de los espesores registrados a intervalos regulares tras la exposición al producto problema en el ensayo ICE. El grado de inflamación de la córnea indica las lesiones que esta ha sufrido. Irritación ocular : Producción de cambios en el ojo, como consecuencia de la aplicación de un producto problema en la superficie anterior del ojo, que son totalmente reversibles en los 21 días siguientes a la aplicación. Intercambiable con “efectos reversibles en el ojo” y con “categoría 2 del SGA de las Naciones Unidas” (4). Tasa de falsos negativos : Proporción de todos los productos positivos identificados erróneamente como negativos por un método de ensayo. Es uno de los indicadores del comportamiento del método de ensayo. Tasa de falsos positivos : Proporción de todos los productos negativos identificados erróneamente como positivos por un método de ensayo. Es uno de los indicadores del comportamiento del método de ensayo. Retención de fluoresceína : Medición subjetiva en el ensayo ICE de la cantidad de fluoresceína sódica retenida por las células epiteliales de la córnea tras la exposición de esta a una sustancia problema. El grado de retención de fluoresceína indica las lesiones que ha sufrido el epitelio de la córnea. Peligro : Propiedad inherente de un agente o situación que tiene capacidad para provocar efectos adversos cuando un organismo, sistema o (sub)población se expone a dicho agente. Efectos irreversibles en el ojo : Véanse “lesiones oculares graves” y “categoría 1 del SGA de las Naciones Unidas”. Mezcla : Mezcla o solución compuesta por dos o más sustancias que no reaccionan (4). Testigo negativo : Muestra replicada no tratada que contiene todos los componentes de un sistema de ensayo. Esta muestra se somete al mismo proceso que las muestras tratadas con producto problema y otras muestras testigo para determinar si el disolvente interactúa con el sistema de ensayo. Sin clasificar : Sustancia que no está clasificada por irritación ocular (categoría 2 del SGA de las Naciones Unidas) ni por lesión ocular grave (categoría 1 del SGA de las Naciones Unidas). Intercambiable con “sin categoría del SGA de las Naciones Unidas”. Testigo positivo : Muestra replicada que contiene todos los componentes de un sistema de ensayo y que se trata con un producto del que se sabe que induce una respuesta positiva. Para asegurar la posibilidad de evaluar la variabilidad de las respuestas del testigo positivo a lo largo del tiempo, no debe ser excesiva la intensidad de la respuesta. Fiabilidad : Medida del grado en que un método de ensayo puede aplicarse de forma reproducible a lo largo del tiempo, en un mismo laboratorio y en distintos laboratorios, utilizando el mismo protocolo. Se evalúa calculando la reproducibilidad intra e interlaboratorios y la repetibilidad intralaboratorios. Efectos reversibles en el ojo : Véanse “irritación ocular” y “categoría 2 del SGA de las Naciones Unidas”. Lesiones oculares graves : Producción de una lesión tisular en el ojo o una degradación física severa de la vista, como consecuencia de la aplicación de un producto problema en la superficie anterior del ojo, y que no es totalmente reversible en los 21 días siguientes a la aplicación. Intercambiable con “efectos irreversibles en el ojo” y con “categoría 1 del SGA de las Naciones Unidas” (4). Microscopio de lámpara de hendidura : Instrumento utilizado para examinar directamente el ojo con el aumento de un microscopio binocular creando una imagen estereoscópica en vertical. En el método de ensayo ICE, este instrumento se utiliza para ver las estructuras anteriores del ojo del pollo, así como para medir objetivamente el espesor de la córnea con ayuda de un medidor de profundidad. Testigo del disolvente/vehículo : Muestra no tratada que contiene todos los componentes de un sistema de ensayo, incluido el disolvente o vehículo, y que se somete al mismo proceso que las muestras tratadas con producto problema y otras muestras testigo a fin de determinar la respuesta de base correspondiente a las muestras tratadas con el producto problema disuelto en el mismo disolvente o vehículo. Cuando se somete a ensayo con un testigo negativo en paralelo, esta muestra pone de manifiesto también si el disolvente o vehículo interactúa con el sistema de ensayo. Sustancia : Elementos químicos y sus compuestos en estado natural u obtenidos mediante cualquier proceso de producción, incluidos los aditivos necesarios para mantener la estabilidad del producto y las impurezas derivadas del proceso empleado, pero excluidos los disolventes que se puedan separar sin afectar a la estabilidad de la sustancia ni cambiar su composición (4). Tensioactivo : También denominado agente tensioactivo, es una sustancia, como un detergente, que puede reducir la tensión superficial de un líquido y, de este modo, permitir que forme espuma o penetre en los sólidos; se conoce también como agente humectante. Enfoque descendente : Enfoque gradual utilizado para un producto del que se sospecha que causa lesiones oculares graves, que comienza con la determinación de los productos que inducen lesiones oculares graves (resultado positivo) frente a otros productos (resultado negativo). Producto problema : Toda sustancia o mezcla sometida a ensayo con este método de ensayo. Estrategia de ensayos escalonados : Estrategia de ensayo por fases, en la que se revisa toda la información existente sobre un producto problema, siguiendo un orden especificado, en un proceso de ponderación de las pruebas en cada escalón, a fin de determinar si se dispone de información suficiente para tomar una decisión sobre la clasificación de un peligro, antes de pasar al escalón siguiente. Si puede establecerse la capacidad de irritación de un producto problema con la información disponible, no hace falta efectuar más ensayos. Si no puede establecerse la capacidad de irritación de un producto problema con la información disponible, se aplica un procedimiento secuencial de ensayos con animales por fases hasta que pueda efectuarse una clasificación inequívoca. Sistema Globalmente Armonizado de clasificación y etiquetado de productos químicos de las Naciones Unidas (SGA de la ONU) : Sistema que propone la clasificación de productos químicos (sustancias y mezclas) según tipos y niveles normalizados de peligros físicos, sanitarios y ambientales, y que hace referencia a los elementos correspondientes de comunicación, como pictogramas, palabras de advertencia, indicaciones de peligro, consejos de prudencia, y fichas de datos de seguridad, a efectos de proporcionar información sobre sus efectos adversos con el fin de proteger a la población (incluidos empresarios, trabajadores, transportistas, consumidores y personal de protección civil) y al medio ambiente (4). Categoría 1 del SGA de las Naciones Unidas : Véase “lesiones oculares graves” o “efectos irreversibles en el ojo”. Categoría 2 del SGA de las Naciones Unidas : Véase “irritación ocular” o “efectos reversibles en el ojo”. Sin categoría del SGA de las Naciones Unidas : Sustancias que no cumplen los requisitos para clasificarse en la categoría 1 o 2 (2A o 2B) del SGA de las Naciones Unidas. Intercambiable con “sin clasificar”. Método de ensayo validado : Método de ensayo sobre el cual se han completado estudios de validación para determinar su pertinencia (incluida su exactitud) y su fiabilidad con un fin específico. Es importante señalar que un método de ensayo validado puede tener un comportamiento insuficiente en términos de exactitud y fiabilidad como para considerarse aceptable a efectos del fin propuesto. Ponderación de las pruebas : Proceso de consideración de los aspectos favorables y desfavorables de los distintos elementos de información a efectos de alcanzar y confirmar una conclusión en cuanto al peligro potencial de un producto. Apéndice 2 PRODUCTOS UTILIZADOS PARA DEMOSTRAR LA APTITUD CON EL MÉTODO DE ENSAYO ICE Antes de proceder al uso sistemático de un método de ensayo que siga este modelo, los laboratorios deben demostrar su aptitud técnica, identificando correctamente la clasificación de los trece productos recomendados del cuadro 1 en cuanto al peligro para los ojos. Estos productos se seleccionaron para representar la gama de respuestas en cuanto a los peligros para los ojos sobre la base de los resultados del ensayo in vivo con ojo de conejo (TG 405) y el sistema de clasificación SGA de las Naciones Unidas (es decir, categorías 1, 2A, 2B, o sin clasificar según dicho sistema) (4). Otros criterios de selección fueron el que los productos estuvieran disponibles en el mercado, que tuvieran datos de referencia in vivo de alta calidad y también datos in vitro de alta calidad obtenidos con el método de ensayo ICE. Se encuentran datos de referencia en el documento SSD (5) y en el documento de revisión de fondo del ICCVAM sobre el método de ensayo ICE (9). Cuadro 1 Productos recomendados para demostrar la aptitud técnica con el método ICE
Apéndice 3 DIAGRAMAS DEL APARATO DE SUPERFUSIÓN Y DEL SOPORTE PARA LOS OJOS (Burton et al. (18) ofrecen descripciones genéricas adicionales del aparato de superfusión y del soporte para los ojos) 
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

 , lo que equivale a 0,095 mm. Los usuarios deben ser conscientes de que con los microscopios de lámpara de hendidura pueden obtenerse diferentes mediciones del espesor de la córnea si son diferentes los ajustes de la anchura de la hendidura.
, lo que equivale a 0,095 mm. Los usuarios deben ser conscientes de que con los microscopios de lámpara de hendidura pueden obtenerse diferentes mediciones del espesor de la córnea si son diferentes los ajustes de la anchura de la hendidura.|
14) |
En la parte B, el capítulo B.49 se sustituye por el texto siguiente: «B.49. Ensayo de micronúcleos en células de mamífero in vitro INTRODUCCIÓN El presente método de ensayo es equivalente a las directrices de ensayo de la OCDE 487 (2016). Forma parte de una serie de métodos de ensayo sobre toxicología genética. Se ha elaborado un documento de la OCDE que aporta información sucinta sobre los ensayos de toxicología genética y una síntesis de los recientes cambios aportados a dichas directrices de ensayo (1). El ensayo de micronúcleos in vitro (MNvit) es un ensayo de genotoxicidad para la detección de micronúcleos (MN) en el citoplasma de células en interfase. Los micronúcleos pueden proceder de fragmentos cromosómicos acéntricos (es decir, que carecen de centrómero) o de cromosomas completos que no pueden migrar a los polos durante la anafase de la división celular. Por tanto, el ensayo MNvit es un método in vitro que proporciona una base completa para investigar in vitro el potencial de causar lesiones cromosómicas porque permite detectar tanto anéugenos como clastógenos (2) (3) en células que han sufrido división celular durante la exposición al producto problema o tras ella (véanse más detalles en el punto 13). Los micronúcleos representan una lesión que se ha transmitido a las células hijas, mientras que las aberraciones cromosómicas registradas en las células en metafase pueden no transmitirse. En ambos casos, los cambios pueden no ser compatibles con la supervivencia de las células. El presente método de ensayo permite el uso de protocolos con y sin citocalasina B (citoB), que es un inhibidor de la polimerización de la actina. La adición de citoB antes de la mitosis provoca la formación de células binucleadas, con lo que permite la identificación y análisis de micronúcleos solo en las células que han completado una única mitosis (4) (5). Este método de ensayo contempla también el uso de protocolos sin bloqueo de la citocinesis, siempre que haya pruebas de que la población celular analizada ha experimentado la mitosis. Además de utilizar el ensayo MNvit para identificar productos que inducen la formación de micronúcleos, el uso del etiquetado inmunoquímico de cinetocoros, o de la hibridación con sondas centroméricas o teloméricas [hibridación fluorescente in situ (FISH)], puede aportar también información adicional sobre los mecanismos del daño cromosómico y de la formación de micronúcleos (6) (7) (8) (9) (10) (11) (12) (13) (14) (15) (16) (17). Dichos procedimientos de etiquetado e hibridación pueden utilizarse cuando hay un aumento en la formación de micronúcleos y el investigador desea determinar si el aumento es consecuencia de fenómenos clastogénicos o aneugénicos. Debido a que los micronúcleos de las células en interfase pueden evaluarse de forma relativamente objetiva, el personal de laboratorio solamente tiene que determinar el número de células binucleadas cuando se emplea citoB y la incidencia de las células micronucleadas en todos los casos. Como resultado, los portaobjetos pueden evaluarse de forma relativamente rápida y el análisis puede automatizarse. Esto hace que sea factible evaluar miles en lugar de cientos de células por tratamiento, lo que aumenta la potencia del ensayo. Finalmente, como los micronúcleos pueden derivarse de cromosomas retardados, existe la posibilidad de detectar agentes inductores de aneuploidía que son difíciles de estudiar en ensayos convencionales de aberraciones cromosómicas como, por ejemplo, el del capítulo B.10 del presente anexo (18). Sin embargo, el ensayo MNvit descrito en el presente método de ensayo no permite diferenciar los productos que inducen cambios en el número de cromosomas o en la ploidía de aquellos que inducen efectos clastogénicos sin recurrir a técnicas especiales como la FISH mencionada en el punto 4. El ensayo MNvit es sólido y puede aplicarse a una diversidad de tipos celulares, y tanto en presencia como en ausencia de citoB. Hay muchos datos que apoyan la validez del ensayo MNvit recurriendo a diferentes tipos de células (cultivos de líneas celulares o cultivos primarios de células) (19) (20) (21) (22) (23) (24) (25) (26) (27) (28) (29) (30) (31) (32) (33) (34) (35) (36). Aquí se incluyen, en particular, los estudios internacionales de validación coordinados por la Société Française de Toxicologie Génétique (SFTG) (19) (20) (21) (22) (23) y los informes del International Workshop on Genotoxicity Testing (5) (17). Los datos disponibles también se han vuelto a evaluar en un estudio de validación retrospectiva con ponderación de los datos, por parte del Centro Europeo para la Validación de Métodos Alternativos (CEVMA) de la Comisión Europea (CE), y el método de ensayo ha sido avalado en cuanto a su validez científica por el Comité Científico Consultivo del CEVMA (ESAC) (37) (38) (39). El ensayo MNvit en células de mamífero puede emplear cultivos de líneas celulares o cultivos primarios de células humanas o de roedores. Como la frecuencia de fondo de la formación de micronúcleos influye en la sensibilidad del ensayo, se recomienda la utilización de tipos celulares con una frecuencia de fondo de micronúcleos que sea estable y definida. Las células utilizadas deben seleccionarse sobre la base de su capacidad de crecer bien en cultivo, la estabilidad de su cariotipo (incluido el número de cromosomas) y su frecuencia espontánea de micronúcleos (40). Por el momento, los datos de que se dispone no permiten formular recomendaciones firmes pero sugieren que es importante, a la hora de evaluar los peligros químicos, examinar la situación de la proteína p53, la estabilidad genética (cariotipo), la capacidad de reparación del ADN y el origen (roedores frente a hombre) de las células elegidas para el ensayo. Se recomienda, pues, a los usuarios de este método de ensayo que consideren la influencia de estas y otras características de las células sobre el comportamiento de una línea celular en cuanto a la detección de la inducción de la formación de micronúcleos, ya que la situación de los conocimientos en este ámbito está evolucionando. En el apéndice 1 se dan las definiciones utilizadas. CONSIDERACIONES INICIALES Y LIMITACIONES Los ensayos realizados in vitro suelen exigir la utilización de una fuente exógena de activación metabólica, salvo que las células sean competentes metabólicamente respecto a los productos problema. El sistema exógeno de activación metabólica no reproduce completamente las condiciones in vivo. Deben evitarse las condiciones que puedan conducir a resultados positivos falsos, que no reflejen la genotoxicidad de los productos problema; tales condiciones incluyen cambios en el pH (41) (42) (43) o en la osmolalidad, la interacción con el medio de cultivo (44) (45) o unos niveles excesivos de citotoxicidad (véase el punto 29). Para analizar la inducción de la formación de micronúcleos, es fundamental que haya habido mitosis en los cultivos tanto tratados como sin tratar. La fase que proporciona más información para evaluar la formación de micronúcleos es la de las células que han completado una sola mitosis durante el tratamiento con el producto problema, o después de este tratamiento. En caso de nanomateriales fabricados, es necesario recurrir a adaptaciones específicas del presente método de ensayo, pero estas no se describen aquí. Antes de la utilización del método de ensayo con una mezcla para obtener datos con fines reglamentarios, debe considerarse si podría proporcionar resultados adecuados a tal fin y, en caso afirmativo, por qué. Tales consideraciones no son necesarias si la reglamentación impone el ensayo de la mezcla. PRINCIPIO DEL ENSAYO Los cultivos celulares de origen humano o de otros mamíferos se exponen al producto problema tanto con fuente exógena de activación metabólica como sin ella, salvo que se utilicen células con una capacidad adecuada de metabolización (véase el punto 19). Durante la exposición al producto problema, o después de ella, las células se cultivan durante un tiempo suficiente para permitir que las lesiones cromosómicas u otros efectos sobre los ciclos o divisiones celulares provoquen la formación de micronúcleos en las células en interfase. Para la inducción de aneuploidía, es necesario normalmente que el producto problema esté presente durante la mitosis. Las células en interfase se recogen, se tiñen y se analizan para detectar la presencia de micronúcleos. Lo ideal es que los micronúcleos se evalúen solamente en las células que hayan completado la mitosis durante la exposición al producto problema o durante el período tras el tratamiento, en su caso. En los cultivos que se hayan tratado con un bloqueante de la citocinesis, esto se consigue fácilmente evaluando solo las células binucleadas. En ausencia de bloqueante de la citocinesis, es importante demostrar la probabilidad de que las células analizadas hayan experimentado la división celular, sobre la base de un aumento de la población celular, durante la exposición al producto problema o tras ella. En relación con todos los protocolos, es importante demostrar que ha habido proliferación celular en los cultivos tanto tratados como de testigo, y el grado de citotoxicidad o citostasis inducido por el producto problema debe estimarse en todos los cultivos que se examinan para evaluar la presencia de micronúcleos. DESCRIPCIÓN DEL MÉTODO Células Pueden utilizarse cultivos primarios de linfocitos de sangre periférica de hombre o de otros mamíferos (7) (20) (46) (47) y una serie de líneas celulares de roedor, tales como las células CHO, V 79, CHL/IU y L5178Y, o líneas celulares humanas tales como TK6 (19) (20) (21) (22) (23) (26) (27) (28) (29) (31) (33) (34) (35) (36) (véase el punto 6). Se han utilizado para el ensayo de micronúcleos otras líneas celulares, tales como las HT29 (48), Caco-2 (49), HepaRG (50) (51), HepG2 (52) (53), A549 y células primarias de embrión de hámster sirio (54), pero por el momento no se han validado ampliamente. Por tanto, el uso de estos tipos y líneas celulares debe justificarse por la demostración de su comportamiento en el ensayo, como se describe en la sección de criterios de aceptabilidad. Se ha informado de que la citoB puede afectar al crecimiento de las células L5178Y, por lo que no se recomienda utilizarla con esta línea celular (23). Cuando se utilizan células primarias, por razones de bienestar animal debe considerarse la utilización de células de origen humano, cuando sea viable y se tomen de conformidad con los principios éticos humanos y la reglamentación. Los linfocitos de sangre periférica humana deben obtenerse de individuos jóvenes (aproximadamente de 18 a 35 años de edad), no fumadores, sin enfermedades conocidas ni exposiciones recientes a agentes genotóxicos (por ejemplo, productos, radiaciones ionizantes) a niveles que puedan aumentar la incidencia de fondo de las células micronucleadas. Esto garantizaría una incidencia de fondo de las células micronucleadas baja y constante. La incidencia de base de las células micronucleadas aumenta con la edad y esta tendencia es más pronunciada en las mujeres que en los varones (55). Si se combinan células procedentes de más de un donante, debe especificarse el número de donantes. Es necesario demostrar que las células se han dividido entre el inicio de la administración del producto problema y la recolección de las células. Los cultivos celulares se mantienen en fase exponencial de crecimiento (líneas celulares) o se estimulan para que se dividan (cultivos primarios de linfocitos), a fin de exponer las células en diferentes fases del ciclo celular, ya que es posible que no se conozca la sensibilidad de las distintas fases celulares a los productos problema. Las células primarias que es preciso estimular con agentes mitogénicos para que se dividan dejan generalmente de estar sincronizadas durante la exposición al producto problema (por ejemplo, los linfocitos humanos tras una estimulación mitogénica de 48 horas). La utilización de células sincronizadas durante el tratamiento con el producto problema no es recomendable, pero puede aceptarse si está justificada. Medios y condiciones de cultivo Para el mantenimiento de los cultivos deben utilizarse medios de cultivo y condiciones de incubación adecuados (recipientes de cultivo, atmósfera humidificada y con 5 % de CO2 en su caso, temperatura de 37 °C). Las líneas celulares deben examinarse sistemáticamente para comprobar la estabilidad del número modal de cromosomas y la ausencia de contaminación por Mycoplasma, y no deben utilizarse las células que se contaminen o que presenten un cambio en el número modal de cromosomas. Debe determinarse la duración del ciclo celular normal de las líneas celulares o cultivos primarios utilizados en el laboratorio de ensayo, la cual debe ser coherente con las características celulares publicadas. Preparación de los cultivos Líneas celulares: las células se propagan a partir de cultivos madre, se siembran en el medio de cultivo a una densidad tal que las células en suspensión o en monocapas sigan creciendo exponencialmente hasta el momento de la recolección (por ejemplo, debe evitarse la confluencia de las células cultivadas en monocapas). Linfocitos: se cultiva (p.ej., durante 48 horas en el caso de los linfocitos humanos) sangre completa tratada con anticoagulante (p. ej., heparina) o bien linfocitos separados en presencia de un mitógeno [p. ej., fitohemaglutinina (PHA) en el caso de los linfocitos humanos] a fin de inducir la división celular antes de la exposición al producto problema y a citoB. Activación metabólica Se debe recurrir a sistemas de metabolización exógenos cuando se utilizan células con una capacidad metabólica endógena inadecuada. El sistema utilizado con más frecuencia que se recomienda por defecto, salvo que se justifique otro sistema, es una fracción postmitocondrial (S9) a la que se añaden cofactores y que se obtiene a partir del hígado de roedores (generalmente ratas) tratados con inductores enzimáticos como el aroclor 1254 (56) (57) o una combinación de fenobarbital y b-naftoflavona (58) (59) (60). Esta última combinación no infringe el Convenio de Estocolmo sobre contaminantes orgánicos persistentes (61), y se ha visto que es tan efectiva como el aroclor 1254 para inducir oxidasas de función mixta (58) (59) (60). La fracción S9 se utiliza normalmente a concentraciones que varían entre el 1 y el 2 % (v/v), pero pueden aumentarse hasta el 10 % (v/v) en el medio de ensayo final. Debe evitarse durante el tratamiento el uso de productos que reduzcan el índice mitótico, especialmente productos que acomplejen el calcio (62). La elección del tipo y de la concentración del sistema exógeno de activación metabólica o del inductor metabólico utilizado puede estar influida por la clase de los productos que se someten a ensayo. Preparación del producto problema Los productos problema sólidos deben prepararse en disolventes adecuados y, si es conveniente, diluirse antes de tratar las células. Los productos problema líquidos pueden añadirse directamente al sistema de ensayo o diluirse antes del tratamiento del sistema de ensayo. Los productos problema gaseosos o volátiles deben someterse a ensayo aplicando modificaciones adecuadas a los protocolos normales, tales como el tratamiento en recipientes de cultivo sellados (63) (64) (65). La preparación del producto problema debe hacerse justo antes del tratamiento, salvo que se cuente con datos de estabilidad que avalen la posibilidad de su conservación. Condiciones del ensayo Disolventes El disolvente debe elegirse para optimizar la solubilidad de los productos problema sin tener impacto negativo en la realización del ensayo, es decir, cambiar el crecimiento celular, afectar a la integridad del producto problema, reaccionar con los recipientes de cultivo, o interferir con el sistema de activación metabólica. Siempre que sea posible, se recomienda considerar en primer lugar la utilización de un disolvente (o medio de cultivo) acuoso. Son disolventes bien establecidos el agua o el dimetilsulfóxido (DMSO). En general, los disolventes orgánicos no deben exceder del 1 % (v/v). Si se disuelve la citoB en DMSO, el conjunto de disolvente orgánico utilizado para el producto problema y para la citoB no debe superar el 1 % (v/v); en caso contrario, deben utilizarse testigos sin tratar, a fin de garantizar que el porcentaje de disolvente orgánico no tiene ningún efecto adverso. Los disolventes acuosos (solución salina o agua) no deben superar el 10 % (v/v) en el medio de tratamiento final. Si se utilizan disolventes distintos de los que están bien establecidos (por ejemplo, etanol o acetona), debe disponerse de datos justificativos que indiquen su compatibilidad con el sistema de ensayo y el producto problema, y su ausencia de toxicidad genética a la concentración utilizada. En ausencia de estos datos justificativos, es importante incluir testigos sin tratar (véase el apéndice 1), así como testigos del disolvente para demostrar que el disolvente elegido no induce efectos nocivos ni cromosómicos (p. ej., aneuploidía o clastogenicidad). Utilización de citoB como bloqueante de la citocinesis Una de las consideraciones más importantes en la realización del ensayo MNvit es la de velar por que las células que se examinen hayan completado la mitosis durante el tratamiento o en el período de incubación tras el mismo, en su caso. Así pues, el recuento de micronúcleos debe limitarse a las células que han experimentado la mitosis durante el tratamiento o después de este. La citoB es el agente más ampliamente utilizado para bloquear la citocinesis, ya que inhibe el ensamblaje de la actina, con lo que impide la separación de las células hijas tras la mitosis, y así se forman células binucleadas (6) (66) (67). El efecto del producto problema sobre la cinética de proliferación celular puede medirse simultáneamente, cuando se utiliza la citoB. La citoB debe utilizarse como bloqueante de la citocinesis cuando se utilizan linfocitos humanos, ya que la duración de los ciclos celulares es variable entre los donantes, y debido a que no todos los linfocitos responden a la estimulación con PHA. El uso de citoB no es obligatorio con los demás tipos de células si se puede establecer que han sufrido división como se describe en el punto 27. Por otra parte, la citoB no se utiliza generalmente cuando se evalúa la presencia de micronúcleos en las muestras utilizando métodos citométricos. La concentración adecuada de citoB debe determinarse en el laboratorio para cada tipo celular a fin de conseguir la frecuencia óptima de células binucleadas en los cultivos de testigo de disolvente y debe demostrarse que proporciona un buen rendimiento de células binucleadas para la evaluación. La concentración adecuada de citoB está generalmente entre 3 y 6 μg/ml (19). Medición de la proliferación celular y de la citotoxicidad, y selección de las concentraciones de tratamiento Al determinar la concentración máxima de producto problema, debe evitarse llegar a concentraciones que puedan producir respuestas positivas falsas, tales como las que provocan una citotoxicidad excesiva (véase el punto 29), precipitación en el medio de cultivo (véase el punto 30), o cambios marcados del pH o de la osmolalidad (véase el punto 9). Si el producto problema provoca un cambio marcado en el pH del medio en el momento de su adición, el pH puede ajustarse amortiguando el medio de tratamiento final para evitar resultados positivos falsos y mantener unas condiciones de cultivo adecuadas. Se mide la proliferación celular para asegurarse de que un número suficiente de células tratadas ha sufrido mitosis durante el ensayo y de que los tratamientos se efectúan a los niveles adecuados de citotoxicidad (véase el punto 29). La citotoxicidad debe determinarse en el experimento principal con y sin activación metabólica, utilizando un indicador adecuado de la muerte y del crecimiento de las células (véanse los puntos 26 y 27). Si bien la evaluación de la citotoxicidad en un ensayo preliminar inicial puede ser útil para definir mejor las concentraciones que deben utilizarse en el experimento principal, no es obligatorio efectuar un ensayo inicial. Si se lleva a cabo, no debe sustituir a la medición de la citotoxicidad en el experimento principal. El tratamiento de los cultivos con citoB, junto con la medición de las frecuencias relativas de células mononucleadas, binucleadas y multinucleadas en el cultivo, constituye un método exacto de cuantificación del efecto sobre la proliferación celular y de la actividad citotóxica o citostática de un tratamiento (6), y garantiza que solamente se evalúan al microscopio las células que se han dividido durante el tratamiento o después de este. Se recomienda utilizar el índice de proliferación con bloqueo citocinético (IPBC) (6) (27) (68) o el índice de replicación (IR) de, al menos, 500 células por cultivo (véanse las fórmulas en el apéndice 2) para estimar la actividad citotóxica y citostática de un tratamiento comparando los valores obtenidos en los cultivos tratados y en los testigos. La evaluación de otros indicadores de citotoxicidad (por ejemplo, integridad celular, apoptosis, necrosis, recuento de las células en metafase, ciclo celular) puede aportar información útil, pero no debe utilizarse en lugar del IPBC o del IR. En los estudios sin citoB, es necesario demostrar que las células en cultivo se han dividido, de modo que una parte sustancial de las células analizadas hayan sufrido división durante el tratamiento con el producto problema o tras él; en caso contrario, pueden obtenerse respuestas negativas falsas. Para evaluar la actividad citotóxica y citostática de un tratamiento, se recomienda recurrir a la medición de la duplicación relativa de la población (DRP) o del aumento relativo del recuento celular (ARRC) (17) (68) (69) (70) (71) (véanse las fórmulas en el apéndice 2). Con unos tiempos de muestreo ampliados (por ejemplo, tratamiento durante 1,5-2 veces la duración del ciclo celular normal y recolección después de otras 1,5-2 veces la duración del ciclo celular normal, lo que llevaría a tiempos de muestreo superiores a 3-4 veces la duración del ciclo celular normal en total, como se describe en los puntos 38 y 39), la DRP podría subestimar la citotoxicidad (71). En tales circunstancias, puede ser preferible medir el ARRC, o puede ser útil evaluar la citotoxicidad tras un tiempo igual a 1,5-2 veces la duración del ciclo celular normal. La evaluación de otros indicadores de citotoxicidad o citostasis (por ejemplo, integridad celular, apoptosis, necrosis, recuento de las células en metafase, índice de proliferación (IP), ciclo celular, puentes nucleoplásmicos o yemas nucleares) puede aportar información útil, pero no debe utilizarse en lugar de la DRP o del ARRC. Deben evaluarse al menos tres concentraciones de ensayo (sin incluir los testigos positivos y de disolvente) que cumplan los criterios de aceptabilidad (citotoxicidad apropiada, número de células, etc.). Cualquiera que sea el tipo de células (líneas celulares o cultivos primarios de linfocitos), pueden utilizarse a cada concentración de ensayo cultivos tratados replicados o sin replicar. Si bien se recomienda el uso de cultivos duplicados, también son aceptables los cultivos sin replicar, siempre que se examine el mismo número total de células, sea en cultivos duplicados o sin replicar. La utilización de cultivos sin replicar es especialmente pertinente cuando se evalúan más de tres concentraciones (véanse los puntos 44-45). Los resultados obtenidos en los cultivos replicados independientes a una concentración determinada se pueden poner en común para el análisis de los datos. En el caso de productos problema que muestren escasa o nula citotoxicidad, normalmente serán adecuados los intervalos de concentración de aproximadamente el doble o el triple. Cuando se produce citotoxicidad, las concentraciones de ensayo seleccionadas deben cubrir una gama a partir de la que provoca citotoxicidad según se describe en el punto 29, con inclusión en particular de las concentraciones a las que existe citotoxicidad moderada y débil o nula. Muchos productos problema presentan curvas de respuesta a la concentración de elevada pendiente y, con el fin de obtener datos a niveles bajos y moderados de citotoxicidad o de estudiar en detalle la relación entre dosis y respuesta, en tales casos es necesario recurrir a concentraciones más próximas entre sí o a más de tres concentraciones (cultivos replicados o sin replicar), en particular en situaciones en que se requiere una repetición del experimento (véase el punto 60). Si la concentración máxima se basa en la citotoxicidad, la concentración más elevada debe aspirar a provocar un 55 ± 5 % de citotoxicidad utilizando los parámetros recomendados de citotoxicidad (es decir, reducción del ARRC y de la DRP con líneas celulares cuando no se utiliza la citoB, y reducción del IPBC o del IR cuando se utiliza la citoB, al 45 ± 5 % del testigo negativo en paralelo) (72). Ha de tenerse cuidado al interpretar resultados positivos que solo se encuentren en el extremo superior de este intervalo de citotoxicidad del 55 ± 5 % (71). Para los productos problema poco solubles que no son citotóxicos a concentraciones inferiores a la concentración mínima insoluble, la mayor concentración analizada debe producir turbidez o precipitado visibles a simple vista o con ayuda de un microscopio invertido al final del tratamiento con el producto problema. Incluso si se produce citotoxicidad por encima de la concentración mínima insoluble, es aconsejable hacer el ensayo a una única concentración que produzca turbidez o precipitado visible porque este puede provocar efectos falsos. A la concentración que produce precipitado, se debe evitar que este interfiera con la realización del ensayo (por ejemplo, con la tinción o el examen celular). Puede ser útil determinar la solubilidad en el medio de cultivo antes de que efectuar el experimento. Si no se observa precipitado ni citotoxicidad limitante, la concentración de ensayo más elevada debe corresponder a la más baja de las siguientes: 10 mM, 2 mg/ml o 2 μl/ml (73) (74) (75). Si el producto problema no tiene una composición definida y se trata, por ejemplo, de sustancias de composición desconocida o variable, productos complejos de reacción o materiales biológicos (UVCB) (76), extractos medioambientales, etc., es posible que la concentración superior tenga que ser mayor (por ejemplo, 5 mg/ml), en ausencia de citotoxicidad suficiente, para aumentar la concentración de cada uno de los componentes. Conviene señalar, no obstante, que estos requisitos pueden variar en caso de medicamentos de uso humano (93). Testigos Se incluirán, para cada período de recolección, testigos negativos en paralelo (véase el punto 21), consistentes en el disolvente solo en el medio de tratamiento y sometidos al mismo proceso que los cultivos tratados. Es necesario disponer testigos positivos en paralelo a fin de demostrar la capacidad del laboratorio para detectar clastógenos y anéugenos en las condiciones establecidas en el protocolo de ensayo utilizado y la efectividad del sistema exógeno de activación metabólica, si procede. En el cuadro 1 a continuación se encuentran ejemplos de testigos positivos. Es posible utilizar como testigos positivos otros productos, si se justifica. Por el momento, no se conoce ningún anéugeno que necesite activación metabólica para su actividad genotóxica (17). Debido a que los ensayos de toxicidad genética con células de mamífero in vitro están suficientemente normalizados respecto a los tratamientos a corto plazo realizados en paralelo, con y sin activación metabólica, utilizando la misma duración del tratamiento, el uso de testigos positivos puede limitarse a un clastógeno que requiera activación metabólica. En este caso, una sola respuesta clastogénica de un testigo positivo demostrará tanto la actividad del sistema de activación metabólica como la sensibilidad del sistema de ensayo. No obstante, los tratamientos a largo plazo (sin fracción S9) deben tener su propio testigo positivo, ya que la duración del tratamiento será diferente de la del ensayo con activación metabólica. Si se selecciona un clastógeno como único testigo positivo en un tratamiento a corto plazo con y sin activación metabólica, debe seleccionarse un anéugeno para el tratamiento a largo plazo sin activación metabólica. Deben utilizarse testigos positivos tanto en cuanto a la clastogenicidad como en cuanto a la aneugenicidad con células competentes metabólicamente que no requieren S9. Cada testigo positivo debe utilizarse a una o varias concentraciones de las que quepa esperar que produzcan incrementos reproducibles y detectables respecto a los valores de fondo para demostrar la sensibilidad del sistema de ensayo (es decir, que los efectos sean claros, pero sin revelar inmediatamente al lector la identidad de los portaobjetos codificados), y la respuesta no debe verse comprometida por una citotoxicidad que exceda de los límites especificados en este método de ensayo. Cuadro 1. Productos de referencia recomendados para evaluar la competencia del laboratorio, y para la selección de los testigos positivos.
PROCEDIMIENTO Pauta de tratamiento Para maximizar la probabilidad de detectar un anéugeno o un clastógeno que actúe en una fase específica del ciclo celular, es importante que se trate con el producto problema un número suficiente de células que representen todas las diferentes fases de sus ciclos celulares. Todos los tratamientos deben iniciarse y terminarse mientras las células están en fase de crecimiento exponencial y las células deben seguir creciendo hasta el momento de la toma de muestras. Por tanto, la pauta de tratamiento de líneas celulares y de cultivos celulares primarios puede diferir en algunos aspectos respecto a la aplicada a los linfocitos que requieren estimulación mitogénica para empezar su ciclo celular (17). En el caso de los linfocitos, el enfoque más eficiente es iniciar el tratamiento con el producto problema a las 44-48 horas tras la estimulación con PHA, cuando las células se estén dividiendo de forma asíncrona (6). Los datos publicados (19) indican que la mayoría de los anéugenos y clastógenos se detectan con un corto período de tratamiento, de entre 3 y 6 horas, en presencia y ausencia de S9, seguido de la eliminación del producto problema, y con la toma de muestras a un tiempo equivalente a unas 1,5-2,0 veces la duración del ciclo celular normal desde el inicio del tratamiento (7). Sin embargo, para una evaluación a fondo, que sería necesaria para deducir un resultado negativo, deben aplicarse las tres condiciones experimentales siguientes, utilizando un tratamiento a corto plazo con y sin activación metabólica y un tratamiento a largo plazo sin activación metabólica (véanse los puntos 56, 57 y 58):
En caso de que alguna de las anteriores condiciones experimentales conduzca a una respuesta positiva, puede no ser necesario investigar los otros regímenes de tratamiento. Si se sabe o se sospecha que el producto problema afecta a la duración del ciclo celular (por ejemplo, cuando se someten a ensayo análogos de nucleósidos), especialmente en el caso de células competentes para p53 (35) (36) (77), los tiempos de toma de muestras o de recuperación pueden ampliarse hasta un nuevo período de 1,5-2,0 veces la duración del ciclo celular normal (es decir, un total de 3,0 a 4,0 veces la duración del ciclo celular desde el inicio de los tratamientos a corto y a largo plazo). Estas opciones se refieren a situaciones en que puede haber preocupación respecto a posibles interacciones entre el producto problema y la citoB. Cuando se utilizan tiempos de muestreo ampliados (es decir, un total de 3,0 a 4,0 veces la duración del ciclo celular), conviene asegurarse de que las células se siguen dividiendo aún activamente. Por ejemplo, en el caso de los linfocitos, el crecimiento exponencial podría declinar a las 96 horas de la estimulación y pueden convertirse en confluentes los cultivos en monocapas. Las pautas sugeridas de tratamiento celular se resumen en el cuadro 2. Estas pautas generales de tratamiento pueden modificarse (lo que debería justificarse) en función de la estabilidad o reactividad del producto problema o de las características particulares de crecimiento de las células utilizadas. Cuadro 2. Tratamiento celular y tiempos de recogida para el ensayo MNvit
En los cultivos de monocapas, es posible que haya células mitóticas (identificables por su forma redondeada y por despegarse de la superficie) al final del tratamiento de 3-6 horas. Como estas células mitóticas se despegan con facilidad, pueden perderse cuando se retira el medio que contiene el producto problema. Si hay pruebas de un aumento sustancial del número de células mitóticas en comparación con los testigos, lo que indica una probable detención mitótica, las células deben recogerse por centrifugación y volver a añadirse al cultivo, para evitar la pérdida de células que estén en mitosis, y que podrían presentar micronúcleos o aberraciones cromosómicas, en el momento de la recolección. Recolección de las células y preparación de los portaobjetos Cada cultivo debe recogerse y manipularse por separado. La preparación de las células puede implicar un tratamiento hipotónico, pero esta fase no es necesaria si se consigue de otra manera una extensión adecuada de las células. Pueden usarse distintas técnicas para la preparación de los portaobjetos, siempre que lleven a la obtención de preparaciones celulares de buena calidad para su evaluación. Deben seleccionarse células con la membrana celular intacta y el citoplasma intacto para poder detectar los micronúcleos y (en el método de bloqueo de la citocinesis) poder identificar de forma fiable las células binucleadas. Las preparaciones pueden teñirse siguiendo varios métodos, tales como la tinción de Giemsa o la aplicación de colorantes fluorescentes específicos del ADN. Si se emplean colorantes fluorescentes apropiados [p. ej., naranja de acridina (78) o Hoechst 33258 más pironina-Y (79)] pueden evitarse algunos de los artefactos que aparecen cuando se utiliza un colorante no específico del ADN. Pueden utilizarse anticuerpos anti-cinetocoros, la técnica FISH con sondas pancentroméricas de ADN, o el etiquetado in situ cebado con cebadores pancentroméricos específicos, junto con una tinción de contraste adecuada del ADN, para determinar el contenido de micronúcleos (se teñirán los cromosomas enteros, mientras que los fragmentos cromosómicos acéntricos quedarán sin teñir) si resulta interesante disponer de información sobre el mecanismo de su formación (16) (17). Pueden utilizarse otros métodos para diferenciar entre clastógenos y anéugenos, siempre que se haya comprobado que son efectivos y están validados. Por ejemplo, en el caso de ciertas líneas celulares, también podrían aportar información útil las mediciones de núcleos sub-2N como fenómenos de hipodiploidía utilizando técnicas tales como el análisis de imágenes, la citometría de barrido por láser o la citometría de flujo (80) (81) (82). Las observaciones morfológicas de los núcleos podrían aportan también indicaciones de posible aneuploidía. Por otra parte, un ensayo de aberraciones cromosómicas en metafase, preferiblemente con el mismo tipo de células y un protocolo de sensibilidad comparable, también podría ser una forma útil de determinar si los micronúcleos se deben a la rotura de cromosomas (sabiendo que la pérdida de cromosomas no se detectaría en el ensayo de aberraciones cromosómicas). Análisis Todos los portaobjetos, incluidos los de los testigos del disolvente, de la muestra sin tratar (en su caso) y los positivos, reciben un código independiente antes de analizar al microscopio las frecuencias de los micronúcleos. Conviene utilizar técnicas adecuadas para controlar todo eventual sesgo o deriva cuando se utilice un sistema de examen automatizado como, por ejemplo, la citometría de flujo, la citometría de barrido por láser o el análisis de imágenes. Con independencia del programa automatizado que se utilice para enumerar los micronúcleos, deben evaluarse de manera simultánea el IPBC, el IR, la DRP o el ARRC. En los cultivos tratados con citoB, deben analizarse las frecuencias de los micronúcleos en al menos 2 000 células binucleadas por cada concentración y testigo (83), divididas a partes iguales entre las réplicas, si se utilizan estas. En caso de realizarse un solo cultivo por dosis (véase el punto 28), deben evaluarse al menos 2 000 células binucleadas en ese cultivo único (83). Si para el examen a cada concentración se dispone de un número sustancialmente inferior a 1 000 células binucleadas por cultivo (en caso de cultivos duplicados), o a 2 000 (en caso de cultivos únicos), y si no se detecta un aumento significativo de la frecuencia de los micronúcleos, debe repetirse el ensayo utilizando más células, o unas concentraciones menos citotóxicas, según sea más conveniente. Ha de procurarse no tener en cuenta para el examen las células binucleadas con formas irregulares o en las que los dos núcleos difieran mucho por su tamaño. Por otra parte, no deben confundirse las células binucleadas con células multinucleadas mal extendidas. Las células que contengan más de dos núcleos principales no deben examinarse en cuanto a los micronúcleos, ya que la frecuencia de micronúcleos de base puede ser más elevada en estas células (84). Es aceptable el examen de células mononucleadas si se demuestra que el producto problema interfiere con la actividad de la citoB. Un segundo ensayo sin citoB podría ser útil en tales casos. El examen de células mononucleadas, además del de las células binucleadas, puede aportar información útil (85) (86), pero no es obligatorio realizarlo. En el caso de las líneas celulares estudiadas sin tratamiento con citoB, deben examinarse los micronúcleos en al menos 2 000 células por cada concentración y testigo (83), divididas a partes iguales entre las réplicas, si se utilizan estas. Cuando se utiliza un solo cultivo por concentración (véase el punto 28), deben examinarse al menos 2 000 células de este cultivo único. Si para el examen a cada concentración se dispone de un número sustancialmente inferior a 1 000 células por cultivo (en caso de cultivos duplicados), o a 2 000 (en caso de cultivos únicos), y si no se detecta un aumento significativo de la presencia de micronúcleos, debe repetirse el ensayo utilizando más células, o unas concentraciones menos citotóxicas, según sea más conveniente. Cuando se utiliza la citoB, para evaluar la proliferación celular hay que determinar el IPBC o el IR (véase el apéndice 2) basándose en un mínimo de 500 células por cultivo. Cuando se hacen los tratamientos en ausencia de citoB, es fundamental aportar pruebas de que las células del cultivo se han dividido, como se indica en los puntos 24-28. Competencia del laboratorio Con el fin de conseguir la suficiente experiencia con el ensayo antes de su uso para los ensayos sistemáticos, el laboratorio debe haber efectuado una serie de experimentos con productos positivos de referencia que actúen a través de mecanismos diferentes (como mínimo, uno con y otro sin activación metabólica, y uno que actúe a través de un mecanismo aneugénico, seleccionados de entre los productos químicos enumerados en el cuadro 1) y con diversos testigos negativos (incluidos los cultivos sin tratar y diferentes disolventes o vehículos). Las respuestas obtenidas con estos testigos positivos y negativos deben ser coherentes con la bibliografía. Esto no es aplicable a los laboratorios que tienen experiencia, esto es, que disponen de una base de datos históricos, según se define en los puntos 49-52. Debe investigarse una selección de productos testigo positivos (véase el cuadro 1) con tratamientos cortos y largos en ausencia de activación metabólica, y también con tratamientos cortos en presencia de activación metabólica, para demostrar su capacidad de detectar productos clastogénicos y aneugénicos, determinar la efectividad del sistema de activación metabólica y demonstrar la adecuación de los procedimientos de examen (análisis visual microscópico, citometría de flujo, citometría de barrido por láser o análisis de imágenes). Debe elegirse una gama de concentraciones de los productos seleccionados de forma que produzcan aumentos sobre el nivel de fondo relacionados con la concentración y reproducibles para demostrar la sensibilidad y el intervalo dinámico del sistema de ensayo. Datos sobre testigos históricos El laboratorio debe determinar:
Cuando se obtengan datos por primera vez en relación con la distribución de testigos negativos históricos, los testigos negativos en paralelo deben ser coherentes con los datos publicados de los testigos negativos, si existen. Según se añadan más datos experimentales sobre la distribución de los testigos, los testigos negativos en paralelo deben situarse idealmente dentro de los límites de control del 95 % de dicha distribución (87) (88). La base de datos de testigos negativos históricos del laboratorio debe constituirse en un principio con un mínimo de 10 experimentos, pero preferiblemente con al menos 20 experimentos realizados en condiciones experimentales comparables. Los laboratorios deben utilizar métodos de control de calidad, como gráficos de control [por ejemplo, gráficos C o gráficos de medias (88)], con el fin de determinar la variabilidad de sus datos sobre los testigos positivos y negativos, y de demostrar que la metodología está «controlada» en su laboratorio (83). En la bibliografía pueden encontrarse más recomendaciones sobre cómo conseguir y utilizar los datos históricos (es decir, criterios de inclusión y exclusión de datos en los datos históricos y criterios de aceptabilidad para un determinado experimento) (87). Cualquier cambio en el protocolo experimental debe considerarse en función de la coherencia de los datos con las bases de datos de testigos históricos existentes del laboratorio. Cualquier incoherencia importante debería dar lugar a la creación de una nueva base de datos de testigos históricos. Los datos sobre los testigos negativos deben consistir en la incidencia de células micronucleadas procedentes de un solo cultivo o de la suma de cultivos replicados como se describe en el punto 28. Lo ideal sería que los testigos negativos en paralelo estuvieran dentro de los límites de control del 95 % de la distribución de la base de datos de testigos negativos históricos del laboratorio (87) (88). Cuando hay datos de los testigos negativos en paralelo que quedan fuera de los límites de control del 95 %, su inclusión en la distribución de testigos históricos puede ser aceptable en la medida en que dichos datos no sean valores discrepantes extremos y haya pruebas de que el sistema de ensayo está «controlado» (véase el punto 50) y pruebas de la ausencia de fallos técnicos o humanos. DATOS E INFORME Presentación de los resultados Si se utiliza la técnica de bloqueo de la citocinesis, para la evaluación de la inducción de micronúcleos se emplearán solo las frecuencias de las células binucleadas con micronúcleos (independientemente del número de micronúcleos por célula). El resultado del número de células con uno, dos o más micronúcleos puede notificarse por separado, lo que puede aportar información útil, pero no es obligatorio hacerlo así. También deben medirse en paralelo la citotoxicidad o la citostasis para todos los cultivos tratados y los testigos negativos y positivos (16). Los valores del IPBC y del IR deben calcularse para todos los cultivos tratados y testigos, como medidas del retraso del ciclo celular cuando se utiliza el método de bloqueo de la citocinesis. En ausencia de citoB, debe utilizarse la DRP o el ARRC (véase el apéndice 2). Deben proporcionarse datos de cada cultivo. Además, se resumirán todos los datos en forma de cuadro. Criterios de aceptabilidad La aceptabilidad de un ensayo se basa en los criterios siguientes:
Evaluación e interpretación de los resultados Siempre que se cumplan todos los criterios de aceptabilidad, se considera que el producto problema es claramente positivo si, en alguna de las condiciones experimentales examinadas (véanse los puntos 36-39):
Cuando se cumplen todos estos criterios, el producto problema se considera capaz de inducir roturas, aumentos o pérdidas de cromosomas en este sistema de ensayo. En la bibliografía se encuentran también recomendaciones sobre los métodos estadísticos más adecuados (90) (91) (92). Siempre que se cumplan todos los criterios de aceptabilidad, se considera que el producto problema es claramente negativo si, en todas las condiciones experimentales examinadas (véanse los puntos 36-39):
El producto problema se considera entonces incapaz de inducir roturas, aumentos o pérdidas de cromosomas en este sistema de ensayo. En la bibliografía se encuentran también recomendaciones sobre los métodos estadísticos más adecuados (90) (91) (92). No se requiere ninguna verificación de una respuesta positiva o negativa clara. En caso de que la respuesta no sea ni claramente positiva ni claramente negativa como se describe más arriba, o a fin de ayudar a determinar la relevancia biológica de un resultado, los datos deben ser evaluados por expertos o mediante más investigaciones. Puede ser útil examinar células adicionales (en su caso) o realizar una repetición del experimento modificando las condiciones experimentales, como, por ejemplo, la separación entre concentraciones u otras condiciones de activación metabólica (es decir, concentración u origen de la fracción S9). En casos raros, incluso después de hacer más investigaciones, el conjunto de datos puede no permitir que se extraiga una conclusión de resultado positivo o negativo, por lo que se considerará dudoso. Los productos problema que inducen micronúcleos en el ensayo MNvit pueden deber esta propiedad a su capacidad de inducir rotura cromosómica, pérdida de cromosomas, o una combinación de ambas. Para determinar si el mecanismo de inducción de micronúcleos se debe a actividad clastogénica y/o aneugénica, pueden realizarse otros análisis utilizando anticuerpos anti-cinetocoro, sondas in situ específicas del centrómero, u otros métodos. Informe del ensayo El informe del ensayo debe incluir la información siguiente:
BIBLIOGRAFÍA
Apéndice 1 DEFINICIONES Anéugeno : Producto o proceso que, por interacción con los componentes del aparato del ciclo de división celular mitótica y meiótica, produce la aneuploidía de células u organismos. Aneuploidía : Desviación respecto al número diploide (o haploide) normal de cromosomas que afecta a uno o varios cromosomas, pero no a dotaciones completas de cromosomas (poliploidía). Apoptosis : Muerte celular programada, caracterizada por una serie de fases que llevan a la desintegración de las células para formar partículas limitadas por membranas que se eliminan a continuación por fagocitosis o por liberación. Proliferación celular : Aumento del número de células como resultado de divisiones celulares mitóticas. Centrómero : Región del ADN del cromosoma en la que se mantienen juntas las dos cromátidas y a la que se unen lateralmente los dos cinetocoros. Producto : Sustancia o mezcla. Concentraciones : Se refiere a las concentraciones del producto problema en el medio de cultivo. Clastógeno : Producto o fenómeno que provoca aberraciones cromosómicas estructurales en poblaciones de células u organismos eucarióticos. Citocinesis : Proceso de división celular que sigue inmediatamente a la mitosis para formar dos células hijas, cada una de las cuales contiene un solo núcleo. Índice de proliferación con bloqueo citocinético (IPBC, o CBPI, por su nombre en inglés) : Relación entre el número de células de segunda división en la población tratada respecto al de células del testigo sin tratar (véase la fórmula en el apéndice 2). Citostasis : Inhibición del crecimiento celular (véase la fórmula en el apéndice 2). Citotoxicidad : Para los ensayos que siguen este método de ensayo y se realizan en presencia de citocalasina B, la citotoxicidad se identifica como una reducción del índice de proliferación con bloqueo citocinético (IPBC) o del índice de replicación (IR) de las células tratadas en comparación con el testigo negativo (véanse el punto 26 y el apéndice 2). Para los ensayos que siguen este método de ensayo y se realizan en ausencia de citocalasina B, la citotoxicidad se identifica como una reducción de la duplicación relativa de la población (DRP) o del aumento relativo del recuento de células (ARRC) de las células tratadas en comparación con el testigo negativo (véanse el punto 27 y el apéndice 2). Genotoxicidad : Término general que engloba todos los tipos de lesión del ADN o de los cromosomas, con inclusión de roturas, deleciones, adiciones, modificaciones y uniones de nucleótidos, reorganizaciones, mutaciones génicas, aberraciones cromosómicas y aneuploidía. No todos los tipos de efectos genotóxicos resultan en mutaciones o en lesiones cromosómicas estables. Células en interfase : Células que no se encuentran en fase mitótica. Cinetocoro : Estructura proteínica que se encuentra en el centrómero del cromosoma y a la cual se asocian las fibras del huso acromático durante la división celular para permitir el movimiento ordenado de los cromosomas hijos hacia los polos de las células hijas. Micronúcleos : Núcleos pequeños, adicionales a los núcleos celulares principales y separados de ellos, y producidos durante la telofase de la mitosis o de la meiosis por fragmentos cromosómicos o cromosomas enteros retardados. Mitosis : División del núcleo celular, generalmente compuesta por profase, prometafase, metafase, anafase y telofase. Índice mitótico : Relación entre el número de células en metafase y el número total de células observadas en una población celular; indica el grado de proliferación celular de dicha población. Mutágeno : Agente que provoca un cambio hereditario en una o varias secuencias de pares de bases del ADN en los genes o en la estructura de los cromosomas (aberraciones cromosómicas). No disyunción : Caso en que las cromátidas emparejadas no se separan ni se segregan adecuadamente para repartirse entre las células hijas en formación, lo que provoca en estas la presencia de unos números anormales de cromosomas. Situación de la proteína p53 : La proteína p53 participa en la regulación del ciclo celular, en la apoptosis y en la reparación del ADN. Las células deficientes en proteína p53 funcional, incapaces de detener el ciclo celular o de eliminar las células dañadas por apoptosis u otros mecanismos (por ejemplo, inducción de la reparación del ADN) en relación con funciones de la proteína p53 en respuesta a las lesiones del ADN, deben estar, en teoría, más expuestas a las mutaciones génicas o a las aberraciones cromosómicas. Poliploidía : Aberraciones cromosómicas numéricas en células u organismos que afectan a dotaciones completas de cromosomas, frente a las aberraciones que afectan solo a cromosomas sueltos (aneuploidía). Índice de proliferación (IP) : Método de medición de la citotoxicidad sin utilización de la citoB (véase la fórmula en el apéndice 2). Aumento relativo del recuento celular (ARRC, o RICC por su nombre en inglés) : Método de medición de la citotoxicidad sin utilización de la citoB (véase la fórmula en el apéndice 2). Duplicación relativa de la población (DRP) : Método de medición de la citotoxicidad sin utilización de la citoB (véase la fórmula en el apéndice 2). Índice de replicación (IR) : Relación entre el número de ciclos de división celular completados en un cultivo tratado y el del testigo sin tratar, durante el tiempo de exposición y la recuperación (véase la fórmula en el apéndice 2). Fracción hepática S9 : Sobrenadante de homogeneizado de hígado después de centrifugación a 9 000 g, es decir, extracto de hígado crudo. Mezcla S9 : Mezcla de la fracción hepática S9 y de cofactores necesarios para la actividad metabólica de las enzimas. Testigo del disolvente : Término general para definir los cultivos testigo que reciben solo el disolvente utilizado para disolver el producto problema. Producto problema : Sustancia o mezcla estudiada con este método de ensayo. Testigos sin tratar : Cultivos que no reciben tratamiento (es decir, ni producto problema ni disolvente), pero que se someten al mismo proceso en paralelo que los cultivos que reciben el producto problema. Apéndice 2 FÓRMULAS PARA LA EVALUACIÓN DE LA CITOTOXICIDAD Cuando se utiliza citoB , la evaluación de la citotoxicidad debe basarse en el índice de proliferación con bloqueo citocinético (IPBC) o en el índice de replicación (IR) (17) (69). El IPBC indica el número medio de núcleos por célula, y puede utilizarse para calcular la proliferación celular. El IR indica el número relativo de ciclos celulares por célula durante el período de exposición a la citoB en los cultivos tratados respecto al de los cultivos testigo y puede utilizarse para calcular el porcentaje de citostasis: % de citostasis = 100 – 100{(IPBCT – 1) ÷ (IPBCC – 1)} y:
donde:
Así pues, un IPBC de 1 (todas las células son mononucleadas) es equivalente a un 100 % de citostasis. Citostasis = 100 – IR
Así pues, un IR del 53 % significa que, respecto al número de células que se han dividido para formar células binucleadas y multinucleadas en el cultivo testigo, solo el 53 % de este número se ha dividido en el cultivo tratado, es decir, hay un 47 % de citostasis. Cuando no se utiliza citoB , se recomienda evaluar la citotoxicidad sobre la base del aumento relativo del recuento celular (ARRC) o de la duplicación relativa de la población (DRP) (69), ya que ambos parámetros tienen en cuenta la proporción de la población celular que se ha dividido.
donde: N.o de duplicaciones de la población = [log (número de células tras el tratamiento ÷ número de células al inicio)] ÷ log 2 Así pues, un ARRC o una DRP del 53 % indica un 47 % de citotoxicidad o citostasis. Si se utiliza el índice de proliferación (IP), es posible evaluar la citotoxicidad mediante el recuento de los clones formados por una célula (cl1), dos células (cl2), de tres a cuatro células (cl4) y de cinco a ocho células (cl8):
El IP se viene utilizando como parámetro válido y fiable de citotoxicidad también con líneas celulares cultivadas in vitro en ausencia de citoB (35) (36) (37) (38) y puede considerarse un parámetro adicional útil. En cualquier caso, el número de células antes del tratamiento debe ser el mismo en el caso de los cultivos testigo tratados y negativos. Si bien el recuento celular relativo (RCR, número de células en los cultivos tratados dividido por el número de células en los cultivos testigo) se había utilizado como parámetro de citotoxicidad en el pasado, ya no se recomienda porque puede subestimar la citotoxicidad. Cuando se utilizan sistemas de examen automático, por ejemplo, la citometría de flujo, la citometría de barrido por láser o el análisis de imágenes, el número de células en la fórmula puede sustituirse por el número de núcleos. En los cultivos de testigos negativos, la duplicación de la población o el índice de replicación deberán ser compatibles con el requisito de muestrear las células después del tratamiento durante un tiempo equivalente a unas 1,5-2,0 veces la duración del ciclo celular normal. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

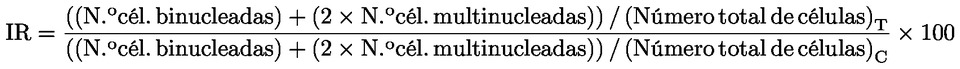

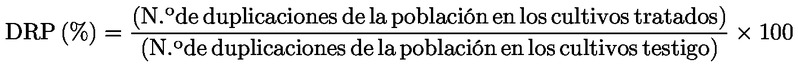

|
15) |
En la parte B se añaden los capítulos siguientes: «B.59. Sensibilización cutánea in chemico: ensayo directo de reactividad peptídica (DPRA) INTRODUCCIÓN El presente método de ensayo es equivalente a las directrices de ensayo de la OCDE TG 442C (2015). Un sensibilizante cutáneo es una sustancia que induce una respuesta alérgica por contacto con la piel, tal como se define en el Sistema Globalmente Armonizado de clasificación y etiquetado de productos químicos de las Naciones Unidas (SGA de la ONU) (1) y en el Reglamento (CE) n.o 1272/2008, sobre clasificación, etiquetado y envasado de sustancias y mezclas (CLP) de la Unión Europea (UE) (18). El presente método de ensayo proporciona un procedimiento in chemico (ensayo directo de reactividad peptídica, DPRA por su nombre en inglés) utilizado para apoyar la discriminación entre los sensibilizantes cutáneos y los no sensibilizantes de conformidad con el SGA de las Naciones Unidas y el CLP. Existe un acuerdo general sobre los principales fenómenos biológicos subyacentes a la sensibilización cutánea. Los actuales conocimientos acerca de los mecanismos biológicos y químicos asociados con la sensibilización cutánea se han resumido en forma de una ruta de resultados adversos (Adverse Outcome Pathway, AOP) (2), desde el fenómeno molecular desencadenante, pasando por los fenómenos intermedios, hasta el efecto adverso, en concreto la dermatitis alérgica de contacto en seres humanos o la hipersensibilidad de contacto en roedores. Dentro de la AOP de la sensibilización cutánea, el fenómeno molecular desencadenante es la unión covalente de sustancias electrofílicas a los centros nucleofílicos de las proteínas de la piel. Tradicionalmente, la evaluación de la sensibilización cutánea se hacía con animales de laboratorio. Los métodos clásicos con cobayas, como el ensayo de maximización con cobayas de Magnusson y Kligman (GMPT) y el ensayo de Buehler [método B.6 (3)], estudian tanto la fase de inducción como la de desencadenamiento de la sensibilización cutánea. Un ensayo con múridos, el ensayo con ganglios linfáticos locales [LLNA, método B.42 (4)], y sus dos modificaciones no radiactivas, el LLNA: DA [método B.50 (5)] y el LLNA: BrdU-ELISA [método B.51 (6)], que evalúan exclusivamente la respuesta de inducción, también han conseguido aceptación porque, sobre los ensayos con cobayas, tienen la ventaja de respetar más el bienestar de los animales y de permitir una medición objetiva de la fase de inducción de la sensibilización cutánea. Más recientemente, algunos métodos farmacodinámicos in chemico e in vitro se han considerado científicamente válidos para la evaluación del peligro de sensibilización cutánea que representan los productos. Sin embargo, será necesario recurrir a combinaciones de métodos alternativos sin animales (in silico, in chemico, in vitro) dentro de un enfoque integrado de pruebas y evaluación (IATA) para poder sustituir por completo los ensayos con animales que se utilizan en la actualidad, habida cuenta de la limitada cobertura en cuanto a los mecanismos de la AOP de cada uno de los métodos de ensayo actualmente disponibles sin animales (2) (7). Se propone el DPRA para abordar el fenómeno molecular desencadenante de la AOP de la sensibilización cutánea, que es la reactividad proteínica, cuantificando la reactividad de los productos problema frente a péptidos sintéticos modelo que contienen lisina o bien cisteína (8). Los valores de reducción porcentual de los péptidos que contienen cisteína y lisina se utilizan para clasificar las sustancias en una de los cuatro clases de reactividad en apoyo de la discriminación entre los sensibilizantes cutáneos y los no sensibilizantes (9). El DPRA se ha evaluado en un estudio de validación dirigido por el laboratorio de referencia de la Unión Europea para las alternativas a los ensayos con animales (LRUE del CEVMA) y sujeto posteriormente a revisión inter pares independiente por el Comité Científico Consultivo del CEVMA (ESAC) (10) y se considera científicamente válido como parte de un enfoque IATA para apoyar la discriminación entre los sensibilizantes cutáneos y los no sensibilizantes con fines de clasificación de peligro y etiquetado. En la bibliografía se recogen ejemplos del uso de datos del DPRA en combinación con otra información (11) (12) (13) (14). En el apéndice I se dan las definiciones pertinentes. CONSIDERACIONES INICIALES, APLICABILIDAD Y LIMITACIONES La correlación de la reactividad proteínica con el potencial de sensibilización cutánea está bien establecida (15) (16) (17). No obstante, dado que la unión a proteínas representa solamente uno de los fenómenos clave, aunque sea el fenómeno molecular desencadenante de la AOP de la sensibilización cutánea, la información sobre la reactividad proteínica obtenida con métodos de ensayo y otros no experimentales puede no ser suficiente por sí sola para llegar a una conclusión sobre la ausencia de potencial de sensibilización cutánea de los productos. Por lo tanto, los datos obtenidos con este método de ensayo deben considerarse en el contexto de enfoques integrados, como los enfoques IATA, combinándolos con otra información complementaria, por ejemplo derivada de ensayos in vitro que aborden otros fenómenos clave de la AOP de la sensibilización cutánea, así como de métodos no experimentales, incluida la extrapolación a partir de análogos químicos. El presente método de ensayo se puede utilizar en combinación con otra información complementaria, para apoyar la discriminación entre sensibilizantes cutáneos (es decir, categoría 1 del SGA de las Naciones Unidas o del CLP) y no sensibilizantes en el contexto de un enfoque IATA. El presente método de ensayo no puede utilizarse por sí solo para clasificar sensibilizantes cutáneos en las subcategorías 1A y 1B definidas por el SGA de las Naciones Unidas o el CLP, ni para predecir la potencia de decisiones sobre evaluación de seguridad. Sin embargo, en función del marco normativo, un resultado positivo con el DPRA podrá utilizarse por sí solo para la clasificación de un producto en la categoría 1 del SGA de las Naciones Unidas o del CLP. Está comprobado que el método de ensayo DPRA es transferible a laboratorios con experiencia en los análisis con cromatografía de líquidos de alta resolución (HPLC). El nivel de reproducibilidad de las predicciones que cabe esperar del método de ensayo es del orden del 85 % dentro de un laboratorio y del 80 % entre laboratorios (10). Los resultados obtenidos en el estudio de validación (18) y en estudios publicados (19) en general indican que la exactitud del DPRA en cuanto a la discriminación de los sensibilizantes (es decir, cat. 1 del SGA de las Naciones Unidas o del CLP) y los no sensibilizantes es del 80 % (N=157) con una sensibilidad del 80 % (88/109) y una especificidad del 77 % (37/48) en comparación con los resultados del LLNA. Es más probable que el DPRA subestime los productos que muestran una potencia de sensibilización cutánea baja o moderada (es decir, subcategoría 1B del SGA de las Naciones Unidas o del CLP) que los productos que presentan una potencia de sensibilización cutánea elevada (es decir, subcategoría 1A del SGA de las Naciones Unidas o del CLP) (18) (19). Sin embargo, los valores de la exactitud aquí indicados para el DPRA utilizado como único método de ensayo solo son indicativos, ya que el método de ensayo debe considerarse en combinación con otras fuentes de información en el contexto de un enfoque IATA y de conformidad con las disposiciones del punto 9 anterior. Además, a la hora de evaluar los métodos de ensayo de la sensibilización cutánea sin animales, debe tenerse en cuenta que el ensayo LLNA y otros ensayos con animales pueden no reflejar plenamente la situación en la especie de interés, es decir, los seres humanos. Sobre la base de los datos disponibles en general, se ha demostrado que el DPRA es aplicable a productos problema que abarcan una gran variedad de grupos funcionales orgánicos, mecanismos de reacción, potencias de sensibilización cutánea (determinada en estudios in vivo) y propiedades fisicoquímicas (8) (9) (10) (19). En conjunto, esta información indica la utilidad del DPRA para contribuir a la identificación del peligro de sensibilización cutánea. El término «producto problema» se utiliza en el presente método de ensayo para referirse a lo que se somete a ensayo y no está relacionado con la aplicabilidad del DPRA al ensayo de sustancias o mezclas. El presente método de ensayo no es aplicable a los compuestos metálicos, de los que se sabe que reaccionan con proteínas por mecanismos distintos de la unión covalente. El producto problema debe ser soluble en un disolvente adecuado a una concentración final de 100 mM (véase el punto 18). No obstante, los productos problema que no sean solubles a dicha concentración pueden someterse a ensayo a concentraciones solubles más bajas. En tal caso, un resultado positivo podría seguir utilizándose para apoyar la identificación del producto problema como sensibilizante cutáneo, pero de un resultado negativo no podría deducirse ninguna conclusión firme sobre la falta de reactividad. Se dispone actualmente de información limitada sobre la aplicabilidad del DPRA a las mezclas de composición conocida (18) (19). Sin embargo, se considera que el DPRA es técnicamente aplicable al ensayo de las sustancias con componentes múltiples y mezclas de composición conocida (véase el punto 18). Antes de la utilización de este método de ensayo con una mezcla para obtener datos con fines reglamentarios, debe considerarse si podría proporcionar resultados adecuados a tal fin y, en caso afirmativo, por qué. Tales consideraciones no son necesarias si la reglamentación impone el ensayo de la mezcla. El actual modelo de predicción no puede utilizarse con mezclas complejas de composición desconocida o con sustancias de composición desconocida o variable, productos complejos de reacción o materiales biológicos (es decir, sustancias UVCB) debido a la proporción molar definida del producto problema y del péptido. Con este fin, deberá desarrollarse un nuevo modelo de predicción basado en un enfoque gravimétrico. En los casos en que haya pruebas sobre la inaplicabilidad del método de ensayo a otras categorías específicas de productos, el método de ensayo no deberá utilizarse con tales categorías específicas de productos. El presente método de ensayo es un método in chemico que no incluye ningún sistema metabólico. Los productos que requieren bioactivation enzimática para realizar su potencial de sensibilización cutánea (es decir, pro-haptenos) no pueden detectarse por este método de ensayo. Se señala que los productos que se hacen sensibilizantes después de una transformación abiótica (es decir, pre-haptenos) son detectados correctamente en algunos casos por este método de ensayo (18). En vista de lo anterior, los resultados obtenidos con el método de ensayo deben interpretarse en el contexto de las limitaciones señaladas y en relación con otras fuentes de información en el marco de un enfoque IATA. Los productos problema que no se unan covalentemente al péptido sino que fomenten su oxidación (es decir, la dimerización de la cisteína) podrían provocar una posible sobreestimación de la reducción del péptido, con el consiguiente riesgo de predicciones positivas falsas o de la adscripción a una clase de reactividad más elevada (véanse los puntos 29 y 30). Tal como se describe, el DPRA apoya la discriminación entre los sensibilizantes cutáneos y los no sensibilizantes. No obstante, podrá contribuir también a la evaluación de la potencia sensibilizante (11) cuando se utilice en enfoques integrados, como los enfoques IATA. Sin embargo, es necesario seguir trabajando, preferentemente a partir de datos humanos, para determinar de qué modo los resultados del DPRA pueden informar sobre la evaluación de la potencia. PRINCIPIO DEL ENSAYO El DPRA es un método in chemico que cuantifica la concentración restante de péptido que contiene cisteína o lisina tras 24 horas de incubación a 25 ± 2,5 °C con el producto problema. Los péptidos sintéticos contienen fenilalanina a fin de ayudar en la detección. La concentración relativa del péptido se mide mediante cromatografía de líquidos de alta resolución (HPLC) con elución en gradiente y detección por UV a 220 nm. Los valores porcentuales de reducción del péptido con cisteína y lisina se calculan a continuación y se utilizan en un modelo de predicción (véase el punto 29) que permite asignar el producto problema a una de las cuatro clases de reactividad utilizadas para apoyar la discriminación entre sensibilizantes y no sensibilizantes. Antes de proceder al uso sistemático del método descrito en el presente método de ensayo, los laboratorios deben demostrar su competencia técnica, utilizando las diez sustancias recomendadas al efecto en el apéndice 2. PROCEDIMIENTO El presente método de ensayo se basa en el protocolo n.o 154 DB-ALM del DPRA (20), que representa el protocolo utilizado en el estudio de validación coordinado por el LRUE del CEVMA. Se recomienda que se siga este protocolo cuando se aplique y utilice el método en el laboratorio. A continuación se recoge una descripción de los principales componentes y procedimientos del DPRA. Si se utiliza una configuración alternativa de HPLC, debe demostrarse su equivalencia con la configuración validada descrita en el protocolo DB-ALM (por ejemplo, sometiendo a ensayo las sustancias para la competencia que figuran en el apéndice 2). Preparación de los péptidos que contienen cisteína o lisina Las soluciones madre de péptidos sintéticos que contienen cisteína (Ac-RFAACAA-COOH) y lisina (Ac-RFAAKAA-COOH), de pureza superior al 85 % y preferiblemente en la gama de 90-95 %, deben prepararse justo antes de su incubación con el producto problema. La concentración final del péptido con cisteína debe ser de 0,667 mM en solución amortiguadora de fosfato de pH 7,5, mientras que la concentración final del péptido con lisina debe ser de 0,667 mM en solución amortiguadora de acetato de amonio de pH 10,2. La secuencia de tandas de HPLC debe establecerse de forma que se mantenga el tiempo de análisis con HPLC por debajo de 30 horas. Con la configuración de HPLC utilizada en el estudio de validación y descrita en este método de ensayo, pueden acogerse en una misma tanda de HPLC hasta 26 muestras analíticas (que incluyen el producto problema, el testigo positivo y el número adecuado de testigos de disolvente en función del número de disolventes distintos utilizados en el ensayo, cada uno ensayado por triplicado). Todas las réplicas analizadas en la misma tanda deben utilizar las misma soluciones madre de péptidos con cisteína y lisina. Se recomienda comprobar los diferentes lotes de péptidos antes de su utilización para garantizar que su solubilidad es adecuada. Preparación del producto problema La solubilidad del producto problema en un disolvente adecuado debe evaluarse antes de realizar el ensayo siguiendo el procedimiento de solubilización descrito en el protocolo DB-ALM del DPRA (20). Un disolvente adecuado debe disolver completamente el producto problema. Dado que en el DPRA se incuba con péptidos de cisteína o de lisina un gran exceso de producto problema, la inspección visual de la formación de una solución clara se considera suficiente para determinar que está disuelto el producto problema (y todos sus componentes en el caso de someter a ensayo una sustancia de componentes múltiples o una mezcla). Son disolventes adecuados el acetonitrilo, el agua, la mezcla 1:1 de agua y acetonitrilo, el isopropanol, la acetona o la mezcla 1:1 de acetona y acetonitrilo. Pueden utilizarse otros disolventes siempre y cuando no afecten a la estabilidad del péptido controlada con testigos de referencia C (es decir, muestras constituidas por el péptido solo, disuelto en el disolvente adecuado; véase el apéndice 3). Como última opción, si el producto problema no es soluble en ninguno de estos disolventes, se debe intentar solubilizarlo en 300 μl de DMSO y diluir la solución resultante con 2 700 μl de acetonitrilo y, si el producto problema no es soluble en esta mezcla, se debe intentar solubilizar la misma cantidad de producto problema en 1 500 μl de DMSO y diluir la solución resultante con 1 500 μl de acetonitrilo. El producto problema debe pesarse previamente en frascos de vidrio y disolverse inmediatamente antes del ensayo en un disolvente apropiado a fin de preparar una solución 100 mM. Con las mezclas y las sustancias de componentes múltiples de composición conocida, deben determinarse una única pureza, sumando la proporción de sus componentes (excepto el agua), y un único peso molecular aparente, considerando los distintos pesos moleculares de cada componente de la mezcla (excepto el agua) y sus respectivas proporciones. La pureza y el peso molecular aparente resultantes deben utilizarse a continuación en el cálculo del peso de producto problema necesario para preparar una solución 100 mM. En el caso de los polímeros de los que no pueda determinarse un peso molecular predominante, podría considerarse el peso molecular del monómero (o el peso molecular aparente de los distintos monómeros que constituyen el polímero) para preparar una solución 100 mM. Sin embargo, cuando se someten a ensayo mezclas, sustancias de componentes múltiples o polímeros de composición conocida, debe considerarse la posibilidad de someter a ensayo también el producto puro. En el caso de los líquidos, el producto puro debe someterse a ensayo tal cual, sin dilución previa, incubándolo a las proporciones molares de 1:10 y 1:50 con los péptidos de cisteína y lisina, respectivamente. En el caso de los sólidos, el producto problema se debe disolver a la máxima concentración soluble en el mismo disolvente utilizado para preparar la solución de concentración aparente 100 mM. Debe entonces someterse a ensayo tal cual, sin diluir más, incubándolo a las proporciones molares de 1:10 y 1:50 con los péptidos de cisteína y lisina, respectivamente. Unos resultados concordantes (reactivos o no reactivos) entre la solución de concentración aparente 100 mM y el producto puro deben permitir la deducción de una conclusión firme sobre el resultado. Preparación del testigo positivo, de los testigos de referencia y de los testigos de coelución El aldehído cinámico (CAS 104-55-2; ≥ 95 % de pureza de grado alimentario) debe utilizarse como testigo positivo (TP) a una concentración de 100 mM en acetonitrilo. Pueden utilizarse otros testigos positivos adecuados que den preferentemente valores de reducción en la gama media, si se dispone de datos históricos para obtener criterios de aceptación de tandas comparables. Por otra parte, también han de incluirse testigos de referencia (es decir, muestras que contengan únicamente el péptido disuelto en el disolvente adecuado) en la secuencia de tandas de HPLC y se utilizan para comprobar la idoneidad del sistema de HPLC antes del análisis (testigos de referencia A), la estabilidad de los testigos de referencia a lo largo del tiempo (testigos de referencia B) y que el disolvente utilizado para disolver el producto problema no afecta a la reducción porcentual del péptido (testigos de referencia C) (véase el apéndice 3). El testigo de referencia apropiado para cada producto se utiliza para calcular la reducción porcentual del péptido correspondiente a ese producto (véase el punto 26). Además, respecto a cada uno de los productos problema analizados, debe incluirse en la secuencia de tandas un testigo de coelución constituido por el producto problema solo, a fin de detectar la posible coelución del producto problema con el péptido de lisina o de cisteína. Incubación del producto problema con las soluciones del péptido de lisina y de cisteína Las soluciones del péptido de cisteína y de lisina deben incubarse en frascos de vidrio del automuestreador con el producto problema a las proporciones de 1:10 y 1:50 respectivamente. Si se observa un precipitado inmediatamente después de la adición de la solución del producto problema a la solución de péptido, debido a la baja hidrosolubilidad del producto problema, no se puede estar seguro de qué proporción del producto problema permanece en la solución para reaccionar con el péptido. Por consiguiente, en ese caso, un resultado positivo puede ser útil todavía, pero un resultado negativo es incierto y debe interpretarse con la debida diligencia (véase también lo dispuesto en el punto 11 sobre el ensayo de productos insolubles hasta una concentración de 100 mM). La solución de reacción debe quedarse en la oscuridad y a 25 ± 2,5 °C durante 24 ± 2 horas antes del análisis con HPLC. Cada producto problema debe analizarse por triplicado con ambos péptidos. Se inspeccionarán visualmente unas muestras antes del análisis con HPLC. Si se observa precipitado o separación de fases, pueden centrifugarse las muestras a baja velocidad (100-400 x g) para llevar el precipitado hasta el fondo del frasco como precaución, ya que la presencia de grandes cantidades de precipitado puede obstruir los tubos o columnas de HPLC. Si se observa precipitación o separación de fases tras el período de incubación, es posible subestimar la reducción del péptido, por lo que en caso de resultado negativo no puede extraerse con la confianza suficiente ninguna conclusión sobre la falta de reactividad. Preparación de la curva patrón de calibración de HPLC Debe obtenerse una curva patrón de calibración para los péptidos, tanto para el de cisteína como para el de lisina. Deben prepararse patrones de los péptidos en una solución del 20 % o 25 % de acetonitrilo:solución amortiguadora, utilizando solución amortiguadora de fosfato (pH 7,5) para el péptido de cisteína y solución amortiguadora de acetato de amonio (pH 10,2) para el péptido de lisina. Utilizando diluciones en serie de la solución madre de péptido (0,667 mM), deben prepararse seis soluciones de calibración para cubrir la gama de 0,534 a 0,0167 mM. Debe incluirse también en la curva patrón de calibración un blanco de la solución amortiguadora de dilución. Las curvas de calibración adecuadas deben tener un valor r2 > 0,99. Preparación de la HPLC y análisis La idoneidad del sistema de HPLC debe verificarse antes de efectuar el análisis. La reducción de los péptidos se determina mediante HPLC combinada con un detector de UV (detector por red de fotodiodos o detector de absorbancia de longitud de onda fija con señal de 220 nm). La columna correspondiente se instala en el sistema de HPLC. La configuración de HPLC descrita en el protocolo validado utiliza como columna preferida una Zorbax SB-C-18 de 2,1 mm × 100 mm × 3,5 micras. Con esta columna de HPLC de fase inversa, todo el sistema debe equilibrarse a 30 °C con un 50 % de fase A (0,1 % (v/v) de ácido trifluoroacético en agua) y un 50 % de fase B (0,085 % (v/v) de ácido trifluoroacético en acetonitrilo) durante al menos 2 horas antes de pasar la tanda. El análisis con HPLC debe efectuarse utilizando un caudal de 0,35 ml/min y un gradiente lineal del 10 % al 25 % de acetonitrilo durante 10 minutos, seguido de un rápido incremento hasta el 90 % de acetonitrilo para eliminar otros materiales. Deben inyectarse volúmenes iguales de cada patrón, muestra y testigo. La columna debe volver a equilibrarse entre inyecciones en las condiciones iniciales durante 7 minutos. Si se utiliza una columna de HPLC de fase inversa diferente, puede ser necesario ajustar los parámetros de configuración descritos anteriormente, para garantizar la adecuada elución e integración de los péptidos de cisteína y lisina, incluido el volumen de inyección, que podrá variar en función del sistema utilizado (normalmente en la banda de 3-10 μl). Si se utiliza una configuración alternativa de HPLC, es importante demostrar su equivalencia con la configuración validada descrita anteriormente (por ejemplo, sometiendo a ensayo las sustancias para la competencia que figuran en el apéndice 2). La absorbancia se mide a 220 nm. En caso de que se utilice un detector por red de fotodiodos, debe registrarse también la absorbancia a 258 nm. Debe señalarse que algunos lotes de acetonitrilo pueden tener un impacto negativo en la estabilidad de los péptidos, y esto ha de evaluarse cuando se utilice un nuevo lote de acetonitrilo. La relación entre el área del pico 220 y el área del pico 258 puede utilizarse como indicador de coelución. Respecto a cada muestra, una relación en la banda de 90 % < relación media (19) de las áreas de las muestras testigo <100 % puede constituir una buena indicación de que no ha habido coelución. Puede haber productos problema que favorezcan la oxidación del péptido de cisteína. El pico del péptido de cisteína dimerizado puede ser objeto de seguimiento visual. En los casos en que resulte que se ha producido dimerización, esto debe indicarse, ya que es posible sobrestimar la reducción porcentual del péptido, lo que llevaría a falsas predicciones positivas o a la asignación a una clase de reactividad más elevada (véanse los puntos 29 y 30). El análisis con HPLC de los péptidos de cisteína y de lisina puede realizarse simultáneamente (si se dispone de dos sistemas de HPLC) o en días distintos. Si el análisis se realiza en días distintos, todas las soluciones del producto problema deben prepararse de nuevo para los dos ensayos cada día. El análisis debe programarse para garantizar que la inyección de la primera muestra comienza entre 22 y 26 horas después de la mezcla del producto problema con la solución de péptido. La secuencia de tandas de HPLC debe establecerse de forma que se mantenga el tiempo de análisis con HPLC por debajo de 30 horas. Con la configuración de HPLC utilizada en el estudio de validación y descrita en este método de ensayo, pueden analizarse hasta 26 muestras en una sola tanda de HPLC (véase también el punto 17). En el apéndice 3 se encuentra un ejemplo de secuencia de análisis con HPLC. DATOS E INFORME Evaluación de los datos La concentración de péptido de cisteína o de lisina se determina por fotometría a 220 nm en cada muestra midiendo el área del pico (área bajo la curva, AUC) de los picos adecuados y se calcula la concentración de péptido utilizando la curva de calibración lineal obtenida con los patrones. La reducción porcentual de los péptidos en cada muestra se determina midiendo el área del pico y dividiéndola por la media del área del pico de los testigos de referencia C pertinentes (véase el apéndice 3), con arreglo a la fórmula descrita a continuación.
Criterios de aceptación Para que una tanda se considere válida, deben cumplirse los siguientes criterios:
En caso de que no se cumpla uno o varios de estos criterios, la tanda debe repetirse. Para que los resultados de un producto problema se consideren válidos, deben cumplirse los siguientes criterios:
Modelo de predicción Se calcula la reducción porcentual media de cisteína y de lisina para cada producto problema. Una reducción negativa se considera «0» al calcular la media. Utilizando el modelo de predicción cisteína 1:10/lisina 1:50 que se muestra en el cuadro 1, ha de aplicarse el umbral del 6,38 % de reducción media de los péptidos para apoyar la discriminación entre los sensibilizantes cutáneos y los no sensibilizantes en el marco de un enfoque IATA. La aplicación del modelo de predicción para asignar un producto problema a una clase de reactividad (es decir, reactividad baja, moderada y elevada) puede quizás resultar útil para informar sobre la evaluación de la potencia en el marco de un enfoque IATA. Cuadro 1: Modelo de predicción (20) cisteína 1:10/lisina 1:50
Puede haber casos en los que el producto problema (la sustancia o uno o varios de los componentes de una sustancia de componentes múltiples o de una mezcla) absorba significativamente a 220 nm y tenga el mismo tiempo de retención que el péptido (coelución). La coelución puede resolverse ajustando ligeramente la configuración de la HPLC para separar más el tiempo de elución del producto problema y el del péptido. Si se utiliza una configuración alternativa de HPLC para resolver la coelución, se debe demostrar su equivalencia con la configuración validada (por ejemplo, sometiendo a ensayo las sustancias para la competencia que figuran en el apéndice 2). Cuando se produce coelución, no puede integrarse el pico del péptido y resulta imposible calcular la reducción porcentual del péptido. Si se produce coelución de tales productos problema tanto con el péptido de cisteína como con el de lisina, el análisis debe considerarse «no concluyente». En los casos en que solo se produce coelución con el péptido de lisina, puede utilizarse el modelo de predicción cisteína 1:10 indicado en el cuadro 2. Cuadro 2: Modelo de predicción (22) cisteína 1:10
Es posible que haya otros casos en los que sea incompleto el solapamiento entre el tiempo de retención del producto problema y el de alguno de los péptidos. En tales casos, pueden calcularse los valores de reducción porcentual de los péptidos, y se pueden utilizar en el modelo de predicción cisteína 1:10/lisina 1:50; sin embargo, la asignación del producto problema a una clase de reactividad no puede efectuarse con exactitud. Debe ser suficiente un único análisis de un producto problema con HPLC tanto para el péptido de cisteína como para el de lisina, si el resultado es inequívoco. Sin embargo, en casos de resultados próximos al umbral utilizado para discriminar entre resultados positivos y negativos (es decir, resultados límite), puede ser necesario efectuar más ensayos. En las situaciones en las que la reducción porcentual media está comprendida en el intervalo del 3 % al 10 % con el modelo de predicción cisteína 1:10/lisina 1:50 o la reducción porcentual de cisteína está comprendida en el intervalo del 9 % al 17 % con el modelo de predicción cisteína 1:10, debe considerarse la conveniencia de efectuar una segunda tanda, así como una tercera en caso de resultados discordantes entre las dos primeras. Informe del ensayo El informe del ensayo debe incluir la información siguiente:
BIBLIOGRAFÍA
Apéndice 1 DEFINICIONES Exactitud : Grado de coincidencia entre los resultados obtenidos con el método de ensayo y los valores de referencia aceptados. Se trata de una medida del comportamiento del método de ensayo y es un aspecto de su »pertinencia«. Este término y el de »concordancia« se suelen usar indistintamente para indicar la proporción de resultados correctos de un método de ensayo (21). Ruta de resultados adversos (AOP, Adverse Outcome Pathway) : Secuencia de fenómenos desde la estructura química de un producto o grupo de productos similares diana, pasando por el fenómeno molecular desencadenante, hasta un resultado in vivo de interés (2). Curva de calibración : Relación entre el valor de la respuesta experimental y la concentración analítica (también denominada curva patrón) de una sustancia conocida. Producto : Sustancia o mezcla. Coeficiente de variación : Medida de la variabilidad que se calcula para un grupo de datos replicados dividiendo la desviación típica por la media. Puede multiplicarse por 100 para expresarlo como porcentaje. Peligro : Propiedad inherente de un agente o situación que tiene capacidad para provocar efectos adversos cuando un organismo, sistema o (sub)población se expone a dicho agente. Enfoque integrado de pruebas y evaluación (IATA, Integrated Approach to Testing and Assessment) : Enfoque estructurado para la identificación del peligro (potencial), caracterización del peligro (potencia) o evaluación de la seguridad (potencial/potencia y exposición) de un producto o grupo de productos, que integra y pondera de forma estratégica todos los datos pertinentes para fundamentar una decisión reglamentaria relativa a posibles peligros o riesgos o a la necesidad de realizar ensayos más específicos y, por tanto, mínimos. Fenómeno molecular desencadenante : Perturbación de un sistema biológico inducida por un producto a nivel molecular y caracterizada como el fenómeno inicial de la ruta de resultados adversos. Mezcla : Mezcla o solución compuesta por dos o más sustancias que no reaccionan en ella (1). Sustancia con un solo componente : Sustancia, definida por su composición cuantitativa, en la que un solo componente principal representa al menos el 80 % (p/p). Sustancia de componentes múltiples : Sustancia, definida por su composición cuantitativa, en la que hay varios componentes principales presentes a una concentración ≥ 10 % (p/p) y < 80 % (p/p). Una sustancia de componentes múltiples es el resultado de un proceso de fabricación. La diferencia entre mezcla y sustancia de componentes múltiples es que una mezcla se obtiene mezclando dos o más sustancias sin reacción química, mientras que una sustancia de componentes múltiples es el resultado de una reacción química. Testigo positivo : Réplica que contiene todos los componentes de un sistema de ensayo y que se trata con una sustancia de la que se sabe que induce una respuesta positiva. Para asegurar la posibilidad de evaluar la variabilidad de las respuestas del testigo positivo a lo largo del tiempo, no debe ser excesiva la magnitud de la respuesta positiva. Testigo de referencia : Muestra no tratada que contiene todos los componentes de un sistema de ensayo, incluido el disolvente o vehículo, y que se somete al mismo proceso que las muestras tratadas con el producto problema y otras muestras testigo a fin de determinar la respuesta de base correspondiente a las muestras tratadas con el producto problema disuelto en el mismo disolvente o vehículo. Cuando se somete a ensayo con un testigo negativo en paralelo, esta muestra pone de manifiesto también si el disolvente o vehículo interactúa con el sistema de ensayo. Pertinencia : Descripción de la relación del ensayo con el efecto de interés y de si es significativo y útil para un objetivo concreto. Es el grado en que el ensayo mide o predice correctamente el efecto biológico de interés. La pertinencia incorpora la consideración de la exactitud (concordancia) de un método de ensayo (21). Fiabilidad : Medida del grado en que un método de ensayo puede aplicarse de forma reproducible a lo largo del tiempo, en un mismo laboratorio y en distintos laboratorios, utilizando el mismo protocolo. Se evalúa calculando la reproducibilidad intra e interlaboratorios y la repetibilidad intralaboratorios (21). Reproducibilidad : Coincidencia entre los resultados obtenidos en el ensayo del mismo producto utilizando el mismo protocolo de ensayo (véase »Fiabilidad«) (21). Sensibilidad : Proporción de todos los productos activos/positivos que se clasifican correctamente mediante el método de ensayo. Es una medida de la exactitud de un método de ensayo que produce resultados categoriales, y un factor importante en la evaluación de la pertinencia de un método de ensayo (21). Especificidad : Proporción de todos los productos inactivos/negativos que se clasifican correctamente mediante el método de ensayo. Es una medida de la exactitud de un método de ensayo que produce resultados categoriales, y un factor importante en la evaluación de la pertinencia de un método de ensayo (21). Sustancia : Elementos químicos y sus compuestos en estado natural u obtenidos mediante cualquier proceso de producción, incluidos los aditivos necesarios para mantener la estabilidad del producto y las eventuales impurezas derivadas del proceso empleado, pero excluidos los disolventes que se puedan separar sin influir en la estabilidad de la sustancia ni cambiar su composición (1). Idoneidad del sistema : Determinación del comportamiento de un instrumento (por ejemplo, sensibilidad) mediante el análisis de un patrón de referencia antes de aplicarlo al lote analítico (22). Producto problema : El término »producto problema« se utiliza para hacer referencia a lo que se somete a ensayo. Sistema Globalmente Armonizado de clasificación y etiquetado de productos químicos de las Naciones Unidas (SGA de la ONU) : Sistema que propone la clasificación de productos químicos (sustancias y mezclas) según tipos y niveles normalizados de peligros físicos, sanitarios y ambientales, y que hace referencia a los elementos correspondientes de comunicación, como pictogramas, palabras de advertencia, indicaciones de peligro, consejos de prudencia, y fichas de datos de seguridad, a efectos de proporcionar información sobre sus efectos adversos con el fin de proteger a la población (incluidos empresarios, trabajadores, transportistas, consumidores y personal de protección civil) y al medio ambiente (1). UVCB : Sustancias de composición desconocida o variable, productos complejos de reacción o materiales biológicos. Método de ensayo válido : Método de ensayo del que se considera que tiene suficiente pertinencia y fiabilidad con un fin específico y que se basa en principios sólidos desde el punto de vista científico. Un método de ensayo nunca es válido en un sentido absoluto, sino únicamente en relación con un fin determinado (21). Apéndice 2 SUSTANCIAS UTILIZADAS PARA DEMOSTRAR LA COMPETENCIA Sensibilización cutánea in chemico: Ensayo directo de reactividad peptídica Antes de proceder al uso sistemático de este método de ensayo, los laboratorios deben demostrar su competencia técnica mediante la correcta obtención de la predicción del DPRA prevista respecto a las diez sustancias recomendadas al efecto en el cuadro 1, y la obtención de valores de reducción de cisteína y lisina que estén dentro de la gama de referencia con ocho de las diez sustancias utilizadas para demostrar la competencia en relación con cada péptido. Estas sustancias para la competencia se han seleccionado a fin de representar la gama de respuestas de los peligros de sensibilización cutánea. Otros criterios de selección son su disponibilidad comercial, la disponibilidad de datos de referencia in vivo de alta calidad y de datos de alta calidad obtenidos in vitro con el DPRA, y que se hayan utilizado en el estudio de validación coordinado por el LRUE del CEVMA para demostrar el éxito de la aplicación del método de ensayo en los laboratorios participantes. Cuadro 1: Sustancias recomendadas para demostrar la competencia técnica respecto al ensayo directo de reactividad peptídica
Apéndice 3 EJEMPLOS DE SECUENCIA DE ANÁLISIS
B.60. Sensibilización cutánea in vitro: Método de ensayo de la luciferasa ARE-Nrf2 INTRODUCCIÓN El presente método de ensayo es equivalente a las directrices de ensayo de la OCDE TG 442D (2015). Un sensibilizante cutáneo es una sustancia que induce una respuesta alérgica por contacto con la piel, tal como se define en el Sistema Globalmente Armonizado de clasificación y etiquetado de productos químicos de las Naciones Unidas (SGA de la ONU) (1) y en el Reglamento (CE) n.o 1272/2008, sobre clasificación, etiquetado y envasado de sustancias y mezclas (CLP) de la Unión Europea (UE) (27). El presente método de ensayo proporciona un procedimiento in vitro (ensayo de la luciferasa ARE-Nrf2) utilizado para apoyar la discriminación entre los sensibilizantes cutáneos y los no sensibilizantes de conformidad con el SGA de la ONU (1) y el CLP. Existe un acuerdo general sobre los principales fenómenos biológicos subyacentes a la sensibilización cutánea. Los actuales conocimientos acerca de los mecanismos biológicos y químicos asociados con la sensibilización cutánea se han resumido en forma de una ruta de resultados adversos (Adverse Outcome Pathway, AOP) (2), que va desde el fenómeno molecular desencadenante, pasando por los fenómenos intermedios, hasta el efecto adverso para la salud, es decir la dermatitis alérgica de contacto en seres humanos o la hipersensibilidad de contacto en roedores (2) (3). El fenómeno molecular desencadenante es la unión covalente de sustancias electrofílicas a los centros nucleofílicos de las proteínas de la piel. El segundo fenómeno clave en esta AOP tiene lugar en los queratinocitos e incluye las respuestas inflamatorias, así como la expresión génica relacionada con rutas específicas de comunicación celular, tales como las rutas dependientes del elemento de respuesta antioxidante/electrófilo (ARE, antioxidant/electrophile response element). El tercer fenómeno clave es la activación de las células dendríticas, normalmente evaluada por la expresión de marcadores específicos de la superficie celular, quimiocinas y citocinas. El cuarto fenómeno clave es la proliferación de linfocitos T, que se evalúa indirectamente en el ensayo con ganglios linfáticos locales de múridos (4). Tradicionalmente, la evaluación de la sensibilización cutánea se hacía con animales de laboratorio. Los métodos clásicos con cobayas, como el ensayo de maximización con cobayas de Magnusson y Kligman (GMPT) y el ensayo de Buehler [método B.6 (5)], estudian tanto la fase de inducción como la de desencadenamiento de la sensibilización cutánea. Un ensayo con múridos, el ensayo con ganglios linfáticos locales [LLNA, método B.42 (4)], y sus dos modificaciones no radiactivas, el LLNA: DA [método B.50 (6)] y el LLNA: BrdU-ELISA [método B.51 (7)], que evalúan exclusivamente la respuesta de inducción, también han conseguido aceptación porque, sobre los ensayos con cobayas, tienen ventaja en relación tanto con el bienestar de los animales como con la medición objetiva de la fase de inducción de la sensibilización cutánea. Más recientemente, algunos métodos farmacodinámicos in chemico e in vitro se han considerado científicamente válidos para la evaluación del peligro de sensibilización cutánea que representan los productos. Sin embargo, será necesario recurrir a combinaciones de métodos alternativos sin animales (in silico, in chemico, in vitro) dentro de un enfoque integrado de pruebas y evaluación (IATA) para poder sustituir por completo los ensayos con animales que se utilizan en la actualidad, habida cuenta de la limitada cobertura en cuanto a los mecanismos de la AOP de cada uno de los métodos de ensayo actualmente disponibles sin animales (2) (3). Se propone el presente método de ensayo (ensayo de la luciferasa ARE-Nrf2) para abordar el segundo fenómeno clave, como se explica en el punto 2. Se ha notificado que los sensibilizantes cutáneos inducen genes que están regulados por el elemento de respuesta antioxidante (ARE) (8) (9). Las pequeñas sustancias electrofílicas tales como los sensibilizantes cutáneos pueden actuar sobre la proteína sensora Keap1 (proteína 1 asociada a ECH y de tipo Kelch) mediante, por ejemplo, la modificación covalente de su residuo de cisteína, lo que da lugar a que se disocie del factor de transcripción Nrf2 (factor nuclear 2 relacionado con el factor eritroide-2). El Nrf2 disociado puede entonces activar genes dependientes del ARE, como los genes que codifican las enzimas de destoxificación de fase II (8) (10) (11). En la actualidad, el único ensayo de la luciferasa ARE-Nrf 2 in vitro correspondiente al presente método de ensayo es el ensayo KeratinoSensTM para el cual se han completado estudios de validación (9) (12) (13), seguidos de una revisión independiente inter pares dirigida por el laboratorio de referencia de la Unión Europea para las alternativas a los ensayos con animales (LRUE del CEVMA) (14). El ensayo KeratinoSensTM fue considerado científicamente válido para utilizarse como parte de un enfoque IATA, en apoyo de la discriminación entre los sensibilizantes cutáneos y los no sensibilizantes con fines de clasificación de peligro y etiquetado (14). Los laboratorios que deseen aplicar el método de ensayo pueden obtener la línea celular recombinante utilizada en el ensayo KeratinoSensTM estableciendo un acuerdo de licencia con el creador del método de ensayo (15). En el apéndice 1 se dan las definiciones pertinentes. CONSIDERACIONES INICIALES, APLICABILIDAD Y LIMITACIONES Dado que la activación de la ruta Keap1-Nrf2-ARE aborda únicamente el segundo fenómeno clave de la AOP de la sensibilización cutánea, resulta poco probable que la información obtenida con los métodos de ensayo basados en la activación de esta ruta sea suficiente por sí sola para permitir una conclusión sobre el potencial de sensibilización cutánea de los productos. Por lo tanto, los datos obtenidos con el presente método de ensayo deben considerarse en el contexto de enfoques integrados, como los enfoques IATA, combinándolos con otra información complementaria, por ejemplo derivada de ensayos in vitro que aborden otros fenómenos clave de la AOP de la sensibilización cutánea, así como de métodos no experimentales, incluida la extrapolación a partir de análogos químicos. En la bibliografía se recogen ejemplos del uso del método de la luciferasa ARE-Nrf2 en combinación con otra información (13) (16) (17) (18) (19). El presente método de ensayo se puede utilizar para apoyar la discriminación entre sensibilizantes cutáneos (es decir, categoría 1 del SGA de la ONU o del CLP) y no sensibilizantes en el contexto de un enfoque IATA. El presente método de ensayo no puede utilizarse por sí solo para subcategorizar los sensibilizantes cutáneos en las subcategorías 1A y 1B definidas por el SGA de la ONU o el CLP, ni para predecir la potencia de las decisiones sobre la evaluación de la seguridad. Sin embargo, en función del marco normativo, un resultado positivo podrá utilizarse por sí solo para la clasificación de un producto en la categoría 1 del SGA de la ONU o del CLP. Sobre la base del conjunto de datos obtenidos del estudio de validación y de ensayos internos utilizados para la revisión inter pares independiente del método de ensayo, el ensayo KeratinoSensTM resultó ser transferible a laboratorios con experiencia en cultivo celular. El nivel de reproducibilidad de las predicciones que cabe esperar del método de ensayo es del orden del 85 % dentro de un laboratorio y entre laboratorios (14). La exactitud (77 % — 155/201), la sensibilidad (78 % — 71/91) y la especificidad (76 % — 84/110) del ensayo KeratinoSensTM para discriminar los sensibilizantes cutáneos (es decir, Cat. 1 del SGA de la ONU o del CLP) de los no sensibilizantes en comparación con los resultados del LLNA se calcularon teniendo en cuenta todos los datos presentados al LRUE del CEVMA para la evaluación y revisión inter pares del método de ensayo (14). Estas cifras son similares a las publicadas recientemente sobre la base de ensayos internos de unas 145 sustancias (77 % de exactitud, 79 % de sensibilidad, 72 % de especificidad) (13). Es más probable que el ensayo KeratinoSensTM subestime los productos que muestran una potencia de sensibilización cutánea baja o moderada (es decir, subcategoría 1B del SGA de la ONU o del CLP) que los productos que presentan una potencia de sensibilización cutánea elevada (es decir, subcategoría 1A del SGA de la ONU o del CLP) (13) (14). En conjunto, esta información indica la utilidad del ensayo KeratinoSensTM para contribuir a la identificación del peligro de sensibilización cutánea. Sin embargo, los valores de la exactitud aquí indicados para el ensayo KeratinoSensTM utilizado como único método de ensayo solo son indicativos, ya que el método de ensayo debe considerarse en combinación con otras fuentes de información en el contexto de un enfoque IATA y de conformidad con las disposiciones del punto 9 anterior. Por otra parte, a la hora de evaluar los métodos de ensayo de la sensibilización cutánea sin animales, debe tenerse en cuenta que el LLNA y otros ensayos con animales pueden no reflejar plenamente la situación en la especie de interés, es decir, los seres humanos. El término «producto problema» se utiliza en el presente método de ensayo para referirse a lo que se somete a ensayo y no está relacionado con la aplicabilidad del método de ensayo de la luciferasa ARE-Nrf2 al ensayo de sustancias o mezclas. Sobre la base de los datos disponibles actualmente, se ha demostrado que el ensayo KeratinoSensTM es aplicable a productos problema que abarcan una variedad de grupos funcionales orgánicos, mecanismos de reacción, potencias de sensibilización cutánea (determinada con estudios in vivo) y propiedades fisicoquímicas (9) (12) (13) (14). Se han sometido a ensayo principalmente sustancias con un solo componente, aunque también existe una cantidad limitada de datos sobre ensayos de mezclas (20). Sin embargo, el método de ensayo es técnicamente aplicable a los ensayos de sustancias de componentes múltiples y de mezclas. No obstante, antes de la utilización de este método de ensayo con una mezcla para obtener datos con fines reglamentarios, debe considerarse si podría proporcionar resultados adecuados a tal fin y, en caso afirmativo, por qué. Tales consideraciones no son necesarias si la reglamentación impone el ensayo de la mezcla. Por otra parte, al ensayar sustancias de componentes múltiples o mezclas, deben tenerse en cuenta las posibles interferencias de componentes citotóxicos con las respuestas obtenidas. El método de ensayo es aplicable a productos problema solubles o que formen una dispersión estable (es decir, un coloide o una suspensión en que el producto problema no sedimente ni se separe del disolvente para formar distintas fases) en agua o en DMSO (incluyendo todos los componentes del producto problema en caso de que se realice el ensayo de una sustancia de componentes múltiples o una mezcla). Los productos problema que no cumplan estas condiciones a la concentración final más elevada requerida de 2 000 μM (véase el punto 22) pueden someterse a ensayo a concentraciones más bajas. En tal caso, los resultados que reúnan los criterios de positividad descritos en el punto 39 pueden utilizarse para apoyar la identificación del producto problema como sensibilizante cutáneo, mientras que un resultado negativo obtenido con concentraciones < 1 000 μM debe considerarse como no concluyente (véase el modelo de predicción en el punto 39). En general, se han sometido a ensayo con éxito sustancias con Log P de hasta 5, mientras que las sustancias extremamente hidrófobas con Log P por encima de 7 se encuentran fuera de la aplicabilidad conocida del método de ensayo (14). Sobre las sustancias cuyo Log P está comprendido entre 5 y 7, solo se dispone de información limitada. Los resultados negativos deben interpretarse con cautela, ya que las sustancias con reactividad exclusiva frente a los residuos de lisina pueden detectarse como negativas por el método de ensayo. Por otra parte, debido a la limitada capacidad metabólica de la línea celular utilizada (21) y debido a las condiciones experimentales, pueden obtenerse también resultados negativos con los pro-haptenos (es decir, productos que requieren activación enzimática, por ejemplo mediante enzimas P450) y pre-haptenos (es decir, productos activados por auto-oxidación), en particular con una baja velocidad de oxidación. Los productos problema que no actúan como sensibilizantes pero que, no obstante, provocan agresión química pueden dar lugar, en cambio, a resultados positivos falsos (14). Por otra parte, no siempre es posible someter a ensayo de forma fiable los productos problema muy citotóxicos. Por último, los productos problema que interfieran con la enzima luciferasa pueden afectar a la actividad de esta en ensayos con células, provocando inhibición o aumento aparentes de la luminiscencia (22). Por ejemplo, se ha informado de que las concentraciones de fitoestrógenos superiores a 1 μM interfieren con las señales de luminiscencia en otros ensayos de luciferasa con gen marcador debido a la sobreactivación del gen marcador de la luciferasa (23). Como consecuencia, debe examinarse detenidamente la expresión de la luciferasa obtenida a altas concentraciones de fitoestrógenos o de productos similares sospechosos de provocar la sobreactivación fitoestrogenoide del gen marcador de la luciferasa (23). En los casos en que haya pruebas sobre la inaplicabilidad del método de ensayo a otras categorías específicas de productos problema, el método de ensayo no deberá utilizarse con tales categorías específicas. Además del apoyo a la discriminación entre los sensibilizantes cutáneos y los no sensibilizantes, el ensayo KeratinoSensTM también ofrece información sobre la relación concentración-respuesta que puede contribuir potencialmente a la evaluación de la potencia de sensibilización cuando se utiliza en enfoques integrados, como los IATA (19). Sin embargo, es necesario seguir trabajando, preferentemente a partir de datos fiables obtenidos con seres humanos, para determinar cómo el ensayo KeratinoSensTM puede contribuir a la evaluación de la potencia (24) y a la subcategorización de los sensibilizantes de acuerdo con el SGA de la ONU o del CLP. PRINCIPIO DEL ENSAYO El método de ensayo de la luciferasa ARE-Nrf2 utiliza una línea celular adherida inmortalizada derivada de queratinocitos humanos HaCaT transinfectados de forma estable con un plásmido seleccionable. La línea celular contiene el gen de la luciferasa bajo el control transcripcional de un promotor constitutivo fusionado con un elemento ARE de un gen conocido por elevar su expresión ante la presencia de sensibilizantes por contacto (25) (26). La señal de la luciferasa refleja la activación, provocada por los sensibilizantes, de los genes dependientes de Nrf2 endógeno, y se ha demostrado que la señal de la luciferasa en la línea celular recombinante depende del Nrf2 (27). Esto permite la medición cuantitativa (mediante detección de la luminiscencia) de la inducción del gen de la luciferasa, utilizando sustratos de luciferasa bien conocidos que producen luz, como indicador de la actividad del factor de transcripción Nrf2 en las células tras su exposición a sustancias electrofílicas. Los productos problema se consideran positivos en el ensayo KeratinoSens™ si provocan de forma estadísticamente significativa la inducción de la actividad de la luciferasa por encima de un determinado umbral (es decir, un factor multiplicador > 1,5 o un incremento del 50 %), por debajo de una concentración definida que no afecte de forma significativa a la viabilidad celular (es decir, de menos de 1 000 μM y a una concentración en la que la viabilidad celular se sitúe por encima del 70 % (9) (12)). Con este fin, se determina el factor multiplicador máximo de la inducción de la actividad de la luciferasa respecto al testigo de disolvente (negativo) (Imáx). Por otra parte, como se exponen las células a una serie de concentraciones de los productos problema, la concentración necesaria para una inducción estadísticamente significativa de la actividad de la luciferasa por encima del umbral (es decir, valor EC1,5) se interpolará a partir de la curva dosis-respuesta (para el cálculo, véase el punto 32). Por último, debe procederse a medir en paralelo la citotoxicidad con el fin de evaluar si se dan niveles de inducción de la actividad de la luciferasa a concentraciones subcitotóxicas. Antes de proceder al uso sistemático del ensayo de la luciferasa ARE-Nrf2 según el presente método de ensayo, los laboratorios deben demostrar su competencia técnica, utilizando las diez sustancias recomendadas al efecto en el apéndice 2. Se dispone de normas de comportamiento (28) para facilitar la validación de métodos de ensayo de la luciferasa ARE-Nrf2 in vitro nuevos o modificados, similares al ensayo KeratinoSens™, las cuales permiten la oportuna modificación del presente método de ensayo para su inclusión. La mutua aceptación de datos (MAD) con arreglo al acuerdo de la OCDE solo estará garantizada respecto a métodos de ensayo validados de conformidad con dichas normas de comportamiento, si estos métodos de ensayo han sido revisados e incluidos en las correspondientes directrices de ensayo por la OCDE. PROCEDIMIENTO En la actualidad, el único método que se acoge al presente método de ensayo es el ensayo KeratinoSensTM científicamente válido (9) (12) (13) (14). Se dispone de procedimientos normalizados de trabajo (Standard Operating Procedures, SOP) para el ensayo KeratinoSensTM y deben emplearse cuando se aplica y utiliza el método de ensayo en el laboratorio (15). Los laboratorios que deseen aplicar el método de ensayo pueden obtener la línea celular recombinante utilizada en el ensayo KeratinoSensTM estableciendo un acuerdo de licencia con el creador del método de ensayo. En los siguientes puntos se ofrece una descripción de los principales componentes y procedimientos del método de ensayo de la luciferasa Are-Nrf 2. Preparación de los cultivos de queratinocitos Debe utilizarse una línea celular transgénica con una inserción estable del gen marcador de la luciferasa bajo el control del elemento ARE (por ejemplo, la línea celular de KeratinoSens™). Una vez recibidas, las células se propagan (por ejemplo, se someten a 2-4 pases) y se conservan congeladas como una población homogénea. Las células de esta población original pueden propagarse hasta un número máximo de pases (que es de 25 en el caso de KeratinoSens™) y se emplean para los ensayos sistemáticos utilizando el medio adecuado de mantenimiento (en el caso de KeratinoSens™, se trata de DMEM con suero y geneticina). Para el ensayo, las células deben tener una confluencia de entre el 80 y el 90 %, y debe velarse por que las células nunca se cultiven hasta su confluencia total. Un día antes del ensayo, se recolectan las células y se distribuyen en placas de 96 pocillos (10 000 células/pocillo en el caso del KeratinoSensTM). Debe prestarse atención para evitar la sedimentación de las células durante la siembra, a fin de garantizar la distribución de un número homogéneo de células en todos los pocillos. Si no se hace así, este paso podría ocasionar una elevada variabilidad entre pocillos. En cada repetición se utilizan tres réplicas para las mediciones de la actividad de la luciferasa, y una réplica en paralelo para el ensayo de la viabilidad celular. Preparación del producto problema y de las sustancias testigo El producto problema y las sustancias testigo se preparan el día del ensayo. Para el ensayo de KeratinoSensTM, se disuelven los productos problema en dimetilsulfóxido (DMSO) a la concentración final deseada (por ejemplo, 200 mM). Las soluciones en DMSO pueden considerarse auto-esterilizantes, por lo que no es necesaria su esterilización por filtración. Los productos problema insolubles en DMSO se disuelven en agua o medio de cultivo estéril, y las soluciones se esterilizan, por ejemplo mediante filtración. En el caso de un producto problema que no tenga peso molecular (PM) definido, se prepara una solución madre a una concentración por defecto (40 mg/ml o 4 % (p/v) en el ensayo de KeratinoSensTM). En caso de que se utilicen disolventes distintos del DMSO, el agua o el medio de cultivo, debe proporcionarse una justificación científica suficiente. A partir de las soluciones madre del producto problema en DMSO, se realizan diluciones seriadas con DMSO para obtener doce concentraciones principales del producto problema (de 0,098 a 200 mM en el ensayo de KeratinoSensTM). En caso de que el producto problema sea insoluble en DMSO, las diluciones para obtener las concentraciones principales se realizan con agua estéril o medio de cultivo estéril. Independiente del disolvente utilizado, las concentraciones principales se vuelven a diluir a continuación con un factor de dilución de 25 en medio de cultivo que contenga suero, y, por último, se utilizan para el tratamiento con un factor de dilución adicional de 4, de forma que las concentraciones finales de producto problema se encuentren en el intervalo de 0,98 a 2 000 μM en el ensayo de KeratinoSensTM. Se pueden aplicar otras concentraciones previa justificación (por ejemplo, en caso de citotoxicidad o escasa solubilidad). El testigo negativo (disolvente) utilizado en el ensayo de KeratinoSensTM es DMSO (no CAS 67-68-5, pureza ≥ 99 %), para el cual se preparan seis pocillos por placa. Se somete a la misma dilución descrita para las concentraciones principales en el punto 22, de modo que la concentración final del testigo negativo (disolvente) sea del 1 %, de la que se sabe que no afecta a la viabilidad de las células y corresponde a la misma concentración de DMSO que se encuentra en el producto problema y en el testigo positivo. En caso de productos problema insolubles en DMSO, cuyas diluciones se han realizado en agua, el nivel de DMSO en todos los pocillos de la solución problema final debe ajustarse al 1 %, como para los demás productos problema y sustancias testigo. El testigo positivo utilizado en el caso del ensayo de KeratinoSensTM es el aldehído cinámico (n.o CAS 14371-10-9, pureza ≥ 98 %), del cual se prepara una serie de cinco concentraciones principales en el intervalo de 0,4 a 6,4 mM en DMSO (a partir de una solución madre de 6,4 mM), que se diluyen como se describe para las concentraciones principales en el punto 22, de forma que la concentración final del testigo positivo se encuentre en el intervalo de 4 a 64 μM. Pueden utilizarse otros testigos positivos adecuados que den preferentemente valores de EC1,5 en la gama media, si se dispone de datos históricos para obtener criterios comparables de aceptación de las tandas. Aplicación del producto problema y de las sustancias testigo Para deducir una predicción (positiva o negativa), con cada producto problema y sustancia testigo positivo es necesario hacer un solo experimento, compuesto al menos de dos repeticiones independientes, con tres réplicas en cada una de ellas (es decir, n = 6). En caso de resultados discordantes entre las dos repeticiones independientes, debe realizarse una tercera repetición con tres réplicas (es decir, n = 9). Cada repetición independiente se realiza un día diferente con nueva solución madre de los productos problema y células recolectadas de forma independiente. No obstante, las células pueden proceder del mismo pase. Después de la siembra, según se describe en el punto 20, las células se cultivan durante 24 horas en placas de microvaloración de 96 pocillos. A continuación se retira el medio y se sustituye por medio de cultivo fresco (150 μl de medio de cultivo con suero pero sin geneticina en el caso de KeratinoSensTM) al que se añaden 50 μl de las soluciones de producto problema y de las sustancias testigo diluidas con un factor de 25. Debe dejarse vacío al menos un pocillo por placa (sin células y sin tratamiento) para evaluar los valores de fondo. A continuación se incuban las placas tratadas, durante unas 48 horas a 37 ± 1 °C en presencia de un 5 % de CO2 en el ensayo de KeratinoSensTM. Deben tomarse precauciones para evitar la evaporación de los productos problema volátiles y la contaminación cruzada entre pocillos por productos problema, por ejemplo cubriendo las placas con una hoja antes de la incubación con los productos problema. Mediciones de la actividad de la luciferasa Tres elementos son fundamentales para garantizar una lectura adecuada de la luminiscencia:
Antes de los ensayos, debe efectuarse un experimento de control, dispuesto tal como se describe en el apéndice 3, para cerciorarse de que se cumplen estos tres puntos. Tras el período de exposición de 48 horas con el producto problema y las sustancias testigo en el ensayo de KeratinoSensTM, las células se lavan con una solución salina amortiguadora de fosfato, y se añade a cada pocillo la solución amortiguadora de lisis correspondiente para las lecturas de luminiscencia durante 20 minutos a temperatura ambiente. A continuación se ponen las placas con el lisado celular en el luminómetro para el proceso de lectura, que en el ensayo de KeratinoSensTM se programa de la forma siguiente: i) se añade el sustrato de la luciferasa a cada pocillo (es decir, 50 μl), ii) se espera 1 segundo, y iii) se integra la actividad de la luciferasa durante 2 segundos. Debe justificarse, en su caso, la utilización de una configuración diferente, por ejemplo en función del modelo de luminómetro utilizado. Además, también puede utilizarse un sustrato brillante, siempre que el experimento de control de la calidad del apéndice 3 se complete con éxito. Evaluación de la citotoxicidad Para el ensayo de viabilidad celular de KeratinoSensTM, después del periodo de exposición de 48 horas se sustituye el medio por medio fresco que contiene MTT [bromuro de 3- (4,5-dimetiltiazol- 2-il)-2,5-difeniltetrazolio, azul de tiazolilo; n.o CAS 298-93-1) y las células se incuban durante 4 horas a 37 °C en presencia de 5 % de CO2. A continuación, se retira el medio con MTT y las células se lisan (p. ej., añadiendo a cada pocillo solución de SDS al 10 %) hasta el día siguiente. Después de agitar, se mide la absorción a 600 nm con un fotómetro. DATOS E INFORME Evaluación de los datos En el ensayo de KeratinoSensTM se calculan los siguientes parámetros:
Ecuación 1:
donde
La EC1,5 se calcula por interpolación lineal según la ecuación 2, y la EC1,5 general se calcula como la media geométrica de las distintas repeticiones. Ecuación 2:
donde
La viabilidad se calcula mediante la ecuación 3: Ecuación 3:
donde
Las IC50 e IC30 se calculan por interpolación lineal según la ecuación 4, y las IC50 e IC30 generales se calculan como la media geométrica de las distintas repeticiones. Ecuación 4:
donde
Para cada concentración que presente un factor de inducción de la actividad de la luciferasa > 1,5, se calcula la significación estadística (por ejemplo, mediante una prueba t de Student de dos colas), comparando los valores de luminiscencia de las tres muestras replicadas con los valores de luminiscencia de los pocillos de testigo de disolvente (negativo) para determinar si la inducción de la actividad de la luciferasa es estadísticamente significativa (p< 0,05). La concentración mínima con factor de inducción de la actividad de la luciferasa > 1,5 es el valor que determina el valor de EC1,5. Se comprueba en cada caso si este valor es inferior al valor de IC30, lo que indica que hay menos del 30 % de reducción de la viabilidad celular a la concentración que determina la EC1,5. Se recomienda comprobar los datos visualmente con ayuda de gráficos. Si no se observa una curva dosis-respuesta clara, o si la curva dosis-respuesta obtenida es bifásica (es decir, cruza dos veces el umbral de 1,5), el experimento debe repetirse para comprobar si esto es específico del producto problema o se debe a un artefacto experimental. En caso de que la respuesta bifásica sea reproducible en un experimento independiente, debe indicarse el menor valor de EC1,5 (la concentración cuando se cruza el umbral de 1,5 por primera vez). En los raros casos en que se observa un factor de inducción estadísticamente no significativo por encima de 1,5, seguido de una concentración más elevada con factor de inducción estadísticamente significativo, los resultados de esta repetición se consideran válidos y positivos solo si el factor de inducción estadísticamente significativo por encima de 1,5 se ha obtenido con una concentración no citotóxica. Por último, en el caso de productos problema que presenten un factor de inducción igual o superior a 1,5 ya a la concentración de ensayo mínima de 0,98 μM, el valor de EC1,5 < 0,98 se establece en función de la inspección visual de la curva dosis-respuesta. Criterios de aceptación Deben cumplirse los siguientes criterios de aceptación cuando se use el ensayo de KeratinoSensTM. En primer lugar, el factor de inducción de la actividad de la luciferasa obtenido con el testigo positivo, el aldehído cinámico, debe estar de forma estadísticamente significativa por encima del umbral de 1,5 (por ejemplo, utilizando una prueba T) a al menos una de las concentraciones ensayadas (de 4 a 64 μM). En segundo lugar, el valor de EC1,5 debe estar situado en el margen de dos desviaciones típicas respecto a la media histórica de la instalación de ensayo (por ejemplo, entre 7 μM y 30 μM sobre la base del conjunto de datos de la validación) y estos valores deben actualizarse periódicamente. Además, el factor medio de inducción en las tres réplicas con aldehído cinámico a 64 μM debe situarse entre 2 y 8. Si este último criterio no se cumple, deberá comprobarse cuidadosamente la relación dosis-respuesta del aldehído cinámico, y los ensayos podrán aceptarse solo si existe una clara relación dosis-respuesta con un aumento de la inducción de la actividad de la luciferasa al incrementar las concentraciones del testigo positivo. Por último, el coeficiente medio de variación de la lectura de la luminiscencia del testigo negativo de DMSO (disolvente) debe ser inferior al 20 % en cada repetición consistente en seis pocillos ensayados por triplicado. Si la variabilidad es superior, los resultados deben desecharse. Interpretación de los resultados y modelo de predicción Una predicción de KeratinoSensTM se considera positiva si las siguientes cuatro condiciones se cumplen en su totalidad en 2 repeticiones de 2 o en las mismas 2 repeticiones de 3; de lo contrario, la predicción de KeratinoSensTM se considera negativa (figura 1):
Si en una determinada repetición se cumplen las tres primeras condiciones, pero no puede observarse una clara relación dosis-respuesta para la inducción de la luciferasa, entonces el resultado de dicha repetición debe considerarse no concluyente y podrá ser necesario proceder a efectuar más ensayos (figura 1). Además, un resultado negativo obtenido con concentraciones < 1 000 μM (o < 200 μg/ml en caso de productos problema sin peso molecular definido) también ha de considerarse no concluyente (véase el punto 11). Figura 1: Modelo de predicción utilizado en el ensayo de KeratinoSensTM. Las predicciones del ensayo de KeratinoSensTM deben tenerse en cuenta en el marco de un enfoque IATA y de conformidad con las disposiciones de los puntos 9 y 11.
En algunos raros casos, los productos problema que inducen la actividad de la luciferasa muy cerca de los niveles citotóxicos pueden ser positivos en algunas repeticiones a niveles no citotóxicos (es decir, a la concentración que determina la EC1,5 por debajo (<) de la IC30), y en otras repeticiones solo a niveles citotóxicos (es decir, a la concentración que determina la EC1,5 por encima (>) de la IC30). Tales productos problema se volverán a someter a ensayo con un análisis de la relación dosis-respuesta más estrecho utilizando un factor de dilución inferior (por ejemplo, factor de dilución de 1,33 o Ö2 (= 1,41) entre pocillos), para determinar si la inducción se ha producido o no a niveles citotóxicos (9). Informe del ensayo El informe del ensayo debe incluir la información siguiente:
BIBLIOGRAFÍA
Apéndice 1 DEFINICIONES Exactitud : Grado de coincidencia entre los resultados obtenidos con el método de ensayo y los valores de referencia aceptados. Se trata de una medida del comportamiento del método de ensayo y es un aspecto de su «pertinencia«. Este término y el de «concordancia« se suelen usar indistintamente para indicar la proporción de resultados correctos de un método de ensayo (29). Ruta de resultados adversos (AOP, Adverse Outcome Pathway) : Secuencia de fenómenos desde la estructura química de un producto o grupo de productos similares diana, pasando por el fenómeno molecular desencadenante, hasta un resultado in vivo de interés (2). ARE : Elemento de respuesta antioxidante (antioxidant response element), también denominado EpRE (electrophile response element, elemento de respuesta electrófilo), es un elemento de respuesta constatado en la región anterior del promotor de muchos genes citoprotectores y de fase II. Cuando está activado por Nfr2, sirve de mediador para la inducción transcripcional de estos genes. Producto : Sustancia o mezcla. Coeficiente de variación : Medida de la variabilidad que se calcula para un grupo de datos replicados dividiendo la desviación típica por la media. Puede multiplicarse por 100 para expresarlo como porcentaje. EC1,5 : Concentración interpolada correspondiente a un factor de inducción de luciferasa igual a 1,5. IC30 : Concentración que provoca la reducción de la viabilidad celular en un 30 %. IC50 : Concentración que provoca la reducción de la viabilidad celular en un 50 %. Peligro : Propiedad inherente de un agente o situación que tiene capacidad para provocar efectos adversos cuando un organismo, sistema o (sub)población se expone a dicho agente. Enfoque integrado de pruebas y evaluación (IATA, Integrated Approach to Testing and Assessment) : Enfoque estructurado para la identificación del peligro (potencial), caracterización del peligro (potencia) o evaluación de la seguridad (potencial/potencia y exposición) de un producto o grupo de productos, que integra y pondera de forma estratégica todos los datos pertinentes para fundamentar una decisión reglamentaria relativa a posibles peligros o riesgos o a la necesidad de realizar ensayos más específicos y, por tanto, mínimos. Imáx : Factor de inducción máximo de la actividad de la luciferasa frente al testigo de disolvente (negativo) medido a cualquier concentración del producto problema. Keap1 : Proteína 1 asociada a ECH y de tipo Kelch (Kelch-like ECH-associated protein 1), es una proteína sensora que puede regular la actividad del Nrf2. En ausencia de inducción, la proteína sensora Keap1 actúa sobre el factor de transcripción Nrf2 para la ubiquitinización y degradación proteolítica en el proteasoma. La modificación covalente de los residuos reactivos de cisteína de Keap1 por pequeñas moléculas puede provocar que el Nrf2 se disocie de Keap1 (8) (10) (11). Mezcla : Mezcla o solución compuesta por dos o más sustancias que no reaccionan en ella (1). Sustancia con un solo componente : Sustancia, definida por su composición cuantitativa, en la que un solo componente principal representa al menos el 80 % (p/p). Sustancia de componentes múltiples : Sustancia, definida por su composición cuantitativa, en la que hay varios componentes principales presentes a una concentración ≥ 10 % (p/p) y < 80 % (p/p). Una sustancia de componentes múltiples es el resultado de un proceso de fabricación. La diferencia entre mezcla y sustancia de componentes múltiples es que una mezcla se obtiene mezclando dos o más sustancias sin reacción química, mientras que una sustancia de componentes múltiples es el resultado de una reacción química. Nrf2 : Factor nuclear 2 similar al factor 2 derivado de eritroide, es un factor de transcripción que participa en la ruta de la respuesta antioxidante. Cuando el Nrf2 no está ubiquitinizado, se concentra en el citoplasma y se transloca al núcleo, donde se combina con el ARE en la región anterior del promotor de muchos genes citoprotectores, con lo que se inicia su transcripción (8) (10) (11). Testigo positivo : Réplica que contiene todos los componentes de un sistema de ensayo y que se trata con una sustancia de la que se sabe que induce una respuesta positiva. Para asegurar la posibilidad de evaluar la variabilidad de las respuestas del testigo positivo a lo largo del tiempo, no debe ser excesiva la magnitud de la respuesta positiva. Pertinencia : Descripción de la relación del ensayo con el efecto de interés y de si es significativo y útil para un objetivo concreto. Es el grado en que el ensayo mide o predice correctamente el efecto biológico de interés. La pertinencia incorpora la consideración de la exactitud (concordancia) de un método de ensayo (29). Fiabilidad : Medida del grado en que un método de ensayo puede aplicarse de forma reproducible a lo largo del tiempo, en un mismo laboratorio y en distintos laboratorios, utilizando el mismo protocolo. Se evalúa calculando la reproducibilidad intra e interlaboratorios y la repetibilidad intralaboratorios (29). Reproducibilidad : Coincidencia entre los resultados obtenidos en el ensayo del mismo producto utilizando el mismo protocolo de ensayo (véase «Fiabilidad«) (29). Sensibilidad : Proporción de todos los productos activos/positivos que se clasifican correctamente mediante el método de ensayo. Es una medida de la exactitud de un método de ensayo que produce resultados categoriales, y un factor importante en la evaluación de la pertinencia de un método de ensayo (29). Testigo del disolvente/vehículo : Réplica que contiene todos los componentes de un sistema de ensayo, a excepción del producto problema, pero incluido el disolvente que se utiliza. Se emplea para determinar la respuesta de base respecto a las muestras tratadas con el producto problema disuelto en el mismo disolvente. Especificidad : Proporción de todos los productos inactivos/negativos que se clasifican correctamente mediante el método de ensayo. Es una medida de la exactitud de un método de ensayo que produce resultados categoriales, y un factor importante en la evaluación de la pertinencia de un método de ensayo (29). Sustancia : Elementos químicos y sus compuestos en estado natural u obtenidos mediante cualquier proceso de producción, incluidos los aditivos necesarios para mantener la estabilidad del producto y las eventuales impurezas derivadas del proceso empleado, pero excluidos los disolventes que se puedan separar sin influir en la estabilidad de la sustancia ni cambiar su composición (1). Producto problema : El término «producto problema« se utiliza para hacer referencia a lo que se somete a ensayo. Sistema Globalmente Armonizado de clasificación y etiquetado de productos químicos de las Naciones Unidas (SGA de la ONU) : Sistema que propone la clasificación de productos químicos (sustancias y mezclas) según tipos y niveles normalizados de peligros físicos, sanitarios y ambientales, y que hace referencia a los elementos correspondientes de comunicación, como pictogramas, palabras de advertencia, indicaciones de peligro, consejos de prudencia, y fichas de datos de seguridad, a efectos de proporcionar información sobre sus efectos adversos con el fin de proteger a la población (incluidos empresarios, trabajadores, transportistas, consumidores y personal de protección civil) y al medio ambiente (1). UVCB : Sustancias de composición desconocida o variable, productos complejos de reacción o materiales biológicos. Método de ensayo válido : Método de ensayo del que se considera que tiene suficiente pertinencia y fiabilidad con un fin específico y que se basa en principios sólidos desde el punto de vista científico. Un método de ensayo nunca es válido en un sentido absoluto, sino únicamente en relación con un fin determinado (29). Apéndice 2 SUSTANCIAS UTILIZADAS PARA DEMOSTRAR LA COMPETENCIA Sensibilización cutánea in vitro: Método de ensayo de la luciferasa ARE-Nrf2 Antes de proceder al uso sistemático de este método de ensayo, los laboratorios deben demostrar su competencia técnica mediante la correcta obtención de la predicción del KeratinoSens™ prevista respecto a las diez sustancias recomendadas al efecto en el cuadro 1, y la obtención de valores de EC1,5 e IC50 que estén dentro de la gama de referencia respectiva con al menos ocho de las diez sustancias utilizadas para demostrar la competencia. Estas sustancias de la prueba de la competencia se han seleccionado a fin de representar la gama de respuestas correspondientes a los peligros de sensibilización cutánea. Otros criterios de selección son la disponibilidad comercial, la disponibilidad de referencias in vivo de alta calidad, y la disponibilidad de datos in vitro de alta calidad obtenidos con el ensayo de KeratinoSensTM. Cuadro 1 Sustancias recomendadas para demostrar la competencia técnica con el ensayo de KeratinoSensTM
Apéndice 3 CONTROL DE CALIDAD DE LAS MEDICIONES DE LUMINISCENCIA Experimento de base para garantizar unas mediciones óptimas de la luminiscencia en el ensayo de KeratinoSensTM Los tres parámetros siguientes son esenciales para garantizar la obtención de resultados fiables con el luminómetro:
Antes del ensayo, se recomienda velar por que las mediciones de luminiscencia sean adecuadas, comprobando una placa testigo dispuesta como se describe más abajo (análisis por triplicado). Disposición de la placa del primer experimento de formación
EGDMA= dimetacrilato de etilenglicol (no CAS: 97-90-5), sustancia fuertemente inductora CA= aldehído cinámico, referencia positiva (no CAS: 104-55-2) El análisis de control de la calidad debe demostrar:
B.61. Método de ensayo de pérdida de fluoresceína para detectar agentes corrosivos e irritantes intensos para los ojos INTRODUCCIÓN El presente método de ensayo es equivalente a las directrices de ensayo (TG) de la OCDE 460 (2012). El método de ensayo de pérdida de fluoresceína (FL, fluorescein leakage) es un procedimiento in vitro que puede utilizarse, en determinadas circunstancias y con limitaciones específicas, para clasificar los productos (sustancias y mezclas) como agentes corrosivos e irritantes intensos para los ojos, según se definen en el Sistema Globalmente Armonizado de clasificación y etiquetado de productos químicos de las Naciones Unidas (SGA de la ONU) (categoría 1), en el Reglamento (CE) n.o 1272/2008, sobre clasificación, etiquetado y envasado de sustancias y mezclas (CLP) (31) (categoría 1), y en la Agencia de Protección del Medio Ambiente de los Estados Unidos (EPA) (categoría I) (1) (2). A efectos de este método de ensayo, se entiende por «agentes irritantes intensos para los ojos» los productos que provocan lesiones tisulares en el ojo como consecuencia de la administración del producto, las cuales no son reversibles en el plazo de 21 días, o una grave reducción física de la vista, mientras que «agentes corrosivos para los ojos» son los productos que causan lesiones tisulares irreversibles en el ojo. Estos productos se clasifican en la categoría 1 del SGA de la ONU, en la categoría 1 del CLP de la UE, o en la categoría I de la EPA. Si bien el método de ensayo FL no se considera válido para sustituir completamente al ensayo in vivo con ojo de conejo, sí se recomienda utilizarlo como parte de una estrategia de ensayos escalonados para la clasificación y el etiquetado reglamentarios. Así pues, se recomienda el método FL como un primer paso dentro de un enfoque descendente para identificar agentes corrosivos o irritantes intensos para los ojos, específicamente para ciertos tipos de productos (es decir, sustancias hidrosolubles y mezclas) (3) (4). Actualmente se reconoce en general que, en un futuro próximo, ningún ensayo de irritación ocular in vitro podrá sustituir por sí solo al ensayo ocular in vivo [método de ensayo B.5 (5)] para hacer predicciones en toda la gama de irritación causada por las distintas clases de productos. Sin embargo, unas combinaciones estratégicas de varios métodos de ensayo alternativos dentro de una estrategia de ensayos (escalonados) sí podrán sustituir al ensayo ocular in vivo (4). El enfoque descendente (4) está diseñado para utilizarse cuando, sobre la base de la información existente, se espera que un producto tenga un alto potencial de irritación. Sobre la base del modelo de predicción detallado en el punto 35, el método FL, dentro de un ámbito de aplicación limitado, puede identificar los productos como agentes corrosivos o irritantes intensos para los ojos (categoría 1 del SGA de la ONU; categoría 1 del CLP de la UE; categoría I de la EPA de los EE.UU.), sin necesidad de realizar más ensayos. Se acepta que lo mismo ocurre con las mezclas, aunque estas no se hayan utilizado en la validación. Por lo tanto, el método de ensayo FL puede utilizarse para determinar la capacidad de irritación/corrosión ocular de los productos, siguiendo la estrategia secuencial de ensayos del método B.5 (5). No obstante, un producto del que el método de ensayo FL no prediga que es corrosivo o irritante intenso para los ojos tendría que someterse a ensayo con uno o varios métodos de ensayo adicionales (in vitro o in vivo) capaces de identificar con exactitud: i) los productos que den resultados falsos negativos in vitro como agentes corrosivos/ irritantes intensos para los ojos con el método FL (categoría 1 del SGA de la ONU; categoría 1 del CLP de la UE; categoría I de la EPA de los EE.UU.); ii) los productos que no estén clasificados por corrosión/irritación ocular (sin categoría del SGA de la ONU; sin categoría del CLP de la UE; categoría IV de la EPA de los EE.UU.); y iii) los productos que sean agentes irritantes oculares leves o moderados (categorías 2A y 2B del SGA de la ONU; categoría 2 del CLP de la UE; categorías II y III de la EPA de los EE.UU.). El objetivo del presente método de ensayo es describir los procedimientos utilizados para evaluar la posible capacidad de corrosión o de irritación intensa para los ojos de un producto problema, medida por su capacidad de provocar lesiones a una monocapa epitelial confluente impermeable. La integridad de la permeabilidad transepitelial es una importante función de los epitelios como los que se encuentran en la conjuntiva y en la córnea. La permeabilidad transepitelial está controlada por diversas uniones intercelulares herméticas. Se ha observado que el aumento de la permeabilidad del epitelio de la córnea in vivo está en correlación con el nivel de inflamación y de lesiones superficiales que se observan cuando se irritan los ojos. En el método de ensayo FL, los efectos tóxicos tras un breve tiempo de exposición al producto problema se miden por el aumento de la permeabilidad a la fluoresceína sódica a través de la monocapa epitelial de células renales de perro de Madin-Darby (MDCK) cultivadas sobre elementos insertados permeables. La cantidad de fluoresceína que se pierde es proporcional a las lesiones provocadas por el producto a las uniones intercelulares herméticas, a las uniones desmosómicas y a las membranas celulares, y se puede utilizar para estimar el potencial de la toxicidad ocular de un producto problema. El apéndice 1 incluye un diagrama de células MDCK cultivadas sobre la membrana de un elemento insertado para el método de ensayo FL. En el apéndice 2 se dan las definiciones pertinentes. CONSIDERACIONES INICIALES Y LIMITACIONES El presente método de ensayo se basa en el Protocolo n.o 71 de INVITTOX (6), que ha sido evaluado en un estudio de validación internacional por el Centro Europeo para la Validación de Métodos Alternativos (CEVMA), en colaboración con el Comité de Coordinación Interagencias sobre la validación de métodos alternativos (Interagency Coordinating Committee on the Validation of Alternative Methods, ICCVAM) de los Estados Unidos, y con el Centro Japonés para la Validación de Métodos Alternativos (Japanese Center for the Validation of Alternative Methods, JaCVAM). El método de ensayo FL no se recomienda para la identificación de productos que deben clasificarse como irritantes leves o moderados o de productos que no deben clasificarse como irritantes oculares (sustancias y mezclas) (es decir, sin categoría o categoría 2A/2B del SGA; sin categoría o categoría 2 del CLP de la UE; categoría II/III/IV de la EPA de los EE.UU.), según el estudio de validación (3) (7). El método de ensayo es aplicable solo a los productos (sustancias y mezclas) hidrosolubles. La capacidad de irritación ocular intensa de los productos hidrosolubles o de aquellos cuyo efecto tóxico no se ve afectado por la dilución es predicha generalmente de forma exacta con el método de ensayo FL (7). Para clasificar un producto como hidrosoluble, en condiciones experimentales, debe ser soluble en solución salina equilibrada de Hank (HBSS) estéril, con calcio (a la concentración de 1,0-1,8 mM), sin rojo de fenol, a una concentración ≥ 250 mg/ml (una dosis por encima del umbral de 100 mg/ml). Sin embargo, si el producto problema es soluble a una concentración inferior a 100 mg/ml, pero a esa concentración ya induce la pérdida de fluoresceína del 20 % (lo que significa FL20 < 100 mg/ml), puede clasificarse en la Cat. 1 del SGA o en la Cat. I de la EPA. Las limitaciones de este método de ensayo que se han detectado excluyen de su ámbito de aplicación a los ácidos y bases fuertes, los fijadores celulares y los productos muy volátiles. Estos productos tienen mecanismos que no se miden con el método de ensayo FL como, por ejemplo, importante coagulación, saponificación o reacciones químicas específicas. Otras limitaciones de este método que se han detectado se basan en los resultados relativos a la capacidad de predicción con productos problema viscosos y de color (7). Parece que estos dos tipos de productos son difíciles de eliminar de la monocapa tras el breve período de exposición y que la capacidad de predicción del método de ensayo podría mejorarse con un mayor número de fases de lavado. Los productos sólidos suspendidos en líquido tienden a separarse por precipitación y puede ser difícil determinar la concentración final en contacto con las células. Si se excluyen de la base de datos los productos de estas clases químicas y físicas, mejora sustancialmente la exactitud del ensayo FL en los sistemas de clasificación de la UE, la EPA y el SGA (7). Teniendo en cuenta la finalidad del presente método de ensayo (es decir, identificar solamente agentes corrosivos/ irritantes intensos para los ojos), las tasas de falsos negativos (véase el punto 13) no son fundamentales, ya que tales productos se someterían a otros ensayos in vitro validados adecuadamente o con conejos, en función de las exigencias normativas, utilizando una estrategia de ensayo secuencial con un enfoque de ponderación de los datos (5) (véanse también los puntos 3 y 4). Otras limitaciones detectadas del método de ensayo FL se refieren a las tasas de falsos positivos y de falsos negativos. Cuando se utilizó como fase inicial dentro de un planteamiento descendente para determinar sustancias y mezclas hidrosolubles corrosivas/ irritantes intensas (categoría 1 del SGA de la ONU; categoría 1 del CLP de la UE; categoría I de la EPA de los EE.UU.), la tasa de falsos positivos del método de ensayo FL osciló entre el 7 % (7/103; SGA de la ONU y CLP de la UE) y el 9 % (9/99; EPA de los EE.UU.) y la tasa de falsos negativos osciló entre el 54 % (15/28; EPA de los EE.UU.) y el 56 % (27/48; SGA de la ONU y CLP de la UE) en comparación con los resultados in vivo. No se definen aquí grupos químicos que presenten falsos resultados positivos o negativos con el método de ensayo FL. Hay ciertas limitaciones técnicas que son específicas del cultivo de células MDCK. Las uniones intercelulares herméticas que impiden el paso del colorante fluoresceína sódica a través de la monocapa se van alterando cada vez más al aumentar el número de pases celulares. La formación incompleta de estas uniones intercelulares herméticas redunda en un aumento de la pérdida de fluoresceína en los testigos sin tratar. Por lo tanto, es importante definir la pérdida máxima permisible en los testigos sin tratar (véase el punto 38: 0 % de pérdida). Como con todos los ensayos in vitro, existe la posibilidad de que las células se transformen a lo largo del tiempo, por lo que es fundamental que se indique la banda del número de pases en relación con los ensayos. El actual ámbito de aplicación podría ampliarse en algunos casos, pero solo después de analizar un conjunto ampliado de datos de productos problema estudiados, obtenido preferentemente mediante ensayos (3). El presente método de ensayo se actualizará en consecuencia a la luz de la nueva información y los datos que se vayan considerando. Cuando algún laboratorio establezca inicialmente este ensayo, debe utilizar los productos de la prueba de la competencia que se indican en el apéndice 3. Los laboratorios pueden utilizar estos productos para demostrar su competencia técnica en la realización del método de ensayo FL antes de presentar, con fines de clasificación normativa de peligros, los datos obtenidos con este ensayo. PRINCIPIO DEL ENSAYO El método FL es un ensayo in vitro que se basa en la citotoxicidad y en la función celular, realizado con una monocapa confluente de células de epitelio tubular MDCK CB997, cultivadas sobre elementos insertados semipermeables, que sirven de modelo del estado no proliferativo del epitelio corneal in vivo. La línea celular MDCK está bien establecida y forma uniones intercelulares herméticas y uniones desmosómicas similares a las que se encuentran en el lado apical de los epitelios conjuntival y corneal. Las uniones intercelulares herméticas y las uniones desmosómicas in vivo evitan que los solutos y las materias extrañas penetren en el epitelio corneal. La pérdida de la impermeabilidad transepitelial, debida a la alteración de las uniones intercelulares herméticas y de las uniones desmosómicas, constituye uno de los primeros fenómenos de la irritación ocular causada por los productos. El producto problema se aplica a la capa confluente de células cultivadas en el lado apical del elemento insertado. Habitualmente se utiliza una breve exposición de un minuto para reflejar la velocidad de eliminación normal en la exposición humana. Una ventaja del breve período de exposición es que las sustancias y mezclas a base de agua pueden someterse a ensayo en estado puro, si pueden retirarse fácilmente después del período de exposición. Esto permite una comparación más directa de los resultados con los efectos de los productos en los seres humanos. El producto problema se retira a continuación y el colorante fluoresceína sódica, que no es tóxico y es muy fluorescente, se añade al lado apical de la monocapa durante 30 minutos. La lesión causada por el producto problema a las uniones intercelulares herméticas se determina por la cantidad de fluoresceína que se pierde a través de la capa celular dentro de un plazo de tiempo determinado. La cantidad de colorante fluoresceína sódica que pasa a través de la monocapa y de la membrana del elemento insertado a un volumen determinado de solución presente en el pocillo (al que llega la fluoresceína sódica perdida) se determina midiendo por espectrofluorometría la concentración de fluoresceína en el pocillo. La cantidad de fluoresceína filtrada (FL) se calcula con referencia a las lecturas de intensidad de la fluorescencia (FI) de dos testigos: un testigo en blanco, y un testigo de pérdida máxima. Se indica, en relación con estos testigos, para cada una de la concentraciones fijadas del producto problema el porcentaje de pérdida y, por lo tanto, la cuantía de la lesión provocada a las uniones intercelulares herméticas. A continuación se calcula la FL20 (es decir, la concentración que provoca un 20 % de pérdida de fluoresceína teniendo en cuenta los valores registrados con la monocapa confluente sin tratar y con los elementos insertados sin células). El valor de FL20 (mg/ml) se utiliza en el modelo de predicción para identificar los agentes corrosivos e irritantes intensos para los ojos (véase el punto 35). La recuperación es una parte importante del perfil de toxicidad de un producto problema que se evalúa también con el ensayo de irritación ocular in vivo. Los análisis preliminares indican que los datos de recuperación (hasta 72 h después de la exposición al producto) podrían incrementar la capacidad predictiva del Protocolo n.o 71 de INVITTOX, pero es necesario proseguir la evaluación, que mejoraría con más datos, adquiridos preferentemente mediante nuevos ensayos (6). El presente método de ensayo se actualizará en consecuencia a la luz de la nueva información y los datos que se vayan considerando. PROCEDIMIENTO Preparación de la monocapa celular La monocapa de células MDCK CB997 se elabora utilizando células subconfluentes cultivadas en matraces de cultivo celular con medio DMEM/Nutrient Mix F12 (1x concentrado con L-glutamina, HEPES 15 mM, calcio (a una concentración de 1,0-1,8 mM) y un 10 % de FCS/FBS inactivado por calor). Es muy importante que todos los medios/soluciones utilizados a lo largo del ensayo FL contengan calcio a una concentración entre 1,8 mM (200 mg/l) y 1,0 mM (111 mg/l) para asegurar la formación e integridad de las uniones intercelulares herméticas. Debe controlarse la banda del número de pases celulares para que la formación de uniones intercelulares herméticas sea homogénea y reproducible. Las células debe estar preferentemente en la banda de 3-30 pases desde la descongelación, ya que las células en esta banda de pases tienen funciones similares, lo que contribuye a la reproducibilidad de los resultados del ensayo. Antes de aplicar el método de ensayo FL, las células se desprenden del matraz por tripsinización y se centrifugan, y un número adecuado de células se siembra en los elementos insertados colocados en placas de 24 pocillos (véase el apéndice 1). Para sembrar las células deben usarse elementos insertados de 12 mm de diámetro, con membrana de mezcla de ésteres de celulosa, un espesor de 80 a 150 μm y un tamaño de poro de 0,45 μm. En el estudio de validación se utilizaron elementos insertados de 12 mm Millicell-HA. Las propiedades del elemento insertado y el tipo de membrana son importantes, puesto que pueden afectar al crecimiento celular y a la unión de los productos. Algunos tipos de productos pueden unirse a la membrana Millicell-HA, lo que puede afectar a la interpretación de los resultados. Si se utilizan otras membranas, para demostrar su equivalencia deben utilizarse productos de la prueba de la competencia (véase el apéndice 3). La unión del producto a la membrana del elemento insertado es más común en caso de productos catiónicos, como el cloruro de benzalconio, que son atraídos por la membrana cargada (7). La unión del producto a la membrana del elemento insertado puede aumentar el período de exposición al producto, dando lugar a una sobreestimación del potencial tóxico del producto, pero también puede reducir físicamente la pérdida de fluoresceína a través del elemento insertado por unión del colorante al producto catiónico unido a la membrana del elemento insertado, dando lugar a una subestimación del potencial tóxico del producto. Esto puede verificarse fácilmente exponiendo la membrana sola a la concentración máxima del producto problema y añadiendo a continuación el colorante fluoresceína sódica a la concentración normal durante el tiempo establecido (testigo sin células). Si se produce unión del colorante de fluoresceína, la membrana del elemento insertado se verá amarilla después de que el producto problema se haya lavado. Por lo tanto, es esencial conocer las propiedades de unión del producto problema para poder interpretar los efectos de este sobre las células. La siembra de las células sobre los elementos insertados debe producir una monocapa confluente en el momento de la exposición al producto. Deben añadirse 1,6 x 105 células por elemento insertado (400 μl de una suspensión celular con una densidad de 4 x 105 células/ml). En estas condiciones, se obtiene una monocapa confluente por lo general al cabo de 96 horas de cultivo. Los elementos insertados deben examinarse visualmente antes de la siembra, con objeto de garantizar que los eventuales daños obervados en el control visual descrito en el punto 30 se deben a la manipulación. Los cultivos de células MDCK deben mantenerse en incubadores con atmósfera humidificada, con un 5 % ± 1 % de CO2 y a 37 °C ± 1 °C. Las células deben estar exentas de contaminación por bacterias, virus, micoplasmas y hongos. Aplicación de los productos problema y testigo Para cada tanda experimental debe prepararse una nueva solución madre de producto problema, la cual se ha de utilizar en el plazo de 30 minutos a partir de su preparación. Los productos problema deben prepararse en HBSS sin rojo de fenol, con calcio (a una concentración de 1,0-1,8 mM), para evitar la unión a las proteínas plasmáticas. Antes del ensayo debe evaluarse la solubilidad del producto a 250 mg/ml en HBSS. Si a esta concentración el producto forma una suspensión o emulsión estable (es decir, mantiene la uniformidad y no sedimenta ni se separa en más de una fase) durante 30 minutos, la HBSS puede utilizarse como disolvente. Sin embargo, si se observa que el producto es insoluble en HBSS a esta concentración, debe considerarse el uso de otros métodos de ensayo en lugar del método FL. El uso de aceite de vaselina fluido como disolvente, en los casos en que se vea que el producto es insoluble en HBSS, debe considerarse con cautela, ya que no se dispone de suficientes datos para llegar a una conclusión sobre el comportamiento del ensayo FL en tales condiciones. Todos los productos problema se preparan en HBSS sin rojo de fenol, con calcio (a una concentración de 1,0-1,8 mM), a partir de la solución madre, diluida a cinco concentraciones fijas, expresadas en peso por volumen: 1, 25, 100, 250 mg/ml y una forma sin diluir o una solución saturada. Al someter a ensayo un producto sólido, debe incluirse una concentración muy alta, de 750 mg/ml. Esta concentración de producto puede tener que aplicarse a las células utilizando una pipeta de desplazamiento positivo. Si se observa que la toxicidad está entre 25 y 100 mg/ml, se someten a ensayo en dos tandas las siguientes concentraciones adicionales: 1, 25, 50, 75, 100 mg/ml. De estas concentraciones debe derivarse el valor de FL20 siempre que se cumplan los criterios de aceptación. Los productos problema se aplican a las monocapas de células confluentes después de retirar el medio de cultivo celular y de lavar dos veces con HBSS sin rojo de fenol, con calcio (a una concentración de 1,0-1,8 mM), estéril y caliente (37 °C). Previamente, los filtros se habrán comprobado visualmente para detectar los eventuales daños preexistentes que pudieran atribuirse erróneamente a posibles incompatibilidades con los productos problema. Deben utilizarse al menos tres réplicas para cada concentración del producto problema y para los testigos en cada tanda. Después de 1 minuto de exposición a temperatura ambiente, el producto problema debe retirarse cuidadosamente por aspiración, la monocapa debe lavarse dos veces con HBSS sin rojo de fenol, con calcio (a una concentración de 1,0-1,8 mM), estéril y caliente (37 °C), y la pérdida de fluoresceína debe medirse de forma inmediata. En cada tanda deben utilizarse testigos negativos (TN) y positivos (TP) en paralelo para demostrar que la integridad de la monocapa (TN) y la sensibilidad de las células (TP) se encuentran dentro de una gama de aceptación definida históricamente. El producto sugerido como TP es Brij 35 (n.o CAS 9002-92-0) a 100 mg/ml. Esta concentración debe dar aproximadamente un 30 % de pérdida de fluoresceína (intervalo aceptable: 20-40 % de pérdida de fluoresceína, sinónimo de lesión de la capa celular). El producto sugerido como TN es HBSS sin rojo de fenol, con calcio (a una concentración de 1,0-1,8 mM) (testigo en blanco, sin tratar). En cada tanda debe incluirse también un testigo de pérdida máxima para poder calcular los valores de FL20. La pérdida máxima se determina utilizando un elemento insertado testigo sin células. Determinación de la permeabilidad a la fluoresceína Inmediatamente después de la eliminación de los productos problema y testigo, se añaden a los elementos insertados (p. ej., Milicell-HA) 400 μl de solución de fluoresceína sódica de 0,1 mg/ml [0,01 % (p/v) en HBSS sin rojo de fenol, con calcio (a una concentración de 1,0-1,8 mM)]. Los cultivos se mantienen durante 30 minutos a temperatura ambiente. Al final de la incubación con fluoresceína, los elementos insertados se retiran cuidadosamente de cada pocillo. Se efectúa una comprobación visual de cada filtro y se registran los eventuales daños que se hayan producido durante la manipulación. La cantidad de fluoresceína que haya pasado a través de la monocapa y del elemento insertado se cuantifica en la solución que haya permanecido en los pocillos después de la retirada de los elementos insertados. Las mediciones se realizan en un espectrofluorímetro a las longitudes de onda de excitación y de emisión de 485 nm y 530 nm, respectivamente. La sensibilidad del espectrofluorímetro debe fijarse de manera que exista la mayor diferencia numérica entre la pérdida máxima de fluoresceína (elemento insertado sin células) y la pérdida mínima de fluoresceína (elemento insertado con monocapa confluente, tratado con el TN). Habida cuenta de las diferencias en el espectrofluorímetro utilizado, se sugiere utilizar una sensibilidad que dé una intensidad de fluorescencia > 4 000 en el testigo de pérdida máxima de fluoresceína. El valor correspondiente a la pérdida máxima de fluoresceína no debe ser superior a 9 999. La intensidad de la fluorescencia correspondiente a la pérdida máxima debe estar dentro del intervalo lineal del espectrofluorímetro utilizado. Interpretación de los resultados y modelo de predicción La cantidad de la pérdida de fluoresceína es proporcional a las lesiones inducidas por el producto en las uniones intercelulares herméticas. El porcentaje de pérdida de fluoresceína a cada concentración estudiada de producto problema se calcula a partir de la valores de pérdida de fluoresceína obtenidos con el producto problema haciendo referencia a los valores de pérdida de fluoresceína del TN (lectura de la monocapa confluente de células tratadas con el TN) y un testigo de pérdida máxima (lectura correspondiente a la cantidad de fluoresceína perdida a través de un elemento insertado sin células). La intensidad media de la fluorescencia correspondiente a la pérdida máxima = x La intensidad media de la fluorescencia correspondiente a la pérdida 0 % (TN) = y La intensidad media correspondiente a la pérdida del 100 % se obtiene restando, de la intensidad media correspondiente a la pérdida máxima, la intensidad media correspondiente a la pérdida del 0 %, es decir, x – y = z El porcentaje de pérdida a cada dosis fijada se obtiene restando el valor correspondiente a la pérdida del 0 % de la intensidad de la fluorescencia media de las lecturas de las tres réplicas (m) y dividiendo este valor por el correspondiente a la pérdida del 100 %, es decir, % FL = [(m – y) / z] × 100 %, donde:
Debe aplicarse la siguiente ecuación para el cálculo de la concentración del producto que provoca una FL del 20 %: FLD = [(A – B) / (C – B)] × (MC – MB) + MB donde:
El valor de corte de FL20 para la predicción de un producto como agente corrosivo o irritante intenso para los ojos se indica a continuación:
El método de ensayo FL solo se recomienda para la identificación de agentes corrosivos e irritantes intensos para los ojos hidrosolubles (categoría 1 del SGA de la ONU, categoría 1 del CLP de la UE, categoría I de la EPA de los EE.UU.) (véanse los puntos 1 y 10). Para identificar un producto (sustancia o mezcla) hidrosoluble (3) (6) (7) como «inductor de lesiones oculares graves» (categoría 1 del SGA de la ONU o del CLP de la UE) o como «corrosivo o irritante intenso para los ojos» (categoría I de la EPA de los EE.UU.), el producto problema debe tener un valor de FL20 ≤ 100 mg/ml. Aceptación de los resultados El valor medio correspondiente a la pérdida máxima de fluoresceína (x) debe ser superior a 4 000 (véase el punto 31), el valor medio correspondiente a la pérdida del 0 % (y) debe ser inferior o igual a 300, y el valor medio correspondiente a la pérdida del 100 % (z) debe estar comprendido entre 3 700 y 6 000. Se considera que un ensayo es aceptable si el testigo positivo provoca entre un 20 % y un 40 % de lesiones a la capa celular (medida como pérdida porcentual de fluoresceína). DATOS E INFORME Datos De cada tanda deben indicarse en forma tabular los datos obtenidos con cada uno de los pocillos replicados (por ejemplo, valores de la intensidad de la fluorescencia y datos calculados de porcentaje de FL para cada producto problema, incluida la clasificación). Además, deben comunicarse las medias ± desviación típica de las mediciones de las distintas réplicas de cada tanda. Informe del ensayo El informe del ensayo debe incluir la información siguiente:
BIBLIOGRAFÍA
Apéndice 1 DIAGRAMA DE CÉLULAS MDCK CULTIVADAS SOBRE LA MEMBRANA DE UN ELEMENTO INSERTADO PARA EL MÉTODO DE ENSAYO FL Se cultiva una capa confluente de células MDCK en la membrana semipermeable de un elemento insertado. Los elementos insertados se ponen en los pocillos de placas de 24 pocillos. 
La figura procede de: Wilkinson, P.J. (2006), Development of an in vitro model to investigate repeat ocular exposure, Ph.D. Thesis, University of Nottingham, UK. Apéndice 2 DEFINICIONES Exactitud : Grado de coincidencia entre los resultados obtenidos con el método de ensayo y los valores de referencia aceptados. Se trata de una medida del comportamiento del método de ensayo y es un aspecto de su» pertinencia«. Este término y el de» concordancia «se suelen usar indistintamente para indicar la proporción de resultados correctos de un método de ensayo. Producto : Sustancia o mezcla. Categoría I de la EPA : Productos que producen corrosión (destrucción irreversible del tejido ocular) o afectación o irritación de la córnea, persistente durante más de 21 días (2). CLP de la UE : (Reglamento (CE) n.o 1272/2008, sobre clasificación, etiquetado y envasado de sustancias y mezclas): Aplica en la Unión Europea (UE) el sistema SGA de la ONU para la clasificación de los productos (sustancias y mezclas). Tasa de falsos negativos : Proporción de todos los productos positivos identificados erróneamente como negativos por un método de ensayo. Es uno de los indicadores del comportamiento del método de ensayo. Tasa de falsos positivos : Proporción de todos los productos negativos identificados erróneamente como positivos por un método de ensayo. Es uno de los indicadores del comportamiento del método de ensayo. FL20 : Puede estimarse mediante la determinación de la concentración a la que el producto problema hace que el 20 % de la fluoresceína pase a través de la capa celular. Pérdida de fluoresceína : Cantidad de fluoresceína que pasa a través de la capa celular, medida por espectrofluorometría. SGA [(Sistema Globalmente Armonizado de clasificación y etiquetado de productos químicos de las Naciones Unidas (ONU)] : Sistema que propone la clasificación de productos químicos (sustancias y mezclas) según tipos y niveles normalizados de peligros físicos, sanitarios y ambientales, y que hace referencia a los elementos correspondientes de comunicación, como pictogramas, palabras de advertencia, indicaciones de peligro, consejos de prudencia, y fichas de datos de seguridad, a efectos de proporcionar información sobre sus efectos adversos con el fin de proteger a la población (incluidos empresarios, trabajadores, transportistas, consumidores y personal de protección civil) y al medio ambiente. Categoría 1 del SGA : Producción de una lesión tisular en el ojo o una degradación física severa de la vista, como consecuencia de la aplicación de un producto problema en la superficie anterior del ojo, y que no es totalmente reversible en los 21 días siguientes a la aplicación. Peligro : Propiedad inherente de un agente o situación que tiene capacidad para provocar efectos adversos cuando un organismo, sistema o (sub)población se expone a dicho agente. Mezcla : Término utilizado en el contexto de la SGA de la ONU como mezcla o solución compuesta por dos o más sustancias que no reaccionan en ella. Testigo negativo : Muestra replicada no tratada que contiene todos los componentes de un sistema de ensayo. Esta muestra se somete al mismo proceso que las muestras tratadas con producto problema y otras muestras testigo para determinar si el disolvente interactúa con el sistema de ensayo. Sin clasificar : Productos que no están clasificados como irritantes oculares en las categorías 1, 2A o 2B del SGA de la ONU; categorías 1 o 2 del CLP de la UE; o categorías I, II o III de la EPA de los EE.UU. Agentes corrosivos para los ojos : a) Productos que provocan una lesión tisular irreversible en los ojos; b) Productos clasificados como irritantes para los ojos en la categoría 1 del SGA de la ONU; categoría 1 del CLP de la UE; o categoría I de la EPA de los EE.UU. Agentes irritantes para los ojos : a) Productos que producen un cambio reversible en el ojo como consecuencia de su aplicación a la superficie anterior de este; b) Productos clasificados como irritantes para los ojos en las categorías 2A o 2B del SGA de la ONU; categoría 2 del CLP de la UE; o categorías II o III de la EPA de los EE.UU. Agentes irritantes intensos para los ojos : a) Productos que provocan lesiones tisulares en los ojos como consecuencia de su aplicación en la superficie anterior del ojo, y que no son reversibles en los 21 días siguientes a la aplicación, o una grave reducción física de la vista; b) Productos clasificados como irritantes para los ojos en la categoría 1 del SGA de la ONU; categoría 1 del CLP de la UE; o categoría I de la EPA de los EE.UU. Testigo positivo : Muestra replicada que contiene todos los componentes de un sistema de ensayo y que se trata con un producto del que se sabe que induce una respuesta positiva. Para asegurar la posibilidad de evaluar la variabilidad de las respuestas del testigo positivo a lo largo del tiempo, no debe ser extrema la magnitud de la respuesta positiva. Productos de la prueba de la competencia : Subconjunto de la lista de productos de referencia que puede ser utilizado por un laboratorio sin experiencia para demostrar su competencia con el método de ensayo de referencia validado. Pertinencia : Descripción de la relación del ensayo con el efecto de interés y de si es significativo y útil para un objetivo concreto. Es el grado en que el ensayo mide o predice correctamente el efecto biológico de interés. La pertinencia incorpora la consideración de la exactitud (concordancia) de un método de ensayo (8). Fiabilidad : Medida del grado en que un método de ensayo puede aplicarse de forma reproducible a lo largo del tiempo, en un mismo laboratorio y en distintos laboratorios, utilizando el mismo protocolo. Se evalúa calculando la reproducibilidad intra e interlaboratorios y la repetibilidad intralaboratorios. Ensayo de sustitución : Ensayo que se diseña para sustituir un ensayo utilizado de forma sistemática y aceptado para la identificación de peligros o la evaluación de riesgos, y del que se ha determinado que proporciona una protección equivalente o mejorada de la salud humana o del medio ambiente, según corresponda, respecto al ensayo aceptado, en relación con todas las situaciones de ensayo y todos los productos posibles. Sensibilidad : Proporción de todos los productos activos/positivos que se clasifican correctamente mediante el ensayo. Es una medida de la exactitud de un método de ensayo que produce resultados categoriales, y un factor importante en la evaluación de la pertinencia de un método de ensayo (8). Lesiones oculares graves : Producción de una lesión tisular en el ojo o una degradación física severa de la vista, como consecuencia de la aplicación de un producto problema en la superficie anterior del ojo, y que no es totalmente reversible en los 21 días siguientes a la aplicación. Testigo del disolvente/vehículo : Muestra no tratada que contiene todos los componentes de un sistema de ensayo, incluido el disolvente o vehículo, y que se somete al mismo proceso que las muestras tratadas con el producto problema y otras muestras testigo a fin de determinar la respuesta de base correspondiente a las muestras tratadas con el producto problema disuelto en el mismo disolvente o vehículo. Cuando se somete a ensayo con un testigo negativo en paralelo, esta muestra pone de manifiesto también si el disolvente o vehículo interactúa con el sistema de ensayo. Especificidad : Proporción de todos los productos inactivos/negativos que se clasifican correctamente mediante el ensayo. Es una medida de la exactitud de un método de ensayo que produce resultados categoriales, y un factor importante en la evaluación de la pertinencia de un método de ensayo. Sustancia : En el contexto del SGA de la ONU tiene el sentido de elementos químicos y sus compuestos en estado natural o en el estado obtenido por algún proceso de producción, incluidos los eventuales aditivos necesarios para conservar su estabilidad y las impurezas resultantes del proceso utilizado, con exclusión de cualquier disolvente que pueda separarse sin afectar a la estabilidad de la sustancia ni cambiar su composición. Producto problema : Sustancia o mezcla estudiada con este método de ensayo. Estrategia de ensayos escalonados : Estrategia de ensayo por fases, en la que se revisa toda la información existente sobre un producto problema, siguiendo un orden especificado, en un proceso de ponderación de las pruebas en cada escalón, a fin de determinar si se dispone de información suficiente para tomar una decisión sobre la clasificación de un peligro, antes de pasar al escalón siguiente. Si puede establecerse la capacidad de irritación de un producto problema con la información disponible, no hace falta efectuar más ensayos. Si no puede establecerse la capacidad de irritación de un producto problema con la información disponible, se aplica un procedimiento secuencial de ensayos con animales por fases hasta que pueda efectuarse una clasificación inequívoca. Método de ensayo validado : Método de ensayo sobre el cual se han completado estudios de validación para determinar su pertinencia (incluida su exactitud) y su fiabilidad con un fin específico. Es importante señalar que un método de ensayo validado podría no tener un comportamiento suficiente en términos de exactitud y fiabilidad como para considerarse aceptable a efectos del fin propuesto (8). Ponderación de las pruebas : Proceso de consideración de los aspectos favorables y desfavorables de los distintos elementos de información a efectos de alcanzar y confirmar una conclusión en cuanto al peligro potencial de un producto. Apéndice 3 PRODUCTOS UTILIZADOS PARA DEMOSTRAR LA COMPETENCIA CON EL MÉTODO DE ENSAYO FL Antes de proceder al uso sistemático de este método de ensayo, los laboratorios deben demostrar su competencia técnica, identificando correctamente la clasificación de los ocho productos recomendados del cuadro 1 en cuanto a su corrosividad ocular. Estos productos se seleccionaron para representar la gama de respuestas en cuanto a la irritación/corrosión local de los ojos, sobre la base de los resultados del ensayo in vivo con ojo de conejo [TG 405, método B.5 (5)] (es decir, categorías 1, 2A, 2B, o sin clasificar según el SGA de la ONU). Sin embargo, considerando la utilidad validada del ensayo FL (es decir, identificar solamente agentes corrosivos/ irritantes intensos para los ojos), solo hay dos resultados del ensayo a efectos de clasificación (agente corrosivo/ irritante intenso o agente no corrosivo / no irritante intenso) para demostrar la competencia con el método. Otros criterios de selección fueron el que los productos estuvieran disponibles en el mercado, que tuvieran datos de referencia in vivo de alta calidad y también datos de alta calidad obtenidos con el método de ensayo FL. Por esta razón, los productos de la prueba de competencia se seleccionaron a partir del documento» Fluorescein Leakage Assay Background Review Document as an Alternative Method for Eye Irritation Testing «(8), utilizado para la validación retrospectiva del método de ensayo FL. Cuadro 1: Productos recomendados para demostrar la competencia técnica con el método FL
B.62. Ensayo del cometa en condiciones alcalinas con mamíferos in vivo INTRODUCCIÓN El presente método de ensayo es equivalente a las directrices de ensayo (TG) de la OCDE 489 (2016). El ensayo del cometa en condiciones alcalinas (electroforesis en gel de células aisladas) (en lo sucesivo, denominado simplemente el ensayo del cometa) se utiliza para la detección de roturas de cadenas de ADN en células o núcleos aislados de múltiples tejidos de animales (por lo general, roedores), que se han visto expuestos a materiales potencialmente genotóxicos. El ensayo del cometa ha sido revisado por distintos grupos de expertos, que han publicado sus recomendaciones (1) (2) (3) (4) (5) (6) (7) (8) (9) (10). El presente método forma parte de una serie de métodos de ensayo sobre toxicología genética. Se ha elaborado un documento de la OCDE que aporta información sucinta sobre los ensayos de toxicología genética y una síntesis de los recientes cambios aportados a dichas directrices de ensayo (11). La finalidad del ensayo del cometa es identificar los productos que causan lesiones del ADN. En condiciones alcalinas (pH > 13), el ensayo del cometa puede detectar roturas de cadenas sencillas y dobles que son resultado, por ejemplo, de interacciones directas con secuencias de ADN lábiles en condiciones alcalinas, o como consecuencia de roturas fugaces de cadenas de ADN resultantes de la reparación del ADN por excisión. Estas roturas de cadenas pueden repararse, con lo que no tendrán efecto persistente, pueden ser letales para las células, o pueden fijarse en una mutación que provoque un cambio viable permanente. También pueden dar lugar a lesiones cromosómicas asociadas con numerosas enfermedades humanas, como el cáncer. En 2006-2012 se llevó a cabo un estudio de validación oficial del ensayo del cometa con roedores in vivo, coordinado por el Centro Japonés para la Validación de Métodos Alternativos (Japanese Center for the Validation of Alternative Methods, JaCVAM), en colaboración con el Centro Europeo para la Validación de Métodos Alternativos (CEVMA), el Comité de Coordinación Interagencias sobre la Validación de Métodos Alternativos (Interagency Coordinating Committee on the Validation of Alternative Methods, ICCVAM) y el Centro Interagencias del NTP para la Evaluación de Métodos Toxicológicos Alternativos (NTP Interagency Center for the Evaluation of Alternative Toxicological Methods, NICEATM) (12). Este método de ensayo incluye el uso recomendado y las limitaciones del ensayo del cometa, y se basa en el protocolo final (12) utilizado en el estudio de validación, y en datos adicionales pertinentes tanto publicados como no publicados (propiedad de los laboratorios). Las definiciones de los términos clave figuran en el apéndice 1. Cabe señalar que pueden utilizarse para este ensayo muchos soportes diferentes (portaobjetos para microscopio, manchas en gel, placas de 96 pocillos, etc.). Por razones prácticas, se emplea en el resto de este documento el término «portaobjetos», pero abarca todos los demás soportes. CONSIDERACIONES INICIALES Y LIMITACIONES El ensayo del cometa es un método para medir las roturas de las cadenas de ADN en células eucarióticas. Con detergente y una elevada concentración de sales, se lisan células o núcleos aislados incorporados en gel de agarosa en un portaobjetos. Esta fase de lisis celular digiere las membranas nucleares y celulares y permite la liberación de bucles helicoidales de ADN, denominados generalmente nucleoides y fragmentos de ADN. La electroforesis a pH elevado resulta en estructuras parecidas a los cometas, que, mediante las adecuadas tinciones fluorescentes, pueden observarse mediante microscopia de fluorescencia; los fragmentos de ADN migran desde la «cabeza» a la «cola» en función de su tamaño, y la intensidad de la cola del cometa respecto a la intensidad total (cabeza y cola) refleja la proporción de la rotura del ADN (13) (14) (15). El ensayo del cometa en condiciones alcalinas in vivo está especialmente indicado para evaluar el peligro genotóxico porque las respuestas del ensayo dependen del ADME (absorción, distribución, metabolismo y excreción) in vivo, y también de los procesos de reparación del ADN. Estos pueden variar según la especie, el tejido y el tipo de lesiones del ADN. Para cumplir los requisitos de bienestar animal, en particular la reducción del uso de animales (principio de las tres eres: reemplazar, reducir y refinar), es posible también integrar este ensayo con otros estudios toxicológicos, como los de toxicidad por administración continuada (10) (16) (17), o combinar su criterio de valoración con otros criterios de genotoxicidad, como el del ensayo de micronúcleos en eritrocitos de mamíferos in vivo (18) (19) (20). El ensayo del cometa se realiza en la mayoría de los casos con roedores, aunque se ha aplicado a otras especies de mamíferos y de animales no mamíferos. El uso de especies distintas de los roedores debe justificarse desde el punto de vista científico y ético en cada caso, y se recomienda vivamente que el ensayo del cometa se realice con especies distintas de los roedores solo como parte de otro estudio de toxicidad y no como ensayo independiente. La vía de exposición y el tejido o tejidos objeto de estudio deben elegirse en función de todos los datos disponibles/existentes sobre los productos problema, tales como la vía prevista o posible de exposición humana, el metabolismo y distribución; la posibilidad de efectos en el punto de contacto, las alertas estructurales, otros datos de toxicidad y genotoxicidad, y el objeto del estudio. Por lo tanto, en su caso, el potencial genotóxico de los productos problema podrá analizarse en el tejido o tejidos diana de los efectos carcinogénicos y/u otros efectos tóxicos. El ensayo también se considera útil para ahondar en el estudio de la genotoxicidad detectada por un sistema in vitro. Es adecuado realizar un ensayo del cometa in vivo en un tejido de interés cuando se pueda esperar razonablemente que este tejido estará convenientemente expuesto. El ensayo se ha validado más ampliamente en tejidos somáticos de ratas machos en estudios colaborativos como el estudio del JaCVAM (12) y en Rothfuss et al., 2010 (10). En el estudio de validación internacional del JaCVAM se utilizaron el hígado y el estómago: el hígado, porque es el órgano más activo en el metabolismo de los productos y también con frecuencia un órgano diana de la carcinogenicidad; el estómago, porque normalmente es el primer punto de contacto con los productos tras la exposición oral, aunque también deben considerarse tejidos del punto de contacto otras zonas del tubo gastrointestinal como el duodeno y el yeyuno, las cuales podrían considerarse más pertinentes para la exposición humana que el estómago glandular de los roedores. Debe velarse por que estos tejidos no se expongan a concentraciones excesivamente elevadas del producto problema (21). La técnica es aplicable, en principio, a cualquier tejido del que puedan obtenerse suspensiones de núcleos o células aisladas analizables. Ciertos datos propiedad de varios laboratorios demuestran el éxito de su aplicación a muchos tejidos diferentes, y existen muchas publicaciones que muestran la aplicabilidad de la técnica a órganos o tejidos distintos de, por ejemplo, hígado y estómago, como el yeyuno (22), el riñón (23) (24), la piel (25) (26), o la vejiga urinaria (27) (28), los pulmones y las células del lavado broncoalveolar (pertinentes para estudios de productos inhalados) (29) (30), y también se han realizado ensayos en múltiples órganos (31) (32). Si bien puede haber interés por determinar los efectos genotóxicos en células germinales, debe señalarse que el ensayo del cometa normal en condiciones alcalinas, tal como se describe en el presente método de ensayo, no se considera adecuado para medir las roturas de las cadenas de ADN en las células germinales maduras. Dado que en un estudio bibliográfico sobre la utilización del ensayo del cometa sobre la genotoxicidad en células germinales se señalaron unos niveles de fondo elevados y variables de las lesiones del ADN (33), se considera necesario aportar modificaciones a los protocolos y mejorar la normalización y los estudios de validación antes de que pueda incluirse en el método de ensayo el ensayo del cometa con células germinales (por ejemplo, esperma). Por otra parte, la pauta de exposición recomendada descrita en el presente método de ensayo no es la óptima y puede ser necesario aplicar otros tiempos más largos de exposición o de muestreo para un análisis significativo de las roturas de las cadenas de ADN en el esperma maduro. En la bibliografía se han descrito efectos genotóxicos, medidos por el ensayo del cometa en células testiculares en diferentes etapas de la diferenciación (34) (35). No obstante, cabe señalar que las gónadas contienen una mezcla de células somáticas y germinales. Por ello, unos resultados positivos en las gónadas completas (testículos) no reflejan necesariamente las lesiones causada en las células germinales; sin embargo, indican que el producto o productos problema o sus metabolitos han alcanzado las gónadas. El entrecruzamiento no puede detectarse de manera fiable en las condiciones experimentales normales del ensayo del cometa. En determinadas condiciones experimentales modificadas podrían detectarse los enlaces cruzados ADN-ADN y ADN-proteína, y otras modificaciones de las bases, como su oxidación (23) (36) (37) (38) (39). Pero sería necesario seguir trabajando para caracterizar adecuadamente las necesarias modificaciones del protocolo. Así pues, la detección de los agentes de entrecruzamiento no es el objetivo principal del ensayo que se describe aquí. El ensayo no es adecuado, ni siquiera con modificaciones, para detectar anéugenos. Debido al estado actual de los conocimientos, el ensayo del cometa in vivo tiene asociadas varias limitaciones adicionales (véase el apéndice 3). Se espera que el método de ensayo se revise en el futuro y, en caso necesario, se modifique a la luz de la experiencia adquirida. Antes de la utilización del método de ensayo con una mezcla para obtener datos con fines reglamentarios, debe considerarse si podría proporcionar resultados adecuados a tal fin y, en caso afirmativo, por qué. Tales consideraciones no son necesarias si la reglamentación impone el ensayo de la mezcla. PRINCIPIO DEL MÉTODO Se exponen los animales al producto problema por una vía adecuada. Una descripción detallada de la administración y del muestreo figura en los puntos 36-40. En el momento o momentos de muestreo seleccionados, se extraen los tejidos de interés y se preparan suspensiones de células/núcleos aislados (puede realizarse perfusión in situ cuando se considere útil, por ejemplo en el hígado), que se incorporan a agar blando para inmovilizarlas en portaobjetos. Las células/núcleos se tratan con solución amortiguadora de lisis para eliminar las membranas celulares y/o nucleares, y se exponen a bases fuertes, por ejemplo a pH ≥ 13, a fin de permitir que se desenrolle el ADN y se liberen los bucles y fragmentos de ADN relajado. El ADN nuclear presente en el agar se somete entonces a electroforesis. Las moléculas de ADN no fragmentado normal permanecen en la posición en la que se encontraba el ADN nuclear en el agar, al tiempo que los eventuales bucles de ADN fragmentado y ADN relajado migran hacia el ánodo. Tras la electroforesis, el ADN se visualiza utilizando una tinción fluorescente apropiada. Las preparaciones deben analizarse utilizando un microscopio y sistemas de análisis de imágenes totalmente automatizados o semiautomatizados. La proporción de ADN que ha migrado durante la electroforesis y la distancia de migración reflejan la cantidad y el tamaño de los fragmentos de ADN. Hay varios criterios de valoración del ensayo del cometa. Para evaluar las lesiones del ADN se ha recomendado el ADN contenido en la cola (porcentaje de ADN de la cola o porcentaje de intensidad de la cola) (12) (40) (41) (42). Tras el análisis de un número suficiente de núcleos, los datos se analizan con métodos adecuados para evaluar los resultados del ensayo. Debe tenerse en cuenta que se ha investigado la modificación de varios aspectos de la metodología, con inclusión de la preparación de las muestras, las condiciones de la electroforesis, los parámetros del análisis visual (por ejemplo, intensidad de la tinción, intensidad de la luz de la bombilla del microscopio, y utilización de filtros de microscopio y ajustes de la cámara) y las condiciones ambientales (p. ej., iluminación de fondo), que pueden afectar a la migración del ADN (43) (44) (45) (46). VERIFICACIÓN DE LA COMPETENCIA DEL LABORATORIO Cada laboratorio debe establecer su competencia experimental con el ensayo del cometa demostrando su capacidad para obtener suspensiones de células o núcleos aislados de calidad suficiente con cada tejido diana de cada una de las especies utilizadas. La calidad de las preparaciones se evaluará en primer lugar observando si el porcentaje de ADN de la cola de animales tratados con un vehículo está en un intervalo bajo y reproducible. Los datos actuales sugieren que la media del grupo del porcentaje de ADN de la cola (sobre la base de la media de las medianas — véanse en el punto 57 detalles de estos términos) en hígado de rata no debe superar preferentemente el 6 %, lo que sería compatible con los valores del estudio de validación del JaCVAM (12) y con otros datos publicados y protegidos. No hay suficientes datos por el momento para hacer recomendaciones sobre intervalos óptimos o aceptables para otros tejidos. Esto no excluye la utilización de otros tejidos, cuando esté justificado. El informe del ensayo debe proporcionar un análisis adecuado del comportamiento del ensayo del cometa con estos tejidos en relación con la bibliografía publicada o con datos protegidos. En primer lugar, es deseable que un intervalo bajo de porcentaje de ADN de cola en los testigos permita un intervalo analítico suficiente para detectar un efecto positivo. En segundo lugar, cada laboratorio debe ser capaz de reproducir las respuestas previstas ante mutágenos directos y promutágenos con distintos modos de acción, tal como se sugiere en el cuadro 1 (punto 29). Pueden seleccionarse sustancias positivas, por ejemplo a partir del estudio de validación del JaCVAM (12) o de otros datos publicados (véase el punto 9), cuando sea pertinente, con justificación, y que demuestren respuestas positivas claras en los tejidos de interés. También debe demostrarse la capacidad de detectar efectos débiles de mutágenos conocidos, como el EMS a dosis bajas, por ejemplo estableciendo relaciones dosis-respuesta con dosis apropiadas en cuanto a su número y al intervalo entre ellas. Los esfuerzos iniciales deben centrarse en establecer la competencia con los tejidos más utilizados como, por ejemplo, el hígado de roedores, para poder efectuar comparaciones con los datos existentes y los resultados previstos (12). Al mismo tiempo podrían recogerse datos de otros tejidos como, por ejemplo, estómago/duodeno/yeyuno, sangre, etc. El laboratorio debe demostrar su competencia con cada tejido de cada especie que tenga intención de estudiar, y debe demostrar que se puede obtener en ese tejido una respuesta positiva aceptable con un mutágeno conocido (por ejemplo, EMS). Deben recogerse datos de testigos negativos o del vehículo con el fin de demostrar la reproducibilidad de las respuestas de datos negativos, y de garantizar que los aspectos técnicos del ensayo se controlan adecuadamente o de sugerir la necesidad de volver a establecer los intervalos de los testigos históricos (véase el punto 22). Conviene señalar que, si bien pueden recogerse en la autopsia y someterse al ensayo del cometa muchos tejidos, el laboratorio tiene que ser competente en la recolección de múltiples tejidos de un solo animal, garantizando así que no se pierde ninguna de las posibles lesiones del ADN y que no se pone en peligro el análisis del cometa. El plazo transcurrido entre la eutanasia y la extracción de los tejidos para su transformación puede ser crítica (véase el punto 44). Debe considerarse el bienestar de los animales al adquirir la competencia con este ensayo, por lo que pueden utilizarse tejidos de animales utilizados en otros ensayos cuando se esté adquiriendo competencia con los diversos aspectos del ensayo. Por otra parte, podrá no ser necesario realizar un estudio completo durante las fases de establecimiento de un nuevo método de ensayo en un laboratorio y podrán utilizarse menos animales o menos concentraciones de ensayo cuando se estén adquiriendo las capacidades necesarias. Datos sobre testigos históricos En el transcurso de las investigaciones de competencia, el laboratorio debe crear una base de datos históricos para establecer los intervalos de los testigos positivos y negativos correspondientes y las distribuciones relativas a los tejidos y especies pertinentes. En la bibliografía pueden encontrarse recomendaciones sobre cómo conseguir y utilizar los datos históricos (es decir, criterios de inclusión y exclusión de datos en los datos históricos y criterios de aceptabilidad para un determinado experimento) (47). Con otros tejidos y otras especies, así como con otros vehículos y vías de administración, pueden obtenerse valores diferentes de porcentaje de ADN de la cola en el testigo negativo. Por lo tanto, es importante establecer los intervalos de los testigos negativos para cada uno de los tejidos y especies. Los laboratorios deben utilizar métodos de control de calidad, como gráficos de control (por ejemplo, gráficos C o gráficos de medias (48)), con el fin de determinar la variabilidad de sus datos y de demostrar que la metodología está «controlada» en su laboratorio. Es posible que para la detección de los efectos débiles también resulte necesario optimizar la selección de las sustancias adecuadas para servir de testigo positivo, los intervalos de dosis y las condiciones experimentales (por ejemplo, las condiciones de la electroforesis) (véase el punto 17). Cualquier cambio en el protocolo experimental debe considerarse en función de su coherencia con las bases de datos de testigos históricos existentes del laboratorio. Cualquier incoherencia importante debería dar lugar a la creación de una nueva base de datos de testigos históricos. DESCRIPCIÓN DEL MÉTODO Preparación Selección de la especie animal Se utilizan normalmente cepas comunes de laboratorio de roedores adultos jóvenes y sanos (6-10 semanas de edad al inicio del tratamiento, aunque también se pueden aceptar animales ligeramente de más edad). La elección de la especie de roedores debe basarse en: i) las especies utilizadas en otros estudios de toxicidad (para poder relacionar los datos y permitir la integración de los estudios), ii) las especies que hayan formado tumores en un estudio de carcinogenicidad (al investigar el mecanismo de la carcinogénesis), o iii) las especies con el metabolismo más pertinente para los seres humanos, si se conoce. En este ensayo se utilizan habitualmente ratas. No obstante, pueden utilizarse otras especies si se justifica desde el punto de vista científico y ético. Condiciones de alojamiento y alimentación de los animales Con roedores, la temperatura en el animalario experimental debe ser idealmente de 22 °C (± 3 °C). Lo ideal es que la humedad relativa sea del 50-60 %, con un mínimo del 30 % y un máximo preferentemente del 70 %, salvo durante la limpieza del local. La iluminación debe ser artificial, con 12 horas de luz y 12 horas de oscuridad. Para la alimentación se pueden utilizar dietas de laboratorio convencionales, con suministro ilimitado de agua para beber. La elección de la dieta puede verse influida por la necesidad de garantizar una mezcla conveniente del producto problema si se administra por esta vía. Los roedores deben alojarse en grupos pequeños (en general, no más de cinco animales juntos) del mismo sexo si no se espera conducta agresiva. Los animales podrán alojarse por separado solo si está justificado científicamente. Debe utilizarse siempre que sea posible un suelo continuo, ya que los suelos de malla pueden provocar lesiones graves (49). Debe proporcionarse enriquecimiento ambiental adecuado. Preparación de los animales Los animales se reparten al azar entre los lotes tratados y los testigo. Se identifica a los animales de forma unívoca y se acostumbran a las condiciones del laboratorio durante al menos cinco días antes del inicio del tratamiento. Debe utilizarse el método menos invasivo para la identificación unívoca de los animales. Entre los métodos adecuados figuran el anillamiento, el marcado, la implantación de un microchip y la identificación biométrica. El grapado a las orejas o los dedos no está justificados científicamente en estos ensayos. Las jaulas deben disponerse de forma que se reduzcan al mínimo los posibles efectos debidos al enjaulamiento. La variación del peso de los animales al empezar el estudio ha de ser mínima y no debe exceder del ± 20 %. Preparación de las dosis Los productos problema sólidos deben disolverse o suspenderse en vehículos adecuados, o mezclarse con la dieta o con el agua de bebida, antes de su administración a los animales. Los productos problema líquidos pueden administrarse directamente o diluirse antes de la administración. En lo que respecta a la exposición por inhalación, los productos problema pueden administrarse en forma de gas, de vapor o de aerosol sólido o líquido, dependiendo de sus propiedades fisicoquímicas (50) (51). Deben utilizarse preparaciones recientes del producto problema, salvo que se cuente con datos de estabilidad que avalen la posibilidad de su conservación y definan las condiciones adecuadas de esta. Condiciones del ensayo Vehículo El vehículo no debe producir efectos tóxicos a los volúmenes de dosis empleados, y no debe haber ninguna sospecha de que reaccione químicamente con los productos problema. Si se emplean vehículos poco conocidos, su inclusión debe estar avalada por información de referencia que indique su compatibilidad en cuanto a los animales de ensayo, la vía de administración y el criterio de valoración. Siempre que sea posible, se recomienda considerar en primer lugar la utilización de un disolvente o vehículo acuoso. Cabe señalar que algunos de los vehículos (sobre todo los vehículos viscosos) pueden provocar inflamación y aumento de los niveles de base de las roturas de las cadenas de ADN en el punto de contacto, en particular en caso de administración múltiple. Testigos Testigos positivos En este momento se considera en principio que debe incluirse en cada ensayo un grupo formado por al menos tres animales analizables de un mismo sexo, o de cada sexo si se utilizan ambos (véase el punto 32), tratados con una sustancia testigo positivo. En el futuro podrá ser posible demostrar que se tiene la competencia adecuada y reducir la necesidad de testigos positivos. Si se utilizan múltiples momentos de muestreo (por ejemplo, con un protocolo de una única administración) solo es necesario incluir testigos positivos en uno de los tiempos de muestreo, pero debe garantizarse un diseño equilibrado (véase el punto 48). No es preciso administrar sustancias testigo positivo en paralelo por la misma vía que el producto problema, aunque es importante utilizar la misma vía cuando se miden los efectos en el punto de contacto. Debe demostrarse que las sustancias testigo positivo inducen la rotura de las cadenas de ADN en todos los tejidos de interés para el producto problema, y resulta probable que el EMS sea el testigo positivo de elección, dado que provoca la rotura de las cadenas de ADN en todos los tejidos que se han estudiado. Las dosis de las sustancias testigo positivo deben seleccionarse para generar efectos moderados que sirvan para evaluar críticamente el comportamiento y la sensibilidad del ensayo, y podrían basarse en las curvas de dosis-respuesta establecidas por el laboratorio durante la demostración de la competencia. El porcentaje de ADN de la cola de animales testigo positivo en paralelo debe ser coherente con el intervalo preestablecido en el laboratorio para cada tejido y tiempo de muestreo de la especie correspondiente (véase el punto 16). En el cuadro 1 se incluyen ejemplos de sustancias testigo positivo y algunos de sus tejidos diana (en roedores). Se pueden seleccionar sustancias distintas de las que figuran en el cuadro 1, cuando esté justificado científicamente. Cuadro 1: Ejemplos de sustancias testigo positivo y algunos de sus tejidos diana Sustancias y CAS RN Metanosulfonato de etilo (CAS RN 62-50-0) en cualquier tejido Etil-nitrosourea (CAS RN 759-73-9) en hígado y estómago, duodeno o yeyuno Metanosulfonato de metilo (CAS RN 66-27-3) en hígado, estómago, duodeno o yeyuno, pulmón y células del lavado broncoalveolar (BAL), riñón, vejiga urinaria, pulmón, testículo y médula ósea o sangre N-Metil-N'-nitro-N-nitrosoguanidina (CAS RN 70-25-7) en estómago, duodeno o yeyuno 1,2-Dimetilhidrazina 2HCl (CAS RN 306-37-6) en hígado e intestino N-Metil-N-nitrosourea (CAS RN 684-93-5) en hígado, médula ósea, sangre, riñón, estómago, yeyuno y cerebro. Testigos negativos Debe incluirse en cada ensayo, respecto a cada tejido y momento de muestreo, un grupo de animales testigo negativo, tratados solo con el vehículo, pero por lo demás sometidos al mismo proceso que los grupos tratados con el producto problema. El porcentaje de ADN de la cola de los animales testigo negativo debe estar dentro del intervalo de fondo preestablecido en el laboratorio para cada tejido y tiempo de muestreo de la especie correspondiente (véase el punto 16). En ausencia de información anterior o publicada sobre testigos que demuestre que no se encuentran efectos nocivos ni genotóxicos inducidos por el vehículo elegido, por el número de administraciones o por la vía de administración, deberán realizarse estudios iniciales antes de llevar a cabo el estudio completo, a fin de establecer que es aceptable el testigo del vehículo. PROCEDIMIENTO Número y sexo de los animales Aunque hay pocos datos sobre hembras para posibilitar la comparación entre sexos en relación con el ensayo del cometa, otras respuestas de genotoxicidad in vivo son similares, en general, entre machos y hembras, por lo que la mayor parte de los estudios podrían realizarse con cualquier sexo. Unos datos que demuestren la existencia de diferencias pertinentes entre machos y hembras (como diferencias de toxicidad sistémica, metabolismo, biodisponibilidad, etc., por ejemplo en un estudio de determinación del intervalo) favorecerán la utilización de ambos sexos. En este caso puede ser conveniente realizar un estudio con ambos sexos, por ejemplo como parte de un estudio de toxicidad por administración continuada. Podría ser adecuado utilizar el diseño factorial en caso de que se utilizaran animales de ambos sexos. En el apéndice 2 se recogen detalles sobre cómo analizar los datos utilizando dicho diseño. El tamaño de los grupos al principio del estudio (y durante la determinación de la competencia) debe establecerse de manera que comprendan un mínimo de cinco animales analizables de un mismo sexo, o de cada sexo si se utilizan ambos, por grupo (menos en el grupo del testigo positivo en paralelo, véase el punto 29). En caso de que la exposición humana a los productos pueda ser específica de un sexo, como sucede con algunos productos farmacéuticos, el ensayo debe realizarse con animales del sexo correspondiente. Como orientación sobre las necesidades máximas típicas de animales, un estudio realizado de acuerdo con los parámetros establecidos en el punto 33 con tres grupos tratados y testigos negativo y positivo en paralelo (cada grupo compuesto por cinco animales de un solo sexo), requeriría entre 25 y 35 animales. PAUTA DE TRATAMIENTO Los animales deben recibir tratamientos diarios durante un período de dos o más días (es decir, dos o más administraciones a intervalos de 24 horas, aproximadamente), y deben recogerse muestras una vez a las 2-6 h (o al Tmax) después del último tratamiento (12). Son aceptables las muestras tomadas de pautas de tratamiento ampliadas (por ejemplo, administración diaria durante 28 días). Se ha demostrado el éxito de la combinación del ensayo del cometa con el de micronúcleos en eritrocitos (10) (19). No obstante, es preciso prestar especial atención a la logística necesaria en relación con el muestreo de tejidos para el análisis de cometa junto con los requisitos del muestreo de tejidos para otros tipos de evaluaciones toxicológicas. La recolección a las 24 horas de la última administración, típica de un estudio de toxicidad general, no es adecuada en la mayoría de los casos (véase el punto 40 sobre el tiempo de muestreo). El uso de otras pautas de tratamiento y de muestreo debe justificarse (véase el apéndice 3). Por ejemplo, podría utilizarse un único tratamiento con múltiples muestreos; debe observarse, no obstante, que serán necesarios más animales para un estudio con una única administración debido a la necesidad de múltiples momentos de muestreo, pero en ocasiones esto puede ser preferible, por ejemplo cuando el producto problema induce toxicidad excesiva tras la administración repetida. Independientemente de cómo se realice el ensayo, es aceptable en la medida en que el producto problema dé una respuesta positiva o, en caso de estudio negativo, siempre que se hayan aportado pruebas directas o indirectas de la exposición del tejido o tejidos diana (o de toxicidad para estos), o si se alcanza la dosis límite (véase el punto 36). Los productos problema también pueden administrarse en dosis divididas (es decir, en dos tratamientos el mismo día separados por no más de 2 o 3 horas) con el fin de facilitar la administración de grandes volúmenes. En estas circunstancias, el tiempo de muestreo debe programarse en función del momento de la última administración (véase el punto 40). Dosis Si se lleva a cabo un estudio previo de determinación del intervalo porque no se dispone de datos adecuados de otros estudios pertinentes para ayudar en la selección de las dosis, ha de hacerse en el mismo laboratorio, con la misma especie, cepa, sexo y pauta de tratamiento que se vayan a utilizar en el estudio principal, según los planteamientos actuales de la realización de estudios de determinación del intervalo (10). El estudio deberá tratar de determinar la dosis máxima tolerada (DMT), definida como la dosis que induce efectos ligeramente tóxicos con respecto a la duración del período de estudio (por ejemplo, signos clínicos claros, tales como comportamiento o reacciones de tipo anormal, pequeña pérdida de peso corporal o citotoxicidad para el tejido diana), pero sin llegar a provocar la muerte ni signos de dolor, sufrimiento o angustia que hagan necesaria la eutanasia. En caso de producto problema no tóxico, con un período de administración de 14 días o más, la dosis máxima (límite) es de 1 000 mg/kg de peso corporal/día. Si el período de administración es inferior a 14 días, la dosis máxima (límite) será de 2 000 mg/kg de peso corporal/día. Estos límites pueden variar con determinados tipos de productos problema (por ejemplo, productos farmacéuticos de uso humano) a los que se apliquen normativas específicas. Los productos que presentan saturación de las propiedades toxicocinéticas, o inducen procesos de destoxificación que puedan dar lugar a una disminución de la exposición después de una administración a largo plazo, pueden constituir excepciones en cuanto a los criterios de establecimiento de la dosis y han de evaluarse caso por caso. Para las versiones del ensayo del cometa sobre toxicidad tanto aguda como subaguda, además de la dosis máxima (DMT, dosis máxima posible, dosis máxima de exposición o dosis límite), debe seleccionarse una secuencia descendente de al menos otras dos dosis espaciadas adecuadamente (de preferencia separadas por menos de 10) para cada tiempo de muestreo a fin de demostrar la relación de las respuestas con la dosis. No obstante, las dosis empleadas también deben abarcar preferentemente el intervalo entre la máxima y una que produzca toxicidad escasa o nula. Si se observa toxicidad para el tejido diana a todas las dosis utilizadas en el ensayo, se recomienda hacer otro estudio a dosis no tóxicas (véanse los puntos 54-55). Los estudios destinados a investigar de forma más completa la forma de la curva dosis-respuesta pueden necesitar más grupos de dosis. Administración de las dosis A la hora de diseñar el ensayo, debe tenerse en cuenta la vía prevista de exposición humana. Por lo tanto, cuando esté justificado, podrán elegirse vías de exposición como los alimentos, el agua de bebida, la tópica, la subcutánea, la intravenosa, la oral forzada (por sonda), la inhalación, la intratraqueal o la implantación. En cualquier caso, la vía debe elegirse de forma que se garantice una exposición adecuada del tejido o tejidos diana. En general no se recomienda la inyección intraperitoneal, ya que no es una vía de exposición humana típica y pertinente, y solo debe utilizarse con justificación específica (por ejemplo, algunas sustancias testigo positivo, a efectos de investigación, o algunos medicamentos que se administran por vía intraperitoneal). El volumen máximo de líquido que puede administrarse de una sola vez por sonda o por inyección depende del tamaño del animal utilizado. Este volumen no debe superar 1 ml/l00 g de peso corporal, salvo en el caso de las soluciones acuosas, en que puede llegar a 2 ml/l00 g de peso corporal. Debe justificarse el uso de volúmenes superiores (en caso de que lo permita la legislación sobre bienestar animal). Siempre que sea posible, las diferentes dosis deben lograrse ajustando la concentración de la formulación administrada para garantizar un volumen constante en relación con el peso corporal a todas las dosis. Momento del muestreo El momento del muestreo es una variable crítica, ya que viene determinado por el plazo necesario para que los productos problema alcancen la concentración máxima en el tejido diana y para que se induzcan las roturas de las cadenas de ADN, pero antes de que tales roturas se supriman, reparen o den lugar a la muerte de las células. La persistencia de algunas de las lesiones que dan lugar a la rotura de las cadenas de ADN detectadas por el ensayo del cometa puede ser muy breve, al menos con algunos productos estudiados in vitro (52) (53). Por consiguiente, en caso de que se sospeche la existencia de tales lesiones transitorias de ADN, deben tomarse medidas para mitigar su pérdida velando por que los tejidos se muestreen antes de que sea demasiado tarde, quizás antes de los plazos previstos que figuran a continuación. El tiempo o tiempos de muestreo óptimos pueden ser específicos del producto o de la vía, lo que puede dar lugar, por ejemplo, a una exposición tisular rápida cuando la administración se hace por vía intravenosa o por inhalación. En consecuencia, cuando sea posible, los tiempos de muestreo deben determinarse a partir de los datos cinéticos (por ejemplo, el tiempo (Tmax) al que se alcance la concentración plasmática o tisular máxima (Cmax), o al que se alcance el estado de equilibrio en caso de administración múltiple). En ausencia de datos cinéticos, un compromiso adecuado para la medición de la genotoxicidad es tomar las muestras a las 2-6 h después de la última administración en caso de dos o más administraciones, o tanto a las 2-6 horas como a las 16-26 horas tras una administración única, aunque debe velarse por que la autopsia se haga a todos los animales al mismo tiempo desde la última dosis (o desde la dosis única). La información sobre la aparición de efectos tóxicos en los órganos diana (si se dispone de ella) también puede utilizarse para seleccionar los tiempos de muestreo adecuados. Observaciones Deben hacerse y registrarse observaciones clínicas generales en relación con la salud de los animales al menos una vez al día, preferentemente a la misma hora u horas cada día y teniendo en cuenta el período de mayor intensidad de los efectos previstos tras la administración (54). Al menos dos veces al día, se debe observar la posible morbilidad y mortalidad de todos los animales. En los estudios a largo plazo, deben pesarse todos los animales al menos una vez por semana y en el momento de la terminación del período del ensayo. El consumo de alimentos debe medirse a cada cambio de alimentos y al menos una vez por semana. Si el producto problema se administra con el agua de bebida, debe medirse el consumo de agua cada vez que se cambie el agua y al menos una vez por semana. Los animales que presenten indicadores de toxicidad excesiva pero no letal deben sacrificarse antes de que termine el período del ensayo, y normalmente no se utilizan para el análisis de cometa. Recogida de tejidos Teniendo en cuenta que es posible estudiar la inducción de la rotura de cadenas de ADN (cometas) en prácticamente cualquier tejido, la justificación de la selección del tejido o tejidos que se van a recoger ha de estar claramente definida y basarse en el motivo de la realización del estudio, junto con los eventuales datos existentes sobre ADME, genotoxicidad, carcinogenicidad u otros tipos de toxicidad en relación con los productos problema sometidos a investigación. Entre los factores importantes que deben considerarse están la vía de administración (sobre la base de la vía o vías probables de exposición humana), la distribución y la absorción tisulares previstas, el papel del metabolismo y el posible mecanismo de acción de los productos problema. El hígado es el tejido estudiado con mayor frecuencia y del que se dispone de más datos. Por lo tanto, a falta de información de fondo, y si no se señala ningún tejido de interés específico, el muestreo del hígado estaría justificado por ser este el principal lugar de metabolismo de las sustancias xenobióticas y estar a menudo muy expuesto tanto a la sustancia original como a sus metabolitos. En algunos casos, puede ser muy pertinente el análisis de un punto de contacto directo (por ejemplo, el estómago glandular o el duodeno/yeyuno en el caso de los productos de administración oral, o los pulmones en el de los productos inhalados). Deben seleccionarse tejidos adicionales o alternativos sobre la base de las razones específicas para la realización del ensayo, pero puede ser útil examinar varios tejidos en los mismos animales, siempre que el laboratorio haya demostrado su competencia con dichos tejidos y con la manipulación de varios tejidos al mismo tiempo. Preparación de las muestras Para los procesos descritos en los puntos siguientes (44-49) es importante que todas las soluciones o suspensiones estables se utilicen antes de su fecha de caducidad, o que se preparen de nuevo en caso necesario. También en los siguientes puntos, se consideran variables críticas (véanse las definiciones del apéndice 1) los momentos elegidos para: i) retirar cada tejido después de la autopsia, ii) procesar cada tejido para convertirlo en suspensiones de núcleos o células, y iii) procesar la suspensión y preparar los portaobjetos, y deben haberse determinado unos plazos aceptables para cada uno de estas fases durante el establecimiento del método y la demostración de la competencia. Los animales se sacrifican, de acuerdo con la legislación sobre bienestar animal y el principio de las tres eres, en el momento o momentos adecuados después del último tratamiento con el producto problema. El tejido o tejidos seleccionados se retiran, se disecan, y una parte se recoge para el ensayo del cometa, y, al mismo tiempo, una sección de la misma parte del tejido debe cortarse y colocarse en solución de formaldehído o en un fijador adecuado para su posible examen histopatológico (véase el punto 55) según métodos normalizados (12). Los tejidos para el ensayo del cometa se colocan en solución amortiguadora de picar, se lavan suficientemente con esta solución en frío para retirar la sangre residual, y se conservan en esta solución enfriada con hielo hasta su transformación. También puede realizarse una perfusión in situ, por ejemplo con el hígado o el riñón. Existen muchos métodos publicados para aislar las células/núcleos. Entre ellos figuran el picado de tejidos tales como el hígado y el riñón, el raspado de mucosas en el caso del tubo gastrointestinal, la homogeneización y la digestión enzimática. El estudio de validación del JaCVAM solo estudiaba células aisladas y, por lo tanto, en términos de establecer el método y poder referirse a los datos del estudio del JaCVAM a efectos de demostración de la competencia, es preferible utilizar células aisladas. Sin embargo, se ha puesto de manifiesto que no existía ninguna diferencia esencial en el resultado del ensayo entre utilizar células aisladas o núcleos aislados (8). También se obtuvieron resultados comparables al utilizar métodos diferentes para aislar las células/núcleos (por ejemplo, homogeneización, picado, digestión enzimática y filtración por malla) (55). Por consiguiente, pueden utilizarse tanto células aisladas como núcleos aislados. Los laboratorios deben evaluar a fondo y validar métodos de aislamiento de núcleos/células, específicos de los distintos tejidos. Como se comenta en el punto 40, la persistencia de algunas de las lesiones que dan lugar a la rotura de las cadenas de ADN detectadas por el ensayo del cometa puede ser muy breve (52) (53). Por lo tanto, cualquiera que sea el método utilizado para preparar las suspensiones de células/núcleos aislados, es importante que los tejidos se procesen tan pronto como sea posible una vez que los animales se hayan sacrificado y se pongan en condiciones que reduzcan la eliminación de las lesiones (por ejemplo, manteniendo el tejido a baja temperatura). Las suspensiones celulares deben mantenerse enfriadas con hielo hasta que estén listas para su uso, de modo que puedan demostrarse la mínima variación entre muestras y unas respuestas adecuadas de los testigos positivos y negativos. PREPARACIÓN DE LOS PORTAOBJETOS La preparación de los portaobjetos debe hacerse tan pronto como sea posible (idealmente en el plazo de una hora) después de la preparación de las células/núcleos aislados, pero la temperatura y el tiempo transcurrido entre la muerte de los animales y la preparación de los portaobjetos deben estar estrictamente controlados y validados con arreglo a las condiciones del laboratorio. El volumen de suspensión celular añadido a agarosa de bajo punto de fusión (generalmente de 0,5 a 1,0 %) para preparar los portaobjetos no debe reducir el porcentaje de la agarosa de bajo punto de fusión a menos del 0,45 %. La densidad celular óptima se determinará mediante el sistema de análisis de imágenes utilizado para examinar los cometas. Lisis Las condiciones de la lisis también son una variable crítica y pueden interferir con las roturas de cadena debidas a tipos específicos de modificaciones del ADN (ciertas alquilaciones del ADN y adiciones de bases). Se recomienda, por lo tanto, que las condiciones de la lisis se mantengan lo más constantes posible para todos los portaobjetos de un experimento. Una vez preparados, los portaobjetos se sumergen en solución de lisis refrigerada durante al menos una hora (o durante una noche) en torno a 2-8 °C, bajo una iluminación tenue, por ejemplo de luz amarilla (o protegidos de la luz) para evitar su exposición a la luz blanca que puede contener componentes UV. Tras este período de incubación, los portaobjetos deben lavarse para eliminar los restos de detergente y sales antes de la fase de desenrollamiento alcalino. Esto puede hacerse utilizando agua purificada o solución amortiguadora de neutralización o de fosfato. También puede utilizarse la solución amortiguadora de electroforesis. Esto mantendría las condiciones alcalinas en la cámara de electroforesis. Desenrollamiento y electroforesis Los portaobjetos se colocan de forma aleatoria en la plataforma de una unidad de electroforesis de tipo submarino con una cantidad suficiente de solución de electroforesis, de modo que la superficie de los portaobjetos esté totalmente recubierta (la profundidad de la cobertura debe también ser coherente de una tanda a otra). En otro tipo de unidades de electroforesis para el ensayo del cometa, es decir, con refrigeración activa, circulación y alimentación eléctrica de elevada capacidad, una cobertura más profunda con la solución dará lugar a mayor intensidad de corriente eléctrica a tensión constante. Debe utilizarse un diseño equilibrado para colocar los portaobjetos en el depósito de electroforesis de forma que se mitiguen los efectos de eventuales tendencias o efectos de borde dentro del depósito y se minimice la variabilidad entre lotes, es decir, en cada tanda de electroforesis debe contarse con el mismo número de portaobjetos de cada animal utilizado en el estudio y deben incluirse muestras de los diferentes grupos tratados y de los testigos negativos y positivos. Los portaobjetos se dejan durante al menos 20 minutos para que se desenrolle el ADN y después se someten a la electroforesis en condiciones controladas que maximicen la sensibilidad y el intervalo analítico del ensayo (es decir, que lleven a unos niveles aceptables de porcentaje de ADN de la cola de los testigos positivos y negativos tales que maximicen la sensibilidad). El grado de migración del ADN está asociado linealmente con la duración de la electroforesis, y también con el potencial (V/cm). Sobre la base del ensayo del JaCVAM, estos valores podrían ser 0,7 V/cm durante al menos 20 minutos. La duración de la electroforesis se considera una variable crítica y debe establecerse el tiempo de electroforesis para optimizar el intervalo analítico. Unos tiempos de electroforesis más largos (por ejemplo, de 30 o 40 minutos para maximizar la sensibilidad) suelen dar lugar a respuestas positivas más fuertes con mutágenos conocidos. Sin embargo, unos tiempos de electroforesis más largos también pueden dar lugar a una migración excesiva en las muestras de los testigos. En cada experimento se debe mantener constante la tensión, y la variabilidad de los demás parámetros debe mantenerse dentro de un intervalo estrecho y especificado, por ejemplo en el ensayo del JaCVAM 0,7 V/cm daban una intensidad de corriente inicial de 300 mA. La profundidad de la solución amortiguadora debe ajustarse para permitir las condiciones exigidas y mantenerse a lo largo de todo el experimento. Debe registrarse la intensidad de la corriente al inicio y al final del período de electroforesis. Por tanto, las condiciones óptimas deben determinarse durante la demostración inicial de la competencia en el laboratorio correspondiente con cada tejido estudiado. La temperatura de la solución de electroforesis a lo largo del proceso de desenrollado y electroforesis debe mantenerse baja, generalmente entre 2 y 10 °C (10). Debe registrarse la temperatura de la solución de electroforesis al principio del desenrollado, al inicio de la electroforesis y al final de la electroforesis. Tras la terminación de la electroforesis, los portaobjetos deben sumergirse en la solución amortiguadora de neutralización (o lavarse con ella) durante al menos 5 minutos. Los geles pueden teñirse y examinarse «en fresco» (por ejemplo, en el plazo de 1 o 2 días) o pueden deshidratarse para su posterior examen (por ejemplo, en el plazo de 1 a 2 semanas después de la tinción) (56). Sin embargo, las condiciones deben validarse durante la demostración de la competencia y deben obtenerse y conservarse datos históricos por separado de cada una de estas condiciones. En caso de que se seleccione la segunda de tales posibilidades, los portaobjetos se deshidratan sumergiéndolos en etanol absoluto durante un mínimo de 5 minutos; después se dejan secar al aire y luego se guardan a temperatura ambiente o en un recipiente en el frigorífico hasta el momento del examen. Métodos de medición Los cometas deben examinarse cuantitativamente utilizando un sistema de análisis de imágenes automatizado o semiautomatizado. Los portaobjetos se tiñen con un colorante fluorescente apropiado como, por ejemplo, SYBR Gold, Green I, yoduro de propidio o bromuro de etidio, y se miden a un aumento adecuado (por ejemplo, 200x) en un microscopio de epifluorescencia provisto de detectores apropiados o de una cámara digital (por ejemplo, DAC). Las células pueden clasificarse en tres categorías, según se describe en el Atlas de imágenes de cometas (57), a saber: evaluables, no evaluables y «erizo» (véase en el punto 56 un análisis más detallado). Para evitar artefactos, el porcentaje de ADN de la cola debe evaluarse solo en las células evaluables (cabeza y cola claramente definidas, sin ninguna interferencia con las células adyacentes). No es necesario informar de la frecuencia de las células no evaluables. La frecuencia de los erizos debe determinarse sobre la base del examen visual (puesto que la ausencia de una cabeza claramente definida significa que no son detectados fácilmente por análisis de imágenes) de al menos 150 células por muestra (véase en el punto 56 una discusión más profunda) y documentarse por separado. Todos los portaobjetos que se hayan de analizar, incluidos los de los testigos positivos y negativos, deben codificarse independientemente y deben examinarse de forma aleatorizada, tal que el examinador no tenga conocimiento de cómo se han tratado. De cada muestra (por tejido y por animal) deben analizarse al menos 150 células (excepto los erizos, véase el punto 56). El examen de 150 células por animal, en al menos 5 animales por dosis (salvo en el testigo positivo en paralelo, véase el punto 29), supone una potencia estadística adecuada según el análisis de Smith et al., 2008 (5). Si se utilizan portaobjetos, esto podría hacerse con el examen de 2 o 3 portaobjetos por muestra cuando se utilizan cinco animales por grupo. Deben observarse varias zonas del portaobjeto con una densidad que garantice que no hay solapamiento de colas. Debe evitarse la evaluación cerca de los bordes de los portaobjetos. Las roturas de las cadenas de ADN en el ensayo del cometa se pueden medir con criterios de valoración independientes, tales como el porcentaje de ADN de la cola, y la longitud y el momento de esta. Pueden hacerse las tres mediciones si se utiliza un programa informático apropiado de análisis de imágenes. No obstante, el porcentaje de ADN de la cola (también conocido como porcentaje de intensidad de la cola) se recomienda para la evaluación e interpretación de los resultados (12) (40) (41) (42), y se determina mediante la intensidad del fragmento de ADN de la cola, expresada como porcentaje de la intensidad total de la célula (13). Lesiones en los tejidos y citotoxicidad Los resultados positivos en el ensayo del cometa pueden no deberse únicamente a la genotoxicidad, sino que la toxicidad para el tejido diana también puede dar lugar a un aumento de la migración del ADN (12) (41). Por el contrario, a menudo se observa baja o moderada citotoxicidad con genotoxinas conocidas (12), lo que indica que no es posible distinguir la migración de ADN inducida por la genotoxicidad frente a la inducida por la citotoxicidad en el ensayo del cometa solo. No obstante, en caso de que se observen aumentos en la migración de ADN, se recomienda efectuar el examen de uno o más indicadores de citotoxicidad, como ayuda en la interpretación de los resultados. Los aumentos en la migración de ADN en presencia de pruebas claras de citotoxicidad deben interpretarse con cautela. Se han propuesto muchas mediciones de la citotoxicidad y de estos cambios histopatológicos, que se consideran una medición pertinente de la toxicidad para los tejidos. Ciertas observaciones, tales como la inflamación, la infiltración celular, o cambios apoptóticos o necróticos, se han asociado con aumentos en la migración del ADN; sin embargo, como se demostró en el estudio de validación del JaCVAM (12), no se dispone de ninguna lista definitiva de cambios histopatológicos que estén siempre asociados a un aumento de la migración del ADN. Los cambios en ciertos parámetros de química clínica (por ejemplo, AST, ALT) también pueden proporcionar información útil sobre las lesiones tisulares y pueden tenerse en cuenta asimismo otros indicadores, tales como la activación de la caspasa, la tinción de TUNEL, la tinción de la anexina V, etc. Sin embargo, hay pocos datos publicados sobre estudios in vivo en los que se hayan utilizado tales indicadores y unos pueden ser menos fiables que otros. Los erizos (o nubes, o células fantasma) son células que presentan una imagen al microscopio compuesta por una cabeza pequeña o inexistente y una cola grande y difusa, y se consideran células muy dañadas, aunque su etiología es incierta (véase el apéndice 3). Debido a su aspecto, son poco fiables las mediciones del porcentaje de ADN de la cola por análisis de imágenes, por lo que los erizos deben evaluarse por separado. La presencia de erizos debe registrarse y comunicarse, y se debe investigar e interpretar con cautela cualquier incremento relevante cuyo origen pueda considerarse que es el producto problema. El conocimiento del posible modo de acción de los productos problema puede ayudar a dichas consideraciones. DATOS E INFORME Tratamiento de los resultados La unidad experimental es el animal y, por lo tanto, se deben presentar en forma tabular tanto los datos relativos a cada animal como un resumen de los resultados. Debido a la naturaleza jerárquica de los datos, se recomienda que se determine la mediana del porcentaje de ADN de la cola para cada portaobjetos y que se calcule la media de los valores de la mediana para cada animal (12). A continuación se determina la media de las medias de cada animal, para dar la media del grupo. Todos estos valores deben incluirse en el informe. Pueden aplicarse planteamientos alternativos (véase el punto 53) cuando estén justificados desde el punto de vista científico y estadístico. El análisis estadístico se puede realizar con una variedad de planteamientos (58) (59) (60) (61). A la hora de decidir qué métodos estadísticos utilizar, debe tenerse en cuenta la necesidad de transformar (por ejemplo, logaritmo o raíz cuadrada) los datos o de añadir un pequeño número (por ejemplo, 0,001) a todos los valores (incluso a los distintos de cero) para mitigar los efectos de los valores celulares iguales a cero, como se indica en las referencias anteriormente mencionadas. En el apéndice 2 se encuentran datos del análisis de las interacciones tratamiento/sexo cuando se utilizan animales de ambos sexos, y del posterior análisis de los datos cuando se encuentran diferencias o no. Deben también consignarse los datos sobre toxicidad y signos clínicos. Criterios de aceptabilidad La aceptabilidad de un ensayo se basa en los criterios siguientes:
Evaluación e interpretación de los resultados Siempre que se cumplan todos los criterios de aceptabilidad, se considera que el producto problema es claramente positivo si:
Cuando se cumplen todos estos criterios, el producto problema se considera capaz de inducir roturas de las cadenas de ADN en los tejidos estudiados en este sistema de ensayo. Si se cumplen solo uno o dos de estos criterios, véase el punto 62. Siempre que se cumplan todos los criterios de aceptabilidad, se considera que el producto problema es claramente negativo si:
El producto problema se considera entonces incapaz de inducir roturas de las cadenas de ADN en los tejidos estudiados en este sistema de ensayo. No se requiere ninguna verificación de una respuesta claramente positiva o negativa. En caso de que la respuesta no sea ni claramente positiva ni claramente negativa (es decir, si no se cumplen todos los criterios incluidos en los puntos 59 o 60) y a fin de ayudar a determinar la importancia biológica de un resultado, los datos deben ser evaluados por expertos o mediante más investigaciones, cuando esté justificado científicamente. Puede ser útil examinar células adicionales (en su caso) o realizar una repetición del experimento quizá optimizando las condiciones experimentales, como, por ejemplo, la separación entre dosis, las vías de administración, el momento de la toma de muestras o los tejidos empleados. En casos raros, incluso después de hacer más investigaciones, el conjunto de datos no permite que se extraiga una conclusión de resultado positivo o negativo, por lo que se considerará dudoso. Para evaluar la importancia biológica de un resultado positivo o dudoso, hay que disponer de información sobre la citotoxicidad en el tejido diana (véanse los puntos 54-55). Cuando se observan resultados positivos o dudosos únicamente en presencia de claros signos de citotoxicidad, se concluirá que el estudio es dudoso en cuanto a la genotoxicidad, a menos que exista suficiente información que apoye una conclusión definitiva. En los casos de resultado negativo de un estudio en que haya signos de toxicidad a todas las dosis sometidas a ensayo, puede ser aconsejable proceder a nuevos estudios a dosis no tóxicas. Informe del ensayo El informe del ensayo debe incluir la información siguiente:
BIBLIOGRAFÍA
Apéndice 1 DEFINICIONES Electroforesis en gel de células aisladas en condiciones alcalinas : Técnica sensible para la detección de lesiones primarias del ADN a nivel de células/núcleos individuales. Producto : Sustancia o mezcla. Cometa : Forma que adoptan los nucleoides después de someterse a un campo electroforético, debido a su similitud con los cometas: la cabeza es el núcleo y la cola está constituida por el ADN que migra afuera del núcleo en el campo eléctrico. Variable/parámetro crítico : Variable del protocolo cuyos pequeños cambios pueden tener una gran incidencia en la conclusión del ensayo. Las variables críticas pueden ser específicas de cada tejido. Las variables críticas no deben alterarse, especialmente dentro de un ensayo, sin tener en cuenta cómo la modificación va a alterar una respuesta del ensayo, por ejemplo según indiquen la magnitud y la variabilidad en los testigos positivos y negativos. El informe del ensayo debe enumerar las alteraciones de variables críticas aportadas durante el ensayo o en comparación con el protocolo de referencia en el laboratorio y proporcionar una justificación de cada alteración. Intensidad de la cola o porcentaje de ADN de la cola : Esto corresponde a la intensidad de la cola del cometa con respecto a la intensidad total (cabeza y cola). Refleja la cantidad de roturas del ADN, y se expresa como porcentaje. Producto problema : Sustancia o mezcla estudiada con este método de ensayo. UVCB : Sustancias de composición desconocida o variable, productos complejos de reacción o materiales biológicos. Apéndice 2 DISEÑO FACTORIAL PARA IDENTIFICAR LAS DIFERENCIAS DE SEXO EN EL ENSAYO DEL COMETA IN VIVO Diseño factorial y su análisis En este diseño, un mínimo de 5 machos y 5 hembras se someten a ensayo a cada concentración, lo que resulta en la utilización de un mínimo de 40 animales (20 machos y 20 hembras, más los testigos positivos pertinentes). Este diseño, que es uno de los diseños factoriales más simples, es equivalente a un análisis de varianza de dos factores, con el sexo y el nivel de concentración como efectos principales. Los datos pueden analizarse con numerosos paquetes de software estadístico estándar tales como SPSS, SAS, STATA, Genstat, así como utilizando R. El análisis divide la variabilidad de la serie de datos en variabilidad entre los sexos, variabilidad entre las concentraciones y variabilidad relativa a la interacción entre los sexos y las concentraciones. Cada uno de los términos se somete a prueba frente a una estimación de la variabilidad entre los animales replicados dentro de los grupos de animales del mismo sexo que han recibido la misma concentración. Todos los detalles de la metodología subyacente se encuentran en muchos manuales estadísticos conocidos (véanse las referencias) y en las funciones de «ayuda» de los paquetes estadísticos. El análisis se lleva a cabo inspeccionando el término de la interacción sexo x concentración en el cuadro de ANOVA (35). A falta de un término de interacción significativo, los valores combinados de distintos sexos o niveles de concentración ofrecen pruebas estadísticas válidas entre los niveles, sobre la base del término de ANOVA de variabilidad intragrupo puesta en común. El análisis continúa dividiendo la estimación de la variabilidad entre concentraciones en contrastes que permiten una prueba de contrastes lineales y cuadráticos de las respuestas a través de los niveles de concentración. Cuando existe un término de interacción significativa sexo x concentración, este término también puede dividirse en contrastes de interacción lineal x sexo y cuadrático x sexo. Estos términos permiten pruebas de si las respuestas a la concentración son paralelas en los dos sexos, o si existe una diferencia en la respuesta entre estos. La estimación de la variabilidad intragrupo puesta en común se puede utilizar para proporcionar pruebas pareadas de la diferencia entre medias. Estas comparaciones podrían realizarse entre las medias para los dos sexos y entre las medias para los diferentes niveles de concentración, así como para las comparaciones con los niveles de los testigos negativos. En los casos en los que existe una interacción significativa pueden hacerse comparaciones entre las medias de concentraciones diferentes dentro de un sexo o entre las medias de los sexos a la misma concentración. Bibliografía Hay muchos manuales estadísticos que debaten la teoría, el diseño, la metodología, el análisis y la interpretación de los diseños factoriales que van desde los análisis bifactoriales más sencillos hasta las formas más complejas utilizadas en la metodología de diseño de experimentos. La lista que sigue no es exhaustiva. Algunos libros proporcionan ejemplos resueltos de diseños comparables, en algunos casos con el código para aplicar los análisis utilizando diversos paquetes de software.
Apéndice 3 LIMITACIONES ACTUALES DEL ENSAYO Debido al estado actual de los conocimientos, el ensayo del cometa in vivo tiene asociadas varias limitaciones. Se espera que estas limitaciones se reduzcan o definan de manera más estricta según se vaya ganando más experiencia con la aplicación del ensayo para responder a cuestiones de seguridad en un contexto reglamentario.
Bibliografía
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||






|
16) |
En la parte B, el capítulo C.13 se sustituye por el texto siguiente: «C.13. Bioacumulación en peces: exposición acuática y alimentaria INTRODUCCIÓN El presente método de ensayo es equivalente a las directrices de ensayo (TG) de la OCDE 305 (2012). El objetivo principal de la presente revisión del método de ensayo es doble. En primer lugar, se propone incorporar un ensayo de bioacumulación alimentaria (36) apropiado para determinar el potencial de bioacumulación de sustancias con hidrosolubilidad muy baja. En segundo lugar, se pretende crear un método de ensayo que, cuando proceda, utilice menos peces por motivos de bienestar de los animales, y que sea económicamente más rentable. En los años transcurridos desde la aprobación del método de ensayo consolidado C.13 (1), se han sometido a ensayo numerosas sustancias, y han acumulado considerable experiencia tanto los laboratorios como las autoridades normativas. Esto ha llevado a la convicción de que la complejidad del ensayo puede reducirse si se cumplen unos criterios específicos (véase el punto 88), y de que es posible un enfoque escalonado. La experiencia también ha demostrado que ciertos factores biológicos, como el crecimiento y el contenido lipídico de los peces, pueden tener un fuerte impacto sobre los resultados y puede ser necesario tenerlos en cuenta. Además, se ha reconocido que puede no ser técnicamente viable el ensayo de sustancias muy poco hidrosolubles. Además, en el caso de las sustancias muy poco hidrosolubles en el medio acuático, la exposición a través del agua puede ser de importancia limitada en comparación con la vía alimentaria. Esto ha dado lugar a la elaboración de un método de ensayo en el que los peces se exponen a través de su alimentación (véanse los puntos 7-14 y a partir del 97). En 2010 se procedió a una validación (prueba interlaboratorios) de la exposición alimentaria (51). Los principales cambios son los siguientes:
Antes de llevar a cabo ninguno de los ensayos de bioacumulación, debe conocerse la siguiente información acerca de la sustancia problema:
Independientemente del método de exposición o de la pauta de muestreo que se hayan elegido, el presente método de ensayo describe un procedimiento para caracterizar el potencial de bioacumulación de las sustancias en los peces. Aunque son muy preferibles los regímenes de ensayos dinámicos, los regímenes semiestáticos son aceptables a condición de que se cumplan los criterios de validez (véanse los puntos 24 y 113). En la vía de exposición alimentaria, el sistema dinámico no es necesario para mantener las concentraciones acuosas de la sustancia problema, pero contribuye a mantener las concentraciones adecuadas de oxígeno disuelto y a mantener el agua limpia y eliminar las influencias de, por ejemplo, los productos de excreción. Independientemente del método de ensayo elegido, en el presente método de ensayo se da suficiente información para realizarlo, dejando bastante libertad para adaptar el diseño experimental a las condiciones específicas de cada laboratorio y para variar las características de las sustancias problema. El ensayo de exposición acuática se aplica de forma más adecuada a las sustancias orgánicas estables con valor de log K OW entre 1,5 y 6,0 (13) pero también es aplicable a las sustancias muy hidrófobas (con log K OW > 6,0), si puede demostrarse una concentración estable y totalmente disuelta de la sustancia problema en agua. Si no puede demostrarse una concentración estable de la sustancia problema en agua, no será adecuado el ensayo acuático; por tanto, será necesario seguir el enfoque alimentario para el ensayo de la sustancia en los peces (aunque la interpretación y el uso de los resultados del ensayo alimentario pueden depender del marco reglamentario). Mediante la ecuación de Bintein et al. (14) se pueden obtener valores pre-estimados del factor de bioconcentración (FBC, a veces expresado como K B) de sustancias orgánicas con valores de log K OW de hasta alrededor de 9,0. El valor pre-estimado del factor de bioconcentración de tales sustancias muy hidrófobas puede ser mayor que el valor del factor de bioconcentración en estado de equilibrio (FBCSS) que se puede esperar en un experimento de laboratorio, especialmente cuando se utiliza un modelo lineal simple para el valor pre-estimado. Entre los parámetros que caracterizan el potencial de bioacumulación se encuentran la constante de la velocidad de absorción (k 1), las constantes de la velocidad de pérdida que incluyen la constante de la velocidad de depuración (k 2), el factor de bioconcentración en el estado de equilibrio (FBCSS), el factor de bioconcentración cinético (FBCK) y el factor de biomagnificación alimentario (FBM) (38). Puede facilitarse el análisis de las muestras de agua, alimentos y peces con ayuda de sustancias problema radiomarcadas, que pueden también servir para determinar si se deben identificar y cuantificar los metabolitos. Si solo se miden los residuos radiactivos totales (por ejemplo, por combustión o solubilización de los tejidos), el FBC o FBM se basa en el total de la sustancia original, en los eventuales metabolitos retenidos y también en el carbono asimilado. Así pues, los valores del FBC o FBM basados en los residuos radiactivos totales no pueden ser directamente comparables a los valores de FBC o FBM derivados de un análisis químico específico solo de la sustancia original. Pueden aplicarse procedimientos de separación, como la TLC, HPLC o GC (39), antes del análisis en estudios radiomarcados para determinar el FBC o el FBM basados en la sustancia original. Cuando se aplican tales técnicas de separación, debe efectuarse la identificación y cuantificación de la sustancia original y de los metabolitos pertinentes (40) (véase el punto 65) si se quiere que el FBC o FBM se base en la concentración de la sustancia original en los peces y no en la del total de residuos radiomarcados. También se puede combinar un estudio del metabolismo de los peces o de la distribución in vivo con un estudio de bioacumulación por análisis e identificación de los residuos en los tejidos. La posibilidad del metabolismo se puede predecir mediante instrumentos adecuados [por ejemplo, herramientas sobre la relación estructura-actividad de la OCDE (OECD QSAR toolbox) (15) y programas de QSAR protegidos]. La decisión de si llevar a cabo un ensayo de exposición acuática o alimentaria, y en qué configuración debe basarse en los factores del punto 3, considerados junto con el marco normativo pertinente. Por ejemplo, en el caso de las sustancias que tienen un valor elevado de log K OW pero presentan hidrosolubilidad apreciable con respecto a la sensibilidad de las técnicas analíticas disponibles, debe considerarse en primera instancia un ensayo de exposición acuática. No obstante, es posible que la información sobre la hidrosolubilidad no sea definitiva para estos tipos de sustancias hidrófobas, por lo que, antes de tomar una decisión sobre qué método de ensayo utilizar, debe investigarse la posibilidad de preparar en agua concentraciones disueltas, estables y mensurables (no se permiten emulsiones estables) aplicables a un estudio de exposición acuática (16). No es posible dar orientaciones normativas exactas en relación con el método que debe usarse, solo sobre la base de criterios de «corte» como la hidrosolubilidad y el coeficiente de reparto octanol/agua, ya que otros factores (técnicas analíticas, degradación, adsorción, etc.) pueden tener una influencia marcada en la aplicabilidad del método por las razones anteriormente expuestas. No obstante, un log K OW superior a 5 y una hidrosolubilidad por debajo de ~ 0,01-0,1 mg/l marcan la gama de sustancias en las que los ensayos por exposición acuática pueden ser cada vez más difíciles. Deben considerarse los demás factores que puedan influir en la elección del ensayo, incluido el potencial de adsorción de la sustancia a los recipientes y aparatos del ensayo, su estabilidad en solución acuosa frente a su estabilidad en los alimentos de los peces (17) (18), etc. Puede encontrarse información sobre estos aspectos prácticos en otros estudios completados en agua. En la bibliografía se dispone de más información sobre la evaluación de los aspectos vinculados al comportamiento de los estudios de bioacumulación (véase, p. ej., (19)). Respecto a aquellas sustancias con las cuales la solubilidad o el mantenimiento de la concentración en el agua, así como el análisis de estas concentraciones, no plantean ninguna restricción a la utilización de un método de exposición acuática, es preferible utilizar este método para determinar el potencial de bioconcentración de la sustancia. En cualquier caso, debe verificarse que la concentración o concentraciones para exposición acuática que deben aplicarse están dentro del margen de solubilidad en el medio de ensayo. Pueden utilizarse diferentes métodos para mantener estable la concentración de la sustancia problema disuelta, como el uso de soluciones madre o de sistemas de administración pasiva (por ejemplo, método de elución en columna), siempre que pueda demostrarse que las concentraciones pueden mantenerse estables y que los medios de ensayo no se alteran respecto a los recomendados en el punto 27. Con sustancias muy hidrófobas (log K OW > 5 y solubilidad inferior a ~ 0,01-0,1 mg/l), los ensayos por exposición acuática pueden ser cada vez más difíciles. Entre los motivos de las restricciones están el que la concentración en el que no pueda mantenerse a un nivel que se considere suficientemente constante (por ejemplo, debido a la sorción al vidrio de los recipientes de exposición o una rápida absorción por los peces) o el que las concentraciones acuosas que se deban aplicar sean tan bajas que se sitúen en el mismo intervalo que el límite analítico de cuantificación o por debajo de él (41). Con estas sustancias muy hidrófobas se recomienda el ensayo alimentario, siempre que el ensayo sea coherente con el marco reglamentario pertinente y las necesidades de evaluación de riesgos. En el caso de los agentes tensioactivos, se debe considerar si es factible el ensayo de bioconcentración acuosa, habida cuenta de las propiedades de la sustancia; en caso contrario, el estudio alimentario es probablemente más adecuado. Los agentes tensioactivos actúan sobre la tensión superficial y reducen la tensión interfacial entre dos líquidos. Su naturaleza anfifílica (es decir, contienen una parte hidrófila y una parte hidrófoba) hace que se acumulen en los interfaces como, por ejemplo, la interfaz agua-aire, la interfaz agua-alimento, y las paredes de vidrio, lo que dificulta la determinación de su concentración en el agua. El ensayo alimentario permite superar algunos de los aspectos de la exposición en caso de mezclas complejas con componentes de diferentes límites de hidrosolubilidad, ya que es más probable conseguir una exposición comparable a todos los componentes de la mezcla que con el método acuático (véase (20)). Debe señalarse que el planteamiento alimentario arroja un factor de biomagnificación alimentario (FBM) en lugar de un factor de bioconcentración (FBC) (42). Se dispone de enfoques para calcular un factor de bioconcentración cinético (FBCK) a partir de datos obtenidos en el estudio alimentario (como se indica en el apéndice 8), pero estos enfoques se deben utilizar con prudencia. En general, asumen una cinética de primer orden, y son aplicables solo a determinados grupos de compuestos. Es poco probable que este tipo de enfoques pueda aplicarse en el caso de los agentes tensioactivos (véase el punto 12). Una configuración minimizada de ensayo de exposición acuática con menos puntos de muestreo para reducir el número de animales o recursos (véase a partir del punto 83) solo debe aplicarse a aquellas sustancias con las que haya motivos para esperar que la absorción y la depuración sigan una cinética aproximadamente de primer orden (es decir, en general, sustancias orgánicas no ionizadas, véase el punto 88). C.13 — I: Ensayo de bioconcentración en peces con exposición acuática PRINCIPIO DEL ENSAYO El ensayo se divide en dos fases: la de exposición (absorción) y la posterior a la exposición (depuración). Durante la fase de absorción, un grupo de peces de una sola especie queda expuesto a la sustancia problema a una o varias concentraciones elegidas, en función de las propiedades de la sustancia problema (véase el punto 49). Luego se transfieren a un medio libre de esta sustancia, para la fase de depuración. Esta última fase es necesaria siempre, excepto cuando la absorción de la sustancia durante la fase de absorción ha sido insignificante. La concentración de la sustancia problema en los peces (o en tejidos específicos) se mide durante las dos fases del ensayo. Además del grupo expuesto a la sustancia problema, se mantiene un grupo testigo de peces en condiciones idénticas, salvo que no se expone a la sustancia problema, para relacionar los posibles efectos nocivos observados en el ensayo de bioconcentración con un grupo testigo correspondiente, y deducir las concentraciones de fondo de la sustancia problema (43). En el ensayo de exposición acuática, la fase de absorción tiene generalmente una duración de 28 días. La duración puede aumentarse en caso necesario (véase el punto 18), o reducirse si se demuestra que se ha alcanzado antes el estado de equilibrio (véase el apéndice 1, Definiciones y unidades). Puede preverse la duración de la fase de absorción y el tiempo necesario para la obtención del estado de equilibrio gracias a las ecuaciones del apéndice 5. El periodo de depuración comienza entonces, cuando los peces dejan de exponerse a la sustancia problema, con la transferencia de los peces a un recipiente limpio con el mismo medio, pero sin la sustancia problema. Cuando sea posible, se calcula el factor de bioconcentración preferentemente como proporción de la concentración en los peces (C f) y en el agua (C w) en el estado de equilibrio (FBCSS; véase la definición en el apéndice 1) y como factor de bioconcentración cinético (FBCK; véanse las definiciones y unidades del apéndice 1), que se calcula como la proporción de las constantes de velocidad de absorción (k 1) y depuración (k 2) suponiendo cinética de primer orden (44). Si no se llega al estado de equilibrio en 28 días, se calcula el FBC utilizando el enfoque cinético (véase el punto 38) o se puede ampliar la fase de absorción. En caso de que para alcanzar el estado de equilibrio sea necesaria una fase de absorción de longitud excesiva en la práctica (véanse los puntos 37 y 38, apéndice 5), será preferible el enfoque cinético. Como alternativa, en el caso de las sustancias muy hidrófobas debe considerarse la realización de un estudio alimentario (45), a condición de que el ensayo alimentario sea compatible con el marco normativo pertinente. La constante de la velocidad de absorción, la constante de la velocidad de depuración (pérdida) (o las constantes, cuando entren en juego modelos más complejos), el factor de bioconcentración (en el estado de equilibrio y/o cinético) y, si es posible, los intervalos de confianza de cada uno de estos parámetros, se calculan a partir del modelo que describa mejor las concentraciones medidas de la sustancia problema en los peces y en el agua (véase el apéndice 5). El aumento de la masa de los peces durante el ensayo se traducirá en una disminución de la concentración de la sustancia problema en los peces en crecimiento (la denominada dilución debida al crecimiento) y, por tanto, el FBC cinético estará subestimado si no se corrige para tener en cuenta este fenómeno (véanse los puntos 72 y 73). El FBC se basará en la concentración total en los peces (es decir, respecto al peso húmedo total de los peces). Sin embargo, para algunos estudios pueden utilizarse tejidos u órganos específicos (por ejemplo, músculos, hígado) si los peces son suficientemente grandes o si es posible separar las partes comestibles (filetes) y no comestibles (vísceras). Como hay una clara relación entre el potencial de bioconcentración y la hidrofobicidad de numerosas sustancias orgánicas, también hay una relación correspondiente entre el contenido lipídico de los peces del ensayo y las bioconcentraciones observadas de tales sustancias. Así pues, para reducir esta fuente de variabilidad en los resultados de los ensayos relativos a las sustancias con fuerte lipofilia (es decir, con log K OW > 3), la bioconcentración debe expresarse normalizada con referencia a un contenido lipídico de los peces del 5 % (sobre la base del peso corporal húmedo entero) además de la obtenida directamente del estudio. Esto es necesario para proporcionar una base sobre la cual puedan compararse entre sí los resultados de las distintas sustancias o con diferentes especies de ensayo. La cifra de 5 % de contenido lipídico es de amplia utilización, ya que se trata de la media del contenido lipídico de los peces utilizados comúnmente en el presente método de ensayo (21). INFORMACIÓN SOBRE LA SUSTANCIA PROBLEMA Además de las propiedades de la sustancia problema que figuran en la introducción (punto 3), será necesario también conocer su toxicidad para la especie de peces utilizada en el ensayo, preferiblemente la LC50 asintótica (es decir, independiente del tiempo) o la toxicidad estimada a partir de ensayos de toxicidad a largo plazo en peces (por ejemplo, métodos C.47 (22), C.15 (23) y C.14 (24)). Debe disponerse de un método analítico adecuado, con una exactitud, una precisión y una sensibilidad conocidas para la cuantificación de la sustancia en las soluciones de ensayo y en el material biológico, así como de datos precisos sobre la preparación y la conservación de las muestras. Es necesario también conocer el límite de cuantificación analítica de la sustancia problema tanto en el agua como en los tejidos de los peces. Si se utiliza una sustancia problema radiomarcada, deberá ser del nivel más alto de pureza (por ejemplo, preferiblemente > 98 %) y deberá conocerse el porcentaje de radiactividad asociada con las impurezas. VALIDEZ DEL ENSAYO Para que el ensayo sea válido han de darse las condiciones siguientes:
SUSTANCIAS DE REFERENCIA El uso de sustancias de referencia de metabolismo bajo y de potencial de bioconcentración conocido podrá ayudar al control del procedimiento experimental, en caso necesario (por ejemplo, cuando un laboratorio no tenga experiencia previa con el ensayo o se hayan modificado las condiciones experimentales). DESCRIPCIÓN DEL MÉTODO Equipo Se han de evitar los materiales que puedan presentar fenómenos de disolución, sorción o lixiviación o tener algún efecto adverso sobre los peces, y esto en todas las partes del equipo. Pueden utilizarse recipientes normales rectangulares o cilíndricos, hechos de material químicamente inerte y de una capacidad adaptada a la tasa de carga (véase el punto 43). Se debe reducir al mínimo el uso de tubos de plástico blando. Deben utilizarse tubos de politetrafluoroetileno, acero inoxidable o vidrio. La experiencia pone de manifiesto que pudiera ser necesario el vidrio silanizado para las sustancias que presentan un fuerte coeficiente de adsorción, como los piretroides sintéticos por ejemplo. Será necesario, en tales casos, deshacerse de los equipos una vez usados. Es preferible exponer los sistemas de ensayo a las concentraciones de la sustancia problema que se vayan a utilizar en el estudio, durante el tiempo que se requiera para demostrar el mantenimiento de unas concentraciones de exposición estables, antes de introducir los organismos de ensayo. Agua Se utiliza generalmente para el ensayo agua natural procedente de una fuente no contaminada y de calidad uniforme. Sin embargo, puede ser más adecuado utilizar agua reconstituida (es decir, agua desmineralizada con nutrientes específicos añadidos en cantidades conocidas) para garantizar la uniformidad de la calidad a lo largo del tiempo. El agua de dilución, que es el agua que se mezcla con la sustancia problema antes de introducirla en el recipiente de ensayo (véase el punto 30), debe ser tal que permita la supervivencia de la especie de peces elegida, durante los periodos de aclimatación y de ensayo, sin que aparezcan ningún comportamiento ni aspecto anormales. Idealmente, se debería demostrar que las especies sometidas al ensayo pueden sobrevivir, crecer y reproducirse en el agua de dilución (por ejemplo, mediante cría en laboratorio o un estudio de toxicidad sobre el ciclo biológico). El agua de dilución se caracterizará al menos mediante su pH, dureza, materia sólida total, carbono orgánico total (COT (47)) pero también, preferentemente, mediante su contenido en amoniaco y nitritos y su alcalinidad, así como, en el caso de las especies marinas, su salinidad. No se conocen perfectamente los parámetros importantes para el bienestar óptimo de los peces, pero el apéndice 2 indica las concentraciones máximas recomendadas de una serie de parámetros relativos al agua de ensayo, tanto dulce como salada. A lo largo de toda la duración de un ensayo, el agua de dilución debe tener calidad constante. El pH se ha de mantener entre 6,0 y 8,5 al inicio del ensayo, pero durante un ensayo dado debe permanecer dentro de un intervalo de ± 0,5 unidades de pH. Para garantizar que el agua de dilución no influye indebidamente en los resultados del estudio (por ejemplo, por complejación de la sustancia problema) ni afecta negativamente al comportamiento de las poblaciones de peces, se deben tomar regularmente muestras para análisis, al menos al inicio y al final del ensayo. Conviene proceder, por ejemplo cada tres meses si se sabe que el agua de dilución es de calidad relativamente constante, a la determinación de los metales pesados (por ejemplo, Cu, Pb, Zn, Hg, Cd y Ni), aniones y cationes principales (por ejemplo, Ca2+, Mg2+, Na+, K+, Cl-, y SO4 2-), plaguicidas (por ejemplo, plaguicidas organofosforados totales y organoclorados totales), carbono orgánico total y sólidos en suspensión. Si se demuestra que la calidad del agua de dilución es constante al menos durante un año, las determinaciones pueden ser menos frecuentes y espaciarse más (por ejemplo, cada seis meses). El contenido en partículas naturales y el carbono orgánico total del agua de dilución deben ser lo más bajos posible para evitar la adsorción de la sustancia problema a la materia orgánica, que podría reducir su biodisponibilidad y con ello dar lugar a una subestimación del FBC. El valor máximo admisible es de 5 mg/l para las partículas (materia seca que no atraviesa un filtro de 0,45 μm) y de 2 mg/l para el carbono orgánico total (véase el apéndice 2). En caso necesario, el agua de dilución se debe filtrar antes de usarse. La contribución al contenido en carbono orgánico del agua del ensayo que aportan los peces (excrementos) y los residuos alimentarios debe mantenerse lo más baja posible (véase el punto 46). Soluciones de ensayo Se prepara una solución madre de la sustancia problema, a una concentración adecuada. La solución madre se prepara preferiblemente por simple mezcla o agitación de la sustancia problema en el agua de dilución. Una alternativa que puede resultar apropiada en algunos casos es la utilización de un sistema de dosificación por desorción de una fase sólida. No se recomienda en general el uso de disolventes o dispersantes (agentes solubilizantes) (véase (25)); sin embargo, la utilización de estos materiales puede ser aceptable para obtener una solución madre de concentración adecuada, pero deben hacerse todos los esfuerzos posibles para minimizar el uso de tales materiales y no debe superarse su concentración micelar crítica (si procede). Los disolventes que pueden utilizarse son la acetona, el etanol, el metanol, la dimetil-formamida y el trietilenglicol; los dispersantes que se han utilizado son el Tween 80, la metilcelulosa al 0,01 % y el HCO-40. La concentración de disolvente en el medio de ensayo final debe ser la misma en todos los tratamientos (es decir, con independencia de la concentración de la sustancia problema) y no debe ser superior a los correspondientes umbrales de toxicidad determinados para el disolvente en las condiciones del ensayo. El nivel máximo es una concentración de 100 mg/l (o 0,1 ml/l). Es poco probable que una concentración de disolvente de 100 mg/l altere de manera significativa la máxima concentración disuelta de la sustancia problema que pueda alcanzarse en el medio (25). Debe conocerse la contribución del disolvente, junto con la de la sustancia problema, al contenido global de carbono orgánico en el agua del ensayo. A lo largo del ensayo, la concentración de carbono orgánico total en los recipientes del ensayo no debe sobrepasar en más de 10 mg/l (± 20 %) la del carbono orgánico procedente de la sustancia problema y, en su caso del disolvente o agente de disolución (48). El contenido de materia orgánica puede afectar significativamente a la cantidad de sustancia problema disuelta libremente durante los ensayos dinámicos con peces, especialmente en el caso de sustancias muy lipófilas. La microextracción en fase sólida (véase el punto 60) puede aportar información importante sobre la relación entre los compuestos disueltos libremente y los ligados, de los que se supone que los primeros representan la fracción biodisponible. La concentración de la sustancia problema debe ser inferior a su límite de solubilidad en los medios de ensayo a pesar de la utilización de un disolvente o agente solubilizante. Deben tomarse precauciones en caso de utilización de disolventes fácilmente biodegradables, ya que estos pueden causar problemas de crecimiento bacteriano en los ensayos dinámicos. Si no es posible preparar una solución madre sin el uso de un agente solubilizante, debe prestarse atención a la adecuación de un estudio de exposición acuática frente a un estudio de exposición alimentaria. Para los ensayos dinámicos se necesitará un sistema que aporte y diluya continuamente la solución madre de la sustancia problema (por ejemplo, bomba dosificadora, diluyente proporcional, sistema saturador) o un sistema de dosificación por desorción de una fase sólida, para conseguir las concentraciones requeridas en los recipientes de ensayo. Es preferible permitir al menos cinco renovaciones del volumen al día en cada recipiente de ensayo. Se preferirá el método dinámico, pero en caso de imposibilidad (por ejemplo, cuando los organismos del ensayo sufran efectos adversos) podrá aplicarse una técnica semiestática si siguen cumpliéndose los criterios de validez (véase el punto 24). Los caudales de las soluciones madre y del agua de dilución se controlarán 48 horas antes del ensayo y luego al menos diariamente durante este. Dicho control incluirá la determinación del caudal en cada recipiente de ensayo y garantizará que no varía en más del 20 % dentro de cada recipiente ni entre recipientes distintos. Selección de las especies Entre los criterios importantes de selección de las especies figuran el que sean de fácil disponibilidad, que puedan obtenerse de los tamaños adecuados y que puedan mantenerse satisfactoriamente en el laboratorio. Otros criterios de selección de las especies de peces son su importancia recreativa, comercial o ecológica, así como una sensibilidad comparable, haber producido buenos resultados anteriormente, etc. Las especies de ensayo recomendadas figuran en el apéndice 3. Pueden utilizarse otras especies, pero es posible entonces tener que adaptar el procedimiento de ensayo para obtener condiciones experimentales convenientes. En tal caso, deben justificarse la elección de la especie y el método experimental. En general, la utilización de especies de peces más pequeños acorta el tiempo necesario para alcanzar el estado de equilibrio, pero puede ser necesario un número superior de peces (muestras) para analizar adecuadamente el contenido lipídico y las concentraciones de la sustancia problema en los peces. Además, es posible que las diferencias en la tasa de respiración y en el metabolismo entre los peces de más edad y los más jóvenes dificulten la comparación de resultados entre diferentes ensayos y especies de ensayo. Cabe señalar que, si los peces utilizados en un ensayo están en una fase de su ciclo vital con crecimiento rápido (juveniles), se puede complicar la interpretación de los datos. Preparación de los peces (pertinente para la exposición alimentaria y acuática) La población inicial de peces debe aclimatarse durante dos semanas al menos en agua (véase el punto 28) a la temperatura del ensayo y alimentarse con una dieta suficiente (véase el punto 45). Tanto el agua como los alimentos deben ser del mismo tipo que los usados en el ensayo. Después de un periodo de adaptación de 48 horas, se registra la mortalidad y se aplican los criterios siguientes:
Los peces utilizados en los ensayos deben estar libres de enfermedades ni anomalías observables. Se deben descartar todos los peces enfermos. Los peces no deben recibir tratamiento terapéutico alguno durante el ensayo ni en las dos semanas anteriores al mismo. REALIZACIÓN DEL ENSAYO Ensayo preliminar Puede ser útil proceder a un experimento preliminar para optimizar las condiciones de realización del ensayo definitivo, como, por ejemplo, la concentración o concentraciones de la sustancia problema, o la duración de las fases de absorción y de depuración, o para determinar si es necesario llevar a cabo un ensayo completo. El diseño del ensayo preliminar debe ser tal que se obtenga la información requerida. Se puede considerar si un ensayo minimizado puede ser suficiente para obtener un FBC, o si es necesario un estudio completo (véanse los puntos 83-95 sobre el ensayo minimizado). Condiciones de exposición Duración de la fase de absorción Es posible obtener una previsión de la duración de la fase de absorción a partir de la experiencia práctica (por ejemplo, a partir de un estudio previo o de un estudio de acumulación de una sustancia estructuralmente relacionada) o a partir de ciertas relaciones empíricas utilizando el dato de la hidrosolubilidad o del coeficiente de reparto octanol/agua de la sustancia problema (siempre que la absorción siga una cinética de primer orden, véase el apéndice 5). La fase de absorción durará 28 días salvo si puede demostrarse que se alcanza antes el estado de equilibrio (véase el apéndice 1, Definiciones y unidades). Se alcanza el estado de equilibrio en la representación gráfica de la concentración de la sustancia problema en los peces (C f) en función del tiempo cuando la curva se hace paralela al eje del tiempo y tres análisis sucesivos de C f realizados con muestras tomadas a intervalos de, al menos, dos días presentan una diferencia máxima del ± 20 % entre sí, y no hay ningún aumento significativo de C f entre el primero y el último de los análisis sucesivos. Cuando se analizan muestras puestas conjuntamente, es necesario proceder al menos a cuatro análisis sucesivos. Si las sustancias problema se absorben con lentitud, se optará preferiblemente por intervalos semanales. Si no se ha alcanzado el estado de equilibrio en 28 días, el FBC se calcula utilizando únicamente el enfoque cinético, que no depende de que se alcance el estado de equilibrio, o bien puede ampliarse la fase de absorción, con la realización de más mediciones, hasta alcanzar el estado de equilibrio, o durante 60 días, si esto supone un plazo menor. Asimismo, es necesario que la concentración de la sustancia problema en los peces al final de la fase de absorción sea lo bastante alta como para permitir una estimación fiable de k 2 de la fase de depuración. Si no se consigue una absorción significativa al cabo de 28 días, el ensayo puede detenerse. Duración de la fase de depuración En el caso de las sustancias que siguen una cinética de primer orden, generalmente es suficiente la mitad de la duración de la fase de absorción para conseguir una reducción conveniente (por ejemplo, del 95 %) de la carga corporal de la sustancia (véanse en el apéndice 5 las explicaciones sobre el cálculo). Si el tiempo necesario para llegar a una pérdida del 95 % es excesivamente largo, sobrepasando por ejemplo dos veces la duración normal de la fase de absorción (es decir, más de 56 días), se podrá utilizar un periodo más corto (p. ej., hasta que la concentración de la sustancia problema sea inferior al 10 % de la concentración en el estado de equilibrio). No obstante, puede ser necesario un periodo de depuración más largo con las sustancias que presentan patrones de absorción y depuración más complejos, que no se ajustan al modelo de peces con un único compartimento, el cual corresponde a una cinética de primer orden. Si se observan o se esperan tales patrones complejos, se recomienda solicitar consejo a un bioestadístico o a un técnico de farmacocinética para garantizar una adecuada configuración del ensayo. Según se amplía el periodo de depuración, el número de peces que ha de muestrearse puede llegar a ser limitante y las diferencias de crecimiento entre los peces pueden influir en los resultados. El periodo será función también del tiempo durante el cual la concentración de la sustancia problema en los peces permanezca por encima del límite analítico de cuantificación. Número de peces del ensayo El número de peces por concentración del ensayo debe elegirse de modo que se disponga de, como mínimo, cuatro peces por cada punto de muestreo. Solo se deben poner en común los peces si no es factible el análisis de uno solo. Si es preciso conseguir una mayor precisión en el ajuste de la curva (y parámetros derivados) o si se requieren estudios del metabolismo (por ejemplo, para distinguir entre los metabolitos y la sustancia original cuando se utilizan sustancias problema radiomarcadas), será necesario disponer de más peces por punto de muestreo. El contenido lipídico debe determinarse con los mismos materiales biológicos que se utilicen para determinar la concentración de la sustancia problema. Si ello no es posible, podrá ser necesario disponer de más peces (véanse los puntos 56 y 57). Si se utilizan peces adultos (es decir, sexualmente maduros), no deben estar en fase de desove ni haberla pasado recientemente (es decir, haber desovado ya) ni antes del ensayo ni durante el mismo. También debe indicarse si en el experimento se utilizan machos o hembras, o los dos sexos. En este último caso, será necesario comprobar (antes del inicio de la exposición) que no hay diferencias significativas de crecimiento y de contenido lipídico entre sexos, en particular si se prevé que va a ser necesario poner en común peces machos y hembras para garantizar que puedan detectarse las concentraciones de la sustancia o el contenido lipídico. En cada uno de los ensayos se elegirán peces de peso similar, de modo que el peso del más pequeño no sea inferior a dos tercios del peso del mayor. Pertenecerán a la misma clase de edad y procederán de la misma fuente. Ya que el peso y la edad de un pez pueden tener efectos notables sobre los valores del FBC (12), estos datos se registrarán con exactitud. Se recomienda pesar poco antes del inicio del ensayo una submuestra de la población de peces para calcular el peso medio (véase el punto 61). Carga Se debe utilizar una relación elevada agua/peces con el fin de minimizar la reducción de la concentración de la sustancia problema en el agua debida a la introducción de los peces al principio del ensayo, y también para evitar el descenso de la concentración de oxígeno disuelto. Es importante que la tasa de carga sea adaptada a la especie utilizada para el ensayo. De todos modos, se recomienda normalmente una tasa de carga peces / agua de 0,1-1,0 g de peces (peso húmedo) por litro de agua al día. Se pueden utilizar tasas de carga más elevadas si se demuestra que la concentración requerida de sustancia problema puede mantenerse en el intervalo del ± 20 %, y que la concentración de oxígeno disuelto no cae por debajo del 60 % del nivel de saturación (véase el punto 24). Se tendrá en cuenta el hábitat normal de la especie de peces al elegir la tasa de carga adecuada. Los peces bentónicos, por ejemplo, pueden necesitar un acuario que tenga más superficie de fondo que las especies pelágicas, para un mismo volumen de agua. Alimentación Durante los periodos de aclimatación y de ensayo, se dará a los peces un alimento conveniente que tenga un contenido conocido en lípidos y proteínas totales, en cantidades suficientes para mantenerlos en buena salud y conservar el peso corporal (se permite cierto crecimiento). Se dará alimento diariamente durante los periodos de aclimatación y de ensayo a una dosis establecida en función de la especie utilizada, de las condiciones experimentales y del valor calorífico del alimento (por ejemplo, en el caso de la trucha arco iris, aproximadamente entre el 1 y el 2 % del peso corporal por día). La dosis alimentaria debe seleccionarse de forma que se evite el crecimiento rápido y el amplio aumento del contenido lipídico. Para mantener la misma dosis alimentaria, debe volver a calcularse la cantidad de alimento según proceda, por ejemplo una vez por semana. Para ese cálculo, el peso de los peces de cada recipiente de ensayo se estima a partir del peso de los peces muestreados más recientemente en ese recipiente. No hay que pesar los peces que permanecen en el recipiente. La comida no consumida y los excrementos deben evacuarse de los recipientes de ensayo mediante sifón cada día poco después de la alimentación (de 30 minutos a una hora). Los recipientes se tendrán lo más limpios posible a lo largo del ensayo, de modo que la concentración de materia orgánica permanezca al nivel más bajo posible (véase el punto 29), puesto que la presencia de carbono orgánico puede limitar la biodisponibilidad de la sustancia problema (12). Como muchos alimentos son derivados de harinas de pescado, debe garantizarse que el alimento no influye en los resultados del ensayo ni provoca efectos adversos, por ejemplo por contener (restos de) plaguicidas, metales pesados o la propia sustancia problema. Luz y temperatura Se recomienda un fotoperiodo de 12 a 16 horas y la temperatura (± 2°C) ha de ser adecuada para la especie utilizada (véase el apéndice 3). Deben conocerse el tipo y las características de la iluminación. Es necesario prestar atención a la posible fototransformación de la sustancia problema en las condiciones de irradiación del estudio. Se ha de utilizar una iluminación adecuada que no exponga a los peces a fotoproductos no naturales. En algunos casos, puede ser conveniente utilizar un filtro para eliminar los rayos UV por debajo de 290 nm. Concentraciones de ensayo El ensayo se concibió inicialmente para las sustancias orgánicas apolares. Para este tipo de sustancias, se prevé que es suficiente la exposición de los peces a una única concentración, dado que no se esperan efectos de concentración, aunque pueden ser necesarias dos concentraciones en virtud del marco normativo pertinente. Si se someten a ensayo sustancias fuera de este ámbito, o se conocen otros datos, como una posible dependencia de la concentración, el ensayo deberá efectuarse con dos o más concentraciones. Si se somete a ensayo una única concentración, deberá justificarse (véase el punto 79). Además, la concentración de ensayo debe ser tan baja como sea técnica o prácticamente posible (es decir, no muy cerca del límite de solubilidad). En algunos casos, puede anticiparse que la bioconcentración de una sustancia depende de la concentración en el agua (por ejemplo, en el caso de los metales, cuya absorción en los peces puede estar regulada al menos en parte). En tal caso, es necesario que se estudien al menos dos, pero preferiblemente más, concentraciones (véase el punto 49) que sean pertinentes desde el punto de vista medioambiental. También en el caso de las sustancias cuyas concentraciones de ensayo tienen que encontrarse cerca del límite de solubilidad por razones prácticas, se recomienda utilizar al menos dos concentraciones de ensayo, ya que esto puede dar una idea de la fiabilidad de las concentraciones de la exposición. La elección de las concentraciones de ensayo debe incorporar la concentración ambientalmente realista, así como la concentración que sea pertinente para los fines de la evaluación específica. La concentración o concentraciones de la sustancia problema deben seleccionarse por debajo de su nivel de efecto crónico o del 1 % de su LC50 aguda asintótica, dentro de un intervalo relevante para el medio ambiente y, como mínimo, un orden de magnitud por encima de su límite de cuantificación en el agua por el método de análisis elegido. La concentración de ensayo permisible más elevada puede establecerse también dividiendo la LC50 aguda de 96 h por una relación aguda/crónica adecuada (p. ej., las relaciones adecuadas de algunas sustancias están alrededor de tres, pero unas cuantas están por encima de 100). Si se utiliza una segunda concentración, debe diferir de la antes citada en un factor de diez. Cuando esto no sea posible por el criterio de toxicidad (que limita la concentración de ensayo superior) y por el límite analítico (que limita la concentración de ensayo inferior), puede utilizarse un factor inferior a diez y debe considerarse el empleo de una sustancia problema radiomarcada (del nivel más alto de pureza; por ejemplo, preferiblemente > 98 %). Debe velarse por que no se utilice ninguna concentración que quede por encima del límite de solubilidad de la sustancia problema en los medios de ensayo. Testigos Además de las series de ensayo, debe estudiarse un testigo del agua de dilución o, en su caso (véanse los puntos 30 y 31), un testigo del disolvente. Frecuencia de la medición de la calidad del agua Durante el ensayo se medirán en todos los recipientes de ensayo y de testigo el oxígeno disuelto, el COT, el pH y la temperatura. En el testigo o testigos y en un recipiente de ensayo se medirán la dureza total y la salinidad (cuando proceda). Si se someten a ensayo dos o más concentraciones, estos parámetros se medirán a la concentración más elevada. Por lo menos, el oxígeno disuelto y la salinidad (cuando proceda) se medirán tres veces —al principio, hacia el medio y al final de la fase de absorción— y una vez por semana durante la fase de depuración. El COT se medirá al principio del ensayo (24 y 48 horas antes del comienzo de la fase de absorción), antes de la adición de los peces y al menos una vez por semana durante las fases de absorción y de depuración. La temperatura se medirá y registrará diariamente, el pH al principio y al final de cada periodo y la dureza una vez en cada ensayo. La temperatura se medirá preferiblemente de forma continua en un recipiente al menos. Muestreo y análisis de los peces y del agua Pauta de muestreo de los peces y del agua Se tomará agua de los recipientes de ensayo para determinar la concentración de la sustancia problema antes de la introducción de los peces y durante las fases de absorción y de depuración. Deben tomarse muestras de agua antes de añadir el alimento, al mismo tiempo que se muestrean los peces. Puede ser útil aumentar la frecuencia de muestreo para garantizar la estabilidad de las concentraciones tras la introducción de los peces. Durante la fase de absorción, deben determinarse las concentraciones de la sustancia problema a fin de comprobar el cumplimiento de los criterios de validez (punto 24). Si los análisis de las muestras de agua al inicio de la fase de depuración ponen de manifiesto que no se detecta la sustancia problema, puede utilizarse este hecho como justificación para no medir la sustancia problema en el agua de ensayo y de los testigos durante el resto de la fase de depuración. Se deben muestrear los peces en al menos cinco ocasiones durante la fase de absorción y en al menos cuatro ocasiones durante la fase de depuración para determinar su contenido de sustancia problema. Como a veces es difícil calcular una estimación razonablemente precisa del FBC a partir de este número de muestras, en particular cuando la cinética de absorción y depuración no es la simple de primer orden, puede ser recomendable muestrear con mayor frecuencia en los dos periodos (véase el apéndice 4). El contenido lipídico debe establecerse con los mismos materiales biológicos que se utilicen para determinar la concentración de la sustancia problema, como mínimo al inicio y al final de la fase de absorción y al final de la fase de depuración. Si ello no es posible, deben muestrearse al menos tres peces independientes para determinar su contenido lipídico en cada uno de esos tres tiempos. El número de peces por recipiente al inicio del experimento debe ajustarse en consecuencia (49). De forma alternativa, en caso de que no se detecten cantidades significativas de la sustancia problema en los peces testigo (es decir, peces de la población inicial), los peces testigo tomados del ensayo pueden analizarse para determinar solo el contenido lipídico, y el análisis de la sustancia problema en el grupo o grupos de ensayo (y la constante de la velocidad de absorción, la constante de la velocidad de depuración y los valores del FBC relacionados) pueden corregirse para tener en cuenta el contenido lipídico del grupo testigo durante el ensayo (50). Los peces muertos o enfermos no deben analizarse en cuanto a la concentración de la sustancia problema ni la de los lípidos. En el apéndice 4 se encuentra un ejemplo de pauta de muestreo aceptable. Pueden establecerse otras pautas fácilmente sobre la base de otros valores supuestos de K OW para calcular el tiempo de exposición que corresponde a una absorción del 95 % (véanse los cálculos en el apéndice 5). Debe continuarse el muestreo durante la fase de absorción hasta que se alcance el estado de equilibrio (véase el apéndice 1, Definiciones y unidades) o se dé por terminada de otra manera la fase de absorción (después de 28 o 60 días, véanse los puntos 37 y 38). Antes de empezar la fase de depuración, los peces deben transferirse a recipientes limpios. Muestreo y preparación de las muestras Deben tomarse muestras de agua para su análisis, por ejemplo mediante sifonado a través de un tubo inerte desde un punto central del recipiente de ensayo. Ni la filtración ni la centrifugación permiten siempre separar la fracción no biodisponible de la sustancia problema respecto a su fracción biodisponible. En caso de que se aplique una técnica de separación, debe facilitarse siempre en el informe de ensayo una justificación o una validación de la técnica aplicada, dadas las dificultades de biodisponibilidad (25). Especialmente en el caso de las sustancias muy hidrófobas (es decir, sustancias con log K OW > 5) (12) (26), que podrían adsorberse a la matriz de filtración o a los recipientes de centrifugación, no deben someterse las muestras a estos tratamientos. En lugar de ello, se deben adoptar medidas para que los recipientes se mantengan lo más limpios posible (véase el punto 46) y debe supervisarse el contenido en carbono orgánico total a lo largo de las fases tanto de absorción como de depuración (véase el punto 53). Para evitar posibles problemas de biodisponibilidad reducida, puede recurrirse al muestreo con técnicas de microextracción en fase sólida en caso de sustancias poco solubles y muy hidrófobas. Los peces muestreados deben sacrificarse inmediatamente, de la manera más adecuada e incruenta posible (para las mediciones de los peces enteros, no deben aplicarse más tratamientos que el lavado con agua (véase el punto 28) y el secado con un pañuelo). Se pesan y se mide la longitud total (51). Respecto a cada pez, deben relacionarse el peso y la longitud medidos con la concentración de la sustancia analizada (y el contenido lipídico, si procede), por ejemplo utilizando un código único de identificación para cada pez muestreado. Es preferible analizar los peces y el agua inmediatamente después del muestreo para evitar toda degradación u otras pérdidas y para calcular aproximadamente las constantes de las velocidades de absorción y de depuración mientras continúa el ensayo. El análisis inmediato evita también retrasos en la determinación del momento en que se alcanza una meseta (estado de equilibrio). Si no se hace el análisis inmediatamente, las muestras deben conservarse según un método conveniente. Antes del principio del estudio, debe conseguirse información sobre el método de conservación adecuado para la sustancia problema correspondiente como, por ejemplo, congelación, mantenimiento a 4 °C, extracción, etc. El periodo de almacenamiento debe seleccionarse para garantizar que la sustancia no se degrade mientras esté almacenada. Calidad del método analítico Como la totalidad del procedimiento se rige principalmente por la exactitud, la precisión y la sensibilidad del método analítico utilizado para la sustancia problema, es necesario comprobar experimentalmente que la exactitud, la precisión y la reproducibilidad del análisis de la sustancia, así como la recuperación de la sustancia problema tanto del agua como de los peces, son satisfactorias con el método particular utilizado. Esto debe formar parte de los ensayos previos. Ha de comprobarse también que la sustancia problema no es detectable en el agua de dilución utilizada. En caso necesario, se corrigen los valores de la concentración de la sustancia problema en el agua y en los peces obtenidos de los ensayos para tener en cuenta las recuperaciones y los valores de fondo de los testigos. Las muestras de peces y de agua se deben manipular en todo momento de forma que se minimicen las contaminaciones y las pérdidas (derivadas, por ejemplo, de la adsorción al equipo de muestreo). Análisis de las muestras de peces Si se utilizan para el ensayo materiales radiomarcados, se puede analizar el marcador radiactivo total (es decir, sustancia original y metabolitos) o se pueden purificar las muestras de modo que sea posible analizar separadamente la sustancia original. Si el FBC se va a basar en la sustancia original, deben caracterizarse los principales metabolitos, como mínimo al final de la fase de absorción (véase el punto 6). Los principales metabolitos serán los que representen ≥ 10 % de los residuos totales en los tejidos de los peces, los que representen ≥ 5 % en dos puntos consecutivos de muestreo, los que muestren un aumento de los niveles a lo largo de toda la fase de absorción, y los de importancia toxicológica conocida. Si el FBC para el pez entero en términos de residuos radiomarcados totales es ≥ 500, puede ser conveniente (y, para algunas categorías de sustancias como los plaguicidas, muy recomendable), identificar y cuantificar los principales metabolitos. La cuantificación de tales metabolitos puede estar impuesta por algunas autoridades normativas. Si se identifican y cuantifican metabolitos que representan ≥ 10 % de los residuos radiomarcados totales en los tejidos de los peces, se recomienda entonces identificar y cuantificar los metabolitos también en el agua del ensayo. En caso de que esto no sea posible, deberá explicarse en el informe. La concentración de la sustancia problema debe determinarse generalmente en cada pez pesado aparte. Si esto no es posible, se podrán poner conjuntamente las muestras de cada momento de muestreo, pero esta mezcla limita los procedimientos estadísticos que pueden aplicarse a los datos, por lo que debe incluirse en el ensayo un número adecuado de peces para conciliar la puesta en común con el procedimiento y la potencia estadísticos deseados. Las referencias (27) y (28) pueden utilizarse como introducción a los procedimientos pertinentes de puesta en común. El FBC debe expresarse normalizado en referencia a peces con un 5 % de contenido lipídico (sobre la base del peso húmedo), además del valor derivado directamente del estudio (véase el punto 21), a menos que pueda justificarse que la sustancia problema no se acumula principalmente en los lípidos. El contenido lipídico de los peces debe determinarse si es posible en cada momento de muestreo, preferiblemente con el mismo extracto que el consagrado al análisis de la sustancia problema, puesto que a menudo los lípidos deben retirarse del extracto antes de que este se pueda analizar por cromatografía. Sin embargo, el análisis de las sustancias problema exige con frecuencia procedimientos de extracción específicos que pueden estar en contradicción con los métodos de ensayo para la determinación de los lípidos. En este caso (hasta que se disponga de métodos instrumentales adecuados no destructivos), se recomienda emplear una estrategia diferente para determinar el contenido lipídico de los peces (véase el punto 56). Deben utilizarse métodos adecuados para la determinación del contenido lipídico (20). La técnica de extracción con cloroformo/metanol (29) puede recomendarse como método normal (30), pero se recomienda como alternativa técnica el método de Smedes (31). Este último método se caracteriza por una eficiencia comparable de extracción, una alta exactitud, el uso de disolventes orgánicos menos tóxicos y la facilidad de ejecución. Pueden utilizarse, en casos debidamente justificados, otros métodos que presenten alguna ventaja en términos de exactitud respecto a los métodos recomendados. Es importante precisar bien el método utilizado. Medición del crecimiento de los peces Al comienzo del ensayo, deben pesarse por separado de cinco a diez peces de la población inicial, y debe medirse su longitud total. Puede tratarse de los mismos peces utilizados para el análisis de lípidos (véase el punto 56). El peso y la longitud de los peces utilizados para cada toma de muestras de los grupos tanto de ensayo como testigo deben medirse antes de proceder al análisis del producto o de los lípidos. Las mediciones de estos peces muestreados se pueden utilizar para estimar el peso y la longitud de los peces que quedan en los recipientes de ensayo y de testigo (véase el punto 45). DATOS E INFORME Tratamiento de los resultados La curva de absorción de la sustancia problema debe obtenerse representando su concentración en los peces (interior o superficie, o tejidos específicos) durante la fase de absorción en función del tiempo, con escala aritmética. Si la curva alcanza una meseta, es decir, adopta aproximadamente una disposición asintótica respecto al eje del tiempo, se calculará del siguiente modo el FBC en el estado de equilibrio (FBCSS):
La evolución de C f puede verse influida por el crecimiento de los peces (véanse los puntos 72 y 73). La concentración de exposición media (Cw ) se ve influida por las variaciones a lo largo del tiempo. Cabe esperar que una concentración media ponderada en función del tiempo sea más pertinente y precisa para los estudios de bioacumulación, incluso aunque la variación se encuentre en el intervalo adecuado de validez (véase el punto 24). La concentración en el agua media ponderada en función del tiempo puede calcularse de conformidad con el apéndice 5, sección 1. El factor de bioconcentración cinético (FBCK) debe determinarse como la relación k 1/k 2, que son las dos constantes de velocidad de primer orden. Las constantes de velocidad k 1 y k 2 y el FBCK pueden obtenerse ajustando simultáneamente la fase de absorción y la fase de depuración. Otra posibilidad consiste en determinar secuencialmente k 1 y k 2 (véase en el apéndice 5 una descripción y comparación de estos métodos). Es posible tener que corregir la constante de la velocidad de depuración (k 2) para tener en cuenta la dilución por el crecimiento (véanse los puntos 72 y 73). Si resulta evidente que la curva de absorción o de depuración no es de primer orden, entonces convendrá utilizar modelos más complejos (véanse las referencias en el apéndice 5) y buscar el consejo de un bioestadístico o de un técnico de farmacocinética. Datos de peso/longitud de los peces Los pesos húmedos y longitudes totales de cada uno de los peces en todos los intervalos de muestreo se tabulan por separado para los grupos de ensayo y testigo durante las fases de absorción (incluida la población inicial para el comienzo de la absorción) y de depuración. Respecto a cada pez, deben relacionarse el peso y la longitud medidos con la concentración del producto analizado, por ejemplo utilizando un código único de identificación para cada pez muestreado. El peso es la medida preferida del crecimiento a efectos de la corrección de los valores del FBC cinético para tener en cuenta la dilución por el crecimiento (véase, en el punto 73 y en el apéndice 5, el método utilizado para corregir los datos en función de esta dilución por el crecimiento). Corrección en función de la dilución por el crecimiento y normalización lipídica El crecimiento de los peces durante la fase de depuración puede reducir las concentraciones medidas del producto en los peces, con el resultado de que la constante de la velocidad de depuración general (k 2) sea mayor que la que se derivaría únicamente de los procesos de eliminación (por ejemplo, respiración, metabolismo, excreción). Los factores de bioconcentración cinéticos deben corregirse para tener en cuenta la dilución por el crecimiento. Los FBCSS también se verá influidos por el crecimiento, pero no se dispone de ningún procedimiento acordado para corregir los FBCSS en función del crecimiento. En los casos de crecimiento significativo, también deben calcularse los FBCK, corregidos en función del crecimiento (FBCKg), ya que pueden ser una medida más pertinente del factor de bioconcentración. El contenido lipídico de los peces del ensayo (que está estrechamente relacionado con la bioacumulación de las sustancias hidrófobas) puede variar en la práctica tanto que haga necesario proceder a la normalización respecto a un contenido lipídico de los peces fijo (5 % p/p) para presentar los factores de bioconcentración, tanto el cinético como el del estado de equilibrio, de modo que tengan sentido, a menos que pueda justificarse que la sustancia problema no se acumula principalmente en los lípidos (por ejemplo, algunas sustancias perfluoradas pueden unirse a las proteínas). Las ecuaciones y ejemplos de estos cálculos figuran en el apéndice 5. Para corregir un FBC cinético para tener en cuenta la dilución debida al crecimiento, la constante de la velocidad de depuración debe corregirse para tener en cuenta el crecimiento. Esta constante de la velocidad de depuración corregida según el crecimiento (k 2g) se calcula restando la constante de la velocidad de crecimiento (k g, obtenida directamente de los datos de pesos medidos) de la constante de la velocidad de depuración general (k 2). A continuación se obtiene el factor de bioconcentración cinético corregido en función del crecimiento dividiendo la constante de la velocidad de absorción (k 1) por la constante de la velocidad de deputación corregida según el crecimiento (k 2g) (véase el apéndice 5). En algunos casos es difícil aplicar este enfoque. Por ejemplo, en el caso de sustancias de depuración muy lenta que se estudien con peces de rápido crecimiento, la constante obtenida k 2g puede ser muy pequeña, con lo que el error en las dos constantes de velocidad utilizadas para calcularla reviste una importancia crítica, y en algunos casos las estimaciones de k g pueden ser mayores que k 2. Un enfoque alternativo que elude la necesidad de corregir según la dilución debida al crecimiento implica la utilización de datos de depuración de masa de sustancia problema por pez (peces enteros) en lugar de los habituales datos de masa de sustancia problema por unidad de masa de peces (concentración). Esto puede conseguirse fácilmente, ya que los ensayos según el presente método deben vincular las concentraciones tisulares registradas con los pesos de cada pez. El sencillo procedimiento correspondiente se describe en el apéndice 5. Obsérvese que sigue siendo necesario comunicar el valor de k 2 aunque se utilice este enfoque alternativo. Los factores de bioconcentración cinético y en estado de equilibrio deben consignarse también en relación con un contenido lipídico de los peces de referencia, del 5 % (p/p), salvo que pueda justificarse que la sustancia problema no se acumula principalmente en los lípidos. Los datos de concentración en peces, o el FBC, se normalizan con arreglo a la relación entre el 5 % y la media real (individual) del contenido lipídico (en % de peso húmedo) (véase el apéndice 5). Si los análisis del producto y de los lípidos se realizan con un mismo pez, entonces, para calcular un FBC normalizado según el contenido lipídico, deben utilizarse los datos normalizados según el contenido lipídico de cada pez. Por otra parte, si es similar el crecimiento de los peces testigo y el de los expuestos al producto, el contenido lipídico de los peces testigo solos podrá utilizarse para la corrección según el contenido lipídico (véase el punto 56). En el apéndice 5 se describe un método de cálculo del FBC normalizado según el contenido lipídico. Interpretación de los resultados Los resultados se interpretarán con precaución cuando las concentraciones medidas de las soluciones de ensayo se encuentren a niveles cercanos al límite de detección del método de análisis. El crecimiento medio tanto en los grupos de ensayo como en los testigos no debe ser, en principio, significativamente diferente, a fin de excluir los efectos tóxicos. Las constantes de la velocidad de crecimiento o las curvas de crecimiento de los dos grupos deben compararse siguiendo un procedimiento adecuado (52). La claridad de definición de las curvas de absorción y de depuración indica la buena calidad de los datos de bioconcentración. Para las constantes de velocidad, el resultado de una prueba χ2 de la bondad del ajuste debe mostrar un buen ajuste (es decir, un pequeño porcentaje de error en la medición (32)) para el modelo de bioacumulación, de forma que las constantes de velocidad puedan considerarse fiables (véase el apéndice 5). Si se utiliza más de una concentración de ensayo, la variación de las constantes de absorción/depuración entre las concentraciones de ensayo debe ser inferior al 20 % (53). En caso contrario, podría indicarse la dependencia de la concentración. Se registrarán y, si es posible, se explicarán las diferencias notables que se observen en las constantes de la velocidad de absorción/depuración entre las concentraciones de ensayo aplicadas. En general, el límite de confianza del 95 % de los FBC obtenidos de estudios bien diseñados se acerca al ± 20 % del FBC derivado. Si se someten a ensayo dos o más concentraciones, los resultados de todas las concentraciones se utilizarán para determinar si los resultados son coherentes y para mostrar si existe dependencia de la concentración. Si se somete a ensayo una sola concentración para reducir la utilización de animales o de recursos, debe darse la justificación correspondiente. El FBCSS resultante es dudoso si el FBCK es considerablemente mayor que el FBCSS, ya que esto puede ser un indicio de que no se ha alcanzado el estado de equilibrio o de que no se han tenido en cuenta los eventuales procesos de pérdida y de dilución por el crecimiento. En los casos en que el FBCSS está muy por encima del FBCK, deberá comprobarse si no se han cometido errores en la obtención de las constantes de velocidad de absorción y de depuración y se evaluarán de nuevo. Un procedimiento diferente de ajuste podría mejorar la estimación de FBCK (véase el apéndice 5). Informe del ensayo Aparte de la información sobre la sustancia problema indicada en el punto 3, el informe del ensayo incluirá la información siguiente:
C.13 — II: Ensayo minimizado en peces con exposición acuática INTRODUCCIÓN La creciente experiencia adquirida con la realización e interpretación del ensayo completo, tanto por los laboratorios como por los organismos normativos, pone de manifiesto que, con algunas excepciones, se aplica la cinética de primer orden para el cálculo de las constantes de las velocidades de absorción y de depuración. Así pues, con un mínimo de puntos de muestreo es posible calcular las constantes de las velocidades de absorción y de depuración, y obtener el FBC cinético. El objetivo inicial del examen de diseños alternativos de los estudios del FBC era desarrollar un pequeño ensayo a fin de utilizarlo en una fase intermedia de ensayo para descartar o confirmar las estimaciones del FBC basadas en el K OW y en la relación QSAR, y eliminar así la necesidad de hacer un estudio completo de muchas sustancias y reducir los costes y el uso de animales a través de la reducción del muestreo y del número de secuencias analíticas efectuadas. Aun siguiendo el diseño principal del método de ensayo anterior para permitir la integración de los resultados del ensayo con los datos sobre FBC ya existentes, y para facilitar la ejecución de los ensayos y la interpretación de los datos, el objetivo era proporcionar estimaciones adecuadas del FBC, con exactitud y precisión adecuadas para poder tomar decisiones de evaluación de riesgos. Son válidas muchas de las consideraciones que se aplican también en el ensayo completo como, por ejemplo, los criterios de validez (véase el punto 24) y la detención del ensayo si se considera insignificante la absorción al final de la fase de absorción (véanse los puntos 16 y 38). Las sustancias que podrían acogerse al diseño del ensayo minimizado deben pertenecer al ámbito general para el que se ha desarrollado este método de ensayo, es decir, el de las sustancias orgánicas apolares (véase el punto 49). Si hay alguna indicación de que la sustancia problema puede mostrar un comportamiento diferente (por ejemplo, una clara desviación respecto a la cinética de primer orden), debe llevarse a cabo un ensayo completo, a efectos de la normativa. Por lo general, el ensayo minimizado no se realiza durante un periodo más corto que el ensayo del FBC normal, pero comprende menos muestreos de peces (véase la justificación en el apéndice 6). No obstante, se puede reducir el periodo de depuración en caso de sustancias que se depuren rápidamente, para evitar que las concentraciones en los peces caigan por debajo del límite de detección/cuantificación antes del final del ensayo. Puede utilizarse un ensayo minimizado con exposición a una única concentración para determinar la necesidad de hacer un ensayo completo, y, si los datos resultantes utilizados para calcular las constantes de velocidad y el FBC son sólidos (véase el punto 93), podrá evitarse hacer el ensayo completo si el FBC resultante está lejos de los valores preocupantes a efectos normativos. En algunos casos, puede ser ventajoso aplicar el diseño del ensayo minimizado con más de una concentración de ensayo como ensayo preliminar para determinar si las estimaciones del FBC de una sustancia dependen de la concentración. Si las estimaciones del FBC a partir del ensayo minimizado indican dependencia de la concentración, será necesario ejecutar el ensayo completo. Si, a la vista de dicho ensayo minimizado, las estimaciones del FBC no dependen de la concentración pero los resultados no se consideran definitivos, cualquier ensayo completo que se realice posteriormente podrá limitarse a una única concentración, reduciendo así el uso de animales en comparación con un ensayo completo, con dos (o más) concentraciones de ensayo. Para poder acogerse al ensayo minimizado, las sustancias deben:
PAUTA DE MUESTREO PARA ESTUDIOS SEGÚN EL DISEÑO MINIMIZADO Muestreo de peces El muestreo de peces se reduce a cuatro puntos de muestreo:
Muestreo de agua Para el diseño minimizado, el agua se muestrea como en el estudio completo (véase el punto 54) o, al menos, cinco veces repartidas equitativamente a lo largo de la fase de absorción, y una vez por semana en la fase de depuración. Modificaciones del diseño Teniendo en cuenta las propiedades de la sustancia problema, las previsiones válidas deducidas de la QSAR y el objetivo específico del estudio, puede considerarse la oportunidad de introducir algunas modificaciones en el diseño del estudio:
Cálculos La justificación de este enfoque consiste en que el factor de bioconcentración puede determinarse en un ensayo completo bien como factor de bioconcentración en estado de equilibrio (FBCSS) calculando la relación entre la concentración de la sustancia problema en los tejidos del pez y la concentración de la sustancia problema en el agua, o bien calculando el factor de bioconcentración cinético (FBCK) como la relación entre la constante de la velocidad de absorción k 1 y la constante de la velocidad de depuración k 2. El FBCK es válido incluso si no se consigue ninguna concentración en estado de equilibrio de la sustancia durante la absorción, siempre que los procesos de absorción y de depuración sigan aproximadamente una cinética de primer orden. Como mínimo absoluto, para calcular las constantes de las velocidades de absorción y de depuración se necesitan dos puntos de datos, uno al final de la fase de absorción (es decir, al inicio de la fase de depuración) y otro al final (o cuando haya pasado una parte significativa) de la fase de depuración. Se recomienda el punto de muestreo intermedio para comprobar la cinética de la absorción y de la depuración (57). Para los cálculos, véanse los apéndices 5 y 6. Interpretación de los resultados Para apreciar la validez y el valor informativo del ensayo, hay que verificar que el periodo de depuración supera a la semivida. Asimismo, el FBCKm (FBC cinético obtenido en un ensayo minimizado) debe compararse con el valor del FBCSS minimizado (que es el FBCSS calculado al final de la fase de absorción, suponiendo que se ha alcanzado el estado de equilibrio. Esto solo puede suponerse, ya que el número de puntos de muestreo no es suficiente para demostrarlo). Si el FBCKm < FBCSS minimizado, debería darse preferencia al valor del FBCSS minimizado. Si el FBCKm es inferior al 70 % del FBCSS minimizado, los resultados no son válidos, y debe llevarse a cabo un ensayo completo. Si el ensayo minimizado proporciona un FBCKm en la región de algún valor preocupante a efectos normativos, debe llevarse a cabo un ensayo completo. Si el resultado está lejos de cualquier valor preocupante a efectos normativos (muy por encima o muy por debajo), puede no ser necesario realizar un ensayo completo, o puede realizarse un ensayo completo con una sola concentración si así lo exige el marco normativo pertinente. Si se ve que es necesario efectuar un ensayo completo después de un ensayo minimizado con una sola concentración, puede hacerse a una segunda concentración. Si los resultados son coherentes, se puede renunciar a efectuar un nuevo ensayo completo a una concentración diferente, ya que no se espera que la bioconcentración de la sustancia dependa de la concentración. En caso de que se haya llevado a cabo el ensayo minimizado con dos concentraciones, y los resultados muestren que no hay dependencia de la concentración, podrá realizarse el ensayo completo con una única concentración (punto 87). Informe del ensayo El informe del ensayo correspondiente al ensayo minimizado debe incluir toda la información solicitada para el ensayo completo (punto 81), con la excepción de lo que no sea posible elaborar (es decir, una curva que indique el tiempo para alcanzar el estado de equilibrio y el factor de bioconcentración en el estado de equilibrio; en lugar de este último debe darse el FBCSS del ensayo minimizado). Además, debe incluir también la justificación de la utilización del ensayo minimizado y el FBCKm resultante. C.13 — III: Ensayo de bioacumulación en peces con exposición alimentaria INTRODUCCIÓN El método descrito en la presente sección debe utilizarse con las sustancias a las que no sea aplicable la metodología de exposición acuática (por ejemplo, porque no puedan mantenerse concentraciones acuáticas estables y mensurables, o no puedan alcanzarse cargas corporales adecuadas en el plazo de 60 días de exposición; véanse las secciones anteriores sobre el método de la exposición acuática). Debe señalarse, sin embargo, que el criterio de valoración de este ensayo es un factor de biomagnificación (FBM) alimentario en lugar de un factor de bioconcentración (FBC) (58). En mayo de 2001 se presentó en la conferencia SETAC Europe celebrada en Madrid un nuevo método para el ensayo de bioacumulación de sustancias orgánicas poco hidrosolubles (36). Este trabajo se basaba en una serie de estudios de bioacumulación notificados en la bibliografía, utilizando un método de dosificación por adición de la sustancia a los alimentos (p. ej., (37)). A principios de 2004 se sometió a un grupo de trabajo sobre sustancias PBT de la UE un proyecto de protocolo (38) diseñado para medir el potencial de bioacumulación de sustancias orgánicas poco hidrosolubles a las que no era aplicable el método de bioconcentración por exposición acuática normal, junto con un documento justificativo de referencia (39). Otra justificación aportada para este método es que la exposición ambiental potencial a tales sustancias poco solubles (es decir, log K OW > 5) puede darse en gran parte a través de la dieta (40) (41) (42) (43) (44). Por esta razón, se hace referencia a los ensayos de exposición alimentaria en algunas normas publicadas sobre sustancias y mezclas químicas (59). No obstante, debe tenerse en cuenta que en el método descrito aquí la exposición a través de la fase acuosa se evita cuidadosamente, por lo que es imposible comparar directamente el valor del FBM obtenido mediante este método de ensayo con un valor del FBM obtenido a partir de un estudio de campo (en el que puede combinarse la exposición alimentaria con la acuática). Esta sección del presente método de ensayo se basa en dicho protocolo (38) y es un nuevo método que no figuraba en la versión anterior del método C.13. Este ensayo alternativo permite investigar directamente la vía de la exposición alimentaria en condiciones de laboratorio controladas. Los investigadores potenciales deben referirse a los puntos 1 a 14 del presente método de ensayo para conseguir información sobre cuándo puede ser preferible el ensayo de exposición alimentaria frente al ensayo de exposición acuática. Se expone información sobre las distintas consideraciones de las sustancias, la cual debe tenerse en cuenta antes de realizar el ensayo. El uso de sustancias problema radiomarcadas puede plantearse con consideraciones similares a las relativas al método de la exposición acuática (véanse los puntos 6 y 65). El método alimentario puede utilizarse para el ensayo de más de una sustancia en un único ensayo, siempre que se cumplan determinadas condiciones; estas se explican más detalladamente en el punto 112. En aras de la simplicidad, la metodología descrita aquí se refiere a un ensayo con una sola sustancia problema. El ensayo alimentario es similar al método de la exposición acuática en muchos aspectos, con la evidente excepción de la vía de exposición. Por lo tanto, muchos aspectos del método aquí descrito se solapan con el método de exposición acuática descrito en la sección anterior. Se hace referencia a los puntos pertinentes de la sección anterior en la medida de lo posible pero, para mantener la legibilidad y comprensión, es inevitable una cierta cantidad de repeticiones. PRINCIPIO DEL ENSAYO Pueden emplearse condiciones dinámicas o semiestáticas (véase el punto 4); se recomiendan las condiciones dinámicas para limitar la exposición potencial de la sustancia problema a través del agua como resultado de una eventual desorción de los alimentos enriquecidos o por las heces. El ensayo se divide en dos fases: absorción (alimentos enriquecidos con la sustancia problema) y depuración (alimentos puros, sin tratar) (véase el punto 16). Durante la fase de absorción, un grupo «de ensayo» de peces recibe diariamente una dieta fija de alimentos comerciales para peces de composición conocida, enriquecidos con la sustancia problema. Lo ideal es que los peces consuman todos los alimentos que se les ofrecen (véase el punto 141). A continuación, durante la fase de depuración se dan a los peces los alimentos comerciales puros, sin tratar. Como en el caso de la exposición acuática, pueden utilizarse varios grupos de ensayo con diferentes concentraciones de la sustancia problema en los alimentos en caso necesario, pero, para la mayoría de las sustancias problema orgánicas muy hidrófobas, es suficiente un solo grupo de ensayo (véanse los puntos 49 y 107). Si se utilizan condiciones semiestáticas, deben trasladarse los peces a un nuevo medio o a un nuevo recipiente de ensayo al final de la fase de absorción (en caso de que el medio o aparato utilizado en la fase de absorción se haya contaminado con la sustancia problema por lixiviación). Las concentraciones de la sustancia problema en los peces se miden durante las dos fases del ensayo. Además del grupo de peces a los que se dan los alimentos enriquecidos con la sustancia problema (grupo de ensayo), se somete a las mismas condiciones un grupo testigo de peces alimentados de forma idéntica, salvo que no se añade la sustancia problema a los alimentos comerciales para peces. Este grupo testigo permite cuantificar los niveles de fondo de la sustancia problema en los peces no expuestos y sirve de comparación en cuanto a los eventuales efectos adversos relacionados con el tratamiento observados en el grupo o grupos de ensayo (60). También permite la comparación de las constantes de la velocidad de crecimiento entre grupos para comprobar que se consumen cantidades similares de los alimentos ofrecidos (debe considerarse también la posibilidad de que haya diferencias de palatabilidad entre los alimentos como explicación de diferencias en las constantes de la velocidad de crecimiento; véase el punto 138). Es importante que durante las fases de absorción y de depuración se den a los grupos de ensayo y testigo alimentos nutricionalmente equivalentes. En general es suficiente que la duración de la fase de absorción sea de 7 a 14 días, según la experiencia adquirida en el desarrollo del método (38) (39). Este intervalo debe reducir al mínimo el coste de llevar a cabo el ensayo, con una exposición suficiente para la mayoría de las sustancias. No obstante, en algunos casos puede ampliarse la fase de absorción (punto 127). Durante la fase de absorción, la concentración de la sustancia en los peces puede no alcanzar el estado de equilibrio, por lo que el tratamiento de datos y los resultados de este método se basan generalmente en un análisis cinético de los residuos tisulares (Nota: Pueden aplicarse aquí ecuaciones para calcular el tiempo necesario para alcanzar el estado de equilibrio, como en el ensayo de exposición acuática; véase el apéndice 5). La fase de depuración comienza cuando se dan a los peces por primera vez alimentos sin enriquecer con la sustancia problema, y normalmente dura hasta 28 días o hasta que la sustancia problema deja de poder cuantificarse en el pez entero, si esta fecha es anterior. La fase de depuración puede reducirse o prolongarse más allá de 28 días, dependiendo de la evolución temporal de las concentraciones medidas del producto y de la talla de los peces. Este método permite la determinación de la semivida específica de la sustancia (t 1/2, a partir de la constante de la velocidad de depuración, k 2), la eficiencia de la asimilación (absorción intestinal; a), el factor de biomagnificación alimentario cinético (FBMK), el factor de biomagnificación alimentario cinético corregido según el crecimiento (FBMKg), y el factor de biomagnificación alimentario cinético corregido según el contenido lipídico (61) (FBMKL) (y/o el factor de biomagnificación alimentario cinético corregido según el crecimiento y el contenido lipídico, FBMKgL) correspondientes a la sustancia problema en los peces. Como con el método de exposición acuática, el aumento de la masa de los peces durante el ensayo se traducirá en una disminución de la concentración de la sustancia problema en los peces en crecimiento y, por tanto, el FBM (cinético) estará subestimado si no se corrige para tener en cuenta este fenómeno (véanse los puntos 162 y 163). Además, si se considera que se ha alcanzado el estado de equilibrio durante la fase de absorción, puede calcularse a título indicativo un FBM en estado de equilibrio. Hay enfoques disponibles que permiten calcular un factor de bioconcentración cinético (FBCK) a partir de los datos obtenidos en el estudio alimentario (p. ej., (44) (45) (46) (47) (48)). En el apéndice 8 se debaten las ventajas e inconvenientes de estos enfoques. El ensayo ha sido diseñado principalmente para sustancias orgánicas apolares poco solubles que siguen aproximadamente una cinética de primer orden en los procesos de absorción y depuración en los peces. En caso de que se someta a ensayo una sustancia que no siga aproximadamente una cinética de absorción y de depuración de primer orden, convendrá utilizar modelos más complejos (véanse las referencias en el apéndice 5) y buscar el consejo de un bioestadístico o de un técnico de farmacocinética. El FBM se determina normalmente mediante el análisis de la sustancia problema en el pez entero (en peso húmedo). Cuando sea pertinente para los objetivos del estudio, pueden muestrearse tejidos específicos (por ejemplo, músculos, hígado) si los peces se dividen en partes comestibles y no comestibles (véase el punto 21). Por otra parte, puede recurrirse a la retirada y al análisis aparte del tubo digestivo para determinar su contribución a las concentraciones en los peces enteros en los puntos de muestreo al final de la fase de absorción y cerca del comienzo de la fase de depuración, o como parte de un enfoque de balance de masas. Debe medirse el contenido lipídico de los peces enteros muestreados, de manera que las concentraciones puedan corregirse en función del contenido lipídico, teniendo en cuenta el contenido lipídico tanto de los peces como de los alimentos (véanse los puntos 56 y 57 y el apéndice 7). Debe medirse y registrarse el peso de los distintos peces muestreados, y relacionarse con la concentración del producto analizado en el pez correspondiente (por ejemplo, indicarse de acuerdo con un código único de identificación para cada pez muestreado), a efectos del cálculo del crecimiento que pueda producirse durante el ensayo. También debe medirse cuando sea posible la longitud total de los peces (62). Los datos de peso son también necesarios para la estimación del FBC a partir de los datos de depuración del ensayo alimentario. INFORMACIÓN SOBRE LA SUSTANCIA PROBLEMA Debe disponerse de la información sobre la sustancia problema que se describe en los puntos 3 y 22. No es generalmente necesario disponer de un método de análisis para las concentraciones de la sustancia problema en el agua; sí se necesitan métodos con la sensibilidad adecuada para medir las concentraciones en los alimentos para peces y en los tejidos de los peces. El método puede utilizarse para el ensayo de más de una sustancia en un único ensayo. No obstante, las sustancias problema deben ser compatibles entre sí, de manera que no interactúen ni cambien su identidad química tras añadirse a los alimentos para peces. El objetivo es que los resultados medidos para cada sustancia ensayada conjuntamente no difieran considerablemente de los resultados que se obtendrían si se hicieran ensayos aparte con cada una de las sustancias problema. Mediante unos trabajos de análisis preliminares debe establecerse que cada sustancia puede recuperarse de una muestra de alimentos a los que se han añadido varias sustancias y de una muestra de tejidos de peces con: i) recuperaciones elevadas (por ejemplo, > 85 % del contenido nominal) y ii) la sensibilidad necesaria para el ensayo. La dosis total de las sustancias sometidas a ensayo conjunto debe estar por debajo de la concentración combinada que pueda causar efectos tóxicos (véase el punto 51). Por otra parte, deben tomarse en consideración en el diseño experimental los posibles efectos adversos en los peces y el potencial de efectos interactivos (por ejemplo, efectos metabólicos) asociados al ensayo simultáneo de varias sustancias. Debe evitarse el ensayo simultáneo de sustancias ionizables. En términos de exposición, el método es también adecuado para mezclas complejas (véase el punto 13, aunque se aplicarán las mismas limitaciones analíticas que en cualquier otro método). VALIDEZ DEL ENSAYO Para que el ensayo sea válido han de darse las condiciones siguientes (véase el punto 24):
SUSTANCIAS DE REFERENCIA Si un laboratorio no ha realizado el ensayo antes o si se han producido cambios importantes (por ejemplo, cambio de cepa o del proveedor de los peces, especie de peces diferente, cambio importante del tamaño de los peces, alimentos para peces o método de enriquecimiento de la sustancia), se aconseja realizar un estudio de competencia técnica, utilizando una sustancia de referencia. La sustancia de referencia se utiliza fundamentalmente para determinar si la técnica del enriquecimiento alimentario es adecuada para garantizar un máximo de homogeneidad y de biodisponibilidad de las sustancias problema. Un ejemplo que se ha utilizado en el caso de las sustancias hidrófobas apolares es el hexaclorobenceno (HCB), pero debe considerarse recurrir a otras sustancias con datos fiables existentes sobre su absorción y biomagnificación, debido a las propiedades peligrosas del HCB (63). En caso de que se utilicen sustancias de referencia, debe recogerse en el informe de ensayo información básica sobre ellas, incluidos el nombre, la pureza, el número CAS, la estructura y datos sobre toxicidad (si se dispone de ellos), como en el caso de las sustancias problema (véanse los puntos 3 y 22). DESCRIPCIÓN DEL MÉTODO Equipo Deben utilizarse materiales y equipos como se describe en el método de la exposición acuática (punto 26). Debe utilizarse un sistema de ensayo dinámico o de renovación semiestática que proporcione un volumen suficiente de agua de dilución a los recipientes del ensayo. Deben registrarse los caudales. Agua Debe utilizarse agua de ensayo como se describe en el método de la exposición acuática (puntos 27-29). El medio de ensayo debe caracterizarse como se describe y su calidad debe permanecer constante durante el ensayo. El contenido natural en partículas y el carbono orgánico total deben ser lo más bajos posible (≤ 5 mg/l de partículas; ≤ 2 mg/l de carbono orgánico total) antes del inicio del ensayo. El COT debe determinarse solamente antes del ensayo en el marco de la caracterización del agua de ensayo (véase el punto 53). Alimentación Se recomienda utilizar alimentos para peces disponibles en el comercio (alimentos granulados que floten o que sedimenten lentamente) que estén caracterizados al menos en cuanto a su contenido de proteínas y de grasas. El alimento debe tener un granulado de tamaño uniforme para aumentar la eficiencia de la exposición alimentaria, es decir, los peces ingerirán más alimento en lugar de ingerir los gránulos más grandes y dejar perderse los más pequeños. Los gránulos deben ser de tamaño adecuado para el tamaño de los peces al principio del ensayo (por ejemplo, pueden emplearse gránulos de 0,6-0,85 mm de diámetro para peces de entre 3 y 7 cm de longitud total, y de 0,85-1,2 mm para peces de entre 6 y 12 cm de longitud total). El tamaño de los gránulos puede ajustarse según el crecimiento de los peces al inicio de la fase de depuración. En el apéndice 7 figura un ejemplo de composición adecuada de un alimento disponible en el comercio. Se han utilizado comúnmente en el desarrollo de este método dietas de ensayo con un contenido lipídico total de entre el 15 y el 20 % (p/p). Es posible que en ciertas regiones no se disponga de alimentos para peces con tan alta concentración lipídica. En estos casos, se pueden realizar estudios con una menor concentración lipídica en el alimento y, en caso necesario, se puede ajustar la dosis alimentaria de forma adecuada para mantener la salud de los peces (sobre la base de pruebas preliminares). El contenido lipídico total de las dietas del grupo de ensayo y del grupo testigo ha de medirse y registrarse antes del inicio del ensayo y al final de la fase de absorción. En el informe del estudio deben presentarse datos analíticos, facilitados por el proveedor comercial de los alimentos, sobre el contenido de nutrientes, humedad, fibra y ceniza y, si es posible, minerales y residuos de plaguicidas (por ejemplo, contaminantes prioritarios «normales»). Al añadir la sustancia problema al alimento, se hará todo lo posible para garantizar que todo el alimento de ensayo es homogéneo. La concentración de la sustancia problema en el alimento para el grupo de ensayo debe seleccionarse teniendo en cuenta la sensibilidad de la técnica analítica, la toxicidad de la sustancia problema (con su NOEC si se conoce) y los datos fisicoquímicos pertinentes. En caso de utilizarse una sustancia de referencia, esta debe incorporarse preferentemente a una concentración de alrededor del 10 % de la de la sustancia problema (o, en cualquier caso, tan baja como sea posible), en función de la sensibilidad del análisis (por ejemplo, para el hexaclorobenceno se ha considerado aceptable una concentración en el alimento de 1-100 μg/g; véase en la referencia (47) más información sobre las eficiencias de asimilación del HCB). La sustancia problema puede añadirse al alimento para peces de varias formas según sus características físicas y su solubilidad (véase en el apéndice 7 más información sobre los métodos de enriquecimiento):
En unos pocos casos, por ejemplo en los de las sustancias problema menos hidrófobas con mayor probabilidad de desorberse del alimento, puede ser necesario recubrir los gránulos de alimento, ya preparados, con una pequeña cantidad de aceite de maíz/pescado (véase el punto 142). En tales casos, el alimento testigo debe tratarse de la misma manera, y para la medición de los lípidos debe utilizarse el alimento preparado final. En caso de que se utilice una sustancia de referencia, los resultados obtenidos con ella deben ser comparables a los datos de los estudios de la bibliografía efectuados en condiciones similares con una dosis alimentaria comparable (véase el punto 45), y los parámetros específicos de la sustancia de referencia deben cumplir los criterios pertinentes del punto 113 (puntos 3.o, 4.o y 5.o). Si un aceite o disolvente sirve como vehículo para la sustancia problema, debe mezclarse una cantidad equivalente del mismo vehículo (excluida la sustancia problema) con el alimento del testigo con el fin de mantener la equivalencia con el alimento enriquecido. Es importante que, durante las fases de absorción y de depuración, se den a los grupos de ensayo y testigo alimentos nutricionalmente equivalentes. El alimento enriquecido debe conservarse en condiciones que preserven la estabilidad de la sustancia problema en la mezcla alimentaria (por ejemplo, refrigeración) y estas condiciones deben notificarse. Selección de la especie de peces Pueden utilizarse las especies de peces señaladas para la exposición acuática (veánse el punto 32 y el apéndice 3). Antes de la publicación del presente método de ensayo se han utilizado comúnmente la trucha arco iris (Oncorhynchus mykiss), la carpa (Cyprinus carpio) y el pez cabeza gorda (Pimephales promelas) en los estudios de bioacumulación alimentaria de sustancias orgánicas. Las especies de ensayo deben tener un comportamiento alimentario que dé lugar al consumo rápido de la ración alimentaria administrada, para garantizar que se reduce al mínimo cualquier factor que influya en la concentración de la sustancia problema en el alimento (por ejemplo, la lixiviación en el agua y la posibilidad de exposición acuática). Deben utilizarse peces en el intervalo recomendado de tamaño/peso (véase el apéndice 3). Los peces no deben ser tan pequeños como para que resulte difícil hacer análisis de cada individuo. Las especies sometidas a ensayo en una etapa de su vida de rápido crecimiento pueden complicar la interpretación de los datos, y unas elevadas velocidades de crecimiento pueden influir en el cálculo de la eficiencia de la asimilación (64). Preparación de los peces Los criterios de aceptación de aclimatación, mortalidad y enfermedad son los mismos que los del método de exposición acuática antes de realizar el ensayo (véanse los puntos 33-35). REALIZACIÓN DEL ENSAYO Trabajo preliminar y ensayo de determinación del intervalo Es necesario proceder a un trabajo analítico preliminar para demostrar la recuperación de la sustancia a partir de los tejidos de los peces y de los alimentos enriquecidos. No siempre es necesario efectuar un ensayo de determinación del intervalo para seleccionar una concentración adecuada en el alimento. A efectos de demostrar que no se observan efectos adversos y de evaluar la palatabilidad del alimento enriquecido, la sensibilidad del método analítico para los tejidos de los peces y los alimentos, y la selección de la dosis alimentaria y los intervalos de muestreo adecuados durante la fase de depuración, etc., podrán llevarse a cabo experimentos preliminares de alimentación, pero no serán obligatorios. Podría ser útil incluir un estudio preliminar para estimar el número de peces necesarios para el muestreo durante la fase de depuración. Esto puede dar lugar a una reducción significativa del número de peces utilizados, especialmente en el caso de las sustancias problema que sean particularmente susceptibles de metabolismo. Condiciones de exposición Duración de la fase de absorción Suele ser suficiente una fase de absorción de 7 a 14 días, durante la cual un grupo de peces recibe el alimento testigo y otro grupo de peces recibe el alimento enriquecido, en una ración diaria fija, dependiente de la especie estudiada y de las condiciones experimentales, por ejemplo, entre el 1 y el 2 % del peso corporal (peso húmedo) en el caso de la trucha arco iris. La dosis alimentaria debe seleccionarse de forma que se evite el crecimiento rápido y el amplio aumento del contenido lipídico. En caso necesario, podrá ampliarse la fase de absorción basándose en la experiencia práctica a partir de estudios anteriores o en el conocimiento de la absorción/depuración de la sustancia problema (o de algún análogo suyo) en los peces. El inicio del ensayo se define como el momento de la primera aportación de alimento enriquecido. Un día experimental va desde el momento de la alimentación hasta poco antes (por ejemplo, una hora) del momento del aporte siguiente de alimento. Así, el primer día experimental de la absorción empieza en el momento de la primera aportación de alimento enriquecido y termina poco antes de la segunda aportación de alimento enriquecido. En la práctica, la fase de absorción termina poco antes (por ejemplo, una hora) de la primera aportación de alimento no enriquecido, ya que los peces seguirán digiriendo el alimento enriquecido y absorbiendo la sustancia problema en ese intervalo de 24 horas. Es importante garantizar que se consigue un nivel suficientemente elevado (no tóxico) de carga corporal de la sustancia problema con respecto al método analítico, de modo que pueda medirse durante la fase de depuración una disminución de al menos un orden de magnitud. En casos especiales se puede ampliar la fase de absorción (hasta 28 días) con muestreo adicional para estudiar mejor la cinética de la absorción. Durante la fase de absorción es posible que la concentración en los peces no alcance el estado de equilibrio. Pueden aplicarse aquí, como en el ensayo de exposición acuática, ecuaciones para calcular el tiempo hasta alcanzar el estado de equilibrio, como indicación del tiempo probablemente necesario para conseguir unas concentraciones apreciables en los peces (véase el apéndice 5). En algunos casos puede saberse que la absorción de la sustancia en los peces durante 7-14 días será insuficiente para que, con la concentración alimentaria utilizada, se alcance una concentración suficientemente elevada en los peces que permita analizar una disminución de al menos un orden de magnitud durante la depuración, bien debido a la escasa sensibilidad analítica o a la baja eficiencia de la asimilación. En tales casos puede resultar conveniente ampliar la fase inicial de alimentación a más de 14 días o, especialmente cuando se trate de sustancias altamente metabolizables, debe considerarse una concentración alimentaria superior. Sin embargo, debe tenerse cuidado para mantener la carga corporal durante la absorción por debajo de la concentración sin efecto crónica (estimada) (NOEC) en los tejidos de los peces (véase el punto 138). Duración de la fase de depuración La depuración normalmente dura hasta 28 días, a contar desde que los peces del grupo de ensayo reciben el alimento puro, sin tratar, después de la fase de absorción. La depuración comienza con la primera aportación de alimento no enriquecido, y no inmediatamente después de la última aportación de alimento enriquecido, ya que los peces siguen digiriendo el alimento y absorbiendo la sustancia problema en ese intervalo de 24 horas, como se indica en el punto 126. Por lo tanto, la primera muestra de la fase de depuración se tomará poco antes de la segunda aportación de alimento no enriquecido. Este periodo de depuración está diseñado para recoger las sustancias con semivida potencial de hasta 14 días, la cual es coherente con la de las sustancias bioacumulativas (65), por lo que el plazo de 28 días comprende dos semividas de dichas sustancias. En los casos de sustancias muy bioacumulativas puede ser ventajoso ampliar la fase de depuración (si así lo indican las pruebas preliminares). Si una sustancia se depura muy lentamente de manera que no puede determinarse en la fase de depuración una semivida exacta, la información puede seguir siendo suficiente a efectos de evaluación para indicar un nivel elevado de bioacumulación. A la inversa, si una sustancia se depura tan deprisa que no pueden obtenerse una concentración fiable a tiempo cero (concentración al final de la absorción o principio de la depuración, C 0,d) ni la k 2, puede hacerse una estimación prudente de k 2 (véase el apéndice 7). Si los análisis de los peces a intervalos anteriores (por ejemplo, a los 7 o 14 días) muestran que la sustancia se ha depurado por debajo de los niveles de cuantificación antes de que se complete el periodo de 28 días, podrá abandonarse el muestreo posterior y darse por concluido el ensayo. En algunos casos puede no haberse producido una absorción mensurable de la sustancia problema al final de la fase de absorción (o con la segunda muestra de depuración). Si puede demostrarse que: i) se cumplen los criterios de validez del punto 113, y ii) la falta de absorción no se debe a alguna otra deficiencia del ensayo (por ejemplo, insuficiencia de la duración de la fase de absorción, deficiencias de la técnica de enriquecimiento del alimento que reducen la biodisponibilidad, falta de sensibilidad del método analítico, falta de consumo del alimento por los peces, etc.), quizá sea posible terminar el estudio sin necesidad de volver a realizarlo con una duración mayor de la absorción. Si los trabajos preliminares han indicado que podría darse esta situación, se podrá recomendar el análisis de las heces, si es posible, para buscar en ellas la sustancia problema no digerida como parte de un enfoque de «balance de masas». Número de peces del ensayo De forma análoga a la del ensayo de exposición acuática, deben seleccionarse peces de peso y longitud similares, de forma que los peces más pequeños no tengan un peso inferior a dos tercios del peso de los mayores (véanse los puntos 40-42). El número total de peces para el estudio debe seleccionarse en función de la pauta de muestreo (un mínimo de una muestra al final de la fase de absorción y de cuatro a seis muestras durante la fase de depuración, pero según la duración de las fases), teniendo en cuenta la sensibilidad de la técnica analítica, la concentración que pueda lograrse al final de la fase de absorción (sobre la base de datos anteriores) y la duración de la depuración (si los datos previos permiten la estimación). En cada punto de muestreo deben tomarse de cinco a diez peces, y se miden los parámetros de crecimiento (peso y longitud total) antes de proceder al análisis del producto o de los lípidos. Debido a la variabilidad intrínseca del tamaño, de la velocidad de crecimiento y de la fisiología de los peces, y a las posibles variaciones de la cantidad de alimento aportado que consume cada pez, en cada punto de muestreo deben tomarse al menos cinco peces del grupo de ensayo y cinco del grupo testigo para determinar de manera adecuada la concentración media y su variabilidad. La variabilidad entre los peces utilizados puede contribuir más a la variabilidad incontrolada general del ensayo que la variabilidad intrínseca de los métodos analíticos empleados, lo que justifica el uso de hasta diez peces por punto de muestreo en algunos casos. Sin embargo, si al inicio de la depuración no son mensurables las concentraciones de fondo de la sustancia problema en los peces testigo, el análisis químico de solo dos o tres peces testigo en el intervalo de muestreo final puede ser suficiente siempre que siga tomándose el resto de los peces testigo en todos los puntos de muestreo para medir su peso y longitud total (de forma que se muestree el mismo número a partir de los grupos de ensayo y testigo en cuanto al crecimiento). Los peces deben almacenarse y pesarse por separado (aun cuando resulte necesario que los resultados de la muestra se combinen posteriormente), y hay que medir su longitud total. Para un ensayo normal con, por ejemplo, una duración de depuración de 28 días, que incluya cinco muestras de depuración, esto significa un total de 59-120 peces tomados de los grupos de ensayo y 50-110 peces de los grupos testigo, suponiendo que la técnica analítica de la sustancia permite el análisis del contenido lipídico en el mismo pez. Si el análisis de los lípidos no puede realizarse con el mismo pez que el análisis del producto, y tampoco es factible utilizar peces testigo solo para el análisis de los lípidos (véase el punto 56), serán necesarios quince peces más (tres de la población inicial al comienzo del ensayo, tres de cada uno de los grupos testigo y de ensayo al inicio de la depuración y tres de cada uno de los grupos testigo y de ensayo al final del experimento). Un ejemplo de pauta de muestreo con el número de peces figura en el apéndice 4. Carga Deben utilizarse elevadas relaciones agua/peces similares a las del método de exposición acuática (puntos 43 y 44). Aunque las tasas de carga peces/agua no tienen efecto sobre las concentraciones de exposición en este ensayo, se recomienda una tasa de carga de 0,1-1,0 g de peces (peso húmedo) por litro de agua al día, para mantener unas concentraciones de oxígeno disuelto adecuadas y reducir al mínimo la agresión para los organismos de ensayo. Alimento del ensayo y su aportación Durante el periodo de aclimatación, debe darse a los peces un alimento adecuado según lo descrito anteriormente (punto 117). Si el ensayo se efectúa en condiciones dinámicas, el flujo debe suspenderse mientras se alimentan los peces. Durante el ensayo, el alimento para el grupo de ensayo debe atenerse al descrito anteriormente (puntos 116 a 121). Además de la consideración de factores específicos de la sustancia, de la sensibilidad analítica, de la concentración prevista en el alimento en condiciones ambientales y los niveles de toxicidad crónica / carga corporal, para la selección de la concentración objetivo en el alimento enriquecido debe tenerse en cuenta la palatabilidad del alimento (de modo que los peces no eviten comerlo). La concentración nominal de la sustancia problema en el alimento enriquecido debe documentarse en el informe. Según la experiencia, unas concentraciones en el alimento enriquecido en el margen de 1-1 000 μg/g proporcionan un intervalo de trabajo práctico para sustancias problema que no presentan un mecanismo tóxico específico. En el caso de las sustancias que actúen a través de un mecanismo inespecífico, los contenidos de residuos tisulares no deben superar los 5 μmol/g de lípidos, ya que los residuos por encima de este nivel pueden presentar efectos crónicos (19) (48) (50) (66). En el caso de las demás sustancias, debe velarse por que no se produzcan efectos adversos debidos a la exposición acumulada (véase el punto 127). Esto es especialmente importante si se somete a ensayo simultáneamente más de una sustancia (véase el punto 112). La cantidad adecuada de la sustancia problema puede añadirse al alimento para peces de una de las tres maneras descritas en el punto 119 y en el apéndice 7. Los métodos y procedimientos para el enriquecimiento de los alimentos deben indicarse en el informe. A los peces testigo se les da alimento sin tratar, que contenga una cantidad equivalente de vehículo oleoso no enriquecido si este se ha utilizado en el alimento enriquecido para la fase de absorción, o que haya sido tratado con disolvente «puro» si se ha utilizado algún disolvente como vehículo para la preparación del alimento del grupo de ensayo. En los alimentos tratado y sin tratar debe determinarse por análisis, al menos por triplicado, la concentración de la sustancia problema antes del inicio y al final de la fase de absorción. Después de la exposición al alimento tratado (fase de absorción), los peces (ambos grupos) reciben el alimento sin tratar (fase de depuración). Se alimenta a los peces con una ración fija (en función de la especie; por ejemplo, aproximadamente el 1-2 % del peso corporal húmedo por día en el caso de la trucha arco iris). La dosis alimentaria debe seleccionarse de forma que se evite el crecimiento rápido y el amplio aumento del contenido lipídico. Debe registrarse la dosis alimentaria exacta utilizada durante el experimento. La alimentación inicial debe basarse en las mediciones de peso programadas de la población inicial justo antes del comienzo del ensayo. La cantidad de alimento debe ajustarse sobre la base del peso húmedo de los peces tomados en cada punto de muestreo para tener en cuenta el crecimiento durante el experimento. Los pesos y longitudes de los peces presentes en los recipientes testigo y de ensayo pueden calcularse a partir del peso y de la longitud total de los peces utilizados en cada toma de muestras; no deben pesarse ni medirse los peces que permanecen en los recipientes testigo y de ensayo. Es importante mantener a lo largo de todo el experimento la misma dosis alimentaria fijada. Deben observarse los peces en el momento de darles el alimento para asegurarse de que consumen visiblemente todo el alimento que se les ofrece, a fin de garantizar que se utilizan en los cálculos las tasas de ingestión correctas. Debe considerarse la realización de experimentos preliminares de alimentación o la experiencia anterior, a la hora de elegir una dosis alimentaria que garantice que se consume todo el alimento aportado una vez al día. En caso de que siempre quede alimento sin comer, puede ser recomendable repartir la dosis con una aportación extra de alimentación cada día experimental (por ejemplo, sustituir la aportación de la dosis una vez al día por la aportación de la mitad de la dosis dos veces al día). Si es necesaria, la segunda aportación debe hacerse a un momento fijado y programado de forma que pase el mayor tiempo posible antes de la toma de muestras de peces (por ejemplo, el momento de la segunda aportación se fija en la primera mitad de un día experimental). Aunque en general los peces consumen el alimento rápidamente, es importante garantizar que la sustancia permanece adsorbida al alimento. Debe procurarse evitar la dispersión de la sustancia problema en el agua, a partir del alimento que haría que los peces quedaran expuestos a concentraciones acuáticas de la sustancia problema, además de la exposición alimentaria. Esto puede lograrse retirando todo el alimento sin comer (y las heces) de los recipientes testigo y de ensayo en el plazo de una hora tras la aportación del alimento, y preferentemente en el plazo de 30 minutos. Además, puede utilizarse un sistema en el que el agua se limpie continuamente a través de un filtro de carbón activo para absorber cualquier contaminante «disuelto». Los sistemas dinámicos pueden contribuir a eliminar rápidamente las partículas de alimento y las sustancias disueltas (67). En algunos casos, una ligera modificación de la técnica de preparación del alimento enriquecido puede contribuir a aliviar este problema (véase el punto 119). Luz y temperatura Como en el método de la exposición acuática (véase el punto 48), se recomienda un fotoperiodo de 12 a 16 horas y una temperatura apropiada (± 2 °C) para la especie de ensayo empleada (vease el apéndice 3). Se definirán y documentarán el tipo y las características de la iluminación. Testigos Se utilizará un grupo testigo con peces alimentados con la misma ración que el grupo de ensayo, pero sin que esté presente en el alimento la sustancia problema. Si se ha utilizado un aceite o un disolvente como vehículo para enriquecer el alimento del grupo de ensayo, el grupo testigo debe tratarse exactamente igual, pero con la ausencia de la sustancia problema, de manera que los alimentos del grupo testigo y del grupo de ensayo sean equivalentes (véanse los puntos 121 y 139). Frecuencia de la medición de la calidad del agua Las condiciones descritas en el método de exposición acuática se aplican también en este caso, salvo que el COT deberá determinarse únicamente antes del ensayo como parte de la caracterización de agua de ensayo (véase el punto 53). Muestreo y análisis de los peces y del alimento Análisis de las muestras de alimento Deben analizarse muestras de los alimentos de ensayo y testigo, al menos por triplicado, para determinar la concentración de la sustancia problema y el contenido lipídico, al menos antes del inicio y al final de la fase de absorción. Deben incluirse en el informe los métodos de análisis y los procedimientos para garantizar la homogeneidad del alimento. Las muestras deben analizarse en cuanto a su contenido de sustancia problema siguiendo el método establecido y validado. Debe efectuarse un trabajo previo al estudio para establecer el límite de cuantificación, el porcentaje de recuperación, las interferencias y la variabilidad analítica en la matriz de la muestra prevista. En caso de que se someta a ensayo un material radiomarcado, deben tenerse en cuenta consideraciones similares a las del método de exposición acuática, sustituyendo el análisis del agua por el análisis del alimento (véase el punto 65). Análisis de los peces En cada punto de muestreo de peces, se toman 5-10 ejemplares de los grupos de ensayo y testigo (en algunos casos puede reducirse el número de peces testigo; véase el punto 134). Los muestreos deben tener lugar al mismo tiempo cada día experimental (en relación con el momento de la aportación de alimento) y deben programarse de manera que se reduzca al mínimo la probabilidad de que queden restos de alimento en el intestino durante la fase de absorción y la primera parte de la fase de depuración, con el fin de evitar las contribuciones espurias a las concentraciones totales de la sustancia problema (es decir, los peces muestreados deben retirarse al final de un día experimental, teniendo en cuenta que el día experimental comienza en el momento de la aportación de alimento y termina en el momento de la comida siguiente, unas 24 horas después. La depuración comienza con la primera aportación de alimento sin enriquecer; véase el punto 128). Es importante la primera muestra de la fase de depuración (tomada poco antes de la segunda aportación de alimento sin enriquecer), ya que la extrapolación de esta medición a un día antes se utiliza para calcular la concentración al tiempo cero (C 0,d, concentración en los peces al final de la absorción / inicio de la depuración). Con carácter opcional, puede extraerse el tubo digestivo de los peces y analizarse por separado al final de la absorción y a los días 1 y 3 de la depuración. En cada toma de muestras, deben retirarse peces de ambos recipientes de ensayo y tratarse de la misma forma que se describe en el método de exposición acuática (véanse los puntos 61-63). Se miden las concentraciones de la sustancia problema en los peces enteros (peso húmedo), al menos al final de la fase de absorción y durante la fase de depuración, tanto en los grupos de ensayo como en los testigos. Durante la fase de depuración, se recomienda utilizar de cuatro a seis puntos de muestreo (por ejemplo, 1, 3, 7, 14 y 28 días). Después de 1-3 días de absorción puede incluirse, de forma opcional, un punto de muestreo adicional para estimar la eficiencia de la asimilación por los peces en la fase lineal de la absorción, todavía cerca del inicio del periodo de exposición. Hay dos desviaciones principales respecto a la pauta prevista: i) si se acude a una fase de absorción ampliada con fines de investigación de la cinética de la absorción, se dispondrán más puntos de muestreo durante la fase de absorción y así tendrán que incluirse más peces (véase el punto 126); ii) si el estudio ha concluido al final de la fase de absorción porque no ha habido absorción mensurable (véase el punto 131). Deben pesarse por separado los distintos peces muestreados (y se debe medir su longitud total) a fin de poder determinar las constantes de la velocidad de crecimiento. Las concentraciones de la sustancia en tejidos específicos de los peces (partes comestibles y no comestibles) también pueden medirse al final de la absorción y a tiempos seleccionados de la depuración. En caso de que se someta a ensayo un material radiomarcado, deben tenerse en cuenta consideraciones similares a las del método de exposición acuática, sustituyendo el análisis del agua por el análisis del alimento (véase el punto 65). En cuanto a la utilización periódica de una sustancia de referencia (véase el punto 25), es preferible medir las concentraciones en el grupo de ensayo al final de la absorción y a todos los tiempos de depuración especificados para la sustancia problema (peces enteros); en el grupo testigo solo es preciso analizar las concentraciones al final de la absorción (peces enteros). En determinadas circunstancias (por ejemplo, si son incompatibles las técnicas de análisis de la sustancia problema y de la sustancia de referencia, de manera que sería necesario contar con más peces para seguir la pauta de muestreo), puede aplicarse el planteamiento siguiente para minimizar el número de peces adicionales necesarios. Las concentraciones de la sustancia de referencia se miden durante la depuración solo en los días 1, 3 y en dos puntos más de muestreo, seleccionados de forma que puedan hacerse estimaciones fiables de la concentración a tiempo cero (C 0,d) y de k 2 de la sustancia de referencia. En la medida de lo posible, debe determinarse el contenido lipídico de los distintos peces en cada punto de muestreo o, como mínimo, al inicio y al final de la fase de absorción y al final de la fase de depuración (véanse los puntos 56 y 57). Dependiendo del método analítico (véanse el punto 67 y el apéndice 4), quizás sea posible usar el mismo pez para la determinación tanto del contenido lipídico como de la concentración de la sustancia problema. Esto es preferible a efectos de reducir al mínimo el número de peces. No obstante, si esto no es posible, puede utilizarse el mismo enfoque descrito en el método de exposición acuática (véase el punto 56 en relación con estas opciones alternativas de medición de los lípidos). El método utilizado para cuantificar el contenido lipídico debe documentarse en el informe. Calidad del método analítico Se deben realizar controles experimentales para garantizar la especificidad, la exactitud, la precisión y la reproducibilidad de la técnica analítica específica de la sustancia, así como la recuperación de la sustancia problema tanto del alimento como de los peces. Medición del crecimiento de los peces Al comienzo del ensayo debe pesarse una muestra de peces de la población inicial, y debe medirse su longitud total. Estos peces deben muestrearse poco antes de la primera aportación de alimento enriquecido (p. ej., una hora), y asignarse al día experimental 0. El número de peces de esta muestra debe ser al menos igual que el de las muestras durante el ensayo. Algunos de estos peces pueden ser los mismos utilizados para el análisis de lípidos antes del comienzo de la fase de absorción (véase el punto 153). En cada punto de muestreo se pesan primero los peces y se mide su longitud. Respecto a cada pez, deben relacionarse el peso y la longitud medidos con la concentración del producto analizado (y el contenido lipídico, si procede), por ejemplo utilizando un código único de identificación para cada pez muestreado. Las mediciones de estos peces muestreados se pueden utilizar para estimar el peso y la longitud de los peces que quedan en los recipientes de ensayo y de testigo. Evaluación experimental Han de realizarse diariamente observaciones de la mortalidad, que se deben registrar. Deben hacerse otras observaciones de efectos adversos, por ejemplo sobre comportamiento o pigmentación anormales, y se registrarán también. Los peces se consideran muertos si no existe ningún movimiento respiratorio y no puede detectarse ninguna reacción a un estímulo mecánico ligero. Los peces muertos o claramente moribundos deben eliminarse. DATOS E INFORME Tratamiento de los resultados Los resultados de los ensayos se utilizan para calcular la constante de la velocidad de depuración (k 2) en función del peso total húmedo de los peces. Se calcula la constante de la velocidad de crecimiento, k g, basada en el aumento medio del peso de los peces, y se utiliza para obtener la constante de la velocidad de depuración corregida según el crecimiento, k 2g, si procede. Además deben indicarse la eficiencia de la asimilación (a; absorción intestinal), el factor de biomagnificación cinético (FBMK) (en caso necesario, corregido según el crecimiento, BMFKg), su valor corregido según el contenido lipídico (FBMKL o BMFKgL, si se corrige según la dilución por el crecimiento) y la dosis alimentaria. Asimismo, si puede estimarse el tiempo necesario para alcanzar el estado de equilibrio en la fase de absorción (p. ej., tiempo al 95 % del estado de equilibrio o t 95 = 3,0/k 2), podrá incluirse una estimación del FBM en el estado de equilibrio (FBMSS) (véanse los puntos 105 y 106, y el apéndice 5) si el valor de t 95 indica que es posible haber alcanzado las condiciones del estado de equilibrio. Debe aplicarse a este FBMSS la misma corrección según el contenido lipídico que al FBM derivado cinéticamente (FBMK) para dar un valor corregido según el contenido lipídico, FBMSSL (téngase en cuenta que no se dispone de ningún procedimiento reconocido para corregir un FBM en estado de equilibrio según la dilución por el crecimiento). En el apéndice 7 se encuentran fórmulas y ejemplos de cálculos. Hay enfoques disponibles que permiten calcular un factor de bioconcentración cinético (FBCK) a partir de los datos obtenidos en el estudio alimentario. Este aspecto se trata en el apéndice 8. Datos de peso/longitud de los peces Los pesos húmedos y las longitudes de los distintos peces correspondientes a todos los puntos temporales se tabulan por separado para los grupos de ensayo y de testigo, en todos los días de muestreo durante la fase de absorción (población inicial para el comienzo de la absorción; grupo de ensayo y grupo testigo para el final de la absorción y, en su caso, la fase primera (por ejemplo, días 1-3 de absorción) y la fase de depuración (p. ej., días 1, 2, 4, 7, 14, 28, para los grupos de ensayo y testigo). El peso es la forma más adecuada de medir el crecimiento a efectos de corrección de la dilución debida al crecimiento. Véase más adelante (puntos 162 y 163) y el apéndice 5 sobre el método o métodos utilizados para corregir los datos según la dilución por el crecimiento. Datos de la concentración de la sustancia problema en los peces Respecto a cada momento de muestreo, se recogen en cuadros las mediciones de residuos de la sustancia problema en cada pez (o en muestras conjuntas de peces si no es posible hacer la medición en cada pez), expresadas en términos de concentración en peso húmedo (p/p), tanto de los peces de ensayo como de los testigos. Si se han realizado análisis de lípidos en cada pez muestreado, pueden obtenerse y recogerse en cuadros las concentraciones individuales corregidas según el contenido lipídico, expresadas en concentración en lípidos (p/p lípidos).
Velocidad de depuración y factor de biomagnificación Para calcular el factor de biomagnificación a partir de los datos, en primer lugar debe obtenerse la eficiencia de la asimilación (absorción de la sustancia problema a través del intestino, α). Con este objetivo debe utilizarse la ecuación A7.1 del apéndice 7, para lo que han de conocerse la concentración derivada en los peces al tiempo cero de la fase de depuración (C 0,d), la constante de la velocidad de depuración (general) (k 2), la concentración en el alimento (C food), la constante de velocidad de ingestión del alimento (I) y la duración de la fase de absorción (t). La pendiente y la ordenada en el origen de la relación lineal entre el ln (concentración) y el tiempo de depuración se comunican como la constante de la velocidad de depuración general (k 2 = pendiente) y la concentración a tiempo cero (C 0,d = eordenada en origen), como arriba. Los valores derivados deben comprobarse en cuanto a su verosimilitud biológica (por ejemplo, la eficiencia de la asimilación como fracción no es superior a 1). (I) se calcula dividiendo la masa de alimentos por la masa de peces alimentados cada día (si la cantidad de alimento que se les da es el 2 % del peso corporal, (I) será 0,02). No obstante, puede ser necesario ajustar la dosis alimentaria utilizada en el cálculo para tener en cuenta el crecimiento de los peces (esto puede hacerse utilizando la constante conocida de la velocidad de crecimiento para estimar el peso de los peces en cada punto temporal durante la fase de absorción; véase el apéndice 7). En los casos en que no pueden derivarse k 2 y C 0,d debido a que, por ejemplo, las concentraciones caen por debajo del límite de detección en la segunda muestra de la fase de depuración, puede hacerse una estimación prudente de k 2 y establecerse un «límite superior» del FBMk (véase el apéndice 7). Una vez obtenida la eficiencia de la asimilación (α), puede calcularse el factor de biomagnificación multiplicando α por la constante de la velocidad de ingestión (I) y dividiendo por la constante de la velocidad de depuración (general) (k 2). El factor de biomagnificación corregido según el crecimiento se calcula del mismo modo pero utilizando la constante de la velocidad de depuración corregida según el crecimiento (k 2g); véanse los puntos 162 y 163. Puede obtenerse una estimación alternativa de la eficiencia de la asimilación si se ha realizado el análisis de tejidos de los peces muestreados en la primera etapa, lineal, de la fase de absorción; véanse el punto 151 y el apéndice 7. Este valor representa una estimación independiente de la eficiencia de la asimilación en el caso de un organismo principalmente no expuesto (es decir, los peces cerca del comienzo de la fase de absorción). Para obtener el FBM se suele utilizar la eficiencia de la asimilación estimada a partir de los datos de depuración. Corrección según el contenido lipídico y corrección según la dilución debida al crecimiento El crecimiento de los peces durante la fase de depuración puede reducir las concentraciones medidas del producto en los peces, con el resultado de que la constante de la velocidad de depuración general (k2 ) sea mayor que la que se derivaría únicamente de los procesos de eliminación (por ejemplo, metabolismo, excreción) (véase el punto 72). Los contenidos lipídicos de los peces del ensayo (que están estrechamente relacionados con la bioacumulación de sustancias hidrófobas) y los contenidos lipídicos de los alimentos pueden variar bastante en la práctica, de tal manera que su corrección es necesaria para presentar los factores de biomagnificación de manera significativa. Debe corregirse el factor de biomagnificación según la dilución debida al crecimiento (como el FBC cinético en el método de exposición acuática) y según el contenido lipídico de los alimentos con respecto al de los peces (factor de corrección por los lípidos). Las ecuaciones y ejemplos de estos cálculos figuran en el apéndice 5 y en el apéndice 7, respectivamente. Para corregir según la dilución debida al crecimiento, debe calcularse la constante de la velocidad de depuración corregida según el crecimiento (k 2g) (véanse las ecuaciones en el apéndice 5). Esta constante de la velocidad de depuración corregida según el crecimiento (k 2g) se utiliza a continuación para calcular el factor de biomagnificación corregido según el crecimiento, como en el punto 73. En algunos casos es imposible aplicar este enfoque. Un enfoque alternativo que elude la necesidad de corregir según la dilución debida al crecimiento implica la utilización de datos de depuración de masa de sustancia problema por pez (peces enteros) en lugar de los habituales datos de masa de sustancia problema por unidad de masa de peces (concentración). Esto puede conseguirse fácilmente, ya que los ensayos según el presente método deben vincular las concentraciones tisulares registradas con los pesos de cada pez. El sencillo procedimiento correspondiente se describe en el apéndice 5. Obsérvese que sigue siendo necesario estimar y comunicar el valor de k 2 aunque se utilice este enfoque alternativo. Para corregir según el contenido lipídico de los peces y de los alimentos cuando no se ha realizado el análisis de los lípidos en todos los peces muestreados, se derivan las fracciones lipídicas medias (p/p) en los peces y en los alimentos (68). El factor de corrección según el contenido lipídico (Lc ) se calcula entonces dividiendo la fracción lipídica media de los peces por la fracción lipídica media de los alimentos. El factor de biomagnificación, corregido según el crecimiento o no, en función del caso, se divide por el factor de corrección según el contenido lipídico para calcular el factor de biomagnificación corregido según el contenido lipídico. Si se han llevado a cabo análisis del producto y de los lípidos con los mismos peces en cada punto de muestreo, los datos tisulares corregidos según el contenido lipídico relativos a cada pez pueden utilizarse para calcular directamente un FBM corregido según el contenido lipídico (véase (37)). La gráfica de los datos de la concentración corregidos según el contenido lipídico proporciona C 0,d referida al contenido lipídico y k 2. Entonces puede procederse al análisis mtemático utilizando las mismas ecuaciones del apéndice 7, pero la eficiencia de la asimilación (a) se calcula utilizando la constante de la velocidad de ingestión de alimentos normalizada según el contenido lipídico (I lipid) y la concentración alimentaria referida al contenido lipídico (C food-lipid). Los parámetros corregidos según el contenido lipídico se utilizan a continuación de forma similar para calcular el FBM (obsérvese que la corrección de la constante de la velocidad de crecimiento debe aplicarse también a la fracción lipídica, en lugar de al peso húmedo de los peces, para calcular el FBMKgL corregido según el contenido lipídico y el crecimiento). Interpretación de los resultados El crecimiento medio tanto en los grupos de ensayo como en los testigos no debe ser, en principio, significativamente diferente, a fin de excluir los efectos tóxicos. Las constantes de la velocidad de crecimiento o las curvas de crecimiento de los dos grupos deben compararse siguiendo un procedimiento adecuado (69). Informe del ensayo Después de terminar el estudio, se preparará un informe final que contenga la información sobre la sustancia problema, la especie de ensayo y las condiciones del ensayo que figuran en el punto 81 (como en el caso del método de la exposición acuática). Además se exigirá la siguiente información:
BIBLIOGRAFÍA
Apéndice 1 DEFINICIONES Y UNIDADES
BIBLIOGRAFÍA
Apéndice 2 CARACTERÍSTICAS QUÍMICAS DE UN AGUA DE DILUCIÓN ACEPTABLE
Apéndice 3 ESPECIES DE PECES RECOMENDADAS PARA EL ENSAYO
Se han utilizado menos ampliamente distintas especies marinas y de estuario como, por ejemplo:
Los peces de agua dulce enumerados en el cuadro anterior son fáciles de criar o están fácilmente disponibles a lo largo del año, mientras que la disponibilidad de las especies marinas o de estuario se limita en parte a los países correspondientes. Pueden reproducirse y desarrollarse en explotaciones piscícolas o en laboratorio, en condiciones donde las enfermedades y los parásitos están bajo control; los animales utilizados serán, pues, sanos y de origen conocido. Estos peces se encuentran en muchas partes del mundo. BIBLIOGRAFÍA
Apéndice 4 PAUTAS DE MUESTREO PARA ENSAYOS CON EXPOSICIÓN ALIMENTARIA Y ACUÁTICA 1. Ejemplo teórico de pauta de muestreo para un ensayo completo de bioconcentración con exposición acuática de una sustancia con log Kow = 4.
2. Ejemplo teórico de pauta de muestreo para un ensayo de bioacumulación alimentaria de la sustancia tras una fase de absorción de 10 días y una fase de depuración de 42 días.
Nota sobre los momentos de las fases y del muestreo: La fase de absorción comienza con el primer aporte del alimento enriquecido. Un día experimental va desde un aporte de alimento hasta poco antes del siguiente, 24 horas después. El primer muestreo (1 en el cuadro) debe efectuarse poco antes del primer aporte de alimento (por ejemplo, una hora). Lo ideal es que el muestreo durante un estudio se lleve a cabo poco antes del aporte de alimento del día siguiente (es decir, unas 23 horas después del aporte de alimento del día de la muestra). La fase de absorción termina poco antes del primer aporte de alimento no enriquecido, cuando comienza la fase de depuración (es probable que los peces del grupo de ensayo estén aún digiriendo alimento enriquecido en el intervalo de 24 horas después del último aporte de alimento enriquecido). Esto significa que la muestra del final de la absorción debe tomarse poco antes del primer aporte de alimento no enriquecido y que la primera muestra de la fase de depuración debe tomarse unas 23 horas después del primer aporte de alimento no enriquecido. Apéndice 5 CÁLCULOS GENERALES
1. INTRODUCCIÓN El modelo general de bioacumulación acuática de los peces puede describirse en términos de procesos de absorción y de pérdida, ignorando la absorción con el alimento. La ecuación diferencial (dC f/dt) que describe la velocidad del cambio de la concentración en los peces (mg·kg– 1·día– 1) viene dada por (1):
Donde:
Para las sustancias bioacumulativas, cabe esperar que una media ponderada en función del tiempo (TWA) sea la concentración en el agua más importante desde el punto de vista de la exposición (Cw ) dentro del margen permitido de fluctuación (véase el punto 24). Se recomienda calcular una concentración TWA en el agua, de acuerdo con el procedimiento establecido en el apéndice 6 del método de ensayo C.20 (2). Conviene señalar que es conveniente utilizar el logaritmo natural de la concentración en el agua cuando se espera una disminución exponencial entre los periodos de renovación, por ejemplo en un diseño de ensayo semiestático. En un sistema dinámico puede que no sea necesario recurrir a los logaritmos naturales de las concentraciones de exposición. En caso de que se obtengan las concentraciones TWA en el agua, deben notificarse y utilizarse en los cálculos posteriores. En un ensayo normal de FBC en peces, la absorción y la depuración de ensayo pueden describirse en términos de dos procesos de cinética de primer orden.
En el estado de equilibrio, suponiendo que el crecimiento y el metabolismo son despreciables (es decir, que los valores de k g y k m no pueden distinguirse de cero), la velocidad de absorción es igual a la velocidad de depuración, por lo que combinando la ecuación A5.2 y la ecuación A5.3 se obtiene la relación siguiente:
Donde:
La relación k 1/k 2 se conoce como el FBC cinético (FBCK) y debe ser igual al FBC en el estado de equilibrio (FBCSS) obtenido de la proporción de la concentración en el estado de equilibrio en los peces respecto a la concentración en el agua, pero pueden darse desviaciones si el estado de equilibrio es dudoso o si se han aplicado al FBC cinético correcciones según el crecimiento. Sin embargo, como k 1 y k 2 son constantes, no es necesario llegar al estado de equilibrio para derivar el valor del FBCK. A partir de estas ecuaciones de primer orden, el presente apéndice 5 incluye los cálculos generales necesarios para los métodos de bioacumulación con exposición tanto alimentaria como acuática. Sin embargo, las secciones 5, 6 y 8 solo son pertinentes para el método con exposición acuática, pero se incluyen aquí por ser técnicas “generales”. Los métodos secuencial (secciones 4 y 5) y simultáneo (sección 6) permiten el cálculo de las constantes de absorción y de depuración que sirven para derivar los FBC cinéticos. El método secuencial para determinar k 2 (sección 4) es importante para el método alimentario, ya que esta constante es necesaria para calcular tanto la eficiencia de la asimilación como el FBM. En el apéndice 7 se detallan los cálculos que son específicos del método alimentario. 2. PREVISIÓN DE LA DURACIÓN DE LA FASE DE ABSORCIÓN Antes de realizar el ensayo, puede obtenerse una estimación de k 2 y, en consecuencia, un porcentaje del tiempo necesario para llegar al estado de equilibrio, a partir de relaciones empíricas entre k 2 y el coeficiente de reparto n-octanol/agua (K OW) o k 1 y el FBC. Hay que tener en cuenta, sin embargo, que las ecuaciones de esta sección solo son aplicables en caso de que la absorción y la depuración sigan una cinética de primer orden. Si claramente esto no es así, se recomienda solicitar consejo a un bioestadístico o a un técnico de farmacocinética, si resulta deseable prever la fase de absorción. Puede obtenerse por diversos métodos una estimación de k 2 (día-1). Por ejemplo, pueden utilizarse en primera instancia las siguientes relaciones empíricas (87):
o bien
W = peso medio de los peces tratados (gramos de peso húmedo) al final de la absorción / inicio de la depuración (88) Para otras relaciones correspondientes, véase (6). Puede ser ventajoso utilizar modelos más complicados en la estimación de k 2 si, por ejemplo, resulta probable que haya un importante metabolismo (7) (8). Sin embargo, al aumentar la complejidad del modelo, debe prestarse más atención a la interpretación de las predicciones. Por ejemplo, la presencia de grupos nitro puede indicar rápido metabolismo, pero esto no es siempre así. El usuario debe, por lo tanto, ponderar los resultados obtenidos con el método predictivo respecto a la estructura química y cualquier otra información pertinente (por ejemplo, estudios preliminares) para programar un estudio. El tiempo necesario para alcanzar un determinado porcentaje del estado de equilibrio puede obtenerse, aplicando la estimación de k 2, a partir de la ecuación cinética general que describe la absorción y la depuración (cinética de primer orden), suponiendo que el crecimiento y el metabolismo son insignificantes. Si se produce crecimiento sustancial durante el estudio, no serán fiables las estimaciones descritas a continuación. En tales casos, es mejor utilizar la k 2g corregida según el crecimiento como se describe más adelante (véase la sección 7 del presente apéndice):
o, si C w es constante:
Cuando se aproxima el estado de equilibrio (t → ∞), la ecuación A5.10 puede reducirse (véase (9) (10)) a:
o bien
Entonces, FBC × C w es una aproximación a la concentración en los peces en el estado de equilibrio (C f-SS). [Nota: El mismo enfoque puede utilizarse en la estimación de un FBM en el estado de equilibrio con el ensayo alimentario. En este caso, en las ecuaciones anteriores el FBC se sustituye por el BMF y C w por C food, concentración en el alimento] La ecuación A5.10 puede transcribirse así:
o bien
Aplicando la ecuación A5.14, puede preverse el tiempo necesario para alcanzar un determinado porcentaje del estado de equilibrio cuando k 2 se ha estimado previamente con ayuda de las ecuaciones A5.5 o A5.6. A título indicativo, la duración estadísticamente óptima de la fase de absorción que permite obtener datos estadísticamente aceptables (FBCK) es el periodo necesario para que la curva del logaritmo de la concentración de la sustancia problema en los peces, representado frente al tiempo lineal, llegue al menos al 50 % del estado de equilibrio (es decir, 0,69/k 2), pero sin sobrepasar el 95 % del estado de equilibrio (es decir, 3,0/k 2) (11). En caso de que la acumulación rebase el 95 % del estado de equilibrio, se hace posible calcular un FBCSS. El tiempo necesario para alcanzar el 80 por ciento del estado de equilibrio es (utilizando la ecuación A5.14):
o bien
Análogamente, el tiempo necesario para alcanzar el 95 % del estado de equilibrio es:
Por ejemplo, la duración de la fase de absorción (es decir, el tiempo para alcanzar un determinado porcentaje del estado de equilibrio, p. ej. t 80 o t 95) en el caso de una sustancia problema con log K OW = 4 sería (utilizando las ecuaciones A5.5, A5.16 y A5.17): logk 2 = 1,47 – 0,414 · 4 k 2 = 0,652 día– 1
o bien Se puede utilizar, como alternativa, la expresión:
para calcular el tiempo de llegada al estado de equilibrio efectivo (t eSS ) (12). En el caso de una sustancia problema con log K OW = 4, el resultado es: 𝑡𝑒 SS = 6,54 · 10– 3 · 104 + 55,31 = 121 horas 3. PREVISIÓN DE LA DURACIÓN DE LA FASE DE DEPURACIÓN Es posible obtener también una previsión del tiempo necesario para reducir la carga corporal hasta un determinado porcentaje de la concentración inicial, a partir de la ecuación general que describe la absorción y la depuración (suponiendo una cinética de primer orden, véase la ecuación A5.9 (1) (13)). Para la fase de depuración, se supone que C w (o C food en caso de ensayo alimentario) es cero. La ecuación puede reducirse entonces a:
o bien
donde C f,0 es la concentración al principio del periodo de depuración. Se llegará así a una depuración del 50 % en el tiempo (t 50):
o bien
Del mismo modo, el 95 % de depuración se alcanzará en el tiempo:
Si se utiliza el 80 % de la absorción para el primer periodo (l,6/k 2) y el 95 % de pérdida en la fase de depuración (3,0/k 2), entonces la fase de depuración durará aproximadamente el doble de la fase de absorción. Téngase en cuenta que estas estimaciones se basan en la hipótesis de que los procesos de absorción y de depuración siguen una cinética de primer orden. Si es evidente que no se sigue una cinética de primer orden, estas estimaciones no son válidas. 4. MÉTODO SECUENCIAL: DETERMINACIÓN DE LA CONSTANTE DE LA VELOCIDAD DE DEPURACIÓN (PÉRDIDA) K 2 Se supone que la mayoría de los datos de bioconcentración se describen “razonablemente” bien con un modelo simple de dos compartimentos / dos parámetros, según indica la curva rectilínea que une aproximadamente los puntos de las concentraciones en los peces (en escala de logaritmos naturales) durante la fase de depuración.
Obsérvese que las desviaciones en relación con esta línea recta pueden indicar un proceso de depuración más complejo que una cinética de primer orden. Puede aplicarse el método gráfico para solucionar los tipos de depuración que se desvíen de la cinética de primer orden. Para calcular k 2 en caso de múltiples puntos temporales (de muestreo), debe realizarse una regresión lineal de ln (concentración) frente al tiempo. La pendiente de la línea de regresión es una estimación de la constante de la velocidad de depuración k 2 (89). De la ordenada en el origen puede calcularse fácilmente la concentración media en los peces al principio de la fase de depuración (C 0,d; que es igual a la concentración media en los peces al final de la fase de absorción) (incluidos los márgenes de error) (89):
Para calcular k 2 cuando solo se dispone de dos puntos temporales (de muestreo) (como en el diseño minimizado), deben sustituirse las dos concentraciones medias en la ecuación siguiente:
Donde ln (C f1) y ln (C f2) son los logaritmos naturales de las concentraciones a los tiempos t 1 y t 2, respectivamente, y t 2 y t 1 son los tiempos a los que se toman las dos muestras respecto al inicio de la depuración (90). 5. MÉTODO SECUENCIAL: DETERMINACIÓN DE LA CONSTANTE DE LA VELOCIDAD DE ABSORCIÓN K 1 (MÉTODO DE EXPOSICIÓN ACUÁTICA ÚNICAMENTE) Para hallar un valor de k1 a partir de un conjunto de datos secuenciales de concentración a lo largo del tiempo durante la fase de absorción, debe utilizarse un programa informático para ajustarse al modelo siguiente:
Donde k 2 viene dada por el cálculo anterior, y C f(t) y C w(t) son las concentraciones en los peces y en el agua, respectivamente, al tiempo t. Para calcular k 1 cuando solo se dispone de dos puntos temporales (de muestreo) (como en el diseño minimizado), debe utilizarse la fórmula siguiente:
Donde k 2 viene dada por el cálculo anterior, C f es la concentración en los peces al inicio de la fase de depuración, y C w es la concentración media en el agua durante la fase de absorción (91). Para evaluar la calidad del ajuste puede recurrirse a la inspección visual de las pendientes k 1 y k 2 en la representación gráfica frente a los datos medidos en los puntos de muestreo. Si resulta que el método secuencial da una estimación deficiente de k 1, debe aplicarse el enfoque simultáneo para calcular k 1 y k 2 (véase la sección 6 siguiente). También aquí, las pendientes resultantes deben compararse con la representación gráfica de los datos medidos para la inspección visual de la calidad del ajuste. Si la calidad del ajuste sigue siendo baja, puede constituir un indicador de que no se aplica una cinética de primer orden y de que deben emplearse otros modelos más complejos. 6. MÉTODO SIMULTÁNEO DE CÁLCULO DE LAS CONSTANTES DE VELOCIDAD DE ABSORCIÓN Y DE DEPURACIÓN (PÉRDIDA) (MÉTODO DE EXPOSICIÓN ACUÁTICA ÚNICAMENTE) Pueden utilizarse programas informáticos para hallar los valores de k 1 y k 2 en función de un conjunto de datos secuenciales de concentración a lo largo del tiempo y del modelo:
donde
Este enfoque ofrece directamente errores típicos de las estimaciones de k 1 y k 2. Cuando k 1/k 2 se sustituye por el FBC (véase la ecuación A5.4) en las ecuaciones A5.25 y A5.26, pueden calcularse también el error típico y el IC del 95 % del FBC. Esto es especialmente útil cuando se comparan distintas estimaciones debido a la transformación de los datos. La variable dependiente (concentración en los peces) puede ajustarse con o sin transformación logarítmica natural, y puede evaluarse la incertidumbre del FBC resultante. Como existe una fuerte correlación entre los dos parámetros k 1 y k 2 si se estiman simultáneamente, puede ser conveniente calcular primero k 2 a partir de los datos de depuración solamente (véase más arriba); k 2 en la mayoría de los casos puede estimarse a partir de la curva de depuración con una precisión relativamente elevada. k 1 puede calcularse posteriormente a partir de los datos de absorción utilizando una regresión no lineal (92). Se aconseja utilizar la misma transformación de los datos cuando se hace el ajuste secuencial. Para evaluar la calidad del ajuste puede recurrirse a la inspección visual de las pendientes resultantes en la representación gráfica frente a los datos medidos en los puntos de muestreo. Si resulta que este método da una estimación deficiente de k 1, puede aplicarse el enfoque simultáneo para calcular k 1 y k 2. También aquí ha de compararse el modelo ajustado frente a los datos medidos representados gráficamente para la inspección visual de la calidad del ajuste y las estimaciones de los parámetros resultantes de k 1, k 2 y FBC resultante y sus errores típicos y/o intervalos de confianza deben compararse entre diferentes tipos de ajuste. Si la calidad del ajuste es baja, puede constituir un indicador de que no se sigue una cinética de primer orden y de que deben emplearse otros modelos más complejos. Una de las dificultades más comunes es el crecimiento de los peces durante el ensayo. 7. CORRECCIÓN SEGÚN LA DILUCIÓN DEBIDA AL CRECIMIENTO PARA CALCULAR EL FBC CINÉTICO Y EL FBM En la presente sección se describe un método de referencia para la corrección del efecto del crecimiento de los peces durante el ensayo (denominado “dilución debida al crecimiento”), que solo es válido cuando se sigue la cinética de primer orden. En caso de que existan indicios de que no se sigue tal cinética, se recomienda solicitar consejo a un bioestadístico sobre la corrección adecuada de la dilución debida al crecimiento o utilizar el enfoque de masa que se describe a continuación. En ciertos casos, este método de corregir la dilución debida al crecimiento adolece de falta de precisión o a veces no funciona (por ejemplo, cuando se estudian con peces de rápido crecimiento sustancias que se depuran muy despacio, la constante de la velocidad de depuración derivada corregida según la dilución debida al crecimiento, k 2g, puede ser muy pequeña, por lo que el error en las dos constantes de velocidad utilizadas para obtenerla se hace crítico, y a veces las estimaciones de k g pueden ser mayores que k 2). En tales casos puede utilizarse un enfoque alternativo (es decir, el enfoque de masa), que también funciona cuando el crecimiento no sigue una cinética de primer orden, lo que evita la necesidad de la corrección. Este enfoque se expone al final de la presente sección. Método de sustracción de la constante de la velocidad de crecimiento para la corrección según el crecimiento Con el método de referencia, todos los datos individuales de peso y longitud se convierten en logaritmos naturales y el ln (peso) o el ln (1/peso) se representa frente al tiempo (días), por separado para los grupos de tratamiento y de testigo. El mismo proceso se lleva a cabo con los datos de las fases de absorción y de depuración por separado. Por lo general, a efectos de corrección de la dilución debida al crecimiento es más apropiado utilizar los datos de peso de todo el estudio para derivar la constante de la velocidad de crecimiento (k g), pero unas diferencias estadísticamente significativas entre las constantes de velocidad de crecimiento derivadas para la fase de absorción y para la fase de depuración pueden indicar que debe utilizarse la constante de velocidad de la fase de depuración. Las velocidades de crecimiento generales procedentes de estudios acuáticos correspondientes a los grupos de ensayo y testigo pueden utilizarse para comprobar si hay efectos relacionados con el tratamiento. Se calcula una correlación lineal de mínimos cuadrados para la gráfica del ln (peso de los peces) (y del ln (1/peso)) frente al tiempo en días para cada grupo (grupos de ensayo y testigos, datos individuales, no valores medios diarios) para todo el estudio, las fases de absorción y de depuración utilizando procedimientos estadísticos normales. Se calculan las varianzas de las pendientes de las rectas y se utilizan para evaluar la significación estadística (p = 0,05) de la diferencia en las pendientes (constantes de la velocidad de crecimiento) utilizando la prueba t de Student (o ANOVA si se somete a ensayo más de una concentración). En general se prefieren los datos de peso a efectos de la corrección según el crecimiento. Los datos de longitud, tratados de la misma forma, pueden ser útiles para comparar los grupos testigo y de ensayo en cuanto a los efectos relacionados con el tratamiento. Si no hay diferencias estadísticamente significativas en el análisis de los datos de peso, podrán mezclarse los datos de los grupos testigo y de ensayo, y calcularse una constante general de la velocidad de crecimiento de los peces para el estudio (k g), como la pendiente general de la correlación lineal. Si se observan diferencias estadísticamente significativas, se presentarán por separado las constantes de la velocidad de crecimiento de cada grupo y/o fase del estudio. A continuación debe utilizarse la constante de velocidad de cada grupo tratado con fines de corrección según la dilución debida al crecimiento de ese grupo. Si se observan diferencias estadísticas entre las constantes de velocidad de la fase de absorción y de la de depuración, deben utilizarse las constantes de velocidad derivadas de la fase de depuración. La constante calculada de la velocidad de crecimiento (k g expresada en día-1) puede sustraerse de la constante general de la velocidad de depuración (k 2) para dar la constante de la velocidad de depuración, k 2g.
La constante de la velocidad de absorción se divide por la constante de la velocidad de depuración corregida según el crecimiento para obtener el FBC cinético corregido según el crecimiento, denominado FBCKg (o FBMKg).
La constante de la velocidad de crecimiento obtenida con un estudio alimentario se utiliza en la ecuación A7.5 para calcular el FBMKg corregido según el crecimiento (véase el apéndice 7). Método de la masa para la corrección según el crecimiento Puede utilizarse como se indica a continuación una alternativa al anterior “método de sustracción de la constante de la velocidad de crecimiento” que evita la necesidad de corregir según el crecimiento. El principio consiste en utilizar los datos de depuración referidos a la masa del pez entero, y no en función de la concentración. Se convierten las concentraciones tisulares de la fase de depuración (masa de sustancia problema / unidad de masa del pez) en masa de sustancia problema /pez: se emparejan las concentraciones y los pesos de cada pez en forma tabular (por ejemplo, con una hoja de cálculo informatizada) y se multiplica cada concentración por el peso total del pez correspondiente para obtener un conjunto de datos de masa de sustancia problema por pez correspondientes a todas las muestras de la fase de depuración. Se representa gráficamente el logaritmo natural de los datos de masa de sustancia resultantes frente al tiempo para el experimento (fase de depuración) tal y como se haría normalmente. Con el método de exposición acuática se deriva normalmente la constante de la velocidad de absorción (véanse las secciones 4 y 6) (obsérvese que el valor de k 2“normal” debe utilizarse en las ecuaciones de ajuste de la curva para k 1) y se deriva la constante de la velocidad de depuración a partir de los datos anteriores. Como el valor resultante de la constante de la velocidad de depuración es independiente del crecimiento, puesto que se ha obtenido en función de la masa por pez entero, debe designarse como k 2g y no como k 2. 8. NORMALIZACIÓN RESPECTO A LOS LÍPIDOS A UN CONTENIDO LIPÍDICO DEL 5 % (MÉTODO DE EXPOSICIÓN ACUÁTICA ÚNICAMENTE) Los resultados de los FBC (cinético y en estado de equilibrio) obtenidos con ensayos de exposición acuática deben consignarse también en relación con un contenido lipídico de los peces de referencia del 5 % en peso húmedo, salvo que pueda justificarse que la sustancia problema no se acumula principalmente en los lípidos (p. ej., algunas sustancias perfluoradas pueden unirse a las proteínas). Los datos de concentración en peces, o el FBC, tienen que convertirse para referirse a un 5 % de contenido lipídico en peso húmedo. Si se utilizan los mismos peces para medir las concentraciones de sustancia y el contenido lipídico en todos los puntos de muestreo, esto requiere que cada concentración medida en los peces se corrija según el contenido lipídico de estos.
donde
Si no se realizan análisis del contenido lipídico en todos los peces muestreados, se utiliza un valor medio del contenido lipídico para normalizar el FBC. Para el FBC en estado de equilibrio, debe utilizarse el valor medio registrado al final de la fase de absorción en el grupo de tratamiento. Para la normalización de un FBC cinético pueden darse casos en los que se justifique un enfoque diferente, por ejemplo si el contenido lipídico cambia notablemente durante la fase de absorción o de depuración. No obstante, debe utilizarse sistemáticamente de todos modos una dosis alimentaria que reduzca al mínimo los cambios radicales del contenido lipídico.
donde
BIBLIOGRAFÍA
Apéndice 6 SECCIÓN DE ECUACIONES DEL ENSAYO DE EXPOSICIÓN ACUÁTICA: DISEÑO DEL ENSAYO MINIMIZADO La justificación de este enfoque consiste en que el factor de bioconcentración puede determinarse en un ensayo completo bien como factor de bioconcentración en estado de equilibrio (FBCSS) calculando la relación entre la concentración de la sustancia problema en los tejidos del pez y la concentración de la sustancia problema en el agua, o bien calculando el factor de bioconcentración cinético (FBCK) como la relación entre la constante de la velocidad de absorción k 1 y la constante de la velocidad de depuración k 2. El FBCK es válido incluso si no se consigue ninguna concentración en estado de equilibrio de la sustancia durante la absorción, siempre que los procesos de absorción y de depuración sigan aproximadamente una cinética de primer orden. Si se mide la concentración tisular de la sustancia (C f1) en el momento en que termina la exposición (t 1) y esta concentración tisular se vuelve a medir (C f2) cuando ha pasado cierto tiempo (t 2), es posible calcular la constante de la velocidad de depuración (k 2) utilizando la ecuación A5.22 del apéndice 5. La constante de la velocidad de absorción, k 1, puede determinarse de forma algebraica utilizando la ecuación A5.23 del apéndice 5 (donde C f equivale a C f1 y t equivale a t 1) (1). Así pues, el factor de bioconcentración cinético con el diseño minimizado (designado como BCFKm para distinguirlo de los factores de bioconcentración cinéticos determinados mediante otros métodos):
Las concentraciones o los resultados deben corregirse según la dilución debida al crecimiento y normalizarse con relación a un contenido lipídico de los peces del 5 %, tal como se describe en el apéndice 5. El FBCSS minimizado es el FBC calculado al final de la fase de absorción, suponiendo que se ha alcanzado el estado de equilibrio. Esto solo puede suponerse, ya que el número de puntos de muestreo no es suficiente para demostrarlo.
Donde: C f-minSS = concentración en los peces en el estado de equilibrio supuesto al final de la absorción (mg·kg-1 peso húmedo). C w-minSS = concentración en el agua en el estado de equilibrio supuesto al final de la absorción (mg·l-1). BIBLIOGRAFÍA
Apéndice 7 SECCIÓN DE ECUACIONES DEL ENSAYO DE EXPOSICIÓN ALIMENTARIA
1. EJEMPLO DE CANTIDADES DE COMPONENTES DE UN ALIMENTO COMERCIAL ADECUADO PARA PECES
2. EJEMPLOS DE TÉCNICAS DE ENRIQUECIMIENTO ALIMENTARIO Aspectos generales Deben prepararse alimentos testigo, exactamente de la misma manera que los alimentos enriquecidos, pero sin la sustancia problema. Para comprobar la concentración del alimento tratado, se toman de él muestras triplicadas con un método adecuado de extracción y se mide la radiactividad o la concentración de la sustancia problema en los extractos. Deben conseguirse unas recuperaciones analíticas elevadas (> 85 %) con escasa variación entre las muestras (las concentraciones de la sustancia en las tres muestras tomadas al inicio del ensayo no deben variar en más del ± 15 % respecto a la media). Durante el ensayo alimentario, deben tomarse tres muestras del alimento el día 0 y al final de la fase de absorción, para determinar con ellas el contenido de la sustancia problema en el alimento. Preparación del alimento de los peces con un material de ensayo líquido (puro) Se fija una concentración de ensayo nominal objetivo en el alimento tratado, por ejemplo 500 μg de sustancia problema /g de alimento. La cantidad adecuada (por masa molar o radiactividad específica) de la sustancia problema pura se añade a una masa conocida de alimento para peces en un tarro de vidrio o en el matraz redondo de un evaporador rotatorio. La masa de alimentos para peces debe ser suficiente para la duración de la fase de absorción (teniendo en cuenta la necesidad de ir aumentando las cantidades en cada aporte de alimento debido al crecimiento de los peces). El alimento para peces debe mezclarse con la sustancia problema durante una noche mediante volteo lento (por ejemplo, utilizando un mezclador de bastidor rotatorio o por rotación si se utiliza el matraz redondo de un evaporador rotatorio). El alimento enriquecido debe conservarse en condiciones que preserven la estabilidad de la sustancia problema en la mezcla alimentaria (por ejemplo, refrigeración) hasta el momento de la utilización. Preparación del alimento de los peces con un vehículo de aceite de pescado o de maíz Las sustancias problema sólidas deben reducirse a polvo fino con un mortero. Las sustancias problema líquidas pueden añadirse directamente al aceite de pescado o maíz. La sustancia problema se disuelve en una cantidad determinada de aceite de pescado o maíz (por ejemplo, 5-15 ml). El aceite enriquecido se transfiere cuantitativamente al matraz redondo de un evaporador rotatorio de tamaño adecuado. El recipiente utilizado para preparar el aceite enriquecido debe lavarse con dos pequeñas alícuotas de aceite, que se añaden al matraz redondo para garantizar que se transfiere toda la sustancia problema disuelta. Para garantizar la completa disolución o dispersión en el aceite (o si se utiliza en el estudio más de una sustancia problema), se añade un micro-agitador, se tapa el matraz y se agita la mezcla deprisa durante una noche. Para el ensayo se añade al matraz redondo una cantidad apropiada de alimento de los peces (normalmente en forma de granulado) y el contenido del matraz redondo se mezcla a fondo volteándolo continuamente durante al menos 30 minutos, pero preferentemente durante toda una noche. A continuación, el alimento enriquecido se almacena adecuadamente (por ejemplo, refrigerado) para garantizar la estabilidad de la sustancia problema en el alimento hasta su utilización. Preparación del alimento de los peces con un disolvente orgánico La cantidad apropiada de sustancia problema (por masa molar o radiactividad específica) suficiente para alcanzar la dosis objetivo se disuelve en un disolvente orgánico apropiado (por ejemplo, ciclohexano o acetona; 10-40 ml, pero puede ser necesario un volumen mayor en función de la cantidad de alimento por enriquecer). Una parte alícuota de esta solución o toda ella (añadida por partes) se mezcla con la masa adecuada de alimento de peces suficiente para que el ensayo alcance el nivel necesario de dosis nominal. El alimento y la sustancia problema pueden mezclarse en una mezcladora de acero inoxidable y el alimento recién enriquecido puede dejarse en la mezcladora, en una campana de laboratorio, durante dos días (agitando de vez en cuando) para permitir que se evapore el disolvente en exceso, o mezclarse en el matraz redondo de un evaporador rotatorio de rotación continua. El exceso de disolvente puede eliminarse arrastrándolo con una corriente de aire o de nitrógeno en caso necesario. Debe velarse por que la sustancia problema no cristalice al eliminar el disolvente. El alimento enriquecido debe conservarse en condiciones (por ejemplo, refrigeración) que preserven la estabilidad de la sustancia problema en la mezcla alimentaria hasta el momento de la utilización. 3. CÁLCULO DE LA EFICIENCIA DE LA ASIMILACIÓN Y DEL FACTOR DE BIOMAGNIFICACIÓN Para calcular la eficiencia de la asimilación, debe estimarse primero la constante de la velocidad de depuración general con arreglo a la sección 4 del apéndice 5 (siguiendo el método secuencial, es decir, una regresión lineal normal) y utilizando las concentraciones medias de las muestras de la fase de depuración. La dosis alimentaria, I, y la duración de la absorción, t, son parámetros conocidos del estudio. C food, la concentración medida media de la sustancia problema en los alimentos es una variable medida en el estudio. C 0,d, la concentración de la sustancia problema en los peces al final de la fase de absorción, se obtiene generalmente de la ordenada en el origen de la gráfica de ln (concentración) frente a los días de depuración. La eficiencia de la asimilación de la sustancia (a, absorción de la sustancia problema en el intestino) se calcula de la siguiente manera:
donde: C 0,d = concentración en los peces derivada en el tiempo cero de la fase de depuración (mg kg-1); k 2 = constante de la velocidad de depuración general (sin corregir según el crecimiento) (día-1), calculada según las ecuaciones del apéndice 5, sección 3; I = velocidad de ingestión de alimento (g alimento g-1 peces día-1); C food = concentración en el alimento (mg kg-1 alimento); t= duración del periodo de alimentación (días) No obstante, es posible que sea necesario ajustar la dosis alimentaria, I, utilizada en el cálculo para tener en cuenta el crecimiento de los peces, a fin de obtener una eficiencia de asimilación exacta, a. En un ensayo en el que los peces crezcan de manera significativa durante la fase de absorción (sin que se haga ninguna corrección de las cantidades de alimento para mantener la dosis alimentaria fijada), la tasa alimentaria efectiva, a medida que avanza la fase de absorción, será inferior a la fijada, lo que resulta en una mayor eficiencia de asimilación «real». (Obsérvese que esto no es importante para el cálculo general del FBM, ya que los términos de I se anulan efectivamente entre la ecuación A7.1 y la ecuación A7.4). La dosis alimentaria media corregida según la dilución debida al crecimiento, I g, puede obtenerse de diversas formas, de las que una directa y rigurosa es utilizar la constante de la velocidad de crecimiento conocida (k g) para estimar los pesos de los peces del ensayo a diversos tiempos de la fase de absorción, es decir:
donde W f(t)= peso medio de los peces el día t de la fase de absorción W f,0 = peso medio de los peces al inicio del experimento De esta manera puede estimarse (al menos) el peso medio de los peces del último día de exposición (W f,end-of-uptake). Como la dosis alimentaria se fija sobre la base del W f,0, la dosis alimentaria efectiva de cada día de la fase de absorción puede calcularse utilizando estos dos valores del peso. La dosis alimentaria corregida según el crecimiento, I g (g alimento g-1 pez día-1), que debe utilizarse en lugar de I en caso de crecimiento rápido durante la fase de absorción, puede calcularse entonces de la manera siguiente:
Una vez obtenida la eficiencia de la asimilación, puede calcularse el FBM multiplicándola por la dosis alimentaria I (o I g, si se utiliza esta para calcular α) y dividiendo el resultado por la constante de la velocidad de depuración general k 2:
El factor de biomagnificación corregido según el crecimiento debe calcularse también de la misma forma, utilizando la constante de la velocidad de depuración corregida según el crecimiento (obtenida con arreglo a la sección 7 del apéndice 5). De nuevo, si se ha utilizado I g para calcular α, debe utilizarse también aquí en lugar de I:
donde: α = eficiencia de la asimilación (absorción de la sustancia problema en el intestino); k 2 = constante de la velocidad de depuración general (sin corregir según el crecimiento) (día-1), calculada según las ecuaciones del apéndice 5, sección 3; k 2g = constante de la velocidad de depuración corregida según el crecimiento (día-1); I = velocidad de ingestión de alimento (g alimento g-1 peces día-1); La semivida (t 1/2) corregida según el crecimiento se calcula de la forma siguiente:
La eficiencia de la asimilación de la sustancia desde el alimento también puede estimarse si se determinan los residuos en los tejidos durante la fase lineal de la absorción (entre los días 1 y 3). En este caso la eficiencia de la asimilación de la sustancia (α) puede determinarse como sigue:
Donde: C fish (t) = concentración de la sustancia problema en los peces al tiempo t (mg kg-1 peso húmedo). 4. CORRECCIÓN SEGÚN EL CONTENIDO LIPÍDICO Si el contenido lipídico se mide en los mismos peces en que se hacen los análisis del producto en todos los intervalos de muestreo, deben corregirse las concentraciones individuales según el contenido lipídico y hay que representar gráficamente el ln (concentración corregida según el contenido lipídico) frente al tiempo de depuración (días) para obtener C 0,d y k 2. A continuación puede calcularse la eficiencia de la asimilación (ecuación A7.1) según el contenido lipídico, utilizando C food según el contenido lipídico (es decir, C food se multiplica por la fracción lipídica media del alimento). Un cálculo utilizando las ecuaciones A7.4 y A7.5 permite entonces obtener directamente el FBM corregido según el contenido lipídico (y según la dilución debida al crecimiento). En caso contrario, la fracción lipídica media (p/p) de los peces y la de los alimentos se derivan tanto respecto a los grupos de tratamiento y a los testigos (para los alimentos y los peces de los grupos testigos suele hacerse a partir de datos medidos al inicio y al final de la exposición; para los peces de los grupos de tratamiento suele hacerse a partir de datos medidos al final de la exposición solo). En algunos estudios puede aumentar notablemente el contenido lipídico de los peces; en tales casos, es más adecuado utilizar una concentración lipídica media de los peces de ensayo calculada a partir de los valores medidos al final de la exposición y al final de la depuración. En general, los datos de los grupos de tratamiento solo deben utilizarse para derivar las dos fracciones lipídicas. El factor de corrección según el contenido lipídico (Lc ) se calcula del siguiente modo:
donde L fish y L food son las fracciones lipídicas medias de los peces y de los alimentos, respectivamente. El factor de corrección según el contenido lipídico se utiliza para calcular el factor de biomagnificación corregido según el contenido lipídico (FBML):
5. EVALUACIÓN DE LAS DIFERENCIAS ENTRE LA CONCENTRACIÓN A TIEMPO CERO MEDIDA (C 0,M) Y LA CONCENTRACIÓN A TIEMPO CERO DERIVADA (C 0,D) Deben compararse la concentración a tiempo cero medida (C 0,m) y la concentración a tiempo cero derivada (C 0,d). Si son muy similares, se confirma el modelo de primer orden utilizado para determinar los parámetros de depuración. En algunos estudios puede haber una marcada diferencia entre el valor a tiempo cero derivado, C 0,d, y la concentración a tiempo cero medida media. C 0,m (véase el último inciso del punto 159 del presente método de ensayo). Si C 0,d es mucho menor que C 0,m (C 0,d << C 0,m), la diferencia puede sugerir la presencia en el intestino de alimento enriquecido sin digerir. Esto puede comprobarse de manera experimental mediante la realización de análisis aparte con el intestino extirpado si se han tomado y conservado muestras adicionales (peces enteros) al final de la fase de absorción. En caso contrario, si una prueba estadísticamente válida de resultados discrepantes se aplica a la regresión lineal de la fase de depuración e indica que la concentración del primer punto de muestreo de la depuración es erróneamente elevada, puede ser adecuado efectuar la regresión lineal para calcular k 2, pero omitiendo el primer punto de concentración de la depuración. En estos casos, si la incertidumbre de la regresión lineal disminuye en gran medida, y es evidente que se cumple una cinética de depuración aproximadamente de primer orden, puede ser conveniente utilizar los valores resultantes de C 0,d y k 2 en el cálculo de la eficiencia de la asimilación. Esto debe justificarse plenamente en el informe. También es posible que en la fase de depuración rija una cinética distinta de la de primer orden. Si esto resulta probable (es decir, si la gráfica de los logaritmos naturales de los datos sigue un trazado en curva en comparación con la línea recta de la gráfica de regresión lineal), no es fácil que resulten válidos los cálculos de k 2 y C 0,d, y debe buscarse el consejo de un bioestadístico. Si C 0,d es mucho más elevado que el valor medido (C 0,d >> C 0,m), esto puede indicar: que la sustancia se depura muy rápidamente (es decir, los puntos de muestreo se acercan al límite de cuantificación del método analítico muy pronto en la fase de depuración, véase la sección 6 a continuación); que hay desviación respecto a una cinética de depuración de primer orden; que la regresión lineal para calcular k 2 y C 0,d es deficiente; o que se ha producido en algunos tiempos de muestreo un problema con las concentraciones medidas en el estudio. En esos casos, debe examinarse la gráfica de la regresión lineal para buscar indicios de muestras en el límite de cuantificación o cerca de este, o de valores anómalos y curvaturas evidentes (sugerentes de que la cinética no es de primer orden), lo cual debe destacarse en el informe. Debe describirse y justificarse la eventual reevaluación posterior de la regresión lineal para mejorar los valores estimados. Si se observa una desviación marcada respecto a la cinética de primer orden, resulta poco probable que sean válidos los cálculos de k 2 y C 0,d, y debe pedirse consejo a un bioestadístico. 6. ORIENTACIONES PARA LAS SUSTANCIAS PROBLEMA DE MUY RÁPIDA DEPURACIÓN Como se comentaba en el punto 129 del método de ensayo, algunas sustancias pueden depurarse tan deprisa que no puede derivarse una concentración a tiempo cero, C 0,d, ni una k 2 fiables porque ya muy al inicio de la fase de depuración (es decir, a partir de la segunda muestra de esta fase) la sustancia ya no puede medirse efectivamente (concentraciones indicadas al límite de cuantificación). Esta situación se ha observado en el ensayo interlaboratorios realizado en apoyo del presente método de ensayo con benzo[a]pireno, y se ha documentado en el informe de validación del método. En esos casos, no puede realizarse una regresión lineal de manera fiable, y es probable que se dé una estimación exageradamente elevada de C 0,d, con el resultado de una eficiencia de asimilación aparente muy superior a 1. En estas circunstancias es posible calcular una estimación prudente de k 2 y un «límite superior» del FBM. Utilizando estos puntos de datos de la fase de depuración en los que se mide la concentración, hasta la primera concentración «no detectada» (concentración fijada al límite de cuantificación) inclusive, puede conseguirse una estimación de k 2 mediante una regresión lineal (utilizando la representación del logaritmo natural de los datos de concentración frente al tiempo). Para este tipo de casos es probable que solo se tengan en cuenta dos puntos de datos (por ejemplo, en los días de muestreo 1 y 2 de la depuración) y entonces puede estimarse k 2 utilizando la ecuación A5.22 del apéndice 5. Esta estimación de k2 puede utilizarse para estimar la eficiencia de la asimilación con arreglo a la ecuación A7.1, sustituyendo el valor de C0,d en la ecuación por la concentración al tiempo cero medida (C0,m) en los casos en que se estima claramente que C0,d es mucho mayor de lo que habría podido alcanzarse en el ensayo. Si C0,m no es mensurable, debe utilizarse el límite de detección en los tejidos de los peces. Si, en algunos casos, ello arroja un valor de α > 1, se supone que la eficiencia de asimilación es igual a 1 en el «peor de los casos». Entonces puede estimarse el FBMK máximo utilizando la ecuación A7.4, y debe indicarse como valor «muy inferior a» (<<). Por ejemplo, si se trata de un estudio llevado a cabo con una dosis alimentaria del 3 % y una semivida de depuración inferior a 3 días, y una α en el «peor de los casos» igual a 1, es probable que el FBMK sea inferior a aproximadamente 0,13. Habida cuenta de la finalidad de esta estimación y del hecho de que los valores son de naturaleza prudente, no es necesario corregirlos según la dilución debida al crecimiento ni según el contenido lipídico de los peces y los alimentos. Apéndice 8 ENFOQUES PARA LA ESTIMACIÓN PROVISIONAL DE LOS FBC A PARTIR DE DATOS RECOGIDOS EN EL ESTUDIO DE EXPOSICIÓN ALIMENTARIA El método alimentario se incluye aquí para el ensayo de bioacumulación de sustancias que en la práctica no pueden someterse a ensayo utilizando el método de exposición acuática. El método de exposición acuática da un factor de bioconcentración, mientras que el método alimentario proporciona directamente información sobre el potencial de biomagnificación por la alimentación. En muchos regímenes de seguridad de los productos químicos se requiere información sobre la bioconcentración acuática (por ejemplo, en la evaluación del riesgo y en el Sistema Globalmente Armonizado de clasificación). Por tanto, es preciso recurrir a los datos obtenidos en un estudio alimentario para estimar un factor de bioconcentración que sea comparable al de los ensayos realizados de conformidad con el método de exposición acuática (94). En esta sección se presentan los enfoques que pueden seguirse para ello, reconociendo a la vez las deficiencias inherentes a las estimaciones. El estudio alimentario mide la depuración para dar una constante de la velocidad de depuración, k 2. Si se puede calcular una constante de la velocidad de absorción con los datos disponibles para la situación en que los peces se exponen a la sustancia problema a través del agua, entonces podría estimarse un FBC cinético. La estimación de una constante de la velocidad de absorción en caso de exposición acuática a una sustancia problema depende de muchas hipótesis, todas las cuales contribuirán a la incertidumbre de la estimación. Por otra parte, este enfoque para estimar un FBC supone que la velocidad general de depuración (incluidos los factores correspondientes, como la distribución en el organismo y los distintos procesos de depuración) es independiente de la técnica de exposición utilizada para producir una carga corporal de la sustancia problema. Las principales hipótesis inherentes al enfoque de la estimación pueden resumirse de la siguiente manera. La depuración tras la absorción alimentaria es la misma que la depuración tras la exposición acuática con una sustancia dada. La absorción desde el agua sigue una cinética de primer orden. Dependiendo del método utilizado para la estimación de la absorción:
la base de datos («conjunto de formación») utilizada para elaborar el método de estimación de la absorción es representativa de la sustancia de que se trate. Varias publicaciones en la bibliografía de acceso libre proporcionan ecuaciones en las que la absorción en los peces desde el agua a través de las branquias se relaciona con las siguientes propiedades: coeficiente de reparto octanol/agua de la sustancia, peso de los peces (1) (2) (3) (4), volumen y/o contenido lipídico, difusión/ permeabilidad por membrana (5) (6), volumen de ventilación de los peces (7), y mediante un enfoque de fugacidad/balance de masas (8) (9) (10). En Crookes & Brooke (11) se da una evaluación detallada de tales métodos en este contexto. También es útil aquí una publicación de Barber (12) centrada en la modelización de la bioacumulación mediante la absorción alimentaria, puesto que incluye contribuciones de los modelos de la velocidad de absorción por las branquias. También se dedicaba a este aspecto una sección del documento de base del protocolo alimentario de 2004 (13). Parece que la mayoría de estos modelos se ha elaborado utilizando bases de datos limitadas. Para los modelos en los que se dispone de los detalles de la base de datos utilizada para construirlos, parece que los tipos de sustancias utilizadas son a menudo de estructura o clase similar (en términos de funcionalidad, por ejemplo compuestos organoclorados). Esto contribuye a la incertidumbre de utilizar un modelo para predecir la constante de la velocidad de absorción para un tipo diferente de sustancia, además de las consideraciones específicas del ensayo, como la especie, la temperatura, etc. Una revisión de las técnicas disponibles (11) destacaba que no hay un método «más correcto» que los otros. Por lo tanto, debe darse una justificación clara del modelo utilizado. En caso de que se disponga de varios métodos cuyo uso se pueda justificar, parece prudente presentar varias estimaciones de k 1 (y también del FBC) o un intervalo de valores de k 1 (y del FBC) con arreglo a varios métodos de estimación de la absorción. Sin embargo, habida cuenta de las diferencias existentes en los tipos de modelo y en los conjuntos de datos usados para elaborarlos, puede que no sea apropiado tomar un valor medio a partir de estimaciones obtenidas de distintas maneras. Algunos investigadores han planteado que las estimaciones del FBC de este tipo requieren una corrección según la biodisponibilidad para tener en cuenta la adsorción de la sustancia al carbono orgánico disuelto (COD) en condiciones de exposición acuática, para situar la estimación en consonancia con los resultados de los estudios de exposición acuática (p. ej., (13) (14)). No obstante, puede que esta corrección no sea adecuada, habida cuenta de los bajos niveles de COD requeridos en un estudio de exposición acuática para hacer una estimación en el «peor de los casos» (es decir, la proporción de sustancia biodisponible respecto a la sustancia medida en la solución). En el caso de las sustancias muy hidrófobas, la absorción en las branquias puede verse limitada por la velocidad de difusión pasiva cerca de la superficie de las branquias; en este caso, es posible que la corrección tenga en cuenta tal efecto, en lugar de aquel para el que se había concebido. Se recomienda centrarse en los métodos que requieren aportaciones para las que se puede disponer fácilmente de datos sobre las sustancias sometidas a ensayo según el estudio alimentario aquí descrito (es decir, log K OW, peso de los peces). Puede aplicarse otros métodos que requieran aportaciones más complejas, pero pueden exigir otras mediciones en el ensayo o un conocimiento detallado de la sustancia problema o de la especie de peces que pueden ser difíciles de obtener. Además, la elección del modelo puede verse influida por el nivel de la validación y el ámbito de aplicabilidad (véase en (11) una revisión y una comparación de métodos diferentes). Debe tenerse en cuenta que la estimación resultante de k 1, y el FBC estimado, son inciertos y puede ser necesario tratarlos según un enfoque de ponderación de las pruebas junto con el FBM derivado y los parámetros de la sustancia (por ejemplo, el tamaño molecular) para conseguir una visión global del potencial de bioacumulación de la sustancia. La interpretación y utilización de estos parámetros puede depender del marco normativo. BIBLIOGRAFÍA
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
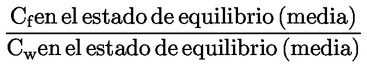


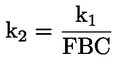

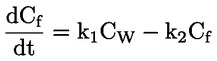
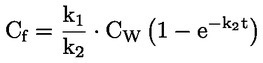
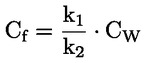
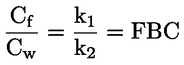






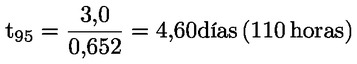









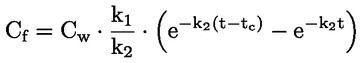

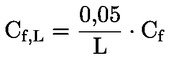

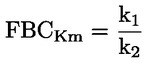
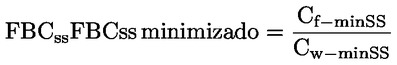
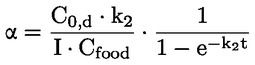
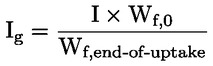
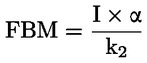





|
17) |
En la parte C, el capítulo C.20 se sustituye por el texto siguiente: «C.20. Ensayo de reproducción en Daphnia magna INTRODUCCIÓN El presente método de ensayo reproduce las directrices de ensayo (TG) 211 de la OCDE (2012). Las directrices de ensayo de la OCDE se revisan periódicamente en función de los avances científicos. Las directrices de ensayo de reproducción 211 tienen su origen en las directrices de ensayo 202, parte II, ensayo de reproducción en Daphnia sp. (1984). En general, se había reconocido que los datos obtenidos con ensayos realizados según las TG 202 podían ser variables. Esto dio lugar a que se dedicara un considerable esfuerzo a la identificación de las razones de esta variabilidad, con objeto de conseguir un método de ensayo mejor. Las directrices de ensayo 211 se basan en el resultado de estas actividades de investigación, estudios interlaboratorios y estudios de validación efectuados en 1992 (1), 1994 (2) y 2008 (3). Las principales diferencias entre la versión inicial (TG 202, 1984) y la segunda versión (TG 211, 1998) de las directrices de ensayo de reproducción son las siguientes:
En el apéndice 1 se dan las definiciones utilizadas. PRINCIPIO DEL ENSAYO La principal finalidad de este ensayo es evaluar el efecto de los productos químicos sobre el resultado reproductor de Daphnia magna. A tal fin, se exponen al producto problema, añadido al agua a una gama de concentraciones diferentes, hembras jóvenes de dafnia (animales parentales), con una edad inferior a 24 horas al principio del ensayo. La duración del ensayo es de 21 días. Al final del ensayo se evalúa el número total de descendientes vivos producidos. El resultado reproductor de los animales parentales se puede expresar de otras formas (por ejemplo, como número de descendientes vivos producidos por animal y por día a partir del primer día en que se observa descendencia), pero estas deben indicarse además del número total de descendientes vivos producidos al final del ensayo. A causa del particular diseño del ensayo semiestático en comparación con otros métodos de ensayo de reproducción de invertebrados, también es posible contar el número de descendientes vivos producidos por cada animal parental. Esto permite que, contrariamente a otros métodos de ensayo de reproducción de invertebrados, si el animal parental muere de forma fortuita o inadvertida durante el período de ensayo, su descendencia producida pueda excluirse de la evaluación de los datos. Por tanto, si se da mortalidad de animales parentales en réplicas expuestas, debe considerarse si la mortalidad sigue o no una pauta de respuesta a la concentración, por ejemplo si hay una regresión significativa de la respuesta frente a la concentración del producto problema con pendiente positiva (podría utilizarse para ello un método estadístico como la prueba de tendencia de Cochran-Armitage). Si la mortalidad no sigue una pauta de respuesta a la concentración, entonces esas réplicas con mortalidad de animales parentales deben excluirse del análisis de los resultados del ensayo. Si la mortalidad sigue una pauta de respuesta a la concentración, la mortalidad parental debe asignarse como efecto del producto problema y las réplicas no deben excluirse del análisis. Si el animal parental muere durante el ensayo de forma fortuita por un trato inadecuado o por accidente, o de forma inadvertida debido a un incidente inexplicado que no está relacionado con el efecto del producto problema, o resulta ser macho, la réplica correspondiente se excluye del análisis (véase el punto 51). El efecto tóxico del producto problema sobre el resultado reproductor se expresa como ECx ajustando los datos a un modelo adecuado mediante regresión no lineal para estimar la concentración que provocaría una reducción del x % en el resultado reproductor, o también el valor de la NOEC/LOEC (4). Es preferible que las concentraciones de ensayo abarquen la más baja de las concentraciones con efecto utilizadas (por ejemplo, EC10), lo que significa que este valor se calcula por interpolación y no por extrapolación. También hay que indicar la supervivencia de los animales parentales y el momento de la producción de la primera nidada. También pueden examinarse otros efectos, relacionados con el producto, sobre parámetros tales como el crecimiento (p. ej., la longitud) y, posiblemente, la tasa intrínseca de aumento de la población (véase el punto 44). INFORMACIÓN SOBRE EL PRODUCTO PROBLEMA Los resultados de un ensayo de toxicidad aguda (véase el capítulo C.2 del presente anexo: Ensayo de inmovilización aguda de Daphnia sp.) realizado con Daphnia magna pueden ser útiles para seleccionar un intervalo adecuado de concentraciones de ensayo para los ensayos de reproducción. Hay que conocer la hidrosolubilidad y la presión de vapor del producto problema; también se debe disponer de un método analítico fiable para cuantificar el producto en las soluciones de ensayo con la eficiencia de recuperación y el límite de determinación establecidos. Entre la información sobre el producto problema que puede ser útil para establecer las condiciones de ensayo se incluyen la fórmula estructural, la pureza del producto, la estabilidad a la luz, la estabilidad en las condiciones de ensayo, el pKa, el Pow y los resultados de un ensayo de biodegradabilidad fácil [véanse los capítulos C.4 (determinación de la biodegradabilidad «fácil») y C.29 (biodegradabilidad fácil — CO2 en recipientes sellados) del presente anexo]. VALIDEZ DEL ENSAYO Para que el ensayo sea válido, los testigos deben cumplir los criterios de comportamiento siguientes:
Nota: El mismo criterio de validez (20 %) puede utilizarse para la mortalidad de animales parentales de forma fortuita e inadvertida en los testigos, así como en cada una de las concentraciones de ensayo. DESCRIPCIÓN DEL MÉTODO Equipo Los recipientes de ensayo y demás instrumentos que hayan de entrar en contacto con las soluciones de ensayo serán íntegramente de vidrio o de otro material químicamente inerte. Los recipientes de ensayo serán normalmente vasos de vidrio. Además, hará falta el siguiente equipo, en todo o en parte:
Organismo de ensayo La especie que debe usarse en el ensayo es Daphnia magna Straus (95). De preferencia, el clon debe estar identificado genotípicamente. Las investigaciones realizadas (1) han demostrado que el comportamiento reproductor del clon A (procedente del IRCHA de Francia) (5) cumple siempre el criterio de validez de una media de ≥ 60 descendientes vivos por animal parental que sobrevive cuando se cría en las condiciones descritas en este método de ensayo. No obstante, son aceptables otros clones, siempre que se demuestre que el cultivo de Daphnia cumple los criterios de validez del ensayo. Al principio del ensayo, los animales tendrán una edad inferior a 24 horas y no deben ser progenie de primera nidada. Han de proceder de una población sana (es decir, sin signos de agresión, tales como mortalidad elevada, presencia de machos y efipios, retraso de la producción de la primera nidada, animales decolorados, etc.). Los animales de la población inicial deben mantenerse en condiciones de cultivo (luz, temperatura, medio, nutrición y número de animales por unidad de volumen) similares a las usadas en el ensayo. Cuando el medio de cultivo de dafnias que vaya a usarse en el ensayo sea distinto del usado en el cultivo habitual de dafnias, es recomendable prever un periodo de aclimatación anterior al ensayo, por lo general de unas 3 semanas de duración (es decir, una generación), para evitar agredir a los animales parentales. Medio de ensayo Es recomendable usar en este ensayo un medio definido completamente. Con ello se evita el uso de aditivos (tales como algas marinas o extracto de suelo) difíciles de caracterizar y se aumentan en consecuencia las oportunidades de normalización entre laboratorios. Se ha observado que los medios Elendt M4 (6) y M7 (véase el apéndice 2) son adecuados para este propósito. No obstante, son aceptables medios distintos [por ejemplo, (7) (8)], siempre que se demuestre que el comportamiento del cultivo de dafnias cumple los criterios de validez del ensayo. Si se usan medios con aditivos sin definir, estos deben especificarse con claridad, y en el informe del ensayo hay que aportar información sobre su composición, en particular el contenido en carbono, pues podría contribuir a la dieta suministrada. Es recomendable determinar en la preparación madre del aditivo orgánico el carbono orgánico total (COT), la demanda química de oxígeno (DQO) o las dos cosas, así como hacer una estimación de la contribución resultante al COT/DQO del medio de ensayo. Conviene también que los niveles de COT en el medio (es decir, antes de incorporar las algas) sean inferiores a 2 mg/l (9). Si se ensayan productos que contengan metales, es importante tener en cuenta que las propiedades del medio de ensayo (por ejemplo, dureza, capacidad quelante) pueden afectar a la toxicidad del producto problema. Por ello es deseable usar un medio totalmente definido. Sin embargo, en este momento, los únicos medios totalmente definidos adecuados para el cultivo prolongado de Daphnia magna que se conocen son los Elendt M4 y M7. Ambos contienen el agente quelante EDTA. Se ha demostrado (2) que la «toxicidad aparente» del cadmio es generalmente inferior cuando el ensayo de reproducción se hace en medios M4 o M7 que cuando se hace en medios exentos de EDTA. Por tanto, M4 y M7 no son recomendables para ensayar productos con metales, y deben evitarse asimismo en estos casos otros medios que contengan agentes quelantes conocidos. En caso de productos que contengan metales, puede ser aconsejable usar otros medios como, por ejemplo, agua dulce dura reconstituida según ASTM (9), que no contiene EDTA. Esta combinación de agua dulce dura reconstituida según ASTM y extracto de algas marinas (10) es adecuada para el cultivo prolongado de Daphnia magna (2). La concentración de oxígeno disuelto debe ser superior a 3 mg/l al comienzo y durante el ensayo. El pH debe estar comprendido entre 6 y 9, y normalmente no debe oscilar en más de 1,5 unidades en ninguno de los ensayos. Se recomienda utilizar una dureza superior a 140 mg/l (expresada como CaCO3). Los ensayos realizados con valores iguales o superiores a este han demostrado un comportamiento reproductor que cumple los criterios de validez (11) (12). Soluciones de ensayo Las soluciones de ensayo de las concentraciones elegidas se preparan generalmente por dilución de una solución madre. Preferentemente, estas soluciones madre deben prepararse, si es posible sin utilizar disolventes ni dispersantes, por mezcla o agitación del producto problema en el medio de ensayo de forma mecánica, como, por ejemplo, con un agitador o ultrasonidos, u otros métodos adecuados. Es preferible exponer los sistemas de ensayo a las concentraciones del producto problema que se vayan a utilizar en el estudio, durante el tiempo que se requiera para demostrar el mantenimiento de unas concentraciones de exposición estables, antes de introducir los organismos de ensayo. Si el producto problema es difícil de disolver en agua, se deben seguir los procedimientos descritos en el documento de orientación de la OCDE sobre sustancias y mezclas difíciles (13). Debe evitarse el uso de disolventes o dispersantes, aunque en algunos casos puede ser necesario utilizarlos para obtener una solución madre con la concentración adecuada para la dosificación. Además de las concentraciones de ensayo, debe utilizarse un testigo del agua de dilución con las réplicas adecuadas y, caso de resultar inevitable el uso de un disolvente, un testigo de este, también con sus réplicas adecuadas. Solo deben utilizarse en el ensayo disolventes o dispersantes de los que se hayan hecho estudios que demuestren que no afectan (o solo mínimamente) a la variable de respuesta. En (13) figuran ejemplos de disolventes (como la acetona, el etanol, el metanol, la dimetilformamida y el trietilenglicol) y dispersantes (como el Cremophor RH40, la metilcelulosa al 0,01 % y el HCO-40) adecuados. Si se utiliza un disolvente o dispersante, su concentración final no deberá exceder de 0,1 ml/l (13) y habrá de ser la misma en todos los recipientes de ensayo, excepto en el testigo del agua de dilución. No obstante, debe hacerse todo lo posible para mantener la concentración de disolvente al mínimo. PROCEDIMIENTO Condiciones de exposición Duración La duración del ensayo es de 21 días. Carga Los animales parentales se mantienen separados, uno por recipiente de ensayo, con un volumen de medio en cada recipiente de 50 a 100 ml (en el caso de Daphnia magna; es posible utilizar volúmenes menores con dáfnidos más pequeños, como Ceriodaphnia dubia), a menos que sea necesario un diseño dinámico para realizar el ensayo. En ocasiones hay que emplear un volumen mayor a fin de satisfacer los requisitos del método analítico usado para determinar la concentración del producto problema, aunque también se permite poner en común las réplicas a efectos de este análisis. Si se usan volúmenes superiores a 100 ml, puede ser necesario aumentar la ración administrada a las dafnias para garantizar una disponibilidad suficiente de alimento y el cumplimiento de los criterios de validez. Animales de experimentación En ensayos semiestáticos, se mantienen por separado al menos 10 animales a cada concentración de ensayo y al menos 10 animales en la serie de testigo. En ensayos dinámicos, se ha demostrado que es adecuado repartir 40 animales en 4 grupos de 10 a cada concentración de ensayo (1). Puede usarse un número inferior de organismos de ensayo, y se recomienda un mínimo de 20 animales por concentración repartidos en 2 o más réplicas de igual número de animales (por ejemplo, 4 réplicas con 5 dáfnidos cada una). Obsérvese que en los ensayos en los cuales se mantengan los animales en grupos, no será posible excluir ninguna descendencia de los análisis estadísticos si se da mortalidad parental fortuita o inadvertida cuando haya comenzado la reproducción y, por consiguiente, en estos casos el resultado reproductor debe expresarse como número total de descendientes vivos producidos por animal parental presente al principio del ensayo. Hay que asignar los tratamientos a los recipientes de ensayo y realizar todas las manipulaciones posteriores de dichos recipientes de forma aleatoria. De otro modo podría introducirse un sesgo susceptible de ser interpretado como efecto de la concentración. En particular, si las unidades experimentales se manipulan en orden de tratamiento o de concentración, determinados efectos vinculados con el tiempo, como la fatiga u otros errores del operador, podrían ejercer un efecto más acusado a concentraciones más elevadas. Además, si es probable que los resultados se vean afectados por una condición inicial o ambiental del ensayo, como la posición en el laboratorio, habrá que pensar en la conveniencia de practicar agrupamientos. Alimentación En ensayos semiestáticos, la alimentación debe ser preferentemente diaria; en cualquier caso, tendrá una frecuencia mínima de 3 veces por semana (es decir, en correspondencia con los cambios de medio). Debe tenerse en cuenta la posible dilución de las concentraciones de exposición debida a la adición de los alimentos, y evitarla en la medida de lo posible utilizando suspensiones de algas concentradas. Deben documentarse las desviaciones con respecto a esta pauta (por ejemplo, ensayos dinámicos). Durante el ensayo, la dieta de los animales parentales debe estar formada preferentemente por algas vivas de uno o varios de los siguientes tipos: Chlorella sp., Pseudokirchneriella subcapitata (antes Selenastrum capricornutum) y Desmodesmus subspicatus (antes Scenedesmus subspicatus). La dieta suministrada se basará en la cantidad de carbono orgánico (C) proporcionada a cada animal parental. Las investigaciones realizadas (14) han demostrado que, en el caso de Daphnia magna, son suficientes raciones comprendidas entre 0,1 y 0,2 mg C/dafnia/día para alcanzar el número de descendientes vivos necesario a fin de cumplir los criterios de validez. La ración puede administrarse a una tasa constante durante todo el período de ensayo o, si se prefiere, a una tasa inferior al principio que se va incrementando en función del crecimiento de los animales parentales. No obstante, en este caso, las raciones deben permanecer en todo momento dentro del intervalo recomendado de 0,1 a 0,2 mg C/dafnia/día. Si es preciso recurrir a parámetros indirectos, como el número de células de algas o la absorbancia luminosa, para determinar la ración adecuada (por razones de comodidad, pues medir el contenido de carbono requiere mucho tiempo), cada laboratorio debe crear su propio nomograma para la medición indirecta del contenido de carbono del cultivo de algas (véanse algunos consejos sobre construcción de nomogramas en el apéndice 3). Los nomogramas deben revisarse al menos una vez al año, y con mayor frecuencia si cambian las condiciones de los cultivos de algas. Se ha observado que la absorbancia luminosa proporciona una indicación del contenido de carbono mejor que el número de células (15). Para reducir al mínimo el volumen de medio de cultivo de algas trasvasado a los recipientes de ensayo, la suspensión de algas administrada a las dafnias como alimento debe ser concentrada. Las algas pueden concentrarse mediante centrifugación seguida de resuspensión en medio de cultivo de dafnias. Iluminación Se aporta luz durante 16 horas a una intensidad no superior a 15-20 μE · m– 2 · s– 1, medida en la superficie del agua del recipiente. Con los instrumentos de medición de la luz calibrados en luxes, una gama equivalente de 1 000-1 500 lux con luz blanca fría corresponde aproximadamente a la intensidad luminosa recomendada de 15-20 μE·m– 2·s– 1.) Temperatura La temperatura de los medios de ensayo debe encontrarse en el intervalo de 18-22 °C. No obstante, en un mismo ensayo, la temperatura no debe variar, a ser posible, más de 2 °C dentro de estos límites (por ejemplo, 18-20, 19-21 o 20-22 °C). Puede ser conveniente utilizar un recipiente de ensayo adicional con el fin de vigilar la temperatura. Aireación Los recipientes de ensayo no deben recibir aireación durante el ensayo. Diseño del ensayo Ensayo de determinación del intervalo Cuando sea necesario, se llevará a cabo un ensayo de determinación del intervalo con, por ejemplo, cinco concentraciones del producto problema y dos réplicas de cada tratamiento y testigo. Para decidir sobre la gama de concentraciones que deba utilizarse en el ensayo de determinación del intervalo puede ser útil disponer de información adicional, procedente de ensayos con productos similares o de la bibliografía, sobre toxicidad aguda para Daphnia y/u otros organismos acuáticos. La duración del experimento de determinación del intervalo será de 21 días, o suficiente para predecir de manera fiable los niveles con efecto. Al final del ensayo se evalúa la reproducción de las dafnias. Debe registrarse el número de animales parentales y la presencia de descendientes. Ensayo definitivo Normalmente se deben preparar al menos cinco concentraciones de ensayo que abarquen la concentración con efecto (p. ej., ECx) y que formen una progresión geométrica con un factor de separación preferentemente no superior a 3,2. Debe utilizarse un número apropiado de réplicas con cada concentración de ensayo (véanse los puntos 24-25). Si se usan menos de cinco concentraciones, habrá que justificarlo. Los productos no deben someterse a ensayo a concentraciones superiores a su límite de solubilidad en el medio de ensayo. Antes de la realización del experimento, se recomienda considerar la potencia estadística del diseño de los ensayos utilizando métodos estadísticos apropiados (4). Al determinar el intervalo de concentraciones, debe tenerse en cuenta lo siguiente:
Si no se observan efectos a la concentración más alta en el ensayo de determinación del intervalo (p. ej., a 10 mg/l), o cuando se considere muy probable que el producto problema sea de toxicidad baja o nula, debido a la falta de toxicidad para otros organismos o a una absorción baja o nula, el ensayo de reproducción podrá realizarse como un ensayo límite, utilizando una concentración de ensayo de, por ejemplo, 10 mg/l y el testigo. Deben utilizarse diez réplicas, tanto para los grupos de tratamiento como para los testigos. Cuando tenga que hacerse un ensayo límite en un sistema dinámico, puede ser adecuado un número menor de réplicas. Un ensayo límite ofrece la oportunidad de demostrar que no existe ningún efecto estadísticamente significativo a la concentración límite, pero si se registra algún efecto habrá que hacer en principio un ensayo completo. Testigos Además de la serie de ensayo, debe realizarse una serie de testigo con el medio de ensayo y, si procede, otra que contenga el disolvente o el dispersante. Cuando se utilice disolvente o dispersante, su concentración ha de ser igual a la usada en los recipientes que contienen el producto problema. Hay que emplear el número adecuado de réplicas (véanse los puntos 23-24). Por lo general, en un ensayo bien ejecutado, el coeficiente de variación con respecto al número medio de descendientes vivos producidos por cada animal parental del testigo o testigos debe ser ≤ 25 %; este valor debe señalarse en caso de diseños de ensayo en los que se usen animales mantenidos por separado. Renovación del medio de ensayo La frecuencia de renovación del medio dependerá de la estabilidad del producto problema, pero debe ser al menos de tres veces por semana. Si de los ensayos de estabilidad preliminares (véase el punto 7) se deduce que la concentración del producto problema no es estable (es decir, si está fuera del intervalo comprendido entre el 80 y el 120 % del valor nominal o si cae por debajo del 80 % de la concentración inicial medida) a lo largo del período de renovación máximo (es decir, tres días), hay que considerar la renovación más frecuente del medio o la realización de un ensayo dinámico. Al renovar el medio en ensayos semiestáticos, se prepara una segunda serie de recipientes de ensayo y los animales parentales se transfieren a estos con ayuda, por ejemplo, de una pipeta de vidrio de diámetro adecuado. El volumen de medio transferido con las dafnias debe reducirse al mínimo. Observaciones Los resultados de las observaciones hechas durante el ensayo deben consignarse en fichas de datos (véanse ejemplos en los apéndices 4 y 5). Si hacen falta otras mediciones (véase el punto 44), puede ser necesario realizar más observaciones. Descendientes Preferiblemente, los descendientes producidos por cada animal parental se retirarán y contarán a diario desde la aparición de la primera nidada para evitar que consuman el alimento destinado a los padres. A los efectos de este método, solo es necesario contar el número de descendientes vivos, aunque debe registrarse la presencia de huevos abortados o de descendientes muertos. Mortalidad La mortalidad de los animales parentales se debe registrar preferiblemente a diario y al menos en las mismas ocasiones en que se cuenten los descendientes. Otros parámetros Aunque este método de ensayo se ha diseñado principalmente para evaluar los efectos sobre el resultado reproductor, quizá sea posible cuantificar otros efectos de forma suficiente para admitir el análisis estadístico. Se puede registrar el resultado reproductor por animal parental superviviente, es decir, el número de descendientes vivos producidos durante el ensayo por animal parental superviviente. Esto puede compararse con la variable de respuesta principal (resultado reproductor por animal parental al comienzo de la prueba que no haya muerto de forma fortuita o inadvertida durante el ensayo). Si se da mortalidad de animales parentales en réplicas expuestas, debe considerarse si la mortalidad sigue o no una pauta de respuesta a la concentración, por ejemplo si hay una regresión significativa de la respuesta frente a la concentración del producto problema con pendiente positiva (podría utilizarse para ello un método estadístico como la prueba de tendencia de Cochran-Armitage). Si la mortalidad no sigue una pauta de respuesta a la concentración, entonces esas réplicas con mortalidad de animales parentales deben excluirse del análisis de los resultados del ensayo. Si la mortalidad sigue una pauta de respuesta a la concentración, la mortalidad parental debe asignarse como efecto del producto problema y las réplicas no deben excluirse del análisis del resultado del ensayo. Las mediciones del crecimiento son muy interesantes, pues aportan información sobre posibles efectos subletales que pueden resultar útiles además de las medidas de la reproducción solas; es recomendable medir la longitud de los animales parentales (longitud del cuerpo sin contar la espina anal) al término del ensayo. Otras variables que pueden medirse o calcularse son el tiempo transcurrido hasta la producción de la primera nidada (y de las siguientes), el número y el tamaño de las nidadas por animal, el número de nidadas abortadas, la presencia de machos recién nacidos (OCDE, 2008) o efipios y, posiblemente, la tasa intrínseca de aumento de la población (véase el apéndice 1 para la definición y el apéndice 7 para la determinación del sexo de los recién nacidos). Frecuencia de las determinaciones analíticas y de las mediciones La concentración de oxígeno, la temperatura, la dureza y el pH deben medirse al menos una vez a la semana, en los medios fresco y viejo, en los testigos y a la concentración más alta del producto problema. Durante el ensayo, las concentraciones del producto problema deben determinarse a intervalos regulares. En los ensayos semiestáticos en los que se espere que la concentración del producto problema se mantenga dentro del ± 20 % de la concentración nominal (es decir, en el intervalo del 80-120 %, véanse los puntos 6, 7 y 39), se recomienda que las concentraciones de ensayo más alta y más baja se analicen recién preparadas y en el momento de su renovación al menos una vez durante la primera semana del ensayo (es decir, deben efectuarse análisis con una muestra de la misma solución, recién preparada y en el momento de su renovación). A partir de este momento, las determinaciones han de repetirse a intervalos al menos semanales. En ensayos en los que no se espere que la concentración del producto problema se mantenga dentro del ± 20 % de la concentración nominal, es preciso analizar todas las concentraciones de ensayo, recién preparadas y en el momento de su renovación. No obstante, en los ensayos en los que la concentración inicial medida del producto problema no esté dentro del ± 20 % de la concentración nominal, pero en los que puedan aportarse pruebas suficientes de que las concentraciones iniciales son repetibles y estables (es decir, que están dentro del intervalo del 80-120 % de la concentración inicial), las determinaciones del producto podrían limitarse en las semanas 2 y 3 del ensayo a las concentraciones más alta y más baja. En todos los casos, la determinación de las concentraciones de producto problema antes de la renovación solo debe realizarse en un recipiente replicado de cada concentración de ensayo. Para los ensayos dinámicos es apropiado un régimen de muestreo similar al descrito para los semiestáticos (si bien en este caso no cabe hacer determinaciones de las soluciones «viejas»). Sin embargo, puede ser aconsejable aumentar el número de ocasiones de muestreo durante la primera semana (tres series de mediciones, por ejemplo) para garantizar que las concentraciones de ensayo permanezcan estables. En esta clase de ensayos, debe verificarse a diario el caudal del diluyente y del producto problema. Si hay indicios de que la concentración de producto problema se ha mantenido satisfactoriamente a lo largo de todo el ensayo dentro del intervalo de ± 20 % de la concentración nominal o de la medida inicialmente, los resultados pueden basarse en los valores nominales o medidos inicialmente. Si la desviación respecto a la concentración nominal o medida inicialmente es superior al ± 20 %, los resultados deben expresarse en términos de media ponderada en función del tiempo (véanse las orientaciones para el cálculo en el apéndice 6). DATOS E INFORME Tratamiento de los resultados El objetivo del presente ensayo es determinar los efectos del producto problema sobre el resultado reproductor. El número total de descendientes vivos por animal parental debe calcularse por recipiente de ensayo (es decir, por cada réplica). Además, la reproducción puede calcularse sobre la base de la producción de descendientes vivos por organismo parental superviviente. Sin embargo, la variable de respuesta más pertinente desde el punto de vista medioambiental es el número total de descendientes vivos producidos por animal parental que no muere de forma fortuita (96) o inadvertida (97) durante el ensayo. Si el animal parental muere de forma fortuita o inadvertida durante el ensayo o resulta ser macho, la réplica correspondiente se excluye del análisis. Por tanto, este se basará en un número reducido de réplicas. Si se da mortalidad de animales parentales en réplicas expuestas, debe considerarse si la mortalidad sigue o no una pauta de respuesta a la concentración, por ejemplo si hay una regresión significativa de la respuesta frente a la concentración del producto problema con pendiente positiva (puede utilizarse para ello un método estadístico como la prueba de tendencia de Cochran-Armitage). Si la mortalidad no sigue una pauta de respuesta a la concentración, entonces esas réplicas con mortalidad de animales parentales deben excluirse del análisis de los resultados del ensayo. Si la mortalidad sigue una pauta de respuesta a la concentración, la mortalidad parental debe asignarse como efecto del producto problema y las réplicas no deben excluirse del análisis del resultado del ensayo. En resumen, cuando la LOEC y la NOEC o la ECx se utilizan para expresar los efectos, se recomienda calcular los efectos sobre la reproducción utilizando las dos variables de respuesta mencionadas, es decir:
y después utilizar como resultado final el valor más bajo de LOEC y NOEC o ECx calculado utilizando cualquiera de estas dos variables de respuesta. Antes de emplear los análisis estadísticos, p. ej. procedimientos de ANOVA, comparación de los grupos de tratamiento con el testigo mediante la prueba t de Stident, la prueba de Dunnett, la prueba de Williams, o la prueba de ajuste secuencial de Jonckheere-Terpstra, se recomienda considerar la transformación de los datos cuando sea necesario para cumplir los requisitos de la prueba estadística de que se trate. Se pueden considerar como alternativas no paramétricas la prueba de Dunn o la de Mann-Whitney. Se calculan los intervalos de confianza del 95 % de las medias de los distintos grupos de tratamiento. El número de animales parentales supervivientes en los testigos sin tratar es un criterio de validez, y debe documentarse y comunicarse. También deben consignarse en el informe final todos los demás efectos nocivos, por ejemplo el comportamiento anormal y las observaciones toxicológicas significativas. ECx Los valores de ECx, junto con sus correspondientes límites de confianza inferior y superior, se calculan utilizando métodos estadísticos apropiados (por ejemplo, función logística o de Weibull, método de Spearman-Karber recortado o interpolación simple). Para calcular la EC10, la EC50 o cualquier otra ECx, debe someterse a análisis de regresión el conjunto completo de datos. NOEC/LOEC Si el análisis estadístico se dirige a determinar la NOEC/LOEC, deben utilizarse métodos estadísticos adecuados de acuerdo con el documento 54 de la OCDE sobre enfoques actuales en el análisis estadístico de los datos de ecotoxicidad: guía de aplicación (4). En general, los efectos adversos del producto problema en comparación con el testigo se investigan utilizando contrastes de hipótesis unilaterales con p ≤ 0,05. La distribución normal y la homogeneidad de la varianza pueden probarse mediante un método estadístico adecuado, p. ej. la prueba de Shapiro-Wilk y la prueba de Levene, respectivamente (p ≤ 0,05). Puede realizarse ANOVA de un factor, seguido de pruebas de comparación múltiple. Las comparaciones múltiples (por ejemplo, prueba de Dunnett) o las pruebas de tendencia de ajuste secuencial (p. ej., prueba de Williams o prueba de ajuste secuencial de Jonckheere-Terpstra) pueden utilizarse para calcular si existen diferencias significativas (p ≤ 0,05) entre los testigos y las diversas concentraciones del producto problema [selección del ensayo recomendado de acuerdo con el documento de orientación 54 de la OCDE (4)]. De lo contrario, para determinar la LOEC y la NOEC pueden utilizarse métodos no paramétricos (p. ej., prueba U de Bonferroni según Holm o prueba de tendencia de Jonckheere-Terpstra). Ensayo límite Si se realiza un ensayo límite (comparación del testigo y de un solo un tratamiento) y se cumplen los requisitos previos de procedimientos de ensayos paramétricos (normalidad, homogeneidad), pueden evaluarse las respuestas métricas con la prueba de Student (prueba t). Si estos requisitos no se satisfacen, puede utilizarse una prueba t de varianzas desiguales (como la prueba de Welch) o una prueba no paramétrica, como la prueba U de Mann-Whitney. Para determinar la existencia de diferencias significativas entre los testigos (testigo y testigo del disolvente o dispersante), las réplicas de cada testigo pueden someterse a ensayo según lo descrito para el ensayo límite. En caso de que tales ensayos no detecten diferencias significativas, pueden agruparse todas las réplicas de testigo y de testigo del disolvente. En caso contrario, todos los tratamientos deben compararse con el testigo del disolvente. Informe del ensayo El informe del ensayo debe incluir la información siguiente:
BIBLIOGRAFÍA
Apéndice 1 DEFINICIONES A efectos del presente método de ensayo, se aplican las siguientes definiciones: Mortalidad fortuita : mortalidad no relacionada con el producto, de causa accidental (es decir, causa conocida). Producto : sustancia o mezcla. ECx : concentración del producto problema disuelto en agua que produce una reducción del x % en la reproducción de Daphnia dentro del período de exposición establecido. Mortalidad inadvertida : mortalidad no relacionada con el producto, sin que se conozca su causa. Tasa intrínseca de aumento de la población : medida del crecimiento de la población que integra el resultado reproductor y la mortalidad específica de la edad (1) (2) (3). En poblaciones en equilibrio estacionario es cero. En poblaciones en crecimiento es positiva, y negativa en las que disminuyen. Obviamente, toda tasa negativa es insostenible y en última instancia conduce a la extinción. Límite de detección : concentración más baja que puede detectarse, aunque no cuantificarse. Límite de determinación : concentración más baja que puede medirse cuantitativamente. Concentración mínima con efecto observado (LOEC) : concentración estudiada mínima a la que se observa que el producto ejerce un efecto estadísticamente significativo en la reproducción y en la mortalidad de los animales parentales (con p < 0,05) cuando se compara con el testigo, dentro de un tiempo de exposición dado. Además, todas las concentraciones de ensayo superiores a la LOEC deben ejercer un efecto nocivo igual o mayor que el observado a dicha concentración. Si no se cumplen estas dos condiciones, es preciso dar una explicación completa sobre cómo se ha elegido la LOEC (y, por tanto, la NOEC). Mortalidad : un animal se considera muerto cuando está inmóvil, es decir, cuando es incapaz de nadar o cuando no se observa movimiento de los apéndices ni de la región posterior del abdomen en el plazo de 15 segundos desde una agitación suave del recipiente de ensayo (si se usa otra definición, debe indicarse junto con la referencia correspondiente). Concentración sin efecto observado (NOEC) : concentración de ensayo inmediatamente inferior a la LOEC que, en comparación con el testigo, no ejerce ningún efecto estadísticamente significativo (p < 0,05) dentro del periodo de exposición establecido. Descendientes : ejemplares de dafnia jóvenes producidos por los animales parentales en el curso del ensayo. Animales parentales : hembras de dafnia presentes al principio del ensayo y cuyo resultado reproductor constituye el objeto de estudio. Resultado reproductor : número de descendientes vivos producidos por los animales parentales dentro del período de ensayo. Producto problema : toda sustancia o mezcla estudiada con este método de ensayo. BIBLIOGRAFÍA
Apéndice 2 PREPARACIÓN DE MEDIOS ELENDT M7 Y M4 TOTALMENTE DEFINIDOS Aclimatación a los medios Elendt M7 y M4 Algunos laboratorios han tenido dificultades para transferir directamente las dafnias a los medios M4 (1) y M7. Sin embargo, se ha obtenido cierto éxito con una aclimatación gradual, esto es, cambiando del medio propio a Elendt al 30 %, luego al 60 % y por último al 100 %. El período de aclimatación necesario puede ser bastante largo, incluso de un mes. Preparación Oligoelementos Se empieza por preparar soluciones madre distintas (I) de cada oligoelemento en agua de pureza apropiada (desionizada, destilada o tratada mediante ósmosis inversa). A partir de estas soluciones madre (I) se prepara una segunda solución madre única (II) que contiene todos los oligoelementos (solución combinada), de la forma siguiente:
Medios M4 y M7 Los medios M4 y M7 se preparan a partir de la solución madre II con los macronutrientes y las vitaminas que siguen:
La solución madre de vitaminas combinadas se conserva congelada en pequeñas partes alícuotas. Las vitaminas se incorporan a los medios poco antes de usarlos.
Apéndice 3 ANÁLISIS DEL CARBONO ORGÁNICO TOTAL (COT) Y ELABORACIÓN DE UN NOMOGRAMA DEL CONTENIDO EN COT DEL ALIMENTO DE ALGAS Es sabido que el contenido en carbono del alimento de algas no suele medirse directamente, sino por medio de correlaciones (es decir, de nomogramas) con medidas indirectas, como el número de células de algas o la absorbancia de luz. El COT debe medirse por oxidación a alta temperatura y no con métodos de UV o de persulfato (véase más información en: The Instrumental Determination of Total Organic Carbon, Total Oxygen Demand and Related Determinands 1979, HMSO 1980; 49 High Holborn, London WC1V 6HB). Para elaborar el nomograma, las algas se separan del medio de crecimiento por centrifugación seguida de resuspensión en agua destilada. Se mide la variable indirecta y la concentración de COT en cada una de las muestras por triplicado. Hay que analizar testigos con agua destilada únicamente y restar su concentración de COT de la observada en la muestra de algas. El nomograma debe ser lineal a lo largo del intervalo necesario de concentraciones de carbono. A continuación se ilustran algunos ejemplos.
Chlorella vulgaris var. viridis (CCAP 211/12). Regresión de mg/l de peso seco frente a mg C/1. Datos de suspensiones concentradas de células cultivadas en lotes semicontinuos, suspendidas de nuevo en agua destilada. Eje de abscisas: mg C/1 de alimento concentrado de algas Eje de ordenadas: mg/1 de peso seco del alimento concentrado de algas Coeficiente corrector -0,980 
Chlorella vulgaris var. viridis (CCAP 211/12). Regresión del número de células frente a mg C/1. Datos de suspensiones concentradas de células cultivadas en lotes semicontinuos, suspendidas de nuevo en agua destilada. Eje de abscisas: mg C/1 de alimento concentrado de algas Eje de ordenadas: N.o de células/1 de alimento concentrado de algas Coeficiente corrector -0,926 
Chlorella vulgaris var. viridis (CCAP 211/12). Regresión de la absorbancia frente a mg C/1 (1 cm de camino óptico). Datos de suspensiones concentradas de células cultivadas en lotes semicontinuos, suspendidas de nuevo en agua destilada. Eje de abscisas: mg C/1 de alimento concentrado de algas Eje de ordenadas: absorbancia a 440 nm de una dilución al 1/10 de alimento concentrado de algas Coeficiente corrector -0,998 Apéndice 4 EJEMPLO DE FICHA DE DATOS RELATIVOS A LA RENOVACIÓN DEL MEDIO, LA VIGILANCIA FISICOQUÍMICA, LA ALIMENTACIÓN, LA REPRODUCCIÓN Y LA MORTALIDAD DE ANIMALES PARENTALES DE DAFNIA
Apéndice 5 EJEMPLO DE FICHA DE DATOS RELATIVOS A LOS RESULTADOS DE LOS ANÁLISIS DEL PRODUCTO a) Concentraciones medidas
b) Concentraciones medidas en porcentaje de la concentración nominal
Apéndice 6 CÁLCULO DE UNA MEDIA PONDERADA EN FUNCIÓN DEL TIEMPO Media ponderada en función del tiempo Puesto que la concentración del producto problema puede disminuir a lo largo del tiempo comprendido entre las sucesivas renovaciones del medio, es preciso considerar cuál es la concentración que debe elegirse como representativa de la gama de concentraciones aplicadas a las dafnias parentales. La elección debe basarse en consideraciones biológicas y estadísticas. Si, por ejemplo, se piensa que la mayor influencia en la reproducción la ejerce la máxima concentración aplicada, debe usarse este valor máximo. En cambio, si se considera más importante el efecto acumulado o a plazo más largo del producto tóxico, será más relevante la concentración media. En este caso, una media apropiada será la concentración media ponderada en función del tiempo, pues tiene en cuenta la variación de la concentración instantánea a lo largo del tiempo. Figura 1 Ejemplo de media ponderada en función del tiempo 
La figura 1 ilustra un ejemplo de ensayo (simplificado) de 7 días de duración en el cual el medio se renueva los días 0, 2 y 4.
La media ponderada en función del tiempo se calcula de manera que el área situada bajo ella sea igual al área situada bajo la curva de concentración. El cálculo correspondiente al ejemplo anterior se ilustra en el cuadro 1. Cuadro 1 Cálculo de una media ponderada en función del tiempo
Días : número de días del periodo de renovación. Conc 0 : concentración medida al principio de cada periodo de renovación. Conc 1 : concentración medida al final de cada periodo de renovación. Ln (conc 0) : logaritmo natural de conc 0. Ln (conc 1) : logaritmo natural de conc 1. Superficie : área comprendida bajo la curva exponencial para cada periodo de renovación. Se calcula como sigue: FOR-L_2017112ES.01000301.notes.0059.xml.jpgLa media ponderada en función del tiempo (Media PT) es igual a la superficie total dividida por los días totales. Naturalmente, en el caso de un ensayo de reproducción de dafnias, el cuadro debe ampliarse para cubrir 21 días. Es obvio que, si solo se hacen observaciones al principio y al final de cada periodo de renovación, será imposible confirmar que el proceso de descomposición sea, en efecto, exponencial. Una curva distinta daría lugar a un cálculo distinto de la superficie. En cualquier caso, la descomposición exponencial es una hipótesis plausible, y esta es probablemente la curva mejor a falta de más datos. No obstante, es preciso hacer una advertencia para el caso de que el análisis del producto no logre detectar ningún producto al final del periodo de renovación. Salvo que se pueda estimar la velocidad a la que el producto desaparece de la solución, será imposible obtener un área bajo la curva realista y, por tanto, obtener una media ponderada en función del tiempo que sea razonable. Apéndice 7 ORIENTACIONES PARA LA IDENTIFICACIÓN DEL SEXO DE LOS RECIÉN NACIDOS Puede darse producción de recién nacidos machos en condiciones ambientales cambiantes, tales como la reducción del fotoperiodo, la temperatura, la reducción de la concentración alimentaria, y el aumento de la densidad de población (Hobaek y Larson, 1990; Bauch et al., 1992). La producción de machos es también una respuesta conocida a ciertos reguladores del crecimiento de los insectos (Oda et al., 2005). En condiciones en que los productos agresores inducen una reducción de la descendencia reproductora de las hembras partenogenéticas, es de esperar un mayor número de machos (OCDE, 2008). Sobre la base de la información disponible, no es posible predecir qué será más sensible, si la proporción de machos y hembras o el parámetro de la reproducción; sin embargo, existen indicios (“informe de validación” de referencia, parte 1) de que este aumento en el número de machos podría ser menos sensible que la disminución de descendencia. Puesto que la finalidad primaria del método de ensayo es evaluar el número de descendientes producidos, la aparición de machos es una observación opcional. Si este parámetro opcional se evalúa en un estudio, debe emplearse el criterio adicional de validez del ensayo de no más del 5 % de machos en los testigos. La forma más práctica y fácil de distinguir el sexo de las dafnias es utilizar sus características fenotípicas, ya que los machos y las hembras son genéticamente idénticos, y su sexo es determinado por los factores ambientales. Los machos y las hembras difieren en la longitud y la morfología de las primeras antenas, que son más largas en los machos que en las hembras (fig. 1). Esta diferencia es reconocible justo después del nacimiento, aunque según crecen se van desarrollando otros caracteres sexuales secundarios (por ejemplo, véase la figura 2 de Olmstead y LeBlanc, 2000). Para observar el sexo morfológico, los animales recién nacidos producidos por cada animal del ensayo deben transferirse con pipeta y colocarse en una placa de Petri con medio de ensayo. El medio se reduce a un mínimo para limitar el movimiento de los animales. La observación de las primeras antenas puede efectuarse en un microscopio estereoscópico ('10-60). Figura 1. Macho (izquierda) y hembra (derecha) de 24 horas de vida de D. magna. Los machos se distinguen de las hembras por la longitud y la morfología de las primeras antenas, como se muestra en los círculos (Tatarazako et al., 2004). 
BIBLIOGRAFÍA Hobaek A and Larson P. 1990. Sex determination in Daphnia magna. Ecology 71: 2255-2268. Kleiven O.T., Larsson P., Hobaek A. 1992. Sexual reproduction in Daphnia magna requires three stimuli. Oikos 65, 197-206. Oda S., Tatarazako N, Watanabe H., Morita M., and Iguchi T. 2005. Production of male neonates in Daphnia magna (Cladocera, Crustacea) exposed to juvenile hormones and their analogs. Chemosphere 61:1168-1174. OCDE, 2008. Validation report for an enhancement of OECD TG 211 Daphnia magna reproduction test. OECD Series on Testing and Assessment, Number 88. Organisation for Economic Co-operation and Development, Paris. Olmstead, A.W., LeBlanc, G.A., 2000. Effects of endocrine-active chemicals on the development characteristics of Daphnia magna. Environmental Toxicology and Chemistry 19:2107-2113. Tatarazako, N., Oda, S., Abe, R., Morita M. and Iguchi T., 2004. Development of a screening method for endocrine disruptors in crustaceans using Daphnia magna (Cladocera, Crustacea). Environmental Science 17, 439-449. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
18) |
En la parte C, capítulo C.29, el punto 66 se sustituye por el texto siguiente:
|
|
19) |
En la parte C, se añaden los siguientes capítulos: «C.47. Ensayo de toxicidad en las primeras fases de vida de los peces INTRODUCCIÓN
PRINCIPIO DEL ENSAYO
INFORMACIÓN SOBRE EL PRODUCTO PROBLEMA
VALIDEZ DEL ENSAYO
DESCRIPCIÓN DEL MÉTODO Recipientes de ensayo
Selección de las especies
Preparación de los peces reproductores
Manipulación de los huevos fecundados, los embriones y las larvas
Agua
Soluciones de ensayo
PROCEDIMIENTO Condiciones de exposición Duración
Carga
Luz y temperatura
Alimentación
Concentraciones de ensayo
Testigos
Frecuencia de las determinaciones analíticas y las mediciones
Observaciones 26. Fase de desarrollo embrionario : al principio de la exposición al producto problema debe comprobarse con tanta precisión como sea posible la fase embrionaria en una muestra representativa de huevos debidamente conservados y limpios. 27. Eclosión y supervivencia : al menos una vez al día deben observarse y contarse las eclosiones y los supervivientes, y anotar estos datos. Si se observan hongos en los huevos al principio de la fase de desarrollo embrionario (p. ej., en el primer o segundo día del ensayo), los huevos afectados deberán contarse y retirarse. Los embriones, las larvas y los peces juveniles muertos deben retirarse en cuanto se localicen, ya que pueden descomponerse rápidamente y pueden deshacerse con las acciones de los demás peces. Dicha retirada ha de hacerse con sumo cuidado para no dañar físicamente los huevos y larvas adyacentes. Los signos para considerar que un organismo está muerto varían según la especie y la fase del desarrollo. Por ejemplo:
28. Aspecto anómalo : el número de larvas o peces juveniles que presenten anomalías corporales debe registrarse a intervalos adecuados con arreglo a la duración del ensayo y la naturaleza de la anomalía observada. Cabe señalar que la aparición de larvas y peces juveniles anómalos ocurre en condiciones normales y que su proporción puede alcanzar varias unidades por cada cien en los testigos de algunas especies. Si las deformaciones y los comportamientos anómalos asociados se consideran tan graves como para que haya un notable sufrimiento del organismo y se ha alcanzado un punto en el que la recuperación no será posible, se podrá retirar el organismo del ensayo. Se deberá practicar la eutanasia a dichos animales y se considerarán como muertes en el posterior análisis de datos. Se ha documentado un desarrollo embrionario normal para la mayoría de las especies recomendadas en este método de ensayo (9) (10) (11) (12). 29. Comportamiento anómalo : las anomalías como la hiperventilación, natación descoordinada, inactividad atípica o comportamiento alimentario atípico deben registrarse a intervalos adecuados según la duración del ensayo (p. ej., una vez al día para las especies de aguas cálidas). Pese a ser difíciles de cuantificar, estos efectos, si se observan, pueden contribuir a interpretar los datos de mortalidad. 30. Peso : al final del ensayo deben pesarse todos los peces supervivientes, al menos por réplicas (registrando el número de animales en cada réplica y el peso medio por animal); se prefiere el peso húmedo (secados con material absorbente), aunque también se podrá registrar el peso seco (13). 31. Longitud : al final del ensayo debe medirse la longitud de cada sujeto. Se recomienda medir la longitud total de los peces; no obstante, si se observa una descomposición de la aleta caudal o un desgaste de las aletas, debe emplearse la longitud estándar. Debe usarse el mismo método para medir todos los peces de un ensayo determinado. La longitud de cada sujeto puede medirse, p. ej., mediante pinzas, una cámara digital o un micrómetro ocular calibrado. En el apéndice 2 se recogen las longitudes mínimas típicas. DATOS E INFORME Tratamiento de los resultados
Informe del ensayo
BIBLIOGRAFÍA
Apéndice 1 DEFINICIONES Longitud furcal (FL): longitud desde la punta del hocico hasta la escotadura de la aleta caudal. Se utiliza en peces en los que es difícil apreciar dónde termina la columna vertebral (www.fishbase.org). Longitud estándar (SL) : longitud de un pez medida desde la punta del hocico hasta el extremo posterior de la última vértebra o hasta el extremo posterior de la parte mediolateral de la placa hipural. Básicamente, en esta medida se excluye la aleta caudal (www.fishbase.org). Longitud total (TL) : longitud desde la punta del hocico hasta la punta del lóbulo más largo de la aleta caudal, normalmente se mide con los lóbulos comprimidos a lo largo de la línea media. Se mide en línea recta, y no siguiendo la curvatura del cuerpo (www.fishbase.org). Figura 1 Descripción de las diferentes longitudes utilizadas Longitud estándar Longitud furcal Longitud total Producto : sustancia o mezcla. ECx : (concentración con efecto del x %); concentración que provoca el x % de un efecto sobre los organismos de ensayo dentro de un determinado periodo de exposición cuando se compara con un testigo. Por ejemplo, una EC50 es una concentración de la que se estima que causa un efecto sobre un parámetro de ensayo en el 50 % de una población a lo largo de un determinado periodo de exposición. Concentración mínima con efecto observado (LOEC) : concentración más baja de producto problema con la que se observa un efecto estadístico significativo (con p < 0,05) en comparación con el testigo. Además, todas las concentraciones de ensayo superiores a la LOEC deben ejercer un efecto nocivo igual o mayor que el observado a dicha concentración. Si no se cumplen estas dos condiciones, es preciso dar una explicación completa sobre cómo se ha elegido la LOEC (y, por tanto, la NOEC). En los apéndices 5 y 6 se recogen las directrices pertinentes. Concentración sin efecto observado (NOEC) : concentración de ensayo inmediatamente inferior a la LOEC que, en comparación con el testigo, no ejerce ningún efecto estadísticamente significativo (p < 0,05) dentro del periodo de exposición establecido. Producto problema : sustancia o mezcla estudiada con este método de ensayo. UVCB : sustancia de composición desconocida o variable, productos de reacción compleja y materiales biológicos. IUPAC : Unión Internacional de Química Pura y Aplicada. SMILES : Sistema Simplificado de Registro de Líneas Moleculares. Apéndice 2 CONDICIONES Y DURACIÓN DEL ENSAYO Y CRITERIOS DE SUPERVIVENCIA PARA LAS ESPECIES RECOMENDADAS
Apéndice 3 DIRECTRICES DE ALIMENTACIÓN Y MANIPULACIÓN DE PECES REPRODUCTORES Y ANIMALES DE ENSAYO PARA LAS ESPECIES RECOMENDADAS
Apéndice 4 CARACTERÍSTICAS QUÍMICAS DE UN AGUA DE DILUCIÓN ADECUADA
Apéndice 5 ORIENTACIONES ESTADÍSTICAS PARA LA DETERMINACIÓN DE LA NOEC Consideraciones generales Los recipientes replicados en paralelo (réplicas) constituyen la unidad de análisis. De este modo, para obtener mediciones continuas, como el tamaño, se deberá calcular la media o mediana de las réplicas y estos valores serán los datos que se usen para el análisis. Habrá que demostrar la potencia de los ensayos utilizados, preferiblemente a partir de una base de datos históricos de cada laboratorio. Para cada parámetro deberá indicarse el efecto sobre el tamaño que pueda detectarse con una potencia del 75-80 % junto con la prueba estadística que se usará. Las bases de datos disponibles en el momento de elaborar el presente método de ensayo indican la potencia que podrá alcanzarse con los procedimientos estadísticos recomendados. Cada laboratorio deberá demostrar su capacidad para cumplir con este requisito de potencia, bien realizando sus propios análisis de potencia o bien probando que el coeficiente de variación (CV) de cada respuesta no sobrepasa el percentil 90 de los CV utilizados para elaborar las directrices de ensayo. El cuadro 1 recoge dichos CV. Si solo se dispone de las medias o medianas de las réplicas, se podrá ignorar el CV intra-réplicas. Cuadro 1 Percentil 90 de los CV de las especies de agua dulce seleccionadas
En casi todas las pruebas estadísticas empleadas para evaluar los estudios de toxicología en laboratorios, las comparaciones relevantes son las que se hacen entre los grupos de tratamiento y el testigo. Por esta razón, no es apropiado exigir una prueba F de ANOVA significativa antes de usar la prueba de Dunnett o de Williams o una prueba de Kruskal-Wallis significativa antes de utilizar la prueba de Jonckheere-Terpstra, Mann-Whitney o Dunn (Hochberg y Tamhane 1987, Hsu 1996, Dunnett 1955, 1964, Williams 1971, 1972, 1975, 1977, Robertson et al. 1988, Jonckheere 1954, Dunn 1964). La prueba de Dunnett incluye un ajuste de multiplicidad y, al emplear la prueba F como filtro, los porcentajes de falsos positivos y falsos negativos podrían verse afectados de forma negativa. Asimismo, las pruebas de ajuste secuencial de Williams y Jonckheere-Terpstra con un nivel de significación del 0,05 en cada paso conservan un porcentaje global de falsos positivos del 5 %. Este porcentaje y la potencia de las pruebas pueden verse afectadas de forma negativa si se usa la prueba F o la prueba de Kruskal-Wallis como filtro. Debe ajustare la multiplicidad de las pruebas de Mann-Whitney y de Dunn; se recomienda el ajuste de Bonferroni-Holm. En las directrices de la OCDE (2006) se recoge una exhaustiva discusión sobre la mayoría de las recomendaciones para las pruebas de hipótesis y la verificación de supuestos en las que se basan estos ensayos; además, incluyen una amplia bibliografía. Tratamiento de los testigos cuando se usa un disolvente Si se usa un disolvente, deberán incluirse un testigo de agua de dilución y un testigo de disolvente. Los dos testigos deben compararse para cada respuesta y, si no se observa una diferencia significativa entre ambos, deberán combinarse para realizar un análisis estadístico. Si se observa una diferencia significativa, el testigo de disolvente deberá utilizarse para determinar la NOEC o estimar la ECx y no se utilizará el testigo del agua. Véanse las restricciones de los criterios de validez en el punto 7. Respecto a la longitud, el peso, la tasa de eclosión de huevos, de mortalidad de larvas o de larvas anómalas y el primero o el último día de eclosión o de la etapa de nadar hacia arriba, se debe realizar una prueba T o una prueba de Mann-Whitney para comparar el testigo de agua de dilución y el testigo de disolvente al nivel de significación del 0,05, ignorando todos los grupos de tratamiento. Los resultados de estas pruebas deberán recogerse en el informe. Mediciones de tamaño (longitud y peso) Los valores de longitud y peso de cada pez pueden tener una distribución normal o una distribución logarítmica normal. En ambos casos, las medias de las réplicas tienden hacia una distribución normal debido al teorema del límite central; esto se confirma con datos de más de 100 estudios de las primeras fases de vida de tres especies de agua dulce. Por otro lado, cuando los datos o las bases de datos históricos sugieran una distribución logarítmica normal de los valores de los tamaños de cada pez, se podrá calcular el logaritmo de la media de las réplicas de los valores de cada pez y los datos del análisis podrán ser los antilogaritmos de dichos logaritmos de la media de las réplicas. Se deberán evaluar los datos en cuanto a la coherencia con una distribución normal y la homogeneidad de la varianza. Para ello, se utilizarán los valores residuales de un modelo ANOVA con la concentración como la única variable de tipo explicativa. Podrá llevarse a cabo una determinación visual a partir de diagramas de dispersión e histogramas o usarse diagramas de tallos y hojas. También se podrá emplear una prueba en regla, como la de Shapiro-Wilk o Anderson-Darling. La coherencia con la homogeneidad de la varianza puede evaluarse mediante un análisis visual del mismo diagrama de dispersión o de manera formal con la prueba de Levene. Solo habrá que evaluar la normalidad o la homogeneidad de la varianza en las pruebas paramétricas (p. ej., la de Williams o Dunnett). Se debe prestar especial atención a los posibles valores anómalos y a sus efectos sobre el análisis. Puede usarse la prueba de valores anómalos de Tukey e inspeccionar de forma visual las mismas representaciones gráficas de los valores residuales mencionados anteriormente. Cabe recordar que los objetos de observación son las réplicas enteras, por lo que solo se deberá omitir un valor anómalo del análisis tras una cuidadosa evaluación. Las pruebas estadísticas que hacen uso de las características del diseño experimental y de la expectativa biológica son las pruebas de ajuste secuencial, como las de Williams y Jonckheere-Terpstra. Estas pruebas asumen una relación concentración-respuesta monótona y deberá evaluarse la coherencia de los datos con dicho supuesto. Puede hacerse de forma visual mediante un diagrama de dispersión de la media de las réplicas frente a la concentración de ensayo. Será útil superponer el diagrama de dispersión con un diagrama lineal por tramos que una las medias de concentración ponderadas por el tamaño de las muestras de las réplicas. Una gran desviación de la monotonicidad de dicho diagrama lineal indicaría la posibilidad de tener que utilizar pruebas que no sean de tendencia. Pueden emplearse también pruebas en regla. Una sencilla prueba en regla consiste en calcular los contrastes lineales y cuadráticos de las medias de la concentración. Si el contraste cuadrático es significativo y el contraste lineal no lo es, puede haber un problema de monotonicidad que debe analizarse en profundidad mediante las representaciones gráficas. Cuando la normalidad o la homogeneidad de la varianza puedan suponer un problema, estos contrastes pueden crearse a partir de datos transformados por orden de rangos. Podrán utilizarse otros procedimientos, como la prueba de monotonicidad de Bartholomew, aunque añadirían complejidad. Figura 2 Diagrama de flujo de la NOEC de las mediciones de tamaño (longitud y peso) 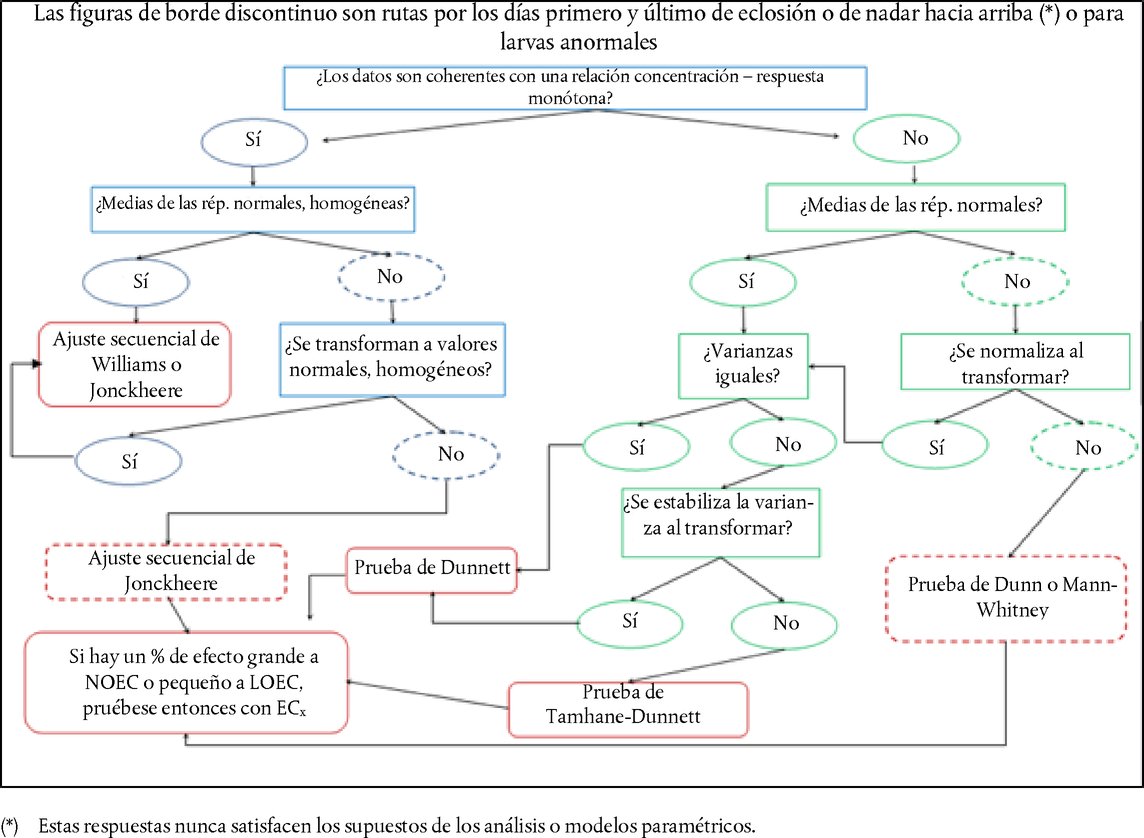
A menos que los datos no sean coherentes con los requisitos de estas pruebas, la NOEC se determinará con una aplicación de ajuste secuencial de la prueba de Williams o de Jonckheere-Terpstra. Las directrices de la OCDE (2006) ofrecen información sobre dichos procedimientos. Cuando los datos no sean coherentes con los requisitos de una prueba de tendencia de ajuste secuencial, se podrá utilizar la prueba de Dunnett o de Tamhane-Dunnett (T3); ambas pruebas incluyen ajustes de multiplicidad. Estas pruebas suponen que hay normalidad y, en el caso de Dunnet, homogeneidad de la varianza. Cuando no se cumplan las condiciones, se podrá utilizar la prueba no paramétrica de Dunn. En las directrices de la OCDE (2006) se incluye información sobre todas estas pruebas. La figura 2 ofrece una visión general para ayudar a elegir una prueba. Tasa de eclosión de los huevos y de supervivencia de las larvas Los datos son las proporciones de huevos que han eclosionado o de larvas que sobreviven en cada réplica. Dichas proporciones deben evaluarse en cuanto a su variación extra-binomial, que suele ser común pero no universal para tales mediciones. El diagrama de flujo de la figura 3 puede servir de orientación a la hora de seleccionar una prueba. En el texto se ofrecen descripciones detalladas. Habitualmente se usan dos pruebas; la prueba de Tarone C(α) (Tarone, 1979) y la prueba de ji cuadrado; se aplica cada una por separado a cada concentración de ensayo. Si se detecta variación extra-binomial incluso en solo una concentración de ensayo, deberán usarse métodos que se adapten a ello. Fórmula 1 Prueba C (α) de Tarone (Tarone 1979)
donde ̂ es la proporción media de una concentración determinada, m es el número de recipientes en paralelo, nj es el número de sujetos en la réplica j, y xj es el número de sujetos en dicha réplica que responden al tratamiento, p. ej., que no han eclosionado o están muertos. Esta prueba se aplica a cada concentración por separado. Aunque puede verse como una prueba de ji cuadrado ajustada, las simulaciones de potencia limitadas efectuadas por Tarone demostraron que es más potente que una prueba de ji cuadrado. Figura 3 Diagrama de flujo de la NOEC de la tasa de eclosión de huevos y de mortalidad de larvas 
Cuando no exista un indicio significativo de variación extra-binomial, podrá usarse la prueba de ajuste secuencial de Cochran-Armitage. Esta prueba ignora las réplicas, por ello cuando existe tal indicio, se recomienda usar el ajuste de Rao-Scott de la prueba de Cochran-Armitage (RSCA), que tiene en cuenta las réplicas, los tamaños de las réplicas y la variación extra-binomial. Pueden utilizarse otras pruebas, como la prueba de ajuste secuencial de Williams y Jonckheere-Terpstra y la prueba de Dunnett, como se describe para las mediciones de los tamaños. Estas pruebas se aplican haya o no variación extra-binomial, pero a veces pueden tener una menor potencia (Agresti 2002, Morgan 1992, Rao y Scott 1992, 1999, Fung et al. 1994, 1996). Primer o último día de eclosión o de la etapa de nadar hacia arriba La respuesta es un número entero, que da el día del ensayo en el que se detecta la observación indicada en un recipiente en paralelo determinado. Por lo general, la gama de valores es muy limitada y suele haber altas proporciones de valores vinculados, p. ej., se observa el mismo primer día de eclosión en todas las réplicas testigo y, quizás, en una o dos de las concentraciones de ensayo bajas. Las pruebas paramétricas, como las de Williams y Dunnett, no resultan adecuadas para estos datos. A menos que se disponga de indicios de una importante ausencia de monotonicidad, puede usarse la prueba de ajuste secuencial de Jonckheere-Terpstra, que es muy potente para detectar los efectos del producto problema. En caso contrario, podrá utilizarse la prueba de Dunn. Anomalías de las larvas La respuesta es el recuento de larvas en las que se ha observado algún aspecto anómalo. Con frecuencia, esta respuesta es de baja incidencia y presenta algunos de los mismos problemas que el primer día de eclosión, ya que a veces puede ser errática en cuanto a la relación concentración-respuesta. Si los datos siguen al menos una forma aproximada de concentración monótona, la prueba de ajuste secuencial de Jonckheere-Terpstra resulta eficaz para detectar los efectos. En caso contrario, podrá utilizarse la prueba de Dunn. BIBLIOGRAFÍA Agresti, A. (2002); Categorical Data Analysis, second edition, Wiley, Hoboken. Dunnett C. W. (1955); A multiple comparison procedure for comparing several treatments with a control, J. American Statistical Association 50, 1096-1121. Dunn O. J. (1964 ); Multiple Comparisons Using Rank Sums, Technometrics 6, 241-252. Dunnett C. W. (1964); New tables for multiple comparisons with a control, Biometrics 20, 482-491. Fung, K.Y., D. Krewski, J.N.K. Rao, A.J. Scott (1994); Tests for Trend in Developmental Toxicity Experiments with Correlated Binary Data, Risk Analysis 14, 639-648. Fung, K.Y, D. Krewski, R.T. Smythe (1996); A comparison of tests for trend with historical controls in carcinogen bioassay, Canadian Journal of Statistics 24, 431-454. Hochberg, Y. and A. C. Tamhane (1987); Multiple Comparison Procedures, Wiley, New York. Hsu, J.C. (1996); Multiple Comparisons: Theory and Methods; Chapman and Hall/CRC Press, Boca Raton. Jonckheere A. R. (1954); A distribution-free k-sample test against ordered alternatives, Biometrika 41, 133. Morgan, B.J.T. (1992); Analysis of Quantal Response Data, Chapman and Hall, London. OCDE (2006). Current approaches in the statistical analysis of ecotoxicity data: A guidance to application. Series on Testing and Assessment, No. 54. Organisation for Economic Co-operation and Development, OECD, Paris.. Rao J.N.K. and Scott A.J. (1992) — A simple method for the analysis of clustered binary data, Biometrics 48, 577-585. Rao J.N.K. and Scott A.J. (1999) — A simple method for analyzing overdispersion in clustered Poisson data, Statistics in Medicine 18, 1373-1385. Robertson, T., Wright F.T. and Dykstra R.L. (1988); Order restricted statistical inference, Wiley. Tarone, R.E. (1979); Testing the goodness of fit of the Binomial distribution, Biometrika 66, 585-590. Williams D.A. (1971); A test for differences between treatment means when several dose levels are compared with a zero dose control, Biometrics 27, 103-117. Williams D.A. (1972); The comparison of several dose levels with a zero dose control, Biometrics 28, 519-531. Williams D. A. (1975); The Analysis of Binary Responses from Toxicological Experiments Involving Reproduction and Teratology, Biometrics 31, 949-952. Williams D.A. (1977); Some inference procedures for monotonically ordered normal means, Biometrika 64, 9-14. Apéndice 6 ORIENTACIONES ESTADÍSTICAS PARA LAS ESTIMACIONES DE LA REGRESIÓN Consideraciones generales Las observaciones utilizadas para ajustar un modelo son las medias de las réplicas (longitud y peso) o las proporciones de las réplicas (eclosión de huevos y mortalidad de larvas) (OCDE 2006). Por lo general, se aconseja la regresión ponderada utilizando como peso el tamaño de las muestras de las réplicas. Se pueden emplear otros esquemas de ponderación, como la ponderación por respuesta media prevista o una combinación de esta ponderación y la del tamaño de las muestras de las réplicas. No se recomienda la ponderación mediante la inversa de la varianza de una misma muestra dentro de la concentración (Bunke et al. 1999, Seber y Wild, 2003, Motulsky y Christopoulos 2004, Huet et al. 2003). Cualquier transformación de las respuestas antes del análisis debe mantener la independencia de las observaciones y la ECx y sus límites de confianza deben expresarse en las unidades originales de medida, en lugar de en las unidades transformadas. Por ejemplo, un cambio del 20 % en el logaritmo de la longitud no equivale a un cambio del 20 % en la longitud (Lyles et al. 2008, Draper y Smith 1999). El diagrama de flujo de la figura 4 ofrece una visión general de las estimaciones de la ECx. Los detalles se describen más adelante. Figura 4 Diagrama de flujo de las estimaciones de la ECx de la longitud media por réplica del peso, o de la proporción de eclosiones de huevos o mortalidad de las larvas (véase el texto para más detalles) 
Consideraciones sobre la tasa de eclosión de huevos y de mortalidad de larvas Para la eclosión de huevos y la mortalidad de larvas, por lo general lo mejor es ajustar un modelo decreciente, a menos que se esté ajustando un modelo de probit, como se describe más adelante. Es decir, conviene crear un modelo para la proporción de huevos que no eclosionan o de larvas que mueren. El motivo de ello es que la ECx se refiere a una concentración a la que hay un cambio igual al x % de la respuesta media del testigo. Si el 5 % de los huevos testigo no consiguen eclosionar y se crea un modelo de la incapacidad de eclosionar, la EC20 se refiere a una concentración a la que se produce una variación del 20 % del 5 % de los huevos testigo no eclosionados, lo que supone una variación de 0,2*0,05 = 0,01 o 1 punto porcentual hasta una tasa de no eclosión de los huevos del 6 %. Una variación tan pequeña no se puede estimar de forma significativa a partir de los datos disponibles y no es biológicamente importante. Mientras que si se crea un modelo de la proporción de huevos que eclosionan, la proporción del testigo será del 95 % en este ejemplo y una reducción del 20 % con respecto a la media del testigo supondría una variación de 0,95*0,2 = 0,18, con lo que la tasa de eclosión pasaría del 95 % al 77 % (= 95-18); la concentración que produce dichos efectos puede estimarse y probablemente sea de más interés. No se trata de un problema con las mediciones de tamaño, aunque unos efectos adversos sobre el tamaño suelen traducirse en una disminución de la talla. Modelos relacionados con el tamaño (longitud o peso) y la tasa de eclosión de huevos o de supervivencia de las larvas Excepto el modelo hormético de Brain-Cousens, todos estos modelos se describen y recomiendan en las directrices de la OCDE (2006). También se discuten los experimentos de ecotoxicidad en Slob de los llamados modelos OCDE 2-5 (2002). Existen, por supuesto, muchos otros modelos que pueden resultar de utilidad. Bunke, et al. (1999) enumeran los muchos modelos que no se incluyen en el presente documento y ofrecen un gran número de referencias a otros modelos. Los modelos indicados a continuación se consideran especialmente adecuados en experimentos de ecotoxicidad y se han usado ampliamente. Con 5 concentraciones de ensayo más un testigo
Con 6 concentraciones de ensayo como mínimo más un testigo
Cuando existen indicios visuales de hormesis (poco probable en caso de éxito en la eclosión de huevos o de supervivencia de las larvas, pero se observa a veces con el tamaño):
Otros modelos relacionados con la tasa de huevos no eclosionados y de mortalidad de larvas
Calidad del ajuste de un solo modelo
Comparación de los modelos
Calidad de la estimación de ECx El intervalo de confianza (CI) de la ECx no debe ser demasiado amplio. Debe aplicarse el juicio estadístico para decidir la amplitud del intervalo de confianza y que la ECx siga siendo útil. Las simulaciones de los modelos de regresión ajustados a los datos relacionados con la eclosión de los huevos y el tamaño muestran que alrededor del 75 % de los intervalos de confianza de la ECx (x = 10, 20 o 30) no abarcan más de dos concentraciones de ensayo. Este hecho nos da una idea general de lo que es aceptable y factible. Numerosos autores han afirmado que es necesario indicar los intervalos de confianza de todos los parámetros del modelo y que, cuando dichos intervalos de confianza son amplios, los modelos son inaceptables (Ott y Longnecker 2008, Alvord y Rossio 1993, Motulsky y Christopoulos 2004, Lyles et al. 2008, Seber y Wild 2003, Bunke et al. 1999, Environnement Canada, 2005). El intervalo de confianza de ECx (o de cualquier otro parámetro del modelo) no debe contener el valor nulo (cero) (Motulsky y Christopoulos 2004). Se trata del equivalente en modelos de regresión a la diferencia significativa mínima que se suele citar en los enfoques de verificación de hipótesis (Wang et al. 2000, por ejemplo). Corresponde igualmente al intervalo de confianza de las respuestas medias a la LOEC que no contienen la media del testigo. Cabe preguntarse si las estimaciones de los parámetros son científicamente plausibles. Por ejemplo, si el intervalo de confianza de y0 es ± 20 %, no es plausible ninguna estimación de EC10. Si el modelo prevé un efecto del 20 % a una concentración C y si el efecto máximo observado a la concentración C y a las concentraciones inferiores es del 10 %, la EC20 no es plausible (Motulsky y Christopoulos 2004, Wang et al. 2000, Environnement Canada, 2005). La ECx no debe extrapolarse fuera de la gama de concentraciones positivas (Draper y Smith 1999, OCDE 2006). Por ejemplo, una orientación general podría ser que la ECx no esté más de un 25 % aproximadamente por debajo de la concentración de ensayo más baja ni por encima de la concentración de ensayo más alta. BIBLIOGRAFÍA Alvord, W.G., Rossio, J.L. (1993); Determining confidence limits for drug potency in immunoassay, Journal of Immunological Methods 157, 155-163. Brain P. and Cousens R. (1989); An equation to describe dose responses where there is stimulation of growth at low doses. Weed res. 29: 93-96. Bunke, O., Droge, B. and Polzehl, J. (1999). Model selection, transformations and variance estimation in nonlinear regression. Statistics 33, 197-240. Collett, D. (2002); Modelling Binary Data, second edition, Chapman and Hall, London. Collett, D. (2003); Modelling Survival Data in Medical Research, second edition, Chapman and Hall, London. Draper, N.R. and Smith, H. (1999); Applied Regression Analysis, third edition. New York: John Wiley & Sons. Environment Canada (2005); Guidance Document on Statistical Methods for Environmental Toxicity Tests, Report EPS 1/RM/46 Huet, S., A. Bouvier, M.-A. Poursat, E. Jolivet (2003); Statistical Tools for Nonlinear Regression: A Practical Guide with S-PLUS and R Examples, Springer Series in Statistics, New York. Lyles, R. H., C. Poindexter, A. Evans, M. Brown, and C.R. Cooper (2008); Nonlinear Model-Based Estimates of IC50 for Studies Involving Continuous Therapeutic Dose-Response Data, Contemp Clin Trials. 2008 November; 29(6): 878-886. Morgan, B.J.T. (1992); Analysis of Quantal Response Data, Chapman and Hall, London. Motulsky, H., A. Christopoulos (2004); Fitting Models to Biological Data Using Linear and Nonlinear Regression: A Practical Guide to Curve Fitting, Oxford University Press, USA. O'Hara Hines, R. J. and J. F. Lawless (1993); Modelling Overdispersion in Toxicological Mortality Data Grouped over Time, Biometrics Vol. 49, pp. 107-121 OCDE (2006); Current approaches in the statistical analysis of ecotoxicity data: A guidance to application. Series Testing and Assessment, No. 54, Organisation for Economic Co-operation and Development, OECD, Paris. Ott, R.L., M.T. Longnecker, An Introduction to Statistical Methods and Data Analysis, sixth edition, 2008, Brooks-Cole, Belmont, CA Ratkowsky, D.A. (1993); Principles of nonlinear regression, Journal of Industrial Microbiology 12, 195-199. Seber, G.A.F., C.J. Wild, Nonlinear Regression, Wiley, 2003 Slob W. (2002); Dose-response modelling of continuous endpoints. Toxicol. Sci., 66, 298-312 Wang, Q., D.L. Denton, and R. Shukla (2000); Applications and Statistical Properties Of Minimum Significant Difference-Based Criterion Testing In a Toxicity Testing Program, Environmental Toxicology and Chemistry, Vol. 19, pp. 113–117, 2000. C.48 Ensayo de reproducción de peces a corto plazo INTRODUCCIÓN
CONSIDERACIONES INICIALES Y LIMITACIONES
PRINCIPIO DEL ENSAYO
CRITERIOS DE ACEPTACIÓN DEL ENSAYO
DESCRIPCIÓN DEL MÉTODO Equipo
Agua
Soluciones de ensayo
Preparación de los peces
Preexposición y selección de los peces
DISEÑO DEL ENSAYO
Selección de las concentraciones de ensayo
PROCEDIMIENTO Selección y pesada de los peces
Condiciones de exposición Duración
Alimentación
Luz y temperatura
Frecuencia de los análisis y las mediciones
Observaciones
Supervivencia
Comportamiento y aspecto
Fecundidad
Sacrificio compasivo de los peces
Observación de los caracteres sexuales secundarios
Vitelogenina (VTG)
Evaluación de la histopatología gonadal
DATOS E INFORME Evaluación de las respuestas de los biomarcadores mediante el análisis de la varianza (ANOVA)
Preparación de informes sobre los resultados del ensayo
ORIENTACIÓN PARA LA INTERPRETACIÓN Y LA ACEPTACIÓN DE LOS RESULTADOS DEL ENSAYO
BIBLIOGRAFÍA
Apéndice 1 ABREVIATURAS Y DEFINICIONES Producto : sustancia o mezcla. CV : coeficiente de variación. ELISA : enzimoinmunoanálisis de adsorción. Eje HPG : eje hipotálamo-hipófisis-gónadas. Tasa de carga : peso húmedo de peces por unidad de volumen de agua. MTC : concentración máxima tolerada, que constituye aproximadamente el 10 % de la LC50. Densidad de población : número de peces por unidad de volumen de agua. Producto problema : sustancia o mezcla estudiada con este método de ensayo. VTG (vitelogenina) : fosfolipoglucoproteína precursora de la proteína de la yema de huevo que normalmente se produce en las hembras sexualmente activas de todas las especies ovíparas. Apéndice 2 CONDICIONES EXPERIMENTALES PARA EL ENSAYO DE CRIBADO ENDOCRINO DE PECES
Apéndice 3 CARACTERÍSTICAS QUÍMICAS DE UN AGUA DE DILUCIÓN ADECUADA
Apéndice 4A SUSTRATO DE DESOVE PARA EL PEZ CEBRA Bandeja de desove : placa de instrumentos de vidrio, por ejemplo, 22 x 15 x 5,5 cm (largo x ancho x fondo), cubierta con una rejilla de alambre de acero inoxidable extraíble (2 mm de abertura de malla). La rejilla debe cubrir la placa de instrumentos por debajo del nivel del borde. 
El sustrato de desove debe estar fijado a la rejilla; debe proporcionar una estructura en la que los peces puedan introducirse. Por ejemplo, son adecuadas las plantas artificiales de acuario hechas de material plástico de color verde (Nota: Debe tenerse en cuenta la posible adsorción del producto problema al material de plástico). El material de plástico debe lixiviarse en un volumen suficiente de agua caliente durante un tiempo para garantizar que no pasan productos al agua del ensayo. Cuando se utilicen materiales de vidrio, hay que asegurarse de que los peces no puedan resultar heridos ni quedar hacinados durante sus acciones vigorosas. La distancia entre la bandeja y los paneles de vidrio debe ser de 3 cm como mínimo para garantizar que el desove no se realice fuera de la bandeja. Los huevos desovados en la bandeja caen a través de la rejilla y se puede recoger una muestra entre 45 y 60 minutos después de activar la iluminación. Los huevos transparentes no se adhieren y se pueden contar fácilmente con luz transversal. Cuando se utilicen cinco hembras por recipiente, una cantidad de hasta veinte huevos diarios puede considerarse una cifra baja, una cantidad de hasta 100 huevos se considera una cifra intermedia y una cantidad superior a 100 se considera una cifra alta. La bandeja de desove se retira para recoger los huevos, y se vuelve a introducir en el recipiente de ensayo, lo más tarde posible por la tarde o muy temprano por la mañana. El tiempo transcurrido hasta la reintroducción no debe superar una hora ya que, de lo contrario, la señal del sustrato de desove puede inducir algún apareamiento y desove a una hora poco habitual. Si la situación requiriera una introducción posterior de la bandeja de desove, debería hacerse al menos nueve horas después de activar la iluminación. A esta última hora del día ya no se induce el desove. Apéndice 4B SUSTRATO DE DESOVE DEL PEZ CABEZA GORDA En cada recipiente de ensayo se colocan dos o tres tejas y bandejas de desove combinadas de plástico/cerámica/vidrio o de acero inoxidable (por ejemplo, un canalón semicircular gris de 80 mm de largo colocado sobre una bandeja con borde de 130 mm de largo) (véase la imagen). Ha quedado demostrado que las tejas de PVC o cerámica bien preparadas resultan adecuadas como sustrato de desove (Thorpe et al., 2007). Se recomienda someter a abrasión las tejas para mejorar la adherencia. La bandeja también debe protegerse para evitar que los peces accedan a los huevos que hayan caído, a menos que haya quedado demostrada la eficiencia de la adherencia de los huevos en el sustrato de desove utilizado. 
La base se diseña para contener los huevos que no se adhieran a la superficie de la teja y que, por lo tanto, caigan al fondo del recipiente (o los huevos que directamente se coloquen en la base de plástico plana). Todos los sustratos de desove deben lixiviarse durante 12 horas como mínimo, con agua de dilución, antes de usarse. Thorpe KL, Benstead R, Hutchinson TH, Tyler CR, 2007. An optimised experimental test procedure for measuring chemical effects on reproduction in the fathead minnow, Pimephales promelas. Aquatic Toxicology, 81, 90–98. Apéndice 5A EVALUACIÓN DE LOS CARACTERES SEXUALES SECUNDARIOS EN EL PEZ CABEZA GORDA PARA LA DETECCIÓN DE DETERMINADOS PRODUCTOS CON ACTIVIDAD ENDOCRINA Introducción Entre las características potencialmente importantes del aspecto físico de los peces cabeza gorda adultos en los ensayos sobre alteradores endocrinos se incluyen el color del cuerpo (es decir, claro u oscuro), los modelos de coloración (es decir, la presencia o ausencia de franjas verticales), la forma del cuerpo (es decir, forma de la zona de la cabeza y del pecho, distensión del abdomen) y caracteres sexuales secundarios específicos (es decir, número y tamaño de tubérculos nupciales, tamaño de la almohadilla dorsal y ovipositor). Los tubérculos nupciales se encuentran en la cabeza (almohadilla dorsal) de los peces cabeza gorda machos con actividad reproductora y, habitualmente, están dispuestos siguiendo un modelo simétrico bilateral (Jensen et al. 2001). Las hembras testigo y los machos y hembras juveniles no presentan desarrollo de tubérculos (Jensen et al. 2001). Puede haber hasta ocho tubérculos distintos alrededor de los ojos y entre las narinas de los machos. Los tubérculos más abundantes y de mayor tamaño se encuentran en dos líneas paralelas que aparecen justo debajo de las narinas y encima de la boca. Muchos peces tienen grupos de tubérculos debajo de la mandíbula inferior. Los que más cerca están de la boca aparecen, en general, en forma de par simple, mientras que los de posición más ventral pueden estar formados hasta por cuatro tubérculos. El número real de tubérculos rara vez supera los 30 (rango, 18-28; Jensen et al. 2001). Los tubérculos predominantes (en términos de número) aparecen como una estructura individual y relativamente redonda, con una altura que equivale aproximadamente al radio. Los machos con mayor actividad reproductora también tienen, al menos algunos, tubérculos que engrosan y protruyen de forma que no pueden diferenciarse como estructuras individuales. Algunos tipos de productos que alteran la actividad endocrina pueden provocar la aparición anómala de determinados caracteres sexuales secundarios en el sexo opuesto; por ejemplo, los agonistas de receptores de andrógenos, como la 17α-metiltestosterona o la 17β-trembolona, pueden provocar que los peces cabeza gorda hembra desarrollen tubérculos nupciales (Smith 1974; Ankley et al. 2001; 2003), mientras que los agonistas de receptores de estrógenos pueden reducir el número o el tamaño de los tubérculos nupciales en los machos (Miles-Richardson et al. 1999; Harries et al. 2000). A continuación se presenta una descripción de la caracterización de los tubérculos nupciales en el pez cabeza gorda, con arreglo a los procedimientos utilizados en el laboratorio de la Agencia de Protección del Medio Ambiente de Estados Unidos, ubicado en Duluth, Minnesota. Algunos productos o equipos específicos se pueden sustituir por materiales comparables disponibles. La mejor visualización se logra con una lupa iluminada o con un microscopio de disección iluminado 3X. Observe los peces en posición dorsal, con la parte anterior hacia delante (con la cabeza orientada hacia el observador).
Recuento y clasificación de los tubérculos Se han determinado seis áreas concretas para evaluar la presencia y el desarrollo de tubérculos en peces cabeza gorda adultos. Se ha elaborado una plantilla para cartografiar la ubicación y la cantidad de los tubérculos presentes (véase el final de este apéndice). Se registra el número de tubérculos y su tamaño puede clasificarse cuantitativamente de la siguiente manera: 0: ausencia; 1: presente; 2: agrandado y 3: pronunciado, para cada organismo (véase la figura 1). Tipo 0: ausencia de tubérculos. Tipo 1: presente, que se asigna cuando algún tubérculo tiene un solo punto cuya altura sea casi equivalente a su radio. Tipo 2: agrandado, que se asigna cuando hay tejido que se parece a un asterisco, habitualmente con una gran base radial con ranuras o surcos que salen del centro. La altura de los tubérculos suele ser más irregular, pero a veces pueden presentar una forma redondeada. Tipo 3: pronunciado; habitualmente son bastante grandes y redondeados con una estructura menos definida. En ocasiones, estos tubérculos se unen y forman una sola masa en torno a una sola área o una combinación de áreas (B, C y D, que se describen a continuación). El color y el diseño son similares a los del tipo 2, pero en ocasiones son bastante indiscriminados. Con este sistema, el resultado será, en general, una clasificación de tubérculos inferior a 50 en un macho testigo normal con un recuento de tubérculos de 18 a 20 (Jensen et al. 2001). Figura 1 
El número real de tubérculos en algunos peces puede ser superior al de los recuadros de la plantilla en un área de clasificación determinada. En tal caso, pueden marcarse otros números de clasificación a la derecha o a la izquierda del recuadro. Por lo tanto, no es necesario que la plantilla sea simétrica. Otra técnica para la cartografía de los tubérculos que se emparejan o unen verticalmente a lo largo del plano horizontal de la boca consiste en el doble marcado de dos puntos de clasificación de los tubérculos en un mismo recuadro. Regiones de cartografía:
BIBLIOGRAFÍA
Apéndice 5B EVALUACIÓN DE LOS CARACTERES SEXUALES SECUNDARIOS EN EL MEDAKA PARA LA DETECCIÓN DE DETERMINADOS PRODUCTOS CON ACTIVIDAD ENDOCRINA A continuación se presenta una descripción de la medición de los tubérculos papilares (*10), que constituyen los caracteres sexuales secundarios del medaka (Oryzias latipes).
Medición
Fig. 1. Diagrama que muestra las diferencias sexuales en la forma y el tamaño de la aleta anal. A, macho; B, hembra. Oka, T. B., 1931. On the processes on the fin rays of the male of Oryzias latipes and other sex characters of this fish. J. Fac. Sci., Tokyo Univ., IV, 2: 209-218. 
Fig. 2. A: Tubérculos en los segmentos del radio de la aleta anal. J.P., segmento; A.S., espacio axial; P., tubérculo. B: Extremo distal del radio de la aleta. En el extremo hay actinotricos (Act.). Oka, T. B., 1931. On the processes on the fin rays of the male of Oryzias latipes and other sex characters of this fish. J. Fac. Sci., Tokyo Univ., IV, 2: 209-218. 
Fig. 3. Fotografía del cuerpo de un pez que muestra el lugar del corte cuando la gónada se fija con una solución de fijación distinta del formol tamponado neutro al 10 %. En ese caso, el resto del cuerpo se cortará entre la región anterior de la aleta anal y la región anal con una cuchilla (línea roja) y la parte de la cabeza del cuerpo del pez se introducirá en la solución de fijación para la gónada y la parte de la cola del cuerpo del pez se introducirá en el formol tamponado neutro al 10 %. 
Apéndice 6 PROCEDIMIENTOS RECOMENDADOS PARA LA RECOGIDA DE MUESTRAS PARA EL ANÁLISIS DE VITELOGENINA Se debe tener cuidado para evitar la contaminación cruzada entre las muestras de VTG de machos y hembras. Procedimiento 1A: Pez cabeza gorda, extracción de sangre de la vena o la arteria caudal Tras anestesiar, el pedúnculo caudal se corta parcialmente con una hoja de bisturí y se extrae sangre de la vena o la arteria caudal con un tubo capilar para microhematocrito heparinizado. Una vez recogida la sangre, el plasma se aísla rápidamente mediante centrifugado durante tres minutos a 15 000g (o, de manera alternativa, durante diez minutos a 15 000g a 4 °C). Si se desea, se puede determinar el hematocrito porcentual después del centrifugado. A continuación, se retira la parte de plasma del tubo para microhematocrito y se conserva en un tubo de centrífuga con 0,13 unidades de aprotinina (inhibidor de la proteasa) a – 80 °C hasta que pueda determinarse la VTG. Según el tamaño del pez cabeza gorda (en función del sexo), los volúmenes de plasma que se pueden recoger oscilan, en general, entre 5 y 60 microlitros por pez (Jensen et al. 2001). Procedimiento 1 B: Pez cabeza gorda, extracción de sangre del corazón También se puede recoger sangre mediante punción cardíaca mediante una jeringa heparinizada (1 000 unidades de heparina por ml). La sangre se transfiere a tubos Eppendorf (que se mantienen en hielo) y, a continuación, se centrifugan (5 min, 7 000g, a temperatura ambiente). El plasma debe transferirse a tubos Eppendorf limpios (en partes alícuotas si el volumen del plasma lo permite) y congelarse de inmediato a -80 °C hasta que se analice (Panter et al., 1998). Procedimiento 2 A: Medaka, extirpación del hígado del medaka Extracción de los peces del ensayo del recipiente de ensayo
Extirpación del hígado
Tras la extirpación del hígado, el cadáver del pez puede utilizarse para la histología gonadal o para medir los caracteres sexuales secundarios. Muestra Guarde las muestras hepáticas recogidas de los peces del ensayo a ≤ – 70 °C si no van a utilizarse en un pretratamiento poco después de la extirpación. Fig. 1 Se realiza una incisión con unas tijeras justo en la zona anterior a las aletas pectorales. 
Fig. 2 Se realiza una incisión en la línea media del abdomen con las tijeras hasta un punto aproximadamente 2 mm anterior al ano. 
Fig. 3 Las paredes abdominales se extienden con unas pinzas para exponer el hígado y otros órganos internos. (Otra posibilidad es sujetar lateralmente las paredes abdominales). La flecha señala el hígado. 
Fig. 4 El hígado se somete a disección roma y se extirpa con pinzas. 
Fig. 5 Se tira levemente con pinzas de los intestinos. 
Fig. 6 Los dos extremos de los intestinos y los eventuales elementos mesentéricos se cortan con unas tijeras. 
Fig. 7 (hembra) El procedimiento es idéntico con la hembra. 
Fig. 8 Una vez finalizado el procedimiento. 
Procedimiento 2 B: Pretratamiento del hígado del medaka (Oryzias latipes) para el análisis de la vitelogenina Tome el frasco de tampón de homogeneización del juego ELISA y enfríelo con hielo picado (temperatura de la solución: ≤ 4°C). Si se utiliza el tampón de homogeneización del sistema ELISA EnBio, descongele la solución a temperatura ambiente y, a continuación, enfríe el frasco con hielo picado. Calcule el volumen del tampón de homogeneización para el hígado con arreglo a su peso (añada 50 μl de tampón de homogeneización por cada mg de peso del hígado). Por ejemplo, si el hígado pesa 4,5 mg, el volumen de tampón de homogeneización para el hígado será de 225 μl. Prepare una lista de volúmenes de tampón de homogeneización para todos los hígados. Preparación del hígado para el pretratamiento
Realización del pretratamiento 1) Adición del tampón de homogeneización Compruebe en la lista el volumen del tampón de homogeneización que debe utilizarse para una muestra concreta de hígado y ajuste la micropipeta (intervalo de volumen: 100 — 1 000 μl) al volumen correspondiente. Coloque una punta limpia en la micropipeta. Tome el tampón de homogeneización del frasco de reactivo y añada el tampón al microtubo de 1,5 ml que contiene el hígado. Añada el tampón de homogeneización a todos los microtubos de 1,5 ml que contienen el hígado siguiendo el procedimiento descrito anteriormente. No es necesario cambiar la punta de la micropipeta por una nueva. No obstante, si la punta se contamina o se cree que puede estar contaminada, debe reemplazarse. 2) Homogeneización del hígado
3) Centrifugado del homogeneizado de hígado suspendido
4) Recogida de los sobrenadantes
Almacenamiento de la muestra Almacene los microtubos de 0,5 ml que contengan el sobrenadante del homogeneizado de hígado a ≤ – 70 °C hasta que se vayan a utilizar en la prueba ELISA. Procedimiento 3 A: Pez cebra, extracción de sangre de la vena o de la arteria caudal Tras anestesiar, el pedúnculo caudal se corta transversalmente y se extrae sangre de la arteria o de la vena caudal con un tubo capilar para microhematocrito heparinizado. El volumen de sangre varía entre 5 y 15 microlitros según el tamaño del pez. Se añade al tubo microcapilar un volumen igual de tampón de aprotinina (6 microgramos/ml en PBS) y el plasma se separa de la sangre por centrifugado (5 minutos a 600 g). El plasma se recoge en los tubos de ensayo y se conserva a -20 °C hasta que se analice para determinar la VTG u otras proteínas de interés. Procedimiento 3 B: Pez cebra, extracción de sangre por punción cardíaca Para evitar la coagulación de la sangre y el deterioro de la proteína, las muestras se recogen en un tampón con solución salina tamponada con fosfato (PBS) que contiene heparina (1 000 unidades/ml) y aprotinina inhibidora de la proteasa (2 TIU/ml). Se recomiendan la sal amónica de la heparina y la aprotinina liofilizada como ingredientes del tampón. Para la recogida de las muestras de sangre se recomienda usar una jeringa (1 ml) con una aguja fina fija (por ejemplo, Braun Omnikan-F). La jeringa debe llenarse previamente con el tampón (aproximadamente 100 microlitros) para eluir completamente los pequeños volúmenes de sangre de cada pez. Las muestras de sangre se toman mediante punción cardíaca. En un primer momento, el pez debe anestesiarse con MS-222 (100 mg/l). El plano anestésico adecuado permite al usuario distinguir el latido del pez cebra. Mientras puncione el corazón, mantenga una ligera presión en el pistón de la jeringa. Los volúmenes de sangre que pueden extraerse oscilan entre los 20 y los 40 microlitros. Tras la punción cardíaca, la mezcla de tampón y sangre debe pasarse al tubo de ensayo. El plasma se separa de la sangre por centrifugado (20 min; 5 000g) y debe conservarse a – 80 °C hasta que se necesite para realizar el análisis. Procedimiento 3C: PNT: Pez cebra, homogeneización de la cabeza y la cola
Apéndice 7 MUESTRAS DE ENRIQUECIMIENTO DE VITELOGENINA Y PATRÓN DE REFERENCIA INTERENSAYOS Cada día que se realicen ensayos de VTG, se analizará una muestra de enriquecimiento preparada con un patrón de referencia interensayos. La VTG utilizada para realizar el patrón de referencia interensayos procederá de un lote distinto del usado para preparar los patrones de calibración del ensayo que se esté llevando a cabo. La muestra de enriquecimiento se elaborará añadiendo una cantidad conocida del patrón interensayos a una muestra de plasma de los machos testigo. La muestra se enriquecerá para lograr una concentración de VTG entre 10 y 100 veces la concentración de vitelogenina esperada en los peces macho testigo. La muestra de plasma de machos testigo que se enriquece puede provenir de un solo pez o puede ser una mezcla procedente de varios peces. Se analizará una submuestra del plasma de los machos del testigo no enriquecido en, como mínimo, dos pocillos duplicados. La muestra enriquecida también se analizará en, como mínimo, dos pocillos duplicados. La cantidad media de vitelogenina de las dos muestras no enriquecidas de plasma de los machos testigo se añadirá a la cantidad calculada de VTG que se haya añadido para enriquecer las muestras a fin de determinar la concentración prevista. La relación entre esta concentración prevista y la concentración medida se indicará junto con los resultados de cada conjunto de ensayos que se haya realizado ese día. Apéndice 8 DIAGRAMA DE FLUJO PARA TOMAR DECISIONES PARA EL ANÁLISIS ESTADÍSTICO 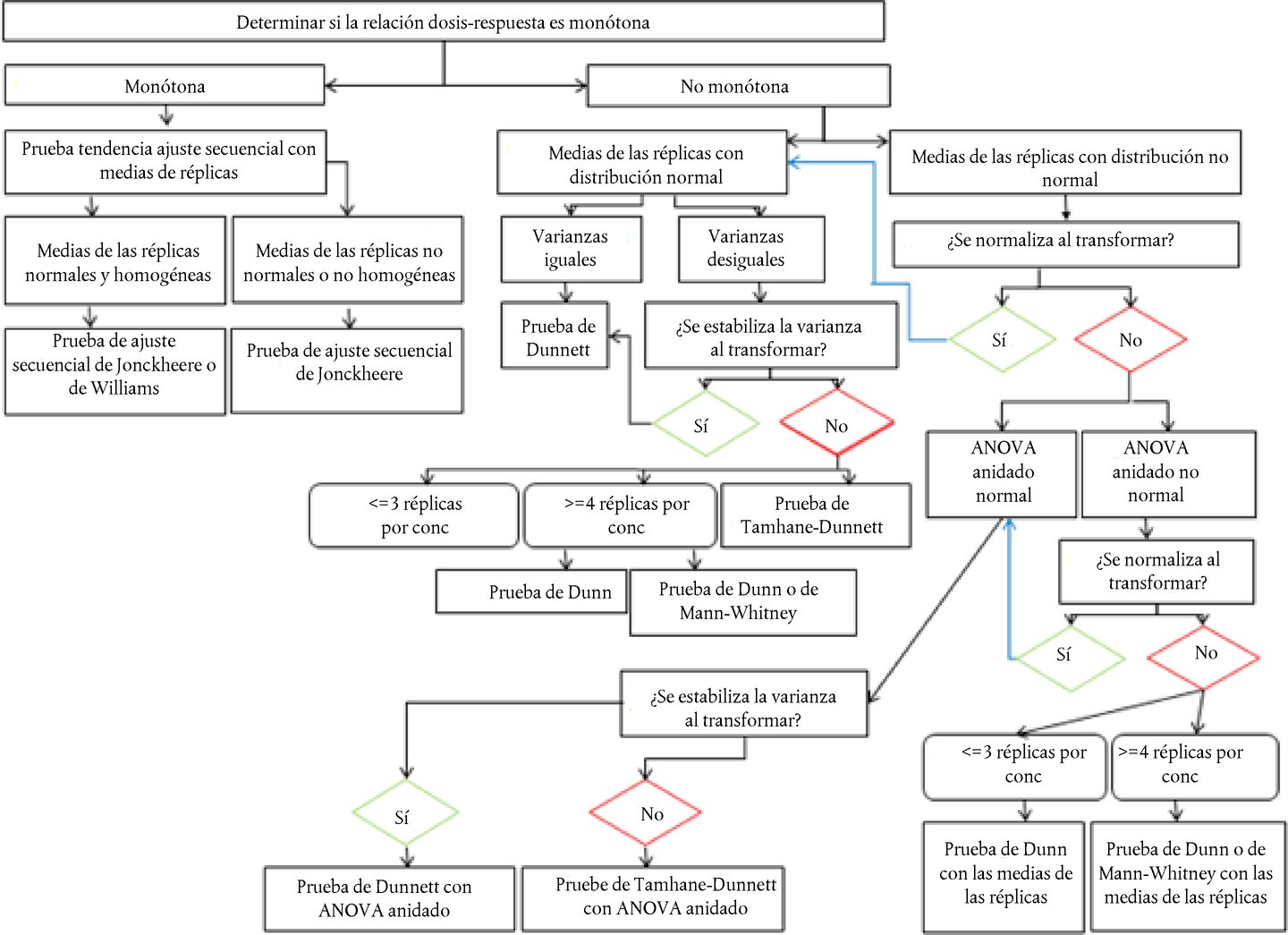
C.49 Ensayo de toxicidad aguda en embriones de pez (FET) INTRODUCCIÓN
PRINCIPIO DEL ENSAYO
CONSIDERACIONES INICIALES
VALIDEZ DEL ENSAYO
DESCRIPCIÓN DEL MÉTODO
Equipo
Recipientes de ensayo
Agua y condiciones del ensayo
Soluciones problema
Mantenimiento de los peces reproductores
Ensayos de competencia
Producción de huevos
Diferenciación de los huevos
PROCEDIMIENTO Condiciones de exposición
Concentraciones de ensayo
Testigos
Inicio de la exposición y duración del ensayo
Distribución de los huevos en las placas de 24 pocillos
Observaciones
Mediciones analíticas
ENSAYO LÍMITE
DATOS E INFORME Tratamiento de los resultados
Informe del ensayo
BIBLIOGRAFÍA
Apéndice 1 DEFINICIONES Parámetro principal : parámetro que causa un efecto a nivel de la población. Blástula : formación celular alrededor del polo animal que cubre una determinada parte del vitelo. Producto : sustancia o mezcla. Epibolia : proliferación masiva de células predominantemente epidérmicas en la fase de gastrulación del embrión y su movimiento del lado dorsal al ventral, mediante el cual las capas de células endodérmicas se internalizan en un proceso similar a la invaginación y el vitelo se incorpora al embrión. Ensayo dinámico : ensayo con un flujo continuo de soluciones de ensayo por el sistema de ensayo durante la duración de la exposición. Testigo interno de la placa : testigo interno que consiste en cuatro pocillos rellenos con agua de dilución por placa de 24 pocillos con el fin de identificar la posible contaminación de las placas por el fabricante o el investigador durante el procedimiento, y cualquier efecto de la placa que pueda influir en el resultado del ensayo (p. ej., gradiente de temperatura). IUPAC : Unión Internacional de Química Pura y Aplicada. Agua de mantenimiento : agua en la que se practica la cría de los peces adultos. Concentración letal mediana (LC50) : concentración de un producto problema que se estima que mata al 50 % de los organismos de ensayo durante la duración del ensayo. Ensayo de renovación semiestático : ensayo con una renovación regular de las soluciones de ensayo después de períodos definidos (p. ej., cada 24 horas). SMILES : Sistema Simplificado de Registro de Líneas Moleculares. Somita : en la fase de desarrollo embrionario de los vertebrados, las somitas son masas del mesodermo que se distribuyen lateralmente respecto al tubo neural y que darán origen a la dermis (dermatoma), los músculos esqueléticos (miotoma) y las vértebras (esclerotoma). Ensayo estático : ensayo durante el cual las soluciones de ensayo permanecen sin cambios durante todo el ensayo. Producto problema : toda sustancia o mezcla estudiada con este método de ensayo. UVCB : sustancia de composición desconocida o variable, productos complejos de reacción y materiales biológicos. Apéndice 2 MANTENIMIENTO, REPRODUCCIÓN Y CONDICIONES TÍPICAS PARA LOS ENSAYOS DE TOXICIDAD AGUDA CON EMBRIONES DE PEZ CEBRA
Apéndice 3 DESARROLLO NORMAL DEL PEZ CEBRA A 26 °C 


Apéndice 4 Fig. 1. Distribución de las placas de 24 pocillos 
Fig. 2. Esquema del procedimiento de ensayo de toxicidad aguda en embriones de pez cebra (de izquierda a derecha): producción de los huevos, recogida de los huevos, exposición previa inmediatamente después de la fecundación en recipientes de vidrio, selección de huevos fecundados con un microscopio invertido o binocular y distribución de los huevos fecundados en placas de 24 pocillos preparadas con las respectivas concentraciones de ensayo/testigos, n = número de huevos requeridos por concentración de ensayo/ testigo (20 aquí), hpf = horas tras la fecundación 
Apéndice 5 ATLAS DE PARÁMETROS LETALES DEL ENSAYO DE TOXICIDAD AGUDA CON EMBRIONES DE PEZ CEBRA Los siguientes parámetros principales indican toxicidad aguda y, por consiguiente, la muerte de los embriones: coagulación de los embriones, no desprendimiento de la cola, ausencia de formación de somitas y ausencia de latido cardíaco. Las siguientes microfotografías se han seleccionado para ilustrar estos parámetros. Fig. 1. Coagulación del embrión: 
Con iluminación a campo claro, los embriones de pez cebra coagulados muestran varias inclusiones opacas. Fig. 2. Ausencia de formación de somitas: 
Aunque tiene un retardo en el desarrollo de unas 10 horas, el embrión de pez cebra de 24 horas de (a) muestra unas somitas bien desarrolladas (→), mientras que el embrión de (b) no da muestras de formación de somitas (→). A pesar de que muestra un pronunciado edema en el saco vitelino (*), el embrión de pez cebra de 48 horas de (c) muestra una evidente formación de somitas (→), mientras que el embrión de 96 horas de (d) no da muestras de formación de somitas (→). Hay que observar además la curvatura vertebral (escoliosis) y el edema pericárdico (*) del embrión mostrado en (d). Fig. 3. No desprendimiento de la cola del embrión en la vista lateral 
Fig. 4. Ausencia de latido cardíaco 
La ausencia de latido cardíaco resulta, por definición, difícil de ilustrar en una microfotografía. Se indica por la falta de convulsión del corazón (flecha doble). La inmovilidad de los glóbulos sanguíneos en, por ejemplo, la aorta abdominal (→ en la ampliación) no constituye un indicador de ausencia de latido cardíaco. Asimismo se observa la ausencia de formación de somitas en este embrión (*, aspecto homogéneo en lugar de segmentario de los tejidos musculares). El tiempo de observación para registrar la ausencia de latido cardíaco debe ser al menos de un minuto, con un aumento mínimo de 80×. C.50. Ensayo de toxicidad sin sedimentos en Myriophyllum spicatum INTRODUCCIÓN
PRINCIPIO DEL ENSAYO
INFORMACIÓN SOBRE EL PRODUCTO PROBLEMA
VALIDEZ DEL ENSAYO
PRODUCTO DE REFERENCIA
DESCRIPCIÓN DEL MÉTODO Equipo
Organismo de ensayo
Cultivo
Medio de ensayo
Soluciones de ensayo
Grupos de ensayo y testigos
Exposición
Condiciones del ensayo
Duración
Mediciones y determinaciones analíticas
Frecuencia de las mediciones y determinaciones analíticas
Ensayo límite
DATOS E INFORME Variables de respuesta
Tasa media de crecimiento específico
Rendimiento
Tiempo de duplicación
Trazado de las curvas concentración-respuesta
Estimación de ECx
Procedimientos estadísticos
Presentación de informes
BIBLIOGRAFÍA
Apéndice 1 DEFINICIONES Biomasa : el peso fresco/seco de materia viva presente en una población. En este ensayo, la biomasa es la suma del brote principal, todas las ramas laterales y todas las raíces. Producto : sustancia o mezcla. Clorosis : cambio de color del verde al amarillento del organismo de ensayo, especialmente de los verticilos. ECx : concentración del producto problema disuelto en el medio de ensayo que produce una reducción del x % (por ejemplo, del 50 %) en el crecimiento de Myriophyllum spicatum dentro de un plazo de exposición definido (que debe indicarse explícitamente si es distinto de la duración completa o normal del ensayo). Para expresar de forma inequívoca el valor de EC obtenido a partir de la tasa de crecimiento o del rendimiento, se usan respectivamente los símbolos» ErC «y» EyC«, seguidos de la variable de medición que se ha utilizado, por ejemplo ErC (longitud del brote principal). Crecimiento : aumento en la variable de medición, como la longitud del brote principal, longitud total de las ramas laterales, longitud total de los brotes, longitud total de las raíces, peso fresco, peso seco y número de verticilos durante el período de ensayo. Tasa de crecimiento (tasa media de crecimiento específico): aumento logarítmico de la variable de medición durante el período de exposición. Nota: Las variables de respuesta relacionadas con la tasa de crecimiento son independientes de la duración del ensayo, siempre que el patrón de crecimiento de los organismos testigo no expuestos sea exponencial. Concentración mínima con efecto observado (LOEC) : concentración estudiada mínima a la que se observa que el producto ejerce un efecto estadísticamente significativo de reducción del crecimiento (con p < 0,05) cuando se compara con el testigo, dentro de un tiempo de exposición dado. Además, todas las concentraciones de ensayo superiores a la LOEC deben ejercer un efecto nocivo igual o mayor que el observado a dicha concentración. Si no se cumplen estas dos condiciones, es preciso dar una explicación completa sobre cómo se ha elegido la LOEC (y, por tanto, la NOEC). Variables de medición : las variables de cualquier tipo que se miden para expresar el parámetro del ensayo utilizando una o más variables de respuesta diferentes. En este método de ensayo son variables de medición la longitud del brote principal, la longitud total de las ramas laterales, la longitud total de los brotes, la longitud total de las raíces, el peso fresco, el peso seco y el número de verticilos. Monocultivo : cultivo con una sola especie vegetal. Necrosis : tejido muerto (es decir, blanco o marrón oscuro) del organismo de ensayo. Concentración sin efecto observado (NOEC) : concentración estudiada que se encuentra inmediatamente por debajo de la LOEC. Variable de respuesta : variable para la estimación de la toxicidad derivada de cualquier variable medida que describa la biomasa por distintos métodos de cálculo. A efectos del presente método de ensayo, la tasa de crecimiento y el rendimiento son variables de respuesta derivadas de variables de medición como la longitud del brote principal, la longitud total de los brotes, el peso fresco, el peso seco o el número de verticilos. Ensayo semiestático (de renovación semiestática) : ensayo en que, a lo largo de su duración, la solución problema se sustituye periódicamente, a intervalos específicos. Ensayo estático : ensayo en que, a lo largo de su duración, no hay renovación de la solución problema. Producto problema : sustancia o mezcla estudiada con este método de ensayo. Parámetro del ensayo : factor general que, como objetivo del ensayo, es modificado por el producto problema respecto al control. En este método el parámetro del ensayo es la inhibición del crecimiento, que se puede expresar mediante diferentes variables de respuesta, basadas en una o más variables de medición. Medio de ensayo : medio de cultivo sintético y completo en que crecen las plantas del ensayo cuando se exponen al producto problema. Este se disuelve en principio en el medio de ensayo. UVCB : sustancia de composición desconocida o variable, producto complejo de reacción y material biológico. Rendimiento : valor de una variable de medición para expresar la biomasa al final del período de exposición menos el valor de la variable de medición al inicio del período de exposición. Nota: Cuando el patrón de crecimiento de los organismos no expuestos sea exponencial, las variables de respuesta basadas en el rendimiento disminuirán con la duración del ensayo. Apéndice 2 MEDIO DE ANDREWS MODIFICADO PARA EL CULTIVO MADRE Y EL PRECULTIVO El medio de Andrews modificado, requerido para el cultivo madre y el precultivo, se preparará a partir de cinco soluciones madre de nutrientes preparadas por separado y a las cuales se añadirá un 3 % de sacarosa. Cuadro 1 Composición de la solución de nutrientes de Andrews: (Norma ASTM E 1913-04)
Las soluciones madre se pueden mantener en un refrigerador durante 6 meses (a 5-10 oC). Solo la solución madre n.o 5 tiene una vida útil reducida (dos meses). Cuadro 2 Producción de la solución madre n.o 3.1 para preparar la solución madre n.o 3
Después de haber preparado la solución madre n.o 3.1 (cuadro 2), congele esta solución en porciones de 11 ml aproximadamente (a -18 oC como mínimo). La vida útil de estas porciones congeladas es de cinco años. Para producir la solución madre n.o 3, descongele la solución madre n.o 3.1, vierta 10 ml de dicha solución en un matraz aforado de 1 l y añada agua ultrapura hasta la marca del matraz. Para obtener el medio de Andrews modificado, ponga aproximadamente 2 500 l de agua ultra pura en un matraz aforado de 5 l. Después de añadir 50 ml de cada solución madre, llene el 90 % del matraz aforado con agua ultrapura y ajuste el pH a 5.8. A continuación, añada 150 g de sacarosa disuelta (3 % por 5 l); luego, llene el matraz aforado con agua ultra pura hasta la marca. Por último, vierta la solución nutritiva en matraces Schott de 1 l y esterilícelos en un autoclave a 121 oC durante 20 minutos. La solución nutritiva que se obtiene así se puede conservar estéril en un refrigerador (entre 5 y 10 oC) durante tres meses. Medio de Andrews modificado para el ensayo de toxicidad sin sedimentos El medio de Andrews modificado concentrado diez veces, necesario para obtener las soluciones de ensayo, se prepara a partir de las cinco soluciones madre nutritivas ya mencionadas en los cuadros 1 y 2, a las que se les añade un 30 % de sacarosa. Para ello, ponga aproximadamente 100 ml de agua ultra pura en un matraz aforado de 1 l. Añada 100 ml de cada solución madre y ajuste el pH a 5,8. A continuación, añada un 30 % de sacarosa disuelta (300 g por 1 000 ml) y llene el matraz aforado con agua ultrapura hasta llegar a la marca. Por último, vierta la solución de nutrientes en matraces Schott de 0,5 l y esterilícelos en un autoclave a 121 oC durante 20 minutos. La solución de nutrientes modificada concentrada diez veces que se obtiene así se puede conservar estéril en un refrigerador (entre 5 y 10 oC) durante tres meses. Apéndice 3 MANTENIMIENTO DEL CULTIVO MADRE En el presente apéndice 3 se describe el cultivo madre de Myriophyllum spicatum L. (103), una especie de planta acuática sumergida dicotiledónea del grupo de las milenrama. Entre junio y agosto produce discretas flores de color rosa y blanco que sobresalen por encima de la superficie del agua. Las plantas enraízan en el suelo mediante un sistema de rizomas robustos y pueden encontrarse en todo el hemisferio norte en aguas eutróficas, (pero no contaminadas) estancadas, muy calcáreas, con sustrato fangoso. Myriophyllum spicatum prefiere el agua dulce, pero también se encuentra en aguas salobres. Para un cultivo madre sin sedimentos en condiciones de laboratorio se requieren plantas estériles. Las plantas estériles están disponibles en el laboratorio de ecotoxicología de la Umweltbundesamt alemana (Agencia Federal Alemana de Medio Ambiente). También es posible preparar los organismos de ensayo a partir de plantas no estériles según la norma ASTM E 1913-04. A continuación se presenta un extracto de la guía de la norma ASTM que indica el procedimiento de cultivo de ejemplares de Myriophyllum sibiricum obtenidos de la naturaleza: “Si optamos por plantas no estériles obtenidas de la naturaleza, hay que recoger turiones de M. sibiricum en el otoño. Los turiones deben colocarse en un acuario de 20 l que contenga 5 cm de sedimento estéril recubierto de arena de sílice o, por ejemplo de Turface® y 18 l de agua de grado reactivo. Airear el acuario y mantenerlo a una temperatura de 15 oC, así como a un caudal de entre 200 y 300 μmol m–2 s–1 durante 16 horas al día. El cultivo de las plantas en el acuario puede proporcionar plantas de repuesto en caso de que los cultivos estériles se destruyan por un mal funcionamiento mecánico del recipiente de cultivo, contaminación u otro motivo. Las plantas cultivadas en el acuario no son estériles y los cultivos estériles no pueden conservarse en un sistema de cultivo en lotes. Para esterilizar el cultivo, hay que retirar las plantas del acuario y enjuagarlas bajo una corriente de agua desionizada durante media hora aproximadamente. A continuación, se desinfectan en condiciones asépticas en una cabina de flujo laminar durante 20 minutos como máximo (hasta que la mayoría del tejido vegetal se haya blanqueado y solo quede de color verde el ápice en crecimiento) en una solución de hipoclorito de sodio al 3 % (p/v) que contenga 0,01 % de un tensoactivo adecuado. Agitar el desinfectante y el material vegetal. Se transfieren los segmentos con varios nodos a tubos de cultivo estériles con 45 ml de medio de Andrews modificado esterilizado y se tapan con cierres de tubo de cultivo corrientes. Solo se coloca un segmento vegetal por recipiente de ensayo. Para garantizar que los recipientes de cultivo están bien cerrados, se sellan con lámina adhesiva de laboratorio. Cuando se ha establecido un cultivo estéril, deben transferirse segmentos vegetales con varios nodos a recipientes de ensayo nuevos que contengan medio nutritivo líquido que debe prepararse de nuevo cada diez o doce días. Como se habrá demostrado mediante cultivos en placas de agar, las plantas deben estar estériles y mantenerse estériles durante ocho semanas antes de iniciar el ensayo”. Dado que el medio modificado de Andrews contiene sacarosa (que estimula el crecimiento de hongos y bacterias), todos los materiales, soluciones y cultivos deben prepararse en condiciones estériles. Todos los líquidos y el equipo deben esterilizarse antes de su uso. Dicha esterilización se lleva a cabo mediante tratamiento con aire caliente (210 oC) durante 4 horas o en autoclave a 121 oC durante 20 minutos. Además, todos los matraces, placas, cubetas y el resto del equipo deben someterse a flameado en una campana estéril antes de su uso. Los cultivos madre pueden mantenerse en condiciones de iluminación reducida y a temperaturas bajas (50 μE m-2 s-1, 20 ± 2 oC) durante tiempos prolongados sin tener que volver a establecerlos de nuevo. El medio de cultivo de Myriophyllum debe ser el mismo que se utilice en los ensayos, pero también pueden utilizarse otros medios ricos en nutrientes para los cultivos madre. Los segmentos vegetales se distribuyen en condiciones axénicas en varios matraces Erlenmeyer de 500 ml o Fernbach de 2 000 ml, cada uno respectivamente con unos 450 ml o 1 000 ml de medio de Andrews modificado. Los matraces se cerrarán en condiciones axénicas con tapones de celulosa. Además, es obligatorio someter el equipo a flameado en una campana de laboratorio estéril justo antes de su uso. En función del número y el tamaño, las plantas se transferirán a la solución nutritiva recién preparada aproximadamente cada tres semanas. Para este cultivo renovado se pueden utilizar ápices o segmentos de la parte central del tallo. El número y el tamaño de las plantas transferidas (o de los segmentos de plantas) dependen del número de plantas necesario. Por ejemplo, pueden transferirse cinco segmentos de brotes a un matraz Fernbach y tres segmentos de brotes a un matraz Erlenmeyer, cada uno con una longitud de 5 cm. Hay que eliminar los segmentos que presenten raíces, flores, zonas muertas u otras partes que llamen la atención. Figura 1 Corte de las plantas para el cultivo madre y el precultivo tras tres semanas de cultivo. 
Las plantas se cultivan en los matraces Erlenmeyer de 500 ml y Fernbach de 2 000 ml en una incubadora de refrigeración a 20 ± 2 oC y con una iluminación continua de aproximadamente 100-150 μE m-2 s-1 o 6 000-9 000 Lux (emitida por iluminación de la cámara con una temperatura de color correspondiente a “luz blanca cálida”). Figura 2 Cultivo de las plantas en una incubadora de refrigeración con iluminación de la cámara. 
Para los cultivos deben utilizarse recipientes estériles de vidrio, químicamente limpios (lavados con ácido); su manipulación se hará asépticamente. En caso de contaminación del cultivo madre, por ejemplo mediante algas, hongos o bacterias, habrá que preparar un nuevo cultivo o utilizar un cultivo madre de otro laboratorio para renovar el cultivo en cuestión. Apéndice 4 MANTENIMIENTO DEL PRECULTIVO Y PREPARACIÓN DEL ORGANISMO DE ENSAYO PARA EL ENSAYO Para obtener un precultivo, hay que cortar los brotes del cultivo madre en segmentos con dos verticilos cada uno. Los segmentos se colocarán en matraces Fernbach con medio de Andrews modificado (con un 3 % de sacarosa). Cada matraz puede contener hasta 50 segmentos de brotes. Hay que asegurarse, no obstante, de que los segmentos están vivos y no tienen raíces, ramas laterales ni yemas (véase la figura 1 del apéndice 3). Los organismos de precultivo se cultivan entre 14 y 21 días en condiciones estériles en una cámara ambiental, alternando 16 horas de luz y 8 de oscuridad. La intensidad luminosa debe estar comprendida entre 100-150 μE m-2 s-1. La temperatura de los recipientes de ensayo debe ser de 23 ± 2 oC. Dado que el medio modificado de Andrews contiene sacarosa (que estimula el crecimiento de algas, hongos y bacterias), las soluciones del producto problema deben prepararse y los cultivos deben realizarse en condiciones estériles. Todos los líquidos y el equipo deben esterilizarse antes de su uso. Dicha esterilización se lleva a cabo mediante tratamiento con aire caliente (210 oC) durante 4 horas o en autoclave a 121 oC durante 20 minutos. Además, todos los matraces, placas, cubetas y el resto del equipo deben someterse a flameado en una campana estéril antes de su uso. Hay que retirar los brotes de los matraces de precultivo en condiciones axénicas, seleccionando el material que sea lo más homogéneo posible. Cada ensayo requiere como mínimo 60 organismos de ensayo (ensayo con ocho concentraciones del producto problema). Para llevar a cabo el ensayo, hay que tomar las ramas laterales frescas de los precultivos, cortarlas a 2,5 cm de la base (se mide con una regla) y transferirlas a un vaso con medio modificado de Andrews estéril. Estas ramas laterales frescas pueden utilizarse para el ensayo de toxicidad sin sedimentos en Myriophyllum spicatum. Figura 2 Corte de las plantas del precultivo para el ensayo de toxicidad sin sedimentos en Myriophyllum spicatum. 
C.51. Ensayo de toxicidad agua-sedimento en Myriophyllum spicatum INTRODUCCIÓN
PRINCIPIO DEL ENSAYO
INFORMACIÓN SOBRE EL PRODUCTO PROBLEMA
VALIDEZ DEL ENSAYO
PRODUCTO DE REFERENCIA
DESCRIPCIÓN DEL MÉTODO Equipos de ensayo
Organismo de ensayo
Cultivo del organismo de ensayo
Sedimento
Medio de ensayo
Diseño experimental
Concentraciones del producto problema y grupos testigo
Ensayo límite
Soluciones de ensayo
PROCEDIMIENTO DE ENSAYO
Fase de implantación
Selección de material vegetal homogéneo
Exposición a través de la fase acuosa
Exposición a través del sedimento
Mantenimiento de los niveles de agua durante el período de ensayo
Condiciones del ensayo
Duración del ensayo
Mediciones y determinaciones analíticas
Frecuencia de las mediciones y determinaciones analíticas
Mediciones analíticas del producto problema
EVALUACIÓN DE LOS DATOS
Variables de respuesta
Tasa media de crecimiento específico
Rendimiento
Trazado de las curvas concentración-respuesta
Estimación de ECx
Procedimientos estadísticos
PRESENTACIÓN DE INFORMES
BIBLIOGRAFÍA
Apéndice 1 COMPOSICIÓN DEL MEDIO DE SMART Y BARKO
Apéndice 2 DEFINICIONES Biomasa : peso fresco/seco de materia viva presente en una población. En este ensayo, la biomasa es la suma del brote principal, todas las ramas laterales y todas las raíces. Producto : sustancia o mezcla. Clorosis : cambio de color del verde al amarillento del organismo de ensayo, especialmente de los verticilos. ECx : concentración del producto problema disuelta en el medio de ensayo que produce una reducción del x % (por ejemplo, del 50 %) en el crecimiento de Myriophyllum spicatum dentro de un plazo de exposición definido (que debe indicarse explícitamente si es distinto de la duración completa o normal del ensayo). Para expresar de forma inequívoca el valor de EC obtenido a partir de la tasa de crecimiento o del rendimiento, se usan respectivamente los símbolos “ErC” y “EyC”, seguidos de la variable de medición que se ha utilizado, por ejemplo ErC (longitud del brote principal). Crecimiento : aumento en la variable de medición, como la longitud del brote principal, longitud total de las ramas laterales, longitud total de los brotes, longitud total de las raíces, peso fresco, peso seco o número de verticilos durante el período de ensayo. Tasa de crecimiento (tasa media de crecimiento específico): aumento logarítmico de la variable de medición durante el período de exposición. Nota: Las variables de respuesta relacionadas con la tasa de crecimiento son independientes de la duración del ensayo, siempre que el patrón de crecimiento de los organismos testigo no expuestos sea exponencial. Concentración mínima con efecto observado (LOEC) : concentración estudiada mínima a la que se observa que el producto ejerce un efecto estadísticamente significativo de reducción del crecimiento (con p < 0,05) cuando se compara con el testigo, dentro de un tiempo de exposición dado. Además, todas las concentraciones de ensayo superiores a la LOEC deben ejercer un efecto nocivo igual o mayor que el observado a dicha concentración. Si no se cumplen estas dos condiciones, es preciso dar una explicación completa sobre cómo se ha elegido la LOEC (y, por tanto, la NOEC). Variables de medición : variables de cualquier tipo que se miden para expresar el parámetro del ensayo utilizando una o más variables de respuesta diferentes. En este método de ensayo son variables de medición la longitud del brote principal, la longitud total de las ramas laterales, la longitud total de los brotes, la longitud total de las raíces, el peso fresco, el peso seco y el número de verticilos. Monocultivo : cultivo con una sola especie vegetal. Necrosis : tejido muerto (es decir, blanco o marrón oscuro) del organismo de ensayo. Concentración sin efecto observado (NOEC) : concentración estudiada que se encuentra inmediatamente por debajo de la LOEC. Variable de respuesta : variable para la estimación de la toxicidad derivada de cualquier variable medida que describa la biomasa por distintos métodos de cálculo. A efectos del presente método de ensayo, la tasa de crecimiento y el rendimiento son variables de respuesta derivadas de variables de medición como la longitud del brote principal, la longitud total de los brotes, el peso fresco, el peso seco o el número de verticilos. Ensayo semiestático (de renovación) : ensayo en que, a lo largo de su duración, la solución de ensayo se sustituye periódicamente, a intervalos específicos. Ensayo estático : ensayo en que, a lo largo de su duración, no hay renovación de la solución de ensayo. Producto problema : sustancia o mezcla estudiada con este método de ensayo. Parámetro del ensayo : factor general que, como objetivo del ensayo, es modificado por el producto problema respecto al testigo. En este método, el parámetro del ensayo es la inhibición del crecimiento, que se puede expresar mediante diferentes variables de respuesta, basadas en una o más variables de medición. Medio de ensayo : medio de cultivo sintético y completo en que crecen las plantas del ensayo cuando se exponen al producto problema. Este se disuelve en principio en el medio de ensayo. UVCB : sustancias de composición desconocida o variable, productos complejos de reacción y materiales biológicos. Rendimiento : valor de una variable de medición para expresar la biomasa al final del período de exposición menos el valor de la variable de medición al inicio del período de exposición. Nota: Cuando el patrón de crecimiento de los organismos no expuestos sea exponencial, las variables de respuesta basadas en el rendimiento disminuirán con la duración del ensayo. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
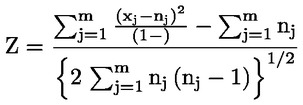
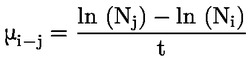
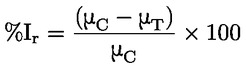
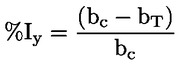

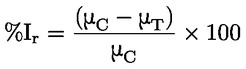

(1) No se dispone de ningún valor a 20 °C, pero puede aceptarse que la variabilidad de los resultados de la medición es superior a la dependencia de la temperatura que cabe esperar.
(2) Reglamento (CE) n.o 1907/2006 del Parlamento Europeo y del Consejo, de 18 de diciembre de 2006, relativo al registro, la evaluación, la autorización y la restricción de las sustancias y mezclas químicas (REACH), por el que se crea la Agencia Europea de Sustancias y Mezclas Químicas, se modifica la Directiva 1999/45/CE y se derogan el Reglamento (CEE) n.o 793/93 del Consejo y el Reglamento (CE) n.o 1488/94 de la Comisión, así como la Directiva 76/769/CEE del Consejo y las Directivas 91/155/CEE, 93/67/CEE, 93/105/CE y 2000/21/CE de la Comisión. (DO L 304 de 22.11.2007, p. 1).
(3) Interagency Coordinating Committee on the Validation of Alternative Methods (Comité de Coordinación Interagencias sobre la Validación de Métodos Alternativos) de Estados Unidos.
(*1) Hay que tomar nota de la superficie de la opacidad de la córnea.
(4) Reglamento (CE) n.o 1272/2008 del Parlamento Europeo y del Consejo, de 16 de diciembre de 2008, sobre clasificación, etiquetado y envasado de sustancias y mezclas, y por el que se modifican y derogan las Directivas 67/548/CEE y 1999/45/CE y se modifica el Reglamento (CE) n.o 1907/2006 (DO L 353 de 31.12.2008, p. 1).
(5) Los estadísticos que siguen un enfoque de modelización como el de usar modelos lineales generales pueden plantear el análisis de forma diferente, pero comparable, sin derivar necesariamente el cuadro tradicional de ANOVA, que se basa en enfoques algorítmicos para calcular las estadísticas elaboradas en una edad pre-informática.
(6) Los estadísticos que siguen un enfoque de modelización como el de usar modelos lineales generales pueden plantear el análisis de forma diferente, pero comparable, sin derivar necesariamente el cuadro tradicional de ANOVA, que se basa en enfoques algorítmicos para calcular las estadísticas elaboradas en una edad pre-informática.
(7) Reglamento (CE) n.o 1272/2008 del Parlamento Europeo y del Consejo, de 16 de diciembre de 2008, sobre clasificación, etiquetado y envasado de sustancias y mezclas, y por el que se modifican y derogan las Directivas 67/548/CEE y 1999/45/CE y se modifica el Reglamento (CE) n.o 1907/2006 (DO L 353 de 31.12.2008, p. 1).
(8) Se han asignado clases químicas a cada producto problema utilizando un sistema de clasificación habitual, basado en el sistema de clasificación de materias médicas de la National Library of Medicine (MeSH) (se puede encontrar en la dirección http//www.nlm.nih.gov/mesh).
(9) Sobre la base de los resultados obtenidos en el ensayo con ojos de conejo in vivo (TG 405 de la OCDE) (17) y utilizando el SGA de las Naciones Unidas (4).
(10) La clasificación como 2A o 2B depende de la interpretación del criterio del SGA de las Naciones Unidas para distinguir entre estas dos categorías, es decir, 1 de 3 frente a 2 de 3 animales que presentan el día 7 efectos necesarios para generar una clasificación de la categoría 2A. El estudio in vivo incluye 3 animales. Todos los parámetros aparte del enrojecimiento de la conjuntiva en un solo animal se recuperan hasta una puntuación de cero para el día 7 o antes. El único animal que no está recuperado completamente para el día 7 tiene una puntuación de 1 en cuanto al enrojecimiento de la conjuntiva (el día 7) que se recupera plenamente el día 10.
(11) Estas dimensiones corresponden al soporte de córnea utilizado para vacas de entre 12 y 60 meses de edad. En caso de que se utilicen animales de entre 6 y 12 meses de edad, puede ser necesario diseñar el soporte de manera que cada cámara albergue un volumen de 4 ml, y cada una de las cámaras internas mida 1,5 cm de diámetro y 2,2 cm de profundidad. Es importante que en todos los soportes de córnea de nuevo diseño la proporción entre la superficie de córnea expuesta y el volumen de la cámara posterior sea la misma que en el soporte de córnea tradicional. Se trata de una condición necesaria para asegurar que los valores de permeabilidad se determinan correctamente a efectos del cálculo de la puntuación IVIS con la fórmula propuesta.
(12) Reglamento (CE) n.o 1272/2008 del Parlamento Europeo y del Consejo, de 16 de diciembre de 2008, sobre clasificación, etiquetado y envasado de sustancias y mezclas, y por el que se modifican y derogan las Directivas 67/548/CEE y 1999/45/CE y se modifica el Reglamento (CE) n.o 1907/2006 (DO L 353 de 31.12.2008, p. 1).
(*2) Puntuación media más elevada observada a cualquiera de los tiempos seleccionados.
(*3) Puntuación media más elevada observada a cualquiera de los tiempos seleccionados (basándose en las puntuaciones de opacidad definidas en el cuadro 1).
(*4) Sobre la base de las puntuaciones definidas en el cuadro 2.
(*5) Combinaciones menos probables.
(13) Se han asignado clases químicas a cada producto problema utilizando un sistema de clasificación habitual, basado en el sistema de clasificación de materias médicas de la National Library of Medicine (MeSH) (se puede encontrar en la dirección http//www.nlm.nih.gov/mesh).
(14) Sobre la base de los resultados obtenidos en el ensayo con ojos de conejo in vivo (TG 405 de la OCDE) y utilizando el SGA de las Naciones Unidas (4) (6).
(15) Sobre la base de los resultados del ICE descritos en el cuadro 6.
(16) Combinación de puntuaciones de ICE distinta de las descritas en el cuadro 6 para la identificación de productos sin categoría 1 del SGA y de la categoría 1 del SGA (véase el cuadro 6).
(17) La clasificación como 2A o 2B depende de la interpretación del criterio del SGA de las Naciones Unidas para distinguir entre estas dos categorías, es decir, 1 de 3 frente a 2 de 3 animales que presentan el día 7 efectos necesarios para generar una clasificación en la categoría 2A. El estudio in vivo incluye 3 animales. Todos los parámetros aparte del enrojecimiento de la conjuntiva en un solo animal se recuperan hasta una puntuación de cero para el día 7 o antes. El único animal que no está recuperado completamente para el día 7 tiene una puntuación de 1 en cuanto al enrojecimiento de la conjuntiva (el día 7) que se recupera plenamente el día 10.
(18) Por media se entiende en todo el documento la media aritmética.
(19) Reglamento (CE) n.o 1272/2008 del Parlamento Europeo y del Consejo, de 16 de diciembre de 2008, sobre clasificación, etiquetado y envasado de sustancias y mezclas, y por el que se modifican y derogan las Directivas 67/548/CEE y 1999/45/CE y se modifica el Reglamento (CE) n.o 1907/2006 (DO L 353 de 31.12.2008, p. 1).
(20) Los números se refieren a valores umbral obtenidos estadísticamente y no están ligados a la precisión de la medición.
(21) Las predicciones del DPRA deben tenerse en cuenta en el marco de un enfoque IATA y de conformidad con las disposiciones de los puntos 9 y 12.
(22) Los números se refieren a valores umbral obtenidos estadísticamente y no están ligados a la precisión de la medición.
(23) Las predicciones del DPRA deben tenerse en cuenta en el marco de un enfoque IATA y de conformidad con las disposiciones de los puntos 9 y 12.
(24) Las predicciones de peligro in vivo y de (potencia) se basan en datos de LLNA (19). La potencia in vivo se obtiene con los criterios propuestos por ECETOC (23).
(25) Las predicciones del DPRA deben tenerse en cuenta en el marco de un enfoque IATA y de conformidad con las disposiciones de los puntos 9 y 11.
(26) Intervalos determinados sobre la base de al menos diez valores de reducción obtenidos por seis laboratorios independientes.
(27) Reglamento (CE) n.o 1272/2008 del Parlamento Europeo y del Consejo, de 16 de diciembre de 2008, sobre clasificación, etiquetado y envasado de sustancias y mezclas, y por el que se modifican y derogan las Directivas 67/548/CEE y 1999/45/CE y se modifica el Reglamento (CE) n.o 1907/2006 (DO L 353 de 31.12.2008, p. 1).
(28) Las predicciones de peligro (y de potencia) in vivo se basan en datos de LLNA (13). La potencia in vivo se obtiene con los criterios propuestos por ECETOC (24).
(29) Las predicciones del ensayo de KeratinoSensTM deben tenerse en cuenta en el marco de un enfoque IATA y de conformidad con las disposiciones de los puntos 9 y 11 del presente método de ensayo.
(30) Sobre la base de los valores observados históricos (12).
(31) Los estadísticos que siguen un enfoque de modelización como el de usar modelos lineales generales pueden plantear el análisis de forma diferente, pero comparable, sin derivar necesariamente el cuadro tradicional de ANOVA, que se basa en enfoques algorítmicos para calcular las estadísticas elaboradas en una época pre-informática.
(32) Se han asignado clases químicas a cada producto problema utilizando un sistema de clasificación habitual, basado en el sistema de clasificación de materias médicas de la National Library of Medicine (MeSH) (se puede encontrar en la dirección http//www.nlm.nih.gov/mesh).
(33) Sobre la base de los resultados obtenidos en el ensayo con ojos de conejo in vivo (TG 405 de la OCDE, método B.5) y utilizando el SGA de la ONU y el CLP de la UE.
(34) Sobre la base de los resultados obtenidos con FL (Protocolo n.o 71 de INVITTOX (6)).
(35) Los estadísticos que siguen un enfoque de modelización como el de usar modelos lineales generales pueden plantear el análisis de forma diferente, pero comparable, sin derivar necesariamente el cuadro tradicional de ANOVA, que se basa en enfoques algorítmicos para calcular las estadísticas elaboradas en una época pre-informática.
(36) Véanse en el apéndice 1 las definiciones y unidades.
(37) Expresado a veces como P OW; se determina por un método de frasco de agitación en el método A.8 (4), un método de HPLC en el método A.24 (5) y un método de agitación lenta en el método A.23 (6). La técnica de la columna generadora se utiliza ocasionalmente para la determinación del log KOW. Se dispone de un número limitado de estudios que hacen uso de esta técnica, principalmente para los clorobifenilos y las dibenzodioxinas cloradas (por ejemplo, Li and Doucette, 1993) (3). En el caso de las sustancias que podrían ionizarse, el log K OW debe referirse a la forma no ionizada.
(38) Véanse en el apéndice 1 las definiciones y unidades.
(39) TLC: cromatografía de capa fina; HPLC: cromatografía de líquidos de alta resolución; CG: cromatografía de gases.
(40) En algunos marcos reguladores, el análisis de los metabolitos puede ser obligatorio cuando se cumplen ciertas condiciones (véase el punto 65).
(41) En general, las concentraciones medidas en agua durante la fase de absorción deben ser, como mínimo, de un orden de magnitud por encima del límite de cuantificación para que pueda medirse más de una semivida de carga corporal en la fase de depuración del estudio.
(42) Véanse en el apéndice 1 las definiciones y unidades.
(43) Con la mayoría de las sustancias problema, no debería detectarse nada en el agua del testigo. Las concentraciones de fondo deben ser pertinentes solo en el caso de los materiales naturales (por ejemplo, algunos metales) y de las sustancias que son ubicuas en el medio ambiente.
(44) Si resulta evidente que no se sigue una cinética de primer orden, entonces convendrá utilizar modelos más complejos (véanse las referencias en el apéndice 5) y buscar el consejo de un bioestadístico.
(45) La absorción puede verse limitada por unas bajas concentraciones de exposición debidas a la escasa hidrosolubilidad en el ensayo de bioconcentración, mientras que con el ensayo alimentario pueden conseguirse concentraciones de exposición mucho mayores.
(46) Para las sustancias de componentes múltiples, sustancias UVCB y mezclas, debe considerarse la hidrosolubilidad de cada uno de los componentes pertinentes para determinar las concentraciones de exposición adecuadas.
(47) El COT incluye el carbono orgánico de las partículas y el carbono orgánico disuelto, es decir, COT = COP + COD.
(48) Aunque no se recomienda en general, si se utiliza un disolvente o un agente solubilizante, el carbono orgánico procedente de este agente debe añadirse al carbono orgánico procedente de la sustancia problema para evaluar la concentración de carbono orgánico en los recipientes de ensayo.
(49) Si el contenido lipídico no se analiza en el mismo pez que la sustancia problema, los peces deben ser como mínimo de peso similar, y (en su caso) del mismo sexo.
(50) Esta alternativa solo es válida si los peces en todos los grupos de ensayo se mantienen en grupos de tamaño similar, se extraen los peces según el mismo patrón y se alimentan de la misma manera. De esta forma se garantiza que el crecimiento de los peces es similar en todos los grupos de ensayo, si la concentración ensayada es inferior al intervalo tóxico. Si el crecimiento es similar, es previsible que también el contenido lipídico sea similar. Un crecimiento diferente en el testigo indicaría un efecto de la sustancia e invalidaría el estudio.
(51) Además del peso, debe registrarse la longitud total, ya que la comparación del grado de aumento de la longitud durante el ensayo es un buen indicador de si se ha producido un efecto adverso.
(52) Puede llevarse a cabo una prueba t de las constantes de la velocidad de crecimiento, para comprobar si el crecimiento presenta diferencias entre los grupos testigo los y de ensayo, o una prueba F en caso de análisis de la varianza. Cuando sea necesario, puede utilizarse una prueba F o prueba de la razón de verosimilitud para contribuir a la elección del modelo de crecimiento adecuado (monografía 54 de la OCDE) (32).
(53) Estos porcentajes suponen que los métodos de análisis son fiables y que la semivida es < 14 días. Si los métodos de análisis son menos fiables o la semivida está (muy) aumentada, estas cifras serán más grandes.
IC: Intervalo de confianza (cuando sea posible calcularlo).
DT: Desviación típica (cuando sea posible calcularla).
(56) El ensayo minimizado puede utilizarse de hecho para demostrar que el metabolismo es rápido cuando se sabe que esto es probable.
(57) Si solo se miden dos puntos de datos, las estimaciones de los límites de confianza para el FBCKm podrán realizarse utilizando métodos de bootstrap. Cuando se disponga también de puntos de datos intermedios, los límites de confianza para el FBCKm podrán calcularse como en el ensayo completo.
(58) Véanse en el apéndice 1 las definiciones y unidades.
(59) A los efectos del Reglamento (CE) n.o 1907/2006, relativo al registro, la evaluación, la autorización y la restricción de las sustancias y mezclas químicas (REACH) (DO L 396 de 30.12.2006, p. 1), esta cuestión se aborda en el documento de orientación sobre los requisitos de información y sobre la valoración de la seguridad química, capítulo R.7c, R.7.10.3.1; R.7.10.4.1; y en la figura R7.10-2.
(60) Con la mayoría de las sustancias problema, no debería detectarse nada en el agua del testigo. Las concentraciones de fondo deben ser pertinentes solo en el caso de los materiales naturales (por ejemplo, algunos metales) y de las sustancias que son ubicuas en el medio ambiente.
(61) Dado que el FBM se define como la relación de la concentración de una sustancia en un organismo frente a la presente en el alimento del organismo en estado de equilibrio, se tienen en cuenta los lípidos mediante la corrección según el contenido lipídico en el organismo y en el alimento, por lo que se describe con mayor exactitud como «corrección». Este enfoque difiere de la «normalización» respecto a un determinado contenido lipídico de un organismo, como se hace en el ensayo de bioconcentración de exposición acuática.
(62) Debe registrarse asimismo la longitud total durante el ensayo, ya que es un buen indicador de si se ha producido algún efecto adverso.
(63) El HCB figura en los anexos A y C del Convenio de Estocolmo, y en los anexos I y III del Reglamento (CE) n.o 850/2004 sobre contaminantes orgánicos persistentes (DO L 158 de 30.4.2004, p. 7).
(64) En caso de rápido crecimiento durante la fase de absorción, la dosis alimentaria real disminuirá por debajo de la establecida al comienzo de la exposición.
(65) En un estudio de exposición acuática, una semivida de 14 días correspondería a un FBC de aproximadamente 10 000 l/kg, utilizando peces de 1 g con una velocidad de absorción correspondiente de unos 500 l/kg/d (según la ecuación de Sijm et al. (46)).
(66) Como las concentraciones internas reales solo se pueden determinar una vez que se haya realizado el ensayo, hay que hacer una estimación de la concentración interna prevista (por ejemplo, sobre la base del FBM previsto y la concentración en el alimento; véase la ecuación A5.8 en el apéndice 5).
(67) Puede no ser totalmente evitable la presencia de la sustancia problema en el medio de ensayo como consecuencia de la excreción de los peces o la lixiviación a partir del alimento. Por lo tanto, una opción es medir la concentración de la sustancia en el agua al final de la fase de absorción, especialmente si se utiliza un sistema semiestático, para ayudar a determinar si se ha producido alguna exposición acuática.
(68) Este enfoque es específico del estudio alimentario, distinto del procedimiento seguido en la exposición acuática, de ahí que se haya utilizado el término «corrección» en lugar de «normalización» a fin de evitar confusiones (véase también la nota a pie de página del punto 106).
(69) Puede llevarse a cabo una prueba t de las constantes de la velocidad de crecimiento, para comprobar si el crecimiento presenta diferencias entre los grupos testigo y de ensayo, o un test F en caso de análisis de la varianza. Cuando sea necesario, puede utilizarse una prueba F o prueba de la razón de verosimilitud para contribuir a la elección del modelo de crecimiento adecuado (monografía 54 de la OCDE) (32).
(70) Técnica de análisis de productos alimenticios para determinar el contenido de proteínas, lípidos, fibra bruta y cenizas; esta información la tiene normalmente el proveedor del alimento.
IC: Intervalo de confianza (cuando sea posible calcularlo).
DT: Desviación típica (cuando sea posible calcularla).
(73) Meyer et al. (1)
(74) Cabe observar que en el propio ensayo se considera preferible el peso como medida para obtener el tamaño y la constante de la velocidad de crecimiento. Sin embargo, se sabe que la longitud es una medida más práctica si los peces han de seleccionarse visualmente al inicio de un experimento (es decir, de entre la población inicial).
(75) Este intervalo de longitud se indica en los métodos de ensayo relativos a las nuevas sustancias químicas, etc. basados en la Ley sobre el control de sustancias químicas de Japón (CSCL).
(76) Los valores entre paréntesis son los números de muestras (agua, peces) que se toman si se procede a un muestreo suplementario.
(77) La estimación previa al ensayo del valor de k 2 con log K OW de 4,0 es 0,652 día– 1. La duración total del experimento se fija en 3 × t SS = 3 × 4,6 días, o sea 14 días. Para la estimación de t SS, consúltese el apéndice 5.
(78) Muestra de agua tomada después de transferir al menos 3 «volúmenes de recipiente».
(79) Estos peces se muestrean de la población inicial.
(80) Si se necesitan estudios sobre el metabolismo o de mayor precisión que requieran más peces, deben tomarse las muestras de peces especialmente al final de las fases de absorción y de depuración (véase el punto 40).
(81) Pueden ser necesarios al menos tres peces adicionales para el análisis del contenido lipídico si no es posible utilizar los mismos peces muestreados para determinar las concentraciones de la sustancia al inicio del ensayo, al final de la fase de absorción y al final de la fase de depuración. Téngase en cuenta que en muchos casos debe ser posible utilizar solo los tres peces testigo (véase el punto 56).
(82) Tres muestras de alimento procedentes tanto del grupo de ensayo como del testigo se analizan para medir las concentraciones de la sustancia problema y el contenido lipídico.
(83) Los peces se muestrean de la población inicial lo más cerca posible del comienzo del estudio; deben muestrearse al menos tres peces de la población inicial al comienzo del ensayo para determinar en ellos el contenido lipídico.
(84) El muestreo (optativo) al inicio de la fase de absorción proporciona datos para calcular la asimilación alimentaria de la sustancia problema que pueda compararse con la eficiencia de la asimilación calculada a partir de los datos de la fase de depuración.
(85) Podrán muestrearse cinco peces adicionales para efectuar análisis específicos de cada tejido.
(86) Pueden ser necesarios al menos tres peces adicionales para el análisis del contenido lipídico si no es posible utilizar los mismos peces muestreados para determinar las concentraciones de la sustancia al inicio del ensayo, al final de la fase de absorción y al final de la fase de depuración. Téngase en cuenta que en muchos casos debe ser posible utilizar solo los tres peces testigo (véanse los puntos 56 y 153).
(87) Tal y como ocurre con cada relación empírica, debe verificarse que la sustancia problema está incluida en el ámbito de aplicabilidad de la relación.
(88) El peso de los peces al final de la fase de absorción puede estimarse a partir de los datos de estudios anteriores o del conocimiento del aumento probable de tamaño de la especie de ensayo a partir del peso típico al inicio del ensayo a lo lago de una duración típica de la absorción (por ejemplo, 28 días).
(89) En la mayoría de programas que permiten realizar la regresión lineal, también se dan los errores típicos y el intervalo de confianza (IC) de las estimaciones como, por ejemplo, en Microsoft Excel utilizando el paquete de herramientas de análisis de datos.
(90) En contraste con el método de la regresión lineal, la utilización de esta fórmula no da un error típico para k2.
(91) En contraste con un método de ajuste lineal, este método generalmente no da un error típico o intervalo de confianza para la estimación de k1.
(92) Hay que tener en cuenta que la incertidumbre en la estimación de k2 no se utiliza correctamente en el modelo de bioacumulación cuando se considera esencialmente como constante al ajustar k1 en el método de ajuste secuencial. La incertidumbre del FBC resultante será, por tanto, diferente con los métodos de ajuste secuencial y simultáneo.
(93) Es posible que en algunas regiones solo se puedan obtener alimentos para peces con una concentración lipídica que se quede muy por debajo de este límite máximo. En estos casos, se deben realizar estudios con la concentración lipídica baja del alimento tal como se suministre, y se ha de ajustar la dosis alimentaria de forma adecuada para mantener la salud de los peces. Los lípidos del alimento no deben aumentarse artificialmente mediante la adición de aceite en exceso.
(94) En la naturaleza, es probable que la ruta que conduzca a la mayor exposición en entornos acuáticos sea a través de la ingestión de sustancias muy hidrófobas, por lo que el FBC estimado no es estrictamente representativo del potencial de bioacumulación de tales sustancias.
(95) Tal y como ocurre con cada relación empírica, debe verificarse que la sustancia problema está incluida en el ámbito de aplicabilidad de la relación.
(96) El peso de los peces al final de la fase de absorción puede estimarse a partir de los datos de estudios anteriores o del conocimiento del aumento probable de tamaño de la especie de ensayo a partir del peso típico al inicio del ensayo a lo lago de una duración típica de la absorción (por ejemplo, 28 días).
(97) En la mayoría de programas que permiten realizar la regresión lineal, también se dan los errores típicos y el intervalo de confianza (IC) de las estimaciones como, por ejemplo, en Microsoft Excel utilizando el paquete de herramientas de análisis de datos.
(*6) Indíquese qué recipiente se ha utilizado para el experimento.
(*7) Indíquense las nidadas abortadas como ‘AB’ en la casilla pertinente.
(*8) Indíquese la mortalidad de los animales parentales como ‘M’ en la casilla pertinente.
(98) A pesar de que la longitud total media mínima típica no constituya un criterio de validez, deberán examinarse cuidadosamente las desviaciones inferiores al valor indicado en relación con la sensibilidad del ensayo. La longitud total media mínima se obtiene de una selección de datos actualizados en el momento actual.
(99) Es posible que deban utilizarse otras temperaturas para la variedad concreta de trucha arcoíris utilizada en el ensayo. Los peces reproductores deben permanecer a la misma temperatura que los huevos. Después de recibir los huevos de un criadero comercial, hay que realizar una breve adaptación (p. ej., 1-2 horas) a la temperatura del ensayo.
(100) Las larvas deben permanecer en la oscuridad hasta una semana después de la eclosión, salvo durante las observaciones. Después, deben estar a luz tenue hasta el final del ensayo (12-16 horas de fotoperiodo) (4).
(101) En cualquiera de las condiciones del ensayo, el régimen de iluminación deberá ser constante.
(102) En cualquier ensayo, se realizará a ± 2 0/00.
(*9) Debe ofrecerse comida hasta que los peces estén saciados. La comida no consumida y los excrementos deberán evacuarse siempre que sea necesario para evitar la acumulación de desechos.
|
FBS |
Artemia salina congelada; adultos de Artemia sp. |
|
BSN |
Nauplios de artemia salina; recién eclosionados |
|
BSN48 |
Nauplios de artemia salina; 48 horas de vida |
(1) No es necesario alimentar a las larvas con saco vitelino.
(2) Filtrado de un cultivo mixto.
(3) Gránulos procedentes de un proceso de fermentación.
(*10) Los tubérculos papilares aparecen normalmente solo en los machos adultos y se encuentran en los radios de las aletas, del segundo al séptimo u octavo contando desde el extremo posterior de la aleta anal (fig. 1 y fig. 2). Sin embargo, en raras ocasiones aparecen tubérculos en el primer radio de la aleta, contando desde el extremo posterior de la aleta anal. Este procedimiento normalizado de trabajo (PNT) abarca la medición de los tubérculos en el primer radio de la aleta (en este PNT el número de radio de la aleta hace referencia al orden desde el extremo posterior de la aleta anal).
(*11) Tampón de homogeneización:
|
— |
[Tris-HCl 50 mM pH 7,4; mezcla inhibidora de la proteasa al 1 % (Sigma)]: 12 ml de Tris-HCl pH 7,4 + 120 μl de mezcla inhibidora de la proteasa. |
|
— |
TRIS: TRIS-ULTRA PURE (ICN); por ejemplo, de Bie & Berntsen, Dinamarca. |
|
— |
Mezcla inhibidora de la proteasa: de Sigma (para tejidos de mamíferos), número de referencia P 8340. |
NOTA: El tampón de homogeneización debe utilizarse el mismo día que se elabore. Colóquelo en hielo durante su uso.
(103) Carl von Linné (* 23 de mayo de 1707, Råshult /Älmhult; † 10 de enero de 1778, Uppsala).
(*12) Agua desmineralizada (es decir, destilada o desionizada)