|
8)
|
I del B ska följande kapitel läggas till:
”B.63 SCREENINGTEST FÖR REPRODUKTIONS-/UTVECKLINGSTOXICITET
INLEDNING
|
1.
|
Denna testmetod motsvarar OECD:s testriktlinje (TG) 421 (2016). OECD:s riktlinjer för kemikalietestning utvärderas med jämna mellanrum som en anpassning till den vetenskapliga utvecklingen. De ursprungliga riktlinjerna för screeningtest (421) antogs 1995, utifrån ett protokoll för ett preliminärt screeningtest för reproduktionstoxicitet som diskuterats vid två expertmöten, i London 1990 (1) och Tokyo 1992 (2).
|
|
2.
|
Denna testmetod har uppdaterats med endpoints som är relevanta för hormonstörande ämnen, och detta är en uppföljning till den högprioriterade verksamhet som OECD inledde 1998 för att revidera befintliga testriktlinjer och ta fram nya riktlinjer för screening och testning av potentiella hormonstörande ämnen (3). OECD TG 407 (28-dagars toxicitetsstudie med upprepad oral dosering på gnagare, kapitel B.7 i denna bilaga) utökades till exempel 2008 med parametrar för att påvisa endokrin aktivitet hos testkemikalier. Syftet med uppdateringen av TG 421 var att inkludera några endpoints som är relevanta för hormonstörande ämnen i riktlinjerna för screeningtester där exponeringsperioderna omfattar några av de känsliga utvecklingsperioderna (pre- eller tidiga postnatalperioder).
|
|
3.
|
De nya utvalda endpoints som är relevanta för hormonstörande ämnen och som också ingår i TG 443 (utökad enkelgenerationsstudie av reproduktionstoxicitet, motsvarande kapitel B.56 i denna bilaga) inkluderades i TG 421 på grundval av en förstudie som behandlade vetenskapliga och tekniska aspekter av deras inkluderande samt nödvändiga anpassningar av testets utformning för att möjliggöra ett inkluderande (4).
|
|
4.
|
Denna testmetod är utformad för att generera begränsad information om en testkemikalies effekter på den hanliga och honliga fortplantningsförmågan, t.ex. beträffande könskörtlarnas funktion, parningsbeteende, befruktning, embryoutveckling och födsel. Den är inte ett alternativ till, eller en ersättning för, de befintliga testmetoderna B.31, B.34, B.35 eller B.56.
|
INLEDANDE ÖVERVÄGANDEN
|
5.
|
Denna screeningtestmetod kan användas för att få en första information om vilka eventuella effekter en kemikalie har på reproduktion och/eller utveckling, antingen på ett tidigt stadium genom bedömning av kemikaliens toxikologiska egenskaper eller genom bedömning av kemikalien i sig. Studien kan också användas som en del av flera screeningtest för att ge en första bedömning av befintliga kemikalier för vilka ingen eller begränsad toxikologisk information finns tillgänglig, eller som en preliminär undersökning inför mer omfattande studier om reproduktion/utveckling eller när det av andra skäl anses relevant. Vid genomförande av studien bör man följa de vägledande principer och överväganden som beskrivs i OECD:s vägledning nr 19 om erkännande, bedömning och användning av kliniska tecken som humana endpoints för försöksdjur som används för säkerhetsbedömningar (5).
|
|
6.
|
Denna testmetod ger ingen komplett information om alla aspekter av reproduktion och utveckling. Den är särskilt begränsad när det gäller att detektera postnatala uttryck av prenatal exponering, eller effekter som kan orsakas av postnatal exponering. Bland annat på grund av det ringa antalet försöksdjur i doseringsgrupperna, selektiviteten hos endpoints samt studiens begränsade varaktighet ger denna metod inga bevis för definitiva påståenden om utebliven effekt. Vid avsaknad av data från andra toxicitetstest om reproduktion/utveckling kan de positiva resultaten dessutom användas för en inledande riskbedömning och bidra till beslutsfattande gällande vilka övriga test som behövs och när.
|
|
7.
|
Resultaten som erhölls genom de endokrinrelaterade parametrarna bör betraktas mot bakgrund av OECD:s begreppsram för provning och bedömning av hormonstörande ämnen (6). Den vidareutvecklade OECD TG 421 ingår i nivå 4 av begreppsramen som ett in vivo-test som genererar data om skadliga effekter för endokrinrelaterade endpoints. En endokrin signal kanske dock inte räcker för att påvisa att testkemikalien är ett hormonstörande ämne.
|
|
8.
|
Vid test enligt denna metod används oral tillförsel av testkemikalien. Om tillförsel sker på annat sätt kan metoden behöva modifieras.
|
|
9.
|
Innan testmetoden används på en blandning i syfte att samla in data för ett lagstadgat ändamål bör man överväga om, och i så fall varför, den kommer att ge godtagbara resultat för det syftet. Sådana överväganden är inte nödvändiga om blandningen måste provas enligt lag.
|
|
10.
|
De definitioner som används finns i tillägg 1.
|
TESTPRINCIP
|
11.
|
Testkemikalien tillförs i graderade doser till flera grupper av hanar och honor. Hanarna ska behandlas i minst fyra veckor, inbegripet dagen före den planerade avlivningen (detta inkluderar minst två veckor före parning, under parningsperioden och ungefär två veckor efter parning). Eftersom hanarna endast behandlas under en begränsad period före parning utgör fertilitet ingen särskilt känslig indikator för testikeltoxicitet. Därför krävs en detaljerad histologisk undersökning av testiklarna. En två veckors doseringsperiod före parning i kombination med efterföljande observationer av parning/fertilitet och en sammanlagd doseringsperiod på minst fyra veckor, följt av en ingående histopatologisk analys av hanarnas könskörtlar, anses vara tillräckligt för att påvisa flertalet effekter på hanarnas fertilitet och spermatogenes.
|
|
12.
|
Honorna bör behandlas under hela studien. Detta inbegriper två veckor före parning (i syfte att täcka minst två kompletta ägglossningscykler), tiden fram till befruktning, dräktighetsperioden samt minst tretton dagar efter födsel, fram till dag för planerad avlivning.
|
|
13.
|
Studiens längd, efter acklimatisering och bedömning av ägglossningscykeln före behandling, beror på honornas kondition och ligger på runt 63 dagar (minst 14 dagar före parning, (upp till) 14 dagars parning, 22 dagars dräktighetsperiod samt 13 dagars laktation).
|
|
14.
|
Under administreringsperioden observeras djuren varje dag noggrant för tecken på toxicitet. Djur som dör eller avlivas under testet obduceras, och i slutet av testet avlivas överlevande djur och obduceras.
|
BESKRIVNING AV TESTMETODEN
Val av djurart
|
15.
|
Denna testmetod är avsedd att användas på råtta. En utförlig motivering ska ges om de parametrar som anges i denna testmetod undersöks hos en annan art av gnagare. I det internationella valideringsprogrammet för att påvisa hormonstörande ämnen i OECD TG 407 (motsvaras av kapitel B.7 i denna bilaga) var råtta den enda art som användes. Stammar med låg fertilitet eller välkänd hög incidens av utvecklingsdefekter ska inte användas. Friska tidigare obefruktade djur, som inte tidigare har använts i försök, ska användas. Försöksdjuren ska karakteriseras efter art, stam, kön, vikt och ålder. Vid testets början bör variationen i vikt mellan djuren vara minimal och får inte överstiga 20 % av vardera könets medelvikt. När en upprepad oral dos utförs som inledning till en långtidsstudie, är det att föredra att djur från samma stam och ursprung används för båda studierna.
|
Inhysning och utfodring
|
16.
|
Alla förfaranden bör överensstämma med lokala normer för laboratoriedjurvård. Temperaturen i försöksdjurens utrymmen bör vara 22 °C (± 3 °). Den relativa fuktigheten bör vara minst 30 % och helst inte överstiga 70 % utom när rummet rengörs, och målet bör vara 50–60 %. Belysningen bör vara artificiell med ljusperioden 12 timmar ljus och 12 timmar mörker. För utfodring kan konventionellt laboratoriefoder användas med obegränsad tillgång till dricksvatten. Valet av foder kan påverkas av behovet av att säkerställa en lämplig blandning av en testkemikalie när den administreras genom denna metod.
|
|
17.
|
Djuren bör inhysas i små grupper med individer av samma kön. Djuren kan inhysas individuellt om det är vetenskapligt motiverat. Vid grupplacering i bur bör inte fler än fem djur inhysas i en och samma bur. För parningsprocedurerna används burar som är lämpliga för ändamålet. Dräktiga honor ska hysas individuellt och förses med bomaterial. Digivande honor ska hysas individuellt med sina ungar.
|
|
18.
|
Fodret bör regelbundet analyseras beträffande främmande ämnen. Ett prov av fodret bör sparas tills rapporten är slutförd.
|
Förberedelse av djuren
|
19.
|
Friska yngre adulta djur fördelas slumpmässigt till kontroll- och behandlingsgrupperna. Burarna bör placeras i sådan ordning att eventuella verkningar på grund av placeringen minimeras. Djuren ska ha en unik identifiering och hållas i sina burar under minst fem dagar innan undersökningen börjar, för att göra det möjligt för dem att acklimatiseras till förhållandena i laboratoriet.
|
Beredning av doser
|
20.
|
Det rekommenderas att testkemikalien administreras oralt såvida inte andra tillförselsätt anses mer lämpliga. Vid oral administrering ges testkemikalien i regel via sond, men testkemikalien kan även ges via fodret eller dricksvattnet.
|
|
21.
|
Vid behov kan testkemikalien upplösas eller suspenderas i en lämplig vehikel. Det rekommenderas, när så är möjligt, att man först använder en vattenbaserad lösning/suspension följt av en lösning/emulsion i olja (t.ex. majsolja) och därefter eventuell lösning i andra vehiklar. För andra vehiklar än vatten bör vehikelns toxiska egenskaper vara kända. Testkemikaliens stabilitet och homogenitet i vehikeln bör bestämmas.
|
FÖRFARANDE
Djurens antal och kön
|
22.
|
Det rekommenderas att varje grupp inledningsvis innehåller minst 10 hanar och 12–13 honor. Honornas ägglossningscykler undersöks före exponeringen, och individer som inte uppvisar typiska cykler på 4–5 dagar ska inte tas med i studien. Det är därför extra honor rekommenderas initialt så att varje grupp innehåller 10 honor. Förutom vid betydande toxiska effekter förväntas detta leda till minst 8 dräktiga honor per grupp, vilket i regel utgör det minsta acceptabla antalet dräktiga honor per grupp. Syftet med detta är att säkerställa ett tillräckligt antal dräktigheter och ungar för att möjliggöra en meningsfull utvärdering av testkemikaliens potential att påverka fertilitet, dräktighet och moderskaps- och dibeteende samt tillväxt och utveckling hos F1-avkomman från befruktning till dag 13 post partum.
|
Dosering
|
23.
|
I allmänhet bör minst tre doseringsgrupper och en kontrollgrupp användas. Dosnivåerna kan baseras på information från akuta toxicitetstester eller på resultat från studier med upprepad dosering. Med undantag av behandlingen med testkemikalien bör djuren i kontrollgruppen hanteras på exakt samma sätt som testgruppen. Om en vehikel används för att tillföra testkemikalien bör kontrollgruppen tillföras vehikeln i den största volym som används.
|
|
24.
|
Dosnivåerna bör väljas med hänsyn till eventuell toxicitetsdata och (toxikologisk) kinetisk data som finns tillgänglig. Hänsyn bör tas till eventuell skillnad i sensitivitet mellan dräktiga och icke-dräktiga djur. Den högsta nivån på dosen bör väljas i syfte att inducera toxiska verkningar, men inte leda till döden eller orsaka svåra lidanden. Därefter bör en fallande nivå på doserna väljas i syfte att påvisa en dosrelaterad respons och ingen påvisbar skadlig effekt vid den lägsta dosnivån (NOAEL). Två- till fyrfaldiga dosintervall är ofta optimala för de fallande doseringsnivåerna, och att lägga till en fjärde testgrupp är att föredra framför att använda mycket stora intervall (t.ex. mer än faktor 10) mellan doseringarna.
|
|
25.
|
Vid observation av tecken på allmän toxicitet (t.ex. minskad kroppsvikt, effekter på lever, hjärta, lungor eller njurar) eller andra förändringar som kanske inte är toxiska reaktioner (t.ex. nedsatt födointag, leverförstoring) ska observerade effekter avseende endokrint känsliga endpoints tolkas med försiktighet.
|
Toleranstest
|
26.
|
Om ett försök med en doseringsnivå om minst 1 000 mg per kg kroppsvikt och dag eller, vid tillförsel via föda eller dricksvatten, en motsvarande andel i födan eller dricksvattnet och med användning av det tillvägagångssätt som beskrivits för denna studie, inte ger några observerbara toxiska effekter och om toxicitet inte förväntas på grundval av uppgifter från strukturellt närbesläktade ämnen, kan en fullständig undersökning med tre dosnivåer anses överflödig. Detta toleranstest är tillämpligt förutom i fall där sannolik exponering på människor indikerar att en högre oral doseringsnivå behövs. För andra former av tillförsel, såsom inandning eller applicering på huden, kan testkemikaliens fysikalkemiska egenskaper ofta utnyttjas för att indikera och begränsa den maximala exponeringsnivån.
|
Administrering av doser
|
27.
|
Försöksdjuren behandlas med testkemikalien dagligen under en vecka. När testkemikalien tillförs genom sondmatning bör detta ske i en engångsdos via magsond eller lämplig intubationskanyl. Den maximala vätskevolymen som kan administreras vid ett tillfälle är beroende av försöksdjurets storlek. Volymen får inte överstiga 1 ml per 100 g kroppsvikt, utom för vattenlösningar där volymen kan uppgå till 2 ml per 100 g kroppsvikt. Med undantag för irriterande eller korrosiva testkemikalier, som normalt kommer att visa stegrade verkningar vid högre koncentrationer, bör variationer av testvolymen minimeras genom justering av koncentrationen för att säkerställa en konstant volym vid alla dosnivåer.
|
|
28.
|
För kemikalier som tillförs via fodret eller dricksvattnet är det viktigt att säkerställa att kvantiteterna av den berörda testkemikalien inte påverkar normal närings- eller vattenbalans. När testkemikalien tillförs via kosten kan antingen en konstant foderkoncentration (PPM) eller en konstant doseringsnivå i förhållande till djurens kroppsvikt användas. Det alternativ som används ska anges. En testkemikalie som tillförs genom sondmatning bör ges vid samma tid varje dag och justeras minst varje vecka så att dosnivån hålls konstant i förhållande till djurens kroppsvikt.
|
Försöksschema
|
29.
|
Behandlingen av båda könen bör påbörjas minst två veckor före parning, efter att de har acklimatiserats under minst fem dagar och efter att honorna har screenats för normala ägglossningscykler (under en tvåveckorsperiod före behandlingen). Studien bör planläggas så att bedömningen av ägglossningscykeln påbörjas kort efter att djuret ifråga blivit könsmoget. Detta kan variera något mellan olika råttstammar i olika laboratorier, t.ex. 10 veckors ålder för Sprague Dawley-råttor och 12 veckors ålder för Wistar-råttor. Moderdjur med ungar bör avlivas på dag 13 post partum, eller kort därefter. Dagen för födseln (dvs. efter avslutad förlossning) definieras som dag 0 post partum. Honor som inte uppvisar några tecken på att ha parat sig ska avlivas 24–26 dagar efter parningsperiodens sista dag. Tillförseln fortsätts för båda könen under parningsperioden. Hanar ska ges fortsatt behandling efter parningsperioden och minst till dess att en minimidoseringsperiod på totalt 28 dagar har uppnåtts. Därefter avlivas de eller hålls kvar och ges fortsatt behandling för en andra parning om så bedöms lämpligt.
|
|
30.
|
Daglig dosering till dräktiga honor och moderdjur ska fortsätta genom hela dräktighetsperioden och minst till och med dag 13 post partum eller dagen före avlivningsdagen. När testkemikalien tillförs via inhalation eller applicering på huden ska doseringen fortsätta minst till och med dräktighetsdag 19, och doseringen bör återupptas så fort som möjligt efter födsel och inte senare än PND 4.
|
|
31.
|
Ett diagram för försöksschemat finns i tillägg 2.
|
Parningsprocedur
|
32.
|
I regel ska 1:1-parningar (en hane per hona) användas i denna studie. Undantagssituationer kan uppstå om hanar dör. Honan ska inhysas med samma hane tills parning bevisligen har skett eller två veckor har förflutit. Honorna ska undersökas varje morgon för närvaron av sperma eller vaginalproppar. Dag 0 av dräktigheten definieras som den dag då bevis på parning bekräftas (fynd av vaginalpropp eller sperma). Om parningen inte lyckas kan man överväga ett nytt parningsförsök där honor sammanförs med hanar med fastställd fertilitet från samma grupp.
|
Kullstorlek
|
33.
|
På dag 4 efter födseln kan storleken på varje kull justeras genom att eliminera extra ungar genom slumpmässigt urval, så att det blir, så nära som möjligt, fyra eller fem ungar per kön per kull, beroende på den normala kullstorleken i den råttstam som använts. Blodprover ska tas på två av överskottsungarna och sparas för bestämning av T4-nivåer i serum. Selektiv eliminering av ungar, t.ex. utifrån kroppsvikt eller anogenitalt avstånd (AGD), är inte lämpligt. När antalet han- eller honungar inte medger fyra eller fem av varje kön per kull, är partiell anpassning acceptabelt (t.ex. sex hanar och fyra honor). Inga ungar ska elimineras när kullstorleken är mindre än det avsedda målet (8 eller 10 ungar/kull). Om det endast finns en överskottsunge utöver det avsedda målet ska endast en unge elimineras och användas för blodinsamling och eventuell bedömning av T4 i serum.
|
|
34.
|
Om kullstorleken inte korrigeras ska två ungar per kull avlivas på dag 4 efter födseln och blodprov tas för att mäta koncentrationen av tyreoideahormoner i serum. Om det är möjligt ska dessa två ungar per kull vara honungar, så att hanungar kan reserveras för senare bedömning av bröstvårteretention, förutom då eliminering av dessa honungar innebär att det inte finns några honor kvar för den slutliga bedömningen. Inga ungar ska elimineras när kullstorleken är mindre än 8 eller 10 ungar/kull (beroende på den normala kullstorleken för den råttstam som använts). Om det endast finns en unge mer än normalt antal ungar per kull ska endast en unge elimineras och användas för blodinsamling och eventuell bedömning av T4 i serum.
|
Observationer av levande djur
Kliniska observationer
|
35.
|
Allmänna kliniska observationer bör göras minst en gång per dag under hela testperioden, och oftare när tecken på toxicitet observeras. Observationerna bör företrädesvis utföras vid samma tidpunkt varje dag och med hänsyn till förväntade toppeffekter efter doseringen. Relevanta beteendeförändringar, tecken på svårartad eller förlängd födsel och alla toxicitetstecken, inbegripet dödlighet, ska registreras. Dessa rapporter ska inkludera information om toxicitetstecknens början, grad och varaktighet.
|
Kroppsvikt samt foder- och vattenkonsumtion
|
36.
|
Hanar och honor ska vägas på doseringens första dag, minst en gång i veckan därefter samt vid testets slut. Under dräktighetstiden ska honorna vägas på dag 0, 7, 14 och 20 samt inom 24 timmar efter födseln (dag 0 eller 1 post partum) och åtminstone på dag 4 och 13 post partum. Dessa observationer bör rapporteras individuellt för varje vuxet djur.
|
|
37.
|
Under tiden före parning, dräktighetsperiod samt laktation ska foderintaget mätas åtminstone en gång i veckan. Det är inte obligatoriskt att mäta foderintaget under parningsperioden. Om testkemikalien tillförs via dricksvattnet bör även vattenkonsumtionen mätas under nämnda perioder.
|
Ägglossningscykler
|
38.
|
Ägglossningscyklerna bör övervakas innan behandlingen startar i syfte att välja ut honor med regelbundna cykler till försöket se punkt 22). Även vaginala utstryk bör övervakas dagligen från behandlingens början till dess att parning bevisligen skett. Om det vid doseringens början föreligger misstankar om akuta stresseffekter som kan störa ägglossningscyklerna kan laboratoriet exponera försöksdjuren under två veckor och därefter samla in vaginala utstryk dagligen i syfte att övervaka ägglossningscyklerna under minst två veckor före parning, med fortsatt övervakning under parningsperioden fram till dess att parning bevisligen skett. När celler från vagina eller livmoderhals samlas in bör försiktighet iakttas för att undvika skador på slemhinnan, eftersom sådana kan framkalla pseudodräktighet (7) (8).
|
Parametrar för avkomman
|
39.
|
Dräktighetsperioden bör registreras och beräknas från dräktighetsdag 0. Varje kull ska undersökas snarast möjligt efter nedkomsten i syfte att fastställa antal och kön för ungar, dödfödda, levande födda, småväxta ungar (avsevärt mindre än motsvarande kontrollungar) samt eventuella större avvikelser.
|
|
40.
|
Levande ungar ska räknas och könsbestämmas, och kullarna ska vägas inom 24 timmar efter födseln (dag 0 eller 1 post partum) samt åtminstone på dag 4 och 13 post partum. Utöver de observationer som beskrivs i punkt 35 ska allt onormalt beteende hos ungarna registreras.
|
|
41.
|
AGD på varje unge ska mätas på samma dag efter födseln, någon gång mellan PND 0 och PND 4. Ungarnas kroppsvikt bör samlas in under dagen för AGD-mätningen, och AGD bör normaliseras till ett mått på ungarnas storlek, företrädesvis kubikroten ur kroppsvikten (9). Antal bröstvårtor/vårtgårdar på hanungarna ska räknas på PND 12 eller 13 enligt rekommendationerna i OECD GD 151 (10).
|
Klinisk biokemi
|
42.
|
Blodprov tas från ett definierat ställe utifrån följande schema:
|
—
|
från minst två ungar per kull på dag 4 efter födseln, om antalet ungar så tillåter (se punkterna 33 och 34),
|
|
—
|
från alla moderdjur och minst två ungar per kull när försöket avslutas på dag 13, och
|
|
—
|
från alla vuxna hanar vid försökets slut.
|
|
Alla blodprov ska förvaras under lämpliga förhållanden. Blodproverna som tas på 13 dagar gamla ungar samt vuxna hanar ska analyseras för tyreoideahormoner (T4) i serum. Ytterligare analys av T4 i blodproven från moderdjuren och de 4 dagar gamla ungarna görs om det bedöms relevant. Om det är relevant kan blodet även analyseras för andra hormoner. Ungarnas blod kan samförvaras per kull för analyser av tyreoideahormoner. Tyreoideahormonerna (T4 och TSH) bör helst mätas som ett totalvärde.
|
43.
|
Följande faktorer kan påverka variationen och de absoluta koncentrationerna för hormonbestämningarna:
|
—
|
Tidpunkt för avlivningen beroende på hormonkoncentrationernas dygnsvariation.
|
|
—
|
Avlivningsmetod för att undvika onödig stress hos djuren som kan påverka hormonkoncentrationerna.
|
|
—
|
Testsatser för hormonbestämningar som kan skilja sig åt genom sina standardkurvor.
|
|
|
44.
|
Plasmaprover särskilt avsedda för hormonbestämning bör tas vid en jämförbar tid på dagen. De numeriska värden som erhölls vid analys av hormonkoncentrationer skiljer sig åt mellan de olika analyssatser som finns i handeln.
|
Patologi
Obduktion
|
45.
|
Vid avlivning eller om djur dör under pågående test ska vuxna djur undersökas makroskopiskt med avseende på avvikelser eller patologiska förändringar. Särskild uppmärksamhet bör ägnas åt organen i reproduktionssystemet. Antal implantationsställen ska registreras. Vaginala utstryk ska undersökas på morgonen samma dag som obduktionen äger rum för att fastställa ägglossningscykelns skede och möjliggöra korrelation med äggstockarnas histopatologi.
|
|
46.
|
På samtliga hanar ska all angränsande vävnad putsas bort från testiklar och bitestiklar liksom från prostata och sädesblåsor med koaguleringskörtlar varefter våtvikten mäts snarast möjligt efter dissektion innan organen hinner torka. Utöver detta kan fler organ vägas, som till exempel muskelkomplexen levator ani och bulbocavernosus, Cowpers körtlar eller ollonet hos hanar samt äggstockspar (våtvikt) och livmoder (inklusive livmoderhals) hos honor. Om dessa organ vägs ska deras vikt registreras snarast möjligt efter dissektion.
|
|
47.
|
Döda ungar och ungar som avlivas på dag 13 post partum, eller kort därefter, ska undersökas noga åtminstone externt för grova avvikelser. Särskild uppmärksamhet bör fästas vid de yttre reproduktionsorganen, som ska undersökas för tecken på avvikande utveckling. På dag 13 ska sköldkörteln från 1 hanunge och 1 honunge per kull bevaras.
|
|
48.
|
Äggstockar, testiklar, accessoriska könsorgan (livmoder och livmoderhals, bitestiklar, sädesblåsor plus koaguleringskörtlar) och sköldkörtlar från samtliga vuxna försöksdjur, inklusive alla organ som uppvisar skador vid makroskopisk undersökning, ska bevaras. Fixering med formalin rekommenderas inte för rutinundersökning av testiklar och bitestiklar. I stället bör Bouins lösning eller modifierat fixeringsmedel enligt Davidson användas för dessa vävnader (11). Tunica albuginea kan punkteras försiktigt och ytligt med en nål på organets båda poler för en snabb inträngning av fixeringsmedlet.
|
Histopatologi
|
49.
|
En detaljerad histologisk undersökning ska genomföras på äggstockar, testiklar och bitestiklar (särskild uppmärksamhet bör fästas vid spermatogenesens stadier samt vid histopatologin hos den interstitala cellstrukturen i testiklarna) på försöksdjuren i den högsta doseringsgruppen samt på djuren i kontrollgruppen. Övriga organ som bevarats inklusive sköldkörtlar från ungar och vuxna djur kan undersökas om det behövs. Sköldkörtelns vikt kan bestämmas efter fixering. Putsning ska också göras väldigt försiktigt, och först efter fixeringen för att undvika vävnadsskador. Vävnadsskador kan påverka den histopatologiska analysen negativt. När förändringar observeras i gruppen med högst dosering kan undersökningen utökas till att inkludera djur från andra doseringsgrupper. Vägledningen om histopatologi (11) omfattar detaljerad information om dissektion, fixering, sektionering och histopatologi av endokrina vävnader.
|
DATA OCH RAPPORTERING
Data
|
50.
|
Data bör rapporteras individuellt per djur. Alla data ska sammanställas i en tabell. Tabellen ska för varje testgrupp visa antalet djur vid testets början, antalet djur som under testets gång har dött eller avlivats av humana skäl, tidpunkten för alla dödsfall eller humana avlivningar, antalet fertila djur, antalet dräktiga honor, antalet djur som uppvisar tecken på toxicitet, en beskrivning av de toxicitetstecken som har observerats (inklusive tidpunkt då dessa först upptäcktes samt verkningarnas varaktighet och allvarlighetsgrad), typerna av histopatologiska förändringar samt all annan relevant data om kullarna. I tillägg 3 finns ett tabellformat som har visat sig vara mycket användbart för att redovisa reproduktiva effekter och utvecklingseffekter.
|
|
51.
|
På grund av denna studies begränsade omfattning har statistiska analyser om ”signifikans” begränsat värde för flera endpoints, särskilt reproduktiva sådana. Om statistiska analyser används ska en lämplig metod för distributionen av den valda variabeln väljas ut innan testet påbörjas. Statistiska analyser av AGD och bröstvårteretention ska baseras på data för varje individuell unge med beaktande av effekter på kullen ifråga. När så är lämpligt ska kullen utgöra analysenheten. Statistiska analyser av ungarnas kroppsvikt ska baseras på data för varje individuell unge med beaktande av kullstorlek. På grund av den ringa gruppstorleken kan det också vara värdefullt att använda historiska kontrolldata (t.ex. för kullstorlek), om sådana finns, som ett stöd för tolkning av studien.
|
Utvärdering av resultaten
|
52.
|
Resultaten bör utvärderas i termer av de observerade effekterna, inklusive obduktion och mikroskopiska iakttagelser. Utvärderingen ska omfatta förhållandet mellan testkemikaliedosen och närvaron eller frånvaron av samt incidensen och allvarlighetsgraden av avvikelser, inbegripet grova skador, identifierade målorgan, infertilitet, kliniska avvikelser, verkningar på reproduktionsförmågan och kullen, avvikelser rörande kroppsvikt, verkningar på mortaliteten samt alla andra toxiska effekter.
|
|
53.
|
Eftersom hanarna endast behandlas under en kort tid bör histopatologin hos testiklar och bitestiklar bedömas tillsammans med fertilitetsdata vid uppskattningen av effekter på reproduktionsförmågan hos hanar. Historiska kontrolldata om reproduktion/utveckling (t.ex. kullstorlek, AGD, bröstvårteretention, T4-nivåer i serum), om sådana finns, kan vara användbara vid tolkningen av studien.
|
|
54.
|
Som kvalitetskontroll föreslås att historiska kontrolldata samlas in och att man för numeriska data beräknar variationskoefficienter, särskilt för de parametrar som kopplas till påvisande av hormonstörande ämnen. Dessa data kan sedan användas för jämförelseändamål när aktuella studier utvärderas.
|
Testrapport
|
55.
|
Testrapporten ska innehålla följande information:
|
|
Testkemikalie:
|
—
|
Källa, satsnummer, sista förbrukningsdag om detta anges.
|
|
—
|
Testkemikaliens stabilitet, om den är känd.
|
|
|
|
Monokomponentämne:
|
—
|
fysikaliskt tillstånd, vattenlöslighet och ytterligare relevanta fysikalisk-kemiska egenskaper.
|
|
—
|
kemisk identifiering, t.ex. med IUPAC- eller CAS-namn, CAS-nummer, Smiles- eller InChI-kod, strukturformel, renhet samt motsvarande identifiering av föroreningar i förekommande fall och om det är praktiskt möjligt.
|
|
|
|
Multikomponentämne, UVCB-ämnen och blandningar:
|
—
|
karakteriseras så långt som möjligt genom beståndsdelarnas kemiska identitet (se ovan), kvantitativa förekomst och relevanta fysikalisk-kemiska egenskaper.
|
|
|
|
Vehikel (i förekommande fall):
|
—
|
Motivering för val av vehikel om annan än vatten.
|
|
|
|
Försöksdjur:
|
—
|
Art och stam som används.
|
|
—
|
Djurens antal, ålder och kön.
|
|
—
|
Ursprung, inhysning, foder osv.
|
|
—
|
Djurens individuella vikt vid testets början.
|
|
—
|
Motivering för val av art, om inte råtta.
|
|
|
|
Testbetingelser:
|
—
|
Grund för valet av dosnivå.
|
|
—
|
Uppgifter om testkemikaliens sammansättning/foderberedning, uppnådd koncentration, preparatets stabilitet och homogenitet.
|
|
—
|
Uppgifter om administreringen av testkemikalien.
|
|
—
|
Omvandling från fodrets/dricksvattnets testkemikaliekoncentration (ppm) till faktisk dos (mg/kg kroppsvikt/dag), i förekommande fall.
|
|
—
|
Uppgifter om foder- och vattenkvalitet.
|
|
—
|
Detaljerad beskrivning av slumpmässighetsförfaranden för att välja ut ungar för avlivning, om avlivning skett.
|
|
|
|
Resultat:
|
—
|
Kroppsvikt/kroppsviktsförändringar.
|
|
—
|
Foderkonsumtion och vattenkonsumtion, i förekommande fall.
|
|
—
|
Uppgifter om toxisk reaktion uppdelade efter kön och dos, inbegripet fertilitet, dräktighet och avkommans livsduglighet.
|
|
—
|
Dräktighetsperiodens längd.
|
|
—
|
Toxiska eller andra verkningar på reproduktion, avkomma, postnatal tillväxt och dylikt.
|
|
—
|
Art, allvarlighetsgrad och varaktighet för kliniska observationer (huruvida de är reversibla eller ej).
|
|
—
|
Antal vuxna honor med normal eller onormal ägglossningscykel samt cykelns varaktighet.
|
|
—
|
Antalet levande födda och postimplantationsförlust.
|
|
—
|
Uppgifter om ungarnas kroppsvikt.
|
|
—
|
AGD på samtliga ungar (och deras kroppsvikt på dagen för mätning av AGD).
|
|
—
|
Bröstvårteretention hos hanungar.
|
|
—
|
Nivåer av tyreoideahormoner hos 13 dagar gamla ungar och vuxna hanar (samt på moderdjur och 4 dagar gamla ungar om detta har uppmätts).
|
|
—
|
Antal ungar med lätt synliga avvikelser, generell bedömning av yttre könsorgan och antal småväxta ungar.
|
|
—
|
Tidpunkt för dödsfall under testet eller huruvida djuren överlevde fram till avslutningen.
|
|
—
|
Antal implantationer, kullstorlek och kullens vikt vid tiden för registrering.
|
|
—
|
Kroppsvikt vid avlivningen samt absolut och relativ organvikt för föräldradjuren.
|
|
—
|
En detaljerad redogörelse av alla histopatologiska fynd.
|
|
—
|
Absorptionsdata (om sådana är kända).
|
|
—
|
Statistisk bearbetning av resultaten, i tillämpliga fall.
|
|
Diskussion av resultaten.
Slutsatser.
|
Tolkning av resultaten
|
56.
|
Denna studie bedömer toxicitetsverkningar på reproduktion/utveckling vid administrering av upprepade doser (se punkterna 5 och 6). Studien kan ge en indikation om behovet av vidare undersökningar och vägledning för hur uppföljande studier bör utformas. OECD:s vägledningsdokument 43 bör konsulteras som stöd vid tolkningen av reproduktions- och utvecklingsresultaten (12). OECD:s vägledningsdokument 106 om histologisk bedömning av endokrina och reproduktiva tester på gnagare (11) ger information om beredning och bedömning av (endokrina) organ och vaginala utstryk som kan vara användbar för denna testmetod.
|
LITTERATURHÄNVISNINGAR
|
(1)
|
OECD (1990). Room Document No 1 for the 14th Joint Meeting of the Chemicals Group and Management Committee. Tillgängligt på förfrågan hos Organisationen för ekonomiskt samarbete och utveckling, Paris.
|
|
(2)
|
OECD (1992). Chairman’s Report of the ad hoc Expert Meeting on Reproductive Toxicity Screening Methods, Tokyo, 27–29 oktober, 1992. Tillgängligt på förfrågan hos Organisationen för ekonomiskt samarbete och utveckling, Paris.
|
|
(3)
|
OECD (1998). Report of the First Meeting of the OECD Endocrine Disrupter Testing and Assessment (EDTA) Task Force, 10th-11th March 1998. Tillgängligt på förfrågan hos Organisationen för ekonomiskt samarbete och utveckling, Paris.
|
|
(4)
|
OECD (2015). Feasibility Study for Minor Enhancements of TG 421/422 with ED Relevant Endpoints. Environmental Health and Safety Publications, Series on Testing and Assessment nr 217, Organisationen för ekonomiskt samarbete och utveckling, Paris.
|
|
(5)
|
OECD (2000). Guidance Document on the Recognition, Assessment, and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluations. Series on Testing and Assessment nr 19, Organisationen för ekonomiskt samarbete och utveckling, Paris.
|
|
(6)
|
OECD (2011). Annex I to Draft Guidance Document on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption. Environmental Health and Safety Publications, Series on Testing and Assessment nr 150, Organisationen för ekonomiskt samarbete och utveckling, Paris.
|
|
(7)
|
Goldman, J.M., Murr A.S., Buckalew A.R., Ferrell J.M. and Cooper R.L. (2007). The Rodent Estrous Cycle: Characterization of Vaginal Cytology and its Utility in Toxicological Studies, Birth Defects Research, Part B, 80 (2), 84-97.
|
|
(8)
|
Sadleir R.M.F.S (1979). Cycles and Seasons, i Auston C.R. and Short R.V. (eds.), Reproduction in Mammals: I. Germ Cells and Fertilization, Cambridge, New York.
|
|
(9)
|
Gallavan R.H. Jr, Holson J.F., Stump D.G., Knapp J.F. and Reynolds V.L. (1999). Interpreting the Toxicologic Significance of Alterations in Anogenital Distance: Potential for Confounding Effects of Progeny Body Weights, Reproductive Toxicology, 13: 383-390.
|
|
(10)
|
OECD (2013). Guidance Document in Support of the Test Guideline on the Extended One Generation Reproductive Toxicity Study. Environmental Health and Safety Publications, Series on Testing and Assessment nr 151, Organisationen för ekonomiskt samarbete och utveckling, Paris.
|
|
(11)
|
OECD (2009). Guidance Document for Histologic Evaluation of Endocrine and Reproductive Tests in Rodents. Environmental Health and Safety Publications, Series on Testing and Assessment nr 106, Organisationen för ekonomiskt samarbete och utveckling, Paris.
|
|
(12)
|
OECD (2008). Guidance Document on Mammalian Reproductive Toxicity Testing and Assessment. Environmental Health and Safety Publications, Series on Testing and Assessment nr 43, Organisationen för ekonomiskt samarbete och utveckling, Paris.
|
Tillägg 1
DEFINITIONER (SE ÄVEN OECD GD 150 (6))
Androgenicitet är förmågan hos en kemikalie att fungera som ett naturligt androgent hormon (t.ex. testosteron) hos ett däggdjur.
Antiandrogenicitet är förmågan hos en kemikalie att undertrycka verkan av ett naturligt androgent hormon (t.ex. testosteron) hos ett däggdjur.
Anti-östrogenicitet är förmågan hos en kemikalie att undertrycka verkan av ett naturligt östrogent hormon (t.ex. 17ß-östradiol) hos ett däggdjur.
Antityreoid aktivitet är förmågan hos en kemikalie att undertrycka verkan av ett naturligt tyreoideahormon (t.ex. T3) hos ett däggdjur.
Kemikalie: ett ämne eller en blandning.
Utvecklingstoxicitet: uttryck för reproduktionstoxicitet i form av pre-, peri- eller postnatala strukturella eller funktionella störningar i avkomman.
Dosering är en allmän term som omfattar dos, dess frekvens samt doseringens varaktighet.
Dos är den mängd testkemikalie som administreras. Dosen uttrycks som testkemikaliens vikt per enhet kroppsvikt av försöksdjur per dag (t.ex. mg/kg kroppsvikt/dag), eller som en konstant kostkoncentration.
Uppenbar toxicitet är en allmän term som beskriver tydliga tecken på toxicitet efter tillförsel av testkemikalien. Dessa bör vara tillräckliga för en riskbedömning och bör vara sådana att en ökning av den tillförda dosen kan förväntas resultera i utvecklingen av allvarliga toxicitetssymptom och sannolikt leda till mortalitet.
Nedsatt fertilitet innebär störningar i fortplantningsförmågan hos hanen eller honan.
Maternell toxicitet: skadliga effekter på dräktiga honor som kan ske antingen specifikt (direkt effekt) eller icke-specifikt (indirekt effekt).
NOAEL är förkortningen för nivån där ingen skadlig effekt observeras. Detta är den högsta dosnivå där inga behandlingsrelaterade fynd observerats på grund av behandling.
Östrogenicitet är förmågan hos en kemikalie att agera som ett naturligt östrogent hormon (t.ex. 17ß-östradiol) hos ett däggdjur.
Reproduktionstoxicitet innebär skadliga effekter på avkomman och/eller en försämring av fortplantningsförmågan hos hanar och honor.
Testkemikalie: varje ämne eller blandning som testas med denna testmetod.
Tyreoid aktivitet är förmågan hos en kemikalie att agera som ett naturligt tyreoideahormon (t.ex. T3) hos ett däggdjur.
Validering är en vetenskaplig process utformad för att karakterisera de operativa kraven och begränsningarna hos en testmetod och visa dess tillförlitlighet och relevans för ett visst ändamål.
Tillägg 2
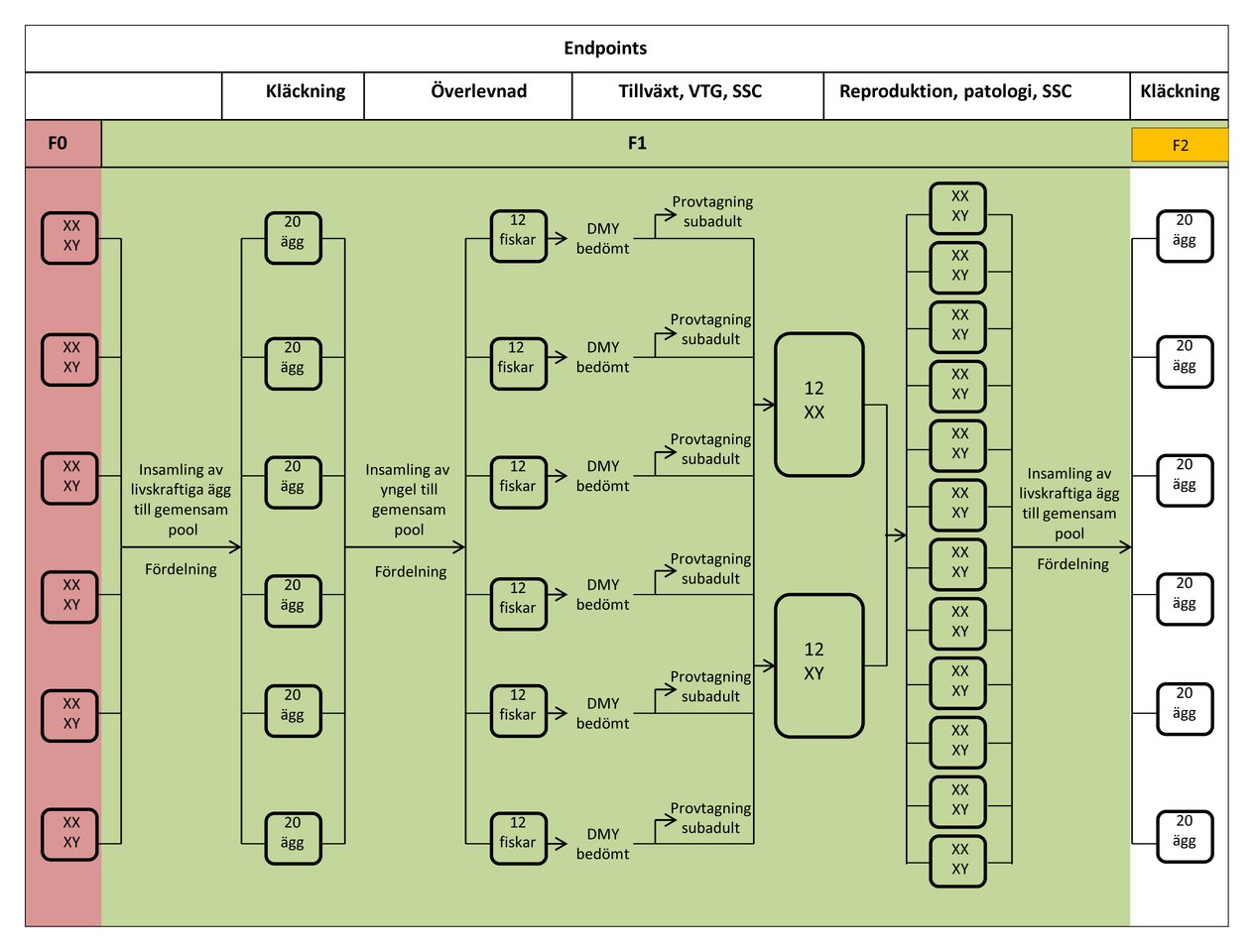
FÖRSÖKSSCHEMA SOM VISAR STUDIENS MAXIMALA VARAKTIGHET, BASERAT PÅ EN 14-DAGARS PARNINGSPERIOD

Tillägg 3
SAMMANFATTNING I TABELLFORM AV EFFEKTER PÅ REPRODUKTION/UTVECKLING
|
IAKTTAGELSER
|
VÄRDEN
|
|
|
|
Dosering (enheter)
|
0 (kontroll)
|
…
|
…
|
…
|
…
|
|
Startade par (N)
|
|
|
|
|
|
|
Ägglossningscykel (för oregelbundna cykler åtminstone medellängd och frekvens)
|
|
|
|
|
|
|
Honor som bevisligen befruktats (N)
|
|
|
|
|
|
|
Honor som blivit dräktiga (N)
|
|
|
|
|
|
|
Befruktningsdag 1–5 (N)
|
|
|
|
|
|
|
Befruktningsdag 6–... (21) (N)
|
|
|
|
|
|
|
Dräktighetsperiod ≤ 21 dagar (N)
|
|
|
|
|
|
|
Dräktighetsperiod = 22 dagar (N)
|
|
|
|
|
|
|
Dräktighetsperiod ≥ 23 dagar (N)
|
|
|
|
|
|
|
Moderdjur med levande ungar (N)
|
|
|
|
|
|
|
Moderdjur med levande ungar på dag 4 post partum (N)
|
|
|
|
|
|
|
Implantationer/moderdjur (medel)
|
|
|
|
|
|
|
Levande ungar/moderdjur vid födseln (medel)
|
|
|
|
|
|
|
Levande ungar/moderdjur på dag 4 (medel)
|
|
|
|
|
|
|
Könsfördelning (m/f) vid födseln (medel)
|
|
|
|
|
|
|
Könsfördelning (m/f) på dag 4 (medel)
|
|
|
|
|
|
|
Kullens vikt vid födseln (medel)
|
|
|
|
|
|
|
Kullens vikt på dag 4 (medel)
|
|
|
|
|
|
|
Ungarnas vikt vid födseln (medel)
|
|
|
|
|
|
|
Ungarnas vikt vid AGD-mätningen (medel – hanungar, medel – honungar)
|
|
|
|
|
|
|
Ungarnas AGD på samma dag efter födseln, från födseln – dag 4 (medel – hanungar, medel – honungar)
|
|
|
|
|
|
|
Ungarnas vikt på dag 4 (medel)
|
|
|
|
|
|
|
Bröstvårteretention hos hanungarna på dag 13 (medel)
|
|
|
|
|
|
|
Ungarnas vikt på dag 13 (medel)
|
|
|
|
|
|
|
|
|
ONORMALA UNGAR
|
|
Moderdjur med 0
|
|
|
|
|
|
|
Moderdjur med 1
|
|
|
|
|
|
|
Moderdjur med ≥ 2
|
|
|
|
|
|
|
|
|
FÖRLORAD AVKOMMA
|
|
|
|
Prenatal/post-implantationer (implantationer minus levande födda)
|
|
Honor med 0
|
|
|
|
|
|
|
Honor med 1
|
|
|
|
|
|
|
Honor med 2
|
|
|
|
|
|
|
Honor med ≥ 3
|
|
|
|
|
|
|
|
|
Postnatal (levande födda minus levande ungar på dag 13)
|
|
Honor med 0
|
|
|
|
|
|
|
Honor med 1
|
|
|
|
|
|
|
Honor med 2
|
|
|
|
|
|
|
Honor med ≥ 3
|
|
|
|
|
|
B.64 TOXICITETSSTUDIE MED UPPREPADE DOSER KOMBINERAD MED SCREENINGTEST FÖR REPRODUKTIONS-/UTVECKLINGSTOXICITET
INLEDNING
|
1.
|
Denna testmetod motsvarar OECD:s testriktlinje (TG) 422 (2016). OECD:s riktlinjer för kemikalietestning utvärderas med jämna mellanrum för att anpassas till den vetenskapliga utvecklingen. De ursprungliga riktlinjerna för screeningtest 422 antogs 1996 utifrån ett protokoll för upprepad dosering kombinerat med reproduktions-/utvecklingstoxicitet som diskuterats vid två expertmöten, i London 1990 (1) och i Tokyo 1992 (2).
|
|
2.
|
I denna testmetod kombineras dels en screeningdel för reproduktions-/utvecklingstoxicitet som bygger på erfarenheter från medlemsländer som använt sig av den ursprungliga metoden på befintliga högvolymkemikalier och som gjort explorativa tester med positiva kontrollämnen (3) (4), dels en del som avser toxicitet vid upprepad dosering, i enlighet med OECD:s testriktlinje 407 (28-dagars toxicitetsstudie med upprepad oral dosering på gnagare, som motsvaras av kapitel B.7 i denna bilaga).
|
|
3.
|
Denna testmetod har uppdaterats med endpoints som är relevanta för hormonstörande ämnen, och detta är en uppföljning till den högprioriterade verksamhet som OECD inledde 1998 för att revidera befintliga testriktlinjer och ta fram nya riktlinjer för screening och testning av potentiella hormonstörande ämnen (5). Inom ramen för denna verksamhet utökades TG 407 (motsvaras av kapitel B.7 i denna bilaga) 2008 med parametrar för att påvisa endokrin aktivitet hos testkemikalier. Syftet med uppdateringen av TG 422 var att inkludera några endpoints som är relevanta för hormonstörande ämnen i riktlinjerna för screeningtester där exponeringsperioderna omfattar några av de känsliga utvecklingsperioderna (pre- eller tidiga postnatalperioder).
|
|
4.
|
De nya utvalda endpoints som är relevanta för hormonstörande ämnen och som också ingår i TG 443 (utökad enkelgenerationsstudie av reproduktionstoxicitet, motsvarande kapitel B.56 i denna bilaga) inkluderades i TG 422 på grundval av en förstudie som behandlade vetenskapliga och tekniska aspekter av deras inkluderande samt nödvändiga anpassningar av testets utformning för att möjliggöra ett inkluderande (6).
|
|
5.
|
Denna testmetod är utformad för att generera begränsad information om en testkemikalies effekter på den hanliga och honliga fortplantningsförmågan, t.ex. beträffande könskörtlarnas funktion, parningsbeteende, befruktning, embryoutveckling och födsel. Den är inte ett alternativ till, eller en ersättning för, de befintliga testmetoderna B.31, B.34, B.35 eller B.56.
|
INLEDANDE ÖVERVÄGANDEN
|
6.
|
Vid bedömningen och utvärderingen av de toxiska egenskaperna hos en testkemikalie kan bestämningen av toxicitet med upprepad oral dosering genomföras efter att inledande information om toxicitet har erhållits genom testning av akut toxicitet. Denna studie ger information om eventuella troliga hälsorisker vid upprepad exponering under en relativt begränsad tidsperiod. Metoden innefattar den grundläggande toxicitetsstudien med upprepad dosering som kan användas för kemikalier för vilka en 90-dagarsstudie inte är motiverad (t.ex. när produktionsvolymen inte överskrider vissa gränser) eller som en inledning till en långtidsstudie. Vid genomförandet av studien bör man alltid följa de vägledande principer och överväganden som beskrivs i OECD:s vägledningsdokument 19 om erkännande, bedömning och användning av kliniska tecken som humana endpoints för försöksdjur som används för säkerhetsbedömningar (7).
|
|
7.
|
Metoden består dessutom av ett screeningtest för reproduktions-/utvecklingstoxicitet och kan därför även användas för att ge preliminär information om möjliga effekter på fortplantningsförmågan hos hanar och honor, t.ex. beträffande könskörtlarnas funktion, parningsbeteende, befruktning, embryoutveckling och förlossning, antingen för en tidig bedömning av testkemikaliers toxikologiska egenskaper eller för bedömning av farliga testkemikalier. Denna testmetod ger ingen komplett information om alla aspekter av reproduktion och utveckling. Metoden ger endast begränsad möjlighet att detektera postnatala uttryck på prenatal exponering, eller effekter som kan ha orsakats av postnatal exponering. Bland annat på grund av selektiviteten hos endpoints samt studiens begränsade varaktighet ger denna metod inga bevis eller definitiva svar om utebliven effekt på reproduktion/utveckling. Vid avsaknad av data från andra toxicitetstest om reproduktion/utveckling kan de positiva resultaten dessutom användas för en inledande riskbedömning och bidra till beslutsfattande gällande vilka övriga test som behövs och när.
|
|
8.
|
Resultaten som erhölls genom de endokrinrelaterade parametrarna bör betraktas mot bakgrund av OECD:s begreppsram för provning och bedömning av hormonstörande ämnen (8). Den vidareutvecklade OECD TG 422 ingår i nivå 4 av begreppsramen som ett in vivo-test som genererar data om skadliga effekter för endokrinrelaterade endpoints. En endokrin signal kanske dock inte räcker för att påvisa att testkemikalien är ett hormonstörande ämne.
|
|
9.
|
Denna metod lägger även vikt vid neurologiska effekter som en särskild endpoint samt behovet av noggranna kliniska observationer av djuren för att erhålla mesta möjliga information. Metoden ska identifiera kemikalier med eventuell neurotoxisk verkan, vilket kan motivera mer ingående undersökningar av den aspekten. Denna metod kan dessutom ge basindikationer på immunologiska effekter.
|
|
10.
|
Med tanke på avsaknaden av data från andra systemtest om reproduktions/utvecklingstoxicitet, neurotoxicitet och/eller immuntoxicitet kan de positiva resultaten användas för en inledande riskbedömning och bidra till beslutsfattande gällande vilka övriga test som behövs och när. Detta test är särskilt användbart som en del av SIDS (OECD Screening Information Data Set) för att bedöma befintliga kemikalier som det finns ringa eller ingen toxikologisk information om, och det kan användas som ett alternativ till genomförande av två separata test: ett för toxicitet vid upprepad dosering (OCD TG 407, motsvaras av kapitel B.7 i denna bilaga) respektive ett för reproduktions-/utvecklingstoxicitet (OECD TG 421, motsvaras av kapitel B.63 i denna bilaga). Det kan även användas som en preliminär undersökning inför mer omfattande studier om reproduktion/utveckling, eller av andra anledningar.
|
|
11.
|
Den allmänna uppfattningen är att det finns skillnader i sensitivitet mellan dräktiga och icke-dräktiga djur. I kombinerade tester kan det därför vara mer komplicerat att fastställa dosnivåer som är korrekta för bedömning av både generell systemtoxicitet och specifik reproduktions-/utvecklingstoxicitet än vad det är i individuella test som utförs separat. Det kan därför vara svårare att tolka testresultaten vad gäller allmän systemtoxicitet än vid genomförande av separata studier med upprepade doser, särskilt när parametrarna för serum och histopatologi inte bedöms samtidigt i studien. På grund av denna tekniska komplexitet krävs det stor erfarenhet av toxicitetstest för att utföra detta kombinerade screeningtest. Å andra sidan kan ett kombinerat test, förutom att det kräver färre försöksdjur, ge bättre möjligheter att skilja direkta effekter på reproduktion/utveckling från effekter som beror på andra (systemiska) effekter.
|
|
12.
|
I det här testet är doseringsperioden längre än i en konventionell 28-dagars studie med upprepade doser. Det kräver dock färre djur av varje kön per grupp jämfört med när en konventionell 28-dagars studie med upprepade doser genomförs som ett komplement till ett screeningtest för reproduktions-/utvecklingstoxicitet.
|
|
13.
|
Vid test enligt denna metod används oral tillförsel av testkemikalien. Om tillförsel sker på annat sätt kan metoden behöva modifieras.
|
|
14.
|
Innan testmetoden används på en blandning i syfte att samla in data för ett lagstadgat ändamål bör man överväga om, och i så fall varför, den kommer att ge godtagbara resultat för det syftet. Sådana överväganden är inte nödvändiga om blandningen måste provas enligt lag.
|
|
15.
|
De definitioner som används finns i tillägg 1.
|
TESTPRINCIP
|
16.
|
Testkemikalien tillförs i graderade doser till flera grupper av hanar och honor. Hanarna ska behandlas i minst fyra veckor, inbegripet dagen före den planerade avlivningen (detta inkluderar minst två veckor före parning, under parningsperioden och ungefär två veckor efter parning). Eftersom hanarna endast behandlas under en begränsad period före parning kan fertilitet inte anses utgöra en särskilt känslig indikator för testikeltoxicitet. Därför krävs en detaljerad histologisk undersökning av testiklarna. En två veckors doseringsperiod före parning i kombination med efterföljande observationer av parning/fertilitet och en sammanlagd doseringsperiod på minst fyra veckor, följt av en ingående histopatologisk analys av hanarnas könskörtlar, anses vara tillräckligt för att påvisa flertalet effekter på hanarnas fertilitet och spermatogenes.
|
|
17.
|
Honorna bör behandlas under hela studien. Detta inbegriper två veckor före parning (i syfte att täcka minst två kompletta ägglossningscykler), tiden fram till befruktning, dräktighetsperioden samt minst tretton dagar efter födsel, fram till dag för planerad avlivning.
|
|
18.
|
Studiens längd, efter acklimatisering och bedömning av ägglossningscykeln före behandling, beror på honornas kondition och ligger på runt 63 dagar (minst 14 dagar före parning, (upp till) 14 dagars parning, 22 dagars dräktighetsperiod samt 13 dagars laktation).
|
|
19.
|
Under administreringsperioden observeras djuren varje dag noggrant för tecken på toxicitet. Djur som dör eller avlivas under testet obduceras, och i slutet av testet avlivas överlevande djur och obduceras.
|
BESKRIVNING AV TESTMETODEN
Val av djurart
|
20.
|
Denna testmetod är avsedd att användas på råtta. Om de parametrar som anges i denna TG 422 undersöks hos en annan gnagarart ska detta motiveras utförligt. I det internationella valideringsprogrammet för att påvisa hormonstörande ämnen i TG 407 var råtta den enda art som användes. Stammar med låg fertilitet eller välkänd hög incidens av utvecklingsdefekter ska inte användas. Friska tidigare obefruktade djur, som inte tidigare har använts i försök, ska användas. Försöksdjuren ska karakteriseras efter art, stam, kön, vikt och ålder. Vid testets början bör variationen i vikt mellan de använda djuren vara minimal och får inte överstiga ± 20 % av vardera könets medelvikt. När en upprepad oral dos utförs som inledning till en långtidsstudie, är det att föredra att djur från samma stam och ursprung används för båda studierna.
|
Inhysning och utfodring
|
21.
|
Alla förfaranden bör överensstämma med lokala normer för laboratoriedjurvård. Temperaturen i försöksdjurens utrymmen bör vara 22 °C (± 3 °). Den relativa fuktigheten bör vara minst 30 % och helst inte överstiga 70 %, utom när rummet rengörs. Belysningen bör vara artificiell med ljusperioden 12 timmar ljus och 12 timmar mörker. För utfodring kan konventionellt laboratoriefoder användas med obegränsad tillgång till dricksvatten. Valet av foder kan påverkas av behovet av att säkerställa en lämplig blandning av en testkemikalie när den administreras genom denna metod.
|
|
22.
|
Djuren bör inhysas i små grupper med individer av samma kön. Djuren kan inhysas individuellt om det är vetenskapligt motiverat. Vid grupplacering i bur bör inte fler än fem djur inhysas i en och samma bur. För parningsprocedurerna används burar som är lämpliga för ändamålet. Dräktiga honor ska hysas individuellt och förses med bomaterial. Digivande honor ska hysas individuellt med sina ungar.
|
|
23.
|
Fodret bör regelbundet analyseras beträffande främmande ämnen. Ett prov av fodret bör sparas tills rapporten är slutförd.
|
Förberedelse av djuren
|
24.
|
Friska yngre adulta djur fördelas slumpmässigt till behandlingsgrupper och burar. Burarna bör arrangeras på ett sådant sätt att eventuella effekter på grund av burarnas placering minimeras. Djuren ska ha en unik identifiering och hållas i sina burar under minst fem dagar innan undersökningen börjar, för att göra det möjligt för dem att acklimatiseras till förhållandena i laboratoriet.
|
Beredning av doser
|
25.
|
Det rekommenderas att testkemikalien administreras oralt såvida inte andra tillförselsätt anses mer lämpliga. Vid oral administrering ges testkemikalien i regel via sond, men testkemikalien kan även ges via fodret eller via dricksvattnet.
|
|
26.
|
Vid behov kan testkemikalien upplösas eller suspenderas i en lämplig vehikel. Det rekommenderas att man, när så är möjligt, i första hand använder en vattenbaserad lösning/suspension, i andra hand en lösning/suspension i olja (t.ex. majsolja) och i tredje hand en lösning i andra vehiklar. För andra vehiklar än vatten bör vehikelns toxiska egenskaper vara kända. Testkemikaliens stabilitet och homogenitet i vehikeln bör bestämmas.
|
FÖRFARANDE
Djurens antal och kön
|
27.
|
Det rekommenderas att varje grupp inledningsvis innehåller minst 10 hanar och 12–13 honor. Honornas ägglossningscykler undersöks före exponeringen, och individer som inte uppvisar typiska cykler på 4–5 dagar ska inte tas med i studien. Det är därför extra honor rekommenderas initialt så att varje grupp innehåller 10 honor. Förutom vid betydande toxiska effekter förväntas detta leda till minst 8 dräktiga honor per grupp, vilket i regel utgör det minsta acceptabla antalet dräktiga honor per grupp. Syftet med detta är att säkerställa ett tillräckligt antal dräktigheter och ungar för att möjliggöra en meningsfull utvärdering av testkemikaliens potential att påverka fertilitet, dräktighet och moderskaps- och dibeteende samt tillväxt och utveckling hos F1-avkomman från befruktning till dag 13 post partum. Om avlivningar är inplanerade under testets gång bör antalet djur ökas med det antal djur som enligt planeringen ska avlivas innan testet avslutas. En extra satellitgrupp på fem djur per kön i kontroll- respektive toppdosgruppen bör övervägas för observation beträffande reversibilitet, persistens eller fördröjda toxiska verkningar under minst 14 dagar efter behandlingen. Djuren i satellitgrupperna ska inte paras och används därför inte för bedömning av reproduktions-/utvecklingstoxicitet.
|
Dosering
|
28.
|
I allmänhet bör minst tre doseringsgrupper och en kontrollgrupp användas. Om det inte finns några lämpliga data tillgängliga kan en preliminär undersökning (djur av samma stam och ursprung) utföras för att underlätta bestämningen av de doser som ska användas. Med undantag av behandlingen med testkemikalien bör djuren i kontrollgruppen hanteras på exakt samma sätt som testgruppen. Om en vehikel används för att tillföra testkemikalien bör kontrollgruppen tillföras vehikeln i den största volym som används.
|
|
29.
|
Dosnivåerna bör väljas med hänsyn till eventuell toxicitetsdata och (toxikologisk) kinetisk data som finns tillgänglig. Hänsyn bör tas till eventuell skillnad i sensitivitet mellan dräktiga och icke-dräktiga djur. Den högsta doseringsnivån bör väljas i syfte att den ska inducera toxiska verkningar, men inte leda till döden eller orsaka uppenbart lidande. Därefter bör en fallande nivå på doserna väljas i syfte att påvisa eventuella dosrelaterade responser och frånvaro av skadliga effekter vid den lägsta dosnivån. Det är ofta optimalt med två till fyra intervall, och tillägg av en fjärde testgrupp är ofta att föredra framför användning av mycket breda intervall (exempelvis mer än faktorn 10) mellan doseringsnivåerna.
|
|
30.
|
Vid observation av tecken på allmän toxicitet (t.ex. minskad kroppsvikt, effekter på lever, hjärta, lungor eller njurar) eller andra förändringar som kanske inte är toxiska reaktioner (t.ex. nedsatt födointag, leverförstoring) ska observerade effekter avseende endokrint känsliga endpoints tolkas med försiktighet.
|
Toleranstest
|
31.
|
Om ett försök med en doseringsnivå om minst 1 000 mg per kg kroppsvikt och dag eller, vid tillförsel i föda eller dricksvatten, en motsvarande andel i födan eller dricksvattnet (baserad på fastställd kroppsvikt) och med användning av det tillvägagångssätt som beskrivits för denna studie, inte ger några observerbara toxiska effekter och om toxicitet inte förväntas på grundval av uppgifter från strukturellt närbesläktade ämnen, kan en fullständig undersökning med flera dosnivåer anses överflödig. Toleranstestet ska tillämpas utom när mänsklig exponering anger att en högre doseringsnivå ska användas. När testkemikalien tillförs på annat sätt, t.ex. via inandning eller applicering på huden, kan dess fysikalisk-kemiska egenskaper ofta användas för att fastställa den maximala exponeringsnivå som går att uppnå.
|
Administrering av doser
|
32.
|
Försöksdjuren behandlas med testkemikalien dagligen under en vecka. När testkemikalien tillförs genom sondmatning bör detta ske i en engångsdos via magsond eller lämplig intubationskanyl. Den maximala vätskevolymen som kan administreras vid ett tillfälle är beroende av försöksdjurets storlek. Volymen får inte överstiga 1 ml per 100 g kroppsvikt, utom för vattenlösningar där volymen kan uppgå till 2 ml per 100 g kroppsvikt. Med undantag för irriterande eller korrosiva testkemikalier, som normalt kommer att visa stegrade verkningar vid högre koncentrationer, bör variationer av testvolymen minimeras genom justering av koncentrationen för att säkerställa en konstant volym vid alla dosnivåer.
|
|
33.
|
För kemikalier som tillförs via fodret eller dricksvattnet är det viktigt att säkerställa att kvantiteterna av testkemikalien ifråga inte påverkar normal närings- eller vattenbalans. När testkemikalien tillförs via kosten kan antingen en konstant foderkoncentration (PPM) eller en konstant doseringsnivå i förhållande till djurens kroppsvikt användas. Det alternativ som används ska anges. En testkemikalie som tillförs genom sondmatning bör ges vid samma tid varje dag och justeras minst varje vecka så att dosnivån hålls konstant i förhållande till djurens kroppsvikt. Om en kombinerad studie används för att förbereda en långtidsstudie eller en fullständig studie avseende reproduktionstoxicitet bör liknande foder användas i båda testerna.
|
Försöksschema
|
34.
|
Behandlingen av båda könen bör påbörjas två veckor före parning, efter att de har acklimatiserats under minst fem dagar och efter att honorna har screenats för normala ägglossningscykler (under en tvåveckorsperiod före behandlingen). Studien bör planläggas så att bedömningen av ägglossningscykeln påbörjas kort efter att djuret ifråga blivit könsmoget. Detta kan variera något mellan olika råttstammar i olika laboratorier, t.ex. 10 veckors ålder för Sprague Dawley-råttor och 12 veckors ålder för Wistar-råttor. Moderdjur med ungar bör avlivas på dag 13 post partum, eller kort därefter. Moderdjur och ungar behöver inte nödvändigtvis avlivas på samma dag, om detta omöjliggör för moderdjuret att fasta dagen före blodprovstagningen (om fastande är att föredra). Dagen för födseln (dvs. efter avslutad förlossning) definieras som dag 0 post partum. Honor som inte uppvisar några tecken på att ha parat sig ska avlivas 24–26 dagar efter parningsperiodens sista dag. Tillförseln fortsätts för båda könen under den två veckor långa parningsperioden. Hanar ska ges fortsatt behandling efter parningsperioden och minst till dess att en minimidoseringsperiod på totalt 28 dagar har uppnåtts. Därefter avlivas de eller hålls kvar och ges fortsatt behandling för en andra parning om så bedöms lämpligt.
|
|
35.
|
Daglig dosering till dräktiga honor och moderdjur ska fortsätta genom hela dräktighetsperioden och minst till och med dag 13 post partum eller dagen före avlivningsdagen. När testkemikalien tillförs via inhalation eller applicering på huden ska doseringen fortsätta minst till och med dräktighetsdag 19, och doseringen bör återupptas så fort som möjligt efter födseln och inte senare än PND 4.
|
|
36.
|
Djur i satellitgrupper för vilka uppföljande observationer planeras, om sådana är inkluderade i testet, ska inte paras. Dessa djur bör hållas i ytterligare minst 14 dagar efter den första planerade avlivningen av moderdjur, utan behandling för att fördröjda eller kvarstående toxiska effekter eller återhämtning från toxiska effekter ska kunna upptäckas.
|
|
37.
|
Ett diagram för försöksschemat finns i tillägg 2.
|
Ägglossningscykler
|
38.
|
Ägglossningscyklerna bör övervakas innan behandlingen startar i syfte att välja ut honor med regelbundna cykler till försöket se punkt 27). Även vaginala utstryk bör övervakas dagligen från behandlingens början till dess att parning bevisligen skett. Om det vid doseringens början föreligger misstankar om akuta stresseffekter som kan störa ägglossningscyklerna kan laboratoriet exponera försöksdjuren under två veckor och därefter samla in vaginala utstryk dagligen i syfte att övervaka ägglossningscyklerna under minst två veckor före parning, med fortsatt övervakning under parningsperioden fram till dess att parning bevisligen skett. När celler från vagina eller livmoderhals samlas in bör försiktighet iakttas för att undvika skador på slemhinnan, eftersom sådana kan framkalla pseudodräktighet (8) (9).
|
Parningsprocedur
|
39.
|
I regel ska 1:1-parningar (en hane per hona) användas i denna studie. Undantagssituationer kan uppstå om hanar dör. Honan ska inhysas med samma hane tills parning bevisligen har skett eller två veckor har förflutit. Honorna ska undersökas varje morgon för närvaron av sperma eller vaginalproppar. Dag 0 av dräktigheten definieras som den dag då bevis på parning bekräftas (fynd av vaginalpropp eller sperma). Om parningen inte lyckas kan man överväga ett nytt parningsförsök där honor sammanförs med hanar med fastställd fertilitet från samma grupp.
|
Kullstorlek
|
40.
|
På dag 4 efter födseln kan storleken på varje kull justeras genom att eliminera extra ungar genom slumpmässigt urval, så att det blir, så nära som möjligt, fyra eller fem ungar per kön per kull, beroende på den normala kullstorleken i den råttstam som använts. Blodprov ska tas från två av överskottsungarna och sedan slås ihop och användas för bestämning av T4-nivåer i serum. Selektiv eliminering av ungar, t.ex. utifrån kroppsvikt eller anogenitalt avstånd (AGD), är inte lämpligt. När antalet han- eller honungar inte medger fyra eller fem av varje kön per kull, är partiell anpassning acceptabelt (t.ex. sex hanar och fyra honor). Inga ungar ska elimineras när kullstorleken är mindre än det avsedda målet (8 eller 10 ungar/kull). Om det endast finns en överskottsunge utöver det avsedda målet ska endast en unge elimineras och användas för blodinsamling och eventuell bedömning av T4 i serum.
|
|
41.
|
Om kullstorleken inte korrigeras ska två ungar per kull avlivas på dag 4 efter födseln och blodprov tas för att mäta koncentrationen av tyreoideahormoner i serum. Om det är möjligt ska dessa två ungar per kull vara honungar, så att hanungar kan reserveras för senare bedömning av bröstvårteretention, förutom då eliminering av dessa honungar innebär att det inte finns några honor kvar för den slutliga bedömningen. Inga ungar ska elimineras när kullstorleken är mindre än 8 eller 10 ungar/kull (beroende på den normala kullstorleken för den råttstam som använts). Om det endast finns en unge mer än normalt antal ungar per kull ska endast en unge elimineras och användas för blodinsamling och eventuell bedömning av T4 i serum.
|
Observationer
|
42.
|
Allmänna kliniska observationer bör göras minst en gång per dag, helst vid samma tidpunkt varje dag med hänsyn till maximalt förväntade effekter efter doseringen. Djurens hälsotillstånd bör registreras. Minst två gånger dagligen bör alla djur observeras beträffande morbiditet och mortalitet.
|
|
43.
|
En gång före den första exponeringen (för att möjliggöra individuella jämförelser), och därefter minst en gång per vecka, ska ingående kliniska observationer göras av samtliga föräldradjur. Dessa observationer bör göras utanför buren på en ”standardarena” och helst vid samma tidpunkt varje gång. Observationerna ska registreras noggrant, helst med hjälp av ett poängsystem som definierats utförligt av testlaboratoriet. Åtgärder bör vidtas för att säkerställa att variationen i testbetingelser är minimal och att observationerna helst utförs av personer som inte känner till behandlingen. Observationerna bör omfatta, men inte begränsas till, förändringar i hud, päls, ögon, slemhinnor, förekomst av sekret och utsöndringar samt autonom aktivitet (t.ex. tårflöde, piloerektion, pupillstorlek, ovanligt andningsmönster). Förändringar i gång, hållning och reaktion på hantering liksom eventuella kramper eller spasmer, stereotypier (t.ex. överdrivet putsande, repetitivt cirklande), svåra eller förlängda förlossningar eller bisarrt beteende (t.ex. självstympning, baklängesgång) ska också registreras (10).
|
|
44.
|
Vid ett tillfälle under studiens gång ska en bedömning göras av djurens sinnesreaktioner på olika sorters stimuli (t.ex. auditiva, visuella och proprioceptiva stimuli) (8) (9) (11), samt av djurens gripstyrka (12) och motorisk aktivitet (13), och för denna bedömning ska fem hanar och fem honor väljas ut slumpmässigt ur varje grupp. Ytterligare uppgifter om de procedurer som kan följas finns i respektive hänvisningar. Dock kan även andra procedurer än de som hänvisas till användas. När det gäller hanarna ska nämnda funktionsobservationer göras mot slutet av doseringsperioden, kort före planerad avlivning men före hematologisk eller klinisk blodprovstagning (se punkterna 53–56 inklusive fotnot 1). Honorna bör befinna sig i ett liknande fysiologiskt tillstånd vid tidpunkten för dessa funktionstester, vilka företrädesvis ska utföras en gång under den sista laktationsveckan (t.ex. LD 6–13), kort före planerad avlivning. Eventuell separation av moderdjur och deras ungar ska minimeras så långt det är möjligt.
|
|
45.
|
Funktionsobservationer som görs en gång mot slutet av studien kan utelämnas om studien utförs som inledning till en subkronisk (90-dagars) eller en långtidsstudie. I så fall bör funktionsobservationerna i stället tas med i nämnda uppföljningsstudie. Å andra sidan kan tillgången på data om funktionsobservationer från studien med upprepad dosering öka möjligheten att välja dosnivåer för en efterföljande subkronisk studie eller långtidsstudie.
|
|
46.
|
I undantagsfall kan funktionsobservationer utlämnas för grupper som annars skulle uppvisa tecken på toxicitet i sådan utsträckning att det väsentligen skulle påverka funktionstestets prestanda.
|
|
47.
|
Dräktighetsperioden bör registreras och beräknas från dräktighetsdag 0. Varje kull ska undersökas snarast möjligt efter nedkomsten i syfte att fastställa antal och kön för ungar, dödfödda, levande födda, småväxta djur (ungar som är avsevärt mindre än motsvarande kontrollungar) samt eventuella större avvikelser.
|
|
48.
|
Levande ungar ska räknas och könsbestämmas, och kullarna ska vägas inom 24 timmar efter födseln (dag 0 eller dag 1 post partum) samt åtminstone på dag 4 och 13 post partum. Utöver observationerna av föräldradjuren (se punkterna 43 och 44) ska allt onormalt beteende hos ungarna registreras.
|
|
49.
|
AGD på varje unge ska mätas på samma dag efter födseln, någon gång mellan PND 0 och PND 4. Ungarnas kroppsvikt bör samlas in under dagen för AGD-mätningen, och AGD bör normaliseras till ett mått på ungarnas storlek, företrädesvis kubikroten ur kroppsvikten (14). Antal bröstvårtor/vårtgårdar på hanungarna ska räknas på PND 12 eller 13 enligt rekommendationerna i OECD GD 151 (15).
|
Kroppsvikt och foder-/vattenkonsumtion
|
50.
|
Hanar och honor ska vägas på behandlingens första dag, minst en gång i veckan därefter samt vid testets slut. Under dräktighetstiden ska honorna vägas på dag 0, 7, 14 och 20 samt inom 24 timmar efter födseln (dag 0 eller 1 post partum) och åtminstone på dag 4 och 13 post partum. Dessa observationer bör rapporteras individuellt för varje vuxet djur.
|
|
51.
|
Under tiden före parning, dräktighetsperiod samt laktation ska foderintaget mätas åtminstone en gång i veckan. Det är inte obligatoriskt att mäta foderintaget under parningsperioden. Om testkemikalien tillförs via dricksvattnet bör även vattenkonsumtionen mätas under nämnda perioder.
|
Hematologi
|
52.
|
Vid ett tillfälle under studiens gång ska följande hematologiska undersökningar göras på fem hanar och fem honor som väljs ut slumpmässigt ur varje grupp: hematokrit, hemoglobinkoncentrationer, räkning av röda blodkroppar, retikulocyter, total- och differentialräkning av vita blodkroppar, blodplättsräkning samt ett mått på koaguleringstid och koaguleringsförmåga. Andra bestämningar som bör utföras, om testkemikalien eller dess förmodade metaboliteter har eller misstänks ha oxiderande egenskaper, inkluderar methemoglobinkoncentration och Heinz-kroppar.
|
|
53.
|
Blodproven ska tas från ett angivet ställe. Honorna bör befinna sig i ett liknande fysiologiskt tillstånd under provtagningen. För att undvika praktiska problem relaterade till skillnad i dräktighetslängd kan blodprovstagningen utföras i slutet av perioden före parning i stället för strax innan, eller i samband med, avlivningen av djuren. Blodproven på hanarna ska helst tas strax innan, eller i samband med, avlivningen av djuren. Blodproven på hanarna kan också tas i slutet av perioden före parning om denna tidpunkt har föredragits för honorna.
|
|
54.
|
Alla blodprov ska förvaras under lämpliga förhållanden.
|
Klinisk biokemi
|
55.
|
En klinisk biokemisk bedömning i syfte att undersöka större toxikologiska effekter på vävnaderna, och i synnerhet på njurar och lever, bör utföras på de blodprover som samlats in från de fem hanar och fem honor som valts ut från varje grupp. Det rekommenderas att djuren fastar natten före blodprovstagningen. (22) Undersökningar av plasma eller serum bör omfatta natrium, kalium, glukos, totalkolesterol, urinämne, kreatinin, totalprotein och albumin, minst två enzymer som indikerar effekter på leverceller (t.ex. alaninaminotransferas, aspartataminotransferas och sorbitoldehydrogenas) samt gallsyror. Mätningar av ytterligare enzymer (från lever eller annat ursprung) och bilirubin kan under vissa omständigheter ge användbar information.
|
|
56.
|
Blodprov tas från ett definierat ställe utifrån följande schema:
|
—
|
från minst två ungar per kull på dag 4 efter födseln, om antalet ungar så tillåter (se punkterna 40 och 41),
|
|
—
|
från alla moderdjur och minst två ungar per kull när försöket avslutas på dag 13, och
|
|
—
|
från alla vuxna hanar, vid försökets slut.
|
Alla blodprov ska förvaras under lämpliga förhållanden. Blodproverna som tas på 13 dagar gamla ungar samt vuxna hanar ska analyseras för tyreoideahormoner (T4) i serum. Ytterligare analys av T4 i blodproven från moderdjuren och de 4 dagar gamla ungarna görs om det bedöms relevant. Om det är relevant kan blodet även analyseras för andra hormoner. Ungarnas blod kan samförvaras per kull för analyser av tyreoideahormoner. Tyreoideahormonerna (T4 och TSH) bör helst mätas som ett totalvärde.
|
|
57.
|
Följande urinanalyser kan utföras på fem slumpmässigt utvalda hanar från varje grupp under studiens sista vecka med hjälp av schemalagd urininsamling: utseende, volym, osmolalitet eller specifik vikt, pH, protein, glukos och blod/blodceller.
|
|
58.
|
Dessutom bör undersökning av serummarkörer av allmänna vävnadsskador övervägas. Andra bestämningar som bör utföras om testkemikaliens kända egenskaper kan, eller misstänks, påverka relaterade metaboliska profiler omfattar kalcium, fosfat, fastevärden av triglycerider och glukos samt specifika hormoner, methemoglobin och kolinesteras. Detta behöver undersökas från fall till fall.
|
|
59.
|
Följande faktorer kan påverka variationen och de absoluta koncentrationerna för hormonbestämningarna:
|
—
|
Tidpunkt för avlivningen beroende på hormonkoncentrationernas dygnsvariation.
|
|
—
|
Avlivningsmetod för att undvika onödig stress hos djuren som kan påverka hormonkoncentrationerna.
|
|
—
|
Testsatser för hormonbestämningar som kan skilja sig åt genom sina standardkurvor.
|
|
|
60.
|
Plasmaprover särskilt avsedda för hormonbestämning bör tas vid en jämförbar tid på dagen. De numeriska värden som erhölls vid analys av hormonkoncentrationer skiljer sig åt mellan de olika analyssatser som finns i handeln.
|
|
61.
|
Om historiska basdata är otillräckliga bör hänsyn tas till hematologiska och kliniska biokemiska variabler innan doseringen påbörjas eller företrädesvis för en uppsättning djur som inte ingår i försöksgrupperna. För honorna ska data samlas in från digivande djur.
|
PATOLOGI
Obduktion
|
62.
|
Alla vuxna djur i undersökningen bör underkastas en fullständig detaljerad obduktion som omfattar en noggrann yttre granskning av kroppen och alla kroppsöppningar liksom av kranium och bröst- och bukhåla samt deras innehåll. Särskild uppmärksamhet bör ägnas åt organen i reproduktionssystemet. Antal implantationsställen ska registreras. Vaginala utstryk ska undersökas på dagen för obduktionen för att fastställa stadium i ägglossningscykeln och möjliggöra korrelation med histopatologin hos honornas fortplantningsorgan.
|
|
63.
|
På samtliga hanar ska all angränsande vävnad putsas bort från testiklar och bitestiklar liksom från prostata och sädesblåsor med koaguleringskörtlar, varefter våtvikten mäts snarast möjligt efter dissektion innan organen hinner torka. Utöver detta kan fler organ vägas, som till exempel muskelkomplexen levator ani och bulbocavernosus, Cowpers körtlar eller ollonet hos hanar samt äggstockspar (våtvikt) och livmoder (inklusive livmoderhals) hos honor. Om dessa organ vägs ska deras vikt registreras snarast möjligt efter dissektion. Äggstockar, testiklar, bitestiklar, accessoriska könsorgan samt alla organ som uppvisar skador vid makroskopisk undersökning ska sparas från samtliga vuxna försöksdjur.
|
|
64.
|
Sköldkörtlar ska sparas från alla vuxna hanar och honor samt från två 13 dagar gamla ungar från varje kull (en hane och en hona) och förvaras i det fixativ som bedöms lämpligast för en senare histopatologisk undersökning. Sköldkörtelns vikt kan bestämmas efter fixering. Putsning ska också göras väldigt försiktigt, och först efter fixeringen för att undvika vävnadsskador. Vävnadsskador kan påverka den histopatologiska analysen negativt. Blodprover bör tas från ett angivet ställe strax före eller i samband med avlivningen av djuren, och förvaras under lämpliga förhållanden (se punkt 56).
|
|
65.
|
För minst fem vuxna hanar och honor som valts ut slumpmässigt från varje grupp (utom sådana som påträffats döende och/eller avlivats före försökets slut) ska all angränsande vävnad putsas bort från lever, njurar, binjurar, bräss, mjälte, hjärna och hjärta, beroende på vad som gäller, och organens våtvikt ska mätas snarast möjligt efter dissektion innan de hinner torka. Följande vävnader och organ bör bevaras i det mest lämpade fixeringsmediet med hänsyn till både typen av vävnad och planerade efterföljande histopatologiska undersökningar: alla större skador, hjärna (representativa områden inklusive cerebrum, cerebellum och pons), ryggmärg, ögon, mage, tunn- och tjocktarm (inklusive Peyers plack), lever, njurar, binjurar, mjälte, hjärta, bräss, luftstrupe och lungor (konserverade genom uppumpning med fixeringsmedel följt av nedsänkning), könskörtlar (testiklar och äggstockar), accessoriska könsorgan (livmoder och livmoderhals, bitestiklar, prostata, sädesblåsor plus koaguleringskörtlar), vagina, urinblåsa, lymfkörtlar (utöver den mest proximala dräneringsnoden bör en annan lymfnod tas i enlighet med laboratoriets erfarenheter (16)), perifer nerv (ischias eller tibia) helst nära muskeln, skelettmuskel och ben med benmärg (sektion eller, alternativt, ett nymonterat benmärgsaspirat). Det rekommenderas att testiklarna fixeras genom nedsänkning i Bouins lösning eller modifierat fixeringsmedel enligt Davidson (16) (17) (18). Fixering med formalin rekommenderas inte för dessa vävnader. Tunica albuginea kan punkteras försiktigt och ytligt med en nål på organets båda poler för en snabb inträngning av fixeringsmedlet. Kliniska och andra fynd kan indikera behovet av att undersöka ytterligare vävnader. Även samtliga organ som sannolikt är målorgan, baserat på kunskap om testkemikaliens egenskaper, ska konserveras.
|
|
66.
|
Följande vävnader kan ge värdefulla tecken på endokrinrelaterade effekter: könskörtlar (äggstockar och testiklar), accessoriska könsorgan (livmoder inklusive livmoderhals, bitestiklar, sädesblåsor med koaguleringskörtlar, dorsolaterala och ventrala prostata), vagina, hypofys, hanliga bröstkörtlar och binjurar). Förändringar i hanliga bröstkörtlar är inte tillräckligt väl dokumenterade, men denna parameter kan vara väldigt känslig för ämnen med östrogen verkan. Observation av organ/vävnader som inte nämns i punkt 65 är valfria.
|
|
67.
|
Döda ungar och ungar som avlivas på dag 13 post partum, eller kort därefter, ska undersökas noga åtminstone externt för grova avvikelser. Särskild uppmärksamhet bör fästas vid de yttre reproduktionsorganen, som ska undersökas för tecken på avvikande utveckling.
|
Histopatologi
|
68.
|
En fullständig histopatologisk analys bör utföras på bevarade organ och vävnader från de utvalda djuren i kontroll- och högdosgrupperna (med särskild tonvikt på spermatogenesens stadier i de hanliga könskörtlarna och histopatologin hos den interstitiala cellstrukturen i testiklarna). Sköldkörtlar från ungar och resterande vuxna djur kan undersökas vid behov. Dessa undersökningar bör utsträckas till djur i alla andra doseringsgrupper, om behandlingsrelaterade förändringar observeras i högdosgruppen. Vägledningen om histopatologi (10) omfattar detaljerad information om dissektion, fixering, sektionering och histopatologi av endokrina vävnader.
|
|
69.
|
Alla stora skador bör undersökas. Målorgan från övriga dosgrupper bör undersökas som ett led i NOAEL-klarläggningsprocessen, och särskilt organ från grupper som påstås uppvisa NOAEL.
|
|
70.
|
Om en satellitgrupp används bör en histopatologisk undersökning utföras på de vävnader och organ på vilka effekter har visats i de behandlade grupperna.
|
DATA OCH RAPPORTERING
Data
|
71.
|
Data bör rapporteras individuellt per djur. Därutöver bör alla data sammanfattas i tabellform, som för varje testgrupp visar antal djur vid testets början, antal djur som hittats döda under testet eller som avlivats av humanitära skäl och tidpunkten för dödsfallen eller avlivningen, antal fertila djur, antal dräktiga honor, antal djur som visar tecken på toxicitet, en beskrivning av de tecken på toxicitet som observerats, inklusive tidpunkt för de första tecknen och deras varaktighet och allvarlighetsgrad, typ av histopatologiska förändringar, samt alla relevanta data om kullarna. I tillägg 3 finns ett tabellformat som har visat sig vara mycket användbart för bedömning av reproduktions- och utvecklingseffekter.
|
|
72.
|
Om möjligt bör numeriska resultat utvärderas genom en lämplig och allmänt vedertagen statistisk metod. Vid jämförelser av effekten längs ett dosintervall bör man undvika användningen av multipla t-tester. Statistiska metoder ska väljas vid utformningen av studien. Statistiska analyser av AGD och bröstvårteretention ska baseras på data för varje individuell unge med beaktande av effekter på kullen ifråga. När så är lämpligt ska kullen utgöra analysenheten. Statistiska analyser av ungarnas kroppsvikt ska baseras på data för varje individuell unge med beaktande av kullstorlek. På grund av denna studies begränsade omfattning har statistiska analyser om ”signifikans” begränsat värde för flera endpoints, särskilt reproduktiva sådana. Några av de vanligaste metoderna, särskilt parametriska tester för mätning av centrala tendenser, är olämpliga. Om statistiska analyser används ska en lämplig metod för distributionen av den valda variabeln väljas ut innan testet påbörjas.
|
Utvärdering av resultaten
|
73.
|
Resultaten bör utvärderas i termer av de observerade effekterna, inklusive obduktion och mikroskopiska iakttagelser. Utvärderingen ska omfatta förhållandet mellan testkemikaliedosen och närvaron eller frånvaron av samt incidensen och allvarlighetsgraden av avvikelser, inbegripet grova skador, identifierade målorgan, infertilitet, kliniska avvikelser, verkningar på reproduktionsförmågan och kullen, avvikelser rörande kroppsvikt, verkningar på mortaliteten samt alla andra toxiska effekter.
|
|
74.
|
Eftersom hanarna endast behandlas under en kort tid bör histopatologin hos testiklar och bitestiklar bedömas tillsammans med fertilitetsdata vid uppskattningen av effekter på hanarnas fortplantningsförmåga. Historiska kontrolldata om reproduktion/utveckling (t.ex. kullstorlek, AGD, bröstvårteretention, T4-nivåer i serum), om sådana finns, kan också vara användbara vid tolkningen av studien.
|
|
75.
|
Som kvalitetskontroll föreslås att historiska kontrolldata samlas in och att man för numeriska data beräknar variationskoefficienter, särskilt för de parametrar som kopplas till påvisande av hormonstörande ämnen. Dessa data kan sedan användas för jämförelseändamål när aktuella studier utvärderas.
|
Testrapport
|
76.
|
Testrapporten ska innehålla följande information:
|
|
Testkemikalie:
|
—
|
Källa, satsnummer, sista förbrukningsdag om detta anges.
|
|
—
|
Testkemikaliens stabilitet, om den är känd.
|
|
|
|
Monokomponentämne:
|
—
|
fysikaliskt tillstånd, vattenlöslighet och ytterligare relevanta fysikalisk-kemiska egenskaper.
|
|
—
|
kemisk identifiering, t.ex. med IUPAC- eller CAS-namn, CAS-nummer, Smiles- eller InChI-kod, strukturformel, renhet samt motsvarande identifiering av föroreningar i förekommande fall och om det är praktiskt möjligt.
|
|
|
|
Multikomponentämne, UVCB-ämnen och blandningar:
|
—
|
karakteriseras så långt som möjligt genom beståndsdelarnas kemiska identitet (se ovan), kvantitativa förekomst och relevanta fysikalisk-kemiska egenskaper
|
. |
|
|
Vehikel (i förekommande fall):
|
—
|
Motivering för val av vehikel, om annan än vatten.
|
|
|
|
Försöksdjur:
|
—
|
Art och stam som används.
|
|
—
|
Djurens antal, ålder och kön.
|
|
—
|
Ursprung, inhysning, foder osv.
|
|
—
|
Djurens individuella vikt vid testets början.
|
|
—
|
Motivering för val av art, om inte råtta.
|
|
|
|
Testbetingelser:
|
—
|
Grund för valet av dosnivå.
|
|
—
|
Uppgifter om testkemikaliens sammansättning/foderberedning, uppnådd koncentration, beredningens stabilitet och homogenitet.
|
|
—
|
Uppgifter om administreringen av testkemikalien.
|
|
—
|
Omvandling från fodrets/dricksvattnets testkemikaliekoncentration (ppm) till faktisk dos (mg/kg kroppsvikt/dag), i förekommande fall.
|
|
—
|
Uppgifter om foder- och vattenkvalitet.
|
|
—
|
Detaljerad beskrivning av slumpmässighetsförfaranden för att välja ut ungar för avlivning, om avlivning skett.
|
|
|
|
Resultat:
|
—
|
Kroppsvikt/kroppsviktsförändringar.
|
|
—
|
Foder- och vattenkonsumtion, i förekommande fall.
|
|
—
|
Uppgifter om toxiska reaktioner uppdelade efter kön och dos, inbegripet fertilitet, dräktighet och andra tecken på
|
|
—
|
Dräktighetsperiodens längd. Toxiska eller andra verkningar på reproduktion, avkomma, postnatal tillväxt etc.
|
|
—
|
Art, allvarlighetsgrad och varaktighet för kliniska observationer (vare sig de är reversibla eller ej).
|
|
—
|
Bedömningar av sensorisk aktivitet, gripstyrka och motorisk aktivitet.
|
|
—
|
Hematologiska tester med relevanta basvärden.
|
|
—
|
Kliniska biokemiska tester med relevanta basvärden.
|
|
—
|
Antal vuxna honor med normal eller onormal ägglossningscykel samt cykelns varaktighet.
|
|
—
|
Antalet levande födda och postimplantationsförlust.
|
|
—
|
Antal ungar med grova synliga avvikelser. En generell bedömning av yttre könsorgan, antalet småväxta ungar.
|
|
—
|
Tidpunkt för dödsfall under testet eller huruvida djuren överlevde fram till avslutningen.
|
|
—
|
Antal implantationer, kullstorlek och kullens vikt vid tiden för registrering.
|
|
—
|
Uppgifter om ungarnas kroppsvikt.
|
|
—
|
AGD på samtliga ungar (och deras kroppsvikt på dagen för mätning av AGD).
|
|
—
|
Bröstvårteretention hos hanungar.
|
|
—
|
Nivåer av tyreoideahormoner hos 13 dagar gamla ungar och vuxna hanar (samt på moderdjur och 4 dagar gamla ungar om?>detta har uppmätts).
|
|
—
|
Kroppsvikt vid avlivningen samt absolut och relativ organvikt för föräldradjuren.
|
|
—
|
En detaljerad redogörelse för alla histopatologiska fynd.
|
|
—
|
Absorptionsdata (om sådana är kända).
|
|
—
|
Statistisk bearbetning av resultaten, i tillämpliga fall.
|
|
|
|
Diskussion av resultaten.
|
|
Tolkning av resultaten
|
77.
|
Denna studie bedömer reproduktions-/utvecklingstoxicitet vid administrering av upprepade doser. Eftersom vikten läggs på endpoints för både allmän toxicitet och reproduktions-/utvecklingstoxicitet, kommer resultaten från studien att göra det möjligt att skilja mellan sådana reproduktions-/utvecklingseffekter som förekommer i frånvaron av allmän toxicitet och sådana som bara uttrycks på nivåer som också är giftiga för föräldradjuret (se punkterna 7–11). Studien kan ge en indikation om behovet av vidare undersökningar och vägledning för hur uppföljande studier bör utformas. OECD:s vägledningsdokument 43 bör konsulteras som stöd vid tolkningen av reproduktions- och utvecklingsresultaten (19). OECD:s vägledningsdokument 106 om histologisk bedömning av endokrina och reproduktiva tester på gnagare (16) ger information om beredning och bedömning av (endokrina) organ och vaginala utstryk som kan vara användbar för denna testmetod.
|
LITTERATURHÄNVISNINGAR
|
(1)
|
OECD (1990). Room Document No 1 for the 14th Joint Meeting of the Chemicals Group and Management Committee. Tillgängligt på förfrågan hos Organisationen för ekonomiskt samarbete och utveckling, Paris
|
|
(2)
|
OECD (1992). Chairman’s Report of the ad hoc Expert Meeting on Reproductive Toxicity Screening Methods, Tokyo, 27–29 oktober, 1992. Tillgängligt på förfrågan hos Organisationen för ekonomiskt samarbete och utveckling, Paris
|
|
(3)
|
Mitsumori K., Kodama Y., Uchida O., Takada K., Saito M. Naito K., Tanaka S., Kurokawa Y., Usami, M., Kawashima K., Yasuhara K., Toyoda K., Onodera H., Furukawa F., Takahashi M. and Hayashi Y. (1994). Confirmation Study, Using Nitro-Benzene, of the Combined Repeat Dose and Reproductive/ Developmental Toxicity Test Protocol Proposed by the Organization for Economic Cooperation and Development (OECD). J. Toxicol Sci., 19, s. 141–149.
|
|
(4)
|
Tanaka S., Kawashima K., Naito K., Usami M., Nakadate M., Imaida K., Takahashi M., Hayashi Y., Kurokawa Y. och Tobe M. (1992). Combined Repeat Dose and Reproductive/Developmental Toxicity Screening Test (OECD): Familiarization Using Cyclophosphamide. Fundam. Appl. Toxicol., kap. 18, s. 89–95.
|
|
(5)
|
OECD (1998). Report of the First Meeting of the OECD Endocrine Disrupter Testing and Assessment (EDTA) Task Force, den 10–11 mars 1998, tillgänglig på förfrågan hos Organisationen för ekonomiskt samarbete och utveckling, Paris.
|
|
(6)
|
OECD (2015). Feasibility Study for Minor Enhancements of TG 421/422 with ED Relevant Endpoints. Environmental Health and Safety Publications, Series on Testing and Assessment nr 217, Organisationen för ekonomiskt samarbete och utveckling, Paris.
|
|
(7)
|
OECD (2000). Guidance Document on the Recognition, Assessment, and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluations, Environment, Health and Safety Publications, Series on Testing and Assessment, (No 19), Organisationen för ekonomiskt samarbete och utveckling, Paris.
|
|
(8)
|
Goldman J.M., Murr A.S., Buckalew A.R., Ferrell J.M. och Cooper R.L. (2007). The Rodent Estrous Cycle: Characterization of Vaginal Cytology and its Utility in Toxicological Studies, Birth Defects Research, Part B, 80 (2), 84-97.
|
|
(9)
|
Sadleir, R.M.F.S. (1979). Cycles and Seasons, i Auston C.R. and Short R.V. (Eds.), Reproduction in Mammals: I. Germ Cells and Fertilization, Cambridge, New York.
|
|
(10)
|
IPCS (1986). Principles and Methods for the Assessment of Neurotoxicity Associated with Exposure to Chemicals. Environmental Health Criteria Document nr 60.
|
|
(11)
|
Moser V.C., McDaniel K.M. and Phillips P.M. (1991). Rat Strain and Stock Comparisons Using a Functional Observational Battery: Baseline Values and Effects of Amitraz. Toxicol. Appl. Pharmacol., 108, s. 267–283.
|
|
(12)
|
Meyer O.A., Tilson H.A., Byrd W.C. och Riley M.T. (1979). A Method for the Routine Assessment of Fore- and Hindlimb Grip Strength of Rats and Mice. Neurobehav. Toxicol., 1, s. 233–236.
|
|
(13)
|
Crofton, K.M., Howard, J.L., Moser, V.C., Gill, M.W., Reiter, L.W., Tilson, H.A., MacPhail, R.C. (1991). Interlaboratory Comparison of Motor Activity Experiments: Implication for Neurotoxicological Assessments. Neurotoxicol. Teratol. 13, 599-609.
|
|
(14)
|
Gallavan R.H. Jr, J.F. Holson, D.G. Stump, J.F. Knapp och V.L. Reynolds. (1999). ”Interpreting the Toxicologic Significance of Alterations in Anogenital Distance: Potential for Confounding Effects of Progeny Body Weights”, Reproductive Toxicology, 13: 383-390.
|
|
(15)
|
OECD (2013). Guidance Document in Support of the Test Guideline on the Extended One Generation Reproductive Toxicity Study. Environment, Health and Safety publications, Series on Testing and Assessment nr 151. Organisationen för ekonomiskt samarbete och utveckling, Paris.
|
|
(16)
|
OECD (2009). Guidance Document for Histologic Evaluation of Endocrine and Reproductive Tests in Rodents. Environment, Health and Safety Publications, Series on Testing and Assessment, nr 106, Organisationen för ekonomiskt samarbete och utveckling, Paris.
|
|
(17)
|
Hess RA och Moore BJ. (1993). Histological Methods for the Evaluation of the Testis. I: Methods in Reproductive Toxicology, Chapin RE and Heindel JJ (eds). Academic Press. San Diego, CA, s. 52–85.
|
|
(18)
|
Latendresse JR, Warbrittion AR, Jonassen H, Creasy DM. (2002). Fixation of Testes and Eyes Using a Modified Davidson’s Fluid: Comparison with Bouin’s Fluid and Conventional Davidson’s fluid. Toxicol. Pathol. 30, s. 524-533.
|
|
(19)
|
OECD (2008). Guidance Document on Mammalian Reproductive Toxicity Testing and Assessment. Environmental Health and Safety Publications, Series on Testing and Assessment nr 43, Organisationen för ekonomiskt samarbete och utveckling, Paris.
|
|
(20)
|
OECD (2011), Guidance Document on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption, nr 150, Organisationen för ekonomiskt samarbete och utveckling, Paris.
|
Tillägg 1
DEFINITIONER (SE ÄVEN (20) OECD GD 150)
Androgenicitet är förmågan hos en kemikalie att fungera som ett naturligt androgent hormon (t.ex. testosteron) hos ett däggdjur.
Antiandrogenicitet är förmågan hos en kemikalie att undertrycka verkan av ett naturligt androgent hormon (t.ex. testosteron) hos ett däggdjur.
Anti-östrogenicitet är förmågan hos en kemikalie att undertrycka verkan av ett naturligt östrogent hormon (t.ex. 17ß-östradiol) hos ett däggdjur.
Antityreoid aktivitet är förmågan hos en kemikalie att undertrycka verkan av ett naturligt tyreoideahormon (t.ex. T3) hos ett däggdjur.
Kemikalie ett ämne eller en blandning.
Utvecklingstoxicitet uttryck för reproduktionstoxicitet i form av pre-, peri- eller postnatala strukturella eller funktionella störningar i avkomman.
Dos är den mängd testkemikalie som administreras. Dosen uttrycks som testkemikaliens vikt per enhet kroppsvikt av försöksdjuret per dag (t.ex. mg/kg kroppsvikt/dag), eller som en konstant kostkoncentration.
Dosering är en allmän term som omfattar dosen, dess frekvens samt doseringens varaktighet.
Uppenbar toxicitet är en allmän term som beskriver tydliga tecken på toxicitet efter tillförsel av testkemikalien. Dessa bör vara tillräckliga för en riskbedömning och bör vara sådana att en ökning av den tillförda dosen kan förväntas resultera i utvecklingen av allvarliga toxicitetssymptom och sannolikt leda till mortalitet.
Nedsatt fertilitet innebär störningar i fortplantningsförmågan hos hanen eller honan.
Maternell toxicitet skadliga effekter på dräktiga honor som kan ske antingen specifikt (direkt effekt) eller icke-specifikt (indirekt effekt) och som är relaterade till det dräktiga tillståndet.
NOAEL är förkortningen för den nivå där ingen skadlig effekt observeras. Detta är den högsta dosnivå där inga behandlingsrelaterade fynd observerats på grund av behandling.
Östrogenicitet är förmågan hos en kemikalie att agera som ett naturligt östrogent hormon (t.ex. 17ß-östradiol) hos ett däggdjur.
Reproduktionstoxicitet innebär skadliga effekter på avkomman och/eller en försämring av fortplantningsförmågan hos hanar och honor.
Testkemikalie varje ämne eller blandning som testas med denna testmetod.
Tyreoid aktivitet är förmågan hos en kemikalie att agera som ett naturligt tyreoideahormon (t.ex. T3) hos ett däggdjur.
Validering är en vetenskaplig process utformad för att karakterisera de operativa kraven och begränsningarna hos en testmetod och visa dess tillförlitlighet och relevans för ett visst ändamål.
Tillägg 2
FÖRSÖKSSCHEMA SOM VISAR STUDIENS MAXIMALA VARAKTIGHET, BASERAT PÅ EN 14-DAGARS PARNINGSPERIOD
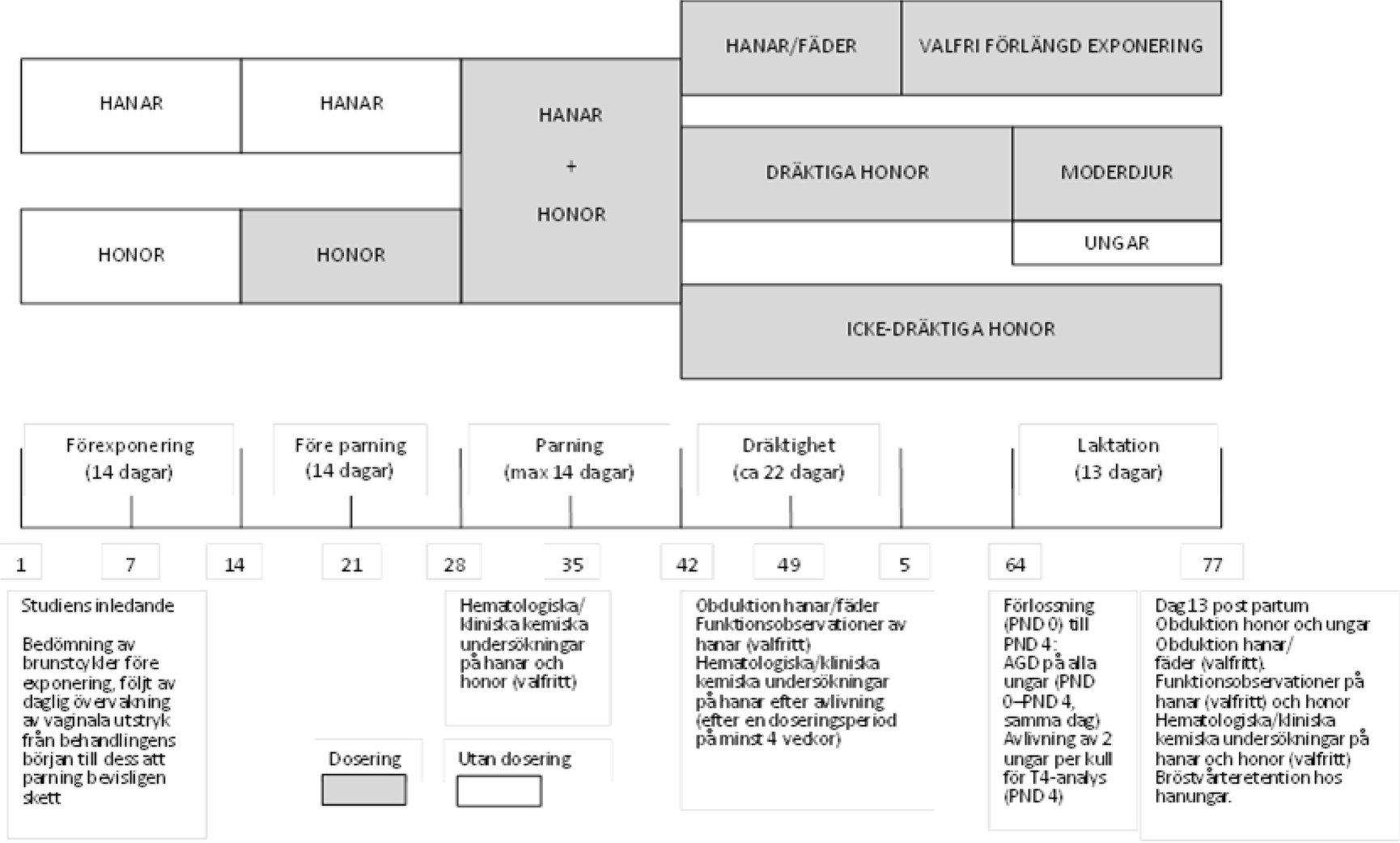

Tillägg 3
SAMMANFATTNING I TABELLFORM AV EFFEKTER PÅ REPRODUKTION/UTVECKLING
|
IAKTTAGELSER
|
VÄRDEN
|
|
Dosering (enheter).......
|
0 (kontroll)
|
…
|
…
|
…
|
…
|
|
Startade par (N)
|
|
|
|
|
|
|
Ägglossningscykel (för oregelbundna cykler åtminstone medellängd och frekvens)
|
|
|
|
|
|
|
Honor som bevisligen befruktats (N)
|
|
|
|
|
|
|
Honor som blivit dräktiga (N)
|
|
|
|
|
|
|
Befruktningsdag 1–5 (N)
|
|
|
|
|
|
|
Befruktningsdag 6–... (23) (N)
|
|
|
|
|
|
|
Dräktighetsperiod ≤ 21 dagar (N)
|
|
|
|
|
|
|
Dräktighetsperiod = 22 dagar (N)
|
|
|
|
|
|
|
Dräktighetsperiod ≥ 23 dagar (N)
|
|
|
|
|
|
|
Moderdjur med levande ungar (N)
|
|
|
|
|
|
|
Moderdjur med levande ungar på dag 4 post partum (N)
|
|
|
|
|
|
|
Implantationer/moderdjur (medel)
|
|
|
|
|
|
|
Levande ungar/moderdjur vid födseln (medel)
|
|
|
|
|
|
|
Levande ungar/moderdjur på dag 4 (medel)
|
|
|
|
|
|
|
Könsfördelning (m/f) vid födseln (medel)
|
|
|
|
|
|
|
Könsfördelning (m/f) på dag 4 (medel)
|
|
|
|
|
|
|
Kullens vikt vid födseln (medel)
|
|
|
|
|
|
|
Kullens vikt på dag 4 (medel)
|
|
|
|
|
|
|
Ungarnas vikt vid födseln (medel)
|
|
|
|
|
|
|
Ungarnas vikt vid tidpunkten för AGD-mätningen (medel – hanungar, medel – honungar)
|
|
|
|
|
|
|
Ungarnas AGD på samma dag efter födseln, från födseln – dag 4 (medel – hanungar, medel – honungar, notera PND)
|
|
|
|
|
|
|
Ungarnas vikt på dag 4 (medel)
|
|
|
|
|
|
|
Ungarnas vikt på dag 13 (medel)
|
|
|
|
|
|
|
Bröstvårteretention hos hanungarna på dag 13 (medel)
|
|
|
|
|
|
|
ONORMALA UNGAR
|
|
Moderdjur med 0
|
|
|
|
|
|
|
Moderdjur med 1
|
|
|
|
|
|
|
Moderdjur med ≥ 2
|
|
|
|
|
|
|
FÖRLORAD AVKOMMA
|
|
Prenatal (implantationer minus levande födda)
|
|
Honor med 0
|
|
|
|
|
|
|
Honor med 1
|
|
|
|
|
|
|
Honor med 2
|
|
|
|
|
|
|
Honor med ≥ 3
|
|
|
|
|
|
|
Postnatal (levande födda minus levande ungar på PND 13)
|
|
Honor med 0
|
|
|
|
|
|
|
Honor med 1
|
|
|
|
|
|
|
Honor med 2
|
|
|
|
|
|
|
Honor med ≥ 3
|
|
|
|
|
|
B.65
IN VITRO-MEMBRANBARRIÄRTEST FÖR HUDKORROSION
INLEDNING
|
1.
|
Denna testmetod motsvarar OECD:s testriktlinje (TG) 435 (2015). Hudkorrosion innebär att det uppstår en irreversibel skada på huden i form av synlig nekros genom epidermis och in i dermis, efter applicering av en testkemikalie enligt definitionen i Förenta nationernas globalt harmoniserade system för klassificering och märkning av kemikalier (GHS) (1) och Europaparlamentets och rådets förordning (EG) nr 1272/2008 om klassificering, märkning och förpackning av ämnen och blandningar (CLP-förordningen) (24). Denna testmetod, som motsvarar OECD:s uppdaterade testriktlinje 435, innefattar ett in vitro-membranbarriärtest som kan användas för att identifiera frätande kemikalier. I testmetoden används ett konstgjort membran som är utformat för att ge en liknande respons på frätande kemikalier som djurhud in situ.
|
|
2.
|
Hudkorrosivitet har traditionellt bedömts genom applicering av testkemikalien på levande djurs hud och bedömning av omfattningen av vävnadsskador efter en fastställd tidsperiod (2). Förutom denna testmetod har ett antal andra in vitro-testmetoder godkänts som alternativ (3)(4) till standardförfarandet för in vivo-test på kaninhud (kapitel B.4 i denna bilaga, motsvarar OECD TG 404) för att identifiera frätande kemikalier (2). Enligt GHS-systemets strategi för stegvis testning och utvärdering för bedömning och klassificering av hudfrätande verkan och OECD:s vägledningsdokument om ett integrerat synsätt på testning och bedömning av hudirritation/hudkorrosion (IATA) (Guidance Document on Integrated Approaches to Testing and Assessment (IATA) for Skin Irritation/Corrosion) rekommenderas användning av validerade och vedertagna in vitro-testmetoder för modulerna 3 och 4 (1)(5). I vägledningsdokumentet om IATA beskrivs flera moduler som grupperar olika informationskällor och analysverktyg, och där ges också i) vägledning om hur tillgängliga testdata och icke testdata kan integreras och användas i bedömningen av kemikaliers potential för hudirritation och hudkorrosion, och ii) förslag på hur man ska gå till väga när det krävs ytterligare testning, inbegripet när negativa resultat erhålls (5). I denna modulmetod kan positiva resultat från in vitro-testmetoder användas för att klassificera en kemikalie som frätande utan behov av djurförsök, och därigenom begränsa och förfina användningen av djur och minska den smärta och ångest som kan uppstå om djur används för detta ändamål.
|
|
3.
|
Valideringsstudier har slutförts för in vitro-membranbarriärmodellen, som finns kommersiellt tillgänglig som Corrositex® (6)(7)(8). De visar en övergripande noggrannhet för att förutsäga hudkorrosivitet på 79 % (128/163), en sensitivitet på 85 % (76/89) och en specificitet på 70 % (52/74) för en databas med 163 ämnen och blandningar (7). Baserat på den bekräftade validiteten har denna validerade referensmetod (VRM) rekommenderats för användning som ett led i en stegvis teststrategi för att bedöma kemikaliers potential att orsaka hudkorrosion (5)(7). Innan en in vitro-membranbarriärmodell för hudkorrosion kan användas för lagstadgade ändamål bör metodens tillförlitlighet, relevans (noggrannhet) och begränsningar för det föreslagna användningsområdet bestämmas för att garantera dess likhet med VRM (9) i enlighet med de på förhand fastställda prestandastandarderna (PS) (10). OECD:s ömsesidiga erkännande av data kan endast garanteras efter det att eventuella föreslagna metoder, nya eller uppdaterade, som följer PS har granskats och inkluderats i motsvarande OECD-testriktlinje. För närvarande omfattas endast en in vitro-metod av OECD:s testriktlinje 435 och denna testmetod, nämligen den kommersiellt tillgängliga modellen Corrositex®.
|
|
4.
|
Andra testmetoder för hudkorrosivitet baseras på användning av rekonstruerad human hud (OECD TG 431) (3) och isolerad råtthud (OECD TG 430) (4). Enligt denna testriktlinje delas frätande kemikalier även in i underkategorier, dvs. de tre GHS-underkategorierna för frätande verkan och FN:s tre transportförpackningsgrupper för faror i samband med frätande ämnen. Denna testriktlinje antogs första gången 2006 och uppdaterades 2015 för att överensstämma med vägledningsdokumentet om IATA och uppdatera förteckningen över kvalifikationsämnen.
|
DEFINITIONER
|
5.
|
Definitioner av begreppen finns i tillägget.
|
INLEDANDE ÖVERVÄGANDEN OCH BEGRÄNSNINGAR
|
6.
|
Det test som beskrivs i denna testmetod gör det möjligt att identifiera frätande testkemikalier och dela in dem i underkategorier enligt GHS/CLP (tabell 1). Testmetoden kan även användas för att fatta beslut om huruvida specifika kemikalieklasser är frätande eller ej, t.ex. organiska och oorganiska syror, syraderivat (25) och baser för vissa transporttestningsändamål (7) (11) (12). Denna testmetod beskriver ett generiskt förfarande som liknar den validerade referenstestmetoden (7). Eftersom denna testmetod inte ger adekvat information om hudirritation bör nämnas att TM B.46 (motsvarande OECD TG 439) är särskilt avsedd för hälsoeffekten hudirritation in vitro (13). För en fullständig bedömning av lokala effekter på huden efter en enstaka dermal exponering se OECD:s vägledningsdokument om IATA (5).
Tabell 1
GHS-kategori och underkategorier för frätande verkan på huden (26)
|
Kategori för frätande verkan (kategori 1) (för myndigheter som inte använder underkategorier)
|
Underkategorier för potentiell frätande verkan (26) (för myndigheter som använder underkategorier, inklusive CLP-förordningen)
|
Frätande på ≥ 1 av 3 djur
|
|
Exponering
|
Anmärkning
|
|
Frätande
|
Frätande, underkategori 1A
|
≤ 3 minuter
|
≤ 1 timme
|
|
Frätande, underkategori 1B
|
> 3 minuter/≤ 1 timme
|
≤ 14 dagar
|
|
Frätande, underkategori 1C
|
> 1 timme/≤ 4 timmar
|
≤ 14 dagar
|
|
|
7.
|
En begränsning med den validerade referensmetoden (7) är att många icke frätande kemikalier och vissa frätande kemikalier kanske inte kommer i fråga för testning baserat på det inledande kompatibilitetstestet (se punkt 13). Vattenbaserade kemikalier med pH-värde i intervallet 4,5–8,5 kommer ofta inte i fråga för testning, men 85 % av de kemikalier som testades inom detta pH-intervall var icke frätande i djurförsök (7). In vitro-membranbarriärmetoden kan användas för att testa fasta ämnen (lösliga eller olösliga i vatten), vätskor (vattenbaserade eller icke vattenbaserade) samt emulsioner. Testkemikalier som inte orsakar en detekterbar förändring i kompatibilitetstestet (dvs. färgändring i det kemiska detektionssystemet för den validerade referenstestmetoden) kan inte testas med membranbarriärmetoden och bör därför testas med andra testmetoder.
|
TESTPRINCIP
|
8.
|
Testsystemet består av två delar: en syntetisk makromolekylär biobarriär och ett kemiskt detektionssystem. I denna testmetod påvisas via det kemiska detektionssystemet membranbarriärskada som orsakas av testkemikalier med frätande verkan efter applicering av testkemikalien på den syntetiska makromolekylära membranbarriärens yta (7), och som kan antas bero på samma frätande mekanism(er) som på levande hud.
|
|
9.
|
Penetrationen av membranbarriären (eller genombrottet) kan mätas med hjälp av ett antal förfaranden eller kemiska detektionssystem, bland annat förändringar av färgen på ett pH-indikatorfärgämne eller andra egenskaper hos indikatorlösningen under barriären.
|
|
10.
|
Det måste fastställas att membranbarriären är giltig, dvs. relevant och tillförlitlig, för den avsedda användningen. Detta kan till exempel göras genom att kontrollera att olika beredningar har likvärdiga barriäregenskaper, t.ex. att de kan upprätthålla en barriär mot icke frätande kemikalier och att kemikaliernas frätande egenskaper kan kategoriseras inom GHS-underkategorierna för frätande verkan (1). Klassificeringen beror på hur lång tid det tar för kemikalien att tränga igenom membranbarriären till indikatorlösningen.
|
DEMONSTRATION AV KOMPETENS
|
11.
|
Före rutinanvändning av in vitro-membranbarriärmetoden bör laboratoriet visa den tekniska kvaliteten genom att korrekt klassificera de tolv kvalifikationsämnen som rekommenderas i tabell 2. När ett förtecknat ämne inte finns tillgängligt eller när så är motiverat kan ett annat ämne för vilket det finns adekvata in vivo- och in vitro-referensdata för (t.ex. från listan över referenskemikalier (10)) användas, förutsatt att samma urvalskriterier tillämpas som i tabell 1.
Tabell 2
Kvalifikationsämnen
(27)
|
Ämne (28)
|
CAS-nr
|
Kemisk klass
|
GHS-underkategori in vivo (29)
|
GHS-underkategori in vitro (29)
|
|
Bortrifluoriddihydrat
|
13319-75-0
|
Oorganiska syror
|
1A
|
1A
|
|
Salpetersyra
|
7697-37-2
|
Oorganiska syror
|
1A
|
1A
|
|
Fosforpentaklorid
|
10026-13-8
|
Prekursorer för oorganiska syror
|
1A
|
1A
|
|
Valerylklorid
|
638-29-9
|
Syraklorider
|
1B
|
1B
|
|
Natriumhydroxid
|
1310-73-2
|
Oorganiska baser
|
1B
|
1B
|
|
1-2-aminoetylpiperazin
|
140-31-8
|
Alifatiska aminer
|
1B
|
1B
|
|
Bensensulfonylklorid
|
98-09-9
|
Syraklorider
|
1C
|
1C
|
|
N,N-dimetylbensylamin
|
103-83-3
|
Aniliner
|
1C
|
1C
|
|
Tetraetylenpentamin
|
112-57-2
|
Alifatiska aminer
|
1C
|
1C
|
|
Eugenol
|
97-53-0
|
Fenoler
|
NC
|
NC
|
|
Nonylakrylat
|
2664-55-3
|
Akrylater/metakrylater
|
NC
|
NC
|
|
Natriumvätekarbonat
|
144-55-8
|
Oorganiska salter
|
NC
|
NC
|
|
FÖRFARANDE
|
12.
|
I följande stycken beskrivs komponenterna i och förfarandena för en testmetod med en konstgjord membranbarriär för bedömning av frätande verkan (7)(15), baserad på den nuvarande validerade referensmetoden, dvs. den kommersiellt tillgängliga Corrositex®. Membranbarriären och kompatibilitets-/indikator- och kategoriseringslösningarna kan konstrueras, beredas eller erhållas kommersiellt, såsom den validerade referensmetoden Corrositex®. Det finns ett exempel på testmetodprotokoll för den validerade referenstestmetoden (7). Provningen bör utföras i rumstemperatur (17–25 °C) och komponenterna bör uppfylla följande villkor:
|
Kompatibilitetstest för testkemikalien
|
13.
|
Innan membranbarriärtestet påbörjas görs ett kompatibilitetstest för att fastställa om det kemiska detektionssystemet kan detektera testkemikalien. Om detektionssystemet inte detekterar testkemikalien är membranbarriärtestmetoden inte lämplig för att utvärdera den aktuella testkemikaliens potentiella frätande verkan, och en annan testmetod bör väljas. Detektionssystemet och de exponeringsförhållanden som används för kompatibilitetstestet bör avspegla exponeringen i membranbarriärtestet.
|
Provning av testkemikaliens tidskategori
|
14.
|
En testkemikalie som är lämplig för testmetoden enligt kompatibilitetstestet bör även genomgå ett tidskategoritest, dvs. ett screeningtest för att skilja mellan svaga och starka syror eller baser. I den validerade referenstestmetoden används till exempel ett tidskategoritest för att avgöra vilken av två tidsskalor som bör användas beroende på om betydande syra- eller basreserver detekteras. Två olika tidsskalor för genombrott för att fastställa frätande verkan och GHS-underkategori för frätande verkan bör användas, baserat på testkemikaliens syra- eller basreserv.
|
MEMBRANBARRIÄRTESTMETODENS KOMPONENTER
Membranbarriär
|
15.
|
Membranbarriären består av två komponenter: en proteinhaltig makromolekylär vattenbaserad gel och ett permeabelt stödjande membran. Den proteinhaltiga gelen bör vara impermeabel för vatten och fasta ämnen, men kan korrodera och bli permeabel. Den färdiga membranbarriären bör förvaras under på förhand fastställda förhållanden som har visat sig förebygga försämringar av gelen, t.ex. att den torkar, att det uppstår mikrobtillväxt eller att den förändras eller spricker, vilket försämrar prestandan. En godtagbar förvaringsperiod bör fastställas, och membranbarriärberedningarna bör inte användas efter den perioden.
|
|
16.
|
Det permeabla stödjande membranet ger mekaniskt stöd till de proteinhaltiga gelen under gelbildningsprocessen och exponeringen för testkemikalien. Det stödjande membranet bör förebygga att gelen sjunker eller skiftar och bör ha hög permeabilitet för alla testkemikalier.
|
|
17.
|
Den proteinhaltiga gelen, som består av protein, t.ex. keratin, kollagen eller blandningar av proteiner som bildar en gelmatris, fungerar som målet för testkemikalien. Det proteinhaltiga materialet placeras på det stödjande membranets yta och får bilda en gel innan membranbarriären placeras ovanpå indikatorlösningen. Den proteinhaltiga gelen bör ha en homogen tjocklek och densitet, utan luftbubblor eller defekter som kan påverka dess funktionella integritet.
|
Kemiskt detektionssystem
|
18.
|
Indikatorlösningen, som är samma lösning som används för kompatibilitetstestet, bör reagera på förekomst av testkemikalien. Ett pH-indikatorfärgämne eller en kombination av sådana färgämnen, t.ex. kresolrött och metylorange som skiftar färg vid förekomst av testkemikalien bör användas. Mätsystemet kan vara visuellt eller elektroniskt.
|
|
19.
|
Relevansen och tillförlitligheten hos detektionssystem som har utformats för att detektera testkemikaliens väg genom barriärmembranet bör bedömas för att fastställa vilka kemikalier som kan detekteras och de kvantitativa detektionsgränserna.
|
TESTPRESTANDA
Montering av testmetodens komponenter
|
20.
|
Membranbarriären placeras i en vial (eller ett provrör) som innehåller indikatorlösningen så att det stödjande membranet har fullständig kontakt med indikatorlösningen och luftbubblor undviks. Det är viktigt att se till att barriärens integritet bibehålls.
|
Applicering av testkemikalien
|
21.
|
En lämplig mängd av testkemikalien, t.ex. 500 μl av en vätska eller 500 mg av ett fast ämne i finfördelad pulverform (7) varvas försiktigt i lager på membranbarriärens övre yta och fördelas jämnt. Ett lämpligt antal replikat, t.ex. fyra (7) bereds för varje testkemikalie och motsvarande kontroller (se punkterna 23–25). Den tid det tar att applicera testkemikalien på membranbarriären registreras. För att se till att korta frätningsperioder registreras korrekt appliceras testkemikalien i replikatbehållarna i omgångar.
|
Mätning av penetration av membranbarriären
|
22.
|
Varje behållare övervakas och tidpunkten för den första förändringen i indikatorlösningen registreras, dvs. när barriären penetreras. Därefter mäts tiden mellan applicering och penetration av membranbarriären.
|
Kontroller
|
23.
|
I tester där en vehikel eller ett lösningsmedel används tillsammans med testkemikalien bör de vara kompatibla med membranbarriärsystemet, dvs. inte ändra dess integritet och testkemikaliens frätande verkan. I tillämpliga fall bör lösningsmedelskontrollen (vehikelkontrollen) testas parallellt med testkemikalien för att visa att lösningsmedlet (vehikeln) är kompatibelt med membranbarriärsystemet.
|
|
24.
|
En positiv (frätande) kontroll med medelhög frätande aktivitet (t.ex. 110 ± 15 mg natriumhydroxid (GHS-kategori för frätande verkan 1B) (7), bör testas parallellt med testkemikalien för att bedöma om testsystemet fungerar på ett godtagbart sätt. En andra positiv kontroll i samma kemiska klass som testkemikalien kan vara användbar för en bedömning av den frätande kemikaliens relativa frätande potential. De valda positiva kontrollerna bör ha en medelhög frätande verkan (t.ex. GHS-underkategori 1B) för att det ska vara möjligt att upptäcka förändringar av penetrationstiden som kan vara oacceptabelt längre eller kortare än det fastställda referensvärdet, eftersom det tyder på att testsystemet inte fungerar ordentligt. För detta ändamål är extremt frätande (GHS-underkategori 1A) eller icke frätande kemikalier av begränsad nytta. En frätande kemikalie i GHS-underkategori 1B gör det möjligt att detektera för snabba eller för långsamma genombrottstider. En svagt frätande kemikalie (GHS-underkategori 1C) kan används som positiv kontroll för att mäta testmetodens förmåga att kontinuerligt skilja mellan svagt frätande och icke frätande kemikalier. Oavsett tillvägagångssätt bör ett godtagbart responsintervall för den positiva kontrollen utvecklas, baserat på det historiska intervallet av genombrottstider för de positiva kontroller som används, t.ex. medelvärdet ± 2–3 standardavvikelser. I varje studie bör den exakta genombrottstiden fastställas för den positiva kontrollen så att avvikelser utanför det godtagbara intervallet kan upptäckas.
|
|
25.
|
En negativ (icke frätande) kontroll, t.ex. 10 % citronsyra, 6 % propionsyra (7), bör också testas parallellt med testkemikalien som en ytterligare kvalitetskontroll för att visa membranbarriärens funktionella integritet.
|
Acceptanskriterier för studien
|
26.
|
Enligt de fastställda tidsparametrarna för varje underkategori för frätande verkan i GHS-systemet används tiden (i minuter) från det att en testkemikalie appliceras på membranbarriären fram till dess att barriären penetreras för att uppskatta testkemikaliens frätande verkan. För att en studie ska anses vara godtagbar bör den parallella positiva kontrollen visa den förväntade responstiden för penetration (t.ex. 8–16 minuters genombrottstid för natriumhydroxid om det används som positiv kontroll). Den parallella negativa kontrollen bör inte visa frätande verkan och den parallella lösningsmedelskontrollen bör i förekommande fall varken visa frätande verkan eller ändra testkemikaliens frätande potential. Innan en metod som följer denna testmetod börjar användas rutinmässigt bör laboratorierna visa att de har teknisk kompetens genom att använda de tolv ämnen som rekommenderas i tabell 2. För nya ”me too”-metoder som utvecklas enligt denna testmetod och som strukturellt och funktionellt sett liknar den validerade referensmetoden (14) bör de på förhand fastställda prestandastandarderna användas för att visa den nya metodens tillförlitlighet och noggrannhet innan den används för lagstadgad testning (10).
|
Tolkning av resultat och korrosivitetsklassificering av testkemikalier
|
27.
|
Tiden (i minuter) från det att testkemikalien appliceras på membranbarriären fram till dess att barriären penetreras används för att klassificera testkemikalien enligt GHS-underkategorierna för frätande kemikalier (1), och i tillämpliga fall FN:s förpackningsgrupper (16). Bryttidsvärdena för var och en av de tre underkategorierna för frätande verkan fastställs för varje föreslagen testmetod. De slutliga besluten om bryttider bör fattas med hänsyn till behovet av att minimera underklassificering av riskerna för frätande verkan (dvs. falska negativa värden). I denna testriktlinje bör bryttiderna för Corrositex® enligt tabell 3 användas, eftersom Corrositex® är den enda testmetod som för närvarande omfattas av testriktlinjen (7).
Tabell 3
Prediktionsmodellen Corrositex®
|
Genomsnittlig bryttid (i minuter)
|
GHS-prediktion (32)
|
|
Testkemikalier i kategori 1 (30) (fastställda med hjälp av metodens kategoriseringstest)
|
Testkemikalier i kategori 2 (31) (fastställda med hjälp av metodens kategoriseringstest)
|
|
0–3 min.
|
0–3 min.
|
Frätande valfri underkategori 1A
|
|
> 3 till 60 min.
|
> 3 till 30 min.
|
Frätande valfri underkategori 1B
|
|
> 60 till 240 min.
|
> 30 till 60 min.
|
Frätande valfri underkategori 1C
|
|
> 240 min.
|
> 60 min.
|
Icke frätande
|
|
DATA OCH RAPPORTERING
Data
|
28.
|
Tiden (i minuter) mellan applicering och barriärpenetration för testkemikalien och positiv(a) kontroll(er) bör rapporteras i tabellform som individuella replikatdata och som medelvärden ± standardavvikelse för varje test.
|
Testrapport
|
29.
|
Testrapporten ska innehålla följande information:
|
|
Testkemikalie och kontrollämnen:
|
—
|
Monokomponentämne: kemisk identifiering, t.ex. med IUPAC- eller CAS-namn, CAS-nummer, Smiles- eller InChI-kod, strukturformel, renhet samt motsvarande identifiering av föroreningar i förekommande fall och om det är praktiskt möjligt.
|
|
—
|
Multikomponentämne, UVCB-ämne och blandning: karakteriseras så långt som möjligt genom beståndsdelarnas kemiska identitet (se ovan), kvantitativa förekomst och relevanta fysikalisk-kemiska egenskaper.
|
|
—
|
Fysikaliskt tillstånd, vattenlöslighet och ytterligare relevanta fysikalisk-kemiska egenskaper.
|
|
—
|
Ursprung, satsnummer om sådant finns.
|
|
—
|
Eventuell behandling av testkemikalien/kontrollämnet före testning (t.ex. uppvärmning, malning).
|
|
—
|
Testkemikaliens stabilitet, sista förbrukningsdatum eller sista datum för upprepad analys, om känt.
|
|
|
|
Vehikel:
|
—
|
Identifieringsdata, koncentration (om tillämpligt), volym som har använts.
|
|
—
|
Motivering för val av vehikel.
|
|
|
|
Använd in vitro-barriärmodell och använt protokoll, med visad noggrannhet och tillförlitlighet.
|
|
|
Testbetingelser:
|
—
|
Beskrivning av använda apparater och rutiner för förberedelse.
|
|
—
|
Källa och sammansättning för den använda in vitro-membranbarriären.
|
|
—
|
Indikatorlösningens sammansättning och egenskaper.
|
|
—
|
Mängder av testkemikalien och kontrollämnena.
|
|
—
|
Beskrivning och motivering av tidskategoriseringstestet.
|
|
—
|
Beskrivning av de utvärderings- och klassificeringskriterier som tillämpats.
|
|
—
|
Demonstrationen av kompetens för utförande av testmetoden ska göras före rutinanvändning, genom testning av kvalifikationsämnen.
|
|
|
|
Resultat:
|
—
|
Tabelluppställning av individuella rådata från individuella test- och kontrollurval för varje replikat.
|
|
—
|
Beskrivning av övriga observerade effekter.
|
|
—
|
Härledda klassificeringar med hänvisning till prediktionsmodell och/eller beslutskriterier.
|
|
Diskussion av resultaten
Slutsatser
|
LITTERATUR
|
(1)
|
United Nations (UN) (2013). Globally Harmonized System of Classification and Labelling of Chemicals (GHS), First Revised Edition, UN New York and Geneva, 2013. Se http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html.
|
|
(2)
|
Kapitel B.4 i denna bilaga, Akut hudirritation/hudkorrosion.
|
|
(3)
|
Kapitel B.40a i denna bilaga, Hudkorrosion in vitro: Testmetod med rekonstruerad human epidermis (RhE).
|
|
(4)
|
Kapitel B.40 i denna bilaga, Hudkorrosion in vitro: Transkutant elektriskt motstånd (TER).
|
|
(5)
|
OECD (2015). Guidance Document on Integrated Approaches to Testing and Assessment of Skin Irritation/Corrosion. Environment, Health and Safety Publications, Series on Testing and Assessment, (No 203). Organisationen för ekonomiskt samarbete och utveckling (OECD), Paris.
|
|
(6)
|
Fentem, J.H., Archer, G.E.B., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Esdaile, D.J., Holzhutter, H.-G. and Liebsch, M. (1998). The ECVAM International Validation Study on In Vitro Tests for Skin Corrosivity. 2. Results and evaluation by the Management Team. Toxicology In Vitro 12, 483–524.
|
|
(7)
|
ICCVAM (1999). Corrositex®. An In Vitro Test Method for Assessing Dermal Corrosivity Potential of Chemicals. The Results of an Independent Peer Review Evaluation Coordinated by ICCVAM, NTP and NICEATM. NIEHS, NIH Publication (nr 99–4495.)
|
|
(8)
|
Gordon V.C., Harvell J.D. och Maibach H.I. (1994). Dermal Corrosion, the Corrositex® System: A DOT Accepted Method to Predict Corrosivity Potential of Test Materials. In vitro Skin Toxicology-Irritation, Phototoxicity, Sensitization. Alternative Methods in Toxicology 10, 37–45.
|
|
(9)
|
OECD (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environmental Health and Safety Publications. Series on Testing and Assessment, nr 34.
|
|
(10)
|
OECD (2014). Performance Standards for the Assessment of Proposed Similar or Modified In Vitro Membrane Barrier Test Method for Skin Corrosion in Relation to TG 435. Organisationen för ekonomiskt samarbete och utveckling (OECD), Paris. Se http://www.oecd.org/chemicalsafety/testing/PerfStand-TG430-June14.pdf.
|
|
(11)
|
ECVAM (2001). Statement on the Application of the CORROSITEX® Assay for Skin Corrosivity Testing. 15th Meeting of ECVAM Scientific Advisory Committee (ESAC), Ispra, Italy. ATLA 29, 96–97.
|
|
(12)
|
U.S. DOT (2002). Exemption DOT-E-10904 (femte översynen). (20 september 2002). Washington, D.C., U.S. DOT.
|
|
(13)
|
Kapitel B.46 i denna bilaga, Hudirritation in vitro: Testmetod med rekonstruerad human epidermis. ICCVAM (2004). ICCVAM Recommended Performance Standards for In Vitro Test Methods for Skin Corrosion. NIEHS, NIH-publikation nr 04-4510. Se http://www.ntp.niehs.nih.gov/iccvam/docs/dermal_docs/ps/ps044510.pdf.
|
|
(14)
|
U.S. EPA (1996). Method 1120, Dermal Corrosion. Se http://www.epa.gov/osw/hazard/testmethods/sw846/pdfs/1120.pdf.
|
|
(15)
|
United Nations (UN) (2013). UN Recommendations on the Transport of Dangerous Goods, Model Regulations, 18th Revised Edition (Part, Chapter 2.8), UN, 2013. Se http://www.unece.org/fileadmin/DAM/trans/danger/publi/unrec/rev18/English/Rev18_Volume1_Part2.pdf.
|
Tillägg
DEFINITIONER
Noggrannhet
: Ett mått på hur väl testmetodresultaten och de godtagna referensvärdena överensstämmer med varandra. Exakthet är ett mått på testmetodens prestanda och en relevansaspekt. Detta begrepp används ofta omväxlande med ”konkordans” för att beskriva andelen korrekta resultat med en testmetod (9).
Kemikalie
: Ett ämne eller en blandning.
Kemiskt detektionssystem
: Ett visuellt eller elektroniskt mätsystem med en indikatorlösning som reagerar på en testkemikalie, t.ex. genom en ändring i ett pH-indikatorfärgämne eller en kombination av sådana färgämnen, som skiftar färg vid förekomst av testkemikalien, eller genom andra typer av kemiska eller elektrokemiska reaktioner.
Konkordans
: ett mått på testmetodens prestanda för sådana testmetoder som ger kategoriska resultat, och en aspekt av relevans. Termen används omväxlande med noggrannhet och definieras som andelen av alla testade kemikalier som klassificeras korrekt som positiva eller negativa (9). Konkordansen är direkt beroende av förekomsten av positiva värden i den testkemikalie som undersöks (9).
GHS (Globally Harmonized System of Classification and Labelling of Chemicals)
: ett system för att klassificera ämnen och blandningar enligt standardiserade typer och nivåer av fysiska risker, hälsorisker och miljörisker och informera om resultaten genom t.ex. piktogram, signalord, faroangivelser, skyddsangivelser och säkerhetsdatablad i syfte att informera om ämnenas skadliga effekter för att skydda personer (inklusive arbetsgivare, arbetstagare, transportörer, konsumenter och personal inom räddningstjänsten) och miljön (1).
IATA
: integrerat synsätt på testning och bedömning (Integrated Approach to Testing and Assessment).
Blandning
: blandning eller lösning som består av två eller flera ämnen.
Monokomponentämne
: Ett ämne, definierat av sin kvantitativa sammansättning, där en huvudsaklig beståndsdel utgör minst 80 viktprocent.
Multikomponentämne
: Ett ämne, definierat av sin kvantitativa sammansättning, där fler än en huvudsaklig beståndsdel förekommer i en koncentration på ≥ 10 viktprocent och < 80 viktprocent. Ett multikomponentämne är resultatet av en tillverkningsprocess. Skillnaden mellan blandningar och multikomponentämnen är att en blandning erhålls genom att två eller flera ämnen blandas utan någon kemisk reaktion. Ett multikomponentämne är resultatet av en kemisk reaktion.
NC
: Icke frätande.
Prestandastandarder
: standarder som baseras på en validerad testmetod och som utgör grunden för utvärdering av jämförbarheten för en föreslagen testmetod som är mekaniskt och funktionellt likartad. Dessa standarder omfattar i) testmetodens grundläggande komponenter, ii) en minimiförteckning med referenskemikalier som valts ut från de kemikalier som använts för att visa att den validerade metoden fungerar tillfredsställande, och iii) de jämförbara nivåer av tillförlitlighet och noggrannhet, på grundval av vad som erhållits för den validerade testmetoden, som man bör kunna påvisa för den föreslagna testmetoden när den utvärderas med användning av minimiförteckningen med referenskemikalier (9).
Relevans
: beskrivning av förhållandet mellan testet och den berörda effekten och huruvida detta förhållande är meningsfullt och användbart för ett visst ändamål. Relevansen är ett mått på hur väl man genom testet korrekt kan mäta eller förutse den biologiska effekt det gäller. Relevans omfattar också överväganden om en testmetods noggrannhet (konkordans) (9).
Tillförlitlighet
: Mått på i vilken utsträckning en testmetod kan reproduceras inom och mellan laboratorier över tid, när den utförs med användning av samma protokoll. Den bedöms genom att man beräknar reproducerbarheten inom och mellan laboratorier (9).
Sensitivitet
: den andel av alla positiva/aktiva testkemikalier som klassificeras korrekt i testet. Det är ett mått på noggrannhet för en testmetod som ger kategoriska resultat och är en viktig faktor vid bedömningen av relevansen för en testmetod (9).
Hudkorrosion in vivo
: en irreversibel skada på huden, i form av synlig nekros genom epidermis och in i dermis, efter applicering av en testkemikalie i upp till fyra timmar. Korrosiva reaktioner karakteriseras av sår, blödning, blodiga sårskorpor och, vid slutet av observationsperioden på 14 dagar, avfärgning genom blekning av huden, hela områden med alopeci (håravfall) och ärr. Histopatologi bör övervägas för att bedöma tveksamma skador.
Specificitet
: den andel av alla negativa/inaktiva testkemikalier som klassificeras korrekt i testet. Det är ett mått på noggrannhet för en testmetod som ger kategoriska resultat och är en viktig faktor vid bedömningen av relevansen för en testmetod (9).
Ämne
: Ett kemiskt grundämne och föreningar av detta grundämne i naturlig eller tillverkad form, inklusive eventuella tillsatser som är nödvändiga för att bevara ämnets stabilitet och sådana föroreningar som härrör från tillverkningsprocessen, men exklusive eventuella lösningsmedel som kan avskiljas utan att det påverkar ämnets stabilitet eller ändrar dess sammansättning.
Testkemikalie
: Alla ämnen eller blandningar som testas med denna testmetod.
UVCB-ämne
: ämne med okänd eller varierande sammansättning, komplexa reaktionsprodukter och biologiskt material.
B.66
IN VITRO-TESTER MED STABILT TRANSFEKTERAD TRANSAKTIVERING FÖR DETEKTION AV ÖSTROGENRECEPTORERS AGONISTER OCH ANTAGONISTER
ALLMÄN INLEDNING
OECD:s prestandabaserade testriktlinje
|
1.
|
Denna testmetod motsvarar OECD:s testriktlinje (TG) 455 (2016). TG 455 är en prestandabaserad testriktlinje (PBTG) som beskriver metoden för in vitro-tester med stabilt transfekterad transaktivering för detektion av östrogenreceptorers agonister och antagonister (ER TA-tester). Den består av flera mekanistiskt och funktionellt liknande testmetoder för att identifiera östrogenreceptorers (dvs. ERα och/eller ERβ) agonister och antagonister, och syftar till att underlätta nya liknande eller modifierade testmetoder enligt de valideringsprinciper som anges i OECD:s vägledningsdokument om validering och internationellt godtagande av nya eller uppdaterade testmetoder för farobedömning (OECD Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment) (1). De fullständigt validerade referenstestmetoder (tilläggen 2 och 3) som ligger till grund för denna prestandabaserade riktlinje är
|
—
|
STTA-testet (stabilt transfekterad transaktivering) (2), som använder cellinjen (h) ERα-HeLa-9903, och
|
|
—
|
VM7Luc ER TA-testet (3), som använder cellinjen VM7Luc4E2 (33) som huvudsakligen uttrycker hERα med visst bidrag från hERββ(4)(5).
|
Prestandastandarder (PS) (6) (7) finns tillgängliga och bör användas vid utveckling och validering av liknande tester för samma effekter. De möjliggör snabba ändringar av PBTG 455, så att nya liknande tester kan läggas till i en uppdaterad PBTG. Liknande tester kommer dock att läggas till först efter det att de har granskats och OECD har bekräftat att prestandastandarderna är uppfyllda. De tester som ingår i TG 455 kan användas obehindrat för att uppfylla medlemsländernas krav på testresultat om transaktivering av östrogenreceptorer och omfattas av OECD:s rådslag om ömsesidigt erkännande av data.
|
Bakgrund och principer för de tester som ingår i denna testmetod
|
2.
|
OECD inledde en högprioriterad verksamhet under 1998 för att se över befintliga, och ta fram nya, testriktlinjer för screening och testning av potentiellt hormonstörande kemikalier. OECD:s begreppsram för provning och bedömning av potentiellt hormonstörande kemikalier reviderades 2012. Det ursprungliga och det reviderade ramverket är bifogade som bilagor till OECD:s vägledningsdokument om standardiserade testriktlinjer för utvärdering av kemikaliers hormonstörande egenskaper (8). Ramen har fem nivåer, vilka motsvarar olika nivåer av biologisk komplexitet. De ER-transaktiveringstester som beskrivs i denna testmetod tillhör nivå 2, som omfattar in vitro-tester som ger data om utvalda endokrinmekanismer/vägar. Denna testmetod består av in vitro-transaktiveringstester (TA) som är utformade för att identifiera östrogenreceptorantagonister och -agonister.
|
|
3.
|
Östrogenernas interaktion med östrogenreceptorer kan påverka transkriptionen av östrogenkontrollerade gener, vilket kan leda till induktion eller inhibition av cellulära processer, även sådana som är nödvändiga för cellförökning, normal fosterutveckling och fortplantningsfunktionen (9)(10)(11). En störning av normala östrogensystem kan leda till skadliga effekter på normal utveckling (ontogenes), reproduktionshälsa och reproduktionssystemets tillstånd.
|
|
4.
|
In vitro-transaktiveringstester grundas på en direkt eller indirekt interaktion mellan ämnena och en specifik receptor som reglerar transkriptionen av en rapportgenprodukt. Sådana tester har i stor utsträckning använts för att utvärdera det genuttryck som regleras av specifika nukleära receptorer, t.ex. östrogenreceptorer (12) (13) (14) (15) (16). De har föreslagits för detektering av östrogen transaktivering som regleras av östrogenreceptorer (17) (18) (19). Det finns minst två stora undertyper av nukleära östrogenreceptorer, α och β, som kodas av olika gener. De respektive proteinerna har olika biologiska funktioner, olika vävnadsdistribution och olika bindningsaffinitet hos liganderna (20) (21) (22) (23) (24) (25) (26). Nukleära ERα medierar den klassiska östrogena responsen (27)(28)(29)(30), och de flesta modeller som för närvarande utvecklas för att mäta aktivering eller inhibering av östrogenreceptorer är därför specifika för ERα. Metoderna används för att identifiera kemikalier som aktiverar (eller inhiberar) östrogenreceptorerna efter ligandbindning, efter vilket receptor–ligand-komplexet binder sig till specifika DNA-responselement och transaktiverar en rapportgen, vilket i sin tur leder till ökat celluttryck hos ett markörprotein. Olika responser från rapportgener kan användas i dessa tester. I luciferasbaserade system omvandlar luciferasenzymet luciferassubstratet till en bioluminiscent produkt som kan mätas kvantitativt med en luminometer. Andra exempel på vanliga rapportgener är fluorescerande protein och LacZ-genen, som kodar för β-galaktosidas, en enzymprodukt som kan omvandla det färglösa substratet X-gal (5-brom-4-klor-indolyl-galaktopyranosid) till en blå produkt som kan kvantifieras med en spektrofotometer. Dessa rapportgener kan utvärderas snabbt och till låg kostnad med kommersiellt tillgängliga testkit.
|
|
5.
|
Valideringsstudier av STTA- och VM7Luc TA-testerna har visat att de är relevanta och tillförlitliga för det avsedda syftet (3) (4) (5) (30). Prestandastandarder för luminiscensbaserade ER TA-tester med användning av bröstcellinjer ingår i ICCVAM Test Method Evaluation Report on the LUMI-CELL® ER (VM7Luc ER TA) Test Method: An In Vitro Assay for Identifying Human Estrogen Receptor Agonist and Antagonist Activity of Chemicals (3). Dessa prestandastandarder har modifierats för att vara tillämpliga på både STTA- och VM7Luc TA-metoderna (2).
|
|
6.
|
Definitioner och förkortningar som används i denna testmetod beskrivs i tillägg 1.
|
Omfattning av och begränsningar hos transaktiveringstester
|
7.
|
Dessa tester föreslås för screening- och prioriteringsändamål, men kan även ge mekanistisk information som kan användas i en sammanvägd bedömning. De behandlar transaktivering som inducerats genom kemisk bindning till östrogenreceptorer i ett in vitro-system. Resultaten bör således inte extrapoleras direkt till det intakta endokrina systemets komplexa signalering och reglering in vivo.
|
|
8.
|
Transaktivering medierad av östrogenreceptorer är en av de viktigaste mekanismerna för hormonstörning, även om hormonstörningar även kan uppstå genom andra mekanismer, exempelvis i) interaktioner med andra receptorer och enzymsystem inom det endokrina systemet, ii) hormonsyntes, iii) metabolisk aktivering och/eller inaktivering av hormoner, iv) fördelningen av hormoner till målvävnader, och v) clearance av hormoner från kroppen. Inget av testerna i denna testmetod behandlar dessa verkningssätt.
|
|
9.
|
Denna testmetod behandlar kemikaliers förmåga att aktivera (dvs. agera som agonister) och även att dämpa (dvs. agera som antagonister) transkription som är beroende av östrogenreceptorer. En del kemikalier kan, beroende på celltyp, uppvisa både agonistisk och antagonistisk aktivitet, och benämns selektiva östrogenreceptormodulatorer (SERM). Kemikalier som visar negativt resultat i dessa tester kan utvärderas i ett test av östrogenreceptorbindning innan slutsatsen dras att kemikalien inte binder till receptorn. Dessutom kommer testerna sannolikt endast att ge information om det ursprungliga ämnet, med tanke på in vitro-cellsystemens begränsade metaboliska kapacitet. Med tanke på att endast enstaka ämnen användes under valideringen har analysens tillämplighet på testblandningar inte behandlats. Testmetoden är emellertid teoretiskt tillämplig på testning av multikomponentämnen, UVCB-ämnen och blandningar. Innan testmetoden används på ett multikomponentämne, UVCB-ämne eller en blandning i syfte att samla in data för ett lagstadgat ändamål bör man överväga om, och i så fall varför, den kan ge godtagbara resultat för det syftet. Sådana överväganden är inte nödvändiga om blandningen måste provas enligt lag.
|
|
10.
|
I informationssyfte innehåller tabell 1 agonist-testresultaten för de 34 ämnen som testades i båda de fullständigt validerade referenstestmetoder som beskrivs i denna testmetod. Av dessa ämnen är 26 klassificerade som definitiva ER-agonister och 8 som negativa baserat på publicerade rapporter, inklusive in vitro-tester av östrogenreceptorbindning och transaktivering och/eller uterotroft test (2) (3) (18) (31) (32) (33) (34). Tabell 2 innehåller antagonist-testresultaten för de 15 ämnen som testades i båda de fullständigt validerade referenstestmetoder som beskrivs i denna testmetod. Av dessa ämnen är 4 klassificerade som definitiva/förmodade ER-agonister och 10 som negativa baserat på publicerade rapporter, inklusive in vitro-tester av östrogenreceptorbindning och transaktivering (2) (3) (18) (31). När det gäller de data som sammanfattas i tabellerna 1 och 2 överensstämde de två referenstestmetoderna till 100 procent i klassificeringarna av alla ämnen utom ett (Mifepriston) för antagonisttest, och varje ämne klassificerades korrekt som ER-agonist/antagonist eller som negativt. Kompletterande information om denna grupp av kemikalier och ytterligare kemikalier som har testats i STTA- och VM7Luc ER TA-testerna under valideringsstudierna ges i prestandastandarderna för ERTA (6)(7), tillägg 2 (tabellerna 1, 2 och 3).
|
Tabell 1
Översikt över resultaten från STTA- och VM7Luc ER TA-testerna för ämnen som testats i de båda agonisttesterna och klassificerats som ER-agonister (POS) eller som negativa (NEG)
|
|
Ämne
|
CAS-nr
|
STTA – test (35)
|
VM7Luc ER TA – test (36)
|
Datakälla för klassificering (38)
|
|
ER TA Aktivitet
|
PC10-värde (M)
|
PC50-värde (2) (M)
|
ER TA-aktivitet
|
EC50-värde (2), (37) (M)
|
Övriga ER TA (34)
|
ER Bindemedel
|
Uterotrof
|
|
1
|
17ß-östradiol (1)
|
50-28-2
|
POS
|
< 1,00 × 10–11
|
< 1,00 × 10–11
|
POS
|
5,63 × 10–12
|
POS (227/227)
|
POS
|
POS
|
|
2
|
17α-östradiol (1)
|
57-91-0
|
POS
|
7,24 × 10–11
|
6,44 × 10–10
|
POS
|
1,40 × 10–9
|
POS (11/11)
|
POS
|
POS
|
|
3
|
17α-etinylöstradiol (1)
|
57-63-6
|
POS
|
< 1,00 × 10–11
|
< 1,00 × 10–11
|
POS
|
7,31 × 10–12
|
POS (22/22)
|
POS
|
POS
|
|
4
|
17β-trenbolon
|
10161-33-8
|
POS
|
1,78 × 10–8
|
2,73 × 10–7
|
POS
|
4,20 × 10–8
|
POS (2/2)
|
NT
|
NT
|
|
5
|
19-nortestosteron (1)
|
434-22-0
|
POS
|
9,64 × 10–9
|
2,71 × 10–7
|
POS
|
1,80 × 10–6
|
POS (4/4)
|
POS
|
POS
|
|
6
|
4-kumylfenol (1)
|
599-64-4
|
POS
|
1,49 × 10–7
|
1,60 × 10–6
|
POS
|
3,20 × 10–7
|
POS (5/5)
|
POS
|
NT
|
|
7
|
4-tert-oktylfenol (1)
|
140-66-9
|
POS
|
1,85 × 10–9
|
7,37 × 10–8
|
POS
|
3,19 × 10–8
|
POS (21/24)
|
POS
|
POS
|
|
8
|
Apigenin (1)
|
520-36-5
|
POS
|
1,31 × 10–7
|
5,71 × 10–7
|
POS
|
1,60 × 10–6
|
POS (26/26)
|
POS
|
NT
|
|
9
|
Atrazin (1)
|
1912-24-9
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (30/30)
|
NEG
|
NT
|
|
10
|
Bisfenol A (1)
|
80-05-7
|
POS
|
2,02 × 10–8
|
2,94 × 10–7
|
POS
|
5,33 × 10–7
|
POS (65/65)
|
POS
|
POS
|
|
11
|
Bisfenol B (1)
|
77-40-7
|
POS
|
2,36 × 10–8
|
2,11 × 10–7
|
POS
|
1,95 × 10–7
|
POS (6/6)
|
POS
|
POS
|
|
12
|
Bensylbutylftalat (1)
|
85-68-7
|
POS
|
1,14 × 10–6
|
4,11 × 10–6
|
POS
|
1,98 × 10–6
|
POS (12/14)
|
POS
|
NEG
|
|
13
|
Kortikosteron (1)
|
50-22-6
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (6/6)
|
NEG
|
NT
|
|
14
|
Kumestrol (1)
|
479-13-0
|
POS
|
1,23 × 10–9
|
2,00 × 10–8
|
POS
|
1,32 × 10–7
|
POS (30/30)
|
POS
|
NT
|
|
15
|
Daidzein (1)
|
486-66-8
|
POS
|
1,76 × 10–8
|
1,51 × 10–7
|
POS
|
7,95 × 10–7
|
POS (39/39)
|
POS
|
POS
|
|
16
|
Dietylstilbestrol (1)
|
56-53-1
|
POS
|
< 1,00 × 10–11
|
2,04 × 10–11
|
POS
|
3,34 × 10–11
|
POS (42/42)
|
POS
|
NT
|
|
17
|
Dibutylftalat
|
84-74-2
|
POS
|
4,09 × 10–6
|
|
POS
|
4,09 × 10–6
|
POS (6/11)
|
POS
|
NEG
|
|
18
|
Etylparaben
|
120-47-8
|
POS
|
5,00 × 10–6
|
(ej PC50)
|
POS
|
2,48 × 10–5
|
POS
|
|
NT
|
|
19
|
Östron (1)
|
53-16-7
|
POS
|
3,02 × 10–11
|
5,88 × 10–10
|
POS
|
2,34 × 10–10
|
POS (26/28)
|
POS
|
POS
|
|
20
|
Genistein (1)
|
446-72-0
|
POS
|
2,24 × 10–9
|
2,45 × 10–8
|
POS
|
2,71 × 10–7
|
POS (100/102)
|
POS
|
POS
|
|
21
|
Haloperidol
|
52-86-8
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (2/2)
|
NEG
|
NT
|
|
22
|
Kaempferol (1)
|
520-18-3
|
POS
|
1,36 × 10–7
|
1,21 × 10–6
|
POS
|
3,99 × 10–6
|
POS (23/23)
|
POS
|
NT
|
|
23
|
Kepon (1)
|
143-50-0
|
POS
|
7,11 × 10–7
|
7,68 × 10–6
|
POS
|
4,91 × 10–7
|
POS (14/18)
|
POS
|
NT
|
|
24
|
Ketokonazol
|
65277-42-1
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (2/2)
|
NEG
|
NT
|
|
25
|
Linuron (1)
|
330-55-2
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (8/8)
|
NEG
|
NT
|
|
26
|
meso-hexestrol (1)
|
84-16-2
|
POS
|
< 1,00 × 10–11
|
2,75 × 10–11
|
POS
|
1,65 × 10–11
|
POS (4/4)
|
POS
|
NT
|
|
27
|
Metyltestosteron (1)
|
58-18-4
|
POS
|
1,73 × 10–7
|
4,11 × 10–6
|
POS
|
2,68 × 10–6
|
POS (5/6)
|
POS
|
NT
|
|
28
|
Morin
|
480-16-0
|
POS
|
5,43 × 10–7
|
4,16 × 10–6
|
POS
|
2,37 × 10–6
|
POS (2/2)
|
POS
|
NT
|
|
29
|
Noretynodrel (1)
|
68-23-5
|
POS
|
1,11 × 10–11
|
1,50 × 10–9
|
POS
|
9,39 × 10–10
|
POS (5/5)
|
POS
|
NT
|
|
30
|
p,p’-metoxiklor (1)
|
72-43-5
|
POS
|
1,23 × 10–6
|
(ej PC50) (2)
|
POS
|
1,92 × 10–6
|
POS (24/27)
|
POS
|
POS
|
|
31
|
Fenobarbital (1)
|
57-30-7
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (2/2)
|
NEG
|
NT
|
|
32
|
Reserpin
|
50-55-5
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (4/4)
|
NEG
|
NT
|
|
33
|
Spironolakton (1)
|
52-01-7
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (4/4)
|
NEG
|
NT
|
|
34
|
Testosteron
|
58-22-0
|
POS
|
2,82 × 10–8
|
9,78 × 10–6
|
POS
|
1,75 × 10–5
|
POS (5/10)
|
POS
|
NT
|
|
Förkortningar: CAS-nummer = nummer i registret som förs av Chemical Abstracts Service, M = molar. EC50 = halva den maximala effektiva koncentrationen av testämnet. NEG = negativ. POS = positiv. NT = Inte testat. PC10 (och PC50) = koncentration av ett testämne vid vilken responsen är 10 % (eller 50 % för PC50) av den respons som orsakas av den positiva kontrollen (E2, 1nM) i varje platta.
|
Tabell 2
Jämförelse mellan resultaten från STTA- och VM7Luc ER TA-testerna för ämnen som testats i båda antagonisttesterna och klassificerats som ER-antagonister (POS) eller som negativa (NEG)
|
|
Ämne (3)
|
CAS-nr
|
ER STTA-test (39)
|
VM7Luc ER TA-test (40)
|
ER STTA kandidat- effekter (42)
|
ICCVAM (43) Överenskommen klassificering
|
MeSH (44) Kemisk klass
|
Produktklass (45)
|
|
ER TA Aktivitet
|
IC50-värde (4) (M)
|
ER TA Aktivitet
|
IC50-värde (4), (41) (M)
|
|
1
|
4-hydroxitamoxifen
|
68047-06-3
|
POS
|
3,97 × 10–9
|
POS
|
2,08 × 10–7
|
måttligt POS
|
POS
|
Kolväte (cykliskt)
|
Läkemedel
|
|
2
|
Dibenz[a,h]antracen
|
53-70-3
|
POS
|
Ej IC50
|
POS
|
Ej IC50
|
POS
|
PP
|
Polycykliska kolväten
|
Laboratoriekemikalie, naturprodukt
|
|
3
|
Mifepriston
|
84371-65-3
|
POS
|
5,61 × 10–6
|
NEG
|
—
|
svagt POS
|
NEG
|
Steroid
|
Läkemedel
|
|
4
|
Raloxifen HCl
|
82640-04-8
|
POS
|
7,86 × 10–10
|
POS
|
1,19 × 10–9
|
måttligt POS
|
POS
|
Kolväte (cykliskt)
|
Läkemedel
|
|
5
|
Tamoxifen
|
10540-29-1
|
POS
|
4,91 × 10–7
|
POS
|
8,17 × 10–7
|
POS
|
POS
|
Kolväte (cykliskt)
|
Läkemedel
|
|
6
|
17β-östradiol
|
50-28-2
|
NEG
|
—
|
NEG
|
—
|
PN
|
PN
|
Steroid
|
Humant/veterinärt läkemedel
|
|
7
|
Apigenin
|
520-36-5
|
NEG
|
—
|
NEG
|
—
|
NEG
|
NEG
|
Heterocyklisk förening
|
Färg, naturprodukt, farmaceutisk mellanprodukt
|
|
8
|
Atrazin
|
1912-24-9
|
NEG
|
—
|
NEG
|
—
|
NEG
|
PN
|
Heterocyklisk förening
|
Herbicid
|
|
9
|
Dibutylftalat
|
84-74-2
|
NEG
|
—
|
NEG
|
—
|
NEG
|
NEG
|
Ester, ftalsyra
|
Ingrediens i kosmetika, industrikemikalie, mjukningsmedel
|
|
10
|
Fenarimol
|
60168-88-9
|
NEG
|
—
|
NEG
|
—
|
ej testat
|
PN
|
Heterocyklisk förening, pyrimidin
|
Svampmedel
|
|
11
|
Flavon
|
525-82-6
|
NEG
|
—
|
NEG
|
—
|
PN
|
PN
|
Flavonoid, heterocyklisk förening
|
Naturprodukt, läkemedel
|
|
12
|
Flutamid
|
13311-84-7
|
NEG
|
—
|
NEG
|
—
|
NEG
|
PN
|
Amid
|
Läkemedel, veterinärmedicin
|
|
13
|
Genistein
|
446-72-0
|
NEG
|
—
|
NEG
|
—
|
PN
|
NEG
|
Flavonoid, heterocyklisk förening
|
Naturprodukt, läkemedel
|
|
14
|
p-n-nonylfenol
|
104-40-5
|
NEG
|
—
|
NEG
|
—
|
ej testat
|
NEG
|
Fenol
|
Kemisk intermediär
|
|
15
|
Resveratrol
|
501-36-0
|
NEG
|
—
|
NEG
|
—
|
PN
|
NEG
|
Kolväte (cykliskt)
|
Naturprodukt
|
|
Förkortningar: CAS-nummer = nummer i registret som förs av Chemical Abstracts Service, M = molar. IC50 = halva den maximala hämmande koncentrationen av testämnet. NEG = negativ. PN = förmodat negativ. POS = positiv. PP = förmodat positiv.
|
KOMPONENTER I ER TA-TESTET
Viktiga testdelar
|
11.
|
Denna testmetod är tillämplig på tester som använder en stabilt transfekterad eller endogen ERα-receptor och en stabilt transfekterad rapportgen, under kontroll av ett eller flera φstrogenresponselement. Andra receptorer sεsom ERβ kan dock förekomma. De är de huvudsakliga testkomponenterna.
|
Kontroller
|
12.
|
Grunden för de föreslagna parallella referensstandarderna för varje typ av agonist- och antagonisttest bör beskrivas. Relevanta parallella kontroller (negativ, lösningsmedel och positiv) anger om testet fungerar under testbetingelserna och utgör underlag för jämförelser mellan försök. De ingår vanligen i acceptanskriterierna för ett visst försök (1).
|
Standardförfaranden för kvalitetskontroll
|
13.
|
Standardförfaranden för kvalitetskontroll bör genomföras enligt beskrivningen för varje test för att säkerställa att cellinjen håller sig stabil under flera passager, förblir fri från mykoplasma (dvs. fri från bakterieinfektion) och behåller förmågan att ge den förväntade ER-medierade responsen över tiden. Cellinjerna bör kontrolleras närmare så att de har rätt identitet och inte innehåller andra föroreningar (t.ex. svamp, jäst och virus).
|
Demonstration av laboratoriets kompetens
|
14.
|
Innan okända kemikalier testas i något av de tester som ingår i denna testmetod bör laboratorierna visa att de har kompetens att använda testet För att visa kompetens bör varje laboratorium testa de 14 kvalifikationsämnena i tabell 3 för agonisttestet och de 10 kvalifikationsämnena i tabell 4 för antagonisttestet. Kompetenstestningen kommer även att bekräfta testsystemets respons. Förteckningen över kvalifikationsämnen är en delmängd av de referensämnen som anges i prestandastandarderna för ER TA-testerna (6). Ämnena finns kommersiellt tillgängliga, motsvarar de klasser av kemikalier som vanligen förknippas med agonistisk eller antagonistisk aktivitet hos östrogenreceptorer, visar ett lämpligt potensintervall som förväntas för östrogenreceptorers agonister/antagonister (dvs. stark till svag) och omfattar negativa resultat. Testningen av kvalifikationsämnen bör upprepas minst två gånger, på olika dagar. Kompetens uppvisas genom korrekt klassificering (positiv/negativ) av alla kvalifikationsämnen. Kompetenstestning bör göras av varje tekniker som lär sig testmetoderna. Beroende på celltyp kan vissa av kvalifikationsämnena bete sig som selektiva östrogenreceptormodulatorer (SERM) och uppvisa aktivitet som både agonister och antagonister. I tabellerna 3 och 4 klassificeras kvalifikationsämnena dock efter sin kända dominerande aktivitet, och detta bör användas i kompetensbedömningen.
|
|
15.
|
För att visa prestanda och för kvalitetskontrolländamål bör laboratorierna sammanställa historiska databaser över data om agonister och antagonister med en referensstandard (t.ex. 17β-östradiol och tamoxifen), positiva och negativa kontrollkemikalier och lösningsmedelskontroll (t.ex. DMSO). Som utgångspunkt bör databasen skapas utifrån minst tio oberoende testomgångar med agonister (t.ex. 17β-östradiol) och tio oberoende testomgångar med antagonister (t.ex. tamoxifen). Resultaten från framtida tester av dessa referensstandarder och lösningsmedelskontroller bör läggas in i databasen så att den utökas för att säkerställa konsekvens och prestanda för det biologiska testet vid laboratoriet över tiden.
|
Tabell 3
Förteckning över (14) kvalifikationsämnen för agonisttest
(53)
|
Nr (52)
|
Ämne
|
CAS-nr
|
Förväntad respons (46)
|
STTA-test
|
VM7Luc ER TA-test
|
Kemisk klass i MeSH (50)
|
Produktklass (51)
|
|
PC10-värde(M) (47)
|
PC50-värde(M) (47)
|
Testkonc.intervall (M)
|
VM7Luc EC50-värde (M) (48)
|
Högsta konc. för förberedande koncentrationsintervalltest (M) (49)
|
|
14
|
Dietylstilbestrol
|
56-53-1
|
POS
|
< 1,00 × 10–11
|
2,04 × 10–11
|
10–14 – 10–8
|
3,34 × 10–11
|
3,73 × 10–4
|
Kolväte (cykliskt)
|
Läkemedel, veterinärmedicin
|
|
12
|
17α-östradiol
|
57-91-0
|
POS
|
4,27 × 10–11
|
6,44 × 10–10
|
10–11 – 10–5
|
1,40 × 10–9
|
3,67 × 10–3
|
Steroid
|
Humant/veterinärt läkemedel
|
|
15
|
meso-hexestrol
|
84-16-2
|
POS
|
< 1,00 × 10–11
|
2,75 × 10–11
|
10–11 – 10–5
|
1,65 × 10–11
|
3,70 × 10–3
|
Kolväte (cykliskt), fenol
|
Humant/veterinärt läkemedel
|
|
11
|
4-tert-oktylfenol
|
140-66-9
|
POS
|
1,85 × 10–9
|
7,37 × 10–8
|
10–11 – 10–5
|
3,19 × 10–8
|
4,85 × 10–3
|
Fenol
|
Kemisk intermediär
|
|
9
|
Genistein
|
446-72-0
|
POS
|
2,24 × 10–9
|
2,45 × 10–8
|
10–11 – 10–5
|
2,71 × 10–7
|
3,70 × 10–4
|
Flavonoid, heterocyklisk förening
|
Naturprodukt, läkemedel
|
|
6
|
Bisfenol A
|
80-05-7
|
POS
|
2,02 × 10–8
|
2,94 × 10–7
|
10–11 – 10–5
|
5,33 × 10–7
|
4,38 × 10–3
|
Fenol
|
Kemisk intermediär
|
|
2
|
Kaempferol
|
520-18-3
|
POS
|
1,36 × 10–7
|
1,21 × 10–6
|
10–11 – 10–5
|
3,99 × 10–6
|
3,49 × 10–3
|
Flavonoid, heterocyklisk förening
|
Naturprodukt
|
|
3
|
Bensylbutylftalat
|
85-68-7
|
POS
|
1,14 × 10–6
|
4,11 × 10–6
|
10–11 – 10–5
|
1,98 × 10–6
|
3,20 × 10–4
|
Karboxylsyra, ester, ftalsyra
|
Mjukningsmedel, industrikemikalie
|
|
4
|
p,p’-metoxiklor
|
72-43-5
|
POS
|
1,23 × 10–6
|
—
|
10–11 – 10–5
|
1,92 × 10–6
|
2,89 × 10–3
|
Kolväte (halogenerat)
|
Bekämpningsmedel, veterinärmedicin
|
|
1
|
Etylparaben
|
120-47-8
|
POS
|
5,00 × 10–6
|
—
|
10–11 – 10–5
|
2,48 × 10–5
|
6,02 × 10–3
|
Karboxylsyra, fenol
|
Läkemedel, konserveringsmedel
|
|
17
|
Atrazin
|
1912-24-9
|
NEG
|
—
|
—
|
10–10–10–4
|
—
|
4,64 × 10–4
|
Heterocyklisk förening
|
Herbicid
|
|
20
|
Spironolakton
|
52-01-7
|
NEG
|
—
|
—
|
10–11 – 10–5
|
—
|
2,40 × 10–3
|
Lakton, steroid
|
Läkemedel
|
|
21
|
Ketokonazol
|
65277-42-1
|
NEG
|
—
|
—
|
10–11 – 10–5
|
—
|
9,41 × 10–5
|
Heterocyklisk förening
|
Läkemedel
|
|
22
|
Reserpin
|
50-55-5
|
NEG
|
—
|
—
|
10–11 – 10–5
|
—
|
1,64 × 10–3
|
Heterocyklisk förening, indol
|
Läkemedel, veterinärmedicin
|
|
Förkortningar: CAS-nummer = nummer i registret som förs av Chemical Abstracts Service, EC50 = halva den maximala effektiva koncentrationen av testämnet. NEG = negativ. POS = positiv. PC10 (och PC50) = koncentration av ett testämne vid vilken responsen är 10 % (eller 50 % för PC50) av den respons som orsakas av den positiva kontrollen (E2, 1nM) i varje platta.
|
Tabell 4
Förteckning över (10) kvalifikationsämnen för antagonisttest
|
|
Ämne (5)
|
CAS-nr
|
ER STTA-test (54)
|
VM7Luc ER TA-test (55)
|
ER STTA (54) kandidateffekter
|
ICCVAM (58) Överenskommen klassificering
|
MeSH (59) Kemisk klass
|
Produktklass (60)
|
|
ER TA-aktivitet
|
IC50 (M)
|
Testkonc.intervall (M)
|
ER TA-aktivitet
|
IC50
(56) (M)
|
Högsta konc. för förberedande test (M) (57)
|
|
1
|
4-hydroxitamoxifen
|
68047-06-3
|
POS
|
3,97 × 10–9
|
10–12 – 10–7
|
POS
|
2,08 × 10–7
|
2,58 × 10–4
|
måttligt POS
|
POS
|
Kolväte (cykliskt)
|
Läkemedel
|
|
2
|
Raloxifen HCl
|
82640-04-8
|
POS
|
7,86 × 10–10
|
10–12 – 10–7
|
POS
|
1,19 × 10–9
|
1,96 × 10–4
|
måttligt POS
|
POS
|
Kolväte (cykliskt)
|
Läkemedel
|
|
3
|
Tamoxifen
|
10540-29-1
|
POS
|
4,91 × 10–7
|
10–10 – 10–5
|
POS
|
8,17 × 10–7
|
2,69 × 10–4
|
POS
|
POS
|
Kolväte (cykliskt)
|
Läkemedel
|
|
4
|
17β-östradiol
|
50-28-2
|
NEG
|
—
|
10–9 – 10–4
|
NEG
|
—
|
3,67 × 10–3
|
förväntas vara negativ (*4)
|
PN
|
Steroid
|
Humant/veterinärt läkemedel
|
|
5
|
Apigenin
|
520-36-5
|
NEG
|
—
|
10–9 – 10–4
|
NEG
|
—
|
3,70 × 10–4
|
NEG
|
NEG
|
Heterocyklisk förening
|
Färg, naturprodukt, farmaceutisk mellanprodukt
|
|
6
|
Dibutylftalat
|
84-74-2
|
NEG
|
—
|
10–8 – 10–3
|
NEG
|
—
|
3,59 × 10–3
|
NEG
|
NEG
|
Ester, ftalsyra
|
Ingrediens i kosmetika, industrikemikalie, mjukningsmedel
|
|
7
|
Flavon
|
525-82-6
|
NEG
|
—
|
10–8 – 10–3
|
NEG
|
—
|
4,50 × 10–4
|
förväntas vara negativ (*4)
|
PN
|
Flavonoid, heterocyklisk förening
|
Naturprodukt, läkemedel
|
|
8
|
Genistein
|
446-72-0
|
NEG
|
—
|
10–9 – 10–4
|
NEG
|
—
|
3,70 × 10–4
|
förväntas vara negativ (*4)
|
NEG
|
Flavonoid, heterocyklisk förening
|
Naturprodukt, läkemedel
|
|
9
|
p-n-nonylfenol
|
104-40-5
|
NEG
|
—
|
10–9 – 10–4
|
NEG
|
—
|
4,54 × 10–4
|
ej testat
|
NEG
|
Fenol
|
Kemisk intermediär
|
|
10
|
Resveratrol
|
501-36-0
|
NEG
|
—
|
10–8 – 10–3
|
NEG
|
—
|
4,38 × 10–4
|
förväntas vara negativ (*4)
|
NEG
|
Kolväte (cykliskt)
|
Naturprodukt
|
|
Förkortningar: CAS-nummer = nummer i registret som förs av Chemical Abstracts Service, M = molar. IC50 = halva den maximala hämmande koncentrationen av testämnet. NEG = negativ. PN = förmodat negativ. POS = positiv.
|
Acceptanskriterier för testomgångar
|
16.
|
Godkännandet eller förkastandet av en testomgång baseras på en utvärdering av de resultat som har erhållits för de referensstandarder och kontroller som använts för varje försök. Värdena för PC50 (EC50) eller IC50 för referensstandarderna bör uppfylla acceptanskriterierna för det valda testet (se tillägg 2 för STTA och tillägg 3 för VM7Luc ER TA), och samtliga positiva/negativa kontroller bör klassificeras korrekt för varje godtaget försök. Laboratoriet bör visa att det har förmåga att utföra testet på ett konsekvent sätt genom att utveckla och underhålla en historisk databas för referensstandarderna och kontrollerna (se punkt 15). Standardavvikelser eller variationskoefficienter för medelvärden av parametrars anpassning till referensstandardkurvan erhållna från flera olika försök kan användas som ett mått på laboratoriets kunnighet att utföra testet. Dessutom bör följande principer för acceptanskriterierna vara uppfyllda:
|
—
|
Laboratoriet bör förfoga över tillräckliga data för att kunna göra en kvantitativ bedömning av östrogenreceptorers aktivering (för agonisttest) eller undertryckning (för antagonisttest) dvs. effektivitet och potens.
|
|
—
|
För att säkerställa tillräcklig sensitivitet bör rapportgenens genomsnittliga aktivitet för referenskoncentrationen av referensöstrogenet vara lägst det värde som anges i metoderna jämfört med vehikelkontrollen (lösningsmedelskontrollen). För STTA- och VM7Luc ER TA-testerna är detta fyra gånger medelvärdet för vehikelkontrollen på varje platta.
|
|
—
|
De testade koncentrationerna bör hållas sig inom testkemikaliernas löslighetsintervall och bör inte uppvisa cytotoxicitet.
|
|
Analys av data
|
17.
|
Det fastställda datatolkningsförfarandet för varje test bör användas för att klassificera positiv och negativ respons.
|
|
18.
|
Om acceptanskriterierna uppfylls (punkt 16) innebär detta att testet fungerar korrekt, men det säkerställer inte att en viss testomgång kommer att ge korrekta data. En upprepning av resultaten från den första testomgången är den bästa indikationen på att korrekta data har erhållits. Om två testomgångar ger reproducerbara resultat (t.ex. resultaten från båda testomgångarna visar att en testkemikalie är positiv) är det inte nödvändigt att utföra en tredje testomgång.
|
|
19.
|
Om två testomgångar inte ger reproducerbara resultat (t.ex. att en testkemikalie är positiv i en testomgång och negativ i nästa), eller om en högre precision krävs för resultatet av testet, bör minst tre oberoende testomgångar utföras. I ett sådant fall baseras klassificeringen på de två samstämmiga resultaten av de tre.
|
Allmänna datatolkningskriterier
|
20.
|
Det finns för närvarande ingen allmänt överenskommen metod för att tolka ER TA-data. Både kvalitativa (t.ex. positiv/negativ) och/eller kvantitativa (t.ex. EC50, PC50, IC50) bedömningar av ER-medierad aktivitet bör dock baseras på empiriska data och välgrundade vetenskapliga bedömningar. Om det är möjligt bör positiva resultat karakteriseras av både effektens omfattning jämfört med vehikelkontrollen (lösningsmedelskontrollen) eller referensöstrogenet och den koncentration vid vilken effekten inträffar (t.ex. en EC50, PC50, RPCMax, IC50 osv.).
|
Testrapport
|
21.
|
Testrapporten ska innehålla följande information:
|
|
Test:
|
—
|
Kontroll/referensstandard/testkemikalie.
|
|
—
|
Källa, satsnummer, sista förbrukningsdag om detta anges,
|
|
—
|
Testkemikaliens stabilitet i sig, om den är känd.
|
|
—
|
Testkemikaliens löslighet och stabilitet i lösningsmedlet, om detta är känt.
|
|
—
|
Mätning av pH, osmolalitet och fällningar i det odlingsmedium till vilket testkemikalien tillsattes, i förekommande fall.
|
|
|
|
Monokomponentämne:
|
—
|
fysikaliskt tillstånd, vattenlöslighet och ytterligare relevanta fysikalisk-kemiska egenskaper.
|
|
—
|
Kemisk identifiering, t.ex. med IUPAC- eller CAS-namn, CAS-nummer, Smiles- eller InChI-kod, strukturformel, renhet samt motsvarande identifiering av föroreningar i förekommande fall och om det är praktiskt möjligt.
|
|
|
|
Multikomponentämne, UVCB-ämnen och blandningar:
|
—
|
karakteriseras så långt som möjligt genom beståndsdelarnas kemiska identitet (se ovan), kvantitativa förekomst och relevanta fysikalisk-kemiska egenskaper.
|
|
|
|
Lösningsmedel/vehikel:
|
—
|
Karakterisering (typ, leverantör och sats).
|
|
—
|
Motivering för val av lösningsmedel/vehikel.
|
|
—
|
Testkemikaliens löslighet och stabilitet i lösningsmedlet/vehikeln, om detta är känt.
|
|
|
|
Celler:
|
—
|
Typ av celler och deras källa.
|
—
|
Har östrogenreceptorn ett endogent uttryck? Om så inte är fallet, vilken eller vilka receptorer transfekterades?
|
|
—
|
Använd(a) rapportgen(er) (inklusive källarter).
|
|
—
|
Urvalsmetod för upprätthållande av stabil transfektion (i förekommande fall).
|
|
—
|
Är transfektionsmetoden relevant för stabila linjer?
|
|
|
—
|
Antal cellpassager (från upptining).
|
|
—
|
Antal passager för celler vid upptining.
|
|
—
|
Metoder för underhåll av cellodlingar.
|
|
|
|
Testbetingelser:
|
—
|
Löslighetsbegränsningar.
|
|
—
|
Beskrivning av använda metoder för viabilitetsbedömning.
|
|
—
|
Mediernas sammansättning, CO2-koncentration.
|
|
—
|
Testkemikaliens koncentrationer.
|
|
—
|
Den tillsatta vehikelns och testkemikaliens volym.
|
|
—
|
Inkubationstemperatur och luftfuktighet.
|
|
—
|
Behandlingens varaktighet,
|
|
—
|
Celltäthet i början av och under behandlingen.
|
|
—
|
Positiva och negativa referensstandarder.
|
|
—
|
Rapportgenreagenser (produktnamn, leverantör och partinummer).
|
|
—
|
Kriterier för när testomgångar ska anses som positiva, negativa eller tvetydiga.
|
|
|
|
Acceptanskontroll:
|
—
|
Induktionsförhållande för varje testplatta och om de uppfyller det minimum som krävs enligt testmetoden baserat på historiska kontroller.
|
|
—
|
Faktiska värden för acceptanskriterier, t.ex. log10EC50, log10PC50, logIC50 och Hill-koefficient (Hillslope) för parallella positiva kontroller/referensstandarder.
|
|
|
|
Resultat:
|
—
|
Rådata och normaliserade data.
|
|
—
|
Maximalt induktionsförhållande.
|
|
—
|
I förekommande fall, lägsta effektiva koncentration (LEC).
|
|
—
|
Värden för RPCMax, PCMax, PC50, IC50 och/eller EC50, i förekommande fall.
|
|
—
|
Koncentration-responsförhållande, där det är möjligt.
|
|
—
|
Eventuella statistiska analyser, tillsammans med fel- och konfidensmått (t.ex. SEM, SD, CV eller 95 % CI) samt en beskrivning av hur dessa värden har erhållits.
|
|
|
LITTERATUR
|
(1)
|
OECD (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environmental Health and Safety Publications,Series on Testing and Assessment (nr 34), Organisationen för ekonomiskt samarbete och utveckling, Paris.
|
|
(2)
|
OECD (2015). Report of the Inter-Laboratory Validation for Stably Transfected Transactivation Assay to detect Estrogenic and Anti-estrogenic Activity. Environmental Health and Safety Publications, Series on Testing and Assessment (nr 225), Organisationen för ekonomiskt samarbete och utveckling, Paris.
|
|
(3)
|
ICCVAM (2011). ICCVAM Test Method Evaluation Report on the LUMI-CELL® ER (BG1Luc ER TA) Test Method, an In Vitro Method for Identifying ER Agonists and Antagonists, National Institute of Environmental Health Sciences: Research Triangle Park, NC.
|
|
(4)
|
Pujol P., et al.(1998). Differential Expression of Estrogen Receptor-Alpha and -Beta Messenger RNAs as a Potential Marker of Ovarian Carcinogenesis, Cancer. Res., 58(23): s. 5367–5373.
|
|
(5)
|
Rogers J.M. och Denison M.S. (2000). Recombinant Cell Bioassays for Endocrine Disruptors: Development of a Stably Transfected Human Ovarian Cell Line for the Detection of Estrogenic and Anti-Estrogenic Chemicals, In Vitro and Molecular Toxicology: Journal of Basic and Applied Research, 13(1): s. 67–82.
|
|
(6)
|
OECD (2012). Performance Standards For Stably Transfected Transactivation In Vitro Assay to Detect Estrogen Receptor Agonists (for TG 455). Environmental Health and Safety Publications, Series on Testing and Assessment (nr 173), Organisationen för ekonomiskt samarbete och utveckling, Paris.
|
|
(7)
|
OECD (2015). Performance Standards For Stably Transfected Transactivation In Vitro Assay to Detect Estrogen Receptor Antagonists. Environmental Health and Safety Publications, Series on Testing and Assessment (nr 174), Organisationen för ekonomiskt samarbete och utveckling, Paris.
|
|
(8)
|
OECD (2012). Guidance Document on Standardized Test Guidelines for Evaluating Chemicals for Endocrine Disruption. Environmental Health and Safety Publications,Series on Testing and Assessment (nr 150), Organisationen för ekonomiskt samarbete och utveckling, Paris.
|
|
(9)
|
Cavailles V. (2002). Estrogens and Receptors: an Evolving Concept. Climacteric, 5 Suppl 2: s. 20–26.
|
|
(10)
|
Welboren W.J. et al. (2009). Genomic Actions of Estrogen Receptor Alpha: What are the Targets and how are they Regulated? Endocr. Relat. Cancer, 16(4): s. 1073–89.
|
|
(11)
|
Younes M. och Honma N. (2011). Estrogen Receptor Beta, Arch. Pathol. Lab. Med., 135(1): s. 63–66.
|
|
(12)
|
Jefferson W.N., et al. (2002). Assessing Estrogenic Activity of Phytochemicals Using Transcriptional Activation and Immature Mouse Uterotrophic Responses, Journal of Chromatography B, 777(1–2): s. 179-189.
|
|
(13)
|
Sonneveld E., et al. (2006). Comparison of In Vitro and In Vivo Screening Models for Androgenic and Estrogenic Activities, Toxicol. Sci., 89(1): s. 173-187.
|
|
(14)
|
Takeyoshi M., et al. (2002). The Efficacy of Endocrine Disruptor Screening Tests in Detecting Anti- Estrogenic Effects Downstream of Receptor-Ligand Interactions, Toxicology Letters, 126(2): s. 91–98.
|
|
(15)
|
Combes R.D. (2000). Endocrine Disruptors: a Critical Review of In Vitro and In Vivo Testing Strategies for Assessing their Toxic Hazard to Humans, ATLA Alternatives to Laboratory Animals,28(1): s. 81-118.
|
|
(16)
|
Escande A., et al. (2006). Evaluation of Ligand Selectivity Using Reporter Cell Lines Stably Expressing Estrogen Receptor Alpha or Beta, Biochem. Pharmacol,71(10): s. 1459–1469.
|
|
(17)
|
Gray, L.E. Jr. (1998). Tiered Screening and Testing Strategy for Xenoestrogens and Antiandrogens, Toxicol. Lett, 102–103, 677–680.
|
|
(18)
|
EDSTAC (1998). Endocrine Disruptor Screening and Testing Advisory Committee (EDSTAC) Final Report.
|
|
(19)
|
ICCVAM (2003). ICCVAM Evaluation of In Vitro Test Methods for Detecting Potential Endocrine Disruptors: Estrogen Receptor and Androgen Receptor Binding and Transcriptional Activation Assays.
|
|
(20)
|
Gustafsson J.Ö. (1999). Estrogen Receptor ß – A New Dimension in Estrogen Mechanism of Action, Journal of Endocrinology, 163(3): s. 379-383.
|
|
(21)
|
Ogawa S. et al. (1998). The Complete Primary Structure of Human Estrogen Receptor ß (hERß) and its Heterodimerization with ERαIn Vivo and In Vitro, Biochemical and Biophysical Research Communications, 243(1): s. 122-126.
|
|
(22)
|
Enmark E. et al. (1997). Human Estrogen Receptor ß-Gene Structure, Chromosomal Localization, and Expression Pattern, Journal of Clinical Endocrinology and Metabolism,82(12): s. 4258-4265.
|
|
(23)
|
Ball L.J. et al. (2009). Cell Type- and Estrogen Receptor-Subtype Specific Regulation of Selective Estrogen Receptor Modulator Regulatory Elements, Molecular and Cellular Endocrinology, 299(2): s. 204-211.
|
|
(24)
|
Barkhem T. et al. (1998). Differential Response of Estrogen Receptor Alpha and Estrogen Receptor Beta to Partial Estrogen Agonists/Antagonists, Mol. Pharmacol, 54(1): s. 105–112.
|
|
(25)
|
Deroo B.J. och Buensuceso A.V. (2010). Minireview: Estrogen Receptor-ß: Mechanistic Insights from Recent Studies, Molecular Endocrinology, 24(9): s. 1703-–1714.
|
|
(26)
|
Harris D.M. et al. (2005). Phytoestrogens Induce Differential Estrogen Receptor Alpha- or Beta- Mediated Responses in Transfected Breast Cancer Cells, Experimental Biology and Medicine, 230(8): s. 558-568.
|
|
(27)
|
Anderson J.N. Clark J.H. och Peck E.J.Jr. (1972). The Relationship Between Nuclear Receptor- Estrogen Binding and Uterotrophic Responses, Biochemical and Biophysical Research Communications, 48(6): s. 1460-1468.
|
|
(28)
|
Toft D. (1972). The Interaction of Uterine Estrogen Receptors with DNA, Journal of Steroid Biochemistry, 3(3): s. 515-522.
|
|
(29)
|
Gorski J. et al. (1968), Hormone Receptors: Studies on the Interaction of Estrogen with the Uterus, Recent Progress in Hormone Research, 24: s. 45-80.
|
|
(30)
|
Jensen E.V., et al. (1967), Estrogen-Receptor Interactions in Target Tissues, Archives d’Anatomie Microscopique et de Morphologie Experimentale, 56(3): s. s. 547–569.
|
|
(31)
|
ICCVAM (2002). Background review document: Estrogen Receptor Transcriptional Activation (TA) Assay. Appendix D, Substances Tested in the ER TA Assay, NIH Publication Report (nr 03-4505.).
|
|
(32)
|
Kanno J. et al. (2001). The OECD Program to Validate the Rat Uterotrophic Bioassay to Screen Compounds for In Vivo Estrogenic Responses: Phase 1, Environ. Health Persp., 109:785–794.
|
|
(33)
|
Kanno J., et al. (2003). The OECD program to validate the rat uterotrophic bioassay: Phase Two – Dose Response Studies. Health Persp., 111:1530–1549.
|
|
(34)
|
Kanno J., et al. (2003), The OECD Program to Validate the Rat Uterotrophic Bioassay: Phase Two – Coded Single-Dose Studies, Environ. Health Persp., 111:1550-1558.
|
|
(35)
|
Geisinger, et al. (1989) Characterization of a human ovarian carcinoma cell line with estrogen and progesterone receptors, Cancer 63, 280–288.
|
|
(36)
|
Baldwin, et al. (1998) BG-1 ovarian cell line: an alternative model for examining estrogen-dependent growth in vitro, In Vitro Cell. Dev. Biol. – Animal, 34, 649–654.
|
|
(37)
|
Li, Y., et al. (2014) Research resource: STR DNA profile and gene expression comparisons of human BG-1 cells and a BG-1/MCF-7 clonal variant, Mol. Endo. 28, s. 2072–2081.
|
|
(38)
|
Rogers, J.M. och Denison, M.S. (2000) Recombinant cell bioassays for endocrine disruptors: development of a stably transfected human ovarian cell line for the detection of estrogenic and anti-estrogenic chemicals, In Vitro & Molec. Toxicol. 13, s. 67-82.
|
Tillägg 1
DEFINITIONER OCH FÖRKORTNINGAR
Acceptanskriterier
: Minimistandarder för experimentets prestanda avseende kontroller och referensstandarder. Alla acceptanskriterier ska uppfyllas för att ett experiment ska anses vara giltigt.
Noggrannhet (konkordans)
: Ett mått på hur väl testresultaten och de godtagna referensvärdena överensstämmer med varandra. Det är en relevansaspekt och ett mått på testets prestanda. Detta begrepp används ofta omväxlande med ”konkordans” för att beskriva andelen korrekta resultat med en testmetod (1).
Agonist
: ett ämne som producerar en respons, t.ex. transkription, när det binds till en viss receptor.
Antagonist
: en typ av receptorligand eller kemikalie som i sig inte utlöser en biologisk respons när den binds till en receptor, utan blockerar eller hämmar agonistmedierad respons.
Antiöstrogen aktivitet
: en kemikalies förmåga att undertrycka aktiviteten hos en 17β-östradiol som medieras genom östrogenreceptorer.
Cellmorfologi
: form och utseende hos en cell som odlats i ett monolager i ett enda hål i en vävnadodlingsplatta. Celler som håller på att torka uppvisar ofta en onormal cellmorfologi.
CF
: The OECD Conceptual Framework for the Testing and Evaluation of Endocrine Disrupters (OECD:s begreppsram för provning och bedömning av hormonstörande ämnen).
Träkols-/dextranbehandling
: behandling av serum som används i en cellodling. Behandling med träkol/dextran (ofta benämnd ”strippning”) eliminerar endogena hormoner och hormonbindande proteiner.
Kemikalie
: Ett ämne eller en blandning.
Cytotoxicitet
: skadliga effekter på cellens struktur eller funktion som i slutändan kan leda till celldöd. Cytotoxicitet kan avspeglas genom ett minskat antal celler i hålet i slutet av exponeringsperioden eller genom minskad kapacitet att mäta cellfunktionen jämfört med den parallella vehikelkontrollen.
CV
: Variationskoefficient (Coefficient of Variation).
DCC-FBS
: dextranbelagt träkolsbehandlat fetalt bovinserum.
DMEM
: Dulbecco’s Modification of Eagle’s Medium.
DMSO
: Dimetylsulfoxid.
E2
: 17β-östradiol.
EC50
: halva den maximala effektiva koncentrationen av testkemikalien.
ED
: hormonstörning.
hERα
: Human östrogenreceptor alfa.
hERß
: human östrogenreceptor beta.
EFM
: östrogenfritt medium. Dulbecco’s Modification of Eagle’s Medium (DMEM), kompletterat med 4,5 % träkols-/dextranbehandlat fetalt bovinserum (FBS), 1,9 % L-glutamin och 0,9 % Pen-Strep.
ER
: Östrogenreceptor.
ERE
: östrogenreceptorelement.
Östrogen aktivitet
: en kemikalies förmåga att imitera 17β-östradiol i dess förmåga att binda sig till och aktivera östrogenreceptorer. Det är möjligt att detektera hERα-medierad östrogen aktivitet med hjälp av denna metod.
ERTA
: transaktivering av östrogenreceptor.
FBS
: fetalt bovinserum.
HeLa
: en odödlig human cervikal cellinje.
HeLa9903
: en subklon av en HeLa-cell, i vilken hERααoch en luciferasrapportgen har transfekterats stabilt.
IC
50
: Halva maximalt effektiva koncentrationen för en inhiberande testkemikalie.
ICCVAM
: The Interagency Coordinating Committee on the Validation of Alternative Methods (i Förenta staterna).
Reproducerbarhet mellan laboratorier
: Ett mått på i hur stor utsträckning olika kvalificerade laboratorier kan producera kvalitativt och kvantitativt liknande resultat, med användning av samma protokoll och samma testämnen. Reproducerbarheten mellan laboratorier fastställs under förvalideringen och valideringen och indikerar i hur stor utsträckning ett test framgångsrikt kan överföras mellan laboratorier, även kallat inter-laborativ reproducerbarhet (1).
Reproducerbarhet inom laboratoriet
: Ett mått på i vilken utsträckning kvalificerade personer inom samma laboratorium framgångsrikt kan upprepa resultat vid olika tidpunkter när de använder ett visst protokoll. Kallas även intra-laborativ reproducerbarhet (1).
LEC
: lägsta effektiva koncentration är den lägsta koncentration av testkemikalien som producerar en respons (dvs. den lägsta kemiska koncentrationen vid vilken induktionsförhållandet statistiskt skiljer sig från den parallella vehikelkontrollen).
Me too-test
: Ett vardagligt uttryck för ett test som strukturellt och funktionellt liknar en validerad och godtagen referenstestmetod. Kallas även ”liknande testmetod” (14).
MT
: metallotionein.
MMTV
: Mouse Mammary Tumor Virus.
OHT
: 4-hydroxitamoxifen
PBTG
: Prestandabaserad testriktlinje (Performance-Based Test Guideline)
PC (positiv kontroll)
: ett ämne med stark aktivitet, företrädesvis 17ß-östradiol, som inkluderas i alla tester för att bidra till att säkerställa att testet fungerar korrekt.
PC
10
: den koncentration av en testkemikalie vid vilken den mätta aktiviteten i ett agonisttest utgör 10 % av den maximala effektivitet som inducerats av den positiva kontrollen (E2 vid 1nM för STTA-testet) i varje platta.
PC
50
: den koncentration av en testkemikalie vid vilken den mätta aktiviteten i ett agonisttest utgör 50 % av den maximala effektivitet som inducerats av den positiva kontrollen (E2 vid den referenskoncentration som anges i testmetoden) i varje platta.
PC
Max
: den koncentration av testkemikalien som inducerar RPCMax.
Prestandastandarder
: standarder som baseras på en validerad testmetod och som utgör grunden för utvärdering av jämförbarheten för en föreslagen testmetod som är mekaniskt och funktionellt likartad. Häri ingår 1) viktiga testdelar, 2) en minimiförteckning med referenskemikalier som valts ut från de kemikalier som har använts för att påvisa att den validerade testmetoden fungerar tillfredsställande och 3) de jämförbara nivåer av noggrannhet och tillförlitlighet, på grundval av vad som erhållits för den validerade testmetoden, som man bör kunna påvisa för det föreslagna testet när det utvärderas med användning av minimiförteckningen med referenskemikalier (1).
Kvalifikationsämnen
: en delmängd av de referensämnen som anges i prestandastandarderna, och som laboratorierna kan använda för att visa teknisk kompetens med en standardiserad testmetod. Bland urvalskriterierna för dessa ämnen ingår oftast att de representerar responsintervallet, är kommersiellt tillgängliga och att referensdata av hög kvalitet finns tillgängliga.
Kompetens
: Påvisad förmåga att korrekt genomföra ett test, innan okända ämnen testas.
Referensöstrogen (positiv kontroll, PC)
: 17β-östradiol (E2, CAS-nr 50-28-2).
Referensstandard
: ett referensämne som används för att visa att en testmetod är lämplig. 17β-östradiol är referensstandarden för STTA- och VM7Luc ERTA-testerna.
Referenstestmetoder
: De tester som PBTG 455 baserar sig på.
Relevans
: Beskrivning av förhållandet mellan testet och den berörda effekten och huruvida detta förhållande är meningsfullt och användbart för ett visst ändamål. Relevansen är ett mått på hur väl man genom testet korrekt kan mäta och prediktera den aktuella biologiska effekten. Relevans omfattar också överväganden om noggrannheten hos ett test (konkordans) (1).
Tillförlitlighet
: Mått på i vilken utsträckning ett test kan reproduceras inom och mellan laboratorier över tid, när den utförs enligt samma protokoll. Den bedöms genom att man beräknar reproducerbarheten inom och mellan laboratorier.
RLU
: relativa ljusenheter.
RNA
: ribonukleinsyra.
RPC
Max
: maximal responsnivå som induceras av en testkemikalie, uttryckt som en procentenhet av den respons som 1 nM E2 inducerar på samma platta.
RPMI
: RPMI 1 640-medium, kompletterat med 0,9 % Pen-Strep och 8,0 % fetalt bovinserum (FBS).
Testomgång
: ett enskilt försök där den kemiska aktiviteten utvärderas i förhållande till testets biologiska utfall. Varje testomgång utgör ett fullständigt försök som utförs på replikathål med celler som samtidigt överförts till platta från en gemensam cellpool.
Oberoende testomgång
: ett separat, oberoende försök där man utvärderar den kemiska aktivitetens inverkan på testets biologiska utfall, med användning av celler från en annan pool och nyligen utspädda kemikalier, som utförts på olika dagar eller på samma dag av olika personer.
SD
: Standardavvikelse (Standard Deviation).
Sensitivitet
: den andel av alla positiva/aktiva ämnen som klassificeras korrekt i testet. Det är ett mått på noggrannhet för en testmetod som ger kategoriska resultat och är en viktig faktor vid bedömningen av relevansen för en testmetod (1).
Specificitet
: den andel av alla negativa/inaktiva ämnen som klassificeras korrekt i testet. Det är ett mått på noggrannhet för en testmetod som ger kategoriska resultat och är en viktig faktor vid bedömningen av relevansen för en testmetod (1).
Stabil transfektion
: när DNA transfekteras in i odlade celler på ett sådant sätt att det stabilt integreras i cellens genom, vilket leder till ett stabilt uttryck av transfekterade gener. Kloner av stabilt transfekterade celler väljs med hjälp av stabila markörer (t.ex. motståndsförmåga mot G418).
STTA-test
: Stably Transfected Transactivation Assay, ERα transkriptionell aktivering med användning av cellinjen HeLa 9 903.
Studie
: hela det försöksarbete som utförs för att utvärdera ett enstaka specifikt ämne med användning av en viss testmetod. En studie omfattar samtliga steg, inklusive test av utspädning av testämnet i testmediet, förberedande test för att bestämma koncentrationsintervall, alla nödvändiga fullständiga testomgångar, dataanalyser, kvalitetssäkring, cytotoxicitetsbedömningar osv. När studien är slutförd är det möjligt att klassificera den använda testkemikaliens aktivitet på toxicitetsmålet (dvs. aktiv, inaktiv eller obestämd) och göra en uppskattning av den positiva referenskemikaliens potens.
Ämne
: enligt Reachförordningen (61) definieras ett ämne som ett kemiskt grundämne och föreningar av detta grundämne i naturlig eller tillverkad form, inklusive de eventuella tillsatser som är nödvändiga för att bevara dess stabilitet och sådana föroreningar som härrör från tillverkningsprocessen, men exklusive eventuella lösningsmedel som kan avskiljas utan att det påverkar ämnets stabilitet eller ändrar dess sammansättning. En mycket liknande definition används i GHS (1).
TA (transaktivering): inledandet av mRNA-syntesen till svar på en specifik kemisk signal, såsom bindning av ett östrogen till östrogenreceptorn.
Test
: inom ramen för denna testmetod är test en av de metoder som är giltiga för att uppfylla de fastställda prestandakriterierna. Komponenter i ett test är exempelvis den specifika cellinjen och dess tillväxtförhållanden, det specifika medium som testet utförs i, förhållanden för beredning av plattor, rutiner för och utspädning av testkemikalier samt eventuella andra nödvändiga kvalitetskontrollåtgärder och relaterade datautvärderingssteg.
Testkemikalie
: Alla ämnen eller blandningar som testas med denna testmetod.
Transkription
: mRNA-syntes.
UVCB-ämne
: kemiskt ämne med okänd eller varierande sammansättning, komplexa reaktionsprodukter och biologiskt material.
Validerad testmetod
: Ett test för vilket valideringsstudier har genomförts för att fastställa relevans (inbegripet noggrannhet) och tillförlitlighet för ett specifikt ändamål. Det är viktigt att notera att en validerad testmetod kanske inte är tillräckligt noggrann och tillförlitlig när det gäller ett visst föreslaget ändamål (1).
Validering
: Processen enligt vilken pålitlighet och relevans för en särskild strategi, metod, ett särskilt test, en särskild process eller bedömning etableras för ett fastställt syfte (1).
VC (vehikelkontroll)
: det lösningsmedel som används för att lösa upp test- och kontrollkemikalierna och som testas ensamt som vehikel, utan den upplösta kemikalien.
VM7
: en odödlig adenokarcinomcell som endogent uttrycker östrogenreceptorn.
VM7Luc4E2
: Cellinjen VM7Luc4E2 härleddes från VM7, immortaliserade humana adenokarcinomceller som endogent uttrycker båda formerna av östrogenreceptorn (ERα och ERβ), och har transfekterats stabilt med plasmiden pGudLuc7.ERE. Denna plasmid innehåller fyra kopior av en syntetisk oligonukleotid, som i sin tur innehåller östrogenreceptorelementet uppströms från MMTV-promotorn och firefly-luciferas-genen.
Svag positiv kontroll
: ett ämne med svag aktivitet som valts från förteckningen över referenskemikalier och inkluderas i alla tester för att bidra till att säkerställa att testmetoden fungerar korrekt.
Tillägg 2
STABILT TRANSFEKTERAD MÄNSKLIG ÖSTROGENRECEPTOR-Α – TRANSAKTIVERINGSANALYS FÖR DETEKTION AV ÖSTROGENAGONISTISK OCH -ANTAGONISTISK AKTIVITET HOS KEMIKALIER MED ANVÄNDNING AV CELLINJEN HERΑ-HELA-9903
INLEDANDE ÖVERVÄGANDEN OCH BEGRÄNSNINGAR (SE ÄVEN ALLMÄN INLEDNING)
|
1.
|
Detta transaktiveringstest (TA) använder cellinjen hERα-HeLa-9 903 för att detektera östrogenagonistisk aktivitet, medierad genom human östrogenreceptor alfa (hERα). Valideringsstudien för stabilt transfekterad transaktivering (Stably Transfected Transactivation) (STTA), som utförts av CERI (Japanese Chemicals Evaluation and Research Institute) med användning av cellinjen hERα-HeLa-9 903 för att detektera östrogenagonistisk och antagonistisk aktivitet medierad genom human östrogenreceptor alfa, visade att testmetoden är relevant och tillförlitlig för det avsedda syftet (1).
|
|
2.
|
Denna testmetod är särskilt utformad för att detektera hERα-medierad transaktivering genom att mäta kemiluminiscens som endpoint. Ej receptormedierade luminiscenssignaler har dock rapporterats vid fytoöstrogenkoncentrationer högre än 1 μM till följd av överaktivering av luciferasrapportgenen (2) (3). Dosresponskurvan visar att den faktiska aktiveringen av östrogenreceptorsystemet inträffar vid lägre koncentrationer, men det luciferasuttryck som erhålls vid höga koncentrationer av fytoöstrogener eller liknande föreningar som misstänks producera fytoöstrogenlik överaktivering av luciferasrapportgenen, måste undersökas noggrant i stabilt transfekterade ER TA-testsystem (tillägg 1).
|
|
3.
|
Avsnitten ”Allmän inledning” och ”Komponenter i ER TA-testet” bör läsas innan denna testmetod används för lagstadgade ändamål. Definitioner och förkortningar som används i denna testriktlinje beskrivs i tillägg 2.1.
|
PRINCIPER FÖR METODEN (SE ÄVEN ALLMÄN INLEDNING)
|
4.
|
Denna testmetod används för att signalera östrogenreceptorns bindning med en ligand. Efter ligandbindning translokerar receptor–ligand-komplexet till kärnan, där det binder specifika DNA-responselement och transaktiverar en firefly-luciferasrapportgen, vilket leder till ökat celluttryck av luciferasenzym. Luciferin är ett substrat som luciferasenzymet omvandlar till en bioluminiscent produkt som kan mätas kvantitativt med en luminometer. Luciferasaktivitet kan utvärderas snabbt och till låg kostnad med ett antal kommersiellt tillgängliga testkit.
|
|
5.
|
Testsystemet använder cellinjen hERα-HeLa-9 903, som härleds från en human cervikal tumör, med två stabilt infogade konstruktioner: i) Konstruktionen med hERαα-uttryck (som kodar för hela den humana receptorn), och ii) en konstruktion med en firefly-luciferas-rapportgen som bär fem tandemupprepningar av ett vitellogenin-östrogenreceptivt element (ERE), som drivs av ett MT-promotor (metallotionein) TATA-element från möss. MT TATA-genkonstruktionen från möss har visat bäst resultat, och används därför utbrett. Denna hERα-HeLa-9 903-cellinje kan således mäta en testkemikalies förmåga att inducera hERα-medierad transaktivering från luciferasgenuttrycket.
|
|
6.
|
I ER-agonisttestet baseras datatolkningen på huruvida den maximala responsnivå som induceras av en agonistrespons motsvarar eller överskrider 10 % av den respons som induceras genom en maximalt inducerande koncentration (1 nM) av den positiva kontrollen (PC) 17β-östradiol (E2) (dvs. PC10). I ER-antagonisttestet baseras datatolkningen på huruvida responsen visar en minskning på minst 30 % i aktiviteten från den respons som induceras genom den spikade kontrollen (25 pM E2) utan cytotoxicitet. Dataanalys och tolkning diskuteras i detalj i punkterna 34–48.
|
FÖRFARANDE
Cellinjer
|
7.
|
Den stabilt transfekterade cellinjen hERα-HeLa-9 903 bör användas i testet. Denna cellinje kan erhållas från cellbanken JRCB (Japanese Collection of Research Bioresources) (62) genom att man ingår ett materialöverföringsavtal (MTA).
|
|
8.
|
Endast celler som karakteriseras som mykoplasmafria bör användas i testningen. RT-PCR (Real Time Polymerase Chain Reaktion) är den valda metoden för en känslig detektion av mykoplasmainfektion (4) (5) (6).
|
Cellinjens stabilitet
|
9.
|
För att övervaka cellinjens stabilitet bör E2, 17α-östradiol, 17α-metyltestosteron och kortikosteron användas som referensstandarder för agonisttest. En fullständig koncentrationsresponskurva i det testkoncentrationsintervall som anges i tabell 1 bör mätas minst en gång varje gång testet utförs, och resultaten bör överensstämma med de resultat som anges i tabell 1.
|
|
10.
|
Vad beträffar antagonisttest bör två fullständiga koncentrationskurvor för två referensstandarder, tamoxifen och flutamid, mätas samtidigt med varje testomgång. Det är viktigt att övervaka en korrekt kvalitativ klassificering som positiv eller negativ för de två kemikalierna.
|
Betingelser för cellodling och överföring till platta
|
11.
|
Cellerna bör förvaras i EMEM (Eagle’s Minimum Essential Medium) utan fenolrött, kompletterat med 60 mg/l antibiotiskt kanamycin och 10 % dextranbelagt träkolsbehandlat fetalt bovinserum (DCC-FBS), i en CO2-inkubator (5 % CO2) vid 37±1°C. När cellerna når 75–90 % konfluens kan de subkultiveras vid 10 ml av 0,4 x 105 – 1 x 105 celler/ml för en cellodlingsskål på 100 mm. Cellerna bör suspenderas med 10 % FBS-EMEM (som är detsamma som EMEM med DCC-FBS) och sedan överföras till hål på en mikrotiterplatta vid en densitet på 1 x 104 celler/(100 μl x hål). Därefter bör cellerna förinkuberas i en 5 % CO2-inkubator vid 37 ± 1 °C i tre timmar före kemisk exponering. Plastvaran bör vara fri från östrogenaktivitet.
|
|
12.
|
För att bibehålla responsens integritet bör cellerna odlas mer än en passage från den frysta stamodlingen i det konditionerade mediet, men bör inte odlas mer än 40 passager. För cellinjen hERα-HeLa-9 903 kommer detta att ta mindre än tre månader. Cellernas prestanda kan dock minskas om de odlas under olämpliga odlingsförhållanden.
|
|
13.
|
DCC-FBS kan beredas enligt beskrivningen i tillägg 2.2, eller erhållas från kommersiella källor.
|
Acceptanskriterier
Positiva och negativa referensstandarder för ER-agonisttest
|
14.
|
Före och under studien bör testsystemets responsförmåga kontrolleras med hjälp av lämpliga koncentrationer av en stark östrogen (E2), en svag östrogen (17α-östradiol), en mycket svag agonist (17α-metyltestosteron) och ett negativt ämne (kortikosteron). Godtagbara intervallvärden som härletts från valideringsstudien (1) anges i tabell 1. Dessa fyra parallella referensstandarder bör ingå i varje försök och resultaten bör falla inom de givna godtagbara gränserna. Om så inte är fallet bör man fastställa orsaken till att acceptanskriterierna inte har uppfyllts (t.ex. hantering av cellerna, serum och antibiotika för kvalitet och koncentration), och därefter upprepa testet. När acceptanskriterierna har uppfyllts är det viktigt att cellodlingsmaterialet används på ett konsekvent sätt för att säkerställa minimal variation av värdena för EC50, PC50 och PC10. De fyra parallella referensstandarderna, som bör ingå i varje försök (som utförs under samma förhållanden, inklusive material, cellernas passagenivå och av samma tekniker), kan säkerställa testets sensitivitet, eftersom PC10 från de tre positiva referensstandarderna bör falla inom ett godtagbart intervall, vilket även gäller PC50 och EC50 om de kan beräknas (se tabell 1).
|
Tabell 1
Godtagbara intervallvärden för de fyra referensstandarderna för ER-agonisttestet
|
Namn
|
logPC50
|
logPC10
|
logEC50
|
Hillslope
|
Testintervall
|
|
17β-östradiol (E2) CAS-nr: 50-28-2
|
–11,4~–10,1
|
< – 11
|
-11,3~–10,1
|
0,7~1,5
|
10-14~10-8M
|
|
17α-östradiol CAS-nr: 57-91-0
|
-9,6~-8,1
|
-10,7~-9,3
|
-9,6~-8,4
|
0,9~2,0
|
10-12~10-6M
|
|
Kortikosteron CAS-nr: 50-22-6
|
—
|
—
|
—
|
—
|
10-10~10-4M
|
|
17α-metyltestosteron CAS-nr: 58-18-4
|
-6,0~-5,1
|
– 8,0~– 6,2
|
—
|
—
|
10-11~10-5M
|
Positiva och negativa referensstandarder för ER-antagonisttestet
|
15.
|
Före och under studien bör testsystemets responsförmåga kontrolleras med hjälp av lämpliga koncentrationer av ett positivt ämne (tamoxifen) och ett negativt ämne (flutamid). Godtagbara intervallvärden som härletts från valideringsstudien (1) anges i tabell 2. Dessa två parallella referensstandarder bör ingå i varje försök och resultaten bör bedömas som korrekta enligt kriterierna. Om så inte är fallet bör man fastställa orsaken till att kriterierna inte har uppfyllts (t.ex. hantering av cellerna, kvalitet och koncentration hos serum och antibiotika), och därefter upprepa testet. Dessutom bör IC50-värden för ett positivt ämne (tamoxifen) beräknas och resultaten bör falla inom givna godtagbara gränser. När acceptanskriterierna har uppfyllts är det viktigt att cellodlingsmaterialet används på ett konsekvent sätt för att säkerställa minimal variation hos IC50-värdena. De två parallella referensstandarderna, som bör ingå i varje försök (som utförs under samma förhållanden, inklusive material, cellernas passagenivå och av samma tekniker), kan säkerställa testets sensitivitet (se tabell 2).
|
Tabell 2
Kriterier och godtagbara intervallvärden för de två referensstandarderna för ER-antagonisttestet
|
Namn
|
Kriterier
|
LogIC50
|
Testintervall
|
|
Tamoxifen CAS-nr: 10 540 -29-1
|
Positivt: IC50 bör beräknas.
|
–5,942~–7,596
|
10-10~10-5M
|
|
Flutamid CAS-nr: 13 311 -84-7
|
Negativt: IC30 bör inte beräknas.
|
—
|
10-10 ~10-5M
|
Positiva kontroller och vehikelkontroller
|
16.
|
Den positiva kontrollen för ER-agonisttest (1 nM av E2) och för ER-antagonisttest (10μM tamoxifen) bör testas i minst tre replikat på varje platta. Den vehikel som används för att lösa upp testkemikalien bör testas som vehikelkontroll i minst tre replikat på varje platta. Om en annan vehikel används i den positiva kontrollen än för testkemikalien, bör en annan vehikelkontroll utöver denna vehikelkontroll testas i minst tre replikat på samma platta med den positiva kontrollen.
|
Kvalitetskriterier för ER-agonisttest
|
17.
|
Medelvärdet för luciferasaktiviteten i den positiva kontrollen (1 nM E2) bör vara minst fyra gånger medelvärdet för den positiva kontrollen på varje platta. Detta kriterium fastställs på grundval av hur tillförlitliga endpoint-värdena från valideringsstudien är (historiskt mellan fyra och trettio gånger).
|
|
18.
|
När det gäller kvalitetskontrollen av testet bör den x-faldiga induktion som motsvarar PC10-värdet för den parallella positiva kontrollen (1 nM E2) vara högre än n 1+2SD av den x-faldiga induktionen (= 1) för den parallella vehikelkontrollen. För prioriteringssyften kan PC10-värdet vara användbart för att förenkla den nödvändiga dataanalysen jämfört med en statistisk analys. En statistisk analys ger visserligen information om signifikans men är inte en kvantitativ parameter för den koncentrationsbaserade potentialen, och är därför mindre användbar i prioriteringssyfte.
|
Kvalitetskriterier för ER-antagonisttest
|
19.
|
Medelvärdet för luciferasaktiviteten i den spikade kontrollen (25 pM E2) bör vara minst fyra gånger medelvärdet för den positiva kontrollen på varje platta. Detta kriterium fastställs på grundval av hur tillförlitliga endpoint-värdena från valideringsstudien är.
|
|
20.
|
Vad gäller kvalitetskontrollen av testet bör den relativa transkriptionella aktiveringen (RTA) av 1 nM E2 vara större än 100 %, RTA av 1μM 4-hydroxitamoxifen (OHT) vara mindre än 40,6 % och RTA av 100 μM Digitonin (Dig) vara mindre än 0 %.
Demonstration av laboratoriets kompetens (se punkt 14 och tabellerna 3 och 4 i ”Komponenter i ER TA-test” i denna testmetod).
|
Vehikel
|
21.
|
Dimetylsulfoxid (DMSO) eller ett lämpligt lösningsmedel vid samma koncentration som används för de olika positiva och negativa kontrollerna samt testkemikalierna bör användas som parallell vehikelkontroll. Testkemikalierna bör lösas upp i ett lämpligt lösningsmedel som är blandbart med cellmediet. Vatten, etanol (renhetsgrad 95 % till 100 %) och DMSO är lämpliga vehiklar. Om DMSO används bör nivån inte överskrida 0,1 % (v/v). För alla vehiklar ska det visas att den maximala volym som används inte är cytotoxisk och inte har en störande verkan på genomförandet av testet.
|
Beredning av testkemikalierna
|
22.
|
Generellt bör testkemikalierna lösas upp i DMSO eller ett annat lämpligt lösningsmedel och spädas ut i omgångar med samma lösningsmedel vid ett gemensamt intervall på 1:10 för att bereda lösningar med utspädning med mediet.
|
Löslighet och cytotoxicitet: överväganden för koncentrationsintervalltest
|
23.
|
Ett förberedande test bör göras för att fastställa lämpligt koncentrationsintervall för den kemikalie som ska testas och för att utröna om testkemikalien har några löslighets- och cytotoxicitetsproblem. Inledningsvis testas kemikalierna upp till den högsta koncentrationen på 1 μl/ml, 1 mg/ml eller 1 mM, beroende på vilken koncentration som är lägst. Baserat på omfattningen av cytotoxicitet eller bristande lösningsförmåga som observerats under det förberedande testet, bör den första definitiva testomgången testa kemikalien vid logaritmisk seriespädning, med utgångspunkt från den högsta godtagbara koncentrationen (t.ex. 1 mM, 100μM, 10μM, osv.). Förekomst av grumlighet, fällningar eller cytotoxicitet ska noteras. Koncentrationerna i den andra och vid behov den tredje testomgången bör om nödvändigt justeras för att bättre karakterisera koncentrationens responskurva och undvika koncentrationer som konstateras vara olösliga eller ger överdriven cytotoxicitet.
|
|
24.
|
För ER-agonister och ER-antagonister kan ökande nivåer av cytotoxicitet avsevärt ändra eller eliminera den typiska sigmoida (S-formiga) responsen, vilket bör vägas in i datatolkningen. Testmetoder för cytotoxicitet som kan ge information om 80 % cellviabilitet bör användas, med hjälp av ett lämpligt test baserat på laboratoriets erfarenhet.
|
|
25.
|
Om resultatet av cytotoxicitetstestet visar att koncentrationen av testkemikalien har minskat cellantalet med 20 % eller mer bör denna koncentration anses vara cytotoxisk, och koncentrationer vid eller över den cytotoxiska koncentrationen bör undantas från utvärderingen.
|
Kemisk exponering och beredning av testplattor
|
26.
|
Förfarandet för kemiska utspädningar (steg-1 och steg-2) samt exponering till cellerna (steg-3) kan utföras enligt följande:
Steg-1: Varje testkemikalie bör spädas ut i omgångar i DMSO eller ett lämpligt lösningsmedel och överföras till hålen i en mikrotiterplatta för att uppnå slutliga seriekoncentrationer enligt det förberedande testet för att bestämma koncentrationsintervall (vanligen i serier av exempelvis 1 mM, 100 μM, 10 μM, 1μM, 100 nM, 10 nM, 1 nM, 100 pM och 10 pM (10-3–10-11 M)) för triplikattestning.
Steg-2: Kemisk utspädning: Späd först ut 1,5 μl av testkemikalien i lösningsmedlet till en volym på 500 μl av mediet.
Steg-3: Kemisk exponering av cellerna: Överför 50 μl spädning till mediet (som förbereddes i steg-2) till en testbrunn som innehåller 104 celler/100 μl/brunn.
Den rekommenderade slutliga volymen av medium som behövs för varje brunn är 150 μl. Testprov och referensstandarder kan fördelas enligt tabellerna 3 och 4.
|
Tabell 3
Exempel på fördelning av koncentrationer av referensstandarderna i testplattan i ER-agonisttestet
|
Rad
|
17α-metyltestosteron
|
Kortikosteron
|
17α-östradiol
|
E2
|
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
konc. 1 (10 μM)
|
→
|
→
|
100 μM
|
→
|
→
|
1 μM
|
→
|
→
|
10 nM
|
→
|
→
|
|
B
|
konc. 2 (1 μM)
|
→
|
→
|
10 μM
|
→
|
→
|
100 nM
|
→
|
→
|
1 nM
|
→
|
→
|
|
C
|
konc. 3 (100 nM)
|
→
|
→
|
1 μM
|
→
|
→
|
10 nM
|
→
|
→
|
100 pM
|
→
|
→
|
|
D
|
konc. 4 (10 nM)
|
→
|
→
|
100 nM
|
→
|
→
|
1 nM
|
→
|
→
|
10 pM
|
→
|
→
|
|
E
|
konc. 5 (1 nM)
|
→
|
→
|
10 nM
|
→
|
→
|
100 pM
|
→
|
→
|
1 pM
|
→
|
→
|
|
F
|
konc. 6 (100 pM)
|
→
|
→
|
1 nM
|
→
|
→
|
10 pM
|
→
|
→
|
0,1 pM
|
→
|
→
|
|
G
|
konc. 7 (10 pM)
|
→
|
→
|
100 pM
|
→
|
→
|
1 pM
|
→
|
→
|
0,01 pM
|
→
|
→
|
|
H
|
VC
|
→
|
→
|
→
|
→
|
→
|
PC
|
→
|
→
|
→
|
→
|
→
|
|
VC: Vehikelkontroll (0,1 % DMSO). PC: Positiv kontroll (1 nM E2)
|
|
27.
|
Referensstandarderna (E2, 17α-östradiol, 17-μετψλτεστοστερονμοχηοκορτικοστερονκκββρρτεσταστιιιαρφεετεστομγΣνγννταβελλτττττHål för positiva kontroller som behandlas med 1 nM av E2 som kan ge maximal induktion av E2 och hål för vehikelkontroll som endast behandlas med DMSO (eller lämpligt lösningsmedel) bör ingå i varje testplatta (tabell 4). Om celler från olika källor (t.ex. olika passageantal, olika partier osv.) används i samma försök bör referensstandarderna testas för varje cellkälla.
|
Tabell 4
Exempel på fördelning av koncentrationer av test- och kontrollkemikalierna i testplattan i ER-agonisttest
|
Rad
|
Testkemikalie 1
|
Testkemikalie 2
|
Testkemikalie 3
|
Testkemikalie 4
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
konc. 1 (10 μM)
|
→
|
→
|
1 mM
|
→
|
→
|
1 μM
|
→
|
→
|
10 nM
|
→
|
→
|
|
B
|
konc. 2 (1 μM)
|
→
|
→
|
100 μM
|
→
|
→
|
100 nM
|
→
|
→
|
1 nM
|
→
|
→
|
|
C
|
konc. 3 (100 nM)
|
→
|
→
|
10 μM
|
→
|
→
|
10 nM
|
→
|
→
|
100 pM
|
→
|
→
|
|
D
|
konc. 4 (10 nM)
|
→
|
→
|
1 μM
|
→
|
→
|
1 nM
|
→
|
→
|
10 pM
|
→
|
→
|
|
E
|
konc. 5 (1 nM)
|
→
|
→
|
100 nM
|
→
|
→
|
100 pM
|
→
|
→
|
1 pM
|
→
|
→
|
|
F
|
konc. 6 (100 pM)
|
→
|
→
|
10 nM
|
→
|
→
|
10 pM
|
→
|
→
|
0,1 pM
|
→
|
→
|
|
G
|
konc. 7 (10 pM)
|
→
|
→
|
1 nM
|
→
|
→
|
1 pM
|
→
|
→
|
0,01 pM
|
→
|
→
|
|
H
|
VC
|
→
|
→
|
→
|
→
|
→
|
PC
|
→
|
→
|
→
|
→
|
→
|
|
VC: Vehikelkontroll (0,1 % DMSO). PC: Positiv kontroll (1 nM E2)
|
Tabell 5
Exempel på fördelning av koncentrationer av referensstandarderna i testplattan i ER-antagonisttest
|
28.
|
För att utvärdera kemikaliers antagonistiska aktivitet bör testhålen i raderna A till G spikas med 25pM E2. Referensstandarderna (tamoxifen och flutamid) bör testas i varje testomgång. Hål för positiva kontroller som behandlats med 1 nM av E2, som kan användas som kvalitetskontroll för cellinjen hERα-HeLa-9 903, hål för vehikelkontroller som behandlats med DMSO (eller lämpligt lösningsmedel), 0,1 % DMSO-hål som behandlats med en tillsats av DMSO till den spikade E2 motsvarande hål med ”spikad kontroll”, hål som behandlats med den slutliga koncentrationen 1 μM OHT och hål som behandlats med 100 μM Dig bör ingå i varje testplatta (tabell 5). De efterföljande testplattorna bör utformas på samma sätt, men utan referensstandardhålen (tabell 6). Om celler från olika källor (t.ex. olika passageantal, olika partier osv.) används i samma försök bör referensstandarderna testas för varje cellkälla.
|
Tabell 6
Exempel på fördelning av koncentrationer av test- och kontrollkemikalierna i testplattan i ER-antagonisttest
|
29.
|
Avsaknaden av kanteffekter bör bekräftas i förekommande fall, och om kanteffekter misstänks bör plattans utformning ändras för att undvika sådana effekter. Man kan till exempel använda en platta utan kanthål.
|
|
30.
|
Efter tillsättningen av kemikalierna bör plattorna inkuberas i en 5 % CO2-inkubator vid 37 ± 1test °C i 20–24 timmar för att inducera rapportgenprodukterna.
|
|
31.
|
Dessa föreningar bör behandlas särskilt försiktigt, eftersom de är mycket flyktiga. I sådana fall kan närliggande kontrollhål skapa falska positiva värden, och detta bör övervägas mot bakgrund av de förväntade och historiska kontrollvärdena. I de enstaka fall där flyktighet kan vara ett problem kan användning av ”plattförseglare” bidra till att effektivt isolera individuella hål under testningen, och rekommenderas därför i dessa fall.
|
|
32.
|
Upprepade definitiva tester av samma kemikalie bör utföras på olika dagar för att säkerställa oberoende.
|
Luciferastest
|
33.
|
En kommersiell luciferastestreagens (t.ex. Steady-Glo® Luciferase Assay System [Promega, E2510 eller motsvarande]) eller ett standardsystem för luciferastest (t.ex. Promega, E1500 eller motsvarande) kan användas för testet så länge acceptanskriterierna är uppfyllda. Testreagenserna bör väljas beroende på sensitiviteten hos den luminometer som ska användas. Om standardsystemet för luciferastest används bör lyseringsreagens för cellodling (t.ex. Promega, E1531 eller motsvarande) användas innan substratet tillförs. Luciferasreagensen bör appliceras enligt tillverkarens anvisningar.
|
DATAANALYS
ER-agonisttest
|
34.
|
För att få fram den relativa transkriptionella aktiviteten jämfört med den positiva kontrollen (1 nM av E2) i ER-agonisttestet kan luminiscenssignalerna från samma platta analyseras enligt följande steg (andra likvärdiga matematiska processer är också godtagbara:
|
Steg 1. Beräkna medelvärdet för vehikelkontrollen.
Steg 2. Subtrahera medelvärdet för vehikelkontrollen från varje hålvärde för att normalisera data.
Steg 3. Beräkna medelvärdet för den normaliserade positiva kontrollen.
Steg 4. Dividera det normaliserade värdet för varje hål i plattan med medelvärdet för den normaliserade positiva kontrollen (PC = 100 %).
Det slutliga värdet för varje hål utgör den relativa transkriptionella aktiviteten för det hålet jämfört med PC-responsen.
Steg 5. Beräkna medelvärdet för den relativa transkriptionella aktiviteten för varje koncentrationsgrupp av testkemikalien. Det finns två dimensioner av responsen: den genomsnittliga transkriptionella aktiviteten (respons) och den koncentration vid vilken responsen inträffar (se följande avsnitt).
Överväganden i fråga om induktion av EC50, PC50 och PC10
|
35.
|
Det krävs en fullständig koncentrationsresponskurva för att beräkna EC50, men detta kanske inte alltid är möjligt eller praktiskt till följd av begränsningar i testkoncentrationsintervallet (till exempel på grund av problem med cytotoxicitet eller löslighet). Eftersom EC50 och högsta induktionsnivå (motsvarande toppvärdet i Hill-ekvationen) är informativa parametrar bör dessa rapporteras om så är möjligt. Lämplig statistisk programvara bör användas för beräkningen av EC50 och högsta induktionsnivå (t.ex. statistikprogramvaran Graphpad Prism). Om Hills logistiska ekvation är tillämplig på koncentrationsresponsdata bör EC50 beräknas enligt följande ekvation (7):
Y = Bottom + (Top-Bottom) / (1+10 exp ((log EC50 -X) x Hill slope)), där
X är koncentrationens logaritm, och
Y är svaret. Y startar i botten och går upp till toppen i en S-kurva. Botten är fastställd till noll i Hills logistiska ekvation.
|
|
36.
|
Följande uppgifter bör lämnas för varje testkemikalie:
RPCMax, som är den maximala responsnivå som induceras av en testkemikalie, uttryckt som en procentenhet av den respons som 1 nM E2 inducerar på samma platta, samt PCMax (den koncentration som sammanhänger med RPCMax), och
för positiva kemikalier, de koncentrationer som inducerar PC10 och, i förekommande fall, PC50.
|
|
37.
|
PCx-värdet kan beräknas genom interpolering mellan två punkter på X-Y-koordinaten, en omedelbart över och en omedelbart under ett PCx-värde. När de datapunkter som ligger omedelbart över och under PCx-värdet har koordinaterna (a,b) respektive (c,d) kan PCx-värdet beräknas med följande ekvation:
log[PCx] = log[c]+(x-d)/(d-b)
|
|
38.
|
PC-värdena beskrivs i figur 1 nedan.
Figur 1
Exempel på härledning av PC-värden PC (1 nM of E2) induceras på varje testplatta.

|
ER-antagonisttest
|
39.
|
För att få fram den relativa transkriptionella aktiviteten (RTA) jämfört med den spikade kontrollen (25 pM av E2) i ER-antagonisttestet kan luminiscenssignalerna från samma platta analyseras enligt följande steg (andra likvärdiga matematiska processer är också godtagbara:
Steg 1. Beräkna medelvärdet för vehikelkontrollen.
Steg 2. Subtrahera medelvärdet för vehikelkontrollen från varje hålvärde för att normalisera data. Steg 3. Beräkna medelvärdet för den normaliserade spikade kontrollen.
Steg 4. Dividera det normaliserade värdet för varje hål i plattan med medelvärdet för den normaliserade spikade kontrollen (spikad kontroll = 100 %).
Det slutliga värdet för varje hål utgör den relativa transkriptionella aktiviteten för det hålet jämfört med responsen på den spikade kontrollen.
Steg 5. Beräkna medelvärdet för den relativa transkriptionella aktiviteten för varje behandling.
|
Överväganden i fråga om induktion av IC30 och IC50
|
40.
|
För positiva kemikalier bör de koncentrationer som inducerar IC30 och i förekommande fall IC50 anges.
|
|
41.
|
ICx-värdet kan beräknas genom interpolering mellan två punkter på X-Y-koordinaten, en omedelbart över och en omedelbart under ett ICx-värde. När de datapunkter som ligger omedelbart över och under ICx-värdet har koordinaterna (c,d) respektive (a,b) kan ICx-värdet beräknas med följande ekvation:
lin ICx = a-(b-(100-x)) (a-c) /(b-d)
Figur 2
Exempel på hur man härleder IC-värden Den spikade kontrollen (25 pM E2) inkluderas på varje testplatta.

RTA: relativ transkriptionsaktivitet.
|
|
42.
|
Resultaten bör baseras på två (eller tre) oberoende testomgångar. Om två testomgångar ger jämförbara och därför reproducerbara resultat är det inte nödvändigt att utföra en tredje testomgång. För att vara godtagbara bör resultaten
|
—
|
uppfylla acceptanskriterierna (se Acceptanskriterier, punkterna 14–20),
|
|
Datatolkningskriterier
Tabell 7
Positiva och negativa beslutskriterier i ER-agonisttest
|
Positiv
|
Om ett RPCMax erhålls som motsvarar eller överskrider 10 % av responsen i den positiva kontrollen i minst två av två eller två av tre testomgångar.
|
|
Negativ
|
Om RPCMax inte når minst 10 % av responsen i den positiva kontrollen i två av två eller två av tre testomgångar.
|
Tabell 8
Positiva och negativa beslutskriterier i ER-antagonisttest
|
Positiv
|
Om IC30 beräknas i minst två av två eller två av tre testomgångar.
|
|
Negativ
|
Om IC30 inte kan beräknas i två av två eller två av tre testomgångar.
|
|
43.
|
Datatolkningskriterierna anges i tabellerna 7 och 8. Positiva resultat karakteriseras av både effektens omfattning och den koncentration vid vilken effekten uppstår. Båda dessa mål uppnås genom att resultaten uttrycks som en koncentration vid vilken 50 % (PC50) eller 10 % (PC10) av PC-värdena uppnås för agonisttestet, och 50 % (IC50) eller 30 % (IC30) av värdet för den spikade kontrollen hämmas för antagonisttestet. Testkemikalien anses dock vara positiv om den maximala respons som induceras av testkemikalien (RPCMax) motsvarar eller överskrider 10 % av den positiva kontrollens respons i minst två av två eller två av tre testomgångar. Testkemikalien anses vara negativ om RPCMax inte uppnår minst 10 % av den positiva kontrollens respons i två av två eller två av tre testomgångar.
|
|
44.
|
Beräkningarna av PC10, PC50 och PCMax i ER-agonisttestet samt IC30 och IC50 i ER-antagonisttestet kan göras med hjälp av ett kalkylblad som finns tillgängligt tillsammans med testriktlinjen på OECD:s offentliga webbplats (63).
|
|
45.
|
Det bör vara tillräckligt att erhålla värden för PC10 eller PC50 respektive IC30 eller IC50 minst två gånger. Om de resulterande basvärdena för data i samma koncentrationsintervall uppvisar variationer med en oacceptabelt hög variationskoefficient (CV, %) kan data anses otillförlitliga. I så fall bör källan till den höga variationen identifieras. Variationskoefficienten av rådatan är tre gånger så hög (dvs. Luminescensintensiteten för de datapunkter som används för beräkningen av PC10 bör vara lägre än 20 %.
|
|
46.
|
Om acceptanskriterierna uppfylls innebär detta att testsystemet fungerar korrekt, men det säkerställer inte att en viss testomgång kommer att ge korrekta data. Att upprepa resultaten från den första testomgången är det bästa sättet att försäkra sig om att korrekta data har erhållits.
|
|
47.
|
När det gäller ER-agonisttest, som kräver mer information utöver screening- och prioriteringsändamålen för denna testriktlinje för positiva testkemikalier, särskilt PC10-PC49-kemikalier och kemikalier som misstänks överstimulera luciferas, kan bekräftelse på att den observerade luciferasaktiviteten endast är en ERα-specifik respons fås med hjälp av en ERα-antagonist (se tillägg 2.1).
|
TESTRAPPORT
|
48.
|
Se punkt 20 i ”Komponenter i ER TA-testet”.
|
LITTERATUR
|
(1)
|
OECD (2015). Report of the Inter-Laboratory Validation for Stably Transfected Transactivation Assay to detect Estrogenic and Anti-estrogenic Activity. Environmental Health and Safety Publications, Series on Testing and Assessment (nr 225), Organisationen för ekonomiskt samarbete och utveckling, Paris.
|
|
(2)
|
Escande A., et al. (2006). Evaluation of Ligand Selectivity Using Reporter Cell Lines Stably Expressing Estrogen Receptor Alpha or Beta, Biochem. Pharmacol., 71, 1 459–1 469.
|
|
(3)
|
Kuiper G.G., et al. (1998). Interaction of Estrogenic Chemicals and Phytoestrogens with Estrogen Receptor Beta, Endocrinol., 139, 4 252–4 263.
|
|
(4)
|
Spaepen M., et al. (1992). Detection of Bacterial and Mycoplasma Contamination in Cell Cultures by Polymerase Chain Reaction, FEMS Microbiol. Lett., 78(1), 89–94.
|
|
(5)
|
Kobayashi H., et al. (1995). Rapid Detection of Mycoplasma Contamination in Cell Cultures by Enzymatic Detection of Polymerase Chain Reaction (PCR) Products, J. Vet. Med. Sci., 57(4), 769–771.
|
|
(6)
|
Dussurget O. och Roulland-Dussoix D. (1994). Rapid, Sensitive PCR-Based Detection of Mycoplasmas in Simulated Samples of Animal Sera, Appl. Environ. Microbiol., 60(3), 953–959.
|
|
(7)
|
De Lean A., Munson P.J. och Rodbard D. (1978). Simultaneous Analysis of Families of Sigmoidal Curves: Application to Bioassay, Radioligand Assay, and Physiological Dose-Response Curves, Am. J. Physiol., 235, E97–El02.
|
Tillägg 2,1
FALSKT POSITIVT VÄRDE: BEDÖMNING AV EJ RECEPTORMEDIERADE LUMINESCENSSIGNALER
|
1.
|
Falska positiva värden i ER-agonisttestet kan genereras av ej ER-medierad aktivering av luciferasgenen eller direkt aktivering av genprodukten eller orelaterad fluorescens. Sådana effekter visar sig genom en ofullständig eller ovanlig dosresponskurva. Om sådana effekter misstänks bör effekten av en ER-antagonist (t.ex. 4-hydroxitamoxifen (OHT) vid icke-toxisk koncentration) på responsen undersökas. Den rena antagonisten ICI 1827 80 är kanske inte lämplig för detta syfte eftersom en tillräcklig koncentration av ICI 1827 80 kan sänka VC-värdet, vilket i sin tur påverkar dataanalysen.
|
|
2.
|
För att säkerställa att detta tillvägagångssätt är giltigt måste följande testas i samma platta:
|
—
|
Den okända kemikaliens agonistiska aktivitet med/utan 10 μM OHT.
|
|
—
|
Vehikelkontroll (i triplikat).
|
|
—
|
1 nM av E2 (i triplikat) som agonistisk positiv kontroll.
|
|
—
|
1 nM av E2 + OHT (i triplikat).
|
|
Datatolkningskriterier
Anmärkning: Samtliga hål bör behandlas med samma koncentration av vehikeln.
|
—
|
Om den okända kemikaliens agonistiska aktivitet INTE påverkas av behandlingen med ER-antagonisten klassificeras den som ”negativ”.
|
|
—
|
I fall där den okända kemikaliens agonistiska aktivitet hämmas fullständigt ska beslutskriterierna tillämpas.
|
|
—
|
Om den agonistiska aktiviteten vid den lägsta koncentrationen motsvarar eller överskrider PC10-responsen hämmas den okända kemikalien i en grad som motsvarar eller överskrider PC10-responsen. Skillnaden i respons mellan de hål som inte har behandlats och de hål som har behandlats med ER-antagonisten beräknas. Denna skillnad bör anses motsvara den verkliga responsen och bör användas för beräkningen av lämpliga parametrar som underlag för ett klassificeringsbeslut.
|
Dataanalys
Kontrollera prestandastandarden.
Kontrollera variationskoefficienten mellan hål som behandlats under samma förhållanden.
|
1.
|
Beräkna variationskoefficientens medelvärde.
|
|
2.
|
Subtrahera variationskoefficientens medelvärde från värdet för varje hål som inte har behandlats med OHT.
|
|
3.
|
Beräkna medelvärdet för OHT.
|
|
4.
|
Subtrahera variationskoefficientens medelvärde från värdet för varje hål som har behandlats med OHT.
|
|
5.
|
Beräkna den positiva kontrollens medelvärde.
|
|
6.
|
Beräkna den relativa transkriptionella aktiviteten för alla andra hål i förhållandet till den positiva kontrollen.
|
Tillägg 2,2
BEREDNING AV SERUM BEHANDLAT MED DEXTRANBELAGT TRÄKOL (DCC)
|
1.
|
Behandling av serum med dextranbelagt träkol (DCC) är en generell metod för att eliminera östrogenföreningar från serum som tillförs cellmedier. Syftet är att utesluta missvisande respons som är förknippad med östrogenrester i serumet. 500 ml fetalt bovinserum (FBS) kan behandlas genom detta förfarande.
|
Komponenter
|
2.
|
Följande material och utrustning kommer att behövas:
|
Material
|
Magnesiumkloridhexahydrat (MgCl2·6H2O).
|
|
1 M HEPES buffertlösning (pH 7.4).
|
|
Ultrarent vatten som producerats från ett filtersystem.
|
Utrustning
Autoklaverad glasbehållare (av lämplig storlek), en vanlig laboratoriecentrifug (som kan hålla en temperatur på 4 oC).
Förfarande
|
3.
|
Följande förfarande har anpassats till användning av 50 ml centrifugprovrör:
[Dag-1] Bered en suspension av dextranbelagt träkol med 1 liter ultrarent vatten som innehåller 1,5 mM MgCl2, 0,25 M sackaros, 2,5 g träkol, 0,25 g dextran och 5 mM HEPES, och rör om vid 4 oC över natten.
[Dag-2] Fördela suspensionen i 50 ml centrifugprovrör och centrifugera vid 10 000 rpm vid 4 oC i 10 minuter. Avlägsna supernatanten och lagra hälften av träkolssedimentet vid 4 oC för användning på dag-3. Suspendera den andra hälften av träkolet med FBS som har tinats något för att undvika fällning och värmeinaktiverats vid 56 oC i 30 minuter, överför sedan träkolet till en autoklaverad glasbehållare, t.ex. en E-kolv. Låt suspensionen stå under omrörning vid 4 oC över natten.
[Dag-3] Fördela suspensionen med FBS i centrifugprovrör för centrifugering vid 10 000 rpm vid 4 oC i 10 minuter. Samla in FBS och överför det till det nya träkolssediment som bereddes och lagrades på dag-2. Suspendera träkolssedimentet och låt suspensionen stå under försiktig omrörning i en autoklaverad glasbehållare vid 4 oC över natten.
[Dag-4] Fördela suspensionen för centrifugering vid 10 000 rpm vid 4 oC i 10 minuter och sterilisera supernatanten genom att filtrera den genom ett 0,2 μm sterilt filter. Denna DCC-behandlade FBS bör lagras vid -20 oC och kan användas i upp till ett år.
|
Tillägg 3
TESTMETOD MED TRANSAKTIVERING AV ÖSTROGENRECEPTORN VM7LUC FÖR IDENTIFIERING AV ÖSTROGENRECEPTORERS AGONISTER OCH ANTAGONISTER
INLEDANDE ÖVERVÄGANDEN OCH BEGRÄNSNINGAR (SE ÄVEN ALLMÄN INLEDNING)
|
1.
|
Denna testmetod använder cellinjen VM7Luc4E2 (64). Den har validerats av National Toxicology Program Interagency Center for the Evaluation of Alternative Toxicological Methods (NICEATM) och Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM) (1). VM7Luc-cellinjerna uttrycker framför allt endogena ERα och en mindre mängd endogent ERβ (2) (3) (4).
|
|
2.
|
Denna metod är tillämplig på många olika ämnen, förutsatt att de är lösliga i dimetylsulfoxid (DMSO, CAS-nr 67-68-5), inte reagerar med DMSO eller cellodlingsmediet och inte är cytotoxiska vid de testade koncentrationerna. Om det inte är möjligt att använda DMSO kan en annan vehikel, såsom etanol eller vatten, användas i stället (se punkt 12). Den demonstrerade prestandan hos (ant)agonistmetoden VM7Luc ER TA tyder på att testet kan ge information om ER-medierade verkningssätt och att det kan övervägas i samband med prioritering av ämnen för ytterligare testning
|
|
3.
|
Denna metod är särskilt utformad för att detektera hERα- och hERß-medierad transaktivering genom att mäta kemiluminiscens som endpoint. Det är vanligt att kemiluminiscens används i biologiska tester, eftersom luminiscens har ett högt signal-brusförhållande. Firefly-luciferasaktiviteten i cellbaserade tester kan dock förväxlas med ämnen som hämmar luciferasenzymet, vilket orsakar både en uppenbar inhibering eller ökad luminiscens på grund av proteinstabilisering. I vissa luciferasbaserade tester med ER-rapportgener har dessutom ej receptormedierade luminiscenssignaler rapporterats vid fytoöstrogena koncentrationer högre än 1 μM till följd av överaktivering av luciferasrapportgenen(9) (11). Dosresponskurvan visar att den faktiska aktiveringen av östrogenreceptorsystemet inträffar vid lägre koncentrationer, men det luciferasuttryck som erhålls vid höga koncentrationer av fytoöstrogener eller liknande föreningar som misstänks producera fytoöstrogenlik överaktivering av luciferasrapportgenen, måste undersökas noggrant i stabilt transfekterade ER TA-testsystem.
|
|
4.
|
Avsnitten ”Allmän inledning” och ”Komponenter i ER TA-testet” bör läsas innan denna testmetod används för lagstadgade ändamål. Definitioner och förkortningar som används i denna testmetod beskrivs i tillägg 1.
|
PRINCIPER FÖR METODEN(SE ÄVEN ALLMÄN INLEDNING)
|
5.
|
Metoden används för att indikera ligandbindning till östrogenreceptorer, följt av translokering av receptorligandkomplexet till kärnan. I kärnan binds receptor–ligand-komplexet till specifika DNA-responselement och transaktiverar rapportgenen (luc). Då produceras luciferas som sedan släpper ut ljus som kan kvantifieras med en luminometer. Luciferasaktivitet kan utvärderas snabbt och till låg kostnad med ett antal kommersiellt tillgängliga kit. VM7Luc ER TA använder VM7, en adenokarcinom cellinje från mänskligt bröst som är mottaglig för östrogenreceptorer, och har transfekterats stabilt med en firefly-luc-rapportgen under kontroll av fyra östrogenresponselement som har placerats uppströms från musens MMTV-promotor för att detektera ämnen med agonistisk eller antagonistisk aktivitet in vitro i östrogenreceptorerna. Denna MMTV-promotor uppvisar endast smärre korsreaktivitet med andra steroida och icke-steroida hormoner (8). Datatolkningskriterierna beskrivs i detalj i punkt 41. Kortfattat identifieras positiv respons genom en koncentrationsresponskurva vid minst tre punkter, utan överlappande felstaplar (medelvärde ± SD), samt en förändring i omfånget (normaliserad relativ ljusenhet [normalised relative light unit RLU]) på minst 20 % av det maximala värdet för referensstandarden (17β-östradiol [E2; CAS-nr 50-28-2] för agonisttestet, raloxifen HCl [Ral, CAS-nr 84 449-90-1]/E2 för antagonisttestet).
|
FÖRFARANDE
Cellinje
|
6.
|
Den stabilt transfekterade cellinjen VM7Luc4E2 bör användas i testet. Cellinjen finns för närvarande endast tillgänglig med ett tekniskt licensavtal från University of California, Davis, Kalifornien, USA (65) och från Xenobiotic Detection Systems Inc., Durham, North Carolina USA (66).
|
Cellinjens stabilitet
|
7.
|
För att bibehålla cellinjens stabilitet och integritet bör cellerna odlas mer än en passage från den frysta stammen i cellunderhållsmediet (se punkt 9). Cellerna bör inte odlas mer än 30 passager. För cellinjen VM7Luc4E2 cell line tar 30 passager omkring tre månader.
|
Betingelser för cellodling och överföring till platta
|
8.
|
De rutiner som anges i vägledningen om god praxis för cellodling (5) (6) bör följas för att säkerställa kvalitet hos alla material och metoder och på så sätt bibehålla arbetets integritet, giltighet och reproducerbarhet.
|
|
9.
|
VM7Luc4E2-cellerna förvaras i ett RPMI 1 640-medium kompletterat med 0,9 % Pen-Strep och 8,0 % fetalt bovinserum (FBS) i en särskild vävnadsodlingsinkubator vid 37 ± 1 °C, 90 % ± 5 % luftfuktighet och 5,0 % ± 1 % CO2/luft.
|
|
10.
|
När VM7Luc4E2-cellerna når ~80 % konfluens subkultiveras och konditioneras de till en östrogenfri miljö i 48 timmar innan cellerna överförs till 96-hålsplattor för exponering för testkemikalierna och analys av östrogenberoende induktion av luciferasaktivitet. Det östrogenfria mediet (EFM) innehåller Dulbecco’s Modification of Eagle’s Medium (DMEM) utan fenolrött, kompletterat med 4,5 % träkols-/dextranbehandlat fetalt bovinserum (FBS), 1,9 % L-glutamin och 0,9 % Pen-Strep. Alla plastvaror bör vara fria från östrogenaktivitet [se detaljerat protokoll (7)].
|
Acceptanskriterier
|
11.
|
Godkännandet eller förkastandet av ett test baseras på en utvärdering av referensstandarden och kontrollresultaten från varje försök, som utförts på en 96-hålsplatta. Varje referensstandard testas i flera koncentrationer och det ska finnas flera prov av varje referens- och kontrollkoncentration. Resultaten jämförs med kvalitetskontrollerna för dessa parametrar som härletts från de historiska databaser över agonistisk och antagonistisk aktivitet som varje laboratorium skapar när de visar sin kompetens. De historiska databaserna uppdateras kontinuerligt med värden för referensstandarden och kontrollerna. Ändringar i utrustning eller laboratorieförhållanden kan göra det nödvändigt att uppdatera de historiska databaserna.
|
Agonisttest
Förberedande test för att bestämma koncentrationsintervall
|
•
|
Induktion: Plattornas induktion bör mätas genom att den genomsnittligt högsta E2-referensstandardens RLU-värde jämförs med det genomsnittliga RLU-värdet för DMSO-kontrollen. Femfaldig induktion uppnås vanligen, men för att godtas måste induktionen vara minst fyrfaldig.
|
|
•
|
DMSO-kontrollresultat: Lösningsmedelskontrollens RLU-värden bör hålla sig inom 2,5 gånger standardavvikelsen för den historiska lösningskontrollens RLU-medelvärde.
|
|
•
|
Försök som inte uppfyller ett av dessa acceptanskriterier bör förkastas och upprepas.
|
Fullständigt test
Det fullständiga testet innefattar acceptanskriterier för det förberedande agonisttestet samt följande:
|
•
|
Resultat för referensstandarden: E2-referensstandardens koncentrationsresponskurva bör vara S-formad och uppvisa minst tre värden inom den linjära delen av koncentrationsresponskurvan.
|
|
•
|
Resultat för positiva kontroller: Metoxiklorkontrollens RLU-värden bör vara högre än DMSO-medelvärdet, plus tre gånger standardavvikelsen från DMSO-medelvärdet.
|
|
•
|
Försök som inte uppfyller ett av acceptanskriterierna bör förkastas och upprepas.
|
Antagonisttest
Förberedande test
|
•
|
Reduktion: Plattornas reduktion mäts genom att den genomsnittligt högsta Ral/E2-referensstandardens RLU-värde jämförs med det genomsnittliga RLU-värdet för DMSO-kontrollen. Femfaldig reduktion uppnås vanligen, men för att godtas måste reduktionen vara minst trefaldig.
|
|
•
|
E2-kontrollresultat. E2-kontrollens RLU-värden bör hålla sig inom 2,5 gånger standardavvikelsen för den historiska E2-kontrollens RLU-medelvärde.
|
|
•
|
DMSO-kontrollresultat: DMSO-kontrollens RLU-värden bör hålla sig inom 2,5 gånger standardavvikelsen för den historiska lösningsmedelskontrollens RLU-medelvärde.
|
|
•
|
Försök som inte uppfyller ett av acceptanskriterierna förkastas och upprepas.
|
Fullständigt test
Det fullständiga testet innefattar acceptanskriterier för det förberedande antagonisttestet samt följande:
|
•
|
Resultat för referensstandarden: Ral/E2-referensstandardens koncentrationsresponskurva bör vara S-formad och uppvisa minst tre värden inom den linjära delen av koncentrationsresponskurvan.
|
|
•
|
Resultat för positiva kontroller: RLU-värdena för tamoxifen-/E2-kontrollen bör vara lägre än E2-kontrollens medelvärde minus tre gånger standardavvikelsen från E2-kontrollens medelvärde.
|
|
•
|
Försök som inte uppfyller ett av acceptanskriterierna förkastas och upprepas.
|
Referensstandarder, positiva kontroller och vehikelkontroller
Vehikelkontroll (agonist- och antagonisttest)
|
12.
|
Den vehikel som används för att lösa upp testkemikalierna bör testas som vehikelkontroll. Den vehikel som användes under valideringen av VM7Luc ER TA-testet var 1 % (v/v) dimetylsulfoxid (DMSO, CAS-nr 67-68-5) (se punkt 24). Om en annan vehikel än DMSO används bör alla referensstandarder, kontroller och testkemikalier i förekommande fall testas i samma vehikel.
|
Referensstandard (förberedande agonisttest)
|
13.
|
Referensstandarden är E2 (CAS-nr 50-28-2). För det förberedande testet består referensstandarden av en seriespädning med fyra koncentrationer av E2 (1,84 x 10-10, 4,59 x 10-11, 1,15 x 10-11 och 2,87 x 10-12 M), och varje koncentration testas i duplikathål.
|
Referensstandard (fullständigt agonisttest)
|
14.
|
E2 för fullständig testning består av en seriespädning på 1:2 med elva koncentrationer (från 3,67 x 10-10 till 3,59 x 10-13 M) E2 i duplikathål.
|
Referensstandard (förberedande antagonisttest)
|
15.
|
Referensstandarden är en kombination av Ral (CAS-nr 84 449-90-1) och E2 (CAS-nr 50-28-2). Ral/E2 för förberedande testning består av en seriespädning med tre koncentrationer av Ral (3,06 x 10-9, 7,67 x 10-10 och 1,92 x 10-10M), plus en fast koncentration (9,18 × 10-11 M) av E2 i duplikathål.
|
Referensstandard (fullständigt antagonisttest)
|
16.
|
Ral/E2 för fullständig testning består av en seriespädning på 1:2 Ral (från 2,45x 10-8 till 9,57 x 10-11M), plus en fast koncentration (9,18 × 10-11 M) av E2 bestående av nio koncentrationer av Ral/E2 i duplikathål.
|
Svag positiv kontroll (agonist)
|
17.
|
Den svaga positiva kontrollen är 9,06 x 10-6 M p,p’-metoxiklor (metoxiklor, CAS-nr 72-43-5) i EFM).
|
Svag positiv kontroll (antagonist)
|
18.
|
Den svaga positiva kontrollen består av tamoxifen (CAS-nr 10 540-29-1) 3.36 x 10-6 M med 9,18 × 10-11 M E2 i EFM.
|
E2-kontroll (endast antagonisttest)
|
19.
|
E2-kontrollerna är 9,18 × 10-11 M E2 i EFM och används som en referens för negativ kontroll.
|
Induktionsförhållande (agonist)
|
20.
|
Induktionen av referensstandardens (E2) luciferasaktivitet mäts genom att E2-referensstandardens högsta genomsnittliga RLU-värde divideras med DMSO-kontrollens genomsnittliga RLU-värde, och resultatet bör inte vara högre än fyrfaldigt.
|
Induktionsreduktion (antagonist)
|
21.
|
Medelvärdet för referensstandardens (Ral/E2) luciferasaktivitet mäts genom att Ral/E2-referensstandardens högsta genomsnittliga RLU-värde divideras med DMSO-kontrollens genomsnittliga RLU-värde, och bör inte vara högre än trefaldigt.
Demonstration av laboratoriets kompetens (se punkt 14 och tabellerna 3 och 4 i ”Komponenter i ER TA-testet” i denna testmetod).
|
Vehikel
|
22.
|
Testkemikalierna bör lösas upp i ett lämpligt lösningsmedel som är blandbart med cellmediet. Vatten, etanol (renhetsgrad 95 % till 100 %) och DMSO är lämpliga vehiklar. Om DMSO används bör nivån inte överskrida 1 % (v/v). För alla vehiklar ska det visas att den maximala volym som används inte är cytotoxisk och inte har en störande verkan på genomförandet av testet. Referensstandarderna och kontrollerna löses upp i 100 % lösningsmedel och späds därefter ned till lämpliga koncentrationer i EFM.
|
Beredning av testkemikalierna
|
23.
|
Testkemikalierna löses upp i 100 % DMSO (eller lämpligt lösningsmedel) och späds därefter ned till lämpliga koncentrationer i EFM. Alla testkemikalier bör få anta rumstemperatur innan de löses upp och späds ut. Nya testkemikalielösningar bör beredas för varje försök. Lösningarna bör inte ha märkbara fällningar eller grumlighet. Referensstandarden och kontrollstammarna kan beredas i bulk. Nya beredningar av den slutliga referensstandarden, kontrollutspädningarna och testkemikalierna bör dock göras för varje försök och användas inom 24 timmar från beredningen.
|
Löslighet och cytotoxicitet: överväganden för koncentrationsintervalltest
|
24.
|
Det förberedande testet består av sju-punkts 1:10 seriespädningar som körs i duplikat. Först testas testkemikalierna upp till den högsta koncentrationen på 1 mg/ml (~1 mM) för agonisttestet och 20 μg/ml (~10 μM) för antagonisttestet. Förberedande test används för att fastställa följande:
|
|
—
|
Startkoncentrationer för testkemikalierna som ska användas under den fullständiga testningen
|
|
—
|
Kemiska utspädningar för testkemikalierna (1:2 eller 1:5) som ska användas under den fullständiga testningen
|
|
25.
|
En bedömning av cellviabilitet/cytotoxicitet ingår i protokollen för agonist- och antagonisttesterna (7) och integreras i de förberedande och fullständiga testerna. Den cytotoxicitetsmetod som användes för att bedöma cellviabilitet under valideringen av VM7Luc ER TA (1) var en skalad kvalitativ visuell observationsmetod. Det är dock även möjligt att använda en kvantitativ metod för att fastställa cytotoxicitet (se protokollet (7)). Data från testkemikaliekoncentrationer som orsakar mer än 20 % reduktion av viabiliteten kan inte användas.
|
Testkemikaliens exponering och beredning av testplattor
|
26.
|
Cellerna räknas och överförs till 96-hålsplattor för vävnadsodling (2 x 105 celler per hål) i EFM och inkuberas i 24 timmar så att cellerna fäster vid plattan. EFM avlägsnas och ersätts med test- och referenskemikalierna i EFM och inkuberas i 19–24 timmar. Det är viktigt att vara särskilt försiktig med mycket flyktiga ämnen, eftersom närliggande kontrollhål kan skapa falska positiva resultat. I sådana fall kan användning av ”plattförseglare” bidra till att effektivt isolera individuella hål under testningen, och rekommenderas därför i dessa fall.
|
Förberedande tester för att bestämma koncentrationsintervall
|
27.
|
Vid den förberedande testningen används alla hål i 96-hålsplattan för att testa kemikalier som sju-punkts 1:10 seriespädningar i duplikat (se figurerna 1 och 2).
|
|
—
|
I den förberedande testningen av agonister används fyra koncentrationer av E2 i duplikat som referensstandard och fyra replikathål för DMSO-kontrollen.
|
|
—
|
I den förberedande testningen av antagonister används tre koncentrationer av Ral/E2 med 9,18 × 10-11 M E2 i duplikat som referensstandard, med tre replikathål för E2- och DMSO-kontrollerna.
|
Figur 1
Utformning av förberedande agonisttest med 96-hålsplatta
Förkortningar: E2-1 till E2-4 = koncentrationer av referensstandarden E2 (från hög till låg). TC1-1 till TC1-7 = koncentrationer (från hög till låg) av testkemikalie 1 (TC1). TC2-1 till TC2-7 = koncentrationer (från hög till låg) av testkemikalie 2 (TC2). TC3-1 till TC3-7 = koncentrationer (från hög till låg) av testkemikalie 3 (TC3). TC4-1 till TC4-7 = koncentrationer (från hög till låg) av testkemikalie 4 (TC4). TC5-1 till TC5-7 = koncentrationer (från hög till låg) av testkemikalie 5 (TC5). TC6-1 till TC6-7 = koncentrationer (från hög till låg) av testkemikalie 6 (TC6). VC = vehikelkontroll (DMSO [1 % v/v EFM.]).
Figur 2
Utformning av förberedande antagonisttest med 96-hålsplatta
Förkortningar: E2 = E2-kontroll. Ral-1 till Ral-3 = koncentrationer av referensstandarden raloxifen/E2 (från hög till låg). TC1-1 till TC1-7 = koncentrationer (från hög till låg) av testkemikalie 1 (TC1). TC2-1 till TC2-7 = koncentrationer (från hög till låg) av testkemikalie 2 (TC2). TC3-1 till TC3-7 = koncentrationer (från hög till låg) av testkemikalie 3 (TC3). TC4-1 till TC4-7 = koncentrationer (från hög till låg) av testkemikalie 4 (TC4). TC5-1 till TC5-7 = koncentrationer (från hög till låg) av testkemikalie 5 (TC5). TC6-1 till TC6-7 = koncentrationer (från hög till låg) av testkemikalie 6 (TC6). VC = vehikelkontroll (DMSO [1 % v/v EFM.]).
Anmärkning: Alla testkemikalier testas i närvaro av 9,18 × 10-11 M E2.
|
28.
|
Den rekommenderade slutliga volymen av medium som behövs för varje brunn är 200 μl. Använd endast testplattor där cellerna i alla hål ger en viabilitet på minst 80 %.
|
|
29.
|
Fastställande av startkoncentrationer för fullständig
agonist
-testning beskrivs i detalj i agonistprotokollet (7). Kortfattat används följande kriterier:
|
|
—
|
Om det inte finns några punkter på testkemikaliens kurva som är högre än medelvärdet plus tre gånger standardavvikelsen för DMSO-kontrollen görs ett fullständigt test med användning av elva-punkts 1:2 seriespädning, med den maximala lösliga koncentrationen som utgångspunkt.
|
|
—
|
Om det inte finns några punkter på testkemikaliens kurva som är högre än medelvärdet plus tre gånger standardavvikelsen för DMSO-kontrollen bör den startkoncentration som används för elva-punkts spädningsschemat i fullständig testning vara en log högre än den koncentration som ger det högsta justerade RLU-värdet i det förberedande koncentrationsintervalltestet. Elva-punktsschemat baseras antingen på en 1:2- eller 1:5-spädning enligt följande kriterier:
|
En elva-punkts 1:2-seriespädning bör användas om det resulterande koncentrationsintervallet omfattar all respons baserat på den koncentrationsresponskurva som fåtts fram genom det förberedande testet. Annars bör en 1:5-spädning användas.
|
|
|
—
|
Om testkemikalien uppvisar en bifasisk koncentrationsresponskurva i det förberedande testet bör båda faserna även upplösas i den fullständiga testningen.
|
|
30.
|
Fastställande av startkoncentrationer för fullständig
antagonist
-testning beskrivs i detalj i antagonistprotokollet (7). Kortfattat används följande kriterier:
|
|
—
|
Om det inte finns några punkter på testkemikaliens koncentrationskurva som är lägre än medelvärdet minus tre gånger standardavvikelsen för E2-kontrollen görs ett fullständigt test med användning av elva-punkts 1:2 seriespädning, med den maximala lösliga koncentrationen som utgångspunkt.
|
|
—
|
Om det inte finns några punkter på testkemikaliens kurva som är lägre än medelvärdet minus tre gånger standardavvikelsen för E2-kontrollen bör den startkoncentration som används för elva-punkts spädningsschemat i fullständig testning vara en av följande:
|
—
|
Den koncentration som ger det lägsta justerade RLU-värdet i det förberedande testet.
|
|
—
|
Den maximala lösliga koncentrationen (se antagonistprotokollet (7), figur 14-2).
|
|
—
|
Den lägsta cytotoxiska koncentrationen (se relaterat exempel i antagonistprotokollet (7), figur 14-3).
|
|
|
—
|
Elva-punktsschemat baseras antingen på en 1:2- eller 1:5-spädning enligt följande kriterier:
|
En elva-punkts 1:2-seriespädning bör användas om det resulterande koncentrationsintervallet omfattar all respons baserat på den koncentrationsresponskurva som fåtts fram genom det förberedande testet. Annars bör en 1:5-spädning användas.
|
|
Fullständiga tester
|
31.
|
Det fullständiga testet består av elva-punkts seriespädningar (antingen 1:2- eller 1:5-seriespädningar baserat på startkoncentrationen för kriterierna för fullständig testning), där varje koncentration testas i tre hål i 96-hålsplattan (se figurerna 3 och 4).
|
|
—
|
I den fullständiga testningen av agonister används elva koncentrationer av E2 i duplikat som referensstandard. Fyra replikathål för DMSO-kontrollen och fyra replikathål för metoxiklorkontrollen (9,06 x 10-6 M) finns med i varje platta.
|
|
—
|
I den fullständiga testningen av antagonister används nio koncentrationer av Ral/E2 med 9,18 × 10-11 M E2 i duplikat som referensstandard, med fyra replikathål för E2-kontrollen med 9,18 x 10-11 M, fyra replikathål för DMSO-kontroller och fyra replikathål för tamoxifen 3,36 x 10-6M.
|
Figur 3
Utformning av fullständigt test av agonister med en 96-hålsplatta

Förkortningar: TC1-1 till TC1-11 = koncentrationer (från hög till låg) av testkemikalie 1. TC2-1 till TC2-11 = koncentrationer (från hög till låg) av testkemikalie 2. E2-1 till E2-11 = koncentrationer av referensstandarden E2 (från hög till låg). Meth = p,p’-metoxiklor, svag positiv kontroll. VC = DMSO (1 % v/v) EFM vehikelkontroll.
Figur 4
Utformning av fullständigt test av antagonister med en 96-hålsplatta
Förkortningar: E2 = E2-kontroll. Ral-1 till Ral-9 = koncentrationer av referensstandarden raloxifen/E2 (från hög till låg). Tam = tamoxifen/E2, svag positiv kontroll. TC1-1 till TC1-11 = koncentrationer (från hög till låg) av testkemikalie 1 (TC1). TC2-1 till TC2-11 = koncentrationer (från hög till låg) av testkemikalie 2 (TC2). VC = vehikelkontroll (DMSO [1 % v/v EFM.]).
Anmärkning: Såsom har påpekats innehåller alla referens- och testhål en fast koncentration av E2 (9,18 x 10-11M).
|
32.
|
Upprepade fullständiga tester av samma kemikalie bör utföras på olika dagar för att säkerställa oberoende. Minst två fullständiga tester bör utföras. Om testresultaten motsäger varandra (t.ex. om ett test är positivt och det andra negativt) eller om ett av testerna är olämpligt bör ett ytterligare tredje test utföras.
|
Mätning av luminescens
|
33.
|
Luminescens mäts i intervallet 300 till 650 nm med användning av en injektionsluminometer och programvara som kontrollerar injiceringsvolymen och mätintervallet (7). Ljusemissionen från varje hål uttrycks som RLU per hål.
|
DATAANALYS
Bestämning av EC50 /IC50
|
34.
|
EC50-värdet (halva den maximala effektiva koncentrationen av testkemikalien [agonister]) och IC50-värdet (halva den maximala hämmande koncentrationen av testkemikalien [antagonister]) fastställs utifrån koncentrationsresponsdata. För testkemikalier som är positiva vid en eller flera koncentrationer beräknas den koncentration av testkemikalien som orsakar halva den maximala responsen (IC50 eller EC50) med hjälp av en Hillfunktionsanalys eller ett lämpligt alternativ. Hillfunktionen är en logistisk matematisk modell med fyra parametrar som kopplar testkemikaliekoncentrationen till responsen (som vanligen följer en S-kurva), med användning av nedanstående ekvation:
|

Där
Y= respons (dvs. RLU-värden),
X= koncentrationens logaritm.
Bottom= lägsta respons.
Top= högsta respons.
lg EC50 (eller lg IC50)= logaritmen för X som responsen halvvägs mellan topp och botten.
Hillslope= kurvans lutning.
Modellen beräknar den bästa passningen för parametrarna Top, Bottom, Hillslope samt IC50 och EC50. Lämplig statistisk programvara bör användas för beräkningen av värdena för EC50 och IC50 (t.ex. den statistiska programvaran Graphpad PrismR).
Bestämning av avvikande värden (”outliers”)
|
35.
|
En god statistisk bedömning kan underlättas genom att inbegripa (men inte begränsat till) Q-testet (se agonist- och antagonistprotokollen (7)) för att bestämma ”oanvändbara” hål som utesluts från dataanalysen.
|
|
36.
|
För replikat av E2-referensstandarden (två per koncentration) betraktas alla justerade RLU-värden för ett replikat vid en given E2-koncentration som avvikande värden om dess värde ligger mer än 20 % över eller under det justerade RLU-värdet för den koncentrationen i den historiska databasen.
|
Insamling och justering av luminometerdata för förberedande koncentrationsintervalltestning
|
37.
|
Rådata från luminometern bör överföras till en kalkylbladsmall som har utformats för testet. Det bör bestämmas om det finns datapunkter som visar avvikande värden som måste tas bort. (Se testacceptanskriterier för de parametrar som fastställs i analyserna.) Följande beräkningar bör göras:
|
Agonist
|
Steg 1
|
Beräkna medelvärdet för DMSO-vehikelkontrollen (VC).
|
|
Steg 2
|
Subtrahera medelvärdet för DMSO-vehikelkontrollen från varje hålvärde för att normalisera data.
|
|
Steg 3
|
Beräkna medelvärdet för referensstandardens induktionsförhållande (E2).
|
|
Steg 4
|
Beräkna medelvärdet för EC50-värdet för testkemikalierna.
|
Antagonist
|
Steg 1
|
Beräkna medelvärdet för DMSO-vehikelkontrollen.
|
|
Steg 2
|
Subtrahera medelvärdet för DMSO-vehikelkontrollen från varje hålvärde för att normalisera data.
|
|
Steg 3
|
Beräkna medelvärdet för referensstandardens reduktionsförhållande (Ral/E2).
|
|
Steg 4
|
Beräkna medelvärdet för E2-referensstandarden.
|
|
Steg 5
|
Beräkna medelvärdet för IC50-värdet för testkemikalierna.
|
Insamling och justering av luminometerdata för fullständig testning
|
38.
|
Rådata från luminometern bör överföras till en kalkylbladsmall som har utformats för testet. Det bör bestämmas om det finns datapunkter som visar avvikande värden som måste tas bort. (Se testacceptanskriterier för de parametrar som fastställs i analyserna.) Följande beräkningar görs:
|
Agonist
|
Steg 1
|
Beräkna medelvärdet för DMSO-vehikelkontrollen.
|
|
Steg 2
|
Subtrahera medelvärdet för DMSO-vehikelkontrollen från varje hålvärde för att normalisera data.
|
|
Steg 3
|
Beräkna medelvärdet för referensstandardens induktionsförhållande (E2).
|
|
Steg 4
|
Beräkna medelvärdet för EC50-värdet för E2 och testkemikalierna.
|
|
Steg 5
|
Beräkna det justerade RLU-medelvärdet för metoxiklor.
|
Antagonist
|
Steg 1
|
Beräkna medelvärdet för DMSO-vehikelkontrollen.
|
|
Steg 2
|
Subtrahera medelvärdet för DMSO-vehikelkontrollen från varje hålvärde för att normalisera data.
|
|
Steg 3
|
Beräkna medelvärdet för referensstandardens induktionsförhållande (Ral/E2).
|
|
Steg 4
|
Beräkna medelvärdet för IC50-värdet för Ral/E2 och testkemikalierna.
|
|
Steg 5
|
Beräkna det justerade RLU-medelvärdet för tamoxifen.
|
|
Steg 6
|
Beräkna medelvärdet för E2-referensstandarden.
|
Datatolkningskriterier
|
39.
|
VM7Luc ER TA är avsett att ingå i en sammanvägd bedömning för att underlätta prioritering av ämnen för ED-testning in vivo. Ett led i detta prioriteringsförfarande är att klassificera testkemikalien som positiv eller negativ för antingen ER-agonistisk eller ER-antagonistisk aktivitet. De positiva och negativa beslutskriterier som används i valideringsstudien för VM7Luc ER TA beskrivs i tabell 1.
|
Tabell 1
Positiva och negativa beslutskriterier
|
|
|
AGONISTISK AKTIVITET
|
|
Positiv
|
|
—
|
Alla testkemikalier som har klassificerats som positiva för ER-agonistisk aktivitet bör ha en koncentrationsresponskurva som består av en baslinje som följs av en positiv lutning och avslutas i en platå eller topp. I vissa fall är det endast möjligt att fastställa två av dessa egenskaper (baslinje–lutning eller lutning–topp).
|
|
—
|
Den linje som anger den positiva lutningen bör innehålla minst tre punkter med ej överlappande felstaplar (medelvärde ± SD). De punkter som formar baslinjen utesluts, men den linjära delen av kurvan kan omfatta toppen eller den första punkten i platån.
|
|
—
|
En positiv klassificering kräver en responsamplitud, skillnaden mellan baslinje och topp, på minst 20 % av det maximala värdet för referensstandarden E2 (dvs. 2 000 RLU eller mer när referensstandardernas [E2] värde justeras upp till 10 000 RLU).
|
|
—
|
Om möjligt bör ett EC50-värde beräknas för varje positiv testkemikalie.
|
|
|
Negativ
|
Det genomsnittliga justerade RLU för en given koncentration motsvarar eller är lägre än RLU-medelvärdet för DMSO-kontrollen plus tre gånger standardavvikelsen för DMSO:s RLU.
|
|
Otillräcklig
|
Data som inte kan tolkas som att de visar närvaro eller avsaknad av aktivitet på grund av betydande kvalitativa eller kvantitativa begränsningar anses vara olämpliga, och kan inte användas för att fastställa om testkemikalien är positiv eller negativ. Kemikalierna bör omtestas.
|
|
|
|
ANTAGONISTISK AKTIVITET
|
|
Positiv
|
|
—
|
Testkemikaliens data skapar en koncentrationsresponskurva som består av en baslinje, följd av en negativ lutning.
|
|
—
|
Den linje som anger den negativa lutningen bör innehålla minst tre punkter med ej överlappande felstaplar. De punkter som formar baslinjen utesluts, men den linjära delen av kurvan kan omfatta den första punkten i platån.
|
|
—
|
Det bör finnas en reduktion på minst 20 % av aktiviteten jämfört med det maximala värdet för referensstandarden Ral/E2 (dvs. 8 000 RLU eller mindre när det maximala värdet för referensstandarden [Ral/E2] justeras till 10 000 RLU).
|
|
—
|
Testkemikaliens högsta icke-cytotoxiska koncentrationer bör vara lägre än eller motsvarande 1 x 10-5 M.
|
|
—
|
Om möjligt bör ett IC50-värde beräknas för varje positiv testkemikalie.
|
|
|
Negativ
|
Alla datapunkter över EC80-värdet (80 % av E2-responsen, eller 8 000 RLU), vid koncentrationer lägre än 1,0 x 10-5 M.
|
|
Otillräcklig
|
Data som inte kan tolkas som att de visar närvaro eller avsaknad av aktivitet på grund av betydande kvalitativa eller kvantitativa begränsningar anses vara olämpliga, och kan inte användas för att fastställa om testkemikalien är positiv eller negativ. Kemikalien bör omtestas.
|
|
40.
|
Positiva resultat karakteriseras av både effektens omfattning och om så är möjligt den koncentration vid vilken effekten uppstår. Exempel på positiva, negativa och otillräckliga data visas i figurerna 5 och 6.
|
Figur 5
Exempel på agonister Positiva, negativa och otillräckliga data
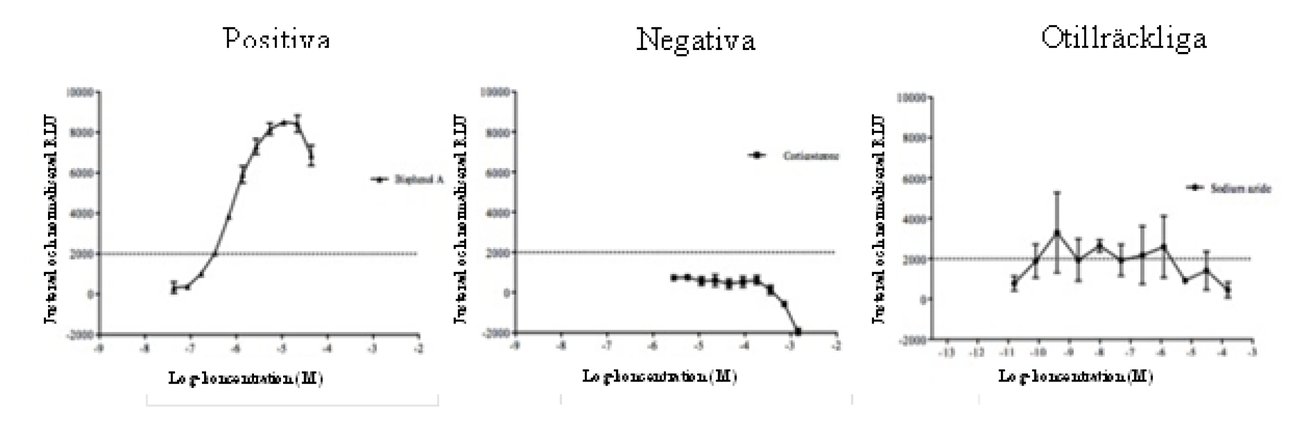
Den streckade linjen anger 20 % av E2-responsen, 2 000 justerade och normaliserade RLU.
Figur 6
Exempel på antagonister Positiva, negativa och otillräckliga data
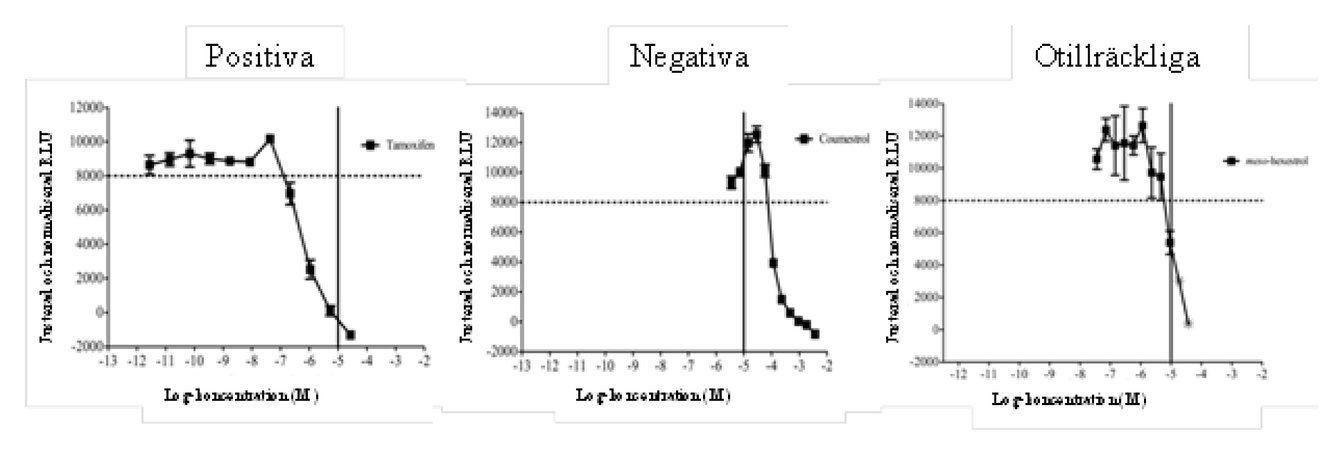
Den streckade linjen anger 80 % av Ral/E2-responsen, 8 000 justerade och normaliserade RLU.
Den heldragna linjen anger 1,00 x 10-5 M. För att en respons ska anses vara positiv bör den ligga under 8 000 RLU-linjen och vid koncentrationer lägre än 1,00 x 10-5M.
Koncentrationer märkta med en asterisk i meso-hexestroldiagrammet anger viabilitetspoäng på 2 eller högre.
Testresultaten för meso-hexestrol anses utgöra olämpliga data, eftersom den enda responsen under 8 000 RLU förekommer vid 1,00 x 10-5M.
|
41.
|
Beräkningarna av EC50 och IC50 kan göras med hjälp av en Hillfunktion med fyra parametrar (se agonist- och antagonistprotokollen för närmare uppgifter (7)). Om acceptanskriterierna uppfylls innebär detta att systemet fungerar korrekt, men det säkerställer inte att en viss testomgång kommer att ge korrekta data. Att upprepa resultaten från den första testomgången är det bästa sättet att försäkra sig om att korrekta data har erhållits (se punkt19 i ”Komponenter i ER TA-testet”).
|
TESTRAPPORT
|
42.
|
Se punkt 20 i ”Komponenter i ER TA-testet”.
|
LITTERATUR
|
(1)
|
ICCVAM (2011). ICCVAM Test Method Evaluation Report on the LUMI-CELL® ER (BG1Luc ER TA) Test Method: An In Vitro Method for Identifying ER Agonists and Antagonists, National Institute of Environmental Health Sciences: Research Triangle Park, NC.
|
|
(2)
|
Monje P., Boland R. (2001). Subcellular Distribution of Native Estrogen Receptor α and β Isoforms in Rabbit Uterus and Ovary, J. Cell Biochem., 82(3): 467-479.
|
|
(3)
|
Pujol P., et al. (1998). Differential Expression of Estrogen Receptor-Alpha and -Beta Messenger RNAs as a Potential Marker of Ovarian Carcinogenesis, Cancer Res., 58(23): 5 367-5 373.
|
|
(4)
|
Weihua Z., et al. (2000). Estrogen Receptor (ER) β, a Modulator of ERα in the Uterus, Proceedings of the National Academy of Sciences of the United States of America 97(11): 936-5 941.
|
|
(5)
|
Balls M., et al. (2006). The Importance of Good Cell Culture Practice (GCCP), ALTEX, 23 (Suppl): s. 270-273.
|
|
(6)
|
Coecke S., et al. (2005). Guidance on Good Cell Culture Practice: a Report of the Second ECVAM Task Force on Good Cell Culture Practice, Alternatives to Laboratory Animals, 33: s. 261-287.
|
|
(7)
|
ICCVAM (2011). ICCVAM Test Method Evaluation Report, The LUMI-CELL® ER (BG1Luc ER TA) Test Method: An In Vitro Assay for Identifying Human Estrogen Receptor Agonist and Antagonist Activity of Chemicals, NIH Publication nr 11–7 850.
|
|
(8)
|
Rogers J.M., Denison M.S. (2000). Recombinant Cell Bioassays for Endocrine Disruptors: Development of a Stably Transfected Human Ovarian Cell Line for the Detection of Estrogenic and Anti-Estrogenic Chemicals, In Vitro Mol. Toxicol.,13(1):67–82.
|
|
(9)
|
Escande A., et al. (2006). Evaluation of Ligand Selectivity Using Reporter Cell Lines Stably Expressing Estrogen Receptor Alpha or Beta, Biochem. Pharmacol., 71(10):1 459–1 469.
|
|
(10)
|
Thorne N., Inglese J., Auld D.S. (2010). Illuminating Insights into Firefly Luciferase and Other Bioluminescent Reporters Used in Chemical Biology, Chemistry and Biology,17(6): 646–657.
|
|
(11)
|
Kuiper G.G., et al. (1998). Interaction of Estrogenic Chemicals and Phytoestrogens with Estrogen Receptor Beta, Endocrinology,139(10):4 252–4 263.
|
|
(12)
|
Geisinger, et al.(1989). Characterization of a human ovarian carcinoma cell line with estrogen and progesterone receptors, Cancer 63, 280–288.
|
|
(13)
|
Baldwin, et.al. (1998). BG-1 ovarian cell line: an alternative model for examining estrogen-dependent growth in vitro, In Vitro Cell. Dev. Biol. – Animal, 34, 649–654.
|
|
(14)
|
Li, Y., et al. (2014). Research resource: STR DNA profile and gene expression comparisons of human BG-1 cells and a BG-1/MCF-7 clonal variant, Mol. Endo. 28, s. 2072–2081.
|
|
(15)
|
Rogers, J.M. och Denison, M.S. (2000). Recombinant cell bioassays for endocrine disruptors:development of a stably transfected human ovarian cell line for the detection of estrogenicand anti-estrogenic chemicals, In Vitro & Molec. Toxicol. 13, s. 67-82.
|
Tillägg 4
STABILT TRANSFEKTERAD HUMAN ÖSTROGENRECEPTOR-Α – TRANSAKTIVERINGSTEST FÖR DETEKTION AV ÖSTROGENAGONISTISK OCH -ANTAGONISTISK AKTIVITET HOS KEMIKALIER MED ANVÄNDNING AV CELLINJEN ERΑ CALUX
INLEDANDE ÖVERVÄGANDEN OCH BEGRÄNSNINGAR (SE ÄVEN ALLMÄN INLEDNING)
|
1.
|
Denna ERα CALUX-transaktiveringstest använder den humana U2OS-cellinjen för att detektera östrogenagonistisk och -antagonistisk aktivitet, medierad genom human östrogenreceptor alfa (hERα). Valideringsstudien av det biologiska testet Stabilt transfekterad ERα CALUX, som utfördes av BioDetection Systems BV (Amsterdam, Nederländerna), visade att denna metod är relevant och tillförlitlig för det avsedda syftet (1). Cellinjen ERα CALUX uttrycker endast stabilt transfekterad human ERα (2) (3).
|
|
2.
|
Denna metod är särskilt utformad för att detektera hERα-medierad transaktivering genom att mäta bioluminiscens som endpoint. Bioluminiscens används ofta i biologiska tester på grund av dess höga signal-brusförhållande (4).
|
|
3.
|
Fytoöstrogenkoncentrationer högre än 1 μM har rapporterats överaktivera luciferasrapportgenen, vilket leder till ej receptormedierad luminescens (5) (6) (7). Högre koncentrationer av fytoöstrogener eller andra liknande föreningar som kan överaktivera luciferasuttrycket måste därför undersökas mycket noggrant i tester med stabilt transfekterad ER-transaktivering (se tillägg 2).
|
|
4.
|
Avsnitten ”Allmän inledning” och ”Komponenter i ER TA-testet” bör läsas innan denna testmetod används för lagstadgade ändamål. Definitioner och förkortningar som används i denna testmetod beskrivs i tillägg 1.
|
PRINCIPER FÖR METODEN(SE ÄVEN ALLMÄN INLEDNING)
|
5.
|
DET biologiska testet används för att bedöma ligandbindning till östrogenreceptorer, följt av translokering av receptor–ligand-komplexet till kärnan. I kärnan binder receptor–ligand-komplexet specifika DNA-responselement och transaktiverar en firefly-luciferasrapportgen, vilket leder till ökat celluttryck hos luciferasenzymen. Efter tillsats av luciferassubstratet luciferin omvandlas luciferinet till en bioluminiscent produkt. Det ljus som alstras kan enkelt upptäckas och kvantifieras med hjälp av en luminometer.
|
|
6.
|
Testsystemet använder stabilt transfekterade ERα CALUX-celler. ERα CALUX-celler med ursprung från cellinjen U2OS från humant osteoblastiskt osteosarkom. Humana U2OS-celler transfekterades stabilt med 3xHRE-TATA-Luc och pSG5-neo-hERα, med användning av metoden för samfällning med kalciumfosfat. U2OS-cellinjen valdes som det den bästa kandidaten för att fungera som östrogenresponsiv (och andra steorida hormoner) rapportcellinje, baserat på iakttagelsen att U2OS-cellinjen uppvisar låg eller ingen endogen receptoraktivitet. Frånvaron av endogena receptorer bedömdes endast med användning av luciferasrapportplasmider, som inte visade någon aktivitet när receptorligander tillsattes. Denna cellinje stödde även starka hormonmedierade responser när kognata receptorer infördes transient (2) (3) (8).
|
|
7.
|
Testningen av kemikaliers östrogena eller antiöstrogena aktivitet med hjälp av cellinjen ERα CALUX omfattar ett förberedande test och fullständiga testomgångar. Under den förberedande testet bestäms löslighet, cytotoxicitet och ett förfinat koncentrationsintervall för testkemikalierna inför den fullständiga testningen. Under de fullständiga testomgångarna testas testkemikaliernas förfinade koncentrationsintervall i ERα CALUX-testerna, för att därefter klassificera testkemikalierna som agonistiska eller antagonistiska.
|
|
8.
|
Datatolkningskriterierna beskrivs i detalj i punkt 59. Kortfattat anses en testkemikalie vara positiv för agonistisk aktivitet om minst två på varandra följande koncentrationer av testkemikalien visar en respons som motsvarar eller är högre än 10 % av den maximala responsen för referensstandarden 17β-östradiol (PC10). En testkemikalie anses vara positiv för antagonistisk aktivitet om minst två på varandra följande koncentrationer av testkemikalien visar en respons som motsvarar eller är lägre än 80 % av den maximala responsen för referensstandarden tamoxifen (PC80).
|
FÖRFARANDE
Cellinjer
|
9.
|
Den stabilt transfekterade cellinjen U2OS ERα CALUX bör användas i testet. Cellinjen kan erhållas från BioDetection Systems BV, Amsterdam, Nederländerna med ett tekniskt licensavtal.
|
|
10.
|
Endast mykoplasmafria cellodlingar bör användas. De använda cellsatserna bör ha certifierats som negativa för mykoplasmaförorening. I annat fall bör ett mykoplasmatest utföras före användning. RT-PCR (Real Time Polymerase Chain Reaktion) bör användas för känslig detektion av mykoplasmainfektion (9).
|
Cellinjens stabilitet
|
11.
|
För att bibehålla CALUX-cellernas stabilitet och integritet bör cellerna lagras i flytande kväve (–80 oC). När cellerna har tinats upp för att starta en ny odling bör de subkultiveras minst två gånger innan de används för att bedöma östrogenagonistisk och -antagonistisk aktivitet hos kemikalier. Cellerna bör inte subkultiveras för mer än 30 passager.
|
|
12.
|
För att övervaka cellinjens stabilitet över tiden bör det agonistiska och antagonistiska testsystemets respons kontrolleras genom en utvärdering av referensstandardens värden EC50 eller IC50. Dessutom bör det positiva och det negativa kontrollurvalets relativa induktion övervakas. Resultaten bör överensstämma med acceptanskriterierna för det agonistiska (tabell 3C) eller det antagonistiska ERα CALUX-biotestet (tabell 4C). Referensstandarder och positiva och negativa kontroller anges i tabellerna 1 och 2 för den agonistiska respektive antagonistiska varianten.
|
Betingelser för cellodling och överföring till platta
|
13.
|
U2OS-cellerna bör odlas i ett odlingsmedium (DMEM/F12 (1:1) med fenolrött som pH-indikator, kompletterat med fetalt bovinserum (7,5 %), icke-essentiella aminosyror (1 %), 10 enheter/ml penicillin, streptomycin och geneticin (G-418) som urvalsmarkör). Cellerna bör placeras i en CO2-inkubator (5 % CO2) vid 37 °C och 100 % luftfuktighet. När cellerna når en konfluens på 85–95 % bör de antingen subkultiveras eller beredas för sådd i 96-håls mikrotiterplattor. I det senare fallet bör cellerna resuspenderas vid 1 x 105 celler/ml i ett östrogenfritt testmedium (DMEM/F12 (1:1) utan fenolrött, kompletterat med dextranbelagt träkol som behandlats med fetalt bovinserum (5 % v/v), icke-essentiella aminosyror (1 %) och 10 enheter/ml penicillin, streptomycin) och överföras till 96-hålsplattorna (100 μl homogeniserad cellsuspension). Cellerna bör förinkuberas i en CO2-inkubator (5 % CO2, 37 °C, 100 % luftfuktighet) i 24 timmar före exponering. Plasten bör vara östrogenfri.
|
Acceptanskriterier
|
14.
|
Testkemikaliernas agonistiska och antagonistiska aktivitet testas i serier. En testserie består av högst sex mikrotiterplattor. Varje testserie består av minst en hel serie av utspädningar av en referensstandard, ett positivt kontrollprov, ett negativt kontrollprov och lösningsmedelskontroller. I figurerna 1 och 2 beskrivs beredningen av plattor för agonistiska och antagonistiska testserier.
|
|
15.
|
Varje utspädning av referensstandarderna, testkemikalierna, alla lösningsmedelskontroller samt de positiva och negativa kontrollerna bör analyseras i triplikat. Varje triplikatanalys bör uppfylla kraven i tabellerna 3A och 4A.
|
|
16.
|
En komplett serie av utspädningar av referensstandarden (17β-östradiol för agonistisk aktivitet, tamoxifen för antagonistisk aktivitet) mäts på den första plattan i varje testserie. För att kunna jämföra analysresultaten för de återstående fem mikrotiterplattorna med den första mikrotiterplattan som innehåller referensstandardens fullständiga koncentrationsresponskurva bör alla plattor innehålla tre kontrollprover: lösningsmedelskontroll, den högsta koncentrationen av den testade referensstandarden och approximativ koncentration av EC50 (agonistisk aktivitet) eller IC50 (antagonistisk aktivitet) för referensstandarden. Förhållandet mellan de genomsnittliga kontrollproverna för den första plattan och de återstående fem plattorna bör uppfylla de krav som anges i tabell 3C (agonistisk aktivitet) eller tabell 4C (antagonistisk aktivitet).
|
|
17.
|
Z-faktorn beräknas för varje mikrotiterplatta inom en testserie (10). Z-faktorn bör beräknas med användning av responsen vid referensstandardens högsta och lägsta koncentration. En mikrotiterplatta anses giltig om den uppfyller kraven i tabell 3C (agonistisk aktivitet) eller tabell 4C (antagonistisk aktivitet).
|
|
18.
|
Referensstandarden bör uppvisa en S-formad dosresponskurva. Det EC50- eller IC50-värde som härleds från responsen på en serie utspädningar av referensstandarden bör uppfylla kraven i tabell 3C (agonistisk aktivitet) eller tabell 4C (antagonistisk aktivitet).
|
|
19.
|
Varje testserie bör innehålla ett positivt och ett negativt kontrollprov. Den beräknade relativa induktionen både för det positiva och det negativa kontrollprovet bör uppfylla kraven i tabell 3C (agonistisk aktivitet) eller tabell 4C (antagonistisk aktivitet).
|
|
20.
|
Under alla mätningar bör induktionsfaktorn för referensstandardens högsta koncentration mätas genom att den genomsnittligt högsta RLU-responsen för referensstandarden 17β-östradiol divideras med referenslösningsmedelskontrollens genomsnittliga RLU-respons. Denna induktionsfaktor bör uppfylla kraven för x-faldig induktion i tabell 3C (agonistisk aktivitet) eller tabell 4C (antagonistisk aktivitet).
|
|
21.
|
Endast mikrotiterplattor som uppfyller samtliga ovannämnda acceptanskriterier anses giltiga och kan användas för att utvärdera testkemikaliernas respons.
|
|
22.
|
Acceptanskriterierna är tillämpliga både på det förberedande testet och de fullständiga testomgångarna.
|
Tabell 1
Koncentrationer av referensstandarden samt positiv kontroll och negativ kontroll för det agonistiska CALUX-testet
|
|
Ämne
|
CAS-nr
|
Testintervall (M)
|
|
Referensstandard
|
17β-östradiol
|
50-28-2
|
1*10–13 – 1*10–10
|
|
Positiv kontroll (PC)
|
17α-metyltestosteron
|
58-18-4
|
3*10–6
|
|
Negativ kontroll (NC)
|
kortikosteron
|
50-22-6
|
1*10-8
|
Tabell 2
Koncentrationer av referensstandarden samt positiv kontroll och negativ kontroll för det antagonistiska CALUX-testet
|
|
Ämne
|
CAS-nr
|
Testintervall (M)
|
|
Referensstandard
|
tamoxifen
|
10540-29-1
|
3*10-9 – 1*10-5
|
|
Positiv kontroll (PC)
|
4-hydroxitamoxifen
|
68047-06-3
|
1*10-9
|
|
Negativ kontroll (NC)
|
resveratrol
|
501-36-0
|
1*10-5
|
Tabell 3
Acceptanskriterier för det agonistiska ERα CALUX-testet
|
A – individuella prov på platta
|
Kriterium
|
|
1
|
Maximal % SD för triplikathål (för negativ kontroll, positiv kontroll, kemikalie och referensstandard, förutom C0)
|
< 15 %
|
|
2
|
Maximal % SD för triplikathål (för referensstandard och testkemikaliens lösningsmedelskontroller (C0, SC))
|
< 30 %
|
|
3
|
Maximalt LDH-läckage, som ett mått på cytotoxicitet.
|
< 120 %
|
|
B – inom en enskild mikrotiterplatta
|
|
|
4
|
Förhållande för referensstandardens lösningsmedelskontroll (C0, platta 1) och testkemikaliens lösningsmedelskontroll (SC, plattorna 2 till x)
|
0,5 till 2,0
|
|
5
|
Förhållande för appr. EC50 och de högsta referensstandardkoncentrationerna på platta 1 samt appr. EC50 och de högsta referensstandardkoncentrationerna på plattorna 2 till x (C4, C8)
|
0,70 till 1,30
|
|
6
|
Z-faktor för varje platta
|
> 0,6
|
|
C – inom en enskild analysserie (samtliga plattor i en serie)
|
|
|
7
|
S-kurva för referensstandarden
|
Ja (17ß-östradiol)
|
|
8
|
EC50-förhållande för referensstandarden 17ß-östradiol
|
4*10-12 – 4*10-11 M
|
|
9
|
Minsta induktionsförhållande för den högsta koncentrationen av 17ß-östradiol med avseende på referensstandardens lösningsmedelskontroll.
|
5
|
|
10
|
Relativ induktion (%) PC.
|
> 30 %
|
|
11
|
Relativ induktion (%) NC.
|
< 10 %
|
Appr.: approximativ. PC: positiv kontroll. NC: negativ kontroll. SC: testkemikaliens lösningsmedelskontroll. C0: referensstandardens lösningsmedelskontroll. SD: standardavvikelse. LDH: laktatdehydrogenas.
Tabell 4
Acceptanskriterier för det antagonistiska ERα CALUX-testet
|
A – individuella prov på platta
|
Kriterium
|
|
1
|
Maximal % SD för triplikathål (för negativ kontroll, positiv kontroll, kemikalie och referensstandard, lösningsmedelskontroll C0))
|
< 15 %
|
|
2
|
Maximal % SD för triplikathål (för vehikelkontroll och högsta referensstandardkoncentration (C8))
|
< 30 %
|
|
3
|
Maximalt LDH-läckage, som ett mått på cytotoxicitet.
|
< 120 %
|
|
B – inom en enskild mikrotiterplatta
|
|
|
4
|
Förhållande för referensstandardens lösningsmedelskontroll (C0, platta 1) och testkemikaliens lösningsmedelskontroll (SC, plattorna 2 till x)
|
0,70 till 1,30
|
|
5
|
Förhållande för appr. referensstandardkoncentrationer för IC50 på platta 1 samt appr. IC50-referensstandardkoncentrationer för IC50 på plattorna 2 till x (C4)
|
0,70 till 1,30
|
|
6
|
Förhållande för de högsta referensstandardkoncentrationerna på platta 1 och de högsta referensstandardkoncentrationerna på plattorna 2 till x (C8)
|
0,50 till 2,0
|
|
7
|
Z-faktor för varje platta
|
> 0,6
|
|
C – inom en enskild analysserie (samtliga plattor i en serie)
|
|
|
8
|
S-kurva för referensstandarden
|
Ja (tamoxifen)
|
|
9
|
IC50-förhållande för referensstandarden (tamoxifen)
|
1*10-8 - 1*10-7 M
|
|
10
|
Minsta induktionsförhållande för referensstandardens lösningsmedelskontroll med avseende på den högsta tamoxifenkoncentrationen.
|
2,5
|
|
11
|
Relativ induktion (%) PC.
|
< 70 %
|
|
12
|
Relativ induktion (%) NC.
|
> 85 %
|
Appr.: approximativ. PC: positiv kontroll. NC: negativ kontroll. VC: vehikelkontroll (lösningsmedelskontroll utan fast koncentration av den agonistiska referensstandarden). SC: testkemikaliens lösningsmedelskontroll. C0: referensstandardens lösningsmedelskontroll. SD: standardavvikelse. LDH: laktatdehydrogenas.
Lösningsmedels-/vehikelkontroll, referensstandarder, positiva kontroller, negativa kontroller
|
23.
|
Samma lösningsmedels-/vehikelkontroll, referensstandarder och positiva och negativa kontroller bör användas både för det förberedande testet och de fullständiga testomgångarna. Dessutom bör koncentrationen av referensstandarder och positiva och negativa kontroller vara desamma.
|
Lösningsmedelskontroll
|
24.
|
Det lösningsmedel som används för att lösa upp testkemikalierna bör användas som lösningsmedelskontroll. Dimetylsulfoxid (DMSO, 1 % (v/v), CAS-nr 67-68-5) användes som vehikel under valideringen av ERα CALUX-testet. Om ett annat lösningsmedel än DMSO används bör alla referensstandarder, kontroller och testkemikalier testas i samma vehikel. Observera att lösningsmedelskontrollen för antagonistiska studier innehåller en fast koncentration av den agonistiska referensstandarden 17β-östradiol (ungefär EC50-koncentration). En vehikelkontroll bör beredas och testas för att testa det lösningsmedel som används för antagonistiska studier.
|
Vehikelkontroll (antagonistisk aktivitet)
|
25.
|
För att testa antagonistisk aktivitet kompletteras testmediet med en fast koncentration av den agonistiska referensstandarden 17β-östradiol (ungefär EC50-koncentration). För att testa det lösningsmedel som använts för att lösa upp testkemikalierna i antagonisttestet, bör ett testmedium utan fast koncentration av den agonistiska referensstandarden 17β-östradiol beredas. Detta kontrollprov utgör vehikelkontrollen. Dimetylsulfoxid (DMSO, 1 % (v/v), CAS-nr 67-68-5) användes som vehikel under valideringen av ERα CALUX-testet. Om ett annat lösningsmedel än DMSO används bör alla referensstandarder, kontroller och testkemikalier testas i samma vehikel.
|
Referensstandarder
|
26.
|
Den agonistiska referensstandarden är 17β-östradiol (tabell 1). Referensstandarderna består av seriespädningar av åtta koncentrationer av 17β-östradiol (1*10-13, 3*10-13, 1*10-12, 3*10-12, 6*10-12, 1*10-11, 3*10-11, 1*10-10 M).
|
|
27.
|
Den antagonistiska referensstandarden är tamoxifen (tabell 2). Referensstandarderna består av seriespädningar av åtta koncentrationer av tamoxifen (3*10-9, 1*10-8, 3*10-8, 1*10-7, 3*10-7, 1*10-6, 3*10-6, 1*10-5 M). Varje koncentration av den agonistiska referensstandarden saminkuberas med en fast koncentration av den agonistiska referensstandarden17β-östradiol (3*10-12 M).
|
Positiv kontroll
|
28.
|
Den positiva kontrollen för agonistiska studier är 17α-metyltestosteron (tabell 1).
|
|
29.
|
Den positiva kontrollen för antagonistiska studier är 4-hydroxitamoxifen (tabell 2). Den antagonistiska positiva kontrollen saminkuberas med en fast koncentration av den agonistiska referensstandarden17β-östradiol (3*10-12 M).
|
Negativ kontroll
|
30.
|
Den negativa kontrollen för agonistiska studier är kortikosteron (tabell 1).
|
|
31.
|
Den negativa kontrollen för antagonistiska studier är resveratrol (tabell 2). Den antagonistiska negativa kontrollen saminkuberas med en fast koncentration av den agonistiska referensstandarden17β-östradiol (3*10-12 M).
|
Demonstration av laboratoriets kompetens (se punkt 14 och tabellerna 3 och 4 i ”Komponenter i ER TA-testet” i denna testmetod)
Vehikel
|
32.
|
Det lösningsmedel som används för att lösa upp testkemikalierna bör lösa upp testkemikalien fullständigt och bör vara blandbart med cellmediet. DMSO, vatten och etanol (renhetsgrad 95 % till 100 %) är lämpliga lösningsmedel. Om DMSO används som lösningsmedel bör den maximala koncentrationen av DMSO under inkubationen inte överskrida 1 % (v/v). Före användning bör lösningsmedlet testas för att kontrollera att det inte är cytotoxiskt och inte stör testet.
|
Beredning av referensstandarder, positiva kontroller, negativa kontroller och testkemikalier
|
33.
|
Referensstandarder, positiva kontroller, negativa kontroller och testkemikalier löses upp i 100 % DMSO (eller ett annat lämpligt lösningsmedel). Lämpliga (serie)spädningar bör sedan beredas i samma lösningsmedel. Innan de löses upp bör alla ämnen få anta rumstemperatur. Färska stamlösningar eller referensstandarder, positiva kontroller, negativa kontroller och testkemikalier bör inte ha märkbara fällningar eller grumlighet. Referensstandarden och kontrollstammarna kan beredas i bulk. Nya stamlösningar av testkemikalierna bör beredas före varje försök. Nya slutliga utspädningar av referensstandarderna, de positiva kontrollerna, de negativa kontrollerna och testkemikalierna bör dock beredas för varje försök och användas inom 24 timmar från beredningen.
|
Löslighet, cytotoxicitet och koncentrationsintervall
|
34.
|
Under det förberedande testet fastställs testkemikaliernas löslighet i det valda lösningsmedlet. En maximal stamkoncentration på 0,1 M bereds. Om denna koncentration visar löslighetsproblem bör lägre stamlösningar beredas till dess att testkemikalierna är fullständigt upplösta. Under det förberedande testet testas seriespädningar på 1:10 av testkemikalien. Den maximala testkoncentrationen för agonistisk eller antagonistisk testning är 1 mM. Efter det förberedande testet härleds ett lämpligt förfinat koncentrationsintervall för testkemikalierna som bör testas under de fullständiga testomgångarna. De utspädningar som används för fullständig testning bör vara 1x, 3x, 10x, 30x, 100x, 300x, 1000x och 3000x.
|
|
35.
|
Cytotoxicitetstestning ingår i protokollet för den agonistiska och den antagonistiska testmetoden (11). Cytotoxicitetstestning ingår dessutom både i det förberedande testet och de fullständiga testomgångarna. Den metod som användes för att bedöma cytotoxicitet under valideringen av ERα CALUX-testet var läckagetestet för laktatdehydrogenas (LDH) i kombination med en kvalitativ visuell inspektion av cellerna (se tillägg 4.1) och följt av exponering för textkemikalierna. Andra kvantitativa metoder kan dock användas för att bestämma cytotoxicitet (t.ex. tetrazoliumbaserat kolorimetriskt test (MTT) eller CALUX-test av cytotoxicitet). Generellt anses testkemikaliekoncentrationer som visar mer än 20 % reduktion av cellviabiliteten vara cytotoxiska och kan därför inte användas för datautvärdering. När det gäller LDH-läckagetest anses testkemikaliens koncentration vara cytotoxisk om procentandelen LDH-läckage är högre än 120 %.
|
Testkemikaliens exponering och beredning av testplattor
|
36.
|
Efter trypsinering av en konfluent kolv med odlade celler resuspenderas cellerna vid 1 x 105 celler/ml i ett östrogenfritt testmedium 100 μl av resuspenderade celler överförs till de inre hålen på en 96-håls mikrotiterplatta. De yttre hålen fylls med 200 μl fosfatbuffrad saltlösning (PBS) (se figurerna 1 och 2). De överförda cellerna förinkuberas i 24 timmar i en CO2-inkubator (5 % CO2, 37 °C, 100 % luftfuktighet).
|
|
37.
|
Efter förinkuberingen inspekteras cellerna för att kontrollera om det finns någon synlig cytotoxicitet (se tillägg 4.1), kontamination eller konfluens. Endast plattor som inte visar någon synlig cytotoxicitet eller kontamination och har minst 85 % konfluens används för testningen. Mediet från de inre hålen avlägsnas försiktigt och ersätts med 200 μl östrogenfritt testmedium som innehåller lämpliga spädningsserier av referensstandarderna, testkemikalierna, de positiva kontrollerna, de negativa kontrollerna och lösningsmedelskontrollerna (tabell 5: agonistiska studier, Tabell 6: antagonistiska studier). Alla referensstandarder, testkemikalier, positiva kontroller, negativa kontroller och lösningsmedelskontroller testas i triplikat. Plattans utformning för agonistisk testning anges i figur 1. Plattans utformning för antagonistisk testning anges i figur 2. Plattorna för förberedande test och fullständig testning utformas på identiskt sätt. För antagonistisk testning innehåller alla inre hål, förutom hålen för vehikelkontroll, även en fast koncentration av den agonistiska referensstandarden 17β-östradiol (3*10-12 M). Observera att referensstandarderna C8 och C4 bör tillsättas varje TC-platta.
|
|
38.
|
När cellerna har exponerats för alla kemikalier bör de 96-håls mikrotiterplattorna inkuberas i ytterligare 24 timmar i en CO2-inkubator (5 % CO2, 37 °C, 100 % luftfuktighet).
|
Figur 1
Utformning av 96-håls mikrotiterplattor för förberedande test och bedömning av agonistisk effekt.
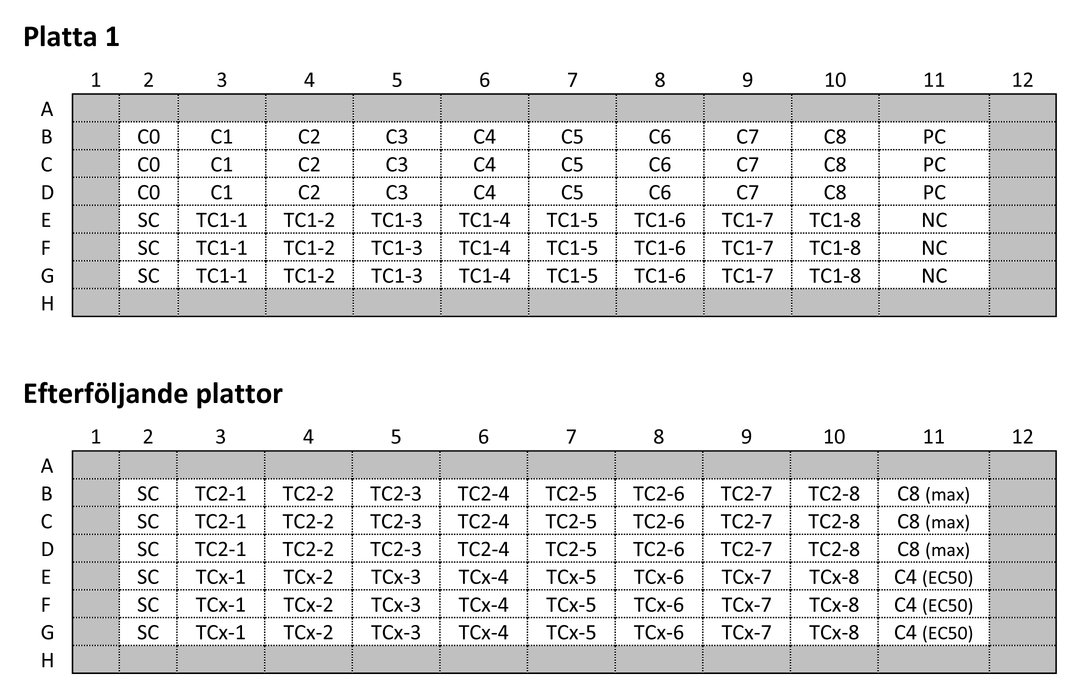
C0 = referensstandardens lösningsmedel.
C(1-8) = referensstandardens spädningsserier (1-8, låga-till-höga koncentrationer).
PC = positiv kontroll.
NC = negativ kontroll.
TCx-(1-8) = spädningar (1-8, låga till höga koncentrationer) av testkemikalien för det förberedande testet samt bedömning av den agonistiska effekten hos testkemikalie x.
SC = lösningsmedelskontroll av testkemikalien (helst samma lösningsmedel som i C0, men eventuellt från en annan sats).
Grå celler: = Yttre hål, fyllda med 200 μl PBS.
Figur 2
Utformning av 96-håls mikrotiterplattor för förberedande test och bedömning av antagonistisk effekt.

C0 = referensstandardens lösningsmedel.
C(1-8) = referensstandardens spädningsserier (1-8, låga-till-höga koncentrationer).
NC = negativ kontroll.
PC = positiv kontroll.
TCx-(1-8) = spädningar (1-8, låga till höga koncentrationer) av testkemikalien för det förberedande testet samt bedömning av den agonistiska effekten hos testkemikalie x.
SC = lösningsmedelskontroll av testkemikalien (helst samma lösningsmedel som i C0, men eventuellt från en annan sats).
VC = vehikelkontroll (lösningsmedelskontroll utan fast koncentration av den agonistiska referensstandarden 17β-östradiol).
Grå celler: = Yttre hål, fyllda med 200 μl PBS.
Anmärkning: alla inre hål, utom hålen för vehikelkontroll, innehåller också en fast koncentration av den agonistiska referensstandarden 17β-östradiol (3,0*10-12 M).
Mätning av luminescens
|
39.
|
Luminiscensmätningen beskrivs i detalj i protokollet för den agonistiska och den antagonistiska testmetoden (10). Mediet bör avlägsnas från hålen och cellerna bör lyseras efter 24 timmars inkubation för att öppna cellmembranet och möjliggöra mätning av luciferasaktiviteten.
|
|
40.
|
För luminescensmätningen kräver detta förfarande en luminometer som är utrustad med två injektorer. Luciferasreaktionen utlöses genom injektion av substratet luciferin. Reaktionen stoppas genom tillsats av 0,2 M NaOH. Reaktionen stoppas för att förebygga att luminescens överförs från ett hål till ett annat.
|
|
41.
|
Det ljus som släpps ut från varje hål uttrycks som relativa ljusenheter (RLU) från varje hål.
|
Förberedande test
|
42.
|
Analysresultaten från det förberedande testet används för att bestämma ett förfinat koncentrationsintervall för testkemikalierna inför den fullständiga testningen. Utvärdering av resultaten från det förberedande testet och bestämning av det förfinade koncentrationsintervallet för testkemikalier för fullständig testning beskrivs detaljerat i protokollet för den agonistiska och den antagonistiska testmetoden (10). Här ges endast en kort sammanfattning av förfarandena för att bestämma testkemikaliernas koncentrationsintervall för agonistisk och antagonistisk testning. Vägledning för utformningen av seriespädningar ges i tabellerna 5 och 6.
|
Val av koncentrationer för bedömning av agonistiska effekter
|
43.
|
Under det förberedande testet bör testkemikalierna testas med användning av de seriespädningar som anges i tabell 5 (agonistisk testning) och 6 (antagonistisk testning). Alla koncentrationer bör testas i triplikathål enligt den utformning av plattorna som ges i figur 1 (agonistisk testning) respektive figur 2 (antagonistisk testning).
|
|
44.
|
Endast analysresultat som uppfyller acceptanskriterierna (tabell 3) anses giltiga och kan användas för att utvärdera testkemikaliernas respons. Om en eller flera mikrotiterplattor i en analysserie inte uppfyller acceptanskriterierna bör de respektive mikrotiterplattorna analyseras igen. Om den första plattan som innehåller den fullständiga serien av spädningar av referensstandarden inte uppfyller acceptanskriterierna måste hela testserien (sex plattor) analyseras igen.
|
|
45.
|
Testkemikaliernas ursprungliga koncentrationsintervall bör justeras och det förberedande testet bör upprepas i följande fall:
|
—
|
Om cytotoxicitet observeras. Det förberedande testet bör upprepas med lägre icke-cytotoxiska koncentrationer av testkemikalien.
|
|
—
|
Om det förberedande testet av testkemikalien inte visar en fullständig dosresponskurva på grund av att de testade kemikalierna genererar maximal induktion. Det förberedande testet bör upprepas med lägre koncentrationer av testkemikalien.
|
|
|
46.
|
När en giltig dosrelaterad respons observeras bör den (lägsta) koncentration vid vilken maximal induktion observeras och inte visar cytotoxicitet väljas. Den högsta koncentrationen av den testkemikalie som ska testas i de fullständiga testomgångarna bör vara tre gånger denna valda koncentration.
|
|
47.
|
En fullständig förfinad spädningsserie av testkemikalien bör beredas med de utspädningssteg som anges i tabell 5, med start vid den högsta koncentration som fastställts enligt ovan.
|
|
48.
|
En testkemikalie som inte framkallar någon agonistisk effekt bör testas i de fullständiga testomgångarna, med start vid den högsta icke-cytotoxiska koncentration som identifierats under det förberedande testet.
|
Val av koncentrationer för bedömning av antagonistiska effekter
|
49.
|
Endast analysresultat som uppfyller acceptanskriterierna (tabell 4) anses giltiga och kan användas för att utvärdera testkemikaliernas respons. Om en eller flera mikrotiterplattor i en analysserie inte uppfyller acceptanskriterierna bör de respektive mikrotiterplattorna analyseras igen. Om den första plattan som innehåller den fullständiga serien av spädningar av referensstandarden inte uppfyller acceptanskriterierna måste hela testserien (sex plattor) analyseras igen.
|
|
50.
|
Testkemikaliernas ursprungliga koncentrationsintervall bör justeras och det förberedande testet bör upprepas i följande fall:
|
—
|
Om cytotoxicitet observeras. Det förberedande testet bör upprepas med lägre icke-cytotoxiska koncentrationer av testkemikalien.
|
|
—
|
Om det förberedande testet av testkemikalien inte visar en fullständig dosresponskurva på grund av att de testade kemikalierna genererar maximal inhibering. Det förberedande testet bör upprepas med lägre koncentrationer av testkemikalien.
|
|
|
51.
|
När en giltig dosrelaterad respons påvisas bör den (lägsta) koncentration vid vilken maximal inhibiering observeras och inte visar cytotoxicitet väljas. Den högsta koncentrationen av den testkemikalie som ska testas i de fullständiga testomgångarna bör vara tre gånger denna valda koncentration.
|
|
52.
|
En fullständig förfinad spädningsserie av testkemikalien bör beredas med de utspädningssteg som anges i tabell 6, med start vid den högsta koncentration som fastställts enligt ovan.
|
|
53.
|
Testkemikalier som inte framkallar några antagonistisk effekter bör testas i de fullständiga testomgångarna, med start vid den högsta icke-cytotoxiska koncentration som testats under det förberedande testet.
|
Fullständiga testomgångar
|
54.
|
När de förfinade koncentrationsintervallen har valts bör testkemikalierna testas fullständigt med användning av de spädningsserier som anges i tabell 5 (antagonistisk analys) och 6 (antagonistisk analys). Alla koncentrationer bör testas i triplikathål enligt den utformning av plattorna som ges i figur 1 (agonistisk testning) respektive figur 2 (antagonistisk testning).
|
|
55.
|
Endast analysresultat som uppfyller acceptanskriterierna (tabellerna 3 och 4) anses giltiga och kan användas för att utvärdera testkemikaliernas respons. Om en eller flera mikrotiterplattor i en analysserie inte uppfyller acceptanskriterierna bör de respektive mikrotiterplattorna analyseras igen. Om den första plattan som innehåller den fullständiga serien av spädningar av referensstandarden inte uppfyller acceptanskriterierna måste hela testserien (sex plattor) analyseras igen.
|
Tabell 5
Koncentrationer och utspädningar av de referensstandarder, kontroller och testkemikalier som används för agonistisk testning
|
Referens 17β-östradiol
|
TCx – förberedande test
|
TCx – fullständig körning
|
Kontroller
|
|
konc. (M)
|
utspädning
|
utspädning
|
konc. (M)
|
|
C0
|
0
|
TCx-1
|
10 000 000 x
|
TCx-1
|
3 000 x
|
PC
|
3*10-6
|
|
C1
|
1*10-13
|
TCx-2
|
1 000 000 x
|
TCx-2
|
1 000 x
|
NC
|
1*10-8
|
|
C2
|
3*10-13
|
TCx-3
|
100 000 x
|
TCx-3
|
300 x
|
C0
|
0
|
|
C3
|
1*10-12
|
TCx-4
|
10 000 x
|
TCx-4
|
100 x
|
SC
|
0
|
|
C4
|
3*10-12
|
TCx-5
|
1 000 x
|
TCx-5
|
30 x
|
|
|
|
C5
|
6*10-12
|
TCx-6
|
100 x
|
TCx-6
|
10 x
|
|
|
|
C6
|
1*10-11
|
TCx-7
|
10 x
|
TCx-7
|
3 x
|
|
|
|
C7
|
3*10-11
|
TCx-8
|
1 x
|
TCx-8
|
1 x
|
|
|
|
C8
|
1*10-10
|
|
|
|
|
|
|
|
TCx – testkemikalie x
PC – positiv kontroll (17α-metyltestosteron)
NC – negativ kontroll (kortikosteron)
C0 – referensstandardens lösningsmedelskontroll
SC – testkemikaliens lösningsmedelskontroll
|
Tabell 6
Koncentrationer och utspädningar av de referensstandarder, kontroller och testkemikalier som används för antagonistisk testning
|
Referenstamoxifen
|
TCx – förberedande test
|
TCx – fullständig körning
|
Kontroller
|
|
konc. (M)
|
utspädning
|
utspädning
|
konc. (M)
|
|
C0
|
0
|
TCx-1
|
10 000 000 x
|
TCx-1
|
3 000 x
|
PC
|
1*10-9
|
|
C1
|
3*10-9
|
TCx-2
|
1 000 000 x
|
TCx-2
|
1 000 x
|
NC
|
1*10-5
|
|
C2
|
1*10-8
|
TCx-3
|
100 000 x
|
TCx-3
|
300 x
|
C0
|
0
|
|
C3
|
3*10-8
|
TCx-4
|
10 000 x
|
TCx-4
|
100 x
|
SC
|
0
|
|
C4
|
1*10-7
|
TCx-5
|
1 000 x
|
TCx-5
|
30 x
|
|
|
|
C5
|
3*10-
|
TCx-6
|
100 x
|
TCx-6
|
10 x
|
Kompletterad agonist
|
|
C6
|
1*10-6
|
TCx-7
|
10 x
|
TCx-7
|
3 x
|
konc. (M)
|
|
C7
|
3*10-6
|
TCx-8
|
1 x
|
TCx-8
|
1 x
|
17β-östradiol
|
3*10-12
|
|
C8
|
1*10-5
|
|
|
|
|
|
|
|
TCx – testkemikalie x
PC – positiv kontroll (4-hydroxitamoxifen)
NC – negativ kontroll (resveratrol)
C0 – referensstandardens lösningsmedelskontroll
SC – testkemikaliens lösningsmedelskontroll
VC – vehikelkontroll (innehåller ingen fast koncentration av den agonistiska referensstandarden 17β-östradiol (3,0*10-12 M)
|
Insamling av data och dataanalys
|
56.
|
Efter det förberedande testet och den fullständiga testomgångarna bör testkemikaliens EC10, EC50, PC10, PC50 och maximala induktion (TCxmax) bestämmas för agonistisk testning. För antagonistisk testning bör IC20, IC50, PC80, PC50 och minimal induktion (TCxmin) beräknas. En grafisk återgivning av dessa parametrar ges i figur 3 (agonistisk aktivitet) och figur 4 (antagonistisk aktivitet). De parametrar som behövs beräknas baserat på varje testkemikalies relativa induktion (i förhållande till referensstandardens maximala induktion (= 100 %)). Icke-linjär regression (variabel lutning, fyra parametrar) bör användas för datautvärderingen, enligt följande ekvation:

Där
X = Log av dos eller koncentration
Y = Respons (relativ induktion (%))
Top = Maximal induktion (%)
Bottom = Minimal induktion (%)
LogEC50 = log av koncentration vid vilken 50 % av den maximala responsen observeras.
Hill-koefficient (Hill slope) = lutningsfaktor eller Hill slope.
|
|
57.
|
Rådata från luminometern, uttryckt som relativa ljusenheter (RLU), bör överföras till det kalkylblad för dataanalysen som har utformats för det förberedande testet och de fullständiga testomgångarna. Rådatan bör uppfylla de acceptanskriterier som anges i tabellerna 3A och 3B (agonistisk analys) respektive tabellerna 4A och 4B (antagonistisk analys). Om rådatan uppfyller acceptanskriterierna utförs följande beräkningssteg för att bestämma de parametrar som behövs:
|
Agonistisk analys
|
—
|
Subtrahera genomsnittlig RLU för referensstandardens lösningsmedelskontroll från varje råanalysdata för referensstandarderna.
|
|
—
|
Subtrahera genomsnittlig RLU för testkemikaliens lösningsmedelskontroll från varje råanalysdata för testkemikalierna.
|
|
—
|
Beräkna den relativa induktionen från varje koncentration av referensstandarden. Sätt induktionen för den högsta koncentrationen av referensstandarden till 100 %.
|
|
—
|
Beräkna den relativa induktionen för varje koncentration av testkemikalien jämfört med den högsta koncentrationen av referensstandarden som 100 %.
|
|
—
|
Utvärdera analysresultaten enligt icke-linjär regression (variabel lutning, fyra parametrar).
|
|
—
|
Bestäm EC50 och EC10 för referensstandarden.
|
|
—
|
Bestäm EC50 och EC10 för testkemikalierna.
|
|
—
|
Bestäm den högsta relativa induktionen för testkemikalien (TCmax).
|
|
—
|
Bestäm PC10 och PC50 för testkemikalierna.
|
För testkemikalier är det kanske inte alltid möjligt att erhålla en fullständig dosresponskurva, t.ex. på grund av problem med cytotoxicitet eller löslighet. Det innebär i sin tur att EC50, EC10 och PC50 inte kan bestämmas. I sådana fall kan endast PC10 och TCmax bestämmas.
Antagonistisk analys
|
—
|
Subtrahera genomsnittlig RLU för den högsta koncentrationen av referensstandarden från varje råanalysdata för referensstandarderna.
|
|
—
|
Subtrahera genomsnittlig RLU för referensstandardens lösningsmedelskontroll från varje råanalysdata för testkemikalierna.
|
|
—
|
Beräkna den relativa induktionen från varje koncentration av referensstandarden. Sätt induktionen för den lägsta koncentrationen av referensstandarden till 100 %.
|
|
—
|
Beräkna den relativa induktionen för varje koncentration av testkemikalien jämfört med den lägsta koncentrationen av referensstandarden som 100 %.
|
|
—
|
Utvärdera analysresultaten enligt icke-linjär regression (variabel lutning, fyra parametrar).
|
|
—
|
Bestäm IC50 och IC20 för referensstandarden.
|
|
—
|
Bestäm IC50 och IC20 för testkemikalierna.
|
|
—
|
Bestäm den lägsta relativa induktionen för testkemikalien (TCmin).
|
|
—
|
Bestäm PC80 och PC50 för testkemikalierna.
|
Figur 3
Översikt över parametrar som bestäms i det agonistiska testet

EC10 = koncentration av ett ämne vid vilken 10 % av dess maximala respons observeras.
EC50 = koncentration av ett ämne vid vilken 50 % av dess maximala respons observeras.
PC10 = koncentration av en testkemikalie vid vilken dess respons motsvarar referensstandardens EC10.
PC50 = koncentration av en testkemikalie vid vilken dess respons motsvarar referensstandardens EC50.
TCxmax = testkemikaliens maximala relativa induktion.
Figur 4
Översikt över parametrar som bestäms i det antagonistiska testet

IC20 = koncentration av ett ämne vid vilken 80 % av dess maximala respons observeras (20 % inhibition).
IC50 = koncentration av ett ämne vid vilken 50 % av dess maximala respons observeras (50 % inhibition).
PC80 = koncentration av en testkemikalie vid vilken dess respons motsvarar referensstandardens IC20.
PC50 = koncentration av en testkemikalie vid vilken dess respons motsvarar referensstandardens IC50.
TCxmin = testkemikaliens minimala relativa induktion.
För testkemikalier är det kanske inte alltid möjligt att erhålla en fullständig dosresponskurva, t.ex. på grund av problem med cytotoxicitet eller löslighet. Följaktligen kan IC50, IC20 och PC50 inte fastställas. I sådana fall kan endast PC20 och TCmin bestämmas.
|
58.
|
Resultaten bör baseras på två (eller tre) oberoende testomgångar. Om två testomgångar ger jämförbara och därför reproducerbara resultat är det inte nödvändigt att utföra en tredje testomgång. För att vara godtagbara bör resultaten
|
—
|
uppfylla acceptanskriterierna (se Acceptanskriterier, punkterna 14–22),
|
|
Datatolkningskriterier
|
59.
|
Följande kriterier ska användas för datatolkning och beslut om huruvida en testkemikalie anses vara positiv eller negativ:
|
För varje fullständig körning anses en testkemikalie vara positiv om
|
1
|
TCmax motsvarar eller överskrider 10 % av referensstandardens maximala respons (REF10),
|
|
2
|
minst två på varandra följande koncentrationer av testkemikalien motsvarar eller överskrider REF10.
|
|
|
För varje fullständig körning anses en testkemikalie vara negativ om
|
1
|
TCmax inte motsvarar eller överskrider 10 % av referensstandardens maximala respons (REF10).
|
|
2
|
mindre än två på varandra följande koncentrationer av testkemikalien motsvarar eller överskrider REF10.
|
|
|
För varje fullständig körning anses en testkemikalie vara positiv om
|
1
|
TCmin motsvarar eller är lägre än 80 % av referensstandardens maximala respons (REF80 = 20 % inhibition).
|
|
2
|
minst två på varandra följande koncentrationer av testkemikalien motsvarar eller är lägre än REF80.
|
|
|
För varje fullständig körning anses en testkemikalie vara negativ om
|
1
|
TCmin överskrider 80 % av referensstandardens maximala respons (REF80 = 20 % inhibition),
|
|
2
|
mindre än två på varandra följande koncentrationer av testkemikalien motsvarar eller är lägre än REF80.
|
|
|
|
60.
|
För att karakterisera potensen hos den positiva responsen från en testkemikalie bör effektens omfattning (agonistisk analys TCmax, antagonistisk analys TCmin) samt den koncentration vid vilken effekten uppstår (agonistisk analys EC10, EC50, PC10, PC50. antagonistisk analys IC20, IC50, PC80, PC50) rapporteras.
|
TESTRAPPORT
|
61.
|
Se punkt 20 i ”Komponenter i ER TA-testet”.
|
LITTERATUR
|
(1)
|
OECD (2016). Draft Validation report of the (anti-) ERα CALUX bioassay – transactivation bioassay for the detection of compounds with (anti)estrogenic potential. Environmental Health and Safety Publications, Series on Testing and Assessment (nr 240). Organisationen för ekonomiskt samarbete och utveckling, Paris
|
|
(2)
|
Sonneveld E., Jansen H.J., Riteco J.A., Brouwer A., van der Burg B. (2005). Development of androgen- and estrogen-responsive bioassays, members of a panel of human cell line-based highly selective steroid-responsive bioassays. Toxicol Sci. 83(1), 136–148.
|
|
(3)
|
Quaedackers M.E., van den Brink C.E., Wissink S., Schreurs R.H.M.M., Gustafsson J.A., van der Saag P.T. och van der Burg B. (2001). 4-Hydroxytamoxifen trans-represses nuclear factor-kB Activity in human osteoblastic U2OS cells through estrogen receptor (ER)α and not through ERβ. Endocrinology 142(3), 1156–1166.
|
|
(4)
|
Thorne N., Inglese J. och Auld D.S. (2010). Illuminating Insights into Firefly Luciferase and Other Bioluminescent Reporters Used in Chemical Biology, Chemistry and Biology17(6):646–57.
|
|
(5)
|
Escande A., Pillon A., Servant N., Cravedi J.P., Larrea F., Muhn P., Nicolas J.C., Cavaillès V. och Balaguer P.. (2006). Evaluation of ligand selectivity using reporter cell lines stably expressing estrogen receptor alpha or beta. Biochem. Pharmacol., 71, 1459–1469.
|
|
(6)
|
Kuiper G.G., Lemmen J.G., Carlsson B., Corton J.C., Safe S.H., van der Saag P.T., van der Burg B. och Gustafsson J.A. (1998). Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinol., 139, 4252–4263.
|
|
(7)
|
Sotoca A.M., Bovee T.F.H., Brand W., Velikova N., Boeren S., Murk A.J., Vervoort J., Rietjens I.M.C.M. (2010). Superinduction of estrogen receptor mediated gene expression in luciferase based reporter gene assays is mediated by a post-transcriptional mechanism. J. Steroid. Biochem. Mol. Biol., 122, 204–211.
|
|
(8)
|
Sonneveld E., Riteco J.A.C., Jansen H.J., Pieterse B., Brouwer A., Schoonen W.G., and van der Burg B. (2006). Comparison of in vitro and in vivo screening models for androgenic and estrogenic activities. Toxicol. Sci., 89(1), 173–187.
|
|
(9)
|
Kobayashi H., Yamamoto K., Eguchi M., Kubo M., Nakagami S., Wakisaka S., Kaizuka M. och Ishii H. (1995). Rapid detection of mycoplasma contamination in cell cultures by enzymatic detection of polymerase chain reaction (PCR) products. J. Vet. Med. Sci., 57(4), 769–771.
|
|
(10)
|
Zhang J-H., Chung T.D.Y. och Oldenburg K.R. (1999). A simple statistical parameter for use in evaluation and validation of high throuphut screening assays. J. Biomol. Scr., 4, 67–73
|
|
(11)
|
Besselink H., Middelhof.I, och Felzel, E. (2014). Transactivation assay for the detection of compounds with (anti)estrogenic potential using ERα CALUX cells. BioDetection Systems BV (BDS). Amsterdam, Nederländerna,
|
Tillägg 4.1
VISUELL INSPEKTION AV CELLVIABILITET
B.67 TEST AV GENMUTATIONER HOS DÄGGDJURSCELLER IN VITRO MED ANVÄNDNING AV TYMIDINKINASGENEN
INLEDNING
|
1.
|
Den här testmetoden motsvarar OECD:s testriktlinje (TG) 490 (2016). Testmetoderna ses regelbundet över och revideras med avseende på den vetenskapliga utvecklingen, bestämmelser samt djurens välbefinnande. Muslymfomtestet (MLA) och TK6-testet med användning av tymidinkinaslokuset (TK) ingick ursprungligen i testmetod B.17. Därefter har MLA-expertarbetsgruppen vid International Workshop for Genotoxicity Testing (IWGT) utvecklat internationellt harmoniserade rekommendationer för acceptanskriterier och datatolkning för MLA (1)(2)(3)(4)(5), och dessa rekommendationer inbegrips i denna nya testmetod B.67. Denna testmetod är utformad för MLA, och även för TK6-testet, eftersom den använder TK-lokus. MLA har använts i stor utsträckning för lagstadgade ändamål, medan TK6 har använts betydligt mindre. Det bör noteras att trots likheterna mellan de två cellinjernas endpoints är de inte ömsesidigt utbytbara och de rättsliga systemen kan mycket väl föredra den ena cellinjen framför den andra för ett visst lagstadgat ändamål. Valideringen av MLA visade exempelvis att metoden inte bara är lämplig för att upptäcka genmutation, utan även en testkemikalies förmåga att inducera strukturell kromosomskada. Denna testmetod är en del av en serie av testmetoder för genetisk toxikologi. OECD har utarbetat ett dokument som ger kortfattad information om tester inom genetisk toxikologi och en översikt över de senaste ändringarna av OECD:s testriktlinjer inom detta område (6).
|
|
2.
|
Syftet med genmutationstesterna för däggdjursceller in vitro är att upptäcka genmutationer som induceras av kemikalier. De cellinjer som används i dessa tester mäter framåtmutationer i rapportgener, särskilt den endogena tymidinkinasgenen (TK för celler från människa och Tk för celler från gnagare, i denna testmetod tillsammans kallade TK). Denna testmetod är avsedd för användning med två cellinjer: L5178Y TK+/––3.7.2C-muslymfomcellinjen (allmänt benämnd L5178Y) och TK6-lymfoblastoidceller från människa (allmänt benämnd TK6). Trots att de två cellinjerna varierar sinsemellan på grund av ursprung, celltillväxt, p53-status osv., kan TK-genmutationstester utföras på liknande sätt för båda celltyperna enligt beskrivningen i denna testmetod.
|
|
3.
|
Tymidinkinasgenens autosomala och heterozygota karaktär möjliggör detektion av viabla kolonier vars celler saknar enzymet tymidinkinas efter mutation från TK+/–
till TK–/–
. Denna brist kan vara följden av genetiska händelser som påverkar TK-genen, och det kan både röra sig om genmutationer (punktmutationer, läsramsmutationer, små deletioner osv.) och kromosomhändelser (stora deletioner, kromosomomlagringar och mitotisk rekombination). De senare händelserna uttrycks som en förlust av heterozygositet, som är en vanlig genetisk förändring hos tumörsuppressorgener i human tumorigenes. Teoretiskt sett kan en förlust av hela den kromosom som bär TK-genen till följd av försämring av kärnspolen och/eller mitotisk icke-disjunktion detekteras i MLA. En kombination av cytogenetisk och molekylär analys visar också tydligt att vissa MLA TK-mutanter är följden av icke-disjunktion. En sammanvägd bedömning visar dock att TK-genmutationstestet inte på ett tillförlitligt sätt kan detektera aneugener vid tillämpning av standardkriterier för cytotoxicitet (enligt beskrivningen i denna testmetod), och det är därför inte lämpligt att använda dessa tester för att detektera aneugener (7)(8)(9).
|
|
4.
|
I TK-genmutationstester genereras två olika fenotypiska klasser av TK-mutanter: mutanter med normal tillväxt, som växer i samma takt som TK-heterozygota celler, och mutanter med långsam tillväxt, som växer med förlängda fördubblingstider. Mutanter med normal tillväxt och långsam tillväxt känns igen som stora och små kolonimutanter i MLA, och som tidigt och sent uppträdande kolonimutanter i TK6. Den molekylära och cytogenetiska naturen hos både stora och små kolonier av MLA-mutanter har undersökts mycket ingående (8) (10) (11) (12) (13). Den molekylära och cytogenetiska naturen hos tidigt och sent uppträdande TK6-mutanter har också undersökts i stor utsträckning (14) (15) (16) (17). Långsamt växande mutanter för båda celltyperna har lidit genetisk skada som involverar förmodat tillväxtreglerande gener nära TK-lokus, vilket leder till förlängda fördubblingstider och bildandet av sent uppträdande eller små kolonier (18). Induktion av mutanter med långsam tillväxt har associerats med kemikalier som inducerar omfattande strukturella förändringar på kromosomnivå. Celler vars skador inte involverar förmodat tillväxtreglerande gener nära TK-lokus växer i en liknande takt som de parentala cellerna och blir normalt växande mutanter. Induktion av huvudsakligen normalt växande mutanter associeras med kemikalier som främst agerar som punktmutagener. Följaktligen är det viktigt att räkna både långsamt och normalt växande mutanter för att återvinna alla mutanter och skapa insikt i de typer av skador (mutagener jämfört med klastogener) som induceras av testkemikalien (10) (12) (18) (19).
|
|
5.
|
Testmetoden har utformats för att ge allmän information som gäller både MLA och TK6 samt specialiserad vägledning för de individuella testerna.
|
|
6.
|
Definitioner av begreppen finns i tillägg 1.
|
INLEDANDE ÖVERVÄGANDEN OCH BEGRÄNSNINGAR
|
7.
|
Tester som utförs in vitro kräver i allmänhet att en exogen källa för metabolisk aktivering används. Det exogena metaboliska aktiveringssystemet efterliknar inte helt förhållandena in vivo.
|
|
8.
|
Åtgärder bör vidtas för att undvika förhållanden som leder till artefaktiska positiva resultat (t.ex. möjlig interaktion med testsystemet) som inte orsakas av interaktion mellan testkemikalien och cellens genetiska material. Sådana förhållanden kan vara ändringar av pH eller osmolalitet, interaktion med mediets komponenter (20) (21) eller alltför hög cytotoxicitet (22)(23)(24). En cytotoxicitet som överskrider de rekommenderade maxnivåerna för cytotoxicitet enligt definitionen i avsnitt 28 anses vara för hög för MLA och TK6. Dessutom bör det noteras att testkemikalier som är tymidinanaloger eller beter sig som tymidinanaloger kan öka mutantfrekvensen genom selektiv tillväxt hos de spontana bakgrundsmutanterna under cellbehandling, vilket kräver ytterligare testmetoder för lämplig utvärdering (25).
|
|
9.
|
Tillverkade nanomaterial kan kräva särskilda anpassningar av testmetoden, men de beskrivs inte här.
|
|
10.
|
Innan testmetoden används för att testa en blandning i syfte att samla in data för ett lagstadgat ändamål bör man överväga om, och i så fall varför, den kommer att ge godtagbara resultat för det syftet. Sådana överväganden är inte nödvändiga om blandningen måste provas enligt lag.
|
|
11.
|
Muterade celler som saknar tymidinkinasenzymaktivitet beroende på mutationen TK
+/– to TK
–/–, är resistenta mot de cytostatiska effekterna av pyrimidinanalogen trifluortymidin (TFT). Celler med fungerande tymidinkinas är känsliga för TFT, eftersom TFT orsakar inhibering av cellmetabolismen och stoppar fortsatt celldelning. Muterade celler kan således utvecklas i närvaro av TFT och bilda synliga kolonier, medan celler som innehåller TK-enzymet inte klarar detta.
|
TESTPRINCIP
|
12.
|
Celler i suspension exponeras för testkemikalien, både med och utan metabolisk aktivering ( se punkt 19), under en lämplig tidsperiod (se punkt 33) och subkultiveras för att bestämma cytotoxiciteten och för att tillåta fenotypisk expression innan mutanturvalet sker. Cytotoxicitet bestäms genom relativ total tillväxt (RTG – se punkt 25) för MLA och genom relativ överlevnad (RS – se punkt 26) för TK6. De behandlade kulturerna förvaras i tillväxtmedium under en tillräckligt lång tid, karakteristisk för varje celltyp (se punkt 37), för att tillåta i det närmaste optimal fenotypisk expression av inducerade mutationer. Efter fenotypisk expression bestäms mutantfrekvensen genom inokulering av ett känt antal celler i ett medium som innehåller det selektiva ämnet för detektion av mutantkolonier, och i ett medium utan selektivt ämne för bestämning av kloningseffektiviteten (viabiliteten). Efter en lämplig inkubationstid räknas kolonierna. Mutantfrekvensen räknas fram utifrån antalet mutantkolonier korrigerat för kloningseffektiviteten vid tidpunkten för mutanturvalet.
|
BESKRIVNING AV TESTMETODEN
Beredningar
Celler
|
13.
|
För MLA: Eftersom MLA utvecklades och karakteriserades med användning av sublinjen TK
+/– –3.7.2C av L5178Y-celler måste denna specifika sublinje användas för MLA. L5178Y-cellinjen härleddes från ett metylkolantren-inducerat tymiskt lymfom från en DBA-2-mus (26). Clive och medarbetare behandlade L5178Y-celler (av Clive benämnda TK+/+ –3) med etylmetansulfonat och isolerade en TK–/–-klon (benämnd TK–/– –3.7) med bromdeoxiuridin som selektivt ämne. Från TK–/–-klonen isolerades och karakteriserades en spontan TK+/–-klon (benämnd TK+/– –3.7.2.) och en subklon (benämnd som TK+/––3.7.2C) för användning i MLA (27). Karyotypen för cellinjen har publicerats (28)(29)(30)(31). Det modala kromosomantalet är 40. Det finns en metacentrisk kromosom (t12;13) som bör räknas som en kromosom. TK-lokus hos möss finns på den distala änden av kromosom 11. Cellinjen L5178Y TK
+/– –3.7.2C har mutationer i båda p53-allelerna och producerar mutant-p53-protein (32) (33). p53-status för cellinjen TK+/––3.7.2C är sannolikt orsaken till att testet kan detektera storskalig skada (17).
|
|
14.
|
För TK6: TK6 är en human lymfoblastoidcellinje. Den parentala cellinjen är cellinjen WI-L2 som transformerats med Epstein-Barr-virus, och som ursprungligen härleddes från en femårig pojke med hereditär sfärocytos. Den första isolerade klonen, HH4, mutageniserades med ICR191 och en TK-heterozygot cellinje, TK6, skapades (34). TK6-cellerna är nästan diploida och den representativa karyotypen är 47, XY, 13+, t(14; 20), t(3; 21) (35). TK-lokus hos människa finns på den långa armen av kromosom 17. TK6 är en p53-kompetent cellinje, eftersom den har en p53-sekvens av vildtyp i båda allelerna och endast uttrycker p53-protein av vildtyp (36).
|
|
15.
|
För både MLA och TK6 gäller att när testlaboratoriet etablerar eller fyller på en mastercellstam bör det försäkra sig om frånvaro av mykoplasmakontamination , karyotypbestämma cellerna eller färga de kromosomer som hyser TK-lokus samt kontrollera populationernas fördubblingstider. Den normala cellcykeltiden för de celler som används i testlaboratoriet bör fastställas och bör överensstämma med kända cellegenskaper (16)(19)(37). Stamodlingen bör lagras vid –150 °C eller lägre och användas för att bereda alla arbetslösningar med celler.
|
|
16.
|
Antingen innan ett stort antal nedfrysta stammar för användning etableras eller precis innan de används i ett försök, kan odlingen behöva rengöras av redan befintliga muterade celler (om inte lösningsmedelskontrollens mutantfrekvens [MF] redan ligger inom det godtagbara intervallet – se tabell 2 för MLA). Detta åstadkoms genom användning av metotrexat (aminopterin) för att selektera mot celler som saknar TK och tillföra tymidin, hypoxantin och glycin (L5178Y) eller 2’-deoxicytidin (TK6) till odlingen för att säkerställa optimal tillväxt hos de TK-kompetenta cellerna (19) (38) (39) samt (40) för TK6). Allmänna råd om god praxis för underhåll av cellodlingar och specifika råd för L5178Y- och TK6-celler finns i (19) (31) (37) (39) (41). För laboratorier som behöver mastercellstammar för att inleda antingen MLA eller TK6 eller för att erhålla nya mastercellstammar finns ett förråd med väl karakteriserade celler tillgängligt (37).
|
Medier och odlingsbetingelser
|
17.
|
För båda testen bör lämpliga odlingsmedier och inkubationsförhållanden (odlingskärl, fuktad atmosfär med 5 % CO2, inkubationstemperatur på 37 °C) väljas för odlingarna. Celler bör alltid odlas under förhållanden som garanterar en logfastillväxt. Det är särskilt viktigt att betingelserna för odlingarna väljs på ett sådant sätt att man säkrar en optimal celltillväxt under expressionsperioden och kloning för både muterade celler och icke-muterade celler. För MLA och TK6 är det också viktigt att odlingsförhållandena säkerställer optimal tillväxt hos både stora kolonier av/tidigt uppträdande TK-mutanter och små kolonier av/sent uppträdande TK-mutanter. Närmare information om odlingsförhållanden, inklusive behovet av att värmeinaktivera hästserum om RPMI-medium används under mutanturvalet, finns i (19) (31) (38) (39) (40) (42).
|
Beredning av odlingarna
|
18.
|
Cellerna dras upp från stamodlingar som är sådda i ett odlingsmedium som har en sådan densitet att suspensionsodlingarna fortsätter att växa exponentiellt genom behandlings- och expressionsperioderna.
|
Metabolisk aktivering
|
19.
|
Exogena metaboliserande system bör användas när L5178Y- och TK6-cellerna inte har tillräcklig endogen metabolisk kapacitet. Det mest använda systemet, som rekommenderas som standard om inte annat kan motiveras, är en kofaktorstödd postmitokondriell fraktion (S9) som beretts av lever från gnagare (vanligen råttor) som behandlats med enzyminducerande ämnen, t.ex. Aroclor 1254 (43) (44) (45) eller en kombination av fenobarbital och β-naftoflavon (46) (47) (48) (49) (50) (51). Den senare kombinationen bryter inte mot Stockholmskonventionen om långlivade organiska föroreningar (52) och har visat sig vara lika effektiv som Aroclor 1254 när det gäller att inducera multifunktionsoxidaser (”mixed-function oxidases”) (45)(46)(47)(48)(49). S9-fraktionen används vanligen i koncentrationer mellan 1–2 volymprocent men kan ökas till 10 volymprocent i det slutliga testmediet. Valet av typ och koncentration av använt exogent metaboliskt aktiveringssystem eller metabolisk induktion kan påverkas av klassen av testkemikalier.
|
Beredning av testkemikalien
|
20.
|
Fasta testkemikalier bör lösas upp i eller blandas med lämpliga lösningsmedel eller vehiklar och vid behov spädas ut innan cellerna behandlas (se punkt 21). Flytande testkemikalier kan tillsättas direkt till testsystemet och/eller spädas ut före behandling av testsystemet. Gaser eller flyktiga testkemikalier bör testas genom lämpliga modifikationer av standardprotokollen, såsom behandling i slutna odlingskärl (53) (54) (55). Testkemikalien bör beredas strax innan behandling om inte stabilitetsdata visar att lagringen är acceptabel.
|
TESTBETINGELSER
Lösningsmedel
|
21.
|
Man bör välja ett lösningsmedel som optimerar testkemikaliens löslighet utan att testet påverkas negativt, t.ex. genom förändringar i celltillväxten, påverkan på testkemikaliens integritet, reaktion med odlingskärlen eller försämrad metabolisk aktivering. När det är möjligt, rekommenderas att man i första hand överväger vattenbaserade lösningsmedel (eller odlingsmedier). Vedertagna lösningsmedel är vatten eller dimetylsulfoxid. Generellt bör organiska lösningsmedel inte överstiga 1 volymprocent och vattenbaserade lösningsmedel (saltlösningar eller vatten) inte överstiga 10 volymprocent i det slutliga behandlingsmediet. Om man använder lösningsmedel som inte är vedertagna (t.ex. etanol eller aceton) bör detta stödjas av data som visar att de är kompatibla med testkemikalierna och testsystemet och att de inte är genotoxiska vid den använda koncentrationen. Om ett sådant underlag saknas är det viktigt att inkludera obehandlade kontroller (se tillägg 1, Definitioner) för att visa att inga skadliga eller mutagena effekter induceras av det valda lösningsmedlet.
|
MÄTNING AV CYTOTOXICITET OCH VAL AV BEHANDLINGSKONCENTRATIONER
|
22.
|
Vid bestämning av maximal testkemikaliekoncentration bör man undvika koncentrationer som kan ge artefaktiska positiva resultat, t.ex. sådana som medför alltför kraftig cytotoxicitet (se punkt 28), fällning i odlingsmediet (se punkt 29) eller betydande ändringar av pH eller osmolalitet (se punkt 8). Om testkemikalien orsakar en betydande ändring av mediets pH vid tillsättningen kan pH justeras genom buffring av det slutliga behandlingsmediet för att undvika artefaktiska positiva resultat och upprätthålla lämpliga odlingsförhållanden.
|
|
23.
|
Valet av koncentrationer baseras på cytotoxicitet och andra överväganden (se punkterna 27-30). Även om det kan vara användbart att utvärdera cytotoxicitet i ett inledande test för att bättre definiera de koncentrationer som ska användas i huvudförsöket är ett inledande test inte obligatoriskt. Även om en inledande cytotoxicitetsbedömning görs måste cytotoxiciteten för varje odling mätas i huvudexperimentet. Om ett förberedande test görs bör det täcka ett stort koncentrationsintervall och kan antingen avslutas på dag 1 efter behandling eller fortsätta genom expressionsperioden på två dagar fram till mutanturvalet (om det står klart att de använda koncentrationerna är lämpliga).
|
|
24.
|
Cytotoxicitet bör fastställas för varje individuell test- och kontrollodling: metoderna för MLA (2) och TK6 (15) definieras genom internationellt vedertagen praxis.
|
|
25.
|
För både agar- och mikrotiterversionerna av MLA: Cytotoxicitet bör utvärderas med användning av relativ total tillväxt (RTG), som ursprungligen definierades av Clive och Spector 1975 (2). Detta mått omfattar relativ suspensionstillväxt (RSG: testodling jämfört med lösningsmedelskontroll) under cellbehandling, expressionstiden samt relativ kloningseffektivitet (RCE: testodling jämfört med lösningsmedelskontroll) vid den tidpunkt då mutanterna väljs (2). Det bör noteras att RSG innefattar eventuell cellförlust som förekommer i testodlingen under behandling (formeln anges i tillägg 2).
|
|
26.
|
För TK6: Cytotoxicitet bör bedömas genom användning av relativ överlevnad (RS), dvs. kloningseffektiviteten för celler som överförts till platta omedelbart efter behandling, och justeras utifrån eventuell cellförlust under behandlingen samt baseras på antal celler jämfört med den negativa kontrollen (som tilldelas en överlevnadskvot på 100 %) (se tillägg 2 för formeln).
|
|
27.
|
Minst tre testkoncentrationer (exklusive lösningsmedel och positiva kontroller) som uppfyller acceptanskriterierna (lämplig cytotoxicitet, antal celler osv.) bör utvärderas. Även om användning av duplikat är att föredra kan både replikat och enskilt behandlade odlingar användas för varje koncentration som testas. De resultat som erhölls för replikatodlingarna vid en given koncentration kan slås ihop för dataanalysen (55). För testkemikalier med låg eller ingen cytotoxicitet är två- till trefaldiga koncentrationsintervall vanligen lämpliga. Om testkemikalien är cytotoxisk bör testkoncentrationerna väljas så att de täcker ett intervall för cytotoxiciteten enligt beskrivningen i punkt 28 och omfatta koncentrationer med måttlig, låg eller ingen cytotoxicitet. Många testkemikalier uppvisar koncentrationsresponskurvor. För att täcka in alla grader av cytotoxicitet eller undersöka koncentrationsresponsen i detalj kan det vara nödvändigt att använda koncentrationer som ligger närmare varandra och fler än fyra koncentrationer, i synnerhet i situationer där försöket måste upprepas (se punkt 70). Användning av mer än fyra koncentrationer kan vara särskilt viktigt vid användande av enstaka odlingar.
|
|
28.
|
Om den maximala koncentrationen baseras på cytotoxicitet bör den högsta koncentrationen syfta till att uppnå mellan 20 och 10 % relativ total tillväxt (RTG) för MLA, och mellan 20 och 10 % relativ överlevnad (RS) för TK6 (punkt 67).
|
|
29.
|
För svårlösliga testkemikalier som inte är cytotoxiska vid koncentrationer som är lägre än den lägsta olösliga koncentrationen bör den högsta analyserade koncentrationen framkalla grumlighet eller en fällning som är synlig med blotta ögat eller med hjälp av ett inverterat mikroskop vid slutet av behandlingen med testkemikalien. Även om cytotoxicitet förekommer över den lägsta olösliga koncentrationen är det lämpligt att testa endast en koncentration som producerar grumlighet eller har en synlig fällning, eftersom fällningen kan ge artefaktiska effekter. MLA ochTK6 använder suspensionsodlingar, och det är därför viktigt att noggrant se till att fällningen inte stör testet. Bestämning av löslighet i odlingsmediet före försöket kan också vara användbart.
|
|
30.
|
Om ingen fällning eller begränsande cytotoxicitet observeras ska de högsta testkoncentrationerna motsvara det lägsta av följande värden: 10 mM, 2 mg/ml eller 2 μl/ml (57) (58). Om testkemikalien inte har en definierad sammansättning, t.ex. ett ämne med okänd eller varierande sammansättning, komplexa reaktionsprodukter eller biologiskt material (dvs. UVCB-ämnen), miljöextrakt osv., kan man behöva använda en högre toppkoncentration (t.ex. 5 mg/ml) om cytotoxiciteten inte är tillräckligt hög, så att koncentrationen av varje komponent ökas. Det bör dock noteras att dessa krav kan skilja sig för humanläkemedel (59).
|
Kontroller
|
31.
|
Parallella negativa kontroller (se punkt 21), bestående av enbart lösningsmedel i behandlingsmediet, och hanterade på samma sätt som behandlingsodlingarna, bör inkluderas för varje testbetingelse.
|
|
32.
|
Parallella positiva kontroller behövs för att visa laboratoriets förmåga att identifiera mutagener enligt villkoren i det använda testprotokollet och, i förekommande fall, effektiviteten hos det exogena systemet för metabolisk aktivering, och för att visa adekvat detektion både av små/sent uppträdande och stora/tidigt uppträdande TK-mutanter. Exempel på positiva kontroller ges i tabell 1 nedan. Alternativa positiva kontrollämnen kan användas om det är motiverat. In vitro-tester av däggdjursceller med avseende på genetisk toxicitet är tillräckligt standardiserade för kortvariga behandlingar (3–4 timmar) som görs parallellt med och utan metabolisk aktivering med samma behandlingslängd. Användningen av positiva kontroller kan därför begränsas till en mutagen som kräver metabolisk aktivering. I detta fall visar en enda positiv kontrollrespons både aktiviteten hos det metaboliska aktiveringssystemet och testsystemets känslighet. Separata positiva kontroller bör dock göras av långvariga behandlingar (dvs. 24 timmar utan S9) om sådana används, eftersom behandlingstiden skiljer sig från testet med metabolisk aktivering. Varje positiv kontroll bör användas för en eller flera koncentrationer som förväntas ge ökningar över bakgrunden som går att reproducera och detektera. Syftet med detta är att visa testsystemets sensitivitet , och responsen får inte äventyras av cytotoxicitet som överskrider de gränser som anges i denna testmetod (se punkt 28).
|
Tabell 1
Rekommenderade referensämnen för utvärdering av laboratoriets kompetens och för val av positiva kontroller
|
Kategori
|
Ämne
|
CAS-nr
|
|
1. Mutagener utan metabolisk aktivering
|
|
Metylmetansulfonat
|
66-27-3
|
|
Mitomycin C
|
50-07-7
|
|
4-nitrokinolin-N-oxid
|
56-57-5
|
|
2. Mutagener som kräver metabolisk aktivering
|
|
Benso(a)pyren
|
50-32-8
|
|
Cyklofosfamid (monohydrat)
|
50-18-0 (6055-19-2)
|
|
7,12-dimetylbenzantracen
|
57-97-6
|
|
3-metylkolantren
|
56-49-5
|
FÖRFARANDE
Behandling med testkemikalien
|
33.
|
Prolifererande celler behandlas med testkemikalien i såväl närvaro som frånvaro av ett system för metabolisk aktivering. Exponering bör ske under en lämplig tidsperiod (normalt är 3 till 4 timmar tillräckligt). Det bör dock noteras att dessa krav kan skilja sig för humanläkemedel (59). I fall där den kortsiktiga behandlingen ger negativa resultat för MLA och det finns information som tyder på att det krävs en längre behandling [t.ex. nukleosida analoger, svårlösliga kemikalier (5)(59)], bör man överväga att utföra testet med längre behandling, dvs. 24 timmar utan S9.
|
|
34.
|
Minimiantalet celler som används i varje testodling (kontrollodlingar och behandlade odlingar) för varje etapp i testet bör baseras på den spontana mutantfrekvensen. En generell regel är att behandla och ympa om ett tillräckligt antal celler i varje försöksodling för att bibehålla minst 10 men helst 100 spontana mutanter i alla faser av testet (behandling, fenotypiskt uttryck och mutanturval) (56).
|
|
35.
|
För MLA är den rekommenderade godtagbara spontana mutantfrekvensen 35–140 × 10–6 (agarversionen) och 50–170 × 10–6 (mikrotiterversionen) (se tabell 2). För att minst 10 och helst 100 spontana mutanter ska överleva behandlingen för varje testodling är det nödvändigt att behandla minst 6 × 106 celler. Om detta antal celler behandlas och ett tillräckligt antal celler bibehålls under expressionsperioden och kloningen för mutanturvalet, ger det ett tillräckligt antal spontana mutanter (tio eller fler) under alla faser i försöket, även för de odlingar som behandlas vid koncentrationer som leder till 90 % cytotoxicitet (mätt med en RTG på 10 %) (19)(38)(39).
|
|
36.
|
För TK6 brukar den spontana mutantfrekvensen ligga på mellan 2 och 10 × 10–6. För att minst 10 spontana mutanter ska överleva behandlingen för varje odling är det nödvändigt att behandla minst 20 × 106 celler. Om detta antal celler behandlas erhålls ett tillräckligt antal spontana mutanter (tio eller fler), även för de odlingar som behandlas vid koncentrationer som leder till 90 % cytotoxicitet under behandling (10 % RS). Dessutom måste ett tillräckligt stort antal celler odlas fram under expressionsperioden och överföras till platta för mutanturvalet (60).
|
Fenotypisk expressionstid och mätning av cytotoxicitet och mutantfrekvens
|
37.
|
I slutet av behandlingsperioden odlas celler under en fastställd tidsperiod så att nära optimal fenotypisk expression av nyligen inducerade mutanter medges, som är specifik för varje cellinje. För MLA är den fenotypiska expressionstiden två dagar. För TK6 är den fenotypiska expressionstiden tre till fyra dagar. Om 24-timmarsbehandling används inleds expressionstiden när behandlingen har avslutats.
|
|
38.
|
Under den fenotypiska expressionsperioden räknas cellerna dagligen. För MLA används den dagliga cellräkningen för att beräkna den dagliga suspensionstillväxten (SG). Efter den två dagar långa expressionsperioden suspenderas cellerna i ett medium med eller utan selektivt ämne för bestämning av antalet mutanter (urvalsplattor) respektive kloningseffektivitet (viabilitetsplattor). För MLA finns det två lika godtagbara metoder för kloning av mutanturvalet: i den ena metoden används mjuk agar och i den andra ett flytande medium i 96-hålsplattor (19) (38) (39). Kloningen i TK6 utförs med ett flytande medium och 96-hålsplattor (16).
|
|
39.
|
Trifluortymidin (TFT) är det enda rekommenderade selektiva ämnet för TK-mutanter (61).
|
|
40.
|
För MLA räknas agarplattor och mikrotiterplattor efter 10–12 dagars inkubation. Vad beträffar TK6 räknas kolonierna i mikrotiterplattorna efter 10–14 för de tidigt uppträdande mutanterna. För att återvinna långsamt växande (sent uppträdande) TK6-mutanter är det nödvändigt att återigen tillföra cellerna tillväxtmedium och TFT efter räkningen av tidigt uppträdande mutanter och därefter inkubera plattorna i ytterligare 7–10 dagar (62). Räkningen av långsamt och normalt växande TK-mutanter diskuteras i punkterna 42 och 44.
|
|
41.
|
Lämpliga beräkningar för de två testerna inklusive de två metoderna (agar och mikrotiterplatta) för MLA anges i tillägg 2. När det gäller agarmetoden för MLA räknas kolonierna och antalet mutantkolonier justeras med kloningseffektiviteten för att beräkna en mutantfrekvens. Vad gäller mikrotiterversionen för MLA och TK6 bestäms kloningseffektiviteten för både urvals- och kloningseffektivitetsplattorna enligt Poissonfördelningen (63). Mutantfrekvensen beräknas utifrån dessa två kloningseffektivitetsvärden.
|
Karakterisering av mutantkolonier
|
42.
|
För MLA bör, om testkemikalien är positiv (se punkterna 62–63), karakteriseringen av kolonier genom kolonistorlek eller tillväxt utföras på minst en av testodlingarna (vanligen den högsta godtagbara positiva koncentrationen) och på de negativa och positiva kontrollerna. Om testkemikalien är negativ (se punkt 64) bör karakteriseringen av mutantkolonier utföras på de negativa och positiva kontrollerna. I mikrotitermetoden för MLA definieras små kolonimutanter som sådana som täcker mindre än 25 % av hålets diameter, och stora kolonimutanter som sådana som täcker mer än 25 % av hålets diameter. För agarmetoden används en automatisk koloniräknare för att räkna mutantkolonierna och bestämma kolonistorlek. Metoder för storleksbestämning av kolonier anges i litteraturen (19) (38) (40). Karakterisering av kolonier för den negativa och positiva kontrollen är nödvändig för att visa att studierna utförs på lämpligt sätt.
|
|
43.
|
Testkemikalien kan inte bestämmas som negativ om varken de stora eller de små kolonimutanterna detekteras korrekt i den positiva kontrollen. Kolonikarakterisering kan användas för att tillhandahålla allmän information om testkemikaliens förmåga att orsaka punktmutationer och/eller kromosomhändelser (punkt 4).
|
|
44.
|
TK6: Normalt växande och långsamt växande mutanter differentieras genom en skillnad i inkubationstid (se punkt 40). För TK6 räknas vanligen både de tidigt och de sent uppträdande mutanterna för samtliga odlingar, inklusive de negativa och positiva kontrollerna. Karakterisering av kolonier för den negativa och positiva kontrollen är nödvändig för att visa att studierna utförs på lämpligt sätt. Testkemikalien kan inte bestämmas som negativ om varken de tidigt eller de sent uppträdande mutanterna detekteras korrekt i den positiva kontrollen. Kolonikarakterisering kan användas för att tillhandahålla allmän information om testkemikaliens förmåga att orsaka punktmutationer och/eller kromosomhändelser (punkt 4).
|
Laboratoriets kompetens
|
45.
|
För att visa att det finns tillräcklig erfarenhet av testet innan det används för rutintestning bör laboratoriet ha utfört en serie försök med positiva referensämnen som agerar via olika mekanismer (minst ett som är aktivt med och ett som är aktivt utan metabolisk aktivering, valt från kemikalierna i tabell 1) och flera negativa kontroller (inklusive obehandlade odlingar och olika lösningsmedel/vehiklar). Dessa positiva och negativa kontrollresponser bör överensstämma med litteraturen. Detta gäller inte laboratorier som redan har erfarenhet, dvs. har en sådan historisk databas som beskrivs i punkterna 47–50. För MLA bör de värden som erhålls för både positiva och negativa kontroller överensstämma med IWGT-rekommendationerna (se tabell 2).
|
|
46.
|
Ett urval av positiva kontrollämnen (se tabell 1) bör undersökas med korta och långa behandlingstider (om långa behandlingstider används) utan metabolisk aktivering, och även med kort behandlingstid med metabolisk aktivering, för att visa att det har kompetens att upptäcka mutagena kemikalier, bestämma effektiviteten hos det metaboliska aktiveringssystemet och visa att det råder lämpliga förhållanden för celltillväxt under behandling, fenotypisk expression och mutanturval samt att räkningsförfarandena är lämpliga. En serie koncentrationer av de valda ämnena bör användas för att ge reproducerbara och koncentrationsrelaterade ökningar över bakgrunden i syfte att visa testsystemets sensitivitet och dynamiska omfång.
|
Historiska kontrolldata
|
47.
|
Laboratoriet bör fastställa följande:
|
—
|
Historiskt positivt kontrollintervall samt fördelning.
|
|
—
|
Historiskt negativt (obehandlad, lösningsmedel) kontrollintervall samt fördelning.
|
|
|
48.
|
Om data för en historisk negativ kontrollfördelning samlas in först bör de parallella negativa kontrollerna överensstämma med publicerade data för negativa kontroller. Allteftersom kontrollfördelningen kompletteras med fler försöksdata bör de parallella negativa kontrollerna företrädesvis ligga inom fördelningens kontrollgräns på 95 % (64) (65).
|
|
49.
|
Laboratoriets historiska databas för negativa kontroller bör inledningsvis baseras på minst 10 försök, men bör helst bygga på minst 20 försök som utförts under jämförbara testbetingelser. Laboratorierna bör använda kvalitetskontrollmetoder såsom kontrolldiagram (t.ex. C-diagram eller X-stapeldiagram (65)), för att fastställa hur variabla deras positiva och negativa kontrolldata är och för att visa att metoden är ”under kontroll” i deras laboratorium (66). Närmare anvisningar och rekommendationer om hur man bygger upp och använder de historiska data finns i litteraturen (64).
|
|
50.
|
Negativ kontrolldata bör bestå av mutantfrekvenser från enstaka eller helst replikata odlingar enligt beskrivningen i punkt 27. Parallella negativa kontroller bör helst ligga inom kontrollgränsen på 95 % för fördelningen av laboratoriets historiska negativa kontrolldatabas. Om negativa kontrolldata faller utanför kontrollgränsen på 95 % kan de ändå ingå i den historiska kontrollfördelningen så länge de inte är extremt avvikande och det finns bevis för att testsystemet är under kontroll (se punkt 49) och att inga tekniska eller mänskliga fel förekommit.
|
|
51.
|
Eventuella ändringar av försöksprotokollen bör övervägas när det gäller datas överensstämmelse med laboratoriets befintliga historiska kontrolldatabaser. Alla större inkonsekvenser bör leda till att en ny historisk kontrolldatabas upprättas.
|
DATA OCH RAPPORTERING
Presentation av resultaten
|
52.
|
Presentationen av data för både MLA och TK6 bör, för både behandlade odlingar och kontrollodlingar, innefatta de data som krävs för beräkning av cytotoxicitet (relativ total tillväxt respektive relativ överlevnad) och mutantfrekvenser, enligt beskrivningen nedan.
|
|
53.
|
För MLA bör data för enskilda odlingar lämnas för relativ suspensionstillväxt, relativ total tillväxt, kloningseffektivitet vid tidpunkten för mutanturvalet samt antal mutantkolonier (för agarversionen) eller antal tomma hål (för mikrotiterversionen). Mutantfrekvensen bör uttryckas som antalet muterade celler per miljon överlevande celler. Om responsen är positiv bör mutantfrekvensen för små och stora kolonier (och/eller procentandelen av den totala mutantfrekvensen) anges för minst en koncentration av testkemikalien (vanligen den högsta positiva koncentrationen) samt de negativa och positiva kontrollerna. I händelse av negativ respons bör mutantfrekvensen för små och stora kolonier anges för den negativa och den positiva kontrollen.
|
|
54.
|
För TK6 bör data för enskilda odlingar anges för relativ överlevnad, kloningseffektivitet vid tidpunkten för mutanturvalet samt antalet tomma hål för tidigt och sent uppträdande mutanter. Mutantfrekvensen bör uttryckas som antalet muterade celler per antalet överlevande celler, och bör inbegripa den totala mutantfrekvensen och mutantfrekvensen (och eller procentandelen av den totala mutantfrekvensen) för tidigt och sent uppträdande mutanter.
|
Acceptanskriterier
|
55.
|
Följande kriterier bör uppfyllas för både MLA och TK6 innan de övergripande resultaten bestäms för en specifik testkemikalie:
|
—
|
Två olika testbetingelser (dvs kort behandlingstid med och utan metabolisk aktivering – se punkt 33) har tillämpats om inte ett av dem gav positiva resultat.
|
|
—
|
Tillräckligt många celler och koncentrationer bör finnas tillgängliga för analys (se punkterna 27 och 34–36).
|
|
—
|
Urvalskriterierna för toppkoncentrationen överensstämmer med de kriterier som beskrivs i punkterna 28–30.
|
|
Acceptanskriterier för negativa och positiva kontroller
|
56.
|
IWGT:s expertarbetsgrupp för MLA genomförde en analys av en stor mängd MLA-data, som ledde till internationell enighet om specifika acceptanskriterier för MLA (1)(2)(3)(4)(5). Denna testmetod innehåller därför särskilda rekommendationer för att fastställa om negativa och positiva kontroller kan godtas och för utvärdering av enskilda ämnesresultat i MLA. TK6 har en mycket mindre databas och har inte utvärderats av en arbetsgrupp.
|
|
57.
|
För MLA bör varje försök utvärderas för att se om den obehandlade kontrollen/lösningsmedelskontrollen uppfyller IWGT MLA-arbetsgruppens acceptanskriterier ((4) och tabell 2 nedan) för 1) mutantfrekvens (observera att de mutantfrekvenser som IWGT anser vara godtagbara skiljer sig åt för agar- och mikrotiterversionerna av MLA), 2) kloningseffektivitet vid tidpunkten för mutanturvalet, och 3) suspensionstillväxt för lösningsmedelskontrollen (formeln anges i tillägg 2).
Tabell 2
Acceptanskriterier för MLA
|
Parameter
|
Mjukagarmetod
|
Mikrotitermetod
|
|
Mutantfrekvens
|
35–140 × 10–6
|
50–170 × 10–6
|
|
Kloningseffektivitet
|
65–120 %
|
65–120 %
|
|
Suspensionstillväxt
|
8–32-faldig (3–4 timmars behandling) 32–180-faldig (24 timmars behandling, om den utförs)
|
8–32-faldig (3–4 timmars behandling) 32–180-faldig (24 timmars behandling, om den utförs)
|
|
58.
|
För MLA bör varje test även utvärderas för att se om de positiva kontrollerna uppfyller minst en av följande två acceptanskriterier som har tagits fram av IWGT-arbetsgruppen:
|
—
|
Den positiva kontrollen bör uppvisa en absolut ökning i total mutantfrekvens, dvs. en ökning över den spontana bakgrundsmutantfrekvensen (inducerad mutantfrekvens [IMF]) på minst 300 × 10–6. Minst 40 % av IMF bör avspeglas i mutantfrekvensen för små kolonier.
|
|
—
|
Den positiva kontrollen ska uppvisa en ökning av mutantfrekvensen i små kolonier på minst 150 × 10–6 över den frekvens som observeras i den parallella obehandlade/lösningsmedelskontrollen (en IMF i små kolonier på 150 × 10–6).
|
|
|
59.
|
För TK6 är ett test godtagbart om den parallella negativa kontrollen anses godtagbar för registrering i laboratoriets historiska databas över negativa kontroller enligt punkterna 48–49. Parallella positiva kontroller (se punkt 32) bör dessutom framkalla reaktioner som överensstämmer med de kontroller som genereras i den historiska databasen över positiva kontroller och ge en statistiskt signifikant ökning jämfört med den parallella negativa kontrollen.
|
|
60.
|
För båda testerna bör den övre cytotoxicitetsgräns som observerats i den positiva kontrollodlingen vara densamma som för försökskulturerna, dvs. relativ total tillväxt/relativ överlevnad (RTG/RS) bör inte vara lägre än 10 %. Det är tillräckligt att använda en enstaka koncentration (eller en av koncentrationerna i de positiva kontrollodlingarna om fler än en koncentration används) för att visa att acceptanskriterierna för den positiva kontrollen har uppfyllts. Den positiva kontrollens mutantfrekvens måste dessutom ligga inom det godtagbara intervall som har fastställts av laboratoriet.
|
|
Utvärdering och tolkning av resultaten
|
61.
|
För MLA har ingående arbete om biologisk relevans och kriterier för positiv respons utförts av IWGT:s expertarbetsgrupp för muslymfom (4). Denna testmetod innehåller därför särskilda rekommendationer för tolkning av testkemikalieresultat från MLA (se punkterna 62–64). TK6 har en mycket mindre databas och har inte utvärderats av en arbetsgrupp. Rekommendationerna för tolkning av data för TK6 är därför mer generellt utformade (se punkterna 65–66). Ytterligare rekommendationer gäller för båda testerna (se punkterna 67–71).
|
MLA
|
62.
|
En metod för att definiera positiva och negativa responser rekommenderas för att säkerställa att den ökade mutantfrekvensen är biologiskt relevant. I stället för de statistiska analyser som generellt används för andra tester bygger denna metod på användningen av en på förhand fastställd inducerad mutantfrekvens (dvs. en ökning av mutantfrekvensen utöver den parallella kontrollen). Den bestämmer den globala utvärderingsfaktorn (GEF), som i sin tur baseras på en analys av distributionen av data om mutantfrekvensen i negativa kontroller från deltagande laboratorier (4). För agarversionen av MLA är GEF 90 × 10–6, och för mikrotiterversionen är GEF 126 × 10–6.
|
|
63.
|
Förutsatt att samtliga acceptanskriterier är uppfyllda anses en testkemikalie vara klart positiv om ökningen av mutantfrekvensen över den parallella bakgrunden överskrider GEF under något av de undersökta testbetingelserna (se punkt 33), och ökningen är koncentrationsrelaterad (t.ex. med användning av ett trendtest). Testkemikalien anses då kunna inducera mutation i detta testsystem.
|
|
64.
|
Förutsatt att samtliga acceptanskriterier är uppfyllda anses en testkemikalie vara klart negativ om det inte förekommer någon koncentrationsrelaterad respons under något av de undersökta testbetingelserna (se punkt 33), eller om det finns en ökning av mutantfrekvensen som inte överskrider GEF. Testkemikalien anses då inte kunna inducera mutationer i detta testsystem.
|
TK6
|
65.
|
Under förutsättning att samtliga acceptanskriterier är uppfyllda anses testkemikalien vara klart positiv om något av följande gäller för någon av de undersökta testbetingelserna (se punkt 33):
|
—
|
Minst en av testkoncentrationerna uppvisar en statistiskt signifikant ökning jämfört med den parallella negativa kontrollen.
|
|
—
|
Det finns ingen koncentrationsrelaterad ökning vid utvärdering med ett lämpligt trendtest (se punkt 33).
|
|
—
|
Något av resultaten ligger utanför fördelningen av historiska negativa kontrolldata (t.ex. Poisson-baserade 95 %-kontrollgränser, se punkt 48).
|
Om alla dessa kriterier är uppfyllda anses testkemikalien kunna inducera mutation i detta testsystem. Rekommendationer om de lämpligaste statistiska metoderna finns i litteraturen (66) (67).
|
|
66.
|
Under förutsättning att samtliga acceptanskriterier är uppfyllda anses testkemikalien vara klart negativ om följande gäller för samtliga undersökta testbetingelser (se punkt 33):
|
—
|
Ingen av testkoncentrationerna uppvisar en statistiskt signifikant ökning jämfört med den parallella negativa kontrollen.
|
|
—
|
Det finns ingen koncentrationsrelaterad ökning vid utvärdering med ett lämpligt trendtest.
|
|
—
|
Alla resultat ligger inom fördelningen av historiska negativa kontrolldata (t.ex. Poisson-baserade 95 %-kontrollgränser, se punkt 48).
|
Testkemikalien anses då inte kunna inducera mutationer i detta testsystem.
|
För både MLA och TK6:
|
67.
|
Om den maximala koncentrationen baseras på cytotoxicitet bör den högsta koncentrationen syfta till att uppnå mellan 20 och 10 % RTG/RS. Det råder samförstånd om att man bör vara försiktig vid tolkning av positiva resultat som endast påvisats mellan 20 och 10 % RTG/RS. Resultatet anses inte vara positivt om ökningen av mutantfrekvensen endast inträffade vid eller under 10 % RTG/RS (om dessa värden utvärderas) (2)(59).
|
|
68.
|
Under vissa omständigheter kan kompletterande information bidra till att fastställa att en testkemikalie inte är mutagen när det inte finns någon odling som visar ett RTG-värde mellan 10–20 % RTG/RS. Dessa situationer kan beskrivas enligt följande: 1) Det finns inga bevis på mutagenicitet (t.ex. ingen dosrespons, inga mutantfrekvenser över de frekvenser som observerats i den parallella negativa kontrollen eller i historiska bakgrundsintervall osv.) i en serie av datapunkter inom 100 % till 20 % RTG/RS och det finns minst en datapunkt mellan 20 och 25 % RTG/RS. 2) Det finns inga bevis på mutagenicitet (t.ex. ingen dosrespons, inga mutantfrekvenser över de frekvenser som observerats i den parallella negativa kontrollen eller i historiska bakgrundsintervall osv.) i en serie av datapunkter mellan 100 % till 25 % RTG/RS och det finns även en negativ datapunkt strax under 10 % RTG/RS. I båda dessa situationer kan testkemikalien anses vara negativ.
|
|
69.
|
Det finns inget krav på verifiering av ett tydligt positivt eller negativt svar.
|
|
70.
|
Om responsen varken är klart negativ eller klart positiv enligt beskrivningen ovan och/eller för att bidra till att fastställa resultatets biologiska relevans bör data utvärderas genom en expertbedömning och/eller ytterligare undersökningar. Det kan även vara användbart att upprepa försöket med ändrade testbetingelser [t.ex. koncentrationsavstånd för att öka sannolikheten för att erhålla datapunkter inom intervallet 10–20 % RTG/RS, användning av andra metaboliska aktiveringsförhållanden (dvs. S9-koncentration eller S9-ursprung) och behandlingstider].
|
|
71.
|
I sällsynta fall är det, trots ytterligare undersökningar, inte möjligt att dra en slutsats om huruvida testkemikalien ger positiva eller negativa resultat. Responsen på testkemikalien ska i sådana fall anses vara tvetydig (vilket innebär att den lika gärna kan vara positiv som negativ).
|
TESTRAPPORT
|
72.
|
Testrapporten ska innehålla följande information:
|
|
Testkemikalie:
|
—
|
källa, satsnummer, sista förbrukningsdag om detta anges,
|
|
—
|
testkemikaliens stabilitet i sig, om den är känd,
|
|
—
|
löslighet och stabilitet av testkemikalien i lösningsmedlet, om detta är känt,
|
|
—
|
Mätning av pH, osmolalitet och fällningar i det odlingsmedium till vilket testkemikalien tillsattes, i förekommande fall.
|
|
|
Monokomponentämne:
|
—
|
fysikaliskt tillstånd, vattenlöslighet och ytterligare relevanta fysikalisk-kemiska egenskaper.
|
|
—
|
kemisk identifiering, t.ex. med IUPAC- eller CAS-namn, CAS-nummer, Smiles- eller InChI-kod, strukturformel, renhet samt motsvarande identifiering av föroreningar i förekommande fall och om det är praktiskt möjligt.
|
|
|
|
Multikomponentämne, UVCB-ämnen och blandningar:
|
—
|
karakteriseras så långt som möjligt genom beståndsdelarnas kemiska identitet (se ovan), kvantitativa förekomst och relevanta fysikalisk-kemiska egenskaper.
|
|
|
|
|
Lösningsmedel:
|
—
|
motivering av valet av lösningsmedel,
|
|
—
|
procentandelen lösningsmedel i den slutliga odlingsmediet.
|
|
|
|
Celler:
|
|
För stamodlingar i laboratorium:
|
—
|
cellernas typ och källa samt historia i det testande laboratoriet,
|
|
—
|
Kromosomernas karyotypiska egenskaper och/eller modala antal.
|
|
—
|
Metoder för underhåll av cellodlingar.
|
|
—
|
frånvaro av mykoplasma,
|
|
|
|
|
Testbetingelser:
|
—
|
grund för val av koncentrationer och antalet cellodlingar, inklusive t.ex. cytotoxicitetsdata och löslighetsbegränsningar,
|
|
—
|
Mediets sammansättning, CO2-koncentration och luftfuktighet.
|
|
—
|
Testkemikaliens koncentration, uttryckt som slutlig koncentration i odlingsmediet (t.ex. μg eller mg/ml eller mM av odlingsmediet).
|
|
—
|
Koncentration (och/eller volym) av det lösningsmedel och den testkemikalie som tillsätts odlingsmediet.
|
|
—
|
behandlingens varaktighet,
|
|
—
|
celldensitet under behandlingen,
|
|
—
|
typ och sammansättning hos det system som används för metabolisk aktivering (S9-källa, beredningsmetod för S9-blandningen, koncentration eller volym för S9-blandningen, S9 i det slutliga odlingsmediet, kvalitetskontroller av S9),
|
|
—
|
positiva och negativa kontrollämnen, slutkoncentrationer för varje behandlingsförhållande,
|
|
—
|
Expressionsperiodens längd (inklusive antal celler som inokulerats samt subkulturer och scheman för näringstillförsel, om detta är tillämpligt).
|
|
—
|
Det selektiva ämnets identitet och koncentration.
|
|
—
|
för MLA bör använd version (agar eller mikrotiterplatta) anges,
|
|
—
|
acceptanskriterier för testerna,
|
|
—
|
Metoder som används för att räkna antalet viabla och muterade celler.
|
|
—
|
Metoder för mätning av cytotoxicitet.
|
|
—
|
All tilläggsinformation som är relevant rörande cytotoxicitet samt använd metod.
|
|
—
|
Inkubationstidens varaktighet efter överföring till plattor.
|
|
—
|
definition av kolonier vars storlek och typ ska beaktas (inklusive kriterier för ”små” och ”stora” kolonier, där detta är tillämpligt),
|
|
—
|
kriterier för att bedöma om testresultaten är positiva, negativa eller tvetydiga,
|
|
—
|
metoder som används för att bestämma pH, osmolalitet, om detta görs och utfällningar, om relevant.
|
|
|
|
Resultat:
|
—
|
antal behandlade celler och antal subkultiverade celler i varje odling,
|
|
—
|
toxicitetsparametrar (RTG för MLA och RS för TK6),
|
|
—
|
tecken på utfällning och tid för bestämningen,
|
|
—
|
Antal celler som överförts till plattor i selektiva och icke-selektiva medier.
|
|
—
|
antal kolonier i icke-selektiva medier och antal resistenta kolonier i selektiva medier, samt relaterad mutantfrekvens,
|
|
—
|
kolonistorlek för de negativa och positiva kontrollerna, och om testkemikalien är positiv, minst en koncentration och relaterade mutantfrekvenser,
|
|
—
|
koncentration-responsförhållande, där det är möjligt,
|
|
—
|
data om parallella negativa (lösningsmedel) och positiva kontroller (koncentrationer och lösningsmedel),
|
|
—
|
historiska negativa (lösningsmedel) och positiva kontrolldata (koncentrationer och lösningsmedel), med variationer, medelvärden och standardavvikelser, antal tester som de historiska testerna baseras på,
|
|
—
|
statistik (för individuella odlingar och sammanslagna replikat, i förkommande fall) samt p-värden om dessa finns, och för MLA, GEF-utvärderingen.
|
|
|
LITTERATUR
|
(1)
|
Moore, M.M., Honma, M. Clements, J. (Rapporteur), Awogi, T., Bolcsfoldi, G., Cole, J., Gollapudi, B., Harrington-Brock, K., Mitchell, A., Muster, W., Myhr, B., O'Donovan, M., Ouldelhkim, M-C., San, R., Shimada, H. and Stankowski, L.F. Jr. (2000). Mouse Lymphoma Thymidine Kinase Locus (TK) Gene Mutation Assay: International Workshop on Genotoxicity Test Procedures (IWGTP) Workgroup Report, Environ. Mol. Mutagen., 35 (3): s. 185-190.
|
|
(2)
|
Moore, M.M., Honma, M., Clements, J., Harrington-Brock, K., Awogi, T., Bolcsfoldi, G., Cifone, M., Collard, D., Fellows, M., Flanders, K., Gollapudi, B., Jenkinson, P., Kirby, P., Kirchner, S., Kraycer, J., McEnaney, S., Muster, W., Myhr, B., O’Donovan, M., Oliver, Ouldelhkim, M-C., Pant, K., Preston, R., Riach, C., San, R., Shimada, H. and Stankowski, L.F. Jr. (2002). Mouse Lymphoma Thymidine Kinase Locus Gene Mutation Assay: Follow-Up International Workshop on Genotoxicity Test Procedures, New Orleans, Louisiana, (april 2000), Environ. Mol. Mutagen., 40 (4): s. 292-299.
|
|
(3)
|
Moore, M.M., Honma, M., Clements, J., Bolcsfoldi, G., Cifone, M., Delongchamp, R., Fellows, M., Gollapudi, B., Jenkinson, P., Kirby, P., Kirchner, S., Muster, W., Myhr, B., O’Donovan, M., Ouldelhkim, M-C., Pant, K., Preston, R., Riach, C., San, R., Stankowski, L.F. Jr., Thakur, A., Wakuri, S. and Yoshimura, I. (2003). Mouse Lymphoma Thymidine Kinase Locus Gene Mutation Assay: International Workshop (Plymouth, UK) on Genotoxicity Test Procedures Workgroup Report, Mutation Res., 540: s. 127-140.
|
|
(4)
|
Moore, M.M., Honma, M., Clements, J., Bolcsfoldi, G., Burlinson, B., Cifone, M., Clarke, J., Delongchamp, R., Durward, R., Fellows, M., Gollapudi, B., Hou, S., Jenkinson, P., Lloyd, M., Majeska, J., Myhr, B., O’Donovan, M., Omori, T., Riach, C., San, R., Stankowski, L.F. Jr., Thakur, A.K., Van Goethem, F., Wakuri, S. and Yoshimura, I. (2006). Mouse Lymphoma Thymidine Kinase Gene Mutation Assay: Follow-Up Meeting of the International Workshop on Genotoxicity Tests – Aberdeen, Scotland, 2003 – Assay Acceptance Criteria, Positive Controls, and Data Evaluation, Environ. Mol. Mutagen., 47 (1): s. 1-5.
|
|
(5)
|
Moore, M.M., Honma, M., Clements, J., Bolcsfoldi, G., Burlinson, B., Cifone, M., Clarke, J., Clay, P., Doppalapudi, R., Fellows, M., Gollapudi, B., Hou, S., Jenkinson, P., Muster, W., Pant, K., Kidd, D.A., Lorge, E., Lloyd, M., Myhr, B., O’Donovan, M., Riach, C., Stankowski, L.F. Jr., Thakur A.K. and Van Goethem, F. (2007). Mouse Lymphoma Thymidine Kinase Mutation Assay: Meeting of the International Workshop on Genotoxicity Testing, San Francisco, 2005, Recommendations for 24-h Treatment, Mutation. Res., 627 (1): s. 36-40.
|
|
(6)
|
OECD (2016). Overview of the set of OECD Genetic Toxicology Test Guidelines and updates performed in 2014–2015. ENV Publications. ENV Publications. Series on Testing and Assessment, nr 234, OECD, Paris.
|
|
(7)
|
Fellows M.D., Luker, T., Cooper, A. och O’Donovan, M.R. (2012). Unusual Structure-Genotoxicity Relationship in Mouse Lymphoma Cells Observed with a Series of Kinase Inhibitors. Mutation, Res., 746 (1): s. 21-28.
|
|
(8)
|
Honma, M., Momose, M., Sakamoto, H., Sofuni, T. och Hayashi, M. (2001). Spindol Poisons Induce Allelic Loss in Mouse Lymphoma Cells Through Mitotic Non-Disjunction. Mutation Res., 493, 1–2: s. 101-114.
|
|
(9)
|
Wang, J., Sawyer, J.R., Chen, L., Chen, T., Honma, M., Mei, N. och Moore, M.M. (2009). The Mouse Lymphoma Assay Detects Recombination, Deletion, and Aneuploidy, Toxicol. Sci.., 109 (1): s. 96-105.
|
|
(10)
|
Applegate, M.L., Moore, M.M., Broder, C.B., Burrell, A. och Hozier, J.C. (1990). Molecular Dissection of Mutations at the Heterozygous Thymidine Kinase Locus in Mouse Lymphoma Cells. Proc. National. Academy. Sci. USA, 87 (1): s. 51-55.
|
|
(11)
|
Hozier, J., Sawyer, J., Moore, M., Howard, B. och Clive, D. (1981). Cytogenetic Analysis of the L5178Y/TK+/– Leads to TK–/– Mouse Lymphoma Mutagenesis Assay System, Mutation Res., 84 (1): s. 169-181.
|
|
(12)
|
Hozier, J., Sawyer, J., Clive, D. och Moore, M.M. (1985). Chromosome 11 Aberrations in Small Colony L5178Y TK–/– Mutants Early in their Clonal History, Mutation Res., 147 (5): s. 237-242.
|
|
(13)
|
Moore, M.M., Clive, D., Hozier, J.C., Howard, B.E., Batson, A.G., Turner, N.T. och Sawyer, J. (1985). Analysis of Trifluorothymidine-Resistant (TFTr) Mutants of L5178Y/TK+/– Mouse Lymphoma Cells. Mutation Res., 151 (1): s. 161-174.
|
|
(14)
|
Liber H.L., Call K.M. och Little J.B. (1987). Molecular and Biochemical Analyses of Spontaneous and X-Ray-Induced Mutants in Human Lymphoblastoid Cells. Mutation Res., 178 (1): s. 143-153.
|
|
(15)
|
Li C.Y., Yandell D.W. och Little J.B. (1992). Molecular Mechanisms of Spontaneous and Induced Loss of Heterozygosity in Human Cells In Vitro. Somat. Cell Mol. Genet., 18 (1): s. 77-87.
|
|
(16)
|
Honma M., Hayashi M. och Sofuni T. (1997). Cytotoxic and Mutagenic Responses to X-Rays and Chemical Mutagens in Normal and P53-Mutated Human Lymphoblastoid Cells. Mutation. Res., 374 (1): s. 89-98.
|
|
(17)
|
Honma, M., Momose, M., Tanabe, H., Sakamoto, H., Yu, Y., Little, J.B., Sofuni, T. och Hayashi, M. (2000). Requirement of Wild-Type P53 Protein for Maintenance of Chromosomal Integrity. Mol. Carcinogen., 28 (4): s. 203–214.
|
|
(18)
|
Amundson S.A. och Liber H.L. (1992). A Comparison of Induced Mutation at Homologous Alleles of the TK Locus in Human Cells. II. Molecular Analysis of Mutants. Mutation Res., 267 (1): s. 89-95.
|
|
(19)
|
Schisler M.R., Moore M.M. och Gollapudi B.B. (2013). In Vitro Mouse Lymphoma (L5178Y TK+/– –3.7.2C) Forward Mutation Assay. In Protocols in Genotoxicity Assessment A. Dhawan and M. Bajpayee (Eds.), Springer Protocols, Humana Press: s. 27-50.
|
|
(20)
|
Long, L.H., Kirkland, D., Whitwell, J. och Halliwell, B. (2007). Different Cytotoxic and Clastogenic Effects of Epigallocatechin Gallate in Various Cell-Culture Media Due to Variable Rates of its Oxidation in the Culture Medium, Mutation Res., 634 (1–2): s. 177-183.
|
|
(21)
|
Nesslany, F., Simar-Meintieres, S., Watzinger, M., Talahari, I.och Marzin, D. (2008). Characterization of the Genotoxicity of Nitrilotriacetic Acid. Environ. Mol. Mutagen., 49 (6): s. 439-452.
|
|
(22)
|
Brusick D. (1986). Genotoxic effects in cultured mammalian cells produced by low pH treatment conditions and increased ion concentrations. Environ. Mutagen., 8 (6): s. 879-886.
|
|
(23)
|
Morita, T., Nagaki, T., Fukuda, I. och Okumura, K. (1992). Clastogenicity of Low pH to Various Cultured Mammalian Cells. Mutation Res., 268 (2): s. 297-305.
|
|
(24)
|
Scott, D., Galloway, S.M., Marshall, R.R., Ishidate, M.Jr, Brusick, D., Ashby, J. och Myhr, B.C. (1991). Genotoxicity under Extreme Culture Conditions. A report from ICPEMC Task Group 9. Mutation Res., 257: s. 147-204.
|
|
(25)
|
Wang J., Heflich R.H. och Moore M.M. (2007). A Method to Distinguish Between the De Novo Induction of Thymidine Kinase Mutants and the Selection of Pre-Existing Thymidine Kinase Mutants in the Mouse Lymphoma Assay. Mutation Res., 626 (1–2): s. 185-190.
|
|
(26)
|
Fischer, G.A. (1958). Studies on the Culture of Leukemic Cells In Vitro. Ann. N.Y. Academy Sci., 76: s. 673-680.
|
|
(27)
|
Clive, D., Johnson, K.O., Spector, J.F.S., Batson, A.G. och Brown, M.M.M. (1979). Validation and Characterization of the L5178Y/TK+/– Mouse Lymphoma Mutagen Assay System. Mutation Res., 59(1): s. 61-108.
|
|
(28)
|
Sawyer, J., Moore, M.M., Clive, D. och Hozier, J. (1985). Cytogenetic Characterization of the L5178Y TK+/– 3.7.2C Mouse Lymphoma Cell Line, Mutation Res., 147 (5): s. 243-253.
|
|
(29)
|
Sawyer J.R., Moore M.M. och Hozier J.C. (1989). High-Resolution Cytogenetic Characterization of the L5178Y TK+/– Mouse Lymphoma Cell Line, Mutation Res., 214 (2): s. 181-193.
|
|
(30)
|
Sawyer, J.R., Binz, R.L., Wang, J. och Moore, M.M. (2006). Multicolor Spectral Karyotyping of the L5178Y TK+/––3.7.2C Mouse Lymphoma Cell Line, Environ. Mol. Mutagen., 47 (2): s. 127-131.
|
|
(31)
|
Fellows, M.D., McDermott, A., Clare, K.R., Doherty, A. och Aardema, M.J. (2014). The Spectral Karyotype of L5178Y TK+/– Mouse Lymphoma Cells Clone 3.7.2C and Factors Affecting Mutant Frequency at the Thymidine Kinase (TK) Locus in the Microtitre Mouse Lymphoma Assay, Environ. Mol. Mutagen., 55 (1): s. 35-42.
|
|
(32)
|
Storer, R.D., Jraynak, A.R., McKelvey, T.W., Elia, M.C., Goodrow, T.L. och DeLuca, J.G. (1997). The Mouse Lymphoma L5178Y TK+/– Cell Line is Heterozygous for a Codon 170 Mutation in the P53 Tumor Suppressor Gene. Mutation. Res., 373 (2): s. 157-165.
|
|
(33)
|
Clark L.S., Harrington-Brock, K., Wang, J., Sargent, L., Lowry, D., Reynolds, S.H. och Moore, M.M. (2004). Loss of P53 Heterozygosity is not Responsible for the Small Colony Thymidine Kinase Mutant Phenotype in L5178Y Mouse Lymphoma Cells. Mutagen., 19 (4): s. 263-268.
|
|
(34)
|
Skopek T.R., Liber, H.L., Penman, B.W. och Thilly, W.G. (1978). Isolation of a Human Lymphoblastoid Line Heterozygous at the Thymidine Kinase Locus: Possibility for a Rapid Human Cell Mutation Assay. Biochem. Biophys. Res. Commun., 84 (2): s. 411-416.
|
|
(35)
|
Honma M. (2005). Generation of Loss of Heterozygosity and its Dependency on P53 Status in Human Lymphoblastoid Cells. Environ. Mol. Mutagen., 45 (2–3): s. 162-176.
|
|
(36)
|
Xia, F., Wang, X., Wang, Y.H., Tsang, N.M., Yandell, D.W., Kelsey, K.T. och Liber, H.L. (1995). Altered P53 Status Correlates with Differences in Sensitivity to Radiation-Induced Mutation and Apoptosis in Two Closely Related Human Lymphoblast Lines. Cancer. Res., 55 (1): s. 12-15.
|
|
(37)
|
Lorge, E., M. Moore, J. Clements, M. O Donovan, M. Honma, A. Kohara, J. van Benthem, S. Galloway, M.J. Armstrong, V. Thybaud, B. Gollapudi, M. Aardema, J. Kim, A. Sutter, D.J. Kirkland (2015). Standardized Cell Sources and Recommendations for Good Cell Culture Practices in Genotoxicity Testing. (Manus under bearbetning.)
|
|
(38)
|
Lloyd M. och Kidd D. (2012). The Mouse Lymphoma Assay. Springer Protocols: Methods in Molecular Biology 817, Genetic Toxicology Principles and Methods, ed. Parry and Parry, Humana Press. ISBN, 978-1-61779-420-9, 35–54.
|
|
(39)
|
Mei N., Guo X. och Moore M.M. (2014). Methods for Using the Mouse Lymphoma Assay to Screen for Chemical Mutagenicity and Photo-Mutagenicity. I: Optimization in Drug Discover: In Vitro Methods: Yan Z. och Caldwell (Eds) , 2nd Edition, GW; Humana Press, Totowa, NJ.
|
|
(40)
|
Liber H.L. and Thilly W.G. (1982). Mutation Assay at the Thymidine Kinase Locus in Diploidhuman Lymphoblasts. Mutation Res., 94 (2): s. 467-485.
|
|
(41)
|
Coecke, S., Balls, M., Bowe, G., Davis, J., Gstraunthaler, G., Hartung, T., Hay, R., Merten, OW., Price, A., Schechtman, L., Stacey, G. och Stokes, W. (2005). Guidance on Good Cell Culture Practice. A Report of the Second ECVAM Task Force on Good Cell Culture Practice. ATLA, 33 (3): s. 261-287.
|
|
(42)
|
Moore M.M. och Howard B.E. (1982). Quantitation of Small Colony Trifluorothymidine-Resistant Mutants of L5178Y/TK+/- Mouse Lymphoma Cells in RPMI-1640 Medium, Mutation Res., 104 (4–5): s. 287-294.
|
|
(43)
|
Ames B.N., McCann J. och Yamasaki E. (1975). Methods for Detecting Carcinogens and Mutagens with the Salmonella/Mammalian Microsome Mutagenicity Test. Mutation Res., 31 (6): s. 347-364.
|
|
(44)
|
Maron D.M. och Ames B.N. (1983). Revised Methods for the Salmonella Mutagenicity Test. Mutation Res., 113 (3–4): s. 173-215.
|
|
(45)
|
Natarajan, A.T., Tates, A.D, Van Buul, P.P.W., Meijers, M. och De Vogel, N. (1976). Cytogenetic Effects of Mutagens/Carcinogens After Activation in a Microsomal System In Vitro, I. Induction of Chromosomal Aberrations and Sister Chromatid Exchanges by Diethylnitrosamine (DEN) and Dimethylnitrosamine (DMN) in CHO Cells in the Presence of Rat-Liver Microsomes. Mutation Res., 37 (1): s. 83-90.
|
|
(46)
|
Matsuoka A., Hayashi M. och Ishidate M. Jr. (1979). Chromosomal Aberration Tests on 29 Chemicals Combined with S9 Mix In Vitro. Mutation Res., 66 (3): s. 277-290.
|
|
(47)
|
Ong T.M., et al. (1980). Differential Effects of Cytochrome P450-Inducers on Promutagen Activation Capabilities and Enzymatic Activities of S-9 from Rat Liver, J. Environ. Pathol. Toxicol., 4 (1): s. 55–65
|
|
(48)
|
Elliott, B.M., Combes, R.D., Elcombe, C.R., Gatehouse, D.G., Gibson, G.G., Mackay, J.M. och Wolf, R.C. (1992). Report of UK Environmental Mutagen Society Working Party. Alternatives to Aroclor 1254-Induced S9 in In Vitro Genotoxicity Assays. Mutagen., 7 (3): s. 175-177.
|
|
(49)
|
Matsushima, T., Sawamura, M., Hara, K. och Sugimura, T. (1976). A Safe Substitute for Polychlorinated Biphenyls as an Inducer of Metabolic Activation Systems. I: In Vitro Metabolic Activation in Mutagenesis Testing. de Serres F.J., et al. (Eds, Elsevier, North-Holland, s. 85–88.
|
|
(50)
|
Galloway S.M., et al.(1994). Report from Working Group on In Vitro Tests for Chromosomal Aberrations. Mutation Res., 312 (3): s. 241-261.
|
|
(51)
|
Johnson, T.E., Umbenhauer, D.R. och Galloway, S.M. (1996). Human Liver S-9 Metabolic Activation: Proficiency in Cytogenetic Assays and Comparison with Phenobarbital/Beta-Naphthoflavone or Aroclor 1254 Induced Rat S-9, Environ. Mol. Mutagen., 28 (1): s. 51-59.
|
|
(52)
|
Unep (2001). Stockholmskonventionen om långlivade organiska föroreningar, FN:s miljöprogram (Unep).
|
|
(53)
|
Krahn, D.F., Barsky, F.C. och McCooey, K.T. (1982). CHO/HGPRT Mutation Assay: Evaluation of Gases and Volatile Liquids. I: Genotoxic Effects of Airborne Agents Tice R.R., Costa D.L. och Schaich K.M. (Eds.). New York, Plenum, s. 91–103.
|
|
(54)
|
Zamora, P.O., Benson, J.M., Li, A.P. och Brooks, A.L. (1983). Evaluation of an Exposure System Using Cells Grown on Collagen Gels for Detecting Highly Volatile Mutagens in the CHO/HGPRT Mutation Assay. Environ. Mutagen., 5 (6): s. 795-801.
|
|
(55)
|
Asakura M., Sasaki T., Sugiyama T., Arito H., Fukushima, S. och Matsushima, T. (2008). An Improved System for Exposure of Cultured Mammalian Cells to Gaseous Compounds in the Chromosomal Aberration Assay. Mutation Res., 652 (2): s. 122-130.
|
|
(56)
|
Arlett C.F., et al. (1989). Mammalian Cell Gene Mutation Assays Based upon Colony Formation. I: Statistical Evaluation of Mutagenicity Test Data, Kirkland, D.J. (Ed.), CambridgeUniversity Press, s. 66–101.
|
|
(57)
|
Morita T., Honma M. och Morikawa K. (2012). Effect of Reducing the Top Concentration Used in the In Vitro Chromosomal Aberration Test in CHL Cells on the Evaluation of Industrial Chemical Genotoxicity. Mutation Res., 741 (1-2): s. 32-56.
|
|
(58)
|
Brookmire L., Chen J.J. och Levy D.D. (2013). Evaluation of the Highest Concentrations Used in the In Vitro Chromosome Aberrations Assay. Environ. Mol. Mutagen., 54 (1): s. 36-43.
|
|
(59)
|
USFDA (2012). International Conference on Harmonisation (ICH) Guidance S2 (R1) on Genotoxicity Testing and Data Interpretation for Pharmaceuticals Intended For Human Use. Se [https://www.federalregister.gov/a/2012-13774].
|
|
(60)
|
Honma M. och Hayashi M. (2011). Comparison of In Vitro Micronucleus and Gene Mutation Assay Results for P53-Competent Versus P53-Deficient Human Lymphoblastoid Cells. Environ. Mol. Mutagen., 52 (5): s. 373-384.
|
|
(61)
|
Moore-Brown, M.M., Clive, D., Howard, B.E., Batson, A.G. och Johnson, K.O. (1981). The Utilization of Trifluorothymidine (TFT) to Select for Thymidine Kinase-Deficient (TK–/–) Mutants from L5178Y/TK+/– Mouse Lymphoma Cells, Mutation Res., 85 (5): s. 363-378.
|
|
(62)
|
Liber H.L., Yandell D.W. och Little J.B. (1989). A Comparison of Mutation Induction at the TK and HRPT Loci in Human Lymphoblastoid Cells; Quantitative Differences are Due to an Additional Class of Mutations at the Autosomal TK locus. Mutation Res., 216 (1): s. 9-17.
|
|
(63)
|
Furth E.E., Thilly, W.G., Penman, B.W., Liber, H.L. och Rand, W.M. (1981). Quantitative Assay for Mutation in Diploid Human Lymphoblasts Using Microtiter Plates. Anal. Biochem., 110 (1): s. 1-8.
|
|
(64)
|
Hayashi, M, Dearfield, K., Kasper, P., Lovell, D., Martus, H. J. och Thybaud, V. (2011). Compilation and Use of Genetic Toxicity Historical Control Data, Mutation Res., 723 (2): s. 87-90.
|
|
(65)
|
Ryan T.P. (2000). Statistical Methods for Quality Improvement. John Wiley and Sons, New York 2nd Edition.
|
|
(66)
|
OECD (2014). Statistical analysis supporting the revision of the genotoxicity Test Guidelines. Environmental Health and Safety Publications,Series on Testing and Assessment (nr 199), Organisationen för ekonomiskt samarbete och utveckling, Paris.
|
|
(67)
|
Fleiss J.L., Levin B. och Paik M.C. (2003). Statistical Methods for Rates and Proportions, Third Edition, New York: John Wiley & Sons.
|
Tillägg 1
DEFINITIONER
Aneugen
: en kemikalie eller en process som genom växelverkan med komponenterna i den mitotiska och meiotiska celldelningscykeln leder till aneuploidi i celler eller organismer.
Aneuploidi
: alla avvikelser från det normala antalet diploida (eller haploida) kromosomer med en eller flera kromosomer, men inte med hela uppsättningar kromosomer (polyploidi).
Mutagener som orsakar punktmutationer
: kemikalier som orsakar utbyte av baspar i DNA.
Kemikalie
: Ett ämne eller en blandning.
Kloningseffektivitet
: Procentandel celler som överförts till plattor vid låg densitet och som kan tillväxa till en koloni som kan räknas.
Klastogen
: alla kemikalier eller processer som orsakar strukturella kromosomavvikelser i populationer av celler eller organismer.
Cytotoxicitet
: för de analyser som omfattas av denna testmetod definieras cytotoxicitet som en reduktion i den relativa totala tillväxten (RTG) eller den relativa överlevnaden (RS) för MLA respektive TK6.
Framåtmutation
: en genmutation från den parentala typen till mutantformen, vilket ger upphov till en förändring eller förlust av den enzymatiska aktiviteten eller funktionen hos det protein som bildas.
Läsramsmutagener
: kemikalier som orsakar addition eller deletion av ett eller flera baspar i DNA-molekylen.
Genotoxisk
: en allmän term som omfattar alla typer av DNA- och kromosomskador, inklusive brott, addukter, omlagringar, mutationer, kromosomavvikelser och aneuploidi. Inte alla typer av genotoxiska effekter leder till mutationer eller stabila kromosomskador.
Mitotisk rekombination
: rekombination mellan homologa kromatider under mitosen, vilket kan resultera i dubbla trådbrott i DNA eller förlust av heterozygositet.
Mutagent ämne
: ämne som producerar en ärftlig ändring av DNA-basparssekvensen (-sekvenserna) i gener eller i kromosomstrukturen (kromosomavvikelser).
Mutantfrekvens
: antalet muterade celler som observerats, delat med antalet viabla celler.
Fenotypisk expressionstid
: den tid efter behandlingen under vilken den genetiska förändringen fixeras i genomet och de ursprungliga genprodukterna försvinner i så hög grad att den fenotypiska egenskapen förändras.
Relativ överlevnad (RS)
: RS används som ett mått på behandlingsrelaterad cytotoxicitet i TK6. RS är den relativa kloningseffektiviteten (CE) för celler som överförts till platta omedelbart efter cellbehandling justerat utifrån eventuell cellförlust under behandlingen och jämfört med kloningseffektiviteten i den negativa kontrollen.
Relativ suspensionstillväxt (RSG)
: för MLA, den relativa totala tvådagars suspensionstillväxten av testodlingen jämförd med den totala tvådagars suspensionstillväxten för den negativa kontrollen/lösningsmedelskontrollen (Clive och Spector, 1975). RSG bör innefatta testodlingens relativa tillväxt jämförd med den negativa kontrollen/lösningsmedelskontrollen under behandlingsperioden.
Relativ total tillväxt (RTG)
: RTG används som ett mått på behandlingsrelaterad cytotoxicitet i MLA. RTG är ett mått på testodlingarnas relativa tillväxt (jämfört med vehikelkontrollen) under behandlingen, expressionsperioden på två dagar och mutanturvalets kloningsfaser i testet. Testodlingens relativa suspensionstillväxt multipliceras med testodlingens relativa kloningseffektivitet vid tidpunkten för mutanturvalet, och uttrycks i förhållande till den negativa kontrollens/lösningsmedelskontrollens kloningseffektivitet (Clive och Spector, 1975).
S9-leverfraktion
: supernatant av leverhomogenat efter centrifugering vid 9 000g, dvs. råleverextrakt.
S9-blandning
: blandning av S9-leverfraktion och kofaktorer som krävs för metabolisk enzymaktivitet.
Suspensionstillväxt
: x-faldig ökning av antalet celler under behandlingens gång och expressionsfaserna i MLA. Suspensionstillväxten beräknas genom att den x-faldiga ökningen på dag 1 multipliceras med den x-faldiga ökningen på dag 2 för den kortvariga behandlingsperioden (tre eller fyra timmar). Om 24-timmarsbehandling används motsvarar suspensionstillväxten den x-faldiga ökningen under 24-timmarsbehandlingen, multiplicerat med de x-faldiga ökningarna under expressionsdagarna 1 och 2.
Lösningsmedelskontroll
: Allmän term för att definiera kontrollodlingar som endast tillsatts det lösningsmedel som används för att lösa upp testkemikalien.
Testkemikalie
: Alla ämnen eller blandningar som testas med denna testmetod.
Obehandlade kontroller
: odlingar som inte behandlas (dvs. varken testkemikalie eller lösningsmedel tillförs), men som behandlas på samma sätt som de odlingar där testkemikalien tillförs.
Tillägg 2
FORMLER
Cytotoxicitet
För båda versionerna (agar och mikrotiterplatta) av MLA
Cytotoxicitet definieras som relativ total tillväxt (RTG), vilket omfattar relativ suspensionstillväxt (RSG) under expressionsperioden på två dagar och relativ kloningseffektivitet (RCE), som erhållits vid tidpunkten för mutanturvalet. RTG, RSG och RCE uttrycks i procent.
Beräkning av RSG: Suspensionstillväxt ett (SG1) är tillväxttakten mellan dag 0 och dag 1 (cellkoncentration på dag 1/cellkoncentration på dag 0), och suspensionstillväxt två (SG2) är tillväxttakten mellan dag 1 och dag 2 (cellkoncentration på dag 2/cellkoncentration på dag 1). RSG är den totala suspensionstillväxten (SG1 x SG2) för den behandlade odlingen jämfört med den obehandlade kontrollen/lösningsmedelskontrollen. Det innebär följande: RSG = [SG1(test) x SG2(test)] / [SG1(kontroll) x SG2(kontroll)]. SG1 bör beräknas från den ursprungliga cellkoncentration som användes i början av cellbehandlingen. Detta förklarar eventuell differentiell cytotoxicitet som förekommer i testodlingarna under cellbehandlingen.
RCE är testodlingens relativa kloningseffektivitet jämfört med kloningseffektiviteten hos den obehandlade kontroll/lösningsmedelskontroll som erhållits vid tidpunkten för mutanturvalet.
Relativ total tillväxt (RTG):
RTG = RSG x RCE
TK6
Relativ överlevnad (RS):
Cytotoxicitet bedöms genom den relativa överlevnaden, dvs. kloningseffektiviteten för celler som överförts till platta omedelbart efter behandling justerat utifrån eventuell cellförlust under behandlingen jämfört med kloningseffektiviteten i negativa kontroller (som tilldelas en överlevnadskvot på 100 %). Justeringen för cellförlust under behandlingen kan beräknas som
RS för celler som behandlats av en testkemikalie beräknas på följande sätt:

Mutantfrekvens för både MLA och TK6
Mutantfrekvensen (MF) är kloningseffektiviteten hos mutantkolonier i ett selektivt medium CEM) justerat med kloningseffektiviteten i ett icke-selektivt medium vid tidpunkten för mutanturvalet (CEV). Det vill säga, MF=CEM/CEV. Beräkningen av kloningseffektivitet beskrivs nedan för kloningsmetoderna med agar och mikrotiterplatta.
Agarversionen av MLA: I mjukagarversionen av MLA erhålls antalet kolonier på plattan för mutanturvalet (CM) och antalet kolonier på plattan för icke utvalda kolonier eller kloningseffektivitet (viabilitetsräkning) (CV) genom att klonerna räknas direkt. När 600 celler överförs för kloningseffektivitet (CE) till plattorna för mutanturvalet (CEM) och plattorna för icke utvalda kolonier eller kloningseffektivitet (viabilitetsräkning) (CEV) och 3 x 106 celler används för mutanturval,
CEM = CM / (3 x 106) = (CM / 3) x 10-6
CEV = CV / 600
Mikrotiterversionen av MLA och TK6:
I mikrotiterversionen av MLA CM och CV bestäms som produkten av det totala antalet hål (TW) och det förmodade antalet kolonier per hål (P) på mikrotiterplattorna.
CM = PM x TWM
CV = PV x TWV
Från nolltermen i Poissonfördelningen (Furth et al., 1981), P ges genom
P = - ln (EW / TW)
där EW är tomma hål och TW totalt antal hål. Därför
CEM = CM / TM = (PM x TWM) / TM
CEV = CV / TV = (PV x TWV) / TV
För mikrotiterversionen av MLA beräknas mutantfrekvenserna för små och stora kolonier på samma sätt, med användning av antalet tomma hål för små och stora kolonier.
För TK6 baseras mutantfrekvenserna för små och stora kolonier på tidigt och sent uppträdande mutanter.
B.68 TESTMETOD FÖR KORTTIDSEXPONERING IN VITRO FÖR IDENTIFIERING AV i) KEMIKALIER SOM ORSAKAR ALLVARLIG ÖGONSKADA OCH ii) KEMIKALIER FÖR VILKA DET INTE KRÄVS KLASSIFICERING FÖR ÖGONIRRITATION ELLER ALLVARLIG ÖGONSKADA
INLEDNING
|
1.
|
Denna testmetod motsvarar OECD:s testriktlinje (TG) 491 (2017). Testmetoden korttidsexponering (STE) är en testmetod in vitro som kan användas under särskilda omständigheter och med specifika begränsningar för faroklassificering och märkning av kemikalier (ämnen och blandningar) som orsakar allvarlig ögonskada och kemikalier för vilka det inte krävs klassificering för ögonirritation eller allvarlig ögonskada, enligt definitionen i FN:s globala harmoniserade system för klassificering och märkning av kemikalier (GHS) (1) och förordning (EG) nr 1272/2008 om klassificering, märkning och förpackning av ämnen och blandningar (CLP) (67).
|
|
2.
|
I många år har kemikaliers eventuella skadliga verkan på ögonen huvudsakligen utvärderats med hjälp av ett in vivo-test på kaninögon (TM B.5 (8), motsvarande OECD TG 405). Det är allmänt vedertaget att inget enskilt alternativt in vitro-test inom överskådlig framtid kommer att fullständigt kunna ersätta in vivo-testet på kaninögon för att förutsäga alla typer av allvarlig ögonskada/ögonirritation för olika kemiska klasser. Emellertid kan strategiska kombinationer av alternativa testmetoder som används i en (stegvis) teststrategi mycket väl fullständigt ersätta testet på kaninögon (2). Top-down-strategin är utformad för testning av kemikalier som baserat på befintlig information kan förväntas vara mycket irriterande eller orsaka allvarlig ögonskada. Bottom-up-strategin är däremot utformad för testning av kemikalier som baserat på befintlig information inte förväntas orsaka tillräcklig ögonirritation för att kräva en klassificering. STE-testmetoden anses inte vara en fullständig ersättning för in vivo-testet på kaninögon, men är lämplig för användning som en del av en stegvis teststrategi för lagstadgad klassificering och märkning, såsom top-down-strategin/bottom-up-strategin, för att utan ytterligare testning identifiera i) kemikalier som inducerar allvarlig ögonskada (GHS/CLP-kategori 1), och ii) för kemikalier (exklusive mycket flyktiga ämnen och alla fasta kemikalier utom ytaktiva ämnen) som inte kräver klassificering för ögonirritation eller allvarlig ögonskada enligt (GHS/CLP Ingen klassificering) (1)(2). Kemikalier som inte förutsägs orsaka allvarlig ögonskada (GHS/CLP-kategori 1) och inte heller inducerar allvarlig ögonskada eller ögonirritation (GHS/CLP Ingen klassificering) enligt STE-testmetoden kräver dock ytterligare testning för att fastställa en definitiv klassificering. Dessutom bör berörda tillsynsmyndigheter rådfrågas innan man använder STE-metoden inom ramen för en bottom-up-strategi enligt andra klassificeringssystem än GHS/CLP. Valet av lämpligaste testmetod och användningen av denna testmetod bör övervägas mot bakgrund av OECD:s vägledningsdokument om ett integrerat synsätt på testning och bedömning av allvarlig ögonskada och ögonirritation (14).
|
|
3.
|
Syftet med denna testmetod är att beskriva de förfaranden som används för att utvärdera om en testkemikalie kan vara skadlig för ögonen, baserat på dess förmåga att inducera cytotoxicitet i testmetoden för korttidsexponering. Kemikaliers cytotoxiska effekt på hornhinnans epitelceller är ett viktigt verkningssätt som leder till skada på hornhinnans epitel och ögonirritation. Cellviabiliteten i STE-testmetoden bedöms, efter extraktion av cellerna, med hjälp av en kvantitativ mätning av blått formazansalt, som produceras av de levande cellerna genom enzymatisk omvandling av vitalfärgämnet MTT (3-(4,5-dimetyltiazol-2-yl)-2,5-difenyltetrazoliumbromid), även benämnt tiazolylblått-tetrazoliumbromid (3). Den cellviabilitet som erhålls jämförs med lösningsmedelskontrollen (relativ viabilitet) och används för att uppskatta testkemikaliens eventuella skadliga effekt på ögonen. En testkemikalie klassificeras som GHS/CLP-kategori 1 när både koncentrationerna på 5 % och 0,05 % leder till en cellviabilitet som är lägre än eller motsvarar (≤) 70 %. Omvänt klassificeras en testkemikalie som GHS/CLP Ingen klassificering när både koncentrationerna på 5 % och 0,05 % leder till en cellviabilitet som är högre än (≤) 70 %.
|
|
4.
|
I denna metod avser begreppet ”testkemikalie” endast testade kemikalier. Frågan om huruvida STE-testmetoden är tillämplig på testning av ämnen och/eller blandningar tas inte upp här. Definitioner av begreppen finns i tillägg 2.
|
INLEDANDE ÖVERVÄGANDEN OCH BEGRÄNSNINGAR
|
5.
|
Denna testmetod är baserad på ett protokoll som utvecklats av Kao Corporation (4), som var föremål för två olika valideringsstudier: den ena av Validation Committee of the Japanese Society for Alternative to Animal Experiments (JSAAE) (5) och den andra av Japanese Center for the Validation of Alternative Methods (JaCVAM) (6). En sakkunnigbedömning utfördes av NICEATM/ICCVAM baserat på rapporterna om valideringsstudierna och bakgrundsgranskningsdokument om testmetoden (7).
|
|
6.
|
Vid identifiering av kemikalier (ämnen och blandningar) som inducerar allvarlig ögonskada (GHS/CLP-kategori 1 (1) visade data som erhållits med hjälp av STE-testmetoden om 125 kemikalier (inklusive både ämnen och blandningar) en övergripande noggrannhet på 83 % (104/125), ett falskt positivt värde på 1 % (1/86), och ett falskt negativt värde på 51 % (20/39) jämfört med in vivo-testet på kaninögon (7). Det falska negativa värde som erhållits är inte kritiskt under nuvarande omständigheter, eftersom alla testkemikalier som inducerar en cellviabilitet på ≤ 70 % vid en koncentration på 5 % och > 70 % vid en koncentration på 0,05 % därefter testas med andra tillbörligt validerade in vitro-testmetoder, eller som ett sista alternativ i in vivo-testet på kaninögon, beroende på kraven i tillämpliga bestämmelser, och enligt strategin med stegvis testning och de metoder för sammanvägd bedömning som för närvarande rekommenderas (1) (8). De ämnen som testades var främst monokomponentämnen, men det finns också vissa data om testning av blandningar. Testmetoden är emellertid tekniskt tillämplig på testning av multikomponentämnen och blandningar. Innan testmetoden används på en blandning i syfte att samla in data för ett lagstadgat ändamål bör man dock överväga om, och i så fall varför, den kommer att ge godtagbara resultat för det syftet. Sådana överväganden är inte nödvändiga om blandningen måste provas enligt lag. STE-testmetoden visade inga andra specifika brister när den användes för att identifiera testkemikalier som tillhörande GHS/CLP-kategori 1. Forskare kan överväga att använda denna testmetod för testkemikalier. I ett sådant fall bör en cellviabilitet på ≤ 70 % vid både koncentrationen 5 % och 0,05 % godtas som en indikation på en respons som inducerar allvarlig ögonskada och kemikalien bör klassificeras som GHS/CLP-kategori 1 utan ytterligare testning.
|
|
7.
|
Vid identifiering av kemikalier (ämnen och blandningar) som inte kräver klassificering för ögonirritation och allvarlig ögonskada (dvs. GHS/CLP Ingen klassificering) visade data som erhållits med hjälp av STE-testmetoden för 130 kemikalier (inklusive både ämnen och blandningar) en övergripande noggrannhet på 85 % (110/130), ett falskt negativt värde på 12 % (9/73), och ett falskt positivt värde på 19 % (11/57) jämfört med in vivo-testet på kaninögon (7). Om mycket flyktiga ämnen och fasta ämnen som inte är ytaktiva ämnen undantas från datamängden förbättras den övergripande noggrannheten till 90 % (92/102), andelen falskt negativa värden till 2 % (1/54), och andelen falskt positiva värden till 19 % (9/48) (7). Detta innebär att de eventuella bristerna hos STE-metoden när den används för att identifiera testkemikalier som inte kräver klassificering för ögonirritation och allvarlig ögonskada (GHS/CLP Ingen klassificering) är en hög andel falskt negativt värden för i) mycket flyktiga ämnen med ett ångtryck över 6 kPa, och ii) andra fasta kemikalier (ämnen och blandningar) än ytaktiva ämnen och blandningar som endast består av ytaktiva ämnen. Sådana kemikalier undantas från tillämpningsområdet för STE-testmetoden (7).
|
|
8.
|
Förutom de kemikalier som nämns i punkterna 6 och 7 innehåller de datamängder som genererats genom STE-testmetoden även interna data om 40 blandningar, som när de jämfördes med Draizes ögontest in vivo, visade en noggrannhet på 88 % (35/40), en andel falskt positiva värden på 50 % (5/10), och en andel falskt negativa värden på 0 % (0/30) för förutsägning av blandningar som inte kräver klassificering enligt GHS/CLP-klassificeringssystemet (9). STE-testmetoden kan därför användas för att identifiera blandningar som tillhörande GHS/CLP Ingen klassificering i en bottom-up-strategi, med undantag för andra fasta blandningar än fasta blandningar som endast består av ytaktiva medel, som en förlängning av metodens begränsningar avseende fasta ämnen. Blandningar som består av ämnen med ett ångtryck högre än 6 kPa bör dessutom utvärderas noggrant för att undvika eventuella underskattningar, och bör motiveras från fall till fall.
|
|
9.
|
STE-testmetoden kan inte användas för att identifiera testkemikalier som tillhörande GHS/CLP-kategori 2 eller kategori 2A (ögonirritation) eller 2B (svag ögonirritation), eftersom många kemikalier i GHS/CLP-kategori 1 underskattas som kategori 2, 2A, eller 2B och kemikalier i GHS/CLP-kategorin Ingen klassificering överskattas som kategori 2, 2A, eller 2B (7). Därför kan det krävas ytterligare testning med en annan passande metod.
|
|
10.
|
STE-testmetoden är lämplig för testkemikalier som löses upp eller suspenderas enhetligt i minst fem minuter i fysiologisk saltlösning, 5 % dimetylsulfoxid (DMSO) i saltlösning eller mineralolja. STE-testmetoden är inte lämplig för testkemikalier som är olösliga eller inte kan suspenderas enhetligt i minst fem minuter i fysiologisk saltlösning, 5 % dimetylsulfoxid (DMSO) i saltlösning eller mineralolja. Det är möjligt att använda mineralolja i STE-testmetoden på grund av den korta tidsexponeringen. STE-testmetoden är därför lämplig för att förutsäga vattenlösliga testkemikaliers skadliga verkan på ögonen (t.ex. långkedjiga fettalkoholer eller ketoner), förutsatt att de är lösliga i minst ett av de tre lösningsmedel som föreslås ovan (4).
|
|
11.
|
I denna metod avser begreppet ”testkemikalie” endast testade kemikalier (68). Frågan om huruvida DPRA är tillämplig på testning av ämnen och/eller blandningar tas inte upp här.
|
TESTPRINCIP
|
12.
|
STE-testmetoden är ett cytotoxicitetsbaserad in vitro-test som utförs på ett konfluent monolager av celler från kaniners hornhinna (SIRC-celler) från Statens Seruminstitut, som odlats på en 96-håls mikrotiterplatta av polykarbonat (4). Efter fem minuters exponering för testkemikalien mäts cytotoxiciteten kvantitativt som SIRC-cellernas relativa viabilitet, med användning av MTT-testet (4). Minskad cellviabilitet används för att förutsäga eventuellt negativa effekter som leder till ögonskador.
|
|
13.
|
Det har rapporterats att 80 % av en lösning som droppas in i ett kaninöga utsöndras via konjunktivalsäcken inom tre till fyra minuter, medan mer än 80 % av en lösning som droppas in i en människa öga utsöndras inom en till två minuter (10). STE-metoden försöker närma sig dessa exponeringstider och använder cytotoxicitet som en endpoint för att bedöma graden av skada på SIRC-cellerna efter fem minuters exponering för testkemikalien.
|
DEMONSTRATION AV KOMPETENS
|
14.
|
Före rutinanvändning av STE-testmetoden enligt beskrivningen i denna testmetod bör laboratoriet visa teknisk kompetens genom att korrekt klassificera de elva ämnen som rekommenderas i tabell 1. Dessa ämnen valdes för att representera hela responsintervallet för allvarlig ögonskada eller ögonirritation baserat på resultat från in vivo-tester på kaninöga (TG 405) och GHS/CLP-klassificeringssystem (1). Till övriga kriterier hör att ämnena ska vara kommersiellt tillgängliga, att det finns tillgång till in vivo-referensdata av hög kvalitet och att det finns in vivo-data av hög kvalitet från STE-testmetoden (3). När ett listat ämne inte finns tillgängligt eller när det är motiverat kan ett annat ämne som det finns adekvata in vivo- och in vitro-referensdata för användas, förutsatt att samma kriterier tillämpas som de som beskrivs här.
Tabell 1
Förteckning över kvalifikationsämnen
|
Ämne
|
CAS-nr
|
Kemisk klass (69)
|
Fysikaliskt tillstånd
|
In vivo GHS/CLP-kategori (70)
|
Lösningsmedel i STE-testet
|
STE GHS/CLP kategori
|
|
Bensalkoniumklorid (10 %, vattenbaserad)
|
8001-54-5
|
Oniumförening
|
Vätska
|
Kategori 1
|
Saltlösning
|
Kategori 1
|
|
Triton X-100 (100 %)
|
9002-93-1
|
Eter
|
Vätska
|
Kategori 1
|
Saltlösning
|
Kategori 1
|
|
Acid Red 92
|
18472-87-2
|
Heterocyklisk förening, bromförening, klorförening
|
I fast form
|
Kategori 1
|
Saltlösning
|
Kategori 1
|
|
Natriumhydroxid
|
1310-73-2
|
Alkali, oorganisk kemikalie
|
I fast form
|
Kategori 1 (71)
|
Saltlösning
|
Kategori 1
|
|
Butyrolakton
|
96-48-0
|
Lakton Heterocyklisk förening
|
Vätska
|
Kategori 2A (kategori 2 i CLP)
|
Saltlösning
|
Ingen prediktion kan göras
|
|
1-Oktanol
|
111-87-5
|
Alkohol
|
Vätska
|
Kategori 2A /B (72) (kategori 2 i CLP)
|
Mineralolja
|
Ingen prediktion kan göras
|
|
Cyklopentanol
|
96-41-3
|
Alkohol, kolväte (cykliskt)
|
Vätska
|
Kategori 2A/B (73) (kategori 2 i CLP)
|
Saltlösning
|
Ingen prediktion kan göras
|
|
2-etoxyetylacetat
|
111-15-9
|
Alkohol, Eter
|
Vätska
|
Ingen klassificering
|
Saltlösning
|
Ingen klassificering
|
|
Dodekan
|
112-40-3
|
Kolväte, acykliskt
|
Vätska
|
Ingen klassificering
|
Mineralolja
|
Ingen klassificering
|
|
Metylisobutylketon
|
108-10-1
|
Keton
|
Vätska
|
Ingen klassificering
|
Mineralolja
|
Ingen klassificering
|
|
1,1-dimetylguanidinsulfat
|
598-65-2
|
Amidin, Svavelförening
|
I fast form
|
Ingen klassificering
|
Saltlösning
|
Ingen klassificering
|
|
Förkortningar: CASRN = Chemical Abstracts Service Registry Number
|
|
FÖRFARANDE
Beredning av cellmonolager
|
15.
|
SIRC, cellinjen från kaniners hornhinna, bör användas i STE-testmetoden. Det rekommenderas att SIRC-cellerna erhålls från en väl kvalificerad cellbank, såsom American Type Culture Collection CCL60.
|
|
16.
|
SIRC-celler odlas vid 37 °C i en fuktad atmosfär med 5 % CO2 i en odlingskolv där odlingsmediet består av Eagle’s minimum essential medium (MEM) kompletterat med 10 % fetalt bovinserum (FBS), 2 mM L-glutamin, 50–100 enheter/ml penicillin och 50–100 μg/ml streptomycin. Celler som har blivit konfluenta i odlingskolven bör avskiljas med användning av etylendiamintetraättiksyralösning, med eller utan cellskrapa. Cellerna förökas (t.ex. 2 till 3 passager) i en odlingskolv innan de används för rutintestning, och bör inte genomgå fler än 25 passager från upptining.
|
|
17.
|
Celler som är färdiga att användas för STE-testet bereds sedan i lämplig täthet och sås i 96-hålsplattor. Rekommenderad täthet för cellsådden är 6,0 × 103 celler per hål om cellerna används fyra dagar efter sådd eller 3,0 × 103 celler per hål när de används fem dagar efter sådd, vid en odlingsvolym på 200 μl. Celler som används för STE-testet och sås i ett odlingsmedium i lämplig täthet kommer att uppnå en konfluens på mer än 80 % vid testningstillfället, dvs. fyra eller fem dagar efter sådd.
|
Applicering av testkemikalier och kontrollämnen
|
18.
|
Förstahandsvalet för lösningsmedel för att lösa upp eller suspendera testkemikalierna är fysiologisk saltlösning. Om testkemikalien uppvisar låg löslighet eller inte kan lösas upp eller suspenderas på ett enhetligt sätt i minst fem minuter i saltlösning används 5 % DMSO (CAS-nr 67-68-5) i saltlösning som andrahandsval. För testkemikalier som inte kan lösas upp eller suspenderas på ett enhetligt sätt i minst fem minuter i varken saltlösning eller 5 % DMSO i saltlösning används mineralolja (CAS-nr 8042-47-5) som tredjehandsval.
|
|
19.
|
Testkemikalier löses upp eller suspenderas enhetligt i det valda lösningsmedlet i en koncentration med ett massförhållande på 5 % och späds ytterligare genom 10-faldig seriespädning till koncentrationer på 0,5 % och 0,05 %. Varje testkemikalie ska testas både vid koncentrationer på 5 % och 0,05 %. De celler som odlats i 96-hålsplattan exponeras för 200 μl/hål av antingen en koncentration på 5 % eller 0,05 % av lösningen (eller suspensionen) av testkemikalien i fem minuter vid rumstemperatur. Testkemikalierna (monokomponentämnen eller multikomponentämnen eller blandningar) anses vara rena och späds eller suspenderas enligt metoden, oavsett renhetsgrad.
|
|
20.
|
Det odlingsmedium som beskrivs i punkt 16 används som mediumkontroll i varje platta av varje repetition. Cellerna ska även exponeras för prover av lösningsmedelskontrollen i varje platta av varje repetition. Det har bekräftats att de lösningsmedel som anges i punkt 18 inte har några negativa effekter på SIRC-cellernas viabilitet.
|
|
21.
|
I STE-testmetoden ska 0,01 % natriumlaurylsulfat (SLS) i saltlösning användas som positiv kontroll i varje platta av varje repetition. För att beräkna cellviabiliteten i den positiva kontrollen måste varje platta av varje repetition även innehålla en lösningsmedelskontroll med saltlösning.
|
|
22.
|
Ett blankprov är nödvändigt för att bestämma kompensationen för den optiska densiteten, och bör utföras på hål som endast innehåller fosfatbuffrad saltlösning, men inte kalcium eller magnesium (PBS-) eller celler.
|
|
23.
|
Varje prov (testkemikalien vid 5 % och 0,05 %, mediumkontroll, lösningsmedelskontroll och positiv kontroll) bör testas i triplikat i varje repetition genom att cellerna exponeras för 200 μl av en lämplig test- eller kontrollkemikalie i fem minuter vid rumstemperatur.
|
|
24.
|
Referensämnen kan användas för utvärdering av förmågan att framkalla ögonirritation hos okända kemikalier tillhörande en specifik kemisk klass eller produktklass, eller för utvärdering av den relativa irritationspotentialen hos ett ögonirriterande ämne inom ett specifikt responsområde.
|
Mätning av cellviabilitet
|
25.
|
Efter exponering tvättas cellerna två gånger med 200 μl PBS och 200 μl MTT-lösning (0,5 mg MTT/ml av odlingsmediet) tillsätts. Efter en reaktionstid på två timmar i en inkubator (37 °C, 5 % CO2) dekanteras MTT-lösningen, MTT-formazan extraheras genom tillsats av 200 μl 0,04 N saltsyra-isopropanol i 60 minuter i mörker vid rumstemperatur, och MTT-formazanlösningens absorbans mäts vid 570 nm med en plattläsare. Testkemikaliernas störningar av MTT-testet (genom färgämnen eller direkt MTT-reduktion) inträffar endast om en betydande mängd av testkemikalien stannar kvar i testsystemet efter sköljning efter exponering, vilket är fallet med 3D-rekonstruerad human hornhinna eller rekonstruerad human epidermisvävnad, men är inte relevant för de 2D-cellodlingar som används för STE-testmetoden.
|
Tolkning av resultaten och prediktionsmodell
|
26.
|
De värden för den optiska densiteten (OD) som erhållits för varje testprov används sedan för att beräkna cellviabilitet i förhållande till lösningsmedelskontrollen, som sätts till 100 %. Relativ cellviabilitet uttrycks som en procentenhet och erhålls genom att testkemikaliens OD divideras med lösningsmedelskontrollens OD efter att blankprovets OD har subtraherats från båda värdena.

Relativ cellviabilitet för varje lösningsmedelskontroll uttrycks likaså som en procentenhet och erhålls genom att varje lösningsmedelskontrolls OD divideras med odlingsmediets OD efter att blankprovets OD har subtraherats från båda värdena.
|
|
27.
|
Tre oberoende repetitioner, som var och en innehåller tre replikathål (dvs., n = 9), bör utföras. Det aritmetiska medelvärdet för de tre hålen för varje testkemikalie och lösningsmedelskontroll i varje oberoende repetition används för att beräkna det aritmetiska medelvärdet för relativ cellviabilitet. Det slutliga aritmetiska medelvärdet för cellviabiliteten beräknas från de tre oberoende repetitionerna.
|
|
28.
|
Cellviabilitetens brytpunktsvärden för att identifiera testkemikalier som framkallar allvarliga ögonskador (GHS/CLP-kategori 1) och testkemikalier som inte kräver klassificering för ögonirritation eller allvarliga ögonskador (GHS/CLP Ingen klassificering) ges nedan:
|
Tabell 2
Prediktionsmodell för STE-testmetoden
|
Cellviabilitet
|
Klassificering enligt GHS/CLP
|
Tillämplighet
|
|
vid 5 %
|
vid 0,05 %
|
|
> 70 %
|
> 70 %
|
Ingen klassificering
|
Ämnen och blandningar, med undantag för i) mycket flyktiga ämnen med ett ångtryck över 6 kPa (74), och ii) fasta kemikalier (ämnen och blandningar), andra än ytaktiva medel och blandningar som endast består av ytaktiva medel.
|
|
≤ 70 %
|
> 70 %
|
Ingen prediktion kan göras
|
Ej tillämpligt
|
|
≤ 70 %
|
≤ 70 %
|
Kategori 1
|
Ämnen och blandningar (75)
|
Acceptanskriterier
|
29.
|
Testresultaten anses vara godtagbara om samtliga följande kriterier är uppfyllda:
|
a)
|
Mediumkontrollens optiska densitet (exponerat för odlingsmediet) bör vara 0,3 eller högre efter det att blankprovets optiska densitet har subtraherats.
|
|
b)
|
Lösningsmedelskontrollens viabilitet bör vara 80 % eller högre i förhållande till mediumkontrollen. Om flera lösningsmedelskontroller används i varje repetition bör var och en av dessa kontroller uppvisa en cellviabilitet större än 80 % för att testkemikalierna ska kunna testas med dessa lösningsmedel.
|
|
c)
|
Den cellviabilitet som erhålls med den positiva kontrollen (0,01 % SLS) bör ligga inom två standardavvikelser från det historiska medelvärdet. De högre och lägre acceptansgränserna för den positiva kontrollen bör uppdateras ofta, dvs. var tredje månad eller varje gång ett godtagbart test utförs i laboratorier där tester utförs sällan (dvs. mindre än en gång i månaden). Om ett laboratorium inte slutför ett tillräckligt antal försök för att kunna fastställa en statistiskt robust fördelning av de positiva kontrollerna är det godtagbart att de högre och lägre acceptansgränser som har fastställts av metodutvecklaren används, dvs. mellan 21,1 % och 62,3 % enligt laboratoriets historiska data, medan en intern fördelning byggs upp under de första rutintesterna.
|
|
d)
|
Standardavvikelsen för den slutliga cellviabilitet som härletts från tre oberoende repetitioner bör vara mindre än 15 % både för koncentrationen på 5 % och 0,05 % av testkemikalien.
Om ett eller flera av dessa kriterier inte är uppfyllt bör resultaten förkastas och ytterligare tre oberoende repetitioner utföras.
|
|
DATA OCH RAPPORTERING
Data
|
30.
|
Data för varje individuellt hål (t.ex. cellviabilitetsvärden) för varje repetition samt totalt medelvärde, SD och klassificering ska rapporteras.
|
Testrapport
|
31.
|
Testrapporten ska innehålla följande information:
|
|
Testkemikalie och kontrollämnen
|
—
|
Monokomponentämne: kemisk identifiering, t.ex. med IUPAC- eller CAS-namn, CAS-nummer, Smiles- eller InChI-kod, strukturformel och/eller andra identifieringar.
|
|
—
|
Multikomponentämne, UVCB-ämne och blandning: karakteriseras så långt som möjligt genom t.ex. beståndsdelarnas kemiska identitet (se ovan), renhet, kvantitativa förekomst och relevanta fysikalisk-kemiska egenskaper (se ovan), i den mån sådana uppgifter finns tillgängliga.
|
|
—
|
Fysikaliskt tillstånd, volatilitet, pH, LogP, molekylvikt, kemisk klass och andra relevanta fysikalisk-kemiska egenskaper med relevans för studiens genomförande, i den utsträckning de finns tillgängliga.
|
|
—
|
Renhet, kemisk identitet för föroreningar i förekommande fall och när så är praktiskt möjligt.
|
|
—
|
Behandling före testning, om detta är tillämpligt (t.ex. uppvärmning, finfördelning).
|
|
—
|
Uppgifter om lagringsförhållanden och stabilitet i den mån sådana uppgifter finns tillgängliga.
|
|
|
|
Testmetodens betingelser och förfaranden
|
—
|
Namn och adress för sponsor, testanläggning och försöksledare.
|
|
—
|
Beskrivning av använd testmetod.
|
|
—
|
Använd cellinje, dess källa, antal passager och konfluens för de celler som använts för testningen.
|
|
—
|
Detaljerade uppgifter om försöksförfarandet.
|
|
—
|
Antal repetitioner och replikat.
|
|
—
|
Använda koncentrationer av testkemikalierna (om andra koncentrationer än de rekommenderade har använts).
|
|
—
|
Motivering för valet av lösningsmedel för varje testkemikalie.
|
|
—
|
Varaktighet för exponeringen för testkemikalien (om en annan exponeringstid än den rekommenderade har använts).
|
|
—
|
Beskrivning av eventuella modifieringar av försöksförfarandet.
|
|
—
|
Beskrivning av utvärderings- och beslutskriterierna.
|
|
—
|
Hänvisning till historiska medelvärden för positiva kontroller samt standardavvikelse (SD).
|
|
—
|
Demonstration av att laboratoriet är kompetent att tillämpa testmetoden (t.ex. genom testning av kvalifikationsämnen) eller för att visa att testmetoden kan reproduceras över tiden.
|
|
|
|
Resultat
|
—
|
För varje testkemikalie och kontrollämne och för varje testad koncentration bör följande anges i tabellform: individuella OD-värden per replikathål, aritmetiskt medelvärde för OD-värdena för varje oberoende repetition, cellviabilitet uttryckt i procent för varje oberoende repetition och det slutliga aritmetiska medelvärdet för cellviabilitet och SD över de tre repetitionerna.
|
|
—
|
Resultat för mediumkontrollen, lösningsmedelskontrollen och den positiva kontrollen, som visar lämpliga acceptanskriterier för studien.
|
|
—
|
Beskrivning av övriga observerade effekter.
|
|
—
|
Övergripande härledda klassificeringar med hänvisning till prediktionsmodell och/eller beslutskriterier.
|
|
|
LITTERATUR
|
(1)
|
Förenta nationerna (FN) (2013). Globally Harmonized System of Classification and Labelling of Chemicals (GHS, det globalt harmoniserade systemet för klassificering och märkning av kemikalier). Fifth revised edition. New York & Geneva: United Nations Publications. ISBN: 978-92-1-117006-1. Se http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html.
|
|
(2)
|
Scott, L., et al. (2010). A proposed Eye Irritation Testing Strategy to Reduce and Replace in vivo Studies Using Bottom-Up and Top-Down Approaches. Toxicol. In Vitro 24, 1-9.
|
|
(3)
|
Mosmann T. (1983). Rapid Colorimetric Assay for Cellular Growth and Survival: Application to 7 Proliferation and Cytotoxicity Assays. J. Immunol. Methods 65, 55-63.
|
|
(4)
|
Takahashi Y., et al. (2008). Development of the Short Time Exposure (STE) Test: an In Vitro Eye Irritation Test Using SIRC Cells. Toxicol. In Vitro 22,760–770.
|
|
(5)
|
Sakaguchi H., et al. (2011). Validation Study of the Short Time Exposure (STE) Test to Assess the Eye Irritation Potential of Chemicals. Toxicol. In Vitro 25,796-809.
|
|
(6)
|
Kojima H., et al. (2013). Second-Phase Validation of Short Time Exposure Tests for Assessment of Eye Irritation Potency of Chemicals. Toxicol. In Vitro 27, s. 1855-1869.
|
|
(7)
|
ICCVAM (2013). Short Time Exposure (STE) Test Method Summary Review Document, NIH. Se [http://www.ntp.niehs.nih.gov/iccvam/docs/ocutox_docs/STE-SRD-NICEATM-508.pdf].
|
|
(8)
|
Kapitel B.5 i denna bilaga, Akut ögonirritation/ögonkorrosion.
|
|
(9)
|
Saito K, et al. (2015). Predictive Performance of the Short Time Exposure Test for Identifying Eye Irritation Potential of Chemical Mixtures.
|
|
(10)
|
Mikkelson T.J., Chrai S.S. och Robinson J.R. (1973). Altered Bioavailability of Drugs in the Eye Due to Drug-Protein Interaction. J. Pharm. Sci.1648-1653.
|
|
(11)
|
ECETOC (1998). Eye Irritation Reference Chemicals Data Bank. Technical Report nr 48. (2)), Bryssel, Belgien
|
|
(12)
|
Gautheron P, et al. (1992). Bovine Corneal Opacity and Permeability Test: an In Vitro Assay of Ocular Irritancy. Fundam Appl Toxicol. 18, s. 442-449.
|
|
(13)
|
OECD (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environmental, Health and Safety Publications, Series on Testing and Assessment (No 34). Organisationen för ekonomiskt samarbete och utveckling (OECD), Paris.
|
|
(14)
|
OECD (2017). Guidance Document on an Integrated Approaches on Testing and Assessment for Serious Eye Damage and Eye irritation. Environmental, Health and Safety Publications, Series on Testing and Assessment (No 263). Organisationen för ekonomiskt samarbete och utveckling (OECD), Paris.
|
Tillägg
DEFINITIONER
Noggrannhet
: Ett mått på hur väl testmetodresultaten och de godtagna referensvärdena överensstämmer med varandra. Exakthet är ett mått på testmetodens prestanda och en relevansaspekt. Detta begrepp används ofta omväxlande med ”konkordans” för att beskriva andelen korrekta resultat med en testmetod (13).
Referensämne
: ett ämne som används som standard för jämförelse med en testkemikalie. Ett referensämne ska ha följande egenskaper: i) En eller flera enhetliga och tillförlitliga källor. ii) Strukturell och funktionell likhet med den ämnesklass som testas iii) Kända fysikaliska/kemiska egenskaper, stödjande data om kända effekter och v) känd styrka inom det önskade responsområdet.
Bottom-up-strategi
: en stegvis metod som tillämpas på testkemikalier som inte tros kräva klassificering för ögonirritation eller allvarlig ögonskada. Det första steget är att skilja kemikalier som inte behöver klassificeras (negativt utfall) från andra kemikalier (positivt utfall).
Kemikalie
: Ett ämne eller en blandning.
Ögonirritation
: ändring som uppstår i ögat efter applicering av en testkemikalie på ögats främre yta och som inte är fullt reversibla inom 21 dagar från appliceringen. Utbytbar med ”reversibla effekter på ögat” och GHS/CLP kategori 2.
Falskt negativt värde
: den andel av alla positiva ämnen som av en testmetod felaktigt identifieras som negativ. Det är en indikator på testmetodens prestanda.
Falskt positivt värde
: den andel av alla negativa (icke-aktiva) ämnen som av en testmetod felaktigt identifieras som positiva. Det är en indikator på testmetodens prestanda.
Fara
: Inneboende egenskap hos ett ämne eller en situation som har potential att orsaka skadliga effekter när en organism, ett system eller en (del-) population exponeras för ämnet.
Mediumkontroll
: ett obehandlat replikat som innehåller alla komponenter i ett testsystem. Detta prov hanteras tillsammans med testkemikaliebehandlade prover och andra kontrollprover för att avgöra huruvida lösningsmedlet påverkar testsystemet.
Blandning
: En blandning eller en lösning som består av två eller flera ämnen.
Monokomponentämne
: Ett ämne, definierat av sin kvantitativa sammansättning, där en huvudsaklig beståndsdel utgör minst 80 viktprocent.
MTT
: 3-(4,5-dimetyltiazol-2-yl)-2,5-difenyltetrazoliumbromid, tiazolylblåtetrazoliumbromid.
Multikomponentämne
: Ett ämne, definierat av sin kvantitativa sammansättning, där fler än en huvudsaklig beståndsdel förekommer i en koncentration på ≥ 10 viktprocent och < 80 viktprocent. Ett multikomponentämne är resultatet av en tillverkningsprocess. Skillnaden mellan blandningar och multikomponentämnen är att en blandning erhålls genom att två eller flera ämnen blandas utan någon kemisk reaktion. Ett multikomponentämne är resultatet av en kemisk reaktion.
OD
: Optisk densitet
Positiv kontroll
: ett replikat som innehåller testsystemets alla komponenter och behandlas med ett ämne som man vet ger positiv respons. För att säkerställa att den positiva kontrollresponsens variationer kan bedömas över tiden, bör den kraftiga irritation som framkallas inte vara överdriven.
Relevans
: Beskrivning av förhållandet mellan testet och den berörda effekten och huruvida testet är meningsfullt och användbart för ett visst ändamål. Relevansen är ett mått på hur väl man genom testet korrekt kan mäta och prediktera den biologiska effekt det gäller. Relevans omfattar också överväganden om en testmetods noggrannhet (konkordans) (10).
Tillförlitlighet
: Mått på i vilken utsträckning en testmetod kan reproduceras inom och mellan laboratorier över tid, när den utförs med användning av samma protokoll. Tillförlitligheten bedöms genom att man beräknar reproducerbarheten inom och mellan laboratorier och repeterbarheten inom laboratorier (13).
Sensitivitet
: den andel av alla positiva/aktiva kemikalier som klassificeras korrekt i testet. Det är ett mått på noggrannhet för en testmetod som ger kategoriska resultat och är en viktig faktor vid bedömningen av relevansen för en testmetod (10).
Allvarliga ögonskador
: vävnadsskada i ögat eller kraftig synnedsättning som uppstår efter applicering av en testkemikalie på ögats främre yta och som inte är fullt reversibel inom 21 dagar från appliceringen (2). Utbytbar med ”irreversibla effekter på ögat” och GHS/CLP kategori 1.
Lösningsmedels-/vehikelkontroll
: ett obehandlat prov som innehåller testsystemets alla komponenter, inbegripet lösningsmedlet eller vehikeln som behandlas tillsammans med testkemikaliebehandlade och andra kontrollprover i syfte att fastställa bakgrundsrespons för de prover som behandlas med testkemikalien upplöst i samma lösningsmedel eller vehikel. När detta prov testas med en parallell mediumkontroll, visar provet också huruvida lösningsmedlet eller vehikeln påverkar testsystemet.
Specificitet
: den andel av alla negativa/inaktiva kemikalier som klassificeras korrekt i testet. Det är ett mått på noggrannhet för en testmetod som ger kategoriska resultat och är en viktig faktor vid bedömningen av relevansen för en testmetod (13).
Ämne
: kemiskt grundämne och föreningar av detta grundämne i naturlig eller tillverkad form, inklusive de eventuella tillsatser som är nödvändiga för att bevara dess stabilitet och sådana föroreningar som härrör från tillverkningsprocessen, men exklusive eventuella lösningsmedel som kan avskiljas utan att det påverkar ämnets stabilitet eller ändrar dess sammansättning.
Ytaktivt ämne
: en kemikalie, såsom ett rengöringsmedel, som kan minska en vätskas ytspänning så att lödder bildas eller fasta ämnen penetrerar. Även kallat tensid eller vätmedel.
Testkemikalie
: alla ämnen eller blandningar som testas med denna testmetod.
Stegvis teststrategi
: en strategi för stegvis testning där all befintlig information om en testkemikalie granskas i en angiven ordning, med användning av förfarandet med sammanvägd bedömning (weight of evidence) för att avgöra om det finns tillräckligt med information till hands för att besluta om faroklassificering innan man går vidare till nästa steg. Om testkemikaliens förmåga att framkalla irritation kan fastställas på grundval av befintlig information behövs ingen ytterligare testning. Om testkemikaliens förmåga att framkalla irritation inte kan fastställas på grundval av befintlig information genomförs ett stegvis sekventiellt djurförsöksförfarande tills att det går att fastställa en otvetydig klassificering.
Top-down-strategi
: stegvis metod som tillämpas på testkemikalier som misstänks orsaka allvarlig ögonskada. Det första steget är att skilja kemikalier som inducerar allvarlig ögonskada (positivt utfall) från andra kemikalier (negativt utfall).
FN:s globalt harmoniserade system för klassificering och märkning av kemikalier (GHS)
: ett system för att klassificera ämnen och blandningar enligt standardiserade typer och nivåer av fysiska risker, hälsorisker och miljörisker och informera om resultaten genom t.ex. piktogram, signalord, faroangivelser, skyddsangivelser och säkerhetsdatablad i syfte att informera om ämnenas skadliga effekter för att skydda personer (inklusive arbetsgivare, arbetstagare, transportörer, konsumenter och personal inom räddningstjänsten) och miljön (1).
GHS/CLP-kategori 1
: se ”allvarliga ögonskador”.
GHS/CLP-kategori 2
: se ”ögonirritation”.
GHS/CLP Ingen klassificering
: Kemikalier som inte är klassificerade enligt GHS/CLP-kategori 1 eller 2 (eller 2A eller 2B).
UVCB-ämne
: Ämne med okänd eller varierande sammansättning, komplexa reaktionsprodukter och biologiskt material.
B.69 TESTMETOD MED REKONSTRUERAT HUMANT HORNHINNELIKT EPITEL (RhCE) FÖR IDENTIFIERING AV KEMIKALIER SOM INTE KRÄVER KLASSIFICERING ELLER MÄRKNING FÖR ÖGONIRRITATION ELLER ALLVARLIG ÖGONSKADA
INLEDNING
|
1.
|
Denna testmetod (TM) motsvarar OECD:s testriktlinje (TG) 492 (2017). Allvarlig ögonskada avser vävnadsskada i ögat eller kraftig synnedsättning som uppstår efter applicering av en testkemikalie på ögats främre yta och som inte är fullt reversibel inom 21 dagar från appliceringen, så som definierat enligt FN:s globalt harmoniserade system för klassificering och märkning av kemikalier (GHS) (1) och förordning (EG) nr 1272/2008 om klassificering, märkning och förpackning av ämnen och blandningar (CLP-förordningen) (76). Likaså avses med ögonirritation, enligt GHS och CLP-förordningen, ändringar som uppstår i ögat efter applicering av en testkemikalie på ögats främre yta och som är fullt reversibla inom 21 dagar från appliceringen. Testkemikalier som orsakar allvarlig ögonskada klassificeras som GHS- och CLP-kategori 1, medan de som orsakar ögonirritation klassificeras som GHS- och CLP-kategori 2. Testkemikalier som inte har klassificerats för ögonirritation eller allvarlig ögonskada definieras som de som inte uppfyller kraven för att klassificeras som GHS- respektive CLP-kategori 1 eller 2 (2A eller 2B), det vill säga de hänvisas till som GHS och CLP Ingen klassificering.
|
|
2.
|
Bedömningen av allvarlig ögonskada/ögonirritation har vanligen inbegripit användningen av försöksdjur (TM B.5 [2]). Valet av den lämpligaste testmetoden och användningen av den här testmetoden bör ses i samband med OECD:s vägledning Integrated Approaches on Testing and Assessment for Serious Eye Damage and Eye Irritation (39).
|
|
3.
|
Denna testmetod beskriver ett in vitro-förfarande med vilket det är möjligt att identifiera kemikalier (ämnen och blandningar) som inte behöver klassificeras och märkas för ögonirritation eller allvarlig ögonskada i enlighet med GHS och CLP-förordningen. I testet använder man sig av rekonstruerat humant hornhinnelikt epitel (Reconstructed human Cornea-like Epithelium, RhCE) som imiterar det humana hornhinneepitelets histologiska, morfologiska, biokemiska och fysiologiska egenskaper. Fyra andra in vitro-testmetoder har validerats, bedömts vara vetenskapligt giltiga och antagits som TM B.47 (3), B.48 (4), B.61 (5) och B.68 (6) för att, med avseende på människors hälsa, undersöka endpointen allvarlig ögonskada/ögonirritation.
|
|
4.
|
Två validerade tester där man använder sig av kommersiellt tillgängliga RhCE-modeller har inkluderats i denna testmetod. Valideringsstudier för att bedöma ögonirritation/allvarlig ögonskada har genomförts (7) (8) (9) (10) (11) (12) (13) med EpiOcular™ Eye Irritation Test (EIT) och SkinEthic™ Human Corneal Epithelium (HCE) Eye Irritation Test (EIT). Båda dessa tester använder kommersiellt tillgängliga RhCE-vävnadsmodeller som testsystem, och omnämns i nedanstående text som de Validerade referensmetoderna – VRM1 respektive VRM2. Utifrån dessa valideringsstudier och den oberoende expertgranskningen (9) (12) fann man att EpiOcular™ EIT och SkinEthic™ HCE EIT korrekt kan identifiera kemikalier (både ämnen och blandningar) som inte kräver klassificering eller märkning för ögonirritation eller allvarlig ögonskada enligt GHS, och testerna rekommenderades som vetenskapligt giltiga för det ändamålet (13).
|
|
5.
|
Det är för närvarande allmänt vedertaget att inom överskådlig framtid kommer ingen enskild in vitro-testmetod helt att kunna ersätta Draizes in vivo-ögontest (2) (14) för att prediktera alla typer av responser i form av allvarlig ögonskada/ögonirritation för olika kemiska klasser. Men strategiska kombinationer av flera olika testmetoder inom ramen för (differentierade) teststrategier, som nedifrån och upp-/uppifrån och ned-strategin, kan eventuellt komma att helt kunna ersätta Draizes ögontest (15). Nedifrån och upp-strategin (15) är utformad för att användas när en kemikalie, utifrån befintlig information, inte förväntas orsaka tillräcklig ögonirritation för att kräva en klassificering, medan uppifrån och ned-strategin (15) är utformad för att användas när en kemikalie, utifrån befintlig information, förväntas orsaka allvarlig ögonskada. EpiOcular™ EIT och SkinEthic™ HCE EIT rekommenderas för att identifiera kemikalier som inte kräver klassificering för ögonirritation eller allvarlig ögonskada enligt GHS/CLP-förordningen (Ingen klassificering) utan vidare tester, inom ramen för en teststrategi som nedifrån och upp eller uppifrån och ned, föreslagen av Scott m.fl., till exempel som ett första steg i en nedifrån och upp-strategi, eller som ett av de sista stegen i en uppifrån och ned-strategi (15). Men EpiOcular™ EIT och SkinEthic™ HCE EIT är inte avsedda att differentiera mellan GHS-/CLP-kategori 1 (allvarlig ögonskada) respektive kategori 2 (ögonirritation). Denna differentiering kommer att behöva undersökas i ytterligare ett steg i teststrategin (15). En testkemikalie som med EpiOcular™ EIT eller SkinEthic™ HCE EIT identifieras kräva klassificering för ögonirritation/allvarlig ögonskada kommer därför att kräva ytterligare tester (in vitro och/eller in vivo) för att nå en definitiv slutsats (GHS-/CLP-kategori Ingen klassificering, kategori 2 eller kategori 1), med användning av till exempel TM B.47, B.48, B.61 eller B.68.
|
|
6.
|
Syftet med denna testmetod är att beskriva det förfarande som används för att utvärdera om en testkemikalie kan vara skadlig för ögonen, utifrån dess förmåga att inducera cytotoxicitet i en RhCE-vävnadsmodell, mätt med MTT-testet (16) (se punkt 21). Viabiliteten i RhCE-vävnaden efter exponering för en testkemikalie fastställs genom jämförelse med vävnader som behandlats med negativt kontrollämne (% viabilitet) och används sedan för att prediktera om testkemikalien kan vara skadlig för ögonen.
|
|
7.
|
Prestandastandarder (17) finns tillgängliga för att underlätta valideringen av nya eller modifierade in vitro-RhCE-baserade tester som liknar EpiOcular™ EIT och SkinEthic™ HCE EIT, i enlighet med principerna i OECD:s vägledning nr 34 (18), och möjliggöra ändring av OECD TG 492 vid lämplig tidpunkt. Ömsesidigt accepterande av data (Mutual Acceptance of Data, MAD) enligt OECD-avtalet garanteras endast för tester som validerats enligt prestandastandarder, om OECD har granskat och infogat dessa tester i motsvarande testriktlinje.
|
DEFINITIONER
|
8.
|
Definitioner av begreppen finns i tillägg 1.
|
INLEDANDE ÖVERVÄGANDEN OCH BEGRÄNSNINGAR
|
9.
|
Denna testmetod bygger på kommersiella tredimensionella RhCE-vävnadsmodeller som framställs från antingen primära humana epidermala keratinocyter (dvs. EpiOcular™ OCL-200) eller humana immortaliserade hornhinne-epitelceller (dvs. SkinEthic™ HCE/S). Dessa RhCE-vävnadsmodeller, EpiOcular™ OCL-200 och SkinEthic™ HCE/S, liknar hornhinnans tredimensionella epitelstruktur in vivo och produceras med hjälp av celler från de arter som är av intresse (19) (20). Vidare mäter testerna direkt cytotoxicitet orsakad av kemikaliens penetration av hornhinnan och framkallandet av cell- och vävnadsskada, den cytotoxiska responsen fastställer då det sammantagna utfallet av den allvarliga ögonskadan/ögonirritationen in vivo. Cellskada kan uppstå genom flera olika verkningsmekanismer (se punkt 20), men cytotoxiciteten spelar en viktig, om inte den främsta, mekanistiska rollen för den sammantagna responsen på en kemikalie vid en allvarlig ögonskada/ögonirritation in vivo främst uttryckt genom hornhinnegrumling, irit, konjunktivarodnad och/eller kemos, oberoende av de fysikalisk-kemiska processer som ger upphov till vävnadsskadan.
|
|
10.
|
En stor mängd kemikalier, omfattande en stor variation i kemisk typ och klass, molekylvikt, LogP-värden, kemisk struktur och så vidare har testats i den valideringsstudie som ligger till grund för denna testmetod. Valideringsdatabasen för EpiOcular™ EIT innehöll totalt 113 kemikalier, omfattande 95 olika organiska funktionella grupper enligt en QSAR-toolbox-analys från OECD (8). Majoriteten av dessa kemikalier utgjordes av monokomponentämnen, men flera multikomponentämnen (inklusive 3 homopolymerer, 5 sampolymerer och 10 kvasipolymerer) inkluderades också i studien. I fråga om fysikaliskt tillstånd och GHS-/CLP-kategorier fördelade sig de 113 testade kemikalierna som följer: 13 vätskor enligt kategori 1, 15 fasta ämnen enligt kategori 1, 6 vätskor enligt kategori 2A, 10 fasta ämnen enligt kategori 2A, 7 vätskor enligt kategori 2B, 7 fasta ämnen enligt kategori 2B, 27 vätskor enligt Ingen klassificering och 28 fasta ämnen enligt Ingen klassificering (8). Valideringsdatabasen för SkinEthic™ HCE EIT innehöll totalt 200 kemikalier, omfattande 165 olika organiska funktionella grupper (8) (10) (11). Majoriteten av dessa kemikalier utgjordes av monokomponentämnen, men flera multikomponentämnen (inklusive 10 polymerer) inkluderades också i studien. I fråga om fysikaliskt tillstånd och GHS-/CLP-kategorier fördelade sig de 200 testade kemikalierna som följer: 27 vätskor enligt kategori 1, 24 fasta ämnen enligt kategori 1, 19 vätskor enligt kategori 2A, 10 fasta ämnen enligt kategori 2A, 9 vätskor enligt kategori 2B, 8 fasta ämnen enligt kategori 2B, 50 vätskor enligt Ingen klassificering och 53 fasta ämnen enligt Ingen klassificering (10) (11).
|
|
11.
|
Denna testmetod kan användas för ämnen och blandningar, för fasta ämnen, vätskor, halvfasta ämnen och vaxer. Vätskorna kan vara vattenbaserade eller inte, fasta ämnen kan vara lösliga eller olösliga i vatten. När det är möjligt bör ämnen i fast form pulveriseras före applicering, ingen annan förbehandling av provet krävs. Gaser och aerosoler har inte bedömts i någon valideringsstudie. Det är tänkbart att de kan testas med RhCE-teknik, men den aktuella testmetoden medger inte tester av gaser och aerosoler.
|
|
12.
|
Testkemikalier som absorberar ljus i samma intervall som MTT-formazan (naturligt eller efter behandling) och testkemikalier som direkt kan reducera färgämnet MTT (till MTT-formazan) kan störa mätningarna av vävnadsviabilitet och kräver anpassade kontroller för korrigering. Den sorts anpassade kontroller som kan komma att krävas varierar med den typ av interferens som testkemikalien orsakar och det förfarande som används för att kvantifiera MTT-formazan (se punkterna 36–42).
|
|
13.
|
Resultat från pre-valideringsstudier (21) (22) och fullständiga valideringsstudier (8) (10) (11) har visat att både EpiOcular™ EIT och SkinEthic™ HCE EIT kan överföras till laboratorier som kan anses vara nya i fråga om analysernas utförande, och att testerna också är reproducerbara inom och mellan laboratorier. Utifrån dessa studier kan den nivå av reproducerbarhet, uttryckt som prediktionskonkordans, som kan förväntas från EpiOcular™ EIT utifrån data om 113 kemikalier vara i storleksordningen 95 % inom laboratorier och 93 % mellan laboratorier. Nivån på reproducerbarhet, uttryckt som prediktionskonkordans, som kan förväntas från SkinEthic™ HCE EIT utifrån data om 120 kemikalier är i storleksordningen 92 % inom laboratorier och 95 % mellan laboratorier.
|
|
14.
|
EpiOcular™ EIT kan användas för att identifiera kemikalier som inte kräver klassificering för ögonirritation eller allvarlig ögonskada enligt GHS- och CLP-klassificeringssystemet. Med tanke på de data som erhölls i valideringsstudien (8), har EpiOcular™ EIT en sammantagen noggrannhet på 80 % (baserat på 112 kemikalier), en känslighet på 96 % (baserat på 57 kemikalier), ett falskt negativt värde på 4 % (baserat på 57 kemikalier), en specificitet på 63 % (baserat på 55 kemikalier) och ett falskt positivt värde på 37 % (baserat på 55 kemikalier) i jämförelse med referensdata från in vivo-kaninögontester (TM B.5) (2) (14) klassificerat enligt GHS- och CLP-klassificeringssystemet. En studie där 97 flytande agrokemiska sammansättningar testades med EpiOcular™ EIT uppvisade resultat för denna typ av blandningar som liknade dem som erhölls i valideringsstudien (23). De 97 sammansättningarna fördelade sig som följer: 21 enligt kategori 1, 19 enligt kategori 2A, 14 enligt kategori 2B och 43 enligt Ingen klassificering, klassificerade enligt GHS-klassificeringssystemet baserat på referensdata från in vivo-kaninögontester (TM B.5) (2) (14). En sammantagen noggrannhet på 82 % (baserat på 97 sammansättningar), känslighet på 91 % (baserat på 54 sammansättningar), falskt negativt värde på 9 % (baserat på 54 sammansättningar), specificitet på 72 % (baserat på 43 sammansättningar) och falskt positivt värde på 28 % (baserat på 43 sammansättningar) erhölls (23).
|
|
15.
|
SkinEthic™ HCE EIT kan användas för att identifiera kemikalier som inte kräver klassificering för ögonirritation eller allvarlig ögonskada enligt GHS och CLP-förordningens klassificeringssystem. Med tanke på de data som erhölls i valideringsstudien (10) (11), har SkinEthic™ HCE en sammantagen noggrannhet på 84 % (baserat på 200 kemikalier), en känslighet på 95 % (baserat på 97 kemikalier), ett falskt negativt värde på 5 % (baserat på 97 kemikalier), en specificitet på 72 % (baserat på 103 kemikalier) och ett falskt positivt värde på 28 % (baserat på 103 kemikalier) i jämförelse med referensdata från in vivo-kaninögontester (TM B.5) (2) (14) klassificerat enligt GHS och CLP-förordningens klassificeringssystem.
|
|
16.
|
De falskt negativa värden som erhölls med båda RhCE-testerna, med antingen ämnen eller blandningar, faller inom de 12 % sammantagen sannolikhet att kemikalier klassificeras antingen som GHS- och CLP-kategori 2 eller GHS- och CLP-kategori Ingen klassificering enligt Draizes in vivo-ögontest, i upprepade tester, detta beror på metodens inneboende inom-test-variation (24). De falskt positiva värdena som erhölls med båda RhCE-testerna med antingen ämnen eller blandningar är inte kritiskt avgörande för denna testmetod eftersom alla testkemikalier som framkallar en vävnadsviabilitet som är lika med eller lägre än de etablerade brytvärdena (se punkt 44) kommer att kräva ytterligare tester med andra in vitro-testmetoder, eller som ett sista alternativ på kaniner, beroende på lagkraven, med hjälp av en strategi med stegvis testning i ett tillvägagångssätt med sammanvägd bedömning. Dessa testmetoder kan användas för alla sorters kemikalier, där ett negativt resultat bör accepteras som underlag för att avstå från att klassificera kemikalien för ögonirritation och allvarlig ögonskada (GHS och CLP Ingen klassificering). Vederbörande tillsynsmyndigheter bör rådfrågas innan EpiOcular™ EIT och SkinEthic™ HCE EIT används för andra klassificeringssystem än för GHS/CLP-förordningen.
|
|
17.
|
En begränsning hos denna testmetod är att den inte gör det möjligt att skilja mellan ögonirritation/reversibla effekter på ögat (kategori 2) och allvarliga ögonskador/irreversibla effekter på ögonen (kategori 1) som de definieras i GHS och CLP-förordningen, och inte heller mellan irriterande för ögonen (valfri kategori 2A) och milt irriterande för ögonen (valfri kategori 2B) som definieras i GHS (1). För dessa syften krävs ytterligare tester med andra in vitro-testmetoder.
|
|
18.
|
Termen ”testkemikalie” används i denna testmetod för att benämna det som testas (77) och är inte kopplad till applicerbarheten hos RhCE-testmetoden för att testa ämnen och/eller blandningar.
|
TESTETS PRINCIP
|
19.
|
Testkemikalien appliceras lokalt på minst två tredimensionella RhCE-vävnadsmodeller och vävnadsviabilitet mäts en tid efter exponering och inkubation. Dessa RhCE-vävnader rekonstrueras från primära humana epidermala keratinocyter eller humana immortaliserade hornhinne-epitelceller, vilka har odlats i flera dagar så att de har bildat ett stratifierat, högdifferentierat skivepitel med en morfologi som liknar den hos human hornhinna. EpiOcular™ RhCE-vävnadsmodellen består av minst 3 viabla cellager och en icke-keratiniserad yta, uppvisande en hornhinnelik struktur, jämförbar med den som ses in vivo. SkinEthic™ HCE RhCE-vävnadsmodellen består av minst 4 viabla cellager, inklusive pelarformade basalceller, övergångsformer av wing-celler och ytliga skivepitelceller som liknar dem hos normalt, humant hornhinneepitel (20) (26).
|
|
20.
|
Kemiskt inducerad allvarlig ögonskada/ögonirritation, vilken in vivo främst yttrar sig som hornhinnegrumling, irit, konjunktivarodnad och/eller kemos, är resultatet av en kaskad av händelser som börjar med penetration av kemikalien genom hornhinnan och/eller bindhinnan och framkallande av cellskada. Cellskada kan uppstå genom flera olika verkningsmekanismer, inklusive lysering av cellmembran (t.ex. av ytaktiva ämnen, organiska lösningsmedel), koagulation av makromolekyler (särskilt proteiner) (t.ex. av ytaktiva ämnen, organiska lösningsmedel, alkalier och syror), saponifiering av lipider (t.ex. av alkalier), och alkylering eller annan kovalent interaktion med makromolekyler (t.ex. av blekmedel, peroxider och alkylerande ämnen) (15) (27) (28). Men det har visats att cytotoxicitet spelar en viktig, om inte den främsta, mekanistiska rollen för den sammantagna responsen på en kemikalie vid en allvarlig ögonskada/ögonirritation, oberoende av de fysikalisk-kemiska processer som ligger till grund för skadan (29) (30). Vidare bestäms kemikaliens potential till allvarlig ögonskada/ögonirritation i princip av den inledande skadans omfattning (31), vilken korrelerar med omfattningen av celldöd (29) och med omfattningen av efterföljande responser och slutligt utfall (32). Därför påverkar lätt irriterande ämnen i allmänhet endast det ytliga hornhinneepitelet, de milt till måttligt irriterande ämnena skadar i princip epitelet och det ytliga stromat och de kraftigt irriterande ämnena skadar epitelet, det djupa stromat och ibland hornhinnans endotel (30) (33). Mätningen av viabilitet hos RhCE-vävnaden efter lokal exponering för en testkemikalie i syfte att identifiera kemikalier som inte kräver klassificering för allvarlig ögonskada/ögonirritation (GHS- och CLP-kategori Ingen klassificering) grundas på antagandet att alla kemikalier som framkallar allvarlig ögonskada eller ögonirritation kommer att framkalla en cytotoxisk verkan i hornhinnans epitel och/eller bindhinna.
|
|
21.
|
Viabiliteten hos RhCE-vävnaden mäts traditionellt genom enzymatisk omvandling av färgämnet MTT (3-[4,5-dimetyltiazol-2-yl]-2,5-difenyltetrazoliumbromid, tiazolylblåtetrazoliumbromid, CAS-nr 298-93-1) av vävnadens viabla celler till ett blått MTT-formazansalt som kvantifieras efter extraktion ur vävnader (16). Kemikalier som inte kräver klassificering och märkning enligt GHS/CLP-förordningen (Ingen klassificering) identifieras som de som inte minskar vävnadsviabiliteten under en definierad tröskel (dvs. vävnadsviabilitet > 60 %, i EpiOcular™ EIT och SkinEthic™ HCE EITL (78), eller > 50 %, i SkinEthic™ HCE EITS (79)) (se punkt 44).
|
DEMONSTRATION AV KOMPETENS
|
22.
|
Inför rutinmässig användning av RhCE-tester för tillsynsändamål ska laboratorier visa sin tekniska kompetens genom att korrekt prediktera de femton kvalifikationskemikalier som anges i tabell 1. Dessa kemikalier har valts ut från de kemikalier som användes i valideringsstudierna av VRM:erna (8) (10) (11). I urvalet ingår, så långt det är möjligt, kemikalier som i) täcker olika fysikaliska tillstånd, ii) täcker hela intervallet av in vivo-responser av allvarlig ögonskada/ögonirritation utifrån högkvalitativa resultat från in vivo-referenstestet på kaninögon (TM B.5) (2) (14) och GHS-klassificeringssystemet (dvs. kategorier 1, 2A, 2B eller Ingen klassificering) (1) och CLP-klassificeringssystemet (dvs. kategorier 1, 2 eller Ingen klassificering), iii) täcker de olika pådrivande in vivo-faktorerna för klassificering (24) (25), iv) är representativa för de kemikalieklasser som används i valideringsstudien (8) (10) (11), v) täcker ett bra, brett urval av organiska funktionella grupper (8) (10) (11), vi) har väl definierade kemiska strukturer (8) (10) (11), vii) är färgade och/eller reducerar MTT direkt, viii) gav reproducerbara resultat med RhCE-testmetoder i samband med deras validering, ix) predikterades korrekt av RhCE-testmetoder i samband med deras validering, x) täcker hela intervallet av in vivo-responser baserat på högkvalitativa RhCE-testmetoddata (0 till 100 % viabilitet), xi) finns kommersiellt tillgängliga, och xii) inte är förknippade med oöverkomliga anskaffnings- och eller bortskaffningskostnader. I situationer där en kemikalie i förteckningen inte finns att tillgå eller inte kan användas av andra giltiga skäl kan en annan kemikalie som uppfyller de ovan beskrivna kriterierna användas, till exempel en av dem som användes i valideringen av VRM. Men sådana avvikelser bör motiveras.
Tabell 1
Förteckning över kvalifikationskemikalier
|
Kemiskt namn
|
CAS-nummer
|
Organisk funktionell grupp (80)
|
Fysikaliskt tillstånd
|
VRM1 viabilitet (%) (81)
|
VRM2 viabilitet (%) (82)
|
VRM-prediktion
|
Reducerar MTT
|
Färginterf.
|
|
Metyltioglykolat
(83)
|
|
|
2365-48-2
|
Karboxylsyraester, tioalkohol
|
Flytande
|
10,9 ± 6,4
|
5,5 ± 7,4
|
Ingen prediktion kan göras
|
J (starkt)
|
N
|
Hydroxietylakrylat
|
|
818-61-1
|
Akrylat,
|
alkohol
|
Flytande
|
7,5 ± 4,7 (84)
|
1,6 ± 1,0
|
Ingen prediktion kan göras
|
N
|
N
|
|
2,5-dimetyl-2,5-hexandiol
|
110-03-2
|
Alkohol
|
Fast
|
2,3 ± 0,2
|
0,2 ± 0,1
|
Ingen prediktion kan göras
|
N
|
N
|
|
Natriumoxalat
|
62-76-0
|
Oxokarboxylsyra
|
Fast
|
29,0 ± 1,2
|
5,3 ± 4,1
|
Ingen prediktion kan göras
|
N
|
N
|
|
In vivo kategori 2A (83)
|
|
|
2,4,11,13-Tetraazatetradekan-diimidamid, N,N″-bis(4-klorfenyl)-3,12-diimino-, di-D-glukonat (20 %, vattenbaserad) (85)
|
18472-51-0
|
Aromatisk heterocyklisk halid, arylhalid, dihydroxylgrupp, guanidin
|
Flytande
|
4,0 ± 1,1
|
1,3 ± 0,6
|
Ingen prediktion kan göras
|
N
|
J (svagt)
|
|
Natriumbensoat
|
532-32-1
|
Aryl, karboxylsyra
|
Fast
|
3,5 ± 2,6
|
0,6 ± 0,1
|
Ingen prediktion kan göras
|
N
|
N
|
|
In vivo Kategori 2B (83)
|
|
|
Dietyltoluamid
|
134-62-3
|
Bensamid
|
Flytande
|
15,6 ± 6,3
|
2,8 ± 0,9
|
Ingen prediktion kan göras
|
N
|
N
|
|
2,2-Dimetyl-3-metylenbicyklo [2.2.1] heptan
|
79-92-5
|
Alkan, grenad med tertiärt kol, alken, bicykloheptan, karbocykliska ämnen med ringbrygga, cykloalkan
|
Fast
|
4,7 ± 1,5
|
15,8 ± 1,1
|
Ingen prediktion kan göras
|
N
|
N
|
|
In vivo Ingen klassificering (83)
|
|
|
1-Etyl-3-metylimidazoliumetylsulfat
|
342573-75-5
|
Alkoxi, ammoniumsalt, aryl, imidazol, sulfat
|
Flytande
|
79,9 ± 6,4
|
79,4 ± 6,2
|
Ingen klassificering
|
N
|
N
|
|
Dikaprylyleter
|
629-82-3
|
Alkoxi, eter
|
Flytande
|
97,8 ± 4,3
|
95,2 ± 3,0
|
Ingen klassificering
|
N
|
N
|
|
Piperonylbutoxid
|
51-03-6
|
Alkoxi, benzodioxol, bensyl, eter
|
Flytande
|
104,2 ± 4,2
|
96,5 ± 3,5
|
Ingen klassificering
|
N
|
N
|
|
Polyetylenglykol (PEG-40) hydrerad ricinolja
|
61788-85-0
|
Acyl-grupp (acylal), alkohol, allyl, eter
|
Viskös
|
77,6 ± 5,4
|
89,1 ± 2,9
|
Ingen klassificering
|
N
|
N
|
|
1-(4-klorfenyl)-3-(3,4-diklorfenyl)urea
|
101-20-2
|
Aromatisk heterocyklisk halid, arylhalid, ureaderivat
|
Fast
|
106,7 ± 5,3
|
101,9 ± 6,6
|
Ingen klassificering
|
N
|
N
|
|
2,2′-Metylen-bis[6-(2H-bensotriazol-2-yl)-4-(1,1,3,3-tetrametylbutyl)-fenol]
|
103597-45-1
|
Alkan grenad med kvartärt kol, fuserat karbocykliskt aromatiskt ämne, fuserade mättade heterocykliska ämnen, prekursorer till kinoida ämnen, tert-butyl
|
Fast
|
102,7 ± 13,4
|
97,7 ± 5,6
|
Ingen klassificering
|
N
|
N
|
|
Kaliumtetrafluorborat
|
14075-53-7
|
Oorganiskt salt
|
Fast
|
88,6 ± 3,3
|
92,9 ± 5,1
|
Ingen klassificering
|
N
|
N
|
|
Förkortningar:
CAS-nummer = nummer i registret som förs av Chemical Abstracts Service, GHS = FN:s globalt harmoniserade system för klassificering och märkning av kemikalier (1), VRM1 = Validerad referensmetod, EpiOcular™ EIT, VRM2 = Validerad referensmetod, SkinEthic™ HCE EIT, Färginterf. = färginterferens med standardabsorbansmätning av MTT-formazan (optisk densitet, OD).
|
|
|
23.
|
Som en del av kompetenstesterna rekommenderas att användarna efter att ha mottagit vävnaderna verifierar deras barriäregenskaper enligt RhCE-modelltillverkarens specifikationer (se punkter 25, 27 och 30). Detta är särskilt viktigt om vävnader transporteras långa sträckor eller om transporten tar lång tid. När ett test framgångsrikt har etablerats och kompetensen i metodanvändningen har påvisats, behöver sådana verifikationer inte göras rutinmässigt. Vid rutinmässig användning av ett test rekommenderas dock att man med jämna mellanrum bedömer barriäregenskaperna.
|
FÖRFARANDE
|
24.
|
De tester som för närvarande omfattas av denna testmetod är de vetenskapligt validerade EpiOcular™ EIT och SkinEthic™ HCE EIT (9) (12) (13), omnämnda som Validerad referensmetod (VRM1 respektive VRM2). Standardförfaranden (Standard Operating Procedures, SOP) för RhCE-testmetoder finns tillgängliga och bör användas när testmetoden genomförs och används i laboratoriet (34) (35). Följande punkter och tillägg 2 beskriver RhCE-testernas huvudsakliga delar och förfaranden.
|
RHCE-TESTMETODENS DELAR
Allmänna betingelser
|
25.
|
Relevanta celler av humant ursprung bör användas för att rekonstruera den hornhinneliknande, tredimensionella epitelvävnaden, vilken bör bestå av progressivt stratifierade, men inte förhornade celler. Själva RhCE-vävnadsmodellen bereds inuti ilägg försedda med ett poröst, syntetiskt membran genom vilka näringsämnen kan passera in till cellerna. Flera lager av viabla, icke-keratiniserade epitelceller bör finnas i det rekonstruerade hornhinnelika epitelet. För att det ska vara möjligt att genomföra lokal exponering av testkemikalier på ett sätt som liknar den exponering hornhinneepitelet skulle utsättas för in vivo bör RhCE-vävnadsmodellens epitelyta ha direktkontakt med luften. Vidare bör RhCE-vävnadsmodellen bilda en funktionell barriär som är tillräckligt robust för att motstå snabb penetrering av cytotoxiska referensämnen, till exempel Triton X-100 eller natriumdodecylsulfat (SDS). Barriärfunktionen bör demonstreras och kan skattas genom att fastställa antingen den exponeringstid som krävs för att minska vävnadens viabilitet med 50 % (ET50) efter applicering av ett referensämne vid en specificerad, fast koncentration (t.ex. 100 μl med 0,3-volymprocentig Triton X-100), eller den koncentration vid vilken ett referensämne minskar vävnadens viabilitet med 50 % (IC50) efter en fast exponeringstid (t.ex. 30 minuters behandling med 50 μl SDS) (se punkt 30). Inneslutningsegenskaperna hos RhCE-vävnadsmodellen ska förhindra att testkemikalie passerar runt kanten på den viabla vävnaden, vilket skulle kunna leda till en bristande modellering av hornhinneexponeringen. De celler av humant ursprung som används för att etablera RhCE-vävnadsmodellen ska vara fria från förorening av bakterier, virus, mykoplasma och svamp. Vävnadens sterila tillstånd bör kontrolleras av leverantören så att den är fri från förorening av svamp och bakterier.
|
Funktionella betingelser
Viabilitet
|
26.
|
MTT-testet används för kvantifiering av vävnadsviabilitet (16). Viabla celler i RhCE-vävnaden reducerar färgämnet MTT till en blå MTT-formazanfällning, vilken sedan extraheras ur vävnaden med isopropanol (eller ett liknande lösningsmedel). Det extraherade MTT-formazanet kan kvantifieras, antingen med en standardabsorbansmätning (optisk densitet, OD) eller ett HPLC-/UPLC-spektrofotometriskt förfarande (36). Extraktionsmedlets OD bör vara tillräcklig låg, det vill säga under 0,1. Användare av RhCE-vävnadsmodellen bör se till att varje sats av RhCE-vävnad som används uppfyller de definierade kriterierna för den negativa kontrollen. Intervallet för acceptabla OD-värden för VRM:ernas negativa kontroller ges i tabell 2. En HPLC-/UPLC-spektrofotometrianvändare bör använda OD-intervallen för negativ kontroll angivna i tabell 2 som acceptanskriterier för den negativa kontrollen. Det bör dokumenteras i testrapporten att de vävnader som behandlas med det negativa kontrollämnet är stabila i odling (ger liknande resultat vid viabilitetsmätning) under testets hela exponeringsperiod. Ett liknande förfarande bör följas av vävnadsproducenten som en del av kvalitetskontroll av vävnadssats inför leverans, men i det fallet kan andra acceptanskriterier än de som specificeras i tabell 2 vara tillämpliga. Utvecklaren/leverantören av RhCE-vävnadsmodellen bör fastställa acceptabla gränsvärden (övre och nedre gräns) för den negativa kontrollens OD-värden (under testmetodens kvalitetskontrollbetingelser).
|
Tabell 2
Acceptansintervall för de negativa kontrollernas optiska densitetsvärden (för testanvändare)
|
Test
|
Acceptansintervallets nedre gräns
|
Acceptansintervallets övre gräns
|
|
EpiOcular™ EIT (OCL-200) – VRM1 (för båda protokollen, för flytande resp. fasta ämnen)
|
> 0,8 (86)
|
< 2,5
|
|
SkinEthic™ HCE EIT (HCE/S) – VRM2 (för båda protokollen, för flytande resp. fasta ämnen)
|
> 1,0
|
≤ 2,5
|
Barriärfunktion
|
27.
|
RhCE-vävnaden bör vara tillräckligt tjock och robust för att stå emot snabb penetrering av cytotoxiska referensämnen, bedömt till exempel med ET50 (Triton X-100) eller IC50 (SDS) (tabell 3). Utvecklaren/återförsäljaren av RhCE-vävnadsmodellen bör demonstrera barriärfunktionen hos varje sats av RhCE-vävnad vid leverans av vävnaden till slutanvändaren (se punkt 30).
|
Morfologi
|
28.
|
Histologisk undersökning av RhCE-vävnaden bör fastställa human, hornhinnelik epitelstruktur (omfattande minst 3 lager av viabla epitelceller och en icke-keratiniserad yta). För VRM:erna har lämplig morfologi fastställts av utvecklaren/leverantören och behöver därför inte fastställas på nytt av testmetodanvändaren för varje sats av vävnad som används.
|
Reproducerbarhet
|
29.
|
Resultaten för testmetodens positiva och negativa kontroller bör visa reproducerbarhet över tid.
|
Kvalitetskontroll
|
30.
|
RhCE-vävnaden bör endast användas om utvecklaren/leverantören har visat att varje sats av RhCE-vävnad motsvarar fastställda leveranskriterier, bland vilka de för viabilitet (punkt 26) och barriärfunktion (se punkt 27) är de mest relevanta. Utvecklaren/leverantören av RhCE-vävnadsmodellen bör fastställa acceptabla gränsvärden (övre och nedre gräns) för barriärfunktioner, uppmätta som ET50 eller IC50 (se punkterna 25 och 26). I tabell 3 anges de acceptabla gränsvärden för ET50 och IC50 som utvecklaren/leverantören använder som kvalitetskriterium för leverans av RhCE-vävnaden (använda i VRM:erna). Utvecklaren/leverantören av RhCE-vävnadsmodellen bör tillhandahålla data som demonstrerar överensstämmelse med alla leveranskriterier till testmetodens användare, så att de kan ta med denna information i testrapporten. Endast resultat från vävnader som uppfyller alla dessa leveranskriterier kan godkännas för tillförlitlig prediktion av kemikalier som, i enlighet med GHS/CLP-förordningen, inte kräver klassificering och märkning för ögonirritation eller allvarlig ögonskada.
|
Tabell 3
Kvalitetskriterium för leverans av en sats
|
Test
|
Acceptansintervallets nedre gräns
|
Acceptansintervallets övre gräns
|
|
EpiOcular™ EIT (OCL-200) – VRM1 (100 μl 0,3-volymprocentig Triton X-100)
|
ET50 = 12,2 min
|
ET50 = 37,5 min
|
|
SkinEthic™ HCE EIT (HCE/S) – VRM2 (30 minuters behandling med 50 μl SDS)
|
IC50 = 1 mg/ml
|
IC50 = 3,2 mg/ml
|
Applicering av testkemikalien och kontrollämnen
|
31.
|
Minst två vävnadsreplikat ska användas per testkemikalie och kontrollämne i varje testomgång. Två olika behandlingsprotokoll används, ett för testkemikalier i flytande form och ett för testkemikalier i fast form (34) (35). I båda metodernas respektive protokoll bör vävnadsmodellens yta fuktas med kalcium- och magnesiumfri Dulbeccos fosfatbuffrade saltlösning (Ca2+/Mg2+-fri DPBS) innan testkemikalierna appliceras, för att imitera det mänskliga ögats våta tillstånd. Behandlingen av vävnaderna inleds med exponering för testkemikalie(r) och kontrollämnen. I båda behandlingsprotokollen i båda VRM:erna ska tillräcklig mängd testkemikalie eller kontrollämne appliceras för att jämnt täcka epitelytan samtidigt som man undviker infinit dos (se punkt 32 och 33) (tillägg 2).
|
|
32.
|
Testkemikalier som kan pipetteras vid 37 °C eller lägre temperaturer (vid behov med hjälp av en positiv deplacementpipett) behandlas som vätskor i VRM:erna, i annat fall ska de behandlas som fasta ämnen (se punkt 33). I VRM:erna sprids de flytande testkemikalierna jämnt över vävnadsytan (t.ex. minst 60 μl/cm2 applicering) (se tillägg 2, [33] [34]). Kapillära effekter (ytspänningseffekter) som kan uppstå på grund av de små volymer som appliceras på ilägget (på vävnadsytan) bör undvikas så långt det är möjligt för att garantera korrekt dosering på vävnaden. Vävnader som behandlas med flytande testkemikalier inkuberas i 30 min. under standardodlingsbetingelser (37 ± 2 °C, 5 ± 1 % CO2, ≥ 95 % relativ luftfuktighet). Vid exponeringstidens slut ska den flytande testkemikalien och kontrollämnena noggrant avlägsnas från vävnadsytan genom grundlig sköljning med Ca2+/Mg2+-fri DPBS vid rumstemperatur. Detta sköljsteg följs efter exponering av en nedsänkning i nytt medium vid rumstemperatur (för att avlägsna eventuella kemikalier som kan ha absorberats in i vävnaden) under en fördefinierad tid, vilken varierar med aktuell VRM. Endast för VMR1 görs en postexponerings-inkubation i nytt medium under standardodlingsbetingelser innan MTT-testet genomförs (se tillägg 2, [34] [35]).
|
|
33.
|
Testkemikalier som inte kan pipetteras vid temperaturer upp till 37 °C behandlas i VRM:erna som fasta ämnen. Mängden testkemikalie som appliceras bör vara tillräcklig för att täcka vävnadens hela yta, det vill säga minst 60 mg/cm2 (tillägg 2). När det är möjligt ska fasta ämnen testas i form av ett fint pulver. Vävnader som behandlas med testkemikalier i fast form inkuberas under en fördefinierad tid (beroende på aktuell VRM) under standardodlingsbetingelser (se tillägg 2 [34] [35]). Vid exponeringstidens slut ska testkemikalien i fast form och kontrollämnena noggrant avlägsnas från vävnadsytan genom grundlig sköljning med Ca2+/Mg2+-fri DPBS vid rumstemperatur. Detta sköljsteg följs efter exponering av en nedsänkning i nytt medium vid rumstemperatur (för att avlägsna eventuella kemikalier som kan ha absorberats in i vävnaden) under en fördefinierad tid, vilken varierar med vald VRM, och en postexponerings-inkubation i nytt medium under standardodlingsbetingelser, innan MTT-testet genomförs (se tillägg 2, [34] [35]).
|
|
34.
|
Parallella negativa och positiva kontroller bör inkluderas i varje testomgång för att kontrollera att vävnadens viabilitet (fastställd med negativ kontroll) och känslighet (fastställd med positiv kontroll) håller sig inom de acceptansintervall som har definierats utifrån historiska data. Den parallella negativa kontrollen ger också ett referensvärde (100 % vävnadsviabilitet) för beräkning av relativ viabilitet i den vävnad som behandlats med testkemikalie (% Viabilitettest). Det ämne som rekommenderas som positivt kontrollämne med VRM:erna är rent metylacetat (CAS-nr 79-20-9, kommersiellt tillgänglig från till exempel Sigma-Aldrich, katalognr 45997, vätska). Rekommenderade ämnen som negativa kontrollämnen i VRM1 och VRM2 är ultrarent H2O respektive Ca2+/Mg2+-fri DPBS. Dessa är de kontrollämnen som användes vid valideringsstudierna av VRM:erna och för vilka det finns mest historiska data. Användningen av lämpliga alternativa positiva eller negativa kontrollämnen bör vara vetenskapligt och adekvat motiverade. Negativa och positiva kontroller bör testas enligt det/de protokoll som används för de testkemikalier vilka ingår i testomgången (dvs. för flytande och/eller fasta ämnen). Denna applicering bör följas av behandlingsexponeringen, sköljning, en nedsänkning efter exponering, och i aktuella fall en postexponerings-inkubation, som beskrivits för kontrollomgångar som görs parallellt med flytande testkemikalier (se punkt 32) eller för kontrollomgångar som görs parallellt med testkemikalier i fast form (se punkt 33), innan MTT-testet görs (se punkt 35) (34) (35). En uppsättning negativa och positiva kontroller är tillräcklig för alla testkemikalier i samma fysikaliska tillstånd (flytande eller fast form) som ingår i samma testomgång.
|
Mätningar av vävnadsviabilitet
|
35.
|
MTT-testet är en standardiserad kvantitativ metod (16) som bör användas för att mäta vävnadsviabiliteten i denna testmetod. Den kan användas på en tredimensionell vävnadsmodell. MTT-testet utförs omedelbart efter postexponerings-inkubationen. I VRM:erna placeras RhCE-vävnadsprovet i 0,3 ml av 1 mg/ml MTT-lösning under 180 ± 15 min. vid standardodlingsbetingelser. Färgämnet MTT reduceras av de viabla cellerna i RhCE-vävnaden till en blå MTT-formazanfällning. Den utfällda blå MTT-formazanprodukten extraheras sedan från vävnaden med lämplig volym isopropanol (eller liknande lösningsmedel) (34) (35). Extraktion av de vävnader som har testats med flytande testkemikalier bör göras från både ovan- och undersidan av vävnaden, medan extraktion av vävnader som testats med fasta kemikalier eller färgade vätskor bör göras endast från vävnadens undersida (för att minimera eventuell kontamination av isopropanol-extraktionslösning med eventuellt kvarvarande testkemikalie på vävnadens yta). För vävnader som testats med flytande testkemikalier som inte enkelt kan tvättas bort kan extraktionen också göras från enbart undersidan. De parallellt testade negativa och positiva kontrollämnena ska behandlas på samma sätt som den testade kemikalien. Det extraherade MTT-formazanet kan kvantifieras antingen genom standardabsorbansmätning (OD) vid 570 nm med hjälp av ett filterbandpass på högst ± 30 nm eller genom att använda ett HPLC-/UPLC-spektrofotometriskt förfarande (se punkt 42) (11) (36).
|
|
36.
|
Optiska egenskaper hos testkemikalien eller dess kemiska verkan på MTT kan störa mätningarna av MTT-formazan och ge felaktiga skattningar av vävnadsviabilitet. Testkemikalier kan störa mätningarna av MTT-formazan genom direkt reduktion av MTT till blått MTT-formazan, och/eller genom färginterferens om testkemikalien absorberar, naturligt eller till följd av behandlingsprocedurer, inom samma OD-intervall som MTT-formazan (dvs. runt 570 nm). Förkontroller bör göras inför testerna för att identifiera möjliga direkt MTT-reducerande ämnen och/eller färginterfererande kemikalier, och ytterligare kontroller bör användas för att detektera och korrigera möjliga störningar från sådana testkemikalier (se punkterna 37–41). Detta är särskilt viktigt när en viss testkemikalie inte har avlägsnats helt från RhCE-vävnaden genom sköljning, eller när den penetrerar det hornhinneliknande epitelet och därför förekommer i RhCE-vävnaden när MTT-testet utförs. För testkemikalier som absorberar ljus i samma intervall som MTT-formazan (naturligt eller efter behandling) och som inte är kompatibla med standardabsorbansmätningen (OD) av MTT-formazan på grund av för stor interferens, det vill säga stark absorption vid 570 ± 30 nm, kan ett HPLC-/UPLC-spektrofotometriskt förfarande för att mäta MTT-formazan användas (se punkterna 41 och 42) (11) (36). En detaljerad beskrivning av hur man detekterar och korrigerar direkt MTT-reduktion och interferenser av färgämnen finns i VRM:ernas standardförfaranden (34) (35). Illustrerande flödesscheman som ger vägledning i hur man i VRM1 respektive VRM2 identifierar och hanterar direkt MTT-reducerande ämnen och/eller färginterfererande kemikalier finns också i tillägg 3 och 4.
|
|
37.
|
För att identifiera möjlig interferens från testkemikalier som absorberar ljus i samma intervall som MTT-formazan (naturligt eller efter behandling) och besluta om behovet av ytterligare kontroller, blandas testkemikalien med vatten och/eller isopropanol och inkuberas en lämplig tid i rumstemperatur (se tillägg 2, [34] [35]). Om testkemikalien, blandad med vatten och/eller isopropanol, absorberar tillräckligt med ljus inom intervallet 570 ± 20 nm för VRM1 (se tillägg 3), eller om en färgad lösning erhålls när testkemikalien blandas med vatten för VRM2 (se tillägg 4), antas testkemikalien störa standardabsorbansmätningen (OD) för MTT-formazan och ytterligare färgkontroller bör göras, alternativt kan ett HPLC-/UPLC-spektrofotometriskt förfarande användas varvid dessa kontroller inte krävs (se punkter 41 och 42 och tillägg 3 och 4) (34) (35). När standardabsorbansmätningen (OD) utförs bör varje interfererande testkemikalie appliceras på minst två viabla vävnadsreplikat, vilka går igenom hela testförfarandet men under MTT-inkubationssteget inkuberas med medium istället för MTT-lösning för att ge en icke-specifik färgkontroll för levande vävnader (NSCliving) (34) (35). Den icke-specifika färgkontrollen NSCliving behöver utföras parallellt med testerna av den färgade testkemikalien, och i händelse av flera tester ska en oberoende NSCliving-kontroll utföras vid varje genomfört test (i varje testomgång) på grund av den inneboende biologiska variationen i levande vävnader. Faktisk vävnadsviabilitet beräknas som följer: den procentuella vävnadsviabilitet som erhålls med levande vävnad som har exponerats för den interfererande testkemikalien och som har inkuberats med MTT-lösning (% Viabilitettest) minus den procentuella icke-specifika färg som erhålls med levande vävnad, vilken har exponerats för den interfererande kemikalien och som har inkuberats med medium utan MTT, parallellt med testet som korrigeras (% NSCliving) det vill säga Faktisk vävnadsviabilitet = [% Viabilitettest] – [% NSCliving].
|
|
38.
|
För att identifiera ämnen som direkt reducerar MTT bör varje testkemikalie blandas med nyblandad MTT-lösning. En lämplig mängd testkemikalie tillsätts till en MTT-lösning och blandningen inkuberas i ungefär 3 timmar under standardodlingsbetingelser (se tillägg 3 och 4) (34) (35). Om MTT-blandningen som innehåller testkemikalien (eller suspensionen, för olösliga testkemikalier) blir blå/lila, antas testkemikalien direkt reducera MTT och vidare funktionskontroller av icke-viabla RhCE-vävnader bör utföras, oberoende av standardabsorbansmätningen (OD) eller ett HPLC-/UPLC-spektrofotometriskt förfarande. Denna ytterligare funktionella kontroll använder sig av avdödad vävnad som endast har metabolisk restaktivitet men absorberar och behåller testkemikalien på ett sätt som liknar levande vävnader. Avdödad vävnad bereds i VRM1 genom exponering för låg temperatur (”frysdödad”). Avdödad vävnad bereds i VRM2 genom förlängd inkubation (t.ex. minst 24 ± 1 timmar) i vatten följd av förvaring vid låg temperatur (”vattendödad”). Varje testkemikalie som reducerar MTT appliceras på minst två avdödade vävnadsreplikat, vilka genomgår hela testförfarandet för att ge en icke-specifik MTT-reduktionskontroll (NSMTT) (34) (35). En enda NSMTT-kontroll per testkemikalie räcker, oavsett antalet oberoende tester/testomgångar som genomförs. Faktisk vävnadsviabilitet beräknas som följer: den procentuella vävnadsviabilitet som erhålls med levande vävnad som exponerats för den MTT-reducerande kemikalien (% Viabilitettest) minus den procentuella icke-specifika MTT-reduktion som erhålls med den avdödade vävnaden, vilken exponerats för samma MTT-reducerande ämne, beräknad i förhållande till den negativa kontroll som har löpt parallellt med det test som korrigeras (% NSMTT) det vill säga Faktisk vävnadsviabilitet = [% Viabilitettest] – [% NSMTT].
|
|
39.
|
Testkemikalier som konstateras både ge färginterferens (se punkt 37) och direkt reducera MTT (se punkt 38) kommer dessutom att kräva en tredje uppsättning kontroller när man utför standardabsorbansmätningen (OD), utöver de NSMTT- och NSCliving-kontroller som beskrivits i föregående punkter. Detta är oftast fallet med mörkt färgade testkemikalier som absorberar ljus i intervallet 570 ± 30 nm (t.ex. blå, lila, svart) eftersom deras färg försvårar skattningen av deras förmåga att direkt reducera MTT, som beskrivits i punkt 38. Detta tvingar fram användningen av NSMTT-kontroller, som standard, tillsammans med NSCliving-kontroller. Testkemikalier för vilka både NSMTT- och NSCliving-kontroller utförs kan absorberas och stanna kvar i både levande och avdödad vävnad. Därför kan NSMTT-kontrollen korrigera, inte bara för testkemikaliens eventuella, direkta MTT-reduktion, utan också för färginterferens som uppstår vid absorption och retention av testkemikalien i avdödad vävnad. Detta skulle kunna leda till dubbel korrigering för färginterferens eftersom NSCliving-kontrollen redan korrigerar för färginterferens som uppstår vid absorption och retention av testkemikalien i levande vävnad. För att undvika möjlig dubbel korrigering för färginterferens behöver en tredje kontroll för icke-specifik färg i avdödad vävnad (NSCkilled) genomföras (se tillägg 3 och 4) (34) (35). I denna ytterligare kontroll behöver testkemikalien appliceras på minst två avdödade vävnadsreplikat, vilka genomgår hela testförfarandet men som inkuberas med medium istället för MTT-lösning under steget med MTT-inkubation. En enda NSCkilled-kontroll per testkemikalie räcker, oavsett antalet oberoende tester/testomgångar som genomförs, men bör genomföras parallellt med NSMTT-kontrollen och med samma sats av vävnader. Faktisk vävnadsviabilitet beräknas som följer: den procentuella vävnadsviabilitet som erhålls med levande vävnad som exponerats för testkemikalien (% Viabilitettest) minus % NSMTT minus % NSCliving
plus den procentuella icke-specifika färg som erhålls med den avdödade vävnaden vilken exponerats för den interfererande testkemikalien och inkuberats med medium utan MTT, beräknad i förhållande till den negativa kontroll som har löpt parallellt med det test som korrigeras (%NSCkilled), det vill säga Faktisk vävnadsviabilitet = [% Viabilitettest] – [% NSMTT] – [% NSCliving] + [% NSCkilled].
|
|
40.
|
Det är viktigt att notera att icke-specifik MTT-reduktion och icke-specifik färginterferens (i samband med standardabsorbansmätningar) kan öka vävnadsextraktets OD över spektrofotometerns linjära intervall och att icke-specifik MTT-reduktion (i samband med HPLC-/UPLC-spektrofotometriska mätningar) också kan öka vävnadsextraktets topparea för MTT-formazan över spektrofotometerns linjära intervall. På grund av detta är det viktigt att varje laboratorium fastställer det linjära intervallet för OD/topparea hos sin spektrofotometer med till exempel MTT-formazan (CAS-nr 57360-69-7), kommersiellt tillgänglig från till exempel Sigma-Aldrich (katalognr M2003), innan testning av testkemikalier påbörjas i tillsynssyfte.
|
|
41.
|
Standardabsorbansmätning (OD) med hjälp av en spektrofotometer är lämplig vid bedömning av direkta MTT-reducerare och färginterfererande testkemikalier när den aktuella störningen av MTT-formazanmätningen inte är för kraftig (dvs. OD-värdena för vävnadsextrakten som erhålls med testkemikalien, utan korrektion för direkt MTT-reduktion och/eller färginterferens, ligger inom spektrofotometerns linjaritetsintervall). Men resultat där en testkemikalie ger % NSMTT och/eller % NSCliving ≥ 60 % (VRM1, och VRM2-protokoll för flytande ämnen) eller 50 % (VRM2-protokoll för fasta ämnen) av den negativa kontrollen bör hanteras med försiktighet eftersom dessa är de etablerade brytvärden som används i VRM:erna för att skilja klassificerade från icke-klassificerade kemikalier (se punkt 44). Men standardabsorbans (OD) kan inte mätas när interferensen med MTT-formazanmätningen är för kraftig (dvs. det leder till att okorrigerade OD-värden för testvävnadsextrakten faller utanför spektrofotometerns linjära intervall). Färgade testkemikalier, eller testkemikalier som blir färgade vid kontakt med vatten eller isopropanol, som interfererar för starkt med standardabsorbansmätningen (OD) för MTT-formazan kan ändå utvärderas med HPLC-/UPLC-spektrofotometri (se tillägg 3 och 4). Anledningen till det är att HPLC-/UPLC-systemet gör det möjligt att skilja MTT-formazanet från testkemikalien innan den kvantifieras (36). Därför krävs inga NSCliving- eller NSCkilled-kontroller när man använder HPLC-/UPLC-spektrofotometri, oberoende av vilken kemikalie som testas. Trots detta bör NSMTT-kontroller användas, om testkemikalien misstänks direkt reducera MTT (enligt förfarande beskrivet i punkt 38). Dessutom bör NSMTT-kontroller också användas tillsammans med testkemikalier som har en färg (inneboende eller som uppkommer i vattenlösning) som hämmar bedömningen av deras kapacitet att direkt reducera MTT så som beskrivits i punkt 38. Vid användning av HPLC/UPLC-spektrofotometri för att mäta MTT-formazan beräknas vävnadens viabilitet som förhållandet (i procent) mellan å ena sidan den topparea för MTT-formazan som erhållits med levande vävnad exponerad för testkemikalien och, å andra sidan, den topparea för MTT-formazan som erhållits med den parallellt utförda negativa kontrollen. För testkemikalier som direkt kan reducera MTT beräknas faktisk vävnadsviabilitet som följer: % viabilitettest
minus % NSMTT, som beskrivs i den sista meningen under punkt 38. Slutligen bör man notera att direkta MTT-reducerare eller direkta MTT-reducerare som också är färginterfererande, vilka stannar kvar i vävnaderna efter behandling och reducerar MTT så kraftigt att de leder till OD-värden (vid standard OD-mätning) eller toppareor (vid UPLC-/HPLC-spektrofotometri) hos de testade vävnadsextrakten som faller utanför spektrofotometerns linjära intervall inte kan bedömas med RhCE-testmetoder, även om de förväntas förekomma väldigt sällan.
|
|
42.
|
HPLC-/UPLC-spektrofotometri kan användas för alla sorters testkemikalier (färgade, icke-färgade, MTT-reducerare och icke-MTT-reducerare) för MTT-formazanmätning (11) (36). På grund av mångfalden av HPLC/UPLC-spektrofotometersystem är det inte möjligt för varje användare att upprätta exakt samma systemförhållanden. Innan det spektrofotometriska HPLC-/UPLC-systemet används för att kvantifiera MTT-formazan från vävnadsextrakt bör dess kvalifikationer påvisas genom att det visas motsvara acceptanskriterierna för en uppsättning standardparametrar för kvalificering, baserade på de parametrar som beskrivs i branschvägledningen från amerikanska Food and Drug Administration för validering av bioanalytiska metoder (36) (38). Dessa nyckelparametrar och deras acceptanskriterier visas i tillägg 5. När de acceptanskriterier som definieras i tillägg 5 har uppfyllts anses det spektrofotometriska HPLC-/UPLC-systemet vara kvalificerat och färdigt att mäta MTT-formazan under de experimentella förhållanden som beskrivs i denna testmetod.
|
Acceptanskriterier
|
43.
|
För varje testomgång där man använder satser av RhCE-vävnad som uppfyllt kvalitetskraven (se punkt 30), ska de vävnadsprover som behandlas med negativt kontrollämne uppvisa OD som motsvarar den kvalitet vävnaderna hade efter frakt, mottagningsrutiner och alla protokollrelaterade procedurer och bör inte ligga utanför de historiskt fastställda gränserna som beskrivs i tabell 2 (se punkt 26). På samma sätt ska vävnader som behandlas med positivt kontrollämne, det vill säga metylacetat, uppvisa genomsnittlig vävnadsviabilitet < 50 % i förhållande till den negativa kontrollen i VRM1, med protokollet för antingen flytande eller fasta ämnen, och ≤ 30 % (protokollet för flytande ämnen) eller ≤ 20 % (protokollet för fasta ämnen) i förhållande till den negativa kontrollen i VRM2 så att de återspeglar vävnadernas förmåga att svara på en irriterande testkemikalie under testmetodens förhållanden (34) (35). Variationen mellan vävnadsreplikat med testkemikalier och kontrollämnen bör falla inom accepterade gränser (dvs. skillnaden i viabilitet mellan två vävnadsreplikat bör vara mindre än 20 %, eller standardavvikelsen [SD] mellan tre vävnadsreplikat bör inte överstiga 18 %). Om antingen den negativa eller positiva kontrollen som ingår i en testomgång faller utanför accepterat intervall anses omgången ”icke-kvalificerad” och bör upprepas. Om variationen mellan vävnadsreplikat av en testkemikalie faller utanför det accepterade intervallet anses testet ”icke-kvalificerat” och testkemikalien bör testas på nytt.
|
Tolkning av resultaten och prediktionsmodell
|
44.
|
De OD-värden/toppareor som erhålls med extrakten från vävnadsreplikaten för varje testkemikalie ska användas för att beräkna genomsnittlig procentuell vävnadsviabilitet (medelvärde mellan vävnadsreplikaten) normaliserad mot negativ kontroll, vilken sätts som 100 %. Brytvärdet för procentuell vävnadsviabilitet som identifierar testkemikalier som inte kräver klassificering för ögonirritation eller allvarlig ögonskada (GHS och CLP Ingen klassificering) ges i tabell 4. Resultat ska då tolkas som följer:
|
—
|
Testkemikalien fastställs till att inte kräva klassificering och märkning enligt GHS och CLP-förordningen (Ingen klassificering) om medelvärdet för procentuell vävnadsviabilitet efter exponering och postexponerings-inkubation är mer än (>) det fastställda brytvärdet för procentuell vävnadsviabilitet, enligt tabell 4. I detta fall krävs ingen ytterligare testning med andra testmetoder.
|
|
—
|
Om medelvärdet för procentuell vävnadsviabilitet efter exponering och postexponerings-inkubation är lika med eller mindre än (≤) det fastställda brytvärdet för procentuell vävnadsviabilitet, kan ingen prediktion göras, som visas i tabell 4. I det här fallet kommer ytterligare testning med andra testmetoder att krävas eftersom RhCE-testmetoder visar ett visst antal falskt positiva resultat (se punkterna 14–15) och inte kan särskilja mellan GHS- och CLP-kategorierna 1 och 2 i (se punkt 17).
|
Tabell 4
Prediktionsmodeller för GHS- och CLP-klassificering
|
VRM
|
Ingen klassificering
|
Ingen prediktion kan göras
|
|
VRM1 – EpiOcular™ EIT (för båda protokollen)
|
Genomsnittlig vävnadsviabilitet > 60 %
|
Genomsnittlig vävnadsviabilitet ≤ 60 %
|
|
VRM2 – SkinEthic™ HCE EIT (för protokollet för flytande ämnen)
|
Genomsnittlig vävnadsviabilitet > 60 %
|
Genomsnittlig vävnadsviabilitet ≤ 60 %
|
|
VRM2 – SkinEthic™ HCE EIT (för protokollet för fasta ämnen)
|
Genomsnittlig vävnadsviabilitet > 50 %
|
Genomsnittlig vävnadsviabilitet ≤ 50 %
|
|
|
45.
|
Ett enskilt test med minst två vävnadsreplikat bör vara tillräckligt för en testkemikalie, förutsatt att resultatet är otvetydigt. Om man får resultat som ligger på gränsen, t.ex. icke-konkordanta replikatmätningar och/eller genomsnittlig procentuell vävnadsviabilitet lika med 60 ± 5 % (VRM1, och VRM2 med protokoll för flytande ämnen) eller 50 ± 5 % (VRM2 med protokoll för fasta ämnen), bör man dock överväga ett andra test, och ett tredje om resultaten mellan de två första testerna är diskordanta.
|
|
46.
|
Olika brytvärden för procentuell vävnadsviabilitet vilka skiljer klassificerade från icke-klassificerade testkemikalier kan övervägas för särskilda sorters blandningar, när så är lämpligt och motiverat, för att öka testmetodens sammantagna prestanda för den sortens blandningar (se punkt 14). Referenskemikalier kan vara värdefulla vid utvärdering av potentialen för allvarlig ögonskada/ögonirritation hos okända testkemikalier eller okänd produktklass, eller för att utvärdera den relativa okulära toxicitetspotentialen hos en klassificerad kemikalie inom ett specifikt intervall av positiva responser.
|
DATA OCH RAPPORTERING
Data
|
47.
|
Data från enskilda vävnadsreplikat i en testomgång (t.ex. OD-värden/MTT-formazantoppareor och beräknade procentuella vävnadsviabilitetsdata för testkemikalien och kontrollerna, och den slutliga prediktionen från RhCE-testmetoden) bör rapporteras i tabellform för varje testkemikalie, inklusive data från upprepade tester, när det är lämpligt. Dessutom bör genomsnittlig procentuell vävnadsviabilitet och skillnad i viabilitet mellan två vävnadsreplikat (om n = 2 vävnadsreplikat) eller SD (om n > 3 vävnadsreplikat) för varje enskild testkemikalie och kontroll rapporteras. Eventuella uppmärksammade interferenser av en testkemikalie med mätningen av MTT-formazan genom direkt MTT-reduktion och/eller färginterferens bör rapporteras för varje testad kemikalie.
|
Testrapport
|
48.
|
Testrapporten ska innehålla följande information:
Testkemikalie
|
|
Monokomponentämne
|
—
|
Kemisk beteckning, såsom IUPAC- eller CAS-namn, CAS-nummer, Smiles- eller InChI-kod, strukturformel och/eller andra beteckningar.
|
|
—
|
Fysikaliskt tillstånd, volatilitet, pH, LogP, molekylvikt, kemisk klass och andra relevanta fysikalisk-kemiska egenskaper med relevans för studiens genomförande, i den utsträckning de finns tillgängliga.
|
|
—
|
Renhet, kemisk identitet för föroreningar i förekommande fall och när så är praktiskt möjligt.
|
|
—
|
Behandling före testning, om detta är tillämpligt (t.ex. uppvärmning, finfördelning).
|
|
—
|
Uppgifter om lagringsförhållanden och stabilitet i den mån sådana uppgifter finns tillgängliga.
|
|
|
|
Multikomponentämne, UVCB och blandning
|
—
|
karakteriseras så långt som möjligt genom t.ex. beståndsdelarnas kemiska identitet (se ovan), renhet, kvantitativa förekomst och relevanta fysikalisk-kemiska egenskaper (se ovan), i den mån sådana uppgifter finns tillgängliga.
|
|
—
|
Fysikaliskt tillstånd och andra relevanta fysikalisk-kemiska egenskaper med relevans för studiens genomförande, i den utsträckning de finns tillgängliga.
|
|
—
|
Renhet, kemisk identitet för föroreningar i förekommande fall och när så är praktiskt möjligt.
|
|
—
|
Behandling före testning, om detta är tillämpligt (t.ex. uppvärmning, finfördelning).
|
|
—
|
Uppgifter om lagringsförhållanden och stabilitet i den mån sådana uppgifter finns tillgängliga.
|
|
Positiva och negativa kontrollämnen
|
—
|
Kemisk beteckning, såsom IUPAC- eller CAS-namn, CAS-nummer, Smiles- eller InChI-kod, strukturformel och/eller andra beteckningar.
|
|
—
|
Fysikaliskt tillstånd, volatilitet, molekylvikt, kemisk klass och andra relevanta fysikalisk-kemiska egenskaper med relevans för studiens genomförande, i den utsträckning de finns tillgängliga.
|
|
—
|
Renhet, kemisk identitet för föroreningar i förekommande fall och när så är praktiskt möjligt.
|
|
—
|
Behandling före testning, om detta är tillämpligt (t.ex. uppvärmning, finfördelning).
|
|
—
|
Uppgifter om lagringsförhållanden och stabilitet i den mån sådana uppgifter finns tillgängliga.
|
|
—
|
Motivering till varför annan negativ kontroll än ultrarent H2O eller Ca2+/Mg2+-fri DPBS använts, om tillämpligt.
|
|
—
|
Motivering till varför annan positiv kontroll än ren metylacetat använts, om tillämpligt.
|
|
—
|
Hänvisning till historiska positiva och negativa kontrollresultat som visar lämpliga acceptanskriterier för testomgångarna.
|
Uppgifter om sponsor och testanläggning
|
—
|
Namn och adress för sponsor, testanläggning och försöksledare.
|
|
—
|
Uppgift om vilken RhCE-vävnadsmodell och vilket protokoll som använts (med motivering till valen, om tillämpligt)
|
Testmetodens betingelser
|
—
|
Använd RhCE-vävnadsmodell, inklusive satsnummer.
|
|
—
|
Våglängd och filterbandpass (om tillämpligt) som använts för kvantifiering av MTT-formazan, och linjärt intervall hos mätanordningen (t.ex. spektrofotometer).
|
|
—
|
Beskrivning av den metod som använts för att kvantifiera MTT-formazan.
|
|
—
|
Beskrivning av det spektrofotometriska HPLC-/UPLC-systemet, om tillämpligt.
|
|
—
|
Fullständig stödjande information för den specifika RhCE-vävnadsmodell som använts, inklusive dess prestanda. Denna ska omfatta men inte vara begränsad till följande:
|
i)
|
Kvalitetskontroll av viabilitet (leverantör).
|
|
ii)
|
Viabilitet under testmetodens betingelser (användare).
|
|
iii)
|
Kvalitetskontroll av barriärfunktion.
|
|
iv)
|
Morfologi, om tillgänglig.
|
|
v)
|
Reproducerbarhet och prediktiv kapacitet.
|
|
vi)
|
Andra kvalitetskontroller (QC) av RhCE-vävnadsmodellen, om tillgängliga.
|
|
|
—
|
Hänvisning till historiska data om RhCE-vävnadsmodellen. Denna ska omfatta men inte vara begränsad till följande: Uppgifter om huruvida kvalitetskontrolldata är godtagbara med avseende på historiska data om respektive sats.
|
|
—
|
Uppgift om att testanläggningen har uppvisat kompetens i användningen av testmetoden inför rutinmässig användning genom ett test av kvalifikationskemikalier.
|
Acceptanskriterier för tester och testomgångar
|
—
|
Medelvärden och acceptansintervall för positiva och negativa kontroller baserade på historiska data.
|
|
—
|
Acceptabel variation mellan vävnadsreplikat för positiva och negativa kontroller.
|
|
—
|
Acceptabel variation mellan vävnadsreplikat för testkemikalien.
|
Testförfarande
|
—
|
Detaljerade uppgifter om testförfarandet.
|
|
—
|
Doser av använda testkemikalier och kontrollämnen.
|
|
—
|
Exponeringens varaktighet och temperatur, varaktighet på nedsänkning efter exponering och postexponerings-inkubation (om tillämpligt).
|
|
—
|
Beskrivning av eventuella modifieringar av testförfarandet.
|
|
—
|
Uppgifter om kontroller som använts för direkt MTT-reduktion och/eller färgade testkemikalier, om tillämpligt.
|
|
—
|
Antal vävnadsreplikat som används per testkemikalie och kontroller (positiv kontroll, negativ kontroll, NSMTT, NSCliving och NSCkilled, om tillämpligt).
|
Resultat
|
—
|
Tabell med data från enskilda testkemikalier och kontrollämnen för varje testomgång (inklusive upprepade experiment om tillämpligt) och mätningar för varje replikat, inklusive OD-värde eller MTT-formazantopparea, procentuell vävnadsviabilitet, genomsnittlig procentuell vävnadsviabilitet, skillnad mellan vävnadsreplikat eller SD och slutlig prediktion.
|
|
—
|
Om tillämpligt, resultat från kontroller som använts för direkt MTT-reduktion och/eller färgade testkemikalier, inklusive OD-värde eller topparea för MTT-formazan, % NSMTT, % NSCliving, % NSCkilled, skillnad mellan vävnadsreplikat eller SD, slutlig korrekt procentuell vävnadsviabilitet och slutlig prediktion.
|
|
—
|
Resultat som erhållits med testkemikalien (testkemikalierna) och kontrollämnena i förhållande till definierade acceptanskriterier för testomgången och testet som helhet.
|
|
—
|
Beskrivning av övriga observerade effekter, till exempel färgning av vävnaderna av en färgad testkemikalie.
|
Diskussion kring resultaten
Slutsats
|
LITTERATUR
|
(1)
|
FN (2015). FN:s globalt harmoniserade system för klassificering och märkning av kemikalier (GHS). ST/SG/AC.10/30/Rev.6, sjätte reviderade upplagan, New York och Genève: FN. Finns på http://www.unece.org/fileadmin/DAM/trans/danger/publi/ghs/ghs_rev06/English/ST-SG-AC10-30-Rev6e.pdf.
|
|
(2)
|
Kapitel B.5 i denna bilaga, Akut ögonirritation/ögonkorrosion.
|
|
(3)
|
Kapitel B.47 i denna bilaga, Testmetoden BCOP (Bovine Corneal Opacity and Permeability) för identifiering av i) kemikalier som orsakar allvarlig ögonskada och ii) kemikalier för vilka det inte krävs klassificering för ögonirritation eller allvarlig ögonskada.
|
|
(4)
|
Kapitel B.48 i denna bilaga, Testmetoden ICE (Isolated Chicken Eye test) för identifiering av i) kemikalier som orsakar allvarlig ögonskada och ii) kemikalier för vilka det inte krävs klassificering för ögonirritation eller allvarlig ögonskada.
|
|
(5)
|
Kapitel B.61 i denna bilaga, Testmetoden fluoresceinläckage för identifiering av ämnen som är frätande och kraftigt irriterande för ögonen.
|
|
(6)
|
Kapitel B.68 i denna bilaga, In vitro-testmetoden korttidsexponering (Short Time Exposure Test) för identifiering av i) kemikalier som orsakar allvarlig ögonskada och ii) kemikalier för vilka det inte krävs klassificering för ögonirritation eller allvarlig ögonskada.
|
|
(7)
|
Freeman, S.J., Alépée N., Barroso, J., Cole, T., Compagnoni, A., Rubingh, C., Eskes, C., Lammers, J., McNamee, P., Pfannenbecker, U., Zuang, V. (2010). Prospective Validation Study of Reconstructed Human Tissue Models for Eye Irritation Testing. ALTEX 27, Special issue 2010,261-266.
|
|
(8)
|
EC EURL ECVAM (2014). The EURL ECVAM - Cosmetics Europe prospective validation study of Reconstructed human Cornea-like Epithelium (RhCE)-based test methods for identifying chemicals not requiring classification and labelling for serious eye damage/eye irritation: Validation Study Report. EUR 28125 EN; doi:10.2787/41680. Finns på http://publications.jrc.ec.europa.eu/repository/handle/JRC100280.
|
|
(9)
|
EURL ECVAM Science Advisory Committee (2014). ESAC Opinion on the EURL ECVAM Eye Irritation Validation Study (EIVS) on EpiOcular™ EIT and SkinEthic™ HCE and a related Cosmetics Europe study on HPLC/UPLC-spectrophotometry as an alternative endpoint detection system for MTT-formazan. ESAC Opinion No 2014-03 of 17 November 2014; EUR 28173 EN; doi: 10.2787/043697. Finns på http://publications.jrc.ec.europa.eu/repository/handle/JRC103702.
|
|
(10)
|
Alépée, N., Leblanc, V., Adriaens, E., Grandidier, M.H., Lelièvre, D, Meloni, M., Nardelli, L., Roper, C.S, Santirocco, E., Toner, F., Van Rompay, A., Vinall, J., Cotovio, J. (2016). Multi-laboratory validation of SkinEthic HCE test method for testing serious eye damage/eye irritation using liquid chemicals. Toxicol. In Vitro 31, 43-53.
|
|
(11)
|
Alépée, N., Adriaens, E., Grandidier, M.H., Meloni, M., Nardelli, L., Vinall, C.J., Toner, F., Roper, C.S, Van Rompay, A.R., Leblanc, V., Cotovio, J. (2016). Multi-laboratory evaluation of SkinEthic HCE test method for testing serious eye damage/eye irritation using solid chemicals and overall performance of the test method with regard to solid and liquid chemicals testing. Toxicol. In Vitro 34, 55-70.
|
|
(12)
|
EURL ECVAM Science Advisory Committee (2016). ESAC Opinion on the SkinEthic™ Human Corneal Epithelium (HCE) Eye Irritation Test (EIT). ESAC Opinion No 2016-02 of 24 June 2016; EUR 28175 EN; doi: 10.2787/390390. Finns på http://publications.jrc.ec.europa.eu/repository/handle/JRC103704.
|
|
(13)
|
EC EURL ECVAM (2016). Recommendation on the Use of the Reconstructed human Cornea-like Epithelium (RhCE) Test Methods for Identifying Chemicals not Requiring Classification and Labelling for Serious Eye Damage/Eye Irritation According to UN GHS. (Manuscript in preparation).
|
|
(14)
|
Draize, J.H., Woodard, G., Calvery, H.O. (1944). Methods for the Study of Irritation and Toxicity of Substances Applied Topically to the Skin and Mucous Membranes. Journal of Pharmacol. and Exp. Therapeutics 82, 377–390.
|
|
(15)
|
Scott, L., Eskes, C., Hoffmann, S., Adriaens, E., Alépée, N., Bufo, M., Clothier, R., Facchini, D., Faller, C., Guest, R., Harbell, J., Hartung, T., Kamp, H., Le Varlet, B., Meloni, M., McNamee, P., Osborne, R., Pape, W., Pfannenbecker, U., Prinsen, M., Seaman, C., Spielman, H., Stokes, W., Trouba, K., Van den Berghe, C., Van Goethem, F., Vassallo, M., Vinardell, P., Zuang, V. (2010). A Proposed Eye Irritation Testing Strategy to Reduce and Replace In Vivo Studies Using Bottom-Up and Top-Down Approaches. Toxicol. In Vitro 24, 1-9.
|
|
(16)
|
Mosmann, T. (1983). Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 65, 55-63.
|
|
(17)
|
OECD (2016). Series on Testing and Assessment No 216: Performance Standards for the Assessment of Proposed Similar or Modified In Vitro Reconstructed Human Cornea-Like Epithelium (RhCE) Test Methods for Identifying Chemicals not Requiring Classification and Labelling for Eye Irritation or Serious Eye Damage, Based on the Validated Reference Methods EpiOcular™ EIT and SkinEthic™ HCE EIT described in TG 492. Organisationen för ekonomiskt samarbete och utveckling (OECD), Paris.
|
|
(18)
|
OECD (2005). Series on Testing and Assessment No 34: Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Organisationen för ekonomiskt samarbete och utveckling (OECD), Paris.
|
|
(19)
|
Kaluzhny, Y., Kandárová, H., Hayden, P., Kubilus, J., d’Argembeau-Thornton, L., Klausner, M. (2011). Development of the EpiOcular™ Eye Irritation Test for Hazard Identification and Labelling of Eye Irritating Chemicals in Response to the Requirements of the EU Cosmetics Directive and REACH Legislation. Altern. Lab. Anim. 39, 339-364.
|
|
(20)
|
Nguyen, D.H., Beuerman, R.W., De Wever, B., Rosdy, M. (2003). Three-dimensional construct of the human corneal epithelium for in vitro toxicology. I: Salem, H., Katz, S.A. (red.), Alternative toxicological methods, CRC Press, s. 147-159.
|
|
(21)
|
Pfannenbecker, U., Bessou-Touya, S., Faller, C., Harbell, J., Jacob, T., Raabe, H., Tailhardat, M., Alépée, N., De Smedt, A., De Wever, B., Jones, P., Kaluzhny, Y., Le Varlet, B., McNamee, P., Marrec-Fairley, M., Van Goethem, F. (2013). Cosmetics Europe multi-laboratory pre-validation of the EpiOcular™ reconstituted Human Tissue Test Method for the Prediction of Eye Irritation. Toxicol. In Vitro 27, 619-626.
|
|
(22)
|
Alépée, N., Bessou-Touya, S., Cotovio, J., de Smedt, A., de Wever, B., Faller, C., Jones, P., Le Varlet, B., Marrec-Fairley, M., Pfannenbecker, U., Tailhardat, M., van Goethem, F., McNamee, P. (2013). Cosmetics Europe Multi-Laboratory Pre-Validation of the SkinEthic™ Reconstituted Human Corneal Epithelium Test Method for the Prediction of Eye Irritation. Toxicol. In Vitro 27, 1476-1488.
|
|
(23)
|
Kolle, S.N., Moreno, M.C.R., Mayer, W., van Cott, A., van Ravenzwaay, B., Landsiedel, R. (2015). The EpiOcular™ Eye Irritation Test is the Method of Choice for In Vitro Eye Irritation Testing of Agrochemical Formulations: Correlation Analysis of EpiOcular™ Eye Irritation Test and BCOP Test Data to UN GHS, US EPA and Brazil ANIVSA Classifications. Altern. Lab. Anim. 43, 1-18.
|
|
(24)
|
Adriaens, E., Barroso, J., Eskes, C., Hoffmann, S., McNamee, P., Alépée, N., Bessou-Touya, S., De Smedt, A., De Wever, B., Pfannenbecker, U., Tailhardat, M., Zuang, V. (2014). Retrospective Analysis of the Draize Test for Serious Eye Damage/Eye Irritation: Importance of Understanding the In Vivo Endpoints Under UN GHS/EU CLP for the Development and Evaluation of In Vitro Test Methods. Arch. Toxicol. 88, 701-723.
|
|
(25)
|
Barroso, J., Pfannenbecker, U., Adriaens, E., Alépée, N., Cluzel, M., De Smedt, A., Hibatallah, J., Klaric, M., Mewes, K.R., Millet, M., Templier, M., McNamee, P. (2017). Cosmetics Europe compilation of historical serious eye damage/eye irritation in vivo data analysed by drivers of classification to support the selection of chemicals for development and evaluation of alternative methods/strategies: the Draize eye test Reference Database (DRD). Arch. Toxicol. 91, 521-547.
|
|
(26)
|
Meloni, M., De Servi, B., Marasco, D., Del Prete, S. (2011). Molecular mechanism of ocular surface damage: Application to an in vitro dry eye model on human corneal epithelium. Molecular Vision 17, 113–126.
|
|
(27)
|
Hackett, R.B., McDonald, T.O. (1991). Eye irritation. I: Advances in Modern Toxicology: Dermatoxicology Marzulli F.N. och Maibach H.I. (red.), fjärde upplagan, s. 749–815. Washington, DC, Förenta staterna: Hemisphere Publishing Corporation.
|
|
(28)
|
Fox, D.A., Boyes, W.K. (2008). Toxic Responses of the Ocular and Visual System. I: Cassaret and Doull’s Toxicology: The Basic Science of Poisons Klaassen C.D. (red.), sjunde upplagan s. 665-697. Withby, ON, Kanada: McGraw-Hill Ryerson.
|
|
(29)
|
Jester, J.V., Li, H.F., Petroll, W.M., Parker, R.D., Cavanagh, H.D., Carr, G.J., Smith, B., Maurer, J.K. (1998). Area and Depth of Surfactant Induced Corneal Injury Correlates with Cell Death. Invest. Ophthalmol. Vis. Sci. 39, 922-936.
|
|
(30)
|
Maurer, J.K., Parker, R.D., Jester, J.V. (2002). Extent of Corneal Injury as the Mechanistic Basis for Ocular Irritation: Key Findings and Recommendations for the Development of Alternative Assays. Reg. Tox. Pharmacol. 36, 106-117.
|
|
(31)
|
Jester, J.V., Li, L., Molai, A., Maurer, J.K. (2001). Extent of Corneal Injury as a Mechanistic Basis for Alternative Eye Irritation Tests. Toxicol. In Vitro 15, 115-130.
|
|
(32)
|
Jester, J.V., Petroll, W.M., Bean, J., Parker, R.D., Carr, G.J., Cavanagh, H.D., Maurer, J.K. (1998). Area and Depth of Surfactant-Induced Corneal Injury Predicts Extent of Subsequent Ocular Responses. Invest. Ophthalmol. Vis. Sci. 39, 2610-2625.
|
|
(33)
|
Jester, J.V. (2006). Extent of Corneal Injury as a Biomarker for Hazard Assessment and the Development of Alternative Models to the Draize Rabbit Eye Test. Cutan. Ocul. Toxicol. 25, 41-54.
|
|
(34)
|
EpiOcular™ EIT SOP, Version 8 (March 05, 2013). EpiOcular™ EIT for the Prediction of Acute Ocular Irritation of Chemicals. Finns på [https://ecvam-dbalm.jrc.ec.europa.eu/beta/index.cfm/methodsAndProtocols/index].
|
|
(35)
|
SkinEthic™ HCE EIT SOP, Version 1. (July 20, 2015). SKINETHIC™ HCE Eye Irritation Test (EITL för flytande ämnen, EITS för fasta ämnen) for the Prediction of Acute Ocular Irritation of Chemicals. Finns på https://ecvam-dbalm.jrc.ec.europa.eu/beta/index.cfm/methodsAndProtocols/index.
|
|
(36)
|
Alépée, N., Barroso, J., De Smedt, A., De Wever, B., Hibatallah, J., Klaric, M., Mewes, K.R., Millet, M., Pfannenbecker, U., Tailhardat, M., Templier, M., McNamee, P. (2015). Use of HPLC/UPLC-Spectrophotometry for Detection of Formazan in In Vitro Reconstructed Human Tissue (RhT)-Based Test Methods Employing the MTT-Reduction Assay to Expand their Applicability to Strongly Coloured Test Chemicals. Toxicol. In Vitro 29, 741-761.
|
|
(37)
|
Kaluzhny, Y., Kandárová, H., Handa, Y., DeLuca, J., Truong, T., Hunter, A., Kearney, P., d’Argembeau-Thornton, L., Klausner, M. (2015). EpiOcular™ Eye Irritation Test (EIT) for Hazard Identification and Labeling of Eye Irritating Chemicals: Protocol Optimization for Solid Materials and Extended Shipment Times. Altern. Lab Anim. 43, 101-127.
|
|
(38)
|
US FDA (2001). Guidance for Industry: Bioanalytical Method Validation. U.S. Department of Health and Human Services, Food and Drug Administration. Maj 2001. Finns på http://www.fda.gov/downloads/Drugs/Guidances/ucm070107.pdf.
|
|
(39)
|
OECD (2017). Guidance Document on an Integrated Approaches on Testing and Assessment for Serious Eye Damage and Eye irritation. Series on Testing and Assessment No 263. ENV Publications, Organisationen för ekonomiskt samarbete och utveckling (OECD), Paris.
|
Tillägg 1
DEFINITIONER
Noggrannhet
: Ett mått på hur väl testmetodresultaten och de godtagna referensvärdena överensstämmer med varandra. Det är en relevansaspekt och ett mått på testmetodens prestanda. Detta begrepp används ofta omväxlande med ”konkordans” för att beskriva andelen korrekta resultat med en testmetod (18).
Referenskemikalie
: En kemikalie som används som en standard för jämförelse med en testkemikalie. En referenskemikalie ska ha följande egenskaper: i) konsekvent och pålitligt medel för dess identifiering och karakterisering, ii) strukturell, funktionell och/eller kemisk likhet eller produktklass-likhet med den/de kemikalie(r) som testas, iii) kända fysikalisk-kemiska egenskaper, iv) stödjande data om kända effekter, och v) känd styrka inom det önskade responsområdet.
Nedifrån och upp-strategi
: Stegvis metod som tillämpas på en testkemikalie som inte tros kräva klassificering och märkning för ögonirritation eller allvarlig ögonskada; det första steget är att skilja kemikalier som inte behöver klassificeras och märkas (negativt utfall) från andra kemikalier (positivt utfall).
Kemikalie
: Ett ämne eller en blandning.
Konkordans
: se ”Noggrannhet”.
Hornhinna (kornea)
: Den transparenta delen av ögonglobens framsida, som täcker iris och pupillen och släpper in ljus i ögat.
CV
: Variationskoefficient (Coefficient of Variation).
Dev
: Avvikelse (deviation).
EIT
: Ögonirritationstest (Eye Irritation Test).
EURL ECVAM
: EU:s referenslaboratorium för alternativ till djurförsök (European Union reference laboratory for alternatives to animal testing).
Ögonirritation
: Förändringar som uppstår i ögat efter applicering av en testkemikalie på ögats främre yta och som är fullt reversibla inom 21 dagar från appliceringen. Utbytbart mot ”Reversibla effekter på ögat” och ”GHS-/CLP-kategori 2”.
ET50
: Den exponeringstid som krävs för att minska vävnadsviabiliteten med 50 % vid applicering av en referenskemikalie med en fastställd, fast koncentration.
Falskt negativt värde
: Den andel av alla positiva ämnen som av en testmetod felaktigt identifieras som negativa. Det är en indikator på testmetodens prestanda.
Falskt positivt värde
: Den andel av alla negativa ämnen som av en testmetod felaktigt identifieras som positiva. Det är en indikator på testmetodens prestanda.
Fara
: Inneboende egenskap hos ett ämne eller en situation som har potential att orsaka skadliga effekter när en organism, ett system eller en (del-) population exponeras för ämnet.
HCE
: SkinEthic™ Human Corneal Epithelium, humant hornhinneepitel.
HPLC
: Högupplösande vätskekromatografi.
IC50
: Koncentration vid vilken en referenskemikalie minskar vävnadens viabilitet med 50 % efter en fast exponeringstid (t.ex. 30 minuters behandling med SDS).
Infinit dos
: En mängd testkemikalie som appliceras på RhCE-vävnaden och som överstiger den mängd som krävs för att helt täcka epitelets yta med ett jämnt lager.
Irreversibla effekter på ögat
: Se ”Allvarlig ögonskada”.
LLOQ
: Nedre kvantifieringsgräns (Lower Limit of Quantification).
LogP
: Logaritmen för fördelningskoefficienten för oktanol-vatten.
Blandning
: En blandning eller en lösning som består av två eller flera ämnen.
Monokomponentämne
: Ett ämne, definierat av sin kvantitativa sammansättning, där en huvudsaklig beståndsdel utgör minst 80 viktprocent.
Multikomponentämne
: Ett ämne, definierat av sin kvantitativa sammansättning, där fler än en huvudsaklig beståndsdel förekommer i en koncentration på ≥ 10 viktprocent och < 80 viktprocent. Ett multikomponentämne är resultatet av en tillverkningsprocess. Skillnaden mellan blandningar och multikomponentämnen är att en blandning erhålls genom att två eller flera ämnen blandas utan någon kemisk reaktion. Ett multikomponentämne är resultatet av en kemisk reaktion.
MTT
: 3-(4,5-dimetyltiazol-2-yl)-2,5-difenyltetrazoliumbromid, tiazolylblåtetrazoliumbromid.
Negativ kontroll
: Ett prov som innehåller ett testsystems alla komponenter och som behandlas med ett ämne känt för att inte inducera positiv respons från testsystemet. Detta prov hanteras tillsammans med testkemikaliebehandlade prover och andra kontrollprover och används för att fastställa 100 % vävnadsviabilitet.
Icke-klassificerad
: Kemikalier som inte är klassificerade som ögonirriterande (GHS/CLP-förordningen kategori 2, GHS kategori 2A eller 2B) eller som orsakande allvarlig ögonskada (GHS/CLP-kategori 1). Utbytbart mot ”GHS-/CLP-kategori Ingen klassificering”.
NSCkilled
: Icke-specifik färg i avdödade vävnader.
NSCliving
: Icke-specifik färg i levande vävnader.
NSMTT
: Icke-specifik MTT-reduktion (Non-Specific MTT).
OD
: Optisk densitet
Prestandastandarder
: Standarder, baserade på en validerad testmetod som ansetts vetenskapligt giltig, vilka utgör grunden för utvärdering av jämförbarheten hos en föreslagen testmetod som är mekanistiskt och funktionellt likartad. Följande ingår: i) testmetodens grundläggande komponenter, ii) en minimiförteckning med referenskemikalier som valts ut från de kemikalier som använts för att påvisa att den validerade testmetoden fungerar tillfredsställande och iii) de jämförbara nivåer av noggrannhet och tillförlitlighet, på grundval av vad som erhållits för den validerade testmetoden, som man bör kunna påvisa för den föreslagna testmetoden när den utvärderas med användning av minimiförteckningen med referenskemikalier (18).
Positiv kontroll
: Ett prov som innehåller ett testsystems alla komponenter och som behandlas med ett ämne känt för att inducera positiv respons från testsystemet. Detta prov hanteras tillsammans med testkemikaliebehandlade prover och andra kontrollprover. För att säkerställa att variationer i den positiva kontrollens respons över tid kan bedömas, bör den positiva responsen inte vara alltför kraftfull.
Relevans
: Beskrivning av förhållandet mellan testet och den berörda effekten och huruvida testet är meningsfullt och användbart för ett visst ändamål. Relevansen är ett mått på hur väl man genom testet korrekt kan mäta och prediktera den biologiska effekt det gäller. Relevans omfattar också överväganden om en testmetods noggrannhet (konkordans) (18).
Tillförlitlighet
: Mått på i vilken utsträckning en testmetod kan reproduceras inom och mellan laboratorier över tid, när den utförs med användning av samma protokoll. Tillförlitligheten bedöms genom att man beräknar reproducerbarheten inom och mellan laboratorier och repeterbarheten inom laboratorier (18).
Ersättningstest
: Ett test utformat för att ersätta ett test som används rutinmässigt och som är godkänt för faroidentifiering och/eller riskbedömning och som har konstaterats ge motsvarande eller bättre skydd av människors eller djurs hälsa eller miljön, enligt det som är tillämpligt, jämfört med det godkända testet, för alla tänkbara testningssituationer och kemikalier (18).
Reproducerbarhet
: Överensstämmelse mellan resultat som erhållits från upprepade tester av samma testkemikalie med användning av samma testprotokoll (se ”Tillförlitlighet”) (18).
Reversibla effekter på ögat
: Se ”Ögonirritation”.
RhCE
: Rekonstruerat humant hornhinnelikt epitel (Reconstructed human Cornea-like Epithelium).
Testomgång
: En testomgång utgörs av att en eller flera testkemikalier testas parallellt med en negativ kontroll och en positiv kontroll.
SD
: Standardavvikelse (Standard Deviation).
Känslighet
: Den andel av alla positiva/aktiva testkemikalier som klassificeras korrekt i testet. Det är ett mått på noggrannheten hos en kategoriserande testmetod och är en viktig faktor vid bedömningen av en testmetods relevans (18).
Allvarlig ögonskada
: Vävnadsskada i ögat eller kraftig synnedsättning som uppstår efter applicering av ett testämne på ögats främre yta och som inte är fullt reversibel inom 21 dagar från appliceringen. Utbytbart mot ”Irreversibla effekter på ögat” och ”GHS/CLP-kategori 1”.
Standardförfaranden
: Formella, skriftliga förfaranden som i detalj beskriver hur särskilda rutiner och testspecifika laboratoriearbeten ska utföras (Standard Operating Procedure, SOP). De är obligatoriska enligt GLP (Good Laboratory Practice).
Specificitet
: Den andel av alla negativa/inaktiva testkemikalier som klassificeras korrekt i testet. Det är ett mått på noggrannheten hos en kategoriserande testmetod och är en viktig faktor vid bedömningen av en testmetods relevans (18).
Ämne
: Ett kemiskt grundämne och föreningar av detta grundämne i naturlig eller tillverkad form, inklusive eventuella tillsatser som är nödvändiga för att bevara ämnets stabilitet och sådana föroreningar som härrör från tillverkningsprocessen, men exklusive eventuella lösningsmedel som kan avskiljas utan att det påverkar ämnets stabilitet eller ändrar dess sammansättning.
Test
: Parallella tester av en enskild testkemikalie på minst två vävnadsreplikat enligt angivelser i relevant standardförfarande.
Vävnadsviabilitet
: Parameter som mäter en cellpopulations totala aktivitet i en rekonstruerad vävnad som dess förmåga att reducera MTT, vilket, beroende på testets utformning och vilken endpoint som mäts, korrelerar till det totala antalet celler och/eller till cellernas viabilitet.
Uppifrån och ned-strategi
: Stegvis metod som tillämpas på kemikalier som misstänks orsaka allvarlig ögonskada; det första steget är att skilja kemikalier som inducerar allvarlig ögonskada (positivt utfall) från andra kemikalier (negativt utfall).
Testkemikalie
: Alla ämnen eller blandningar som testas med denna testmetod.
Stegvis teststrategi
: En sekventiell teststrategi som använder sig av olika testmetoder efter varandra. All befintlig information om en testkemikalie granskas vid varje steg, med användning av förfarandet med sammanvägd bedömning (weight of evidence) för att avgöra om det finns tillräckligt med information till hands för att besluta om faroklassificering innan man går vidare till nästa steg i strategin. Om testkemikaliens potential/styrka att framkalla skada kan fastställas utifrån befintlig information vid ett givet steg behövs ingen ytterligare testning (18).
ULOQ
: Övre kvantifieringsgräns (Upper Limit of Quantification).
FN:s globalt harmoniserade system för klassificering och märkning av kemikalier (GHS)
: Ett system för att klassificera kemikalier (ämnen och blandningar) enligt standardiserade typer och nivåer av fysiska risker, hälsorisker och miljörisker och informera om resultaten genom till exempel piktogram, signalord, faroangivelser, skyddsangivelser och säkerhetsdatablad i syfte att informera om ämnenas skadliga effekter för att skydda personer (inklusive arbetsgivare, arbetstagare, transportörer, konsumenter och personal inom räddningstjänsten) och miljön (1).
GHS-/CLP-kategori 1
: Se ”Allvarlig ögonskada”.
GHS-/CLP-kategori 2
: Se ”Ögonirritation”.
GHS-/CLP-kategori Ingen klassificering
: Kemikalier som inte uppfyller kraven för att klassificeras enligt GHS-/CLP-kategori 1 eller 2 (eller GHS-kategori 2A eller 2B). Utbytbart mot Icke-klassificerad.
UPLC
: Ultra-högupplösande vätskekromatografi.
UVCB-ämne
: Ämne med okänd eller varierande sammansättning, komplexa reaktionsprodukter och biologiskt material.
Giltig testmetod
: En testmetod som anses vara tillräckligt relevant och tillförlitlig för ett särskilt ändamål och är grundad på vetenskapligt sunda principer. En testmetod är aldrig giltig i absolut bemärkelse, utan endast i förhållande till ett fastställt ändamål (18).
Validerad testmetod
: En testmetod för vilken valideringsstudier har genomförts för att fastställa relevans (inklusive noggrannhet) och tillförlitlighet för ett specifikt ändamål. Det är viktigt att notera att en validerad testmetod kanske inte är tillräckligt noggrann och tillförlitlig när det gäller ett visst föreslaget ändamål (18).
VRM
: Validerad referensmetod.
VRM1
: EpiOcular™ EIT anges som Validerad referensmetod 1.
VRM2
: SkinEthic™ HCE EIT anges som Validerad referensmetod 2.
Sammanvägd bedömning
: Ett förfarande där man bedömer styrkor och svagheter hos flera uppgifter för att nå fram till och motivera en slutsats huruvida ett testämne kan vara skadligt.
Tillägg 2
HUVUDSAKLIGA DELAR I DE RHCE-TESTER SOM HAR VALIDERATS FÖR IDENTIFIERING AV KEMIKALIER SOM INTE KRÄVER KLASSIFICERING ELLER MÄRKNING FÖR ÖGONIRRITATION ELLER ALLVARLIG ÖGONSKADA
|
Testdelar
|
EpiOcular™ EIT
(VRM1)
|
SkinEthic™ HCE EIT
(VRM2)
|
|
Protokoll
|
Vätskor
(pipetterbara vid 37 ± 1°C eller lägre temperaturer under 15 min.)
|
Ämnen i fast form
(ej pipetterbara)
|
Vätskor och viskösa ämnen
(pipetterbara)
|
Ämnen i fast form
(ej pipetterbara)
|
|
Modellyta
|
0,6 cm2
|
0,6 cm2
|
0,5 cm2
|
0,5 cm2
|
|
Antal vävnadsreplikat
|
Minst två
|
Minst två
|
Minst två
|
Minst två
|
|
Förkontroll av eventuell färginterferens
|
50 μl + 1 ml H2O i 60 min. vid 37 ± 2 °C, 5 ± 1 % CO2, ≥ 95 % relativ luftfuktighet (ofärgade testkemikalier), eller 50 μl + 2 ml isopropanol blandade i 2–3 timmar vid rumstemperatur (färgade testkemikalier)
|
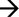 om testkemikaliens OD vid 570 ± 20 nm, efter subtraktion av OD för isopropanol eller vatten är > 0,08 (vilket motsvarar ungefär 5 % av den negativa kontrollens genomsnittliga OD-värde), bör anpassade kontroller med levande vävnad genomföras. om testkemikaliens OD vid 570 ± 20 nm, efter subtraktion av OD för isopropanol eller vatten är > 0,08 (vilket motsvarar ungefär 5 % av den negativa kontrollens genomsnittliga OD-värde), bör anpassade kontroller med levande vävnad genomföras.
|
|
50 mg + 1 ml H2O i 60 min. vid 37 ± 2 °C, 5 ± 1 % CO2, ≥ 95 % relativ luftfuktighet
(ofärgade testkemikalier), och/eller
50 mg + 2 ml isopropanol blandade i 2–3 timmar vid rumstemperatur (färgade och ofärgade testkemikalier)
|
 om testkemikaliens OD vid 570 ± 20 nm, efter subtraktion av OD för isopropanol eller vatten, är > 0,08 (vilket motsvarar ungefär 5 % av den negativa kontrollens genomsnittliga OD-värde), bör anpassade kontrolltester med levande vävnad genomföras. om testkemikaliens OD vid 570 ± 20 nm, efter subtraktion av OD för isopropanol eller vatten, är > 0,08 (vilket motsvarar ungefär 5 % av den negativa kontrollens genomsnittliga OD-värde), bör anpassade kontrolltester med levande vävnad genomföras.
|
|
10 μl + 90 μl H2O blandade i 30 ± 2 min. vid rumstemperatur (RT, 18–28 °C)
|
 om testkemikalien är färgad, bör anpassade kontroller med levande vävnad genomföras om testkemikalien är färgad, bör anpassade kontroller med levande vävnad genomföras
|
|
10 mg + 90 μl H2O blandade i 30 ± 2 min. vid rumstemperatur
|
 om testkemikalien är färgad, bör anpassade kontroller med levande vävnad genomföras om testkemikalien är färgad, bör anpassade kontroller med levande vävnad genomföras
|
|
|
Förkontroll av eventuell direkt MTT-reduktion
|
50 μl + 1 ml av 1 mg/ml MTT-lösning under 180 ± 15 min. vid 37 ± 2 °C, 5 ± 1 % CO2, ≥ 95 % relativ luftfuktighet
|
 om lösningen blir blå/lila, bör anpassade kontroller med frysdödad vävnad utföras om lösningen blir blå/lila, bör anpassade kontroller med frysdödad vävnad utföras
(50 μl sterilt avjoniserat vatten i MTT-lösning används då som negativ kontroll)
|
|
50 mg + 1 ml av 1 mg/ml MTT-lösning under 180 ± 15 min. vid 37 ± 2 °C, 5 ± 1 % CO2, ≥ 95 % relativ luftfuktighet
|
 om lösningen blir blå/lila, bör anpassade kontrolltester med frysdödad vävnad utföras om lösningen blir blå/lila, bör anpassade kontrolltester med frysdödad vävnad utföras
(50 μl sterilt avjoniserat vatten i MTT-lösning används då som negativ kontroll)
|
|
30 μl + 300 μl av 1 mg/ml MTT-lösning i 180 ± 15 min. vid 37 ± 2 °C, 5 ± 1 % CO2, ≥ 95 % relativ luftfuktighet
|
 om lösningen blir blå/lila, bör anpassade kontroller med vattendödad vävnad utföras om lösningen blir blå/lila, bör anpassade kontroller med vattendödad vävnad utföras
(30 μl sterilt avjoniserat vatten i MTT-lösning används då som negativ kontroll)
|
|
30 mg + 300 μl av 1 mg/ml MTT-lösning i 180 ± 15 min. vid 37 ± 2 °C, 5 ± 1 % CO2, ≥ 95 % relativ luftfuktighet
|
 om lösningen blir blå/lila, bör anpassade kontroller med vattendödad vävnad utföras om lösningen blir blå/lila, bör anpassade kontroller med vattendödad vävnad utföras
(30 μl sterilt avjoniserat vatten i MTT-lösning används då som negativ kontroll)
|
|
|
Förbehandling
|
20 μl Ca2+/Mg2+-fri DPBS
i 30 ± 2 min. vid 37 ± 2 oC, 5 ± 1 % CO2, ≥ 95 % relativ luftfuktighet, skyddad från ljus.
|
20 μl Ca2+/Mg2+-fri DPBS
i 30 ± 2 min. vid 37 ± 2 °C, 5 ± 1 % CO2, ≥ 95 % relativ luftfuktighet, skyddad från ljus.
|
—
|
—
|
|
Behandlingsdoser och applicering
|
50 μl (83,3 μl/cm2)
|
50 mg (83,3 mg/cm2)
|
10 μl Ca2+/Mg2+-fri DPBS + 30 ± 2 μl (60 μl/cm2)
För viskösa ämnen, använd ett nylonnät
|
30 μl Ca2+/Mg2+-fri DPBS + 30 ± 2 mg (60 mg/cm2)
|
|
Exponering, tid och temperatur
|
30 min. (± 2 min.)
i odlingsmedium
vid 37 ± 2 °C, 5 ± 1 % CO2, ≥ 95 % relativ luftfuktighet
|
6 timmar (± 0,25 timmar)
i odlingsmedium
vid 37 ± 2 °C, 5 ± 1 % CO2, ≥ 95 % relativ luftfuktighet
|
30 min. (± 2 min.)
i odlingsmedium
vid 37 ± 2 °C, 5 ± 1 % CO2, ≥ 95 % relativ luftfuktighet
|
4 timmar (± 0,1 timmar)
i odlingsmedium
vid 37 ± 2 °C, 5 ± 1 % CO2, ≥ 95 % relativ luftfuktighet
|
|
Sköljning vid rumstemperatur
|
3 gånger i 100 ml Ca2+/Mg2+-fri DPBS
|
3 gånger i 100 ml Ca2+/Mg2+-fri DPBS
|
20 ml Ca2+/Mg2+-fri DPBS
|
25 ml Ca2+/Mg2+-fri DPBS
|
|
Nedsänkning efter exponering
|
12 min. (± 2 min.) vid rumstemperatur i odlingsmedium
|
25 min. (± 2 min.) vid rumstemperatur i odlingsmedium
|
30 min. (± 2 min.) vid 37 °C, 5 % CO2, 95 % relativ luftfuktighet, i odlingsmedium
|
30 min. (± 2 min.) vid rumstemperatur i odlingsmedium
|
|
Inkubation efter exponering
|
120 min. (± 15 min.) i odlingsmedium vid 37 ± 2 °C, 5 ± 1 % CO2, ≥ 95 % relativ luftfuktighet
|
18 timmar (± 0,25 timmar) i odlingsmedium vid 37 ± 2 °C, 5 ± 1 % CO2, ≥ 95 % relativ luftfuktighet
|
Ingen
|
18 timmar (± 0,5 h) i odlingsmedium vid 37 ± 2 °C, 5 ± 1 % CO2, ≥ 95 % relativ luftfuktighet
|
|
Negativ kontroll
|
50 μl H2O
Testas parallellt
|
50 μl H2O
Testas parallellt
|
30 ± 2 μl Ca2+/Mg2+-fri DPBS
Testas parallellt
|
30 ± 2 μl Ca2+/Mg2+-fri DPBS
Testas parallellt
|
|
Positiv kontroll
|
50 μl metylacetat
Testas parallellt
|
50 μl metylacetat
Testas parallellt
|
30 ± 2μl metylacetat
Testas parallellt
|
30 ± 2μl metylacetat
Testas parallellt
|
|
MTT-lösning
|
300 μl 1 mg/ml
|
300 μl 1 mg/ml
|
300 μl 1 mg/ml
|
300 μl 1 mg/ml
|
|
MTT-inkubation, tid och temperatur
|
180 min. (± 15 min.) vid 37 ± 2 °C, 5 ± 1 % CO2, ≥ 95 % relativ luftfuktighet
|
180 min. (± 15 min.) vid 37 ± 2 °C, 5 ± 1 % CO2, ≥ 95 % relativ luftfuktighet
|
180 min. (± 15 min.) vid 37 ± 2 °C, 5 ± 1 % CO2, ≥ 95 % relativ luftfuktighet
|
180 min. (± 15 min.) vid 37 ± 2 °C, 5 ± 1 % CO2, ≥ 95 % relativ luftfuktighet
|
|
Extraktionsmedel
|
2 ml isopropanol
(extraktion från iläggets topp och botten genom att sticka hål på vävnaden)
|
2 ml isopropanol
(extraktion av iläggets botten genom att sticka hål på vävnaden)
|
1,5 ml isopropanol
(extraktion från iläggets topp och botten)
|
1,5 ml isopropanol
(extraktion av iläggets botten)
|
|
Extraktion, tid och temperatur
|
2–3 timmar med skak (~120 rpm) vid rumstemperatur eller över natt vid 4–10 °C
|
2–3 timmar med skak (~120 rpm) vid rumstemperatur eller över natt vid 4–10 °C
|
4 timmar med skak (~120 rpm) vid rumstemperatur eller åtminstone över natt utan skak vid 4–10 °C
|
Minst 2 timmar med skak (~120 rpm) vid rumstemperatur
|
|
Avläsning av optisk densitet
|
570 nm (550–590 nm)
utan referensfilter
|
570 nm (550–590 nm)
utan referensfilter
|
570 nm (540–600 nm)
utan referensfilter
|
570 nm (540–600 nm)
utan referensfilter
|
|
Kvalitetskontroll av vävnaden
|
Behandling med 100 μl 0,3-volymprocentig Triton X-100
12,2 min. ≤ ET50 ≤ 37,5 min.
|
Behandling med 100 μl 0,3-volymprocentig Triton X-100
12,2 min. ≤ ET50 ≤ 37,5 min.
|
30 min. behandling med SDS (50 μl)
1,0 mg/ml ≤ IC50 ≤ 3,5 mg/ml
|
30 min. behandling med SDS (50 μl)
1,0 mg/ml ≤ IC50 ≤ 3,2 mg/ml
|
|
Acceptanskriterier
|
|
1.
|
Genomsnittligt OD-värde för de vävnadsreplikat som behandlats med negativ kontroll ska vara > 0,8 och < 2,5.
|
|
2.
|
Genomsnittlig viabilitet hos de vävnadsreplikat som exponerats för positiv kontroll under 30 min, uttryckt i procent av negativ kontroll, ska vara < 50 %.
|
|
3.
|
Skillnaden i viabilitet mellan två vävnadsreplikat ska vara mindre än 20 %.
|
|
|
1.
|
Genomsnittligt OD-värde för de vävnadsreplikat som behandlats med negativ kontroll ska vara > 0,8 och < 2,5.
|
|
2.
|
Genomsnittlig viabilitet hos de vävnadsreplikat som exponerats för positiv kontroll under 6 timmar, uttryckt i procent av negativ kontroll, ska vara < 50 %.
|
|
3.
|
Skillnaden i viabilitet mellan två vävnadsreplikat ska vara mindre än 20 %.
|
|
|
1.
|
Genomsnittligt OD-värde för de vävnadsreplikat som behandlats med negativ kontroll ska vara > 1,0 och ≤ 2,5.
|
|
2.
|
Genomsnittlig viabilitet hos de vävnadsreplikat som exponerats för positiv kontroll under 30 min, uttryckt i procent av negativ kontroll, ska vara ≤ 30 %.
|
|
3.
|
Skillnaden i viabilitet mellan två vävnadsreplikat ska vara mindre än 20 %.
|
|
|
1.
|
Genomsnittligt OD-värde för de vävnadsreplikat som behandlats med negativ kontroll ska vara > 1,0 och ≤ 2,5.
|
|
2.
|
Genomsnittlig viabilitet hos de vävnadsreplikat som exponerats för positiv kontroll under 4 timmar, uttryckt i procent av negativ kontroll, ska vara ≤ 20 %.
|
|
3.
|
Skillnaden i viabilitet mellan två vävnadsreplikat ska vara mindre än 20 %.
|
|
Tillägg 3
ILLUSTRERANDE FLÖDESSCHEMA SOM GER VÄGLEDNING I HUR MAN IDENTIFIERAR OCH HANTERAR DIREKT MTT-REDUCERANDE ÄMNEN OCH/ELLER FÄRGINTERFERERANDE KEMIKALIER, BASERAT PÅ STANDARDFÖRFARANDET FÖR VRM1
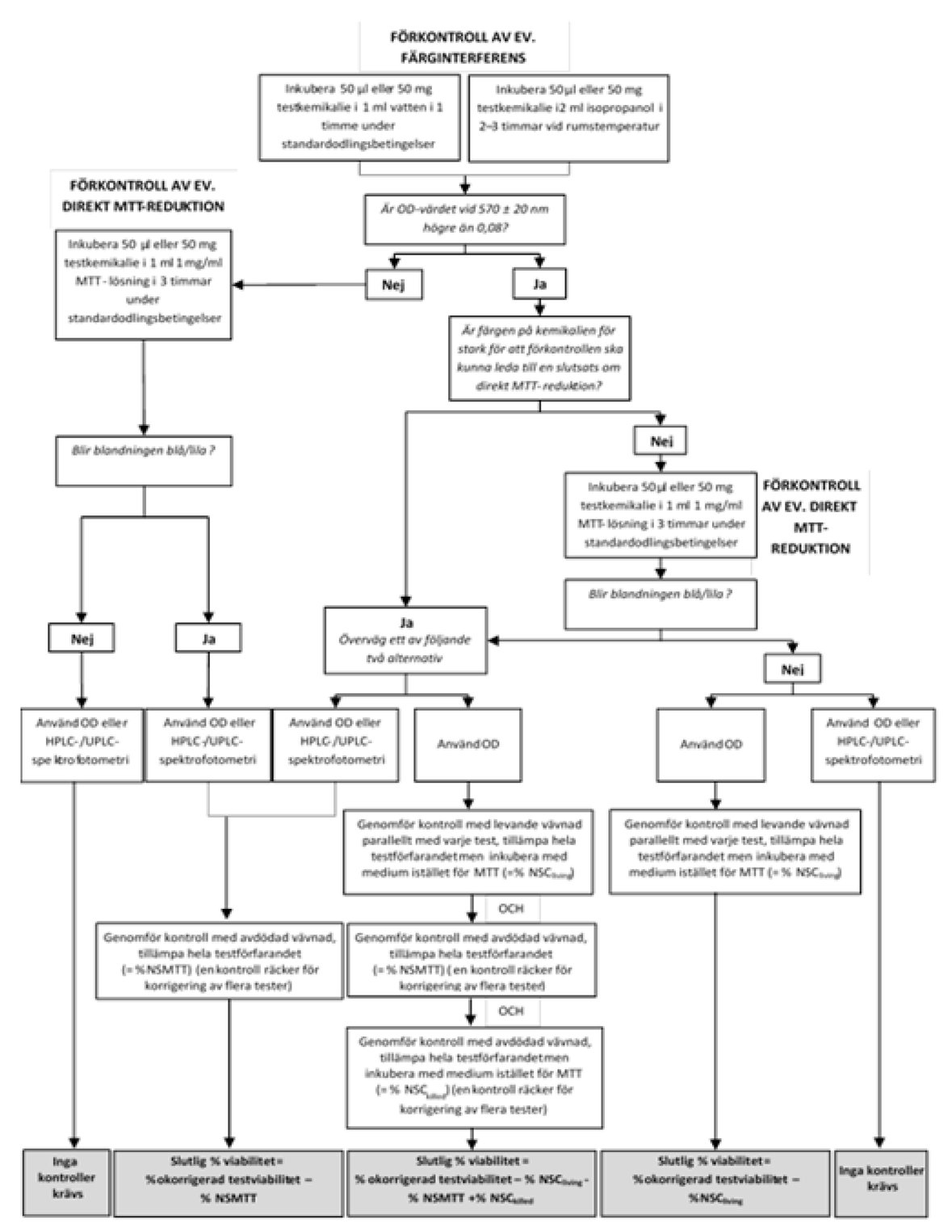
Tillägg 4
ILLUSTRERANDE FLÖDESSCHEMA SOM GER VÄGLEDNING I HUR MAN IDENTIFIERAR OCH HANTERAR DIREKT MTT-REDUCERANDE ÄMNEN OCH/ELLER FÄRGINTERFERERANDE KEMIKALIER, BASERAT PÅ STANDARDFÖRFARANDET FÖR VRM2

Tillägg 5
NYCKELPARAMETRAR OCH ACCEPTANSKRITERIER FÖR KVALIFICERING AV ETT SPEKTROFOTOMETRISKT HPLC-/UPLC-SYSTEM FÖR MÄTNING AV MTT-FORMAZAN EXTRAHERAD FRÅN RHCE-VÄVNAD
|
Parameter
|
Protokoll baserat på FDA-vägledning (36) (38)
|
Acceptanskriterier
|
|
Selektivitet
|
Analys av isopropanol, levande blankprov (isopropanolextrakt från levande RhCE-vävnad utan någon behandling), dött blankprov (isopropanolextrakt från avdödad RhCE-vävnad utan någon behandling), och av ett färgämne (t.ex. metylenblått)
|
Areainterference ≤ 20 % av areaLLOQ
(87)
|
|
Precision
|
Kvalitetskontroller (dvs. MTT-formazan vid 1,6 μg/ml, 16 μg/ml och 160 μg/ml) i isopropanol (n = 5)
|
CV ≤ 15 % eller ≤ 20 % för LLOQ
|
|
Noggrannhet
|
Kvalitetskontroller i isopropanol (n = 5)
|
% Dev ≤ 15 % eller ≤ 20 % för LLOQ
|
|
Matriseffekt
|
Kvalitetskontroller i levande blankprov (n = 5)
|
85 % ≤ % matriseffekt ≤ 115 %
|
|
Överföring
|
Analys av isopropanol mot en ULOQ (88)-standard
|
Areainterference ≤ 20 % av areaLLOQ
|
|
Reproducerbarhet (inom en dag)
|
Tre oberoende kalibreringskurvor (baserade på 6 konsekutiva 1/3-spädningar av MTT-formazan i isopropanol med början vid ULOQ, dvs. 200 μg/ml);
Kvalitetskontroller i isopropanol (n = 5)
|
Kalibreringskurvor: % Dev ≤ 15 % eller ≤ 20 % för LLOQ
Kvalitetskontroller: % Dev ≤ 15 % och CV ≤ 15 %
|
|
Reproducerbarhet (mellan dagar)
|
Dag 1: En kalibreringskurva och kvalitetskontroller i isopropanol (n = 3)
Dag 2: En kalibreringskurva och kvalitetskontroller i isopropanol (n = 3)
Dag 3: En kalibreringskurva och kvalitetskontroller i isopropanol (n = 3)
|
|
Kortidsstabilitet för MTT-formazan i RhCE-vävnadsextrakt
|
Kvalitetskontroller i levande blankprov (n = 3), analyserade på beredningsdagen och efter 24 timmars förvaring i rumstemperatur
|
% Dev ≤ 15 %
|
|
Långtidsstabilitet för MTT-formazan i RhCE-vävnadsextrakt, om det krävs
|
Kvalitetskontroller i levande blankprov (n = 3), analyserade på beredningsdagen och efter några dygns förvaring i –20 °C
|
% Dev ≤ 15 %
|
B.70
IN VITRO-TESTER MED HUMAN REKOMBINANT ÖSTROGENRECEPTOR (hrER) FÖR ATT DETEKTERA KEMIKALIER MED BINDNINGSAFFINITET TILL ÖSTROGENRECEPTOR
ALLMÄN INLEDNING
OECD:s prestandabaserade testriktlinje
|
1.
|
Denna testmetod motsvarar OECD:s testriktlinje (TG) 493 (2015). Testriktlinje 493 är en prestandabaserad testriktlinje (PBTG) som beskriver metodologin för in vitro-tester med humant rekombinant material för att detektera ämnen med bindningsaffinitet till östrogenreceptor (hrER-bindningstester). Den består av två mekanistiskt och funktionellt likartade tester för identifiering av ämnen som binder till östrogenreceptor α (dvs. ERα) och ska underlätta utvecklingen av nya, liknande eller modifierade tester som överensstämmer med de principer för validering som fastställts i OECD:s vägledningsdokument Guidance document on the validation and international acceptance of new or updated test methods for hazard assessment (1). De fullständigt validerade referenstestmetoder (tilläggen 2 och 3) som ligger till grund för denna prestandabaserade riktlinje är
|
—
|
Freyberger-Wilsons (FW) in vitro-test av östrogenreceptorbindning (östrogenreceptor, ER) med human, rekombinant fullängds-ERα (2), och
|
|
—
|
Chemical Evaluation and Research Institutes (CERI) in vitro-test av östrogenreceptorbindning med humant, rekombinant, ligandbindande domänprotein (2).
|
Prestandastandarder (PS) (3) finns tillgängliga för att underlätta utvecklingen och valideringen av liknande testmetoder för samma faro-endpoint och möjliggöra tillägg till PBTG 493 vid lämplig tidpunkt så att nya liknande tester kan fogas till en uppdaterad PBTG. Men liknande tester kommer att läggas till först efter OECD:s granskning och godkännande av att prestandastandarderna uppfylls. De tester som ingår i TG 493 kan användas utan förbehåll för att bemöta OECD-medlemsländers krav på testresultat för östrogenreceptorbindning genom att dra nytta av OECD:s ömsesidiga acceptans av data.
|
Bakgrund och principer för de tester som ingår i denna testmetod
|
2.
|
OECD inledde en högprioriterad verksamhet under 1998 för att se över befintliga, och ta fram nya, testriktlinjer för screening och testning av potentiellt hormonstörande kemikalier. OECD:s begreppsram för provning och bedömning av potentiellt hormonstörande kemikalier reviderades 2012. De ursprungliga och reviderade ramarna ingår som bilagor i Guidance document on standardised test guidelines for evaluating chemicals for endocrine disruption (4). Ramen har fem nivåer, vilka motsvarar olika nivåer av biologisk komplexitet. De ER-bindningstester som beskrivs i denna testmetod ligger på nivå 2, vilken omfattar in vitro-tester som ger data om utvalda hormonella mekanism(er)/signalväg(ar) Denna testmetod är avsedd för in vitro-tester av receptorbindning som är utformade för att identifiera ligander till den humana östrogenreceptorn alfa (ERα).
|
|
3.
|
Relevansen hos in vitro-ER-bindningstestet för biologisk funktion har påvisats tydligt. ER-bindningstester är utformade för att identifiera kemikalier som har potential att störa östrogenhormonsignalvägen och har använts i stor omfattning under de två senaste decennierna för att karakterisera ER-distributionen i olika vävnader samt för att identifiera ER-agonister/antagonister. Dessa tester återspeglar ligand–receptorinteraktionen som är det inledande steget i östrogensignalvägen och är avgörande för reproduktiva funktioner hos alla ryggradsdjur.
|
|
4.
|
Interaktionen mellan östrogener och östrogenreceptorer kan påverka transkriptionen av östrogenkontrollerade gener och inducera icke-genomiska effekter, vilka kan leda till induktion eller inhibering av cellulära processer, inklusive de som behövs för celldelning, normal fosterutveckling och reproduktion (5) (6) (7). En störning av normala östrogensystem kan leda till skadliga effekter på normal utveckling (ontogenes), reproduktionshälsa och reproduktionssystemets tillstånd. Olämplig ER-signalering kan leda till sådana effekter som ökad risk för hormonberoende cancer, nedsatt fertilitet samt förändringar i fostertillväxt och fosterutveckling (8).
|
|
5.
|
In vitro-bindningstester baseras på en direkt interaktion mellan ett ämne och en specifik, gentranskriptionsreglerande ligandbindningsplats på receptorn. Den viktigaste delen av östrogenreceptor alfa-bindningstestet med humant rekombinant östrogen (hrERα) mäter förmågan hos radioaktivt inmärkt ligand ([3H]-17β-östradiol) att binda till ER i närvaron av ökande koncentrationer av testkemikalie (dvs. konkurrerande ligand). Testkemikalier med hög affinitet för ER konkurrerar med den radioaktivt inmärkta liganden vid en lägre koncentration än de kemikalier som har lägre affinitet för receptorn. Detta test består av två huvuddelar: ett mättnadsbindningsexperiment för att karakterisera receptor–ligandinteraktionsparametrar och fastställa ER-specificitet, följt av ett kompetitivt bindningsexperiment som karakteriserar konkurrensen mellan en testkemikalie och en radioaktivt inmärkt ligand vid inbindning till ER.
|
|
6.
|
Valideringsstudier av CERI- och FW-bindningstesterna har visat att de är relevanta och tillförlitliga för det avsedda syftet (2).
|
|
7.
|
Definitioner och förkortningar som används i denna testmetod beskrivs i tillägg 1.
|
Omfattning och begränsningar för receptorbindningstester
|
8.
|
Dessa tester föreslås för screening- och prioriteringssyften men kan också ge information för en molekylär inledande händelse (Molecular Initiation Event, MIE) som kan användas vid en strategi med sammanvägd bedömning. Med hjälp av dem kan kemisk bindning till den ERα-ligandbindande domänen undersökas i ett in vitro-system. Därför ska resultat inte direkt extrapoleras till det intakta hormonella systemets komplexa signalering och reglering in vivo.
|
|
9.
|
Inbindningen av den naturliga liganden, 17β-östradiol, är det inledande steget i en serie molekylära händelser som aktiverar transkription av målgener och i slutänden kulminerar i fysiologiska förändringar (9). Därför betraktas bindning till den ERα-ligandbindande domänen som en av nyckelmekanismerna i ER-medierad hormonell störning (Endocrine Disruption, ED), även om det finns andra mekanismer genom vilka hormonell störning kan uppstå, inklusive i) interaktioner med andra platser på ERα än den ligandbindande fickan, ii) interaktioner med andra receptorer som är relevanta för östrogensignalering, ER-β och G-proteinkopplad östrogenreceptor, andra receptorer och enzymatiska system inom det hormonella systemet, iii) hormonsyntes, iv) metabol aktivering och/eller deaktivering av hormoner, v) transport av hormoner till målvävnader och vi) utsöndring av hormoner från kroppen. Inget av testerna i denna testmetod fokuserar på någon av dessa verkningsmekanismer.
|
|
10.
|
Denna testmetod är inriktad på olika ämnens förmåga att binda till human ERα och skiljer inte mellan agonister respektive antagonister till ERα. Dessa tester är inte inriktade på vare sig vidare nedströms händelser, som gentranskription, eller fysiologiska förändringar. Med hänsyn till att endast monokomponentämnen användes i valideringen har inte möjligheten att testa blandningar inte behandlats. Teoretiskt är testerna emellertid tillämpliga på testning av multikomponentämnen och blandningar. Innan testmetoden används på en blandning i syfte att samla in data för ett tillsynsändamål bör man överväga om, och i så fall varför, den kommer att ge godtagbara resultat för det syftet. Sådana överväganden är inte nödvändiga om blandningen måste provas enligt lag.
|
|
11.
|
Cellfria receptorsystem har ingen inneboende metabol förmåga och har inte validerats tillsammans med metabola enzymsystem. Men det kan vara möjligt att utforma en studie som inkorporerar metabol aktivitet, det fordrar dock ytterligare valideringsarbete.
|
|
12.
|
Kemikalier som kan denaturera proteinet (dvs. receptorproteinet), som ytaktiva ämnen eller kemikalier som kan ändra testbuffertens pH, kan eventuellt inte testas eller kan endast testas vid koncentrationer där sådan interaktion inte äger rum. I övrigt begränsas koncentrationsintervallet inom vilket testkemikalien kan testas av kemikaliens löslighet i testbufferten.
|
|
13.
|
Notera att i tabell 1 anges testresultaten för de 24 ämnen som testades i de båda fullständigt validerade testerna som beskrivs i denna testmetod. Av dessa ämnen har 17 klassificerats som ER-bindande och 6 som icke-bindande utifrån publicerade rapporter, inklusive in vitro-tester för ER-transkriptionsaktivering och/eller det uterotrofa testet (9) (10) (11) (12) (13) (14) (15). För de data som sammanfattas i tabell 1 var det nästan 100 % överensstämmelse mellan de två testerna kring klassificeringen av alla ämnen upp till 10–4 M, och varje ämne identifierades korrekt som antingen ER-bindande eller icke ER-bindande. Ytterligare information om denna grupp av ämnen, liksom andra ämnen som testats i ER-bindningstesterna i valideringsstudierna ges i prestandastandarderna för hrER-bindningstestet (3) tillägg 2 (tabell 1, 2 och 3).
Tabell 1
Indelning av ämnen som ER-bindande eller icke ER-bindande, efter att de testats i FW- och CERI-hrER-bindningstesterna med jämförelse av förväntad respons
|
|
Ämnesnamn
|
CAS-nummer
|
Förväntad respons
|
FW-test
|
CERI-test
|
MESH, kemikalie-klass
|
Produktklass
|
|
|
Koncentrations- intervall (M)
|
Klassificering
|
Koncentrations- intervall (M)
|
Klassificering
|
|
1
|
17β-östradiol
|
50-28-2
|
Bindare
|
1 × 10–11–1 × 10–6
|
Bindare
|
1 × 10–11–1 × 10–6
|
Bindare
|
Steroid
|
Humant/veterinärt läkemedel
|
|
2
|
Noretynodrel
|
68-23-5
|
Bindare
|
3 × 10–9–30 × 10–4
|
Bindare
|
3 × 10–9–30 × 10–4
|
Bindare
|
Steroid
|
Humant/veterinärt läkemedel
|
|
3
|
Noretindron
|
68-22-4
|
Bindare
|
3 × 10–9–30 × 10–4
|
Bindare
|
3 × 10–9–30 × 10–4
|
Bindare
|
Steroid
|
Humant/veterinärt läkemedel
|
|
4
|
Di-n-butylftalat
|
84-74-2
|
Icke-bindare (*5)
|
1 × 10–10–1 × 10–4
|
Icke-bindare (*6)
(1)
|
1 × 10–10–1 × 10–4
|
Icke-bindare (*6)
(1)
|
Kolväte (cykliskt), ester
|
Mjukgörare, kemisk intermediär
|
|
5
|
DES (dietylstilbestrol)
|
56-53-1
|
Bindare
|
1 × 10–10–1 × 10–3
|
Bindare
|
1 × 10–10–1 × 10–3
|
Bindare
|
Kolväte (cykliskt), fenol
|
Humant/veterinärt läkemedel
|
|
6
|
17α-etynylöstradiol
|
57-63-6
|
Bindare
|
1 × 10–10–1 × 10–3
|
Bindare
|
1 × 10–10–1 × 10–3
|
Bindare
|
Steroid
|
Humant/veterinärt läkemedel
|
|
7
|
Meso-hexestrol
|
84-16-2
|
Bindare
|
1 × 10–10–1 × 10–3
|
Bindare
|
1 × 10–10–1 × 10–3
|
Bindare
|
Kolväte (cykliskt), fenol
|
Humant/veterinärt läkemedel
|
|
8
|
Genistein
|
446-72-0
|
Bindare
|
1 × 10–10–1 × 10–3
|
Bindare
|
1 × 10–10–1 × 10–3
|
Bindare
|
Kolväte (heterocykliskt), flavonoid
|
Naturprodukt
|
|
9
|
Equol
|
531-95-3
|
Bindare
|
1 × 10–10–1 × 10–3
|
Bindare
|
1 × 10–10–1 × 10–3
|
Bindare
|
Fytoöstrogenmetabolit
|
Naturprodukt
|
|
10
|
Butylparaben (n-butyl-4-hydroxibensoat)
|
94-26-8
|
Bindare
|
1 × 10–10–1 × 10–3
|
Bindare
|
1 × 10–10–1 × 10–3
|
Bindare
|
Paraben
|
Konserveringsmedel
|
|
11
|
Nonylfenol (blandning)
|
84852-15-3
|
Bindare
|
1 × 10–10–1 × 10–3
|
Bindare
|
1 × 10–10–1 × 10–3
|
Bindare
|
Alkylfenol
|
Intermediär
|
|
12
|
o,p′-DDT
|
789-02-6
|
Bindare
|
1 × 10–10–1 × 10–3
|
Bindare
|
1 × 10–10–1 × 10–3
|
Bindare
|
Organisk klorförening
|
Insekticid
|
|
13
|
Kortikosteron
|
50-22-6
|
Icke-bindare (*5)
|
1 × 10–10–1 × 10–4
|
Icke-bindare
|
1 × 10–10–1 × 10–4
|
Icke-bindare
|
Steroid
|
Naturprodukt
|
|
14
|
Zearalenon
|
17924-92-4
|
Bindare
|
1 × 10–10–1 × 10–3
|
Bindare
|
1 × 10–10–1 × 10–3
|
Bindare
|
Kolväte (heterocykliskt), lakton
|
Naturprodukt
|
|
15
|
Tamoxifen
|
10540-29-1
|
Bindare
|
1 × 10–10–1 × 10–3
|
Bindare
|
1 × 10–10–1 × 10–3
|
Bindare
|
Kolväte (cykliskt)
|
Humant/veterinärt läkemedel
|
|
16
|
5α-dihydrotestosteron
|
521-18-6
|
Bindare
|
1 × 10–10–1 × 10–3
|
Bindare
|
1 × 10–10–1 × 10–3
|
Bindare
|
Steroid, icke-fenolisk
|
Naturprodukt
|
|
17
|
Bisfenol A
|
80-05-7
|
Bindare
|
1 × 10–10–1 × 10–3
|
Bindare
|
1 × 10–10–1 × 10–3
|
Bindare
|
Fenol
|
Kemisk intermediär
|
|
18
|
4-n-heptylfenol
|
1987-50-4
|
Bindare
|
1 × 10–10–1 × 10–3
|
Tvetydig (6)
|
1 × 10–10–1 × 10–3
|
Bindare
|
Alkylfenol
|
Intermediär
|
|
19
|
Kepon (klordekon)
|
143-50-0
|
Bindare
|
1 × 10–10–1 × 10–3
|
Bindare
|
1 × 10–10–1 × 10–3
|
Bindare
|
Kolväten (halogenerad)
|
Bekämpningsmedel
|
|
20
|
Bens(a)antracen
|
56-55-3
|
Icke-bindare
|
1 × 10–10–1 × 10–3
|
Icke-bindare (7)
|
1 × 10–10–1 × 10–3
|
Icke-bindare (7)
|
Aromatiskt kolväte
|
Intermediär
|
|
21
|
Enterolakton
|
78473-71-9
|
Bindare
|
1 × 10–10–1 × 10–3
|
Bindare
|
1 × 10–10–1 × 10–3
|
Bindare
|
Fytoöstrogen
|
Naturprodukt
|
|
22
|
Progesteron
|
57-83-0
|
Icke-bindare (*5)
|
1 × 10–10–1 × 10–4
|
Icke-bindare
|
1 × 10–10–1 × 10–4
|
Icke-bindare
|
Steroid
|
Naturprodukt
|
|
23
|
Oktyltrietoxisilan
|
2943-75-1
|
Icke-bindare
|
1 × 10–10–1 × 10–3
|
Icke-bindare
|
1 × 10–10–1 × 10–3
|
Icke-bindare
|
Silan
|
Ytbehandlingsmedel
|
|
24
|
Atrazin
|
1912-24-9
|
Icke-bindare (*5)
|
1 × 10–10–1 × 10–4
|
Icke-bindare
|
1 × 10–10–1 × 10–4
|
Icke-bindare
|
Heterocykliskt ämne
|
Ogräsbekämpningsmedel
|
|
hrER-BINDNINGSTESTETS DELAR
Viktiga testdelar
|
14.
|
Denna testmetod gäller tester som använder en ER-receptor och en lämpligt stark receptorligand som kan användas som en markör/spårsubstans i testet och kan trängas bort vid högre koncentrationer av en testkemikalie. I bindningstester ingår två huvuddelar: 1) mättnadsbindning och 2) kompetitiv bindning. Mättnadsbindningstestet används för att bekräfta receptorberedningarnas specificitet och aktivitet, medan den kompetitiva delen av experimentet används för att utvärdera testkemikaliens förmåga att binda till hrER.
|
Kontroller
|
15.
|
Grunden för föreslagna parallella östrogenreferenser och kontroller bör beskrivas. Parallellt löpande kontroller (lösningsmedel [vehikel], positiv [ER-bindare, stark resp. svag affinitet], negativ [icke-bindare]), utifrån omständigheter, tjänar som en indikation på att testet fungerar under testbetingelserna och utgör en grund för jämförelser mellan experiment, vanligen ingår de bland acceptanskriterierna för ett visst experiment (1). Fullständiga koncentrationskurvor för östrogenreferens och kontroller (dvs. svag bindare resp. icke-bindare) ska placeras i en platta vid varje testomgång. Alla andra plattor ska innehålla: 1) en hög koncentration (ungefär fullständig bortträngning av radioaktivt inmärkt ligand) och en mellankoncentration (ungefär IC50) av E2 respektive svag bindare i tripletter, 2) lösningsmedelskontroll och icke-specifik bindning, båda som tripletter.
|
Rutinmässig kvalitetskontroll
|
16.
|
Rutinmässiga kvalitetskontroller ska utföras som beskrivits för varje test för att säkerställa att receptorerna är aktiva, kemikalier har rätt koncentrationer, toleransgränser förblir stabila genom flera upprepningar och för att bibehålla förmågan till att ge förväntade ER-bindningsresponser över tid.
|
Demonstration av laboratoriets kompetens
|
17.
|
Innan okända kemikalier kan testas med något av testerna i denna testmetod ska varje laboratorium uppvisa kompetens i att använda testet genom att göra mättnadstester som bekräftar ER-beredningens aktivitet och specificitet, samt kompetitiva bindningstester med östrogenreferensen och kontroller (svag bindare och icke-bindare). Laboratoriet ska utifrån 3–5 oberoende experiment som genomförts under skilda dagar upprätta en historisk databas med resultat för östrogenreferensen och kontrollerna. Dessa experiment kommer att utgöra grunden för laboratoriets östrogenreferenser och historiska kontroller och kommer att användas som en delbedömning för godkännande av senare testomgångar.
|
|
18.
|
Testsystemets responsivitet kommer också att bekräftas genom tester av de kvalifikationsämnen som anges i tabell 2. Förteckningen över kvalifikationsämnen är en undergrupp av de referensämnen som anges i prestandastandarderna för ER-bindningstester (3). Dessa ämnen är kommersiellt tillgängliga, representerar de klasser av kemikalier som vanligen förknippas med ER-bindande aktivitet och bildar ett lämpligt intervall av förväntad styrka på ER-bindning (dvs. stark till svag) och icke-bindning (dvs. negativa svar). Varje kvalifikationsämne ska testas i koncentrationer vilka täcker intervallet som anges i tabell 2. Minst tre experiment ska genomföras för varje ämne och resultaten ska överensstämma med förväntad kemisk aktivitet. Varje experiment ska utföras oberoende (dvs. med färska spädningar av receptor, kemikalier och reagens), med tre replikat för varje koncentration. Kompetens uppvisas genom korrekt klassificering (positiv/negativ) av alla kvalifikationsämnen. Kompetenstesterna ska utföras av varje laboratorietekniker i samband med att han/hon lär sig testförfarandet.
Tabell 2
Förteckning över kontroller och kvalifikationsämnen för kompetitiva bindningstester med hrER
(89)
|
Nr
|
Ämnesnamn
|
CAS-nummer (90)
|
Förväntad respons (91)
(92)
|
Testkoncentrationsintervall (M)
|
MeSH kemisk klass (93)
|
Produktklass (94)
|
|
Kontroller (östrogenreferens, svag bindare, icke-bindare)
|
|
1
|
17–öστραδιολ
|
50-28-2
|
Bindare
|
1 × 10–11–1 × 10–6
|
Steroid
|
Humant/veterinärt läkemedel
|
|
2
|
Noretynodrel (eller) Noretindron
|
68-23-5 (eller) 68-22-4
|
Bindare
|
3 × 10–9–30 × 10–6
|
Steroid
|
Humant/veterinärt läkemedel
|
|
3
|
Oktyltrietoxisilan
|
2943-75-1
|
Icke-bindare
|
1 × 10–10–1 × 10–3
|
Silan
|
Ytbehandlingsmedel
|
|
Kvalifikationsämnen (94)
|
|
4
|
Dietylstilbestrol
|
56-53-1
|
Bindare
|
1 × 10–11–1 × 10–6
|
Kolväte (cykliskt), fenol
|
Humant/veterinärt läkemedel
|
|
5
|
17α-etynylöstradiol
|
57-63-6
|
Bindare
|
1 × 10–11–1 × 10–6
|
Steroid
|
Humant/veterinärt läkemedel
|
|
6
|
Meso-hexestrol
|
84-16-2
|
Bindare
|
1 × 10–11–1 × 10–6
|
Kolväte (cykliskt), fenol
|
Humant/veterinärt läkemedel
|
|
7
|
Tamoxifen
|
10540-29-1
|
Bindare
|
1 × 10–11–1 × 10–6
|
Kolväte (cykliskt)
|
Humant/veterinärt läkemedel
|
|
8
|
Genistein
|
446-72-0
|
Bindare
|
1 × 10–10–1 × 10–3
|
Heterocykliskt ämne, flavonoid
|
Naturprodukt
|
|
9
|
Bisfenol A
|
80-05-7
|
Bindare
|
1 × 10–10–1 × 10–3
|
Fenol
|
Kemisk intermediär
|
|
10
|
Zearalonon
|
17924-92-4
|
Bindare
|
1 × 10–11–1 × 10–3
|
Heterocykliskt ämne, lakton
|
Naturprodukt
|
|
11
|
Butylparaben
|
94-26-8
|
Bindare
|
1 × 10–11–1 × 10–3
|
Karboxylsyra, fenol
|
Konserveringsmedel
|
|
12
|
Atrazin
|
1912-24-9
|
Icke-bindare
|
1 × 10–11–1 × 10–6
|
Heterocykliskt ämne
|
Ogräsbekämpningsmedel
|
|
13
|
Di-n-butylftalat (DBP) (95)
|
84-74-2
|
Icke-bindare (96)
|
1 × 10–10–1 × 10–4
|
Kolväte (cykliskt), ester
|
Mjukgörare, kemisk intermediär
|
|
14
|
Kortikosteron
|
50-22-6
|
Icke-bindare
|
1 × 10–11–1 × 10–4
|
Steroid
|
Naturprodukt
|
|
Löslighetstester och fastställande av koncentrationsintervall för testkemikalier
|
19.
|
Ett preliminärt test bör genomföras för att fastställa löslighetsgränsen för varje testkemikalie och för att identifiera lämpligt koncentrationsintervall att använda sig av under testet. Samtliga testkemikaliers respektive löslighetsgräns i lösningsmedlet ska fastställas initialt och vidare bekräftas under testbetingelser. Slutkoncentrationen i testet bör inte överstiga 1 mM. Lämpligt intervall söks med tester som utgörs av en lösningsmedelskontroll och åtta logaritmiska seriespädningar, där man börjar med maximal acceptabel koncentration (t.ex. 1 mM eller lägre, baserat på löslighetsgräns), och närvaron av grumlighet eller fällning. Koncentrationer i det andra och tredje experimenten bör anpassas för en bättre karakterisering av koncentration–responskurvan.
|
Acceptanskriterier för testomgångar
|
20.
|
Godkännande eller avvisande av en testomgång baseras på utvärderingen av resultaten för den östrogenreferens och kontroll som används i varje experiment. För det första, för platta 1 ska kompletta koncentrationskurvor för referenskontrollerna från varje experiment motsvara prestandamätningarna med kurvanpassningsparametrar (t.ex. IC50 och Hill-koefficient [Hill slope]) utifrån resultaten som rapporteras för respektive protokoll för CERI- och FW-testerna (tillägg 2 och 3) och historiska kontrolldata från laboratoriet som genomför testet. Alla kontroller (östrogenreferens, svag bindare och icke-bindare) ska klassificeras korrekt vid varje experiment. För det andra ska man skatta överensstämmelsen hos kontrollerna på alla påföljande plattor med dem på platta 1. Vidare ska man använda ett tillräckligt intervall av testkemikaliens koncentration för att klart kunna beskriva toppen på kurvan för kompetitiv bindning. Variationen mellan replikat vid varje koncentration av testkemikalien, liksom mellan de tre oberoende testomgångarna, ska vara rimlig och vetenskapligt försvarbar. Förmågan att genomföra testet konsekvent ska uppvisas genom upprättande och underhåll av en historisk databas för östrogenreferensen och kontroller. Standardavvikelser (SD) eller variationskoefficienter (CV) för medelvärdena för referensöstrogen och kurvanpassningsparametrar för kontrollkurvor för svaga bindare från flera experiment kan användas som ett mått på reproducerbarhet inom laboratoriet. Yrkesmässigt omdöme ska tillämpas vid granskning av kontrollresultaten på plattan, från varje testomgång, liksom för varje testkemikalie.
Vidare ska följande principer kring acceptanskriterier tillämpas:
|
—
|
Data ska vara tillräckliga för en kvantitativ skattning av ER-bindning.
|
|
—
|
Testkoncentrationerna ska vara inom testkemikaliens löslighetsintervall.
|
|
Dataanalys
|
21.
|
Det fastställda förfarandet för dataanalys av mättnad respektive kompetitiv bindning ska följa huvudprinciperna för karakterisering av receptor–ligand-interaktioner. Vanligen analyseras mättnadsbindningsdata med en icke-linjär regressionsmodell som tar hänsyn till total och icke-specifik bindning. En korrigering för ligandförbrukning (t.ex. Swillens, 1995 [19]) kan behövas vid fastställande av Bmax och Kd. Data från tester av kompetitiv bindning omvandlas oftast (t.ex. procent specifik bindning och testkemikaliekoncentration [log M]). Skattningar av log(IC50)-värdet för varje testkemikalie ska göras med hjälp av en lämplig programvara för icke-linjär kurvanpassning med anpassning till en fyraparameters Hill-ekvation. Efter en inledande analys ska kurvanpassningsparametrarna för bindningsdata tas fram samt en visuell granskning göras av hur väl den upprättade kurvan över kompetitiv bindning speglar bindningsdata. I en del fall kan ytterligare analys krävas för att erhålla bästa kurvanpassning (t.ex. genom att begränsa [constrain] kurvans topp och/eller botten, använda 10-procentsregeln, se tillägg 4 och referens 2 (avsnitt III.A.2).
|
|
22.
|
Om testsystemet motsvarar acceptanskriterierna (punkt 20) är det en indikation på att testsystemet fungerar korrekt, men det säkerställer inte att ett specifikt test ger korrekta data. En upprepning av det första testets korrekta resultat är den bästa indikationen på att producerade data är korrekta.
|
Generella kriterier för tolkning av data
|
23.
|
För närvarande finns ingen allmänt överenskommen metod för att tolka ER-bindningsdata. Men både kvalitativa (t.ex. bindare/icke-bindare) och/eller kvantitativa skattningar (t.ex. log[IC50]-värde, relativ bindningsaffinitet [RBA] osv.) av hrER-medierad aktivitet bör baseras på empiriska data och gott vetenskapligt omdöme.
|
Testrapport
|
24.
|
Testrapporten ska innehålla följande information:
|
|
Kontroll/referens/testkemikalie
|
—
|
Källa, satsnummer, sista förbrukningsdag, om detta anges.
|
|
—
|
Testkemikaliens stabilitet i sig, om den är känd.
|
|
—
|
Löslighet och stabilitet av testkemikalien i lösningsmedlet, om detta är känt.
|
|
—
|
Mätning av pH, osmolalitet och utfällningar i det odlingsmedium till vilket testkemikalien tillsattes, i förekommande fall.
|
|
|
|
Monokomponentämne
|
—
|
fysikaliskt tillstånd, vattenlöslighet och ytterligare relevanta fysikalisk-kemiska egenskaper.
|
|
—
|
Kemiska beteckningar, såsom IUPAC- eller CAS-namn, CAS-nummer, Smiles- eller InChI-kod, strukturformel, renhet, kemisk identitet för föroreningar i förekommande fall och när så är praktiskt möjligt.
|
|
|
|
Multikomponentämne, UVCB-ämnen och blandningar
|
—
|
karakteriseras så långt som möjligt genom beståndsdelarnas kemiska identitet (se ovan), kvantitativa förekomst och relevanta fysikalisk-kemiska egenskaper.
|
|
|
|
Lösningsmedel/vehikel
|
—
|
Karakterisering (typ, leverantör och sats).
|
|
—
|
Motivering för val av lösningsmedel/vehikel.
|
|
—
|
Testkemikaliens löslighet och stabilitet i lösningsmedlet/vehikeln, om dessa är kända.
|
|
|
|
Receptorer
|
—
|
Källa till receptorer (leverantör, katalognr, sats, receptortyp, koncentration av aktiv receptor enligt leverantören, certifikat från leverantören).
|
|
—
|
Karakterisering av receptorer (inklusive mättnadsbindningsresultat): Kd, Bmax.
|
|
—
|
Förvaring av receptorer.
|
|
—
|
Radioaktivt inmärkt ligand.
|
|
—
|
Leverantör, katalognr, sats, specifik aktivitet.
|
|
|
|
Testbetingelser
|
—
|
Löslighetsbegränsningar under testbetingelser.
|
|
—
|
Bindningsbuffertens sammansättning.
|
|
—
|
Koncentration av spårsubstans (dvs. radioaktivt inmärkt ligand).
|
|
—
|
Testkemikaliens koncentration.
|
|
—
|
Procent vehikel i slutligt test.
|
|
—
|
Inkubationstemperatur och tid.
|
|
—
|
Separationsmetod bunden/fri.
|
|
—
|
Positiva och negativa kontroller/referensämnen.
|
|
—
|
Kriterier för när tester ska anses som positiva, negativa eller tvetydiga.
|
|
|
|
Acceptanskontroll
|
—
|
Faktiska värden för IC50 och Hill-koefficient (Hill slope) för parallella positiva kontroller/referensämnen.
|
|
|
|
Resultat
|
—
|
Rådata och data för bunden/fri.
|
|
—
|
Bekräftelsekontroll av denaturering, om tillämpligt.
|
|
—
|
Om det finns tillgängligt, lägsta effektiva koncentration (LEC).
|
|
—
|
RBA och/eller IC50-värden, där det är lämpligt.
|
|
—
|
Koncentration–responsförhållande, där det är möjligt.
|
|
—
|
Eventuella statistiska analyser tillsammans med fel- och konfidensmått (t.ex. SEM, SD, CV eller 95 % CI) samt en beskrivning av hur dessa värden har erhållits.
|
|
|
|
Diskussion kring resultaten
|
—
|
Applicering av 10-procentsregeln.
|
|
Slutsats
|
LITTERATUR
|
(1)
|
OECD (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environmental, Health and Safety Publications, Series on Testing and Assessment (No 34), Organisationen för ekonomiskt samarbete och utveckling (OECD), Paris.
|
|
(2)
|
OECD (2015). Integrated Summary Report: Validation of Two Binding Assays Using Human Recombinant Estrogen Receptor Alpha (hrERα), Health and Safety Publications, Series on Testing and Assessment (No 226), Organisationen för ekonomiskt samarbete och utveckling (OECD), Paris.
|
|
(3)
|
OECD (2015). Performance Standards for Binding Assays Using Human Recombinant Estrogen Receptor Alpha (hrERα), Health and Safety Publications, Series on Testing and Assessment (No 222), Organisationen för ekonomiskt samarbete och utveckling (OECD), Paris.
|
|
(4)
|
OECD (2012). Guidance Document on Standardized Test Guidelines for Evaluating Chemicals for Endocrine Disruption. Environmental, Health and Safety Publications, Series on Testing and Assessment (No 150), Organisationen för ekonomiskt samarbete och utveckling (OECD), Paris.
|
|
(5)
|
Cavailles V. (2002). Estrogens and Receptors: an Evolving Concept, Climacteric, 5 Suppl 2: s. 20-6.
|
|
(6)
|
Welboren W.J., m.fl. (2009). Genomic Actions of Estrogen Receptor Alpha: What are the Targets and How are they Regulated? Endocr. Relat. Cancer., 16(4): s. 1073–89.
|
|
(7)
|
Younes M. och Honma N. (2011). Estrogen Receptor Beta, Arch. Pathol. Lab. Med., 135(1): s. 63-6.
|
|
(8)
|
Diamanti-Kandarakis m.fl. (2009). Endocrine-Disrupting Chemicals: an Endocrine Society Sci. Statement, Endo Rev 30(4):293-342.
|
|
(9)
|
ICCVAM (2002). Background Review Document. Current Status of Test Methods for Detecting Endocrine Disruptors: In Vitro Estrogen Receptor Binding Assays. (NIH-publikation nr 03-4504). National Institute of Environmental Health Sciences, Research Triangle Park, NC.
|
|
(10)
|
ICCVAM (2003). ICCVAM Evaluation of In Vitro Test Methods for Detecting Potential Endocrine Disruptors: Estrogen Receptor and Androgen Receptor Binding and Transcriptional Activation Assays.
|
|
(11)
|
ICCVAM (2006). ICCVAM Evaluation of In Vitro Test Methods for Detecting Potential Endocrine Disruptors: Estrogen Receptor and Androgen Receptor Binding and Transcriptional Activation Assays.
|
|
(12)
|
Akahori Y. et al. (2008). Relationship Between the Results of In Vitro Receptor Binding Assay to Human Estrogen Receptor Alpha and In Vivo Uterotrophic Assay: Comparative Study with 65 Selected Chemicals, Toxicol. In Vitro, 22(1): 225-231.
|
|
(13)
|
OECD (2007). Additional Data Supporting the Test Guideline on the Uterotrophic Bioassay in Rodents, Environment, Health and Safety Publications, Series on Testing and Assessment (No 67), Organisationen för ekonomiskt samarbete och utveckling (OECD), Paris.
|
|
(14)
|
Takeyoshi, M. (2006). Draft Report of Pre-validation and Inter-laboratory Validation For Stably Transfected Transcriptional Activation (TA) Assay to Detect Estrogenic Activity - The Human Estrogen Receptor Alpha Mediated Reporter Gene Assay Using hER-HeLa-9903 Cell Line, Chemicals Evaluation and Research Institute (CERI): Japan. s. 1–188.
|
|
(15)
|
Yamasaki, K; Noda, S; Imatanaka, N; Yakabe, Y. (2004). Comparative Study of the Uterotrophic Potency of 14 Chemicals in a Uterotrophic Assay and their Receptor-Binding Affinity, Toxicol. Letters, 146: 111-120.
|
|
(16)
|
Kummer V; Maskova, J; Zraly, Z; Neca, J; Simeckova, P; Vondracek, J; Machala, M. (2008). Estrogenic Activity of Environmental Polycyclic Aromatic Hydrocarbons in Uterus of Immature Wistar Rats. Toxicol. Letters, 180: 213-221.
|
|
(17)
|
Gozgit, JM; Nestor, KM; Fasco, MJ; Pentecost, BT; Arcaro, KF. (2004). Differential Action of Polycyclic Aromatic Hydrocarbons on Endogenous Estrogen-Responsive Genes and on a Transfected Estrogen-Responsive Reporter in MCF-7 Cells. Toxicol. and Applied Pharmacol., 196: 58-67.
|
|
(18)
|
Santodonato, J. (1997). Review of the Estrogenic and Antiestrogenic Activity of Polycyclic Aromatic Hydrocarbons: Relationship to Carcinogenicity. Chemosphere, 34: 835-848.
|
|
(19)
|
Swillens S (1995). Interpretation of Binding Curves Obtained with High Receptor Concentrations: Practical Aid for Computer Analysis, Mol Pharmacol 47(6):1197-1203.
|
Tillägg 1
DEFINITIONER OCH FÖRKORTNINGAR
10-procentsregeln: Möjlighet att från analyserna utesluta datapunkter där replikatens medelvärde för procent specifikt bunden [3H]-17β-östradiol är 10 % eller mer över det observerade medelvärdet vid en lägre koncentration (se tillägg 4).
Acceptanskriterier: Minimistandarder för experimentets prestanda avseende kontroller och referensstandarder. Alla acceptanskriterier ska uppfyllas för att ett experiment ska anses vara giltigt.
Noggrannhet (konkordans): Ett mått på hur väl testresultaten och de godtagna referensvärdena överensstämmer med varandra. Det är en relevansaspekt och ett mått på testets prestanda. Detta begrepp används ofta omväxlande med ”konkordans” för att beskriva andelen korrekta resultat med ett test (1).
CF: The OECD Conceptual Framework for the Testing and Evaluation of Endocrine Disrupters (OECD:s begreppsram för provning och bedömning av hormonstörande ämnen).
Kemikalie: Ett ämne eller en blandning.
CV: Variationskoefficient (Coefficient of Variation).
E2: 17β-östradiol.
ED.: hormonstörning.
hERα: Human östrogenreceptor alfa.
ER: Östrogenreceptor.
Östrogen aktivitet: Kapaciteten hos en kemikalie att imitera 17β-östradiols förmåga att binda östrogenreceptorer. Bindning till hERα kan undersökas med denna testmetod.
IC50
: Halva maximalt effektiva koncentrationen för en inhiberande testkemikalie.
ICCVAM: The Interagency Coordinating Committee on the Validation of Alternative Methods (i Förenta staterna).
Reproducerbarhet mellan laboratorier: Ett mått på i hur stor utsträckning olika kvalificerade laboratorier kan producera kvalitativt och kvantitativt liknande resultat, med användning av samma protokoll och samma testämnen. Reproducerbarheten mellan laboratorier fastställs under förvalideringen och valideringen och indikerar i hur stor utsträckning ett test framgångsrikt kan överföras mellan laboratorier, även kallat inter-laborativ reproducerbarhet (1).
Reproducerbarhet inom laboratoriet: Ett mått på i vilken utsträckning kvalificerade personer inom samma laboratorium framgångsrikt kan upprepa resultat vid olika tidpunkter när de använder ett visst protokoll. Kallas även intra-laborativ reproducerbarhet (1).
LEC: Lägsta effektiva koncentration (Lowest Effective Concentration) är den lägsta koncentration av testkemikalie som ger en respons (dvs. den lägsta koncentration av testkemikalie vid vilken induktionen statistiskt skiljer sig från parallell vehikelkontroll).
Me too-test: Ett vardagligt uttryck för ett test som strukturellt och funktionellt liknar en validerad och godtagen referenstestmetod. Kallas även ’liknande testmetod’.
PBTG: Prestandabaserad testriktlinje (Performance-Based Test Guideline)
Prestandastandarder: Standarder som baseras på en validerad testmetod och som utgör grunden för utvärdering av jämförbarheten hos ett föreslaget, mekanistiskt och funktionellt likartat test. Häri ingår 1) viktiga testdelar, 2) en minimiförteckning med referenskemikalier som valts ut från de kemikalier som har använts för att påvisa att den validerade testmetoden fungerar tillfredsställande och 3) de jämförbara nivåer av noggrannhet och tillförlitlighet, på grundval av vad som erhållits för den validerade testmetoden, som man bör kunna påvisa för det föreslagna testet när det utvärderas med användning av minimiförteckningen med referenskemikalier (1).
Kvalifikationsämnen: En undergrupp av referensämnen som ingår i prestandastandarderna som kan användas av laboratorier för att påvisa teknisk färdighet med en standardiserad testmetod. Bland urvalskriterierna för dessa ämnen ingår oftast att de representerar responsintervallet, är kommersiellt tillgängliga och att referensdata av hög kvalitet finns tillgängliga.
Kompetens: Påvisad förmåga att korrekt genomföra ett test, innan okända ämnen testas.
Referensöstrogen: 17ß-östradiol (E2, CAS 50-28-2).
Referenstestmetod: De tester som PBTG 493 baserar sig på.
RBA: Relativ bindningsaffinitet. Ett ämnes RBA beräknas som andelen i procent av ämnets log(IC50)-värde i förhållande till 17β-östradiols log(IC50)-värde.
Relevans: Beskrivning av förhållandet mellan testet och den berörda effekten och huruvida detta förhållande är meningsfullt och användbart för ett visst ändamål. Relevansen är ett mått på hur väl man genom testet korrekt kan mäta och prediktera den aktuella biologiska effekten. Relevans omfattar också överväganden om noggrannheten hos ett test (konkordans) (1).
Tillförlitlighet: Mått på i vilken utsträckning ett test kan reproduceras inom och mellan laboratorier över tid, när den utförs enligt samma protokoll. Den bedöms genom att man beräknar reproducerbarheten inom och mellan laboratorier.
SD: Standardavvikelse (Standard Deviation).
Testkemikalie: Alla ämnen eller blandningar som testas med denna testmetod.
Validerad testmetod: Ett test för vilket valideringsstudier har genomförts för att fastställa relevans (inbegripet noggrannhet) och tillförlitlighet för ett specifikt ändamål. Det är viktigt att notera att en validerad testmetod kanske inte är tillräckligt noggrann och tillförlitlig när det gäller ett visst föreslaget ändamål (1).
Validering: Processen enligt vilken pålitlighet och relevans för en särskild strategi, metod, ett särskilt test, en särskild process eller bedömning etableras för ett fastställt syfte (1).
Tillägg 2
FREYBERGER-WILSONS IN VITRO-TESTER AV MÄTTNAD HOS RESPEKTIVE KOMPETITIV BINDNING TILL ÖSTROGENRECEPTOR (ERΑ) MED REKOMBINANT FULLÄNGDS-ERΑ
INLEDANDE ÖVERVÄGANDEN OCH BEGRÄNSNINGAR (SE ÄVEN ALLMÄN INLEDNING)
|
1.
|
I detta in vitro-test av mättnad hos och kompetitiv bindning till östrogenreceptor (ERα) används human fullängds-ERαα-receptor (hrERα) som produceras i och isoleras från bakulovirusinfekterade insektsceller. Protokollet, som utvecklades av Freyberger och Wilson, prövades i en internationell valideringsstudie som involverade flera laboratorier (2) och vilken visade dess relevans och pålitlighet för det avsedda syftet.
|
|
2.
|
Detta test är ett screeningförfarande för att identifiera ämnen som kan binda till fullängds-hrERα. Det används för att fastställa en testkemikalies förmåga att konkurrera med 17β-östradiol om att binda till hrERα. Kvantitativa testresultat kan omfatta IC50 (ett mått på den koncentration av testkemikalie som behövs för att tränga bort hälften av [3H]-17β-östradiolen från hrERα) och testkemikaliernas relativa bindningsaffiniteter för hrERα, jämförda med den hos 17β-östradiol. För kemisk screening kan godtagbara kvalitativa testresultat omfatta klassificeringar av testkemikalier som antingen hrERα-bindare, icke-bindare eller tvetydiga, utifrån de kriterier som beskrivits för bindningskurvorna.
|
|
3.
|
Testet använder en radioaktiv ligand, vilket kräver att laboratoriet har licens för att hantera radioaktiva material. Allt handhavande av radioisotoper och farliga kemikalier ska följa de regelverk och rutiner som föreskrivs av nationell lagstiftning.
|
|
4.
|
Läs ”ALLMÄN INLEDNING” och ”hrER-BINDNINGSTESTETS DELAR” innan testet används för tillsynsändamål. Definitioner och förkortningar som används i denna testriktlinje beskrivs i tillägg 1.
|
TESTETS PRINCIP (SE ÄVEN ALLMÄN INLEDNING)
|
5.
|
Bindningstestet med hrERα mäter förmågan hos radioaktivt inmärkt ligand ([3H]-17β-östradiol) att binda till ER i närvaron av ökande koncentrationer av testkemikalie (dvs. konkurrerande ligand). Testkemikalier med hög affinitet för ER konkurrerar med den radioaktivt inmärkta liganden vid en lägre koncentration än de kemikalier som har lägre affinitet för receptorn.
|
|
6.
|
Detta test består av två huvuddelar: ett mättnadsbindningsexperiment för att karakterisera receptor–ligandinteraktionsparametrar, följt av ett kompetitivt bindningsexperiment som karakteriserar konkurrensen mellan en testkemikalie och en radioaktivt inmärkt ligand vid inbindning till ER.
|
|
7.
|
Mättnadsbindningsexperimentets syfte är att inför det kompetitiva bindningsexperimentet karakterisera en receptorsats med avseende på bindningsaffinitet och antal receptorer. Mättnadsbindningsexperimentet mäter, vid jämviktsförhållanden och en fast receptorkoncentration, östrogenreceptorns affinitet för dess naturliga ligand (mätt som dissociationskonstant, Kd) och koncentrationen av aktiva bindningsplatser (Bmax).
|
|
8.
|
Det kompetitiva bindningsexperimentet mäter benägenheten hos ett ämne att konkurrera med [3H]-17β-östradiol om att binda till ER. Affiniteten kvantifieras genom koncentrationen av testkemikalie som, vid jämvikt, hämmar 50 % av den specifika bindningen av [3H]-17β-östradiol (kallad hämmande koncentration 50 %, eller IC50). Detta kan också utvärderas med hjälp av den relativa bindningsaffiniteten (RBA, affiniteten jämförd med IC50 för östradiol som mäts separat i samma testomgång). Det kompetitiva bindningsexperimentet mäter bindningen av [3H]-17β-östradiol vid en fast koncentration i närvaron av ett stort intervall (åtta tiopotenser) av testkemikaliekoncentrationer. Data anpassas sedan, där så är möjligt, till en form av Hill-ekvationen (Hill, 1910) som beskriver bortträngningen av den radioaktivt märkta liganden av en kompetitiv bindare som binder till en bindningsplats. Den utsträckning i vilken den radioaktivt inmärkta östradiolen trängs bort vid jämvikt används för att karakterisera testkemikalien som en bindare, en icke-bindare eller med tvetydig respons.
|
FÖRFARANDE
Fastställande av acceptabel proteinfunktion hos hrERα
|
9.
|
Innan rutinmässiga tester av mättnad och kompetitiv bindning genomförs ska varje ny sats av hrERα visas fungera korrekt i det laboratorium där den ska användas. En tvåstegsprocedur bör användas för att kontrollera prestanda. Dessa steg är som följer:
|
—
|
Utför ett mättnadsbindningstest med [3H]-17β-östradiol för att fastställa specificitet och mättnad hos hrERα. Icke-linjär regressionsanalys av dessa data (t.ex. BioSoft, McPherson, 1985, Motulsky, 1995) och påföljande Scatchard-diagram ska dokumentera [3H]-17β-östradiolens bindningsaffinitet för hrERα (Kd) samt antalet receptorer (Bmax) för varje sats av hrERα.
|
|
—
|
Genomför ett kompetitivt bindningstest med hjälp av kontrollämnen (referensöstrogen (17β-östradiol)), en svag bindare (t.ex. noretynodrel eller noretindron) och en icke-bindare (oktyltrietoxisilan, OTES). Varje laboratorium ska upprätta en historisk databas för att dokumentera överensstämmelsen hos IC50 och andra relevanta värden för referensöstrogenet och svaga bindare, mellan olika experiment och olika hrERα-satser. Parametrarna för kontrollämnenas kurvor över kompetitiv bindning ska vara inom gränserna för 95-procentigt konfidensintervall (se tabell 1), vilka har tagits fram med data från laboratorier som deltog i valideringsstudien för detta test (2).
|
|
Tabell 1
Prestandakriterier framtagna för referensöstrogen och svag bindare, FW-hrER-bindningstest
|
Ämne
|
Parameter
|
Medelvärde (8)
|
Standardavvikelse (n)
|
95 % konfidensintervall (9)
|
|
Nedre gräns
|
Övre gräns
|
|
17β-östradiol.
|
Topp (%)
|
100,44
|
10,84 (67)
|
97,8
|
103,1
|
|
Botten (%)
|
0,29
|
1,25 (67)
|
–0,01
|
0,60
|
|
Hill-koefficient (Hill slope)
|
–1,06
|
0,20 (67)
|
–1,11
|
–1,02
|
|
Log(IC50) (M)
|
–8,92 (10)
|
0,18 (67)
|
–8,97
|
–8,88
|
|
Noretynodrel
|
Topp (%)
|
99,42
|
8,90 (68)
|
97,27
|
101,60
|
|
Botten (%)
|
2,02
|
3,42 (68)
|
1,19
|
2,84
|
|
Hill-koefficient (Hill slope)
|
–1,01
|
0,38 (68)
|
–1,10
|
–0,92
|
|
Log(IC50) (M)
|
–6,39
|
0,27 (68)
|
–6,46
|
–6,33
|
|
Noretindron (10)
|
Topp (%)
|
96,14
|
8,44 (27)
|
92,80
|
99,48
|
|
Botten (%)
|
2,38
|
5,02 (27)
|
0,40
|
4,37
|
|
Hill-koefficient (Hill slope)
|
–1,41
|
0,32 (27)
|
–1,53
|
–1,28
|
|
Log(IC50 ) (M)
|
–5,73
|
0,27 (27)
|
–5,84
|
–5,62
|
Demonstration av laboratoriets kompetens
|
10.
|
Se punkterna 17 och 18 och tabell 2 i ”hrER-BINDNINGSTESTETS DELAR” i denna testmetod. Varje test (mättnad och kompetitiv bindning) ska bestå av tre oberoende testomgångar (dvs. med nya spädningar av receptor, kemikalier och reagens) på olika dagar och varje omgång ska innehålla tre replikat.
|
Bestämning av hrERα-koncentration
|
11.
|
Koncentrationen av aktiv receptor varierar något mellan satser och med förvaringsförhållanden. Därför ska koncentrationen av aktiv receptor som erhållits från leverantören fastställas. Detta ger tillämplig koncentration av aktiv receptor när testomgången ska göras.
|
|
12.
|
Under förhållanden som motsvarar kompetitiv bindning (dvs. 1 nM [3H]-östradiol), ska nominella koncentrationer om 0,25, 0,5, 0,75, respektive 1 nM receptor inkuberas i frånvaro (total bindning) och närvaro (icke-specifik bindning) av 1 μM ej inmärkt östradiol. Specifik bindning, beräknad som differensen av total och icke-specifik bindning, ritas i ett diagram mot den nominella receptorkoncentrationen. Den receptorkoncentration som ger specifika bindningsvärden motsvarande 20 % av tillsatt radioaktivt inmärkt ligand relateras till motsvarande nominella receptorkoncentration, och denna ska användas vid tester av mättnad och kompetitiv bindning. Ofta överensstämmer en slutlig hrER-koncentration på 0,5 nM med detta förhållande.
|
|
13.
|
Om 20-procentskriteriet vid upprepade försök inte kan uppfyllas ska experimentets uppställning kontrolleras för möjliga fel. Svårigheter att uppnå 20-procentskriteriet kan indikera att det finns väldigt lite aktiv receptor i satsen av rekombinant receptor och man bör då överväga att använda en annan sats.
|
Mättnadstest
|
14.
|
Åtta ökande koncentrationer av [3H]-17β-östradiol ska utvärderas i tripletter, enligt följande tre betingelser (se tabell 2):
|
—
|
I frånvaron av ej inmärkt 17β-östradiol och närvaro av ER. Här fastställs total bindning genom att mäta radioaktiviteten i de brunnar som enbart innehåller [3H]-17β-östradiol.
|
|
—
|
I närvaron av 1 000 gångers koncentrationsöverskott av ej inmärkt 17β-östradiol över inmärkt 17β-östradiol och närvaro av ER. Avsikten med denna betingelse är att mätta de aktiva bindningsplatserna med ej inmärkt 17β-östradiol och, genom att mäta radioaktiviteten i brunnarna, fastställa den icke-specifika bindningen. Eventuellt kvarvarande radioaktiv östradiol som kan binda till receptorn anses binda till en icke-specifik bindningsplats eftersom den tillsatta icke-radioaktiva östradiolen ska ha så hög koncentration att den har bundit till alla specifika bindningsplatser på receptorn.
|
|
—
|
I frånvaro av ej inmärkt 17β-östradiol och frånvaro av ER (fastställande av total radioaktivitet).
|
|
Beredning av lösningar med [3H]-17β-östradiol respektive ej inmärkt 17β-östradiol
|
15.
|
Spädningar av [3H]-17β-östradiol ska beredas genom att tillsätta testbuffert till en 12 nM stamlösning av [3H]-17β-östradiol för att få koncentrationer som initialt sträcker sig från 0,12 nM till 12 nM. Genom att tillsätta 40 μl av dessa lösningar till respektive testbrunnar i en 96-hålsmikrotiterplatta (till en slutvolym på 160 μl) erhålls slutliga testkoncentrationer på 0,03–3,0 nM. Beredning av testbuffert, stamlösning med [3H]-17β-östradiol samt spädningar och fastställande av koncentrationer beskrivs i detalj i FW-protokollet (2).
|
|
16.
|
Spädningar av etanolinnehållande 17β-östradiollösningar ska beredas genom att tillsätta testbuffert och på så sätt erhålla åtta ökande koncentrationer, initialt från 0,06 μM till 6 μM. Genom att tillsätta 80 μl av dessa lösningar till respektive testbrunn i en 96-hålsmikrotiterplatta (till en slutvolym på 160 μl) erhålls slutliga testkoncentrationer på 0,03–3 μM. Slutkoncentrationen av ej inmärkt 17β-östradiol i de olika icke-specifika bindningstestbrunnarna bör vara 1 000 gånger högre än koncentrationen av [3H]-17β-östradiol. Beredning av spädningar av ej inmärkt 17β-östradiol beskrivs i detalj i FW-protokollet (2).
|
|
17.
|
Den nominella receptorkoncentration som ger specifik bindning av 20 ± 5 % bör användas (se punkterna 12–13). Lösningen med hrERα ska beredas omedelbart före användning.
|
|
18.
|
Förbered 96-hålsmikrotiterplattorna enligt schema i tabell 2, med tre replikat per koncentration. Exempel på fördelning över plattan av koncentrationer och volymer av [3H]-17β-östradiol, ej inmärkt 17β-östradiol, buffert och receptor ges i tillägg 2.2.
|
Tabell 2
Upplägg för mikrotiterplatta till mättnadsbindningstest
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
|
A
|
0,03 nM [3H]E2 + ER
|
0,06 nM [3H]E2 + ER
|
0,08 nM [3H]E2 + ER
|
0,10 nM [3H]E2 + ER
|
Total bindning (lösningsmedel)
|
|
B
|
0,30 nM [3H]E2 + ER
|
0,60 nM [3H]E2 + ER
|
1,0 nM [3H]E2 + ER
|
3,0 nM [3H]E2 + ER
|
|
|
C
|
|
|
|
|
|
|
D
|
0,03 nM [3H]E2 + ER + 0,03 μM E2
|
0,06 nM [3H]E2 + ER + 0,06 μM E2
|
0,08 nM [3H]E2 + ER + 0,08 μM E2
|
0,10 nM [3H]E2 + ER + 0,10 μM E2
|
Icke-specifik bindning
|
|
E
|
0,30 nM [3H]E2 + ER + 0,30 μM E2
|
0,60 nM [3H]E2 + ER + 0,60 μM E2
|
1,0 nM [3H]E2 + ER + 1,0 μM E2
|
3,0 nM [3H]E2 + ER + 3,0 μM E2
|
|
|
F
|
|
|
|
|
|
|
G
|
|
|
|
|
|
|
H
|
|
|
|
|
|
|
[3H]E2: [3H]-17β-östradiol.
ER: östrogenreceptor.
E2: ej inmärkt 17β-östradiol.
|
|
19.
|
Testmikrotiterplattor bör inkuberas på horisontellt cirkulerande skak vid 2–8 °C i 16–20 timmar.
|
Mätning av [3H]-17β-östradiol bunden till hrERα
|
20.
|
Den [3H]-17β-östradiol som är bunden till hrERα ska separeras från fri [3H]-17β-östradiol genom tillsats av 80 μl kall DCC-suspension till varje brunn, plattorna skakas sedan i 10 minuter och centrifugeras i 10 minuter med ungefär 2 500 varv per minut. För att minimera dissociationen av den bundna [3H]-17β-östradiolen från hrERα under denna procedur är det extremt viktigt att buffertarna och testbrunnarna håller en temperatur mellan 2 och 8 °C och att varje steg utförs snabbt. En skakapparat för mikrotiterplattor är nödvändig för snabb och effektiv hantering av plattorna.
|
|
21.
|
Sedan tas 50 μl supernatant innehållande den hrERα-bundna [3H]-17β-östradiolen, men extremt försiktigt så att man inte råkar förorena brunnarna genom att vidröra DCC, och placeras i en andra mikrotiterplatta.
|
|
22.
|
Sedan tillsätts 200 μl scintillationsvätska, som kan omvandla den nukleära strålningens rörelseenergi till ljus, till varje brunn (A1–B12 och D1–E12). Brunnarna G1–H12 (kvantifieras i totala dpm) utgör en seriespädning av [3H]-17β-östradiol (40 μl) som ska överföras direkt till scintillationsvätskan i mätplattans brunnar enligt tabell 3, dvs. dessa brunnar innehåller endast 200 μl scintillationsvätska och korrekt spädning av [3H]-17β-östradiol. Dessa mätningar visar, i dpm, hur mycket [3H]-17β-östradiol som tillsattes till varje uppsättning brunnar för total bindning och icke-specifik bindning.
|
Tabell 3
Upplägg av mikrotiterplatta vid mättnadsbindningstest, mätning av radioaktivitet
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
|
A
|
0,03 nM [3H]E2 + ER
|
0,06 nM [3H]E2 + ER
|
0,08 nM [3H]E2 + ER
|
0,10 nM [3H]E2 + ER
|
Total bindning (lösningsmedel)
|
|
B
|
0,30 nM [3H]E2 + ER
|
0,60 nM [3H]E2 + ER
|
1,0 nM [3H]E2 + ER
|
3,0 nM [3H]E2 + ER
|
|
C
|
|
|
|
|
|
|
D
|
0,03 nM [3H]E2 + ER + 0,03 μM E2
|
0,06 nM [3H]E2 + ER + 0,06 μM E2
|
0,08 nM [3H]E2 + ER + 0,08 μM E2
|
0,10 nM [3H]E2 + ER + 0,10 μM E2
|
Icke-specifik bindning
|
|
E
|
0,30 nM [3H]E2 + ER + 0,30 μM E2
|
0,60 nM [3H]E2 + ER + 0,60 μM E2
|
1,0 nM [3H]E2 + ER + 1,0 μM E2
|
3,0 nM [3H]E2 + ER + 3,0 μM E2
|
|
F
|
|
|
|
|
|
|
G
|
0,03 nM [3H] E2 (totala dpm)
|
0,06 nM [3H] E2
|
0,08 nM [3H] E2
|
0,10 nM [3H] E2
|
Totala dpm (*7)
|
|
H
|
0,30 nM [3H]E2
|
0,60 nM [3H]E2
|
1,0 nM [3H]E2
|
3,0 nM [3H]E2
|
|
[3H]E2: [3H]-17β-östradiol.
ER: östrogenreceptor.
E2: ej inmärkt 17β-östradiol.
dpm: sönderfall per minut (disintegrations per minute).
|
|
23.
|
Mätningarna ska starta med en fördröjning om minst 2 timmar och varje brunn ska mätas i 40 minuter. En scintillationsräknare för mikrotiterplattor bör användas för att fastställa dpm/brunn med korrigering för quenching. Om en scintillationsräknare för mikrotiterplattor inte finns att tillgå kan proverna mätas i en traditionell räknare. Under sådan förhållanden kan man överväga en kortare mättid.
|
Kompetitivt bindningstest
|
24.
|
Det kompetitiva bindningstestet mäter bindningen av [3H]-17β-östradiol vid en viss koncentration i närvaro av ökande koncentration av testkemikalie. Tre parallella replikat bör användas för varje koncentration i varje testomgång. Dessutom bör tre icke-parallella testomgångar utföras för varje testad kemikalie. Testet bör utföras i en eller flera 96-hålsmikrotiterplattor.
|
Kontroller
|
25.
|
Varje experiment ska inkludera parallella lösningsmedelprover och kontroller (dvs. referensöstrogen, svag bindare och icke-bindare). Fullständiga koncentrationskurvor för östrogenreferens och kontroller (dvs. svag bindare resp. icke-bindare) ska placeras i en platta vid varje testomgång. Alla andra plattor ska innehålla: i) en hög (maximal bortträngning) respektive medelhög (ungefär IC50) koncentration av E2 respektive svag bindare i tripletter, ii) lösningsmedelskontroll och icke-specifik bindning, båda som tripletter. Förfaranden för beredning av testbuffert, kontroller [3H]-17β-östradiol, hrERα och testkemikalielösningar beskrivs i referens 2 (bilaga K, se FW Assay Protocol).
|
Lösningsmedelskontroll
|
26.
|
Lösningsmedelskontrollen indikerar att lösningsmedlet inte interagerar med testsystemet och mäter också total bindning (TB). Etanol är det mest lämpliga lösningsmedlet. Men om testkemikaliens högsta koncentration inte är löslig i etanol kan DMSO användas. Koncentrationen av etanol, eller DMSO om det används, blir 1,5 % i det slutliga testbrunnarna och får inte överstiga 2 %.
|
Buffertkontroll
|
27.
|
Buffertkontrollen (BC) ska inte innehålla vare sig lösningsmedel eller testkemikalie men testets alla övriga ingående delar. Resultaten för buffertkontrollen jämförs med lösningsmedelskontrollen för att säkerställa att det valda lösningsmedlet inte påverkar testsystemet.
|
Stark bindare (referensöstrogen)
|
28.
|
Den endogena liganden är 17β-östradiol (CAS-nr 50-28-2) och den binder med hög affinitet till alfa-subtypen av ER. En standardkurva med ej inmärkt 17β-östradiol ska upprättas för varje kompetitivt hrERα-bindningstest, för att möjliggöra en bedömning av variationen när testet, över tid, upprepas i samma laboratorium. Åtta lösningar av ej inmärkt 17β-östradiol ska beredas i etanol, med koncentrationer i testbrunnarna som sträcker sig från 100 nM–10 pM (–7[logM] till –11[logM]), med följande spridning (–7[logM], –8[logM], –8,5[logM], –9[logM], –9,5[logM], –10[logM], –11[logM]). Den högsta koncentrationen av ej inmärkt 17β-östradiol (1 μM) indikerar också icke-specifik bindning. Denna koncentration markeras med märkningen ”NSB” i tabell 4 även om den också är en del av standardkurvan.
|
Svag bindare
|
29.
|
En svag bindare (noretynodrel [CAS-nr 68-23-5] eller noretindron [CAS-nr 68-22-4]) bör inkluderas för att påvisa varje experiments känslighet och möjliggöra en bedömning av variation när testet används längre perioder. Åtta lösningar av den svaga bindaren ska beredas i etanol, med koncentrationer i testbrunnarna som sträcker sig från 3 nM till 30 μM (–8,5[logM] till –4,5logM]), med följande spridning: –4,5[logM], –5[logM], –5,5[logM], –6[logM], –6,5[logM], –7[logM], –7,5[logM], –8,5[logM].
|
Icke-bindare
|
30.
|
Oktyltrietoxisilan (OTES, CAS-nr 2943-75-1) ska användas som negativ kontroll (icke-bindare). Den ger återkoppling om att testet, så som det utförs, detekterar när testkemikalier inte binder till hrERα. Åtta lösningar av icke-bindaren ska beredas i etanol, med koncentrationer som i testbrunnarna sträcker sig, i tiopotenssteg, från 0,1 nM–1 000 μM (–10[logM] till –3[logM]). Di-n-butylftalat (DBP) kan användas som en alternativ icke-bindare-kontroll. Dess maximala löslighet har visats vara –4[logM].
|
hrERα-koncentration
|
31.
|
Den mängd receptor som ger en specifik bindning på 20 ± 5 % av 1 nM radioaktivt märkt ligand bör användas (se punkterna 12–13 i tillägg 2). Lösningen med hrERα ska beredas omedelbart före användning.
|
[3H]-17β-östradiol.
|
32.
|
Koncentrationen för [3H]-17β-östradiol i testbrunnarna bör vara 1,0 nM.
|
Testkemikalier
|
33.
|
Vid det första tillfället är det nödvändigt att genomföra ett löslighetstest för att fastställa löslighetsgränsen för varje testkemikalie och för att identifiera lämpligt koncentrationsintervall att använda sig av under testet. Samtliga testkemikaliers respektive löslighetsgräns i lösningsmedlet ska fastställas initialt och vidare bekräftas under testbetingelser. Den slutkoncentration som används i testet ska inte överstiga 1 mM. Intervallsökningen görs med en lösningsmedelskontroll och åtta logaritmiska seriespädningar, där man börjar med maximal acceptabel koncentration (t.ex. 1 mM eller lägre, baserat på löslighetsgräns), och närvaron av grumlighet eller fällning (se också punkt 35). Testkemikalien ska testas med 8 koncentrationskurvor med logaritmisk spridning utifrån fynden i det föregående intervallsökningstestet. Koncentrationer i det andra och tredje experimenten bör anpassas för en bättre karakterisering av koncentration–responskurvan.
|
|
34.
|
Spädningar av testkemikalien ska beredas i lämpligt lösningsmedel (se punkt 26 i tillägg 2). Om testkemikaliens högsta koncentration inte är löslig i vare sig etanol eller DMSO, och en tillsats av mer lösningsmedel skulle göra att koncentrationen av lösningsmedel i det slutliga provröret överskred den godtagbara gränsen, kan den högsta koncentrationen sänkas till nästa lägre koncentration. I sådana fall kan ytterligare en koncentration läggas till i den lägre änden av koncentrationsserien. Övriga koncentrationer i serien bör förbli oförändrade.
|
|
35.
|
Testkemikalielösningarna ska iakttas noggrant vid tillsatsen till testbrunnen, eftersom testkemikalien kan fälla ut i detta steg. Data för alla brunnar som innehåller en fällning ska uteslutas från kurvanpassning och anledningen till uteslutningen av data noteras.
|
|
36.
|
Om det sedan tidigare från andra källor finns information som tillhandahåller ett log(IC50)-värde för en testkemikalie, kan det vara lämpligt att geometriskt sprida spädningarna (dvs. 0,5 log-enheter runt det förväntade log[IC50]-värdet). Slutresultatet bör återspegla tillräcklig spridning av koncentrationer på båda sidor av log(IC50)-värdet, inklusive ”topp” och ”botten”, så att bindningskurvan kan karakteriseras tillfredsställande.
|
Organisering av testplattor
|
37.
|
Märkta mikrotiterplattor ska beredas för sexfaldiga inkubationer med koder för lösningsmedelskontrollen, den högsta koncentrationen av referensöstrogen som också tjänar som indikator på icke-specifik bindning (NSB), och buffertkontrollen, samt för inkubation i tripletter med koder för var och en av de åtta koncentrationerna för icke-bindande kontroll (oktyltrietoxisilan), de sju lägre koncentrationerna av referensöstrogen, de åtta koncentrationsnivåerna för svag bindare och de åtta koncentrationerna av varje testkemikalie (TC). Ett exempel på upplägget av plattschemat för fullständiga koncentrationskurvor för referensöstrogen och kontroll ges nedan i tabell 4. Ytterligare mikrotiterplattor används för testkemikalierna och ska inkludera plattkontroller, dvs. 1) en hög koncentration (maximal bortträngning) och en mellankoncentration (ungefär IC50) av vardera E2 och svag bindare i tripletter, 2) lösningsmedelskontroll och icke-specifik bindning, båda sexfalt (tabell 5). Ett exempel på plattupplägg för ett kompetitivt test med tre okända testkemikalier ges i tillägg 2.3. De koncentrationer som anges i tabellerna 4 och 5 är testets slutkoncentrationer. Maximal koncentration för E2 ska vara 1 × 10-7 M och för den svaga bindaren bör den högsta koncentrationen som används för den svaga bindaren på platta 1 användas. Laboratoriet ska fastställa IC50 med hjälp av sin historiska kontrolldatabas. Detta värde kan förväntas vara jämförbart med det som iakttagits i valideringsstudierna (se tabell 1).
|
Tabell 4
Upplägg för mikrotiterplatta till kompetitivt bindningstest, fullständiga koncentrationskurvor för referensöstrogen och kontroller (platta 1)
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
TB (endast lösningsmedel)
|
TB (endast lösningsmedel)
|
NSB
|
NSB
|
|
B
|
E2 (1 × 10-7)
|
E2 (1 × 10-8)
|
E2 (1 × 10-8,5)
|
E2 (1 × 10-9)
|
|
C
|
E2 (1 × 10-9,5)
|
E2 (1 × 10-10)
|
E2 (1 × 10-11)
|
Blankprov (*8)
|
|
D
|
NE (1 × 10-4,5)
|
NE (1 × 10-5)
|
NE (1 × 10-5,5)
|
NE (1 × 10-6)
|
|
E
|
NE (1 × 10-6,5)
|
NE (1 × 10-7)
|
NE (1 × 10-7,5)
|
NE (1 × 10-8,5)
|
|
F
|
OTES (1 × 10-3)
|
OTES (1 × 10-4)
|
OTES (1 × 10-5)
|
OTES (1 × 10-6)
|
|
G
|
OTES (1 × 10-7)
|
OTES (1 × 10-8)
|
OTES (1 × 10-9)
|
OTES (1 × 10-10)
|
|
H
|
Blankprov (radioaktivitet) (*9)
|
Blankprov (radioaktivitet) (*9)
|
Buffertkontroll
|
Buffertkontroll
|
|
I detta exempel är den svaga bindaren noretinodrel (NE).
|
Tabell 5
Upplägg för mikrotiterplatta till kompetitivt bindningstest, fullständiga koncentrationskurvor för testkemikalier och plattkontroller
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
TB (endast lösningsmedel)
|
TB (endast lösningsmedel)
|
NSB
|
NSB
|
|
B
|
TC1 (1 × 10-3)
|
TC1 (1 × 10-4)
|
TC1 (1 × 10-5)
|
TC1 (1 × 10-6)
|
|
C
|
TC1 (1 × 10-7)
|
TC1 (1 × 10-8)
|
TC1 (1 × 10-9)
|
TC1 (1 × 10-10)
|
|
D
|
TC2 (1 × 10-3)
|
TC2 (1 × 10-4)
|
TC2 (1 × 10-5)
|
TC2 (1 × 10-6)
|
|
E
|
TC2 (1 × 10-7)
|
TC2 (1 × 10-8)
|
TC2 (1 × 10-9)
|
TC2 (1 × 10-10)
|
|
F
|
TC3 (1 × 10-3)
|
TC3 (1 × 10-4)
|
TC3 (1 × 10-5)
|
TC3 (1 × 10-6)
|
|
G
|
TC3 (1 × 10-7)
|
TC3 (1 × 10-8)
|
TC3 (1 × 10-9)
|
TC3 (1 × 10-10)
|
|
H
|
NE (IC50)
|
NE (1 × 10-4,5)
|
E2 (IC50)
|
E2 (1 × 10-7)
|
|
I detta exempel är den svaga bindaren noretinodrel (NE).
|
Slutförande av kompetitivt bindningstest
|
38.
|
Som visas i tabell 6, ska 80 μl av lösningsmedelskontrollen, buffertkontrollen, östrogenreferensen, den svaga bindaren, icke-bindaren och testkemikalierna som har beretts i testbuffert tillsättas till brunnarna. Sedan ska 40 μl av en 4 nM [3H]-17β-östradiollösning tillsättas till varje brunn. Efter mild rotation i 10–15 minuter vid 2–8 °C, ska 40 μl hrERα-lösning tillsättas. Testmikrotiterplattor bör inkuberas på roterande skak vid 2–8 °C i 16–20 timmar.
|
Tabell 6
Tillsatsvolymer av beståndsdelar till kompetitivt bindningstest för hrER, i mikrotiterplattor
|
Volym (μl)
|
Beståndsdel
|
|
80
|
Ej inmärkt 17β-östradiol, noretynodrel, OTES, testkemikalier, lösningsmedel eller buffert
|
|
40
|
4 nM [3H]-17β-östradiollösning
|
|
40
|
hrERα-lösning, enligt fastställd koncentration
|
|
160
|
Total volym i varje testbrunn
|
|
39.
|
Kvantifieringen av [3H]-17β-östradiol bundet till hrERα, efter separation av [3H]-17β-östradiol bundet till hrERα från fritt [3H]-17β-östradiol genom tillsats av 80 μl av kall DCC-suspension till varje brunn, bör sedan utföras enligt instruktioner för mättnadsbindningstestet, i punkterna 20–23.
|
|
40.
|
Brunnarna H1–6 (identifierade som blankprov [radioaktivitet] i tabell 4) anger dpm för [3H]-inmärkt östradiol i 40 μl. Dosen om 40 μl bör överföras direkt till scintillationsvätskan i brunnarna H1–H6.
|
Acceptanskriterier
Mättnadsbindningstest
|
41.
|
Den specifika bindningskurvan bör nå en platå med ökande koncentrationer av [3H]-17β-östradiol, indikerande mättnad av hrERα med ligand.
|
|
42.
|
Den specifika bindningen vid 1 nM av [3H]-17β-östradiol bör vara inom acceptansintervallet 15–25 % för genomsnittet av uppmätt total tillsatt radioaktivitet från de olika testomgångarna. Enstaka mindre avvikelser utanför detta intervall kan godtas, men om testomgångar konsekvent faller utanför intervallet eller om en viss omgång signifikant faller utanför intervallet bör proteinkoncentrationen justeras och mättnadstestet upprepas.
|
|
43.
|
Data bör bilda ett linjärt Scatchard-diagram.
|
|
44.
|
Den icke-specifika bindningen bör inte vara överdrivet stor. Storleken på den icke-specifika bindningen bör normalt vara < 35 % av den totala bindningen. Men kvoten kan emellanåt överskrida denna gräns vid mätningar av mycket låga dpm-tal för den lägsta koncentrationen av testad radioaktivt inmärkt 17β-östradiol.
|
Kompetitivt bindningstest
|
45.
|
Ökande koncentrationer av ej inmärkt 17β-östradiol bör tränga bort [3H]-17β-östradiol från receptorn på ett sätt som överensstämmer med kompetitiv bindning till en bindningsplats.
|
|
46.
|
Den hämmande koncentrationen, IC50, för referensöstrogenet (dvs. 17β-östradiol) bör ungefär motsvara den molära koncentrationen för [3H]-17β-östradiol plus Kd-värdet, som fastställts vid mättnadsbindningstestet.
|
|
47.
|
Den totala specifika bindningen bör genomgående hamna inom acceptansintervallet på 20 ± 5 % när den genomsnittliga uppmätta totala radioaktiva koncentrationen, som har tillsatts till varje brunn, har varit 1 nM vid flera testomgångar. Enstaka mindre avvikelser utanför detta intervall kan godtas, men om testomgångar konsekvent faller utanför intervallet eller om en viss omgång signifikant hamnar utanför intervallet bör proteinkoncentrationen justeras.
|
|
48.
|
Lösningsmedlet bör inte påverka vare sig testets känslighet eller reproducerbarhet. Resultaten för lösningsmedelskontrollen (TB-brunnar) jämförs med buffertkontrollen för att säkerställa att det valda lösningsmedlet inte påverkar testsystemet. Om lösningsmedlet inte har någon effekt på testet bör resultaten för TB och buffertkontroll vara jämförbara.
|
|
49.
|
Icke-bindaren bör inte tränga bort mer än 25 % av [3H]-17β-östradiol från hrERα när den testas upp till 10–3 M (OTES) eller 10–4 M (DBP).
|
|
50.
|
Prestandakriterier utarbetades för referensöstrogenet och två svaga bindare (t.ex. noretynodrel, noretindron) med hjälp av data från valideringsstudien av FW-hrER-bindningstestet (bilaga N till referens 2). I valideringsstudien uppges 95-procentiga konfidensintervall för genomsnittet (n) ± SD för deltagande laboratoriers alla kontrollomgångar. De 95-procentiga konfidensintervallen beräknades för kurvanpassningsparametrarna (dvs. topp, botten, Hill-koefficient [Hill slope], log[IC50]-värde) för referensöstrogenet och de svaga bindarna, och för log10RBA-värdet för de svaga bindarna jämförda med referensöstrogenet, och anges som prestandakriterier för de positiva kontrollerna. Tabell 1 anger förväntade intervall för kurvanpassningsparametrarna, vilka kan användas som prestandakriterier. I praktiken kan intervallet för IC50 variera något med Kd för receptorberedningen och ligandkoncentrationen.
|
|
51.
|
Inga prestandakriterier för testkemikaliernas kurvanpassningsparametrar utarbetades på grund av den stora mängden möjliga testkemikalier och deras variation i möjliga affiniteter och utfall (t.ex. fullständig kurva, delkurva, ingen kurvanpassning). Men yrkesmässigt omdöme ska tillämpas vid granskning av resultaten från en testkemikalies alla testomgångar. Vidare ska man använda ett tillräckligt intervall av testkemikaliekoncentration för att klart kunna beskriva toppen (t.ex. 90–100 % bindning) på den kompetitiva bindningskurvan. Variationen mellan replikat vid varje koncentration av testkemikalie, liksom mellan de tre oberoende testomgångarna, ska vara rimlig och vetenskapligt försvarbar. Kontroller från en testkemikalies alla testomgångar bör hålla sig nära de prestandamätningar som har rapporterats för detta FW-test och överensstämma med historiska kontrolldata från respektive laboratorium.
|
DATAANALYS
Mättnadsbindningstest
|
52.
|
Både total och icke-specifik bindning mäts. Från dessa värden beräknas specifik bindning av ökande koncentrationer av [3H]-17β-östradiol under jämviktsförhållanden genom att subtrahera icke-specifik från total. Ett diagram över specifik bindning mot [3H]-17β-östradiol-koncentration bör nå en platå för maximal specifik bindning, vilken indikerar ett hrERα mättat av [3H]-17β-östradiol. Dessutom bör dataanalysen klargöra inbindning av [3H]-17β-östradiol till en enda bindningsplats med hög affinitet. Icke-specifik, total och specifik bindning bör illustreras med en mättnadsbindningskurva. Vidare analys av dessa data bör använda sig av icke-linjär regressionsanalys (t.ex. BioSoft, McPherson, 1985, Motulsky, 1995) med en slutlig dataredovisning i form av ett Scatchard-diagram.
|
|
53.
|
Dataanalysen bör fastställa Bmax och Kd enbart utifrån totala bindningsdata, med antagandet att den icke-specifika bindningen är linjär, om inte en motivering ges för att använda en annan metod. Dessutom ska robust regression användas i fastställande av bästa anpassning, om inte annat val motiveras. Val av robust regressionsmetod ska anges. Korrigering för liganduttömning (t.ex. med hjälp av Swillens metod 1995) bör alltid användas i fastställande av Bmax och Kd utifrån mättnadsbindningsdata.
|
Kompetitivt bindningstest
|
54.
|
Kurvan över kompetitiv bindning ritas som specifik [3H]-17β-östradiol-bindning mot koncentrationen av konkurrerande ligand (log10-enheter). Den koncentration av testkemikalie som hämmar 50 % av den maximala specifika [3H]-17β-östradiol-bindningen är IC50-värdet.
|
|
55.
|
Uppskattningar av log(IC50)-värden för de positiva kontrollerna (t.ex. referensöstrogen och svag bindare) ska fastställas med hjälp av lämplig programvara för icke-linjär kurvanpassning så att de passar en fyraparameters Hill-ekvation (t.ex. BioSoft, McPherson, 1985, Motulsky, 1995). Kurvans topp, botten, lutning och log(IC50)-värde ska i allmänhet lämnas utan restriktion (unconstrained) vid anpassning av dessa kurvor. Robust regression ska användas i fastställande av bästa anpassning, om inte annat val motiveras. Ingen korrigering för liganduttömning ska göras. Efter inledande analys ska varje bindningskurva granskas för att säkerställa korrekt anpassning till modellen. Den svaga bindarens relativa bindningsaffinitet (RBA) beräknas som procentuell andel av dess log(IC50)-värde i förhållande till 17β-östradiols log(IC50)-värde. Resultat från de positiva kontrollerna och den icke-bindande kontrollen ska utvärderas genom att använda måtten på testprestanda i punkterna 45–50 i detta tillägg 2.
|
|
56.
|
Data för alla testkemikalier ska analyseras med en stegvis strategi för att säkerställa att data analyseras korrekt och att varje kurva för kompetitiv bindning klassificeras korrekt. Det är lämpligt att en testkemikalies alla testomgångar initialt underkastas en standardiserad dataanalys som är identisk med den som använts för referensöstrogen och kontroller med svaga bindare (se punkt 55 ovan). När den är klar bör en teknisk granskning av kurvanpassningsparametrarna göras, liksom en visuell granskning av hur väl data stämmer med den genererade kurvan över kompetitiv bindning, för varje testomgång. Under denna tekniska granskning är följande iakttagelser goda indikationer på att testet och dataanalyserna genomförts korrekt: en koncentrationsberoende minskning i procentuell, specifikt bunden [3H]-17β-östradiol, låg variation mellan de tekniska replikaten för varje kemisk koncentration och överensstämmelse mellan anpassningsparametrarna från de tre testomgångarna.
|
Tolkning av data
|
57.
|
Förutsatt att samtliga acceptanskriterier är uppfyllda anses en testkemikalie binda till hrERα om data kan anpassas till en bindningskurva och den lägsta punkten på responskurvan, inom dataintervallet, är mindre än 50 % (figur 1).
|
|
58.
|
Förutsatt att samtliga acceptanskriterier är uppfyllda, anses en testkemikalie inte binda till hrERα om
|
—
|
data kan anpassas till en bindningskurva och den lägsta punkten på responskurvan, inom dataintervallet, är över 75 %, eller
|
|
—
|
data inte kan anpassas till en bindningskurva och den lägsta, outjämnade genomsnittliga procentuella bindningen inom datamängdens koncentrationsgrupper är över 75 %.
|
|
|
59.
|
Testkemikalier anses tvetydiga om inget av villkoren ovan uppfylls (t.ex. den lägsta punkten på den anpassade responskurvan ligger mellan 76 % och 51 %).
|
Tabell 7
Kriterier för klassificering utifrån en testkemikalies kurva över kompetitiv bindning
|
Klassificering
|
Kriterier
|
|
Bindarea
|
Data kan anpassas till en bindningskurva.
Den lägsta punkten på responskurvan, inom dataintervallet, är mindre än 50 %.
|
|
Icke-bindareb
|
Om data kan anpassas till en bindningskurva och
den lägsta punkten på den anpassade responskurvan, inom dataintervallet, är över 75 %.
Om data inte kan anpassas till en bindningskurva och den
lägsta, outjämnade genomsnittliga procentuella bindningen inom datamängdens koncentrationsgrupper är över 75 %.
|
|
Tvetydigc
|
Varje mätbar testomgång som varken är en bindare eller en icke-bindare
(t.ex. den lägsta punkten längs den anpassade kurvan ligger mellan 76–51 %).
|
Figur 1
Exempel på klassificeringar av testkemikalier med hjälp av kurva för kompetitiv bindning
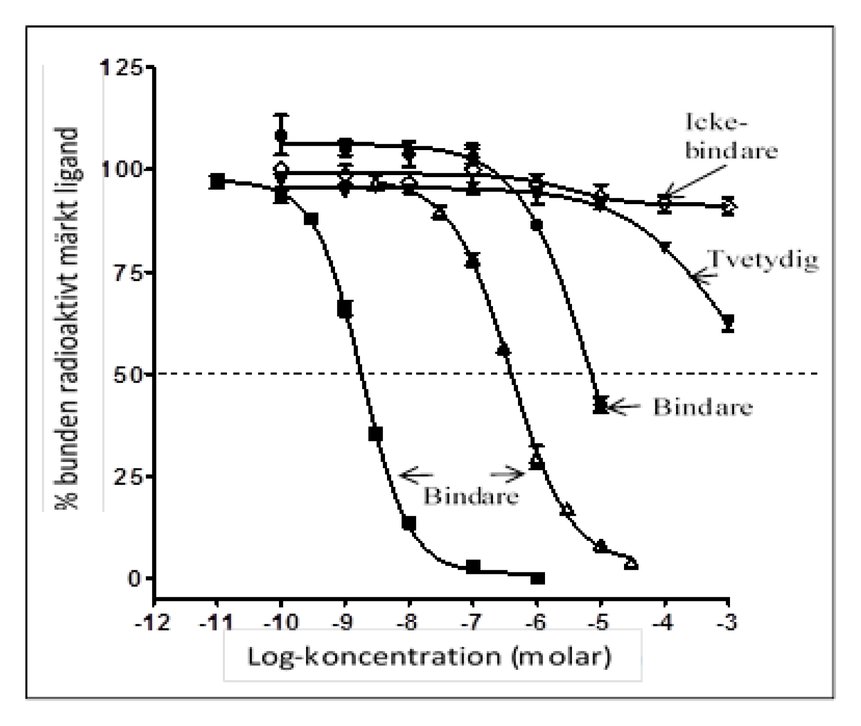
|
60.
|
Flera testomgångar genomförda med en viss testkemikalie, i samma laboratorium, kombineras genom att tilldela numeriska värden till varje omgång och ta fram ett medelvärde enligt tabell 8. Resultaten för de kombinerade testomgångarna inom varje laboratorium jämförs med den förväntade klassificeringen för varje testkemikalie.
|
Tabell 8
Metod för klassificering av testkemikalier med användning av flera testomgångar inom ett laboratorium
|
Tilldelning av numeriska värden för enskilda testomgångar:
|
|
|
Klassificering
|
Numeriskt värde
|
|
Bindare
|
2
|
|
Tvetydig
|
1
|
|
Icke-bindare
|
0
|
|
Klassificering av genomsnittligt numeriskt värde vid flera testomgångar:
|
|
|
Klassificering
|
Numeriskt värde
|
|
Bindare
|
Genomsnitt ≥ 1,5
|
|
Tvetydig
|
0,5 ≤ genomsnitt < 1,5
|
|
Icke-bindare
|
Genomsnitt < 0,5
|
TESTRAPPORT
|
61.
|
Se punkt 24 i ”hrER-BINDNINGSTESTETS DELAR” i denna testmetod.
|
Tillägg 2.1
TERMER
[3H]E2: 17β-östradiol radioaktivt inmärkt med tritium.
DCC: Dextran-täckt träkol (Dextran-Coated Charcoal).
E2: Ej inmärkt 17β-östradiol (inert).
Testbuffert: 10 mM tris, 10 mg/ml bovint serumalbumin, 2 mM DTT, 10 % glycerol, 0,2 mM leupeptin, pH 7,5.
hrERα: Human rekombinant östrogenreceptor alfa.
Replikat: En av flera brunnar med samma innehåll och koncentrationer vilka testas parallellt inom en enskild testomgång. I detta protokoll testas varje koncentration av testkemikalie i tripletter, det betyder att tre replikat testas samtidigt vid varje koncentration av testkemikalie.
Testomgång: En fullständig uppsättning av parallellt hanterade brunnar i mikrotiterplattor, vilka ger all nödvändig information för att karakterisera en testkemikalies bindning till hrERα (dvs. totalt [3H]-17β-östradiol tillsatt till testbrunnen, maximal bindning hos [3H]-17β-östradiol till hrERα, icke-specifik bindning och total bindning vid olika koncentrationer av testkemikalie). En testomgång kan bestå av så lite som en testbrunn (dvs. ett replikat) per koncentration, men eftersom protokollet kräver tripletter, består en testomgång av tre testbrunnar per koncentration. Dessutom kräver detta protokoll tre oberoende (dvs. icke-parallella) testomgångar per kemikalie.
Tillägg 2.2
REPRESENTATIVT [3H]-17Β-ÖSTRADIOL-MÄTTNADSTEST MED REPLIKAT I TRE BRUNNAR
|
Representativt [3H]-17β-östradiol-mättnadstest med replikat i tre brunnar
|
|
Position
|
Replikat
|
Kod för brunnstyp
|
Initial radioaktiv E2-koncentration (nM)
|
Radioaktiv E2-volym (μl)
|
Slutlig radioaktiv E2-koncentration (nM)
|
Initial icke-radioaktiv E2-koncentration (μM)
|
Icke-radioaktiv E2-volym (μl)
|
Slutlig icke-radioaktiv E2-koncentration (μM)
|
Buffertvolym (μl)
|
Receptorvolym (μl)
|
Total volym i brunnarna (μl)
|
|
A1
|
1
|
H
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A2
|
2
|
H
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A3
|
3
|
H
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A4
|
1
|
H
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A5
|
2
|
H
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A6
|
3
|
H
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A7
|
1
|
H
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A8
|
2
|
H
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A9
|
3
|
H
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A10
|
1
|
H
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A11
|
2
|
H
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A12
|
3
|
H
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B1
|
1
|
H
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B2
|
2
|
H
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B3
|
3
|
H
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B4
|
1
|
H
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B5
|
2
|
H
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B6
|
3
|
H
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B7
|
1
|
H
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B8
|
2
|
H
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B9
|
3
|
H
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B10
|
1
|
H
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B11
|
2
|
H
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B12
|
3
|
H
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
D1
|
1
|
HC
|
0,12
|
40
|
0,03
|
0,06
|
80
|
0,03
|
—
|
40
|
160
|
|
D2
|
2
|
HC
|
0,12
|
40
|
0,03
|
0,06
|
80
|
0,03
|
—
|
40
|
160
|
|
D3
|
3
|
HC
|
0,12
|
40
|
0,03
|
0,06
|
80
|
0,03
|
—
|
40
|
160
|
|
D4
|
1
|
HC
|
0,24
|
40
|
0,06
|
0,12
|
80
|
0,06
|
—
|
40
|
160
|
|
D5
|
2
|
HC
|
0,24
|
40
|
0,06
|
0,12
|
80
|
0,06
|
—
|
40
|
160
|
|
D6
|
3
|
HC
|
0,24
|
40
|
0,06
|
0,12
|
80
|
0,06
|
—
|
40
|
160
|
|
D7
|
1
|
HC
|
0,32
|
40
|
0,08
|
0,16
|
80
|
0,08
|
—
|
40
|
160
|
|
D8
|
2
|
HC
|
0,32
|
40
|
0,08
|
0,16
|
80
|
0,08
|
—
|
40
|
160
|
|
D9
|
3
|
HC
|
0,32
|
40
|
0,08
|
0,16
|
80
|
0,08
|
—
|
40
|
160
|
|
D10
|
1
|
HC
|
0,40
|
40
|
0,10
|
0,2
|
80
|
0,1
|
—
|
40
|
160
|
|
D11
|
2
|
HC
|
0,40
|
40
|
0,10
|
0,2
|
80
|
0,1
|
—
|
40
|
160
|
|
D12
|
3
|
HC
|
0,40
|
40
|
0,10
|
0,2
|
80
|
0,1
|
—
|
40
|
160
|
|
E1
|
1
|
HC
|
1,20
|
40
|
0,30
|
0,6
|
80
|
0,3
|
—
|
40
|
160
|
|
E2
|
2
|
HC
|
1,20
|
40
|
0,30
|
0,6
|
80
|
0,3
|
—
|
40
|
160
|
|
E3
|
3
|
HC
|
1,20
|
40
|
0,30
|
0,6
|
80
|
0,3
|
—
|
40
|
160
|
|
E4
|
1
|
HC
|
2,40
|
40
|
0,60
|
1,2
|
80
|
0,6
|
—
|
40
|
160
|
|
E5
|
2
|
HC
|
2,40
|
40
|
0,60
|
1,2
|
80
|
0,6
|
—
|
40
|
160
|
|
E6
|
3
|
HC
|
2,40
|
40
|
0,60
|
1,2
|
80
|
0,6
|
—
|
40
|
160
|
|
E7
|
1
|
HC
|
4,00
|
40
|
1,00
|
2
|
80
|
1
|
—
|
40
|
160
|
|
E8
|
2
|
HC
|
4,00
|
40
|
1,00
|
2
|
80
|
1
|
—
|
40
|
160
|
|
E9
|
3
|
HC
|
4,00
|
40
|
1,00
|
2
|
80
|
1
|
—
|
40
|
160
|
|
E10
|
1
|
HC
|
12,00
|
40
|
3,00
|
6
|
80
|
3
|
—
|
40
|
160
|
|
E11
|
2
|
HC
|
12,00
|
40
|
3,00
|
6
|
80
|
3
|
—
|
40
|
160
|
|
E12
|
3
|
HC
|
12,00
|
40
|
3,00
|
6
|
80
|
3
|
—
|
40
|
160
|
|
G1
|
1
|
Radioaktiv
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G2
|
2
|
Radioaktiv
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G3
|
3
|
Radioaktiv
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G4
|
1
|
Radioaktiv
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G5
|
2
|
Radioaktiv
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G6
|
3
|
Radioaktiv
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G7
|
1
|
Radioaktiv
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G8
|
2
|
Radioaktiv
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G9
|
3
|
Radioaktiv
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G10
|
1
|
Radioaktiv
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G11
|
2
|
Radioaktiv
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G12
|
3
|
Radioaktiv
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H1
|
1
|
Radioaktiv
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H2
|
2
|
Radioaktiv
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H3
|
3
|
Radioaktiv
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H4
|
1
|
Radioaktiv
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H5
|
2
|
Radioaktiv
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H6
|
3
|
Radioaktiv
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H7
|
1
|
Radioaktiv
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H8
|
2
|
Radioaktiv
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H9
|
3
|
Radioaktiv
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H10
|
1
|
Radioaktiv
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H11
|
2
|
Radioaktiv
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H12
|
3
|
Radioaktiv
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
Obs: brunnarna som märkts med ”radioaktiv” är tomma under inkubationen. Dessa 40 μl tillsätts endast i scintillationsräkningssyfte.
|
Tillägg 2.3
UPPLÄGG FÖR BRUNNAR TILL TEST AV KOMPETITIV BINDNING
|
Platta
|
Position
|
Replikat
|
Brunnstyp
|
Brunnskod
|
Koncentrationskod
|
Inledande koncentr. av konkurrerande ligand (M)
|
hrER-stamlösning (μl)
|
Buffertvolym (μl)
|
Volym spårsubstans (radioaktiv E2) (μl)
|
Volym från spädningsplatta (μl)
|
Slutvolym (μl)
|
Slutkoncentration av konkurrerande ligand (M)
|
|
Fast
|
A1
|
1
|
Total bindning
|
TB
|
TB1
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
Fast
|
A2
|
2
|
Total bindning
|
TB
|
TB2
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
Fast
|
A3
|
3
|
Total bindning
|
TB
|
TB3
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
Fast
|
A4
|
1
|
Total bindning
|
TB
|
TB4
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
Fast
|
A5
|
2
|
Total bindning
|
TB
|
TB5
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
Fast
|
A6
|
3
|
Total bindning
|
TB
|
TB6
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
Fast
|
A7
|
1
|
Icke-radioaktiv E2 (hög)
|
NSB
|
S0
|
2,00 × 10-6
|
40
|
—
|
40
|
80
|
160
|
1,0 × 10-6
|
|
Fast
|
A8
|
2
|
Icke-radioaktiv E2 (hög)
|
NSB
|
S0
|
2,00 × 10-6
|
40
|
—
|
40
|
80
|
160
|
1,0 × 10-6
|
|
Fast
|
A9
|
3
|
Icke-radioaktiv E2 (hög)
|
NSB
|
S0
|
2,00 × 10-6
|
40
|
—
|
40
|
80
|
160
|
1,0 × 10-6
|
|
Fast
|
A10
|
1
|
Icke-radioaktiv E2 (hög)
|
NSB
|
S0
|
2,00 × 10-6
|
40
|
—
|
40
|
80
|
160
|
1,0 × 10-6
|
|
Fast
|
A11
|
2
|
Icke-radioaktiv E2 (hög)
|
NSB
|
S0
|
2,00 × 10-6
|
40
|
—
|
40
|
80
|
160
|
1,0 × 10-6
|
|
Fast
|
A12
|
3
|
Icke-radioaktiv E2 (hög)
|
NSB
|
S0
|
2,00 × 10-6
|
40
|
—
|
40
|
80
|
160
|
1,0 × 10-6
|
|
Fast
|
B1
|
1
|
Icke-radioaktiv E2
|
Fast
|
S1
|
2,00 × 10-7
|
40
|
—
|
40
|
80
|
160
|
1,0 × 10-7
|
|
Fast
|
B2
|
2
|
Icke-radioaktiv E2
|
Fast
|
S1
|
2,00 × 10-7
|
40
|
—
|
40
|
80
|
160
|
1,0 × 10-7
|
|
Fast
|
B3
|
3
|
Icke-radioaktiv E2
|
Fast
|
S1
|
2,00 × 10-7
|
40
|
—
|
40
|
80
|
160
|
1,0 × 10-7
|
|
Fast
|
B4
|
1
|
Icke-radioaktiv E2
|
Fast
|
S2
|
2,00 × 10-8
|
40
|
—
|
40
|
80
|
160
|
1,0 × 10-8
|
|
Fast
|
B5
|
2
|
Icke-radioaktiv E2
|
Fast
|
S2
|
2,00 × 10-8
|
40
|
—
|
40
|
80
|
160
|
1,0 × 10-8
|
|
Fast
|
B6
|
3
|
Icke-radioaktiv E2
|
Fast
|
S2
|
2,00 × 10-8
|
40
|
—
|
40
|
80
|
160
|
1,0 × 10-8
|
|
Fast
|
B7
|
1
|
Icke-radioaktiv E2
|
Fast
|
S3
|
6,00 × 10-9
|
40
|
—
|
40
|
80
|
160
|
3,0 × 10-9
|
|
Fast
|
B8
|
2
|
Icke-radioaktiv E2
|
Fast
|
S3
|
6,00 × 10-9
|
40
|
—
|
40
|
80
|
160
|
3,0 × 10-9
|
|
Fast
|
B9
|
3
|
Icke-radioaktiv E2
|
Fast
|
S3
|
6,00 × 10-9
|
40
|
—
|
40
|
80
|
160
|
3,0 × 10-9
|
|
Fast
|
B10
|
1
|
Icke-radioaktiv E2
|
Fast
|
S4
|
2,00 × 10-9
|
40
|
—
|
40
|
80
|
160
|
1,0 × 10-9
|
|
Fast
|
B11
|
2
|
Icke-radioaktiv E2
|
Fast
|
S4
|
2,00 × 10-9
|
40
|
—
|
40
|
80
|
160
|
1,0 × 10-9
|
|
Fast
|
B12
|
3
|
Icke-radioaktiv E2
|
Fast
|
S4
|
2,00 × 10-9
|
40
|
—
|
40
|
80
|
160
|
1,0 × 10-9
|
|
Fast
|
C1
|
1
|
Icke-radioaktiv E2
|
Fast
|
S5
|
6,00 × 10-10
|
40
|
—
|
40
|
80
|
160
|
3,0 × 10-10
|
|
Fast
|
C2
|
2
|
Icke-radioaktiv E2
|
Fast
|
S5
|
6,00 × 10-10
|
40
|
—
|
40
|
80
|
160
|
3,0 × 10-10
|
|
Fast
|
C3
|
3
|
Icke-radioaktiv E2
|
Fast
|
S5
|
6,00 × 10-10
|
40
|
—
|
40
|
80
|
160
|
3,0 × 10-10
|
|
Fast
|
C4
|
1
|
Icke-radioaktiv E2
|
Fast
|
S6
|
2,00 × 10-10
|
40
|
—
|
40
|
80
|
160
|
1,0 × 10-10
|
|
Fast
|
C5
|
2
|
Icke-radioaktiv E2
|
Fast
|
S6
|
2,00 × 10-10
|
40
|
—
|
40
|
80
|
160
|
1,0 × 10-10
|
|
Fast
|
C6
|
3
|
Icke-radioaktiv E2
|
Fast
|
S6
|
2,00 × 10-10
|
40
|
—
|
40
|
80
|
160
|
1,0 × 10-10
|
|
Fast
|
C7
|
1
|
Icke-radioaktiv E2
|
Fast
|
S7
|
2,00 × 10-11
|
40
|
—
|
40
|
80
|
160
|
1,0 × 10-11
|
|
Fast
|
C8
|
2
|
Icke-radioaktiv E2
|
Fast
|
S7
|
2,00 × 10-11
|
40
|
—
|
40
|
80
|
160
|
1,0 × 10-11
|
|
Fast
|
C9
|
3
|
Icke-radioaktiv E2
|
Fast
|
S7
|
2,00 × 10-11
|
40
|
—
|
40
|
80
|
160
|
1,0 × 10-11
|
|
Fast
|
C10
|
1
|
Blankprov
|
Blankprov
|
B1
|
—
|
—
|
160
|
—
|
—
|
160
|
—
|
|
Fast
|
C11
|
2
|
Blankprov
|
Blankprov
|
B2
|
—
|
—
|
160
|
—
|
—
|
160
|
—
|
|
Fast
|
C12
|
3
|
Blankprov
|
Blankprov
|
B3
|
—
|
—
|
160
|
—
|
—
|
160
|
—
|
|
Fast
|
D1
|
1
|
Noretynodrel
|
NE
|
WP1
|
6,00 × 10-5
|
40
|
—
|
40
|
80
|
160
|
3,0 × 10-5
|
|
Fast
|
D2
|
1
|
Noretynodrel
|
NE
|
WP1
|
6,00 × 10-5
|
40
|
—
|
40
|
80
|
160
|
3,0 × 10-5
|
|
Fast
|
D3
|
1
|
Noretynodrel
|
NE
|
WP1
|
6,00 × 10-5
|
40
|
—
|
40
|
80
|
160
|
3,0 × 10-5
|
|
Fast
|
D4
|
1
|
Noretynodrel
|
NE
|
WP2
|
2,00 × 10-5
|
40
|
—
|
40
|
80
|
160
|
1,0 × 10-5
|
|
Fast
|
D5
|
1
|
Noretynodrel
|
NE
|
WP2
|
2,00 × 10-5
|
40
|
—
|
40
|
80
|
160
|
1,0 × 10-5
|
|
Fast
|
D6
|
1
|
Noretynodrel
|
NE
|
WP2
|
2,00 × 10-5
|
40
|
—
|
40
|
80
|
160
|
1,0 × 10-5
|
|
Fast
|
D7
|
1
|
Noretynodrel
|
NE
|
WP3
|
6,00 × 10-6
|
40
|
—
|
40
|
80
|
160
|
3,0 × 10-6
|
|
Fast
|
D8
|
1
|
Noretynodrel
|
NE
|
WP3
|
6,00 × 10-6
|
40
|
—
|
40
|
80
|
160
|
3,0 × 10-6
|
|
Fast
|
D9
|
1
|
Noretynodrel
|
NE
|
WP3
|
6,00 × 10-6
|
40
|
—
|
40
|
80
|
160
|
3,0 × 10-6
|
|
Fast
|
D10
|
1
|
Noretynodrel
|
NE
|
WP4
|
2,00 × 10-6
|
40
|
—
|
40
|
80
|
160
|
1,0 × 10-6
|
|
Fast
|
D11
|
1
|
Noretynodrel
|
NE
|
WP4
|
2,00 × 10-6
|
40
|
—
|
40
|
80
|
160
|
1,0 × 10-6
|
|
Fast
|
D12
|
1
|
Noretynodrel
|
NE
|
WP4
|
2,00 × 10-6
|
40
|
—
|
40
|
80
|
160
|
1,0 × 10-6
|
|
Fast
|
E1
|
1
|
Noretynodrel
|
NE
|
WP
|
6,00 × 10-7
|
40
|
|
40
|
80
|
160
|
3,0 × 10-7
|
|
Fast
|
E2
|
2
|
Noretynodrel
|
NE
|
WP
|
6,00 × 10-7
|
40
|
|
40
|
80
|
160
|
3,0 × 10-7
|
|
Fast
|
E3
|
3
|
Noretynodrel
|
NE
|
WP
|
6,00 × 10-7
|
40
|
|
40
|
80
|
160
|
3,0 × 10-7
|
|
Fast
|
E4
|
1
|
Noretynodrel
|
NE
|
WP
|
2,00 × 10-7
|
40
|
|
40
|
80
|
160
|
1,0 × 10-7
|
|
Fast
|
E5
|
2
|
Noretynodrel
|
NE
|
WP
|
2,00 × 10-7
|
40
|
|
40
|
80
|
160
|
1,0 × 10-7
|
|
Fast
|
E6
|
3
|
Noretynodrel
|
NE
|
WP
|
2,00 × 10-7
|
40
|
|
40
|
80
|
160
|
1,0 × 10-7
|
|
Fast
|
E7
|
1
|
Noretynodrel
|
NE
|
WP
|
6,00 × 10-8
|
40
|
—
|
40
|
80
|
160
|
3,0 × 10-8
|
|
Fast
|
E8
|
2
|
Noretynodrel
|
NE
|
WP
|
6,00 × 10-8
|
40
|
—
|
40
|
80
|
160
|
3,0 × 10-8
|
|
Fast
|
E9
|
3
|
Noretynodrel
|
NE
|
WP
|
6,00 × 10-8
|
40
|
—
|
40
|
80
|
160
|
3,0 × 10-8
|
|
Fast
|
E10
|
1
|
Noretynodrel
|
NE
|
WP
|
6,00 × 10-9
|
40
|
—
|
40
|
80
|
160
|
3,0 × 10-9
|
|
Fast
|
E11
|
2
|
Noretynodrel
|
NE
|
WP
|
6,00 × 10-9
|
40
|
—
|
40
|
80
|
160
|
3,0 × 10-9
|
|
Fast
|
E12
|
3
|
Noretynodrel
|
NE
|
WP
|
6,00 × 10-9
|
40
|
—
|
40
|
80
|
160
|
3,0 × 10-9
|
|
Fast
|
F1
|
1
|
OTES
|
N
|
OTES
|
2,00 × 10-3
|
40
|
—
|
40
|
80
|
160
|
1,0 × 10-3
|
|
Fast
|
F2
|
2
|
OTES
|
N
|
OTES
|
2,00 × 10-3
|
40
|
—
|
40
|
80
|
160
|
1,0 × 10-3
|
|
Fast
|
F3
|
3
|
OTES
|
N
|
OTES
|
2,00 × 10-3
|
40
|
—
|
40
|
80
|
160
|
1,0 × 10-3
|
|
Fast
|
F4
|
1
|
OTES
|
N
|
OTES
|
2,00 × 10-4
|
40
|
—
|
40
|
80
|
160
|
1,0 × 10-4
|
|
Fast
|
F5
|
2
|
OTES
|
N
|
OTES
|
2,00 × 10-4
|
40
|
—
|
40
|
80
|
160
|
1,0 × 10-4
|
|
Fast
|
F6
|
3
|
OTES
|
N
|
OTES
|
2,00 × 10-4
|
40
|
—
|
40
|
80
|
160
|
1,0 × 10-4
|
|
Fast
|
F7
|
1
|
OTES
|
N
|
OTES
|
2,00 × 10-5
|
40
|
—
|
40
|
80
|
160
|
3,0 × 10-5
|
|
Fast
|
F8
|
2
|
OTES
|
N
|
OTES
|
2,00 × 10-5
|
40
|
—
|
40
|
80
|
160
|
3,0 × 10-5
|
|
Fast
|
F9
|
3
|
OTES
|
N
|
OTES
|
2,00 × 10-5
|
40
|
—
|
40
|
80
|
160
|
3,0 × 10-5
|
|
Fast
|
F10
|
1
|
OTES
|
N
|
OTES
|
2,00 × 10-6
|
40
|
—
|
40
|
80
|
160
|
1,0 × 10-6
|
|
Fast
|
F11
|
2
|
OTES
|
N
|
OTES
|
2,00 × 10-6
|
40
|
—
|
40
|
80
|
160
|
1,0 × 10-6
|
|
Fast
|
F12
|
3
|
OTES
|
N
|
OTES
|
2,00 × 10-6
|
40
|
—
|
40
|
80
|
160
|
1,0 × 10-6
|
|
Fast
|
G1
|
1
|
OTES
|
N
|
OTES
|
2,00 × 10-7
|
40
|
—
|
40
|
80
|
160
|
3,0 × 10-7
|
|
Fast
|
G2
|
2
|
OTES
|
N
|
OTES
|
2,00 × 10-7
|
40
|
—
|
40
|
80
|
160
|
3,0 × 10-7
|
|
Fast
|
G3
|
3
|
OTES
|
N
|
OTES
|
2,00 × 10-7
|
40
|
—
|
40
|
80
|
160
|
3,0 × 10-7
|
|
Fast
|
G4
|
1
|
OTES
|
N
|
OTES
|
2,00 × 10-8
|
40
|
—
|
40
|
80
|
160
|
1,0 × 10-8
|
|
Fast
|
G5
|
2
|
OTES
|
N
|
OTES
|
2,00 × 10-8
|
40
|
—
|
40
|
80
|
160
|
1,0 × 10-8
|
|
Fast
|
G6
|
3
|
OTES
|
N
|
OTES
|
2,00 × 10-8
|
40
|
—
|
40
|
80
|
160
|
1,0 × 10-8
|
|
Fast
|
G7
|
1
|
OTES
|
N
|
OTES
|
2,00 × 10-9
|
40
|
—
|
40
|
80
|
160
|
1,0 × 10-9
|
|
Fast
|
G8
|
2
|
OTES
|
N
|
OTES
|
2,00 × 10-9
|
40
|
—
|
40
|
80
|
160
|
1,0 × 10-9
|
|
Fast
|
G9
|
3
|
OTES
|
N
|
OTES
|
2,00 × 10-9
|
40
|
—
|
40
|
80
|
160
|
1,0 × 10-9
|
|
Fast
|
G10
|
1
|
OTES
|
N
|
OTES
|
2,00 × 10-10
|
40
|
—
|
40
|
—
|
160
|
1,0 × 10-10
|
|
Fast
|
G11
|
2
|
OTES
|
N
|
OTES
|
2,00 × 10-10
|
40
|
—
|
40
|
—
|
160
|
1,0 × 10-10
|
|
Fast
|
G12
|
3
|
OTES
|
N
|
OTES
|
2,00 × 10-10
|
40
|
—
|
40
|
—
|
160
|
1,0 × 10-10
|
|
Fast
|
H1
|
1
|
Radioaktiv
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
Fast
|
H2
|
1
|
Radioaktiv
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
Fast
|
H3
|
1
|
Radioaktiv
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
Fast
|
H4
|
1
|
Radioaktiv
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
Fast
|
H5
|
1
|
Radioaktiv
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
Fast
|
H6
|
1
|
Radioaktiv
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
Fast
|
H7
|
1
|
Buffertkontroll
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
Fast
|
H8
|
1
|
Buffertkontroll
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
Fast
|
H9
|
1
|
Buffertkontroll
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
Fast
|
H10
|
1
|
Buffertkontroll
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
Fast
|
H11
|
1
|
Buffertkontroll
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
Fast
|
H12
|
1
|
Buffertkontroll
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
Obs: brunnarna som märkts med ”radioaktiv” är tomma under inkubationen. Dessa 40 μl tillsätts endast i scintillationsräkningssyfte.
|
Upplägg för brunnar till test av kompetitiv bindning
|
|
Platta
|
Position
|
Replikat
|
Brunnstyp
|
Brunnskod
|
Koncentrationskod
|
Inledande koncentr. av konkurrerande ligand (M)
|
hrER-stamlösning (μl)
|
Buffertvolym (μl)
|
Volym spårsubstans (radioaktiv E2) (μl)
|
Volym från spädningsplatta (μl)
|
Slutvolym (μl)
|
Slutkoncentration av konkurrerande ligand (M)
|
|
P1
|
A1
|
1
|
Total bindning
|
TB
|
TBB1B1
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A2
|
2
|
Total bindning
|
TB
|
TB2
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A3
|
3
|
Total bindning
|
TB
|
TB3
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A4
|
1
|
Total bindning
|
TB
|
TB4
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A5
|
2
|
Total bindning
|
TB
|
TB5
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A6
|
3
|
Total bindning
|
TB
|
TB6
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A7
|
1
|
Icke-radioaktiv E2 (hög)
|
NSB
|
S0
|
2,00 × 10-6
|
40
|
—
|
40
|
80
|
160
|
1,0 × 10-6
|
|
P1
|
A8
|
2
|
Icke-radioaktiv E2 (hög)
|
NSB
|
S0
|
2,00 × 10-6
|
40
|
—
|
40
|
80
|
160
|
1,0 × 10-6
|
|
P1
|
A9
|
3
|
Icke-radioaktiv E2 (hög)
|
NSB
|
S0
|
2,00 × 10-6
|
40
|
—
|
40
|
80
|
160
|
1,0 × 10-6
|
|
P1
|
A10
|
1
|
Icke-radioaktiv E2 (hög)
|
NSB
|
S0
|
2,00 × 10-6
|
40
|
—
|
40
|
80
|
160
|
1,0 × 10-6
|
|
P1
|
A11
|
2
|
Icke-radioaktiv E2 (hög)
|
NSB
|
S0
|
2,00 × 10-6
|
40
|
—
|
40
|
80
|
160
|
1,0 × 10-6
|
|
P1
|
A12
|
3
|
Icke-radioaktiv E2 (hög)
|
NSB
|
S0
|
2,00 × 10-6
|
40
|
—
|
40
|
80
|
160
|
1,0 × 10-6
|
|
P1
|
B1
|
1
|
Testkemikalie 1
|
TC1
|
1
|
2,00 × 10-3
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-3
|
|
P1
|
B2
|
2
|
Testkemikalie 1
|
TC1
|
1
|
2,00 × 10-3
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-3
|
|
P1
|
B3
|
3
|
Testkemikalie 1
|
TC1
|
1
|
2,00 × 10-3
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-3
|
|
P1
|
B4
|
1
|
Testkemikalie 1
|
TC1
|
2
|
2,00 × 10-4
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-4
|
|
P1
|
B5
|
2
|
Testkemikalie 1
|
TC1
|
2
|
2,00 × 10-4
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-4
|
|
P1
|
B6
|
3
|
Testkemikalie 1
|
TC1
|
2
|
2,00 × 10-4
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-4
|
|
P1
|
B7
|
1
|
Testkemikalie 1
|
TC1
|
3
|
2,00 × 10-5
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-5
|
|
P1
|
B8
|
2
|
Testkemikalie 1
|
TC1
|
3
|
2,00 × 10-5
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-5
|
|
P1
|
B9
|
3
|
Testkemikalie 1
|
TC1
|
3
|
2,00 × 10-5
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-5
|
|
P1
|
B10
|
1
|
Testkemikalie 1
|
TC1
|
4
|
2,00 × 10-6
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-6
|
|
P1
|
B11
|
2
|
Testkemikalie 1
|
TC1
|
4
|
2,00 × 10-6
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-6
|
|
P1
|
B12
|
3
|
Testkemikalie 1
|
TC1
|
4
|
2,00 × 10-6
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-6
|
|
P1
|
C1
|
1
|
Testkemikalie 1
|
TC1
|
5
|
2,00 × 10-7
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-7
|
|
P1
|
C2
|
2
|
Testkemikalie 1
|
TC1
|
5
|
2,00 × 10-7
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-7
|
|
P1
|
C3
|
3
|
Testkemikalie 1
|
TC1
|
5
|
2,00 × 10-7
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-7
|
|
P1
|
C4
|
1
|
Testkemikalie 1
|
TC1
|
6
|
2,00 × 10-8
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-8
|
|
P1
|
C5
|
2
|
Testkemikalie 1
|
TC1
|
6
|
2,00 × 10-8
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-8
|
|
P1
|
C6
|
3
|
Testkemikalie 1
|
TC1
|
6
|
2,00 × 10-8
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-8
|
|
P1
|
C7
|
1
|
Testkemikalie 1
|
TC1
|
7
|
2,00 × 10-9
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-9
|
|
P1
|
C8
|
2
|
Testkemikalie 1
|
TC1
|
7
|
2,00 × 10-9
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-9
|
|
P1
|
C9
|
3
|
Testkemikalie 1
|
TC1
|
7
|
2,00 × 10-9
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-9
|
|
P1
|
C10
|
1
|
Testkemikalie 1
|
TC1
|
8
|
2,00 × 10-10
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-10
|
|
P1
|
C11
|
2
|
Testkemikalie 1
|
TC1
|
8
|
2,00 × 10-10
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-10
|
|
P1
|
C12
|
3
|
Testkemikalie 1
|
TC1
|
8
|
2,00 × 10-10
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-10
|
|
P1
|
D1
|
1
|
Testkemikalie 2
|
TC2
|
1
|
2,00 × 10-3
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-3
|
|
P1
|
D2
|
2
|
Testkemikalie 2
|
TC2
|
1
|
2,00 × 10-3
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-3
|
|
P1
|
D3
|
3
|
Testkemikalie 2
|
TC2
|
1
|
2,00 × 10-3
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-3
|
|
P1
|
D4
|
1
|
Testkemikalie 2
|
TC2
|
2
|
2,00 × 10-4
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-4
|
|
P1
|
D5
|
2
|
Testkemikalie 2
|
TC2
|
2
|
2,00 × 10-4
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-4
|
|
P1
|
D6
|
3
|
Testkemikalie 2
|
TC2
|
2
|
2,00 × 10-4
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-4
|
|
P1
|
D7
|
1
|
Testkemikalie 2
|
TC2
|
3
|
2,00 × 10-5
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-5
|
|
P1
|
D8
|
2
|
Testkemikalie 2
|
TC2
|
3
|
2,00 × 10-5
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-5
|
|
P1
|
D9
|
3
|
Testkemikalie 2
|
TC2
|
3
|
2,00 × 10-5
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-5
|
|
P1
|
D10
|
1
|
Testkemikalie 2
|
TC2
|
4
|
2,00 × 10-6
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-6
|
|
P1
|
D11
|
2
|
Testkemikalie 2
|
TC2
|
4
|
2,00 × 10-6
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-6
|
|
P1
|
D12
|
3
|
Testkemikalie 2
|
TC2
|
4
|
2,00 × 10-6
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-6
|
|
P1
|
E1
|
1
|
Testkemikalie 2
|
TC2
|
5
|
2,00 × 10-7
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-7
|
|
P1
|
E2
|
2
|
Testkemikalie 2
|
TC2
|
5
|
2,00 × 10-7
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-7
|
|
P1
|
E3
|
3
|
Testkemikalie 2
|
TC2
|
5
|
2,00 × 10-7
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-7
|
|
P1
|
E4
|
1
|
Testkemikalie 2
|
TC2
|
6
|
—
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-8
|
|
P1
|
E5
|
2
|
Testkemikalie 2
|
TC2
|
6
|
—
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-8
|
|
P1
|
E6
|
3
|
Testkemikalie 2
|
TC2
|
6
|
—
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-8
|
|
P1
|
E7
|
1
|
Testkemikalie 2
|
TC2
|
7
|
2,00 × 10-6
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-9
|
|
P1
|
E8
|
2
|
Testkemikalie 2
|
TC2
|
7
|
2,00 × 10-6
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-9
|
|
P1
|
E9
|
3
|
Testkemikalie 2
|
TC2
|
7
|
2,00 × 10-6
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-9
|
|
P1
|
E10
|
1
|
Testkemikalie 2
|
TC2
|
8
|
2,00 × 10-6
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-10
|
|
P1
|
E11
|
2
|
Testkemikalie 2
|
TC2
|
8
|
2,00 × 10-6
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-10
|
|
P1
|
E12
|
3
|
Testkemikalie 2
|
TC2
|
8
|
2,00 × 10-6
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-10
|
|
P1
|
F1
|
1
|
Testkemikalie 3
|
TC3
|
1
|
2,00 × 10-3
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-3
|
|
P1
|
F2
|
2
|
Testkemikalie 3
|
TC3
|
1
|
2,00 × 10-3
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-3
|
|
P1
|
F3
|
3
|
Testkemikalie 3
|
TC3
|
1
|
2,00 × 10-3
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-3
|
|
P1
|
F4
|
1
|
Testkemikalie 3
|
TC3
|
2
|
2,00 × 10-4
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-4
|
|
P1
|
F5
|
2
|
Testkemikalie 3
|
TC3
|
2
|
2,00 × 10-4
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-4
|
|
P1
|
F6
|
3
|
Testkemikalie 3
|
TC3
|
2
|
2,00 × 10-4
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-4
|
|
P1
|
F7
|
1
|
Testkemikalie 3
|
TC3
|
3
|
2,00 × 10-5
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-5
|
|
P1
|
F8
|
2
|
Testkemikalie 3
|
TC3
|
3
|
2,00 × 10-5
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-5
|
|
P1
|
F9
|
3
|
Testkemikalie 3
|
TC3
|
3
|
2,00 × 10-5
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-5
|
|
P1
|
F10
|
1
|
Testkemikalie 3
|
TC3
|
4
|
2,00 × 10-6
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-6
|
|
P1
|
F11
|
2
|
Testkemikalie 3
|
TC3
|
4
|
2,00 × 10-6
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-6
|
|
P1
|
F12
|
3
|
Testkemikalie 3
|
TC3
|
4
|
2,00 × 10-6
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-6
|
|
P1
|
G1
|
1
|
Testkemikalie 3
|
TC3
|
5
|
2,00 × 10-7
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-7
|
|
P1
|
G2
|
2
|
Testkemikalie 3
|
TC3
|
5
|
2,00 × 10-7
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-7
|
|
P1
|
G3
|
3
|
Testkemikalie 3
|
TC3
|
5
|
2,00 × 10-7
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-7
|
|
P1
|
G4
|
1
|
Testkemikalie 3
|
TC3
|
6
|
2,00 × 10-8
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-8
|
|
P1
|
G5
|
2
|
Testkemikalie 3
|
TC3
|
6
|
2,00 × 10-8
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-8
|
|
P1
|
G6
|
3
|
Testkemikalie 3
|
TC3
|
6
|
2,00 × 10-8
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-8
|
|
P1
|
G7
|
1
|
Testkemikalie 3
|
TC3
|
7
|
2,00 × 10-9
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-9
|
|
P1
|
G8
|
2
|
Testkemikalie 3
|
TC3
|
7
|
2,00 × 10-9
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-9
|
|
P1
|
G9
|
3
|
Testkemikalie 3
|
TC3
|
7
|
2,00 × 10-9
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-9
|
|
P1
|
G10
|
1
|
Testkemikalie 3
|
TC3
|
8
|
2,00 × 10-10
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-10
|
|
P1
|
G11
|
2
|
Testkemikalie 3
|
TC3
|
8
|
2,00 × 10-10
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-10
|
|
P1
|
G12
|
3
|
Testkemikalie 3
|
TC3
|
8
|
2,00 × 10-10
|
40
|
0
|
40
|
80
|
160
|
1,0 × 10-10
|
|
P1
|
H1
|
1
|
Noretynodrel
|
NE
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H2
|
2
|
Noretynodrel
|
NE
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H3
|
3
|
Noretynodrel
|
NE
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H4
|
1
|
Noretynodrel
|
NE
|
|
1,00 × 10-4,5
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H5
|
2
|
Noretynodrel
|
NE
|
|
1,00 × 10-4,5
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H6
|
3
|
Noretynodrel
|
NE
|
|
1,00 × 10-4,5
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H7
|
1
|
Icke-radioaktiv E2 S
|
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H8
|
2
|
Icke-radioaktiv E2 S
|
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H9
|
3
|
Icke-radioaktiv E2 S
|
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H10
|
1
|
Icke-radioaktiv E2 S
|
|
|
1,00 × 10-7
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H11
|
2
|
Icke-radioaktiv E2 S
|
|
|
1,00 × 10-7
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H12
|
3
|
Icke-radioaktiv E2 S
|
|
|
1,00 × 10-7
|
40
|
0
|
40
|
80
|
160
|
|
Tillägg 3
CHEMICAL EVALUATION AND RESEARCH INSTITUTES (CERI) IN VITRO-TEST AV ÖSTROGENRECEPTORBINDNING MED HUMANT, REKOMBINANT, LIGANDBINDANDE ERΑ-DOMÄNPROTEIN
INLEDANDE ÖVERVÄGANDEN OCH BEGRÄNSNINGAR (SE ÄVEN ALLMÄN INLEDNING)
|
1.
|
Detta in vitro-test av mättnad hos och kompetitiv bindning till östrogenreceptor (ERα) använder sig av den ligandbindande domänen (LBD) hos det humana ERα (hrERα). Denna proteinmodell har framställts av Chemicals evaluation research institute (CERI), Japan, och är ett glutation-S-transferas- (GST) fusionsprotein som uttrycks i E. coli. Protokollet prövades i en internationell valideringsstudie som involverade flera laboratorier (2) och vilken visade dess relevans och pålitlighet för det avsedda syftet.
|
|
2.
|
Detta test är ett screeningförfarande för identifiering av ämnen som kan binda till hrERα. Det används för att fastställa en testkemikalies förmåga att konkurrera med 17β-östradiol i bindning till hrERα:s ligandbindande domän. Kvantitativa testresultat kan omfatta IC50 (ett mått på den koncentration av testkemikalie som behövs för att tränga bort hälften av [3H]-17β-östradiolen från hrERα) och testkemikaliernas relativa bindningsaffiniteter för hrERα, jämförda med den hos 17β-östradiol. För kemisk screening kan godtagbara kvalitativa testresultat omfatta klassificeringar av testkemikalier som antingen hrERα-bindare, icke-bindare eller tvetydiga, utifrån de kriterier som beskrivits för bindningskurvorna.
|
|
3.
|
Testet använder en radioaktiv ligand, vilket kräver att laboratoriet har licens för att hantera radioaktiva material. Allt handhavande av radioisotoper och farliga kemikalier ska följa de regelverk och rutiner som föreskrivs av nationell lagstiftning.
|
|
4.
|
Läs ”ALLMÄN INLEDNING” och ”hrER-BINDNINGSTESTETS DELAR” innan testet används för tillsynsändamål. Definitioner och förkortningar som används i denna testriktlinje beskrivs i tillägg 1.
|
TESTETS PRINCIP (SE ÄVEN ALLMÄN INLEDNING)
|
5.
|
Bindningstestet med hrERα mäter förmågan hos radioaktivt inmärkt ligand ([3H]-17β-östradiol) att binda till ER i närvaron av ökande koncentrationer av testkemikalie (dvs. konkurrerande ligand). Testkemikalier med hög affinitet för ER konkurrerar med den radioaktivt inmärkta liganden vid en lägre koncentration än de kemikalier som har lägre affinitet för receptorn.
|
|
6.
|
Detta test består av två huvuddelar: ett mättnadsbindningsexperiment för att karakterisera receptor–ligandinteraktionsparametrar, följt av ett kompetitivt bindningsexperiment som karakteriserar konkurrensen mellan en testkemikalie och en radioaktivt inmärkt ligand vid inbindning till ER.
|
|
7.
|
Mättnadsbindningsexperimentets syfte är att inför det kompetitiva bindningsexperimentet karakterisera en receptorsats med avseende på bindningsaffinitet och antal receptorer. Mättnadsbindningsexperimentet mäter, vid jämviktsförhållanden och en fast receptorkoncentration, östrogenreceptorns affinitet för dess naturliga ligand (mätt som dissociationskonstant, Kd) och koncentrationen av aktiva bindningsplatser (Bmax).
|
|
8.
|
Det kompetitiva bindningsexperimentet mäter benägenheten hos ett ämne att konkurrera med [3H]-17β-östradiol om att binda till ER. Affiniteten kvantifieras genom koncentrationen av testkemikalie som, vid jämvikt, hämmar 50 % av den specifika bindningen av [3H]-17β-östradiol (kallad hämmande koncentration 50 %, eller IC50). Detta kan också utvärderas med hjälp av den relativa bindningsaffiniteten (RBA, affiniteten jämförd med IC50 för östradiol som mäts separat i samma testomgång). Det kompetitiva bindningsexperimentet mäter bindningen av [3H]-17β-östradiol vid en fast koncentration i närvaron av ett stort intervall (åtta tiopotenser) av testkemikaliekoncentrationer. Data anpassas sedan, där så är möjligt, till en form av Hill-ekvationen (Hill, 1910) som beskriver bortträngningen av den radioaktivt märkta liganden av en kompetitiv bindare som binder till en bindningsplats. Den utsträckning i vilken den radioaktivt inmärkta östradiolen trängs bort vid jämvikt används för att karakterisera testkemikalien som en bindare, en icke-bindare eller med tvetydig respons.
|
FÖRFARANDE
Fastställande av acceptabel proteinfunktion hos hrERα
|
9.
|
Innan rutinmässiga tester av mättnad och kompetitiv bindning genomförs ska varje ny sats av hrERα visas fungera korrekt i det laboratorium där den ska användas. En tvåstegsprocedur bör användas för att kontrollera prestanda. Dessa steg är som följer:
|
—
|
Utför ett mättnadsbindningstest med [3H]-17β-östradiol för att fastställa specificitet och mättnad hos hrERα. Icke-linjär regressionsanalys av dessa data (t.ex. BioSoft, McPherson, 1985, Motulsky, 1995) och påföljande Scatchard-diagram ska dokumentera [3H]-17β-östradiolens bindningsaffinitet för hrERα (Kd) samt antalet receptorer (Bmax) för varje sats av hrERα.
|
|
—
|
Genomför ett kompetitivt bindningstest med hjälp av kontrollämnen (referensöstrogen, 17β-östradiol), en svag bindare (t.ex. noretynodrel eller noretindron) och en icke-bindare (oktyltrietoxisilan, OTES). Varje laboratorium ska upprätta en historisk databas för att dokumentera överensstämmelsen hos IC50 och andra relevanta värden för referensöstrogenet och svaga bindaren, mellan olika experiment och olika hrERα-satser. Dessutom ska parametrarna för kontrollämnenas kurvor över kompetitiv bindning vara inom gränserna för 95-procentigt konfidensintervall (se tabell 1), vilka har tagits fram med data från laboratorier som deltog i valideringsstudien för detta test (2).
|
Tabell 1
Prestandakriterier framtagna för referensöstrogen och svag bindare, CERI-hrER-bindningstest
|
Ämne
|
Parameter
|
Medelvärde (11)
|
Standardavvikelse (n)
|
95 % konfidensintervall (12)
|
|
Nedre gräns
|
Övre gräns
|
|
17β-östradiol.
|
Topp
|
104,74
|
13,12 (70)
|
101,6
|
107,9
|
|
Botten
|
0,85
|
2,41 (70)
|
0,28
|
1,43
|
|
Hill-koefficient (Hill slope)
|
–1,22
|
0,20 (70)
|
–1,27
|
–1,17
|
|
Log(IC50)-värde
|
–8,93
|
0,23 (70)
|
–8,98
|
–8,87
|
|
Noretynodrel
|
Topp
|
101,31
|
10,55 (68)
|
98,76
|
103,90
|
|
Botten
|
2,39
|
5,01 (68)
|
1,18
|
3,60
|
|
Hill-koefficient (Hill slope)
|
–1,04
|
0,21 (68)
|
–1,09
|
–0,99
|
|
Log(IC50)-värde
|
–6,19
|
0,40 (68)
|
–6,29
|
–6,10
|
|
Noretindron (13)
|
Topp
|
92,27
|
7,79 (23)
|
88,90
|
95,63
|
|
Botten
|
16,52
|
10,59 (23)
|
11,94
|
21,10
|
|
Hill-koefficient (Hill slope)
|
–1,18
|
0,32 (23)
|
–1,31
|
–1,04
|
|
Log(IC50)-värde
|
–6,01
|
0,54 (23)
|
–6,25
|
–5,78
|
|
Demonstration av laboratoriets kompetens
|
10.
|
Se punkterna 17 och 18 och tabell 2 i ”hrER-BINDNINGSTESTETS DELAR” i denna testmetod. Varje test (mättnad och kompetitiv bindning) ska bestå av tre oberoende testomgångar (dvs. med nya spädningar av receptor, kemikalier och reagens) på olika dagar och varje omgång ska innehålla tre replikat.
|
Bestämning av hrERα-koncentration
|
11.
|
Koncentrationen av aktiv receptor varierar något mellan satser och med förvaringsförhållanden. Därför ska koncentrationen av aktiv receptor som erhållits från leverantören fastställas. Detta ger tillämplig koncentration av aktiv receptor när testomgången ska göras.
|
|
12.
|
Under förhållanden som motsvarar kompetitiv bindning (dvs. 0,5 nM [3H]-inmärkt östradiol), ska nominella koncentrationer om 0,1, 0,2, 0,4 och 0,6 nM receptor inkuberas i frånvaro (total bindning) och närvaro (icke-specifik bindning) av 1 μM ej inmärkt östradiol. Specifik bindning, beräknad som differensen av total och icke-specifik bindning, ritas i ett diagram mot den nominella receptorkoncentrationen. Den receptorkoncentration som ger specifika bindningsvärden motsvarande 40 % av tillsatt radioaktivt inmärkt ligand relateras till motsvarande receptorkoncentration, och denna receptorkoncentration ska användas för tester av mättnad och kompetitiv bindning. Ofta överensstämmer en slutlig hrER-koncentration på 0,2 nM med detta förhållande.
|
|
13.
|
Om 40-procentskriteriet vid upprepade försök inte kan uppfyllas ska experimentets uppställning kontrolleras för möjliga fel. Svårigheter att uppnå 40-procentskriteriet kan indikera att det finns väldigt lite aktiv receptor i satsen med rekombinant receptor och man bör då överväga att använda en annan sats.
|
Mättnadstest
|
14.
|
Åtta ökande koncentrationer av [3H]-17β-östradiol ska utvärderas i tripletter, enligt följande tre betingelser (se tabell 2):
|
a.
|
I frånvaron av ej inmärkt 17β-östradiol och närvaro av ER. Här fastställs total bindning genom att mäta radioaktiviteten i de brunnar som enbart innehåller [3H]-17β-östradiol.
|
|
b.
|
I närvaron av 2000 gångers koncentrationsöverskott av ej inmärkt 17β-östradiol över inmärkt 17β-östradiol och närvaro av ER. Avsikten med denna betingelse är att mätta de aktiva bindningsplatserna med ej inmärkt 17β-östradiol och, genom att mäta radioaktiviteten i brunnarna, fastställa den icke-specifika bindningen. Eventuellt kvarvarande radioaktiv östradiol som kan binda till receptorn anses binda till en icke-specifik bindningsplats eftersom den tillsatta icke-radioaktiva östradiolen ska ha så hög koncentration att den har bundit till alla specifika bindningsplatser på receptorn.
|
|
c.
|
I frånvaro av ej inmärkt 17β-östradiol och frånvaro av ER (fastställande av total radioaktivitet).
|
Beredning av lösningar med [3H]-17β-östradiol respektive ej inmärkt 17β-östradiol och hrERα
|
15.
|
En 40 nM lösning av [3H]-17β-östradiol ska beredas från en 1 μM stamlösning av [3H]-17β-östradiol i DMSO, genom att tillsätta DMSO (för att bereda 200 nM) och testbuffert vid rumstemperatur (för att bereda 40 nM). Med hjälp av denna 40 nM-lösning, bereds seriespädningen av [3H]-17β-östradiol i koncentrationer från 0,313 nM till 40 nM med rumstempererad testbuffert (som angivet i kolumn 12 i tabell 2). De slutliga testkoncentrationerna, från 0,0313 nM till 4,0 nM, erhålls genom att tillsätta 10 μl av dessa lösningar till respektive testbrunnar i en 96-hålsmikrotiterplatta (se tabellerna 2 och 3) Beredning av testbuffert, beräkning av ursprunglig stamlösning av [3H]-17β-östradiol utifrån dess specifika aktivitet, beredning av spädningar och fastställande av koncentrationer beskrivs i detalj i CERI-protokollet (2).
|
|
16.
|
Spädningar av ej inmärkta 17β-östradiollösningar ska beredas från en 1 nM 17β-östradiol-stamlösning genom att tillsätta testbuffert och på så sätt erhålla åtta ökande koncentrationer, initialt från 0,625 μM till 80 μM. De slutliga testkoncentrationerna, från 0,0625 till 8 μM, erhålls genom att tillsätta 10 μl av dessa lösningar till respektive testbrunnar i en 96-hålsmikrotiterplatta som reserverats för att mäta icke-specifik bindning (se tabellerna 2 och 3). Beredning av spädningar av ej inmärkt 17β-östradiol beskrivs i detalj i CERI-protokollet (2).
|
|
17.
|
Den receptorkoncentration som ger specifik bindning av 40 ± 10 % bör användas (se punkterna 12–13). Lösningen med hrERα ska beredas med iskall testbuffert omedelbart före dess användning, det vill säga efter att alla brunnar för total bindning, icke-specifik bindning och enbart radioaktiv ligand har beretts.
|
|
18.
|
Förbered 96-hålsmikrotiterplattorna enligt schema i tabell 2, med 3 replikat per [3H]-17β-östradiolkoncentration. Fördelning över plattan av volymer av [3H]-17β-östradiol, ej inmärkt 17β-östradiol, buffert och receptor ges i tabell 3.
Tabell 2
Upplägg för mikrotiterplatta till mättnadsbindningstest
|
|
1 (*10)
|
2 (*10)
|
3 (*10)
|
4 (*10)
|
5 (*10)
|
6 (*10)
|
7 (*10)
|
8 (*10)
|
9 (*10)
|
10
|
11 (*11)
|
12 (*11)
|
|
För mätning av TB
|
För mätning av NSB
|
För kvantitativ analys av enbart radioaktiv ligand
|
|
Ej inmärkta E2-spädningar för plattkolumn 4–6
|
[3H]E2-spädningar för plattkolumn 1–9
|
|
A
|
0,0313 nM [3H]E2 + ER
|
0,0313 nM [3H]E2 + 0,0625 μM E2 + ER
|
0,0313 nM
|
|
0,625 μM
|
0,313 nM
|
|
B
|
0,0625 nM [3H]E2+ ER
|
0,0625 nM [3H]E2+ 0,125 μM E2+ ER
|
0,0625 nM
|
|
1,25 μM
|
0,625 nM
|
|
C
|
0,125 nM [3H]E2+ ER
|
0,125 nM [3H]E2+ 0,25 μM E2+ ER
|
0,125 nM
|
|
2,5 μM
|
1,25 nM
|
|
D
|
0,250 nM [3H]E2+ ER
|
0,250 nM [3H]E2+ 0,5 μM E2+ ER
|
0,250 nM
|
|
5 μM
|
2,5 nM
|
|
E
|
0,50 nM [3H]E2+ ER
|
0,50 nM [3H]E2+ 1 μM E2+ ER
|
0,50 nM
|
|
10 μM
|
5 nM
|
|
F
|
1,00 nM [3H]E2+ ER
|
1,00 nM [3H]E2+ 2 μM E2+ ER
|
1,00 nM
|
|
20 μM
|
10 nM
|
|
G
|
2,00 nM [3H]E2+ ER
|
2,00 nM [3H]E2+ 4 μM E2+ ER
|
2,00 nM
|
|
40 μM
|
20 nM
|
|
H
|
4,00 nM [3H]E2+ ER
|
4,00 nM [3H]E2+ 8 μM E2+ ER
|
4,00 nM
|
|
80 μM
|
40 nM
|
|
TB
|
:
|
Total bindning.
|
|
NSB
|
:
|
Icke-specifik bindning.
|
|
[3H]E2
|
:
|
[3H]-17β-östradiol.
|
|
E2
|
:
|
ej inmärkt 17β-östradiol.
|
|
Tabell 3
Volymer för reagenser till mättnadstest i mikrotiterplatta
|
Kolumn-nummer
|
1
|
2
|
3
|
4
|
5
|
6
|
7 (*12)
|
8 (*12)
|
9 (*12)
|
|
Beredningssteg
|
TB-brunnar
|
NSB-brunnar
|
Endast radioaktivt märkt ligand
|
|
Volymer för beståndsdelar till ovannämnda reaktionsbrunnar och turordning för tillsats
|
Buffert
|
60 μl
|
50 μl
|
90 μl
|
|
Ej inmärkt E2 från kolumn 11 i tabell 2
|
–
|
10 μl
|
–
|
|
[3H]E2 från kolumn 12 i tabell 2
|
10 μl
|
10 μl
|
10 μl
|
|
hrERα
|
30 μl
|
30 μl
|
–
|
|
Total reaktionsvolym
|
100 μl
|
100 μl
|
100 μl
|
|
Inkubation
|
EFTER 2 TIMMARS INKUBATION
|
Kvantifiering av radioaktiviteten precis efter beredning. Ingen inkubation
|
|
Behandling med 0,4 % DCC
|
Ja
|
Ja
|
Nej
|
|
Volym av 0,4 % DCC
|
100 μl
|
100 μl
|
–
|
|
Filtrering
|
Ja
|
Ja
|
Nej
|
|
DPM-MÄTNING
|
|
Kvantifieringsvolym att tillsätta till scintillationsblandningen
|
100 μl (*13)
|
100 μl (*13)
|
50 μl
|
|
|
19.
|
Testmikrotiterplattor avsedda för bestämning av total bindning och icke-specifik bindning bör inkuberas vid rumstemperatur (22 °C till 28 °C) i två timmar.
|
|
Mätning av [3H]-17β-östradiol bunden till hrERα
|
20.
|
Efter två timmars inkubation ska den [3H]-17β-östradiol som bundit till hrERα separeras från obunden [3H]-17β-östradiol genom tillsats av 100 μl iskall 0,4 % DCC-suspension till brunnarna. Plattorna ska då placeras på is i 10 minuter och reaktionsblandningen och DCC-suspensionen filtreras, genom att överföras till ett mikrotiterplattefilter, för att avlägsna DCC. Därefter ska 100 μl av filtratet tillsättas till scintillationsvätskan i scintillationsvialer för bestämning av sönderfall per minut (disintegrations per minute, dpm) i varje vial med hjälp av vätskescintillationsräknare.
|
|
21.
|
Alternativt, om mikrotiterplattefilter inte finns att tillgå, kan DCC avlägsnas genom centrifugering. Sedan tas 50 μl supernatant, innehållande den hrERα-bundna [3H]-17β-östradiolen, extremt försiktigt så att man inte råkar förorena brunnarna genom att vidröra DCC och används för scintillationsräkning.
|
|
22.
|
Betingelsen med enbart radioaktivt märkt ligand används för att fastställa sönderfall per minut (dpm) hos den [3H]-17β-östradiol som har tillsatts till testbrunnarna. Radioaktiviteten ska kvantifieras precis efter beredning. Dessa brunnar ska inte inkuberas och inte heller behandlas med DCC-suspension utan tillsättas direkt till scintillationsvätskan. Dessa mätningar visar, i dpm, hur mycket [3H]-17β-östradiol som tillsattes till varje uppsättning brunnar för total bindning och icke-specifik bindning.
|
Kompetitivt bindningstest
|
23.
|
Det kompetitiva bindningstestet mäter bindningen av [3H]-17β-östradiol vid en viss koncentration i närvaro av ökande koncentration av testkemikalie. Tre parallella replikat bör användas för varje koncentration i varje testomgång. Dessutom bör tre icke-parallella testomgångar utföras för varje testad kemikalie. Testet bör utföras i en eller flera 96-hålsmikrotiterplattor.
|
Kontroller
|
24.
|
Varje experiment ska inkludera parallella lösningsmedelprover och kontroller (dvs. referensöstrogen, svag bindare och icke-bindare). Fullständiga koncentrationskurvor för östrogenreferens och kontroller (dvs. svag bindare resp. icke-bindare) ska placeras i en platta vid varje testomgång. Alla andra plattor ska innehålla: i) en hög koncentration (maximal bortträngning, dvs. ungefär fullständig bortträngning av radioaktivt inmärkt ligand) och en mellankoncentration (ungefär IC50) av E2 respektive svag bindare i tripletter, ii) lösningsmedelskontroll och icke-specifik bindning, båda som tripletter. Förfaranden för beredning av testbuffert [3H]-17β-östradiol, hrERα och testkemikalielösningar beskrivs ingående i CERI-protokollet (2).
|
Lösningsmedelskontroll
|
25.
|
Lösningsmedelskontrollen indikerar att lösningsmedlet inte interagerar med testsystemet och mäter också total bindning (TB). Det mest lämpliga lösningsmedlet är DMSO. Men om testkemikaliens högsta koncentration inte är löslig i DMSO kan etanol användas. Koncentrationen av DMSO i de slutliga testbrunnarna ska vara 2,05 % och kan ökas till 2,5 % om testkemikalien visar bristande löslighet. Koncentrationer av DMSO över 2,5 % ska inte användas eftersom högre koncentrationer av lösningsmedel interfererar med testet. Till testkemikalier som inte är lösliga i DMSO men lösliga i etanol kan ett maximum av 2 % etanol användas i testet utan interferens.
|
Buffertkontroll
|
26.
|
Buffertkontrollen (BC) ska inte innehålla vare sig lösningsmedel eller testkemikalie men testets alla övriga ingående delar. Resultaten för buffertkontrollen jämförs med lösningsmedelskontrollen för att säkerställa att det valda lösningsmedlet inte påverkar testsystemet.
|
Stark bindare (referensöstrogen)
|
27.
|
Den endogena liganden är 17β-östradiol (CAS-nr 50-28-2) och den binder med hög affinitet till alfa-subtypen av ER. En standardkurva med ej inmärkt 17β-östradiol ska upprättas för varje kompetitivt hrERα-bindningstest, för att möjliggöra en bedömning av variationen när testet, över tid, upprepas i samma laboratorium. Åtta lösningar av ej inmärkt 17β-östradiol ska beredas i DMSO och testbuffert för att användas till standardkurvan, med slutlig koncentration i testbrunnarna och spridning som följer: 10–6, 10–7, 10–8, 10–8,5, 10–9, 10–9,5, 10–10, 10–11 M. Den högsta koncentrationen av ej inmärkt 17β–östradiol (1 μM) bör tjäna som indikator för icke-specifik bindning. Denna koncentration markeras med märkningen ”NSB” i tabell 4 även om den också är en del av standardkurvan.
|
Svag bindare
|
28.
|
En svag bindare (noretynodrel [CAS-nr 68-23-5] eller alternativt noretindron [CAS-nr 68-22-4]) bör inkluderas för att påvisa varje experiments känslighet och möjliggöra en bedömning av variation när testet används längre perioder. Åtta lösningar av den svaga bindaren ska beredas i DMSO och testbuffert för att användas till standardkurvan, med slutlig koncentration i testbrunnarna som följer: 10–4,5, 10–5,5, 10–6, 10–6,5, 10–7, 10–7,5, 10–8 och 10–9 M.
|
Icke-bindare
|
29.
|
Oktyltrietoxisilan (OTES, CAS-nr 2943-75-1) ska användas som negativ kontroll (icke-bindare). Den ger återkoppling om att testet, så som det utförs, kommer att detektera testkemikalier som inte binder till hrERα. Åtta lösningar av icke-bindaren ska beredas i DMSO och testbuffert för att användas till standardkurvan, med slutlig koncentration i testbrunnarna som följer: 10–3,10–4, 10–5, 10–6, 10–7, 10–8, 10–9, 10–10 M. Di-n-butylftalat (DBP, CAS-nr 84-72-2) kan användas som alternativ icke-bindare, men är endast testad upp till 10–4 M. Maximal löslighet för DBP i testet har visats vara 10–4 M.
|
hrERα-koncentration
|
30.
|
Den mängd receptor som ger en specifik bindning på 40 ± 10 % bör användas (se punkterna 12–13 i tillägg 3). Lösningen med hrERα ska beredas genom spädning av funktionellt hrERα i iskall testbuffert, omedelbart före användning.
|
[3H]-17β-östradiol
|
31.
|
Slutkoncentrationen av [3H]-17β-östradiol i testbrunnarna bör vara 0,5 nM.
|
Testkemikalier
|
32.
|
Vid det första tillfället är det nödvändigt att genomföra ett löslighetstest för att fastställa löslighetsgränsen för varje testkemikalie och för att identifiera lämpligt koncentrationsintervall att använda sig av under testet. Samtliga testkemikaliers respektive löslighetsgräns i lösningsmedlet ska fastställas initialt och vidare bekräftas under testbetingelser. Den i testet testade slutkoncentrationen bör inte överstiga 1 mM. Intervallsökning omfattar en lösningsmedelskontroll och minst åtta logaritmiska seriespädningar, som börjar vid maximal acceptabel koncentration (t.ex. 1 mM eller lägre, baserat på löslighetsgräns), och närvaron av iakttagen grumlighet eller fällning (se även punkt 35 i tillägg 3). När koncentrationsintervallet för testet har fastställts, ska en testkemikalie testas med åtta logaritmiska koncentrationer, lämpligt utspridda enligt definitionen i det föregående intervallsökningstestet. Koncentrationer i det andra och tredje experimenten bör, vid behov, anpassas ytterligare för en bättre identifiering av koncentration–responskurvan.
|
|
33.
|
Spädningar av testkemikalien ska beredas i lämpligt lösningsmedel (se punkt 25 i tillägg 3). Om testkemikaliens högsta koncentration inte är löslig i vare sig DMSO eller etanol, och en tillsats av mer lösningsmedel skulle göra att koncentrationen av lösningsmedel i det slutliga provröret överskred den godtagbara gränsen, kan den högsta koncentrationen sänkas till nästa lägre koncentration. I sådana fall kan ytterligare en koncentration läggas till i den lägre änden av koncentrationsserien. Övriga koncentrationer i serien bör förbli oförändrade.
|
|
34.
|
Testkemikalielösningarna ska iakttas noggrant vid tillsatsen till testbrunnen, eftersom testkemikalien kan fälla ut i detta steg. Data för alla brunnar som innehåller en fällning ska uteslutas från kurvanpassning och anledningen till uteslutningen av data noteras.
|
|
35.
|
Om det sedan tidigare från andra källor finns information som tillhandahåller ett log(IC50)-värde för en testkemikalie, kan det vara lämpligt att geometriskt sprida spädningarna närmre runt det förväntade log(IC50)-värdet (dvs. 0,5 log-enheter). Slutresultaten bör återspegla tillräcklig spridning av koncentrationer på båda sidor om log(IC50)-värdet, inklusive ”topp” och ”botten”, så att bindningskurvan kan karakteriseras tillfredsställande.
|
Organisering av testplattor
|
36.
|
Märkta mikrotiterplattor ska beredas för sexfaldiga inkubationer med lösningsmedelskontrollen, den högsta koncentrationen av referensöstrogen (E2) som också tjänar som indikator på icke-specifik bindning (NSB), buffertkontrollen, och trefaldiga inkubationer för var och en av de åtta koncentrationerna av icke-bindande kontroll (oktyltrietoxisilan), de sju lägre koncentrationerna av referensöstrogenet (E2), de åtta koncentrationerna av svag bindare (noretynodrel eller noretindron) och de åtta koncentrationerna av varje testkemikalie (TC). Ett exempel på upplägget med plattschema över fullständiga koncentrationskurvor för referensöstrogenet och kontroller ges nedan i tabell 4. Ytterligare mikrotiterplattor används för testkemikalierna och ska inkludera plattkontroller, det vill säga i) en hög koncentration (maximal bortträngning) och en mellankoncentration (ungefär IC50) av vardera E2 och svag bindare i triplikat, ii) lösningsmedelskontroll (som total bindning) och icke-specifik bindning, båda sexfaldiga (tabell 5). Ett exempel på plattupplägg för ett kompetitivt test med tre okända testkemikalier ges i tillägg 3.3. De koncentrationer som anges i tabellen, samt i tabeller 4 och 5, avser slutkoncentrationer i testbrunnarna. Maximal koncentration för E2 ska vara 1 × 10-7 M och för den svaga bindaren bör den högsta koncentrationen som används för den svaga bindaren på platta 1 användas. Laboratoriet ska fastställa IC50 med hjälp av sin historiska kontrolldatabas. Detta värde kan förväntas vara jämförbart med det som iakttagits i valideringsstudierna (se tabell 1).
|
Tabell 4
Upplägg för mikrotiterplatta till kompetitivt bindningstest
(97)
(98)
, fullständiga koncentrationskurvor för referensöstrogen och kontroller (platta 1)
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
Buffertkontroll och positiv kontroll (E2)
|
Svagt positiv (Noretynodrel)
|
Negativ kontroll (OTES)
|
TB och NSB
|
|
A
|
Blankprov (*14)
|
1 × 10–9 M
|
1 × 10–10 M
|
TB (lösningsmedelskontroll) (2,05 % DMSO)
|
|
B
|
E2 (1 × 10–11 M)
|
1 × 10–8 M
|
1 × 10–9 M
|
|
C
|
E2 (1 × 10–10 M)
|
1 × 10–7,5 M
|
1 × 10–8 M
|
NSB (10–6 M E2)
|
|
D
|
E2 (1 × 10–9,5 M)
|
1 × 10–7 M
|
1 × 10–7 M
|
|
E
|
E2 (1 × 10–9 M)
|
1 × 10–6,5 M
|
1 × 10–6 M
|
Buffertkontroll
|
|
F
|
E2 (1 × 10–8,5 M)
|
1 × 10–6 M
|
1 × 10–5 M
|
|
G
|
E2 (1 × 10–8 M)
|
1 × 10–5,5 M
|
1 × 10–4 M
|
Blankprov (radioaktivitet) (*15)
|
|
H
|
E2 (1 × 10–7 M)
|
1 × 10–4,5 M
|
1 × 10–3 M
|
Tabell 5
Upplägg för mikrotiterplatta till kompetitivt bindningstest, tillkommande plattor för testkemikalier (TC) och plattkontroller
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
|
Testkemikalie 1 (TC-1)
|
Testkemikalie-2 (TC-2)
|
Testkemikalie-3 (TC-3)
|
Kontroller
|
|
A
|
TC-1 (1 × 10–10 M)
|
TC-2 (1 × 10–10 M)
|
TC-3 (1 × 10–10 M)
|
E2 (1 × 10–7 M)
|
|
B
|
TC-1 (1 × 10–9 M)
|
TC-2 (1 × 10–9 M)
|
TC-3 (1 × 10–9 M)
|
E2 (IC50)
|
|
C
|
TC-1 (1 × 10–8 M)
|
TC-2 (1 × 10–8 M)
|
TC-3 (1 × 10–8 M)
|
NE (1 × 10
–4,5
M)
|
|
D
|
TC-1 (1 × 10–7 M)
|
TC-2 (1 × 10–7 M)
|
TC-3 (1 × 10–7 M)
|
NE (IC50)
|
|
E
|
TC-1 (1 × 10–6 M)
|
TC-2 (1 × 10–6 M)
|
TC-3 (1 × 10–6 M)
|
NSB (10–6 M E2)
|
|
F
|
TC-1 (1 × 10–5 M)
|
TC-2 (1 × 10–5 M)
|
TC-3 (1 × 10–5 M)
|
|
G
|
TC-1 (1 × 10–4 M)
|
TC-2 (1 × 10–4 M)
|
TC-3 (1 × 10–4 M)
|
(TB) (Lösningsmedelskontroll)
|
|
H
|
TC-1 (1 × 10–3 M)
|
TC-2 (1 × 10–3 M)
|
TC-3 (1 × 10–3 M)
|
|
I detta exempel är den svaga bindaren noretinodrel (NE).
|
Slutförande av kompetitivt bindningstest
|
37.
|
Med undantag för brunnarna för total bindning och blankprov (för radioaktiv märkning), så som visas i tabell 6, ska 50 μl av testbufferten överföras till varje brunn och blandas med 10 μl lösningsmedelskontroll, referensöstrogen (E2), svag bindare, icke-bindare eller testkemikalier och 10 μl av en 5 nM [3H]-17β-östradiollösning. Sedan tillsätts 30 μl iskall receptorlösning till varje platta och blandas försiktigt. Lösningen med hrERα ska vara den sista reagens som tillsätts till brunnarna. Testmikrotiterplattorna inkuberas i rumstemperatur (22–28 °C) i 2 timmar.
Tabell 6
Tillsatsvolymer av beståndsdelar till kompetitivt bindningstest för hrER, i mikrotiterplattor
|
Beredningssteg
|
Andra än TB-brunnar
|
TB-brunnar
|
Blankprov (för radioaktiv märkning)
|
|
Volymer för beståndsdelar till ovannämnda reaktionsbrunnar och turordning för tillsats
|
Rumstempererad testbuffert
|
50 μl
|
60 μl
|
90 μl
|
|
Ej inmärkt E2, svag bindare, icke-bindare, lösningsmedel och testkemikalier (*16)
|
10 μl
|
—
|
—
|
|
[3H]-17β-östradiol för att ge en slutkoncentration på 0,5 nM (dvs. 5 nM)
|
10 μl
|
10 μl
|
10 μl
|
|
Fastställd hrERα-koncentration (se punkterna 12–13)
|
30 μl
|
30 μl
|
–
|
|
Total volym i varje testbrunn
|
100 μl
|
100 μl
|
100 μl
|
|
|
38.
|
Kvantifieringen av [3H]-17β-östradiol bunden till hrERα, efter separation av [3H]-17β-östradiol bunden till hrERα från fri [3H]-17β-östradiol genom tillsats av 100 μl av iskall DCC-suspension till varje brunn, bör sedan utföras enligt instruktioner för mättnadsbindningstestet, i punkterna 21–23 i tillägg 3.
|
|
39.
|
Brunnarna G10–12 och H10–12 (identifierade som blankprov [för radioaktiv märkning] i tabell 4) anger dpm för [3H]-inmärkt östradiol i 10 μl. Dosen om 10 μl bör överföras direkt till scintillationsvätskan.
|
Acceptanskriterier
Mättnadsbindningstest
|
40.
|
Den specifika bindningskurvan bör nå en platå med ökande koncentrationer av [3H]-17β-östradiol, indikerande mättnad av hrERα med ligand.
|
|
41.
|
Den specifika bindningen vid 0,5 nM av [3H]-17β-östradiol bör vara inom acceptansintervallet 30–50 % för genomsnittet av uppmätt total tillsatt radioaktivitet från de olika testomgångarna. Enstaka mindre avvikelser utanför detta intervall kan godtas, men om testomgångar konsekvent faller utanför intervallet eller om en viss omgång signifikant faller utanför intervallet bör proteinkoncentrationen justeras och mättnadstestet upprepas.
|
|
42.
|
Data bör bilda ett linjärt Scatchard-diagram.
|
|
43.
|
Den icke-specifika bindningen bör inte vara överdrivet stor. Storleken på den icke-specifika bindningen bör normalt vara < 35 % av den totala bindningen. Men kvoten kan emellanåt överskrida denna gräns vid mätningar av mycket låga dpm-tal för den lägsta koncentrationen av testad radioaktivt inmärkt 17β-östradiol.
|
Kompetitivt bindningstest
|
44.
|
Ökande koncentrationer av ej inmärkt 17β-östradiol bör tränga bort [3H]-17β-östradiol från receptorn på ett sätt som överensstämmer med kompetitiv bindning till en bindningsplats.
|
|
45.
|
Den hämmande koncentrationen, IC50, för referensöstrogenet (dvs. 17β-östradiol) bör ungefär motsvara den molära koncentrationen för [3H]-17β-östradiol plus Kd-värdet, som fastställts vid mättnadsbindningstestet.
|
|
46.
|
Den totala specifika bindningen bör genomgående hamna inom acceptansintervallet på 40 ± 10 % när den genomsnittliga uppmätta totala radioaktiva koncentrationen, som har tillsatts till varje brunn, har varit 0,5 nM vid flera testomgångar. Enstaka mindre avvikelser utanför detta intervall kan godtas, men om testomgångar konsekvent faller utanför intervallet eller om en viss omgång signifikant hamnar utanför intervallet bör proteinkoncentrationen justeras.
|
|
47.
|
Lösningsmedlet bör inte påverka vare sig testets känslighet eller reproducerbarhet. Resultaten för lösningsmedelskontrollen (TB-brunnar) jämförs med buffertkontrollen för att säkerställa att det valda lösningsmedlet inte påverkar testsystemet. Om lösningsmedlet inte har någon effekt på testet bör resultaten för TB och buffertkontroll vara jämförbara.
|
|
48.
|
Icke-bindaren bör inte tränga bort mer än 25 % av [3H]-17β-östradiolen från hrERα när den testas upp till 10–3 M (OTES) eller 10–4 M (DBP).
|
|
49.
|
Prestandakriterier utarbetades för referensöstrogenet och två svaga bindare (t.ex. noretynodrel, noretindron) med hjälp av data från valideringsstudien av CERI-hrER-bindningstestet (bilaga N i referens 2). I valideringsstudien uppges 95-procentiga konfidensintervall för genomsnittet ± SD (n) för de deltagande fyra laboratoriernas alla kontrollomgångar. De 95-procentiga konfidensintervallen beräknades för kurvanpassningsparametrarna (dvs. topp, botten, Hill-koefficient [Hill slope], log[IC50]-värde) för referensöstrogenet och de svaga bindarna, och log10RBA-värdet för de svaga bindarna jämförda med referensöstrogenet. Tabell 1 anger förväntade intervall för kurvanpassningsparametrarna, vilka kan användas som prestandakriterier. I praktiken kan intervallet för IC50 variera något med det experimentellt erhållna Kd-värdet för den receptorberedning och ligandkoncentration som använts i testet.
|
|
50.
|
Inga prestandakriterier för testkemikaliernas kurvanpassningsparametrar utarbetades på grund av den stora mängden möjliga testkemikalier och deras variation i möjliga affiniteter och utfall (t.ex. fullständig kurva, delkurva, ingen kurvanpassning). Men yrkesmässigt omdöme ska tillämpas vid granskning av resultaten från en testkemikalies alla testomgångar. Vidare ska man använda ett tillräckligt intervall av testkemikaliekoncentration för att klart kunna beskriva toppen (t.ex. 90–100 % bindning) på den kompetitiva bindningskurvan. Variationen mellan replikat vid varje koncentration av testkemikalie, liksom mellan de tre oberoende testomgångarna, ska vara rimlig och vetenskapligt försvarbar. Kontroller från en testkemikalies alla testomgångar bör hålla sig nära de prestandamätningar som har rapporterats för detta CERI-test och överensstämma med historiska kontrolldata från respektive laboratorium.
|
DATAANALYS
Mättnadsbindningstest
|
51.
|
Både total och icke-specifik bindning mäts. Från dessa värden beräknas specifik bindning av ökande koncentrationer av [3H]-17β-östradiol under jämviktsförhållanden genom att subtrahera icke-specifik från total. Ett diagram över specifik bindning mot [3H]-17β-östradiol-koncentration bör nå en platå för maximal specifik bindning, vilken indikerar ett hrERα mättat av [3H]-17β-östradiol. Dessutom bör dataanalysen klargöra inbindning av [3H]-17β-östradiol till en enda bindningsplats med hög affinitet. Icke-specifik, total och specifik bindning bör illustreras med en mättnadsbindningskurva. Vidare analys av dessa data bör använda sig av icke-linjär regressionsanalys (t.ex. BioSoft, McPherson, 1985, Motulsky, 1995) med en slutlig dataredovisning i form av ett Scatchard-diagram.
|
|
52.
|
Dataanalysen bör fastställa Bmax och Kd enbart utifrån totala bindningsdata, med antagandet att den icke-specifika bindningen är linjär, om inte en motivering ges för att använda en annan metod. Dessutom ska robust regression användas i fastställande av bästa anpassning, om inte annat val motiveras. Val av robust regressionsmetod ska anges. Korrigering för liganduttömning (t.ex. med hjälp av Swillens metod 1995) bör alltid användas i fastställande av Bmax och Kd utifrån mättnadsbindningsdata.
|
Kompetitivt bindningstest
|
53.
|
Kurvan över kompetitiv bindning ritas som specifik [3H]-17β-östradiol-bindning mot koncentrationen av konkurrerande ligand (log10-enheter). Den koncentration av testkemikalie som hämmar 50 % av den maximala specifika [3H]-17β-östradiol-bindningen är IC50-värdet.
|
|
54.
|
Uppskattningar av log(IC50)-värden för de positiva kontrollerna (t.ex. referensöstrogen och svag bindare) ska fastställas med hjälp av lämplig programvara för icke-linjär kurvanpassning så att de passar en fyraparameters Hill-ekvation (t.ex. BioSoft, McPherson, 1985, Motulsky, 1995). Kurvans topp, botten, lutning och log(IC50)-värde ska i allmänhet lämnas utan restriktion (unconstrained) vid anpassning av dessa kurvor. Robust regression ska användas i fastställande av bästa anpassning, om inte annat val motiveras. Ingen korrigering för liganduttömning ska göras. Efter inledande analys ska varje bindningskurva granskas för att säkerställa korrekt anpassning till modellen. Den svaga bindarens relativa bindningsaffinitet (RBA) beräknas som procentuell andel av dess log(IC50)-värde i förhållande till 17β-östradiols log(IC50)-värde. Resultat från de positiva kontrollerna och den icke-bindande kontrollen ska utvärderas genom att använda måtten på testprestanda i punkterna 44–49 i detta tillägg 3.
|
|
55.
|
Data för alla testkemikalier ska analyseras med en stegvis strategi för att säkerställa att data analyseras korrekt och att varje kurva för kompetitiv bindning klassificeras korrekt. Det är lämpligt att en testkemikalies alla testomgångar initialt underkastas en standardiserad dataanalys som är identisk med den som använts för referensöstrogen och kontroller med svaga bindare (se punkt 54 i detta tillägg 3). När den är klar bör en teknisk granskning av kurvanpassningsparametrarna göras, liksom en visuell granskning av hur väl data stämmer med den genererade kurvan över kompetitiv bindning, för varje testomgång. Under denna tekniska granskning är följande iakttagelser goda indikationer på att testet och dataanalyserna har genomförts korrekt: en koncentrationsberoende minskning i procentuell, specifikt bunden [3H]-17β-östradiol, låg variation mellan de tekniska replikaten för varje kemisk koncentration och överensstämmelse mellan anpassningsparametrarna från de tre testomgångarna.
|
Tolkning av data
|
56.
|
Förutsatt att samtliga acceptanskriterier är uppfyllda anses en testkemikalie binda till hrERα om data kan anpassas till en bindningskurva och den lägsta punkten på responskurvan, inom dataintervallet, är mindre än 50 % (figur 1).
|
|
57.
|
Förutsatt att samtliga acceptanskriterier är uppfyllda, anses en testkemikalie inte binda till hrERα om
|
—
|
data kan anpassas till en bindningskurva och den lägsta punkten på responskurvan, inom dataintervallet, är över 75 %, eller
|
|
—
|
data inte kan anpassas till en bindningskurva och den lägsta, outjämnade genomsnittliga procentuella bindningen inom datamängdens koncentrationsgrupper är över 75 %.
|
|
|
58.
|
Testkemikalier anses tvetydiga om inget av villkoren ovan uppfylls (t.ex. den lägsta punkten på den anpassade responskurvan ligger mellan 76 % och 51 %).
|
Tabell 7
Kriterier för klassificering utifrån en testkemikalies kurva över kompetitiv bindning
|
Klassificering
|
Kriterier
|
|
Bindarea
|
Data kan anpassas till en bindningskurva.
Den lägsta punkten på responskurvan, inom dataintervallet, är mindre än 50 %.
|
|
Icke-bindareb
|
Om data kan anpassas till en bindningskurva och den
lägsta punkten på den anpassade responskurvan, inom dataintervallet, är över 75 %.
Om data inte kan anpassas till en bindningskurva och den
lägsta, outjämnade genomsnittliga procentuella bindningen inom datamängdens koncentrationsgrupper är över 75 %.
|
|
Tvetydigc
|
Varje mätbar testomgång som varken är en bindare eller en icke-bindare
(t.ex. den lägsta punkten längs den anpassade kurvan ligger mellan 76–51 %).
|
Figur 1
Exempel på klassificeringar av testkemikalier med hjälp av kurva för kompetitiv bindning.

|
59.
|
Flera testomgångar genomförda med en viss testkemikalie, i samma laboratorium, kombineras genom att tilldela numeriska värden till varje testomgång och ta fram ett medelvärde enligt tabell 8. Resultaten för de kombinerade testomgångarna inom varje laboratorium jämförs med den förväntade klassificeringen för varje testkemikalie.
|
Tabell 8
Metod för klassificering av testkemikalier med användning av flera testomgångar inom ett laboratorium
|
Tilldelning av numeriska värden för enskilda testomgångar:
|
|
Klassificering
|
Numeriskt värde
|
|
Bindare
|
2
|
|
Tvetydig
|
1
|
|
Icke-bindare
|
0
|
|
Klassificering av genomsnittligt numeriskt värde vid flera testomgångar:
|
|
Klassificering
|
Numeriskt värde
|
|
Bindare
|
Genomsnitt ≥ 1,5
|
|
Tvetydig
|
0,5 ≤ Genomsnitt < 1,5
|
|
Icke-bindare
|
Genomsnitt < 0,5
|
TESTRAPPORT
|
60.
|
Se punkt 24 i ”hrER-BINDNINGSTESTETS DELAR” i denna testmetod.
|
Tillägg 3.1
TERMER
[3H]E2
: 17β-östradiol radioaktivt inmärkt med tritium.
DCC
: Dextran-täckt träkol (Dextran-Coated Charcoal).
E2
: Ej inmärkt 17β-östradiol (inert).
Testbuffert
: 10 mM Tris-HCl, pH 7,4, innehållande 1 mM EDTA, 1 mM EGTA, 1 mM NaVO3, 10 % glycerol, 0,2 mM leupeptin, 1 mM ditiotreitol och 10 mg/ml bovint serumalbumin.
hrERα
: Human rekombinant östrogenreceptor alfa (ligandbindande domän).
Replikat
: En av flera brunnar med samma innehåll och koncentrationer vilka testas parallellt inom en enskild testomgång. I detta protokoll testas varje koncentration av testkemikalie i tripletter, det betyder att tre replikat testas samtidigt vid varje koncentration av testkemikalie.
Testomgång
: En fullständig uppsättning av parallellt hanterade brunnar i mikrotiterplattor, vilka ger all nödvändig information för att karakterisera en testkemikalies bindning till hrERα (dvs. totalt [3H]-17β-östradiol tillsatt till testbrunnen, maximal bindning hos [3H]-17β-östradiol till hrERα, icke-specifik bindning och total bindning vid olika koncentrationer av testkemikalie). En testomgång kan bestå av så lite som en testbrunn (dvs. ett replikat) per koncentration, men eftersom protokollet kräver tripletter, består en omgång av tre testbrunnar per koncentration. Dessutom kräver detta protokoll tre oberoende (dvs. icke-parallella) testomgångar per kemikalie.
Tillägg 3.2
UPPLÄGG FÖR BRUNNAR TILL TEST AV KOMPETITIV BINDNING
|
Platta
|
Position
|
Replikat
|
Brunnstyp
|
Brunnskod
|
Koncentrationskod
|
Inledande koncentr. av konkurrerande ligand (M)
|
hrER-stamlösning (μl)
|
Buffertvolym (μl)
|
Volym spårsubstans (radioaktiv E2) (μl)
|
Volym från spädningsplatta (μl)
|
Slutvolym (μl)
|
Slutkoncentration av konkurrerande ligand (M)
|
|
Fast
|
A1
|
1
|
Blankprov
|
BK
|
BK1
|
—
|
—
|
—
|
—
|
—
|
—
|
—
|
|
Fast
|
A2
|
2
|
Blankprov
|
BK
|
BK2
|
—
|
—
|
—
|
—
|
—
|
—
|
—
|
|
Fast
|
A3
|
3
|
Blankprov
|
BK
|
BK3
|
—
|
—
|
—
|
—
|
—
|
—
|
—
|
|
Fast
|
B1
|
1
|
Icke-radioaktiv E2
|
Fast
|
S1
|
1,00 × 10-10
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-11
|
|
Fast
|
B2
|
2
|
Icke-radioaktiv E2
|
Fast
|
S1
|
1,00 × 10-10
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-11
|
|
Fast
|
B3
|
3
|
Icke-radioaktiv E2
|
Fast
|
S1
|
1,00 × 10-10
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-11
|
|
Fast
|
C1
|
1
|
Icke-radioaktiv E2
|
Fast
|
S2
|
1,00 × 10-9
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-10
|
|
Fast
|
C2
|
2
|
Icke-radioaktiv E2
|
Fast
|
S2
|
1,00 × 10-9
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-10
|
|
Fast
|
C3
|
3
|
Icke-radioaktiv E2
|
Fast
|
S2
|
1,00 × 10-9
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-10
|
|
Fast
|
D1
|
1
|
Icke-radioaktiv E2
|
Fast
|
S3
|
3,16 × 10-9
|
30
|
50
|
10
|
10
|
100
|
3,2 × 10-10
|
|
Fast
|
D2
|
2
|
Icke-radioaktiv E2
|
Fast
|
S3
|
3,16 × 10-9
|
30
|
50
|
10
|
10
|
100
|
3,2 × 10-10
|
|
Fast
|
D3
|
3
|
Icke-radioaktiv E2
|
Fast
|
S3
|
3,16 × 10-9
|
30
|
50
|
10
|
10
|
100
|
3,2 × 10-10
|
|
Fast
|
E1
|
1
|
Icke-radioaktiv E2
|
Fast
|
S4
|
1,00 × 10-8
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-9
|
|
Fast
|
E2
|
2
|
Icke-radioaktiv E2
|
Fast
|
S4
|
1,00 × 10-8
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-9
|
|
Fast
|
E3
|
3
|
Icke-radioaktiv E2
|
Fast
|
S4
|
1,00 × 10-8
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-9
|
|
Fast
|
F1
|
1
|
Icke-radioaktiv E2
|
Fast
|
S5
|
3,16 × 10-8
|
30
|
50
|
10
|
10
|
100
|
3,2 × 10-9
|
|
Fast
|
F2
|
2
|
Icke-radioaktiv E2
|
Fast
|
S5
|
3,16 × 10-8
|
30
|
50
|
10
|
10
|
100
|
3,2 × 10-9
|
|
Fast
|
F3
|
3
|
Icke-radioaktiv E2
|
Fast
|
S5
|
3,16 × 10-8
|
30
|
50
|
10
|
10
|
100
|
3,2 × 10-9
|
|
Fast
|
G1
|
1
|
Icke-radioaktiv E2
|
Fast
|
S6
|
1,00 × 10-7
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-8
|
|
Fast
|
G2
|
2
|
Icke-radioaktiv E2
|
Fast
|
S6
|
1,00 × 10-7
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-8
|
|
Fast
|
G3
|
3
|
Icke-radioaktiv E2
|
Fast
|
S6
|
1,00 × 10-7
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-8
|
|
Fast
|
H1
|
1
|
Icke-radioaktiv E2
|
Fast
|
S7
|
1,00 × 10-6
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-7
|
|
Fast
|
H2
|
2
|
Icke-radioaktiv E2
|
Fast
|
S7
|
1,00 × 10-6
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-7
|
|
Fast
|
H3
|
3
|
Icke-radioaktiv E2
|
Fast
|
S7
|
1,00 × 10-6
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-7
|
|
Fast
|
A4
|
1
|
Noretynodrel
|
NE
|
WP1
|
1,00 × 10-8
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-9
|
|
Fast
|
A5
|
2
|
Noretynodrel
|
NE
|
WP1
|
1,00 × 10-8
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-9
|
|
Fast
|
A6
|
3
|
Noretynodrel
|
NE
|
WP1
|
1,00 × 10-8
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-9
|
|
Fast
|
B4
|
1
|
Noretynodrel
|
NE
|
WP2
|
1,00 × 10-7
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-8
|
|
Fast
|
B5
|
2
|
Noretynodrel
|
NE
|
WP2
|
1,00 × 10-7
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-8
|
|
Fast
|
B6
|
3
|
Noretynodrel
|
NE
|
WP2
|
1,00 × 10-7
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-8
|
|
Fast
|
C4
|
1
|
Noretynodrel
|
NE
|
WP3
|
3,16 × 10-7
|
30
|
50
|
10
|
10
|
100
|
3,2 × 10-8
|
|
Fast
|
C5
|
2
|
Noretynodrel
|
NE
|
WP3
|
3,16 × 10-7
|
30
|
50
|
10
|
10
|
100
|
3,2 × 10-8
|
|
Fast
|
C6
|
3
|
Noretynodrel
|
NE
|
WP3
|
3,16 × 10-7
|
30
|
50
|
10
|
10
|
100
|
3,2 × 10-8
|
|
Fast
|
D4
|
1
|
Noretynodrel
|
NE
|
WP4
|
1,00 × 10-6
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-7
|
|
Fast
|
D5
|
2
|
Noretynodrel
|
NE
|
WP4
|
1,00 × 10-6
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-7
|
|
Fast
|
D6
|
3
|
Noretynodrel
|
NE
|
WP4
|
1,00 × 10-6
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-7
|
|
Fast
|
E4
|
1
|
Noretynodrel
|
NE
|
WP5
|
3,16 × 10-6
|
30
|
50
|
10
|
10
|
100
|
3,2 × 10-7
|
|
Fast
|
E5
|
2
|
Noretynodrel
|
NE
|
WP5
|
3,16 × 10-6
|
30
|
50
|
10
|
10
|
100
|
3,2 × 10-7
|
|
Fast
|
E6
|
3
|
Noretynodrel
|
NE
|
WP5
|
3,16 × 10-6
|
30
|
50
|
10
|
10
|
100
|
3,2 × 10-7
|
|
Fast
|
F4
|
1
|
Noretynodrel
|
NE
|
WP6
|
1,00 × 10-5
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-6
|
|
Fast
|
F5
|
2
|
Noretynodrel
|
NE
|
WP6
|
1,00 × 10-5
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-6
|
|
Fast
|
F6
|
3
|
Noretynodrel
|
NE
|
WP6
|
1,00 × 10-5
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-6
|
|
Fast
|
G4
|
1
|
Noretynodrel
|
NE
|
WP7
|
3,16 × 10-5
|
30
|
50
|
10
|
10
|
100
|
3,2 × 10-6
|
|
Fast
|
G5
|
2
|
Noretynodrel
|
NE
|
WP7
|
3,16 × 10-5
|
30
|
50
|
10
|
10
|
100
|
3,2 × 10-6
|
|
Fast
|
G6
|
3
|
Noretynodrel
|
NE
|
WP7
|
3,16 × 10-5
|
30
|
50
|
10
|
10
|
100
|
3,2 × 10-6
|
|
Fast
|
H4
|
1
|
Noretynodrel
|
NE
|
WP8
|
3,16 × 10-4
|
30
|
50
|
10
|
10
|
100
|
3,2 × 10-5
|
|
Fast
|
H5
|
2
|
Noretynodrel
|
NE
|
WP8
|
3,16 × 10-4
|
30
|
50
|
10
|
10
|
100
|
3,2 × 10-5
|
|
Fast
|
H6
|
3
|
Noretynodrel
|
NE
|
WP8
|
3,16 × 10-4
|
30
|
50
|
10
|
10
|
100
|
3,2 × 10-5
|
|
Fast
|
A7
|
1
|
OTES
|
N
|
OTES1
|
1,00 × 10-9
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-10
|
|
Fast
|
A8
|
2
|
OTES
|
N
|
OTES1
|
1,00 × 10-9
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-10
|
|
Fast
|
A9
|
3
|
OTES
|
N
|
OTES1
|
1,00 × 10-9
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-10
|
|
Fast
|
B7
|
1
|
OTES
|
N
|
OTES2
|
1,00 × 10-8
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-9
|
|
Fast
|
B8
|
2
|
OTES
|
N
|
OTES2
|
1,00 × 10-8
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-9
|
|
Fast
|
B9
|
3
|
OTES
|
N
|
OTES2
|
1,00 × 10-8
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-9
|
|
Fast
|
C7
|
1
|
OTES
|
N
|
OTES3
|
1,00 × 10-7
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-8
|
|
Fast
|
C8
|
2
|
OTES
|
N
|
OTES3
|
1,00 × 10-7
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-8
|
|
Fast
|
C9
|
3
|
OTES
|
N
|
OTES3
|
1,00 × 10-7
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-8
|
|
Fast
|
D7
|
1
|
OTES
|
N
|
OTES4
|
1,00 × 10-6
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-7
|
|
Fast
|
D8
|
2
|
OTES
|
N
|
OTES4
|
1,00 × 10-6
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-7
|
|
Fast
|
D9
|
3
|
OTES
|
N
|
OTES4
|
1,00 × 10-6
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-7
|
|
Fast
|
E7
|
1
|
OTES
|
N
|
OTES5
|
1,00 × 10-5
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-6
|
|
Fast
|
E8
|
2
|
OTES
|
N
|
OTES5
|
1,00 × 10-5
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-6
|
|
Fast
|
E9
|
3
|
OTES
|
N
|
OTES5
|
1,00 × 10-5
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-6
|
|
Fast
|
F7
|
1
|
OTES
|
N
|
OTES6
|
1,00 × 10-4
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-5
|
|
Fast
|
F8
|
2
|
OTES
|
N
|
OTES6
|
1,00 × 10-4
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-5
|
|
Fast
|
F9
|
3
|
OTES
|
N
|
OTES6
|
1,00 × 10-4
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-5
|
|
Fast
|
G7
|
1
|
OTES
|
N
|
OTES7
|
1,00 × 10-3
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-4
|
|
Fast
|
G8
|
2
|
OTES
|
N
|
OTES7
|
1,00 × 10-3
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-4
|
|
Fast
|
G9
|
3
|
OTES
|
N
|
OTES7
|
1,00 × 10-3
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-4
|
|
Fast
|
H7
|
1
|
OTES
|
N
|
OTES8DBP7
|
1,00 × 10-2
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-3
|
|
Fast
|
H8
|
2
|
OTES
|
N
|
OTES88
|
1,00 × 10-2
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-3
|
|
Fast
|
H9
|
3
|
OTES
|
N
|
OTES8
|
1,00 × 10-2
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-3
|
|
Fast
|
A10
|
1
|
Total bindning
|
TB
|
TB1
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
Fast
|
A11
|
2
|
Total bindning
|
TB
|
TB2
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
Fast
|
A12
|
3
|
Total bindning
|
TB
|
TB3
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
Fast
|
B10
|
4
|
Total bindning
|
TB
|
TB4
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
Fast
|
B11
|
5
|
Total bindning
|
TB
|
TB5
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
Fast
|
B12
|
6
|
Total bindning
|
TB
|
TB6
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
Fast
|
C10
|
1
|
Icke-radioaktiv E2 (hög)
|
NSB
|
S1
|
1,00 × 10-5
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-6
|
|
Fast
|
C11
|
2
|
Icke-radioaktiv E2 (hög)
|
NSB
|
S2
|
1,00 × 10-5
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-6
|
|
Fast
|
C12
|
3
|
Icke-radioaktiv E2 (hög)
|
NSB
|
S3
|
1,00 × 10-5
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-6
|
|
Fast
|
D10
|
4
|
Icke-radioaktiv E2 (hög)
|
NSB
|
S4
|
1,00 × 10-5
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-6
|
|
Fast
|
D11
|
5
|
Icke-radioaktiv E2 (hög)
|
NSB
|
S5
|
1,00 × 10-5
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-6
|
|
Fast
|
D12
|
6
|
Icke-radioaktiv E2 (hög)
|
NSB
|
S6
|
1,00 × 10-5
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-6
|
|
Fast
|
E10
|
1
|
Buffertkontroll
|
BC
|
BC1
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
Fast
|
E11
|
2
|
Buffertkontroll
|
BC
|
BC2
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
Fast
|
E12
|
3
|
Buffertkontroll
|
BC
|
BC3
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
Fast
|
F10
|
4
|
Buffertkontroll
|
BC
|
BC4
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
Fast
|
F11
|
5
|
Buffertkontroll
|
BC
|
BC5
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
Fast
|
F12
|
6
|
Buffertkontroll
|
BC
|
BC6
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
Fast
|
G10 (*17)
|
1
|
Blankprov (för radioaktiv märkning)
|
Radioaktiv
|
H1
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
Fast
|
G11 (*17)
|
2
|
Blankprov (för radioaktiv märkning)
|
Radioaktiv
|
H2
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
Fast
|
G12 (*17)
|
3
|
Blankprov (för radioaktiv märkning)
|
Radioaktiv
|
H3
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
Fast
|
H10 (*17)
|
4
|
Blankprov (för radioaktiv märkning)
|
Radioaktiv
|
H4
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
Fast
|
H11 (*17)
|
5
|
Blankprov (för radioaktiv märkning)
|
Radioaktiv
|
H5
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
Fast
|
H12
|
6
|
Blankprov (för radioaktiv märkning)
|
Radioaktiv
|
H6
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
P1
|
A1
|
1
|
Okänd 1
|
U1
|
1
|
1,00 × 10-9
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-10
|
|
P1
|
A2
|
2
|
Okänd 1
|
U1
|
1
|
1,00 × 10-9
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-10
|
|
P1
|
A3
|
3
|
Okänd 1
|
U1
|
1
|
1,00 × 10-9
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-10
|
|
P1
|
B1
|
1
|
Okänd 1
|
U1
|
2
|
1,00 × 10-8
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-9
|
|
P1
|
B2
|
2
|
Okänd 1
|
U1
|
2
|
1,00 × 10-8
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-9
|
|
P1
|
B3
|
3
|
Okänd 1
|
U1
|
2
|
1,00 × 10-8
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-9
|
|
P1
|
C1
|
1
|
Okänd 1
|
U1
|
3
|
1,00 × 10-7
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-8
|
|
P1
|
C2
|
2
|
Okänd 1
|
U1
|
3
|
1,00 × 10-7
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-8
|
|
P1
|
C3
|
3
|
Okänd 1
|
U1
|
3
|
1,00 × 10-7
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-8
|
|
P1
|
D1
|
1
|
Okänd 1
|
U1
|
4
|
1,00 × 10-6
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-7
|
|
P1
|
D2
|
2
|
Okänd 1
|
U1
|
4
|
1,00 × 10-6
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-7
|
|
P1
|
D3
|
3
|
Okänd 1
|
U1
|
4
|
1,00 × 10-6
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-7
|
|
P1
|
E1
|
1
|
Okänd 1
|
U1
|
5
|
1,00 × 10-5
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-6
|
|
P1
|
E2
|
2
|
Okänd 1
|
U1
|
5
|
1,00 × 10-5
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-6
|
|
P1
|
E3
|
3
|
Okänd 1
|
U1
|
5
|
1,00 × 10-5
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-6
|
|
P1
|
F1
|
1
|
Okänd 1
|
U1
|
6
|
1,00 × 10-4
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-5
|
|
P1
|
F2
|
2
|
Okänd 1
|
U1
|
6
|
1,00 × 10-4
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-5
|
|
P1
|
F3
|
3
|
Okänd 1
|
U1
|
6
|
1,00 × 10-4
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-5
|
|
P1
|
G1
|
1
|
Okänd 1
|
U1
|
7
|
1,00 × 10-3
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-4
|
|
P1
|
G2
|
2
|
Okänd 1
|
U1
|
7
|
1,00 × 10-3
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-4
|
|
P1
|
G3
|
3
|
Okänd 1
|
U1
|
7
|
1,00 × 10-3
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-4
|
|
P1
|
H1
|
1
|
Okänd 1
|
U1
|
8
|
1,00 × 10-2
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-3
|
|
P1
|
H2
|
2
|
Okänd 1
|
U1
|
8
|
1,00 × 10-2
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-3
|
|
P1
|
H3
|
3
|
Okänd 1
|
U1
|
8
|
1,00 × 10-2
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-3
|
|
P1
|
A4
|
1
|
Okänd 2
|
U2
|
1
|
1,00 × 10-9
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-10
|
|
P1
|
A5
|
2
|
Okänd 2
|
U2
|
1
|
1,00 × 10-9
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-10
|
|
P1
|
A6
|
3
|
Okänd 2
|
U2
|
1
|
1,00 × 10-9
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-10
|
|
P1
|
B4
|
1
|
Okänd 2
|
U2
|
2
|
1,00 × 10-8
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-9
|
|
P1
|
B5
|
2
|
Okänd 2
|
U2
|
2
|
1,00 × 10-8
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-9
|
|
P1
|
B6
|
3
|
Okänd 2
|
U2
|
2
|
1,00 × 10-8
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-9
|
|
P1
|
C4
|
1
|
Okänd 2
|
U2
|
3
|
1,00 × 10-7
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-8
|
|
P1
|
C5
|
2
|
Okänd 2
|
U2
|
3
|
1,00 × 10-7
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-8
|
|
P1
|
C6
|
3
|
Okänd 2
|
U2
|
3
|
1,00 × 10-7
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-8
|
|
P1
|
D4
|
1
|
Okänd 2
|
U2
|
4
|
1,00 × 10-6
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-7
|
|
P1
|
D5
|
2
|
Okänd 2
|
U2
|
4
|
1,00 × 10-6
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-7
|
|
P1
|
D6
|
3
|
Okänd 2
|
U2
|
4
|
1,00 × 10-6
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-7
|
|
P1
|
E4
|
1
|
Okänd 2
|
U2
|
5
|
1,00 × 10-5
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-6
|
|
P1
|
E5
|
2
|
Okänd 2
|
U2
|
5
|
1,00 × 10-5
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-6
|
|
P1
|
E6
|
3
|
Okänd 2
|
U2
|
5
|
1,00 × 10-5
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-6
|
|
P1
|
F4
|
1
|
Okänd 2
|
U2
|
6
|
1,00 × 10-4
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-5
|
|
P1
|
F5
|
2
|
Okänd 2
|
U2
|
6
|
1,00 × 10-4
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-5
|
|
P1
|
F6
|
3
|
Okänd 2
|
U2
|
6
|
1,00 × 10-4
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-5
|
|
P1
|
G4
|
1
|
Okänd 2
|
U2
|
7
|
1,00 × 10-3
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-4
|
|
P1
|
G5
|
2
|
Okänd 2
|
U2
|
7
|
1,00 × 10-3
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-4
|
|
P1
|
G6
|
3
|
Okänd 2
|
U2
|
7
|
1,00 × 10-3
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-4
|
|
P1
|
H4
|
1
|
Okänd 2
|
U2
|
8
|
1,00 × 10-2
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-3
|
|
P1
|
H5
|
2
|
Okänd 2
|
U2
|
8
|
1,00 × 10-2
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-3
|
|
P1
|
H6
|
3
|
Okänd 2
|
U2
|
8
|
1,00 × 10-2
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-3
|
|
P1
|
A7
|
1
|
Okänd 3
|
U3
|
1
|
1,00 × 10-9
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-10
|
|
P1
|
A8
|
2
|
Okänd 3
|
U3
|
1
|
1,00 × 10-9
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-10
|
|
P1
|
A9
|
3
|
Okänd 3
|
U3
|
1
|
1,00 × 10-9
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-10
|
|
P1
|
B7
|
1
|
Okänd 3
|
U3
|
2
|
1,00 × 10-8
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-9
|
|
P1
|
B8
|
2
|
Okänd 3
|
U3
|
2
|
1,00 × 10-8
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-9
|
|
P1
|
B9
|
3
|
Okänd 3
|
U3
|
2
|
1,00 × 10-8
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-9
|
|
P1
|
C7
|
1
|
Okänd 3
|
U3
|
3
|
1,00 × 10-7
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-8
|
|
P1
|
C8
|
2
|
Okänd 3
|
U3
|
3
|
1,00 × 10-7
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-8
|
|
P1
|
C9
|
3
|
Okänd 3
|
U3
|
3
|
1,00 × 10-7
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-8
|
|
P1
|
D7
|
1
|
Okänd 3
|
U3
|
4
|
1,00 × 10-6
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-7
|
|
P1
|
D8
|
2
|
Okänd 3
|
U3
|
4
|
1,00 × 10-6
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-7
|
|
P1
|
D9
|
3
|
Okänd 3
|
U3
|
4
|
1,00 × 10-6
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-7
|
|
P1
|
E7
|
1
|
Okänd 3
|
U3
|
5
|
1,00 × 10-5
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-6
|
|
P1
|
E8
|
2
|
Okänd 3
|
U3
|
5
|
1,00 × 10-5
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-6
|
|
P1
|
E9
|
3
|
Okänd 3
|
U3
|
5
|
1,00 × 10-5
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-6
|
|
P1
|
F7
|
1
|
Okänd 3
|
U3
|
6
|
1,00 × 10-4
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-5
|
|
P1
|
F8
|
2
|
Okänd 3
|
U3
|
6
|
1,00 × 10-4
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-5
|
|
P1
|
F9
|
3
|
Okänd 3
|
U3
|
6
|
1,00 × 10-4
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-5
|
|
P1
|
G7
|
1
|
Okänd 3
|
U3
|
7
|
1,00 × 10-3
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-4
|
|
P1
|
G8
|
2
|
Okänd 3
|
U3
|
7
|
1,00 × 10-3
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-4
|
|
P1
|
G9
|
3
|
Okänd 3
|
U3
|
7
|
1,00 × 10-3
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-4
|
|
P1
|
H7
|
1
|
Okänd 3
|
U3
|
8
|
1,00 × 10-2
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-3
|
|
P1
|
H8
|
2
|
Okänd 3
|
U3
|
8
|
1,00 × 10-2
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-3
|
|
P1
|
H9
|
3
|
Okänd 3
|
U3
|
8
|
1,00 × 10-2
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-3
|
|
P1
|
A10
|
1
|
Kontroll E2 (max)
|
Fast
|
E2max1
|
1,00 × 10-6
|
30
|
50
|
10
|
10
|
100
|
1,00 × 10-7
|
|
P1
|
A11
|
2
|
Kontroll E2 (max)
|
Fast
|
E2max2
|
1,00 × 10-6
|
30
|
50
|
10
|
10
|
100
|
1,00 × 10-7
|
|
P1
|
A12
|
3
|
Kontroll E2 (max)
|
Fast
|
E2max3
|
1,00 × 10-6
|
30
|
50
|
10
|
10
|
100
|
1,00 × 10-7
|
|
P1
|
B10
|
1
|
Kontroll E2 (IC50)
|
Fast
|
E2IC501
|
E2IC50 × 10
|
30
|
50
|
10
|
10
|
100
|
E2IC50
|
|
P1
|
B11
|
2
|
Kontroll E2 (IC50)
|
Fast
|
E2IC502
|
E2IC50 × 10
|
30
|
50
|
10
|
10
|
100
|
E2IC50
|
|
P1
|
B12
|
3
|
Kontroll E2 (IC50)
|
Fast
|
E2IC503
|
E2IC50 × 10
|
30
|
50
|
10
|
10
|
100
|
E2IC50
|
|
P1
|
C10
|
1
|
Kontroll NE (max)
|
Fast
|
Nemax1
|
1,00 × 10-3,5
|
30
|
50
|
10
|
10
|
100
|
1,00 × 10-4,5
|
|
P1
|
C11
|
2
|
Kontroll NE (max)
|
Fast
|
Nemax2
|
1,00 × 10-3,5
|
30
|
50
|
10
|
10
|
100
|
1,00 × 10-4,5
|
|
P1
|
C12
|
3
|
Kontroll NE (max)
|
Fast
|
Nemax3
|
1,00 × 10-3,5
|
30
|
50
|
10
|
10
|
100
|
1,00 × 10-4,5
|
|
P1
|
D10
|
1
|
Kontroll NE (IC50)
|
Fast
|
NEIC501
|
NEIC50 × 10
|
30
|
50
|
10
|
10
|
100
|
NEIC50
|
|
P1
|
D11
|
2
|
Kontroll NE (IC50)
|
Fast
|
NEIC502
|
NEIC50 × 10
|
30
|
50
|
10
|
10
|
100
|
NEIC50
|
|
P1
|
D12
|
3
|
Kontroll NE (IC50)
|
Fast
|
NEIC503
|
NEIC50 × 10
|
30
|
50
|
10
|
10
|
100
|
NEIC50
|
|
P1
|
E10
|
1
|
Icke-radioaktiv E2 (hög)
|
NSB
|
S1
|
1,00 × 10-5
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-6
|
|
P1
|
E11
|
2
|
Icke-radioaktiv E2 (hög)
|
NSB
|
S2
|
1,00 × 10-5
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-6
|
|
P1
|
E12
|
3
|
Icke-radioaktiv E2 (hög)
|
NSB
|
S3
|
1,00 × 10-5
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-6
|
|
P1
|
F10
|
4
|
Icke-radioaktiv E2 (hög)
|
NSB
|
S4
|
1,00 × 10-5
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-6
|
|
P1
|
F11
|
5
|
Icke-radioaktiv E2 (hög)
|
NSB
|
S5
|
1,00 × 10-5
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-6
|
|
P1
|
F12
|
6
|
Icke-radioaktiv E2 (hög)
|
NSB
|
S6
|
1,00 × 10-5
|
30
|
50
|
10
|
10
|
100
|
1,0 × 10-6
|
|
P1
|
G10
|
1
|
Total bindning
|
TB
|
TB1
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
G11
|
2
|
Total bindning
|
TB
|
TB2
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
G12
|
3
|
Total bindning
|
TB
|
TB3
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
H10
|
4
|
Total bindning
|
TB
|
TB4
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
H11
|
5
|
Total bindning
|
TB
|
TB5
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
H12
|
6
|
Total bindning
|
TB
|
TB6
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
Tillägg 4
ÖVERVÄGANDEN INFÖR ANALYS AV DATA FRÅN KOMPETITIVT BINDNINGSTEST FÖR HRER
|
1.
|
Det kompetitiva bindningstestet för hrERα mäter bindningen av [3H]-17β-östradiol vid en viss koncentration i närvaro av ökande koncentrationer av en testkemikalie. Kurvan över kompetitiv bindning ritas som specifik [3H]-17β-östradiol-bindning mot koncentrationen av konkurrerande ligand (log10-enheter). Den koncentration av testkemikalie som hämmar 50 % av den maximala specifika [3H]-17β-östradiol-bindningen är IC50.
|
Analys av data för referensöstrogen och svag bindare (1)
|
2.
|
Data från kontrollomgångarna omvandlas (dvs. procentuell, specifik [3H]-17β-östradiol-bindning och log-koncentrationen av kontrollkemikalien) för vidare analys. Uppskattningar av log(IC50)-värden för de positiva kontrollerna (t.ex. referensöstrogen och svag bindare) ska fastställas med hjälp av lämplig programvara för icke-linjär kurvanpassning så att de passar en fyraparameters Hill-ekvation (t.ex. BioSoft, GraphPad Prism) (2). Kurvans topp, botten, lutning och log(IC50)-värde kan i allmänhet lämnas utan restriktion (unconstrained) vid anpassning av dessa kurvor. Robust regression ska användas i fastställande av bästa anpassning, om inte annat val motiveras. Val av robust regressionsmetod ska anges. Korrigering av liganduttömning behövdes inte för FW- eller CERI-hrER-tester, men kan vid behov övervägas. Efter inledande analys ska varje bindningskurva granskas för att säkerställa korrekt anpassning till modellen. Den svaga bindarens relativa bindningsaffinitet (RBA) kan beräknas procentuellt som dess log(IC50)-värde i förhållande till log(IC50)-värdet för 17β-östradiol. Resultat för de positiva kontrollerna och den icke-bindande kontrollen bör utvärderas med mått på testprestanda och acceptanskriterier som beskrivits i denna testmetod (punkt 20), tillägg 2 (FW-test, punkterna 41–51) och tillägg 3 (CERI-test, punkterna 41–51). Exempel på 3 testomgångar för referensöstrogen och svag bindare visas i figur 1.
|
Figur 1
Exempel på kurvor över kompetitiv bindning för referensöstrogen och kontrollen med svag bindare

Dataanalys för testkemikalier
|
3.
|
Data för alla testkemikalier ska analyseras med en stegvis strategi för att säkerställa att data analyseras korrekt och att varje kurva för kompetitiv bindning klassificeras korrekt. Det är lämpligt att en testkemikalies alla testomgångar initialt underkastas en standardiserad dataanalys som är identisk med den som använts för referensöstrogen och kontroller med svaga bindare. När den är klar bör en teknisk granskning av kurvanpassningsparametrarna göras, liksom en visuell granskning av hur väl data stämmer med den genererade kurvan över kompetitiv bindning, för varje testomgång. Under denna tekniska granskning är följande iakttagelser goda indikationer på att testet och dataanalyserna har genomförts korrekt: en koncentrationsberoende minskning i procentuell, specifikt bunden [3H]-17β-östradiol, låg variation mellan de tekniska replikaten för varje kemisk koncentration och överensstämmelse mellan anpassningsparametrarna från de tre testomgångarna. Yrkesmässigt omdöme ska tillämpas vid granskning av resultaten från en testkemikalies alla testomgångar och de data som används för att klassificera respektive testkemikalie som bindare eller icke-bindare ska vara vetenskapligt försvarbara.
|
|
4.
|
Emellanåt kan det finnas data som kräver ytterligare beaktande för att man ska kunna analysera och tolka hrER-bindningsdata korrekt. Olika studier har uppvisat fall där analys och tolkning av kompetitiva receptorbindningsdata kan kompliceras av en uppgång i procentuell specifik bindning när kemikalier testas vid sina högsta koncentrationer (figur 2). Detta är ett välkänt fenomen som har påträffats vid användning av protokoll för ett antal olika kompetitiva receptorbindningstester (3). I dessa fall observeras en koncentrationsberoende respons vid lägre koncentrationer, men när testkemikaliens koncentration närmar sig löslighetsgränsen minskar inte längre bortträngningen av [3H]-17β-östradiol. I dessa fall indikerar data för de högre koncentrationerna att man har nått testets biologiska gräns. Till exempel är detta fenomen många gånger förknippat med kemisk olöslighet och utfällning vid höga koncentrationer, alternativ återspeglar det att man har överskridit det dextran-täckta träkolets förmåga att binda den icke-bundna, radioaktivt inmärkta liganden under separationssteget, vid den högsta kemiska koncentrationen. Att lämna kvar sådana datapunkter vid anpassning av kompetitiva bindningsdata till en sigmoidal kurva kan ibland leda till en felklassificering av testkemikaliens ER-bindningspotential (figur 2). För att undvika detta ingår det i FW- och CERI-hrER-bindningstesterna en möjlighet att från analyserna utesluta datapunkter där medelvärdet för replikaten när det gäller andelen specifikt bunden [3H]-17β-östradiol är 10 % eller mer över det observerade medelvärdet vid en lägre koncentration (detta betecknas ofta som 10-procentsregeln). Denna regel kan endast användas en gång för varje enskild kurva, och det måste finnas kvar data från minst sex koncentrationer för att kurvan ska kunna klassificeras korrekt.
|
Figur 2
Exempel på kompetitiva bindningskurvor med och utan användning av 10-procentsregeln

|
5.
|
Lämplig användning av 10-procentsregeln för att korrigera dessa kurvor ska övervägas noga och förbehållas de fall där det finns starka indikationer på att ämnet är en hrER-bindare. I samband med utförande av experimenten för valideringsstudien av FW-hrER-bindningstestet noterade att 10-procentsregeln ibland ledde till en oavsiktlig och oförutsedd konsekvens. Kemikalier som inte interagerade med receptorn (dvs. faktiska icke-bindare) visade ofta en variation runt 100 % bunden radioaktivt märkt ligand som var större än 10 % över det testade koncentrationsintervallet. Om det lägsta värdet råkade vara vid en låg koncentration riskerade data från alla högre koncentrationer att uteslutas från analysen genom användning av 10-procentsregeln, även om dessa koncentrationer kunde vara användbara för att fastställa att kemikalien är en icke-bindare. Figur 3 visar exempel där det är olämpligt att använda 10-procentsregeln.
|
Figur 3
Exempel, kompetitiva bindningsdata där det är olämpligt att använda 10-procentsregeln

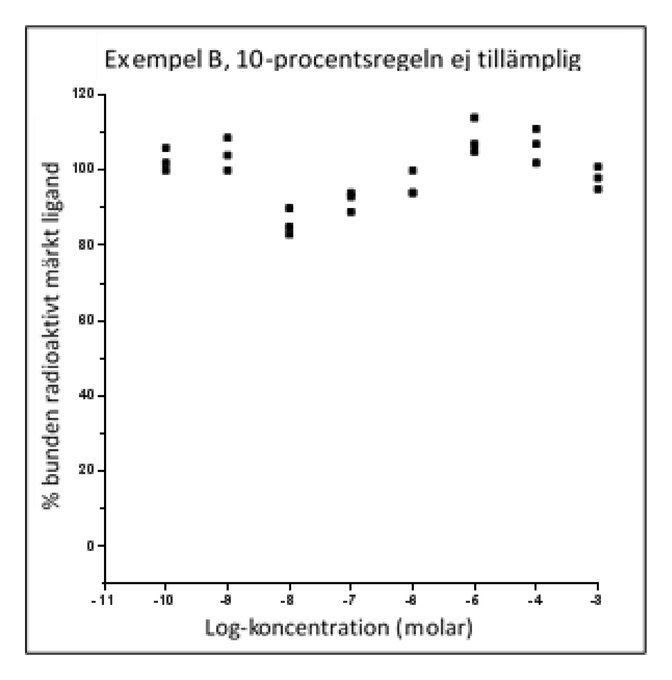
Litteratur
|
(1)
|
OECD (2015). Integrated Summary Report: Validation of Two Binding Assays Using Human Recombinant Estrogen Receptor Alpha (hrERα), Health and Safety Publications, Series on Testing and Assessment (No 226), Organisationen för ekonomiskt samarbete och utveckling (OECD), Paris.
|
|
(2)
|
Motulsky H. och Christopoulos A. (2003). The law of mass action, I: Fitting Models to Biological Data Using Linear and Non-linear Regression. GraphPad Software Inc., San Diego, CA, s. 187-191. Www.graphpad.com/manuals/Prism4/RegressionBook.pdf
|
|
(3)
|
Laws SC, Yavanhxay S, Cooper RL, Eldridge JC. (2006). Nature of the Binding Interaction for 50 Structurally Diverse Chemicals with Rat Estrogen Receptors. Toxicological Sci. 94(1):46-56.
|
B.71
IN VITRO-TESTER AVSEENDE HUDSENSIBILISERING SOM BEHANDLAR NYCKELHÄNDELSEN AKTIVERING AV DENDRITISKA CELLER INOM AOP:N FÖR HUDSENSIBILISERING
ALLMÄN INLEDNING
Testmetod baserad på nyckelhändelsen aktivering av dendritiska celler
|
1.
|
Hudsensibiliserande ämnen är ämnen som leder till en allergisk reaktion efter hudkontakt enligt definitionen i FN:s globalt harmoniserade system för klassificering och märkning av kemikalier (GHS) (1) och förordning (EG) nr 1272/2008 om klassificering, märkning och förpackning av ämnen och blandningar (CLP-förordningen) (99). Det råder allmän enighet om de viktigaste biologiska händelserna (nyckelhändelserna) som utlöser hudsensibilisering. Den befintliga kunskapen om de kemiska och biologiska mekanismer som är förknippade med hudsensibilisering har sammanfattats i form av en AOP (Adverse Outcome Pathway) under OECD:s AOP-program (2), från den inledande molekylära händelsen, via de mellanliggande händelserna och fram till den skadliga effekten, närmare bestämt allergisk kontaktdermatit. I det här fallet är den inledande molekylära händelsen (det vill säga den första nyckelhändelsen) den kovalenta bindningen av elektrofila ämnen till nukleofila centrum i hudproteiner. Den andra nyckelhändelsen i AOP:n sker i keratinocyterna och innefattar inflammatoriska responser samt förändringar i genuttryck kopplade till specifika cellsignaleringsvägar, till exempel ARE-beroende vägar (Antioxidant/Electrophile Response Element). Den tredje nyckelhändelsen är aktiveringen av dendritiska celler, som vanligen bedöms utifrån uttrycket av specifika cellytemarkörer, kemokiner och cytokiner. Den fjärde nyckelhändelsen är aktiveringen och spridningen av T-celler, som bedöms indirekt genom LLNA-testet på mus (Local Lymph Node Assay) (3).
|
|
2.
|
Den här testmetoden (TM) motsvarar OECD:s testriktlinje (TG) 442E (2017). Den beskriver in vitro-tester gällande mekanismer som beskrivs under nyckelhändelsen aktivering av dendritiska celler inom AOP:n för hudsensibilisering (2). Testmetoden utgörs av tester som används för att understödja uppdelningen mellan hudsensibiliserande ämnen och icke-hudsensibiliserande ämnen i enlighet med GHS och CLP-förordningen.
|
|
Testerna som beskrivs i denna testmetod är följande:
|
—
|
h-CLAT (Human Cell Line Activation Test).
|
|
—
|
U-SENS™ (aktiveringstest för cellinjen U937).
|
|
—
|
IL-8 Luc-testet (Interleukin-8 Reporter Gene Assay).
|
|
|
|
3.
|
Testerna som ingår i denna testmetod och motsvarande OECD-testriktlinje kan skilja sig åt i fråga om förfarandet som används för att generera data och de avläsningar som uppmäts, men de kan användas utan åtskillnad för att uppfylla ländernas krav på testresultat avseende nyckelhändelsen aktivering av dendritiska celler inom AOP:n för hudsensibilisering, samtidigt som de omfattas av OECD:s rådslag om ömsesidigt erkännande av data (MAD).
|
Bakgrund och principer för testerna som ingår i den nyckelhändelsebaserade testmetoden
|
4.
|
Bedömning av hudsensibilisering har normalt inbegripit användning av försöksdjur. I de klassiska metoder som använder sig av marsvin – Magnusson och Kligmans GPMT-test (Guinea Pig Maximisation Test) och Buehler-testet (TM B.6) (4) – bedöms både induktions- och eliciteringsfaserna i hudsensibiliseringen. I testerna på mus, LLNA (TM B.42) (3) och de båda icke-radioaktiva modifieringarna LLNA: DA (TM B.50) (5) och LLNA: BrdU-ELISA (TM B.51) (6), bedöms endast induktionsresponsen och dessa tester har också vunnit erkännande eftersom de har djurskyddsmässiga fördelar framför marsvinstester och ger en objektiv mätning av hudsensibiliseringens induktionsfas.
|
|
5.
|
På senare tid har mekanistiskt baserade in chemico- och in vitro-testmetoder som behandlar den första nyckelhändelsen (TM B.59; DPRA [Direct Peptide Reactivity Assay] [7]) och den andra nyckelhändelsen (TM B.60; testmetoden ARE-Nrf2-luciferas [8]) i AOP:n för hudsensibilisering antagits för att bidra till utvärderingen av kemikaliers hudsensibiliseringspotential.
|
|
6.
|
Testerna som beskrivs i den här testmetoden kvantifierar antingen förändringar i uttrycket av cellytemarkörer (en eller flera) som är kopplade till aktiveringsprocessen för monocyter och dendritiska celler efter exponering för sensibiliserande ämnen (till exempel CD54 eller CD86) eller förändringar i uttrycket av IL-8, som är en cytokin kopplad till aktiveringen av dendritiska celler. Hudsensibiliserande ämnen har rapporterats ge upphov till uttryck av cellmembranmarkörer, som CD40, CD54, CD80, CD83 och CD86, i tillägg till pro-inflammatoriska cytokiner, som IL-1β och TNF-α, och åtskilliga kemokiner, inklusive IL-8 (CXCL8) och CCL3, (9) (10) (11) (12) som är kopplade till aktiveringen av dendritiska celler (2).
|
|
7.
|
Eftersom aktivering av dendritiska celler endast utgör en av nyckelhändelserna inom AOP:n för hudsensibilisering (2) (13), kan det emellertid hända att information som genereras genom tester som mäter markörer för aktivering av dendritiska celler inte på egen hand räcker för att man ska kunna avgöra huruvida kemikalier har eller inte har hudsensibiliseringspotential. Av den anledningen är data som genereras genom de tester som beskrivs i denna testmetod tänkta att understödja uppdelningen mellan hudsensibiliserande ämnen (det vill säga ämnen enligt kategori 1 i GHS/CLP-förordningen) och icke-hudsensibiliserande ämnen när de används inom ramen för en IATA (Integrated Approach to Testing and Assessment), tillsammans med annan relevant kompletterande information, som exempelvis erhållits genom in vitro-tester som studerar andra nyckelhändelser inom AOP:n för hudsensibilisering och metoder som inte bygger på testning, vilket innefattar jämförelser med kemiskt analoga ämnen (13). Exempel på användning av data som genererats genom dessa tester inom ramen för definierade arbetssätt (Defined Approaches), det vill säga arbetsmetoder som är standardiserade både i fråga om de informationskällor som används och i fråga om förfarandet som tillämpas för att härleda prediktioner från dessa data, har publicerats (13) och kan användas som användbara element inom aktuell IATA.
|
|
8.
|
Testerna som beskrivs inom denna testmetod kan inte användas som fristående tester, vare sig för att dela in hudsensibiliserande ämnen i underkategorierna 1A och 1B enligt definitionerna i GHS/CLP-förordningen, för myndigheter som använder sig av dessa båda frivilliga underkategorier, eller för att förutsäga styrkan när det handlar om säkerhetsbedömningsbeslut. Beroende på det tillämpliga regelverket kan dock positiva resultat som erhållits med dessa metoder användas fristående för att klassificera kemikalier i kategori 1 enligt GHS/CLP-förordningen.
|
|
9.
|
I denna testmetod används begreppet testkemikalie endast för att syfta på det som testas (100) och begreppet berör inte testernas tillämplighet för att testa monokomponentämnen, multikomponentämnen och/eller blandningar. För närvarande finns endast begränsad information om testernas tillämplighet för multikomponentämnen/blandningar (14) (15). Testerna är inte desto mindre tekniskt tillämpbara för att testa multikomponentämnen och blandningar. Innan testmetoden används för en blandning i syfte att samla in data för ett avsett tillsynsändamål bör man dock överväga om, och i så fall varför, testmetoden kommer att ge godtagbara resultat för detta ändamål (101). Sådana överväganden är inte nödvändiga om blandningen måste testas enligt lag. Vid testning av multikomponentämnen eller blandningar bör man även ta hänsyn till eventuella störningar orsakade av cytotoxiska beståndsdelar i de observerade responserna.
|
LITTERATUR
|
(1)
|
Förenta nationerna (FN) (2015). Globally Harmonized System of Classification and Labelling of Chemicals (GHS, det globalt harmoniserade systemet för klassificering och märkning av kemikalier). Sjätte reviderade utgåvan. New York och Genève: United Nations Publications. ISBN: 978-92-1-117006-1. Finns på https://www.unece.org/trans/danger/publi/ghs/ghs_rev06/06files_e.html.
|
|
(2)
|
OECD (2012). The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins. Part 1: Scientific Evidence. Series on Testing and Assessment No. 168. Finns på http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=ENV/JM/MONO(2012)10/PART1&docLanguage=En.
|
|
(3)
|
Kapitel B.42 i denna bilaga: Hudsensibilisering: LLNA-metoden (Local Lymph Node Assay).
|
|
(4)
|
Kapitel B.6 i denna bilaga: Hudsensibilisering.
|
|
(5)
|
Kapitel B.50 i denna bilaga: Hudsensibilisering: LLNA-metoden (Local Lymph Node Assay): DA.
|
|
(6)
|
Kapitel B.51 i denna bilaga: Hudsensibilisering: LLNA-metoden (Local Lymph Node Assay): BrdU-ELISA.
|
|
(7)
|
Kapitel B.59 i denna bilaga: Hudsensibilisering in chemico: Direct Peptide Reactivity Assay (DPRA).
|
|
(8)
|
Kapitel B.60 i denna bilaga: Hudsensibilisering in vitro: Testmetoden ARE-Nrf2-luciferas.
|
|
(9)
|
Steinman RM. (1991). The dendritic cell system and its role in immunogenicity. Annu Rev Immunol 9:271-96.
|
|
(10)
|
Caux C, Vanbervliet B, Massacrier C, Azuma M, Okumura K, Lanier LL and Banchereau J. (1994). B70/B7-2 is identical to CD86 and is the major functional ligand for CD28 expressed on human dendritic cells. J Exp Med 180:1841-7.
|
|
(11)
|
Aiba S, Terunuma A, Manome H and Tagami H. (1997). Dendritic cells differently respond to haptens and irritants by their production of cytokines and expression of co-stimulatory molecules. Eur J Immunol 27:3031-8.
|
|
(12)
|
Aiba S, Manome H, Nakagawa S, Mollah ZU, Mizuashi M, Ohtani T, Yoshino Y and Tagami H. (2003). p38 mitogen-activated protein kinase and extracellular signal-regulated kinases play distinct roles in the activation of dendritic cells by two representative haptens, NiCl2 and DNCB. J Invest Dermatol 120:390-8.
|
|
(13)
|
OECD (2016). Series on Testing & Assessment No 256: Guidance Document On The Reporting Of Defined Approaches And Individual Information Sources To Be Used Within Integrated Approaches To Testing And Assessment (IATA) For Skin Sensitisation, Annex 1 and Annex 2, ENV/JM/HA(2016)29, Organisationen för ekonomiskt samarbete och utveckling (OECD), Paris. Finns på https://community.oecd.org/community/iatass.
|
|
(14)
|
Ashikaga T, Sakaguchi H, Sono S, Kosaka N, Ishikawa M, Nukada Y, Miyazawa M, Ito Y, Nishiyama N, Itagaki H. (2010). A comparative evaluation of in vitro skin sensitisation tests: the human cell-line activation test (h-CLAT) versus the local lymph node assay (LLNA). Altern. Lab. Anim. 38, 275-284.
|
|
(15)
|
Piroird, C., Ovigne, J.M., Rousset, F., Martinozzi-Teissier, S., Gomes, C., Cotovio, J., Alépée, N. (2015). The Myeloid U937 Skin Sensitization Test (U-SENS) addresses the activation of dendritic cell event in the adverse outcome pathway for skin sensitization. Toxicol. In Vitro 29, 901-916.
|
Tillägg 1
IN VITRO-TEST AVSEENDE HUDSENSIBILISERING: H-CLAT (HUMAN CELL LINE ACTIVATION TEST)
INLEDANDE ÖVERVÄGANDEN OCH BEGRÄNSNINGAR
|
1.
|
h-CLAT-testet kvantifierar förändringar i uttrycket av cellytemarkörer som är kopplade till aktiveringsprocessen för monocyter och dendritiska celler (det vill säga CD86 och CD54) i den humana cellinjen THP-1 (med ursprung i monocytisk leukemi) efter exponering för sensibiliserande ämnen (1) (2). De uppmätta uttrycksnivåerna för cellytemarkörerna CD86 och CD54 används sedan för att understödja uppdelningen mellan hudsensibiliserande ämnen och icke-hudsensibiliserande ämnen.
|
|
2.
|
h-CLAT-testet har utvärderats i en valideringsstudie samordnad av Europeiska unionens referenslaboratorium för alternativ till djurförsök (EURL ECVAM) med efterföljande oberoende kollegial granskning av EURL ECVAM:s rådgivande vetenskapliga kommitté (ESAC, EURL ECVAM Scientific Advisory Committee). Med beaktande av alla tillgängliga bevis och inkomna åsikter från tillsynsmyndigheter och intressenter, rekommenderades h-CLAT-testet av EURL ECVAM (3) att användas som en del av en IATA för att understödja uppdelningen mellan hudsensibiliserande ämnen och icke-hudsensibiliserande ämnen för faroklassificering och -märkning. Exempel på användning av data från h-CLAT-test i kombination med annan information finns redovisade i litteraturen (4) (5) (6) (7) (8) (9) (10) (11).
|
|
3.
|
h-CLAT-testet har visat sig vara överförbart till laboratorier som har erfarenhet av cellodlingstekniker och flödescytometrisk analys. Testets förväntade reproducerbarhet i fråga om prediktioner är i storleksordningen 80 % inom och mellan laboratorier (3) (12). De erhållna resultaten i valideringsstudien (13) och andra publicerade studier (14) visar sammantaget att jämfört med LLNA-resultat är noggrannheten när det gäller att skilja mellan hudsensibiliserande ämnen (det vill säga kategori 1 enligt GHS/CLP-förordningen) och icke-hudsensibiliserande ämnen 85 % (N = 142), med en sensitivitet på 93 % (94/101) och en specificitet på 66 % (27/41) (baserat på en ny analys av EURL ECVAM [12] med beaktande av alla befintliga data och utan hänsyn tagen till negativa resultat för kemikalier med ett log Kow-värde över 3,5, enligt beskrivningen i punkt 4). Det är mer sannolikt att falska negativa prediktioner med h-CLAT-testet gäller kemikalier som uppvisar en låg till måttlig hudsensibiliserande styrka (det vill säga underkategori 1B enligt GHS/CLP-förordningen) än kemikalier som uppvisar en hög hudsensibiliserande styrka (det vill säga underkategori 1A enligt GHS/CLP-förordningen) (4) (13) (15). Sammantaget visar informationen att h-CLAT-testet på ett användbart sätt bidrar till identifieringen av risker för hudsensibilisering. De noggrannhetsvärden som anges här för h-CLAT-testet som ett fristående test är dock endast vägledande, eftersom testet bör beaktas i kombination med andra informationskällor inom ramen för en IATA och i enlighet med bestämmelserna i punkterna 7 och 8 i Allmän inledning. Vid utvärdering av tester utan djurförsök gällande hudsensibilisering bör man dessutom tänka på att såväl LLNA-testet som andra djurförsök eventuellt inte avspeglar situationen för människor fullt ut.
|
|
4.
|
h-CLAT-testet har, baserat på de uppgifter som finns tillgängliga för närvarande, visat sig vara tillämpligt för testkemikalier som omfattar en rad olika organiska funktionella grupper, reaktionsmekanismer, hudsensibiliseringsstyrkor (vilket har fastställts i in vivo-undersökningar) och fysikalisk-kemiska egenskaper (3) (14) (15). h-CLAT-testet är tillämpligt för testkemikalier som är lösliga eller som bildar en stabil dispersion (det vill säga en kolloid eller en suspension i vilken testkemikalien inte sjunker till botten eller separeras från lösningsmedlet/vehikeln till olika faser) i ett lämpligt lösningsmedel/en lämplig vehikel (se punkt 14). Testkemikalier som har ett log Kow-värde över 3,5 tenderar att ge falska negativa resultat (14). Negativa resultat som erhålls med testkemikalier med ett log Kow-värde över 3,5 ska därför inte tas i beaktande. Positiva resultat som erhålls med testkemikalier med ett log Kow-värde över 3,5 kan dock fortfarande användas som stöd för att identifiera testkemikalien som ett hudsensibiliserande ämne. På grund av den begränsade metaboliska kapaciteten hos den använda cellinjen (16) och testbetingelserna kan dessutom prohaptener (det vill säga ämnen som kräver enzymatisk aktivering, till exempel via P450-enzymer) och prehaptener (det vill säga ämnen som aktiveras av oxidation), i synnerhet om de har en långsam oxidationshastighet, också ge negativa resultat i h-CLAT-testet (15). Fluorescerande testkemikalier kan bedömas med h-CLAT-testet (17), men det bör noteras att kraftigt fluorescerande testkemikalier som sänder ut ljus med samma våglängd som fluoresceinisotiocyanat (FITC) eller propidiumjodid (PI) kommer att störa den flödescytometriska detekteringen och därför inte kan bedömas korrekt med användning av FITC-konjugerade antikroppar eller PI. I sådana fall kan andra fluorokrommärkta antikroppar respektive andra markörer för cytotoxicitet användas, så länge det kan visas att de ger liknande resultat som FITC-märkta antikroppar (se punkt 24) eller PI (se punkt 18), till exempel genom test av kvalifikationsämnena i tillägg 1.2. Mot bakgrund av det ovanstående bör negativa resultat tolkas utifrån de fastställda begränsningarna och tillsammans med andra informationskällor, inom ramen för en IATA. Om det finns bevis för att h-CLAT-testet inte är tillämpligt för andra specifika kategorier av testkemikalier, bör det inte användas för dessa specifika kategorier.
|
|
5.
|
Som beskrivs ovan kan h-CLAT-testet användas för att understödja uppdelningen mellan hudsensibiliserande ämnen och icke-hudsensibiliserande ämnen. Eventuellt kan testet även bidra till bedömningen av sensibiliseringens styrka (4) (5) (9) när det används i integrerade arbetssätt, exempelvis en IATA. Det krävs dock ytterligare arbete, företrädesvis baserat på data gällande människor, för att avgöra hur h-CLAT-resultat skulle kunna ligga till grund för en bedömning av sensibiliseringens styrka.
|
|
6.
|
Definitioner av begrepp finns i tillägg 1.1.
|
TESTETS PRINCIP
|
7.
|
h-CLAT-testet är ett in vitro-test som kvantifierar förändringar i uttrycket av cellytemarkörer (det vill säga CD86 och CD54) hos celler från den humana cellinjen THP-1 (med ursprung i monocytisk leukemi), efter 24 timmars exponering för testkemikalien. Dessa ytmolekyler är typiska markörer för monocytisk THP-1-aktivering och kan efterlikna aktiveringen av dendritiska celler, som spelar en avgörande roll vid priming av T-celler. Förändringarna i uttrycket av cellytemarkörer mäts genom flödescytometri efter infärgning av cellerna med fluorokrommärkta antikroppar. Mätningar av cytotoxiciteten utförs också parallellt för att bedöma huruvida uppregleringar av uttrycket av cellytemarkörer förekommer vid subcytotoxiska koncentrationer. Den relativa fluorescensintensiteten hos ytmarkörerna jämfört med lösningsmedels-/vehikelkontrollen beräknas och används i prediktionsmodellen (se punkt 26) för att understödja uppdelningen mellan hudsensibiliserande ämnen och icke-hudsensibiliserande ämnen.
|
DEMONSTRATION AV KOMPETENS
|
8.
|
Innan testet som beskrivs i detta tillägg till testmetod B.71 används rutinmässigt, bör laboratoriet visa sin tekniska kompetens med användning av de tio kvalifikationsämnen som anges i tillägg 1.2. Dessutom bör användare av testet föra en historisk databas över data som erhålls genom reaktivitetskontrollerna (se punkt 11) och genom de positiva kontrollerna och lösningsmedels-/vehikelkontrollerna (se punkterna 20–22) och använda dessa data för att styrka att testets reproducerbarhet bibehålls i laboratoriet över tid.
|
FÖRFARANDE
|
9.
|
Det här testet är baserat på h-CLAT DB-ALM-protokoll (DataBase service on ALternative Methods to animal experimentation) nr 158 (18), som är det protokoll som användes för den EURL ECVAM-samordnade valideringsstudien. Det rekommenderas att detta protokoll tillämpas när h-CLAT-testet införs och används i laboratoriet. Nedan följer en beskrivning av de viktigaste delarna i och förfarandena för h-CLAT-testet, som utgörs av två steg: ett dosfinnande test och en mätning av CD86/CD54-uttryck.
|
Förberedelse av celler
|
10.
|
Den humana cellinjen THP-1 (med ursprung i monocytisk leukemi) ska användas vid utförandet av h-CLAT-testet. Det rekommenderas att celler (TIB-202™) erhålls från en välmeriterad cellbank, exempelvis American Type Culture Collection.
|
|
11.
|
THP-1-celler odlas, vid 37°C i en fuktad atmosfär med 5 % CO2, i RPMI-1640-medium som har kompletterats med 10 % fetalt bovint serum (FBS), 0,05 mM 2-merkaptoetanol, 100 enheter/ml penicillin och 100 μg/ml streptomycin. Om så önskas går det att utelämna användningen av penicillin och streptomycin i odlingsmediet. I sådana fall ska dock användarna verifiera att frånvaron av antibiotika i odlingsmediet inte har någon inverkan på resultaten, till exempel genom att testa de kvalifikationsämnen som anges i tillägg 1.2. För att minimera risken för föroreningar ska, under alla omständigheter, goda rutiner för cellodling följas, oberoende av om antibiotika ingår i cellodlingsmediet eller inte. THP-1-celler sås regelbundet ut, varannan till var tredje dag, med en densitet på 0,1 till 0,2 × 106 celler/ml. Cellernas densitet bör hållas mellan 0,1 och 1,0 × 106 celler/ml. Innan de används för testning bör cellerna genomgå en kvalificering i form av en reaktivitetskontroll. Reaktivitetskontrollen av cellerna bör utföras med användning av de positiva kontrollerna 2,4-dinitroklorbensen (DNCB) (CAS-nr 97-00-7, ≥ 99 % renhet) och nickelsulfat (NiSO4) (CAS-nr 10101-97-0, ≥ 99 % renhet) och den negativa kontrollen mjölksyra (LA) (CAS-nr 50-21-5, ≥ 85 % renhet), vilket ska ske två veckor efter upptining. Såväl DNCB som NiSO4 ska ge positiv respons för både cellytemarkören CD86 och cellytemarkören CD54, och LA ska ge negativ respons för både cellytemarkören CD86 och cellytemarkören CD54. Endast celler som har klarat reaktivitetskontrollen ska användas för testet. Celler kan förökas upp till två månader efter upptining. Antalet passager bör inte överstiga 30. Reaktivitetskontrollen bör utföras i enlighet med de förfaranden som beskrivs i punkterna 20–24.
|
|
12.
|
För testet sås THP-1-celler ut med en densitet på antingen 0,1 × 106 celler/ml eller 0,2 × 106 celler/ml, varefter de förodlas i odlingsflaskor under 72 timmar respektive 48 timmar. Det är viktigt att celldensiteten i odlingsflaskan direkt efter förodlingsperioden är så likartad som möjligt för varje experiment (genom användning av en av de båda förodlingsbetingelser som beskrivs ovan), eftersom celldensiteten i odlingsflaskan direkt efter förodlingen kan påverka det allergeninducerade uttrycket av CD86/CD54 (19). På testdagen resuspenderas de skördade cellerna från odlingsflaskorna med nytt odlingsmedium, så att en koncentration på 2 × 106 celler/ml erhålls. Därefter fördelas cellerna i en flatbottnad 24-hålsplatta (500 μl per brunn, 1 × 106 celler/brunn) eller en flatbottnad 96-hålsplatta (80 μl per brunn, 1,6 × 105 celler/brunn).
|
Dosfinnande test
|
13.
|
Ett dosfinnande test utförs för att fastställa CV75-värdet, som är den koncentration av testkemikalien som resulterar i 75 % cellviabilitet (CV) jämfört med lösningsmedels-/vehikelkontrollen. CV75-värdet används för att fastställa testkemikaliekoncentrationen för mätningen av CD86/CD54-uttrycket (se punkterna 20–24).
|
Beredning av testkemikalier och kontrollämnen
|
14.
|
Testkemikalierna och kontrollämnena bereds på testdagen. För h-CLAT-testet löses testkemikalierna upp eller dispergeras i en stabil dispersion (se även punkt 4) i saltlösning eller medium som primärt lösningsmedels-/vehikelalternativ eller dimetylsulfoxid (DMSO, ≥ 99 % renhet) som sekundärt lösningsmedels-/vehikelalternativ om testkemikalien inte är löslig eller inte bildar en stabil dispersion i de tidigare båda lösningsmedlen/vehiklarna, till en slutlig koncentration på 100 mg/ml (i saltlösning eller medium) eller 500 mg/ml (i DMSO). Andra lösningsmedel/vehiklar än de som beskrivs ovan kan användas om en tillfredsställande vetenskaplig motivering tillhandahålls. Testkemikaliens stabilitet i det slutliga lösningsmedlet/den slutliga vehikeln ska tas i beaktande.
|
|
15.
|
Med utgångspunkt i testkemikaliernas stamlösningar på 100 mg/ml (i saltlösning eller medium) eller 500 mg/ml (i DMSO) ska följande utspädningssteg utföras:
|
—
|
För saltlösning eller medium som lösningsmedel/vehikel: Åtta stamlösningar (åtta koncentrationer) bereds, genom tvåfaldiga serieutspädningar med användning av motsvarande lösningsmedel/vehikel. Dessa stamlösningar späds sedan vidare 50-faldigt i odlingsmedium (arbetslösningar). Om den högsta slutliga koncentrationen i plattan på 1 000 μg/ml är icke-toxisk, ska den maximala koncentrationen fastställas på nytt genom att ett nytt cytotoxicitetstest utförs. Den slutliga koncentrationen i plattan bör inte överstiga 5 000 μg/ml för testkemikalier som är upplösta eller dispergerade i en stabil dispersion i saltlösning eller medium.
|
|
—
|
För DMSO som lösningsmedel/vehikel: Åtta stamlösningar (åtta koncentrationer) bereds, genom tvåfaldiga serieutspädningar med användning av motsvarande lösningsmedel/vehikel. Dessa stamlösningar späds sedan vidare 250-faldigt i odlingsmedium (arbetslösningar). Den slutliga koncentrationen i plattan bör inte överstiga 1 000 μg/ml, inte ens om denna koncentration är icke-toxisk.
|
Arbetslösningarna används slutligen för exponeringen genom att en lika stor volym arbetslösning som volymen THP-1-cellsuspension tillsätts till plattan (se även punkt 17), vilket ger en ytterligare tvåfaldig spädning (normalt är det slutliga intervallet av koncentrationer i plattan 7,81–1 000 μg/ml).
|
|
16.
|
Lösningsmedels-/vehikelkontrollen som används i h-CLAT-testet utgörs av odlingsmedium (för testkemikalier som är upplösta eller dispergerade i en stabil dispersion [se punkt 4] i antingen medium eller saltlösning) eller DMSO (för testkemikalier som är upplösta eller dispergerade i en stabil dispersion i DMSO) som testas vid en enda slutlig koncentration i plattan på 0,2 %. Kontrollen genomgår samma utspädning som beskrivs för arbetslösningarna i punkt 15.
|
Applicering av testkemikalier och kontrollämnen
|
17.
|
Odlingsmediet eller arbetslösningarna som beskrivs i punkterna 15 och 16 blandas 1:1 (volymförhållande) med de cellsuspensioner som beretts i den flatbottnade 24- eller 96-hålsplattan (se punkt 12). De behandlade plattorna inkuberas därefter i 24 ± 0,5 timmar vid 37°C i en atmosfär med 5 % CO2. Se till att flyktiga testkemikalier inte avdunstar och att det inte sker någon korskontaminering mellan testkemikalier i olika brunnar, till exempel genom att plattan förseglas innan testkemikalierna inkuberas (20).
|
Infärgning med propidiumjodid (PI)
|
18.
|
Efter 24 ± 0,5 timmars exponering överförs cellerna till provrör och samlas upp genom centrifugering. Supernatanterna kasseras och de kvarvarande cellerna resuspenderas med 200 μl (vid användning av en 96-hålsplatta) eller 600 μl (vid användning av en 24-hålsplatta) av en fosfatbuffrad saltlösning som innehåller 0,1 % bovint serumalbumin (infärgningsbuffert). 200 μl cellsuspension överförs till en rundbottnad 96-hålsplatta (vid användning av en 96-hålsplatta) eller mikrorör (vid användning av en 24-hålsplatta) och tvättas två gånger med 200 μl (vid användning av en 96-hålsplatta) eller 600 μl (vid användning av en 24-hålsplatta) infärgningsbuffert. Slutligen resuspenderas cellerna i infärgningsbuffert (till exempel 400 μl) varpå PI-lösning (till exempel 20 μl) tillsätts (den slutliga koncentrationen PI kan till exempel vara 0,625 μg/ml). Andra cytotoxicitetsmarkörer, som 7-aminoaktinomycin D (7-AAD), trypanblått eller annan markör, kan användas om det kan visas att den alternativa infärgningen ger liknande resultat som PI, till exempel genom test av kvalifikationsämnena i tillägg 1.2.
|
Cytotoxicitetsmätning genom flödescytometri och uppskattning av CV75-värdet
|
19.
|
Upptaget av PI analyseras med hjälp av flödescytometri, med användning av fluorescenskanalen FL-3. Totalt 10 000 levande celler (PI-negativa) mäts. Cellviabiliteten kan beräknas av cytometriundersökningens analysprogram med användning av nedanstående formel. Om cellviabiliteten är låg bör upp till 30 000 celler, inklusive döda celler, mätas. Alternativt kan data samlas in under en minut från det att analysen påbörjas.

CV75-värdet (se punkt 13), det vill säga en koncentration som uppvisar 75 % överlevnad hos THP-1-cellerna (25 % cytotoxicitet), beräknas genom log-linjär interpolering med användning av följande formel:

där
a är minimivärdet för en cellviabilitet över 75 %,
c är maximivärdet för en cellviabilitet under 75 %,
b och d är koncentrationerna som ger upphov till cellviabilitetsvärdena a respektive c.
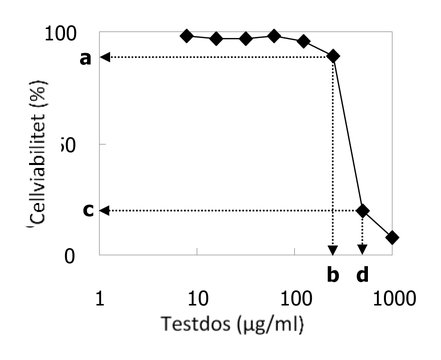
Andra metoder för att få fram CV75-värdet kan användas så länge det kan visas att detta inte har någon inverkan på resultaten (till exempel genom test av kvalifikationsämnena).
|
Mätning av CD86/CD54-uttryck
Beredning av testkemikalier och kontrollämnen
|
20.
|
Lämpligt lösningsmedel/lämplig vehikel (saltlösning, medium eller DMSO, se punkt 14) används för att lösa upp testkemikalierna eller dispergera dem i en stabil dispersion. Testkemikalierna späds först till den koncentration som motsvarar 100-faldig (för saltlösning eller medium) eller 500-faldig (för DMSO) koncentration i förhållande till 1,2 × CV75-värdet som fastställdes i det dosfinnande testet (se punkt 19). Om CV75-värdet inte kan fastställas (det vill säga om tillräcklig cytotoxicitet inte kan observeras i det dosfinnande testet), ska den högsta lösliga eller stabilt dispergerade koncentrationen av testkemikalien som beretts med varje lösningsmedel/vehikel användas som startkoncentration. Observera att den slutliga koncentrationen i plattan inte bör överstiga 5 000 μg/ml (för saltlösning eller medium) eller 1 000 μg/ml (för DMSO). Därefter görs 1,2-faldiga serieutspädningar med användning av motsvarande lösningsmedel/vehikel för att erhålla de stamlösningar (åtta koncentrationer som sträcker sig från 100 × 1,2 × CV75 till 100 × 0,335 × CV75 [för saltlösning eller medium] eller från 500 × 1,2 × CV75 till 500 × 0,335 × CV75 [för DMSO]) som ska testas med h-CLAT-testet (se DB ALM-protokoll nr 158 för ett exempel på doseringsprocedur). Stamlösningarna späds sedan vidare 50-faldigt (för saltlösning eller medium) eller 250-faldigt (för DMSO) i odlingsmediet (arbetslösningar). Dessa arbetslösningar används slutligen för exponeringen med en ytterligare avslutande tvåfaldig utspädningsfaktor i plattan. Om resultaten inte uppfyller acceptanskriterierna som beskrivs i punkterna 29 och 30 i fråga om cellviabilitet, kan det dosfinnande testet upprepas för att fastställa ett mer exakt CV75-värde. Observera att endast 24-hålsplattor kan användas för mätningen av CD86/CD54-uttryck.
|
|
21.
|
Lösningsmedels-/vehikelkontrollen bereds enligt beskrivningen i punkt 16. Den positiva kontroll som används i h-CLAT-testet utgörs av DNCB (se punkt 11), för vilken stamlösningar bereds i DMSO och späds enligt beskrivningen för stamlösningar i punkt 20. DNCB bör användas som positiv kontroll för mätning av CD86/CD54-uttryck i en enda slutlig koncentration i plattan (normalt 4,0 μg/ml). För att uppnå en koncentration på 4,0 μg/ml DNCB i plattan bereds en stamlösning med 2 mg/ml DNCB i DMSO, som sedan späds vidare 250-faldigt med odlingsmedium till en arbetslösning på 8 μg/ml. Alternativt kan även CV75-värdet för DNCB, som fastställs inom varje testanläggning, användas som koncentration för positiv kontroll. Andra lämpliga positiva kontroller kan användas om historiska data finns att tillgå för att härleda jämförbara acceptanskriterier. För positiva kontroller bör den slutliga och enda koncentrationen i plattan inte överstiga 5 000 μg/ml (för saltlösning eller medium) eller 1 000 μg/ml (för DMSO). Acceptanskriterierna är desamma som de som beskrivs för testkemikalien (se punkt 29), förutom vad gäller det sista acceptanskriteriet eftersom den positiva kontrollen testas vid endast en koncentration.
|
Applicering av testkemikalier och kontrollämnen
|
22.
|
För varje testkemikalie och kontrollämne krävs ett experiment för att erhålla en prediktion. Varje experiment består av åtminstone två oberoende testomgångar för mätning av CD86/CD54-uttryck (se punkterna 26–28). De oberoende testomgångarna ska utföras på olika dagar, alternativt på samma dag under förutsättning att följande gäller för varje testomgång: a) oberoende färska stamlösningar och arbetslösningar av testkemikalien och antikroppslösningar bereds och b) oavhängigt skördade celler används (det vill säga celler hämtas från olika odlingsflaskor); cellerna får dock komma från samma passage. Testkemikalier och kontrollämnen som beretts som arbetslösningar (500 μl) blandas med 500 μl suspenderade celler (1 × 106 celler) i förhållandet 1:1, varpå cellerna inkuberas i 24 ± 0,5 timmar enligt beskrivningen i punkterna 20 och 21. För varje testomgång räcker det med ett enda replikat för varje koncentration av testkemikalien och kontrollämnet, eftersom en prediktion erhålls från åtminstone två oberoende testomgångar.
|
Cellinfärgning och analys
|
23.
|
Efter 24 ± 0,5 timmars exponering överförs cellerna från 24-hålsplattan till provrör, samlas upp genom centrifugering och tvättas sedan två gånger med 1 ml infärgningsbuffert (vid behov kan ytterligare tvättsteg genomföras). Efter tvättningen blockeras cellerna med 600 μl blockeringslösning (infärgningsbuffert innehållande 0,01 % [vikt/volym] globulin [Cohn-fraktion II, III från människa; SIGMA, nr G2388-10G eller motsvarande]) och inkuberas vid 4°C i 15 minuter. Efter blockeringen delas cellerna upp i tre alikvoter om 180 μl i en rundbottnad 96-hålsplatta eller mikrorör.
|
|
24.
|
Efter centrifugering färgas cellerna med 50 μl FITC-märkta antikroppar av typ anti-CD86, anti-CD54 eller IgG1 från mus (isotyp) vid 4°C under 30 minuter. Antikropparna som beskrivs i h-CLAT DB-ALM-protokoll nr 158 (18) bör användas genom utspädning 3:25 (volymförhållande) (för CD86 [BD-PharMingen, nr 5556 57; klon: Fun-1]) eller 3:50 (volymförhållande) (för CD54 [DAKO, nr F7143; klon: 6.5B5] och IgG1 [DAKO, nr X0927]) med infärgningsbuffert. Dessa utspädningsfaktorer för antikropparna har definierats av testutvecklarna som de som ger det bästa signal-brusförhållandet. Enligt testutvecklarnas erfarenhet är fluorescensintensiteten hos antikropparna vanligtvis likartad mellan olika satser. Användarna kan dock överväga att titrera antikropparna under de egna laboratorieförhållandena för att få fram de bästa användningskoncentrationerna. Andra fluorokrommärkta antikroppar av typ anti-CD86 och/eller anti-CD54 kan användas om det kan visas att de ger liknande resultat som FITC-konjugerade antikroppar, till exempel genom test av kvalifikationsämnena i tillägg 1.2. Det bör noteras att byte av klon eller leverantör av antikropparna, enligt beskrivningen i h-CLAT DB-ALM-protokoll nr 158 (18), kan påverka resultaten. Efter två eller flera tvättar med 150 μl infärgningsbuffert resuspenderas cellerna i infärgningsbuffert (till exempel 400 μl) och PI-lösningen (till exempel 20 μl för att erhålla en slutlig koncentration på 0,625 μg/ml) eller en annan lösning med en cytotoxicitetsmarkör (se punkt 18) tillsätts. Uttrycksnivåerna för CD86 och CD54 och cellviabiliteten analyseras med hjälp av flödescytometri.
|
DATA OCH RAPPORTERING
Utvärdering av data
|
25.
|
Uttrycket av CD86 och CD54 analyseras genom flödescytometri med användning av fluorescenskanalen FL-1. Baserat på det geometriska medelvärdet för fluorescensintensiteten (MFI) beräknas den relativa fluorescensintensiteten (RFI) för CD86 och CD54 för celler använda för positiv kontroll och kemikaliebehandlade celler enligt följande formel:

Cellviabiliteten för isotyp-kontrollcellerna (som är färgade med IgG1-antikroppar från mus [isotyp]) beräknas också enligt den formel som beskrivs i punkt 19.
|
Prediktionsmodell
|
26.
|
För mätningen av CD86/CD54-uttryck testas varje testkemikalie i minst två oberoende testomgångar för att få fram en enda prediktion (POSITIV eller NEGATIV). En h-CLAT-prediktion anses vara POSITIV om åtminstone ett av följande villkor uppfylls i två av två, eller i åtminstone två av tre, oberoende testomgångar; i annat fall anses h-CLAT-prediktionen vara NEGATIV (figur 1):
|
—
|
RFI-värdet för CD86 är lika med eller större än 150 % vid någon testad koncentration (med cellviabilitet ≥ 50 %).
|
|
—
|
RFI-värdet för CD54 är lika med eller större än 200 % vid någon testad koncentration (med cellviabilitet ≥ 50 %).
|
|
|
27.
|
Baserat på ovanstående gäller att om båda de första två testomgångarna är positiva för CD86 och/eller för CD54 anses h-CLAT-prediktionen vara POSITIV och någon tredje testomgång behöver inte genomföras. På motsvarande sätt gäller att om de första två testomgångarna är negativa för båda markörerna anses h-CLAT-prediktionen vara NEGATIV (med hänsyn tagen till bestämmelserna i punkt 30) utan att någon tredje testomgång behöver genomföras. Om de första två testomgångarna emellertid inte ger samma resultat för minst en av markörerna (CD54 eller CD86), krävs en tredje testomgång och den slutliga prediktionen baseras då på majoritetsresultatet för de tre enskilda testomgångarna (det vill säga två av tre). I detta sammanhang bör det noteras att om två oberoende testomgångar genomförs och en endast är positiv för CD86 (nedan kallat P1
) och den andra endast är positiv för CD54 (nedan kallat P2
) krävs en tredje testomgång. Om denna tredje testomgång är negativ för båda markörerna (nedan kallat N) anses h-CLAT-prediktionen vara NEGATIV. Om å andra sidan den tredje testomgången är positiv för någon av markörerna (P1 eller P2) eller för båda markörerna (nedan kallat P12
) anses h-CLAT-prediktionen vara POSITIV.

Figur 1: Prediktionsmodell som används för h-CLAT-testet. En h-CLAT-prediktion bör beaktas inom ramen för en IATA och i enlighet med bestämmelserna i punkterna 7 och 8 i Allmän inledning.
P1: testomgång med endast CD86 positiv, P2: testomgång med endast CD54 positiv, P12: testomgång med både CD86 och CD54 positiva, N: testomgång med varken CD86 eller CD54 positiva.
* Rutorna visar de relevanta kombinationerna av resultat från de första två testomgångarna, oberoende av i vilken ordning de kan ha erhållits.
# Rutorna visar de relevanta kombinationerna av resultat från de tre testomgångarna baserat på resultaten från de första två testomgångarna i rutan ovanför, men återspeglar inte den ordning i vilken de kan ha erhållits.
|
|
28.
|
För testkemikalier som predikteras vara POSITIVA med h-CLAT-testet kan, om så önskas, två EC-värden (effektiv koncentration) fastställas, EC150 för CD86 och EC200 för CD54, det vill säga de koncentrationer vid vilka testkemikalierna ger upphov till ett RFI-värde på 150 eller 200. Dessa EC-värden kan potentiellt bidra till bedömningen av sensibiliseringsstyrka (9) när de används i integrerade arbetssätt, som exempelvis en IATA (4) (5) (6) (7) (8). De kan beräknas med hjälp av följande formler:
EC 150 (för CD86) = Bkonc
+ [(150 - BRFI
)/ARFI - BRFI
) × (Akonc - Bkonc
)]
EC 200 (för CD86) = Bkonc
+ [(200 - BRFI
)/ARFI - BRFI
) × (Akonc - Bkonc
)]
där
Akonc är den lägsta koncentration i μg/ml vid vilken RFI > 150 (CD86) eller 200 (CD54),
Bkonc är den högsta koncentration i μg/ml vid vilken RFI < 150 (CD86) eller 200 (CD54),
ARFI är RFI-värdet vid den lägsta koncentration vid vilken RFI > 150 (CD86) eller 200 (CD54),
BRFI är RFI-värdet vid den högsta koncentration vid vilken RFI < 150 (CD86) eller 200 (CD54).
För att få fram mer exakta EC150- och EC200-värden kan tre oberoende testomgångar för mätning av CD86/CD54-uttryck krävas. De slutliga EC150- och EC200-värdena fastställs sedan som medianvärdet för EC-värdena som räknats fram utifrån de tre oberoende testomgångarna. Om endast två av tre oberoende testomgångar uppfyller kriterierna för positivt resultat (se punkterna 26–27) används det högre EC150- eller EC200-värdet av de två beräknade värdena.
|
Acceptanskriterier
|
29.
|
Följande acceptanskriterier ska vara uppfyllda vid användning av h-CLAT-testet (22) (27):
|
—
|
Cellviabiliteten ska vara högre än 90 % för medium- och lösningsmedels-/vehikelkontrollerna.
|
|
—
|
I lösningsmedels-/vehikelkontrollen får inte RFI-värdena för vare sig CD86 eller CD54 överstiga kriterierna för positivt resultat (CD86 RFI ≥ 150 % och CD54 RFI ≥ 200 %). RFI-värdena för lösningsmedels-/vehikelkontrollen beräknas med användning av formeln som beskrivs i punkt 25 (”MFI för kemikalie” ska bytas ut mot ”MFI för lösningsmedel/vehikel” och ”MFI för lösningsmedel/vehikel” ska bytas ut mot ”MFI för [medium-] kontroll”).
|
|
—
|
För såväl medium- som lösningsmedels-/vehikelkontrollerna ska MFI-förhållandet för både CD86 och CD54 gentemot isotypkontrollen vara > 105 %.
|
|
—
|
I den positiva kontrollen (DNCB) ska RFI-värdena för både CD86 och CD54 uppfylla kriterierna för positivt resultat (CD86 RFI ≥ 150 och CD54 RFI ≥ 200) och cellviabiliteten ska vara högre än 50 %.
|
|
—
|
För testkemikalien ska cellviabiliteten vara högre än 50 % i minst fyra testade koncentrationer i varje testomgång.
|
|
|
30.
|
Negativa resultat godtas endast för testkemikalier som uppvisar en cellviabilitet som är lägre än 90 % vid den högsta testade koncentrationen (det vill säga 1,2 × CV75 enligt den procedur för serieutspädning som beskrivs i punkt 20). Om cellviabiliteten vid 1,2 × CV75 är lika med eller högre än 90 % ska det negativa resultatet förkastas. När så är fallet rekommenderas det att man försöker förfina dosvalet genom att upprepa fastställandet av CV75-värdet. Det bör noteras att när 5 000 μg/ml i saltlösning (eller medium eller andra lösningsmedel/vehiklar), 1 000 μg/ml i DMSO eller den högsta lösliga koncentrationen används som maximal testkoncentration för en testkemikalie är ett negativt resultat godtagbart även om cellviabiliteten är över 90 %.
|
Testrapport
|
31.
|
Testrapporten ska innehålla följande information:
|
Testkemikalie
|
Monokomponentämne:
|
Kemisk identifiering, t.ex. med IUPAC- eller CAS-namn, CAS-nummer, Smiles- eller InChI-kod, strukturformel och/eller andra identifieringar.
|
|
Fysikaliskt tillstånd, log Kow, vattenlöslighet, löslighet i DMSO, molekylvikt och andra relevanta fysikalisk-kemiska egenskaper, i den mån sådana uppgifter finns tillgängliga.
|
|
Renhet, kemisk identitet för föroreningar i förekommande fall och när så är praktiskt möjligt.
|
|
Behandling före testning, om detta är tillämpligt (t.ex. uppvärmning eller finfördelning).
|
|
Testad koncentration/testade koncentrationer.
|
|
Uppgifter om lagringsförhållanden och stabilitet i den mån sådana uppgifter finns tillgängliga.
|
|
Motivering för valet av lösningsmedel/vehikel för varje testkemikalie.
|
|
|
Multikomponentämne, UVCB och blandning:
|
Karakteriseras så långt det är möjligt genom exempelvis kemisk identitet (se ovan), renhet, kvantitativ förekomst och relevanta fysikalisk-kemiska egenskaper (se ovan) hos beståndsdelarna, i den mån sådana uppgifter finns tillgängliga.
|
|
Fysikaliskt tillstånd, vattenlöslighet, löslighet i DMSO och andra relevanta fysikalisk-kemiska egenskaper, i den mån sådana uppgifter finns tillgängliga.
|
|
Molekylvikt eller uppskattad molekylvikt för blandningar/polymerer med kända sammansättningar eller annan information som är relevant för utförandet av undersökningen.
|
|
Behandling före testning, om detta är tillämpligt (t.ex. uppvärmning eller finfördelning).
|
|
Testad koncentration/testade koncentrationer.
|
|
Uppgifter om lagringsförhållanden och stabilitet i den mån sådana uppgifter finns tillgängliga.
|
|
Motivering för valet av lösningsmedel/vehikel för varje testkemikalie.
|
|
|
|
Kontroller
|
Positiv kontroll:
|
Kemisk identifiering, t.ex. med IUPAC- eller CAS-namn, CAS-nummer, Smiles- eller InChI-kod, strukturformel och/eller andra identifieringar.
|
|
Fysikaliskt tillstånd, log Kow, vattenlöslighet, löslighet i DMSO, molekylvikt och andra relevanta fysikalisk-kemiska egenskaper, i den mån sådana uppgifter finns tillgängliga och när så är tillämpligt.
|
|
Renhet, kemisk identitet för föroreningar i förekommande fall och när så är praktiskt möjligt.
|
|
Behandling före testning, om detta är tillämpligt (t.ex. uppvärmning eller finfördelning).
|
|
Testad koncentration/testade koncentrationer.
|
|
Uppgifter om lagringsförhållanden och stabilitet i den mån sådana uppgifter finns tillgängliga.
|
|
Hänvisning till tidigare resultat för positiv kontroll som visar på lämpliga acceptanskriterier för testomgångar, om detta är tillämpligt.
|
|
|
|
Negativ kontroll och lösningsmedels-/vehikelkontroll:
|
Kemisk identifiering, t.ex. med IUPAC- eller CAS-namn, CAS-nummer, Smiles- eller InChI-kod, strukturformel och/eller andra identifieringar.
|
|
Renhet, kemisk identitet för föroreningar i förekommande fall och när så är praktiskt möjligt.
|
|
Fysikaliskt tillstånd, molekylvikt och andra relevanta fysikalisk-kemiska egenskaper om annat lösningsmedel/annan vehikel för kontroll än de som anges i testriktlinjen används, i den mån sådana uppgifter finns tillgängliga.
|
|
Uppgifter om lagringsförhållanden och stabilitet i den mån sådana uppgifter finns tillgängliga.
|
|
Motivering för valet av lösningsmedel/vehikel för varje testkemikalie.
|
|
|
Testbetingelser
|
Namn och adress för sponsor, testanläggning och försöksledare.
|
|
Beskrivning av använt test.
|
|
Använd cellinje samt dess lagringsförhållanden och källa (till exempel den anläggning varifrån cellerna erhölls).
|
|
Använd utrustning för flödescytometri (till exempel modell), inklusive använda instrumentinställningar, använt globulin och använda antikroppar och cytotoxicitetsmarkörer.
|
|
Det förfarande som använts för att visa att laboratoriet är kvalificerat att utföra testet, genom test av kvalifikationsämnen, samt det förfarande som använts för att visa att testet kan reproduceras över tid, till exempel historiska kontrolldata och/eller historiska data för reaktivitetskontroller.
|
|
|
Testets acceptanskriterier
|
Erhållna värden för cellviabilitet, MFI och RFI med lösningsmedels-/vehikelkontrollen jämfört med de godkända intervallen.
|
|
Erhållna värden för cellviabilitet och RFI med den positiva kontrollen jämfört med de godkända intervallen.
|
|
Cellviabiliteten för alla testade koncentrationer av den testade kemikalien.
|
|
|
Testförfarande
|
Antal utförda testomgångar.
|
|
Testkemikaliens koncentrationer, applicering och exponeringstid (vid avvikelser från rekommendationerna).
|
|
Beskrivning av de utvärderings- och beslutskriterier som använts.
|
|
Beskrivning av eventuella modifieringar av testförfarandet.
|
|
|
Resultat
|
Tabelluppställning av erhållna data, inklusive CV75 (om detta är tillämpligt), individuella geometriska MFI-, RFI- och cellviabilitetsvärden samt EC150/EC200-värden (om detta är tillämpligt), för testkemikalien och för den positiva kontrollen i varje testomgång, samt en indikering av testkemikaliens klassificering enligt prediktionsmodellen.
|
|
Beskrivning av andra relevanta observationer, i förekommande fall.
|
|
|
Diskussion kring resultaten
|
Diskussion kring de resultat som har erhållits med h-CLAT-testet.
|
|
Beaktande av testets resultat inom ramen för en IATA, om annan relevant information finns att tillgå.
|
|
|
LITTERATUR
|
(1)
|
Ashikaga T, Yoshida Y, Hirota M, Yoneyama K, Itagaki H, Sakaguchi H, Miyazawa M, Ito Y, Suzuki H, Toyoda H. (2006). Development of an in vitro skin sensitization test using human cell lines: The human Cell Line Activation Test (h-CLAT) I. Optimization of the h-CLAT protocol. Toxicol. In Vitro 20, s. 767–773.
|
|
(2)
|
Miyazawa M, Ito Y, Yoshida Y, Sakaguchi H, Suzuki H. (2007). Phenotypic alterations and cytokine production in THP-1 cells in response to allergens. Toxicol. In Vitro 21, s. 428–437.
|
|
(3)
|
EC EURL-ECVAM (2013). Recommendation on the human Cell Line Activation Test (h-CLAT) for skin sensitisation testing. Finns på https://eurl-ecvam.jrc.ec.europa.eu/eurl-ecvam-recommendations
|
|
(4)
|
Takenouchi O, Fukui S, Okamoto K, Kurotani S, Imai N, Fujishiro M, Kyotani D, Kato Y, Kasahara T, Fujita M, Toyoda A, Sekiya D, Watanabe S, Seto H, Hirota M, Ashikaga T, Miyazawa M. (2015). Test battery with the human cell line activation test, direct peptide reactivity assay and DEREK based on a 139 chemical data set for predicting skin sensitizing potential and potency of chemicals. J Appl Toxicol. 35, s. 1318–1332.
|
|
(5)
|
Hirota M, Fukui S, Okamoto K, Kurotani S, Imai N, Fujishiro M, Kyotani D, Kato Y, Kasahara T, Fujita M, Toyoda A, Sekiya D, Watanabe S, Seto H, Takenouchi O, Ashikaga T, Miyazawa M. (2015). Evaluation of combinations of in vitro sensitization test descriptors for the artificial neural network-based risk assessment model of skin sensitization. J Appl Toxicol. 35, s. 1333–1347.
|
|
(6)
|
Bauch C, Kolle SN, Ramirez T, Fabian E, Mehling A, Teubner W, van Ravenzwaay B, Landsiedel R. (2012). Putting the parts together: combining in vitro methods to test for skin sensitizing potencials. Regul Toxicol Parmacol. 63, s. 489–504.
|
|
(7)
|
Van der Veen JW, Rorije E, Emter R, Natch A, van Loveren H, Ezendam J. (2014). Evaluating the performance of integrated approaches for hazard identification of skin sensitizing chemicals. Regul Toxicol Pharmacol. 69, s. 371–379.
|
|
(8)
|
Urbisch D, Mehling A, Guth K, Ramirez T, Honarvar N, Kolle S, Landsiedel R, Jaworska J, Kern PS, Gerberick F, Natsch A, Emter R, Ashikaga T, Miyazawa M, Sakaguchi H. (2015). Assessing skin sensitization hazard in mice and men using non-animal test methods. Regul Toxicol Parmacol. 71, s. 337–351.
|
|
(9)
|
Jaworska JS, Natsch A, Ryan C, Strickland J, Ashikaga T, Miyazawa M. (2015). Bayesian integrated testing strategy (ITS) for skin sensitization potency assessment: a decision support system for quantitative weight of evidence and adaptive testing strategy. Arch Toxicol. 89, s. 2355–2383.
|
|
(10)
|
Strickland J, Zang Q, Kleinstreuer N, Paris M, Lehmann DM, Choksi N, Matheson J, Jacobs A, Lowit A, Allen D, Casey W. (2016). Integrated decision strategies for skin sensitization hazard. J Appl Toxicol. DOI 10.1002/jat.3281.
|
|
(11)
|
Nukada Y, Ashikaga T, Miyazawa M, Hirota M, Sakaguchi H, Sasa H, Nishiyama N. (2012). Prediction of skin sensitization potency of chemicals by human Cell Line Activation Test (h-CLAT) and an attempt at classifying skin sensitization potency. Toxicol. In Vitro 26, s. 1150–1160.
|
|
(12)
|
EC EURL ECVAM (2015). Re-analysis of the within and between laboratory reproducibility of the human Cell Line Activation Test (h-CLAT). Finns på https://eurl-ecvam.jrc.ec.europa.eu/eurl-ecvam-recommendations/eurl-ecvam-recommendation-on-the-human-cell-line-activation-test-h-clat-for-skin-sensitisation-testing
|
|
(13)
|
EC EURL ECVAM (2012). human Cell Line Activation Test (h-CLAT) Validation Study Report. Finns på https://eurl-ecvam.jrc.ec.europa.eu/eurl-ecvam-recommendations
|
|
(14)
|
Takenouchi O, Miyazawa M, Saito K, Ashikaga T, Sakaguchi H. (2013). Predictive performance of the human Cell Line Activation Test (h-CLAT) for lipophilic with high octanol-water partition coefficients. J. Toxicol. Sci. 38, s. 599–609.
|
|
(15)
|
Ashikaga T, Sakaguchi H, Sono S, Kosaka N, Ishikawa M, Nukada Y, Miyazawa M, Ito Y, Nishiyama N, Itagaki H. (2010). A comparative evaluation of in vitro skin sensitisation tests: the human cell-line activation test (h-CLAT) versus the local lymph node assay (LLNA). Altern. Lab. Anim. 38, s. 275–284.
|
|
(16)
|
Fabian E., Vogel D., Blatz V., Ramirez T., Kolle S., Eltze T., van Ravenzwaay B., Oesch F., Landsiedel R. (2013). Xenobiotic metabolizin enzyme activities in cells used for testing skin sensitization in vitro. Arch Toxicol 87, s. 1683–1969.
|
|
(17)
|
Okamoto K, Kato Y, Kosaka N, Mizuno M, Inaba H, Sono S, Ashikaga T, Nakamura T, Okamoto Y, Sakaguchi H, Kishi M, Kuwahara H, Ohno Y. (2010). The Japanese ring study of a human Cell Line Activation Test (h-CLAT) for predicting skin sensitization potential (6th report): A study for evaluating oxidative hair dye sensitization potential using h-CLAT. ATLA 15, s. 81–88.
|
|
(18)
|
DB-ALM (INVITTOX) (2014). Protocol 158: human Cell Line Activation Test (h-CLAT), s. 23. Finns på http://ecvam-dbalm.jrc.ec.europa.eu/
|
|
(19)
|
Mizuno M, Yoshida M, Kodama T, Kosaka N, Okamato K, Sono S, Yamada T, Hasegawa S, Ashikaga T, Kuwahara H, Sakaguchi H, Sato J, Ota N, Okamoto Y, Ohno Y. (2008). Effects of pre-culture conditions on the human Cell Line Activation Test (h-CLAT) results; Results of the 4th Japanese inter-laboratory study. ATLA 13, s. 70–82.
|
|
(20)
|
Sono S, Mizuno M, Kosaka N, Okamoto K, Kato Y, Inaba H, Nakamura T, Kishi M, Kuwahara H, Sakaguchi H, Okamoto Y, Ashikaga T, Ohno Y. (2010). The Japanese ring study of a human Cell Line Activation Test (h-CLAT) for predicting skin sensitization potential (7th report): Evaluation of volatile, poorly soluble fragrance materials. AATEX 15, s. 89–96.
|
|
(21)
|
OECD (2005). Guidance Document No 34 on The Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. OECD Series on Testing and Assessment. Organisationen för ekonomiskt samarbete och utveckling (OECD), Paris, Frankrike, 2005, s. 96.
|
|
(22)
|
OECD (2012). The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins. Part 1: Scientific Evidence. Series on Testing and Assessment No 168. Finns på http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=ENV/JM/MONO(2012)10/PART1&docLanguage=En
|
|
(23)
|
Förenta nationerna (FN) (2013). Globally Harmonized System of Classification and Labelling of Chemicals (GHS, det globalt harmoniserade systemet för klassificering och märkning av kemikalier). Femte reviderade utgåvan. New York och Genève: United Nations Publications. ISBN: 978-92-1-117006-1. Finns på http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html
|
|
(24)
|
ECETOC (2003). Contact sensitization: Classification according to potency. European Centre for Ecotoxicology & Toxicology of Chemicals (Technical Report No 87).
|
|
(25)
|
Ashikaga T, Sakaguchi H, Okamoto K, Mizuno M, Sato J, Yamada T, Yoshida M, Ota N, Hasegawa S, Kodama T, Okamoto Y, Kuwahara H, Kosaka N, Sono S, Ohno Y. (2008). Assessment of the human Cell Line Activation Test (h-CLAT) for Skin Sensitization; Results of the First Japanese Inter-laboratory Study. AATEX 13, s. 27–35.
|
Tillägg 1.1
DEFINITIONER
Noggrannhet
: Anger hur väl testresultaten och de godtagna referensvärdena överensstämmer med varandra. Det är ett mått på testets prestanda och en relevansaspekt. Detta begrepp används ofta omväxlande med ”konkordans” för att beskriva andelen korrekta resultat med ett test (21).
AOP (Adverse Outcome Pathway)
: Sekvens av händelser, från den kemiska strukturen hos en målkemikalie eller en grupp av liknande kemikalier, genom den molekylära inledande händelsen och fram till ett in vivo-resultat av intresse (22).
Kemikalie
: Ett ämne eller en blandning.
CV75
: Den uppskattade koncentration som uppvisar 75 % cellviabilitet.
EC150
: Koncentrationer som uppvisar RFI-värden på 150 för CD86-uttryck.
EC200
: Koncentrationer som uppvisar RFI-värden på 200 för CD54-uttryck.
Flödescytometri
: En cytometrisk teknik där celler som är suspenderade i en vätska en i taget flödar genom ett fokuserat exciterande ljus, som sprids i mönster som är kännetecknande för cellerna och deras komponenter; celler märks ofta med fluorescerande markörer, så att ljus först absorberas och sedan emitteras med ändrad frekvens.
Fara
: Inneboende egenskap hos ett ämne eller en situation som har potential att orsaka skadliga effekter när en organism, ett system eller en (del-)population exponeras för ämnet.
IATA (Integrated Approach to Testing and Assessment)
: En strukturerad strategi som används för identifiering av fara (risk), karakterisering av fara (styrka) och/eller säkerhetsbedömning (risk/styrka och exponering) för en kemikalie eller en grupp av kemikalier, som strategiskt integrerar och väger in alla relevanta data som underlag för tillsynsbeslut om potentiell fara och/eller risk och/eller om behovet av ytterligare riktad och därigenom minimerad testning.
Mediumkontroll
: Ett obehandlat replikat som innehåller alla komponenter i ett testsystem. Detta prov hanteras tillsammans med testkemikaliebehandlade prover och andra kontrollprover för att avgöra huruvida lösningsmedlet/vehikeln interagerar med testsystemet.
Blandning
: En blandning eller en lösning som består av två eller flera ämnen.
Monokomponentämne
: Ett ämne, definierat av sin kvantitativa sammansättning, där en huvudsaklig beståndsdel utgör minst 80 viktprocent.
Multikomponentämne
: Ett ämne, definierat av sin kvantitativa sammansättning, där fler än en huvudsaklig beståndsdel förekommer i en koncentration på ≥ 10 viktprocent och < 80 viktprocent. Ett multikomponentämne är resultatet av en tillverkningsprocess. Skillnaden mellan blandningar och multikomponentämnen är att en blandning erhålls genom att två eller flera ämnen blandas utan någon kemisk reaktion. Ett multikomponentämne är resultatet av en kemisk reaktion.
Positiv kontroll
: Ett replikat som innehåller testsystemets alla komponenter och som behandlats med ett ämne som man vet ger en positiv respons. För att säkerställa att variationer i den positiva kontrollens respons över tid kan bedömas, bör den positiva responsen inte vara alltför kraftfull.
Prehaptener
: Kemikalier som blir sensibiliserande ämnen genom abiotisk omvandling.
Prohaptener
: Kemikalier som kräver enzymatisk aktivering för att deras hudsensibiliserande potential ska aktiveras.
Relativ fluorescensintensitet (RFI)
: Relativa värden avseende MFI-värdet (det geometriska medelvärdet för fluorescensintensiteten) för kemikaliebehandlade celler jämfört med MFI-värdet för lösningsmedels-/vehikelbehandlade celler.
Relevans
: Beskrivning av förhållandet mellan testet och den berörda effekten och huruvida testet är meningsfullt och användbart för ett visst ändamål. Relevansen är ett mått på hur pass väl man genom testet korrekt kan mäta eller prediktera den biologiska effekt det gäller. Relevansen omfattar även överväganden om ett tests noggrannhet (konkordans) (21).
Tillförlitlighet
: Mått på i vilken utsträckning ett test kan reproduceras inom och mellan laboratorier över tid, när det utförs med användning av samma protokoll. Tillförlitligheten bedöms genom att man beräknar reproducerbarheten inom och mellan laboratorier och repeterbarheten inom laboratorier (21).
Testomgång
: En testomgång utgörs av att en eller flera testkemikalier testas parallellt med en lösningsmedels-/vehikelkontroll och en positiv kontroll.
Sensitivitet
: Den andel av alla positiva/aktiva kemikalier som klassificeras korrekt av testet. Känsligheten är ett mått på noggrannheten hos ett test som ger kategoriserande resultat och är en viktig faktor vid bedömningen av ett tests relevans (21).
Infärgningsbuffert
: En fosfatbuffrad saltlösning som innehåller 0,1 % bovint serumalbumin.
Lösningsmedels-/vehikelkontroll
: Ett obehandlat prov som innehåller alla komponenter i ett testsystem förutom testkemikalien, men med det lösningsmedel/den vehikel som används. Lösningsmedels-/vehikelkontrollen används för att fastställa grundresponsen för de prover som behandlas med testkemikalien som är upplöst eller dispergerad i en stabil dispersion i samma lösningsmedel/vehikel. Vid test med en parallell mediumkontroll, visar provet också huruvida lösningsmedlet/vehikeln interagerar med testsystemet.
Specificitet
: Den andel av alla negativa/inaktiva kemikalier som klassificeras korrekt av testet. Specificiteten är ett mått på noggrannheten hos ett test som ger kategoriserande resultat och är en viktig faktor vid bedömningen av ett tests relevans (21).
Ämne
: Ett kemiskt grundämne och föreningar av detta grundämne i naturlig eller tillverkad form, inklusive eventuella tillsatser som är nödvändiga för att bevara ämnets stabilitet och sådana föroreningar som härrör från tillverkningsprocessen, men exklusive eventuella lösningsmedel som kan avskiljas utan att det påverkar ämnets stabilitet eller ändrar dess sammansättning.
Testkemikalie
: Alla ämnen eller blandningar som testas med detta test.
FN:s globalt harmoniserade system för klassificering och märkning av kemikalier (GHS)
: Ett system för att klassificera kemikalier (ämnen och blandningar) utifrån standardiserade typer och nivåer av fysiska risker, hälsorisker och miljörisker och informera om resultaten genom exempelvis piktogram, signalord, faroangivelser, skyddsangivelser och säkerhetsdatablad, i syfte att informera om kemikaliernas skadliga effekter för att skydda människor (inklusive arbetsgivare, arbetstagare, transportörer, konsumenter och personal inom räddningstjänsten) och miljön (23).
UVCB-ämnen
: Ämnen med okänd eller varierande sammansättning, komplexa reaktionsprodukter eller biologiska material.
Giltigt test
: Ett test som anses vara tillräckligt relevant och tillförlitligt för ett särskilt ändamål och som är baserat på vetenskapligt sunda principer. Ett test är aldrig giltigt i absolut bemärkelse, utan endast i förhållande till ett fastställt ändamål (21).
Tillägg 1.2
KVALIFIKATIONSÄMNEN
Innan testet som beskrivs i detta tillägg till testmetod B.71 används rutinmässigt, bör laboratoriet styrka sin tekniska kompetens genom att korrekt erhålla den förväntade h-CLAT-prediktionen för de tio ämnen som rekommenderas i tabell 1 och genom att erhålla CV75-, EC150- och EC200-värden som håller sig inom respektive referensintervall för minst åtta av de tio kvalifikationsämnena. Dessa kvalifikationsämnen har valts ut för att representera hela skalan av responser gällande risker för hudsensibilisering. Övriga urvalskriterier var att ämnena ska vara kommersiellt tillgängliga, att det ska finnas in vivo-referensdata av hög kvalitet och att det ska finnas in vitro-data av hög kvalitet genererade med h-CLAT-testet. Det finns dessutom publicerade referensdata att tillgå för h-CLAT-testet (3) (14).
Tabell 1
Rekommenderade ämnen för att påvisa teknisk kompetens i fråga om h-CLAT-testet
|
Kvalifikationsämne
|
CAS-nr
|
Fysikaliskt tillstånd
|
In vivo-prediktion (102)
|
CV75, referensintervall i μg/ml (103)
|
h-CLAT-resultat för CD86 (EC150-referensintervall i μg/ml) (103)
|
h-CLAT-resultat för CD54 (EC200-referensintervall i μg/ml) (103)
|
|
2,4-dinitroklorbensen
|
97-00-7
|
Fast
|
Sensibiliserande (extremt)
|
2–12
|
Positivt (0,5–10)
|
Positivt (0,5–15)
|
|
4-fenylendiamin
|
106-50-3
|
Fast
|
Sensibiliserande (starkt)
|
5–95
|
Positivt (< 40)
|
Negativt (> 1,5) (104)
|
|
Nickelsulfat
|
10101-97-0
|
Fast
|
Sensibiliserande (måttligt)
|
30–500
|
Positivt (< 100)
|
Positivt (10–100)
|
|
2-merkaptobensotiazol
|
149-30-4
|
Fast
|
Sensibiliserande (måttligt)
|
30–400
|
Negativt (> 10) (104)
|
Positivt (10–140)
|
|
R(+)-limonen
|
5989-27-5
|
Flytande
|
Sensibiliserande (svagt)
|
> 20
|
Negativt (> 5) (104)
|
Positivt (< 250)
|
|
Imidazolidinylurea
|
39236-46-9
|
Fast
|
Sensibiliserande (svagt)
|
25–100
|
Positivt (20–90)
|
Positivt (20–75)
|
|
Isopropanol
|
67-63-0
|
Flytande
|
Ej sensibiliserande
|
> 5000
|
Negativt (> 5000)
|
Negativt (> 5000)
|
|
Glycerol
|
56-81-5
|
Flytande
|
Ej sensibiliserande
|
> 5000
|
Negativt (> 5000)
|
Negativt (> 5000)
|
|
Mjölksyra
|
50-21-5
|
Flytande
|
Ej sensibiliserande
|
1500–5000
|
Negativt (> 5000)
|
Negativt (> 5000)
|
|
4-aminobensoesyra
|
150-13-0
|
Fast
|
Ej sensibiliserande
|
> 1000
|
Negativt (> 1000)
|
Negativt (> 1000)
|
|
Förkortningar: CAS-nr = nummer i registret som förs av Chemical Abstracts Service.
|
Tillägg 2
IN VITRO-TEST AVSEENDE HUDSENSIBILISERING: U-SENS™ (AKTIVERINGSTEST FÖR CELLINJEN U937)
INLEDANDE ÖVERVÄGANDEN OCH BEGRÄNSNINGAR
|
1.
|
U-SENS™-testet kvantifierar förändringen i uttrycket av en cellytemarkör som är kopplad till aktiveringsprocessen för monocyter och dendritiska celler (det vill säga CD86) hos den humana cellinjen U937 (med ursprung i histiocytiskt lymfom) efter exponering för sensibiliserande ämnen (1). De uppmätta uttrycksnivåerna för cellytemarkören CD86 i cellinjen U937 används sedan för att understödja uppdelningen mellan hudsensibiliserande ämnen och icke-hudsensibiliserande ämnen.
|
|
2.
|
U-SENS™-testet har utvärderats i en valideringsstudie (2) samordnad av L’Oreal med efterföljande oberoende kollegial granskning av EURL ECVAM:s rådgivande vetenskapliga kommitté (ESAC) (3). Med beaktande av alla tillgängliga bevis och inkomna åsikter från tillsynsmyndigheter och intressenter, rekommenderades U-SENS™-testet av EURL ECVAM (4) att användas som en del av en IATA för att understödja uppdelningen mellan hudsensibiliserande ämnen och icke-hudsensibiliserande ämnen för faroklassificering och -märkning. I sitt vägledande dokument om redovisning av strukturerade arbetssätt för dataintegrering och individuella informationskällor använda inom ramen för en IATA för hudsensibilisering, diskuterar OECD för närvarande ett antal fallstudier som beskriver olika teststrategier och prediktionsmodeller. Ett av de definierade arbetssätten är baserat på U-SENS™-testet (5). Exempel på användning av data från U-SENS™-test i kombination med annan information, däribland historiska data och befintliga giltiga data gällande människor (6), finns även redovisade på andra ställen i litteraturen (4) (5) (7).
|
|
3.
|
U-SENS™-testet har visat sig vara överförbart till laboratorier som har erfarenhet av cellodlingstekniker och flödescytometrisk analys. Testets förväntade reproducerbarhet i fråga om prediktioner är i storleksordningen 90 % inom laboratorier respektive 84 % mellan laboratorier (8). De erhållna resultaten i valideringsstudien (8) och andra publicerade studier (1) visar sammantaget att jämfört med LLNA-resultat är noggrannheten när det gäller att skilja mellan hudsensibiliserande ämnen (det vill säga kategori 1 enligt GHS/CLP-förordningen) och icke-hudsensibiliserande ämnen 86 % (N = 166), med en sensitivitet på 91 % (118/129) och en specificitet på 65 % (24/37). Jämfört med resultat på människa är noggrannheten när det gäller att skilja mellan hudsensibiliserande ämnen (det vill säga kategori 1 enligt GHS/CLP-förordningen) och icke-hudsensibiliserande ämnen 77 % (N = 101) med en sensitivitet på 100 % (58/58) och en specificitet på 47 % (20/43). Det är mer sannolikt att falska negativa prediktioner med U-SENS™-testet, jämfört med LLNA-resultat, gäller kemikalier som uppvisar en låg till måttlig hudsensibiliserande styrka (det vill säga underkategori 1B enligt GHS/CLP-förordningen) än kemikalier som uppvisar en hög hudsensibiliserande styrka (det vill säga underkategori 1A enligt GHS/CLP-förordningen) (1) (8) (9). Sammantaget visar informationen att U-SENS™-testet på ett användbart sätt bidrar till identifieringen av risker för hudsensibilisering. De noggrannhetsvärden som anges här för U-SENS™-testet som ett fristående test är dock endast vägledande, eftersom testet bör beaktas i kombination med andra informationskällor inom ramen för en IATA och i enlighet med bestämmelserna i punkterna 7 och 8 i Allmän inledning. Vid utvärdering av tester utan djurförsök gällande hudsensibilisering bör man dessutom tänka på att såväl LLNA-testet som andra djurförsök eventuellt inte avspeglar situationen för människor fullt ut.
|
|
4.
|
U-SENS™-testet har, baserat på de uppgifter som finns tillgängliga för närvarande, visat sig vara tillämpligt för testkemikalier (inklusive beståndsdelar i kosmetika, till exempel konserveringsmedel, ytaktiva ämnen, aktiva substanser och färgämnen) som omfattar en rad olika organiska funktionella grupper, fysikalisk-kemiska egenskaper och hudsensibiliseringsstyrkor (vilket har fastställts i in vivo-undersökningar), samt spektrumet av reaktionsmekanismer som är kända för att vara associerade med hudsensibilisering (det vill säga Michaeladdition, Schiff-basbildning, acylering, bimolekylär nukleofil substitution [SN2] eller nukleofil aromatisk substitution [SNAr]) (1) (8) (9) (10). U-SENS™-testet är tillämpligt för testkemikalier som är lösliga eller som bildar en stabil dispersion (det vill säga en kolloid eller en suspension i vilken testkemikalien inte sjunker till botten eller separeras från lösningsmedlet/vehikeln till olika faser) i ett lämpligt lösningsmedel/en lämplig vehikel (se punkt 13). Kemikalier i datamängden som rapporteras vara prehaptener (det vill säga ämnen som aktiveras genom oxidation) eller prohaptener (det vill säga ämnen som kräver enzymatisk aktivering, till exempel via P450-enzymer) har kunnat predikteras korrekt med U-SENS™-testet (1) (10). Membranförstörande ämnen kan leda till falska positiva resultat till följd av en icke-specifik ökning av CD86-uttrycket, vilket visas av att tre av sju falska positiva svar i förhållande till in vivo-referensklassificeringen var ytaktiva ämnen (1). Av den anledningen bör positiva resultat med ytaktiva ämnen betraktas med försiktighet, medan negativa resultat med ytaktiva ämnen fortfarande kan användas som stöd för identifieringen av testkemikalien som ett icke-sensibiliserande ämne. Fluorescerande testkemikalier kan bedömas med U-SENS™-testet (1), men det bör noteras att kraftigt fluorescerande testkemikalier som sänder ut ljus med samma våglängd som fluoresceinisotiocyanat (FITC) eller propidiumjodid (PI) kommer att störa den flödescytometriska detekteringen och därför inte kan bedömas korrekt med användning av FITC-konjugerade antikroppar (potential för falska negativa resultat) eller PI (viabiliteten är inte mätbar). I sådana fall kan andra fluorokrommärkta antikroppar respektive andra markörer för cytotoxicitet användas, så länge det kan visas att de ger liknande resultat som FITC-märkta antikroppar eller PI (se punkt 18), till exempel genom test av kvalifikationsämnena i tillägg 2.2. Mot bakgrund av det ovanstående bör positiva resultat med ytaktiva ämnen och negativa resultat med kraftigt fluorescerande testkemikalier tolkas utifrån de fastställda begränsningarna och tillsammans med andra informationskällor, inom ramen för en IATA. Om det finns bevis för att U-SENS™-testet inte är tillämpligt för andra specifika kategorier av testkemikalier, bör det inte användas för dessa specifika kategorier.
|
|
5.
|
Som beskrivs ovan kan U-SENS™-testet användas för att understödja uppdelningen mellan hudsensibiliserande ämnen och icke-hudsensibiliserande ämnen. Eventuellt kan testet även bidra till bedömningen av sensibiliseringens styrka när det används i integrerade arbetssätt, exempelvis en IATA. Det krävs dock ytterligare arbete, företrädesvis baserat på data gällande människor, för att avgöra hur U-SENS™-resultat skulle kunna ligga till grund för en bedömning av sensibiliseringens styrka.
|
|
6.
|
Definitioner av begrepp finns i tillägg 2.1.
|
TESTETS PRINCIP
|
7.
|
U-SENS™-testet är ett in vitro-test som kvantifierar förändringar i uttrycket av CD86-cellytemarkör hos celler från den humana cellinjen U937 (med ursprung i histiocytiskt lymfom), efter 45 ± 3 timmars exponering för testkemikalien. CD86-cellytemarkören är en typisk markör för U937-aktivering. Det är allmänt känt att CD86 är en samstimulerande molekyl som kan efterlikna monocytisk aktivering, som spelar en avgörande roll vid priming av T-celler. Förändringarna i uttrycket av CD86-cellytemarkören mäts genom flödescytometri efter infärgning av cellerna, normalt med FITC-märkta antikroppar. Mätningar av cytotoxiciteten utförs också parallellt (till exempel med användning av PI) för att bedöma huruvida uppregleringar av uttrycket av CD86-cellytemarkören förekommer vid subcytotoxiska koncentrationer. Stimulationsindexet (SI) för CD86-cellytemarkören jämfört med lösningsmedels-/vehikelkontrollen beräknas och används i prediktionsmodellen (se punkt 19) för att understödja uppdelningen mellan hudsensibiliserande ämnen och icke-hudsensibiliserande ämnen.
|
DEMONSTRATION AV KOMPETENS
|
8.
|
Innan testet som beskrivs i detta tillägg till testmetod B.71 används rutinmässigt, bör laboratoriet styrka sin tekniska kompetens med användning av de tio kvalifikationsämnen som anges i tillägg 2.2, i enlighet med god praxis för in vitro-metoder (11). Dessutom bör användare av testet föra en historisk databas över data som erhålls genom reaktivitetskontrollerna (se punkt 11) och genom de positiva kontrollerna och lösningsmedels-/vehikelkontrollerna (se punkterna 15–16) och använda dessa data för att styrka att testets reproducerbarhet bibehålls i laboratoriet över tid.
|
FÖRFARANDE
|
9.
|
Det här testet är baserat på U-SENS™ DB-ALM-protokoll (DataBase service on ALternative Methods to animal experimentation) nr 183 (12). Standardförfaranden (SOP, Standard Operating Procedures) bör användas när U-SENS™-testet införs och används i laboratoriet. Ett automatiserat system för genomförande av U-SENS™-testet kan användas om det kan styrkas att det ger liknande resultat, till exempel genom test av kvalifikationsämnena i tillägg 2.2. Nedan följer en beskrivning av de viktigaste delarna i och förfarandena för U-SENS™-testet.
|
Förberedelse av celler
|
10.
|
Den humana cellinjen U937 (med ursprung i histiocytiskt lymfom) (13) ska användas vid utförandet av U-SENS™-testet. Celler (av klon CRL1593.2) bör erhållas från en välmeriterad cellbank, exempelvis American Type Culture Collection.
|
|
11.
|
U937-celler odlas, vid 37 °C i en fuktad atmosfär med 5 % CO2, i RPMI-1 640-medium som har kompletterats med 10 % fetalt kalvserum (FCS), 2 mM L-glutamin, 100 enheter/ml penicillin och 100 μg/ml streptomycin (nedan kallat komplett medium). U937-celler ska regelbundet genomgå cellpassage, varannan till var tredje dag, med en densitet på 1,5 respektive 3 × 105 celler/ml. Celldensiteten bör inte överstiga 2 × 106 celler/ml och cellviabiliteten, uppmätt genom infärgning av döda celler med trypanblått, bör vara ≥ 90 % (gäller inte vid den första cellpassagen efter upptining). Innan de används för testning bör varje sats med celler, FCS eller antikroppar genomgå en kvalificering i form av en reaktivitetskontroll. Reaktivitetskontrollen av cellerna bör utföras med användning av den positiva kontrollen trinitrobensensulfonsyra (2,4,6-trinitrobensensulfonsyra: TNBS) (CAS-nr 2 508-19-2, ≥ 99 % renhet) och den negativa kontrollen mjölksyra (CAS-nr 50-21-5, ≥ 85 % renhet), när det har gått åtminstone en vecka efter upptining. För reaktivitetskontrollen bör sex slutliga koncentrationer testas för var och en av de båda kontrollerna (TNBS: 1, 12,5, 25, 50, 75 och 100 μg/ml och mjölksyra: 1, 10, 20, 50, 100 och 200 μg/ml). TNBS löst i komplett medium bör ge en positiv och koncentrationsrelaterad respons för CD86 (till exempel när en positiv koncentration, CD86 SI ≥ 150, följs av en koncentration med ett högre CD86 SI-värde) och mjölksyra löst i komplett medium bör ge en negativ respons för CD86 (se punkt 21). Endast sådana satser med celler som har klarat reaktivitetskontrollen två gånger ska användas för testet. Celler kan förökas upp till sju veckor efter upptining. Antalet passager bör inte överstiga 21. Reaktivitetskontrollen bör utföras i enlighet med de förfaranden som beskrivs i punkterna 18–22.
|
|
12.
|
För testet sås U937-celler ut med en densitet på antingen 3 × 105 celler/ml eller 6 × 105 celler/ml, varefter de förodlas i odlingsflaskor under två dygn respektive ett dygn. Andra förodlingsförhållanden än de som beskrivs ovan kan användas om en tillfredsställande vetenskaplig motivering tillhandahålls och om det kan styrkas att liknande resultat uppnås, till exempel genom test av kvalifikationsämnena i tillägg 2.2. På testdagen resuspenderas de skördade cellerna från odlingsflaskorna med nytt odlingsmedium, så att en koncentration på 5 × 105 celler/ml erhålls. Därefter fördelas cellerna i en flatbottnad 96-hålsplatta med 100 μl per brunn (vilket ger en slutlig celldensitet på 0,5 × 105 celler/brunn).
|
Beredning av testkemikalier och kontrollämnen
|
13.
|
Lösligheten bedöms före testet. För detta ändamål löses testkemikalierna upp eller dispergeras i en stabil dispersion, med koncentrationen 50 mg/ml, i komplett medium som primärt lösningsmedelsalternativ eller dimetylsulfoxid (DMSO, ≥ 99 % renhet) som sekundärt lösningsmedels-/vehikelalternativ om testkemikalien inte är löslig i lösningsmedlet/vehikeln komplett medium. För själva testet löses testkemikalien till en slutlig koncentration på 0,4 mg/ml i komplett medium, om kemikalien är löslig i detta lösningsmedel/denna vehikel. Om kemikalien endast är löslig i DMSO, löses kemikalien till en koncentration på 50 mg/ml. Andra lösningsmedel/vehiklar än de som beskrivs ovan kan användas om en tillfredsställande vetenskaplig motivering tillhandahålls. Testkemikaliens stabilitet i det slutliga lösningsmedlet/den slutliga vehikeln ska tas i beaktande.
|
|
14.
|
Testkemikalierna och kontrollämnena bereds på testdagen. Eftersom något dosfinnande test inte genomförs bör, för den första testomgången, sex slutliga koncentrationer testas (1, 10, 20, 50, 100 och 200 μg/ml), utspädda i motsvarande lösningsmedel/vehikel, antingen komplett medium eller 0,4 % DMSO i medium. För de efterföljande testomgångarna ska minst fyra arbetslösningar (det vill säga minst fyra koncentrationer), med start från en testkemikalielösning på 0,4 mg/ml i komplett medium eller 50 mg/ml i DMSO, beredas med användning av motsvarande lösningsmedel/vehikel. Arbetslösningarna används slutligen för behandlingen genom att en lika stor volym U937-cellsuspension (se punkt 11 ovan) som volymen arbetslösning tillsätts till plattan, vilket ger en ytterligare tvåfaldig spädning (12). Koncentrationerna (minst fyra koncentrationer) för eventuella ytterligare testomgångar väljs baserat på de enskilda resultaten från alla tidigare testomgångar (8). De användbara slutliga koncentrationerna är 1, 2, 3, 4, 5, 7,5, 10, 12,5, 15, 20, 25, 30, 35, 40, 45, 50, 60, 70, 80, 90, 100, 120, 140, 160, 180 och 200 μg/ml. Den maximala slutliga koncentrationen är 200 μg/ml. Om ett CD86-positivt värde observeras vid 1 μg/ml ska man göra ett test vid 0,1 μg/ml för att hitta den koncentration av testkemikalien som inte ger upphov till en CD86-respons över gränsen för positivt resultat. För varje testomgång beräknas EC150-värdet (den koncentration vid vilken en kemikalie uppnår det CD86-positiva gränsvärdet 150 %, se punkt 19), om en CD86-positiv koncentrationsrespons observeras. Om testkemikalien ger upphov till en positiv CD86-respons som inte är koncentrationsrelaterad är det eventuellt inte relevant att beräkna EC150-värdet, enligt beskrivningen i U-SENS™ DB-ALM-protokoll nr 183 (12). För varje testomgång beräknas CV70-värdet (den koncentration vid vilken en kemikalie uppnår cytotoxicitetsgränsvärdet på 70 %, se punkt 19), om detta är möjligt (12). För att undersöka den koncentrationsrelaterade responsen vid ökat CD86-värde, bör valfria koncentrationer bland de användbara koncentrationerna väljas ut, jämnt spridda mellan EC150 (eller den högsta CD86-negativa icke-cytotoxiska koncentrationen) och CV70 (eller den högsta tillåtna koncentrationen, det vill säga 200 μg/ml). Minst fyra koncentrationer bör testas per testomgång, där åtminstone två koncentrationer är gemensamma med den föregående testomgången/de föregående testomgångarna, för jämförelse.
|
|
15.
|
Lösningsmedels-/vehikelkontrollen som används i U-SENS™-testet utgörs av komplett medium (för testkemikalier som är upplösta eller dispergerade i en stabil dispersion i komplett medium) (se punkt 4) eller 0,4 % DMSO i komplett medium (för testkemikalier som är upplösta eller dispergerade i en stabil dispersion i DMSO).
|
|
16.
|
Den positiva kontrollen som används i U-SENS™-testet utgörs av TNBS (se punkt 11), som har beretts i komplett medium. TNBS bör användas som positiv kontroll för mätning av CD86-uttryck i en enda slutlig koncentration i plattan (50 μg/ml) som ger > 70 % cellviabilitet. För att uppnå en koncentration på 50 μg/ml TNBS i plattan bereds en 1 M (det vill säga 293 mg/ml) stamlösning av TNBS i komplett medium, som sedan späds vidare 2 930-faldigt med komplett medium till en arbetslösning på 100 μg/ml. Mjölksyra (CAS-nr 50-21-5) bör användas som negativ kontroll med koncentrationen 200 μg/ml löst i komplett medium (från en stamlösning på 0,4 mg/ml). I varje platta för varje testomgång bereds tre replikat med obehandlad kontroll (komplett medium), lösningsmedels-/vehikelkontroll, negativ kontroll och positiv kontroll (12). Andra lämpliga positiva kontroller kan användas om historiska data finns att tillgå för att härleda jämförbara acceptanskriterier. Acceptanskriterierna är desamma som de som beskrivs för testkemikalien (se punkt 12).
|
Applicering av testkemikalier och kontrollämnen
|
17.
|
Lösningsmedels-/vehikelkontrollen eller arbetslösningarna som beskrivs i punkterna 14–16 blandas 1:1 (volymförhållande) med de cellsuspensioner som beretts i den flatbottnade 96-hålsplattan (se punkt 12). De behandlade plattorna inkuberas därefter i 45 ± 3 timmar vid 37 °C i en atmosfär med 5 % CO2. Innan inkuberingen förseglas plattorna med semipermeabla membran, för att förhindra avdunstning av flyktiga testkemikalier och korskontaminering mellan celler behandlade med testkemikalier (12).
|
Cellinfärgning
|
18.
|
Efter 45 ± 3 timmars exponering överförs cellerna till en mikrotiterplatta med brunnar med V-formad botten och samlas upp genom centrifugering. Störd löslighet definieras som kristaller eller droppar som kan observeras under mikroskop 45 ± 3 timmar efter behandling (innan cellinfärgningen). Supernatanterna kasseras och de kvarvarande cellerna tvättas en gång med 100 μl iskall fosfatbuffrad saltlösning (PBS, Phosphate Buffered Saline) som innehåller 5 % fetalt kalvserum (infärgningsbuffert). Efter centrifugeringen resuspenderas cellerna med 100 μl infärgningsbuffert och färgas med 5 μl (till exempel 0,25 μg) FITC-märkta antikroppar av typ anti-CD86 eller IgG1 från mus (isotyp) vid 4 °C under 30 minuter, skyddat från ljus. De antikroppar som beskrivs i U-SENS™ DB-ALM-protokoll nr 183 (12) ska användas (för CD86: BD-PharMingen nr 5556 57, klon: Fun-1, eller Caltag/Invitrogen nr MHCD8601, klon: BU63; och för IgG1: BD-PharMingen nr 5557 48 eller Caltag/Invitrogen nr GM4992). Enligt testutvecklarnas erfarenhet är fluorescensintensiteten hos antikropparna vanligtvis likartad mellan olika satser. Antikroppar av andra kloner eller från andra leverantörer som har klarat reaktivitetskontrollen får användas för testet (se punkt 11). Användarna kan dock överväga att titrera antikropparna under de egna laboratorieförhållandena för att få fram den bästa användningskoncentrationen. Andra detekteringssystem, som fluorokrommärkta antikroppar av typ anti-CD86, kan användas om det kan visas att de ger liknande resultat som FITC-konjugerade antikroppar, till exempel genom av test av kvalifikationsämnena i tillägg 2.2. Efter två tvättar med 100 μl infärgningsbuffert och en tvätt med 100 μl iskall PBS resuspenderas cellerna i iskall PBS (till exempel 125 μl för prover som ska analyseras manuellt rör för rör eller 50 μl vid användning av en autosampler-platta) och PI-lösning tillsätts (med en slutlig koncentration på 3 μg/ml). Andra cytotoxicitetsmarkörer, som 7-aminoaktinomycin D (7-AAD) eller trypanblått, kan användas om det kan visas att den alternativa infärgningen ger liknande resultat som PI, till exempel genom test av kvalifikationsämnena i tillägg 2.2.
|
Analys genom flödescytometri
|
19.
|
Uttrycksnivån för CD86 och cellviabiliteten analyseras med hjälp av flödescytometri. Cellerna visas i ett punktdiagram med logaritmisk skala utifrån storlek (FSC) och granularitet (SSC), i syfte att tydligt identifiera populationen inom en första avgränsning R1 och eliminera skräp från döda celler. Ett målantal på 10 000 celler inom avgränsning R1 mäts för varje brunn. Celler från samma R1-avgränsning visas i ett FL3 eller FL4/SSC-punktdiagram. Viabla celler avskiljs genom en andra avgränsning R2, som väljer ut populationen av propidiumjodid-negativa celler (kanal FL3 eller FL4). Cellviabiliteten kan beräknas av cytometriundersökningens analysprogram med användning av nedanstående formel. Om cellviabiliteten är låg bör upp till 20 000 celler, inklusive döda celler, mätas. Alternativt kan data samlas in under en minut från det att analysen påbörjas.

Procentandelen FL1-positiva celler mäts sedan bland dessa viabla celler som avgränsats genom R2 (inom R1). Cellytornas uttryck av CD86 analyseras i ett FL1/SSC-punktdiagram som avgränsats till viabla celler (R2).
För brunnarna med komplett medium/IgG1 sätts analysmarkören nära huvudpopulationen, så att kontrollerna för komplett medium har IgG1 inom målzonen 0,6 % till 0,9 %.
Färginterferens definieras som en skiftning hos punktdiagrammet för FITC-märkt IgG1 (SI [geometriskt medelvärde] för IgG1 FL1 ≥ 150 %)
Stimulationsindexet (SI) för CD86 för kontrollceller (obehandlade eller i 0,4 % DMSO) och kemikaliebehandlade celler beräknas enligt följande formel:.

% IgG1+ obehandlade kontrollceller: avser procentandelen FL1-positiva IgG1-celler som definierats med analysmarkören (godkänt intervall ≥ 0,6 % och < 1,5 %, se punkt 22) bland de viabla obehandlade cellerna.
% IgG1+/CD86+ kontrollceller/behandlade celler: avser procentandelen FL1-positiva IgG1/CD86-celler uppmätta utan förflyttning av analysmarkören bland de viabla kontrollcellerna/behandlade cellerna.
|
DATA OCH RAPPORTERING
Utvärdering av data
|
20.
|
Följande parametrar beräknas i U-SENS™-testet: CV70-värdet, det vill säga den koncentration som uppvisar 70 % överlevnad hos U937-cellerna (30 % cytotoxicitet), och EC150-värdet, det vill säga den koncentration vid vilken testkemikalierna ger upphov till ett CD86-stimulationsindex (SI) på 150 %.
CV70-värdet beräknas genom log-linjär interpolering med användning av följande formel:
CV70 = C1 + [(V1 – 70) / (V1 – V2) × (C2 – C1)]
där
V1 är minimivärdet för en cellviabilitet över 70 %,
V2 är maximivärdet för en cellviabilitet under 70 %,
C1 och C2 är koncentrationerna som ger upphov till cellviabilitetsvärdena V1 respektive V2.
Andra metoder för att få fram CV70-värdet kan användas så länge det kan visas att detta inte har någon inverkan på resultaten (till exempel genom test av kvalifikationsämnena).
EC150-värdet beräknas genom log-linjär interpolering med användning av följande formel:
EC150 = C1 + [(150 – SI 1) / (SI 2 – SI 1) × (C2 – C1)]
där
C1 är den högsta koncentrationen i μg/ml med ett CD86 SI-värde < 150 % (SI 1),
C2 är den lägsta koncentrationen i μg/ml med ett CD86 SI-värde ≥ 150 % (SI 2).

EC150- och CV70-värdena beräknas enligt följande:
|
—
|
För varje enskild testomgång: De enskilda EC150- och CV70-värdena används som verktyg för att undersöka den koncentrationsrelaterade responsen vid ökat CD86-värde (se punkt 14).
|
|
—
|
Det övergripande CV70-värdet fastställs baserat på de genomsnittliga viabiliteterna (12).
|
|
—
|
Det övergripande EC150-värdet fastställs för en testkemikalie som predikterats som POSITIV med U-SENS™-testet baserat på genomsnittligt SI för CD86 (se punkt 21) (12).
|
|
Prediktionsmodell
|
21.
|
För mätningen av CD86-uttryck testas varje testkemikalie i minst fyra koncentrationer och i minst två oberoende testomgångar (som utförs på olika dagar) för att få fram en enda prediktion (NEGATIV eller POSITIV).
|
—
|
Det enskilda resultatet för en U-SENS™-testomgång anses vara negativt (nedan kallat N) om SI-värdet för CD86 är lägre än 150 % vid alla icke-cytotoxiska koncentrationer (cellviabilitet ≥ 70 %) och om ingen interferens observeras (cytotoxicitet, löslighet: se punkt 18 eller färg: se punkt 19, oberoende av de icke-cytotoxiska koncentrationer vid vilka interferensen detekteras). I alla övriga fall: när SI-värdet för CD86 är högre än eller lika med 150 % och/eller interferens observeras, anses det enskilda resultatet för en U-SENS™-testomgång vara positivt (nedan kallat P).
|
|
—
|
En U-SENS™-prediktion anses vara NEGATIV om åtminstone två oberoende testomgångar är negativa (N) (se figur 1). Om de första två testomgångarna båda är negativa (N) anses U-SENS™-prediktionen vara NEGATIV och någon tredje testomgång behöver inte utföras.
|
|
—
|
En U-SENS™-prediktion anses vara POSITIV om åtminstone två oberoende testomgångar är positiva (P) (se figur 1). Om de första två testomgångarna båda är positiva (P) anses U-SENS™-prediktionen vara POSITIV och någon tredje testomgång behöver inte utföras.
|
|
—
|
Eftersom något dosfinnande test inte utförs gäller ett undantag om SI-värdet för CD86 i den första testomgången är högre än eller lika med 150 % enbart vid den högsta icke-cytotoxiska koncentrationen. Testomgången anses då vara EJ AVGÖRANDE (NC, NOT CONCLUSIVE) och ytterligare koncentrationer (mellan den högsta icke-cytotoxiska koncentrationen och den lägsta cytotoxiska koncentrationen – se punkt 20) bör testas i ytterligare testomgångar. Om en testomgång identifieras som NC ska åtminstone två ytterligare testomgångar genomföras, samt en fjärde testomgång om testomgångarna 2 och 3 inte är samstämmiga (den ena är N och den andra är P) (se figur 1). Uppföljande testomgångar anses positiva även om endast en icke-cytotoxisk koncentration ger ett CD86-värde som är lika med eller högre än 150 %, eftersom koncentrationsvärdena har justerats för den specifika testkemikalien. Den slutliga prediktionen ska baseras på majoritetsresultatet för de tre eller fyra enskilda testomgångarna (det vill säga två av tre eller två av fyra) (se figur 1).
|

Figur 1: Prediktionsmodell som används för U-SENS™-testet. En U-SENS™-prediktion bör beaktas inom ramen för en IATA och i enlighet med bestämmelserna i punkt 4 samt punkterna 7, 8 och 9 i Allmän inledning.
N: Testomgång utan positivt CD86-resultat och utan observerad interferens.
P: Testomgång med positivt CD86-resultat och/eller observerad interferens.
NC: Ej avgörande. Den första testomgången räknas som Ej avgörande när CD86-resultatet endast är positivt vid den högsta icke-cytotoxiska koncentrationen.
#: Ett NC-resultat (ej avgörande), vilket endast är aktuellt för den första testomgången, medför automatiskt att det krävs en tredje testomgång för att uppnå en majoritet positiva (P) eller negativa (N) resultat, det vill säga samma resultat för minst två av tre oberoende testomgångar.
$: Rutorna visar de relevanta kombinationerna av resultat från de tre testomgångarna, baserat på resultaten från de första två testomgångarna i rutan ovanför.
°: Rutorna visar de relevanta kombinationerna av resultat från de fyra testomgångarna, baserat på resultaten från de första tre testomgångarna i rutan ovanför.
|
Acceptanskriterier
|
22.
|
Följande acceptanskriterier ska vara uppfyllda vid användning av U-SENS™-testet (12):
|
—
|
I slutet av exponeringsperioden på 45 ± 3 timmar måste den genomsnittliga viabiliteten för de tre replikaten med obehandlade U937-celler vara > 90 % och ingen drift av CD86-uttrycket får observeras. Det grundläggande CD86-uttrycket för obehandlade U937-celler måste ligga inom intervallet ≥ 2 % och ≤ 25 %.
|
|
—
|
När DMSO används som lösningsmedel bedöms giltigheten för DMSO-vehikelkontrollen genom beräkning av ett DMSO SI-värde som jämförs med motsvarande värde för obehandlade celler, och den genomsnittliga viabiliteten för de tre replikaten med celler måste vara > 90 %. DMSO-vehikelkontrollen är giltig om genomsnittet för de tre replikatens CD86 SI-värden är mindre än 250 % av genomsnittet för CD86 SI-värdena för de tre replikaten med obehandlade U937-celler.
|
|
—
|
Testomgångarna anses giltiga om åtminstone två av tre IgG1-värden för obehandlade U937-celler hamnar inom intervallet ≥ 0,6 % och < 1,5 %.
|
|
—
|
Den parallellt testade negativa kontrollen (mjölksyra) anses giltig om åtminstone två av de tre replikaten är negativa (CD86 SI < 150 %) och icke-cytotoxiska (cellviabilitet ≥ 70 %).
|
|
—
|
Den positiva kontrollen (TNBS) anses giltig om åtminstone två av de tre replikaten är positiva (CD86 SI ≥ 150 %) och icke-cytotoxiska (cellviabilitet ≥ 70 %).
|
|
Testrapport
|
23.
|
Testrapporten ska innehålla följande information:
|
Testkemikalie
Monokomponentämne:
|
Kemisk identifiering, t.ex. med IUPAC- eller CAS-namn, CAS-nummer, Smiles- eller InChI-kod, strukturformel och/eller andra identifieringar.
|
|
Fysikaliskt tillstånd, löslighet i komplett medium, löslighet i DMSO, molekylvikt och andra relevanta fysikalisk-kemiska egenskaper, i den mån sådana uppgifter finns tillgängliga.
|
|
Renhet, kemisk identitet för föroreningar i förekommande fall och när så är praktiskt möjligt.
|
|
Behandling före testning, om detta är tillämpligt (t.ex. uppvärmning eller finfördelning).
|
|
Testad koncentration/testade koncentrationer.
|
|
Uppgifter om lagringsförhållanden och stabilitet i den mån sådana uppgifter finns tillgängliga.
|
|
Motivering för valet av lösningsmedel/vehikel för varje testkemikalie.
|
Multikomponentämne, UVCB och blandning:
|
Karakteriseras så långt det är möjligt genom exempelvis kemisk identitet (se ovan), renhet, kvantitativ förekomst och relevanta fysikalisk-kemiska egenskaper (se ovan) hos beståndsdelarna, i den mån sådana uppgifter finns tillgängliga.
|
|
Fysikaliskt tillstånd, löslighet i komplett medium, löslighet i DMSO och andra relevanta fysikalisk-kemiska egenskaper, i den mån sådana uppgifter finns tillgängliga.
|
|
Molekylvikt eller uppskattad molekylvikt för blandningar/polymerer med kända sammansättningar eller annan information som är relevant för utförandet av undersökningen.
|
|
Behandling före testning, om detta är tillämpligt (t.ex. uppvärmning eller finfördelning).
|
|
Testad koncentration/testade koncentrationer.
|
|
Uppgifter om lagringsförhållanden och stabilitet i den mån sådana uppgifter finns tillgängliga.
|
|
Motivering för valet av lösningsmedel/vehikel för varje testkemikalie.
|
|
|
Kontroller
Positiv kontroll:
|
Kemisk identifiering, t.ex. med IUPAC- eller CAS-namn, CAS-nummer, Smiles- eller InChI-kod, strukturformel och/eller andra identifieringar.
|
|
Fysikaliskt tillstånd, löslighet i DMSO, molekylvikt och andra relevanta fysikalisk-kemiska egenskaper, i den mån sådana uppgifter finns tillgängliga och när så är tillämpligt.
|
|
Renhet, kemisk identitet för föroreningar i förekommande fall och när så är praktiskt möjligt.
|
|
Behandling före testning, om detta är tillämpligt (t.ex. uppvärmning eller finfördelning).
|
|
Testad koncentration/testade koncentrationer.
|
|
Uppgifter om lagringsförhållanden och stabilitet i den mån sådana uppgifter finns tillgängliga.
|
|
Hänvisning till tidigare resultat för positiv kontroll som visar på lämpliga acceptanskriterier för testomgångar, om detta är tillämpligt.
|
Negativ kontroll och lösningsmedels-/vehikelkontroll:
|
Kemisk identifiering, t.ex. med IUPAC- eller CAS-namn, CAS-nummer, Smiles- eller InChI-kod, strukturformel och/eller andra identifieringar.
|
|
Renhet, kemisk identitet för föroreningar i förekommande fall och när så är praktiskt möjligt.
|
|
Fysikaliskt tillstånd, molekylvikt och andra relevanta fysikalisk-kemiska egenskaper om annat lösningsmedel/annan vehikel för kontroll än de som anges i testriktlinjen används, i den mån sådana uppgifter finns tillgängliga.
|
|
Uppgifter om lagringsförhållanden och stabilitet i den mån sådana uppgifter finns tillgängliga.
|
|
Motivering för valet av lösningsmedel/vehikel för varje testkemikalie.
|
|
|
Testbetingelser
|
Namn och adress för sponsor, testanläggning och försöksledare.
|
|
Beskrivning av testet som använts.
|
|
Använd cellinje samt dess lagringsförhållanden och källa (till exempel den anläggning varifrån cellerna erhölls).
|
|
Använd flödescytometri (till exempel modell), inklusive använda instrumentinställningar, antikroppar och cytotoxicitetsmarkörer.
|
|
Det förfarande som använts för att visa att laboratoriet är kvalificerat att utföra testet, genom test av kvalifikationsämnen, samt det förfarande som använts för att visa att testet kan reproduceras över tid, till exempel historiska kontrolldata och/eller historiska data för reaktivitetskontroller.
|
|
|
Testets acceptanskriterier
|
Erhållna värden för cellviabilitet och CD86 SI med lösningsmedels-/vehikelkontrollen jämfört med de godkända intervallen.
|
|
Erhållna värden för cellviabilitet och SI med den positiva kontrollen jämfört med de godkända intervallen.
|
|
Cellviabiliteten för alla testade koncentrationer av den testade kemikalien.
|
|
|
Testförfarande
|
Antal utförda testomgångar.
|
|
Testkemikaliens koncentrationer, applicering och exponeringstid (vid avvikelser från rekommendationerna).
|
|
Beskrivning av de utvärderings- och beslutskriterier som använts.
|
|
Beskrivning av eventuella modifieringar av testförfarandet.
|
|
|
Resultat
|
Tabelluppställning av erhållna data, inklusive CV70 (om detta är tillämpligt), SI, cellviabilitetsvärden och EC150-värden (om detta är tillämpligt), för testkemikalien och för den positiva kontrollen i varje testomgång, samt en indikering av testkemikaliens klassificering enligt prediktionsmodellen.
|
|
Beskrivning av andra relevanta observationer, i förekommande fall.
|
|
|
Diskussion kring resultaten
|
Diskussion kring de resultat som har erhållits med U-SENS™-testet.
|
|
Beaktande av testets resultat inom ramen för en IATA, om annan relevant information finns att tillgå.
|
|
Slutsatser
|
LITTERATUR
|
(1)
|
Piroird, C., Ovigne, J.M., Rousset, F., Martinozzi-Teissier, S., Gomes, C., Cotovio, J., Alépée, N. (2015). The Myeloid U937 Skin Sensitization Test (U-SENS) addresses the activation of dendritic cell event in the adverse outcome pathway for skin sensitization. Toxicol. In Vitro 29, s. 901–916.
|
|
(2)
|
EURL ECVAM (2017). The U-SENS™ test method Validation Study Report. Finns på http://ihcp.jrc.ec.europa.eu/our_labs/eurl-ecvam/eurl-ecvam-recommendations.
|
|
(3)
|
EC EURL ECVAM (2016). ESAC Opinion No 2016-03 on the L’Oréal-coordinated study on the transferability and reliability of the U-SENS™ test method for skin sensitisation testing. EUR 28 178 EN; doi 10.2787/815737. Finns på [http://publications.jrc.ec.europa.eu/repository/handle/JRC103705].
|
|
(4)
|
EC EURL ECVAM (2017). EURL ECVAM Recommendation on the use of non-animal approaches for skin sensitisation testing. EUR 28 553 EN; doi 10.2760/588955. Finns på https://ec.europa.eu/jrc/en/publication/eur-scientific-and-technical-research-reports/eurl-ecvam-recommendation-use-non-animal-approaches-skin-sensitisation-testing.
|
|
(5)
|
Steiling, W. (2016). Safety Evaluation of Cosmetic Ingredients Regarding their Skin Sensitization Potential. doi:10.3390/cosmetics3020014. Cosmetics 3, s. 14.
|
|
(6)
|
OECD (2016). Guidance Document on The Reporting of Defined Approaches and Individual Information Sources to be Used Within Integrated Approaches to Testing and Assessment (IATA) For Skin Sensitisation, Series on Testing & Assessment No 256, ENV/JM/MONO(2016)29. Organisationen för ekonomiskt samarbete och utveckling (OECD), Paris. Finns på http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(7)
|
Urbisch, D., Mehling, A., Guth, K., Ramirez, T., Honarvar, N., Kolle, S., Landsiedel, R., Jaworska, J., Kern, P.S., Gerberick, F., Natsch, A., Emter, R., Ashikaga, T., Miyazawa, M., Sakaguchi, H. (2015). Assessing skin sensitization hazard in mice and men using non-animal test methods. Regul. Toxicol. Pharmacol. 71, s. 337–351.
|
|
(8)
|
Alépée, N., Piroird, C., Aujoulat, M., Dreyfuss, S., Hoffmann, S., Hohenstein, A., Meloni, M., Nardelli, L., Gerbeix, C., Cotovio, J. (2015). Prospective multicentre study of the U-SENS test method for skin sensitization testing. Toxicol In Vitro 30, s. 373–382.
|
|
(9)
|
Reisinger, K., Hoffmann, S., Alépée, N., Ashikaga, T., Barroso, J., Elcombe, C., Gellatly, N., Galbiati, V., Gibbs, S., Groux, H., Hibatallah, J., Keller, D., Kern, P., Klaric, M., Kolle, S., Kuehnl, J., Lambrechts, N., Lindstedt, M., Millet, M., Martinozzi-Teissier, S., Natsch, A., Petersohn, D., Pike, I., Sakaguchi, H., Schepky, A., Tailhardat, M., Templier, M., van Vliet, E., Maxwell, G. (2014). Systematic evaluation of non-animal test methods for skin sensitisation safety assessment. Toxicol. In Vitro 29, s. 259–270.
|
|
(10)
|
Fabian, E., Vogel, D., Blatz, V., Ramirez, T., Kolle, S., Eltze, T., van Ravenzwaay, B., Oesch, F., Landsiedel, R. (2013). Xenobiotic metabolizin enzyme activities in cells used for testing skin sensitization in vitro. Arch. Toxicol. 87, s. 1683–1696.
|
|
(11)
|
OECD. (2018). Draft Guidance document: Good In Vitro Method Practices (GIVIMP) for the Development and Implementation of In Vitro Methods for Regulatory Use in Human Safety Assessment. Organisationen för ekonomiskt samarbete och utveckling (OECD), Paris. Finns på http://www.oecd.org/env/ehs/testing/OECD Final Draft GIVIMP.pdf.
|
|
(12)
|
DB-ALM (2016). Protocol no 183: Myeloid U937 Skin Sensitization Test (U-SENS™), s. 33. Finns på http://ecvam-dbalm.jrc.ec.europa.eu/.
|
|
(13)
|
Sundström, C., Nilsson, K. (1976). Establishment and characterization of a human histiocytic lymphoma cell line (U-937). Int. J. Cancer 17, s. 565–577.
|
|
(14)
|
OECD (2005). Series on Testing and Assessment No. 34: Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Organisationen för ekonomiskt samarbete och utveckling (OECD), Paris. Finns på http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(15)
|
Förenta nationerna (FN) (2015). Globally Harmonized System of Classification and Labelling of Chemicals (GHS, det globalt harmoniserade systemet för klassificering och märkning av kemikalier). ST/SG/AC.10/30/Rev.6, sjätte reviderade utgåvan, New York och Genève: United Nations Publications. Finns på http://www.unece.org/fileadmin/DAM/trans/danger/publi/ghs/ghs_rev06/English/ST-SG-AC10-30-Rev6e.pdf.
|
|
(16)
|
OECD (2012). Series on Testing and Assessment No 168: The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins. Part 1: Scientific Evidence. Organisationen för ekonomiskt samarbete och utveckling (OECD), Paris. Finns på http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(17)
|
ECETOC (2003). Technical Report No 87: Contact sensitization: Classification according to potency. European Centre for Ecotoxicology & Toxicology of Chemicals, Bryssel. Finns på https://ftp.cdc.gov/pub/Documents/OEL/06.%20Dotson/References/ECETOC_2003-TR87.pdf.
|
Tillägg 2.1
DEFINITIONER
Noggrannhet
: Anger hur väl testresultaten och de godtagna referensvärdena överensstämmer med varandra. Det är ett mått på testets prestanda och en relevansaspekt. Detta begrepp används ofta omväxlande med ”konkordans” för att beskriva andelen korrekta resultat med ett test (14).
AOP (Adverse Outcome Pathway)
: Sekvens av händelser, från den kemiska strukturen hos en målkemikalie eller en grupp av liknande kemikalier, genom den molekylära inledande händelsen och fram till ett in vivo-resultat av intresse (15).
Koncentrationsrelaterad respons för CD86
: Det råder ett koncentrationsberoende (eller en koncentrationsrelaterad respons) när en positiv koncentration (CD86 SI ≥ 150) följs av en koncentration med ett ökande CD86 SI-värde.
Kemikalie
: Ett ämne eller en blandning.
CV70
: Den uppskattade koncentration som uppvisar 70 % cellviabilitet.
Drift
: Definitionen av drift är att i) det korrigerade % CD86+-värdet för det obehandlade kontrollreplikatet med nr 3 är mindre än 50 % av genomsnittet för de korrigerade % CD86+-värdena för de obehandlade kontrollreplikaten med nr 1 och 2, och ii) det korrigerade % CD86+-värdet för det negativa kontrollreplikatet med nr 3 är mindre än 50 % av genomsnittet för de korrigerade % CD86+-värdena för de negativa kontrollreplikaten med nr 1 och 2.
EC150
: De uppskattade koncentrationer som uppvisar ett SI-värde på 150 % för CD86-uttryck.
Flödescytometri
: En cytometrisk teknik där celler som är suspenderade i en vätska en i taget flödar genom ett fokuserat exciterande ljus, som sprids i mönster som är kännetecknande för cellerna och deras komponenter; celler märks ofta med fluorescerande markörer, så att ljus först absorberas och sedan emitteras med ändrad frekvens.
Fara
: Inneboende egenskap hos ett ämne eller en situation som har potential att orsaka skadliga effekter när en organism, ett system eller en (del-)population exponeras för ämnet.
IATA (Integrated Approach to Testing and Assessment)
: En strukturerad strategi som används för identifiering av fara (risk), karakterisering av fara (styrka) och/eller säkerhetsbedömning (risk/styrka och exponering) för en kemikalie eller en grupp av kemikalier, som strategiskt integrerar och väger in alla relevanta data som underlag för tillsynsbeslut om potentiell fara och/eller risk och/eller om behovet av ytterligare riktad och därigenom minimerad testning.
Blandning
: En blandning eller en lösning som består av två eller flera ämnen.
Monokomponentämne
: Ett ämne, definierat av sin kvantitativa sammansättning, där en huvudsaklig beståndsdel utgör minst 80 viktprocent.
Multikomponentämne
: Ett ämne, definierat av sin kvantitativa sammansättning, där fler än en huvudsaklig beståndsdel förekommer i en koncentration på ≥ 10 viktprocent och < 80 viktprocent. Ett multikomponentämne är resultatet av en tillverkningsprocess. Skillnaden mellan blandningar och multikomponentämnen är att en blandning erhålls genom att två eller flera ämnen blandas utan någon kemisk reaktion. Ett multikomponentämne är resultatet av en kemisk reaktion.
Positiv kontroll
: Ett replikat som innehåller testsystemets alla komponenter och som behandlats med ett ämne som man vet ger en positiv respons. För att säkerställa att variationer i den positiva kontrollens respons över tid kan bedömas, bör den positiva responsen inte vara alltför kraftfull.
Prehaptener
: Kemikalier som blir sensibiliserande ämnen genom abiotisk omvandling, till exempel genom oxidering.
Prohaptener
: Kemikalier som kräver enzymatisk aktivering för att deras hudsensibiliserande potential ska aktiveras.
Relevans
: Beskrivning av förhållandet mellan testet och den berörda effekten och huruvida det är meningsfullt och användbart för ett visst ändamål. Relevansen är ett mått på hur pass väl man genom testet korrekt kan mäta eller prediktera den biologiska effekt det gäller. Relevansen omfattar även överväganden om ett tests noggrannhet (konkordans) (14).
Tillförlitlighet
: Mått på i vilken utsträckning ett test kan reproduceras inom och mellan laboratorier över tid, när det utförs med användning av samma protokoll. Tillförlitligheten bedöms genom att man beräknar reproducerbarheten inom och mellan laboratorier och repeterbarheten inom laboratorier (14).
Testomgång
: En testomgång utgörs av att en eller flera testkemikalier testas parallellt med en lösningsmedels-/vehikelkontroll och en positiv kontroll.
Sensitivitet
: Den andel av alla positiva/aktiva kemikalier som klassificeras korrekt av testet. Känsligheten är ett mått på noggrannheten hos ett test som ger kategoriserande resultat och är en viktig faktor vid bedömningen av ett tests relevans (14).
SI
: Stimulationsindex. Relativa värden avseende MFI-värdet (det geometriska medelvärdet för fluorescensintensiteten) för kemikaliebehandlade celler jämfört med MFI-värdet för lösningsmedels-/vehikelbehandlade celler.
Lösningsmedels-/vehikelkontroll
: Ett obehandlat prov som innehåller alla komponenter i ett testsystem förutom testkemikalien, men med det lösningsmedel/den vehikel som används. Lösningsmedels-/vehikelkontrollen används för att fastställa grundresponsen för de prover som behandlas med testkemikalien som är upplöst eller dispergerad i en stabil dispersion i samma lösningsmedel/vehikel. Vid test med en parallell mediumkontroll, visar provet också huruvida lösningsmedlet/vehikeln interagerar med testsystemet.
Specificitet
: Den andel av alla negativa/inaktiva kemikalier som klassificeras korrekt av testet. Specificiteten är ett mått på noggrannheten hos ett test som ger kategoriserande resultat och är en viktig faktor vid bedömningen av ett tests relevans (14).
Infärgningsbuffert
: En fosfatbuffrad saltlösning som innehåller 5 % fetalt kalvserum.
Ämne
: Ett kemiskt grundämne och föreningar av detta grundämne i naturlig eller tillverkad form, inklusive eventuella tillsatser som är nödvändiga för att bevara ämnets stabilitet och sådana föroreningar som härrör från tillverkningsprocessen, men exklusive eventuella lösningsmedel som kan avskiljas utan att det påverkar ämnets stabilitet eller ändrar dess sammansättning.
Testkemikalie
: Alla ämnen eller blandningar som testas med detta test.
FN:s globalt harmoniserade system för klassificering och märkning av kemikalier (GHS)
: Ett system för att klassificera kemikalier (ämnen och blandningar) utifrån standardiserade typer och nivåer av fysiska risker, hälsorisker och miljörisker och informera om resultaten genom exempelvis piktogram, signalord, faroangivelser, skyddsangivelser och säkerhetsdatablad, i syfte att informera om kemikaliernas skadliga effekter för att skydda människor (inklusive arbetsgivare, arbetstagare, transportörer, konsumenter och personal inom räddningstjänsten) och miljön (16).
UVCB-ämnen
: Ämnen med okänd eller varierande sammansättning, komplexa reaktionsprodukter eller biologiska material.
Giltigt test
: Ett test som anses vara tillräckligt relevant och tillförlitligt för ett särskilt ändamål och som är baserat på vetenskapligt sunda principer. Ett test är aldrig giltigt i absolut bemärkelse, utan endast i förhållande till ett fastställt ändamål (14).
Tillägg 2.2
KVALIFIKATIONSÄMNEN
Innan testet som beskrivs i detta tillägg till testmetod B.71 används rutinmässigt, bör laboratoriet styrka sin tekniska kompetens genom att korrekt erhålla den förväntade U-SENS™-prediktionen för de tio ämnen som rekommenderas i tabell 1 och genom att erhålla CV70- och EC150-värden som håller sig inom respektive referensintervall för minst åtta av de tio kvalifikationsämnena. Dessa kvalifikationsämnen har valts ut för att representera hela skalan av responser gällande risker för hudsensibilisering. Övriga urvalskriterier var att ämnena ska vara kommersiellt tillgängliga, att det ska finnas in vivo-referensdata av hög kvalitet och att det ska finnas in vitro-data av hög kvalitet genererade med U-SENS™-testet. Det finns dessutom publicerade referensdata att tillgå för U-SENS™-testet (1) (8).
Tabell 1
Rekommenderade ämnen för att påvisa teknisk kompetens i fråga om U-SENS™-testet
|
Kvalifikationsämne
|
CAS-nr
|
Fysikaliskt tillstånd
|
In vivo-prediktion (105)
|
U-SENS Lösningsmedel/vehikel
|
U-SENS CV70-referensintervall i μg/ml (106)
|
U-SENS EC150-referensintervall i μg/ml (106)
|
|
4-fenylendiamin
|
106-50-3
|
Fast
|
Sensibiliserande (starkt)
|
Komplett medium (107)
|
< 30
|
Positivt (≤ 10)
|
|
Trinitrobensensulfonsyra
|
2508-19-2
|
Flytande
|
Sensibiliserande (starkt)
|
Komplett medium
|
> 50
|
Positivt (≤ 50)
|
|
Dietylmaleat
|
141-05-9
|
Flytande
|
Sensibiliserande (måttligt)
|
DMSO
|
10–100
|
Positivt (≤ 20)
|
|
Resorcinol
|
108-46-3
|
Fast
|
Sensibiliserande (måttligt)
|
Komplett medium
|
> 100
|
Positivt (≤ 50)
|
|
Kanelalkohol
|
104-54-1
|
Fast
|
Sensibiliserande (svagt)
|
DMSO
|
> 100
|
Positivt (10–100)
|
|
4-allylanisol
|
140-67-0
|
Flytande
|
Sensibiliserande (svagt)
|
DMSO
|
> 100
|
Positivt (< 200)
|
|
Sackarin
|
81-07-2
|
Fast
|
Ej sensibiliserande
|
DMSO
|
> 200
|
Negativt (> 200)
|
|
Glycerol
|
56-81-5
|
Flytande
|
Ej sensibiliserande
|
Komplett medium
|
> 200
|
Negativt (> 200)
|
|
Mjölksyra
|
50-21-5
|
Flytande
|
Ej sensibiliserande
|
Komplett medium
|
> 200
|
Negativt (> 200)
|
|
Salicylsyra
|
69-72-7
|
Fast
|
Ej sensibiliserande
|
DMSO
|
> 200
|
Negativt (> 200)
|
|
Förkortningar: CAS-nr = nummer i registret som förs av Chemical Abstracts Service.
|
Tillägg 3
IN VITRO-TEST AVSEENDE HUDSENSIBILISERING: IL-8 LUC-TESTET
INLEDANDE ÖVERVÄGANDEN OCH BEGRÄNSNINGAR
|
1.
|
Till skillnad från tester som analyserar uttrycket av cellytemarkörer, kvantifierar IL-8 Luc-testet förändringar i uttrycket av IL-8, som är en cytokin kopplad till aktiveringen av dendritiska celler. I den på THP-1 baserade IL-8-reporter-cellinjen (THP-G8, etablerad från den humana cellinjen THP-1 med ursprung i akut monocytisk leukemi) mäts IL-8-uttrycket efter exponering för sensibiliserande ämnen (1). Uttrycket av luciferas används sedan för att understödja uppdelningen mellan hudsensibiliserande ämnen och icke-hudsensibiliserande ämnen.
|
|
2.
|
IL-8 Luc-testet har utvärderats i en valideringsstudie (2) som genomförts av Japanese Centre for the Validation of Alternatives Methods (JaCVAM), Ministry of Economy, Trade and Industry (METI) och Japanese Society for Alternatives to Animal Experiments (JSAAE) med efterföljande oberoende kollegial granskning (3) på uppdrag av JaCVAM och Ministry of Health, Labour and Welfare (MHLW) med stöd av International Cooperation on Alternative Test Methods (ICATM). Med beaktande av alla tillgängliga bevis och inkomna åsikter från tillsynsmyndigheter och intressenter, anses IL-8 Luc-testet vara en användbar del av en IATA för att skilja mellan hudsensibiliserande ämnen och icke-hudsensibiliserande ämnen för faroklassificering och -märkning. Exempel på användning av data från IL-8 Luc-test i kombination med annan information finns redovisade i litteraturen (4) (5) (6).
|
|
3.
|
IL-8 Luc-testet har visat sig vara överförbart till laboratorier som har erfarenhet av cellodling och luciferasmätning. Inom och mellan laboratorier var reproducerbarheten 87,7 % respektive 87,5 % (2). Data som erhållits från valideringsstudien (2) och andra publicerade arbeten (1) (6) visar att jämfört med LLNA-tester bedömde IL-8 Luc-testet 118 av 143 kemikalier som positiva eller negativa och 25 kemikalier som oklara, och noggrannheten hos IL-8 Luc-testet i fråga om att skilja mellan hudsensibiliserande ämnen (kategori 1 enligt GHS/CLP-förordningen) och icke-hudsensibiliserande ämnen (ingen kategori enligt GHS/CLP-förordningen) uppgår till 86 % (101/118) med en sensitivitet på 96 % (92/96) och en specificitet på 41 % (9/22). Borträknat de ämnen som ligger utanför det tillämpningsområde som beskrivs nedan (punkt 5), bedömde IL-8 Luc-testet 113 av 136 kemikalier som positiva eller negativa och 23 kemikalier som oklara, och noggrannheten hos IL-8 Luc-testet uppgår då till 89 % (101/113) med en sensitivitet på 96 % (92/96) och en specificitet på 53 % (9/17). Vid användning av data från människa enligt Urbisch m.fl. (7) bedömde IL-8 Luc-testet 76 av 90 kemikalier som positiva eller negativa och 14 kemikalier som oklara, och noggrannheten uppgår till 80 % (61/76) med en sensitivitet på 93 % (54/58) och en specificitet på 39 % (7/18) Borträknat de ämnen som ligger utanför tillämpningsområdet, bedömde IL-8 Luc-testet 71 av 84 kemikalier som positiva eller negativa och 13 kemikalier som oklara, och noggrannheten uppgår till 86 % (61/71) med en sensitivitet på 93 % (54/58) och en specificitet på 54 % (7/13). Det är mer sannolikt att falska negativa prediktioner med IL-8 Luc-testet gäller kemikalier som uppvisar en låg/måttlig hudsensibiliserande styrka (underkategori 1B enligt GHS/CLP-förordningen) än kemikalier som uppvisar en hög hudsensibiliserande styrka (underkategori 1A enligt GHS/CLP-förordningen) (6). Sammantaget visar informationen att IL-8 Luc-testet har en uppgift att fylla i identifieringen av risker för hudsensibilisering. Noggrannheten som anges för IL-8 Luc-testet som ett fristående test är endast vägledande, eftersom testet bör beaktas i kombination med andra informationskällor inom ramen för en IATA och i enlighet med bestämmelserna i punkterna 7 och 8 i Allmän inledning. Vid utvärdering av tester utan djurförsök gällande hudsensibilisering bör man dessutom komma ihåg att såväl LLNA-testet som andra djurförsök eventuellt inte avspeglar situationen för människor fullt ut.
|
|
4.
|
IL-8 Luc-testet har, baserat på de uppgifter som finns tillgängliga för närvarande, visat sig vara tillämpligt för testkemikalier som omfattar en rad olika organiska funktionella grupper, reaktionsmekanismer, hudsensibiliseringsstyrkor (vilket har fastställts i in vivo-undersökningar) och fysikalisk-kemiska egenskaper (2) (6).
|
|
5.
|
Även om IL-8 Luc-testet använder X-VIVO™ 15 som lösningsmedel har det korrekt kunnat bedöma kemikalier med ett log Kow-värde > 3,5 och kemikalier med en löslighet i vatten på runt 100 μg/ml, enligt beräkning med EPI Suite™, och testets förmåga att detektera sensibiliserande ämnen med låg löslighet i vatten är bättre än den för IL-8 Luc-test där dimetylsulfoxid (DMSO) används som lösningsmedel (2). Testkemikalier som inte löses upp vid 20 mg/ml kan dock ge falska negativa resultat, till följd av deras oförmåga att lösas upp i X-VIVO™ 15. Av detta skäl bör negativa resultat för dessa kemikalier inte tas i beaktande. En hög andel falska negativa resultat upptäcktes för anhydrider i valideringsstudien. Dessutom kan, till följd av den begränsade metaboliska kapaciteten hos cellinjen (8) och testets betingelser, prohaptener (ämnen som kräver metabolisk aktivering) och prehaptener (ämnen som aktiveras genom oxidering i luft) ge negativa resultat i testet. Även om negativa resultat för misstänkta prehaptener/prohaptener ska tolkas med försiktighet, lyckades dock IL-8 Luc-testet korrekt bedöma 11 av 11 prehaptener, 6 av 6 prohaptener och 6 av 8 pre/pro-haptener i IL-8 Luc-testets datamängd (2). Baserat på en nyligen genomförd omfattande genomgång av tre tester utan djurförsök (DPRA, KeratinoSens™ och h-CLAT) med avseende på detektering av pre- och prohaptener (9), och med utgångspunkt i det faktum att THP-G8-cellerna som används i IL-8 Luc-testet tillhör en cellinje som härrör från THP-1 som används i h-CLAT-testet, kan IL-8 Luc-testet även bidra till att öka sensitiviteten hos tester utan djurförsök vad gäller att detektera pre- och prohaptener, i kombination med andra tester. De ytaktiva ämnen som har testats hittills gav (falska) positiva resultat, oberoende av typ (till exempel katjoniska, anjoniska eller icke-joniska). Slutligen kan kemikalier som interfererar med luciferas störa ämnets aktivitet/mätning, vilket kan orsaka skenbar inhibition eller ökad luminiscens (10). Till exempel har fytoöstrogenkoncentrationer som är högre än 1 μM rapporterats störa luminiscenssignaler i andra luciferasbaserade reportergentester till följd av överaktivering av luciferasreportergenen. Därför krävs en noggrann undersökning av luciferasuttryck som erhålls vid höga koncentrationer av fytoöstrogener eller föreningar som misstänks producera en fytoöstrogenliknande aktivering av luciferasreportergenen (11). Till följd av det ovanstående ligger ytaktiva ämnen, anhydrider och kemikalier som interfererar med luciferas utanför tillämplighetsområdet för detta test. Om det finns bevis för att IL-8 Luc-testet inte är tillämpligt för andra specifika kategorier av testkemikalier, bör det inte användas för dessa specifika kategorier.
|
|
6.
|
Som beskrivs ovan kan IL-8 Luc-testet användas för att understödja uppdelningen mellan hudsensibiliserande ämnen och icke-hudsensibiliserande ämnen. Ytterligare arbete, företrädesvis baserat på data gällande människor, krävs för att avgöra huruvida IL-8 Luc-resultat skulle kunna bidra till bedömningar av sensibiliseringens styrka, när de beaktas i kombination med andra informationskällor.
|
|
7.
|
Definitioner av begrepp finns i tillägg 3.1.
|
TESTETS PRINCIP
|
8.
|
För IL-8 Luc-testet används en human cellinje med ursprung i monocytisk leukemi, THP-1, som har erhållits från American Type Culture Collection (Manassas, Virginia, Förenta staterna). Med användning av denna cellinje har avdelningen för dermatologi vid Tohoku University School of Medicine tagit fram en THP-1-deriverad IL-8-reporter-cellinje, THP-G8, som innehåller luciferasgenerna SLO (Stable Luciferase Orange) och SLR (Stable Luciferase Red) som styrs av IL-8- respektive GAPDH-promotorer (glyceraldehyd-3-fosfatdehydrogenas) (1). Detta möjliggör kvantitativ mätning av luciferasgenens induktion genom detektering av luminiscensen från väletablerade ljusproducerande luciferassubstrat, som en indikator på IL-8- och GAPDH-promotorernas aktivitet i celler efter exponering för sensibiliserande kemikalier.
|
|
9.
|
Testsystemet med dubbla färger baseras på ett luciferasenzym som sänder ut orangefärgat ljus (SLO, λmax = 580 nm) (12) för IL-8-promotorns genuttryck och ett luciferasenzym som sänder ut rött ljus (SLR, λmax = 630 nm) (13) för GAPDH-promotorns genuttryck (promotorn för intern kontroll). De båda luciferasenzymerna sänder ut ljus med olika färger när de reagerar med D-luciferin (firefly) och deras luminiscens mäts parallellt i en enstegsreaktion genom att ljuset från testblandningen delas upp med ett optiskt filter (14) (se tillägg 3.2).
|
|
10.
|
THP-G8-celler behandlas under 16 timmar med testkemikalien, varefter SLO-luciferasaktiviteten (SLO-LA), som återspeglar IL-8-promotorns aktivitet, och SLR-luciferasaktiviteten (SLR-LA), som återspeglar GAPDH-promotorns aktivitet, mäts. För att göra förkortningarna enkla att förstå kallas SLO-LA och SLR-LA för IL8LA respektive GAPLA nedan. I tabell 1 finns en beskrivning av de termer som är associerade med luciferasaktiviteten i IL-8 Luc-testet. De uppmätta värdena används för att beräkna normaliserad IL8LA (nIL8LA), som är förhållandet mellan IL8LA och GAPLA, induktionen av nIL8LA (Ind-IL8LA), som är förhållandet mellan de aritmetiska medelvärdena för fyrfaldigt uppmätta nIL8LA-värden för THP-G8-celler behandlade med en testkemikalie och nIL8LA-värdena för obehandlade THP-G8-celler, samt inhiberingen av GAPLA (Inh-GAPLA), som är förhållandet mellan de aritmetiska medelvärdena för fyrfaldigt uppmätta GAPLA-värden för THP-G8-celler behandlade med en testkemikalie och GAPLA-värdena för obehandlade THP-G8-celler, som används som en indikator på cytotoxicitet.
Tabell 1
Beskrivning av termer som är associerade med luciferasaktiviteten i IL-8 Luc-testet
|
Förkortning
|
Definition
|
|
GAPLA
|
SLR-luciferasaktivitet som återspeglar GAPDH-promotorns aktivitet
|
|
IL8LA
|
SLO-luciferasaktivitet som återspeglar IL-8-promotorns aktivitet
|
|
nIL8LA
|
IL8LA/GAPLA
|
|
Ind-IL8LA
|
nIL8LA för THP-G8-celler behandlade med kemikalier/nIL8LA för obehandlade celler
|
|
Inh-GAPLA
|
GAPLA för THP-G8-celler behandlade med kemikalier/GAPLA för obehandlade celler
|
|
CV05
|
Den lägsta koncentration av kemikalien vid vilken Inh-GAPLA blir < 0,05
|
|
|
11.
|
Prestandastandarder (PS) (15) finns tillgängliga för att underlätta valideringen av modifierade in vitro-tester avseende IL-8-luciferas som liknar IL-8 Luc-testet, och för att möjliggöra snabba ändringar av OECD:s testriktlinje 442E för att inbegripa dessa. Ömsesidigt erkännande av data (MAD) enligt OECD-avtalet garanteras endast för tester som validerats enligt prestandastandarderna och om de har granskats och inbegripits i testriktlinje 442E av OECD (16).
|
DEMONSTRATION AV KOMPETENS
|
12.
|
Innan testet som beskrivs i detta tillägg till testmetod B.71 används rutinmässigt, bör laboratoriet styrka sin tekniska kompetens med användning av de tio kvalifikationsämnen som anges i tillägg 3.3, i enlighet med god praxis för in vitro-metoder (17). Dessutom bör användare av testet föra en historisk databas över data som erhålls genom reaktivitetskontrollerna (se punkt 15) och genom de positiva kontrollerna och lösningsmedels-/vehikelkontrollerna (se punkterna 21–24) och använda dessa data för att styrka att testets reproducerbarhet bibehålls i laboratoriet över tid.
|
FÖRFARANDE
|
13.
|
Ett standardförfarande (SOP) för IL-8 Luc-testet finns att tillgå och bör användas när testet utförs (18). Laboratorier som vill utföra testet kan erhålla den rekombinanta THP-G8-cellinjen från GPC Lab. Co. Ltd., Tottori, Japan, efter att ha undertecknat ett avtal om materialöverföring (Material Transfer Agreement, MTA) i enlighet med OECD-mallens villkor. I de följande punkterna ges en beskrivning av de viktigaste delarna i och förfarandena för testet.
|
Förberedelse av celler
|
14.
|
THP-G8-cellinjen från GPC Lab. Co. Ltd., Tottori, Japan, ska användas vid utförandet av IL-8 Luc-testet (se punkterna 8 och 13). När cellerna har tagits emot förökas de (två till fyra passager) och lagras frysta som en enhetlig stam. Celler från denna stam kan förökas genom maximalt tolv passager eller under maximalt sex veckor. Odlingsmediet som ska användas för förökningen är RPMI-1640-medium innehållande 10 % fetalt bovint serum (FBS), lösning med antibiotika/antimykotika (100 enheter/ml penicillin G, 100 μg/ml streptomycin och 0,25 μg/ml amfotericin B i 0,85-procentig saltlösning) (till exempel GIBCO katalognr 15240-062), 0,15 μg/ml Puromycin (till exempel CAS-nr 58-58-2) och 300 μg/ml G418 (till exempel CAS-nr 108321-42-2).
|
|
15.
|
Innan cellerna används för testning bör de genomgå en kvalificering i form av en reaktivitetskontroll. Denna kontroll bör utföras 1–2 veckor eller 2–4 passager efter upptining med användning av den positiva kontrollen 4-nitrobensylbromid (4-NBB) (CAS-nr 100-11-8, ≥ 99 % renhet) och den negativa kontrollen mjölksyra (CAS-nr 50-21-5, ≥ 85 % renhet). 4-NBB ska ge en positiv respons för Ind-IL8LA (≥ 1,4) medan mjölksyra ska ge en negativ respons för Ind-IL8LA (< 1,4). Endast celler som har klarat reaktivitetskontrollen ska användas för testet. Kontrollen bör utföras i enlighet med de förfaranden som beskrivs i punkterna 22–24.
|
|
16.
|
För testet sås THP-G8-celler ut med en densitet på 2 till 5 × 105 celler/ml, varefter de förodlas i odlingsflaskor under 48 till 96 timmar. På testdagen tvättas skördade celler från odlingsflaskan med RPMI-1640 innehållande 10 % FBS men utan innehåll av antibiotika och resuspenderas sedan i RPMI-1640 innehållande 10 % FBS men utan innehåll av antibiotika till densiteten 1 × 106 celler/ml. Därefter fördelas cellerna i en flatbottnad svart 96-hålsplatta (till exempel Costar katalognr 3603) med 50 μl per brunn (5 × 104 celler/brunn).
|
Beredning av testkemikalie och kontrollämnen
|
17.
|
Testkemikalien och kontrollämnena bereds på testdagen. För IL-8 Luc-testet löses testkemikalien upp i X-VIVO™ 15, som är ett kommersiellt tillgängligt serumfritt medium (Lonza, 04-418Q), till en slutlig koncentration på 20 mg/ml. X-VIVO™ 15 tillsätts till 20 mg av testkemikalien (oavsett kemikaliens löslighet) i ett mikrocentrifugrör till en total volym på 1 ml, varefter röret roteras kraftigt och skakas i en rotor med en maximal hastighet på 8 varv per minut under 30 minuter vid en rumstemperatur på ungefär 20 °C. Om fasta kemikalier fortfarande är olösta ska röret dessutom ultraljudsbehandlas till dess att kemikalien har lösts upp helt och hållet eller är dispergerad i en stabil dispersion. För testkemikalier som är lösliga i X-VIVO™ 15 späds lösningen med en faktor 5 med X-VIVO™ 15 och används som en X-VIVO™ 15-stamlösning av testkemikalien (4 mg/ml). För testkemikalier som inte är lösliga i X-VIVO™ 15 roteras blandningen igen under minst 30 minuter och centrifugeras sedan vid 15000 varv per minut (≈ 20000 g) under 5 minuter; den resulterande supernatanten används som en X-VIVO™ 15-stamlösning av testkemikalien. För användning av andra lösningsmedel, som DMSO, vatten eller odlingsmedium, ska en vetenskaplig motivering tillhandahållas. Det detaljerade förfarandet för att lösa upp kemikalier beskrivs i tillägg 3.5. X-VIVO™ 15-lösningarna som beskrivs i punkterna 18–23 blandas 1:1 (volymförhållande) med de cellsuspensioner som beretts i en flatbottnad svart 96-hålsplatta (se punkt 16).
|
|
18.
|
Den första testomgången syftar till att fastställa den cytotoxiska koncentrationen och att undersöka hudsensibiliseringspotentialen hos kemikalier. Med användning av X-VIVO™ 15 görs serieutspädningar av X-VIVO™ 15-stamlösningarna av testkemikalierna med en utspädningsfaktor 2 (se tillägg 3.5), och för detta används en 96-hålsplatta (till exempel Costar katalognr EW-01729-03). Därefter tillsätts 50 μl utspädd lösning per brunn till 50 μl cellsuspension i en flatbottnad svart 96-hålsplatta. För testkemikalier som är lösliga i X-VIVO™ 15 sträcker sig därför de slutliga koncentrationerna av testkemikalierna från 0,002 till 2 mg/ml (se tillägg 3.5). För testkemikalier som inte är lösliga i X-VIVO™ 15 vid 20 mg/ml fastställs endast utspädningsfaktorer som sträcker sig från 2 till 210, även om de faktiska slutliga koncentrationerna av testkemikalierna förblir osäkra och är beroende av den mättade koncentrationen av testkemikalierna i X-VIVO™ 15-stamlösningen.
|
|
19.
|
För de efterföljande testomgångarna (det vill säga de andra, tredje och fjärde replikaten) bereds X-VIVO™ 15-stamlösningen vid en koncentration som är fyra gånger högre än koncentrationen CV05 (cellviabilitet 05, den lägsta koncentration vid vilken Inh-GAPLA blir < 0,05) i det första experimentet. Om Inh-GAPLA inte sjunker under 0,05 vid den högsta koncentrationen i den första testomgången, bereds X-VIVO™ 15-stamlösningen vid den första testomgångens högsta koncentration. Koncentrationen CV05 beräknas genom att koncentrationen hos stamlösningen i den första testomgången divideras med utspädningsfaktorn för CV05 (X) (utspädningsfaktorn för CV05 [X] är den utspädningsfaktor som krävs för att späda stamlösningen till CV05) (se tillägg 3.5). För testkemikalier som inte är lösliga i X-VIVO™ 15 vid 20 mg/ml fastställs CV05 utifrån koncentrationen hos stamlösningen × 1/X. För testomgångarna två till fyra bereds en andra stamlösning med koncentrationen 4 × CV05 (se tillägg 3.5).
|
|
20.
|
Serieutspädningarna av denna andra X-VIVO™ 15-stamlösning görs med en utspädningsfaktor på 1,5 med användning av en 96-hålsplatta. Därefter tillsätts 50 μl utspädd lösning per brunn till 50 μl cellsuspension i brunnarna i en flatbottnad svart 96-hålsplatta. Varje koncentration av varje testkemikalie ska testas i fyra brunnar. Proverna blandas sedan med en plattskakare och inkuberas under 16 timmar vid 37 °C i en atmosfär med 5 % CO2, varefter luciferasaktiviteten mäts enligt beskrivningen nedan.
|
|
21.
|
Lösningsmedelskontrollen är en blandning av 50 μl/brunn med X-VIVO™ 15 och 50 μl/brunn med cellsuspension i RPMI-1640 innehållande 10 % FBS.
|
|
22.
|
Den rekommenderade positiva kontrollen är 4-NBB. 20 mg 4-NBB bereds i ett mikrocentrifugrör på 1,5 ml, till vilket X-VIVO™ 15 tillsätts till en total volym på 1 ml. Röret roteras kraftigt och skakas i en rotor med en maximal hastighet på 8 varv per minut under minst 30 minuter. Efter centrifugering vid 20000 g under fem minuter späds supernatanten med en faktor 4 med X-VIVO™ 15, varpå 500 μl av den utspädda supernatanten överförs till en brunn i en 96-hålsplatta. Den utspädda supernatanten späds ytterligare med X-VIVO™ 15 med faktorerna 2 och 4, varefter 50 μl av lösningen tillsätts till 50 μl THP-G8-cellsuspension i brunnarna på en flatbottnad svart 96-hålsplatta (se tillägg 3.6). Varje koncentration av den positiva kontrollen ska testas i fyra brunnar. Plattan skakas i en plattskakare och inkuberas i en CO2-inkubator under 16 timmar (37 °C, 5 % CO2), varefter luciferasaktiviteten mäts enligt beskrivningen i punkt 29.
|
|
23.
|
Den rekommenderade negativa kontrollen är mjölksyra. 20 mg mjölksyra bereds i ett mikrocentrifugrör på 1,5 ml, till vilket X-VIVO™ 15 tillsätts till en total volym på 1 ml (20 mg/ml). Mjölksyralösningen på 20 mg/ml späds sedan med en faktor 5 med X-VIVO™ 15 (till 4 mg/ml), varpå 500 μl av denna mjölksyralösning på 4 mg/ml överförs till en brunn i en 96-hålsplatta. Denna lösning späds med en faktor 2 med X-VIVO™ 15 och därefter igen med en faktor 2, för att ge lösningar på 2 mg/ml och 1 mg/ml. 50 μl av dessa tre lösningar och vehikelkontrollen (X-VIVO™ 15) tillsätts till 50 μl THP-G8-cellsuspension i brunnarna på en flatbottnad svart 96-hålsplatta. Varje koncentration av den negativa kontrollen testas i fyra brunnar. Plattan skakas i en plattskakare och inkuberas i en CO2-inkubator under 16 timmar (37 °C, 5 % CO2), varefter luciferasaktiviteten mäts enligt beskrivningen i punkt 29.
|
|
24.
|
Andra lämpliga positiva eller negativa kontroller kan användas om historiska data finns att tillgå för att härleda jämförbara acceptanskriterier.
|
|
25.
|
Se till att flyktiga testkemikalier inte avdunstar och att det inte sker någon korskontaminering mellan testkemikalier i olika brunnar, till exempel genom att plattan förseglas innan testkemikalierna inkuberas.
|
|
26.
|
Testkemikalierna och lösningsmedelskontrollen kräver två till fyra testomgångar för att få fram en positiv eller negativ prediktion (se tabell 2). Alla testomgångar ska utföras på olika dagar med färska X-VIVO™ 15-stamlösningar av testkemikalierna och oavhängigt skördade celler. Cellerna får dock komma från samma passage.
|
Mätning av luciferasaktiviteten
|
27.
|
Luminiscensen mäts med användning av en luminometer för 96-hålsmikrotiterplattor som är försedd med optiska filter, till exempel Phelios (ATTO, Tokyo, Japan), Tristan 941 (Berthold, Bad Wildbad, Tyskland) eller ARVO-serien (PerkinElmer, Waltham, Massachusetts, Förenta staterna). Luminometern måste kalibreras inför varje test för att säkerställa reproducerbarheten (19). Rekombinanta luciferaser som avger orange- och rödfärgat ljus finns att tillgå för denna kalibrering.
|
|
28.
|
100 μl förvärmt Tripluc®-reagens för luciferastest (Tripluc) överförs till varje brunn i plattan som innehåller cellsuspension, oavsett om den är behandlad med kemikalien eller inte. Plattan skakas under tio minuter vid en rumstemperatur på ungefär 20 °C. Därefter placeras plattan i luminometern för mätning av luciferasaktiviteten. Bioluminiscensen mäts utan (F0) respektive med (F1) det optiska filtret, under tre sekunder vardera. Om alternativa inställningar eller metoder används, till exempel till följd av den använda luminometermodellen, ska detta motiveras.
|
|
29.
|
Parametrar för varje koncentration beräknas utifrån de uppmätta värdena, till exempel IL8LA, GAPLA, nIL8LA, Ind-IL8LA, Inh-GAPLA, genomsnittlig ±SD (standardavvikelse) för IL8LA, genomsnittlig ±SD för GAPLA, genomsnittlig ±SD för nIL8LA, genomsnittlig ±SD för Ind-IL8LA, genomsnittlig ±SD för Inh-GAPLA och det 95-procentiga konfidensintervallet för Ind-IL8LA. Definitioner av parametrarna som används i denna punkt finns i tilläggen 3.1 och 3.4.
|
|
30.
|
Innan mätningen startar görs vanligtvis, för reportertester med flera olika färger, en färgseparering med hjälp av detektorer (luminometer och plattavläsare) som har försetts med optiska filter, till exempel filter som blockerar korta eller långa våglängder (långpass- eller kortpassfilter) eller bandpassfilter. Transmissionskoefficienterna för filtren för varje bioluminiscens-signalfärg bör kalibreras innan testet utförs, enligt tillägg 3.2.
|
DATA OCH RAPPORTERING
Utvärdering av data
|
31.
|
Kriterierna för en positiv/negativ bedömning är, för varje testomgång, enligt följande:
|
—
|
En prediktion i ett IL-8 Luc-test bedöms som positiv om en testkemikalie har ett Ind-IL8LA-värde ≥ 1,4 och den nedre gränsen för det 95-procentiga konfidensintervallet för Ind-IL8LA är ≥ 1,0.
|
|
—
|
En prediktion i ett IL-8 Luc-test bedöms som negativ om en testkemikalie har ett Ind-IL8LA-värde < 1,4 och/eller den nedre gränsen för det 95-procentiga konfidensintervallet för Ind-IL8LA är < 1,0.
|
|
Prediktionsmodell
|
32.
|
Testkemikalier som uppvisar två positiva resultat bland testomgångarna 1, 2, 3 och 4 identifieras som positiva, medan de som uppvisar tre negativa resultat bland testomgångarna 1, 2, 3 och 4 identifieras som förmodat negativa (se tabell 2). Bland de förmodat negativa kemikalierna bedöms kemikalier som löses upp vid koncentrationen 20 mg/ml i X-VIVO™ 15 som negativa, medan kemikalier som inte löses upp vid koncentrationen 20 mg/ml i X-VIVO™ 15 inte ska tas i beaktande (se figur 1).
Tabell 2
Kriterier för att identifiera positivt resultat och förmodat negativt resultat
|
Testomgång 1
|
Testomgång 2
|
Testomgång 3
|
Testomgång 4
|
Slutlig prediktion
|
|
Positivt
|
Positivt
|
—
|
—
|
Positivt
|
|
Negativt
|
Positivt
|
—
|
Positivt
|
|
Negativt
|
Positivt
|
Positivt
|
|
Negativt
|
Förmodat negativt
|
|
Negativt
|
Positivt
|
Positivt
|
—
|
Positivt
|
|
Negativt
|
Positivt
|
Positivt
|
|
Negativt
|
Förmodat negativt
|
|
Negativt
|
Positivt
|
Positivt
|
Positivt
|
|
Negativt
|
Förmodat negativt
|
|
Negativt
|
—
|
Förmodat negativt
|
|
Figur 1
Prediktionsmodell för slutlig bedömning

Acceptanskriterier
|
33.
|
Följande acceptanskriterier ska vara uppfyllda vid användning av IL-8 Luc-testet:
|
—
|
Ind-IL8LA ska vara högre än 5,0 för åtminstone en av koncentrationerna av den positiva kontrollen, 4-NBB, i varje testomgång.
|
|
—
|
Ind-IL8LA ska vara lägre än 1,4 för varje koncentration av den negativa kontrollen, mjölksyra, i varje testomgång.
|
|
—
|
Data från plattor för vilka GAPLA-värdet för kontrollbrunnar med celler och Tripluc men utan testkemikalier är mindre än 5 gånger värdet för brunnar som endast innehåller testmedium (50 μl/hål med RPMI-1640 innehållande 10 % FBS och 50 μl/brunn med X-VIVO™ 15) ska kasseras.
|
|
—
|
Data från plattor för vilka Inh-GAPLA-värdet för alla koncentrationer av test- eller kontrollkemikalierna är lägre än 0,05 ska kasseras. När så är fallet ska det första testet upprepas, så att den högsta slutliga koncentrationen för det upprepade testet är den lägsta slutliga koncentrationen för det föregående testet.
|
|
Testrapport
|
34.
|
Testrapporten ska innehålla följande information:
|
Testkemikalier
Monokomponentämne:
|
Kemisk identifiering, t.ex. med IUPAC- eller CAS-namn, CAS-nummer, Smiles- eller InChI-kod, strukturformel och/eller andra identifieringar.
|
|
Fysikaliskt tillstånd, vattenlöslighet, molekylvikt och andra relevanta fysikalisk-kemiska egenskaper, i den mån sådana uppgifter finns tillgängliga.
|
|
Renhet, kemisk identitet för föroreningar i förekommande fall och när så är praktiskt möjligt.
|
|
Behandling före testning, om detta är tillämpligt (t.ex. uppvärmning eller finfördelning).
|
|
Löslighet i X-VIVO™ 15. För kemikalier som är olösliga i X-VIVO™ 15, huruvida fällning eller flotation observeras efter centrifugering.
|
|
Testad koncentration/testade koncentrationer.
|
|
Uppgifter om lagringsförhållanden och stabilitet i den mån sådana uppgifter finns tillgängliga.
|
|
Motivering för valet av lösningsmedel/vehikel för varje testkemikalie om X-VIVO™ 15 inte har använts.
|
Multikomponentämne, UVCB och blandning:
|
Karakteriseras så långt det är möjligt genom exempelvis kemisk identitet (se ovan), renhet, kvantitativ förekomst och relevanta fysikalisk-kemiska egenskaper (se ovan) hos beståndsdelarna, i den mån sådana uppgifter finns tillgängliga.
|
|
Fysikaliskt tillstånd, vattenlöslighet och andra relevanta fysikalisk-kemiska egenskaper, i den mån sådana uppgifter finns tillgängliga.
|
|
Molekylvikt eller uppskattad molekylvikt för blandningar/polymerer med kända sammansättningar eller annan information som är relevant för utförandet av undersökningen.
|
|
Behandling före testning, om detta är tillämpligt (t.ex. uppvärmning eller finfördelning).
|
|
Löslighet i X-VIVO™ 15. För kemikalier som är olösliga i X-VIVO™ 15, huruvida fällning eller flotation observeras efter centrifugering.
|
|
Testad koncentration/testade koncentrationer.
|
|
Uppgifter om lagringsförhållanden och stabilitet i den mån sådana uppgifter finns tillgängliga.
|
|
Motivering för valet av lösningsmedel/vehikel för varje testkemikalie om X-VIVO™ 15 inte har använts.
|
|
|
Kontroller
Positiv kontroll:
|
Kemisk identifiering, t.ex. med IUPAC- eller CAS-namn, CAS-nummer, Smiles- eller InChI-kod, strukturformel och/eller andra identifieringar.
|
|
Fysikaliskt tillstånd, vattenlöslighet, molekylvikt och andra relevanta fysikalisk-kemiska egenskaper, i den mån sådana uppgifter finns tillgängliga och när så är tillämpligt.
|
|
Renhet, kemisk identitet för föroreningar i förekommande fall och när så är praktiskt möjligt.
|
|
Behandling före testning, om detta är tillämpligt (t.ex. uppvärmning eller finfördelning).
|
|
Testad koncentration/testade koncentrationer.
|
|
Uppgifter om lagringsförhållanden och stabilitet i den mån sådana uppgifter finns tillgängliga.
|
|
Hänvisning till tidigare resultat för positiv kontroll som visar på lämpliga acceptanskriterier, om detta är tillämpligt.
|
Negativ kontroll:
|
Kemisk identifiering, t.ex. med IUPAC- eller CAS-namn, CAS-nummer och/eller andra identifieringar.
|
|
Renhet, kemisk identitet för föroreningar i förekommande fall och när så är praktiskt möjligt.
|
|
Fysikaliskt tillstånd, molekylvikt och andra relevanta fysikalisk-kemiska egenskaper om andra negativa kontroller än de som anges i testriktlinjen används och i den mån sådana uppgifter finns tillgängliga.
|
|
Uppgifter om lagringsförhållanden och stabilitet i den mån sådana uppgifter finns tillgängliga.
|
|
Motivering för valet av lösningsmedel för varje testkemikalie.
|
|
|
Testbetingelser
|
Namn och adress för sponsor, testanläggning och försöksledare.
|
|
Beskrivning av testet som använts.
|
|
Använd cellinje samt dess lagringsförhållanden och källa (till exempel den anläggning varifrån cellinjen erhölls).
|
|
Partinummer och ursprung för FBS, namn på leverantör, partinummer för flatbottnad svart 96-hålsplatta och partinummer för Tripluc-reagens.
|
|
Passageantal och celldensitet som används för testet.
|
|
Cellräkningsmetod som använts för utsådden före testet, samt vidtagna åtgärder för att säkerställa en homogen fördelning av celler.
|
|
Använd luminometer (till exempel modell), inklusive instrumentinställningar och använt luciferassubstrat, samt ett uppvisande av korrekta luminiscensmätningar baserat på det kontrolltest som beskrivs i tillägg 3.2.
|
|
Det förfarande som använts för att visa att laboratoriet är kvalificerat att utföra testet (till exempel genom test av kvalifikationsämnen) eller för att visa att testet kan reproduceras över tid.
|
|
|
Testförfarande
|
Antal replikat och genomförda testomgångar.
|
|
Testkemikaliens koncentrationer, appliceringsförfarande och exponeringstid (vid avvikelse från rekommendationerna).
|
|
Beskrivning av de utvärderings- och beslutskriterier som använts.
|
|
Beskrivning av de använda acceptanskriterierna för undersökningen.
|
|
Beskrivning av eventuella modifieringar av testförfarandet.
|
|
|
Resultat
|
Mätresultat för IL8LA och GAPLA.
|
|
Beräkningar av nIL8LA, Ind-IL8LA och Inh-GAPLA.
|
|
Det 95-procentiga konfidensintervallet för Ind-IL8LA.
|
|
Ett diagram som visar dos-effektkurvorna för induktionen av luciferasaktivitet samt viabilitet.
|
|
Beskrivning av andra relevanta observationer, i förekommande fall.
|
|
|
Diskussion kring resultaten
|
Diskussion kring de resultat som har erhållits med IL-8 Luc-testet.
|
|
Beaktande av testets resultat inom ramen för en IATA, om annan relevant information finns att tillgå.
|
|
Slutsats
|
LITTERATUR
|
(1)
|
Takahashi T, Kimura Y, Saito R, Nakajima Y, Ohmiya Y, Yamasaki K and Aiba S. (2011). An in vitro test to screen skin sensitizers using a stable THP-1-derived IL-8 reporter cell line, THP-G8. Toxicol Sci 124:359-69.
|
|
(2)
|
OECD (2017). Validation report for the international validation study on the IL-8 Luc assay as a test evaluating the skin sensitizing potential of chemicals conducted by the IL-8 Luc Assay. Series on Testing and Assessment No 267, ENV/JM/MONO(2017)19. Organisationen för ekonomiskt samarbete och utveckling (OECD), Paris. Finns på http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(3)
|
OECD (2017). Report of the Peer Review Panel for the IL-8 Luciferase (IL-8 Luc) Assay for in vitro skin sensitisation. Series on Testing and Assessment No 258, ENV/JM/MONO(2017)20. Organisationen för ekonomiskt samarbete och utveckling (OECD), Paris. Finns på http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(4)
|
OECD (2016) Guidance Document On The Reporting Of Defined Approaches And Individual Information Sources To Be Used Within Integrated Approaches To Testing And Assessment (IATA) For Skin Sensitisation, Series on Testing & Assessment No 256, ENV/JM/MONO(2016)29. Organisationen för ekonomiskt samarbete och utveckling (OECD), Paris. Finns på http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(5)
|
van der Veen JW, Rorije E, Emter R, Natsch A, van Loveren H and Ezendam J. (2014). Evaluating the performance of integrated approaches for hazard identification of skin sensitizing chemicals. Regul Toxicol Pharmacol 69:371-9.
|
|
(6)
|
Kimura Y, Fujimura C, Ito Y, Takahashi T, Nakajima Y, Ohmiya Y and Aiba S. (2015). Optimization of the IL-8 Luc assay as an in vitro test for skin sensitization. Toxicol In Vitro 29:1816-30.
|
|
(7)
|
Urbisch D, Mehling A, Guth K, Ramirez T, Honarvar N, Kolle S, Landsiedel R, Jaworska J, Kern PS, Gerberick F m.fl. (2015). Assessing skin sensitization hazard in mice and men using non-animal test methods. Regul Toxicol Pharmacol 71:337-51.
|
|
(8)
|
Ashikaga T, Sakaguchi H, Sono S, Kosaka N, Ishikawa M, Nukada Y, Miyazawa M, Ito Y, Nishiyama N and Itagaki H. (2010). A comparative evaluation of in vitro skin sensitisation tests: the human cell-line activation test (h-CLAT) versus the local lymph node assay (LLNA). Alternatives to laboratory animals: ATLA 38:275-84.
|
|
(9)
|
Patlewicz G, Casati S, Basketter DA, Asturiol D, Roberts DW, Lepoittevin J-P, Worth A and Aschberger K (2016). Can currently available non-animal methods detect pre and pro haptens relevant for skin sensitisation? Regul Toxicol Pharmacol, 82:147-155.
|
|
(10)
|
Thorne N, Inglese J and Auld DS. (2010). Illuminating insights into firefly luciferase and other bioluminescent reporters used in chemical biology. Chem Biol 17:646-57.
|
|
(11)
|
OECD (2016). Test No 455: Performance-Based Test Guideline for Stably Transfected Transactivation In Vitro Assays to Detect Estrogen Receptor Agonists and Antagonists, OECD Publishing, Paris. http://dx.doi.org/10.1787/9789264265295-en.
|
|
(12)
|
Viviani V, Uchida A, Suenaga N, Ryufuku M and Ohmiya Y. (2001). Thr226 is a key residue for bioluminescence spectra determination in beetle luciferases. Biochem Biophys Res Commun 280:1286-91.
|
|
(13)
|
Viviani VR, Bechara EJ and Ohmiya Y. (1999). Cloning, sequence analysis, and expression of active Phrixothrix railroad-worms luciferases: relationship between bioluminescence spectra and primary structures. Biochemistry 38:8271-9.
|
|
(14)
|
Nakajima Y, Kimura T, Sugata K, Enomoto T, Asakawa A, Kubota H, Ikeda M and Ohmiya Y. (2005). Multicolor luciferase assay system: one-step monitoring of multiple gene expressions with a single substrate. Biotechniques 38:891-4.
|
|
(15)
|
OECD (2017). Ska publiceras – Performance Standards for the assessment of proposed similar or modified in vitro skin sensitisation IL-8 luc test methods. OECD Environment, Health and Safety Publications, Series on Testing and Assessment. OECD, Paris, Frankrike.
|
|
(16)
|
OECD (2005). Guidance Document the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. OECD Environment, Health and Safety publications, OECD Series on Testing and Assessment No 34. OECD, Paris, Frankrike.
|
|
(17)
|
OECD (2018). Draft Guidance document: Good In Vitro Method Practices (GIVIMP) for the Development and Implementation of In Vitro Methods for Regulatory Use in Human Safety Assessment. Organisationen för ekonomiskt samarbete och utveckling (OECD), Paris. Finns på http://www.oecd.org/env/ehs/testing/OECD Final Draft GIVIMP.pdf.
|
|
(18)
|
JaCVAM (2016). IL-8 Luc assay protocol. Finns på http://www.jacvam.jp/en_effort/effort02.html.
|
|
(19)
|
Niwa K, Ichino Y, Kumata S, Nakajima Y, Hiraishi Y, Kato D, Viviani VR and Ohmiya Y. (2010). Quantum yields and kinetics of the firefly bioluminescence reaction of beetle luciferases. Photochem Photobiol 86:1046-9.
|
|
(20)
|
OECD (2012). The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins, Part 1: Scientific Evidence. OECD Environment, Health and Safety Publications Series on Testing and Assessment No. 168. OECD, Paris, Frankrike.
|
|
(21)
|
Förenta nationerna (2015). Globally Harmonized System of Classification and Labelling of Chemicals (GHS, det globalt harmoniserade systemet för klassificering och märkning av kemikalier). Sjätte reviderade utgåvan. New York och Genève: United Nations Publications. ISBN: 978-92-1-117006-1. Finns på http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html.
|
Tillägg 3.1
DEFINITIONER
Noggrannhet
: Anger hur väl testresultaten och de godtagna referensvärdena överensstämmer med varandra. Det är ett mått på testets prestanda och en relevansaspekt. Detta begrepp används ofta omväxlande med ”konkordans” för att beskriva andelen korrekta resultat med ett test (16).
AOP (Adverse Outcome Pathway)
: Sekvens av händelser, från den kemiska strukturen hos en målkemikalie eller en grupp av liknande kemikalier, genom den molekylära inledande händelsen och fram till ett in vivo-resultat av intresse (20).
Kemikalie
: Ett ämne eller en blandning.
CV05
: Cellviabilitet 05, det vill säga den lägsta koncentration vid vilken kemikalier uppvisar ett Inh-GAPLA-värde som är lägre än 0,05.
FInSLO-LA
: Förkortning som används i valideringsrapporten och i tidigare publikationer gällande IL-8 Luc-testet med samma betydelse som Ind-IL8LA. Se Ind-IL8LA för närmare definition.
GAPLA
: SLR-luciferasaktivitet (λmax = 630 nm) som styrs av GAPDH-promotorn och som indikerar cellviabiliteten och antalet viabla celler.
Fara
: Inneboende egenskap hos ett ämne eller en situation som har potential att orsaka skadliga effekter när en organism, ett system eller en (del-)population exponeras för ämnet.
IATA (Integrated Approach to Testing and Assessment)
: En strukturerad strategi som används för identifiering av fara (risk), karakterisering av fara (styrka) och/eller säkerhetsbedömning (risk/styrka och exponering) för en kemikalie eller en grupp av kemikalier, som strategiskt integrerar och väger in alla relevanta data som underlag för tillsynsbeslut om potentiell fara och/eller risk och/eller om behovet av ytterligare riktad och därigenom minimerad testning.
II-SLR-LA
: Förkortning som används i valideringsrapporten och i tidigare publikationer gällande IL-8 Luc-testet med samma betydelse som Inh-GAPLA. Se Inh-GAPLA för närmare definition.
IL-8 (Interleukin-8)
: En cytokin som erhålls från endotelceller, fibroblaster, keratinocyter, makrofager och monocyter som orsakar kemotaxis hos neutrofiler och T-lymfocyter.
IL8LA
: SLO-luciferasaktivitet (λmax = 580 nm) som styrs av IL-8-promotorn.
Ind-IL8LA
: Induktionsförhållande för nIL8LA. Värdet erhålls genom att nIL8LA-värdet för THP-G8-celler behandlade med kemikalier divideras med motsvarande värde för icke-stimulerade THP-G8-celler och representerar induktionen av IL-8-promotoraktivitet till följd av kemikalier.
Inh-GAPLA
: Inhibering av GAPLA. Värdet erhålls genom att GAPLA-värdet för THP-G8-celler behandlade med kemikalier divideras med GAPLA-värdet för obehandlade THP-G8-celler och representerar kemikaliernas cytotoxicitet.
Lägsta gränsvärde för induktion (MIT, Minimum Induction Threshold)
: Den lägsta koncentration vid vilken en kemikalie uppfyller kriteriet för positivt resultat.
Blandning
: En blandning eller en lösning som består av två eller flera ämnen.
Monokomponentämne
: Ett ämne, definierat av sin kvantitativa sammansättning, där en huvudsaklig beståndsdel utgör minst 80 viktprocent.
Multikomponentämne
: Ett ämne, definierat av sin kvantitativa sammansättning, där fler än en huvudsaklig beståndsdel förekommer i en koncentration på ≥ 10 viktprocent och < 80 viktprocent. Ett multikomponentämne är resultatet av en tillverkningsprocess. Skillnaden mellan blandningar och multikomponentämnen är att en blandning erhålls genom att två eller flera ämnen blandas utan någon kemisk reaktion. Ett multikomponentämne är resultatet av en kemisk reaktion.
nIL8LA
: SLO-luciferasaktiviteten som återspeglar IL-8-promotorns aktivitet (IL8LA) normaliserad genom SLR-luciferasaktiviteten som återspeglar GAPDH-promotorns aktivitet (GAPLA). Värdet representerar IL-8-promotorns aktivitet efter beaktande av cellviabilitet eller cellantal.
nSLO-LA
: Förkortning som används i valideringsrapporten och i tidigare publikationer gällande IL-8 Luc-testet med samma betydelse som nIL8LA. Se nIL8LA för närmare definition.
Positiv kontroll
: Ett replikat som innehåller testsystemets alla komponenter och som behandlats med ett ämne som man vet ger en positiv respons. För att säkerställa att variationer i den positiva kontrollens respons över tid kan bedömas, bör den positiva responsen inte vara alltför kraftfull.
Prehaptener
: Kemikalier som blir sensibiliserande ämnen genom abiotisk omvandling.
Prohaptener
: Kemikalier som kräver enzymatisk aktivering för att deras hudsensibiliserande potential ska aktiveras.
Relevans
: Beskrivning av förhållandet mellan testet och den berörda effekten och huruvida det är meningsfullt och användbart för ett visst ändamål. Relevansen är ett mått på hur pass väl man genom testet korrekt kan mäta eller prediktera den biologiska effekt det gäller. Relevansen omfattar även överväganden om ett tests noggrannhet (konkordans) (16).
Tillförlitlighet
: Mått på i vilken utsträckning ett test kan reproduceras inom och mellan laboratorier över tid, när det utförs med användning av samma protokoll. Tillförlitligheten bedöms genom att man beräknar reproducerbarheten inom och mellan laboratorier och repeterbarheten inom laboratorier (16).
Testomgång
: En testomgång utgörs av att en eller flera testkemikalier testas parallellt med en lösningsmedels-/vehikelkontroll och en positiv kontroll.
Sensitivitet
: Den andel av alla positiva/aktiva kemikalier som klassificeras korrekt av testet. Känsligheten är ett mått på noggrannheten hos ett test som ger kategoriserande resultat och är en viktig faktor vid bedömningen av ett tests relevans (16).
SLO-LA
: Förkortning som används i valideringsrapporten och i tidigare publikationer gällande IL-8 Luc-testet med samma betydelse som IL8LA. Se IL8LA för närmare definition.
SLR-LA
: Förkortning som används i valideringsrapporten och i tidigare publikationer gällande IL-8 Luc-testet med samma betydelse som GAPLA. Se GAPLA för närmare definition.
Lösningsmedels-/vehikelkontroll
: Ett obehandlat prov som innehåller alla komponenter i ett testsystem förutom testkemikalien, men med det lösningsmedel/den vehikel som används. Lösningsmedels-/vehikelkontrollen används för att fastställa grundresponsen för de prover som behandlas med testkemikalien som är upplöst eller dispergerad i en stabil dispersion i samma lösningsmedel/vehikel. Vid test med en parallell mediumkontroll, visar provet också huruvida lösningsmedlet/vehikeln interagerar med testsystemet.
Specificitet
: Den andel av alla negativa/inaktiva kemikalier som klassificeras korrekt av testet. Specificiteten är ett mått på noggrannheten hos ett test som ger kategoriserande resultat och är en viktig faktor vid bedömningen av ett tests relevans (16).
Ämne
: Ett kemiskt grundämne och föreningar av detta grundämne i naturlig eller tillverkad form, inklusive eventuella tillsatser som är nödvändiga för att bevara ämnets stabilitet och sådana föroreningar som härrör från tillverkningsprocessen, men exklusive eventuella lösningsmedel som kan avskiljas utan att det påverkar ämnets stabilitet eller ändrar dess sammansättning.
Ytaktivt ämne
: Ett ämne, exempelvis ett rengöringsmedel, som kan minska en vätskas ytspänning så att vätskan kan löddra eller penetrera fasta ämnen; kallas även tensid eller vätmedel. (Testriktlinje 437)
Testkemikalie
: Alla ämnen eller blandningar som testas med detta test.
THP-G8
: En IL-8-reportercellinje som används i IL-8 Luc-testet. Den utgår från den humana makrofagliknande cellinjen THP-1 som sedan transfekterats med luciferasgenerna SLO och SLR, styrda av promotorerna IL-8 respektive GAPDH.
FN:s globalt harmoniserade system för klassificering och märkning av kemikalier (GHS)
: Ett system för att klassificera kemikalier (ämnen och blandningar) utifrån standardiserade typer och nivåer av fysiska risker, hälsorisker och miljörisker och informera om resultaten genom exempelvis piktogram, signalord, faroangivelser, skyddsangivelser och säkerhetsdatablad, i syfte att informera om kemikaliernas skadliga effekter för att skydda människor (inklusive arbetsgivare, arbetstagare, transportörer, konsumenter och personal inom räddningstjänsten) och miljön (21).
UVCB-ämnen
: Ämnen med okänd eller varierande sammansättning, komplexa reaktionsprodukter eller biologiska material.
Giltigt test
: Ett test som anses vara tillräckligt relevant och tillförlitligt för ett särskilt ändamål och som är baserat på vetenskapligt sunda principer. Ett test är aldrig giltigt i absolut bemärkelse, utan endast i förhållande till ett fastställt ändamål.
Tillägg 3.2
PRINCIP FÖR MÄTNING AV LUCIFERASAKTIVITET OCH FASTSTÄLLANDE AV TRANSMISSIONSKOEFFICIENTER FÖR OPTISKA FILTER FÖR SLO OCH SLR
Multireporter-testsystemet Tripluc (MultiReporter Assay System – Tripluc) kan användas med en luminometer för mikrotiterplattor som har ett flerfärgsdetektorsystem som går att förse med ett optiskt filter (till exempel Phelios AB-2350 [ATTO], ARVO [PerkinElmer] eller Tristar LB941 [Berthold]). Det optiska filter som används vid mätningen är ett lång- eller kortpassfilter på 600–620 nm eller ett bandpassfilter på 600–700 nm.
Mätning av luciferas med två färger med hjälp av ett optiskt filter
Nedan beskrivs ett exempel med användning av Phelios AB-2350 (ATTO). Denna luminometer är försedd med ett långpassfilter på 600 nm (R60 HOYA Co., 600 nm LP, filter 1) för att dela upp luminiscensen mellan SLO (λmax = 580 nm) och SLR (λmax = 630 nm).
För att fastställa transmissionskoefficienterna för långpassfiltret på 600 nm behöver man, med användning av renade SLO- och SLR-luciferasenzymer, i) mäta SLO- och SLR-bioluminiscensens intensitet utan filter (F0), ii) mäta SLO- och SLR-bioluminiscensens intensitet efter att ljuset har passerat genom långpassfiltret på 600 nm (filter 1) och iii) beräkna transmissionskoefficienterna för långpassfiltret på 600 nm för SLO och SLR, enligt instruktionerna nedan.
|
Transmission coefficients
|
Abbreviation
|
Definition
|
|
SLO
|
Filter 1 Transmission coefficients
|
=κOR60
|
The filter’s transmission coefficient for the SLO
|
|
SLR
|
Filter 1 Transmission coefficients
|
κRR60
|
The filter’s transmission coefficient for the SLR
|
När intensiteten för SLO och SLR i provet definieras som O respektive R beskrivs i) intensiteten hos ljuset utan filter (helt optiskt), F0, och ii) intensiteten hos ljuset som passerar genom långpassfiltret på 600 nm (filter 1), F1, enligt nedan.
F0 = O + R
F1 = κOR60 × O + κRR60 × R
Dessa formler kan även formuleras om på följande sätt:
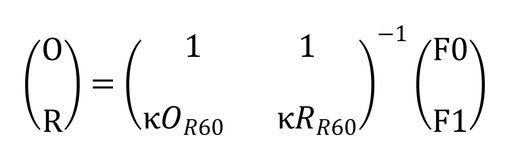
Med användning av de beräknade transmissionsfaktorerna (κOR60 och κRR60) och de uppmätta värdena F0 och F1 går det sedan att beräkna O- och R-värdena enligt följande:

Material och metoder för att fastställa transmissionsfaktorer
|
1)
|
Reagenser
Enkla renade luciferasenzymer:
|
Frystorkat renat SLO-enzym
|
|
Frystorkat renat SLR-enzym
|
|
(som för valideringsarbetet erhölls från GPC Lab. Co. Ltd., Tottori, Japan, med användning av THP-G8-cellinjen)
|
Testreagens:
|
Tripluc®-reagens för luciferastest (till exempel från TOYOBO, katalognr MRA-301)
|
Medium: för luciferastest (30 ml, förvarat vid 2–8 °C)
|
|
Reagens
|
Konc.
|
Slutlig konc. i medium
|
Mängd som behövs
|
|
RPMI-1640
|
—
|
—
|
27 ml
|
|
FBS
|
—
|
10 %
|
3 ml
|
|
2)
|
Beredning av enzymlösning:
Lös upp frystorkat renat luciferasenzym i ett provrör genom att tillsätta 200 μl 10–100 mM Tris-HCl eller Hepes-HCl (pH 7,5–8,0) som har kompletterats med 10 % (vikt/volym) glycerol, dela upp enzymlösningen i alikvoter om 10 μl i engångsprovrör på 1,5 ml och förvara dem i frys vid –80 °C. Den frysta enzymlösningen kan användas i upp till sex månader. Vid användning tillsätts 1 ml medium för luciferastest (RPMI-1640 med 10 % FBS) till varje provrör som innehåller enzymlösning (utspädd enzymlösning), varpå provrören förvaras på is för att förhindra deaktivering.
|
|
3)
|
Mätning av bioluminiscens
Tina upp Tripluc®-reagens för luciferastest (Tripluc) och förvara reagensen i rumstemperatur, antingen i ett vattenbad eller på en plats där den omgivande luften har rumstemperatur. Sätt på luminometern 30 minuter innan mätningen ska börja, så att fotomultiplikatorn får tid att stabiliseras. Överför 100 μl av den utspädda enzymlösningen till en svart 96-hålsplatta (flatbottnad) (SLO-referensprov till #B1, #B2 och #B3, och SLR-referensprov till #D1, #D2 och #D3). Överför därefter 100 μl förvärmd Tripluc till var och en av brunnarna i plattan som innehåller utspädd enzymlösning, med hjälp av en pipett. Skaka plattan under 10 minuter vid rumstemperatur (ungefär 25 °C) med hjälp av en plattskakare. Avlägsna eventuella bubblor från lösningarna i brunnarna. Placera plattan i luminometern för att mäta luciferasaktiviteten. Bioluminiscensen mäts utan (F0) respektive med (F1) det optiska filtret, under tre sekunder vardera.
Transmissionskoefficienten för det optiska filtret beräknas enligt följande:
Transmissionskoefficient (SLO [κOR60]) = (#B1 vid F1 + #B2 vid F1 + #B3 vid F1) / (#B1 vid F0 + #B2 vid F0 + #B3 vid F0)
Transmissionskoefficient (SLR [κOR60]) = (#D1 vid F1 + #D2 vid F1 + #D3 vid F1) / (#D1 vid F0 + #D2 vid F0 + #D3 vid F0)
De beräknade transmissionsfaktorerna används för alla mätningar som utförs med användning av samma luminometer.
|
Kvalitetskontroll för utrustning
De förfaranden som beskrivs i IL-8 Luc-protokollet bör användas (18).
Tillägg 3.3
KVALIFIKATIONSÄMNEN
Innan testet som beskrivs i detta tillägg till testmetod B.71 används rutinmässigt, bör laboratoriet styrka sin tekniska kompetens genom att erhålla den förväntade IL-8 Luc-prediktionen för de nio ämnen som rekommenderas i tabell 1 och genom att erhålla värden som håller sig inom respektive referensintervall för minst åtta av de nio kvalifikationsämnena (som är utvalda för att representera hela skalan av responser gällande risker för hudsensibilisering). Övriga urvalskriterier var att ämnena ska vara kommersiellt tillgängliga, att det ska finnas in vivo-referensdata av hög kvalitet och att det ska finnas in vitro-data av hög kvalitet genererade med IL-8 Luc-testet. Det finns dessutom publicerade referensdata att tillgå för IL-8 Luc-testet (1) (6).
Tabell 1
Rekommenderade ämnen för att påvisa teknisk kompetens i fråga om IL-8 Luc-testet
|
Kvalifikationsämne
|
CAS-nr
|
Fysikaliskt tillstånd
|
Löslighet i X-VIVO™ 15 vid 20 mg/ml
|
In vivo-prediktion (108)
|
IL-8 Luc-prediktion (109)
|
Referensintervall (μg/ml) (110)
|
|
|
CV05 (111)
|
IL-8 Luc MIT (112)
|
|
2,4-dinitroklorbensen
|
97-00-7
|
Fast
|
Olösligt
|
Sensibiliserande (extremt)
|
Positiv
|
2,3–3,9
|
0,5–2,3
|
|
Formaldehyd
|
50-00-0
|
Flytande
|
Lösligt
|
Sensibiliserande (starkt)
|
Positiv
|
9–30
|
4–9
|
|
2-merkaptobensotiazol
|
149-30-4
|
Fast
|
Olösligt
|
Sensibiliserande (måttligt)
|
Positiv
|
250–290
|
60–250
|
|
Etylendiamin
|
107-15-3
|
Flytande
|
Lösligt
|
Sensibiliserande (måttligt)
|
Positiv
|
500–700
|
0,1–0,4
|
|
Etylenglykoldimetakrylat
|
97-90-5
|
Flytande
|
Olösligt
|
Sensibiliserande (svagt)
|
Positiv
|
> 2000
|
0,04–0,1
|
|
4-allylanisol (Estragol)
|
140-67-0
|
Flytande
|
Olösligt
|
Sensibiliserande (svagt)
|
Positiv
|
> 2000
|
0,01–0,07
|
|
Streptomycinsulfat
|
3810-74-0
|
Fast
|
Lösligt
|
Ej sensibiliserande
|
Negativ
|
> 2000
|
> 2000
|
|
Glycerol
|
56-81-5
|
Flytande
|
Lösligt
|
Ej sensibiliserande
|
Negativ
|
> 2000
|
> 2000
|
|
Isopropanol
|
67-63-0
|
Flytande
|
Lösligt
|
Ej sensibiliserande
|
Negativ
|
> 2000
|
> 2000
|
|
Förkortningar: CAS-nr = nummer i registret som förs av Chemical Abstracts Service.
|
Tillägg 3.4
INDEX OCH BEDÖMNINGSKRITERIER
nIL8LA (nSLO-LA)
Den j:te repetitionen (j = 1–4) för den i:te koncentrationen (i = 0–11) mäts för IL8LA (SLO-LA) respektive GAPLA (SLR-LA). Normaliserad IL8LA, som kallas för nIL8LA (nSLO-LA), definieras enligt följande:
nIL8LAij = IL8LAij/GAPLAij
Det här är den grundläggande måttenheten i detta test.
Ind-IL8LA (FInSLO-LA)
Ind-IL8LA, som är den X-faldiga ökningen av det genomsnittliga nIL8LA-värdet (nSLO-LA) för de upprepade mätningarna av den i:te koncentrationen i förhållande till motsvarande värde för nollkoncentrationen, är det primära mätvärdet i detta test. Detta förhållande skrivs med följande formel:

Huvudlaboratoriet (laboratoriet som styrde valideringsarbetet) har föreslagit att ett värde på 1,4 ska motsvara ett positivt resultat för den testade kemikalien. Detta värde är baserat på det huvudlaboratoriets undersökning av historiska data. Datahanteringsgruppen har sedan använt detta värde under alla faser av valideringsstudien. Det primära resultatet, Ind-IL8LA, är förhållandet mellan två aritmetiska genomsnitt, vilket visas i formeln.
95-procentigt konfidensintervall (95 % CI)
Det 95-procentiga konfidensintervallet (95 % CI) för det nyss nämnda förhållandet kan uppskattas för att visa exaktheten i det primära resultatmåttet. Den nedre gränsen för 95 % CI ≥ 1 anger att nIL8LA-värdet med den i:te koncentrationen är betydligt högre än motsvarande värde med lösningsmedelskontroll. Det finns flera olika sätt att fastställa det 95-procentiga konfidensintervallet. Vi har använt oss av metoden som kallas Fiellers teorem i den här studien. Detta teorem för det 95-procentiga konfidensintervallet använder sig av följande formel:

där .



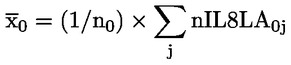



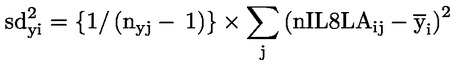
är den 97,5:e percentilen för den centrala t-fördelningen med frihetsgraden ν, där

Inh-GAPLA (II-SLR-LA)
Inh-GAPLA är förhållandet mellan det genomsnittliga GAPLA-värdet (SLR-LA) för de upprepade mätningarna av den i:te koncentrationen jämfört med motsvarande värde för lösningsmedelskontrollen, vilket skrivs på följande sätt:

Eftersom GAPLA-värdet är nämnare vid uträkningen av nIL8LA, orsakar ett extremt litet värde en stor variation i nIL8LA-värdet. Av den anledningen kan Ind-IL8LA-värden med ett extremt litet värde för Inh-GAPLA (mindre än 0,05) anses ha en dålig precision.
Tillägg 3.5
SCHEMATISK BESKRIVNING AV DE METODER SOM ANVÄNDS FÖR ATT LÖSA UPP KEMIKALIER FÖR IL-8 LUC-TESTET
|
a)
|
För kemikalier som är lösliga i X-VIVOTM 15 vid koncentrationen 20 mg/ml

|
|
b)
|
För kemikalier som är olösliga i X-VIVOTM 15 vid koncentrationen 20 mg/ml

|
Tillägg 3.6
SCHEMATISK BESKRIVNING AV DEN METOD SOM ANVÄNDS FÖR ATT LÖSA UPP 4-NBB FÖR IL-8 LUC-TESTETS POSITIVA KONTROLL

” |