BILAGA II
VETENSKAPLIGA KRAV FÖR RISKBEDÖMNINGEN AV GENETISKT MODIFIERADE LIVSMEDEL OCH FODER
I. INLEDNING
1. DEFINITIONER
I denna bilaga avses med
1. faroidentifiering: identifiering av biologiska, kemiska eller fysikaliska agenser som kan ha negativa hälsoeffekter och som kan finnas i ett visst livsmedel och foder eller i en livsmedels- och fodergrupp,
2. farokarakterisering: kvalitativ och/eller kvantitativ utvärdering av vilka slags negativa hälsoeffekter som kopplas till biologiska, kemiska eller fysikaliska agenser som kan finnas i livsmedel och foder,
3. riskkarakterisering: kvalitativ och/eller kvantitativ uppskattning av sannolikheten för att det förekommer kända eller potentiella negativa hälsoeffekter i en viss befolkningsgrupp samt av deras allvarlighet, inbegripet därmed förknippade osäkerheter, på grundval av faroidentifiering, farokarakterisering och exponeringsbedömning.
II. SÄRSKILDA FAKTORER ATT BEAKTA
2.1 Införande av markörgener och andra nukleinsyrasekvenser som inte är nödvändiga för att uppnå den önskade egenskapen
För att underlätta riskbedömningen ska sökanden sträva efter att minimera förekomsten av införda nukleinsyrasekvenser som inte är nödvändiga för att uppnå den önskade egenskapen.
Under den genetiska modifieringen av växter och andra organismer används ofta markörgener för att underlätta urvalet och identifieringen av genetiskt modifierade celler, som innehåller den gen som införts i värdorganismens genom, i den stora merparten av oförändrade celler. Sökanden ska välja sådana markörgener med omsorg och med iakttagande av artikel 4.2 i direktiv 2001/18/EG. Mot den bakgrunden ska sökanden därför ha som mål att utveckla genetiskt modifierade organismer utan att använda gener för antibiotikaresistens som markörgener.
2.2 Riskbedömning av genetiskt modifierade livsmedel och foder som innehåller staplade transformationshändelser (stacked events)
Vid riskbedömningen av genetiskt modifierade livsmedel och foder som innehåller staplade transformationshändelser, som erhållits genom en konventionell korsning av genetiskt modifierade växter som innehåller en eller flera transformationshändelser, ska sökanden tillhandahålla en riskbedömning för varje enskild transformationshändelse eller, i enlighet med artikel 3.6 i denna förordning, hänvisa till en eller flera ansökningar som redan lämnats in. Riskbedömningen av genetiskt modifierade livsmedel och foder som innehåller staplade transformationshändelser ska dessutom innehålla en bedömning av följande:
|
a) |
Transformationshändelsernas stabilitet. |
|
b) |
Transformationshändelsernas uttryck. |
|
c) |
Potentiellt sam- eller motverkande effekter som är en följd av att transformationshändelser kombinerats ska bedömas i enlighet med avsnitten 1.4 (Toxikologi), 1.5 (Allergiframkallande egenskaper) och 1.6 (Näringsbedömning). |
För genetiskt modifierade livsmedel och foder som innehåller, består av eller har framställts av genetiskt modifierade växter, vars odling är förknippad med framställningen av genetiskt modifierat material som innehåller olika klyvningsprodukter av transformationshändelser (klyvande grödor), ska ansökan inbegripa alla klyvningsprodukter, oberoende av ursprung, som ännu inte har godkänts. I sådana fall ska sökanden tillhandahålla en vetenskaplig grund som styrker att några experimentella data inte behöver tillhandahållas för de berörda klyvningsprodukterna eller, om en sådan vetenskaplig grund saknas, tillhandahålla experimentella data.
För genetiskt modifierade livsmedel och foder som innehåller, består av eller har framställts av genetiskt modifierade växter, vars odling inte leder till framställning av genetiskt modifierat material som innehåller olika kombinationer av transformationshändelser (icke klyvande grödor), ska ansökan endast omfatta den kombination som ska släppas ut på marknaden.
Bestämmelserna i detta avsnitt gäller också för transformationshändelser som kombineras på annat sätt, såsom samtidig transformation och retransformation.
II. VETENSKAPLIGA KRAV
1. FAROIDENTIFIERING OCH FAROKARAKTERISERING
1.1 Uppgifter om mottagar- eller (i förekommande fall) moderväxter
|
1.1.1 |
Sökanden ska lämna omfattande uppgifter om mottagar- eller (i förekommande fall) moderväxter för att
|
|
1.1.2 |
För de ändamål som avses i punkt 1.1.1 ska sökanden lämna följande uppgifter:
|
1.2 Molekylär karakterisering
1.2.1 Uppgifter om den genetiska modifieringen
Sökanden ska lämna tillräckliga uppgifter om den genetiska modifieringen
|
a) |
för att identifiera de nukleinsyror som är avsedda för transformation och relaterade vektorsekvenser som potentiellt har överförts till mottagarväxten, |
|
b) |
för att beskriva de nukleinsyror som faktiskt har införts i växten. |
1.2.1.1 Beskrivning av de metoder som använts för den genetiska modifieringen
Sökanden ska lämna uppgifter om följande:
|
a) |
Metod för genetisk transformation inklusive relevanta referenser. |
|
b) |
Mottagarväxtmaterial. |
|
c) |
Arten och stammen av Agrobacterium och andra mikrober, om de använts under den genetiska transformationsprocessen. |
|
d) |
Hjälpplasmider, om sådana använts under den genetiska transformationsprocessen. |
|
e) |
Ursprung för bäraren av nukleinsyrorna, om den använts under den genetiska transformationsprocessen. |
1.2.1.2 Den använda vektorns beskaffenhet och ursprung
Sökanden ska lämna följande uppgifter:
|
a) |
En fysisk karta över de funktionella elementen och andra beståndsdelar i plasmiden/vektorn tillsammans med de relevanta uppgifter som behövs för att tolka den molekylära analysen (såsom restriktionsställen, placering av primrar som använts i en polymeraskedjereaktion (PCR), placering av sonder som använts i en Southern-analys). Det område som är avsett att införas ska markeras tydligt. |
|
b) |
En tabell som identifierar varje beståndsdel i plasmiden/vektorn (inbegripet det område som är avsett att införas), dess storlek och avsedda funktion. |
1.2.1.3 Ursprung för de nukleinsyror som använts för transformation, storlek och avsedd funktion för alla beståndsdelar i det område som är avsett att införas
Sökanden ska lämna uppgifter om givarorganismerna och om de nukleinsyrasekvenser som är avsedda att införas, för att avgöra om givarorganismernas eller nukleinsyrasekvensernas beskaffenhet kan utgöra en säkerhetsrisk.
Uppgifterna om funktion för de nukleinsyraområden som är avsedda att införas ska omfatta följande:
|
a) |
De fullständiga nukleinsyrasekvenser som är avsedda att införas, inbegripet uppgifter om eventuella avsiktliga förändringar i motsvarande sekvenser i givarorganismerna. |
|
b) |
Tidigare säker användning av genprodukter som härrör från de områden som är avsedda att införas. |
|
c) |
Uppgifter om genprodukternas möjliga samband med kända toxiner, antinutritionella ämnen och allergener. |
Uppgifterna om varje givarorganism ska omfatta följande:
|
— |
Taxonomisk klassificering. |
|
— |
Tidigare användning avseende livsmedels- och fodersäkerhet. |
1.2.2 Uppgifter om den genetiskt modifierade växten
1.2.2.1 Allmän beskrivning av de egenskaper som har införts eller modifierats
Uppgifter som lämnas under denna punkt kan begränsas till en allmän beskrivning av de införda egenskaperna och de ändringar som dessa orsakat i växtens fenotyp och metabolism.
Om den införda egenskapen är herbicidtolerans ska sökanden t.ex. lämna uppgifter om den aktiva substansens verkningssätt och dess metabolism i växten.
1.2.2.2 Uppgifter om de sekvenser som faktiskt har införts/tagits bort
Sökanden ska lämna följande uppgifter:
|
a) |
Storleken på och antal kopior av alla detekterbara införda sekvenser, både fullständiga och partiella. Detta fastställts normalt genom en Southern-analys. De kombinationer av sonder/restriktionsenzymer som används för detta ändamål ska helt täcka de sekvenser som skulle kunna införas i den genetiskt modifierade växten, såsom delar av plasmiden/vektorn, bärare eller främmande nukleinsyror som finns kvar i den genetiskt modifierade växten. Southern-analysen ska omfatta de fullständiga transgena lokusen samt flankerande sekvenser och inbegripa alla lämpliga kontroller. För att fastställa antalet kopior av den införda sekvensen kan även kompletterande metoder användas (såsom realtids-PCR). |
|
b) |
Det införda genetiska materialets organisering och sekvens på varje införingsställe i standardiserad elektronisk form, i syfte att identifiera förändringar av de införda sekvenserna i förhållande till den sekvens som var avsedd att införas. |
|
c) |
När områden tagits bort, dessa områdens storlek och funktion där så är möjligt. |
|
d) |
De införda sekvensernas placering i cellen (cellkärna, kloroplaster, mitokondrier eller bevarad i icke-integrerad form) och metoderna för bestämning av dem. |
|
e) |
Sekvensuppgifter i standardiserad elektronisk form för både 5’- och 3’-flankerande områden vid varje införingsställe, i syfte att identifiera avbrott i kända gener. Bioinformatikanalyser ska genomföras med aktuella databaser för att söka efter likheter både inom och mellan arterna. För genetiskt modifierade växter som innehåller staplade transformationshändelser ska säkerheten beträffande potentiell interaktion mellan eventuella oavsiktliga modifieringar vid varje införingsställe bedömas. |
|
f) |
Öppna läsramar (Open Reading Frames, nedan kallade ORF och definieras som en nukleotidsekvens som innehåller en följd av kodon som inte avbryts av ett stoppkodon i samma läsram) som skapats till följd av en genetisk modifiering antingen vid föreningspunkter med genomiskt DNA eller till följd av strukturella förändringar i de införda sekvenserna. ORF ska analyseras mellan stoppkodoner som inte begränsar deras längd. Bioinformatikanalyser ska genomföras med aktuella databaser för att undersöka möjliga likheter med kända toxiner eller allergener. Databasernas egenskaper och versioner ska tillhandahållas. Beroende på vilka uppgifter som har samlats in kan fler analyser (t.ex. en transkriptionsanalys) behövas för att komplettera riskbedömningen. |
1.2.2.3 Uppgifter om de införda sekvensernas uttryck
Sökanden ska lämna uppgifter
|
— |
för att visa om den införda/modifierade sekvensen leder till avsiktliga ändringar på protein-, RNA- och/eller metabolitnivå, |
|
— |
för att beskriva potentiellt oavsiktliga uttryck av nya ORF, som identifierats utgöra en säkerhetsrisk i enlighet med punkt 1.2.2.2 f. |
För dessa ändamål ska sökanden lämna följande uppgifter:
|
a) |
Vilka metoder som använts för analys av genuttryck tillsammans med deras prestandaegenskaper. |
|
b) |
Uppgifter om den införda sekvensens utvecklingsmässiga uttryck under växtens livscykel. Kravet på uppgifter om utvecklingsmässigt uttryck ska beaktas från fall till fall med hänsyn till vilken promotor som använts, modifieringens avsedda effekter och ansökans omfattning. |
|
c) |
Delar av växten där de införda/modifierade sekvenserna uttrycks. |
|
d) |
Potentiellt oavsiktliga uttryck av nya ORF som identifierats enligt punkt 1.2.2.2 f och som utgör en säkerhetsrisk. |
|
e) |
Uppgifter om proteiners uttryck, inklusive rådata, som erhållits från fältförsök och som har anknytning till grödans odlingsvillkor. Uppgifter om uttrycksnivåer från de delar av växten som används till livsmedel och foder ska lämnas i samtliga fall. Dessutom ska uppgifter lämnas om målgenernas uttryck i andra delar av växten om vävnadsspecifika promotorer har använts och detta är relevant för säkerhetsbedömningen. Minimikravet för proteiners uttryck ska vara att uppgifter lämnas från tre odlingsplatser eller från en plats under tre säsonger. Permutationer av platser och säsonger ska vara godtagbara under förutsättning att minimikravet är uppfyllt. När det är motiverat med hänsyn till den införda sekvensens beskaffenhet (vid exempelvis inaktivering eller när de biokemiska vägarna avsiktligt har modifierats) ska specifika RNA-molekyler eller metaboliter analyseras. För inaktivering genom RNAi-uttryck bör potentiella gener som inte är målgener sökas genom in silico-analys för att bedöma om den genetiska modifieringen kan påverka andra geners uttryck på ett sätt som medför säkerhetsrisker. |
|
f) |
För transformationshändelser som staplats (stacked events) genom konventionell korsning ska uppgifter om uttryck lämnas för att bedöma potentiell interaktion mellan händelserna som kan medföra ytterligare säkerhetsrisker när det gäller proteinernas och egenskapernas uttryck jämfört med en enda transformationshändelse. Jämförelsen ska göras med uppgifter som erhålls från växter som odlats i samma fältförsök. Kompletterande uppgifter kan behövas i vissa fall och där farhågor uppstår. |
1.2.2.4 Den införda sekvensens genetiska stabilitet och den genetiskt modifierade växtens fenotypiska stabilitet
Sökanden ska lämna uppgifter
|
a) |
för att visa genetisk stabilitet för de transgena lokusen och fenotypisk stabilitet och arvsmönster för de införda egenskaperna, |
|
b) |
för staplade transformationshändelser, för att visa att varje transformationshändelse som staplats i växten har samma molekylära egenskaper och utmärkande drag som i växter med en enda transformationshändelse. |
För dessa ändamål ska sökanden lämna uppgifter som styrker stabilitet under flera (normalt sett fem) generationer eller växtcykler för växter med en transformationshändelse. Uppgifter från den första och den sista generationen av växtcykler är tillräckligt. Ursprunget för det material som använts i analysen ska anges. Uppgifterna ska analyseras med lämpliga statistiska metoder.
För staplade transformationshändelser ska jämförelser mellan de ursprungliga och de staplade transformationshändelserna göras med växtmaterial som är representativt för sådant växtmaterial som är avsett för kommersiell framställning. Sökanden ska motivera det använda växtmaterialet på lämpligt sätt. Jämförelserna ska omfatta jämförelser av införda sekvenser och flankerande områden i genetiskt modifierade växter som innehåller en händelse och växter som innehåller staplade transformationshändelser.
För att bedöma transformationshändelsernas genetiska stabilitet ska sökanden använda de lämpliga molekylära metoder som anges i avsnitt 1.2.2.2.
1.2.2.5 Potentiell risk vid horisontell genöverföring
Sökanden ska bedöma sannolikheten för horisontell genöverföring från produkten till människor, djur och mikroorganismer samt potentiella risker i samband med detta när intakta och funktionella nukleinsyror finns kvar i genetiskt modifierade livsmedel och foder.
1.2.3 Slutsatser efter den molekylära karakteriseringen
Den molekylära karakteriseringen ska ge uppgifter om de införda sekvensernas struktur och uttryck samt de avsedda egenskapernas stabilitet. Detta ska också gälla i de situationer där transformationshändelser har staplats genom konventionell förädling.
Det ska särskilt anges om det i den molekylära karakteriseringen av de genetiska modifieringarna framkommer säkerhetsrisker avseende avbrott i endogena gener eller reglerande sekvenser.
Den molekylära karakteriseringen ska också syfta till att fastställa om de genetiska modifieringarna eventuellt kan leda till att andra proteiner/ämnen än de avsedda framställs och då särskilt nya toxiner eller allergener.
De potentiella oavsiktliga ändringar som identifieras i detta avsnitt ska tas upp i relevanta kompletterande delar av säkerhetsbedömningen.
1.3 Jämförande analys
Den jämförande analysen av sammansättning samt av odlingsegenskaper och fenotypiska egenskaper ska tillsammans med den molekylära karaktäriseringen vara utgångspunkten för att strukturera och genomföra en riskbedömning av nya genetiskt modifierade livsmedel och foder.
Den ska syfta till att identifiera likheter och skillnader vad gäller följande:
|
a) |
Sammansättning, odlingsutfall och fenotypiska egenskaper (avsiktliga och oavsiktliga förändringar) för den genetiskt modifierade växten och dess konventionella motsvarighet. |
|
b) |
Sammansättning för det genetiskt modifierade livsmedlet och fodret samt dess konventionella motsvarighet. |
Om ingen lämplig konventionell motsvarighet kan identifieras kan inte en jämförande säkerhetsbedömning göras, och följaktligen ska en säkerhets- och näringsbedömning av det genetiskt modifierade livsmedlet eller fodret utföras som för nya livsmedel som omfattas av Europaparlamentets och rådets förordning (EG) nr 258/97 (1) och inte har konventionella motsvarigheter (såsom när det genetiskt modifierade livsmedlet eller fodret inte är nära besläktat med ett livsmedel eller foder med tidigare säker användning eller när en eller flera specifika egenskaper införs i syfte att åstadkomma komplexa ändringar i det genetiskt modifierade livsmedlets eller fodrets sammansättning).
1.3.1 Val av konventionell motsvarighet och kompletterande jämförelsematerial
För grödor som förökas vegetativt ska den konventionella motsvarigheten i princip vara den i det närmaste isogena sort som använts för att utveckla den transgena linjen.
För grödor som förökas sexuellt ska den konventionella motsvarigheten ha en genetisk bakgrund som är jämförbar med den genetiskt modifierade växten. När den genetiskt modifierade växten har utvecklats genom återkorsning ska en konventionell motsvarighet med en genetisk bakgrund som är så nära den genetiskt modifierade växten som möjligt väljas.
Dessutom kan sökanden inkludera ett jämförelsematerial vars genetiska bakgrund är närmare den genetiskt modifierade växten än den konventionella motsvarigheten (såsom en klyvningsprodukt utan transgener).
För att bedöma om de förväntade jordbruksmetoderna påverkar de studerade parametrarnas (endpoints) uttryck i genetiskt modifierade växter som är herbicidtoleranta ska tre testmaterial jämföras: den genetiskt modifierade växten som utsatts för den avsedda herbiciden, den konventionella motsvarigheten som behandlats med konventionella metoder för ogräsbekämpning och den genetiskt modifierade växten som behandlats med samma konventionella metoder för ogräsbekämpning.
För staplade transformationshändelser går det inte alltid att använda en konventionell motsvarighet med en genetisk bakgrund som ligger så nära den genetiskt modifierade växten som den konventionella motsvarighet som normalt används för en transformationshändelse. Om så är fallet ska sökanden lämna en motivering med angivna skäl till valet av konventionell motsvarighet och bedöma dess begränsningar för riskbedömningen. Som kompletterande jämförelsematerial kan sökanden dessutom inkludera genetiskt modifierade föräldralinjer med en transformationshändelse, genetiskt modifierade linjer som innehåller en klyvningsprodukt av de staplade transformationshändelser för vilka en ansökan har lämnats in eller klyvningsprodukter utan transgener som härrör från dessa genetiskt modifierade linjer. Sökanden ska lämna närmare uppgifter som motiverar valet av kompletterande jämförelsematerial.
Sökanden ska alltid lämna uppgifter om förädlingsprogrammet (stam) för den genetiskt modifierade växten, den konventionella motsvarigheten och, i förekommande fall, kompletterande jämförelsematerial tillsammans med en lämplig motivering av varför de valts. Tidigare säker användning av den konventionella motsvarigheten ska styrkas på lämpligt sätt med både kvalitativa och kvantitativa data.
Närmare vägledning för tillämpning av kraven i detta avsnitt finns i Efsas vetenskapliga yttrande Guidance on selection of comparators for the risk assessment of genetically modified plants and derived food and feed (2).
1.3.2 Försöksplan och statistisk analys av fältförsöksdata för den jämförande analysen
1.3.2.1 Beskrivning av protokoll för försöksplanen
a) Försöksplanens principer
Fältförsök som används för att ta fram material för den jämförande analysen ska utföras för att fastställa om den genetiskt modifierade växten och/eller det genetiskt modifierade livsmedlet och fodret skiljer sig från sina konventionella motsvarigheter och/eller är ekvivalenta med de icke genetiskt modifierade referenssorterna med tidigare säker användning.
För varje parameter ska den jämförande analysen omfatta följande två angreppssätt:
|
i) |
Ett test av skillnad – för att kontrollera om den genetiskt modifierade växten skiljer sig från sin konventionella motsvarighet och därför kan anses utgöra en fara beroende på typ av identifierad skillnad samt storlek på och typ av exponering. |
|
ii) |
Ett ekvivalenstest – för att kontrollera om den genetiskt modifierade växten är ekvivalent eller inte med icke genetiskt modifierade referenssorter, bortsett från de införda egenskaperna. |
Vid test av skillnad ska nollhypotesen vara att det inte finns någon skillnad mellan den genetiskt modifierade organismen och dess konventionella motsvarighet, medan alternativhypotesen ska vara att det finns en skillnad.
När kompletterande jämförelsematerial används för riskbedömningen ska ett test av skillnad utföras mellan den genetiskt modifierade växten och varje kompletterande jämförelsematerial i enlighet med kraven i avsnitt 1.3.2.2 för testet av skillnaden mellan den genetiskt modifierade växten och dess konventionella motsvarighet.
Vid ekvivalenstest ska nollhypotesen vara att skillnaden mellan den genetiskt modifierade organismen och referenssorterna minst uppgår till en angiven minimistorlek (se avsnitt 1.3.2.2), medan alternativhypotesen ska vara att det inte finns någon skillnad mellan den genetiskt modifierade organismen och referenssorterna eller att skillnaden är mindre än den angivna minimistorleken.
Det krävs att nollhypotesen förkastas för att dra slutsatsen att den genetiskt modifierade organismen och referenssorterna är otvetydigt ekvivalenta för den aktuella parametern. De ekvivalensgränser som används i ekvivalenstestet ska på lämpligt sätt återge det intervall med naturliga variationer som förväntas för referenssorter med tidigare säker användning.
b) Särskilda protokoll för försöksplanen
Naturlig variation kan ha flera orsaker, t.ex. uppstår variation inom en sort på grund av miljöfaktorer och variation mellan sorter på grund av en kombination av både genetiska faktorer och miljöfaktorer. För att identifiera och bedöma skillnader som beror endast på genotyper är det av största vikt att variationer i miljön kontrolleras. Därför ska icke genetiskt modifierade referenssorter inkluderas i fältförsökens försöksplan i tillräckligt antal för att säkerställa en fullgod bedömning av variationen, vilket är nödvändigt för att fastställa ekvivalensgränserna. Allt testmaterial som består av genetiskt modifierade växter, konventionell motsvarighet, referenssorter och, i förekommande fall, kompletterande jämförelsematerial, ska randomiseras till ytor inom ett och samma fält på varje plats, vanligtvis i en fullständigt randomiserad försöksplan eller i randomiserade block. De olika platser som valts för fältförsöken ska motsvara de olika väder- och jordbruksförhållanden som råder där grödan ska odlas. Valet ska uttryckligen motiveras. Valet av icke genetiskt modifierade referenssorter ska vara lämpligt för de valda platserna och ska uttryckligen motiveras. Om platserna täcker ett begränsat urval av odlingsförhållanden ska sökanden replikera fältförsöken under mer än ett år.
På varje plats ska testmaterialen, som består av genetiskt modifierade växter, konventionell motsvarighet och i förekommande fall kompletterande jämförelsematerial, vara identiska för alla replikat. Om det inte finns tydliga skäl för ett annat förfarande ska det dessutom på varje plats finnas minst tre lämpliga icke genetiskt modifierade referenssorter av grödan som har en dokumenterad tidigare säker användning, vilka även ska vara identiska mellan alla replikat. Replikationen på varje plats är antalet resultat som erhålls för varje testmaterial och replikationen bör aldrig vara mindre än fyra på varje plats. Om bara två lämpliga referenssorter är tillgängliga på en viss plats ska dock replikationen vara sex på den platsen, och om bara en referenssort är tillgänglig ska replikationen vara åtta.
Varje fältförsök ska replikeras på minst åtta platser, som valts för att vara representativa för de olika mottagande miljöer där växten sannolikt ska odlas. Fältförsöken kan utföras under ett och samma år eller spridas över flera år. De icke genetiskt modifierade referenssorterna får variera mellan de olika platserna och totalt ska minst sex olika referenssorter användas i de olika fältförsöken.
När den genetiskt modifierade växten testas tillsammans med andra genetiskt modifierade växter av samma art (såsom Zea mays) kan materialet för den jämförande bedömningen av dessa olika genetiskt modifierade växter framställas samtidigt på samma plats och inom samma fältförsök genom att de olika genetiskt modifierade växterna och lämpligt jämförelsematerial till dessa placeras i samma randomiserade block. För detta ska följande två stränga villkor gälla:
|
i) |
Den konventionella motsvarigheten och, i förekommande fall, kompletterande jämförelsematerial ska alltid förekomma tillsammans med den genetiskt modifierade växten i samma block. |
|
ii) |
Alla olika genetiskt modifierade växter, deras jämförelsematerial och alla icke genetiskt modifierade referenssorter som används för att testa ekvivalens med dessa genetiskt modifierade växter ska vara fullständigt randomiserade inom varje block. |
Om det krävs fler än 16 ytor per block för ett sådant fältförsök kan ett delvis balanserat ofullständigt blockförsök användas för att minska antalet ytor per block, genom att vissa av de genetiskt modifierade växterna och lämpligt jämförelsematerial till dessa utesluts från varje block. För detta ska följande två stränga villkor gälla:
|
i) |
Den konventionella motsvarigheten ska alltid förekomma tillsammans med sin bestämda genetiskt modifierade växt i samma block. |
|
ii) |
Alla icke genetiskt modifierade referenssorter ska förekomma i varje ofullständigt block och vara fullständigt randomiserade med växterna och deras jämförelsematerial. |
Fältförsöken ska beskrivas på lämpligt sätt, med uppgifter om viktiga parametrar såsom hantering av fältet innan sådd, datum för sådd, jordtyp, herbicidanvändning, klimatförhållanden och andra odlings-/miljöförhållanden under odlingen och skörden, samt det skördade materialets lagringsförhållanden.
Närmare vägledning för tillämpning av kraven i detta avsnitt finns i Efsas yttrande Statistical considerations for the safety evaluation of GMOs (3).
1.3.2.2 Statistisk analys
Dataanalyser ska presenteras i ett tydligt format, med standardiserade vetenskapliga enheter. De rådata och den programmeringskod som använts för den statistiska analysen ska lämnas i redigerbar form.
Det kan vara nödvändigt att tranformera data för att säkerställa normalfördelning och få en lämplig skala där statistiska effekter är additiva. För många parametrars responsvariabler kan en logaritmisk transformation vara lämplig. I sådana fall ska eventuella skillnader mellan det genetiskt modifierade materialet och övrigt testmaterial tolkas som en kvot på den naturliga skalan. Men om logaritmiska transformationer inte ger lämpliga resultat ska den naturliga skalan eller en annan skala övervägas.
Den totala variationen för varje parameter som observerats under fältförsöken ska beräknas och fördelas med lämpliga statistiska modeller för att erhålla två konfidensgränser och fastställa en nedre och en övre ekvivalensgräns på grundval av den variation som observerats bland referenssorterna. En av konfidensgränserna ska användas för testet av skillnad medan den andra konfidensgränsen och ekvivalensgränserna ska användas för ekvivalenstestet.
En blandad linjär statistisk modell ska användas för att beräkna konfidensgränserna för båda testerna (dvs. testet av skillnad och ekvivalenstestet) och en något annorlunda modell ska användas för att beräkna ekvivalensgränserna som ska användas i ekvivalenstestet.
En indikatorvariabel (icke-centrerad i den blandade modellen) betecknas med I så att I = 1 betecknar en fältyta med någon av de icke genetiskt modifierade referenssorterna, och I = 0 betecknar övriga fall. Därefter ska de slumpmässiga faktorerna för modell 1 vara, men inte nödvändigtvis begränsas till, de faktorer som representerar variationen i) mellan testmaterialen (den genetiskt modifierade växten, dess konventionella motsvarighet, alla icke genetiskt modifierade referenssorter och eventuella kompletterande jämförelsematerial), ii) i interaktionen mellan testmaterialen och variabel I, iii) mellan platserna och iv) mellan blocken inom platserna. Modell 2 bör vara identisk med modell 1, förutom att den slumpmässiga faktor som representerar interaktionen mellan testmaterialen och variabel I utgår.
Den oberoende faktorn för båda modeller bör ha lika många nivåer som antalet testmaterial och representera kontrasterna mellan medelvärdena för testmaterialen. Testmaterialen är följande enligt definitionen ovan: den genetiska modifierade växten, dess konventionella motsvarighet, de icke genetiskt modifierade referenssorterna och eventuella kompletterande testmaterial. De icke genetiskt modifierade referenssorterna ska betraktas som en nivå i den oberoende faktorn. För testet av skillnad är komponenten i den relevanta oberoende faktorn kontrasten i en modell med en frihetsgrad mellan den genetiskt modifierade växten och dess konventionella motsvarighet. För ekvivalenstestet är komponenten i den relevanta oberoende faktorn kontrasten i en modell med en frihetsgrad mellan den genetiskt modifierade växten och de icke genetiskt modifierade referenssorterna.
Både testet av skillnad och ekvivalenstestet ska genomföras så att sambandet mellan hypotesprövningen och konfidensgränserna utnyttjas. För ekvivalenstest ska en metod med två ensidiga test (TOST, two one-sided tests) följas genom att nollhypotesen om icke-ekvivalens förkastas när båda konfidensgränserna faller inom ekvivalensgränserna. Valet av en konfidensgräns på 90 procent motsvarar den sedvanliga nivån på 95 procent för statistiska ekvivalenstester.
Resultaten från testet av skillnad och ekvivalenstestet ska återges visuellt för alla parametrar samtidigt, i ett eller ett fåtal diagram.
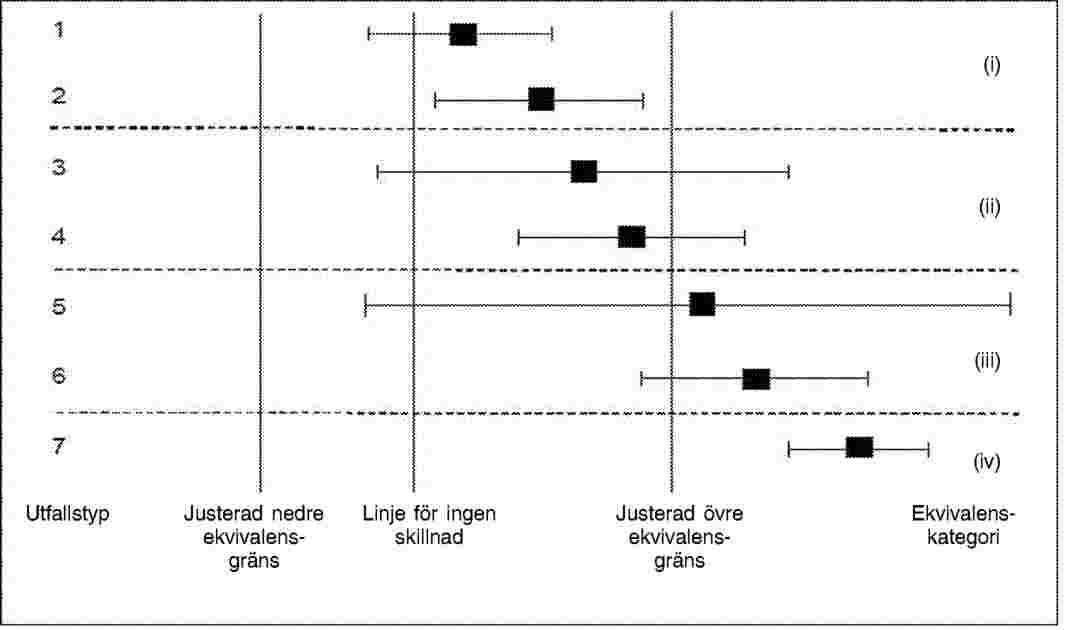
I diagrammen ska det finnas en linje för ingen skillnad mellan det genetiskt modifierade materialet och dess konventionella motsvarighet och för varje parameter ska följande anges: de justerade nedre och övre ekvivalensgränserna, den genomsnittliga skillnaden mellan det genetiskt modifierade materialet och dess konventionella motsvarighet och konfidensgränserna för denna skillnad (de olika möjliga utfallen för en enskild parameter visas i diagrammet i figur 1).
Om man förutom den konventionella motsvarigheten använder annat testmaterial som jämförelsematerial ska den genomsnittliga skillnaden mellan det genetiskt modifierade materialet och jämförelsematerialet, dess konfidensgräns och dess justerade ekvivalensgränser anges i diagrammen för varje kompletterande jämförelsematerial, genom att detta hänförs till samma nollbaslinje som för den konventionella motsvarigheten. Linjen för ingen skillnad på den logaritmiska skalan motsvarar den multiplikativa faktorn ett på den naturliga skalan. Den horisontella axeln ska ha värden som anger förändringen på den naturliga skalan. Vid logaritmisk transformation kommer förändringar på 2x och ½x att visas med lika stort mellanrum på vardera sidan om linjen för ingen skillnad.
Även om man kan förvänta sig en viss andel felaktiga signifikanta skillnader ska sökanden rapportera och diskutera alla signifikanta skillnader som observerats mellan den genetiskt modifierade grödan, dess konventionella motsvarighet och, i förekommande fall, annat testmaterial, med fokus på deras biologiska relevans (se avsnitt 3 om riskkarakterisering).
Vid rapportering ska fullständiga uppgifter lämnas om följande för varje analyserad parameter:
|
a) |
Vilka antaganden som ligger till grund för analysen. |
|
b) |
Fullständig specifikation av valda blandade modeller, inbegripet oberoende och slumpmässiga effekter. |
|
c) |
Resultat från eventuella interaktionstest mellan testmaterialen och platserna. |
|
d) |
Oberoende effekter, tillsammans med en lämplig beräknad residualvarians som de jämförs med, och varianskomponenter för de slumpmässiga faktorerna. |
|
e) |
Beräknade frihetsgrader. |
|
f) |
Övrig relevant statistik. |
En diskussion om sannolik effekt under andra odlingsförhållanden, som inte testats i fältförsöket, ska tillhandahållas.
Figur 1: Förenklad version av ett diagram för jämförande bedömning, som visar de sju möjliga utfallstyperna för varje enskild parameter. När ekvivalensgränserna har justerats kan en enda konfidensgräns (för skillnaden) bidra visuellt till att bedöma utfallet i båda testerna (skillnad och ekvivalens). Här beaktas endast den justerade övre ekvivalensgränsen. Följande visas i diagrammet: medelvärdet för den genetiskt modifierade grödan på en lämplig skala (låda), konfidensgränserna (morrhår/whiskers) för skillnaden mellan den genetiskt modifierade grödan och dess konventionella motsvarighet (den horisontella linjen visar konfidensintervallet), en vertikal linje för ingen skillnad (för test av skillnad) och vertikala linjer som anger de justerade ekvivalensgränserna (för ekvivalenstest). För utfallstyperna 1, 3 och 5 kan inte nollhypotesen om att det inte finns någon skillnad förkastas. För utfallstyperna 2, 4, 6 och 7 skiljer sig den genetiskt modifierade grödan från sin konventionella motsvarighet. För tolkning av ekvivalensen kan fyra kategorier i–iv urskiljas: i kategori i förkastas nollhypotesen om icke-ekvivalens till förmån för ekvivalens, i kategorierna ii, iii och iv kan icke-ekvivalens inte förkastas.
|
A. |
När det gäller test av skillnad ska varje utfall i diagrammet kategoriseras enligt följande, och en lämplig slutsats ska dras för respektive utfall.
|
|
B. |
När det gäller ekvivalenstest ska varje utfall i diagrammet kategoriseras enligt följande, och en lämplig slutsats ska dras för respektive utfall.
Vid signifikanta skillnader och/eller bristande ekvivalens för en viss parameter ska kompletterande statistiska analyser utföras för att bedöma om det förekommer interaktion mellan några av testmaterialen och platserna, eventuellt med en enkel standarmetod för variansanalys (ANOVA). Oavsett vilken metod som används ska man för varje analyserad parameter ange a) vilka antaganden som ligger till grund för analysen och, i tillämpliga fall, b) frihetsgrader, c) beräknad residualvariation för varje variationskälla och för varianskomponenterna samt d) övrig relevant statistik. Dessa kompletterande analyser syftar till att underlätta tolkningen av eventuella signifikanta skillnader som identifierats och att studera potentiella interaktioner mellan testmaterial och andra faktorer. Närmare vägledning för tillämpningen av kraven i detta avsnitt finns i Efsas yttrande Statistical considerations for the safety evaluation of GMOs (4). |
1.3.3 Val av material och kemiska föreningar för analys
Det är viktigt att analysera växtmaterialets sammansättning när det genetiskt modifierade livsmedlet och fodret jämförs med sin konventionella motsvarighet. Det material som ska användas för den jämförande bedömningen ska väljas med hänsyn till den genetiskt modifierade växtens användningsområden och den genetiska modifieringens beskaffenhet. För genetiskt modifierade växter som är herbicidtoleranta ska tre testmaterial användas: den genetiskt modifierade växten som utsatts för den avsedda herbiciden, den konventionella motsvarigheten som behandlats med konventionella metoder för ogräsbekämpning och den genetiskt modifierade växten som behandlats med samma konventionella metoder för ogräsbekämpning. Om inte annat vederbörligen har motiverats ska analysen utföras på jordbruksråvaran, eftersom det vanligtvis är genom den som materialet kommer in i framställnings- och bearbetningskedjan för livsmedel och foder. Kompletterande analyser av bearbetade produkter (såsom livsmedel och foder, livsmedelsingredienser, foderråvaror, livsmedels- och fodertillsatser eller livsmedelsaromer) ska utföras i de fall där så är lämpligt (se även avsnitt 1.3.6). Provtagning, analys och beredning av det testade materialet ska utföras enligt lämpliga kvalitetsstandarder.
1.3.4 Jämförande analys av sammansättning
Utöver analysen av de nya proteinerna som uttrycks (se avsnitt 1.2.2.3) ska analysen av sammansättningen omfatta ett lämpligt antal kemiska föreningar. I varje fall ska sökanden tillhandahålla åtminstone analyser av proximater (bl.a. vattenhalt och total askhalt), viktiga makro- och mikronäringsämnen, antinutritionella ämnen, naturliga toxiner och redan identifierade allergener samt andra sekundära växtmetaboliters egenskaper för en viss växtart, enligt Organisationen för ekonomiskt samarbete och utvecklings (OECD) konsensusdokument om hänsynstaganden vad gäller nya växtsorters sammansättning (nedan kallat OECD:s konsensusdokument) (5). De vitaminer och mineraler som väljs ut för analys ska vara sådana som finns i näringsmässigt signifikanta halter och/eller ger näringsmässigt signifikanta bidrag till kosten i den omfattning som växten konsumeras. Vilka särskilda analyser som krävs beror på vilken växtsort som undersöks, men de ska innehålla en mer detaljerad bedömning som är lämplig med hänsyn till den genetiska modifieringens avsedda effekt samt växtens näringsvärde och användning. Sökanden ska lägga särskild vikt vid viktiga näringsämnen såsom proteiner, kolhydrater, lipider/fetter, fibrer, vitaminer och mineraler. Det ska exempelvis ingå en fettsyreprofil (huvudsakliga enskilda mättade, enkelomättade och fleromättade fettsyror) för oljerika växter och en aminosyreprofil (enskilda aminosyror som förekommer i proteiner och huvudsakliga aminosyror som inte förekommer i proteiner) för växter som är viktiga proteinkällor. Analys av beståndsdelar i växtens cellvägg krävs också för de växtdelar som används som foder.
Sökanden ska dessutom tillhandahålla en analys av viktiga toxiner som finns naturligt i mottagarväxten och som kan påverka människors/djurs hälsa negativt beroende på deras toxiska styrka och halter. Koncentrationerna av sådana kemiska föreningar ska bedömas med hänsyn till växtsort samt livsmedels- och foderproduktens avsedda användning. På liknande sätt ska antinutritionella ämnen, såsom inhibitorer av matspjälkningsenzymer, och redan identifierade allergener undersökas.
Särdragen hos den införda egenskapen kan leda till kompletterande analyser av specifika kemiska föreningar, bl.a. metaboliter i potentiellt modifierade ämnesomsättningsvägar. Sökanden ska i förekommande fall överväga att ta med andra kemiska föreningar än de viktiga näringsämnen, viktiga toxiner, antinutritionella ämnen och allergener som anges i OECD:s konsensusdokument, och motivera varför dessa kemiska föreningar har valts.
1.3.5 Jämförande analys av odlingsegenskaper och fenotypiska egenskaper
Sökanden ska tillhandahålla en jämförelse mellan den genetiskt modifierade växten och dess konventionella motsvarighet. Jämförelsen ska göra det möjligt för sökanden att identifiera oavsiktliga effekter till följd av den genetiska modifieringen och ska även ta upp växtens biologi och odlingsegenskaper, bl.a. vanliga förädlingsparametrar (såsom avkastning, växtens morfologi, tidpunkt för blomning, graddagar till mognad, hur länge pollen är livskraftigt, respons på växtpatogener och insektbekämpningsmedel och känslighet för abiotisk stress). Protokollen från dessa fältförsök ska följa specifikationerna i avsnitt 1.3.2.
Om transformationshändelser staplas genom konventionell korsning kan även odlingsegenskaperna och de fenotypiska egenskaperna ändras. Möjliga skillnader som avser fenotypiska egenskaper och odlingsegenskaper vid staplade transformationshändelser ska bedömas i fältförsök. I förekommande fall ska sökanden lämna kompletterande uppgifter om de staplade transformationshändelsernas odlingsegenskaper från kompletterande fältförsök.
1.3.6 Bearbetningens effekter
Sökanden ska bedöma om den teknik som används för bearbetning och/eller bevaring eventuellt kan ändra egenskaperna hos de genetiskt modifierade slutprodukterna jämfört med deras respektive konventionella motsvarighet. Sökanden ska tillhandahålla en tillräckligt detaljerad beskrivning av de olika bearbetningsteknikerna, med särskilt fokus på moment som kan leda till betydande förändringar av produktens innehåll, kvalitet eller renhet.
Genetisk modifiering kan sikta in sig på ämnesomsättningsvägarna, vilket leder till ändringar i koncentrationen av andra ämnen än proteiner eller i nya metaboliter (såsom i näringsberikade livsmedel). Bearbetade produkter kan antingen bedömas samtidigt med säkerhetsbedömningen av den genetiskt modifierade växten eller bedömas separat. Sökanden ska tillhandahålla den vetenskapliga grunden för riskbedömning av dessa produkter. Sökanden ska från fall till fall överväga att lämna in kompletterande experimentella data.
I förekommande fall, beroende på produkt, är det nödvändigt med uppgifter om sammansättning, halt av icke önskvärda ämnen, näringsvärde, metabolism och avsedd användning.
I förekommande fall, beroende på beskaffenhet hos de nya proteiner som uttrycks, är det nödvändigt att bedöma i vilken omfattning bearbetningsmomenten leder till att dessa proteiner koncentreras eller elimineras, denatureras och/eller bryts ned i slutprodukten.
1.3.7 Slutsats
I den jämförande analysens slutsats ska följande anges tydligt:
|
a) |
Om den genetiskt modifierade växtens odlingsegenskaper och fenotypiska egenskaper, förutom de införda egenskaperna, skiljer sig från de egenskaper som dess konventionella motsvarighet har och/eller är ekvivalenta med referenssorterna, med hänsyn till naturliga variationer. |
|
b) |
Om egenskaper hos det genetiskt modifierade livsmedlets och fodrets sammansättning, med hänsyn till naturliga variationer, skiljer sig från de egenskaper som dess konventionella motsvarighet har och/eller är ekvivalenta med referenssorterna, förutom de införda egenskaperna. |
|
c) |
De egenskaper för vilka den genetiskt modifierade växten eller det genetiskt modifierade livsmedlet och fodret skiljer sig från dess konventionella motsvarighet och/eller är icke-ekvivalenta med referenssorterna, med hänsyn till naturliga variationer, och som kräver kompletterande utredning. |
|
d) |
Om det vid transformationshändelser som staplats genom konventionell korsning finns tecken på interaktioner mellan de kombinerade transformationshändelserna. |
1.4 Toxikologi
Den toxikologiska effekten av ändringar av hela det genetiskt modifierade livsmedlet/fodret till följd av den genetiska modifieringen, t.ex. införande av nya gener, inaktivering av gener eller överuttryck av en endogen gen, ska bedömas.
En toxikologisk bedömning ska göras i följande syfte:
|
a) |
Visa att den genetiska modifieringens avsiktliga effekter inte har några negativa effekter på människors och djurs hälsa. |
|
b) |
Visa att de genetiska modifieringarnas oavsiktliga effekter, som har identifierats eller förutsätts ha inträffat baserat på de tidigare jämförande molekylära analyserna eller analyserna av sammansättning eller fenotyp, inte har några negativa effekter på människors och djurs hälsa. |
|
c) |
Identifiera potentiella negativa effekter av nya beståndsdelar och bestämma de maximala dosnivåer som inte orsakar negativa effekter. Människors acceptabla dagliga intag (ADI) av enskilda kemiska föreningar kan härledas från data från lämpliga djurförsök genom att osäkerhets- eller säkerhetsfaktorer som tar hänsyn till skillnader mellan försöksdjurarter och människor samt variationer mellan enskilda människor används. |
|
d) |
Identifiera potentiella negativa effekter hos hela det genetiskt modifierade livsmedlet/fodret eller behandla återstående osäkerhetsfaktorer genom att utföra 90-dagars utfodringsstudier. |
Sökanden ska överväga vilken typ av toxikologisk testning som ska utföras på nya beståndsdelar och på hela det genetiskt modifierade livsmedlet/fodret på grundval av resultaten från de molekylära och jämförande analyser som avses i avsnitten 1.2 och 1.3, nämligen de skillnader som identifierats mellan den genetiskt modifierade produkten och dess konventionella motsvarighet, inbegripet såväl avsiktliga som oavsiktliga ändringar. Sökanden ska också bedöma resultaten av de toxikologiska tester som utförts för att överväga behovet av att genomföra kompletterande testning på nya beståndsdelar eller på hela det genetiskt modifierade livsmedlet/fodret i enlighet med avsnitten 1.4.4.2 och 1.4.4.3.
Sökanden ska ta hänsyn till förekomsten av nya proteiner som uttrycks, potentiell förekomst av andra nya beståndsdelar och/eller eventuellt ändrade halter av naturliga beståndsdelar utöver normal variation. Krav på särskilda uppgifter och teststrategier fastställs i avsnitten 1.4.1–1.4.4.
För ansökningar som omfattar eller är begränsade till genetiskt modifierade livsmedel och foder som framställts av genetiskt modifierade växter, ska toxikologiska studier av de bearbetade produkterna tillhandahållas, utom när sökanden tillhandahåller en riskbedömning av den genetiskt modifierade växten (eller relevanta delar av den) som visar att den är säker och det inte finns några tecken på att de bearbetade genetiskt modifierade livsmedlen och fodren skulle skilja sig från sin respektive konventionella motsvarighet. Sökanden ska motivera detta på lämpligt sätt.
Toxikologiska studier som ska utvärdera risker för människors och/eller djurs hälsa ska komplettera varandra. De flesta studier som krävs för att bedöma genetiskt modifierade livsmedels säkerhet är även giltiga för bedömning av genetiskt modifierade foder.
Förutom människors och djurs exponering genom intag av livsmedel och foder ska sökanden rapportera eventuella negativa effekter på individer som kan bero på exponering för genetiskt modifierade livsmedels- och foderråvaror genom deras yrkesverksamhet, t.ex. jordbruk eller bearbetning av utsäde. Lämpliga studier ska genomföras för att beskriva sådana tecken på potentiella negativa effekter mer ingående.
Sökanden ska använda internationellt överenskomna protokoll och testmetoder för toxicitetstestning (se tabellerna 1 och 2 i avsnitt 1.7). Om dessa protokoll anpassas eller om metoder som skiljer sig från dessa protokoll används ska detta motiveras i ansökan.
1.4.1 Testning av nya proteiner som uttrycks
Sökanden ska tillhandahålla en utvärdering av alla nya proteiner som uttrycks. De studier som krävs för att utreda den potentiella toxiciteten hos ett nytt protein som uttrycks ska väljas från fall till fall, beroende på vad man känner till om proteinets ursprung, funktion eller aktivitet och människors eller djurs tidigare konsumtion. För proteiner som uttrycks i den genetiskt modifierade växten ska det inte krävas någon särskild toxicitetstestning enligt detta avsnitt om det finns en väldokumenterad tidigare säker användning av livsmedlet och/eller fodret för både växten och de nya proteiner som uttrycks. I sådana fall ska sökanden lämna nödvändiga uppgifter om tidigare säker användning av proteinerna.
Om särskild testning krävs ska det testade proteinet vara ekvivalent med det nya proteinet så som det uttrycks i den genetiskt modifierade växten. Om ett protein framställt av mikroorganismer används på grund av att det inte finns tillräckligt med testmaterial från växten, ska strukturell, biokemisk och funktionell ekvivalens med det nya växtproteinet som uttrycks kunna påvisas för det mikrobiella substitutet. I synnerhet behövs jämförelser av molekylvikt, aminosyrasekvens, posttranslationell modifiering, immunologisk reaktivitet och, för enzymer, enzymaktivitet för att bevisa ekvivalensen. Om det finns skillnader mellan proteinet som växten uttrycker och dess mikrobiella substitut ska skillnadernas betydelse för säkerhetsstudierna utvärderas.
För att visa att de nya proteiner som uttrycks är säkra ska sökanden tillhandahålla följande:
|
a) |
En molekylär och biokemisk karakterisering av det nya proteinet som uttrycks, inbegripet en bestämning av primär struktur, molekylvikt (t.ex. genom masspektrometri), studier av posttranslationella modifieringar och en beskrivning av dess funktion. För nya enzymer som uttrycks ska dessutom uppgifter lämnas om enzymaktivitet, inbegripet temperatur och pH-intervall för optimal aktivitet, substratspecificitet och möjliga reaktionsprodukter. Potentiell interaktion med växtens andra beståndsdelar ska också utvärderas. |
|
b) |
En aktuell homologisökning som avser proteiner som är kända för att orsaka negativa effekter, såsom toxiska proteiner. En homologisökning som avser proteiner som har en normal metabolism eller strukturell funktion kan också bidra med värdefulla uppgifter. De databaser och den metod som använts för sökningen ska anges. |
|
c) |
En beskrivning av proteinets stabilitet under de relevanta förhållanden som råder vid livsmedlets och fodrets bearbetning, lagring och förväntade hantering. Påverkan från ändrade temperaturer och pH-värden ska också undersökas, och potentiella modifieringar av proteinerna (såsom denaturering) och/eller framställning av stabila proteinfragment genom sådana behandlingar ska beskrivas. |
|
d) |
Uppgifter om resistensen hos det nya proteinet som uttrycks mot nedbrytning av proteaser (såsom pepsin), t.ex. genom undersökningar in vitro med lämpliga och standardiserade tester. Stabila nedbrytningsprodukter ska beskrivas och utvärderas med hänsyn till potentiella negativa hälsoeffekter som kan kopplas till deras biologiska aktivitet. |
|
e) |
Ett 28-dagars toxicitetstest med upprepat oralt intag av det nya proteinet som uttrycks på gnagare. Beroende på resultaten från 28-dagars toxicitetstestet ska i lämpliga fall kompletterande riktade undersökningar tillhandahållas, bl.a. en analys av immunotoxiciteten. |
Test av akut toxicitet av de genetiskt modifierade växternas nya proteiner som uttrycks har litet mervärde för riskbedömningen av människors och djurs upprepade intag av genetiskt modifierade livsmedel och foder och ska inte tillhandahållas som en del av de studier som utförs enligt denna punkt.
Sökanden ska utföra studier med kombinerad administrering av proteiner när den genetiska modifieringen leder till att två eller flera proteiner uttrycks i den genetiskt modifierade växten och när man på grundval av vetenskapliga rön identifierar möjliga sam- eller motverkande interaktioner som kan utgöra säkerhetsrisker.
1.4.2 Testning av andra nya beståndsdelar än proteiner
Sökanden ska tillhandahålla en riskbedömning av andra nya beståndsdelar än proteiner som identifierats. Den ska i förekommande fall omfatta en utvärdering av deras toxiska styrka och behovet av toxikologisk testning, samt en bestämning av deras koncentration i det genetiskt modifierade livsmedlet och fodret. För att fastställa säkerheten för nya beståndsdelar när det inte finns någon tidigare säker användning av livsmedlet och fodret ska sökanden lämna uppgifter som är jämförbara med de som beskrivs i Guidance for submissions for food additive evaluations by the EFSA Panel on Food Additives and Nutrient Sources added to Food av den 16 augusti 2012 (6) och i kommissionens förordning (EG) nr 429/2008 av den 25 april 2008 om tillämpningsföreskrifter för Europaparlamentets och rådets förordning (EG) nr 1831/2003 avseende utformning och presentation av ansökningar samt bedömning och godkännande av fodertillsatser (7). Detta ska omfatta att lämna uppgifter om ett antal viktiga studier, bl.a. om metabolism/toxikokinetik, subkronisk toxicitet, genotoxicitet, kronisk toxicitet, cancerogenitet samt reproduktions- och utvecklingstoxicitet, åtföljda av andra lämpliga studier. Särskild vägledning för djurförsök finns i tabell 1 i avsnitt 1.7 i denna bilaga. Testprotokoll för genotoxicitet anges i tabell 2 i avsnitt 1.7 i denna bilaga.
1.4.3 Uppgifter om ändrade halter av beståndsdelar i livsmedel och foder
Detta avsnitt ska endast tillämpas när den avsiktliga eller oavsiktliga effekten av den genetiska modifieringen leder till ändrade halter av beståndsdelar i livsmedel och foder utöver den naturliga variationen.
För att påvisa säkerheten för ändrade halter av beståndsdelar i livsmedlet och fodret t.ex. makro- och mikronäringsämnen, antinutritionella ämnen, naturliga toxiner och andra sekundära växtmetaboliter, ska sökanden lämna en detaljerad riskbedömning baserad på kunskap om dessa beståndsdelars fysiologiska funktion och/eller toxiska egenskaper.
Resultatet av riskbedömningen ska avgöra om och i vilken omfattning sökanden ska tillhandahålla toxikologiska tester för valda beståndsdelar i livsmedlet och fodret för att komplettera 90-dagars utfodringsstudien på gnagare med hela det genetiskt modifierade livsmedlet/fodret.
1.4.4 Testning av hela det genetiskt modifierade livsmedlet och fodret
Sökandens riskbedömning av det genetiskt modifierade livsmedlet fodret ska i första hand bygga på en molekylär karaktärisering, en jämförande odlingsanalys och fenotypisk analys, en omfattande analys av sammansättningen och en toxikologisk utvärdering av identifierade avsiktliga och oavsiktliga effekter, inklusive en 90-dagars utfodringsstudie på gnagare med hela det genetiskt modifierade livsmedlet/fodret i enlighet med avsnitt 1.4.4.1. Under de omständigheter som anges i punkterna 1.4.4.2 och 1.4.4.3 i detta avsnitt ska kompletterande specifika toxikologiska studier med hela det genetiskt modifierade livsmedlet och fodret utföras.
1.4.4.1 En 90-dagars utfodringsstudier på gnagare med hela det genetiskt modifierade livsmedlet/fodret:
Sökanden ska inkludera en 90-dagars utfodringsstudie med hela livsmedel och foder på gnagare för bedömningen av livsmedel och foder som innehåller, består av eller har framställts av genetiskt modifierade växter med en transformationshändelse eller med staplade transformationshändelser som inte erhålls genom konventionell korsning av genetiskt modifierade växter som innehåller en transformationshändelse.
För staplade transformationshändelser som erhålls genom konventionell korsning av genetiskt modifierade växter som innehåller en eller flera transformationshändelser ska en 90-dagars utfodringsstudie med hela livsmedel och foder på gnagare inkluderas för var och en av de genetiskt modifierade växter med en transformationshändelse som använts. En kompletterande 90-dagars utfodringsstudie med hela livsmedel och foder på gnagare med den genetiskt modifierade växten med de staplade transformationshändelserna ska inkluderas om man ser tecken på potentiella negativa effekter under bedömningen av i) de införda sekvensernas stabilitet, ii) de införda sekvensernas uttryck och iii) de potentiella sam- eller motverkande effekter som beror på kombinationen av transformationshändelser.
Toxicitetsstudien med genetiskt modifierade livsmedel och foder bör utföras i enlighet med den modell för ”test avseende subkronisk oral toxicitet (90-dagars upprepat oraltest på gnagare)” som anges i tabell 1 enligt ett anpassat protokoll. I princip bör minst två testdoser och en negativ kontroll användas. Den högsta dosen ska vara den som maximalt kan uppnås utan att orsaka näringsmässig obalans. Den lästa dosen ska innehålla det testade livsmedlet/fodret i en mängd som alltid är högre än det förväntade intaget för människor/målintaget för djur. Det genetiskt modifierade livsmedlet och fodret som analyseras bör vara relevant för den produkt som ska konsumeras. För genetiskt modifierade växter som är herbicidtoleranta bör det material som testas komma från genetiskt modifierade växter som utsatts för den avsedda herbiciden. Alltid när det är möjligt ska uppgifter om testparametrarnas naturliga variation härledas från tidigare bakgrundsdata snarare än från inkludering av referenssorter, bestående av kommersiellt tillgängliga livsmedel och foder framställda av icke genetiskt modifierade växter med en tidigare säker användning, i experimenten. Den statistiska analysen ska inriktas på detektion av eventuella skillnader mellan testmaterialet och dess kontroll. En styrkeanalys för att uppskatta en provsstorlek som kan påvisa en på förhand fastställd biologiskt relevant effektstorlek med en angiven styrke- och signifikansnivå bör användas. Närmare vägledning för genomförandet av denna studie finns i Efsas riktlinjer för genomförande av 90-dagars toxicitetstest med upprepat oralt intag av hela livsmedel/foder på gnagare (8).
1.4.4.2 Djurförsök för testning av reproduktions- och utvecklingstoxicitet
När de uppgifter om genetiskt modifierade livsmedel och foder som krävs i avsnitten 1.4.1, 1.4.2 och 1.4.3 tyder på en potentiell reproduktionstoxicitet, utvecklingstoxicitet eller kronisk toxicitet, eller om det i en 90-dagars utfodringsstudie på gnagare finns tecken på negativa effekter (såsom funktionella och/eller histologiska ändringar av neurologiska, endokrina, reproduktiva eller immunologiska vävnader/organ), ska lämplig testning utföras. Protokollen för testning av reproduktionstoxicitet, utvecklingstoxicitet och kronisk toxicitet (se tabell 1 i avsnitt 1.7) får anpassas i syfte att testa hela det genetiskt modifierade livsmedlet och fodret.
Eftersom 90-dagars utfodringsstudien på gnagare har utformats enbart för att upptäcka effekter på reproduktiva organs vikter och histopatologi hos vuxna djur och eftersom man med denna metod inte kan upptäcka andra reproduktions- eller utvecklingseffekter, ska tester på hela livsmedel och foder utföras utöver en 90-dagars utfodringsstudie på gnagare om sådana faror har identifierats.
1.4.4.3 Andra djurförsök för att undersöka genetiskt modifierade livsmedels och foders säkerhet och egenskaper (se även avsnitten 1.6.1 och 1.6.2)
Utfordringsstudier med djurarter från målgruppen ska lämnas in om indikationer på negativa effekter har noterats i de uppgifter om genetiskt modifierade livsmedel och foder som krävs enligt avsnitten 1.4.1, 1.4.2 och 1.4.3 alternativt utifrån resultaten av en 90-dagars utfodringsstudie på gnagare. De ska fokusera på de nya beståndsdelarnas säkerhet (nya proteiner som uttrycks och andra nya beståndsdelar), identifiering och karakterisering av oavsiktliga effekter samt den näringsmässiga effekten av avsiktliga och omfattande ändringar av den genetiskt modifierade växtens sammansättning (se även avsnitt 1.6).
Sådana studier ska begränsas till växtmaterial som är lämpliga att inkluderas i deras kost och som näringsmässigt kan matchas av en lämplig kontrollkost.
1.4.4.4 Tolkning av djurförsökens relevans
Relevanta effekter som observeras under djurförsöken ska utvärderas för att identifiera potentiella konsekvenser för människors och djurs hälsa och för att bedöma deras relevans för de genetiskt modifierade livsmedlens och fodrens säkerhet. Utvärderingen kan stödjas av kompletterande uppgifter och hänsynstaganden. Det bör uppmärksammas att vissa effekter kan vara specifika för försöksdjuren, men inte för människor på grund av skillnader mellan arterna.
Sökanden ska i synnerhet beakta samband mellan dos och respons för de parametrar som har ändrats (dvs. förändringarna ökar proportionellt vid ökad dos) eftersom de är tydliga tecken på att den testade kemiska föreningen har en effekt. Om en skillnad endast noteras vid den högsta dosen ska andra faktorer beaktas för att fastställa om det finns ett samband med behandlingen. Sökanden kan hämta uppgifter om en viss parameters bakgrundsvariation från data som avser andra djur av samma art/stam som testats i samma eller andra experiment, eller från internationellt harmoniserade databaser.
I tester där djur av båda könen används kan även ändringar som endast förekommer i djur av det ena könet vara relevanta tecken på en effekt, beroende på vilken parameter som ändras och vilken mekanism som har orsakat ändringen. Djur av det ena könet kan exempelvis vara mer benägna till ändringar som orsakats av en viss beståndsdel än djur av det andra könet, eller till och med särskilt benägna till ändringar, såsom är fallet för endokrina effekter.
Sökanden ska även identifiera möjliga inbördes förhållanden mellan observerade ändringar av enskilda parametrar som kan stärka tecknen på att en effekt har uppstått. Exempelvis kan en leverskada observeras i själva levern som en förändring i histopatologi, makroskopisk patologi och organvikt, men även påvisas genom ändrade halter av vissa kemiska föreningar som härrör från levern, såsom enzymer eller bilirubin i serum.
När det gäller den potentiella orsaken till en observerad effekt ska hänsyn tas till sannolikheten för orsakssamband, inte bara för den testade kemiska föreningen utan även för andra faktorer som också kan ha påverkat resultaten (såsom minskad kroppsvikt till följd av minskat intag av en mindre välsmakande kost). Uppgifter som stödjer en hypotes om orsakssamband mellan den testade kemiska föreningen och effekter på försöksdjur kan exempelvis vara prediktionsvärden för sannolika effekter från in vitro- och in silico-försök och samband mellan dos och respons som observerats vid djurförsök.
1.4.5 Slutsatser efter den toxikologiska bedömningen
I slutsatserna efter den toxikologiska bedömningen ska det anges huruvida
|
a) |
de potentiella negativa effekter som identifieras i andra delar av säkerhetsbedömningen har bekräftats eller förkastats, |
|
b) |
de tillgängliga uppgifterna om nya proteiner som uttrycks och andra nya beståndsdelar som är en följd av den genetiska modifieringen tyder på potentiella negativa effekter, och i synnerhet om och vid vilka dosnivåer de negativa effekterna identifierats i särskilda studier, |
|
c) |
uppgifterna om de naturliga beståndsdelar för vilka halterna skiljer sig från den konventionella motsvarigheten tyder på potentiella negativa effekter, och i synnerhet om och vid vilka dosnivåer de negativa effekterna identifierats i särskilda studier, |
|
d) |
negativa effekter har identifierats i de studier som gjorts av hela det genetiskt modifierade livsmedlet och fodret samt vid vilka dosnivåer. |
Sökanden ska utvärdera resultatet av den toxikologiska bedömningen mot bakgrund av förväntat intag av det genetiskt modifierade livsmedlet och fodret (se avsnitt 2).
1.5 Allergiframkallande egenskaper
Livsmedelsallergi är en negativ reaktion på livsmedel och är ett stort folkhälsoproblem. Livsmedelsallergi är inte detsamma som toxiska reaktioner och överkänslighet. Allergi är en patologisk avvikelse i immunsvaret på ett visst ämne, som endast påverkar vissa individer där en kombinerad effekt av variationer i miljön och genetiska anlag har orsakat en allergisk sensibilisering.
För allergiska individer kan ibland mycket små mängder av ett livsmedel, som den stora merparten av befolkningen tål, orsaka allvarliga symtom och dödsfall. Det är inte allergenet i sig som orsakar den negativa hälsoeffekten, utan den allergiska personens avvikande reaktion på allergenet.
Livsmedelsallergi kan orsakas av olika immunmekanismer. IgE-förmedlad livsmedelsallergi är dock den huvudsakliga formen av livsmedelsallergi som orsakar de allvarligaste reaktionerna och är den enda formen som orsakar livshotande reaktioner. Den IgE-förmedlade livsmedelsallergin har varit i fokus vid riskbedömning av genetiskt modifierade organismers allergiframkallande egenskaper. Livsmedelsallergi består huvudsakligen av två skilda faser: först sensibilisering där inga symtom förekommer men immunsystemets förmåga att reagera ökar drastiskt och sedan elicitering (provokation) med kliniska symtom.
Vid intag av allergener, dvs. sensibiliserande livsmedel eller livsmedelsbeståndsdelar, bryts de i viss mån ned av matspjälkningsenzymer, absorberas av tarmarnas slemhinnor (i små mängder även av munnens slemhinna), bearbetas i immunsystemets specialiserade celler och presenteras sedan för de reaktiva immuncellerna som utlöser ett immunsvar. Sensibilisering kan också inträffa vid hudkontakt med eller inandning av livsmedelsallergenet.
En majoritet av de beståndsdelar som gör att livsmedel och pollen är allergiframkallande är proteiner. Vissa proteinnedbrytningsprodukter, nämligen peptidfragment, kan bevara delar av det nativa proteinets allergiframkallande egenskaper och kan alltså anses vara allergener.
De genetiskt modifierade organismernas särskilda allergirisk är kopplad till i) exponering för nya proteiner som uttrycks och som kan förekomma i ätbara växtdelar eller i pollen, vilket har att göra med transgenens biologiska ursprung, och ii) ändringar av de allergiframkallande egenskaperna i hela växten och dess produkter, t.ex. till följd av naturliga endogena allergeners överuttryck som en oavsiktlig effekt av den genetiska modifieringen, vilket har att göra med själva mottagarväxtens biologi.
Närmare vägledning för tillämpning av kraven i detta avsnitt finns i Efsas vetenskapliga yttrande om bedömning av allergiframkallande egenskaper i genetiskt modifierade växter och mikroorganismer samt livsmedel och foder som härrör från dessa, som antogs den 30 juni 2010 (9).
1.5.1 Bedömning av de allergiframkallande egenskaperna hos det nya proteinet som uttrycks
Allergiframkallande förmåga är inte en inneboende och helt förutsägbar egenskap hos ett visst protein, det är snarare en biologisk aktivitet som kräver interaktion med individer med genetiska anlag för allergi. Därför är den allergiframkallande förmågan beroende av atopiska människors genetiska mångfald och variation. De allergiska reaktionernas frekvens, allvarlighet och specificitet beror även på geografiska faktorer och miljöfaktorer. Eftersom de inte helt kan förutsägas är det nödvändigt att ta hänsyn till flera aspekter vid bedömningen av de allergiframkallande egenskaperna för att samla underlag som minimerar osäkerheten kring proteinerna i fråga.
När de strukturella, biologiska och fysikalisk-kemiska egenskaperna studeras för ett nytt protein som uttrycks är det viktigt att det testade proteinet är ekvivalenta i fråga om struktur och aktivitet med det nya proteinet som uttrycks i den genetiskt modifierade växten. Studier som utförs med renade målproteiner som beretts genom uttryck i organismer såsom Escherichia coli ska vara godtagbara under förutsättning att det mikrobiella substitutets egenskaper är identiska med egenskaperna hos det protein som uttrycks i växten och alltså tar hänsyn till alla posttranslationella modifieringar som specifikt förekommer i växten.
Sökanden ska kontrollera om transgenens ursprung är allergiframkallande. När det införda genetiska materialet erhålls från vete, råg, korn, havre eller liknande spannmål ska sökanden även bedöma om de nya proteiner som uttrycks har en möjlig roll vid elicitering av celiaki eller andra enteropatier som inte är IgE-förmedlade. När transformationshändelser har staplats ska sökanden i förekommande fall tillhandahålla en bedömning om detta potentiellt kan öka den allergiframkallande förmågan i människor och djur. Dessa potentiella effekter kan härröra från genproduktens additiva, samverkande eller motverkande effekter.
Sökanden ska göra en integrerad bedömning av de enskilda fallen, dvs. en sammanvägd bedömning av möjliga allergiframkallande egenskaper hos nya proteiner som uttrycks. Bedömningen ska innehålla följande:
|
a) |
En jämförelse av aminosyrors sekvenshomologi i det nya proteinet som uttrycks och i kända allergener I samtliga fall ska man söka efter sekvenshomologi och/eller strukturella likheter mellan proteinet som uttrycks och kända allergener för att identifiera potentiell IgE-korsreaktivitet mellan det nya proteinet som uttrycks och kända allergener. Sökanden ska se till att databasens kvalitet och omfattning motsvarar den aktuella kunskapsnivån. Vid en bioinformatisk jämförelse mellan aminosyrasekvenser anses ett minsta krav vara 35 procent sekvensidentitet med ett känt allergen över ett sammanhängande område av åtminstone 80 aminosyror. Alla parametrar för sekvensinpassning som använts i analysen ska tillhandahållas, inbegripet beräkning av procent identitet (PID). Beräkningen av PID ska utföras över ett sammanhängande område av åtminstone 80 aminosyror med luckor så att de införda luckorna hanteras som bristande överensstämmelse. I vissa fall kan man vid bedömning av korta peptidfragment, såsom ORF, söka efter sammanhängande sekvenser av identiska eller kemiskt liknande aminosyrarester. Denna sökning ska dock inte utföras rutinmässigt för att identifiera potentiella linjära IgE-bindande epitoper på grund av dess låga känslighet eller specificitet. |
|
b) |
Specifik serumscreening När det finns tecken på sekvenshomologi eller liknande struktur baseras ett viktigt förfarande för att bedöma om exponering för nya proteiner som uttrycks potentiellt kan framkalla en allergisk reaktion hos individer som redan sensibiliserats för korsreaktiva proteiner på in vitro-tester som mäter förmågan hos ett specifikt IgE från allergiska patienters serum att binda testproteinerna. Olika människors IgE-svar kan variera i specificitet och affinitet. I synnerhet kan IgE-antikropparnas specificitet för olika allergener i ett visst livsmedel eller en viss källa och/eller för olika epitoper i ett visst protein variera mellan allergiska individer. För att optimera testets känslighet ska individuella sera från välbeskrivna allergiska individer användas. Sökanden ska utföra specifik serumscreening i följande fall:
Specifik serumscreening ska utföras med individuella sera från individer med en påvisad och välbeskriven allergi mot källan eller mot det potentiellt korsreagerande allergenet med hjälp av relevanta immunokemiska tester. Lämpliga metoder är IgE-bindningsanalyser, såsom radioallergosorbenttest (Rast), enzymatisk bestämning i fastfassystem (EAST, Enzyme Allergosorbent Test), enzymkopplad immunadsorberande analys (Elisa) och elektrofores följt av immunoblotting med specifika sera som innehåller IgE. |
|
c) |
Prövning för resistens mot pepsinnedbrytning och in vitro-test av digererbarheten Allergiframkallande proteiner har länge ansetts vara stabila mot nedbrytning av proteaser. Även om det har fastställts att det inte finns något absolut samband är proteiners resistens mot pepsinnedbrytning ytterligare ett kriterium som ska beaktas i den sammanvägda bedömningen av de allergiframkallande egenskaperna. Prövning för resistens mot pepsinnedbrytning görs i allmänhet under relativt standardiserade förhållanden, med låga pH-värden och stor andel pepsin i förhållande till protein. Det är allmänt erkänt att en prövning för resistens mot pepsinnedbrytning inte avspeglar matspjälkningens fysiologiska förhållanden. Digererbarheten hos de nya proteinerna som uttrycks i specifika befolkningssegment, såsom spädbarn och individer med nedsatt matspjälkning, kan bedömas med hjälp av in vitro-tester av digererbarheten vid olika förhållanden. Eftersom det protein som kodats av de nya gener som införts kommer att finnas i produkten som en komplex matris, ska vid kompletterande in vitro-tester av digererbarheten hänsyn dessutom tas till effekten av en möjlig interaktion mellan proteinet och de övriga beståndsdelarna i matrisen samt bearbetningens effekter. Beroende på resultatet av in vitro-testet av digererbarheten ska en jämförelse av IgE-bindningen hos intakta, värmedenaturerade och pepsinnedbrutna proteiner bedömas, eftersom en ändrad digererbarhet kan påverka de allergiframkallande egenskaperna hos det nya proteinet som uttrycks. |
|
d) |
Kompletterande tester Även om kompletterande tester som cellbaserade analyser in vitro eller djurförsök in vivo hittills inte har validerats i regleringssyfte, kan de ge kompletterande användbara uppgifter, t.ex. om potentialen hos de nya proteinerna som uttrycks att orsaka de novo-sensibilisering. |
1.5.2 Bedömning av allergiframkallande egenskaper i det genetiskt modifierade livsmedlet eller fodret
När mottagarväxt har kända allergiframkallande egenskaper ska sökanden bedöma potentiella ändringar av de allergiframkallande egenskaperna i det genetiskt modifierad livsmedlet eller fodret genom att jämföra dess allergener med de som finns i den konventionella motsvarigheten. I synnerhet ska potentiella överuttryck av naturliga endogena allergener i den genetiskt modifierade växten utredas.
Sökandens tillvägagångssätt ska fastställas från fall till fall beroende på vilka uppgifter som är tillgängliga om mottagarväxtens allergiframkallande potential. I allmänhet används analysmetoder såsom proteomik i samband med att sera från allergiska människor används som sonder. Sera från kliniskt välbeskrivna allergiska individer som är referensmaterial för studier av IgE-bindningen kan finnas i begränsade antal och kvantiteter. För att minimera användningen av sera från människor kan man få preliminär viktig information om sannolikheten för en oavsiktlig ändring av den genetiskt modifierade växtens totala allergiframkallande egenskaper genom att använda sera från djur som utsatts för experimentell sensibilisering under väldefinierade förhållanden och genom att inkludera relevanta identifierade endogena allergener i den jämförande analysen av sammansättningen.
Dessutom ska sökanden lämna uppgifter om allergiprevalensen hos personer som arbetar med, kommer i kontakt med eller kommer i närheten av odling av genetiskt modifierade växter, om sådana uppgifter finns tillgängliga.
1.5.3 Adjuvansförmåga
Adjuvanser är ämnen som när de administreras tillsammans med en antigen ökar immunsvaret på antigenen och därför även kan öka den allergiska reaktionen. I de fall kända funktionella aspekter av det nya proteinet som uttrycks eller en strukturell likhet med kända starka adjuvanser tyder på en möjlig adjuvansaktivitet, ska sökanden bedöma proteinernas möjliga roll som adjuvanser. Precis som för allergener kan interaktioner med de övriga beståndsdelarna i livsmedelsmatrisen och/eller bearbetning ändra adjuvansens struktur och biotillgänglighet, och alltså ändra dess biologiska aktivitet.
1.5.4 Slutsatser efter bedömningen av de allergiframkallande egenskaperna
I slutsatserna efter bedömningen av de allergiframkallande egenskaperna ska följande anges:
|
a) |
Om de nya proteinerna sannolikt är allergiframkallande. |
|
b) |
Om det genetiskt modifierade livsmedlet eller fodret sannolikt är mer allergiframkallande än dess konventionella motsvarighet. |
Om det genetiskt modifierade livsmedlet eller fodret sannolikt är mer allergiframkallande till följd av den genetiska modifieringen ska det beskrivas mer ingående mot bakgrund av dess förväntade intag (se avsnitt 2). Sökanden ska föreslå lämpliga villkor för utsläppande på marknaden (såsom övervakning efter utsläppandet på marknaden och märkning).
1.6 Näringsbedömning
1.6.1 Näringsbedömningens syfte
Sökanden ska tillhandahålla en näringsbedömning för att visa följande:
|
a) |
Att ett marknadsinförande av det genetiskt modifierade livsmedlet och fodret inte är näringsmässigt ofördelaktigt för människor och djur. Bedömningen ska omfatta den näringsmässiga relevansen av nya proteiner som uttrycks och andra nya beståndsdelar, ändringar av beståndsdelarnas halter i livsmedlet och fodret samt potentiella ändringar av människors eller djurs totala kost. |
|
b) |
Att de genetiska modifieringarnas oavsiktliga effekter, som har identifierats eller kan förutsättas ha inträffat på grundval av de tidigare jämförande molekylära analyserna eller analyserna av sammansättning eller fenotyp i enlighet med avsnitt 1.2 och 1.3, inte har några negativa effekter på det genetiskt modifierade livsmedlets och fodrets näringsvärde. |
För staplade transformationshändelser som kombinerats genom konventionell korsning ska sökanden tillhandahålla en bedömning av de potentiella ändringar av näringsvärdet som kan härröra från genprodukternas sam- eller motverkande effekter, inbegripet ändringar av sammansättningen. Detta kan vara särskilt relevant om det kombinerade uttrycket av de nya generna som införts har oväntade effekter på de biokemiska vägarna.
1.6.2 Faktorer att beakta vid näringsbedömningen av genetiskt modifierade livsmedel och foder
Vid näringsbedömningen av genetiskt modifierade livsmedel och foder ska följande beaktas:
|
a) |
Det genetiskt modifierade livsmedlets och fodrets sammansättning när det gäller halter av näringsämnen och antinutritionella ämnen (se studier av sammansättningen i avsnitt 1.3). |
|
b) |
Biotillgänglighet och biologisk effektivitet för näringsämnen i livsmedlet och fodret med hänsyn till potentiell påverkan av transport, lagring och förväntad hantering av livsmedlet och fodret. |
|
c) |
Det förväntade kostintag av livsmedlet och fodret (se avsnitt 2), och näringsmässiga effekter av detta. |
När man genom den jämförande analysen har identifierat egenskaper i det genetiskt modifierade livsmedlets och fodrets sammansättning som skiljer sig från dess konventionella motsvarighet och/eller inte är ekvivalenta med referenssorternas egenskaper, ska deras näringsmässiga relevans utvärderas på grundval av aktuella vetenskapliga rön. Om man i bedömningen drar slutsatsen att det genetiskt modifierade livsmedlet och fodret är näringsmässigt ekvivalent med den konventionella motsvarigheten ska inga kompletterande studier utföras. Om man på grundval av bedömningen av uppgifterna från den jämförande analysen inte kan dra slutsatser om näringsmässig ekvivalens ska däremot kompletterande näringsstudier utföras. Jämförande tillväxtstudier ska utföras med unga snabbväxande djurarter (t.ex. broilerkyckling som djurmodell för andra djur än idisslare, lamm för idisslare, eller andra snabbväxande arter).
1.6.3 Näringsstudier av genetiskt modifierade livsmedel
Sökanden ska fastställa behovet och utformningen av näringsstudier på grundval av de införda egenskaperna, resultaten av den jämförande analysen och, i förekommande fall, en 90-dagars utfodringsstudie. Kompletterande uppgifter om näringsvärde kan erhållas från jämförande tillväxtstudier som utförs med andra djurarter, t.ex. broilerkycklingar, där man gör en näringsbedömning av genetiskt modifierade foder. Om näringsstudier utförs ska den konventionella motsvarigheten och i förekommande fall kompletterande jämförelsematerial ingå i kontrollkosten. För genetiskt modifierade växter som är herbicidtoleranta bör det material som testas komma från genetiskt modifierade växter som utsatts för den avsedda herbiciden.
Genetiskt modifierade livsmedel som ändrats för att ge konsumenten ytterligare hälsovinster jämfört med konventionella livsmedel kan gagna specifika befolkningsgrupper eller delar av dessa medan samma livsmedel kan utgöra en risk för andra. I fall där en ändrad biotillgänglighet måste fastställas och kan utgöra en risk för delar av vissa befolkningsgrupper ska näringsämnets halt i livsmedlet fastställas, med hänsyn till den kemiska föreningens alla olika former. Metoderna för att testa biotillgängligheten ska väljas från fall till fall beroende på näringsämne eller annan beståndsdel, livsmedel som innehåller dessa beståndsdelar samt hälsa, näringsmässig status och matvanor hos de specifika befolkningsgrupper som förväntas konsumera livsmedlet.
1.6.4 Näringsstudier av genetiskt modifierade foder
Sökanden ska fastställa behovet och utformningen av kompletterande näringsstudier på grundval av de införda egenskaperna, resultaten av den jämförande analysen och, i förekommande fall, en 90-dagars utfodringsstudie. Kompletterande uppgifter om näringsvärde kan erhållas från jämförande tillväxtstudier som utförs med andra djurarter, t.ex. broilerkycklingar, där man gör en näringsbedömning av genetiskt modifierade foder. Om näringsstudier utförs ska den konventionella motsvarigheten och i förekommande fall kompletterande jämförelsematerial ingå i kontrollkosten.
För genetiskt modifierade foder med förbättrade näringsmässiga egenskaper ska utfordringsstudier utföras med livsmedelsproducerande djurarter från målgruppen för att bedöma fodrets effekter. För genetiskt modifierade växter som modifierats för att förbättra näringsinnehållet och näringsämnenas biotillgänglighet ska studier utföras med livsmedelsproducerande djurarter från målgruppen för att fastställa enskilda näringsämnens biotillgänglighet i den genetiskt modifierade växten jämfört med i dess konventionella motsvarighet. I de fall genetiskt modifierade växters egenskaper specifikt har ändrats för att öka djurens produktivitet genom ökad näringstäthet (såsom ökat innehåll av olja) eller en ökad halt av ett visst näringsämne (såsom en essentiell aminosyra eller ett vitamin) ska en lämplig kontrollkost utformas genom att den konventionella motsvarigheten kompletteras med det specifika näringsämnet i samma omfattning som det ändrats i den genetiskt modifierade växten. Biprodukter (såsom oljeväxtmjöl), från vilka den beståndsdel som den genetiska modifieringen berör har extraherats, kan jämföras med biprodukter framställda från den konventionella motsvarigheten.
Utfodringsstudier med djur från målgruppen ska omfatta antingen tillväxtperioden och/eller slutperioden före slakt för kycklingar, grisar och slaktnöt eller en större del av laktationsperioden för mjölkkor eller värpperioden för värphöns eller vaktlar. För foder som endast är avsett för vattenbruk ska tillväxtstudier med vattenlevande arter såsom karp, mal, lax eller typiska växtätare väljas.
I tillämpliga fall ska tester med olika försöksplaner tillhandahållas för att visa att den genetiskt modifierade växt som förbättrats näringsmässigt uppfyller det förväntade näringsvärdet. Hur exakt försöksplanen utformas och vilka statistiska metoder som används för utfodringsstudier på livsmedelsproducerande djur för att testa näringsvärdet i det genetiskt modifierade foder som modifierats för att förbättra dess näringsmässiga egenskaper ska bero på vilken djurart i målgruppen som avses, vilken typ av växtegenskaper som studeras och den förväntade effektens omfattning. Den experimentella kosten ska utformas så att de huvudsakliga uppmätta parametrarna påverkas av en skillnad i det aktuella näringsämnets mängd och/eller tillgänglighet. Hur parametrarna mäts ska variera beroende på vilken djurart i målgruppen som används i studien, men måtten ska inkludera foderintag, kroppsvikt, djurens produktivitet och näringsämnenas biotillgänglighet.
Närmare vägledning för tillämpning av kraven i detta avsnitt finns i rapporten från Efsas GMO-panels arbetsgrupp för djurutfodringsstudier (10).
1.6.5 Slutsatser efter näringsbedömningen
I slutsatserna efter näringsbedömningen av genetiskt modifierade livsmedel och foder ska man ange om det genetiskt modifierade livsmedlet och fodret, med hänsyn till naturliga variationer, är näringsmässigt ekvivalent med dess konventionella motsvarighet.
Sökanden ska utvärdera näringsbedömningens resultat mot bakgrund av förväntat intag av det genetiskt modifierade livsmedlet och fodret (se avsnitt 2).
1.7 Standardiserade riktlinjer för toxicitetstester
För toxicitetstestning ska sökanden använda de internationellt överenskomna riktlinjer och testmetoder som beskrivs i kommissionens förordning (EG) nr 440/2008 av den 30 maj 2008 om testmetoder enligt Europaparlamentets och rådets förordning (EG) nr 1907/2006 om registrering, utvärdering, godkännande och begränsning av kemikalier (Reach) (11) (se tabellerna 1 och 2). En icke uttömmande förteckning över validerade testmetoder som vid behov ska användas för toxikologisk testning av genetiskt modifierade organismer, eventuellt i en anpassad form, finns i tabellerna 1 och 2.
Testmetodernas prestanda beror på typ av genetiskt modifierade livsmedel och foder, typ av genetisk modifiering och därav följande avsiktliga och oavsiktliga förändringar, avsedd användning och exponering/intag samt tillgänglig kunskap. Vissa av testerna har utvecklats för att bedöma arbetsplatsrisker (se avsnitten 1.4 och 1.5).
Tabell 1
Icke uttömmande förteckning över de validerade testmetoder för kemikalier som anges i förordning (EG) nr 440/2008 och som får användas för toxikologisk testning av genetiskt modifierade organismer, eventuellt i en anpassad form
|
Namn |
Metodens referensbeteckning i del B i bilagan till förordning (EG) nr 440/2008 |
|
AKUT TOXICITET (DERMALT) |
B.3 |
|
HUDSENSIBILISERING |
B.6 |
|
28 DAGARS STUDIE AV TOXICITET MED UPPREPAD ORAL DOSERING |
B.7 |
|
TOXICITET VID UPPREPAD DOSERING (28 DAGAR, DERMALT) |
B.9 |
|
TEST AVSEENDE SUBKRONISK ORAL TOXICITET (90-DAGARS UPPREPAT ORALTEST PÅ GNAGARE) |
B.26 |
|
TEST AVSEENDE KRONISK TOXICITET |
B.30 |
|
TEST AVSEENDE CANCERGENICITET |
B.32 |
|
KOMBINERAT TEST AVSEENDE KRONISK TOXICITET OCH CANCERGENICITET |
B.33 |
|
REPRODUKTIONSTOXICITETSTEST PÅ EN GENERATION |
B.34 |
|
TEST AVSEENDE REPRODUKTIONSTOXICITET PÅ TVÅ GENERATIONER |
B.35 |
|
TOXIKOKINETIK |
B.36 |
|
TEST AVSEENDE NEUROTOXICITET PÅ GNAGARE |
B.43 |
Tabell 2
Genotoxicitetstest som anges i förordning (EG) nr 440/2008
|
Namn |
Metodens referensbeteckning i del B i bilagan till förordning (EG) nr 440/2008 |
|
MUTAGENICITET – TEST IN VIVO AV KROMOSOMAVVIKELSER I BENMÄRG HOS DÄGGDJUR |
B.11 |
|
MUTAGENICITET – TEST AV MIKROKÄRNOR IN VIVO I ERYTROCYTER HOS DÄGGDJUR |
B.12 |
|
MUTAGENICITET – TEST AV ÅTERMUTATION HOS BAKTERIER |
B.13/14 |
|
MUTAGENICITETSTEST OCH SCREENING FÖR CANCERGENICITET GENMUTATION – SACCHAROMYCES CEREVISIAE |
B.15 |
|
MITOTISK REKOMBINATION – SACCHAROMYCES CEREVISIAE |
B.16 |
|
DNA-SKADA OCH DNA-REPARATION – FELAKTIG DNA-SYNTES – DÄGGDJURSCELLER IN VITRO |
B.18 |
|
MUTAGENICITET – TEST AV GENMUTATIONER HOS DÄGGDJURSCELLER IN VITRO |
B.17 |
|
TEST AVSEENDE SYSTERKROMATIDSBYTEN IN VITRO |
B.19 |
|
CELLTRANSFORMATIONSTEST PÅ DÄGGDJURSCELLER IN VITRO |
B.21 |
|
SPERMATOGENIAL TEST AV KROMOSOMAVVIKELSER HOS DÄGGDJUR |
B.23 |
2. EXPONERINGSBEDÖMNING – FÖRVÄNTAT INTAG/ANVÄNDNINGENS OMFATTNING
En uppskattning av det förväntade intaget ska vara en väsentlig del i riskbedömningen av genetiskt modifierade livsmedel och foder, och ska även krävas för näringsbedömningen. Sökanden ska lämna uppgifter om avsedd funktion, roll i kosten och förväntad användningsnivå för det genetiskt modifierade livsmedlet och fodret i EU. Sökanden ska också lämna uppgifter om det förväntade koncentrationsintervallet för nya proteiner som framställts eller befintliga växtproteiner som avsiktligt modifierats i de genetiskt modifierade livsmedel och foder som ska släppas ut på marknaden.
På grundval av representativa konsumtionsdata för produkter som framställs från de respektive konventionella växterna ska sökanden uppskatta det förväntade genomsnittliga och maximala intaget av det genetiskt modifierade livsmedlet och fodret. Probabilistiska metoder kan användas för att fastställa intervall med troliga värden snarare än enskilda värden eller punktskattningar. Sökanden ska identifiera och beakta specifika grupper i EU-befolkningen som kan förväntas ha en högre exponering, och ska beakta denna högre exponering inom ramen för riskbedömningen. Alla antaganden som görs i exponeringsbedömningen ska anges. Den senaste metodutvecklingen ska tillämpas och lämpliga konsumtionsdata ska användas. Data om mängden som importeras och framställs kan ge kompletterande uppgifter för intagsbedömningen.
Sökanden ska med lämpliga metoder fastställa koncentrationerna av nya proteiner som uttrycks, andra nya beståndsdelar och endogena beståndsdelar i livsmedel och foder vars halter har ändrats till följd av den genetiska modifieringen (t.ex. till följd av ändrade ämnesomsättningsvägar) i de delar av den genetiskt modifierade växten som är avsedda att användas som livsmedel eller foder. Det förväntade intaget av dessa beståndsdelar ska uppskattas med hänsyn till hur de påverkas av bearbetning, lagring och förväntad hantering av livsmedlet och fodret i fråga, t.ex. potentiell ackumulering eller minskning. I de fall där den genetiska modifieringen har lett till ändrade halter av en naturlig beståndsdel, eller om en ny beståndsdel förekommer naturligt i andra livsmedels- och foderprodukter, ska den förväntade förändringen av beståndsdelens totala intag bedömas med hänsyn till både realistiska och värsta tänkbara intagsscenarier.
Sökanden ska lämna uppgifter om människors/djurs kända eller förväntade intag av liknande genetiskt modifierade livsmedel och foder samt om andra exponeringsvägar för de nya respektive naturliga beståndsdelarna, inklusive mängd, frekvens och andra faktorer som påverkar exponeringen.
3. RISKKARAKTERISERING
3.1 Inledning
Sökanden ska basera sin riskkarakterisering av genetiskt modifierade växter, livsmedel och foder på uppgifter från en faroidentifiering och en farokarakterisering samt på uppgifter om exponering/intag. Sökanden ska se till att riskkarakteriseringen är omfattande genom att hänsyn tas till alla tillgängliga underlag från olika analyser, bl.a. molekylär analys, analyser av fenotyp, odlingsanalys och analys av sammansättning samt testning av toxicitet och allergiframkallande egenskaper. Om det i riskkarakteriseringen framkommer tecken på att det kan behövas särskild övervakning av det genetiskt modifierade livsmedlet och fodret efter utsläppandet på marknaden ska sökanden beakta dessa.
Under riskkarakteriseringen ska sökanden påvisa att faroidentifieringen och farokarakteriseringen är fullständiga. Sökanden ska diskutera kvaliteten på befintliga data och uppgifter. I diskussionen ska sökanden tydligt ange hur denna informationsmängd har beaktats vid fastställandet av den slutliga riskkarakteriseringen.
Sökanden ska tillhandahålla uppskattningar av de osäkerhetsfaktorer som är förbundna med varje test och med riskbedömningens olika stadier. Sökanden ska i så stor utsträckning som möjligt kvantifiera dem. Åtskillnad ska göras mellan osäkerhetsfaktorer som avspeglar naturliga variationer i biologiska parametrar (inbegripet variationer som avser populationers känslighet) och variationer mellan olika arters respons.
Beroende på vilken fråga som utreds och vilka data som finns tillgängliga ska sökanden göra en kvalitativ och, där så är möjligt, en kvantitativ riskkarakterisering. Omständigheterna kring den bedömda risken och därmed förenade osäkerhetsfaktorer ska vara så exakta som möjligt.
3.2 Frågor som ska beaktas vid riskkarakterisering
I tillämpliga fall och beroende på typ av genetisk modifiering ska sökanden utföra en riskbedömning av de genetiskt modifierade växterna på ett integrerat sätt i enlighet med avsnitt 3.1. Riskbedömningen ska utföras utifrån det enskilda fallet beroende på den modifierade växten och typen av genetisk modifiering, odlingsmetoderna för den genetiskt modifierade växten och användningsområdena för det genetiskt modifierade livsmedlet och fodret. Sökanden ska ta hänsyn till de olika frågor som beaktats under faroidentifierings-, farokarakteriserings- och exponeringsmomenten. Sökanden ska göra en samlad bedömning av slutsatserna i dessa frågor under riskkarakteriseringsmomentet. Förteckningen över frågor som finns i detta avsnitt är inte uttömmande.
3.2.1 Molekylär karakterisering
En utvärdering av givar- och mottagarväxtens egenskaper och tidigare användning är viktigt för att identifiera behovet av särskilda analyser, såsom förekomsten av specifika toxiner eller allergener i den omodifierade mottagarväxten som oavsiktligt kan komma att ökas till följd av den genetiska modifieringen.
Transformationsprotokoll, strategier för molekylär karakterisering samt de använda metodernas specificitet och känslighet ska diskuteras av sökanden i förhållande till gensekvensernas avsiktliga och eventuellt oavsiktliga införande och uttryck.
Om man genom en sekvensanalys har identifierat en potentiell fara ska sökanden visa hur metoder såsom bioinformatikanalys, analys av sammansättning/odlingsanalys och möjligen djurutfodringsstudier med hela det genetiskt modifierade livsmedlet och fodret bidrar till säkerhetsbedömningen. De erhållna resultatens värde ska utvärderas mot bakgrund av tillgänglig kunskap om hur genomdatabaserna för den aktuella grödan eller besläktade arter är strukturerade och fungerar.
För genetiskt modifierade växter som innehåller staplade transformationshändelser ska de ytterligare risker som kan uppstå på grund av de kombinerade effekterna av de staplade generna utvärderas.
3.2.2 Jämförande analys
Det första syftet med den jämförande analysen är att identifiera eventuella skillnader mellan den genetiskt modifierade växten och dess konventionella motsvarighet och, i förekommande fall, det kompletterande jämförelsematerialet. Det andra syftet med den jämförande analysen är att identifiera eventuell bristande ekvivalens mellan den genetiskt modifierade växten och dess referenssorter. Dessa skillnader och/eller denna bristande ekvivalens bör bedömas med hänsyn till deras eventuella effekt på livsmedlets och fodrets säkerhet och näringsegenskaper, med beaktande av den naturliga variationen. Den uppskattade risken och de osäkerhetsfaktorer som är förbundna med denna bör vara så exakta som möjligt och bör beaktas.
Sökanden ska visa att den jämförande analysen av odlingsegenskaper och morfologiska egenskaper samt sammansättning i den genetiskt modifierade växten respektive dess konventionella motsvarighet har utförts i enlighet med kraven i denna förordning. Valet av konventionell motsvarighet och, i förekommande fall, kompletterande jämförelsematerial ska motiveras.
3.2.3 Livsmedels- och fodersäkerhet vid intag
Sökanden ska utvärdera de uppgifter som genereras för att uppskatta möjliga kort- och långsiktiga risker för människors och djurs hälsa som är kopplade till konsumtion av genetiskt modifierade livsmedel eller foder, med hänsyn till nya proteiners/metaboliters uttryck och avsevärt ändrade halter av de ursprungliga växtproteinerna/växtmetaboliterna i genetiskt modifierade livsmedel/foder. I utvärderingen ska det ingå en noggrann analys av relevans och begränsningar för varje test och för alla uppgifter.
Sökanden ska beakta de olika halter som observerats för kemiska föreningar som man vet förekommer i den konventionella motsvarigheten och referenssorterna. Denna variation kan orsakas av skillnader som är beroende av genotyp eller miljö, eller orsakas av genotyp x:s interaktioner med miljön. Dessutom kan man ta hänsyn till de olika halter som observeras i ett brett spektrum av livsmedel och foder som är representativa för människors och djurs kost, under förutsättning att detta avspeglar konsumenternas exponeringsnivåer för den specifika kemiska föreningen.
Om enskilda beståndsdelar och/eller hela genetiskt modifierade livsmedel och foder visar sig ge negativa effekter i specifika studier ska sökanden lämna in uppgifter om samband mellan dos och respons, gränsvärden, fördröjd debut för de negativa effekterna, risker för vissa befolkningsgrupper samt användning av osäkerhetsfaktorer när uppgifter som avser djur extrapoleras till människor.
Sökanden ska beakta uppgifter om egenskaperna hos de nya kemiska föreningar som förekommer i den genetiskt modifierade växten, inbegripet potentiella biologiska effekter på människor och djur. Om de kemiska föreningarna har kända negativa hälsoeffekter och det i specifik lagstiftning har fastställts maxhalter för dessa kemiska föreningars förekomst i växten eller dess produkter, ska hänsyn tas till dessa maxhalter. Annars ska referensvärden för acceptabla eller tolerabla intagsnivåer, såsom acceptabelt dagligt intag (ADI) eller tolerabel övre intagsnivå (UL), beaktas i förhållande till det förväntade intaget. Om det finns tidigare säker användning av den kemiska föreningen i livsmedel ska konsumenternas intagsnivåer vid en traditionell kost anses vara säkra.
Sökanden ska utvärdera uppgifterna om bearbetningens effekter på de nya kemiska föreningarna. Potentiell ackumulering/minskning i livsmedels- och foderprodukter som ingår i människors eller djurs kost ska beaktas. Sökanden ska också utvärdera relevansen av de skillnader som uppstår till följd av kända kemiska reaktioner vid de förhållanden som råder vid bearbetningen.
I de fall där mer komplexa genetiska modifieringar framställs, t.ex. genom överföring av flera gener i en konstruktion, retransformation av befintliga genetiskt modifierade linjer eller stapling av transformationshändelser genom traditionell förädling av genetiskt modifierade moderväxter, ska sökanden diskutera strategier för bedömning av de eventuella risker som är förenade med möjliga interaktioner mellan de nya proteinerna som uttrycks, de nya metaboliterna och de ursprungliga växtbeståndsdelarna. I bedömningen ska alla tillgängliga uppgifter beaktas, inklusive verkningssättet hos de nya proteinerna som uttrycks, den genetiskt modifierade växtens molekylära egenskaper och odlingsegenskaper eller sammansättning samt resultaten från toxicitetsstudier på djur och djurutfodringsstudier.
Sökanden ska utvärdera de uppgifter som erhållits för att bedöma den allergiframkallande potentialen hos de nya proteiner som uttrycks i genetiskt modifierade växter, med hänsyn till att nya allergiframkallande proteiner införs i livsmedels- och foderväxter och att allergiska reaktioner eventuellt kan provoceras fram hos känsliga individer, samt de uppgifter som visar att den genetiska modifieringen inte leder till oavsiktliga ändringar av de endogena allergiframkallande proteinernas egenskaper hos och/eller uttrycksnivåer i genetiskt modifierade livsmedel. Valet av testmodeller ska i synnerhet motiveras med hänsyn till specificitet, förutsägbarhet och valideringsstatus.
När det gäller uppskattat intag av genetiskt modifierade livsmedel ska sökanden utvärdera de använda metoderna vad gäller den osäkerhet som kan kopplas till uppskattningen av långsiktigt intag. Särskild uppmärksamhet ska ägnas åt de genetiskt modifierade växter som syftar till att ändra livsmedlets och fodrets näringsmässiga egenskaper. För sådana genetiskt modifierade produkter ska kravet på övervakning efter utsläppandet på marknaden diskuteras som en mekanism för att fastställa hur det totala kostintaget av det genetiskt modifierade livsmedlet faktiskt har ändrats, i vilken omfattning detta har inträffat och huruvida produkten orsakar kända (bi-)effekter eller oväntade bieffekter. Om det bedöms vara nödvändigt med övervakning efter utsläppandet på marknaden ska de föreslagna metodernas tillförlitlighet, känslighet och specificitet anges.
3.3 Riskkarakteriseringens resultat
I enlighet med kraven i artiklarna 4 och 16 i förordning (EG) nr 1829/2003 ska sökanden se till att den slutliga riskkarakteriseringen tydligt visar följande:
|
a) |
Att det genetiskt modifierade livsmedlet och fodret inte har några negativa effekter på människors och djurs hälsa. |
|
b) |
Att det genetiskt modifierade livsmedlet inte skiljer sig från det livsmedel det är avsett att ersätta i sådan utsträckning att en normal konsumtion av det skulle vara näringsmässigt ofördelaktig för konsumenten. |
|
c) |
Att det genetiskt modifierade livsmedlet inte vilseleder konsumenten. |
|
d) |
Att det genetiskt modifierade fodret inte skadar eller vilseleder konsumenten genom att försämra animalieprodukternas utmärkande egenskaper. |
|
e) |
Att det genetiskt modifierade fodret inte skiljer sig från det foder det är avsett att ersätta i sådan utsträckning att en normal konsumtion av det skulle vara näringsmässigt ofördelaktig för djur eller människor. |
Sökanden ska tydligt ange vilka antaganden som har gjorts under riskbedömningen för att förutsäga sannolikheten för negativa effekter i en viss population och för hur kraftiga dessa effekter är samt typ av och storlek på den osäkerhet som förknippas med fastställandet av dessa risker.
Sökanden ska också inkludera detaljerade uppgifter som motiverar varför eller varför inte ansökan innehåller ett förslag till märkning i enlighet med artiklarna 13.2 a, 13.3, 25.2 c och 25.3 i förordning (EG) nr 1829/2003.
(1) EGT L 43, 14.2.1997, s. 1.
(2) The EFSA Journal, vol. 9(2011):5, artikelnr 2149.
(3) The EFSA Journal, vol. 8(2010):1, artikelnr 1250.
(4) The EFSA Journal, vol. 8(2010):1, artikelnr 1250.
(5) http://www.oecd.org/document/15/0,3746,en_2649_34385_46726799_1_1_1_1,00.html
(6) http://www.efsa.europa.eu/en/efsajournal/pub/2760.htm
(7) EUT L 133, 22.5.2008, s. 1.
(8) ”Guidance on conducting repeated-dose 90-day oral toxicity study in rodents on whole food/feed”, The EFSA Journal, vol. 9(2011):12, artikelnr 2438.
(9) The EFSA Journal, vol. 8(2010):7, artikelnr 1700.
(10) Report of the EFSA GMO Panel Working Group on Animal Feeding Trials, ”Safety and nutritional assessment of GM plants and derived food and feed: The role of animal feeding trials”, Food Chem. Toxicol., vol. 46, 2008, s. S2–S70.