
1991R2568 — SL — 01.01.2014 — 025.001
Ta dokument je mišljen zgolj kot dokumentacijsko orodje in institucije za njegovo vsebino ne prevzemajo nobene odgovornosti
|
UREDBA KOMISIJE (EGS) št. 2568/91 z dne 11. julija 1991 o značilnostih oljčnega olja in olja iz oljčnih tropin ter o ustreznih analiznih metodah (UL L 248 5.9.1991, str. 1) |
spremenjena z:
|
|
|
Uradni list |
||
|
No |
page |
date |
||
|
L 349 |
36 |
18.12.1991 |
||
|
L 150 |
17 |
2.6.1992 |
||
|
L 176 |
27 |
30.6.1992 |
||
|
L 199 |
18 |
18.7.1992 |
||
|
L 327 |
28 |
13.11.1992 |
||
|
L 22 |
58 |
30.1.1993 |
||
|
L 66 |
29 |
18.3.1993 |
||
|
L 24 |
33 |
29.1.1994 |
||
|
COMMISSION REGULATION (EC) No 2632/94 of 28 October 1994 (*) |
L 280 |
43 |
29.10.1994 |
|
|
L 69 |
1 |
29.3.1995 |
||
|
COMMISSION REGULATION (EC) No 2527/95 of 27 October 1995 (*) |
L 258 |
49 |
28.10.1995 |
|
|
COMMISSION REGULATION (EC) No 2472/97 of 11 December 1997 (*) |
L 341 |
25 |
12.12.1997 |
|
|
L 28 |
5 |
4.2.1998 |
||
|
COMMISSION REGULATION (EC) No 2248/98 of 19 October 1998 (*) |
L 282 |
55 |
20.10.1998 |
|
|
L 46 |
15 |
20.2.1999 |
||
|
L 65 |
9 |
7.3.2001 |
||
|
COMMISSION REGULATION (EC) No 2042/2001 of 18 October 2001 (*) |
L 276 |
8 |
19.10.2001 |
|
|
L 128 |
8 |
15.5.2002 |
||
|
L 295 |
57 |
13.11.2003 |
||
|
L 161 |
11 |
22.6.2007 |
||
|
L 178 |
11 |
5.7.2008 |
||
|
L 23 |
1 |
27.1.2011 |
||
|
IZVEDBENA UREDBA KOMISIJE (EU) št. 661/2012 z dne 19. julija 2012 |
L 192 |
3 |
20.7.2012 |
|
|
IZVEDBENA UREDBA KOMISIJE (EU) št. 299/2013 z dne 26. marca 2013 |
L 90 |
52 |
28.3.2013 |
|
popravljena z:
|
(*) |
Ta akt ni bil nikoli objavljen v slovenščini. |
UREDBA KOMISIJE (EGS) št. 2568/91
z dne 11. julija 1991
o značilnostih oljčnega olja in olja iz oljčnih tropin ter o ustreznih analiznih metodah
KOMISIJA EVROPSKIH SKUPNOSTI JE
ob upoštevanju Pogodbe o ustanovitvi Evropske gospodarske skupnosti,
ob upoštevanju Uredbe Sveta št. 136/66/EGS z dne 22. septembra 1966 o vzpostavitvi skupne ureditve trga za olja in masti ( 1 ), nazadnje spremenjene z Uredbo (EGS) št. 3577/90 ( 2 ), in zlasti člena 35a Uredbe,
ker Priloga k Uredbi št. 136/66/EGS vsebuje opise in opredelitve pojmov za oljčno olje in olje iz oljčnih tropin, ki je v prometu znotraj vsake države članice, v trgovini v Skupnosti in v trgovini s tretjimi državami;
ker naj bi se z namenom razločevanja med različnimi vrstami olja, opredelile fizikalne in kemijske lastnosti vsake od njih ter senzorične značilnosti deviškega olja, zato da se zagotovita pristnost in kakovost zadevnih proizvodov, ne da bi se posegalo v druge obstoječe določbe;
ker naj bi bile značilnosti različnih vrst olj enotno opredeljene po vsej Skupnosti; ker naj bi bile s tem namenom vzpostavljene metode kemijske analize in senzorične ocene Skupnosti; ker naj bi bila v prehodnem obdobju dovoljena uporaba drugih analiznih metod, ki se uporabljajo v državah članicah pod pogojem, da če se rezultati razlikujejo, imajo odločilen vpliv rezultati, dobljeni z uporabo navedenih metod Skupnosti;
ker opredelitev fizikalnih in kemijskih značilnosti oljčnega olja in analiznih metod povzročijo spremembo dodatnih opomb k Poglavju 15 kombinirane nomenklature;
ker metoda za ocenjevanje senzoričnih značilnosti deviškega olja vključuje ustanovitev ocenjevalnih skupin; izbranih in usposobljenih pokuševalcev; ker naj bi se zato določilo obdobje, potrebno za vzpostavitev takšne strukture; ker naj bi bilo glede na težave, s katerimi se bodo soočale nekatere države članice pri ustanavljanju skupin pokuševalcev, odobri se uporaba skupin v drugih državah članicah;
ker naj se za zagotovitev pravilnega delovanja sistema prelevmanov pri uvozu oljčnih tropin, določi enotna metoda za določanje vsebnosti olja v teh proizvodih;
da se ne bi škodilo trgovanju, je treba olje, ki je bilo predpakirano pred začetkom veljavnosti te uredbe, prodati v omejenem roku;
ker je treba razveljaviti Uredbo Komisije (EGS) št. 1058/77 ( 3 ), nazadnje spremenjeno z Uredbo (EGS) št. 1858/88 ( 4 );
ker Upravljalni odbor za olja in masti ni podal svojega mnenja v roku, ki ga je določil njegov predsedujoči,
SPREJELA NASLEDNJO UREDBO:
Člen 1
1. Olja, katerih značilnosti so skladne z značilnostmi iz točk 1 in 2 Priloge I k tej uredbi, se štejejo za deviška oljčna olja v smislu točke 1(a) in (b) Priloge k Uredbi št. 136/66/EGS.
2. Olje, katerega značilnosti so skladne z značilnostmi iz točke 3 Priloge I k tej uredbi, se šteje za lampante oljčno olje v smislu točke 1(c) Priloge k Uredbi št. 136/66/EGS.
3. Olje, katerega značilnosti so skladne z značilnostmi iz točke 4 Priloge I k tej uredbi, se šteje za rafinirano oljčno olje v smislu točke 2 Priloge k Uredbi št. 136/66/EGS.
4. Olje, katerega značilnosti so skladne z značilnostmi iz točke 5 Priloge I k tej uredbi, se šteje za oljčno olje, sestavljeno iz rafiniranih oljčnih olj in deviških oljčnih olj v smislu točke 3 Priloge k Uredbi št. 136/66/EGS.
5. Olje, katerega značilnosti so skladne z značilnostmi iz točke 6 Priloge I k tej uredbi, se šteje za surovo olje iz oljčnih tropin v smislu točke 4 Priloge k Uredbi št. 136/66/EGS.
6. Olje, katerega značilnosti so skladne z značilnostmi iz točke 7 Priloge I k tej uredbi, se šteje za rafinirano olje iz oljčnih tropin v smislu točke 5 Priloge k Uredbi št. 136/66/EGS.
7. Olje, katerega značilnosti so skladne z značilnostmi iz točke 8 Priloge I k tej uredbi, se šteje za olje iz oljčnih tropin v smislu točke 6 Priloge k Uredbi št. 136/66/EGS.
Člen 2
1. Značilnosti olj, določenih v Prilogi I, se določijo v skladu z naslednjimi analiznimi metodami:
— za določanje prostih maščobnih kislin, izraženih v masnem odstotnem deležu oleinske kisline, metoda, določena v Prilogi II,
— za določanje peroksidnega števila, metoda, določena v Prilogi III,
— for determination of the wax content, the method given in Annex IV,
— za določanje vsebnosti sterolov, metoda, določena v Prilogi V,
— za določanje eritrodiola in uvaola, metoda, določena v Prilogi VI,
— pri določanju odstotnega deleža 2-gliceril monopalmitata, metoda, določena v Prilogi VII,
▼M20 —————
— za spektrofotometrično analizo, metoda, določena v Prilogi IX,
— za določanje sestave maščobnih kislin, metoda, določena v Prilogi X A in X B,
— za določanje hlapnih halogeniranih topil, metoda, določena v Prilogi XI,
— za določanje senzoričnih značilnosti deviškega oljčnega olja, metoda, določena v Prilogi XII,
▼M20 —————
— za določanje stigmastadienov metoda iz Priloge XVII,
— for determining the content of triglycerides with ECN42, the method set out in Annex XVIII,
— for determination of the aliphatic alcohol content, the method given in Annex XIX,
— za določanje vsebnosti voskov, metilnih estrov maščobnih kislin in etilnih estrov maščobnih kislin s kapilarno plinsko kromatografijo, metoda, določena v Prilogi XX.
2. Verification by national authorities or their representatives of the organoleptic characteristics of virgin oils shall be effected by tasting panels approved by the Member States.
The organoleptic characteristics of an oil as referred to in the first subparagraph shall be deemed consonant with the category declared if a panel approved by the Member State confirms the grading.
Should the panel not confirm the category declared as regards the organoleptic characteristics, at the interested party's request the national authorities or their representatives shall have two counter-assessments carried out by other approved panels, at least one by a panel approved by the producer Member State concerned. The characteristics concerned shall be deemed consonant with the characteristics declared if at least two of the counter-assessments confirm the declared grade. If that is not the case, the interested party shall be responsible for the cost of the counter-assessments.
3. When the national authorities or their representatives verify the characteristics of the oil as provided for in paragraph 1, samples shall be taken in accordance with international standards EN ISO 661 on the preparation of test samples and EN ISO 5555 on sampling. However, notwithstanding point 6.8 of standard EN ISO 5555, in the case of batches of such oils in immediate packaging not exceeding 100 litres, the sample shall be taken in accordance with Annex Ia to this Regulation.
Without prejudice to standard EN ISO 5555 and Chapter 6 of standard EN ISO 661, the samples taken shall be put in a dark place away from strong heat as quickly as possible and sent to the laboratory for analysis no later than:
— the tenth working day after they are taken, during the period from October to May, and
— the fifth working day after they are taken, during the period from June to September.
4. ►M20 Za namene preverjanja iz odstavka 3 se analize, navedene v prilogah II, III, IX, X in XII, in, kjer je primerno, vse nasprotne analize, ki jih zahteva nacionalna zakonodaja, izvedejo pred iztekom minimalnega roka trajanja. Če se vzorčenje izvede več kot štiri mesece pred iztekom minimalnega roka trajanja, se analize izvedejo najpozneje štiri mesece po mesecu odvzema vzorca. Za druge analize iz zadevne uredbe ne veljajo nobeni roki. ◄
Unless the sample was taken less than one month before the minimum durability date, if the results of the analyses do not match the characteristics of the category of olive oil or olive-residue oil declared, the party concerned shall be notified no later than one month before the end of the period laid down in the first subparagraph.
5. For the purpose of determining the characteristics of olive oils by the methods provided for in paragraph 1, the analysis results shall be directly compared with the limits laid down in this Regulation.
Člen 2a
1. Za namene tega člena „oljčno olje, dano na trg“ pomeni skupno količino oljčnega olja in olja iz oljčnih tropin posamezne države članice, ki se porabi v navedeni državi članici ali se iz nje izvozi.
2. Države članice zagotovijo, da se na podlagi analize tveganja selektivno in dovolj pogosto opravijo preverjanja skladnosti, in tako zagotovijo, da je oljčno olje, dano na trg, skladno z deklarirano kategorijo.
3. Merila za ocenjevanje tveganja lahko vključujejo:
(a) kategorijo olja, čas proizvodnje, ceno olja v primerjavi z drugimi rastlinskimi olji, mešanje in pakiranje, skladiščne prostore in razmere, državo porekla, namembno državo, prevozno sredstvo ali količino serije;
(b) položaj izvajalcev v tržni verigi, količino in/ali vrednost, ki jo tržijo, razpon kategorij olja, ki jih tržijo, vrsto dejavnosti, ki se izvajajo, kot so mletje, skladiščenje, rafiniranje, mešanje, pakiranje ali prodaja na drobno;
(c) ugotovitve iz prejšnjih pregledov, vključno s številom in vrsto ugotovljenih pomanjkljivosti, običajno kakovost olja, danega na trg, učinkovitost uporabljene tehnične opreme;
(d) zanesljivost sistemov izvajalcev za zagotavljanje kakovosti ali samonadzor v zvezi s skladnostjo s tržnimi standardi;
(e) mesto, kjer se opravi preverjanje, zlasti, če je to mesto prvega vstopa v Unijo, mesto zadnjega izstopa iz Unije, ali mesto, kjer se olje proizvaja, pakira, natovarja ali prodaja končnemu potrošniku;
(f) kakršne koli informacije, ki bi lahko nakazovale tveganje za neskladnost.
4. Države članice vnaprej določijo:
(a) merila za ocenjevanje tveganja za neskladnost serij;
(b) na podlagi analize tveganja za vsako kategorijo tveganja najmanjše število izvajalcev ali serij in/ali količin, pri katerih se preverja skladnost.
Vsako leto je treba izvesti vsaj eno preverjanje skladnosti na tisoč ton oljčnega olja, danega na trg v državi članici.
5. Države članice preverjajo skladnost:
(a) z izvedbo analiz, po katerem koli vrstnem redu, iz Priloge I, ali
(b) z upoštevanjem vrstnega reda iz Priloge Ib o drevesu odločanja, dokler se ne sprejme ena od odločitev iz drevesa odločanja.
▼M19 —————
Člen 3
Kadar se ugotovi, da olje ne ustreza opisu svoje kategorije, mora zadevna država članica, brez poseganja v kakršne koli druge kazni, uporabiti učinkovite, sorazmerne in odvračilne kazni, ki se določijo glede na resnost odkrite nepravilnosti.
Če se pri pregledih odkrijejo večje nepravilnosti, države članice opravljajo pogostejše preglede v zvezi s fazo trženja, kategorijo olja, poreklom ali drugimi merili.
Člen 4
1. The Member States may approve assessment panels so that national authorities or their representatives can assess and verify organoleptic characteristics.
The terms of approval shall be set by Member States and ensure that:
— the requirements of Annex XII.4 are met,
— the panel head is given training recognised for this purpose by the Member State,
— continued approval depends on performance in annual checks arranged by the Member State.
Member States shall notify to the Commission a list of approved panels and the action taken under this paragraph.
2. Če države članice naletijo na težave pri ustanavljanju skupin ocenjevalcev na svojem ozemlju, se lahko obrnejo na skupino ocenjevalcev, potrjeno v drugi državi članici.
3. Vsaka država članica sestavi seznam skupin ocenjevalcev, ki so jih ustanovile strokovne ali medpanožne organizacije v skladu s pogoji, določenimi v odstavku 1, in zagotovi, da so ti pogoji izpolnjeni.
▼M19 —————
Člen 6
1. Vsebnost olja v oljni pogači in drugi ostanki, ki nastanejo pri pridobivanju oljčnega olja (oznaki KN 2306 90 11 in 2306 90 19 ) se določijo z uporabo metode, predpisane v Prilogi XV.
2. Vsebnost olja, navedenega v odstavku 1, je izražena v masnem odstotnem deležu olja na maso suhe snovi.
Člen 7
Uporabljajo se določbe Skupnosti o prisotnosti onesnaževalcev.
Kar zadeva halogenirana topila so mejne vrednosti za vse kategorije oljčnih olj naslednje:
— največji delež vsakega zaznanega halogeniranega topila: 0,1 mg/kg,
— največji skupni delež zaznanih halogeniranih topil: 0,2 mg/kg.
Člen 7a
Fizične ali pravne osebe in skupine oseb, ki imajo v lasti oljčno olje in olje iz oljčnih tropin od faze pridobivanja olja v oljarni do ustekleničenja, v kakršne koli poslovne ali komercialne namene, morajo za vsako kategorijo teh olj voditi evidence dobav in izdanih količin.
Države članice zagotovijo, da je obveznost iz prvega odstavka ustrezno izpolnjena.
Člen 8
1. Države članice morajo obvestiti Komisijo o ukrepih, sprejetih za izvajanje te uredbe. Komisijo obvestijo o kakršnih koli naknadnih spremembah.
2. Države članice morajo na začetku vsakega polletja poslati Komisiji poročilo o analiznih podatkih v zvezi s preskusi, opravljenimi v teku predhodne polovice leta.
Upravljalni odbor za olja in masti mora proučiti rezultate v skladu s postopkom, predpisanim v členu 39 Uredbe št. 136/66/EGS.
3. Obvestila iz te uredbe se predložijo v skladu z Uredbo Komisije (ES) št. 792/2009 ( 5 ).
Člen 9
Uredba (EGS) št. 1058/77 se s tem razveljavi.
Člen 10
1. Ta uredba začne veljati tretji dan po objavi v Uradnem listu Evropskih skupnosti.
Vendar se metoda, predpisana v Prilogi XII, uporablja od ►M1 1. november 1992 ◄ , razen če ne zadeva postopka sistema intervencij.
Ta metoda se ne uporablja za deviško olje, ki je bilo pripravljeno za trg pred 1. novembrom 1992.
2. Ta uredba se ne uporablja za oljčno olje in oljčne tropine, predpakirane pred začetkom veljavnosti te uredbe in so v prometu do 31. oktobra 1992.
Ta uredba je v celoti zavezujoča in se neposredno uporablja v vseh državah članicah.
PRILOGE
Vsebina
|
Priloga I: |
Značilnosti oljčnega olja |
|
Priloga IA: |
Vzorčenje oljčnega olja ali olja iz oljčnih tropin, dostavljenega v izvirnem pakiranju, ki ne presega 100 litrov |
|
Priloga IB: |
Drevo odločanja |
|
Priloga II: |
►M21 Določevanje prostih maščobnih kislin, metoda v hladnem ◄ |
|
Priloga III: |
Določanje peroksidnega števila |
|
Priloga IV: |
►M6 Določanje vsebnosti voskov z uporabo kapilarne plinsko-tekočinske kromatografije ◄ |
|
Priloga V: |
Določanje sestave in vsebnosti sterolov s kapilarno plinsko kromatografijo |
|
Priloga VI: |
Določanje eritrodiola in uvaola |
|
Priloga VII: |
►M21 Določevanje odstotnega deleža 2-gliceril monopalmitata ◄ |
|
▼M20 ————— |
|
|
Priloga IX: |
Spektrofotometrično merjenje na UV-območju |
|
Priloga XA: |
Plinsko kromatografska analiza metilnih estrov maščobnih kislin: |
|
Annex XB: |
Preparation of the fatty acid methyl esters from olive oil and olive-pomace oil |
|
Priloga XI: |
Določanje hlapnih halogeniranih topil v oljčnem olju |
|
Annex XII: |
Organoleptic assessment of virgin olive oils |
|
▼M20 ————— |
|
|
▼M19 ————— |
|
|
Priloga XV: |
Vsebnost olja v oljčnih tropinah |
|
Priloga XVI: |
Določanje jodnega števila |
|
Priloga XVII: |
Določanje stigmastadienov v rastlinskih oljih |
|
Priloga XVIII: |
Določanje razlike med dejansko in teoretično vsebnostjo triacilglicerolov z ekvivalentnim ogljikovim številom (ECN) 42 |
|
Annex XIX: |
Method for determining aliphatic alcohol content |
|
Priloga XX: |
Metoda za določanje vsebnosti voskov, metilnih estrov maščobnih kislin in etilnih estrov maščobnih kislin s kapilarno plinsko kromatografijo |
|
Priloga XXI: |
Rezultati preverjanj skladnosti oljčnega olja iz člena 8(2) |
PRILOGA I
ZNAČILNOSTI OLJČNEGA OLJA
|
Kategorija |
Metilni estri maščobnih kislin (FAME) in etilni estri maščobnih kislin (FAEE) |
Vsebnost kislin (%) (*) |
►C1 Peroksidno število mekv O2/kg (*) ◄ |
Voski mg/kg (**) |
2 gliceril monopalmitat (%) |
Stigmastadien mg/kg (1) |
Razlika: ECN42 (HPLC) in ECN42 (teoretičen izračun) |
K232 (*) |
K270 (*) |
Delta-K (*) |
Organoleptično ocenjevanje Mediana napake (Md) (*) |
Organoleptično ocenjevanje Mediana sadežnosti (Mf) |
|
1. Ekstra deviško oljčno olje |
Σ FAME + FAEE ≤ 75 mg/kg ali 75 mg/kg < Σ FAME + FAEE ≤ 150 mg/kg in (FAEE/FAME) ≤ 1,5 |
≤ 0,8 |
≤ 20 |
≤ 250 |
≤ 0,9 če je skupni % palmitinske kisline ≤ 14 % |
≤ 0,10 |
≤ 0,2 |
≤ 2,50 |
≤ 0,22 |
≤ 0,01 |
Md = 0 |
Mf > 0 |
|
≤ 1,0 če je skupni % palmitinske kisline > 14 % |
||||||||||||
|
2. Deviško oljčno olje |
— |
≤ 2,0 |
≤ 20 |
≤ 250 |
≤ 0,9 če je skupni % palmitinske kisline ≤ 14 % |
≤ 0,10 |
≤ 0,2 |
≤ 2,60 |
≤ 0,25 |
≤ 0,01 |
Md ≤ 3,5 |
Mf > 0 |
|
≤ 1,0 če je skupni % palmitinske kisline > 14 % |
||||||||||||
|
3. Lampante oljčno olje |
— |
> 2,0 |
— |
≤ 300 (3) |
≤ 0,9 če je skupni % palmitinske kisline ≤ 14 % |
≤ 0,50 |
≤ 0,3 |
— |
— |
— |
Md > 3,5 (2) |
— |
|
≤ 1,1 če je skupni % palmitinske kisline > 14 % |
||||||||||||
|
4. Rafinirano oljčno olje |
— |
≤ 0,3 |
≤ 5 |
≤ 350 |
≤ 0,9 če je skupni % palmitinske kisline ≤ 14 % |
— |
≤ 0,3 |
— |
≤ 1,10 |
≤ 0,16 |
— |
— |
|
≤ 1,1 če je skupni % palmitinske kisline > 14 % |
||||||||||||
|
5. Oljčno olje - mešanica rafiniranega oljčnega olja in deviškega oljčnega olja |
— |
≤ 1,0 |
≤ 15 |
≤ 350 |
≤ 0,9 če je skupni % palmitinske kisline ≤ 14 % |
— |
≤ 0,3 |
— |
≤ 0,90 |
≤ 0,15 |
— |
— |
|
≤ 1,0 če je skupni % palmitinske kisline > 14 % |
||||||||||||
|
6. Surovo olje iz oljčnih tropin |
— |
— |
— |
> 350 (4) |
≤ 1,4 |
— |
≤ 0,6 |
— |
— |
— |
— |
— |
|
7. Rafinirano olje iz oljčnih tropin |
— |
≤ 0,3 |
≤ 5 |
> 350 |
≤ 1,4 |
— |
≤ 0,5 |
— |
≤ 2,00 |
≤ 0,20 |
— |
— |
|
8. Olje iz oljčnih tropin |
— |
≤ 1,0 |
≤ 15 |
> 350 |
≤ 1,2 |
— |
≤ 0,5 |
— |
≤ 1,70 |
≤ 0,18 |
— |
— |
|
Kategorija |
Vsebnost kislin (1) |
Transoleinski izomerji skupaj (%) |
Translinolni + translinolenski izomerji skupaj (%) |
Sestava sterolov |
Steroli skupaj (mg/kg) |
Eritrodiol in uvaol (%) (**) |
||||||||||
|
Miristinska (%) |
Linolenska (%) |
Arašidova (%) |
Eikozanojska (%) |
Behenska (%) |
Lignocerinska (%) |
Holesterol (%) |
Brassikasterol (%) |
Campesterol (%) |
Stigmasterol (%) |
Betasitosterol (%) (2) |
Delta-7- stigmastenol (%) |
|||||
|
1. Ekstra deviško oljčno olje |
≤ 0,05 |
≤ 1,0 |
≤ 0,6 |
≤ 0,4 |
≤ 0,2 |
≤ 0,2 |
≤ 0,05 |
≤ 0,05 |
≤ 0,5 |
≤ 0,1 |
≤ 4,0 |
< Camp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 000 |
≤ 4,5 |
|
2. Deviško oljčno olje |
≤ 0,05 |
≤ 1,0 |
≤ 0,6 |
≤ 0,4 |
≤ 0,2 |
≤ 0,2 |
≤ 0,05 |
≤ 0,05 |
≤ 0,5 |
≤ 0,1 |
≤ 4,0 |
< Camp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 000 |
≤ 4,5 |
|
3 Lampante oljčno olje |
≤ 0,05 |
≤ 1,0 |
≤ 0,6 |
≤ 0,4 |
≤ 0,2 |
≤ 0,2 |
≤ 0,10 |
≤ 0,10 |
≤ 0,5 |
≤ 0,1 |
≤ 4,0 |
— |
≥ 93,0 |
≤ 0,5 |
≥ 1 000 |
≤ 4,5 (3) |
|
4 Rafinirano oljčno olje |
≤ 0,05 |
≤ 1,0 |
≤ 0,6 |
≤ 0,4 |
≤ 0,2 |
≤ 0,2 |
≤ 0,20 |
≤ 0,30 |
≤ 0,5 |
≤ 0,1 |
≤ 4,0 |
< Camp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 000 |
≤ 4,5 |
|
5 Oljčno olje - mešanica rafin. oljč. olja in deviškega oljčnega olja |
≤ 0,05 |
≤ 1,0 |
≤ 0,6 |
≤ 0,4 |
≤ 0,2 |
≤ 0,2 |
≤ 0,20 |
≤ 0,30 |
≤ 0,5 |
≤ 0,1 |
≤ 4,0 |
< Camp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 000 |
≤ 4,5 |
|
6 Surovo olje iz oljčnih tropin |
≤ 0,05 |
≤ 1,0 |
≤ 0,6 |
≤ 0,4 |
≤ 0,3 |
≤ 0,2 |
≤ 0,20 |
≤ 0,10 |
≤ 0,5 |
≤ 0,2 |
≤ 4,0 |
— |
≥ 93,0 |
≤ 0,5 |
≥ 2 500 |
> 4,5 (4) |
|
7 Rafinirano olje iz oljčnih tropin |
≤ 0,05 |
≤ 1,0 |
≤ 0,6 |
≤ 0,4 |
≤ 0,3 |
≤ 0,2 |
≤ 0,40 |
≤ 0,35 |
≤ 0,5 |
≤ 0,2 |
≤ 4,0 |
< Camp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 800 |
> 4,5 |
|
8 Olje iz oljčnih tropin |
≤ 0,05 |
≤ 1,0 |
≤ 0,6 |
≤ 0,4 |
≤ 0,3 |
≤ 0,2 |
≤ 0,40 |
≤ 0,35 |
≤ 0,5 |
≤ 0,2 |
≤ 4,0 |
< Camp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 600 |
> 4,5 |
PRILOGA IA
VZORČENJE OLJČNEGA OLJA ALI OLJA IZ OLJČNIH TROPIN, DOSTAVLJENEGA V IZVIRNEM PAKIRANJU, KI NE PRESEGA 100 LITROV
Ta metoda vzorčenja se uporablja za dobave oljčnega olja ali olja iz oljčnih tropin, ki ne presegajo 125 000 litrov, v izvirnem pakiranju, ki ne presega 100 litrov.
Če dobava presega 125 000 litrov, jo je treba razdeliti v serije po 125 000 litrov ali manj. Če znaša dobava manj kot 125 000 litrov, sestavlja eno serijo. Zadevna metoda se nato uporabi za vsako serijo.
Najmanjše število primarnih vzorcev, ki jih je treba vzeti, se določi na podlagi velikosti serije v skladu s tabelo iz točke 1.
Velikost primarnega vzorca se določi na podlagi prostornine izvirnega pakiranja v skladu s tabelo iz točke 2.1.
Opredelitve dobave, primarnega vzorca in laboratorijskega vzorca so navedene v standardu EN ISO 5555.
Serija‘ pomeni skupek prodajnih enot, ki se proizvajajo, izdelujejo in pakirajo v takih okoliščinah, da se olje, ki ga vsebuje vsaka prodajna enota, šteje za homogeno v smislu vseh analitičnih značilnosti.
1. ŠTEVILO PRIMARNIH VZORCEV, KI JIH JE TREBA VZETI
Najmanjše število primarnih vzorcev, ki jih je treba vzeti, se določi na podlagi velikosti serije v skladu z naslednjo tabelo:
|
Velikost serije (v litrih), ki znaša manj kot |
Najmanjše število primarnih vzorcev |
|
7 500 |
2 |
|
25 000 |
3 |
|
75 000 |
4 |
|
125 000 |
5 |
Izvirna pakiranja, ki sestavljajo primarni vzorec, morajo biti izbrana med sosednjimi pakiranji v partiji.
V primeru dvoma države članice povečajo število primarnih vzorcev, ki jih je treba vzeti.
2. VSEBINA PRIMARNIH VZORCEV
|
2.1 |
Primarni vzorci morajo vsebovati naslednje:
|
|
2.2 |
Primarne vzorce je treba hraniti v izvirnem pakiranju do časa analize. Olje v primarnih vzorcih se nato, kakor je ustrezno, razdeli v tri laboratorijske vzorce za izvedbo: (a) analiz iz prilog II, III, IX in X, (a) analize iz Priloge XII, (c) drugih analiz. |
|
2.3 |
Pakiranja, ki sestavljajo primarni vzorec, se razdelijo v skladu s postopki kontrole, ki jih določa nacionalna zakonodaja. |
3. ANALIZE IN REZULTATI
(a) Vsak primarni vzorec iz točke 1 se razdeli v laboratorijske vzorce v skladu s točko 2.5 standarda EN ISO 5555 in analizira na naslednji način:
— določanje prostih maščobnih kislin iz prve alinee člena 2(1),
— določanje peroksidnega števila iz druge alinee člena 2(1),
— spektrofotometrična analiza iz osme alinee člena 2(1),
— določanje sestave maščobnih kislin iz devete alinee člena 2(1),
(b) Če eden od rezultatov analiz iz točke (a) za najmanj en primarni vzorec, vzet iz iste serije, ni v skladu z značilnostmi deklarirane kategorije olja, je treba celotno zadevno serijo deklarirati kot neskladno.
Če rezultati analiz iz točke (a) za vsak primarni vzorec, vzet iz iste partije, niso enotni, ob upoštevanju značilnosti ponovljivosti zadevnih metod, je treba celotno serijo deklarirati kot neenotno in vsak primarni vzorec podvreči drugi zahtevani analizi. V nasprotnem primeru je treba en primarni vzorec iz serije podvreči drugi zahtevani analizi.
(c) Če eden od rezultatov analiz iz drugega odstavka točke (b) ni v skladu z značilnostmi deklarirane kategorije olja, je treba celotno zadevno serijo deklarirati kot neskladno.
Če so vsi rezultati analiz iz drugega odstavka točke (b) v skladu z značilnostmi deklarirane kategorije olja, je treba celotno zadevno serijo deklarirati kot skladno.
PRILOGA IB
DREVO ODLOČANJA ZA PREVERJANJE, ALI JE VZOREC OLJČNEGA OLJA SKLADEN Z DEKLARIRANO KATEGORIJO
Analiza za preverjanje, ali je oljčno olje ali olje iz oljčnih tropin skladno z deklarirano kategorijo, se lahko opravi:
(a) bodisi z izvedbo, po naključnem vrstnem redu, analiz, predvidenih za namen preverjanja skladnosti z značilnostmi iz Priloge I; bodisi
(b) z izvedbo, po vrstnem redu iz drevesa odločanja, analiz, ki so v njem opredeljene, dokler se ne sprejme ena od odločitev iz drevesa odločanja.
Analize, ki se nanašajo na onesnaževalce in so potrebne za preverjanje skladnosti s standardi Evropske skupnosti, je treba opraviti ločeno.
Drevo odločanja se uporablja za vse kategorije oljčnega olja in olja iz oljčnih tropin. Sestavljeno je iz tabel, oštevilčenih od 1 do 11, ki jim je treba slediti na podlagi deklarirane kategorije zadevnega olja po vrstnem redu, določenem v splošni tabeli.
Legenda splošne tabele in tabel od 1 do 11:
— dvojna črta (=) označuje smer, ki ji je treba slediti v primeru skladnosti (pozitiven odgovor) z merili, navedenimi v predhodnem okencu. Pikčasta črta (…) označuje nadomestno smer, ki ji je treba slediti v primeru neskladnosti,
— naslovi v okencih v tabelah 1 do 11 se nanašajo na analize iz te uredbe na podlagi tabele ekvivalentnosti iz Dodatka 1 k tej prilogi,
— naslovi v oklepajih, ki se pojavljajo v krogih z negativnimi odločitvami v tabelah 1 do 11, se sklicujejo na okvirne informacije iz Dodatka 2 k tej prilogi. Črke same po sebi ne zavezujejo k izvedbi analiz ali označujejo verodostojnost navedenih predpostavk.

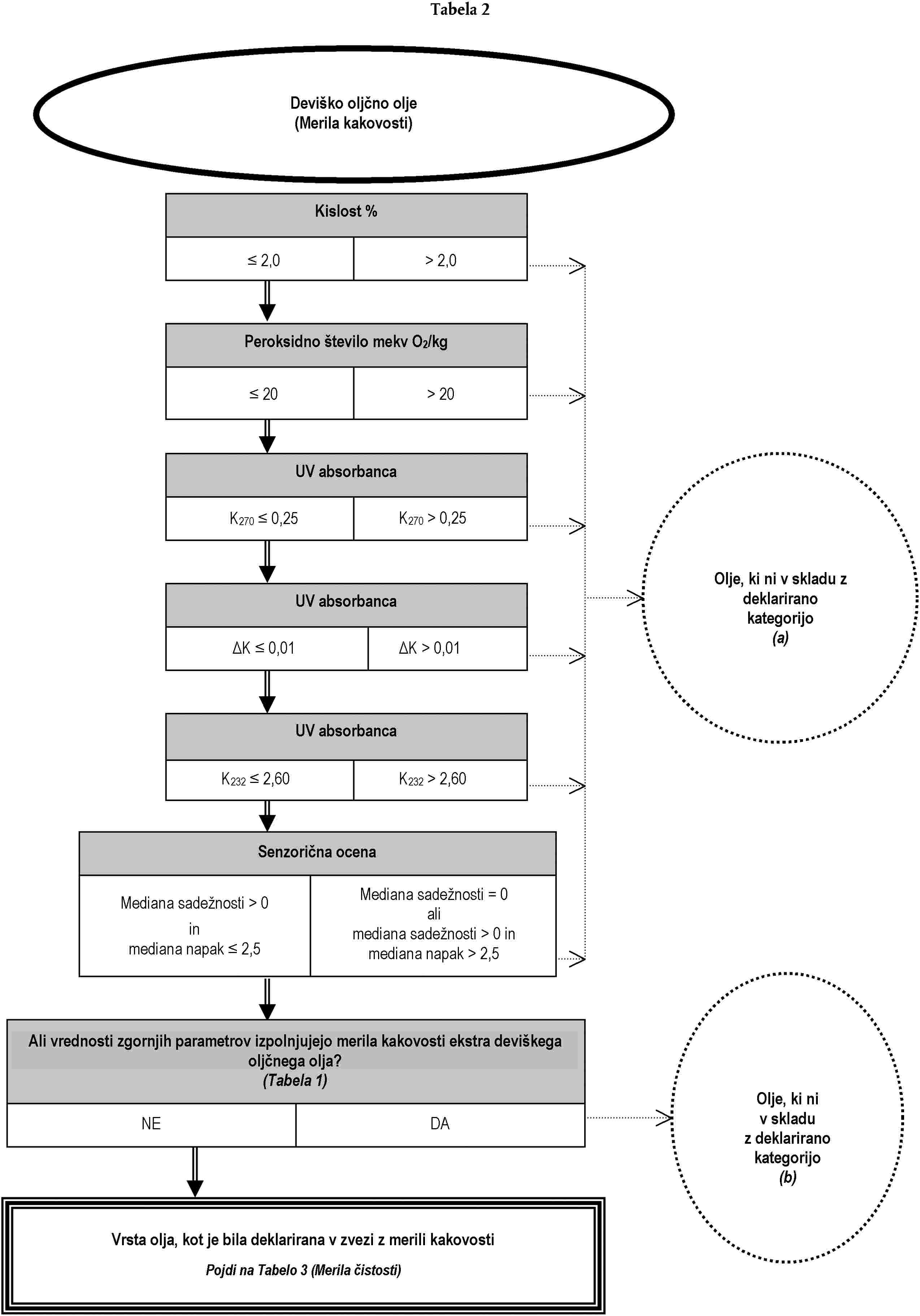


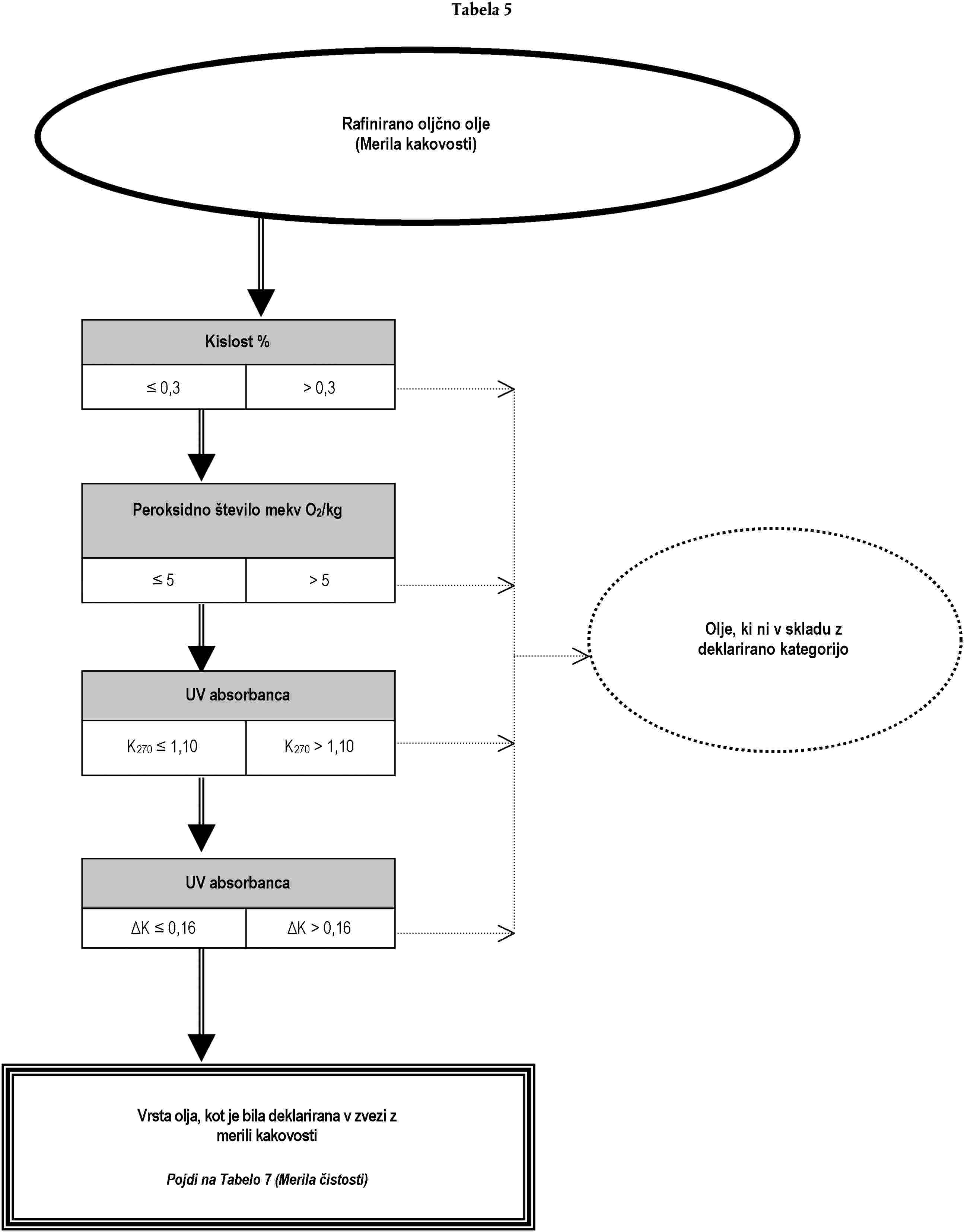
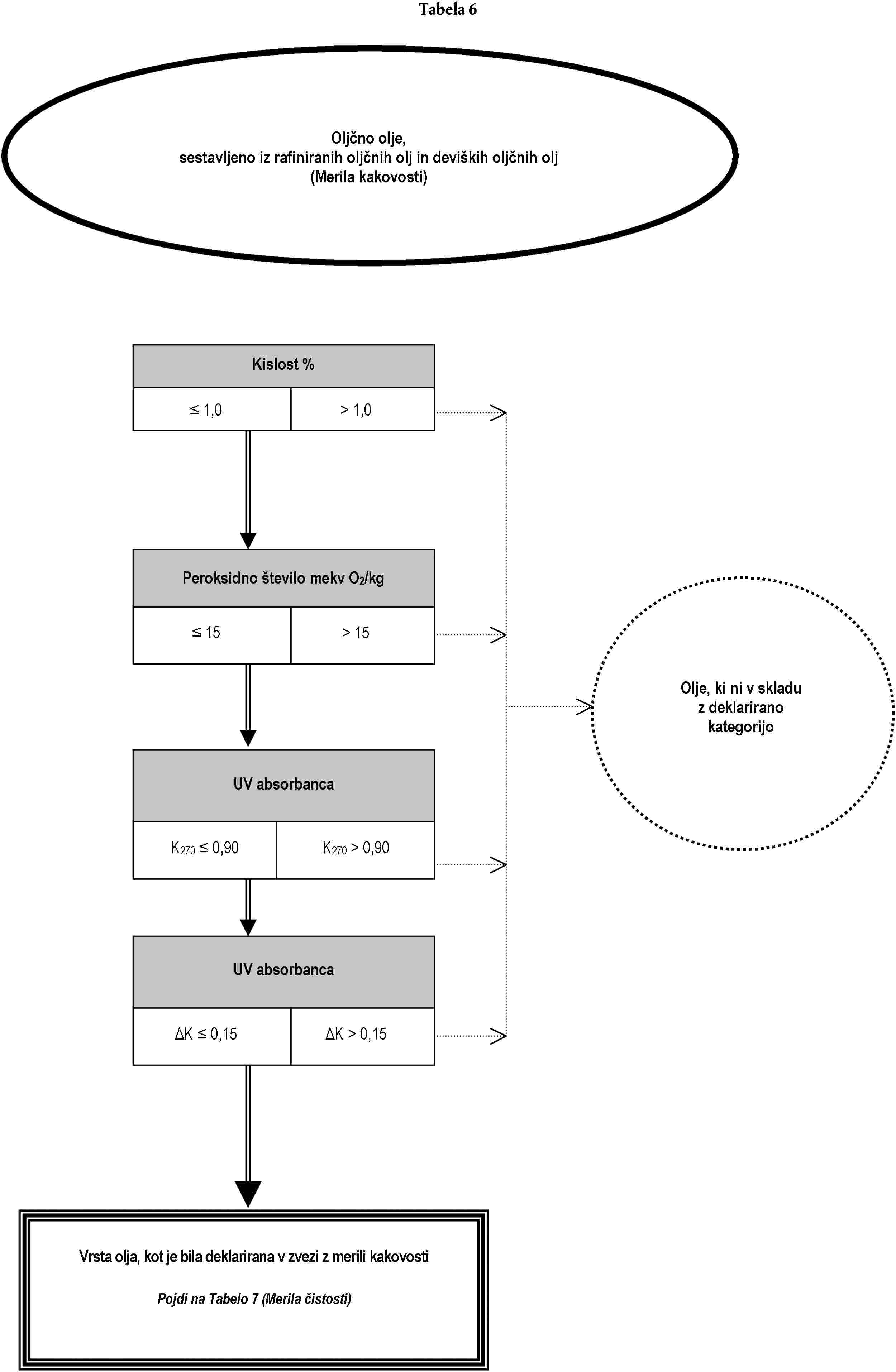



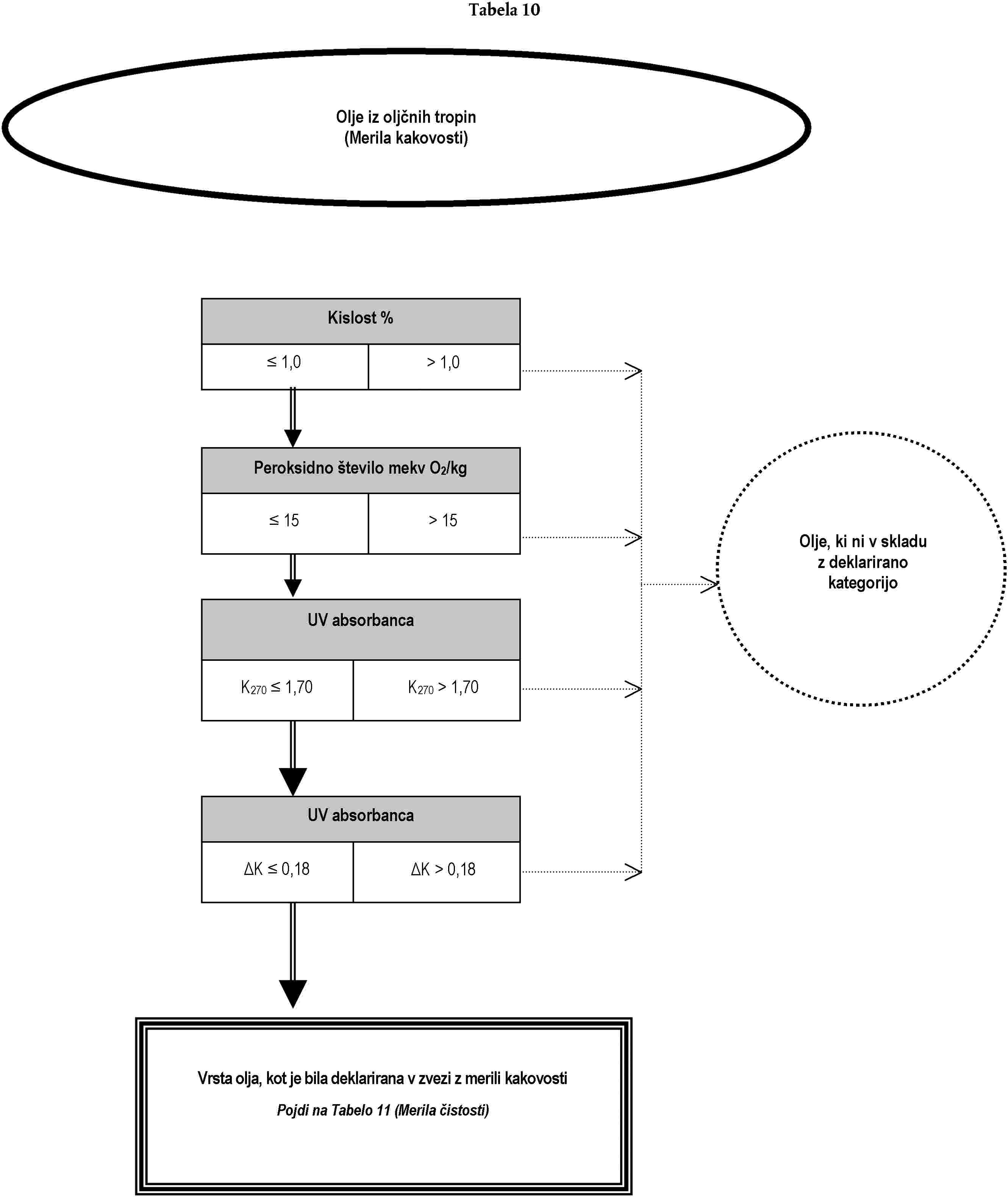

DODATEK 1
Tabela ekvivalentnosti med prilogami k tej uredbi in analizami iz drevesa odločanja
|
— Kislost |
Priloga II |
Določevanje prostih maščobnih kislin, metoda v hladnem |
|
— Peroksidno število |
Priloga III |
Določanje peroksidnega števila |
|
— UV absorbanca |
Priloga IX |
Spektrofotometrična analiza |
|
— Senzorična ocena |
Priloga XII |
Senzorična ocena deviškega oljčnega olja |
|
— 3,5-stigmastadieni |
Priloga XVII |
Metoda določanja stigmastadienov v rastlinskih oljih |
|
— Trans izomer maščobnih kislin |
Priloga Xa in |
Analiza metilnih estrov maščobnih kislin s plinsko kromatografijo |
|
Priloga Xb |
Priprava metilnih estrov maščobnih kislin |
|
|
— Delež maščobnih kislin |
Priloga Xa in |
Analiza metilnih estrov maščobnih kislin s plinsko kromatografijo |
|
Priloga Xb |
Priprava metilnih estrov maščobnih kislin |
|
|
— DECN42 |
Priloga XVIII |
Določanje sestave trigliceridov z ECN42 (razlika med podatki HPCL in teoretičnim deležem) |
|
— Sestava sterolov in skupni steroli |
Priloga V |
Določanje sestave in deleža sterolov s kapilarno plinsko kromatografijo |
|
— Eritrodiol in uvaol |
Priloga VI |
Določanje eritrodiola in uvaola |
|
— Voski |
Priloga IV |
Določanje deleža voskov s kapilarno plinsko kromatografijo |
|
— Alifatski alkoholi |
Priloga XIX |
Določanje deleža alifatskih alkoholov s kapilarno plinsko kromatografijo |
|
— Nasičene maščobne kisline na položaju 2 |
Priloga VII |
Določevanje odstotnega deleža 2-gliceril monopalmitata |
DODATEK 2
Tabela 1
(a) Glej deviško ali lampante oljčno olje (Merila kakovosti Tabela 2 ali Merila kakovosti in čistosti Tabela 4)
(b) Glej lampante oljčno olje (Merila kakovosti in čistosti Tabela 4)
Tabela 2
(a) Glej lampante oljčno olje (Merila kakovosti in čistosti Tabela 4)
(b) Glej ekstra deviško oljčno olje (Merila kakovosti Tabela 1)
Tabela 3
(a) Prisotnost rafiniranega olja (oljčnega ali drugih)
(b) Prisotnost olja iz oljčnih tropin
Tabela 4
(a) Glej ekstra deviško oljčno olje in deviško oljčno olje (Merila kakovosti Tabela 1 in Tabela 2)
(b) Prisotnost rafiniranega olja (oljčnega ali drugih)
(c) Prisotnost olja iz oljčnih tropin
(d) Prisotnost esterificiranih olj
Tabela 7
(a) Prisotnost olja iz oljčnih tropin
(b) Prisotnost esterificiranih olj
Tabela 8
(a) Prisotnost rafiniranega olja (oljčnega ali drugih)
(b) Glej lampante oljčno olje (Merila kakovosti in čistosti Tabela 4)
(c) Prisotnost esterificiranih olj
Tabela 11
(a) Prisotnost esterificiranih olj
PRILOGA II
DOLOČEVANJE PROSTIH MAŠČOBNIH KISLIN, METODA V HLADNEM
1. DOLOČANJE KISLOSTI
Določanje prostih maščobnih kislin v oljčnih oljih. Vsebnost prostih maščobnih kislin je dogovorjeno izražena kot izračunana kislost.
1.1. Princip
Vzorec se raztopi v mešanici topil in obstoječe proste maščobne kisline, se titrira s pomočjo etanolne raztopine kalijevega hidroksida
1.2. Reagenti
Vsi reagenti naj bodo priznane analitske kakovosti in uporabljena voda naj bo destilirana ali enakovredne čistoče.
|
1.2.1. |
Dietilni eter; 95 % etanol (v/v), mešanica enakih volumenskih delov. Opomba:Dietilni eter je zelo vnetljiv in lahko tvori eksplozivne perokside. Pri njegovi uporabi je treba biti posebno pazljiv. Nevtalizirajte natančno v trenutku uporabe z raztopino kalijevega hidroksida (1.2.2), z dodatkom 0,3 ml fenolftaleinove raztopine (1.2.3) na 100 ml mešanice. Opomba: Če ni mogoče uporabiti dietilnega etra, se lahko uporabi mešanica topil, ki vsebujejo etanol in toluen. Če je treba, se etanol lahko nadomesti s 2-propanolom. |
|
1.2.2. |
Kalijev hidroksid, standardizirana etanolna raztopina, c(KOH), približno 0,1 mol/1 ali, če je potrebno, c(KOH), približno 0,5 mol/1. Tik pred uporabo moramo poznati in preveriti točno koncentracijo etanolne raztopine kalijevega hidroksida. Uporabite raztopino, ki je bila pripravljena najmanj pet dni pred uporabo in prelita v rjavo steklenico z gumijastim zamaškom. Raztopina mora biti brezbarvna ali barve slame. Opomba: Stabilno brezbarvno raztopino kalijevega hidroksida lahko pripravimo kot sledi. Zavremo 1 000 ml etanola z 8 g kalijevega hidroksida in 0,5 aluminijevih ostružkov in nadaljujemo z vrenjem pod povratnim hladilnikom eno uro. Takoj destiliramo. Raztopimo v destilatu zahtevano količino kalijevega hidroksida. Pustimo mirovati nekaj dni in z dekantiranjem ločimo bistro raztopino od oborine kalijevega karbonata. Raztopino lahko brez destilacije pripravimo kot sledi: k 1 000 ml etanola dodamo 4 ml aluminijevega butilata in mešanico pustimo nekaj dni. V destilatu raztopimo zahtevano količino kalijevega hidroksida. Raztopina je pripravljena za uporabo. |
|
1.2.3. |
Raztopina 10 g/l fenolftaleina v 95 do 96 % etanolu (v/v) ali raztopina 20 g/l alkalno modrega (v primeru močno obarvanih maščob) v 95 do 96 % etanolu (v/v). |
1.3. Oprema
Običajna laboratorijska oprema, vključno z:
|
1.3.1. |
analitsko tehtnico; |
|
1.3.2. |
250-ml erlenmajerico; |
|
1.3.3. |
10-ml bireta, z razdelkom 0,05 ml. |
1.4. Postopek
|
1.4.1. |
Priprava vzorca za preskus (Preskus izvajajte na filtriranem vzorcu. Kadar so vlaga in nečistoče skupaj nižje od 1 %, uporabite vzorec brez nadaljnje obdelave; kadar presegajo 1 %, ga je treba filtrirati.) |
|
1.4.2. |
Odvzem vzorca Odvisno od predvidenega kislinskega števila natehtamo vzorec v skladu z naslednjo tabelo:
Vzorec tehtamo v erlenmajerici (1.3.2). |
|
1.4.3 |
Določanje Raztopimo vzorec (1.4.2) v 50- do 150-ml v predhodno nevtralizirani mešanici dietilnega etra in etanola (1.2.1). Med mešanjem titriramo z 0,1 mol/l raztopino kalijevega hidroksida (1.2.2) (glej Opombo 2) do preskoka barve indikatorja.(rožnata obarvanost fenolftaleina je obstojna najmanj 10 sekund). Opomba 1. Opomba 1. Standardizirano etanolno raztopino kalijevega hidroksida (1.2.2) se lahko nadomesti z vodno raztopino kalijevega ali natrijevega hidroksida pod pogojem, da količina vnešene vode ne sproži faznega ločevanja. Opomba 2. Opomba 2. Če zahtevana količina 0,1 mol/l raztopine kalijevega hidroksida preseže 10 ml, uporabite 0,5 mol/1raztopino. Opomba 3. Opomba 3. Če raztopina med titracijo postane motna, dodajte dovolj topil (1.2.1), da dobite prozorno raztopino. |
1.5 Kislost: izražena v masnem odstotnem deležu oleinske kisline
Kislost v utežnih o dstotkih je enaka:
![]()
kjer je:
|
V |
= |
volumen uporabljene standardizirane raztopine kalijevega hidroksida, v mililitrih; |
|
c |
= |
točna koncentracija v molih na liter uporabljene titrirane raztopine kalijevega hidroksida; |
|
M |
= |
molska masa uporabljene kisline za izražanje rezultata (= 282); |
|
m |
= |
masa vzorca v gramih. |
Za rezultat je treba vzeti aritmetično povprečje ►M6 dveh določitev ◄ .
PRILOGA III
DOLOČANJE PEROKSIDNEGA ŠTEVILA
1. NAMEN
Ta standard opisuje metodo za določanje peroksidnega števila olj in masti.
2. PODROČJE UPORABE
Ta standard se uporablja za olja in masti živalskega in rastlinskega izvora.
3. DEFINICIJA POJMOV
Peroksidno število je količina tistih snovi v vzorcu, izražena v miliekvivalentih aktivnega kisika na kilogram, ki oksidira kalijev jodid v opisanih delovnih pogojih.
4. PRINCIP
Reakcija preskusnega deleža vzorca v raztopini ocetne kisline in kloroforma, z raztopino kalijevega jodida. Titracija sproščenega joda s standardizirano raztopino natrijevega tiosulfata.
5. OPREMA
Vsa uporabljena oprema je brez reducirajočih ali oksidirajočih snovi.
Opomba: Brušenih površin ne namažite.
|
5.1 |
3-ml steklena ladjica. |
|
5.2 |
Bučke z obrusom kapacitete približno 250 ml predhodno osušimo z inertnim plinom (dušikom, ali še bolje, z ogljikovim dioksidom). |
|
5.3 |
25-ml ali 50-ml bireta z razdelkom 0,1 ml. |
6. REAGENTI
|
6.1 |
Kloroform, analitske čistoče, očiščen kisika s prepihovanjem s čistim, suhim inertnim plinom. |
|
6.2 |
Ledocet, analitske čistoče, očiščen kisika s prepihovanjem s čistim, suhim inertnim plinom. |
|
6.3 |
Nasičena vodna raztopina kalijevega jodida, sveže pripravljena, brez joda in jodatov. |
|
6.4 |
Tik pred uporabo točno standardizirana 0,01 ali 0,002 mol/L vodna raztopina natrijevega tiosulfata. |
|
6.5 |
Škrobovica, 10g/l vodna suspenzija, sveže pripravljena iz naravnega topnega škroba. |
7. VZOREC
Pazimo, da vzorec jemljemo in skladiščimo v temnem, hranimo na hladnem v popolnoma zaprtih steklenih posodah. Posode so hermetično zatesnjene z zamaški z obrusom ali iz plute.
8. POSTOPEK
Preskus se izvede pri difuzni dnevni svetlobi ali pri umetni svetlobi. V skladu s pričakovanim peroksidnim številom, natehtamo na stekleni ladjici (5.1) ali,če vam to ne uspe, v bučki (5.2) s točnostjo 1 mg.
|
Pričakovano peroksidno število (mekv) |
Masa vzorca (g) |
|
0 do 12 |
5,0 do 2,0 |
|
12 do 20 |
2,0 do 1,2 |
|
20 do 30 |
1,2 do 0,8 |
|
30 do 50 |
0,8 do 0,5 |
|
50 do 90 |
0,5 do 0,3 |
V bučko prenesemo stekleno ladjico z vzorcem. Dodamo 10 ml kloroforma (6.1). Vzorec hitro raztopimo z mešanjem. Dodamo 15 ml ocetne kisline (6.2), nato 1 ml raztopine kalijevega jodida (6.3). Hitro zamašimo z zamaškom, stresamo eno minuto in pustimo točno pet minut, v temnem prostoru na temperaturi od 15 do 25 °C.
Dodamo približno 75 ml destilirane vode. Titriramo sproščeni jod z raztopino natrijevega tiosulfata (6.4) (za pričakovana peroksidna števila, nižja od 12 titriramo z 0,002 mol/L raztopino in z 0,01 mol/L raztopino za pričakovana števila, nad 12), močno tresemo, uporabimo škrobovico (6.5) kot indikator.
Na istem preskusnem vzorcu opravimo dve določitvi.
Istočasno izvedemo slepi preskus. Če rezultat presega 0,05 ml 0,01 mol/L raztopine natrijevega tiosulfata (6.4), zamenjamo reagente s čistejšimi.
9. IZRAŽANJE REZULTATOV
Peroksidno število (PV), izraženo v miliekvivalentih aktivnega kisika na kilogram, se izrazi s formulo:
![]()
kjer je:
|
V |
= |
volumen raztopine standardiziranega natrijevega tiosulfata v ml (6.4), z upoštevanim volumnom slepega preskusa; |
|
T |
= |
točna molarnost uporabljene raztopine natrijevega tiosulfata (6.4); |
|
m |
= |
masa vzorca v gramih. |
Za rezultat se vzame aritmetično povprečje dveh določitev.
PRILOGA IV
DOLOČEVANJE VSEBNOSTI VOSKOV S KAPILARNO PLINSKO KROMATOGRAFIJO
1. NAMEN
Ta metoda opisuje postopek za določitev vsebnosti voskov v oljčnem olju. Voski se ločijo glede na število ogljikovih atomov. Metodo lahko uporabimo zlasti za razlikovanje med oljčnim oljem, pridobljenim s stiskanjem, in oljčnim oljem, pridobljenim z ekstrakcijo (olje iz oljčnih tropin).
2. PRINCIP
Maščobo, ki smo ji dodali ustrezen interni standard, na z vodo omočenem silikagelu frakcioniramo s kolonsko kromatografijo; eluirano frakcijo najprej ujamemo pri pogojih preskušanja (katere polarnost je manjša kot pri trigliceridih), nato jo neposredno analiziramo s kapilarno plinsko kromatografijo.
3. OPREMA
|
3.1 |
Erlenmajerica prostornine 25 ml. |
|
3.2 |
Steklena kolona za plinsko kromatografijo, z notranjim premerom 15,0 mm, dolžine 30 do 40 cm in opremljena s petelinčkom. |
|
3.3 |
Plinski kromatograf, primeren za uporabo s kapilarno kolono in opremljen s sistemom za neposredno injiciranje v kolono, ki obsega:
|
|
3.4 |
10 μl-mikrobrizgalka s kaljeno iglo in z možnostjo neposrednega injiciranja v kolono. |
|
3.5 |
Električni vibrator. |
|
3.6 |
Rotavapor. |
|
3.7 |
Žarilna peč. |
|
3.8 |
Analitska tehtnica, ki zagotavlja ± 0,1 mg natančnost. |
|
3.9 |
Običajna laboratorijska steklovina. |
4. REAGENTI
|
4.1 |
Silikagel z velikostjo delcev med 60 in 200 μm. Silikagel vsaj 4 ure žarimo pri 500 °C. Nato ga ohladimo in dodamo 2 % vode glede na količino silikagela. Dobro premešamo, da se zmes homogenizira. Pred uporabo hranimo vsaj 12 ur v temi. |
|
4.2 |
n-heksan, kromatografske čistoče. |
|
4.3 |
Dietil eter, kromatografske čistoče. |
|
4.4 |
n-heptan, kromatografske čistoče. |
|
4.5 |
Standardna raztopina lauril arahidata koncentracije 0,1 % (m/V) v heksanu (interni standard). (Lahko uporabimo tudi -palmitil palmitat ali miristil stearat.)
|
|
4.6 |
Nosilni plin: vodik ali helij, čist, plinsko-kromatografske čistoče. |
|
4.7 |
Pomožni plini: — vodik, čist, plinsko-kromatografske čistoče, — zrak, čist, plinsko-kromatografske čistoče. |
5. POSTOPEK
5.1 Priprava kromatografske kolone
V n-heksanu (4.2) suspendiramo 15 g silikagela (4.1) in napolnimo kolono (3.2). Počakamo, da se posede. Homogenost posedanja, ki poveča homogenost kromatografskih pasov, lahko dosežemo z električnim vibratorjem (3.5). Spiramo s 30 ml n-heksana, da odstranimo vse morebitne nečistoče. S tehtnico (3.8) v erlenmajerico prostornine 25 ml (3.1) natehtamo natanko 500 mg vzorca in dodamo ustrezno količino internega standarda (4.5), ki je odvisna od pričakovane vsebnosti voskov. Na primer, pri oljčnem olju dodamo 0,1 mg, pri olju iz oljčnih tropin pa 0,25 do 0,5 mg lauril arahidata. Tako pripravljen vzorec prenesemo v kromatografsko kolono, in sicer z dvema 2-ml odmerkoma n-heksana (4.2).
Topilo izpuščamo iz kolone tako dolgo, da doseže 1 mm nad zgornjim robom absorbenta, zatem spiramo z dodatnimi 70 ml n-heksana, da odstranimo morebitne naravno prisotne n-alkane. Nato začnemo kromatografsko eluiranje, in sicer zberemo 180 ml mešanice n-heksan/etilni eter v razmerju 99:1 in pri pretoku približno 15 kapljic na 10 sekund. Eluiranje vzorca mora potekati pri sobni temperaturi 22 ± 4 °C.
Opombe:
— Dnevno je treba pripraviti svežo mešanico n-heksana/etilnega etra (99:1).
— Za opazovanje pravilnega eluiranja voskov lahko vzorcu v raztopini dodamo 100 μl 1-odstotnega sudana v elucijski mešanici. Ker ima barvilo vmesno retencijo med voski in trigliceridi, je treba elucijo suspendirati, ko obarvanje doseže dno kromatografske kolone, ker so se eluirali vsi voski.
Iz tako dobljene frakcije na rotavaporju (3.6) odparimo skoraj vse topilo. Preostala 2 ml topila odstranimo z uporabo blagega toka dušika, nato dodamo 2-4 ml n-heptana.
5.2 Plinsko-kromatografska analiza
5.2.1 Predpriprava
Kolono pritrdimo na plinski kromatograf (3.3), in sicer vstopni del na injektor, izhodnega pa na detektor. Izvedemo splošno preverjanje plinskega kromatografa (tesnjenje plinskih povezav, pravilno delovanje detektorja in rekorderja itd.).
Če kolono uporabljamo prvič, je priporočljivo, da jo kondicioniramo. Pri majhnem pretoku plina skozi kolono vključimo plinski kromatograf. Postopoma segrevamo, tako da po približno 4 urah dosežemo temperaturo 350 °C. Pri tej temperaturi kolono kondicioniramo vsaj dve uri, nato naravnamo instrument v skladu s pogoji delovanja (naravnamo pretok, prižgemo plamen, povežemo z elektronskim rekorderjem (3.3.4), naravnamo delovno temperaturo peči za kolono, naravnamo detektor itd.). Pri občutljivosti, ki je vsaj dvakrat večja od delovne, posnamemo bazno linijo. Le-ta mora biti linearna, brez kakršnih koli vrhov in odklonov.
Negativen premočrtni odklon kaže na slabo tesnjenje med kolono in instrumentom, pozitiven odklon pa na slabo kondicionirano kolono.
5.2.2 Izbira delovnih pogojev
Splošni delovni pogoji, ki jih je treba upoštevati, so naslednji:
— temperatura kolone:
—
|
|
20 °C/minuto |
|
5 °C/minuto |
|
20 °C/minuto |
|
|
na začetku 80 °C (1′) |
→ |
240 °C |
→ |
325 °C (6′) |
→ |
340 °C (10′) |
— temperatura detektorja: 350 °C,
— količina injicirane snovi: 1 μl raztopine (2–4 ml) n-heptana,
— nosilni plin: helij ali vodik z optimalno linearno hitrostjo izbranega plina (glej Dodatek),
— občutljivost instrumenta: taka, da izpolnjuje spodnje pogoje:
Te pogoje lahko spremenimo glede na značilnosti kolone in plinskega kromografa, da bi dosegli ločevanje vseh voskov, zadostno resolucijo vrhov (glej sliko) in retencijski čas internega standarda C32, ki mora biti 18 ± 3 minute. Najznačilnejši vrh voskov mora biti izmerjen najmanj 60 % od spodnjega dela skale.
Integracijske parametre moramo določiti tako, da dobimo pravilno vrednotenje površin vrhov.
Opomba:Glede na visoko končno temperaturo je dovoljen pozitivni odklon, ki pa ne sme biti večji od 10 % od spodnjega dela skale.
5.3 Izvedba analize
Z 10-μl mikrobrizgalko odvzamemo 1 μl raztopine; bat izvlečemo, da se igla izprazni. Iglo vbodemo v injektor in po eni ali dveh sekundah hitro vbrizgnemo. Po približno petih sekundah iglo pazljivo izvlečemo.
Kromatogram snemamo, dokler ne eluirajo vsi voski.
Bazna linija mora ves čas izpolnjevati zahtevane pogoje.
5.4 Identifikacija vrhov
Različne vrhove identificiramo na podlagi retencijskih časov in s primerjavo med mešanicami voskov z znanimi retencijskimi časi, ki so bile analizirane pri enakih pogojih.
Slika prikazuje kromatogram voskov deviškega oljčnega olja.
5.5 Kvantitativna analiza
Z integratorjem izračunamo površine vrhov alifatskih estrov od C40 do C46 in površino vrha internega standarda.
Na podlagi naslednje formule izračunamo vsebnost voskov posameznega estra v mg/kg maščobe:
![]()
Kjer je:
|
Ax |
= |
površina vrha posameznega estra, v kvadratnih milimetrih; |
|
As |
= |
površina vrha internega standarda, v kvadratnih milimetrih; |
|
ms |
= |
masa dodanega internega standarda, v miligramih; |
|
m |
= |
masa odvzetega vzorca za določitev, v gramih. |
6. PODAJANJE REZULTATOV
Navedemo vsoto vsebnosti posameznih sestavin različnih voskov od C40 do C46, v mg/kg maščobe (ppm).
Opomba:Spojine, ki jih je treba količinsko določiti, se nanašajo na vrhove s sodim številom ogljikovih atomov med estri C40 do C46, tako kot je to prikazano na kromatograma voskov oljčnega olja. Če sta za ester C46 dva vrhova, je za njegovo identifikacijo priporočljiva analiza frakcije voskov olja iz oljčnih tropin, kjer je vrh C46 dobro viden, ker je razločno večji.
Rezultate je treba izraziti na eno decimalno mesto natančno.
Slika
Kromatogram voskov v oljčnem olju ( 6 )

Legenda:
|
I.S. |
= |
lauril arahidat |
|
1. |
= |
diterpenski estri |
|
2 + 2′ |
= |
estri C40 |
|
3 + 3′ |
= |
estri C42 |
|
4 + 4′ |
= |
estri C44 |
|
5. |
= |
estri C46 |
|
6. |
= |
sterolni estri in triterpenski alkoholi. |
DODATEK
Določanje linearne hitrosti plina
V plinski kromatograf, naravnan na običajne delovne pogoje, injiciramo 1 do 3 μl metana (ali propana). Merimo čas, ki ga plin potrebuje za pot skozi kolono od trenutka vbrizga do trenutka, ko se pojavi vrh (tM).
Linearna hitrost v cm/s je izražena s formulo L/tM, kjer je L dolžina kolone v cm, tM pa izmerjeni čas v sekundah.
PRILOGA V
DOLOČANJE SESTAVE IN VSEBNOSTI STEROLOV S KAPILARNO PLINSKO KROMATOGRAFIJO
1. NAMEN
Ta metoda opisuje postopek za določanje vsebnosti posameznih in celokupnih sterolov v maščobah.
2. PRINCIP METODE
Maščoba z dodanim β-holestanolom kot internim standardom, se umili s kalijevim hidroksidom in etanolno raztopino in potem se neumiljive snovi ekstrahirajo z dietilnim etrom.
Sterolna frakcija se loči od neumiljivega ekstrakta s kromatografijo na z lugom obdelani silikagelni plošči. Steroli, dobljeni iz silikagela, se pretvorijo v trimetilsililne etre in se jih analizira s kapilarno plinsko kromatografijo.
3. OPREMA
|
3.1 |
250-ml steklenica, s povratnim hladilnikom, s priključki iz steklenih obrusov. |
|
3.2 |
500-ml liji ločniki. |
|
3.3 |
250-ml bučke. |
|
3.4 |
Celotna oprema za analizo s tankoplastno kromatografijo s steklenimi ploščami 20 × 20 cm. |
|
3.5 |
Ultravijolična svetilka z valovno dolžino 366 ali 254 nm. |
|
3.6 |
100-μl in 500-μl mikrobrizgalke. |
|
3.7 |
Cilindrični filtrirni liji s porozno frito G3 (poroznosti 15 do 40 μm), premera približno 2 cm in globine približno 5 cm, z nastavkom, primernim za filtriranje v vakuumu in priključek 12/21 z zunanjim obrusom. |
|
3.8 |
50-ml presesalna erlenmajerica z 12/21 priključkom z notranjim obrusom za uporabo s filtrirnim lijem (3.7). |
|
3.9 |
Epruveta kapacitete 10 ml s koničnim dnom in zamaškom. |
|
3.10 |
Plinski kromatograf, primeren za uporabo s kapilarno kolono, opremljen s sistemom deljenja, ki ga sestavljajo:
|
|
3.11 |
Steklena ali kvarčna kapilarna kolona, dolžine 20 do 30 m, notranjega premera 0,25 do 0,32 mm, popolnoma prekrita s stacionarno fazo SE-52 ali SE-54 ali podobno, enakomerne debeline med 0,10 in 0,30 μm. |
|
3.12 |
10-μl mikrosiringa za plinsko kromatografijo s konico iz kaljenega jekla. |
4. REAGENTI
|
4.1 |
Kalijev hidroksid, približno 2 mol/L etanolna raztopina: Raztopite 130 g kalijevega hidroksida (minimalne koncentracije 85 %) s hlajenjem, v 200 ml distilirane vode in potem do enega litra dodajte etanol. Raztopino hranite v dobro zamašenih temnih steklenicah. |
|
4.2 |
Dietilni eter, čistoče za analizo. |
|
4.3 |
Brezvodni natrijev sulfat, čistoče za analizo. |
|
4.4 |
Steklene plošče, premazane s silikagelom, brez indikatorja fluorescence, debeline 0,25 mm (ki se jih dobi v prodaji, pripravljene za uporabo). |
|
4.5 |
Kalijev hidroksid, približno 0,2 mol/L etanolna raztopina; Raztopite 13 g kalijevega hidroksida v 20 ml distilirane vode in do enega litra dodajte etanol. |
|
4.6 |
Benzen, kromatografske čistoče (Glej 5.2.2). |
|
4.7 |
Aceton, kromatografske čistoče. (Glej 5.2.2) |
|
4.8 |
Heksan, kromatografske čistoče. (Glej 5.2.2) |
|
4.9 |
Dietilni eter, kromatografske čistoče. (Glej 5.2.2) |
|
4.10 |
Kloroform,, kromatografske čistoče. (Glej 5.2.2) |
|
4.11 |
Referenčna raztopina za tankoplastno kromatografijo: holesterol ali fitosteroli, ►M6 2 % ◄ raztopina v kloroformu. |
|
4.12 |
2,7-diklorofluorescein, 0,2-% etanolna raztopina. Naredite rahlo bazično z dodatkom nekaj kapelj alkoholne raztopine 2 mol/L kalijevega hidroksida. |
|
4.13 |
Brezvodni piridin, kromatografske čistoče. |
|
4.14 |
Hexametil disilazan. |
|
4.15 |
Trimetilklorosilan. |
|
4.16 |
Referenčne raztopine sterolnih trimetilsililnih etrov. Pripravimo jih tik pred uporabo iz čistih sterolov ali iz mešanic sterolov iz olj, ki jih vsebujejo. |
|
4.17 |
β-holestanol, 0,2 % raztopina (m/V) v kloroformu (interni standard). |
|
4.18 |
Nosilni plin: vodik ali helij, plinsko-kromatografske čistoče. |
|
4.19 |
Pomožni plini: — hydrogen, gas-chromatographic purity,– vodik, plinsko-kromatografske čistoče, — air, gas-chromatographic purity.– zrak, plinsko-kromatografske čistoče. |
5. POSTOPEK
|
5.1 |
Priprava neumiljivih snovi.
|
|
5.2 |
Ločevanje sterolne frakcije.
|
|
5.3 |
Priprava trimetilsililnih etrov.
|
|
5.4 |
Plinsko kromatografska analiza
|
6. IZRAŽANJE REZULTATOV
|
6.1 |
Zabeležite posamezne koncentracije sterola kot mg/100 g maščobe in njihovo vsoto kot „celokupni steroli“. |
|
6.2 |
Izračunajte odstotek za vsak posamezni sterol iz razmerja med ustrezno površino vrha in površino vseh sterolnih vrhov.
kjer je:
|
DODATEK
Določanje linearne hitrosti plina
Pri nastavitvi plinskega kromatografa na normalne delovne pogoje injicirajte 1 do 3 μl metana (ali propana) in izmerite čas, ki je potreben, da se plin pretoči skozi kolono, od trenutka injiciranja do trenutka, ko se pojavi vrh (tM).
Linearna hitrost v cm/s je podana z L/tM, pri čemer je L dolžina kolone v centimetrih in tM izmerjeni čas v sekundah.
Tabela 1
Relativni retenzijski časi sterolov
|
Pik |
Identifikacija |
Relativni retenzijski čas |
||
|
SE 54 kolona |
SE 52 kolona |
|||
|
1 |
holesterol |
Δ-5-holesten-3ß-ol |
0,67 |
0,63 |
|
2 |
holestanol |
5α-holestan-3ß-ol |
0,68 |
0,64 |
|
3 |
Brasikasterol |
[24S]-24-metil-Δ-5,22-holestadien-3ß-o1 |
0,73 |
0,71 |
|
4 |
24-metilen-holesterol |
24-metilen-Δ-5,24-holesten-3ß-ol |
0,82 |
0,80 |
|
5 |
kampesterol |
[24R]-24-metil-Δ-5-holesten-3ß-o1 |
0,83 |
0,81 |
|
6 |
kampestanol |
[24R]-24-metil-holestan-3ß-ol |
0,85 |
0,82 |
|
7 |
stigmasterol |
[24R]-24-etil-Δ-5,22-holestadien-3ß-o1 |
0,88 |
0,87 |
|
8 |
Δ-7-kampesterol |
[24R]-24-metil-Δ-7-holesten-3ß-o1 |
0,93 |
0,92 |
|
9 |
Δ-5,23-stigmastadienol |
[24R,S]-24-etil-Δ-5,23-holestadien-3ß-o1 |
0,95 |
0,95 |
|
10 |
klerosterol |
[24S]-24-etil-Δ-5,25-holastadien-3ß-ol |
0,96 |
0,96 |
|
11 |
β-sitosterol |
[24R]-24-etil-Δ-5-holestan-3ß-ol |
1,00 |
1,00 |
|
12 |
sitostanol |
24-etil-holestan-3ß-ol |
1,02 |
1,02 |
|
13 |
Δ-5-avenasterol |
[24Z]-24-etiliden-5-holesten-3ß-o1 |
1,03 |
1,03 |
|
14 |
Δ-5,24-stigmastadienol |
[24R,S]-24-etil-Δ-5,24-holestadien-3ß-o1 |
1,08 |
1,08 |
|
15 |
Δ-7-stigmastenol |
[24R,S]-24-etil-Δ-7,24-holestadien-3ß-ol |
1,12 |
1,12 |
|
16 |
Δ-7-avenasterol |
[24Z]-24-etiliden-Δ-7-holesten-3ß-o1 |
1,16 |
1,16 |
 Slika 1
Plinski kromatogram sterolne frakcije nerafiniranega oljčnega olja.
Slika 1
Plinski kromatogram sterolne frakcije nerafiniranega oljčnega olja.
 Slika 2
Plinski kromatogram sterolne frakcije rafiniranega oljčnega olja.
Slika 2
Plinski kromatogram sterolne frakcije rafiniranega oljčnega olja.
PRILOGA VI
DOLOČANJE ERITRODIOLA IN UVAOLA
UVOD
Eritrodiol (ki se ga običajno podaja kot vsoto glikolov eritrodiola in uvaola) je sestavina neumiljive frakcije, značilna za nekatere vrste maščob. V bistveno višjih koncentracijah se nahaja v oljčnem olju, ekstrahiranem s topili, kot pa v drugih oljih, kot sta prešano oljčno olje in olje iz grozdnih pešk, ki ga tudi vsebujeta in tako njegova prisotnost lahko potrdi prisotnost oljčnega olja, ekstrahiranega s topili.
1. NAMEN
Ta metoda opisuje postopek za določanje eritrodiola v maščobah.
2. PRINCIP METODE
Maščobo umilimo z etanolno raztopino kalijevega hidroksida. Neumiljivo frakcijo, potem ekstrahiramo z dietilnim etrom in očistimo s prehodom skozi kolono, napolnjeno z aluminijevim oksidom.
Neumiljive snovi tretiramo s tankoplastno kromatografijo na silikagelni plošči, dokler se ne ločijo pasovi, ki ustrezajo frakcijam sterola in eritrodiola. Steroli in eritrodiol, dobljeni s plošče, se pretvorijo v trimetilsililne etre in mešanico se analizira s plinsko kromatografijo.
Rezultati so izraženi v odstotnem deležu eritrodiola in mešanice eritrodiola in sterolov.
3. OPREMA
|
3.1 |
Oprema, kot je specificirana v Prilogi V (določanje vsebnosti sterolov). |
4. REAGENTI
|
4.1 |
Reagenti, kot so specificirani v Prilogi V (določanje vsebnosti sterolov). |
|
4.2 |
Referenčna raztopina eritrodiola, 0,5-% raztopina v kloroformu. |
5. POSTOPEK
5.1 Priprava neumiljivih snovi.
Kot je opisano v odstavku 5.1.2 Priloge V.
5.2 Ločevanje eritrodiola in sterolov.
|
5.2.1 |
Glej odstavek 5.2.1 Priloge V. |
|
5.2.2 |
Glej odstavek 5.2.2 Priloge V. |
|
5.2.3 |
Pripravite 5-% raztopino neumiljivih snovi v kloroformu. Z 0,1 μl mikrobrizgalko nanesite na kromatografsko ploščo 0,3 ml raztopine, približno 1,5 cm od spodnjega robu v progi, ki je čimbolj tanka in homogena. Na en konec plošče nanesite nekaj mikrolitrov raztopin holesterola in eritrodiola, ki naj služita kot referenca. |
|
5.2.4 |
Ploščo vložite v razvijalno komoro, pripravljeno kot je navedeno v 5.2.1. Temperatura okolja mora biti približno 20 °C. Takoj zaprite komoro s pokrovom in pustite, da poteka eluiranje, dokler fronta topila ne doseže razdalje 1 cm od vrhnjega roba plošče. Vzemite ploščo iz razvijalne komore in odparite topilo pod tokom vročega zraka. |
|
5.2.5 |
Rahlo in enokomerno napršite ploščo z alkoholno raztopino 2,7-diklorofluorosceina. Če se ploščo opazuje pod ultravijolično svetlobo, se pasova sterola in eritrodiola lahko identificirata tako, da se ju poravna z referencami. Označite s piko izven robov fluorescence. |
|
5.2.6 |
S kovinsko žličko odstrgajte silikagel v označenih območjih. Material s plošče dajte v 50-ml bučko. Dodajte 15 ml vročega kloroforma, dobro pretresite in filtrirajte skozi lij s ploščo iz sintranega stekla, tako da silikagel prenesete na filter. Trikrat sperite z vročim kloroformom (vsakokrat z 10 ml), tako da zberete filtrat v 100-ml bučki. Odparite filtrat na približno 4 do 5 ml volumna, prenesite v kalibrirano 10-ml epruveto za centrifugiranje s konusnim dnom, posušite z rahlim segrevanjem v toku dušika in stehtajte. |
5.3 Priprava trimetilsililnih estrov
Kot je opisano v odstavku 5.3 Priloge V.
5.4 Plinsko kromatografska analiza
Kot je opisano v odstavku 5.4 zgoraj navedenega postopka. Delovni pogoji plinskega kromatografa morajo biti pri analizi takšni, da se izvede analiza sterolov in loči TMSE od eritrodiola in uvaola.
Potem, ko je bil vzorec vbrizgan, nadaljujte z beleženjem, dokler prisotni steroli, eritrodiol in uvaol ne eluirajo. Potem identificirajte vrhove (retenzijska časa za eritrodiol in uvaol glede na ß-sitosterol sta okrog 1,45 oziroma 1,55) in površine izračunajte kot za sterole.
6. IZRAŽANJE REZULTATOV
![]()
kjer je:
|
A1 |
= |
površina vrha eritrodiola ►M6 ————— ◄ ; |
|
A2 |
= |
površina vrha uvaola ►M6 ————— ◄ ; |
|
Σ Asteroli |
= |
vsota površin vrhov sterolov ►M6 ————— ◄ . |
Rezultat se izrazi na eno decimalno mesto.
PRILOGA VII
DOLOČANJE ODSTOTNEGA DELEŽA 2-GLICERIL MONOPALMITATA
1. PREDMET UREJANJA IN PODROČJE UPORABE
S to metodo je opisan analitski postopek za določevanje odstotnega deleža palmitinske kisline na položaju 2 v trigliceridih z ovrednotenjem 2-gliceril monopalmitata.
To metodo uporabljamo za rastlinska olja, tekoča pri sobni temperaturi (20 °C).
2. PRINCIP
Po pripravi pustimo, da na vzorec olja učinkuje pankreatična lipaza: poteče delna hidroliza, specifična za položaja 1 in 3 v molekuli triglicerida, ki povzroči nastanek 2-monoacilglicerolov. Odstotni delež 2-gliceril monopalmitata v monoacilglicerolni frakciji se po sililiranju določi s kapilarno plinsko kromatografijo.
3. APARATURE IN OBIČAJNA LABORATORIJSKA OPREMA
|
3.1 |
Erlenmajerica, 25 ml |
|
3.2 |
Čaše 100, 250 in 300 ml |
|
3.3 |
Steklena kromatografska kolona z notranjim premerom 21–23 mm, dolžine 400 mm, opremljena s sintrano stekleno ploščico in petelinčkom |
|
3.4 |
Merilni valji prostornine 10, 50, 100 in 200 ml |
|
3.5 |
Bučke prostornine 100 in 250 ml |
|
3.6 |
Rotavapor |
|
3.7 |
Epruvete za centrifugiranje prostornine 10 ml s koničnim dnom in obrušenim zamaškom |
|
3.8 |
Centrifuga za epruvete prostornine 10 in 100 ml |
|
3.9 |
Termostat, ki omogoča vzdrževanje temperature pri 40 °C ± 0,5 °C |
|
3.10 |
Merilni pipeti prostornine 1 in 2 ml |
|
3.11 |
Brizgalka prostornine 1 ml |
|
3.12 |
Mikrobrizgalka prostornine 100 μl |
|
3.13 |
Lij-ločnik, 1 000 ml |
|
3.14 |
Plinski kromatograf za kapilarne kolone, opremljen z injicirnim sistemom za hladno injiciranje vzorca neposredno v kolono in pečjo, ki lahko vzdržuje izbrano temperaturo znotraj 1 °C |
|
3.15 |
Injektor za hladno injiciranje vzorca neposredno v kolono |
|
3.16 |
Plamensko ionizacijski detektor in elektrometer |
|
3.17 |
Rekorder-integrator, kompatibilen z elektrometrom, z odzivnim časom, manjšim od 1 sekunde, in nastavljivo hitrostjo pomika papirja |
|
3.18 |
Steklena ali kvarčna kapilarna kolona dolžine 8 do 12 m, z notranjim premerom 0,25 do 0,32 mm, pokrita z metilpolisiloksanom ali 5-odstotnim fenil metilpolisiloksanom, debeline 0,10–0,30 μm, ki jo je mogoče uporabiti pri 370 °C |
|
3.19 |
Mikrobrizgalka prostornine 10 μl z nesnemljivo, vsaj 7,5 cm dolgo iglo, za neposredno vbrizganje na začetek kolone |
4. REAGENTI
|
4.1 |
Silikagel z velikostjo delcev med 0,063 in 0,200 mm (70/280 mesh), pripravljen na naslednji način: silikagel damo v porcelansko posodico, ga pri 160 °C 4 ure sušimo v sušilniku, nato pa pustimo, da se na sobni temperaturi ohladi v eksikatorju. Nato dodamo količino vode, ki ustreza 5 % teže silikagela: v erlenmajerico prostornine 500 ml natehtamo 152 g silikagela in dodamo 8 g destilirane vode, zamašimo ter homogeniziramo. Pred uporabo pustimo mirovati vsaj 12 ur. |
|
4.2 |
n-heksan (kromatografske čistoče) |
|
4.3 |
Izopropanol |
|
4.4 |
Izpropanol, vodna raztopina 1/1 (V/V) |
|
4.5 |
Pankreatična lipaza. Aktivnost uporabljene lipaze mora biti med 2,0 in 10 lipaznimi enotami na mg (V prodaji so pankreatične lipaze z aktivnostjo med 2 in 10 enotami na mg encima.) |
|
4.6 |
Pufrska raztopina tris-hidroksi-metilaminometana: 1 M vodna raztopina, ki ji s koncentrirano HCI (1/1 V/V) uravnamo pH na vrednost 8 (preverimo s pH-metrom) |
|
4.7 |
Natrijev holat, encimske čistosti, 0,1-odstotna vodna raztopina (to raztopino je treba uporabiti v petnajstih dneh po pripravi) |
|
4.8 |
Kalcijev klorid, 22-odstotna vodna raztopina |
|
4.9 |
Dietil eter kromatografske čistoče |
|
4.10 |
Elucijsko topilo: mešanica n-heksana/dietilnega etra (87/13) (V/V) |
|
4.11 |
Natrijev hidroksid, 12-odstotna raztopina v masnih odstotkih |
|
4.12 |
Fenolftalein, 1-odstotna raztopina v etanolu |
|
4.13 |
Nosilni plin: vodik ali helij, za plinsko kromatografijo |
|
4.14 |
Pomožna plina: vodik, najmanj 99-odstoten, brez vlage in organskih snovi, in zrak, za plinsko kromatografijo in enake čistoče |
|
4.15 |
Regent za silaniziranje: mešanica piridina, heksametildisilazana, trimetilklorosilana v razmerju 9:3:1 (V/V/V) (V prodaji so raztopine, pripravljene za uporabo. Uporabimo lahko tudi druge reagente za silaniziranje, zlasti bis-trimetilsilil trifluoroacetamid + 1-odstoten trimetilklorosilan, razredčen z enako količino brezvodnega piridina.) |
|
4.16 |
Referenčni vzorci: čisti monogliceridi ali mešanice monogliceridov, za katere je znano, da imajo podobno odstotkovno sestavo kot vzorec. |
5. POSTOPEK
5.1 Priprava vzorca
|
5.1.1 |
Olj z deležem prostih kislin, manjšim od 3 %, pred kolonsko kromatografijo ni treba nevtralizirati s silikagelom. Olja, katerih delež prostih kislin je večji od 3 %, je treba nevtralizirati v skladu s točko 5.1.1.1.
|
|
5.1.2 |
V erlenmajerico prostornine 25 ml damo 1,0 g olja, pripravljenega po spodnjih navodilih (3.1), in ga raztopimo v 10 ml razvijalne mešanice (4.10). Pred kolonsko kromatografijo s silikagelom raztopino pustimo mirovati najmanj 15 minut. Če je raztopina motna, jo centrifugiramo, da zagotovimo optimalne pogoje za kromatografijo. (Uporabimo lahko komercialne 500-mg silikagelne kartuše SPE.) |
|
5.1.3 |
Priprava kromatografske kolone V kolono (3.3) vlijemo približno 30 m razvijalnega topila (4.10), s stekleno paličko v spodnji del kolone vstavimo košček vate; stisnemo, da odstranimo zrak. V čaši pripravimo raztopino 25 g silikagela (4.1) v približno 80 ml razvijalne raztopine in jo z lijakom prelijemo v kolono. Preverimo, da smo v kolono dali ves silikagel; speremo z elucijskim topilom (4.10), odpremo petelinček in pustimo, da raven tekočine seže približno 2 mm nad zgornjo raven silikagela. |
|
5.1.4 |
Kolonska kromatografija V erlenmajerico prostornine 25 ml (3.1.) natehtamo natanko 1,0 g vzorca, ki smo ga pripravili v skladu s točko 5.1. Vzorec raztopimo v 10 ml elucijskega topila (4.10). Raztopino prelijemo v kromatografsko kolono, ki smo jo pripravili v skladu s točko 5.1.3. Pazimo, da ne premešamo površine kolone. Odpremo ventil in pustimo raztopino vzorca odtekati, dokler ne doseže ravni silikagela. Eluiramo s 150 ml razvijalnega topila. Količino pretoka nastavimo na 2 ml/min (tako da 150 ml odteče v kolono v približno 60–70 minutah). Eluat zberemo v 250-mililitrsko bučko z znano maso. V vakuumu odparimo topilo in njegove zadnje sledi odstranimo s tokom dušika. Bučko stehtamo in izračunamo pridobljeni ekstrakt (Če uporabljamo silikagelne kartuše SPE, storimo naslednje: v kartuše, ki smo jih predhodno kondicionirali s 3 ml n-heksana, vlijemo 1 ml raztopine (5.1.2). Po filtraciji raztopine razvijemo s 4 ml n-heksana/dietilnega etra 9/1 (V/V). Eluat zberemo v 10-mililitrsko epruveto in ga z uvajanjem toka dušika odparimo do suhega. Na suhem preostanku pustimo učinkovati pankreatično lipazo (5.2). Ključno je, da pred in po uporabi kartuše SPE preverimo sestavo maščobnih kislin. |
5.2 Hidroliza s pankreatično lipazo
|
5.2.1 |
V epruveto centrifuge natehtamo 0,1 g olja, pripravljenega v skladu s točko 5.1. Dodamo 2 ml pufrske raztopine (4.6), 0,5 ml raztopine natrijevega holata (4.7) in 0,2 ml raztopine kalcijevega klorida, pri čemer po vsakem dodajanju dobro pretresemo. Epruveto zapremo z obrušenim zamaškom in jo namestimo v termostat, naravnan na 40 ± 0,5 °C. |
|
5.2.2 |
Dodamo 20 mg lipaze, previdno pretresemo (pazimo, da ne zmočimo zamaška) in damo epruveto za natanko 2 minuti v termostat, nato jo damo ven, jo natanko 1 minuto močno stresamo in pustimo, da se ohladi. |
|
5.2.3 |
Dodamo 1 ml dietilnega etra, zamašimo in močno stresamo, nato centrifugiramo ter raztopino etra z mikrobrizgalko prenesemo v čisto in suho epruveto. |
5.3 Priprava silaniziranih derivatov in plinska kromatografija
|
5.3.1 |
Z mikrobrizgalko prenesemo 100 μl raztopine (5.2.3) v epruveto prostornine 10 ml s koničastim dnom. |
|
5.3.2 |
Topilo odstranimo z uvajanjem rahlega toka dušika, dodamo 200 μl reagenta za silaniziranje (4.15), zamašimo epruveto in pustimo mirovati 20 minut. |
|
5.3.3 |
Po 20 minutah dodamo 1 do 5 ml n-heksana (odvisno od kromatografskih pogojev): raztopina, ki jo dobimo, je nared za plinsko kromatografijo. |
5.4 Plinska kromatografija
Pogoji za postopek so naslednji:
— temperatura injektorja (injektor za vbrizgavanje v kolono) mora biti nižja od temperature vrelišča topila (68 °C),
— temperatura detektorja: 350 °C,
— temperatura kolone: programiranje temperature peči: 1 minuto pri 60 °C, s hitrostjo segrevanja 15 °C na minuto, dokler ne dosežemo temperature 180 °C, nato s hitrostjo 5 °C na minuto, dokler ne dosežemo temperature 340 °C, nato 13 minut pri 340 °C,
— nosilni plin: vodik ali helij, nastavljen na ustrezno linearno hitrost, da dosežemo resolucijo, navedeno na Sliki 1. Retencijski čas triglicerida C54 mora biti 40 + 5 minut (glej Sliko 2); (Pogoji za spodaj navedene postopke so okvirni. Vsak izvajalec jih mora optimizirati, da doseže želeno resolucijo. Najmanjša višina vrha za 2-gliceril monopalmitat mora biti enaka 10 % polne skale rekorderja.),
— količina injicirane snovi: 0,5-1 μl raztopine (5 ml) n-heksana (5.3.3).
5.4.1 Identifikacija vrhov
Posamezne monoacilglicerole identificiramo na podlagi dobljenih retencijskih časov in glede na čase, dobljene za standardne mešanice monogliceridov, analizirane pri enakih pogojih.
5.4.2 Kvantitativna analiza
Površina vsakega vrha se izračuna z elektronskim integratorjem.
6. PODAJANJE REZULTATOV
Odstotek gliceril monopalmitata izračunamo iz razmerja med ustrezno površino vrha in vsoto površin vrhov vseh monoacilglicerolov (glej Sliko 2), in sicer na podlagi formule:
gliceril monopalmitat (%):

kjer je:
|
Ax |
= |
površina vrha gliceril monopalmitata |
|
ΣA |
= |
vsota površin vseh vrhov monoacilglicerolov |
Rezultat je treba podati na eno decimalko natančno.
7. POROČILO O ANALIZI
V poročilu o analizi je treba podrobno navesti:
— sklicevanje na to metodo,
— vse informacije, potrebne za popolno identifikacijo vzorca,
— rezultat analize,
— vsako odstopanje od te metode, ne glede na to, ali gre za odločitev zadevnih oseb ali zaradi kakega drugega razloga,
— podrobne podatke o laboratoriju, datum analize in podpis njenih odgovornih oseb.
Slika 1
Kromatogram proizvodov reakcije silaniziranja, dobljenih z delovanjem lipaze na rafiniranem oljčnem olju, ki mu je dodanih 20 % 100 % zaestrenega olja

Legenda: Acides gras libres = proste maščobne kisline Huile d’olive raffinée + 20 % huile estérifiée = rafinirano oljčno olje + 20 % zaestrenega olja 1-2 monopalmitate = 1-2 monopalmitat 1-2 monopalmitoléine = 1-2 monopalmitolein 1-2 mono C18 insat. = 1-2 mono C18 nenasič. squalène = skvalen.
Slika 2
Kromatogram
|
(A) |
nezaestrenega oljčnega olja po delovanju lipaze, in silaniziranega; pri teh pogojih (kapilarna kolona 8–12 m) frakcija voskov eluira hkrati s frakcijo diacilglicerolov ali malo zatem. Vsebnost triacilglicerolov po lipazi ne sme preseči 15 %. 
Legenda:
|
Kromatogram:
|
(B) |
zaestrenega olja po delovanju lipaze; po silaniziranju; pri teh pogojih (kapilarna kolona 8–12 m) frakcija voskov eluira hkrati s frakcijo diglicerida ali malo zatem. Vsebnost trigliceridov po lipazi ne sme preseči 15 % 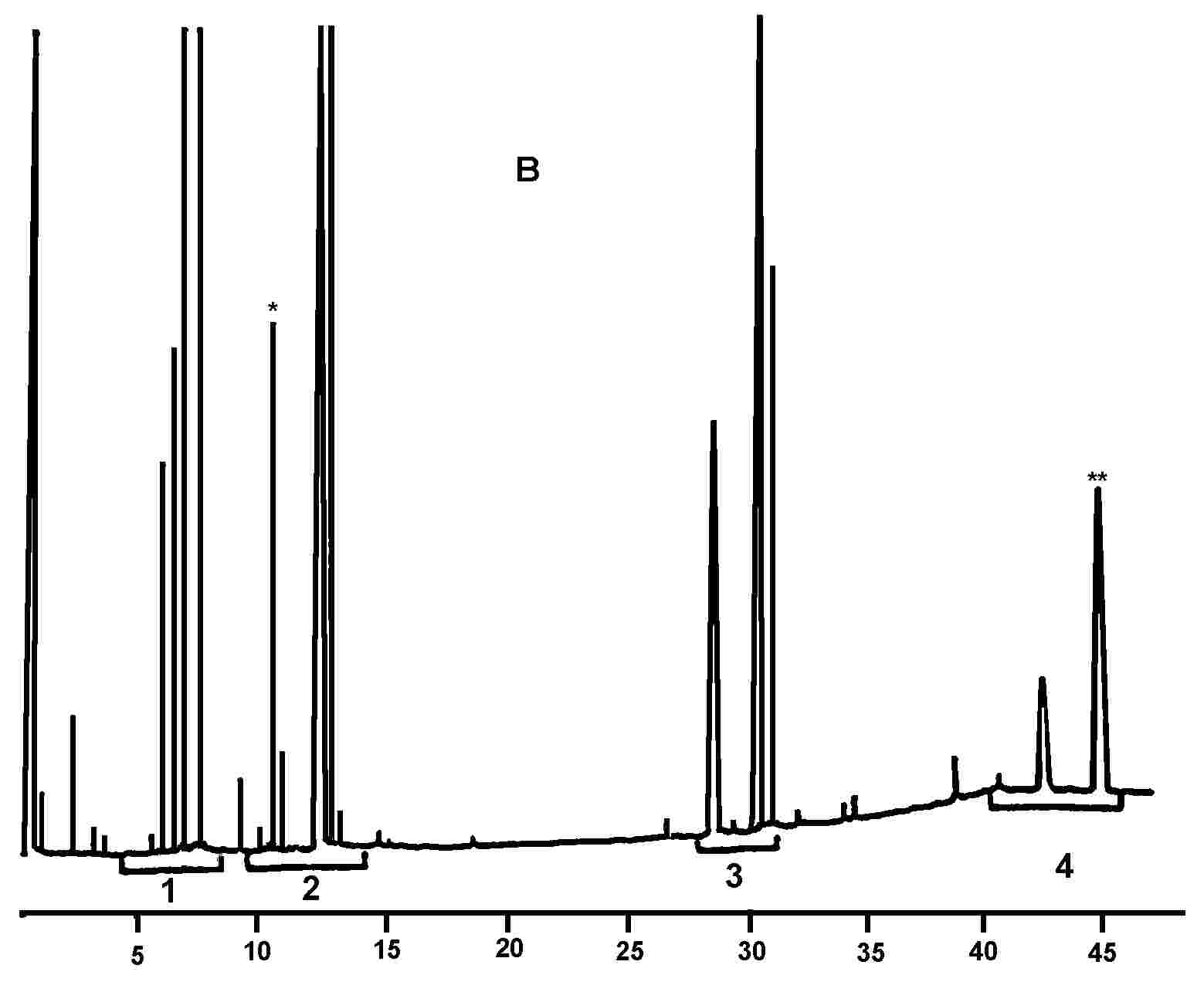
Legenda:
|
8. OPOMBE
PRIPRAVA LIPAZE
Lipaze z zadostno aktivnostjo so komercialno dostopne. Lahko jih pripravimo tudi v laboratoriju, in sicer na naslednji način:
5 kg sveže prašičje trebušne slinavke ohladimo na 0 °C. Odstranimo okoliško trdo maščevje in vezno tkivo ter jo zmeljemo v mlinčku z rezili, da dobimo kašasto tekočino. To tekočino 4 do 6 ur mešamo z 2,5 litra brezvodnega acetona in nato centrifugiramo. Izvleček naredimo še trikrat z enako količino acetona, nato dvakrat z mešanico acetona/dietilnega etra (1/1) (V/V) in dvakrat z dietilnim etrom.
Preostanek 48 ur sušimo v vakuumu, da dobimo stabilen prah, ki ga je treba dolgo časa hraniti v hladilniku in pred vlago.
PREVERJANJE AKTIVNOSTI LIPAZE
Oljno emulzijo pripravimo na naslednji način:
V mešalniku 10 minut mešamo mešanico 165 ml raztopine gumarabikuma (100 g/l), 15 g zdrobljenega ledu in 20 ml predhodno nevtraliziranega oljčnega olja.
V čašo prostornine 50 ml zaporedoma damo 10 ml te emulzije, nato 0,3 ml raztopine natrijevega holata (0,2 g/ml) in 20 ml destilirane vode.
Čašo damo v termostat, naravnan na 37 °C; namestimo elektrode pH metra in spiralni mešalnik.
Z bireto po kapljicah dodajamo raztopino natrijevega hidroksida 0,1 N, dokler ne dobimo pH vrednosti 8,3.
Dodamo ustrezno količino v vodi raztopljene lipaze v prahu (0,1 g/ml lipaze). Takoj ko pH meter pokaže pH 8,3, sprožimo štoparico in po kapljah dodajamo raztopino natrijevega hidroksida, in sicer s tako hitrostjo, da ohranimo pH vrednost 8,3. Vsako minuto odčitamo volumen porabljene raztopine.
Podatke zabeležimo v sistem koordinatnih osi, in sicer odčitke časa navedemo kot absciso, kot ordinato pa ml alkalne raztopine 0,1 N, ki smo jih porabili za ohranitev konstantnega pH. Dobiti moramo linearen graf.
Aktivnost lipaze, izmerjena v lipaznih enotah na mg, je izražena z naslednjo formulo:
![]()
kjer je:
|
A |
aktivnost v lipaznih enotah/mg |
|
V |
število ml raztopine natrijevega hidroksida 0,1 N na minuto (izračunano iz grafa) |
|
N |
normalnost raztopine natrijevega hidroksida |
|
m |
masa vzorca lipaze v mg. |
Lipazna enota je opredeljena kot količina encima, ki sprosti 10 mikro-ekvivalentov kisline na minuto.
▼M20 —————
PRILOGA IX
SPEKTROFOTOMETRIČNO MERJENJE NA UV-OBMOČJU
PREDGOVOR
Spektrofotometrično merjenje na UV-območju lahko da podatke o kakovosti maščobe, njenem stanju ohranitve in spremembah, ki so jih v njej povzročili tehnološki procesi.
Valovne dolžine, določene v postopku, se absorbirajo zaradi prisotnosti konjugiranih dienov in trienov. Te absorpcije so izražene kot specifične ekstinkcije E 1 % 1 cm (ekstinkcija 1-odstotne raztopine maščobe v specificiranem topilu, v debelini 1 cm), konvencionalno označene s K (imenuje se tudi „ekstinkcijski koeficient“).
1. PODROČJE UPORABE
Ta metoda opisuje postopek za izvajanje spektrofotometrične preiskave oljčnega olja (opisane v Dodatku) na UV-območju.
2. PRINCIP METODE
Zadevna maščoba se raztopi v zahtevanem topilu in ekstinkcija raztopine se potem določi pri specifični valovni dolžini glede na čisto topilo. Specifične ekstinkcije se izračunajo iz spektrofotometričnih odčitkov. Izračuna se specifična absorbanca pri 232 nm in 268 nm v izo-oktanu ali pri 232 nm in 270 nm v cikloheksanu za koncentracijo 1 g/100 ml v 10-milimetrski kiveti.
3. OPREMA
|
3.1 |
Spektrofotometer za merjenje ekstinkcije na UV-območju med 220 in 360 nm z možnostjo odčitavanja posameznih nanometrskih enot. Pred uporabo je priporočljivo preveriti lestvice valovnih dolžin in absorbanc spektrometra, kot sledi. 3.1.1 Lestvica valovnih dolžin: preveriti jo je mogoče z referenčnim materialom, ki ga sestavlja filter iz optičnega stekla s holmijevim oksidom, ki ima izrazite absorpcijske pasove. Referenčni material je namenjen preverjanju in kalibriranju lestvic valovnih dolžin spektrofotometrov za vidno in ultravijolično svetlobo z nominalnimi spektralnimi širinami 5 nm ali manj. Izmeri se absorbanca steklenega filtra s holmijevim oksidom proti zraku kot slepemu vzorcu v razponu valovnih dolžin od 640 do 240 nm. Za vsako spektralno širino (0,10 – 0,25 – 0,50 – 1,00 – 1,50 – 2,00 in 3,00) se izvede korekcija bazne linije s praznim nosilcem kivete. Valovne dolžine spektralne širine so navedene v certifikatu referenčnega materiala v standardu ISO 3656. 3.1.2 Lestvica absorbance: preveriti jo je mogoče z referenčnim materialom, ki ga sestavljajo štiri raztopine kalijevega dikromata v perklorovi kislini, zaprtega v štirih kvarčnih kivetah za UV-območje, da se izmeri referenčna vrednost linearnosti in fotometrične natančnosti na UV-območju. Kivete, napolnjene s kalijevim dikromatom (40 mg/ml, 60 mg/ml, 80 mg/ml in 100 mg/ml), se izmerijo proti slepemu vzorcu perklorove kisline. Neto vrednosti absorbance so navedene v certifikatu referenčnega materiala v standardu ISO 3656. |
|
3.2 |
Pravokotne kvarčne kivete, s pokrovčki, z optično dolžino 1 cm. Če jih napolnimo z vodo ali drugim primernim topilom, ne smejo kazati razlik med njimi za več kot 0,01 ekstinkcijske enote. |
|
3.3 |
25-mililitrske merilne bučke. |
|
3.4 |
Analitska tehtnica, ki omogoča tehtanje do 0,0001 g natančno. |
4. REAGENTI
Uporabljajte samo reagente priznane analitske čistosti, razen če ni navedeno drugače.
Topilo: izo-oktan (2,2,4-trimetilpentan) za merjenje pri 232 nm in 268 nm ali cikloheksan za merjenje pri 232 nm in 270 nm z absorbanco, manjšo od 0,12 pri 232 nm in manjšo od 0,05 pri 250 nm, proti destilirani vodi, izmerjeno v 10-milimetrski kiveti.
5. POSTOPEK
|
5.1 |
Obravnavani vzorec mora biti popolnoma homogen in brez dvomljivih nečistoč. Olja, ki so pri sobni temperaturi tekoča, je treba prefiltrirati skozi papir pri temperaturi približno 30 °C, trdne maščobe je treba homogenizirati in prefiltrirati pri temperaturi, ki ni več kot 10 °C nad tališčem. |
|
5.2 |
Točno natehtajte približno 0,25 g (na 1 mg natančno) tako pripravljenega vzorca v 25-mililitrsko merilno bučko, dopolnite do označbe specificirano topilo in homogenizirajte. Tako pripravljena raztopina mora biti popolnoma čista. Če je raztopina opalescentna ali motna, hitro prefiltrirajte skozi papir. |
|
5.3 |
Napolnite kvarčno kiveto s pripravljeno raztopino in izmerite ekstinkcijo pri ustrezni valovni dolžini med 232 in 276 nm, s tem da topilo uporabite kot referenco. Izmerjene ekstinkcijske vrednosti morajo biti v razponu od 0,1 do 0,8. Če niso, je treba meritve ponoviti z ustreznimi bolj koncentriranimi ali bolj razredčenimi raztopinami. Opomba: Ni vedno nujno izmeriti absorbance za celoten razpon valovnih dolžin. |
6. IZRAŽANJE REZULTATOV
|
6.1 |
Zabeležite specifične ekstinkcije (ekstinkcijske koeficiente) pri različnih valovnih dolžinah, izračunane kot sledi:
kjer je:
Rezultati se izrazijo na dve decimalni mesti natančno. |
|
6.2 |
Variacija specifične ekstinkcije (ΔΚ)
Spektrofotometrična analiza oljčnega olja v skladu z uradno metodo iz zakonodaje Unije vključuje tudi določitev variacije absolutne vrednosti specifične ekstinkcije (ΔΚ), ki je podana kot:
kjer je Km specifična ekstinkcija pri valovni dolžini m, pri čemer je valovna dolžina za maksimalno absorpcijo odvisna od uporabljenega topila: 270 za cikloheksan in 268 za izo-oktan. |
Dodatek
ZNAČILNOSTI OLJČNEGA OLJA
|
Kategorija |
Metilni estri maščobnih kislin (FAME) in etilni estri maščobnih kislin (FAEE) |
Vsebnost kislin (%) (*) |
Peroksidno število mEq 02/kg (*) |
Voski mg/kg (**) |
2-gliceril monopalmitat (%) |
Stigmastad ien mg/kg (1) |
Razlika: ECN42 (HPLC) in ECN42 (teoretični izračun) |
K232 (*) |
K270 (*) „K 270 ali K 268 (5)“ |
Delta-K (*) (5) |
Organoleptično ocenjevanje Mediana napake (Md) (*) |
Organoleptično ocenjevanje Mediana sadežnosti (Mf) (*) |
|
|
1. |
Ekstra deviško oljčno olje |
Σ FAME + FAEE ≤75 mg/kg ali 75 mg/kg <Σ FAME + FAEE ≤150 mg/kg in (FAEE/FAME) ≤1,5 |
≤ 0,8 |
≤ 20 |
≤ 250 |
≤ 0,9 če je skupni % palmitinske kisline ≤ 14 % |
≤ 0,10 |
≤ 0,2 |
≤ 2,50 |
≤ 0,22 |
≤ 0,01 |
Md = 0 |
Mf > 0 |
|
≤ 1,0 če je skupni % palmitinske kisline > 14 % |
|||||||||||||
|
2. |
Deviško oljčno olje |
— |
≤ 2,0 |
≤ 20 |
≤ 250 |
≤ 0,9 če je skupni % palmitinske kisline ≤ 14 % |
≤ 0,10 |
≤ 0,2 |
≤ 2,60 |
≤ 0,25 |
≤ 0,01 |
Md ≤ 3,5 |
Mf > 0 |
|
≤ 1,0 če je skupni % palmitinske kisline > 14 % |
|||||||||||||
|
3. |
Lampante oljčno olje |
— |
> 2,0 |
— |
≤ 300 (3) |
≤ 0,9 če je skupni % palmitinske kisline ≤ 14 % |
≤ 0,50 |
≤ 0,3 |
— |
— |
— |
Md > 3,5 (2) |
— |
|
≤ 1,1 če je skupni % palmitinske kisline > 14 % |
|||||||||||||
|
4. |
Rafinirano oljčno olje |
— |
≤ 0,3 |
≤ 5 |
≤ 350 |
≤ 0,9 če je skupni % palmitinske kisline ≤ 14 % |
— |
≤ 0,3 |
— |
≤ 1,10 |
≤ 0,16 |
— |
— |
|
≤ 1,1 če je skupni % palmitinske kisline> 14 % |
|||||||||||||
|
5. |
Oljčno olje – mešanica rafiniranega in deviškega oljčnega olja |
— |
≤ 1,0 |
≤ 15 |
≤ 350 |
≤ 0,9 če je skupni % palmitinske kisline ≤ 14 % |
— |
≤ 0,3 |
— |
≤ 0,90 |
≤ 0,15 |
— |
— |
|
≤ 1,0 če je skupni % palmitinske kisline > 14 % |
|||||||||||||
|
6. |
Surovo olje iz oljčnih tropin |
— |
— |
— |
> 350 (4) |
≤ 1,4 |
— |
≤ 0,6 |
— |
— |
— |
— |
— |
|
7. |
Rafinirano olje iz oljčnih tropin |
— |
≤ 0,3 |
≤ 5 |
> 350 |
≤ 1,4 |
— |
≤ 0,5 |
— |
≤ 2,00 |
≤ 0,20 |
— |
— |
|
8. |
Olje iz oljčnih tropin |
|
≤ 1,0 |
≤ 15 |
> 350 |
≤ 1,2 |
— |
≤ 0,5 |
— |
≤ 1,70 |
≤ 0,18 |
— |
— |
|
(1) Vsota izomerov, ki jih lahko (ali ne) ločimo s kapilarno kolono. (2) Ali če je mediana napake manjša ali enaka 3,5 in je mediana sadežnosti 0. (3) Olja z vsebnostjo voskov med 300 mg/kg in 350 mg/kg se uvrščajo med lampante oljčna olja, če je skupna vsebnost alifatskih alkoholov manjša ali enaka 350 mg/kg ali če je delež eritrodiola in uvaola manjši ali enak 3,5 %. (4) Olja z vsebnostjo voskov med 300 mg/kg in 350 mg/kg se uvrščajo med surova olja iz oljčnih tropin, če je skupna vsebnost alifatskih alkoholov večja od 350 mg/kg in če je delež eritrodiola in uvaola večji od 3,5 %. (5) K 270, če je topilo cikloheksan, K 268, če je topilo izo-oktan. |
|||||||||||||
PRILOGA X A
PLINSKOKROMATOGRAFSKA ANALIZA METILNIH ESTROV MAŠČOBNIH KISLIN
1. NAMEN
Ta metoda daje splošne smernice za uporabo plinske kromatografije, z uporabo polnjenih ali kapilarnih kolon, za določanje kvalitativne in kvantitativne sestave mešanice metilnih estrov maščobnih kislin, dobljenih po postopku, določenem v Prilogi X B.
Metoda se ne uporablja za polimerizirane maščobne kisline.
2. REAGENTI
2.1 Nosilni plin
Inertni plin (dušik, helij, argon, vodik itd.) popolnoma posušen in z vsebnostjo kisika, manjšo od 10 mg/kg.
Opomba 1.
Vodik, ki se kot inertni plin uporablja samo pri kapilarnih kolonah, lahko hitrost analize podvoji, vendar je nevaren. Varnostne naprave so na voljo.
2.2 Pomožni plini:
|
2.2.1 |
Vodik (čistoče ≥ 99,9 %), brez organskih nečistoč. |
|
2.2.2 |
Zrak ali kisik, brez organskih nečistoč. |
2.3 Referenčni standard
Mešanica metilnih estrov čistih maščobnih kislin, ali metilnih estrov maščobe poznane sestave, najraje podobne maščobi, ki jo je treba analizirati.
Treba je paziti, da se prepreči oksidacija polinenasičenih maščobnih kislin.
3. OPREMA
Dana navodila se nanašajo na običajno opremo, ki se uporablja za plinsko kromatografijo, z uporabo polnjene in/ali kapilarne kolone in plamensko ionizacijskega detektorja. Primerne so vse aparature, ki dajo učinkovitost in resulocijo, specificirano v 4.1.2.
3.1 Plinski kromatograf
Plinski kromatograf vključuje naslednje elemente:
3.1.1 Injicirni sistem
Uporabite injicirni sistem:
(a) s polnjenimi kolonami, z najmanjšim možnim mrtvim volumnom (v tem primeru je injicirni sistem možno segreti na temperaturo, ki je 20 do 50 oC višja od temperature kolone); ali
(b) s kapilarnimi kolonami, v tem primeru je injicirni sistem posebej izdelan za uporabo s takšnimi kolonami. Lahko je tip injektorja z deljenjem vzorca ali tip injektorja brez deljenja vzorca v koloni.
Opomba 2.V odsotnosti maščobnih kislin z manj kot 16 ogljikovimi atomi, se lahko uporabi injektor s pomično iglo.
3.1.2 Peč
Peč je takšna, da lahko segreje kolono na temperaturo najmanj 260 oC in vzdržuje željeno temperaturo znotraj 1 oC v polnjeni koloni in znotraj 0,1 oC v kapilarni koloni. Zadnja zahteva je zlasti pomembna kadar se uporablja kapilarna kolona.
Uporabo programiranega temperaturnega gretja priporočamo v vseh primerih in zlasti za maščobne kisline z manj kot 16 ogljikovimi atomi.
3.1.3 Polnjena kolona
|
3.1.3.1 |
Kolona, iz materiala, inertnega na snovi, ki naj bi jih analizirali (t.j– steklo ali jeklo), naslednjih dimenzij: (a) dolžina: 1 do 3 m. Kadar so prisotne dolgoverižne maščobne kisline (nad C20) je treba uporabiti relativno kratko kolono. Kadar analizirate kisline s 4 ali 6 ogljikovimi atomi, je priporočljivo, da uporabite kolono dolžine 2 m; (b) notranji premer: 2 do 4 mm. Opomba 3. Če so prisotne polinenasičene komponente z več kot tremi dvojnimi vezmi, se v jekleni koloni lahko razgradijo. Opomba 4.Lahko se uporabi sistem z dvema identičnima polnjenima kolonama |
|
3.1.3.2 |
Polnilo, ki vključuje naslednje elemente: (a) nosilec: kislinsko obdelana in silanizirana diatomejska zemlja, ali drugi primeren inertni nosilec z ozkim območjem velikosti delcev (25 μm območje med 125 do 200 μm), tako da je povprečna velikost delcev povezana z notranjim premerom in dolžino kolone; (b) stacionarna faza: polarna topila poliestrskega tipa (npr. dietilenglikol polisukcinat, butandiol polisukcinat, etilenglikol poliadipat, itd.) cianosilikoni ali katera koli druga tekočina, ki omogoča zahtevano kromatografsko ločevanje (glej klavzulo 4). Vsebnost stacionarne faze mora biti 5 do 20 % (m/m) polnila. Za nekatere analize se lahko uporabi nepolarna stacionarna faza. |
|
3.1.3.3 |
Kondicioniranje kolone Ob odklopljeni koloni, če je mogoče, od detektorja, postopoma segrejte peč na 185 oC in uvajajte tok inertnega plina skozi sveže pripravljeno kolono s hitrostjo 20 do 60 ml/min najmanj 16 ur pri tej temperaturi in nadaljni 2 uri pri 195 oC. |
3.1.4 Kapilarna kolona
|
3.1.4.1 |
Kolona, iz materiala, inertnega na snovi, ki naj bi jih analizirali (običajno steklo ali kvarčno steklo). Notranji premer mora biti med 0,2 in 0,8 mm. Notranja površina mora biti ustrezno obdelana (npr. površinska obdelava, pasiviranje) pred nanosom stacionarne faze. V večini primerov zadošča dolžina 25 mm. |
|
3.1.4.2 |
Stacionarna faza, običajno poliglikolnega tipa (polietilenglikol 20 000 ), poliester (butandiol polisukcinat) ali polarni polisiloksan (cianosilikoni). Primerne so kolone, kjer je stacionarna faza kemijsko vezana (zamrežena) na nosilni material. Opomba 5. Obstaja nevarnost, da polarni polisiloksani povzročijo težave pri identifikaciji in ločevanju linolenske kisline ali pri kislinah C20. Nanosi stacionarne faze so tanki, t.j. 0,1 do 0,2 μm. |
|
3.1.4.3 |
Namestitev in kondicioniranje kolone Upoštevajte običajne varnostne ukrepe za montažo kapilarnih kolon, t.j.namestitev kolone v peči (nosilec), izbira in montaža priključkov (tesnenje), pozicioniranje končnih mest kolone v injektorju in na detektorju (zmanjšanje mrtvih volumnov). Pretok nosilnega plina za kolono dolžine 25 mm in notranjega premera 0,3 mm naj bo 0,3 bar (30 kPa). Kondicionirajte kolono s temperaturnim programiranjem peči s hitrostjo 3 °C/min od temperature okolja na temperaturo 10 oC pod temperaturo razpada stacionarne faze. Vzdržujte tako temperaturo peči eno uro do stabilizacije bazne linije. Ohladite jo na 180 °C in nadaljujte pri izotermičnih pogojih. Opomba 6.Ustrezno predhodno kondicionirane kolone se dobijo v prodaji. |
|
3.1.5 |
Detektor, po možnosti tak, ki ga je mogoče segreti na temperaturo, višjo od temperature kolone. |
3.2 Brizgalka
Brizgalka je maksimalne kapacitete 10 μl z razdelki na 0,1 μl.
3.3 Rekorder
Če se bo krivuljo rekorderja uporabilo za izračun sestave analizirane mešanice, je potreben elektronski rekorder visoke natančnosti, kompatibilen z uporabljenimi aparaturami. Rekorder ima naslednje karakteristike:
(a) odzivna hitrost pod 1,5 s, če je mogoče 1 s (odzivna hitrost je čas, ki ga potrebuje pisalo rekorderja, da naredi prehod od 0 do 90 % po nenadnem vstopu 100 % signala);
(b) širina papirja najmanj 20 cm;
(c) hitrost papirja, ki jo je mogoče prilagoditi na vrednosti med 0,4 in 2,5 cm/min.
3.4 Integrator
Hitri in točni izračuni se lahko opravijo z elektronskim integratorjem. Ta daje linearni odziv z ustrezno občutljivostjo, in korekcija deviacij bazne linije mora biti zadovoljiva.
4. POSTOPEK
Postopki, opisani v 4.1 do 4.3, se nanašajo na uporabo plamensko ionizacijskega detektorja.
Kot alternativa se lahko uporabi plinski kromatograf s katarometrskim detektorjem (ki deluje na principu spremembe toplotne prevodnosti). Delovni pogoji se potem prilagodijo, kot je opisano v klavzuli 6.
4.1 Preskusni pogoji
|
4.1.1 |
Izbor optimalnih delovnih pogojev
|
|
4.1.2 |
Določanje števila teoretskih prekatov (učinkovitost) in resolucija (glej Sliko 1) Naredite analizo mešanice metilnega stearata in metilnega oleata v približno ekvivalentnih razmerjih (na primer, metilni estri iz kakavovega masla). Izberite temperaturo kolone in pretok nosilnega plina, tako da je maksimalni vrh metilnega stearata zabeležen približno 15 minut po vrhu topila. Uporabite tako količino mešanice metilnih estrov, da bo vrh metilnega stearata dosegel približno tri-četrtine celotne skale. Izračunajte število teoretskih prekatov, n (učinkovitost), z uporabo formule:
in resolucijo, R, z uporabo enačbe:
kjer je:
in indeks resolucije, lr, z uporabo formule
pri čemer je
Izbrati moramo take delovne pogoje, ki dajo najmanj 2 000 teoretskih prekatov na meter kolone stearat in resolucijo najmanj 1,25 za metilni stearat. |
4.2 Količina vzorca
S pomočjo brizgalke (3.2) vzemite 0,1 do 2 μl raztopine metilnih estrov, pripravljenih v skladu s Prilogo X B in jih vbrizgajte v kolono.
Če nimate že pripravljene raztopine estrov, si jih pripravite v koncentraciji 100 mg/ml heptana kromatografske čistoče in vbrizgajte 0,1 do 1 ml te raztopine.
Če se analizira sestavine, prisotne samo v sledovih, se količina vbrizganega vzorca lahko poveča (do 10-krat).
4.3 Analiza
Splošni delovni pogoji so določeni v 4.1.1.
Vendar je možno delati pri nižji temperaturi kolone, kadar se zahteva določanje maščobnih kislin z manj kot 12 ogljikovimi atomi, ali pri višji temperaturi, za določanje maščobnih kislin z več kot 20 ogljikovimi atomi. Včasih je možno v obeh primerih uporabiti temperaturno programiranje. Na primer, če vzorec vsebuje metilne estre maščobnih kislin, z manj kot 12 ogljikovimi atomi, injicirajte vzorec pri 100 oC (ali pri 50 do 60 oC, če je prisotna butirična kislina) in takoj, s hitrostjo 4 do 8 oC/min, dvignite temperaturo na optimalno. V določenih primerih se lahko oba postopka kombinirata.
Po programiranem segrevanju nadaljujte z eluiranjem pri konstantni temperaturi dokler niso vse sestavine eluirane. Če instrument nima programiranega gretja, ga uporabite na dveh nespremenljivih temperaturah med 100 in 195 oC.
Če je potrebno, je priporočljivo, da se analiza izvaja na dveh nespremenljivih fazah z različnima polaritetama, da se preveri odsotnost skritih vrhov, na primer v primeru hkratne prisotnosti konjugiranih C18:3 in C20:0 ali C18:3 in C18:2 .
4.4 Priprava referenčnega kromatograma in referenčnih grafov
Analizirajte referenčno standardno mešanico (2.3) z uporabo istih delovnih pogojev kot so bili tisti, uporabljeni za vzorec ter izmerite retenzijske čase ali retenzijske razdalje za osnovne maščobne kisline. Na semi logaritemski papir za vsako stopnjo nenasičenosti naredite grafe, ki prikazujejo logaritem retenzijskega časa ali razdalje kot enačbo števila ogljikovih atomov. V izotermnih pogojih bi morali biti grafi za nerazvejane kisline iste stopnje nenasičenosti ravne črte. Te črte bi morale biti približno vzporedne.
Treba se je izogibati pogojev pri katerih lahko pride do skritih vrhov t.j., takrat, ko je resolucija nezadostna za ločevanje dveh sestavin.
5. IZRAŽANJE REZULTATOV
5.1 Kvalitativna analiza
Identificirajte vrhove metilnih estrov v vzorcu iz grafov, pripravljenih v 4.4, z interpolacijo, če je potrebno.
5.2 Kvantitativna analiza
5.2.1 Določanje sestave
Razen izjemnih primerov, uporabite interni normalizacijski postopek, t.j., predpostavite, da so vse sestavine vzorca zastopane na kromatogramu, tako da celotno področje, ki ga pokrivajo vrhovi, predstavlja 100 % sestavin (celokupno ločevanje).
Če oprema vključuje integrator, uporabite tam dobljene podatke. Če ne, določite površino vrha tako, da pomnožite višino vrha z njegovo širino na sredini višine in morebitne različne stopnje atenuacije, uporabljene med beleženjem.
5.2.2 IZRAČUN
|
5.2.2.1 |
Splošni primer Izračunajte vsebnost dane komponente i, izražene kot masni odstotni delež metilnih estrov tako da določite površinski delež danega vrha glede na površino vseh vrhov, z uporabo naslednje enačbe:
kjer je:
Izračunajte rezultat na eno decimalko Opomba 7: V tem splošnem primeru se šteje, da rezultat izračunan na podlagi relativnih površin predstavlja masni odstotek. Za primere, pri katerih ta predpostavka ni dovoljena, glej 5.2.2.2. |
|
5.2.2.2 |
Uporaba korekcijskih faktorjev V določenih primerih, na primer ob prisotnosti maščobnih kislin z manj kot osmimi ogljikovimi atomi ali kislinami s sekundarnimi skupinami, če se uporabljajo detektorji toplotne prevodnosti ali kadar je izrecno zahtevana najvišja stopnja točnosti, je treba uporabiti korekcijske faktorje za pretvorbo površin vrhov v masne odstotne deleže sestavin. Določite korekcijske faktorje s pomočjo kromatograma iz referenčne mešanice metilnih estrov znane sestave, analizirane v delovnih pogojih, identičnih tistim, ki so bili uporabljeni za vzorec. Za to referenčno mešanico je masni odstotni delež sestavine i podan z enačbo:
kjer je:
Iz kromatograma referenčne mešanice (4.4) izračunajte delež (površina/površina) sestavine i tako:
kjer je:
Korekcijski faktor se potem izračuna kot:
Običajno so korekcijski faktorji izraženi v odnosu na KC16, tako da relativni faktorji postanejo:
Za vzorec je vsebina vsake sestavine i, izražena v masnem odstotnem deležu metilnih estrov:
Izračunajte rezultat na eno decimalko. |
|
5.2.2.3 |
Uporaba internega standarda Pri nekaterih analizah (na primer, kjer vse maščobne kisline niso kvantificirane, kot na primer, v prisotnosti kislin s štirimi ali šestimi ogljiki ob kislinah s 16 ali 18 ogljikovimi atomi, ali kadar je treba določiti absolutno količino maščobne kisline v vzorcu) je treba uporabiti interni standard. Pogosto se uporabljajo maščobne kisline s petimi, 15 ali 17 ogljikovimi atomi. Treba je določiti (morebitni) korekcijski faktor za interni standard. Masni odstotni delež sestavine i, izražen kot metilni estri, je potem podan z enačbo:
kjer je:
Izračunajte rezultate na eno decimalko. |
6. POSEBEN PRIMER – DOLOČANJE TRANS-IZOMEROV
Vsebnost trans-izomerov maščobnih kislin s številom ogljikovih atomov med 10 in 24 se ugotavlja z ločevanjem njihovih metilnih estrov s pomočjo kromatografskih kapilarnih kolon s specifično polarnostjo.
|
6.1. |
Kvarčna kolona z notranjim premerom med 0,25 mm in 0,32 mm in dolžino 50 m, prekrita z notranjim nanosom cioanopropilsilikona, debeline med 0,1 in 0,3 μm (tip SP 2380, C.P. sil 88, silor 10 in podobni tipi). |
|
6.2. |
Metilni estri se pripravijo v skladu s postopkom B, navedenim v Prilogi XB. Vse maščobe z deležem prostih maščobnih kislin, ki je večji od 3 %, je treba predhodno nevtralizirati v skladu s točko 5.1.1 Priloge VII. |
|
6.3. |
Delovni pogoji pri plinski kromatografiji so na splošno naslednji: — temperatura kolone, nastavljena med 150 °C in 230 °C (na primer 15 minut pri 165 °C, nakar se s hitrostjo 5 °C na minuto povišuje do 200 °C); — temperatura injektorja: 250 °C, če se uporablja split-splittless injektor, ali kar začetna temperatura kolone, če se uporablja sistem neposrednega injiciranja na kolono; — temperatura detektorja: 260 °C; — pretok nosilnega plina (helij in vodik): 1,2 ml na minuto. Vbrizgana količina mora biti taka, da je pri delovni občutljivosti višina vrha, ki ustreza metilnemu estru arašidove kisline enaka ali večja od 20 % uporabljene skale. |
|
6.4. |
Identifikacija različnih metilnih estrov se opravi na podlagi retencijskih časov, ki se jih primerja s časi za referenčne mešanice (kot je navedeno v točki 2.3). Estri trans maščobnih kislin eluirajo pred ustreznimi cis-izomeri. Primer kromatograma je podan na Sliki 2.
|
|
6.5. |
Učinkovitost, ki je določena skladno s točko 4.1.2. mora biti taka, da omogoči ločevanje nekaterih kritičnih dvojic, npr. dvojico gruče trans-izoleinskih kislin in vrha oleinske kisline, trans C18:1/cis C18:1, z resolucijo večjo od 2. |
|
6.6. |
Odstotni delež posameznih trans maščobnih kislin se izračuna na osnovi razmerja med površino posameznega vrha in vsoto površin vseh prisotnih vrhov. Odstotne deleže: — trans oktadecenojskih kislin (T 18:1), ki je v Prilogi I te uredbe označen kot vsota transoleinskih izomerov; — cis-trans in trans-cis oktadekadienojskih kislin [(CT/TC) 18:2], ki je v Prilogi I te uredbe označen kot vsota translinolenskih izomerov — trans-cis-trans, cis-cis-trans, cis-trans-cis, trans-cis-cis oktadekatrienojskih kislin [(TCT + CCT + CTC + TCC) 18:3], ki je v Prilogi I te uredbe označen kot vsota translinolenskih izomerov se računa. Opomba 8:Upoštevaje značilnosti te metode podajamo rezultate na dve decimalni mesti točno. |
7. POSEBEN PRIMER – UPORABA KATAROMETRSKEGA DETEKTORJA (KI DELUJE NA PRINCIPU SPREMEMB TOPLOTNE PREVODNOSTI)
Plinski kromatograf z uporabo detektorja, ki deluje na osnovi sprememb toplotne prevodnosti (katarometer), se lahko uporabi za določanje kvantitativne in kvalitativne sestave mešanice metilnih estrov maščobnih kislin. Če se ga uporabi, je treba pogoje, določene v klavzuli 3 in klavzuli 4, prilagoditi tako, kot je to prikazano v Tabeli 3.
Za kvantitativno analizo uporabite korekcijske faktorje, določene v 5.2.2.2.
Tabela 3
|
Spremenljivka |
Stanje vrednosti |
|
Kolona |
Dolžina: 2 do 4 m Notranji premer: 4 mm |
|
Nosilec |
Velikost delcev med 160 in 200 μm |
|
Koncentracija stacionarne faze |
15 do 25 % (m/m) |
|
Nosilni plin |
Helij ali, če tega ni, vodik, z najmanjšo možno vsebnostjo kisika |
|
Pomožni plini |
Noben |
|
Temperatura injektorja |
Od 40 do 60 °C nad temperaturo kolone |
|
Temperature kolone |
180 do 200 °C |
|
Tok nosilnega plina |
Običajno med 60 in 80 ml/min |
|
Velikost injiciranega vzorca |
Običajno med 0.5 in 2 μl |
8. POROČILO O PRESKUSU
V poročilu o preskusu se navedejo metode, uporabljene za pripravo metilnih estrov, za analizo s plinsko kromatografijo ter za dobljene rezultate. Navedejo se tudi vse delovne podrobnosti, ki v tem Mednarodnem standardu niso določene,ali veljajo za neobvezne, skupaj s podrobnostmi kakršnega koli dogodka, ki bi lahko vplival na rezultate.
Poročilo o preskusu vključuje vse potrebne podatke za popolno identificiranje vzorca.
ANNEX X B
PREPARATION OF THE FATTY ACID METHYL ESTERS FROM OLIVE OIL AND OLIVE-POMACE OIL
The following two methods are recommended for preparing the fatty acid methyl esters from olive oils and olive-pomace oils:
|
Method A |
: |
Trans-esterification with cold methanolic solution of potassium hydroxide |
|
Method B |
: |
Methylation by heating with sodium methylate in methanol followed by esterification in acid medium. |
Each method will be applied according to the analytical parameter to be determined and the oil category as indicated below:
(a) determination of difference between actual and theoretical content of triglycerides with ECN42 (ΔECN42):
— method A will be applied to samples of all the oil categories after purification of the oil by passing it through a silica gel column;
(b) determination of the fatty acid composition:
— method A will be applied directly to samples of the following oil categories:
—
— virgin olive oils with an acidity of less than 3,3 %,
— refined olive oil,
— olive oil (blend of virgin olive oils and refined olive oil),
— refined olive-pomace oil,
— olive-pomace oil (blend of virgin olive oils and refined olive-pomace oil);
— method B will be applied directly to samples of the following oil categories:
—
— virgin olive oil with an acidity of more than 3,3 %,
— crude olive-pomace oil;
(c) determination of trans-isomers of fatty acids:
— method A will be applied directly to samples of the following oil categories:
—
— virgin olive oils with an acidity of less than 3,3 %,
— refined olive oil,
— olive oil (blend of virgin olive oils and refined olive oil),
— refined olive-pomace oil,
— olive-pomace oil (blend of virgin olive oils and refined olive-pomace oil);
— method A will be applied to the following categories of oils after purification of the oil by passing it through a silica gel column:
—
— virgin olive oil with an acidity of more than 3,3 %,
— crude olive-pomace oil.
PURIFICATION OF OIL SAMPLES
When necessary, the samples will be purified by passing the oil through a silica gel column, eluting with hexane/diethyl ether (87:13, v/v) as described in IUPAC method 2.507.
Alternatively, solid-phase extraction on silica gel phase cartridges can be used. A silica gel cartridge (1 g, 6 ml) is placed in a vacuum elution apparatus and washed with 6 ml of hexane. The vacuum is released to prevent the column from becoming dry and then a solution of the oil (0,12 g approximately) in 0,5 ml of hexane is loaded into the column and vacuum is applied. The solution is pulled down and then eluted with 10 ml of hexane/diethyl ether (87:13 v/v) under vacuum. The combined eluates are homogenised and divided in two similar volumes. An aliquot is evaporated to dryness in a rotary evaporator under reduced pressure at room temperature. The pomace is dissolved in 1 ml of heptane and the solution is ready for fatty acid analysis by GC. The second aliquot is evaporated and the pomace is dissolved in 1 ml of acetone for triglyceride analysis by HPLC, if necessary.
METHODS FOR PREPARING THE FATTY ACID METHYL ESTERS
1. Method A: Trans-esterification with cold methanolic solution of potassium hydroxide
1.1. Purpose
This rapid method is applicable to olive oils and olive-pomace oils with a free fatty acid content of less than 3,3 %. Free fatty acids are not esterified by potassium hydroxide. Fatty acid ethyl esters are trans-esterified at a lower rate than glyceridic esters and may be only partially methylated.
1.2. Principle
Methyl esters are formed by trans-esterification with methanolic potassium hydroxide as an intermediate stage before saponification takes place (title 5 in ISO-5509:2000, title 5 in IUPAC method 2.301).
1.3. Reagents
Methanol containing not more than 0,5 % (m/m) water.
Heptane, chromatographic quality.
Potassium hydroxide, approximately 2 N methanolic solution: dissolve 11,2 g of potassium hydroxide in 100 ml of methanol.
1.4. Apparatus
Screw-top test tubes (5 ml volume) with cap fitted with a PTFE joint.
Graduated or automatic pipettes, 2 ml and 0,2 ml
1.5. Procedure
In a 5 ml screw-top test tube weigh approximately 0,1 g of the oil sample. Add 2 ml of heptane, and shake. Add 0,2 ml of 2 N methanolic potassium hydroxide solution, put on the cap fitted with a PTFE joint, tighten the cap, and shake vigorously for 30 seconds. Leave to stratify until the upper solution becomes clear. Decant the upper layer containing the methyl esters. The heptane solution is suitable for injection into the gas chromatograph. It is advisable to keep the solution in the refrigerator until gas chromatographic analysis. Storage of the solution for more than 12 hours is not recommended.
2. Method B: Methylation by heating with sodium methylate in methanol followed by esterification in acid medium
2.1. Purpose
This method is applicable to olive oils and olive-pomace oils with a free fatty acid content of more than 3,3 %.
2.2. Principle
Neutralisation of the free fatty acids and alkaline methanolysis of the glycerides, followed by esterification of the fatty acids in acid medium (title 4.2. in IUPAC method 2.301).
2.3. Reagents
— heptane, chromatographic quality,
— methanol containing not more than 0,05 % (m/m) water,
— sodium methylate, 0,2 N methanolic solution: dissolve 5 g of sodium in 1 000 ml of methanol (this may be prepared from commercial solutions),
— phenolphthalein, 0,2 % methanolic solution,
— sulphuric acid, 1 N in methanolic solution: add 3 ml of 96 % sulphuric acid to 100 ml of methanol,
— saturated solution of sodium chloride in water.
2.4. Apparatus
— 50 ml flat-bottomed volumetric flask with long, narrow, ground neck,
— reflux condenser: air condenser (1 m long) with ground joint appropriate to the neck of the flask,
— boiling chips,
— glass funnel.
2.5. Procedure
Transfer about 0,25 g of the oil sample into a 50 ml ground-necked volumetric flask. With the aid of a funnel, add 10 ml of 0,2 N sodium methylate in methanol and the boiling chips. Fit a reflux condenser, shake, and bring to the boil. The solution should become clear, which usually occurs in about 10 minutes. The reaction is complete after 15 minutes. Remove the flask from the source of heat, wait until the reflux stops, remove the condenser, and add two drops of phenolphthalein solution. Add a few ml of 1 N sulphuric acid in methanol solution until the solution becomes colourless and then add 1 ml in excess. Fit the condenser and boil again for 20 minutes. Withdraw from the source of heat and cool the flask under running water. Remove the condenser, add 20 ml of saturated sodium chloride solution, and shake. Add 5 ml of heptane, plug the flask, and shake vigorously for 15 seconds.
Leave to settle until the two phases have separated. Add saturated sodium chloride solution again until the aqueous layer reaches the lower end of the flask neck. The upper layer containing the methyl esters fills the flask neck. This solution is ready to be injected in the GC.
Caution: Methylation by method B must be done under a hood.
2.6. Alternatives to methylation Method B
2.6.1. Method C
2.6.1.1. Principle
The fatty matter undergoing analysis is treated with methanol-hydrochloric acid, in a sealed vial, at 100 °C.
2.6.1.2. Apparatus
— Strong glass vial of a capacity of about 5 ml (height 40 to 45 mm, diameter 14 to 16 mm).
— 1 and 2 ml graduated pipettes.
2.6.1.3. Reagents
Solution of hydrochloric acid in 2 % methanol. This is prepared from gaseous hydrochloric acid and anhydrous methanol (Note 1).
Hexane, chromatographic quality.
Commercial solutions of hydrogen chloride in methanol can be used. Small amounts of gaseous hydrochloric acid can easily be prepared in the laboratory by simple displacement from the commercial solution (p = 1,18) by dripping concentrated sulphuric acid. Since hydrochloric acid is very rapidly absorbed by methanol, it is advisable to take the usual precautions when dissolving it, e.g. introduce the gas through a small inverted funnel with the rim just touching the surface of the liquid. Large quantities of methanolic hydrochloric acid solution can be prepared in advance, as it keeps perfectly in glass-stoppered bottles stored in the dark. Alternatively, this reagent can be prepared by dissolution of acetyl chloride in anhydrous methanol.
2.6.1.4. Procedure
— Place in the glass vial 0,2 g of the fatty matter, which has previously been dried out on sodium sulphate and filtered, and 2 ml of hydrochloric acid-methanol solution. Heat seal the vial.
— Immerse the vial at 100 °C for 40 minutes.
— Cool the vial under running water, open, add 2 ml of distilled water and 1 ml of hexane.
— Centrifuge and remove the hexane phase, which is ready for use.
2.6.2. Method D
2.6.2.1. Principle
The fatty matter undergoing analysis is heated under reflux with methanol-hexane-sulphuric acid. The methyl esters obtained are extracted with petroleum ether.
2.6.2.2. Apparatus
— Test tube of a capacity of about 20 ml, fitted with an air reflux condenser approximately 1 m in length, with ground glass joints.
— 5 ml graduated pipette.
— 50 ml separating funnel.
— 10 ml and 25 ml measuring beakers.
— 15 ml test tube with conical base.
2.6.2.3. Reagents
— Methylation reagent: anhydrous methanol-hexane-concentrated sulphuric acid (p = 1,84) in the ratio 75:25:1 (V/V/V).
— 40 to 60 °C petroleum ether.
— Anhydrous sodium sulphate.
2.6.2.4. Procedure
Place 0,1 g of oil in the 20 ml test tube and add 5 ml of methylation reagent.
Fit the reflux condenser and heat for 30 minutes in a boiling water bath (Note 2).
Transfer quantitatively the mixture into a 50 ml separating funnel, with the aid of 10 ml distilled water and 10 ml petroleum ether. Shake vigorously, and allow the phases to separate, remove the aqueous phase and wash the ether layer twice with 20 ml distilled water. Add to the separating funnel a small quantity of anhydrous sodium sulphate, shake, allow to settle for a few minutes and filter, collecting the filtrate in a 15 ml test tube with a conical base.
Evaporate the solvent over a water bath in a current of nitrogen.
Note 2: To control boiling, insert a glass rod into the test tube and limit the temperature of the water bath to 90 °C.
3. Precision parameters
The statistical evaluation of the precision of methods A and B was published by the International Olive Oil Council in its method COI/T.20/CO. No 24.
RECOMMENDATIONS FOR GAS CHROMATOGRAPHIC ANALYSIS OF THE FATTY ACID ESTERS FROM OLIVE OIL AND OLIVE-POMACE OIL
1. Procedure
The gas chromatographic analysis of solutions of fatty esters in heptane is to be carried out according to standard ISO-5508 using a capillary column (50 m length x 0,25 or 0,32 mm i.d.) impregnated with cyanopropylsilicone phase as indicated for the determination of fatty acid trans-isomers (COI/T.20/Doc. no. 17).
Figure 1 gives the typical gas chromatographic profile of an olive-pomace oil containing methyl and ethyl esters of fatty acids, and trans-isomers of methyl esters.
2. Calculations
|
2.1. |
For the calculation of the fatty acid composition and ΔECN42, all the following fatty acids will be taken into account: Myristic (C14:0). Palmitic (C16:0). Sum of the areas of the peaks corresponding to the methyl and ethyl esters. Palmitoleic (C16:1). Sum of the areas of the peaks corresponding to the ω9 and ω7 isomers of the methyl ester. Margaric (C17:0). Margaroleic (C17:1). Stearic (C18:0). Oleic (C18:1). Sum of the areas of the peaks corresponding to the ω9 and ω7 isomers of the methyl ester, ethyl ester, and trans-isomers of the methyl ester. Linoleic (C18:2). Sum of the areas of the peaks corresponding to the methyl and ethyl esters, and the trans-isomers of the methyl ester. Arachidic (C20:0). Linolenic (C18:3). Sum of the areas of the methyl ester and the trans-isomers of the methyl ester. Eicosenoic (C20:1). Behenic (C22:0). Lignoceric (C24:0). Squalene will not be taken into account for the calculation of the total area. |
|
2.2. |
For the calculation of the percentage of trans-C18:1 the peak corresponding to the methyl esters of this fatty acid is to be used. For the sum [trans-C18:2 + trans-C18:3], all the peaks corresponding to the trans-isomers of these two fatty acids are to be added together. For the calculation of the total area, all the peaks mentioned in 2.1. are to be taken into account (see COI/T.20/Doc. No. 17). The calculation of the percentage of each fatty acid will be carried out according to the formula:
|
PRILOGA XI
DOLOČANJE HLAPNIH HALOGENIRANIH TOPIL V OLJČNEM OLJU
1. METODA
Analiza s plinsko kromatografijo z uporabo tehnike „head space“.
2. OPREMA
|
2.1 |
Plinski kromatograf, opremljen z detektorjem na zajemanje elektronov (ECD). |
|
2.2 |
Naprava „head space“. |
|
2.3 |
Steklena kolona za plinski kromatograf, 2 m dolžine in z notranjim premerom 2 mm, z 10 % stacionarne faze OV101 (ali enakovredne) na nosilcu iz kalcinirane, kislinsko sprane in silanizirane diatomejske zemlje, velikosti delcev 80 do 100 mesh. |
|
2.4 |
Prenosni in pomožni plin: dušik za plinsko kromatografijo, primeren za detekcijo na zajemanje elektronov. |
|
2.5 |
Steklene fiole, 10- do 15-ml, s teflonsko prevleko in aluminijastim zamaškom z napravo za brizgalko. |
|
2.6 |
Objemke, ki hermetično zatesnijo. |
|
2.7 |
Steklena brizgalka 0,5- do 2-ml. |
3. REAGENTI
Standard: halogenirana topila, plinsko-kromatografske čistoče.
4. POSTOPEK
|
4.1 |
Točno natehtajte približno 3 g olja v fiolo (ni za ponovno uporabo); hermetično jo zaprite. Položite jo v termostat na 70 °C za eno uro. Z brizgalko previdno odstranite 0,2 do 0,5 ml parne faze, ki je v ravnotežju s tekočino (head space). Injicirajte to v kolono plinskega kromatografa, ki je nastavljen sledeče: — temperatura injektorja: 150 oC, — temperatura kolone: 70 do 80 oC, — temperatura detektorja: 200 do 250 oC, Lahko se uporabi druge temperature, pod pogojem, da ostanejo rezultati ekvivalentni. |
|
4.2 |
Referenčne raztopine: pripravite standardne raztopine z uporabo rafiniranega oljčnega olja, ki je brez sledu topil, s koncentracijami med 0,05 do 1 ppm (mg/kg) in ustreza domnevni vsebnosti vzorca. Halogenirana topila se lahko razredči z uporabo pentana. |
|
4.3 |
Kvantitativna določitev: naredite korelacijo med površino ali višino vrhov vzorca in standardne raztopine za koncentracijo, ki je domnevno najbližja. Če je deviacija večja od 10 %, je treba analizo ponoviti v primerjavi z drugo standardno raztopino, dokler deviacija ni znotraj 10 %. Deviacija se določi na podlagi povprečja začetnih injiciranj. |
|
4.4 |
Izračun rezultatov: v ppm (mg/kg). Meja detekcije za postopek je 0,01 mg/kg. |
PRILOGA XII
METODA MEDNARODNEGA SVETA ZA OLJČNO OLJE ZA SENZORIČNO OCENJEVANJE DEVIŠKEGA OLJČNEGA OLJA
1. NAMEN IN PODROČJE UPORABE
Ta metoda temelji na odločitvi Mednarodnega sveta za oljčno olje št. DEC-21/95-V/2007 z dne 16. novembra 2007 o spremenjeni metodi za senzorično ocenjevanje deviškega oljčnega olja. Njen namen je določiti postopek za ocenjevanje senzoričnih značilnosti deviškega oljčnega olja v smislu točke 1 Priloge XVI k Uredbi (ES) št. 1234/2007 in na podlagi teh značilnosti določiti način za razvrščanje olj. Metoda vsebuje tudi navedbe za neobvezno etiketiranje.
Opisana metoda se uporablja samo za deviško oljčno olje izključno za razvrščanje ali etiketiranje glede na intenzivnost odkritih napak, sadežnost in ostale pozitivne lastnosti, ki jih določi komisija izbranih, usposobljenih in preverjenih pokuševalcev, ki sestavljajo ocenjevalno komisijo.
2. SPLOŠNO
Kar zadeva osnovni splošni besednjak, degustacijsko sobo in kozarce za degustacijo olja ter vsa ostala vprašanja, vezana na to metodo, je priporočeno upoštevati zahteve Mednarodnega sveta za oljčno olje, zlasti odločitev št. DEC-21/95-V/2007 z dne 16. novembra 2007 o spremenjeni metodi za senzorično ocenjevanje deviškega oljčnega olja.
3. POSEBNI BESEDNJAK
3.1 Pozitivne lastnosti
Sadežno :
skupek vonja, odvisen od sorte oljk, ki izvira iz zdravih in svežih, zelenih ali zrelih sadežev, ki se zazna neposredno in/ali retronazalno.
Lastnost sadežno se označi za zeleno, če vonj spominja na zelene sadeže in značilnosti olja izvirajo iz zelenih sadežev.
Lastnost sadežno se označi za zrelo, če vonj spominjajo na zrele sadeže in značilnosti olja izvirajo iz zelenih in zrelih sadežev.
Grenko : značilen osnovni okus olja, pridobljenega iz zelenih oljk ali oljk na stopnji zorenja; okus se zaznava s papilami, ki so na korenu jezika v obliki črke V.
Pikantno : skeleč občutek, značilen za olje, proizvedeno na začetku letine, v glavnem iz še zelenih oljk, ki se lahko okuša v celotni ustni votlini in zlasti v grlu.
3.2 Negativne lastnosti
Pregreto/morklja : značilna aroma olja iz oljk, ki so bile zložene ali skladiščene v takšnih pogojih, da so dosegle visoko stopnjo anaerobne fermentacije, ali olja, ki je ob pretakanju v rezervoarjih ali sodih ostalo v stiku z usedlinami, pri katerih je prav tako prišlo do anaerobne fermentacije.
Plesnivo-vlažno : značilna aroma olja, pridobljenega iz oljk, na katerih so se zaradi večdnevnega skladiščenja v vlagi razvile plesni in kvasovke.
Zakisano/kiselkasto : značilna aroma olja, ki spominja na vino ali kis. Aroma nastane zaradi aerobne fermentacije oljk ali ostankov oljčne mase v nepravilno opranih slojnicah, kar povzroči nastanek ocetne kisline, etilacetata in etanola.
Kovinsko : aroma, ki spominja na kovino. Značilna je za olje, ki je bilo med mletjem, mešanjem, stiskanjem ali skladiščenjem dolgo v stiku s kovinskimi površinami.
Žarko : aroma olja, pri katerem je prišlo do intenzivne oksidacije.
Segreto ali zažgano : značilna aroma olja, ki so ga med predelavo preveč in/ali predolgo segrevali, zlasti med termičnim mešanjem oljčne drozge pod neustreznimi temperaturnimi pogoji.
Seno-les : značilna aroma nekaterih olj iz suhih oljk.
Grobo : grob in gost občutek pri nekaterih starih oljih, ki ob pokušanju obložijo ustno votlino.
Strojno olje : aroma olja, ki spominja na dizel, maščobe ali mineralna olja.
Rastlinska voda : aroma, ki jo olje pridobi ob daljšem stiku s fermentirano rastlinsko vodo.
Slanica : aroma olja iz oljk, hranjenih v slanici.
Športa : značilna aroma olja iz oljk, stiskanih v novih športah. Razlikuje se glede na to, ali so slojnice izdelane iz zelenega ali posušenega materiala.
Zemlja : značilna aroma olja iz oljk, ki so bile pobrane umazane z zemljo ali blatom in niso bile oprane.
Črvivo : aroma olja iz oljk, ki so jih napadle ličinke oljčne muhe (Bactrocera Oleae).
Kumara : aroma olja, ki nastane, kadar je olje predolgo neprepustno zaprto, zlasti v pločevinkah, in jo pripisujejo nastanku 2,6-nonadienala.
Vlažen les : aroma, značilna za olje iz oljčnih plodov, ki so na drevju zmrznili.
3.3 Neobvezna terminologija za etiketiranje
Vodja ocenjevalne komisije lahko na zahtevo potrdi, da so ocenjena olja skladna z opredelitvami in intervali, ki glede intenzivnosti in zaznavanja lastnosti ustrezajo sledečim izrazom:
(a) za vsako izmed pozitivnih lastnosti iz točke 3.1 (sadežno (po potrebi skupaj z izrazom zeleno ali zrelo), pikantno in grenko):
(i) izraz „intenzivno“ se lahko uporabi, če je mediana zadevne lastnosti višja od 6;
(ii) izraz „srednje“ se lahko uporabi, če je mediana zadevne lastnosti med 3 in 6;
(iii) izraz „lahko“ se lahko uporabi, če je mediana zadevne lastnosti nižja od 3;
(iv) zadevne lastnosti se lahko uporabijo brez sklica na izraze iz točk (i) do (iii), če je mediana zadevne lastnosti višja ali enaka 3;
(b) izraz „uravnoteženo“ se lahko uporabi za olje, ki ni neuravnoteženo. Izraz neuravnoteženo pomeni vonj, okus in občutek olja, pri katerem je mediana lastnosti grenko in/ali pikantno za več kot 2 točki višja od mediane lastnosti sadežno;
(c) izraz „blago olje“ se lahko uporabi za olje, pri katerem je mediana lastnosti grenko in pikantno nižja ali enaka 2.
4. OCENJEVALNA KOMISIJA
Ocenjevalna komisija je sestavljena iz vodje komisije ter osmih do dvanajstih pokuševalcev.
Vodja ocenjevalne komisije mora biti primerno usposobljen in biti poznavalec ter izkušen strokovnjak za različne vrste olja. Odgovoren je za komisijo, njeno organizacijo in delovanje, pripravo, označevanje vzorcev in predstavitev vzorcev pokuševalcem, pa tudi za zbiranje podatkov in njihovo statistično obdelavo.
Vodja ocenjevalne komisije izbere pokuševalce in skrbi za njihovo usposabljanje ter nadzira njihovo delo, s čimer zagotavlja, da so njihove sposobnosti na primerni ravni.
Pokuševalce za senzorična preverjanja oljčnega olja je treba izbrati in usposabljati na podlagi njihove sposobnosti za razlikovanje med podobnimi vzorci v skladu z navodili Mednarodnega sveta za oljčno olje za izbiro, usposabljanje in preverjanje kvalificiranih pokuševalcev deviških oljčnih olj.
Ocenjevalne komisije se morajo obvezati, da bodo sodelovale pri predvidenih senzoričnih ocenjevanjih na nacionalni ali mednarodni ravni ali na ravni Skupnosti za redno preverjanje in usklajevanje meril zaznavanja. Poleg tega morajo v primeru ocenjevalnih komisij, ki so odobrene v skladu z določbami člena 4(1) te uredbe, letno predložiti zadevni državi članici vse podatke o svoji sestavi in številu ocenjevanj, ki so jih opravile kot odobrene ocenjevalne komisije.
5. POSTOPEK ZA SENZORIČNO OCENJEVANJE IN RAZVRŠČANJE
5.1 Uporaba ocenjevalnega lista s strani pokuševalca
Dodatek A te metode vsebuje ocenjevalni list, ki ga uporablja pokuševalec.
Vsak pokuševalec iz komisije povonja in nato pokusi olje, ki ga ocenjuje. Nato v ocenjevalni list v 10-centimetrsko lestvico vnese intenzivnost zaznave vsake od negativnih in pozitivnih lastnosti ( 7 ). Če pri lastnosti sadežno zazna zelenost ali zrelost, odkljuka ustrezno polje na ocenjevalnem listu.
Če pokuševalec zazna negativne lastnosti, ki niso navedene na ocenjevalnem listu, jih navede v rubriki „Drugo“, pri čemer za njihov opis uporabi izraz ali izraze, ki so najbližji opredeljenim izrazom.
5.2 Uporaba podatkov s strani vodje ocenjevalne komisije
Vodja komisije ocenjevalcev zbere ocenjevalne liste, ki jih je izpolnil vsak izmed pokuševalcev, in preveri intenzivnost, ki je določena posameznim lastnostim; če ugotovi nepravilnost, zahteva od pokuševalca, da ponovno pregleda svoj ocenjevalni list in po potrebi ponovi pokušnjo.
Vodja ocenjevalne komisije lahko podatke vsakega pokuševalca vnese v računalniški program v skladu z metodo statističnega izračuna mediane iz dodatka B. Vnos podatkov za en vzorec se izvede s pomočjo matrike z 9 stolpci, ki ustrezajo 9 senzoričnim lastnostim, in s toliko vrsticami, kot je pokuševalcev ocenjevalne komisije.
Če v rubriki „Drugo“ vsaj 50 % ocenjevalne komisije zabeleži neko negativno lastnost, se izračuna mediana te napake in olje ustrezno razvrsti.
Vodja ocenjevalne komisije lahko potrdi, da ocenjevano olje izpolnjuje pogoje iz točke 3.3.a glede izrazov „zeleno“ in „zrelo“ samo v primeru, če pri lastnosti sadežno vsaj 50 % ocenjevalne komisije zazna zelenost ali zrelost.
Pri analizah, izvedenih v okviru preverjanja skladnosti s standardi, se opravi preskus. Če pride do ponovne analize, vodja ocenjevalne komisije zagotovi, da se analiza dvakrat ponovi. V primeru razveljavljenih analiz se ocena opravi na podlagi treh ocenjevanj. V tem primeru se mediana lastnosti izračuna na podlagi povprečja median. Vse ponovitve analiz morajo biti izvršene ob različnih pokušnjah.
5.3 Razvrstitev olj
Olje se razvrsti v spodaj navedene kategorije na podlagi mediane napak in mediane lastnosti sadežno. Mediana napak je mediana napake, ki se zazna z največjo intenzivnostjo. Madiana napak in mediana sadežnosti sta izraženi z enim decimalnim mestom, vrednost grobega koeficienta variacije, ki ju določa, pa mora biti enaka ali nižja od 20 %
Razvrstitev olja se izvede s primerjavo vrednosti mediane napak in mediane sadežnosti z referenčnimi intervali, navedenimi spodaj. Ker so bile meje intervalov določene ob upoštevanju napak metode, veljajo za absolutne. Programska oprema omogoča vidno razvrščanje v razpredelnico statističnih podatkov ali v grafični prikaz.
|
(a) |
Ekstra deviško oljčno olje : mediana napak je enaka 0, mediana lastnosti sadežno je višja od 0; |
|
(b) |
Deviško oljčno olje : mediana napak je višja od 0 in enaka ali nižja od 3,5, mediana lastnosti sadežno je višja od 0; |
|
(c) |
Lampante oljčno olje : mediana napak je višja od 3,5; ali mediana napak je enaka ali nižja od 3,5, mediana lastnosti sadežno pa je enaka 0. |
5.4 Posebni primer
Če je mediana pozitivne lastnosti, ki ni „sadežno“, višja od 5,0, vodja ocenjevalne komisije to navede v potrdilu o analizi olja.
Dodatek A
Ocenjevalni list za deviško oljčno olje

Dodatek B
METODA IZRAČUNA MEDIANE IN INTERVALOV ZAUPANJA
Mediana
![]()
Mediana je realno število Xm, za katerega je značilno, da je verjetnost (P), da so vrednosti distribucije (X) nižje od tega števila (Xm), enaka ali manjša od 0,5 in da je hkrati verjetnost (P), da so vrednosti distribucije (X) enake ali nižje od Xm, enaka ali višja od 0,5. Po drugi opredelitvi je mediana 50. percentil distribucije števil, urejenih po naraščajočem vrstnem redu. Enostavneje to pomeni, da predstavlja mediana osrednjo vrednost urejenega niza lihih števil ali povprečje dveh osrednjih vrednosti urejenega niza sodih števil.
Grobi standardni odklon
Za zanesljivo oceno variabilnosti, ki nastane okrog mediane, je treba upoštevati grobi standardni odklon po Stuartu in Kendallu. Spodnja formula izraža asimptotičen standardni odklon, to je grobo oceno variabilnosti upoštevanih podatkov, kjer je N število opažanj in IQR interkvartilni interval, ki zajema točno 50 % primerov kakršne koli distribucije verjetnosti.
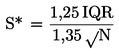
Izračun interkvartilnega intervala se izvaja tako, da se izračuna velikost razpona med 75. in 25. percentilom.
IQR = 75. percentil – 25. percentil
Percentil je vrednost Xpc, za katero je značilno, da je verjetnost (P), da so vrednosti distribucije nižje od Xpc, enaka ali manjša od določene stotine in da je hkrati verjetnost (P), da so vrednosti distribucije nižje ali enake Xpc, enaka ali višja od navedene stotine. Stotina označuje upoštevani delež distribucije. V primeru mediane je slednji enak 50/100.
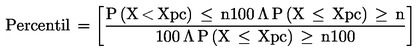
V praksi je percentil vrednost distribucije, ki ustreza določenemu območju, zarisanemu od krivulje distribucije ali gostote. Tako na primer 25. percentil predstavlja vrednost distribucije, ki ustreza območju 0,25 ali 25/100.
Grobi koeficient variacije %
CVr% predstavlja čisto število, torej brez dimenzije, ki označuje delež variabilnosti analiziranega niza števil. Zato je ta koeficient zelo koristen za preverjanje zanesljivosti članov ocenjevalne komisije.
![]()
95-odstotni intervali zaupanja pri mediani
95-odstotni intervali zaupanja (vrednost napake prve vrste je enaka 0,05 ali 5 %) predstavljajo interval, v katerem bi se vrednost mediane lahko spreminja, če bi bilo mogoče preskus ponoviti neštetokrat. V praksi ta interval označuje interval variabilnosti preskusa pod preskusnimi pogoji ob predpostavki, da se preskus lahko ponovi večkrat. Kot pri CVr% interval pomaga oceniti zanesljivost preskusa.
ICsup = Me + (c.S*)
ICinf = Me – (c.S*)
Pri tem je c v primeru intervala zaupanja pri 0,95 enak 1,96.
▼M20 —————
▼M19 —————
PRILOGA XV
1. VSEBNOST OLJA V OLJČNIH TROPINAH
1.1 Oprema
— primerna aparatura za ekstrakcijo z 200–250-ml bučko z okroglim dnom,
— električno greta kopel (na primer peščena ali vodna kopel) ali grelna plošča,
— analitska tehtnica,
— sušilnik, nastavljen na temperaturo največ 80 °C
— električno gret sušilnik s termostatom, nastavljen na 103 ± 2 °C, z možnostjo prepihovanja z zrakom pri znižanem tlaku,
— mehanski mlin, ki se lahko čisti in v katerem je možno zmleti oljčne tropine brez zvišanja njihove temperature ali kakršne koli znatne spremembe vsebnosti vlage, hlapnih snovi ali snovi, ki se lahko ekstrahirajo s heksanom,
— kartuša in vata ali filtrirni papir, s katerega so tik pred uporabo odstranjene snovi, ki se lahko ekstrahirajo s heksanom,
— eksikator,
— sito z odprtinami 1 mm,
— majhni koščki predhodno osušenega plovca.
1.2 Reagenti
Normalni heksan, v katerem po popolnem sušenje suhi ostanek ne sme biti višji od 0,002 g v 100 ml.
2. POSTOPEK
2.1 Priprava vzorca
Če je potrebno, s predhodno dobro očiščenim mehanskim mlinom zmeljemo laboratorijski vzorec do velikosti delcev, ki v celoti preidejo skozi sito.
S približno dvanajstino vzorca očistimo mlin, zmleti material zavržemo, zmeljemo preostali del vzorca in ga takoj analiziramo.
2.2 Vzorec za pokušanje
Takoj po končanem mletju natehtamo približno 10 g vzorca s točnostjo 0,01 g.
2.3 Priprava kartuše
Natehtani vzorec prenesemo v kartušo, ki jo pokrijemo z vato. Pri uporabi filtrirnega papirja vzorec v papir zavijemo.
2.4 Predhodno sušenje
Če so oljčne tropine zelo vlažne (to je, če je vsebnost hlapnih snovi višja od 10 %), izvedemo predhodno sušenje tako, da napolnjeno kartušo (ali filtrirni papir) postavimo v sušilnik, v katerem temperatura ni višja od 80 °C, da vsebnost vlage in hlapnih snovi znižamo na manj od 10 %.
2.5 Priprava bučke z okroglim dnom
S točnostjo 1 mg stehtamo bučko, v kateri se nahaja en do dva koščka plovca, ki smo ga predhodno sušili pri 103 ± 2 °C in v eksikatorju hladili eno uro.
2.6 Začetna ekstrakcija
V aparaturo za ekstrakcijo vstavimo kartušo (ali filtrirni papir) z vzorcem. V bučko vlijemo zahtevano količino heksana. Bučko spojimo z aparaturo za ekstrakcijo in jo postavimo na električno greto kopel. Hitrost gretja nastavimo tako, da je hitrost refluksa najmanj tri kaplje na sekundo (srednje, ne burno vretje). Po štirih urah ohladimo. Kartušo vzamemo iz aparature za ekstrakcijo in s tokom zraka odstranimo večji del topila.
2.7 Druga ekstrakcija
Vsebino iz kartuše prenesemo v mikro drobilec in jo čim bolje zdrobimo. Zdrobljeno mešanico kvantitativno vrnemo v kartušo, ki jo ponovno vstavimo v aparaturo za ekstrakcijo.
Nadaljujemo z ekstrakcijo nadaljnji dve uri ob uporabi iste bučke z okroglim dnom, v kateri se nahaja ekstrakt iz začetne ekstrakcije.
Dobljena raztopina v ekstrakcijski bučki mora biti bistra. Če ni, jo filtriramo skozi filtrirni papir in originalno bučko ter filtrirni papir nekolikokrat speremo s heksanom. Filtrat in raztopino za izpiranje lovimo v drugo predhodno osušeno bučko, stehtano s točnostjo 1 mg.
2.8 Odstranjevanje topila in tehtanje ekstrakta
Večino topila odstranimo z destilacijo na električno greti kopeli. Zadnje ostanke topila odstranimo s segrevanjem bučke v sušilniku pri 103 ± 2 °C 20 minut. Odstranjevanje topila lahko pospešimo s prepihovanjem z zrakom ali, še bolje, z inertnim plinom v časovnih presledkih ali pri znižanem tlaku.
Bučko hladimo v eksikatorju najmanj eno uro in jo nato stehtamo s točnostjo 1 mg.
Pod enakimi pogoji ponovno grejemo 10 minut, ohladimo v eksikatorju in ponovno tehtamo.
Razlika med obema tehtanjema ne sme biti večja od 10 mg. Če je večja, ponovno grejemo 10 minut, ohladimo in stehtamo ter postopek ponavljamo, dokler razlika med dvema tehtanjema ni manjša od 10 mg. Zabeležimo zadnjo maso bučke.
Izvedemo dve vzporedni določitvi.
3. IZRAŽANJE REZULTATOV
3.1 Metoda izračuna in formula
(a) Ekstrakt, izražen kot odstotni masni delež glede na izdelek, kot ga je prejel laboratorij:
![]()
|
kjer je: |
S = odstotni masni delež ekstrakta v izdelku, kot ga je prejel laboratorij, m0 = masa preiskovanega vzorca, v g, m1 = masa ekstrakta po sušenju, v g. |
Rezultat je aritmetična sredina dveh vzporednih določitev, pod pogojem, da so izpolnjeni kriteriji ponovljivosti.
Rezultat izrazimo z enim decimalnim mestom.
(b) Ekstrakt glede na suho snov izrazimo po formuli:
![]()
|
kjer je |
: |
S = odstotni masni delež ekstrakta izdelka, kot ga je prejel laboratorij (glej a), U = vsebnost vlage in hlapnih snovi. |
3.2 Ponovljivost
Razlika med dvema vzporednima določitvama, ki jih izvede isti analitik istočasno ali v kratkem časovnem intervalu, ne sme biti večja od 0,2 g heksanskega ekstrakta v 100 g vzorca.
Če ta pogoj ni izpolnjen, analizo ponovimo z dvema drugima deloma vzorca. Če je tudi v tem primeru razlika večja od 0,2 g, je rezultat srednja vrednost štirih določitev.
Priloga XVI
DOLOČANJE JODNEGA ŠTEVILA
1. NAMEN
V tem mednarodnem standardu je opisana metoda za določanje jodnega števila v maščobah in oljih živalskega in rastlinskega izvora, v nadaljevanju maščobah.
2. DEFINICIJA
Za namene tega mednarodnega standarda se uporablja naslednja opredelitev:
|
2.1 |
Jodno število. Jodno število je masa joda, ki jo absorbira vzorec pri pogojih dela, kot so opisani v tem mednarodnem standardu. Jodno število izrazimo v gramih joda na 100 g vzorca. |
3. PRINCIP
Vzorec raztopimo v topilu ob dodatku Wijs-ovega regenta. Po določenem času dodamo raztopino kalijevega jodida v vodi in sproščeni jod titriramo z raztopino natrijevega tiosulfata.
4. REAGENTI
Vsi reagenti morajo biti priznane čistoče za analizo.
|
4.1 |
Voda, stopnja 3, mora ustrezati zahtevam ISO 3969. |
|
4.2 |
Kalijev jodid, 100g/L, brez jodata ali prostega joda. |
|
4.3 |
Raztopina škroba. 5 g škroba zmešamo s 3 ml vode in to mešanico vlijemo v 1 000 ml vrele vode ter pustimo vreti tri minute. Ohladimo. |
|
4.4 |
Natrijev tiosulfat, standardna raztopina, c (Na2S2O3.5H2O) = 0,1 mol/l, pred uporabo standardizirana (manj kot sedem dni). |
|
4.5 |
Topilo, pripravljeno z mešanjem enakih delov cikloheksana in ocetne kisline. |
|
4.6 |
Wijsov reagent, ki vsebuje jodov monoklorid v ocetni kislini. Uporabljati moramo komercialno dostopen reagent. |
5. OPREMA
Običajna laboratorijska oprema, posebno:
|
5.1 |
Steklene ladjice za tehtanje, primerne za količino vzorca in za prenos vzorca v bučko (6.2). |
|
5.2 |
Erlenmajerice, 500-ml, z brušenim zamaškom, popolnoma suhe. |
6. PRIPRAVA VZORCA ZA ANALIZO
Homogeniziran vzorec osušimo nad natrijevim sulfatom in filtriramo.
7. POSTOPEK
|
7.1 |
Vzorec za analizo Masa vzorca za analizo je odvisna od pričakovanega jodnega števila, kot je prikazano v tabeli 1.
Tabela 1
Vzorec natehtamo v stekleno ladjico s točnostjo 0,1 mg. |
|
7.2 |
Določitev Vzorec za analizo prenesemo v 500-ml erlenmajerico (6.2). Dodamo 20 ml topila (4.5), da raztopimo maščobo. Dodamo točno 25 ml Wijs-ovega reagenta (4.6), zamašimo, premešamo vsebino in erlenmajerico postavimo na temno. Za Wijs-ov regent ne smemo uporabiti ustne pipete! Na enak način pripravimo s topilom in reagentom slepo probo brez vzorca. Vzorce z jodnim številom nižjim od 150 pustimo stati na temnem eno uro; vzorce z jodnim številom višjim od 150, polimerizirane izdelke in znatno oksidirane izdelke pustimo na temnem dve uri. Po pretečenem času dodamo v vsako od erlenmajeric po 20 ml raztopine joda (4.2) in 150 ml vode (4.1). Titriramo s standardno raztopino natrijevega tiosulfata (4.4), dokler ne izgine rumena barva joda. Dodamo nekaj kapljic raztopine škroba (4.3) in nadaljujemo s titracijo, dokler po močnem stresanju ne izgine modra barva. Opomba: Dopustna je potenciometrična titracija. |
|
7.3 |
Število določitev Na istem vzorcu izvedemo dve določitvi. |
8. IZRAŽANJE REZULTATOV
Jodno število podaja izraz
![]()
kjer je:
|
c |
= |
numerična vrednost uporabljene standardne raztopine natrijevega tiosulfata (4.4) s točno koncentracijo v mol/L; |
|
V1 |
= |
volumen standardne raztopine natrijevega tiosulfata (4.4), porabljen pri slepi probi, v ml; |
|
V2 |
= |
volumen standardne raztopine natrijevega tiosulfata (4.4), porabljen pri določitvi, v ml; |
|
m |
= |
masa vzorca, v g (7.1). |
Rezultat je aritmetična sredina dveh določitev, pod pogojem, da so izpolnjeni pogoji ponovljivosti (9.2).
PRILOGA XVII
METODA ZA DOLOČANJE STIGMASTADIENOV V RASTLINSKIH OLJIH
1. NAMEN
Določitev stigmastadienov v rastlinskih oljih z nizko vsebnostjo teh ogljikovodikov, predvsem v deviškem oljčnem olju in v surovem olju iz oljčnih tropin.
2. PODROČJE
Metodo lahko uporabljamo za vsa rastlinska olja, čeprav so določitve zanesljive le v primerih, ko je vsebnost teh ogljikovodikov med 0,01 in 4,0 mg/kg. Metoda je še posebej primerna za ugotavljanje prisotnosti rafiniranih rastlinskih olj (oljčnih, oljčnih tropin, sončničnih, palmovih itd.) v deviških oljčnih oljih, kajti rafinirana olja vsebujejo stigmastadiene, deviška olja pa ne.
3. PRINCIP
Izolacija neumiljivih snovi. Ločitev steroidne ogljikovodične frakcije s kolonsko kromatografijo na silikagelu ter analiza s kapilarno plinsko kromatografijo.
4. OPREMA
|
4.1 |
250-ml bučke za povratni hladilnik. |
|
4.2 |
500-ml liji ločniki. |
|
4.3 |
100-ml bučke z okroglim dnom. |
|
4.4 |
Rotavapor. |
|
4.5 |
Steklena kromatografska kolona (notranjega premera 1,5 do 2,0 cm in dolžine 50 cm) s teflonskim petelinčkom in kosmom steklene volne ali sintrano stekleno ploščico na dnu. Za uporabo jo pripravimo tako, da vanjo vlijemo toliko heksana, da sega 5 cm od njenega dna, nato pa jo napolnimo s suspenzijo silikagela v heksanu (15 g v 40 ml) s pomočjo dodatkov heksana. Počakamo, da se silikagel usede, zatem pa z uporabo rahlih vibracij silikagel dokončno posedemo. Dodamo brezvodni natrijev sulfat do višine približno 0,5 cm in končno izpustimo odvečni heksan. |
|
4.6 |
Plinski kromatograf, opremljen s plamensko ionizacijskim detektorjem, s split injektorjem ali injektorjem, ki omogoča neposredne vbrizge na začetek (hladne) kolone (cold-on-column) in z možnostjo temperaturne nastavitve na ± 1 °C. |
|
4.7 |
Kvarčna kapilarna kolona za plinsko kromatografijo (z notranjim premerom 0,25 ali 0,32 mm in dolžino 25 m), z notranjim nanosom 5 % fenilmetilsilikonske faze v debelini 0,25 mm. Opomba 1: Uporabimo lahko tudi kolone s podobno ali manjšo polarnostjo. |
|
4.8 |
Integrator-rekorder z možnostjo uporabe integracijskega načina dolina-dolina. |
|
4.9 |
5 do 10-μl mikrobrizgalke za plinsko kromatografijo z nesnemljivo iglo. |
|
4.10 |
Električni grelni plašč ali grelna površina. |
5. REAGENTI
Vsi reagenti morajo biti analizne čistoče, razen če ni drugače določeno. Voda, ki se uporabi, mora biti destilirana ali vsaj enakovredne čistoče.
|
5.1 |
Heksan ali mešanica alkanov z intervalom vrelišč 65 do 70 °C, destiliran z ločevalno kolono. Opomba 2: Topilo moramo destilirati, da odstranimo nečistoče. |
|
5.2 |
96 % v/v etanol. |
|
5.3 |
Brezvodni natrijev sulfat. |
|
5.4 |
10 % alkoholna raztopina kalijevega hidroksida. K 50 g kalijevega hidroksida dodamo 10 ml vode in mešanico raztopimo v etanolu, da je skupni volumen 500 ml. Opomba 3: Etanolni kalijev hidroksid sčasoma porjavi, zato ga moramo pripravljati dnevno in hraniti v dobro zaprtih temnih steklenicah. |
|
5.5 |
Silikagel 60 za kolonsko kromatografijo, 70 do 239 mesh, (Merck, kat. št. 7734 ali podoben). Opomba 4: Ponavadi je silikagel primeren za takojšnjo uporabo. Včasih pa lahko s svojo šibko aktivnostjo vpliva na poslabšanje kromatografskega ločevanja. V takih primerih ga je treba obdelati na naslednji način: aktiviramo ga tako, da ga grejemo vsaj štiri ure pri 550 °C. Po segrevanju damo silikagel v eksikator medtem, ko se hladi, in ga nato prenesemo v bučko z obrusom. Dodamo 2 % vode ter zmes tako premešamo, da v njej ni več grudic in da je sipka. Če se kromatografski vrhovi prekrivajo, moramo silikagel aktivirati na zgoraj opisani način. Kot alternativa se lahko uporabi posebno čist silikagel 60 (Merck, kat. št. 7754). |
|
5.6 |
Koncentrirana raztopina (200 ppm) holesta-3,5-diena (Sigma, čistoče 99 %) v heksanu (10 mg v 50 ml). |
|
5.7 |
Standardna raztopina holesta-3,5-diena v heksanu koncentracije 20 ppm, ki jo dobimo z redčenjem gornje raztopine. Opomba 5: Raztopini iz 5.6 in 5.7 sta stabilni vsaj štiri mesece, če ju hranimo pri temperaturi manj kot 4 °C. |
|
5.8 |
Raztopina n-nonakozana v heksanu koncentracije približno 100 ppm. |
|
5.9 |
Nosilni plin za kromatografijo: helij ali vodik, čistoče 99,9990 %. |
|
5.10 |
Pomožna plina za plamensko ionizacijski detektor: vodik čistoče 99,9990 % in očiščen zrak. |
6. POSTOPEK
6.1 Priprava neumiljivih snovi
|
6.1.1 |
V 250-ml bučko (4.1) natehtamo 20 ± 0, 1 g olja, dodamo 1 ml standardne raztopine holesta-3,5-diena (20 μg), 75 ml 10 % alkoholne raztopine kalijevega hidroksida, jo povežemo s povratnim hladilnikom ter ob šibkem vrenju refluktiramo 30 minut. Odstranimo bučko, ki vsebuje vzorec, in rahlo ohladimo (ne preveč, sicer se vzorec usede). Dodamo 100 ml vode in raztopino s pomočjo 100 ml heksana prelijemo v lij ločnik (4.2). Mešanico močno stresamo 30 sekund in počakamo, da se fazi ločita. Opomba 6: Če se tvori emulzija, ki ne izgine hitro, dodamo manjše količine etanola. |
|
6.1.2 |
Spodnjo vodno fazo prenesemo v drug lij ločnik in jo ponovno ekstrahiramo s 100 ml heksana. Spet odločimo vodno fazo in speremo heksanska ekstrakta (združena v drugem liju ločniku) trikrat s po 100 ml mešanice etanol-voda (1:1) dokler ne dosežemo nevtralnega pH. |
|
6.1.3 |
Heksansko raztopimo osušimo tako, da jo prelijemo prek 50 g brezvodnega natrijevega sulfata, ki ga speremo z 20 ml heksana in nato na rotavaporju pri 30 °C odparimo ves heksan. |
6.2 Ločitev steroidne ogljikovodične frakcije
|
6.2.1 |
S pomočjo dveh 1-ml dodatkov heksana prenesemo suhi ostanek v kolono, damo vzorec v kolono tako, da raven raztopine pade na vrh natrijevega sulfata in začnemo s kromatografsko elucijo s heksanom pri pretoku približno 1 ml/min. Zavržemo prvih 25 do 30 ml eluata in nato zberemo naslednjo 40-ml frakcijo. Po zbiranju prestavimo to frakcijo v 100-ml bučko z okroglim dnom (4.3). Opomba 7: Prva frakcija vsebuje nasičene ogljikovodike (slika 1a) in druga steroidne ogljikovodike. Preostale frakcije pa vsebujejo še skvalen in skvalenu sorodne spojine. Če hočemo dobro ločiti steroidne ogljikovodike od nasičenih, moramo poskrbeti za optimizacijo frakcijskih volumnov. Volumen prve frakcije mora biti tolikšen, da so vrhovi nasičenih ogljikovodikov pri analizi druge frakcije nizki (glej sliko 1c). Če le-teh ni, intenziteta vrha internega standarda pa je nizka, moramo volumen zmanjšati. Popolna ločitev med spojinami iz prve in druge frakcije ni potrebna; kajti vrhovi se med GC analizo ne prekrivajo, če so pogoji GC prilagojeni, kot je navedeno v odstavku 6.3.1. Optimizacija volumna druge frakcije v glavnem ni potrebna, saj obstaja dobra ločitev z nadaljnjimi komponentami. Kljub temu pa je prisotnost velikega vrha skvalena s približno 1,5 minute krajšim retencijskim časom od retencijskega časa vrha internega standarda dokaz za slabo ločevanje. |
|
6.2.2 |
Drugo frakcijo odparimo na rotavaporju pri 30 °C pod zmanjšanim pritiskom in preostanek takoj raztopimo v 0,2 ml heksana. Raztopino hranimo v hladilniku do analize. Opomba 8: Preostankov iz 6.1.3 in 6.2.2 ne smemo hraniti na sobni temperaturi v suhi obliki. Takoj ko ju pridobimo, jima moramo dodati topilo in raztopino shraniti v hladilniku. |
6.3 Plinska kromatografija
|
6.3.1 |
Delovni pogoji za split injiciranje: — temperatura injektorja: 300 °C, — temperatura detektorja: 320 °C, — integrator-rekorder: integracijski parametri morajo biti takšni, da omogočajo točno določitev površin. Priporočljiva je integracija dolina-dolina (valley-valley), — občutljivost: 16-kratna najmanjša atenuacija, — količina injicirane raztopine: 1 μl, — programiranje temperature peči: začetna temperatura 235 °C za šest minut in nato segrevanje s hitrostjo 2 °C/minuto do 285 °C, — injektor z 1: 15 razdelilcem pretoka, — nosilni plin: helij ali vodik s pritiskom približno 120 kPa. Te pogoje lahko prilagodimo v skladu z značilnostmi kromatografa in kolone tako, da dobimo kromatograme, ki ustrezajo naslednjim merilom: retencijski čas internega standarda se ne sme razlikovati za več kot pet minut od časa, ki je podan v 6.3.2; višina vrha internega standarda mora segati vsaj do 80 % višine polne skale. Plinsko kromatografski sistem moramo preveriti še z injiciranjem mešanice koncentrirane raztopine holestadiena (5.6) in raztopine n-nonakozana (5.8). Retencijski čas holesta-3,5-diena mora biti manjši od retencijskega časa n-nonakozana (slika 1c). Če temu ni tako, lahko to popravimo na dva načina: ali znižamo temperaturo kolone in/ali uporabimo manj polarno kolono. |
|
6.3.2 |
Identifikacija vrhov Vrh internega standarda se pojavlja na približno 19 minut, relativni retencijski čas 3,5-stigmastadiena pa je približno 1,29 (glej sliko 1b). 3,5-stigmastadien se pojavlja v spremstvu manjših količin svojega izomera; oba ponavadi eluirata kot en kromatografski vrh. Če pa je kromatografska kolona preveč polarna ali pa zelo dobro ločuje, se lahko izomer pojavi kot majhen vrh pred in blizu stigmasta-3,5-diena (slika 2). Da se zagotovi, da so stigmastadieni eluirani kot en vrh, je priporočljivo kolono zamenjati z manj polarno ali pa s tako, ki ima večji notranji premer. Opomba 9: Referenčne stigmastadiene lahko dobimo z analizo rafiniranega rastlinskega olja z uporabo manjše količine vzorca (1 do 2 g). Stigmastadieni tvorijo značilen in dobro viden vrh. |
|
6.3.3 |
Kvantitativna analiza Vsebina stigmastadienov se določi v skladu s formulo: mg/kg stigmastadienov =
Meja detekcije: približno 0,01 mg/kg. |

 Slika 1
Slika 1
Plinski kromatogrami vzorcev oljčnega olja, analizirani na kapilarni koloni (z notranjim premerom 0,25 mm, dolžine 25 m) z notranjim nanosom 5 % -fenilmetilsilikonske faze v debelini 0,25 μm).
(a) Prva frakcija (30 ml) iz deviškega olja z dodanim standardom.
(b) Druga frakcija (40 ml) iz oljčnega olja z vsebnostjo 0,10 mg/kg stigmastadienov.
(c) Druga frakcija (40 ml), ki vsebuje nizek delež prve frakcije.
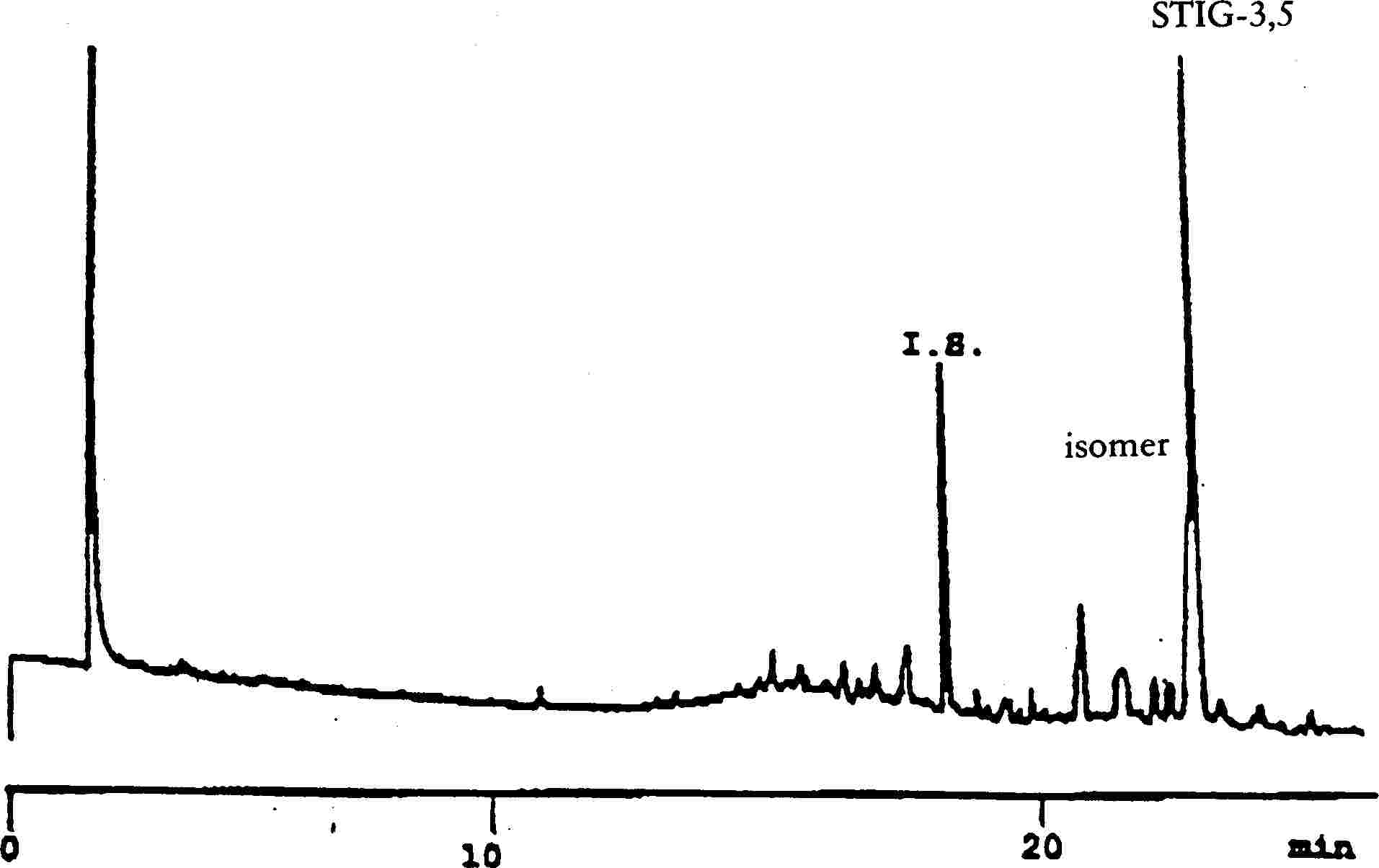 Slika 2
Slika 2
Plinski kromatogram, pridobljen iz vzorca rafiniranega oljčnega olja, analiziranega na DB-5 koloni, kaže izomer 3,5-stigmastadiena.
PRILOGA XVIII
DOLOČANJE RAZLIKE MED DEJANSKO IN TEORETIČNO VSEBNOSTJO TRIACILGLICEROLOV Z EKVIVALENTNIM OGLJIKOVIM ŠTEVILOM (ECN) 42
1. NAMEN
Določanje absolutne razlike med eksperimentalnimi vrednostmi triacilglicerolov (TAG) z ekvivalentnim ogljikovim številom 42 (ECN42HPLC) v olju, dobljenimi s tekočinsko kromatografijo visoke ločljivosti, in teoretično vrednostjo triacilglicerolov z ekvivalentnim ogljikovim številom 42 (ECN 42teoretično), izračunano na podlagi sestave maščobnih kislin.
2. PODROČJE UPORABE
Standard se uporablja za oljčna olja. Metoda se uporablja za odkrivanje prisotnosti majhnih količin semenskih olj (bogatih z linolno kislino) v vsaki posamezni kategoriji oljčnih olj.
3. PRINCIP
Vsebnost triacilglicerolov z ECN 42, določena z analizo tekočinske kromatografije visoke ločljivosti (HPLC), in teoretična vsebnost triacilglicerolov z ECN 42 (izračunana na podlagi določitve sestave maščobnih kislin s plinsko-tekočinsko kromatografijo) sta v okviru določene mejne vrednosti za pristna oljčna olja. Če je razlika večja od vrednosti, sprejetih za posamezno vrsto olja, to pomeni, da olje vsebuje semenska olja.
4. METODA
Metoda za izračun teoretične vsebnosti triacilglicerolov z ECN 42 in razlike glede na podatke tekočinske kromatografije visoke ločljivosti se v bistvu izvaja z uskladitvijo analitičnih podatkov, dobljenih z drugimi metodami. Razlikovati je mogoče med tremi fazami: določanjem sestave maščobnih kislin s kapilarno plinsko kromatografijo, izračunom teoretične sestave triacilglicerolov z ECN 42 in določanjem triacilglicerolov z ECN 42 s tekočinsko kromatografijo visoke ločljivosti.
4.1 Oprema
|
4.1.1 |
Bučke z okroglim dnom, 250 in 500 ml. |
|
4.1.2 |
Čaše, 100 ml. |
|
4.1.3 |
Steklena kromatografska kolona z notranjim premerom 21 mm, dolžine 450 mm, s petelinčkom in standardiziranim konusom (z notranjim obrusom) na vrhu. |
|
4.1.4 |
Liji ločniki, 250 ml, s standardiziranim konusom (z zunanjim obrusom) na dnu, primerni za pritrditev na vrh kolone. |
|
4.1.5 |
Steklena paličica dolžine 600 mm. |
|
4.1.6 |
Stekleni lij s premerom 80 mm. |
|
4.1.7 |
Merilne bučke, 50 ml. |
|
4.1.8 |
Merilne bučke, 20 ml. |
|
4.1.9 |
Rotacijski izparilnik. |
|
4.1.10 |
Kromatograf za tekočinsko kromatografijo visoke ločljivosti, ki omogoča termostatski nadzor temperature kolone. |
|
4.1.11 |
Injektorji za vnos 10 μl. |
|
4.1.12 |
Detektor: diferencialni refraktometer. Občutljivost celotne skale mora biti najmanj 10–4 enote lomnega količnika. |
|
4.1.13 |
Kolona: jeklena cevka, dolga 250 mm in z notranjim premerom 4,5 mm, polnjena z delci silicijevega dioksida premera 5 μm, z 22 do 23 % ogljika v obliki oktadecilsilana. |
|
4.1.14 |
Programska oprema za obdelavo podatkov. |
|
4.1.15 |
Stekleničke (viale) s prostornino približno 2 ml, s teflonskimi tesnili in navojnimi zamaški. |
4.2 Reagenti
Reagenti morajo biti analitsko čisti. Elucijska topila morajo biti razplinjena in se jih lahko večkrat reciklira brez vpliva na ločevanje.
|
4.2.1 |
Petroleter 40–60 °C za kromatografijo ali heksan. |
|
4.2.2 |
Sveže destilirani etil eter brez peroksidov. |
|
4.2.3 |
Elucijsko topilo za čiščenje olja s kolonsko kromatografijo, mešanica petroletra/etil etra 87/13 (v/v). |
|
4.2.4 |
Silikagel, 70–230 mesh, tip Merck 7734, s standardizirano 5-odstotno vsebnostjo vode (w/w/). |
|
4.2.5 |
Steklena volna. |
|
4.2.6 |
Aceton za HPLC. |
|
4.2.7 |
Acetonitril ali propionitril za HPLC. |
|
4.2.8 |
Elucijsko topilo za HPLC: acetonitril + aceton (treba je prilagoditi deleže, da se doseže želeno ločevanje; začnite z mešanico 50: 50) ali propionitril. |
|
4.2.9 |
Topilo za raztapljanje vzorca: aceton. |
|
4.2.10 |
Referenčni trigliceridi: uporabijo se lahko komercialni trigliceridi (tripalmitin, triolein itd.) in se potem retencijski časi zapisujejo glede na ekvivalentno ogljikovo število; ali alternativno, referenčni kromatogram, dobljen s sojinim oljem, mešanico 30: 70 sojino olje – oljčno olje in čistim oljčnim oljem (glej opombi 1 in 2 ter slike 1 do 4). |
|
4.2.11 |
6-mililitrska kolona za ekstrakcijo v trdni fazi, s fazo 1 g silikagela. |
4.3 Priprava vzorcev
Ker lahko zaradi številnih motečih snovi dobimo lažno pozitivne rezultate, je treba vzorec vedno očistiti po metodi IUPAC št. 2507, ki se uporablja za določanje polarnih spojin v maščobah za cvrtje.
4.3.1 Priprava kromatografske kolone
V kolono (4.1.3) vlijemo približno 30 ml elucijskega topila (4.2.3), nato v kolono vstavimo nekaj steklene volne (4.2.5) in jo s stekleno paličico potisnemo na dno (4.1.5).
V 100-mililitrski čaši raztopimo 25 g silikagela (4.2.4) v 80 ml elucijske mešanice (4.2.3.), nato jo skozi stekleni lij prenesemo v kolono (4.1.6).
Da zagotovimo popoln prenos silikagela v kolono, speremo čašo z elucijsko mešanico in tudi to tekočino prenesemo v kolono.
Odpremo petelinček in pustimo, da topilo eluira iz kolone, dokler ne sega približno 1 cm nad silikagel.
4.3.2 Kolonska kromatografija
Z natančnostjo 0,001 g natehtamo 2,5 ± 0,1 g olja, po potrebi predhodno prefiltriranega, homogeniziranega in dehidriranega, v 50-mililitrsko merilno bučko (4.1.7).
Raztopimo ga v približno 20 ml elucijskega topila (4.2.3). Po potrebi nekoliko segrejemo, da pospešimo raztapljanje. Ohladimo pri sobni temperaturi in volumen prilagodimo z elucijskim topilom.
Z merilno pipeto vnesemo 20 ml raztopine v kolono, pripravljeno v skladu s točko 4.3.1, odpremo petelinček in pustimo, da topilo eluira do višine plasti silikagela.
Nato eluiramo s 150 ml elucijskega topila (4.2.3.), pri čemer hitrost pretoka topila prilagodimo na približno 2 ml/min (150 ml bo šlo skozi kolono v približno 60–70 minutah).
Eluat se zbira v 250-mililitrski bučki z okroglim dnom (4.1.1), ki je bila predhodno tarirana v peči in natančno stehtana. Topilo odstranimo pri znižanem tlaku v rotacijskem izparilniku (4.1.9) in stehtamo ostanek, ki se bo uporabil za pripravo raztopine za analizo HPLC in pripravek metil estra.
Izkoristek vzorca iz kolone mora biti najmanj 90-odstoten za kategorije oljčnega olja ekstra deviško, deviško, navadno in rafinirano, ter najmanj 80-odstoten za lampante oljčno olje in olje iz oljčnih tropin.
4.3.3 Čiščenje z ekstrakcijo v trdni fazi
Kolona s silikagelom za ekstrakcijo v trdni fazi se aktivira s 6 ml heksana (4.2.3) v vakuumu, pri čemer je treba preprečiti izsušitev.
Z natančnostjo 0,001 g natehtamo 0,12 g v 2-mililitrsko vialo (4.1.15) in razredčimo z 0,5 ml heksana (4.2.3).
Raztopino vnesemo v kolono za ekstrakcijo v trdni fazi in eluiramo z 10 ml heksana/dietil etra (87: 13 v/v) (4.2.3) v vakuumu.
Zbrana frakcija se v rotacijskem izparilniku (4.1.9) odpari do suhega pod znižanim tlakom pri sobni temperaturi. Ostanek se raztopi v 2 ml acetona (4.2.6) za analizo triacilglicerolov (TAG).
4.4 Analiza HPLC
4.4.1 Priprava vzorcev za kromatografsko analizo
5-odstotno raztopino vzorca za analizo pripravimo tako, da v 10-mililitrsko merilno bučko natehtamo 0,5 ± 0,001 g vzorca in dolijemo topilo do oznake 10 ml (4.2.9).
4.4.2 Postopek
Pripravimo kromatografski sistem. Elucijsko topilo (4.2.8) črpamo s hitrostjo 1,5 ml/min, da očistimo celotni sistem. Počakamo, da dobimo stabilno bazno linijo.
Vbrizgamo 10 μl vzorca, pripravljenega v skladu s točko 4.3.
4.4.3 Izračun in izražanje rezultatov
Uporabimo metodo normalizacije površine, tj. predpostavimo, da je vsota površin vrhov, ki ustreza triacilglicerolom z ekvivalentnim ogljikovim številom od ECN 42 do ECN 52, enaka 100 %.
Izračunamo relativni odstotni delež posameznega triglicerida z uporabo enačbe:
![]()
.
Rezultate je treba podati na vsaj dve decimalni mesti natančno.
Glej opombe od 1 do 4.
4.5 Izračun sestave triacilglicerolov (mol %) na podlagi podatkov o sestavi maščobnih kislin (površinski %)
4.5.1 Določanje sestave maščobnih kislin
Sestava maščobnih kislin se določi na podlagi standarda ISO 5508 s kapilarno kolono. Metil estri se pripravijo v skladu z dokumentom Mednarodnega sveta za oljčno olje COI/T.20/Dok. št. 24.
4.5.2 Maščobne kisline za izračun
Gliceridi se razvrstijo glede na ekvivalentno ogljikovo število (ECN), pri čemer se upoštevajo naslednje ekvivalence med ECN in maščobnimi kislinami. Upoštevane so bile samo maščobne kisline s 16 in 18 ogljikovimi atomi, ker so samo te pomembne za oljčno olje. Maščobne kisline je treba normalizirati do 100 %.
|
Maščobna kislina (MK) |
Kratica |
Molekulska masa (M) |
ECN |
|
Palmitinska kislina |
P |
256,4 |
16 |
|
Palmitoleinska kislina |
Po |
254,4 |
14 |
|
Stearinska kislina |
S |
284,5 |
18 |
|
Oleinska kislina |
O |
282,5 |
16 |
|
Linolna kislina |
L |
280,4 |
14 |
|
Linolenska kislina |
Ln |
278,4 |
12 |
4.5.3 Pretvorba površinskega % v mole za vse maščobne kisline (1)
|
|
|
|
|
|
|
|
4.5.4 Normalizacija maščobnih kislin do 100 % (2)
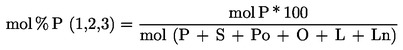





Rezultat pomeni odstotni delež posamezne maščobne kisline v mol % na vseh (1, 2, 3) položajih triacilglicerolov.
Nato so izračunata vsoti nasičenih maščobnih kislin P in S (NMK) ter nenasičenih maščobnih kislin Po, O, L in Ln (NEMK)(3):
![]()
![]()
4.5.5 Izračun sestave maščobnih kislin na položajih 2 ter 1 in 3 triacilglicerolov
Maščobne kisline so razdeljene na naslednje tri skupine: ena za položaj 2 in dve enaki za položaja 1 in 3, z različnimi koeficienti za nasičene (P in S) in nenasičene kisline (Po, O, L in Ln).
4.5.5.1 Nasičene maščobne kisline na položaju 2 [P(2) in S(2)] (4)
![]()
![]()
4.5.5.2 Nenasičene maščobne kisline na položaju 2 [Po(2), O(2), L(2) in Ln(2)] (5):
![]()
![]()
![]()
![]()
4.5.5.3 Maščobne kisline na položajih 1 in 3 [P(1,3), S(1,3), Po(1,3), O(1,3), L(1,3) in Ln(1,3)] (6):
![]()
![]()
![]()
![]()
![]()
![]()
4.5.6 Izračun triacilglicerolov
4.5.6.1 TAG s samo eno maščobno kislino (AAA, tukaj LLL, PoPoPo) (7)
![]()
4.5.6.2 TAG z dvema maščobnima kislinama (AAB, tukaj PoPoL, PoLL) (8)
![]()
![]()
4.5.6.3 TAG s tremi različnimi maščobnimi kislinami (ABC, tukaj OLLn, PLLn, PoOLn, PPoLn) (9)
![]()
![]()
![]()
4.5.6.4 Triacilgliceroli z ekvivalentnim ogljikovim številom (ECN 42)
Triacilgliceroli z ECN 42 se izračunajo po enačbah 7, 8 in 9, nato pa se navedejo v vrstnem redu pričakovane elucije v HPLC (običajno samo trije vrhovi).
LLL
PoLL in položajni izomer LPoL
OLLn ter položajna izomera OLnL in LnOL
PoPoL in položajni izomer PoLPo
PoOLn ter položajna izomera OPoLn in OLnPo
PLLn ter položajna izomera LLnP in LnPL
PoPoPo
SLnLn in položajni izomer LnSLn
PPoLn ter položajna izomera PLnPo in PoPLn
Triacilglicerole z ECN 42 dobimo z vsoto devetih triacilglicerolov, vključno z njihovimi položajnimi izomeri. Rezultate je treba podati na vsaj dve decimalni mesti natančno.
5. VREDNOTENJE REZULTATOV
Primerjata se izračunana teoretična vsebnost in vsebnost, določena z analizo HPLC. Če je absolutna razlika, ki jo dobimo, ko od podatkov HPLC odštejemo teoretične podatke, večja od vrednosti, navedenih za ustrezno kategorijo olja v standardu, vzorec vsebuje semensko olje.
Rezultati so podani na dve decimalni mesti natančno.
6. PRIMER (ŠTEVILKE SE NANAŠAJO NA ODDELKE V BESEDILU METODE)
— 4.5.1 Izračun mol % maščobnih kislin na podlagi podatkov plinsko-tekočinske kromatografije (normaliziran površinski %)
Glede sestave maščobnih kislin s plinsko-tekočinsko kromatografijo dobimo naslednje podatke:
|
MK |
P |
S |
Po |
O |
L |
Ln |
|
M |
256,4 |
284,5 |
254,4 |
282,5 |
280,4 |
278,4 |
|
Površinski % |
10,0 |
3,0 |
1,0 |
75,0 |
10,0 |
1,0 |
— 4.5.3 Pretvorba površinskega % v mole za vse maščobne kisline (glej enačbo 1)
|
mol P |
= |
|
|
mol S |
= |
|
|
mol Po |
= |
|
|
mol O |
= |
|
|
mol L |
= |
|
|
mol Ln |
= |
|
|
Skupaj |
= |
0,35821 mol TAG |
— 4.5.4 Normalizacija molov maščobnih kislin do 100 % (glej enačbo 2)
|
mol % P(1,2,3) |
= |
|
|
mol % S(1,2,3) |
= |
|
|
mol % Po(1,2,3) |
= |
|
|
mol % O(1,2,3) |
= |
|
|
mol % L(1,2,3) |
= |
|
|
mol % Ln(1,2,3) |
= |
|
|
Skupaj mol % |
= |
100 % |
Vsota nasičenih in nenasičenih maščobnih kislin na položajih 1, 2 in 3 TAG (glej enačbo 3):
![]()
![]()
— 4.5.5 Izračun sestave maščobnih kislin na položajih 2 ter 1 in 3 TAG
— 4.5.5.1 Nasičene maščobne kisline na položaju 2 [P(2) in S(2)] (glej enačbo 4)
![]()
![]()
— 4.5.5.2 Nenasičene maščobne kisline na položaju 2 [Po(1,3), O(1,3), L(1,3) in Ln(1,3)] (glej enačbo 5)

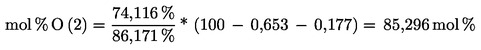


— 4.5.5.3 Maščobne kisline na položajih 1 in 3 [P(1,3), S(1,3), Po(1,3), O(1,3), L(1,3) in Ln(1,3)] (glej enačbo 6)
![]()
![]()
![]()
![]()
![]()
![]()
— 4.5.6 Izračun triacilglicerolov
Iz izračunane sestave maščobnih kislin na položajih sn-2 in sn-1,3:
|
MK in |
Položaja 1 in 3 |
Položaj 2 |
|
P |
16,004 % |
0,653 % |
|
S |
4,325 % |
0,177 % |
|
Po |
1,015 % |
1,262 % |
|
O |
68,526 % |
85,296 % |
|
L |
9,204 % |
11,457 % |
|
Ln |
0,927 % |
1,153 % |
|
Vsota |
100,0 % |
100,0 % |
se izračunajo naslednji triacilgliceroli:
LLL
PoPoPo
PoLL z enim položajnim izomerom
SLnLn z enim položajnim izomerom
PoPoL z enim položajnim izomerom
PPoLn z dvema položajnima izomeroma
OLLn z dvema položajnima izomeroma
PLLn z dvema položajnima izomeroma
PoOLn z dvema položajnima izomeroma
— 4.5.6.1 TAG z eno maščobno kislino (LLL, PoPoPo) (glej enačbo 7)
![]() = 0,09706 mol LLL
= 0,09706 mol LLL
![]()
— 4.5.6.2 TAG z dvema maščobnima kislinama (PoLL, SLnLn, PoPoL) (glej enačbo 8)
![]()
![]()
0,03210 mol PoLL
![]()
![]()
0,00094 mol SLnLn
![]()
![]()
0,00354 mol PoPoL
— 4.5.6.3 TAG s tremi različnimi maščobnimi kislinami (PoPLn, OLLn, PLLn, PoOLn) Glej enačbo 9
![]()
![]()
![]()
0,00761 mol PPoLn
![]()
![]()
![]()
0,43655 mol OLLn
![]()
![]()
![]()
0,06907 mol PLLn
![]()
![]()
![]()
0,04812 mol PoOLn
ECN 42 = 0,69512 mol TAG
Opomba 1: Vrstni red elucije se lahko določi z izračunom ekvivalentnih ogljikovih števil, pogosto definiranih z odnosom ![]() , kjer je CN ogljikovo število in n število dvojnih vezi; veliko bolj natančno jo je mogoče izračunati z upoštevanjem izvora dvojnih vezi. Če so no, nl in nln števila dvojnih vezi, ki se pripisujejo oleinski, linolni oziroma linolenski kislini, se ekvivalentno ogljikovo število lahko izračuna s pomočjo relacije v enačbi:
, kjer je CN ogljikovo število in n število dvojnih vezi; veliko bolj natančno jo je mogoče izračunati z upoštevanjem izvora dvojnih vezi. Če so no, nl in nln števila dvojnih vezi, ki se pripisujejo oleinski, linolni oziroma linolenski kislini, se ekvivalentno ogljikovo število lahko izračuna s pomočjo relacije v enačbi:
![]()
kjer se koeficienti do, dl in dln lahko izračunajo s pomočjo referenčnih trigliceridov. Pod pogoji, določenimi v tem postopku, bo dobljena relacija približno:
![]()
Opomba 2: Z več referenčnimi trigliceridi je mogoče izračunati tudi resolucijo glede na triolein:
![]()
trioleinz uporabo reduciranega retenzijskega časa
![]()
Graf log α kot funkcija f (število dvojnih vezi) omogoča, da se določijo retenzijske vrednosti za vse trigliceride maščobnih kislin, vsebovanih v referenčnih trigliceridih – glej sliko 1.
Opomba 3: Učinkovitost kolone mora omogočati jasno ločevanje vrha trilinoleina od vrhov trigliceridov s sosednjim retenzijskim časom. Elucija se izvede do vrha ECN 52.
Opomba 4: Pravilna meritev površin vseh vrhov, upoštevnih za to določanje, je zagotovljena, če drugi vrh za ECN 50 pokriva 50 % celotne lestvice rekorderja.
Slika 1
Graf log α kot funkcija f (število dvojnih vezi)
Število dvojnih vezi
La: lavrinska kislina; My: miristinska kislina; P: palmitinska kislina; S: stearinska kislina; O: oleinska kislina; L: linolna kislina; Ln: linolenska kislina
Slika 2
Oljčno olje z nizko vsebnostjo linolne kisline
(a)

S topilom: aceton/acetonitril.
PROFIL a: Glavne sestavine kromatografskih vrhov: ECN42: (1) LLL + PoLL; (2) OLLn + PoOLn; (3) PLLn; ECN44: (4) OLL + PoOL; (5) OOLn + PLL; (6) POLn + PPoPo; (7) OOL + PoOO; ECN46: (8) OOL + LnPP; (9) PoOO; (10) SLL + PLO; (11) PoOP + SPoL + SOLn + SPoPo; (12) PLP; ECN48: (13) OOO + PoPP; (14 + 15) SOL + POO; (16) POP; ECN50: (17) SOO; (18) POS + SLS.
(b)

S topilom: propionitril
PROFIL b: Glavne sestavine kromatografskih vrhov: ECN42: (1) LLL; (2) OLLn + PoLL; (3) PLLn; ECN44: (4) OLL; (5) OOLn + PoOL; (6) PLL + PoPoO; (7) POLn + PPoPo + PPoL; ECN46: (8) OOL + LnPP; (9) PoOO; (10) SLL + PLO; (11) PoOP + SPoL + SOLn + SPoPo; (12) PLP; ECN48: (13) OOO + PoPP; (14) SOL; (15) POO; (16) POP; ECN50: (17) SOO; (18) POS + SLS
Slika 3
Oljčno olje z visoko vsebnostjo linolne kisline
(a)

S topilom: aceton/acetonitril (50:50).
Profil a: Glavne sestavine kromatografskih vrhov: ECN42: (1‘) LLL + PoLL; (2‘) OLLn + PoOLn; (3‘) PLLn; ECN44: (4‘) OLL + PoOL; (5‘) OOLn + PLL; (6‘) POLn + PPoPo; ECN46: (7‘) OOL + PoOO; (8‘) PLO + SLL + PoOP; (9‘) PLP + PoPP; ECN48: (10‘) OOO; (11‘) POO + SLL + PPoO; (12‘) POP + PLS; ECN50: (13‘) SOO; (14‘) POS + SLS
(b)

S topilom: propionitril.
Profil b: Glavne sestavine kromatografskih vrhov: ECN42: (1) LLL; (2 + 2‘) OLLn + PoLL; (3) PLLn; ECN44: (4) OLL; (5) OOLn + PoOL; (6) PLL + PoPoO; (7) POLn + PPoPo + PPoL; ECN46: (8) OOL + LnPP; (9) PoOO; (10) SLL + PLO; (11) PoOP + SPoL + SOLn + SPoPo; ECN48: (12) PLP; (13) OOO + PoPP; (14) SOL; (15) POO; (16) POP; ECN50: (17) SOO; (18) POS + SLS; ECN52: (19) AOO.
ANNEX XIX
DETERMINATION OF ALIPHATIC ALCOHOLS CONTENT BY CAPILLARY GAS CHROMATOGRAPHY
1. OBJECT
The procedure describes a method for the determination of aliphatic alcohols content in oils and fats.
2. PRINCIPLE OF THE METHOD
The fatty substance, with 1-eicosanol added as internal standard, is saponified with ethanolic potassium hydroxide and then the unsaponifiable matter extracted with ethyl ether. The alcoholic fraction is separated from the unsaponifiable matter by chromatography on a basic silica gel plate; the alcohols recovered from the silica gel are transformed into trimethylsilyl ethers and analysed by capillary gas chromatography.
3. EQUIPMENT
3.1. 250 ml round-bottomed flask fitted with a reflux condenser having ground-glass joints.
3.2. 500 ml separating funnel.
3.3. 250 ml round-bottomed flasks.
3.4. Chromatographic tank for thin-layer chromatographic analysis, for glass plates of dimensions 20 x 20 cm.
3.5. Ultraviolet lamp having a wavelength of 366 or 254 nm.
3.6. 100 μl and 500 μl microsyringes.
3.7. A cylindrical filter funnel with a G3 porous septum (porosity 15 to 40 μm) of diameter approximately 2 cm and a depth of some 5 cm, with an attachment suitable for filtration under vacuum and a 12/21 male ground glass joint.
3.8. 50 ml vacuum conical flask with a 12/21 ground-glass female joint which can be fitted to the filter funnel (3.7).
3.9. A 10 ml test tube with a tapering bottom and a sealing stopper.
3.10. Gas chromatograph for use with a capillary column, and provided with a splitting system composed of:
3.10.1. Thermostatic chamber for columns (column oven) to hold the temperature desired with a precision of ± 1 °C.
3.10.2. A temperature-adjustable injection unit with a persilanised glass vaporising element.
3.10.3. A flame ionisation detector and converter-amplifier.
3.10.4. Recorder-integrator for operation with the converter-amplifier (3.10.3), with response time not exceeding one second and with variable paper-speed.
3.11. Glass or fused silica capillary column, of length 20 to 30 m, internal diameter 0,25 to 0,32 mm, with SE-52 or SE-54 liquid phase or equivalent, with a film thickness between 0,10 and 0,30 μm.
3.12. Microsyringe for gas chromatography, of 10 μl capacity with hardened needle.
3.13. Analytical balance sensitive to 1 mg (with 0,1 mg display).
4. REAGENTS
4.1. Potassium hydroxide, approximately 2 N ethanolic solution: 130 g potassium hydroxide (minimum concentration 85 %) is dissolved, with cooling, in 200 ml distilled water and then made up to one litre with ethanol. The solution should be stored in a well-stoppered opaque glass bottle.
4.2. Ethyl ether, pure for analysis.
4.3. Anhydrous sodium sulphate, analytical purity.
4.4. Glass plates coated with silica gel, without fluorescence indicator, thickness 0,25 mm (commercially available ready for use).
4.5. Potassium hydroxide, approximately 0,2 N ethanolic solution; 13 g of potassium hydroxide are dissolved in 20 ml of distilled water and made up to one litre with ethanol.
4.6. Benzene, for chromatography (see 5.2.2).
4.7. Acetone, for chromatography (See 5.2.2).
4.8. Hexane, for chromatography (see 5.2.2).
4.9. Ethyl ether, for chromatography (see 5.2.2).
4.10. Chloroform, for chromatography.
4.11. Reference solution for thin-layer chromatography: cholesterol or phytosterols, 5 % solution in chloroform.
4.12. 0,2 % solution of 2', 7'-dichlorofluorescein in ethanol. Make slightly basic by adding a few drops of 2 N alcoholic potassium hydroxide solution.
4.13. Anhydrous pyridine, for chromatography.
4.14. Hexamethyl disilazane.
4.15. Trimethylchlorosilane.
4.16. Standard solutions of trimethylsilyl ethers of aliphatic alcohols from C20 to C28. They may be prepared from mixtures of pure alcohols at the time they are required for use.
4.17. A 0,1 % (m/v) solution of 1-eicosanol in chloroform (internal standard).
4.18. Carrier gas: hydrogen or helium, gas-chromatographic purity.
4.19. Auxiliary gas: nitrogen, gas-chromatographic purity.
5. PROCEDURE
5.1. Preparation of the unsaponifiables
|
5.1.1. |
Using a 500 μl microsyringe place, into a 250 ml round-bottom flask, a volume of 0,1 % 1-eicosanol solution in chloroform (4.17) containing a quantity of 1-eicosanol approximately equal to 10 % of the aliphatic alcohols content in that portion of sample to be taken for analysis. For example, to 5 g of sample add 250 μl of the 0,1 % 1-eicosanol solution if olive oil and 1 500 μl if olive pomace oil. Evaporate to dryness in current of nitrogen and then weigh accurately 5 g of the dry filtered sample into the same flask. |
|
5.1.2. |
Add 50 ml of 2 N potassium hydroxide ethanolic solution, fit the reflux condenser and heat the apparatus to slight boiling on a steam bath, stirring continuously throughout the heating process until saponification has taken place (the solution becomes clear). Continue heating for a further 20 minutes and then add 50 ml of distilled water through the condenser. The condenser is then disconnected and the flask cooled to approximately 30 °C. |
|
5.1.3. |
The contents of the flask are quantitatively transferred to a separating funnel of 500 ml capacity by adding distilled water several times, using a total of around 50 ml distilled water. Add approximately 80 ml of ethyl ether, shake vigorously for approximately 30 seconds and allow to settle (Note 1). Separate off the lower aqueous phase collecting it in a second separating funnel. Two further extractions are effected on the aqueous phase, in the same manner, using each time 60 to 70 ml ethyl ether. Note 1: Emulsions may be eliminated by adding, using as a spray, small quantities of ethyl alcohol or methyl alcohol. |
|
5.1.4. |
The ethyl ether extracts are combined in a separating funnel and washed with distilled water (50 ml at a time) until the washing water gives a neutral reaction. Discard the washing water, dry with anhydrous sodium sulphate and filter, into a flask of 250 ml capacity which has been weighed beforehand, the funnel and filter being washed with small quantities of ethyl ether which are added to the total. |
|
5.1.5. |
Distil the ether down to a few ml, then bring to dryness under a slight vacuum or in a current of nitrogen, completing drying in an oven at 100 °C for approximately a quarter of an hour, and then weigh after cooling in a desiccator. |
5.2. Separation of alcoholic fractions
|
5.2.1. |
Preparation of basic TLC plates: the silica gel plates (4.4) are immersed completely, in 0,2 N potassium hydroxide solution (4.5) for 10 seconds, and then left to dry for two hours under an extractor hood and finally placed in an oven at 100 °C for one hour. Remove from the oven and keep in a calcium chloride desiccator until required for use (plates treated in this way must be used within 15 days). Note 2: When basic silica gel plates are used to separate the alcoholic fraction there is no need to treat the unsaponifiables with alumina. It follows that all acid compounds (fatty acids and others) are retained at the origin thereby obtaining both aliphatic alcohol and terpenic alcohol bands which are both separated distinctly from the sterol band. |
|
5.2.2. |
Place a 65/35 by volume hexane/ethyl ether mixture in the plate-developing chamber to a depth of approximately 1 cm ( 8 ). Close the chamber using an appropriate cover and leave for half an hour to allow equilibration between vapour and liquid. Strips of filter paper dipping into the eluent may be affixed to the inside surfaces of the tank to reduce the development time by approximately one third and obtain more uniform, regular elution of the components. Note 3: The developing solution must be replaced for each analysis in order to obtain reproducible developing conditions. |
|
5.2.3. |
An approximately 5 % solution of unsaponifiable matter (5.1.5) in chloroform is prepared and 0,3 ml of the solution is streaked as a uniform strip of minimum thickness, using the 100 μl microsyringe, on a TLC plate at approximately 2 cm from the bottom of the TLC plate. Aligned with the origin, 2 to 3 μl of the aliphatic alcohols reference solution (4.11) are spotted for the identification of the aliphatic alcohols band after development has been completed. |
|
5.2.4. |
Place the plate inside the development tank as stated in 5.2.2. The ambient temperature should be maintained between 15 and 20 °C. Immediately close the chamber with the cover and allow to elute until the solvent front reaches approximately 1 cm from the upper edge of the plate. The plate is then removed from the development chamber and the solvent evaporated under a hot air current or the plate is left for a while under the extractor hood. |
|
5.2.5. |
The plate is sprayed lightly and evenly with the solution of 2', 7'-dichlorofluorescein when the plate is observed under ultra violet light. The aliphatic alcohols band can be identified through being aligned with the stain obtained from the reference solution: mark the limits of the band with a black pencil; outlining the band of aliphatic alcohols and the band immediately above that, which is the terpenic alcohols band, together. Note 4: The aliphatic alcohols band and the terpenic alcohols band are to be grouped together in view of the possible migration of some aliphatic alcohols into the triterpenic alcohols band. |
|
5.2.6. |
Using a metal spatula scrape off the silica gel in the marked area. Place the finely comminuted material removed into the filter funnel (3.7). Add 10 ml of hot chloroform, mix carefully with the metal spatula and filter under vacuum, collecting the filtrate in the conical flask (3.8) attached to the filter funnel. Wash the pomace in the flask three times with ethyl ether (approximately 10 ml each time) collecting the filtrate in the same flask attached to the funnel. Evaporate the filtrate to a volume of 4 to 5 ml, transfer the residual solution to the previously weighed 10 ml test tube (3.9), evaporate to dryness by mild heating in a gentle flow of nitrogen, make up again using a few drops of acetone, evaporate again to dryness, place in an oven at 105 °C for approximately 10 minutes and then allow to cool in a desiccator and weigh. The pomace inside the test tube is composed of the alcoholic fraction. |
5.3. Preparation of the trimethylsilyl ethers
|
5.3.1. |
The reagent for silylation, consisting of a mixture of 9:3:1 by volume (Note 5) of pyridine-hexamethyldisilazane-trimethylchlorosilane in the proportion of 50 μl for each milligram of aliphatic alcohols, is added to the test tube containing the alcoholic fraction, avoiding all absorption of moisture (Note 6). Note 5: Solutions which are ready for use are available commercially. Other silanising reagents such as, for example, bis-trimethylsilyl, trifluor acetamide + 1 % trimethyl chlorosilane, which has to be diluted with an equal volume of anhydrous pyridine, are also available. Note 6: The slight opalescence which may form is normal and does not cause any interference. The formation of a white floc or the appearance of a pink colour are indicative of the presence of moisture or deterioration of the reagent. If these occur the test must be repeated. |
|
5.3.2. |
Stopper the test tube, shake carefully (without overturning) until the aliphatic alcohols are completely dissolved. Stand for at least 15 minutes at ambient temperature and then centrifuge for a few minutes. The clear solution is ready for gas chromatographic analysis. |
5.4. Gas chromatography analysis
5.4.1. Preliminary operations, column packing
|
5.4.1.1. |
Fit the column in the gas chromatograph, attaching the inlet end to the injector connected to the splitting system and the outlet end to the detector. Carry out a general check of the gas chromatography assembly (tightness of gas fittings, efficiency of the detector, efficiency of the splitting system and of the recording system, etc.). |
|
5.4.1.2. |
If the column is being used for the first time it is recommended that it should be subjected to conditioning. A little carrier gas is passed through the capillary column and then the gas chromatography assembly is switched on and gradually heated until a temperature not less than 20 °C above the operating temperature (see Note 7) is attained. That temperature is held for not less than two hours and then the assembly is brought to the operating conditions (regulation of gas flow, split flame ignition, connection to the electronic recorder, adjustment of the temperature of the capillary column oven, the detector and the injector, etc.) and the signal is adjusted to a sensitivity not less than twice the highest level contemplated for the execution of the analysis. The course of the base line must be linear, without peaks of any kind, and must not drift. A negative straight-line drift indicates leakage from the column connections; a positive drift indicates inadequate conditioning of the column. Note 7: The conditioning temperature shall be at least 20 °C less than the maximum temperature contemplated for the liquid phase employed. |
5.4.2. Choice of operating conditions
5.4.2.1. The guideline operating conditions are as follows:
— column temperature: the initial isotherm is set at 180 °C for eight minutes and then programmed at 5 °C/minute to 260 °C and a further 15 minutes at 260 °C,
— temperature of evaporator: 280 °C,
— temperature of detector: 290 °C,
— linear velocity of carrier gas: helium 20 to 35 cm/s, hydrogen 30 to 50 cm/s,
— splitting ratio: 1:50 to 1:100,
— sensitivity of instrument: 4 to 16 times the minimum attenuation,
— sensitivity of recording: 1 to 2 mV fs,
— paper speed: 30 to 60 cm/h,
— quantity of substance injected: 0,5 to 1 μl of TMSE solution.
The above conditions may be modified according to the characteristics of the column and of the gas chromatograph to obtain chromatograms satisfying the following conditions:
— alcohol C26 retention time shall be 18 ± 5 minutes,
— the alcohol C22 peak shall be 80 ± 20 % of the full scale value for olive oil and 40 ± 20 % of the full scale value for seed oil.
5.4.2.2. The above requirements are checked by repeated injection of the standard TMSE mixture of alcohols and the operating conditions are adjusted to yield the best possible results.
5.4.2.3. The parameters for the integration of peaks shall be set so that a correct appraisal of the areas of the peaks considered is obtained.
5.4.3. Analytical procedure
5.4.3.1. Using the microsyringe of 10 μl capacity draw in 1 μl of hexane followed by 0,5 μl of air and subsequently 0,5 to 1 μl of the sample solution; raise the plunger of the syringe further so the needle is emptied. Push the needle through the membrane of the injection unit and after one to two seconds inject rapidly, then slowly remove the needle after some five seconds.
5.4.3.2. Recording is effected until the TMSE of the aliphatic alcohols present have been eluted completely. The base line shall always correspond to the requirements of 5.4.1.2.
5.4.4. Peak identification
The identification of individual peaks is effected according to the retention times and by comparison with the standard TMSE mixture, analysed under the same conditions.
A chromatogram of the alcoholic fraction of a virgin olive oil is shown in Figure 1.
5.4.5. Quantitative evaluation
5.4.5.1. The peak areas of 1-eicosanol and of the aliphatic alcohols C22, C24, C26 and C28 are calculated by electronic integration.
5.4.5.2. The contents of each aliphatic alcohol, expressed in mg/1 000 g fatty substance, are calculated as follows:
![]()
where:
|
Ax |
= |
area of the alcohol peak x |
|
As |
= |
area of 1-eicosanol |
|
ms |
= |
mass of 1-eicosanol in milligrams |
|
m |
= |
mass of sample drawn for determination, in grams. |
6. EXPRESSION OF THE RESULTS
The contents of the individual aliphatic alcohols in mg/1 000 g of fatty substance and the sum of the ‘total aliphatic alcohols’ are reported.
APPENDIX
Determination of the linear velocity of the gas
1 to 3 μl of methane or propane are injected into the gas chromatograph set at normal operating conditions and the time taken for the methane or propane to flow through the column from the instant of injection to the instant the peak elutes (tM) is measured using a stop clock.
The linear velocity in cm/s is given by L/tM, where L is the length of the column in centimetres and tM is the measured time in seconds.
 Figure 1 — Chromatogram of the alcoholic fraction of virgin oil
Figure 1 — Chromatogram of the alcoholic fraction of virgin oil
|
1 |
= |
Eicosanol |
|
2 |
= |
Decosanol |
|
3 |
= |
Tricosanol |
|
4 |
= |
Tetracosanol |
|
5 |
= |
Pentacosanol |
|
6 |
= |
Hexacosanol |
|
7 |
= |
Heptacosanol |
|
8 |
= |
Octacosanol |
PRILOGA XX
Metoda za določanje vsebnosti voskov, metilnih estrov maščobnih kislin in etilnih estrov maščobnih kislin s kapilarno plinsko kromatografijo
1. NAMEN
Metoda je namenjena določanju vsebnosti voskov, metilnih estrov maščobnih kislin in etilnih estrov maščobnih kislin v oljčnih oljih. Posamezni voski in alkilni estri se ločijo glede na število atomov ogljika. Metoda se priporoča kot orodje za ločevanje med oljčnim oljem in oljem iz oljčnih tropin ter kot parameter kakovosti za ekstra deviška oljčna olja, s katerim je mogoče ugotavljati neprave mešanice ekstra deviških oljčnih olj z olji slabše kakovosti, torej z deviškimi, lampante ali nekaterimi razdišavljenimi olji.
2. PRINCIP
Olje, ki smo mu dodali ustrezne interne standarde, na z vodo omočenem silikagelu frakcioniramo s kolonsko kromatografijo. Ujamemo eluirano frakcijo najprej pri pogojih preskušanja (katere polarnost je manjša kot pri trigliceridih) in neposredno analiziramo s pomočjo kapilarne plinske kromatografije.
3. APARAT
|
3.1. |
Erlenmajerica prostornine 25 ml. |
|
3.2. |
Steklena kolona za tekočinsko kromatografijo z notranjim premerom 15 mm, dolžine 30 do 40 cm in opremljena z ustreznim ventilom. |
|
3.3. |
Plinski kromatograf, primeren za uporabo s kapilarno kolono, opremljen z injicirnim sistemom za injiciranje vzorca neposredno v kolono, ki zajema:
|
|
3.4. |
Mikrobrizgalka, 10 μl, z utrjeno iglo, za neposredno vbrizganje na začetek kolone. |
|
3.5. |
Električni vibrator. |
|
3.6. |
Rotavapor. |
|
3.7. |
Žarilna peč. |
|
3.8. |
Analitska tehtnica, ki zagotavlja ± 0,1 mg natančnost. |
|
3.9. |
Običajna laboratorijska steklovina. |
4. REAGENTI
|
4.1. |
Silikagel, 60–200 μm mesh. Silikagel vsaj 4 ure žarimo v žarilni peči pri 500 °C. Pustimo, da se ohladi, in potem dodamo 2 % vode glede na količino uporabljenega silikagela. Dobro premešamo, da se suspenzija homogenizira, in pred uporabo hranimo vsaj 12 ur v eksikatorju. |
|
4.2. |
►M24
N-heksan, kromatografske stopnje ali stopnje ostanka (čistost je treba preveriti). OPOZORILO – Hlapi se lahko vnamejo. Ne približujte virom toplote, iskram ali neposrednemu ognju. Prepričajte se, da so steklenice vedno dobro zaprte. Med uporabo zagotovite ustrezno prezračevanje. Preprečiti je treba zbiranje hlapov in odstraniti vsa možna tveganja za izbruh ognja, ki jih povzročajo na primer grelniki ali električni aparati, ki niso proizvedeni iz nevnetljivih materialov. Škodljivi pri vdihavanju, ker lahko poškodujejo živčne celice. Ne vdihavajte hlapov. Če je potrebno, za dihanje uporabite ustrezen pripomoček. Preprečite stik z očmi ali poškodovano kožo. ◄ |
|
4.3. |
Etil eter, za kromatografijo.
OPOZORILO – Zelo vnetljiv in zmerno toksičen. Draži kožo. Škodljiv pri vdihavanju. Lahko poškoduje oči. Lahko učinkuje s časovnim zamikom. Formira lahko eksplozivne perokside. Hlapi se lahko vnamejo. Ne približujte virom toplote, iskram ali neposrednemu ognju. Prepričajte se, da so steklenice vedno dobro zaprte. Med uporabo zagotovite ustrezno prezračevanje. Preprečiti je treba zbiranje hlapov in odstraniti vsa možna tveganja za izbruh ognja, ki jih povzročajo na primer grelniki ali električni aparati, ki niso proizvedeni iz nevnetljivih materialov. Ne izparevajte do suhega ali skoraj suhega. Dodajanje vode ali ustreznega reducenta lahko reducira oblikovanje peroksida. Ne pijte. Ne vdihavajte hlapov. Izogibajte se daljšemu ali večkratnemu stiku s kožo. |
|
4.4. |
►C1
N-heptan za kromatografijo ali izo-oktan. ◄OPOZORILO – Vnetljiv. Škodljiv pri vdihavanju. Ne približujte virom toplote, iskram ali neposrednemu ognju. Prepričajte se, da so steklenice vedno dobro zaprte. Med uporabo zagotovite ustrezno prezračevanje. Ne vdihavajte hlapov. Izogibajte se daljšemu ali večkratnemu stiku s kožo. |
|
4.5. |
Standardna raztopina lauril arahidata (opomba 3) koncentracije 0,05 % (m/V) v heptanu (interni standard za voske). Opomba 3: Uporabijo se lahko tudi palmitil palmitat, miristil stearat ali arahidil laureat. |
|
4.6. |
Standardna raztopina metilnega heptadekanoata koncentracije 0,02 % (m/V) v heptanu (interni standard za metilne in etilne estre). |
|
4.7. |
Sudan 1 (1-fenilazo-2-naftol). |
|
4.8. |
►C1
Nosilni plin: vodik ali helij, čist, za plinsko kromatografijo. ◄OPOZORILO Vodik. Pod pritiskom zelo vnetljiv. Ne približujte virom toplote, iskram, neposrednemu ognju ali električnim aparatom, ki niso proizvedeni iz nevnetljivih materialov. Prepričajte se, da je ventil steklenice zaprt, kadar se vodik ne uporablja. Vedno uporabljajte z reducentom pritiska. Preden odprete ventil steklenice, sprostite napetost vzmeti reducenta. Pri odpiranju ventila ne stojte pred odprtino steklenice. Med uporabo zagotovite ustrezno prezračevanje. Ne pretakajte vodika iz ene steklenice v drugo. V steklenici ne mešajte plina. Prepričajte se, da steklenic ni mogoče prevrniti. Ne hranite jih na svetlobi in ob virih toplote. Hranite jih v okolju, ki je odporno na korozijo. Ne uporabljajte poškodovanih steklenic ali steklenic brez nalepke. Helij. Stisnjen plin pod visokim pritiskom. Zmanjša količino kisika na voljo za dihanje. Steklenica naj bo zaprta. Med uporabo zagotovite ustrezno prezračevanje. Ne vstopajte v prostore za hranjenje, razen če so ustrezno prezračeni. Vedno uporabljajte z reducentom pritiska. Preden odprete ventil steklenice, sprostite napetost vzmeti reducenta. Ne pretakajte plina iz ene steklenice v drugo. Prepričajte se, da steklenic ni mogoče prevrniti. Pri odpiranju ventila ne stojte pred odprtino steklenice. Ne hranite jih na svetlobi in ob virih toplote. Hranite jih v okolju, ki je odporno na korozijo. Ne uporabljajte poškodovanih steklenic ali steklenic brez nalepke. Ne vdihavajte. Uporabljajte samo za tehnične namene. |
|
4.9. |
Pomožna plina:
— Vodik, čist, za plinsko kromatografijo. — Zrak, čist, za plinsko kromatografijo. OPOZORILO Zrak. Stisnjen plin pod visokim pritiskom. Uporabljajte previdno v prisotnosti vnetljivih snovi, saj je temperatura, potrebna za samovžig večine organskih sestavin zraka pod visokim pritiskom znatno nižja. Prepričajte se, da je ventil steklenice zaprt, kadar se vodik ne uporablja. Vedno uporabljajte reducent pritiska. Preden odprete ventil steklenice, sprostite napetost vzmeti reducenta. Pri odpiranju ventila ne stojte pred odprtino steklenice. Ne pretakajte plina iz ene steklenice v drugo. V steklenici ne mešajte plina. Prepričajte se, da steklenic ni mogoče prevrniti. Ne hranite jih na svetlobi in ob virih toplote. Hranite jih v okolju, ki je odporno na korozijo. Ne uporabljajte poškodovanih steklenic ali steklenic brez nalepke. Zraka za tehnične namene se ne sme uporabljati za vdihavanje ali dihalne naprave. |
5. POSTOPEK
5.1. Priprava kromatografske kolone
V n-heksanu (točka 4.2) suspendiramo 15 g silikagela (točka 4.1) in napolnimo kolono (točka 3.2). Počakamo, da se posede. Homogenost posedanja, ki omogoči homogene kromatografske pasove, lahko dosežemo z uporabo električnega vibratorja. Spiramo s 30 ml n-heksana, da odstranimo vse morebitne nečistoče. V 25-ml bučko (točka 3.1) z analitsko tehtnico (točka 3.8) natehtamo natanko 500 mg vzorca in dodamo ustrezno količino internega standarda (točka 4.5), ki je odvisna od pričakovane vsebnosti voskov, npr. pri oljčnem olju dodamo 0,1 mg lauril arahidata, pri olju iz oljčnih tropin 0,25 do 0,50 mg in 0,05 mg metil heptadekanoata pri oljčnih oljih (točka 4.6).
Tako pripravljen vzorec prenesemo v kromatografsko kolono s pomočjo dveh 2-ml odmerkov n-heksana (točka 4.2).
Topilo izpuščamo iz kolone tako dolgo, da doseže 1 mm nad zgornjim robom absorbenta. Nadalje spiramo z n-heksanom/etilnim etrom (99:1) in zberemo 220 ml pri pretoku približno 15 kapljic vsakih 10 sekund. (Ta frakcija vsebuje metilne in etilne estre ter voske). (opomba 4) (opomba 5).
Opomba 4: Vsak dan je treba pripraviti svežo mešanico n-heksana/etilnega etra (99:1).
Opomba 5: Za opazovanje pravilnega eluiranja voskov lahko vzorcu v raztopini dodamo 100 μl 1-odstotnega sudana I v elucijski mešanici.
Barvilo ima vmesno retencijo med voski in trigliceridi. Zato je treba elucijo suspendirati, ko obarvanje doseže dno kromatografske kolone, ker so se eluirali vsi voski.
Iz tako dobljene frakcije na rotavaporju odparimo skoraj vse topilo. Preostala 2 ml odstranimo s pomočjo blagega toka dušika. Zbrano frakcijo, ki vsebuje metilne in etilne estre, razredčimo z 2 do 4 ml n-heptana ali izo-oktana.
5.2. Plinsko kromatografska analiza
5.2.1. Postopek
Kolono pritrdimo na plinski kromatograf (točka 3.3), in sicer vstopni del na injektor, izhodnega pa na detektor. Preverimo plinski kromatograf (plinske povezave, pravilno delovanje detektorja in rekorderja itd.).
Če kolono uporabljamo prvič, jo je priporočljivo kondicionirati. Pri majhnem pretoku plina skozi kolono vključimo plinski kromatograf. Postopoma segrevamo, tako da po približno 4 urah dosežemo temperaturo 350 °C.
Pri tej temperaturi kolono kondicioniramo vsaj 2 uri, nato uravnamo instrument v skladu s pogoji delovanja (uravnamo pretok plina, prižgemo plamen, povežemo z elektronskim rekorderjem (točka 3.3.4), naravnamo delovno temperaturo peči, naravnamo detektor itd.). Pri občutljivosti, ki je vsaj dvakrat večja od delovne, posnamemo bazno linijo. Le-ta mora biti linearna, brez vrhov in brez kakršnih koli odstopanj.
Negativno premočrtni odklon kaže na slabo tesnjenje med kolono in instrumentom, pozitiven odklon pa na slabo kondicionirano kolono.
5.2.2. Izbira delovnih pogojev za voske ter metilne in etilne estre (opomba 6).
Splošni delovni pogoji, ki jih je treba upoštevati, so naslednji:
|
— |
Temperatura kolone : 20 °C/min 5 °C/min 80 °C najprej (1′) |
|
— |
Temperatura detektorja : 350 °C. |
|
— |
Brizg : 1 μl raztopine (2–4 ml) n-heptana. |
|
— |
Nosilni plin : helij ali vodik z optimalno linearno hitrostjo izbranega plina (glej Dodatek A). |
|
— |
Občutljivost instrumenta : primerna za izpolnjevanje zgoraj navedenih pogojev. |
Opomba 6: Zaradi visoke končne temperature je pozitivni odklon dovoljen, a ne sme preseči 10 % polne vrednosti skale.
Te pogoje lahko spremenimo, da bi zadostili značilnostim kolone in plinskega kromatografa, ločili vse voske ter metilne in etilne estre maščobnih kislin ter dosegli zadovoljivo najvišjo separacijo (glej slike 2, 3 in 4) in retencijski čas, ki za interni standard lauril arahidata znaša 18 ± 3 minute. Najznačilnejši vrh voskov mora biti najmanj 60 % od spodnjega dela skale, interni standard metilnega heptadekanoata za metilne in etilne estre pa mora doseči polno vrednost skale.
Vrhove integracijskih parametrov moramo določiti na tak način, da bomo dobili pravilno vrednotenje zadevnih površin vrhov.
5.3. Izvedba analize
Z 10-μl mikrobrizgalko vzamemo 10 μl raztopine; bat potegnemo nazaj, da se igla izprazni. Iglo vbodemo v injektor in po eni ali dveh sekundah hitro vbrizgnemo. Iglo po približno petih sekundah nežno izvlečemo.
Kromatogram snemamo, dokler ne eluirajo vsi voski oziroma stigmastadieni, odvisno od frakcije, ki jo analiziramo.
Bazna linija mora ves čas izpolnjevati zahtevane pogoje.
5.4. Identifikacija vrhov
Vrhove identificiramo s pomočjo retencijskih časov s primerjavo med mešanicami voskov z znanimi retencijskimi časi, ki so bili analizirani pri istih pogojih. Alkilni estri se določijo iz mešanic metilnih in etilnih estrov glavnih maščobnih kislin v oljčnih oljih (palmitinskih in oleinskih).
Slika 1 prikazuje kromatogram voskov deviškega oljčnega olja. Sliki 2 in 3 prikazujeta kromatograma dveh ekstra deviških oljčnih olj v prodaji, od katerih eno vsebuje metilne in etilne estre, drugo pa ne. Slika 4 prikazuje kromatogram za ekstra deviško oljčno olje najvišje kakovosti in isto olje, mešano z 20 % razdišavljenega olja.
5.5. Kvantitativna analiza voskov
S pomočjo integratorja določimo površine vrhov alifatskih estrov od C40 do C46 in površino vrhov internega standarda lauril arahidata.
Skupno vsebnost voskov določimo z dodajanjem vsakega voska posebej v mg/kg maščobe, kot sledi:
![]()
kjer je:
|
Ax |
= |
površina vrha za posamezni ester z računalniškim preštevanjem; |
|
As |
= |
površina vrha za interni standard lauril arahidata ester z računalniškim preštevanjem; |
|
ms |
= |
masa dodanega internega standarda lauril arahidata, v miligramih; |
|
m |
= |
masa vzorca za določanje, v gramih. |
5.5.1. Kvantitativna analiza metilnih in etilnih estrov
S pomočjo integratorja določimo površine vrhov internega standarda metil heptadekanoata, maščobnih kislin metilnih estrov C16 in C18 ter maščobnih kislin etilnih estrov C16 in C18.
Določimo vsebnost vsakega alkilnega estra v mg/kg maščobe, kot sledi
![]()
kjer je:
|
Ax |
= |
površina vrha za posamezni ester C16 in C18 z računalniškim preštevanjem; |
|
As |
= |
površina vrha za interni standard metil heptadekanoata z računalniškim preštevanjem; |
|
ms |
= |
masa dodanega internega standarda metil heptadekanoata, v miligramih; |
|
m |
= |
masa vzorca za določanje, v gramih. |
6. PODAJANJE REZULTATOV
Podamo vsoto vsebnosti različnih voskov od C40 do C46 (opomba 7) v miligramih na kilogram maščobe.
Podamo vsoto vsebnosti metilnih estrov in etilnih estrov od C16 do C18 ter vsoto obeh skupaj.
Rezultate izrazimo do najbližjega mg/kg.
Opomba 7: Spojine, ki jih je treba količinsko določiti, se nanašajo na vrhove s sodim številom ogljikovih atomov med estri C40 do C46, tako kot je to prikazano na primeru kromatograma voskov oljčnega olja v priloženi sliki. ►C1 Za namene določanja se v primeru, da je ester C46 razdeljen, priporoča, da se analizira frakcija voskov olja iz oljčnih tropin, pri čemer je vrh C46 razločen, ker je jasno prevladuje. ◄
Podamo razmerje med etilnimi estri in metilnimi estri.
Slika 1
Primer plinskega kromatograma frakcije voskov oljčnega olja ( 9 )

Vrhovi z retencijo med 5 in 8 min metilnih in etilnih estrov maščobnih kislin
Ključ:
|
I.S. |
= |
lauril arahidat |
|
1 |
= |
diterpenski estri |
|
2+2′ |
= |
estri C40 |
|
3+3′ |
= |
estri C42 |
|
4+4′ |
= |
estri C44 |
|
5 |
= |
estri C46 |
|
6 |
= |
sterolni estri in triterpenski alkoholi |
Slika 2
Metilni estri, etilni estri in voski v deviškem oljčnem olju
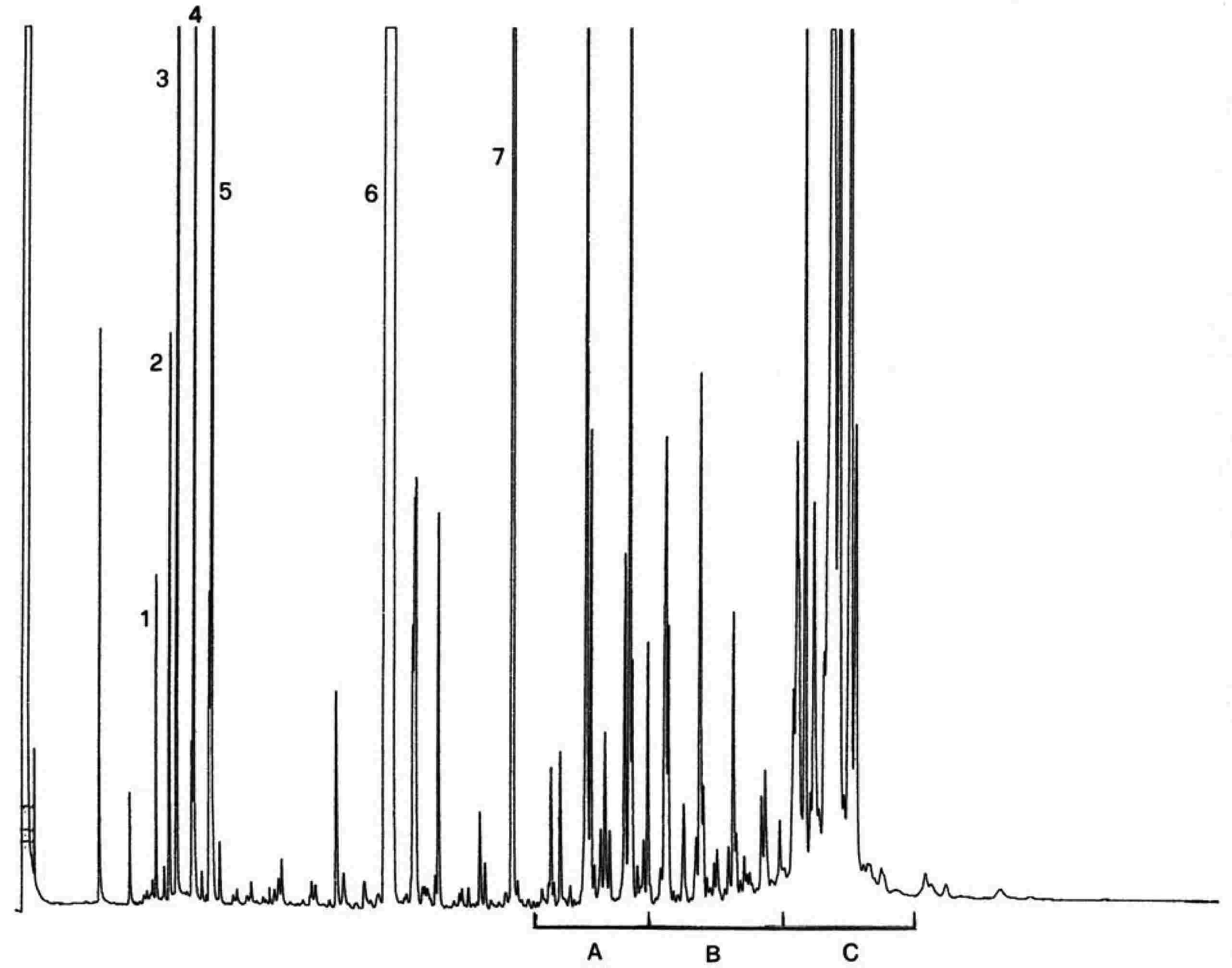
Ključ:
|
1 |
– |
metil C16 |
|
2 |
– |
etil C16 |
|
3 |
– |
IS metil heptadekanoat |
|
4 |
– |
metil C18 |
|
5 |
– |
etil C18 |
|
6 |
– |
skvalen |
|
7 |
– |
I.S. lauril arahidata |
|
A |
– |
diterpenski estri |
|
B |
– |
voski |
|
C |
– |
sterolni estri in triterpenski estri |
Slika 3
Metilni estri, etilni estri in voski v ekstra deviškem oljčnem olju

Ključ:
|
1 |
– |
IS metil heptadekanoat |
|
2 |
– |
metil C18 |
|
3 |
– |
etil C18 |
|
4 |
– |
skvalen |
|
5 |
– |
I.S. lauril arahidata |
|
A |
– |
diterpenski estri |
|
B |
– |
voski |
|
C |
– |
sterolni estri in triterpenski estri |
Slika 4
Del kromatograma za ekstra deviško oljčno olje in isto olje, mešano z razdišavljenim oljem
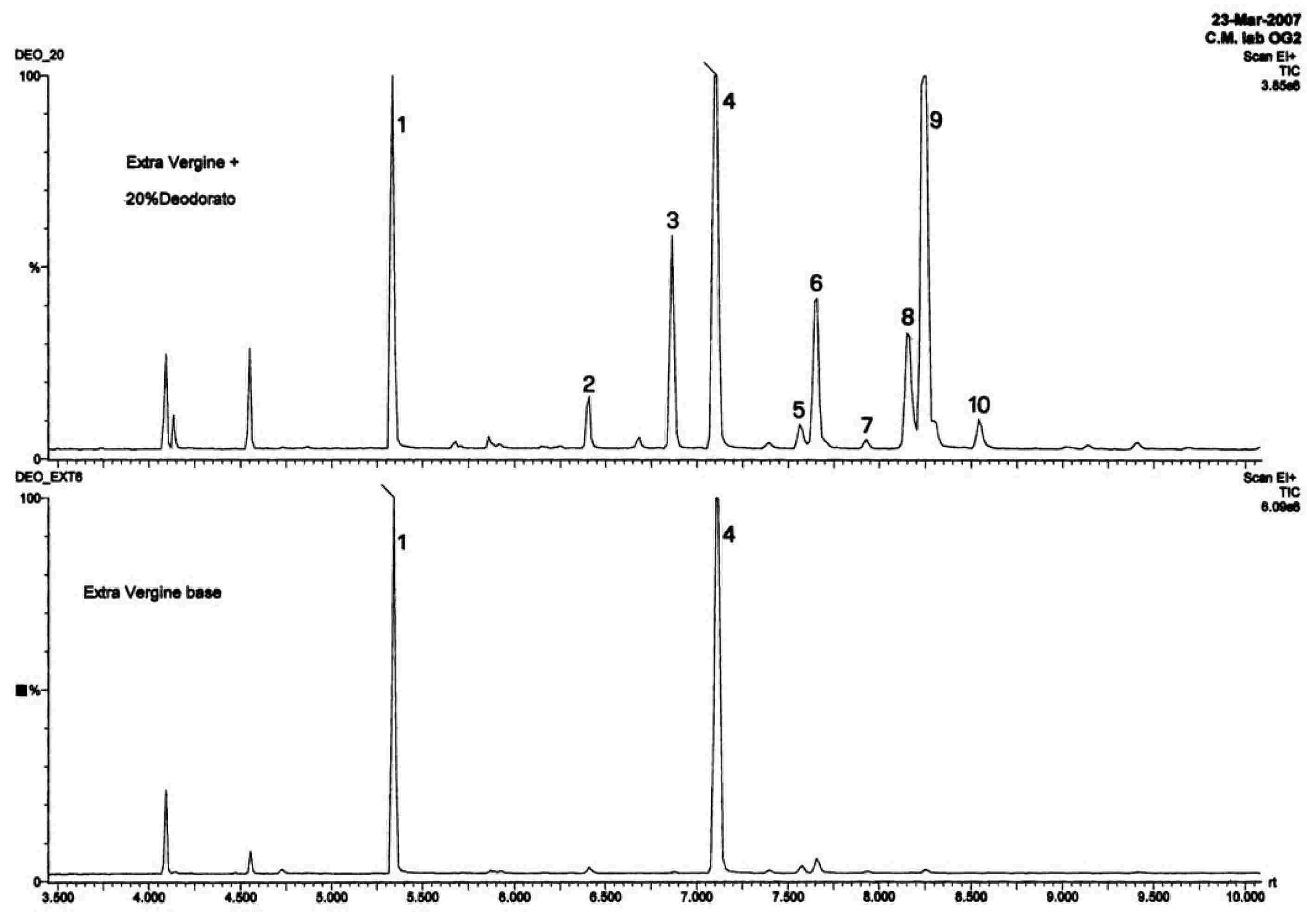
Ključ:
|
1 |
– |
IS metil miristat |
|
2 |
– |
metil palmitat |
|
3 |
– |
etil palmitat |
|
4 |
– |
IS metil heptadekanoat |
|
5 |
– |
metil linoleat |
|
6 |
– |
metil oleat |
|
7 |
– |
metil stearat |
|
8 |
– |
etil linoleat |
|
9 |
– |
etil oleat |
|
10 |
– |
etil stearat |
Dodatek A
Določitev linearne hitrosti plina
V plinski kromatograf, ki je naravnan na delovne pogoje, vbrizgnemo 1:3 μl metana (propana). Merimo čas, ki ga plin potrebuje za pot skozi kolono od trenutka vbrizga do trenutka, ko se pojavi vrh (tM).
Linearna hitrost v cm/s je podana z L/tM, pri čemer je L dolžina kolone v centimetrih in tM izmerjeni čas v sekundah.
PRILOGA XXI
Rezultati preverjanj skladnosti oljčnega olja iz člena 8(2)
|
|
Označevanje |
Kemijski parametri |
Senzorične značilnosti (4) |
Končni sklep |
|||||||||||||
|
Vzorec |
Kategorija |
Država porekla |
Kraj inšpekcijskega pregleda (1) |
Uradno ime |
Označba porekla |
Pogoji skladiščenja |
Napačni podatki |
Čitljivost |
S/NS (3) |
Parametri zunaj mejnih vrednosti DA/NE |
Če je tako, navedite, kateri (2) |
S/NS (3) |
Mediana napak |
Mediana sadežnosti |
S/NS (3) |
Zahtevan ukrep |
Kazen |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(1) Notranji trg (oljarna, polnilci, faza prodaje na drobno), izvoz, uvoz. (2) Vsaka značilnost oljčnega olja iz Priloge I ima oznako. (3) Skladno/neskladno. (4) Ne zahteva se za oljčno olje in olje iz oljčnih tropin. |
|||||||||||||||||
( 1 ) UL 172, 30.9.1966, str. 3025/66.
( 2 ) UL L 353, 17.12.1990, str. 23.
( 3 ) UL L 128, 24.5.1977, str. 6.
( 4 ) UL L 166, 1.7.1988, str. 10.
( 5 ) UL L 228, 1.9.2009, str. 3.
( 6 ) Po eluciji sterolnih estrov kromatografska linija ne sme imeti značilnih vrhov (trigliceridi).
( 7 ) Pokuševalec lahko preneha s pokušanjem olja, če pri neposredni vohalni zaznavi odkrije izjemno intenzivno negativno lastnost. Na ocenjevalni list vpiše to izjemno okoliščino.
( 8 ) In these cases in particular, a 95/5 by volume benzene/acetone eluent mixture must be used to obtain distinct band separation.
( 9 ) Po eluciji sterolnih estrov kromatogram ne sme imeti izpostavljenih vrhov (trigliceroli).


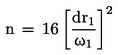


 Figure 1: Gas chromatographic profile obtained by the cold methylation method from olive-pomace oil. The chromatographic peaks correspond to the methyl and ethyl esters except where otherwise indicated.
Figure 1: Gas chromatographic profile obtained by the cold methylation method from olive-pomace oil. The chromatographic peaks correspond to the methyl and ethyl esters except where otherwise indicated.