

EUR-Lex Access to European Union law
This document is an excerpt from the EUR-Lex website
Document 02009R0152-20170426
Commission Regulation (EC) No 152/2009 of 27 January 2009 laying down the methods of sampling and analysis for the official control of feed (Text with EEA relevance)
Consolidated text: Uredba Komisije (ES) št. 152/2009 z dne 27. januarja 2009 o določitvi metod vzorčenja in analitskih metod za uradni nadzor krme (Besedilo velja za EGP)
Uredba Komisije (ES) št. 152/2009 z dne 27. januarja 2009 o določitvi metod vzorčenja in analitskih metod za uradni nadzor krme (Besedilo velja za EGP)

02009R0152 — SL — 26.04.2017 — 005.001
To besedilo je zgolj informativne narave in nima pravnega učinka. Institucije Unije za njegovo vsebino ne prevzemajo nobene odgovornosti. Verodostojne različice zadevnih aktov, vključno z uvodnimi izjavami, so objavljene v Uradnem listu Evropske unije. Na voljo so na portalu EUR-Lex. Uradna besedila so neposredno dostopna prek povezav v tem dokumentu
|
UREDBA KOMISIJE (ES) št. 152/2009 z dne 27. januarja 2009 o določitvi metod vzorčenja in analitskih metod za uradni nadzor krme (UL L 054 26.2.2009, str. 1) |
spremenjena z:
|
|
|
Uradni list |
||
|
št. |
stran |
datum |
||
|
L 91 |
8 |
29.3.2012 |
||
|
L 20 |
33 |
23.1.2013 |
||
|
L 197 |
1 |
20.7.2013 |
||
|
L 188 |
1 |
27.6.2014 |
||
|
L 92 |
35 |
6.4.2017 |
||
UREDBA KOMISIJE (ES) št. 152/2009
z dne 27. januarja 2009
o določitvi metod vzorčenja in analitskih metod za uradni nadzor krme
(Besedilo velja za EGP)
Člen 1
Vzorčenje za uradni nadzor krme, zlasti v zvezi z določanjem sestavin, vključno s snovmi, ki vsebujejo, sestojijo iz ali so proizvedene iz gensko spremenjenih organizmov (GSO), krmnih dodatkov, kakor so opredeljeni v Uredbi (ES) št. 1831/2003 Evropskega parlamenta in Sveta ( 1 ), in nezaželenih snovi, kakor so opredeljene v Direktivi 2002/32/ES Evropskega Parlamenta in Sveta ( 2 ), se izvaja v skladu z metodami iz Priloge I.
Metoda vzorčenja iz Priloge I se uporablja za nadzor krme v zvezi z določanjem ostankov pesticidov, kakor so opredeljeni v Uredbi (ES) št. 396/2005 Evropskega parlamenta in Sveta ( 3 ), in nadzor skladnosti z Uredbo (EU) št. 619/2011.
Člen 2
Priprava vzorcev za analizo in prikaz rezultatov se izvajata v skladu z metodami iz Priloge II.
Člen 3
Analiza za uradni nadzor krme se izvaja z uporabo metod iz Priloge III (Analitske metode za nadzor sestave posamičnih krmil in krmnih mešanic), Priloge IV (Analitske metode za nadzor ravni dovoljenih krmnih dodatkov), Priloge V (Analitske metode za nadzor nezaželenih snovi v krmi) in Priloge VI (Analitske metode za določitev sestavin živalskega izvora za uradni nadzor krme).
Člen 4
Energijska vrednost krmnih mešanic za perutnino se izračuna v skladu s Prilogo VII.
Člen 5
Za potrditev se uporabljajo analitske metode za nadzor nedovoljenih krmnih dodatkov iz Priloge VIII.
Člen 6
Direktive 71/250/EGS, 71/393/EGS, 72/199/EGS, 73/46/EGS, 76/371/EGS, 76/372/EGS, 78/633/EGS, 81/715/EGS, 84/425/EGS, 86/174/EGS, 93/70/EGS, 93/117/ES, 98/64/ES, 1999/27/ES, 1999/76/ES, 2000/45/ES, 2002/70/ES in 2003/126/ES se razveljavijo.
Sklici na razveljavljene direktive se upoštevajo kot sklici na to uredbo in se berejo v skladu s korelacijsko tabelo iz Priloge IX.
Člen 7
Ta uredba začne veljati dvajseti dan po objavi v Uradnem listu Evropske unije.
Uporablja se od 26. avgusta 2009
Ta uredba je v celoti zavezujoča in se neposredno uporablja v vseh državah članicah.
PRILOGA I
METODE VZORČENJA
1. NAMEN IN PODROČJE UPORABE
Vzorci za uradni nadzor krme se jemljejo v skladu s spodaj opisanimi metodami. Tako dobljeni vzorci se obravnavajo kot reprezentativni za vzorčene deleže.
Namen reprezentativnega vzorčenja je pridobiti majhen delež iz serije, pri čemer opredelitev katere koli značilnosti tega deleža pomeni srednjo vrednost značilnosti serije. Serija se vzorči tako, da se večkrat odvzamejo posamezni vzorci na različnih mestih v seriji. Posamezni vzorci se z mešanjem združijo v zbirni vzorec, iz katerega se pripravijo reprezentativni končni vzorci z reprezentativno delitvijo.
Če se na podlagi vizualnega pregleda deležev krme za vzorčenje ugotovi, da se ti po kakovosti razlikujejo od preostale krme iz iste serije, se taki deleži ločijo od preostale krme in obravnavajo kot ločena podserija. Če krme ni mogoče razdeliti v ločene podserije, se vzorči kot ena serija. V takih primerih je to treba navesti v zapisniku o vzorčenju.
Če se ugotovi, da krma, ki je vzorčena v skladu z določbami te uredbe, del serije krme iz iste kategorije ali z istim opisom za katerega je ugotovljeno da ne izpolnjuje zahtev EU, se domneva, da je enako neustrezna vsa krma v zadevni seriji, razen če po podrobni oceni ni nobenega dokaza, da preostanek serije ne izpolnjuje zahtev EU.
2. OPREDELITEV POJMOV
|
— |
Lot (ali serija) : določena količina krme s skupnimi značilnostmi, kot so izvor, sorta, vrsta pakiranja, izvajalec pakiranja, pošiljatelj ali označevanje, in v primeru proizvodnega postopka enota proizvodnje iz posameznega obrata, pri kateri so uporabljeni enotni proizvodni parametri, ali več takšnih enot, kadar se proizvedejo v zaporednem vrstnem redu in se skupaj skladiščijo. |
|
— |
Vzorčeni delež : serija ali opredeljeni del serije oziroma podserije. |
|
— |
Zapečateni vzorec : vzorec, ki je zapečaten na način, ki preprečuje kakršen koli dostop do vzorca, pri katerem se pečat ne bi zlomil ali odstranil. |
|
— |
Posamezni vzorec : količina, ki se odvzame z ene točke vzorčenega deleža. |
|
— |
Zbirni vzorec : zbirek posameznih vzorcev, odvzetih iz istega vzorčenega deleža. |
|
— |
Zmanjšani vzorec : del zbirnega vzorca, pridobljen z reprezentativnim zmanjšanjem istega vzorca. |
|
— |
Končni vzorec : del zmanjšanega vzorca ali homogeniziranega zbirnega vzorca. |
|
— |
Laboratorijski vzorec : vzorec, namenjen za laboratorij (kakor ga sprejme laboratorij), in je lahko končni vzorec, zmanjšan vzorec oziroma zbirni vzorec. |
3. SPLOŠNE DOLOČBE
— Osebje, ki jemlje vzorce: vzorce jemljejo osebe, ki jih za to pooblasti pristojni organ.
— Vzorec mora biti zapečaten tako, da se vanj ne more poseči, ne da bi se pri tem zlomil ali odstranil pečat. Oznaka na pečatu mora biti jasno razpoznavna in vidna. Lahko pa se vzorec položi tudi v sprejemno posodo, ki se lahko zapre tako, da je ni mogoče odpreti, ne da bi se pri tem nepopravljivo poškodovala zbiralna posoda ali posoda, s čimer se prepreči ponovna uporaba zbiralne posode ali posode.
— Identifikacija vzorca: vzorec mora biti neizbrisno označen in identificiran tako, da je nedvoumno povezan z zapisnikom o vzorčenju.
— Iz vsakega zbirnega vzorca se odvzameta najmanj dva končna vzorca: vsaj en vzorec za nadzor (uradni nadzor) in en vzorec za nosilca dejavnosti poslovanja s krmo (dopolnilno izvedensko mnenje). Lahko se odvzame tudi en končni vzorec za referenčne namene. Če je ves zbirni vzorec homogeniziran, se končni vzorci odvzamejo iz homogeniziranega zbirnega vzorca, če tak postopek ni v nasprotju s pravili držav članic o pravicah nosilca dejavnosti poslovanja s krmo.
4. OPREMA
|
4.1 |
Oprema za vzorčenje mora biti izdelana iz snovi, ki ne morejo onesnažiti proizvodov, katerih vzorci se jemljejo. Oprema, ki je namenjena večkratni uporabi, mora biti preprosta za čiščenje, da se prepreči navzkrižno onesnaženje. |
|
4.2 |
Priporočena oprema za vzorčenje trdne krme
4.2.1 Ročno vzorčenje 4.2.1.1 Lopatka z ravnim dnom in navpičnimi stranicami.
4.2.2 Mehansko vzorčenje Za vzorčenje premikajoče se krme se lahko uporablja ustrezna mehanska oprema. Ustrezna pomeni, da se vzorči najmanj ves del pretoka. Vzorčenje premikajoče se krme (pri visokih hitrostih pretoka) se lahko opravi z avtomatskimi napravami za vzorčenje. 4.2.3 Razdelilnik Če je to mogoče in primerno, se mora naprava, namenjena za delitev vzorca na približno enake dele, uporabiti za pripravo reprezentativnih zmanjšanih vzorcev. |
5. KOLIČINSKE ZAHTEVE GLEDE ŠTEVILA POSAMEZNIH VZORCEV
— Količinske zahteve v točkah 5.1 in 5.2 glede števila posameznih vzorcev veljajo za velikosti vzorčenih deležev do največ 500 ton, ki se lahko reprezentativno vzorčijo. Opisani postopek vzorčenja velja tudi za količine, večje od predpisane največje velikosti vzorčenega deleža, če se ne upošteva največje število posameznih vzorcev, navedeno v spodnjih preglednicah, pri čemer se število posameznih vzorcev določi s formulo kvadratnega korena, navedeno v ustreznem delu postopka (glej točko 5.3), najmanjša velikost zbirnega vzorca pa se sorazmerno poveča. To ne onemogoča delitve večje serije v manjše podserije, pri čemer se vsaka podserija vzorči v skladu s postopkom iz točk 5.1 in 5.2.
— Velikost vzorčenega deleža mora biti takšna, da se lahko vzorči vsak njegov sestavni del.
— Za zelo velike serije ali podserije (> 500 ton) in za serije, ki se prevažajo ali skladiščijo tako, da ni mogoče opraviti vzorčenja v skladu s postopkom vzorčenja iz točk 5.1 in 5.2 tega poglavja, se uporabi postopek vzorčenja iz točke 5.3.
— Če zakonodaja določa, da mora nosilec dejavnosti poslovanja s krmo ravnati v skladu s to uredbo v okviru obveznega sistema spremljanja, lahko nosilec dejavnosti poslovanja s krmo zaradi upoštevanja operativnih značilnosti odstopa od količinskih zahtev iz tega poglavja, pod pogojem, da pristojnemu organu predloži dokaz o enakovrednosti postopka vzorčenja, s katerim se pristojni organ strinja.
— V izjemnih primerih, če ni mogoče izvesti metode vzorčenja z opredeljenimi količinskimi zahtevami zaradi nesprejemljivih poškodb serije pri trženju (zaradi oblik pakiranj, prevoznih sredstev, načina skladiščenja itd.), se lahko uporabi alternativna metoda vzorčenja, pod pogojem, da je kar najbolj reprezentativna ter obširno opisana in dokumentirana.
5.1 Količinske zahteve glede posameznih vzorcev pri nadzoru snovi ali proizvodov, ki so enakomerno porazdeljeni po krmi
5.1.1 Razsuta trdna krma
|
Velikost vzorčenega deleža |
Najmanjše število posameznih vzorcev |
|
≤ 2,5 tone |
7 |
|
> 2,5 tone |
√20-kratnik mase vzorčenega deleža (1) v tonah, do največ 40 posameznih vzorcev |
|
(*1) Kadar število ni celo število, se zaokroži navzgor na naslednje celo število. |
|
5.1.2 Razsuta tekoča krma
|
Velikost vzorčenega deleža |
Najmanjše število posameznih vzorcev |
|
≤ 2,5 tone ali ≤ 2 500 litrov |
4 (1) |
|
> 2,5 tone ali > 2 500 litrov |
7 (1) |
|
(*1) Če tekočine ni mogoče homogenizirati, je treba povečati število posameznih vzorcev. |
|
5.1.3 Pakirana krma
Krma (trdna in tekoča) je lahko pakirana v torbe, vreče, pločevinke, sode itd., ki so v preglednicah navedeni kot enote. Velike enote (≥ 500 kg ali l) je treba vzorčiti v skladu z določbami za razsuto krmo (glej točki 5.1.1 in 5.1.2)
|
Velikost vzorčenega deleža |
Najmanjše število enot, iz katerih je treba vzeti (najmanj) en posameznih vzorec (1) |
|
od 1 do 20 enot |
1 enota (2) |
|
od 21 do 150 enot |
3 enote (2) |
|
od 151 do 400 enot |
5 enot (2) |
|
> 400 enot |
¼ √število enot vzorčenega deleža (3), do 40 enot |
|
(*1) Če bi lahko odprtje enote vplivalo na analizo (na primer pokvarljiva mokra krma), se kot posamezen vzorec vzame neodprta enota. (*2) Pri enotah, katerih vsebina ne presega 1 kg oziroma 1 l, je vsebina posameznega vzorca ena izvirna enota. (*3) Kadar dobljeno število ni celo število, se zaokroži navzgor na naslednje celo število. |
|
5.1.4 Krmni bloki in mineralni lizalni kamni
Vzorči se najmanj en blok ali lizalni kamen na vzorčeni delež s 25 enotami, do največ štirih blokov ali lizalnih kamnov.
Pri blokih ali lizalnih kamnih, ki posamično ne tehtajo več kot 1 kg, je vsebina posameznega vzorca en blok ali en kamen.
5.1.5 Voluminozna krma/sveža krma
|
Velikost vzorčenega deleža |
Najmanjše število posameznih vzorcev (1) |
|
≤ 5 ton |
5 |
|
> 5 ton |
√5-kratnik mase vzorčenega deleža (2) v tonah, do največ 40 posameznih vzorcev |
|
(*1) Ugotovljeno je, da v nekaterih okoliščinah (na primer silaža) ni mogoče vzeti zahtevanih posameznih vzorcev, ne da bi pri tem nesprejemljivo poškodovali serijo. V takih primerih se lahko uporabi druga metoda vzorčenja, smernice za vzorčenje takih serij pa bodo pripravljene pred začetkom veljavnosti te uredbe. (*2) Kadar dobljeno število ni celo število, se zaokroži navzgor na naslednje celo število. |
|
5.2 Količinske zahteve glede posameznih vzorcev pri nadzoru sestavin ali snovi, za katere obstaja verjetnost, da so neenakomerno porazdeljene po krmi
Te količinske zahteve glede posameznih vzorcev se morajo uporabljati v naslednjih okoliščinah:
— nadzor nad aflatoksini, rženimi rožički, drugimi mikotoksini in škodljivimi botaničnimi nečistočami v posamičnih krmilih,
— nadzor nad navzkrižnim onesnaženjem s sestavino, vključno z gensko spremenjenimi snovmi, ali snovjo, za katero obstaja verjetnost, da je neenakomerno porazdeljena po krmi.
Če nadzorni organ utemeljeno sumi, da velja taka neenakomerna porazdelitev tudi v primeru navzkrižnega onesnaženja s sestavino ali snovjo v krmni mešanici, se lahko uporabijo količinske zahteve iz spodnje preglednice.
|
Velikost vzorčenega deleža |
Najmanjše število posameznih vzorcev |
|
< 80 ton |
Glej količinske zahteve iz točke 5.1. Število posameznih vzorcev, ki jih je treba vzeti, je treba pomnožiti z 2,5. |
|
≥ 80 ton |
100 |
5.3 Količinske zahteve glede posameznih vzorcev v primeru zelo velikih serij
V primeru velikih vzorčenih deležev (vzorčeni deleži > 500 ton) je treba vzeti 40 posameznih vzorcev + √ton pri nadzoru snovi ali proizvodov, ki so enakomerno porazdeljeni po krmi, ali 100 posameznih vzorcev + √ton pri nadzoru sestavin ali snovi, za katere obstaja verjetnost, da so neenakomerno porazdeljene po krmi.
6. KOLIČINSKE ZAHTEVE GLEDE ZBIRNEGA VZORCA
|
Zahtevan je en zbirni vzorec na vzorčeni delež. |
||
|
|
Vrsta krme |
|
|
6.1. |
Razsuta krma |
4 kg |
|
6.2. |
Pakirana krma |
4 kg (3) |
|
6.3. |
Tekoča ali poltekoča krma |
4 litri |
|
6.4. |
Krmni bloki ali mineralni lizalni kamni |
|
|
6.4.1. |
mase več kot 1 kg po kosu |
4 kg |
|
6.4.2. |
mase do 1 kg po kosu |
masa štirih izvirnih blokov ali lizalnih kamnov |
|
6.5. |
Voluminozna krma/sveža krma |
4 kg (4) |
|
(*1) Če je vzorčena krma velike vrednosti, se lahko odvzame manjša količina zbirnega vzorca, pri čemer se to opiše in dokumentira v zapisniku o vzorčenju. (*2) V skladu z določbami Uredbe Komisije (EU) št. 619/2011 z dne 24. junija 2011 o določitvi metod vzorčenja in analitskih metod za uradni nadzor krme glede prisotnosti gensko spremenjene snovi, za katero je bil začet postopek odobritve ali je odobritev zanjo potekla (UL L 166, 25.6.2011, str. 9), mora zbirni vzorec za nadzor glede prisotnosti gensko spremenjene snovi vsebovati najmanj 35 000 zrn/semen. To pomeni, da mora biti velikost zbirnega vzorca pri koruzi najmanj 10,5 kg, pri soji pa najmanj 7 kg. Pri drugih zrnih in semenih, kot so ječmen, proso, oves, riž, rž, pšenica in seme oljne ogrščice, velikost zbirnega vzorca 4 kg ustreza več kot 35 000 zrn/semen. (*3) Pri pakirani krmi včasih prav tako ni mogoče doseči velikosti zbirnega vzorca 4 kg, kar je odvisno od velikosti posamezne enote. (*4) Pri voluminozni krmi ali sveži krmi z majhno specifično težo (npr. seno, slama) mora biti najmanjša velikost zbirnega vzorca 1 kg. |
||
7. KOLIČINSKE ZAHTEVE GLEDE KONČNIH VZORCEV
Končni vzorci
Zahtevana je analiza vsaj enega končnega vzorca. Količina končnega vzorca za analizo ne sme biti manjša od:
|
trdna krma |
|
|
tekoča ali poltekoča krma |
500 ml (1) |
|
(*1) V skladu z določbami Uredbe Komisije (EU) št. 619/2011 mora končni vzorec za nadzor glede prisotnosti gensko spremenjene snovi vsebovati najmanj 10 000 zrn/semen. To pomeni, da mora biti velikost končnega vzorca pri koruzi najmanj 3 000 g, pri soji pa najmanj 2 000 g. Pri drugih zrnih in semenih, kot so ječmen, proso, oves, riž, rž, pšenica in seme oljne ogrščice, velikost končnega vzorca 500 g ustreza več kot 10 000 zrn/semen. (*2) Če je velikost končnega vzorca precej manjša od 4 kg oziroma litrov (glej opombe pod točko 6), se lahko odvzame tudi manjša količina končnega vzorca, pod pogojem, da je to opisano in dokumentirano v zapisniku o vzorčenju. (*3) Pri vzorčenju stročnic, žitnih zrn in lupinarjev za ugotavljanje ostankov pesticidov je najmanjša velikost končnega vzorca v skladu z določbami Direktive Komisije 2002/63/ES (UL L 187, 16.7.2002, str. 30) 1 kg. |
|
8. METODA VZORČENJA ZA ZELO VELIKE SERIJE ALI SERIJE, KI SE SKLADIŠČIJO ALI PREVAŽAJO TAKO, DA NI MOGOČE VZORČENJE PO CELOTNI SERIJI
8.1 Splošna načela
Če način prevoza ali skladiščenja serije ne omogoča odvzema posameznih vzorcev po celotni seriji, se vzorčenje takih serij po možnosti opravi ob pretoku serije.
V primeru velikih skladišč, namenjenih skladiščenju krme, je treba nosilce dejavnosti spodbujati, da v skladišče vgradijo opremo, ki omogoča (avtomatsko) vzorčenje celotne skladiščene serije.
Če se uporabljajo postopki vzorčenja, določeni v tem poglavju, se o postopku vzorčenja obvesti nosilca dejavnosti poslovanja s krmo. Če nosilec dejavnosti poslovanja s krmo ali njegov predstavnik izrazi dvom o postopku vzorčenja, pristojnemu organu omogoči, da na njegove stroške vzorči celotno serijo.
8.2 Velike serije, ki se prevažajo z ladjo
8.2.1 Dinamično vzorčenje velikih serij, ki se prevažajo z ladjo
Vzorčenje velikih serij na ladjah se po možnosti izvaja med samim pretokom proizvoda (dinamično vzorčenje).
Vzorčenje se izvaja po posameznih ladijskih skladiščih (fizično ločenih enotah). Ladijska skladišča se sicer praznijo drugo za drugo tako, da po prenosu v skladiščne prostore prvotne fizične ločitve ni več. Vzorčenje se zato lahko izvede na začetku, ko je proizvod še fizično ločen ali po izvedenem prenosu v skladiščne prostore.
Raztovarjanje ladje lahko traja več dni. Običajno je treba vzorčenje izvesti v rednih presledkih med celotnim trajanjem raztovarjanja. Vendar ni vedno mogoče ali primerno, da je uradni inšpektor prisoten za vzorčenje med celotnim trajanjem raztovarjanja. Zato je dopustno, da se vzorčenje opravi za del (vzorčeni delež) celotne serije. Število posameznih vzorcev se določi ob upoštevanju velikosti vzorčenega deleža.
V primeru vzorčenja dela serije krme iz iste kategorije ali z istim opisom,za katerega je ugotovljeno, da ne izpolnjuje zahtev EU, se domneva, da je enako neustrezna vsa krma v zadevni seriji, razen če po podrobni oceni ni nobenega dokaza, da preostanek serije ne izpolnjuje zahtev EU.
Inšpektor mora biti prisoten tudi, če se avtomatsko odvzame uradni vzorec. Pri avtomatskem vzorčenju s prednastavljenimi parametri, ki jih ni mogoče spremeniti med vzorčenjem, in če se posamezni vzorci zbirajo v zapečateni zbiralni posodi, da se preprečijo morebitne goljufije, mora biti inšpektor prisoten le na začetku vzorčenja, vsakokrat, ko je treba zamenjati posodo za zbiranje vzorcev, in na koncu vzorčenja.
8.2.2 Statično vzorčenje serij, ki se prevažajo z ladjo
Če se izvaja statično vzorčenje, je treba uporabiti enak postopek, kot je določen za skladiščne prostore (silose), ki so dostopni od zgoraj (glej točko 8.4.1).
Vzorčenje je treba izvajati na dostopnem delu (od zgoraj) serije/ladijskega skladišča. Število posameznih vzorcev se določi ob upoštevanju velikosti vzorčenega deleža. V primeru vzorčenja dela serije krme iz iste kategorije ali z istim opisom, za katerega je ugotovljeno,, da ne izpolnjuje zahtev EU, se domneva, da je enako neustrezna vsa krma v zadevni serije, razen če po podrobni oceni ni nobenega dokaza, da preostanek serije ne izpolnjuje zahtev EU.
8.3 Vzorčenje velikih serij, ki se skladiščijo v skladiščih
Vzorčenje je treba izvajati na dostopnem delu serije. Število posameznih vzorcev se določi ob upoštevanju velikosti vzorčenega deleža. V primeru vzorčenja dela serije krme iz iste kategorije ali z istim opisom za katerega je ugotovljeno, da ne izpolnjuje zahtev EU, se domneva, da je enako neustrezna vsa krmo v zadevni seriji, razen če po podrobni oceni ni nobenega dokaza, da preostanek serije ne izpolnjuje zahtev EU.
8.4 Vzorčenje skladiščnih prostorov (silosov)
8.4.1 Vzorčenje silosov, (zlahka) dostopnih od zgoraj
Vzorčenje je treba izvajati na dostopnem delu serije. Število posameznih vzorcev se določi ob upoštevanju velikosti vzorčenega deleža. V primeru vzorčenja dela serije krme iz iste kategorije ali z istim opisom za katerega je ugotovljeno, da ne izpolnjuje zahtev EU, se domneva, da je enako neustrezna vsa krma v zadevni seriji, razen če po podrobni oceni ni nobenega dokaza, da preostanek serije ne izpolnjuje zahtev EU.
8.4.2 Vzorčenje silosov, ki niso dostopni od zgoraj (zaprti silosi)
8.4.2.1
Krme, ki se skladišči v takih silosih, ni mogoče statično vzorčiti. Če je treba krmo v silosu vzorčiti in pošiljke ni mogoče premakniti, se je treba z nosilcem dejavnosti dogovoriti, da inšpektorja obvesti, kdaj se bo silos razkladal, da se omogoči vzorčenje med pretokom krme.
8.4.2.2
Postopek vzorčenja vključuje prenos od 50 do 100 kg krme v zbiralno posodo in odvzem vzorca iz nje. Velikost zbirnega vzorca ustreza celotni seriji, število posameznih vzorcev pa je povezano s količino iz silosa, preneseno v zbiralno posodo za vzorčenje. V primeru vzorčenja dela serije krme iz iste kategorije ali z istim opisom za katerega je ugotovljeno, da ne izpolnjuje zahtev EU, se domneva, da je enako neustrezna vsa krma v zadevni seriji, razen če po podrobni oceni ni nobenega dokaza, da preostanek serije ne izpolnjuje zahtev EU.
8.5 Vzorčenje razsute krme v velikih zaprtih zabojnikih
Take serije se pogosto lahko vzorčijo šele, ko se razkladajo. V nekaterih primerih ni mogoče opraviti raztovarjanja na točki uvoza ali kontrolni točki, zato se mora vzorčenje izvesti, ko se taki zabojniki razkladajo.
9. NAVODILA ZA JEMANJE, PRIPRAVO IN PAKIRANJE VZORCEV
9.1 Splošno
Vzorci se morajo jemati in pripravljati brez nepotrebnega odlašanja, pri tem pa se morajo upoštevati vsi potrebni previdnostni ukrepi za preprečitev spremembe ali onesnaženja proizvoda. Instrumenti ter površine in posode, namenjeni za sprejem vzorcev, morajo biti čisti in suhi.
9.2 Posamezni vzorci
Posamezne vzorce je treba vzeti naključno iz celotnega vzorčenega deleža in morajo biti približno enako veliki.
Velikost posameznega vzorca je najmanj 100 g oziroma 25 g v primeru voluminozne krme ali sveže krme z majhno specifično težo.
Če je treba v skladu s pravili za postopek vzorčenja iz točke 8 vzeti manj kot 40 posameznih vzorcev, se velikost posameznih vzorcev določi glede na zahtevano velikost zbirnega vzorca (glej točko 6).
Če se vzorčijo majhne serije pakirane krme, pri katerih je treba v skladu s količinskimi zahtevami vzeti omejeno število posameznih vzorcev, je vsebina posameznega vzorca ena izvirna enota, ki ne presega 1 kg ali 1 litra.
Če se vzorči pakirana krma, ki je sestavljena iz majhnih enot (npr. < 250 g), je velikost posameznega vzorca odvisna od velikosti enote.
9.2.1 Razsuta krma
Po potrebi se lahko vzorčenje opravi med premikanjem (nakladanjem ali raztovarjanjem) vzorčenega deleža.
9.2.2 Pakirana krma
Po izbiri zahtevanega števila enot za vzorčenje, kot je navedeno v poglavju 5, se del vsebine vsake enote odstrani s sondo ali lopatko. Po potrebi se enote izpraznijo ločeno in šele nato odvzamejo vzorci.
9.2.3 Homogenizirana ali za homogeniziranje primerna tekoča ali poltekoča krma
Po izbiri zahtevanega števila enot za vzorčenje, kot je navedeno v poglavju 5, se vsebina po potrebi homogenizira in iz vsake enote se odvzame določena količina.
Posamezni vzorci se lahko odvzamejo, ko se enota izprazni.
9.2.4 Tekoča ali poltekoča krma, ki ni primerna za homogeniziranje
Po izbiri zahtevanega števila enot za jemanje vzorcev, kot je navedeno v poglavju 5, se vzorci odvzamejo z različnih ravni.
Vzorci se lahko odvzamejo tudi, ko se enota izprazni, vendar se prvi deli zavržejo.
Vsa odvzeta količina v nobenem primeru ne sme biti manjša od 10 litrov.
9.2.5 Krmni bloki in mineralni lizalni kamni
Po izbiri zahtevanega števila blokov ali lizalnih kamnov za vzorčenje, kot je navedeno v poglavju 5, se odvzame del vsakega bloka ali lizalnega kamna. Če obstaja sum, da je blok ali lizalni kamen nehomogeniziran, se lahko kot vzorec vzame cel blok ali lizalni kamen.
Pri blokih ali lizalnih kamnih, ki posamično ne tehtajo več kot 1 kg, je vsebina posameznega vzorca en blok ali en kamen.
9.3 Priprava zbirnih vzorcev
Posamezni vzorci se zmešajo, da sestavljajo zbirni vzorec.
9.4 Priprava končnih vzorcev
Material v zbirnem vzorcu se previdno zmeša ( 4 ).
— Vsak vzorec se shrani v ustrezno posodo/zbiralno posodo. Sprejeti je treba vse potrebne previdnostne ukrepe, da se med prevozom ali shranjevanjem prepreči kakršna koli sprememba sestave vzorca, onesnaženje ali mešanje.
— Pri nadzoru sestavin ali snovi, ki so enakomerno porazdeljene po krmi, se lahko zbirni vzorec reprezentativno zmanjša na najmanj 2,0 kg ali 2,0 l(zmanjšan vzorec) ( 5 ), po možnosti z uporabo mehanskega ali avtomatskega razdelilnika. Za nadzor nad vsebnostjo ostankov pesticidov v stročnicah, žitnih zrnih in lupinarjih je najmanjša velikost zmanjšanega vzorca 3 kg. Če zaradi vrste krme ni mogoče uporabiti razdelilnika ali če ta ni na voljo, se lahko vzorec zmanjša z razdelitvijo na četrtine. Iz zmanjšanih vzorcev se nato pripravijo končni vzorci (za uradni nadzor, dopolnilno izvedensko mnenje in referenčne namene) približno enake količine, ki izpolnjujejo količinske zahteve iz poglavja 7. Pri nadzoru sestavin, vključno z gensko spremenjenimi snovmi, ali snovi, za katere obstaja verjetnost, da so neenakomerno porazdeljene po krmi, je zbirni vzorec:
—
— popolnoma homogeniziran in nato razdeljen v končne vzorce ali
— zmanjšan na najmanj 2 kg ali 2 l ( 6 ) z uporabo mehanskega ali avtomatskega razdelilnika. Le v primeru, če zaradi vrste krme ni mogoče uporabiti razdelilnika, se lahko vzorec po potrebi zmanjša z razdelitvijo na četrtine. Pri nadzoru glede prisotnosti gensko spremenjenih snovi v okviru Uredbe (EU) št. 619/2011 mora zmanjšan vzorec vsebovati najmanj 35 000 zrn/semen, da se lahko pridobijo končni vzorci za uradni nadzor, dopolnilno izvedensko mnenje in referenčne namene, ki vsebujejo najmanj 10 000 zrn/semen (glej opombo (**) v poglavju 6 in opombo (*) v poglavju 7).
9.5 Pakiranje vzorcev
Posode ali paketi se zapečatijo in označijo z etiketami tako, da jih ni mogoče odpreti, ne da bi se pri tem poškodoval pečat. Celotna etiketa mora biti vključena v pečat.
9.6 Pošiljanje vzorcev v laboratorij
Vzorec se brez nepotrebnega odlašanja pošlje v pooblaščeni laboratorij skupaj z vsemi informacijami, ki jih potrebuje oseba, ki izvaja analizo.
10. EVIDENTIRANJE VZORČENJA
Ob odvzemu vzorca se napiše zapisnik o vzorčenju, ki omogoča nedvoumno prepoznavanje vsakega vzorčenega deleža in njegove velikosti.
V zapisniku je treba navesti tudi kakršno koli odstopanje od postopka vzorčenja, določenega v tej uredbi.
Izvod zapisnika prejme laboratorij za uradni nadzor ter nosilec dejavnosti poslovanja s krmo in/ali laboratorij, ki ga določi nosilec dejavnosti poslovanja s krmo.
PRILOGA II
SPLOŠNE DOLOČBE O ANALITSKIH METODAH ZA KRMO
A. PRIPRAVA VZORCEV ZA ANALIZO
1. Namen
Spodaj navedeni postopki opisujejo pripravo vzorcev za analizo, ki se pošljejo nadzornim laboratorijem po vzorčenju v skladu z določbami iz Priloge I.
Laboratorijski vzorci se morajo pripraviti tako, da so natehtane količine, določene za analitske metode, homogene in reprezentativne za končne vzorce.
2. Previdnostni ukrepi
Postopek za pripravo vzorca je odvisen od uporabljene analitske metode in od sestavin ali snovi, ki se nadzirajo. Zato je zelo pomembno zagotoviti, da je postopek za pripravo vzorca primeren za uporabljeno analitsko metodo in sestavine ali snovi, ki se nadzirajo.
Vse potrebne operacije se morajo izvesti tako, da se čim bolj preprečita onesnaženje vzorca in sprememba njegove sestave.
Mletje, mešanje in sejanje se izvedejo brez odlašanja, tako da je vzorec čim manj izpostavljen zraku in svetlobi. Uporaba mlinov in drobilcev, pri katerih se vzorec močno segreje, ni dovoljena.
Krmo, ki je posebej občutljiva na toploto, je priporočljivo drobiti ročno. Pri delu z opremo se zagotovi, da ta ni vir onesnaženja.
Če pri pripravi ni mogoče preprečiti bistvene spremembe vsebnosti vlage v vzorcu, je treba v skladu z metodo iz dela A Priloge III določiti vsebnost vlage pred pripravo in po njej.
3. Postopek
3.1 Splošni postopek
Iz končnega vzorca se odvzame testni alikvot. Stožčasta delitev vzorca ali delitev vzorca v četrtine nista priporočljivi, ker se lahko pojavijo hude napake v delitvi testnih alikvotov.
3.1.1
— Presejani končni vzorec se premeša in zbere v primerno čisto in suho posodo z neprepustnim zamaškom. Tik preden se natehta količina za analizo (testni alikvot), se vzorec še enkrat premeša, da se zagotovi popolna homogenizacija.
3.1.2
— Če v analitski metodi ni navedeno drugače, se končni vzorec v skladu s postopkom za predhodno sušenje iz točke 4.3 metode za določanje vsebnosti vlage iz dela A Priloge III suši toliko časa, da se vsebnost vlage zniža na 8–12 %. Nato se nadaljuje, kakor je navedeno v oddelku 3.1.1.
3.1.3
— Končni vzorec se zbere v primerno čisto in suho posodo z neprepustnim zamaškom. Tik preden se natehta količina za analizo (testni alikvot), se vzorec še enkrat dobro premeša, da se zagotovi popolna homogenizacija.
3.1.4
— Končni vzorci, ki se ne morejo pripraviti v skladu z enim izmed zgoraj navedenih postopkov, se obdelajo po katerem koli drugem postopku, ki zagotavlja, da bo količina vzorca, natehtana za analizo (testni alikvot), homogena in reprezentativna za končne vzorce.
3.2 Poseben postopek v primeru vizualnega pregleda ali mikroskopije ali kadar je homogeniziran celoten zbirni vzorec
— V primeru vizualnega pregleda (brez uporabe mikroskopa) se za pregled uporabi celoten laboratorijski vzorec.
— V primeru mikroskopske preiskave lahko laboratorij zmanjša zbirni vzorec ali dodatno zmanjša zmanjšani vzorec. Končni vzorci za dopolnilno izvedensko mnenje in morebitne referenčne namene se odvzamejo v skladu s postopkom, ki je enakovreden postopku za jemanje končnega vzorca za uradni nadzor.
— Če je celoten zbirni vzorec homogeniziran, se končni vzorci odvzamejo iz homogeniziranega zbirnega vzorca.
4. Shranjevanje vzorcev
Vzorci se morajo hraniti pri temperaturi, ki ne bo spremenila njihove sestave. Vzorci, namenjeni za analizo vitaminov ali snovi, ki so posebno občutljive na svetlobo, se hranijo pod pogoji, ki preprečujejo škodljiv učinek svetlobe na vzorec.
B. DOLOČBE O REAGENTIH IN OPREMI, KI SE UPORABLJAJO PRI ANALITSKIH METODAH
1. Če v analitskih metodah ni posebej določeno, morajo biti vsi reagenti za analizo analitsko čisti (p.a.). Pri analiziranju sledov se mora čistost reagentov preveriti s slepim preizkusom. Glede na dobljene rezultate je morda potrebno dodatno čiščenje reagentov.
2. Pri vseh postopkih analitskih metod, ki vključujejo pripravo raztopin, razredčevanje, spiranje ali izpiranje in pri katerih vrsta uporabljenega topila ali razredčila ni navedena, se mora uporabiti voda. Praviloma je voda demineralizirana ali destilirana. V posameznih primerih, ki so posebej navedeni v analitskih metodah, jo je treba prečistiti s posebnimi postopki.
3. Ob upoštevanju opreme, ki je po navadi v kontrolnih laboratorijih, so pri analitskih metodah navedeni le posebni instrumenti in naprave ali tisti, ki zahtevajo posebno uporabo. Biti morajo čisti, zlasti pri določanju zelo majhnih količin snovi.
C. UPORABA ANALITSKIH METOD IN PRIKAZ REZULTATOV
1. Postopek ekstrakcije
Pri številnih metodah je določen poseben postopek ekstrakcije. Praviloma se lahko uporabijo postopki ekstrakcije, ki se razlikujejo od postopka iz metode, če je za uporabljeni postopek učinkovitost ekstrakcije za analiziran matriks dokazano enakovredna postopku iz metode.
2. Postopek čiščenja
Pri številnih metodah je določen poseben postopek čiščenja. Praviloma se lahko uporabijo drugi postopki čiščenja, ki se razlikujejo od postopka iz metode, če je za uporabljeni postopek analitska učinkovitost čiščenja za analiziran matriks dokazano enakovredna postopku iz metode.
3. Število določanj
Če je pri analizi nezaželenih snovi rezultat prvega določanja bistveno (> 50 %) nižji od specifikacije, ki se nadzira, dodatna določanja niso potrebna, če so bili uporabljeni ustrezni postopki kakovosti. V drugih primerih je potrebna dvakratna analiza (drugo določanje), da se izključi možnost notranjega navzkrižnega onesnaženja ali naključne zamenjave vzorcev. Srednja vrednost obeh določanj se ob upoštevanju merilne negotovosti uporablja za potrditev skladnosti.
Pri nadzoru navedene vsebnosti snovi ali sestavine, če rezultat prvega določanja potrdi navedeno vsebnost, tj. rezultat analize je znotraj sprejemljivih meja odstopanja navedene vsebnosti, dodatna določanja niso potrebna, če so bili uporabljeni ustrezni postopki kakovosti. V drugih primerih je potrebna dvakratna analiza (drugo določanje), da se izključi možnost notranjega navzkrižnega onesnaženja ali naključne zamenjave vzorcev. Srednja vrednost obeh določanj se ob upoštevanju merilne negotovosti uporablja za potrditev skladnosti.
V nekaterih primerih je ta sprejemljiva meja odstopanja opredeljena v zakonodaji, kot je Uredba (ES) št. 767/2009 Evropskega parlamenta in Sveta z dne 13. julija 2009 o dajanju krme v promet in njeni uporabi, spremembi Uredbe (ES) št. 1831/2003 Evropskega parlamenta in Sveta in razveljavitvi Direktive Sveta 79/373/EGS, Direktive Komisije 80/511/EGS, direktiv Sveta 82/471/EGS, 83/228/EGS, 93/74/EGS, 93/113/ES in 96/25/ES ter Odločbe Komisije 2004/217/ES ( 7 ).
4. Poročanje o uporabljeni analitski metodi
V zapisniku o analizi se navede uporabljena analitska metoda.
5. Poročanje o rezultatih analize
Rezultat analize se prikaže tako, kakor je navedeno v analitski metodi, na primerno število decimalk, in se po potrebi popravi glede na vsebnost vlage v končnem vzorcu pred pripravo.
6. Merilna negotovost in popravek za izkoristek pri analizi nezaželenih snovi
V zvezi z nezaželenimi snovmi po Direktivi 2002/32/ES se proizvod, namenjen za krmo, šteje za neskladnega z določeno najvišjo vsebnostjo, če rezultat analize za krmo z 12 % vsebnostjo vlage presega najvišjo vsebnost ob upoštevanju razširjene merilne negotovosti in popravka za izkoristek. Za oceno skladnosti se uporabi koncentracija, popravljena za izkoristek, od katere se odšteje razširjena merilna negotovost. Ta postopek se uporabi le v primerih, kadar analitska metoda omogoča oceno merilne negotovosti in popravka za izkoristek (npr. to ni možno v primeru mikroskopske analize).
O rezultatu analize se poroča (če uporabljena analitska metoda omogoča oceno merilne negotovosti in popravka za izkoristek) na naslednji način:
(a) s popravkom za izkoristek, pri čemer se navede raven izkoristka. Popravka za izkoristek ni treba uporabiti, če je izkoristek med 90–110 %;
(b) kot „x +/– U“, pri čemer je x rezultat analize in U razširjena merilna negotovost, ob uporabi faktorja pokritja 2, zaradi katerega je stopnja zanesljivosti približno 95 %.
Vendar če je rezultat analize bistveno (> 50 %) nižji od specifikacije, ki se nadzira, in je bil uporabljen ustrezni postopek kakovosti ter je namen analize le preverjanje skladnosti z zakonodajnimi določbami, se lahko o rezultatu analize poroča brez popravka za izkoristek, popravek za izkoristek in merilna negotovost pa se lahko v teh primerih izpustita.
PRILOGA III
ANALITSKE METODE ZA NADZOR SESTAVE POSAMIČNIH KRMIL IN KRMNIH MEŠANIC
À. DOLOČANJE VLAGE
1. Namen in področje uporabe
Ta metoda omogoča določanje vsebnosti vlage v krmi. Kadar krma vsebuje hlapne snovi, kot so organske kisline, je treba upoštevati, da se pri določanju vlage zajame tudi bistvena količina hlapnih snovi.
Ta metoda ne zajema analizo mlečnih izdelkov kot posamičnih krmil, analizo mineralnih snovi, analizo mineralnih mešanic, sestavljenih predvsem iz mineralnih snovi, analizo živalskih in rastlinskih maščob in olj ali analizo oljnih semen in oljnatega sadja.
2. Načelo
Vzorec se osuši pri določenih pogojih, ki se spreminjajo glede na vrsto krme. Izguba mase se določi s tehtanjem. Pri trdni krmi z visoko vsebnostjo vlage je treba izvesti predhodno sušenje.
3. Oprema
3.1 Drobilec iz materiala, ki ne absorbira vlage, in se lahko enostavno čisti, omogoča hitro in enakomerno drobljenje brez pregretja, v največji možni meri preprečuje stik z zunanjim zrakom ter ustreza zahtevam iz 4.1.1 in 4.1.2 (npr. udarni ali vodno hlajeni mikrodrobilci, konusni mlini, počasni drobilniki ali drobilniki na zobata kolesa).
3.2 Analitska tehtnica, natančna na 1 mg
3.3 Suhe posode iz nerjaveče kovine ali stekla s pokrovi, ki omogočajo zapiranje, neprepustno za zrak; delovna površina, ki omogoča razprostrtje vzorca na približno 0,3 g/cm2.
3.4 Električno ogrevan izotermičen sušilnik (± 2 oC) s primerno ventilacijo in ki zagotavlja hitro uravnavanje temperature ( 8 ).
3.5 Nastavljiv električni vakuumski sušilnik, opremljen z oljno črpalko in mehanizmom za dovajanje suhega vročega zraka ali s sušilnim sredstvom (npr. kalcijevim oksidom).
3.6 Eksikator z debelo kovinsko ali porcelansko perforirano ploščo, ki vsebuje učinkovito sušilno sredstvo.
4. Postopek
|
Opomba |
: |
Postopki, opisani v tem oddelku, se morajo izvajati takoj po odprtju posod z vzorci. Analiza se mora izvesti najmanj v dveh vzporednih določitvah. |
4.1 Priprava
4.1.1
Vzame se najmanj 50 g vzorca. Po potrebi se zdrobi ali razdeli tako, da se prepreči sprememba vsebnosti vlage (glej točko 6).
4.1.2
Vzame se najmanj 50 g vzorca. Zdrobi se tako, da najmanj 50 % delcev preide skozi sito z odprtinami velikosti 0,5 mm in jih na situ z okroglimi odprtinami velikosti 1 mm ostane največ 10 %.
4.1.3
Približno 25 g vzorca se natehta na 10 mg natančno, doda se primerna količina brezvodnega peska, stehtanega na 10 mg natančno, in se meša do nastanka homogene mase.
4.2 Sušenje
4.2.1
Posoda (3.3) s pokrovom se stehta na 1 mg natančno. V stehtano posodo se natehta 5 g vzorca na 1 mg natančno, ki se enakomerno porazdeli. Posoda brez pokrova se postavi v sušilnik, segret na 103 oC. Posoda se postavi v sušilnik v najkrajšem možnem času, da se prepreči prevelik padec temperature. Sušenje traja štiri ure od trenutka, ko je temperatura v sušilniku ponovno 103 oC. Posoda se pokrije s pokrovom, vzame iz sušilnika, pusti v eksikatorju (3.6) 30 do 45 minut, da se ohladi, in stehta na 1 mg natančno.
Krma, sestavljena zlasti iz olj in maščob, se suši v sušilniku nadaljnjih 30 minut pri 130 oC. Razlika med obema tehtanjema ne sme presegati 0,1 % vlage.
4.2.2
Posoda (3.3) s pokrovom se stehta na 0,5 mg natančno. V stehtano posodo se natehta približno 5 g zdrobljenega vzorca na 1 mg natančno, ki se enakomerno porazdeli. Posoda brez pokrova se postavi v sušilnik, segret na 130 oC. Posoda se postavi v sušilnik v najkrajšem možnem času, da se prepreči prevelik padec temperature. Sušenje traja 2 uri od trenutka, ko je temperatura v sušilniku ponovno 130 oC. Posoda se pokrije s pokrovom, vzame iz sušilnika, pusti v eksikatorju (3.6) 30 do 45 minut, da se ohladi, in stehta na 1 mg natančno.
4.2.3 Krmne mešanice, ki vsebujejo več kot 4 % saharoze ali laktoze: posamična krmila, kot so rožiči, hidrolizirani žitni proizvodi, fermentirana semena, posušena mleta pesa, ribe in raztopine sladkorja; krmne mešanice, ki vsebujejo več kot 25 % mineralnih soli, vključno s kristalno vodo.
Posoda (3.3) s pokrovom se stehta na 0,5 mg natančno. V stehtano posodo se natehta približno 5 g vzorca na 1 mg natančno, ki se enakomerno porazdeli. Posoda brez pokrova se postavi v vakuumski sušilnik (3.5), segret na 80–85 oC. Posoda se postavi v sušilnik v najkrajšem možnem času, da se prepreči prevelik padec temperature.
Počaka se, da tlak naraste do 100 torov in pri tem tlaku sušenje traja štiri ure na vročem suhem zraku ali s sušilnim sredstvom (približno 300 g za 20 vzorcev). V zadnjem primeru se vakuumska črpalka izklopi, ko je želeni tlak dosežen. Čas sušenja se računa od trenutka, ko je temperatura v sušilniku ponovno 80–85 oC. Tlak v sušilniku se previdno izenači z atmosferskim. Sušilnik se odpre, posoda se takoj pokrije s pokrovom in vzame iz peči, pusti se 30 do 45 minut v eksikatorju (3.6), da se ohladi, in stehta na 1 mg natančno. Suši se nadaljnjih 30 minut v vakuumskem sušilniku pri 80–85 oC in ponovno stehta. Razlika med obema tehtanjema ne sme presegati 0,1 % vlage.
4.3 Predhodno sušenje
4.3.1
Trdna krma z visoko vsebnostjo vlage, ki jo je težko drobiti, mora biti predhodno sušena na naslednji način:
v primerno posodo (npr. aluminijast krožnik velikosti 20 × 12 cm in z robom 0,5 cm) se natehta 50 g nezdrobljenega vzorca na 10 mg natančno (stisnjeno ali zgoščeno krmo se lahko po potrebi na grobo razdeli). V sušilniku se suši pri 60–70 oC, dokler se vsebnost vlage ne zniža na 8–12 %. Posoda se vzame iz sušilnika in eno uro ohlaja v laboratoriju brez pokrova, nato pa stehta na 10 mg natančno. Krma se takoj zdrobi, kakor je navedeno v točki 4.1.1, in osuši, kakor je navedeno v točki 4.2.1 ali 4.2.3 glede na vrsto krme.
4.3.2
Zrna z vsebnostjo vlage nad 17 % morajo biti predhodno sušena na naslednji način:
v primerno posodo (npr. aluminijast krožnik velikosti 20 × 12 cm in z robom 0,5 cm) se natehta 50 g nezdrobljenega zrna na 10 mg natančno. Suši se 5 do 7 minut v sušilniku pri 130 oC. Posoda se vzame iz sušilnika in 2 uri ohlaja v laboratoriju brez pokrova ter nato stehta na 10 mg natančno. Krma se takoj zmelje, kakor je navedeno v točki 4.1.2, in osuši, kakor je navedeno v točki 4.2.2.
5. Izračun rezultatov
Vsebnost vlage (X), izražena kot masni delež vzorca, se izračuna po naslednji formuli:
5.1 Sušenje brez predhodnega sušenja

pri čemer je:
|
m |
= |
začetna masa vzorčenega deleža v gramih; |
|
m0 |
= |
masa suhega vzorčenega deleža v gramih. |
5.2 Sušenje s predhodnim sušenjem
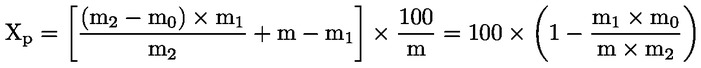
pri čemer je:
|
m |
= |
začetna masa vzorčenega deleža v gramih; |
|
m1 |
= |
masa vzorčenega deleža po predhodnem sušenju v gramih; |
|
m2 |
= |
masa vzorčenega deleža po drobljenju ali mletju v gramih; |
|
m0 |
= |
masa suhega vzorčenega deleža v gramih. |
5.3 Ponovljivost
Razlika med rezultatoma dveh vzporednih določanj, izvedenih na istem vzorcu, ne presega 0,2 % absolutne vrednosti vlage.
6. Opomba
Če se izkaže, da je potrebno drobljenje, in če se izkaže, da se pri tem spremeni vsebnost vlage v proizvodu, se morajo rezultati analize sestavin krme popraviti glede na vsebnost vlage v vzorcu v prvotnem stanju.
B. DOLOČANJE VLAGE V MAŠČOBAH IN OLJIH ŽIVALSKEGA IN RASTLINSKEGA IZVORA
1. Namen in področje uporabe
Ta metoda se uporablja za določanje vsebnosti vode in hlapnih snovi v maščobah in oljih živalskega in rastlinskega izvora.
2. Načelo
Vzorec se posuši do konstantne mase (izguba mase med dvema zaporednima tehtanjema ne sme presegati 1 mg) pri 103 oC. Izguba mase se določi s tehtanjem.
3. Oprema
3.1 Posoda z ravnim dnom iz nerjavečega materiala s premerom 8 do 9 cm in višino približno 3 cm
3.2 Termometer z utrjeno bučko in podaljškom na zgornjem delu, s skalo od približno 80 oC do vsaj 110 oC in dolžino približno 10 cm
3.3 Peščena kopel ali električna plošča
3.4 Eksikator, ki vključuje učinkovito sušilo
3.5 Analitska tehtnica
4. Postopek
Približno 20 g homogeniziranega vzorca se natehta na 1 mg natančno in prenese v suho, stehtano posodo (3.1), v kateri je termometer (3.2). Ob nenehnem mešanju s termometrom se segreva na peščeni kopeli ali vroči plošči (3.3) tako, da temperatura doseže 90 oC v približno 7 minutah.
Temperatura se zniža, pri čemer se opazuje pogostost dviganja mehurčkov z dna posode. Temperatura ne sme preseči 105 oC. Nadaljuje se z mešanjem in pri tem se drgne dno posode, dokler mehurčki ne prenehajo izhajati.
Da se zagotovi popolna odstranitev vlage, se večkrat segreje na 103 ± 2 oC ob ohlajevanju na 93 oC med zaporednimi segrevanji. Nato se pusti, da se vzorec v eksikatorju (3.4) ohladi na sobno temperaturo, in se stehta. Postopek se ponavlja, dokler izguba mase med dvema zaporednima tehtanjema ni manjša od 2 mg.
|
Opomba |
: |
Povečanje mase vzorca po ponovljenem segrevanju kaže na oksidacijo maščobe, v tem primeru je treba upoštevati rezultat tehtanja, izvedenega neposredno pred začetkom povečevanja mase. |
5. Izračun rezultatov
Masni delež vlage (X) v vzorcu se izračuna po naslednji formuli:

pri čemer je:
|
m |
= |
masa vzorčenega deleža v gramih; |
|
m1 |
= |
masa posode z vsebino pred segrevanjem v gramih; |
|
m2 |
= |
masa posode z vsebino po segrevanju v gramih. |
Rezultati, nižji od 0,05 %, se navedejo kot „manj kot 0,05 %“.
Ponovljivost
Razlika med rezultatoma dveh vzporednih določanj, izvedenih na istem vzorcu, ne sme presegati 0,05 % absolutne vrednosti.
C. DOLOČANJE VSEBNOSTI SUROVIH BELJAKOVIN
1. Namen in področje uporabe
Ta metoda se uporablja za določanje vsebnosti surovih beljakovin v krmi na podlagi vsebnosti dušika, določenega po Kjeldahlovi metodi.
2. Načelo
Vzorec se razkroji z žveplovo kislino v prisotnosti katalizatorja. Raztopina natrijevega hidroksida se doda kisli raztopini, da postane bazična. Amonijak se destilira in zbere v odmerjeno količino žveplove kisline, katere presežek se titrira s standardno raztopino natrijevega hidroksida.
Sproščeni amonijak se lahko destilira v presežek raztopine borove kisline ter nato titrira z raztopino klorovodikove ali žveplove kisline.
3. Reagenti
3.1 Kalijev sulfat
3.2 Katalizator: bakrov (II) oksid CuO ali bakrov (II) sulfat pentahidrat, CuSO4 5H2O
3.3 Cink v zrncih
3.4 Žveplova kislina, ρ20 = 1,84 g/ml
3.5 Žveplova kislina, standardna volumetrična raztopina, c(H2SO4) = 0,25 mol/l
3.6 Žveplova kislina, standardna volumetrična raztopina, c(H2SO4) = 0,10 mol/l
3.7 Žveplova kislina, standardna volumetrična raztopina, c(H2SO4) = 0,05 mol/l
3.8 Indikator metil rdeče; 300 mg metil rdečega se raztopi v 100 ml etanola, σ = 95–96 % (v/v)
3.9 Raztopina natrijevega hidroksida (lahko tehnična) β = 40 g/100 ml (m/v: 40 %)
3.10 Žveplova kislina, standardna volumetrična raztopina, c(NaOH) = 0,25 mol/l
3.11 Žveplova kislina, standardna volumetrična raztopina, c(NaOH) = 0,10 mol/l
3.12 Zdrobljen plovec, opran v klorovodikovi kislini in prežaren
3.13 Acetanilid (tališče = 114 oC, N-vsebnost = 10,36 %)
3.14 Saharoza (brez dušika)
3.15 Borova kislina (H3BO3)
3.16 Raztopina indikatorja metil rdeče: 100 mg metil rdečega se raztopi v 100 ml etanola ali metanola.
3.17 Raztopina bromokrezol zelenega: 100 mg bromokrezol zelenega se raztopi v 100 ml etanola ali metanola.
3.18 Raztopina borove kisline (10–40 g/l glede na uporabljeno opremo)
Pri določanju s kolorimetrično ekvivalentno točko se morata raztopinam borove kisline dodati indikatorja metil rdeče in bromokrezol. Pri pripravi 1 litra raztopine borove kisline se pred uravnavanjem volumna doda 7 ml raztopine indikatorja metil rdeče (3.16) in 10 ml raztopine bromokrezol zelenega (3.17).
Glede na uporabljeno vodo se lahko pH raztopine borove kisline med serijami razlikuje. Pogosto je za doseganje pozitivnega slepega vzorca treba dodati majhen volumen baze.
|
Opomba |
: |
3 do 4 ml NaOH (3.11) v 1 liter 10 g/l borove kisline ponavadi zadošča. Raztopina se shrani pri sobni temperaturi ter zaščiti pred svetlobo in viri amonijevih hlapov. |
3.19 Klorovodikova kislina, standardna volumetrična raztopina, c(HCl) = 0,10 mol/l
|
Opomba |
: |
Druge koncentracije volumetričnih raztopin (3.5, 3.6, 3.7, 3.10, 3.11 in 3.19) se lahko uporabljajo, če se upoštevajo v izračunih. Koncentracije se vedno izrazijo na štiri decimalke natančno. |
4. Oprema
Naprava, primerna za izvajanje razklopa, destilacije in titracije po Kjeldahlovi metodi.
5. Postopek
5.1 Razklop
1 g vzorca se natehta na 0,001 g natančno in se prenese v bučko naprave za razklop. Doda se 15 g kalijevega sulfata (3.1), ustrezna količina katalizatorja (3.2) (0,3 –0,4 g bakrovega (II) oksida ali 0,9 –1,2 g bakrovega (II) sulfat pentahidrata), 25 ml žveplove (VI) kisline (3.4) in po potrebi nekaj zrnc plovca (3.12) ter vse skupaj premeša.
Bučka se najprej zmerno segreva in po potrebi občasno obrne, dokler masa ne zogleni in pena ne izgine; nato se močneje segreva do enakomernega vretja tekočine. Segrevanje je ustrezno, če se vrela kislina kondenzira na stenah bučke. Paziti je treba, da se stene bučke ne pregrejejo in da se organski delci ne sprimejo nanje.
Ko se raztopina zbistri in obarva rahlo zeleno, se pusti vreti še 2 uri, nato pa pusti, da se ohladi.
5.2 Destilacija
Previdno se doda toliko vode, da se sulfati povsem raztopijo. Pusti se, da se ohladi, nato pa se po potrebi doda nekaj zrnc cinka (3.3). Nadaljuje se v skladu s točko 5.2.1 ali 5.2.2.
5.2.1
V zbirno bučko naprave za destilacijo se doda natančno 25 ml žveplove kisline (3.5) ali (3.7) glede na pričakovano vsebnost dušika. Doda se nekaj kapljic indikatorja metil rdeče (3.8).
Bučka za razklop se poveže s kondenzatorjem naprave za destilacijo in konica kondenzatorja se potopi v tekočino v zbirni bučki vsaj do globine 1 cm (glej opombo 8.3). V bučko za razklop se počasi vlije 100 ml raztopine natrijevega hidroksida (3.9) tako, da ne pride do izgube amonijaka (glej opombo 8.1). Bučka se segreva do popolne destilacije amonijaka.
5.2.2.
Pri ročni titraciji amonijaka iz destilata se uporablja spodaj navedeni postopek. Kadar je destilacijska enota v celoti avtomatizirana in vključuje titracijo amonijaka iz destilata, se upoštevajo navodila proizvajalca za upravljanje z destilacijsko enoto.
Zbirna bučka s 25–30 ml raztopine borove kisline (3.18) se postavi pod izhodno odprtino kondenzatorja tako, da je dovodna cev pod površino presežka raztopine borove kisline. Destilacijska enota se nastavi tako, da se dovaja 50 ml raztopine natrijevega hidroksida (3.9). Destilacijska enota se upravlja v skladu z navodili proizvajalca, sproščeni amonijak pa se destilira z raztopino natrijevega hidroksida. Destilat se zbere v raztopino borove kisline. Na količino destilata (čas parne destilacije) vpliva količina dušika v vzorcu. Upoštevati je treba navodila proizvajalca.
|
Opomba |
: |
V polavtomatski destilacijski enoti sta dodajanje presežka natrijevega hidroksida in parna destilacija avtomatizirana postopka. |
5.3 Titracija
Nadaljuje se v skladu s točko 5.3.1 ali 5.3.2.
5.3.1
Presežek žveplove kisline v zbirni bučki se titrira do ekvivalentne točke z raztopino natrijevega hidroksida (3.10 ali 3.11) glede na koncentracijo uporabljene žveplove kisline.
5.3.2
Vsebina zbirne bučke se titrira s standardno volumetrično raztopino klorovodikove kisline (3.19) ali standardno volumetrično raztopino žveplove kisline (3.6), pri čemer se uporabi bireta in odčita količina uporabljene titracijske snovi.
Pri določanju s kolorimetrično ekvivalentno točko je ekvivalentna točka dosežena pri prvem znaku rožnatega obarvanja vsebine. Volumen birete se odčita na 0,05 ml natančno. Magnetna mešalna plošča z osvetlitvijo ali fotometrični detektor lahko pripomoreta k določanju ekvivalentne točke.
Parni destilator z avtomatično titracijo lahko to opravi avtomatično.
Upoštevati je treba navodila proizvajalca za upravljanje specifičnega destilatorja ali destilatorja/titratorja.
|
Opomba |
: |
Pri avtomatičnem sistemu titriranja se titracija začne takoj po začetku destilacije, uporabi pa se 1-odstotna raztopina borove kisline (3.18). Pri popolnoma avtomatizirani destilacijski enoti se lahko izvaja avtomatična titracija amonijaka, ekvivalentna točka pa se lahko določa s potenciometričnim pH sistemom. V tem primeru se uporablja avtomatični titrator s pH metrom. Ta pH meter se ustrezno umeri med pH 4 in pH 7 z običajnimi laboratorijskimi postopki za umerjanje pH. Ekvivalentna točka pH pri titraciji je dosežena pri pH 4,6 , ko je naklon titracijske krivulje največji (točka pregiba). |
5.4 Slepi preskus
Slepi preskus (razklop, destilacija in titracija), v katerem se namesto vzorca uporabi 1 g saharoze (3.14), se izvede za potrditev, da reagenti ne vsebujejo dušika.
6. Izračun rezultatov
Izračuni se izvedejo v skladu s točko 6.1 ali 6.2.
6.1 Izračun za titracijo v skladu s točko 5.3.1
Vsebnost surovih beljakovin, izražena kot masni delež, se izračuna po naslednji formuli:

pri čemer je:
|
Vo |
= |
volumen (ml) NaOH (3.10 ali 3.11), uporabljen pri slepem preskusu; |
|
V1 |
= |
volumen (ml) NaOH (3.10 ali 3.11), uporabljen pri titraciji vzorca; |
|
c |
= |
koncentracija (mol/l) natrijevega hidroksida (3.10 ali 3.11); |
|
m |
= |
masa (g) vzorca. |
6.2 Izračun za titracijo v skladu s točko 5.3.2
6.2.1
Vsebnost surovih beljakovin, izražena kot masni delež, se izračuna po naslednji formuli:

pri čemer je:
|
m |
= |
masa (g) vzorčenega deleža; |
|
c |
= |
koncentracija (mol/l) standardne volumetrične raztopine klorovodikove kisline (3.19); |
|
V0 |
= |
volumen (ml) klorovodikove kisline, uporabljen pri slepem preskusu; |
|
V1 |
= |
volumen (ml) klorovodikove kisline, uporabljen v vzorčenem deležu. |
6.2.2
Vsebnost surovih beljakovin, izražena kot masni delež, se izračuna po naslednji formuli:

pri čemer je:
|
m |
= |
masa (g) vzorčenega deleža; |
|
c |
= |
koncentracija (mol/l) standardne volumetrične raztopine žveplove kisline (3.6); |
|
V0 |
= |
volumen (ml) žveplove kisline (3.6), uporabljen v slepem preskusu; |
|
V1 |
= |
volumen (ml) žveplove kisline (3.6), uporabljen v vzorčenem deležu. |
7. Preverjanje metode
7.1 Ponovljivost
Razlika med rezultatoma dveh vzporednih določanj, izvedenih na istem vzorcu, ne sme presegati:
— 0,2 % absolutne vrednosti za vsebnosti surovih beljakovin pod 20 %,
— 1,0 % višje vrednosti za vsebnosti surovih beljakovin 20–40 %,
— 0,4 % absolutne vrednosti za vsebnosti surovih beljakovin nad 40 %.
7.2 Točnost
Za analizo (razklop, destilacija, titracija) se uporabi 1,5 do 2,0 g acetanilida (3.13) ob navzočnosti 1 g saharoze (3.14); za 1 g acetanilida se porabi 14,80 ml žveplove kisline (3.5). Izkoristek mora biti najmanj 99-odstoten.
8. Opombe
8.1 Naprava je lahko ročna, polavtomatska ali avtomatska. Če je pri napravi med razklopom in destilacijo potreben prenos, se ta prenos mora izvesti brez kakršne koli izgube. Če bučka naprave za destilacijo ni opremljena s kapalnim lijakom, se natrijev hidroksid doda tik pred povezavo bučke s kondenzatorjem, pri čemer se tekočina vliva počasi ob steni bučke.
8.2 Če se vsebina bučke strjuje, se določanje začne znova, vendar z večjo količino žveplove kisline (3.4), kakor je navedeno zgoraj.
8.3 Pri proizvodih z nizko vsebnostjo dušika se lahko volumen žveplove kisline (3.7), ki se doda v zbirno bučko, po potrebi zmanjša na 10 ali 15 ml, bučka pa se dopolni do 25 ml z vodo.
8.4 Pri redni analizi se lahko za določanje surovih beljakovin uporabi nadomestna analitska metoda, vendar je Kjeldahlova metoda, opisana v tem delu C, referenčna metoda. Za vsako matrico posebej je treba dokazati, da so rezultati, pridobljeni z nadomestno metodo (npr. DUMAS), enakovredni rezultatom, pridobljenim z referenčno metodo. V poročilu o analizi je treba navesti analitsko metodo, uporabljeno za določanje surovih beljakovin, saj lahko rezultati, pridobljeni z nadomestno metodo, kljub potrditvi enakovrednosti, odstopajo od rezultatov, pridobljenih z referenčno metodo.
D. DOLOČANJE SEČNINE
1. Namen in področje uporabe
Ta metoda omogoča določanje ravni sečnine v krmi.
2. Načelo
Vzorec se suspendira v vodi s snovjo za zbistritev. Suspenzija se filtrira. Doda se 4-dimetilaminobenzaldehid (4-DMAB), nato pa se določi vsebnost sečnine v filtratu z merjenjem optične gostote pri valovni dolžini 420 nm.
3. Reagenti
3.1 Raztopina 4-dimetilaminobenzaldehida: 1,6 g 4-DMAB se raztopi v 100 ml 96-odstotnega etanola in doda 10 ml klorovodikove kisline (ρ201,19 g/ml). Reagent se lahko hrani največ dva tedna.
3.2 Raztopina Carrez I: 21,9 g cinkovega acetata Zn(CH3COO)2 2H2O in 3 g ledoceta se raztopi v vodi. Dopolni se z vodo do 100 ml.
3.3 Raztopina Carrez II: 10,6 g kalijevega ferocianida K4 Fe (CN)6 3H2O se raztopi v vodi. Dopolni se z vodo do 100 ml.
3.4 Aktivno oglje, ki ne absorbira sečnine (treba je preveriti).
3.5 Sečnina, 0,1 -odstotna raztopina (m/v)
4. Oprema
4.1 Mešalnik: približno 35 do 40 obr./min.
4.2 Epruvete: 160 × 16 mm z brušenimi zamaški
4.3 Spektrofotometer
5. Postopek
5.1 Analiza vzorca
2 g vzorca se natehta na 1 mg natančno in skupaj z 1 g aktivnega oglja (3.4) prenese v 500-mililitrsko merilno bučko. Doda se 400 ml vode in 5 ml raztopine Carrez I (3.2), meša se približno 30 sekund in doda 5 ml raztopine Carrez II (3.3). Meša se 30 minut v mešalniku. Dopolni se z vodo do oznake, pretrese in filtrira.
5 ml prozornega brezbarvnega filtrata se odstrani in prenese v epruvete z brušenimi zamaški, doda se 5 ml raztopine 4-DMAB (3.1) ter premeša. Epruvete se postavijo v kopel z vodo pri 20 oC (+/- 4 oC). Po 15 minutah se izmeri optična gostota raztopine vzorca s spektrofotometrom pri 420 nm. Rezultat se primerja z raztopino reagentov iz slepega preskusa.
5.2 Umeritvena krivulja
V 100-mililitrske merilne bučke se prenese 1, 2, 4, 5 in 10 ml raztopine sečnine (3.5), ki se dopolnijo z vodo do oznake. Od vsake raztopine se odpipetira 5 ml, vsaki se doda 5 ml raztopine 4-DMAB (3.1), homogenizira se in izmeri optična gostota, kakor je opisano zgoraj s primerjavo s kontrolno raztopino, ki vsebuje 5 ml 4-DMAB in 5 ml vode brez sečnine. Pripravi se umeritvena krivulja.
6. Izračun rezultatov
Količina sečnine v vzorcu se določi iz umeritvene krivulje.
Rezultat se izrazi kot masni delež vzorca.
7. Opombe
7.1 Če vsebnost sečnine presega 3 %, se vzorec zmanjša na 1 g ali se prvotna raztopina razredči, tako da v 500 ml ni več kot 50 mg sečnine.
7.2 Če je vsebnost sečnine nizka, se vzorec poveča, dokler je filtrat še prozoren in brezbarven.
7.3 Če vzorec vsebuje preproste dušikove spojine, kot so aminokisline, se optična gostota meri pri 435 nm.
E. DOLOČANJE HLAPNIH DUŠIKOVIH BAZ
I. Z MIKRODIFUZIJO
1. Namen in področje uporabe
Ta metoda se uporablja za določanje vsebnosti hlapnih dušikovih baz v krmi, izraženih kot amonijak.
2. Načelo
Vzorec se ekstrahira z vodo, raztopina pa zbistri in filtrira. Hlapne dušikove baze se ločijo z mikrodifuzijo z uporabo raztopine kalijevega karbonata, zberejo v raztopino borove kisline in titrirajo z žveplovo kislino.
3. Reagenti
3.1 Trikloroocetna kislina, 20-odstotna raztopina (m/v)
3.2 Indikator: 33 mg bromokrezol zelenega in 65 mg metil rdečega se raztopi v 100 ml 95–96-odstotnega (v/v) etanola.
3.3 Raztopina borove kisline: v litrski merilni bučki se v 200 ml 95–96-odstotnega (v/v) etanola in 700 ml vode raztopi 10 g borove kisline. Doda se 10 ml indikatorja (3.2). Premeša se, po potrebi pa se barva raztopine spremeni v svetlo rdečo z dodajanjem raztopine natrijevega hidroksida. 1 ml te raztopine ustreza največ 300 μg NH3.
3.4 Nasičena raztopina kalijevega karbonata: v 100 ml vrele vode se raztopi 100 g kalijevega karbonata. Pusti se, da se ohladi, ter nato filtrira.
3.5 Žveplova kislina, 0,01 mol/l
4. Oprema
4.1 Mešalec: približno 35 do 40 obr./min.
4.2 Steklene ali plastične Conwayeve celice (glej diagram)
4.3 Mikrobirete z označenim merilom 1/100 ml
5. Postopek
10 g vzorca se natehta na 1 mg natančno in se s 100 ml vode prenese v 200-mililitrsko merilno bučko. V mešalcu se meša ali stresa 30 minut. Doda se 50 ml raztopine trikloroocetne kisline (3.1), dopolni z vodo do oznake, močno pretrese in filtrira skozi nagubani filter.
S pipeto se doda 1 ml raztopine borove kisline (3.3) v srednji del Conwayeve celice in 1 ml filtrata vzorca v zgornji del celice. Delno se pokrije z namaščenim pokrovom. V zgornji del celice se hitro nakapa 1 ml nasičene raztopine kalijevega karbonata (3.4), nato se neprepustno zapre. Celica se pazljivo obrača v vodoravni ravnini, da se reagenta premešata. Pusti se najmanj štiri ure pri sobni temperaturi ali eno uro pri temperaturi 40 oC.
Z mikrobireto (4.3) se hlapne baze v raztopini borove kisline titrirajo z žveplovo kislino (3.5).
Slepi preskus se izvede po istem postopku, vendar brez vzorca, ki se analizira.
6. Izračun rezultatov
1 ml H2SO40,01 mol/l ustreza 0,34 mg amonijaka.
Rezultat se izrazi kot masni delež vzorca.
Ponovljivost
Razlika med rezultatoma dveh vzporednih določanj, izvedenih na istem vzorcu, ne presega:
— 10 % relativne vrednosti, če je vsebnost amonijaka nižja od 1,0 %,
— 0,1 % absolutne vrednosti, če je vsebnost amonijaka 1,0 % ali več.
7. Opomba
Če vsebnost amonijaka v vzorcu presega 0,6 %, se začetni filtrat razredči.

II. Z DESTILACIJO
1. Namen in področje uporabe
Ta metoda se uporablja za določanje vsebnosti hlapnih dušikovih baz, izraženih kot amonijak, v ribji moki, ki praktično ne vsebuje sečnine. Uporablja se le za vsebnost amonijaka, ki je nižja od 0,25 %.
2. Načelo
Vzorec se ekstrahira z vodo, raztopina pa zbistri in filtrira. Hlapne dušikove baze se izločijo pri temperaturi vrelišča z dodatkom magnezijevega oksida in zberejo v določeno količino žveplove kisline, katere presežek se titrira z raztopino natrijevega hidroksida.
3. Reagenti
3.1 Trikloroocetna kislina, 20-odstotna raztopina (m/v)
3.2 Magnezijev oksid
3.3 Emulzija proti penjenju (npr. silikon)
3.4 Žveplova kislina, 0,05 mol/l
3.5 Raztopina natrijevega hidroksida, 0,1 mol/l
3.6 0,3 -odstotna raztopina metil rdečega v 95–96-odstotnem (v/v) etanola
4. Oprema
4.1 Mešalec: približno 35 do 40 obr./min.
4.2 Naprava za destilacijo po Kjeldahlu
5. Postopek
10 g vzorca se natehta na 1 mg natančno in s 100 ml vode prenese v 200-mililitrsko merilno bučko. V mešalcu se meša ali stresa 30 minut. Doda se 50 ml raztopine trikloroocetne kisline (3.1), dopolni z vodo do oznake, močno pretrese in filtrira skozi nagubani filter.
Odpipetira se volumen bistrega filtrata, ki ustreza pričakovani vsebnosti hlapnih dušikovih baz (ponavadi zadošča 100 ml). Razredči se na 200 ml, doda se 2 g magnezijevega oksida (3.2) in nekaj kapljic emulzije proti penjenju (3.3). Raztopina mora biti alkalna pri preskusu z lakmusovim papirjem, če ni, se doda magnezijev oksid (3.2). Nadaljevati je treba v skladu s točkama 5.2 in 5.3 dela C te priloge, tj. Analitske metode za določanje vsebnosti surovih beljakovin.
Slepi preskus se izvede po istem postopku, vendar brez vzorca, ki se analizira.
6. Izračun rezultatov
1 ml H2SO40,05 mol/l ustreza 1,7 mg amonijaka.
Rezultat se izrazi kot masni delež vzorca.
Ponovljivost
Relativna razlika med rezultatoma dveh vzporednih določitev, izvedenih na istem vzorcu, ne presega 10 % amonijaka.
F. DOLOČANJE AMINOKISLIN (RAZEN TRIPTOFANA)
1. Namen in področje uporabe
Ta metoda se uporablja za določanje prostih (sintetičnih in naravnih) in celotnih (vezanih s peptidi in prostih) aminokislin v krmi z uporabo analizatorja aminokislin. Uporablja se za naslednje aminokisline: cist(e)in, metionin, lizin, treonin, alanin, arginin, aspartat, glutaminsko kislino, glicin, histidin, izolevcin, levcin, fenilalanin, prolin, serin, tirozin in valin.
S metodo ni možno ločiti soli aminokislin ter D in L oblik aminokislin. Ne uporablja se za določanje triptofana ali hidroksi-analogov aminokislin.
2. Načelo
2.1 Proste aminokisline
Proste aminokisline se ekstrahirajo z razredčeno klorovodikovo kislino. Soekstrahirane dušikove makromolekule se oborijo s sulfosalicilno kislino in odstranijo s filtriranjem. pH filtrirane raztopine se uravna na 2,20 . Aminokisline se ločijo z ionsko izmenjevalno kromatografijo in določijo z reakcijo z ninhidrinom s fotometrično detekcijo pri 570 nm.
2.2 Celotne aminokisline
Postopek se izbere glede na preiskovane aminokisline. Cist(e)in in metionin se morata pred hidrolizo oksidirati v cisteinsko kislino in metionin-sulfon. Tirozin se mora določiti v hidrolizatih neoksidiranih vzorcev. Vse druge aminokisline, navedene v točki 1, se lahko določajo v oksidiranem ali neoksidiranem vzorcu.
Oksidacija se izvede pri 0 oC z mešanico peroksimravljične kisline in fenola. Presežek oksidacijskega reagenta se razgradi z natrijevim disulfitom. Oksidiran ali neoksidiran vzorec se 23 ur hidrolizira s klorovodikovo kislino (3.20). pH hidrolizata se uravna na 2,20 . Aminokisline se ločijo z ionsko izmenjevalno kromatografijo in določijo z reakcijo z ninhidrinom s fotometrično detekcijo pri 570 nm (440 nm za prolin).
3. Reagenti
Uporabiti je treba dvojno destilirano vodo ali vodo enakovredne kakovosti (prevodnost < 10 μS)
3.1 Vodikov peroksid, m (m/m) = 30 %
3.2 Mravljična kislina, m (m/m) = 98–100 %
3.3 Fenol
3.4 Natrijev disulfit
3.5 Natrijev hidroksid
3.6 5-sulfosalicilat dihidrat
3.7 Klorovodikova kislina, približno 1,18 g/ml
3.8 Tri-natrijev citrat dihidrat
3.9 2,2 '-tiodietanol (tiodiglikol)
3.10 Natrijev klorid
3.11 Ninhidrin
3.12 Petroleter, vrelišče 40–60 oC
3.13 Norlevcin ali druga spojina, primerna za uporabo kot notranji standard
3.14 Plinasti dušik (< 10 ppm kisika)
3.15 1-oktanol
3.16 Aminokisline
3.16.1 Standardne snovi iz točke 1. Čiste spojine, ki ne vsebujejo kristalne vode. Pred uporabo se suši 1 teden pod vakuumom s P2O5 ali H2SO4.
3.16.2 Cisteinska kislina
3.16.3 Metionin-sulfon
3.17 Raztopina natrijevega hidroksida, c = 7,5 mol/l:
300 g NaOH (3.5) se raztopi v vodi in dopolni z vodo do 1 litra.
3.18 Raztopina natrijevega hidroksida, c = 1 mol/l:
40 g NaOH (3.5) se raztopi v vodi in dopolni z vodo do 1 litra.
3.19 Mravljična kislina – raztopina fenola:
889 g mravljične kisline (3.2) se zmeša s 111 g vode ter se doda 4,73 g fenola (3.3).
3.20 Hidrolizna mešanica, c = 6 mol HCl/l, ki vsebuje 1 g fenola/l:
1 g fenola (3.3) se doda k 492 ml HCl (3.7) in dopolni z vodo do 1 litra.
3.21 Mešanica ekstrakcije, c = 0,1 mol HCl/l, ki vsebuje 2 % tiodiglikola: 8,2 ml HCl (3.7) se razredči s približno 900 ml vode, doda se 20 ml tiodiglikola (3.9) in dopolni z vodo do 1 litra (3.7 in 3.9 se ne smeta zmešati neposredno).
3.22 5-sulfosalicilna kislina, ß = 6 %:
60 g 5-sulfosalicilne kisline (3.6) se raztopi v vodi in dopolni z vodo do 1 litra.
3.23 Oksidacijska mešanica (peroksimravljična kislina – fenol):
0,5 ml vodikovega peroksida (3.1) se zmeša s 4,5 ml raztopine mravljične kisline in fenola (3.19) v majhni čaši. Inkubira se 1 uro pri 20–30 oC, da nastane peroksimravljična kislina, nato se ohladi v ledeni kopeli (15 min), preden se doda vzorcu.
Opozorilo: Izogibati se je treba stiku s kožo in nositi je treba zaščitna oblačila.
3.24 Citratni pufer, c = 0,2 mol Na+/l, pH 2,20 :
v približno 800 ml vode se raztopi 19,61 g natrijevega citrata (3.8), 5 ml tiodiglikola (3.9), 1 g fenola (3.3) in 16,50 ml HCl (3.7). pH se uravna na 2,20 . Dopolni se z vodo do 1 litra.
3.25 Pufri za eluiranje, pripravljeni v skladu z navodili za uporabljeni analizator (4.9)
3.26 Reagent ninhidrin, pripravljen v skladu z navodili za uporabljeni analizator (4.9)
3.27 Standardne raztopine aminokislin. Te raztopine se hranijo pri temperaturi do 5 oC.
3.27.1 Osnovna standardna raztopina aminokislin (3.16.1).
c = 2,5 μmol/ml za vsako v klorovodikovi kislini.
Lahko se priskrbi iz proste prodaje.
3.27.2 Osnovna standardna raztopina cisteinske kisline in metionin-sulfona, c = 1,25 μmol/ml.
V litrski merilni bučki se raztopi 0,2115 g cisteinske kisline (3.16.2) in 0,2265 g metionin-sulfona (3.16.3) v citratnem pufru (3.24) ter dopolni s citratnim pufrom do oznake. Hrani se največ 12 mesecev pri temperaturi do 5 oC. Ta raztopina se ne uporablja, če osnovna standardna raztopina (3.27.1) vsebuje cisteinsko kislino in metionin-sulfon.
3.27.3 Osnovna standardna raztopina notranjega standarda, npr. norlevcina, c = 20 μmol/ml.
V merilni bučki se raztopi 0,6560 g norlevcina (3.13) v citratnem pufru (3.24) in dopolni s citratnim pufrom do 250 ml. Hrani se največ 6 mesecev pri temperaturi do 5 oC.
3.27.4 Raztopina za umerjanje standardnih aminokislin, ki se uporablja s hidrolizati, c = 5 nmol/50 μl za cisteinsko kislino in metionin-sulfon ter c = 10 nmol/50 μl za druge aminokisline. V 100-mililitrski čaši se raztopi 2,2 g natrijevega klorida (3.10) s 30 ml citratnega pufra (3.24). Dodajo se 4,00 ml osnovne standardne raztopine aminokisline (3.27.1), 4,00 ml osnovne standardne raztopine cisteinske kisline in metionin-sulfona (3.27.2) ter 0,50 ml osnovne standardne raztopine notranjega standarda (3.27.3), če se uporablja. pH se uravna na 2,20 z natrijevim hidroksidom (3.18).
Vsebina se prenese v 50-mililitrsko merilno bučko, dopolni se s citratnim pufrom (3.24) do oznake in premeša.
Hrani se največ 3 mesece pri temperaturi do 5 oC.
Glej tudi opombo 9.1.
3.27.5 Raztopina za umerjanje standardnih aminokislin, ki se uporablja s hidrolizati, se pripravi v skladu s točko 5.3.3.1, za ekstrakte pa v skladu s točko 5.2. Raztopina za umerjanje se pripravi v skladu s točko 3.27.4, vendar brez natrijevega klorida.
Hrani se največ 3 mesece pri temperaturi do 5 oC.
4. Oprema
4.1 100- ali 250-mililitrska bučka z okroglim dnom in kondenzatorjem povratnega toka
4.2 100-mililitrska borosilikatna steklena posoda z zamaškom z navojem in ovita v gumo/teflon (npr. Duran, Schott) za uporabo v sušilniku
4.3 Sušilnik s prezračevanjem z umetnim dotokom zraka in regulatorjem temperature z natančnostjo, boljšo od ± 2 oC
4.4 pH meter (na tri decimalke)
4.5 Membranski filter (0,22 μm)
4.6 Centrifuga
4.7 Rotacijski vakuumski uparjalnik
4.8 Mehanski stresalnik ali magnetno mešalo
4.9 Aminokislinski analizator ali oprema za HPLC s kolono z ionskim izmenjevalcem, pripravo za ninhidrin, derivatizacijo po koloni in fotometričnim detektorjem
Kolona se napolni s smolo sulfoniranega polistirena, ki lahko loči aminokisline med seboj in od drugih materialov, ki vsebujejo ninhidrin. Črpalki s stabilnim tokom ±0,5 % zagotavljata tok v pufrski in ninhidrinski črti pri standardnem umerjanju in analizi vzorca.
Za nekatere aminokisline se lahko uporabi hidroliza v analizatorju, pri čemer je koncentracija natrija v hidrolizatu c = 0,8 mol/l in v hidrolizatu je vsa mravljična kislina, ki nastane pri oksidaciji. Druge metode ne zagotavljajo zadovoljive ločitve nekaterih aminokislin, če je v hidrolizatu preveč mravljične kisline in/ali visoka koncentracija natrijevih ionov. V tem primeru se volumen kisline po hidrolizi in pred uravnavanjem pH zmanjša z uparevanjem na približno 5 ml. Uparevanje se izvede pod vakuumom pri največ 40 oC.
5. Postopek
5.1 Priprava vzorca
Vzorec se zdrobi, tako da preide skozi 0,5 mm sito. Vzorci z visoko vlago se morajo pred drobljenjem osušiti na zraku pri temperaturi, ki ne presega 50 oC, ali osušiti z zmrzovanjem. Vzorci z visoko vsebnostjo maščobe se pred drobljenjem ekstrahirajo s petroletrom (3.12).
5.2 Določanje prostih aminokislin v krmi in premiksih
Primerna količina (1–5 g) pripravljenega vzorca (5.1) se natehta na 0,2 mg natančno in prenese v erlenmajerico, temu pa se doda 100,0 ml mešanice ekstrakcije (3.21). Z mehanskim stresalnikom ali magnetnim mešalom (4.8) se mešanica stresa 60 minut. Pusti se, da se usedlina usede na dno, in odpipetira 10,0 ml supernatanta v 100-mililitrsko čašo.
Med mešanjem se doda 5,0 ml raztopine sulfosalicilne kisline (3.22) in 5 minut nadaljuje z mešanjem s pomočjo magnetnega mešala. Supernatant se filtrira ali centrifugira, da se odstrani ves precipitat. 10,0 ml te raztopine se prenese v 100-mililitrsko čašo in pH uravnava na 2,20 z raztopino natrijevega hidroksida (3.18), s citratnim pufrom (3.24) se prenese v merilno bučko ustreznega volumna in dopolni z raztopino pufra (3.24) do oznake.
Če se uporablja notranji standard, se za vsakih 100 ml končne raztopine doda 1,00 ml notranjega standarda (3.27.3) ter dopolni z raztopino pufra (3.24) do oznake.
Nadaljuje se s kromatografijo v skladu s točko 5.4.
Če se ekstrakti ne pregledajo isti dan, se morajo hraniti pri temperaturi do 5 oC.
5.3 Določanje celotnih aminokislin
5.3.1
0,1 –1 g pripravljenega vzorca (5.1) se natehta na 0,2 mg natančno in prenese v:
— 100-mililitrsko bučko z okroglim dnom (4.1) (za odprto hidrolizo 5.3.2.3) ali
— 250-mililitrsko bučko z okroglim dnom (4.1), če je zahtevana nizka koncentracija natrija (5.3.3.1), ali
— 100-mililitrsko steklenico z zamaškom z navojem (4.2) (za zaprto hidrolizo 5.3.2.4).
V natehtanem vzorčenem deležu mora biti vsebnost dušika približno 10 mg, vsebnost vlage pa ne sme preseči 100 mg.
Bučka/steklenica se postavi v ledeno kopel in ohladi na 0 oC, doda se 5 ml oksidacijske mešanice (3.23) in meša s stekleno lopatico z ukrivljeno konico. Bučka/steklenica z lopatico se nepredušno zapre s filmom, ledena kopel, ki vsebuje nepredušno zaprto posodo, pa se postavi v hladilnik s temperaturo 0 oC in pusti 16 ur. Po 16 urah se posoda vzame iz hladilnika, presežni oksidacijski reagent pa se razgradi z dodatkom 0,84 g natrijevega disulfita (3.4).
Nadaljuje se s točko 5.3.2.1.
5.3.2
5.3.2.1
Oksidiranemu vzorcu, pripravljenemu v skladu s točko 5.3.1, se doda 25 ml hidrolizne mešanice (3.20), pri čemer je treba pazljivo sprati kakršne koli ostanke vzorca s stene posode in lopatice.
Nadaljuje se v skladu s točko 5.3.2.3 ali 5.3.2.4 odvisno od uporabljenega postopka hidrolize.
5.3.2.2
V 100- ali 250-mililitrsko bučko z okroglim dnom (4.1) ali 100-mililitrsko steklenico z zamaškom z navojem (4.2) se natehta 0,1 –1 g pripravljenega vzorca (5.1) na 0,2 mg natančno. V natehtanem vzorčenem deležu mora biti vsebnost dušika približno 10 mg. Pazljivo se doda 25 ml hidrolizne mešanice (3.20) in zmeša z vzorcem. Nadaljuje se v skladu s točko 5.3.2.3 ali 5.3.2.4.
5.3.2.3
Mešanici v bučki (pripravljeni v skladu s točko 5.3.2.1 ali 5.3.2.2) se dodajo 3 steklene kroglice, nato se pusti vreti z neprestanim mehurčenjem pri povratnem toku 23 ur. Po zaključku hidrolize se kondenzator spere s 5 ml citratnega pufra (3.24). Bučka se odstrani in ohladi v ledeni kopeli.
Nadaljuje se v skladu s točko 5.3.3.
5.3.2.4
Steklenica, ki vsebuje mešanico, pripravljeno v skladu s točko 5.3.2.1 ali 5.3.2.2, se postavi v sušilnik (4.3) pri 110 oC. V prvi uri se za preprečitev povečanja tlaka (zaradi nastanka plinastih snovi) in eksplozije zamašek z navojem postavi čez vrh steklenice. Steklenica se ne sme zapreti z zamaškom. Po eni uri se steklenica zapre z zamaškom in pusti v sušilniku (4.3) 23 ur. Po zaključku hidrolize se steklenica odstrani iz sušilnika, zamašek steklenice se pazljivo odpre, steklenica pa položi v ledeno kopel. Pusti se, da se ohladi.
Odvisno od postopka za uravnavanje pH (5.3.3) se vsebina steklenice z uporabo citratnega pufra (3.24) prenese v 250-mililitrsko čašo ali 250-mililitrsko bučko z okroglim dnom.
Nadaljuje se v skladu s točko 5.3.3.
5.3.3
Glede na odstopanje analizatorja aminokislin (4.9) za natrij se za uravnavanje pH nadaljuje v skladu točko 5.3.3.1 ali 5.3.3.2.
5.3.3.1
Ko se uporabljajo analizatorji aminokislin, pri katerih je zahtevana nizka koncentracija natrija (ko je treba zmanjšati volumen kisline), je priporočljivo uporabiti raztopino notranjega osnovnega standarda (3.27.3).
V tem primeru se hidrolizatu pred uparevanjem dodata 2,00 ml raztopine notranjega osnovnega standarda (3.27.3).
Hidrolizatu, pridobljenemu v skladu s točko 5.3.2.3 ali 5.3.2.4, se dodata 2 kaplji 1-oktanola (3.15).
Z rotacijskim uparilnikom (4.7) se pod vakuumom pri 40 oC volumen zmanjša na 5–10 ml. Če se volumen nenamerno zmanjša na manj kot 5 ml, se mora hidrolizat izločiti in analiza začeti znova.
pH se uravna na 2,20 z raztopino natrijevega hidroksida (3.18), nato pa se nadaljuje v skladu s točko 5.3.4.
5.3.3.2
Hidrolizati, pridobljeni v skladu s točko 5.3.2.3 ali 5.3.2.4, se delno nevtralizirajo, tako da se med mešanjem pazljivo doda 17 ml raztopine natrijevega hidroksida (3.17), pri čemer se mora ohranjati temperatura pod 40 oC.
Pri sobni temperaturi se pH uravna na 2,20 z raztopino natrijevega hidroksida (3.17) in nato raztopino natrijevega hidroksida (3.18). Nadaljuje se v skladu s točko 5.3.4.
5.3.4
Hidrolizat (5.3.3.1 ali 5.3.3.2), uravnan na pH, se s citratnim pufrom (3.24) prenese v 200-mililitrsko merilno bučko in dopolni s pufrom (3.24) do oznake.
Če notranji standard še ni bil uporabljen, se dodata 2,00 ml notranjega standarda (3.27.3), nato pa se dopolni s citratnim pufrom (3.24) do oznake. Temeljito se premeša.
Nadaljuje se s kromatografijo (5.4).
Če se raztopine vzorca ne pregledajo še isti dan, se morajo shraniti pri temperaturi pod 5 oC.
5.4 Kromatografija
Pred kromatografijo mora biti temperatura ekstrakta (5.2) ali hidrolizata (5.3.4) enaka sobni temperaturi. Mešanica se stresa, primerna količina pa se filtrira skozi membranski filter 0,22 μm (4.5). S to bistro raztopino se opravi ionska izmenjevalna kromatografija z analizatorjem aminokislin (4.9).
Vbrizgavanje je lahko ročno ali avtomatično. Pomembno je, da se ista količina raztopine (± 0,5 %) doda koloni za analizo standardov in vzorcev, razen ko se uporablja notranji standard, ter da je razmerje med natrijem in aminokislino v raztopinah standarda in vzorca takšno, kot se uporablja ponavadi.
Na splošno je pogostost umerjanja odvisna od stabilnosti reagenta ninhidrina in analitskega sistema. Standard ali vzorec se razredči s citratnim pufrom (3.24), da se doseže površina vrha standarda, ki je 30–200 % površine vrha aminokislin v vzorcu.
Kromatografija aminokislin se nekoliko razlikuje glede na vrsto uporabljenega analizatorja in smole. Izbrani sistem mora imeti možnost ločitve aminokisline med seboj in od materialov, ki vsebujejo ninhidrin. V delovnem območju se mora kromatografski sistem odzivati linearno na spremembe količin aminokislin, dodanih koloni.
Pri kromatografiji se uporabljajo spodaj navedena razmerja med najnižjimi in najvišjimi vrednostmi, ko se analizira istomolarna raztopina (aminokislin, ki se določajo). Istomolarna raztopina mora vsebovati vsaj 30 % največje vsebnosti vsake aminokisline, ki se lahko natančno izmeri s sistemom za analizo aminokislin (4.9).
Za ločevanje treonina od serina razmerje med najnižjo in najvišjo vrednostjo za aminokislino z nižjo vrednostjo izmed dveh prekrivajočih se aminokislin ne sme presegati 2:10. (Če se določajo le cistein, metionin, treonin in lizin, bo neustrezna ločitev od sosednjih vrhov negativno vplivala na določanje.) Za vse druge aminokisline mora biti ločevanje boljše od 1:10.
Sistem mora zagotavljati, da se lizin loči od „lizinskih artefaktov“ in ornitina.
6. Izračun rezultatov
Za vsako posamezno aminokislino se izračuna površina vzorca in najvišje vrednosti standarda ter količina (X), izražena v g aminokislin na kg vzorca.
Če se uporablja notranji standard, se pomnoži z:
![]()
|
A |
= |
površina vrha hidrolizata ali ekstrakta; |
|
B |
= |
površina vrha standardne raztopine za umerjanje; |
|
C |
= |
površina vrha hidrolizata ali ekstrakta kot notranjega standarda; |
|
D |
= |
površina vrha standardne raztopine za umerjanje kot notranjega standarda; |
|
M |
= |
molska masa aminokisline, ki se določa; |
|
c |
= |
koncentracija standarda v μmol/ml; |
|
m |
= |
masa vzorca v gramih (popravljena na prvotno maso, če je vzorec posušen ali razmaščen); |
|
V |
= |
ml celotnega hidrolizata (5.3.4) ali izračunani celotni volumen razredčitve ekstrakta (6.1) v ml. |
Cistin in cistein se določata kot cisteinska kislina v hidrolizatih oksidiranega vzorca, vendar se izračunata kot cistin (C6H12N2O4S2, M 240,30 g/mol) z uporabo molske mase 120,15 g/mol (= 0,5 x 240,30 g/mol).
Metionin se določa kot metionin-sulfon v hidrolizatih oksidiranega vzorca, vendar se izračuna kot metionin, pri čemer se uporabi molska masa metionina: 149,21 g/mol.
Dodani prosti metionin se določa po ekstrakciji kot metionin, za izračun pa se uporabi ista molska masa.
6.1 Celotni volumen razredčitve ekstraktov (F) za določanje prostih aminokislin (5.2) se izračuna po naslednji formuli:

|
V |
= |
volumen končnega ekstrakta. |
7. Ocena metode
Metoda je bila preskušena v mednarodni medlaboratorijski primerjavi, ki je bila izvedena leta 1990 s štirimi različnimi vrstami krme (mešana prašičja krma, mešanica za pitovne piščance, beljakovinski koncentrat, premiks). Rezultati, brez izjem, s srednjimi vrednostmi in standardnimi odmiki so prikazani v preglednici v tej točki:
Srednje vrednosti v g/kg
|
Referenčni material |
Aminokislina |
|||
|
Treonin |
Cist(e)in |
Metionin |
Lizin |
|
|
Mešana prašičja krma |
6,94 n = 15 |
3,01 n = 17 |
3,27 n = 17 |
9,55 n = 13 |
|
Mešanica za pitovne piščance |
9,31 n = 16 |
3,92 n = 18 |
5,08 n = 18 |
13,93 n = 16 |
|
Beljakovinski koncentrat |
22,32 n = 16 |
5,06 n = 17 |
12,01 n = 17 |
47,74 n = 15 |
|
Premiks |
58,42 n = 16 |
— |
90,21 n = 16 |
98,03 n = 16 |
|
n = število sodelujočih laboratorijev. |
||||
7.1 Ponovljivost
Ponovljivost zgoraj navedene primerjave, izražena kot „standardni odmik v laboratoriju“, je prikazana v spodnji preglednici:
Standardni odmik (Sr) v laboratoriju, izražen v g/kg
|
Referenčni material |
Aminokislina |
|||
|
Treonin |
Cist(e)in |
Metionin |
Lizin |
|
|
Mešana prašičja krma |
0,13 n = 15 |
0,10 n = 17 |
0,11 n = 17 |
0,26 n = 13 |
|
Mešanica za pitovne piščance |
0,20 n = 16 |
0,11 n = 18 |
0,16 n = 18 |
0,28 n = 16 |
|
Beljakovinski koncentrat |
0,48 n = 16 |
0,13 n = 17 |
0,27 n = 17 |
0,99 n = 15 |
|
Premiks |
1,30 n = 16 |
— |
2,19 n = 16 |
2,06 n = 16 |
|
n = število sodelujočih laboratorijev. |
||||
Koeficient variacije (%) standardnega odmika (Sr) v laboratoriju
|
Referenčni material |
Aminokislina |
|||
|
Treonin |
Cist(e)in |
Metionin |
Lizin |
|
|
Mešana prašičja krma |
1,9 n = 15 |
3,3 n = 17 |
3,4 n = 17 |
2,8 n = 13 |
|
Mešanica za pitovne piščance |
2,1 n = 16 |
2,8 n = 18 |
3,1 n = 18 |
2,1 n = 16 |
|
Beljakovinski koncentrat |
2,7 n = 16 |
2,6 n = 17 |
2,2 n = 17 |
2,4 n = 15 |
|
Premiks |
2,2 n= 16 |
— |
2,4 n = 16 |
2,1 n = 16 |
|
n = število sodelujočih laboratorijev. |
||||
7.2 Obnovljivost
Rezultati zgoraj navedene primerjave, izraženi kot standardni odmik med laboratoriji, so prikazani v spodnji preglednici:
Standardni odmik (SR) med laboratoriji, izražen v g/kg
|
Referenčni material |
Aminokislina |
|||
|
Treonin |
Cist(e)in |
Metionin |
Lizin |
|
|
Mešana prašičja krma |
0,28 n = 15 |
0,30 n = 17 |
0,23 n = 17 |
0,30 n = 13 |
|
Mešanica za pitovne piščance |
0,48 n = 16 |
0,34 n = 18 |
0,55 n = 18 |
0,75 n = 16 |
|
Beljakovinski koncentrat |
0,85 n = 16 |
0,62 n = 17 |
1,57 n = 17 |
1,24 n = 15 |
|
Premiks |
2,49 n = 16 |
— |
6,20 n = 16 |
6,62 n = 16 |
|
n = število sodelujočih laboratorijev. |
||||
Koeficient variacije (%) standardnega odmika (SR) med laboratoriji
|
Referečni material |
Amino Acid |
|||
|
Treonin |
Cist(e)in |
Metionin |
Lizin |
|
|
Mešana prašičja krma |
4,1 n = 15 |
9,9 n = 17 |
7,0 n = 17 |
3,2 n = 13 |
|
Mešanica za pitovne piščance |
5,2 n = 16 |
8,8 n = 18 |
10,9 n = 18 |
5,4 n = 16 |
|
Beljakovinski koncentrat |
3,8 n = 16 |
12,3 n = 17 |
13,0 n = 17 |
3,0 n = 15 |
|
Premiks |
4,3 n = 16 |
— |
6,9 n = 16 |
6,7 n = 16 |
|
n = število sodelujočih laboratorijev. |
||||
8. Uporaba referenčnih materialov
Pravilna uporaba metode se preveri s ponovljenimi merjenji na potrjenih referenčnih materialih, kadar so na voljo. Priporočeno je umerjanje s potrjeno raztopino za umerjanje aminokislin.
9. Opombe
9.1 Zaradi razlik med analizatorji aminokislin se kot smernica obravnavajo končne koncentracije raztopin standardnih aminokislin za umerjanje (glej 3.27.4 in 3.27.5) in hidrolizata (glej 5.3.4).
Za vse aminokisline je treba preveriti območje naprave, v katerem je odziv linearen.
Standardna raztopina se razredči s citratnim pufrom, da je površina vrha na sredini tega območja.
9.2 Kadar se za analizo hidrolizatov uporablja oprema za tekočinsko kromatografijo visoke ločljivosti, se morajo eksperimentalni pogoji optimizirati v skladu s priporočili proizvajalca.
9.3 Pri uporabi metode za krmo, ki vsebuje več kot 1 % klorida (koncentrat, mineralna krma, nadomestna krma), se lahko pojavi prenizka vrednost vsebnosti metionina in je zato potrebna posebna obdelava.
G. DOLOČANJE TRIPTOFANA
1. Namen in področje uporabe
Ta metoda omogoča določanje celotnega in prostega triptofana v krmi. Ne omogoča pa ločevanja med oblikama D in L.
2. Načelo
Za določanje celotnega triptofana se vzorec hidrolizira pri bazičnih pogojih z nasičeno raztopino barijevega hidroksida in 20 ur segreva do 110 oC. Po hidrolizi se doda notranji standard.
Za določanje prostega triptofana se vzorec ekstrahira pri rahlo kislih pogojih v prisotnosti notranjega standarda.
Triptofan in notranji standard v hidrolizatu ali ekstraktu se določata s HPLC s fluorescenčno detekcijo.
3. Reagenti
3.1 Uporabljati je treba dvakrat destilirano vodo ali vodo enake kakovosti (prevodnost < 10 μS/cm).
3.2 Standardna snov: triptofan (čistoča/vsebnost ≥ 99 %), osušen pod vakuumom nad fosforjevim pentoksidom
3.3 Snov, ki je notranji standard: α-metil-triptofan (čistoča/vsebnost ≥ 99 %), osušen pod vakuumom nad fosforjevim pentoksidom.
3.4 Barijev hidroksid oktahidrat (paziti je treba, da Ba(OH)2 .8 H2O ni pretirano izpostavljen zraku, da se ne bi tvoril BaCO3, ki lahko moti določanje) (glej opombo 9.3).
3.5 Natrijev hidroksid
3.6 Ortofosforna kislina, m (m/m) = 85 %
3.7 Klorovodikova kislina, ρ201,19 g/ml
3.8 Metanol, za HPLC
3.9 Petroleter, vrelišče 40–60 oC
3.10 Raztopina natrijevega hidroksida, c = 1 mol/l:
40,0 g NaOH (3.5) se raztopi v vodi in dopolni z vodo do 1 litra (3.1).
3.11 Klorovodikova kislina, c = 6 mol/l:
492 ml HCl (3.7) se dopolni z vodo do 1 litra.
3.12 Klorovodikova kislina, c = 1 mol/l:
82 ml HCl (3.7) se dopolni z vodo do 1 litra.
3.13 Klorovodikova kislina, c = 0,1 mol/l:
8,2 ml HCl (3.7) se dopolni z vodo do 1 litra.
3.14 Ortofosforna kislina, c = 0,5 mol/l:
34 ml ortofosforne kisline (3.6) se dopolni z vodo (3.1) do 1 litra.
3.15 Koncentrirana raztopina triptofana (3.2), c = 2,50 μmol/ml:
v 500-mililitrski merilni bučki se raztopi 0,2553 g triptofana (3.2) v klorovodikovi kislini (3.13) in dopolni s klorovodikovo kislino (3.13) do oznake. Hrani se največ 4 tedne pri –18 oC.
3.16 Koncentrirana raztopina notranjega standarda, c = 2,50 μmol/ml:
v 500-mililitrski merilni bučki se raztopi 0,2728 g α-metil-triptofana (3.3) v klorovodikovi kislini (3.13) in dopolni se s klorovodikovo kislino (3.13) do oznake. Pri –18 oC se shrani največ 4 tedne.
3.17 Standardna raztopina triptofana in notranjega standarda za umerjanje:
pripravi se 2,00 ml koncentrirane raztopine triptofana (3.15) in 2,00 ml koncentrirane raztopine notranjega standarda (α-metil-triptofan) (3.16). Razredči se z vodo (3.1) in metanolom (3.8) na približno enak volumen in približno enako koncentracijo metanola (10–30 %) kot pri končnem hidrolizatu.
Ta raztopina se mora pripraviti sveža pred uporabo.
Med pripravo jo je treba zaščititi pred neposredno sončno svetlobo.
3.18 Ocetna kislina
3.19 1,1,1-trikloro-2-metil-2-propanol
3.20 Etanolamin, m (m/m) > 98 %
3.21 1 g 1,1,1-trikloro-2-metil-2-propanola (3.19) se raztopi v 100 ml metanola (3.8).
3.22 Mobilna faza za HPLC: 3,00 g ocetne kisline (3.18) + 900 ml vode (3.1) +50,0 ml raztopine (3.21) 1,1,1-trikloro-2-metil-2-propanola (3.19) v metanolu (3.8) (1 g/100 ml). pH se uravna na 5,00 z etanolaminom (3.20). Dopolni se z vodo do 1 000 ml (3.1).
4. Oprema
4.1 Oprema HPLC s spektrofluorometričnim detektorjem
4.2 Kolona za tekočinsko kromatografijo, 125 mm x 4 mm C18, polnitev 3 μm ali enakovredna
4.3 pH meter
4.4 Bučka iz polipropilena, volumna 125 ml, s širokim vratom in zamaškom z navojem
4.5 Membranski filter, 0,45 μm
4.6 Avtoklav, 110 (± 2) oC, 1,4 (±0,1 ) bar
4.7 Mehanski stresalnik ali magnetno mešalo
4.8 Vorteks mešalnik
5. Postopek
5.1 Priprava vzorcev
Vzorec se zdrobi, tako da preide skozi 0,5 mm sito. Vzorce z visoko vsebnostjo vlage je treba pred drobljenjem osušiti na zraku pri temperaturi do 50 oC ali pa osušiti z zmrzovanjem. Vzorci z visoko vsebnostjo maščobe se pred drobljenjem ekstrahirajo s petroletrom (3.9).
5.2 Določanje prostega triptofana (ekstrakt)
Primerna količina (1–5 g) pripravljenega vzorca (5.1) se natehta na 1 mg natančno in prenese v erlenmajerico. Doda se 100,0 ml klorovodikove kisline (3.13) in 5,00 ml koncentrirane raztopine notranjega standarda (3.16). Z mehanskim stresalnikom ali magnetnim mešalom (4.7) se stresa ali meša 60 minut. Pusti se, da se usedlina usede na dno, nato pa se odpipetira 10,0 ml supernatanta v čašo. Doda se 5 ml ortofosforne kisline (3.14). pH raztopine se z natrijevim hidroksidom (3.10) uravna na 3. Doda se dovolj metanola (3.8), da je v končnem volumnu koncentracija metanola 10–30 %. Prenese se v merilno bučko primernega volumna in razredči z vodo na volumen, potreben za kromatografijo (približno enak volumen kot pri standardni raztopini za umerjanje (3.17)).
Pred vbrizganjem na HPLC kolono se nekaj ml raztopine prefiltrira skozi 0,45 μm membranski filter (4.5). Nadaljuje se s kromatografijo v skladu s točko 5.4.
Standardna raztopina in ekstrakti se zaščitijo pred neposredno sončno svetlobo. Če ni možno analizirati ekstraktov istega dne, se lahko hranijo največ 3 dni pri 5 oC.
5.3 Določanje celotnega triptofana (hidrolizat)
0,1 –1 g pripravljenega vzorca (5.1) se natehta na 0,2 mg natančno in prenese v bučko iz polipropilena (4.4). Vsebnost dušika v natehtanem delu vzorca je približno 10 mg. Doda se 8,4 mg barijevega hidroksid oktahidrata (3.4) in 10 ml vode. Meša se na vorteks mešalniku (4.8) ali z magnetnim mešalom (4.7). S teflonom prevlečeni magnet se pusti v mešanici. Stene posode se sperejo s 4 ml vode. Zapre se z zamaškom z navojem, bučka pa se rahlo zapre. Prenese se v avtoklav (4.6), kjer je vrela voda, in pusti v pari 30–60 minut. Zapre se avtoklav in 20 ur avtoklavira pri 110 (± 2) oC.
Preden se avtoklav odpre, se temperatura zmanjša nekoliko pod 100 oC. Da se prepreči kristalizacija Ba(OH)2 ×8 H2O, se topli mešanici doda 30 ml vode sobne temperature. Rahlo se pretresa ali meša. Doda se 2,00 ml koncentrirane raztopine notranjega standarda (α-metil-triptofana) (3.16). Posode se 15 minut hladijo na vodni/ledeni kopeli.
Nato se doda 5 ml ortofosforne kisline (3.14). Posoda se zadrži v hladilni kopeli, med mešanjem nevtralizira s HCl (3.11), pH pa se s HCl (3.12) uravna na 3,0 . Doda se dovolj metanola, da je koncentracija metanola v končnem volumnu 10–30 %. Prenese se v merilno bučko primernega volumna in razredči z vodo do volumna, potrebnega za kromatografijo (na primer 100 ml). Dodatek metanola ne povzroči obarjanja.
Pred vbrizganjem na HPLC kolono se nekaj ml raztopine prefiltrira skozi 0,45 μm membranski filter (4.5). Nadaljuje se s kromatografijo v skladu s točko 5.4.
Standardna raztopina in hidrolizati se zaščitijo pred neposredno sončno svetlobo. Če ni možno analizirati ekstraktov istega dne, se lahko hranijo največ 3 dni pri 5 oC.
5.4 Določanje s HPLC
Za izokratsko eluiranje se lahko kot smernica uporabljajo naslednji pogoji; lahko se uporabljajo tudi drugi pogoji, če zagotavljajo enakovredne rezultate (glej tudi opombi 9.1 in 9.2).
|
Kolona za tekočinsko kromatografijo (4.2): |
125 mm × 4 mm, C18, polnitev 3 μm ali enakovredna |
|
Temperatura kolone: |
sobna temperatura |
|
Mobilna faza (3.22): |
3,00 g ocetne kisline (3.18) + 900 ml vode (3.1) +50,0 ml raztopine (3.21) 1,1,1-trikloro-2-metil-2-propanola (3.19) v metanolu (3.8) (1 g/100 ml). pH raztopine se z etanolaminom (3.20) uravna na 5,00 . Dopolni se z vodo (3.1) do 1 000 ml. |
|
Hitrost pretoka: |
1 ml/min |
|
Skupni čas izvedbe: |
približno 34 min |
|
Detekcijska valovna dolžina: |
ekscitacija: 280 nm, emisija: 356 nm. |
|
Volumen, ki se vbrizga: |
20 μl |
6. Izračun rezultatov
Količina triptofana (X), izražena v g na 100 g vzorca, se izračuna po naslednji formuli:
|
A |
= |
površina vrha notranjega standarda, standardne raztopina za umerjanje (3.17); |
|
B |
= |
površina vrha triptofana, ekstrakta (5.2) ali hidrolizata (5.3); |
|
V1 |
= |
volumen, v ml (2 ml), koncentrirane raztopine triptofana (3.15), dodane raztopini za umerjanje (3.17); |
|
c |
= |
koncentracija, v μmol/ml (= 2,50 ), koncentrirane raztopine triptofana (3.15), dodane raztopini za umerjanje (3.17); |
|
V2 |
= |
volumen, v ml, koncentrirane raztopine notranjega standarda (3.16), dodane pri ekstrakciji (5.2) (= 5,00 ml) ali hidrolizatu (5.3) (= 2,00 ml); |
|
C |
= |
površina vrha notranjega standarda, ekstrakta (5.2) ali hidrolizata (5.3); |
|
D |
= |
površina vrha triptofana, standardne raztopine za umerjanje (3.17); |
|
V3 |
= |
volumen, v ml (= 2,00 ml), koncentrirane raztopine notranjega standarda (3.16), dodane standardni raztopini za umerjanje (3.17); |
|
m |
= |
masa vzorca v g (popravljena na prvotno maso, če je vzorec posušen in/ali razmaščen); |
|
M |
= |
molska masa triptofana (= 204,23 g/mol). |
7. Ponovljivost
Razlika med rezultatoma dveh vzporednih določanj, izvedenih na istem vzorcu, ne sme presegati 10 % najvišjega rezultata.
8. Rezultati medlaboratorijske študije
Organizirana je bila medlaboratorijska študija ES (četrta medlaboratorijska primerjava), pri kateri je 12 laboratorijev analiziralo 3 vzorce, da se potrdi metoda za hidrolizo. Vsak vzorec je bil analiziran petkrat. Rezultati so prikazani v spodnji preglednici:
|
|
Vzorec 1 Prašičja krma |
Vzorec 2 Prašičja krma z dodanim L-triptofanom |
Vzorec 3 Koncentrat prašičje krme |
|
L |
12 |
12 |
12 |
|
n |
50 |
55 |
50 |
|
Srednja vrednost [g/kg] |
2,42 |
3,40 |
4,22 |
|
Sr [g/kg] |
0,05 |
0,05 |
0,08 |
|
r [g/kg] |
0,14 |
0,14 |
0,22 |
|
CVr [ %] |
1,9 |
1,6 |
1,9 |
|
SR [g/kg] |
0,15 |
0,20 |
0,09 |
|
R [g/kg] |
0,42 |
0,56 |
0,25 |
|
CVR [ %] |
6,3 |
6,0 |
2,2 |
|
L |
= |
število laboratorijev, ki so predložili rezultate; |
|
n |
= |
število uporabljenih posameznih rezultatov, razen izjem (identificiranih s testom za izjeme po Cochranu in Dixonu); |
|
Sr |
= |
standardni odmik ponovljivosti; |
|
SR |
= |
standardni odmik obnovljivosti; |
|
r |
= |
ponovljivost; |
|
R |
= |
obnovljivost; |
|
CVr |
= |
koeficient variacije ponovljivosti v %; |
|
CVR |
= |
koeficient variacije obnovljivosti v %. |
Organizirana je bila še ena medlaboratorijska študija ES (tretja medlaboratorijska primerjava), v kateri je 13 laboratorijev analiziralo 2 vzorca, da se potrdi metoda za ekstrakcijo prostega triptofana. Vsak vzorec je bil analiziran petkrat. Rezultati so prikazani v spodnji preglednici:
|
|
Vzorec 4 Mešanica pšenice in soje |
Vzorec 5 Mešanica pšenice in soje (= vzorec 4) z dodanim triptofanom (0,457 g/kg1) |
|
L |
12 |
12 |
|
n |
55 |
60 |
|
Srednja vrednost [g/kg] |
0,391 |
0,931 |
|
Sr [g/kg] |
0,005 |
0,012 |
|
r [g/kg] |
0,014 |
0,034 |
|
CVr [ %] |
1,34 |
1,34 |
|
SR [g/kg] |
0,018 |
0,048 |
|
R [g/kg] |
0,050 |
0,134 |
|
CVR [ %] |
4,71 |
5,11 |
|
L |
= |
število laboratorijev, ki so predložili rezultate; |
|
n |
= |
število uporabljenih posameznih rezultatov, razen izjem (identificiranih s testom za izjeme po Cochranu in Dixonu); |
|
Sr |
= |
standardni odmik ponovljivosti; |
|
SR |
= |
standardni odmik obnovljivosti; |
|
r |
= |
ponovljivost; |
|
R |
= |
obnovljivost; |
|
CVr |
= |
koeficient variacije ponovljivosti v %; |
|
CVR |
= |
koeficient variacije obnovljivosti v %. |
Organizirana je bila še ena medlaboratorijska študija ES, v kateri je do 7 laboratorijev analiziralo 4 vzorce, da se potrdi metoda za hidrolizo triptofana. Rezultati so prikazani spodaj. Vsak vzorec je bil analiziran petkrat.
|
|
Vzorec 1 Mešana prašičja krma (CRM 117) |
Vzorec 2 Ribja moka z nizko vsebnostjo maščob (CRM 118) |
Vzorec 3 Sojina moka (CRM 119) |
Vzorec 4 Posneto mleko v prahu (CRM 120) |
|
L |
7 |
7 |
7 |
7 |
|
n |
25 |
30 |
30 |
30 |
|
Srednja vrednost [g/kg] |
2,064 |
8,801 |
6,882 |
5,236 |
|
Sr [g/kg] |
0,021 |
0,101 |
0,089 |
0,040 |
|
r [g/kg] |
0,059 |
0,283 |
0,249 |
0,112 |
|
CVr [ %] |
1,04 |
1,15 |
1,30 |
0,76 |
|
SR [g/kg] |
0,031 |
0,413 |
0,283 |
0,221 |
|
R [g/kg] |
0,087 |
1,156 |
0,792 |
0,619 |
|
CVR [ %] |
1,48 |
4,69 |
4,11 |
4,22 |
|
L |
= |
število laboratorijev, ki so predložili rezultate; |
|
n |
= |
število uporabljenih posameznih rezultatov, po izločitvi izjem (identificiranih s testom za izjeme po Cochranu in Dixonu); |
|
Sr |
= |
standardni odmik ponovljivosti; |
|
SR |
= |
standardni odmik obnovljivosti; |
|
r |
= |
ponovljivost; |
|
R |
= |
obnovljivost; |
|
CVr |
= |
koeficient variacije ponovljivosti v %; |
|
CVR |
= |
koeficient variacije obnovljivosti v %. |
9. Opombe
9.1 Ločevanje triptofana in α-metil-triptofana lahko izboljšajo naslednji posebni kromatografski pogoji.
Izokratsko eluiranje, ki mu sledi gradientno čiščenje kolone:
|
Kolona za tekočinsko kromatografijo: |
125 mm x 4 mm, C18, polnitev 5 μm ali enakovredna |
||
|
Temperatura kolone: |
32 oC |
||
|
Mobilna faza: |
A. 0,01 mol/l KH2PO4/Metanol, 95+5 (V+V). B: Metanol |
||
|
Gradientni program: |
0 min |
100 % A |
0 % B |
|
|
15 min |
100 % A |
0 % B |
|
|
17 min |
60 % A |
40 % B |
|
|
19 min |
60 % A |
40 % B |
|
|
21 min |
100 % A |
0 % B |
|
|
33 min |
100 % A |
0 % B |
|
Hitrost pretoka: |
1,2 ml/min |
||
|
Skupni čas izvedbe: |
približno 33 min |
||
9.2 Kromatografija se spreminja glede na vrsto HPLC in uporabljeni material za polnitev kolone. Izbrani sistem mora zagotavljati ločevanje bazne linije triptofana in notranjega standarda. Poleg tega je pomembno, da se produkti razgradnje dobro ločijo od triptofana in notranjega standarda. Izvede se postopek s hidrolizatom brez notranjega standarda, da se preveri bazna linija notranjega standarda za nečistoče. Pomembno je, da je čas izvedbe dovolj dolg, da se eluirajo vsi produkti razgradnje, sicer lahko zapozneli vrhovi eluiranja motijo nadaljnje kromatografiranje.
Kromatografski sistem se v delovnem območju odziva linearno. Linearni odziv se izmeri s konstantno (običajno) koncentracijo notranjega standarda in različnimi koncentracijami triptofana. Zelo pomembno je, da so velikosti vrhov triptofana in notranjega standarda v linearnem območju HPLC/fluorescenčnega sistema. Če so vrhovi triptofana in/ali notranjega standarda prenizki ali previsoki, se analiza ponovi z drugačno količino vzorca in/ali drugačnim končnim volumnom.
9.3 Barijev hidroksid
Topnost barijevega hidroksida se s staranjem zmanjša. Posledica tega je motna raztopina za določitev HPLC, kar lahko povzroči nizke rezultate za triptofan.
H. DOLOČANJE SUROVIH OLJ IN MAŠČOB
1. Namen in področje uporabe
Ta metoda omogoča določanje vsebnosti surovih olj in maščob v krmi. Ne omogoča analize oljnih semen in oljnatega sadja.
Vrsta in sestava krme ter namena izvajanja analize vplivata na uporabo spodaj opisanih postopkov.
1.1 Postopek A – Surova olja in maščobe, ki se ekstrahirajo neposredno
Ta metoda se uporablja za krmo rastlinskega izvora, razen tiste, ki je zajeta v področju uporabe postopka B.
1.2 Postopek B – Celotna surova olja in maščobe
Ta metoda se uporablja za krmo živalskega izvora in za vse krmne mešanice. Uporablja se za vso krmo, iz katere ni mogoče povsem ekstrahirati olj in maščob brez predhodne hidrolize (npr. gluteni, kvas, krompirjeve beljakovine in izdelki, pri katerih se izvaja izrivanje, kosmičenje in segrevanje).
1.3 Razlaga rezultatov
Vedno kadar je rezultat pri postopku B višji od rezultata pri postopku A, se kot prava vrednost sprejme rezultat, dobljen pri postopku B.
2. Načelo
2.1 Postopek A
Vzorec se ekstrahira s petroletrom. Topilo se oddestilira, ostanek pa osuši in stehta.
2.2 Postopek B
Vzorec se med segrevanjem obdela s klorovodikovo kislino. Mešanica se ohladi in filtrira. Ostanek se spere in osuši, nato pa se izvede določanje v skladu s postopkom A.
3. Reagenti
3.1 Petroleter, vrelišče 40–60 oC. Bromno število mora biti nižje od 1 in ostanek pri uparevanju nižji od 2 mg/100 ml.
3.2 Natrijev sulfat, brezvodni
3.3 Klorovodikova kislina, c = 3 mol/l
3.4 Filtracijsko sredstvo, npr. Kieselguhr, Hyflo-supercel
4. Oprema
4.1 Naprava za ekstrakcijo. Če je opremljena s sifonom (naprava Soxhlet), je hitrost povratnega toka približno 10 ciklov na uro; če ni opremljena s sifonom, je hitrost povratnega toka približno 10 ml na minuto.
4.2 Kartuše za ekstrakcijo, ki ne vsebujejo materialov, topnih v petroletru, in s poroznostjo, ki je v skladu z zahtevami iz točke 4.1.
4.3 Sušilnik, vakuumski, nastavljen na 75 oC ± 3 oC, ali zračni, nastavljen na 100 oC ± 3 oC.
5. Postopek
5.1 Postopek A (glej točko 8.1)
5 g vzorca se natehta na 1 mg natančno, prenese v kartušo za ekstrakcijo (4.2) in pokrije s čepkom iz vate, ki ne vsebuje maščobe.
Kartuša se postavi v ekstraktor (4.1) in šest ur ekstrahira s petroletrom (3.1). Ekstrakt petroletra se zbere v suho stehtano bučko, v kateri so delci plovca ( 9 ).
Topilo se oddestilira. Ostanek se osuši tako, da se bučka za uro in pol postavi v sušilnik (4.3). Pusti se, da se ohladi v eksikatorju, in stehta. Ponovno se suši 30 minut, da se zagotovi ohranitev konstantne mase olj in maščob (izguba mase med dvema zaporednima tehtanjema ne sme presegati 1 mg).
5.2 Postopek B
2,5 g vzorca se natehta na 1 mg natančno (glej točko 8.2), prenese v 400-mililitrsko čašo ali 300-mililitrsko erlenmajerico in ter doda 100 ml klorovodikove kisline (3.3) in delci plovca. Čaša se pokrije z urnim steklom ali pa se erlenmajerica spoji s kondenzatorjem povratnega toka. Pri nizkem plamenu ali na električnem kuhalniku se mešanica počasi zavre in se pusti rahlo vreti eno uro. Paziti je treba, da se vzorec ne oprijema sten posode.
Ohladi se in doda toliko filtracijskega sredstva (3.4), da med filtracijo ne pride do izgub olja in maščob. Filtrira se skozi navlažen, dvojni filtrirni papir, ki ne vsebuje maščob. Ostanek se spere s hladno vodo, dokler filtrat ni nevtralen. Prepričati se je treba, da filtrat ne vsebuje nobenih olj ali maščob. Njihova prisotnost pomeni, da se mora vzorec pred hidrolizo ekstrahirati s petroletrom po postopku A.
Dvojni filtrirni papir, na katerem je ostanek, se prenese na urno steklo in se uro in pol suši v sušilniku (4.3) pri 100 oC ± 3 oC.
Dvojni filtrirni papir s suhim ostankom se prenese v kartušo za ekstrahiranje (4.2) in pokrije s čepkom iz vate, ki ne vsebuje maščob. Kartuša se postavi v ekstraktor (4.1), nato pa se nadaljuje, kakor je navedeno v drugem in tretjem odstavku točke 5.1.
6. Prikaz rezultatov
Masa ostanka se izrazi kot masni delež vzorca.
7. Ponovljivost
Razlika med rezultatoma dveh vzporednih določanj, ki jih na istem vzorcu izvaja isti analitik, ne presega:
— 0,2 % absolutne vrednosti za vsebnosti surovih olj in maščobe, nižje od 5 %,
— 4,0 % višjega rezultata za vsebnosti 5–10 %,
— 0,4 % absolutne vrednosti za vsebnosti nad 10 %.
8. Opombe
8.1 Pri proizvodih z visoko vsebnostjo olj in maščob, ki jih je težko zdrobiti ali so neprimerni za pripravo homogenega zmanjšanega skupnega preskusnega vzorca, se uporabi naslednji postopek.
20 g vzorca se natehta na 1 mg natančno in zmeša z 10 g ali več brezvodnega natrijevega sulfata (3.2). Ekstrahira se s petroletrom (3.1), kakor je navedeno v točki 5.1. Ekstrakt se dopolni s petroletrom do 500 ml (3.1) in premeša. 50 ml raztopine se prenese v majhno suho stehtano bučko, v kateri so delci plovca. Oddestilira se topilo, se osuši, nato pa se nadaljuje, kakor je določeno v zadnjem odstavku točke 5.1.
Iz ostanka po ekstrakciji v kartuši se odstrani topilo, ostanek se zdrobi do velikosti delcev 1 mm, vrne v kartušo za ekstrakcijo (natrijev sulfat se ne doda), nato pa se nadaljuje, kakor je določeno v drugem in tretjem odstavku točke 5.1.
Vsebnost olj in maščob kot masni delež vzorca se izračuna po naslednji formuli:
(10m1 + m2) × 5
pri čemer je:
|
m1 |
= |
masa ostanka po prvi ekstrakciji (alikvot ekstrakta) v gramih; |
|
m2 |
= |
masa ostanka po drugi ekstrakciji v gramih. |
8.2 Pri proizvodih z nizko vsebnostjo olj in maščob se lahko preskusni vzorec poveča na 5 g.
8.3 Hrano za hišne živali, ki vsebuje veliko vode, je treba včasih pred hidrolizo in ekstrakcijo po postopku B zmešati z brezvodnim natrijevim sulfatom.
8.4 Pri točki 5.2 je za spiranje ostanka po filtraciji morda učinkoviteje uporabiti vročo vodo namesto hladne.
8.5 Pri nekateri krmi je morda treba podaljšati čas sušenja na več kot uro in pol. Odvečnemu sušenju se izogiba, saj lahko povzroči nizke rezultate. Uporabi se lahko tudi mikrovalovna pečica.
8.6 Če je vsebnost surovih olj/maščob višja od 15 %, se pred hidrolizo po postopku A priporoča predhodna ekstrakcija, pri postopku B pa ponovna ekstrakcija. Do določene mere je to odvisno od vrste krme ter vrste olj in maščob v krmi.
I. DOLOČANJE SUROVIH VLAKEN
1. Namen in področje uporabe
Ta metoda omogoča določanje nemastnih organskih snovi v krmi, ki so netopne v kislih in baznih medijih ter so ponavadi poimenovane surova vlakna.
2. Načelo
Vzorec se po potrebi razmasti in zaporedno obdela s specificiranimi koncentracijami vrelih raztopin žveplove kisline in kalijevega hidroksida. Ostanek se loči s filtracijo skozi filter iz sintranega stekla, spere, posuši, stehta in upepeli pri 475–500 oC. Izguba teže pri upepeljevanju ustreza vsebnosti surovih vlaken v preskusnem vzorcu.
3. Reagenti
3.1 Žveplova kislina, c = 0,13 mol/l
3.2 Sredstvo proti penjenju (npr. n-oktanol)
3.3 Filtrirno sredstvo (Celite 545 ali enakovredno), segreto na 500 oC za štiri ure (8.6)
3.4 Aceton
3.5 Petroleter, vrelišče 40–60 oC
3.6 Klorovodikova kislina, c = 0,5 mol/l
3.7 Raztopina kalijevega hidroksida, c = 0,23 mol/l
4. Oprema
4.1 Enota za segrevanje za razklop z žveplovo kislino in raztopino kalijevega hidroksida, opremljena z dodatkom za filtrirni lonček (4.2) in odtočno cevjo, ki je preko zamaška priklopljena na tekočino in vakuum, po možnosti s stisnjenim zrakom. Pred vsako uporabo se enota pet minut segreva z vrelo vodo.
4.2 Stekleni filtrirni lonček z nameščeno filtrirno ploščo iz sintranega stekla in velikosti por 40–90 μm. Pred prvo uporabo ga je treba za nekaj minut segreti na 500 oC in ohladiti (8.6).
4.3 Valj, volumna vsaj 270 ml, s kondenzatorjem povratnega toka, primernim za vretje
4.4 Sušilnik s termostatom
4.5 Žarilna peč s termostatom
4.6 Enota za ekstrakcijo, sestavljena iz podporne plošče za filtrirni lonček (4.2) in odtočne cevi, ki je preko zamaška priklopljena na vakuum in tekočino.
4.7 Obročki, ki povežejo enoto za segrevanje (4.1), lonček (4.2) in valj (4.3) ter enoto za hladno ekstrakcijo (4.6) in lonček.
5. Postopek
1 g pripravljenega vzorca se natehta na 1 mg natančno in prenese v lonček (4.2) (glej opombe 8.1, 8.2 in 8.3), čemur se doda 1 g filtrirnega sredstva (3.3).
Najprej se povežeta enota za segrevanje (4.1) in filtrirni lonček (4.2), nato pa valj (4.3) in lonček. V povezana valj in lonček se nalije 150 ml vrele žveplove kisline (3.1) in po potrebi nekaj kapljic sredstva proti penjenju (3.2).
Tekočina se zavre v 5 ± 2 minutah ter pusti močno vreti natančno 30 minut.
Odpre se zamašek na odtočni cevi (4.1), žveplova kislina se pod vakuumom filtrira skozi filtrirni lonček, ostanek pa se trikrat spere s po 30 ml vrele vode, da je filtrirni ostanek po vsakem spiranju suh.
Odtočni zamašek se zapre, v povezana valj in lonček se nalije 150 ml vrele raztopine kalijevega hidroksida (3.7), nato pa se doda nekaj kapljic sredstva proti penjenju (3.2). Tekočina se zavre v 5 ± 2 minutah ter pusti močno vreti natančno 30 minut. To se filtrira, postopek spiranja pa se ponovi, kakor je bil uporabljen za žveplovo kislino.
Po končnem spiranju in sušenju se lonček z vsebino odklopi in priklopi na enoto za hladno ekstrakcijo (4.6). Ostanek v lončku se pod vakuumom trikrat spere s po 25 ml acetona (3.4), da je filtrirni ostanek po vsakem spiranju suh.
Lonček se suši v sušilniku pri 130 oC do konstantne teže. Po vsakem sušenju se ohladi v eksikatorju in hitro stehta. Lonček se postavi v žarilno peč in pri 475–500 oC žari najmanj 30 minut do konstantne teže (izguba teže med dvema zaporednima tehtanjema ne sme presegati 2 mg).
Po vsakem segrevanju in pred merjenjem se lonček najprej ohladi v peči, nato pa še v eksikatorju.
Slepi preskus se izvede brez vzorca. Izguba teže pri žarjenju ne sme presegati 4 mg.
6. Izračun rezultatov
Vsebnost surovih vlaken, izražena kot masni delež vzorca, se izračuna po naslednji formuli:

pri čemer je:
|
m |
= |
masa vzorca v gramih; |
|
m0 |
= |
izguba mase po žarjenju pri določanju v gramih; |
|
m1 |
= |
izguba mase po žarjenju pri slepem preskusu v gramih. |
7. Ponovljivost
Razlika med rezultatoma dveh vzporednih določanj, izvedenih na istem vzorcu, ne sme presegati:
— 0,6 % absolutne vrednosti za vsebnosti surovih vlaken, manjše od 10 %,
— 6 % višje vrednosti za vsebnosti surovih vlaken 10 % ali več.
8. Opombe
8.1 Krmo, ki vsebuje več kakor 10 % surovih maščob, je treba pred analizo razmastiti s petroletrom (3.5). Filtrirni lonček (4.2) z vsebino se poveže z enoto za hladno ekstrakcijo (4.6), ostanek pa se pod vakuumom trikrat spere s po 30 ml petroletra, da je ostanek suh. Lonček z vsebino se poveže z enoto za segrevanje (4.1), nadaljuje pa se v skladu s točko 5.
8.2 Krmo, ki vsebuje maščobe, ki se ne morejo neposredno ekstrahirati s petroletrom (3.5), je treba razmastiti, kakor je navedeno v točki 8.1, in ponovno razmastiti po vretju s kislino. Po vretju s kislino in nadaljnjim spiranjem se lonček z vsebino poveže z enoto za hladno ekstrakcijo (4.6) ter trikrat spere s po 30 ml acetona in trikrat s po 30 ml petroletra. Filtrira se pod vakuumom do suhega stanja, nadaljuje pa se, kakor je navedeno v točki 5, pri čemer se začne z dodajanjem kalijevega hidroksida.
8.3 Če krma vsebuje več kot 5 % karbonatov, izraženih kot kalcijev karbonat, se lonček (4.2) z natehtanim vzorcem poveže z enoto za segrevanje (4.1). Vzorec se trikrat spere s po 30 ml klorovodikove kisline (3.6). Po vsakem dodajanju je treba vzorec pustiti stati približno eno minuto, preden se filtrira. Ponovno se spere s 30 ml vode, nato pa se nadaljuje, kakor je navedeno v točki 5.
8.4 Če se uporablja naprava v obliki stojala (številni lončki, povezani na isto enoto za segrevanje), se v isti seriji ne smeta opraviti posamezni določanji na istem vzorcu za analizo.
8.5 Če je po vretju težko filtrirati kisle in bazične raztopine, se uporabi stisnjen zrak skozi odtočno cev enote za segrevanje in se nato nadaljuje s filtriranjem.
8.6 Temperatura žarjenja ne presega 500 oC, da se podaljša obstojnost steklenih filtrirnih lončkov. Potrebna je previdnost, da se preprečijo odvečni toplotni šoki med segrevanjem ali hlajenjem.
J. DOLOČANJE SLADKORJA
1. Namen in področje uporabe
Ta metoda omogoča določanje količine reducirajočih sladkorjev in celotnih sladkorjev po inverziji, izraženih kot glukoza ali, kadar je to primerno, saharoza s faktorjem preračunavanja 0,95 . Uporablja se za krmne mešanice. Za drugo krmo se uporabljajo posebne metode. Kadar je primerno, se laktoza izmeri ločeno in upošteva pri izračunu rezultata.
2. Načelo
Sladkorji se ekstrahirajo z razredčenim etanolom in raztopina se zbistri z raztopinama Carrez I in II. Po odstranitvi etanola se določijo količine pred inverzijo in po njej po Luff-Schoorlovi metodi.
3. Reagenti
3.1 Etanol, 40-odstotna raztopina (v/v), gostota: 0,948 g/ml pri 20 oC, nevtraliziran proti fenolftaleinu
3.2 Raztopina Carrez I: 21,9 g cinkovega acetata Zn (CH3COO)2 × 2H2O in 3 g ledoceta se raztopijo v vodi. Dopolni se z vodo do 100 ml.
3.3 Raztopina Carrez II: 10,6 g kalijevega ferocianida K4 Fe (CN)6 × 3H2O se raztopi v vodi. Dopolni se z vodo do 100 ml.
3.4 Metiloranž 0,1 -odstotna raztopina (m/v)
3.5 Klorovodikova kislina, 4 mol/l
3.6 Klorovodikova kislina, 0,1 mol/l
3.7 Raztopina natrijevega hidroksida, 0,1 mol/l
3.8 Luff-Schoorlov reagent:
Med previdnim mešanjem se v raztopimo natrijevega karbonata (3.8.3) vlije raztopina citronske kisline (3.8.2). Doda se raztopina bakrovega sulfata (3.8.1), nato pa se dopolni z vodo do 1 litra. Pusti se čez noč in nato filtrira.
Preveri se koncentracija tako dobljenega reagenta (Cu 0,05 mol/l; Na2 CO3 1 mol/l), glej zadnji odstavek točke 5.4. pH raztopine je približno 9,4 .
3.8.1 Raztopina bakrovega sulfata: v 100 ml vode se raztopi 25 g bakrovega sulfata (Cu SO4 5H2O), ki ne vsebuje železa.
3.8.2 Raztopina citronske kisline: v 50 ml vode se raztopi 50 g citronske kisline (C6H8O7.H2O).
3.8.3 Raztopina natrijevega karbonata: v približno 300 ml tople vode se raztopi 143,8 g brezvodnega natrijevega karbonata. Pusti se, da se ohladi.
3.9 Raztopina natrijevega tiosulfata, 0,1 mol/l.
3.10 Raztopina škroba: v liter vrele vode se doda suspenzija 5 g topnega škroba v 30 ml vode. Pusti se vreti 3 minute, nato se ohladi in po potrebi doda 10 mg živosrebrovega jodida kot konzervansa.
3.11 Žveplova kislina, 3 mol/l
3.12 Kalijev jodid, 30-odstotna raztopina (m/v)
3.13 Zdrobljen plovec, prevret v klorovodikovi kislini, opran z vodo in osušen
3.14 3-metilbutan-l-ol
4. Oprema
Mešalnik: približno 35 do 40 obr./min.
5. Postopek
5.1 Ekstrakcija vzorca
2,5 g vzorca se natehta na 1 mg natančno in prenese v 250-mililitrsko merilno bučko. Doda se 200 ml etanola (3.1) in v mešalniku meša 1 uro. Doda se 5 ml raztopine Carrez I (3.2) in meša približno 30 sekund. Doda se 5 ml raztopine Carrez II (3.3) in ponovno meša 1 minuto. Dopolni se z etanolom (3.1) do oznake, homogenizira in filtrira. Odstrani se 200 ml filtrata in upari približno do polovice, da se odstrani skoraj ves etanol. Ostanek po uparevanju se s toplo vodo prenese v 200-mililitrsko merilno bučko, ohladi se, dopolni z vodo do oznake, homogenizira in po potrebi filtrira. Ta raztopina se uporabi za določanje količine reducirajočih sladkorjev, po inverziji pa celotnih sladkorjev.
5.2 Določanje reducirajočih sladkorjev
Odpipetira se največ 25 ml raztopine, ki vsebuje manj kot 60 mg reducirajočih sladkorjev, izraženih kot glukoza. Po potrebi se dopolni z destilirano vodo do 25 ml in določi vsebnost reducirajočih sladkorjev po Luff-Schoorlovi metodi. Rezultat se izrazi kot masni delež glukoze v vzorcu.
5.3 Določanje celotnih sladkorjev po inverziji
50 ml raztopine se odpipetira v 100-mililitrsko merilno bučko. Doda se nekaj kapljic raztopine metiloranža (3.4), nato pa se previdno in ob neprestanem mešanju doda klorovodikova kislina (3.5), dokler tekočina na postane močno rdeča. Doda se 15 ml klorovodikove kisline (3.6), bučka se potopi v skoraj vrelo vodno kopel in v njej pusti 30 minut. Hitro se ohladi na približno 20 oC, nato pa se doda 15 ml raztopine natrijevega hidroksida (3.7). Dopolni se z vodo do 100 ml in homogenizira. Odpipetira se največ 25 ml raztopine, ki vsebuje manj kot 60 mg reducirajočih sladkorjev, izraženih kot glukoza. Po potrebi se dopolni z destilirano vodo do 25 ml in določi vsebnost reducirajočih sladkorjev po Luff-Schoorlovi metodi. Rezultat se izrazi kot masni delež glukoze ali, kadar je to primerno, saharoze tako, da se pomnoži s faktorjem 0,95 .
5.4 Titracija po Luff-Schoorlovi metodi
25 ml Luff-Schoorlovega reagenta (3.8) se odpipetira v 300-mililitrsko erlenmajerico; doda se natančno 25 ml zbistrene raztopine sladkorja. Dodata se 2 zrni plovca (3.13) in med ročnim mešanjem se segreva nad prostim plamenom srednje višine tako, da raztopina zavre v približno dveh minutah. Erlenmajerica se takoj postavi na žično mrežico, prevlečeno z azbestom, ki ima odprtino s premerom 6 cm, pod katero je prižgan plamen. Plamen se naravna tako, da se segreva le dno erlenmajerice. Na erlenmajerico se namesti kondenzator povratnega toka. Pusti se vreti natančno 10 minut. Takoj se ohladi v mrzli vodi in čez približno pet minut titrira na naslednji način:
doda se 10 ml raztopine kalijevega jodida (3.12) in takoj nato (previdno zaradi možnosti močnega penjenja) 25 ml žveplove kisline (3.11). Z raztopino natrijevega tiosulfata (3.9) se titrira do svetlorumene barve, doda se indikator škrob (3.10), nato pa se titracija zaključi.
Enaka titracija se izvede brez vretja z natančno izmerjeno mešanico 25 ml Luff-Schoorlovega reagenta (3.8) in 25 ml vode, potem ko se ji doda 10 ml raztopine kalijevega jodida (3.12) in 25 ml žveplove kisline (3.11).
6. Izračun rezultatov
S pomočjo preglednice se določi količina glukoze v mg, ki ustreza razliki med vrednostma obeh titracij, izraženi v mg natrijevega tiosulfata 0,1 mol/l. Rezultat se izrazi kot masni delež vzorca.
7. Posebni postopki
7.1 Pri krmi, ki vsebuje veliko melase, in drugi krmi, ki ni zelo homogena, se natehta 20 g vzorca in s 500 ml vode prenese v litrsko merilno bučko. V mešalniku se meša 1 uro. Raztopina se zbistri z uporabo reagentov Carrez I (3.2) in II (3.3), kakor je opisano v točki 5.1, vendar se uporabi štirikratna količina vsakega reagenta. Bučka se dopolni z 80-odstotnim etanolom (v/v) do oznake.
Homogenizira se in filtrira. Etanol se odstrani, kakor je opisano v točki 5.1. Če ni dekstriniranega škroba, se dopolni do oznake z destilirano vodo.
7.2 V primeru melas in posamične krme, ki vsebujejo veliko sladkorja in so skoraj brez škroba (rožiči, posušene rezine pese itd.), se natehta 5 g in prenese v 250-mililitrsko merilno bučko, doda se 200 ml destilirane vode in eno uro ali po potrebi več meša v mešalniku. Raztopina se zbistri z uporabo reagentov Carrez I (3.2) in II (3.3), kakor je opisano v točki 5.1. Dopolni se z mrzlo vodo do oznake, homogenizira in filtrira. Za določanje količine celotnih sladkorjev se nadaljuje, kakor je opisano v točki 5.3.
8. Opombe
8.1 Za preprečevanje penjenja je priporočljivo, da se pred vretjem z Luff-Schoorlovim reagentom doda (ne glede na volumen) približno 1 ml 3-metilbutan-l-ola (3.14).
8.2 Razlika med vsebnostjo celotnih sladkorjev po inverziji, izraženih kot glukoza, in vsebnostjo reducirajočih sladkorjev, izraženih kot glukoza, pomnoženo z 0,95 , je masni deleži saharoze.
8.3 Za določanje vsebnosti reducirajočih sladkorjev, razen laktoze, se lahko uporabljata dve metodi:
8.3.1 za približen izračun se vsebnost laktoze, določena z drugo analitsko metodo, pomnoži z 0,675 , dobljeni rezultat pa se odšteje od vsebnosti reducirajočih sladkorjev;
8.3.2 za natančen izračun reducirajočih sladkorjev, razen laktoze, je treba za končno določitev uporabiti isti vzorec. Ena od analiz se izvede na delu raztopine, ki nastane pri postopku iz točke 5.1, druga pa na delu raztopine, ki nastane pri določanju laktoze z metodo, določeno za navedeni namen (po fermentiranju drugih vrst sladkorja in zbistritve).
V obeh primerih se količina sladkorja določi z Luff-Schoorlovo metodo in izračuna v mg glukoze. Ena vrednost se odšteje od druge in razlika se izrazi kot masni delež vzorca.
Primer:
Za vsako določanje oba uporabljena volumna ustrezata 250 mg vzorca.
V prvem primeru se porabi 17 ml raztopine natrijevega tiosulfata 0,1 mol/l, kar ustreza 44,2 mg glukoze, v drugem pa 11 ml, kar ustreza 27,6 mg glukoze.
Razlika je 16,6 mg glukoze.
Vsebnost reducirajočih sladkorjev (razen laktoze), izračunana kot glukoza, je zato:
Preglednica vrednosti za 25 ml Luff-Schoorlovega reagenta
ml Na2 S2 O30,1 mol/l, 2 minuti segrevanja, 10 minut vretja
|
Na2S2O3 0,1 mol/l |
Glukoza, fruktoza, sladkorji po inveriziji C6H12O6 |
Laktoza C12H22O11 |
Maltoza C12H22O11 |
Na2S2O3 0,1 mol/l |
|||
|
ml |
mg |
razlika |
mg |
razlika |
mg |
razlika |
ml |
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
2,4 4,8 7,2 9,7 12,2 14,7 17,2 19,8 22,4 25,0 27,6 30,3 33,0 35,7 38,5 41,3 44,2 47,1 50,0 53,0 56,0 59,1 62,2 |
2,4 2,4 2,5 2,5 2,5 2,5 2,6 2,6 2,6 2,6 2,7 2,7 2,7 2,8 2,8 2,9 2,9 2,9 3,0 3,0 3,1 3,1 |
3,6 7,3 11,0 14,7 18,4 22,1 25,8 29,5 33,2 37,0 40,8 44,6 48,4 52,2 56,0 59,9 63,8 67,7 71,7 75,7 79,8 83,9 88,0 |
3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,8 3,8 3,8 3,8 3,8 3,8 3,9 3,9 3,9 4,0 4,0 4,1 4,1 4,1 |
3,9 7,8 11,7 15,6 19,6 23,5 27,5 31,5 35,5 39,5 43,5 47,5 51,6 55,7 59,8 63,9 68,0 72,2 76,5 80,9 85,4 90,0 94,6 |
3,9 3,9 3,9 4,0 3,9 4,0 4,0 4,0 4,0 4,0 4,0 4,1 4,1 4,1 4,1 4,1 4,2 4,3 4,4 4,5 4,6 4,6 |
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
K. DOLOČANJE LAKTOZE
1. Namen in področje uporabe
Ta metoda omogoča določanje ravni laktoze v krmi, ki vsebuje več kot 0,5 % laktoze.
2. Načelo
Sladkorji se raztopijo v vodi. Raztopina se fermentira s kvasovkami vrste Saccharomyces cerevisiae, ki ne spreminjajo laktoze. Po zbistritvi in filtriranju se vsebnost laktoze v filtratu določi po Luff-Schoorlovi metodi.
3. Reagenti
3.1 Suspenzija kvasovk vrste Saccharomyces cerevisiae: 25 g svežih kvasovk se raztopi v 100 ml vode. Suspenzija je ob hranjenju v hladilniku uporabna največ en teden.
3.2 Raztopina Carrez I: 21,9 g cinkovega acetata Zn (CH3 COO)2 2H2O in 3 g ledoceta se raztopijo v vodi. Dopolni se z vodo do 100 ml.
3.3 Raztopina Carrez II: 10,6 g kalijevega ferocianida K4 Fe (CN)6 3H2O se raztopi v vodi. Dopolni se z vodo do 100 ml.
3.4 Luff-Schoorlov reagent:
med previdnim mešanjem se v raztopino natrijevega karbonata (3.4.3) vlije raztopina citronske kisline (3.4.2). Doda se raztopina bakrovega sulfata (3.4.1), nato pa se dopolni z vodo do 1 litra. Pusti se čez noč in nato filtrira. Preveri se koncentracija tako dobljenega reagenta (Cu 0,05 mol/l, Na2 CO3 1 mol/l). pH raztopine je približno 9,4 .
3.4.1 Raztopina bakrovega sulfata: v 100 ml vode se raztopi 25 g bakrovega sulfata (Cu SO4 × 5H2O), ki ne vsebuje železa.
3.4.2 Raztopina citronske kisline: v 50 ml vode se raztopi 50 g citronske kisline (C6H8O7×H2O).
3.4.3 Raztopina natrijevega karbonata: v približno 300 ml tople vode se raztopi 143,8 g brezvodnega natrijevega karbonata. Pusti se, da se ohladi.
3.5 Zdrobljen plovec, prevret v klorovodikovi kislini, opran z vodo in osušen
3.6 Kalijev jodid, 30-odstotna raztopina (m/v)
3.7 Žveplova kislina, 3 mol/l
3.8 Raztopina natrijevega tiosulfata, 0,1 mol/l
3.9 Raztopina škroba: v liter vrele vode se doda suspenzija 5 g topnega škroba v 30 ml vode. Pusti se vreti 3 minute, nato pa se ohladi in po potrebi doda 10 mg živosrebrovega jodida kot konzervansa.
4. Oprema
Vodna kopel s termostatom, nastavljenim na 38–40 oC
5. Postopek
1 g vzorca se natehta na 1 mg natančno in prenese v 100-mililitrsko merilno bučko. Doda se 25–30 ml vode. Bučka se za 30 minut postavi v vrelo vodno kopel, nato ohladi na približno 35 oC. Doda se 5 ml suspenzije kvasovk (3.1) in homogenizira. Bučka se pusti 2 uri v vodni kopeli pri temperaturi 38–40 oC. Ohladi se na približno 20 oC.
Doda se 2,5 ml raztopine Carrez I (3.2) in meša 30 sekund, nato se doda 2,5 ml raztopine Carrez II (3.3) in ponovno meša 30 sekund. Dopolni se z vodo do 100 ml, premeša in filtrira. Del filtrata, ki ne presega 25 ml in ki priporočljivo vsebuje 40 do 80 mg laktoze, se odpipetira in prenese v 300-mililitrsko erlenmajerico. Po potrebi se dopolni z vodo do 25 ml.
Slepi preskus s 5 ml suspenzije kvasovk (3.1) se izvede pri enakih pogojih. Vsebnost laktoze se določi po Luff-Schoorlovi metodi na naslednji način: doda se natančno 25 ml Luff-Schoorlovega reagenta (3.4) in 2 zrni plovca (3.5). Med ročnim mešanjem se segreva nad prostim plamenom srednje jakosti tako, da tekočina zavre v približno 2 minutah. Erlenmajerica se takoj postavi na žično mrežico, prevlečeno z azbestom, ki ima odprtino s premerom 6 cm, pod katero je prižgan plamen. Plamen se naravna tako, da se segreva le dno erlenmajerice. Na erlenmajerico se namesti kondenzator povratnega toka. Pusti se vreti natančno 10 minut. Takoj se ohladi v mrzli vodi in čez približno 5 minut titrira na naslednji način:
doda se 10 ml raztopine kalijevega jodida (3.6) in takoj nato (previdno zaradi možnosti močnega penjenja) 25 ml žveplove kisline (3.7). Z raztopino natrijevega tiosulfata (3.8) se titrira do svetlorumene barve, doda se indikator škrob (3.9), nato pa se titracija zaključi.
Enaka titracija se izvede brez vretja z natančno izmerjeno mešanico 25 ml Luff-Schoorlovega reagenta (3.4) in 25 ml vode, potem ko se ji doda 10 ml raztopine kalijevega jodida (3.6) in 25 ml žveplove kisline (3.7).
6. Izračun rezultatov
S pomočjo priložene preglednice se določi količina laktoze v mg, ki ustreza razliki med rezultatoma dveh titracij, izraženih v ml natrijevega tiosulfata 0,1 mol/l.
Rezultat se izrazi kot masni delež brezvodne laktoze v vzorcu.
7. Opomba
Pri proizvodih, ki vsebujejo več kot 40 % fermentiranega sladkorja, se uporabi več kot 5 ml suspenzije kvasovk (3.1).
Preglednica vrednosti za 25 ml Luff-Schoorlovega reagenta
ml Na2 S2 O30,1 mol/l, 2 minuti segrevanja, 10 minut vretja
|
Na2S2O3 0,1 mol/l |
Glukoza, fruktoza, sladkorji po inveriziji C6H12O6 |
Laktoza C12H22O11 |
Maltoza C12H22O11 |
Na2S2O3 0,1 mol/l |
|||
|
ml |
mg |
razlika |
mg |
razlika |
mg |
razlike |
ml |
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
2,4 4,8 7,2 9,7 12,2 14,7 17,2 19,8 22,4 25,0 27,6 30,3 33,0 35,7 38,5 41,3 44,2 47,1 50,0 53,0 56,0 59,1 62,2 |
2,4 2,4 2,5 2,5 2,5 2,5 2,6 2,6 2,6 2,6 2,7 2,7 2,7 2,8 2,8 2,9 2,9 2,9 3,0 3,0 3,1 3,1 |
3,6 7,3 11,0 14,7 18,4 22,1 25,8 29,5 33,2 37,0 40,8 44,6 48,4 52,2 56,0 59,9 63,8 67,7 71,7 75,7 79,8 83,9 88,0 |
3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,8 3,8 3,8 3,8 3,8 3,8 3,9 3,9 3,9 4,0 4,0 4,1 4,1 4,1 |
3,9 7,8 11,7 15,6 19,6 23,5 27,5 31,5 35,5 39,5 43,5 47,5 51,6 55,7 59,8 63,9 68,0 72,2 76,5 80,9 85,4 90,0 94,6 |
3,9 3,9 3,9 4,0 3,9 4,0 4,0 4,0 4,0 4,0 4,0 4,1 4,1 4,1 4,1 4,1 4,2 4,3 4,4 4,5 4,6 4,6 |
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
L. DOLOČANJE ŠKROBA
POLARIMETRIČNA METODA
1. Namen in področje uporabe
Ta metoda omogoča določanje ravni škroba in visokomolekularnih produktov razgradnje škroba v krmi za preverjanje skladnosti z označeno energijsko vrednostjo (določbe iz Priloge VII) in Direktivo Sveta 96/25/ES ( 10 ).
2. Načelo
Metoda vključuje dve določanji. Pri prvi se vzorec obdela z razredčeno klorovodikovo kislino. Po zbistritvi in filtriranju se izmeri optična rotacija raztopine s polarimetrijo.
Pri drugi se vzorec ekstrahira s 40-odstotnim etanolom. Po nakisanju filtrata s klorovodikovo kislino ter zbistritvi in filtriranju se izmeri optična rotacija tako kot pri prvem določanju.
Razlika med dvema meritvama, pomnožena z znanim faktorjem, je vsebnost škroba v vzorcu.
3. Reagenti
3.1 Klorovodikova kislina, 25-odstotna raztopina (m/v), gostota: 1,126 g/ml
3.2 Trikloroocetna kislina, 1,13 -raztopina raztopina (m/v)
Koncentracija se mora preveriti s titracijo z 0,1 mol/l raztopino natrijevega hidroksida v prisotnosti 0,1 -odstotnega (m/v) metil rdečega v 94-odstotnem (v/v) etanolu. Za nevtralizacijo 10 ml je potrebno 30,94 ml NaOH 0,1 mol/l.
3.3 Raztopina Carrez I: 21,9 g cinkovega acetata Zn(CH3COO)2 2H2O in 3 g ledoceta se raztopi v vodi. Dopolni se z vodo do 100 ml.
3.4 Raztopina Carrez II: 10,6 g kalijevega ferocianida K4(Fe(CN)6 3H2O se raztopi v vodi. Dopolni se z vodo do 100 ml.
3.5 Etanol, 40-odstotna raztopina (v/v), gostota: 0,948 g/ml pri 20 oC
4. Oprema
4.1 250-mililitrska erlenmajerica s standardnim brušenim vratom in kondenzatorjem povratnega toka
4.2 Polarimeter ali saharometer
5. Postopek
5.1 Priprava vzorca
Vzorec se zdrobi tako, da vsi delci preidejo skozi sito z okroglimi luknjicami premera 0,5 mm.
5.2 Določanje celotne optične rotacije (P ali S) (glej opombo 7.1)
2,5 g zdrobljenega vzorca se natehta na 1 mg natančno in prenese v 100-mililitrsko merilno bučko. Doda se 25 ml klorovodikove kisline (3.2), pretrese se, da nastane enakomerna porazdelitev preskusnega vzorca, nato pa se doda nadaljnjih 25 ml klorovodikove kisline (3.2). Bučka se potopi v vrelo vodno kopel in prve 3 minute močno in enakomerno stresa, da se prepreči nastanek aglomeratov. V vodni kopeli mora biti toliko vode, da kopel ostane na točki vrelišča, ko se vanjo potopi bučka. Bučke se med stresanjem ne sme odstraniti iz kopeli. Po natanko 15 minutah se bučka odstrani iz kopeli, doda se 30 ml hladne vode, nato pa se takoj ohladi na 20 oC.
Doda se 5 ml Raztopine Carrez I (3.3) in stresa približno 30 sekund. Nato se doda 5 ml raztopine Carrez II (3.4) in ponovno stresa 30 sekund. Dopolni se z vodo do oznake, premeša in filtrira. Če filtrat ni popolnoma bister (kar je redko), se ponovi določanje z uporabo večje količine raztopin Carrez I in II, na primer 10 ml.
Optična rotacija raztopine v 200-milimetrski epruveti se izmeri s polarimetrom ali saharimetrom.
5.3 Določanje optične rotacije (P' ali S') snovi, ki so topne v 40-odstotnem etanolu
5 g vzorca se natehta na 1 mg natančno, prenese v 100-mililitrsko merilno bučko in doda se približno 80 ml etanola (3.5) (glej opombo 7.2). Bučka se pusti stati 1 uro na sobni temperaturi; v tem času se šestkrat močno pretrese, da se preskusni vzorec temeljito premeša z etanolom. Dopolni se z etanolom (3.5) do oznake, premeša in filtrira.
50 ml filtrata (kar ustreza 2,5 g vzorca) se odpipetira v 250-mililitrsko erlenmajerico, doda se 2,1 ml klorovodikove kisline (3.1) in močno pretrese. Erlenmajerica se spoji s kondenzatorjem povratnega toka in potopi v vrelo vodno kopel. Po natanko 15 minutah se erlenmajerica odstrani iz kopeli, vsebina se prenese v 100-mililitrsko merilno bučko, spere se z malo hladne vode in ohladi na 20 oC.
Za zbistritev se uporabita raztopini Carrez I (3.3.) in II (3.4), dopolni se z vodo do oznake, premeša in filtrira, nato pa se izmeri optična rotacija, kakor je navedeno v drugem in tretjem odstavku točke 5.2.
6. Izračun rezultatov
Vsebnost škroba (%) se izračuna na naslednji način:
6.1 meritve s polarimetrom

|
P |
= |
celotna optična rotacija v kotnih stopinjah; |
|
P' |
= |
optična rotacija snovi, ki so topne v 40-odstotnem (V/V) etanolu, v kotnih stopinjah; |
|
|
= |
specifična optična rotacija čistega škroba. Vrednosti, D ki se ponavadi uporabljajo za ta faktor, so:
|
6.2 meritve s saharimetrom
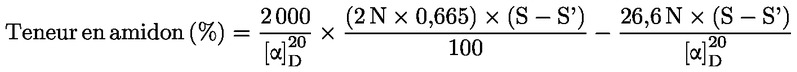
|
S |
= |
celotna optična rotacija v stopinjah saharimetra; |
|
S' |
= |
optična rotacija snovi, ki so topne v 40-odstotnem (v/v) etanolu, v stopinjah saharimetra; |
|
N |
= |
masa (g) saharoze v 100 ml vode, pri kateri je optična rotacija 100 saharimetrskih stopinj, kadar se meri z uporabo 200-milimetrske epruvete; 16,29 g za francoske saharimetre; 26,00 g za nemške saharimetre; 20,00 g za mešane saharimetre; |
|
|
= |
specifična optična rotacija čistega škroba (glej 6.1). |
6.3 Ponovljivost
Razlika med rezultatoma dveh vzporednih določanj, izvedenih na istem vzorcu, ne sme presegati 0,4 absolutne vrednosti za vsebnost škroba, ki je nižja od 40 %, in 1 % relativne vrednosti za vsebnost škroba, ki je enaka ali višja od 40 %.
7. Opombe
7.1 Če vzorec vsebuje več kot 6 % karbonatov, izračunanih v obliki kalcijevega karbonata, se jih mora pred določanjem celotne optične rotacije razkrojiti z obdelavo z natančno primerno količino razredčene žveplove kisline.
7.2 V primeru proizvodov, ki imajo visoko vsebnost laktoze, kot je serum mleka v prahu ali posneto mleko v prahu, se po dodatku 80 ml etanola (3.5) izvaja naslednje: bučka se spoji s kondenzatorjem povratnega toka in se za 30 minut potopi v vodno kopel pri 50 oC. Pusti se, da se ohladi, in nadaljuje z analizo, kakor je navedeno v točki 5.3.
7.3 Naslednja posamična krmila, kadar so prisotna v znatni količini v krmi, povzročajo motnje pri določanju vsebnosti škroba s polarimetrično metodo, zaradi česar so lahko rezultati nepravilni:
— proizvodi iz (sladkorne) pese, kot so pulpa (sladkorne) pese, melasa (sladkorne) pese, melasirana pulpa (sladkorne) pese, usedlina pri destilaciji (sladkorne) pese, (pesni) sladkor,
— pulpa agrumov,
— laneno seme; laneno seme z nižjo vsebnostjo maščobe; ekstrahirano laneno seme,
— seme oljne ogrščice; oljna ogrščica z nižjo vsebnostjo maščobe; ekstrakt oljne ogrščice; luščine oljne ogrščice,
— sončnično seme; ekstrahirano sončnično seme; sončnično seme, delno oluščeno, ekstrahirano,
— kopra z nižjo vrednostjo maščobe; ekstrakt kopre,
— krompirjeva pulpa,
— suhi kvas,
— proizvodi z visoko vsebnostjo inulina (npr. koščki in moka topinamburja),
— ocvirki.
M. DOLOČANJE SUROVEGA PEPELA
1. Namen in področje uporabe
Ta metoda omogoča določanje vsebnosti surovega pepela v krmi.
2. Načelo
Vzorec se upepeli pri 550 oC; ostanek se stehta.
3. Reagenti
Amonijev nitrat, 20-odstotna raztopina (m/v)
4. Oprema
4.1 Grelna plošča
4.2 Električna žarilna peč s termostatom
4.3 Žarilni lončki za upepeljevanje iz kremena, porcelana ali platine, pravokotni (približno 60 × 40 × 25 mm) ali okrogli (premer: 60 do 75 mm, višina: 20 do 40 mm)
5. Postopek
Približno 5 g vzorca se natehta na 1 mg natančno (2,5 g pri proizvodih, ki lahko nabreknejo) in prenese v žarilni lonček za upepelitev, ki se predhodno segreje na 550 oC, ohladi in tarira. Lonček se postavi na grelno ploščo in postopoma segreva, da snov zogleni. Upepeli se v skladu s točko 5.1 ali 5.2.
5.1 Lonček se postavi v žarilno peč, segreto na 550 oC. Pri tej temperaturi se pusti, dokler ne nastane bel, svetlosiv ali rdečkast pepel, ki je videti, da ne vsebuje ogljikovih delcev. Lonček se postavi v eksikator, pusti se, da se ohladi, in takoj stehta.
5.2 Lonček se postavi v žarilno peč, segreto na 550 oC. V peči se pusti 3 ure. Lonček se postavi v eksikator, pusti se, da se ohladi, in takoj stehta. Ponovno se postavi v žarilno peč za 30 minut, pri čemer mora biti masa pepela konstantna (izguba mase med dvema zaporednima tehtanjema ne sme presegati 1 mg).
6. Izračun rezultatov
Masa ostanka se izračuna tako, da se odšteje tara.
Rezultat se izrazi kot masni delež vzorca.
7. Opombe
7.1 Snovi, ki se težko upepelijo, je treba najprej najmanj tri ure upepeljevati, nato ohladiti in jim dodati nekaj kapljic 20-odstotne raztopine amonijevega nitrata ali vode (pazljivo, da se pepel ne razprši ali da ne nastanejo grude). Po sušenju v sušilniku se nadaljuje z žarjenjem. Postopek se po potrebi ponovi, dokler upepelitev ni popolna.
7.2 Pri snoveh, ki so odporne na obdelavo iz točke 7.1, se nadaljuje na naslednji način: po triurni upepelitvi se pepel prenese v vročo vodo in filtrira skozi majhen filter brez pepela. Filter in njegova vsebina se upepelita v istem lončku. Filtrat se prenese v ohlajen lonček, upari do suhega, upepeli in stehta.
7.3 Pri oljih in maščobah se 25 g vzorca natančno natehta v lonček primerne velikosti. Upepeli se tako, da se snov zažge s trakom iz filtrirnega papirja brez pepela. Po sežigu se navlaži s čim manj vode. Osuši in upepeli se, kakor je opisano v točki 5.
N. DOLOČANJE PEPELA, NETOPNEGA V KLOROVODIKOVI KISLINI
1. Namen in področje uporabe
Ta metoda omogoča določanje ravni mineralnih snovi v krmi, netopnih v klorovodikovi kislini. Glede na naravo vzorca se lahko uporabljata dve metodi.
1.1 Metoda A: uporablja se za organska posamična krmila in za večino krmnih mešanic;
1.2 Metoda B: uporablja se za mineralne krmne mešanice in krmne mešanice, ki vsebujejo več kot 1 % snovi, netopnih v klorovodikovi kislini in določenih po metodi A.
2. Načelo
2.1 Metoda A: vzorec se upepeli, pepel se raztopi v vreli klorovodikovi kislini, netopni ostanek pa filtrira in stehta.
2.2 Metoda B: vzorec se obdela s klorovodikovo kislino. Raztopina se filtrira, ostanek se upepeli, tako dobljeni pepel pa se obdela v skladu z metodo A.
3. Reagenti
3.1 Klorovodikova kislina, 3 mol/l
3.2 Trikloroocetna kislina, 20-odstotna raztopina (m/v)
3.3 Trikloroocetna kislina, 1-odstotna raztopina (m/v)
4. Oprema
4.1 Grelna plošča
4.2 Električna žarilna peč s termostatom
4.3 Žarilni lončki za upepeljevanje iz kremena, porcelana ali platine, pravokotni (približno 60 × 40 × 25 mm) ali okrogli (premer: 60 do 75 mm, višina: 20 do 40 mm)
5. Postopek
5.1 Metoda A
Vzorec se upepeli po metodi za določanje surovega pepela. Lahko se uporabi pepel, dobljen pri navedeni analizi.
Pepel se prenese v 250- do 400-mililitrsko čašo s 75 ml klorovodikove kisline (3.1). Počasi se segreje do vretja in pusti rahlo vreti petnajst minut. Topla raztopina se filtrira skozi filtrirni papir brez pepela, ostanek pa se spira s toplo vodo, dokler kisla reakcija ne preneha. Filter z ostankom in pepelom se posuši v tariranem žarilnem lončku pri 550–700 oC. Ohladi se v eksikatorju in stehta.
5.2 Metoda B
5 g vzorca se natehta na 1 mg natančno in prenese v 250- do 400-mililitrsko čašo. Doda se 25 ml vode in nato 25 ml klorovodikove kisline (3.1), premeša se, nato pa se počaka, da se reakcija umiri. Doda se še 50 ml klorovodikove kisline (3.1). Počaka se, da se preneha izločati plin, nato pa se čaša postavi v vrelo vodno kopel za 30 minut ali po potrebi dlje, da se popolnoma hidrolizira kakršen koli škrob, ki je lahko prisoten. Vsebina, ki je še topla, se filtrira skozi filter brez pepela, filter pa se spere s 50 ml tople vode (glej opombo 7). Filter, ki vsebuje ostanek, se prenese v žarilni lonček, posuši in upepeli pri 550–700 oC. Pepel se prenese v 250- do 400-mililitrsko čašo s 75 ml klorovodikove kisline (3.1); nadaljuje se, kakor je opisano v drugem pododstavku točke 5.1.
6. Izračun rezultatov
Masa ostanka se izračuna tako, da se odšteje tara. Rezultat se izrazi kot masni delež vzorca.
7. Opomba
Če pri filtriranju nastopijo težave, se analiza ponovi, pri čemer se 50 ml klorovodikove kisline (3.1) nadomesti s 50 ml 20-odstotne trikloroocetne kisline (3.2), filter pa se spere s toplo raztopino 1-odstotne trikloroocetne kisline (3.3).
O. DOLOČANJE KARBONATOV
1. Namen in področje uporabe
Ta metoda omogoča določanje količine karbonatov v večini krme, ponavadi izraženih kot kalcijev karbonat.
Vendar je treba v nekaterih primerih (na primer pri železovem karbonatu) uporabiti posebna metoda.
2. Načelo
Karbonati se razkrojijo s klorovodikovo kislino; sproščeni ogljikov dioksid se zbere v bireto, nastala prostornina pa se primerja s prostornino, ki se pod istimi pogoji sprosti iz znane količine kalcijevega karbonata.
3. Reagenti
3.1 Klorovodikova kislina, gostota 1,10 g/ml
3.2 Kalcijev karbonat
3.3 Žveplova kislina, približno 0,05 mol/l, obarvana z metil rdečim
4. Oprema
Scheibler-Dietrichova naprava (glej sliko) ali ustrezna druga naprava
5. Postopek
Glede na vsebnost karbonata v vzorcu se natehta del vzorca, kakor je navedeno spodaj:
— 0,5 g pri proizvodih, ki vsebujejo 50–100 % karbonatov, izraženih kot kalcijev karbonat,
— 1 g pri proizvodih, ki vsebujejo 40–50 % karbonatov, izraženih kot kalcijev karbonat,
— 2 do 3 g za druge proizvode.
Natehtani del vzorca se prenese v posebno bučko (4) naprave, povezano z majhno epruveto iz nezdrobljivega materiala, ki vsebuje 10 ml klorovodikove kisline (3.1), bučka pa se poveže z napravo. Trojni petelinček (5) se obrne tako, da je bireta (1) odprta navzven. S pomočjo pomične cevi (2), napolnjene z obarvano žveplovo kislino (3.3) in povezane z bireto (1), se nivo tekočine uravna na oznako nič. Petelinček (5) se obrne tako, da se bireta (1) in cev (3) povežeta, nato pa se preveri, ali je raven na oznaki nič.
Z nagibanjem bučke (4) se klorovodikova kislina (3.1) počasi spušča prek vzorca. Tlaka se izenačita tako, da se cev (2) spušča. Bučka (4) se stresa, dokler se ogljikov dioksid popolnoma ne preneha izločati.
Tlaka se izenačita tako, da se izenačita ravni tekočin v bireti (1) in cevi (2). Rezultat se odčita po nekaj minutah, ko se volumen plina ustali.
Pod enakimi pogoji se izvede kontrolni preskus z 0,5 g kalcijevega karbonata (3.2).
6. Izračun rezultatov
Vsebnost karbonatov, izraženih kot kalcijev karbonat, se izračuna po naslednji formuli:
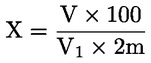
pri čemer je:
|
X |
= |
% (m/m) karbonatov v vzorcu, izraženih kot kalcijev karbonat; |
|
V |
= |
ml sproščenega CO2 iz dela vzorca; |
|
V1 |
= |
ml sproščenega CO2 iz 0,5 g CaCO3; |
|
m |
= |
masa dela vzorca v gramih. |
7. Opombe
7.1 Ko je masa dela vzorca večja od 2 g, se najprej v bučko (4) nalije 15 ml destilirane vode in se pred začetkom preskusa premeša. Enaka količina vode se uporabi za kontrolni preskus.
7.2 Če se volumen naprave, ki se uporablja, razlikuje od volumna Scheibler-Dietrichove naprave, je treba ustrezno prilagoditi količino dela vzorca in kontrolne snovi ter izračun rezultata.

P. DOLOČANJE CELOTNEGA FOSFORJA
FOTOMETRIČNA METODA
1. Namen in področje uporabe
Ta metoda omogoča določanje celotne vsebnosti fosforja v krmi. Zlasti primerna je za analizo proizvodov z nizko vsebnostjo fosforja. V nekaterih primerih (pri proizvodih, ki vsebujejo veliko fosforja) se lahko uporabi gravimetrična metoda.
2. Načelo
Vzorec se mineralizira s suhim sežigom (pri organski krmi) ali z raztapljanjem s kislino (pri mineralnih mešanicah in tekoči krmi) ter prenese v kislo raztopino. Raztopini se doda molibdovanadatni reagent. Optična gostota rumene raztopine, ki pri tem nastane, se meri s spektrofotometrom pri 430 nm.
3. Reagenti
3.1 Kalcijev karbonat
3.2 Klorovodikova kislina, ρ20 = 1,10 g/ml (približno 6 mol/l)
3.3 Dušikova kislina, ρ20 = 1,045 g/ml
3.4 Dušikova kislina, ρ20 = 1,38 do 1,42 g/ml
3.5 Žveplova kislina, ρ20 = 1,84 g/ml
3.6 Molibdovanadatni reagent: v litrski merilni bučki se zmeša 200 ml raztopine amonijevega heptamolibdata (3.6.1), 200 ml raztopine amonijevega monovanadata (3.6.2) in 134 ml dušikove kisline (3.4). Dopolni se z vodo do oznake.
3.6.1 Raztopina amonijevega heptamolibdata: v vroči vodi se raztopi 100 g amonijevega heptamolibdata ((NH4) 6Mo7O24×4H2O). Doda se 10 ml amonijaka (gostota 0,91 g/ml) in dopolni z vodo do 1 litra.
3.6.2 Raztopina amonijevega monovanadata: v 400 ml vroče vode se raztopi 2,35 g amonijevega monovanadata (NH4VO3). Med neprestanim mešanjem se počasi doda 20 ml razredčene dušikove kisline (7 ml HNO3 (3.4) + 13 ml H2O) in dopolni z vodo do 1 litra.
3.7 Standardna raztopina 1 mg fosforja na ml: v vodi se raztopi 4,387 g kalijevega dihidrogen fosfata (KH2PO4). Dopolni se z vodo do 1 litra.
4. Oprema
4.1 Žarilni lončki iz kremena, porcelana ali platine
4.2 Električna žarilna peč s termostatom, nastavljenim na 550 oC
4.3 250-mililitrska Kjeldahlova bučka
4.4 Merilne bučke in precizijske pipete
4.5 Spektrofotometer
4.6 Epruvete premera približno 16 mm, z brušenimi zamaški premera 14,5 mm, volumna 25–30 ml
5. Postopek
5.1 Priprava raztopine
Glede na naravo vzorca se raztopina pripravi, kakor je navedeno v točki 5.1.1 ali 5.1.2.
5.1.1
1 g vzorca ali več se natehta na 1 mg natančno. Vzorec se prenese v Kjeldahlovo bučko, doda se 20 ml žveplove kisline (3.5), pretrese se, da se vzorec popolnoma prepoji s kislino in da se ne sprijema sten bučke, nato se segreje in pusti vreti 10 minut. Pusti se, da se nekoliko ohladi, dodata se 2 ml dušikove kisline (3.4), zmerno se segreje, pusti se, da se nekoliko ohladi, doda se še nekaj dušikove kisline (3.4) in ponovno segreje do vretja. Postopek se ponavlja do nastanka brezbarvne raztopine. Ohladi se, doda nekaj vode, tekočina se prelije v 500-mililitrsko merilno bučko, Kjeldahlova bučka pa se spere z vročo vodo. Pusti se, da se ohladi, dopolni se z vodo do oznake, homogenizira in filtrira.
5.1.2
V žarilni lonček se natehta približno 2,5 g vzorca na 1 mg natančno. Doda se 1 g kalcijevega karbonata (3.1) in meša, da se snovi popolnoma premešata. Žari se v pečici pri 550 oC, da nastane bel ali siv pepel (majhna količina oglja ni problematična). Pepel se prenese v 250-mililitrsko čašo. Doda se 20 ml vode, nato pa se dodaja klorovodikova kislina (3.2), dokler ne prenehajo izhajati mehurčki. Doda se nadaljnjih 10 ml klorovodikove kisline (3.2). Čaša se postavi na peščeno kopel in pusti uparevati do suhega, da kremen postane netopen. Ostanek se raztopi v 10 ml dušikove kisline (3.3) in se na peščeni kopeli ali vroči plošči pusti vreti 5 minut, raztopina pa ne sme upariti v celoti. Tekočina se prelije v 500-mililitrsko merilno bučko, čaša pa se večkrat spere z vročo vodo. Pusti se, da se ohladi, dopolni se z vodo do oznake, homogenizira in filtrira.
5.2 Nastanek barve in merjenje optične gostote
Alikvot filtrata iz točke 5.1.1 ali 5.1.2 se razredči do koncentracije fosforja največ 40 μg/ml. 10 ml te raztopine se prenese v epruveto (4.6) in doda 10 ml molibdovanadatnega reagenta (3.6). Homogenizira se in pusti stati najmanj 10 minut pri 20 oC. Izmeri se optična gostota s spektrofotometrom pri 430 nm raztopine, ki se dobi z dodajanjem 10 ml molibdovanadatnega reagenta (3.6) v 10 ml vode.
5.3 Umeritvena krivulja
Iz standardne raztopine (3.7) se pripravijo raztopine, ki vsebujejo 5, 10, 20, 30 in 40 μg fosforja na ml. V 10 ml vsake izmed teh raztopin se doda 10 ml molibdovanadatnega reagenta (3.6). Homogenizira se in pusti stati najmanj 10 minut pri 20 oC. Izmeri se optična gostota, kakor je navedeno v točki 5.2. Umeritvena krivulja se pripravi tako, da se v diagram vnesejo izmerjene optične gostote ustreznih količin fosforja. Za koncentracije 0–40 μg/ml bo krivulja linearna.
6. Izračun rezultatov
Količina fosforja v preskusnem vzorcu se določi iz umeritvene krivulje.
Rezultat se izrazi kot masni delež vzorca.
Ponovljivost
Razlika med rezultatoma dveh vzporednih določanj, izvedenih na istem vzorcu, ne presega:
— 3 % višjega rezultata, če je vsebnost fosforja nižja od 5 %,
— 0,15 % absolutne vrednosti, če je vsebnost fosforja 5 % ali več.
Q. DOLOČANJE KLORA IZ KLORIDOV
1. Namen in področje uporabe
Ta metoda omogoča določanja količine klora v kloridih, topnih v vodi, ki so ponavadi izraženi kot natrijev klorid. Uporablja se za vso krmo.
2. Načelo
Kloridi se raztopijo v vodi. Če proizvod vsebuje organske snovi, se zbistri. Raztopina se rahlo nakisa z dušikovo kislino in kloridi se oborijo v obliki srebrovega klorida z raztopino srebrovega nitrata. Presežek srebrovega nitrata se titrira z raztopino amonijevega tiocianata po Volhardovi metodi.
3. Reagenti
3.1 Raztopina amonijevega tiocianata, 0,1 mol/liter
3.2 Raztopina srebrovega nitrata, 0,1 mol/l
3.3 Nasičena raztopina amonij-železovega sulfata (NH4)Fe(SO4)2
3.4 Dušikova kislina, gostota: 1,38 g/ml
3.5 Dietileter
3.6 Aceton
3.7 Raztopina Carrez I: 21,9 g cinkovega acetata Zn (CH3COO)2·2H2O in 3 g ledoceta se raztopijo v vodi. Dopolni se z vodo do 100 ml.
3.8 Raztopina Carrez II: 10,6 g kalijevega ferocianida K4Fe(CN)6·3H2O se raztopi v vodi. Dopolni se z vodo do 100 ml.
3.9 Aktivno oglje, ki ne vsebuje kloridov in jih ne absorbira.
4. Oprema
Mešalnik: približno 35 do 40 obr./min.
5. Postopek
5.1 Priprava raztopine
Glede na naravo vzorca se raztopina pripravi, kakor je navedeno v točki 5.1.1, 5.1.2 ali 5.1.3.
Hkrati se opravi slepi preskus brez vzorca, ki se analizira.
5.1.1
Največ 10 g vzorca, ki vsebuje največ 3 g klora v obliki kloridov, se natehta na 1 mg natančno. S 400 ml vode se prenese v 500-mililitrsko merilno bučko pri približno 20 oC. V mešalniku se meša 30 minut, dopolni do oznake, homogenizira in filtrira.
5.1.2
Približno 5 g vzorca se natehta na 1 mg natančno in se skupaj z 1 g aktivnega oglja prenese v 500-mililitrsko merilno bučko. Doda se 400 ml vode s temperaturo približno 20 oC in 5 ml raztopine Carrez I (3.7), meša se 30 sekund in nato doda 5 ml raztopine Carrez II (3.8). V mešalniku se meša 30 minut, dopolni do oznake, homogenizira in filtrira.
5.1.3
Raztopina se pripravi, kakor je opisano v točki 5.1.2, vendar se ne filtrira. Dekantira se (po potrebi centrifugira), 100 ml supernatanta pa se odstrani in prenese v 200-mililitrsko merilno bučko. Zmeša se z acetonom (3.6) in z njim dopolni do oznake, homogenizira in filtrira.
5.2 Titracija
S pipeto se v erlenmajerico prenese 25–100 ml filtrata (glede na pričakovano vsebnost klora), ki nastane pri postopku iz točke 5.1.1, 5.1.2 ali 5.1.3. Alikvot ne sme vsebovati več kot 150 mg klora (Cl). Po potrebi se z vodo razredči na najmanj 50 ml, doda se 5 ml dušikove kisline (3.4), 20 ml nasičene raztopine amonij-železovega sulfata (3.3) in 2 kapljici raztopine amonijevega tiocianata (3.1), ki se doda iz birete, napolnjene do oznake nič. Z bireto se doda raztopina srebrovega nitrata (3.2), tako da nastane 5 ml presežka. Doda se 5 ml dietiletra (3.5) in dobro se pretrese, da oborina koagulira. Presežek srebrovega nitrata se titrira z raztopino amonijevega tiocianata (3.1), dokler ne nastane rdečkastorjavi odtenek, ki je obstojen 1 minuto.
6. Izračun rezultatov
Količina klora (X), izražena kot masni delež natrijevega klorida, se izračuna po naslednji formuli:

pri čemer je:
|
V1 |
= |
ml dodane raztopine srebrovega nitrata 0,1 mol/l; |
|
V2 |
= |
ml za titracijo uporabljene raztopine amonijevega tiocianata 0,1 mol/l; |
|
m |
= |
masa vzorca. |
Če je pri slepem preskusu uporabljen srebrov nitrat 0,1 mol/l, se ta vrednost odšteje od volumna (V1 – V2).
7. Opombe
7.1 Titracija se lahko izvede tudi s potenciometrom.
7.2 Proizvodi, ki vsebujejo veliko olj in maščob, se najprej razmastijo z dietiletrom ali petroletrom.
7.3 Pri ribji moki se titracija lahko izvede z Mohrovo metodo.
PRILOGA IV
ANALITSKE METODE ZA NADZOR RAVNI DOVOLJENIH KRMNIH DODATKOV
A. DOLOČANJE VITAMINA A
1. Namen in področje uporabe
Ta metoda omogoča določanje ravni vitamina A (retinola) v krmi in premiksih. Vitamin A vključuje celoten trans-retinil alkohol in njegove cis izomere, ki se določajo s to metodo. Vsebnost vitamina A se izrazi v mednarodnih enotah (IU) na kg. Ena IU ustreza aktivnosti 0,300 μg celotnega trans-vitamin A alkohola ali 0,344 μg acetata celotnega trans-vitamina A ali 0,550 μg palmitata celotnega trans-vitamina A.
Meja kvantifikacije je 2 000 IU vitamina A/kg.
2. Načelo
Vzorec se hidrolizira z raztopino etanolnega kalijevega hidroksida, vitamin A pa se ekstrahira v petroleter. Topilo se odstrani z uparevanjem, ostanek pa raztopi v metanolu in po potrebi razredči do zahtevane koncentracije. Vsebnost vitamina A se določi s plinsko kromatografijo visoke ločljivosti z reverzno fazo (RP–HPLC) z uporabo UV- ali fluorescenčnega detektorja. Kromatografski parametri se izberejo tako, da se celoten trans-vitamin A alkohol in njegovi cis izomeri ne ločijo.
3. Reagenti
3.1 Etanol, σ = 96 %
3.2 Petroleter, vrelišče 40–60 oC
3.3 Metanol
3.4 Raztopina kalijevega hidroksida, c = 50 g/100 ml
3.5 Raztopina natrijevega askorbata, c = 10 g/100 ml (glej opombo 7.7)
3.6 Natrijev sulfid, Na2S· x H2O (x = 7–9)
3.6.1 Raztopina natrijevega sulfida, c = 0,5 mol/l v glicerolu, ß = 120 g/l (za x = 9) (glej opombo 7.8)
3.7 Raztopina fenolftaleina, c = 2 g/100 ml v etanolu (3.1)
3.8 2-propanol
3.9 Mobilna faza za HPLC: mešanica metanola (3,3 ) in vode, npr. 980 + 20 (v + v). Točno razmerje bo določeno glede na značilnosti uporabljene kolone.
3.10 Dušik, brez kisika
3.11 Acetat celotnega trans-vitamina A, posebne čistosti, certificirane aktivnosti, npr. 2,80 × 106 IU/g
3.11.1 Osnovna raztopina acetata celotnega trans-vitamina A: 50 mg acetata vitamina A (3.11) se natehta na 0,1 mg natančno in prenese v 100-mililitrsko merilno bučko. Raztopi se v 2-propanolu (3.8) in dopolni do oznake z istim topilom. Nominalna koncentracija te raztopine je 1 400 IU vitamina A na ml. Točno vsebnost je treba določiti v skladu s točko 5.6.3.1.
3.12 Palmitat celotnega trans-vitamina A, posebne čistosti, certificirane aktivnosti, npr. 1,80 × 106 IU/g
3.12.1 Osnovna raztopina palmitata celotnega trans-vitamina A: 80 mg palmitata vitamina A (3.12) se natehta na 0,1 mg natančno in prenese v 100-mililitrsko merilno bučko. Raztopi se v 2-propanolu (3.8) in dopolni do oznake z istim topilom. Nominalna koncentracija te raztopine je 1 400 IU vitamina A na ml. Točno vsebnost je treba določiti v skladu s točko 5.6.3.2.
3.13 2,6-Di-tert-butil-4-metilfenol (BHT) (glej opombo 7.5)
4. Oprema
4.1 Rotacijski vakuumski uparjalnik
4.2 Temna steklovina
4.2.1 Bučke z ravnim dnom ali erlenmajerice, 500-mililitrske, z brušenim vratom
4.2.2 Merilne bučke z zamaški iz brušenega stekla, z ozkim vratom, 10, 25, 100 in 500-mililitrske
4.2.3 Liji ločniki, konusni, 1 000 -mililitrski, z zamaški iz brušenega stekla
4.2.4 Hruškaste bučke, 250-mililitrske, z zamaški iz brušenega stekla
4.3 Allihnov kondenzator, z dolžino obroča 300 mm s spojem iz brušenega stekla in z nastavkom za dovodno cev plina
4.4 Naguban filtrirni papir za ločevanje faz, premera 185 mm (npr. Schleicher & Schuell 597 HY 1/2)
4.5 Oprema HPLC s sistemom vbrizgavanja
4.5.1 Kolona za tekočinsko kromatografijo, 250 mm x 4 mm, C18, polnitev 5 ali 10 μm ali enakovredna (kriterij za zmogljivost: samo en vrh za vse izomere retinola pod pogoji kromatografije HPLC)
4.5.2 UV- ali fluorescenčni detektor s spremenljivo nastavitvijo valovne dolžine
4.6 Spektrofotometer z 10 mm kvarcnimi kivetami
4.7 Vodna kopel z magnetnim mešalnikom
4.8 Naprava za ekstrakcijo (glej sliko 1), sestavljena iz:
4.8.1 steklenega valja z volumnom l litra, vratom iz brušenega stekla in zamaškom;
4.8.2 vložka iz brušenega stekla s stransko ročico in nastavljivo cevjo, ki gre skozi središče. Nastavljiva cev ima spodnji del v obliki črke U in vbrizg na nasprotnem koncu, tako da vrhnja plast tekočine v valju lahko preide v lij ločnik.
5. Postopek
|
Opomba |
: |
Vitamin A je občutljiv na (UV) svetlobo in oksidacijo. Vsi postopki se izvajajo brez svetlobe (z uporabo temne steklovine ali steklene posode, zaščitene v aluminijasto folijo) in kisika (spiranje z dušikom). Med ekstrakcijo se zrak nad tekočino zamenja z dušikom (previsokemu tlaku se izogne z občasnim popuščanjem zamaška). |
5.1 Priprava vzorca
Vzorec se zdrobi tako, da preide skozi 1 mm sito, pri čemer se je treba izogibati nastanku toplote. Drobljenje se mora izvesti neposredno pred tehtanjem in umiljenjem, drugače lahko pride do izgube vitamina A.
5.2 Umiljenje
Glede na vsebnost vitamina A se natehta 2–25 g vzorca na 1 mg natančno in prenese v 500-mililitrsko bučko z ravnim dnom ali erlenmajerico (4.2.1). Med obračanjem se postopoma dodajo 130 ml etanola (3.1), približno 100 mg BHT (3.13), 2 ml raztopine natrijevega askorbata (3.5) in 2 ml raztopine natrijevega sulfida (3.6). Na bučko se namesti kondenzator (4.3), bučka pa se potopi v vodno kopel z magnetnim mešalnikom (4.7). Segreje se do vretja in 5 minut pusti pod kondenzatorjem povratnega toka. Nato se skozi kondenzator doda 25 ml raztopine kalijevega hidroksida (3.4) in pusti pod kondenzatorjem (4.3) povratnega toka nadaljnjih 25 minut, z mešanjem pod rahlim pretokom dušika. Nato se kondenzator spere s približno 20 ml vode, vsebina bučke pa ohladi na sobno temperaturo.
5.3 Ekstrakcija
Z dekantiranjem se raztopina za umiljenje kvantitativno prenese v 1 000 -mililitrski lij ločnik (4.2.3) ali napravo za ekstrakcijo (4.8), tako da se spere s skupno 250 ml vode. Nato se bučka za umiljenje spere s 25 ml etanola (3.1) in 100 ml petroletra (3.2), raztopine za spiranje pa se prenesejo v lij ločnik ali napravo za ekstrakcijo. Razmerje vode in etanola v združenih raztopinah mora biti približno 2:1. 2 minuti se močno stresa in nato 2 minuti pusti, da se umiri.
5.3.1
Ko so plasti ločene (glej opombo 7.3), se plast petroletra prenese v drug lij ločnik (4.2.3). Ekstrakcija se ponovi dvakrat s 100 ml petroletra (3.2) in dvakrat s 50 ml petroletra (3.2).
Združene ekstrakte v liju ločniku se med rahlim pretresanjem (da se prepreči nastanek emulzij) dvakrat spere s po 100 ml vode in nato s ponavljajočim se stresanjem ponovno s po 100 ml vode, dokler voda pri dodajanju raztopine fenolftaleina (3.7) ne ostane brezbarvna (navadno zadošča štirikratno spiranje). Sprani ekstrakt se filtrira skozi suh naguban filter za ločevanje faz (4.4) v 500-mililitrsko merilno bučko (4.2.2), da se odstrani suspendirana voda. Lij ločnik in filter se spereta s 50 ml petroletra (3.2), dopolni se do oznake s petroletrom (3.2) in dobro premeša.
5.3.2
Ko se plasti ločijo (glej opombo 7.3), se zamašek steklenega valja (4.8.1) zamenja z vložkom iz brušenega stekla (4.8.2), spodnji del nastavljive cevi pa se namesti v obliki črke U tik nad raven vmesnika. S tlakom, ki ga tvori dušik v stranski ročici, se zgornja plast petroletra prenese v 1 000 -mililitrski lij ločnik (4.2.3). V stekleni valj se doda 100 ml petroletra (3.2), zamaši in dobro pretrese. Počaka se, da se plasti ločijo, zgornja plast pa se prenese v lij ločnik tako kot prej. Postopek ekstrakcije se ponovi s 100 ml petroletra (3.2), nato dvakrat s po 50 ml petroletra (3.2), plasti petroletra pa se dodajo v lij ločnik.
Združeni ekstrakti petroletra se sperejo, kakor je opisano v točki 5.3.1, nato pa se nadaljuje, kakor je navedeno v navedeni točki.
5.4 Priprava raztopine vzorca za HPLC
Alikvot raztopine petroletra (iz 5.3.1 ali 5.3.2) se odpipetira v 250-mililitrsko hruškasto bučko (4.2.4). Topilo se upari skoraj do suhega v rotacijskem uparjalniku (4.1) pri znižanem tlaku in temperaturi kopeli do 40 oC. Atmosferski tlak se vzpostavi z dovajanjem dušika (3.10), bučka pa se odstrani iz rotacijskega uparjalnika. Preostalo topilo se odstrani v toku dušika (3.10), ostanek pa se takoj raztopi v znanem volumnu (10–100 ml) metanola (3.3) (koncentracija vitamina A mora biti 5–30 IU/ml).
5.5 Določanje s HPLC
Vitamin A se loči na koloni C18 z reverzno fazo (4.5.1), koncentracija pa se izmeri z UV-detektorjem (325 nm) ali fluorescenčnim detektorjem (ekscitacija: 325 nm, emisija: 475 nm) (4.5.2).
Vbrizga se alikvot (npr. 20 μl) raztopine metanola, ki nastane pri točki 5.4, in eluira z mobilno fazo (3.9). Izračuna se srednja vrednost višine (površine) vrhov za več vbrizgov iste raztopine vzorca in srednje vrednosti višin (površin) vrhov za več vbrizgov raztopin za umerjanje (5.6.2).
Naslednji pogoji se lahko uporabljajo kot smernica; lahko se uporabljajo tudi drugi pogoji, če zagotavljajo enakovredne rezultate.
|
Kolona za tekočinsko kromatografijo (4.5.1): |
250 mm × 4 mm, C18, polnitev 5 ali 10 μm ali enakovredna |
|
Mobilna faza (3.9): |
Mešanica metanol (3.3) in vode, npr. 980 + 20 (v + v) |
|
Hitrost pretoka: |
1–2 ml/min |
|
Detektor (4.5.2): |
UV-detektor (325 nm) ali fluorescenčni detektor (ekscitacija: 325 nm/emisija: 475 nm) |
5.6 Umerjanje
5.6.1
20 ml osnovne raztopine acetata vitamina A (3.11.1) ali 20 ml osnovne raztopine palmitata vitamina A (3.12.1) se odpipetira v 500-mililitrsko bučko z ravnim dnom ali erlenmajerico (4.2.1) ter hidrolizira, kakor je navedeno v točki 5.2, vendar brez dodatka BHT. Nato se ekstrahira s petroletrom (3.2) v skladu s točko 5.3 in dopolni do 500 ml s petroletrom (3.2). 100 ml tega ekstrakta se upari v rotacijskem uparjalniku (glej 5.4) skoraj do suhega, odstrani se preostalo topilo s tokom dušika (3.10), ostanek pa se ponovno raztopi v 10,0 ml metanola (3.3). Nominalna koncentracija te raztopine je 560 IU vitamina A na ml. Točno vsebnost je treba določiti v skladu s točko 5.6.3.3. Delovno standardno raztopino je treba sveže pripraviti pred uporabo.
2,0 ml te delovne standardne raztopine se odpipetira v 20-mililitrsko merilno bučko, dopolni se z metanolom (3.3) do oznake in premeša. Nominalna koncentracija te razredčene delovne standardne raztopine je 56 IU vitamina A na ml.
5.6.2
1,0 , 2,0 , 5,0 in 10,0 ml razredčene delovne standardne raztopine se prenese v serijo 20-mililitrskih merilnih bučk, dopolni se z metanolom (3.3) do oznake in premeša. Nominalne koncentracije teh raztopin so 2,8 , 5,6 , 14,0 in 28,0 IU vitamina A na ml.
Večkrat se vbrizga 20 μl vsake od raztopin za umerjanje in določijo se srednje vrednosti višin (površin) vrhov. Iz srednjih vrednosti višin (površin) vrhov se pripravi umeritvena krivulja ob upoštevanju rezultatov UV-kontrole (5.6.3.3).
5.6.3
5.6.3.1
2,0 ml osnovne raztopine acetata vitamina A (3.11.1) se odpipetira v 50 ml merilno bučko (4.2.2) in dopolni z 2-propanolom (3.8) do oznake. Nominalna koncentracija te raztopine je 56 IU vitamina A na ml. 3,0 ml te razredčene raztopine acetata vitamina A se odpipetira v 25-mililitrsko merilno bučko in dopolni z 2-propanolom (3.8) do oznake. Nominalna koncentracija te raztopine je 6,72 IU vitamina A na ml. Izmeri se UV-spekter te raztopine proti 2-propanolu (3.8) v spektrofotometru (4.6) med 300 in 400 nm. Maksimalna ekstinkcija mora biti med 325 in 327 nm.
Izračun vsebnosti vitamina A:
IU vitamina A/ml = E326 × 19,0
(
![]()
za acetat vitamina A = 1 530 pri 326 nm v 2-propanolu)
5.6.3.2
2,0 ml osnovne raztopine palmitata vitamina A (3.12.1) se odpipetira v 50-mililitrsko merilno bučko (4.2.2) in dopolni z 2-propanolom (3.8) do oznake . Nominalna koncentracija te raztopine je 56 IU vitamina A na ml. 3,0 ml te razredčene raztopine palmitata vitamina A se odpipetira v 25-mililitrsko merilno bučko in dopolni z 2-propanolom (3.8) do oznake. Nominalna koncentracija te raztopine je 6,72 IU vitamina A na ml. Izmeri se UV-spekter te raztopine proti 2-propanolu (3.8) v spektrofotometru (4.6) med 300 in 400 nm. Maksimalna ekstinkcija mora biti med 325 in 327 nm.
Izračun vsebnosti vitamina A:
IU vitamina A/ml = E326 × 19,0
(
![]()
za palmitat vitamina A = 957 pri 326 nm v 2-propanolu)
5.6.3.3
3,0 ml nerazredčene delovne standardne raztopine vitamina A, pripravljene v skladu s točko 5.6.1, se odpipetira v 50-mililitrsko merilno bučko (4.2.2) in dopolni z 2-propanolom (3.8) do oznake. 5,0 ml te raztopine se odpipetira v 25-mililitrsko merilno bučko in dopolni z 2-propanolom (3.8) do oznake . Nominalna koncentracija te raztopine je 6,72 IU vitamina A na ml. Izmeri se UV-spekter te raztopine proti 2-propanolu (3.8) v spektrofotometru (4.6) med 300 in 400 nm. Maksimalna ekstinkcija mora biti med 325 in 327 nm.
Izračun vsebnosti vitamina A:
IU vitamina A/ml = E325 × 18,3
(
![]()
za vitamin A alkohol = 1 821 pri 325 nm v 2-propanolu)
6. Izračun rezultatov
Iz srednje vrednosti višin (površin) vrhov vitamina A iz raztopine vzorca se s pomočjo umeritvene krivulje (5.6.2) določi koncentracija raztopine vzorca v IU/ml.
Vsebnost vitamina A (w) v vzorcu, izražena v IU/kg, se izračuna po naslednji formuli:

[IU/kg]
pri čemer je:
|
c |
= |
koncentracija vitamina A (5.4) v IU/ml v raztopini vzorca; |
|
V1 |
= |
volumen raztopine vzorca (5.4) v ml; |
|
V2 |
= |
volumen alikvota, uporabljenega pri točki 5.4, v ml; |
|
m |
= |
masa preskusnega vzorca v gramih. |
7. Opombe
7.1 Pri vzorcih z nizko koncentracijo vitamina A se ekstrakte petroletra iz dveh umilitvenih šarž (natehtana količina: 25 g) lahko združi v eno raztopino vzorca za določanje s HPLC.
7.2 Masa vzorca za analizo ne vsebuje več kot 2 g maščobe.
7.3 Če se fazi ne ločita, se doda približno 10 ml etanola (3.1) za razbitje emulzije.
7.4 Pri ribjem olju in drugih čistih maščobah se čas umiljenja podaljša na 45–60 minut.
7.5 Namesto BHT se lahko uporablja hidrokinon.
7.6 Z običajno fazno kolono je možno ločiti izomere retinola. Vendar je pri tem treba za izračune zabeležiti vse višine (površine) vrhov vseh cis in trans izomerov.
7.7 Namesto raztopine natrijevega askorbata se lahko uporabi približno 150 mg askorbinske kisline.
7.8 Namesto raztopine natrijevega sulfida se lahko uporabi približno 50 mg EDTA.
7.9 Pri analizi vitamina A v mlečnih nadomestkih je treba paziti:
— pri umiljenju (5.2): zaradi količine maščobe v vzorcu je lahko potrebna večja količina raztopine kalijevega hidroksida (3.4),
— pri ekstrakciji (5.3): zaradi prisotnosti emulzij je morda potrebna sprememba razmerja voda/etanol 2:1.
Da se preveri, ali uporabljena analitska metoda zagotavlja zanesljive rezultate glede te specifične matrice (mlečni nadomestek), je treba na dodatnem preskusnem vzorcu izvesti preskus izkoristka. Če je izkoristek nižji od 80 %, se pri analitskem rezultatu uporabi popravek za izkoristek.
8. Ponovljivost
Razlika med rezultatoma dveh vzporednih določanj, izvedenih na istem vzorcu, ne sme presegati 15 % najvišjega rezultata.
9. Rezultati medlaboratorijske študije ( 11 )
|
|
Premiks |
Krma in premiksi |
Mineralni koncentrat |
Beljakovinska krma |
Krma za prašiče |
|
L |
13 |
12 |
13 |
12 |
13 |
|
n |
48 |
45 |
47 |
46 |
49 |
|
Srednja vrednost [IU/kg] |
17,02 x 106 |
1,21 x 106 |
537 100 |
151 800 |
18 070 |
|
Sr [IU/kg] |
0,51 x 106 |
0,039 x 106 |
22 080 |
12 280 |
682 |
|
r [IU/kg] |
1,43 x 106 |
0,109 x 106 |
61 824 |
34 384 |
1 910 |
|
CVr [ %] |
3,0 |
3,5 |
4,1 |
8,1 |
3,8 |
|
SR [IU/kg] |
1,36 x 106 |
0,069 x 106 |
46 300 |
23 060 |
3 614 |
|
R [IU/kg] |
3,81 x 106 |
0,193 x 106 |
129 640 |
64 568 |
10 119 |
|
CVR [ %] |
8,0 |
6,2 |
8,6 |
15 |
20 |
|
L |
: |
število laboratorijev; |
|
n |
: |
število posameznih vrednosti; |
|
Sr |
: |
standardni odmik ponovljivosti; |
|
SR |
: |
standardni odmik obnovljivosti; |
|
r |
: |
ponovljivost; |
|
R |
: |
obnovljivost; |
|
CVr |
: |
koeficient variacije ponovljivosti; |
|
CVR |
: |
koeficient variacije obnovljivosti; |

B. DOLOČANJE VITAMINA E
1. Namen in področje uporabe
Ta metoda omogoča določanje ravni vitamina E v krmi in premiksih. Vsebnost vitamina E se izrazi v mg acetata DL-α-tokoferola na kg. 1 mg acetata DL-α-tokoferola ustreza 0,91 mg DL-α-tokoferola (vitamina E).
Meja kvantifikacije je 2 mg vitamina E/kg. Ta meja kvantifikacije se lahko doseže le s fluorescenčnim detektorjem. Meja kvantifikacije za UV-detektor je 10 mg/kg.
2. Načelo
Vzorec se hidrolizira z raztopino etanolnega kalijevega hidroksida, vitamin E pa se ekstrahira v petroleter. Topilo se odstrani z uparevanjem, ostanek pa raztopi v metanolu in po potrebi razredči do zahtevane koncentracije. Vsebnost vitamina E se določi s tekočinsko kromatografijo visoke ločljivosti z reverzno fazo (RP–HPLC) z uporabo UV- ali fluorescenčnega detektorja.
3. Reagenti
3.1 Etanol, σ = 96 %
3.2 Petroleter, vrelišče 40–60 oC
3.3 Metanol
3.4 Raztopina kalijevega hidroksida, c = 50 g/100 ml
3.5 Raztopina natrijevega askorbata, c = 10 g/100 ml (glej opombo 7.7)
3.6 Natrijev sulfid, Na2S· x H2O (x = 7–9)
3.6.1 Raztopina natrijevega sulfida, c = 0,5 mol/l v glicerolu, β = 120 g/l (za x = 9) (glej opombo 7.8)
3.7 Raztopina fenolftaleina, c = 2 g/100 ml v etanolu (3.1)
3.8 Mobilna faza za HPLC: mešanica metanola (3.3) in vode, npr. 980 + 20 (v + v). Točno razmerje bo določeno glede na značilnosti uporabljene kolone.
3.9 Dušik, brez kisika
3.10 Acetat DL-α-tokoferola, posebne čistosti, certificirane aktivnosti
3.10.1 Osnovna raztopina acetata DL-α-tokoferola: 100 mg acetata DL-α-tokoferola (3.10) se natehta na 0,1 mg natančno in prenese v 100-mililitrsko merilno bučko. Raztopi se v etanolu (3.1) in dopolni z istim topilom do oznake. 1 ml te raztopine vsebuje 1 mg acetata DL-α-tokoferola. (Za UV-kontrolo glej točko 5.6.1.3, za stabilizacijo pa opombo 7.4.)
3.11 DL-α-tokoferol, posebne čistosti, certificirane aktivnosti
3.11.1 Osnovna raztopina acetata DL-α-tokoferola: 100 mg DL-α-tokoferola (3.10) se natehta na 0,1 mg natančno in prenese v 100-mililitrsko merilno bučko. Raztopi se v etanolu (3.1) in dopolni z istim topilom do oznake. 1 ml te raztopine vsebuje 1 mg DL-α-tokoferola. (Za UV-kontrolo glej točko 5.6.2.3, za stabilizacijo pa opombo 7.4.)
3.12 2,6-Di-tert-butil-4-metilfenol (BHT) (glej opombo 7.5)
4. Oprema
4.1 Rotacijski uparjalnik
4.2 Temna steklovina
4.2.1 Bučke z ravnim dnom ali erlenmajerice, 500-mililitrske, z brušenim vratom
4.2.2 Merilne bučke z zamaški iz brušenega stekla, z ozkim vratom, 10-, 25-, 100- in 500-mililitrske
4.2.3 Liji ločniki, konusni, 1 000 -mililitrski, z zamaški iz brušenega stekla
4.2.4 Hruškaste bučke, 250-mililitrske, z zamaški iz brušenega stekla
4.3 Allihnov kondenzator, dolžina obroča 300 mm, s spojem iz brušenega stekla in nastavkom za dovodno cev plina
4.4 Naguban filtrirni papir za ločevanje faz, premera 185 mm (npr. Schleicher & Schuell 597 HY 1/2)
4.5 Oprema za kromatografijo HPLC s sistemom vbrizgavanja
4.5.1 Kolona za tekočinsko kromatografijo, 250 mm x 4 mm C18, polnitev 5 ali 10 μm ali enakovredna
4.5.2 Fluorescenčni ali UV-detektor, s spremenljivo nastavitvijo valovne dolžine
4.6 Spektrofotometer z 10 mm kvarcnimi kivetami
4.7 Vodna kopel z magnetnim mešalnikom
4.8 Naprava za ekstrakcijo (glej sliko 1), sestavljena iz:
4.8.1 steklenega valja z volumnom l litra, vratom iz brušenega stekla in zamaškom;
4.8.2 vložka iz brušenega stekla s stransko ročico in nastavljivo cevjo, ki gre skozi središče. Nastavljiva cev ima spodnji del v obliki črke U in vbrizg na nasprotnem koncu, tako da vrhnja plast tekočine v valju lahko preide v lij ločnik.
5. Postopek
|
Opomba |
: |
Vitamin E je občutljiv na (UV) svetlobo in oksidacijo. Vsi postopki se izvajajo brez svetlobe (z uporabo temne steklovine ali steklene posode, zaščitene v aluminijasto folijo) in kisika (spiranje z dušikom). Med ekstrakcijo se zrak nad tekočino zamenja z dušikom (previsokemu tlaku se izogne z občasnim rahljanjem zamaška). |
5.1 Priprava vzorca
Vzorec se zdrobi tako, da preide skozi 1 mm sito, pri čemer se je treba izogibati nastanku toplote. Drobljenje se mora izvesti neposredno pred tehtanjem in umiljenjem, drugače lahko pride do izgube vitamina E.
5.2 Umiljenje
Glede na vsebnost vitamina E se natehta 2–25 g vzorca na 0,01 g natančno in prenese v 500-mililitrsko bučko z ravnim dnom ali erlenmajerico (4.2.1). Med obračanjem se postopoma dodajo 130 ml etanola (3.1), približno 100 mg BHT (3.12), 2 ml raztopine natrijevega askorbata (3.5) in 2 ml raztopine natrijevega sulfida (3.6). Na bučko se namesti kondenzator (4.3), bučka pa se potopi v vodno kopel z magnetnim mešalnikom (4.7). Segreje se do vretja in 5 minut pusti pod kondenzatorjem povratnega toka. Nato se skozi kondenzator (4.3) doda 25 ml raztopine kalijevega hidroksida (3.4) in z mešanjem pod rahlim pretokom dušika pusti pod kondenzatorjem povratnega toka nadaljnjih 25 minut. Nato se kondenzator spere s približno 20 ml vode, vsebina bučke pa ohladi na sobno temperaturo.
5.3 Ekstrakcija
Z dekantiranjem se raztopina za umiljenje kvantitativno prenese v 1 000 -mililitrski lij ločnik (4.2.3) ali napravo za ekstrakcijo (4.8), tako da se spere s skupno 250 ml vode. Nato se bučka za umiljenje spere s 25 ml etanola (3.1) in 100 ml petroletra (3.2), raztopine za spiranje pa se prenesejo v lij ločnik ali napravo za ekstrakcijo. Razmerje vode in etanola v združenih raztopinah mora biti približno 2:1. 2 minuti se močno stresa in nato 2 minuti pusti, da se umiri.
5.3.1
Ko so plasti ločene (glej opombo 7.3), se plast petroletra prenese v drug lij ločnik (4.2.3). Ekstrakcija se ponovi dvakrat s 100 ml petroletra (3.2) in dvakrat s 50 ml petroletra (3.2).
Združene ekstrakte v liju ločniku se med rahlim pretresanjem (da se prepreči nastanek emulzij) dvakrat spere s po 100 ml vode in nato s ponavljajočim se stresanjem ponovno s po 100 ml vode, dokler voda pri dodajanju raztopine fenolftaleina (3.7) ne ostane brezbarvna (navadno zadošča štirikratno spiranje). Sprani ekstrakt se filtrira skozi suh naguban filter za ločevanje faz (4.4) v 500-mililitrsko merilno bučko (4.2.2), da se odstrani suspendirana voda. Lij ločnik in filter se spereta s 50 ml petroletra (3.2), dopolni se do oznake s petroletrom (3.2) in dobro premeša.
5.3.2
Ko se plasti ločijo (glej opombo 7.3), se zamašek steklenega valja (4.8.1) zamenja z vložkom iz brušenega stekla (4.8.2), spodnji del nastavljive cevi pa se namesti v obliki črke U tik nad raven vmesnika. S tlakom, ki ga tvori dušik v stranski ročici, se zgornja plast petroletra prenese v 1 000 -mililitrski lij ločnik (4.2.3). V stekleni valj se doda 100 ml petroletra (3.2), zamaši se in dobro pretrese. Počaka se, da se plasti ločijo, zgornja plast pa se prenese v lij ločnik tako kot prej. Postopek ekstrakcije se ponovi s 100 ml petroletra (3.2), nato dvakrat s po 50 ml petroletra (3.2), plasti petroletra pa se dodajo v lij ločnik.
Združeni ekstrakti petroletra se sperejo, kakor je opisano v točki 5.3.1, nato pa se nadaljuje, kakor je navedeno v navedeni točki.
5.4 Priprava raztopine vzorca za HPLC
Alikvot raztopine petroletra (iz točke 5.3.1 ali 5.3.2) se odpipetira v 250-mililitrsko hruškasto bučko (4.2.4). Topilo se upari skoraj do suhega v rotacijskem uparjalniku (4.1) pri znižanem tlaku in temperaturi kopeli do 40 oC. Atmosferski tlak se vzpostavi z dovajanjem dušika (3.9), bučka pa se odstrani iz rotacijskega uparjalnika. Preostalo topilo se odstrani v toku dušika (3.9), ostanek pa se takoj raztopi v znanem volumnu (10–100 ml) metanola (3.3) (koncentracija DL-α-tokoferola mora biti 5–30 μg/ml).
5.5 Določanje s HPLC
Vitamin E se loči na koloni C18 z reverzno fazo (4.5.1), koncentracija pa se izmeri s fluorescenčnim detektorjem (ekscitacija: 295 nm, emisija: 330 nm) ali UV-detektorjem (292 nm) (4.5.2).
Vbrizga se alikvot (npr. 20 μl) raztopine metanola, ki nastane pri točki 5.4, in eluira se z mobilno fazo (3.8). Izračunajo se srednje vrednosti višin (površin) vrhov za več vbrizgov iste raztopine vzorca in srednje vrednosti višin (površin) vrhov za več vbrizgov raztopin za umerjanje (5.6.2).
Naslednji pogoji se lahko uporabljajo kot smernica; lahko se uporabljajo tudi drugi pogoji, če zagotavljajo enakovredne rezultate.
|
Kolona za tekočinsko kromatografijo (4.5.1): |
250 mm × 4 mm, C18, polnitev 5 ali 10 μm ali enakovredna |
|
Mobilna faza (3.8): |
Mešanica metanola (3.3) in vode, npr. 980 + 20 (v + v). |
|
Hitrost pretoka: |
1–2 ml/min |
|
Detektor (4.5.2): |
Fluorescečni detektor: (ekscitacija: 295 nm/emisija: 330 nm) ali UV-detektor (292 nm) |
5.6 Umerjanje (acetat DL-α-tokoferola ali DL-α-tokoferol)
5.6.1
5.6.1.1
25 ml osnovne raztopine acetata DL-α-tokoferola (3.10.1) se odpipetira v 500-mililitrsko bučko z ravnim dnom ali erlenmajerico (4.2.1) in hidrolizira, kakor je opisano v točki 5.2. Nato se ekstrahira s petroletrom (3.2) v skladu s točko 5.3 in dopolni do 500 ml s petroletrom. 25 ml tega ekstrakta se upari v rotacijskem uparjalniku (glej 5.4) skoraj do suhega, odstrani se preostalo topilo s tokom dušika (3.9), ostanek pa se ponovno raztopi v 25,0 ml metanola (3.3). Nominalna koncentracija te raztopine je 45,5 μg DL-α-tokoferola na ml, kar je enakovredno 50 μg acetata DL-α-tokoferola na ml. Delovno standardno raztopino je treba pripraviti svežo pred uporabo.
5.6.1.2
1,0 , 2,0 , 4,0 in 10,0 ml delovne standardne raztopine se prenese v serijo 20-mililitrskih merilnih bučk, dopolni se z metanolom (3.3) do oznake in premeša. Nominalne koncentracije teh raztopin so 2,5 , 5,0 , 10,0 in 25,0 μg/ml acetata DL-α-tokoferola, tj. 2,28 , 4,55 , 9,10 in 22,8 μg/ml DL-α-tokoferola.
Večkrat se vbrizga 20 μl vsake raztopine za umerjanje, nato pa se določijo srednje vrednosti višin (površin) vrhov. Iz srednjih vrednosti višin (površin) vrhov se pripravi umeritvena krivulja.
5.6.1.3
5,0 ml osnovne raztopine acetata DL-α-tokoferola (3.10.1) se razredči na 25,0 ml z etanolom, UV-spekter te raztopine pa se izmeri proti etanolu (3.1) v spektrofotometru (4.6) med 250 in 320 nm.
Maksimalna absorpcija je pri 284 nm:
![]()
= 43,6 pri 284 nm v etanolu
Pri tem razredčenju mora biti vrednost ekstinkcije 0,84 do 0,88 .
5.6.2
5.6.2.1
2 ml osnovne raztopine DL-α-tokoferola (3.11.1) se odpipetira v 50-mililitrsko merilno bučko, raztopi v metanolu (3.3) in dopolni z metanolom do oznake. Nominalna koncentracija te raztopine je 40 μg DL-α-tokoferola na ml, kar je enakovredno 44,0 μg acetata DL-α-tokoferola na ml. Delovno standardno raztopino je treba pripraviti svežo pred uporabo.
5.6.2.2
1,0 , 2,0 , 4,0 in 10,0 ml delovne standardne raztopine se prenese v serijo 20-mililitrskih merilnih bučk, dopolni se do oznake z metanolom (3.3) in premeša. Nominalne koncentracije teh raztopin so 2,0 , 4,0 , 8,0 in 20,0 μg/ml DL-α-tokoferola, tj. 2,20 , 4,40 , 8,79 in 22,0 μg/ml acetata DL-α-tokoferola.
Večkrat se vbrizga 20 μl vsake raztopine za umerjanje, nato pa se določijo srednje vrednosti višin (površin) vrhov. Iz srednjih vrednosti višin (površin) vrhov se pripravi umeritvena krivulja.
5.6.2.3
2,0 ml osnovne raztopine DL-α-tokoferola (3.11.1) se razredči na 25,0 ml z etanolom, UV-spekter te raztopine pa se izmeri proti etanolu (3.1) v spektrofotometru (4.6) med 250 in 320 nm. Maksimalna absorpcija je pri 292 nm:
![]()
= 75,8 pri 292 nm v etanolu
Pri tem razredčenju mora biti vrednost ekstinkcije 0,6 .
6. Izračun rezultatov
Iz srednje vrednosti višine (površine) vrhov vitamina E iz raztopine vzorca se s pomočjo umeritvene krivulje (5.6.1.2 ali 5.6.2.2) določi koncentracija raztopine vzorca v μg/ml (izračunana kot acetat DL-α-tokoferola).
Vsebnost vitamina E (w) v mg/kg v vzorcu se izračuna po naslednji formuli:

[mg/kg]
pri čemer je:
|
c |
= |
koncentracija vitamina E (kot acetata α-tokoferola) v raztopini vzorca (5.4) v μg/ml; |
|
V1 |
= |
volumen raztopine vzorca (5.4) v ml; |
|
V2 |
= |
volumen alikvota, uporabljenega pri točki 5.4, v ml; |
|
m |
= |
masa preskusnega vzorca v gramih. |
7. Opombe
7.1 Pri vzorcih z nizko koncentracijo vitamina E se ekstrakti petroletra iz dveh umilitvenih šarž (natehtana množina: 25 g) združijo v eno raztopino vzorca za določanje s HPLC.
7.2 Masa vzorca za analizo ne vsebuje več kot 2 g maščobe.
7.3 Če se fazi ne ločita, se doda približno 10 ml etanola (3.1) za razbitje emulzije.
7.4 Po spektrofotometrični meritvi raztopine acetata DL-α-tokoferola ali raztopine DL-α-tokoferola v skladu s točko 5.6.1.3 ali pa 5.6.2.3 se doda približno 10 mg BHT (3.12) v raztopino (3.10.1 ali 3.10.2), raztopina pa se hrani v hladilniku (hrani se lahko največ 4 tedne).
7.5 Namesto BHT se lahko uporablja hidrokinon.
7.6 Z običajno fazno kolono je možno ločiti α-, β-, γ- in δ-tokoferol.
7.7 Namesto raztopine natrijevega askorbata se lahko uporabi približno 150 mg askorbinske kisline.
7.8 Namesto raztopine natrijevega sulfida se lahko uporabi približno 50 mg EDTA.
7.9 Acetat vitamina E se v bazičnih pogojih hidrolizira zelo hitro, zato je zelo občutljiv na oksidacijo, zlasti v navzočnosti elementov v sledovih, kot sta železo in baker. Če raven vitamina E v premiksih presega 5 000 mg/kg, se vitamin E pri določanju lahko razgradi. Zato je za potrditev priporočena metoda HPLC z encimsko razgradnjo strukture vitamina E brez bazičnega umiljenja.
8. Ponovljivost
Razlika med rezultatoma dveh vzporednih določanj, izvedenih na istem vzorcu, ne sme presegati 15 % najvišjega rezultata.
9. Rezultati medlaboratorijske študije ( 12 )
|
|
Premiks |
Krma in premiksi |
Mineralni koncentrat |
Beljakovinska krma |
Krma za prašiče |
|
L |
12 |
12 |
12 |
12 |
12 |
|
n |
48 |
48 |
48 |
48 |
48 |
|
Srednja vrednost [mg/kg] |
17 380 |
1 187 |
926 |
315 |
61,3 |
|
Sr [mg/kg] |
384 |
45,3 |
25,2 |
13,0 |
2,3 |
|
r [mg/kg] |
1 075 |
126,8 |
70,6 |
36,4 |
6,4 |
|
CVr [ %] |
2,2 |
3,8 |
2,7 |
4,1 |
3,8 |
|
SR mg/kg] |
830 |
65,0 |
55,5 |
18,9 |
7,8 |
|
R [mg/kg] |
2 324 |
182,0 |
155,4 |
52,9 |
21,8 |
|
CVR [ %] |
4,8 |
5,5 |
6,0 |
6,0 |
12,7 |
|
L |
: |
število laboratorijev; |
|
n |
: |
število posameznih vrednosti; |
|
Sr |
: |
standardni odmik ponovljivosti; |
|
SR |
: |
standardni odmik obnovljivosti; |
|
r |
: |
ponovljivost; |
|
R |
: |
obnovljivost; |
|
CVr |
: |
koeficient variacije ponovljivosti; |
|
CVR |
: |
koeficient variacije obnovljivosti. |

C. DOLOČANJE ŽELEZA, BAKRA, MANGANA IN CINKA V SLEDOVIH
1. Namen in področje uporabe
Ta metoda omogoča določanje železa, bakra, mangana in cinka v sledovih v krmi. Meje za določanje so:
— železo (Fe): 20 mg/kg,
— baker (Cu): 10 mg/kg,
— mangan (Mn): 20 mg/kg,
— cink (Zn): 20 mg/kg.
2. Načelo
Po razkroju morebitnih organskih snovi se vzorec raztopi v klorovodikovi kislini. Po primernem razredčenju se z atomsko absorpcijsko spektrometrijo določijo železo, baker, mangan in cink.
3. Reagenti
Uvodne opombe
Za pripravo reagentov in raztopin za analizo se uporabi voda brez kationov, ki jih je treba določiti, ki se pripravi z dvojnim destiliranjem vode v destilatorju iz borosilikatnega ali kremenovega stekla ali z dvojno ionsko izmenjavo.
Reagenti morajo biti najmanj analitske čistoče. Odsotnost elementa, ki se določa, se preveri s slepim preskusom. Po potrebi se morajo reagenti dodatno prečistiti.
Namesto spodaj navedenih standardnih raztopin se lahko uporabijo komercialne standardne raztopine, če imajo jamstvo in so bile pred uporabo preverjene.
3.1 Klorovodikova kislina (g: 1,19 g/ml)
3.2 Klorovodikova kislina (6 mol/l)
3.3 Klorovodikova kislina (0,5 mol/l)
3.4 38–40-odstotna (v/v) klorovodikova kislina z vsebnostjo železa (Fe) manj kot 1 mg/l in z manj kot 10 mg/l ostanka (v obliki sulfata) po uparevanju
3.5 Žveplova kislina (g: 1,84 g/ml)
3.6 Vodikov peroksid a.č. (približno 100 volumnov kisika (30 % po masi))
3.7 Standardna raztopina železa (1 000 μg Fe/ml), pripravljena na naslednji način, ali enakovredna komercialno dostopna raztopina: 1 g železne žice se raztopi v 200 ml klorovodikove kisline 6 mol/l (3.2), doda se 16 ml vodikovega peroksida (3.6) in dopolni z vodo do 1 litra.
3.7.1 Standardna delovna raztopina železa (100 μg Fe/ml), pripravljena z redčenjem 1 dela standardne raztopine (3.7) z 9 deli vode.
3.8 Standardna raztopina bakra (1 000 μg Cu/ml), pripravljena na naslednji način, ali enakovredna komercialno dostopna raztopina:
— 1 g bakra v prahu se raztopi v 25 ml klorovodikove kisline 6 mol/l (3.2), doda se 5 ml vodikovega peroksida (3.6) in dopolni z vodo do 1 litra.
3.8.1 Standardna delovna raztopina bakra (10 μg Cu/ml), pripravljena z redčenjem 1 dela standardne raztopine (3.8) z 9 deli vode, nato pa z redčenjem 1 dela te raztopine z 9 deli vode.
3.9 Standardna raztopina mangana (1 000 μg Mn/ml), pripravljena na naslednji način, ali enakovredna komercialno dostopna raztopina:
— 1 g mangana v prahu se raztopi v 25 ml klorovodikove kisline 6 mol/l (3.2) in dopolni z vodo do 1 litra.
3.9.1 Standardna delovna raztopina mangana (10 μg Mn/ml), pripravljena z redčenjem 1 dela standardne raztopine (3.9) z 9 deli vode, nato pa z redčenjem 1 dela te raztopine z 9 deli vode.
3.10 Standardna raztopina cinka (1 000 μg Zn/ml), pripravljena na naslednji način, ali enakovredna komercialno dostopna raztopina:
— 1 g cinka v foliji ali granulah se raztopi v 25 ml klorovodikove kisline 6 mol/l (3.2) in dopolni z vodo do 1 litra.
3.10.1 Standardna delovna raztopina cinka (10 μg Zn/ml), pripravljena z redčenjem 1 dela standardne raztopine (3.10) z 9 deli vode, nato pa z redčenjem 1 dela te raztopine z 9 deli vode.
3.11 Raztopina lantanovega klorida: 12 g lantanovega klorida se raztopi v 150 ml vode, doda se 100 ml klorovodikove kisline 6 mol/l (3.2) in dopolni z vodo do 1 litra.
4. Oprema
4.1 Žarilna peč z regulacijo temperature in po možnosti rekorderjem
4.2 Stekleni deli morajo biti iz odpornega borosilikata, priporočljiva pa je uporaba opreme, ki je namenjena izključno določanju elementov v sledovih.
4.3 Atomski absorpcijski spektrofotometer, ki ustreza zahtevam metode glede občutljivosti in natančnosti zahtevanega območja.
5. Postopek ( 13 )
5.1 Vzorci, ki vsebujejo organske snovi
5.1.1 ( 14 )
5.1.1.1 5–10 g vzorca se natehta na 0,2 mg natančno in prenese v žarilni lonček iz kvarca ali platine (glej opombo (b)), osuši v sušilniku pri 105 oC in postavi v hladno žarilno peč (4.1). Peč se zapre (glej opombo (c)), temperatura pa se približno 90 minut postopoma viša na 450–475 oC. Ta temperatura se ohranja 4–16 ur (npr. čez noč), da se odstranijo zoglenele snovi, nato pa se peč odpre in ohladi (glej opombo (d)).
Pepel se navlaži z vodo in prenese v 250-mililitrsko čašo. Lonček se spere s skupaj približno 5 ml klorovodikove kisline (3.1), vsebina pa se počasi in previdno prenese v čašo (lahko pride do burne reakcije zaradi nastanka CO2). Po kapljicah se dodaja klorovodikova kislina (3.1), meša pa se, dokler penjenje ne poneha. Upareva se do suhega, pri čemer se občasno premeša s stekleno palčko.
Nato se ostanku doda 15 ml klorovodikove kisline 6 mol/l (3.2) in približno 120 ml vode. Premeša se s stekleno paličico, ki se pusti v čaši, čaša pa se pokrije z urnim steklom. Počasi se segreva do vretja in pusti vreti do popolne raztopitve pepela. Skozi filter, ki ne vsebuje pepela, se filtrira v 250-mililitrsko merilno bučko. Čaša in filter se spereta s 5 ml vroče klorovodikove kisline 6 mol/l (3.2) in dvakrat z vrelo vodo. Merilna bučka se napolni z vodo do oznake (koncentracija HCl približno 0,5 mol/l).
5.1.1.2 Če je ostanek na filtru videti črn (ogljik), se ponovno postavi v peč in upepeli pri 450–475 oC. Ta upepelitev, ki zahteva le nekaj ur (približno 3–5 ur), je končana, ko je pepel videti bel ali skoraj bel. Ostanek se raztopi s približno 2 ml klorovodikove kisline (3.1), upari do suhega in doda 5 ml klorovodikove kisline 6 mol/l (3.2). Segreje se, raztopina se filtrira v merilno bučko in dopolni z vodo do oznake (koncentracija HCl približno 0,5 mol/l).
Opombe:
(a) Pri določanju elementov v sledovih je pomembno paziti zaradi tveganja onesnaženja, predvsem s cinkom, bakrom in železom. Zato oprema, ki se uporablja pri pripravi vzorcev, ne sme vsebovati teh kovin.
Za zmanjšanje splošnega tveganja onesnaženja je treba delati v brezprašnem prostoru s popolnoma čisto opremo in dobro pomitimi steklenimi posodami. Določanje cinka je posebej občutljivo na več vrst onesnaženja, npr. s stekleno posodo, reagenti, prahom itd.
(b) Masa vzorca, ki se upepeli, se izračuna iz približne vsebnosti elementov v sledovih v krmi glede na občutljivost uporabljenega spektrofotometra. Pri nekateri krmi z nizko vsebnostjo elementov v sledovih je morda potrebno začeti z 10–20 g vzorca ter končno raztopino dopolniti do samo 100 ml.
(c) Upepeljevanje se mora izvajati v zaprti peči brez dovajanja zraka ali kisika.
(d) Temperatura na pirometru ne sme preseči 475 oC.
5.1.2
5.1.2.1
Za vsak element, ki se določa, se iz standardnih delovnih raztopin iz točk 3.7.1, 3.8.1, 3.9.1 in 3.10.1 pripravi serija raztopin za umerjanje, pri čemer je koncentracija HCl v vsaki približno 0,5 mol/l (in (pri železu, manganu in cinku) koncentracija lantanovega klorida, ki ustreza 0,1 % La (m/v)).
Izbrane koncentracije elementov v sledovih morajo ustrezati območju občutljivosti uporabljenega spektrofotometra. V spodnjih preglednicah so kot primer navedena tipična območja raztopin za umerjanje glede na vrsto in občutljivost uporabljenega spektrofotometra, zato je morda treba izbrati drugačne koncentracije.
|
μg Fe/ml |
0 |
0,5 |
1 |
2 |
3 |
4 |
5 |
|
ml delovne standardne raztopine (3.7.1) (1 ml = 100 μg Fe) |
0 |
0,5 |
1 |
2 |
3 |
4 |
5 |
|
ml HCl (3.2) |
7 |
7 |
7 |
7 |
7 |
7 |
7 |
|
+ 10 ml raztopine lantanovega klorida (3.11) in dopolniti z vodo do 100 ml |
|||||||
|
μg Cu/ml |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
ml delovne standardne raztopine (3.8.1) (1 ml = 10 μg Cu) |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
|
ml HCl (3.2) |
8 |
8 |
8 |
8 |
8 |
8 |
8 |
|
μg Mn/ml |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
ml delovne standardne raztopine (3.9.1) (1 ml = 10 μg Mn) |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
|
ml HCl (3.2) |
7 |
7 |
7 |
7 |
7 |
7 |
7 |
|
+ 10 ml raztopine lantanovega klorida (3.11) in dopolniti z vodo do 100 ml |
|||||||
|
μg Zn/ml |
0 |
0,05 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
|
ml delovne standardne raztopine (3.10.1) (1 ml = 10 μg Zn) |
0 |
0,5 |
1 |
2 |
4 |
6 |
8 |
|
ml HCl (3.2) |
7 |
7 |
7 |
7 |
7 |
7 |
7 |
|
+ 10 ml raztopine lantanovega klorida (3.11) in dopolniti z vodo do 100 ml |
|||||||
5.1.2.2
Za določanje bakra se lahko raztopina, pripravljena v skladu s točko 5.1.1, ponavadi uporabi neposredno. Če je treba koncentracijo prilagoditi območju umeritve, se ustrezen alikvot odpipetira v 100-mililitrsko merilno bučko in dopolni s klorovodikovo kislino 0,5 mol/l (3.3) do oznake.
Za določanje železa, mangana in cinka se odpipetira ustrezen alikvot raztopine, pripravljene v skladu s točko 5.1.1, v 100-mililitrsko merilno bučko, doda se 10 ml raztopine lantanovega klorida (3.11) in dopolni do oznake s klorovodikovo kislino 0,5 mol/l (3.3) (glej tudi točko 8 „Opomba“).
5.1.2.3
Slepi preskus mora vključevati vse predpisane korake postopka, le da se izpusti vzorec. Raztopina za umerjanje „0“ se ne sme uporabljati kot slepa raztopina.
5.1.2.4
Atomska absorpcija raztopin za umerjanje in raztopine, ki se analizira, se izmeri z uporabo oksidnega plamena zrak-acetilen pri naslednjih valovnih dolžinah:
Fe: 248,3 nm
Cu: 324,8 nm
Mn: 279,5 nm
Zn: 213,8 nm
Vsako merjenje se izvede štirikrat.
5.2 Mineralna krma
Če vzorec ne vsebuje organskih snovi, predhodno upepeljevanje ni potrebno. Nadaljuje se v skladu z drugim odstavkom točke 5.1.1.1. Uparevanje s klorovodikovo kislino se lahko izpusti.
6. Izračun rezultatov
Koncentracija elementa v sledovih v raztopini, ki se analizira, se izračuna s pomočjo umeritvene krivulje, rezultat pa se izrazi v miligramih elementa v sledovih na kilogram vzorca (ppm).
7. Ponovljivost
Razlika med rezultatoma dveh vzporednih določanj, ki jih na istem vzorcu izvaja isti analitik, ne presega:
— 5 mg/kg absolutne vrednosti za vsebnosti zadevnega elementa v sledovih do 50 mg/kg,
— 10 % višjega rezultata za vsebnosti zadevnega elementa v sledovih 50–100 mg/kg,
— 10 mg/kg absolutne vrednosti za vsebnosti zadevnega elementa v sledovih 100–200 mg/kg,
— 5 % višjega rezultata za vsebnosti zadevnega elementa v sledovih nad 200 mg/kg.
8. Opomba
Če je količina prisotnih fosfatov visoka, lahko moti določanje železa, mangana in cinka. To se mora popraviti z dodajanjem raztopine lantanovega klorida (3.11). Če pa je masno razmerju v vzorcu (Ca + Mg/P) > 2, se lahko izpusti dodajanje raztopine lantanovega klorida (3.11) raztopini za analizo in raztopinam za umerjanje.
D. DOLOČANJE HALOFUGINONA
DL-trans-7-bromo-6-kloro-3-[3-(3-hidroksi-2-piperidil)acetonil]-kvinazolin-4-(3H)-ena hidrobromid
1. Namen in področje uporabe
Ta metoda omogoča določanje ravni halofuginona v krmi. Meja kvantifikacije je 1 mg/kg.
2. Načelo
Po obdelavi z vročo vodo se halofuginon ekstrahira v obliki proste baze v etilni acetat in se nato kot hidroklorid loči v vodno raztopino kisline. Ekstrakt se prečisti z ionsko izmenjevalno kromatografijo. Vsebnost halofuginona se določi s tekočinsko kromatografijo visoke ločljivosti (HPLC) z reverzno fazo in z UV-detektorjem.
3. Reagenti
3.1 Acetonitril, za HPLC
3.2 Izmenjevalec Amberlite XAD-2
3.3 Amonijev acetat
3.4 Etilni acetat
3.5 Ledocetna kislina
3.6 Standardna snov halofuginon (DL-trans-7-bromo-6-kloro-3-[3-hidroksi-2-piperidil)acetonil]-kvinazolin-4-(3H)-ena hidrobromid, E 764)
3.6.1
50 mg halofuginona (3.6) se natehta na 0,1 mg natančno in prenese v 500-mililitrsko merilno bučko ter raztopi v pufrski raztopini amonijevega acetata (3.18), dopolni se s pufrsko raztopino do oznake in premeša. Ta raztopina je stabilna tri tedne pri 5 oC, če se hrani na temnem.
3.6.2
V serijo 100-mililitrskih merilnih bučk se prenese 1,0 , 2,0 , 3,0 , 4,0 in 6,0 ml osnovne standardne raztopine (3.6.1). Dopolni se z mobilno fazo (3.21) do oznake in premeša. V teh raztopinah je koncentracija halofuginona 1,0 , 2,0 , 3,0 , 4,0 in 6,0 μg/ml. Te raztopine je treba pripraviti sveže pred uporabo.
3.7 Klorovodikova kislina, (ρ20 približno 1,16 g/ml)
3.8 Metanol
3.9 Srebrov nitrat
3.10 Natrijev askorbat
3.11 Natrijev karbonat
3.12 Natrijev klorid
3.13 EDTA (etilendiamintetraocetna kislina, dinatrijeva sol)
3.14 Voda, za HPLC
3.15 Raztopina natrijevega karbonata, c = 10 g/100 ml
3.16 Raztopina natrijevega klorida in nasičenega natrijevega karbonata, c = 5 g/100 ml
50 g natrijevega karbonata (3.11) se raztopi v vodi, razredči do 1 litra, nato pa se doda natrijev klorid (3.12), dokler raztopina ni nasičena.
3.17 Klorovodikova kislina, približno 0,1 mol/l
10 ml HCl (3.7) se razredči z vodo do 1 litra.
3.18 Pufrska raztopina amonijevega acetata, približno 0,25 mol/l
19,3 g amonijevega acetata (3.3) in 30 ml ocetne kisline (3.5) se raztopi v vodi (3.14) ter razredči do 1 litra.
3.19 Priprava izmenjevalca Amberlite XAD-2
Ustrezna količina Amberlita (3.2) se spira z vodo, dokler niso odstranjeni vsi kloridni ioni, kar dokazuje preskus s srebrovim nitratom (3.20) v zavrženi vodni fazi. Nato se izmenjevalec spere s 50 ml metanola (3.8), metanol se zavrže, izmenjevalec pa hrani v svežem metanolu.
3.20 Raztopina srebrovega nitrata, približno 0,1 mol/l
0,17 g srebrovega nitrata (3.9) se raztopi v 10 ml vode.
3.21 Mobilna faza HPLC
500 ml acetonitrila (3.1) se zmeša s 300 ml pufrske raztopine amonijevega acetata (3.18) in 1 200 ml vode (3.14). pH se uravna na 4,3 z ocetno kislino (3.5). Filtrira se skozi filter 0,22 μm (4.8) in raztopina se razplini (npr. 10 minut v ultrazvočni kopeli). Ta raztopina je stabilna en mesec, če se hrani na temnem v zaprti posodi.
4. Oprema
4.1 Ultrazvočna kopel
4.2 Rotacijski uparjalnik
4.3 Centrifuga
4.4 HPLC-oprema z ultravijoličnim detektorjem z nastavljivo valovno dolžino ali detektorjem s serijo diod
4.4.1 Kolona za tekočinsko kromatografijo, 300 mm × 4 mm, C18, polnitev 10 μm ali enakovredna kolona
4.5 Steklena kolona (300 mm × 10 mm) s filtrom iz sintranega stekla in petelinčkom
4.6 Filtri iz steklenih vlaken, premera 150 mm
4.7 Membranski filtri, 0,45 μm
4.8 Membranski filtri, 0,22 μm
5. Postopek
|
Opomba |
: |
Halofuginon kot prosta baza je nestabilen v bazičnih in etilacetatnih raztopinah. V etilacetatu se ne pusti več kot 30 minut. |
5.1 Splošno
5.1.1 Z analizo slepe krme se preveri, da niso prisotni niti halofuginon niti moteče snovi.
5.1.2 Preskus izkoristka se izvede z analizo slepe krme, ki se ji doda količina halofuginona, podobna količini v vzorcu. Za koncentracijo 3 mg/kg se k 10 g slepe krme doda 300 μl osnovne standardne raztopine (3.6.1), premeša se in počaka 10 minut, preden se začne postopek ekstrakcije (5.2).
|
Opomba |
: |
Pri tej metodi je vrsta slepe krme podobna vrsti vzorca, pri analizi pa se dokaže odsotnost halofuginona. |
5.2 Ekstrakcija
10 g pripravljenega vzorca se natehta na 0,1 g natančno in prenese v 200-mililitrsko centrifugirno epruveto, doda se 0,5 g natrijevega askorbata (3.10), 0,5 g EDTA (3.13) in 20 ml vode ter premeša. Epruveta se postavi za 5 min v vodno kopel (80 oC). Po ohladitvi na sobno temperaturo se doda 20 ml raztopine natrijevega karbonata (3.15) in premeša. Takoj se doda 100 ml etilacetata (3.4) in 15 sekund ročno močno stresa. Nato se epruveta postavi za 3 minute v ultrazvočno kopel (4.1), zamašek pa zrahlja. Centrifugira se 2 minuti, faza etilacetata pa se oddekantira skozi filter iz steklenih vlaken (4.6) v 500-mililitrski lij ločnik. Ekstrakcija vzorca se ponovi z nadaljnjimi 100 ml etilacetata. Sestavljeni ekstrakti se spirajo 1 minuto s 50 ml raztopine nasičenega natrijevega klorida-natrijevega karbonata (3.16), vodna plast pa se zavrže.
Organska plast se ekstrahira 1 minuto s 50 ml klorovodikove kisline (3.17). Spodnja kislinska plast se prenese v 250-mililitrski lij ločnik. Organska plast se ponovno ekstrahira 1,5 minute z nadaljnjimi 50 ml klorovodikove kisline, ekstrakt pa se združi s prvim ekstraktom. Združena ekstrakta se približno 10 sekund spirata z 10 ml etilacetata (3.4) z vrtenjem.
Vodna plast se prenese v 250-mililitrsko bučko z okroglim dnom, organska faza pa se zavrže. Ves preostali etilacetat iz kisle raztopine se upari v rotacijskem uparjalniku (4.2). Temperatura vodne kopeli ne sme preseči 40 oC. Ves preostali etilacetat se odstrani v 5 minutah v vakuumu pri približno 25 milibarih in 38 oC.
5.3 Čiščenje
5.3.1
Kolona XAD-2 se pripravi za vsak ekstrakt vzorca. 10 g pripravljenega Amberlita (3.19) se z metanolom (3.8) prenese v stekleno kolono (4.5). Na vrh posteljice izmenjevalca se položi majhen zamašek iz steklene volne. Metanol se izpusti iz kolone, izmenjevalec pa se spere s 100 ml vode tako, da se tok vode ustavi, ko vrh posteljice doseže izmenjevalec. Pred uporabo se pusti 10 minut, da se kolona uravnoteži. Kolona se ne sme nikoli izsušiti.
5.3.2
Ekstrakt (5.2) se prenese na vrh pripravljene kolone z Amberlitom (5.3.1) in eluira, eluat pa se zavrže. Hitrost eluiranja ne sme biti večja od 20 ml/min. Bučka z okroglim dnom se spere z 20 ml klorovodikove kisline (3.17) in vsebina se uporabi za spiranje izmenjalne kolone. Kakršna koli preostala raztopina kisline se prepiha z zrakom. Ostanek po izpiranju se zavrže. V kolono se doda 100 ml metanola (3.8), 5–10 ml se eluira, eluat pa se zbere v 250-mililitrsko bučko z okroglim dnom. Preostali metanol se pusti 10 minut, da se uravnoteži z izmenjevalcem, eluiranje se nadaljuje s hitrostjo, ki ne presega 20 ml/min, eluat pa se zbira v isto bučko z okroglim dnom. Metanol se upari na rotacijskem uparjalniku (4.2), pri čemer temperatura vodne kopeli ne sme preseči 40 oC. Ostanek se z mobilno fazo (3.21) prenese v 10-mililitrsko merilno bučko. Dopolni se z mobilno fazo do oznake in premeša. Alikvot se prefiltrira skozi membranski filter (4.7). Raztopina se shrani za določanje s HPLC (5.4).
5.4 Določanje s HPLC
5.4.1
Naslednji pogoji se lahko uporabljajo kot smernice, lahko pa se uporabljajo drugi pogoji, če zagotavljajo enakovredne rezultate.
Kolona za tekočinsko kromatografijo (4.4.1)
Mobilna faza HPLC (3.21)
Hitrost pretoka: 1,5 –2 ml/min
Detekcijska valovna dolžina: 243 nm
Volumen, ki se vbrizga: 40 do 100 μl
Stabilnost kromatografskega sistema se preveri z večkratnim vbrizgavanjem raztopine za umerjanje s koncentracijo 3,0 μg/ml (3.6.2), dokler niso dosežene konstantne višine (ali površine) vrhov in konstantni retencijski časi.
5.4.2
Vsaka raztopina za umerjanje (3.6.2) se vbrizga večkrat, za vsako koncentracijo pa se izmerijo srednje vrednosti višin (površin) vrhov. Umeritvena krivulja se pripravi tako, da se na ordinato nanesejo srednje vrednosti višin ali površin vrhov raztopin za umerjanje, na absciso pa ustrezne koncentracije v μg/ml.
5.4.3
Ekstrakt vzorca (5.3.2) se vbrizga večkrat, pri čemer se uporabijo enaki volumni kot za raztopino za umerjanje, nato pa se določi srednja vrednost višin (površin) vrhov halofuginona.
6. Izračun rezultatov
Koncentracija raztopine v μg/ml se določi iz srednjih vrednosti višin (površin) vrhov halofuginona v raztopini vzorca iz umeritvene krivulje (5.4.2).
Vsebnost halofuginona (w) (mg/kg) v vzorcu se izračuna po naslednji formuli:

pri čemer je:
|
c |
: |
koncentracija halofuginona v raztopini vzorca v μg/ml; |
|
m |
: |
masa preskušanega vzorca v gramih. |
7. Potrditev rezultatov
7.1 Identiteta
Identiteta analita se lahko potrdi z dodatno kromatografijo ali z detektorjem s serijo diod, s katerim se primerjata spekter ekstrakta vzorca in spekter raztopine za umerjanje (3.6.2) s koncentracijo 6,0 μg/ml.
7.1.1
Ekstraktu vzorca se doda ustrezna količina raztopine za umerjanje (3.6.2). Količina dodanega halofuginona mora biti podobna ocenjeni količini halofuginona v ekstraktu vzorca.
Povišan je le vrh halofuginona ob upoštevanju dodane količine in razredčitve ekstrakta. Širina vrha na polovici maksimalne višine mora biti znotraj ± 10 % prvotne širine.
7.1.2
Rezultati se ocenijo glede na naslednja merila:
(a) valovni dolžini maksimalne absorpcije spektra vzorca in spektra standarda, zabeleženi na najvišji točki vrha kromatograma, morata biti isti znotraj meja, določenih z ločljivostjo sistema za detekcijo. Za detekcijo s serijo diodo je to ponavadi znotraj ± 2 nm;
(b) med 225 in 300 nm se spekter vzorca in spekter standarda, zabeležena na najvišji točki kromatografskega vrha, za navedene dele spektra ne smeta razlikovati znotraj območja 10–100 % relativne absorbance. To merilo je izpolnjeno, ko so prisotne iste najvišje vrednosti in ko odmik med dvema spektroma na nobeni opazovani točki ne preseže 15 % absorbance standardnega analita;
(c) med 225 in 300 nm se spektri naraščanja, najvišje točke vrha in padanja vrha, pridobljeni iz ekstrakta vzorca, za navedene dele spektra ne smejo razlikovati znotraj območja 10–100 % relativne absorbance. To merilo je izpolnjeno, ko so prisotne iste najvišje točke in ko odmik med spektri na nobeni od opazovanih točk ne preseže 15 % absorbance spektra najvišje točke.
Če eno od teh meril ni izpolnjeno, prisotnost analita ni potrjena.
7.2 Ponovljivost
Razlika med rezultatoma dveh vzporednih določanj, izvedenih na istem vzorcu, ne sme presegati 0,5 mg/kg za vsebnost halofuginona do 3 mg/kg.
7.3 Izkoristek
Za slepi vzorec z dodatkom je izkoristek najmanj 80 %.
8. Rezultati medlaboratorijske študije
Organizirana je bila medlaboratorijska študija ( 15 ), v kateri je osem laboratorijev analiziralo tri vzorce.
Rezultati
|
|
Vzorec A (slepi) ob prejemu |
Vzorec B (v prahu) |
Vzorec C (v briketih) |
||
|
|
|
ob prejemu |
po 2 mesecih |
ob prejemu |
po 2 mesecih |
|
Srednja vrednost [mg/kg] |
ND |
2,80 |
2,42 |
2,89 |
2,45 |
|
SR [mg/kg] |
— |
0,45 |
0,43 |
0,40 |
0,42 |
|
CVR [ %] |
— |
16 |
18 |
14 |
17 |
|
Rec. [ %] |
|
86 |
74 |
88 |
75 |
|
ND |
= |
ni zaznano; |
|
SR |
= |
standardni odmik obnovljivosti; |
|
CVR |
= |
koeficient variacije obnovljivosti (%); |
|
Rec. |
= |
izkoristek (%). |
E. DOLOČANJE ROBENIDINA
1,3-bis [(4-klorobenziliden)amino]gvanidin – hidroklorid
1. Namen in področje uporabe
Ta metoda omogoča določanje ravni robenidina v krmi. Meja kvantifikacije je 5 mg/kg.
2. Načelo
Vzorec se ekstrahira z nakisanim metanolom. Ekstrakt se osuši in alikvot se prečisti na koloni iz aluminijevega oksida. Robenidin se eluira s kolone z metanolom, koncentrira in dopolni z mobilno fazo do ustreznega volumna. Vsebnost robenidina se določi s tekočinsko kromatografijo visoke ločljivosti (HPLC) z reverzno fazo in UV-detektorjem.
3. Reagenti
3.1 Metanol
3.2 Nakisani metanol
4,0 ml klorovodikove kisline (ρ20 = 1,18 g/ml) se prenese v 500-mililitrsko merilno bučko, dopolni se do oznake z metanolom (3.1) in premeša. Ta raztopina se pred vsako uporabo sveže pripravi.
3.3 Acetonitril, za HPLC
3.4 Molekularno sito
Tip 3A, kroglice 8 do 12 meš (kroglice, premera 1,6 – 2,5 mm, kristalinični aluminijev silikat, premer por 0,3 mm)
3.5 Stopnja kislinske aktivnosti I aluminijevega oksida, za kolonsko kromatografijo
V primerno posodo se prenese 100 g aluminijevega oksida in doda 2,0 ml vode. Zamaši se in stresa približno 20 minut. Hrani se v dobro zaprti posodi.
3.6 Raztopina kalijevega dihidrogenfosfata, c = 0,025 mol/l
3,40 g kalijevega dihidrogenfosfata se raztopi v vodi (za HPLC) v 1 000 -mililitrski merilni bučki, dopolni se do oznake in premeša.
3.7 Raztopina dinatrijevega hidrogenfosfata, c = 0,025 mol/l
3,55 g brezvodnega dinatrijevega hidrogenfosfata (ali 4,45 g dihidrata ali 8,95 g dodekahidrata) se raztopi v vodi (za HPLC) v 1 000 -mililitrski merilni bučki, dopolni se do oznake in premeša.
3.8 HPLC-mobilna faza
Zmešajo se naslednji reagenti:
650 ml acetonitrila (3.3),
250 ml vode (za HPLC),
50 ml raztopine kalijevega dihidrogenfosfata (3.6),
50 ml raztopine dinatrijevega hidrogenfosfata (3.7).
Filtrira se skozi filter 0,22 μm (4,6 ), raztopina pa se nato razplini (npr. 10 minut v ultrazvočni kopeli).
3.9 Standardna snov
Čisti robenidin: 1,3 -bis[4-klorobenziliden)amino] gvanidin hidroklorid
3.9.1
30 mg standardnega robenidina (3.9) se natehta na 0,1 mg natančno. Raztopi se v nakisanem metanolu (3.2) v 100-mililitrski merilni bučki, dopolni se z istim topilom do oznake in premeša. Bučka se ovije v aluminijasto folijo in hrani v temi.
3.9.2
10,0 ml osnovne standardne raztopine robenidina (3.9.1) se prenese v 250-mililitrsko merilno bučko, dopolni se z mobilno fazo (3.8) do oznake in premeša. Bučka se ovije v aluminijasto folijo in hrani v temi.
3.9.3
V serijo 50-mililitrskih merilnih bučk se prenese 5,0 , 10,0 , 15,0 , 20,0 in 25,0 ml vmesne standardne raztopine (3.9.2). Dopolni se z mobilno fazo (3.8) do oznake in premeša. Te raztopine ustrezajo koncentracijam robenidina 1,2 , 2,4 , 3,6 , 4,8 in 6,0 μg/ml. Te raztopine je treba pripraviti sveže pred uporabo.
3.10 Voda za HPLC
4. Oprema
4.1 Steklena kolona
Iz temnega stekla, s petelinčkom in rezervoarjem z volumnom približno 150 ml, notranjega premera 10–15 mm, dolžine 250 mm
4.2 Mehanski stresalnik ali magnetno mešalo
4.3 Rotacijski uparjalnik
4.4 Oprema za HPLC z ultravijoličnim detektorjem ali detektorjem s serijo diod, z nastavljivo valovno dolžino v območju 250–400 nm.
4.4.1 Kolona za tekočinsko kromatografijo: 300 mm × 4 mm, C18, polnitev 10 μm ali enakovredna
4.5 Filtrski papir iz steklenih vlaken (Whatman GF/A ali enakovreden)
4.6 Membranski filtri, 0,22 μm
4.7 Membranski filtri, 0,45 μm
5. Postopek
|
Opomba |
: |
Robenidin je občutljiv na svetlobo. Pri vseh postopkih se uporablja temna steklovina. |
5.1 Splošno
5.1.1 Z analizo slepe krme se zagotovi, da niso prisotni niti robenidin niti moteče snovi.
5.1.2 Preskus izkoristka se izvede z analizo slepe krme (5.1.1), ki se ji doda količina amproliuma, podobna količini v vzorcu. Za koncentracijo 60 mg/kg se 3,0 ml osnovne standardne raztopine (3.9.1) prenese v 250-mililitrsko erlenmajerico. Raztopina se upari v toku dušika do približno 0,5 ml. Doda se 15 g slepe krme, premeša, pusti 10 minut in nadaljuje z ekstrakcijo (5.2).
|
Opomba |
: |
Pri tej metodi je vrsta slepe krme podobna vrsti vzorca in pri analizi se dokaže odsotnost robenidina. |
5.2 Ekstrakcija
Približno 15 g vzorca se natehta na 0,01 g natančno. Prenese se v 250-mililitrsko erlenmajerico, doda se 100,0 ml nakisanega metanola (3.2), zamaši se in stresa eno uro v stresalniku (4.2). Raztopina se filtrira skozi filtrirni papir iz steklenih vlaken (4.5) v 150-mililitrsko erlenmajerico. Doda se 7,5 g molekularnega sita (3.4), zamaši se in stresa pet minut. Takoj se filtrira skozi filtrirni papir iz steklenih vlaken. Ta raztopina se shrani za fazo čiščenja (5.3).
5.3 Čiščenje
5.3.1
V spodnji del steklene kolone se vstavi majhen zamašek iz steklene volne (4.1), ki se potlači s stekleno palčko. 11,0 g pripravljenega aluminijevega oksida (3.5) se natehta in prenese v kolono. Pazi se, da je pri tem postopku izpostavljenost zraku čim manjša. Rahlo se udarja po spodnjem delu polne kolone, da se aluminijev oksid sesede.
5.3.2
Na kolono se s pipeto prenese 5,0 ml ekstrakta vzorca, pripravljenega v (5.2). Pipeta se nasloni na steno kolone, nato pa se pusti, da se raztopina absorbira na aluminijev oksid. Robenidin se eluira iz kolone s 100 ml metanola (3.1) s hitrostjo pretoka 2–3 ml/min, eluat pa se zbere v 250-mililitrsko bučko z okroglim dnom. Raztopina metanola se upari do suhega pri znižanem tlaku in pri 40 oC v rotacijskem uparjalniku (4.3). Ostanek se raztopi v 3–4 ml mobilne faze (3.8) in prenese v 10-mililitrsko merilno bučko. Bučka se nekajkrat spere z 1–2 ml mobilne faze, izpirki pa se prenesejo v merilno bučko. Dopolni se do oznake z istim topilom in premeša. Alikvot se prefiltrira skozi membranski filter 0,45 μm (4.7). Ta raztopina se shrani za določanje s HPLC (5.4).
5.4 Določanje s HPLC
5.4.1
Naslednji pogoji se lahko uporabljajo kot smernice, lahko pa se uporabljajo drugi pogoji, če zagotavljajo enakovredne rezultate:
kolona za tekočinsko kromatografijo (4.4.1),
mobilna faza HPLC (3.8),
hitrost pretoka: 1,5 –2 ml/min,
valovna dolžina detekcije: 317 nm,
volumen, ki se vbrizga: 20–50 μl.
Stabilnost kromatografskega sistema se preveri z večkratnim vbrizgavanjem raztopine za umerjanje (3.9.3) s koncentracijo 3,6 μg/ml, dokler se ne dosežejo konstantne višine vrhov in konstantni retencijski časi.
5.4.2
Vsaka raztopina za umerjanje (3.9.3) se večkrat vbrizga, za vsako koncentracijo pa se izmerijo višine (površine) vrhov. Umeritvena krivulja se pripravi tako, da se na ordinato nanesejo srednje vrednosti višin ali površin vrhov raztopin za umerjanje, na absciso pa ustrezne koncentracije v μg/ml.
5.4.3
Večkrat se vbrizga ekstrakt vzorca (5.3.2) z enakimi volumni, kakor se uporabljajo pri raztopini za umerjanje, nato pa se določijo srednje vrednosti višin (površin) vrhov robenidina.
6. Izračun rezultatov
Koncentracija raztopine vzorca v μg/ml se določi iz srednje vrednosti višin (površin) vrhov robenidina iz umeritvene krivulje (5.4.2).
Vsebnost robenidina (w) (mg/kg) v vzorcu se izračuna po naslednji formuli:
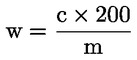
pri čemer je:
|
c |
= |
koncentracija robenidina v raztopini vzorca v μg/ml; |
|
m |
= |
masa preskušanega vzorca v gramih. |
7. Potrditev rezultatov
7.1 Identiteta
Identiteta analita se lahko potrdi z dodatno kromatografijo ali z detektorjem s serijo diod, s katerim se primerjata spekter ekstrakta vzorca in spekter raztopine za umerjanje (3.9.3) s koncentracijo 6,0 μg/ml.
7.1.1
Ekstraktu vzorca se doda primerna količina raztopine za umerjanje (3.9.3). Količina dodanega robenidina mora biti podobna ocenjeni količini robenidina v ekstraktu vzorca.
Povišan je le vrh robenidina ob upoštevanju dodane količine in razredčitve ekstrakta. Širina vrha na polovici njegove največje višine mora biti v mejah približno 10 % prvotne širine.
7.1.2
Rezultati se ocenijo glede na naslednja merila:
(a) valovni dolžini maksimalne absorpcije spektra vzorca in spektra standarda, zabeleženi na najvišji točki vrha kromatograma, morata biti isti znotraj meja, določenih z ločljivostjo sistema za detekcijo. Za detekcijo s serijo diod je to ponavadi v območju približno 2 nm;
(b) med 250 in 400 nm se spekter vzorca in spekter standarda na najvišji točki kromatografskega vrha za navedene dele spektra ne smeta razlikovati znotraj območja 10–100 % relativne absorbance. To merilo je izpolnjeno, ko so prisotne iste najvišje vrednosti in ko odmik med dvema spektroma na nobeni opazovani točki ne preseže 15 % absorbance standardnega analita;
(c) med 250 in 400 nm se spektri naraščanja, najvišje točke vrha in padanja vrha, pridobljeni iz ekstrakta vzorca, za navedene dele spektra ne smejo razlikovati znotraj območja 10–100 % relativne absorbance. To merilo je izpolnjeno, ko so prisotne iste najvišje točke in ko odmik med spektri na nobeni od opazovanih točk ne preseže 15 % absorbance spektra najvišje točke.
Če eno od teh meril ni izpolnjeno, prisotnost analita ni potrjena.
7.2 Ponovljivost
Razlika med rezultatoma dveh vzporednih določanj, izvedenih na istem vzorcu, ne sme presegati 10 % višjega rezultata za vsebnost robenidina nad 15 mg/kg.
7.3 Izkoristek
Za slepi vzorec z dodatkom je izkoristek najmanj 85 %.
8. Rezultati medlaboratorijske študije
ES je organizirala medlaboratorijsko študijo, pri kateri je 12 laboratorijev analiziralo štiri vzorce perutninske in kunčje krme v prahu in briketih. Vsak vzorec je bil analiziran dvakrat. Rezultati so zbrani v spodnji preglednici:
|
|
Perutninska krma |
Kunčja krma |
||
|
|
v prahu |
v briketih |
v prahu |
v briketih |
|
Srednja vrednost [mg/kg] |
27,00 |
27,99 |
43,6 |
40,1 |
|
Sr [mg/kg] |
1,46 |
1,26 |
1,44 |
1,66 |
|
CVr [ %] |
5,4 |
4,5 |
3,3 |
4,1 |
|
SR [mg/kg] |
4,36 |
3,36 |
4,61 |
3,91 |
|
CVR [ %] |
16,1 |
12,0 |
10,6 |
9,7 |
|
Recovery [ %] |
90,0 |
93,3 |
87,2 |
80,2 |
|
Sr |
= |
standardni odmik ponovljivosti; |
|
CVr |
= |
koeficient variacije ponovljivosti v %; |
|
SR |
= |
standardni odmik obnovljivosti; |
|
CVR |
= |
koeficient variacije obnovljivosti v %. |
F. DOLOČANJE DIKLAZURILA
(+)-4-klorfenil [2,6-dikloro-4-(2,3,4,5-tetrahidro-3,5-diokso-1,2,4-triazin-2-il) fenil] acetonitril
1. Namen in področje uporabe
Ta metoda omogoča določanje ravni diklazurila v krmi in premiksih. Meja določanja je 0,1 mg/kg, meja kvantifikacije pa 0,5 mg/kg.
2. Načelo
Po dodatku notranjega standarda se vzorec ekstrahira z nakisanim metanolom. Pri krmi se alikvot ekstrakta prečisti na kartuši C18 za ekstrakcijo v trdni fazi. Diklazuril se eluira iz kartuše z mešanico nakisanega metanola in vode. Po uparevanju se ostanek raztopi v raztopini DMF in vode. Pri premiksih se ekstrakt upari in nato se ostanek raztopi v raztopini DMF in vode. Vsebnost diklazurila se določi s tekočinsko kromatografijo visoke ločljivosti (HPLC) z reverzno fazo s ternarnim gradientom, z uporabo UV-detektorja.
3. Reagenti
3.1 Voda, za HPLC
3.2 Amonijev acetat
3.3 Tetrabutilamonijev hidrogen sulfat (TBHS)
3.4 Acetonitril, za HPLC
3.5 Metanol, za HPLC
3.6 N, N-dimetilformamid (DMF)
3.7 Klorovodikova kislina, ρ20 = 1,19 g/ml
3.8 Standardna snov: diklazuril II-24: (+)-4-klorfenil [2,6-diklor-4-(2,3,4,5-tetrahidro-3,5-diokso-1,2,4-triazin-2-il) fenil] acetonitril, zajamčene čistosti, E771.
3.8.1
25 mg standardne snovi diklazurila (3.8) se natehta na 0,1 mg natančno in prenese v 50-mililitrsko merilno bučko. Raztopi se v DMF (3.6), bučka se dopolni z DMF (3.6) do oznake in premeša. Bučka se ovije v aluminijasto folijo ali pa se uporabi rjava bučka ter shrani v hladilniku. Pri temperaturi ≤ 4 oC je raztopina stabilna 1 mesec.
3.8.2
5,00 ml osnovne standardne raztopine (3.8.1) se prenese v 50-mililitrsko merilno bučko, dopolni se z DMF (3.6) do oznake in premeša. Bučka se ovije v aluminijasto folijo ali pa se uporabi rjava bučka ter shrani v hladilniku. Pri temperaturi ≤ 4 oC je raztopina stabilna 1 mesec.
3.9 Snov, ki je notranji standard: 2,6 dikloro-α-(4-klorofenil)-4-(4,5 dihidro-3,5-diokso-1,2,4-triazin-2 (3H) – il) α-metilbenzen-acetonitril.
3.9.1
25 mg notranjega standarda (3.9) se natehta na 0,1 mg natančno in prenese v 50-mililitrsko merilno bučko. Raztopi se v DMF (3.6), bučka se dopolni z DMF (3.6) do oznake in premeša. Bučka se ovije v aluminijasto folijo ali se pa uporabi rjava bučka ter shrani v hladilniku. Pri temperaturi ≤ 4 oC je raztopina stabilna 1 mesec.
3.9.2
5,00 ml osnovne raztopine notranjega standarda (3.9.1) se prenese v 50-mililitrsko merilno bučko, dopolni se z DMF (3.6) do oznake in premeša. Bučka se ovije v aluminijasto folijo ali pa se uporabi rjava bučka ter shrani v hladilniku. Pri temperaturi ≤ 4 oC je raztopina stabilna 1 mesec.
3.9.3
(p = nominalna vsebnost diklazurila v mg/kg v premiksu)
p/10 mg notranjega standarda se natehta na 0,1 mg natančno in prenese v 100-mililitrsko merilno bučko, raztopi se v DMF (3.6) v ultrazvočni kopeli (4.6), dopolni se z DMF do oznake in premeša. Bučka se ovije v aluminijasto folijo ali pa se uporabi rjava bučka ter shrani v hladilniku. Pri temperaturi ≤ 4 oC je raztopina stabilna 1 mesec.
3.10 Raztopina za umerjanje, 2 μg/ml
2,00 ml standardne raztopine diklazurila (3.8.2) in 2,00 ml raztopine notranjega standarda (3.9.2) se odpipetira v 50-mililitrsko merilno bučko. Doda se 16 ml DMF (3.6), bučka se dopolni z vodo do oznake in premeša. To raztopino je treba pripraviti svežo pred uporabo.
3.11 Ekstrakcijska kartuša za trdno fazo C18, npr. Bond Elut, velikost: 1 cc, masa absorbenta: 100 mg
3.12 Ekstrakcijsko topilo: nakisani metanol
5,0 ml klorovodikove kisline (3.7) se odpipetira v 1 000 ml metanola (3.5) in premeša.
3.13 Mobilna faza za HPLC
3.13.1 Eluent A: raztopina amonijevega acetata in tetrabutilamonijevega hidrogen sulfata
5 g amonijevega acetata (3.2) in 3,4 g TBHS (3.3) se raztopi v 1 000 ml vode (3.1) ter premeša.
3.13.2 Eluent B: acetonitril (3.4)
3.13.3 Eluent C: metanol (3.5)
4. Oprema
4.1 Mehanski stresalnik
4.2 Oprema za HPLC s terciarnim gradientom
4.2.1 Kolona za tekočinsko kromatografsko kolono, Hypersil ODS, polnitev 3 μm, 100 mm x 4,6 mm ali enakovredna
4.2.2 UV-detektor z nastavljivo valovno dolžino ali detektor s serijo diod.
4.3 Rotacijski uparjalnik
4.4 Membranski filter, 0,45 μm
4.5 Vakuumski ventil
4.6 Ultrazvočna kopel
5. Postopek
5.1 Splošno
5.1.1
Z analizo slepe krme se zagotovi, da niso prisotni niti diklazuril niti moteče snovi. Vrsta slepe krme je podobna vrsti vzorca in z analizo se dokaže odsotnost diklazurila ali motečih substanc.
5.1.2
Preskus izkoristka se izvede tako, da se analizira slepa krma, ki se ji doda toliko diklazurila, kolikor ga je v vzorcu. Za koncentracijo l mg/kg se doda 0,1 ml osnovne standardne raztopine (3.8.1) v 50 g slepe krme, dobro se premeša in pusti 10 minut, pred nadaljevanjem (5.2) pa se večkrat ponovno premeša.
Če pa vrsta slepe krme, podobna vrsti vzorca, ni na voljo (glej 5.1.1), se lahko preskus izkoristka izvede po metodi standardnega dodatka. V tem primeru se vzorcu za analizo doda količina diklazurila, podobna tisti v vzorcu. Ta vzorec se analizira skupaj z vzorcem brez dodanega diklazurila, izkoristek pa se izračuna z odštevanjem.
5.2 Ekstrakcija
5.2.1
Približno 50 g vzorca se natehta na 0,01 g natančno. Prenese se v 500-mililitrsko erlenmajerico, doda se 1,00 ml raztopine notranjega standarda (3.9.2) in 200 ml ekstrakcijskega topila (3.12), nato pa se erlenmajerica zamaši. Mešanica se prek noči stresa v stresalniku (4.1). Pusti se 10 minut, da se usede. 20-mililitrski alikvot supernatanta se prenese v primerno stekleno posodo in razredči z 20 ml vode. Ta raztopina se prenese na ekstrakcijsko kartušo (3.11), skoznjo pa se spusti z vakuumom (4.5). Kartuša se spere s 25 ml mešanice ekstrakcijskega topila (3.12) in vode, 65 + 35 (V + V). Zbrane frakcije se zavržejo, spojine pa se eluirajo s 25 ml mešanice ekstrakcijskega topila (3.12) in vode, 80 + 20 (V + V). Ta frakcija se upari v rotacijskem uparjalniku (4.3) pri 60 oC do prve sušine. Ostanek se raztopi v 1,0 ml DMF (3.6), doda se 1,5 ml vode (3.1) in premeša. Filtrira se skozi membranski filter (4.4). Nadaljuje se z določanjem s HPLC (5.3).
5.2.2
Približno 1 g vzorca se natehta na 0,001 g natančno. Prenese se v 500-mililitrsko erlenmajerico, doda se 1,00 ml raztopine notranjega standarda (3.9.3) in 200 ml ekstrakcijskega topila (3.12), nato pa se erlenmajerica zamaši. Mešanica se prek noči stresa v stresalniku (4.1). Pusti se 10 minut, da se usede. Alikvot (10 000 /p ml, p = nominalna vsebnost diklazurila v mg/kg v premiksu) supernatanta se prenese v primerno veliko bučko z okroglim dnom. Do prve sušine se upari pri znižanem tlaku in 60 oC v rotacijskem uparjalniku (4.3). Ostanek se ponovno raztopi v 10,0 ml DMF (3.6), doda se 15,0 ml vode (3.1) in premeša. Nadaljuje se z določanjem s HPLC (5.3).
5.3 Določanje s HPLC
5.3.1
Naslednji pogoji se lahko uporabljajo kot smernice, lahko pa se uporabljajo drugi pogoji, če zagotavljajo enakovredne rezultate.
|
Kolona za tekočinsko kromatografijo (4.2.1): |
100 mm × 4,6 mm, Hypersil ODS, polnitev 3 μm ali enakovredna |
|
|
Mobilna faza: |
Eluent A (3.13.1): |
vodna raztopina amonijevega acetata in tetrabutil amonijevega hidrogen sulfata |
|
|
Eluent B (3.13.2): |
acetonitril |
|
|
Eluent C (3.13.3): |
metanol |
|
Način eluiranja: |
— linearni gradient — začetni pogoji: A + B + C = 60 + 20 + 20 (V + V + V) — po 10 minutah se elucijski gradient v 30 minutah spremeni na: A + B + C = 45 + 20 + 35 (V + V + V). V 10 minutah se spere z B. |
|
|
Hitrost pretoka: |
1,5 –2 ml/min |
|
|
Volumen, ki se vbrizga: |
20 μl |
|
|
Valovna dolžina detekcije: |
280 nm |
|
Stabilnost kromatografskega sistema se preveri z večkratnim vbrizgavanjem raztopine za umerjanje s koncentracijo 2 μg/ml (3.10), dokler se ne dosežejo konstantne višine vrhov in konstantni retencijski časi.
5.3.2
Večkrat se vbrizga 20 μl raztopine za umerjanje (3.10) ter določi srednja vrednost višine (površine) vrhov diklazurila in vrhov raztopine notranjega standarda.
5.3.3
Večkrat se vbrizga 20 μl raztopine vzorca (5.2.1 ali 5.2.2) ter določi srednja vrednost višine (površine) vrha diklazurila in raztopine notranjega standarda.
6. Izračun rezultatov
6.1 Krma
Vsebnost diklazurila (w) (mg/kg) v vzorcu se izračuna po naslednji formuli:

[mg/kg]
pri čemer je:
|
hd, s |
= |
višina (površina) vrha diklazurila v raztopini vzorca (5.2.1); |
|
hi, s |
= |
višina (površina) vrha notranjega standarda v raztopini vzorca (5.2.1); |
|
hd, c |
= |
višina (površina) vrha diklazurila v raztopini za umerjanje (3.10); |
|
hi, c |
= |
višina (površina) vrha notranjega standarda v raztopini za umerjanje (3.10); |
|
cd, c |
= |
koncentracija diklazurila v raztopini za umerjanje v μg/ml (3.10); |
|
m |
= |
masa preskusnega vzorca v gramih; |
|
V |
= |
volumen ekstrakta vzorca v skladu s točko 5.2.1 (tj. 2,5 ml). |
6.2 Premiksi
Vsebnost diklazurila (w) (mg/kg) v vzorcu se izračuna po naslednji formuli:

[mg/kg]
pri čemer je:
|
hd, c |
= |
višina (površina) vrha diklazurila v raztopini za umerjanje (3.10); |
|
hi, c |
= |
višina (površina) vrha notranjega standarda v raztopini za umerjanje (3.10); |
|
hd, s |
= |
višina (površina) vrha diklazurila v raztopini vzorca (5.2.2); |
|
hi, s |
= |
višina (površina) vrha notranjega standarda v raztopini vzorca (5.2.2); |
|
cd, c |
= |
koncentracija diklazurila v raztopini za umerjanje v μg/ml (3.10); |
|
m |
= |
masa preskusnega vzorca v gramih; |
|
V |
= |
volumen ekstrakta vzorca v skladu s točko 5.2.2 (tj. 25 ml); |
|
p |
= |
nominalna vsebnost diklazurila v mg/kg v premiksu. |
7. Potrditev rezultatov
7.1 Identiteta
Identiteta analita se lahko potrdi z dodatno kromatografijo ali z detektorjem s serijo diod, s katerim se primerjata spekter ekstrakta vzorca (5.2.1 ali 5.2.2) in spekter raztopine za umerjanje (3.10).
7.1.1
Ekstraktu vzorca (5.2.1 ali 5.2.2) se doda primerna količina raztopine za umerjanje (3.10). Količina dodanega diklazurila mora biti podobna količini diklazurila v ekstraktu vzorca.
Povišana sta le vrh diklazurila in vrh notranjega standarda ob upoštevanju dodane količine in razredčitve ekstrakta. Širina vrha na polovici višine mora biti znotraj ± 10 % prvotne širine vrha diklazurila ali notranjega standardnega vrha v vzorcu, ki mu diklazuril ni bil dodan.
7.1.2
Rezultati se ocenijo glede na naslednja merila:
(a) valovni dolžini maksimalne absorpcije spektra vzorca in spektra standarda, zabeleženi na najvišji točki vrha na kromatogramu, morata biti isti znotraj meja, določenih z ločljivostjo sistema za detekcijo. Pri detekciji s serijo diod je to ponavadi ± 2 nm;
(b) med 230 in 320 nm se spekter vzorca in spekter standarda, zabeležena na najvišji točki vrha na kromatogramu, za navedene dele spektra ne smeta razlikovati v območju 10–100 % relativne absorbance. To merilo je izpolnjeno, ko so prisotne iste najvišje vrednosti in ko odmik med dvema spektroma na nobeni opazovani točki ne preseže 15 % absorbance standardnega analita;
(c) med 230 in 320 nm se spektri naraščanja, najvišje točke vrha in padanja vrha, pridobljeni iz ekstrakta vzorca, za navedene dele spektra ne smejo razlikovati v območju 10–100 % relativne absorbance. To merilo je izpolnjeno, ko so prisotne iste najvišje točke in ko odmik med spektri na nobeni od opazovanih točk ne preseže 15 % absorbance spektra najvišje točke.
Če eno od teh meril ni izpolnjeno, prisotnost analita ni potrjena.
7.2 Ponovljivost
Razlika med rezultatoma dveh vzporednih določanj, izvedenih na istem vzorcu, ne sme presegati:
— 30 % višje vrednosti za vsebnosti diklazurila 0,5 –2,5 mg/kg,
— 0,75 mg/kg za vsebnosti diklazurila 2,5 –5 mg/kg,
— 15 % višje vrednosti za vsebnosti diklazurila nad 5 mg/kg.
7.3 Izkoristek
Pri vzorcu z dodatkom (slepem vzorcu) je izkoristek najmanj 80 %.
8. Rezultati medlaboratorijske študije
Organizirana je bila medlaboratorijska študija, pri kateri je 11 laboratorijev analiziralo 5 vzorcev. Te vzorce sta sestavljala dva premiksa; eden je bil mešan z organsko matrico (O 100), drug pa z anorgansko matrico (A 100). Teoretična vsebnost je 100 mg diklazurila na kg. Trije različni proizvajalci (NL) (L1/Z1/K1) so proizvedli 3 mešane perutninske krme. Teoretična vsebnost je 1 mg diklazurila na kg. Laboratorijem je bilo naročeno, naj vsak vzorec analizirajo enkrat ali dvakrat. (Podrobnejše informacije o navedeni medlaboratorijski študiji so v Journal of AOAC International, knjiga 77, št. 6, 1994, str. 1359–1361). Rezultati so navedeni v spodnji preglednici.
|
|
Vzorec 1 A 100 |
Vzorec 2 O 100 |
Vzorec 3 L1 |
Vzorec 4 Z1 |
Vzorec 5 K1 |
|
L |
11 |
11 |
11 |
11 |
6 |
|
n |
19 |
18 |
19 |
19 |
12 |
|
Srednja vrednost |
100,8 |
103,5 |
0,89 |
1,15 |
0,89 |
|
Sr (mg/kg) |
5,88 |
7,64 |
0,15 |
0,02 |
0,03 |
|
CVr (%) |
5,83 |
7,38 |
17,32 |
1,92 |
3,34 |
|
SR (mg/kg) |
7,59 |
7,64 |
0,17 |
0,11 |
0,12 |
|
CVR (%) |
7,53 |
7,38 |
18,61 |
9,67 |
13,65 |
|
Nominalna vsebnost (mg/kg) |
100 |
100 |
1 |
1 |
1 |
|
L |
= |
število laboratorijev; |
|
n |
= |
število posameznih vrednosti; |
|
Sr |
= |
standardni odmik ponovljivosti; |
|
CVr |
= |
koeficient variacije ponovljivosti; |
|
SR |
= |
standardni odmik obnovljivosti; |
|
CVR |
= |
koeficient variacije obnovljivosti. |
9. Opomba
Predhodno je treba dokazati, da je odziv diklazurila linearen v območju koncentracij, ki se merijo.
G. DOLOČANJE LASALOCID NATRIJA
Natrijeva sol polieter monokarboksilne kisline, proizvedena s Streptomyces lasaliensis
1. Namen in področje uporabe
Ta metoda omogoča določanje ravni lasalocid natrija v krmi in premiksih. Meja določanja je 5 mg/kg, meja kvantifikacije pa 10 mg/kg.
2. Načelo
Lasalocid natrij se ekstrahira iz vzorca v nakisani metanol in določi s tekočinsko kromatografijo visoke ločljivosti (HPLC) z reverzno fazo z uporabo spektrofluorimetričnega detektorja.
3. Reagenti
3.1 Kalijev dihidrogen fosfat (KH2PO4)
3.2 Ortofosforna kislina, m (m/m) = 85 %
3.3 Raztopina ortofosforne kisline, c = 20 %
23,5 ml ortofosforne kisline (3.2) se razredči s 100 ml vode.
3.4 6-metil-2-heptilamin (1,5 -dimetilheksilamin), m (m/m) = 99 %
3.5 Metanol, za HPLC
3.6 Klorovodikova kislina, gostota = 1,19 g/ml
3.7 Raztopina fosfatnega pufra, c = 0,01 mol/l
1,36 g KH2PO4 (3.1) se raztopi v 500 ml vode (3.11), doda se 3,5 ml ortofosforne kisline (3.2) in 10,0 ml 6-metil-2-heptilamina (3.4). pH se uravna na 4,0 z raztopino ortofosforne kisline (3.3) in razredči z vodo (3.11) do 1 000 ml.
3.8 Nakisani metanol
5,0 ml klorovodikove kisline (3.6) se prenese v litrsko merilno bučko, dopolni se z metanolom (3.5) do oznake in premeša. To raztopino je treba pripraviti svežo pred uporabo.
3.9 Mobilna faza za tekočinsko kromatografijo visoke ločljivosti (HPLC), raztopina fosfatnega pufra in metanola 5 + 95 (V + V)
5 ml raztopine fosfatnega pufra (3.7) se zmeša s 95 ml metanola (3.5).
3.10 Lasalocid natrij standardna snov, zajamčene čistosti, C34H53O8Na (natrijeva sol polieter monokarboksilne kisline, proizvedena s Streptomyces lasaliensis), E763
3.10.1
50 mg lasalocid natrija (3.10) se natehta na 0,1 mg natančno in prenese v 100-mililitrsko merilno bučko, raztopi se v nakisanem metanolu (3.8) ter z istim topilom dopolni do oznake in premeša. To raztopino je treba pripraviti svežo pred uporabo.
3.10.2
10,0 ml osnovne standardne raztopine (3.10.1) se odpipetira v 100-mililitrsko merilno bučko, z nakisanim metanolom (3.8) se dopolni do oznake in premeša. Ta raztopina se mora pripraviti sveža pred uporabo.
3.10.3
V serijo 50-mililitrskih merilnih bučk se prenese 1,0 , 2,0 , 4,0 , 5,0 in 10,0 ml vmesne standardne raztopine (3.10.2). Z nakisanim metanolom se dopolni do oznake (3.8) in premeša. Te raztopine ustrezajo 1,0 , 2,0 , 4,0 , 5,0 in 10,0 μg lasalocid natrija na ml. Te raztopine se morajo pripraviti sveže pred uporabo.
3.11 Voda, za HPLC
4. Oprema
4.1 Ultrazvočna kopel (ali stresalna vodna kopel) z nadzorom temperature
4.2 Membranski filtri, 0,45 μm
4.3 Oprema za tekočinsko kromatografijo visoke ločljivosti (HPLC) s sistemom za vbrizgavanje, primernim za vbrizgavanje volumnov 20 μl
4.3.1 Kolona za tekočinsko kromatografijo 125 mm x 4 mm, reverzna faza C18, polnitev 5 μm ali enakovredna
4.3.2 Spektrofluorimeter s spremenljivo nastavitvijo valovnih dolžin ekscitacije in emisije
5. Postopek
5.1 Splošno
5.1.1
Za učinkovitost preskusa izkoristka (5.1.2) se z analizo slepe krme zagotovi, da niso prisotni niti lasalocid natrij niti moteče snovi. Slepa krma je podobne vrste kot vzorec in dokaže se odsotnost lasalocid natrija ali motečih snovi.
5.1.2
Preskus izkoristka se izvede z analiziranjem slepe krme, ki se ji doda količina lasalocid natrija, podobna količini v vzorcu. Za koncentracijo 100 mg/kg se 10,0 ml osnovnega standarda (3.10.1) prenese v 250-mililitrsko erlenmajerico, raztopina pa se upari do približno 0,5 ml. Doda se 50 g slepe krme, dobro se premeša in pusti 10 minut, preden se nadaljuje s postopkom ekstrakcije (5.2) pa se nekajkrat ponovno premeša.
Če pa vrsta slepe krme, podobna vrsti vzorca, ni na voljo (glej točko 5.1.1), se lahko preskus izkoristka izvede po metodi standardnega dodatka. V tem primeru se vzorcu za analizo doda količina lasalocid natrija, podobna tisti v vzorcu. Ta vzorec se analizira skupaj z vzorcem brez dodanega lasalocid natrija, izkoristek pa se izračuna z odštevanjem.
5.2 Ekstrakcija
5.2.1
5–10 g vzorca se natehta na 0,01 g natančno in prenese v 250-mililitrsko erlenmajerico z zamaškom. S pipeto se doda 100,0 ml nakisanega metanola (3.8). Zamašek se rahlo zapre, erlenmajerica pa se zavrti, da se delci razpršijo. Erlenmajerica se postavi v ultrazvočno kopel (4.1) pri približno 40 oC za 20 minut, nato se odstrani iz kopeli in ohladi na sobno temperaturo. Pusti se stati približno 1 uro, dokler se suspenzija ne usede, nato pa se alikvot filtrira skozi 0,45 μm membranski filter (4.2) v primerno posodo. Nadaljuje se z določanjem s HPLC (5.3).
5.2.2
Približno 2 g nezmletega premiksa se natehta na 0,001 g natančno in prenese v 250-mililitrsko merilno bučko. Doda se 100,0 ml nakisanega metanola (3.8) in bučka se zavrti, da se delci razbijejo. Erlenmajerica z vsebino se postavi v ultrazvočno kopel (4.1) pri približno 40 oC za 20 minut, nato se odstrani iz kopeli in ohladi na sobno temperaturo. Z nakisanim metanolom (3.8) se razredči do oznake in dobro premeša. Pusti se stati 1 uro, dokler se suspenzija ne obori, nato se alikvot filtrira skozi 0,45 μm membranski filter (4.2). Primeren volumen čistega filtrata se razredči z nakisanim metanolom (3.8), da nastane končna preskusna raztopina s koncentracijo približno 4 μg/ml lasalocid natrija. Nadaljuje se z določanjem s HPLC (5.3).
5.3 Določanje s HPLC
5.3.1
Naslednji pogoji se lahko uporabljajo kot smernice; lahko se uporabljajo tudi drugi pogoji, če zagotavljajo enakovredne rezultate:
|
Kolona za tekočinsko kromatografijo (4.3.1): |
125 mm × 4 mm, reverzna faza C18, polnitev 5 μm ali enakovredna |
|
Mobilna faza (3.9): |
Mešanica raztopine fosfatnega pufra (3.7) in metanola (3.5), 5+95 (V+V) |
|
Hitrost pretoka: |
1,2 ml/min |
|
Detekcijska valovna dolžina: |
|
|
ekscitacija: |
310 nm |
|
emisija |
419 nm |
|
Volumen, ki se vbrizga: |
20 μl |
Stabilnost kromatografskega sistema se preveri z večkratnim vbrizgavanjem raztopine za umerjanja s koncentracijo 4,0 μg/ml (3.10.3), dokler niso dosežene konstantne višine (ali površine) vrhov in konstantni retencijski časi.
5.3.2
Vsaka raztopina za umerjanje (3.10.3) se večkrat vbrizga in za vsako koncentracijo se določijo srednje vrednosti višin (površin) vrhov. Umeritvena krivulja se pripravi tako, da se na ordinato nanesejo srednje vrednosti višin (površin) vrhov, na absciso pa ustrezne koncentracije v μg/ml.
5.3.3
Večkrat se vbrizgajo ekstrakti vzorca, ki nastanejo pri točki 5.2.1 ali 5.2.2, pri čemer se uporabijo enaki volumni kot pri raztopinah za umerjanje ter določijo srednje vrednosti višin (površin) vrhov lasalocid natrija.
6. Izračun rezultatov
Iz srednjih vrednosti višin (površin) vrhov, ki so nastali z vbrizganjem raztopine vzorca (5.3.3), se določi koncentracija lasalocid natrija (μg/ml) iz umeritvene krivulje.
6.1 Krma
Vsebnost lasalocid natrija (w) (mg/kg) v vzorcu se izračuna po naslednji formuli:

[mg/kg]
pri čemer je:
|
c |
= |
koncentracija lasalocid natrija v raztopini vzorca (5.2.1) v μg/ml; |
|
V1 |
= |
volumen ekstrakta vzorca v skladu s točko 5.2.1 v ml (tj. 100); |
|
m |
= |
masa preskusnega vzorca v gramih. |
6.2 Premiksi
Vsebnost lasalocid natrija (w) (mg/kg) v vzorcu se izračuna po naslednji formuli:

[mg/kg]
pri čemer je:
|
c |
= |
koncentracija lasalocid natrija v raztopini vzorca (5.2.2) v μg/ml; |
|
V2 |
= |
volumen ekstrakta vzorca v skladu s točko 5.2.2 v ml (tj. 250); |
|
f |
= |
faktor razredčitve v skladu s 5.2.2; |
|
m |
= |
masa preskusnega vzorca v gramih. |
7. Potrditev rezultatov
7.1 Identiteta
Metode, ki temeljijo na spektrofluorometriji, so manj izpostavljene motnjam kot metode, pri katerih se uporablja UV-detekcija. Identiteta analita se lahko potrdi z dodatno kromatografijo.
7.1.1
Ekstraktu vzorca (5.2.1 ali 5.2.2) se doda ustrezna količina raztopine za umerjanje (3.10.3). Količina dodanega lasalocid natrija mora biti podobna količini lasalocid natrija v ekstraktu vzorca. Le vrh lasalocid natrija je povišan ob upoštevanju količine dodanega lasalocid natrija in razredčitve ekstrakta. Širina vrha na polovici višine mora biti znotraj ± 10 % prvotne širine vrha, ki nastane pri ekstraktu vzorca brez dodanega lasalocid natrija.
7.2 Ponovljivost
Razlika med rezultatoma dveh vzporednih določanj, izvedenih na istem vzorcu, ne sme presegati:
— 15 % višje vrednosti za vsebnosti lasalocid natrija 30–100 mg/kg,
— 15 mg/kg za vsebnosti lasalocid natrija 100–200 mg/kg,
— 7,5 % višje vrednosti za vsebnosti lasalocid natrija več kot 200 mg/kg.
7.3 Izkoristek
Za krmo z dodatkom (slepo krmo) je izkoristek najmanj 80 %. Za vzorce premiksov z dodatkom je izkoristek najmanj 90 %.
8. Rezultati medlaboratorijske študije
Organizirana je bila medlaboratorijska študija ( *1 ), v kateri je 12 laboratorijev analiziralo 2 premiksa (vzorca 1 in 2) in 5 vzorcev krme (vzorci 3–7). Vsak vzorec je bil analiziran dvakrat. Rezultati so navedeni v spodnji preglednici:
|
|
Vzorec 1 Premiks za piščance |
Vzorec 2 Premiks za purane |
Vzorec 3 Briketi za purane |
Vzorec 4 Drobtine za piščance |
Vzorec 5 Krma za purane |
Vzorec 6 Krma za perutnino A |
Vzorec 7 Krma za perutnino B |
|
L |
12 |
12 |
12 |
12 |
12 |
12 |
12 |
|
n |
23 |
23 |
23 |
23 |
23 |
23 |
23 |
|
Srednja vrednost [mg/kg] |
5 050 |
16 200 |
76,5 |
78,4 |
92,9 |
48,3 |
32,6 |
|
Sr [mg/kg] |
107 |
408 |
1,71 |
2,23 |
2,27 |
1,93 |
1,75 |
|
CVr [ %] |
2,12 |
2,52 |
2,24 |
2,84 |
2,44 |
4,00 |
5,37 |
|
SR [mg/kg] |
286 |
883 |
3,85 |
7,32 |
5,29 |
3,47 |
3,49 |
|
CVR [ %] |
5,66 |
5,45 |
5,03 |
9,34 |
5,69 |
7,18 |
10,70 |
|
Nominalna vsebnosti [mg/kg] |
5 000 (*1) |
16 000 (*1) |
80 (*1) |
105 (*1) |
120 (*1) |
50 (*2) |
35 (*2) |
|
(*1) Vsebnost, ki jo navede proizvajalec; (*2) Krma, pripravljena v laboratoriju. |
|||||||
|
L |
= |
število laboratorijev; |
|
n |
= |
število posameznih vrednosti; |
|
Sr |
= |
standardni odmik ponovljivosti; |
|
SR |
= |
standardni odmik obnovljivosti; |
|
CVr |
= |
koeficient variacije ponovljivosti v %; |
|
CVR |
= |
koeficient variacije obnovljivosti v %; |
PRILOGA V
ANALITSKE METODE ZA NADZOR NEZAŽELENIH SNOVI V KRMI
A. DOLOČANJE PROSTEGA IN CELOTNEGA GOSIPOLA
1. Namen in področje uporabe
Ta metoda omogoča določanje ravni prostega in celotnega gosipola ter kemijsko sorodnih snovi v semenu, moki in tropinah bombaževca ter v krmnih mešanicah, ki vsebujejo ta posamična krmila, kadar je koncentracija prostega in celotnega gosipola ter kemijsko sorodnih snovi višja od 20 mg/kg.
2. Načelo
Gosipol se pri določanju prostega gosipola ekstrahira v navzočnosti 3-aminopropan-1-ola z mešanico propan-2-ola in heksana, pri določanju celotnega gosipola pa v navzočnosti dimetilformamida. Gosipol se z anilinom pretvori v gosipol-dianilin, katerega optična gostota se izmeri pri 440 nm.
3. Reagenti
3.1 Mešanica propan-2-ola in heksana: 60 volumskih delov propan-2-ola se zmeša s 40 volumskimi deli n-heksana.
3.2 Topilo A: v litrsko merilno bučko se doda 500 ml mešanice propan-2-ola in heksana (3.1), 2 ml 3-aminopropan-1-ola, 8 ml of ledocetne kisline in 50 ml vode. Z mešanico propan-2-ola in heksana (3.1) se dopolni do oznake. Ta reagent je obstojen en teden.
3.3 Topilo B: v 100-mililitrsko merilno bučko se odpipetira 2 ml 3-aminopropan-1-ola in 10 ml ledocetne kisline. Ohladi se na sobno temperaturo in dopolni z N, N-dimetilformamidom do oznake. Ta reagent je obstojen en teden.
3.4 Anilin: če optična gostota v slepem preskusu presega 0,022 , se anilin destilira prek cinka v prahu, pri čemer se zavrže prvih in zadnjih 10 % destilata. Če se reagent hrani v hladilniku v rjavi, zaprti stekleni posodi, je obstojen več mesecev.
3.5 Standardna raztopina gosipola A: v 250-mililitrsko merilno bučko se da 27,9 mg gosipol acetata. Raztopi se in dopolni s topilom A (3.2) do oznake. 50 ml te raztopine se odpipetira v 250-mililitrsko merilno bučko in dopolni s topilom A do oznake. Koncentracija gosipola v tej raztopini je 0,02 mg/ml. Pred uporabo se raztopina pusti eno uro na sobni temperaturi.
3.6 Standardna raztopina gosipola B: v 50-mililitrsko merilno bučko se da 27,9 mg gosipol acetata. Raztopi se in dopolni s topilom B (3.3) do oznake. Koncentracija gosipola v tej raztopini je 0,5 mg/ml.
Če se standardni raztopini gosipola A in B zaščitita pred svetlobo, sta obstojni 24 ur.
4. Oprema
4.1 Mešalnik: približno 35 obr./min.
4.2 Spektrofotometer
5. Postopek
5.1 Preskusni vzorec
Količina uporabljenega preskusnega vzorca je odvisna od pričakovane vsebnosti gosipola v vzorcu. Primerneje je uporabiti majhen preskusni vzorec in relativno velik alikvot filtrata, da nastane dovolj gosipola za natančne fotometrične meritve. Pri določanju prostega gosipola v semenu, moki in tropinah bombaževca količina preskusnega vzorca ne presega 1 g; za krmno mešanica pa je količina lahko do 5 g. 10-mililitrski alikvot filtrata je primeren v večini primerov; vsebuje 50–100 μg gosipola. Pri določanju celotnega gosipola je količina preskusnega vzorca 0,5 –5 g, zato da 2-mililitrski alikvot filtrata vsebuje 40–200 μg gosipola.
Analiza se izvede pri sobni temperaturi približno 20 oC.
5.2 Določanje prostega gosipola
Preskusni vzorec se prenese v 250-mililitrski bučko z brušenim vratom, katere dno je prekrito z drobci stekla. S pipeto se doda 50 ml topila A (3.2), bučka se zamaši, nato pa se z mešalnikom meša eno uro. Filtrira se skozi suhi filter v majhno bučko z brušenim vratom. Med filtriranjem se lijak prekrije z urnim steklom.
V vsako od dveh 25-mililitrskih merilnih bučk (A in B) se odpipetirajo enaki alikvoti filtrata, ki vsebujejo 50–100 μg gosipola. Po potrebi se s topilom A (3.2) dopolni do 10 ml. Z mešanico propan-2-ola in heksana (3.1) se bučka (A) dopolni do oznake. Ta raztopina se uporabi kot referenčna raztopina za merjenje raztopine vzorca.
V vsako od dveh 25-mililitrskih merilni bučk (C in D) se odpipetira 10 ml topila A (3.2). Z mešanico propan-2-ola in heksana (3.1) se bučka (C) dopolni do oznake. Ta raztopina se uporabi kot referenčna raztopina za merjenje raztopine vzorca.
V bučki (D) in (B) se doda 2 ml anilina (3.4). 30 minut se segrevata nad vrelo vodno kopeljo, da se obarvata. Ohladita se na sobno temperaturo, dopolnita z mešanico propan-2-ola in heksana (3.1) do oznake, homogenizirata in pustita stati eno uro.
V spektrofotometru se pri 440 nm in z 1-centimetrsko kiveto določi optična gostota raztopine iz slepega preskusa (D) tako, da se primerja z referenčno raztopino (C), optična gostota raztopine vzorca (B) pa se primerja z referenčno raztopino (A).
Optična gostota raztopine iz slepega preskusa se odšteje od optične gostote raztopine vzorca (= popravljena optična gostota). Vsebnost prostega gosipola se s pomočjo te vrednosti izračuna, kakor je navedeno v točki 6.
5.3 Določanje celotnega gosipola
Preskusni vzorec z 1–5 mg gosipola se prenese v 50-mililitrsko merilno bučko ter doda 10 ml topila B (3.3). Hkrati se pripravi slepi preskus, tako da se v drugo 50-mililitrsko merilno bučko prenese 10 ml topila B (3.3). Obe bučki se segrevata 30 minut nad vrelo vodno kopeljo. Ohladita se na sobno temperaturo, obe bučki pa se dopolnita z mešanico propan-2-ola in heksana (3.1) do oznake. Homogenizira se in pusti 10–15 minut, da se usede, nato se filtrira v bučko z brušenim vratom.
V dve 25-mililitrski merilni bučki se odpipetira po 2 ml filtrata vzorca, v drugi dve 25-mililitrski merilni bučki pa po 2 ml filtrata slepega preskusa. Ena bučka iz vsake serije se dopolni do 25 ml z mešanico propan-2-ola in heksana (3.1). Te raztopine se uporabijo kot referenčne raztopine.
V vsako od drugih dveh bučk se dodata 2 ml anilina (3.4). 30 minut se segrevata nad vrelo vodno kopeljo, da se obarvata. Ohladita se na sobno temperaturo, dopolnita z mešanico propan-2-ola in heksana (3.1) do 25 ml, homogenizirata in pustita stati eno uro.
Optična gostota prostega gosipola se določi, kakor je opisano v točki 5.2. Vsebnost celotnega gosipola se s pomočjo te vrednosti izračuna, kakor je navedeno v točki 6.
6. Izračun rezultatov
Rezultati se lahko izračunajo s pomočjo specifične optične gostote (6.1) ali iz umeritvene krivulje (6.2).
6.1 S specifično optično gostoto
Specifične optične gostote, pri opisanih pogojih, so naslednje:
|
Prosti gosipol |
|
|
Celotni gosipol |
|

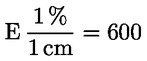
Vsebina prostega ali celotnega gosipola v vzorcu se izračuna po naslednji formuli:

pri čemer je:
|
E |
= |
popravljena optična gostota, ki se določi v skladu s točko 5.2; |
|
p |
= |
masa preskusnega vzorca v gramih; |
|
a |
= |
alikvot filtrata v mililitrih. |
6.2 Iz umeritvene krivulje
6.2.1
Pripravita se 2 seriji petih 25-mililitrskih merilnih bučk. V vsako serijo bučk se odpipetirajo 2,0 , 4,0 , 6,0 , 8,0 in 10,0 -mililitrski alikvoti standardne raztopine gosipola A (3.5). S topilom A (3.2) se dopolni do 10 ml. Zadnja 25-mililitrska merilna bučka v vsaki seriji vsebuje le 10 ml topila A (3.2) (slepi preskus).
Bučke iz prve serije (vključno z bučko za slepi preskus) se dopolnijo do 25 ml z mešanico propan-2-ola in heksana (3.1) (referenčna serija).
V vsako bučko iz druge serije (vključno z bučko za slepi preskus) se dodata 2 ml anilina (3.4). 30 minut se segrevajo nad vrelo vodno kopeljo, da se obarvajo. Ohladijo se na sobno temperaturo, dopolnijo z mešanico propan-2-ola in heksana (3.1) do oznake, homogenizirajo in pustijo stati eno uro (standardna serija).
Optična gostota raztopin iz standardne serije in ustreznih raztopin iz referenčne serije se določi, kakor je navedeno v točki 5.2. Umeritvena krivulja se pripravi tako, da se v diagram vnesejo izmerjene optične gostote glede na količine gosipola (v μg).
6.2.2
Pripravi se šest 50-mililitrskih merilnih bučk. V prvo bučko se prenese 10 ml topila B (3.3), v druge pa 2,0 , 4,0 , 6,0 , 8,0 in 10,0 ml standardne raztopine gosipola B (3.6). Vsaka bučka se dopolni do 10 ml s topilom B (3.3). Segrevajo se 30 minut nad vrelo vodno kopeljo. Ohladijo se na sobno temperaturo, dopolnijo z mešanico propan-2-ola in heksana (3.1) do oznake in homogenizirajo.
2,0 ml vsake od teh raztopin se prenese v vsako od dveh serij šestih 25-mililitrskih merilnih bučk. Bučke iz prve serije se dopolnijo do 25 ml z mešanico propan-2-ola in heksana (3.1) (referenčna serija).
V vsako bučko iz druge serije se dodata 2 ml anilina (3.4). Segrevajo se 30 minut nad vrelo vodno kopeljo. Ohladijo se na sobno temperaturo, dopolnijo z mešanico propan-2-ola in heksana (3.1) do oznake, homogenizirajo in pustijo stati eno uro (standardna serija).
Optična gostota raztopin iz standardne serije in ustreznih raztopin iz referenčne serije se določi, kakor je navedeno v točki 5.2. Umeritvena krivulja se pripravi tako, da se v diagram vnesejo izmerjene optične gostote glede na količine gosipola (v μg).
6.3 Ponovljivost
Razlika med rezultatoma dveh vzporednih določanj, izvedenih na istem vzorcu, ne sme presegati:
— 15 % višje vrednosti za vsebnosti gosipola, nižje od 500 ppm,
— 75 ppm absolutno za vsebnosti gosipola 500–750 ppm,
— 10 % višje vrednosti za vsebnosti gosipola, višje od 750 ppm.
B. DOLOČANJE VSEBNOSTI DIOKSINOV (PCDD/PCDF) IN PCB
POGLAVJE I
Metode vzorčenja in razlaga rezultatov analize
1. Namen in področje uporabe
Vzorci za uradni nadzor vsebnosti polikloriranih dibenzo-p-dioksinov (PCDD), polikloriranih dibenzofuranov (PCDF), dioksinom podobnih polikloriranih bifenilov (PCB) ( 16 ) in dioksinom nepodobnih PCB v krmi se odvzamejo v skladu z določbami iz Priloge I. Upoštevajo se kvantitativne zahteve za nadzor snovi ali proizvodov, enakomerno razporejenih po krmi, kot je določeno v točki 5.1 Priloge I. Tako pridobljeni sestavljeni vzorci se obravnavajo kot reprezentativni za serije ali podserije, iz katerih so odvzeti. Skladnost z mejnimi vrednostmi, določenimi v Direktivi 2002/32/ES, se ugotavlja na podlagi vrednosti, ugotovljenih v laboratorijskih vzorcih.
V tem delu B se uporabljajo opredelitve pojmov iz Priloge I k Odločbi Komisije 2002/657/ES ( 17 ).
Poleg navedenih opredelitev se v tem delu B uporabljajo tudi naslednje opredelitve pojmov:
„Presejalne metode“ pomenijo metode za izbiro tistih vzorcev, katerih vsebnosti PCDD/F in dioksinom podobnih PCB presegajo mejne vrednosti ali pragove ukrepanja. Omogočati morajo stroškovno učinkovito veliko prepustnost vzorcev in tako povečati možnosti za odkrivanje novih incidentov z veliko izpostavljenostjo in nevarnostjo za zdravje potrošnikov. Presejalne metode temeljijo na bioanalitskih metodah ali metodah GC-MS. Rezultati vzorcev, ki pri preverjanju skladnosti z mejno vrednostjo presegajo izločitveno vrednost, se preverijo s popolno ponovno analizo izvornega vzorca s potrditveno metodo.
„Potrditvene metode“ pomenijo metode, ki zagotavljajo celovite ali dopolnilne informacije o PCDD/F in dioksinom podobnih PCB ter njihovo natančno količinsko določitev pri mejni vrednosti ali po potrebi pragu za ukrepanje. Take metode uporabljajo plinsko kromatografijo/masno spektrometrijo visoke ločljivosti (GC-HRMS) ali plinsko kromatografijo/tandemsko masno spektrometrijo (GC-MS/MS).
2. Skladnost serije ali podserije z mejno vrednostjo
2.1 Glede dioksinom nepodobnih PCB
Serija je v skladu z mejno vrednostjo, če rezultat analize ne presega mejne vrednosti dioksinom nepodobnih PCB iz Direktive 2002/32/ES ob upoštevanju merilne negotovosti.
Serija ni v skladu z mejno vrednostjo, če rezultat analize na zgornji meji ( 18 ), potrjen z dvakratno analizo ( 19 ), presega mejno vrednost iz Direktive 2002/32/ES ob upoštevanju merilne negotovosti. Povprečje dveh določanj se ob upoštevanju merilne negotovosti uporablja za preverjanje skladnosti.
Merilna negotovost se upošteva po enem od naslednjih pristopov:
— z izračunom razširjene nezanesljivosti ob uporabi faktorja zajetja 2, ki zagotavlja 95-odstotno stopnjo zaupanja. Serija ali podserija ni skladna, če je izmerjena vrednost minus U nad mejno vrednostjo,
— z določitvijo odločitvene meje (CCα) v skladu s točko 3.1.2.5 Priloge I k Odločbi 2002/657/ES. Serija ali podserija ni skladna, če je izmerjena vrednost enaka ali večja od CCα.
Odstavki 1, 2 in 3 se uporabljajo za rezultat analize, pridobljen na vzorcu za uradni nadzor. Pri analizi za dopolnilno izvedensko mnenje ali referenčne namene se uporabljajo nacionalna pravila.
2.2 Glede PCDD/F in dioksinom podobnih PCB
Serija je v skladu z mejnimi vrednostmi, če rezultat posamezne analize,
— opravljene s presejalno metodo, pri kateri je delež lažno skladnih rezultatov manjši od 5 %, pokaže, da vsebnost ne presega zadevne mejne vrednosti PCDD/PCDF ter vsote PCDD/PCDF in dioksinom podobnih PCB iz Direktive 2002/32/ES,
— opravljene s potrditveno metodo, ne presega zadevne mejne vrednosti PCDD/PCDF ter vsote PCDD/PCDF in dioksinom podobnih PCB iz Direktive 2002/32/ES ob upoštevanju merilne negotovosti.
Za presejalne teste se določi izločitvena vrednost za odločitev o skladnosti vzorca z zadevnimi mejnimi vrednostmi, določenimi za PCDD/PCDF ali za vsoto PCDD/PCDF in dioksinom podobnih PCB.
Serija ni v skladu z mejno vrednostjo, če rezultat analize na zgornji meji ( 20 ), pridobljen s potrditveno metodo in potrjen z dvakratno analizo, presega mejno vrednost iz Direktive 2002/32/ES ob upoštevanju merilne negotovosti ( 21 ). Povprečje dveh določanj se ob upoštevanju merilne negotovosti uporablja za preverjanje skladnosti.
Merilna negotovost se upošteva po enem od naslednjih pristopov:
— z izračunom razširjene nezanesljivosti ob uporabi faktorja zajetja 2, ki zagotavlja 95-odstotno stopnjo zaupanja. Serija ali podserija ni skladna, če je izmerjena vrednost minus U nad mejno vrednostjo. Pri ločenem preskušanju za določitev PCDD/PCDF in dioksinom podobnih PCB se vsota ocenjene razširjene negotovosti ločenih rezultatov analize vsebnosti PCDD/PCDF in dioksinu podobnih PCB uporablja za vsoto PCDD/PCDF in dioksinom podobnih PCB,
— z določitvijo odločitvene meje (CCα) v skladu s točko 3.1.2.5 Priloge I k Odločbi 2002/657/ES. Serija ali podserija ni skladna, če je izmerjena vrednost enaka ali večja od CCα.
Odstavki od 1 do 4 se uporabljajo za rezultat analize, pridobljen na vzorcu za uradni nadzor. Pri analizi za dopolnilno izvedensko mnenje ali referenčne namene se uporabljajo nacionalna pravila.
3. Rezultati, ki presegajo pragove ukrepanja iz Priloge II k Direktivi 2002/32/ES
Pragovi ukrepanja so orodje za izbiro vzorcev v tistih primerih, ko je treba določiti vir kontaminacije in sprejeti ukrepe za njeno zmanjšanje ali odpravo. S presejalnimi metodami se določijo ustrezne izločitvene vrednosti za izbiro navedenih vzorcev. Če so za določitev vira kontaminacije in zmanjšanje ali odpravo kontaminacije potrebna precejšnja prizadevanja, je morda prekoračitev pragov ukrepanja primerno potrditi z dvakratno analizo s potrditveno metodo ob upoštevanju merilne negotovosti ( 22 ).
POGLAVJE II
Priprava vzorca in zahteve za analitske metode pri uradnem nadzoru vsebnosti dioksinov (PCDD/PCDF) in dioksinom podobnih PCB v krmi
1. Področje uporabe
Zahteve iz tega poglavja se uporabijo, kadar se krma analizira za uradni nadzor vsebnosti 2,3,7,8-substituiranih polikloriranih dibenzo-p-dioksinov in polikloriranih dibenzofuranov (PCDD/F) ter dioksinom podobnih polikloriranih bifenilov (dioksinom podobnih PCB) in za druge regulativne namene.
Prisotnost PCDD/F in dioksinom podobnih PCB v krmi se lahko spremlja z dvema različnima vrstama analitskih metod:
(a) Presejalne metode
Cilj presejalnih metod je izbrati tiste vzorce, katerih vsebnosti PCDD/F in dioksinom podobnih PCB presegajo mejne vrednosti ali pragove ukrepanja. Presejalne metode morajo omogočati stroškovno učinkovito veliko prepustnost vzorcev in tako povečati možnosti za odkrivanje novih incidentov z veliko izpostavljenostjo in nevarnostjo za zdravje potrošnikov. Z njihovo uporabo naj bi se izognili lažno skladnim rezultatom. Lahko vključujejo bioanalitske metode in metode GC-MS.
Potrditvene metode primerjajo rezultat analize z izločitveno vrednostjo in omogočajo jasno odločitev o morebitni prekoračitvi mejne vrednosti ali praga ukrepanja. Koncentracijo PCDD/F ter vsoto PCDD/F in dioksinom podobnih PCB v vzorcih, za katere se domneva, da niso v skladu z mejno vrednostjo, je treba določiti/potrditi s potrditveno metodo.
Poleg tega lahko presejalne metode nakažejo vsebnost PCDD/F in dioksinom podobnih PCB v vzorcu. V primeru uporabe bioanalitskih presejalnih metod se rezultat izrazi v bioanalitskih ekvivalentih (BEQ), v primeru uporabe fizikalno-kemijskih metod GC-MS pa v toksičnih ekvivalentih (TEQ). Številčno izraženi rezultati presejalnih metod so primerni za dokaz skladnosti ali sumljive neskladnosti ali prekoračitve pragov ukrepanja in omogočajo oceno območja vsebnosti v primeru nadaljnjih testov s potrditvenimi metodami. Niso primerni za namene, kot so ocenjevanje osnovnih vrednosti, ocenjevanje vnosa, spremljanje časovnih trendov vsebnosti ali ponovno ocenjevanje pragov ukrepanja in mejnih vrednosti.
(b) Potrditvene metode
Potrditvene metode omogočajo nedvoumno identifikacijo in kvantifikacijo PCDD/F in dioksinom podobnih PCB, prisotnih v vzorcu, in zagotavljajo celovite informacije na ravni kongenera. Zato te metode omogočajo nadzor mejnih vrednosti in pragov ukrepanja, vključno s potrditvijo rezultatov, pridobljenih s presejalnimi metodami. Poleg tega se rezultati lahko uporabljajo za druge namene, na primer za določitev nizkih osnovnih vsebnosti v krmi po spremljanje časovnih gibanj, oceno izpostavljenosti ter vzpostavitev zbirke podatkov za morebitno ponovno oceno pragov ukrepanja in mejnih vrednosti. Pomembne so tudi za določitev vzorcev kongenerov za ugotovitev vira mogoče kontaminacije. Take metode uporabljajo GC-HRMS. Za določitev skladnosti ali neskladnosti z mejno vrednostjo se lahko uporabi tudi GC-MS/MS.
2. Ozadje
Za izračun koncentracij toksičnega ekvivalenta (TEQ) se koncentracije posameznih snovi v danem vzorcu pomnožijo z njihovim ustreznim faktorjem toksične ekvivalentnosti (TEF, glej opombo (16) iz poglavja I), in se nato seštejejo, da se dobi skupna koncentracija dioksinom podobnih spojin, izraženih kot toksični ekvivalenti (TEQ).
V tem delu B sprejemljiva specifična meja določanja posameznega kongenera pomeni najnižjo vsebnost analita, ki se lahko izmeri s sprejemljivo statistično gotovostjo, ki izpolnjuje merila za identifikacijo, kot so opisana v mednarodno priznanih standardih, na primer v standardu EN 2012: 16215 (Krma – Določanje dioksinov in dioksinom podobnih PCB z GC-HRMS in indikatorjev PCB z GC-HRMS) in/ali v metodah EPA 1613 in 1668, kakor so bile revidirane.
Meja količinskega določanja posameznega kongenera se lahko opredeli kot:
(a) koncentracija analita v ekstraktu vzorca, ki ustvari instrumentalni odziv pri sledenju dveh različnih ionov, ki ju je treba spremljati z razmerjem med signalom in šumom vsaj S/N 3: 1 pri manj občutljivem signalu neobdelanih podatkov, ali
(b) točka najnižje koncentracije na umeritveni krivulji, ki omogoča sprejemljivo (≤ 30 %) in dosledno (merjeno vsaj na začetku in na koncu analitične serije vzorcev) odstopanje od povprečnega faktorja relativnega odziva, izračunanega za vse točke na umeritveni krivulji v vsaki seriji vzorcev, če zaradi tehničnih razlogov izračun signal/šum ne zagotovi zanesljivih rezultatov. LOQ se izračuna iz točke najnižje koncentracije ob upoštevanju izkoristka internih standardov in količine vzorca.
Bioanalitske presajalne metode ne dajejo rezultatov na ravni kongenerov, temveč zgolj nakažejo ( 23 ) vrednosti toksičnih ekvivalentov (TEQ), izražene v bioanalitskih ekvivalentih (BEQ), saj vse spojine, ki so prisotne v ekstraktu vzorca in pokažejo odziv pri testiranju, morda ne izpolnjujejo vseh zahtev načela TEQ.
Presejalne in potrditvene metode se lahko uporabijo za nadzor določene matrice le, če so metode dovolj občutljive, da zanesljivo odkrijejo vsebnosti pri pragu ukrepanja ali mejni vrednosti.
3. Zahteve za zagotavljanje kakovosti
|
3.1 |
Sprejeti je treba ukrepe za preprečevanje navzkrižne kontaminacije na vseh stopnjah vzorčenja in analitskega postopka. |
|
3.2 |
Vzorci se hranijo in prevažajo v steklenih, aluminijastih, polipropilenskih ali polietilenskih posodah, ki so primerne za shranjevanje in ne vplivajo na vsebnost PCDD/PCDF in dioksinom podobnih PCB v vzorcih. Iz posode za vzorce se odstranijo sledi papirnega prahu. |
|
3.3 |
Hramba in prevoz vzorcev se izvedeta tako, da se ohrani neoporečnost vzorca krme. |
|
3.4 |
Če je ustrezno, se vsak laboratorijski vzorec drobno zmelje in temeljito premeša po postopku, s katerim se dokazano doseže popolna homogenizacija (npr. zmleto tako, da se preseje z 1-milimetrskim sitom). Če je vsebnost vlage previsoka, se vzorci pred mletjem posušijo. |
|
3.5 |
Reagenti, steklovina in oprema se nadzorujejo zaradi mogočega vpliva na rezultate, izražene v TEQ ali BEQ. |
|
3.6 |
Slepa analiza se opravi z izvedbo celotnega analitskega postopka, iz katerega se izpusti le vzorec. |
|
3.7 |
Pri bioanalitskih metodah se testirata steklovina in topilo, ki se uporabljata za analizo, da ne vsebujeta spojin, ki vplivajo na ugotavljanje ciljnih spojin v delovnem območju. Steklovina se spere s topili ali segreje do temperatur, primernih za odstranitev sledi PCDD/PCDF, dioksinom podobnih spojin in motečih spojin s površine. |
|
3.8 |
Količina vzorca, uporabljenega za ekstrakcijo, zadostuje za izpolnjevanje zahtev glede dovolj nizkega delovnega območja, vključno s koncentracijami mejnih vrednosti ali praga ukrepanja. |
|
3.9 |
Posebni postopki za pripravo vzorca, ki se uporabljajo za obravnavane proizvode, so skladni z mednarodno sprejetimi smernicami. |
4. Zahteve za laboratorije
|
4.1 |
V skladu z določbami Uredbe (ES) št. 882/2004 laboratorije akreditirajo priznani organi, ki delujejo v skladu z zahtevami ISO Guide 58, s čimer je zagotovljeno, da izvajajo analitsko zagotavljanje kakovosti. Laboratoriji se akreditirajo po standardu EN ISO/IEC 17025. |
|
4.2 |
Sposobnost laboratorijev se dokazuje z nenehnim uspešnim sodelovanjem v medlaboratorijskih študijah za določanje PCDD/PCDF in dioksinom podobnih PCB v ustreznih matricah krme in območjih koncentracij. |
|
4.3 |
Laboratoriji, ki izvajajo presejalne metode za rutinski nadzor vzorcev, vzpostavijo tesno sodelovanje z laboratoriji, ki izvajajo potrditveno metodo, da se zagotovi kakovost in potrdijo rezultati analize sumljivih vzorcev. |
5. Osnovne zahteve za analitske postopke za dioksine (PCDD/PCDF) in dioksinom podobne PCB
5.1 Nizko delovno območje in meje določanja
Za PCDD/PCDF so zaradi izredne toksičnosti nekaterih od teh spojin zaznavne količine v zgornjem femtogramskem območju (10–15 g). Za večino kongenerov PCB je meja določanja v nanogramskem območju (10–9 g) že zadostna. Za merjenje bolj toksičnih dioksinom podobnih kongenerov PCB (zlasti neorto substituiranih kongenerov) spodnji del delovnega območja dosega spodnje pikogramsko območje (10–12 g). Za vse druge kongenere PCB zadostuje meja določanja v nanogramskem območju (10–9 g).
5.2 Velika selektivnost (specifičnost)
|
5.2.1 |
Razlikovati je treba med PCDD/PCDF in dioksinom podobnimi PCB ter številnimi drugimi spojinami, ki se ekstrahirajo hkrati in so morda moteče ter so prisotne v koncentracijah, ki so do več velikostnih razredov višje od predpisanih analitov. Za metode GC-MS je treba razlikovati med različnimi kongeneri, na primer med toksičnimi (npr. 17 2,3,7,8-substituiranih PCDD/PCDF in 12 dioksinom podobnih PCB) in drugimi kongeneri. |
|
5.2.2 |
Bioanalitske metode omogočajo ugotavljanje ciljnih spojin kot vsote PCDD/PCDF in/ali dioksinom podobnih PCB. Vzorec se očisti, da se odstranijo spojine, ki povzročajo lažno neskladne rezultate, ali spojine, ki lahko zmanjšajo odziv in povzročijo lažno skladne rezultate. |
5.3 Velika točnost (pravilnost in natančnost, navidezni izkoristek pri bioloških testih)
|
5.3.1 |
Pri metodah GC-MS določanje zagotovi veljavno oceno pravih koncentracij v vzorcu. Zahteva se velika točnost, da se prepreči zavrnitev rezultata analize vzorca zaradi slabe zanesljivosti ugotovljene vrednosti TEQ. Točnost se izrazi kot pravilnost (razlika med izmerjeno srednjo vrednostjo analita v certificiranem materialu in njegovo certificirano vrednostjo, izraženo kot delež te vrednosti) in natančnost (natančnost RSDR se izračuna kot relativni standardni odmik od rezultatov, pridobljenih pri vnaprej določenih pogojih obnovljivosti). |
|
5.3.2 |
Pri bioanalitskih metodah se določi navidezni izkoristek pri bioloških testih. Navidezni izkoristek pri bioloških testih pomeni vrednost BEQ, izračunano iz umeritvene krivulje na podlagi TCDD ali PCB 126, popravljene za slepe vrednosti in nato deljene z vrednostjo TEQ, določeno s potrditveno metodo. Njegov namen je popraviti vplive, kot so izguba PCDD/PCDF in dioksinom podobnih spojin med postopki ekstrakcije in čiščenja, soekstrahirane spojine, ki povečujejo ali zmanjšujejo odziv (agonistični in antagonistični učinki), kakovost prilagajanja krivulje ali razlike med vrednostmi faktorja toksične ekvivalentnosti (TEF) in relativne učinkovitosti (REP). Navidezni izkoristek pri bioloških testih se izračuna iz ustreznih referenčnih vzorcev, ki imajo reprezentativno porazdelitev kongenerov v območju predpisane vrednosti. |
5.4 Validacija v območju mejne vrednosti in splošni ukrepi za zagotavljanje kakovosti
|
5.4.1 |
Laboratoriji dokažejo zmogljivost metode v območju mejne vrednosti, npr. 0,5-kratna, 1-kratna in 2-kratna mejna vrednost s sprejemljivim koeficientom variacije za ponovljene analize, med postopkom validacije in rutinsko analizo. |
|
5.4.2 |
Redne slepe kontrole in preskusi z dodatkom ali analize kontrolnih vzorcev (če je na voljo, je zaželen certificiran referenčni material) se izvajajo kot notranji nadzorni ukrepi za zagotavljanje kakovosti. Za slepe kontrole, preskuse z dodatkom ali analize kontrolnih vzorcev se vodijo in preverjajo kontrolne karte, s čimer se zagotovi, da je zmogljivost analiz v skladu z zahtevami. |
5.5 Meja določanja
|
5.5.1 |
Za bioanalitsko presejalno metodo določitev meje določanja (LOQ) ni nujna zahteva, vendar je treba dokazati, da lahko metoda razlikuje med slepo vrednostjo in izločitveno vrednostjo. Pri določanju vrednosti BEQ se določi prag poročanja zaradi obravnave vzorcev, pri katerih je odziv pod tem pragom. Za prag poročanja je treba dokazati, da se razlikuje najmanj za trikrat od postopka s slepimi vzorci, pri katerih je odziv pod delovnim območjem. Zato se izračuna na podlagi vzorcev, ki vsebujejo ciljne spojine blizu zahtevane najnižje vrednosti, ne pa na podlagi razmerja med signalom in šumom ali slepega testa. |
|
5.5.2 |
Meja določanja za potrditveno metodo mora biti približno pri petini mejne vrednosti. |
5.6 Merila za analizo
Za zanesljive rezultate potrditvenih ali presejalnih metod so izpolnjena naslednja merila v območju mejne vrednosti ali praga ukrepanja za vrednost TEQ ali BEQ, ki je določena kot celotna vrednost TEQ (kot vsota PCDD/PCDF in dioksinom podobnih PCB) ali posebej za PCDD/PCDF in dioksinom podobne PCB:
|
|
Presejanje z bioanalitskimi ali fizikalno-kemijskimi metodami |
Potrditvene metode |
|
Delež lažno skladnih rezultatov (1) |
< 5 % |
|
|
Pravilnost |
|
– 20 % do + 20 % |
|
Ponovljivost (RSDr) |
< 20 % |
|
|
Interna laboratorijska obnovljivost (RSDR) |
< 25 % |
< 15 % |
|
(1) Glede na mejne vrednosti. |
||
5.7 Posebne zahteve za presejalne metode
|
5.7.1 |
Pri presejanju se lahko uporabijo metode GC-MS in bioanalitske metode. Metode GC-MS izpolnjujejo zahteve iz točke 6. Za bioanalitske metode na celični osnovi so posebne zahteve določene v točki 7. |
|
5.7.2 |
Laboratoriji, ki izvajajo presejalne metode za rutinski nadzor vzorcev, vzpostavijo tesno sodelovanje z laboratoriji, ki izvajajo potrditveno metodo. |
|
5.7.3 |
Med rutinsko analizo je treba opraviti preverjanje zmogljivosti presejalne metode z analitskim zagotavljanjem kakovosti in nenehno validacijo metod. Nenehno se izvaja program nadzora nad skladnimi rezultati. |
|
5.7.4 |
Preverjanje mogočega zmanjšanja celične odzivnosti in citotoksičnosti: 20 % ekstraktov vzorcev se izmeri med rutinskim presejanjem brez 2,3,7,8-TCDD in z dodanim 2,3,7,8-TCDD, ki ustreza mejni vrednosti ali pragu ukrepanja, da se preveri, ali je morda odziv zmanjšan zaradi motečih snovi v ekstraktu vzorca. Izmerjena koncentracija vzorca z dodatkom se primerja z vsoto koncentracije ekstrakta brez dodatka in koncentracije z dodatkom. Če je ta izmerjena koncentracija za več kot 25 % nižja od izračunane (vsote) koncentracije, to kaže na mogoče zmanjšanje signala, zato se za zadevni vzorec opravi potrditvena analiza GC-HRMS. Rezultati se spremljajo v kontrolnih kartah. |
|
5.7.5 |
Zagotavljanje kakovosti skladnih vzorcev: Približno od 2 % do 10 % skladnih vzorcev, odvisno od matrice vzorca in laboratorijskih izkušenj, je treba potrditi z GC-HRMS. |
|
5.7.6 |
Določitev deleža lažno skladnih rezultatov na podlagi podatkov o zagotavljanju kakovosti: Določi se delež lažno skladnih rezultatov pri presejanju vzorcev pod mejno vrednostjo ali pragom ukrepanja in nad njima. Dejanski delež lažno skladnih rezultatov je manjši kot 5 %. Ko je pri zagotavljanju kakovosti skladnih vzorcev na voljo najmanj 20 potrjenih rezultatov na matrico/skupino matric, se na podlagi te zbirke podatkov pripravijo ugotovitve o deležu lažno skladnih rezultatov. Rezultati vzorcev, analiziranih s primerjalnimi preskusi ali med kontaminacijami, ki zajemajo območje koncentracije do na primer dvakratne mejne vrednosti, so prav tako lahko vključeni v kvoto 20 rezultatov za oceno deleža lažno skladnih rezultatov. Vzorci zajemajo najpogostejše vzorce kongenerov, ki predstavljajo različne vire. Čeprav je glavni namen presejalnih testov odkrivanje vzorcev, ki presegajo prag ukrepanja, je merilo za določanje deleža lažno skladnih rezultatov mejna vrednost, ob upoštevanju merilne negotovosti potrditvene metode. |
|
5.7.7 |
Morebitni neskladni vzorci pri presajanju se vedno preverijo s popolno ponovno analizo izvornega vzorca s potrditveno analitsko metodo Ti vzorci se lahko uporabijo tudi za oceno deleža lažno neskladnih rezultatov. Pri presejalnih metodah je delež lažno neskladnih rezultatov delež rezultatov, za katere je s potrditveno analizo potrjeno, da so skladni, čeprav je bilo pri predhodnem presejanju ugotovljeno, da je vzorec morda neskladen. Ocena koristnosti presejalne metode temelji na primerjavi lažno neskladnih vzorcev in celotnega števila pregledanih vzorcev. Ta delež je dovolj majhen, da je uporaba presejalnega orodja koristna. |
|
5.7.8 |
Bioanalitske metode vsaj pri pogojih validacije veljavno pokažejo vrednost TEQ, ki je izračunana in izražena kot BEQ. Tudi pri bioanalitskih metodah, opravljenih pri pogojih ponovljivosti, je interna laboratorijska ponovljivost (RSDr) običajno manjša od obnovljivosti (RSDr). |
6. Posebne zahteve za metode gc-ms za presejalne in potrditvene namene
6.1 Sprejemljive razlike med rezultati WHO-TEQ na zgornji in spodnji meji
Razlika med vrednostmi na zgornji in spodnji meji ne presega 20 % za potrditev prekoračitve mejne vrednosti ali v primeru potrebe po pragovih ukrepanja.
6.2 Nadzor izkoristka
|
6.2.1 |
Dodajanje s 13C-označenih 2,3,7,8-klor substituiranih internih standardov za PCDD/PCDF in s 13C-označenih internih standardov za dioksinom podobne PCB se izvede na samem začetku analitske metode, na primer pred ekstrakcijo, da se validira analitski postopek. Doda se vsaj en kongener za vsako od tetra- do okta-kloriranih homolognih skupin za PCDD/PCDF in vsaj en kongener za vsako od homolognih skupin za dioksinom podobne PCB (ali vsaj en kongener za vsak masno spektrometričen izbran ion s funkcijo evidentiranja, ki se uporablja za spremljanje PCDD/PCDF in dioksinom podobnih PCB). Pri potrditvenih metodah se uporablja vseh 17 s 13C-označenih 2,3,7,8-klor substituiranih internih standardov za PCDD/PCDF in vseh 12 s 13C-označenih internih standardov za dioksinom podobne PCB. |
|
6.2.2 |
Prav tako se določijo relativni odzivni faktorji za tiste kongenere, za katere ni dodan noben od s 13C-označenih analogov z uporabo ustreznih raztopin za umerjanje. |
|
6.2.3 |
Za krmo rastlinskega in živalskega izvora, ki vsebuje manj kot 10 % maščob, je dodatek internih standardov pred ekstrakcijo obvezen. Za krmo živalskega izvora, ki vsebujejo več kot 10 % maščob, se lahko interni standardi dodajo pred ekstrakcijo maščob ali po njej. Izvede se ustrezna validacija učinkovitosti ekstrakcije, ki je odvisna od faze, pri kateri se dodajo interni standardi, in od tega, ali se o rezultatih poroča na podlagi proizvoda ali maščob. |
|
6.2.4 |
Pred analizo GC-MS se dodata 1 ali 2 (nadomestna) standarda za izračun izkoristka. |
|
6.2.5 |
Zahteva se nadzor izkoristka. Za potrditvene metode so izkoristki posameznih internih standardov v območju od 60 % do 120 %. Manjši ali večji izkoristki za posamezne kongenere, še zlasti za nekatere hepta- in okta-klorirane dibenzo-p-dioksine in dibenzofurane, so sprejemljivi, če njihov prispevek k vrednosti TEQ ne presega 10 % celotne vrednosti TEQ (na podlagi vsote PCDD/PCDF in dioksinom podobnih PCB). Pri presejalnih metodah GC-MS so izkoristki v območju 30 % do 140 %. |
6.3 Odstranitev motečih snovi
— PCDD/PCDF se od motečih kloriranih spojin, kot so dioksinom nepodobni PCB in klorirani difenil etri, ločijo z ustreznimi kromatografskimi tehnikami (prednost imajo florisil, koloni iz aluminijevega oksida in/ali ogljikovi koloni).
— Plinskokromatografsko ločevanje izomerov je < 25 % od vrha do vrha med 1,2,3,4,7,8-HxCDF in 1,2,3,6,7,8-HxCDF.
6.4 Umerjanje s standardno krivuljo
Obseg umeritvene krivulje zajema ustrezno območje mejne vrednosti ali pragov ukrepanja.
6.5 Posebna merila za potrditvene metode
— Za GC-HRMS:
—
V HRMS je ločljivost tipično večja od ali enaka 10 000 za celotni masni razpon pri 10 % doline.
Izpolnjevanje nadaljnjih meril za identifikacijo in potrditev, kot so opisana v mednarodno priznanih standardih, na primer v standardu EN 2012: 16215 (Krma – Določanje dioksinov in dioksinom podobnih PCB z GC/HRMS in indikatorjev PCB z GC/HRMS) in/ali v metodah EPA 1613 in 1668, kakor so bile revidirane.
— Za GC-MS/MS:
—
Spremljanje najmanj dveh posebnih starševskih ionov, vsakega z enim posebnim ustreznim prehodnim produktnim ionom za vse označene in neoznačene analite v okviru analize.
Maksimalna dovoljena toleranca relativnega ionskega odziva je ± 15 % za izbrane prehodne produktne ione v primerjavi z izračunanimi ali izmerjenimi vrednostmi (povprečje umeritvenih standardov), ki se uporablja za identične pogoje MS/MS, zlasti energijo trka in trka pod plinskim tlakom, za vsak prehod analita.
Ločljivost vsakega kvadripola se določi pri isti ali večji ločljivosti kot ločljivost masne enote (ločljivost masne enote: ustrezna ločljivost za ločitev dveh vrhov iste masne enote) za zmanjšanje morebitnih motečih vplivov na predpisane analite.
Izpolnjevanje nadaljnjih meril, kot so opisana v mednarodno priznanih standardih, na primer v standardu EN 2012: 16215 (Krma – Določanje dioksinov in dioksinom podobnih PCB z GC/HRMS in indikatorjev PCB z GC/HRMS) in/ali v metodah EPA 1613 in 1668, kakor so bile revidirane.
7. Posebne zahteve za bioanalitske metode
Bioanalitske metode so metode, ki temeljijo na uporabi bioloških načel, kot so testi na celični osnovi, testi na osnovi receptorjev ali imunološki testi. V tej točki 7 so določene zahteve za bioanalitske metode na splošno.
S presejalno metodo se načeloma vzorec razvrsti kot skladen ali vzorec s sumom na neskladnost. V ta namen se izračunana vrednost BEQ primerja z izločitveno vrednostjo (glej 7.3). Vzorci pod izločitveno vrednostjo se ugotovijo za skladne, za tiste na tej meji ali nad njo pa se sumi, da niso skladni, zato jih je treba analizirati s potrditveno metodo. V praksi se vrednost BEQ, ki ustreza 2/3 mejne vrednosti, lahko uporabi kot izločitvena vrednost, če je zagotovljeno, da je delež lažno skladnih rezultatov nižji od 5 %, delež lažno neskladnih rezultatov pa sprejemljiv. Ker so mejne vrednosti ločene za PCDD/F ter za vsoto PCDD/F in dioksinom podobnih PCB, preverjanje skladnosti vzorcev brez frakcioniranja zahteva ustrezne izločitvene vrednosti za PCDD/F pri bioloških testih. Za preverjanje vzorcev, ki presegajo pragove ukrepanja, bi bil kot izločitvena vrednost primeren ustrezen delež posameznih pragov ukrepanja.
Poleg tega se lahko pri nekaterih bioanalitskih metodah za vzorce v delovnem območju, ki presegajo prag poročanja, navede okvirna vrednost, izražena v BEQ (glej 7.1.1 in 7.1.6).
7.1 Ocena odzivnosti preskusa
7.1.1 Splošne zahteve
— Kadar se koncentracije izračunajo iz umeritvene krivulje TCDD, vrednosti na spodnjem in zgornjem koncu krivulje pokažejo veliko razliko (visok koeficient variacije, KV). Delovno območje je območje, v katerem je KV manjši kot 15 %. Spodnji del delovnega območja (prag poročanja) se določi najmanj trikrat višje kot postopek s slepimi vzorci. Zgornji del delovnega območja običajno predstavlja vrednost EC70 (70 % največje učinkovite koncentracije), vendar je ta nižje, če je KV v tem območju večji kot 15 %. Delovno območje se določi med validacijo. Izločitvene vrednosti (glej točko 7.3) so znotraj delovnega območja.
— Standardne raztopine in ekstrakti vzorcev se preskusijo vsaj z dvakratno analizo. Kadar se uporabijo dvakratne analize, standardne raztopine ali ekstrakti kontrolnih vzorcev, testirani v 4 do 6 jamicah, razporejenih po plošči, pokažejo odziv ali koncentracijo (mogoče le v delovnem območju) na podlagi KV < 15 %.
7.1.2 Umerjanje
7.1.2.1 Umerjanje s standardno krivuljo
— Vrednosti v vzorcih se ocenijo tako, da se primerja odzivnost preskusa z umeritveno krivuljo TCDD (ali PCB 126 ali standardna mešanica PCDD/PCDF/dioksinom podobnih PCB) za izračun vrednosti BEQ v ekstraktu in pozneje v vzorcu.
— Umeritvena krivulja vsebuje 8 do 12 koncentracij (vsaj v dvojnikih), z zadostnimi koncentracijami v spodnjem delu krivulje (delovno območje). Posebna pozornost se nameni kakovosti prilagajanja krivulje v delovnem območju. Tako ima vrednost R2 malo ali sploh nikakršnih koristi za ocenjevanje ustreznosti prileganja pri nelinearni regresiji. Boljše prilagajanje se doseže z zmanjšanjem razlike med izračunanimi in ugotovljenimi vrednostmi v delovnem območju krivulje (na primer z zmanjšanjem vsote kvadratov ostankov).
— Ocenjena vrednost v ekstraktu vzorca se nato popravi glede na vrednost BEQ, izračunano za slepi vzorec matrice/topila (da se upoštevajo nečistoče iz uporabljenih topil in kemikalij), in navidezen izkoristek (izračunan iz vrednosti BEQ ustreznih referenčnih vzorcev z reprezentativnimi vzorci kongenerov blizu mejne vrednosti ali praga ukrepanja). Za popravek izkoristka je navidezni izkoristek vedno v zahtevanem območju (glej točko 7.1.4). Referenčni vzorci, ki se uporabijo za popravek izkoristka, izpolnjujejo zahteve iz točke 7.2.
7.1.2.2 Umerjanje z referenčnimi vzorci
Druga možnost je, da se uporabi umeritvena krivulja, pripravljena iz vsaj štirih referenčnih vzorcev (glej točko 7.2.4: ena slepa matrica ter trije referenčni vzorci z 0,5-kratno, 1-kratno in 2-kratno mejno vrednostjo ali prag ukrepanja), zaradi česar popravek slepih vrednosti in izkoristka ni več potreben. V tem primeru se lahko odzivnost preskusa, ki ustreza 2/3 mejne vrednosti (glej točko 7.3), izračuna neposredno iz teh vzorcev in uporabi kot izločitvena vrednost. Za preverjanje vzorcev, ki presegajo pragove ukrepanja, bi bil kot izločitvena vrednost primeren ustrezen delež teh pragov ukrepanja.
7.1.3 Ločeno določanje PCDD/PCDF in dioksinom podobnih PCB
Ekstrakti se lahko razdelijo v frakcije, ki vsebujejo PCDD/PCDF in dioksinom podobne PCB, kar omogoča ločeno navedbo vrednosti TEQ za PCDD/PCDF in dioksinom podobne PCB (v BEQ). Po možnosti se uporabi standardna umeritvena krivulja PCB 126 za oceno rezultatov za frakcijo, ki vsebuje dioksinom podobne PCB.
7.1.4 Navidezni izkoristki pri bioloških testih
„Navidezni izkoristek pri bioloških testih“ se izračuna iz ustreznih referenčnih vzorcev z reprezentativnimi vzorci kongenerov blizu mejne vrednosti ali praga ukrepanja in se izrazi kot delež vrednosti BEQ v primerjavi z vrednostjo TEQ. Odvisno od vrste testa in uporabljenih TEF ( 24 ) lahko razlike med faktorji TEF in REP za dioksinom podobne PCB povzročijo majhne navidezne izkoristke za dioksinom podobne PCB v primerjavi s PCDD/PCDF. Če se izvaja ločeno določanje PCDD/PCDF in dioksinom podobnih PCB, navidezni izkoristki pri bioloških testih znašajo: za dioksinom podobne PCB 20 % do 60 %, za PCDD/PCDF 50 % do 130 % (območja veljajo za umeritveno krivuljo TCDD). Ker se lahko prispevek dioksinom podobnih PCB k vsoti PCDD/PCDF in dioksinom podobnih PCB razlikuje pri različnih matricah in vzorcih, se pri navideznih izkoristkih pri bioloških testih za vsoto PCDD/PCDF in dioksinom podobnih PCB upoštevajo ta območja in znašajo 30 % do 130 %. Pri kakršni koli bistveni spremembi vrednosti TEF za zakonodajo Unije za PCDD/PCDF in dioksinom podobne PCB je treba spremeniti ta območja.
7.1.5 Nadzor nad izkoristkom pri čiščenju
Izguba spojin med čiščenjem se preveri med validacijo. Za slepi vzorec z dodatkom mešanice različnih kongenerov se opravi čiščenje (najmanj n = 3), nato pa se s potrditveno metodo preverita izkoristek in variabilnost. Izkoristek znaša 60 % do 120 %, zlasti za kongenere, ki k vrednosti TEQ v različnih mešanicah prispevajo več kot 10 %.
7.1.6 Prag poročanja
Za poročanje o vrednostih BEQ se prag poročanja določi na podlagi ustreznih vzorcev matric, ki vključujejo značilne vzorce kongenerov, ne pa na podlagi umeritvene krivulje standardov zaradi manjše natančnosti v njenem spodnjem delu. Upoštevajo se učinki ekstrakcije in čiščenja. Prag poročanja se določi najmanj trikrat višje kot postopek s slepimi vzorci.
7.2 Uporaba referenčnih vzorcev
|
7.2.1 |
Referenčni vzorci predstavljajo vzorce matric, vzorce kongenerov ter območja koncentracij za PCDD/PCDF in dioksinom podobne PCB blizu mejne vrednosti ali praga določanja. |
|
7.2.2 |
V vsako preskusno serijo se vključi slepa matrica, in kadar to ni mogoče, postopek s slepimi vzorci, ter referenčen vzorec pri mejni vrednosti ali pragu ukrepanja. Ti vzorci se ekstrahirajo in preskušajo hkrati in pod enakimi pogoji. Referenčni vzorec pokaže izrazito večji odziv v primerjavi s slepim, kar zagotavlja ustreznost preskusa. Ti vzorci se lahko uporabijo za popravek slepih vrednosti in izkoristka. |
|
7.2.3 |
Referenčni vzorci, ki se izberejo za popravek izkoristka, so reprezentativni za preskusne vzorce, kar pomeni, da vzorci kongenerov ne povzročijo prenizke ocene vrednosti. |
|
7.2.4 |
Vključijo se lahko dodatni referenčni vzorci z na primer 0,5-kratno in 2-kratno mejno vrednostjo ali pragom ukrepanja, da se dokaže primerna uspešnost preskusa v območju predpisane vrednosti za nadzor mejne vrednosti ali praga ukrepanja. Ti vzorci se lahko skupaj uporabijo za izračun vrednosti BEQ v preskusnih vzorcih (glej točko 7.1.2.2). |
7.3 Določitev izločitvenih vrednosti
Določi se razmerje med bioanalitskimi rezultati v BEQ in rezultati potrditvene metode v TEQ, na primer z umeritvenimi preskusi v matrici, ki vključujejo referenčne vzorce z dodatkom pri 0, 0,5-kratni, 1-kratni in 2-kratni mejni vrednosti s šestimi ponovitvami na vsaki ravni (n = 24). Na podlagi tega razmerja se lahko ocenijo korekcijski faktorji (za slepe vrednosti in izkoristek), vendar se preverijo v skladu s točko 7.2.2.
Izločitvene vrednosti se določijo za sprejetje odločitev o skladnosti vzorca z mejnimi vrednostmi ali za nadzor pragov ukrepanja, če je to relevantno, glede na zadevne mejne vrednosti ali prag določanja, določen posebej za PCDD/PCDF in za dioksinom podobne PCB ali za vsoto PCDD/PCDF in dioksinom podobnih PCB. Prikazuje jih spodnja končna točka porazdelitve bioanalitskih rezultatov (s popravkom slepih vrednosti in izkoristka), ki ustreza odločitveni meji potrditvene metode na podlagi 95-odstotne stopnje zaupanja, kar pomeni, da je delež lažno skladnih rezultatov < 5 %, in na podlagi RSDR < 25 %. Odločitvena meja potrditvene metode je mejna vrednost, pri čemer se upošteva merilna negotovost.
Izločitvena vrednost (v BEQ) se lahko izračuna po enem od pristopov iz točk 7.3.1, 7.3.2 in 7.3.3 (glej sliko 1).
|
7.3.1 |
Uporaba spodnjega razpona 95-odstotnega napovednega intervala pri odločitveni meji potrditvene metode:
pri čemer je:
|


|
7.3.2 |
Izračun iz bioanalitskih rezultatov (s popravkom slepih vrednosti in izkoristka) večkratnih analiz vzorcev (n ≥ 6), kontaminiranih pri odločitveni meji potrditvene metode, kot spodnja končna točka porazdelitve podatkov pri ustrezni srednji vrednosti BEQ: Izločitvena vrednost = BEQDL – 1,64 × SDR pri čemer je:
|
|
7.3.3 |
Izračun kot srednja vrednost bioanalitskih rezultatov (v BEQ, s popravkom slepih vrednosti in izkoristka) iz večkratnih analiz vzorcev (n ≥ 6), kontaminiranih pri 2/3 mejne vrednosti ali praga ukrepanja, na podlagi ugotovitve, da bo ta vrednost blizu izločitvene vrednosti iz točke 7.3.1 ali 7.3.2:  Slika 1:
Slika 1:
Izračun izločitvenih vrednosti na podlagi 95-odstotne stopnje zaupanja, kar pomeni, da je delež lažno skladnih rezultatov < 5 %, in na podlagi RSDR < 25 %: 1. iz spodnjega razpona 95-odstotnega napovednega intervala pri odločitveni meji potrditvene metode; 2. iz večkratnih analiz vzorcev (n ≥ 6), kontaminiranih pri odločitveni meji potrditvene metode, kot spodnja končna točka porazdelitve podatkov (na sliki prikazana s krivuljo v obliki zvonca) pri ustrezni srednji vrednosti BEQ. |
|
7.3.4 |
Omejitve izločitvenih vrednosti Izločitvene vrednosti na podlagi BEQ, izračunane iz RSDR, doseženega med validacijo z uporabo omejenega števila vzorcev z različnimi vzorci matric/kongenerov, so lahko višje od mejnih vrednosti ali pragov odločanja na podlagi TEQ zaradi večje natančnosti, kot jo je mogoče doseči rutinsko, kadar je treba nadzirati neznani spekter mogočih vzorcev kongenerov. V takih primerih se izločitvene vrednosti izračunajo iz RSDR = 25 % ali pa se da prednost dvema tretjinama mejne vrednosti ali praga določanja. |
7.4 Značilnosti zmogljivosti metode
|
7.4.1 |
Ker pri bioanalitskih metodah ni mogoče uporabiti internih standardov, se preskusi ponovljivosti bioanalitskih metod izvajajo za pridobitev informacij o standardnem odmiku znotraj serij poskusov in med njimi. Ponovljivost je pod 20 %, interna laboratorijska obnovljivost pa pod 25 %. Pri tem se izhaja iz izračunanih vrednosti v BEQ po popravku slepih vrednosti in izkoristka. |
|
7.4.2 |
V postopku validacije se dokaže, da preskus razlikuje med slepim vzorcem in vsebnostjo pri izločitveni vrednosti ter omogoča ugotovitev vzorcev nad ustrezno izločitveno vrednostjo (glej točko 7.1.2). |
|
7.4.3 |
Opredelijo se ciljne spojine, možni moteči vplivi in mejne sprejemljive slepe vrednosti. |
|
7.4.4 |
Odstotni standardni odmik v odzivu ali koncentracija, izračunana iz odziva (mogoče le v delovnem območju), pri trikratnem določanju ekstrakta vzorca ne presega 15 %. |
|
7.4.5 |
Nepopravljeni rezultati referenčnih vzorcev, izraženi v BEQ (slepa vrednost in mejna vrednost ali prag določanja), se uporabijo za oceno zmogljivosti bioanalitske metode v stalnem časovnem presledku. |
|
7.4.6 |
Za postopke s slepimi vzorci in vsako vrsto referenčnega vzorca se vodijo in preverjajo kontrolne karte, s čimer se zagotovi, da je zmogljivost analiz v skladu z zahtevami, zlasti pri postopkih s slepimi vzorci glede zahtevane najmanjše razlike do spodnjega dela delovnega območja in pri referenčnih vzorcih glede interne laboratorijske obnovljivosti. Postopki s slepimi vzorci se nadzorujejo tako, da se je mogoče izogibati lažno skladnim rezultatom pri odštevanju. |
|
7.4.7 |
Rezultati sumljivih vzorcev, pridobljeni s potrditvenimi metodami, in 2 % do 10 % skladnih vzorcev (najmanj 20 vzorcev na matrico) se zberejo in uporabijo za oceno zmogljivosti presejalne metode ter razmerja med BEQ in TEQ. Ta zbirka podatkov se lahko uporabi za ponovno oceno izločitvenih vrednosti, ki se uporabljajo za rutinske vzorce pri validiranih matricah. |
|
7.4.8 |
Zmogljivost metode se lahko dokaže tudi s sodelovanjem v primerjalnih preskusih. Rezultati iz vzorcev, analiziranih s primerjalnimi preskusi, ki zajemajo območje koncentracije do na primer dvakratne mejne vrednosti, se lahko vključijo v oceno deleža lažno skladnih rezultatov, če laboratorij lahko dokaže svojo zmogljivost. Vzorci zajemajo najpogostejše vzorce kongenerov, ki predstavljajo različne vire. |
|
7.4.9 |
Med incidenti se lahko znova ocenijo izločitvene vrednosti, pri čemer se upoštevajo posebni vzorci matric in kongenerov v zadevnem incidentu. |
8. Poročanje o rezultatih
8.1 Potrditvene metode
|
8.1.1 |
Kolikor uporabljeni analitski postopki to omogočajo, analitski rezultati vsebujejo vrednosti posameznih PCDD/PCDF in kongenerov dioksinom podobnih PCB, sporočijo pa se na spodnji meji, zgornji meji in srednji meji, da se v poročilo o rezultatih vključi kar največ podatkov in tako omogoči razlaga rezultatov glede na posebne zahteve. |
|
8.1.2 |
Poročilo vključuje tudi metodo, uporabljeno za ekstrakcijo PCDD/PCDF in dioksinom podobnih PCB. |
|
8.1.3 |
Izkoristki posameznih internih standardov so na voljo, če so zunaj območja iz točke 6.2.5, če je presežena mejna vrednost (v tem primeru izkoristki za eno od dveh dvakratnih analiz) in na zahtevo tudi v drugih primerih. |
|
8.1.4 |
Ker je treba pri odločanju o skladnosti vzorca upoštevati merilno negotovost, je ta parameter na voljo. Rezultat analize je zato izražen kot x +/– U, pri čemer je x rezultat analize, U pa razširjena merilna negotovost, kar pri faktorju zajetja 2 pomeni približno 95-odstotno stopnjo zaupanja. Pri ločenem preskušanju za določitev PCDD/PCDF in dioksinom podobnih PCB je treba vsoto ocenjene razširjene negotovosti ločenih rezultatov analize vsebnosti PCDD/PCDF in dioksinu podobnih PCB uporabiti za vsoto PCDD/PCDF in dioksinom podobnih PCB. |
|
8.1.5 |
Če se merilna negotovost upošteva z uporabo CCα (kot je opisana v točki 2.2 poglavja I tega dela B), se navede ta parameter. |
|
8.1.6 |
Rezultati analize mejne vrednosti se izrazijo z istimi enotami in zaokrožijo (vsaj) na enako število decimalnih mest, kakor je določeno v Direktivi 2002/32/ES. |
8.2 Bioanalitske presejalne metode
|
8.2.1 |
Rezultat presejanja se navede kot „skladen“ ali „s sumom na neskladnost“ („sumljiv“). |
|
8.2.2 |
Poleg tega se lahko navede rezultat za PCDD/PCDF in/ali dioksinom podobne PCB, izražen v BEQ in ne v TEQ. |
|
8.2.3 |
Za vzorce, pri katerih je odziv pod pragom poročanja, se navede, da so „pod pragom poročanja“. |
|
8.2.4 |
Za vsako vrsto matrice vzorca se v poročilu navede mejna vrednost ali prag ukrepanja, na katerem temelji ocena. |
|
8.2.5 |
V poročilu se navedejo vrsta uporabljenega preskusa, osnovno načelo preskusa in vrsta umerjanja. |
|
8.2.6 |
Poročilo vključuje tudi metodo, uporabljeno za ekstrakcijo PCDD/PCDF in dioksinom podobnih PCB. |
|
8.2.7 |
Kadar se sumi, da vzorci niso skladni, mora biti v poročilu naveden ukrep, ki se sprejme. Koncentracijo PCDD/F ter vsoto PCDD/F in dioksinom podobnih PCB v navedenih vzorcih z zvišanimi vsebnostmi je treba določiti/potrditi s potrditveno metodo. |
POGLAVJE III
Priprava vzorca in zahteve za analitske metode za uradni nadzor vsebnosti dioksinom nepodobnih PCB (PCB # 28, 52, 101, 138, 153, 180)
1. Področje uporabe
Zahteve iz tega poglavja se uporabijo, kadar se krma analizira za uradni nadzor vsebnosti dioksinom nepodobnih polikloriranih bifenilov (dioksinom nepodobnih PCB) in za regulativne namene.
2. Metode ugotavljanja, ki se uporabljajo
Plinska kromatografija/detekcija zajetja elektronov (GC-ECD), GC-LRMS, GC-MS/MS, GC-HRMS ali enakovredne metode.
3. Določitev in potrditev predpisanih analitov
|
3.1 |
Relativni retencijski čas glede na interne standarde ali referenčne standarde (sprejemljivi odmik +/– 0,25 %). |
|
3.2 |
Plinsko kromatografsko ločevanje vseh šestih indikatorskih PCB (PCB 28, PCB 52, PCB 101, PCB 138, PCB 153 in PCB 180) od motečih snovi, zlasti sočasno eluiranih PCB, še posebej če so vzorci v območju zakonsko dovoljenih mej in je treba potrditi neskladnost. [Kongeneri, za katere je pogosto ugotovljeno, da se sočasno eluirajo, so na primer PCB 28/31, PCB 52/69 in PCB 138/163/164. Pri GC-MS se upoštevajo tudi mogoči moteči vplivi fragmentov višjih kloriranih kongenerov.] |
|
3.3 |
Zahteve za tehnike GC-MS Spremljanje najmanj: (a) dveh specifičnih ionov za HRMS; (b) dveh specifičnih ionov z m/z > 200 ali treh specifičnih ionov z m/z > 100 za LRMS; (c) enega starševskega in dveh hčerinskih produktnih ionov za MS-MS. Največja dovoljena odstopanja za odzive izbranih masnih fragmentov: relativni odmik intenzitete izbranih masnih fragmentov od teoretičnega odziva ali umeritveni standard za izbrani ciljni ion (to je ion z najmočnejšim odzivom, ki se spremlja) in potrditvene ione:
|
||||||||||||||||||
|
3.4 |
Zahteve za tehnike GC-ECD Rezultati, ki presegajo dopustne meje, se potrdijo z dvema kolonama GC s stacionarnimi fazami različne polarnosti. |
4. Dokazovanje zmogljivosti metode
Zmogljivost metode se validira v območju mejne vrednosti (0,5-kratna do 2-kratna mejna vrednost) s sprejemljivim koeficientom variacije za ponovljene analize (glej zahteve za srednjo natančnost v točki 9).
5. Meja določanja
Slepe vrednosti ne presegajo 30-odstotne ravni kontaminacije, ki ustreza mejni vrednosti ( 25 ).
6. Zagotavljanje kakovosti
Redne slepe kontrole, analize vzorcev z dodatkom, analize kontrolnih vzorcev, sodelovanje v medlaboratorijskih študijah z različnimi matricami vzorcev.
7. Nadzor izkoristka
|
7.1 |
Uporabljajo se primerni interni standardi s fizikalno-kemijskimi lastnostmi, primerljivimi s predpisanimi analiti. |
|
7.2 |
Dodajanje internih standardov: dodajanje vzorcem proizvodov (pred ekstrakcijo in čiščenjem). |
|
7.3 |
Zahteve za metode, pri katerih se uporablja vseh šest izotopsko označenih kongenerov indikatorskih PCB: (a) rezultati se popravijo glede izkoristka internih standardov; (b) izkoristek izotopsko označenih internih standardov je med 50 in 120 %; (c) sprejemljivi so manjši ali večji izkoristki za posamezne kongenere z manj kot 10-odstotnim prispevkom k vsoti šestih indikatorskih PCB. |
|
7.4 |
Zahteve za metode, pri katerih se ne uporablja vseh šest izotopsko označenih internih standardov ali drugih internih standardov: (a) izkoristek internih standardov se nadzoruje za vsak vzorec; (b) izkoristek internih standardov je med 60 in 120 %; (c) rezultati se popravijo glede izkoristka internih standardov. |
|
7.5 |
Izkoristek neoznačenih kongenerov se preveri z analizo vzorcev z dodatkom ali kontrolnih vzorcev s koncentracijami v območju mejne vrednosti. Izkoristek teh kongenerov se šteje za sprejemljiv, če je med 70 in 120 %. |
8. Zahteve za laboratorije
V skladu z določbami Uredbe (ES) št. 882/2004 laboratorije akreditirajo priznani organi, ki delujejo v skladu z zahtevami ISO Guide 58, s čimer je zagotovljeno, da izvajajo analitsko zagotavljanje kakovosti. Laboratoriji se akreditirajo po standardu EN ISO/IEC 17025.
9. Značilnosti zmogljivosti metode: merila za vsoto šestih indikatorskih PCB pri mejni vrednosti
|
Pravilnost |
– 30 % do +30 % |
|
Srednja natančnost (RSD %) |
≤ 20 % |
|
Razlika med izračunom na zgornji in spodnji meji |
≤ 20 % |
10. Poročilo o rezultatih
|
10.1 |
Če uporabljeni analitski postopki to omogočajo, analitski rezultati vsebujejo vrednosti posameznih kongenerov PCB in se navajajo na spodnji meji, zgornji meji in srednji meji, da se v poročilo o rezultatih vključi kar največ podatkov in tako omogoči razlago rezultatov glede na posebne zahteve. |
|
10.2 |
Poročilo vključuje metodo, uporabljeno za ekstrakcijo PCB in maščob. |
|
10.3 |
Izkoristki posameznih notranjih standardov so na voljo, če so zunaj območja iz točke 7, če je presežena mejna vrednost in na zahtevo tudi v drugih primerih. |
|
10.4 |
Ker je treba pri odločanju o skladnosti vzorca upoštevati merilno negotovost, mora biti na voljo tudi ta parameter. Rezultat analize je zato izražen kot x +/– U, pri čemer je x rezultat analize, U pa razširjena merilna negotovost, kar pri faktorju zajetja 2 pomeni približno 95-odstotno stopnjo zaupanja. |
|
10.5 |
Če se merilna negotovost upošteva z uporabo CCα (kot je opisana v točki 2.1 poglavja I), se navede ta parameter. |
|
10.6 |
Rezultati se izrazijo z istimi enotami in zaokrožijo (vsaj) na enako število decimalnih mest, kakor je določeno v Direktivi 2002/32/ES. |
PRILOGA VI
ANALITSKE METODE ZA DOLOČANJE SESTAVIN ŽIVALSKEGA IZVORA ZA URADNI NADZOR KRME
1. NAMEN IN PODROČJE UPORABE
Sestavine živalskega izvora v krmi se določajo s svetlobno mikroskopijo ali verižno reakcijo s polimerazo (PCR) v skladu z določbami iz te priloge.
Ti metodi omogočata ugotavljanje prisotnosti sestavin živalskega izvora v posamičnih krmilih in krmnih mešanicah, ne pa tudi njihove natančne količine v posamičnih krmilih in krmnih mešanicah. Meja zaznavnosti pri obeh metodah je pod 0,1 % (m/m).
Metoda PCR omogoča določanje taksonomske skupine sestavin živalskega izvora, prisotnih v posamičnih krmilih in krmnih mešanicah.
Ti metodi se uporabljata za nadzor izvajanja prepovedi iz člena 7(1) in Priloge IV k Uredbi (ES) št. 999/2001 ter iz člena 11(1) Uredbe (ES) št. 1069/2009.
Glede na vrsto krme, ki se testira, se ti metodi lahko uporabita v okviru enega samega delovnega protokola bodisi samostojno ali skupaj v skladu s standardnimi operativnimi postopki (SOP), ki jih je določil in na svoji spletni strani ( 26 ) objavil referenčni laboratorij EU za živalske beljakovine v krmi (EURL-AP).
2. METODE
2.1 Svetlobna mikroskopija
2.1.1 Načelo
Sestavine živalskega izvora, ki so lahko prisotne v posamičnih krmilih in krmnih mešanicah, poslanih v analizo, se določijo na podlagi tipičnih, mikroskopsko prepoznavnih značilnosti, kot so mišična vlakna in drugi mesni delci, hrustanec, kost, roževina, dlake, ščetine, kri, perje, jajčne lupine, ribje kosti in luskine).
2.1.2 Reagenti in oprema
2.1.2.1 Reagenti
2.1.2.1.1 Sredstvo za koncentriranje
2.1.2.1.1.1 Tetrakloroetilen (specifična teža 1,62)
2.1.2.1.2 Reagent za obarvanje
|
2.1.2.1.2.1 |
Raztopina rdečega alizarina (2,5 ml 1M klorovodikove kisline se raztopi v 100 ml vode in tej raztopini se doda 200 mg rdečega alizarina.) |
2.1.2.1.3 Sredstva za pripravo preparatov
2.1.2.1.3.1 Lug (NaOH 2,5 % m/v ali KOH 2,5 % m/v)
2.1.2.1.3.2 Glicerol (nerazredčen, viskoznost: 1 490 cP)
2.1.2.1.3.3 Norland Optical Adhesive 65® (viskoznost: 1 200 cP) ali smola z enakovrednimi lastnostmi za pripravo trajnih preparatov
2.1.2.1.4 Sredstva za pripravo preparatov z lastnostmi obarvanja
|
2.1.2.1.4.1 |
Lugolova raztopina (2 g kalijevega jodida se raztopi v 100 ml vode in med pogostim stresanjem se doda 1 g joda) |
|
2.1.2.1.4.2 |
Cistinski reagent (2 g svinčevega acetata, 10 g NaOH/100 ml vode) |
|
2.1.2.1.4.3 |
Fehlingov reagent (pripravi se pred uporabo iz enakih delov (1/1) osnovnih raztopin A in B. Raztopina A: 6,9 g bakrovega (II) sulfata pentahidrata se raztopi v 100 ml vode. Raztopina B: 34,6 g kalij-natrijevega tartrata tetrahidrata in 12 g NaOH se raztopi v 100 ml vode) |
|
2.1.2.1.4.4 |
Tetrametilbenzidin/vodikov peroksid (1 g 3,3’,5,5’-tetrametilbenzidina (TMB) se raztopi v 100 ml ledocetne kisline in 150 ml vode. Pred uporabo se štirje deli te raztopine TMB zmešajo z enim delom 3 % vodikovega peroksida.) |
2.1.2.1.5 Sredstva za izpiranje
2.1.2.1.5.1 Etanol ≥ 96 % (tehnično čist)
2.1.2.1.5.2 Aceton (tehnično čist)
2.1.2.1.6 Reagent za beljenje
2.1.2.1.6.1 Komercialna raztopina natrijevega hipoklorita (9–14 % aktivnega klora)
2.1.2.2 Oprema
2.1.2.2.1 Analitska tehtnica, natančna na 0,001 g
2.1.2.2.2 Oprema za mletje: mlin ali terilnica
2.1.2.2.3 Siti z mrežico s kvadratnimi odprtinami širine 0,25 mm in 1 mm
|
2.1.2.2.4 |
Konični stekleni lij ločnik s prostornino 250 ml in teflonskim petelinčkom ali petelinčkom iz brušenega stekla na dnu konusa. Premer odprtine petelinčka mora biti ≥ 4 mm. Namesto tega se lahko uporabi konična čaša za usedlino, če laboratorij dokaže, da je meja zaznavnosti enakovredna tisti pri uporabi koničnega steklenega lija ločnika. 
|
|
2.1.2.2.5 |
Stereomikroskop, ki zajema vsaj končno območje povečave 6,5× do 40×. |
|
2.1.2.2.6 |
Sestavljeni mikroskop, ki zajema vsaj končno območje povečave 100× do 400× in ima svetlo polje za presevno osvetlitev. Dodatno se lahko uporabita polarizirana svetloba in diferenčni interferenčni kontrast. |
|
2.1.2.2.7 |
Standardna laboratorijska steklovina |
|
2.1.2.2.8 |
Oprema za pripravo preparata: klasična mikroskopska objektna stekelca, stekelca z vdolbino, krovna stekelca (20 × 20 mm), pincete, fina lopatica |
2.1.3 Vzorčenje in priprava vzorcev
2.1.3.1 Vzorčenje
Uporabi se reprezentativni vzorec, ki je bil odvzet v skladu z določbami iz Priloge I.
2.1.3.2 Previdnostni ukrepi
Za preprečitev navzkrižnega onesnaženja je treba laboratorijsko opremo za večkratno uporabo temeljito očistiti pred uporabo. Dele lija ločnika je treba pred čiščenjem razstaviti. Sestavine lija ločnika in vso drugo steklovino je treba najprej očistiti ročno in nato še v pomivalnem stroju. Sita je treba očistiti s krtačo s trdimi sintetičnimi ščetinami. Končno čiščenje sit z acetonom in stisnjenim zrakom se priporoča po sejanju mastnega materiala, kot je ribja moka.
2.1.3.3 Priprava vzorcev, razen vzorcev, ki vsebujejo maščobe ali olja
|
2.1.3.3.1 |
Sušenje vzorca: vzorci z vsebnostjo vlage več kot 14 % se pred obdelavo posušijo. |
|
2.1.3.3.2 |
Predhodno presejanje vzorca: priporoča se, da se krma v briketih in zrnu predhodno preseje na 1 mm, nato pa se dobljeni frakciji analizirata kot ločena vzorca. |
|
2.1.3.3.3 |
Podvzorčenje in mletje: iz najmanj 50 g vzorca se pripravi podvzorec za analizo, ki se nato zmelje. |
|
2.1.3.3.4 |
Ekstrakcija in priprava usedline: 10-gramski vzorec (na 0,01 g natančno) iz zmletega podvzorca se prenese v lij ločnik ali konično čašo za usedlino in se mu doda 50 ml tetrakloroetilena. Vzorec, ki se prenese v lij, tehta največ 3 g, kadar vzorec tvorijo ribja moka ali drugi čisti živalski proizvodi, mineralne sestavine ali premiksi, ki tvorijo več kot 10 % usedline. Mešanica naj se močno stresa vsaj 30 sekund in nato previdno doda še najmanj 50 ml tetrakloroetilena med spiranjem notranje površine lija za odstranitev vseh pritrjenih delcev. Tako dobljena mešanica se pusti stati vsaj 5 minut, nato se usedlino loči z odprtjem petelinčka. Če se uporabi konična čaša za usedlino, se mešanica močno meša vsaj 15 sekund in vsi delci, ki se držijo čaše, se skrbno sperejo po notranji površini z najmanj 10 ml čistega tetrakloroetilena. Mešanica se pusti stati 3 minute in nato se ponovno meša 15 sekund, vsi delci, ki se držijo čaše, pa se skrbno sperejo po notranji površini z najmanj 10 ml čistega tetrakloroetilena. Tako dobljena mešanica se pusti stati vsaj 5 minut, nato se tekoča frakcija pazljivo dekantira in zavrže, da se usedlina v celoti ohrani. Usedlina se posuši in nato stehta (na 0,001 g natančno). Če več kot 5 % usedline sestavljajo delci velikosti > 0,50 mm, se preseje skozi sito 0,25 mm, tako pridobljeni frakciji se nato analizirata. |
|
2.1.3.3.5 |
Ekstrakcija in priprava flotata: po pridobitvi usedline z opisano metodo ostaneta v liju ločniku dve fazi: tekoča faza, ki jo tvori tetrakloroetilen, in trdna faza, ki jo tvori plavajoči material. Ta trdna faza je flotat, ki se izolira tako, da se tetrakloroetilen v celoti odlije iz lija z odprtjem petelinčka. Z obrnitvijo lija ločnika na glavo se flotat prenese v veliko petrijevko in posuši na zraku v digestoriju. Če več kot 5 % flotata sestavljajo delci velikosti > 0,50 mm, se preseje skozi sito 0,25 mm, tako pridobljeni frakciji se nato analizirata. |
|
2.1.3.3.6 |
Priprava osnovnega materiala: pripravi se vzorec iz najmanj 5 g zmletega podvzorca. Če več kot 5 % materiala sestavljajo delci velikosti > 0,50 mm, se preseje skozi sito 0,25 mm, tako pridobljeni frakciji se nato analizirata. |
2.1.3.4 Priprava vzorcev, ki vsebujejo maščobe ali olja
Za pripravo vzorcev, ki vsebujejo maščobe ali olja, se uporabi naslednji protokol:
— če je maščoba trdna, se segreje v pečici, dokler ne postane tekoča,
— 40 ml maščobe ali olja z dna vzorca se odpipetira v centrifugirko,
— centrifugira se 10 minut pri 4 000 obr./min.,
— če je maščoba po centrifugiranju trdna, se segreje v pečici, dokler ne postane tekoča,
— ponovno se centrifugira 5 minut pri 4 000 obr./min.,
— z majhno žličko ali lopatico se polovica dekantirane nečistoče prenese na mikroskopska objektna stekelca za pregled, kot sredstvo za pripravo preparata pa se priporoča glicerol,
— preostale nečistoče se uporabijo za pripravo usedline, kot je opisano v točki 2.1.3.3.
2.1.3.5 Uporaba reagentov za obarvanje
Da se omogoči pravilna identifikacija sestavin živalskega izvora, lahko izvajalec med pripravo vzorca uporabi reagente za obarvanje v skladu s smernicami, ki jih je izdal in na svoji spletni strani objavil EURL-AP.
Če se za barvanje usedline uporabi raztopina rdečega alizarina, se uporabi naslednji protokol:
— posušena usedlina se prenese v stekleno epruveto in dvakrat spere s približno 5 ml etanola (vsakič se uporabi vorteks mešalnik za 30 sekund, topilo se pusti stati približno 1 minuto in 30 sekund ter se nato odtoči),
— usedlina se pobeli z dodatkom najmanj 1 ml raztopine natrijevega hipoklorita. Čas reakcije je 10 minut. Epruveta se napolni z vodo, usedlina se pusti stati 2 do 3 minute, voda in suspendirani delci pa se previdno odtočijo,
— usedlina se dvakrat spere z 10 ml vode (vsakič se za 30 sekund uporabi vorteks mešalnik, pusti se stati in odtoči se voda),
— doda se 2 do 10 kapljic raztopine rdečega alizarina in mešanica se premeša v vorteks mešalniku. Čas reakcije je 30 sekund, obarvana usedlina se dvakrat spere s približno 5 ml etanola, nato pa enkrat z acetonom (vsakič se uporabi vorteks mešalnik za 30 sekund, topilo se pusti stati približno 1 minuto ter se nato odtoči),
— obarvana usedlina se posuši.
2.1.4 Mikroskopski pregled
2.1.4.1 Priprava preparata
Mikroskopski preparat se pripravi iz sedimenta in, odvisno od izbire izvajalca, flotata ali osnovnega materiala. Če je bilo med pripravo vzorca uporabljeno sejanje, se pripravita obe tako pridobljeni frakciji (fina in groba). Testni vzorci frakcij, ki se nanesejo na objektna stekelca, so reprezentativni za celotno frakcijo.
Pripraviti je treba zadostno število preparatov, da se lahko zagotovi izvedba celotnega protokola pregleda, kot je določen v točki 2.1.4.2.
Mikroskopski preparati se pripravijo s primernim sredstvom za pripravo preparata v skladu s standardnimi operativnimi postopki, ki jih je določil in na svoji spletni strani objavil EURL-AP. Preparati se pokrijejo s krovnimi stekelci.
2.1.4.2 Protokol pregleda za določanje živalskih delcev v krmnih mešanicah in posamičnih krmilih
Pripravljeni mikroskopski preparati se pregledajo v skladu s protokolom pregleda, prikazanim v diagramu 1 za krmne mešanice in posamična krmila, razen za čisto ribjo moko, za katero je prikazan v diagramu 2.
Mikroskopski preparat sedimenta in, odvisno od izbire izvajalca, flotata ali osnovnega materiala se pregleda s sestavljenim mikroskopom. Za grobe frakcije se lahko poleg sestavljenega mikroskopa uporabi stereomikroskop. Vsak preparat se pregleda v celoti pri različnih povečavah.
Pri vsakem koraku protokola pregleda je treba pregledati določeno minimalno število preparatov, razen če celotna frakcija materiala ne omogoča, da se pripravi zahtevano število preparatov. Pri vsakem določanju se pregleda največ šest preparatov.
Za lažjo identifikacijo narave in izvora delcev lahko izvajalec uporabi podporne elemente, na primer sisteme za podporo pri odločanju, slikovne knjižnice in referenčne vzorce.


2.1.4.3 Število določanj
Če pri prvem določanju, izvedenem v skladu s protokolom iz diagrama 1 ali diagrama 2, ni ugotovljen noben živalski delec danega izvora (tj. od kopenskih živali ali rib), ni potrebno dodatno določanje, rezultate analize pa je treba sporočiti z uporabo terminologije iz točke 2.1.5.1.
Če je bilo pri prvem določanju, izvedenem v skladu s protokolom iz diagrama 1 ali diagrama 2, skupno ugotovljenih od 1 do 5 živalskih delcev danega izvora (tj. od kopenskih živali ali rib), se izvede drugo določanje na novem 50-gramskem podvzorcu. Če je bilo pri drugem določanju ugotovljenih od 0 do 5 živalskih delcev danega izvora, je treba rezultat analize sporočiti z uporabo terminologije iz točke 2.1.5.2., sicer se izvede tretje določanje na novem 50-gramskem podvzorcu. Če pa je po prvem in drugem določanju vsota ugotovljenih delcev danega izvora višja od 15, dodatno določanje ni potrebno, rezultate analize pa je treba neposredno sporočiti z uporabo terminologije iz točke 2.1.5.3. Če je po tretjem določanju vsota ugotovljenih živalskih delcev danega izvora višja od 15, je treba rezultate analize sporočiti z uporabo terminologije iz točke 2.1.5.3. Sicer je treba rezultate analize sporočiti z uporabo terminologije iz točke 2.1.5.2.
Če se pri prvem določanju, izvedenem v skladu s protokolom pregleda iz diagrama 1 ali diagrama 2, ugotovi več kot 5 živalskih delcev danega izvora (tj. od kopenskih živali ali rib), je treba rezultat analize sporočiti z uporabo terminologije iz točke 2.1.5.3.
2.1.5 Prikaz rezultatov
V poročilu o rezultatih laboratoriji navedejo, na kateri vrsti materiala je bila opravljena analiza (usedlina, flotat ali osnovni material) in koliko določanj je bilo izvedenih.
Laboratorijsko poročilo vsebuje vsaj informacije o prisotnosti sestavin, ki izvirajo od kopenskih živali in rib.
O različnih primerih se poroča na naslednje načine:
|
2.1.5.1 |
Noben ugotovljen živalski delec danega izvora: — kolikor je bilo razvidno iz pregleda s svetlobnim mikroskopom, v predloženem vzorcu niso bili ugotovljeni nobeni delci, ki izvirajo od kopenskih živali, — kolikor je bilo razvidno iz pregleda s svetlobnim mikroskopom, v predloženem vzorcu niso bili ugotovljeni nobeni delci, ki izvirajo od rib. |
|
2.1.5.2 |
V povprečju ugotovljenih od 1 do 5 živalskih delcev danega izvora: — kolikor je bilo razvidno iz pregleda s svetlobnim mikroskopom, v predloženem vzorcu v povprečju na določanje ni bilo ugotovljenih več kot pet delcev, ki izvirajo od kopenskih živali. Delci so bili opredeljeni kot … [kost, hrustanec, mišičevje, dlaka, roževina…]. Ta nizka stopnja prisotnosti, ki je pod mejo določanja z mikroskopsko metodo, pomeni, da ni mogoče izključiti tveganja za lažno pozitiven rezultat. Ali, če je primerno, — kolikor je bilo razvidno iz pregleda s svetlobnim mikroskopom, v predloženem vzorcu v povprečju na določanje ni bilo ugotovljenih več kot pet delcev, ki izvirajo od rib. Delci so bili opredeljeni kot … [ribja kost, ribja luskina, hrustanec, mišičevje, otolit, škrga…]. Ta nizka stopnja prisotnosti, ki je pod mejo določanja z mikroskopsko metodo, pomeni, da ni mogoče izključiti tveganja za lažno pozitiven rezultat. V primeru predhodnega presejanja vzorca mora biti v laboratorijskem poročilu navedeno, v kateri frakciji (presejana frakcija, frakcija iz briketov ali zrna) so bili ugotovljeni živalski delci, če je določitev živalskih delcev samo v presejani frakciji, je to lahko znak okoljskega onesnaženja. |
|
2.1.5.3 |
V povprečju ugotovljenih več kot 5 živalskih delcev danega izvora — kolikor je bilo razvidno iz pregleda s svetlobnim mikroskopom, je bilo v predloženem vzorcu v povprečju na določanje ugotovljenih več kot pet delcev, ki izvirajo od kopenskih živali. Delci so bili opredeljeni kot … [kost, hrustanec, mišičevje, dlaka, roževina…]. Ali, če je primerno, — kolikor je bilo razvidno iz pregleda s svetlobnim mikroskopom, je bilo v predloženem vzorcu v povprečju na določanje ugotovljenih več kot pet delcev, ki izvirajo od rib. Delci so bili opredeljeni kot … [ribja kost, ribja luskina, hrustanec, mišičevje, otolit, škrga…]. V primeru predhodnega presejanja vzorca mora biti v laboratorijskem poročilu navedeno, v kateri frakciji (presejana frakcija, frakcija iz briketov ali zrna) so bili ugotovljeni živalski delci, če je določitev živalskih delcev samo v presejani frakciji, je to lahko znak okoljskega onesnaženja. |
2.2 PCR
2.2.1 Načelo
Fragmenti dezoksiribonukleinske kisline (DNK) živalskega izvora, ki so lahko prisotni v posamičnih krmilih in krmnih mešanicah, se ugotavljajo z metodo PCR (tehnika za pomnoževanje genskega materiala), s katero se ugotavljajo posebne sekvence DNK, značilne za vrsto.
Pri metodi PCR se najprej opravi ekstrakcija DNK. Pomnoževalni del metode se nato izvede na tako dobljenem ekstraktu DNK, da se ugotovi živalska vrsta, na katero se pregleduje.
2.2.2 Reagenti in oprema
2.2.2.1 Reagenti
2.2.2.1.1 Reagenti za ekstrakcijo DNK
Uporabljajo se le reagenti, ki jih je odobril in na svoji spletni strani objavil EURL-AP.
2.2.2.1.2 Reagenti za pomnožitev genskega materiala
2.2.2.1.2.1 Začetni oligonukleotidi in sonde
Uporabljajo se samo začetni oligonukleotidi in sonde z zaporedji oligonukleotidov, ki jih je validiral EURL-AP ( 27 ).
2.2.2.1.2.2 Osnovna zmes
Uporabljajo se samo raztopine osnovne zmesi, ki ne vsebujejo reagentov, ki lahko povzročijo lažne rezultate zaradi prisotnosti živalske DNK ( 28 ).
2.2.2.1.2.3 Reagenti za dekontaminacijo
2.2.2.1.2.3.1 Raztopina klorovodikove kisline (0,1M)
2.2.2.1.2.3.2 Belila (raztopina natrijevega hipoklorita z 0,15 % aktivnega klora)
2.2.2.1.2.3.3 Nejedki reagenti za dekontaminacijo dragih naprav, kot so analitske tehtnice (npr. DNA EraseTM od MP Biomedicals).
2.2.2.2 Oprema
2.2.2.2.1 Analitska tehtnica, natančna na 0,001 g
2.2.2.2.2 Oprema za mletje
2.2.2.2.3 Naprava za ciklično termostatiranje, ki omogoča PCR v realnem času
2.2.2.2.4 Mikrocentrifuga za mikrocentrifugirke
2.2.2.2.5 Sklop mikropipet, ki omogočajo pipetiranje od 1 μl do 1 000 μl
|
2.2.2.2.6 |
Standardna plastična oprema za molekularno biologijo: mikrocentrifugirke, plastični nastavki s filtri za mikropipete, plošče, primerne za napravo za ciklično termostatiranje. |
|
2.2.2.2.7 |
Zamrzovalniki za shranjevanje vzorcev in reagentov |
2.2.3 Vzorčenje in priprava vzorcev
2.2.3.1 Vzorčenje
Uporabi se reprezentativni vzorec, ki je bil odvzet v skladu z določbami iz Priloge I.
2.2.3.2 Priprava vzorca
Pri pripravi laboratorijskih vzorcev do ekstrakcije DNK je treba upoštevati zahteve iz Priloge II. Iz najmanj 50 g vzorca se pripravi podvzorec za analizo, ki se nato zmelje.
V skladu s standardom ISO 24276 se priprava vzorca izvede v drugem prostoru kot ekstrakcija DNK in pomnožitev genskega materiala.
Pripravita se dva testna vzorca z najmanj 100 mg.
2.2.4 Ekstrakcija DNK
Ekstrakcija DNK se opravi na vsakem pripravljenem testnem vzorcu v skladu s standardnimi operativnim postopki, ki jih je določil in na svoji spletni strani objavil EURL-AP.
Za vsako serijo ekstrakcije DNK se pripravita dva kontrolna vzorca, kot je opisano v standardu ISO 24276:
— slepi kontrolni vzorec na stopnji ekstrakcije DNK,
— pozitivni kontrolni vzorec na stopnji ekstrakcije DNK.
2.2.5 Pomnožitev genskega materiala
Genski material se pomnoži z metodami, ki so bile validirane za vsako vrsto, na katero se pregleduje. Te metode so določene v standardnih operativnih postopkih, ki jih je določil in na svoji spletni strani objavil EURL-AP. Vsak ekstrakt DNK se analizira pri najmanj dveh različnih razredčitvah, da se oceni učinek zaviranja.
Za vsako ciljno vrsto se pripravita dva kontrolna vzorca, ki se pomnožita, kot je opisano v standardu ISO 24276:
— pozitivni kontrolni vzorec s ciljno DNK se uporabi za vsako ploščo ali serijo analiz PCR,
— kontrolni vzorec reagenta za pomnoževanje (imenovan tudi kontrolni vzorec brez matrice) se uporabi za vsako ploščo ali serijo analiz PCR.
2.2.6 Razlaga in prikaz rezultatov
V poročilu o rezultatih mora laboratorij navesti vsaj maso uporabljenega testnega vzorca, uporabljeno tehniko za ekstrakcijo, število izvedenih določanj in mejo zaznavnosti metode.
Rezultati se ne smejo razlagati in poročati, če pozitivni kontrolni vzorec na stopnji ekstrakcije DNK in pozitivni kontrolni vzorec s ciljno DNK ne omogočata določitve ciljne vrste pri analizi, medtem ko je rezultat pri kontrolnem vzorcu reagenta za pomnoževanje negativen.
Če se rezultata dveh testnih vzorcev ne ujemata, je treba ponoviti vsaj korak pomnožitve genskega materiala. Če laboratorij domneva, da so razlog za neujemanje lahko ekstrakti DNK, je treba pred razlago rezultatov opraviti novo ekstrakcijo DNK in pomnožitev genskega materiala.
Pri končnem prikazu rezultatov je treba upoštevati smernice o vključevanju in razlagi rezultatov dveh testnih vzorcev v skladu s standardnimi operativnimi postopki, kot jih je določil in na svoji spletni strani objavil EURL-AP.
2.2.6.1 Negativen rezultat
O negativnem rezultatu se poroča na naslednji način:
V predloženem vzorcu ni bila ugotovljena DNK iz X (X pomeni živalsko vrsto ali skupino živalskih vrst, na katere se pregleduje).
2.2.6.2 Pozitiven rezultat
O pozitivnem rezultatu se poroča na naslednji način:
V predloženem vzorcu je bila ugotovljena DNK iz X (X pomeni živalsko vrsto ali skupino živalskih vrst, na katere se pregleduje).
PRILOGA VII
METODA IZRAČUNA ENERGIJSKE VREDNOSTI KRME ZA PERUTNINO
1. Metoda izračuna in prikaza energijske vrednosti
Energijska vrednost krme za perutnino mora biti izračunana v skladu s formulo, določeno v nadaljnjem besedilu na podlagi odstotkov nekaterih analitskih sestavin krme. Ta vrednost se izrazi v megajoulih (MJ) presnovne energije (ME), popravljeno za dušik, na kilogram krmne mešanice:
MJ/kg ME = 0,1551 × % surovih beljakovin + 0,3431 × % surovih maščob + 0,1669 × % škroba + 0,1301 × % celotnega sladkorja (izraženega kot saharoza).
2. Odstopanja, ki se uporabljajo za označene vrednosti
Če se v okviru uradnega pregleda odkrije razlika (povečana ali zmanjšana energijska vrednost krme) med rezultatom pregleda in označeno energijsko vrednostjo, je dovoljeno minimalno odstopanje 0,4 MJ/kg ME.
3. Prikaz rezultatov
Po uporabi zgoraj navedene formule je treba dobljeni rezultat izraziti z eno decimalko.
4. Metode vzorčenja in analitske metode
Vzorčenje krmnih mešanic in določanje vsebnosti analitskih sestavin, navedenih v metodi izračuna, je treba izvesti v skladu z metodami Skupnosti za vzorčenje ali analitskimi metodami Skupnosti za uradni pregled krme.
Uporablja se naslednje:
— za določanje vsebnosti surove maščobe: postopek B metode za določanje surovih olj in maščob iz dela H Priloge III,
— za določanje vsebnosti škroba: polarimetrična metoda iz dela L Priloge III.
PRILOGA VIII
ANALITSKE METODE ZA NADZOR KRMNIH DODATKOV, KI NISO VEČ DOVOLJENI
Pomembno obvestilo:
Za določanje krmnih dodatkov, ki niso več dovoljeni, se lahko uporabljajo analitske metode, ki so občutljivejše od analitskih metod, navedenih v tej prilogi.
Za potrditev se uporabljajo analitske metode iz te priloge.
A. DOLOČANJE METIL BENZOKVATA
7-benziloksi-6-butil-3-metoksikarbonil-4-kinolon
1. Namen in področje uporabe
Ta metoda omogoča določanje ravni metil benzokvata v krmi. Meja kvantifikacije je 1 mg/kg.
2. Načelo
Metil benzokvat se ekstrahira iz vzorca z raztopino metanol metansulfonske kisline. Ekstrakt se prečisti z diklorometanom z ionsko izmenjevalno kromatografijo in nato ponovno z diklorometanom. Vsebnost metil benzokvata se določi s tekočinsko kromatografijo visoke ločljivosti (HPLC) z reverzno fazo in UV-detektorjem.
3. Reagenti
3.1 Diklorometan
3.2 Metanol, za HPLC
3.3 Mobilna faza HPLC
mešanica metanola (3.2) in vode (za HPLC) 75 + 25 (v + v)
Filtrira se skozi filter 0,22 μm (4,5 ), raztopina pa se nato razplini (npr. 10 minut v ultrazvočni kopeli).
3.4 Raztopina metansulfonske kisline, c = 2 %
20,0 ml metansulfonske kisline se razredči z metanolom (3.2) do 1 000 ml.
3.5 Raztopina klorovodikove kisline, c = 10 %
100 ml klorovodikove kisline (ρ201,18 g/ml) se razredči z vodo do 1 000 ml.
3.6 Kationsko izmenjevalna smola Amberlite CG-120 (Na), 100 do 200 meš
Smola se pred uporabo obdela. 100 g smole se suspendira v 500 ml raztopine klorovodikove kisline (3.5) in se med neprestanim mešanjem segreva na vroči plošči do vretja. Ohladi se in kislina se oddekantira. Filtrira se skozi papirni filter pod vakuumom. Smola se dvakrat spere s po 500 ml vode, nato pa še z 250 ml metanola (3.2). Nato se še enkrat spere s 250 ml metanola in osuši z zrakom, ki prehaja skozi filtrirano pogačo. Suha smola se hrani v zaprti steklenici.
3.7 Standardna snov: čisti metil benzokvat (7-benziloksi-6-butil-3-metoksikarbonil-4-kinolon)
3.7.1
50 mg standardne snovi (3.7) se natehta na 0,1 mg natančno in raztopi v raztopini metansulfonske kisline (3.4) v 100-mililitrski merilni bučki, dopolni se do oznake in premeša.
3.7.2
5,0 ml osnovne standardne raztopine metil benzokvata (3.7.1) se prenese v 50-mililitrsko merilno bučko, dopolni do oznake z metanolom (3.2) in premeša.
3.7.3
V serijo 25-mililitrskih merilnih bučk se prenese 1,0 , 2,0 , 3,0 , 4,0 in 5,0 ml vmesne standardne raztopine metil benzokvata (3.7.2). Dopolni se do oznake z mobilno fazo (3.3) in premeša. Koncentracija metil benzokvata v teh raztopinah je 2,0 , 4,0 , 6,0 , 8,0 in 10,0 μg/ml. Te raztopine je treba pripraviti sveže pred uporabo.
4. Oprema
4.1 Laboratorijski stresalnik
4.2 Rotacijski uparjalnik
4.3 Steklena kolona (250 mm × 15 mm) s petelinčkom in kapaciteto rezervoarja približno 200 ml
4.4 Oprema za HPLC z ultravijoličnim detektorjem z nastavljivo valovno dolžino ali detektorjem s serijo diod
4.4.1 Kolona za tekočinsko kromatografijo: 300 mm × 4 mm, C18, polnitev 10 μm ali enakovredna
4.5 Membranski filtri, 0,22 μm
4.6 Membranski filtri, 0,45 μm
5. Postopek
5.1 Splošno
5.1.1 Z analizo slepe krme se zagotovi, da niso prisotni niti metil benzokvat niti moteče snovi.
5.1.2 Preskus izkoristka se izvede z analizo slepe krme, ki se ji doda količina metil benzokvata, podobna količini v vzorcu. Za koncentracijo 15 mg/kg se k 20 g slepe krme doda 600 μl osnovne standardne raztopine (3.7.1), premeša se in počaka 10 minut, preden se začne postopek ekstrakcije (5.2).
Opomba: Pri tej metodi je vrsta slepe krme podobna vrsti vzorca in pri analizi se dokaže odsotnost metil benzikvata.
5.2 Ekstrakcija
Približno 20 g pripravljenega vzorca se natehta na 0,01 g natančno in prenese v 250-mililitrsko erlenmajerico. Doda se 100,0 ml raztopine metansulfonske kisline (3.4) in mehansko stresa (4.1) 30 minut. Raztopina se filtrira skozi papirni filter, filtrat pa se shrani za postopek ločevanja tekočina-tekočina (5.3).
5.3 Ločevanje tekočina-tekočina
V 500-mililitrski lij ločnik, v katerem je 100 ml raztopine klorovodikove kisline (3.5), se prenese 25,0 ml filtrata, ki nastane pri postopku iz točke 5.2. V lij se doda 100 ml diklorometana (3.1) in stresa eno minuto. Počaka se, da se plasti ločijo, spodnja (diklorometanska) plast pa se odlije v 500-mililitrsko bučko z okroglim dnom. Ekstrakcija vodne faze se dvakrat ponovi s po 40 ml diklorometana, ki se priključijo prvemu ekstraktu v bučki z okroglim dnom. Ekstrakt diklorometana se upari do suhega v rotacijskem uparjalniku (4.2) pri znižanem tlaku in 40 oC. Ostanek se raztopi v 20–25 ml metanola (3.2), bučka se zamaši, celotni ekstrakt pa se shrani za ionsko izmenjevalno kromatografijo (5.4).
5.4 Ionska izmenjevalna kromatografija
5.4.1
V spodnji del steklene kolone (4.3) se vstavi zamašek iz steklene volne. Pripravi se suspenzija s 5,0 g obdelane kationske izmenjevalne smole (3.6) in 50 ml klorovodikove kisline (3.5), nato pa se vlije v stekleno kolono in počaka, da se usede. Presežek kisline se odstrani do višine tik nad površino smole, nato pa se kolona spira z vodo do nevtralne reakcije eluata na lakmusov papir. 50 ml metanola (3.2) se prenese v kolono in pusti, da prodre na površino smole.
5.4.2
Nastali ekstrakt (5.3) se pazljivo odpipetira na kolono. Bučka z okroglim dnom se dvakrat spere s po 5–10 ml metanola (3.2), izpirka pa se preneseta na kolono. Ekstrakt se spusti do površine smole, kolona pa se spere s 50 ml metanola, pri čemer hitrost pretoka ne sme preseči 5 ml na minuto. Eluat se zavrže. Metil benzokvat se iz kolone eluira s 150 ml raztopine metansulfonske kisline (3.4), eluat s kolone pa se zbere v 250-mililitrsko erlenmajerico.
5.5 Ločevanje tekočina-tekočina
Eluat, ki nastane pri postopku iz točke 5.4.2, se prenese v litrski lij ločnik. Erlenmajerica se spere s 5–10 ml metanola (3.2), izpirek pa se združi z vsebino v liju ločniku. Doda se 300 ml raztopine klorovodikove kisline (3.5) in 130 ml diklorometana (3.1). Stresa se 1 minuto in pusti, da se fazi ločita. Spodnja (diklorometanska) plast se odtoči v 500-mililitrsko bučko z okroglim dnom. Ekstrakcija vodne faze se dvakrat ponovi s po 70 ml diklorometana, oba ekstrakta pa se združita s prvim v bučki z okroglim dnom.
Ekstrakt diklorometana se upari do suhega v rotacijskem uparjalniku (4.2) pod znižanim tlakom ter pri 40 oC. Ostanek v bučki se raztopi v približno 5 ml metanola (3.2), raztopina pa se prenese v 10-mililitrsko merilno bučko. Bučka z okroglim dnom se dvakrat spere s po 1–2 ml metanola, izpirka pa se preneseta v merilno bučko. Dopolni se z metanolom do oznake in premeša. Alikvot se filtrira skozi membranski filter (4.6). Ta raztopina se shrani za določanje s HPLC (5.6).
5.6 Določanje s HPLC
5.6.1
Naslednji pogoji se lahko uporabljajo kot smernica, lahko pa se uporabljajo drugi pogoji, če zagotavljajo enakovredne rezultate:
— kolona za tekočinsko kromatografijo (4.4.1),
— HPLC-mobilna faza: mešanica metanola in vode (3.3),
— hitrost pretoka: 1–1,5 ml/min,
— valovna dolžina detekcije: 265 nm,
— volumen, ki se vbrizga: 20–50 μl.
Stabilnost kromatografskega sistema se preveri z večkratnim vbrizganjem raztopine za umerjanje (3.7.3) s koncentracijo 4 μg/ml, dokler se ne dosežejo konstantne višine (ali površine) vrhov in konstantni retencijski časi.
5.6.2
Vsaka raztopina za umerjanje (3.7.3) se večkrat vbrizga in za vsako koncentracijo se izmerijo višine (površine) vrhov. Umeritvena krivulja se pripravi tako, da se na ordinato nanesejo srednje vrednosti višin ali površin vrhov raztopin za umerjanje, na absciso pa ustrezne koncentracije v μg/ml.
5.6.3
Ekstrakt vzorca (5.5) se večkrat vbrizga, pri tem se uporabijo enaki volumni kakor pri raztopini za umerjanje, ter določijo srednje vrednosti višin (površin) vrhov metil benzokvata.
6. Izračun rezultatov
Koncentracija raztopine vzorca v μg/ml se določi iz srednjih vrednosti vrhov (površin) metil benzokvata iz umeritvene krivulje (5.6.2).
Vsebnost metil benzokvata (w) (mg/kg) v vzorcu se izračuna po naslednji formuli:

pri čemer je:
|
c |
= |
koncentracija metil benzokvata v raztopini vzorca v μg/ml; |
|
m |
= |
masa preskušanega vzorca v gramih. |
7. Potrditev rezultatov
7.1 Identiteta
Identiteta analita se lahko potrdi z dodatno kromatografijo ali z detektorjem s serijo diod, s katerim se primerjata spekter ekstrakta vzorca in spekter raztopine za umerjanje (3.7.3) s koncentracijo 10 μg/ml.
7.1.1
Ekstraktu vzorca se doda ustrezna količina vmesne standardne raztopine (3.7.2). Količina dodanega metil benzokvata mora biti podobna ocenjeni količini metil benzokvata v ekstraktu vzorca.
Povišan je le vrh metil benzokvata ob upoštevanju dodane količine in razredčitve ekstrakta. Širina vrha na polovici njegove največje višine mora biti v mejah približno 10 % prvotne širine.
7.1.2
Rezultati se ocenijo glede na naslednja merila:
(a) valovni dolžini maksimalne absorpcije spektra vzorca in spektra standarda, zabeleženi na najvišji točki vrha kromatograma, morata biti enaki znotraj meja, določenih z ločljivostjo sistema za detekcijo. Za detekcijo s serijo diod je to ponavadi v območju približno 2 nm;
(b) med 220 in 350 nm se spekter vzorca in spekter standarda na najvišji točki kromatografskega vrha za navedene dele spektra ne smeta razlikovati znotraj območja od 10 do 100 % relativne absorbance. To merilo je izpolnjeno, ko so prisotne iste najvišje vrednosti in ko odmik med dvema spektroma na nobeni opazovani točki ne preseže 15 % absorbance standardnega analita;
(c) med 220 in 350 nm se spektri naraščanja, najvišje točke vrha in padanja vrha, pridobljeni iz ekstrakta vzorca, za navedene dele spektra ne smejo razlikovati med seboj v območju 10 do 100 % relativne absorbance. To merilo je izpolnjeno, ko so prisotne iste najvišje točke in ko odmik med spektri na nobeni od opazovanih točk ne preseže 15 % absorbance spektra najvišje točke.
Če eno od teh meril ni izpolnjeno, prisotnost analita ni potrjena.
7.2 Ponovljivost
Razlika med rezultatoma dveh vzporednih določanj, izvedenih na istem vzorcu, ne sme presegati 10 % višjega rezultata za vsebnosti metil benzokvata 4–20 mg/kg.
7.3 Izkoristek
Za slepi vzorec z dodatkom je izkoristek najmanj 90-odstoten.
8. Rezultati medlaboratorijske študije
10 laboratorijev je analiziralo 5 vzorcev. Vsak vzorec je bil analiziran dvakrat.
|
|
Slepi preskus |
Obrok 1 |
Briketi 1 |
Obrok 2 |
Briketi 2 |
|
Srednja vrednost [mg/kg] |
ND |
4,50 |
4,50 |
8,90 |
8,70 |
|
Sr [mg/kg] |
— |
0,30 |
0,20 |
0,60 |
0,50 |
|
CVr [ %] |
— |
6,70 |
4,40 |
6,70 |
5,70 |
|
SR [mg/kg] |
— |
0,40 |
0,50 |
0,90 |
1,00 |
|
CVR [ %] |
— |
8,90 |
11,10 |
10,10 |
11,50 |
|
Izkoristek [ %] |
— |
92,00 |
93,00 |
92,00 |
89,00 |
|
ND |
= |
ni bilo ugotovljeno; |
|
Sr |
= |
standardni odmik ponovljivosti; |
|
CVr |
= |
koeficient variacije ponovljivosti v %; |
|
SR |
= |
standardni odmik obnovljivosti; |
|
CVR |
= |
koeficient variacije obnovljivosti v %. |
B. DOLOČANJE OLAKVINDOKSA
2-[N-2'-(hidroksietil)karbamoil]-3-metilkvinoksalin-N1,N4-dioksid
1. Namen in področje uporabe
Ta metoda omogoča določanje ravni olakvindoksa v krmi. Meja kvantifikacije je 5 mg/kg.
2. Načelo
Vzorec se ekstrahira z mešanico vode in metanola. Vsebnost olakvindoksa se določi s tekočinsko kromatografijo visoke ločljivosti (HPLC) z reverzno fazo z uporabo UV-detektorja.
3. Reagenti
3.1 Metanol
3.2 Metanol, za HPLC
3.3 Voda, za HPLC
3.4 Mobilna faza za HPLC
Mešanica vode (3.3) in metanola (3.2), 900 + 100 (V + V)
3.5 Standardna snov: čisti olakvindoks 2-[N-2'-(hidroksietil)karbamoil]-3-metilkvinoksalin-N1,N4-dioksid, E 851
3.5.1
50 mg olakvindoksa (3.5) se natehta na 0,1 mg natančno in prenese v 200-mililitrsko merilno bučko, nato pa se doda približno 190 ml vode. Bučka se nato postavi za 20 minut v ultrazvočno kopel (4.1). Po ultrazvočni obdelavi se raztopina segreje na sobno temperaturo, dopolni z vodo do oznake in premeša. Bučka se ovije v aluminijasto folijo in shrani v hladilniku. To raztopino je treba pripraviti svežo vsak mesec.
3.5.2
10,0 ml osnovne standardne raztopine (3.5.1) se prenese v 100-mililitrsko merilno bučko, dopolni z mobilno fazo (3.4) do oznake in premeša. Bučka se ovije v aluminijasto folijo in shrani v hladilniku. To raztopino je treba pripraviti svežo vsak dan.
3.5.3
V serijo 50-mililitrskih merilnih bučk se prenese 1,0 , 2,0 , 5,0 , 10,0 , 15,0 in 20,0 ml vmesne standardne raztopine (3.5.2). Dopolni se z mobilno fazo (3.4) do oznake in premeša. Bučke se ovije v aluminijasto folijo. Te raztopine ustrezajo 0,5 , 1,0 , 2,5 , 5,0 , 7,5 in 10,0 μg olakvindoksa na ml.
Te raztopine je treba pripraviti sveže vsak dan.
4. Oprema
4.1 Ultrazvočna kopel
4.2 Mehanski stresalnik
4.3 Oprema za HPLC z detektorjem ultravijolične svetlobe z nastavljivo valovno dolžino ali detektorjem s serijo diod
4.3.1 Kolona za tekočinsko kromatografijo, 250 mm × 4 mm C18, 10 μm polnitev ali enakovredna
4.4 Membranski filtri, 0,45 μm
5. Postopek
|
Opomba: |
Olakvindoks je občutljiv na svetlobo. Vsi postopki se izvajajo v temnem prostoru ali pa se uporabijo posode iz rjavega stekla. |
5.1 Splošno
5.1.1 Z analizo slepe krme se zagotovi, da niso prisotni niti olakvindoks niti moteče snovi.
5.1.2 Preskus izkoristka se izvede z analizo slepe krme, ki se ji doda količina olakvindoksa, podobna količini v vzorcu. Za koncentracijo 50 mg/kg se 10,0 ml osnovne standardne raztopine (3.5.1) prenese v 250-mililitrsko erlenmajerico, raztopina pa se upari do približno 0,5 ml. Doda se 50 g slepe krme, dobro se premeša in pusti 10 minut ter vmes nekajkrat ponovno premeša, nato pa se nadaljuje s postopkom ekstrakcije (5.2).
|
Opomba: |
Pri tej metodi je vrsta slepe krme podobna tisti iz vzorca in pri analizi se dokaže odsotnost olakvindoksa. |
5.2 Ekstrakcija
Približno 50 g vzorca se natehta na 0,01 g natančno. Prenese se v 1 000 -mililitrsko erlenmajerico, doda se 100 ml metanola (3.1), erlenmajerica pa se postavi za 5 minut v ultrazvočno kopel (4.1). Doda se 410 ml vode in se pusti še 15 minut v ultrazvočni kopeli. Bučka se odstrani iz ultrazvočne kopeli, 30 minut se stresa s stresalnikom (4.2), nato pa se filtrira skozi naguban filter. 10,0 ml filtrata se prenese v 20-mililitrsko merilno bučko, dopolni se z vodo do oznake in premeša. Alikvot se filtrira skozi membranski filter (4.4) (glej točko 9 Opomba). Nadaljuje se z določanjem s HPLC (5.3).
5.3 Določanje s HPLC
5.3.1
Naslednji pogoji se lahko uporabljajo kot smernice, lahko pa se uporabljajo drugi pogoji, če zagotavljajo enakovredne rezultate.
|
Analitska kolona (4.3.1) |
|
|
Mobilna faza (3.4): |
mešanica vode (3.3) in metanola (3.2), 900 + 100 (V + V) |
|
Hitrost pretoka: |
1,5 –2 ml/min |
|
Detekcijska valovna dolžina: |
380 nm |
|
Volumen, ki se vbrizga: |
20–100 μl |
Stabilnost kromatografskega sistema se preveri z večkratnim vbrizgavanjem raztopine za umerjanje (3.5.3) s koncentracijo 2,5 μg/ml, dokler se ne dosežejo konstantne višine vrhov in konstantni retencijski časi.
5.3.2
Za vsako koncentracijo se vsaka raztopina za umerjanje (3.5.3) večkrat vbrizga, nato pa se določijo se srednje vrednosti višin (površin) vrhov. Umeritvena krivulja se pripravi tako, da se na ordinato nanesejo srednje vrednosti višin (površin) vrhov raztopin za umerjanje, na absciso pa ustrezne koncentracije v μg/ml.
5.3.3
Ekstrakt vzorca (5.2) se vbrizga večkrat, pri čemer se uporabijo enaki volumni kakor pri raztopini za umerjanje, nato pa se določijo srednje vrednosti višin (površin) vrhov olakvindoksa.
6. Izračun rezultatov
Koncentracija raztopine vzorca v μg/ml se določi iz srednje vrednosti višin (površin) vrhov olakvindoksa iz umeritvene krivulje (5.3.2).
Vsebnost olakvindoksa v vzorcu (w) v mg/kg se izračuna po naslednji formuli:

pri čemer je:
|
c |
= |
koncentracija olakvindoksa v ekstraktu vzorca (5.2) v μg/ml; |
|
m |
= |
masa preskusnega vzorca v g (5.2). |
7. Potrditev rezultatov
7.1 Identiteta
Identiteta analita se lahko potrdi z dodatno kromatografijo ali z detektorjem s serijo diod, s katerim se primerjata spekter ekstrakta vzorca (5.2) in spekter raztopine za umerjanje (3.5.3) s koncentracijo 5,0 μg/ml.
7.1.1
Ekstraktu vzorca (5.2) se doda ustrezna količina raztopine za umerjanje (3.5.3). Količina dodanega olakvindoksa mora biti podobna količini olakvindoksa v ekstraktu vzorca.
Povišan je le vrh olakvindoksa ob upoštevanju dodane količine in razredčitve ekstrakta. Širina vrha na polovici maksimalne višine mora biti znotraj ± 10 % prvotne širine vrha, ki nastane pri ekstraktu vzorca brez dodanega olakvindoksa.
7.1.2
Rezultati se ocenijo glede na naslednja merila:
(a) valovni dolžini maksimalne absorpcije spektra vzorca in spektra standarda, zabeleženi na najvišji točki vrha na kromatogramu, morata biti znotraj meja, določenih z ločljivostjo sistema za detekcijo. Pri detekciji s serijo diod je to ponavadi ± 2 nm;
(b) med 220 in 400 nm se spekter vzorca in spekter standarda, zabeležena na najvišji točki vrha na kromatogramu, za navedene dele spektra ne smeta razlikovati v območju 10–100 % relativne absorbance. To merilo je izpolnjeno, ko so prisotne iste najvišje vrednosti in ko odmik med dvema spektroma na nobeni opazovani točki ne preseže 15 % absorbance standardnega analita;
(c) med 220 in 400 nm se spektri naraščanja, najvišje točke vrha in padanja vrha, pridobljeni iz ekstrakta vzorca, za navedene dele spektra ne smejo razlikovati med seboj v območju 10–100 % relativne absorbance. To merilo je izpolnjeno, ko so prisotne iste najvišje točke in ko odmik med spektri na nobeni od opazovanih točk ne preseže 15 % absorbance spektra najvišje točke.
Če eno od teh meril ni izpolnjeno, prisotnost analita ni potrjena.
7.2 Ponovljivost
Razlika med rezultatoma dveh vzporednih določanj, izvedenih na istem vzorcu, ne sme presegati 15 % višjega rezultata za vsebnosti olakvindoksa 10–200 mg/kg.
7.3 Izkoristek
Za slepi vzorec z dodatkom je izkoristek najmanj 90-odstoten.
8. Rezultati medlaboratorijske študije
Organizirana je bila medlaboratorijska študija, pri kateri je 13 laboratorijev analiziralo 4 vzorce krme v briketih, vključno z enim slepim vzorcem. Rezultati so prikazani v spodnji preglednici:
|
|
Vzorec 1 |
Vzorec 2 |
Vzorec 3 |
Vzorec 4 |
|
L |
13 |
10 |
11 |
11 |
|
n |
40 |
40 |
44 |
44 |
|
Srednja vrednost [mg/kg] |
— |
14,6 |
48,0 |
95,4 |
|
Sr [mg/kg] |
— |
0,82 |
2,05 |
6,36 |
|
SR [mg/kg] |
— |
1,62 |
4,28 |
8,42 |
|
CVr [ %] |
— |
5,6 |
4,3 |
6,7 |
|
CVR [ %] |
— |
11,1 |
8,9 |
8,8 |
|
Nominalna vsebnost |
|
|
|
|
|
[mg/kg] |
— |
15 |
50 |
100 |
|
Izkoristek % |
— |
97,3 |
96,0 |
95,4 |
|
L |
: |
število laboratorijev; |
|
n |
: |
število posameznih vrednosti; |
|
Sr |
: |
standardni odmik ponovljivosti; |
|
SR |
: |
standardni odmik obnovljivosti; |
|
CVr |
: |
koeficient variacije ponovljivosti; |
|
CVR |
: |
koeficient variacije obnovljivosti. |
9. Opomba
Čeprav metoda ni potrjena za krmo, ki vsebuje več kot 100 mg/kg olakvindoksa, je mogoče dobiti zadovoljive rezultate tako, da se uporabi manjša masa vzorca in/ali razredčitev ekstrakta (5.2), da se doseže koncentracija v območju umeritvene krivulje (5.3.2).
C. DOLOČANJE AMPROLIUMA
1-[(4-amino-2-propilpirimidin-5-il)metil]-2-metil-piridin klorid hidroklorid
1. Namen in področje uporabe
Ta metoda omogoča določanje ravni amproliuma v krmi in premiksih. Meja določanja je 1 mg/kg, meja kvantifikacije pa 5 mg/kg.
2. Načelo
Vzorec se ekstrahira z mešanico metanola in vode. Po redčenju z mobilno fazo in membranski filtraciji se vsebnost amproliuma določi s kationsko izmenjevalno tekočinsko kromatografijo visoke ločljivosti (HPLC) z uporabo UV-detektorja.
3. Reagenti
3.1 Metanol
3.2 Acetonitril, za HPLC
3.3 Voda, za HPLC
3.4 Raztopina natrijevega dihidrogen fosfata, c = 0,1 mol/l
V 1 000 -mililitrski merilni bučki se v vodi (3.3) raztopi 13,80 g natrijevega dihidrogen fosfat monohidrata, dopolni se z vodo (3.3) do oznake in premeša.
3.5 Raztopina natrijevega perklorata, c = 1,6 mol/l
V 1 000 -mililitrski merilni bučki se v vodi (3.3) raztopi 224,74 g natrijevega perklorat monohidrata, dopolni se z vodo (3.3) do oznake in premeša.
3.6 Mobilna faza za HPLC (glej opombo 9.1).
Mešanica acetonitrila (3.2), raztopine natrijevega dihidrogen fosfata (3.4) in raztopine natrijevega perklorata (3.5), 450+450+100 (v+v+v). Pred uporabo se filtrira z membranskim filtrom 0,22 μm (4.3), raztopina pa se razplini (npr. najmanj 15 minut v ultrazvočni kopeli (4.4)).
3.7 Standardna snov: čisti amprolium, l-[(4-amino-2-propilpirimidin-5-il)metil]-2-metil-piridin klorid hidroklorid, E 750 (glej opombo 9.2)
3.7.1
50 mg amproliuma (3.7) se natehta na 0,1 mg natančno in prenese v 100-mililitrsko merilno bučko ter raztopi v 80 ml metanola (3.1), bučka pa se za 10 minut postavi v ultrazvočno kopel (4.4). Po ultrazvočni obdelavi se raztopina segreje na sobno temperaturo, dopolni se z vodo do oznake in premeša. Pri temperaturi ≤ 4 oC je raztopina stabilna 1 mesec.
3.7.2
5,0 ml osnovne standardne raztopine (3.7.1) se odpipetira v 50-mililitrsko merilno bučko, dopolni se z ekstrakcijskim topilom (3.8) do oznake in premeša. Pri temperaturi ≤ 4 oC je raztopina stabilna 1 mesec.
3.7.3
V serijo 50-mililitrskih merilnih bučk se prenese 0,5 , 1,0 , in 2,0 ml vmesne standardne raztopine (3.7.2). Dopolni se z mobilno fazo (3.6) do oznake in premeša. Te raztopine ustrezajo 0,5 , 1,0 in 2,0 μg amproliuma na ml. Te raztopine je treba pripraviti sveže pred uporabo.
3.8 Ekstrakcijsko topilo
Mešanica metanola (3.1) in vode 2+1 (v+v)
4. Oprema
4.1 Oprema za HPLC s sistemom za vbrizgavanje, primernim za vbrizgavanje volumnov 100 μl
4.1.1 Kolona za tekočinsko kromatografijo, 125 mm × 4 mm, kationski izmenjevalec Nucleosil 10 SA, polnitev 5 ali 10 μm ali enakovredna
4.1.2 UV-detektor z nastavljivo valovno dolžino ali detektor s serijo diod
4.2 Membranski filter, material PTFE, 0,45 μm
4.3 Membranski filter, 0,22 μm
4.4 Ultrazvočna kopel
4.5 Mehanski stresalnik ali magnetno mešalo
5. Postopek
5.1 Splošno
5.1.1
Za učinkovitost preskusa izkoristka (5.1.2) se z analizo slepe krme zagotovi, da niso prisotni niti amprolium niti moteče snovi. Vrsta slepe krme je podobna vrsti vzorca in dokaže se odsotnost amproliuma ali motečih snovi.
5.1.2
Preskus izkoristka se izvede z analizo slepe krme, ki se ji doda količina amproliuma, podobna količini v vzorcu. Za koncentracijo 100 mg/kg se 10,0 ml osnovne standardne raztopine (3.7.1) prenese v 250-mililitrsko erlenmajerico, raztopina pa se upari do približno 0,5 ml. Doda se 50 g slepe krme, dobro se premeša in pusti 10 minut ter vmes nekajkrat ponovno premeša, nato pa se nadaljuje s postopkom ekstrakcije (5.2).
Če pa vrsta slepe krme, podobna vrsti vzorca, ni na voljo (glej točko 5.1.1), se lahko preskus izkoristka izvede po metodi standardnega dodatka. V tem primeru se vzorcu za analizo doda toliko amproliuma, kolikor ga je v vzorcu. Ta vzorec se analizira skupaj z vzorcem brez dodanega amproliuma, izkoristek pa se izračuna z odštevanjem.
5.2 Ekstrakcija
5.2.1
V 500-mililitrsko erlenmajerico se natehta na 0,01 g natančno 5–40 g vzorca, odvisno od vsebnosti amproliuma, in doda 200 ml ekstrakcijskega topila (3.8). Erlenmajerica se postavi v ultrazvočno kopel (4.4) in se pusti 15 minut. Odstrani se iz ultrazvočne kopeli in 1 uro stresa s stresalnikom ali meša z magnetnim mešalom (4.5). Alikvot ekstrakta se razredči z mobilno fazo (3.6), da nastane vsebnost amproliuma 0,5 –2 μg/ml, ter premeša (glej opombo 9.3). 5–10 ml tako razredčene raztopine se filtrira skozi membranski filter (4.2). Nadaljuje se z določanjem s HPLC (5.3).
5.2.2
V 500-mililitrsko erlenmajerico se na 0,001 g natančno natehta 1–4 g premiksa, odvisno od vsebnosti amproliuma, in doda 200 ml ekstrakcijskega topila (3.8). Erlenmajerica se postavi v ultrazvočno kopel (4.4) in se pusti 15 minut. Odstrani se iz ultrazvočne kopeli in 1 uro stresa s stresalnikom ali meša z magnetnim mešalom (4.5). Alikvot ekstrakta se razredči z mobilno fazo (3.6), da nastane vsebnost amproliuma 0,5 –2 μg/ml, ter premeša. 5–10 ml tako razredčene raztopine se filtrira skozi membranski filter (4.2). Nadaljuje se z določanjem s HPLC (5.3).
5.3 Določanje s HPLC
5.3.1
Naslednji pogoji se lahko uporabljajo kot smernice, lahko pa se uporabljajo drugi pogoji, če zagotavljajo enakovredne rezultate.
|
Kolona za tekočinsko |
|
|
kromatografijo (4.1.1): |
125 mm × 4 mm, kationski izmenjevalec Nucleosil 10 SA, polnitev 5 ali 10 μm ali enakovredna |
|
Mobilna faza (3.6): |
Mešanica acetonitrila (3.2), raztopine natrijevega dihidrogen fosfata (3.4) in raztopine natrijevega perklorata (3.5), 450+450+100 (v+v+v) |
|
Hitrost pretoka: |
0,7 –1 ml/min |
|
Detekcijska valovna dolžina: |
264 nm |
|
Volumen, ki se vbrizga: |
100 μl |
Stabilnost kromatografskega sistema se preveri z večkratnim vbrizgavanjem raztopine za umerjanje (3.7.3) s koncentracijo 1,0 μg/ml, dokler se ne dosežejo konstantne višine vrhov in konstantni retencijski časi.
5.3.2
Za vsako koncentracijo se vsaka raztopina za umerjanje (3.7.3) vbrizga večkrat, nato pa se določijo srednje vrednosti višin (površin) vrhov. Umeritvena krivulja se pripravi tako, da se na ordinato nanesejo srednje vrednosti višin (površin) vrhov raztopin za umerjanje, na absciso pa ustrezne koncentracije v μg/ml.
5.3.3
Ekstrakt vzorca (5.2) se večkrat vbrizga, pri čemer se uporabijo enaki volumni kakor pri raztopini za umerjanje, nato pa se določijo srednje vrednosti višin (površin) vrhov amproliuma.
6. Izračun rezultatov
Koncentracija raztopine vzorca v μg/ml se določi iz srednje vrednosti višin (površin) vrhov amproliuma iz umeritvene krivulje (5.3.2).
Vsebnost amproliuma (w) v mg/kg vzorca se izračuna po naslednji formuli:

[mg/kg]
pri čemer je:
|
V |
= |
volumen ekstrakcijskega topila (3.8) v ml v skladu s točko 5.2 (tj. 200 ml); |
|
c |
= |
koncentracija amproliuma v ekstraktu vzorca (5.2) v μg/ml; |
|
f |
= |
faktor razredčitve v skladu s točko 5.2; |
|
m |
= |
masa preskusnega vzorca v gramih. |
7. Potrditev rezultatov
7.1 Identiteta
Identiteta analita se lahko potrdi z dodatno kromatografijo ali z detektorjem s serijo diod, s katerim se primerjata spekter ekstrakta vzorca (5.2) in spekter raztopine za umerjanje (3.7.3) s koncentracijo 2,0 μg/ml.
7.1.1
Ekstraktu vzorca (5.2) se doda primerna količina raztopine za umerjanje (3.7.3). Količina dodanega amproliuma mora biti podobna količini amproliuma v ekstraktu vzorca.
Povišan je le vrh amproliuma ob upoštevanju dodane količine in razredčitve ekstrakta. Širina vrha na polovici maksimalne višine mora biti znotraj ± 10 % prvotne širine vrha, ki nastane pri ekstraktu vzorca brez dodanega amproliuma.
7.1.2
Rezultati se ocenijo glede na naslednja merila:
(a) valovni dolžini maksimalne absorpcije spektra vzorca in spektra standarda, zabeleženi na najvišji točki vrha na kromatogramu, morata biti znotraj meja, določenih z ločljivostjo sistema za detekcijo. Pri določanju s serijo diod je to ponavadi ± 2 nm;
(b) med 210 in 320 nm se spekter vzorca in spekter standarda, zabeležena na najvišji točki vrha na kromatogramu, za navedene dele spektra ne smeta razlikovati v območju 10–100 % relativne absorbance. To merilo je izpolnjeno, ko so prisotne iste najvišje vrednosti in ko odmik med dvema spektroma na nobeni opazovani točki ne preseže 15 % absorbance standardnega analita;
(c) med 210 in 320 nm se spektri naraščanja, najvišje točke vrha in padanja vrha, pridobljeni iz ekstrakta vzorca, za navedene dele spektra ne smejo razlikovati med seboj v območju 10–100 % relativne absorbance. To merilo je izpolnjeno, ko so prisotne iste najvišje točke in ko odmik med spektri na nobeni od opazovanih točk ne preseže 15 % absorbance spektra najvišje točke.
Če eno izmed teh meril ni izpolnjeno, prisotnost analita ni potrjena.
7.2 Ponovljivost
Razlika med rezultatoma dveh vzporednih določitev na istem vzorcu ne sme presegati:
— 15 % višje vrednosti za vsebnosti amproliuma 25–500 mg/kg,
— 75 mg/kg za vsebnosti amproliuma 500–1 000 mg/kg,
— 7,5 % višje vrednosti za vsebnosti amproliuma nad 1 000 mg/kg.
7.3 Izkoristek
Pri vzorcu z dodatkom (slepem vzorcu) je izkoristek najmanj 90-odstoten.
8. Rezultati medlaboratorijske študije
Organizirana je bila medlaboratorijska študija, pri kateri so bili analizirani trije vzorci perutninske krme (vzorci 1–3), en vzorec mineralne krme (vzorec 4) in en vzorec premiksa (vzorec 5). Rezultati so navedeni v spodnji preglednici:
|
|
Vzorec 1 (slepa krma) |
Vzorec 2 |
Vzorec 3 |
Vzorec 4 |
Vzorec 5 |
|
L |
14 |
14 |
14 |
14 |
15 |
|
n |
56 |
56 |
56 |
56 |
60 |
|
Srednja vrednost [mg/kg] |
— |
45,5 |
188 |
5 129 |
25 140 |
|
Sr [mg/kg] |
— |
2,26 |
3,57 |
178 |
550 |
|
CVr [ %] |
— |
4,95 |
1,90 |
3,46 |
2,20 |
|
SR [mg/kg] |
— |
2,95 |
11,8 |
266 |
760 |
|
CVR [ %] |
— |
6,47 |
6,27 |
5,19 |
3,00 |
|
Nominalna vsebnost [mg/kg] |
— |
50 |
200 |
5 000 |
25 000 |
|
L |
: |
število laboratorijev; |
|
n |
: |
število posameznih vrednosti; |
|
Sr |
: |
standardni odmik ponovljivosti; |
|
CVr |
: |
koeficient variacije ponovljivosti; |
|
SR |
: |
standardni odmik obnovljivosti; |
|
CVR |
: |
koeficient variacije obnovljivosti. |
9. Opombe
9.1 Če vzorec vsebuje tiamin, se vrh tiamina v kromatogramu pojavi malo pred vrhom amproliuma. Pri tej metodi se morata amprolium in tiamin ločiti. Če se amprolium in tiamin ne ločita s kolono (4.1.1), ki se uporablja pri tej metodi, se do 50 % acetonitrila v mobilni fazi (3.6) zamenja z metanolom.
9.2 Po britanski farmakopeji sta najvišji točki spektra raztopine amproliuma (c = 0,02 mol/l) v klorovodikovi kislini (c = 0,1 mol/l) pri 246 nm in 262 nm. Absorbanca znaša 0,84 pri 246 nm in 0,80 pri 262 nm.
9.3 Ekstrakt se mora vedno razredčiti z mobilno fazo, ker se sicer retencijski čas vrha amproliuma bistveno premakne zaradi sprememb v ionski moči.
D. DOLOČANJE KARBADOKSA
Metil 3-(2-kvinoksalinilmetilen)karbazat N 1 ,N 4 -dioksid
1. Namen in področje uporabe
Ta metoda omogoča določanje ravni karbadoksa v krmi, premiksih in pripravkih. Meja določanja je 1 mg/kg, meja kvantifikacije pa 5 mg/kg.
2. Načelo
Vzorec se uravnoteži z vodo in ekstrahira z metanol-acetonitrilom. Pri krmi se alikvot filtriranega ekstrakta prečisti na koloni iz aluminijevega oksida. Pri premiksih in pripravkih se alikvot filtriranega ekstrakta z vodo, metanolom in acetonitrilom razredči do primerne koncentracije. Vsebnost karbadoksa se določi s tekočinsko kromatografijo visoke ločljivosti (HPLC) z reverzno fazo z uporabo UV-detektorja.
3. Reagenti
3.1 Metanol
3.2 Acetonitril, za HPLC
3.3 Ocetna kislina, m = 100 %
3.4 Aluminijev oksid: nevtralni, stopnja aktivnosti I
3.5 Metanol-acetonitril 1 + 1 (v + v)
500 ml metanola (3.1) se zmeša s 500 ml acetonitrila (3.2).
3.6 Ocetna kislina, σ = 10 %
10 ml ocetne kisline (3.3) se razredči z vodo do 100 ml.
3.7 Natrijev acetat
3.8 Voda, za HPLC
3.9 Raztopina acetatnega pufra, c = 0,01 mol/l, pH = 6,0
0,82 g natrijevega acetata (3.7) se raztopi v 700 ml vode (3.8), pH pa se z ocetno kislino (3.6) uravna na 6,0 . Prenese se v 1 000 -mililitrsko merilno bučko, dopolni se z vodo (3.8) do oznake in premeša.
3.10 Mobilna faza za HPLC
825 ml raztopine acetatnega pufra (3.9) se zmeša s 175 ml acetonitrila (3.2).
Filtrira se skozi filter 0,22 μm (4.5), raztopina pa se razplini (npr. 10 minut v ultrazvočni kopeli).
3.11 Standardna snov
Čisti karbadoks: metil 3-(2-kvinoksalinilmetilen)karbazat N1,N4-dioksid, E 850
3.11.1 Osnovna standardna raztopina karbadoksa, 100 μg/ml (glej točko 5 Postopek):
25 mg standardne snovi karbadoksa (3.11) se natehta na 0,1 mg natančno in prenese v 250-mililitrsko merilno bučko. V ultrazvočni kopeli (4.7) se raztopi v metanol-acetonitrilu (3.5). Po ultrazvočni obdelavi se raztopina segreje na sobno temperaturo, dopolni se z metanol-acetonitrilom (3.5) do oznake in premeša. Bučka se ovije v aluminijasto folijo ali pa se uporabi rjava bučka ter shrani v hladilniku. Pri temperaturi ≤ 4 oC je raztopina stabilna 1 mesec.
3.11.2
2,0 , 5,0 , 10,0 in 20,0 ml osnovne standardne raztopine (3.11.1) se prenese v serijo 100-mililitrskih merilnih bučk. Doda se 30 ml vode, dopolni se z metanol-acetonitrilom (3.5) do oznake in premeša. Bučke se ovijejo v aluminijasto folijo. Te raztopine ustrezajo koncentracijam karbadoksa 2,0 , 5,0 , 10,0 , in 20,0 μg/ml.
Raztopine za umerjanje je treba pripraviti sveže pred uporabo.
|
Opomba: |
Za določanje karbadoksa v krmi, ki vsebuje manj kot 10 mg/kg karbadoksa, se morajo pripraviti raztopine za umerjanje s koncentracijo, nižjo od 2,0 μg/ml. |
3.12 Mešanica voda-[metanol-acetonitril] (3.5), 300 + 700 (v + v)
300 ml vode se zmeša s 700 ml mešanice metanola in acetonitrila (3.5).
4. Oprema
4.1 Laboratorijski stresalnik ali magnetno mešalo
4.2 Filtrski papir iz steklenih vlaken (Whatman GF/A ali enakovreden)
4.3 Steklena kolona (dolžina 300 do 400 mm, notranji premer približno 10 mm) s sintrano stekleno frito in odtočnim ventilom
|
Opomba: |
Lahko se uporablja tudi steklena kolona s petelinčkom ali zamaškom; v tem primeru se v spodnji konec vstavi majhen zamašek iz steklene volne in se potlači s stekleno palčko. |
4.4 Oprema za HPLC s sistemom za vbrizgavanje, primernim za vbrizgavanje volumnov 20 μl
4.4.1 Kolona za tekočinsko kromatografijo: 300 mm x 4 mm, C18, polnitev 10 μm ali enakovredna
4.4.2 UV-detektor z nastavljivo valovno dolžino ali detektor s serijo diod v območju 225 do 400 nm
4.5 Membranski filter, 0,22 μm
4.6 Membranski filter, 0,45 μm
4.7 Ultrazvočna kopel
5. Postopek
|
Opomba: |
Karbadoks je občutljiv na svetlobo. Vsi postopki se izvajajo pri zmanjšani svetlobi ali pa se uporabi posoda iz rjavega stekla ali se bučka ovije v aluminijasto folijo. |
5.1 Splošno
5.1.1
Za učinkovitost preskusa izkoristka (5.1.2) se z analizo slepe krme zagotovi odsotnost karbadoksa in motečih snovi. Vrsta slepe krme je podobna vrsti vzorca in pri analizi se dokaže odsotnost karbadoksa ali motečih snovi.
5.1.2
Preskus izkoristka se izvede z analizo slepe krme (5.1.1) z dodatkom količine karbadoksa, podobne količini v vzorcu. Za koncentracijo 50 mg/kg se 5,0 ml osnovne standardne raztopine (3.11.1) prenese v 200-mililitrsko erlenmajerico. Raztopina se upari na približno 0,5 ml v toku dušika. Doda se 10 g slepe krme, premeša se in pusti 10 minut, nato pa se nadaljuje z ekstrakcijo (5.2).
Če pa vrsta slepe krme, podobna vrsti vzorca, ni na voljo (glej točko 5.1.1), se lahko preskus izkoristka izvede po metodi standardnega dodatka. V tem primeru se vzorcu za analizo doda količina karbadoksa, podobna količini v vzorcu. Ta vzorec se analizira skupaj z vzorcem brez dodanega karbadoksa, izkoristek pa se izračuna z odštevanjem.
5.2 Ekstrakcija
5.2.1
10 g vzorca se natehta na 0,01 g natančno in prenese v 200-mililitrsko erlenmajerico. Doda se 15,0 ml vode, premeša se in pusti 5 minut, da se uravnoteži. Doda se 35,0 ml metanol-acetonitrila (3.5), zamaši se in stresa 30 minut na stresalniku ali meša z magnetnim mešalnikom (4.1). Raztopina se filtrira skozi filtrirni papir iz steklenih vlaken (4.2). Ta raztopina se shrani za fazo čiščenja (5.3).
5.2.2
1 g nezdrobljenega vzorca se natehta na 0,001 g natančno in prenese v 200-mililitrsko erlenmajerico. Doda se 15,0 ml vode, premeša se in pusti 5 minut, da se uravnoteži. Doda se 35,0 ml metanol-acetonitrila (3.5), zamaši se in stresa 30 minut na stresalniku ali meša z magnetnim mešalnikom (4.1). Raztopina se filtrira skozi filtrirni papir iz steklenih vlaken (4.2).
Alikvot filtrata se odpipetira v 50-mililitrsko merilno bučko. Doda se 15,0 ml vode, dopolni se z metanol-acetonitrilom (3.5) do oznake in premeša. Koncentracija karbadoksa v končni raztopini je približno 10 μg/ml. Alikvot se filtrira skozi filter 0,45 μm (4.6).
Nadaljuje se z določanjem s HPLC (5.4).
5.2.3
0,2 g nezdrobljenega vzorca se natehta na 0,001 g natančno in prenese v 250-mililitrsko erlenmajerico. Doda se 45,0 ml vode, premeša se in pusti 5 minut, da se uravnoteži. Doda se 105,0 ml metanol-acetonitrila (3.5), zamaši se in homogenizira. Vzorec se pusti v ultrazvočni kopeli (4.7) 15 minut, nato pa se 15 minut stresa ali meša (4.1). Raztopina se filtrira skozi filtrirni papir iz steklenih vlaken (4.2).
Alikvot filtrata se razredči z mešanico vode, metanola in acetonitrila (3.12), da je končna koncentracija karbadoksa 10–15 μg/ml (za 10-odstotni preparat je faktor razredčitve 10). Alikvot se filtrira skozi filter 0,45 μm (4.6).
Nadaljuje se z določanjem s HPLC (5.4).
5.3 Čiščenje
5.3.1
Natehta se 4 g aluminijevega oksida (3.4) in prenese v stekleno kolono (4.3).
5.3.2
15 ml filtriranega ekstrakta (5.2.1) se nanese na kolono iz aluminijevega oksida, prva 2 ml eluata pa se zavržeta. Zbere se naslednjih 5 ml, alikvot pa se filtrira skozi filter 0,45 μm (4.6).
Nadaljuje se z določanjem s HPLC (5.4).
5.4 Določanje s HPLC
5.4.1
Naslednji pogoji se lahko uporabljajo kot smernice, lahko pa se uporabljajo drugi pogoji, če zagotavljajo enakovredne rezultate:
|
Kolona za tekočinsko |
|
|
kromatografijo (4.4.1): |
300 mm × 4 mm, C18, polnitev 10 μm ali enakovredna |
|
Mobilna faza (3.10): |
Mešanica raztopine acetatnega pufra (3.9) in acetonitrila (3.2), 825 + 175 (v+v) |
|
Hitrost pretoka: |
1,5 –2 ml/min |
|
Detekcijska valovna dolžina: |
365 nm |
|
Volumen, ki se vbrizga: |
20 μl |
Stabilnost kromatografskega sistema se preveri z večkratnim vbrizgavanjem raztopine za umerjanje (3.11.2) s koncentracijo 5,0 μg/ml, dokler se ne dosežejo konstantne višine vrhov (površin) in konstantni retencijski časi.
5.4.2
Vsaka raztopina za umerjanje (3.11.2) se večkrat vbrizga in za vsako koncentracijo se določijo srednje vrednosti višin (površin) vrhov. Umeritvena krivulja se pripravi tako, da se na ordinato nanesejo srednje vrednosti višin ali površin vrhov raztopin za umerjanje, na absciso pa ustrezne koncentracije v μg/ml.
5.4.3
Ekstrakt vzorca [(5.3.2) za krmo, (5.2.2) za premikse in (5.2.3) za pripravke] se vbrizga večkrat, nato pa se določijo srednje vrednosti višin (površin) vrhov karbadoksa.
6. Izračun rezultatov
Koncentracija karbadoksa v raztopini vzorca v μg/ml se določi iz srednje vrednosti višin (površin) vrhov karbadoksa iz umeritvene krivulje (5.4.2).
6.1 Krma
Vsebnost karbadoksa (w) (mg/kg) v vzorcu se izračuna po naslednji formuli:

[mg/kg]
pri čemer je:
|
c |
= |
koncentracija karbadoksa v ekstraktu vzorca (5.3.2) v μg/ml; |
|
V1 |
= |
volumen ekstrakta v ml (tj. 50); |
|
m |
= |
masa preskusnega vzorca v gramih. |
6.2 Premiksi in pripravki
Vsebnost karbadoksa (w) (mg/kg) v vzorcu se izračuna po naslednji formuli:

[mg/kg]
pri čemer je:
|
c |
= |
koncentracija karbadoksa v ekstraktu vzorca (5.2.2 ali 5.2.3) v μg/ml; |
|
V2 |
= |
volumen ekstrakta v ml (tj. 50 za premikse; 150 za pripravke); |
|
f |
= |
faktor razredčitve v skladu s točko 5.2.2 (premiksi) ali 5.2.3 (pripravki); |
|
m |
= |
masa preskusnega vzorca v gramih. |
7. Potrditev rezultatov
7.1 Identiteta
Identiteta analita se lahko potrdi z dodatno kromatografijo ali detektorjem s serijo diod, s katerim se primerjata spekter ekstrakta vzorca in spekter raztopine za umerjanje (3.11.2) s koncentracijo 10,0 μg/ml.
7.1.1
Ekstraktu vzorca se doda primerna količina raztopine za umerjanje (3.11.2). Količina dodanega karbadoksa mora biti podobna ocenjeni količini karbadoksa v ekstraktu vzorca.
Povišan je le vrh karbadoksa ob upoštevanju dodane količine in razredčitve ekstrakta. Širina vrha na polovici njegove največje višine mora biti v mejah približno 10 % prvotne širine.
7.1.2
Rezultati se ocenijo glede na naslednja merila:
(a) valovni dolžini maksimalne absorpcije spektra vzorca in spektra standarda, zabeleženi na najvišji točki vrha na kromatogramu, morata biti znotraj meja, določenih z ločljivostjo sistema za detekcijo. Za detekcijo s serijo diod je to ponavadi znotraj ± 2 nm;
(b) med 225 in 400 nm se spekter vzorca in spekter standarda na najvišji točki kromatografskega vrha za navedene dele spektra ne smeta razlikovati znotraj območja 10–100 % relativne absorbance. To merilo je izpolnjeno, ko so prisotne iste najvišje vrednosti in ko odmik med dvema spektroma na nobeni opazovani točki ne preseže 15 % absorbance standardnega analita;
(c) med 225 in 400 nm se spektri naraščanja, najvišje točke vrha in padanja vrha, pridobljeni iz ekstrakta vzorca, za navedene dele spektra ne smejo razlikovati med seboj v območju 10–100 % relativne absorbance. To merilo je izpolnjeno, ko so prisotni iste najvišje točke in ko odmik med spektri na nobeni od opazovanih točk ne preseže 15 % absorbance spektra najvišje točke.
Če eno od teh meril ni izpolnjeno, prisotnost analita ni potrjena.
7.2 Ponovljivost
Razlika med rezultatoma dveh vzporednih določanj, izvedenih na istem vzorcu, za vsebnosti 10 mg/kg in več ne sme presegati 15 % višjega rezultata.
7.3 Izkoristek
Pri vzorcu z dodatkom (slepem vzorcu) je izkoristek najmanj 90-odstoten.
8. Rezultati medlaboratorijske študije
Organizirana je bila medlaboratorijska študija, pri kateri je 8 laboratorijev analiziralo 6 vzorcev krme, 4 vzorce premiksov in 3 vzorce pripravkov. Vsak vzorec je bil analiziran dvakrat. (Za natančnejše informacije o medlaboratorijski študiji glej Journal of AOAC, knjiga 71, št. 1988, 484, str. 484–490.) Rezultati (razen izjem) so prikazani v spodnji preglednici:
Preglednica 1.
Rezultati medlaboratorijske študije za krmo
|
|
Vzorec 1 |
Vzorec 2 |
Vzorec 3 |
Vzorec 4 |
Vzorec 5 |
Vzorec 6 |
|
L |
8 |
8 |
8 |
8 |
8 |
8 |
|
n |
15 |
14 |
15 |
15 |
15 |
15 |
|
Srednja vrednost (mg/kg) |
50,0 |
47,6 |
48,2 |
49,7 |
46,9 |
49,7 |
|
Sr (mg/kg) |
2,90 |
2,69 |
1,38 |
1,55 |
1,52 |
2,12 |
|
CVr (%) |
5,8 |
5,6 |
2,9 |
3,1 |
3,2 |
4,3 |
|
SR (mg/kg) |
3,92 |
4,13 |
2,23 |
2,58 |
2,26 |
2,44 |
|
CVR (%) |
7,8 |
8,7 |
4,6 |
5,2 |
4,8 |
4,9 |
|
Nominalna vsebnost (mg/kg) |
50,0 |
50,0 |
50,0 |
50,0 |
50,0 |
50,0 |
Preglednica 2.
Rezultati medlaboratorijske študije za premikse in pripravke
|
|
Premiksi |
Pripravki |
|||||
|
A |
B |
C |
D |
A |
B |
C |
|
|
L |
7 |
7 |
7 |
7 |
8 |
8 |
8 |
|
n |
14 |
14 |
14 |
14 |
16 |
16 |
16 |
|
Srednja vrednost (g/kg) |
8,89 |
9,29 |
9,21 |
8,76 |
94,6 |
98,1 |
104 |
|
Sr (g/kg) |
0,37 |
0,28 |
0,28 |
0,44 |
4,1 |
5,1 |
7,7 |
|
CVr (%) |
4,2 |
3,0 |
3,0 |
5,0 |
4,3 |
5,2 |
7,4 |
|
SR (g/kg) |
0,37 |
0,28 |
0,40 |
0,55 |
5,4 |
6,4 |
7,7 |
|
CVR (%) |
4,2 |
3,0 |
4,3 |
6,3 |
5,7 |
6,5 |
7,4 |
|
Nominalna vsebnost (g/kg) |
10,0 |
10,0 |
10,0 |
10,0 |
100 |
100 |
100 |
|
L |
: |
število laboratorijev; |
|
n |
: |
število posameznih vrednosti; |
|
Sr |
: |
standardni odmik ponovljivosti; |
|
CVr |
: |
koeficient variacije ponovljivosti; |
|
SR |
: |
standardni odmik obnovljivosti; |
|
CVR |
: |
koeficient variacije obnovljivosti. |
PRILOGA IX
KORELACIJSKE TABELE IZ ČLENA 6
1. Direktiva 71/250/EGS
|
Direktiva 71/250/EGS |
Ta uredba |
|
Člen 1, prvi pododstavek |
Člen 3 |
|
Člen 1, drugi pododstavek |
Člen 2 |
|
Člen 2 |
— |
|
Člen 3 |
— |
|
Priloga, del 1 |
Priloga II |
|
Priloga, del 2 |
— |
|
Priloga, del 3 |
— |
|
Priloga, del 4 |
Priloga III, del O |
|
Priloga, del 5 |
Priloga III, del M |
|
Priloga, del 6 |
Priloga III, del N |
|
Priloga, del 7 |
Priloga III, del Q |
|
Priloga, del 9 |
Priloga III, del K |
|
Priloga, del 10 |
— |
|
Priloga, del 11 |
— |
|
Priloga, del 12 |
Priloga III, del J |
|
Priloga, del 14 |
Priloga III, del D |
|
Priloga, del 16 |
— |
2. Direktiva 71/393/EGS
|
Direktiva 71/393/EGS |
Ta uredba |
|
Člen 1 |
Člen 3 |
|
Člen 2 |
— |
|
Člen 3 |
— |
|
Priloga, del I |
Priloga III, del A |
|
Priloga, del II |
Priloga III, del E |
|
Priloga, del III |
Priloga III, del P |
|
Priloga, del IV |
Priloga III, del H |
3. Direktiva 72/199/EGS
|
Direktiva 72/199/EGS |
Ta uredba |
|
Člen 1 |
Člen 3 |
|
Člen 2 |
— |
|
Člen 3 |
— |
|
Člen 4 |
— |
|
Priloga I, del 1 |
Priloga III, del L |
|
Priloga I, del 2 |
Priloga III, del C |
|
Priloga I, del 3 |
— |
|
Priloga I, del 4 |
— |
|
Priloga I, del 5 |
Priloga V, del A |
|
Priloga II |
— |
4. Direktiva 73/46/EGS
|
Direktiva 73/46/EGS |
Ta uredba |
|
Člen 1 |
Člen 3 |
|
Člen 3 |
— |
|
Člen 4 |
— |
|
Priloga I, del 1 |
Priloga III, del B |
|
Priloga I, del 2 |
— |
|
Priloga I, del 3 |
Priloga III, del I |
5. Direktiva 76/371/EGS
|
Direktiva 76/371/EGS |
Ta uredba |
|
Člen 1 |
Člen 1 |
|
Člen 2 |
— |
|
Člen 3 |
— |
|
Priloga |
Priloga I |
6. Direktiva 76/372/EGS
|
Direktiva 76/372/EGS |
Ta uredba |
|
Člen 1 |
— |
|
Člen 2 |
— |
|
Člen 3 |
— |
|
Priloga |
— |
7. Direktiva 78/633/EGS
|
Direktiva 78/633/EGS |
Ta uredba |
|
Člen 1 |
Člen 3 |
|
Člen 2 |
— |
|
Člen 3 |
— |
|
Priloga, del 1 |
— |
|
Priloga, del 2 |
— |
|
Priloga, del 3 |
Priloga IV, del C |
8. Direktiva 81/715/EGS
|
Direktiva 81/715/EGS |
Ta uredba |
|
Člen 1 |
— |
|
Člen 2 |
— |
|
Člen 3 |
— |
|
Priloga |
— |
9. Direktiva 84/425/EGS
|
Direktiva 84/425/EGS |
Ta uredba |
|
Člen 1 |
— |
|
Člen 2 |
— |
|
Člen 3 |
— |
|
Priloga |
— |
10. Direktiva 86/174/EGS
|
Direktiva 86/174/EGS |
Ta uredba |
|
Člen 1 |
Člen 4 |
|
Člen 2 |
— |
|
Člen 3 |
— |
|
Priloga |
Priloga VII |
11. Direktiva 93/70/EGS
|
Direktiva 93/70/EGS |
Ta uredba |
|
Člen 1 |
Člen 3 |
|
Člen 2 |
— |
|
Člen 3 |
— |
|
Priloga |
Priloga IV, del D |
12. Direktiva 93/117/ES
|
Direktiva 93/117/ES |
Ta uredba |
|
Člen 1 |
Člena 3 in 5 |
|
Člen 2 |
— |
|
Člen 3 |
— |
|
Priloga, del 1 |
Priloga IV, del E |
|
Priloga, del 2 |
Priloga VIII, del A |
13. Direktiva 98/64/ES
|
Direktiva 98/64/ES |
Ta uredba |
|
Člen 1 |
Člena 3 in 5 |
|
Člen 2 |
— |
|
Člen 3 |
— |
|
Člen 4 |
— |
|
Priloga, del A |
Priloga III, del F |
|
Priloga, del C |
Priloga VIII, del B |
14. Direktiva 1999/27/ES
|
Direktiva 1999/27/ES |
Ta uredba |
|
Člen 1 |
Člena 3 in 5 |
|
Člen 2 |
— |
|
Člen 3 |
— |
|
Člen 4 |
— |
|
Člen 5 |
— |
|
Člen 6 |
— |
|
Člen 7 |
— |
|
Priloga, del A |
Priloga VIII, del C |
|
Priloga, del B |
Priloga IV, del F |
|
Priloga, del C |
Priloga VIII, del D |
15. Direktiva 1999/76/ES
|
Direktiva 1999/76/ES |
Ta uredba |
|
Člen 1 |
Člen 3 |
|
Člen 2 |
— |
|
Člen 3 |
— |
|
Člen 4 |
— |
|
Priloga |
Priloga IV, del G |
16. Direktiva 2000/45/ES
|
Direktiva 2000/45/ES |
Ta uredba |
|
Člen 1 |
Člen 3 |
|
Člen 2 |
— |
|
Člen 3 |
— |
|
Člen 4 |
— |
|
Priloga, del A |
Priloga IV, del A |
|
Priloga, del B |
Priloga IV, del B |
|
Priloga, del C |
Priloga III, del G |
17. Direktiva 2002/70/ES
|
Direktiva 2002/70/ES |
Ta uredba |
|
Člen 1 |
Člen 1 |
|
Člen 2 |
Člena 2 in 3 |
|
Člen 3 |
— |
|
Člen 4 |
— |
|
Člen 5 |
— |
|
Priloga I |
Priloga I in Priloga V, del B (I) |
|
Priloga II |
Priloga II in Priloga V, del B (II) |
18. Direktiva 2003/126/ES
|
Direktiva 2003/126/ES |
Ta uredba |
|
Člen 1 |
Člen 3 |
|
Člen 2 |
— |
|
Člen 3 |
— |
|
Člen 4 |
— |
|
Člen 5 |
— |
|
Člen 6 |
— |
|
Priloga |
Priloga VI |
( 1 ) UL L 268, 18.10.2003, str. 29.
( 2 ) UL L 140, 30.5.2002, str. 10.
( 3 ) UL L 70, 16.3.2005, str. 1.
( 4 ) Morebitne grude se razbijejo (po potrebi z ločevanjem in vrnitvijo v vzorec).
( 5 ) Razen v primeru voluminozne krme ali vlaknin z majhno specifično težo.
( 6 ) Razen v primeru voluminozne krme ali vlaknin z majhno specifično težo.
( 7 ) UL L 229, 1.9.2009, str. 1.
( 7 ) Za sušenje žita, moke, drobljencev in zdroba mora imeti sušilnik takšno toplotno kapaciteto, da se bo, ko je predhodno nastavljen na 131 oC, vrnil na to temperaturo v manj kot 45 minutah, potem ko je bilo vanj za sušenje hkrati postavljeno največje možno število vzorcev. Ventilacija mora biti takšna, da se, ko se v sušilniku dve uri suši največje možno število vzorcev pšenice, rezultati razlikujejo za manj kot 0,15 % od tistih, ki so bili sušeni 4 ure.
( 7 ) Kadar je treba kakovost olja ali maščob naknadno še preskušati, se delci plovca zamenjajo s steklenimi kroglicami.
( 7 ) UL L 125, 23.5.1996, str. 35.
( 7 ) Izvedla delovna skupina za krmo Verband Deutscher Landwirtschaftlicher Untersuchungs- und Forschungsanstalten (VDLUFA).
( 7 ) Izvedla delovna skupina za krmo Verband Deutscher Landwirtschaftlicher Untersuchungs- und Forschungsanstalten (VDLUFA).
( 7 ) Lahko se uporabljajo druge metode za razklop, če dokazano zagotavljajo podobne rezultate (kot je npr. mikrovalovni razklop).
( 7 ) V zeleni krmi (sveži ali posušeni) je lahko velika količina kremena rastlinskega izvora, v katerem so lahko elementi v sledovih in ga je treba odstraniti. Pri vzorcih takšne krme je treba izvajati naslednji spremenjeni postopek. Postopki iz točke 5.1.1.1 se izvedejo do filtracije. Filtrirni papir, ki vsebuje netopen ostanek, se dvakrat spere z vrelo vodo in prenese v žarilni lonček iz kvarca ali platine. Prežari se v žarilni peči (4.1) pri temperaturi pod 550 oC, dokler ves zogleneli material popolnoma ne izgine. Pusti se ohladiti, doda se nekaj kapljic vode in 10–15 ml klorovodikove kisline (3.4), nato pa se upari do suhega pri približno 150 oC. Če v ostanku ostane kremen, se ponovno raztopi v nekaj mililitrih klorovodikove kisline (3.4) in upari do suhega. Doda se pet kapljic žveplove kisline (3.5) in segreva do prenehanja izhajanja belih hlapov. Po dodatku 5 ml klorovodikove kisline 6 mol/l (3.2) in približno 30 ml vode se segreva, raztopina se filtrira v 250-mililitrsko merilno bučko ter dopolni z vodo do oznake (koncentracija HCl približno 0,5 mol/l). Nadaljuje se z določanjem v skladu s točko 5.1.2.
( 7 ) The Analyst 108, 1983, str. 1252–1256.
( *1 ) Analyst, 1995, 120, 2175-2180;
( 8 ) Razpredelnica TEF (faktorjev toksične ekvivalentnosti) za dioksine, furane in dioksinom podobne PCB:
Faktorji toksične ekvivalentnosti (TEF) Svetovne zdravstvene organizacije (SZO) za oceno tveganja za ljudi, ki temeljijo na sklepih Svetovne zdravstvene organizacije – strokovnega srečanja Mednarodnega programa za kemijsko varnost (IPCS) junija 2005 v Ženevi (Martin van den Berg in sod., Ponovna ocena faktorjev toksične ekvivalentnosti za dioksine in dioksinom podobne spojine pri ljudeh in sesalcih, Svetovna zdravstvena organizacija, 2005 (The 2005 World Health Organization Re-evaluation of Human and Mammalian Toxic Equivalency Factors for Dioxins and Dioxin-like Compounds). Toxicological Sciences 93(2), str. 223–241 (2006)).
|
Kongener |
Vrednost faktorja toksične ekvivalentnosti (TEF) |
Kongener |
Vrednost faktorja toksične ekvivalentnosti (TEF) |
|
Dibenzo-p-dioksini („PCDD“) in dibenzo-p-furani („PCDF“) |
Dioksinom podobni neorto PCB + monoorto PCB |
||
|
2,3,7,8-TCDD |
1 |
|
|
|
1,2,3,7,8-PeCDD |
1 |
Neorto PCB |
|
|
1,2,3,4,7,8-HxCDD |
0,1 |
PCB 77 |
0,0001 |
|
1,2,3,6,7,8-HxCDD |
0,1 |
PCB 81 |
0,0003 |
|
1,2,3,7,8,9-HxCDD |
0,1 |
PCB 126 |
0,1 |
|
1,2,3,4,6,7,8-HpCDD |
0,01 |
PCB 169 |
0,03 |
|
OCDD |
0,0003 |
|
|
|
|
|
Monoorto PCB |
|
|
2,3,7,8-TCDF |
0,1 |
PCB 105 |
0,00003 |
|
1,2,3,7,8-PeCDF |
0,03 |
PCB 114 |
0,00003 |
|
2,3,4,7,8-PeCDF |
0,3 |
PCB 118 |
0,00003 |
|
1,2,3,4,7,8-HxCDF |
0,1 |
PCB 123 |
0,00003 |
|
1,2,3,6,7,8-HxCDF |
0,1 |
PCB 156 |
0,00003 |
|
1,2,3,7,8,9-HxCDF |
0,1 |
PCB 157 |
0,00003 |
|
2,3,4,6,7,8-HxCDF |
0,1 |
PCB 167 |
0,00003 |
|
1,2,3,4,6,7,8-HpCDF |
0,01 |
PCB 189 |
0,00003 |
|
1,2,3,4,7,8,9-HpCDF |
0,01 |
|
|
|
OCDF |
0,0003 |
|
|
Uporabljene okrajšave:„T“ = tetra; „Pe“ = penta; „Hx“ = heksa; „Hp“ = hepta; „O“ = okta; „CDD“ = klorodibenzodioksin; „CDF“ = klorodibenzofuran; „CB“ = klorobifenil.
( 9 ) Odločba Komisije 2002/657/ES z dne 14. avgusta 2002 o izvajanju Direktive Sveta 96/23/ES glede opravljanja analitskih metod in razlage rezultatov (UL L 221, 17.8.2002, str. 8).
( 10 ) Pojem „na zgornji meji“ zahteva, da se za izračun prispevka vsakega kongenera, ki ni količinsko določen, uporablja vrednost meje količinskega določanja. Pojem „na spodnji meji“ zahteva, da se za izračun prispevka vsakega kongenera, ki ni določen, uporablja vrednost nič. Pojem „na srednji meji“ zahteva, da se za izračun prispevka vsakega kongenera, ki ni količinsko določen, uporablja polovična vrednost meje količinskega določanja.
( 11 ) Na splošno se uporabljajo zahteve za dvakratno analizo iz točke 3 poglavja C Priloge II. Vendar je za potrditvene metode, pri katerih se za ustrezne analite uporablja s 13C označen interni standard, dvakratna analiza potrebna le, če rezultat prvega določanja z uporabo take potrditvene metode ni skladen. Dvakratna analiza je potrebna za izključitev možnosti notranje navzkrižne kontaminacije ali naključne zamenjave vzorcev. Če se analiza izvaja zaradi kontaminacije, se lahko izpusti potrjevanje z dvakratno analizo, če so vzorci, izbrani za analizo, s sledljivostjo povezani s kontaminacijo, ugotovljena vsebnost pa bistveno presega mejno vrednost.
( 12 ) Pojem „na zgornji meji“ zahteva, da se za izračun prispevka k toksičnemu ekvivalentu (TEQ) vsakega kongenera, ki ni količinsko določen, uporablja vrednost meje količinskega določanja. Pojem „na spodnji meji“ zahteva, da se za izračun prispevka k toksičnemu ekvivalentu (TEQ) vsakega kongenera, ki ni določen, uporablja vrednost nič. Pojem „na srednji meji“ zahteva, da se za izračun prispevka k toksičnemu ekvivalentu (TEQ) vsakega kongenera, ki ni določen, uporablja polovična vrednost meje določanja.
( 13 ) Na splošno se uporabljajo zahteve za dvakratno analizo iz točke 3 poglavja C Priloge II. Vendar je za potrditvene metode, pri katerih se za ustrezne analite uporablja s 13C označen interni standard, dvakratna analiza potrebna le, če rezultat prvega določanja z uporabo takih potrditvenih metod ni skladen. Dvakratna analiza je potrebna za izključitev možnosti notranje navzkrižne kontaminacije ali naključne zamenjave vzorcev. Če se analiza izvaja zaradi kontaminacije, se lahko izpusti potrjevanje z dvakratno analizo, če so vzorci, izbrani za analizo, s sledljivostjo povezani s kontaminacijo, ugotovljena vsebnost pa bistveno presega mejno vrednost.
( 14 ) Enaka razlaga in zahteve za dvakratno analizo za nadzor pragov ukrepanja kot v opombi (20) za mejne vrednosti.
( 15 ) Bioanalitske metode niso specifične za tiste kongenere, ki so vključeni v shemo TEF. Druge strukturno povezane spojine, ki se vežejo na aromatski ogljikovodikov receptor (AhR), so lahko prisotne v ekstraktu vzorca, kar prispeva k celotnemu odzivu. Bioanalitski rezultati zato ne morejo dati ocene vsebnosti, temveč le nakažejo vsebnost TEQ v vzorcu.
( 16 ) Sedanje zahteve temeljijo na TEF, ki so objavljeni v: M. Van den Berg in sod., Toxicol Sci 93 (2), str. 223–241 (2006).
( 17 ) Zelo priporočljivo je, da je prispevek slepega reagenta k vsebnosti onesnaževala v vzorcu manjši. Laboratorij je odgovoren za nadzor spreminjanja slepih vrednosti, zlasti če se slepe vrednosti odštejejo.
( 18 ) http://eurl.craw.eu/
( 19 ) Seznam teh začetnih oligonukleotidov in sond za vsako živalsko vrsto, na katero se pregleduje, je na voljo na spletni strani EURL-AP.
( 20 ) Primeri osnovnih zmesi, ki so funkcionalne. so na voljo na spletni strani EURL-AP.