

EUR-Lex Access to European Union law
This document is an excerpt from the EUR-Lex website
Document 01991R2568-20191020
Commission Regulation (EEC) No 2568/91 of 11 July 1991 on the characteristics of olive oil and olive-residue oil and on the relevant methods of analysis
Consolidated text: Nariadenie Komisie (EHS) č. 2568/91 z 11. júla 1991, o charakteristikách olivového oleja a oleja z olivových zvyškov a o príslušných analytických metódach
Nariadenie Komisie (EHS) č. 2568/91 z 11. júla 1991, o charakteristikách olivového oleja a oleja z olivových zvyškov a o príslušných analytických metódach
 No longer in force
No longer in force
01991R2568 — SK — 20.10.2019 — 032.001
Tento text slúži výlučne ako dokumentačný nástroj a nemá žiadny právny účinok. Inštitúcie Únie nenesú nijakú zodpovednosť za jeho obsah. Autentické verzie príslušných aktov vrátane ich preambúl sú tie, ktoré boli uverejnené v Úradnom vestníku Európskej únie a ktoré sú dostupné na portáli EUR-Lex. Tieto úradné znenia sú priamo dostupné prostredníctvom odkazov v tomto dokumente
|
NARIADENIE KOMISIE (EHS) č. 2568/91 z 11. júla 1991, (Ú. v. ES L 248 5.9.1991, s. 1) |
Zmenené a doplnené:
Opravené a doplnené:
|
(*) |
Tento akt nebol zatiaľ uverejnený v slovenčine. |
NARIADENIE KOMISIE (EHS) č. 2568/91
z 11. júla 1991,
o charakteristikách olivového oleja a oleja z olivových zvyškov a o príslušných analytických metódach
Článok 1
1. Oleje, ktorých charakteristiky sa zhodujú s údajmi uvedenými v bodoch 1 a 2 prílohy I k tomuto nariadeniu, sa považujú za panenské olivové oleje v zmysle bodu 1 písm. a) a b) prílohy k nariadeniu č. 136/66/EHS.
2. Olej, ktorého charakteristiky sa zhodujú s údajmi uvedenými v bode 3 prílohy I k tomuto nariadeniu, sa považuje za lampantový olivový olej v zmysle bodu 1 písm. c) prílohy k nariadeniu č. 136/66/EHS.
3. Olej, ktorého charakteristiky sa zhodujú s údajmi uvedenými v bode 4 prílohy I k tomuto nariadeniu, sa považuje za rafinovaný olivový olej v zmysle bodu 2 prílohy k nariadeniu č. 136/66/EHS.
4. Olej, ktorého charakteristiky sa zhodujú s údajmi uvedenými v bode 5 prílohy I k tomuto nariadeniu, sa považuje za olivový olej zmiešaný z rafinovaných olivových olejov a panenských olivových olejov v zmysle bodu 3 prílohy k nariadeniu č. 136/66/EHS.
5. Olej, ktorého charakteristiky sa zhodujú s údajmi uvedenými v bode 6 prílohy I k tomuto nariadeniu, sa považuje za surový zvyškový olivový olej v zmysle bodu 4 prílohy k nariadeniu č. 136/66/EHS.
6. Olej, ktorého charakteristiky sa zhodujú s údajmi uvedenými v bode 7 prílohy I k tomuto nariadeniu, sa považuje za rafinovaný zvyškový olivový olej v zmysle bodu 5 prílohy k nariadeniu č. 136/66/EHS.
7. Olej, ktorého charakteristiky sa zhodujú s údajmi uvedenými v bode 8 prílohy I k tomuto nariadeniu, sa považuje za zvyškový olivový olej v zmysle bodu 6 prílohy k nariadeniu č. 136/66/EHS.
Článok 2
1. Vlastnosti olejov uvedené v prílohe I sa stanovujú v súlade s týmito analytickými metódami:
a) na stanovenie voľných mastných kyselín vyjadrených ako percentuálny obsah kyseliny olejovej – metóda uvedená v prílohe II;
b) na stanovenie peroxidového čísla – metóda uvedená v prílohe III;
c) na stanovenie obsahu voskov – metóda uvedená v prílohe IV;
d) na stanovenie zloženia a obsahu sterolov a triterpénových diolov kapilárnou plynovou chromatografiou – metóda uvedená v prílohe V;
e) na stanovenie percentuálneho obsahu 2-glyceril monopalmitátu – metóda uvedená v prílohe VII;
f) na spektrofotometrickú analýzu – metóda uvedená v prílohe IX;
g) na stanovenie zloženia mastných kyselín – metóda uvedená v prílohe X;
h) na stanovenie prchavých halogénovaných rozpúšťadiel – metóda uvedená v prílohe XI;
i) na hodnotenie organoleptických vlastností panenského olivového oleja – metóda uvedená v prílohe XII;
j) na stanovenie stigmastadiénov – metóda uvedená v prílohe XVII;
k) na stanovenie obsahu triacylglycerolov prostredníctvom ECN42 – metóda uvedená v prílohe XVIII;
l) na stanovenie zloženia a obsahu sterolov a stanovenie alkoholových zlúčenín kapilárnou plynovou chromatografiou – metóda uvedená v prílohe XIX;
m) na stanovenie obsahu voskov, metylesterov mastných kyselín a etylesterov mastných kyselín – metóda uvedená v prílohe XX.
▼M28 —————
2. Overovanie organoleptických vlastností panenských olejov uskutočňujú vnútroštátne orgány alebo ich zástupcovia prostredníctvom degustačných porôt schválených členskými štátmi.
Organoleptické vlastnosti olejov, ako sa uvádza v prvom pododseku, sa považujú za zhodné s deklarovanou kategóriou, ak degustačná porota schválená príslušným členským štátom toto zatriedenie potvrdila.
V prípade, že by degustačná porota nepotvrdila deklarovanú kategóriu, pokiaľ ide o organoleptické vlastnosti, vnútroštátne orgány alebo ich zástupcovia na žiadosť zainteresovanej strany bezodkladne zadajú vykonanie dvoch kontrolných posúdení iným schváleným porotám. Aspoň jedna z týchto porôt je porotou schválenou dotknutým členským štátom vyrábajúcim olivový olej. Príslušné vlastnosti sa považujú za zhodné s deklarovanými vlastnosťami, ak dané dve kontrolné posúdenia potvrdia deklarované zatriedenie. Ak sa tak nestane, bez ohľadu na typ defektorov zistených pri kontrolných posúdeniach, zatriedenie sa vyhlási za nezhodujúce sa s predmetnými charakteristikami a zainteresovaná strana znáša náklady na kontrolné posúdenia.
3. Na overovanie vlastností oleja podľa odseku 1 vnútroštátnymi orgánmi alebo ich zástupcami sa vzorky odoberajú v súlade s medzinárodnými normami EN ISO 661 o príprave vzoriek na analýzu a EN ISO 5555 o odbere vzoriek. Bez ohľadu na bod 6.8 normy EN ISO 5555 sa však odber vzorky v prípade dávok takýchto olejov vo vnútornom obale uskutoční v súlade s prílohou Ia k tomuto nariadeniu. Odber vzoriek olejov v prepravných obaloch, v prípade ktorých odber nemožno uskutočniť podľa normy EN ISO 5555, sa uskutočňuje v súlade s pokynmi príslušných orgánov členského štátu.
Bez toho, aby bola dotknutá norma EN ISO 5555 a kapitola 6 normy EN ISO 661, sa odobraté vzorky čo najskôr umiestnia na tmavé miesto, mimo silného zdroja tepla, a zašlú do laboratória na analýzu, a to najneskôr piaty pracovný deň po odobratí, inak sa vzorky udržiavajú spôsobom, aby sa ich kvalita neznížila alebo aby sa neznehodnotili počas prepravy alebo skladovania pred odoslaním do laboratória.
4. Na účely overovania stanoveného v odseku 3 sa analýzy uvedené v prílohách II, III, IX, XII a XX a všetky kontrolné analýzy vyžadované podľa vnútroštátnych zákonov, ak také sú, vykonávajú v prípade balených výrobkov pred uplynutím minimálnej doby trvanlivosti. Pokiaľ ide o odber vzoriek olejov v prepravných obaloch, uvedené analýzy sa vykonajú najneskôr šesť mesiacov po mesiaci odberu vzorky.
Na ostatné analýzy stanovené v tomto nariadení sa neuplatňuje žiadne časové obmedzenie.
Ak výsledky analýz nezodpovedajú deklarovaným vlastnostiam kategórie olivového oleja alebo oleja z olivových výliskov, táto skutočnosť sa oznámi dotknutej strane najneskôr mesiac pred koncom lehoty stanovenej v prvom pododseku, a to okrem prípadov, keď sa vzorka odobrala menej ako dva mesiace pred dátumom minimálnej trvanlivosti.
5. Na účely určenia vlastností olivových olejov metódami stanovenými v odseku 1 prvom pododseku sa výsledky analýzy priamo porovnajú s medznými hodnotami stanovenými v tomto nariadení.
Článok 2a
1. Na účely tohto článku označuje „predávaný olivový olej“ celkové množstvo olivového oleja a zvyškového olivového oleja príslušného členského štátu, ktorý sa v tomto štáte spotrebuje alebo sa z neho vyvezie.
2. Členské štáty zabezpečia selektívne a primerane časté vykonávanie kontrol zhody na základe analýzy rizík, aby sa zaistilo, že predávaný olivový olej je v súlade s deklarovanou kategóriou.
3. Kritéria na posúdenie rizík môžu zahŕňať aj:
a) kategóriu oleja, obdobie výroby, cenu oleja vzhľadom na cenu iných rastlinných olejov, postupy miešania a balenia, skladovacie zariadenia a podmienky, krajinu pôvodu, krajinu určenia, spôsob prepravy alebo objem zásielky;
b) pozíciu subjektov v obchodnom reťazci, objem a/alebo hodnotu oleja, ktorý predávajú, kategórie oleja, ktoré predávajú, druh vykonávanej podnikateľskej činnosti, ako je mletie, skladovanie, rafinovanie, miešanie, balenie alebo maloobchodný predaj;
c) zistenia z predchádzajúcich kontrol vrátane počtu a druhu zistených nedostatkov, obvyklej kvality predávaného oleja, úrovne používaného technického vybavenia;
d) spoľahlivosť systémov zaistenia kvality alebo systémov samokontroly subjektu týkajúcich sa dodržiavania obchodných noriem;
e) miesto vykonávania kontrol, najmä či ide o prvé miesto vstupu do Únie, posledné miesto výstupu z Únie alebo miesto výroby, balenia, nakladania alebo predaja oleja koncovému spotrebiteľovi;
f) akékoľvek ďalšie informácie, ktoré môžu poukazovať na riziko nedodržiavania noriem.
4. Členské štáty vopred ustanovia:
a) kritériá posúdenia rizika nedodržiavania obchodných noriem pri zásielkach;
b) na základe analýzy rizík pre každú rizikovú kategóriu minimálny počet subjektov alebo zásielok a/alebo množstiev, pri ktorých sa vykonajú kontroly zhody.
Ročne sa vykoná minimálne jedna kontrola zhody na tisíc ton predávaného olivového oleja v členskom štáte.
5. Členské štáty overujú súlad:
a) vykonaním analýz stanovených v prílohe I v ľubovoľnom poradí alebo
b) postupne v poradí stanovenom v prílohe Ib vo vývojovom diagrame, až kým nedosiahnu jedno z riešení nachádzajúcich sa vo vývojovom diagrame.
▼M19 —————
Článok 3
Ak sa zistí, že určitý olej nezodpovedá jeho opisu kategórie, dotknutý členský štát uplatňuje bez toho, aby boli dotknuté akékoľvek iné pokuty, účinné, primerané a odrádzajúce sankcie, ktoré treba stanoviť vzhľadom na závažnosť zistených nezrovnalostí.
Pokiaľ sa pri kontrolách zistia výrazné nezrovnalosti, zvýšia členské štáty periodicitu kontrol, ktoré sa týkajú stupňa uvedenia na trh, kategórie oleja, pôvodu alebo iných kritérií.
Článok 4
1. The Member States may approve assessment panels so that national authorities or their representatives can assess and verify organoleptic characteristics.
The terms of approval shall be set by Member States and ensure that:
— the requirements of Annex XII.4 are met,
— the panel head is given training recognised for this purpose by the Member State,
— continued approval depends on performance in annual checks arranged by the Member State.
Member States shall notify to the Commission a list of approved panels and the action taken under this paragraph.
2. Ak členské štáty narazia pri ustanovovaní degustačných porôt na svojom území na ťažkosti, môžu sa obrátiť na degustačnú porotu schválenú v inom členskom štáte.
3. Každý členský štát vypracuje zoznam degustačných porôt ustanovených odbornými alebo medziodvetvovými organizáciami v súlade s podmienkami uvedenými v odseku 1 a zabezpečí dosiahnutie súladu s týmito podmienkami.
▼M19 —————
Článok 6
1. Obsah oleja v pokrutinách a ostatných olejových zvyškov, ktoré vznikajú pri extrakcii olivového oleja (číselné znaky KN 2306 90 11 a 2306 90 19 ), sa stanoví použitím metódy uvedenej v prílohe XV.
2. Obsah oleja uvedený v odseku 1 sa vyjadrí ako percento hmotnosti oleja ku hmotnosti sušiny.
Článok 7
Uplatňujú sa ustanovenia spoločenstva týkajúce sa prítomnosti nečistôt.
Pokiaľ ide o halogénované rozpúšťadlá, pre všetky kategórie olivových olejov platia nasledovné limity:
— maximálny objem každého detekovaného halogénovaného rozpúšťadla: 0,1 mg/kg,
— maximálny celkový objem detekovaných halogénovaných rozpúšťadiel: 0,2 mg/kg.
Článok 7a
Fyzické alebo právnické osoby a skupiny osôb, ktoré skladujú olivový olej a zvyškový olivový olej od štádia extrakcie v mlyne až po štádium plnenia do fliaš na akékoľvek odborné alebo komerčné účely, sú povinné viesť evidenciu o vstupe a výstupe každej kategórie tohto oleja.
Členský štát zaistí riadne dodržiavanie povinnosti ustanovenej v prvom odseku.
Článok 8
1. Členské štáty Komisii oznámia opatrenia na vykonávanie tohto nariadenia. Komisiu informujú o ich akýchkoľvek následných zmenách.
2. Najneskôr do 31. mája každého roku členské štáty predložia Komisii správu o zavádzaní tohto nariadenia za predchádzajúci kalendárny rok. Správa obsahuje minimálne výsledky kontrol zhody vykonaných na olivovom oleji podľa šablón stanovených v prílohe XXI.
3. Oznámenia uvedené v tomto nariadení sa podávajú podľa nariadenia Komisie (ES) č. 792/2009 ( 1 ).
Článok 9
Nariadenie EHS č. 1058/77 sa týmto zrušuje.
Článok 10
1. Táto smernica nadobúda účinnosť tretí deň od jej uverejnenia v Úradnom vestníku Európskych spoločenstiev.
Postup uvedený v prílohe XII sa však bude uplatňovať od ►M1 1. novembra 1992 ◄ , okrem postupov týkajúcich sa systému intervencie.
Daná metóda sa neuplatňuje na panenský olivový olej pripravený na trh pred 1. novembrom 1992.
2. Toto nariadenie sa nebude uplatňovať na olivový olej a olej z olivových zvyškov naplnený do obalov pred tým, ako toto nariadenie nadobudlo účinnosť, a na oleje predávané do 31. októbra 1992.
Toto nariadenie je záväzné vo svojej celistvosti a priamo uplatniteľné vo všetkých členských štátoch.
PRÍLOHY
OBSAH
|
Príloha I |
Charakteristiky olivových olejov |
|
Príloha Ia |
Odber vzoriek olivového oleja alebo oleja z olivových výliskov dodávaných v bezprostrednom obale |
|
Príloha Ib |
Vývojový diagram na overenie súladu vzorky olivového oleja s deklarovanou kategóriou |
|
Príloha II |
Stanovenie voľných mastných kyselín, studená metóda |
|
Príloha III |
Stanovenie peroxidového čísla |
|
Príloha IV |
Stanovenie obsahu voskov kapilárnou plynovou chromatografiou |
|
Príloha VII |
Stanovenie percentuálneho obsahu 2-glyceryl-monopalmitátu |
|
Príloha IX |
Spektrofotometrické skúmanie v ultrafialovej oblasti |
|
Príloha X |
Stanovenie metylesterov mastných kyselín plynovou chromatografiou |
|
Príloha XI |
Stanovenie prchavých halogenovaných rozpúšťadiel v olivovom oleji |
|
Príloha XII |
Metóda medzinárodnej rady pre olivy na organoleptické hodnotenie panenských olivových olejov |
|
Príloha XV |
Obsah oleja v olivových zvyškoch |
|
Príloha XVII |
Stanovenie jódového čísla |
|
Príloha XVII |
Metóda stanovenia stigmastadiénov v rastlinných olejoch |
|
Príloha XVIII |
Stanovenie rozdielu medzi skutočným a teoretickým obsahom triglyceridov s ECN 42 |
|
Príloha XIX |
Stanovenie skladby a obsahu sterolov a alkoholových zlúčenín kapilárnou plynovou chromatografiou |
|
Príloha XX |
Metóda na stanovenie obsahu voskov, metylesterov mastných kyselín a etylesterov mastných kyselín kapilárnou plynovou chromatografiou |
|
Príloha XXI |
Výsledky kontrol zhody vykonaných na olivovom oleji podľa článku 8 ods. 2 |
PRÍLOHA I
CHARAKTERISTIKY OLIVOVÝCH OLEJOV
Kvalitatívne znaky
|
Kategória |
Kyslosť (%) (*) |
Peroxidové číslo (mEq O2/kg) |
K232 |
K268 alebo K270 |
Delta-K |
Organoleptické hodnotenie |
Etylestery mastných kyselín (mg/kg) |
|
|
Medián defektoru (Md) (*) |
Medián ovocnosti (Mf) |
|||||||
|
1. Extra panenský olivový olej |
≤ 0,80 |
≤ 20,0 |
≤ 2,50 |
≤ 0,22 |
≤ 0,01 |
Md = 0,0 |
Mf > 0,0 |
≤ 35 |
|
2. Panenský olivový olej |
≤ 2,0 |
≤ 20,0 |
≤ 2,60 |
≤ 0,25 |
≤ 0,01 |
Md ≤ 3,5 |
Mf > 0,0 |
— |
|
3. Lampantový olivový olej |
> 2,0 |
— |
— |
— |
— |
Md > 3,5 (1) |
— |
— |
|
4. Rafinovaný olivový olej |
≤ 0,30 |
≤ 5,0 |
— |
≤ 1,25 |
≤ 0,16 |
|
— |
— |
|
5. Olivový olej zložený z rafinovaných a panenských olivových olejov |
≤ 1,00 |
≤ 15,0 |
— |
≤ 1,15 |
≤ 0,15 |
|
— |
— |
|
6. Surový olej z olivových výliskov |
— |
— |
— |
— |
— |
|
— |
— |
|
7. Rafinovaný olej z olivových výliskov |
≤ 0,30 |
≤ 5,0 |
— |
≤ 2,00 |
≤ 0,20 |
|
— |
— |
|
8. Olej z olivových výliskov |
≤ 1,00 |
≤ 15,0 |
— |
≤ 1,70 |
≤ 0,18 |
|
— |
— |
|
(1) Medián defektoru môže byť 3,5 alebo nižší ak medián ovocnosti je rovný 0,0. |
||||||||
Znaky čistoty
|
Kategória |
Zloženie mastných kyselín (1) |
Celkový obsah trans- izomérov kyseliny olejovej (%) |
Celkový obsah trans- izomérov kyseliny linolovej + kyseliny linolénovej (%) |
Stigmastadiény (mg/kg) (2) |
Rozdiel: ECN42 (HPLC) a ECN42 (teoretický výpočet) |
2-glyceryl monopalmitát (%) |
|||||
|
Myristová (%) |
Linolénová (%) |
Arachidová (%) |
Eikosenová (%) |
Behenová (%) |
Lignocerová (%) |
||||||
|
1. Extra panenský olivový olej |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,50 |
≤ 0,20 |
≤ 0,20 |
≤ 0,05 |
≤ 0,05 |
≤ 0,05 |
≤ |0,20| |
≤ 0,9, ak je % kyseliny palmitovej celkovo ≤ 14,00 % |
|
≤ 1,0, ak je % kyseliny palmitovej celkovo > 14,00 % |
|||||||||||
|
2. Panenský olivový olej |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,50 |
≤ 0,20 |
≤ 0,20 |
≤ 0,05 |
≤ 0,05 |
≤ 0,05 |
≤ |0,20| |
≤ 0,9, ak je % kyseliny palmitovej celkovo ≤ 14,00 % |
|
≤ 1,0, ak je % kyseliny palmitovej celkovo > 14,00 % |
|||||||||||
|
3. Lampantový olivový olej |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,50 |
≤ 0,20 |
≤ 0,20 |
≤ 0,10 |
≤ 0,10 |
≤ 0,50 |
≤ |0,30| |
≤ 0,9, ak je % kyseliny palmitovej celkovo ≤ 14,00 % |
|
≤ 1,1 ak je % kyseliny palmitovej celkovo > 14,00 % |
|||||||||||
|
4. Rafinovaný olivový olej |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,50 |
≤ 0,20 |
≤ 0,20 |
≤ 0,20 |
≤ 0,30 |
— |
≤|0,30| |
≤ 0,9, ak je % kyseliny palmitovej celkovo ≤ 14,00 % |
|
≤ 1,1, ak je % kyseliny palmitovej celkovo > 14,00 % |
|||||||||||
|
5. Olivový olej zložený z rafinovaných a panenských olivových olejov |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,50 |
≤ 0,20 |
≤ 0,20 |
≤ 0,20 |
≤ 0,30 |
— |
≤ |0,30| |
≤ 0,9, ak je % kyseliny palmitovej celkovo ≤ 14,00 % |
|
≤ 1,0, ak je % kyseliny palmitovej celkovo > 14,00 % |
|||||||||||
|
6. Surový olej z olivových výliskov |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,50 |
≤ 0,30 |
≤ 0,20 |
≤ 0,20 |
≤ 0,10 |
— |
≤ |0,60| |
≤ 1,4 |
|
7. Rafinovaný olej z olivových výliskov |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,50 |
≤ 0,30 |
≤ 0,20 |
≤ 0,40 |
≤ 0,35 |
— |
≤ |0,50| |
≤ 1,4 |
|
8. Olej z olivových výliskov |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,50 |
≤ 0,30 |
≤ 0,20 |
≤ 0,40 |
≤ 0,35 |
— |
≤ |0,50| |
≤ 1,2 |
|
(1) Obsah ostatných mastných kyselín (%): palmitovej: 7,50 – 20,00; palmitoolejovej: 0,30 – 3,50; heptadekánovej: ≤ 0,40; heptadecénovej: ≤ 0,60; stearovej: 0,50 – 5,00; olejovej: 55,00 – 83,00; linolovej: 2,50 – 21,00. (2) Celkový obsah izomérov, ktoré by sa (ne-)dali separovať pomocou kapilárnej kolóny. |
|||||||||||
|
Kategória |
Skladba sterolov |
Celkový obsah sterolov (mg/kg) |
Erytrodiol a uvaol (%) (**) |
Vosky (mg/kg) (**) |
|||||
|
Cholesterol (%) |
Brasikasterol (%) |
Kampesterol (1) (%) |
Stigmasterol (%) |
App beta-sitosterol (2) (%) |
Delta-7-stigmastenol (1) (%) |
||||
|
1. Extra panenský olivový olej |
≤ 0,5 |
≤ 0,1 |
≤ 4,0 |
< kamp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 000 |
≤ 4,5 |
C42 + C44 + C46 ≤ 150 |
|
2. Panenský olivový olej |
≤ 0,5 |
≤ 0,1 |
≤ 4,0 |
< kamp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 000 |
≤ 4,5 |
C42 + C44 + C46 ≤ 150 |
|
3. Lampantový olivový olej |
≤ 0,5 |
≤ 0,1 |
≤ 4,0 |
— |
≥ 93,0 |
≤ 0,5 |
≥ 1 000 |
≤ 4,5 (3) |
C40 + C42 + C44 + C46 ≤ 300 (3) |
|
4. Rafinovaný olivový olej |
≤ 0,5 |
≤ 0,1 |
≤ 4,0 |
< kamp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 000 |
≤ 4,5 |
C40 + C42 + C44 + C46 ≤ 350 |
|
5. Olivový olej zložený z rafinovaných a panenských olivových olejov |
≤ 0,5 |
≤ 0,1 |
≤ 4,0 |
< kamp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 000 |
≤ 4,5 |
C40 + C42 + C44 + C46 ≤ 350 |
|
6. Surový olej z olivových výliskov |
≤ 0,5 |
≤ 0,2 |
≤ 4,0 |
— |
≥ 93,0 |
≤ 0,5 |
≥ 2 500 |
> 4,5 (4) |
C40 + C42 + C44 + C46 > 350 (4) |
|
7. Rafinovaný olej z olivových výliskov |
≤ 0,5 |
≤ 0,2 |
≤ 4,0 |
< kamp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 800 |
> 4,5 |
C40 + C42 + C44 + C46 > 350 |
|
8. Olej z olivových výliskov |
≤ 0,5 |
≤ 0,2 |
≤ 4,0 |
< kamp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 600 |
> 4,5 |
C40 + C42 + C44 + C46 > 350 |
|
(1) Pozri dodatok k tejto prílohe. (2) App beta-sitosterol: Delta-5,23-stigmastadienol+klerosterol+beta-sitosterol+sitostanol+delta-5-avenasterol+delta-5,24-stigmastadienol. (3) Oleje s obsahom voskov od 300 mg/kg do 350 mg/kg sa považujú za lampantový olivový olej, ak celkovo obsahujú maximálne 350 mg/kg alifatických alkoholov alebo ak obsahujú maximálne 3,5 % erytrodiolu a uvaolu. (4) Oleje s obsahom voskov od 300 mg/kg do 350 mg/kg sa považujú za surový olej z olivových výliskov, ak celkovo obsahujú viac ako 350 mg/kg alifatických alkoholov a ak je obsah erytrodiolu a uvaolu vyšší ako 3,5 %. |
|||||||||
Poznámky:
a) Výsledky analýz sa musia uvádzať s rovnakým počtom desatinných miest, aký je určený pre jednotlivé charakteristiky. Číslica na poslednom desatinnom mieste sa musí zaokrúhliť smerom nahor, ak je nasledujúca číslica väčšia ako 4.
b) Ak hoci len jediná charakteristika nezodpovedá určeným hodnotám, tento olivový olej možno zaradiť do inej kategórie alebo sa olej označí za nespĺňajúci predpisy na účely tohto nariadenia.
c) V prípade lampantového olivového oleja sa oba kvalitatívne znaky označené hviezdičkou (*) môžu odlišovať od limitov stanovených pre predmetnú kategóriu súčasne.
d) Ak je znak označený dvoma hviezdičkami (**), znamená to, že v prípade surového oleja z olivových výliskov sa oba príslušné limity môžu líšiť od stanovených hodnôt súčasne. V prípade olivového oleja z olivových výliskov a rafinovaného olivového oleja z olivových výliskov sa jeden z relevantných limitov môže líšiť od stanovených hodnôt.
Dodatok
Rozhodovacie stromy
Rozhodovací strom pre kampesterol v panenských a extra panenských olivových olejoch:

Ostatné parametre musia spĺňať limity stanovené v tomto nariadení.
Rozhodovací strom pre delta-7-stigmastenol:
— Extra panenské a panenské olivové oleje
—

Ostatné parametre musia spĺňať limity stanovené v tomto nariadení.
— Oleje z olivových výliskov (surové a rafinované)
—
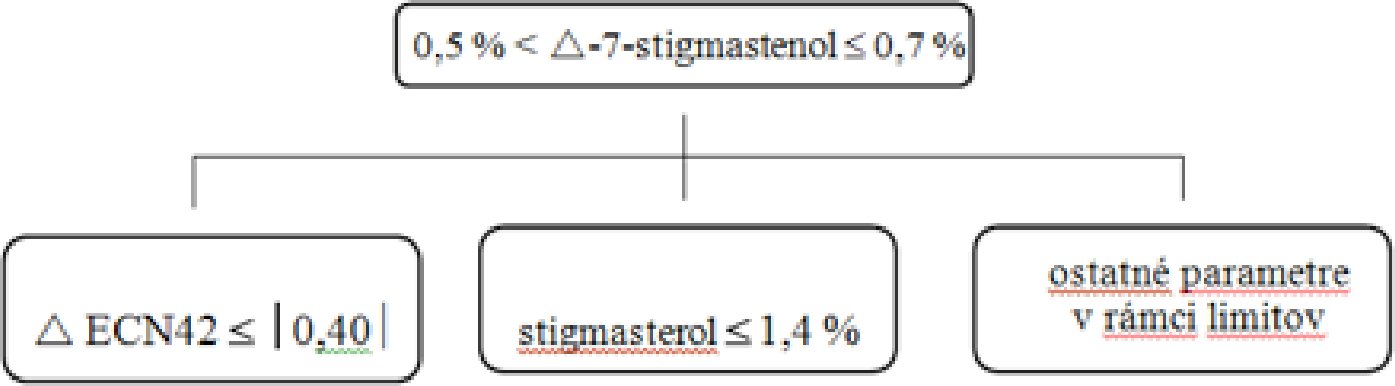
Ostatné parametre musia spĺňať limity stanovené v tomto nariadení.
PRÍLOHA Ia
ODBER VZORIEK OLIVOVÉHO OLEJA ALEBO OLEJA Z OLIVOVÝCH VÝLISKOV DODÁVANÝCH V BEZPROSTREDNOM OBALE
Táto metóda odberu vzoriek sa uplatňuje na dávky olivového oleja alebo oleja z olivových výliskov umiestnené v bezprostrednom obale. V závislosti od toho, či bezprostredný obal presahuje 5 litrov alebo nie, sa uplatňujú rôzne metódy odberu.
„Dávka“ je sada predajných jednotiek, ktoré sa produkujú, vyrábajú a balia v takých podmienkach, že olej nachádzajúci sa v každej predajnej jednotke sa považuje za homogénny, pokiaľ ide o všetky analytické vlastnosti. Individualizácia dávky sa musí uskutočniť v súlade so smernicou Európskeho parlamentu a Rady 2011/91/EÚ ( 2 ).
„Čiastková vzorka“ je množstvo oleja v bezprostrednom obale odobraného z náhodného bodu dávky.
1. OBSAH PRIMÁRNEJ VZORKY
1.1. Bezprostredný obal nepresahujúci 5 litrov
„Primárna vzorka“ v prípade bezprostredného obalu nepresahujúceho 5 litrov je počet čiastkových vzoriek odobraných z dávky, a to v súlade s tabuľkou 1.
Tabuľka 1
Minimálna veľkosť primárnej vzorky musí pozostávať z nasledovného
|
Pokiaľ má bezprostredný obal objem |
Primárna vzorka sa skladá z oleja |
|
a) aspoň 1 liter |
a) z 1 bezprostredného obalu |
|
b) menej ako 1 liter |
b) z minimálneho počtu balení s celkovým objemom aspoň 1,0 litra |
Každý členský štát môže zvýšiť počet balení, ktorý sa uvádza v tabuľke 1 a ktorý predstavuje primárnu vzorku, podľa vlastných potrieb (napr. na účely organoleptického hodnotenia iným laboratóriom, ako tým, ktoré vykonalo chemickú analýzu, kontrolné analýzy atď.).
1.2. Bezprostredný obal presahujúci 5 litrov
„Primárna vzorka“ v prípade bezprostredného obalu presahujúceho 5 litrov je reprezentatívna časť všetkých čiastkových vzoriek získaná ich delením, a to v súlade s tabuľkou 2. Primárna vzorka sa musí skladať z rôznych ukážok.
„Ukážka“ primárnej vzorky je každé balenie, z ktorého prvotná vzorka pozostáva.
Tabuľka 2
Minimálny počet čiastkových vzoriek, ktoré sa majú vybrať
|
Počet balení v dávke |
Minimálny počet čiastkových vzoriek, ktoré sa majú vybrať |
|
Do 10 |
1 |
|
Od … 11 do 150 |
2 |
|
Od … 151 do 500 |
3 |
|
Od … 501 do 1 500 |
4 |
|
Od … 1 501 do 2 500 |
5 |
|
> 2 500 na 1 000 balení |
1 čiastková vzorka naviac |
S cieľom zmenšiť objem vzoriek z bezprostredných obalov sa obsah čiastkových vzoriek homogenizuje na účely prípravy primárnej vzorky. Obsah jednotlivých čiastkových vzoriek sa naleje do spoločnej nádoby, kde sa miešaním zhomogenizuje, pričom sa čo najviac chráni pred prevzdušnením.
Celý obsah primárnej vzorky sa rozleje do série obalov s minimálnym objemom 1,0 litra, z ktorých každý predstavuje ukážku primárnej vzorky.
Každý členský štát môže zvýšiť počet primárnych vzoriek podľa vlastných potrieb (napr. na účely organoleptického hodnotenia iným laboratóriom, ako tým, ktoré vykonalo chemickú analýzu, kontrolné analýzy atď.).
Každý obal sa musí naplniť spôsobom, pri ktorom je vrstva vzduchu nad povrchom vzorky minimálna, a následne vhodne uzavrieť a utesniť, aby sa zabezpečil proti nevhodnej manipulácii.
Uvedené ukážky sa musia označiť, aby sa zabezpečila ich správna identifikácia.
2. ANALÝZA A VÝSLEDKY
2.1. Každá primárna vzorka sa musí v súlade s bodom 2.5 normy EN ISO 5555 rozdeliť na laboratórne vzorky, ktoré sa musia analyzovať v poradí podľa vývojového diagramu uvedeného v prílohe Ib alebo v akomkoľvek inom náhodnom poradí.
2.2. Pokiaľ sú všetky výsledky analýzy v súlade s vlastnosťami deklarovanej kategórie oleja, celá dávka sa vyhlási za vyhovujúcu.
Pokiaľ jediný výsledok analýzy nie je v súlade s vlastnosťami deklarovanej kategórie oleja, celá dávka sa vyhlási za nevyhovujúcu.
3. OVEROVANIE KATEGÓRIE DÁVKY
3.1. S cieľom overiť kategóriu dávky môže príslušný orgán zvýšiť počet primárnych vzoriek odobraných v rôznych bodoch dávky podľa tejto tabuľky:
Tabuľka 3
Počet primárnych vzoriek určený podľa veľkosti dávky
|
Veľkosť dávky (v litroch) |
Počet primárnych vzoriek |
|
menej ako 7 500 |
2 |
|
od 7 500 do menej ako 25 000 |
3 |
|
od 25 000 do menej ako 75 000 |
4 |
|
od 75 000 do menej ako 125 000 |
5 |
|
najmenej 125 000 |
6 + 1 na každých ďalších 50 000 litrov |
Každá čiastková vzorka tvoriaca primárnu vzorku sa musí odobrať z kontinuálneho miesta dávky, pričom je nevyhnutné uviesť do poznámky miesto odberu každej primárnej vzorky a jednoznačne ju identifikovať.
Zostavenie každej primárnej vzorky sa musí uskutočniť v súlade s postupmi uvedenými v bodoch 1.1 a 1.2.
Každá primárna vzorka sa následne podrobí analýze uvedenej v článku 2 ods. 1.
3.2. Pokiaľ jeden z výsledkov analýzy uvedenej v článku 2 ods. 1 týkajúcej sa aspoň jednej primárnej vzorky nie je v súlade s vlastnosťami deklarovanej kategórie oleja, celá dávka, z ktorej sa vzorka odobrala, sa vyhlási za nevyhovujúcu.
PRÍLOHA Ib
VÝVOJOVÝ DIAGRAM NA OVERENIE SÚLADU VZORKY OLIVOVÉHO OLEJA S DEKLAROVANOU KATEGÓRIOU
Všeobecná tabuľka

Tabuľka 1 – extra panenský olivový olej – kritériá kvality

Tabuľka 2 – panenský olivový olej – kritériá kvality

Tabuľka 3 – extra panenský olivový olej a panenský olivový olej – kritériá čistoty

Tabuľka 4 – lampantový olivový olej – kritériá čistoty

Tabuľka 5 – rafinovaný olivový olej – kritériá kvality

Tabuľka 6 – olivový olej (zložený z rafinovaných a panenských olivových olejov) – kritériá kvality

Tabuľka 7 – rafinovaný olivový olej a olivový olej zložený z rafinovaných a panenských olivových olejov – kritériá čistoty

Tabuľka 8 – surový olej z olivových výliskov – kritériá čistoty

Tabuľka 9 – rafinovaný olej z olivových výliskov – kritériá kvality

Tabuľka 10 – olej z olivových výliskov – kritériá kvality

Tabuľka 11 – rafinovaný olej z olivových výliskov a olej z olivových výliskov – kritériá čistoty

PRÍLOHA II
STANOVENIE VOĽNÝCH MASTNÝCH KYSELÍN, STUDENÁ METÓDA
1. ROZSAH A OBLASŤ POUŽITIA
Táto metóda opisuje stanovenie voľných mastných kyselín v olivových olejoch a olejoch z olivových výliskov. Obsah voľných mastných kyselín sa vyjadrí ako kyslosť vypočítaná ako percento kyseliny olejovej.
2. PRINCÍP
Vzorka sa rozpustí v zmesi rozpúšťadiel a prítomné voľné mastné kyseliny sa stitrujú roztokom hydroxidu draselného alebo hydroxidu sodného.
3. ČINIDLÁ
Všetky činidlá (reagenty) by mali byť schválenej analytickej kvality a mala by sa použiť buď destilovaná voda, alebo voda rovnakej čistoty.
|
3.1. |
Dietyléter; 95 % etanol (v/v), zmes rovnakých objemov. Počas použitia ho neutralizujte roztokom hydroxidu draselného (3.2), na 100 ml zmesi pridajte 0,3 ml roztoku fenolftaleínu (3.3). Poznámka 1: Dietyléter je vysoko horľavý a môže tvoriť výbušné peroxidy. Treba s ním narábať opatrne. Poznámka 2: Ak sa nedá použiť dietyléter, je ho možné nahradiť zmesou rozpúšťadiel obsahujúcou etanol a toluén. V prípade potreby sa etanol môže nahradiť propán-2-olom. |
|
3.2. |
Hydroxid draselný alebo hydroxid sodný; titrovaný etanolový alebo vodný roztok, c(KOH) [alebo c(NaOH)] približne 0,1 mol/l alebo v prípade potreby c(KOH) [alebo c(NaOH)] približne 0,5 mol/l. Komerčné roztoky sú k dispozícii. Musí byť známa presná koncentrácia roztoku hydroxidu draselného (alebo roztoku hydroxidu sodného) a treba ju skontrolovať bezprostredne pred použitím. Používajte roztok pripravený najmenej päť dní pred termínom použitia, uschovaný v tmavej sklenenej fľaši s gumovou zátkou. Roztok by mal byť bezfarebný alebo vo farbe slamy. Ak pri použití vodného roztoku hydroxidu draselného (alebo hydroxidu sodného) dochádza k rozdeleniu fáz, nahraďte vodný roztok etanolovým roztokom. Poznámka 3: Stabilný bezfarebný roztok hydroxidu draselného (alebo hydroxidu sodného) možno pripraviť takto: priveďte do varu 1 000 ml etanolu alebo vody s 8 g hydroxidu draselného (alebo hydroxidu sodného) a 0,5 g hliníkových hoblín a nechajte vrieť pod spätným chladičom jednu hodinu. Ihneď oddestilujte. V destiláte rozpustite požadované množstvo hydroxidu draselného (alebo hydroxidu sodného). Nechajte niekoľko dní odstáť a zlejte čistý supernatant zo zrazeniny uhličitanu draselného (alebo uhličitanu sodného). Roztok možno pripraviť aj bez destilácie takto: do 1 000 ml etanolu (alebo vody) pridajte 4 ml butylátu hlinitého a nechajte zmes niekoľko dní odstáť. Odlejte supernatant a rozpustite požadované množstvo hydroxidu draselného (alebo hydroxidu sodného). Roztok je pripravený na použitie. |
|
3.3. |
Fenolftaleín, roztok s koncentráciou 10 g/l v 95 až 96 % etanole (v/v), alebo alkalická modrá 6B či tymolftaleín, roztok s koncentráciou 20 g/l v 95 až 96 % etanole (v/v). V prípade silne sfarbených olejov treba použiť alkalickú modrú alebo tymolftaleín. |
4. PRÍSTROJOVÉ VYBAVENIE
Bežné laboratórne vybavenie zahŕňajúce:
4.1. analytické váhy;
4.2. 250 ml kónickú banku;
4.3. 10 ml byretu triedy A, delenú po 0,05 ml, alebo rovnocennú automatickú byretu.
5. POSTUP
5.1. Príprava skúšobnej vzorky
Ak je vzorka kalná, treba ju prefiltrovať.
5.2. Skúšobná dávka
Odoberte vzorku v závislosti od predpokladanej kyslosti v súlade s touto tabuľkou:
|
Predpokladaná kyslosť (olejová kyslosť g/100g) |
Hmotnosť vzorky (g) |
Presnosť váženia (g) |
|
0 až 2 |
10 |
0,02 |
|
> 2 až 7,5 |
2,5 |
0,01 |
|
> 7,5 |
0,5 |
0,001 |
Vzorku odvážte v kónickej banke (4.2).
5.3. Stanovenie
Vzorku (5.2) rozpustite v 50 až 100 ml predtým zneutralizovanej zmesi dietyléteru a etanolu (3.1).
Titrujte počas zmiešania s 0,1 mol/l roztokom hydroxidu draselného (alebo hydroxidu sodného) (3.2) (pozri pozn. 4) až do zmeny zafarbenia indikátora (farba zafarbeného indikátora vydrží aspoň 10 sekúnd).
Poznámka 4: Ak spotrebované množstvo 0,1 mol/l roztoku hydroxidu draselného (alebo hydroxidu sodného) presiahne 10 ml, použite 0,5 ml/l roztok alebo zmeňte hmotnosť vzorky podľa predpokladanej voľnej kyslosti a navrhovanej tabuľky.
Poznámka 5: Ak sa roztok počas titrácie zakalí, pridajte dostatočné množstvo rozpúšťadiel (3.1), aby sa roztok vyčíril.
Vykonajte druhé stanovenie kyslosti len vtedy, ak je prvý výsledok vyšší než špecifikovaná hranica pre príslušnú kategóriu oleja.
6. VYJADRENIE VÝSLEDKOV
Kyslosť vyjadrená ako hmotnostný zlomok kyseliny olejovej sa rovná:
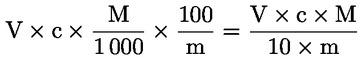
kde:
|
V |
= |
objem použitého titračného roztoku hydroxidu draselného (alebo hydroxidu sodného) v ml, |
|
c |
= |
presná koncentrácia použitého titračného roztoku hydroxidu draselného alebo hydroxidu sodného v mol/l, |
|
M |
= |
282 g/mol, mólová hmotnosť v g/mol kyseliny olejovej, |
|
m |
= |
hmotnosť vzorky v g. |
Olejová kyslosť sa vykazuje takto:
a) na dve desatinné miesta pre hodnoty od 0 do 1 vrátane;
b) na dve desatinné miesta pre hodnoty od 1 do 100 vrátane.
PRÍLOHA III
STANOVENIE PEROXIDOVÉHO ČÍSLA
1. Oblasť
Táto príloha opisuje metódu stanovenia peroxidového čísla živočíšnych a rastlinných olejov a tukov.
2. Definícia
Peroxidové číslo je množstvo tých zložiek vo vzorke, ktoré sú vyjadrené ako miliekvivalent aktívneho kyslíka na kilogram a ktoré oxidujú jodid draselný za uvedených prevádzkových podmienok.
3. Princíp
Úprava roztoku, ktorý tvorí odobratá časť vzorky, kyselina octová a chloroform, roztokom jodidu draselného. Titrácia uvoľneného jódu štandardizovaným roztokom tiosíranu sodného.
4. Prístrojové vybavenie
Všetky použité prístroje musia byť bez redukujúcich alebo oxidujúcich látok.
Poznámka 1: Nemastite zábrusové povrchy.
4.1. 3 ml sklenená navažovacia lodička.
4.2. Banky so zábrusovými hrdlami a zátkami, približne 250 ml objemu, predsušené a naplnené čistým suchým inertným plynom (dusíkom alebo, pokiaľ možno, oxidom uhličitým).
4.3. 5, 10 alebo 25 ml byreta, delená po najmenej 0,05 ml, pokiaľ možno s automatickým nastavením nuly alebo rovnocenná automatická byreta.
4.4. Analytické váhy.
5. Činidlá
5.1. Chloroform, p.a., zbavený kyslíka prebublávaním prúdom čistého, suchého inertného plynu.
5.2. Ľadová kyselina octová, p.a., zbavená kyslíka prebublávaním prúdom čistého, suchého inertného plynu.
5.3. Jodid draselný, nasýtený vodný roztok, čerstvo pripravený, neobsahujúci jód ani jodičnany. Rozpustite približne 14 g jodidu draselného v približne 10 ml vody pri teplote miestnosti.
5.4. Tiosíran sodný (sírnatan), 0,01 mol/l (= 0,01 N) presný štandardizovaný vodný roztok, štandardizovaný tesne pred použitím.
Čerstvo pripravte denne 0,01 mol/l roztoku tiosíranu sodného z 0,1 mol/l štandardného roztoku tiosíranu sodného pred použitím alebo stanovte presnú molaritu. Skúsenosti dokazujú, že stabilita je obmedzená a závisí od hodnoty pH a obsahu voľného oxidu uhličitého. Na zriedenie použite len čerstvo prevarenú vodu, prípadne prepláchnutú dusíkom.
Na stanovenie presnej molarity roztoku tiosíranu sodného sa odporúča tento postup:
Odvážte s presnosťou na 0,001 g 0,27 g až 0,33 g jodičnanu draselného (mKIO3) do odmernej banky (250 ml alebo 500 ml) a zrieďte po značku čerstvo prevarenou vodou (V2), ochladenou na izbovú teplotu. Pomocou pipety preneste 5 ml alebo 10 ml tohto roztoku jodičnanu draselného (V1) do 250 ml Erlenmeyerovej banky. Pridajte 60 ml čerstvo prevarenej vody, 5 ml 4 mol/l kyseliny chlorovodíkovej a 25 mg až 50 mg jodidu draselného alebo 0,5 ml nasýteného roztoku jodidu draselného. Tento roztok stitrujte roztokom tiosíranu sodného (V3) na stanovenie presnej molarity roztoku tiosíranu sodného.

kde:
|
mKIO3 |
= |
hmotnosť jodičnanu draselného v gramoch; |
|
V1 |
= |
objem roztoku jodičnanu draselného v mililitroch (5 ml alebo 10 ml); |
|
V2 |
= |
celkový objem roztoku jodičnanu draselného v mililitroch (250 ml alebo 500 ml); |
|
V3 |
= |
objem roztoku tiosíranu sodného v mililitroch; |
|
wKIO3 |
= |
čistota jodičnanu draselného v g/100 g; |
|
MKIO3 |
= |
molekulárna hmotnosť jodičnanu draselného (214 g/mol); |
|
T |
= |
presná molarita roztoku tiosíranu sodného (mol/l). |
5.5. Roztok škrobu, vodná disperzia s koncentráciou 10 g/l, čerstvo pripravená z prírodného rozpustného škrobu. Možno použiť aj rovnocenné činidlá.
6. Vzorka
Dbajte o to, aby bola vzorka odoberaná a skladovaná bez prístupu svetla, udržiavaná v chlade a v úplne (až povrch) naplnených sklenených zásobníkoch (dózach), hermeticky uzavretých zábrusnými alebo korkovými zátkami.
7. Postup
Test sa musí vykonať pri rozptýlenom dennom svetle alebo pri umelom svetle. V sklenenej navažovačke (4.1) alebo prípadne v banke (4.2) odvážte s presnosťou na 0,001 g množstvo vzorky v súlade s nasledujúcou tabuľkou a podľa predpokladaného peroxidového čísla:
|
Predpokladané peroxidové číslo (meq) |
Hmotnosť navážky g) |
|
0 až 12 |
5,0 až 2,0 |
|
12 až 20 |
2,0 až 1,2 |
|
20 až 30 |
1,2 až 0,8 |
|
30 až 50 |
0,8 až 0,5 |
|
50 až 90 |
0,5 až 0,3 |
Odzátkujte banku (4.2) a vložte sklenenú navažovačku s odváženou časťou testovanej vzorky. Pridajte 10 ml chloroformu (5.1). Miešaním rýchlo rozpustite testovanú vzorku. Pridajte 15 ml kyseliny octovej (5.2), potom 1 ml roztoku jodidu draselného (5.3). Rýchlo zazátkujte, pretrepávajte 1 minútu a nechajte 5 minúť stáť v tme pri teplote 15 až 25 °C.
Pridajte približne 75 ml destilovanej vody. Uvoľnený jód stitrujte roztokom tiosíranu sodného (5.4), dôkladne pretrepávajte, ako indikátor použite roztok škrobu (5.5).
Vykonajte dve zistenia z jednej testovanej vzorky.
Zároveň vykonajte slepý pokus. Ak výsledok slepého pokusu prekročí 0,05 ml 0,01 N roztoku tiosíranu sodného (5.4), vymeňte znečistené činidlá.
8. Vyjadrenie výsledkov
Peroxidové číslo (PV), vyjadrené v miliekvivalentoch aktívneho kyslíka na kilogram, sa udáva podľa vzorca:

kde:
|
V |
= |
objem (vyjadrený v ml) štandardizovaného roztoku tiosíranu sodného (5.4), použitého pri teste, korigovaného výsledkom slepého pokusu; |
|
T |
= |
presná molarita použitého roztoku tiosíranu sodného (5.4) v mol/l; |
|
m |
= |
hmotnosť navážky vzorky v g). |
Za výsledok sa bude považovať aritmetický priemer z dvoch výpočtov.
Uveďte výsledok stanovenia na jedno desatinné miesto.
PRÍLOHA IV
STANOVENIE OBSAHU VOSKOV POMOCOU KAPILÁRNEJ PLYNOVEJ CHROMATOGRAFIE
1. PREDMET
Táto metóda opisuje postup stanovenia obsahu voskov v olivových olejoch. Vosky sa separujú v závislosti od počtu atómov uhlíka. Môže sa používať najmä na rozlíšenie medzi olivovým olejom získaným lisovaním a olejom získaným extrakciou (olej z výliskov).
2. PRINCÍP
Pridanie vhodného vnútorného štandardu k tuku alebo oleju, potom frakcionácia chromatografiou na kolóne s hydratovaným silikagélom. Získanie frakcie eluovanej pri testovacích podmienkach ako prvej (ktorej polarita je nižšia ako polarita frakcie triglyceridov), potom priama analýza kapilárnou plynovou chromatografiou.
3. MATERIÁL
|
3.1. |
25 ml Erlenmeyerova banka. |
|
3.2. |
Sklená kolóna na plynovú chromatografiu, s vnútorným priemerom 15,0 mm, výškou 30 až 40 cm a vybavená kohútikom. |
|
3.3. |
Plynový chromatograf vhodný na fungovanie s kapilárnou kolónou, vybavený systémom na priame zavádzanie do kolóny s týmito zložkami:
|
|
3.4. |
Mikrostriekačka s kapacitou 10 μl na priamy nástrek do kolóny, vybavená cementovanou ihlou. |
|
3.5. |
Elektrický vibrátor. |
|
3.6. |
Rotačná odparka. |
|
3.7. |
Mufľová pec. |
|
3.8. |
Analytické váhy s garantovanou presnosťou merania na ± 0,1 mg. |
|
3.9. |
Bežné laboratórne sklené pomôcky. |
4. CHEMICKÉ ČINIDLÁ
|
4.1. |
Silikagél s granulometriou medzi 60 a 200 μm. Silikagél vložte do pece aspoň na štyri hodiny pri teplote 500 °C. Po vychladnutí pridajte 2 % vody vzhľadom na množstvo odobraného silikagélu. Dobre pretrepte, aby sa hmota homogenizovala. Najmenej 12 hodín pred použitím uchovávajte v tme. |
|
4.2. |
n-hexán na chromatografiu. |
|
4.3. |
Etyléter na chromatografiu. |
|
4.4. |
n-heptán na chromatografiu. |
|
4.5. |
Štandardný roztok laurylarašidátu, 0,1 % (hm/obj) v hexáne (vnútorný štandard). [Je tiež možné použiť palmityl-palmitát (hexadecyl-palmitát) alebo myristyl-stearát (teradecyl-oktadekanoát)].
|
|
4.6. |
Nosný plyn: vodík alebo čisté hélium na plynovú chromatografiu. |
|
4.7. |
Pomocné plyny: — čistý vodík na plynovoú chromatografiu, — čistý vzduch na plynovú chromatografiu. |
5. POSTUP
5.1. Príprava chromatografickej kolóny
Suspenzujte 15 g silikagélu (4.1) v n-hexáne (4.2) a zaveďte do kolóny (3.2). Spontánne usadzovanie dokončite pomocou elektrickej trepačky (3.5) s cieľom dosiahnuť homogénnejšiu chromatografickú vrstvu. Perkolujte 30 ml n-hexánu s cieľom eliminovať všetky prípadné nečistoty. Presne odvážte pomocou váh (3.8). Vložte 500 mg vzorky do 25 ml Erlenmeyerovej banky (3.1), pridajte primerané množstvo vnútorného štandardu (4.5) vzhľadom na predpokladaný obsah voskov. Napríklad: v prípade olivového oleja pridajte 0,1 mg laurylarašidátu a v prípade oleja z olivových výliskov 0,25 až 0,5 mg. Takto pripravenú vzorku preneste do chromatografickej kolóny s pomocou dvoch 2 ml dielov n-hexánu (4.2).
Napustite rozpúšťadlo až do výšky 1 mm nad hornú úroveň absorbentu, potom perkolujte ďalších 70 ml n-hexánu, aby sa eliminovali prirodzene prítomné n-alkány. Začnite chromatografickú elúciu zozbieraním 180 ml zmesi n-hexánu/etyléteru v pomere 99:1 pri zachovaní prietoku približne 15 kvapiek každých 10 sekúnd. Elúcia vzorky sa má uskutočniť pri okolitej teplote 22 °C ± 4 °C.
Poznámky:
— Zmes n-hexánu/etyléteru (99:1) sa musí pripravovať denne.
— Na umožnenie vizuálnej kontroly správnej elúcie voskov je možné pridať do vzorky roztoku 100 μl 1 %-ného sudánu v elučnej zmesi. Farbivo má retenciu na rozhraní voskov a triglyceridov, preto keď zafarbenie dosiahne dno chromatografickej kolóny, je potrebné elúciu zastaviť, lebo všetky vosky boli eluované.
Takto získanú frakciu sušte v rotačnej odparke (3.6) až do takmer úplnej eliminácie rozpúšťadla. Posledné 2 ml rozpúšťadla eliminujte pomocou slabého prúdu azotu; potom pridajte 2 – 4 ml n-heptánu.
5.2. Analýza plynovou chromatografiou
5.2.1. Predbežné úkony
Namontujte kolónu do plynového chromatografu (3.3), pričom vstupný port sa pripojí k systému na kolóne a výstupný port k detektoru. Skontrolujte bežným spôsobom prístrojové vybavenie na plynovú chromatografiu (činnosť uzavretých plynových obvodov, účinnosť detektora a zapisovača atď.).
Pokiaľ sa kolóna používa po prvý raz, je potrebné ju kondicionovať. Zaveďte do kolóny mierny prietok plynu, potom zapnite prístroj na plynovú chromatografiu. Postupne zahrievajte, až kým sa približne po 4 hodinách dosiahne teplota 350 °C. Túto teplotu udržiavajte najmenej počas dvoch hodín, potom prístroj nastavte na prevádzkové podmienky [nastavte prietok plynu, zapáľte plamienok, pripojte k elektrickému zapisovaču (3.3.4), nastavte teplotu komory pre kolónu, nastavte detektor atď.]. Zaznamenajte signál pri citlivosti najmenej dvakrát vyššej, ako sa vyžaduje na vykonanie analýzy. Základná čiara musí byť lineárna, nesmie obsahovať píky akéhokoľvek druhu a nesmie vykazovať žiadnu odchýlku.
Negatívna priamočiara odchýlka ukazuje, že kolóna je nesprávne pripojená; pozitívna odchýlka znamená, že kolóna nebola dostatočne kondicionovaná.
5.2.2. Voľba prevádzkových podmienok
Prevádzkové podmienky, ktoré treba pozorovať, sú spravidla tieto:
— teplota kolóny:
—
|
|
20 °C/minúta |
|
5 °C/minúta |
|
20 °C/minúta |
|
|
na začiatku 80 °C (1′) |
→ |
240 °C |
→ |
325 °C (6′) |
→ |
340 °C (10′) |
— teplota detektora: 350 °C,
— množstvo vstreknutej látky: 1 μl roztoku (2 – 4 ml) n-heptánu,
— nosný plyn: hélium alebo vodík s optimálnou lineárnou rýchlosťou pre zvolený plyn (pozri dodatok),
— citlivosť prístroja: taká, aby zodpovedala nižšie uvedeným podmienkam:
Uvedené podmienky môžu byť upravené v závislosti od vlastností kolóny a prístroja na plynovú chromatografiu tak, aby sa dosiahla separácia všetkých voskov, postačujúce rozlíšenie píkov (pozri obrázok) a retenčný čas vnútorného štandardu C32 bol 18 ± 3 minúty. Najreprezentatívnejší pík vosku by mal dosahovať aspoň 60 % rozsahu stupnice.
Parametre integrácie píkov sa stanovia takým spôsobom, aby sa dosiahlo správne vyhodnotenie príslušných plôch píkov.
Poznámka:Vzhľadom na vysokú konečnú teplotu sa pripúšťa kladná odchýlka, ktorá nesmie prekročiť 10 % rozsahu stupnice.
5.3. Vykonanie analýzy
Naberte 1 μl roztoku pomocou 10 μl mikrostriekačky; natiahnite piestik dozadu, až kým ihla nebude prázdna. Ihlu zaveďte do injekčného systému a po uplynutí jednej až dvoch sekúnd rýchlo vstreknite; po uplynutí asi piatich sekúnd ihlu pomaly vytiahnite.
Záznam nechajte prebiehať dovtedy, až kým neprebehne úplná elúcia voskov.
Základná čiara musí vždy zodpovedať požadovaným podmienkam.
5.4. Identifikácia píkov
Jednotlivé píky identifikujte z retenčných časov a porovnaním so zmesami voskov so známymi retenčnými časmi, analyzovanými pri rovnakých podmienkach.
Na obrázku je znázornený chromatogram voskov panenského olivového oleja.
5.5. Kvantitatívne hodnotenie
Integrátorom vypočítajte plochy píkov interného štandardu a alifatických esterov od C40 do C46.
Vypočítajte obsah voskov každého esteru v mg/kg tuku podľa príslušného vzorca:

kde:
|
Ax |
= |
plocha píku každého esteru v milimetroch štvorcových; |
|
As |
= |
plocha píku vnútorného štandardu v milimetroch štvorcových, |
|
ms |
= |
množstvo pridaného vnútorného štandardu v miligramoch; |
|
m |
= |
množstvo vzorky odobratej na stanovenie v gramoch. |
6. VYJADRENIE VÝSLEDKOV
Uveďte celkové obsahy jednotlivých voskov od C40 do C46 v mg/kg tuku (ppm).
Poznámka:Zložky, ktoré treba kvantifikovať, sa stanovujú vzhľadom na susediace píky esterov s počtom uhlíkov medzi C40 a C46 podľa príkladu chromatogramu voskov olivového oleja znázorneného na nasledujúcom obrázku. Ak sa ester C46 ukáže dvojmo, odporúča sa analyzovať na jeho identifikáciu frakciu voskov oleja z olivových výliskov, v ktorom je pík esteru C46 ľahko identifikovateľný, pretože výrazne prevažuje.
Výsledky sa uvádzajú na jedno desatinné miesto.
Obrázok
Chromatogram voskov istého olivového oleja ( 3 )

Vysvetlivky:
|
I.S. |
= |
laurylarašidát |
|
1. |
= |
diterpénové estery |
|
2 + 2′ |
= |
estery C40 |
|
3 + 3′ |
= |
estery C42 |
|
4 + 4′ |
= |
estery C44 |
|
5. |
= |
estery C46 |
|
6. |
= |
sterol-estery a triterpénový alkohol. |
Dodatok
Stanovenie lineárnej rýchlosti plynu
Vstreknite do prístroja na plynovú chromatografiu nastaveného na bežné prevádzkové podmienky 1 až 3 μl metánu (alebo propánu). Odmerajte čas, ktorý potrebuje plyn na prechod cez kolónu od momentu jeho zavedenia až dovtedy, kým sa neobjaví pík (tM).
Lineárna rýchlosť v cm/sek je daná vzorcom L/tM, pričom L je dĺžka kolóny v cm a tM je čas nameraný v sekundách.
▼M32 —————
▼M26 —————
PRÍLOHA VII
STANOVENIE PERCENTUÁLNEHO OBSAHU 2-GLYCERIL MONOPALMITÁTU
1. ÚČEL A OBLASŤ POUŽITIA
Touto metódou sa popisuje analytický postup na stanovenie percentuálneho obsahu kyseliny palmitovej v pozícii 2 triglyceridov prostredníctvom hodnotenia 2-glyceril monopalmitátu.
Táto metóda je použiteľná na tekuté rastlinné oleje pri temperovanej okolitej teplote (20 °C).
2. PRINCÍP
Po príprave sa vzorka oleja podrobí procesu pankreatickej lipázy: čiastočná a špecifická hydrolýza v pozíciách 1 a 3 molekuly triglyceridu vyvolá objavenie sa monoglyceridov v pozícii 2. Percentuálny obsah 2-glyceril monopalmitátu v monoglyceridovej frakcii sa stanovuje po silanizácii kapilárnou plynovou chromatografiou.
3. PRÍSTROJOVÉ VYBAVENIE A BEŽNÝ LABORATÓRNY MATERIÁL
|
3.1. |
25 ml Erlenmeyerova banka |
|
3.2. |
100, 250 a 300 ml kadičky |
|
3.3. |
Sklená kolóna na chromatografiu, s vnútorným priemerom 21 – 23 mm, dĺžkou 400 mm, vybavená fritovým kotúčom a kohútikom |
|
3.4. |
Odmerné valce s objemom 10, 50, 100 a 200 ml |
|
3.5. |
100 a 250 ml banky |
|
3.6. |
Rotačná odparka |
|
3.7. |
10 ml centrifugačná skúmavka s kónickým dnom so zábrusovou zátkou |
|
3.8. |
Odstredivka pre 10 a 100 ml skúmavky |
|
3.9. |
Termostat s možnosťou udržiavania teploty na 40 °C ± 0,5 °C |
|
3.10. |
Odmerné pipety s objemom 1 a 2 ml |
|
3.11. |
1 ml podkožná striekačka |
|
3.12. |
100 μl mikrostriekačka |
|
3.13. |
1 000 ml lievik |
|
3.14. |
Plynový chromatograf pre kapilárne kolóny, vybavený injekčným systémom „on column“ za studena na priame zavedenie vzorky do kolóny a pecou umožňujúcou udržiavať nastavenú teplotu s presnosťou na 1 °C |
|
3.15. |
Studený injektor „on column“ na priame zavedenie vzorky do kolóny |
|
3.16. |
Plameňovoionizačný detektor a elektrometer |
|
3.17. |
Zapisovač-integrátor prispôsobený elektrometru s rýchlosťou odozvy najviac 1 sekundu a s meniteľnou rýchlosťou posunu papiera |
|
3.18. |
Kapilárna kolóna sklená alebo silikagélová, s dĺžkou 8 až 12 metrov, vnútorným priemerom 0,25 až 0,32 mm, pokrytá metylpolysiloxánom alebo 5 % fenyl metylpolysiloxánom, s hrúbkou 0,10 – 0,30 μm, použiteľná pri teplote 370 °C |
|
3.19. |
10 μl mikrostriekačka vybavená cementovou ihlou dlhou aspoň 7,5 cm na priame vstreknutie na kolónu. |
4. ČINIDLÁ
|
4.1. |
Silikagél s granulometriou medzi 0,063 a 0,200 mm (70/280 mesh) pripravený takto: Silikagél vložte do porcelánovej kapsule, sušte aspoň štyri hodiny v peci pri teplote 160 °C. Nechajte vychladnúť v exsikátore pri temperovanej teplote. Pridajte objem vody, ktorý zodpovedá 5 % hmotnosti silikagélu, takto: odvážte 152 g silikagélu do Erlenmeyerovej banky, pridajte 8 g destilovanej vody, zazátkujte a jemne pretrepte, aby sa voda rovnomerne rozdelila. Pred použitím nechajte aspoň 12 hodín stáť. |
|
4.2. |
n-hexán (na chromatografiu). Hexán možno nahradiť izo-oktánom (2,2,4-trimetylpentánom na chromatografiu) pod podmienkou dosahovania porovnateľných hodnôt presnosti. |
|
4.3. |
Izopropanol |
|
4.4. |
Izopropanol, vodný roztok v objemovom pomere 1/1 |
|
4.5. |
Pankreatická lipáza. Použitá lipáza má mať aktivitu medzi 2,0 a 10 jednotkami lipázy na mg (V obchodnej sieti dostať pankreatické lipázy s aktivitou medzi 2 a 10 jednotkami na mg enzýmu.) |
|
4.6. |
Tlmiaci roztok tri-hydroxy-metylaminometánu: vodný roztok 1 M upravený až na pH = 8 (kontrolované potenciometrom) pridaním koncentrovanej kyseliny chlorovodíkovej (v objemovom pomere 1/1) |
|
4.7. |
Cholát sodný enzymatickej kvality, 0,1 % vodný roztok (tento roztok sa musí použiť do 15 dní od jeho prípravy) |
|
4.8. |
Chlorid vápenatý, 22 % vodný roztok |
|
4.9. |
Dietyléter na chromatografiu |
|
4.10. |
Vyvíjacie rozpúšťadlo: zmes n-hexánu/dietyléteru (v objemovom pomere 87/13) |
|
4.11. |
12 % hmotnostných roztoku hydroxidu sodného |
|
4.12. |
Fenolftaleín, 1 % roztok v etanole |
|
4.13. |
Nosný plyn: vodík alebo hélium na plynovú chromatografiu |
|
4.14. |
Pomocné plyny: vodík, minimálne 99 %, zbavený vlhkosti a organických látok – a vzduch rovnakej čistoty na plynovú chromatografiu |
|
4.15. |
Činidlá na silanizáciu: zmes pyridínu/hexametyldisilazánu, trimetylchlórsilánu v objemovom pomere 9/3/1 (Hotové roztoky dostať v obchodnej sieti. Na silanizáciu sa môžu použiť aj ďalšie činidlá, konkrétne bis-trimetylsilyl trifluóracetamid + 1 % trimetylchlorosilán, zriedené rovnakým objemom bezvodého pyridínu.) |
|
4.16. |
Referenčné vzorky: čisté monoglyceridy alebo zmesi monoglyceridov so známym percentuálnym zložením podobným vzorke. |
5. POSTUP
5.1. Príprava vzorky
|
5.1.1. |
Oleje s obsahom voľných kyselín menej ako 3 % sa nemusia pred chromatografiou na silikagélovej kolóne neutralizovať. Oleje s obsahom voľných kyselín viac ako 3 % sa musia neutralizovať v súlade s bodom 5.1.1.1.
|
|
5.1.2. |
Vložte 1,0 g oleja pripraveného podľa vyššie uvedených indikácií do 25 ml (3.1) Erlenmeyerovej banky a rozpustite v 10 ml vyvíjacej zmesi (4.10). Nechajte roztok postáť aspoň 15 minút pred chromatografiou na silikagélovej kolóne. Ak je roztok zakalený, odstreďte ho, aby boli zabezpečené optimálne podmienky na chromatografiu. (Môžu sa použiť aj už hotové 500 g silikagélové patróny SPE – na extrakciu na tuhých fázach). |
|
5.1.3. |
Príprava chromatografickej kolóny Nalejte do kolóny (3.3) približne 30 ml vyvíjacieho rozpúšťadla (4.10), pomocou sklenenej tyčinky vložte do spodnej časti kolóny kúsok bavlny; stlačte, aby sa odstránil vzduch. V kadičke pripravte suspenziu z 25 g silikagélu (4.1) v približne 80 ml vyvíjacieho rozpúšťadla a pomocou lievika ju vlejte do kolóny. Skontrolujte, či bol do kolóny zavedený všetok silikagél; premyte vyvíjacím rozpúšťadlom (4.10), otvorte kohútik a nechajte, aby hladina tekutiny dosiahla približne 2 mm nad vrchnou úrovňou silikagélu. |
|
5.1.4. |
Chromatografia na kolóne Do 25 ml Erlenmeyerovej banky (3.1) odvážte presne 1,0 g vzorky pripravenej podľa bodu 5.1. Rozpustite vzorku v 10 ml vyvíjacieho roztoku (4.10). Nalejte roztok do chromatografickej kolóny pripravenej podľa bodu 5.1.3. Nepohnite povrchom kolóny. Otvorte kohútik a nechajte roztok vzorky tiecť, až kým nedosiahne úroveň silikagélu. Vyvíjajte so 150 ml vyvíjacieho rozpúšťadla. Upravte prietok na 2 ml/min (tak, aby 150 ml pretieklo do kolóny približne za 60 – 70 minút). Odoberte eluát do 250 ml banky, ktorú ste predtým odvážili. Odparte rozpúšťadlo vo vákuu a odstráňte jeho posledné stopy pod prúdom dusíka. Odvážte banku a vypočítajte množstvo získaného extrátu. [V prípade použitia už hotových silikagélových patrónov SPE postupujte takto: zaveďte 1 ml roztoku (5.1.2) do vopred pripravených patrónov s 3 ml n-hexánu.] Po perkolovaní roztoku vyvíjajte so 4 ml n-hexánu/dietyléteru v objemovom pomere 9/1. Odoberte eluát do 10 ml skúmavky a odparujte pod prúdom dusíka až do vysušenia. Suché rezíduum podrobte pankreatickej lipáze (5.2). Základom je overiť zloženie mastných kyselín pred a po prechode patrónom SPE. |
5.2. Hydrolýza pankreatickou lipázou
|
5.2.1. |
Do centrifugačnej skúmavky odvážte 0,1 g oleja pripraveného podľa bodu 5.1. Pridajte 2 ml tlmiaceho roztoku (4.6), 0,5 ml roztoku cholátu sodného (4.7) a 0,2 ml roztoku chloridu vápenatého, pričom po každom pridaní riadne pretrepte. Uzatvorte skúmavku zábrusovou zátkou a umiestnite ju do termostatu pri teplote 40 ± 0,5 °C. |
|
5.2.2. |
Pridajte 20 mg lipázy, opatrne pretrepte (tak, aby ste nezmáčali zátku) a dajte skúmavku do termostatu presne na 2 minúty, potom ju vyberte, počas 1 minúty dôkladne pretrepávajte a nechajte vychladnúť. |
|
5.2.3. |
Pridajte 1 ml dietyléteru, zazátkujte a dôkladne pretrepte, potom odstreďte a pomocou mikrostriekačky preneste éterový roztok do čistej a suchej skúmavky. |
5.3. Príprava silanizovaných derivátov a plynová chromatografia
|
5.3.1. |
Pomocou mikrostriekačky zaveďte 100 μl roztoku (5.2.3) do 10 ml skúmavky s kónickým dnom. |
|
5.3.2. |
Eliminujte rozpúšťadlo pod miernym prúdom dusíka, pridajte 200 μl činidla na silanizáciu (4.15), uzavrite skúmavku a nechajte 20 minút odstáť. |
|
5.3.3. |
Po 20 minútach pridajte 1 až 5 ml n-hexánu (v závislosti od chromatografických podmienok): výsledný roztok je pripravený na plynovú chromatografiu. |
5.4. Plynová chromatografia
Hlavné operačné podmienky sú tieto:
— teplota injektora (injektor „on column“) nižšia ako teplota varu rozpúšťadla (68 °C),
— teplota detektora: 350 °C,
— teplota kolóny: nastavenie teploty pece: 60 °C počas 1 minúty, každú minútu zvýšte o 15 °C až do dosiahnutia 180 °C, potom o 5 °C za minútu až do 340 °C, ďalej udržiavajte 340 °C počas 13 minút,
— nosný plyn: vodík alebo hélium nastavené na lineárnu rýchlosť adekvátnu na dosiahnutie rozlíšenia znázorneného na obrázku 1. Retenčný čas triglyceridu C54 má byť 40 ± 5 minút (pozri obrázok 2). (Vyššie uvedené podmienky postupu sa uvádzajú len orientačne. Každý subjekt ich musí optimalizovať s cieľom dosiahnuť požadované rozlíšenie. Výška píku zodpovedajúca 2-glyceril monopalmitátu musí dosiahnuť aspoň 10 % rozsahu stupnice zapisovača.),
— Množstvo vstreknutej látky: 0,5 – 1 μl roztoku (5 ml) n-hexánu (5.3.3).
5.4.1. Identifikácia píkov
Jednotlivé monoglyceridy sa identifikujú v závislosti od získaných retenčných časov a vzhľadom na tie, ktoré boli získané so štandardami zmesí monoglyceridov analyzovaných pri rovnakých podmienkach.
5.4.2. Kvantitatívne hodnotenie
Plocha každého píku sa vypočíta pomocou elektronického integrátora.
6. VYJADRENIE VÝSLEDKOV
Percentuálny obsah glyceryl monopalmitátu sa vypočíta na základe vzťahu medzi plochou zodpovedajúceho píku a súčtom plôch píkov všetkých monoglyceridov (pozri obrázok 2) podľa vzorca:
glycéryl monopalmitate (%):
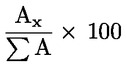
kde:
|
Ax |
= |
plocha píku, ktorý zodpovedá glyceril monopalmitátu, |
|
ΣA |
= |
súčet plôch všetkých píkov, ktoré zodpovedajú monoglyceridom. |
Výsledok sa uvádza s presnosťou na jedno desatinné miesto.
7. SPRÁVA O ANALÝZE
V správe z analýzy sa má konkrétne uviesť:
— odkaz na túto metódu,
— všetky údaje potrebné na úplnú identifikáciu vzorky,
— výsledok analýzy,
— každé odklonenie sa od tejto metódy, či už vplyvom rozhodnutia dotknutých strán alebo z iného dôvodu,
— podrobné identifikačné údaje o laboratóriu, dátum uskutočnenia analýzy a podpis zodpovedných za analýzu.
Obrázok 1
Chromatogram výsledných produktov reakcie silanizácie, ktoré sa získali lipázou na rafinovanom olivovom oleji s pridaním 20 % esterifikovaného oleja (100 %)

Vysvetlivky:„acides gras libres“ = voľné mastné kyseliny; „huile d’olive rafinée + 20 % huile estérifiée“ = rafinovaný olivový olej s pridaním 20 % esterifikovaného oleja; „1-2 monopalmitoléine“ = 1-2 monopalmitín; „1-2 mono C18 insat.“ = nesaturovaný 1-2 mono C18
Obrázok 2
Chromatogram:
|
A) |
neesterifikovaného olivového oleja, po lipáze; po silanizácii; za týchto podmienok (kapilárna kolóna 8 – 12 m) je vosková frakcia eluovaná v rovnakom čase ako frakcia diglyceridu alebo krátko potom. Po lipáze by obsah triglyceridov nemal prekročiť 15 %. 
Vysvetlivky:
|
Chromatogram:
|
B) |
esterifikovaného oleja po lipáze; po silanizácii; za týchto podmienok (kapilárna kolóna 8 – 12 m) je vosková frakcia eluovaná v rovnakom čase ako frakcia diglyceridu alebo krátko potom. Po lipáze by obsah triglyceridov nemal prekročiť 15 %. 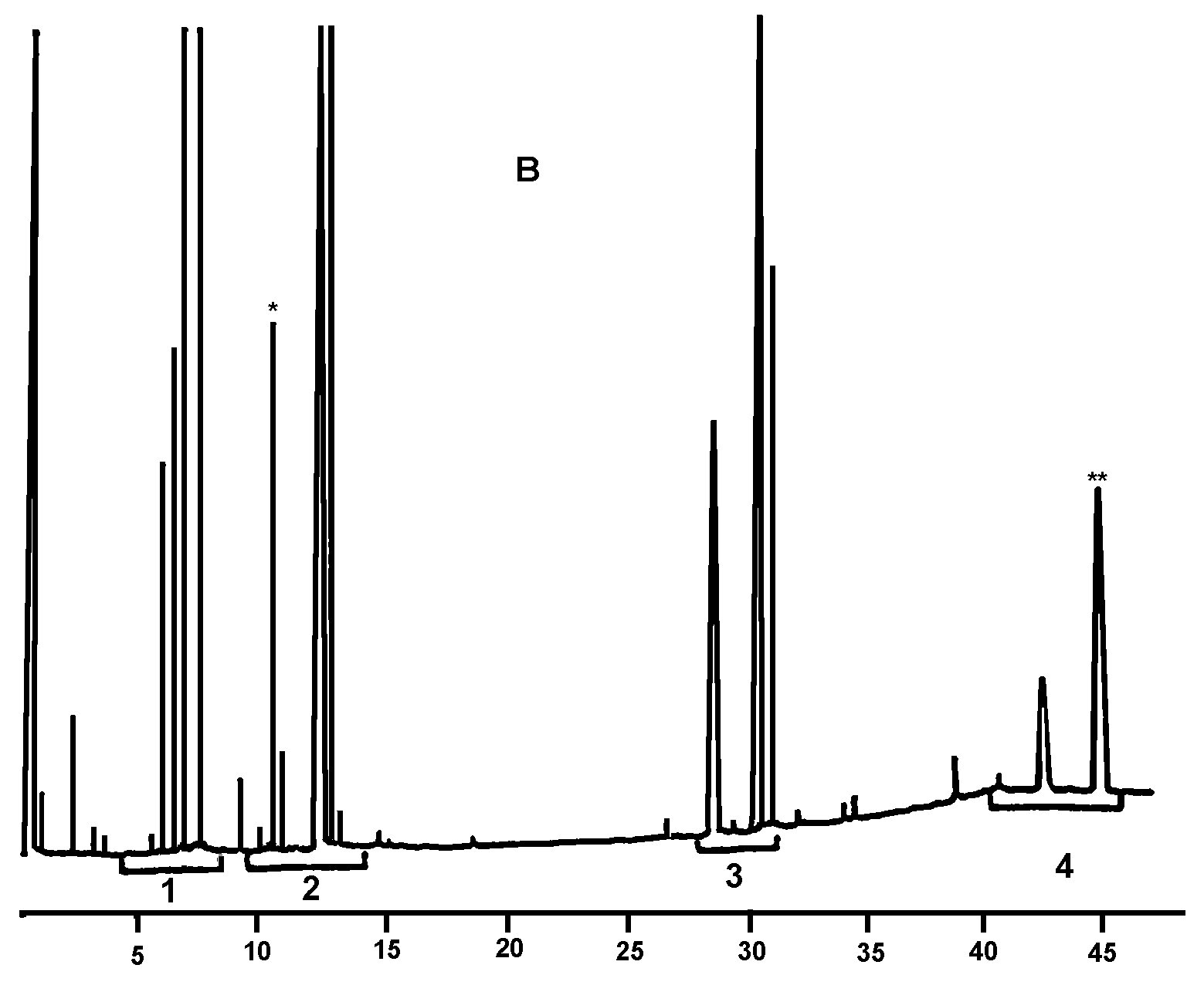
Vysvetlivky:
|
8. POZNÁMKY
PRÍPRAVA LIPÁZY
V obchodnej sieti sú k dispozícii lipázy s dostatočnou aktivitou. V laboratóriu sa tiež dajú pripraviť takto:
5 kg čerstvých pankreasov z ošípaných ochlaďte na 0 °C. Odstráňte tuhý okolitý tuk a spojivové tkanivo a rozomeľte v mlynčeku s lamelami tak, aby ste získali tekutú hmotu. Pretrepávajte túto hmotu 4 až 6 hodín spolu s 2,5 litra bezvodého acetónu a potom odstreďte. Ešte trikrát extrahujte zvyšok s rovnakým objemom bezvodého acetónu, potom ďalších dvakrát so zmesou acetónu/dietyléteru v objemovom pomere (1/1) a dvakrát s dietyléterom.
Sušte zvyšok 48 hodín vo vákuu, aby ste získali stabilný prášok, ktorý bude dlhodobo uskladniteľný v chladničke chránený pred vlhkosťou.
KONTROLA AKTIVITY LIPÁZY
Pripravte emulziu olivového oleja takto:
Počas 10 minút pretrepávajte v miešači zmes zloženú zo 165 ml roztoku arabskej gumy s koncentráciou 100g/l, 15 g roztlčeného ľadu a 20 ml vopred zneutralizovaného oleja.
Postupne zaveďte do 50 ml banky 10 ml tejto emulzie, ďalej 0,3 ml roztoku cholátu sodného s koncentráciou 0,2 g/ml a 20 ml destilovanej vody.
Umiestnite banku do termostatu nastaveného na 37 °C; zaveďte elektródy pH-metra a špirálové miešadlo.
Pomocou byrety pridávajte po kvapkách roztok hydroxidu sodného 0,1 N, až kým nedosiahnete pH 8,3.
Pridajte objem suspenzie prášku lipázy vo vode (0,1 g/ml lipázy). Keď pH-meter ukáže pH 8,3, uveďte chronometer do činnosti a pridávajte po kvapkách roztok hydroxidu sodného takým tempom, aby sa hodnota pH udržiavala na 8,3. Zaznamenajte každú minútu objem spotrebovaného roztoku.
Preneste údaje do grafového systému súradníc tak, že os nezávisle premenných (x-ová os) ponesie časové údaje a na osi závisle premenných (y-ová os) uvediete počet mililitrov alkalického roztoku 0,1 N spotrebovaného na udržanie konštantného pH. Mali by ste získať lineárny graf.
Aktivita lipázy meraná v jednotkách lipázy na mg sa vypočíta na základe vzorca:

kde:
|
A |
je aktivita vyjadrená v jednotkách lipázy/mg |
|
V |
je počet mililitrov roztoku hydroxidu sodného 0,1 N za minútu (vypočítané na základe grafu) |
|
N |
je molarita roztoku hydroxidu sodného |
|
m |
je množstvo testovacej lipázy v mg. |
Jednotka lipázy je definovaná ako množstvo enzýmu, ktoré uvoľní 10 mikro-ekvivalentov kyseliny za minútu.
▼M20 —————
PRÍLOHA IX
SPEKTROFOTOMETRICKÉ SKÚMANIE V ULTRAFIALOVEJ OBLASTI
PREDSLOV
Spektrofotometrické skúmanie v ultrafialovej oblasti môže poskytnúť informácie o kvalite tuku, jeho stave uchovania a o zmenách, ktoré nastali počas technologických procesov. Absorpcia pri vlnových dĺžkach špecifikovaných v metóde je spôsobená prítomnosťou konjugovaných diénových a triénových systémov, ktoré sú výsledkom oxidácie a/alebo rafinovania. Tieto absorpcie sú vyjadrené ako špecifické extinkcie
![]()
(extinkcia 1 % roztoku tuku v špecifikovanom rozpúšťadle, v kyvete hrubej 10 mm) obvykle určené ako K (uvádzané tiež ako „extinkčný koeficient“).
1. OBLASŤ
Táto príloha opisuje postup vykonania spektrofotometrického testovania olivového oleja v ultrafialovej oblasti.
2. PRINCÍP METÓDY
Vzorka sa rozpustí v požadovanom rozpúšťadle a absorbancia roztoku sa meria pri špecifikovaných vlnových dĺžkach vzhľadom na čisté rozpúšťadlo.
Špecifické extinkcie pri 232 nm a 268 nm v izo-oktáne alebo 232 nm a 270 nm v cyklohexáne sa vypočítavajú pri koncentrácii 1 % roztoku v kyvete hrúbky 10 mm.
3. VYBAVENIE
|
3.1. |
Spektrofotometer vhodný na merania pri ultrafialových vlnových dĺžkach (220 nm až 360 nm) s možnosťou odčítavania jednotlivých nanometrických jednotiek. Odporúča sa pravidelná kontrola, pokiaľ ide o presnosť a reprodukovateľnosť mierok vlnovej dĺžky a absorbancie, ako aj čo sa týka rozptýleného svetla.
|
|
3.2. |
Pravouhlé kremenné kyvety, s krytom, vhodné na merania ultrafialových vlnových dĺžok (220 až 360 nm) s optickou dĺžkou 10 mm. Keď sa kyvety naplnia vodou alebo iným vhodným rozpúšťadlom, nemali by medzi sebou vykazovať rozdiel väčší ako 0,01 jednotky extinkcie. |
|
3.3. |
Odmerné banky s jednou značkou s kapacitou 25 ml triedy A. |
|
3.4. |
Analytické váhy s presnosťou 0,0001 g. |
4. ČINIDLÁ
Ak nie je stanovené inak, používajte počas analýzy len činidlá uznanej analytickej kvality a destilovanú alebo demineralizovanú vodu alebo vodu ekvivalentnej čistoty.
Rozpúšťadlo: izo-oktán (2,2,4-trimetylpentán) na merania pri 232 nm a 268 nm alebo cyklohexán na merania pri 232 nm a 270 nm, s absorbanciou menej ako 0,12 pri 232 nm a menej ako 0,05 pri 270 nm oproti destilovanej vode, merané v kyvete hrúbky 10 mm.
5. POSTUP
|
5.1. |
Vzorka musí byť dokonale homogénna a bez podozrivých nečistôt. Ak nie je, musí sa prefiltrovať cez papier pri teplote približne 30 °C. |
|
5.2. |
Presne odvážte približne 0,25 g (s presnosťou na 1 mg) takto pripravenej vzorky do 25 ml odmernej banky, doplňte určeným rozpúšťadlom po značku a zhomogenizujte. Výsledný roztok musí byť úplne priezračný. Ak je prítomná opalescencia alebo zákal, rýchlo roztok prefiltrujte cez papier. POZNÁMKA: Na merania absorbancie panenských a extra panenských olivových olejov pri 268 nm a 270 nm spravidla stačí 0,25 – 0,30 g. Na merania pri 232 nm treba zvyčajne 0,05 g vzorky, takže obyčajne sa pripravujú dva rozdielne roztoky. Vzhľadom na ich vyššiu absorbanciu treba na merania olivového oleja z olivových výliskov, rafinovaného olivového oleja a znehodnoteného olivového oleja zvyčajne nižšie množstvo vzorky, napr. 0,1 g. |
|
5.3. |
V prípade potreby upravte základ (220 – 290 nm) pomocou roztoku v oboch kremenných kyvetách s testovacím roztokom a odmerajte extinkcie pri 232, 268 alebo 270 nm oproti roztoku použitému ako referencia. Zaznamenané hodnoty extinkcie musia byť v rozmedzí 0,1 až 0,8 alebo v rozsahu linearity spektrofotometra, ktorú treba overiť. Ak nie sú, merania sa musia opakovať s použitím koncentrovanejších, prípadne redších roztokov. |
|
5.4. |
Po odmeraní absorbancie pri 268 alebo 270 nm odmerajte absorbanciu pri λmax, λmax + 4 a λmax – 4. Tieto hodnoty absorbancie sa používajú na určenie rozdielu špecifickej extinkcie (ΔΚ). POZNÁMKA: Za λmax sa považuje 268 nm pre izo-oktán použitý ako rozpúšťadlo a 270 nm pre cyclohexán. |
6. VYJADRENIE VÝSLEDKOV
|
6.1. |
Zaznamenajte špecifické extinkcie (extinkčné koeficienty) pri rôznych vlnových dĺžkach, ktoré sa vypočítajú takto:
kde:
vyjadrené na dve desatinné miesta. |

|
6.2. |
Rozdiel špecifickej extinkcie (ΔΚ) Rozdiel absolútnej hodnoty extinkcie (ΔΚ), ktorý je vyjadrený vzorcom:
kde Km je špecifická extinkcia pri vlnovej dĺžke pre maximálnu absorpciu pri 270 nm a 268 nm v závislosti od použitého rozpúšťadla. Výsledky vyjadrené na dve desatinné miesta. |
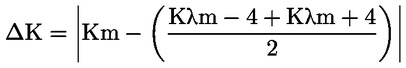
PRÍLOHA X
STANOVOVANIE METYLESTEROV MASTNÝCH KYSELÍN PLYNOVOU CHROMATOGRAFIOU
1. OBLASŤ
Táto príloha poskytuje usmernenia týkajúce sa chromatografického stanovenia voľných a viazaných mastných kyselín v rastlinných tukoch a olejoch po ich konverzii na metylestery mastných kyselín (fatty acid methyl esters, FAME).
Viazané mastné kyseliny triglyceridov (TAG) a v závislosti od metódy esterifikácie voľné mastné kyseliny (free fatty acids, FFA) sa konvertujú na metylestery mastných kyselín, ktoré sa stanovujú kapilárnou plynovou chromatografiou.
Metóda opísaná v tejto prílohe umožňuje stanovenie FAME z C12 až C24 vrátane metylesterov nasýtených, cis- a trans-mononenasýtených a cis- a trans-polynenasýtených mastných kyselín.
2. PRINCÍP
Plynová chromatografia (gas chromatography, GC) sa používa na kvantitatívnu analýzu FAME. FAME sa pripravujú podľa časti A. Potom sa vstreknú do injektora a odparia sa v ňom. Oddelenie FAME sa vykonáva na analytických kolónach so špecifickou polaritou a dĺžkou. Na stanovenie FAME sa používa plameňovoionizačný detektor (Flame Ionisation Detector, FID). Podmienky analýzy sú uvedené v časti B.
V rámci plynovej chromatografie FAME s FID sa ako nosný plyn môžu použiť vodík alebo hélium (mobilná fáza). Vodík zrýchľuje oddeľovanie a zabezpečuje ostrejšie píky. Stacionárna fáza je mikroskopická vrstva tenkého tekutého povlaku na inertnom pevnom povrchu z kremenného skla.
Analyzované odparené zlúčeniny reagujú pri prechode kapilárnou kolónou so stacionárnou fázou, ktorá sa nachádza na vnútornom povrchu kolóny. V dôsledku tohto rozdielneho vzájomného pôsobenia rôznych zlúčenín sa eluujú v rozličnom čase, ktorý sa nazýva retenčný čas zlúčeniny pre daný súbor parametrov analýzy. Porovnanie retenčných časov sa používa na určenie jednotlivých zlúčenín.
ČASŤ A
PRÍPRAVA METYLESTEROV MASTNÝCH KYSELÍN Z OLIVOVÉHO OLEJA A OLEJA Z OLIVOVÝCH VÝLISKOV
1. OBLASŤ
V tejto časti je uvedená príprava metylesterov mastných kyselín. Zahŕňa metódy prípravy metylesterov mastných kyselín z olivového oleja a oleja z olivových výliskov.
2. PÔSOBNOSŤ
Príprava metylesterov mastných kyselín z olivového oleja a oleja z olivových výliskov sa vykonáva transesterifikáciou s metanolovým roztokom hydroxidu draselného pri izbovej teplote. Nevyhnutnosť purifikácie vzorky pred transesterifikáciou závisí od obsahu voľných mastných kyselín vo vzorke a od parametra, ktorý sa má určiť v rámci analýzy, rozhodnúť sa dá podľa tejto tabuľky:
|
Kategória oleja |
Metóda |
|
Panenský olivový olej s kyslosťou ≤ 2,0 % |
1. Mastné kyseliny 2. trans-mastné kyseliny 3. ΔECN42 (po purifikácii so silika gelom SPE) |
|
Rafinovaný olivový olej |
|
|
Olivový olej zložený z rafinovaných a panenských olivových olejov |
|
|
Rafinovaný olej z olivových výliskov |
|
|
Olej z olivových výliskov |
|
|
Panenský olivový olej s kyslosťou > 2,0 % Surový olej z olivových výliskov |
1. Mastné kyseliny (po purifikácii so silika gelom SPE) 2. trans-mastné kyseliny (po purifikácii so silika gelom SPE) 3. ΔECN42 (po purifikácii so silika gelom SPE) |
3. METODIKA
3.1. Transesterifikácia s metanolovým roztokom hydroxidu draselného pri izbovej teplote.
3.1.1. Princíp
Metylestery sa vytvárajú transesterifikáciou s metanolovým hydroxidom draselným ako medzistupeň pred zmydelnením.
3.1.2. Činidlá
|
3.1.2.1. |
Metanol obsahujúci maximálne 0,5 % (m/m) vody. |
|
3.1.2.2. |
Hexán, chromatografickej kvality. |
|
3.1.2.3. |
Heptán, chromatografickej kvality. |
|
3.1.2.4. |
Dietyléter, stabilizovaný na analýzu. |
|
3.1.2.5. |
Acetón, chromatografickej kvality. |
|
3.1.2.6. |
Elučné rozpúšťadlo na purifikáciu oleja prostredníctvom chromatografie s použitím kolóny/SPE, zmes hexán/dietyléter 87/13 (v/v). |
|
3.1.2.7. |
Hydroxid draselný, približne 2M metanolový roztok: rozpustite 11,2 g hydroxidu draselného v 100 ml metanolu. |
|
3.1.2.8. |
Silikagélové patróny, 1 g (6 ml), na extrakciu tuhou fázou. |
3.1.3. Prístrojové vybavenie
|
3.1.3.1. |
Skúmavky so závitovým uzáverom (objem 5 ml) s PTFE spojom. |
|
3.1.3.2. |
Odmerné alebo automatické pipety, 2 ml a 0,2 ml. |
3.1.4. Purifikácia vzoriek olejov
V prípade potreby sa vzorky vyčistia prechodom oleja cez silikagelové patróny na extrakciu tuhou fázou. Silikagelová patróna (3.1.2.8) sa vloží do vákuového elučného zariadenia a premyje sa 6 ml hexánu (3.1.2.2); premytie sa vykonáva bez vákua. Roztok oleja (približne 0,12 g) v 0,5 ml hexánu (3.1.2.2) sa zavedie do kolóny. Po prechode cez kolónu sa následne nechá eluovať pomocou 10 ml zmesi hexán/dietyléter (87:13 v/v) (3.1.2.6). Kombinované eluáty sa homogenizujú a rozdelia na dva podobné objemy. Alikvotná časť sa odparí dosucha v rotačnej odparke pod nižším tlakom pri izbovej teplote. Zvyšok sa rozpustí v 1 ml heptánu a roztok je pripravený na analýzu mastných kyselín prostredníctvom GC. Prípadne sa odparí druhá alikvotná časť a zvyšok sa rozpustí v 1 ml acetónu na analýzu triglyceridov prostredníctvom HPLC.
3.1.5. Postup
V 5 ml skúmavke so závitovým uzáverom (3.1.3.1) odvážte približne 0,1 g vzorky oleja. Pridajte 2 ml heptánu (3.1.2.2) a pretrepávajte. Pridajte 0,2 ml metanolového roztoku hydroxidu draselného (3.1.2.7), nasaďte uzáver s PTFE spojom, uzáver zatiahnite a 30 sekúnd dôkladne pretrepávajte. Nechajte odstáť, kým sa vrchná vrstva nevyčíri. Zlejte vrchnú vrstvu obsahujúcu metylestery. Heptánový roztok je pripravený na vstreknutie do plynového chromatografu. Roztok by sa mal do analýzy plynovou chromatografiou uchovávať v chladničke. Skladovanie roztoku viac než 12 hodín sa neodporúča.
ČASŤ B
ANALÝZA METYLESTEROV MASTNÝCH KYSELÍN PLYNOVOU CHROMATOGRAFIOU
1. OBLASŤ
Táto časť poskytuje všeobecné usmernenia týkajúce sa používania kapilárnej plynovej chromatografie na určenie kvalitatívneho a kvantitatívneho zloženia zmesi metylesterov mastných kyselín získaných v súlade s metódou uvedenou v časti A.
Táto časť sa na nevzťahuje na polymerizované mastné kyseliny.
2. ČINIDLÁ
2.1. Nosný plyn
Inertný plyn (hélium alebo vodík), dokonale vysušený a s obsahom kyslíka menším ako 10 mg/kg.
Poznámka 1: Vodík môže dvojnásobne urýchliť analýzu, ale je nebezpečný. Sú k dispozícii bezpečnostné zariadenia.
2.2. Pomocné plyny
|
2.2.1. |
Vodík (čistota ≥ 99,9 %), bez organických nečistôt. |
|
2.2.2. |
Vzduch alebo kyslík, bez organických nečistôt. |
|
2.2.3. |
Dusík (čistota > 99 %). |
2.3. Referenčný štandard
Zmes metylesterov čistých mastných kyselín alebo metylesterov tuku známeho zloženia, najlepšie podobného zloženia ako tuková látka na analýzu. Na určenie trans-izomérov nenasýtených kyselín sú užitočné cis- a trans-izoméry metylesterov kyseliny oktadecénovej, oktadekadiénovej a oktadekatriénovej.
Je potrebné dbať na to, aby sa zabránilo oxidácii polynenasýtených mastných kyselín.
3. PRÍSTROJOVÉ VYBAVENIE
Uvedené pokyny sa týkajú bežného vybavenia používaného v plynovej chromatografii, využívajúceho kapilárne kolóny a plameňovoionizačný detektor.
3.1. Plynový chromatograf
Plynový chromatograf sa skladá z týchto prvkov:
3.1.1. Injekčný systém
Použite niektorý z injekčných systémov s kapilárnymi kolónami, v tomto prípade by mal byť injekčný systém určený iba na použitie s takýmito kolónami. Môže to byť split (s delením) typ alebo splitless (bez odfuku) on-column injektor.
3.1.2. Pec
Pec musí byť schopná vyhrievať kapilárnu kolónu na teplotu aspoň 260 °C a udržiavať požadovanú teplotu v rozmedzí 0,1 °C. Táto druhá požiadavka je obzvlášť dôležitá pri trubiciach z kremenného skla.
Vo všetkých prípadoch sa odporúča teplotne programovateľné vyhrievanie a to najmä pre mastné kyseliny s menej ako 16 uhlíkovými atómami.
3.1.3. Kapilárna kolóna
|
3.1.3.1. |
Trubica vyrobená z materiálu nereagujúceho s látkami, ktoré sa majú analyzovať (obvykle zo skla alebo z kremenného skla). S vnútorným priemerom 0,2 až 0,32 mm. Vnútorný povrch musí pred nanesením stacionárnej fázy prejsť náležitou úpravou (napr. preparáciou povrchu, deaktiváciou). Pre mastné kyseliny a cis- a trans-izoméry mastných kyselín je dostatočná dĺžka 60 m. |
|
3.1.3.2. |
Stacionárna fáza, vhodné sú kolóny z polárneho polysiloxánu (kyanopropylsilikónu) s krížom viazanými väzbami. Poznámka 2: Existuje riziko, že polárne polysiloxány môžu spôsobiť ťažkosti pri určovaní a oddeľovaní kyseliny linolénovej a C20 kyselín. Nanesené vrstvy musia byť tenké, t. j. 0,1 až 0,2 μm. |
|
3.1.3.3. |
Montáž a kondiciovanie kolóny Pri montáži kapilárnych kolón dodržte obvyklé opatrenia, t. j. umiestnenie kolóny v peci (držiak), výber a montáž spojov (tesnosť), umiestnenie koncov kolóny v injektore a detektore (redukcia mŕtvych objemov). Uložte kolónu pod prietok nosného plynu [napr. 0,3 bar (30 kPa), v prípade kolóny dĺžky 25 mm a s vnútorným priemerom 0,3 mm]. Kondiciujte kolónu v peci s teplotou nastavovanou na 3 °C/min od teploty prostredia až na teplotu 10 °C pod limitom dekompozície stacionárnej fázy. Udržujte pec na tejto teplote jednu hodinu až do stabilizácie základu. Vráťte teplotu na 180 °C, aby ste pracovali v izotermálnych podmienkach. Poznámka 3: Vhodné pre-kondiciované kolóny sú dostupné v obchodnej sieti. |
3.1.4. Plameňovoionizačný detektor a prevodník-zosilňovač
3.2. Striekačka
Striekačka má maximálnu kapacitu 10 μl, delenú po 0,1 μl.
3.3. Systém získavania údajov
Systém získavania údajov spojený online s detektormi a používaný so softvérovým programom vhodným na integráciu a normalizáciu píkov.
4. POSTUP
Operácie opísané v bodoch 4.1 až 4.3 sa vzťahujú na používanie plameňovoionizačného detektora.
4.1. Podmienky testovania
4.1.1. Výber optimálnych prevádzkových podmienok pre kapilárne kolóny
Vzhľadom na účinnosť a priepustnosť kapilárnych kolón závisí oddeľovanie zložiek a trvanie analýzy vo veľkej miere od prietoku nosného plynu cez kolónu. Preto bude potrebné optimalizovať prevádzkové podmienky úpravou tohto parametra (alebo straty na vstupe kolóny) v závislosti od toho, či je cieľom zlepšenie oddeľovania alebo zrýchlenie analýzy.
Tieto podmienky sa ukázali byť vhodné pre oddelenie FAME (C4 až C26). Príklady chromatogramov sú uvedené v dodatku B:
|
Teplota injektora: |
250 °C |
|
Teplota detektora: |
250 °C |
|
Teplota pece: |
165 °C (8 min) až 210 °C pri 2 °C/min |
|
Nosný plyn – vodík: |
tlak na vstupe kolóny 179 kPa |
|
Celkový prietok: |
154,0 ml/min; |
|
Deliaci pomer: |
1:100 |
|
Objem nástreku: |
1 μl |
4.1.2. Stanovenie rozlíšenia (pozri dodatok A)
Vypočítajte rozlíšenie, R, dvoch susediacich píkov I a II s použitím vzorca:
R = 2 × ((dr ( II ) – d r(I))/(ω(I) + ω(II))) alebo R = 2 × ((tr ( II ) – t r(I))/(ω(I) + ω(II))) (USP) (liekopis USA)
alebo
R = 1,18 × ((tr ( II ) – tr(I))/( ω0,5(I) + ω0,5(II))) (EP, BP, JP, DAB) JP (japonský liekopis), EP (európsky liekopis), BP (liekopis UK)
kde:
d r(I) je retenčná vzdialenosť píku I;
d r(II) je retenčná vzdialenosť píku II;
t r(I) je retenčný čas píku I;
t r(II) je retenčný čas píku II;
ω(I) je šírka základu píku I;
ω(II) je šírka základu píku II;
ω0,5 je šírka píku špecifikovanej zlúčeniny, pri strednej výške píku;
Ak ω(I) ≈ ω(II), vypočítajte R s použitím týchto vzorcov:
R = (dr ( II ) – d r(I))/ω = (dr ( II ) – d r(I))/4σ
kde:
σ je štandardná odchýlka (pozri dodatok A, obrázok 1).
Ak sa vzdialenosť dr medzi dvoma píkmi d r(II) – d r(I) rovná 4σ, faktor rozlíšenia R = 1.
Ak sa dva píky úplne neoddelia, tangenty inflexných bodov dvoch píkov sa pretínajú v bode C. Aby sa oba píky úplne oddelili, vzdialenosť medzi nimi musí byť:
d r(II) – d r(I) = 6 σ odkiaľ R = 1,5 (pozri dodatok A, obrázok 3).
5. VYJADRENIE VÝSLEDKOV
5.1. Kvalitatívna analýza
Určite píky metylesterov vzorky z chromatogramu v dodatku B (obrázok 1), podľa potreby interpoláciou alebo porovnaním s píkmi referenčných zmesí metylesterov (ako je opísané v bode 2.3).
5.2. Kvantitatívna analýza
5.2.1. Stanovenie zloženia
Vypočítajte hmotnostný zlomok w i jednotlivých metylesterov mastných kyselín, vyjadrený ako percento hmotnosti metylesterov, takto:
5.2.2. Metóda výpočtu
5.2.2.1. Všeobecný prípad
Vypočítajte obsah danej zložky i, vyjadrený ako percento hmotnosti metylesterov, stanovením percenta predstavujúceho plochu príslušného píku vzťahujúceho sa k sume plôch všetkých píkov, pomocou nasledujúceho vzorca:
wi = (Ai/ΣA) × 100
kde:
Ai je plocha pod píkom individuálneho metylesteru mastnej kyseliny i;
ΣA je suma plôch pod všetkými píkmi všetkých individuálnych metylesterov mastných kyselín.
Výsledky sú vyjadrené na dve desatinné miesta.
Poznámka 4: Pokiaľ ide o tuky a oleje, hmotnostný zlomok metylesterov mastných kyselín sa rovná hmotnostnému zlomku triglyceridov v gramoch na 100 g. V prípade, keď tento predpoklad nie je povolený, pozri bod 5.2.2.2.
5.2.2.2. Použitie korekčných faktorov
V niektorých prípadoch, napríklad za prítomnosti mastných kyselín s menej ako 8 atómami uhlíka alebo kyselín so sekundárnou skupinou plochy, by sa plochy mali upraviť špecifickými korekčnými faktormi (Fci). Tieto faktory sa určia pre každý jeden prístroj. Na tento účel sa použijú vhodné referenčné materiály s certifikovaným zložením mastných kyselín v zodpovedajúcom rozsahu.
Poznámka 5: Tieto korekčné faktory nie sú identické s teoretickými korekčnými faktormi FID, ktoré sú uvedené v dodatku A, keďže zahŕňajú aj výkonnosť injekčného systému atď. V prípade väčších rozdielov by sa však mala kontrolovať výkonnosť celého systému.
Pri tejto porovnávacej zmesi je hmotnostné percento FAME i vyjadrené vzorcom:
wi = (mi /Σm) × 100
kde
m i je hmotnosť FAME i v porovnávacej zmesi;
Σm je súčet hmotností rôznych zložiek ako FAME v porovnávacej zmesi.
Z chromatogramu porovnávacej zmesi vypočítajte percento plochy pre FAME i takto:
wi = (Ai/ΣA) × 100
kde:
Ai je plocha FAME i v porovnávacej zmesi;
ΣA je súčet všetkých plôch všetkých FAME porovnávacej zmesi.
Korekčný faktor Fc je potom
Fc = (mi × ΣA)/(Ai/Σm)
Pokiaľ ide o vzorku, hmotnostné percento každého FAME i je:
wi = (Fi × Ai)/Σ (Fi × Ai)
Výsledky sú vyjadrené na dve desatinné miesta.
Poznámka 6: Vypočítaná hodnota zodpovedá hmotnostnému percentu jednotlivých mastných kyselín vypočítanému ako triglyceridy na 100 g tuku.
5.2.2.3. Použitie vnútorného štandardu
Pri niektorých analýzach (napr. ak nie sú kvantifikované všetky mastné kyseliny, keď sú napríklad spolu s kyselinami so 16 alebo 18 uhlíkmi prítomné kyseliny so štyrmi alebo šiestimi uhlíkmi alebo keď je potrebné vo vzorke určiť absolútne množstvo mastnej kyseliny) je potrebné použiť vnútorný štandard. Často sa používajú mastné kyseliny s 5, 15 alebo so 17 uhlíkmi. Mal by sa určiť korekčný faktor (podľa potreby) pre vnútorný štandard.
Hmotnostné percento zložky i, vyjadrené ako metylester, je potom dané vzorcom:
wi = (mIS × Fi × Ai)/(m × FIS × AIS)
kde:
A i je plocha FAME i;
A IS je plocha vnútorného štandardu;
F i je korekčný faktor i mastnej kyseliny, vyjadrený ako FAME;
F IS je korekčný faktor vnútorného štandardu;
m je hmotnosť testovanej vzorky, v miligramoch;
m IS je hmotnosť vnútorného štandardu, v miligramoch.
Výsledky sú vyjadrené na dve desatinné miesta.
6. SPRÁVA O VÝSLEDKU TESTU
V správe o výsledku testu sa uvádzajú metódy použité na prípravu metylesterov a na plynovú chromatografickú analýzu. Uvádzajú sa v nej aj všetky podrobnosti o pracovnom postupe, ktoré neboli uvedené v tejto štandardnej metóde alebo sa považovali za nepovinné, spolu s podrobnosťami o akomkoľvek sprievodnom jave, ktorý mohol ovplyvniť výsledky.
Správa o výsledku testu zahŕňa všetky informácie potrebné na úplnú identifikáciu vzorky.
7. PRESNOSŤ
7.1. Výsledky medzilaboratórnej skúšky
Podrobnosti medzilaboratórnej skúšky týkajúce sa presnosti metódy sú uvedené v prílohe C k norme IOC/T.20/Doc. No 33. Hodnoty odvodené z tejto medzilaboratórnej skúšky sa nesmú vzťahovať na iné rozsahy a matrice koncentrácie než tie, ktoré sú dané.
7.2. Opakovateľnosť
Absolútny rozdiel medzi dvoma nezávislými výsledkami jednotlivých testov získanými s použitím rovnakej metódy na rovnakom skúšobnom materiáli v rovnakom laboratóriu rovnakým analytikom s použitím rovnakého vybavenia v rámci krátkeho časového intervalu nie je vo viac ako 5 % prípadov väčší ako r uvedené v tabuľkách 1 až 14 v prílohe C k norme IOC/T.20/Doc. No 33.
7.3. Reprodukovateľnosť
Absolútny rozdiel medzi dvoma nezávislými výsledkami jednotlivých testov získanými s použitím rovnakej metódy na rovnakom skúšobnom materiáli v inom laboratóriu iným analytikom s použitím iného vybavenia nie je vo viac ako 5 % prípadov väčší ako R uvedené v tabuľkách 1 až 14 v prílohe C k norme IOC/T.20/Doc. No 33.
Dodatok A
Obrázok 1

So šírkou ω0,5 v polovičnej výške trojuholníka (ABC) a šírkou b v polovičnej výške trojuholníka (NPM).
|
Obrázok 2 |
Obrázok 3 |
|
|
|

Dodatok B
Obrázok 1
Plynovo chromatografický profil získaný metódou studenej metylácie z oleja z olivových výliskov.

Chromatografické píky zodpovedajú metylesterom a etylesterom okrem prípadov, keď je uvedené inak.
PRÍLOHA XI
STANOVENIE OBSAHU PRCHAVÝCH HALOGENOVANÝCH ROZPÚŠŤADIEL V OLIVOVOM OLEJI
1. METÓDA
Analýza plynovou chromatografiou s použitím head space techniky.
2. ZARIADENIE
|
2.1. |
Plynový chromatograf vybavený detektorom elektrónového záchytu (ECD) |
|
2.2. |
Head space zariadenie |
|
2.3. |
Plynovochromatografická kolóna, sklenená, 2 m dlhá a s priemerom 2 mm, so stacionárnou fázou. OV 101 10 % alebo ekvivalent, impregnovaná kalcinovaná rozsievková zemina, kyslo praná a silanizovaná s časticami veľkosti od 80 do 100 mesh |
|
2.4. |
Nosný a pomocný plyn: dusík na plynovú chromatografiu, vhodný na detekciu elektrónovým záchytom |
|
2.5. |
Sklenené banky, od 10 do 15 ml, potiahnuté teflónom a s hliníkovou zátkou s otvormi pre striekačku |
|
2.6. |
Hermeticky uzatvárateľné svorky |
|
2.7. |
Plynotesné striekačky 0,5 až 2ml |
3. ČINIDLÁ
Štandard: halogenované rozpúšťadlá, stupňa čistoty vyhovujúceho plynovej chromatografii
4. POSTUP
|
4.1. |
Presne odvážte 3 g oleja, dajte do sklenenej banky (nepoužitej) a hermeticky ju uzatvorte. Vložte ju do termostatu nastaveného na 70 oC na jednu hodinu. Opatrne pomocou striekačky odoberte 0,2 až 0,5 ml horného priestoru (head space). Nadávkujte do kolóny plynového chromatografu nastaveného nasledujúco: — teplota injektora: 150 oC, — teplota kolóny: 70 až 80 oC, — detektor teploty: 200 až 250 oC. Môžu sa použiť aj iné teploty za predpokladu, že sa s tým nezmenia výsledky. |
|
4.2. |
Porovnávacie roztoky: pripravte roztoky štandardov pomocou rafinovaného olivového oleja so stopami rozpúšťadla v koncentráciách v rozsahu 0.05 až do 1 ppm (mg/kg), zodpovedajúce odhadovanému obsahu vo vzorke. Halogenované rozpúšťadlá sa dajú zriediť a pentánom. |
|
4.3. |
Kvantitatívne vyhodnotenie: uveďte do vzájomného vzťahu plochy alebo výšky píkov vzorky a štandartného roztoku s najbližšou predpokladanou. Ak je odchýlka väčšia ako 10 %, analýza sa musí zopakovať štandartným spôsobom, až kým odchýlka nie je do 10 %. Obsah je stanovený na základe priemeru jednotlivých analýz. |
|
4.4. |
Vyjadrenie výsledkov: v ppm (mg/kg). Detekčný limit pre túto metódu je 0,01 mg/kg. |
PRÍLOHA XII
METÓDA MEDZINÁRODNEJ RADY PRE OLIVY NA ORGANOLEPTICKÉ HODNOTENIE PANENSKÝCH OLIVOVÝCH OLEJOV
1. ÚČEL A ROZSAH PÔSOBNOSTI
Účelom medzinárodnej metódy opísanej v tejto prílohe je stanoviť postup hodnotenia organoleptických vlastností panenských olivových olejov v zmysle časti VIII bodu 1 prílohy VII k nariadeniu Európskeho parlamentu a Rady (EÚ) č. 1308/2013 ( 4 ) a zaviesť metódu ich klasifikácie na základe uvedených vlastností. Touto metódou sa poskytujú aj pokyny týkajúce sa nepovinného označovania.
Opísaná metóda sa vzťahuje iba na panenské olivové oleje a na ich klasifikáciu alebo označovanie podľa intenzity vnímaných nedostatkov a ovocnosti, ako ich stanovila skupina vybraných vyškolených a odskúšaných degustátorov tvoriacich porotu.
Normy IOC uvedené v tejto prílohe sa používajú v ich poslednom znení, ktoré je k dispozícii.
2. VŠEOBECNÝ ZÁKLADNÝ SLOVNÍK SENZORICKEJ ANALÝZY
Pozri normu IOC/T.20/Doc. č. 4 „Senzorická analýza: všeobecný základný slovník.“
3. ŠPECIFICKÝ SLOVNÍK
3.1. Negatívne atribúty
Stuchnutý/po kalových usadeninách characteristická príchuť oleja získaného z olív, ktoré sa hromadili alebo skladovali v takých podmienkach, že prešli do pokročilého stavu anaeróbnej fermentácie, alebo oleja, ktorý sa ponechal v kontakte s kalmi usadzujúcimi sa v podzemných kadiach a nádržiach, ktoré takisto prešli anaeróbnou fermentáciou.
Zatuchnutý-vlhký-zemitý characteristická príchuť olejov získaných z ovocia, v ktorom sa v dôsledku niekoľkodňového skladovania vo vlhkom prostredí vyvinulo veľké množstvo plesní a kvasiniek, alebo oleja získaného z olív, ktoré boli pri zbere zablatené alebo pokryté bahnom a neboli umyté.
Vínový-octový-kyslý-kyslastý charakteristická príchuť niektorých olejov, ktorá pripomína víno alebo ocot. Táto príchuť je spôsobená hlavne aeróbnou fermentáciou olív alebo olivovej hmoty na lisovacích diskoch, ktoré neboli dobre očistené, v dôsledku čoho dochádza k tvorbe kyseliny octovej, etylacetátu a etanolu.
Zožltnutý príchuť olejov, kotré prešli intenzívnym procesom oxidácie.
Po omrznutých olivách (po vlhkom dreve) charakteristická príchuť niektorých olejov extrahovaných z olív, ktoré boli poškodené mrazom ešte na strome.
3.1.1. Ďalšie negatívne atribúty
|
Prehriaty alebo pripálený |
charakteristická príchuť olejov spôsobená príliš silným alebo dlhým zahrievaním počas spracovania, najmä ak sa pasta mieša za tepla a za nevhodných tepelných podmienok. |
|
Po sene–dreve |
charakteristická príchuť určitých olejov vyrobených z vyschnutých olív. |
|
Drsný |
hutný, pastovitý chuťový vnem vyvolaný určitými starými olejmi. |
|
Mastný |
príchuť pripomínajúca motorovú naftu, vazelínu alebo minerálny olej. |
|
Po rastlinných šťavách |
príchuť, ktorú olej získava v dôsledku predĺženého styku s rastlinnými šťavami, ktoré prešli procesom fermentácie. |
|
Slaný |
príchuť oleja extrahovaného z olív konzervovaných v slanom náleve. |
|
Kovový |
príchuť pripomínajúca kovy. Je charakteristická pre olej, ktorý bol dlho v kontakte s kovovými povrchmi počas drvenia, miešania, lisovania alebo skladovania. |
|
Esparto |
charakteristická príchuť oleja získaného z olív lisovaných na nových diskoch z trávy esparto. Táto príchuť sa môže líšiť v závislosti od toho, či ide o disky vyrobené zo zeleného esparta alebo zo suchého esparta. |
|
Po larvách |
príchuť oleja získaného z olív silne napadnutých larvami muchy olivovej (Bactrocera oleae). |
|
Po uhorkách |
príchuť, ktorá vzniká v oleji príliš dlho hermeticky uzavretom najmä v plechovkách a ktorá sa spája so vznikom 2,6-nonadienalu. |
3.2. Pozitívne atribúty
|
Ovocný |
súbor čuchových vnemových vlastností oleja, ktoré závisia od odrody a pochádzajú z nepoškodených, čerstvých, zrelých alebo nezrelých olív. Pociťuje sa priamo a/alebo retronazálne. |
|
Horký |
charakteristická primárna chuť oleja získaného zo zelených olív alebo olív začínajúcich meniť farbu. Pociťuje sa hradenými papilami jazyka v oblasti okolo brázdy v tvare „V“. |
|
Štipľavý |
ostrá taktilná vnemová vlastnosť olejov vyrobených začiatkom zberovej sezóny primárne zo zatiaľ nedozretých olív. Možno ju vnímať v celej ústnej dutine, hlavne v hrdle. |
3.3. Nepovinná terminológia na účely označovania
Predseda poroty môže na požiadanie potvrdiť, že hodnotené oleje spĺňajú vymedzenia a rozsahy zodpovedajúce výlučne týmto výrazom v závislosti od intenzity a vnímania atribútov:
Pozitívne atribúty (ovocný, horký a štipľavý): Podľa intenzity vnímania:
— robustný, ak je medián atribútu väčší ako 6,0,
— stredný, ak je medián atribútu viac než 3 a minimálne 6,0,
— lahodný, ak je medián atribútu maximálne 3,0.
|
Ovocnosť |
súbor čuchových vnemových vlastností oleja, ktoré závisia od odrody olív a pochádzajú z nepoškodených, čerstvých olív, v ktorých nedominuje zelená ani zrelá ovocnosť. Pociťuje sa priamo a/alebo retronazálne. |
|
Zelená ovocnosť |
súbor čuchových vnemových vlastností oleja, ktoré pripomínajú zelené ovocie, závisia od odrody olív a pochádzajú zo zelených, nepoškodených, čerstvých olív. Pociťuje sa priamo a/alebo retronazálne. |
|
Zrelá ovocnosť |
súbor čuchových vnemových vlastností oleja, ktoré pripomínajú zrelé ovocie, závisia od odrody olív a pochádzajú z nepoškodených, čerstvých olív. Pociťuje sa priamo a/alebo retronazálne. |
|
Dobre vyvážený |
olej nevykazujúci žiadnu nevyváženosť, ktorou sa myslí čuchovo-chuťový a taktilný vnem a pri ktorej je medián atribútu horkosti a medián atribútu štipľavosti maximálne o 2,0 bodu vyšší ako medián ovocnosti. |
|
Jemný olej |
olej, ktorého medián atribútov horkosti a štipľavosti je maximálne 2,0. |
Zoznam výrazov podľa intenzity vnímania:
|
Výrazy, ktoré treba použiť v protokole o organoleptickej skúške |
Medián atribútu |
|
Ovocnosť |
— |
|
Zrelá ovocnosť |
— |
|
Zelená ovocnosť |
— |
|
Lahodná ovocnosť |
≤ 3,0 |
|
Stredná ovocnosť |
3,0 < Me ≤ 6,0 |
|
Robustná ovocnosť |
> 6,0 |
|
Lahodná zrelá ovocnosť |
≤ 3,0 |
|
Stredná zrelá ovocnosť |
3,0 < Me ≤ 6,0 |
|
Robustná zrelá ovocnosť |
> 6,0 |
|
Lahodná zelená ovocnosť |
≤ 3,0 |
|
Stredná zelená ovocnosť |
3,0 < Me ≤ 6,0 |
|
Robustná zelená ovocnosť |
> 6,0 |
|
Lahodná horkosť |
≤ 3,0 |
|
Stredná horkosť |
3,0 < Me ≤ 6,0 |
|
Robustná horkosť |
> 6,0 |
|
Lahodná štipľavosť |
≤ 3,0 |
|
Stredná štipľavosť |
3,0 < Me ≤ 6,0 |
|
Robustná štipľavosť |
> 6,0 |
|
Dobre vyvážený olej |
Medián atribútu horkosti a medián atribútu štipľavosti je maximálne o 2,0 bodu vyšší ako medián ovocnosti. |
|
Jemný olej |
Medián atribútu horkosti a medián atribútu štipľavosti je maximálne 2. |
4. POHÁR NA DEGUSTÁCIU OLEJA
Pozri normu IOC/T.20/Doc. č. 5, „Pohár na degustáciu oleja“.
5. TESTOVACIA MIESTNOSŤ
Pozri normu IOC/T.20/Doc. č. 6, „Usmernenie pre zriadenie testovacej miestnosti“.
6. PRÍSLUŠENSTVO
Každá kabína sa musí vybaviť tak, aby mali degustátori na riadne vykonávanie svojej úlohy na dosah toto príslušenstvo:
— (normalizované) číselným znakom označené poháre obsahujúce vzorky, prikryté hodinovým sklíčkom a udržiavané pri teplote 28 °C ± 2 °C,
— profilový hárok (pozri obr. 1) v tlačenej alebo elektronickej podobe za predpokladu, že podmienky uvedené na profilovom liste sú splnené, spolu s prípadnými pokynmi na jeho použitie,
— pero alebo nezmazateľný atrament,
— podnosy s plátkami jabĺk a/alebo vodou, sýtenou vodou a/alebo suchármi,
— pohár vody izbovej teploty,
— hárok so všeobecnými pravidlami uvedenými v oddieloch 8.4 a 9.1.1,
— pľuvadlá.
7. PREDSEDA POROTY A DEGUSTÁTORI
7.1. Predseda poroty
Predseda poroty musí byť vhodne vyškolenou osobou s odbornými znalosťami olejov jednotlivých druhov, ktoré sa budú vyskytovať počas jeho práce. Je kľúčovou postavou poroty a je zodpovedný za jej organizáciu a fungovanie.
Práca predsedu poroty si vyžaduje základnú odbornú prípravu v oblasti nástrojov senzorickej analýzy, zručnosti v oblasti senzorickej analýzy, dôslednosť pri príprave, organizovaní a vykonávaní skúšok a zručnosť a trpezlivosť pri vedeckom plánovaní a vykonávaní skúšok.
Je jedinou osobou zodpovednou za výber, vyškolenie a monitorovanie degustátorov s cieľom zaistiť adekvátnu úroveň ich schopností. Je zodpovedný za hodnotenie degustátorov, ktoré musí byť vždy objektívne a ku ktorému musí vypracovať osobitné postupy na základe skúšok a pevne stanovených kritérií prijímania a vyradzovania. Pozri normu IOC/T.20/Doc. č. 14, „Usmernenia pre výber, výcvik a monitorovanie odborných degustátorov olivového oleja“.
Predseda poroty je zodpovedný za výkon poroty, a preto aj za jej hodnotenie, ku ktorému musí predložiť spoľahlivý a objektívny dôkaz. V každom prípade musí kedykoľvek preukázať, že metóda aj degustátori sú pod kontrolou. Odporúča sa pravidelná kalibrácia poroty (IOC/T.20/Doc. č. 14, časť 5).
Takisto nesie aj konečnú zodpovednosť za vedenie záznamov poroty. Tieto záznamy sa musia dať kedykoľvek vysledovať. Musia byť v súlade s požiadavkami na zabezpečenie a kvalitu stanovenými v medzinárodných normách senzorickej analýzy a celý čas sa v nich musí zaisťovať anonymita vzoriek.
Predseda poroty zodpovedá za inventarizáciu prístrojov a zariadenia potrebných na splnenie špecifikácií uvedenej metódy a zabezpečuje ich riadne čistenie a údržbu, o čom uchováva písomný dôkaz, ako aj dôkaz o splnení podmienok skúšania.
Zodpovedá za príjem a uskladnenie vzoriek pri ich príchode do laboratória, ako aj za ich skladovanie po vykonaní skúšok. Pritom zabezpečuje, že vzorky zostanú po celý čas anonymné a že sa správne skladujú. Na tieto účely písomne vypracuje postup, aby sa zabezpečilo, že celý proces je vysledovateľný a poskytuje príslušné záruky.
Okrem toho je zodpovedný za prípravu, pridelenie číselného znaku a prezentáciu vzoriek degustátorom podľa vhodného návrhu skúšky v súlade s vopred pripravenými protokolmi, ako aj za zostavenie a štatistické spracovanie údajov, ktoré degustátori získali.
Má na starosti vypracovanie a načrtnutie všetkých ďalších postupov, ktoré môžu byť nevyhnutné na doplnenie tejto normy a na zabezpečenie riadneho fungovania poroty.
Musí hľadať spôsoby porovnávania výsledkov dotknutej poroty s výsledkami, ktoré získali iné poroty vykonávajúce analýzy panenských olivových olejov, aby sa ubezpečil, že dotknutá porota pracuje správne.
Povinnosťou predsedu poroty je motivovať členov poroty povzbudzovaním ich záujmov, zvedavosti a súťaživého ducha. Na uvedené účely sa mu dôrazne odporúča, aby zabezpečil bezproblémový obojsmerný tok informácií s členmi poroty tým, že ich priebežne informuje o všetkých vykonávaných úlohách a dosiahnutých výsledkoch. Okrem toho drží v tajnosti vlastný názor a v prípade potreby priebojnejším členom poroty bráni v tom, aby ostatným degustátorom vnucovali vlastné kritériá.
Degustátorov zvoláva v dostatočnom predstihu a odpovedá na všetky otázky týkajúce sa vykonávania skúšok, zdržiava sa však vyjadrovania akéhokoľvek názoru na vzorky.
7.1.1. Podpredseda poroty
Predseda poroty môže byť v odôvodnených prípadoch nahradený podpredsedom poroty, ktorý ho môže zastupovať, pokiaľ ide o povinnosti týkajúce sa vykonávania skúšok. Tento náhradník musí mať všetky potrebné zručnosti, ktoré musí mať predseda poroty.
7.2. Degustátori
Osoby, ktoré pôsobia ako degustátori pri organoleptickom skúšaní olivových olejov, nimi musia byť dobrovoľne. Preto sa odporúča, aby kandidáti predkladali žiadosti v písomnej forme. Kandidátov vyberá, odborne pripravuje a ich činnosť monitoruje predseda poroty v súlade s ich zručnosťami pri rozoznávaní podobných vzoriek; pri tom by mal mať na zreteli, že ich presnosť sa bude precvičovaním zlepšovať.
Degustátori musia konať ako skutoční zmysloví pozorovatelia, nesmú sa nechať ovplyvniť vlastnými preferenciami a musia oznamovať jedine vnemy, ktoré pociťujú. Z uvedeného dôvodu musia vždy pracovať v tichosti, uvoľneným a nenáhlivým spôsobom, pričom musia čo možno najsústredenejšie zamerať svoju vnemovú pozornosť na vzorku, ktorú degustujú.
Pri každej skúške sa vyžaduje prítomnosť 8 až 12 degustátorov, hoci je vhodné mať v rezerve niekoľkých ďalších degustátorov pre prípadnú neprítomnosť.
8. PODMIENKY DEGUSTÁCIE
8.1. Podávanie vzorky
Vzorka oleja na analýzu sa podáva v normalizovaných pohároch na degustáciu v súlade s normou IOC/T.20/Doc. č. 5, „Pohár na degustáciu oleja“.
Pohár musí obsahovať od 14 do 16 ml oleja, resp. od 12,8 do 14,6 g, ak sa vzorky majú vážiť, a prikryje sa hodinovým sklíčkom.
Jednotlivé poháre sa označujú náhodne zvolenými kódmi skladajúcimi sa z čísel alebo z kombinácie písmen a čísel. Označenie kódom nesmie zanechať nijaký zápach.
8.2. Degustácia a teplota vzorky
Vzorky oleja určené na degustáciu sa počas celej degustácie v pohároch udržiavajú pri teplote 28 °C ± 2 °C. Táto teplota bola stanovená, lebo pri izbovej teplote sa ľahšie určujú organoleptické rozdiely, ako aj preto, že pri nižších teplotách aromatické uhľovodíky charakteristické pre tieto oleje nedostatočne prchajú, zatiaľ čo vyššie teploty vedú k tvorbe prchavých zlúčenín charakteristických pre zahrievané oleje. Pozri normu IOC/T.20/Doc. č. 5, „Pohár na degustáciu oleja“, pokiaľ ide o metódu, ktorá sa má použiť na zohrievanie vzoriek v pohári.
Teplota v testovacej miestnosti musí byť od 20 ° do 25 °C (pozri IOC/T.20/Doc. č. 6).
8.3. Čas degustácie
Najlepší čas na degustáciu olejov je ráno. Je dokázané, že existujú obdobia optimálneho vnímania chuti a čuchu počas dňa. Čuchová a chuťová citlivosť narastá pred jedlom, zatiaľ čo po ňom klesá.
Pri zohľadňovaní tohto faktoru by sa však nemalo zachádzať do krajnosti, keďže hlad môže degustátorov rozptýliť a znížiť tak ich posudzovacie schopnosti. Preto sa odporúča degustáciu uskutočňovať medzi desiatou hodinou ráno a poludním.
8.4. Degustátori: všeobecné pravidlá profesionálnej etiky
Počas výkonu úloh degustátora sa uplatňujú tieto odporúčania:
Potom, čo predseda poroty vyzve degustátorov, aby sa zúčastnili na organoleptickom teste, mali by sa dostaviť vo vopred stanovenom čase a dodržiavať tieto pravidlá:
— Aspoň 30 minút pred časom určeným na degustáciu nesmú fajčiť ani piť kávu.
— Nesmú používať parfumy, kozmetické prípravky ani mydlá, ktorých vôňa by mohla pretrvať až do času degustácie. Tak často, ako je to potrebné, si musia na odstránenie akejkoľvek vône alebo zápachu umývať ruky, pričom používajú neparfumované mydlo, ktoré musia dokonale zmyť, a ruky si následne musia osušiť.
— Aspoň jednu hodinu pred začiatkom degustácie nesmú jesť.
— Ak sa degustátori necítia v dobrej fyzickej kondícii, najmä ak má tento stav vplyv na čuchové alebo chuťové vnímanie, alebo ak sú v akomkoľvek psychickom stave, ktorý im nedovoľuje sústrediť sa na prácu, musia sa zdržať degustácie a náležite informovať predsedu poroty.
— Ak degustátori splnili všetky uvedené podmienky, potichu a disciplinovane zaujmú miesto v kabínke, ktorá im bola pridelená.
— Pozorne si prečítajú inštrukcie uvedené na profilovom hárku a vzorku začnú posudzovať až vtedy, keď sú úplne pripravení vykonať pridelenú úlohu (uvoľnene a nenáhlivo). V prípade pochybností by sa degustátori mali v súkromí obrátiť na predsedu poroty.
— Pridelené úlohy musia vykonávať potichu.
— Celý čas musia mať vypnutý mobilný telefón, aby sa nerozptyľovali a nerušili kolegov pri práci.
9. POSTUP ORGANOLEPTICKÉHO HODNOTENIA A KLASIFIKÁCIE PANENSKÉHO OLIVOVÉHO OLEJA
9.1. Technika degustácie
9.1.1. Degustátori zdvihnú pohár, pričom ho držia zakrytý hodinovým sklíčkom a mierne naklonený, v tejto polohe ním začnú otáčať tak, aby sa čo najviac navlhčili vnútorné strany. Hneď po skončení tejto fázy odstránia hodinové sklíčko a ovoniavajú vzorku pomalými a hlbokými nádychmi, aby posúdili olej. Ovoniavanie by nemalo trvať dlhšie ako 30 sekúnd. Ak v danom čase nedospejú k rozhodnutiu, mali by si pred ďalším skúšaním urobiť malú prestávku.
Po vykonaní čuchového testu degustátori posúdia chuť (celkové retronazálne čuchové, chuťové a taktilné vnemy). S týmto cieľom si dajú malý dúšok oleja, približne 3 ml. Je veľmi dôležité, aby sa olej rozšíril po celej ústnej dutine, od prednej časti úst a jazyka pozdĺž strán do zadnej časti až po podnebie a do hrdla, keďže intenzita chuťových a taktilných vnemov je rozdielna v závislosti od miesta jazyka, podnebia a hrdla.
Treba zdôrazniť, že je veľmi dôležité, aby sa dostatočné množstvo oleja veľmi pomaly rozšírilo na zadnú časť jazyka smerom k podnebiu a hrdlu, pričom degustátor sa sústreďuje na poradie, v ktorom sa objaví horkosť a štipľavosť. V opačnom prípade sa môže v prípade niektorých olejov stať, že obidva tieto vnemy uniknú pozornosti, alebo že horkosť sa zastrie štipľavosťou.
Krátke, za sebou nasledujúce nádychy vháňajúce vzduch cez ústa nielen umožnia degustátorovi rozptýliť vzorku po celých ústach, ale aj vnímať prchavé aromatické uhľovodíky cez zadnú časť nosa vynúteným použitím tohto kanálu.
Poznámka: Ak degustátori nevnímajú ovocnosť vo vzorke a intenzita klasifikačného negatívneho atribútu je najviac 3,5, predseda poroty môže rozhodnúť, aby degustátori zopakovali analýzu vzorky pri teplote okolia (COI/T.20/Doc. č. 6/Rev. 1, september 2007, oddiel 3 – Všeobecné ustanovenia pre zriadenie testovacej miestnosti), pričom špecifikuje kontext a koncept teploty okolia. Keď vzorka dosiahne izbovú teplotu, degustátori by mali zopakovať hodnotenie s cieľom overiť výlučne, či je vnímaná ovocnosť. Ak tomu tak je, mali by označiť intenzitu na stupnici.
Malo by sa zohľadniť aj taktilné vnímanie štipľavosti. Na tento účel je vhodné olej prehltnúť.
9.1.2. Odporúča sa, aby sa pri organoleptickom hodnotení panenského olivového oleja posudzovali v každej skúške maximálne ŠTYRI VZORKY a aby sa uskutočnili maximálne tri skúšky za deň, a tak sa zabránilo kontrastnému účinku, ktorý by mohol vzniknúť, ak by sa vzorky ochutnávali bezprostredne jedna za druhou.
Keďže za sebou nasledujúce ochutnávanie vedie k únave alebo strate citlivosti spôsobeným predchádzajúcimi vzorkami, je potrebné vhodným spôsobom odstrániť z úst zvyšky olivového oleja z predchádzajúceho hodnotenia.
Odporúča sa použiť malý kúsok jablka, ktoré sa po požuvaní vypľuje do odpľúvadla. Následne sa ústa vypláchnu malým množstvom vody izbovej teploty. Medzi koncom jednej skúšky a začiatkom ďalšej má uplynúť najmenej 15 minút.
9.2. Použitie profilového hárku degustátormi
Profilový hárok, ktorý majú použiť degustátori, sa nachádza na obrázku 1 tejto prílohy.
Každý degustátor v porote ovonia a potom ochutná ( 5 ) posudzovaný olej. Degustátori následne uvedú na 10-cm stupnici v profilovom hárku intenzitu, s akou vnímajú každý z negatívnych a pozitívnych atribútov.
Ak degustátori vnímajú akékoľvek negatívne atribúty neuvedené v oddiele 4, zaznamenajú ich v položke „ostatné“, pričom použijú termín alebo termíny, ktoré atribúty najpresnejšie opisujú.
9.3. Použitie údajov predsedami poroty
Predseda poroty zozbiera profilové hárky vyplnené jednotlivými degustátormi a skontroluje intenzitu zaznamenanú pri jednotlivých atribútoch. Ak zistí akúkoľvek anomáliu, vyzve degustátora, aby svoj profilový hárok zrevidoval a v prípade potreby skúšku zopakoval.
Predseda poroty zadá údaje z hodnotenia získané od jednotlivých členov poroty do počítačového programu podobného tomu, ktorý stanovuje norma IOC/T.20/Doc. No 15 na účely štatistického výpočtu výsledkov analýzy na základe výpočtu ich mediánu. Pozri bod 9.4 a dodatok k tejto prílohe. Údaje týkajúce sa danej vzorky sa zaznamenajú na matici z deviatich stĺpcov, ktoré zodpovedajú deviatim organoleptickým vlastnostiam a z n riadkov, ktoré zodpovedajú n počtu členov poroty.
Ak aspoň 50 % členov poroty zaznamená vnímaný defektor do položky „ostatné“, predseda poroty vypočíta medián tohto defektoru a vykoná príslušnú klasifikáciu.
Hodnota robustného variačného koeficientu, ktorý vyjadruje klasifikáciu (defektor s najsilnejšou intenzitou a atribútom ovocnosti), musí byť nižšia alebo rovná 20 %.
V opačnom prípade musí predseda poroty hodnotenie konkrétnej vzorky zopakovať pri inej degustačnej skúške.
Ak k takejto situácii dochádza často, odporúča sa, aby predseda poroty poskytol degustátorom osobitnú dodatočnú odbornú prípravu (IOC/T.20/Doc. No 14, § 5) a posúdil výkonnosť degustátorov na základe indexu opakovateľnosti a indexu odchýlky (IOC/T.20/Doc. No 14, § 6).
9.4. Klasifikácia oleja
Olej sa klasifikuje do niektorej z ďalej uvedených kategórií podľa mediánu defektorov a mediánu atribútu ovocnosti. Medián defektorov sa vymedzuje ako medián defektoru vnímaného s najväčšou intenzitou. Medián defektorov a medián atribútu ovocnosti sa uvádzajú s presnosťou na jedno desatinné číslo.
Olej sa klasifikuje na základe porovnania hodnoty mediánu defektorov a mediánu atribútu ovocnosti s ďalej uvedenými referenčnými intervalmi. Pri stanovovaní hraníc týchto intervalov sa zohľadnila chybnosť metódy, a preto sa tieto hranice považujú za absolútne. Počítačové programy umožňujú vizuálne zobrazenie klasifikácie vo forme tabuľky alebo grafu.
a) Extra panenský olivový olej: medián defektorov je 0 a medián atribútu ovocnosti je vyšší ako 0,0;
b) Panenský olivový olej: medián defektorov je vyšší ako 0,0, ale nie vyšší ako 3,5 a medián atribútu ovocnosti je vyšší ako 0,0;
c) Lampantový olivový olej: medián defektoru je vyšší ako 3,5 alebo medián defektoru je nižší alebo rovný 3,5 a medián ovocnosti je rovný 0,0.
Poznámka 1: Ak je medián atribútu horkosti a/alebo atribútu štipľavosti vyšší ako 5,0, predseda poroty túto skutočnosť uvedie v protokole o skúške.
V prípade hodnotení určených na monitorovanie súladu sa musí vykonať jedna skúška. V prípade kontrolných hodnotení sa analýza musí opakovane vykonať v ďalšej degustácii. Výsledky opakovanej analýzy musia byť štatisticky homogénne. (Pozri bod 9.5.) Ak nie sú, analýza vzorky sa ešte raz zopakuje. Konečná hodnota mediánu klasifikačných atribútov sa vypočíta na základe priemeru oboch mediánov.
9.5. Kritériá prijateľnosti a zamietnutia opakovaných meraní
Normalizovaná chyba, vymedzená nižšie, sa použije na určenie toho, či oba výsledky opakovanej analýzy sú homogénne alebo štatisticky prijateľné:

Kde Me1 a Me2 sú mediány dvoch opakovaných meraní (prvá a druhá analýza) a U1 a U2 sú rozšírené neistoty merania získané pre obe hodnoty vypočítané podľa dodatku takto:
U
1 = c × s* and

Pre rozšírenú neistotu merania, c = 1,96; teda:
U1 = 0,0196 × CVr × Me1
kde CVr je robustný variačný koeficient.
Na to, aby bolo možné konštatovať, že tieto dve získané hodnoty nie sú štatisticky odlišné, En musí byť rovnaké alebo menšie ako 1,0.
Obrázok 1
PROFILOVÝ HÁROK PANENSKÉHO OLIVOVÉHO OLEJA
|
Intenzita vnímania defektorov |
||||
|
Stuchnutý/po kalových usadeninách |
|
|
||
|
Zatuchnutý/vlhký/zemitý |
|
|
||
|
Vínový/octový Kyslý/kyslastý |
|
|
||
|
Po omrznutých olivách (po vlhkom dreve) |
|
|
||
|
Zožltnutý |
|
|
||
|
Ďalšie negatívne atribúty: |
|
|
||
|
Deskriptor: |
Kovový □ Po sene □ Po larvách □ Drsný□ Slaný □ Prehriaty alebo pripálený □ Po rastlinných šťavách□ Esparto □ Po uhorkách □ Mastný□ |
|||
|
Intenzita vnímania pozitívnych atribútov |
||||
|
Ovocný |
|
|
||
|
|
Zelený□ |
Zrelý□ |
||
|
Horký |
|
|
||
|
Štipľavý |
|
|
||
|
|
|
|
||
|
Meno degustátora: |
|
Kód degustátora: |
||
|
Kód vzorky: |
Podpis: |
|||
|
Dátum: |
||||
|
Pripomienky: |
||||
Dodatok
Metóda výpočtu mediánu a intervaly spoľahlivosti
Medián
![]()
Medián je definovaný ako reálne číslo Xm, ktoré charakterizuje skutočnosť, že pravdepodobnosť (p) toho, že hodnoty rozdelenia (X) sú nižšie ako toto číslo (Xm), je nižšia alebo rovná 0,5, a zároveň pravdepodobnosť (p) toho, že hodnoty rozdelenia (X) sú nižšie alebo rovné Xm, je vyššia alebo rovná 0,5. Podľa praktickejšej definície je medián je 50. percentil rozdelenia čísel vo vzostupnom poradí. Jednoduchšie povedané, ide o prostrednú hodnotu usporiadaného súboru nepárnych čísel alebo priemer dvoch prostredných hodnôt usporiadaného súboru párnych čísel.
Robustná štandardná odchýlka
Aby sa zostavil spoľahlivý odhad variability okolo strednej hodnoty, je potrebné použiť robustnú štandardnú odchýlku podľa Stuarta a Kendalla (4). Vzorec udáva asymptotickú robustnú štandardnú odchýlku, t. j. robustný odhad variability posudzovaných údajov, kde N je počet pozorovaní a IQR je medzikvartilové rozpätie, ktoré zahŕňa presne 50 % prípadov daného rozdelenia pravdepodobnosti:

Pri výpočte medzikvartilového rozpätia sa vypočítava veľkosť rozdielu medzi 75. a 25. percentilom.
![]()
Percentil je hodnota Xpc, ktorú charakterizuje skutočnosť, že pravdepodobnosť (p) toho, že hodnoty rozdelenia sú nižšie ako Xpc, je nižšia ako konkrétna stotina alebo sa jej rovná, a zároveň pravdepodobnosť (p) toho, že hodnoty rozdelenia sú nižšie alebo rovné Xpc, je vyššia ako konkrétna stotina alebo sa jej rovná. Stotina udáva vybraný fraktil rozdelenia. V prípade mediánu sa rovná 50/100.
V praxi predstavuje percentil hodnotu rozdelenia, ktorá zodpovedá určitej ploche vyznačenej od krivky rozdelenia alebo hustoty. Napríklad 25. percentil predstavuje hodnotu rozdelenia zodpovedajúcu ploche, ktorá sa rovná 0,25 alebo 25/100.
Pri tejto metóde sa percentily vypočítavajú na základe reálnych hodnôt, ktoré sa vyskytujú v matici údajov (postup výpočtu percentilov).
Robustný variačný koeficient (%)
CVr% predstavuje čisté číslo, ktoré udáva percento variability analyzovaného súboru čísel. Z tohto dôvodu je veľmi užitočný na overenie spoľahlivosti členov poroty.

95 % intervaly spoľahlivosti mediánu
95% intervaly spoľahlivosti (hodnota chyby prvého druhu sa rovná 0,05 alebo 5 %) predstavujú interval, v ktorom by hodnota mediánu mohla kolísať za predpokladu, že by bolo možné skúšanie donekonečna opakovať. V praxi udáva interval variability skúšky za prijatých prevádzkových podmienok vychádzajúc z predpokladu, že skúšku je možné veľakrát opakovať. Rovnako ako v prípade CVr%, interval je užitočný pri hodnotení spoľahlivosti skúšky.

kde C = 1,96 pre 95 % interval spoľahlivosti.
Príklad výpočtového hárku je uvedený v prílohe I normy IOC/T 20/Doc. č. 15.
Odkazy
(1) Wilkinson, L. 1990. Systat: The system for statistics. Evanston, IL.SYSTAT Inc.
(2) Cicchitelli, G. 1984. Probabilità e Statistica. Maggioli Editore, Rimini.
(3) Massart, D.L.; Vandeginste, B.G.M.; Deming, Y.; Michotte, L. 1988. Chemometrics. A textbook. Elsevier. Amsterdam.
(4) Kendall, M.G.; Stuart, A. 1967. The advanced theory of statistics. Vol. 1. Hafner Publishing Co.
(5) McGill, R.; Tukey, J.W.; Larsen, W.A. 1978. Variation of Box Plots. The American Statistician, 32, (2), 12-16.
(6) IOC/T.28/Doc. č. 1 september 2007, Guidelines for the accreditation of sensory testing laboratories with particular reference to virgin olive oil according to standard ISO/IEC 17025:2005.
(7) IOC/T.20/Doc. č. 14.
(8) IOC/T.20/Doc. č. 15.
(9) ISO/IEC 1702505).
▼M20 —————
▼M19 —————
PRÍLOHA XV
1. OBSAH OLEJA V OLIVOVÝCH ZVYŠKOCH
1.1. Prístrojové vybavenie
— vhodný extrakčný prístroj vybavený s 200 až 250 ml bankou s guľatým dnom,
— elektricky vyhrievaný kúpeľ (napr. pieskový kúpeľ, vodný kúpeľ),
— analytické váhy,
— sušička nastaviteľná na maximálne 80 oC,
— elektricky vyhrievaná pec s termostatom nastaviteľným na 103 ± 2 oC a ktorú možno vyfukovať prúdom vzduchu alebo pracujúca za zníženého tlaku,
— mechanický mlyn, ľahko udržiavateľný, ktorý umožňuje pomletie olivových zvyškov bez zvýšenia ich teploty alebo nejakej znateľnej zmeny v obsahu vlhkosti, prchavej podstaty alebo látok extrahovateľných hexánom,
— extrakčná patróna a vata alebo filtračný papier, z ktorých už boli odstránené látky extrahovateľné hexánom,
— exsikátor,
— sito s okami s priemerom 1 mm,
— malé kúsky vysušenej pemzy.
1.2. Činidlo
Normálny technický hexán, po ktorom musí po kompletnom odparení zostať zvyšok menej ako 0,002 g na 100 ml.
2. POSTUP
2.1. Príprava testovanej vzorky
Ak je to nutné, použite mechanický mlyn, ktorý ste predtým dokonale vyčistili, na rozomletie laboratórnej vzorky, aby sa zmenšila veľkosť častíc tak, že úplne prejdú sitom.
Použite jednu dvadsatinu vzorky na úplné vyčistenie mlynu, odhoďte zomletý materiál, zvyšok vzorky zomeľte a zozbierajte, opatrne zmiešajte a analyzujte bez odkladu.
2.2. Veľkosť navážky
Hneď po skončení mletia odvážte asi 10 g vzorky s presnosťou na 0,01 g na vykonanie testu.
2.3. Príprava extrakčnej patróny
Naváženú vzorku vložte do patróny, zazátkujte vatou. Ak použijete filtračný papier, navážku vzorky zabaľte do neho.
2.4. Predsušenie
Ak sú olivové zvyšky veľmi vlhké (t. j. obsah vlhkosti a prchavých látok je viac ako 10 %), vykonajte predsušenie tak, že vložíte naplnenú patrónu (alebo filtračný papier) do pece zahriatej na potrebný čas pri teplote nie viac ako 80 oC, aby sa znížil obsah vlhkosti a prchavých látok na menej ako 10 %.
2.5. Príprava banky s okrúhlym dnom
Banku obsahujúcu jeden alebo dva kúsky pemzy odvážte s presnosťou na 1 mg, ktorú ste predtým vysušili v peci pri teplote 103 ± 2 oC a chladili v exsikátore aspoň jednu hodinu.
2.6. Prvotná extrakcia
Do extrakčného prístroja zasuňte extrakčnú patrónu (alebo filtračný papier) s naváženou vzorkou. Do banky vlejte požadované množstvo hexánu. Upevnite banku na extrakčný prístroj a celú ju ponorte do elektricky vyhrievaného kúpeľa. Nastavte vyhrievanie tak, aby nebol refluxný pomer viac ako tri kvapky za sekundu (mierny, nie búrlivý var). Po štyroch hodinách extrahovania nechajte vychladnúť. Vyberte patrónu z extrakčného prístroja a dajte ju do prúdu vzduchu, aby sa odstránila väčšina nasiaknutého rozpúšťadla.
2.7. Druhá extrakcia
Vyprázdnite obsah patróny do mikromlyna a zomeľte čo najjemnejšie. Vráťte zomletú zmes do patróny a umiestnite ju späť do extrakčného prístroja.
Pokračujte v extrakcii ďalšie dve hodiny, použite takú istú banku s guľatým dnom, obsahujúcu prvý extrakt.
Výsledný roztok v extrakčnej banke musí byť číry. Ak nie je, prefiltrujte ho cez filtračný papier a niekoľkokrát vymyte pôvodnú banku a filtračný papier hexánom. Zbierajte filtrát a rozpúšťadlo použité na oplachovanie v druhej banke s okrúhlym dnom, ktorú ste predtým vysušili a odvážili s presnosťou na 1 mg.
2.8. Odstránenie rozpúšťadla a váženie extraktu
Odstráňte väčšiu časť rozpúšťadla oddestilovaním v elektricky vyhrievanom kúpeli. Posledné stopy rozpúšťadla odstráňte zahrievaním banky v peci pri teplote 103 ± 2 oC po dobu 20 minút. Procesu eliminácie rozpúšťadla napomôžte buď privádzaním vzduchu, alebo lepšie inertného plynu, alebo použitím zníženého tlaku.
Nechajte banku chladiť v exsikátore aspoň jednu hodinu a potom ju odvážte s presnosťou na 1 mg.
Znova ju za rovnakých podmienok zahrejte na 10 minút, vychlaďte v exsikátore a prevážte.
Rozdiel medzi dvomi váženiami by nemal presiahnuť 10 mg. Ak je rozdiel vyšší, znova zahrejte po dobu 10 minút, potom ďalej ochladzujte a odvážte, kým nie je rozdiel 10 g alebo nižší.
Vykonajte paralelné stanovenia testovanej vzorky.
3. VYJADRENIE VÝSLEDKOV
3.1. Metóda výpočtu a vzorec:
a) získaný extrakt sa vyjadrí ako percento hmotnosti produktu a rovná sa:

|
kde: |
S je percento hmotnosti získaného extraktu z produktu, m0 je hmotnosť naváženej vzorky, v gramoch, m1 je hmotnosť extraktu po vysušení, v gramoch. |
Výsledkom bude aritmetický priemer dvoch paralelných stanovení za predpokladu, že sa splnili podmienky pre opakovateľnosť.
Výsledok udajte na jedno desatinné miesto.
b) Extrakt sa vyjadrí na základe sušiny použitím vzorca:

|
kde |
: |
S = je percento hmotnosti získaného extraktu z produktu (pozri a)) U = je obsah vlhkosti a prchavých látok |
3.2. Opakovateľnosť
Rozdiel medzi dvomi paralelnými stanoveniami vykonanými naraz alebo v rýchlom slede za sebou jedným pracovníkom nesmie prekročiť 0,2 g hexánového extraktu na 100 g vzorky.
Ak táto podmienka nie je splnená, zopakujte analýzu na dvoch ďalších navážkach vzorky. Ak aj v tomto prípade rozdiel presiahne 0,2 g, ako výsledok sa zoberie aritmetický priemer všetkých štyroch stanovení.
PRÍLOHA XVI
STANOVENIE JÓDOVÉHO ČÍSLA
1. OBLASŤ
Táto medzinárodná technická norma špecifikuje metódu stanovenia jódového čísla živočíšnych a rastlinných tukov a olejov, neskôr uvádzaných ako tuky.
2. DEFINÍCIA
Na účely tejto medzinárodnej technickej normy platia nasledujúce definície:
|
2.1. |
jódové číslo: hmotnosť jódu absorbovaná vzorkou za pracovných podmienok špecifikovaných v tejto medzinárodnej technickej norme. Jódové číslo sa vyjadruje v gramoch jódu na 100 g vzorky. |
3. PODSTATA
Rozpustite navážku vzorky v rozpúšťadle a pridajte činidlo podľa Wijsa. Po určitej dobe pridajte roztok jodidu draselného a vodu a uvoľnený jód stitrujte roztokom tiosíranu sodného.
4. ČINIDLÁ
Všetky činidlá by mali mať známu čistotu:
|
4.1. |
voda, spĺňajúca požiadavky normy ISO 3696, stupeň 3; |
|
4.2. |
jodid draselný, roztok 100 g/l, neobsahujúci jodičnany alebo voľný jód; |
|
4.3. |
škrob, roztok. Rozmiešajte 5 g rozpustného škrobu v 30 ml vody a pridajte túto zmes do 1 000 ml vriacej vody, povarte 3 minúty a nechajte vychladnúť; |
|
4.4. |
tiosíran sodný, štandardný odmerný roztok c(Na2S2O3.5H2O) = 0,1 mol/l, štandardizovaný (ofaktorovaný) nie skôr ako sedem dní pred použitím; |
|
4.5. |
rozpúšťadlo: pripravené zmiešaním rovnakých objemov cyklohexánu a kyseliny octovej; |
|
4.6. |
Wijsove činidlo: obsahujúce chlorid jodný v kyseline octovej. Je možné použiť aj obchodne dostupné Wijsove činidlo. |
5. PRÍSTROJOVÉ VYBAVENIE
Obvyklé laboratórne zariadenia, najmä tieto:
|
5.1. |
sklenené navažovacie lodičky, vhodné na navážku a vsunutie do banky (6.2); |
|
5.2. |
kužeľové banky, 500 ml objemu, vybavené zábrusnými zátkami a úplne suché. |
6. PRÍPRAVA TESTOVANEJ VZORKY
Zhomogenizovaná vzorka sa vysuší tiosíranom sodným a prefiltruje.
7. POSTUP
|
7.1 |
Veľkosť navážky Hmotnosť navážky vzorky sa mení podľa predpokladaného jódového čísla uvedeného v tabuľke 1.
Tabuľke 1
V sklenenej navažovacej lodičke (5.1) odvážte navážku vzorky s presnosťou na 0,1 mg. |
|
7.2. |
Stanovenie Navážku vložte do 500 ml banky (6.2). Pridajte 20 ml rozpúšťadla, aby sa rozpustil tuk. Pridajte presne 25 ml Wijsovho činidla, banku zazátkujte, premiešajte jej obsah a uložte ju do tmy. Nepipetujte Wijsovo činidlo ústami. Podobne pripravte slepý pokus s rozpúšťadlom a činidlom, ale vynechajte navážku vzorky. Banky obsahujúce vzorky s jódovým číslom pod 150 ponechajte v tme jednu hodinu a banky so vzorkami s jódovým číslom nad 150 a s polymerizovanými produktmi alebo produktmi oxidovanými do vysokého stupňa, ponechajte v tme dve hodiny. Po uplynutí tohto času do každej banky pridajte 20 ml roztoku jodidu draselného (4.2) a 150 ml vody (4.1). Titrujte štandardným odmerným roztokom tiosíranu sodného (4.4), až kým sa takmer úplne nestratí žlté zafarbenie spôsobené jódom. Poznámka: Potenciometrické stanovenie konca titrácie je prípustné. |
|
7.3. |
Počet stanovení Vykonajte dve stanovania na tej istej vzorke. |
8. VYJADRENIE VÝSLEDKOV
Jódové číslo je vyjadrené výrazom:
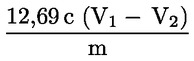
kde:
|
c |
= |
je číselná hodnota presnej koncentrácie v móloch na liter použitého štandardizovaného odmerného roztoku tiosíranu sodného (4.4) |
|
V1 |
= |
je číselná hodnota objemu štandardizovaného odmerného roztoku tiosíranu sodného (4.4), použitého na slepý pokus, v mililitroch |
|
V2 |
= |
je číselná hodnota objemu štandardizovaného odmerného roztoku tiosíranu sodného (4.4), použitého pri stanovení, v mililitroch |
|
m |
= |
je číselná hodnota hmotnosti navážky vzorky (7.1), v gramoch. |
Za výsledok sa bude považovať aritmetický priemer dvoch stanovení, za predpokladu, že opakovateľnosť (9.2) je uspokojivá.
PRÍLOHA XVII
METÓDY STANOVENIA STIGMASTADIÉNOV V RASTLINNÝCH OLEJOCH
1. ÚČEL
Stanovenie stigmastadiénov v rastlinných olejoch obsahujúcich nízke koncentrácie týchto uhľovodíkov, a to najmä v panenskom olivovom oleji a v surovom oleji z olivových zvyškov.
2. ROZSAH
Norma sa môže uplatňovať na všetky rastlinné oleje, hoci merania sú spoľahlivé iba v prípade, keď obsah týchto uhľovodíkov sa pohybuje v rozmedzí 0,01 až 4,0 mg/kg. Táto metóda je obzvlášť vhodná na zistenie prítomnosti rafinovaných rastlinných olejov (olivový olej, olej z olivových zvyšky, slnečnicový olej, palmový olej atď.) v panenskom olivovom oleji, nakoľko rafinované oleje obsahovali stigmastadiény a panenské oleje ich neobsahujú.
3. POSTUP
Izolácia nezmydelniteľnej látky. Oddelenie frakcie steroidných uhľovodíkov pomocou stĺpcovej chromatografie na silikagély a analyzovanie pomocou kapilárnej plynovej chromatografie.
4. APARATÚRA
|
4.1. |
250 ml banky vhodné na použitie so spätným chladičom. |
|
4.2. |
Oddeľovacie lieviky o objeme 500 ml. |
|
4.3. |
100 ml banky s guľatým dnom. |
|
4.4. |
Rotačná odparka. |
|
4.5. |
Sklená chromatografická kolóna (vnútorný priemer 1,5 až 2,0 cm, dĺžka 50 cm) s teflónovým kohútikom a so zátkou z vlákien sklenej vlny alebo s fritovým kotúčom na dne. S cieľom pripraviť silikagélovú kolónu nalejte do chromatografickej kolóny hexán do výšky približne 5 cm a potom ju naplňte suspenziou silikagélu v hexáne (15 g v 40 ml) pomocou dávok hexánu. Nechajte usadiť a usadzovanie ukončite aplikovaním slabých vibrácií. Pridajte bezvodý síran sodný do výšky približne 0,5 cm a nakoniec prebytočný hexán eluujte. |
|
4.6. |
Plynový chromatograf s plameňovým ionizačným detektorom, deleným alebo studeným injektorom na kolóne a pecou programovateľnou s presnosťou ± 1 °C. |
|
4.7. |
Kapilárna kolóna z taveného kremeňa určená na plynovú chromatografiu (vnútorný priemer 0,25 alebo 0,32 mm a dĺžka 25 m) potiahnutá fázou 5 % fenylmetylsilikónu, s hrúbkou filmu 0,25 mm. Poznámka 1: Môžu sa používať aj iné kolóny s podobnou alebo nižšou polaritou. |
|
4.8. |
Integrátor-zapisovač s možnosťou pracovať v integračnom režime údolie-údolie. |
|
4.9. |
5 až 10 ml mikrostriekačka na plynovú chromatografiu s cementovanou ihlou. |
|
4.10. |
Plášť s elektrickým ohrevom alebo zahrievacia platnička. |
5. ČINIDLÁ
Všetky činidlá musia mať analytickú kvalitu, pokiaľ nie je stanovené inak. Musí sa používať destilovaná voda alebo voda minimálne ekvivalentnej čistoty.
|
5.1. |
Hexán alebo zmes alkánov s rozmedzím bodov varu 65 až 70 °C, destilované v rektifikačnej kolóne. Hexán možno nahradiť izo-oktánom (2,2,4-trimetylpentánom na chromatografiu) pod podmienkou dosahovania porovnateľných hodnôt presnosti. Rezíduum po odparení 100 ml rozpúšťadla možno kontrolovať. Rozpúšťadlá s vyššou teplotou varu než n-hexán sa odparujú dlhšie. Uprednostňujú sa však z dôvodu toxicity hexánu. |
|
5.2. |
etanol 96 obj /obj. |
|
5.3. |
Bezvodý síran sodný. |
|
5.4. |
Alkoholický roztok hydroxidu draselného s koncentráciou 10 %. Pridajte 10 ml vody k 50 g hydroxidu draselného, zamiešajte a zmes potom rozpustite v etanole na celkový objem 500 ml. Poznámka 3: Alkoholický hydroxid draselný pri státí hnedne. Musí sa pripravovať čerstvý každý deň a uchovávať v tesne zazátkovaných fľašiach z tmavého skla. |
|
5.5. |
Silikagél 60 pre stĺpcovú chromatografiu, 70 až 230 meš (Merck, referencia 7734 alebo podobne). Poznámka 4: Silikagél sa môže obvykle použiť priamo z kontejnera bez akejkoľvek ďalšej úpravy. Niektoré šarže silikagélu môžu vykazovať nízku aktivitu, čo bude mať za následok zlé chromatografické oddelenie. Za takýchto okolností sa musí silikagél upraviť nasledujúcim spôsobom: Aktivujte silikagél zahrievaním po dobu najmenej štyroch hodín pri teplote 550 °C. Po zahriatí vložte silikagél do exsikátora, pričom sa silikagél ochladí a potom ho preneste do zazátkovanej fľaše. Pridajte 2 % vody a trepte dovtedy, až kým nebudú viditeľné žiadne hrudky a prášok bude voľne prúdiť. Ak niektoré šarže silikagélu budú mať za následok chromatogramy s interferujúcimi píkmi, tak silikagél sa musí upraviť spôsobom opísaným vyššie. Alternatívou môže byť použitie extra čistého silikagélu 60 (Merck, referencia 7754). |
|
5.6. |
Zásobný roztok (200 ppm) cholesta-3, 5-diénu (Sigma, čistota 99 %) v hexáne (10 mg v 50 ml). |
|
5.7. |
Štandardný roztok cholesta-3, 5-diénu v hexáne pri koncentrácii 20 ppm, získaný zriedením roztoku uvedeného vyššie. Poznámka 5: Roztoky 5.6 a 5.7 sú stále po dobu najmenej štyroch mesiacov, ak sa uchovávajú pri teplote nižšej ako 4 °C. |
|
5.8. |
Roztok n-nonakozánu v hexáne pri koncentrácii približne100 ppm. |
|
5.9. |
Nosný plyn pre chromatografiu: hélium alebo vodík o čistote 99,9990 %. |
|
5.10. |
Pomocné plyny pre plameňový ionizačný detektor: vodík o čistote 99,9990 % a čistený vzduch. |
6. POSTUP
6.1. Prípravanezmydelniteľnej látky
|
6.1.1. |
Navážte 20 ± 0, 1 g oleja do 250 ml banky (4.1), pridajte 1 ml štandardného roztoku cholesta-3, 5-diénu (20 mg) a 75 ml 10 % alkoholického hydroxidu draselného, namontujte spätný chladič a zahrievajte do mierneho varu po dobu 30 minút. Vyberte banku obsahujúcu vzorku zo zdroja tepla a nechajte roztok trochu ochladnúť (nenechajte roztok úplne vychladnúť, nakoľko vzorka by stuhla). Pridajte 100 ml vody a preneste roztok do oddeľovacieho lievika (4.2) pomocou 100 ml hexánu. Zmesou 30 sekúnd intenzívne trepte a potom ju nechajte oddeliť sa. Poznámka 6: Pokiaľ by sa vytvorila emulzia, ktorá by sa rýchle nerozplynula, tak pridajte etanol v malom množstve. |
|
6.1.2. |
Preneste spodnú vodnú fázu do druhého oddeľovacieho lievika a znovu extrahujte s použitím 100 ml hexánu. Spodnú fázu znovu vypustite a hexánové extrakty premyte (spojené v ďalšom oddeľovacom lieviku) trikrát, pričom zakaždým použijete 100 ml zmesi etanol-voda (1:1), až kým sa nedosiahne neutrálne pH. |
|
6.1.3. |
Nechajte prejsť roztok hexánu cez bezvodý síran sodný (50 g), premyte s 20 ml hexánu a nechajte odparovať v rotačnej odparke pri 30 °C pri zníženom tlaku až do vysušenia. |
6.2. Oddeleniefrakcie steroidných uhľovodíkov
|
6.2.1. |
Preneste zvyšok do frakcionačnej kolóny pomocou dvoch 1-ml dávok hexánu, nechajte vzorku pretekať kolónou a umožnite, aby hladina roztoku poklesla k hornej časti síranu sodného, a začnite chromatografické eluovanie pomocou hexánu s rýchlosťou prietoku približne 1 ml/min. Prvých 25 až 30 ml eluátu vyraďte a nasledujúcich 40 ml frakcie zachyťte. Po zachytení preneste túto frakciu do 100-ml banky s guľatým dnom. (4.3) Poznámka 7: Prvá frakcia obsahuje nasýtené uhľovodíky (obr. 1 a) a druhá frakcia steroidné uhľovodíky. Ďalšie eluovanie poskytne skvalen a príbuzné zlúčeniny. Ak sa má dosiahnuť dobré oddelenie nasýtených a steroidných uhľovodíkov, tak sa vyžaduje optimalizácie objemov frakcie. Za týmto účelom sa musí objem prvej frakcie upraviť tak, aby pri analýze druhej frakcie, píky predstavujúce nasýtené uhľovodíky boli nízke (pozri obr. 1 c); ak sa neobjavia, ale intenzita štandardného píku je pritom nízka, objem sa musí znížiť. V každom prípade, úplné oddelenie zložiek prvej a druhej frakcie nie je nevyhnutné, nakoľko počas GC (plynovo-chromatografickej) analýzy nedochádza k prekrývaniu píkov, pokiaľ sú podmienky plynovej chromatografie upravené tak, ako je uvedené v 6.3.1. Optimalizácia objemu druhej frakcie nie je spravidla potrebná, nakoľko dochádza k dobrému oddeleniu od ostatných komponentov. Tak či onak, prítomnosť veľkého píku pri približne o 1,5 minútu kratšom retenčnom čase v porovnaní so štandardom je zapríčinená skvalénom a svedčí o zlom oddelení. |
|
6.2.2. |
Odparujte druhú frakciu v rotačnej odparke pri 30 °C pri zníženom tlaku až do vysušenia a zvyšok ihneď rozpustite v 0,2 ml hexánu. Roztok uchovávajte až do analýzy v chladničke. Poznámka 8: Zvyšky 6.1.3 a 6.2.2 sa nesmú držať na suchom mieste a pri izbovej teplote. Ihneď po ich získaní sa musí pridať rozpúšťadlo a roztoky sa musia uchovávať v chladničke. |
6.3. Plynová chromatografia
|
6.3.1. |
Pracovné podmienky deleného vstrekovania — teplota injektora: 300 °C, — teplota detektora: 320 °C, — integrátor-zapisovač: parametre integrácie sa musia stanoviť tak, aby sa dosiahlo správne vyhodnotenie plôch. Odporúča sa integračný režim údolie-údolie. — citlivosť: približne 16 násobok minimálneho tlmenia, — množstvo vstreknutého roztoku: 1 ml, — teploty programovania pece: spočiatku 235 °C po dobu šesť minút a následné zvyšovanie rýchlosťou 2 °C/minúta na teplotu 285 °C, — injektor s deličom prietoku 1: 15, — nosný plyn: hélium alebo vodík pri tlaku približne 120 kPa. Tieto podmienky sa môžu upravovať v súlade s charakteristikami chromatografu a kolóny tak, aby sa získali chromatogramy spĺňajúce nasledujúce požiadavky: pík vnútorného štandardu do približne 5 minút času uvedeného v 6.3.2; pík vnútorného štandardu musí predstavovať aspoň 80 % rozsahu stupnice. Systém na plynovú chromatografiu sa musí skontrolovať vstreknutím zmesi zásobného roztoku cholestadiénu (5.6) a roztoku n- nonakozánu (5.8). Pík cholesta-3, 5-diénu sa musí objaviť pred píkom n-nonakozánu (obr. 1c); pokiaľ sa neobjaví, tak možno vykonať dva kroky: znížiť teplotu pece a/alebo menej polárnu kolónu. |
|
6.3.2. |
Identifikácia píku Pík vnútorného štandardu sa objaví približne za 19 minút a pík 3,5-stigmastadiénu s relatívnym retenčným časom približne 1,29 (pozri obr. 1b). 3,5-stigmastadién sa vyskytuje s malými množstvami izoméru a obvykle sa obidva eluujú spolu ako jeden chromatografický pík. Ak však bude kolóna príliš polárna alebo sa bude vyznačovať vysokou rozlišovacou schopnosťou, izomér sa môže objaviť ako malý pík skôr tesne pred píkom stigmasta-3, 5-diénu (obr. 2). S cieľom zabezpečiť, aby sa stigmastadiény eluovali ako jeden pík, je rozumné nahradiť kolónu kolónou, ktorá je buď menej polárna, alebo má väčší vnútorný priemer. Poznámka 9: Stigmastadiény určené ako referencia sa môžu získať analýzou rafinovaného rastlinného oleja s použitím menšieho množstva vzorky (1až 2 g). Stigmastadiény vytvárajú prominentný a ľahko identifikovateľný pík. |
|
6.3.3. |
Kvantitatívna analýza Obsah stigmastadiénov sa stanoví podľa vzorca: mg/kg stigmastadiénov =
Hranica postrehu: približne 0,01 mg/kg. Poznámka 10: Ak sa stigmastidiény objavia v koncentráciách vyšších než 4 mg/kg a vyžaduje sa kvantifikácia, musí sa uplatniť metóda Medzinárodnej rady pre olivy na určenie sterénov v rafinovaných olejoch. |


 Obrázok 1
Obrázok 1
Plynové chromatogramy získané zo vzoriek olivového oleja analyzovaných na kremennej kapilárnej kolóne (vnútorný priemer 0,25 mm a dĺžka 25 m), potiahnutej 5 % fenylmetylsilikónom s hrúbkou filmu 0,25 mm.
a) Prvá frakcia (30 ml) z panenského oleja, s prídavkom štandardu.
b) Druhá frakcia (40 ml) z olivového oleja obsahujúceho 0,10 mg/kg stigmastadiénov.
c) Druhá frakcia (40 ml) obsahujúca malý podiel prvej frakcie.
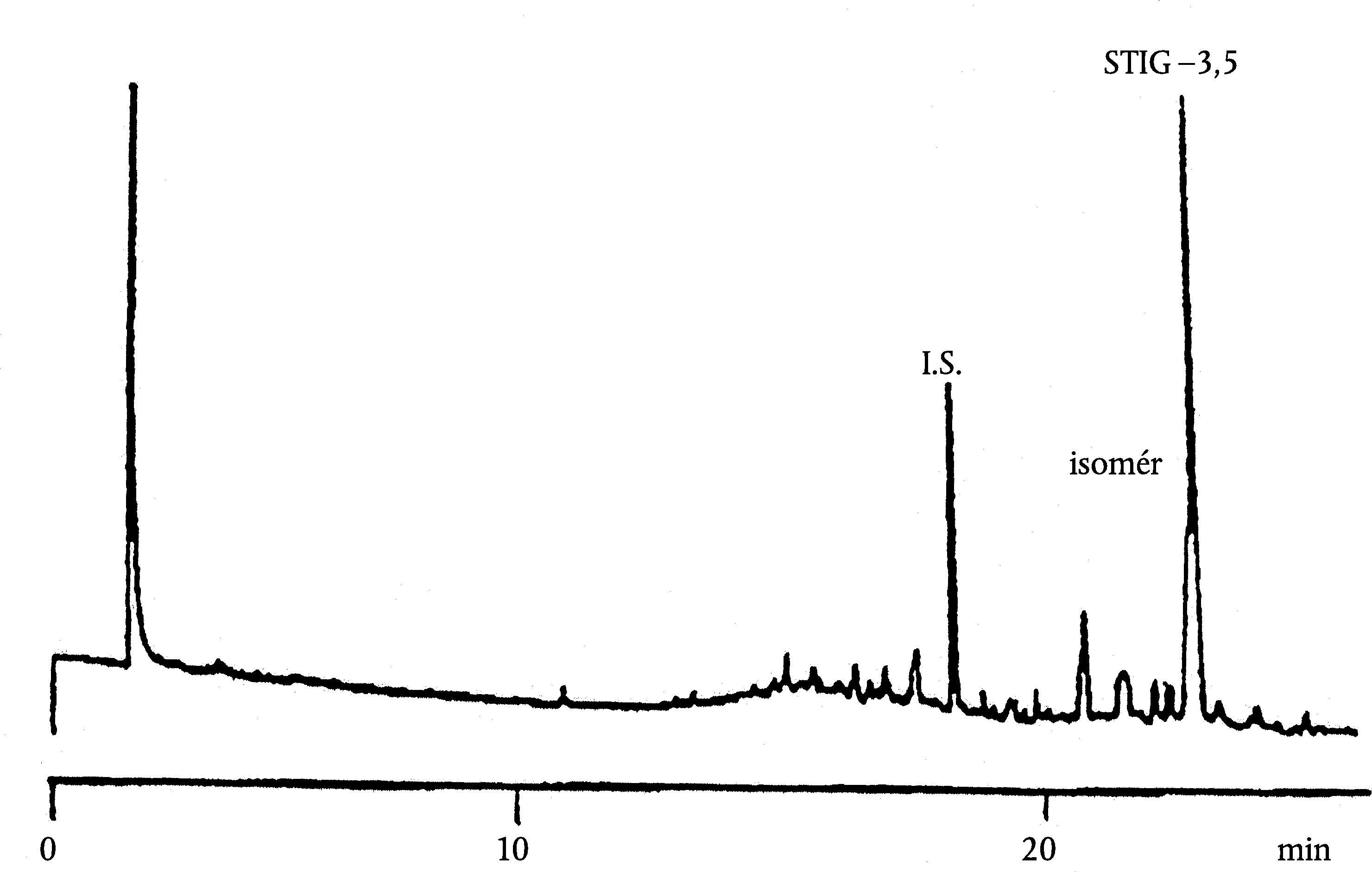 Obrázok 2
Obrázok 2
Plynový chromatogram získaný zo vzorky rafinovaného olivového oleja analyzovaného na DB-5-kolóne vykazujúci izomér 3,5-stigmastadiénu.
PRÍLOHA XVIII
STANOVENIE ROZDIELU MEDZI SKUTOČNÝM A TEORETICKÝM OBSAHOM TRIGLYCERIDOV S ECN 42
1. ROZSAH PÔSOBNOSTI
Stanovenie absolútneho rozdielu medzi experimentálnymi hodnotami triglyceridov (TAG) s ekvivalentným počtom uhlíkov 42 (ECN42HPLC) získanými vysokoúčinnou kvapalinovou chromatografiou v oleji a teoretickou hodnotou TAG s ekvivalentným počtom uhlíkov 42 (ECN 42teoretické) vypočítanou zo zloženia mastných kyselín.
2. OBLASŤ POUŽITIA
Táto technická norma sa vzťahuje na olivové oleje. Metóda sa uplatňuje pri zisťovaní prítomnosti malých množstiev olejov zo semien (bohatých na kyselinu linolovú) vo všetkých triedach olivového oleja.
3. ZÁSADA
Obsah triglyceridov s ECN 42 určený analýzou HPLC a teoretický obsah triglyceridov s ECN 42 (vypočítaný na základe určenia zloženia mastných kyselín pomocou chromatografie GLC) v určitom limite zodpovedá pravému olivovému oleju. Rozdiel väčší než schválené hodnoty pre jednotlivé typy poukazuje na to, že olej obsahuje oleje zo semien.
4. METÓDA
Metóda výpočtu teoretického obsahu triglyceridov s ECN 42 a rozdiel vzhľadom na údaje získané pri HPLC v podstate vychádza z koordinácie analytických údajov získaných prostredníctvom iných metód. Možno rozlišovať tri fázy: stanovenie zloženia mastných kyselín kapilárnou plynovou chromatografiou, výpočet teoretického zloženia triglyceridov s ECN 42, stanovenie triglyceridov s ECN 42 analýzou HPLC.
4.1. Vybavenie
|
4.1.1. |
Banky s guľatým dnom, 250 ml a 500 ml. |
|
4.1.2. |
Kadičky, 100 ml. |
|
4.1.3. |
Sklenená chromatografická kolóna s 21 mm vnútorným priemerom, 450 mm dlhá, navrchu s kohútikom a normalizovaným vnútorným (typu „female“) kónusom. |
|
4.1.4. |
250 ml oddeľovacie lieviky s normalizovaným vonkajším (typu „male“) kónusom na spodku, vhodné na pripojenie k vrchnej časti kolóny. |
|
4.1.5. |
600 mm sklenená tyčinka. |
|
4.1.6. |
Sklenený lievik priemeru 80 mm. |
|
4.1.7. |
Odmerné banky, 50 ml. |
|
4.1.8. |
Odmerné banky, 20 ml. |
|
4.1.9. |
Rotačná odparka. |
|
4.1.10. |
Vysokoúčinný kvapalinový chromatograf s možnosťou termostatickej kontroly teploty kolóny. |
|
4.1.11. |
Dávkovacie zariadenia s 10 μl dávkovaním. |
|
4.1.12. |
Detektor: diferenciálny refraktometer. Citlivosť na celý rozsah by mala byť aspoň 10–4 jednotiek indexu lomu. |
|
4.1.13. |
Kolóna: 250 mm dlhá rúrka z antikorovej ocele s vnútorným priemerom 4,5 mm, naplnená časticami oxidu kremičitého s priemerom 5 μm a s 22 – 23 % uhlíka vo forme oktadecysilánu. |
|
4.1.14. |
Softvér na spracovanie údajov. |
|
4.1.15. |
Ampulky s objemom približne 2 ml, teflónovými priehradkami a skrutkovacími uzávermi. |
4.2. Činidlá
Činidlá by mali byť čistoty p. a. (pre analýzu). Elučné rozpúšťadlá by mali byť odplynené a možno ich použiť niekoľkokrát bez vplyvu na separáciu.
|
4.2.1. |
Petroléter 40 – 60 °C chromatografickej kvality alebo hexán. Hexán možno nahradiť izo-oktánom (2,2,4-trimetylpentánom na chromatografiu) pod podmienkou dosahovania porovnateľných hodnôt presnosti. Rozpúšťadlá s vyššou teplotou varu než n-hexán sa odparujú dlhšie. Uprednostňujú sa však z dôvodu toxicity hexánu. |
|
4.2.2. |
Etyléter čerstvo destilovaný bez obsahu peroxidu. |
|
4.2.3. |
Elučné rozpúšťadlo na prečistenie oleja zmesou stĺpcovej chromatografie petroléter/etyléter v pomere objemov 87/13 (v/v). |
|
4.2.4. |
Silikagél 70 – 230 meš, Merck 7734 so štandardizovaným obsahom vody 5 % (hm). |
|
4.2.5. |
Sklená vata. |
|
4.2.6. |
Acetón na HPLC. |
|
4.2.7. |
Acetonitril alebo propionitril na HPLC. |
|
4.2.8. |
Elučné rozpúšťadlo HPLC: acetonitril + acetón (pomery sa prispôsobia vzhľadom na získanie požadovanej separácie; začnite so zmesou 50: 50) alebo propionitril. |
|
4.2.9. |
Solubilizačné rozpúšťadlo: acetón. |
|
4.2.10. |
Referenčné triglyceridy: je možné použiť obchodné triglyceridy (tripalmitín, trioleín atď.) a získané retenčné časy podľa ekvivalentného počtu uhlíkov alebo alternatívne porovnávacie chromatogramy získané zo sójového oleja, zmesi sójového a olivového oleja 30: 70 a čistého olivového oleja (pozri poznámky 1 a 2 a obrázky 1 – 4). |
|
4.2.11. |
Extrakčná kolóna s pevnou fázou so silikagélom 1 g, 6 ml. |
|
4.2.12. |
Heptán, chromatografickej kvality. Heptán možno nahradiť izo-oktánom (2,2,4-trimetylpentánom na chromatografiu). |
4.3. Príprava vzorky
Keďže množstvo interferujúcich látok môže spôsobiť nesprávne pozitívne výsledky, vzorku je nutné vždy prečistiť podľa metódy IUPAC č. 2.507, ktorá sa používa na stanovenie polárnych zlúčenín v tukoch na vyprážanie.
4.3.1. Príprava chromatografickej kolóny
Naplňte kolónu (4.1.3.) približne 30 ml elučného rozpúšťadla (4.2.3.), potom do kolóny vložte malé množstvo sklenej vaty (4.2.5.) a sklenenou tyčkou (4.1.5.) ju zatlačte na dno kolóny.
V 100 ml kadičke suspendujte 25 g silikagélu (4.2.4) v 80 ml elučnej zmesi (4.2.3) a pomocou skleneného lievika (4.1.6) ju vlejte do kolóny.
Na zabezpečenie úplného preliatia silikagélu do kolóny umyte kadičku elučnou zmesou a do kolóny vylejte aj dávky z umývania.
Otvorte kohútik a nechajte rozpúšťadlo eluovať, kým jeho hladina nedosahuje približne 1 cm nad silikagél.
4.3.2. Stĺpcová chromatografia
S presnosťou na 0,001 g navážte do banky s objemom 50 ml (4.1.7.) 2,5 ± 0,1 g prefiltrovaného, homogenizovaného a v prípade potreby odvodneného oleja.
Rozpustite ho približne v 20 ml elučného rozpúšťadla (4.2.3). Ak je to nutné, mierne ho zahrejte, aby sa uľahčilo rozpúšťanie. Ochlaďte na teplotu miestnosti a pomocou elučného rozpúšťadla upravte objem.
Odmernou pipetou vlejte do kolóny 20 ml roztoku pripraveného podľa bodu 4.3.1., otvorte kohútik a nechajte rozpúšťadlo eluovať na úroveň vrstvy silikagélu.
Potom eluujte so 150 ml elučného rozpúšťadla (4.2.3.) s upraveným prienikom rozpúšťadla približne 2 ml/min (prienik 150 ml cez kolónu bude trvať približne 60 – 70 minút).
Eluát odoberte do 250 ml banky s okrúhlym dnom (4.1.1.) vysušenej v sušičke a presne odváženej. Rozpúšťadlo eliminujte pri zníženom tlaku v rotačnej odparke (4.1.9) a odvážte zvyšok, ktorý sa použije na prípravu roztoku na analýzu HPLC a na prípravu metylesteru.
Vzorka získaná z kolóny musí v prípade kategórií extra panenského, panenského, bežného, rafinovaného a olivového oleja dosahovať minimálne 90 % a v prípade lampantového oleja a zvyškového oleja minimálne 80 %.
4.3.3. Prečistenie SPE
Silikagelová kolóna SPE sa aktivuje priechodom 6 ml hexánu (4.2.3) vo vákuu, aby sa zabránilo vysušeniu.
S presnosťou 0,001 g navážte do 2 ml ampulky (4.1.15) 0,12 g a rozpustite v 0,5 ml hexánu (4.2.3).
Vlejte roztok do kolóny SPE a eluujte vo vákuu s 10 ml hexán-dietyléteru (v objemovom pomere 87: 13) (4.2.3).
Zachytenú frakciu odparte dosucha v rotačnej odparke (4.1.9) pri zníženom tlaku pri izbovej teplote. Zvyšok rozpustite v 2 ml acetónu (4.2.6) na analýzu triglyceridov.
4.4. Analýza HPLC
4.4.1. Príprava vzoriek na chromatografickú analýzu
5 % roztok vzorky, ktorá sa bude analyzovať, pripravíte tak, že odvážite 0,5 ± 0,001 g vzorky do 10 ml odmernej banky a doplníte na objem 10 ml solubilizačným rozpúšťadlom (4.2.9.).
4.4.2. Postup
Nastavte podmienky na chromatografickom systéme. Pumpujte mobilnú fázu (elučné rozpúšťadlo) (4.2.8) v pomere 1,5 ml/min, aby sa prečistil celý systém. Počkajte, kým nedosiahnete stabilnú základnú líniu.
Nadávkujte 10 μl vzorky pripravenej tak, ako je opísane v bode 4.3.
4.4.3. Výpočet a vyjadrenie výsledkov
Použite metódu normalizácie plochy, t. j. predpokladá sa, že suma plôch píkov zodpovedajúcich triglyceridom s ECN 42 – ECN 52 sa rovná 100 %.
Vypočítajte pomerné percentá pre každý triglycerid pomocou tohto vzorca:
![]()
.
Výsledky sa uvádzajú najmenej na dve desatinné miesta.
Pozri poznámky č. 1 – 4.
4.5. Výpočet zloženia triglyceridov (% mol) z údajov o zložení mastných kyselín (% plochy)
4.5.1. Stanovenie zloženia mastných kyselín
Zloženie mastných kyselín sa stanovuje podľa normy ISO 5508 kapilárnou kolónou. Metylestery sa pripravujú podľa COI/T.20/č. dok. 24.
4.5.2. Vypočítavané mastné kyseliny
Glyceridy sa zoskupujú podľa svojho ekvivalentného počtu uhlíkov (ECN), pričom sa berie do úvahy nasledujúca ekvivalencia medzi ECN a mastnými kyselinami. Do úvahy sa berú len mastné kyseliny so 16 a 18 atómami uhlíka, keďže len tieto kyseliny sú významné pre olivový olej. Mastné kyseliny sa normalizujú na 100 %.
|
Mastná kyselina (FA) |
Skratka |
Molekulová hmotnosť (MW) |
ECN |
|
Kyselina palmitová |
P |
256,4 |
16 |
|
Kyselina palmitolejová |
Po |
254,4 |
14 |
|
Kyselina stearová |
S |
284,5 |
18 |
|
Kyselina olejová |
O |
282,5 |
16 |
|
Kyselina linolová |
L |
280,4 |
14 |
|
Kyselina linolénová |
Ln |
278,4 |
12 |
4.5.3. Prepočet % plochy na moly pre mastné kyseliny (1)
|
|
|
|
|
|
|
|
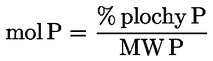





4.5.4. Normalizácia molov mastných kyselín na 100 % (2)





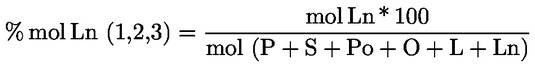
Výsledok vyjadruje percentuálny obsah mastných kyselín v molárnych percentách vo všetkých polohách (1, 2, 3) TAG.
Suma nasýtených mastných kyselín P a S (SFA) a nenasýtených mastných kyselín Po, O, L a Ln (UFA) sa potom vypočíta (3):
![]()
![]()
4.5.5. Výpočet zloženia mastných kyselín v polohách 2 a 1, 3 triglyceridov
Mastné kyseliny sa rozdeľujú do týchto troch skupín: jedna skupina v polohe 2 a dve rovnaké skupiny v polohách 1 a 3 s rozličnými koeficientmi nasýtených (P a S) a nenasýtených kyselín (Po, O, L a Ln).
4.5.5.1. Nasýtené mastné kyseliny v polohe 2 [P(2) a S(2)] (4):
![]()
![]()
4.5.5.2. Nenasýtené mastné kyseliny v polohe 2 [Po(2), O(2), L(2) a Ln(2)] (5):


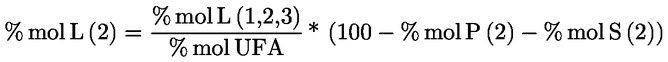

4.5.5.3. Mastné kyseliny v polohách 1,3 [P(1,3), S(1,3), Po(1,3), O(1,3), L(1,3) a Ln(1,3)] (6):



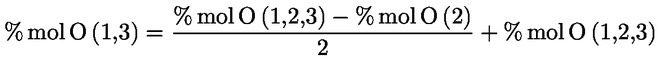


4.5.6. Výpočet triglyceridov
4.5.6.1. TAG s jednou mastnou kyselinou (AAA, tu LLL, PoPoPo) (7):
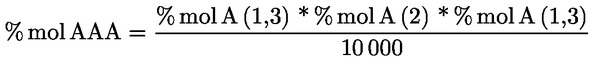
4.5.6.2. TAG s dvoma mastnými kyselinami (AAB, tu PoPoL, PoLL) (8):


4.5.6.3. TAG s troma rôznymi mastnými kyselinami (ABC, tu OLLn, PLLn, PoOLn, PPoLn) (9):



4.5.6.4. Triglyceridy s ECN42
Triglyceridy ECN42 sa vypočítajú podľa rovníc 7, 8 a 9 a sú dané poradím predpokladanej elúcie v HPLC (zvyčajne len tri píky).
LLL
PoLL a polohový izomér LPoL
OLLn a polohové izoméry OLnL a LnOL
PoPoL a polohový izomér PoLPo
PoOLn a polohové izoméry OPoLn a OLnPo
PLLn a polohové izoméry LLnP a LnPL
PoPoPo
SLnLn a polohový izomér LnSLn
PPoLn a polohové izoméry PLnPo a PoPLn
Triglyceridy s ECN42 sú dané sumou deviatich triglyceridov vrátane ich polohových izomérov. Výsledky sa uvádzajú najmenej na dve desatinné miesta.
5. VYHODNOTENIE VÝSLEDKOV
Porovnáva sa vypočítaný teoretický obsah a obsah stanovený analýzou HPLC. Ak je absolútna hodnota rozdielu údajov z analýzy HPLC po odpočítaní teoretických údajov vyššia ako medzné hodnoty pre príslušnú kategóriu oleja uvedené v norme, vzorka obsahuje olej zo semien.
Výsledky sa uvádzajú na dve desatinné miesta.
6. PRÍKLAD (ČÍSLA SA VZŤAHUJÚ NA ČASTI TEXTU OPISU METÓDY)
— 4.5.1. Výpočet molárnych percent mastných kyselín z údajov GLC (% normalizovanej plochy)
Metódou GLC sa získali tieto údaje o zložení mastných kyselín:
|
Mastná kyselina |
P |
S |
Po |
O |
L |
Ln |
|
MW |
256,4 |
284,5 |
254,4 |
282,5 |
280,4 |
278,4 |
|
% plochy |
10,0 |
3,0 |
1,0 |
75,0 |
10,0 |
1,0 |
— 4.5.3 Prepočet % plochy na moly pre mastné kyseliny [pozri vzorec (1)]
|
mol P |
= |
|

|
mol S |
= |
|

|
mol Po |
= |
|

|
mol O |
= |
|

|
mol L |
= |
|

|
mol Ln |
= |
|

|
Spolu |
= |
0,35821 mol TAG |
— 4.5.4 Normalizácia molov mastných kyselín na 100 % [pozri vzorec (2)]
|
% mol P(1,2,3) |
= |
|

|
% mol S(1,2,3) |
= |
|

|
% mol Po(1,2,3) |
= |
|

|
% mol O(1,2,3) |
= |
|

|
% mol L(1,2,3) |
= |
|

|
% mol Ln(1,2,3) |
= |
|

|
Spolu % mol |
= |
100 % |
Suma nasýtených a nenasýtených mastných kyselín v polohe 1,2,3 TAG [pozri vzorec (3)]:
![]()
![]()
— 4.5.5 Výpočet zloženia mastných kyselín v polohe 2 a polohe 1,3 TAG
— 4.5.5.1 Nasýtené mastné kyseliny v polohe 2 [P(2) a S(2)] [pozri vzorec (4)]
![]()
![]()
— 4.5.5.2 Nenasýtené mastné kyseliny v polohe 2 [Po(1,3), O(1,3), L(1,3) a Ln(1,3)] [pozri vzorec (5)]




— 4.5.5.3 Mastné kyseliny v polohách 1,3 [P(1,3), S(1,3), Po(1,3), O(1,3), L(1,3) a Ln(1,3)] [pozri vzorec (6)]
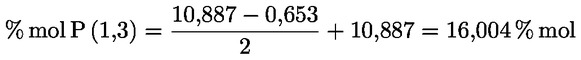





— 4.5.6. Výpočet triglyceridov
Výpočet zloženia mastných kyselín (FA) v polohách sn-2 a sn-1,3
|
FA v |
pol 1,3 |
pol 2 |
|
P |
16,004 % |
0,653 % |
|
S |
4,325 % |
0,177 % |
|
Po |
1,015 % |
1,262 % |
|
O |
68,526 % |
85,296 % |
|
L |
9,204 % |
11,457 % |
|
Ln |
0,927 % |
1,153 % |
|
Spolu |
100,0 % |
100,0 % |
vypočítavajú sa tieto triglyceridy:
LLL
PoPoPo
PoLL s 1 polohovým izomérom
SLnLn s 1 polohovým izomérom
PoPoL s 1 polohovým izomérom
PPoLn s 2 polohovými izomérmi
OLLn s 2 polohovými izomérmi
PLLn s 2 polohovými izomérmi
PoOLn s 2 polohovými izomérmi
— 4.5.6.1. TAG s jednou mastnou kyselinou (LLL, PoPoPo) [pozri vzorec (7)]
 , = 0,09706 mol LLL
, = 0,09706 mol LLL

— 4.5.6.2 TAG s dvoma mastnými kyselinami (PoLL, SLnLn, PoPoL) [pozri vzorec (8)]
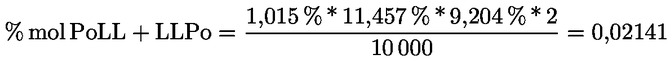

0,03210 mol PoLL


0,00094 mol SLnLn


0,00354 mol PoPoL
— 4.5.6.3 TAG s troma rôznymi mastnými kyselinami (PoPLn, OLLn, PLLn, PoOLn) pozri vzorec (9)



0,00761 mol PPoLn



0,43655 mol OLLn
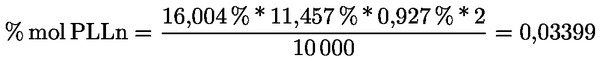

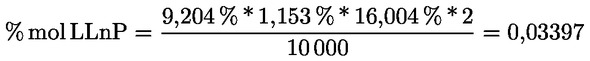
0,06907 mol PLLn
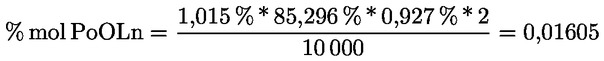

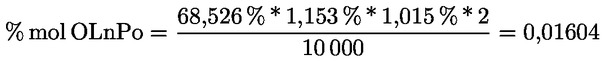
0,04812 mol PoOLn
ECN42 = 0,69512 mol TAG
Poznámka 1: elučné poradie sa môže stanoviť vypočítaním ekvivalentných uhlíkových čísel, často definovaných vzorcom
![]() , kde KN je počet uhlíkov a n je počet dvojitých väzieb; oveľa presnejšie sa poradie vypočíta, ak sa do výpočtu zahrnie počet pôvodných dvojitých väzieb. Ak no, nl a nln sú počty dvojitých väzieb vlastných kyselinám olejovej, linolovej, prípadne linolénovej, ekvivalentný počet uhlíkov je možné vypočítať pomocou vzorca:
, kde KN je počet uhlíkov a n je počet dvojitých väzieb; oveľa presnejšie sa poradie vypočíta, ak sa do výpočtu zahrnie počet pôvodných dvojitých väzieb. Ak no, nl a nln sú počty dvojitých väzieb vlastných kyselinám olejovej, linolovej, prípadne linolénovej, ekvivalentný počet uhlíkov je možné vypočítať pomocou vzorca:
![]()
kde koeficienty do, dl a dln sa môžu vypočítať pomocou referenčných triglyceridov. Podľa podmienok presne vymedzených v tejto metóde sa získaná väzba bude približovať k:
![]()
Poznámka 2: pri niekoľkých referenčných triglyceridoch je možné vypočítať rozlíšenie vzhľadom na trioleín:
![]()
trioleínpoužitím redukovaného retenčného času
![]()
Graf log a oproti f (počet dvojitých väzieb) umožňuje stanoviť retenčné hodnoty pre všetky triglyceridy mastných kyselín obsiahnuté v referenčných triglyceridoch – pozri obrázok 1.
Poznámka 3: účinnosť kolóny by mala umožniť jasnú separáciu píku trilinoleínu od píkov triglyceridov s blízkym retenčným časom RT. Elúcia sa vykonáva po pík ECN 52.
Poznámka 4: správnosť merania plôch všetkých píkov aktuálneho stanovenia je zabezpečená vtedy, ak druhý pík zodpovedajúci ECN 50 dosahuje 50 % celej stupnice zapisovača.
Obrázok 1
Graf log a voči f (počet dvojitých väzieb)

Počet dvojitých väzieb
La: kyselina laurová; My: kyselina myristová; P: kyselina palmitová; S: kyselina stearová; O: kyselina olejová; L: kyselina linolová; Ln: kyselina linolénová
Obrázok 2
Olivový olej s nízkym obsahom kyseliny linolovej
a

S rozpúšťadlom: acetón/acetonitril
PROFIL a: hlavné zložky chromatografických píkov: ECN42: (1) LLL + PoLL; (2) OLLn + PoOLn; (3) PLLn; ECN44: (4) OLL + PoOL; (5) OOLn + PLL; (6) POLn + PPoPo; (7) OOL + PoOO; ECN46: (8) OOL + LnPP; (9) PoOO; (10) SLL + PLO; (11) PoOP + SPoL + SOLn + SPoPo; (12) PLP; ECN48: (13) OOO + PoPP; (14 + 15) SOL + POO; (16) POP; ECN50: (17) SOO; (18) POS + SLS.
b
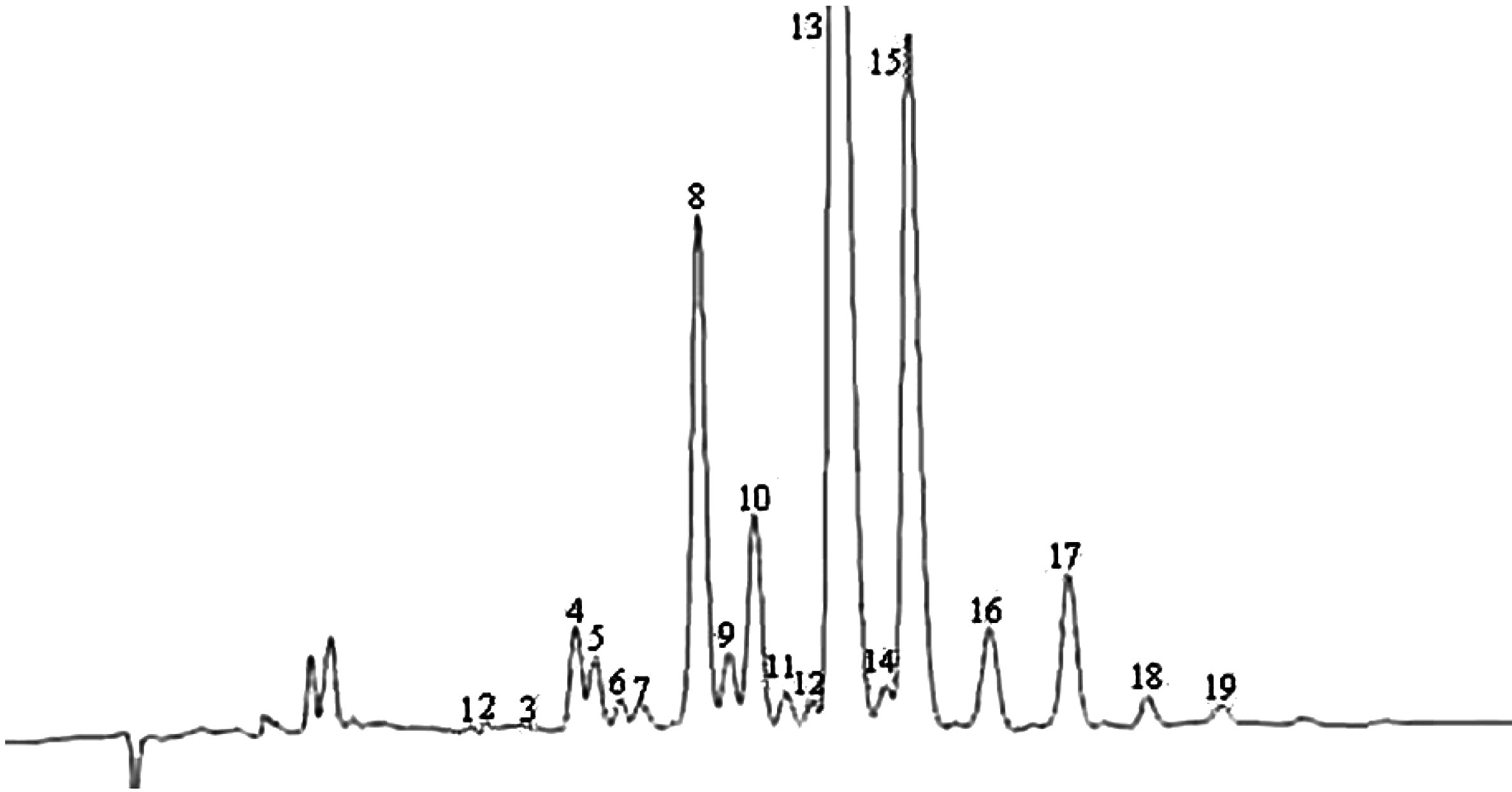
S rozpúšťadlom: propionitril
PROFIL b: hlavné zložky chromatografických píkov: ECN42: (1) LLL; (2) OLLn + PoLL; (3) PLLn; ECN44: (4) OLL; (5) OOLn + PoOL; (6) PLL + PoPoO; (7) POLn + PPoPo + PPoL; ECN46: (8) OOL + LnPP; (9) PoOO; (10) SLL + PLO; (11) PoOP + SPoL + SOLn + SPoPo; (12) PLP; ECN48: (13) OOO + PoPP; (14) SOL; (15) POO; (16) POP; ECN50: (17) SOO; (18) POS + SLS.
Obrázok 3
Olivový olej s vysokým obsahom kyseliny linolovej
a

S rozpúšťadlom: acetón/acetonitril (50: 50)
Profil a: hlavné zložky chromatografických píkov: ECN42: (1‘) LLL + PoLL; (2‘) OLLn + PoOLn; (3‘) PLLn; ECN44: (4‘) OLL + PoOL; (5‘) OOLn + PLL; (6‘) POLn + PPoPo; ECN46: (7‘) OOL + PoOO; (8‘) PLO + SLL + PoOP; (9‘) PLP + PoPP; ECN48: (10‘) OOO; (11‘) POO + SLL + PPoO; (12‘) POP + PLS; ECN50: (13‘) SOO; (14‘) POS + SLS.
b

S rozpúšťadlom: propionitril
Profil b: hlavné zložky chromatografických píkov: ECN42: (1) LLL; (2 + 2‘) OLLn + PoLL; (3) PLLn; ECN44: (4) OLL; (5) OOLn + PoOL; (6) PLL + PoPoO; (7) POLn + PPoPo + PPoL; ECN46: (8) OOL + LnPP; (9) PoOO; (10) SLL + PLO; (11) PoOP + SPoL + SOLn + SPoPo; ECN48: (12) PLP; (13) OOO + PoPP; (14) SOL; (15) POO; (16) POP; ECN50: (17) SOO; (18) POS + SLS; ECN52: (19) AOO.
PRÍLOHA XIX
STANOVENIE SKLADBY A OBSAHU STEROLOV A ALKOHOLOVÝCH ZLÚČENÍN KAPILÁRNOU PLYNOVOU CHROMATOGRAFIOU
1. PREDMET ÚPRAVY
Táto metóda opisuje postup stanovovania obsahu jednotlivých alkoholových zlúčenín a ich celkového obsahu v olivových olejoch a olejoch z olivových výliskov, ako aj v zmesiach týchto dvoch druhov olejov.
K alkoholovým zlúčeninám v olivových olejoch a olejoch z olivových výliskov patria alifatické alkoholy, steroly a triterpénové dioly.
2. PODSTATA METÓDY
Oleje s pridaným α-cholestanolom a 1-eikosanolom ako vnútorným štandardom sa zmydelní etanolovým roztokom hydroxidu draselného a nezmydelniteľná látka sa extrahuje etyléterom.
Rôzne frakcie alkoholových zlúčenín sa separujú od nezmydelniteľnej látky buď tenkovrstvovou chromatografiou na základnej silikagélovej platni (referenčná metóda) alebo HPLC so silikagélovou kolónou. Frakcie získané zo separácie silikagélom sa transformujú na trimetylsilylétery a následne sa analyzujú kapilárnou plynovou chromatografiou.
ČASŤ 1
PRÍPRAVA NEZMYDELNITEĽNEJ LÁTKY
1. PREDMET ÚPRAVY
V tejto časti sa opisuje príprava a extrakcia nezmydelniteľnej látky. Zahŕňa prípravu a extrakciu nezmydelniteľnej látky z olivových olejov a olejov z olivových výliskov.
2. PODSTATA METÓDY
Skúšobná dávka sa zmydelní varom pod spätným chladičom s etanolovým roztokom hydroxidu draselného. Nezmydelniteľná látka sa extrahuje dietyléterom.
3. LABORATÓRNA TECHNIKA
Bežné laboratórne vybavenie, a najmä:
3.1. 250 ml banka s guľatým dnom so spätným chladičom a zábrusovými sklenenými spojmi.
3.2. 500 ml oddeľovací lievik.
3.3. 250 ml banky.
3.4. Mikrostriekačky s objemom 100 μl a 500 μl.
3.5. Valcovitý filtračný lievik s fritou G3 (pórovitosť 15 – 40 μm) s priemerom približne 2 cm a hĺbkou 5 cm, vhodný na filtrovanie vo vákuu, s vonkajším zábrusom.
3.6. 50 ml kónická banka s vnútorným zábrusom, vhodná na použitie s uvedeným filtračným lievikom (bod 3.5).
3.7. 10 ml skúmavka kónickým dnom a so zabrúsenou sklenenou zátkou.
3.8. Exsikátor s chloridom vápenatým.
4. ČINIDLÁ
4.1. Hydroxid draselný s minimálnou koncentráciou stanovenou titrovaním 85 %.
4.2. Hydroxid draselný, etanolový roztok približne 2 M.
130 g hydroxidu draselného (bod 4.1) sa rozpustí za chladenia v 200 ml destilovanej vody a potom sa doplní etanolom na objem 1 litra (4.7). Roztok sa môže skladovať v dobre uzatvorenej fľaši z tmavého skla najviac dva dni.
4.3. Dietyléter, analytickej kvality.
4.4. Bezvodý síran sodný, analytickej kvality.
4.5. Acetón, chromatografickej kvality.
4.6. Dietyléter, chromatografickej kvality.
4.7. Etanol, analytickej kvality.
4.8. Etylacetát, analytickej kvality.
4.9. Vnútorný štandard, α-cholestanol, s čistotou viac ako 99 % (čistota sa musí skontrolovať prostredníctvom plynovochromatografickej analýzy).
4.10. α-cholestanol, vnútorný štandardný roztok, 0,2 % roztok (m/V) v etylacetáte (bod 4.8).
4.11. Fenolftaleínový roztok, 10 g/l v etanole (bod 4.7).
4.12. 0,1 % (m/V) roztok 1-eikosanolu v etylacetáte (vnútorný štandard).
5. POSTUP
Pomocou 500 μl mikrostriekačky (bod 3.4) sa do 250 ml banky (bod 3.1) zavedie taký objem vnútorného štandardného roztoku α-cholestanolu (bodu 4.10) a objem 1-eikosanolu (bod 4.12), ktorý obsahuje množstvo cholestanolu a eikosanolu zodpovedajúce približne 10 % obsahu sterolov a alkoholov vo vzorke. Napr. v prípade 5 g vzorky olivového oleja sa pridá 500 μl roztoku α-cholestanolu (bod 4.10) a 250 μL roztoku 1-eikosanolu (bod 4.12). V prípade olejov z olivových výliskov sa pridá 1500 μl roztoku α-cholestanolu (bod 4.10) a 1500 μl 1-eikosanolu (bod 4.12). V teplom vodnom kúpeli sa roztok pomocou jemného prúdu dusíka odparí dosucha. Po vychladení banky sa do tej istej banky naváži 5,00 ± 0,01 g suchej prefiltrovanej vzorky.
Poznámka 1: Živočíšne alebo rastlinné oleje a tuky s obsahom zistiteľných množstiev cholesterolu môžu vykazovať píky s retenčným časom identickým ako pri cholestanole. V takom prípade sa sterolová frakcia musí analyzovať raz s vnútorným štandardom a raz bez vnútorného štandardu.
Pridá sa 50 ml 2M etanolového roztoku hydroxidu draselného (bod 4.2) a trochu pemzy, nasadí sa spätný chladič a zariadenie sa zahrieva do mierneho varu, až kým neprebehne zmydelnenie (roztok sa vyčíri). Zahrieva sa ešte ďalších 20 minút a potom sa cez chladič prileje 50 ml destilovanej vody, chladič sa odpojí a banka sa ochladí na teplotu približne 30 °C.
Obsah banky sa pomocou niekoľkých dávok destilovanej vody (50 ml) kvantitatívne prenesie do 500 ml oddeľovacieho lievika (bod 3.2). Pridá sa približne 80 ml dietyléteru (bod 4.6) a obsah sa silno pretrepáva približne 60 sekúnd, pričom sa tlak pravidelne uvoľňuje prevracaním oddeľovacieho lievika a otváraním kohútika. Nechá sa stáť do úplného oddelenia oboch fáz (poznámka 2). Následne sa mydlový roztok čo najdokonalejšie oddelí do druhého oddeľovacieho lievika. Rovnakým spôsobom sa vykonajú dve ďalšie extrakcie z vodno-alkoholovej fázy, pričom sa použije 60 až 70 ml dietyléteru (bod 4.6).
Poznámka 2: Akákoľvek emulzia sa dá odstrániť pridaním malých množstiev etanolu (bod 4.7).
Získané tri éterové extrakty sa skombinujú v jednom oddeľovacom lieviku, ktorý obsahuje 50 ml vody. Extrakt sa opakovane premýva vodou (50 ml) dovtedy, kým sa premývacia voda po pridaní kvapky roztoku fenolftaleínu (bod 4.11) neprestane sfarbovať do ružova. Po odstránení premývacej vody sa zostávajúca vrstva prefiltruje cez bezvodý síran sodný (bod 4.4) do predtým odváženej 250 ml banky, pričom sa lievik a filter prepláchne malými množstvami dietyléteru (bod 4.6).
Rozpúšťadlo sa odparí destilovaním v rotačnej vákuovej odparke pri 30 °C. Pridá sa 5 ml acetónu (bod 4.5) a prchavé rozpúšťadlo sa úplne odstráni miernym prúdom dusíka. Zvyšok sa vysuší v peci pri 103 ± 2 °C počas 15 min. Nechá sa vychladiť v exsikátore a odváži sa s presnosťou na 0,1 mg.
ČASŤ 2
SEPARÁCIA FRAKCIÍ ALKOHOLOVÝCH ZLÚČENÍN
1. PREDMET ÚPRAVY
Nezmydelniteľná látka pripravená podľa postupu v časti 1 sa frakcionuje na rôzne alkoholové zlúčeniny, alifatické alkoholy, steroly a triterpénové dioly (erytrodiol + uvaol).
2. PODSTATA METÓDY
Nezmydelniteľnú látku možno frakcionovať pomocou tenkovrstvovej chromatografie na základnej platni (referenčná metóda), zviditeľniť a zodpovedajúce pásma možno zoškrabať a extrahovať. Alternatívnou metódou separácie je HPLC s použitím silikagélovej kolóny a detektora UV a zber jednotlivých frakcií. Alifatické a triterpénové alkoholy, ako aj steroly a triterpénové dioly sa izolujú spoločne.
3. LABORATÓRNA TECHNIKA
Bežné laboratórne vybavenie, a najmä:
3.1. Kompletná zostava na analýzu tenkovrstvovou chromatografiou so sklenenými platňami s rozmermi 20 × 20 cm.
3.2. UV lampa s vlnovou dĺžkou 366 alebo 254 nm.
3.3. Mikrostriekačky s objemom 100 μl a 500 μl.
3.4. Valcovitý filtračný lievik s fritou G3 (pórovitosť 15 – 40 μm) s priemerom približne 2 cm a hĺbkou 5 cm, vhodný na filtrovanie vo vákuu, s vonkajším zábrusom.
3.5. 50 ml kónická banka s vnútorným zábrusom, vhodná na použitie s uvedeným filtračným lievikom (bod 3.4).
3.6. 10 ml skúmavka kónickým dnom a so zabrúsenou sklenenou zátkou.
3.7. Exsikátor s chloridom vápenatým.
3.8. Systém HPLC, ktorý tvorí:
3.8.1. Binárne čerpadlo.
3.8.2. Manuálny alebo automatický injektor vybavený 200 μl dávkovacou slučkou.
3.8.3. Integrovaný odplyňovač.
3.8.4. Detektor UV-VIS alebo IR.
3.9. Kolóna HPLC (25 cm × 4 mm vnútorný priemer) so silikagélom 60 (veľkosť častíc 5 μm).
3.10. Injekčný filter, 0,45 μm.
3.11. 25 ml kónická banka.
4. ČINIDLÁ
4.1. Hydroxid draselný s minimálnou koncentráciou stanovenou titrovaním 85 %.
4.2. Hydroxid draselný, etanolový roztok približne 2 M.
130 g hydroxidu draselného (bod 4.1) sa rozpustí za chladenia v 200 ml destilovanej vody a potom sa doplní etanolom na objem 1 litra (bod 4.9). Roztok sa môže skladovať v dobre uzatvorenej fľaši z tmavého skla najviac dva dni.
4.3. Dietyléter, analytickej kvality.
4.4. Hydroxid draselný, etanolový roztok približne 0,2 M.
13 g hydroxidu draselného (bod 4.1) sa rozpustí v 20 ml destilovanej vody a potom sa doplní etanolom na objem 1 litra (bod 4.9).
4.5. Silikagélové tenkovrstvé sklenené platne (20 × 20 cm), bez fluorescenčného detektora, s hrúbkou 0,25 mm (komerčne dostupné a vhodné na okamžité použitie).
4.6. Acetón, chromatografickej kvality.
4.7. n-hexán, chromatografickej kvality.
4.8. Dietyléter, chromatografickej kvality.
4.9. Etanol, analytickej kvality.
4.10. Etylacetát, analytickej kvality.
4.11. Referenčný roztok na tenkovrstvovú chromatografiu: cholesterol, fytosteroly, alkoholy a 5 % roztok erytrodiolu v etylacetáte (bod 4.10).
4.12. 0,2 % roztok 2,7-dichlórfluóresceínu v etanolovom roztoku. Slabo alkalický roztok získame pridaním niekoľkých kvapiek 2 M alkoholového roztoku hydroxidu draselného (bod 4.2).
4.13. n-hexán (bod 4.7)/dietyléter (bod 4.8) – zmes v pomere 65:35 (V/V).
4.14. mobilná fáza HPLC, n-hexán (bod 4.7)/dietyléter (bod 4.8) (pomer 1:1) (V/V).
5. REFERENČNÁ METÓDA: SEPARÁCIA ALKOHOLOVÝCH ZLÚČENÍN POMOCOU TENKOVRSTVOVEJ CHROMATOGRAFIE NA ZÁKLADNEJ PLATNI
Príprava základných platní pre tenkovrstvovú chromatografiu. Silikagélové platne (bod 4.5) sa ponoria asi 4 cm na 10 sekúnd do 0,2 M etanolového roztoku hydroxidu draselného (bod 4.4), potom sa nechajú dve hodiny sušiť v digestore a nakoniec sa umiestnia na jednu hodinu do pece vyhriatej na 100 °C.
Platne sa po vybratí z pece uložia až do ďalšieho použitia do eksikátora s chloridom vápenatým (bod 3.7) (takto ošetrené platne sa musia použiť do 15 dní).
Do vyvíjacej komory sa naleje zmes hexánu/dietyléteru (bod 4.13) (Poznámka 3) do výšky asi 1 cm. Komora sa uzatvorí vhodným krytom a nechá sa aspoň pol hodinu na chladnom mieste tak, aby sa dosiahla rovnováha nasýtených pár nad hladinou. Na vnútorné povrchy komory možno umiestniť prúžky filtračného papiera siahajúceho do eluentu (vyvíjacej zmesi). Tým sa zníži elučný čas približne o tretinu a elúcia zložiek bude jednotnejšia a pravidelnejšia.
Poznámka 3: Elučná zmes by sa mala vymeniť pri každej skúške, aby sa dosiahli dokonale reprodukovateľné elučné podmienky. Možno pri tom použiť aj alternatívne rozpúšťadlo, a to zmes n-hexánu/dietyléteru v pomere 50:50 (V/V).
Pripraví sa približne 5 % roztok nezmydelniteľnej látky pripravenej podľa postupu v časti 1 v etylacetáte (bod 4.10) a pomocou 100 μl mikrostriekačky (bod 3.3) sa na spodný okraj (2 cm) chromatografickej platne (bod 4.5) nanesie 0,3 ml roztoku v úzkom a rovnomernom prúžku. V línii s týmto prúžkom sa nanesú 2 až 3 μl referenčného roztoku látky (bod 4.11) tak, aby sa pásmo sterolov, triterpénových diolov a alkoholov dalo po vyvolaní identifikovať.
Platňa sa umiestni do vyvíjacej komory (bod 3.1). Teplota okolia by sa mala udržiavať v rozmedzí 15 až 20 °C (poznámka 4). Komora sa ihneď uzatvorí krytom a vzorka sa nechá eluovať, kým čelo rozpúšťadla nedosiahne vzdialenosť približne 1 cm od horného okraja platne. Potom sa platňa vyberie z vyvíjacej komory a rozpúšťadlo sa nechá odpariť prúdom horúceho vzduchu alebo krátkym ponechaním platne v digestore.
Poznámka 4: Pri vyššej teplote sa separácia môže zhoršovať.
Platňa sa zľahka rovnomerne postrieka roztokom 2,7-dichlórfluóresceínu (bod 4.12) a nechá sa vyschnúť. Keď sa platňa sleduje pod ultrafialovou lampou (bod 3.2), pásma sterolov, triterpénových diolov a alkoholov sa dajú identifikovať podľa škvŕn získaných z referenčného roztoku, s ktorými sú zarovno (bod 4.11). Hranice pásiem sa označia čiernou ceruzkou pozdĺž okrajov fluorescencie (pozri TLC platňu na obrázku 1).
Pomocou kovovej škrabky (kopistky) sa silikagél zoškriabe z označenej plochy. Následne získaný jemne rozotretý materiál sa umiestni do filtračného lievika (3.4). Pridá sa 10 ml horúceho etylacetátu (bod 4.10), opatrne sa premieša kovovou kopistkou a prefiltruje sa (v prípade potreby vo vákuu), pričom sa filtrát zbiera do kónickej banky (bod 3.5) pripojenej k filtračnému lieviku.
Zvyšok v banke sa trikrát premyje dietyléterom (bod 4.3) (použije sa približne 10 ml pri každom oplachovaní), pričom sa filtrát zbiera do tej istej banky pripojenej k lieviku, filtrát sa odparí na objem 4 až 5 ml, zvyšný roztok sa prenesie do predtým odváženej 10 ml skúmavky (bod 3.6), odparí sa dosucha miernym teplom pomocou jemného prúdu dusíka, následne sa opäť skvapalní pridaním niekoľkých kvapiek acetónu (bod 4.6) a opäť sa nechá dosucha odpariť. Zvyšok vo vnútri skúmavky pozostáva z frakcie sterolov a triterpénových diolov alebo frakcie alkoholov a triterpénových alkoholov.
6. SEPARÁCIA FRAKCIE ALKOHOLOV POMOCOU HPLC
Nezmydelniteľná látka získaná podľa postupu v časti 1 sa rozpustí v 3 ml mobilnej fázy (bod 4.14), roztok sa filtruje pomocou injekčného filtra (bod 3.10) a zachová.
Do HPLC (bod 3.8) sa vstrekne 200 μl prefiltrovaného nezmydelniteľného roztoku.
Nechá sa prebehnúť separácia v HPLC pri toku 0,8 ml/min, do úvahy sa neberie prvých päť minúť a do 25 ml kónických baniek (bod 3.11) sa zachytávajú alifatické a triterpénové alkoholy odseparované medzi 5. a 10. minútou a steroly a erytrodiol a uvaol odseparované medzi 11. a 25. minútou (poznámka 5).
Separáciu možno monitorovať pomocou detektoru UV pri vlnovej dĺžke 210 mm alebo detektoru indexu lomu (pozri obrázok 6).
Frakcie sa odparia do sucha a pripravia na chromatografickú analýzu.
Poznámka 5: Je potrebné starostlivo kontrolovať tlak čerpadla HPLC, pretože dietyléter môže spôsobiť zvýšenie tlaku, tok sa musí prispôsobiť, aby sa tlak udržal pod kontrolou.
ČASŤ 3
PLYNOVOCHROMATOGRAFICKÁ ANALÝZA FRAKCIÍ ALKOHOLOVÝCH ZLÚČENÍN
1. PREDMET ÚPRAVY
Táto časť poskytuje všeobecné usmernenia týkajúce sa používania kapilárnej plynovej chromatografie na určenie kvalitatívneho a kvantitatívneho zloženia alkoholových zlúčenín izolovaných metódou uvedenou v časti 2.
2. PODSTATA METÓDY
Frakcie získané z nezmydelniteľnej látky pomocou TLC alebo HPLC sa derivatizujú na trimetylsilylétery a analyzujú pomocou kapilárnej plynovej chromatografie s injekčným systémom s deličom a plameňovým ionizačným detektorom.
3. LABORATÓRNA TECHNIKA
Bežné laboratórne vybavenie, a najmä:
3.1. 10 ml skúmavka kónickým dnom a so zabrúsenou sklenenou zátkou.
3.2. Plynový chromatograf vhodný na použitie s kapilárnou kolónou vybavený injekčným systémom s deličom a pozostávajúci z/zo:
3.2.1. termostatickej komory pre kolóny schopnej udržiavať požadovanú teplotu s presnosťou ± 1 °C,
3.2.2. dávkovacej jednotky s nastaviteľnou teplotou so silylovaným skleneným odparovacím prvkom a deličom,
3.2.3. plameňovoionizačného detektora (FID),
3.2.4. systému získavania údajov vhodného na použitie s detektorom FID (bod 3.10.3) s možnosťou manuálnej integrácie.
3.3. Kapilárna kolóna z taveného kremeňa s dĺžkou 20 až 30 m, s vnútorným priemerom 0,25 až 0,32 mm, potiahnutá vrstvou skladajúcou sa z 5 % difenylu a 95 % dimetylpolysiloxánu (SE-52 alebo SE-54 stacionárnou fázou alebo ekvivalentnou fázou) s jednotnou hrúbkou filmu od 0,10 do 0,30 μm.
3.4. Mikrostriekačka s kapacitou 10 μl na plynovú chromatografiu s cementovanou ihlou vhodnou pre injekčný systém s deličom.
4. ČINIDLÁ
4.1. Bezvodný pyridín, chromatografickej kvality.
4.2. Hexametyldisilazán, analytickej kvality.
4.3. Trimetylchlórsilán, analytickej kvality.
4.4. Skúšobné roztoky sterol trimetylsilyléterov. Pripraví sa priamo pred použitím zo sterolov a erytrodiolu získaných z olejov, ktoré ich obsahujú.
4.5. Štandardné roztoky trimetylsilyléterov a alifatických alkoholov od C20 po C28. Môžu sa pripraviť zo zmesí čistých alkoholov priamo pred použitím.
4.6. Nosný plyn: vodík alebo hélium, čistoty vhodnej na plynovú chromatografiu.
4.7. Pomocné plyny: vodík, hélium, dusík a vzduch, čistoty vhodnej na plynovú chromatografiu.
4.8. Silylačné činidlo, pozostávajúce zo zmesi pyridínu/hexametyldisilazánu/trimetylchlórsilánu v pomere 9:3:1 (V/V/V).
4.9. n-hexán, chromatografickej kvality.
5. PRÍPRAVA TRIMETYLSILYLÉTEROV
Do skúmavky (bod 3.1), v ktorej sa nachádza frakcia alkoholovej zlúčeniny, sa pridá silylačné činidlo (bod 4.8) (poznámka 6) v pomere 50 μl na každý miligram alkoholovej zlúčeniny, pričom sa bráni navlhnutiu (poznámka 7).
Poznámka 6: Hotové roztoky sú komerčne dostupné. Dostupné sú takisto iné silylačné činidlá, ako napr. bis-trimetylsilyl trifluóracetamid + 1 % trimetylchlórsilán, ktorý sa musí riediť rovnakým objemom bezvodého pyridínu. Pyridín možno nahradiť rovnakým množstvom acetonitrilu.
Poznámka 7: Slabá opalescencia, ktorá sa prípadne vytvorí, je bežný jav a nespôsobuje žiadnu anomáliu. Vznik bielych vločiek alebo ružového zafarbenia sú znaky prítomnosti vlhkosti alebo znehodnotenia činidla. Ak sa vyskytnú, skúška sa musí zopakovať (jedine, ak sa použil hexametyldisilazán/trimetylchlórsilán).
Skúmavka (bod 3.1) sa zazátkujte a opatrne pretrepáva (bez obracania), až kým sa zlúčeniny úplne nerozpustia. Potom sa skúmavka nechá aspoň 15 minút stáť pri izbovej teplote a následne sa niekoľko minút odstreďuje. Číry roztok je pripravený na plynovochromatografickú analýzu.
6. PLYNOVOCHROMATOGRAFICKÁ ANALÝZA
6.1. Prípravné práce, kondiciovanie kapilárnej kolóny.
Kolóna (bod 3.3) sa upevní do plynového chromatografu tak, že vstupný koniec kolóny sa pripevní k injektoru s deličom a výstupný koniec k detektoru.
Vykoná sa celková kontrola jednotky plynového chromatografu (netesnosti v plynových okruhoch, výkonnosť detektora, výkonnosť deliaceho systému a zapisovača atď.).
Ak sa kolóna používa prvýkrát, odporúča sa jej kondiciovanie: cez samotnú kolónu sa púšťa jemný prúd plynu a následne sa prepne na plynovochromatografickú jednotku a postupne sa zahrieva na teplotu aspoň o 20 °C vyššiu, ako je prevádzková teplota (poznámka 8). Táto teplota sa udržiava aspoň dve hodiny, potom sa jednotka nastaví na prevádzkový režim (upravia sa prietoky plynov a delič, zapáli sa plamienok, pripojí sa počítačový systém, upraví sa teplota kolóny, detektora a injektora atď.) a nakoniec sa zaznamená signál s citlivosťou, ktorá je aspoň dvakrát vyššia ako citlivosť určená pre analýzu. Základná línia musí byť lineárna, bez akýchkoľvek píkov, a nesmie vykazovať žiadne výkyvy (drifty). Negatívny priamočiary drift základnej línie znamená nedokonalú tesnosť kolónových spojov, kým pozitívny drift znamená nedostatočné kondiciovanie kolóny.
Poznámka 8: Teplota kondiciovania musí byť vždy aspoň o 20 °C nižšia, ako je maximálna teplota špecifikovaná pre používanú stacionárnu fázu.
6.2. Prevádzkové podmienky
Optimalizovať teplotný program a tok nosného plynu tak, aby sa získavali chromatogramy podobné zobrazeniam na obrázkoch 3 až 6.
Preskúmali sa a ako užitočné sa zistili tieto parametre:
6.2.1. Alifatické alkoholy
|
Program pece |
180 °C (8 min.) → 260 °C (pri 5 °C/min) → 260 °C (15 min.) |
|
Teplota injektora |
280 °C |
|
Teplota detektora |
290 °C |
|
Lineárna rýchlosť nosného plynu |
hélium (20 až 30 cm/s), vodík (30 až 50 cm/s) |
|
Deliaci pomer |
1:50 až 1:100 |
|
Objem nástreku |
0,5 až 1 μl TMSE roztoku |
6.2.2. Sterol a triterpénové dioly
|
Program pece |
260 ± 5 °C izotermický |
|
Teplota injektora |
280 – 300 °C |
|
Teplota detektora |
280 – 300 °C |
|
Lineárna rýchlosť nosného plynu |
hélium (20 až 30 cm/s), vodík (30 až 50 cm/s) |
|
Deliaci pomer |
1:50 až 1:100 |
|
Objem nástreku |
0,5 až 1 μl TMSE roztoku |
Tieto podmienky možno upraviť podľa vlastností kolóny a plynového chromatografu tak, aby sa získali chromatogramy spĺňajúce tieto požiadavky:
— retenčný čas alkoholu C26 má byť 18 ± 5 minút.
— pík alkoholu C22 má predstavovať 80 ± 20 % hodnoty celého rozsahu v prípade olivového oleja a 40 ± 20 % hodnoty celého rozsahu v prípade oleja z olivových výliskov.
— retenčný čas píku ß-sitosterolu by mal byť pri 20 ± 5 min.
— pík kampesterolu by mal byť: v prípade olivového oleja (priemerný obsah 3 %) 20 ± 5 % celého rozsahu.
— všetky prítomné steroly sa musia separovať. Okrem toho, že píky musia byť oddelené, musia byť aj úplne rozlíšené, t. j. záznam píku by sa mal vrátiť na základnú líniu pred vykreslením ďalšieho píku. Neúplné rozlíšenie sa však toleruje, pod podmienkou, že pík možno pri relatívnom retenčnom čase 1,02 (sitostanol) kvantifikovať použitím kolmice.
6.3. Postup analýzy
Do 10 μl mikrostriekačky (bod 3.4) sa natiahne 1 μl hexánu, potom sa natiahne 0,5 μl vzduchu a následne 0,5 až 1 μl roztoku vzorky. Piest mikrostriekačky sa vytiahne natoľko, aby sa ihla vyprázdnila. Ihla sa zavedie cez membránu injektora a po jednej až dvoch sekundách sa roztok rýchlo vstrekne a ihla sa približne po piatich sekundách pomaly vytiahne. Namiesto striekačky možno použiť aj automatický dávkovač.
Zaznamenávanie sa uskutočňuje do úplnej eluácie TMSE zodpovedajúcich prítomných alkoholových zlúčenín. Základná línia musí naďalej spĺňať zodpovedajúce prevádzkové podmienky (bod 6.2.1 alebo 6.2.2).
6.4. Identifikácia píku
Identifikácia jednotlivých píkov sa vykoná podľa retenčných časov a porovnaním so zmesou TSME alifatických a triterpénových alkoholov alebo sterolov a triterpénových diolov analyzovanou za rovnakých podmienok. Chromatogram frakcie alifatických a triterpénových alkoholov je na obrázku 3 a zodpovedajúce chromatogramy sterolov a triterpénových diolov na obrázku 2.
Alifatické alkoholy sa eluujú v tomto poradí: C20-ol (V.S.), C22-ol, C23-ol, C24-ol, C25-ol, C26-ol, C27-ol a C28-ol.
Steroly a triterpénové dioly sa eluujú v tomto poradí: cholesterol, brasikasterol, ergosterol, 24-metylén-cholesterol, kampesterol, kampestanol, stigmasterol, Δ7-kampesterol, Δ5,23-stigmastadienol, klerosterol, β-sistosterol, sitostanol, Δ5-avenasterol, Δ5,24-stigmastadienol, Δ7-stigmastenol, Δ7-avenasterol, erytrodiol a uvaol.
6.5. Kvantitatívne hodnotenie
Plocha píku 1-eikosanolu a alifatických alkoholov C22, C24, C26, C28 sa počíta pomocou systému získavania údajov. Odozvový faktor 1-eikosanolu by sa mal považovať za rovný 1.
Pomocou počítačového systému sa vypočítajú plochy píkov α-cholestanolu, sterolov a triterpénových diolov. Vynechajú sa píky všetkých zlúčenín, ktoré nie sú zahrnuté (ergosterol sa nemusí počítať) medzi zlúčeninami uvedenými v tabuľke 1. Odozvový faktor α-cholestanolu by sa mal považovať za rovný 1.
Koncentrácia každej jednotlivej alkoholovej zlúčeniny vyjadrená v mg/kg tukových látok sa vypočíta takto:

kde:
|
Ax |
= |
plocha píku alkoholovej zlúčeniny x vypočítaná počítačovým systémom. |
|
As |
= |
plocha píku 1-eikosanolu/α-cholestanolu vypočítaná počítačovým systémom. |
|
ms |
= |
hmotnosť dodaného 1-eikosanolu/α-cholestanolu v miligramoch. |
|
m |
= |
hmotnosť vzorky použitej pri stanovení v gramoch. |
7. VYJADRENIE VÝSLEDKOV
Uvedú sa jednotlivé koncentrácie alifatických a triterpénových alkoholov vyjadrené v mg/kg tukových látok a ich súčet ako „obsah alifatického alkoholu spolu“. Celkový obsah je súčet C22, C24, C26 a C28.
Skladba každej jednotlivej alkoholovej zlúčeniny by sa mala vyjadriť na jedno desatinné miesto.
Celková koncentrácia sterolov by sa mala vyjadriť v celých číslach.
Percentuálny obsah jednotlivých sterolov sa vypočíta z pomeru plochy príslušného píku k celkovej ploche píkov sterolov:
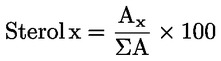
kde:
|
Ax |
= |
plocha píku sterolu x. |
|
ΣA |
= |
celková plocha píkov sterolov. |
Zdanlivý β-sitosterol: Δ5,23-stigmastadienol + klerosterol + β-sitosterol + sitostanol + Δ5-avenasterol + Δ5,24-stigmastadienol.
Percentuálny obsah erytrodiolu a uvaolu sa vypočíta:

kde:
|
AEr |
= |
plocha píku erytrodiolu vypočítaná počítačovým systémom. |
|
AUv |
= |
plocha píku uvaolu vypočítaná počítačovým systémom. |
|
Σ AT |
= |
súčet plôch píkov sterolov + erytrodiolu + uvaolu vypočítaných počítačovým systémom. |
Okrem výpočtu relatívneho percentuálneho podielu jednotlivých sterolov a triterpénových diolov a celkovej koncentrácie sterolov sa musí vypočítať aj koncentrácia erytrodiolu a uvaolu a ich súčet v mg/kg tukových látok, a to podľa týchto vzorov:
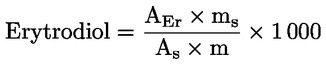
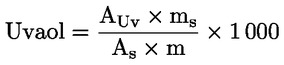
kde:
|
AEr |
= |
plocha píku erytrodiolu vypočítaná počítačovým systémom. |
|
AUv |
= |
plocha píku uvaolu vypočítaná počítačovým systémom. |
|
As |
= |
plocha píku α-cholestanolu vypočítaná počítačovým systémom. |
|
ms |
= |
hmotnosť α-cholestanolu v miligramoch. |
|
m |
= |
hmotnosť vzorky použitej pri stanovení v gramoch. |
Dodatok
|
|
1 Uhľovodíky 2 α-Tokoferol 3 Prenoly 4 Triterpénové alkoholy 5 Alifatické alkoholy 6 Metylsteroly 7 Steroly 8 Triterpénové dioly |

Obrázok 1 – TLC nezmydelniteľnej látky z oleja z olivových výliskov eluovanej dvakrát s hexánom:dietyléterom (65:35), vyvinutej so SO4H2 (50 %) a zahriatej. Pásma, ktoré treba zoškrabať, sú označené v obdĺžniku, 1 sú pásma alifatických alkoholov a 2 sterolov a triterpénových diolov.
Tabuľka I – Relatívne retenčné časy sterolov
|
Pík |
Identifikácia |
Relatívny retenčný čas |
||
|
SE 54 kolóna |
SE 52 kolóna |
|||
|
1 |
Cholesterol |
Δ-5-cholestén-3ß-ol |
0,67 |
0,63 |
|
2 |
Cholestanol |
5α-cholestan-3ß-ol |
0,68 |
0,64 |
|
3 |
Brasikasterol |
[24S]-24-metyl-Δ-5,22-cholestadién-3β-ol |
0,73 |
0,71 |
|
* |
Ergosterol |
[24S]-24-metyl-Δ-5,7,22 cholestatrién -3β-ol |
0,78 |
0,76 |
|
4 |
24-metylén-cholesterol |
24-metylén-Δ-5,24-cholestadién-3ß-o1 |
0,82 |
0,80 |
|
5 |
Kampesterol |
(24R)-24-metyl-Δ-5-cholestén-3ß-ol |
0,83 |
0,81 |
|
6 |
Kampestanol |
(24R)-24-metyl-cholestan-3ß-ol |
0,85 |
0,82 |
|
7 |
Stigmasterol |
(24S)-24-etyl-Δ-5,22-cholestadién-3ß-ol |
0,88 |
0,87 |
|
8 |
Δ-7-kampesterol |
(24R)-24-metyl-Δ-7-cholestén-3ß-ol |
0,93 |
0,92 |
|
9 |
Δ-5,23-stigmastadienol |
(24R,S)-24-etyl-Δ-5,23-cholestadién-3ß-ol |
0,95 |
0,95 |
|
10 |
Klerosterol |
(24S)-24-etyl-Δ-5,25-cholestadién-3ß-ol |
0,96 |
0,96 |
|
11 |
ß-sitosterol |
(24R)-24-etyl-Δ-5-cholestén-3ß-ol |
1,00 |
1,00 |
|
12 |
Sitostanol |
24-etyl-cholestan-3ß-ol |
1,02 |
1,02 |
|
13 |
Δ-5-avenasterol |
(24Z)-24-etylidén-Δ-cholestén-3ß-ol |
1,03 |
1,03 |
|
14 |
Δ-5,24-stigmastadienol |
(24R,S)-24-etyl-Δ-5,24-cholestadién-3ß-ol |
1,08 |
1,08 |
|
15 |
Δ-7-stigmastenol |
(24R,S)-24-etyl-Δ-7-cholestén-3ß-ol |
1,12 |
1,12 |
|
16 |
Δ-7-avenasterol |
(24Z)-24-etylidén-Δ-7-cholestén-3ß-ol |
1,16 |
1,16 |
|
17 |
Erytrodiol |
5α-oleán-12-én-3β,28-diol |
1,41 |
1,41 |
|
18 |
Uvaol |
Δ12-ursen-3β,28-diol |
1,52 |
1,52 |
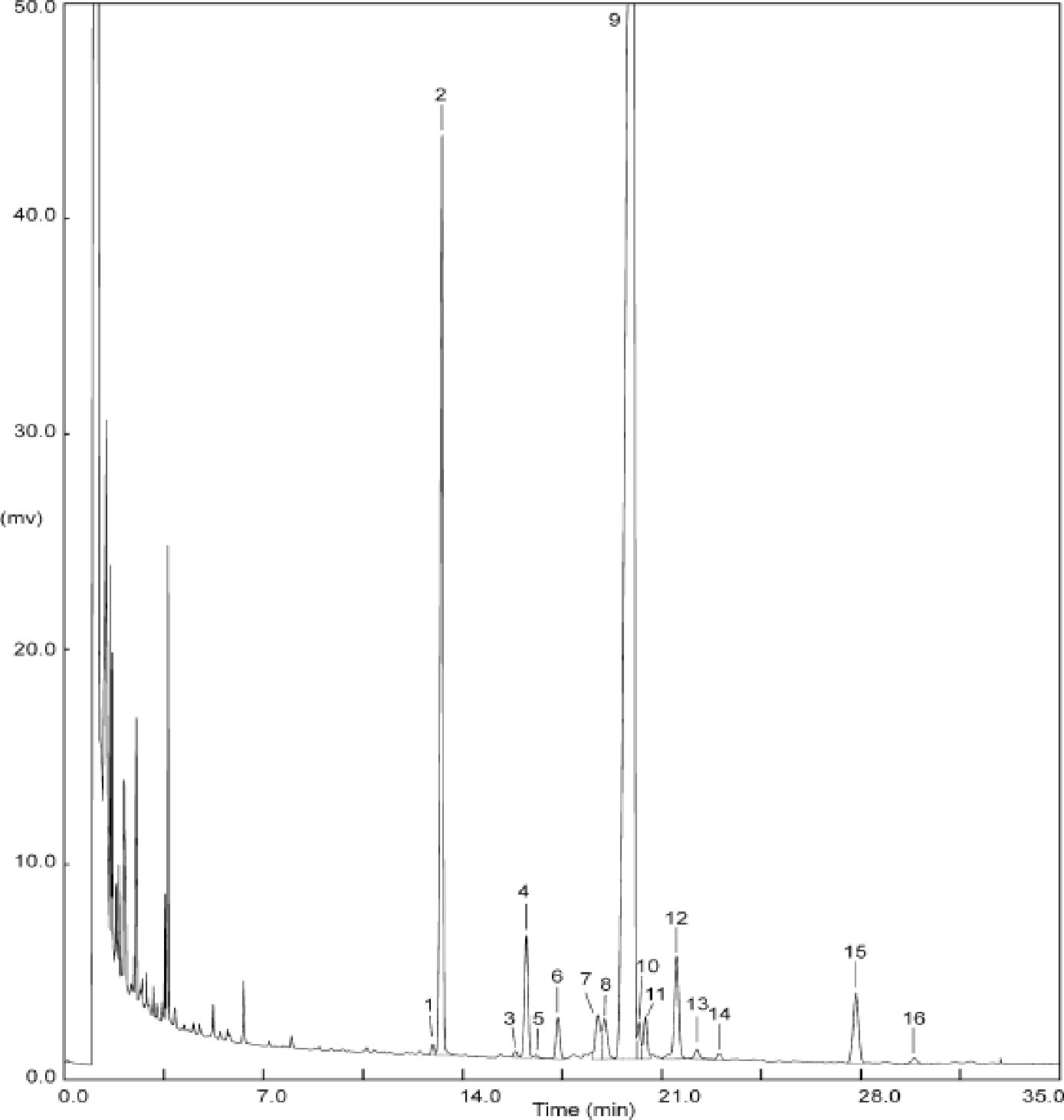
Obrázok 2 – GC-FID chromatografický profil sterolu a triterpénových diolov rafinovaného olivového oleja. (1) Cholesterol, (2) α-cholestanol (V.S.), (3) 24-metylén-cholesterol, (4) kampesterol, (5) kampestanol, (6) stigmasterol, (7) Δ5,23-stigmastadienol, (8) klerosterol, (9) β-sitosterol, (10) sitostanol, (11) Δ5-avenasterol, (12) Δ5,24-stigmastadienol, (13) Δ7-stigmastenol, (14) Δ7-avenasterol, (15) erytrodiol, (16) uvaol.

Obrázok 3 – GC-FID chromatografický profil sterolu triterpénových diolov lampantového olivového oleja. (1) Cholesterol, (2) α-cholestanol, (3) brasikasterol, (4) 24-metylén-cholesterol, (5) kampesterol, (6) kampestanol, (7) stigmasterol, (8) Δ7-kampesterol, (9) Δ5,23-stigmastadienol, (10) klerosterol, (11) β-sitosterol, (12) sitostanol, (13) Δ5-avenasterol, (14) Δ5,24-stigmastadienol, (15) Δ7-stigmastenol, (16) Δ7-avenasterol, (17) erytrodiol, (18) uvaol.
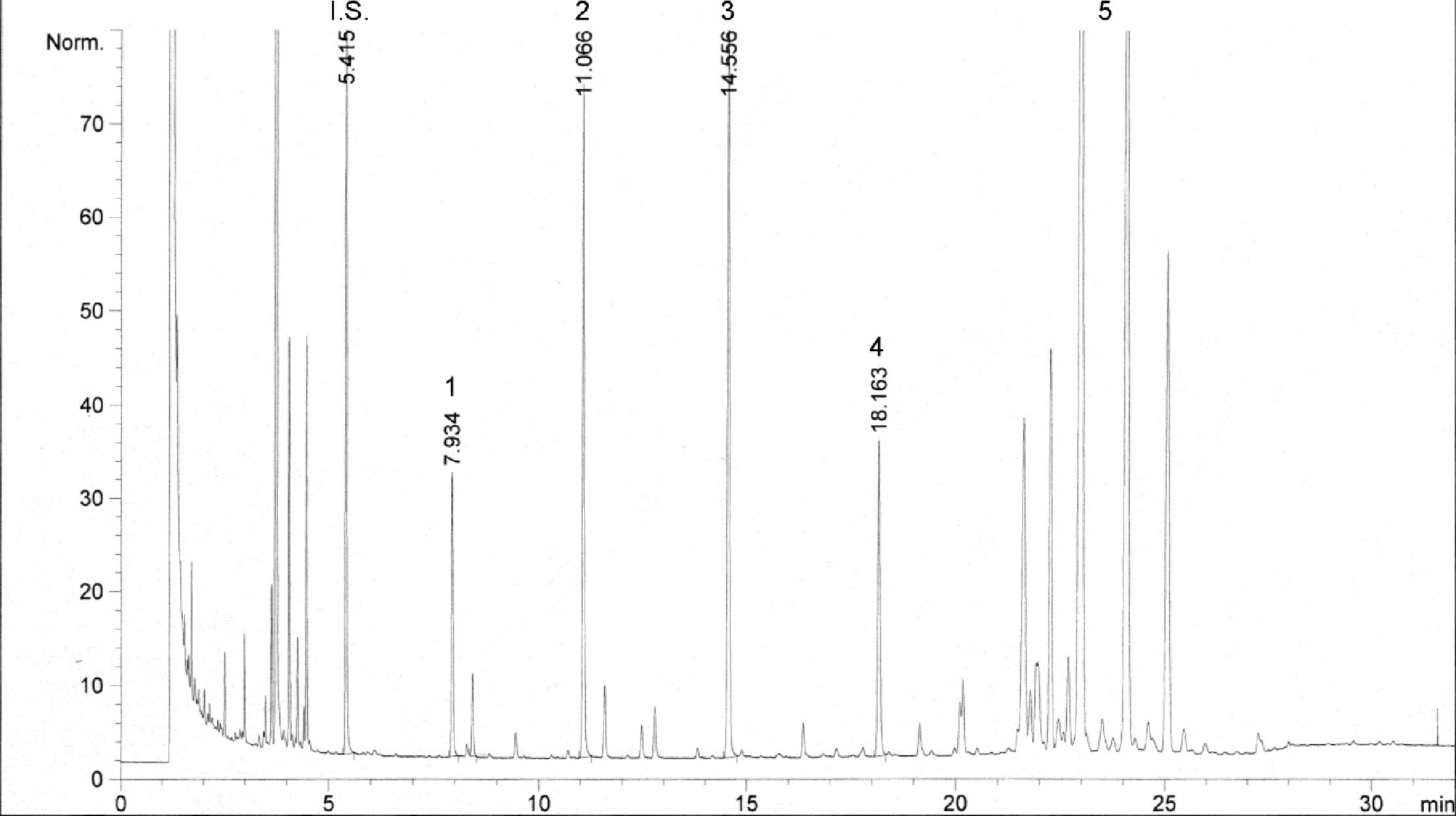
Obrázok 4 – GC-FID chromatografický profil alifatických alkoholov a triterpénových alkoholov olivového oleja. (V.S.) C20-ol, (1) C22-ol, (2) C24-ol, (3) C26-ol, (4) C28-ol, (5) triterpenové alkoholy.

Obrázok 5 – GC-FID chromatografický profil alifatických alkoholov a triterpénových alkoholov rafinovaného olivového oleja a olivového oleja po druhom odstredení. (V.S.) C20-ol, (1) C22-ol, (2) C24-ol, (3) C26-ol, (4) C28-ol, (5) triterpenové alkoholy.

Obrázok 6 – HPLC Chromatogram nezmydelniteľných látok olivového oleja separovaných HPLC s použitím detektoru UV. (1) Alifatické a triterpénové alkoholy, (2) Steroly a triterpénové dioly.
PRÍLOHA XX
Metóda na stanovenie obsahu voskov, metylesterov mastných kyselín a etylesterov mastných kyselín kapilárnou plynovou chromatografiou
1. ÚČEL
Účelom tejto metódy je stanovenie obsahu voskov, metylesterov mastných kyselín a etylesterov mastných kyselín v olivových olejoch. Jednotlivé vosky a alkylestery sa oddelia podľa počtu atómov uhlíka. Metóda sa odporúča ako nástroj na rozlíšenie olivového oleja a oleja z olivových výliskov, ako aj kvalitatívny parameter extra panenských olivových olejov, ktorým sa umožňuje detekcia falšovanej zmesi extra panenských olivových olejov s olejmi nižšej kvality, či už sú to panenské, lampantové oleje alebo niektoré oleje zbavené zápachu.
2. PRINCÍP METÓDY
Pridanie vhodného vnútorného štandardu k oleju a následná frakcionácia chromatografiou na kolóne s hydratovaným silikagélom. Získanie frakcie eluovanej za skúšobných podmienok (ktorej polarita je nižšia ako polarita triglyceridov) a následná priama analýza kapilárnou plynovou chromatografiou.
3. PRÍSTROJE
|
3.1. |
Erlenmeyerova banka, 25 ml. |
|
3.2. |
Sklená kolóna na kvapalinovú chromatografiu, s vnútorným priemerom 15 mm, dĺžkou 30 až 40 cm vybavená vhodným kohútikom. |
|
3.3. |
Plynový chromatograf vhodný na použitie s kapilárnou kolónou, vybavený systémom priamej injektáže na kolónu (tzv. technika on-column), ktorého súčasťou sú:
|
|
3.4. |
Mikrostriekačka s kapacitou 10 μl na priamu injektáž na kolónu, vybavená cementovanou ihlou. |
|
3.5. |
Elektrická miešačka. |
|
3.6. |
Rotačná odparka. |
|
3.7. |
Mufľová pec. |
|
3.8. |
Analytické váhy s presnosťou merania na ± 0,1 mg. |
|
3.9. |
Bežné laboratórne sklo. |
4. ČINIDLÁ
|
4.1. |
Silikagél, veľkosť zrna 60 až 200 μm. Vložte silikagél do mufľovej pece na 500 °C aspoň na 4 hodiny. Po vychladnutí pridajte 2 % vody vzhľadom na množstvo použitého silikagélu. Dobre pretrepte, aby sa hmota homogenizovala, a pred použitím uchovávajte v sušičke najmenej počas 12 hodín. |
|
4.2. |
n-hexán, na chromatografiu alebo analýzu zvyškov. Hexán možno nahradiť izo-oktánom (2,2,4-trimetylpentánom na chromatografiu) pod podmienkou dosahovania porovnateľných hodnôt presnosti. Rozpúšťadlá s vyššou teplotou varu než n-hexán sa odparujú dlhšie. Uprednostňujú sa však z dôvodu toxicity hexánu. Čistota musí byť overená; napríklad možno kontrolovať rezíduum po odparení 100 ml rozpúšťadla. VAROVANIE – Výpary sa môžu vznietiť. Chráňte pred zdrojom tepla, iskrami alebo otvoreným ohňom. Uistite sa, že fľaše sú vždy dobre uzatvorené. Počas použitia zaistite riadnu ventiláciu. Zabráňte hromadeniu výparov a odstráňte všetky príčiny možného vzniku požiaru, ako sú ohrievacie telesá alebo elektrické zariadenia, ktoré nie sú vyrobené z nehorľavého materiálu. Vdýchnutie je škodlivé, lebo môže spôsobiť poškodenie nervových buniek. Zabráňte vdýchnutiu výparov. V prípade potreby použite vhodný dýchací prístroj. Zabráňte kontaktu s očami a pokožkou. Izo-oktán je horľavá kvapalina, ktorá predstavuje nebezpečenstvo vzniku požiaru. Limit výbušnosti vo vzduchu je 1,1 % až 6,0 % (objemový zlomok). Je toxický pri požití a vdýchnutí. Pri práci s týmto rozpúšťadlom používajte dobre fungujúci ventilovaný digestor. |
|
4.3. |
Dietyléter, na chromatografiu.
VAROVANIE – Vysokohorľavý a mierne toxický. Dráždi pokožku. Vdýchnutie je škodlivé. Môže spôsobiť poškodenie očí. Môže mať oneskorené účinky. Môže vytvárať výbušné peroxidy. Výpary sa môžu vznietiť. Chráňte pred zdrojom tepla, iskrami alebo otvoreným ohňom. Uistite sa, že fľaše sú vždy dobre uzatvorené. Počas použitia zaistite riadnu ventiláciu. Zabráňte hromadeniu výparov a odstráňte všetky príčiny možného vzniku požiaru, ako sú ohrievacie telesá alebo elektrické zariadenia, ktoré nie sú vyrobené z nehorľavého materiálu. Neodparujete dosucha ani takmer dosucha. Pridaním vody alebo vhodného redukčného činidla možno znížiť tvorbu peroxidov. Nepite. Zabráňte vdýchnutiu výparov. Zabráňte dlhotrvajúcemu alebo opakovanému kontaktu s pokožkou. |
|
4.4. |
n-heptán, na chromatografiu alebo izo-oktán. VAROVANIE – Vznetlivý. Vdýchnutie je škodlivé. Chráňte pred zdrojom tepla, iskrami alebo otvoreným ohňom. Uistite sa, že fľaše sú vždy dobre uzatvorené. Počas použitia zaistite riadnu ventiláciu. Zabráňte vdýchnutiu výparov. Zabráňte dlhotrvajúcemu alebo opakovanému kontaktu s pokožkou. |
|
4.5. |
Štandardný roztok laurylarašidátu (poznámka 3) s koncentráciou 0,05 % (m/V) v heptáne (vnútorný štandard pre vosky). Poznámka 3: Možno použiť aj palmityl-palmitát, myristyl-stearát alebo aršidyl-laureát. |
|
4.6. |
Štandardný roztok metyl-heptadekanoátu s koncentráciou 0,02 % (m/V) v heptáne (vnútorný štandard pre metyl- a etylestery). |
|
4.7. |
Sudán 1 [1-(fenyldiazenyl)naftalén-2-ol]. |
|
4.8. |
Nosný plyn: vodík alebo hélium, čistý, na plynovú chromatografiu. VAROVANIE Vodík. Vysokohorľavý, pod tlakom. Chráňte pred zdrojom tepla, iskrami, otvoreným ohňom alebo elektrickými zariadeniami, ktoré nie sú vyrobené z nehorľavého materiálu. Uistite sa, že ventil fľaše je uzavretý, keď sa nepoužíva. Vždy používajte s redukčným ventilom. Uvoľnite napätie redukčnej pružiny pred otvorením ventilu fľaše. Pri otváraní ventilu nestojte pred výpustným otvorom fľaše. Počas použitia zaistite riadnu ventiláciu. Neprepúšťajte vodík z jednej fľaše do druhej. Nemiešajte plyn vo fľaši. Uistite sa, že fľaše sa nemôžu prevrhnúť. Chráňte ich pred slnečným žiarením a zdrojmi tepla. Skladujte v nekorozívnom prostredí. Nepoužívajte poškodené alebo neoznačené fľaše. Hélium. Vysokostlačený plyn. Znižuje množstvo kyslíka, ktoré je k dispozícii na dýchanie. Fľaše udržiavajte uzatvorené. Počas použitia zaistite riadnu ventiláciu. Nevstupujte do skladovacích priestorov, ak nie sú riadne vetrané. Vždy používajte s redukčným ventilom. Uvoľnite napätie redukčnej pružiny pred otvorením ventilu fľaše. Neprepúšťajte plyn z jednej fľaše do druhej. Uistite sa, že fľaše sa nemôžu prevrhnúť. Pri otváraní ventilu nestojte pred výpustným otvorom fľaše. Chráňte ich pred slnečným žiarením a zdrojmi tepla. Skladujte v nekorozívnom prostredí. Nepoužívajte poškodené alebo neoznačené fľaše. Nevdychujte. Používajte výhradne na technické účely. |
|
4.9. |
Pomocné plyny: — Vodík, čistý, na plynovú chromatografiu. — Vzduch, čistý, na plynovú chromatografiu. VAROVANIE Vzduch. Vysokostlačený plyn. V prítomnosti horľavých látok používajte opatrne, keďže teplota samovznietenia väčšiny organických zložiek vzduchu je výrazne nižšia pod vysokým tlakom. Uistite sa, že ventil fľaše je uzavretý, keď sa nepoužíva. Vždy používajte s redukčným ventilom. Uvoľnite napätie redukčnej pružiny pred otvorením ventilu fľaše. Pri otváraní ventilu nestojte pred výpustným otvorom fľaše. Neprepúšťajte plyn z jednej fľaše do druhej. Nemiešajte plyn vo fľaši. Uistite sa, že fľaše sa nemôžu prevrhnúť. Chráňte ich pred slnečným žiarením a zdrojmi tepla. Skladujte v nekorozívnom prostredí. Nepoužívajte poškodené alebo neoznačené fľaše. Vzduch na technické účely sa nesmie používať na vdychovanie ani v dýchacích prístrojoch. |
5. POSTUP
5.1. Príprava chromatografickej kolóny
Preveďte do suspenzie 15 g silikagélu (bod 4.1) v n-hexáne (bod 4.2) a zaveďte do kolóny (bod 3.2). Nechajte samovoľne usadiť. Usadzovanie dokončite pomocou elektrickej trepačky s cieľom dosiahnuť homogénnejšiu chromatografickú vrstvu. Premyte 30 ml n-hexánu s cieľom odstrániť všetky prípadné nečistoty. Analytickými váhami (bod 3.8) navážte presne 500 mg vzorky do 25 ml banky (bod 3.1) a pridajte vhodné množstvo vnútorného štandardu (bod 4.5) v závislosti od predpokladaného obsahu vosku, napríklad pridajte 0,1 mg laurylarašidátu v prípade olivového oleja, 0,25 až 0,50 mg v prípade oleja z olivových výliskov a 0,05 mg metyl-heptadekanoátu v prípade olivových olejov (bod 4.6).
Pripravenú vzorku preneste do chromatografickej kolóny pomocou dvoch 2-ml dávok n-hexánu (bod 4.2).
Napustite rozpúšťadlo do výšky 1 mm nad hornou úrovňou absorbentu. Premyte ďalším roztokom n-hexánu/dietyléteru v pomere 99:1 a zachyťte 220 ml roztoku pri prietoku približne 15 kvapiek každých 10 sekúnd. (Táto frakcia obsahuje metyl- a etylestery a vosky). (Poznámka 4) (Poznámka 5).
Poznámka 4: Zmes n-hexánu/dietyléteru (99:1) by sa mala každý deň čerstvo pripraviť.
Poznámka 5: Na umožnenie vizuálnej kontroly správnej elúcie voskov možno do vzorky roztoku pridať 100 μl 1 % farbiva Sudan I v elučnej zmesi.
Retenčný čas uvedeného farbiva sa nachádza medzi retenčným časom voskov a triglyceridov. Preto keď farbivo dosiahne dno chromatografickej kolóny, je potrebné elúciu zastaviť, lebo všetky vosky už boli eluované.
Sušte výslednú frakciu v rotačnej odparke až do odstránenia takmer celého rozpúšťadla. Odstráňte posledné 2 ml pomocou slabého prúdu dusíka. Frakcia obsahujúca metyl- a etylestery sa zachytáva v 2 až 4 ml n-heptánu alebo izo-oktánu ako rozpúšťadla.
5.2. Plynová chromatografická analýza
5.2.1. Postup prípravy
Namontujte kolónu do plynového chromatografu (bod 3.3), pričom vstupný port sa pripojí k systému na kolóne a výstupný port k detektoru. Skontrolujte prístroj na plynovú chromatografiu (činnosť uzavretých plynových obvodov, účinnosť detektora a zapisovača atď.).
Pokiaľ sa kolóna používa po prvýkrát, je potrebné ju kondicionovať. Zaveďte mierny prietok plynu cez kolónu a potom zapnite prístroj na plynovú chromatografiu. Postupne zahrievajte, až kým sa približne po 4 hodinách dosiahne teplota 350 °C.
Túto teplotu udržiavajte najmenej počas 2 hodín, potom prístroj nastavte na prevádzkové podmienky [nastavte prietok plynu, zapáľte plamienok, pripojte k elektrickému zapisovaču (bod 3.3.4), nastavte teplotu pece pre kolónu, nastavte detektor atď.]. Zapíšte signál pri citlivosti najmenej dvakrát vyššej, ako sa vyžaduje na vykonanie analýzy. Základná línia by mala byť lineárna, nesmie obsahovať píky žiadneho druhu a nesmie vykazovať žiadnu nestabilitu (drift).
Záporný priamočiary drift indikuje nedokonalú tesnosť spojov kolóny, kým kladný drift indikuje nedostatočné kondiciovanie kolóny.
5.2.2. Výber prevádzkových podmienok v prípade voskov a metyl- a etylesterov (poznámka 6)
Prevádzkové podmienky sú spravidla tieto:
|
— |
Teplota kolóny : 20 °C/min 5 °C/min 80 °C počiatočná teplota (1′) |
|
— |
Teplota detektora : 350 °C. |
|
— |
Injektované množstvo : 1 μl roztoku n-heptánu (2 až 4 ml). |
|
— |
Nosný plyn : hélium alebo vodík s optimálnou lineárnou rýchlosťou pre zvolený plyn (pozri dodatok A). |
|
— |
Citlivosť prístroja : vhodná na splnenie uvedených podmienok. |
Poznámka 6: Vzhľadom na vysokú konečnú teplotu sa pripúšťa kladný drift, ktorý nesmie prekročiť 10 % rozsahu stupnice.
Uvedené podmienky možno upraviť tak, aby vyhovovali charakteristikám kolóny a prístroju na plynovú chromatografiu, a to s cieľom oddeliť všetky vosky a metylestery mastných kyselín a etylestery mastných kyselín, ako aj získať uspokojivú separáciu píkov (pozri obrázky 2, 3 a 4) a retenčný čas 18 ± 3 min. pre vnútorný štandard laurylarašidátu. Najreprezentatívnejší pík voskov sa musí nachádzať nad 60 % rozsahu stupnice, zatiaľ čo pík metyl-heptadekanoátu – vnútorný štandard pre metyl- a etylestery musí dosiahnuť plný rozsah stupnice.
Parametre integrácie píku by sa mali určiť tak, aby sa dosiahlo správne vyhodnotenie príslušných plôch píkov.
5.3. Vykonanie analýzy
Pomocou 10 μl mikrostriekačky naberte 10 μl roztoku, pričom piestik ťahajte dozadu až dovtedy, kým ihla nebude prázdna. Ihlu zaveďte do injekčného systému a po uplynutí 1 až 2 sekúnd rýchlo vstreknite. Po asi 5 sekundách ihlu jemne vytiahnite.
Zápis nechajte prebiehať dovtedy, kým vosky alebo stigmastadiény nebudú úplne eluované v závislosti od analyzovanej frakcie.
Základná línia musí vždy zodpovedať požadovaným podmienkam.
5.4. Identifikácia píku
Píky identifikujte z retenčných časov ich porovnaním so zmesami voskov so známymi retenčnými časmi analyzovanými za rovnakých podmienok. Alkylestery sa stanovujú zo zmesí metylesterov a etylesterov hlavných mastných kyselín v olivových olejoch (palmitovej a olejovej).
Na obrázku 1 je znázornený chromatogram voskov panenského olivového oleja. Na obrázkoch 2 a 3 sú znázornené chromatogramy dvoch maloobchodne predávaných extra panenských olivových olejov, jedného s metylestermi a etylestermi a druhého bez nich. Na obrázku 4 sú znázornené chromatogramy v prípade extra panenského olivového oleja najvyššej kvality a toho istého oleja, ku ktorému sa primiešalo 20 % oleja zbaveného zápachu.
5.5. Kvantitatívna analýza voskov
Pomocou integrátora určíte plochy píkov zodpovedajúce vnútornému štandardu laurylarašidátu a alifatickým esterom od C40 do C46.
Určíte obsah všetkých voskov pripočítaním každého jednotlivého vosku v mg/kg tuku podľa tohto vzorca:
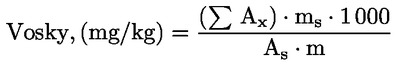
kde:
|
Ax |
= |
plocha zodpovedajúca píku pre jednotlivé estery, vypočítaná pomocou počítača |
|
As |
= |
plocha zodpovedajúca píku pre vnútorný štandard laurylarašidátu, vypočítaná pomocou počítača |
|
ms |
= |
hmotnosť pridaného vnútornému štandardu laurylarašidátu, v miligramoch |
|
m |
= |
hmotnosť vzorky odobratej na stanovenie, v gramoch. |
5.5.1. Kvantitatívna analýza metylesterov a etylesterov
Pomocou integrátora určíte plochy píkov zodpovedajúce vnútornému štandardu metyl-heptadekanoátu, metylesterov mastných kyselín C16 a C18 a etylesterov mastných kyselín C16 a C18.
Obsah alkylesterov v mg/kg tuku určíte takto:

kde:
|
Ax |
= |
plocha zodpovedajúca píku pre jednotlivé estery C16 a C18, vypočítaná pomocou počítača |
|
As |
= |
plocha zodpovedajúca píku pre vnútorný štandard metyl-heptadekanoátu, vypočítaná pomocou počítača |
|
ms |
= |
hmotnosť pridaného vnútornému štandardu metyl-heptadekanoátu, v miligramoch |
|
m |
= |
hmotnosť vzorky odobratej na stanovenie, v gramoch. |
6. VYJADRENIE VÝSLEDKOV
V správe uveďte súčet obsahov rôznych voskov od C40 do C46 (poznámka 7) v miligramoch na kilogram tuku.
V správe uveďte súčet obsahov metylesterov a etylesterov od C16 do C18 a celkový súčet oboch.
Výsledky by sa mali vyjadriť v najbližších mg/kg.
Poznámka 7: Prvky kvantitatívneho hodnotenia sa vzťahujú na píky esterov s párnym počtom atómov uhlíka medzi C40 a C46 v súlade so vzorom chromatogramu voskov olivového oleja znázorneným na pripojenom obrázku. Pokiaľ sa ester C46 rozštiepi, na účely identifikácie sa odporúča analyzovať frakciu vosku oleja z olivových výliskov, kde možno pík C46 rozlíšiť, lebo zreteľne prevláda.
V správe uveďte pomer obsahov etylesterov a metylesterov
Obrázok 1
Príklad plynového chromatogramu frakcie voskov v olivovom oleji ( 6 )

Píky s retenčným časom 5 až 8 min. metyl- a etylesterov mastných kyselín
Legenda:
|
I.S. |
= |
(vnútorný štandard – V.S.) laurylarašidát |
|
1 |
= |
diterpénové estery |
|
2+2′ |
= |
estery C40 |
|
3+3′ |
= |
estery C42 |
|
4+4′ |
= |
estery C44 |
|
5 |
= |
estery C46 |
|
6 |
= |
sterol-estery a triterpénové alkoholy |
Obrázok 2
Metylestery, etylestery a vosky v panenskom olivovom oleji

Legenda:
|
1 |
– |
Metyl C16 |
|
2 |
– |
Etyl C16 |
|
3 |
– |
Metyl-heptadekanoát V.S. |
|
4 |
– |
Metyl C18 |
|
5 |
– |
Etyl C18 |
|
6 |
– |
Skvalén |
|
7 |
– |
Laurylarašidát V.S. |
|
A |
– |
Diterpénové estery |
|
B |
– |
Vosky |
|
C |
– |
Sterol-estery a triterpénové estery |
Obrázok 3
Metylestery, etylestery a vosky v extra panenskom olivovom oleji

Legenda:
|
1 |
– |
Metyl-heptadekanoát V.S. |
|
2 |
– |
Metyl C18 |
|
3 |
– |
Etyl C18 |
|
4 |
– |
Skvalén |
|
5 |
– |
Laurylarašidát V.S. |
|
A |
– |
Diterpénové estery |
|
B |
– |
Vosky |
|
C |
– |
Sterol-estery a triterpénové estery |
Obrázok 4
Časť chromatogramu extra panenského olivového oleja a toho istého oleja, ku ktorému sa primiešal olej zbavený zápachu

Legenda:
|
1 |
– |
Metyl-myristát V.S. |
|
2 |
– |
Metyl-palmitát |
|
3 |
– |
Etyl-palmitát |
|
4 |
– |
Metyl-heptadekanoát V.S. |
|
5 |
– |
Metyl-linoleát |
|
6 |
– |
Metyl-oleát |
|
7 |
– |
Metyl-stearát |
|
8 |
– |
Etyl-linoleát |
|
9 |
– |
Etyl oleát |
|
10 |
– |
Etyl-stearát |
Dodatok A
Stanovenie lineárnej rýchlosti plynu
Vstreknite 1:3 μl metánu (alebo propánu) do plynového chromatografu po jeho nastavení na bežné prevádzkové podmienky. Odmerajte čas, ktorý potrebuje plyn na prechod cez kolónu od momentu jeho vstreknutia až do času, keď sa objaví pík (tM).
Lineárna rýchlosť v cm/s je daná vzorcom L/tM, kde L je dĺžka kolóny v cm a tM je čas nameraný v sekundách.
▼M28 —————
PRÍLOHA XXI
Výsledky kontrol zhody vykonaných na olivovom oleji podľa článku 8 ods. 2
|
|
Označovanie |
Chemické parametre |
Organoleptické vlastnosti (4) |
Záverečné poznámky |
|||||||||||||
|
Vzorka |
Kategória |
Krajina pôvodu |
Miesto kontroly (1) |
Oficiálny názov |
Označenie pôvodu |
Podmienky skladovania |
Chybné informácie |
Čitateľnosť |
C/NC (3) |
Parametre mimo medzných hodnôt Á/N |
Ak áno, uveďte ktoré (2) |
C/NC (3) |
Medián defektov |
Medián ovocnosti |
C/NC (3) |
Požadované opatrenia |
Sankcie |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(1) Vnútorný trh (mlyn, plnenie do fliaš, maloobchodný predaj), vývoz, dovoz. (2) Každá charakteristika olivového oleja stanovená v prílohe I má kód. (3) Spĺňa/nespĺňa. (4) Nevyžaduje sa pre olivový olej a zvyškový olej. |
|||||||||||||||||
( 1 ) Ú. v. EÚ L 228, 1.9.2009, s. 3.
( 2 ) Smernica Európskeho parlamentu a Rady 2011/91/EÚ z 13. decembra 2011 o identifikácii alebo rozlíšení dávky, do ktorej potraviny patria (Ú. v. EÚ L 334, 16.12.2011, s. 1).
( 3 ) Po elúcii sterol-esterov sa na chromatografickom zázname nemajú ukázať výrazné píky (triglyceridy).
( 4 ) Nariadenie Európskeho parlamentu a Rady (EÚ) č. 1308/2013 zo 17. decembra 2013, ktorým sa vytvára spoločná organizácia trhov s poľnohospodárskymi výrobkami, a ktorým sa zrušujú nariadenia Rady (EHS) č. 922/72, (EHS) č. 234/79, (ES) č. 1037/2001 a (ES) č. 1234/2007 (Ú. v. EÚ L 347, 20.12.2013, s. 671).
( 5 ) Ak pri priamom ovoniavaní zaznamenajú, že niektorý z negatívnych atribútov je extrémne intenzívny, olej nemusia ochutnávať, a v tom prípade zaznamenajú túto výnimočnú okolnosť na profilový hárok.
( 6 ) Po elúcii sterol-esterov by chromatografický záznam nemal vykazovať žiadne významné píky (triglyceridy).