|
3.
|
Dopĺňajú sa tieto kapitoly:
„B.49.
IN VITRO TEST MIKROJADIER BUNIEK CICAVCOV
ÚVOD
|
1.
|
In vitro skúška mikrojadier (MNvit) je genotoxický test, ktorého cieľom je zistiť prítomnosť mikrojadier (MN) v cytoplazme buniek v interfáze. Mikrojadrá môžu pochádzať z acentrických fragmentov chromozómov (t. j. bez centroméry) alebo z celých chromozómov, ktoré počas anafázy delenia bunky nie sú schopné migrovať k pólom. Skúška zisťuje aktivitu klastogénnych a aneugénnych chemikálií (látok a zmesí) (1) (2) v bunkách, ktoré prešli bunkovým delením počas expozície testovanej látke alebo po nej. Táto testovacia metóda (TM) umožňuje využívať protokoly s inhibítorom polymerizácie aktínu, ktorý sa nazýva cytochalasín B (cytoB), a bez neho. Pridanie cytoB pred cieľovou mitózou umožňuje identifikovať a selektívne analyzovať frekvenciu mikrojadier v bunkách, ktoré ukončili jednu mitózu, keďže tieto bunky sú dvojjadrové (3) (4). Táto testovacia metóda tiež umožňuje využiť protokoly bez blokovania cytokinézy, ak existuje dôkaz, že populácia analyzovaných buniek prešla mitózou.
|
|
2.
|
Okrem použitia skúšky MNvit na identifikáciu chemikálií (látok a zmesí), ktoré indukujú mikrojadrá, môže použitie inhibítorov cytokinézy, imunochemického označovania kinetochorov alebo hybridizácia s centromérovými/telomérovými sondami [fluorescenčná hybridizácia in situ (FISH)] tiež poskytnúť informácie o mechanizmoch poškodenia chromozómov a tvorbe mikrojadier (5) (6) (7) (8) (9) (10) (11) (12) (13) (14) (15) (16). Postupy označovania a hybridizácie možno použiť, ak dôjde k zvýšenej miere tvorby mikrojadier a výskumník chce určiť, či je tento nárast výsledkom klastogénnych a/alebo aneugénnych procesov.
|
|
3.
|
Mikrojadrá predstavujú poškodenie, ktoré sa prenáša na dcérske bunky, zatiaľ čo chromozómové odchýlky zaznamenané v bunkách v metafáze nemožno preniesť. Keďže mikrojadrá v bunkách v interfáze možno hodnotiť relatívne objektívne, pracovníci laboratória musia určiť len to, či bunky prešli, alebo neprešli delením a koľko buniek obsahuje mikrojadro. Následne možno preparáty pomerne rýchlo vyhodnotiť a analýzu možno vykonať automaticky. Takto sa počas jedného ošetrenia môžu vyhodnotiť prakticky tisíce, a nielen stovky buniek, čím sa zvyšuje účinnosť skúšky. Nakoniec keďže mikrojadrá môžu vzniknúť v dôsledku spomalenia vzniku chromozómov, existuje možnosť zistiť činitele indukujúce aneuploidy, ktoré je zložité skúmať pomocou bežných testov chromozómových odchýlok, napr. usmernenie OECD o vykonávaní testov 473 (kapitola B.10 tejto prílohy) (17). Skúška MNvit však neumožňuje rozlišovať chemikálie indukujúce vznik polyploidov od tých chemikálií, ktoré indukujú klastogenicitu bez použitia špeciálnych postupov, ako napríklad fluorescenčnej hybridizácie in situ (FISH) opísanej v odseku 2.
|
|
4.
|
Skúška MNvit je metóda in vitro, ktorá bežne využíva kultivované ľudské bunky alebo bunky hlodavcov. Poskytuje komplexný základ skúmania potenciálu poškodenia chromozómov in vitro, keďže možno zistiť aneugény, ako aj klastogény.
|
|
5.
|
Skúška MNvit je spoľahlivá a účinná v prípade rôznych druhov buniek, a to s použitím alebo bez použitia cytoB. Platnosť skúšky MNvit podporuje množstvo údajov získaných z rôznych bunkových línií hlodavcov (CHO, V79, CHL/IU, a L5178Y) a ľudských lymfocytov (18) (19) (20) (21) (22) (23) (24) (25) (26) (27) (28) (29) (30) (31). Tieto údaje zahŕňajú najmä medzinárodné validačné štúdie, ktoré koordinovalo združenie Société Française de Toxicologie Génétique (SFTG) (18) (19) (20) (21) (22), a správy z medzinárodného seminára o testovaní genotoxicity (International Workshop on Genotoxicity Testing) (4) (16). Dostupné údaje boli opätovne prehodnotené vzhľadom na závažnosť dôkazov retrospektívnej validačnej štúdie, ktorú uskutočnilo Európske centrum pre validáciu alternatívnych metód (ECVAM) Európskej komisie, pričom táto testovacia metóda bola Vedeckým poradným výborom ECVAM (ESAC) schválená ako vedecky platná (32) (33) (34). Použitie ľudskej lymfoblastoidnej bunkovej línie TK6 (35), buniek HepG2 (36) (37) a primárnych embryonálnych buniek škrečka sýrskeho (38) bolo síce opísané, neboli však použité vo validačných štúdiách.
|
VYMEDZENIE POJMOV
|
6.
|
Použité definície sú uvedené v dodatku 1.
|
ÚVODNÉ ÚVAHY
|
7.
|
Testy, ktoré sa vykonávajú in vitro, si zvyčajne vyžadujú použitie exogénneho zdroja metabolickej aktivácie okrem prípadov, keď sú bunky v súvislosti s testovanými látkami metabolicky spôsobilé. Exogénny systém metabolickej aktivácie napodobňuje v plnej miere podmienky in vivo. Rovnako je potrebné predchádzať podmienkam, ktoré by viedli k falošným pozitívnym výsledkom, ktoré neodrážajú vnútornú mutagenitu a môžu vzniknúť v dôsledku takých faktorov, ako napríklad výrazné zmeny v hodnotách pH alebo osmolality alebo z dôvodu vysokej úrovne cytotoxicity (39) (40) (41). Ak testovaná chemikália spôsobí zmenu v hodnote pH média v čase pridania, je potrebné pH upraviť tlmením zásobného roztoku tak, aby všetky objemy pri všetkých testovacích koncentráciách a pri všetkých kontrolách ostali rovnaké.
|
|
8.
|
Pri analýze indukcie mikrojadier je dôležité, aby mitóza prebehla v ošetrených aj v neošetrených kultúrach. Najinformatívnejšie štádium pre hodnotenie mikrojadier je v bunkách, ktoré ukončili jednu mitózu počas ošetrenia testovanou látky alebo po ošetrení.
|
PRINCÍP TESTU
|
9.
|
Bunkové kultúry ľudského pôvodu alebo pochádzajúce z cicavcov sú vystavené testovanej látke s exogénnym zdrojom metabolickej aktivácie aj bez neho okrem prípadu, ak sú použité bunky s dostatočnou schopnosťou metabolizácie. Súčasťou každého testu sú chemikálie na súbežné kontroly s rozpúšťadlom/nosičom (VC) a chemikálie na pozitívnu kontrolu (PC).
|
|
10.
|
Počas expozície testovanej látke alebo po expozícii sa bunky pestujú dostatočne dlho na to, aby sa umožnilo, že poškodenie chromozómov alebo deliaceho vretienka povedie k vytvoreniu mikrojadier v bunkách v interfáze. Na indukciu aneuploídie je zvyčajne potrebné, aby bola testovaná látka prítomná počas mitózy. Získané a zafarbené bunky v interfáze sa analyzujú na prítomnosť mikrojadier. V ideálnom prípade by sa mali mikrojadrá hodnotiť len v tých bunkách, ktoré počas expozície testovanej látke alebo v čase po expozícii ukončili mitózu. Ak boli kultúry ošetrené blokátorom cytokinézy, rovnaký výsledok sa dosiahne hodnotením len dvojjadrových buniek. Ak sa blokátor cytokinézy nepoužije, je dôležité preukázať, že analyzované bunky pravdepodobne prešli delením počas expozície testovanej látke alebo po nej. Pri všetkých protokoloch je dôležité preukázať, že došlo k proliferácii buniek v kontrolných, ako aj v ošetrených kultúrach, a rozsah cytotoxicity alebo cytostázy, ktorú vyvolala testovaná látka, je potrebné posúdiť v rámci kultúr (alebo v súbežných kultúrach), ktoré sa hodnotia z hľadiska mikrojadier.
|
OPIS SKÚŠKY
Prípravy
|
11.
|
Môžu sa použiť kultivované primárne ľudské periférne krvné lymfocyty (5) (19) (42) (43) a rôzne bunkové línie hlodavcov, ako napríklad bunky CHO, V79, CHL/IU a L5178Y (18) (19) (20) (21) (22) (25) (26) (27) (28) (30). Použitie iných bunkových línií a typov je potrebné zdôvodniť na základe ich preukázanej výkonnosti v skúške, ako sa uvádza v oddiele Kritériá prijateľnosti. Keďže pozaďová frekvencia mikrojadier ovplyvní citlivosť skúšky, odporúča sa použiť druhy buniek, ktoré majú nízku, stabilnú pozaďovú frekvenciu tvorby mikrojadier.
|
|
12.
|
Ľudské periférne krvné lymfocyty je potrebné získať od mladých (od 18 do 35 rokov) zdravých nefajčiarov, ktorí v poslednom čase neboli vystavení genotoxickým chemikáliám alebo radiácii. Ak sa pre jedno použitie spoja bunky od viacerých darcov, mal by sa uviesť ich počet. Frekvencia mikrojadier sa s vekom zvyšuje, pričom tento trend je viac viditeľný u žien než u mužov (44) a treba to zohľadniť pri výbere darcovských buniek na spájanie.
|
Médiá a podmienky kultivácie
|
13.
|
Na udržiavanie kultúr sú potrebné vhodné kultivačné médiá a inkubačné podmienky (kultivačné nádoby, koncentrácia CO2, teplota a vlhkosť). Získané bunkové línie a kmene by sa mali pravidelne kontrolovať, pokiaľ ide o stabilitu modálneho počtu chromozómov a o to, či nedošlo ku kontaminácii mykoplazmou. V prípade kontaminácie alebo zmeny modálneho počtu chromozómov by sa nemali používať. Mal by byť známy čas bežného bunkového cyklu pre podmienky použitých kultúr v testovacom laboratóriu. Ak sa použije metóda blokovania cytokinézy, je potrebné optimalizovať koncentráciu inhibítora cytokinézy vzhľadom na príslušný druh bunky a mala by vykazovať tvorbu dostatočného počtu dvojjadrových buniek na hodnotenie.
|
Príprava kultúr
|
14.
|
Zistené bunkové línie a kmene: bunky sa odoberú z uskladnených kultúr, zapustia sa do kultivačného média v takej hustote, pri ktorej kultúry nedosiahnu zhluk v monovrstvách, a suspenzné kultúry nedosiahnu nadmernú hustotu pred časom zberu a inkubujú sa pri teplote 37 °C.
|
|
15.
|
Lymfocyty: celá krv ošetrená antikoagulantom (napr. heparínom) alebo oddelené lymfocyty sa pred expozíciou testovanej látke a cytoB kultivujú v prítomnosti mitogénu (napr. fytohemaglutinínu).
|
Metabolická aktivácia
|
16.
|
Ak sa použijú bunky s nedostatočnou endogénnou metabolickou kapacitou, je potrebné použiť exogénny systém metabolickej aktivácie. Najpoužívanejším systémom je postmitochondriálna frakcia podporená spolufaktorom (S9), pripravená z pečene hlodavcov ošetrených činidlami na indukciu enzýmov, ako je napríklad Aroclor 1254 (45) (46) alebo zmes fenobarbitonu a β-naftoflavónu (46) (47) (48) (49). Druhá kombinácia nie je v rozpore so Štokholmským dohovorom o perzistentných organických látkach (50) ani nariadením (ES) č. 850/2004 o perzistentných organických znečisťujúcich látkach (66), pričom sa pri indukovaní oxidáz so zmiešanými funkciami preukázala ako rovnako účinná ako Aroclor 1254 (46) (47) (48) (49). Frakcia S9 sa v konečnom testovacom médiu zvyčajne používa v koncentráciách v rozpätí od 1 % – 10 % (objemové percentá). Stav systému metabolickej aktivácie môže závisieť od triedy testovanej chemikálie. V niektorých prípadoch môže byť vhodné využiť viac než jednu koncentráciu S9.
|
|
17.
|
Geneticky získané bunkové línie vyjadrujúce špecifické ľudské aktivačné enzýmy alebo aktivačné enzýmy hlodavcov môžu znížiť potrebu exogénneho systému metabolickej aktivácie a možno ich použiť ako testovacie bunky. V takýchto prípadoch by mal byť výber použitých bunkových línií vedecky podložený, napr. relevantnosťou oxidáz so zmiešanými funkciami pre metabolizmus testovanej látky (51) a ich schopnosťou odozvy na známe klastogény a aneugény (pozri samostatný oddiel Kritériá prijateľnosti). Je potrebné uviesť, že testovanú látku nemusí(-ia) nevyhnutne metabolizovať vyjadrená(-é) oxidáza(-y) so zmiešanými funkciami. V tomto prípade by negatívne výsledky nesvedčili o tom, že testovaná látka nie je schopná indukovať mikrojadrá.
|
Príprava testovanej látky
|
18.
|
Chemikálie v tuhom skupenstve by sa pred ošetrením buniek mali rozpustiť v príslušných rozpúšťadlách alebo nosičoch a v prípade potreby rozriediť. Kvapalné chemikálie sa pred ošetrením môžu pridať priamo do testovacích systémov a/alebo rozriediť. Plyny alebo prchavé chemikálie by sa mali testovať pomocou vhodných úprav štandardných protokolov, ako napríklad ošetrenie v uzavretých nádobách (52) (53). Používajú sa čerstvé preparáty testovanej látky okrem prípadu, keď údaje o stabilite preukazujú prijateľnosť uskladnenia.
|
Podmienky testu
Rozpúšťadlá/nosiče
|
19.
|
Rozpúšťadlo/nosič by nemal reagovať s testovanou látkou a nemal by byť ani nekompatibilný s prežitím buniek alebo so zachovaním aktivity S9 pri použitej koncentrácii. Ak sa používajú iné ako riadne zaužívané rozpúšťadlá/nosiče (napríklad voda, bunkové kultivačné médium, dimetylsulfoxid), ich použitie by malo byť podložené údajmi označujúcimi ich kompatibilitu s testovanou látkou a absenciu genetickej toxicity. Odporúča sa, aby sa vždy, keď je to možné, najskôr zvážilo použitie vodného rozpúšťadla/nosiča.
|
Použitie cytoB ako blokátora cytokinézy
|
20.
|
Jedným z najdôležitejších faktorov pri skúške MNvit je zaistenie toho, aby hodnotené bunky prešli mitózou počas ošetrenia alebo v inkubačnej dobe po ošetrení, ak sa použije. CytoB je najbežnejšie používané činidlo na blokovanie cytokinézy, pretože tlmí vytváranie aktínu, a tým zabraňuje rozdeľovaniu dcérskych buniek po mitóze, čo vedie k vytváraniu dvojjadrových buniek (5) (54) (55). Hodnotenie mikrojadier preto možno obmedziť len na tie bunky, ktoré prešli mitózou počas ošetrenia alebo po ňom. Zároveň je možné merať účinok testovanej látky na kinetiku proliferácie buniek. CytoB sa používa na blokovanie cytokinézy pri použití ľudských lymfocytov, pretože v rámci kultúr, ako aj medzi rôznymi darcami sú dĺžky bunkových cyklov nepravidelné a nie všetky lymfocyty reagujú na PHA. Pri testovaní bunkových línií s cieľom stanoviť, či sa hodnotené bunky delili, boli použité iné metódy, ktoré sú uvedené nižšie (pozri odsek 26).
|
|
21.
|
Vhodnú koncentráciu cytoB pre každý druh buniek by malo stanoviť laboratórium a tak sa dosiahne optimálna frekvencia dvojjadrových buniek v kontrolných kultúrach s rozpúšťadlom/nosičom. Vhodná koncentrácia cytoB je zvyčajne medzi 3 až 6 μg/ml.
|
Meranie proliferácie buniek a cytotoxicity a výber expozičných koncentrácií
|
22.
|
Pri stanovovaní najvyššej koncentrácie testovanej látky je potrebné vyhýbať sa koncentráciám, ktoré majú schopnosť vyvolať falošné pozitívne odozvy, ako napríklad vytváranie nadmernej cytotoxicity, zrážanie kultivačného média a výrazné zmeny v hodnote pH alebo osmolality (39) (40) (41).
|
|
23.
|
Vykonávajú sa merania proliferácie buniek, aby sa zaistilo, že ošetrené bunky prešli mitózou počas skúšky a že ošetrenia sa vykonali na vhodných úrovniach cytotoxicity (pozri odsek 29). Cytotoxicita by sa mala stanoviť pomocou metabolickej aktivácie a bez nej v bunkách, ktoré si vyžadujú metabolickú aktiváciu s použitím relatívneho zvýšenia počtu buniek (RICC) alebo relatívneho zdvojenia populácie (RPD) (pozri vzorce v dodatku 2), okrem prípadov, keď sa použije cytoB. Ak sa použije cytoB, možno stanoviť cytotoxicitu pomocou indexu replikácie (RI) (pozri vzorec v dodatku 2).
|
|
24.
|
Ošetrenie kultúr pomocou cytoB a meranie relatívnych frekvencií jednojadrových, dvojjadrových a viacjadrových buniek v kultúre predstavuje presnú metódu kvantifikácie účinku na proliferáciu buniek a cytotoxickej alebo cytostatickej aktivity ošetrenia (5) a zabezpečuje, že sú hodnotené len tie bunky, ktoré sa delili počas ošetrenia alebo po ňom.
|
|
25.
|
V rámci štúdií využívajúcich cytoB možno kvantifikovať cytostázu/cytotoxicitu pomocou proliferačného indexu blokovania cytokinézy (CBPI) (5) (26) (56) alebo ich možno odvodiť pomocou indexu replikácie (RI) z minimálneho počtu 500 buniek na kultúru (pozri vzorce v dodatku 2). Ak sa cytoB použije na posúdenie proliferácie buniek, CBPI alebo RI by sa mali stanoviť z minimálneho počtu 500 buniek na kultúru. Tieto merania možno okrem iného použiť na odhadovanie cytotoxicity, a to porovnaním hodnôt ošetrených a kontrolných kultúr. Posúdenie iných markerov cytotoxicity (napr. zhluky, počet buniek, apoptóza, nekróza, počet metafáz) môže poskytnúť užitočné informácie.
|
|
26.
|
V rámci štúdií, ktoré nevyužívajú cytoB, je potrebné preukázať, že bunky, ktoré sa v kultúre vyhodnocujú, prešli delením počas ošetrenia testovanou látkou alebo po ošetrení, v opačnom prípade môžu vzniknúť falošné negatívne odozvy. Metódy, ktoré sa využívajú na zabezpečenie toho, aby sa hodnotili delené bunky, zahŕňajú začlenenie a následnú detekciu brómdeoxyuridínu (BrdU) na identifikáciu replikovaných buniek (57), tvorbu klonov, keď sa bunky z permanentných bunkových línií ošetrili a vyhodnotili in situ na mikroskopickom sklíčku [proliferačný index (PI)] (25) (26) (27) (28), alebo meranie relatívneho zdvojenia populácie (RPD), relatívneho zvýšenia počtu buniek (RICC) alebo iné validované metódy (16) (56) (58) (59) (pozri vzorce v dodatku 2). Posúdenie iných markerov cytotoxicity alebo cytostázy (napr. zhluky, počet buniek, apoptóza, nekróza, počet metafáz) môže poskytnúť užitočné informácie.
|
|
27.
|
Mali by sa hodnotiť minimálne tri analyzovateľné testovacie koncentrácie. Na tento účel bude možno potrebné vykonať pokus s použitím väčšieho počtu koncentrácií s tesným odstupom a analyzovať tvorbu mikrojadier v týchto koncentráciách, ktoré poskytujú vhodný rozsah cytotoxicity. Alternatívne možno vykonať predbežný test cytotoxicity s cieľom zúžiť rozsah pre konečné testovanie.
|
|
28.
|
Najvyššia koncentrácia by mala vyvolať 55 ± 5 % cytotoxicitu. Vyššie úrovne môžu ako vedľajší účinok cytotoxicity indukovať poškodenie chromozómov (60). Pri výskyte cytotoxicity by vybrané testované koncentrácie mali pokrývať rozsah od koncentrácie, ktorá vyvolá 55 ± 5 % cytotoxicitu, až po koncentrácie, ktoré vyvolajú malú alebo žiadnu toxicitu.
|
|
29.
|
Ak nie je pozorovaná žiadna cytotoxicita ani zrazenina, mala by sa najvyššia testovaná koncentrácia rovnať 0,01 M, 5 mg/mL alebo 5 μl/mL, podľa toho, ktorá je najnižšia. Medzi koncentráciami vybranými na analýzu by vo všeobecnosti mal byť odstup, ktorý nie je väčší ako 10. Pri testovaných látkach, ktoré sa vyznačujú strmou krivkou odozvy na koncentráciu, by odstup medzi koncentráciami testovaných látok mal byť menší, aby bolo možné hodnotiť aj kultúry s miernym a malým rozsahom toxicity.
|
|
30.
|
Ak je obmedzujúcim faktorom rozpustnosť, mala by byť maximálnou koncentráciou – ak nie je obmedzená cytotoxicitou – najnižšia koncentrácia, pri ktorej je v kultúrach pozorovateľné minimálne zrážanie za predpokladu, že nenarúša hodnotenie. Hodnotenie zrážania by sa malo vykonávať pomocou metód, ako je napríklad svetelná mikroskopia, pričom sa zaznamenáva pretrvávajúca zrazenina alebo zrazenina, ktorá sa objaví počas kultivácie (na konci ošetrenia).
|
Kontroly
|
31.
|
Každý pokus má obsahovať súbežné pozitívne kontroly a kontroly s rozpúšťadlom/nosičom s metabolickou aktiváciou i bez nej.
|
|
32.
|
Pozitívne kontroly sú potrebné na preukázanie schopnosti použitých buniek a testovacieho protokolu identifikovať klastogény a aneugény a potvrdiť metabolickú schopnosť preparátu S9. Pozitívne kontroly by mali využívať známe látky, ktoré indukujú tvorbu mikrojadier pri koncentráciách, pri ktorých sa očakáva malý, ale reprodukovateľný nárast na pozadí, a ktoré preukážu citlivosť testovacieho systému. Koncentrácie pozitívnej kontroly by sa mali voliť tak, aby boli účinky jasné, ale aby pozorovateľovi neodhaľovali ihneď identitu kódovaných sklíčok.
|
|
33.
|
Klastogén, ktorý si vyžaduje metabolickú aktiváciu (napríklad cyklofosfamid; benzo[a]pyrén), by sa mal použiť na preukázanie metabolickej schopnosti, ako aj schopnosti testovacieho systému detekovať klastogény. V prípade opodstatnenosti možno použiť aj iné pozitívne kontroly. Keďže niektoré pozitívne kontroly, ktoré si vyžadujú metabolickú aktiváciu, môžu byť za určitých podmienok ošetrovania alebo v určitých bunkových líniách aktívne aj bez exogénnej metabolickej aktivácie, mala by sa otestovať potreba metabolickej aktivácie a aktivita preparátu S9 vo vybranej bunkovej línii a pri vybraných koncentráciách.
|
|
34.
|
V súčasnosti nie sú známe žiadne aneugény, ktoré si vyžadujú metabolickú aktiváciu svojej genotoxickej aktivity (16). Momentálne akceptované pozitívne kontroly pre aneugenickú aktivitu sú napríklad: kolchicín a vinblastín. Iné chemikálie sa môžu použiť, ak indukujú tvorbu mikrojadier iba pomocou aneugenickej aktivity alebo predovšetkým pomocou nej. Aby sa predišlo potrebe použiť dve pozitívne kontroly (pre klastogenicitu a aneugenicitu) bez metabolickej aktivácie, môže kontrola pre aneugenicitu slúžiť ako pozitívna kontrola bez S9 a kontrola pre klastogenicitu ako test primeranosti použitého systému metabolickej aktivácie. Pozitívna kontrola pre klastogenicitu, ako aj aneugenicitu by sa mala použiť pri tých bunkách, ktoré si nevyžadujú použitie S9. Navrhované pozitívne kontroly sú uvedené v dodatku 3.
|
|
35.
|
Použitie pozitívnych kontrol týkajúcich sa chemických tried možno zvážiť, ak sú dostupné vhodné chemikálie. Všetky použité pozitívne kontroly by mali byť vhodné pre konkrétny bunkový typ a podmienky aktivácie.
|
|
36.
|
Do každého odberu by sa mali zahrnúť kontroly s rozpúšťadlom/nosičom. Zároveň by sa mali použiť aj neošetrené negatívne kontroly (bez rozpúšťadla/nosiča) okrem prípadov, keď existujú zverejnené alebo laboratórne historické kontrolné údaje, ktoré potvrdzujú, že vybrané rozpúšťadlo pri použitej koncentrácii nemá genotoxické ani iné škodlivé účinky.
|
TESTOVACÍ POSTUP
Harmonogram ošetrovania
|
37.
|
S cieľom maximalizovať pravdepodobnosť detekcie aktivity aneugénu alebo klastogénu účinkujúceho v konkrétnom štádiu bunkového cyklu je dôležité, aby počas všetkých štádií bunkového cyklu bol ošetrený dostatočný počet buniek testovanou látkou. Harmonogram ošetrovania bunkových línií a primárnych bunkových kultúr sa teda môže mierne odlišovať od harmonogramu ošetrovania lymfocytov, ktoré si vyžadujú mitogénnu stimuláciu na to, aby sa začal ich bunkový cyklus, a tieto sú uvedené v odsekoch 41 – 43 (16).
|
|
38.
|
Teoretické úvahy, ako aj zverejnené údaje (18) naznačujú, že väčšinu aneugénov a klastogénov možno určiť na základe krátkodobého ošetrenia počas 3 až 6 hodín s použitím S9 a bez použitia S9, po ktorom nasleduje odobranie testovanej látky a rastové obdobie trvajúce 1,5 – 2,0-násobok bunkových cyklov (6). Z buniek sa odoberajú vzorky v čase približne 1,5 – 2,0-násobku dĺžky bežného (t. j. neošetreného) bunkového cyklu, a to na začiatku alebo na konci ošetrenia (pozri tabuľku 1). Čas odberu vzoriek alebo čas regenerácie možno predĺžiť, ak je známe alebo ak existuje podozrenie, že testovaná látka ovplyvňuje čas bunkového cyklu (napríklad pri testovaní nukleozidových analógov).
|
|
39.
|
Kvôli možnej cytotoxicite preparátu S9 pre kultivované bunky cicavcov sa predĺžené expozičné ošetrenie počas 1,5 – 2,0-násobku bežného bunkového cyklu použije len bez S9. Pri predĺženom ošetrení možno ošetriť bunky testovanou chemikáliou s použitím alebo bez použitia cytoB. Tieto možnosti sa vzťahujú na situácie, pri ktorých môže existovať obava, že dôjde k možným interakciám medzi testovanou látkou a cytoB.
|
|
40.
|
Navrhovaný harmonogram ošetrovania buniek je uvedený v tabuľke 1. Tento všeobecný harmonogram ošetrovania možno upraviť v závislosti od stability alebo reaktivity testovanej látky alebo od konkrétnych charakteristík rastu použitých buniek. Všetky ošetrenia by sa mali začať a ukončiť počas exponenciálneho rastu buniek. Harmonogram je detailnejšie opísaný v odsekoch 41 – 47.
Tabuľka 1
Čas ošetrovania a zberu buniek pri skúške MNvit
|
Lymfocyty, primárne bunky a bunkové línie ošetrené cytoB
|
+ S9
|
ošetrujte 3 – 6 hodín s použitím S9;
odstráňte S9 a ošetrovacie médium;
pridajte čerstvé médium a cytoB;
bunky odoberte po 1,5 – 2,0-násobku bežného bunkového cyklu
|
|
– S9
krátka expozícia
|
ošetrujte 3 – 6 hodín;
odstráňte ošetrovacie médium;
pridajte čerstvé médium a cytoB;
bunky odoberte po 1,5 – 2,0-násobku bežného bunkového cyklu
|
|
– S9
predĺžená expozícia
|
možnosť A: ošetrujte 1,5 – 2,0-násobok bežného bunkového cyklu s použitím cytoB;
bunky odoberte na konci expozičného času;
možnosť B: ošetrujte 1,5 – 2,0-násobok bežného bunkového cyklu;
odstráňte testovanú látku;
pridajte čerstvé médium a cytoB;
bunky odoberte po 1,5 – 2,0-násobku bežného bunkového cyklu
|
|
Bunkové línie ošetrené cytoB
(Rovnaký postup ako pri harmonograme ošetrovania uvedenom vyššie s tou výnimkou, že sa nepridáva cytoB)
|
|
Lymfocyty, primárne bunky a bunkové línie s použitím cytoB
|
41.
|
Najúčinnejší prístup pri lymfocytoch je začať expozíciu testovanej látke 44 – 48 hodín po stimulácii PHA, keď sa synchronizácia cyklu stratí (5). V počiatočnej skúške sa bunky ošetrujú 3 až 6 hodín testovanou látkou bez S9 a s použitím S9. Ošetrovacie médium sa odstráni a nahradí čerstvým médiom, ktoré obsahuje cytoB, a bunky sa odoberú po 1,5 – 2-násobku bežného bunkového cyklu.
|
|
42.
|
Ak sú obidva počiatočné testy krátkeho ošetrenia (3 – 6 hodín) negatívne alebo nejasné, v následnom predĺženom expozičnom ošetrení sa S9 nepoužije. K dispozícií sú dve možnosti ošetrenia a sú rovnako prijateľné. Môže byť však vhodnejšie postupovať pri stimulácii lymfocytov podľa možnosti A, kde sa exponenciálny rast môže 96 hodín po stimulácii znižovať. Okrem toho by kultúry buniek do finálneho odberu vzoriek v možnosti B nemali dosiahnuť zhluk.
|
—
|
Možnosť A: Bunky sú ošetrované testovanou látkou počas 1,5 – 2,0-násobku bežného bunkového cyklu a odobrané na konci obdobia ošetrenia.
|
|
—
|
Možnosť B: Bunky sú ošetrované testovanou látkou počas 1,5 – 2,0-násobku bežného bunkového cyklu. Ošetrovacie médium sa odstráni a nahradí čerstvým médiom a bunky sa odoberú po ďalšom 1,5 – 2-násobku bežného bunkového cyklu.
|
|
|
43.
|
Primárne bunky a bunkové línie by sa mali ošetriť podobným spôsobom ako lymfocyty s výnimkou toho, že stimulácia PHA 44 – 48 hodín nie je potrebná. Iné bunky ako lymfocyty by mali byť vystavené tak, aby sa bunky v čase ukončenia štúdie stále nachádzali v log fáze rastu.
|
Bunkové línie bez cytoB
|
44.
|
Bunky by sa mali ošetriť počas 3 – 6 hodín s použitím aj bez použitia S9. Ošetrovacie médium sa odstráni a nahradí čerstvým médiom a bunky sa odoberú po 1,5 – 2-násobku bežného bunkového cyklu.
|
|
45.
|
Ak sú obidva počiatočné testy krátkeho ošetrenia (3 – 6 hodín) negatívne alebo nejasné, nasleduje predĺžené expozičné ošetrenie (bez S9). K dispozícií sú dve možnosti ošetrenia, z ktorých obidve sú rovnako prijateľné:
|
—
|
Možnosť A: Bunky sú ošetrované testovanou látkou počas 1,5 – 2,0-násobku bežného bunkového cyklu a odobrané na konci obdobia ošetrenia.
|
|
—
|
Možnosť B: Bunky sú ošetrované testovanou látkou počas 1,5 – 2,0-násobku bežného bunkového cyklu. Ošetrovacie médium sa odstráni a nahradí čerstvým médiom a bunky sa odoberú po ďalšom 1,5 – 2-násobku bežného bunkového cyklu.
|
|
|
46.
|
V monovrstvách môžu byť na konci 3 – 6-hodinového ošetrenia prítomné mitotické bunky (možno ich identifikovať ako okrúhle a oddeľujúce sa od povrchu). Keďže tieto mitotické bunky sa dajú jednoducho oddeliť, je možné, že sa pri odstraňovaní média obsahujúceho testovanú látku stratia. Preto ich pri umývaní kultúr treba opatrne odobrať a následne vrátiť ku kultúram s cieľom predísť strate buniek v mitóze a riziku pre mikrojadrá v čase zberu.
|
Počet kultúr
|
47.
|
Duplicitné kultúry by sa mali použiť pre každú koncentráciu testovanej látky a pre kultúry s rozpúšťadlom/nosičom a kultúry na negatívnu kontrolu. Ak z historických laboratórnych údajov možno preukázať minimálnu variáciu medzi duplicitnými kultúrami, môže byť prijateľné použitie jednotlivých kultúr. Ak sú použité jednotlivé kultúry, odporúča sa, aby sa analyzoval zvýšený počet koncentrácií.
|
Odber buniek a príprava sklíčok
|
48.
|
Každá kultúra sa odoberá a spracúva samostatne. Príprava buniek môže zahŕňať hypotonické ošetrenie, ale tento krok nie je potrebný, ak sa primerané šírenie buniek dosiahne iným spôsobom. Pri príprave sklíčok možno použiť rôzne techniky za predpokladu, že sa získajú vysokokvalitné bunkové preparáty. Cytoplazma bunky by sa mala zachovať, aby bola možná detekcia mikrojadier a (pri metóde blokovania cytokinézy) spoľahlivá identifikácia dvojjadrových buniek.
|
|
49.
|
Sklíčka možno zafarbiť rôznymi metódami, napríklad farbivom Giemsa alebo fluorescenčným farbivom špecifickým pre DNA (59). Použitie špecifického farbiva DNA [napríklad akridínová oranžová (61) alebo Hoechst 33258 plus pyronín-Y (62)] môže eliminovať niektoré umelo vzniknuté štruktúry buniek spojené s používaním farbiva nešpecifického pre DNA. Protilátky kinetochoru, FISH (fluorescenčná hybridizácia in situ) s pan-centromerérovými sondami DNA alebo značenie in situ pomocou primerov špecifických pre pancentroméru spolu s vhodným odfarbovacím roztokom DNA možno použiť na identifikáciu obsahov mikrojadier (chromozómy/fragmenty chromozómov), ak sú predmetom záujmu mechanické informácie o ich tvorbe (15) (16). Na rozlišovanie medzi klastogénmi a aneugénmi možno použiť aj iné metódy, ak sa ukázalo, že sú účinné.
|
Analýza
|
50.
|
Všetky sklíčka vrátane sklíčok s rozpúšťadlom/nosičom a kontrolami by sa pred mikroskopickou analýzou mali nezávisle označiť kódmi. Prípadne sa kódované vzorky môžu analyzovať pomocou validovaného systému na automatickú tokovú cytometrickú alebo vizuálnu analýzu.
|
|
51.
|
V kultúrach ošetrených cytoB by sa frekvencie mikrojadier mali analyzovať aspoň v 2 000 dvojjadrových bunkách na jednu koncentráciu (aspoň 1 000 dvojjadrových buniek na kultúru; dve kultúry na jednu koncentráciu). Ak sa použijú jednotlivé kultúry, malo by sa z jednej kultúry vyhodnotiť aspoň 2 000 dvojjadrových buniek na jednu koncentráciu. Ak je na hodnotenie k dispozícii podstatne menej ako 1 000 dvojjadrových buniek na kultúru alebo menej ako 2 000 dvojjadrových buniek, ak sa použije jediná kultúra a ak sa nezistí významné zvýšenie mikrojadier, test by sa mal zopakovať s použitím viacerých buniek alebo pri koncentráciách s menšou toxicitou, podľa toho, ktorá možnosť je vhodná. Malo by sa dbať na to, aby sa nehodnotili dvojjadrové bunky nepravidelného tvaru ani bunky, ktorých jadrá sa výrazne odlišujú veľkosťou. Malo by sa dbať aj na to, aby nedošlo k zámene dvojjadrových buniek za mnohojadrové bunky, ktorých šírenie bolo nedostatočné. Bunky obsahujúce viac ako dve hlavné jadrá by sa nemali analyzovať na prítomnosť mikrojadier, keďže základná frekvencia mikrojadra môže byť v týchto bunkách vyššia (63) (64). Hodnotenie jednojadrových buniek je prijateľné v prípade, ak testovaná látka interferuje s aktivitou cytoB.
|
|
52.
|
V bunkových líniách analyzovaných bez ošetrenia cytoB by sa mikrojadrá mali hodnotiť aspoň v 2 000 bunkách na jednu koncentráciu (aspoň v 1 000 bunkách na kultúru, dve kultúry na jednu koncentráciu). Ak sa použije iba jedna kultúra na koncentráciu, malo by sa z nej vyhodnotiť aspoň 2 000 buniek.
|
|
53.
|
Pri použití cytoB by sa mal určiť index CBPI alebo RI, aby sa posúdila proliferácia bunky (pozri dodatok 2) s použitím aspoň 500 buniek na kultúru. Pri ošetrení bez použitia cytoB je nevyhnutné poskytnúť dôkaz, že vyhodnocované bunky proliferovali, ako sa uvádza v odsekoch 24 – 27.
|
Kritériá prijateľnosti
|
54.
|
Laboratórium, ktoré navrhuje použiť skúšku MNvit opísanú v tejto testovacej metóde, by malo preukázať svoju schopnosť spoľahlivo a presne zisťovať chemikálie so známou aneugénnou a klastogénnou aktivitou, či už s metabolickou aktiváciou, alebo bez nej, ako aj známe negatívne chemikálie pomocou referenčných chemikálií uvedených v dodatku 3. Ako dôkaz schopnosti správne vykonať túto testovaciu metódu by laboratórium malo poskytnúť dôkaz, že bunky, v ktorých sa hodnotí tvorba mikrojadra, ukončili jedno jadrové delenie, ak sa test vykonáva bez použitia cytoB.
|
|
55.
|
Chemikálie uvedené v dodatku 3 sa odporúčajú na použitie ako referenčné chemikálie. Náhradné alebo doplňujúce chemikálie možno zahrnúť, ak je ich aktivita známa, ak vytvárajú mikrojadrá rovnakými mechanizmami pôsobenia a ak sa ukážu ako relevantné v porovnaní s chemikáliami, ktoré sa budú testovať postupom MNvit. Odôvodnenie by mohlo zahŕňať validačnú štúdiu, ktorá zapája veľké množstvo látok alebo sa sústredí na užšie spektrum založené na štúdii chemickej triedy testovanej látky alebo mechanizmu poškodenia.
|
|
56.
|
Kontrolné kultúry s rozpúšťadlom/nosičom a neošetrené kultúry by mali mať z hľadiska reprodukovateľnosti a konzistentnosti nízke frekvencie mikrojadier (zvyčajne 5 – 25 mikrojadier/1 000 buniek, ktoré patria do druhov buniek uvedených v odseku 11). Ostatné druhy buniek môžu mať rôzny rozsah odoziev, čo by sa malo určiť pri validácii ich vhodnosti na použitie v skúške MNvit. Údaje z negatívnej kontroly, kontroly s rozpúšťadlom a z pozitívnej kontroly by sa mali použiť na stanovenie historických kontrolných rozsahov. Tieto hodnoty by sa mali použiť pri rozhodovaní o primeranosti súbežnej pozitívnej/negatívnej kontroly v pokuse.
|
|
57.
|
Ak sa navrhnú menšie zmeny protokolu skúšky (napr. použitie automatických namiesto manuálnych techník hodnotenia či použitie nového druhu bunky), je potrebné preukázať účinnosť zmeny skôr, ako takto upravený protokol možno považovať za prijateľný na použitie. Preukázanie účinnosti zahŕňa preukázanie schopnosti zisťovať hlavné mechanizmy chromozómového zlomu, nadbytku alebo straty a dosiahnuť vhodné pozitívne a negatívne výsledky pre triedu jednotlivej látky alebo pre celý rad látok, ktoré sa majú testovať.
|
ÚDAJE A PODÁVANIE SPRÁV
Spracovanie výsledkov
|
58.
|
Ak sa použije metóda blokovania cytokinézy, pri hodnotení indukcie mikrojadier sa používajú iba frekvencie dvojjadrových buniek s mikrojadrami (bez ohľadu na počet mikrojadier v bunke). Hodnotenie počtu buniek s jedným mikrojadrom, s dvoma alebo viacerými mikrojadrami by mohlo poskytnúť užitočné informácie, ale nie je povinné.
|
|
59.
|
Mali by sa určiť súbežné opatrenia v súvislosti s cytotoxicitou a/alebo cytostázou pre všetky ošetrené kultúry a kontrolné kultúry s rozpúšťadlom/nosičom (58). Pri všetkých ošetrených a kontrolných kultúrach by sa mali vypočítať indexy CBPI alebo RI, ktoré merajú oneskorenie bunkového cyklu pri použití metódy blokovania cytokinézy. V prípade, že sa nepoužíva cytoB, mali by sa použiť RDP alebo RICC, alebo index PI (pozri dodatok 2).
|
|
60.
|
Mali by sa uvádzať údaje o jednotlivých kultúrach. Okrem toho by sa všetky údaje mali uvádzať v tabuľkovej podobe.
|
|
61.
|
Chemikálie, ktoré indukujú mikrojadrá v skúške MNvit, tak môžu urobiť, pretože indukujú chromozómový zlom, stratu chromozómu alebo ich kombináciu. Ďalšiu analýzu, ktorá využíva protilátky kinetochoru, sondy in situ špecifické pre centroméry alebo iné metódy, možno použiť na určenie toho, či mechanizmus indukcie mikrojadra je následkom klastogénnej a/alebo aneugénnej aktivity.
|
Vyhodnocovanie a interpretácia výsledkov
|
62.
|
Neexistuje požiadavka na overenie jasnej pozitívnej alebo negatívnej odozvy ďalším testovaním. Nejasné výsledky možno objasniť analýzou ďalších 1 000 buniek zo všetkých kultúr s cieľom zabrániť strate slepých testov. Ak sa týmto prístupom výsledok neobjasní, treba vykonať ďalšie testy. Modifikácia parametrov štúdie z dôvodu rozšírenia alebo zúženia rozsahu podmienok by sa mala zvážiť pri následných pokusoch, ak je to vhodné. Parametre štúdie, ktoré by sa mohli modifikovať, zahŕňajú odstup medzi testovanými koncentráciami, načasovanie ošetrenia a zber buniek a/alebo podmienky metabolickej aktivácie.
|
|
63.
|
Na stanovenie pozitívneho výsledku existuje niekoľko kritérií, ako napríklad nárast súvisiaci s koncentráciou alebo štatisticky významné zvýšenie počtu buniek obsahujúcich mikrojadrá. Najskôr sa zvažuje biologická relevantnosť výsledkov. Zváženie toho, či pozorované hodnoty sú v rozsahu historickej kontroly alebo mimo neho, môže poskytnúť usmernenie pri hodnotení biologického významu odozvy. Ako pomôcky pri hodnotení výsledkov testov možno používať primerané štatistické metódy (65). Výsledky štatistických testov by sa však mali hodnotiť vzhľadom na vzťah medzi dávkou a odozvou. Mala by sa zohľadniť aj reprodukovateľnosť a historické údaje.
|
|
64.
|
Hoci väčšina pokusov poskytne jasne pozitívne alebo negatívne výsledky, v niektorých prípadoch súbor údajov vopred vylúči jednoznačné posúdenie aktivity testovanej látky. Tieto nejasné alebo spochybniteľné odozvy môžu nastať bez ohľadu na počet opakovaní pokusu.
|
|
65.
|
Pozitívne výsledky skúšky MNvit naznačujú, že testovaná látka vyvoláva chromozómový zlom alebo stratu chromozómu v kultivovaných bunkách cicavcov. Negatívne výsledky naznačujú, že testovaná látka nevyvoláva v testovacích podmienkach chromozómové zlomy a/ani ich nadbytok či stratu v kultivovaných bunkách cicavcov.
|
Správa o teste
|
66.
|
Správa o teste by mala obsahovať aspoň tieto informácie, ak sú relevantné pre uskutočnenie štúdie:
|
|
Testovaná chemikália:
|
—
|
identifikačné údaje a registračné číslo CAS (Chemical Abstract Services) a číslo EC,
|
|
—
|
fyzikálna povaha a čistota,
|
|
—
|
fyzikálno-chemické vlastnosti relevantné pre uskutočnenie štúdie,
|
|
—
|
reaktivita testovanej chemikálie v kultivačnom médiu s rozpúšťadlom/nosičom alebo v bunkovom kultivačnom médiu.
|
|
|
|
Rozpúšťadlo/nosič:
|
—
|
zdôvodnenie výberu rozpúšťadla/nosiča,
|
|
—
|
rozpustnosť a stabilita testovanej látky v rozpúšťadle/nosiči.
|
|
|
|
Bunky:
|
—
|
typ a zdroj použitých buniek,
|
|
—
|
vhodnosť použitého typu bunky,
|
|
—
|
neprítomnosť mykoplazmy, ak je to vhodné,
|
|
—
|
informácie o dĺžke bunkového cyklu, násobení času alebo proliferačnom indexe,
|
|
—
|
pri použití lymfocytov pohlavie, vek a počet darcov krvi, ak je to vhodné,
|
|
—
|
pri použití lymfocytov informácia o tom, či je vystavená celá krv, alebo oddelené lymfocyty,
|
|
—
|
počet prechodov, ak je to vhodné,
|
|
—
|
metódy udržiavania bunkových kultúr, ak je to vhodné,
|
|
—
|
modálny počet chromozómov,
|
|
—
|
trvanie bežného bunkového cyklu (negatívna kontrola).
|
|
|
|
Podmienky testovania:
|
—
|
identita látky blokujúcej cytokinézu (napr. cytoB), ak je použitá, jej koncentrácia a trvanie expozície bunky,
|
|
—
|
zdôvodnenie výberu koncentrácií a počtu kultúr vrátane údajov o cytotoxicite a obmedzeniach rozpustnosti, ak sú dostupné,
|
|
—
|
skladba média, koncentrácia CO2, ak je to vhodné,
|
|
—
|
koncentrácie testovanej látky,
|
|
—
|
koncentrácia (a/alebo objem) nosiča a pridanej testovanej látky,
|
|
—
|
inkubačná teplota a inkubačná doba,
|
|
—
|
hustota buniek pri zapustení, ak je to vhodné,
|
|
—
|
typ a zloženie systému metabolickej aktivácie vrátane kritérií prijateľnosti,
|
|
—
|
chemikálie na pozitívne kontroly a negatívne kontroly,
|
|
—
|
použité metódy prípravy sklíčok a metóda farbenia,
|
|
—
|
kritériá na identifikáciu mikrojadier,
|
|
—
|
počet analyzovaných buniek,
|
|
—
|
metódy merania cytoxicity,
|
|
—
|
všetky doplňujúce informácie týkajúce sa cytotoxicity,
|
|
—
|
kritériá hodnotenia štúdií ako pozitívnych, negatívnych alebo nejasných,
|
|
—
|
metóda(-y) použitej štatistickej analýzy,
|
|
—
|
metódy, ako napríklad použitie protilátky kinetochoru s cieľom charakterizovať, či mikrojadrá obsahujú celé chromozómy, alebo fragmenty chromozómov, ak je to vhodné.
|
|
|
|
Výsledky:
|
—
|
meranie použitej cytotoxicity, napr. CBPI alebo RI, v prípade použitia metódy blokovania cytokinézy, RICC, RPD alebo PI, keď sa metódy blokovania cytokinézy nepoužívajú, ďalšie pozorovania, ak je to vhodné, napr. zhluk buniek, apoptóza, nekróza, počítanie metafáz, frekvencia dvojjadrových buniek,
|
|
—
|
údaje o hodnotách pH a osmolality ošetrovacieho média, ak sú stanovené,
|
|
—
|
definovanie buniek prijateľných na analýzu,
|
|
—
|
distribúcia jednojadrových, dvojjadrových a viacjadrových buniek pri použití metódy blokovania cytokinézy,
|
|
—
|
počet buniek s mikrojadrami uvedený oddelene pre každú ošetrenú a kontrolnú kultúru a stanovenie, či pochádzajú z dvojjadrových, alebo jednojadrových buniek, ak je to potrebné,
|
|
—
|
vzťah medzi koncentráciou a odozvou, ak ho možno určiť,
|
|
—
|
údaje o chemikáliách na súbežné negatívne (rozpúšťadlo/nosič) a pozitívne kontroly (koncentrácie a rozpúšťadlá),
|
|
—
|
historické údaje o chemikáliách na negatívne (rozpúšťadlo/nosič) a pozitívne kontroly obsahujúce rozsahy, stredné hodnoty a štandardnú odchýlku a stupeň spoľahlivosti (napr. 95 %),
|
|
—
|
štatistická analýza, p-hodnoty, ak existujú.
|
|
|
LITERATÚRA
|
1.
|
Kirsch-Volders, M. (1997), Towards a validation of the micronucleus test. Mutation Res., 392, 1 – 4.
|
|
2.
|
Parry, J. M. and Sors, A. (1993), The detection and assessment of the aneugenic potential of environmental chemicals: the European Community aneuploidy project, Mutation Res., 287, 3 – 15.
|
|
3.
|
Fenech, M. and Morley, A. A. (1985), Solutions to the kinetic problem in the micronucleus assay, Cytobios., 43, 233 – 246.
|
|
4.
|
Kirsch-Volders, M., Sofuni, T., Aardema, M., Albertini, S., Eastmond, D., Fenech, M., Ishidate, M. Jr, Lorge, E., Norppa, H., Surralles, J., von der Hude, W. and Wakata, A. (2000), Report from the In Vitro Micronucleus Assay Working Group, Environ. Mol. Mutagen., 35, 167 – 172.
|
|
5.
|
Fenech, M. (2007), Cytokinesis-block micronucleus cytome assay, Nature Protocols, 2(5), 1084 – 1104.
|
|
6.
|
Fenech, M. and Morley, A. A. (1986), Cytokinesis-block micronucleus method in human lymphocytes: effect of in-vivo ageing and low dose X-irradiation, Mutation Res., 161, 193 – 198.
|
|
7.
|
Eastmond, D. A. and Tucker, J. D. (1989), Identification of aneuploidy-inducing agents using cytokinesis-blocked human lymphocytes and an antikinetochore antibody, Environ. Mol. Mutagen., 13, 34 – 43.
|
|
8.
|
Eastmond, D. A. and Pinkel, D. (1990), Detection of aneuploidy and aneuploidy-inducing agents in human lymphocytes using fluorescence in-situ hybridisation with chromosome-specific DNA probes, Mutation Res., 234, 9 – 20.
|
|
9.
|
Miller, B. M., Zitzelsberger, H. F., Weier, H. U. and Adler, I. D. (1991), Classification of micronuclei in murine erythrocytes: immunofluorescent staining using CREST antibodies compared to in situ hybridization with biotinylated gamma satellite DNA, Mutagenesi., 6, 297 – 302.
|
|
10.
|
Farooqi, Z., Darroudi, F. and Natarajan, A. T. (1993), The use of fluorescence in-situ hybridisation for the detection of aneugens in cytokinesis-blocked mouse splenocytes, Mutagenesi., 8, 329 – 334.
|
|
11.
|
Migliore, L., Bocciardi, R., Macri, C. and Lo Jacono, F. (1993), Cytogenetic damage induced in human lymphocytes by four vanadium compounds and micronucleus analysis by fluorescence in situ hybridization with a centromeric probe, Mutation Res., 319, 205 – 213.
|
|
12.
|
Norppa, H., Renzi, L. and Lindholm, C. (1993), Detection of whole chromosomes in micronuclei of cytokinesis-blocked human lymphocytes by antikinetochore staining and in situ hybridization, Mutagenesi., 8, 519 – 525.
|
|
13.
|
Eastmond, D. A, Rupa, D. S. and Hasegawa, L. S. (1994), Detection of hyperdiploidy and chromosome breakage in interphase human lymphocytes following exposure to the benzene metabolite hydroquinone using multicolor fluorescence in situ hybridization with DNA probes, Mutation Res., 322, 9 – 20.
|
|
14.
|
Marshall, R. R., Murphy, M., Kirkland, D. J. and Bentley, K. S. (1996), Fluorescence in situ hybridisation (FISH) with chromosome-specific centromeric probes: a sensitive method to detect aneuploidy, Mutation Res., 372, 233 – 245.
|
|
15.
|
Zijno, P., Leopardi, F., Marcon, R. and Crebelli, R. (1996), Analysis of chromosome segregation by means of fluorescence in situ hybridization: application to cytokinesis-blocked human lymphocytes, Mutation Res., 372, 211 – 219.
|
|
16.
|
Kirsch-Volders, M., Sofuni, T., Aardema, M., Albertini, S., Eastmond, D., Fenech, M., Ishidate Jr., M., Lorge, E., Norppa, H., Surrallés, J., von der Hude, W. and Wakata, A. (2003), Report from the in vitro micronucleus assay working group. Mutation Res., 540, 153 – 163.
|
|
17.
|
OECD (1997), In Vitro Mammalian Chromosome Aberration Tes., Test Guideline No 473, OECD Guidelines for Testing of Chemicals, OECD, Paris. K dispozícii na stránke: [www.oecd.org/env/testguidelines].
|
|
18.
|
Lorge, E., Thybaud, V., Aardema, M. J., Oliver, J., Wakata, A., Lorenzon G. and Marzin, D. (2006), SFTG International collaborative Study on in vitro micronucleus test. I. General conditions and overall conclusions of the study, Mutation Res., 607, 13 – 36.
|
|
19.
|
Clare, G., Lorenzon, G., Akhurst, L. C., Marzin, D., van Delft, J., Montero, R., Botta, A., Bertens, A., Cinelli, S., Thybaud, V. and Lorge, E. (2006), SFTG International collaborative study on the in vitro micronucleus test. II. Using human lymphocytes, Mutation Res., 607, 37 – 60.
|
|
20.
|
Aardema, M. J., Snyder, R. D., Spicer, C., Divi, K., Morita, T., Mauthe, R. J., Gibson, D. P., Soelter, S., Curry, P. T., Thybaud, V., Lorenzon, G., Marzin, D. and Lorge, E. (2006), SFTG International collaborative study on the in vitro micronucleus test, III. Using CHO cells, Mutation Res., 607, 61 – 87.
|
|
21.
|
Wakata, A., Matsuoka, A., Yamakage, K., Yoshida, J., Kubo, K., Kobayashi, K., Senjyu, N., Itoh, S., Miyajima, H., Hamada, S., Nishida, S., Araki, H., Yamamura, E., Matsui, A., Thybaud, V., Lorenzon, G., Marzin, D. and Lorge, E. (2006), SFTG International collaborative study on the in vitro micronucleus test, IV. Using CHO/IU cells, Mutation Res., 607, 88 – 124.
|
|
22.
|
Oliver, J., Meunier, J.-R., Awogi, T., Elhajouji, A., Ouldelhkim, M.-C., Bichet, N., Thybaud, V., Lorenzon, G., Marzin, D. and Lorge, E. (2006), SFTG International collaborative study on the in vitro micronucleus test, V. Using L5178Y cells, Mutation Res., 607, 125 – 152.
|
|
23.
|
Albertini, S., Miller, B., Chetelat, A. A. and Locher, F. (1997), Detailed data on in vitro MNT and in vitro CA: industrial experience, Mutation Res., 392, 187 – 208.
|
|
24.
|
Miller, B., Albertini, S., Locher, F., Thybaud, V. and Lorge, E. (1997), Comparative evaluation of the in vitro micronucleus test and the in vitro chromosome aberration test: industrial experience, Mutation Res., 392, 45 – 59.
|
|
25.
|
Miller, B., Potter-Locher, F., Seelbach, A., Stopper, H., Utesch, D. and Madle, S. (1998), Evaluation of the in vitro micronucleus test as an alternative to the in vitro chromosomal aberration assay: position of the GUM Working Group on the in vitro micronucleus test. Gesellschaft fur Umwelt-Mutations-forschung, Mutation Res., 410, 81 – 116.
|
|
26.
|
Kalweit, S., Utesch, U., von der Hude, W. and Madle, S. (1999), Chemically induced micronucleus formation in V79 cells – comparison of three different test approaches, Mutation Res. 439, 183 – 190.
|
|
27.
|
Kersten, B., Zhang, J., Brendler Schwaab, S. Y., Kasper, P. and Müller, L. (1999), The application of the micronucleus test in Chinese hamster V79 cells to detect drug-induced photogenotoxicity, Mutation Res. 445, 55 – 71.
|
|
28.
|
von der Hude, W., Kalweit, S., Engelhardt, G., McKiernan, S., Kasper, P., Slacik-Erben, R., Miltenburger, H. G., Honarvar, N., Fahrig, R., Gorlitz, B., Albertini, S., Kirchner, S., Utesch, D., Potter-Locher, F., Stopper, H. and Madle, S. (2000), In vitro micronucleus assay with Chinese hamster V79 cells – results of a collaborative study with in situ exposure to 26 chemical substances, Mutation Res., 468, 137 – 163.
|
|
29.
|
Garriott, M. L., Phelps, J. B. and Hoffman, W. P. (2002), A protocol for the in vitro micronucleus test, I. Contributions to the development of a protocol suitable for regulatory submissions from an examination of 16 chemicals with different mechanisms of action and different levels of activity, Mutation Res., 517, 123 – 134.
|
|
30.
|
Matsushima, T., Hayashi, M., Matsuoka, A., Ishidate, M. Jr., Miura, K. F., Shimizu, H., Suzuki, Y., Morimoto, K., Ogura, H., Mure, K., Koshi, K. and Sofuni, T. (1999), Validation study of the in vitro micronucleus test in a Chinese hamster lung cell line (CHL/IU), Mutagenesi., 14, 569 – 580.
|
|
31.
|
Elhajouji, A., and Lorge, E. (2006), Special Issue: SFTG International collaborative study on in vitro micronucleus test, Mutation Res., 607, 1 – 152.
|
|
32.
|
ECVAM (2006), Statement by the European Centre for the Validation of Alternative Methods (ECVAM) Scientific Advisory Committee (ESAC) on the scientific validity of the in vitro micronucleus test as an alternative to the in vitro chromosome aberration assay for genotoxicity testing. 25. stretnutie ESAC, 16. a 17. novembra 2006. K dispozícii na stránke: [http://ecvam.jrc.it/index.htm].
|
|
33.
|
ESAC (2006), ECVAM Scientific Advisory Committee (ESAC) Peer Review, Retrospective Validation of the In Vitro Micronucleus Test, Summary and Conclusions of the Peer Review Panel. K dispozícii na stránke: [http://ecvam.jrc.it/index.htm].
|
|
34.
|
Corvi, R., Albertini, S., Hartung, T., Hoffmann, S., Maurici, D., Pfuhler, S, van Benthem, J., Vanparys P. (2008), ECVAM Retrospective Validation of in vitro Micronucleus Test (MNT), Mutagenesi., 23, 271 – 283.
|
|
35.
|
Zhang, L. S., Honma, M., Hayashi, M., Suzuki, T., Matsuoka, A. and Sofuni, T. (1995), A comparative study of TK6 human lymphoblastoid and L5178Y mouse lymphoma cell lines in the in vitro micronucleus test, Mutation Res., 347, 105 – 115.
|
|
36.
|
Ehrlich, V., Darroudi, F., Uhl, M., Steinkellner, S., Zsivkovits, M. and Knasmeuller, S. (2002), Fumonisin B1 is genotoxic in human derived hepatoma (HepG2) cells, Mutagenesi., 17, 257 – 260.
|
|
37.
|
Knasmüller, S., Mersch-Sundermann, V., Kevekordes, S., Darroudi, F., Huber, W. W., Hoelzl, C., Bichler, J. and Majer, B. J. (2004), Use of human-derived liver cell lines for the detection of environmental and dietary genotoxicants; current state of knowledge, Toxicol., 198, 315 – 328.
|
|
38.
|
Gibson, D. P., Brauninger, R., Shaffi, H. S., Kerckaert, G. A., LeBoeuf, R. A., Isfort, R. J. and Aardema, M. J. (1997), Induction of micronuclei in Syrian hamster embryo cells: comparison to results in the SHE cell transformation assay for National Toxicology Program test chemicals, Mutation Res., 392, 61 – 70.
|
|
39.
|
Scott, D., Galloway, S. M., Marshall, R. R., Ishidate, M. Jr., Brusick, D., Ashby, J. and Myhr, B. C. (1991), International Commission for Protection Against Environmental Mutagens and Carcinogens, Genotoxicity under extreme culture conditions. A report from ICPEMC Task Group 9, Mutation Res., 257, 147 – 205.
|
|
40.
|
Morita, T., Nagaki, T., Fukuda, I. and Okumura, K. (1992), Clastogenicity of low pH to various cultured mammalian cells, Mutation Res., 268, 297 – 305.
|
|
41.
|
Brusick, D. (1986), Genotoxic effects in cultured mammalian cells produced by low pH treatment conditions and increased ion concentrations, Environ. Mutagen., 8, 789 – 886.
|
|
42.
|
Fenech, M. and Morley, A. A. (1985), Measurement of micronuclei in lymphocytes, Mutation Res., 147, 29 – 36.
|
|
43.
|
Fenech, M. (1997), The advantages and disadvantages of cytokinesis-blood micronucleus method, Mutation Res., 392, 11 – 18.
|
|
44.
|
Bonassi, S., Fenech, M., Lando, C., Lin, Y. P., Ceppi, M., Chang, W. P., Holland, N., Kirsch-Volders, M., Zeiger, E., Ban, S., Barale, R., Bigatti, M. P., Bolognesi, C., Jia, C., Di Giorgio, M., Ferguson, L. R., Fucic, A., Lima, O. G., Hrelia, P., Krishnaja, A. P., Lee, T. K., Migliore, L., Mikhalevich, L., Mirkova, E., Mosesso, P., Muller, W. U., Odagiri, Y., Scarffi, M. R., Szabova, E., Vorobtsova, I., Vral, A. and Zijno, A. (2001), HUman MicroNucleus Project: international database comparison for results with the cytokinesis-block micronucleus assay in human lymphocytes, I. Effect of laboratory protocol, scoring criteria and host factors on the frequency of micronuclei, Environ. Mol. Mutagen. 37, 31 – 45.
|
|
45.
|
Maron, D. M. and Ames, B. N. (1983), Revised methods for the Salmonella mutagenicity test, Mutation Res., 113, 173 – 215.
|
|
46.
|
Ong, T.-M., Mukhtar, M., Wolf, C. R. and Zeiger, E. (1980), Differential effects of cytochrome P450-inducers on promutagen activation capabilities and enzymatic activities of S-9 from rat liver, J. Environ. Pathol. Toxicol., 4, 55 – 65.
|
|
47.
|
Elliott, B. M., Combes, R. D., Elcombe, C. R., Gatehouse, D. G., Gibson, G. G., Mackay, J. M. and Wolf, R. C. (1992), Alternatives to Aroclor 1254-induced S9 in in-vitro genotoxicity assays. Mutagenesis, 7, 175 – 177.
|
|
48.
|
Matsushima, T., Sawamura, M., Hara, K. and Sugimura, T. (1976), A safe substitute for Polychlorinated Biphenyls as an Inducer of Metabolic Activation Systems, In: de Serres, F. J., Fouts, J. R., Bend, J. R. and Philpot, R. M. (eds), In Vitro Metabolic Activation in Mutagenesis Testing, Elsevier, North-Holland, s. 85 – 88.
|
|
49.
|
Johnson, T. E., Umbenhauer, D. R. and Galloway, S. M. (1996), Human liver S-9 metabolic activation: proficiency in cytogenetic assays and comparison with phenobarbital/beta-naphthoflavone or Aroclor 1254 induced rat S-9, Environ. Mol. Mutagen., 28, 51 – 59.
|
|
50.
|
UNEP (2001), Štokholmský dohovor o perzistentných organických látkach, Program OSN pre životné prostredie (UNEP). K dispozícii na stránke: [http://www.pops.int/].
|
|
51.
|
Doherty, A. T., Ellard, S., Parry, E. M. and Parry, J. M. (1996), An investigation into the activation and deactivation of chlorinated hydrocarbons to genotoxins in metabolically competent human cells, Mutagenesi., 11, 247 – 274.
|
|
52.
|
Krahn, D. F., Barsky, F. C. and McCooey, K. T. (1982), CHO/HGPRT Mutation Assay: Evaluation of Gases and Volatile Liquids, In: Tice, R. R., Costa, D. L. and Schaich, K. M. (eds), Genotoxic Effects of Airborne Agents. New York, Plenum, s. 91 – 103.
|
|
53.
|
Zamora, P. O., Benson, J. M., Li, A. P. and Brooks, A. L. (1983), Evaluation of an exposure system using cells grown on collagen gels for detecting highly volatile mutagens in the CHO/HGPRT mutation assay, Environ. Mutagenesis 5, 795 – 801.
|
|
54.
|
Fenech, M. (1993), The cytokinesis-block micronucleus technique: a detailed description of the method and its application to genotoxicity studies in human populations, Mutation Res., 285, 35 – 44.
|
|
55.
|
Phelps, J. B., Garriott, M. L., and Hoffman, W. P. (2002), A protocol for the in vitro micronucleus test. II. Contributions to the validation of a protocol suitable for regulatory submissions from an examination of 10 chemicals with different mechanisms of action and different levels of activity, Mutation Res., 521, 103 – 112.
|
|
56.
|
Kirsch-Volders, M., Sofuni, T., Aardema, M., Albertini, S., Eastmond, D., Fenech, M., Ishidate, M. Jr., Kirchner, S., Lorge, E., Morita, T., Norppa, H., Surralles, J., Vanhauwaert, A. and Wakata, A. (2004), Corrigendum to ‚Report from the in vitro micronucleus assay working group‘, Mutation Res., 564, 97 – 100.
|
|
57.
|
Pincu, M., Bass, D. and Norman, A. (1984), An improved micronuclear assay in lymphocytes, Mutation Res., 139, 61 – 65.
|
|
58.
|
Lorge, E., Hayashi, M., Albertini, S. and Kirkland, D. (2008), Comparison of different methods for an accurate assessment of cytotoxicity in the in vitro micronucleus test. I. Theoretical aspects, Mutation Res., 655, 1 – 3.
|
|
59.
|
Surralles, J., Xamena, N., Creus, A., Catalan, J., Norppa, H. and Marcos, R. (1995), Induction of micronuclei by five pyrethroid insecticides in whole-blood and isolated human lymphocyte cultures, Mutation Res., 341, 169 – 184.
|
|
60.
|
Galloway, S. (2000), Cytotoxicity and chromosome aberrations in vitro: Experience in industry and the case for an upper limit on toxicity in the aberration assay, Environ. Molec. Mutagenesis 35, 191 – 201.
|
|
61.
|
Hayashi, M., Sofuni, T., and Ishidate, M. Jr. (1983), An Application of Acridine Orange Fluorescent Staining to the Micronucleus Test, Mutation Res., 120, 241 – 247.
|
|
62.
|
MacGregor, J. T., Wehr, C. M., and Langlois, R. G. (1983), A Simple Fluorescent Staining Procedure for Micronuclei and RNA in Erythrocytes Using Hoechst 33258 and Pyronin Y, Mutation Res., 120, 269 – 275.
|
|
63.
|
Hayashi, M., Sofuni, T. and Ishidate, M. Jr. (1983), An application of acridine orange fluorescent staining to the micronucleus test, Mutation Res., 120, 241 – 247.
|
|
64.
|
Fenech, M., Chang, W. P., Kirsch-Volders, M., Holland, N., Bonassi, S. and Zeiger, E. (2003), HUMN project: detailed description of the scoring criteria for the cytokinesis-block micronucleus assay using isolated human lymphocyte cultures, Mutation Res., 534, 65 – 75.
|
|
65.
|
Hoffman, W. P., Garriott, M. L. and Lee, C. (2003), In vitro micronucleus test, In: Encyclopedia of Biopharmaceutical Statistics, Second edition. S. Chow (ed.), Marcel Dekker, Inc. New York, NY, s. 463 – 467.
|
|
66.
|
Nariadenie Európskeho parlamentu a Rady (ES) č. 850/2004 z 29. apríla 2004 o perzistentných organických znečisťujúcich látkach, ktorým sa mení a dopĺňa smernica 79/117/EHS (Ú. v. EÚ L 229, 30.4.2004, s. 5).
|
Dodatok 1
Vymedzenie pojmov
Aneugén: každá látka alebo proces, ktorý vzájomne pôsobí so zložkami cyklu mitotického a meiotického delenia buniek, a tým vedie k aneuploidii v bunkách alebo organizmoch.
Aneuploidia: akákoľvek odchýlka od bežného diploidného (alebo haploidného) počtu chromozómov vo forme jedného alebo viacerých chromozómov, ktoré netvoria celý(-é) súbor(-y) chromozómov (polyploidia).
Apoptóza: programovaná bunková smrť charakterizovaná sériou krokov, ktoré vedú k rozpadu buniek na častice spojené membránou, ktoré sú následne pohltené fagocytózou alebo uvoľnené.
Proliferácia buniek: zvýšenie počtu buniek v dôsledku mitotického delenia bunky.
Centroméra: oblasť chromozómu DNA, kde sa spájajú obe chromatídy a kde sú vedľa seba viazané oba kinetochory.
Klastogén: akákoľvek látka alebo proces, ktoré spôsobujú štrukturálne chromozómové odchýlky v populáciách buniek alebo organizmov.
Cytokinéza: proces delenia bunky, ktorý nasleduje ihneď po mitóze a v ktorom vznikajú dve dcérske bunky – každá s jedným jadrom.
Proliferačný index blokovania cytokinézy (CBPI): pomer druhého delenia buniek v ošetrenej populácii vo vzťahu k neošetrenej kontrole (pozri vzorec v dodatku 2).
Cytostáza: inhibícia rastu buniek (pozri vzorec v dodatku 2).
Cytotoxicita: škodlivé účinky na štruktúru alebo funkciu bunky, ktoré postupne spôsobia jej smrť.
Genotoxický: všeobecný výraz, ktorý zahŕňa všetky typy poškodenia DNA alebo chromozómov vrátane zlomov, aduktov, zmien usporiadania, mutácií, chromozómových odchýlok a aneuploidie. Nie všetky typy genotoxických účinkov majú za následok mutácie alebo trvalé poškodenie chromozómu.
Bunky v interfáze: bunky, ktoré nie sú v štádiu mitotického delenia.
Kinetochor: štruktúra obsahujúca bielkoviny, ktorá sa zhromažďuje na centromére chromozómu, na ktorú sa počas delenia buniek viažu vlákna deliaceho vretienka umožňujúce riadny pohyb dcérskych chromozómov k pólom dcérskych buniek.
Mikrojadrá: samostatné malé jadrá doplňujúce hlavné jadrá buniek, ktoré sa vytvárajú počas telofázy mitózy alebo meiózy zo zaostávajúcich chromozómových fragmentov alebo celých chromozómov.
Mitóza: rozdelenie bunkového jadra, ktoré sa zvyčajne delí na profázu, prometafázu, metafázu, anafázu a telofázu.
Mitotický index: pomer buniek v metafáze delený celkovým počtom buniek pozorovaných v populácii buniek; náznak stupňa proliferácie buniek tejto populácie.
Mutagénny: produkujúci dedičnú zmenu série(-í) bázových párov DNA v génoch alebo štruktúry chromozómov (chromozómové odchýlky).
Nerozpojenie: chyba pri rozdeľovaní spárovaných chromatíd, ktorá spôsobí, že vznikajúce dcérske bunky sa riadne neoddelia, následkom čoho vznikajú dcérske bunky s abnormálnym počtom chromozómov.
Polyploidia: početné chromozómové odchýlky v bunkách alebo organizmoch, ktoré zahŕňajú celý(-é) súbor(-y) chromozómov na rozdiel od jednotlivého chromozómu alebo chromozómov (aneuploidia).
Proliferačný index (PI): metóda merania cytotoxicity, pri ktorej sa nepoužíva cytoB (pozri vzorec v dodatku 2).
Relatívne zvýšenie počtu buniek (RICC): metóda merania cytotoxicity, pri ktorej sa nepoužíva cytoB (pozri vzorec v dodatku 2).
Relatívne zdvojenie populácie (RPD): metóda merania cytotoxicity, pri ktorej sa nepoužíva cytoB (pozri vzorec v dodatku 2).
Index replikácie (RI): pomer cyklov delenia buniek, ktoré boli dokončené v ošetrenej kultúre, vo vzťahu k neošetrenej kontrole počas obdobia expozície a regenerácie (pozri vzorec v dodatku 2).
Testovaná chemikália (tiež sa nazýva testovaná látka): Akákoľvek látka alebo zmes testovaná pomocou tejto testovacej metódy.
Dodatok 2
Vzorce na hodnotenie cytotoxicity
|
1.
|
Pri použití cytoB by hodnotenie cytotoxicity malo byť založené na proliferačnom indexe blokovania cytokinézy (CBPI) alebo na indexe replikácie (RI) (16) (58). CBPI naznačuje priemerný počet bunkových cyklov na bunku počas obdobia expozície cytoB a môže sa použiť na výpočet proliferácie buniek. RI indikuje relatívny počet jadier v ošetrených kultúrach v porovnaní s kontrolnými kultúrami a možno ho použiť na percentuálny výpočet cytostázy:
Cytostáza % = 100 – 100{(CBPIT – 1) ÷ (CBPIC – 1)}
pričom:
|
T
|
=
|
kultúra ošetrená testovanou chemikáliou
|
|
C
|
=
|
kontrolná kultúra s nosičom,
|
kde:

Preto ak CBPI je 1 (všetky bunky sú jednojadrové), rovná sa 100 % cytostáze.
Cytostáza = 100 – RI
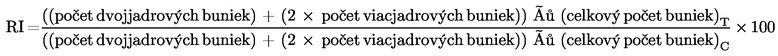
|
T
|
=
|
ošetrené kultúry
|
|
C
|
=
|
kontrolné kultúry.
|
|
|
2.
|
Preto ak RI je 53 %, znamená to, že v porovnaní s počtom buniek, ktorých delením vznikli dvojjadrové a viacjadrové bunky v kontrolnej kultúre, len 53 % tohto počtu sa delilo v ošetrenej kultúre, t. j. cytostáza je 47 %.
|
|
3.
|
Bez použitia cytoB sa odporúča hodnotenie cytotoxicity založené na relatívnom zvýšení počtu buniek (RICC) alebo na relatívnom zdvojení populácie (RPD) (58), pretože v obidvoch prípadoch sa zohľadňuje pomer populácie delených buniek.


kde:
Zdvojenie populácie = [log (počet buniek po ošetrení ÷ počiatočný počet buniek)] ÷ log 2.
|
|
4.
|
Preto 53 % RICC alebo RDP indikuje 47 % cytotoxicitu/cytostázu.
|
|
5.
|
Pri použití indexu proliferácie (PI) môže byť cytotoxicita hodnotená pomocou počítania počtu klonov obsahujúcich jednu bunku (cl1), 2 bunky (cl2), 3 až 4 bunky (cl4) a 5 až 8 buniek (cl8)
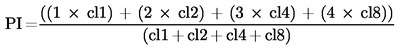
|
|
6.
|
PI sa používa ako hodnotný a spoľahlivý parameter cytotoxicity aj pri bunkových líniách kultivovaných in situ bez použitia cytoB (25) (26) (27) (28).
|
Dodatok 3
Referenčné chemikálie odporúčané na hodnotenie výkonnosti
(16)
|
Kategória
|
Chemikália
|
Číslo CAS
|
Číslo EC
|
| 1. Klastogény aktívne bez metabolickej aktivácie
|
|
|
cytozín arabinozid
|
147-94-4
|
205-705-9
|
|
|
mitomycín C
|
50-07-7
|
200-008-6
|
| 2. Klastogény vyžadujúce metabolickú aktiváciu
|
|
|
benzo(a)pyrén
|
50-32-8
|
200-028-5
|
|
|
cyklofosfamid
|
50-18-0
|
200-015-4
|
| 3. Aneugény
|
|
|
kolchicín
|
64-86-8
|
200-598-5
|
|
|
vinblastín
|
143-67-9
|
205-606-0
|
| 4. Negatívne látky
|
|
|
di(2-etylhexyl)ftalát
|
117-81-7
|
204-211-0
|
|
|
kyselina nalidixová
|
389-08-2
|
206-864-7
|
|
|
pyrén
|
129-00-0
|
204-927-3
|
|
|
chlorid sodný
|
7647-14-5
|
231-598-3
|
B. 50. SENZIBILIZÁCIA KOŽE: LOKÁLNA SKÚŠKA LYMFATICKÝCH UZLÍN: DA
ÚVOD
|
1.
|
Usmernenia Organizácie pre hospodársku spoluprácu a rozvoj (OECD) na testovanie chemikálií a testovacie metódy EÚ sa pravidelne revidujú vzhľadom na vedecký pokrok, zmeny regulačných potrieb a dobré životné podmienky zvierat. Prvá testovacia metóda (TM) (B.42) na stanovenie senzibilizácie kože u myší, tzv. lokálna skúška lymfatických uzlín (LLNA; usmernenie OECD na vykonávanie testov 429), bola zrevidovaná (1). Podrobnosti validácie LLNA a revízia prác v tejto oblasti boli uverejnené (2) (3) (4) (5) (6) (7) (8) (9). Pri LLNA sa na meranie proliferácie lymfocytov používa rádioizotopický tymidín alebo jód, a preto má táto skúška obmedzené použitie v tých prípadoch, kde je problematické získavanie, použitie alebo likvidácia rádioaktívnych látok. LLNA: DA (vyvinutá spoločnosťou Daicel Chemical Industries, Ltd.) je nerádioaktívnou modifikáciou LLNA, ktorá kvantifikuje obsah adenozín-trifosfátu (ATP) prostredníctvom bioluminiscencie, ktorá je ukazovateľom proliferácie lymfocytov. LLNA: Testovacia metóda DA bola validovaná a zrevidovaná a odporúča ju medzinárodný panel pre partnerské preskúmanie ako užitočnú metódu na identifikáciu chemikálií spôsobujúcich alebo nespôsobujúcich senzibilizáciu kože s určitými obmedzeniami (10) (11) (12) (13). Táto testovacia metóda je vytvorená na posúdenie potenciálu chemikálií (látok a zmesí) spôsobiť senzibilizáciu kože u zvierat. Metóda opísaná v kapitole B.6 tejto prílohy a usmernení OECD na vykonávanie testov 406 využíva testy s morčatami, konkrétne maximalizačný test morčiat a Buehlerov test (14). LLNA (kapitola B.42 tejto prílohy; usmernenie OECD na vykonávanie testov 429) a dve nerádioaktívne modifikácie LLNA: DA (kapitola B.50 tejto prílohy; usmernenie OECD na vykonávanie testov 442 A) a LLNA: BrdU-ELISA (kapitola B.51 tejto prílohy; usmernenie OECD na vykonávanie testov 442 B) poskytujú výhodu v porovnaní s testami na morčatách opísanými v kapitole B.6 a usmernení OECD na vykonávanie testov 406 (14), a to pokiaľ ide o obmedzenie a zdokonalenie využívania zvierat.
|
|
2.
|
Podobne ako LLNA, LNNA: DA skúma indukčnú fázu senzibilizácie kože a poskytuje kvantitatívne údaje vhodné na posúdenie odozvy na dávku. Okrem toho schopnosť identifikovať látky spôsobujúce senzibilizáciu kože bez potreby používania rádioaktívneho značenia DNA znižuje potenciál expozície rádioaktivite pri práci a problémy v súvislosti s likvidáciou odpadu. To môže následne umožniť zvýšené využívanie myší na identifikáciu látok spôsobujúcich senzibilizáciu kože, čo by ďalej mohlo znížiť využívanie morčiat na testovanie potenciálu senzibilizácie kože (t. j. B.6; usmernenie OECD na vykonávanie testov 406) (14).
|
VYMEDZENIE POJMOV
|
3.
|
Použité definície sú uvedené v dodatku 1.
|
ÚVODNÉ ÚVAHY A OBMEDZENIA
|
4.
|
LLNA: DA je upravenou metódou LLNA na identifikáciu potenciálu chemikálií spôsobiť senzibilizáciu kože, ktorá má určité obmedzenia. Neznamená to nevyhnutne, že vo všetkých prípadoch sa má použiť LLNA: DA namiesto testu LLNA alebo testov s morčatami (t. j. metóda B.6; usmernenie OECD na vykonávanie testov 406) (14), ale skôr to, že táto skúška je rovnako hodnotná a môže sa použiť ako alternatíva, v ktorej pozitívne a negatívne výsledky vo všeobecnosti nevyžadujú ďalšie potvrdenie (10) (11). Testovacie laboratórium by malo posúdiť všetky dostupné informácie o testovanej látke pred uskutočnením štúdie. Takéto informácie budú obsahovať identitu a chemickú štruktúru testovanej látky, jej fyzikálno-chemické vlastnosti, výsledky všetkých ostatných in vitro alebo in vivo testov toxicity s testovanou látkou a toxikologické údaje o štruktúrne príbuzných chemikáliách. Tieto informácie treba vziať do úvahy pri určovaní, či LLNA: DA je pre testovanú látku vhodná [vzhľadom na nezlučiteľnosť obmedzených druhov chemikálií s LLNA: DA (pozri odsek 5)], a ako pomoc pri výbere dávok.
|
|
5.
|
LLNA: DA je metóda in vivo, a to znamená, že pri hodnotení aktivity alergickej kontaktnej senzibilizácie sa nevylučuje používanie zvierat. Má však potenciál obmedziť používanie zvierat na tento účel pri porovnaní s testami na morčatách (B.6; usmernenie OECD na vykonávanie testov 406) (14). Okrem toho LLNA: DA ponúka značné zjemnenie spôsobu, akým sa zvieratá používajú na testy alergickej kontaktnej senzibilizácie (znižuje bolesť a utrpenie zvierat), keďže na rozdiel od metódy B.6 a usmernenia OECD na vykonávanie testov 406 LLNA: DA nevyžaduje, aby sa vyvolali reakcie dermálnej hypersenzitivity. Napriek výhodám, ktoré má LLNA: DA v porovnaní s metódou B.6 a usmernením OECD na vykonávanie testov 406 (14), existujú určité obmedzenia, ktoré by si mohli vyžadovať použiť metódu B.6 alebo usmernenie OECD na vykonávanie testov 406 [napr. testovanie určitých kovov, falošné pozitívne zistenia s určitými kožnými dráždivými látkami (ako napríklad niektoré povrchovo aktívne látky) (6) (1 a kapitola B.42 tejto prílohy), rozpustnosť testovanej látky]. Okrem toho chemické triedy alebo látky, ktoré obsahujú potenciálne zmätočné funkčné skupiny (16), si tiež môžu vyžadovať použitie testov s morčatami [t. j. B.6, usmernenie OECD na vykonávanie testov 406 (14)]. Obmedzenia, ktoré boli identifikované pre LLNA (1 a kapitola B.42 tejto prílohy), sa odporúčajú aj pre LLNA: DA (10). Okrem toho použitie LLNA: DA nemusí byť vhodné na testovanie látok, ktoré ovplyvňujú úrovne ATP (napr. látky, ktoré fungujú ako inhibítory ATP), alebo látok, ktoré ovplyvňujú presné meranie vnútrobunkového ATP (napr. prítomnosť enzýmov spôsobujúcich degradáciu ATP, prítomnosť ATP v mimobunkovom priestore lymfatickej uzliny). Okrem takto identifikovaných obmedzení by sa LLNA: DA mala uplatňovať pri testovaní všetkých látok okrem prípadov, keď tieto látky majú vlastnosti, ktoré by mohli narúšať presnosť LLNA: DA. Okrem toho by sa mala zvážiť možnosť hraničných pozitívnych výsledkov v prípade, keď sa dosiahnu hodnoty stimulačného indexu (SI) od 1,8 do 2,5 (pozri odseky 31 a 32). To je založené na validačnej databáze 44 látok s použitím SI ≥ 1,8 (pozri odsek 6), pre ktoré LLNA: DA správne identifikovala 32 látok spôsobujúcich senzibilizáciu podľa LLNA, ale nesprávne identifikovala tri z 12 látok nespôsobujúcich senzibilizáciu podľa LLNA s hodnotami SI od 1,8 do 2,5 (t. j. hraničný pozitívny výsledok) (10). Keďže sa však na stanovenie hodnôt SI a výpočet predpokladaných vlastností testu použil rovnaký súbor údajov, stanovené údaje môžu byť nadhodnotením skutočných prediktívnych vlastností.
|
PRINCÍP TESTOVACEJ METÓDY
|
6.
|
Základným princípom, na ktorom sa LLNA: DA zakladá, je, že senzibilizátory vyvolávajú proliferáciu lymfocytov v lymfatickej uzline v drénovanom mieste aplikácie testovanej látky. Táto proliferácia je úmerná dávke a účinnosti aplikovaného alergénu a poskytuje jednoduchý prostriedok na získanie kvantitatívneho merania senzibilizácie. Proliferácia sa meria porovnávaním priemeru proliferácie v každej pokusnej skupine s priemerom proliferácie v kontrolnej skupine, ktorej sa podáva nosič (VC). Určí sa pomer priemernej proliferácie v každej ošetrenej skupine a priemernej proliferácie v súbežnej kontrolnej skupine s nosičom, ktorý sa označuje ako stimulačný index (SI) a mal by mať hodnotu ≥ 1,8 pred ďalším prípadným vyhodnotením testovanej látky ako potenciálneho senzibilizátora kože. Uvedené metódy sú založené na meraní obsahu ATP pomocou bioluminiscencie (ktoré koreluje s počtom živých buniek) (17) na preukázanie proliferácie zvýšeného počtu buniek v drénovaných ušných lymfatických uzlinách (18) (19). Metóda bioluminiscencie využíva enzým luciferáza na katalýzu tvorby svetla z ATP a luciferínu podľa tejto reakcie:
ATP + luciferin + O
2
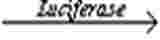 oxyluciferin + AMP + PPi
+ CO
2 + svetlo
oxyluciferin + AMP + PPi
+ CO
2 + svetlo
Intenzita vyžarovaného svetla priamo súvisí s koncentráciou ATP a meria sa pomocou luminometra. Skúška luciferínu – luciferázy je citlivá metóda kvantifikácie ATP, ktorá sa používa v mnohých rôznych aplikáciách (20).
|
OPIS SKÚŠKY
Výber druhov zvierat
|
7.
|
Vybraným druhom pre tento test je myš. Validačné štúdie LLNA: DA sa vykonávali výlučne s myšami kmeňa CBA/J, ktorý sa preto považuje za preferovaný kmeň (12) (13). Použijú sa mladé dospelé negravidné samice myši, ktoré nikdy nevrhli mláďatá. Na začiatku štúdie by zvieratá mali byť vo veku 8 – 12 týždňov a rozdiel medzi hmotnosťami zvierat má byť minimálny a nemá byť vyšší ako 20 % priemernej hmotnosti. Alternatívne sa môžu použiť iné kmene a samce, ak sa na základe údajov dá uspokojivo dokázať, že neexistujú významné odlišnosti špecifické pre kmeň a/alebo pohlavie v odozve na LLNA: DA.
|
Podmienky umiestnenia a kŕmenia
|
8.
|
Myši by sa mali umiestniť v skupinách (21), pokiaľ sa neposkytne primerané vedecké odôvodnenie ich umiestnenia jednotlivo. Teplota miestnosti pre pokusné zvieratá by mala byť 22 ± 3 °C. Aj keď relatívna vlhkosť by mala byť aspoň 30 % a pokiaľ možno by nemala prevyšovať 70 % okrem obdobia počas čistenia miestnosti, cieľom je, aby relatívna vlhkosť dosahovala 50 % – 60 %. Osvetlenie by malo byť umelé a malo by sa striedať 12 hodín svetla a 12 hodín tmy. Na kŕmenie sa môže používať bežné laboratórne krmivo s neobmedzenou dodávkou pitnej vody.
|
Príprava zvierat
|
9.
|
Zvieratá sa vyberú náhodne a sú označené tak, aby sa umožnila jednotlivá identifikácia (ale nie formou ušných značiek), a držia sa v klietkach aspoň 5 dní pred začiatkom podávania dávok, aby sa aklimatizovali na laboratórne podmienky. Pred začatím ošetrenia sa všetky zvieratá vyšetria, aby sa zaistilo, že nemajú žiadne viditeľné kožné poranenia.
|
Príprava dávok
|
10.
|
Chemikálie v tuhom skupenstve by sa pred podaním dávky do ucha myši mali rozpustiť alebo suspendovať v rozpúšťadlách alebo nosičoch, a ak je to vhodné, rozriediť. Kvapalné chemikálie možno podávať priamo alebo pred podaním rozriediť. V prípade nerozpustných chemikálií, ako napríklad tých, ktoré sa vo všeobecnosti používajú ako zdravotnícke pomôcky, by sa pred podaním dávky do ucha myši mala uskutočniť zvýšená extrakcia vo vhodnom rozpúšťadle, aby sa odhalili všetky extrahovateľné zložky, ktoré sa budú testovať. Testované látky sa pripravujú každý deň okrem prípadu, keď údaje o stabilite preukazujú prijateľnosť uskladnenia.
|
Kontrola spoľahlivosti
|
11.
|
Riadne vykonanie skúšky sa dokazuje pomocou chemikálií na pozitívne kontroly (PC), ktoré reagujú primeranou a reprodukovateľnou citlivosťou na senzibilizujúcu testovanú látku, pre ktorú je dobre charakterizovaná šírka odozvy. Odporúča sa zahrnúť súbežnú pozitívnu kontrolu, pretože to dokazuje kompetenciu laboratória úspešne vykonať každú skúšku a umožňuje posúdenie vnútrolaboratórnej a medzilaboratórnej reprodukovateľnosti a porovnateľnosti. Pozitívnu kontrolu v každej štúdii vyžadujú aj niektoré regulačné orgány, a preto by používatelia mali pred vykonaním LLNA: DA konzultovať s príslušnými orgánmi. Rutinné použitie súbežnej pozitívnej kontroly sa podporuje aj preto, aby sa predišlo potrebe ďalšieho testovania na zvieratách, s cieľom splniť také požiadavky, ktoré by mohli vyplynúť z použitia periodickej pozitívnej kontroly (pozri odsek 12). Pozitívna kontrola by mala spôsobiť pozitívnu odozvu na LLNA: DA pri úrovni expozície, pri ktorej sa očakáva nárast stimulačného indexu (SI) > 1,8 v porovnaní s negatívnou kontrolnou skupinou. Dávka chemickej látky na pozitívnu kontrolu by sa mala zvoliť tak, aby nespôsobila nadmerné podráždenie kože ani systémovú toxicitu a aby indukcia bola reprodukovateľná, ale nie nadmerná (napr. SI > 10 by bol nadmerný). Vhodnými chemikáliami na pozitívnu kontrolu sú 25 % 3-fenyl-2-hexylpropanál (číslo CAS 101-86-0) a 25 % eugenol (číslo CAS 97-53-0) v acetóne: olivový olej (4: 1, objemové percentá). Môžu sa vyskytnúť okolnosti, keď sa v riadne zdôvodnených prípadoch môžu použiť iné pozitívne kontroly, ktoré spĺňajú uvedené kritériá.
|
|
12.
|
Hoci sa odporúča súbežná pozitívna kontrolná skupina, môžu sa vyskytnúť situácie, keď môže byť vhodné periodické testovanie (t. j. v intervaloch najviac 6 mesiacov) pozitívnych kontrol v prípade tých laboratórií, ktoré vykonávajú LLNA: DA pravidelne (t. j. LLNA: DA vykonávajú v priebehu jedného mesiaca aspoň raz) a ktoré majú zavedenú historickú databázu pozitívnych kontrol, ktorá preukazuje schopnosť laboratória dosahovať reprodukovateľné a presné výsledky s pozitívnymi kontrolami. Primeranú odbornosť s použitím LLNA: DA možno úspešne preukázať dosiahnutím konzistentných pozitívnych výsledkov s pozitívnou kontrolou najmenej v 10 nezávislých testoch uskutočnených v určitom primeranom období (t. j. v období kratšom ako jeden rok).
|
|
13.
|
Súbežná pozitívna kontrolná skupina by sa mala zahrnúť vždy, keď dôjde k procedurálnej zmene v súvislosti s LLNA: DA (napr. k zmene vyškoleného personálu, zmene materiálov a/alebo reagentov používaných pri testovacej metóde, zmene nástrojov používaných pri testovacej metóde, zmene zdroja testovaných zvierat), a takéto zmeny je potrebné zaznamenať v laboratórnych správach. Je potrebné zvážiť vplyv týchto zmien na primeranosť predtým zriadenej historickej databázy pri určovaní potreby zriadenia novej historickej databázy na zaznamenávanie konzistentnosti výsledkov pozitívnej kontroly.
|
|
14.
|
Výskumní pracovníci by si mali uvedomiť, že rozhodnutie vykonať štúdiu pozitívnych kontrol na periodickom základe namiesto súbežných pozitívnych kontrol môže mať následky na primeranosť a prijateľnosť výsledkov negatívnej štúdie získaných bez súbežnej pozitívnej kontroly počas intervalu medzi každou periodickou štúdiou pozitívnej kontroly. Ak sa dosiahne napríklad falošný negatívny výsledok v periodickej štúdii pozitívnej kontroly, negatívne výsledky získané s testovanou látkou v intervale medzi poslednou prijateľnou periodickou štúdiou pozitívnej kontroly a neprijateľnou periodickou štúdiou pozitívnej kontroly možno spochybniť. Dôsledky týchto výsledkov treba dôkladne zvážiť pri určovaní toho, či vykonať súbežné pozitívne kontroly alebo iba periodické pozitívne kontroly. Treba tiež zvážiť použitie menšieho počtu zvierat v súbežnej pozitívnej kontrolnej skupine, ak je to vedecky odôvodnené a ak laboratórium na základe konkrétnych laboratórnych historických údajov preukáže, že možno použiť menší počet myší (22).
|
|
15.
|
Aj keď by sa pozitívna kontrola mala testovať v nosiči, o ktorom je známe, že vyvoláva konzistentnú odozvu (napr. acetón: olivový olej v pomere 4: 1, objemové percentá), môžu sa vyskytnúť určité regulačné situácie, v ktorých bude potrebné vykonať aj testovanie v neštandardnom nosiči (klinicky/chemicky zodpovedajúcom roztoku) (23). Ak sa súbežná pozitívna kontrola testuje v odlišnom nosiči ako testovaná látka, potom treba do testu súbežnej pozitívnej kontroly zahrnúť samostatnú kontrolnú skupinu, ktorej sa podáva nosič.
|
|
16.
|
V prípadoch hodnotenia látok, ktoré patria do konkrétnej chemickej triedy alebo ktoré zastupujú konkrétny rozsah odoziev, môžu byť tiež užitočné referenčné látky, aby sa preukázalo, že testovacia metóda na zisťovanie potenciálu týchto druhov látok spôsobiť senzibilizáciu kože riadne funguje. Vhodné referenčné látky majú mať tieto vlastnosti:
|
—
|
štrukturálnu a funkčnú podobnosť s triedou testovanej látky, ktorá je predmetom testovania,
|
|
—
|
známe fyzikálno-chemické vlastnosti,
|
|
—
|
podporné údaje z LLNA: DA,
|
|
—
|
podporné údaje z ďalších zvieracích a/alebo ľudských modelov.
|
|
TESTOVACÍ POSTUP
Počet zvierat a veľkosť dávky
|
17.
|
Na dávkovú skupinu sa používajú aspoň štyri zvieratá, aspoň tri koncentrácie testovanej látky a k tomu súbežná negatívna kontrolná skupina, ktorej sa podáva iba nosič testovanej látky, a pozitívna kontrola (súbežná alebo nedávna, podľa laboratórnych pravidiel s prihliadnutím na odseky 11 – 15). Treba zvážiť testovanie viacerých dávok pozitívnej kontroly, najmä ak sa testuje na prerušovanej báze. So zvieratami v kontrolných skupinách by sa malo zaobchádzať rovnakým spôsobom ako s pokusnými zvieratami okrem prípadov, keď sa ošetrovanie testovanou látkou nevykonáva.
|
|
18.
|
Výber dávky a nosiča by mal byť založený na odporúčaniach uvedených v odkazoch (2) a (24). Následné dávky sa zvyčajne vyberajú z vhodných sérií koncentrácií, napr. 100 %, 50 %, 25 %, 10 %, 5 %, 2,5 %, 1 %, 0,5 % atď. Výber zo sérií koncentrácií je potrebné primerane vedecky zdôvodniť. Všetky existujúce informácie o toxicite (napr. akútnej toxicite a podráždení kože), ako aj štrukturálne a fyzikálno-chemické informácie o predmetnej testovanej látke (a/alebo štrukturálne príbuzných látkach) by sa mali prípadne posúdiť výberom troch po sebe idúcich koncentrácií tak, aby najvyššia koncentrácia maximalizovala expozíciu, pričom by sa však zabránilo systémovej toxicite a/alebo nadmernému lokálnemu podráždeniu kože (24) (25). Ak tieto informácie nie sú k dispozícii, možno bude potrebné vykonať predbežný skríningový test (pozri odseky 21 – 24).
|
|
19.
|
Nosič by nemal ovplyvňovať výsledok testu a mal by sa vybrať na základe maximálnej rozpustnosti s cieľom získať najvyššiu dosiahnuteľnú koncentráciu, pričom vznikne roztok/suspenzia vhodná na aplikáciu testovanej látky. Odporúčané nosiče sú acetón: olivový olej (4: 1, objemové percentá), N,N-dimetylformamid, metyletylketón, propylénglykol a dimetylsulfoxid (6), ale môžu sa použiť aj iné nosiče, ak sa uvedie uspokojivé vedecké zdôvodnenie. V určitých situáciách môže byť potrebné použiť ako ďalšiu kontrolu klinicky vhodný roztok alebo komerčný prípravok, v ktorom sa testovaná látka predáva. Osobitnú pozornosť je potrebné venovať zabezpečeniu, aby sa hydrofilné testované látky začlenili do systému nosiča, ktorý zvlhčuje pokožku, a rýchlo neunikli, a to začlenením vhodných rozpúšťadiel (napr. 1 % Pluronic® L92). Preto je potrebné vyhýbať sa použitiu čisto vodných nosičov.
|
|
20.
|
Spracovanie lymfatických uzlín jednotlivých myší umožňuje posúdiť variabilitu medzi zvieratami a štatisticky porovnať rozdiel medzi meraniami testovanej látky a kontrolnej skupiny, ktorej sa podáva nosič (pozri odsek 33). Okrem toho sa zníženie počtu myší v pozitívnej kontrolnej skupine môže prehodnotiť len po poskytnutí informácií o jednotlivých zvieratách (22). Navyše niektoré regulačné orgány požadujú zber údajov o jednotlivých zvieratách. Pravidelný zber údajov o jednotlivých zvieratách poskytuje výhodu, pokiaľ ide o dobré životné podmienky zvierat, pretože sa predchádza duplicitnému testovaniu, ktoré by bolo potrebné v prípade, ak by výsledky testovanej látky pôvodne získané jedným spôsobom (napr. prostredníctvom spojených údajov o zvieratách) museli neskôr posúdiť regulačné orgány podľa iných požiadaviek (napr. prostredníctvom údajov o jednotlivých zvieratách).
|
Predbežný skríningový test
|
21.
|
V prípade, ak nie sú k dispozícii informácie na stanovenie najvyššej dávky testovanej látky (pozri odsek 18), treba vykonať predbežný skríningový test s cieľom stanoviť primeranú úroveň testovanej dávky na použitie v LLNA: DA. Cieľom predbežného skríningového testu je poskytnúť usmernenie na výber maximálnej úrovne dávky na použitie v hlavnej štúdii LLNA: DA v prípade, že nie sú dostupné informácie o koncentrácii, ktorá spôsobuje systémovú toxicitu (pozri odsek 24) a/alebo nadmerné lokálne podráždenie kože (pozri odsek 23). Maximálna úroveň testovanej dávky by mala obsahovať 100 % testovanej látky v prípade kvapalín alebo maximálnu možnú koncentráciu v prípade tuhých látok alebo suspenzií.
|
|
22.
|
Predbežný skríningový test sa vykonáva za podmienok, ktoré sú identické s hlavnou štúdiou LLNA: DA s tým rozdielom, že sa neposudzuje proliferácia lymfatickej uzliny a môže sa použiť menší počet zvierat na dávkovú skupinu. Navrhuje sa jedno alebo dve zvieratá na dávkovú skupinu. U všetkých myší sa každý deň pozorujú klinické príznaky systémovej toxicity alebo lokálneho podráždenia v mieste aplikácie. Telesné hmotnosti zvierat sa zaznamenajú na začiatku testu a pred usmrtením (deň 8). Obidve uši každej myši sa vyšetria, či nemajú príznaky erytému (sčervenania kože), a vyhodnotia sa podľa tabuľky 1 (25). Hrúbka ucha sa odmeria pomocou meradla hrúbky (napr. digitálnym mikrometrom alebo meradlom hrúbky Peacock Dial) v deň 1 (pred aplikáciou dávky), v deň 3 (približne 48 hodín po prvej dávke), v deň 7 (24 hodín pred usmrtením) a v deň 8. Okrem toho možno hrúbku ucha v deň 8 určiť na základe meraní vážením vzoriek uší, ktoré by sa malo vykonať po humánnom usmrtení zvierat. Nadmerné lokálne podráždenie kože indikuje hodnotenie erytému ≥ 3 a/alebo zvýšenie hrúbky ucha o ≥ 25 % v ktorýkoľvek deň merania (26) (27). Najvyššia dávka vybraná pre hlavnú štúdiu LLNA: DA bude najbližšia nižšia dávka z predbežných skríningových sérií koncentrácií (pozri odsek 18), ktorá nespôsobuje systémovú toxicitu a/ani nadmerné lokálne podráždenie kože.
Tabuľka 1
Hodnotenia erytému
|
Pozorovanie
|
Hodnotenie
|
|
Žiadny erytém
|
0
|
|
Veľmi slabý erytém (sotva pozorovateľný)
|
1
|
|
Jasne ohraničený erytém
|
2
|
|
Mierny až silný erytém
|
3
|
|
Výrazný erytém (tmavočervený) až vytvorenie chrasty zabraňujúcej posúdeniu erytému
|
4
|
|
|
23.
|
Okrem 25 % zvýšenia hrúbky ucha (26) (27) sa štatisticky významné zvýšenie hrúbky ucha v testovanej skupine myší v porovnaní s kontrolnou skupinou myší použilo aj na identifikáciu dráždivých látok v LLNA (28) (29) (30) (31) (32) (33) (34). Štatisticky významné zvýšenie hrúbky ucha môže nastať aj vtedy, keď dôjde k zvýšeniu menšiemu ako 25 %, nespája sa však konkrétne s nadmerným podráždením kože (30) (31) (32) (33) (34).
|
|
24.
|
Nasledujúce klinické pozorovania môžu indikovať systémovú toxicitu (35), ak sú súčasťou integrovaného posudzovania, to znamená, že môžu indikovať maximálnu úroveň dávky na použitie v hlavnej štúdii LLNA: DA: zmeny funkcie nervového systému (napr. piloerekcia, ataxia, triaška a kŕče); zmeny správania (napr. agresia, zmena pri čistení, výrazná zmena stupňa aktivity); zmena dýchania (t. j. zmena frekvencie alebo intenzity dýchania, ako napríklad zadúšanie, dýchavičnosť a šelest) a zmeny spotreby potravy a vody. Okrem toho treba v hodnotení zvážiť príznaky letargie a/alebo apatie a akékoľvek klinické príznaky silnejšej alebo dlhodobejšej bolesti a utrpenia alebo viac ako 5 % zníženie telesnej hmotnosti medzi dňom 1 a 8 a úmrtnosť. Umierajúce zvieratá alebo zvieratá, ktoré majú zjavne silné bolesti alebo vykazujú príznaky silného utrpenia, by sa mali humánnym spôsobom usmrtiť (36).
|
Harmonogram pokusov v hlavnej štúdii
|
25.
|
Harmonogram pokusov v rámci skúšky je takýto:
— Deň 1: Označte každé zviera a zaznamenajte jeho hmotnosť a akékoľvek klinické pozorovania. Aplikujte 1 % vodný roztok laurylsulfátu sodného (SLS) na zadnú stranu každého ucha štyrmi až piatimi ťahmi kefky namočenej v roztoku SLS tak, aby bola pokrytá celá zadná strana každého ucha. Hodinu po aplikácii SLS začnite s aplikáciou 25 μL z príslušného zriedenia testovanej látky, samotného nosiča alebo pozitívnej kontroly (súbežnej alebo nedávnej, podľa laboratórnych pravidiel s prihliadnutím na odseky 11 – 15) na zadnú stranu každého ucha.
— Dni 2, 3 a 7: Zopakujte predbežné ošetrenie 1 % vodným roztokom SLS a postup aplikácie testovanej látky vykonaný v prvom dni.
— Dni 4, 5 a 6: Žiadne ošetrenie.
— Deň 8: Zaznamenajte hmotnosť každého zvieraťa a akékoľvek klinické príznaky. Približne 24 až 30 hodín po začatí aplikácie v deň 7 sa zvieratá humánne usmrtia. Drénované ušné lymfatické uzliny sa odstránia z každého ucha myši a jednotlivo sa spracujú vo fosfátom tlmenom fyziologickom roztoku (PBS) pre každé zviera. Údaje a diagramy identifikácie a disekcie lymfatických uzlín možno nájsť v odkaze (22). Pri ďalšom monitorovaní lokálnej odozvy kože v hlavnej štúdii možno do protokolu štúdie zahrnúť ďalšie parametre, ako napríklad hodnotenie erytému ucha alebo merania hrúbky ucha (získané pomocou meradla hrúbky alebo vážením vzoriek uší pri pitve).
|
Príprava bunkových suspenzií
|
26.
|
Jednobunková suspenzia buniek lymfatických uzlín (LNC) odobraná bilaterálne z každej myši sa pripravuje tak, že lymfatické uzliny sa uložia medzi dve podložné sklíčka, ktoré sa mierne stlačia, aby sa uzliny rozdrvili. Presvedčte sa, že tkanivo je rovnomerne rozložené, a potom podložné sklíčka oddeľte. Rozpustite tkanivo na oboch sklíčkach vo fosfátom tlmenom fyziologickom roztoku; každé sklíčko držte v určitom uhle nad Petriho miskou a opláchnite roztokom PBS, pričom súbežne škrabkou na bunky odoberte tkanivo zo sklíčka. Navyše keďže lymfatické uzliny zvierat v negatívnej kontrole sú malé, dôležité je opatrné zaobchádzanie, aby sa predišlo umelým účinkom na hodnoty SI. Na opláchnutie obidvoch sklíčok by sa mal použiť celkový objem 1 ml roztoku PBS. Suspenzia buniek lymfatických uzlín v Petriho miske by sa mala mierne homogenizovať škrabkou na bunky. Následne sa zo suspenzie buniek lymfatických uzlín mikropipetou odoberie alikvotná časť 20 μl, pričom sa dbá na to, aby sa neodobrala okom viditeľná membrána, a táto alikvotná časť sa zmieša s 1,98 ml roztoku PBS tak, aby sa získali 2 ml vzorky. Druhá 2 ml vzorka sa potom pripraví rovnakým postupom, takže pre každé zviera sa pripravia dve vzorky.
|
Stanovenie proliferácie buniek (meranie obsahu ATP v lymfocytoch)
|
27.
|
Zvýšenie obsahu ATP v lymfatických uzlinách sa meria metódou, ktorá využíva luciferín/luciferázu a súpravu na meranie ATP, ktorá meria bioluminiscenciu v jednotkách relatívnej luminiscencie (RLU). Čas skúšky od času usmrtenia zvieraťa po meranie obsahu ATP má byť jednotný u každého zvieraťa a má trvať približne 30 minút, pretože obsah ATP sa zrejme po usmrtení zvieraťa postupne znižuje (12). Preto by sa séria postupov od odobrania ušných lymfatických uzlín až po meranie ATP mala dokončiť do 20 minút podľa vopred určeného časového plánu, ktorý je rovnaký pre každé zviera. Luminiscencia ATP by sa mala merať v každej 2 ml vzorke tak, aby sa získali dva výsledky meraní ATP pre každé zviera. Následne sa určí priemerná luminiscencia ATP, ktorá sa použije v ďalších výpočtoch (pozri odsek 30).
|
POZOROVANIA
Klinické pozorovania
|
28.
|
Každá myš sa starostlivo vyšetrí aspoň jedenkrát denne na všetky klinické príznaky, či už ide o lokálne podráždenie v mieste aplikácie, alebo o systémovú toxicitu. Všetky pozorovania sa systematicky zaznamenávajú, pričom každá myš má individuálny záznam. Plány na monitorovanie by mali zahŕňať kritériá na rýchlu identifikáciu tých myší, ktoré vykazujú systémovú toxicitu, nadmerné lokálne podráždenie alebo poleptanie kože a ktoré majú byť humánne usmrtené (36).
|
Telesné hmotnosti
|
29.
|
Ako sa uvádza v odseku 25, telesné hmotnosti jednotlivých zvierat by sa mali odmerať na začiatku testu a pri naplánovanom usmrtení zvierat.
|
VÝPOČET VÝSLEDKOV
|
30.
|
Výsledky v každej pokusnej skupine sa vyjadrujú ako priemerná hodnota stimulačného indexu (SI). SI sa získa vydelením priemernej hodnoty RLU/myš v rámci každej skupiny, ktorej sa podávala testovaná látka, a pozitívnej kontrolnej skupiny priemernou hodnotou RLU/myš v kontrolnej skupine, ktorej sa podávalo rozpúšťadlo/nosič. Priemerná hodnota SI kontrolných skupín, ktorým sa podával nosič, je potom 1.
|
|
31.
|
V rozhodovacom procese sa za pozitívny výsledok považuje stimulačný index ≥ 1,8 (10). Pri určovaní toho, či hraničný výsledok (t. j. hodnota SI v rozmedzí od 1,8 do 2,5) je pozitívny, sa však môže použiť aj sila vzťahu medzi dávkou a odozvou, štatistický význam a konzistentnosť rozpúšťadla/nosiča a odoziev pozitívnej kontroly (2) (3) (37).
|
|
32.
|
Pri hraničnej pozitívnej odozve, keď sa SI pohybuje medzi 1,8 a 2,5, môžu používatelia na potvrdenie toho, že takéto výsledky sú pozitívne, zvážiť doplňujúce informácie, ako napríklad vzťah medzi dávkou a odozvou, dôkazy systémovej toxicity alebo nadmerného podráždenia a v prípade potreby štatistický význam spolu s hodnotami SI (10). Pozornosť by sa mala venovať aj rôznym vlastnostiam testovanej látky vrátane toho, či má štrukturálny vzťah k látkam, ktoré sú známymi senzibilizátormi kože, či spôsobuje nadmerné podráždenie kože myši, a vrátane charakteru pozorovanej odozvy na dávku. Tieto a ostatné hľadiská sa podrobne rozoberajú inde (4).
|
|
33.
|
Zber údajov na úrovni jednotlivých myší umožní vykonať štatistickú analýzu prítomnosti a úrovne vzťahu medzi dávkou a odozvou v údajoch. Každé štatistické posúdenie by mohlo zahŕňať hodnotenie vzťahu medzi dávkou a odozvou, ako aj vhodne upravené porovnávania testovaných skupín (napr. porovnávania v pároch skupiny, ktorej sa podáva dávka, a súbežnej kontrolnej skupiny s rozpúšťadlom/nosičom). Štatistické analýzy môžu zahŕňať napr. lineárnu regresiu alebo Williamsov test na posúdenie trendov vo vzťahu medzi dávkou a odozvou a Dunnettov test na porovnávanie v pároch. Pri výbere vhodnej metódy štatistickej analýzy by si výskumný pracovník mal uvedomovať možné nerovnosti rozdielov a ďalšie súvisiace problémy, ktoré si môžu vyžadovať premenu údajov alebo štatistickú analýzu založenú na iných ako parametrických údajoch. Výskumný pracovník bude možno v každom prípade musieť vykonať výpočty SI a štatistické analýzy s určitými dátovými bodmi a bez nich (niekedy sa tieto údaje nazývajú ‚extrémne hodnoty‘).
|
ÚDAJE A PODÁVANIE SPRÁV
Údaje
|
34.
|
Údaje by sa mali zhrnúť vo forme tabuľky, v ktorej sa uvádzajú hodnoty RLU jednotlivých zvierat, skupinová priemerná hodnota RLU/zviera, súvisiaci rozsah chyby (napr. SD, SEM) a stredná hodnota SI pre každú dávkovú skupinu v porovnaní so súbežnou kontrolnou skupinou s rozpúšťadlom/nosičom.
|
Správa o teste
|
35.
|
Správa o teste by mala obsahovať tieto informácie:
|
|
Testované a kontrolné chemické látky:
|
—
|
identifikačné údaje (napr. čísla CAS, čísla EC, ak sú k dispozícii, zdroj, čistota, známe nečistoty, číslo šarže),
|
|
—
|
fyzikálny charakter a fyzikálno-chemické vlastnosti (napr. prchavosť, stabilita, rozpustnosť),
|
|
—
|
v prípade zmesi zloženie a relatívny percentuálny podiel zložiek.
|
|
|
|
Rozpúšťadlo/nosič:
|
—
|
identifikačné údaje (čistota, v prípade potreby koncentrácia, použitý objem),
|
|
—
|
zdôvodnenie voľby nosiča.
|
|
|
|
Testovacie zvieratá:
|
—
|
mikrobiologický stav zvierat, ak je známy,
|
|
—
|
zdroj zvierat, podmienky umiestnenia, krmivo atď.
|
|
|
|
Podmienky testu:
|
—
|
zdroj, číslo šarže a kontrolné údaje výrobcu o kvalite/presnosti súpravy ATP,
|
|
—
|
informácie o príprave a aplikácii testovanej látky,
|
|
—
|
zdôvodnenie výberu dávky (vrátane výsledkov z predbežného skríningového testu, ak sa uskutočnil),
|
|
—
|
použité koncentrácie nosiča a testovanej látky a celkové množstvo aplikovanej testovanej látky,
|
|
—
|
údaje o kvalite krmiva a vody (vrátane druhu/zdroja krmiva, zdroja vody),
|
|
—
|
podrobný opis harmonogramu podávania dávok a odberu vzoriek,
|
|
—
|
metódy merania toxicity,
|
|
—
|
kritériá na hodnotenie štúdií ako pozitívnych alebo negatívnych,
|
|
—
|
informácie o akýchkoľvek odchýlkach od protokolu a vysvetlenie, ako odchýlky ovplyvňujú návrh a výsledky štúdie.
|
|
|
|
Kontrola spoľahlivosti:
|
—
|
súhrn výsledkov poslednej kontroly spoľahlivosti vrátane informácií o testovanej látke, koncentrácii a použitom nosiči,
|
|
—
|
súbežné a/alebo historické kontrolné údaje o pozitívnej a súbežnej negatívnej kontrole (s rozpúšťadlom/nosičom) pre testovacie laboratórium,
|
|
—
|
ak nebola zahrnutá súbežná pozitívna kontrola, dátum a laboratórna správa o najaktuálnejšej periodickej pozitívnej kontrole a správa s historickými údajmi o pozitívnej kontrole, ktorými laboratórium zdôvodňuje nevykonanie súbežnej pozitívnej kontroly.
|
|
|
|
Výsledky:
|
—
|
hmotnosti jednotlivých myší na začiatku podávania dávky a pri naplánovanom usmrtení, ako aj priemerný a súvisiaci rozsah chyby (napr. SD a SEM) pre každú pokusnú skupinu,
|
|
—
|
časový priebeh nástupu príznakov toxicity u každého zvieraťa vrátane podráždenia kože v mieste podania látky, ak sa vyskytnú,
|
|
—
|
čas usmrtenia zvieraťa a čas merania ATP u každého zvieraťa,
|
|
—
|
tabuľka hodnôt RLU jednotlivých myší a hodnoty SI každej pokusnej skupiny s dávkou,
|
|
—
|
priemerný a súvisiaci rozsah chyby (napr. SD, SEM) v súvislosti s hodnotami RLU/myš každej pokusnej skupiny a výsledky analýzy extrémnej hodnoty v každej pokusnej skupine,
|
|
—
|
výpočet SI a vhodné opatrenie variability, ktoré berie do úvahy variabilitu medzi zvieratami v skupinách, ktorým sa podáva testovaná látka, a v kontrolných skupinách,
|
|
—
|
vzťah medzi dávkou a odozvou,
|
|
—
|
v prípade potreby štatistické analýzy.
|
|
|
|
Rozbor výsledkov:
|
—
|
stručný komentár v súvislosti s výsledkami, analýza odozvy na dávku, štatistické analýzy, ak existujú, s uvedením záveru, či by sa testovaná látka mala považovať za látku spôsobujúcu senzibilizáciu kože.
|
|
|
LITERATÚRA
|
1.
|
Chamberlain, M. and Basketter, D. A. (1996), The local lymph node assay: status of validation. Food Chem, Toxicol., 34, 999 – 1002.
|
|
2.
|
OECD (2010), Skin Sensitisation: Local Lymph Node Assay, Test Guideline No 429, Guidelines for the Testing of Chemicals, OECD, Paris. K dispozícii na stránke: [http://www.oecd.org/env/testguidelines].
|
|
3.
|
Basketter, D. A., Gerberick, G. F., Kimber, I. and Loveless, S. E. (1996), The local lymph node assay: A viable alternative to currently accepted skin sensitisation tests. Food Chem, Toxicol., 34, 985 – 997.
|
|
4.
|
Basketter, D. A., Gerberick, G. F. and Kimber, I. (1998), Strategies for identifying false positive responses in predictive sensitisation tests. Food Chem. Toxicol., 36, 327 – 333.
|
|
5.
|
Van Och, F. M. M., Slob, W., De Jong, W. H., Vandebriel, R. J. and Van Loveren, H. (2000), A quantitative method for assessing the sensitising potency of low molecular weight chemicals using a local lymph node assay: employment of a regression method that includes determination of uncertainty margins. Toxicol., 146, 49 – 59.
|
|
6.
|
ICCVAM (1999), The murine local lymph node Assay: A test method for assessing the allergic contact dermatitis potential of chemicals/compounds: The results of an independent peer review evaluation coordinated by the Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM) and the National Toxicology Program Center for the Evaluation of Alternative Toxicological Methods (NICETAM). NIH Publication No 99-4494. Research Triangle Park, N.C. K dispozícii na stránke: [http://iccvam.niehs.nih.gov/docs/immunotox_docs/llna/llnarep.pdf].
|
|
7.
|
Dean, J. H., Twerdok, L. E., Tice, R. R., Sailstad, D. M., Hattan, D. G., Stokes, W. S. (2001), ICCVAM evaluation of the murine local lymph node assay: II. Conclusions and recommendations of an independent scientific peer review panel. Reg. Toxicol. Pharmacol. 34, 258 – 273.
|
|
8.
|
Haneke, K. E., Tice, R. R., Carson, B. L., Margolin, B. H., Stokes, W. S. (2001), ICCVAM evaluation of the murine local lymph node assay: III. Data analyses completed by the national toxicology program interagency center for the evaluation of alternative toxicological methods. Reg. Toxicol. Pharmacol. 34, 274 – 286.
|
|
9.
|
Sailstad, D. M., Hattan, D., Hill, R. N., Stokes, W. S. (2001), ICCVAM evaluation of the murine local lymph node assay: I. The ICCVAM review process.Reg. Toxicol. Pharmacol. 34, 249 – 257.
|
|
10.
|
ICCVAM (2010), ICCVAM Test Method Evaluation Report. Nonradioactive local lymph node assay: modified by Daicel Chemical Industries, Ltd., based on ATP content test method protocol (LLNA: DA). NIH Publication No 10-7551A/B. Research Triangle Park, NC: National Institute of Environmental Health Sciences. K dispozícii na stránke: [http://iccvam.niehs.nih.gov/methods/immunotox/llna-DA/TMER.htm].
|
|
11.
|
ICCVAM (2009), Independent Scientific Peer Review Panel Report: Updated validation status of new versions and applications of the murine local lymph node assay: a test method for assessing the allergic contact dermatitis potential of chemicals and products. Research Triangle Park, NC: National Institute of Environmental Health Sciences. K dispozícii na stránke: [http://iccvam.niehs.nih.gov/docs/immunotox_docs/LLNAPRPRept2009.pdf].
|
|
12.
|
Idehara, K., Yamagishi, G., Yamashita, K. and Ito, M. (2008), Characterization and evaluation of a modified local lymph node assay using ATP content as a non-radio isotopic endpoint.J. Pharmacol. Toxicol. Meth., 58, 1 – 10.
|
|
13.
|
Omori, T., Idehara, K., Kojima, H., Sozu, T., Arima, K., Goto, H., Hanada, T., Ikarashi, Y., Inoda, T., Kanazawa, Y., Kosaka, T., Maki, E., Morimoto, T., Shinoda, S., Shinoda, N., Takeyoshi, M., Tanaka, M., Uratani, M., Usami, M., Yamanaka, A., Yoneda, T., Yoshimura, I. and Yuasa, A. (2008), Interlaboratory validation of the modified murine local lymph node assay based on adenosine triphosphate measurement. J. Pharmacol. Toxicol. Meth., 58, 11 – 26.
|
|
14.
|
OECD (1992), Skin Sensitisation, Test Guideline No 406, Guidelines for Testing of Chemicals, OECD, Paris. K dispozícii na stránke: [http://www.oecd.org/env/testguidelines].
|
|
15.
|
Kreiling, R., Hollnagel, H. M., Hareng, L., Eigler, L., Lee, M. S., Griem, P., Dreessen, B., Kleber, M., Albrecht, A., Garcia, C. and Wendel, A. (2008), Comparison of the skin sensitising potential of unsaturated compounds as assessed by the murine local lymph node assay (LLNA) and the guinea pig maximization test (GPMT). Food Chem. Toxicol., 46, 1896 – 1904.
|
|
16.
|
Basketter, D., Ball, N., Cagen, S., Carrillo, J. C., Certa, H., Eigler, D., Garcia, C., Esch, H., Graham, C., Haux, C., Kreiling, R. and Mehling, A. (2009), Application of a weight of evidence approach to assessing discordant sensitisation datasets: implications for REACH. Reg. Toxicol. Pharmacol., 55, 90 – 96.
|
|
17.
|
Crouch, S. P., Kozlowski, R., Slater, K. J. and Fletcher J. (1993), The use of ATP bioluminescence as a measure of cell proliferation and cytotoxicity. J. Immunol. Meth., 160, 81 – 88.
|
|
18.
|
Ishizaka, A., Tono-oka, T. and Matsumoto, S. (1984), Evaluation of the proliferative response of lymphocytes by measurement of intracellular ATP. J. Immunol. Meth., 72, 127 – 132.
|
|
19.
|
Dexter, S. J., Cámara, M., Davies, M. and Shakesheff, K. M. (2003), Development of a bioluminescent ATP assay to quantify mammalian and bacterial cell number from a mixed population. Biomat., 24, 27 – 34.
|
|
20.
|
Lundin A. (2000), Use of firefly luciferase in ATP-related assays of biomass, enzymes, and metabolites. Meth. Enzymol., 305, 346 – 370.
|
|
21.
|
ILAR (1996), Institute of Laboratory Animal Research (ILAR) Guide for the Care and Use of Laboratory Animals. 7th ed. Washington, DC: National Academies Press.
|
|
22.
|
ICCVAM (2009), Recommended Performance Standards: Murine Local Lymph Node Assay, NIH Publication Number 09-7357, Research Triangle Park, NC: National Institute of Environmental Health Science. K dispozícii na stránke: [http://iccvam.niehs.nih.gov/docs/immunotox_docs/llna-ps/LLNAPerfStds.pdf].
|
|
23.
|
McGarry, H. F. (2007), The murine local lymph node assay: regulatory and potency considerations under REACH. Toxicol., 238, 71 – 89.
|
|
24.
|
Kimber, I., Dearman, R. J., Scholes, E. W., and Basketter, D. A. (1994), The local lymph node assay: developments and applications. Toxicol., 93, 13 – 31.
|
|
25.
|
OECD (2002), Acute Dermal Irritation/Corrosion, Test Guideline No 404, Guidelines for Testing of Chemicals, OECD, Paris. K dispozícii na stránke: [http://www.oecd.org/env/testguidelines].
|
|
26.
|
Reeder, M. K., Broomhead, Y. L., DiDonato, L. and DeGeorge, G. L. (2007), Use of an enhanced local lymph node assay to correctly classify irritants and false positive substances. Toxicologist, 96, 235.
|
|
27.
|
ICCVAM (2009), Nonradioactive Murine Local Lymph Node Assay: Flow Cytometry Test Method Protocol (LLNA: BrdU-FC) Revised Draft Background Review Document. Research Triangle Park, NC: National Institute of Environmental Health Sciences. K dispozícii na stránke: [http://iccvam.niehs.nih.gov/methods/immunotox/fcLLNA/BRDcomplete.pdf].
|
|
28.
|
Hayes, B. B., Gerber, P. C., Griffey, S. S. and Meade, B. J. (1998), Contact hypersensitivity to dicyclohexylcarbodiimide and diisopropylcarbodiimide in female B6C3F1 mice. Drug. Chem. Toxicol., 21, 195 – 206.
|
|
29.
|
Homey, B., von Schilling, C., Blumel, J., Schuppe, H. C., Ruzicka, T., Ahr, H. J., Lehmann, P., and Vohr, V. W. (1998), An integrated model for the differentiation of chemical-induced allergic and irritant skin reactions. Toxicol. Appl. Pharmacol., 153, 83 – 94.
|
|
30.
|
Woolhiser, M. R., Hayes, B. B., and Meade, B. J. (1998), A combined murine local lymph node and irritancy assay to predict sensitisation and irritancy potential of chemicals. Toxicol. Meth., 8, 245 – 256.
|
|
31.
|
Hayes, B. B., and Meade, B. J. (1999), Contact sensitivity to selected acrylate compounds in B6C3F1 mice: relative potency, cross reactivity, and comparison of test methods. Drug Chem. Toxicol., 22, 491 – 506.
|
|
32.
|
Ehling, G., Hecht, M., Heusener, A., Huesler, J., Gamer, A. O., van Loveren, H., Maurer, T., Riecke, K., Ullmann, L., Ulrich, P., Vandebriel, R., and Vohr, H.W. (2005), A European inter-laboratory validation of alternative endpoints of the murine local lymph node assay: first round. Toxicol., 212, 60 – 68.
|
|
33.
|
Vohr, H. W., and Ahr, H. J. (2005), The local lymph node assay being too sensitive? Arch. Toxicol., 79, 721 – 728.
|
|
34.
|
Patterson, R. M., Noga, E., and Germolec, D. (2007), Lack of evidence for contact sensitisation by Pfiesteria extract. Environ. Health Perspect., 115, 1023 – 1028.
|
|
35.
|
ICCVAM (2009), Report on the ICCVAM-NICEATM/ECVAM/JaCVAM Scientific Workshop on Acute Chemical Safety Testing: Advancing In Vitro Approaches and Humane Endpoints for Systemic Toxicity Evaluations. Research Triangle Park, NC: National Institute of Environmental Health Sciences. K dispozícii na stránke: [http://iccvam.niehs.nih.gov/methods/acutetox/Tox_workshop.htm].
|
|
36.
|
OECD (2000), Guidance Document on the Recognition, Assessment and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluation, Environmental Health and Safety Monograph Series on Testing and Assessment No 19, ENV/JM/MONO(2000)7, OECD, Paris. K dispozícii na stránke: [http://www.oecd.org/env/testguidelines].
|
|
37.
|
Kimber, I., Hilton, J., Dearman, R. J., Gerberick, G. F., Ryan, C. A., Basketter, D. A., Lea, L., House, R. V., Ladies, G. S., Loveless, S. E. and Hastings, K. L. (1998), Assessment of the skin sensitisation potential of topical medicaments using the local lymph node assay: An interlaboratory exercise. J. Toxicol. Environ. Healt., 53 563 – 579.
|
|
38.
|
OECD (2005), Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessmen., Environment, Health and Safety Monograph Series on Testing and Assessment No 34, ENV/JM/MONO(2005)14, OECD, Paris. K dispozícii na stránke: [http://www.oecd.org/env/testguidelines].
|
Dodatok 1
VYMEDZENIE POJMOV
Presnosť: Miera zhody výsledkov testovacej metódy s akceptovanými referenčnými hodnotami. Ide o opatrenie výkonnosti testovacej metódy a jeden z aspektov relevantnosti. Tento pojem sa často zamieňa s pojmom ‚zhoda‘ a označuje podiel správnych výsledkov testovacej metódy (38).
Referenčná látka: Látka spôsobujúca alebo nespôsobujúca senzibilizáciu, ktorá sa používa ako štandardná látka na porovnávanie s testovanou látkou. Referenčná látka by mala mať tieto vlastnosti: i) konzistentný(-é) a spoľahlivý(-é) zdroj(-e); ii) štrukturálnu a funkčnú podobnosť s triedou látok, ktoré sa budú testovať; iii) známe fyzikálno-chemické vlastnosti; iv) podporné údaje o známych účinkoch a v) známu potenciu v rozsahu želanej odozvy.
Falošný negatívny výsledok: Látka je testovanou metódou nesprávne identifikovaná ako negatívna alebo neaktívna, pričom v skutočnosti je pozitívna alebo aktívna.
Falošný pozitívny výsledok: Látka je testom nesprávne identifikovaná ako pozitívna alebo aktívna, pričom v skutočnosti je negatívna alebo neaktívna.
Nebezpečnosť: Potenciál škodlivého účinku na zdravie alebo životné prostredie. Škodlivý účinok sa prejavuje až pri dostatočnej hladine expozície.
Medzilaboratórna reprodukovateľnosť: Miera, do akej rôzne kvalifikované laboratóriá, ktoré používajú rovnaký protokol a testujú rovnaké testovacie látky, dokážu dosiahnuť kvalitatívne a kvantitatívne podobné výsledky. Medzilaboratórna reprodukovateľnosť sa určuje počas postupov pred validáciou a v rámci postupov validácie a znamená mieru, do akej možno test úspešne prenášať medzi laboratóriami. Takisto je známa ako reprodukovateľnosť medzi laboratóriami (38).
Vnútrolaboratórna reprodukovateľnosť: Určenie miery, do akej kvalifikovaní pracovníci v rámci jedného laboratória dokážu úspešne opakovať výsledky s použitím konkrétneho protokolu v rôznom čase. Takisto je známa ako reprodukovateľnosť v rámci laboratória (38).
Extrémna hodnota: Extrémna hodnota je pozorovanie, ktoré je značne odlišné od ostatných hodnôt v náhodnej vzorke z populácie.
Zabezpečovanie kvality: Riadiaci proces, v ktorom jednotlivci nezávislí od osôb vykonávajúcich testy posudzujú súlad s laboratórnymi testovacími normami, požiadavkami a postupmi vedenia záznamov, ako aj presnosť prenosu údajov.
Spoľahlivosť: Meria rozsah, v rámci ktorého sa môže testovacia metóda počas istého časového obdobia opakovane vykonávať v rámci laboratórií a medzi nimi navzájom, pričom sa používa ten istý protokol. Posudzuje sa vypočítaním vnútrolaboratórnej a medzilaboratórnej reprodukovateľnosti (38).
Senzibilizácia kože: Imunologický proces, ku ktorému dochádza po lokálnom vystavení náchylného jednotlivca chemickému alergénu, ktorý vyvoláva kožnú imunitnú reakciu, ktorá môže viesť k vytvoreniu kontaktnej senzibilizácie.
Stimulačný index (SI): Hodnota vypočítaná na posúdenie potenciálu testovanej látky spôsobiť senzibilizáciu kože; ide teda o pomer proliferácie v pokusných skupinách a proliferácie v súbežných kontrolných skupinách, ktorým sa podáva nosič.
Testovaná látka (tiež sa nazýva testovaná chemikália): Akákoľvek látka alebo zmes testovaná pomocou tejto testovacej metódy.
B. 51. SENZIBILIZÁCIA KOŽE: LOKÁLNA SKÚŠKA LYMFATICKÝCH UZLÍN: BRDU-ELISA
ÚVOD
|
1.
|
Usmernenia Organizácie pre hospodársku spoluprácu a rozvoj (OECD) na testovanie chemikálií a testovacie metódy EÚ sa pravidelne revidujú vzhľadom na vedecký pokrok, zmeny regulačných potrieb a dobré životné podmienky zvierat. Prvá testovacia metóda (TM) (B.42) na stanovenie senzibilizácie kože u myší, tzv. lokálna skúška lymfatických uzlín (LLNA; usmernenie OECD na vykonávanie testov 429) bola zrevidovaná (1 a kapitola B.42 v tejto prílohe). Podrobnosti validácie LLNA a revízia prác v tejto oblasti boli uverejnené (2) (3) (4) (5) (6) (7) (8) (9). Pri LLNA sa na meranie proliferácie lymfocytov používa rádioizotopický tymidín alebo jód, a preto má táto skúška obmedzené použitie v tých prípadoch, kde je problematické získavanie, použitie alebo likvidácia rádioaktívnych látok. LLNA: BrdU-ELISA [enzýmová imunoskúška] je nerádioaktívna modifikácia testovacej metódy LLNA, ktorá na meranie proliferácie lymfocytov využíva 5-bróm-2-deoxyuridín (BrdU) (číslo CAS 59-14-3) v systéme testovania založenom na ELISA. LLNA: BrdU-ELISA bola validovaná a zrevidovaná a odporúča ju medzinárodný nezávislý vedecký panel pre partnerské preskúmanie ako užitočnú metódu na identifikáciu chemikálií spôsobujúcich alebo nespôsobujúcich senzibilizáciu kože s určitými obmedzeniami (10) (11) (12). Táto testovacia metóda je vytvorená na posúdenie potenciálu chemikálií (látok a zmesí) spôsobiť senzibilizáciu kože u zvierat. Metóda opísaná v kapitole B.6 tejto prílohy a usmernení OECD na vykonávanie testov 406 využíva testy s morčatami, a to maximalizačný test morčiat a Buehlerov test (13). LLNA (kapitola B.42 tejto prílohy; usmernenie OECD na vykonávanie testov 429) a dve nerádioaktívne modifikácie LLNA: BrdU-ELISA (kapitola B.51 tejto prílohy; usmernenie OECD na vykonávanie testov 442 B) a LLNA: DA (kapitola B.50 tejto prílohy; usmernenie OECD na vykonávanie testov 442 A) poskytujú výhodu v porovnaní s testami na morčatách opísanými v kapitole B.6 a usmernení OECD na vykonávanie testov 406 (13), a to pokiaľ ide o obmedzenie a zdokonalenie využívania zvierat.
|
|
2.
|
Podobne ako LLNA, LNNA: BrdU-ELISA skúma indukčnú fázu senzibilizácie kože a poskytuje kvantitatívne údaje vhodné na posúdenie odozvy na dávku. Okrem toho schopnosť identifikovať látky spôsobujúce senzibilizáciu kože bez potreby používania rádioaktívneho značenia DNA znižuje potenciál expozície rádioaktivite pri práci a problémy v súvislosti s likvidáciou odpadu. To môže následne umožniť zvýšené využívanie myší na identifikáciu látok spôsobujúcich senzibilizáciu kože, čo by ďalej mohlo znížiť využívanie morčiat na testovanie potenciálu senzibilizácie kože (t. j. B.6; usmernenie OECD na vykonávanie testov 406) (13).
|
VYMEDZENIE POJMOV
|
3.
|
Použité definície sú uvedené v dodatku 1.
|
ÚVODNÉ ÚVAHY A OBMEDZENIA
|
4.
|
LLNA: BrdU-ELISA je upravená metóda LLNA na identifikáciu potenciálu chemikálií spôsobiť senzibilizáciu kože, ktorá má určité obmedzenia. Neznamená to nevyhnutne, že vo všetkých prípadoch sa má použiť LLNA: BrdU-ELISA namiesto testu LLNA alebo testov s morčatami (t. j. metóda B.6; usmernenie OECD na vykonávanie testov 406) (13), ale skôr to, že táto skúška je rovnako hodnotná a môže sa použiť ako alternatíva, v ktorej pozitívne a negatívne výsledky vo všeobecnosti nevyžadujú ďalšie potvrdenie. Testovacie laboratórium by malo posúdiť všetky dostupné informácie o testovanej látke pred uskutočnením štúdie. Takéto informácie budú obsahovať identitu a chemickú štruktúru testovanej látky, jej fyzikálno-chemické vlastnosti, výsledky všetkých ostatných in vitro alebo in vivo testov toxicity s testovanou látkou a toxikologické údaje o štruktúrne príbuzných chemikáliách. Tieto informácie treba vziať do úvahy pri určovaní, či LLNA: BrdU-ELISA je pre testovanú látku vhodná [vzhľadom na nezlučiteľnosť obmedzených druhov chemikálií s LLNA: BrdU-ELISA (pozri odsek 5)] a ako pomoc pri výbere dávok.
|
|
5.
|
LLNA: BrdU-ELISA je metóda in vivo, a to znamená, že pri posudzovaní aktivity alergickej kontaktnej senzibilizácie sa nevylučuje používanie zvierat. Má však potenciál obmedziť používanie zvierat na tento účel pri porovnaní s testami na morčatách (B.6; usmernenie OECD na vykonávanie testov 406) (13). Okrem toho LLNA: BrdU-ELISA ponúka značné zjemnenie spôsobu, akým sa zvieratá používajú na testy alergickej kontaktnej senzibilizácie, keďže na rozdiel od metódy B.6 a usmernenia OECD na vykonávanie testov 406 LLNA: BrdU-ELISA nevyžaduje, aby sa vyvolali reakcie dermálnej hypersenzitivity. Okrem toho LLNA: BrdU-ELISA nevyžaduje použitie adjuvansu ako v prípade maximalizačného testu morčiat (kapitola B.6 tejto prílohy, 13). Testovacia metóda LLNA: BrdU-ELISA tak znižuje utrpenie zvierat. Napriek výhodám, ktoré LLNA: BrdU-ELISA má v porovnaní s metódou B.6 a usmernením OECD na vykonávanie testov 406 (13), existujú určité obmedzenia, pre ktoré možno bude potrebné používať B.6 alebo usmernenie OECD na vykonávanie testov 406 [napr. testovanie určitých kovov, falošné pozitívne zistenia s určitými kožnými dráždivými látkami (ako napríklad niektoré povrchovo aktívne látky) (6) (1 a kapitola B.42 tejto prílohy) alebo rozpustnosť testovanej látky]. Okrem toho chemické triedy alebo látky, ktoré obsahujú potenciálne zmätočné funkčné skupiny (15), si tiež môžu vyžadovať použitie testov s morčatami (t. j. B.6; usmernenie OECD na vykonávanie testov 406) (13). Obmedzenia, ktoré boli identifikované pre LLNA (1 a kapitola B.42 tejto prílohy), sa odporúčajú aj pre LLNA: BrdU-ELISA (10). Okrem takto identifikovaných obmedzení by sa LLNA: BrdU-ELISA mala uplatňovať pri testovaní akýchkoľvek chemikálií okrem prípadu, keď tieto chemikálie majú vlastnosti, ktoré by mohli narúšať presnosť LLNA: BrdU-ELISA. Okrem toho by sa mala zvážiť možnosť hraničných pozitívnych výsledkov v prípade, že sa dosiahnu hodnoty stimulačného indexu (SI) od 1,6 do 1,9 (pozri odseky 31 a 32). To je založené na validačnej databáze 43 látok s použitím SI ≥ 1,6 (pozri odsek 6), pre ktoré LLNA: BrdU-ELISA správne identifikovala všetkých 32 látok spôsobujúcich senzibilizáciu podľa LLNA, ale nesprávne identifikovala dve z 11 látok nespôsobujúcich senzibilizáciu podľa LLNA s hodnotami SI od 1,6 do 1,9 (t. j. hraničný pozitívny výsledok) (10). Keďže sa však na stanovenie hodnôt SI a výpočet predpokladaných vlastností testu použil rovnaký súbor údajov, stanovené údaje môžu byť nadhodnotením skutočných prediktívnych vlastností.
|
PRINCÍP TESTOVACEJ METÓDY
|
6.
|
Základným princípom, na ktorom sa LLNA: BrdU-ELISA zakladá, je, že senzibilizátory vyvolávajú proliferáciu lymfocytov v lymfatickej uzline v drénovanom mieste aplikácie testovanej látky. Táto proliferácia je úmerná dávke a účinnosti aplikovaného alergénu a poskytuje jednoduchý prostriedok na získanie kvantitatívneho merania senzibilizácie. Proliferácia sa meria porovnávaním priemernej proliferácie v každej pokusnej skupine s priemernou proliferáciou v kontrolnej skupine, ktorej sa podáva nosič. Určí sa pomer priemernej proliferácie v každej ošetrenej skupine a priemernej proliferácie v súbežnej kontrolnej skupine s nosičom, ktorý sa označuje ako stimulačný index (SI), a mal by mať hodnotu ≥ 1,6 pred ďalším prípadným vyhodnotením testovanej látky ako potenciálneho senzibilizátora kože. Uvedené metódy sú založené na meraní obsahu BrdU na preukázanie proliferácie zvýšeného počtu buniek v drénovaných ušných lymfatických uzlinách. BrdU je podobný tymidínu a je podobne začlenený do DNA proliferujúcich buniek. Začlenenie BrdU sa meria metódou ELISA, ktorá využíva peroxidázou označenú protilátku špecifickú pre BrdU. Peroxidáza po pridaní substrátu reaguje so substrátom produkujúcim sfarbený produkt, ktorý sa kvantifikuje pri konkrétnej absorbancii na mikrotitračnej platničke.
|
OPIS SKÚŠKY
Výber druhov zvierat
|
7.
|
Vybraným druhom pre tento test je myš. Validačné štúdie LLNA: BrdU-ELISA sa vykonávali výlučne s myšami kmeňa CBA/JN, ktorý sa preto považuje za preferovaný kmeň (10) (12). Použijú sa mladé dospelé negravidné samice myši, ktoré nikdy nevrhli mláďatá. Na začiatku štúdie by zvieratá mali byť vo veku 8 – 12 týždňov a rozdiel medzi hmotnosťami zvierat má byť minimálny a nemá byť vyšší ako 20 % priemernej hmotnosti. Alternatívne sa môžu použiť iné kmene a samce, ak sa na základe údajov dá uspokojivo dokázať, že neexistujú významné odlišnosti špecifické pre kmeň a/alebo pohlavie v odozve na LLNA: BrdU-ELISA.
|
Podmienky umiestnenia a kŕmenia
|
8.
|
Myši by sa mali umiestniť v skupinách (16) okrem prípadu, ak sa poskytne primerané vedecké odôvodnenie ich umiestnenia jednotlivo. Teplota miestnosti pre pokusné zvieratá by mala byť 22 ± 3 °C. Aj keď by relatívna vlhkosť mala byť aspoň 30 % a pokiaľ možno by nemala prevyšovať 70 % okrem obdobia počas čistenia miestnosti, cieľom je, aby relatívna vlhkosť dosahovala 50 % – 60 %. Osvetlenie by malo byť umelé a malo by sa striedať 12 hodín svetla a 12 hodín tmy. Na kŕmenie sa môže používať bežné laboratórne krmivo s neobmedzenou dodávkou pitnej vody.
|
Príprava zvierat
|
9.
|
Zvieratá sa vyberú náhodne a sú označené tak, aby sa umožnila jednotlivá identifikácia (ale nie formou ušných značiek), a držia sa v klietkach aspoň 5 dní pred začiatkom podávania dávok, aby sa aklimatizovali na laboratórne podmienky. Pred začatím ošetrenia sa všetky zvieratá vyšetria, aby sa zaistilo, že nemajú žiadne viditeľné kožné poranenia.
|
Príprava dávok
|
10.
|
Chemikálie v tuhom skupenstve by sa pred podaním dávky do ucha myši mali rozpustiť alebo suspendovať v rozpúšťadlách alebo nosičoch, a ak je to vhodné, rozriediť. Kvapalné chemikálie možno podávať priamo alebo pred podaním rozriediť. V prípade nerozpustných chemikálií, ako napríklad tých, ktoré sa vo všeobecnosti používajú ako zdravotnícke pomôcky, by sa pred podaním dávky do ucha myši mala uskutočniť zvýšená extrakcia vo vhodnom rozpúšťadle, aby sa odhalili všetky extrahovateľné zložky, ktoré sa budú testovať. Testované látky sa pripravujú každý deň okrem prípadu, keď údaje o stabilite preukazujú prijateľnosť uskladnenia.
|
Kontrola spoľahlivosti
|
11.
|
Riadne vykonanie skúšky sa dokazuje pomocou pozitívnych kontrolných chemikálií, ktoré reagujú primeranou a reprodukovateľnou citlivosťou ako senzibilizujúce testované látky, pre ktoré je dobre charakterizovaná šírka odozvy. Odporúča sa zahrnúť súbežnú pozitívnu kontrolu, pretože to dokazuje kompetenciu laboratória úspešne vykonať každú skúšku a umožňuje posúdenie vnútrolaboratórnej a medzilaboratórnej reprodukovateľnosti a porovnateľnosti. Pozitívnu kontrolu v každej štúdii vyžadujú aj niektoré regulačné orgány, a preto by používatelia mali pred vykonaním LLNA: BrdU-ELISA konzultovať s príslušnými orgánmi. Rutinné použitie súbežnej pozitívnej kontroly sa podporuje aj preto, aby sa predišlo potrebe ďalšieho testovania na zvieratách s cieľom splniť také požiadavky, ktoré by mohli vzniknúť z použitia periodickej pozitívnej kontroly (pozri odsek 12). Pozitívna kontrola by mala spôsobiť pozitívnu odozvu na LLNA: BrdU-ELISA pri úrovni expozície, pri ktorej sa očakáva nárast stimulačného indexu (SI) > 1,6 v porovnaní s negatívnou kontrolnou (NC) skupinou. Dávka chemickej látky na pozitívnu kontrolu by sa mala zvoliť tak, aby nespôsobila nadmerné podráždenie kože ani systémovú toxicitu a aby indukcia bola reprodukovateľná, ale nie nadmerná (napr. SI > 14 by bol nadmerný). Vhodnými chemikáliami na pozitívnu kontrolu sú: 25 % 3-fenyl-2-hexylpropanál (číslo CAS 101-86-0) a 25 % eugenol (číslo CAS 97-53-0) v acetóne: olivový olej (4: 1, objemové percentá). Môžu sa vyskytnúť okolnosti, keď sa v riadne zdôvodnených prípadoch môžu použiť iné pozitívne kontroly, ktoré spĺňajú uvedené kritériá.
|
|
12.
|
Hoci sa odporúča zahrnutie súbežnej pozitívnej kontrolnej skupiny, môžu sa vyskytnúť situácie, keď periodické testovanie (v intervaloch ≤ 6 mesiacov) s pozitívnou kontrolou môže byť vhodné v prípade tých laboratórií, ktoré vykonávajú LLNA: BrdU-ELISA pravidelne (t. j. vykonávajú LLNA: BrdU-ELISA v priebehu jedného mesiaca aspoň raz) a ktoré majú zavedenú historickú databázu pozitívnych kontrol, ktorá preukazuje schopnosť laboratória dosahovať reprodukovateľné a presné výsledky v pozitívnych kontrolách. BrdU-ELISA možno úspešne preukázať dosiahnutím konzistentných pozitívnych výsledkov v pozitívnej kontrole najmenej v 10 nezávislých testoch uskutočnených v určitom primeranom období (t. j. v období kratšom ako jeden rok).
|
|
13.
|
Súbežná pozitívna kontrolná skupina sa má zahrnúť vždy, keď dôjde k procedurálnej zmene v súvislosti s LLNA: BrdU-ELISA (napr. k zmene vyškoleného personálu, zmene materiálov a/alebo reagentov používaných pri testovacej metóde, zmene nástrojov používaných pri testovacej metóde alebo zmene zdroja testovaných zvierat), a takéto zmeny je potrebné zaznamenať v laboratórnych správach. Je potrebné zvážiť vplyv týchto zmien na primeranosť predtým zriadenej historickej databázy pri určovaní potreby zriadenia novej historickej databázy na zaznamenávanie konzistentnosti výsledkov pozitívnej kontroly.
|
|
14.
|
Výskumní pracovníci by si mali uvedomiť, že rozhodnutie vykonať štúdiu pozitívnych kontrol na periodickom základe namiesto súbežných pozitívnych kontrol môže mať následky na primeranosť a prijateľnosť výsledkov negatívnej štúdie získaných bez súbežnej pozitívnej kontroly počas intervalu medzi každou periodickou štúdiou pozitívnej kontroly. Ak sa dosiahne napríklad falošný negatívny výsledok v periodickej štúdii pozitívnej kontroly, negatívne výsledky získané s testovanou látkou v intervale medzi poslednou prijateľnou periodickou štúdiou pozitívnej kontroly a neprijateľnou periodickou štúdiou pozitívnej kontroly možno spochybniť. Dôsledky týchto výsledkov treba dôkladne zvážiť pri určovaní toho, či vykonať súbežné pozitívne kontroly, alebo iba periodické pozitívne kontroly. Treba tiež zvážiť použitie menšieho počtu zvierat v súbežnej pozitívnej kontrolnej skupine, ak je to vedecky odôvodnené a ak laboratórium na základe konkrétnych laboratórnych historických údajov preukáže, že možno použiť menší počet myší (17).
|
|
15.
|
Aj keď by sa pozitívna kontrola mala testovať v nosiči, o ktorom je známe, že vyvoláva konzistentnú odozvu (napr. acetón: olivový olej v pomere 4: 1, objemové percentá), môžu sa vyskytnúť určité regulačné situácie, v ktorých bude potrebné vykonať aj testovanie v neštandardnom nosiči (klinicky/chemicky zodpovedajúcom roztoku) (18). Ak sa súbežná pozitívna kontrola testuje v odlišnom nosiči ako testovaná látka, potom treba do testu súbežnej pozitívnej kontroly zahrnúť samostatnú kontrolnú skupinu, ktorej sa podáva nosič.
|
|
16.
|
V prípadoch hodnotenia testovaných látok, ktoré patria do konkrétnej chemickej triedy, alebo rozsahu odoziev môžu byť užitočné aj referenčné látky, aby sa preukázalo, že testovacia metóda na zisťovanie potenciálu týchto druhov testovaných látok spôsobiť senzibilizáciu kože funguje riadne. Vhodné referenčné látky majú mať tieto vlastnosti:
|
—
|
štrukturálnu a funkčnú podobnosť s triedou testovanej látky, ktorá je predmetom testovania,
|
|
—
|
známe fyzikálno-chemické vlastnosti,
|
|
—
|
podporné údaje z LLNA: BrdU-ELISA,
|
|
—
|
podporné údaje z ďalších zvieracích a/alebo ľudských modelov.
|
|
TESTOVACÍ POSTUP
Počet zvierat a veľkosť dávky
|
17.
|
Na dávkovú skupinu sa používajú aspoň štyri zvieratá, aspoň tri koncentrácie testovanej látky a k tomu súbežná negatívna kontrolná skupina, ktorej sa podáva iba nosič pre testovanú látku, a pozitívna kontrolná skupina (súbežná alebo súčasná, podľa laboratórnych pravidiel v súvisiacich odsekoch 11 – 15). Treba zvážiť testovanie viacerých dávok pozitívnej kontroly, najmä ak sa testuje na prerušovanej báze. So zvieratami v kontrolných skupinách by sa malo zaobchádzať rovnakým spôsobom ako s pokusnými zvieratami okrem prípadov, keď sa ošetrovanie testovanou látkou nevykonáva.
|
|
18.
|
Výber dávky a nosiča by mal byť založený na odporúčaniach uvedených v odkazoch 2 a 19. Následné dávky sa zvyčajne vyberajú z vhodných sérií koncentrácií, napr. 100 %, 50 %, 25 %, 10 %, 5 %, 2,5 %, 1 %, 0,5 % atď. Výber zo sérií koncentrácií je potrebné primerane vedecky zdôvodniť. Všetky existujúce informácie o toxicite (napr. akútna toxicita a podráždenie kože), ako aj štrukturálne a fyzikálno-chemické informácie o testovanej látke, ktorá je predmetom záujmu (a/alebo štrukturálne príbuzných látkach), ak sú k dispozícii, by sa mali posúdiť výberom troch po sebe idúcich koncentrácií tak, aby najvyššia koncentrácia maximalizovala expozíciu, pričom by sa však zabránilo systémovej toxicite a/alebo nadmernému lokálnemu podráždeniu kože (19) (20 a kapitola B.4 tejto prílohy). Ak tieto informácie nie sú k dispozícii, možno bude potrebné vykonať predbežný skríningový test (pozri odseky 21 – 24).
|
|
19.
|
Nosič by nemal ovplyvňovať výsledok testu a mal by sa vybrať na základe maximálnej rozpustnosti s cieľom získať najvyššiu dosiahnuteľnú koncentráciu, pričom vznikne roztok/suspenzia vhodná na aplikáciu testovanej látky. Odporúčané nosiče sú: acetón: olivový olej (4: 1, objemové percentá), N,N-dimetylformamid, metyletylketón, propylénglykol a dimetylsulfoxid (6), ale môžu sa použiť aj iné, ak sa uvedie uspokojivé vedecké zdôvodnenie. V určitých situáciách môže byť potrebné použiť ako ďalšiu kontrolu klinicky vhodný roztok alebo komerčný prípravok, v ktorom sa testovaná látka predáva. Osobitnú pozornosť je potrebné venovať zabezpečeniu toho, aby sa hydrofilné testované látky začlenili do systému nosiča, ktorý zvlhčuje pokožku, a rýchlo neunikli, a to začlenením vhodných rozpúšťadiel (napr. 1 % Pluronic® L92). Preto je potrebné vyhýbať sa použitiu čisto vodných nosičov.
|
|
20.
|
Spracovanie lymfatických uzlín jednotlivých myší umožňuje posúdiť variabilitu medzi zvieratami a štatisticky porovnať rozdiel medzi meraniami testovanej látky a kontrolnej skupiny, ktorej sa podáva nosič (pozri odsek 33). Okrem toho sa zníženie počtu myší v pozitívnej kontrolnej skupine môže prehodnotiť len po poskytnutí informácií o jednotlivých zvieratách (17). Navyše niektoré regulačné orgány požadujú zber údajov o jednotlivých zvieratách. Pravidelný zber údajov o jednotlivých zvieratách poskytuje výhodu, pokiaľ ide o dobré životné podmienky zvierat, pretože sa predchádza duplicitnému testovaniu, ktoré by bolo potrebné v prípade, ak by výsledky testovanej látky pôvodne získané jedným spôsobom (napr. prostredníctvom spojených údajov o zvieratách) museli neskôr posúdiť regulačné orgány podľa iných požiadaviek (napr. prostredníctvom údajov o jednotlivých zvieratách).
|
Predbežný skríningový test
|
21.
|
V prípade, ak nie sú k dispozícii informácie na stanovenie najvyššej dávky testovanej látky (pozri odsek 18), treba vykonať predbežný skríningový test s cieľom stanoviť primeranú úroveň testovanej dávky na použitie v LLNA: BrdU-ELISA. Cieľom predbežného skríningového testu je poskytnúť usmernenie na výber maximálnej úrovne dávky na použitie v hlavnej štúdii LLNA: BrdU-ELISA v prípade, že nie sú dostupné informácie o koncentrácii, ktorá spôsobuje systémovú toxicitu (pozri odsek 24) a/alebo nadmerné lokálne podráždenie kože (pozri odsek 23). Maximálna úroveň testovanej dávky by mala obsahovať 100 % testovanej látky v prípade kvapalín alebo maximálnu možnú koncentráciu v prípade tuhých látok alebo suspenzií.
|
|
22.
|
Predbežný skríningový test sa vykonáva za podmienok, ktoré sú identické s hlavnou štúdiou LLNA: BrdU-ELISA, s tým rozdielom, že sa neposudzuje proliferácia lymfatickej uzliny a môže sa použiť menší počet zvierat na dávkovú skupinu. Navrhuje sa jedno alebo dve zvieratá na dávkovú skupinu. U všetkých myší sa každý deň pozorujú klinické príznaky systémovej toxicity alebo lokálneho podráždenia v mieste aplikácie. Telesné hmotnosti zvierat sa zaznamenajú na začiatku testu a pred usmrtením (deň 6). Obidve uši myší sa vyšetria, či nemajú príznaky erytému (sčervenania kože), a vyhodnotia sa podľa tabuľky 1 (20 a kapitola B.4 tejto prílohy). Hrúbka ucha sa meria pomocou meradla hrúbky (napr. digitálnym mikrometrom alebo meradlom hrúbky Peacock Dial) v deň 1 (pred aplikáciou dávky), v deň 3 (približne 48 hodín po prvej dávke) a v deň 6. Okrem toho možno hrúbku ucha v deň 6 určiť na základe meraní vážením vzoriek uší, ktoré by sa malo vykonať po humánnom usmrtení zvierat. Nadmerné lokálne podráždenie kože indikuje hodnotenie erytému ≥ 3 a/alebo zvýšenie hrúbky ucha o ≥ 25 % v ktorýkoľvek deň merania (21) (22). Najvyššia dávka vybraná pre hlavnú štúdiu LLNA: BrdU-ELISA bude najbližšia nižšia dávka z predbežných skríningových sérií koncentrácií (pozri odsek 18), ktorá nespôsobuje systémovú toxicitu a/ani nadmerné lokálne podráždenie kože.
Tabuľka 1
Hodnotenia erytému
|
Pozorovanie
|
Hodnotenie
|
|
Žiadny erytém
|
0
|
|
Veľmi slabý erytém (sotva pozorovateľný)
|
1
|
|
Jasne ohraničený erytém
|
2
|
|
Mierny až silný erytém
|
3
|
|
Výrazný erytém (tmavočervený) až vytvorenie chrasty zabraňujúcej posúdeniu erytému
|
4
|
|
|
23.
|
Okrem 25 % zvýšenia hrúbky ucha (21) (22) sa štatisticky významné zvýšenie hrúbky ucha v testovanej skupine myší v porovnaní s kontrolnou skupinou myší tiež použilo na identifikáciu dráždivých látok v skúške LLNA (22) (23) (24) (25) (26) (27) (28). Kým štatisticky významné zvýšenia môžu nastať, aj keď dôjde k zvýšeniu hrúbky ucha menšiemu ako 25 %, nie sú však konkrétne spájané s nadmerným podráždením kože (25) (26) (27) (28) (29).
|
|
24.
|
Nasledujúce klinické pozorovania môžu indikovať systémovú toxicitu (30), ak sú súčasťou integrovaného posudzovania, to znamená, že môžu indikovať maximálnu dávku na použitie v hlavnej LLNA: BrdU-ELISA: zmeny funkcie nervového systému (napr. piloerekcia, ataxia, triaška a kŕče), zmeny správania (napr. agresia, zmena pri čistení, výrazná zmena stupňa aktivity), zmena dýchania (t. j. zmena frekvencie alebo intenzity dýchania, ako napríklad zadúšanie, dýchavičnosť a šelest) a zmeny spotreby potravy a vody. Okrem toho treba v hodnotení zvážiť príznaky letargie a/alebo apatie a akékoľvek klinické príznaky silnejšej alebo dlhodobejšej bolesti a utrpenia alebo viac ako 5 % zníženie telesnej hmotnosti medzi dňom 1 a 6 a úmrtnosť. Umierajúce zvieratá alebo zvieratá, ktoré majú zjavne silné bolesti alebo vykazujú príznaky silného utrpenia, by sa mali humánnym spôsobom usmrtiť (31).
|
Harmonogram pokusov v hlavnej štúdii
|
25.
|
Harmonogram pokusov v rámci skúšky je takýto:
— Deň 1: Označte každé zviera a zaznamenajte jeho hmotnosť a akékoľvek klinické pozorovania. Začnite s aplikáciou 25 μl z príslušného zriedenia testovanej látky, samotného nosiča alebo pozitívnej kontroly (súbežnej alebo nedávnej, podľa laboratórnych pravidiel s prihliadnutím na odseky 11 – 15) na zadnú stranu každého ucha.
— Dni 2 a 3: Zopakujte aplikačný postup uskutočnený v deň 1.
— Deň 4: Žiadne ošetrenie.
— Deň 5: Intraperitoneálnou injekciou aplikujte 0,5 ml (5 mg/myš) roztoku BrdU (10 mg/ml).
— Deň 6: Zaznamenajte hmotnosť každého zvieraťa a akékoľvek klinické príznaky. Približne o 24 hodín po injekčnej aplikácii roztoku BrdU sa zvieratá humánne usmrtia. Drénované ušné lymfatické uzliny sa odstránia z každého ucha myši a jednotlivo sa spracujú vo fosfátom tlmenom fyziologickom roztoku (PBS) pre každé zviera. Údaje a diagramy identifikácie lymfatických uzlín a disekcie možno nájsť v odkaze (17). Pri ďalšom monitorovaní lokálnej odozvy kože v hlavnej štúdii možno do protokolu štúdie zahrnúť ďalšie parametre, ako napríklad hodnotenie erytému ucha alebo merania hrúbky ucha (získané pomocou meradla hrúbky alebo vážením vzoriek uší pri pitve).
|
Príprava bunkových suspenzií
|
26.
|
Jednobunková suspenzia buniek lymfatických uzlín (LNC) odobraná bilaterálne z každej myši sa pripravuje jemným mechanickým pretlačením cez sitko z nerezovej ocele so šírkou oka 200 μm alebo inou prijateľnou technikou na prípravu jednobunkovej suspenzie (napr. pomocou plastového mažiara na jedno použitie, ktorým sa lymfatické uzliny rozdrvia a následne prepasírujú cez nylonové sitko #70.) Postup na prípravu suspenzie buniek lymfatických uzlín je v tejto skúške veľmi dôležitý, a preto by každý odborník mal byť v tomto postupe vopred vyškolený. Navyše keďže lymfatické uzliny zvierat v negatívnej kontrole sú malé, dôležité je opatrné zaobchádzanie, aby sa predišlo umelým účinkom na hodnoty SI. V každom prípade by sa cieľový objem suspenzie buniek lymfatických uzlín mal prispôsobiť stanovenému optimalizovanému objemu (približne 15 ml). Optimalizovaný objem je založený na dosiahnutí priemernej absorbancie v negatívnej kontrolnej skupine v rozmedzí 0,1 – 0,2.
|
Stanovenie proliferácie buniek (meranie obsahu BrdU v DNA lymfocytov)
|
27.
|
BrdU sa meria metódou ELISA pomocou komerčnej testovacej súpravy (napr. Roche Applied Science, Mannheim, Nemecko, katalógové číslo 11 647 229 001). V skratke 100 μl suspenzie buniek lymfatických uzlín sa trikrát pridá do jamiek mikroplatničky. Po fixácii a denaturácii buniek lymfatických uzlín sa do každej jamky pridá protilátka BrdU a čaká sa na reakciu. Následne sa protilátka BrdU odstráni umytím, pridá sa substrátový roztok a čaká sa na tvorbu chromogénu. Odmeria sa absorbancia 370 nm s referenčnou vlnovou dĺžkou 492 nm. Vo všetkých prípadoch by sa testovacie podmienky skúšky mali optimalizovať (pozri odsek 26).
|
POZOROVANIA
Klinické pozorovania
|
28.
|
Každá myš sa starostlivo vyšetrí aspoň jedenkrát denne na všetky klinické príznaky, či už ide o lokálne podráždenie v mieste aplikácie, alebo systémovú toxicitu. Všetky pozorovania sa systematicky zaznamenávajú, pričom každá myš má individuálny záznam. Plány na monitorovanie by mali zahŕňať kritériá na rýchlu identifikáciu tých myší, ktoré vykazujú systémovú toxicitu, nadmerné lokálne podráždenie alebo poleptanie kože a ktoré majú byť humánne usmrtené (31).
|
Telesné hmotnosti
|
29.
|
Ako sa uvádza v odseku 25, hmotnosti jednotlivých zvierat by sa mali odmerať na začiatku testu a pri naplánovanom humánnom usmrtení zvierat.
|
VÝPOČET VÝSLEDKOV
|
30.
|
Výsledky v každej pokusnej skupine sa vyjadrujú ako priemerná hodnota stimulačného indexu (SI). SI sa vypočíta vydelením priemerného indexu označenia BrdU/myš v rámci každej skupiny, ktorej sa podáva testovaná látka, a pozitívnej kontrolnej skupiny priemerným indexom označenia BrdU v kontrolnej skupine s rozpúšťadlom/nosičom. Priemerná hodnota SI kontrolných skupín, ktorým sa podával nosič, je potom 1.
Index označenia BrdU sa definuje ako:
Index označenia BrdU = (ABSem – ABS prázdnyem) – (ABSref – ABS prázdnyref),
kde: em = emisná vlnová dĺžka a ref = referenčná vlnová dĺžka.
|
|
31.
|
V rozhodovacom procese sa za pozitívny výsledok považuje stimulačný index ≥ 1,6 (10). Pri určovaní toho, či hraničný výsledok (napr. hodnota SI v rozmedzí od 1,6 do 1,9) je pozitívny, sa však môže použiť aj sila vzťahu medzi dávkou a odozvou, štatistický význam a konzistentnosť rozpúšťadla/nosiča a odoziev pozitívnej kontroly (3) (6) (32).
|
|
32.
|
Pri hraničnom výsledku SI od 1,6 do 1,9 môžu používatelia na potvrdenie toho, že také výsledky sú pozitívne, zvážiť doplňujúce informácie, ako napríklad vzťah medzi dávkou a odozvou, dôkazy systémovej toxicity alebo nadmerného podráždenia a v prípade potreby štatistický význam spolu s hodnotami SI (10). Pozornosť by sa mala venovať aj rôznym vlastnostiam testovanej látky vrátane toho, či má štrukturálny vzťah k látkam, ktoré sú známymi senzibilizátormi, či spôsobuje nadmerné podráždenie kože, a vrátane charakteru pozorovanej odozvy na dávku. Tieto a ostatné hľadiská sa podrobne rozoberajú inde (4).
|
|
33.
|
Zber údajov na úrovni jednotlivých myší umožní vykonať štatistickú analýzu prítomnosti a úrovne vzťahu medzi dávkou a odozvou v údajoch. Každé štatistické posúdenie by mohlo zahŕňať hodnotenie vzťahu medzi dávkou a odozvou, ako aj vhodne upravené porovnávania testovaných skupín (napr. porovnávania v pároch skupiny, ktorej sa podáva dávka, a súbežnej kontrolnej skupiny s rozpúšťadlom/nosičom). Štatistické analýzy môžu zahŕňať napr. lineárnu regresiu alebo Williamsov test na posúdenie trendov vo vzťahu medzi dávkou a odozvou a Dunnettov test na porovnávanie v pároch. Pri výbere vhodnej metódy štatistickej analýzy by si výskumný pracovník mal uvedomovať možné nerovnosti rozdielov a ďalšie súvisiace problémy, ktoré si môžu vyžadovať premenu údajov alebo štatistickú analýzu založenú na iných ako parametrických údajoch. Výskumný pracovník bude možno v každom prípade musieť vykonať výpočty SI a štatistické analýzy s určitými dátovými bodmi a bez nich (niekedy sa tieto údaje nazývajú ‚extrémne hodnoty‘).
|
ÚDAJE A PODÁVANIE SPRÁV
Údaje
|
34.
|
Údaje by sa mali zhrnúť vo forme tabuľky, v ktorej sa uvádzajú hodnoty indexu označenia BrdU jednotlivých zvierat, priemerný skupinový index označenia BrdU/zviera, súvisiaci rozsah chyby (napr. SD, SEM) a stredná hodnota SI pre každú skupinu s dávkou v porovnaní so súbežnou kontrolnou skupinou s rozpúšťadlom/nosičom.
|
Správa o teste
|
35.
|
Správa o teste by mala obsahovať tieto informácie:
|
|
Testované a kontrolné chemické látky:
|
—
|
identifikačné údaje (napr. čísla CAS, čísla EC, ak sú k dispozícii, zdroj, čistota, známe nečistoty, číslo šarže),
|
|
—
|
fyzikálny charakter a fyzikálno-chemické vlastnosti (napr. prchavosť, stabilita, rozpustnosť),
|
|
—
|
v prípade zmesi zloženie a relatívny percentuálny podiel zložiek.
|
|
|
|
Rozpúšťadlo/nosič:
|
—
|
identifikačné údaje (čistota, v prípade potreby koncentrácia, použitý objem),
|
|
—
|
zdôvodnenie voľby nosiča.
|
|
|
|
Testovacie zvieratá:
|
—
|
mikrobiologický stav zvierat, ak je známy,
|
|
—
|
zdroj zvierat, podmienky umiestnenia, krmivo atď.
|
|
|
|
Podmienky testu:
|
—
|
zdroj, číslo šarže a kontrolné údaje výrobcu o kvalite/presnosti (citlivosť a špecifickosť protilátky a limit detekcie) pre súpravu ELISA,
|
|
—
|
informácie o príprave a aplikácii testovanej látky,
|
|
—
|
zdôvodnenie výberu dávky (vrátane výsledkov z predbežného skríningového testu, ak sa uskutočnil),
|
|
—
|
použité koncentrácie nosiča a testovanej látky a celkové množstvo aplikovanej testovanej látky,
|
|
—
|
údaje o kvalite krmiva a vody (vrátane druhu/zdroja krmiva, zdroja vody),
|
|
—
|
podrobný opis harmonogramu podávania dávok a odberu vzoriek,
|
|
—
|
metódy merania toxicity,
|
|
—
|
kritériá na hodnotenie štúdií ako pozitívnych alebo negatívnych,
|
|
—
|
informácie o akýchkoľvek odchýlkach od protokolu a vysvetlenie, ako odchýlky ovplyvňujú návrh a výsledky štúdie.
|
|
|
|
Kontrola spoľahlivosti:
|
—
|
súhrn výsledkov poslednej kontroly spoľahlivosti vrátane informácií o testovanej látke, koncentrácii a použitom nosiči,
|
|
—
|
súbežné a/alebo historické kontrolné údaje o pozitívnej a súbežnej negatívnej kontrole (s rozpúšťadlom/nosičom) pre testovacie laboratórium,
|
|
—
|
ak nebola zahrnutá súbežná pozitívna kontrola, dátum a laboratórna správa o najaktuálnejšej periodickej pozitívnej kontrole a správa s historickými údajmi o pozitívnej kontrole, ktorými laboratórium zdôvodňuje nevykonanie súbežnej pozitívnej kontroly.
|
|
|
|
Výsledky:
|
—
|
hmotnosti jednotlivých myší na začiatku podávania dávky a pri naplánovanom humánnom usmrtení, ako aj priemerný a súvisiaci rozsah chyby (napr. SD a SEM) pre každú pokusnú skupinu,
|
|
—
|
časový priebeh nástupu príznakov toxicity u každého zvieraťa vrátane podráždenia kože v mieste podania látky, ak sa vyskytnú,
|
|
—
|
tabuľka indexov označenia BrdU u jednotlivých myší a hodnoty SI v každej pokusnej skupine,
|
|
—
|
priemerný a súvisiaci rozsah chyby (napr. SD, SEM) v súvislosti s indexom označenia BrdU/myš v každej pokusnej skupine a výsledky analýzy extrémnej hodnoty v každej pokusnej skupine,
|
|
—
|
výpočet SI a vhodné opatrenie variability, ktoré berie do úvahy variabilitu medzi zvieratami v skupinách, ktorým sa podáva testovaná látka, a v kontrolných skupinách,
|
|
—
|
vzťah medzi dávkou a odozvou,
|
|
—
|
v prípade potreby štatistické analýzy.
|
|
|
|
Rozbor výsledkov:
|
—
|
stručný komentár v súvislosti s výsledkami, analýza odozvy na dávku, štatistické analýzy, ak existujú, s uvedením záveru, či by sa testovaná látka mala považovať za látku spôsobujúcu senzibilizáciu kože.
|
|
|
LITERATÚRA
|
1.
|
OECD (2010), Skin Sensitisation: Local Lymph Node Assay, Test Guideline No 429, Guidelines for the Testing of Chemicals, OECD, Paris. K dispozícii na stránke: [http://www.oecd.org/env/testguidelines].
|
|
2.
|
Chamberlain, M. and Basketter, D. A. (1996), The local lymph node assay: status of validation. Food Chem. Toxicol., 34, 999 – 1002.
|
|
3.
|
Basketter, D. A., Gerberick, G. F., Kimber, I. and Loveless, S. E. (1996), The local lymph node assay: A viable alternative to currently accepted skin sensitisation tests. Food Chem. Toxicol., 34, 985 – 997.
|
|
4.
|
Basketter, D. A., Gerberick, G. F. and Kimber, I. (1998), Strategies for identifying false positive responses in predictive sensitisation tests. Food Chem. Toxicol., 36, 327 – 333.
|
|
5.
|
Van Och, F. M. M., Slob, W., De Jong, W. H., Vandebriel, R. J. and Van Loveren, H. (2000), A quantitative method for assessing the sensitising potency of low molecular weight chemicals using a local lymph node assay: employment of a regression method that includes determination of uncertainty margins. Toxicol., 146, 49 – 59.
|
|
6.
|
ICCVAM (1999), The murine local lymph node Assay: A test method for assessing the allergic contact dermatitis potential of chemicals/compounds: The results of an independent peer review evaluation coordinated by the Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM) and the National Toxicology Program Center for the Evaluation of Alternative Toxicological Methods (NICETAM). NIH Publication No 99-4494. Research Triangle Park, N.C. K dispozícii na stránke: [http://iccvam.niehs.nih.gov/docs/immunotox_docs/llna/llnarep.pdf].
|
|
7.
|
Dean, J. H., Twerdok, L. E., Tice, R. R., Sailstad, D. M., Hattan, D. G., Stokes, W. S. (2001), ICCVAM evaluation of the murine local lymph node assay: II. Conclusions and recommendations of an independent scientific peer review panel. Reg. Toxicol. Pharmacol., 34(3), 258 – 273.
|
|
8.
|
Haneke, K. E., Tice, R. R., Carson, B. L., Margolin, B. H., Stokes, W. S. (2001), ICCVAM evaluation of the murine local lymph node assay: III. Data analyses completed by the national toxicology program interagency center for the evaluation of alternative toxicological methods. Reg. Toxicol. Pharmacol., 34(3), 274 – 286.
|
|
9.
|
Sailstad, D. M., Hattan, D., Hill, R. N., Stokes, W. S. (2001), ICCVAM evaluation of the murine local lymph node assay: I. The ICCVAM review process. Reg. Toxicol. Pharmacol., 34(3), 249 – 257.
|
|
10.
|
ICCVAM (2010), ICCVAM Test Method Evaluation Report. Nonradioactive local lymph node assay: BrdU-ELISA Test Method Protocol (LLNA: BrdU-ELISA). NIH Publication No 10-7552A/B. Research Triangle Park, NC: National Institute of Environmental Health Sciences. K dispozícii na stránke: [http://iccvam.niehs.nih.gov/methods/immunotox/llna-ELISA/TMER.htm].
|
|
11.
|
ICCVAM (2009), Independent Scientific Peer Review Panel Report: Updated validation status of new versions and applications of the murine local lymph node assay: a test method for assessing the allergic contact dermatitis potential of chemicals and products. Research Triangle Park, NC: National Institute of Environmental Health Sciences. K dispozícii na stránke: [http://iccvam.niehs.nih.gov/docs/immunotox_docs/LLNAPRPRept2009.pdf].
|
|
12.
|
Takeyoshi, M., Iida, K., Shiraishi, K. and Hoshuyama, S. (2005), Novel approach for classifying chemicals according to skin sensitising potency by non-radioisotopic modification of the local lymph node assay. J. Appl. Toxicol., 25, 129 – 134.
|
|
13.
|
OECD (1992), Skin Sensitisation, Test Guideline No 406, Guidelines for Testing of Chemicals, OECD, Paris. K dispozícii na stránke: [http://www.oecd.org/env/testguidelines].
|
|
14.
|
Kreiling, R., Hollnagel, H. M., Hareng, L., Eigler, L., Lee, M. S., Griem, P., Dreessen, B., Kleber, M., Albrecht, A., Garcia, C. and Wendel, A. (2008), Comparison of the skin sensitising potential of unsaturated compounds as assessed by the murine local lymph node assay (LLNA) and the guinea pig maximization test (GPMT). Food Chem. Toxicol., 46, 1896 – 1904.
|
|
15.
|
Basketter, D., Ball, N., Cagen, S., Carrilo, J. C., Certa, H., Eigler, D., Garcia, C., Esch, H., Graham, C., Haux, C., Kreiling, R. and Mehling, A. (2009), Application of a weight of evidence approach to assessing discordant sensitisation datasets: implications for REACH. Reg. Toxicol. Pharmacol., 55, 90 – 96.
|
|
16.
|
ILAR (1996), Institute of Laboratory Animal Research (ILAR) Guide for the Care and Use of Laboratory Animals. 7th ed. Washington, DC: National Academies Press.
|
|
17.
|
ICCVAM (2009), Recommended Performance Standards: Murine Local Lymph Node Assay. NIH Publication Number 09-7357. Research Triangle Park, NC: National Institute of Environmental Health Sciences. K dispozícii na stránke: [http://iccvam.niehs.nih.gov/docs/immunotox_docs/llna-ps/LLNAPerfStds.pdf].
|
|
18.
|
McGarry, H. F. (2007), The murine local lymph node assay: regulatory and potency considerations under REACH. Toxicol., 238, 71 – 89.
|
|
19.
|
Kimber, I., Dearman, R. J., Scholes E. W. and Basketter, D. A. (1994), The local lymph node assay: developments and applications. Toxicol., 93, 13 – 31.
|
|
20.
|
OECD (2002), Acute Dermal Irritation/Corrosion, Test Guideline No 404, Guidelines for Testing of Chemicals, OECD, Paris. K dispozícii na stránke: [http://www.oecd.org/env/testguidelines].
|
|
21.
|
Reeder, M. K., Broomhead, Y. L., DiDonato, L., and DeGeorge, G. L. (2007), Use of an enhanced local lymph node assay to correctly classify irritants and false positive substances. Toxicologist, 96, 235.
|
|
22.
|
ICCVAM (2009), Nonradioactive Murine Local Lymph Node Assay: Flow Cytometry Test Method Protocol (LLNA: BrdU-FC) Revised Draft Background Review Document. Research Triangle Park, NC: National Institute of Environmental Health Sciences. K dispozícii na stránke: [http://iccvam.niehs.nih.gov/methods/immunotox/fcLLNA/BRDcomplete.pdf].
|
|
23.
|
Hayes, B. B., Gerber, P. C., Griffey, S. S., and Meade, B. J. (1998), Contact hypersensitivity to dicyclohexylcarbodiimide and diisopropylcarbodiimide in female B6C3F1 mice. Drug Chem. Toxicol., 21, 195 – 206.
|
|
24.
|
Homey, B., von Schilling, C., Blumel, J., Schuppe, H. C., Ruzicka, T., Ahr, H. J., Lehmann, P. and Vohr, V. W. (1998), An integrated model for the differentiation of chemical-induced allergic and irritant skin reactions. Toxicol. Appl. Pharmacol., 153, 83 – 94.
|
|
25.
|
Woolhiser, M. R., Hayes, B. B., and Meade, B. J. (1998), A combined murine local lymph node and irritancy assay to predict sensitisation and irritancy potential of chemicals. Toxicol. Meth., 8, 245 – 256.
|
|
26.
|
Hayes, B. B., and Meade, B. J. (1999), Contact sensitivity to selected acrylate compounds in B6C3F1 mice: relative potency, cross reactivity, and comparison of test methods. Drug. Chem. Toxicol., 22, 491 – 506.
|
|
27.
|
Ehling, G., Hecht, M., Heusener, A., Huesler, J., Gamer, A. O., van Loveren, H., Maurer, T., Riecke, K., Ullmann, L., Ulrich, P., Vandebriel, R., and Vohr, H. W. (2005), A European inter- laboratory validation of alternative endpoints of the murine local lymph node assay: first round. Toxicol., 212, 60 – 68.
|
|
28.
|
Vohr, H. W. and Ahr, H. J. (2005), The local lymph node assay being too sensitive? Arch. Toxicol., 79, 721 – 728.
|
|
29.
|
Patterson, R. M., Noga, E. and Germolec D. (2007), Lack of evidence for contact sensitisation by Pfiesteria extract. Environ. Health Perspect., 115, 1023 – 1028.
|
|
30.
|
ICCVAM (2009), Report on the ICCVAM-NICEATM/ECVAM/JaCVAM Scientific Workshop on Acute Chemical Safety Testing: Advancing In Vitro Approaches and Humane Endpoints for Systemic Toxicity Evaluations. Research Triangle Park, NC: National Institute of Environmental Health Sciences. K dispozícii na stránke: [http://iccvam.niehs.nih.gov/methods/acutetox/Tox_workshop.htm].
|
|
31.
|
OECD (2000), Guidance Document on the Recognition, Assessment and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluation, Environmental Health and Safety Monograph Series on Testing and Assessment No 19, ENV/JM/MONO(2000)7, OECD, Paris. K dispozícii na stránke: [http://www.oecd.org/env/testguidelines].
|
|
32.
|
Kimber, I., Hilton, J., Dearman, R. J., Gerberick, G. F., Ryan, C. A., Basketter, D. A., Lea, L., House, R. V., Ladies, G. S., Loveless, S. E., and Hastings, K. L. (1998), Assessment of the skin sensitisation potential of topical medicaments using the local lymph node assay: An interlaboratory exercise. J. Toxicol. Environ.l Health, 53, 563 – 579.
|
|
33.
|
OECD (2005), Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment, Environment, Health and Safety Monograph Series on Testing and Assessment No 34, ENV/JM/MONO(2005)14, OECD, Paris. K dispozícii na stránke: [http://www.oecd.org/env/testguidelines].
|
DODATOK 1
VYMEDZENIE POJMOV
Presnosť: Miera zhody výsledkov testovacej metódy s akceptovanými referenčnými hodnotami. Ide o opatrenie výkonnosti testovacej metódy a jeden z aspektov relevantnosti. Tento pojem sa často používa ako synonymum s pojmom ‚zhoda‘ a označuje podiel správnych výsledkov testovacej metódy (33).
Referenčná látka: Látka spôsobujúca alebo nespôsobujúca senzibilizáciu, ktorá sa používa ako štandardná látka na porovnávanie s testovanou látkou. Referenčná látka by mala mať tieto vlastnosti: i) konzistentný(-é) a spoľahlivý(-é) zdroj(-e); ii) štrukturálnu a funkčnú podobnosť s triedou látok, ktoré sa budú testovať; iii) známe fyzikálno-chemické vlastnosti; iv) podporné údaje o známych účinkoch a v) známu potenciu v rozsahu želanej odozvy.
Falošný negatívny výsledok: Testovaná látka je testovanou metódou nesprávne identifikovaná ako negatívna alebo neaktívna, pričom v skutočnosti je pozitívna alebo aktívna (33).
Falošný pozitívny výsledok: Testovaná látka je testom nesprávne identifikovaná ako pozitívna alebo aktívna, pričom v skutočnosti je negatívna alebo neaktívna (33).
Nebezpečnosť: Potenciál škodlivého účinku na zdravie alebo životné prostredie. Škodlivý účinok sa prejavuje až pri dostatočnej hladine expozície.
Medzilaboratórna reprodukovateľnosť: Miera, do akej rôzne kvalifikované laboratóriá, ktoré používajú rovnaký protokol a testujú rovnaké testovacie látky, dokážu dosiahnuť kvalitatívne a kvantitatívne podobné výsledky. Medzilaboratórna reprodukovateľnosť sa určuje počas postupov pred validáciou a v rámci postupov validácie a znamená mieru, do akej možno test úspešne prenášať medzi laboratóriami. Takisto je známa ako reprodukovateľnosť medzi laboratóriami (33).
Vnútrolaboratórna reprodukovateľnosť: Určenie miery, do akej kvalifikovaní pracovníci v rámci jedného laboratória dokážu úspešne opakovať výsledky s použitím konkrétneho protokolu v rôznom čase. Takisto je známa ako reprodukovateľnosť v rámci laboratória (33).
Extrémna hodnota: Extrémna hodnota je pozorovanie, ktoré je značne odlišné od ostatných hodnôt v náhodnej vzorke z populácie.
Zabezpečovanie kvality: Riadiaci proces, v ktorom jednotlivci nezávislí od osôb vykonávajúcich testy posudzujú súlad s laboratórnymi testovacími normami, požiadavkami a postupmi vedenia záznamov, ako aj presnosť prenosu údajov.
Spoľahlivosť: Meria rozsah, v rámci ktorého sa môže testovacia metóda počas istého časového obdobia opakovane vykonávať v rámci laboratórií a medzi nimi navzájom, pričom sa používa ten istý protokol. Posudzuje sa vypočítaním vnútrolaboratórnej a medzilaboratórnej reprodukovateľnosti (33).
Senzibilizácia kože: Imunologický proces, ku ktorému dochádza po lokálnom vystavení náchylného jednotlivca chemickému alergénu, ktorý vyvoláva kožnú imunitnú odozvu, ktorá môže viesť k vzniku kontaktnej senzibilizácie.
Stimulačný index (SI): Hodnota vypočítaná na posúdenie potenciálu testovanej látky spôsobiť senzibilizáciu kože; ide teda o pomer proliferácie v pokusných skupinách a proliferácie v súbežných kontrolných skupinách, ktorým sa podáva nosič.
Testovaná látka (tiež sa nazýva testovaná chemikália): Akákoľvek látka alebo zmes testovaná pomocou tejto testovacej metódy.
“ |


 In force
In force