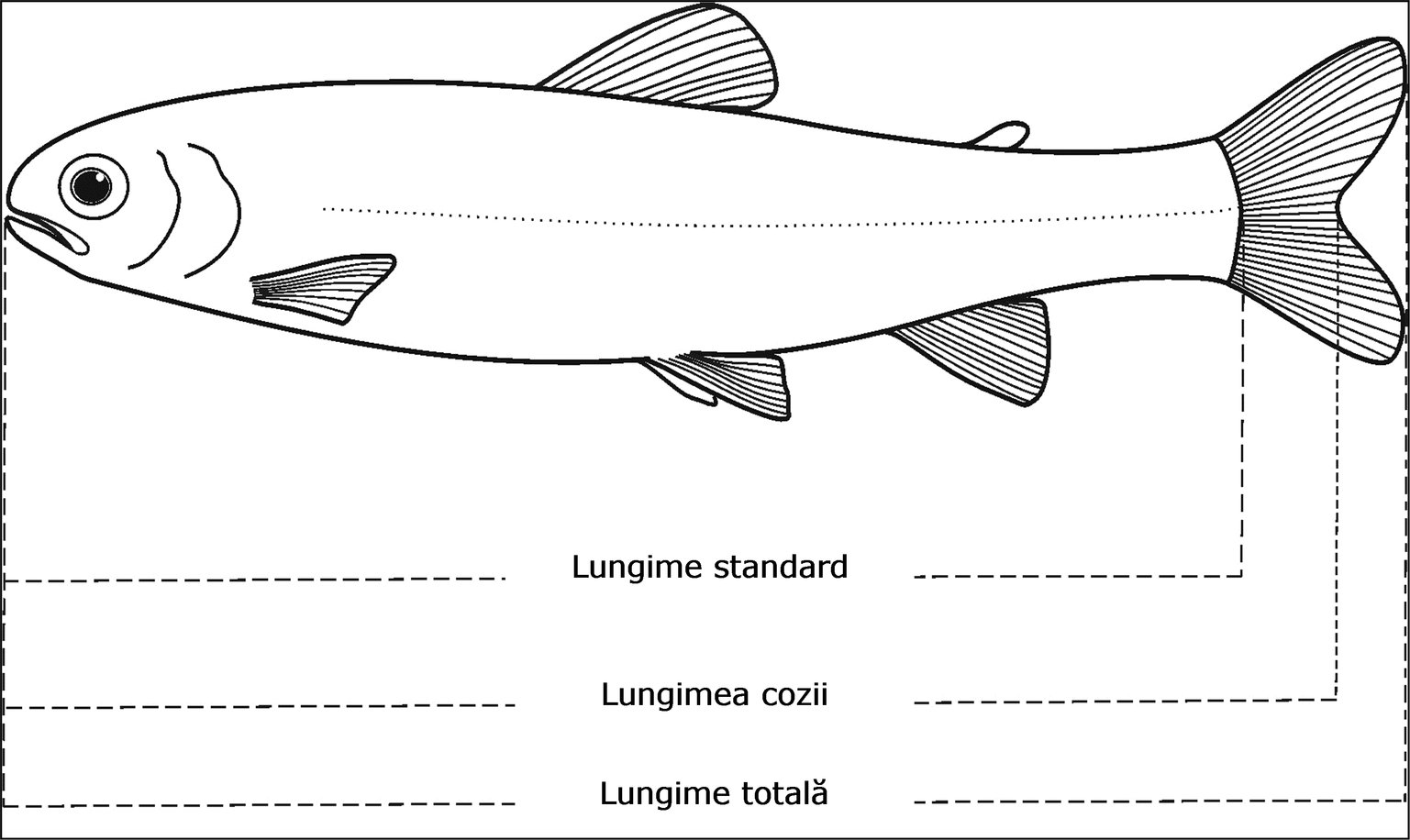
|
(8)
|
În partea B, se adaugă următoarele capitole:
„B.63 TESTUL DE DEPISTARE A TOXICITĂȚII PENTRU REPRODUCERE/DEZVOLTARE
INTRODUCERE
|
1.
|
Această metodă de testare este echivalentă cu Orientarea OCDE privind testarea (TG) nr. 421 (2016). Orientările OCDE pentru testarea substanțelor chimice sunt revizuite periodic în contextul evoluțiilor științifice. Orientarea inițială nr. 421 privind testarea pentru depistare a fost adoptată în 1995, pe baza unui protocol pentru un „Test preliminar de depistare a toxicității pentru reproducere” discutat în cadrul a două reuniuni ale experților, la Londra, în 1990 (1) și la Tokyo în 1992 (2).
|
|
2.
|
Această metodă de testare a fost actualizată cu puncte finale relevante privind perturbatorii endocrini, ca o continuare a activității cu prioritate ridicată inițiată de OCDE în 1998 în vederea revizuirii orientărilor existente privind testarea și a elaborării unor noi orientări privind testarea pentru depistarea și testarea unor potențiali perturbatori endocrini (3). Orientarea nr. 407 a OCDE privind testarea (studiul de toxicitate orală cu doză repetată la rozătoare, de 28 de zile, capitolul B.7 din prezenta anexă), de exemplu, a fost îmbunătățită în 2008 cu parametri adecvați pentru detectarea activității endocrine a substanțelor chimice de testat. În cadrul actualizării Orientării nr. 421 privind testarea, obiectivul a fost acela de a include câteva puncte finale relevante pentru perturbatori endocrini în orientările privind testele pentru depistare în cazul în care perioadele de expunere acoperă unele dintre perioadele sensibile din procesul de dezvoltare (perioadele prenatale sau postnatale timpurii).
|
|
3.
|
Punctele finale relevante suplimentare selectate pentru perturbatorii endocrini fac parte, de asemenea, din TG nr. 443 privind testarea (Studiu extins de toxicitate pentru reproducere la o singură generație, capitolul B.56 din prezenta anexă) au fost incluse în TG nr. 421 pe baza unui studiu de fezabilitate care abordează aspecte științifice și tehnice legate de includerea acestora, precum și de posibile adaptări ale modelului testului, care sunt necesare pentru includerea acestora (4).
|
|
4.
|
Această metodă de testare este concepută pentru a genera informații limitate cu privire la efectele unei substanțe chimice de testat asupra performanței reproductive a masculilor și femelelor, cum ar fi funcția gonadelor, comportamentul de împerechere, concepția, dezvoltarea produsului de concepție și parturiția. Aceasta nu este o alternativă la metodele de testare existente B.31, B.34, B.35 sau B.56 și nici nu le înlocuiește.
|
CONSIDERAȚII INIȚIALE
|
5.
|
Această metodă de testare pentru depistare poate fi utilizată pentru a furniza informații inițiale cu privire la posibile efecte asupra reproducerii și/sau dezvoltării, fie într-un stadiu incipient de evaluare a proprietăților toxicologice ale substanțelor chimice, fie asupra substanțelor chimice de interes. Aceasta poate fi utilizată, de asemenea, în cadrul unui set de teste inițiale pentru depistare în cazul substanțelor chimice existente pentru care sunt disponibile informații toxicologice puține sau pentru care nu sunt disponibile astfel de informații, ca un studiu pentru stabilirea intervalului de dozare pentru studii mai ample privind reproducerea/dezvoltarea sau pentru alte cazuri considerate relevante. În desfășurarea studiului, ar trebui să se respecte principiile directoare și considerentele prezentate în documentul de orientare nr. 19 al OCDE privind recunoașterea, evaluarea și utilizarea semnelor clinice ca puncte finale fără suferință pentru animalele de experiență utilizate în evaluări ale siguranței (5).
|
|
6.
|
Această metodă de testare nu oferă informații complete cu privire la toate aspectele legate de reproducere și dezvoltare. În special, aceasta oferă doar mijloace limitate de detectare a manifestărilor postnatale ale expunerii prenatale sau a efectelor care pot fi induse în timpul expunerii postnatale. Printre altele, din cauza numărului relativ mic de animale din grupurile de dozare, a selectivității punctelor finale și a duratei scurte a studiului, această metodă nu va furniza dovezi pentru concluzii sigure privind inexistența efectelor. În plus, în absența datelor provenite din alte teste de toxicitate pentru reproducere/dezvoltare, rezultatele pozitive sunt utile pentru evaluarea inițială a pericolelor și contribuie la luarea deciziilor cu privire la necesitatea și calendarul testelor suplimentare.
|
|
7.
|
Rezultatele obținute prin parametrii endocrini conecși ar trebui să fie evaluate în contextul „Cadrului conceptual al OCDE pentru testarea și evaluarea substanțelor chimice care perturbă procesele endocrine” (6). În acest cadru conceptual, orientarea nr. 421 a OCDE privind testarea îmbunătățită se regăsește la nivelul 4 ca test in vivo care asigură date privind efectele adverse asupra punctelor finale relevante ale sistemului endocrin. Însă este posibil ca un semnal endocrin să nu fie considerat drept o dovadă suficientă în sine pentru faptul că substanța chimică de testat este un perturbator endocrin.
|
|
8.
|
Această metodă de testare presupune administrarea substanței chimice de testat pe cale orală. Dacă se utilizează alte căi de expunere, este posibil să fie necesare modificări.
|
|
9.
|
Înainte de utilizarea metodei de testare pe un amestec pentru a genera date pentru un obiectiv de reglementare prevăzut, se ia în considerare dacă și, în caz afirmativ, din ce cauză aceasta poate oferi rezultate adecvate pentru respectivul scop. Astfel de considerații nu sunt necesare atunci când există o cerință normativă pentru testarea amestecului.
|
|
10.
|
Definițiile utilizate sunt prezentate în apendicele 1.
|
PRINCIPIUL TESTULUI
|
11.
|
Substanța chimică de testat se administrează în doze gradate mai multor grupuri de masculi și femele. Doza pentru masculi ar trebui să fie administrată pentru o perioadă de cel puțin patru săptămâni și până în ziua dinaintea sacrificării planificate inclusiv (aceasta include cel puțin două săptămâni înainte de împerechere, perioada de împerechere și aproximativ două săptămâni după împerechere). Având în vedere perioada de dozare limitată de pre-împerechere a masculilor, este posibil ca fertilitatea să nu fie un indicator deosebit de sensibil al toxicității testiculare. Prin urmare, este esențială o examinare histologică detaliată a testiculelor. Combinația dintre o perioadă de dozare de pre-împerechere de două săptămâni și de formulare ulterioară a observațiilor privind împerecherea/fertilitatea și o perioadă de dozare generală de cel puțin patru săptămâni, urmată de histopatologia detaliată a gonadelor de sex masculin, este considerată suficientă pentru a permite detectarea majorității efectelor asupra fertilității masculine și a spermatogenezei.
|
|
12.
|
Femelelor ar trebui să li se administreze doze pe toată durata studiului. Aceasta include două săptămâni înainte de împerechere (cu obiectivul de a acoperi cel puțin două cicluri estrale complete), timpul variabil până la concepție, durata sarcinii și cel puțin 13 zile după parturiție, până în ziua dinaintea eutanasierii planificate inclusiv.
|
|
13.
|
Durata studiului, în urma aclimatizării și evaluării ciclului estral înainte de dozare, depinde de performanța femelei și este de aproximativ 63 de zile, [cel puțin 14 zile înainte de împerechere, (până la) 14 zile de împerechere, 22 de zile de gestație, 13 zile de lactație].
|
|
14.
|
În perioada de administrare, animalele sunt examinate cu atenție zilnic, în vederea identificării unor eventuale semne de toxicitate. Animalele care au murit sau care au fost eutanasiate în perioada de testare sunt supuse unei autopsii, iar cele care supraviețuiesc până la sfârșitul testului sunt eutanasiate și autopsiate.
|
DESCRIEREA METODEI
Selecția speciilor de animale
|
15.
|
Această metodă de testare este concepută pentru a fi utilizată la șobolan. În cazul în care parametrii specificați în cadrul acestei metode de testare sunt investigați la o altă specie de rozătoare, ar trebui să se prezinte o justificare detaliată. În cadrul programului de validare internațional pentru detectarea perturbatorilor endocrini în TG nr. 407 din OCDE (care corespunde capitolului B.7 din prezenta anexă), șobolanul a fost singura specie folosită. Nu ar trebui să se utilizeze sușe cu fecunditate scăzută sau cu o incidență a defectelor de dezvoltare cunoscută a fi ridicată. Ar trebui să se folosească animale virgine sănătoase, care nu au fost supuse anterior unor proceduri experimentale. Animalele de experiență ar trebui să fie caracterizate în ceea ce privește specia, sușa, sexul, greutatea și vârsta. La începutul studiului, diferențele de greutate între animalele folosite ar trebui să fie minime și să nu depășească 20 % din greutatea medie pentru fiecare sex. În cazul în care studiul este realizat cu titlu de studiu preliminar pentru un studiu pe termen lung sau pentru o întreagă generație, este de preferat să se utilizeze, în ambele studii, animale din aceeași sușă și din aceeași sursă.
|
Adăpostire și hrănire
|
16.
|
Toate procedurile ar trebui să fie în conformitate cu standardele locale privind îngrijirea animalelor de laborator. Temperatura camerei în care se află animalele de experiență ar trebui să fie de 22 °C (± 3 °). Cu toate că umiditatea relativă ar trebui să fie de cel puțin 30 % și, de preferință, să nu depășească 70 % decât în timpul operațiunilor de curățare a sălii, obiectivul de umiditate este de 50-60 %. Iluminatul ar trebui să fie artificial, alternând perioade de 12 ore de lumină cu 12 ore de întuneric. Pentru alimentație, se poate utiliza hrană convențională de laborator, cu asigurarea unei cantități nelimitate de apă potabilă. Alegerea hranei poate fi influențată de necesitatea de a asigura un amestec adecvat al unei substanțe chimice de testat atunci când aceasta se administrează utilizând această metodă.
|
|
17.
|
Animalele ar trebui să fie adăpostite în grupuri mici de același sex; animalele pot fi adăpostite individual dacă există o justificare științifică. În cazul cuștilor colective nu ar trebui să se depășească numărul de cinci animale în fiecare cușcă. Procedurile de împerechere se desfășoară în cuști adecvate acestui scop. Femelele gestante ar trebui să fie plasate în cuști individuale și prevăzute cu materiale pentru cuibărire. Femelele aflate în lactație vor fi plasate în cuști individuale împreună cu progeniturile acestora.
|
|
18.
|
Hrana ar trebui să fie analizată în mod regulat în vederea identificării contaminanților. Ar trebui să se rețină un eșantion din hrană până la finalizarea raportului.
|
Pregătirea animalelor
|
19.
|
Animale adulte, tinere și sănătoase sunt distribuite în mod aleatoriu în grupuri martor și grupuri de tratament. Cuștile ar trebui să fie dispuse astfel încât să se reducă la minim eventualele efecte datorate amplasării cuștii. Animalele prezintă o identificare unică și sunt ținute în cuștile lor timp de minimum cinci zile înainte de începerea studiului, pentru a permite aclimatizarea acestora la condițiile de laborator.
|
Prepararea dozelor
|
20.
|
Se recomandă administrarea orală a substanței chimice de testat, cu excepția cazului în care se consideră că sunt adecvate alte căi de administrare. Atunci când se alege calea orală, substanța chimică de testat se administrează, de obicei, prin gavaj; însă, în mod alternativ, substanțele chimice de testat pot fi administrate împreună cu hrana sau cu apa potabilă.
|
|
21.
|
Dacă este necesar, substanța chimică de testat se dizolvă sau se suspendă într-un vehicul adecvat. Se recomandă ca, ori de câte ori este posibil, să se ia în considerare în primul rând utilizarea unei soluții/suspensii apoase, apoi a unei soluții/emulsii în ulei (de exemplu, ulei de porumb) și ulterior a unei soluții posibile în alte vehicule. Pentru alte vehicule decât apa, ar trebui să fie cunoscute caracteristicile toxice ale vehiculului respectiv. Ar trebui să se determine stabilitatea și omogenitatea substanței chimice de testat în vehicul.
|
PROCEDURA
Numărul și sexul animalelor
|
22.
|
Se recomandă ca, la început, fiecare grup să aibă cel puțin 10 masculi și 12-13 femele. Femelele vor fi evaluate înainte de expunere pentru ciclicitatea estrală, iar animalele la care nu se produc cicluri tipice de 4-5 zile nu vor fi incluse în studiu; prin urmare, se recomandă femele suplimentare pentru a obține 10 femele într-un grup. Cu excepția cazului în care apar efecte toxice pronunțate, se preconizează că acesta va asigura cel puțin 8 femele gestante în fiecare grup, care reprezintă, în mod normal, numărul minim acceptabil de femele gestante într-un grup. Obiectivul este să se asigure un număr suficient de sarcini și pui pentru a asigura o evaluare semnificativă a potențialului substanței chimice de testat de a afecta fertilitatea, sarcina și comportamentul matern și de supt, precum și creșterea și dezvoltarea puilor F1, de la concepție până în ziua 13 a perioadei post-partum.
|
Dozare
|
23.
|
În general, ar trebui să se utilizeze cel puțin trei grupuri de testare tratate și un grup martor. Nivelurile de dozare pot fi stabilite în funcție de informațiile obținute în urma testelor de toxicitate acută sau de rezultatele studiilor cu doze repetate. Cu excepția tratării cu substanța chimică de testat, animalele din grupul martor ar trebui să fie tratate în mod identic cu animalele din grupul de testare. Dacă pentru administrarea substanței chimice de testat se folosește un vehicul, acesta se administrează grupului martor în volumul maxim utilizat.
|
|
24.
|
Dozele ar trebui să fie selectate ținându-se cont de toxicitatea existentă și de datele toxico-cinetice disponibile. De asemenea, ar trebui să se țină seama de faptul că ar putea exista diferențe de sensibilitate între animalele gestante și cele negestante. Ar trebui să fie aleasă doza cea mai mare pentru a induce efecte toxice, însă nu moartea sau suferințe grave. În continuare, ar trebui să se scadă nivelul dozelor pentru a se pune în evidență un eventual răspuns legat de doză și concluzia că nu există efecte adverse observate (NOAEL) la cel mai redus nivel al dozei. Intervalul optim pentru determinarea nivelurilor descrescătoare ale dozelor este frecvent un factor de doi sau patru și, deseori, este de preferat să se adauge un al patrulea grup de testare, în loc să se utilizeze intervale foarte mari (de exemplu, un factor mai mare de 10) între doze.
|
|
25.
|
În prezența toxicității generale observate (de exemplu, greutate corporală redusă, efecte asupra ficatului, plămânilor sau rinichilor etc.) sau a altor schimbări care pot să nu constituie răspunsuri toxice (de exemplu, consum de hrană redus, mărirea ficatului), efectele observate asupra punctelor finale endocrine sensibile ar trebui să fie interpretate cu precauție.
|
Testul valorilor-limită
|
26.
|
Dacă un studiu oral efectuat la un dozaj de cel puțin 1 000 mg/kg greutate corporală/zi sau, în cazul administrării împreună cu hrana sau cu apă potabilă, într-un procent echivalent în hrană sau în apa potabilă, folosind procedurile descrise pentru acest studiu, nu produce efecte toxice detectabile și dacă nu se anticipează toxicitatea pe baza datelor despre substanțe cu structuri asemănătoare, atunci este posibil să nu fie considerată necesară realizarea unui studiu complet cu mai multe niveluri de dozare. Testul valorilor-limită este valabil, cu excepția cazului în care expunerea umană indică faptul că trebuie să se administreze o doză mai mare pe cale orală. Pentru alte tipuri de administrare, cum sunt inhalarea sau aplicarea dermică, este posibil ca proprietățile fizice și chimice ale substanțelor chimice de testat să determine deseori nivelul maxim posibil al concentrației.
|
Administrarea dozelor
|
27.
|
Animalelor li se administrează substanța chimică de testat zilnic, timp de 7 zile pe săptămână. Când substanța chimică de testat este administrată prin gavaj, administrarea ar trebui să fie în doză unică la animale, folosind o sondă gastrică sau o canulă de intubare adecvată. Volumul maxim de lichid care poate fi administrat o dată depinde de dimensiunea animalului de experiență. Volumul nu ar trebui să depășească 1 ml/100 g greutate corporală, exceptând cazul soluțiilor apoase, când se pot utiliza 2 ml/100 g greutate corporală. Cu excepția substanțelor chimice de testat iritante sau corozive care vor produc, de regulă, efecte exacerbate la concentrații mai mari, variabilitatea volumului de testat ar trebui să fie redusă la minim prin ajustarea concentrației, astfel încât să se asigure un volum constant la toate nivelurile de dozare.
|
|
28.
|
Pentru substanțele chimice de testat administrate în hrană sau în apa potabilă, este important să se asigure că respectivele cantități de substanță chimică de testat implicate nu influențează nutriția normală sau echilibrul hidric. Dacă substanța chimică de testat se administrează în hrană, aceasta poate fi administrată fie într-o concentrație constantă în alimentație (ppm), fie la o doză constantă în raport cu greutatea corporală a animalelor; alternativa utilizată ar trebui precizată. În cazul unei substanțe chimice de testat administrate prin gavaj, doza ar trebui să se administreze la aproximativ aceeași oră în fiecare zi și să fie ajustată cel puțin săptămânal pentru a menține un nivel constant al dozei în funcție de greutatea corporală a animalului.
|
Schema de tratament
|
29.
|
Administrarea dozelor la ambele sexe ar trebui să înceapă cu cel puțin 2 săptămâni înainte de împerechere, după ce acestea au fost aclimatizate timp de cel puțin cinci zile, iar femelele au fost supuse unui control privind ciclurile estrale normale (într-o perioadă de pre-tratament de 2 săptămâni). Studiul ar trebui să fie planificat în așa fel încât evaluarea ciclului estral să înceapă de îndată ce animalele au ajuns la maturitatea sexuală deplină. Aceasta ar putea varia ușor între diferite sușe de șobolani din diferite laboratoare, de exemplu, la șobolanii Sprague Dawley aceasta este la vârsta de 10 săptămâni, la șobolanii Wistar la vârsta de aproximativ 12 săptămâni. Femelele cu pui ar trebui să fie eutanasiate în ziua 13 post-partum sau imediat după aceea. Ziua nașterii (și anume, atunci când s-a încheiat parturiția) este definită ca fiind ziua 0 post-partum. Femelele care nu prezintă semne ale copulației sunt eutanasiate după 24-26 zile de la ultima zi a perioadei de împerechere. Administrarea dozelor continuă la ambele sexe în perioada de împerechere. Masculilor ar trebui să li se administreze în continuare doze după perioada de împerechere cel puțin până la încheierea perioadei de dozare minime totale de 28 de zile. Apoi aceștia sunt eutanasiați sau, alternativ, sunt reținuți pentru administrarea în continuare a dozelor pentru o posibilă desfășurare a unei a doua împerecheri, dacă se consideră că este cazul.
|
|
30.
|
Administrarea de doze zilnice la femelele parentale ar trebui să continue pe întreaga durată a sarcinii și cel puțin până în ziua 13 post-partum inclusiv sau cu o zi înainte de sacrificare. În cazul studiilor în care substanța chimică de testat se administrează prin inhalare sau subcutanat, dozele ar trebui să fie administrate în continuare cel puțin până în ziua 19 de sarcină inclusiv și administrate din nou cât mai curând posibil, dar nu mai târziu de ziua post-fătare (PND) 4.
|
|
31.
|
În apendicele 2 este prezentată o diagramă a schemei de tratament.
|
Procedura de împerechere
|
32.
|
De regulă, în prezentul studiu ar trebui utilizate împerecheri 1:1 (un mascul la o femelă). Pot exista excepții în cazul deceselor ocazionale ale masculilor. Femela ar trebui să fie plasată împreună cu același mascul până când se observă semne ale copulației sau au trecut două săptămâni. În fiecare dimineață, femelele ar trebui să fie examinate pentru a se constata prezența spermei sau a dopului vaginal. Ziua 0 a gestației este definită ca fiind ziua în care se confirmă dovada împerecherii (se constată existența unui dop vaginal sau a spermei). În cazul în care împerecherea eșuează, se poate lua în calcul reîmperecherea femelelor în cauză cu masculi din același grup care și-au dovedit capacitatea reproductivă.
|
Dimensiunea seriei de pui
|
33.
|
În ziua 4 de după naștere, dimensiunea fiecărei serii de pui poate fi ajustată prin eliminarea puilor suplimentari printr-o selecție aleatorie pentru a obține, pe cât posibil, patru sau cinci pui per sex per serie, în funcție de dimensiunea normală a seriei la sușa de șobolani utilizați. Ar trebui să se colecteze probe de sânge de la doi dintre puii în exces, care să fie grupate și utilizate pentru determinarea nivelurilor serului T4. Eliminarea selectivă a puilor, de exemplu pe baza greutății corporale sau a distanței anogenitale (AGD) nu este adecvată. Ori de câte ori numărul de pui masculi sau femele împiedică obținerea unui număr de patru sau cinci exemplare din fiecare sex per serie, se acceptă ajustarea parțială (de exemplu, șase masculi și patru femele). Nu se vor elimina pui dacă dimensiunea seriei va scădea sub ținta de sacrificare (8 sau 10 de pui/serie). Dacă există un singur pui peste obiectivul de sacrificare, se va elimina doar un singur pui, acesta fiind utilizat pentru recoltarea de sânge pentru posibile evaluări ale serului T4.
|
|
34.
|
Dacă dimensiunea seriei nu este ajustată, se sacrifică doi pui per serie în ziua 4, după naștere, și se iau probe de sânge pentru măsurarea concentrațiilor de hormoni tiroidieni din ser. Dacă este posibil, cei doi pui per serie ar trebui să fie pui de sex feminin pentru a rezerva puii masculi în vederea evaluării retenției mamelonului, cu excepția cazului în care în urma îndepărtării puilor respectivi nu mai rămân femele pentru evaluare la încheierea procesului. Nu se vor elimina pui în cazul în care dimensiunea seriei va scădea sub 8 sau 10 pui/serie (în funcție de dimensiunea normală a seriei în sușa de șobolani utilizată). Dacă există un singur pui peste dimensiunea normală a seriei, se va elimina doar un singur pui, acesta fiind utilizat pentru recoltarea de sânge pentru posibile evaluări ale serului T4.
|
Observații în-viață
Observații clinice
|
35.
|
Pe parcursul întregii perioade de testare, ar trebui să se formuleze observații clinice generale cel puțin o dată pe zi și mai frecvent atunci când se observă semne de toxicitate. Acestea ar trebui să fie efectuate, de preferat, la aceeași oră (aceleași ore) în fiecare zi, luând în considerare perioada de vârf a efectelor anticipate după administrarea dozelor. Ar trebui să se înregistreze modificări comportamentale semnificative, semnele unei parturiții dificile sau prelungite și toate semnele de toxicitate, inclusiv mortalitatea. Aceste evidențe ar trebui să includă momentul apariției, gradul și durata semnelor de toxicitate.
|
Greutate corporală și consumul de hrană/apă
|
36.
|
Masculii și femelele ar trebui să fie cântărite în prima zi a administrării dozelor, apoi cel puțin săptămânal și la sfârșit. În timpul sarcinii, femelele ar trebui să fie cântărite în zilele 0, 7, 14 și 20 și în termen de 24 de ore de la parturiție (în ziua 0 sau 1 post-partum) și cel puțin în zilele 4 și 13 post-partum. Aceste observații ar trebui să fie consemnate individual pentru fiecare animal adult.
|
|
37.
|
În perioada de pre-împerechere, de sarcină și de alăptare, consumul de alimente ar trebui să fie măsurat cel puțin o dată pe săptămână. Măsurarea consumului de hrană în timpul împerecherii este opțională. Pe parcursul acestor perioade, consumul de apă ar trebui, de asemenea, să fie măsurat atunci când substanța chimică de testat este administrată împreună cu apa potabilă.
|
Cicluri estrale
|
38.
|
Înainte de a începe tratamentul, ar trebui să se monitorizeze ciclurile estrale pentru femelele studiate cu ciclicitate regulate (a se vedea punctul 22). De asemenea, frotiul vaginal ar trebui să fie monitorizat zilnic de la începutul perioadei de tratament, până la obținerea dovezii împerecherii. În cazul în care există motive de îngrijorare cu privire la efecte de criză acută care ar putea modifica ciclurile estrale odată cu inițierea administrării dozelor, laboratoarele pot să expună animalele de experiență timp de două săptămâni, apoi să colecteze frotiuri vaginale zilnic pentru a monitoriza ciclul estral timp de cel puțin două săptămâni în perioada de pre-împerechere, cu o continuare a monitorizării în perioada de împerechere până când există dovezi ale împerecherii. Atunci când se obțin celule vaginale/cervicale, ar trebui să se evite orice perturbare a mucoasei, care ar putea induce pseudogestația (7) (8).
|
Parametrii puilor
|
39.
|
Durata gestației ar trebui să fie înregistrată, calculându-se din ziua 0 de gestație. Fiecare serie de pui ar trebui să fie examinată cât mai curând posibil după naștere, pentru a se stabili numărul și sexul puilor, puii născuți morți, puii vii, puii subdezvoltați (puii care sunt semnificativ mai mici decât puii corespunzători din grupul martor) și prezența unor anomalii macroscopice.
|
|
40.
|
În termen de 24 de ore de la parturiție, puii vii ar trebui să fie numărați și separați pe sexe, iar seriile de pui cântărite (în ziua 0 sau 1 post-partum) și cel puțin în zilele 4 și 13 post-partum. Pe lângă observațiile descrise la punctul 35, ar trebui să fie consemnat orice comportament anormal al puiului.
|
|
41.
|
Este necesar ca AGD a fiecărui pui să fie măsurată în aceeași zi post-natală de la PND 0 până la PND 4. Greutatea corporală a puilor ar trebui să fie consemnează în ziua în care se măsoară AGD, aceasta trebuind să fie normalizată la o valoare aferentă dimensiunii puiului, de preferat, rădăcina cubică a greutății corporale (9). Numărul sfârcurilor/aureolelor la puii masculi ar trebui să fie calculat în PND 12 sau 13, astfel cum se recomandă în Orientarea nr. 151 a OCDE (10).
|
Biochimia clinică
|
42.
|
Sunt prelevate probe de sânge dintr-un anumit loc pe baza următorului calendar:
|
—
|
de la cel puțin doi pui pe serie în ziua 4, după naștere, în cazul în care numărul puilor permite acest lucru (a se vedea punctele 33-34)
|
|
—
|
de la toate femelele și de la cel puțin doi pui pe serie la încheiere, în ziua 13, și
|
|
—
|
de la toți masculii adulți, la încheiere,
|
|
Toate probele de sânge sunt păstrate în condiții adecvate. Sunt analizate probe de sânge de la pui din ziua 13 și de la masculii adulți pentru identificarea nivelurilor de hormoni tiroidieni din ser (T4). Dacă se consideră că este relevant, sunt efectuate analize suplimentare ale T4 din probele de sânge de la femele și de la puii din ziua 4. Opțional, dacă se consideră relevant, ar putea fi măsurați și alți hormoni. Probele de sânge ale puilor pot fi grupate pe serii pentru analize ale hormonilor tiroidieni. Hormonii tiroidieni (T4 și TSH) ar trebui, de preferință, să fie măsurați ca „valoare totală”.
|
43.
|
Următorii factori pot influența variabilitatea și concentrațiile absolute ale valorilor hormonale determinate:
|
—
|
momentul eutanasierii, din cauza variației diurne a concentrațiilor hormonale
|
|
—
|
metoda de eutanasiere pentru a evita stresul nejustificat exercitat asupra animalelor, care poate afecta concentrațiile hormonale;
|
|
—
|
seturile de testare pentru determinările hormonale, care pot varia prin curbele lor standard.
|
|
|
44.
|
Probele de plasmă prevăzute în mod specific pentru determinarea hormonală ar trebui să fie obținute la momente comparabile din zi. Valorile numerice obținute la analiza concentrațiilor hormonale diferă în funcție de diferitele seturi de testare comerciale.
|
Patologie
Autopsie macroscopică
|
45.
|
La momentul sacrificării sau al decesului pe durata studiului, animalele adulte ar trebui să fie examinate macroscopic pentru determinarea oricăror anomalii sau modificări patologice. Se acordă o atenție deosebită organelor sistemului de reproducere. Ar trebui să se înregistreze numărul de locuri de implantare. Ar trebui să se examineze frotiurile vaginale în dimineața zilei autopsiei pentru a determina stadiul ciclului estral și pentru a permite corelarea cu histopatologia ovarelor.
|
|
46.
|
Testiculele și epididimitele, precum și prostata și veziculele seminale cu glande coagulante în ansamblu, ale tuturor masculilor adulți ar trebui să fie curățate de țesuturile aderente, după caz, și greutatea umedă a acestora înregistrată cât mai curând posibil după disecție, pentru a se evita deshidratarea. În plus, greutățile opționale ale organelor ar putea include complexul muscular levatorul ani plus bulbocavernos, glandele Cowper și glandul penisului la masculi și ovarele (greutate umedă) cuplate cu uterul (inclusiv colul uterin) la femele; dacă sunt incluse, aceste greutăți ar trebui să fie colectate cât mai curând posibil după disecție.
|
|
47.
|
Puii morți și puii eutanasiați în ziua 13 post-partum, sau imediat după aceea, ar trebui să fie cel puțin examinați cu atenție la exterior pentru identificarea anomaliilor macroscopice. Ar trebui să se acorde o atenție deosebită organelor genitale de reproducere externe, care ar trebui să fie examinate pentru identificarea unor modificări în dezvoltare. În ziua 13, ar trebui să fie păstrată tiroida de la un mascul pui și o femelă pui per serie.
|
|
48.
|
Ar trebui să se păstreze ovarele, testiculele, organele sexuale anexe (uter și col uterin, epididimite, prostată, vezicule seminale plus glande coagulante), tiroida și toate organele care prezintă leziuni macroscopice la toate animalele adulte. Fixarea formaldehidei nu este recomandată în cazul examinării de rutină a testiculelor și a epididimitelor. O metodă acceptabilă este utilizarea fixatorului Bouin sau Davidson modificat pentru aceste țesuturi (11). Tunica albuginea ar putea fi perforată ușor și superficial cu un ac la ambii poli ai organului pentru a permite pătrunderea rapidă a fixatorului.
|
Histopatologie
|
49.
|
Ar trebui să se efectueze un examen histologic detaliat al ovarelor, testiculelor și epididimitelor (se pune un accent deosebit pe fazele spermatogenezei și pe histopatologia structurii celulare testiculare interstițiale) a animalelor din grupul tratat cu doza maximă și din grupul martor. Ar putea fi examinate și celelalte organe păstrate, inclusiv tiroida de la pui și animalele adulte, dacă este necesar. După fixare ar putea fi determinată greutatea tiroidei. Curățarea ar trebui să fie făcută, de asemenea, foarte atent și doar după fixare, pentru a evita deteriorarea țesuturilor. Deteriorarea țesuturilor ar putea compromite analiza histopatologică. Examinările ar trebui să fie extinse la animalele din grupurile tratate cu alte doze, atunci când se constată modificări în grupul tratat cu doza cea mai mare. Ghidul privind histopatologia (11) prezintă detalii suplimentare privind disecția, fixarea, secționarea și histopatologia țesuturilor endocrine.
|
DATE ȘI RAPORTARE
Date
|
50.
|
Ar trebui să fie furnizate date despre fiecare animal. În plus, toate datele ar trebui să fie sintetizate în format tabelar, prezentându-se, pentru fiecare grup de testare, numărul de animale la începutul testului, numărul de animale găsite moarte în cursul testului sau eutanasiate, data decesului sau a eutanasierii, numărul de animale fertile, numărul de femele gestante, numărul de animale care prezintă semne de toxicitate, o descriere a semnelor de toxicitate observate, inclusiv momentul declanșării, durata și gravitatea efectelor toxice, tipul de modificări histopatologice și toate datele relevante privind seriile. În apendicele 3 este prezentat un format tabelar de raport de sinteză care s-a dovedit a fi foarte util pentru evaluarea efectului privind reproducerea/dezvoltarea.
|
|
51.
|
Din cauza dimensiunilor limitate ale studiului, analizele statistice sub formă de teste de „semnificație” au o valoare limitată pentru multe puncte finale, în special punctele finale privind reproducerea. În cazul în care se utilizează analize statistice, atunci metoda aleasă ar trebui să fie adecvată pentru distribuirea variabilei examinate și să fie aleasă înainte de începerea studiului. Analiza statistică a AGD și a retenției sfârcurilor ar trebui să se bazeze pe date individuale despre pui, fiind luate în considerare efectele asupra seriei. Dacă este cazul, seria constituie unitatea de analiză. Analiza statistică a greutății corporale a puilor ar trebui să se bazeze pe date individuale despre pui, fiind luată în considerare dimensiunea seriei. Din cauza dimensiunii reduse a grupului, utilizarea datelor anterioare privind martorii (de exemplu, pentru dimensiunea seriei), dacă există, ar putea, de asemenea, să ajute la interpretarea studiului.
|
Evaluarea rezultatelor
|
52.
|
Constatările acestui studiu de toxicitate ar trebui să fie evaluate sub aspectul efectelor observate, al autopsiei și al constatărilor microscopice. Evaluarea va include relația dintre doza de substanță chimică de testat și prezența sau absența, incidența și gravitatea unor anomalii, inclusiv leziuni macroscopice, organe țintă identificate, efecte asupra fertilității, anomalii clinice, efecte asupra performanței de reproducere și a seriei, modificări ale greutății corporale, efecte asupra mortalității și orice alte efecte toxice.
|
|
53.
|
Din cauza perioadei scurte de tratament a masculului, histopatologia testiculelor și a epididimitelor ar trebui luată în considerare împreună cu datele privind fertilitatea la evaluarea efectelor de reproducere la masculi. Utilizarea datelor istorice privind martorii cu privire la reproducere/dezvoltare (de exemplu, pentru dimensiunea seriei, AGD, retenția sfârcurilor, nivelurile T4 din ser), dacă există, ar putea fi utile, de asemenea, pentru interpretarea studiului.
|
|
54.
|
Pentru controlul calității, se propune colectarea datelor anterioare privind martorii și calcularea coeficienților de variație pentru datele numerice, în special pentru parametrii corelați cu detectarea perturbatorilor endocrini. Aceste date pot fi folosite în scop de comparație la evaluarea studiilor curente.
|
Raportul testului
|
55.
|
Raportul testului ar trebui să includă următoarele informații:
|
|
Substanța chimică de testat:
|
—
|
sursa, numărul lotului, data limită de utilizare, dacă sunt disponibile
|
|
—
|
stabilitatea substanței chimice de testat, dacă se cunoaște.
|
|
|
|
Substanță mono-constituent:
|
—
|
aspectul fizic, solubilitatea în apă și alte proprietăți fizico-chimice relevante;
|
|
—
|
identificarea chimică, precum denumirea IUPAC sau CAS, numărul CAS sau codul său InChI, formula structurală, puritatea, identitatea chimică a impurităților după cum este necesar și fezabil din punct de vedere practic etc.
|
|
|
|
Substanță multicomponentă, UVCB-uri și amestecuri:
|
—
|
în măsura în care este posibil, caracterizată prin identitatea chimică (a se vedea mai sus), apariția cantitativă și proprietățile fizico-chimice relevante ale componentelor.
|
|
|
|
Vehicul (dacă este cazul):
|
—
|
justificarea alegerii vehiculului dacă este altul decât apa.
|
|
|
|
Animale de testare:
|
—
|
numărul, vârsta și sexul animalelor;
|
|
—
|
sursa, condițiile de adăpost, regimul alimentar etc.;
|
|
—
|
greutatea fiecărui animal la începutul testului.
|
|
—
|
justificarea, în cazul în care se folosesc alte specii decât șobolanul
|
|
|
|
Condiții de testare:
|
—
|
justificarea selecției nivelului dozei administrate;
|
|
—
|
detalii privind formularea substanței chimice de testat/prepararea hranei, concentrația obținută, stabilitatea și omogenitatea preparatului;
|
|
—
|
detalii privind administrarea substanței chimice de testat;
|
|
—
|
conversia concentrației substanței chimice de testat (ppm) în hrană/apa potabilă în doza reală (mg/kg greutate corporală/zi), dacă este cazul;
|
|
—
|
detalii cu privire la calitatea hranei și a apei;
|
|
—
|
descriere detaliată a procedurii de randomizare pentru selecția puilor pentru sacrificare, dacă aceștia sunt sacrificați.
|
|
|
|
Rezultate:
|
—
|
greutatea corporală/modificările în greutatea corporală;
|
|
—
|
consumul de hrană și apă, dacă este cazul;
|
|
—
|
date privind răspunsul toxic pe sexe și doză, inclusiv fertilitatea, gestația și orice alte semne de toxicitate;
|
|
—
|
efecte toxice sau de altă natură asupra reproducerii, puilor, creșterii postnatale etc.;
|
|
—
|
natura, gravitatea și durata observațiilor clinice (reversibile sau nu);
|
|
—
|
numărul de femele adulte cu ciclu estral normal sau anormal și durata ciclului;
|
|
—
|
numărul de pui născuți vii și de pierderi după implantare;
|
|
—
|
date privind greutatea corporală a puilor;
|
|
—
|
AGD a tuturor puilor (și greutatea corporală în ziua măsurării AGD)
|
|
—
|
retenția sfârcurilor la puii masculi;
|
|
—
|
nivelurile hormonilor tiroidieni la pui din ziua 13 și masculi adulți (și femele și pui din ziua 4, dacă se măsoară)
|
|
—
|
numărul de pui cu anomalii extrem de vizibile, o evaluare brută a organelor genitale externe, numărul de pui subdezvoltați;
|
|
—
|
momentul decesului în timpul studiului sau indicarea faptului că animalele au supraviețuit experimentului;
|
|
—
|
numărul de implantări, dimensiunea seriei și valori ale greutății seriei la momentul înregistrării;
|
|
—
|
greutatea corporală la sacrificare și date privind greutatea organelor în cazul animalelor părinți;
|
|
—
|
o descriere detaliată a constatărilor histopatologice;
|
|
—
|
date privind absorbția (dacă există);
|
|
—
|
tratarea statistică a rezultatelor, dacă este cazul.
|
|
Discutarea rezultatelor.
Concluzii.
|
Interpretarea rezultatelor
|
56.
|
Studiul va furniza evaluări ale toxicității pentru reproducere/dezvoltare, prin corelare cu administrarea de doze repetate (a se vedea punctele 5 și 6). Acesta ar putea indica dacă este necesar să se efectueze investigații suplimentare și ar putea oferi îndrumări în proiectarea studiilor ulterioare. Ar trebui să se consulte Documentul de orientare nr. 43 al OCDE pentru ajutor în interpretarea rezultatelor privind reproducerea și dezvoltarea (12). Documentul de orientare nr. 106 al OCDE privind evaluarea histologică a testelor endocrine și a testelor de reproducere la rozătoare (11) furnizează informații cu privire la pregătirea și evaluarea organelor (endocrine) și a frotiurilor vaginale care pot fi utile pentru această orientare privind testarea.
|
REFERINȚE BIBLIOGRAFICE
|
(1)
|
OCDE (1990). Room Document No 1 for the 14th Joint Meeting of the Chemicals Group and Management Committee. Disponibil, la cerere, la Organizația pentru Cooperare și Dezvoltare Economică, Paris.
|
|
(2)
|
OCDE (1992). Chairman’s Report of the ad hoc Expert Meeting on Reproductive Toxicity Screening Methods, Tokyo, 27-29 octombrie 1992. Disponibil, la cerere, la Organizația pentru Cooperare și Dezvoltare Economică, Paris.
|
|
(3)
|
OCDE (1998). Report of the First Meeting of the OECD Endocrine Disrupter Testing and Assessment (EDTA) Task Force, 10-11 martie 1998. Disponibil, la cerere, la Organizația pentru Cooperare și Dezvoltare Economică, Paris.
|
|
(4)
|
OCDE (2015). Feasibility Study for Minor Enhancements of TG 421/422 with ED Relevant Endpoints. Environmental Health and Safety Publications, Series on Testing and Assessment, (nr. 217), Organizația pentru Cooperare și Dezvoltare Economică, Paris.
|
|
(5)
|
OCDE (2000). Guidance Document on the Recognition, Assessment, and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluations. Series on Testing and Assessment, (nr. 19), Organizația pentru Cooperare și Dezvoltare Economică, Paris.
|
|
(6)
|
OCDE (2011). Guidance Document on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption. Environment, Health and Safety Publications, Series on Testing and Assessment (nr. 150), Organizația pentru Cooperare și Dezvoltare Economică, Paris.
|
|
(7)
|
Goldman, J.M., Murr A.S., Buckalew A.R., Ferrell J.M. și Cooper R.L. (2007). The Rodent Estrous Cycle: Characterization of Vaginal Cytology and its Utility in Toxicological Studies, Birth Defects Research, partea B, 80 (2), 84-97.
|
|
(8)
|
Sadleir R.M.F.S (1979). Cycles and Seasons, în Auston C.R. and Short R.V. (editori), Reproduction in Mammals: I. Germ Cells and Fertilization, Cambridge, New York.
|
|
(9)
|
Gallavan R.H. Jr, Holson J.F., Stump D.G., Knapp J.F. și Reynolds V.L. (1999). Interpreting the Toxicologic Significance of Alterations in Anogenital Distance: Potential for Confounding Effects of Progeny Body Weights, Reproductive Toxicology, 13: 383-390.
|
|
(10)
|
OCDE (2013). Guidance Document in Support of the Test Guideline on the Extended One Generation Reproductive Toxicity Study. Environmental Health and Safety Publications, Series on Testing and Assessment, (nr. 151), Organizația pentru Cooperare și Dezvoltare Economică, Paris.
|
|
(11)
|
OCDE (2009). Guidance Document for Histologic Evaluation of Endocrine and Reproductive Tests in Rodents. Environmental Health and Safety Publications, Series on Testing and Assessment, (nr. 106), Organizația pentru Cooperare și Dezvoltare Economică, Paris.
|
|
(12)
|
OCDE (2008). Guidance Document on Mammalian Reproductive Toxicity Testing and Assessment. Environmental Health and Safety Publications, Series on Testing and Assessment, (nr. 43), Organizația pentru Cooperare și Dezvoltare Economică, Paris.
|
Apendicele 1
DEFINIȚII [A SE VEDEA, DE ASEMENEA, ORIENTAREA NR. 150 A OCDE (6)]
Androgenicitatea este capacitatea unei substanțe chimice de a acționa ca hormon androgen natural (de exemplu, testosteron) în organismul unui mamifer.
Antiandrogenicitatea este capacitatea unei substanțe chimice de a suprima acțiunea unui hormon androgen natural (de exemplu, testosteron) în organismul unui mamifer.
Antiestrogenicitatea este capacitatea unei substanțe chimice de a suprima acțiunea unui hormon estrogen natural (de exemplu, estradiol 17ß) în organismul unui mamifer.
Activitatea antitiroidiană este capacitatea unei substanțe chimice de a suprima acțiunea unui hormon tiroidian natural (de exemplu, estradiol T3) în organismul unui mamifer.
Substanță chimică înseamnă o substanță sau un amestec.
Toxicitate pentru dezvoltare: manifestarea toxicității pentru reproducere, reprezentând afecțiuni prenatale, perinatale, postnatale, structurale sau funcționale ale progeniturii.
Dozarea este un termen general care cuprinde doza, frecvența și durata administrării dozei.
Doză înseamnă cantitatea de substanță chimică de testat administrată. Doza este exprimată ca greutate a substanței chimice de testat pe unitatea de greutate corporală a animalului de experiență pe zi (de exemplu, mg/kg greutate corporală/zi) sau ca o concentrație constantă în regimul alimentar.
Toxicitatea evidentă este un termen general care descrie semne clare de toxicitate în urma administrării unei substanțe chimice de testat. Aceste semne ar trebui să fie suficiente pentru a permite o evaluare a pericolelor și ar trebui să fie de așa natură încât o creștere a dozei administrate să poată fi prevăzută a declanșa apariția unor semne de toxicitate severe și probabil moartea.
Afectarea fertilității reprezintă tulburări ale funcțiilor sau capacității de reproducere masculine sau feminine.
Toxicitate maternă: efecte adverse asupra femelelor gestante, care apar fie în mod specific (efect direct), fie în mod nespecific (efect indirect).
NOAEL este abrevierea pentru nivelul la care nu se observă niciun efect advers. Acesta este cel mai ridicat nivel de dozare la care nu se observă efecte adverse din cauza tratamentului.
Estrogenicitatea este capacitatea unei substanțe chimice de a acționa ca hormon estrogen natural (de exemplu, estradiol 17ß) în organismul unui mamifer.
Toxicitate pentru reproducere reprezintă efecte dăunătoare asupra descendenților și/sau afectarea funcțiilor sau a capacității de reproducere masculine și feminine.
Substanță chimică de testat înseamnă orice substanță sau amestec de substanțe testat(ă) utilizând această metodă de testare.
Activitatea tiroidiană este capacitatea unei substanțe chimice de a acționa ca hormon tiroidian natural (de exemplu, estradiol T3) în organismul unui mamifer.
Validare înseamnă un proces științific conceput pentru a caracteriza cerințele și limitările operaționale ale unei metode de testare și pentru a demonstra fiabilitatea și relevanța acesteia pentru un anumit scop.
Apendicele 2
DIAGRAMĂ A SCHEMEI DE TRATAMENT CARE INDICĂ DURATA MAXIMĂ A STUDIULUI, PE BAZA UNEI PERIOADE DE ÎMPERECHERE COMPLETE DE 14 ZILE

Apendicele 3
RAPORT DE SINTEZĂ TABELAR CU EFECTELE ASUPRA REPRODUCERII/DEZVOLTĂRII
|
OBSERVAȚII
|
VALORI
|
|
|
|
Dozaj (unități)
|
0 (martor)
|
…
|
…
|
…
|
…
|
|
Perechi începute (N)
|
|
|
|
|
|
|
Ciclul estral (cel puțin durata medie și frecvența ciclurilor neregulate)
|
|
|
|
|
|
|
Femele la care apare dovada copulației (N)
|
|
|
|
|
|
|
Femele care au ajuns în gestație (N)
|
|
|
|
|
|
|
Zile de concepție 1-5 (N)
|
|
|
|
|
|
|
Zilele de concepție 6-...(
(21)
) (N)
|
|
|
|
|
|
|
Sarcină ≤ 21 de zile (N)
|
|
|
|
|
|
|
Sarcină = 22 de zile (N)
|
|
|
|
|
|
|
Sarcină ≥ 23 de zile (N)
|
|
|
|
|
|
|
Femele cu pui născuți vii (N)
|
|
|
|
|
|
|
Femele cu pui născuți vii în ziua 4 pp (N)
|
|
|
|
|
|
|
Implanturi/femelă (media)
|
|
|
|
|
|
|
Pui vii/femelă la naștere (media)
|
|
|
|
|
|
|
Pui vii/femelă în ziua 4 (media)
|
|
|
|
|
|
|
Raportul dintre sexe (m/f) la naștere (media)
|
|
|
|
|
|
|
Raportul dintre sexe (m/f) în ziua 4 (media)
|
|
|
|
|
|
|
Greutatea seriei la naștere (media)
|
|
|
|
|
|
|
Greutatea seriei în ziua 4 (media)
|
|
|
|
|
|
|
Greutatea puilor la naștere (media)
|
|
|
|
|
|
|
Greutatea puilor în momentul măsurării AGD (media la masculi, media la femele)
|
|
|
|
|
|
|
AGD a puilor în aceeași zi postnatală, la naștere – ziua 4 (media la masculi, media la femele, se notează PND)
|
|
|
|
|
|
|
Greutatea puilor în ziua 4 (media)
|
|
|
|
|
|
|
Retenția sfârcurilor la pui masculi în ziua 13 (media)
|
|
|
|
|
|
|
Greutatea puilor în ziua 13 (media)
|
|
|
|
|
|
|
|
|
PUI ANORMALI
|
|
Femele cu 0
|
|
|
|
|
|
|
Femele cu 1
|
|
|
|
|
|
|
Femele cu ≥ 2
|
|
|
|
|
|
|
|
|
PIERDEREA PUILOR
|
|
|
|
Implantații prenatale/postnatale (implantații minus născuți vii)
|
|
Femele cu 0
|
|
|
|
|
|
|
Femele cu 1
|
|
|
|
|
|
|
Femele cu 2
|
|
|
|
|
|
|
Femele cu ≥ 3
|
|
|
|
|
|
|
|
|
Postnatal (pui născuți vii minus pui vii în ziua postnatală 13)
|
|
Femele cu 0
|
|
|
|
|
|
|
Femele cu 1
|
|
|
|
|
|
|
Femele cu 2
|
|
|
|
|
|
|
Femele cu ≥ 3
|
|
|
|
|
|
B.64 STUDIU DE TOXICITATE CU DOZĂ REPETATĂ COMBINAT CU TESTUL DE DEPISTARE A TOXICITĂȚII PENTRU REPRODUCERE/DEZVOLTARE
INTRODUCERE
|
1.
|
Această metodă de testare este echivalentă cu Orientarea OCDE privind testarea (TG) nr. 422 (2016). Orientările OCDE pentru testarea substanțelor chimice sunt revizuite periodic în contextul evoluțiilor științifice. Orientarea inițială nr. 422 privind testarea pentru depistare a fost adoptată în 1996, pe baza unui protocol pentru un „Test combinat cu doză repetată și de depistare a toxicității pentru reproducere/dezvoltare” discutat în cadrul a două reuniuni ale experților, la Londra, în 1990 (1) și la Tokyo în 1992 (2).
|
|
2.
|
Această metodă de testare combină o componentă de depistare a toxicității pentru reproducere/dezvoltare, care se bazează pe experiența dobândită în statele membre ca urmare a utilizării metodei inițiale pe substanțele chimice existente în mare volum de producție și în cadrul unor teste de explorare cu substanțe martor pozitive (3) (4), precum și pe o componentă cu repetarea dozei de toxicitate, în concordanță cu orientarea nr. 407 a OCDE privind testarea (Studiu de toxicitate orală cu doză repetată de 28 de zile la rozătoare, corespunzător capitolului B.7 din prezenta anexă).
|
|
3.
|
Această metodă de testare a fost actualizată cu puncte finale relevante privind perturbatorii endocrini, ca o continuare a activității cu prioritate ridicată inițiată de OCDE în 1998 în vederea revizuirii orientărilor existente privind testarea și a elaborării unor noi orientări privind testarea pentru depistarea și testarea unor potențiali perturbatori endocrini (5). În acest context orientarea nr. 407 privind testarea (corespunzătoare capitolului B.7 din prezenta anexă), a fost îmbunătățită în 2008 prin parametri adecvați pentru detectarea activității endocrine a substanțelor chimice de testat. În cadrul actualizării Orientării nr. 422 privind testarea, obiectivul a fost acela de a include câteva puncte finale relevante pentru perturbatori endocrini în orientările privind testele pentru depistare în cazul în care perioadele de expunere acoperă unele dintre perioadele sensibile din procesul de dezvoltare (perioadele prenatale sau postnatale timpurii).
|
|
4.
|
Punctele finale relevante suplimentare selectate pentru perturbatorii endocrini fac parte, de asemenea, din TG nr. 443 privind testarea (Studiu extins de toxicitate pentru reproducere la o singură generație, corespunzător capitolului B.56 din prezenta anexă) au fost incluse în TG nr. 422 pe baza unui studiu de fezabilitate care abordează aspecte științifice și tehnice legate de includerea acestora, precum și de posibile adaptări ale modelului testului, care sunt necesare pentru includerea acestora (6).
|
|
5.
|
Această metodă de testare este concepută pentru a genera informații limitate cu privire la efectele unei substanțe chimice de testat asupra performanței reproductive a masculilor și femelelor, cum ar fi funcția gonadelor, comportamentul de împerechere, concepția, dezvoltarea produsului de concepție și parturiția. Aceasta nu este o alternativă la metodele de testare existente B.31, B.34, B.35 sau B.56 și nici nu le înlocuiește.
|
CONSIDERAȚII INIȚIALE
|
6.
|
În analiza și evaluarea caracteristicilor toxice ale unei substanțe chimice de testat, determinarea toxicității orale folosind doze repetate se poate realiza după ce au fost obținute informațiile inițiale privind toxicitatea din teste de toxicitate acută. Acest studiu oferă informații cu privire la posibilele pericole pentru sănătate care pot apărea în urma expunerii repetate pe o perioadă de timp relativ limitată. Metoda cuprinde studiul de bază de toxicitate la doză repetată, care se poate folosi pentru substanțe chimice pentru care nu este justificat un studiu de 90 de zile (de exemplu, în cazul în care volumul de producție nu depășește anumite limite) sau ca studiu preliminar în cazul unui studiu pe termen lung. În desfășurarea studiului, ar trebui să se respecte principiile directoare și considerentele prezentate în documentul de orientare nr. 19 al OCDE privind recunoașterea, evaluarea și utilizarea semnelor clinice ca puncte finale fără suferință pentru animalele de experiență utilizate în evaluări ale siguranței (7).
|
|
7.
|
De asemenea, acesta cuprinde un test de depistare a toxicității pentru reproducere/dezvoltare și, prin urmare, poate fi utilizat, de asemenea, pentru a furniza informații inițiale cu privire la posibile efecte asupra performanțelor de reproducere ale masculilor și femelelor, cum ar fi funcția gonadală, comportamentul de împerechere, concepția, dezvoltarea produsului de concepție și parturiția, fie într-o etapă timpurie de evaluare a proprietăților toxicologice ale substanțelor chimice de testat, fie cu privire la substanțe chimice de testat de interes. Această metodă de testare nu oferă informații complete cu privire la toate aspectele legate de reproducere și dezvoltare. În special, aceasta oferă doar mijloace limitate de detectare a manifestărilor postnatale ale expunerii prenatale sau a efectelor care pot fi induse în timpul expunerii postnatale. Printre altele, din cauza selectivității punctelor finale și a duratei scurte a studiului, această metodă nu va furniza dovezi pentru concluzii sigure privind inexistența efectelor pentru reproducere/dezvoltare. În plus, în absența datelor provenite din alte teste de toxicitate pentru reproducere/dezvoltare, rezultatele pozitive sunt utile pentru evaluarea inițială a pericolelor și contribuie la luarea deciziilor cu privire la necesitatea și calendarul testelor suplimentare.
|
|
8.
|
Rezultatele obținute prin parametrii endocrini conecși ar trebui să fie evaluate în contextul „Cadrului conceptual al OCDE pentru testarea și evaluarea substanțelor chimice care perturbă procesele endocrine” (8). În acest cadru conceptual, orientarea nr. 422 a OCDE privind testarea îmbunătățită se regăsește la nivelul 4 ca test in vivo care asigură date privind efectele adverse asupra punctelor finale relevante ale sistemului endocrin. Însă este posibil ca un semnal endocrin să nu fie considerat drept o dovadă suficientă în sine pentru faptul că substanța chimică de testat este un perturbator endocrin.
|
|
9.
|
Această metodă de testare pune accentul, de asemenea, pe efectele neurologice ca punct final specific, precum și pe nevoia unor observații clinice riguroase, pentru a se obține cât mai multe informații posibil. Metoda ar trebui să presupună identificarea substanțelor chimice cu potențial neurotoxic, care ar putea justifica desfășurarea în continuare a unor studii mai aprofundate cu privire la acest aspect. În plus, metoda poate oferi, de asemenea, o indicație elementară a efectelor imunologice.
|
|
10.
|
În absența datelor provenite din alte studii de toxicitate sistemică, de toxicitate pentru reproducere/dezvoltare, de neurotoxicitate și/sau imunotoxicitate, rezultatele pozitive sunt utile pentru evaluarea inițială a pericolelor și contribuie la luarea deciziilor cu privire la necesitatea și calendarul testelor suplimentare. Testul poate fi deosebit de util ca parte a setului de date cu informații de depistare al OCDE (SIDS) pentru evaluarea substanțelor chimice existente pentru care există puține informații toxicologice sau pentru care nu există astfel de informații, și care poate servi drept alternativă la efectuarea a două teste separate pentru toxicitatea la doze repetate (TG nr. 407 a OCDE, corespunzătoare capitolului B.7 din prezenta anexă) și pentru toxicitate pentru reproducere/dezvoltare (TG nr. 421 a OCDE, corespunzătoare capitolului B.63 din prezenta anexă). Acesta poate fi utilizat, de asemenea, ca studiu pentru stabilirea dozelor pentru studii mai ample privind reproducerea/dezvoltarea sau în alte cazuri considerate relevante.
|
|
11.
|
În general, se presupune că există diferențe de sensibilitate între animalele gestante și animalele negestante. Prin urmare, ar putea fi mai complicat să se stabilească niveluri ale dozelor în cadrul acestui test combinat, care să fie adecvate pentru evaluarea toxicității sistemice generale și a toxicității specifice pentru reproducere/dezvoltare, decât în cazul în care sunt efectuate teste individuale separat. În plus, interpretarea rezultatelor testelor în ceea ce privește toxicitatea sistemică generală ar putea fi mai dificilă decât atunci când se realizează un studiu cu doză repetată separat, în special atunci când parametrii serului și ai histopatologiei nu sunt evaluați în același timp în cadrul studiului. Din cauza acestor complicații tehnice, este necesară o experiență considerabilă în ceea ce privește testarea toxicității pentru efectuarea prezentului test combinat de depistare. Pe de altă parte, în afara numărului mai mic de animale implicate, testul combinat poate oferi mijloace mai bune de diferențiere a efectelor directe asupra reproducerii/dezvoltării de cele care sunt secundare altor efecte (sistemice).
|
|
12.
|
În cadrul acestui test, perioada de dozare este mai lungă decât cea din cadrul unui studiu convențional cu doză repetată de 28 de zile. Însă, acesta utilizează mai puține animale de fiecare sex pe grup, în comparație cu situația în care se efectuează un test convențional cu doze repetate de 28 de zile pe lângă un test de depistare a toxicității pentru reproducere/dezvoltare.
|
|
13.
|
Această metodă de testare presupune administrarea substanței chimice de testat pe cale orală. Dacă se utilizează alte căi de expunere, este posibil să fie necesare modificări.
|
|
14.
|
Înainte de utilizarea metodei de testare pe un amestec pentru a genera date pentru un obiectiv de reglementare prevăzut, se ia în considerare dacă și, în caz afirmativ, din ce cauză aceasta poate oferi rezultate adecvate pentru respectivul scop. Astfel de considerații nu sunt necesare atunci când există o cerință normativă pentru testarea amestecului.
|
|
15.
|
Definițiile utilizate sunt prezentate în apendicele 1.
|
PRINCIPIUL TESTULUI
|
16.
|
Substanța chimică de testat se administrează în doze gradate mai multor grupuri de masculi și femele. Doza pentru masculi ar trebui să fie administrată pentru o perioadă de cel puțin patru săptămâni până în ziua dinaintea sacrificării planificate inclusiv (aceasta include cel puțin două săptămâni înainte de împerechere, perioada de împerechere și aproximativ două săptămâni după împerechere). Având în vedere perioada de dozare limitată de pre-împerechere a masculilor, este posibil ca fertilitatea să nu fie un indicator deosebit de sensibil al toxicității testiculare. Prin urmare, este esențială o examinare histologică detaliată a testiculelor. Combinația dintre o perioadă de dozare de pre-împerechere de două săptămâni și de formulare ulterioară a observațiilor privind împerecherea/fertilitatea și o perioadă de dozare generală de cel puțin patru săptămâni, urmată de histopatologia detaliată a gonadelor de sex masculin, este considerată suficientă pentru a permite detectarea majorității efectelor asupra fertilității masculine și a spermatogenezei.
|
|
17.
|
Femelelor ar trebui să li se administreze doze pe toată durata studiului. Aceasta include două săptămâni înainte de împerechere (cu obiectivul de a acoperi cel puțin două cicluri estrale complete), timpul variabil până la concepție, durata sarcinii și cel puțin 13 zile după parturiție, până în ziua dinaintea eutanasierii planificate inclusiv.
|
|
18.
|
Durata studiului, în urma aclimatizării și evaluării ciclului estral înainte de dozare, depinde de performanța femelei și este de aproximativ 63 de zile, [cel puțin 14 zile înainte de împerechere, (până la) 14 zile de împerechere, 22 de zile de gestație, 13 zile de lactație].
|
|
19.
|
În perioada de administrare, animalele sunt examinate cu atenție zilnic, în vederea identificării unor eventuale semne de toxicitate. Animalele care au murit sau care au fost eutanasiate în timpul testului sunt supuse unei autopsii, iar cele care supraviețuiesc până la sfârșitul testului sunt eutanasiate și autopsiate.
|
DESCRIEREA METODEI
Selecția speciilor de animale
|
20.
|
Această metodă de testare este concepută pentru a fi utilizată la șobolan. În cazul în care parametrii specificați în prezenta orientare TG nr. 422 privind testarea sunt investigați la o altă specie de rozătoare, ar trebui să se prezinte o justificare detaliată. În cadrul programului de validare internațional pentru detectarea factorilor perturbatori endocrini cu privire la TG nr. 407, șobolanul a fost singura specie folosită. Nu ar trebui să se utilizeze sușe cu fecunditate scăzută sau cu o incidență a defectelor de dezvoltare cunoscută a fi ridicată. Ar trebui să se folosească animale virgine sănătoase, care nu au fost supuse anterior unor proceduri experimentale. Animalele de experiență ar trebui să fie caracterizate în ceea ce privește specia, sușa, sexul, greutatea și vârsta. La începutul studiului, diferențele de greutate între animalele folosite ar trebui să fie minime și să nu depășească 20 % din greutatea medie pentru fiecare sex. În cazul în care studiul este realizat cu titlu de studiu preliminar pentru un studiu pe termen lung sau pentru o întreagă generație, este de preferat să se utilizeze, în ambele studii, animale din aceeași sușă și din aceeași sursă.
|
Adăpostire și hrănire
|
21.
|
Toate procedurile ar trebui să fie în conformitate cu standardele locale privind îngrijirea animalelor de laborator. Temperatura în spațiul de adăpostire a animalelor de experiență ar trebui să fie de 22 °C (± 3 °C). Umiditatea relativă ar trebui să fie de cel puțin 30 % și să nu depășească, de preferință, 70 %, cu excepția momentelor în care se curăță spațiul. Iluminatul ar trebui să fie artificial, alternând perioade de 12 ore de lumină cu 12 ore de întuneric. Pentru alimentație, se poate utiliza hrană convențională de laborator, cu asigurarea unei cantități nelimitate de apă potabilă. Alegerea hranei poate fi influențată de necesitatea de a asigura un amestec adecvat al unei substanțe chimice de testat atunci când aceasta se administrează utilizând această metodă.
|
|
22.
|
Animalele ar trebui să fie adăpostite în grupuri mici de același sex; animalele pot fi adăpostite individual dacă există o justificare științifică. În cazul cuștilor colective nu ar trebui să se depășească numărul de cinci animale în fiecare cușcă. Procedurile de împerechere se desfășoară în cuști adecvate acestui scop. Femelele gestante ar trebui să fie plasate în cuști individuale și prevăzute cu materiale pentru cuibărire. Femelele aflate în lactație vor fi plasate în cuști individuale împreună cu progeniturile acestora.
|
|
23.
|
Hrana ar trebui să fie analizată în mod regulat în vederea identificării contaminanților. Ar trebui să se rețină un eșantion din hrană până la finalizarea raportului.
|
Pregătirea animalelor
|
24.
|
Animalele adulte, tinere și sănătoase sunt distribuite în mod aleatoriu în grupuri și cuști de tratament. Cuștile ar trebui să fie dispuse astfel încât să se reducă la minim eventualele efecte datorate deplasărilor acestora. Animalele prezintă o identificare unică și sunt ținute în cuștile lor timp de minimum cinci zile înainte de începerea studiului, pentru a permite aclimatizarea acestora la condițiile de laborator.
|
Prepararea dozelor
|
25.
|
Se recomandă administrarea orală a substanței chimice de testat, cu excepția cazului în care se consideră că sunt adecvate alte căi de administrare. Atunci când se alege calea orală, substanța chimică de testat se administrează, de obicei, prin gavaj; însă, în mod alternativ, substanțele chimice de testat pot fi administrate, de asemenea, împreună cu hrana sau cu apa potabilă.
|
|
26.
|
Dacă este necesar, substanța chimică de testat se dizolvă sau se suspendă într-un vehicul adecvat. Se recomandă ca, ori de câte ori este posibil, să se aibă în considerare, în primul rând, utilizarea unei soluții/suspensii apoase, apoi a unei soluții/suspensii în ulei (de exemplu, ulei de porumb) și apoi a unei alte soluții posibile în alte vehicule. Pentru vehicule neapoase, ar trebui să fie cunoscute caracteristicile toxice ale vehiculului respectiv. Ar trebui să se determine stabilitatea și omogenitatea substanței chimice de testat în vehicul.
|
PROCEDURA
Numărul și sexul animalelor
|
27.
|
Se recomandă ca, la început, fiecare grup să aibă cel puțin 10 masculi și 12-13 femele. Femelele vor fi evaluate înainte de expunere pentru ciclicitatea estrală, iar animalele la care nu se produc cicluri tipice de 4-5 zile nu vor fi incluse în studiu; prin urmare, se recomandă femele suplimentare pentru a obține 10 femele într-un grup. Cu excepția cazului în care apar efecte toxice pronunțate, se preconizează că acesta va asigura cel puțin 8 femele gestante în fiecare grup, care reprezintă, în mod normal, numărul minim acceptabil de femele gestante într-un grup. Obiectivul este să se asigure un număr suficient de sarcini și pui pentru a asigura o evaluare semnificativă a potențialului substanței chimice de testat de a afecta fertilitatea, sarcina și comportamentul matern și de supt, precum și creșterea și dezvoltarea puilor F1, de la concepție până în ziua 13 a perioadei post-partum. Dacă sunt planificate eutanasieri intermediare, numărul ar trebui suplimentat cu numărul de animale planificate să fie eutanasiate înainte de încheierea studiului. În plus, ar trebui să se aibă în vedere un grup satelit suplimentar de cinci animale pe grup pe sex în grupul martor și în grupul cu doza maximă, pentru observații privind reversibilitatea, persistența sau apariția tardivă a efectelor sistemice toxice pe parcursul a minimum 14 zile după tratament. Animalele din grupurile satelit nu vor fi împerecheate și, în consecință, nu sunt utilizate pentru evaluarea toxicității pentru reproducere/dezvoltare.
|
Dozare
|
28.
|
În general, ar trebui să se utilizeze cel puțin trei grupuri de testare tratate și un grup martor. Dacă nu există date generale adecvate privind toxicitatea, se realizează un studiu de stabilire a intervalului (animale din aceeași sușă și sursă), pentru a contribui la determinarea dozelor care vor fi administrate. Cu excepția tratării cu substanța chimică de testat, animalele din grupul martor ar trebui să fie tratate în mod identic cu animalele din grupul de testare. Dacă pentru administrarea substanței chimice de testat se folosește un vehicul, acesta se administrează grupului martor în volumul maxim utilizat.
|
|
29.
|
Dozele ar trebui să fie selectate ținându-se cont de toxicitatea existentă și de datele toxico-cinetice disponibile. De asemenea, ar trebui să se țină seama de faptul că ar putea exista diferențe de sensibilitate între animalele gestante și cele negestante. Ar trebui să fie aleasă doza cea mai mare pentru a induce efecte toxice, însă nu moartea sau o suferință evidentă. Ulterior, ar trebui să se selecteze o serie de doze descrescătoare pentru a demonstra orice răspuns legat de doză și faptul că nu există efecte adverse la doza minimă. Cele mai bună soluții constau de multe ori în alegerea a două sau patru intervale, iar adăugarea unui al patrulea grup de testare este adesea preferabilă utilizării unor intervale foarte mari (de exemplu, care depășesc factorul 10) între dozaje.
|
|
30.
|
În prezența toxicității generale observate (de exemplu, greutate corporală redusă, efecte asupra ficatului, plămânilor sau rinichilor etc.) sau a altor schimbări care pot să nu constituie răspunsuri toxice (de exemplu, consum de hrană redus, mărirea ficatului), efectele observate asupra punctelor finale endocrine sensibile ar trebui să fie interpretate cu precauție.
|
Testul valorilor-limită
|
31.
|
Dacă un studiu oral efectuat pe baza procedurilor descrise în prezentul studiu, la o doză de cel puțin 1 000 mg/kg greutate corporală/zi sau, în cazul administrării împreună cu hrana, la un procent echivalent în hrană sau în apa de băut (în funcție de valorile greutății corporale), nu produce efecte toxice detectabile și dacă datele despre substanțe cu structuri asemănătoare nu indică toxicitatea, atunci este posibil să nu fie necesar să se realizeze un studiu complet cu mai multe doze. Testul la valori-limită este justificat, cu excepția cazului în care condițiile de expunere umană impun utilizarea unei doze mai ridicate. Pentru alte tipuri de administrare, cum sunt inhalarea sau aplicarea dermică, este posibil ca proprietățile fizice și chimice ale substanțelor chimice de testat să determine deseori nivelul maxim posibil al expunerii.
|
Administrarea dozelor
|
32.
|
Animalelor li se administrează substanța chimică de testat zilnic, timp de 7 zile pe săptămână. Când substanța chimică de testat este administrată prin gavaj, administrarea ar trebui să fie în doză unică la animale, folosind o sondă gastrică sau o canulă de intubare adecvată. Volumul maxim de lichid care poate fi administrat o dată depinde de dimensiunea animalului de experiență. Volumul nu ar trebui să depășească 1 ml/100 g greutate corporală, exceptând cazul soluțiilor apoase, când se pot utiliza 2 ml/100 g greutate corporală. Cu excepția substanțelor chimice de testat iritante sau corozive care vor produc, de regulă, efecte exacerbate la concentrații mai mari, variabilitatea volumului de testat ar trebui să fie redusă la minim prin ajustarea concentrației, astfel încât să se asigure un volum constant la toate nivelurile de dozare.
|
|
33.
|
Pentru substanțele chimice de testat administrate în hrană sau în apa potabilă, este important să se asigure că respectivele cantități de substanță chimică de testat implicate nu influențează nutriția normală sau echilibrul hidric. Dacă substanța chimică de testat se administrează în hrană, aceasta poate fi administrată fie într-o concentrație constantă în alimentație (ppm), fie la o doză constantă în raport cu greutatea corporală a animalelor; alternativa utilizată ar trebui precizată. În cazul unei substanțe chimice de testat administrate prin gavaj, doza ar trebui să se administreze la aproximativ aceeași oră în fiecare zi și să fie ajustată cel puțin săptămânal pentru a menține un nivel constant al dozei în funcție de greutatea corporală a animalului. În cazul în care studiul combinat este utilizat cu titlu preliminar unui studiu de toxicitate pentru reproducere pe termen lung sau complet, în ambele studii ar trebui să se utilizeze o alimentație similară.
|
Schema de tratament
|
34.
|
Administrarea dozelor la ambele sexe ar trebui să înceapă cu două săptămâni înainte de împerechere, după ce acestea au fost aclimatizate timp de cel puțin cinci zile, iar femelele au fost supuse unui control privind ciclurile estrale normale (într-o perioadă de pre-tratament de 2 săptămâni). Studiul ar trebui să fie planificat în așa fel încât evaluarea ciclului estral să înceapă de îndată ce animalele au ajuns la maturitatea sexuală deplină. Aceasta ar putea varia ușor între diferite sușe de șobolani din diferite laboratoare, de exemplu, la șobolanii Sprague Dawley aceasta este la vârsta de 10 săptămâni, la șobolanii Wistar la vârsta de aproximativ 12 săptămâni. Femelele cu pui ar trebui să fie eutanasiate în ziua 13 post-partum sau imediat după aceea. Pentru a permite nealimentarea pe timpul nopții a femelelor înainte de recoltarea sângelui (dacă se preferă această opțiune), nu este necesar ca femelele și puii lor să fie eutanasiați în aceeași zi. Ziua nașterii (și anume, atunci când s-a încheiat parturiția) este definită ca fiind ziua 0 post-partum. Femelele care nu prezintă semne ale copulației sunt eutanasiate după 24-26 zile de la ultima zi a perioadei de împerechere. Administrarea dozelor continuă la ambele sexe în perioada de împerechere. Masculilor ar trebui să li se administreze în continuare doze după perioada de împerechere cel puțin până la încheierea perioadei de dozare minime totale de 28 de zile. Apoi aceștia sunt eutanasiați sau, alternativ, sunt reținuți pentru administrarea în continuare a dozelor pentru o posibilă desfășurare a unei a doua împerecheri, dacă se consideră că este cazul.
|
|
35.
|
Administrarea de doze zilnice la femelele parentale ar trebui să continue pe întreaga durată a sarcinii și cel puțin până în ziua 13 post-partum inclusiv sau cu o zi înainte de sacrificare. În cazul studiilor în care substanța chimică de testat se administrează prin inhalare sau subcutanat, dozele ar trebui să fie administrate în continuare cel puțin până în ziua 19 de sarcină inclusiv și administrate din nou cât mai curând posibil, dar nu mai târziu de ziua post-fătare (PND) 4.
|
|
36.
|
Animalele dintr-un grup satelit planificate pentru observații ulterioare, dacă acestea sunt incluse, nu sunt împerecheate. Acestea ar trebui păstrate timp de cel puțin încă 14 zile după prima eutanasiere programată a femelelor, fără a se aplica un tratament pentru a detecta apariția tardivă sau persistența efectelor toxice ori recuperarea de pe urma acestora.
|
|
37.
|
În apendicele 2 este prezentată o diagramă a schemei de tratament.
|
Cicluri estrale
|
38.
|
Înainte de a începe tratamentul, ar trebui să se monitorizeze ciclurile estrale pentru femelele studiate cu ciclicitate regulate (a se vedea punctul 27). De asemenea, frotiul vaginal ar trebui să fie monitorizat zilnic de la începutul perioadei de tratament, până la obținerea dovezii împerecherii. În cazul în care există motive de îngrijorare cu privire la efecte de criză acută care ar putea modifica ciclurile estrale odată cu inițierea administrării dozelor, laboratoarele pot să expună animalele de experiență timp de două săptămâni, apoi să colecteze frotiuri vaginale zilnic pentru a monitoriza ciclul estral timp de cel puțin două săptămâni în perioada de pre-împerechere, cu o continuare a monitorizării în perioada de împerechere până când există dovezi ale împerecherii. Atunci când se obțin celule vaginale/cervicale, ar trebui să se evite orice perturbare a mucoasei, care ar putea induce pseudogestația (8) (9).
|
Procedura de împerechere
|
39.
|
De regulă, în prezentul studiu ar trebui utilizate împerecheri 1:1 (un mascul la o femelă). Pot exista excepții în cazul deceselor ocazionale ale masculilor. Femela ar trebui să fie plasată împreună cu același mascul până când se observă semne ale copulației sau au trecut două săptămâni. În fiecare dimineață, femelele ar trebui să fie examinate pentru a se constata prezența spermei sau a dopului vaginal. Ziua 0 a gestației este definită ca fiind ziua în care se confirmă dovada împerecherii (se constată existența unui dop vaginal sau a spermei). În cazul în care împerecherea a eșuat, se poate lua în calcul reîmperecherea femelelor în cauză cu masculi din același grup care și-au dovedit capacitatea reproductivă.
|
Dimensiunea seriei de pui
|
40.
|
În ziua 4 de după naștere, dimensiunea fiecărei serii de pui poate fi ajustată prin eliminarea puilor suplimentari printr-o selecție aleatorie pentru a obține, pe cât posibil, patru sau cinci pui per sex per serie, în funcție de dimensiunea normală a seriei la sușa de șobolani utilizați. Ar trebui să se recolteze probe de sânge de la doi dintre puii în exces, care să fie grupate și utilizate pentru determinarea nivelurilor de T4 din ser. Eliminarea selectivă a puilor, de exemplu, pe baza greutății corporale sau a distanței anogenitale (AGD) nu este adecvată. Ori de câte ori numărul de pui masculi sau femele împiedică obținerea unui număr de patru sau cinci exemplare din fiecare sex per serie, se acceptă ajustarea parțială (de exemplu, șase masculi și patru femele). Nu se vor elimina pui dacă dimensiunea seriei va scădea sub ținta de sacrificare (8 sau 10 de pui/serie). Dacă există un singur pui peste obiectivul de sacrificare, se va elimina doar un singur pui, acesta fiind utilizat pentru recoltarea de sânge pentru posibile evaluări ale serului T4.
|
|
41.
|
Dacă dimensiunea seriei nu este ajustată, se sacrifică doi pui per serie în ziua 4, după naștere, și se iau probe de sânge pentru măsurarea concentrațiilor de hormoni tiroidieni din ser. Dacă este posibil, cei doi pui per serie ar trebui să fie pui de sex feminin pentru a rezerva puii masculi în vederea evaluării retenției mamelonului, cu excepția cazului în care în urma îndepărtării puilor respectivi nu mai rămân femele pentru evaluare la încheierea procesului. Nu se vor elimina pui în cazul în care dimensiunea seriei va scădea sub 8 sau 10 pui/serie (în funcție de dimensiunea normală a seriei în sușa de șobolani utilizată). Dacă există un singur pui peste dimensiunea normală a seriei, se va elimina doar un singur pui, acesta fiind utilizat pentru recoltarea de sânge pentru posibile evaluări ale serului T4.
|
Observații
|
42.
|
Ar trebui să se formuleze observații clinice generale cel puțin o dată pe zi, de preferat la aceeași oră (aceleași ore) în fiecare zi, luând în considerare perioada de vârf a efectelor anticipate după administrare. Ar trebui să se consemneze starea de sănătate a animalelor. Toate animalele sunt examinate cel puțin de două ori pe zi, pentru a se determina morbiditatea sau mortalitatea.
|
|
43.
|
Toate animalele parentale ar trebui să fie supuse unor observații clinice detaliate înaintea primei expuneri (pentru a permite comparații la nivelul aceluiași individ) și cel puțin o dată pe săptămână în perioada următoare. Aceste observații ar trebui să fie formulate în afara cuștii animalelor, într-o incintă standard, și, de preferință, la aceeași oră în fiecare zi. Acestea ar trebui să fie înregistrate cu atenție; de preferat, utilizând sisteme de punctare definite în mod explicit de laboratorul de testare. Ar trebui să se depună eforturi pentru a se asigura un nivel minim al variațiilor de la nivelul condițiilor de laborator, iar observațiile sunt formulate, de preferință, de către observatori care nu sunt informați asupra tratamentului. Simptomele consemnate ar trebui să includă, printre altele, modificări la nivelul pielii, blănii, ochilor, membranelor mucoase, apariția unor secreții și excreții și activitatea reflexă (de exemplu, lăcrimarea, erecția piloasă, mărimea pupilei, respirație anormală). Ar trebui să se consemneze, de asemenea, toate modificările de comportament, modificările posturii sau ale reacțiilor la manipulare, precum și apariția unor mișcări clonice sau tonice, a unor comportamente stereotipe (de exemplu, curățare excesivă, mișcări circulare repetate), parturiția dificilă sau prelungită sau orice comportament bizar (automutilare, mers cu spatele) (10).
|
|
44.
|
O singură dată în timpul studiului, ar trebui să fie efectuată reactivitatea senzorială la diferiți stimuli (de exemplu, stimuli auditivi, vizuali și proprioceptivi) (8) (9) (11), evaluarea forței de prindere (12) și evaluarea activității motorii (13) la cinci masculi și cinci femele, selectați în mod aleatoriu din fiecare grup. Detalii suplimentare referitoare la procedurile care ar putea fi urmate sunt prezentate în referințele bibliografice respective. Însă, ar putea fi utilizate, de asemenea, proceduri alternative celor menționate. La masculi, aceste observații funcționale ar trebui să fie formulate către sfârșitul perioadei de administrare a dozelor, cu puțin timp înainte de momentul programat pentru eutanasiere, dar înainte de recoltarea de sânge pentru analize hematologice sau chimice clinice (a se vedea punctele 53-56, inclusiv nota de subsol 1). Femelele ar trebui să fie într-o stare similară din punct de vedere fiziologic în cursul acestor teste funcționale și ar trebui, de preferință, să fie testate o singură dată în ultima săptămână de lactație (de exemplu, zilele de lactație LD 6-13), cu puțin timp înainte de ziua planificată pentru eutanasiere. În măsura în care este posibil, se reduc la minimum perioadele de separare a femelelor de pui.
|
|
45.
|
Observațiile funcționale formulate o singură dată spre sfârșitul studiului ar putea fi omise atunci când studiul este realizat cu titlu preliminar unui studiu subcronic (de 90 de zile) unui studiu pe termen lung. În acest caz, observațiile funcționale ar trebui să facă parte din acest studiu realizat în continuare. Pe de altă parte, disponibilitatea datelor despre observații funcționale formulate ca urmare a acestui studiu cu administrare de doze repetate ar putea spori capacitatea de a alege doze pentru un studiu ulterior subcronic sau pe termen lung.
|
|
46.
|
În mod excepțional, observațiile funcționale pot fi, de asemenea, omise pentru grupuri care prezintă, de altfel, simptome de toxicitate în asemenea măsură încât ar influența semnificativ desfășurarea testului funcțional.
|
|
47.
|
Durata gestației ar trebui să fie înregistrată, calculându-se din ziua 0 de gestație. Fiecare serie de pui ar trebui să fie examinată cât mai curând posibil după naștere, pentru a se stabili numărul și sexul puilor, puii născuți morți, puii vii, puii subdezvoltați (puii care sunt semnificativ mai mici decât puii corespunzători din grupul martor) și prezența unor anomalii macroscopice.
|
|
48.
|
În termen de 24 de ore de la parturiție, puii vii ar trebui să fie numărați și separați pe sexe, iar seriile de pui cântărite (în ziua 0 sau 1 post-partum) și cel puțin în zilele 4 și 13 post-partum. Pe lângă observațiile privind animalele părinți (a se vedea punctele 43 și 44), ar trebui să fie consemnat orice comportament anormal al puiului.
|
|
49.
|
Este necesar ca AGD a fiecărui pui să fie măsurată în aceeași zi post-natală de la PND 0 până la PND 4. Greutatea corporală a puilor ar trebui să fie consemnează în ziua în care se măsoară AGD, aceasta trebuind să fie normalizată la o valoare aferentă dimensiunii puiului, de preferat, rădăcina cubică a greutății corporale (14). Numărul sfârcurilor/aureolelor la puii masculi ar trebui să fie calculat în PND 12 sau 13, astfel cum se recomandă în Orientarea nr. 151 a OCDE (15).
|
Greutate corporală și consumul de hrană/apă
|
50.
|
Masculii și femelele ar trebui să fie cântărite în prima zi a administrării dozelor, apoi cel puțin săptămânal și la sfârșit. În timpul sarcinii, femelele ar trebui să fie cântărite în zilele 0, 7, 14 și 20 și în termen de 24 de ore de la parturiție (în ziua 0 sau 1 post-partum) și cel puțin în zilele 4 și 13 post-partum. Aceste observații ar trebui să fie consemnate individual pentru fiecare animal adult.
|
|
51.
|
În perioada de pre-împerechere, de sarcină și de alăptare, consumul de alimente ar trebui să fie măsurat cel puțin o dată pe săptămână. Măsurarea consumului de hrană în timpul împerecherii este opțională. Pe parcursul acestor perioade, consumul de apă ar trebui, de asemenea, să fie măsurat atunci când substanța chimică de testat este administrată în mediul respectiv.
|
Hematologie
|
52.
|
O dată pe parcursul studiului, următoarele examinări hematologice ar trebui să fie efectuate la cinci masculi și cinci femele selectate aleatoriu din fiecare grup: hematocritul, concentrațiile de hemoglobină, numărul de eritrocite, reticulocitele, numărul total de leucocite și formula leucocitară diferențiată, numărul de trombocite și timpul/potențialul de coagulare. Printre alte determinări care ar trebui să fie efectuate, în cazul în care substanța chimică de testat sau eventualii săi metaboliți au sau se presupune că au proprietăți oxidante, se numără concentrația de methemoglobină și corpii Heinz.
|
|
53.
|
Probele de sânge ar trebui să fie prelevate dintr-un loc desemnat. În timpul eșantionării, femelele ar trebui să se afle într-o stare similară din punct de vedere fiziologic. Pentru a evita dificultățile practice legate de variabilitatea declanșării gestației, recoltarea sângelui la femele se poate face la sfârșitul perioadei de pre-împerechere, ca alternativă la eșantionare imediat înainte de procedura de eutanasiere a animalelor sau în cadrul acesteia. Probele de sânge de la masculi ar trebui să fie prelevate, de preferință, înainte de sau în cadrul procedurii de eutanasiere a animalelor. În mod alternativ, recoltarea de sânge la masculi poate fi realizată, de asemenea, la sfârșitul perioadei de pre-împerechere, atunci când acest moment a fost preferat pentru femele.
|
|
54.
|
Probele de sânge ar trebui să fie păstrate în condiții adecvate.
|
Biochimia clinică
|
55.
|
Determinările de biochimie clinică în scopul de a investiga efectele toxice majore asupra țesuturilor și, în special, efectele asupra rinichiului și a ficatului, ar trebui să fie realizate pe probe de sânge prelevate de la cinci masculi și cinci femele selectați din fiecare grup. Se recomandă ca animalele să nu primească hrană în noaptea dinaintea recoltării (22). Investigațiile plasmei sau ale serului ar trebui să includă analizele pentru sodiu, potasiu, glucoză, colesterolul total, uree, creatinină, proteine totale și albumină, cel puțin pentru două enzime revelatoare ale efectelor hepatocelulare (cum ar fi alanin aminotransferaza, aspartat aminotransferaza și sorbitol dezhidrogenaza) și acizii biliari. În anumite împrejurări, valorile de măsurare a unor enzime suplimentare (de origine hepatică sau de altă origine) și a bilirubinei pot furniza informații utile.
|
|
56.
|
Sunt prelevate probe de sânge dintr-un anumit loc pe baza următorului calendar:
|
—
|
de la cel puțin doi pui pe serie în ziua 4, după naștere, în cazul în care numărul puilor permite acest lucru (a se vedea punctele 40-41)
|
|
—
|
de la toate femelele și de la cel puțin doi pui pe serie la încheiere, în ziua 13, și
|
|
—
|
de la toți masculii adulți, la încheiere
|
Toate probele de sânge sunt păstrate în condiții adecvate. Sunt analizate probe de sânge de la pui din ziua 13 și de la masculii adulți pentru identificarea nivelurilor de hormoni tiroidieni din ser (T4). Dacă se consideră că este relevant, sunt efectuate analize suplimentare ale T4 din probele de sânge de la femele și de la puii din ziua 4. Opțional, dacă se consideră relevant, ar putea fi măsurați și alți hormoni. Probele de sânge ale puilor pot fi grupate pe serii pentru analize ale hormonilor tiroidieni. Hormonii tiroidieni (T4 și TSH) ar trebui, de preferință, să fie măsurați ca „valoare totală”.
|
|
57.
|
Opțional, ar putea fi efectuate următoarele analize de urină la cinci masculi selectați aleatoriu din fiecare grup în ultima săptămână a studiului, cu recoltarea urinei într-un interval de timp; aspect, volum, osmolalitate sau densitate specifică, pH, proteinurie, glucozurie, hematurie.
|
|
58.
|
În plus, ar trebui să fie avute în vedere investigații asupra markerilor din ser care indică lezarea generală a țesuturilor. Alte determinări care ar trebui să fie efectuate dacă proprietățile cunoscute ale substanței chimice de testat pot sau sunt susceptibile de a afecta profilurile metabolice includ măsurarea calciului, a fosfatului, a trigliceridelor măsurate după perioada fără hrană, a glucozei după perioada fără hrană, a hormonilor specifici, a methemoglobinei și a colinesterazei. Acestea trebuie să fie identificate de la caz la caz.
|
|
59.
|
Următorii factori pot influența variabilitatea și concentrațiile absolute ale valorilor hormonale determinate:
|
—
|
momentul eutanasierii, din cauza variației diurne a concentrațiilor hormonale
|
|
—
|
metoda de eutanasiere pentru a evita stresul nejustificat exercitat asupra animalelor, care poate afecta concentrațiile hormonale;
|
|
—
|
seturile de testare pentru determinările hormonale, care pot varia prin curbele lor standard.
|
|
|
60.
|
Probele de plasmă prevăzute în mod specific pentru determinarea hormonală ar trebui să fie obținute la momente comparabile din zi. Valorile numerice obținute la analiza concentrațiilor hormonale diferă în funcție de diferitele seturi de testare comerciale.
|
|
61.
|
Dacă informațiile de referință istorice nu sunt adecvate, ar trebui să se aibă în vedere determinarea variabilelor hematologice și biochimice clinice înaintea începerii administrării dozelor sau, de preferință, la un grup de animale neincluse în grupurile experimentale. În cazul femelelor, datele trebuie să fie asigurate de la animale în lactație.
|
PATOLOGIE
Autopsie macroscopică
|
62.
|
Toate animalele adulte cuprinse în studiu ar trebui să fie supuse unei autopsii generale complete și detaliate, care cuprinde examinarea atentă a suprafeței externe a corpului, a tuturor orificiilor, a cavității craniene, toracice și abdominale, precum și a conținutului acestora. Se acordă o atenție deosebită organelor sistemului de reproducere. Ar trebui să se înregistreze numărul de locuri de implantare. Ar trebui să se examineze frotiurile vaginale în ziua autopsiei pentru a determina stadiul ciclului estral și pentru a permite corelarea cu histopatologia organelor de reproducere feminine.
|
|
63.
|
Testiculele și epididimitele, precum și prostata și veziculele seminale cu glande coagulante în ansamblu, ale tuturor masculilor adulți ar trebui să fie curățate de țesuturile aderente, după caz, și greutatea umedă a acestora înregistrată cât mai curând posibil după disecție, pentru a se evita deshidratarea. În plus, greutățile opționale ale organelor ar putea include complexul muscular levatorul ani plus bulbocavernos, glandele Cowper și glandul penisului la masculi și ovarele (greutate umedă) cuplate cu uterul (inclusiv colul uterin) la femele; dacă sunt incluse, aceste greutăți ar trebui să fie colectate cât mai curând posibil după disecție. Ar trebui să se păstreze ovarele, testiculele, epididimurile, organele sexuale conexe și toate organele care prezintă leziuni macroscopice la toate animalele adulte.
|
|
64.
|
De la toți masculii și femelele adulte și de la un pui mascul și un pui femelă din ziua 13, din fiecare serie, ar trebui să se păstreze glandele tiroide în cel mai adecvat mediu de fixare pentru examenul histopatologic prevăzut ulterior. După fixare ar putea fi determinată greutatea tiroidei. Curățarea ar trebui să fie făcută, de asemenea, foarte atent și doar după fixare, pentru a evita deteriorarea țesuturilor. Deteriorarea țesuturilor ar putea compromite analiza histopatologică. Ar trebui să se recolteze probe de sânge dintr-un loc desemnat imediat înainte de procedura de eutanasiere a animalelor sau în cadrul acesteia, care să fie păstrate în condiții adecvate (a se vedea punctul 56).
|
|
65.
|
În plus, pentru cel puțin cinci masculi și femele adulte, care sunt selectați aleatoriu din fiecare grup (în afară de cele muribunde și/sau eutanasiate înainte de încheierea studiului), ar trebui să se curețe ficatul, rinichii, glandele suprarenale, timusul, splina, creierul și inima de orice țesuturi aderente, după caz, și să se consemneze greutatea umedă a acestora cât mai repede posibil după disecție, pentru a se evita deshidratarea. Țesuturile următoare ar trebui să fie păstrate în cel mai adecvat mediu de fixare atât pentru tipul de țesut, cât și pentru examenul histopatologic prevăzut ulterior: toate țesuturile care prezintă leziuni macroscopice, encefalul (regiuni reprezentative: creierul mare, cerebelul și puntea), măduva spinării, ochiul, stomacul, intestinul subțire, intestinul gros (inclusiv plăcile Peyer), ficatul, rinichii, glandele suprarenale, splina, inima, timusul, tiroida, traheea și plămânii (conservați prin umflare cu fixativ și imersie ulterioară), gonadele (testiculele și ovarele), organele genitale anexe (de exemplu, uterul și colul, epididimul, prostata + veziculele seminale cu glandele coagulante), vaginul, vezica urinară, ganglionii limfatici [în afară de cel mai apropiat ganglion de drenare, ar trebui să fie prelevat încă un ganglion limfatic, conform experienței laboratorului (16)], nervul periferic (sciatic sau tibial), de preferință foarte aproape de mușchi, mușchiul scheletic și osul cu măduvă osoasă (secțiune sau, ca alternativă, aspirat de măduvă osoasă proaspăt fixat pe lamă). Se recomandă fixarea testiculelor prin imersie în fixator Bouin sau Davidson modificat (16) (17) (18); pentru aceste țesuturi nu se recomandă utilizarea formolului. Tunica albuginea ar putea fi perforată ușor și superficial cu un ac la ambii poli ai organului pentru a permite pătrunderea rapidă a fixatorului. Observațiile clinice și cele de altă natură pot sugera necesitatea examinării unor țesuturi suplimentare. Ar trebui conservate și alte organe considerate posibile organe-țintă pentru substanța chimică de testat, având în vedere proprietățile cunoscute ale acesteia.
|
|
66.
|
Următoarele țesuturi pot furniza indicații valoroase privind efectele asupra sistemului endocrin: gonadele (ovarele și testiculele), organele sexuale accesorii (uterul, inclusiv colul uterin, epididimul, veziculele seminale cu glandele coagulante, prostata dorso-laterală și ventrală), vaginul, hipofiza, glanda mamară masculină și glanda suprarenală. Modificările glandelor mamare masculine nu au fost suficient documentate dar acest parametru poate fi foarte sensibil la substanțe cu acțiune estrogenică. Observarea organelor/țesuturilor care nu sunt enumerate la punctul 65 este opțională.
|
|
67.
|
Puii morți și puii eutanasiați în ziua 13 post-partum, sau imediat după aceea, ar trebui să fie cel puțin examinați cu atenție la exterior pentru identificarea anomaliilor macroscopice. Ar trebui să se acorde o atenție deosebită organelor genitale de reproducere externe, care ar trebui să fie examinate pentru identificarea unor modificări în dezvoltare.
|
Histopatologie
|
68.
|
Ar trebui să se efectueze un examen histopatologic complet al organelor și țesuturilor conservate de la animalele selectate din grupurile martor și grupurile tratate cu doza maximă (se pune un accent deosebit pe fazele spermatogenezei la gonadele masculine și pe histopatologia structurii celulare testiculare interstițiale). Ar putea fi examinată, dacă este necesar, glanda tiroidă la pui la pui și la animalele adulte rămase. Aceste examene ar trebui să fie extinse la animalele din grupuri tratate cu alte doze, dacă se constată modificări legate de tratament la grupul tratat cu doză mare. Ghidul privind histopatologia (10) prezintă detalii suplimentare privind disecția, fixarea, secționarea și histopatologia țesuturilor endocrine.
|
|
69.
|
Ar trebui să se examineze toate leziunile macroscopice. Pentru a contribui la identificarea NOAEL, ar trebui să se examineze organele-țintă din alte grupuri de dozare, în special din grupuri pentru care s-a susținut existența NOAEL.
|
|
70.
|
Atunci când se folosește un grup satelit, ar trebui să se efectueze un examen histopatologic al țesuturilor și organelor la care au fost identificate efecte în grupurile tratate.
|
DATE ȘI RAPORTARE
Date
|
71.
|
Ar trebui să fie furnizate date despre fiecare animal. În plus, toate datele ar trebui să fie sintetizate în format tabelar, prezentându-se, pentru fiecare grup de testare, numărul de animale la începutul testului, numărul de animale găsite moarte în cursul testului sau eutanasiate, data decesului sau a eutanasierii, numărul de animale fertile, numărul de femele gestante, numărul de animale care prezintă semne de toxicitate, o descriere a semnelor de toxicitate observate, inclusiv momentul declanșării, durata și gravitatea efectelor toxice, tipul de modificări histopatologice și toate datele relevante privind seriile. În apendicele 3 este prezentat un format tabelar de raport de sinteză care s-a dovedit a fi foarte util pentru evaluarea efectelor privind reproducerea/dezvoltarea.
|
|
72.
|
Dacă este posibil, rezultatele numerice ar trebui să fie evaluate cu ajutorul unei metode statistice adecvate și general acceptate. Pentru comparațiile efectelor pe un interval de dozare ar trebui să se evite recurgerea la teste t multiple. Metodele statistice ar trebui să fie alese în faza de proiectare a studiului. Analiza statistică a AGD și a retenției sfârcurilor ar trebui să se bazeze pe date individuale despre pui, fiind luate în considerare efectele asupra seriei. Dacă este cazul, seria constituie unitatea de analiză. Analiza statistică a greutății corporale a puilor ar trebui să se bazeze pe date individuale despre pui, fiind luată în considerare dimensiunea seriei. Din cauza dimensiunilor limitate ale studiului, analizele statistice sub formă de teste de „semnificație” au o valoare limitată pentru multe puncte finale, în special punctele finale privind reproducerea. Unele dintre cele mai utilizate metode, în special testele parametrice pentru măsurarea tendinței centrale, nu sunt adecvate. În cazul în care se utilizează analize statistice, atunci metoda aleasă ar trebui să fie adecvată pentru distribuirea variabilei examinate și să fie aleasă înainte de începerea studiului.
|
Evaluarea rezultatelor
|
73.
|
Constatările acestui studiu de toxicitate ar trebui să fie evaluate sub aspectul efectelor observate, al autopsiei și al constatărilor microscopice. Evaluarea va include relația dintre doza de substanță chimică de testat și prezența sau absența, incidența și gravitatea unor anomalii, inclusiv leziuni macroscopice, organe țintă identificate, efecte asupra fertilității, anomalii clinice, efecte asupra performanței de reproducere și a seriei, modificări ale greutății corporale, efecte asupra mortalității și orice alte efecte toxice.
|
|
74.
|
Din cauza perioadei scurte de tratament a masculului, histopatologia testiculelor și a epididimitelor ar trebui luată în considerare împreună cu datele privind fertilitatea la evaluarea efectelor de reproducere la masculi. Utilizarea datelor istorice privind martorii cu privire la reproducere/dezvoltare (de exemplu, pentru dimensiunea seriei, AGD, retenția sfârcurilor, nivelurile T4 din ser), dacă există, ar putea fi utile, de asemenea, pentru interpretarea studiului.
|
|
75.
|
Pentru controlul calității, se propune colectarea datelor anterioare privind martorii și calcularea coeficienților de variație pentru datele numerice, în special pentru parametrii corelați cu detectarea perturbatorilor endocrini. Aceste date pot fi folosite în scop de comparație la evaluarea studiilor curente.
|
Raportul testului
|
76.
|
Raportul testului ar trebui să includă următoarele informații:
|
|
Substanța chimică de testat:
|
—
|
sursa, numărul lotului, data limită de utilizare, dacă sunt disponibile
|
|
—
|
stabilitatea substanței chimice de testat, dacă se cunoaște.
|
|
|
|
Substanță mono-constituent:
|
—
|
aspectul fizic, solubilitatea în apă și alte proprietăți fizico-chimice relevante;
|
|
—
|
identificarea chimică, precum denumirea IUPAC sau CAS, numărul CAS sau codul său InChI, formula structurală, puritatea, identitatea chimică a impurităților după cum este necesar și fezabil din punct de vedere practic etc.
|
|
|
|
Substanță multicomponentă, UVCB-uri și amestecuri:
|
—
|
în măsura în care este posibil, caracterizată prin identitatea chimică (a se vedea mai sus), apariția cantitativă și proprietățile fizico-chimice relevante ale componentelor
|
. |
|
|
Vehicul (dacă este cazul):
|
—
|
justificarea alegerii vehiculului dacă este altul decât apa.
|
|
|
|
Animale de testare:
|
—
|
numărul, vârsta și sexul animalelor;
|
|
—
|
sursa, condițiile de adăpost, regimul alimentar etc.;
|
|
—
|
greutatea fiecărui animal la începutul testului.
|
|
—
|
justificarea, în cazul în care se folosesc alte specii decât șobolanul
|
|
|
|
Condiții de testare:
|
—
|
justificarea selecției nivelului dozei administrate;
|
|
—
|
detalii privind formularea substanței chimice de testat/prepararea hranei, concentrația obținută, stabilitatea și omogenitatea preparatului;
|
|
—
|
detalii privind administrarea substanței chimice de testat;
|
|
—
|
conversia concentrației substanței chimice de testat (ppm) în hrană/apa potabilă în doza reală (mg/kg greutate corporală/zi), dacă este cazul;
|
|
—
|
detalii cu privire la calitatea hranei și a apei;
|
|
—
|
descriere detaliată a procedurii de randomizare pentru selecția puilor pentru sacrificare, dacă aceștia sunt sacrificați.
|
|
|
|
Rezultate:
|
—
|
greutatea corporală/modificările în greutatea corporală;
|
|
—
|
consumul de hrană și apă, dacă este cazul;
|
|
—
|
date privind răspunsul toxic pe sexe și doză, inclusiv fertilitatea, gestația și orice alte semne de
|
|
—
|
durata gestației; efecte toxice sau de altă natură asupra reproducerii, puilor, creșterii postnatale etc.;
|
|
—
|
natura, gravitatea și durata observațiilor clinice (reversibile sau nu);
|
|
—
|
evaluările activității senzoriale, a forței de prindere și a activității motorii;
|
|
—
|
examenele hematologice și valorile de referință relevante;
|
|
—
|
examenele de biochimie clinică și valorile de referință relevante;
|
|
—
|
numărul de femele adulte cu ciclu estral normal sau anormal și durata ciclului;
|
|
—
|
numărul de pui născuți vii și de pierderi după implantare;
|
|
—
|
numărul de pui cu anomalii vizibile macroscopice; evaluarea brută a organelor genitale externe, a numărului de pui subdezvoltați;
|
|
—
|
momentul decesului în timpul studiului sau indicarea faptului că animalele au supraviețuit experimentului;
|
|
—
|
numărul de implantări, dimensiunea seriei și valori ale greutății seriei la momentul înregistrării;
|
|
—
|
date privind greutatea corporală a puilor;
|
|
—
|
AGD a tuturor puilor (și greutatea corporală în ziua măsurării AGD)
|
|
—
|
retenția sfârcurilor la puii masculi;
|
|
—
|
nivelurile hormonilor tiroidieni la pui din ziua 13 și masculi adulți (și femele și pui din ziua 4, dacă se măsoară)
|
|
—
|
greutatea corporală la sacrificare și date privind greutatea organelor în cazul animalelor părinți;
|
|
—
|
o descriere detaliată a constatărilor histopatologice;
|
|
—
|
date privind absorbția (dacă există);
|
|
—
|
tratarea statistică a rezultatelor, dacă este cazul.
|
|
|
Interpretarea rezultatelor
|
77.
|
Studiul va furniza evaluări ale toxicității pentru reproducere/dezvoltare, prin corelare cu administrarea de doze repetate. În mod specific, întrucât accentul este plasat atât pe punctele finale de toxicitate generală, cât și pe cele de toxicitate pentru reproducere/dezvoltare, rezultatele studiului vor permite diferențierea între efectele asupra reproducerii/dezvoltării care se produc în absența toxicității generale și cele care se manifestă doar la niveluri care sunt toxice, de asemenea, pentru animalele părinți (a se vedea punctele 7-11). Acesta ar putea indica dacă este necesar să se efectueze investigații suplimentare și ar putea oferi îndrumări în proiectarea studiilor ulterioare. Ar trebui să se consulte Documentul de orientare nr. 43 al OCDE pentru ajutor în interpretarea rezultatelor privind reproducerea și dezvoltarea (19). Documentul de orientare nr. 106 al OCDE privind evaluarea histologică a testelor endocrine și a testelor de reproducere la rozătoare (16) furnizează informații cu privire la pregătirea și evaluarea organelor (endocrine) și a frotiurilor vaginale care pot fi utile pentru această metodă de testare.
|
REFERINȚE BIBLIOGRAFICE
|
(1)
|
OCDE (1990). Room Document No 1 for the 14th Joint Meeting of the Chemicals Group and Management Committee. Disponibil, la cerere, la Organizația pentru Cooperare și Dezvoltare Economică, Paris
|
|
(2)
|
OCDE (1992). Chairman’s Report of the ad hoc Expert Meeting on Reproductive Toxicity Screening Methods, Tokyo, 27-29 octombrie 1992. Disponibil, la cerere, la Organizația pentru Cooperare și Dezvoltare Economică, Paris
|
|
(3)
|
Mitsumori K., Kodama Y., Uchida O., Takada K., Saito M. Naito K., Tanaka S., Kurokawa Y., Usami, M., Kawashima K., Yasuhara K., Toyoda K., Onodera H., Furukawa F., Takahashi M. și Hayashi Y. (1994). Confirmation Study, Using Nitro-Benzene, of the Combined Repeat Dose and Reproductive/ Developmental Toxicity Test Protocol Proposed by the Organization for Economic Cooperation and Development (OECD). J. Toxicol, Sci., 19, 141-149.
|
|
(4)
|
Tanaka S., Kawashima K., Naito K., Usami M., Nakadate M., Imaida K., Takahashi M., Hayashi Y., Kurokawa Y. și Tobe M. (1992). Combined Repeat Dose and Reproductive/Developmental Toxicity Screening Test (OECD): Familiarization Using Cyclophosphamide. Fundam. Appl. Toxicol., 18, 89-95.
|
|
(5)
|
OCDE (1998). Report of the First Meeting of the OECD Endocrine Disrupter Testing and Assessment (EDTA) Task Force, 10-11 martie 1998, Dispponibil, la cerere, la Organizația pentru Cooperare și Dezvoltare Economică, Paris
|
|
(6)
|
OCDE (2015). Feasibility Study for Minor Enhancements of TG 421/422 with ED Relevant Endpoints. Environmental Health and Safety Publications, Series on Testing and Assessment, (nr. 217), Organizația pentru Cooperare și Dezvoltare Economică, Paris.
|
|
(7)
|
OCDE (2000). Guidance Document on the Recognition, Assessment, and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluations, Environment, Health and Safety Publications, Series on Testing and Assessment, (nr. 19), Organizația pentru Cooperare și Dezvoltare Economică, Paris.
|
|
(8)
|
Goldman J.M., Murr A.S., Buckalew A.R., Ferrell J.M. și Cooper R.L. (2007). The Rodent Estrous Cycle: Characterization of Vaginal Cytology and its Utility in Toxicological Studies, Birth Defects Research, partea B, 80 (2), 84-97.
|
|
(9)
|
Sadleir R.M.F.S. (1979). Cycles and Seasons, în Auston C.R. and Short R.V. (editori), Reproduction in Mammals: I. Germ Cells and Fertilization, Cambridge, New York.
|
|
(10)
|
IPCS (1986). Principles and Methods for the Assessment of Neurotoxicity Associated with Exposure to Chemicals. Environmental Health Criteria Document (nr. 60).
|
|
(11)
|
Moser V.C., McDaniel K.M. și Phillips P.M. (1991). Rat Strain and Stock Comparisons Using a Functional Observational Battery: Baseline Values and Effects of Amitraz. Toxicol. Appl. Pharmacol., 108, 267-283.
|
|
(12)
|
Meyer O.A., Tilson H.A., Byrd W.C. și Riley M.T. (1979). A Method for the Routine Assessment of Fore- and Hindlimb Grip Strength of Rats and Mice. Neurobehav. Toxicol., 1, 233-236.
|
|
(13)
|
Crofton K.M., Howard J.L., Moser V.C., Gill M.W., Reiter L.W., Tilson H.A., MacPhail R.C. (1991). Interlaboratory Comparison of Motor Activity Experiments: Implication for Neurotoxicological Assessments. Neurotoxicol. Teratol. 13, 599-609.
|
|
(14)
|
Gallavan R.H. Jr, J.F. Holson, D.G. Stump, J.F. Knapp și V.L. Reynolds. (1999). „Interpreting the Toxicologic Significance of Alterations in Anogenital Distance: Potential for Confounding Effects of Progeny Body Weights”, Reproductive Toxicology, 13: 383-390.
|
|
(15)
|
OCDE (2013). Guidance Document in Support of the Test Guideline on the Extended One Generation Reproductive Toxicity Study. Environment, Health and Safety Publications, Series on Testing and Assessment (nr. 151). Organizația pentru Cooperare și Dezvoltare Economică, Paris.
|
|
(16)
|
OCDE (2009). Guidance Document for Histologic Evaluation of Endocrine and Reproductive Tests in Rodents. Environmental Health and Safety Publications, Series on Testing and Assessment, (nr. 106) Organizația pentru Cooperare și Dezvoltare Economică, Paris.
|
|
(17)
|
Hess RA și Moore BJ. (1993). Histological Methods for the Evaluation of the Testis. În: Methods in Reproductive Toxicology, Chapin RE și Heindel JJ (editori). Academic Press: San Diego, CA, pp. 52-85.
|
|
(18)
|
Latendresse JR, Warbrittion AR, Jonassen H, Creasy DM. (2002). Fixation of Testes and Eyes Using a Modified Davidson’s Fluid: Comparison with Bouin’s Fluid and Conventional Davidson’s fluid. Toxicol. Pathol. 30, 524-533.
|
|
(19)
|
OCDE (2008). Guidance Document on Mammalian Reproductive Toxicity Testing and Assessment. Environmental Health and Safety Publications, Series on Testing and Assessment, (nr. 43), Organizația pentru Cooperare și Dezvoltare Economică, Paris.
|
|
(20)
|
OCDE (2011), Guidance Document on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption (nr. 150), Organizația pentru Cooperare și Dezvoltare Economică, Paris.
|
Apendicele 1
DEFINIȚII [A SE VEDEA, DE ASEMENEA, ORIENTAREA NR. 150 A OCDE PUNCTUL (20)]
Androgenicitatea este capacitatea unei substanțe chimice de a acționa ca hormon androgen natural (de exemplu, testosteron) în organismul unui mamifer.
Antiandrogenicitatea este capacitatea unei substanțe chimice de a suprima acțiunea unui hormon androgen natural (de exemplu, testosteron) în organismul unui mamifer.
Antiestrogenicitatea este capacitatea unei substanțe chimice de a suprima acțiunea unui hormon estrogen natural (de exemplu, estradiol 17ß) în organismul unui mamifer.
Activitatea antitiroidiană este capacitatea unei substanțe chimice de a suprima acțiunea unui hormon tiroidian natural (de exemplu, estradiol T3) în organismul unui mamifer.
Substanță chimică înseamnă o substanță sau un amestec.
Toxicitate pentru dezvoltare manifestarea toxicității pentru reproducere, reprezentând afecțiuni prenatale, perinatale, postnatale, structurale sau funcționale ale progeniturii.
Doză înseamnă cantitatea de substanță chimică de testat administrată. Doza este exprimată ca greutate a substanței chimice de testat pe unitatea de greutate corporală a animalului de experiență pe zi (de exemplu, mg/kg greutate corporală/zi) sau ca o concentrație constantă în regimul alimentar.
Dozarea este un termen general care cuprinde doza, frecvența și durata administrării dozei.
Toxicitatea evidentă este un termen general care descrie semne clare de toxicitate în urma administrării unei substanțe chimice de testat. Aceste semne ar trebui să fie suficiente pentru a permite o evaluare a pericolelor și ar trebui să fie de așa natură încât o creștere a dozei administrate să poată fi prevăzută a declanșa apariția unor semne de toxicitate severe și probabil moartea.
Afectarea fertilității reprezintă tulburări ale funcțiilor sau capacității de reproducere masculine sau feminine.
Toxicitate maternă efecte adverse asupra femelelor gestante, care apar fie în mod specific (efect direct), fie în mod nespecific (efect indirect) și sunt legate de situația de gestație.
NOAEL este abrevierea pentru nivelul la care nu se observă niciun efect advers. Acesta este cel mai ridicat nivel de dozare la care nu se observă efecte adverse din cauza tratamentului.
Estrogenicitatea este capacitatea unei substanțe chimice de a acționa ca hormon estrogen natural (de exemplu, estradiol 17ß) în organismul unui mamifer.
Toxicitate pentru reproducere reprezintă efecte dăunătoare asupra descendenților și/sau afectarea funcțiilor sau a capacității de reproducere masculine și feminine.
Substanță chimică de testat înseamnă orice substanță sau amestec testat(ă) utilizând această metodă de testare.
Activitatea tiroidiană este capacitatea unei substanțe chimice de a acționa ca hormon tiroidian natural (de exemplu, estradiol T3) în organismul unui mamifer.
Validare înseamnă un proces științific conceput pentru a caracteriza cerințele și limitările operaționale ale unei metode de testare și pentru a demonstra fiabilitatea și relevanța acesteia pentru un anumit scop.
Apendicele 2
DIAGRAMĂ A SCHEMEI DE TRATAMENT CARE INDICĂ DURATA MAXIMĂ A STUDIULUI, PE BAZA UNEI PERIOADE DE ÎMPERECHERE COMPLETE DE 14 ZILE


Apendicele 3
RAPORT DE SINTEZĂ TABELAR CU EFECTELE ASUPRA REPRODUCERII/DEZVOLTĂRII
|
OBSERVAȚII
|
VALORI
|
|
Dozaj (unități) ..........
|
0 (martor)
|
…
|
…
|
…
|
…
|
|
Perechi începute (N)
|
|
|
|
|
|
|
Ciclul estral (cel puțin durata medie și frecvența ciclurilor neregulate)
|
|
|
|
|
|
|
Femele la care apare dovada copulației (N)
|
|
|
|
|
|
|
Femele care au ajuns în gestație (N)
|
|
|
|
|
|
|
Zile de concepție 1-5 (N)
|
|
|
|
|
|
|
Zilele de concepție 6-...([1]
(23)
) (N)
|
|
|
|
|
|
|
Sarcină≤21 de zile (N)
|
|
|
|
|
|
|
Sarcină = 22 de zile (N)
|
|
|
|
|
|
|
Sarcină ≥ 23 de zile (N)
|
|
|
|
|
|
|
Femele cu pui născuți vii (N)
|
|
|
|
|
|
|
Femele cu pui născuți vii în ziua 4 pp (N)
|
|
|
|
|
|
|
Implanturi/femelă (media)
|
|
|
|
|
|
|
Pui vii/femelă la naștere (media)
|
|
|
|
|
|
|
Pui vii/femelă în ziua 4 (media)
|
|
|
|
|
|
|
Raportul dintre sexe (m/f) la naștere (media)
|
|
|
|
|
|
|
Raportul dintre sexe (m/f) în ziua 4 (media)
|
|
|
|
|
|
|
Greutatea seriei la naștere (media)
|
|
|
|
|
|
|
Greutatea seriei în ziua 4 (media)
|
|
|
|
|
|
|
Greutatea puilor la naștere (media)
|
|
|
|
|
|
|
Greutatea puilor în momentul măsurării AGD (media la masculi, media la femele)
|
|
|
|
|
|
|
AGD a puilor în aceeași zi postnatală, la naștere - ziua 4 (media la masculi, media la femele, se notează PND)
|
|
|
|
|
|
|
Greutatea puilor în ziua 4 (media)
|
|
|
|
|
|
|
Greutatea puilor în ziua 13 (media)
|
|
|
|
|
|
|
Retenția sfârcurilor la pui masculi în ziua 13 (media)
|
|
|
|
|
|
|
PUI ANORMALI
|
|
Femele cu 0
|
|
|
|
|
|
|
Femele cu 1
|
|
|
|
|
|
|
Femele cu ≥ 2
|
|
|
|
|
|
|
PIERDEREA PUILOR
|
|
Prenatal (implantații minus născuți vii)
|
|
Femele cu 0
|
|
|
|
|
|
|
Femele cu 1
|
|
|
|
|
|
|
Femele cu 2
|
|
|
|
|
|
|
Femele cu ≥ 3
|
|
|
|
|
|
|
Postnatal (pui născuți vii minus pui vii în ziua postnatală 13)
|
|
Femele cu 0
|
|
|
|
|
|
|
Femele cu 1
|
|
|
|
|
|
|
Femele cu 2
|
|
|
|
|
|
|
Femele cu ≥ 3
|
|
|
|
|
|
B.65 METODA DE TESTARE IN VITRO PRIN BARIERĂ CU MEMBRANĂ PENTRU COROZIUNEA CUTANATĂ
INTRODUCERE
|
1.
|
Această metodă de testare este echivalentă cu Orientarea OCDE privind testarea (TG) nr. 435 (2015). Corodarea pielii înseamnă producerea de leziuni ireversibile ale pielii, care se manifestă ca o necroză vizibilă ce pătrunde în epidermă, ajungând în dermă, în urma aplicării unei substanțe chimice de testat [astfel cum este definită de Sistemul global armonizat de clasificare și etichetare a substanțelor chimice (GHS) al Organizației Națiunilor Unite (ONU) (1) și în Regulamentul (CE) nr. 1272/2008 privind clasificarea, etichetarea și ambalarea substanțelor și a amestecurilor (CLP) (24)]. Această metodă de testare, care este echivalentă cu orientarea nr. 435 a OCDE privind testarea actualizată asigură o metod de testare in vitro prin barieră de membrană care poate fi utilizată pentru identificarea substanțelor chimice corozive. Metoda de testare utilizează o membrană artificială prevăzută a răspunde la substanțe chimice corozive în mod similar pielii de animal in situ.
|
|
2.
|
În mod tradițional, corozivitatea cutanată a fost evaluată prin aplicarea substanței chimice de testat pe pielea animalelor vii și prin evaluarea gradului de lezare a țesuturilor după o perioadă de timp determinată (2). În afară de prezenta metodă de testare, au fost adoptate o serie de alte metode de testare in vitro ca alternative (3) (4) la procedura standard in vivo pe piele de iepure (capitolul B.4 din prezenta anexă, echivalentă cu Orientarea TG nr. 404 a OCDE) utilizată pentru identificarea substanțelor chimice corozive (2). Strategia de testare și evaluare secvențială a GHS al ONU pentru evaluarea și clasificarea corozivității cutanate și Documentul de orientare al OCDE privind metodele integrate de testare și evaluare (IATA) pentru iritație/coroziune cutanată recomandă utilizarea unor metode de testare in vitro validate și acceptate în cadrul modulelor 3 și 4 (1) (5). IATA descrie mai multe module care grupează surse de informații și instrumente de analiză și (i) oferă îndrumări cu privire la modul de integrare și utilizare a datelor existente privind testarea și alte date decât cele privind testarea pentru evaluarea potențialului de iritație cutanată și de coroziune cutanată al substanțelor chimice și (ii) propune o abordare atunci când sunt necesare teste suplimentare, inclusiv atunci când se identifică rezultate negative (5). În această abordare modulară, rezultatele pozitive ale metodelor de testare in vitro pot fi utilizate pentru clasificarea unei substanțe chimice ca fiind corozivă fără necesitatea testării pe animale, astfel fiind redusă și adaptată utilizarea animalelor în acest scop și evitându-se durerea și suferința care ar putea apărea la animale dacă acestea ar fi utilizate în acest scop.
|
|
3.
|
Au fost realizate studii de validare pentru modelul de barieră cu membrană in vitro, care disponibil în comerț sub denumirea de Corrositex® (6) (7) (8), care indică o precizie generală de estimare a corozivității cutanate de 79 % (128/163), o sensibilitate de 85 % (76/89) și o specificitate de 70 % (52/74) pentru o bază de date cu 163 de substanțe și amestecuri (7). Pe baza valabilității sale recunoscute, această metodă de referință validată (MRV) a fost recomandată pentru utilizare în cadrul unei strategii de testare stratificate pentru evaluarea potențialului de pericol de coroziune cutanată al substanțelor chimice (5) (7). Înainte ca un model de barieră cu membrană in vitro pentru coroziunea cutanată să poată fi utilizat în scopuri de reglementare, ar trebui să se stabilească fiabilitatea, relevanța (precizia) și limitările acestuia pentru scopul propus, pentru a asigura faptul că acesta este similar MRV (9), în conformitate cu standardele de performanță prestabilite (SP) (10). Sistemul de acceptare reciprocă a datelor al OCDE va fi garantat numai după revizuirea și includerea în orientarea OCDE echivalentă privind testarea a oricăror metode de testare noi sau actualizate propuse în urma standardelor de performanță. În prezent, doar o singură metodă in vitro este vizată de orientarea nr. 435 a OCDE privind testarea și prezenta metodă de testare, modelul disponibil în comerț Corrositex®.
|
|
4.
|
Alte metode de testare pentru testarea corozivității cutanate se bazează pe utilizarea pielii umane reconstituite (orientarea TG nr. 431 a OCDE) (3) și a pielii izolate de șobolan (orientarea TG nr. 430 a OCDE) (4). Această orientare privind testarea prevede, de asemenea, subclasificarea substanțelor chimice corozive în cele trei subcategorii de corozivitate din GHS al ONU și în cele trei grupuri de ambalaje pentru transport ale ONU pentru pericole de corozivitate. Această orientare OCDE privind testarea a fost adoptată inițial în 2006 și actualizată în 2015 pentru a face trimitere la documentul de orientare IATA și a actualiza lista substanțelor de verificare.
|
DEFINIȚII
|
5.
|
Definițiile utilizate sunt prezentate în apendice.
|
CONSIDERAȚII PRELIMINARE ȘI LIMITĂRI
|
6.
|
Testul descris în prezenta metodă de testare permite identificarea substanțelor chimice corozive și permite subclasificarea substanțelor chimice de testat corozive în conformitate cu GHS al ONU/Regulamentul CLP (tabelul 1). În plus, o astfel de metodă de testare poate fi utilizată pentru a lua decizii privind caracterul coroziv și necoroziv al anumitor clase de substanțe chimice, de exemplu, acizi organici și anorganici, derivați ai acizilor (25) și baze pentru anumite scopuri de testare la transport (7) (11) (12). Prezenta metodă de testare descrie o procedură generică similară cu metoda de testare de referință validată (7). Deși această metodă de testare nu oferă informații adecvate privind iritația cutanată, ar trebui remarcat faptul că MT B.46 (echivalentă cu orientarea TG nr. 439 a OCDE) abordează în mod specific efectul iritației cutanate in vitroasupra sănătății (13). Pentru o evaluare completă a efectelor cutanate locale după o singură expunere a dermei, ar trebui să se consulte Documentul de orientare al OCDE privind metodele integrate de testare și evaluare (5).
Tabelul 1
Categoria și subcategoriile de substanțe corozive pentru piele din GHS al ONU (26)
|
Categoria de substanțe corozive (categoria 1) (pentru autoritățile care nu utilizează subcategorii)
|
Subcategorii de substanțe potențial corozive (26) (pentru autoritățile care utilizează subcategorii, inclusiv Regulamentul CLP)
|
Coroziv la ≥ 1 din 3 animale
|
|
Expunere
|
Observație
|
|
Coroziv
|
Subcategoria de substanțe corozive 1A
|
≤ 3 minute
|
≤ 1 oră
|
|
Subcategoria de substanțe corozive 1B
|
> 3 minute/≤ 1 oră
|
≤ 14 zile
|
|
Subcategoria de substanțe corozive 1C
|
> 1 oră/≤ 4 ore
|
≤ 14 zile
|
|
|
7.
|
O limitare a metodei de referință validate (7) este aceea că este posibil ca multe substanțe chimice necorozive și anumite substanțe chimice corozive să nu îndeplinească cerințele pentru a fi testate, pe baza rezultatelor testului de compatibilitate inițial (a se vedea punctul 13). Substanțele chimice apoase cu un pH cuprins în intervalul 4,5-8,5 nu îndeplinesc, de cele mai multe ori, cerințele pentru testare; însă, 85 % dintre substanțele chimice de testat în acest interval de pH au fost necorozive la testele pe animale (7). Metoda barierei cu membrană in vitro poate fi utilizată pentru testarea substanțelor solide (solubile sau insolubile în apă) și lichide (apoase sau neapoase) și a emulsiilor. Însă, substanțele chimice de testat care nu cauzează o modificare detectabilă în cadrul testului de compatibilitate (și anume, modificarea culorii în sistemul de detectare a substanțelor chimice (CDS) al metodei de testare de referință validate) nu pot fi testate prin metoda barierei cu membrană și ar trebui să fie testate folosind alte metode de testare.
|
PRINCIPIUL TESTULUI
|
8.
|
Sistemul de testare cuprinde două componente: o biobarieră macromoleculară sintetică și un sistem de detectare a substanțelor chimice (CDS); prin această metodă de testare se detectează, prin intermediul barierei de membrană CDS, leziunile provocate de substanțele chimice de testat corozive după aplicarea substanței chimice de testat pe suprafața barierei de membrană macromoleculară sintetică (7), probabil prin același aceleași mecanism(e) de coroziune care funcționează pe piele vie.
|
|
9.
|
Penetrarea (sau perforarea) barierei de membrană ar putea fi măsurată printr-o serie de proceduri sau CDS, inclusiv o schimbare a culorii unui colorant indicator al pH-ului sau a unei alte proprietăți a soluției indicatoare sub barieră.
|
|
10.
|
Bariera de membrană ar trebui să fie determinată ca fiind validă, și anume, relevantă și fiabilă pentru scopul prevăzut. Aceasta include asigurarea faptului că diferite preparate sunt consecvente în ceea ce privește proprietățile de barieră, de exemplu, că acestea sunt capabile să mențină o barieră în calea substanțelor chimice necorozive, să clasifice proprietățile corozive ale substanțelor chimice în diferitele subcategorii de corozivitate din GHS al ONU (1). Clasificarea atribuită se bazează pe timpul în care o substanță chimică pătrunde prin bariera de membrană în soluția indicatoare.
|
DEMONSTRAREA PERFORMANȚEI
|
11.
|
Înainte de utilizarea sistematică a metodei cu barieră de membrană in vitro, cu respectarea acestei metode de testare, laboratoarele ar trebui să demonstreze performanțele tehnice prin clasificarea corectă a celor douăsprezece substanțe de verificare recomandate în tabelul 2. În situațiile în care o substanță inclusă pe listă nu este disponibilă sau în cazul în care acest lucru se poate justifica, se poate utiliza o altă substanță pentru care sunt disponibile date de referință in vivo și in vitro adecvate [de exemplu, din lista de substanțe chimice de referință (10)], cu condiția să se aplice aceleași criterii de selecție, astfel cum sunt descrise în tabelul 1.
Tabelul 2
Substanțe de verificare
(27)
|
Substanță (28)
|
NR CAS
|
Clasă chimică
|
Subcategoria din GHS al ONU in vivo
(29)
|
Subcategoria din GHS al ONU in vitro
(29)
|
|
Trifluorură de bor, dihidrat
|
13319-75-0
|
Acizi anorganici
|
1A
|
1A
|
|
Acid nitric
|
7697-37-2
|
Acizi anorganici
|
1A
|
1A
|
|
Pentaclorură de fosfor
|
10026-13-8
|
Precursori ai acizilor anorganici
|
1A
|
1A
|
|
Clorură de valeril
|
638-29-9
|
Cloruri de acil
|
1B
|
1B
|
|
Hidroxid de sodiu
|
1310-73-2
|
Baze anorganice
|
1B
|
1B
|
|
1-(2-aminoetil)piperazină
|
140-31-8
|
Amine alifatice
|
1B
|
1B
|
|
Clorură de benzensulfonil
|
98-09-9
|
Cloruri de acil
|
1C
|
1C
|
|
N, N-Dimetil-benzilamină
|
103-83-3
|
Aniline
|
1C
|
1C
|
|
Tetraetilenpentamină
|
112-57-2
|
Amine alifatice
|
1C
|
1C
|
|
Eugenol
|
97-53-0
|
Fenoli
|
NC
|
NC
|
|
Acrilat de nonil
|
2664-55-3
|
Acrilați/metacrilați
|
NC
|
NC
|
|
Bicarbonat de sodiu
|
144-55-8
|
Săruri anorganice
|
NC
|
NC
|
|
PROCEDURA
|
12.
|
Punctele următoare descriu componentele și procedurile unei metode de testare cu barieră de membrană artificială pentru evaluarea corozivității (7) (15), pe baza actualei MRV, și anume, Corrositex®, care este disponibilă în comerț. Bariera de membrană și soluțiile de compatibilitate/indicare și clasificare pot fi proiectate, preparate sau obținute din comerț, cum ar fi în cazul MRV Corrositex®. Există un model de protocol al metodei de testare pentru metoda de testare de referință validată (7). Testele ar trebui să fie efectuate la temperatura ambiantă (17-25 °C), iar componentele ar trebui să respecte următoarele condiții.
|
Testul de compatibilitate al substanței chimice de testat
|
13.
|
Înainte de efectuarea testului cu barieră de membrană, se efectuează un test de compatibilitate pentru a determina dacă substanța chimică de testat poate fi detectată de CDS. În cazul în care CDS nu detectează substanța chimică de testat, metoda de testare cu barieră de membrană nu este adecvată pentru evaluarea corozivității potențiale a respectivei substanțe chimice de testat respective și ar trebui să fie utilizată o altă metodă de testare. CDS și condițiile de expunere utilizate pentru testul de compatibilitate ar trebui să reflecte expunerea în cadrul testului ulterior cu barieră de membrană.
|
Testul de clasificare a substanțelor chimice după timp
|
14.
|
Dacă este cazul, pentru metoda de testare, o substanță chimică de testat care a fost calificată în urma testului de compatibilitate ar trebui să fie supusă unui test de clasificare după timp, și anume, un test de depistare care face diferența între acizi sau baze slabe și puternice. De exemplu, în cadrul metodei de testare de referință validate se utilizează un test de clasificare după timp pentru indicarea uneia dintre cele două durate care ar trebui folosită, în funcție de detectarea unei rezerve acide sau alcaline semnificative. Ar trebui să se utilizeze doi timpi diferiți de pătrundere pentru determinarea corozivității și a subcategoriei de corozivitate pentru piele a GHS al ONU, în funcție de rezerva acidă sau alcalină a substanței chimice de testat.
|
COMPONENTELE METODEI DE TESTARE CU BARIERĂ DE MEMBRANĂ
Bariera de membrană
|
15.
|
Bariera de membrană cuprinde două componente: un gel apos macromolecular proteinic și o membrană de susținere permeabilă. Gelul proteinic ar trebui să fie impermeabil la lichide și solide, dar poate să fie corodat și să devină permeabil. Bariera de membrană construită integral ar trebui să fie păstrată în condiții prestabilite care sunt dovedite a împiedica deteriorarea gelului, de exemplu, uscarea, creșterea microbiană, schimbarea, crăparea, care ar degrada performanțele acesteia. Ar trebui să se stabilească perioada de păstrare acceptabilă, iar preparatele barierei de membrană nu nu ar trebui să fie utilizate după această perioadă.
|
|
16.
|
Membrana de susținere permeabilă asigură o susținere mecanică pentru gelul proteinic în timpul procesului de coagulare și de expunere la substanța chimică de testat. Membrana de susținere ar trebui să împiedice acumularea sau deplasarea gelului și să fie ușor permeabilă la toate substanțele chimice de testat.
|
|
17.
|
Gelul proteinic, care este compus din proteine, de exemplu, cheratină, colagen sau amestecuri de proteine, și care formează o matrice de gel, servește drept obiectiv pentru substanța chimică de testat. Materialul proteinic este plasat pe suprafața membranei de susținere și lăsat să se coaguleze înainte de a așeza bariera de membrană peste soluția indicator. Gelul proteinic ar trebui să aibă o grosime și o densitate uniformă, fără bule de aer sau defecte care ar putea afecta integritatea sa funcțională.
|
Sistemul de detectare a substanțelor chimice (CDS)
|
18.
|
Soluția indicator, care este aceeași soluție utilizată pentru testul de compatibilitate, ar trebui să reacționeze la prezența unei substanțe chimice de testat. Ar trebui să se utilizeze un colorant sau o combinație de coloranți indicatori ai pH-ului, de exemplu, roșu crezol și portocaliu de metil, care vor prezenta o modificare a culorii, ca răspuns la prezența substanței chimice de testat. Sistemul de măsurare poate fi vizual sau electronic.
|
|
19.
|
Sistemele de detectare care sunt dezvoltate pentru detectarea trecerii substanței chimice de testat prin bariera de membrană ar trebui să fie evaluate pentru relevanță și fiabilitate în vederea demonstrării gamei de substanțe chimice care pot fi detectate și a limitelor cantitative de detecție.
|
PERFORMANȚA TESTULUI
Asamblarea componentelor metodei de testare
|
20.
|
Bariera de membrană este introdusă într-un flacon (sau o eprubetă) care conține soluția indicator, astfel încât membrana de susținere să fie cu totul în contact cu soluția indicator și să nu se formeze bule de aer. Ar trebui să se acorde atenție pentru a asigura menținerea integrității barierei.
|
Aplicarea substanței chimice de testat
|
21.
|
O cantitate adecvată de substanță chimică de testat, de exemplu, 500 μl dintr-un lichid sau 500 mg de substanță solidă sub formă de pulbere (7), se așează cu atenție pe suprafața superioară a barierei de membrană și se distribuie uniform. Pentru fiecare substanță chimică de testat și martorii aferenți se pregătește un număr adecvat de duplicate, de exemplu patru (7) (a se vedea punctele 23-25). Se înregistrează timpul de aplicare a substanței chimice de testat pe bariera de membrană. Pentru a asigura înregistrarea cu precizie a timpilor scurți de coroziune, timpii de aplicare a substanței chimice de testat în flacoanele duplicate sunt stabiliți separat.
|
Măsurarea gradului de pătrundere în bariera de membrană
|
22.
|
Fiecare flacon este monitorizat în mod corespunzător și se înregistrează timpul primei modificări a soluției indicator, și anume, pătrunderea în barieră, determinându-se timpul scurs între aplicare și pătrunderea în bariera de membrană.
|
Martori
|
23.
|
La testele care implică utilizarea unui vehicul sau a unui solvent cu substanța chimică de testat, vehiculul sau solventul respectiv ar trebui să fie compatibil cu sistemul barierei de membrană, și anume, să nu modifice integritatea acestui sistem, și nu ar trebui să modifice corozivitatea substanței chimice de testat. După caz, martorul tratat cu solvent (sau cu vehicul) ar trebui să fie testat concomitent cu substanța chimică de testat pentru a demonstra compatibilitatea solventului cu sistemul barierei de membrană.
|
|
24.
|
Concomitent cu substanța chimică de testat ar trebui să se testeze un martor pozitiv (coroziv) cu o activitate de corozivitate intermediară, de exemplu, 110 ± 15 mg hidroxid de sodiu (subcategoria de substanțe corozive 1B din GHS al ONU) (7) pentru a evalua dacă sistemul de testare funcționează într-un mod acceptabil. Un al doilea martor pozitiv care este din aceeași clasă chimică ca și substanța chimică de testat poate fi util pentru evaluarea potențialului de corozivitate relativă al unei substanțe chimice de testat corozive. Ar trebui să se aleagă un martor(i) pozitiv(i) cu o corozivitate intermediară (de exemplu, din subcategoria 1B din GHS al ONU) pentru a detecta modificările apărute în timpul de pătrundere care ar putea fi inacceptabil de prelungit sau mai scurt decât valoarea de referință stabilită, ceea ce indică faptul că sistemul de testare nu funcționează în mod corect. În acest scop, substanțele chimice extrem de corozive (subcategoria 1A din GHS al ONU) sau cele necorozive au o utilitate limitată. O substanță chimică corozivă din subcategoria 1B din GHS al Națiunilor Unite ar permite detectarea unui timp de pătrundere prea scurt sau prea mare. O substanță slab corozivă (din subcategoria 1C din GHS al ONU) ar putea fi utilizată ca martor pozitiv pentru măsurarea capacității metodei de testare de a face distincția în mod consecvent între substanțele chimice slab corozive și cele necorozive. Indiferent de metoda utilizată, ar trebui să se dezvolte un interval de răspuns acceptabil al martorilor pozitivi pe baza intervalului istoric al timpilor de pătrundere pentru martorul (martorii) pozitiv(i) utilizați, cum ar fi media ± 2-3 abateri standard. Ar trebui să se determine, în cadrul fiecărui studiu, timpul exact de pătrundere pentru martorul pozitiv, astfel încât să se poată detecta abaterile din afara intervalului acceptabil.
|
|
25.
|
De asemenea, ar trebui să se testeze, concomitent cu substanța chimică de testat, un martor negativ (necoroziv), de exemplu, 10 % acid citric, 6 % acid propionic (7), ca o altă măsură de control al calității pentru a demonstra integritatea funcțională a barierei de membrană.
|
Criterii de acceptare a studiului
|
26.
|
Conform parametrilor de timp stabiliți pentru fiecare subcategorie de corozivitate din GHS al ONU, timpul (în minute) care s-a scurs între aplicarea unei substanțe chimice de testat pe bariera de membrană și pătrunderea în barieră se folosește pentru a prezice corosivitatea substanței chimice de testat. Pentru ca un studiu să fie considerat acceptabil, martorul pozitiv testat în paralel ar trebui să indice timpul de răspuns prevăzut pentru pătrundere (de exemplu, un timp de pătrundere de 8-16 minute pentru hidroxidul de sodiu dacă acesta este utilizat ca martor pozitiv), martorul negativ testat în paralel nu ar trebui să fie coroziv și, dacă este inclus, martorul tratat cu solvent testat în paralel nu ar trebui nici să fie coroziv, nici să modifice potențialul de corozivitate al substanței chimice de testat. Înainte de utilizarea sistematică a unei metode care respectă prezenta metodă de testare, laboratoarele ar trebui să demonstreze performanțele sale tehnice, utilizând cele douăsprezece substanțe recomandate în tabelul 2. Pentru metode noi de tipul „me-too” dezvoltate în cadrul acestei metode de testare, care sunt similare din punct de vedere structural și funcțional cu metoda de referință validată (14), standardele de performanță predefinite ar trebui să fie utilizate pentru a demonstra fiabilitatea și precizia noii metode înainte ca aceasta să fie utilizată pentru testarea de reglementare (10).
|
Interpretarea rezultatelor și clasificarea substanțelor chimice de testat în funcție de corozivitate
|
27.
|
Timpul (în minute) scurs între aplicarea substanței chimice de testat pe bariera de membrană și pătrunderea acesteia în barieră este utilizat pentru a clasifica substanța chimică de testat în subcategoriile de substanțe corozive din GHS al ONU (1) și, dacă este cazul, în grupul de ambalaje al ONU (16). Valoril timpului-limită pentru fiecare dintre cele trei subcategorii de substanțe corozive sunt stabilite pentru fiecare metodă de testare propusă. Deciziile finale privind timpul-limită ar trebui să ia în considerare necesitatea de a reduce la minimum subclasificarea pericolului coroziv (și anume, valorile fals negative). În prezenta orientare privind testarea, ar trebui să fie utilizate valorile timpului-limită ale Corrositex®, astfel cum sunt descrise în tabelul 3, deoarece aceasta constituie singura metodă de testare care se încadrează în prezent în orientarea privind testare (7).
Tabelul 3
Modelul predictiv Corrositex®
|
Timpul mediu de pătrundere (min.)
|
Predicția GHS al ONU (32)
|
|
Substanțele chimice de testat din categoria 1 (30) (determinate prin testul de clasificare al metodei)
|
Substanțele chimice de testat din categoria 2 (31) (determinate prin testul de clasificare al metodei)
|
|
0-3 min.
|
0-3 min.
|
Coroziv subcategoria 1A opțională
|
|
> 3-60 min.
|
> 3-30 min.
|
Coroziv subcategoria 1B opțională
|
|
> 60-240 min.
|
> 30-60 min.
|
Coroziv subcategoria 1C opțională
|
|
> 240 min.
|
> 60 min.
|
Necoroziv
|
|
DATE ȘI RAPORTARE
Date
|
28.
|
Timpul (în minute) scurs între aplicare și pătrunderea în barieră, în cazul substanței chimice de testat și al martorului (martorilor) pozitiv(i), ar trebui să fie consemnat într-un tabel, ca date individuale duplicate, precum și ca medie ± abaterea standard pentru fiecare test.
|
Raport testului
|
29.
|
Raportul testului ar trebui să includă următoarele informații:
|
|
Substanțe chimice de testat și substanțe martor
|
—
|
Substanță mono-constituent: identificarea chimică, precum denumirea IUPAC sau CAS, numărul CAS sau codul său InChI, formula structurală, puritatea, identitatea chimică a impurităților după cum este necesar și fezabil din punct de vedere practic etc.;
|
|
—
|
Substanța multicomponentă, UVCB și amestec: în măsura în care este posibil, caracterizată prin identitatea chimică (a se vedea mai sus), apariția cantitativă și proprietățile fizico-chimice relevante ale componentelor;
|
|
—
|
aspectul fizic, solubilitatea în apă și alte proprietăți fizico-chimice relevante;
|
|
—
|
sursa, numărul lotului, dacă există;
|
|
—
|
tratamentul substanțelor chimice de testat/martor înainte de efectuarea testului, după caz (de exemplu, încălzire, măcinare);
|
|
—
|
stabilitatea substanței chimice de testat, data limită de utilizare sau data pentru reanalizare, dacă se cunoaște;
|
|
—
|
condiții de depozitare.
|
|
|
|
Vehicul:
|
—
|
identificare, concentrație (dacă este cazul), volumul utilizat;
|
|
—
|
justificarea alegerii vehiculului.
|
|
|
|
Modelul barierei de membrană in vitro și protocolul utilizat, inclusiv precizia și fiabilitatea demonstrate
|
|
|
Condiții de testare:
|
—
|
descrierea aparaturii și a procedurilor de preparare utilizate;
|
|
—
|
sursa și compoziția barierei de membrană in vitro utilizate;
|
|
—
|
compoziția și proprietățile soluției indicator;
|
|
—
|
cantitățile de substanțe chimice de testat și de substanțe martor;
|
|
—
|
descrierea și justificarea testului de clasificare după timp;
|
|
—
|
descrierea criteriilor de evaluare și de clasificare aplicate;
|
|
—
|
demonstrarea competenței de desfășurare a metodei de testare înainte de utilizarea sistematică prin testarea substanțelor chimice de verificare.
|
|
|
|
Rezultate:
|
—
|
prezentarea sub formă tabelară a datelor brute individuale obținute din probe individuale de testare și martori pentru fiecare duplicat;
|
|
—
|
descrierea altor efecte observate;
|
|
—
|
clasificarea derivată, raportată la modelul predictiv/criteriile de decizie utilizate.
|
|
Discutarea rezultatelor
Concluzii
|
REFERINȚE BIBLIOGRAFICE
|
(1)
|
Organizația Națiunilor Unite (ONU) (2013). Globally Harmonized System of Classification and Labelling of Chemicals (GHS), a cincea ediție revizuită, ONU New York și Geneva, 2013. Publicație disponibilă la adresa: http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html.
|
|
(2)
|
Capitolul B.4 din prezenta anexă, Iritație/coroziune cutanată acută.
|
|
(3)
|
Capitolul B.40bis din prezenta anexă, Coroziune cutanată in vitro: metoda de testare pe epidermă umană reconstruită (RHE).
|
|
(4)
|
Capitolul B.40 din prezenta anexă, Coroziune cutanată in vitro: Rezistența electrică transcutanată (RET).
|
|
(5)
|
OCDE (2015). Guidance Document on Integrated Approaches to Testing and Assessment of Skin Irritation/Corrosion. Environment, Health and Safety Publications, Series on Testing and Assessment (nr. 203). Organizația pentru Cooperare și Dezvoltare Economică, Paris.
|
|
(6)
|
Fentem, J.H., Archer, G.E.B., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Esdaile, D.J., Holzhutter, H.-G. și Liebsch, M. (1998). The ECVAM International Validation Study on In Vitro Tests for Skin Corrosivity. 2. Results and Evaluation by the Management Team. Toxicology In Vitro 12, 483-524.
|
|
(7)
|
ICCVAM (1999). Corrositex®. An In Vitro Test Method for Assessing Dermal Corrosivity Potential of Chemicals. The Results of an Independent Peer Review Evaluation Coordinated by ICCVAM, NTP and NICEATM. NIEHS, NIH Publication (nr. 99-4495.)
|
|
(8)
|
Gordon V.C., Harvell J.D. și Maibach H.I. (1994). Dermal Corrosion, the Corrositex® System: A DOT Accepted Method to Predict Corrosivity Potential of Test Materials. In vitro Skin Toxicology-Irritation, Phototoxicity, Sensitization. Alternative Methods in Toxicology 10, 37-45.
|
|
(9)
|
OCDE (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environmental, Health and Safety Publications. Series on testing and Assessment (nr. 34).
|
|
(10)
|
OCDE (2014). Performance Standards for the Assessment of Proposed Similar or Modified In Vitro Membrane Barrier Test Method for Skin Corrosion in Relation to TG 435. Organizația pentru Cooperare și Dezvoltare Economică, Paris. Publicație disponibilă la adresa: http://www.oecd.org/chemicalsafety/testing/PerfStand-TG430-June14.pdf.
|
|
(11)
|
ECVAM (2001). Statement on the Application of the CORROSITEX® Assay for Skin Corrosivity Testing. 15th Meeting of ECVAM Scientific Advisory Committee (ESAC), Ispra, Italia. ATLA 29, 96-97.
|
|
(12)
|
U.S. DOT (2002). Exemption DOT-E-10904 (a cincea revizie). (20 septembrie 2002). Washington, D.C., U.S. DOT.
|
|
(13)
|
Capitolul B.46 din prezenta anexă, Iritația cutanată in vitro: Metoda de testare pe epidermă umană reconstruită. ICCVAM (2004). ICCVAM Recommended Performance Standards for In Vitro Test Methods for Skin Corrosion. NIEHS, NIH Publication nr. 04-4510. Publicație disponibilă la adresa: http://www.ntp.niehs.nih.gov/iccvam/docs/dermal_docs/ps/ps044510.pdf.
|
|
(14)
|
U.S. EPA (1996). Method 1120, Dermal Corrosion. Publicație disponibilă la adresa: http://www.epa.gov/osw/hazard/testmethods/sw846/pdfs/1120.pdf.
|
|
(15)
|
Organizația Națiunilor Unite (ONU) (2013). UN Recommendations on the Transport of Dangerous Goods, Model Regulations, a 18-a ediție revizuită (partea, capitolul 2.8), UN, 2013. Publicație disponibilă la adresa: http://www.unece.org/fileadmin/DAM/trans/danger/publi/unrec/rev18/English/Rev18_Volume1_Part2.pdf.
|
Anexă
DEFINIȚII
Acuratețe
: Gradul de apropiere dintre rezultatele metodei de testare și valorile de referință acceptate. Aceasta constituie una dintre caracteristicile de performanță ale metodei de testare și unul dintre aspectele relevanței acesteia. Termenul este adesea utilizat în paralel cu termenul sinonim «concordanță», pentru a desemna proporția de rezultate corecte ale unei metode de testare (9).
Substanță chimică
: O substanță sau un amestec.
Sistemul de detectare a substanțelor chimice (CDS)
: Un sistem de măsurare vizuală sau electronică cu o soluție indicator care răspunde la prezența unei substanțe chimice de testat, de exemplu, printr-o modificare a unui colorant indicator de pH sau a unei combinații de coloranți, care va indica o schimbare de culoare ca răspuns la prezența substanței chimice de testat sau ca urmare a altor tipuri de reacții chimice sau electrochimice.
Concordanță
: Este un indicator al performanței metodei de testare pentru metodele de testare care oferă un rezultat categoric și este unul dintre aspectele relevante ale acesteia. Termenul este uneori utilizat în paralel cu termenul „acuratețe” și este definit ca proporție a tuturor substanțelor chimice testate și clasificate corect ca fiind pozitive sau negative. Concordanța depinde foarte mult de prevalența pozitivelor în tipurile de substanțe chimice de testat supuse examinării (9).
GHS (Sistemul global armonizat de clasificare și etichetare a substanțelor chimice):: Un sistem care propune clasificarea substanțelor chimice (substanțe și amestecuri) în funcție de tipuri și niveluri standardizate de pericole fizice, pericole pentru sănătate și pericole pentru mediu și care elaborează elemente de comunicare corespunzătoare, precum pictograme, cuvinte de avertizare, declarații cu privire la pericole, declarații de securitate și fișe tehnice de securitate pentru a transmite informații privind efectele adverse ale acestora în scopul protejării populației (inclusiv angajatorii, lucrătorii, transportatorii, consumatorii și cei care se ocupă de serviciile de urgență) și a mediului (1).
IATA
: Strategie integrată pentru testare și evaluare.
Amestec
: Un amestec sau o soluție compus/ă din două sau mai multe substanțe.
Substanță monocomponentă
: O substanță definită prin compoziția sa cantitativă, în care un singur component principal este prezent în proporție de cel puțin 80 % (g/g).
Substanță multicomponentă
: O substanță definită prin compoziția sa cantitativă, în care mai mult de un singur component principal este prezent într-o concentrație ≥ 10 % (g/g) și < de 80 % (g/g). O substanță multicomponentă este rezultatul unui proces de fabricație. Diferența dintre un amestec și o substanță multicomponentă este faptul că un amestec este obținut prin combinarea a două sau mai multe substanțe chimice fără o reacție chimică. O substanță multicomponentă este rezultatul unei reacții chimice.
NC
: Necoroziv.
Standarde de performanță
: Standarde bazate pe o metodă de referință validată, care permit evaluarea comparabilității unei metode de testare propuse similare din punct de vedere al mecanismului și din punct de vedere funcțional. Sunt incluse (i) componentele esențiale ale metodei de testare; (ii) o listă minimală de substanțe chimice de referință selectate dintre substanțele chimice utilizate pentru a demonstra performanțele acceptabile ale metodei de referință validate; și (iii) niveluri similare de fiabilitate și acuratețe, bazate pe datele obținute pentru metoda de testare validată, pe care metoda de testare propusă ar trebui să le demonstreze în momentul evaluării acesteia utilizându-se lista minimă de substanțe chimice de referință (9).
Relevanță
: Descrierea relației dintre metoda de testare și efectul de interes și dacă aceasta este relevantă și utilă pentru un anumit scop. Aceasta reprezintă măsura în care metoda de testare măsoară sau prezice în mod corect efectul biologic de interes. Relevanța include luarea în considerare a acurateței (conformității) unei metode de testare (9).
Fiabilitate
: Măsuri în care o metodă de testare poate fi reprodusă în mod reproductibil, în același laborator sau în laboratoare diferite în timp, atunci când sunt efectuate utilizând același protocol. Aceasta se evaluează prin calcularea reproductibilității intra-laborator sau inter-laborator (9).
Sensibilitate
: Proporția tuturor substanțelor chimice pozitive/active clasificate în mod corect prin metoda de testare. Acesta permite măsurarea acurateței unei metode de testare care conduce la rezultate clasificabile în categorii și constituie un aspect important în evaluarea relevanței unei metode de testare (9).
Coroziunea cutanată in vivo
: Producerea unor leziuni cutanate ireversibile; și anume, necroză vizibilă prin epidermă și în dermă, ca urmare a aplicării unei substanțe chimice de testat timp de până la patru ore. Reacțiile cutanate se manifestă, în general, prin ulcere, sângerare, cruste sângerânde, și, spre sfârșitul perioadei de observație de 14 zile, o decolorare datorată albirii pielii, zone de alopecie completă și cicatrice. Pentru evaluarea leziunilor incerte ar trebui să se aibă în vedere efectuarea unui examen histopatologic.
Specificitate
: Proporția tuturor substanțelor chimice negative/inactive clasificate în mod corect prin metoda de testare. Acesta permite măsurarea acurateței unei metode de testare care conduce la rezultate clasificabile în categorii și constituie un aspect important în evaluarea relevanței unei metode de testare (9).
Substanță
: un element chimic și compușii săi în stare naturală sau obținuți prin orice proces de producție, inclusiv orice aditiv necesar pentru a-i păstra stabilitatea și orice impuritate care derivă din procesul utilizat, însă excluzând orice solvent care poate fi separat fără a afecta stabilitatea substanței sau fără a-i schimba compoziția.
Substanța chimică de testat
: Orice substanță sau amestec testat utilizând prezenta metodă de testare.
UVCB
: Substanțe cu compoziție necunoscută sau variabilă, produși de reacție complexă sau materiale biologice.
B.66 TESTE IN VITRO DE TRANSACTIVARE CU TRANSFECTARE STABILĂ PENTRU DETECTAREA AGONIȘTILOR ȘI ANTAGONIȘTILOR RECEPTORILOR DE ESTROGEN
INTRODUCERE GENERALĂ
Orientarea OCDE privind testarea pe baza performanței
|
1.
|
Această metodă de testare este echivalentă cu Orientarea OCDE privind testarea (TG) nr. 455 (2016). TG nr. 455 este o orientare privind testarea pe baza performanței (PBTG), care descrie metodologia testelor in vitro de transactivare cu transfectare stabilă pentru detectarea agoniștilor și a antagoniștilor receptorilor de estrogen (testele ER TA). Aceasta cuprinde mai multe metode de testare similare din punct de vedere mecanicist și funcțional pentru identificarea agoniștilor și antagoniștilor receptorilor de estrogen (și anume, ERα și/sau ERβ) și ar trebui să faciliteze dezvoltarea unor metode de testare similare noi sau modificate în conformitate cu principiile de validare prevăzute în documentul de orientare al OCDE privind validarea și acceptarea internațională a metode de testare noi și actualizate pentru evaluarea riscurilor (1). Metodele de testare de referință validate integral (apendicele 2 și apendicele 3), care servesc drept bază pentru această PBTG, sunt:
|
—
|
Testul AT cu transfectare stabilă (STTA) (2) care utilizează linia celulară (h) ERα-HeLa-9903; și
|
|
—
|
Testul VM7Luc ER AT (3), care utilizează linia celulară VM7Luc4E2 (33), care exprimă predominant hERα cu o oarecare contribuție din partea hERβ(4) (5).
|
Pentru dezvoltarea și validarea unor teste similare pentru același punct final de pericol există standarde de performanță (SP) (6) (7) care ar trebui să fie utilizate. Acestea permit modificarea prpomptă a PBTG nr. 455, astfel încât să se poată adăuga teste similare noi la o PBTG actualizată; însă testele similare vor fi adăugate doar după revizuirea și confirmarea de către OCDE a respectării standardelor de performanță. Testele incluse în TG nr. 455 pot fi utilizate fără nicio deosebire pentru a răspunde cerințelor statelor membre ale OCDE pentru rezultatele testelor privind transactivarea receptorilor de estrogen, beneficiind în același timp de sistemul de acceptare reciprocă a datelor al OCDE.
|
Context și principii ale testelor incluse prezenta metodă de testare
|
2.
|
În 1998, OCDE a inițiat o activitate cu un nivel ridicat de prioritate pentru a revizui orientările existente privind testarea și a elabora noi orientări pentru depistarea și testarea substanțelor chimice cu potențial de perturbare a sistemului endocrin. Cadrul conceptual al OCDE (CC) pentru testarea și evaluarea substanțelor chimice cu potențial de perturbare a sistemului endocrin a fost revizuit în 2012. Cadrele conceptuale inițiale și revizuite sunt incluse ca anexe la documentul de orientare al OCDE privind orientările de testare standardizate pentru evaluarea substanțelor chimice în vederea identificării perturbatorilor endocrini (8). CC cuprinde cinci niveluri, fiecare nivel corespunzând unui nivel diferit de complexitate biologică. Testele de transactivare ER (AT) descrise în această metodă de testare sunt de nivelul 2, care include teste in vitro care furnizează date cu privire la (un) anumit(e) mecanism(e)/o cale endocrină/anumite căi endocrine. Această metodă de testare este pentru teste in vitro de transactivare (AT) concepute pentru a identifica agoniștii și antagoniștii receptorilor de estrogen (ER).
|
|
3.
|
Interacțiunea estrogenilor cu ER poate afecta transcrierea genelor controlate de estrogen, ceea ce poate conduce la inducerea sau inhibarea proceselor celulare, inclusiv a celor necesare pentru proliferare celulară, dezvoltare normală a fătului și funcția de reproducere (9) (10) (11). Perturbarea sistemelor estrogenice normale poate avea potențialul de a declanșa efecte adverse asupra dezvoltării normale (ontogeneză), a sănătății reproductive și a integrității sistemului de reproducere.
|
|
4.
|
Testele TA in vitro se bazează pe o interacțiune directă sau indirectă a substanțelor cu un receptor specific care reglează transcrierea unui produs genetic reporter. Astfel de teste au fost utilizate pe scară largă pentru a evalua expresia genică reglementată de anumiți receptori nucleari, cum ar fi ER (12) (13) (14) (15) (16). Acestea au fost propuse pentru detectarea transactivării estrogenice reglementate de ER (17) (18) (19). Există cel puțin două subtipuri majore de ER nucleari, α și β, care sunt codificați prin gene distincte. Proteinele respective au diferite funcții biologice, precum și diferite distribuții de țesuturi și afinități de legare a liganzilor (20) (21) (22) (23) (24) (25) (26). ERα nuclear mediază răspunsul estrogenic clasic (27) (28) (29) (30) și, prin urmare, cele mai multe modele în curs de elaborare pentru a măsura activarea sau inhibarea ER sunt specifice ERα. Testele sunt utilizate pentru a identifica substanțele chimice care activează (sau inhibă) ER în urma legării lianzilor, după care complexul receptor-ligand se leagă de anumite elemente de răspuns ADN specifice și transactivează o genă reporter, ceea ce determină creșterea expresiei celulare a unei proteine marker. În aceste teste pot fi utilizate diferite răspunsuri ale reporterilor. În sistemele bazate pe luciferază, enzima luciferază transformă substratul luciferină într-un produs bioluminiscent care poate fi măsurat cantitativ cu un luminometru. Alte exemple de reporteri comuni sunt proteina fluorescentă și gena LacZ, care codifică β-galactozidaza, o enzimă care poate transforma substratul incolor X-gal (5-bromo-4-cloro-indol-galactopiranozidă) într-un produs de culoare albastră care poate fi cuantificat cu un spectrofotometru. Acești reporteri pot fi evaluați rapid și cu costuri reduse cu ajutorul unor truse de testare disponibile în comerț.
|
|
5.
|
Studiile de validare a testelor STTA și VM7Luc TA și-au demonstrat relevanța și fiabilitatea acestora pentru scopul prevăzut (3) (4) (5) (30). Standardele de performanță pentru testele ER TA bazate pe luminiscență, care folosesc linii celulare din sân, sunt incluse în raportul de evaluare a metodei de testare ICCVAM privind metoda de testare LUMI-CELL® ER (VM7Luc ER TA): Un test in vitro pentru identificarea activității substanțelor chimice ca agoniști sau antagoniști ai receptorilor de estrogen (An In Vitro Assay for Identifying Human Estrogen Receptor Agonist and Antagonist Activity of Chemicals (3). Aceste standarde de performanță au fost modificate pentru a fi aplicabile în cazul testelor STTA și VM7Luc TA (2).
|
|
6.
|
Definițiile și abrevierile utilizate în prezenta metodă de testare sunt descrise în apendicele 1.
|
Domeniul de aplicare și limitările aferente testelor TA
|
7.
|
Aceste teste sunt propuse pentru depistare și stabilirea priorităților, dar pot furniza, de asemenea, informații mecaniciste care pot fi utilizate în cadrul unei abordări bazate pe dovezi. Acestea abordează TA indusă de legarea substanței chimice de ER într-un sistem in vitro. Astfel, rezultatele nu ar trebui să fie extrapolate în mod direct cu semnalizarea și reglarea complexă a sistemului endocrin in vivo.
|
|
8.
|
TA mediată de ER este considerată a fi unul dintre principalele mecanisme de perturbare a sistemului endocrin (ED), deși există și alte mecanisme prin care se poate produce ED, inclusiv (i) interacțiunile cu alți receptori și sisteme enzimatice din sistemul endocrin, (ii) sinteza hormonilor, (iii) activarea metabolică și/sau inactivarea hormonilor, (iv) distribuirea hormonilor la țesuturile țintă și (v) eliminarea hormonilor din corp. Niciunul dintre testele efectuate în cadrul acestei metode de testare nu abordează aceste moduri de acțiune.
|
|
9.
|
Această metodă de testare abordează capacitatea substanțelor chimice de a activa (și anume, de a acționa ca agoniștii) și, de asemenea, de a suprima (și anume, de a acționa ca antagoniști) transcrierea dependentă de ER. Unele substanțe chimice pot, într-o manieră dependentă de tipurile de celule, să afișeze atât activitatea agonistă, cât și activitatea antagonistă și sunt cunoscute sub denumirea de modulatori de receptori de estrogen selectivi (SERM). Substanțele chimice negative din aceste teste ar putea fi evaluate în cadrul unui test de legare de ER înainte de a concluziona că substanța chimică nu se leagă de receptor. În plus, este posibil ca testele să ofere informații numai despre activitatea moleculei mamă, ținând seama de capacitatea de metabolizare limitată a sistemelor de celule in vitro. Având în vedere că, în timpul validării, au fost utilizate numai substanțe unice, aplicabilitatea pentru amestecuri de testare nu a fost abordată. Metoda de testare este totuși aplicabilă din punct de vedere teoretic pentru testarea substanțelor multicomponente, a UVCB și a amestecurilor. Înainte de utilizarea metodei de testare pe o substanță multicomponentă, UVCB sau un amestec pentru a genera date pentru un obiectiv de reglementare prevăzut, se ia în considerare dacă și, în caz afirmativ, din ce cauză aceasta poate oferi rezultate adecvate pentru respectivul scop. Astfel de considerații nu sunt necesare atunci când există o cerință normativă pentru testarea amestecului.
|
|
10.
|
În scop informativ, tabelul 1 prezintă rezultatele testului de agoniști pentru cele 34 de substanțe care au fost testate în cadrul ambelor metode de testare de referință validate complet descrise în prezenta metodă de testare. Dintre aceste substanțe, 26 sunt clasificate ca fiind agoniști ER definitivi și 8 negative pe baza rapoartelor publicate, inclusiv a testelor in vitro pentru legarea de ER și TA, precum și/sau a testului uterotrophic (2) (3) (18) (31) (32) (33) (34). Tabelul 2 prezintă rezultatele testului de antagoniști pentru cele 15 substanțe care au fost testate în cadrul ambelor metode de testare de referință validate complet descrise în prezenta metodă de testare. Dintre aceste substanțe, 4 sunt clasificate ca fiind antagoniști ER definitivi/presupuși și 10 negative pe baza rapoartelor publicate, inclusiv a testelor in vitro pentru legarea de ER și TA (2) (3) (18) (31). În ceea ce privește datele sintetizate în tabelele 1 și 2, s-a ajuns la un acord de 100 % între cele două metode de testare de referință privind clasificările tuturor substanțelor, cu excepția unei substanțe (Mifepristonă) pentru testul antagonist și fiecare substanță a fost clasificată în mod corect ca agonist/antagonist ER sau negativă. Informații suplimentare privind acest grup de substanțe chimice, precum și alte substanțe chimice testate în cadrul testelor STTA și VM7Luc ER TA în timpul studiilor de validare sunt prezentate în standardele de performanță pentru ERTA (6) (7), apendicele 2 (tabelele 1, 2 și 3).
|
Tabelul 1
Prezentarea rezultatelor testelor STTA și VM7Luc ER TA pentru substanțele testate în cadrul ambelor teste privind agoniștii și clasificate ca reprezentând agoniști ER (POS) sau negative (NEG)
|
|
Substanță
|
NR CAS
|
Testul STTA (35)
|
Testul VM7Luc ER TA (36)
|
Sursa datelor pentru clasificare (38)
|
|
Activitatea ER TA
|
Valoare PC10 (M)
|
Valoare PC50
(2) (M)
|
Activitatea ER TA
|
Valoarea EC50
(2), (37) (M)
|
Alte ER TA (34)
|
Legare de ER
|
Uterotrofică
|
|
1
|
17ß-estradiol (1)
|
50-28-2
|
POS
|
< 1,00 × 10-11
|
< 1,00 × 10-11
|
POS
|
5,63 × 10-12
|
POS (227/227)
|
POS
|
POS
|
|
2
|
17α-estradiol (1)
|
57-91-0
|
POS
|
7,24 × 10-11
|
6,44 × 10-10
|
POS
|
1,40 × 10-9
|
POS(11/11)
|
POS
|
POS
|
|
3
|
17α-etinil estradiol (1)
|
57-63-6
|
POS
|
< 1,00 × 10-11
|
< 1,00 × 10-11
|
POS
|
7,31 × 10-12
|
POS(22/22)
|
POS
|
POS
|
|
4
|
17β-trenbolon
|
10161-33-8
|
POS
|
1,78 × 10-8
|
2,73 × 10-7
|
POS
|
4,20 × 10-8
|
POS (2/2)
|
NT
|
NT
|
|
5
|
19-nortestosteron (1)
|
434-22-0
|
POS
|
9,64 × 10-9
|
2,71 × 10-7
|
POS
|
1,80 × 10-6
|
POS(4/4)
|
POS
|
POS
|
|
6
|
4-cumilfenol (1)
|
599-64-4
|
POS
|
1,49 × 10-7
|
1,60 × 10-6
|
POS
|
3,20 × 10-7
|
POS(5/5)
|
POS
|
NT
|
|
7
|
4-terț-octilfenol (1)
|
140-66-9
|
POS
|
1,85 × 10-9
|
7,37 × 10-8
|
POS
|
3,19 × 10-8
|
POS(21/24)
|
POS
|
POS
|
|
8
|
Apigenină (1)
|
520-36-5
|
POS
|
1,31 × 10-7
|
5,71 × 10-7
|
POS
|
1,60 × 10-6
|
POS(26/26)
|
POS
|
NT
|
|
9
|
Atrazină (1)
|
1912-24-9
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (30/30)
|
NEG
|
NT
|
|
10
|
Bisfenol A (1)
|
80-05-7
|
POS
|
2,02 × 10-8
|
2,94 × 10-7
|
POS
|
5,33 × 10-7
|
POS (65/65)
|
POS
|
POS
|
|
11
|
Bisfenol B (1)
|
77-40-7
|
POS
|
2,36 × 10-8
|
2,11 × 10-7
|
POS
|
1,95 × 10-7
|
POS(6/6)
|
POS
|
POS
|
|
12
|
Ftalat de butil benzil (1)
|
85-68-7
|
POS
|
1,14 × 10-6
|
4,11 × 10-6
|
POS
|
1,98 × 10-6
|
POS(12/14)
|
POS
|
NEG
|
|
13
|
Corticosteron (1)
|
50-22-6
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG(6/6)
|
NEG
|
NT
|
|
14
|
Cumestrol (1)
|
479-13-0
|
POS
|
1,23 × 10-9
|
2,00 × 10-8
|
POS
|
1,32 × 10-7
|
POS(30/30)
|
POS
|
NT
|
|
15
|
Daidzeină (1)
|
486-66-8
|
POS
|
1,76 × 10-8
|
1,51 × 10-7
|
POS
|
7,95 × 10-7
|
POS(39/39)
|
POS
|
POS
|
|
16
|
Dietilstilbestrol (1)
|
56-53-1
|
POS
|
< 1,00 × 10-11
|
2,04 × 10-11
|
POS
|
3,34 × 10-11
|
POS(42/42)
|
POS
|
NT
|
|
17
|
Ftalat de di-n-butil
|
84-74-2
|
POS
|
4,09 × 10-6
|
|
POS
|
4,09 × 10-6
|
POS(6/11)
|
POS
|
NEG
|
|
18
|
Etilparaben
|
120-47-8
|
POS
|
5,00 × 10-6
|
(no PC50)
|
POS
|
2,48 × 10-5
|
POS
|
|
NT
|
|
19
|
Estronă (1)
|
53-16-7
|
POS
|
3,02 × 10-11
|
5,88 × 10-10
|
POS
|
2,34 × 10-10
|
POS(26/28)
|
POS
|
POS
|
|
20
|
Genisteină (1)
|
446-72-0
|
POS
|
2,24 × 10-9
|
2,45 × 10-8
|
POS
|
2,71 × 10-7
|
POS(100/102)
|
POS
|
POS
|
|
21
|
Haloperidol
|
52-86-8
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (2/2)
|
NEG
|
NT
|
|
22
|
Kemferol (1)
|
520-18-3
|
POS
|
1,36 × 10-7
|
1,21 × 10-6
|
POS
|
3,99 × 10-6
|
POS(23/23)
|
POS
|
NT
|
|
23
|
Keponă (1)
|
143-50-0
|
POS
|
7,11 × 10-7
|
7,68 × 10-6
|
POS
|
4,91 × 10-7
|
POS(14/18)
|
POS
|
NT
|
|
24
|
Ketoconazol
|
65277-42-1
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (2/2)
|
NEG
|
NT
|
|
25
|
Linuron (1)
|
330-55-2
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (8/8)
|
NEG
|
NT
|
|
26
|
meso-Hexestrol (1)
|
84-16-2
|
POS
|
< 1,00 × 10-11
|
2,75 × 10-11
|
POS
|
1,65 × 10-11
|
POS(4/4)
|
POS
|
NT
|
|
27
|
Metiltestosteron (1)
|
58-18-4
|
POS
|
1,73 × 10-7
|
4,11 × 10-6
|
POS
|
2,68 × 10-6
|
POS(5/6)
|
POS
|
NT
|
|
28
|
Morin
|
480-16-0
|
POS
|
5,43 × 10-7
|
4,16 × 10-6
|
POS
|
2,37 × 10-6
|
POS(2/2)
|
POS
|
NT
|
|
29
|
Noretinodrel (1)
|
68-23-5
|
POS
|
1,11 × 10-11
|
1,50 × 10-9
|
POS
|
9,39 × 10-10
|
POS(5/5)
|
POS
|
NT
|
|
30
|
p,p’-metoxiclor (1)
|
72-43-5
|
POS
|
1,23 × 10-6
|
(fără PC50) (2)
|
POS
|
1,92 × 10-6
|
POS(24/27)
|
POS
|
POS
|
|
31
|
Fenobarbital (1)
|
57-30-7
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG(2/2)
|
NEG
|
NT
|
|
32
|
Reserpină
|
50-55-5
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG(4/4)
|
NEG
|
NT
|
|
33
|
Spironolactonă (1)
|
52-01-7
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG(4/4)
|
NEG
|
NT
|
|
34
|
Testosteron
|
58-22-0
|
POS
|
2,82 × 10-8
|
9,78 × 10-6
|
POS
|
1,75 × 10-5
|
POS(5/10)
|
POS
|
NT
|
|
Abrevieri: Nr. CAS = Număr de înregistrare al Chemical Abstracts Service; M = molar; EC50 = jumătate din concentrația maximă efectivă a substanței de testat; NEG = negativ; POS = pozitiv; NT = Netestat; PC10 (și PC50) = concentrația unei substanțe de testat la care răspunsul este de 10 % (sau 50 % pentru PC50) din răspunsul indus de martorul pozitiv (E2, 1nM) pe fiecare placă.
|
Tabelul 2
Compararea rezultatelor testelor STTA și VM7Luc ER TA pentru substanțele testate în cadrul ambelor teste privind antagoniștii și clasificate ca reprezentând antagoniști ER (POS) sau negative (NEG)
|
|
Substanță (3)
|
NR CAS
|
Testul ER STTA (40)
|
Testul VM7Luc ER TA (41)
|
ER STTA efecte candidate (43)
|
ICCVAM (44) Clasificare prin consens
|
MeSH (45) Clasă chimică
|
Clasă de produse (46)
|
|
Activitatea ER TA
|
Valoarea IC50
(39) (M)
|
Activitatea ER TA
|
Valoarea IC50
(39), (42) (M)
|
|
1
|
4-hidroxi-tamoxifen
|
68047-06-3
|
POS
|
3,97 × 10-9
|
POS
|
2,08 × 10-7
|
valoare POS moderată
|
POS
|
Hidrocarbură (ciclică)
|
Produs farmaceutic
|
|
2
|
Dibenzo[a.h]antracen
|
53-70-3
|
POS
|
Fără IC50
|
POS
|
Fără IC50
|
POS
|
PP
|
Compus policiclic
|
Substanță chimică de laborator, produs natural
|
|
3
|
Mifepristonă
|
84371-65-3
|
POS
|
5,61 × 10-6
|
NEG
|
—
|
ușor POS
|
NEG
|
Steroid
|
Produs farmaceutic
|
|
4
|
Raloxifen HCl
|
82640-04-8
|
POS
|
7,86 × 10-10
|
POS
|
1,19 × 10-9
|
valoare POS moderată
|
POS
|
Hidrocarbură (ciclică)
|
Produs farmaceutic
|
|
5
|
Tamoxifen
|
10540-29-1
|
POS
|
4,91 × 10-7
|
POS
|
8,17 × 10-7
|
POS
|
POS
|
Hidrocarbură (ciclică)
|
Produs farmaceutic
|
|
6
|
17β-estradiol
|
50-28-2
|
NEG
|
—
|
NEG
|
—
|
PN
|
PN
|
Steroid
|
Produs farmaceutic, agent veterinar
|
|
7
|
Apigenină
|
520-36-5
|
NEG
|
—
|
NEG
|
—
|
NEG
|
NEG
|
Compus heterociclic
|
Colorant, produs natural, produs farmaceutic intermediar
|
|
8
|
Atrazină
|
1912-24-9
|
NEG
|
—
|
NEG
|
—
|
NEG
|
PN
|
Compus heterociclic
|
Erbicid
|
|
9
|
Ftalat de di-n-butil
|
84-74-2
|
NEG
|
—
|
NEG
|
—
|
NEG
|
NEG
|
Ester, acid ftalic
|
Ingredient cosmetic, substanță chimică industrială, plastifiant
|
|
10
|
Fenarimol
|
60168-88-9
|
NEG
|
—
|
NEG
|
—
|
netestat
|
PN
|
Compus heterociclic, pirimidină
|
Fungicid
|
|
11
|
Flavonă
|
525-82-6
|
NEG
|
—
|
NEG
|
—
|
PN
|
PN
|
Flavonoid, compus heterociclic
|
Produs natural, farmaceutic
|
|
12
|
Flutamidă
|
13311-84-7
|
NEG
|
—
|
NEG
|
—
|
NEG
|
PN
|
Amidă
|
Produs farmaceutic, agent veterinar
|
|
13
|
Genisteină
|
446-72-0
|
NEG
|
—
|
NEG
|
—
|
PN
|
NEG
|
Flavonoid, compus heterociclic
|
Produs natural, farmaceutic
|
|
14
|
p-n-nonilfenol
|
104-40-5
|
NEG
|
—
|
NEG
|
—
|
netestat
|
NEG
|
Fenol
|
Substanță chimică intermediară
|
|
15
|
Resveratrol
|
501-36-0
|
NEG
|
—
|
NEG
|
—
|
PN
|
NEG
|
Hidrocarbură (ciclică)
|
Produs natural
|
|
Abrevieri: Nr. CAS = Număr de înregistrare al Chemical Abstracts Service; M = molar; IC50 = jumătate din concentrația maximă de inhibare a substanței de testat; NEG = negativ; PN = presupus negativă; POS = pozitiv; PP = presupus pozitivă.
|
COMPONENTELE TESTULUI ER TA
Componentele esențiale ale testului
|
11.
|
Prezenta metodă de testare se aplică pentru testele care utilizează un receptor ERα transfectat stabil sau endogen și o structură de gene reporter transfectate stabil sub controlul unuia sau mai multor elemente de răspuns estrogen; însă, pot fi prezenți și alți receptori precum ERβ. Acestea sunt componente esențiale ale tstului.
|
Martori
|
12.
|
Ar trebui să se descrie baza pentru standardele de referință propuse în paralel pentru fiecare test agonist și antagonist. Martorii testați în paralel (negativi, tratați cu solvent și pozitivi), după caz, servesc drept indicatori pentru operativitatea testului în condițiile de testare și furnizează o bază pentru comparații între experimente; acestea fac parte, de obicei, din criteriile de acceptabilitate pentru un anumit experiment) (1).
|
Proceduri standard de control al calității
|
13.
|
Ar trebui să se desfășoare proceduri standard de control al calității, astfel cum este descris pentru fiecare test, pentru a se asigura faptul că linia celulară rămâne stabilă în urma mai multor treceri, că rămâne fără micoplasme (și anume, fără contaminare bacteriană) și că își menține capacitatea de a furniza în timp răspunsurile mediate de ER preconizate. Ar trebui să se verifice în continuare identitatea corectă a liniilor celulare, precum și a altor contaminanți (de exemplu, fungi, drojdie și virusuri).
|
Demonstrarea competenței laboratorului
|
14.
|
Înainte de a testa substanțe chimice necunoscute prin oricare dintre testele realizate în cadrul acestei metode de testare, fiecare laborator ar trebui să își demonstreze capacitatea de a utiliza testul. Pentru a-și demonstra competența, fiecare laborator ar trebui să testeze cele 14 substanțe de verificare enumerate în tabelul 3 pentru testul agoniștilor și 10 substanțe de verificare din tabelul 4 pentru testul antagoniștilor. Această testare a competenței va confirma, de asemenea, capacitatea de răspuns a sistemului de testare. Lista substanțelor de verificare constituie un subset al substanțelor de referință prevăzute în standardele de performanță pentru testele ER TA (6). Aceste substanțe sunt disponibile în comerț, reprezintă clasele de substanțe chimice asociate în mod obișnuit cu activitatea agonistă sau antagonistă ER, prezintă un interval adecvat de putere preconizată pentru agoniștii/antagoniștii ER (și anume, puternice sau slabe) și includ substanțe negative. Testarea substanțelor de verificare ar trebui să fie duplicate cel puțin de două ori, în zile diferite. Performanța este demonstrată prin clasificarea corectă (pozitiv/negativ) a fiecărei substanțe de verificare. Testarea competenței ar trebui să fie repetată de către fiecare tehnician atunci când învață testele. În funcție de tipul de celule, unele dintre aceste substanțe de verificare ar putea să se comporte ca SERM și să manifeste o activitate atât ca agoniști, cât și ca antagoniști. Însă, substanțele de verificare sunt clasificate în tabelele 3 și 4 după activitatea lor predominantă cunoscută care ar trebui utilizată pentru evaluarea competenței.
|
|
15.
|
Pentru a demonstra performanța și pentru controlul calității, fiecare laborator ar trebui să compileze baze de date istorice despre agoniști și antagoniști cu standard de referință (de exemplu, 17β-estradiol și tamoxifen) și date despre substanțe chimice martor pozitive și negative și martori tratați cu solvent (de exemplu, DMSO). Pentru început, baza de date ar trebui să fie generată în urma a cel puțin 10 teste independente privind agoniștii (de exemplu, 17β-estradiol) și 10 teste independente privind antagoniștii (de exemplu, tamoxifen). Ar trebui să se adauge rezultatele analizelor viitoare privind aceste standarde de referință și martori tratați cu solvent pentru a extinde baza de date și pentru a asigura consecvența și performanța testului biologic de către laborator în timp.
|
Tabelul 3
Lista (14) substanțelor de verificare pentru testul agoniștilor
(47)
|
Nr. (54)
|
Substanță
|
NR CAS
|
Răspuns preconizat (48)
|
Testul STTA
|
Testul VM7Luc ER TA
|
Clasa de substanțe chimice MeSH (52)
|
Clasă de produse (53)
|
|
Valoarea PC10 (M) (49)
|
Valoarea PC50 (M) (49)
|
Interval de concentr. de testare (M)
|
Valoarea VM7Luc EC50 (M) (50)
|
Cea mai mare concentr. pentru identificatorul de intervale (M) (51)
|
|
14
|
Dietilstilbestrol
|
56-53-1
|
POS
|
< 1,00 × 10-11
|
2,04 × 10-11
|
10-14 - 10-8
|
3,34 × 10-11
|
3,73 × 10-4
|
Hidrocarbură (ciclică)
|
Agent veterinar, farmaceutic
|
|
12
|
17α-estradiol
|
57-91-0
|
POS
|
4,27 × 10-11
|
6,44 × 10-10
|
10-11 - 10-5
|
1,40 × 10-9
|
3,67 × 10-3
|
Steroid
|
Produs farmaceutic, agent veterinar
|
|
15
|
meso-Hexestrol
|
84-16-2
|
POS
|
< 1,00 × 10-11
|
2,75 × 10-11
|
10-11 - 10-5
|
1,65 × 10-11
|
3,70 × 10-3
|
Hidrocarbură (ciclică), fenol
|
Produs farmaceutic, agent veterinar
|
|
11
|
4-terț-octilfenol
|
140-66-9
|
POS
|
1,85 × 10-9
|
7,37 × 10-8
|
10-11 - 10-5
|
3,19 × 10-8
|
4,85 × 10-3
|
Fenol
|
Substanță chimică intermediară
|
|
9
|
Genisteină
|
446-72-0
|
POS
|
2,24 × 10-9
|
2,45 × 10-8
|
10-11 - 10-5
|
2,71 × 10-7
|
3,70 × 10-4
|
Flavonoid, compus heterociclic
|
Produs natural, farmaceutic
|
|
6
|
Bisfenol A
|
80-05-7
|
POS
|
2,02 × 10-8
|
2,94 × 10-7
|
10-11 - 10-5
|
5,33 × 10-7
|
4,38 × 10-3
|
Fenol
|
Substanță chimică intermediară
|
|
2
|
Kaempferol
|
520-18-3
|
POS
|
1,36 × 10-7
|
1,21 × 10-6
|
10-11 - 10-5
|
3,99 × 10-6
|
3,49 × 10-3
|
Flavonoid, compus heterociclic
|
Produs natural
|
|
3
|
Ftalat de butil benzil
|
85-68-7
|
POS
|
1,14 × 10-6
|
4,11 × 10-6
|
10-11 - 10-5
|
1,98 × 10-6
|
3,20 × 10-4
|
Acid carboxilic, ester, acid ftalat
|
Plastifiant, substanță chimică industrială
|
|
4
|
p,p’- Metoxiclor
|
72-43-5
|
POS
|
1,23 × 10-6
|
—
|
10-11 - 10-5
|
1,92 × 10-6
|
2,89 × 10-3
|
Hidrocarbură (halogenată)
|
Pesticid, agent veterinar
|
|
1
|
Etilparaben
|
120-47-8
|
POS
|
5,00 × 10-6
|
—
|
10-11 - 10-5
|
2,48 × 10-5
|
6,02 × 10-3
|
Acid carboxilic, fenol
|
Produs farmaceutic, conservant
|
|
17
|
Atrazină
|
1912-24-9
|
NEG
|
—
|
—
|
10-10 - 10-4
|
—
|
4,64 × 10-4
|
Compus heterociclic
|
Erbicid
|
|
20
|
Spironolactonă
|
52-01-7
|
NEG
|
—
|
—
|
10-11 - 10-5
|
—
|
2,40 × 10-3
|
Lactonă, steroid
|
Produs farmaceutic
|
|
21
|
Ketoconazol
|
65277-42-1
|
NEG
|
—
|
—
|
10-11 - 10-5
|
—
|
9,41 × 10-5
|
Compus heterociclic
|
Produs farmaceutic
|
|
22
|
Reserpină
|
50-55-5
|
NEG
|
—
|
—
|
10-11 - 10-5
|
—
|
1,64 × 10-3
|
Compus heterociclic, indol
|
Produs farmaceutic, agent veterinar
|
|
Abrevieri: Nr. CAS = Număr de înregistrare al Chemical Abstracts Service; EC50 = jumătate din concentrația maximă efectivă a substanței de testat; NEG = negativ; POS = pozitiv; PC10 (și PC50) = concentrația unei substanțe de testat la care răspunsul este de 10 % (sau 50 % pentru PC50) din răspunsul indus de martorul pozitiv (E2, 1nM) pe fiecare placă.
|
Tabelul 4
Lista (10) substanțelor de verificare pentru testul antagoniștilor
|
|
Substanță (4)
|
NR CAS
|
Testul ER STTA (55)
|
Testul VM7Luc ER TA (56)
|
Efectele candidate ER STTA (55)
|
ICCVAM (59) Clasificare prin consens
|
MeSH (60) Clasă chimică
|
Clasă de produse (61)
|
|
Activitatea ER TA
|
IC50 (M)
|
Interval de concentr. de testare (M)
|
Activitatea ER TA
|
IC50
(57) (M)
|
Cea mai mare concentr. pentru identificatorul de intervale (M) (58)
|
|
1
|
4-hidroxi-tamoxifen
|
68047-06-3
|
POS
|
3,97 × 10-9
|
10-12 – 10-7
|
POS
|
2,08 × 10-7
|
2,58 × 10-4
|
valoare POS moderată
|
POS
|
Hidrocarbură (ciclică)
|
Produs farmaceutic
|
|
2
|
Raloxifen HCl
|
82640-04-8
|
POS
|
7,86 × 10-10
|
10-12 – 10-7
|
POS
|
1,19 × 10-9
|
1,96 × 10-4
|
valoare POS moderată
|
POS
|
Hidrocarbură (ciclică)
|
Produs farmaceutic
|
|
3
|
Tamoxifen
|
10540-29-1
|
POS
|
4,91 × 10-7
|
10-10 – 10-5
|
POS
|
8,17 × 10-7
|
2,69 × 10-4
|
POS
|
POS
|
Hidrocarbură (ciclică)
|
Produs farmaceutic
|
|
4
|
17β-estradiol
|
50-28-2
|
NEG
|
—
|
10-9 – 10-4
|
NEG
|
—
|
3,67 × 10-3
|
spre negativ (*4)
|
PN
|
Steroid
|
Produs farmaceutic, agent veterinar
|
|
5
|
Apigenină
|
520-36-5
|
NEG
|
—
|
10-9 – 10-4
|
NEG
|
—
|
3,70 × 10-4
|
NEG
|
NEG
|
Compus heterociclic
|
Colorant, produs natural, produs farmaceutic intermediar
|
|
6
|
Ftalat de di-n-butil
|
84-74-2
|
NEG
|
—
|
10-8 – 10-3
|
NEG
|
—
|
3,59 × 10-3
|
NEG
|
NEG
|
Ester, acid ftalic
|
Ingredient cosmetic, substanță chimică industrială, plastifiant
|
|
7
|
Flavonă
|
525-82-6
|
NEG
|
—
|
10-8 – 10-3
|
NEG
|
—
|
4,50 × 10-4
|
spre negativ (*4)
|
PN
|
Flavonoid, compus heterociclic
|
Produs natural, farmaceutic
|
|
8
|
Genisteină
|
446-72-0
|
NEG
|
—
|
10-9 – 10-4
|
NEG
|
—
|
3,70 × 10-4
|
spre negativ (*4)
|
NEG
|
Flavonoid, compus heterociclic
|
Produs natural, farmaceutic
|
|
9
|
p-n-nonilfenol
|
104-40-5
|
NEG
|
—
|
10-9 – 10-4
|
NEG
|
—
|
4,54 × 10-4
|
netestat
|
NEG
|
Fenol
|
Substanță chimică intermediară
|
|
10
|
Resveratrol
|
501-36-0
|
NEG
|
—
|
10-8 – 10-3
|
NEG
|
—
|
4,38 × 10-4
|
spre negativ (*4)
|
NEG
|
Hidrocarbură (ciclică)
|
Produs natural
|
|
Abrevieri: Nr. CAS = Număr de înregistrare al Chemical Abstracts Service; M = molar; IC50 = jumătate din concentrația maximă de inhibare a substanței de testat; NEG = negativ; PN = presupus negativă; POS = pozitiv.
|
Criteriile de acceptabilitate a unui test
|
16.
|
Acceptarea sau respingerea unui test se bazează pe evaluarea rezultatelor obținute pentru standardele și martorii de referință utilizați pentru fiecare experiment. Valorile pentru PC50 (EC50) sau IC50 pentru standardele de referință ar trebui să îndeplinească criteriile de acceptabilitate prevăzute pentru testul selectat (pentru STTA, a se vedea apendicele 2, pentru VM7Luc ER TA, a se vedea apendicele 3) și toți martorii pozitivi/negativi ar trebui să fie clasificați în mod corect pentru fiecare experiment acceptat. Capacitatea de a efectua în mod consecvent testul ar trebui să fie demonstrată prin dezvoltarea și întreținerea unei baze de date istorice pentru standardele de referință și martori (a se vedea punctul 15). Ca o măsură a reproductibilității în cadrul laboratorului, pot fi utilizate abateri standard (SD) sau coeficienți de variație (CV) pentru mediile de pe curba standardelor de referință care corelează parametrii din cadrul mai multor experimente. În plus, ar trebui să fie îndeplinite următoarele principii referitoare la criteriile de acceptabilitate:
|
—
|
Datele ar trebui să fie suficiente pentru o evaluare cantitativă a activării ER (pentru testul de agonist) sau a suprimării ER (pentru testul de antagonist) (și anume, eficacitate și putere).
|
|
—
|
Activitatea medie a reporterilor pentru concentrația de referință a estrogenului de referință ar trebui să fie cel puțin egală cu valoarea minimă specificată în teste, raportat la valoarea martorului tratat cu vehicul (solvent) pentru a asigura o sensibilitate adecvată. Pentru testele STTA și VM7Luc ER TA, aceasta este de patru ori mai mare decât media martorului tratat cu vehicul de pe fiecare placă.
|
|
—
|
Concentrațiile testate ar trebui să rămână în intervalul de solubilitate al substanțelor chimice de testat și să nu demonstreze citotoxicitate.
|
|
Analiza datelor
|
17.
|
Ar trebui să se recurgă la procedura specifică de interpretare a datelor pentru fiecare test pentru clasificarea unui răspuns pozitiv și negativ.
|
|
18.
|
Îndeplinirea criteriilor de acceptabilitate (punctul 16) indică faptul că testul funcționează în mod corespunzător, dar nu asigură faptul că orice testare specifică va genera date precise. Reproducerea rezultatelor primei testări indică cel mai bine faptul că au fost generate date precise. Dacă sunt generate rezultate reproductibile în urma a două testări (de exemplu, rezultatele ambelor testări indică faptul că o substanță chimică de testat este pozitivă), nu este necesar să se efectueze o a treia testare.
|
|
19.
|
Dacă două testări nu generează rezultate reproductibile (de exemplu, o substanță chimică de testat este pozitivă într-un test și negativă în celălalt test) sau în cazul în care este necesar un grad mai mare de certitudine în ceea ce privește rezultatul acestui test, ar trebui să fie efectuate cel puțin trei testări independente. În acest caz, clasificarea se bazează pe cele două rezultate concordante din trei.
|
Criterii generale de interpretare a datelor
|
20.
|
În prezent, nu există nicio metodă convenită în mod universal pentru interpretarea datelor ER TA. Însă ambele evaluări calitative (de exemplu, pozitiv/negativ) și/sau cantitative (de exemplu, EC50, PC50, IC50) ale activității mediate de ER ar trebui să fie bazate pe date empirice și un raționament științific solid. Dacă este posibil, rezultatele pozitive ar trebui să fie caracterizate atât de magnitudinea efectului față de martorul tratat cu vehicul (solvent) sau de estrogenul de referință, cât și de concentrația la care se produce efectul (de exemplu, EC50, PC50, RPCMax, IC50 etc.).
|
Raport testului
|
21.
|
Raportul testului ar trebui să includă următoarele informații:
|
|
Test:
|
—
|
martor/standard de referință/substanță chimică de testat;
|
|
—
|
sursa, numărul lotului, data limită de utilizare, dacă sunt disponibile;
|
|
—
|
stabilitatea substanței chimice de testat, dacă se cunoaște;
|
|
—
|
solubilitatea și stabilitatea substanței chimice de testat în solvent, dacă se cunosc;
|
|
—
|
măsurarea pH-ului, a osmolalității și a precipitatului în mediul de cultură în care s-a adăugat substanța chimică de testat, după caz.
|
|
|
|
Substanță mono-constituent:
|
—
|
aspectul fizic, solubilitatea în apă și alte proprietăți fizico-chimice relevante;
|
|
—
|
identificarea chimică, precum denumirea IUPAC sau CAS, numărul CAS sau codul său InChI, formula structurală, puritatea, identitatea chimică a impurităților după cum este necesar și fezabil din punct de vedere practic etc.
|
|
|
|
Substanță multicomponentă, UVCB și amestecuri:
|
—
|
în măsura în care este posibil, caracterizată prin identitatea chimică (a se vedea mai sus), apariția cantitativă și proprietățile fizico-chimice relevante ale componentelor.
|
|
|
|
Solvent/vehicul:
|
—
|
caracterizare (natură, furnizor și lot);
|
|
—
|
justificarea alegerii solventului/vehiculului;
|
|
—
|
solubilitatea și stabilitatea substanței chimice de testat în solvent/vehicul, dacă se cunoaște.
|
|
|
|
Celule:
|
—
|
tipul și sursa celulelor:
|
—
|
Este ER exprimat din punct de vedere endogen? Dacă nu, ce receptor(i) a/au fost transfectat (transfectați)?
|
|
—
|
structura (structurile) de reporter utilizată (utilizate) (inclusiv specii sursă);
|
|
—
|
metoda de transfectare;
|
|
—
|
metoda de selecție pentru menținerea stabilității transfectării (dacă este cazul);
|
|
—
|
Metoda de transfectare este relevantă pentru liniile stabile?
|
|
|
—
|
numărul trecerilor de celule (de la decongelare);
|
|
—
|
numărul trecerilor de celule la decongelare;
|
|
—
|
metode de întreținere a culturilor de celule.
|
|
|
|
Condiții de testare:
|
—
|
limite de solubilitate;
|
|
—
|
descrierea metodelor de evaluare a viabilității aplicate;
|
|
—
|
compoziția mediilor, concentrația CO2;
|
|
—
|
concentrațiile substanței chimice de testat;
|
|
—
|
volumul vehiculului și al substanței chimice de testat adăugate;
|
|
—
|
temperatura și umiditatea de incubație;
|
|
—
|
densitatea celulară la începutul și în timpul tratamentului;
|
|
—
|
standarde de referință pozitive și negative;
|
|
—
|
reactivii reporter (denumirea produsului, furnizor și lot);
|
|
—
|
criterii utilizate la aprecierea testelor ca fiind pozitive, negative sau echivoce.
|
|
|
|
Verificarea de acceptabilitate:
|
—
|
inducții multiple pentru fiecare placă de testare și dacă acestea respectă cerințele minime ale testului pe baza martorilor testați anterior;
|
|
—
|
valorile efective pentru criteriile de acceptabilitate, de exemplu, log10EC50, log10PC50, logIC50 și valorile Hillslope, pentru martorii pozitivi testați în paralel/standardele de referință.
|
|
|
|
Rezultate:
|
—
|
date brute și normalizate;
|
|
—
|
nivelul maxim de multiplicare a inducției;
|
|
—
|
date privind citotoxicitatea;
|
|
—
|
dacă există, cea mai redusă valoare a concentrației efective (LEC);
|
|
—
|
valorile RPCMax, PCMax, PC50, IC50 și/sau EC50, după caz;
|
|
—
|
relația concentrație-răspuns, dacă este posibil;
|
|
—
|
analize statistice, dacă există, împreună cu o măsurare a erorii și a încrederii (de exemplu, SEM, SD, CV sau 95 % CI) și o descriere a modului în care au fost obținute aceste valori.
|
|
|
REFERINȚE BIBLIOGRAFICE
|
(1)
|
OCDE (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environmental Health and Safety Publications, Series on Testing and Assessment, (nr. 34), Organizația pentru Cooperare și Dezvoltare Economică, Paris.
|
|
(2)
|
OCDE (2015). Report of the Inter-Laboratory Validation for Stably Transfected Transactivation Assay to detect Estrogenic and Anti-estrogenic Activity. Environmental Health and Safety Publications, Series on Testing and Assessment, (nr. 225), Organizația pentru Cooperare și Dezvoltare Economică, Paris.
|
|
(3)
|
ICCVAM (2011). ICCVAM Test Method Evaluation Report on the LUMI-CELL® ER (BG1Luc ER TA) Test Method, an In Vitro Method for Identifying ER Agonists and Antagonists, National Institute of Environmental Health Sciences: Research Triangle Park, NC.
|
|
(4)
|
Pujol P. et al. (1998). Differential Expression of Estrogen Receptor-Alpha and -Beta Messenger RNAs as a Potential Marker of Ovarian Carcinogenesis, Cancer. Res., 58(23): p. 5367-73.
|
|
(5)
|
Rogers J.M. și Denison M.S. (2000). Recombinant Cell Bioassays for Endocrine Disruptors: Development of a Stably Transfected Human Ovarian Cell Line for the Detection of Estrogenic and Anti-Estrogenic Chemicals, In Vitro and Molecular Toxicology: Journal of Basic and Applied Research, 13(1): p. 67-82.
|
|
(6)
|
OCDE (2012). Performance Standards For Stably Transfected Transactivation In Vitro Assay to Detect Estrogen Receptor Agonists (for TG 455). Environmental Health and Safety Publications, Series on Testing and Assessment, (nr. 173), Organizația pentru Cooperare și Dezvoltare Economică, Paris.
|
|
(7)
|
OCDE (2015). Performance Standards For Stably Transfected Transactivation In Vitro Assay to Detect Estrogen Receptor Antagonists. Environmental Health and Safety Publications, Series on Testing and Assessment, (nr. 174), Organizația pentru Cooperare și Dezvoltare Economică, Paris.
|
|
(8)
|
OCDE (2012). Guidance Document on Standardized Test Guidelines for Evaluating Chemicals for Endocrine Disruption. Environmental Health and Safety Publications, Series on Testing and Assessment, (nr. 150), Organizația pentru Cooperare și Dezvoltare Economică, Paris.
|
|
(9)
|
Cavailles V. (2002). Estrogens and Receptors: an Evolving Concept. Climacteric, 5 Suppl 2: p. 20-6.
|
|
(10)
|
Welboren W.J. et al. (2009). Genomic Actions of Estrogen Receptor Alpha: What are the Targets and how are they Regulated? Endocr. Relat. Cancer, 16(4): p. 1073-89.
|
|
(11)
|
Younes M. și Honma N. (2011). Estrogen Receptor Beta, Arch. Pathol. Lab. Med., 135(1): p. 63-6.
|
|
(12)
|
Jefferson W.N., et al. (2002). Assessing Estrogenic Activity of Phytochemicals Using Transcriptional Activation and Immature Mouse Uterotrophic Responses, Journal of Chromatography B, 777(1-2): p. 179-189.
|
|
(13)
|
Sonneveld E. et al. (2006). Comparison of In Vitro and In Vivo Screening Models for Androgenic and Estrogenic Activities, Toxicol. Sci., 89(1): p. 173-187.
|
|
(14)
|
Takeyoshi M. et al. (2002). The Efficacy of Endocrine Disruptor Screening Tests in Detecting Anti- Estrogenic Effects Downstream of Receptor-Ligand Interactions, Toxicology Letters, 126(2): p. 91- 98.
|
|
(15)
|
Combes R.D. (2000). Endocrine Disruptors: a Critical Review of In Vitro and In Vivo Testing Strategies for Assessing their Toxic Hazard to Humans, ATLA Alternatives to Laboratory Animals, 28(1): p. 81-118.
|
|
(16)
|
Escande A. et al. (2006). Evaluation of Ligand Selectivity Using Reporter Cell Lines Stably Expressing Estrogen Receptor Alpha or Beta, Biochem. Pharmacol, 71(10): p. 1459-69.
|
|
(17)
|
Gray L.E. Jr. (1998). Tiered Screening and Testing Strategy for Xenoestrogens and Antiandrogens, Toxicol. Lett, 102-103, 677-680.
|
|
(18)
|
EDSTAC (1998). Endocrine Disruptor Screening and Testing Advisory Committee (EDSTAC) Final Report.
|
|
(19)
|
ICCVAM (2003). ICCVAM Evaluation of In Vitro Test Methods for Detecting Potential Endocrine Disruptors: Estrogen Receptor and Androgen Receptor Binding and Transcriptional Activation Assays.
|
|
(20)
|
Gustafsson J.Ö. (1999). Estrogen Receptor ß - A New Dimension in Estrogen Mechanism of Action, Journal of Endocrinology, 163(3): p. 379-383.
|
|
(21)
|
Ogawa S. et al. (1998). The Complete Primary Structure of Human Estrogen Receptor ß (hERß) and its Heterodimerization with ERα In Vivo and In Vitro, Biochemical and Biophysical Research Communications, 243(1): p. 122-126.
|
|
(22)
|
Enmark E. et al. (1997). Human Estrogen Receptor ß-Gene Structure, Chromosomal Localization, and Expression Pattern, Journal of Clinical Endocrinology and Metabolism, 82(12): p. 4258-4265.
|
|
(23)
|
Ball L.J. et al. (2009). Cell Type- and Estrogen Receptor-Subtype Specific Regulation of Selective Estrogen Receptor Modulator Regulatory Elements, Molecular and Cellular Endocrinology, 299(2): p. 204-211.
|
|
(24)
|
Barkhem T. et al. (1998). Differential Response of Estrogen Receptor Alpha and Estrogen Receptor Beta to Partial Estrogen Agonists/Antagonists, Mol. Pharmacol, 54(1): p. 105-12.
|
|
(25)
|
Deroo B.J. și Buensuceso A.V. (2010). Minireview: Estrogen Receptor-ß: Mechanistic Insights from Recent Studies, Molecular Endocrinology, 24(9): p. 1703-1714.
|
|
(26)
|
Harris D.M. et al. (2005). Phytoestrogens Induce Differential Estrogen Receptor Alpha- or Beta- Mediated Responses in Transfected Breast Cancer Cells, Experimental Biology and Medicine, 230(8): p. 558-568.
|
|
(27)
|
Anderson J.N. Clark J.H. și Peck E.J.Jr. (1972). The Relationship Between Nuclear Receptor- Estrogen Binding and Uterotrophic Responses, Biochemical and Biophysical Research Communications, 48(6): p. 1460-1468.
|
|
(28)
|
Toft D. (1972). The Interaction of Uterine Estrogen Receptors with DNA, Journal of Steroid Biochemistry, 3(3): p. 515-522.
|
|
(29)
|
Gorski J. et al. (1968), Hormone Receptors: Studies on the Interaction of Estrogen with the Uterus, Recent Progress in Hormone Research, 24: p. 45-80.
|
|
(30)
|
Jensen E.V. et al. (1967), Estrogen-Receptor Interactions in Target Tissues, Archives d’Anatomie Microscopique et de Morphologie Experimentale, 56(3):p. 547-569.
|
|
(31)
|
ICCVAM (2002). Background Review Document: Estrogen Receptor Transcriptional Activation (TA) Assay. Apendicele D, Substances Tested in the ER TA Assay, NIH Publication Report (nr. 03-4505.).
|
|
(32)
|
Kanno J. et al. (2001). The OECD Program to Validate the Rat Uterotrophic Bioassay to Screen Compounds for In Vivo Estrogenic Responses: Phase 1, Environ. Health Persp., 109:785-94.
|
|
(33)
|
Kanno J. et al. (2003). The OECD Program to Validate the Rat Uterotrophic Bioassay: Phase Two Dose -Response Studies, Environ. Health Persp., 111:1530-1549.
|
|
(34)
|
Kanno J. et al. (2003), The OECD Program to Validate the Rat Uterotrophic Bioassay: Phase Two – Coded Single-Dose Studies, Environ. Health Persp., 111:1550-1558.
|
|
(35)
|
Geisinger et al. (1989) Characterization of a human ovarian carcinoma cell line with estrogen and progesterone receptors, Cancer 63, 280-288.
|
|
(36)
|
Baldwin et al. (1998) BG-1 ovarian cell line: an alternative model for examining estrogen-dependent growth in vitro, In Vitro Cell. Dev. Biol. – Animal, 34, 649-654.
|
|
(37)
|
Li, Y. et al. (2014) Research resource: STR DNA profile and gene expression comparisons of human BG-1 cells and a BG-1/MCF-7 clonal variant, Mol. Endo. 28, 2072-2081.
|
|
(38)
|
Rogers, J.M. și Denison, M.S. (2000) Recombinant cell bioassays for endocrine disruptors: development of a stably transfected human ovarian cell line for the detection of estrogenic and anti-estrogenic chemicals, In Vitro & Molec. Toxicol. 13, 67-82.
|
Apendicele 1
DEFINIȚII ȘI ABREVIERI
Criterii de acceptabilitate
: Standarde minime pentru efectuarea de controale experimentale și standarde de referință. Toate criteriile de acceptabilitate ar trebui să fie îndeplinite pentru ca un experiment să fie considerat valabil.
Precizie (concordanță)
: Gradul de concordanță între rezultatele testului și valorile de referință acceptate. Aceasta măsoară performanța testului și unul dintre aspectele relevanței acestuia. Termenul este adesea utilizat în mod interschimbabil, iar concordanța înseamnă proporția de rezultate corecte ale unui test (1).
Agonist
: O substanță care produce un răspuns, de exemplu, transcriere, atunci când se leagă de un anumit receptor.
Antagonist
: Un tip de receptor ligand sau substanță chimică ce nu provoacă un răspuns biologic ea însăși la legarea de un receptor, însă blochează sau atenuează răspunsurile mediate de agonist.
Activitate antiestrogenică
: capacitatea unei substanțe chimice de a suprima acțiunea substanței 17β-estradiol mediate prin receptori estrogeni.
Morfologia celulei
: forma și aspectul celulelor cultivate într-un monostrat într-un singur godeu de pe placa de cultură a țesutului. Celulele care mor prezintă adesea o morfologie celulară anormală.
CF
: Cadrul conceptual al OCDE pentru testarea și evaluarea disruptorilor endocrini.
Tratare cu cărbune/dextran
: Tratarea serului utilizat în cultura celulară. Tratarea cu cărbune/dextran (denumită adesea „stripare”) elimină hormonii endogeni și proteinele care leagă hormonii.
Substanță chimică
: O substanță sau un amestec.
Citotoxicitate
: Efecte dăunătoare asupra structurii sau funcției celulelor care pot provoca, în cele din urmă, moartea celulelor și se pot reflecta printr-o reducere a numărului de celule prezente în godeu la sfârșitul perioadei de expunere sau o reducere a capacității de măsurare a funcției celulare comparativ cu martorul tratat cu vehicul testat în paralel.
CV
: Coeficient de variație
DCC-FBS
: Ser fetal bovin tratat cu cărbune acoperit cu dextran.
DMEM
: Modificarea de către Dulbecco a mediului lui Eagle
DMSO
: Dimetil sulfoxid
E2
: 17β-estradiol
EC50
: Jumătate din concentrația maximă efectivă a unei substanțe chimice de testat.
ED
: Perturbare endocrină
hERα
: Receptor alfa de estrogen uman
hERß
: Receptor beta de estrogen uman
EFM
: Mediu fără estrogen. Modificarea de către Dulbecco a mediului lui Eagle (DMEM) suplimentată cu soluție 4,5 % FBS tratată cu cărbune/dextran, 1,9 % L-glutamină și 0,9 % Pen-Strep.
ER
: Receptor de estrogen
ERE
: Element de răspuns estrogen
Activitate estrogenă
: Capacitatea unei substanțe chimice de a imita 17β-estradiol în capacitatea sa de a se lega de receptorii de estrogen și de a-i activa. Cu această metodă de testare se poate detecta activitatea estrogenă mediată de hERα.
ERTA
: Transactivare a receptorului de estrogen
FBS
: Ser fetal bovin
HeLa
: O linie celulară cervicală umană nemuritoare
HeLa9903
: O subclonă de celule HeLa în care au fost transfectate în mod stabil hERαα și o genă luciferază reporter
IC
50
: Jumătate din concentrația maximă efectivă a unei substanțe chimice de testat inhibitoare.
ICCVAM
: Comitetul de coordonare între agenții cu privire la validarea metodelor alternative (The Interagency Coordinating Committee on the Validation of Alternative Methods).
Reproductibilitatea între laboratoare
: Un indicator al măsurii în care diferite laboratoare calificate, utilizând același protocol și testând aceleași substanțe, pot produce rezultate similare din punct de vedere calitativ și cantitativ. Reproductibilitatea între laboratoare este determinată în timpul proceselor de pre-validare și de validare și indică măsura în care un test poate fi transferat cu succes între laboratoare, denumită de asemenea, reproductibilitate între laboratoare (1).
Reproductibilitatea în cadrul laboratorului
: O determinare a măsurii în care persoane calificate din cadrul aceluiași laborator pot repeta cu succes rezultatele la momente diferite, utilizând un protocol specific. Aceasta este denumită, de asemenea, reproductibilitate în cadrul laboratorului (1).
LEC
: Concentrația efectivă cea mai scăzută este concentrația cea mai scăzută a substanței chimice de testat care produce un răspuns (și anume, cea mai scăzută concentrație a substanței chimice de testat la care inducția multiplă este statistic diferită de martorul tratat cu vehicul testat în paralel).
Testul „me-too”
: O expresie colocvială utilizată pentru un test care este, din punct de vedere structural și funcțional, similar cu o metodă de testare de referință validată și acceptată. Utilizată alternativ cu metoda de testare similară.
MT
: Metalotioneină
MMTV
: Virusul tumorilor mamare la șoareci
OHT
: 4-hidroxi-tamoxifen
PBTG
: Orientarea privind testarea pe baza performanței
MP (Martor pozitiv)
: o substanță puternic activă, de preferință 17ß-estradiol, care este inclusă în toate testele pentru a contribui la buna funcționare a testului.
PC
10
: concentrația unei substanțe chimice de testat la care activitatea măsurată într-un test de agonist este de 10 % din activitatea maximă indusă de MP (E2 la 1nM pentru testul STTA) pe fiecare placă.
PC
50
: concentrația unei substanțe chimice de testat la care activitatea măsurată într-un test de agonist este de 50 % din activitatea maximă indusă de MP (E2 la concentrația de referință specificată în metoda de testare) pe fiecare placă.
PC
Max
: concentrația unei substanțe chimice de testat care induce valoarea RPCMax
Standarde de performanță
: Standarde bazate pe un test validat, care asigură baza pentru evaluarea comparabilității unui test propus similar din punct de vedere al mecanismului și funcțional. Sunt incluse (1) componente esențiale ale testului; (2) o listă minimală de substanțe chimice de referință selectate dintre substanțele chimice utilizate pentru a demonstra performanța acceptabilă a metodei de testare validate; și (3) nivelurile comparabile de precizie și fiabilitate, pe baza rezultatelor obținute în cazul metodei de testare validate, care ar trebui să fie demonstrate prin testul propus la evaluarea acesteia pe baza listei minimale de substanțe chimice de referință (1).
Substanțe de verificare
: Un subset de substanțe de referință incluse în standardele de performanță, care pot fi utilizate de către laboratoare pentru a demonstra competența tehnică cu o metodă de testare standardizată. Criteriile de selecție pentru aceste substanțe includ, de regulă, faptul că acestea reprezintă intervalul de răspunsuri, sunt disponibile în comerț și pentru acestea există date de referință de înaltă calitate.
Competență
: Capacitatea demonstrată de a efectua în mod corespunzător un test înainte de a testa substanțe necunoscute.
Estrogen de referință (martor pozitiv, MP)
: 17β-estradiol (E2, CAS 50-28-2).
Standard de referință
: O substanță de referință utilizată pentru a demonstra caracterul adecvat al unui test. 17β-estradiol este standardul de referință pentru testele STTA și VM7Luc ER TA.
Metode de testare de referință
: Testele pe care se bazează PBTG 455.
Relevanță
: Descrierea relației dintre un test și efectul de interes și dacă aceasta este relevantă și utilă pentru un anumit scop. Aceasta reprezintă măsura în care testul măsoară sau prezice în mod corect efectul biologic de interes. Relevanța include analiza preciziei (a concordanței) unui test (1).
Fiabilitate
: Un indicator al faptului că un test poate fi efectuat în mod reproductibil, în același laborator și în laboratoare diferite în timp, atunci când este efectuat utilizând același protocol. Aceasta se evaluează prin calcularea reproductibilității în cadrul aceluiași laborator și între laboratoare.
RLU
: Unități de lumină relative
ARN
: Acid ribonucleic
RPC
Max
: nivelul maxim de răspuns indus de o substanță chimică de testat, exprimat ca procent din răspunsul indus de 1 nM E2 pe aceeași placă
RPMI
: Un mediu RPMI 1 640 completat cu soluție de 0,9 % Pen-Strep și 8,0 % ser fetal bovin (FBS)
Test
: Un experiment individual care evaluează acțiunea chimică asupra rezultatului biologic al testului. Fiecare test constituie un experiment complet realizat în godeuri duplicate de celule așezate în placă dintr-o rezervă comună de celule în același timp.
Test independent
: Un experiment separat, independent, care evaluează acțiunea chimică asupra rezultatului biologic al testului, utilizând celule dintr-o rezervă diferită, substanțe chimice diluate recent, și care este realizat în zile diferite sau în aceeași zi de către persoane diferite.
SD
: Abatere standard.
Sensibilitate
: Proporția tuturor substanțelor pozitive/active clasificate corect în urma testului. Aceasta măsoară precizia în cazul unui test care generează rezultate clasificabile în categorii și constituie un aspect important în evaluarea relevanței unui test (1).
Specificitate
: Proporția tuturor substanțelor negative/inactive clasificate corect în urma testului. Aceasta măsoară precizia în cazul unui test care generează rezultate clasificabile în categorii și constituie un aspect important în evaluarea relevanței unui test (1).
Transfectare stabilă
: Atunci când ADN-ul este transfectat în celule cultivate, astfel încât este integrat în mod stabil în genomul celulelor, ceea ce conduce la manifestarea stabilă a genelor transfectate. Clonele celulelor transfectate stabil sunt selectate prin markeri stabili (de exemplu, rezistența la G418).
Testul STTA
: Test de transactivare cu transfectare stabilă, test de activare transcripțională ERα utilizând linia celulară HeLa 9 903.
Studiu
: Întreaga serie de lucrări experimentale realizate pentru evaluarea unei singure substanțe specifice, utilizând un anumit test. Un studiu cuprinde toate etapele, inclusiv testele de diluare a substanței de testat în mediile de testare, testele preliminare de identificare a intervalelor, toate testele complexe necesare, analize ale datelor, asigurarea calității, evaluările ale citotoxicității etc. Finalizarea unui studiu permite clasificarea activității substanței chimice de testat asupra obiectivului de toxicitate (și anume, activă, inactivă sau neconcludentă) care este evaluat prin testul utilizat și o estimare a puterii cu raportare la substanța chimică de referință pozitivă.
Substanță
: În cadrul REACH (62), o substanță este definită ca fiind un element chimic și compușii săi în stare naturală sau obținuți prin orice proces de producție, inclusiv orice aditiv necesar pentru păstrarea stabilității acesteia și orice impurități care derivă din procesul utilizat, cu excepția oricărui solvent care poate fi separat fără a influența stabilitatea substanței sau fără a-i schimba compoziția. O definiție foarte similară este utilizată în contextul GHS al ONU (1).
TA (Transactivare): Inițierea sintezei mRNA ca răspuns la semnalul unei anumite substanțe chimice, cum ar fi legarea unui estrogen de receptorul de estrogen
Test
: În contextul prezentei metode de testare, un test constituie una dintre metodologiile acceptate ca fiind valabile pentru îndeplinirea criteriilor de performanță prezentate. Componentele testului includ, de exemplu, linia celulară specifică, cu condiții de creștere asociate, medii specifice în care se desfășoară testul, condiții de pregătire a plăcii, dispunerea și diluțiile substanțelor chimice de testat, împreună cu orice alte măsuri de control al calității impuse și etapele conexe de evaluare a datelor.
Substanța chimică de testat
: Orice substanță sau amestec testat utilizând prezenta metodă de testare.
Transcriere
: sinteza mARN
UVCB
: substanțe chimice cu o compoziție necunoscută sau variabilă, produși de reacție complexă și materiale biologice
Metodă de testare validată
: Un test pentru care au fost realizate studii de validare pentru determinarea relevanței (inclusiv a preciziei) și a fiabilității pentru un anumit scop. Este important de precizat faptul că este posibil ca performanțele unei metode de testare validate să nu fie suficiente din punct de vedere al preciziei și fiabilității astfel încât aceasta să fie considerată acceptabilă pentru scopul propus (1).
Validare
: Procesul prin care se stabilește fiabilitatea și relevanța unei anumite abordări, metode, test, proces sau evaluări specifice pentru un scop definit (1).
VC (Martor tratat cu vehicul)
: Solventul utilizat pentru dizolvarea substanțelor chimice de testat și a celor martor este testat numai ca vehicul, fără substanța chimică dizolvată.
VM7
: O celulă de adenocarcinom imortalizat care exprimă în mod endogen receptorul de estrogen.
VM7Luc4E2
: Linia celulară VM7Luc4E2 a fost derivată din celule de adenocarcinom VM7 imortalizate de la om care exprimă în mod endogen ambele forme ale receptorului de estrogen (ERα și ERβ) și au fost transfectate stabil cu plasmidă pGudLuc7.ERE. Această plasmidă conține patru copii ale unei oligonucleotide sintetice care conține elementul de răspuns estrogen în amonte de promotorul virusului tumoral mamar (MMTV) la șoareci și de gena luciferazei la licurici.
Martor slab pozitiv
: O substanță slab activă selectată din lista substanțelor chimice de referință, care este inclusă în toate testele pentru a contribui la buna funcționare a testului.
Apendicele 2
TESTUL DE TRANSACTIVARE A RECEPTORULUI Α DE ESTROGEN UMAN TRANSFECTAT STABIL PENTRU DETECTAREA ACTIVITĂȚII AGONISTE ȘI ANTAGONISTE A SUBSTANȚELOR CHIMICE CARE UTILIZEAZĂ LINIA CELULARĂ HERΑ-HELA-9903
CONSIDERAȚII ȘI LIMITĂRI INIȚIALE (A SE VEDEA, DE ASEMENEA, INTRODUCERE GENERALĂ)
|
1.
|
Acest test de transactivare (TA) utilizează linia celulară hERα-HeLa-9 903 pentru a detecta activitatea agonistă estrogenă mediată prin receptorul alfa de estrogen uman (hERα). Studiul de validare a testului de transactivare cu transafectare stabilă (STTA) realizat de către Institutul Japonez de Cercetare și Evaluare a Substanțelor Chimice (CERI) folosind linia celulară hERα-HeLa-9 903 pentru a detecta activitatea agonistă și antagonistă estrogenă mediată prin receptorul de estrogen uman (hERα) a demonstrat relevanța și fiabilitatea testului pentru scopul prevăzut (1).
|
|
2.
|
Acest test este conceput special pentru a detecta TA mediată de hERα prin măsurarea chemiluminiscenței ca punct final. Însă au fost raportate semnalele de luminescență nemediate de receptori la concentrații de fitoestrogen mai mari de 1 μM din cauza supraactivării genei luciferază reporter (2) (3). În timp ce curba doză-răspuns indică faptul că o reală activare a sistemului ER se produce la concentrații mai mici, expresia luciferazei obținută la concentrații ridicate de fitoestrogeni sau compuși similari susceptibili de a produce o supra-activare a genei luciferază reporter similară fitoestrogenilor trebuie să fie atent examinată în cadrul sistemelor de testare ER TA transfectat stabil (apendicele 1).
|
|
3.
|
Ar trebui să se parcurgă secțiunile „INTRODUCERE GENERALĂ” și „COMPONENTE ALE ANALIZEI ER TA” înainte de utilizarea acestei analize în scopuri de reglementare. Definițiile și abrevierile utilizate în prezenta orientare privind testarea sunt descrise în apendicele 2.1.
|
PRINCIPIILE TESTULUI (A SE VEDEA, DE ASEMENEA, INTRODUCERE GENERALĂ)
|
4.
|
Testul este utilizat pentru a indica legarea receptorului de estrogen cu un ligand. După legarea cu ligand, complexul receptor-ligand circulă spre nucleu unde leagă anumite elemente de răspuns ADN și transactivează o genă luciferază reporter de licurici, ceea ce determină o expresie celulară crescută a enzimei luciferază. Luciferina este un substrat care este transformat de enzima luciferază într-un produs de bioluminiscență care poate fi măsurat cantitativ cu un luminometru. Activitatea luciferazei poate fi evaluată rapid și cu costuri reduse cu ajutorul unor truse de testare disponibile în comerț.
|
|
5.
|
Sistemul de testare utilizează linia celulară hERα-HeLa-9 903, care provine de la o tumoră cervicală umană, cu două structuri inserate în mod stabil: (i) structura expresiei hERαα (codificând receptorul uman complet) și (ii) o structură reporter de luciferază de la licurici care poartă cinci repetiții în tandem de un element de vitelogenină reactiv la estrogen (ERE) activat de un element TATA promotor de metalotioneină (MT) de la șoarece. S-a demonstrat că structura genei TATA MT de la șoarece are cele mai bune performanțe și, prin urmare, este folosită în mod frecvent. În consecință, această linie celulară hERα-HeLa-9 903 poate măsura capacitatea unei substanțe chimice de testat de a induce transactivarea mediată de hERα a expresiei genei luciferază.
|
|
6.
|
În cazul testului de agonist ER, interpretarea datelor se bazează pe problema dacă nivelul maxim de răspuns indus de o substanță chimică de testat este mai mare decât sau egală cu răspunsul unui agonist echivalent cu 10 % din răspunsul indus de o concentrație cu inducere maximă (1 nM) a martorului pozitiv (MP) 17β-estradiol (E2) (și anume, PC10). În cazul testului de antagonist al ER, interpretarea datelor se bazează pe problema dacă răspunsul indică cel puțin o reducere de 30 % a activității pe baza răspunsului indus de creșterea bruscă a concentrației martorului (25 pM din E2) fără citotoxicitate. Analiza datelor și interpretarea sunt discutate în detaliu la punctele 34-48.
|
PROCEDURA
Linii celulare
|
7.
|
Pentru test ar trebui să se utilizeze linia celulară hERα-HeLa-9 903 transfectată stabil. Linia celulară poate fi obținută din colecția japoneză Japanese Collection of Research Bioresources (JCRB) Cell Bank (63), la semnarea unui acord de transfer de material (MTA).
|
|
8.
|
Pentru testare ar trebui să se utilizeze numai celule caracterizate ca fiind fără micoplasme. RT-PCR (reacție de polimerizare în lanț în timp real) reprezintă metoda aleasă pentru detectarea sensibilă a infecției cu micoplasme (4) (5) (6).
|
Stabilitatea liniei celulare
|
9.
|
Pentru a monitoriza stabilitatea liniei celulare, E2, ar trebui să se utilizeze 17α-estradiol, 17α-metiltestosteron și corticosteron ca standarde de referință pentru testul agonist și ar trebui să se măsoare o curbă concentrație-răspuns completă în intervalul de concentrații de testare prevăzut în tabelul 1 cel puțin o dată la fiecare testare, iar rezultatele ar trebui să fie consecvente cu rezultatele prezentate în tabelul 1.
|
|
10.
|
În cazul testului antagonist, ar trebui să se măsoare simultan, la fiecare testare, curbele complete ale concentrației pentru două standarde de referință, tamoxifen și flutamidă. Ar trebui să se monitorizeze clasificarea calitativă corectă ca fiind pozitivă sau negativă pentru cele două substanțe chimice.
|
Cultura celulară și condiții de aranjare pe placă
|
11.
|
Celulele ar trebui să fie păstrate în mediul esențial minim al lui Eagle (EMEM) fără roșu de fenol, suplimentat cu 60 mg/l de antibiotic kanamicină și 10 % ser fetal bovin tratat cu cărbune și acoperit cu dextran (DCC-FBS), într-un incubator CO2 ( 5 % CO2) la 37 ± 1°C. La atingerea unei confluențe de 75-90 %, celulele pot fi subcultivate la 10 din 0,4 x 105 – 1 x 105 celule/ml pentru un vas de cultură a celulelor de 100 mm. Celulele ar trebui să fie suspendate în soluție de 10 % FBS-EMEM (care este identic cu EMEM cu DCC-FBS) și apoi aranjate în godeurile unei microplăci la o densitate de 1 x 104 celule/(100 μl x godeu). Apoi, celulele ar trebui să fie preincubate într-un incubator cu 5 % CO2 la 37° ± 1 °C timp de 3 ore înainte de expunerea la substanțe chimice. Materialul din plastic ar trebui să fie fără activitate estrogenă.
|
|
12.
|
Pentru a menține integritatea răspunsului, celulele ar trebui să fie cultivate pentru mai multe pasaje din stocul congelat în mediu condiționat, dar nu pentru mai mult de 40 de pasaje. Pentru linia celulară hERα-HeLa-9 903, această perioadă va fi mai mică de trei luni. Însă performanța celulelor poate fi redusă dacă acestea sunt cultivate în condiții de cultură necorespunzătoare.
|
|
13.
|
Soluția DCC-FBS poate fi preparată conform descrierii din apendicele 2.2 sau poate fi obținută din surse comerciale.
|
Criterii de acceptabilitate
Standarde de referință pozitive și negative pentru testul de agonist ER
|
14.
|
Înaintea și în timpul studiului, capacitatea de răspuns a sistemului de testare ar trebui să fie verificată utilizând concentrațiile adecvate ale unui estrogen puternic: E2, un estrogen slab (17α-estradiol), un agonist foarte slab (17α-metiltestosteron) și o substanță negativă (corticosteron). Valorile din intervalul acceptabil derivate din studiul de validare (1) sunt date în tabelul 1. Aceste patru standarde de referință concurente ar trebui să fie incluse în fiecare experiment, iar rezultatele ar trebui să se încadreze în limitele acceptabile date. În caz contrar, ar trebui să se identifice cauza neîndeplinirii criteriilor de acceptabilitate (de exemplu, manipularea celulelor, precum și serul și antibioticele pentru calitate și concentrație), iar testul se repetă. Odată ce au fost îndeplinite criteriile de acceptabilitate, pentru a se asigura variabilitatea minimă a valorilor EC50, PC50 and PC10, este esențială utilizarea consecventă a materialelor pentru cultura celulară. Cele patru standarde de referință concurente, care ar trebui să fie incluse în fiecare experiment (realizat în aceleași condiții, inclusiv cu aceleași materiale, nivel de trecere a celulelor și tehnicieni) pot asigura sensibilitatea testului, deoarece valorile PC10 dintre cele trei standarde de referință pozitive ar trebui să se încadreze în intervalul acceptabil, la fel ca și valorile PC50și EC50, în cazul în care acestea pot fi calculate (a se vedea tabelul 1).
|
Tabelul 1
Valori din intervalul acceptabil ale celor patru standarde de referință pentru testul de agonist ER
|
Denumire
|
logPC50
|
logPC10
|
logEC50
|
Panta Hill
|
Interval de testare
|
|
17β-estradiol (E2) CAS nr.: 50-28-2
|
-11,4 ~ -10,1
|
< -11
|
-11,3 ~ -10,1
|
0,7 ~ 1,5
|
10-14 ~ 10-8M
|
|
17α-estradiol CAS nr.: 57-91-0
|
-9,6 ~ -8,1
|
-10,7 ~ -9,3
|
-9,6 ~ -8,4
|
0,9 ~ 2,0
|
10-12 ~ 10-6M
|
|
Corticosteron CAS nr.: 50-22-6
|
–
|
–
|
–
|
–
|
10-10 ~ 10-4M
|
|
17α-metiltestosteron CAS nr.: 58-18-4
|
-6,0 ~ -5,1
|
-8,0 ~ -6,2
|
–
|
–
|
10-11 ~ 10-5M
|
Standarde de referință pozitive și negative pentru testul de antagonist ER
|
15.
|
Înaintea și în timpul studiului, capacitatea de răspuns a sistemului de testare ar trebui să fie verificată utilizând concentrațiile adecvate ale unui substanțe pozitive (tamoxifen) și ale unei substanțe negative (flutamidă). Valorile din intervalul acceptabil derivate din studiul de validare (1) sunt date în tabelul 2. Aceste două standarde de referință concurente ar trebui să fie incluse în fiecare experiment, iar rezultatele ar trebui să fie evaluate ca fiind corecte conform criteriilor. În caz contrar, ar trebui să se identifice cauza neîndeplinirii criteriilor (de exemplu, manipularea celulelor, precum și serul și antibioticele pentru calitate și concentrație), iar testul se repetă. În plus, ar trebui să fie calculate valorile IC50 pentru o substanță pozitivă (tamoxifen), iar rezultatele ar trebui să se încadreze în limitele acceptabile date. Odată ce au fost îndeplinite criteriile de acceptabilitate, pentru a se asigura variabilitatea minimă a valorilor IC50, este esențială utilizarea consecventă a materialelor pentru cultura celulară. Cele două standarde de referință concurente care trebuie incluse în fiecare experiment (realizat în aceleași condiții, inclusiv cu aceleași materiale, nivel de trecere a celulelor și tehnicieni) pot asigura sensibilitatea testului (a se vedea tabelul 2).
|
Tabelul 2
Criterii și valori din intervalul acceptabil ale celor două standarde de referință pentru testul de antagonist ER
|
Denumire
|
Criterii
|
LogIC50
|
Interval de testare
|
|
Tamoxifen CAS nr.: 10 540 -29-1
|
Pozitiv: Ar trebui să se calculeze IC50
|
-5,942~-7,596
|
10-10~10-5 M
|
|
Flutamidă CAS nr.: 13 311 -84-7
|
Negativ: Nu ar trebui să se calculeze IC30
|
—
|
10-10~10-5 M
|
Martori pozitivi și martori tratați cu vehicule
|
16.
|
Martorul pozitiv (MP) pentru testul de agonist ER (1 nM din E2) și pentru testul de antagonist ER (10 μM TAM) ar trebui să fie testat de cel puțin trei ori pe fiecare placă. Vehiculul care se utilizează pentru a dizolva o substanță chimică de testat ar trebui să fie testat ca martor tratat cu vehicul (VC) de cel puțin trei ori pe fiecare placă. în plus față de acest VC, dacă MP utilizează un vehicul diferit de cel al substanței chimice de testat, un alt VC ar trebui testat de cel puțin trei ori pe aceeași placă cu MP.
|
Criterii de calitate pentru testul de agonist ER
|
17.
|
Activitatea medie a luciferazei la martorul pozitiv (1 nM E2) ar trebui să fie de cel puțin patru ori mai mare decât media VC pe fiecare placă. Acest criteriu se stabilește pe baza fiabilității valorilor punctelor finale din studiul de validare (istoric de patru până la 30 de ori).
|
|
18.
|
În ceea ce privește controlul calității testului, inducția multiplicată corespunzătoare valorii PC10 a martorului MP testat în paralel (1 nM E2) ar trebui să fie mai mare de 1 + 2 SD din valoarea inducției multiplicate (= 1) a martorului VC testat în paralel. Pentru stabilirea priorităților, valoarea PC10 poate fi utilă pentru a simplifica analiza datelor, care este necesară în comparație cu o analiză statistică. Deși o analiză statistică oferă informații privind semnificația, o astfel de analiză nu constituie un parametru cantitativ în ceea ce privește potențialul bazat pe concentrație, și astfel este mai puțin utilă în scopul stabilirii priorităților.
|
Criterii de calitate pentru testul de antagonist ER
|
19.
|
Activitatea medie a luciferazei la creșterea bruscă a concentrației martorului (25 pM E2) ar trebui să fie de cel puțin patru ori mai mare decât media VC pe fiecare placă. Acest criteriu se stabilește pe baza fiabilității valorilor punctelor finale din studiul de validare.
|
|
20.
|
În ceea ce privește controlul calității testului, activarea transcipțională relativă (RTA) de 1 nM E2 ar trebui să fie mai mare de 100 %, RTA de 1μM 4-hidroxitamoxifen (OHT) ar trebui să fie mai mică de 40,6 % , iar RTA de 100 μM digitonină (Dig) ar trebui să fie mai mică de 0 %.
Demonstrarea performanțelor laboratorului (a se vedea punctul 14 și tabelele 3 și 4 de la secțiunea «COMPONENTELE TESTULUI ER TA» din prezenta metodă de testare).
|
Vehicul
|
21.
|
Ca și VC testat în paralel ar trebui să se utilizeze dimetilsulfoxid (DMSO) sau un solvent adecvat, la aceeași concentrație utilizată pentru diferiții martori pozitivi și negativi, iar substanțele chimice de testat ar trebui să fie utilizate ca VC testat în paralel. Substanțele chimice de testat ar trebui să se dizolve într-un solvent care solubilizează substanța chimică de testat și este miscibil cu mediul celular. Vehicule adecvate sunt apa, etanolul (puritate de 95-100 %) și DMSO. În cazul în care se utilizează DMSO, nivelul nu ar trebui să depășească 0,1 % (v/v). Pentru orice vehicul, ar trebui să se demonstreze că volumul maxim utilizat nu este citotoxic și nu interferează cu performanțele testului.
|
Prepararea substanțelor chimice de testat
|
22.
|
În general, substanțele chimice de testat ar trebui să fie dizolvate în DMSO sau într-un alt solvent adecvat și diluate în serie cu același solvent la un raport comun de 1:10 în vederea preparării soluțiilor pentru diluare cu medii.
|
Solubilitate și citotoxicitate: Considerații pentru identificarea intervalului.
|
23.
|
Ar trebui efectuat un test preliminar pentru a determina intervalul adecvat de concentrații pentru substanța chimică ce urmează să fie testată și pentru a confirma dacă substanța chimică de testat ar putea avea probleme de solubilitate și citotoxicitate. Inițial, substanțele chimice sunt testate până la concentrația maximă de 1 μl/ml, 1 mg/ml sau 1 mM, luându-se în considerare cea mai mică valoare. În funcție de gradul de citotoxicitate sau de lipsa solubilității observată la testul preliminar, prima testare specifică ar trebui să testeze substanța chimică la diluții log în serie, începând de la concentrația maximă acceptabilă (de exemplu, 1 mM, 100 μM, 10 μM etc.) și se consemnează prezența aspectului cețos sau a precipitatului sau citotoxicitatea. Concentrațiile în cadrul celei de a doua și, dacă este necesar, celei de a treia testări ar trebui să fie ajustate, după caz, pentru a caracteriza mai bine curba concentrație-răspuns și pentru a evita concentrațiile care se dovedesc a fi insolubile sau a induce o citotoxicitate excesivă.
|
|
24.
|
Agoniștii și antagoniștii de ER și prezența unor niveluri tot mai mari ale citotoxicității pot să modifice în mod semnificativ sau să elimine răspunsul sigmoidal tipic și ar trebui să fie luate în considerare la interpretarea datelor. Ar trebui să se folosească metode de testare a citotoxicității care pot oferi informații cu privire la viabilitatea celulară de 80 %, utilizând un test adecvat bazat pe experiența de laborator.
|
|
25.
|
În cazul în care rezultatele testului de citotoxicitate arată că concentrația substanței chimice de testat a redus numărul celulelor cu 20 % sau mai mult, această concentrație ar trebui să fie considerată ca fiind citotoxică, iar concentrațiile la nivelul concentrației citotoxice sau peste aceasta ar trebui să fie excluse de la evaluare.
|
Expunerea la substanțe chimice și organizarea plăcilor pentru testare
|
26.
|
Procedura pentru diluții chimice (etapele 1 și 2) și expunerea la celule (etapa 3) pot fi realizate după cum urmează:
|
Etapa 1: Fiecare substanță chimică de testat ar trebui să fie diluată în serie în DMSO sau înr-un solvent adecvat și adăugată în godeurile unei plăci de microtitrare pentru a realiza concentrații în serie finale, astfel cum sunt determinate prin testul preliminar de identificare a intervalului [de regulă, într-o serie, de exemplu, de 1 mM, 100 μM, 10 μM, 1μM, 100 nM, 10 nM, 1 nM, 100 pM, and 10 pM (10-3-10-11 M)] pentru teste triple.
Etapa 2: Diluție chimică: Mai întâi se diluează 1,5 μl din substanța chimică de testat în solvent la un volum de 500 μl de medii.
Etapa 3: Expunerea celulelor la substanțe chimice: Se adaugă 50 μl de diluție cu medii (preparată în etapa 2) într-un godeu de analiză care conține 104 celule/100 μl/godeu.
Volumul final recomandat de medii necesar pentru fiecare godeu este de 150 μl. Eșantioanele de testare și standardele de referință pot fi distribuite astfel cum este indicat în tabelele 3 și 4.
Tabelul 3
Exemplu de distribuire pe concentrații pe placă a standardelor de referință pe placa de testare în cadrul testului de agonist ER
|
Rând
|
17α-metiltestosteron
|
Corticosteron
|
17α-estradiol
|
E2
|
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
conc 1 (10 μM)
|
→
|
→
|
100 μM
|
→
|
→
|
1 μM
|
→
|
→
|
10 nM
|
→
|
→
|
|
B
|
conc 2 (1 μM)
|
→
|
→
|
10 μM
|
→
|
→
|
100 nM
|
→
|
→
|
1 nM
|
→
|
→
|
|
C
|
conc 3 (100 nM)
|
→
|
→
|
1 μM
|
→
|
→
|
10 nM
|
→
|
→
|
100 pM
|
→
|
→
|
|
D
|
conc 4 (10 nM)
|
→
|
→
|
100 nM
|
→
|
→
|
1 nM
|
→
|
→
|
10 pM
|
→
|
→
|
|
E
|
conc 5 (1 nM)
|
→
|
→
|
10 nM
|
→
|
→
|
100 pM
|
→
|
→
|
1 pM
|
→
|
→
|
|
F
|
conc 6 (100 pM)
|
→
|
→
|
1 nM
|
→
|
→
|
10 pM
|
→
|
→
|
0,1 pM
|
→
|
→
|
|
G
|
conc 7 (10 pM)
|
→
|
→
|
100 pM
|
→
|
→
|
1 pM
|
→
|
→
|
0,01 pM
|
→
|
→
|
|
H
|
VC
|
→
|
→
|
→
|
→
|
→
|
MP
|
→
|
→
|
→
|
→
|
→
|
|
VC: Martor cu vehicul (0,1 % DMSO); MP: Martor pozitiv (1 nM E2)
|
|
27.
|
Standardele de referință (E2, 17α-estradiol, 17-μετιλμτεστοστερονττιιχορτιχοστερονχχαρατρεβυιτσ©©φιεφτεστατεττννχαδρυλχφιεχ©ρειρτεστ©ριρρταβελυλαααααGodeurile cu MP tratați cu 1 nM de E2 și care pot produce o inducție maximă a E2 și VC tratați doar cu DMSO (sau un solvent adecvat) ar trebui să fie incluse pe fiecare placă de testare din cadrul analizei (tabelul 4). Dacă se utilizează celule din surse diferite (de exemplu, un număr de trecere diferit, un lot diferit etc.) în cadrul aceluiași experiment, ar trebui să se testeze standardele de referință pentru fiecare sursă celulară.
|
Tabelul 4
Exemplu de distribuire pe concentrații pe placă a substanțelor chimice martor pe placa de testare în cadrul analizei de agonist ER
|
Rând
|
Substanța chimică de testat 1
|
Substanța chimică de testat 2
|
Substanța chimică de testat 3
|
Substanța chimică de testat 4
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
conc 1 (10 μM)
|
→
|
→
|
1 mM
|
→
|
→
|
1 μM
|
→
|
→
|
10 nM
|
→
|
→
|
|
B
|
conc 2 (1 μM)
|
→
|
→
|
100 μM
|
→
|
→
|
100 nM
|
→
|
→
|
1 nM
|
→
|
→
|
|
C
|
conc 3 (100 nM)
|
→
|
→
|
10 μM
|
→
|
→
|
10 nM
|
→
|
→
|
100 pM
|
→
|
→
|
|
D
|
conc 4 (10 nM)
|
→
|
→
|
1 μM
|
→
|
→
|
1 nM
|
→
|
→
|
10 pM
|
→
|
→
|
|
E
|
conc 5 (1 nM)
|
→
|
→
|
100 nM
|
→
|
→
|
100 pM
|
→
|
→
|
1 pM
|
→
|
→
|
|
F
|
conc 6 (100 pM)
|
→
|
→
|
10 nM
|
→
|
→
|
10 pM
|
→
|
→
|
0,1 pM
|
→
|
→
|
|
G
|
conc 7 (10 pM)
|
→
|
→
|
1 nM
|
→
|
→
|
1 pM
|
→
|
→
|
0,01 pM
|
→
|
→
|
|
H
|
VC
|
→
|
→
|
→
|
→
|
→
|
MP
|
→
|
→
|
→
|
→
|
→
|
|
VC: Martor cu vehicul (0,1 % DMSO); MP: Martor pozitiv (1 nM E2)
|
Tabelul 5
Exemplu de distribuire pe concentrații pe placă a standardelor de referință pe placa de testare în cadrul testului de antagonist ER
|
28.
|
Pentru a evalua activitatea de antagonist a substanțelor chimice, în godeurile de analiză de pe rândurile A-G ar trebui să se introducă 25 pm E2. Standardele de referință (tamoxifen și flutamidă) ar trebui să fie testate la fiecare testare. Godeurile cu MP tratate cu 1 nM de E2 care pot fi utilizate în vederea controlului calității liniei celulare hERα-HeLa-9 903, godeurile cu VC tratate cu DMSO (sau un solvent adecvat), godeurile cu 0,1 % DMSO tratate cu adăugare de DMSO la E2 completat corespunzător cu „Spike-in-control” , godeurile tratate cu o concentrație finală de 1 μM OHT și godeuri tratate cu 100 μM Dig ar trebui să fie adăugate la fiecare placă de testare (tabelul 5). Placa de testare ulterioară ar trebui să urmeze același plan de aranjare a plăcii fără godeuri standard de referință (tabelul 6). Dacă se utilizează celule din surse diferite (de exemplu, un număr de trecere diferit, un lot diferit etc.) în cadrul aceluiași experiment, ar trebui să se testeze standardele de referință pentru fiecare sursă celulară.
|
Tabelul 6
Exemplu de distribuire pe concentrații pe placă a substanțelor chimice martor pe placa de testare în cadrul analizei de antagonist ER
|
29.
|
Lipsa efectelor extreme ar trebui să fie confirmată, după caz, iar dacă se suspectează prezența acestora, modul de aranjare pe placă ar trebui să fie schimbat pentru a evita astfel de efecte. De exemplu, se poate utiliza un mod de aranjare pe placă care să excludă godeurile de pe margini.
|
|
30.
|
După adăugarea substanțelor chimice, plăcile de testare ar trebui să fie incubate într-un incubator de 5 % CO2 la temperatura de 37 ± 1 °C pentru 20-24 de ore pentru a induce produsele genei reporter.
|
|
31.
|
Pentru compușii care sunt foarte volatili va fi necesar să se aplice considerații speciale. În astfel de cazuri, godeurile cu martori din apropiere pot genera valori fals pozitive și acest lucru ar trebui să fie analizat în contextul valorilor estimate și a celor istorice ale martorilor. În cele câteva cazuri în care volatilitatea poate fi o problemă, ar putea fi util să se recurgă la „agenți de etanșeizare a plăcii” pentru a izola în mod eficace godeurile individuale în timpul testării și, prin urmare, aceștia sunt recomandați în astfel de cazuri.
|
|
32.
|
Ar trebui să se efectueze teste definitive repetate pentru aceeași substanță chimică în zile diferite, pentru asigurarea independenței.
|
Analiza luciferazei
|
33.
|
Pentru efectuarea analizei poate fi utilizat un reactiv de analiză a luciferazei din comerț [de exemplu, Steady-Glo® Luciferase Assay System (Promega, E2510 sau un produs echivalent)] sau un sistem de analiză a luciferazei standard (de exemplu, Promega, E1500 sau un produs echivalent), atâta timp cât sunt îndeplinite criteriile de acceptabilitate. Reactivii de analiză ar trebui să fie selectați pe baza sensibilității luminometrului care va fi utilizat. Atunci când se utilizează sistemul de analiză a luciferazei standard, ar trebui să se utilizeze Cell Culture Lysis Reagent (de exemplu, Promega, E1531 sau un produs echivalent) înainte de adăugarea substratului. Reactivul de luciferază ar trebui să fie aplicat conform instrucțiunilor producătorului.
|
ANALIZA DATELOR
Testul de agonist ER
|
34.
|
În cazul testului de agonist ER, pentru a obține activitatea transcripțională relativă pe MP (1 nM de E2), se pot analiza semnalele de luminiscență de la aceeași placă cu respectarea următoarelor etape (sunt acceptabile, de asemenea, alte procese matematice echivalente):
|
Etapa 1. Se calculează valoarea medie pentru VC.
Etapa 2. Se scade valoarea medie a VC din fiecare valoare a godeului pentru a normaliza datele.
Etapa 3. Se calculează media valorii MP normalizate.
Etapa 4. Se împarte valoarea normalizată a fiecărui godeu din placă la media valorii MP normalizate (MP = 100 %).
Valoarea finală a fiecărui godeu constituie activitatea transcripțională relativă pentru godeul respectiv în comparație cu răspunsul MP.
Etapa 5. Se calculează valoarea medie a activității transcripționale relative pentru fiecare grup de concentrații al substanței chimice de testat. Există două dimensiuni la răspuns: media activității transcripționale (răspuns) și concentrația la care se produce răspunsul (a se vedea secțiunea următoare).
Considerații privind inducția EC50, PC50 și PC10
|
35.
|
Curba concentrație-răspuns completă este necesară pentru calcularea EC50, dar este posibil ca acest lucru să nu fie întotdeauna realizabil sau practic din cauza limitărilor intervalului de concentrații de testare (de exemplu, din cauza unor probleme de citotoxicitate sau de solubilitate). Însă, întrucât EC50 și nivelul maxim de inducție (corespunzător valorii superioare a ecuației Hill) sunt parametri informativi, acești parametri ar trebui să fie consemnați, dacă este posibil. Pentru calcularea EC50 și a nivelului maxim de inducție ar trebui să se utilizeze un software adecvat de statistică (de exemplu, software-ul statistic Graphpad Prism). În cazul în care ecuația logistică a lui Hill este aplicabilă pentru datele de răspuns la concentrații, valoarea EC50 ar trebui să fie calculată prin următoarea ecuație (7):
Y=Inferior+ (Superior-Inferior) / (1+10 exp ((log EC50 -X) x panta Hill)) unde:
X este logaritmul concentrației; și,
Y este răspunsul și Y pornește din partea inferioară și ajunge în partea superioară într-o curbă sigmoidă. Partea inferioară este fixată la zero în ecuația logistică a lui Hill.
|
|
36.
|
Pentru fiecare substanță chimică de testat, ar trebui să se prezinte:
valoarea RPCMax care este nivelul maxim de răspuns indus de o substanță chimică de testat, exprimată ca procent din răspunsul indus de 1 nM E2 de pe aceeași placă, precum și PCMax (concentrație asociată cu RPCMax); și
În cazul substanțelor chimice pozitive, concentrațiile care induc PC10 și, dacă este cazul, PC50.
|
|
37.
|
Valoarea PCx poate fi calculată prin interpolarea dintre 2 puncte pe axa coordonatelor X-Y, unul situat imediat deasupra și altul imediat sub o valoare PCx. În cazul în care punctele de date situate imediat deasupra și sub valoarea PCx au coordonatele (a, b) și, respectiv, (c, d), atunci valoarea PCx poate fi calculată folosind următoarea ecuație:
log[PCx] = log[c]+(x-d)/(d-b)
|
|
38.
|
Descrierile valorilor PC sunt prezentate în figura 1 de mai jos.
Figura 1
Exemplu de metodă de obținere a valorilor PC. PC (1 nM de E2) se adaugă pe fiecare placă de testare.
|
Testul de antagonist ER
|
39.
|
În cazul testului de antagonist ER, pentru a obține activitatea transcripțională relativă (RTA) pentru adăugarea de martor (25 pM de E2), se pot analiza semnalele de luminiscență de la aceeași placă cu respectarea următoarelor etape (sunt acceptabile, de asemenea, alte procese matematice echivalente):
Etapa 1. Se calculează valoarea medie pentru VC.
Etapa 2. Se scade valoarea medie a VC din fiecare valoare a godeului pentru a normaliza datele. Etapa 3. Se calculează media martorului de adăugat normalizat.
Etapa 4. Se împarte valoarea normalizată a fiecărui godeu din placă la media valorii martorului de adăugat normalizat (martor de adăugat = 100 %).
Valoarea finală a fiecărui godeu constituie activitatea transcripțională relativă pentru godeul respectiv în comparație cu răspunsul martorului de adăugat.
Etapa 5. Se calculează valoarea medie a activității transcripționale relative pentru fiecare tratament.
|
Considerații privind inducția IC30 și IC50
|
40.
|
În cazul substanțelor chimice pozitive, ar trebui să se prezinte concentrațiile care induc IC30 și, dacă este cazul, IC50.
|
|
41.
|
Valoarea ICx poate fi calculată prin interpolarea dintre 2 puncte pe axa coordonatelor X-Y, unul situat imediat deasupra și altul imediat sub o valoare ICx. În cazul în care punctele de date situate imediat deasupra și sub valoarea ICx au coordonatele (c, d) și, respectiv, (a, b), atunci valoarea ICx poate fi calculată folosind următoarea ecuație:
în ICx = a-[b-(100-x)] (a-c) /(b-d)
Figura 2
Exemplu de metodă de obținere a valorilor IC. Martorul de adăugat (25 nM de E2) se adaugă pe fiecare placă de testare.
RTA: activitate transcripțională relativă
|
|
42.
|
Rezultatele ar trebui să fie bazate pe două (sau trei) testări independente. Dacă două testări generează rezultate comparabile și, prin urmare, reproductibile, nu este necesar să se efectueze o a treia testare. Pentru ca rezultatele să fie acceptabile, acestea ar trebui:
|
—
|
să îndeplinească criteriile de acceptabilitate (a se vedea Criteriile de acceptabilitate de la punctele 14-20),
|
|
Criterii de interpretare a datelor
Tabelul 7
Criterii de decizie pozitivă și negativă în testul de agonist ER
|
Pozitivă
|
În cazul în care se obține o valoare RPCMax mai mare decât sau egală cu 10 % din răspunsul martorului pozitiv în cel puțin două din două sau două din trei testări.
|
|
Negativă
|
În cazul în care valoarea RPCMax nu ajunge la cel puțin 10 % din răspunsul martorului pozitiv în două din două sau două din trei testări.
|
Tabelul 8
Criterii de decizie pozitivă și negativă în testul de antagonist ER
|
Pozitivă
|
Dacă valoarea IC30 este calculată în cel puțin două din două sau două din trei testări.
|
|
Negativă
|
Dacă valoarea IC30 nu este calculată în două din două sau două din trei testări.
|
|
43.
|
Criteriile de interpretare a datelor sunt prezentate în tabelele 7 și 8. Rezultatele pozitive vor fi caracterizate atât prin amploarea efectului, cât și prin concentrația la care se produc efecte. Exprimarea rezultatelor ca o concentrație la care se atinge 50 % (PC50) sau 10 % (PC10) din valorile PC pentru testul de agonist, iar 50 % (IC50) sau 30 % (IC30) din valoarea martorului de adăugat este inhibată pentru testul antagonist, înseamnă că sunt îndeplinite ambele obiective. Cu toate acestea, o substanță chimică de testat este considerată a fi pozitivă dacă răspunsul maxim indus de substanța chimică de testat (RPCMax) este mai mare decât sau egal cu 10 % din PC în cel puțin două din două sau două din trei testări, iar o substanță chimică de testat este considerată a fi negativă în cazul în care RPCMax nu reușește să atingă cel puțin 10 % din răspunsul martorului pozitiv în două din două sau două din trei teste.
|
|
44.
|
Calculele pentru PC10, PC50 and PCMax în testul de agonist ER și IC30 și IC50 în testul antagonist ER al ER pot fi realizate utilizând o fișă disponibilă împreună cu Orientarea privind testarea pe site-ul public al OCDE (64).
|
|
45.
|
Ar trebui să fie suficient să se obțină valori ale PC10 sau PC50 și IC30 sau IC50 cel puțin de două ori. Însă, în cazul în care valoarea de referință rezultată pentru date din același interval de concentrații indică o variabilitate cu un coeficient inacceptabil de ridicat de variație (CV; %), este posibil ca datele să nu fie considerate fiabile și ar trebui să se identifice sursa variabilității ridicate. Valoarea CV a datelor brute triple (și anume, date privind intensitatea luminiscenței) ale punctelor de date care sunt utilizate pentru calcularea PC10 ar trebui să fie sub 20 %.
|
|
46.
|
Îndeplinirea criteriilor de acceptabilitate indică faptul că sistemul de analiză funcționează corect, dar nu asigură faptul că orice testare specifică va genera date precise. Duplicarea rezultatelor primei testări reprezintă cea mai bună asigurare a faptului că au fost generate date precise.
|
|
47.
|
În cazul testului de agonist ER, unde sunt necesare mai multe informații, în plus față de scopurile de depistare și stabilire a priorităților ale prezentei TG pentru substanțele chimice pozitive, în special pentru substanțele chimice PC10-PC49, precum și pentru substanțe chimice suspectate a supra-stimulata luciferaza, se poate confirma că activitatea de luciferază observată este doar un răspuns specific ERα, folosind un antagonist ERα (a se vedea apendicele 2.1).
|
RAPORTUL TESTULUI
|
48.
|
A se vedea punctul 20 din „COMPONENTELE ANALIZEI ER TA”.
|
REFERINȚE BIBLIOGRAFICE
|
(1)
|
OCDE (2015). Report of the Inter-Laboratory Validation for Stably Transfected Transactivation Assay to detect Estrogenic and Anti-estrogenic Activity. Environmental Health and Safety Publications, Series on Testing and Assessment, (nr. 225), Organizația pentru Cooperare și Dezvoltare Economică, Paris.
|
|
(2)
|
Escande A., et al. (2006). Evaluation of Ligand Selectivity Using Reporter Cell Lines Stably Expressing Estrogen Receptor Alpha or Beta, Biochem. Pharmacol., 71, 1 459-1 469.
|
|
(3)
|
Kuiper G.G., et al. (1998). Interaction of Estrogenic Chemicals and Phytoestrogens with Estrogen Receptor Beta, Endocrinol., 139, 4 252-4 263.
|
|
(4)
|
Spaepen M., et al. (1992). Detection of Bacterial and Mycoplasma Contamination in Cell Cultures by Polymerase Chain Reaction, FEMS Microbiol. Lett., 78(1), 89-94.
|
|
(5)
|
Kobayashi H., et al. (1995). Rapid Detection of Mycoplasma Contamination in Cell Cultures by Enzymatic Detection of Polymerase Chain Reaction (PCR) Products, J. Vet. Med. Sci., 57(4), 769- 71.
|
|
(6)
|
Dussurget O. și Roulland-Dussoix D. (1994). Rapid, Sensitive PCR-Based Detection of Mycoplasmas in Simulated Samples of Animal Sera, Appl. Environ. Microbiol., 60(3), 953-9.
|
|
(7)
|
De Lean A., Munson P.J. și Rodbard D. (1978). Simultaneous Analysis of Families of Sigmoidal Curves: Application to Bioassay, Radioligand Assay, and Physiological Dose-Response Curves, Am. J. Physiol., 235, E97-El02.
|
Apendicele 2.1
REZULTATE FALS POZITIVE: EVALUAREA SEMNALELOR DE LUMINISCENȚĂ CARE NU SUNT MEDIATE DE RECEPTORI
|
1.
|
Rezultatele pozitive false din analiza activității agoniste a ER ar putea fi generate prin activarea nemediată de ER a genei luciferază sau prin activarea directă a produsului genei sau prin fluorescență nespecifică. Astfel de efecte sunt indicate printr-o curbă incomplete doză-răspuns incompletă sau neobișnuită. Dacă se suspectează existența unor astfel de efecte, ar trebui să se examineze efectul unui antagonist al ER (de exemplu, 4-hidroxitamoxifen (OHT) la o concentrație netoxică) asupra răspunsului. Este posibil ca antagonistul ICI 1827 80 pur să nu fie adecvat pentru acest scop, deoarece o concentrație suficientă de ICI 1827 80 ar putea diminua valoarea VC, iar acest lucru va afecta analiza datelor.
|
|
2.
|
Pentru a asigura validitatea acestei abordări, următoarele trebuie să fie testate pe aceeași placă:
|
—
|
Activitatea agonistă a substanței chimice necunoscute cu/fără 10 μM de OHT
|
|
—
|
1 nM de E2 (în trei probe) ca și agonist al PC
|
|
—
|
1 nM de E2 + OHT (în trei probe)
|
|
Criterii de interpretare a datelor
Notă: Toate godeurile ar trebui să fie tratate cu aceeași concentrație de vehicul.
|
—
|
Dacă activitatea agonistă a substanței chimice necunoscute NU este afectată de tratamentul cu antagonistul ER, aceasta este clasificată ca fiind „Negativă”.
|
|
—
|
Dacă activitatea agonistă a substanței chimice necunoscute este complet inhibată, se aplică criteriile de decizie.
|
|
—
|
Dacă activitatea agonistă la cea mai scăzută concentrație este egală cu sau mai mare decât răspunsul PC10, substanța chimică necunoscută este inhibată în valoare egală cu sau mai mare decât răspunsul PC10. Se calculează diferența dintre răspunsurile dintre godeurile netratate și cele tratate cu antagonistul ER, iar această diferență ar trebui să fie considerată ca fiind răspunsul real și ar trebui utilizată pentru calcularea parametrilor adecvați pentru a permite luarea unei decizii de clasificare.
|
Analiza datelor
Se verifică standardul de performanță.
Se verifică valorile VC dintre godeurile tratate în aceleași condiții.
|
1.
|
Se calculează media VC
|
|
2.
|
Se scade media VC din valoarea fiecărui godeu care nu este tratat cu OHT
|
|
3.
|
Se calculează media OHT
|
|
4.
|
Se scade media VC din valoarea fiecărui godeu care este tratat cu OHT
|
|
5.
|
Se calculează media PC
|
|
6.
|
Se calculează activitatea transcripțională relativă a tuturor celorlalte godeuri în raport cu PC.
|
Apendicele 2.2
PREPARAREA SERULUI TRATAT CU CĂRBUNE ACOPERIT CU DEXTRAN (DCC)
|
1.
|
Tratarea serului cu cărbune acoperit cu dextran (DCC) este o metodă generală de îndepărtare a compușilor estrogeni din serul care se adaugă în mediul celular, pentru a exclude un răspuns subiectiv asociat cu estrogeni reziduali din ser. un volum de 500 ml de ser fetal bovin (FBS) poate fi tratat prin această procedură.
|
Componente
|
2.
|
Vor fi necesare următoarele materiale și aparate:
|
Materiale
|
Clorură de magneziu hexahidrat (MgCl2·6H2O)
|
|
1 M soluție tampon HEPES (pH 7,4)
|
|
Apa ultrapură obținută printr-un sistem de filtrare
|
Aparatură
Un recipient din sticlă autoclavizat (dimensiunea ar trebui să fie adaptată, după caz) General Laboratory Centrifuge (care poate stabili temperatura la 4 °C)
Procedura
|
3.
|
Următoarea procedură este adaptată pentru utilizarea de eprubete pentru centrifugă de 50 ml:
[Ziua 1] Se prepară suspensia din cărbune acoperit cu dextran cu 1 l de apă ultrapură care conține 1,5 mM de MgCl2, 0,25 M zaharoză, 2,5 g de cărbune, 0,25 g de dextran și 5 mM de HEPES și se agită la 4 °C peste noapte.
[Ziua 2] Se golește suspensia într-o eprubetă pentru centrifugă de 50 ml și se centrifughează la 10 000 rpm la 4 °C timp de 10 minute. Se îndepărtează supranatantul și se păstrează jumătate din sedimentul cu cărbune la 4 °C pentru a fi utilizat în Ziua 3. Cealaltă jumătate din cantitatea de cărbune cu FBS care a fost decongelată încet pentru a se evita precipitarea și inactivată termic la 56 °C timp de 30 de minute se suspendă, apoi se transferă într-un recipient din sticlă autoclavizat, cum ar fi un balon Erlenmeyer. Se agită ușor această suspensie la 4 °C, peste noapte.
[Ziua 3] Se golește suspensia cu FBS în eprubete pentru centrifugă pentru centrifugare la 10 000 rpm la 4 °C timp de 10 minute. FBS este colectat și transferat în noul sediment de cărbune preparat și păstrat în Ziua 2. Se suspendă sedimentul cu cărbune și se agită ușor această suspensie într-un recipient din sticlă autoclavizat la 4 °C, peste noapte.
[Ziua 4] Se golește suspensia pentru centrifugare la 10 000 rpm la 4 °C timp de 10 minute și se sterilizează supernatantul prin filtrare printr-un filtru steril de 0,2 μm. Acest FBS tratat cu DCC ar trebui să fie păstrat la o temperatură de -20 °C și poate fi utilizat pe o perioadă de maxim un an.
|
Apendicele 3
ANALIZA DE TRANSACTIVARE A RECEPTORULUI DE ESTROGEN VM7LUC PENTRU IDENTIFICAREA AGONIșTILOR șI ANTAGONIșTILOR RECEPTORILOR DE ESTROGEN
CONSIDERAȚII ȘI LIMITĂRI INIȚIALE (A SE VEDEA, DE ASEMENEA, INTRODUCERE GENERALĂ)
|
1.
|
Această analiză utilizează linia celulară VM7Luc4E2 (65). Aceasta a fost validată de National Toxicology Program Interagency Center for the Evaluation of Alternative Toxicological Methods (NICAETM) și de Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM) (1). Liniile celulare VM7Luc exprimă în mod predominant o valoare ERα endogenă și o cantitate redusă de ERβ endogenă (2) (3) (4).
|
|
2.
|
Această analiză este aplicabilă pentru o gamă largă de substanțe, cu condiția ca acestea să poată fi dizolvate în dimetilsulfoxid (DMSO; CASRN 67-68-5), să nu reacționeze cu DMSO sau cu mediul de cultură a celulelor și să nu fie citotoxice la concentrațiile care sunt testate. Dacă nu este posibilă utilizarea DMSO, se poate utiliza un alt vehicul cum ar fi etanolul sau apa (a se vedea punctul 12). Performanța demonstrată a analizei activității agoniste (antagoniste) VM7Luc ER TA sugerează faptul că datele generate prin această analiză ar putea oferi informații cu privire la mecanismele de acțiune mediate de ER și ar putea fi luate în considerare la stabilirea priorității substanțelor prevăzute pentru teste suplimentare.
|
|
3.
|
Această analiză este concepută special pentru a detecta TA mediată de hERα și hERß prin măsurarea chemiluminiscenței ca punct final. Utilizarea chemiluminiscenței în bio-analize este larg răspândită, deoarece luminiscența are un raport semnal/fond ridicat. Însă activitatea luciferazei de licurici în analizele bazate pe celule poate fi confundată de substanțe care inhibă enzima luciferază, cauzând inhibarea aparentă sau luminiscența crescută ca urmare a stabilizării proteinelor. În plus, în unele analize pe gene reporter ER bazate pe luciferază au fost raportate semnale de luminescență nemediate de receptori la concentrații de fitoestrogen mai mari de 1 μM din cauza supraactivării genei luciferază reporter. În timp ce curba doză-răspuns indică faptul că o reală activare a sistemului ER se produce la concentrații mai mici, expresia luciferazei obținută la concentrații ridicate de fitoestrogeni sau compuși similari susceptibili de a produce o supra-activare a genei luciferază reporter similară fitoestrogenilor trebuie să fie atent examinată în cadrul sistemelor de testare ER TA transfectat stabil.
|
|
4.
|
Ar trebui să se parcurgă secțiunile „INTRODUCERE GENERALĂ” și „COMPONENTE ALE ANALIZEI ER TA” înainte de utilizarea acestei analize în scopuri de reglementare. Definițiile și abrevierile utilizate în prezenta metodă de testare sunt descrise în apendicele 1.
|
PRINCIPIILE TESTULUI (A SE VEDEA, DE ASEMENEA, INTRODUCERE GENERALĂ)
|
5.
|
Analiza este utilizată pentru a indica legarea ligand de ER, urmată de translocarea complexului receptor-ligand în nucleu. În nucleu, complexul receptor-ligand se leagă de aumite elemente de răspuns ADN și transactivează gena reporter (luc), determinând producerea luciferazei și emisia ulterioară de lumină, care poate fi cuantificată cu ajutorul unui luminometru. Activitatea luciferazei poate fi evaluată rapid și cu costuri reduse cu ajutorul unei serii de truse de testare disponibile în comerț. Analiza VM7Luc ER TA utilizează o linie celulară de adenocarcinom de sân umană reactivă, VM7, care a fost transfectată în mod stabil cu o structură de reporter luc de licurici sub controlul a patru elemente de răspuns estrogen așezate în amonte de promotorul virusului tumoral mamar la șoarece (MMTV) pentru a detecta substanțe cu activitate agonistă sau antagonistă ER in vitro. Acest promotor MMTV prezintă doar o reactivitate încrucișată minoră cu alți hormoni steroizi și nesteroizi (8). Criteriile pentru interpretarea datelor sunt descrise în detaliu la punctul 41. Pe scurt, un răspuns pozitiv este identificat prin curba concentrație-răspuns care conține cel puțin trei puncte cu bare de eroare care nu se suprapun (media ± SD), precum și o variație a amplitudinii (unitate de lumină relativă normalizată [RLU]) de cel puțin 20 % din valoarea maximă pentru standardul de referință (17β-estradiol [E2; CASRN 50-28-2] pentru analiza activității agoniste, raloxifen HCl [Ral; CASRN 84 449-90-1]/E2 pentru analiza activității antagoniste).
|
PROCEDURA
Linia celulară
|
6.
|
Pentru analiză ar trebui să se utilizeze linia celulară VM7Luc4E2 transfectată stabil. În prezent, linia celulară este disponibilă doar prin intermediul unui acord tehnic de licență de la Universitatea din California, Davis, California, SUA (66) și de la Xenobiotic Detection Systems Inc., Durham, North Carolina, USA (67).
|
Stabilitatea liniei celulare
|
7.
|
Pentru a menține stabilitatea și integritatea liniei celulare, celulele ar trebui să fie cultivate pentru mai multe pasaje din stocul congelat în mediu de păstrare a celulelor (a se vedea punctul 9). Celulele nu ar trebui să fie cultivate pentru mai mult de 30 de pasaje. Pentru linia celulară VM7Luc4E2, 30 de pasaje vor însemna o durată de aproximativ trei luni.
|
Cultura celulară și condiții de aranjare pe placă
|
8.
|
Ar trebui să fie urmate procedurile prevăzute în Orientarea privind bunele practici de cultură a celulelor (5) (6) pentru a asigura calitatea tuturor materialelor și metodelor pentru a menține integritatea, valabilitatea și reproductibilitatea oricărei activități desfășurate.
|
|
9.
|
Celulele VM7Luc4E2 sunt menținute în mediu RPMI 1 640, la care se adaugă 0,9 % Pen-Strep și 8,0 % ser fetal bovin (FBS) într-un incubator de cultură tisulară specific la temperatura de 37 °C ± 1°C, 90 % ± 5 % umiditate și 5,0 % ± 1 % CO2/aer.
|
|
10.
|
La atingerea unei confluențe de aproximativ 80 %, celulele VM7Luc4E2 sunt subcultivate și condiționate într-un mediu fără estrogen pentru o perioadă de 48 de ore înainte de introducerea celulelor în plăci cu 96 de godeuri pentru expunerea la substanțe chimice de testat și analiza inducției dependente de estrogen a activității luciferazei. Mediul fără estrogen (EFM) conține modificarea de către Dulbecco a mediului lui Eagle (DMEM) fără roșu de fenol, la care se adaugă soluție 4,5 % FBS tratat cu cărbune/dextran, 1,9 % L-glutamină și 0,9 % Pen-Strep. Toate articolele din plastic ar trebui să fie fără activitate estrogenă [a se vedea protocolul detaliat (7)].
|
Criterii de acceptabilitate
|
11.
|
Acceptarea sau respingerea unui test depinde de evaluarea rezultatelor standardelor de referință și ale martorilor din cadrul fiecărui experiment realizat pe o placă cu 96 de godeuri. Fiecare standard de referință este testat în concentrații multiple și există mai multe eșantioane din fiecare concentrație de referință și martor. Rezultatele sunt comparate cu valorile de control al calității (CC) pentru acești parametri care au fost derivați din bazele de date istorice de agoniști și antagoniști generate de fiecare laborator cu ocazia demonstrării competenței. Bazele de date istorice sunt actualizate permanent cu valori ale standardelor de referință și ale martorilor. Modificările legate de echipamentele sau condițiile de laborator ar putea necesita generarea de baze de date istorice actualizate.
|
Testarea activității agoniste
Testul pentru identificarea intervalului
|
•
|
Inducție: Inducția plăcii ar trebui să fie măsurată prin împărțirea valorii medii a celei mai mari unități de lumină relativă standard de referință (RLU) E2 la valoarea medie a RLU cu martor DMSO. De obicei, se obține o inducție multiplicată de cinci ori, dar, în scopul acceptării, inducția ar trebui să fie mai mare decât sau egală cu valoarea multiplicată de patru ori.
|
|
•
|
Rezultatele martorului DMSO: Valorile RLU ale martorului tratat cu solvent ar trebui să fie în limita de 2,5 ori abaterea standard a valorii medii istorice RLU a martorului tratat cu solvent.
|
|
•
|
Un experiment care nu îndeplinește niciunul dintre criteriile de acceptare ar trebui să fie eliminat și repetat.
|
Testul complex
Acesta include criterii de acceptabilitate din testul de identificare a intervalului de activitate agonistă și următoarele:
|
•
|
Rezultatele standardelor de referință: Curba standard de referință E2 concentrație-răspuns ar trebui să aibă o formă sigmoidală și cel puțin trei valori în porțiunea lineară a curbei concentrație-răspuns.
|
|
•
|
Rezultatele martorului pozitiv: Valorile RLU ale martorului metoxiclor ar trebui să fie peste media DMSO plus de trei ori abaterea standard de la media DMSO.
|
|
•
|
Un experiment care nu îndeplinește oricare dintre criteriile de acceptare ar trebui să fie eliminat și repetat.
|
Testul activității antagoniste
Testul pentru identificarea intervalului
|
•
|
Reducție: Reducția plăcii este măsurată prin împărțirea valorii medii RLU standard de referință Ral/E2 la valoarea medie a RLU cu martor DMSO. De obicei, se obține o reducție multiplicată de cinci ori, dar, în scopul acceptării, reducția ar trebui să fie mai mare decât sau egală cu valoarea multiplicată de trei ori.
|
|
•
|
Rezultatele martorului E2: Valorile RLU ale martorului E2 ar trebui să fie în limita de 2,5 ori abaterea standard a valorii medii istorice RLU a martorului E2.
|
|
•
|
Rezultatele martorului DMSO: Valorile RLU ale martorului DMSO ar trebui să fie în limita de 2,5 ori abaterea standard a valorii medii istorice RLU a martorului tratat cu solvent.
|
|
•
|
Un experiment care nu îndeplinește oricare dintre criteriile de acceptare va fi eliminat și repetat.
|
Testul complex
Acesta include criterii de acceptare din testul de identificare a intervalului de activitate antagonistă și următoarele:
|
•
|
Rezultatele standardelor de referință: Curba standard de referință Ral/E2 concentrație-răspuns ar trebui să aibă o formă sigmoidală și cel puțin trei valori în porțiunea lineară a curbei concentrație-răspuns.
|
|
•
|
Rezultatele martorului pozitiv: Valorile RLU ale martorului tamoxifen/E2 ar trebui să fie sub media martorului E2 minus de trei ori abaterea standard de la media martorului E2.
|
|
•
|
Un experiment care nu îndeplinește oricare dintre criteriile de acceptare va fi eliminat și repetat.
|
Standarde de referință, martori pozitivi și martori tratați cu vehicule
Martor tratat cu vehicul (analize ale activității agoniste și antagoniste)
|
12.
|
Vehiculul care se utilizează pentru a dizolva substanțele chimice de testat ar trebui să fie testat ca martor tratat cu vehicul. Vehiculul utilizat în timpul validării analizei VM7Luc ER TA a fost de 1 % (v/v) dimetilsulfoxid (DMSO, CASRN 67-68-5) (a se vedea punctul 24). Dacă se utilizează un alt vehicul decât DMSO, toate standardele de referință, martorii și substanțele chimice de testat ar trebui să fie testați în același vehicul, dacă este cazul.
|
Standard de referință (identificatorul intervalului pentru activitate agonistă)
|
13.
|
Standardul de referință este E2 (CASRN 50-28-2). Pentru testul de identificare a intervalului, standardul de referință constă într-o diluție în serie de patru concentrații de E2 (1,84 x 10-10, 4,59 x 10-11, 1,15 x 10-11 și 2,87 x 10-12 M), fiecare concentrație fiind testată în godeuri duplicate.
|
Standard de referință (testul complex pentru activitate agonistă)
|
14.
|
E2 pentru testare complexă cuprinde o diluție în serie de 1:2 constând în 11 concentrații (cuprinse între 3,67 x 10-10 și 3,59 x 10-13 M) de E2 în godeuri duplicate.
|
Standard de referință (identificatorul intervalului pentru activitate antagonistă)
|
15.
|
Standardul de referință reprezintă o combinație de Ral (CASRN 84 449-90-1) și E2 (CASRN 50-28-2). Ral/E2 pentru testul de identificare a intervalului cuprinde o diluție în serie din trei concentrații de Ral (3,06 x 10-9, 7,67 x 10-10 și 1,92 x 10-10M) plus o concentrație fixă (9,18 × 10-11 M) de E2 în godeuri duplicate.
|
Standard de referință (testul complex pentru activitate antagonistă)
|
16.
|
Ral/E2 pentru testul complex cuprinde o diluție în serie 1:2 de Ral (între 2,45x 10-8 și 9,57 x 10-11M) plus o concentrație fixă (9,18 × 10-11 M) de E2 care cuprinde nouă concentrații de Ral/E2 în godeuri duplicate.
|
Martor slab pozitiv (agonist)
|
17.
|
Martorul slab pozitiv este 9,06 x 10-6 M p,p’-metoxiclor (metoxiclor; CASRN 72-43-5) în EFM.
|
Martor slab pozitiv (antagonist)
|
18.
|
Martorul slab pozitiv constă în tamoxifen (CASRN 10 540-29-1) 3,36 x 10-6 M cu 9,18 × 10-11 M E2 în EFM.
|
Martorul E2 (doar analiza activității antagoniste)
|
19.
|
Martorul E2 este 9,18 × 10-11 M E2 din EFM și este utilizat ca martor negativ de referință.
|
Inducție multiplicată (agonist)
|
20.
|
Inducția activității luciferazei la standardul de referință (E2) se măsoară prin împărțirea mediei celei mai mari valori RLU a standardului de referință E2 la valoarea medie RLU a martorului DMSO, iar rezultatul ar trebui să fie mai mare decât valoarea multiplicată de patru ori.
|
Inducție multiplicată (antagonist)
|
21.
|
Activitatea medie a luciferazei la standardul de referință (Ral/E2) se măsoară prin împărțirea mediei celei mai mari valori RLU a standardului de referință Ral/E2 la valoarea medie RLU a martorului DMSO și ar trebui să fie mai mare decât valoarea multiplicată de trei ori.
Demonstrarea performanțelor laboratorului (a se vedea punctul 14 și tabelele 3 și 4 de la secțiunea « COMPONENTELE ANALIZEI ER TA» din prezenta metodă de testare).
|
Vehicul
|
22.
|
Substanțele chimice de testat ar trebui să se dizolve într-un solvent care solubilizează substanța chimică de testat și este miscibil cu mediul celular. Vehicule adecvate sunt apa, etanolul (puritate de 95-100 %) și DMSO. În cazul în care se utilizează DMSO, nivelul nu ar trebui să depășească 1 % (v/v). Pentru orice vehicul, ar trebui să se demonstreze că volumul maxim utilizat nu este citotoxic și nu interferează cu performanțele testului. Standardele de referință și martorii sunt dizolvați în solvent 100 %, apoi diluați în concentrații corespunzătoare în EFM.
|
Prepararea substanțelor chimice de testat
|
23.
|
Substanțele chimice de testat sunt dizolvate în DMSO 100 % (sau un solvent adecvat), apoi diluate în concentrații corespunzătoare în EFM. Toate substanțele chimice de testat ar trebui să fie lăsate să se echilibreze la temperatura camerei înainte de a fi dizolvate și diluate. Soluțiile cu substanțe chimice de testat ar trebui să fie proaspăt preparate pentru fiecare experiment. Soluțiile nu ar trebui să prezinte precipitat sau tulburare vizibilă. Stocurile de standarde de referință și de martori pot fi preparate în cantitate mare; însă, standardul de referință final, diluțiile cu martori și substanțele chimice de testat ar trebui să fie proaspăt preparate pentru fiecare experiment și utilizate în termen de 24 de ore de la preparare.
|
Solubilitate și citotoxicitate: Considerații pentru identificarea intervalului
|
24.
|
Testul de identificare a intervalului constă în diluții în serie de 1:10 în șapte puncte testate în duplicat. Inițial, substanțele chimice de testat sunt testate până la concentrația maximă de 1 mg/ml (~ 1 mM) pentru testarea activității agoniste și de 20 μg/ml (~10μM) pentru testarea activității antagoniste. Sunt utilizate experimente de identificare a intervalului pentru a determina următoarele:
|
|
—
|
Concentrațiile inițiale ale substanței chimice de testat care urmează să fie utilizate în timpul testării complexe
|
|
—
|
Diluțiile substanței chimice de testat (1:2 sau 1:5) care urmează să fie utilizate în timpul testării complexe
|
|
25.
|
O evaluare a viabilității/citotoxicității celulare este inclusă în protocoalele de analiză a activității agoniste și antagoniste (7) și este integrată în testul de identificare a intervalului și în testul complex. Metoda de citotoxicitate folosită pentru a evalua viabilitatea celulară în timpul validării VM7Luc ER TA (1) a fost o metodă de observare vizuală calitativă dimensionată; însă, se poate utiliza o metodă cantitativă pentru determinarea citotoxicității [a se vedea protocolul (7)]. Datele privind concentrațiile substanțelor chimice de testat care provoacă o reducere mai mare de 20 % a viabilității nu pot fi utilizate.
|
Expunerea la substanțe chimice de testat și organizarea plăcilor pentru testare
|
26.
|
Celulele sunt numărate și așezate pe plăcile de cultură tisulară cu 96 de godeuri (2 x 105 de celule pe godeu) în EFM și incubate timp de 24 de ore pentru a permite celulelor să se prindă de placă. EFM este eliminat și înlocuit cu substanțe chimice de testat și de referință în EFM și incubat timp de 19-24 de ore. Va fi necesar să se acorde o atenție specială substanțelor extrem de volatile, întrucât godeurile cu martori din apropiere ar putea genera rezultate fals pozitive. În astfel de cazuri, „agenții de etanșeizare a plăcii” ar putea ajuta la izolarea godeurilor individuale în timpul testării și, prin urmare, se recomandă.
|
Testele pentru identificarea intervalului
|
27.
|
În testul pentru identificarea intervalului sunt folosite toate cele 96 de godeuri de pe placă pentru a testa până la șase substanțe chimice de testat ca diluții de 1:10 în serie în șapte puncte în duplicat (a se vedea figurile 1 și 2).
|
|
—
|
Testul pentru identificarea intervalului agonistului folosește patru concentrații ale E2 în duplicat ca standard de referință și patru godeuri duplicate pentru martorul DMSO.
|
|
—
|
Testul pentru identificarea intervalului antagonistului folosește trei concentrații ale Ral/E2 cu 9,18 × 10-11 M E2 în duplicat ca standard de referință și trei godeuri duplicate pentru martorii E2 și DMSO.
|
Figura 1
Distribuția pe placa de 96 de godeuri în testul pentru identificarea intervalului agonistului

Abrevieri: E2-1-E2-4 = concentrațiile standardului de referință E2 (în ordine descrescătoare); TC1-1-TC1-7 = concentrațiile, în ordine descrescătoare, ale substanței chimice de testat 1 (TC1); TC2-1-TC2-7 = concentrațiile, în ordine descrescătoare, ale substanței chimice de testat 2 (TC2); TC3-1-TC3-7 = concentrațiile, în ordine descrescătoare, ale substanței chimice de testat 3 (TC3); TC4-1-TC4-7 = concentrațiile, în ordine descrescătoare, ale substanței chimice de testat 4 (TC4); TC5-1-TC5-7 = concentrațiile, în ordine descrescătoare, ale substanței chimice de testat 5 (TC5); TC6-1-TC6-7 = concentrațiile, în ordine descrescătoare, ale substanței chimice de testat 6 (TC6); VC = martor tratat cu vehicul (DMSO [1 % v/v EFM.]).
Figura 2
Distribuția pe placa de 96 de godeuri în testul pentru identificarea intervalului antagonistului

Abrevieri: E2 = martorul E2; Ral-1-Ral-3 = concentrațiile standardului de referință raloxifen/E2 (în ordine descrescătoare); TC1-1-TC1-7 = concentrațiile, în ordine descrescătoare, ale substanței chimice de testat 1 (TC1); TC2-1-TC2-7 = concentrațiile, în ordine descrescătoare, ale substanței chimice de testat 2 (TC2); TC3-1-TC3-7 = concentrațiile, în ordine descrescătoare, ale substanței chimice de testat 3 (TC3); TC4-1-TC4-7 = concentrațiile, în ordine descrescătoare, ale substanței chimice de testat 4 (TC4); TC5-1-TC5-7 = concentrațiile, în ordine descrescătoare, ale substanței chimice de testat 5 (TC5); TC6-1-TC6-7 = concentrațiile, în ordine descrescătoare, ale substanței chimice de testat 6 (TC6); VC = martor tratat cu vehicul (DMSO [1 % v/v EFM.]).
Notă: Toate substanțele chimice de testat sunt testate în prezența M E2 9,18 × 10-11.
|
28.
|
Volumul final recomandat de medii necesar pentru fiecare godeu este de 200 μl. Se utilizează numai plăci de testare în care celulele din toate godeurile prezintă o viabilitate de cel puțin 80 %.
|
|
29.
|
Determinarea concentrațiilor inițiale pentru testul complex al
agonistului
este descrisă în detaliu în protocolul privind agonistul (7). Pe scurt, se utilizează următoarele criterii:
|
|
—
|
Dacă nu există puncte pe curba concentrațiilor substanței chimice de testat care sunt mai mari decât media plus de trei ori abaterea standard a martorului DMSO, va fi efectuat un test complex utilizând o diluție în serie 1:2 în 11 puncte pornind de la concentrația maximă solubilă.
|
|
—
|
Dacă există puncte pe curba concentrațiilor substanței chimice de testat care sunt mai mari decât media plus de trei ori abaterea standard a martorului DMSO, concentrația inițială care trebuie utilizată pentru sistemul de diluare în 11 puncte, în cadrul testului complex, ar trebui să fie cu un log mai mare decât concentrația care indică cea mai mare valoare RLU ajustată în testul de identificare a intervalului. Sistemul de diluare în 11 puncte se va baza pe diluția 1:2 sau 1:5 în conformitate cu următoarele criterii:
|
Ar trebui să fie utilizată o diluție de 1:2 în serie în 11 puncte dacă intervalul de concentrații rezultant va cuprinde întreaga serie de răspunsuri pe baza curbei concentrație-răspuns generate în cadrul testului de identificare a intervalului. În caz contrar, se utilizează o diluție de 1:5.
|
|
|
—
|
În cazul în care o substanță chimică de testat prezintă o curbă concentrație-răspuns bifazică în cadrul testului de identificare a intervalului, ambele faze ar trebui să fie abordate, de asemenea, în cadrul testului complex.
|
|
30.
|
Determinarea concentrațiilor inițiale pentru testul complex al
antagonistului
este descrisă în detaliu în protocolul privind antagonistul (7). Pe scurt, se utilizează următoarele criterii:
|
|
—
|
Dacă nu există puncte pe curba concentrațiilor substanței chimice de testat care sunt mai mici decât media minus de trei ori abaterea standard a martorului E2, va fi efectuat un test complex utilizând o diluție în serie 1:2 în 11 puncte pornind de la concentrația maximă solubilă.
|
|
—
|
Dacă există puncte pe curba concentrațiilor substanței chimice de testat care sunt mai mici decât media minus de trei ori abaterea standard a martorului E2, concentrația inițială care trebuie utilizată pentru sistemul de diluare în 11 puncte, în cadrul testului complex, ar trebui să fie una dintre următoarele:
|
—
|
concentrația care prezintă cea mai mică valoare RLU ajustată în cadrul testului de stabilire a intervalului
|
|
—
|
concentrația maximă solubilă [a se vedea Protocolul privind antagonistul (7), figura 14-2]
|
|
—
|
concentrația citotoxică minimă [a se vedea Protocolul privind antagonistul (7), figura 14-3 pentru un exemplu aferent].
|
|
|
—
|
Sistemul de diluare în 11 puncte se va baza pe o diluție 1:2 sau 1:5 în serie sau pe o diluție care respectă următoarele criterii:
|
Ar trebui să fie utilizată o diluție de 1:2 în serie în 11 puncte dacă intervalul de concentrații rezultant va cuprinde întreaga serie de răspunsuri pe baza curbei concentrație-răspuns generate în cadrul testului de identificare a intervalului. În caz contrar, ar trebui să se utilizeze o diluție de 1:5.
|
|
Teste complexe
|
31.
|
Testul complex constă în diluții în serie în 11 puncte (diluții în serie de 1:2 sau 1:5 pe baza concentrației inițiale pentru criteriile de testare complexă), fiecare concentrație fiind testată în trei godeuri de pe placa cu 96 de godeuri (a se vedea figurile 3 și 4).
|
|
—
|
Testul complex privind agonistul utilizează 11 concentrații de E2 în duplicat ca standard de referință. Pe fiecare placă sunt incluse patru godeuri replicate pentru martorul DMSO și patru godeuri replicate pentru martorul metoxiclor (9,06 x 10-6 M).
|
|
—
|
Testul complex al antagonistului utilizează nouă concentrații de Ral/E2 cu 9,18 × 10-11 M E2 în duplicat ca standard de referință, cu patru godeuri replicate pentru martorul E2 9,18 x 10-11 M, patru godeuri replicate pentru martorii DMSO și patru godeuri replicate pentru tamoxifen 3,36 x 10-6M.
|
Figura 3
Distribuția pe placa de 96 de godeuri în testul complex al agonistului
Abrevieri: TC1-1 - TC1-11 = concentrațiile, în ordine descrescătoare, ale substanței chimice de testat 1; TC2-1-TC2-11 = concentrațiile, în ordine descrescătoare, ale substanței chimice de testat 2; E2-1-E2-11 = concentrațiile standardului de referință E2 (în ordine descrescătoare); Meth = martor slab pozitiv metoxiclor p,p’; VC = martor DMSO tratat cu vehicul EFM (1 % v/v)
Figura 4
Distribuția pe placa de 96 de godeuri în testul complex al antagonistului

Abrevieri: E2 = martorul E2; Ral-1-Ral-9 = concentrațiile standardului de referință raloxifen/E2 (în ordine descrescătoare); Tam = Martor slab pozitiv tamoxifen/E2; TC1-1-TC1-11 = concentrațiile, în ordine descrescătoare, ale substanței chimice de testat 1 (TC1); TC2-1-TC2-11 = concentrațiile, în ordine descrescătoare, ale substanței chimice de testat 2 (TC2); VC = martor tratat cu vehicul (DMSO [1 % v/v EFM.]).
Notă: După cum s-a menționat, toate godeurile de referință și de testare conțin o concentrație fixă de E2 (9,18 x 10-11M).
|
32.
|
Ar trebui să se efectueze teste complexe repetate pentru aceeași substanță chimică în zile diferite, pentru asigurarea independenței. Ar trebui să se efectueze cel puțin două teste complexe. Dacă rezultatele testelor sunt contradictorii (de exemplu, un test este pozitiv, celălalt negativ) sau dacă unul dintre teste este inadecvat, ar trebui efectuat un al treilea test suplimentar.
|
Măsurarea luminiscenței
|
33.
|
Luminiscența este măsurată în intervalul 300-650 nm, cu ajutorul unui luminometru cu injectare și al unui software care controlează volumul de injecție și intervalul de măsurare (7). Emisia de lumină din fiecare godeu este exprimată în RLU pe godeu.
|
ANALIZA DATELOR
Determinarea EC50 /IC50
|
34.
|
Valoarea EC50 (jumătate din concentrația maximă efectivă a unei substanțe chimice de testat [agoniști)] și valoarea IC50 (jumătate din concentrația de inhibare maximă a unei substanțe chimice de testat [antagoniști]) sunt determinate din datele concentrație-răspuns. Pentru substanțele chimice de testat care sunt pozitive la una sau mai multe concentrații, concentrația substanței chimice de testat care provoacă un răspuns semi-maximal (IC50 sau EC50) se calculează utilizând o analiză a funcției Hill sau o alternativă adecvată. Funcția Hill este un model matematic logistic cu patru parametri care raportează concentrația substanței chimice de testat la răspuns (de regulă, pe baza unei curbe sigmoidale) cu ajutorul ecuației de mai jos:
|

Unde:
Y= răspuns (și anume, valori RLU);
X= logaritmul concentrației;
Nivel inferior= răspunsul minim;
Nivel superior= răspunsul maxim;
lg EC50 (sau lg IC50)= logaritmul X ca mediana răspunsului între partea superioară și cea inferioară;
Hillslope= caracterul abrupt al curbei.
Modelul calculează cea mai bună încadrare pentru nivelul superior, nivelul inferior, Hillslope și parametrii IC50 și EC50. Pentru calcularea valorilor EC50 și IC50, ar trebui să se utilizeze un software statistic adecvat (de exemplu, software-ul statistic Graphpad PrismR).
Determinarea valorilor neconforme
|
35.
|
Un bun raționament statistic ar putea fi facilitat prin includerea, printre altele, a testului Q (a se vedea protocoalele privind agonistul și antagonistul (7) pentru determinarea godeurilor „inutilizabile” care vor fi excluse din analiza datelor.
|
|
36.
|
Pentru replicile standardului de referință E2 (mărime eșantion la două replici), orice valoare RLU ajustată pentru o replică la o anumită concentrație de E2 este considerată a fi o valoare neconformă dacă valoarea acesteia este peste sau sub 20 % din valoarea RLU ajustată pentru concentrația respectivă în baza de date istorice.
|
Colectarea și adaptarea datelor luminometrului pentru testul de stabilire a intervalului
|
37.
|
Datele brute obținute din luminometru ar trebui să fie transferate pe o fișă model prevăzută pentru analiză. Ar trebui să se stabilească dacă există puncte de date neconforme care ar trebui eliminate. (a se vedea criteriile de acceptare a testului pentru parametrii determinați în cadrul analizelor.) Ar trebui să se efectueze următoarele calcule:
|
Agonistul
|
Etapa 1
|
Se calculează valoarea medie pentru martorul cu vehicul DMSO (VC).
|
|
Etapa 2
|
Se scade valoarea medie a VC DMSO din fiecare valoare a godeului pentru a normaliza datele.
|
|
Etapa 3
|
Se calculează inducția multiplicată medie pentru standardul de referință (E2).
|
|
Etapa 4
|
Se calculează media valorii EC50 pentru substanțele chimice de testat.
|
Antagonistul
|
Etapa 1
|
Se calculează valoarea medie pentru VC DMSO.
|
|
Etapa 2
|
Se scade valoarea medie a VC DMSO din fiecare valoare a godeului pentru a normaliza datele.
|
|
Etapa 3
|
Se calculează reducția multiplicată medie pentru standardul de referință (Ral/E2).
|
|
Etapa 4
|
Se calculează valoarea medie a standardului de referință E2.
|
|
Etapa 5
|
Se calculează media valorii IC50 pentru substanțele chimice de testat.
|
Colectarea și adaptarea datelor luminometrului pentru testul complex
|
38.
|
Datele brute obținute din luminometru ar trebui să fie transferate pe o fișă model prevăzută pentru analiză. Ar trebui să se stabilească dacă există puncte de date neconforme care ar trebui eliminate. (a se vedea criteriile de acceptare a testului pentru parametrii determinați în cadrul analizelor.) Sunt efectuate următoarele calcule:
|
Agonistul
|
Etapa 1
|
Se calculează valoarea medie pentru VC DMSO.
|
|
Etapa 2
|
Se scade valoarea medie a VC DMSO din fiecare valoare a godeului pentru a normaliza datele.
|
|
Etapa 3
|
Se calculează inducția multiplicată medie pentru standardul de referință (E2).
|
|
Etapa 4
|
Se calculează media valorii EC50 pentru E2 și substanțele chimice de testat.
|
|
Etapa 5
|
Se calculează media valorii ajustate RLU pentru metoxiclor.
|
Antagonistul
|
Etapa 1
|
Se calculează valoarea medie pentru VC DMSO.
|
|
Etapa 2
|
Se scade valoarea medie a VC DMSO din fiecare valoare a godeului pentru a normaliza datele.
|
|
Etapa 3
|
Se calculează inducția multiplicată medie pentru standardul de referință (Ral/E2).
|
|
Etapa 4
|
Se calculează media valorii IC50 pentru Ral/E2 și substanțele chimice de testat.
|
|
Etapa 5
|
Se calculează media valorii ajustate RLU pentru tamoxifen.
|
|
Etapa 6
|
Se calculează valoarea medie a standardului de referință E2.
|
Criterii de interpretare a datelor
|
39.
|
Testul VM7Luc ER TA este prevăzut în cadrul abordării bazate pe dovezi, pentru a contribui la stabilirea priorității substanțelor la testarea ED in vivo. Din această procedură de stabilire a priorităților va face parte clasificarea substanței chimice de testat ca fiind pozitivă sau negativă pentru activitatea agonistă sau antagonistă a ER. Criteriile de decizie pozitivă și negativă utilizate în studiul de validare VM7Luc ER AT sunt descrise în tabelul 1.
|
Tabelul 1
Criterii de decizie pozitivă și negativă
|
|
|
ACTIVITATEA AGONISTĂ
|
|
Pozitivă
|
|
—
|
Toate substanțele chimice de testat clasificate ca fiind pozitive pentru activitatea agonistă a ER ar trebui să prezinte o curbă concentrație-răspuns care să cuprindă o bază de referință, să urmeze o pantă pozitivă și să culmineze cu un platou sau un vârf. În unele cazuri, pot fi definite numai două dintre aceste caracteristici (bază de referință-pantă sau pantă-vârf).
|
|
—
|
Linia care definește panta pozitivă ar trebui să conțină cel puțin trei puncte cu bare de eroare care nu se suprapun (media ± SD). Sunt excluse punctele care alcătuiesc baza de referință, dar porțiunea liniară a curbei poate include vârful sau primul punct al platoului.
|
|
—
|
O clasificare pozitivă necesită o amplitudine a răspunsului, diferența dintre baza de referință și vârf de cel puțin 20 % din valoarea maximă a standardului de referință E2 [și anume, 2 000 RLU sau mai mult atunci când valoarea maximă de răspuns a standardelor de referință (E2) este ajustată la 10 000 RLU].
|
|
—
|
Dacă este posibil, ar trebui să se calculeze o valoare EC50 pentru fiecare substanță chimică de testat pozitivă.
|
|
|
Negativă
|
Valoarea medie ajustată a RLU pentru o concentrație dată este egală cu sau sub valoarea medie RLU a DMSO plus de trei ori abaterea standard a RLU DMSO.
|
|
Inadecvată
|
Datele care nu pot fi interpretate ca fiind valabile pentru a demonstra prezența sau absența activității din cauza limitărilor calitative sau cantitative majore sunt considerate a fi inadecvate și nu pot fi utilizate pentru a determina dacă substanța chimică de testat este pozitivă sau negativă. Substanțele chimice ar trebui să fie testate din nou.
|
|
|
|
ACTIVITATEA ANTAGONISTĂ
|
|
Pozitivă
|
|
—
|
Datele despre substanțele chimice de testat generează o curbă concentrație-răspuns care constă într-o bază de referință, urmată de o pantă negativă.
|
|
—
|
Linia care definește panta negativă ar trebui să conțină cel puțin trei puncte cu bare de eroare care nu se suprapun; sunt excluse punctele care alcătuiesc baza de referință, dar porțiunea liniară a curbei poate include primul punct al platoului.
|
|
—
|
Ar trebui să existe o reducere de cel puțin 20 % a activității de la valoarea maximă pentru standardul de referință Ral/E2 [și anume, cel mult 8 000 RLU atunci când valoarea maximă de răspuns a standardului de referință (Ral/E2) este ajustată la 10 000 RLU].
|
|
—
|
Cele mai mari concentrații necitotoxice ale substanței chimice de testat ar trebui să fie mai mici decât sau egale cu 1 x 10-5 M.
|
|
—
|
Dacă este posibil, ar trebui să se calculeze o valoare IC50 pentru fiecare substanță chimică de testat pozitivă.
|
|
|
Negativă
|
Toate punctele de date sunt peste valoarea EC80 (80 % din răspunsul E2 sau 8 000 RLU) la concentrații sub 1,0 x 10-5 M.
|
|
Inadecvată
|
Datele care nu pot fi interpretate ca fiind valabile pentru a demonstra prezența sau absența activității din cauza limitărilor calitative sau cantitative majore sunt considerate a fi inadecvate și nu pot fi utilizate pentru a determina dacă substanța chimică de testat este pozitivă sau negativă. Substanța chimică ar trebui să fie testată din nou.
|
|
40.
|
Rezultatele pozitive vor fi caracterizate atât prin amploarea efectului, cât și prin concentrația la care se produc efecte, dacă este posibil. Exemple de date pozitive, negative și inadecvate sunt prezentate în figurile 5 și 6.
|
Figura 5
Exemple de agoniști: Date pozitive, negative și inadecvate

Linia întreruptă indică 20 % din răspunsul E2, valoarea de 2 000 RLU ajustată și normalizată.
Figura 6
Exemple de antagonist: Date pozitive, negative și inadecvate
Linia întreruptă indică 80 % din răspunsul Ral/E2, valoarea de 8 000 RLU ajustată și normalizată.
O linie continuă indică 1,00 x 10-5 M. Pentru ca un răspuns să fie considerat pozitiv, acesta ar trebui să se situeze sub valoarea de 8 000 RLU și la concentrații mai mici de 1,00 x 10-5M.
Concentrațiile marcate cu asterisc în graficul pentru mezo-hexestrol indică punctaje de viabilitate de „2” sau mai mari.
Rezultatele testelor pentru mezo-hexestrol sunt considerate date inadecvate deoarece singurul răspuns care se situează sub 8 000 RLU se produce la 1,00 x 10-5M.
|
41.
|
Calculele EC50 și IC50 pot fi efectuate utilizând o funcție Hill cu patru parametri [a se vedea Protocolul privind agonistul și protocolul privind antagonistul pentru mai multe detalii (7)]. Îndeplinirea criteriilor de acceptabilitate indică faptul că sistemul funcționează corect, dar nu asigură faptul că orice testare specifică va genera date precise. Duplicarea rezultatelor primei testări reprezintă cea mai bună asigurare a faptului că au fost generate date precise (a se vedea punctul 19 din „COMPONENTELE ANALIZEI ER TA”).
|
RAPORTUL TESTULUI
|
42.
|
A se vedea punctul 20 din „COMPONENTELE ANALIZEI ER TA”.
|
REFERINȚE BIBLIOGRAFICE
|
(1)
|
ICCVAM. (2011). ICCVAM Test Method Evaluation Report on the LUMI-CELL® ER (BG1Luc ER TA) Test Method: An In Vitro Method for Identifying ER Agonists and Antagonists, National Institute of Environmental Health Sciences: Research Triangle Park, NC.
|
|
(2)
|
Monje P., Boland R. (2001). Subcellular Distribution of Native Estrogen Receptor α and β Isoforms in Rabbit Uterus and Ovary, J. Cell Biochem., 82(3): 467-479.
|
|
(3)
|
Pujol P., et al. (1998). Differential Expression of Estrogen Receptor-Alpha and -Beta Messenger RNAs as a Potential Marker of Ovarian Carcinogenesis, Cancer Res., 58(23): 5 367-5 373.
|
|
(4)
|
Weihua Z., et al. (2000). Estrogen Receptor (ER) β, a Modulator of ERα in the Uterus, Proceedings of the National Academy of Sciences of the United States of America 97(11): 936-5 941.
|
|
(5)
|
Balls M., et al. (2006). The Importance of Good Cell Culture Practice (GCCP), ALTEX, 23(Suppl): p. 270-273.
|
|
(6)
|
Coecke S., et al. (2005). Guidance on Good Cell Culture Practice: a Report of the Second ECVAM Task Force on Good Cell Culture Practice, Alternatives to Laboratory Animals, 33: p. 261-287.
|
|
(7)
|
ICCVAM (2011). ICCVAM Test Method Evaluation Report, The LUMI-CELL® ER (BG1Luc ER TA) Test Method: An In Vitro Assay for Identifying Human Estrogen Receptor Agonist and Antagonist Activity of Chemicals, NIH Publication nr. 11-7 850.
|
|
(8)
|
Rogers J.M., Denison M.S. (2000). Recombinant Cell Bioassays for Endocrine Disruptors: Development of a Stably Transfected Human Ovarian Cell Line for the Detection of Estrogenic and Anti-Estrogenic Chemicals, In Vitro Mol. Toxicol., 13(1):67-82.
|
|
(9)
|
Escande A., et al. (2006). Evaluation of Ligand Selectivity Using Reporter Cell Lines Stably Expressing Estrogen Receptor Alpha or Beta, Biochem. Pharmacol., 71(10):1 459-69.
|
|
(10)
|
Thorne N., Inglese J., Auld D.S. (2010). Illuminating Insights into Firefly Luciferase and Other Bioluminescent Reporters Used in Chemical Biology, Chemistry and Biology, 17(6):646-57.
|
|
(11)
|
Kuiper G.G, et al. (1998). Interaction of Estrogenic Chemicals and Phytoestrogens with Estrogen Receptor Beta, Endocrinology, 139(10):4 252-63.
|
|
(12)
|
Geisinger, et al. (1989). Characterization of a human ovarian carcinoma cell line with estrogen and progesterone receptors, Cancer 63, 280-288.
|
|
(13)
|
Baldwin, et al. (1998). BG-1 ovarian cell line: an alternative model for examining estrogen-dependent growth in vitro, In Vitro Cell. Dev. Biol. – Animal, 34, 649-654.
|
|
(14)
|
Li, Y., et al. (2014). Research resource: STR DNA profile and gene expression comparisons of human BG-1 cells and a BG-1/MCF-7 clonal variant, Mol. Endo. 28, 2072-2081.
|
|
(15)
|
Rogers, J.M. și Denison, M.S. (2000). Recombinant cell bioassays for endocrine disruptors:development of a stably transfected human ovarian cell line for the detection of estrogenicand anti-estrogenic chemicals, In Vitro & Molec. Toxicol. 13, 67-82.
|
Apendicele 4
TESTUL DE TRANSACTIVARE A RECEPTORULUI Α DE ESTROGEN UMAN TRANSFECTAT STABIL PENTRU DETECTAREA ACTIVITĂȚII AGONISTE ȘI ANTAGONISTE A SUBSTANȚELOR CHIMICE CARE UTILIZEAZĂ LINIA CELULARĂ ERΑ CALUX
CONSIDERAȚII ȘI LIMITĂRI INIȚIALE (A SE VEDEA, DE ASEMENEA, INTRODUCERE GENERALĂ)
|
1.
|
Analiza de transactivare hERα CALUX utilizează linia celulară umană U2OS pentru a detecta activitatea agonistă și antagonistă estrogenă mediată prin receptorul alfa de estrogen uman (hERα). Studiul de validare a bioanalizei cu transfectare stabilă a ERα CALUX al BioDetection Systems BV (Amsterdam, Țările de Jos) a demonstrat relevanța și fiabilitatea analizei pentru scopul său prevăzut (1). Linia celulară ERα CALUX exprimă doar ERα uman transfectat stabil (2) (3).
|
|
2.
|
Această analiză este concepută special pentru a detecta transactivarea mediată de hERα prin măsurarea bioluminiscenței ca punct final. Bioluminiscența este utilizată în mod frecvent în bioanalize datorită ratei semnal-zgomot ridicate (4).
|
|
3.
|
Au fost raportate concentrații de fitoestrogen mai mari de 1 μM pentru supra-activarea genei luciferază reporter, determinând o luminescență nemediată de receptor (5) (6) (7). Prin urmare, trebuie să se examineze cu atenție concentrații mai mari de fitoestrogeni sau alți compuși similari care pot să supra-activeze expresia luciferazei în cadrul analizelor de transactivare a ER transfectat stabil (a se vedea apendicele 2).
|
|
4.
|
Ar trebui să se parcurgă secțiunile „INTRODUCERE GENERALĂ” și „COMPONENTE ALE ANALIZEI ER TA” înainte de utilizarea acestei analize în scopuri de reglementare. Definițiile și abrevierile utilizate în prezenta metodă de testare sunt descrise în apendicele 1.
|
PRINCIPIILE TESTULUI (A SE VEDEA, DE ASEMENEA, INTRODUCERE GENERALĂ)
|
5.
|
Bioanaliza este utilizată pentru a indica legarea ligand de ER, urmată de translocarea complexului receptor-ligand în nucleu. În nucleu, complexul receptor-ligand leagă anumite elemente de răspuns ADN și transactivează o genă luciferază reporter de licurici, ceea ce determină o expresie celulară crescută a enzimei luciferază. Ca urmare a adaosului de luciferină ca substrat pentru luciferază, luciferina se transformă într-un produs bioluminiscent. Lumina produsă poate fi detectată și cuantificată cu ușurință cu ajutorul unui luminometru.
|
|
6.
|
Sistemul de testare utilizează celule ERα CALUX transfectate stabil. Celulele ERα CALUX au provenit de la linia celulară umană de osteosarcom osteoblastic U2OS. Celulele U2OS umane au fost transfectate stabil cu 3 x HRE-TATA-Luc și pSG5-neo-hERα, utilizând metoda coprecipitării fosfatului de calciu. Linia celulară U2OS a fost selectată drept cel mai bun candidat pentru a servi ca linie celulară reporter reactivă la estrogen (și alți hormoni steroizi), pe baza observației conform căreia linia celulară U2OS a indicat o activitate redusă a receptorului endogen sau nu a manifestat nicio astfel de activitate. Absența receptorilor endogeni a fost evaluată folosind exclusiv plasmide reporter pentru luciferază, nefiind indicată nicio activitate atunci când au fost adăugați liganzi de receptor. De asemenea, această linie celulară a susținut răspunsuri puternice mediate de hormoni atunci când au fost introduși în mod tranzitoriu receptori conecși (2) (3) (8).
|
|
7.
|
Substanțele chimice de testat pentru activitatea estrogenă sau anti-estrogenă, care utilizează linia celulară ERα CALUX, includ un test de depistare prealabilă și teste complexe. În timpul testului de depistare prealabilă, se determină solubilitatea, citotoxicitatea și un interval ajustat de concentrații ale substanțelor chimice de testat pentru testul complex. În timpul testelor complexe, intervalele de concentrații ajustate ale substanțelor chimice de testat sunt testate în cadrul bioanalizelor ERα CALUX, urmate de clasificarea substanțelor chimice de testat pentru agonism sau antagonism.
|
|
8.
|
Criteriile pentru interpretarea datelor sunt descrise în detaliu la punctul 59. Pe scurt, o substanță chimică de testat este considerată pozitivă pentru agonism în cazul în care cel puțin două concentrații consecutive ale substanței chimice de testat indică un răspuns egal cu sau mai mare de 10 % din răspunsul maxim al standardului de referință 17β-estradiol (PC10). O substanță chimică de testat este considerată pozitivă pentru antagonism în cazul în care cel puțin două concentrații consecutive ale substanței chimice de testat indică un răspuns egal cu sau mai mic de 80 % din răspunsul maxim al standardului de referință tamoxifen (PC80).
|
PROCEDURA
Linii celulare
|
9.
|
Pentru analiză ar trebui să se utilizeze linia celulară ERα CALUX U2OS transfectată stabil. Linia celulară poate fi obținută de la BioDetection Systems BV, Amsterdam, Țările de Jos, împreună cu un acord tehnic de licență.
|
|
10.
|
Ar trebui să se utilizeze numai culturi de celule fără micoplasme. Loturile de celule utilizate ar trebui să fie certificate ca fiind negative pentru contaminare cu micoplasmă sau ar trebui să se efectueze un test pentru micoplasme înainte de utilizare. Ar trebui să se recurgă la RT-PCR (reacție de polimerizare în lanț în timp real) pentru detectarea sensibilă a infecției cu micoplasme (9).
|
Stabilitatea liniei celulare
|
11.
|
Pentru a menține stabilitatea și integritatea celulelor CALUX, celulele ar trebui să fie păstrate în azot lichid (-800C). După decongelarea celulelor pentru începerea unei noi culturi, celulele ar trebui să fie subcultivate cel puțin de două ori înainte de a fi utilizate pentru evaluarea activității agoniste și antagoniste estrogene a substanțelor chimice. Celulele nu ar trebui să fie subcultivate pentru mai mult de 30 de pasaje.
|
|
12.
|
Pentru a monitoriza stabilitatea liniei celulare în timp, capacitatea de răspuns a sistemului de testare agonistă și antagonistă ar trebui verificată prin evaluarea EC50 sau IC50 a standardului de referință. În plus, ar trebui să fie monitorizată inducția relativă a eșantionului de martor pozitiv (MP) și a eșantionului de martor negativ (MN). Rezultatele ar trebui să fie în concordanță cu criteriile de acceptare pentru bioanaliza agonistă (tabelul 3C) sau antagonistă ERα CATLUX (tabelul 4C). Standardele de referință, martorii pozitivi și negativi sunt prezentați în tabelele 1 și 2 pentru modul agonist și, respectiv, antagonist.
|
Cultura celulară și condiții de aranjare pe placă
|
13.
|
Celulele U2OS ar trebui să fie cultivate în mediu de creștere [DMEM/F12 (1:1) cu roșu de fenol ca indicator al pH-ului, completat cu ser fetal bovin (7,5 %), aminoacizi neesențiale (1 %), 10 unități/ml de penicilină, streptomicină și geneticină (G-418) ca marker de selecție]. Celulele ar trebui să fie introduse într-un incubator cu CO2 (5 % CO2) la 370C și umiditate de 100 %. Atunci când celulele ating o confluență de 85-95 %, acestea ar trebui să fie subcultivate sau pregătite pentru însămânțare în plăci de microtitrare cu 96 de godeuri. În cazul acestora din urmă, celulele ar trebui să fie resuspendate într-o cantitate de 1 x 105 celule/ml în mediu de testare fără estrogen [mediu fără roșu de fenol DMEM/F12 (1:1), la care se adaugă un ser fetal bovin tratat cu dextran și cărbune acoperit (5 % v/v), aminoacizi neesențiali (1 % v/v), 10 unități/ml de penicilină și streptomicină] și introduse în plăcile de microtitrare de 96 de godeuri fiecare (100 μl de suspensie celulară omogenizată). Celulele ar trebui să fie incubate în prealabil într-un incubator cu CO2 (5 % CO2, 370C, umiditate de 100 %) timp de 24 de ore înainte de expunere. Articolele din plastic ar trebui să nu conțină estrogen.
|
Criterii de acceptabilitate
|
14.
|
Activitatea agonistă și cea antagonistă a substanței (substanțelor) chimice de testat sunt testate în cadrul unei serii de teste. O serie de teste cuprinde maxim șase plăci de microtitrare. Fiecare serie de teste conține cel puțin o serie completă de diluții ale unui standard de referință, un eșantion de martor pozitiv, un eșantion de martor negativ și martori tratați cu solvent. Figurile 1 și 2 prezintă configurația plăcii pentru seriile de teste agoniste și antagoniste.
|
|
15.
|
Fiecare diluție a standardelor de referință, a substanțelor chimice de testat, toți martorii tratați cu solvent, precum și martorii pozitivi și negativi ar trebui să fie analizați în trei replici. Fiecare analiză a datelor efectuată în trei replici ar trebui să îndeplinească cerințele prevăzute în tabelele 3A și 4A.
|
|
16.
|
Se măsoară o serie completă de diluții ale standardului de referință (17β-estradiol pentru agonism; tamoxifen pentru antagonism) pe prima placă în cadrul fiecărei serii de teste. Pentru a putea compara rezultatele analizelor aferente celor cinci plăci de microtitrare rămase cu prima placă de microtitrare care conține curba concentrație-răspuns completă a standardului de referință, toate plăcile ar trebui să conțină trei eșantioane de martori: martor tratat cu solvent, cea mai mare concentrație a standardului de referință testat și concentrația aproximativă EC50 (agonism) sau IC50 (antagonism) a standardului de referință. Raportul dintre valorile medii ale eșantioanelor de martori de pe prima placă și cele cinci plăci rămase ar trebui să îndeplinească cerințele indicate în tabelul 3C (agonism) sau în tabelul 4C (antagonism).
|
|
17.
|
Pentru fiecare dintre plăcile de microtitrare din cadrul unei serii de teste, se calculează factorul z (10). Factorul z ar trebui să fie calculat utilizând răspunsurile la concentrația cea mai mare și cea mai mică a standardului de referință. O placă de microtitrare este considerată valabilă în cazul în care îndeplinește cerințele menționate în tabelul 3C (agonism) sau în tabelul 4C (antagonism).
|
|
18.
|
Standardul de referință ar trebui să demonstreze o curbă doză-răspuns sigmoidală. Valoarea EC50 sau IC50 derivată din răspunsul seriei de diluții a standardului de referință ar trebui să îndeplinească cerințele indicate în tabelul 3C (agonism) sau în tabelul 4C (antagonism).
|
|
19.
|
Fiecare serie de teste ar trebui să conțină un eșantion de martor pozitiv și un eșantion de martor negativ. Inducția relativă calculată a eșantionului de martor pozitiv și a celui de martor negativ ar trebui să îndeplinească cerințele indicate în tabelul 3C (agonism) sau în tabelul 4C (antagonism).
|
|
20.
|
În timpul tuturor măsurătorilor, factorul de inducție al celei mai mari concentrații a standardului de referință ar trebui să fie măsurat prin împărțirea mediei celor mai mari valori ale răspunsului ca unitate de lumină relativă (RLU) al standardului de referință 17β-estradiol la media valorilor răspunsului RLU al martorului tratat cu solvent de referință. Acest factor de inducție ar trebui să îndeplinească cerințele minime pentru inducția multiplicată, astfel cum este indicat în tabelul 3C (agonism) sau în tabelul 4C (antagonism).
|
|
21.
|
Numai plăcile de microtitrare care îndeplinesc toate criteriile de acceptare menționate mai sus sunt considerate valabile și pot fi utilizate pentru a evalua răspunsul substanțelor chimice de testat.
|
|
22.
|
Criteriile de acceptare sunt aplicabile atât pentru testele de depistare prealabilă, cât și pentru cele complexe.
|
Tabelul 1
Concentrațiile standardului de referință, ale martorului pozitiv (MP) și ale martorului negativ (MN) pentru bioanaliza agonistă CALUX
|
|
Substanță
|
NR. CAS
|
Interval de testare (M)
|
|
Standard de referință
|
17β-estradiol
|
50-28-2
|
1*10-13 - 1*10-10
|
|
Martor pozitiv (MP)
|
17α-metiltestosteron
|
58-18-4
|
3*10-6
|
|
Martor negativ (MN)
|
corticosteron
|
50-22-6
|
1*10-8
|
Tabelul 2
Concentrațiile standardului de referință, ale martorului pozitiv (MP) și ale martorului negativ (MN) pentru bioanaliza antagonistă CALUX
|
|
Substanță
|
NR. CAS
|
Interval de testare (M)
|
|
Standard de referință
|
tamoxifen
|
10540-29-1
|
3*10-9 - 1*10-5
|
|
Martor pozitiv (MP)
|
4-hidroxi-tamoxifen
|
68047-06-3
|
1*10-9
|
|
Martor negativ (MN)
|
resveratrol
|
501-36-0
|
1*10-5
|
Tabelul 3
Criterii de acceptare pentru bioanaliza agonistă ERα CALUX
|
A - eșantioane individuale pe o placă
|
Criterii
|
|
1
|
Maxim % SD din godeurile replicate de trei ori (pentru MP, MN, fiecare diluție a substanței chimice de testat și a standardului de referință, cu excepția C0)
|
< 15 %
|
|
2
|
Maxim % SD din godeurile replicate de trei ori [pentru standardul de referință și martorii tratați cu solvent ai substanțelor chimice de testat (C0, SC)]
|
< 30 %
|
|
3
|
Scăpare LDH maximă, ca măsură a citotoxicității.
|
< 120 %
|
|
B - în cadrul unei singure plăci de microtitrare
|
|
|
4
|
Raportul dintre martorul standard de referință tratat cu solvent (C0; placa 1) și martorul tratat cu solvent al substanței chimice de testat (SC; plăci 2-x)
|
0,5-2,0
|
|
5
|
Rata concentrației aproximative EC50 și a celei mai mari concentrații standard de referință pe placa 1 și a concentrației aproximative EC50 și a celei mai mari concentrații standard de referință pe plăcile 2-x (C4, C8)
|
0,70-1,30
|
|
6
|
Factorul Z pentru fiecare placă
|
> 0,6
|
|
C - în cadrul unei singure serii de analize (toate plăcile în cadrul unei serii)
|
|
|
7
|
Curba sigmoidală a standardului de referință
|
Da (17ß-estradiol)
|
|
8
|
Standardul de referință pentru intervalul EC50 17ß-estradiol
|
4*10-12 – 4*10-11 M
|
|
9
|
Inducția multiplicată minimă a celei mai mari concentrații de 17ß-estradiol, în ceea ce privește martorul tratat cu solvent standard de referință.
|
5
|
|
10
|
Inducție relativă (%) MP.
|
> 30 %
|
|
11
|
Inducție relativă (%) MN
|
< 10 %
|
Aprox.: aproximativ; MP: martor pozitiv; MN: martor negativ; SC: martor tratat cu solvent al substanței chimice de testat; C0: martor standard de referință tratat cu solvent; SD: abatere standard; LDH: lactat dehidrogenază
Tabelul 4
Criterii de acceptare pentru bioanaliza antagonistă ERα CALUX
|
A - eșantioane individuale pe o placă
|
Criterii
|
|
1
|
Maxim % SD din godeurile replicate de trei ori [pentru MN, MP, fiecare diluție a substanței chimice de testat și a standardului de referință, a martorului tratat cu solvent (C0)]
|
< 15 %
|
|
2
|
Maxim % SD din godeurile replicate de trei ori [pentru martorul tratat cu vehicul (VC) și cea mai mare concentrație standard de referință (C8)]
|
< 30 %
|
|
3
|
Scăpare LDH maximă, ca măsură a citotoxicității.
|
< 120 %
|
|
B - în cadrul unei singure plăci de microtitrare
|
|
|
4
|
Raportul dintre martorul standard de referință tratat cu solvent (C0; placa 1) și martorul tratat cu solvent al substanței chimice de testat (SC; plăci 2-x)
|
0,70-1,30
|
|
5
|
Rata concentrațiilor aproximative standard de referință IC50 pe placa 1 și a concentrațiilor aproximative standard de referință IC50 pe plăcile 2-x (C4)
|
0,70-1,30
|
|
6
|
Raportul dintre cele mai mari concentrații standard de referință de pe placa 1 și cele mai mari concentrații standard de referință de pe plăcile 2-x (C8)
|
0,50-2,0
|
|
7
|
Factorul Z pentru fiecare placă
|
> 0,6
|
|
C - în cadrul unei singure serii de analize (toate plăcile în cadrul unei serii)
|
|
|
8
|
Curba sigmoidală a standardului de referință
|
Da (tamoxifen)
|
|
9
|
Standardul de referință pentru intervalul IC50 (tamoxifen)
|
1*10-8 - 1*10-7 M
|
|
10
|
Inducția minimă multiplicată a martorului tratat cu solvent standard de referință față de cea mai mare concentrație a tamoxifenului.
|
2,5
|
|
11
|
Inducție relativă (%) MP.
|
< 70 %
|
|
12
|
Inducție relativă (%) MN
|
> 85 %
|
Aprox.: aproximativ; MP: martor pozitiv; MN: martor negativ; VC: martorul tratat cu vehicul (martorul tratat cu solvent fără concentrație fixă a standardului de referință agonist); SC: martor tratat cu solvent al substanței chimice de testat; C0: martor standard de referință tratat cu solvent; SD: abatere standard; LDH: lactat dehidrogenază
Martor tratat cu solvent/vehicul, standarde de referință, martori pozitivi, martori negativi
|
23.
|
Pentru testul de depistare prealabilă și pentru testele complexe ar trebui să se utilizeze același martor tratat cu solvent/vehicul, aceleași standarde de referință și aceiași martori pozitivi și martori negativi. În plus, concentrația standardelor de referință, a martorilor pozitivi și a martorilor negativi ar trebui să fie aceeași.
|
Martorul tratat cu solvent
|
24.
|
Solventul utilizat pentru a dizolva substanțele chimice de testat ar trebui să fie testat ca martor tratat cu solvent. Dimetilsulfoxidul [DMSO, 1 % (v/v); CASRN 67-68-5] a fost utilizat ca vehicul în timpul validării bioanalizei ERα CALUX. Dacă se utilizează un alt solvent decât DMSO, toate standardele de referință, martorii și substanțele chimice de testat ar trebui să fie testați în același vehicul. Este de remarcat faptul că martorul tratat cu solvent pentru studiile antagoniste conține o concentrație fixă a standardului de referință agonist 17β-estradiol (aproximativ concentrația EC50). Pentru a testa solventul utilizat pentru studii antagoniste, ar trebui să se prepare și să se testeze un martor tratat cu vehicul.
|
Martor tratat cu vehicul (antagonism)
|
25.
|
Pentru testarea antagonismului, mediul de analiză este suplimentat cu o concentrație fixă a standardului de referință agonist 17β-estradiol (aproximativ concentrația EC50). Pentru a testa solventul utilizat pentru a dizolva substanțele chimice de testat pentru antagonism, ar trebui preparat un mediu de analiză fără o concentrație fixă a standardului de referință agonist 17β-estradiol. Acest eșantion de martor este indicat ca fiind martorul tratat cu vehicul. Dimetilsulfoxidul [DMSO, 1 % (v/v); CASRN 67-68-5] a fost utilizat ca vehicul în timpul validării bioanalizei ERα CALUX. Dacă se utilizează un alt solvent decât DMSO, toate standardele de referință, martorii și substanțele chimice de testat ar trebui să fie testați în același vehicul.
|
Standarde de referință
|
26.
|
Standardul de referință agonist este 17β-estradiol (tabelul 1). Standardele de referință cuprind o serie de diluții de opt concentrații de 17β-estradiol (1*10-13, 3*10-13, 1*10-12, 3*10-12, 6*10-12, 1*10-11, 3*10-11, 1*10-10 M).
|
|
27.
|
Standardul de referință antagonist este tamoxifenul (tabelul 2). Standardele de referință cuprind o serie de diluții de opt concentrații de tamoxifen (3*10-9, 1*10-8, 3*10-8, 1*10-7, 3*10-7, 1*10-6, 3*10-6, 1*10-5 M). Fiecare dintre concentrațiile standardului de referință antagonist este coincubat cu o concentrație fixă a standardului de referință agonist 17β-estradiol (3*10-12 M).
|
Martor pozitiv
|
28.
|
Martorul pozitiv pentru studii agoniste este 17α-metiltestosteron (tabelul 1).
|
|
29.
|
Martorul pozitiv pentru studii antagoniste este 4-hidroxitamoxifen (tabelul 2). Martorul pozitiv antagonist este coincubat cu o concentrație fixă a standardului de referință agonist 17β-estradiol (3*10-12 M).
|
Martor negativ
|
30.
|
Martorul negativ pentru studii agoniste este corticosteronul (tabelul 1).
|
|
31.
|
Martorul negativ pentru studii antagoniste este resveratrolul (tabelul 2). Martorul negativ antagonist este coincubat cu o concentrație fixă a standardului de referință agonist 17β-estradiol (3*10-12 M).
|
Demonstrarea performanțelor laboratorului (a se vedea punctul 14 și tabelele 3 și 4 de la secțiunea „COMPONENTELE ANALIZEI ER TA” din prezenta metodă de testare)
Vehicul
|
32.
|
Solventul utilizat pentru dizolvarea substanței chimice de testat ar trebui să solubilizeze complet substanța chimică de testat și să fie miscibil cu mediul celular. DMSO, apa și etanolul (puritate de 95 %-100 %) sunt solvenți adecvați. În cazul în care se folosește DMSO ca solvent, concentrația maximă de DMSO în timpul incubației nu ar trebui să depășească 1 % (v/v). Înainte de utilizare, solventul ar trebui să fie testat pentru absența citotoxicității și interferența cu analizele de performanță.
|
Pregătirea standardelor de referință, a martorilor pozitivi, a martorilor negative și a substanțelor chimice de testat
|
33.
|
Standardele de referință, martorii pozitivi, martorii negativi și substanțele chimice de testat se dizolvă în DMSO 100 % (sau un solvent adecvat). Apoi ar trebui să se prepare diluții (în serie) adecvate în același solvent. Înainte de a fi dizolvate, toate substanțele ar trebui să fie lăsate să se echilibreze la temperatura camerei. Soluțiile mamă proaspăt preparate ale standardelor de referință, ale martorilor pozitivi, ale martorilor negativi și ale substanțelor chimice de testat nu ar trebui să prezinte precipitat sau tulburare vizibilă. Stocurile de standarde de referință și de martori pot fi preparate în cantitate mare. Soluțiile mamă ale substanțelor chimice de testat ar trebui să fie proaspăt preparate înainte de fiecare experiment. Diluțiile finale ale standardelor de referință, martorii pozitivi, martorii negativi și substanțele chimice de testat ar trebui să fie proaspăt preparate pentru fiecare experiment și utilizate în termen de 24 de ore de la preparare.
|
Solubilitate, citotoxicitate și stabilirea intervalului
|
34.
|
În timpul testului de depistare prealabilă, se stabilește solubilitatea substanțelor chimice de testat în solventul ales. Se prepară o concentrație maximă a stocului de 0,1 M. În cazul în care această concentrație indică probleme de solubilitate, ar trebui să fie preparate soluții mamă cu concentrații mai mici până la solubilizarea completă a substanței chimice de testat. În timpul testului de depistare prealabilă, se testează diluții de 1: 10 în serie ale substanței chimice de testat. Concentrația maximă de testare pentru testarea agonistă sau antagonistă este de 1 mM. În urma testului de depistare prealabilă, se obține un interval al concentrațiilor ajustat corespunzător pentru substanțele chimice de testat care ar trebui să fie testate în cadrul testelor complexe. Diluțiile utilizate pentru testul complex ar trebui să fie de 1x, 3x, 10x, 30x, 100x, 300x, 1000x și 3000x.
|
|
35.
|
Testarea citotoxicității este inclusă în protocolul de testare agonistă și antagonistă (11). Testarea citotoxicității este inclusă atât în testul de depistare prealabilă, cât și în testele complexe. Metoda utilizată pentru a evalua citotoxicitatea în timpul validării bioanalizei ERα CALUX a fost testul de scăpare a lactatului dehidrogenază (LDH) în combinație cu inspecția vizuală calitativă a celulelor (a se vedea apendicele 4.1) în urmare expunerii la substanțele chimice de testat. Însă pot fi utilizate alte metode cantitative pentru determinarea citotoxicității [de exemplu, analiza colorimetrică (MTT) bazată pe tetrazolif (MTT) sau bioanaliza citotoxicității CALUX]. În general, concentrațiile substanțelor chimice de testat care indică o reducere mai mare de 20 % a viabilității celulare sunt considerate citotoxice și, prin urmare, nu pot fi utilizate pentru evaluarea datelor. În ceea ce privește analiza scăpărilor de LDH, concentrația substanței chimice de testat este considerată citotoxică atunci când procentul de scăpare a LDH este mai mare de 120 %.
|
Expunerea la substanța chimică de testat și organizarea plăcilor pentru testare
|
36.
|
În urma tripsinizării unui balon confluent cu celule cultivate, celulele se suspendă din nou la 1x105 celule/ml în mediu de analiză fără estrogen. În godeurile interioare de pe o placă de microtitrare cu 96 de godeuri se introduc o sută de μl de celule re-suspendate. Godeurile exterioare sunt umplute cu 200 μl de soluție salină tamponată de fosfat (PBS) (a se vedea figurile 1 și 2). Celulele introduse sunt incubate în prealabil, timp de 24 de ore, într-un incubator cu CO2 (5 % CO2, 37°C, umiditate de 100 %).
|
|
37.
|
După pre-incubare, plăcile sunt inspectate pentru citotoxicitate vizuală (a se vedea apendicele 4.1), contaminare și confluență. Sunt utiliate pentru testare doar plăcile care nu prezintă vizual citotoxicitate sau contaminare și au o confluență de cel puțin 85 %. Mediul din godeurile interioare se îndepărtează cu grijă și se înlocuiește cu 200 μl de mediu de testare fără estrogen conținând serii de diluții adecvate de standarde de referință, substanțe chimice de testat, martori pozitivi, martori negativi și martori tratați cu solvent (tabelul 5: studii agoniste; Tabelul 6: studii antagoniste). Toate standardele de referință, substanțele chimice de testat, martorii pozitivi, martorii negativi și martorii tratați cu solvent sunt testați în trei replici. Figura 1 prezintă distribuția pe placă pentru testarea agonistă. Figura 2 prezintă distribuția pe placă pentru testarea antagonistă. Distribuția pe placă pentru testul de depistare prealabilă și testul complex este identică. Pentru testul antagonist, toate godeurile interioare, cu excepția godeurilor cu martor tratat cu vehicul (VC) conțin, de asemenea, o concentrație fixă de standard de referință agonist 17β-estradiol (3,0*10-12 M). Este de remarcat faptul că standardele de referință C8 și C4 ar trebui să fie adăugate pe fiecare placă cu TC.
|
|
38.
|
După expunerea celulelor la toate substanțele chimice, plăcile de microtitrare cu 96 de godeuri ar trebui să fie incubate timp de încă 24 de ore într-un incubator cu CO2 (5 % CO2, 37°C, umiditate de 100 %).
|
Figura 1
Configurația plăcii de microtitrare cu 96 de godeuri pentru depistare prealabilă și evaluarea efectului agonist.

C0 = solvent standard de referință
C(1-8) = serii de diluții (1-8, concentrații în ordine crescătoare) ale standardului de referință.
MP = martor pozitiv.
MN = martor negativ.
TCx-(1-8) = diluții (1-8, concentrații în ordine crescătoare) ale substanței chimice de testat pentru testul de depistare prealabilă și evaluarea efectului agonist al substanței chimice de testat x.
SC = martor tratat cu solvent al substanței chimice de testat (în mod optim, același solvent ca și în C0, dar, posibil, din alt lot).
Celule de culoare gri: = Godeuri exterioare, umplute cu 200 μl de PBS.
Figura 2
Configurația plăcii de microtitrare cu 96 de godeuri pentru depistarea prealabilă antagonistă și evaluarea efectului antagonist.
C0 = solvent standard de referință
C(1-8) = serii de diluții (1-8, concentrații în ordine crescătoare) ale standardului de referință.
MN = martor negativ.
MP = martor pozitiv.
TCx-(1-8) = diluții (1-8, concentrații în ordine crescătoare) ale substanței chimice de testat pentru testul de depistare prealabilă și evaluarea efectului agonist al substanței chimice de testat x.
SC = martor tratat cu solvent al substanței chimice de testat (în mod optim, același solvent ca și în C0, dar, posibil, din alt lot).
VC = martor tratat cu vehicul (martorul tratat cu solvent fără concentrație fixă a standardului de referință agonist).
Celule de culoare gri: = Godeuri exterioare, umplute cu 200 μl de PBS.
Notă: toate godeurile interioare, cu excepția godeurilor cu martor tratat cu vehicul (VC) conțin, de asemenea, o concentrație fixă de standard de referință agonist 17β-estradiol (3,0*10-12 M)
Măsurarea luminiscenței
|
39.
|
Măsurarea luminiscenței este descrisă în detaliu în protocolul privind analiza agonistă și antagonistă (10). Mediul din godeuri ar trebui să fie eliminat, iar celulele ar trebui să fie lizate după 24 de ore de incubare pentru a deschide membrana celulară și a permite măsurarea activității luciferazei.
|
|
40.
|
Pentru măsurarea luminiscenței, această procedură necesită un luminometru prevăzut cu două injectoare. Reacția luciferazei se pornește prin injectarea substratului luciferină. Reacția se oprește prin adăugarea a 0,2 M NaOH. Reacția este oprită pentru a împiedica transmiterea luminescenței de la un godeu la altul.
|
|
41.
|
Lumina emisă din fiecare godeu este exprimată în unități relative de lumină (RLU) pentru fiecare godeu.
|
Testul de depistare prealabilă
|
42.
|
Rezultatele analizei de depistare prealabilă sunt utilizate pentru a determina un interval de concentrații a substanțelor chimice de testat ajustat pentru testarea complexă. Evaluarea rezultatelor analizei de depistare prealabilă și determinarea intervalului de concentrații de substanțe chimice de testat ajustat pentru testul complex sunt descrise în detaliu în protocolul privind analiza agonistă și antagonistă (10). Aici se prezintă un scurt rezumat al procedurilor de determinare a intervalului de concentrații al substanțelor chimice de testat pentru testele agoniste și antagoniste. A se vedea tabelele 5 și 6 pentru îndrumări privind conceperea diluției în serie.
|
Selecția concentrațiilor pentru evaluarea efectelor agoniste
|
43.
|
În timpul testului de depistare prealabilă, substanțele chimice de testat ar trebui să fie testate utilizând seriile de diluții indicate în tabelele 5 (agonism) și 6 (antagonism). Toate concentrațiile ar trebui să fie testate în trei godeuri replicate de trei ori conform configurației plăcii indicate în figura 1 (agonism) sau 2 (antagonism).
|
|
44.
|
Numai rezultatele analizei care îndeplinesc criteriile de acceptare (tabelul 3) sunt considerate valabile și pot fi utilizate pentru a evalua răspunsul substanțelor chimice de testat. În cazul în care una sau mai multe plăci de microtitrare dintr-o serie de analize nu îndeplinesc (îndeplinește) criteriile de acceptare, respectivele microplăci ar trebui să fie analizate din nou. În cazul în care prima placă care conține seriile complete de diluții ale standardului de referință nu respectă criteriile de acceptare, întreaga serie de teste (6 plăci) trebuie să fie analizate din nou.
|
|
45.
|
Ar trebui să se ajusteze intervalele inițiale ale concentrațiilor substanțelor chimice de testat, iar testul de depistare prealabilă ar trebui să fie repetat dacă:
|
—
|
se observă citotoxicitatea. Procedura de depistare prealabilă ar trebui să fie repetată folosind concentrații necitotoxice ale substanței chimice de testat mai scăzute.
|
|
—
|
testul de depistare prealabilă asupra substanței chimice de testat nu prezintă o curbă doză-răspuns completă, deoarece concentrațiile testate generează o inducție maximă. Testul de depistare prealabilă ar trebui să fie repetat folosind concentrații ale substanței chimice de testat mai scăzute.
|
|
|
46.
|
Atunci când se observă un răspuns valabil privind doza, ar trebui să se selecteze concentrația (cea mai scăzută) la care se observă o inducție maximă și care nu prezintă citotoxicitate. Cea mai mare concentrație a substanței chimice de testat în cadrul testelor complexe ar trebui să fie de trei ori această concentrație selectată.
|
|
47.
|
Ar trebui să se prepare o serie completă de diluții ajustate ale substanței chimice de testat cu respectarea etapelor de diluție indicate în tabelul 5, începând cu cea mai mare concentrație, astfel cum este stabilită mai sus.
|
|
48.
|
O substanță chimică de testat care nu prezintă un efect agonist ar trebui testată în cadrul testelor complexe începând cu cea mai mare concentrație necitotoxică identificată în timpul testului de depistare prealabilă.
|
Selecția concentrațiilor pentru evaluarea efectelor antagoniste
|
49.
|
Numai rezultatele analizei care îndeplinesc criteriile de acceptare (tabelul 4) sunt considerate valabile și pot fi utilizate pentru a evalua răspunsul substanțelor chimice de testat. În cazul în care una sau mai multe plăci de microtitrare dintr-o serie de analize nu îndeplinesc (îndeplinește) criteriile de acceptare, respectivele microplăci ar trebui să fie analizate din nou. În cazul în care prima placă care conține seriile complete de diluții ale standardului de referință nu respectă criteriile de acceptare, întreaga serie de teste (6 plăci) trebuie să fie analizate din nou.
|
|
50.
|
Ar trebui să se ajusteze intervalele inițiale ale concentrațiilor substanțelor chimice de testat, iar testul de depistare prealabilă ar trebui să fie repetat dacă:
|
—
|
se observă citotoxicitatea. Procedura de depistare prealabilă ar trebui să fie repetată folosind concentrații necitotoxice ale substanței chimice de testat mai scăzute.
|
|
—
|
testul de depistare prealabilă asupra substanței chimice de testat nu prezintă o curbă doză-răspuns completă, deoarece concentrațiile testate generează o inhibiție maximă. Testul de depistare prealabilă ar trebui să fie repetat folosind concentrații ale substanței chimice de testat mai scăzute.
|
|
|
51.
|
Atunci când se identifică un răspuns valabil privind doza, ar trebui să se selecteze concentrația (cea mai scăzută) la care se observă o inhibiție maximă și care nu prezintă citotoxicitate. Cea mai mare concentrație a substanței chimice de testat în cadrul testelor complexe ar trebui să fie de trei ori această concentrație selectată.
|
|
52.
|
Ar trebui să se prepare o serie completă de diluții ajustate ale substanței chimice de testat cu respectarea etapelor de diluție indicate în tabelul 6, începând cu cea mai mare concentrație, astfel cum este stabilită mai sus.
|
|
53.
|
Substanțele chimice de testat care nu prezintă efecte antagoniste ar trebui testate în cadrul testelor complexe începând cu cea mai mare concentrație necitotoxică verificată prin testul de depistare prealabilă.
|
Teste complexe
|
54.
|
În urma selecției intervalelor de concentrații ajustate, substanțele chimice de testat ar trebui să fie testate în mod cuprinzător utilizând seriile de diluții indicate în tabelele 5 (agonism) și 6 (antagonism). Toate concentrațiile ar trebui să fie testate în trei godeuri replicate de trei ori conform configurației plăcii indicate în figura 1 (agonism) sau 2 (antagonism).
|
|
55.
|
Numai rezultatele analizei care îndeplinesc criteriile de acceptare (tabelele 3 și 4) sunt considerate valabile și pot fi utilizate pentru a evalua răspunsul substanțelor chimice de testat. În cazul în care una sau mai multe plăci de microtitrare dintr-o serie de analize nu îndeplinesc (îndeplinește) criteriile de acceptare, respectivele microplăci ar trebui să fie analizate din nou. În cazul în care prima placă care conține seriile complete de diluții ale standardului de referință nu respectă criteriile de acceptare, întreaga serie de teste (6 plăci) trebuie să fie analizate din nou.
|
Tabelul 5
Concentrația și diluțiile standardelor de referință, ale martorilor și substanțelor chimice de testat utilizate pentru testul agonist
|
17β-estradiol de referință
|
TCx - testul de depistare prealabilă
|
TCx - testul complex
|
Martori
|
|
conc. (M)
|
diluție
|
diluție
|
conc. (M)
|
|
C0
|
0
|
TCx-1
|
10 000 000 x
|
TCx-1
|
3000 x
|
MP
|
3*10-6
|
|
C1
|
1*10-13
|
TCx-2
|
1 000 000 x
|
TCx-2
|
1000 x
|
MN
|
1*10-8
|
|
C2
|
3*10-13
|
TCx-3
|
100 000 x
|
TCx-3
|
300 x
|
C0
|
0
|
|
C3
|
1*10-12
|
TCx-4
|
10 000 x
|
TCx-4
|
100 x
|
SC
|
0
|
|
C4
|
3*10-12
|
TCx-5
|
1 000 x
|
TCx-5
|
30 x
|
|
|
|
C5
|
6*10-12
|
TCx-6
|
100 x
|
TCx-6
|
10 x
|
|
|
|
C6
|
1*10-11
|
TCx-7
|
10 x
|
TCx-7
|
3 x
|
|
|
|
C7
|
3*10-11
|
TCx-8
|
1 x
|
TCx-8
|
1 x
|
|
|
|
C8
|
1*10-10
|
|
|
|
|
|
|
|
TCx - substanța chimică de testat x
MP - martor pozitiv (17α-metiltestosteron)
MN - martor negativ (corticosteron)
C0 - martor standard de referință tratat cu solvent
SC - martor tratat cu solvent al substanței chimice de testat
|
Tabelul 6
Concentrația și diluțiile standardelor de referință, ale martorilor și substanțelor chimice de testat utilizate pentru testul antagonist
|
Tamoxifen de referință
|
TCx - testul de depistare prealabilă
|
TCx - testul complex
|
Martori
|
|
conc. (M)
|
diluție
|
diluție
|
conc. (M)
|
|
C0
|
0
|
TCx-1
|
10 000 000 x
|
TCx-1
|
3.000 x
|
MP
|
1*10-9
|
|
C1
|
3*10-9
|
TCx-2
|
1 000 000 x
|
TCx-2
|
1.000 x
|
MN
|
1*10-5
|
|
C2
|
1*10-8
|
TCx-3
|
100 000 x
|
TCx-3
|
300 x
|
C0
|
0
|
|
C3
|
3*10-8
|
TCx-4
|
10 000 x
|
TCx-4
|
100 x
|
SC
|
0
|
|
C4
|
1*10-7
|
TCx-5
|
1 000 x
|
TCx-5
|
30 x
|
|
|
|
C5
|
3*10-
|
TCx-6
|
100 x
|
TCx-6
|
10 x
|
Agonist suplimentat
|
|
C6
|
1*10-6
|
TCx-7
|
10 x
|
TCx-7
|
3 x
|
conc. (M)
|
|
C7
|
3*10-6
|
TCx-8
|
1 x
|
TCx-8
|
1 x
|
17β-estradiol
|
3*10-12
|
|
C8
|
1*10-5
|
|
|
|
|
|
|
|
TCx - substanța chimică de testat x
MP - martor pozitiv (4-hidroxitamoxifen)
MN - martor negativ (resveratrol)
C0 - martor standard de referință tratat cu solvent
SC - martor tratat cu solvent al substanței chimice de testat
VC - martor tratat cu vehicul (nu conține o concentrație fixă a standardului de referință agonist 17β-estradiol (3,0*10-12 M)
|
Colectarea datelor și analiza datelor
|
56.
|
În urma testului de depistare prealabilă și a testului complex, ar trebui să se determine valorile EC10, EC50, PC10, PC50 și inducția maximă (TCxmax) ale unei substanțe chimice de testat pentru testarea agonistă. Pentru testarea antagonistă, ar trebui să se calculeze valorile IC20, IC50, PC80, PC50 și inducția minimă (TCxmin). Figura 3 (agonism) și 4 (antagonism) cuprinde o reprezentare grafică a acestor parametri. Parametrii necesari se calculează pe baza inducției relative a fiecărei substanțe chimice de testat [în raport cu inducția maximă a standardului de referință (= 100 %)]. Regresia nelineară (panta variabilă, 4 parametri) ar trebui să fie utilizată pentru evaluarea datelor conform următoarei ecuații:

Unde:
X = Log al dozei sau concentrației
Y = Răspuns [inducție relativă (%)]
Partea superioară = Inducție maximă (%)
Partea inferioară = Inducție minimă (%)
LogEC50 = Valoare log a concentrației la care se observă 50 % din răspunsul maxim
HillSlope = Factorul de pantă sau panta Hill
|
|
57.
|
Datele brute obținute de la luminometru, exprimate ca unități relative de lumină (RLU), ar trebui să fie transferate în fișa de analiză a datelor concepută pentru teste de depistare prealabilă și teste complexe. Datele brute ar trebui să respecte criteriile de acceptare indicate în tabelele 3A și 3B (agonism) sau 4A și 4B (și antagonism). În cazul în care datele brute respectă criteriile de acceptare, se parcurg următoarele etape de calcul pentru determinarea parametrilor necesari:
|
Agonism
|
—
|
Se scade media RLU a martorului tratat cu solvent al standardelor de referință din fiecare dintre datele de analiză brute ale standardelor de referință.
|
|
—
|
Se scade media RLU a martorului tratat cu solvent al substanței chimice de testat din fiecare dintre datele de analiză brute ale substanțelor chimice de testat.
|
|
—
|
Se calculează inducția relativă a fiecărei concentrații a standardului de referință. Se setează inducția celei mai mari concentrații a standardului de referință la 100 %.
|
|
—
|
Se calculează inducția relativă a fiecărei concentrații a substanței chimice de testat în comparație cu cea mai mare concentrație a standardului de referință de 100 %.
|
|
—
|
Se evaluează rezultatele analizei în urma regresiei neliniare (pantă variabilă, 4 parametri).
|
|
—
|
Se determină valorile EC50 și EC10 ale standardului de referință.
|
|
—
|
Se determină valorile EC50 și EC10 ale substanțelor chimice de testat.
|
|
—
|
Se determină inducția relativă maximă a substanței chimice de testat (TCmax).
|
|
—
|
Se determină valorile PC10 și PC50 ale substanțelor chimice de testat.
|
Pentru substanțele chimice de testat, este posibil să nu se obțină întotdeauna o curbă doză-răspuns completă, de exemplu, din cauza unor probleme de citotoxicitate sau de solubilitate. Prin urmare, nu se pot determina valorile EC50, EC10 și PC50. Într-un astfel de caz, se pot determina doar valorile PC10 și TCmax.
Antagonism
|
—
|
Se scade media RLU a celei mai mari concentrații standard de referință din fiecare dintre datele de analiză brute ale standardelor de referință.
|
|
—
|
Se scade media RLU a celei mai mari concentrații standard de referință din fiecare dintre datele de analiză brute ale substanțelor chimice de testat.
|
|
—
|
Se calculează inducția relativă a fiecărei concentrații a standardului de referință. Se setează inducția celei mai mici concentrații a standardului de referință la 100 %.
|
|
—
|
Se calculează inducția relativă a fiecărei concentrații a substanței chimice de testat în comparație cu cea mai scăzută concentrație a standardului de referință de 100 %.
|
|
—
|
Se evaluează rezultatele analizei în urma regresiei neliniare (pantă variabilă, 4 parametri).
|
|
—
|
Se determină valorile IC50 și IC20 ale standardului de referință.
|
|
—
|
Se determină valorile IC50 și IC20 ale substanțelor chimice de testat.
|
|
—
|
Se determină inducția relativă minimă a substanței chimice de testat (TCmin).
|
|
—
|
Se determină valorile PC80 și PC50 ale substanțelor chimice de testat.
|
Figura 3
Prezentarea parametrilor determinați în analiza agonistă
EC10 = concentrația unei substanțe la care se observă 10 % din răspunsul maxim al acesteia
EC50 = concentrația unei substanțe la care se observă 50 % din răspunsul maxim al acesteia
PC10 = concentrația unei substanțe chimice de testat la care răspunsul acesteia este egal cu valoarea EC10 a standardului de referință.
PC50 = concentrația unei substanțe chimice de testat la care răspunsul acesteia este egal cu valoarea EC50 a standardului de referință.
TCxmax = inducția relativă maximă a substanței chimice de testat.
Figura 4
Prezentarea parametrilor determinați în analiza antagonistă
IC20 = concentrația unei substanțe la care se observă 80 % din răspunsul maxim al acesteia (inhibiție de 20 %).
IC50 = concentrația unei substanțe la care se observă 50 % din răspunsul maxim al acesteia (inhibiție de 50 %).
PC80 = concentrația unei substanțe chimice de testat la care răspunsul acesteia este egal cu valoarea IC20 a standardului de referință.
PC50 = concentrația unei substanțe chimice de testat la care răspunsul acesteia este egal cu valoarea IC50 a standardului de referință.
TCxmin = inducția relativă minimă a substanței chimice de testat.
Pentru substanțele chimice de testat, este posibil să nu se obțină întotdeauna o curbă doză-răspuns completă, de exemplu, din cauza unor probleme de citotoxicitate sau de solubilitate. Prin urmare, nu se pot determina valorile IC50, IC20 și PC50. Într-un astfel de caz, se pot determina doar valorile PC20 și TCmin.
|
58.
|
Rezultatele ar trebui să fie bazate pe două (sau trei) testări independente. Dacă două testări generează rezultate comparabile și, prin urmare, reproductibile, nu este necesar să se efectueze o a treia testare. Pentru ca rezultatele să fie acceptabile, acestea ar trebui:
|
—
|
să îndeplinească criteriile de acceptabilitate (a se vedea Criteriile de acceptabilitate de la punctele 14-22),
|
|
Criterii de interpretare a datelor
|
59.
|
Pentru interpretarea datelor și stabilirea deciziei dacă o substanță chimică de testat este considerată pozitivă sau negativă, se utilizează următoarele criterii:
|
Pentru fiecare test complex, o substanță chimică de testat este considerată pozitivă în cazul în care:
|
1
|
Valoarea TCmax este egală cu sau mai mare decât 10 % din răspunsul maxim al standardului de referință (REF10).
|
|
2
|
Cel puțin două concentrații consecutive ale substanței chimice de testat sunt egale cu sau depășesc valoarea REF10.
|
|
|
Pentru fiecare test complex, o substanță chimică de testat este considerată negativă în cazul în care:
|
1
|
Valoarea TCmax nu depășește rata de 10 % din răspunsul maxim al standardului de referință (REF10).
|
|
2
|
Mai puțin de două concentrații ale substanței chimice de testat sunt egale cu sau depășesc valoarea REF10.
|
|
|
Pentru fiecare test complex, o substanță chimică de testat este considerată pozitivă în cazul în care:
|
1
|
Valoarea TCmin este egală cu sau mai mică decât 80 % din răspunsul maxim al standardului de referință (REF80 = inhibiție de 20 %).
|
|
2
|
Cel puțin două concentrații consecutive ale substanței chimice de testat sunt egale cu sau mai mici decât valoarea REF80.
|
|
|
Pentru fiecare test complex, o substanță chimică de testat este considerată negativă în cazul în care:
|
1
|
Valoarea TCmin depășește 80 % din răspunsul maxim al standardului de referință (REF80 = inhibiție de 20 %).
|
|
2
|
Mai puțin de două concentrații ale substanței chimice de testat sunt egale cu sau mai mici decât valoarea REF80.
|
|
|
|
60.
|
Pentru a caracteriza puterea răspunsului pozitiv al unei substanțe chimice de testat, ar trebui să se consemneze magnitudinea efectului (agonism: TCmax; antagonism: TCmin) și concentrația la care se produce efectul (agonism: EC10, EC50, PC10, PC50; antagonism: IC20, IC50, PC80, PC50).
|
RAPORTUL TESTULUI
|
61.
|
A se vedea punctul 20 din „COMPONENTELE ANALIZEI ER TA”
|
REFERINȚE BIBLIOGRAFICE
|
(1)
|
OCDE (2016). Draft Validation report of the (anti-) ERα CALUX bioassay - transactivation bioassay for the detection of compounds with (anti)estrogenic potential. Environmental Health and Safety Publications, Series on Testing and Assessment (No 240). Organizația pentru Cooperare și Dezvoltare Economică, Paris.
|
|
(2)
|
Sonneveld E, Jansen HJ, Riteco JA, Brouwer A, van der Burg B. (2005). Development of androgen- and estrogen-responsive bioassays, members of a panel of human cell line-based highly selective steroid-responsive bioassays. Toxicol Sci. 83(1), 136-148.
|
|
(3)
|
Quaedackers ME, van den Brink CE, Wissink S, Schreurs RHMM, Gustafsson JA, van der Saag PT și van der Burg B. (2001). 4-Hydroxytamoxifen trans-represses nuclear factor-kB Activity in human osteoblastic U2OS cells through estrogen receptor (ER)α and not through ERβ. Endocrinology 142(3), 1156-1166.
|
|
(4)
|
Thorne N, Inglese J și Auld DS. (2010). Illuminating Insights into Firefly Luciferase and Other Bioluminescent Reporters Used in Chemical Biology, Chemistry and Biology17(6):646-57.
|
|
(5)
|
Escande A, Pillon A, Servant N, Cravedi JP, Larrea F, Muhn P, Nicolas JC, Cavaillès V și Balaguer P. (2006). Evaluation of ligand selectivity using reporter cell lines stably expressing estrogen receptor alpha or beta. Biochem. Pharmacol., 71, 1459-1469.
|
|
(6)
|
Kuiper GG, Lemmen JG, Carlsson B, Corton JC, Safe SH, van der Saag PT, van der Burg B și Gustafsson JA. (1998). Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinol., 139, 4252-4263.
|
|
(7)
|
Sotoca AM, Bovee TFH, Brand W, Velikova N, Boeren S, Murk AJ, Vervoort J, Rietjens IMCM. (2010). Superinduction of estrogen receptor mediated gene expression in luciferase based reporter gene assays is mediated by a post-transcriptional mechanism. J. Steroid. Biochem. Mol. Biol., 122, 204–211.
|
|
(8)
|
Sonneveld E, Riteco JAC, Jansen HJ, Pieterse B, Brouwer A, Schoonen WG și van der Burg B. (2006). Comparison of in vitro and in vivo screening models for androgenic and estrogenic activities. Toxicol. Sci., 89(1), 173-187.
|
|
(9)
|
Kobayashi H, Yamamoto K, Eguchi M, Kubo M, Nakagami S, Wakisaka S, Kaizuka M și Ishii H. (1995). Rapid detection of mycoplasma contamination in cell cultures by enzymatic detection of polymerase chain reaction (PCR) products. J. Vet. Med. Sci., 57(4), 769-771.
|
|
(10)
|
Zhang J-H, Chung TDY și Oldenburg KR. (1999). A simple statistical parameter for use in evaluation and validation of high throuphut screening assays. J. Biomol. Scr., 4, 67-73
|
|
(11)
|
Besselink H, Middelhof I și Felzel, E. (2014). Transactivation assay for the detection of compounds with (anti)estrogenic potential using ERα CALUX cells. BioDetection Systems BV (BDS). Amsterdam, Țările de Jos.
|
Apendicele 4.1:
INSPECțIA VIZUALĂ A VIABILITĂțII CELULARE
B.67 TESTE IN VITRO DE MUTAȚII GENICE PE CELULE DE MAMIFERE FOLOSIND GENA TIMIDIN-KINAZĂ
INTRODUCERE
|
1.
|
Această metodă de testare (MT) este echivalentă cu Orientarea OCDE nr. 490 privind testarea (2016). Metodele de testare sunt revizuite periodic și modificate în contextul progreselor științifice, al necesităților de reglementare și al bunăstării animalelor. Analiza pe limfom de șoarece (MLA) și testul TK6 care utilizează locus de timidin-kinază (TK) au fost incluse inițial în metoda de testare B.17. Ulterior, Grupul de lucru al experților MLA din cadrul Atelierului internațional pentru testarea genotoxicității (International Workshop for Genotoxicity Testing - IWGT) a elaborat recomandări armonizate la nivel internațional privind criteriile de acceptare a analizei și interpretarea datelor pentru MLA (1) (2) (3) (4) (5), iar aceste recomandări sunt integrate în această nouă metodă de testare B.67. Această metodă de testare este dezvoltată pentru MLA și, deoarece utilizează, de asemenea, locusul TK, pentru testul TK6. Deși MLA a fost utilizată pe scară largă în scopuri de reglementare, TK6 a fost utilizat mult mai puțin frecvent. Ar trebui remarcat faptul că, în ciuda similitudinii dintre punctele finale, cele două linii celulare nu sunt interschimbabile, iar programele de reglementare pot să își exprime în mod valabil preferința pentru una față de cealaltă pentru un anumit scop de reglementare. De exemplu, validarea MLA demonstrează caracterul adecvat al acesteia pentru detectarea nu doar a mutației genice, ci și a capacității unei substanțe chimice de testat de a induce daune cromozomiale structurale. Această metodă de testare face parte dintr-o serie de metode de testare cu privire la toxicologia genetică. OCDE a elaborat un document care oferă informații succinte privind testarea pentru toxicologia genetică și o prezentare a modificărilor efectuate recent asupra orientărilor OCDE privind testarea pentru toxicologia genetică (6).
|
|
2.
|
Scopul testelor in vitro de mutații genice pe celule de mamifere este detectarea mutațiilor genice induse de substanțe chimice. Liniile celulare utilizate în aceste teste măsoară mutațiile directe ale genelor reporter, în special a genei endogene timidin-kinază (TK pentru celule umane și Tk pentru celule de rozătoare, denumite în continuare, în mod colectiv, TK în prezenta metodă de testare). Această metodă de testare este prevăzută a fi utilizată pe două linii celulare: linia celulară L5178Y TK+/--3.7.2C de limfom la șoarece (denumită, în general, L5178Y) și linia celulară umană limfoblastoidă TK6 (denumită, în general, TK6). Deși cele două linii celulare diferă din cauza originii lor, a creșterii celulelor, a statutului p53 etc., testele privind mutațiile genice TK pot fi desfășurate într-un mod similar pe ambele tipuri de celule, astfel cum se descrie în prezenta metodă de testare.
|
|
3.
|
Natura autosomală și heterozigotă a genei timidin-kinază permite detectarea de colonii viabile ale căror celule sunt deficitare în enzima timidin-kinază ca urmare a mutației de la TK+/-
la TK-/-
. Această deficiență poate să apară ca urmare a unor evenimente genetice care afectează gena TK, incluzând atât mutații genice (mutații punctiforme, mutații care alterează cadrul de citire, mici deleții etc.), cât și evenimente cromozomiale (deleții semnificative, rearanjări ale cromozomilor și recombinarea mitotică). Aceste din urmă evenimente sunt exprimate ca fiind o pierdere de heterozigozitate, care reprezintă o modificare genetică răspândită a genelor de suprimare a tumorilor în tumorigeneza la om. Teoretic, pierderea întregului cromozom care poartă gena TK ca urmare a deteriorării fusului și/sau a non-disjuncției mitotice poate fi detectată în MLA. Într-adevăr, o combinație de analiză citogenetică și moleculară arată în mod clar că anumite gene mutante TK MLA reprezintă rezultatul non-disjuncției. Însă, dovezile arată că testele privind mutațiile genetice TK nu pot detecta în mod fiabil aneugenii atunci când sunt aplicate criteriile standard de citotoxicitate (astfel cum este descris în prezenta metodă) și, prin urmare, nu este adecvat să se recurgă la aceste teste pentru detectarea aneugenilor (7)(8)(9).
|
|
4.
|
În cadrul testelor privind mutații ale genei TK, sunt generate două clase fenotipice distincte de gene mutante TK; genele mutante normale care cresc în același ritm ca și celulele heterozigote TK și genele mutante care cresc lent, cu perioade de dublare prelungite. Genele mutante care cresc normal și cele care cresc lent sunt recunoscute ca fiind gene mutante din colonii mari și din colonii mici în MLA și ca gene mutante din colonii care se formează timpuriu și din colonii care se formează tardiv în TK6. Natura moleculară și citogenică atât a genelor mutante din colonii mari, cât și a genelor mutante din coloniile mici, în cadrul MLA, a fost examinată în detaliu (8) (10) (11) (12) (13). Natura moleculară și citogenică atât a genelor mutante care apar timpuriu, cât și a genelor mutante care apar tardiv, în cadrul TK6, a fost supusă unei examinări ample (14) (15) (16) (17). Genele mutante care cresc lent, pentru ambele tipuri de celule, au suferit deteriorări genetice care implică o genă (gene) presupusă (presupuse) a regla creșterea în apropierea locusului TK, ceea ce determină perioade de dublare prelungite și formarea de colonii care apar tardiv sau mici (18). Inducția genelor mutante care cresc lent a fost asociată cu substanțe chimice care induc schimbări structurale brute la nivel de cromozomi. Celulele a căror deteriorare nu implică o genă (gene) presupusă (presupuse) a regla creșterea în apropierea locusului TK cresc într-un ritm similar celui al celulelor parentale și devin gene mutante care cresc normal. Inducerea genelor mutante care cresc normal în principal este asociată cu substanțe chimice care acționează în principal ca agenți mutageni punctuali. În consecință, este esențial să se numere atât genele mutante care cresc lent, cât și genele mutante care cresc normal pentru a recupera toate genele mutante și pentru a prezenta o imagine de ansamblu a tipului (tipurilor) de deteriorări (mutageni/clastogeni) induse de substanța chimică de testat (10) (12) (18) (19).
|
|
5.
|
Metoda de testare este organizată astfel încât să furnizeze informații generale care se aplică atât pentru MLA, cât și pentru TK6, precum și indicații de specialitate pentru testele individuale.
|
|
6.
|
Definițiile utilizate sunt furnizate în apendicele 1.
|
CONSIDERAȚII PRELIMINARE ȘI LIMITĂRI
|
7.
|
Testele realizate in vitro necesită, în general, utilizarea unei surse exogene de activare metabolică. Sistemul exogen de activare metabolică nu reproduce integral condițiile in vivo.
|
|
8.
|
Ar trebui să se acorde atenție evitării condițiilor care ar putea conduce la rezultate pozitive artificiale (și anume, o posibilă interacțiune cu sistemul de testare), care nu sunt cauzate de interacțiunea dintre substanța chimică de testat și materialul genetic al celulei; astfel de condiții includ modificări ale pH-ului sau ale osmolalității, interacțiunea cu componentele de mediu (20) (21) sau niveluri excesive ale citotoxicității (22) (23) (24). Citotoxicitatea care depășește nivelurile recomandate de citotoxicitate de vârf, astfel cum sunt definite la punctul 28, este considerată excesivă pentru testul MLA și TK6. În plus, ar trebui remarcat faptul că substanțele chimice de testat care sunt analoge timidinei sau care se comportă ca analoge timidinei pot crește frecvența genelor mutante prin creșterea selectivă a genelor mutante spontane de fond în timpul tratamentului celular și necesită metode de testare suplimentare pentru o evaluare adecvată (25).
|
|
9.
|
În cazul nanomaterialelor fabricate, pot fi necesare adaptări specifice ale acestei metode de testare, dar care nu sunt descrise în prezenta metodă de testare.
|
|
10.
|
Înainte de utilizarea metodei de testare pentru a testa un amestec în vederea generării de date pentru un obiectiv de reglementare prevăzut, ar trebui să se analizeze dacă și, în caz afirmativ, din ce cauză aceasta poate oferi rezultate adecvate pentru respectivul scop. Astfel de considerații nu sunt necesare atunci când există o cerință de reglementare pentru testarea amestecului.
|
|
11.
|
Celulele mutante care nu pot desfășura activitatea enzimei timidin-kinază din cauza unei mutații TK
+/- în TK
-/- sunt rezistente la efectele citostatice ale trifluridinei (TFT) analoge pirimidinei. Celulele cu performanță în TK sunt sensibile la TFT, ceea ce produce inhibarea metabolismului celular și întrerupe continuarea diviziunii celulare. Astfel, celulele mutante pot să prolifereze în prezența TFT și să formeze colonii vizibile, însă celulele care conțin enzima TK nu pot face acest lucru.
|
PRINCIPIUL TESTULUI
|
12.
|
Celulele în suspensie sunt expuse la substanța chimică de testat cu și în absența unei surse exogene de activare metabolică (a se vedea punctul 19), pentru o perioadă adecvată de timp (a se vedea punctul 33) și apoi subcultivate pentru determinarea citotoxicității și pentru a permite manifestarea fenotipică înainte de selectarea mutantului. Citotoxicitatea este determinată prin creștere totală relativă (RTG - a se vedea punctul 25) pentru MLA și prin supraviețuire relativă (RS - a se vedea punctul 26) pentru TK6. Culturile tratate sunt menținute în mediul de creștere pentru un timp suficient, specific fiecărui tip de celulă (a se vedea punctul 37), pentru a permite o expresie fenotipică aproape optimă a mutațiilor induse. În urma manifestării fenotipice, frecvența mutanților se determină prin însămânțarea unui număr cunoscut de celule într-un mediu ce conține agentul selectiv pentru a detecta coloniile mutante și într-un mediu fără agent selectiv pentru a determina eficacitatea clonării (viabilitatea). După un timp corespunzător de incubare, se numără coloniile. Frecvența genelor mutante se calculează pe baza numărului de colonii de gene mutante, corectat prin eficacitatea clonării la momentul selecției genelor mutante.
|
DESCRIEREA METODEI
Pregătiri
Celule
|
13.
|
Pentru MLA: Întrucât MLA a fost dezvoltată și caracterizată folosind sublinia de celule L5178Y TK ± 3.7.2C, această sublinie specifică trebuie să fie utilizată pentru MLA. Linia celulară L5178Y a fost obținută dintr-un limfom timic indus de metilcolantren de la un șoarece DBA-2 (26). Clive și colaboratorii au tratat celulele L5178Y (denumite de Clive TK+/+ -3) cu sulfonat de etil metan și au izolat o clonă TK-/- (denumită TK-/- -3.7) folosind bromodeoxiuridină ca agent selectiv. Din clona TK-/- a fost izolată o clonă spontană TK+/- (denumită TK+/- -3.7.2.) și o subclonă (denumită TK+/--3.7.2C), care au fost caracterizate pentru a fi utilizate în MLA (27). Cariotipul pentru linia celulară a fost publicat (28) (29) (30) (31). Numărul modal de cromozomi este 40. Există un cromozom metacentric (t12;13) care ar trebui să fie considerat drept un singur cromozom. Locusul TK la șoarece este situat la capătul distal al cromozomului 11. Linia celulară L5178Y TK
+/- -3.7.2C suferă mutații la nivelul ambelor alele ale p53 și produce proteina mutantă p53 (32) (33). Statutul p53 al liniei celulare TK+/--3.7.2C este susceptibil de a fi responsabil pentru capacitatea testului de a detecta deteriorări la scară largă (17).
|
|
14.
|
Pentru TK6: TK6 este o linie celulară limfoblastoidă umană. Linia celulară parentală este o linie de celule transformată de virusul Epstein-Barr, WI-L2, care a fost inițial derivată de la un exemplar mascul de 5 ani cu sferocitoză ereditară. Prima clonă izolată, HH4, a fost mutagenizată cu ICR191 și a fost generată o linie celulară TK heterozigotă, TK6 (34). Celulele TK6 sunt aproape diploide și cariotipul reprezentantiv este 47, XY, 13 +, t(14; 20), t(3; 21) (35). Locusul TK la om este situat pe brațul lung al cromozomului 17. TK6 este o linie celulară cu proteină p53 competentă, deoarece prezintă o secvență de p53 de tip „wild” pe ambele alele și exprimă numai proteina p53 de tip „wild” (36).
|
|
15.
|
Pentru MLA și TK6, atunci când se stabilește sau se completează pentru prima dată un stoc principal, se recomandă ca laboratorul de testare să asigure absența contaminării cu micoplasmă, să determine cariotipul celulelor sau să vopsească cromozomii care adăpostesc locusul TK și să verifice perioadele de dublare a populației. Ar trebui să se stabilească durata normală a ciclului celular în cazul celulelor utilizate în laboratorul de testare, care să fie conform caracteristicilor celulare publicate (16) (19) (37). Acest stoc principal ar trebui să fie păstrat la -150o C sau la o temperatură mai mică și utilizat pentru a pregăti toate stocurile de celule de lucru.
|
|
16.
|
Înainte de a stabili un număr mare de stocuri de lucru păstrate în condiții criogenice sau chiar înainte de a fi utilizată într-un experiment, este posibil să fie necesară curățarea culturii de celulele mutante preexistente [cu excepția cazului în care frecvența genei mutante din martorul tratat cu solvent (MF) se află deja în intervalul acceptabil - a se vedea tabelul 2 pentru MLA)]. Aceasta se realizează folosind metotrexat (aminopterină) pentru selectarea și eliminarea celulelor deficiente de TK și adăugarea de timidină, hipoxantină și glicină (L5178Y) sau 2-deoxicitidină (TK6) în cultură pentru a asigura o creștere optimă a celulelor competente TK (19) (38) (39) și (40) pentru TK6. Îndrumări generale cu privire la bune practici pentru întreținerea culturilor celulare, precum și îndrumări specifică pentru celulele L5178Y și TK6 pot fi găsite în referințele (19) (31) (37) (39) (41). Pentru laboratoarele care necesită stocuri de celule principale pentru a iniția MLA sau TK6 sau pentru a obține stocuri de celule principale noi, este disponibil un depozit al celulelor care cuprinde celule bine caracterizate (37).
|
Condiții pentru medii și cultură
|
17.
|
Pentru ambele teste ar trebui să fie asigurate condiții adecvate pentru mediul de cultură și incubare (de exemplu, vase de cultură, umiditate atmosferică de 5 % CO2, temperatură de incubare de 37 °C) pentru menținerea culturilor. Culturile de celule ar trebui să fie menținute întotdeauna în condiții care să asigure creșterea acestora în etapa de înregistrare. Este deosebit de important să se aleagă condiții de medii și de cultură astfel încât să asigure creșterea optimă a celulelor în perioada de manifestare și clonarea atât pentru celulele mutante, cât și pentru cele nemutante. Pentru MLA și TK6, este important, de asemenea, să se asigure, prin condițiile de cultură, creșterea optimă atât a genelor mutante TK din coloniile mari/care se formează timpuriu, cât și a celor din coloniile mici/care se formează tardiv. Mai multe detalii privind cultura, inclusiv cu privire la necesitatea de a inactiva la căldură în mod corespunzător serul de cal dacă se utilizează mediul RPMI în timpul selecției genelor mutante, se pot găsi în referințele (19) (31) (38) (39) (40) (42).
|
Pregătirea culturilor
|
18.
|
Celulele se propagă din culturi mamă, se însămânțează în mediul de cultură la o anumită densitate astfel încât celulele în suspensie să continue să crească exponențial pe durata tratamentului și în perioadele de manifestare.
|
Activare metabolică
|
19.
|
Ar trebui să se utilizeze sisteme endogene de activare metabolică atunci când se utilizează celule L5178Y și TK6, deoarece acestea prezintă o capacitate metabolică endogenă inadecvată. Sistemul utilizat cel mai frecvent și care este recomandat implicit, cu excepția cazului în care se justifică altfel, constă într-o fracție postmitocondrială îmbogățită cu un cofactor (S9) preparată din ficat provenit de la rozătoare (în general, șobolani) tratat cu agenți de inducție enzimatică, de exemplu Aroclor 1254 (43) (44) (45) sau un amestec de fenobarbital și β-naftoflavonă (46) (47) (48) (49) (50) (51). Acest din urmă amestec nu este contrar Convenției de la Stockholm privind poluanții organici persistenți (52) și s-a dovedit a fi la fel de eficient ca și Aroclor 1254 pentru inducerea oxidazelor cu funcție mixtă (45) (46) (47) (48) (49). Fracția S9 este utilizată, în general, la concentrații pornind de la 1-2 % (v/v), dar poate fi crescută la 10 % (v/v) în mediul de testare final. Alegerea tipului și a concentrației sistemului exogen de activare metabolică sau a inductorului metabolic utilizat poate fi influențată de clasa de substanțe chimice de testat.
|
Prepararea substanțelor chimice de testat
|
20.
|
Substanțele chimice de testat în stare solidă ar trebui să fie preparate în solvenți adecvați și diluate, dacă este cazul, înainte de tratamentul aplicat celulelor (a se vedea punctul 21). Substanțele chimice de testat în stare lichidă pot fi adăugate direct în sistemul de testare și/sau diluate anterior tratamentului sistemului de testare. Substanțele chimice de testat gazoase sau volatile ar trebui să fie testate cu modificarea adecvată a protocoalelor standard, de exemplu tratarea în de vase de cultură închise ermetic (53) (54) (55). Preparatele cu substanța chimică de testat ar trebui să fie realizate anterior tratamentului, cu excepția cazului în care există date care să demonstreze stabilitatea acestora în ceea ce privește acceptabilitatea condițiilor de stocare.
|
CONDIȚII DE TESTARE
Solvenți
|
21.
|
Solventul ar trebui ales pentru a optimiza solubilitatea substanței chimice de testat fără a afecta negativ desfășurarea testului, de exemplu, modificarea creșterii celulare, afectarea integrității substanței chimice de testat, reacția cu vasele de cultură, afectarea sistemului de activare metabolică. Se recomandă ca, în măsura în care este posibil, să se ia în considerare în primă instanță utilizarea unui solvent apos (sau a unui mediu de cultură). Solvenții bine stabiliți sunt apa sau dimetilsulfoxidul. În general, solvenții organici nu ar trebui să depășească 1 % (v/v) și solvenții apoși (soluție salină sau apă) nu ar trebui să depășească 10 % (v/v) în mediul final de tratament. În cazul în care nu se utilizează solvenți bine stabiliți (de exemplu, etanol sau acetonă), utilizarea acestora se justifică prin date care să indice compatibilitatea acestora cu substanțele chimice de testat, cu sistemul de testare, precum și absența genotoxicității la concentrația utilizată. În lipsa unor astfel de date justificative, este important să se includă martori netratați (a se vedea apendicele 1, Definiții) pentru a demonstra că solventul ales nu induce efecte vătămătoare sau mutagene.
|
MĂSURAREA CITOTOXICITĂȚII ȘI ALEGEREA CONCENTRAȚIILOR DE TRATAMENT
|
22.
|
În momentul determinării celei mai mari concentrații a substanței chimice de testat, ar trebui să se evite concentrațiile susceptibile de a produce răspunsuri pozitive date de artefacte, cum ar fi cele care produc citotoxicitate excesivă (a se vedea punctul 28), precipitate (a se vedea punctul 29) în mediul de cultură sau modificări importante de pH sau osmolalitate (a se vedea punctul 8). În cazul în care substanța chimică de testat provoacă o modificare importantă a pH-ului mediului la momentul adăugării acesteia, pH-ul ar putea fi ajustat prin tamponarea mediului final de tratament astfel încât să se evite rezultate pozitive date de artefacte și să se mențină condiții de cultură corespunzătoare.
|
|
23.
|
Selecția concentrației este efectuată în funcție de citotoxicitate și pe baza altor considerente (a se vedea punctele 27-30). Chiar dacă evaluarea citotoxicității în cadrul unui test inițial poate fi utilă pentru o mai bună definire a concentrațiilor care urmează să fie utilizate în experimentul principal, efectuarea unui test inițial nu este obligatorie. Chiar dacă se realizează o evaluare inițială a citotoxicității, măsurarea citotoxicității pentru fiecare cultură este totuși necesară în cadrul experimentului principal. În cazul în care se realizează un experiment de stabilire a intervalului, acesta ar trebui să acopere o gamă largă de concentrații și poate fie să fie încheiat în ziua 1 după tratament sau continuat în ziua 2 de manifestare, până la momentul selecției genelor mutante (în cazul în care este evident caracterul adecvat al concentrațiilor).
|
|
24.
|
Citotoxicitatea ar trebui să fie determinată pentru fiecare cultură de testare și cultură martor: metodele pentru MLA (2) și TK6 (15) sunt definite prin practicile convenite la nivel internațional.
|
|
25.
|
În cazul versiunilor cu agar-agar și micro-godeuri ale MLA: ar trebui să se evalueze citotoxicitatea folosind creșterea relativă totală (RTG), care a fost definită inițial de Clive și Spector în 1975 (2). Această măsură include creșterea relativă a suspensiei (RSG: cultură de testare/martor tratat cu solvent) în timpul tratamentului celulelor, timpul de manifestare și eficacitatea clonării relative (RCE: cultură de testare/martor tratat cu solvent) la momentul selecției genelor mutante (2). Ar trebui remarcat faptul că RSG include orice pierdere de celule care se produce în cultura de testare în timpul tratamentului (a se vedea formulele din apendicele 2).
|
|
26.
|
Pentru TK6: Citotoxicitatea ar trebui să fie evaluată folosind indicele de supraviețuire relativă (RS), și anume, eficacitatea clonării celulelor placate imediat după tratament, ajustată pentru orice pierdere de celule în timpul tratamentului, pe baza numărului de celule, în comparație cu martorul negativ (se atribuie o rată de supraviețuire de 100 %) (a se vedea formula din apendicele 2).
|
|
27.
|
Ar trebui să se evalueze cel puțin patru concentrații de testat (fără a include martorii cu solvent și cei pozitivi) care îndeplinesc criteriile de acceptabilitate (citotoxicitate adecvată, număr de celule etc.). Chiar dacă se recomandă utilizarea culturilor duplicate, se poate utiliza fie cultura duplicat, fie cultura tratată ca fiind unică în cadrul fiecărei concentrații testate. Rezultatele obținute la culturile replicate la o concentrație dată ar trebui să fie consemnate separat, însă pot fi grupate pentru analiza datelor (55). În cazul substanțelor chimice de testat cu citotoxicitate ușoară sau fără citotoxicitate, sunt adecvate, de regulă, intervale de concentrație de aproximativ 2 până la 3 ori mai mari. Dacă apare citotoxicitatea, ar trebui să se selecteze concentrații care să acopere intervalul de citotoxicitate de la valoarea care produce citotoxicitate, astfel cum este descris la punctul 28, incluzând concentrații la care gradul de citotoxicitate este moderat și redus sau zero. Multe substanțe chimice de testat prezintă curbe concentrație-răspuns cu pante abrupte, iar pentru a acoperi întreagul interval de valori de citotoxicitate sau pentru a studia în detaliu relația concentrație-răspuns, ar putea fi necesară utilizarea concentrațiilor cu spațiu de separare redus și mai mult de patru concentrații, în special în situațiile în care este necesară repetarea experimentului (a se vedea punctul 70). Utilizarea a peste patru concentrații poate fi deosebit de importantă atunci când se utilizează culturi unice.
|
|
28.
|
În cazul în care concentrația maximă se bazează pe citotoxicitate, cea mai mare concentrație ar trebui să vizeze obținerea unei valori cuprinse între 20 și 10 % RTG pentru MLA și între 20 și 10 % RS pentru TK6 (punctul 67).
|
|
29.
|
În cazul substanțelor chimice de testat cu solubilitate redusă care nu sunt citotoxice la concentrații mai mici decât concentrația minimă de insolubilitate, cea mai mare concentrație analizată produce o turbiditate sau un precipitat vizibil cu ochiul liber sau cu ajutorul unui microscop inversat la sfârșitul tratamentului cu substanța chimică de testat. Chiar dacă apare citototoxicitate peste nivelul cel mai scăzut de concentrația de insolubilitate, se recomandă testarea doar la o singură concentrație care produce turbiditate sau cu un precipitat vizibil deoarece precipitatul ar putea genera efecte artificiale. Deoarece MLA și TK6 utilizează culturi în suspensie, ar trebui să se acorde atenție pentru a se asigura faptul că precipitatul nu interferează cu desfășurarea testului. Ar putea fi utilă, de asemenea, determinarea solubilității în mediul de cultură înainte de experiment.
|
|
30.
|
În cazul în care se constată inexistența precipitatului sau o citotoxicitate limitată, concentrația maximă de testare ar tregbui să corespundă cu 10 mm, 2 mg/ml sau 2 μl/ml, fiind reținută cea mai mică valoare (57) (58). În cazul în care substanța chimică de testat nu are o compoziție definită, de exemplu, o substanță cu o compoziție necunoscută sau variabilă, produse cu reacție complexă sau materiale biologice [și anume, substanțele chimice cu o compoziție necunoscută sau variabilă (UVCB)], extracte din mediu etc., este posibil să fie necesară o concentrație de vârf mai mare (de exemplu, 5 mg/ml), în absența citotoxicității suficiente, pentru a crește concentrația fiecăreia dintre componente. Cu toate acestea, ar trebui remarcat faptul că aceste cerințe pot fi diferite de cele pentru produsele farmaceutice umane (59).
|
Martori
|
31.
|
Pentru fiecare condiție experimentală ar trebui să se includă martori negativi testați concomitent (a se vedea punctul 21) constituiți doar dintr-un solvent în mediul de tratare și tratați în aceleași condiții precum culturile de tratare.
|
|
32.
|
Martorii pozitivi testați în paralel sunt necesari pentru a demonstra capacitatea laboratorului de a identifica genele mutagene în condițiile prevăzute în protocolul de testare utilizat și eficacitatea sistemului exogen de activare metabolică (după caz) și a demonstra detectarea adecvată atât a genelor mutante TK mici/care se formează tardiv, cât și a celor mari/care se formează timpuriu. Tabelul 1 de mai jos prezintă exemple de martori pozitivi. Pot fi utilizate substanțe martor pozitive alternative, dacă acest lucru este justificat. Întrucât testele in vitro de genotoxicitate pe celule de mamifere sunt suficient de standardizate pentru tratamentele pe termen scurt (3-4 ore) efectuate concomitent cu și fără activare metabolică utilizând aceeași durată a tratamentului, utilizarea de martori pozitivi poate fi limitată la un mutagen care necesită activare metabolică. În acest caz, acest răspuns al unicului martor pozitiv va demonstra atât activitatea sistemului de activare metabolică, cât și capacitatea de răspuns a sistemului de testare. Dacă este utilizat, tratamentul de lungă durată (și anume, 24 de ore fără S9) necesită propriul martor poziti, deoarece durata tratamentului va fi diferită de testul care utilizează activarea metabolică. Fiecare martor pozitiv ar trebui să fie utilizat la una sau mai multe concentrații prevăzute a genera creșteri reproductibile și detectabile față de cele de fond pentru a demonstra sensibilitatea sistemului de testare, iar răspunsul nu ar trebui să fie compromis de citotoxicitate care depășește limitele specificate în prezenta MT (a se vedea punctul 28).
|
Tabelul 1
Substanțe de referință recomandate pentru evaluarea competenței de laborator și pentru selectarea de martori pozitivi
|
Categorie
|
martor
|
NR CAS
|
|
1. Mutageni activi fără activare metabolică
|
|
Metansulfonat de metil
|
66-27-3
|
|
Mitomicină C
|
50-07-7
|
|
4-nitrochinolină-N oxid
|
56-57-5
|
|
2. Mutageni care necesită activare metabolică
|
|
Benzo(a)piren
|
50-32-8
|
|
Ciclofosfamidă (monohidrat)
|
50-18-0 (6055-19-2)
|
|
7,12-dimetilbenzantracen
|
57-97-6
|
|
3-metilclorantren
|
56-49-5
|
PROCEDURA
Tratamentul cu substanța chimică de testat
|
33.
|
Celulele în stadiu de proliferare se tratează cu substanța chimică de testat atât în prezența, cât și în absența unui sistem de activare metabolică. Timpul de expunere ar trebui să fie corespunzător (de obicei, un timp de 3 până la 4 ore este suficient). Cu toate acestea, ar trebui remarcat faptul că aceste cerințe pot fi diferite de cele pentru produsele farmaceutice umane (59). Pentru MLA, în cazul în care tratamentul pe termen scurt generează rezultate negative și există informații care sugerează necesitatea unui tratament prelungit [de exemplu, analogi nucleoside, substanțele chimice cu solubilitate redusă, (5) (59)], ar trebui avută în vedere efectuarea testului cu tratamentul prelungit, și anume, 24 de ore fără S9.
|
|
34.
|
Numărul minim de celule utilizate pentru fiecare cultură de testare (martor și tratată) în fiecare etapă a testului ar trebui să depindă de frecvența genelor mutante spontane. Ca și orientare generală, se tratează și se trec celule suficiente în fiecare cultură experimentală, astfel încât să se mențină cel puțin 10, dar, în mod ideal, 100 de gene mutante spontane în toate etapele testului (tratament, expresie fenotipică și selecția genelor mutante) (56).
|
|
35.
|
Pentru MLA, frecvența recomandată acceptabilă a genelor mutante spontane este cuprinsă între 35-140 × 10-6 (versiunea cu agar-agar) și 50-170 × 10-6 (versiunea cu micro-godeuri) (a se vedea tabelul 2). Pentru a avea cel puțin 10 și, în mod ideal, 100 de gene mutante spontane care supraviețuiesc tratamentului pentru fiecare cultură de testare, este necesar să se trateze cel puțin 6 × 106 de celule. Tratarea acestui număr de celule și menținerea unui număr suficient de celule în timpul expresiei și al clonării în scopul selectării genelor mutante asigură un număr suficient de gene mutante spontane (10 sau mai multe) în toate etapele experimentului, chiar și pentru culturile tratate la concentrații care conduc la o citotoxicitate de 90 % (astfel cum este măsurată prin RTG de 10 %) (19) (38) (39).
|
|
36.
|
Pentru TK6, frecvența genelor mutante spontane se situează, în general, între 2 și 10 × 10-6. Pentru a avea cel puțin 10 mutanți spontani care supraviețuiesc tratamentului pentru fiecare cultură, este necesar să se trateze cel puțin 20 × 106 de celule. Tratarea acestui număr de celule asigură un număr suficient de mutanți spontani (10 sau mai multe) chiar și pentru culturile tratate la concentrații care provoacă o citotoxicitate de 90 % în timpul tratamentului (10 % RS). În plus, în perioada de manifestare, trebuie cultivate un număr suficient de celule care să fie placate pentru selecția mutanților (60).
|
Timpul de expresie fenotipică și măsurarea citotoxicității și a frecvenței mutanților
|
37.
|
La sfârșitul perioadei de tratament, celulele sunt cultivate pentru un anumit timp pentru a permite expresia fenotipică aproape optimă a mutanților nou induși; specific fiecărei linii celulare. În cazul MLA, perioada de expresie fenotipică este de 2 zile. În cazul TK6, perioada de expresie fenotipică este de 3-4 zile. În cazul în care se folosește un tratament de 24 h, perioada de expresie începe după încheierea tratamentului.
|
|
38.
|
În timpul perioadei de expresie fenotipică, celulele sunt enumerate zilnic. În cazul MLA, numărul zilnic de celule este utilizat pentru a calcula creșterea zilnică a suspensiei (SG). După perioada de două zile de expresie, celulele sunt suspendate în mediu cu și fără agent selectiv pentru determinarea numărului de mutanți (plăci de selecție) și, respectiv, pentru eficacitatea clonării (plăci de viabilitate). În cazul MLA, există două metode la fel de acceptabile pentru clonarea la selecția mutantului; una care utilizează agar-agar moale și cealaltă care utilizează mediu lichid în plăci cu 96 de godeuri (19) (38) (39). Clonarea în TK6 este realizată utilizând medii lichide și plăci cu 96 de godeuri (16).
|
|
39.
|
Triflurotimidina (TFT) este singurul agent selectiv recomandat pentru mutanții TK (61).
|
|
40.
|
Pentru MLA, plăcile cu agar-agar și plăcile cu micro-godeuri sunt numărate după 10-12 zile de incubare. În cazul TK6, coloniile din plăcile cu micro-godeuri sunt marcate după 10-14 zile pentru mutanții care se formează timpuriu. Pentru a recupera mutanții TK6 care cresc lent (se formează tardiv), celulel trebuie să fie realimentate cu mediu de creștere și TFT după numărarea mutanților timpurii și apoi să se incubează plăcile timp de încă 7-10 zile (62). A se vedea punctele 42 & 44 pentru o discuție referitoare la enumerarea mutanților TK care cresc lent și a celor care cresc normal.
|
|
41.
|
Calculele corespunzătoare pentru cele două teste, inclusiv cele două metode (cu agar-agar și micro-godeuri) pentru MLA sunt date în apendicele 2. Pentru metoda cu agar-agar a MLA, se numără coloniile, iar numărul de colonii de mutanți este ajustat prin eficacitatea clonării pentru calcularea MF. Pentru versiunea cu micro-godeuri a MLA și a TK6, eficacitatea clonării atât în cazul plăcilor de selecție, cât și al celor de eficacitate a clonării este determinată conform distribuției Poisson (63). MF se calculează din aceste două valori ale eficacității clonării.
|
Caracterizarea coloniilor de mutanți
|
42.
|
Pentru MLA, în cazul în care substanța chimică de testat este pozitivă (a se vedea punctele 62-63), coloniile ar trebui să fie caracterizate în funcție de dimensiunea sau creșterea acestora la cel puțin una dintre culturile de testare (în general, cea mai mare concentrație pozitivă acceptabilă) și la martorii negativi și pozitivi. În cazul în care substanța chimică de testat este negativă (a se vedea punctul 64), caracterizarea coloniilor de mutanți ar trebui să fie efectuată la martorii negativi și pozitivi. Pentru metoda cu micro-godeuri a MLA, mutanții din colonii mici sunt definiți a fi cei care acoperă mai puțin de 25 % din diametrul godeului, iar mutanții din colonii mari cei care acoperă mai mult de 25 % din diametrul godeului. Pentru metoda agar-agar, se utilizează un contor de colonii automat pentru numărarea și dimensionarea coloniilor de mutanți. Metodele utilizate pentru dimensionarea coloniilor sunt prezentate în detaliu în literatură (19) (38) (40). Caracterizarea coloniilor la martorii negativi și pozitivi este necesară pentru a demonstra că studiile sunt realizate în mod adecvat.
|
|
43.
|
Substanța chimică de testat nu poate fi determinată ca fiind negativă dacă mutanții, atât cei din coloniile mari, cât și cei din coloniile mici nu sunt detectați corespunzător în martorul pozitiv. Caracterizarea coloniilor poate fi utilizată pentru a furniza informații generale privind capacitatea substanței chimice de testat de a cauza mutații punctiforme și/sau evenimente cromozomiale (punctul 4).
|
|
44.
|
TK6: Diferența dintre mutanții care cresc normal și cei care cresc lent este indicată prin diferența de la nivelul timpului de incubare (a se vedea punctul 40). Pentru TK6, se punctează, în general, atât mutanții care se formează timpuriu, cât și cei care se formează tardiv, pentru toate culturile, inclusiv martorii negativi și pozitivi. Caracterizarea coloniilor la martorii negativi și pozitivi este necesară pentru a demonstra că studiile sunt realizate în mod adecvat. Substanța chimică de testat nu poate fi determinată ca fiind negativă dacă atât mutanții care se formează timpuriu, cât și cei care se formează tardiv nu sunt detectați corespunzător în martorul pozitiv. Caracterizarea coloniilor poate fi utilizată pentru a furniza informații generale privind capacitatea substanței chimice de testat de a cauza mutații punctiforme și/sau evenimente cromozomiale (punctul 4).
|
Competența de laborator
|
45.
|
Pentru a demonstra o experiență suficientă în materie de testare înainte de utilizarea acestuia pentru efectuarea testelor de rutină, laboratorul ar trebui să fi efectuat o serie de experiențe cu substanțe pozitive de referință care acționează prin diferite mecanisme (cel puțin una cu și una fără activare metabolică, alese dintre substanțele enumerate în tabelul 1) și diferiți martori negativi (inclusiv culturi netratate și diferiți solvenți/diferite vehicule). Aceste răspunsuri ale martorilor pozitivi și negativi ar trebui să fie în concordanță cu referințele bibliografice. Această cerință nu se aplică laboratoarelor care au experiență, și anume, cele care au o bază de date anterioare disponibilă, astfel cum este definit la punctele 47-50. În cazul MLA, valorile obținute pentru martorii pozitivi și cei negativi ar trebui să fie în concordanță cu recomandările IWGT (a se vedea tabelul 2).
|
|
46.
|
În absența activării metabolice ar trebui să se examineze o selecție de substanțe martor pozitive (a se vedea tabelul 1) în cadrul unor tratamente pe termen scurt și lung (dacă se utilizează tratamente pe termen lung) și, de asemenea, în cadrul tratamentelor pe termen scurt, în prezența activării metabolice, pentru a demonstra capacitatea de a detecta substanțele chimice mutagene, pentru a determina eficacitatea sistemului de activare metabolică și pentru a demonstra caracterul adecvat al condițiilor de creștere a celulelor în timpul tratamentului, al manifestării fenotipice și al selecției celulelor mutante, precum și al procedurilor de punctare. Ar trebui să se aleagă o plajă de valori ale concentrațiilor substanțelor chimice selectate astfel încât să se obțină creșteri reproductibile și legate de concentrație peste nivelul de fond pentru a demonstra sensibilitatea și gama dinamică a sistemului de testare.
|
Date anterioare privind martorii testați
|
47.
|
Laboratorul ar trebui să stabilească:
|
—
|
o plajă de valori și o distribuție a martorilor pozitivi testați anterior,
|
|
—
|
o plajă de valori și o distribuție a martorilor negativi (netratați, solvenți) testați anterior.
|
|
|
48.
|
La obținerea primelor date pentru o distribuție a martorilor negativi testați anterior, martorii negativi testați în paralel trebuie să fie în concordanță cu datele publicate referitoare la martori negativi. Pe măsură ce se adaugă mai multe date experimentale la distribuția martorilor, martorii negativi testați în paralel ar trebui să se încadreze, în mod ideal, în limita de 95 % a martorilor din distribuția respectivă (64) (65).
|
|
49.
|
Baza de date privind martori negativi testați anterior a laboratorului ar trebui să fie construită inițial cu cel puțin 10 experimente, dar ar fi de preferat să conțină cel puțin 20 de experimente efectuate în conformitate cu condiții experimentale comparabile. Laboratoarele ar trebui să utilizeze metode de control al calității precum grafice de control [de exemplu, diagrame C sau X (65)] pentru a identifica gradul de variabilitate al datelor privind martorii negativi și pozitivi ale acestora și pentru a dovedi faptul că metodologia se află „sub control” în cadrul respectivului laborator (66). Referințele bibliografice prezintă detalii și recomandări suplimentare privind modul de dezvoltare și utilizare a datele istorice (64).
|
|
50.
|
Datele privind martorii negativi ar trebui să constea în frecvențe ale celulelor mutante dintr-o cultură unică sau, de preferință, din culturi duplicate, astfel cum este descris la punctul 27. Martorii negativi testați în paralel ar trebui să se încadreze, în mod ideal, în limita de 95 % a martorilor din distribuția bazei de date privind martorii negativi testați în paralel a laboratorului. În cazul în care datele privind martorii negativi se situează în afara limitei de 95 % a martorilor, acestea pot fi acceptabile pentru a fi incluse în distribuția martorilor testați anterior, în măsura în care datele respective nu reprezintă valori aberante extreme, există dovezi că sistemul de testare se află „sub control” (a se vedea punctul 49) și nu există nicio dovadă a unor erori tehnice sau umane.
|
|
51.
|
Orice modificare a protocolului experimental ar trebui să fie avută în vedere sub aspectul coerenței datelor cu bazele de date existente ale laboratorului cu privire la martorii testați anterior. Orice inconsecvență majoră ar trebui să conducă la stabilirea unei noi baze de date privind martorii testați anterior.
|
DATE ȘI RAPORTARE
Prezentarea rezultatelor
|
52.
|
Prezentarea datelor atât pentru MLA, cât și pentru TK6 ar trebui să includă, atât pentru culturile tratate, cât și pentru cele martor, datele necesare pentru calcularea citotoxicității (RTG sau, respectiv, RS) și frecvențele mutanților, conform descrierii de mai jos.
|
|
53.
|
În cazul MLA, ar trebui să se furnizeze date cu privire la fiecare cultură pentru RSG, RTG, eficacitatea clonării în momentul selecției mutanților și numărul de colonii de mutanți (pentru versiunea cu agar-agar) sau numărul de godeuri goale (pentru versiunea cu micro-godeuri). MF ar trebui exprimat ca număr de celule mutante la un milion de celule supraviețuitoare. Dacă răspunsul este pozitiv, valorile MF pentru colonii mici și mari (și/sau procentul MF total) ar trebui să fie date pentru cel puțin o concentrație a substanței chimice de testat (în general, cea mai mare concentrație pozitivă) și a martorilor negativi și pozitivi. În cazul unui răspuns negativ, ar trebui să fie furnizate valorile MF pentru coloniile mici și cele mari pentru martorul negativ și cel pozitiv.
|
|
54.
|
În cazul TK6, ar trebui să fie furnizate date despre fiecare cultură pentru RS, eficacitatea clonării în momentul selecției mutanților și numărul godeurilor goale pentru mutanții care se formează timpuriu și cei care se formează tardiv. MF ar trebui să fie exprimată ca număr de celule mutante pe număr de celule supraviețuitoare și ar trebui să includă valoarea MF totală, precum și MF (și/sau procentul MF total) pentru mutații care se formează timpuriu și cei care se formează tardiv.
|
Criterii de acceptabilitate
|
55.
|
Atât pentru MLA, cât și pentru TK6, ar trebui să fie îndeplinite următoarele criterii înainte de a determina rezultatele globale pentru o anumită substanță chimică de testat:
|
—
|
au fost aplicate două condiții experimentale (tratament scurt cu și fără activare metabolică - a se vedea punctul 33), cu excepția cazului în care una a generat rezultate pozitive.
|
|
—
|
Ar trebui să se poată analiza un număr adecvat de celule și concentrații (a se vedea punctele 27, 34-36).
|
|
—
|
Criteriile de selecție a concentrației maxime sunt conforme cu cele descrise la punctele 28-30.
|
|
Criterii de acceptabilitate pentru martorii negativi și pozitivi
|
56.
|
Analiza Grupului de lucru de experți MLA din cadrul IWGT asupra unei cantități mari de date MLA a culminat cu un consens internațional cu privire la criteriile de acceptabilitate specifice pentru MLA (1) (2) (3) (4) (5). Prin urmare, această metodă de testare oferă recomandări specifice pentru determinarea acceptabilității martorilor negativi și pozitivi și pentru evaluarea rezultatelor fiecărei substanțe în cadrul MLA. TK6 are o bază de date mult mai mică și nu a făcut obiectul unei evaluări de către un grup de lucru.
|
|
57.
|
În cazul MLA, fiecare experiment ar trebui să fie evaluat cu privire la problema dacă martorul netratat/tratat cu solvent îndeplinește criteriile de acceptare ale grupului de lucru MLA din cadrul IWGT [punctul (4) și tabelul 2 de mai jos] pentru: (1) MF (este de remarcat faptul că valorile acceptabile ale IWGT sunt diferite de versiunile cu agar-agar și cu micro-godeuri ale MLA), (2) eficacitatea clonării (CE) în momentul selectării mutanților și (3) creșterea suspensiei (SG) pentru martorul tratat cu solvent (a se vedea formulele din apendicele 2).
Tabelul 2
Criterii de acceptabilitate pentru MLA
|
Parametru
|
Metoda agar-agar moale
|
Metoda cu micro-godeuri
|
|
Frecvența mutanților
|
35-140 × 10-6
|
50-170 × 10-6
|
|
Eficacitatea clonării
|
65-120 %
|
65-120 %
|
|
Creșterea suspensiei
|
de 8-32 de ori (3-4 ore de tratament) de 32-180 de ori (24 de ore de tratament, dacă este efectuat)
|
de 8-32 de ori (3-4 ore de tratament) de 32-180 de ori (24 de ore de tratament, dacă este efectuat)
|
|
|
58.
|
În cazul MLA, fiecare test ar trebui să fie evaluat, de asemenea, cu privire la problema dacă martorul (martorii) pozitiv(i) respectă cel puțin unul dintre următoarele două criterii de acceptare elaborate de grupul de lucru din cadrul IWGT:
|
—
|
Martorul pozitiv ar trebui să demonstreze o creștere absolută a valorii MF totale, și anume, o creștere peste fondul spontan MF [o valoare MF (IMF) indusă] de cel puțin 300 × 10-6. Cel puțin 40 % din valoarea IMF ar trebui să se reflecte în MF aferent coloniilor mici.
|
|
—
|
Un martor pozitiv înregistrează o creștere a valorii MF a coloniei mici cu cel puțin 150 × 10-6 peste valoarea înregistrată la martorul netratat/tratat cu solvent testat în paralel (o valoare IMF a coloniei mici de 150 × 10-6).
|
|
|
59.
|
Pentru TK6, va fi acceptabil un test dacă martorul negativ testat în paralel este considerat acceptabil pentru a fi adăugat în baza de date privind martorii negativi testați anterior a laboratorului, astfel cum este descris la punctele 48-49. În plus, martorii pozitivi testați în paralel (a se vedea punctul 32) ar trebui să inducă răspunsuri compatibile cu cele provenite din baza de date privind martorii pozitivi testați anterior și să producă o creștere semnificativă din punct de vedere statistic în comparație cu martorul negativ testat în paralel.
|
|
60.
|
Pentru ambele teste, limita superioară de citotoxicitate observată la cultura de martori pozitivi ar trebui să fie aceeași cu cea a culturilor experimentale. Adică RTG/RS nu ar trebui să fie sub 10 %. Este suficient să se utilizeze o singură concentrație (sau una dintre concentrațiile culturilor de martori pozitivi în cazul în care se utilizează mai multe concentrații) pentru a demonstra că au fost îndeplinite criteriile de acceptare pentru martorul pozitiv. În plus, valoarea MF a martorului pozitiv trebuie să se situeze în intervalul acceptabil stabilit pentru laborator.
|
Evaluarea și interpretarea rezultatelor
|
61.
|
În cazul MLA, grupul de lucru de experți privind limfomul la șoarece (The Mouse Lymphoma Expert Workgroup) din cadrul IWGT a desfășurat o activitate intensă cu privire la relevanța biologică și criteriile pentru un răspuns pozitiv. Prin urmare, prezenta metodă de testare oferă recomandări specifice pentru interpretarea rezultatelor substanțelor chimice de testat din MLA (a se vedea punctele 62-64). TK6 are o bază de date mult mai mică și nu a făcut obiectul unei evaluări de către un grup de lucru. Prin urmare, recomandările pentru interpretarea datelor pentru TK6 sunt prezentate în termeni mai generali (a se vedea punctele 65-66). Pentru ambele teste se aplică recomandări suplimentare (a se vedea punctele 67-71).
|
MLA
|
62.
|
Se recomandă o abordare pentru definirea răspunsurilor pozitive și negative, pentru a se asigura relevanța biologică a creșterii MF. În locul analizei statistice utilizate, în general, pentru alte teste, aceasta se bazează pe utilizarea unei frecvențe predefinite a mutanților (și anume, o creștere a MF peste cea a martorului testat în paralel), care a fost denumită factor de evaluare globală (GEF) și care se bazează pe analiza distribuției datelor privind MF a martorul negativ din laboratoarele participante (4). Pentru versiunea cu agar-agar a MLA, GEF este 90 × 10-6 și pentru versiunea cu micro-godeuri a MLA, GEF este 126 × 10-6.
|
|
63.
|
Cu condiția ca toate criteriile de acceptabilitate să fie îndeplinite, o substanță chimică de testat este considerată a fi în mod clar pozitivă dacă, în oricare dintre condițiile experimentale examinate (a se vedea punctul 33), creșterea MF peste fondul testat în paralel depășește GEF, iar creșterea este legată de concentrație (de exemplu, prin utilizarea unui test al tendințelor). Atunci substanța chimică de testat este considerată a fi capabilă de a induce mutații în cadrul acestui sistem de testare.
|
|
64.
|
Cu condiția ca toate criteriile de acceptabilitate să fie îndeplinite, o substanță chimică de testat este considerată a fi în mod clar negativă dacă, în toate condițiile experimentale examinate (a se vedea punctul 33) nu există niciun răspuns legat de concentrare sau, dacă există o creștere a MF, aceasta nu depășește GEF. Atunci substanța chimică de testat este considerată a fi incapabilă de a induce mutații în cadrul acestui sistem de testare.
|
TK6
|
65.
|
Cu condiția ca toate criteriile de acceptabilitate să fie îndeplinite, o substanță chimică de testat este considerată a fi în mod clar pozitivă dacă, în oricare dintre condițiile experimentale examinate (a se vedea punctul 33):
|
—
|
cel puțin una dintre concentrațiile de testare prezintă o creștere semnificativă din punct de vedere statistic în comparație cu martorul negativ testat în paralel
|
|
—
|
atunci când este evaluată prin intermediul unui test de stabilire a tendinței corespunzător, creșterea depinde de concentrație (a se vedea punctul 33)
|
|
—
|
Oricare dintre aceste rezultate se situează în afara distribuției de date cu privire la martorii negativi testați anterior (de exemplu, limita de control de 95 % pe baza distribuției Poisson; a se vedea punctul 48).
|
La îndeplinirea tuturor acestor criterii, se consideră că substanța chimică de testat poate produce mutații în cadrul acestui sistem de testare. Referințele bibliografice prezintă recomandări pentru cele mai adecvate metode statistice (66) (67).
|
|
66.
|
Cu condiția ca toate criteriile de acceptabilitate să fie îndeplinite, o substanță chimică de testat este considerată a fi în mod clar negativă în cazul în care, în toate condițiile experimentale examinate (a se vedea punctul 33):
|
—
|
niciuna dintre concentrațiile de testare nu prezintă o creștere semnificativă din punct de vedere statistic în comparație cu martorul negativ testat în paralel,
|
|
—
|
atunci când este evaluată prin intermediul unui test de stabilire a tendinței, nu se înregistrează nicio creștere legată de concentrație,
|
|
—
|
toate rezultatele se situează în interiorul distribuției de date cu privire la martorii negativi testați anterior (de exemplu, limita de control de 95 % pe baza distribuției Poisson; a se vedea punctul 48).
|
Atunci substanța chimică de testat este considerată a fi incapabilă de a induce mutații în cadrul acestui sistem de testare.
|
Pentru MLA și TK6:
|
67.
|
În cazul în care concentrația maximă se bazează pe citotoxicitate, cea mai mare concentrație ar trebui să ajungă la valori ale RTG/RS situate între 20 și 10 %. Consensul este că ar trebui să se acorde atenție atunci când se interpretează rezultate pozitive aflate numai între 20 și 10 % RTG/RS, iar rezultatul nu ar fi considerat pozitiv în cazul în care creșterea MF s-ar produce doar la 10 % RTG/RS sau sub această valoare (dacă este evaluată) (2) (59).
|
|
68.
|
Există anumite circumstanțe în care este posibil să se stabilească, prin informații suplimentare, dacă o substanță chimică de testat nu este mutagenă atunci când nu există o cultură care să indice o valoare RTG între 10-20 % RTG/RS. Aceste situații sunt prezentate în continuare: (1) Nu există nicio dovadă de mutagenicitate (de exemplu, absența unui răspuns la doză, nicio frecvență a mutanților peste celor observate la martorii negativi testați în paralel sau în intervalele de fond istorice etc.) într-o serie de puncte de date în intervalul 100 %-20 % RTG/RS și există cel puțin un punct de date între 20 și 25 % RTG/RS. (2) Nu există nicio dovadă de mutagenicitate (de exemplu, absența unui răspuns la doză, nicio frecvență a mutanților peste celor observate la martorii negativi testați în paralel sau în intervalele de fond istorice etc.) într-o serie de puncte de date în intervalul 100 %-20 % RTG/RS și există, de asemenea, un punct de date negativ sub 10 % RTG/RS. În ambele situații, se poate concluziona că substanța chimică de testat este negativă.
|
|
69.
|
Nu există nicio cerință referitoare la verificarea unui răspuns clar pozitiv sau negativ.
|
|
70.
|
În cazul în care răspunsul nu este nici în mod clar negativ, nici în mod clar pozitiv, astfel cum este descris mai sus și/sau pentru a ajuta la stabilirea relevanței biologice a unui rezultat, datele ar trebui să fie evaluate de către experți și/sau prin intermediul unor investigații suplimentare. Ar putea fi utilă repetarea unui experiment, eventual folosind condiții experimentale modificate [de exemplu, stabilirea unei distanța între concentrații pentru a crește probabilitatea de a atinge puncte de date în intervalul 10-20 % RTG/RS, utilizând alte condiții de activare metabolică (și anume, concentrație S9 sau origine S9) și durata tratamentului].
|
|
71.
|
În cazuri rare, chiar și după efectuarea de investigații suplimentare, setul de date va exclude posibilitatea stabilirii unei concluzii pentru rezultate pozitive sau negative. Prin urmare, răspunsul substanței chimice de testat ar trebui să fie concluzionat ca fiind echivoc (interpretat cu aceeași probabilitate de a fi pozitiv sau negativ).
|
RAPORT TESTULUI
|
72.
|
Raportul testului ar trebui să includă următoarele informații:
|
|
Substanța chimică de testat:
|
—
|
sursa, numărul lotului, data limită de utilizare, dacă sunt disponibile;
|
|
—
|
stabilitatea substanței chimice de testat, dacă se cunoaște;
|
|
—
|
solubilitatea și stabilitatea substanței chimice de testat în solvent, dacă se cunosc;
|
|
—
|
măsurarea pH-ului, a osmolalității și a precipitatului în mediul de cultură în care s-a adăugat substanța chimică de testat, după caz.
|
|
|
Substanță mono-constituent:
|
—
|
aspectul fizic, solubilitatea în apă și alte proprietăți fizico-chimice relevante;
|
|
—
|
identificarea chimică, precum denumirea IUPAC sau CAS, numărul CAS sau codul său InChI, formula structurală, puritatea, identitatea chimică a impurităților după cum este necesar și fezabil din punct de vedere practic etc.
|
|
|
|
Substanță multicomponentă, UVCB și amestecuri:
|
—
|
în măsura în care este posibil, caracterizată prin identitatea chimică (a se vedea mai sus), apariția cantitativă și proprietățile fizico-chimice relevante ale componentelor.
|
|
|
|
|
Solvent:
|
—
|
justificarea alegerii solventului;
|
|
—
|
procentul de solvent în mediul final de cultură.
|
|
|
|
Celule:
|
|
Pentru culturile principale din laborator:
|
—
|
tipul și sursa celulelor, precum și datele anterioare din laboratorul de testare;
|
|
—
|
caracteristicile cariotipului și/sau numărul modal de cromozomi;
|
|
—
|
metode de întreținere a culturilor de celule;
|
|
—
|
perioade de dublare a celulelor.
|
|
|
|
|
Condiții de testare:
|
—
|
justificarea alegerii concentrațiilor și a numărului de culturi celulare; inclusiv, de exemplu, datele de citotoxicitate și limitele de solubilitate;
|
|
—
|
compoziția mediilor de cultură, concentrația de CO2, nivelul de umiditate;
|
|
—
|
concentrația substanței chimice de testat, exprimată în concentrație finală în mediu de cultură (de exemplu, μg sau mg/ml sau mM din mediul de cultură);
|
|
—
|
concentrația (și/sau volumul) de solvent și substanță chimică de testat adăugate în mediul de cultură;
|
|
—
|
temperatura de incubare;
|
|
—
|
densitatea celulară în timpul tratamentului;
|
|
—
|
tipul și compoziția sistemului de activare metabolică (sursa fracției S9, metoda de preparare a amestecului S9, concentrația și volumul amestecului S9 și fracția S9 în mediul final de cultură, controalele de calitate ale fracției S9);
|
|
—
|
substanțe martor pozitive și negative, concentrații finale pentru fiecare condiție de tratament;
|
|
—
|
timpul de manifestare (inclusiv numărul de celule însămânțate, subculturile și programele de alimentare, dacă este cazul);
|
|
—
|
identitatea agentului selectiv și a concentrației acestuia;
|
|
—
|
pentru MLA, ar trebui să se indice versiunea utilizată (cu agar-agar sau micro-godeuri).
|
|
—
|
criterii de acceptabilitate a testelor;
|
|
—
|
metodele utilizate pentru numărarea celulelor viabile și mutante;
|
|
—
|
metodele utilizate pentru măsurarea citotoxicității;
|
|
—
|
orice alte informații suplimentare cu privire la citotoxicitate și metoda utilizată;
|
|
—
|
durata perioadelor de incubare după placare;
|
|
—
|
definirea coloniilor pentru care se ține seama de dimensiuni și de tip (inclusiv criteriile pentru coloniile "mici" și "mari", dacă este cazul).
|
|
—
|
criteriile conform cărora un studiu poate fi considerat ca fiind pozitiv, negativ sau echivoc;
|
|
—
|
metode utilizate pentru a determina pH-ul, osmolalitatea, dacă este testată, și precipitarea, dacă este relevant.
|
|
|
|
Rezultate:
|
—
|
numărul de celule tratate și numărul de celule subcultivate pentru fiecare cultură;
|
|
—
|
parametrii de toxicitate (RTG pentru MLA și RS pentru TK6);
|
|
—
|
semne de precipitare și ora determinării;
|
|
—
|
numărul de celule placată în mediu selectiv și neselectiv;
|
|
—
|
numărul de colonii în mediul neselectiv și numărul de colonii rezistente în mediu selectiv și frecvențele celulelor mutante aferente;
|
|
—
|
dimensionarea coloniilor pentru martorii negativi și pozitivi și dacă substanța chimică de testat este pozitivă, cel puțin o concentrație, precum și frecvențele aferente ale mutanților;
|
|
—
|
relația concentrație-răspuns, dacă este posibil;
|
|
—
|
date (concentrație și solvenți) privind martorii negativi (solvent) și pozitivi testați în paralel;
|
|
—
|
date cu privire la martorii negativi (tratați cu solvent) și pozitivi testați anterior (concentrații și solvenți) cu intervale, valori medii și abateri standard; numărul de teste pe care se bazează martorii testați anterior;
|
|
—
|
analize statistice (pentru culturi individuale și duplicate grupate, dacă este cazul) și valori p, dacă există; și pentru MLA, evaluarea GEF.
|
|
|
REFERINȚE BIBLIOGRAFICE
|
(1)
|
Moore, M.M., Honma, M. Clements, J. (Rapporteur), Awogi, T., Bolcsfoldi, G., Cole, J., Gollapudi, B., Harrington-Brock, K., Mitchell, A., Muster, W., Myhr, B., O’Donovan, M., Ouldelhkim, M-C., San, R., Shimada, H. și Stankowski, L.F. Jr. (2000). Mouse Lymphoma Thymidine Kinase Locus (TK) Gene Mutation Assay: International Workshop on Genotoxicity Test Procedures (IWGTP) Workgroup Report, Environ. Mol. Mutagen., 35 (3): 185-190.
|
|
(2)
|
Moore, M.M., Honma, M., Clements, J., Harrington-Brock, K., Awogi, T., Bolcsfoldi, G., Cifone, M., Collard, D., Fellows, M., Flanders, K., Gollapudi, B., Jenkinson, P., Kirby, P., Kirchner, S., Kraycer, J., McEnaney, S., Muster, W., Myhr, B., O’Donovan, M., Oliver, Ouldelhkim, M-C., Pant, K., Preston, R., Riach, C., San, R., Shimada, H. și Stankowski, L.F. Jr. (2002). Mouse Lymphoma Thymidine Kinase Locus Gene Mutation Assay: Follow-Up International Workshop on Genotoxicity Test Procedures, New Orleans, Louisiana, (April 2000), Environ. Mol. Mutagen., 40 (4): 292-299.
|
|
(3)
|
Moore, M.M., Honma, M., Clements, J., Bolcsfoldi, G., Cifone, M., Delongchamp, R., Fellows, M., Gollapudi, B., Jenkinson, P., Kirby, P., Kirchner, S., Muster, W., Myhr, B., O’Donovan, M., Ouldelhkim, M-C., Pant, K., Preston, R., Riach, C., San, R., Stankowski, L.F. Jr., Thakur, A., Wakuri, S. and Yoshimura, I. (2003). Mouse Lymphoma Thymidine Kinase Locus Gene Mutation Assay: International Workshop (Plymouth, UK) on Genotoxicity Test Procedures Workgroup Report, Mutation Res., 540: 127-140.
|
|
(4)
|
Moore, M.M., Honma, M., Clements, J., Bolcsfoldi, G., Burlinson, B., Cifone, M., Clarke, J., Delongchamp, R., Durward, R., Fellows, M., Gollapudi, B., Hou, S., Jenkinson, P., Lloyd, M., Majeska, J., Myhr, B., O’Donovan, M., Omori, T., Riach, C., San, R., Stankowski, L.F. Jr., Thakur, A.K., Van Goethem, F., Wakuri, S. și Yoshimura, I. (2006). Mouse Lymphoma Thymidine Kinase Gene Mutation Assay: Follow-Up Meeting of the International Workshop on Genotoxicity Tests – Aberdeen, Scotland, 2003 – Assay Acceptance Criteria, Positive Controls, and Data Evaluation, Environ. Mol. Mutagen., 47 (1): 1-5.
|
|
(5)
|
Moore, M.M., Honma, M., Clements, J., Bolcsfoldi, G., Burlinson, B., Cifone, M., Clarke, J., Clay, P., Doppalapudi, R., Fellows, M., Gollapudi, B., Hou, S., Jenkinson, P., Muster, W., Pant, K., Kidd, D.A., Lorge, E., Lloyd, M., Myhr, B., O’Donovan, M., Riach, C., Stankowski, L.F. Jr., Thakur A.K. și Van Goethem, F. (2007). Mouse Lymphoma Thymidine Kinase Mutation Assay: Meeting of the International Workshop on Genotoxicity Testing, San Francisco, 2005, Recommendations for 24-h Treatment, Mutation. Res., 627 (1): 36-40.
|
|
(6)
|
OCDE (2016). Overview of the set of OECD Genetic Toxicology Test Guidelines and updates performed in 2014-2015. ENV Publications. Series on Testing and Assessment, nr. 234, OCDE, Paris.
|
|
(7)
|
Fellows M.D., Luker, T., Cooper, A. și O’Donovan, M.R. (2012). Unusual Structure-Genotoxicity Relationship in Mouse Lymphoma Cells Observed with a Series of Kinase Inhibitors. Mutation, Res., 746 (1): 21-28.
|
|
(8)
|
Honma, M., Momose, M., Sakamoto, H., Sofuni, T. și Hayashi, M. (2001). Spindol Poisons Induce Allelic Loss in Mouse Lymphoma Cells Through Mitotic Non-Disjunction. Mutation Res., 493 (1-2): 101-114.
|
|
(9)
|
Wang, J., Sawyer, J.R., Chen, L., Chen, T., Honma, M., Mei, N. și Moore, M.M. (2009). The Mouse Lymphoma Assay Detects Recombination, Deletion, and Aneuploidy, Toxicol. Sci.., 109 (1): 96-105.
|
|
(10)
|
Applegate, M.L., Moore, M.M., Broder, C.B., Burrell, A. și Hozier, J.C. (1990). Molecular Dissection of Mutations at the Heterozygous Thymidine Kinase Locus in Mouse Lymphoma Cells. Proc. National. Academy. Sci. SUA, 87 (1): 51-55.
|
|
(11)
|
Hozier, J., Sawyer, J., Moore, M., Howard, B. și Clive, D. (1981). Cytogenetic Analysis of the L5178Y/TK+/- Leads to TK-/- Mouse Lymphoma Mutagenesis Assay System, Mutation Res., 84 (1): 169-181.
|
|
(12)
|
Hozier, J., Sawyer, J., Clive, D. și Moore, M.M. (1985). Chromosome 11 Aberrations in Small Colony L5178Y TK-/- Mutants Early in their Clonal History, Mutation Res., 147 (5): 237-242.
|
|
(13)
|
Moore, M.M., Clive, D., Hozier, J.C., Howard, B.E., Batson, A.G., Turner, N.T. și Sawyer, J. (1985). Analysis of Trifluorothymidine-Resistant (TFTr) Mutants of L5178Y/TK+/- Mouse Lymphoma Cells. Mutation Res., 151 (1): 161-174.
|
|
(14)
|
Liber H.L., Call K.M. și Little J.B. (1987). Molecular and Biochemical Analyses of Spontaneous and X-Ray-Induced Mutants in Human Lymphoblastoid Cells. Mutation Res., 178 (1): 143-153.
|
|
(15)
|
Li C.Y., Yandell D.W. și Little J.B. (1992). Molecular Mechanisms of Spontaneous and Induced Loss of Heterozygosity in Human Cells In Vitro. Somat. Cell Mol. Genet., 18 (1): 77-87.
|
|
(16)
|
Honma M., Hayashi M. și Sofuni T. (1997). Cytotoxic and Mutagenic Responses to X-Rays and Chemical Mutagens in Normal and P53-Mutated Human Lymphoblastoid Cells. Mutation. Res., 374 (1): 89-98.
|
|
(17)
|
Honma, M., Momose, M., Tanabe, H., Sakamoto, H., Yu, Y., Little, J.B., Sofuni, T. și Hayashi, M. (2000). Requirement of Wild-Type P53 Protein for Maintenance of Chromosomal Integrity. Mol. Carcinogen., 28 (4): 203-14.
|
|
(18)
|
Amundson S.A. și Liber H.L. (1992). A Comparison of Induced Mutation at Homologous Alleles of the TK Locus in Human Cells. II. Molecular Analysis of Mutants. Mutation Res., 267 (1): 89-95.
|
|
(19)
|
Schisler M.R., Moore M.M. și Gollapudi B.B. (2013). In Vitro Mouse Lymphoma (L5178Y TK+/- -3.7.2C) Forward Mutation Assay. In Protocols in Genotoxicity Assessment A. Dhawan and M. Bajpayee (Eds.), Springer Protocols, Humana Press: 27-50.
|
|
(20)
|
Long, L.H., Kirkland, D., Whitwell, J. și Halliwell, B. (2007). Different Cytotoxic and Clastogenic Effects of Epigallocatechin Gallate in Various Cell-Culture Media Due to Variable Rates of its Oxidation in the Culture Medium, Mutation Res., 634 (1-2): 177-183.
|
|
(21)
|
Nesslany, F., Simar-Meintieres, S., Watzinger, M., Talahari, I. și Marzin, D. (2008). Characterization of the Genotoxicity of Nitrilotriacetic Acid. Environ. Mol. Mutagen., 49 (6): 439-452.
|
|
(22)
|
Brusick D. (1986). Genotoxic Effects in Cultured Mammalian Cells Produced by Low pH Treatment Conditions and Increased Ion Concentrations. Environ. Mutagen., 8 (6): 879-886.
|
|
(23)
|
Morita, T., Nagaki, T., Fukuda, I. și Okumura, K. (1992). Clastogenicity of Low pH to Various Cultured Mammalian Cells. Mutation Res., 268 (2): 297-305.
|
|
(24)
|
Scott, D., Galloway, S.M., Marshall, R.R., Ishidate, M.Jr, Brusick, D., Ashby, J. și Myhr, B.C. (1991). Genotoxicity under Extreme Culture Conditions. A report from ICPEMC Task Group 9. Mutation Res., 257: 147-204.
|
|
(25)
|
Wang J., Heflich R.H. și Moore M.M. (2007). A Method to Distinguish Between the De Novo Induction of Thymidine Kinase Mutants and the Selection of Pre-Existing Thymidine Kinase Mutants in the Mouse Lymphoma Assay. Mutation Res., 626 (1-2): 185-190.
|
|
(26)
|
Fischer, G.A. (1958). Studies on the Culture of Leukemic Cells In Vitro. Ann. N.Y. Academy Sci., 76: 673-680.
|
|
(27)
|
Clive, D., Johnson, K.O., Spector, J.F.S., Batson, A.G. și Brown, M.M.M. (1979). Validation and Characterization of the L5178Y/TK+/– Mouse Lymphoma Mutagen Assay System. Mutation Res., 59(1): 61-108.
|
|
(28)
|
Sawyer, J., Moore, M.M., Clive, D. și Hozier, J. (1985). Cytogenetic Characterization of the L5178Y TK+/- 3.7.2C Mouse Lymphoma Cell Line, Mutation Res., 147 (5): 243-253.
|
|
(29)
|
Sawyer J.R., Moore M.M. și Hozier J.C. (1989). High-Resolution Cytogenetic Characterization of the L5178Y TK+/- Mouse Lymphoma Cell Line, Mutation Res., 214 (2): 181-193.
|
|
(30)
|
Sawyer, J.R., Binz, R.L., Wang, J. și Moore, M.M. (2006). Multicolor Spectral Karyotyping of the L5178Y TK+/--3.7.2C Mouse Lymphoma Cell Line, Environ. Mol. Mutagen., 47 (2): 127-131.
|
|
(31)
|
Fellows, M.D., McDermott, A., Clare, K.R., Doherty, A. și Aardema, M.J. (2014). The Spectral Karyotype of L5178Y TK+/- Mouse Lymphoma Cells Clone 3.7.2C and Factors Affecting Mutant Frequency at the Thymidine Kinase (TK) Locus in the Microtitre Mouse Lymphoma Assay, Environ. Mol. Mutagen., 55 (1): 35-42.
|
|
(32)
|
Storer, R.D., Jraynak, A.R., McKelvey, T.W., Elia, M.C., Goodrow, T.L. și DeLuca, J.G. (1997). The Mouse Lymphoma L5178Y TK+/- Cell Line is Heterozygous for a Codon 170 Mutation in the P53 Tumor Suppressor Gene. Mutation. Res., 373 (2): 157-165.
|
|
(33)
|
Clark L.S., Harrington-Brock, K., Wang, J., Sargent, L., Lowry, D., Reynolds, S.H. și Moore, M.M. (2004). Loss of P53 Heterozygosity is not Responsible for the Small Colony Thymidine Kinase Mutant Phenotype in L5178Y Mouse Lymphoma Cells. Mutagen., 19 (4): 263-268.
|
|
(34)
|
Skopek T.R., Liber, H.L., Penman, B.W. și Thilly, W.G. (1978). Isolation of a Human Lymphoblastoid Line Heterozygous at the Thymidine Kinase Locus: Possibility for a Rapid Human Cell Mutation Assay. Biochem. Biophys. Res. Commun., 84 (2): 411-416.
|
|
(35)
|
Honma M. (2005). Generation of Loss of Heterozygosity and its Dependency on P53 Status in Human Lymphoblastoid Cells. Environ. Mol. Mutagen., 45 (2-3): 162-176.
|
|
(36)
|
Xia, F., Wang, X., Wang, Y.H., Tsang, N.M., Yandell, D.W., Kelsey, K.T. și Liber, H.L. (1995). Altered P53 Status Correlates with Differences in Sensitivity to Radiation-Induced Mutation and Apoptosis in Two Closely Related Human Lymphoblast Lines. Cancer. Res., 55 (1): 12-15.
|
|
(37)
|
Lorge, E., M. Moore, J. Clements, M. O Donovan, M. Honma, A. Kohara, J. van Benthem, S. Galloway, M.J. Armstrong, V. Thybaud, B. Gollapudi, M. Aardema, J. Kim, A. Sutter, D.J. Kirkland (2015). Standardized Cell Sources and Recommendations for Good Cell Culture Practices in Genotoxicity Testing. (Manuscript in preparation).
|
|
(38)
|
Lloyd M. și Kidd D. (2012). The Mouse Lymphoma Assay. Springer Protocols: Methods in Molecular Biology 817, Genetic Toxicology Principles and Methods, ed. Parry and Parry, Humana Press. ISBN, 978-1-61779-420-9, 35-54.
|
|
(39)
|
Mei N., Guo X. și Moore M.M. (2014). Methods for Using the Mouse Lymphoma Assay to Screen for Chemical Mutagenicity and Photo-Mutagenicity. În: Optimization in Drug Discover: In Vitro Methods: Yan Z și Caldwell(Eds), a doua ediție, GW; Humana Press, Totowa, NJ.
|
|
(40)
|
Liber H.L. și Thilly W.G. (1982). Mutation Assay at the Thymidine Kinase Locus in Diploidhuman Lymphoblasts. Mutation Res., 94 (2): 467-485.
|
|
(41)
|
Coecke, S., Balls, M., Bowe, G., Davis, J., Gstraunthaler, G., Hartung, T., Hay, R., Merten, OW., Price, A., Schechtman, L., Stacey, G. și Stokes, W. (2005). Guidance on Good Cell Culture Practice. A Report of the Second ECVAM Task Force on Good Cell Culture Practice. ATLA, 33 (3): 261-287.
|
|
(42)
|
Moore M.M. și Howard B.E. (1982). Quantitation of Small Colony Trifluorothymidine-Resistant Mutants of L5178Y/TK+/- Mouse Lymphoma Cells in RPMI-1640 Medium, Mutation Res., 104 (4-5): 287-294.
|
|
(43)
|
Ames B.N., McCann J. și Yamasaki E. (1975). Methods for Detecting Carcinogens and Mutagens with the Salmonella/Mammalian Microsome Mutagenicity Test. Mutation Res., 31 (6): 347-364.
|
|
(44)
|
Maron D.M. și Ames B.N. (1983). Revised Methods for the Salmonella Mutagenicity Test. Mutation Res., 113 (3-4): 173-215.
|
|
(45)
|
Natarajan, A.T., Tates, A.D, Van Buul, P.P.W., Meijers, M. și De Vogel, N. (1976). Cytogenetic Effects of Mutagens/Carcinogens After Activation in a Microsomal System In Vitro, I. Induction of Chromosomal Aberrations and Sister Chromatid Exchanges by Diethylnitrosamine (DEN) and Dimethylnitrosamine (DMN) in CHO Cells in the Presence of Rat-Liver Microsomes. Mutation Res., 37 (1): 83-90.
|
|
(46)
|
Matsuoka A., Hayashi M. și Ishidate M. Jr. (1979). Chromosomal Aberration Tests on 29 Chemicals Combined with S9 Mix In Vitro. Mutation Res., 66 (3): 277-290.
|
|
(47)
|
Ong T.M., et al. (1980). Differential Effects of Cytochrome P450-Inducers on Promutagen Activation Capabilities and Enzymatic Activities of S-9 from Rat Liver, J. Environ. Pathol. Toxicol., 4 (1): 55-65
|
|
(48)
|
Elliott, B.M., Combes, R.D., Elcombe, C.R., Gatehouse, D.G., Gibson, G.G., Mackay, J.M. și Wolf, R.C. (1992). Report of UK Environmental Mutagen Society Working Party. Alternatives to Aroclor 1254-Induced S9 in In Vitro Genotoxicity Assays. Mutagen., 7 (3): 175-177.
|
|
(49)
|
Matsushima, T., Sawamura, M., Hara, K. și Sugimura, T. (1976). A Safe Substitute for Polychlorinated Biphenyls as an Inducer of Metabolic Activation Systems. În: In Vitro Metabolic Activation in Mutagenesis Testing. de Serres F.J., et al. (Eds, Elsevier, North-Holland, pp. 85-88.
|
|
(50)
|
Galloway S.M., et al. (1994). Report from Working Group on In Vitro Tests for Chromosomal Aberrations. Mutation Res., 312 (3): 241-261.
|
|
(51)
|
Johnson T.E., Umbenhauer D.R. și Galloway S.M. (1996). Human Liver S-9 Metabolic Activation: Proficiency in Cytogenetic Assays and Comparison with Phenobarbital/Beta-Naphthoflavone or Aroclor 1254 Induced Rat S-9, Environ. Mol. Mutagen., 28 (1): 51-59.
|
|
(52)
|
UNEP (2001). Stockholm Convention on Persistent Organic Pollutants, Programul Organizației Națiunilor Unite pentru Mediu (UNEP).
|
|
(53)
|
Krahn D.F., Barsky F.C. și McCooey K.T. (1982). CHO/HGPRT Mutation Assay: Evaluation of Gases and Volatile Liquids. În: Genotoxic Effects of Airborne Agents Tice R.R., Costa D.L.și Schaich K.M. (Eds.). New York, Plenum, pp. 91-103.
|
|
(54)
|
Zamora, P.O., Benson, J.M., Li, A.P. și Brooks, A.L. (1983). Evaluation of an Exposure System Using Cells Grown on Collagen Gels for Detecting Highly Volatile Mutagens in the CHO/HGPRT Mutation Assay. Environ. Mutagen., 5 (6): 795-801.
|
|
(55)
|
Asakura M., Sasaki T., Sugiyama T., Arito H., Fukushima, S. și Matsushima, T. (2008). An Improved System for Exposure of Cultured Mammalian Cells to Gaseous Compounds in the Chromosomal Aberration Assay. Mutation Res., 652 (2): 122-130.
|
|
(56)
|
Arlett C.F., et al. (1989). Mammalian Cell Gene Mutation Assays Based upon Colony Formation. În: Statistical Evaluation of Mutagenicity Test Data, Kirkland, D.J. (Ed.), Cambridge University Press, pp. 66-101.
|
|
(57)
|
Morita T., Honma M. și Morikawa K. (2012). Effect of Reducing the Top Concentration Used in the In Vitro Chromosomal Aberration Test in CHL Cells on the Evaluation of Industrial Chemical Genotoxicity. Mutation Res., 741 (1-2): 32-56.
|
|
(58)
|
Brookmire L., Chen J.J. și Levy D.D. (2013). Evaluation of the Highest Concentrations Used in the In Vitro Chromosome Aberrations Assay. Environ. Mol. Mutagen., 54 (1): 36-43.
|
|
(59)
|
USFDA (2012). International Conference on Harmonisation (ICH) Guidance S2 (R1) on Genotoxicity Testing and Data Interpretation for Pharmaceuticals Intended For Human Use. Publicație disponibilă la adresa: [https://www.federalregister.gov/a/2012-13774].
|
|
(60)
|
Honma M. și Hayashi M. (2011). Comparison of In Vitro Micronucleus and Gene Mutation Assay Results for P53-Competent Versus P53-Deficient Human Lymphoblastoid Cells. Environ. Mol. Mutagen., 52 (5): 373-384.
|
|
(61)
|
Moore-Brown, M.M., Clive, D., Howard, B.E., Batson, A.G. și Johnson, K.O. (1981). The Utilization of Trifluorothymidine (TFT) to Select for Thymidine Kinase-Deficient (TK-/-) Mutants from L5178Y/TK+/- Mouse Lymphoma Cells, Mutation Res., 85 (5): 363-378.
|
|
(62)
|
Liber H.L., Yandell D.W. and Little J.B. (1989). A Comparison of Mutation Induction at the TK and HRPT Loci in Human Lymphoblastoid Cells; Quantitative Differences are Due to an Additional Class of Mutations at the Autosomal TK locus. Mutation Res., 216 (1): 9-17.
|
|
(63)
|
Furth E.E., Thilly, W.G., Penman, B.W., Liber, H.L. și Rand, W.M. (1981). Quantitative Assay for Mutation in Diploid Human Lymphoblasts Using Microtiter Plates. Anal. Biochem., 110 (1): 1-8.
|
|
(64)
|
Hayashi, M, Dearfield, K., Kasper, P., Lovell, D., Martus, H. J. și Thybaud, V. (2011). Compilation and Use of Genetic Toxicity Historical Control Data, Mutation Res., 723 (2): 87-90.
|
|
(65)
|
Ryan T.P. (2000). Statistical Methods for Quality Improvement. John Wiley and Sons, New York, a doua ediție.
|
|
(66)
|
OCDE (2014). Statistical analysis supporting the revision of the genotoxicity Test Guidelines. Environmental Health and Safety Publications, Series on Testing and Assessment, (nr. 199), Organizația pentru Cooperare și Dezvoltare Economică, Paris.
|
|
(67)
|
Fleiss J.L., Levin B. și Paik M.C. (2003). Statistical Methods for Rates and Proportions, a treia ediție, New York: John Wiley & Sons.
|
Apendicele 1
DEFINIȚII
Aneugen
: orice substanță sau proces chimic care, prin interacțiunea cu componentele ciclului celular de diviziune mitotică și meiotică, conduce la apariția aneuploidiei la celule sau organisme.
Aneuploidie
: orice abatere de la numărul diploid (sau haploid) normal de cromozomi cu unul sau mai mulți cromozomi, dar nu cu un set (seturi) întreg (întregi) de cromozomi (poliploidie).
Mutageni care provoacă substituția perechilor de bază
: substanțe chimice care provoacă substituția perechilor de bază în ADN.
Substanță chimică
: O substanță sau un amestec.
Eficacitatea clonării
: Procentul de celule placate la o densitate scăzută, care pot crește într-o colonie care poate fi numărată.
Clastogen
: orice substanță sau proces chimic care cauzează aberații cromozomiale structurale la populații de celule sau organisme.
Citotoxicitate
: pentru analizele incluse în prezenta metodă de testare, citotoxicitatea este identificată ca o reducere a creșterii relative totale (RSG) sau a supraviețuirii relative (RS) pentru MLA și, respectiv, TK6.
Mutație directă
: o mutație a genelor de la forma parentală la forma mutantă care generează o modificare sau o pierdere a activității enzimatice sau a funcției proteinei codificate.
Mutageni de decalare a cadrului de citire
: substanțe chimice care provoacă adăugarea sau ștergerea uneia sau a mai multor perechi de bază din molecula de ADN.
Genotoxic
: termen general cuprinzând toate tipurile de leziuni ale ADN-ului sau ale cromozomilor, inclusiv ruperi ale ADN-ului, aducții, rearanjări, mutații, aberații cromozomiale și aneuploidie. Nu toate tipurile de efecte genotoxice au drept rezultat mutații sau leziuni cromozomiale stabile.
Recombinare mitotică
: în timpul mitozei, recombinarea între cromatide omoloage, ceea ce ar putea conduce la inducerea unor rupturi duble ale șirului ADN sau la pierderea heterozigozității.
Mutagen
: produce o modificare ereditară a uneia sau mai multor secvențe de perechi de bază de ADN la gene sau a structurii cromozomilor (aberații cromozomiale).
Frecvența mutanților (FM)
: numărul de celule mutante observate împărțit la numărul de celule viabile.
Timpul de expresie fenotipică
: timpul după tratament, în care modificarea genetică se fixează în interiorul genomului și orice produse genetice preexistente sunt epuizate până la modificarea trăsăturii fenotipice.
Supraviețuire relativă (RS)
: RS este utilizat ca indicator al citotoxicității legate de tratament în TK6. Aceasta este eficacitatea relativă a clonării (CE) a celulelor așezate pe placă imediat după tratamentul celular, ajustată cu orice pierdere de celule în timpul tratamentului, în raport cu eficacitatea clonării la martorul negativ.
Creștere relativă în suspensie (RSG)
: Pentru MLA, creșterea relativă totală în suspensie, în două zile, a culturii de testare în raport cu creșterea totală în suspensie, în două zile, a martorului negativ/tratat cu solvent (Clive și Spector, 1975). RSG ar trebui să includă creșterea relativă a culturii de testare în raport cu martorul negativ/tratat cu solvent în perioada tratamentului.
Creștere relativă totală (RTG)
: Valoarea RTG este utilizată ca indicator al citotoxicității legate de tratament în MLA. Aceasta reprezintă un indicator al creșterii relative (la martorul tratat cu vehicul) a culturilor de testare în timpul tratamentului, al expresiei de două zile și în etapele de clonare a selecției mutanților ale testului. Valoarea RSG pentru fiecare cultură de testare se înmulțește cu eficacitatea relativă a clonării culturii de testare în momentul selecției mutanților și este exprimată în raport cu eficacitatea clonării martorului negativ/solvent (Clive și Spector, 1975).
Fracția S9 a ficatului
: supernatant din ficat omogenizat după centrifugare la 9 000 g, și anume extract din ficat crud.
Amestec S9
: amestecul dintre fracția S9 a ficatului și cofactorii necesari pentru activitatea metalică a enzimelor.
Creșterea în suspensie (SG)
: creșterea multiplicată a numărului de celule pe durata tratamentului și în fazele de expresie ale MLA. SG se calculează prin înmulțirea creșterii multiplicate în ziua 1 cu creșterea multiplicată din ziua 2 pentru tratamentul scurt (de 3 sau 4 ore). În cazul în care se folosește un tratament de 24 de ore, SG reprezintă creșterea multiplicată în timpul tratamentului de 24 de ore, înmulțită cu creșterile multiplicate din zilele de expresie 1 și 2.
Martor tratat cu solvent
: Termen general pentru a defini culturile martor care primesc doar solventul, care este utilizat pentru a dizolva substanța chimică de testat.
Substanța chimică de testat
: Orice substanță sau amestec testat utilizând prezenta metodă de testare.
Martori netratați
: Martorii netratați sunt culturi care nu beneficiază de niciun tratament (și anume, nici cu substanță chimică de testat, nici cu solvent), dar care sunt prelucrate în mod simultan culturilor care beneficiază de tratament cu substanță chimică de testat.
Apendicele 2
FORMULE
Citotoxicitate
În cazul ambelor versiuni (cu agar-agar și micro-godeuri) ale MLA
Citotoxicitatea este definită ca fiind creșterea relativă totală (RSG), care include creșterea relativă în suspensie (RSG) în perioada de expresie de două zile și eficacitatea relativă a clonării (RCE) obținută în momentul selecției mutanților. Valorile RTG, RSG și RCE sunt toate exprimate ca procente.
Calcularea RSG: Creșterea în suspensie 1 (SG1) constituie rata de creștere între ziua 0 și ziua 1 (concentrația celulelor în ziua 1/concentrația celulelor în ziua 0) și creșterea în suspensie 2 (SG2) reprezintă rata de creștere între ziua 1 și ziua 2 (concentrația celulelor în ziua 2/concentrația celulelor în ziua 1). RSG reprezintă SG totală (SG1 x SG2) pentru cultura tratată comparativ cu martorul netratat/tratat cu solvent. Și anume: RSG = [SG1(test) x SG2(test)] / [SG1(martor) x SG2(martor)] Valoarea SG1 ar trebui să fie calculată de la concentrația inițială de celule utilizate la începutul tratamentului aplicat celulelor. Aceasta reflectă orice citotoxicitate diferențială care apare la cultura (culturile) de testare în timpul tratamentului celulelor.
RCE reprezintă eficacitatea relativă a clonării culturilor de testare comparativ cu eficacitatea relativă a clonării la martorul netratat/tratat cu solvent obținută la momentul selecției mutanților.
Creștere relativă totală (RTG):
RTG = RSG x RCE
TK6
Supraviețuirea relativă (RS):
Citotoxicitatea este evaluată prin supraviețuirea relativă, și anume, eficacitatea clonării (EC) celulelor placate imediat după tratament, ajustată prin orice pierdere de celule în timpul tratamentului, în comparație cu eficacitatea clonării la martorii negativi (se atribuie o rată de supraviețuire de 100 %). Ajustarea pentru pierderea de celule în timpul tratamentului poate fi calculată după cum urmează:

RS pentru o cultură tratată cu o substanță chimică de testat se calculează după cum urmează:
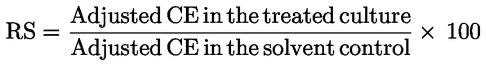
Frecvența mutanților pentru MLA și TK6
Frecvența mutanților (MF) reprezintă eficacitatea clonării coloniilor de mutanți în mediu selectiv (CEM) ajustată la eficacitatea clonării în mediu neselectiv la momentul selecției mutanților (CEV). Și anume, MF = CEM/CEV. Calculul pentru aceste două valori de eficacitate a clonării este descris mai jos pentru metodele de clonare cu agar-agar și cu micro-godeuri.
Versiunea MLA cu agar-agar: În versiunea MLA cu agar-agar, numărul de colonii de pe placa de selecție a mutanților (CM) și numărul de colonii de pe placa fără selecție sau cu eficacitate a clonării (număr fiabil) (CV) se obțin prin numărarea directă a clonelor. Atunci când sunt așezate pe placă 600 de celule pentru plăcile cu eficacitatea clonării (CE) în vederea selecției mutanților (CEM) și plăcile fără selecție sau cu eficacitatea clonării (număr viabil) (CEV) și 3 x 106, celulele sunt utilizate pentru selecția mutanților,
CEM = CM/(3 x 106) = (CM/3) x 10-6
CEV = CV/600
Versiunea MLA și TK6 cu micro-godeuri:
În versiunea MLA cu micro-godeuri, CM și CV sunt determinate ca fiind produsul numărului total de micro-godeuri (TW) și al numărului probabil de colonii pe godeu (P) pe plăcile cu micro-godeuri.
CM = PM x TWM
CV = PV x TWV
Pornind de la termenul zero al distribuției Poisson (Furth et al., 1981), P este dat de
P = - ln (EW / TW)
Unde EW reprezintă godeurile goale, iar TW reprezintă numărul total de godeuri. Prin urmare,
CEM = CM /TM = (PM x TWM)/TM
CEV = CV /TV = (PV x TWV)/TV
În cazul versiunii MLA cu micro-godeuri, frecvențele mutanților din colonii mici și mari vor fi calculate în mod identic, utilizându-se numărul relevant de godeuri goale pentru coloniile mici și mari.
În cazul TK6, frecvențele mutanților din coloniile mici și mari se bazează pe mutanții care se formează timpuriu și cei care se formează tardiv.
B.68 METODĂ DE TESTARE IN VITRO PRIN EXPUNERE PE TERMEN SCURT PENTRU IDENTIFICAREA i) SUBSTANȚELOR CHIMICE CARE PRODUC LEZIUNEA OCULARĂ GRAVĂ ȘI A ii) SUBSTANȚELOR CHIMICE CARE NU NECESITĂ CLASIFICARE PENTRU IRITAȚIA OCULARĂ SAU LEZIUNEA OCULARĂ GRAVĂI
INTRODUCERE
|
1.
|
Această metodă de testare (MT) este echivalentă cu Orientarea OCDE privind testarea (TG) nr. 491 (2017). Metoda de testare prin expunere pe termen scurt (STE) este o metodă in vitro care poate fi utilizată în anumite circumstanțe și cu limitări specifice pentru clasificarea și etichetarea pericolelor pe care le prezintă substanțele chimice (substanțe și amestecuri) care provoacă leziunea oculară gravă, precum și cele care nu necesită o clasificare pentru leziunea oculară gravă sau pentru iritația oculară, astfel cum este definită de Sistemul global armonizat de clasificare și etichetare a substanțelor chimice (GHS) al Organizației Națiunilor Unite (ONU) (1) și de Regulamentul (CE) nr. 1272/2008 privind clasificarea, etichetarea și ambalarea substanțelor și a amestecurilor (CLP) (68).
|
|
2.
|
Potențialul de pericol pentru ochi al substanțelor chimice a fost evaluat timp de mulți ani, în primul rând prin utilizarea unui test in vivo pe ochi de iepure [MT B.5 (8), echivalent cu OCDE TG nr. 405]. Este general acceptat faptul că, în viitorul apropiat, niciun test alternativ in vitro nu va putea înlocui în totalitate testul in vivo pe ochi de iepure pentru a estima întreaga gamă de răspunsuri privind leziunea oculară gravă/iritația oculară pentru diferite clase de substanțe chimice. Cu toate acestea, combinațiile strategice de metode de testare alternative utilizate în cadrul unei strategii de testare (etapizate) ar putea înlocui foarte bine pe deplin testul pe ochi de șoarece (2). Abordarea descendentă este concepută pentru testarea substanțelor chimice care pot fi prevăzute, pe baza informațiilor existente, a avea un potențial iritant ridicat sau a provoacă leziunea oculară gravă. În schimb, abordarea ascendentă este concepută pentru testarea substanțelor chimice care pot fi prevăzute, pe baza informațiilor existente, a nu provoca o iritație oculară suficientă pentru a impune o clasificare. În timp ce metoda de testare STE nu este considerată a fi o înlocuire completă a testului in vivo pe ochi de iepure, aceasta este adecvată pentru a fi utilizată în cadrul unei strategii de testare etapizate pentru clasificarea și etichetarea reglementară, cum ar fi metoda descendentă/ascendentă, pentru a identifica fără testare suplimentară (i) substanțele chimice care provoacă leziunea oculară gravă (categoria 1 din GHS al ONU/Regulamentul CLP) și (ii) substanțele chimice (cu excepția substanțelor foarte volatile și a tuturor substanțelor chimice solide, altele decât surfactanții) care nu necesită clasificare pentru iritația oculară sau leziunea oculară gravă (fără clasificare în GHS al ONU/Regulamentul CLP) (1) (2). Cu toate acestea, o substanță chimică care nu este nici prevăzută a provoca leziunea oculară gravă (categoria 1 din GHS al ONU/Regulamentul CLP) și nici încadrată într-o categorie din GHS al ONU/Regulamentul CLP (nu provoacă nici leziunea oculară gravă și nici iritația oculară) prin metoda de testare STE ar trebui să fie supusă unor teste suplimentare pentru stabilirea unei clasificări definitive. În plus, ar trebui să se consulte autoritățile de reglementare competente înainte de utilizarea STE în cadrul unei abordări ascendente în conformitate cu alte scheme de clasificare decât GHS al ONU/Regulamentul CLP. Alegerea celei mai adecvate metode de testare și utilizarea acestei metode de testare ar trebui să fie avută în vedere în contextul documentului de orientare al OCDE referitor la o strategie integrată privind testarea și evaluarea pentru leziunea oculară gravă și iritația oculară (14).
|
|
3.
|
Scopul acestei metode de testare este de a descrie procedurile utilizate pentru a evalua potențialul de pericol pentru ochi al unei substanțe chimice de testat, pe baza capacității acesteia de a induce citotoxicitatea în cadrul metodei de testare prin expunere pe termen scurt. Efectul citotoxic al substanțelor chimice asupra celulelor epiteliale corneene este un mod important de acțiune (MOA) care determină lezarea epiteliului cornean și iritația oculară. Viabilitatea celulară este evaluată în cadrul metodei de testare STE prin măsurarea cantitativă, după extragerea din celule a sării de formazan de culoare albastră produsă de celulele vii prin conversia enzimatică a colorantului vital MTT [bromură de 3-(4,5-dimetiltiazol-2-il)-2,5-difeniltetrazoliu], cunoscut și sub denumirea de bromură de tiazolil albastru tetrazoliu (3). Viabilitatea celulară obținută este comparată cu martorul tratat cu solvent (viabilitate relativă) și se utilizează pentru estimarea potențialului de pericol pentru ochi al substanței chimice de testat. O substanță chimică de testat este încadrată în categoria 1 din GHS al ONU/Regulamentul CLP atunci când ambele concentrații 5 % și 0,05 % determină o viabilitate celulară mai mică decât sau egală cu (≤) 70 %. Invers, o substanță chimică este prevăzută a fi neîncadrată în nicio categorie din GHS al ONU/Regulamentul CLP atunci când ambele concentrații de 5 % și 0,05 % determină o viabilitate celulară mai mare decât (>) 70 %.
|
|
4.
|
Termenul „substanță chimică de testat” este utilizat în prezenta metodă de testare pentru a face referire la ceea ce este supus testării și nu are legătură cu aplicabilitatea metodei de testare STE pentru testarea substanțelor și/sau a amestecurilor. Definițiile sunt prezentate în apendice.
|
CONSIDERAȚII PRELIMINARE ȘI LIMITĂRI
|
5.
|
Această metodă de testare se bazează pe un protocol elaborat de Kao Corporation (4), care a făcut obiectul a două studii de validare diferite: unul realizat de către Comitetul de validare al Societății japoneze pentru alternative la experimentele pe animale (JSAAE) (5) și altul de către Centrul japonez pentru validarea metodelor alternative (JaCVAM) (6). O evaluare inter pares a fost realizată de NICEATM/ICCVAM pe baza rapoartelor studiilor de validare și a documentelor de analiză de fond privind metoda de testare (7).
|
|
6.
|
Atunci când sunt utilizate pentru a identifica substanțele chimice (substanțe și amestecuri) care provoacă leziunea oculară gravă (categoria 1 din GHS al ONU/Regulamentul CLP (1), datele obținute prin metoda de testare STE cu privire la 125 de substanțe chimice (inclusiv substanțe și amestecuri) au arătat o precizie generală de 83 % (104/125), o rată de rezultate fals pozitive de 1 % (1/86 ) și o rată de rezultate fals negative de 51 % (20/39) în comparație cu testul in vivo pe ochi de iepure (7). Rata de rezultate fals negative obținută nu este esențială în contextul de față, întrucât toate substanțele chimice de testat care induc o viabilitate celulară de ≤ 70 % la o concentrație de 5 % și > 70 % la o concentrație de 0,05 % ar fi testate ulterior cu alte metode de testare in vitro validate în mod adecvat sau, ca o ultimă opțiune, în cadrul testului in vivo pe ochi de iepure, în funcție de cerințele de reglementare și în conformitate cu strategia de testare secvențială și cu abordările bazate pe dovezi recomandate în prezent (1) (8). Au fost testate, în principal, substanțe monocomponente, deși există, de asemenea, o cantitate limitată de date privind testarea amestecurilor. Metoda de testare este totuși aplicabilă din punct de vedere tehnic pentru testarea substanțelor multicomponente și a amestecurilor. Cu toate acestea, înainte de utilizarea acestei metode de testare pe un amestec pentru a genera date pentru un obiectiv de reglementare specific, ar trebui să se analizeze dacă și, în caz afirmativ, din ce cauză aceasta poate oferi rezultate adecvate în scopul respectiv. Astfel de considerații nu sunt necesare atunci când există o cerință de reglementare pentru testarea amestecului. Metoda de testare STE nu a prezentat alte deficiențe specifice atunci când a fost utilizată pentru identificarea substanțelor chimice de testat ca fiind încadrate în categoria 1 din GHS al ONU/Regulamentul CLP. Cercetătorii ar putea lua în calcul utilizarea acestei metode de testare pentru substanțe chimice de testat; prin aceasta, viabilitatea celulară de ≤ 70 % la concentrații de 5 % și 0,05 % ar trebui să fie acceptată ca indicator al unui răspuns care induce leziunea oculară gravă care ar trebui să fie încadrată în categoria 1 din GHS al ONU/Regulamentul CLP fără teste suplimentare.
|
|
7.
|
Atunci când sunt utilizate pentru a identifica substanțele chimice (substanțe și amestecuri) care nu necesită clasificarea pentru iritația oculară și leziunea oculară gravă (și anume, categoria 1 din GHS al ONU/Regulamentul CLP (1), datele obținute prin metoda de testare STE cu privire la 130 de substanțe chimice (inclusiv substanțe și amestecuri) au arătat o precizie generală de 85 % (110/130), o rată de rezultate fals negative de 12 % (9/73) și o rată de rezultate fals pozitive de 19 % (11/57) în comparație cu testul in vivo pe ochi de iepure (7). În cazul în care sunt excluse din setul de date substanțele puternic volatile și substanțele solide, altele decât surfactanții, precizia totală se îmbunătățește, ajungând la 90 % (92 /102), rata de rezultate fals negative la 2 % (1/54) și rata de rezultate fals pozitive la 19 % (9/48) (7). În consecință, eventualele deficiențe ale metodei de testare STE, atunci când aceasta este utilizată pentru a identifica substanțe chimice de testat care nu necesită clasificare pentru iritația oculară și leziunea oculară gravă (fără încadrare în GHS al ONU/Regulamentul CLP), reprezintă o rată ridicată de rezultate fals negative pentru i) substanțe foarte volatile, cu o presiune a vaporilor peste 6 kPa și (ii) substanțe chimice solide (substanțe și amestecuri), altele decât surfactanții și amestecurile alcătuite exclusiv din surfactanți. Aceste substanțe chimice sunt excluse din domeniul de aplicabilitate al metodei de testare STE (7).
|
|
8.
|
În plus față de substanțele chimice menționate la punctele (6) și (7), setul de date generat prin metoda STE conține, de asemenea, date interne privind 40 de amestecuri, care, atunci când au fost comparate cu testul Draize in vivo pe ochi, au arătat o precizie de 88 % (35/40), o rată de rezultate fals pozitive de 50 % (5/10) și o rată de rezultate fals negative de 0 % (0/30) pentru estimarea amestecurilor care nu necesită încadrare în sistemele de clasificare GHS al ONU/Regulamentul CLP (9). Prin urmare, metoda de testare STE poate fi aplicată pentru a identifica amestecuri ca nefiind încadrate în nicio categorie din GHS al ONU/Regulamentul CLP, în cadrul unei abordări ascendente, cu excepția amestecurilor solide, altele decât cele care sunt compuse doar din surfactanți, ca o extindere a limitării sale la substanțe solide. În plus, amestecurile care conțin substanțe cu o presiune a vaporilor mai mare de 6 kPa ar trebui evaluate cu atenție pentru a se evita posibile previziuni insuficiente, acestea trebuind să fie justificate de la caz la caz.
|
|
9.
|
Metoda de testare STE nu poate fi utilizată pentru identificarea substanțelor chimice de testat ca fiind încadrate în categoria 2 din GHS al ONU/Regulamentul CLP sau în categoria 2A (iritație oculară) sau 2B (iritație oculară ușoară), având în vedere numărul considerabil de substanțe chimice din categoria 1 din GHS al ONU/Regulamentul CLP care sunt estimate insuficient ca fiind încadrate în categoria 2, 2A sau 2B, precum și de substanțe chimice neîncadrate în nicio categorie din GHS al ONU/Regulamentul CLP, care sunt estimate insuficient ca fiind încadrate în categoria 2, 2A sau 2B (7). În acest scop, ar putea fi necesare teste suplimentare utilizând o altă metodă adecvată.
|
|
10.
|
Metoda de testare STE este adecvată pentru substanțele chimice de testat care sunt dizolvate sau suspendate uniform pentru cel puțin 5 minute într-o salină fiziologică, cu 5 % dimetilsulfoxid (DMSO) în soluție salină sau ulei mineral. Metoda de testare STE nu este adecvată pentru substanțele chimice de testat care sunt insolubile sau nu pot fi suspendate uniform pentru cel puțin 5 minute într-o salină fiziologică, cu 5 % DMSO în soluție salină sau ulei mineral. Utilizarea de ulei mineral în metoda de testare STE este posibilă datorită expunerii pe termen scurt. Așadar, metoda de testare STE este adecvată pentru estimarea potențialului de pericol pentru ochi al substanțelor chimice de testat insolubile în apă (de exemplu, alcooli grași sau cetone cu lanț lung), cu condiția ca acestea să fie miscibile în cel puțin unul dintre cei trei solvenți propuși mai sus (4).
|
|
11.
|
Termenul „substanță chimică de testat” este utilizat în prezenta metodă de testare pentru a face referire la ceea ce este supus testării (69) și nu are legătură cu aplicabilitatea metodei de testare STE pentru testarea substanțelor și/sau a amestecurilor.
|
PRINCIPIUL TESTULUI
|
12.
|
Metoda de testare STE este o analiză in vitro bazată pe citotoxicitate, care este efectuată pe un monostrat confluent de celule Statens Serumaninstitut Rabbit Cornea (SIRC) cultivate pe o microplacă din policarbonat cu 96 de godeuri (4). După o expunere de cinci minute la o substanță chimică de testat, citotoxicitatea este măsurată cantitativ ca viabilitate relativă a celulelor SIRC prin analiza MTT (4). Viabilitatea celulară scăzută este utilizată pentru a estima eventualele efecte adverse care determină leziuni oculare.
|
|
13.
|
S-a raportat că 80 % dintr-o soluție care intră în ochiul unui iepure este excretată prin intermediul sacului conjunctival în trei sau patru minute, în timp ce peste 80 % dintr-o soluție care intră în ochiul uman este excretată în unu sau două minute (10). Metoda de testare STE încearcă să aproximeze acești timpi de expunere și utilizează citotoxicitatea ca punct final pentru a evalua gradul de lezare a celulelor SIRC ca urmare a unei expuneri de cinci minute la substanța chimică de testat.
|
DEMONSTRAREA PERFORMANȚEI
|
14.
|
Înainte de utilizarea sistematică a metodei de testare STE descrise în prezenta metodă de testare, laboratoarele ar trebui să demonstreze performanțele tehnice prin clasificarea corectă a celor unsprezece substanțe recomandate în tabelul 1. Aceste substanțe au fost selectate pentru a reprezenta întreaga gamă de răspunsuri pentru leziunea oculară gravă sau iritația oculară pe baza rezultatelor testelor in vivo pe ochi de iepure (TG 405) și a sistemului de clasificare GHS al ONU/Regulamentul CLP (1). Alte criterii de selecție au inclus faptul că substanțele ar trebui să fie disponibile în comerț, că ar trebui să fie disponibile date de referință in vivo de înaltă calitate și că ar trebui să fie disponibile date in vitro de înaltă calitate din metoda de testare STE (3). În situațiile în care o substanță inclusă pe listă nu este disponibilă sau în cazul în care acest lucru se poate justifica, se poate utiliza o altă substanță pentru care sunt disponibile date de referință in vivo și in vitro adecvate, cu condiția să se aplice aceleași criterii, astfel cum sunt descrise aici.
Tabelul 1
Lista substanțelor de verificare
|
Substanță
|
NR CAS
|
Clasa chimică (70)
|
Stare fizică
|
In Vivo- Cat. (71) din GHS al ONU/CLP
|
Solvent în testul STE
|
STE - cat. din GHS al ONU/CLP
|
|
Clorură de benzalconiu (10 %, apoasă)
|
8001-54-5
|
Compus de tip onium
|
Lichid
|
Categoria 1
|
Salin
|
Categoria 1
|
|
Triton X-100 (100 %)
|
9002-93-1
|
Eter
|
Lichid
|
Categoria 1
|
Salin
|
Categoria 1
|
|
Acid Red 92
|
18472-87-2
|
Compus heterociclic; Compus de brom; Compus de clor
|
Solid
|
Categoria 1
|
Salin
|
Categoria 1
|
|
Hidroxid de sodiu
|
1310-73-2
|
Alcali; Substanță chimică anorganică
|
Solid
|
Categoria 1 (72)
|
Salin
|
Categoria 1
|
|
Butirlactonă
|
96-48-0
|
Lactonă; Compus heterociclic
|
Lichid
|
Categoria 2A (Categoria 2 din CLP)
|
Salin
|
Nu se poate estima
|
|
1-octanol
|
111-87-5
|
Alcool
|
Lichid
|
Categoria 2A/B (73)(Categoria 2 din CLP)
|
Ulei mineral
|
Nu se poate estima
|
|
Ciclopentanol
|
96-41-3
|
Alcool; Hidrocarbură, ciclică
|
Lichid
|
Categoria 2A/B (74) (Categoria 2 din CLP)
|
Salin
|
Nu se poate estima
|
|
Acetat de 2-etoxietil
|
111-15-9
|
Alcool; Eter
|
Lichid
|
Neclasificată
|
Salin
|
Neclasificată
|
|
Dodecan
|
112-40-3
|
Hidrocarbură, aciclică
|
Lichid
|
Neclasificată
|
Ulei mineral
|
Neclasificată
|
|
Metilizobutilcetonă
|
108-10-1
|
Cetonă
|
Lichid
|
Neclasificată
|
Ulei mineral
|
Neclasificată
|
|
sulfat de 1,1-dimetilguanidină
|
598-65-2
|
amidină; compus de sulf
|
Solid
|
Neclasificată
|
Salin
|
Neclasificată
|
|
Abrevieri: NR CAS = Număr de înregistrare al Chemical Abstracts Service
|
|
PROCEDURA
Prepararea monostratului celular
|
15.
|
Pentru realizarea metodei de testare STE ar trebui să se utilizeze linia celulară din cornee de iepure SIRC. Se recomandă ca celulele SIRC să fie obținute de la o bancă de celule calificată, cum ar fi American Type Culture Collection CCL60.
|
|
16.
|
Celulele SIRC sunt cultivate la 37 °C în atmosferă de 5 % CO2 și de umiditate în balon de cultură conținând un mediu de cultură ce cuprinde mediul esențial minim al lui Eagle (MEM), completat cu 10 % ser fetal bovin (FBS), 2 mM L-glutamină, 50-100 de unități/ml de penicilină și 50-100 μg/ml de streptomicină. Celulele care au devenit confluente în balonul de cultură ar trebui să fie separate folosind soluție tripsină-acid etilendiaminotetraacetic, cu sau fără utilizarea unei raclete de celule. Celulele sunt propagate (de exemplu, 2-3 treceri) într-un balon de cultură înainte de a fi utilizate pentru testele de rutină și ar trebui să fie supuse unui număr maxim de 25 de treceri de la decongelare.
|
|
17.
|
Apoi, celulele gata de utilizat pentru testul STE sunt pregătite la densitatea corespunzătoare și însămânțate în plăci cu 96 de godeuri. Densitatea celulară recomandată pentru însămânțare este de 6,0 × 103 de celule pe godeu atunci când celulele sunt utilizate la patru zile după însămânțare, sau 3,0 × 103 de celule per godeu atunci când celulele sunt utilizate la cinci zile după însămânțare, la un volum de cultură de 200 μl. Celulele utilizate pentru testul STE, care sunt însămânțate într-un mediu de cultură la o densitate adecvată, vor atinge o confluență de peste 80 % la momentul testării, și anume la patru sau cinci zile după însămânțare.
|
Aplicarea substanțelor chimice de testat și a substanțelor martor
|
18.
|
Prima alegere a solventului pentru dizolvarea sau suspendarea substanțelor chimice de testat este soluția fiziologică salină. În cazul în care substanța chimică de testat demonstrează o solubilitate scăzută sau nu poate fi dizolvată sau suspendată uniform timp de cel puțin cinci minute în soluție salină, ca opțiune secundară de solvent se utilizează soluția cu 5 % DMSO (CAS nr. 67-68-5) în soluție salină. Pentru substanțele chimice de testat care nu pot fi dizolvate sau suspendate în mod uniform pentru cel puțin cinci minute în soluție salină sau în soluție salină cu 5 % DMSO, ca opțiune terță de solvent se utilizează uleiul mineral (CAS nr. 8042-47-5).
|
|
19.
|
Substanțele chimice de testat se dizolvă sau se suspendă în mod uniform în solventul selectat la concentrația de 5 % (g/g) și apoi se diluează cu o diluție în serie multiplicată de 10 ori la o concentrație de 0,5 % și 0,05 %. Fiecare substanță chimică de testat trebuie să fie testată atât la concentrația de 5 %, cât și la cea de 0,05 %. Celulele cultivate pe placa cu 96 de godeuri sunt expuse la o cantitate de 200 μl/godeu, la o concentrație de 5 % sau 0,05 % din soluția (sau suspensia) cu substanță chimică de testat timp de cinci minute la temperatura camerei. Substanțele chimice de testat (substanțe monocomponente, multicomponente sau amestecuri) sunt considerate substanțe pure și diluate sau suspendate conform metodei, indiferent de puritatea lor.
|
|
20.
|
Mediul de cultură descris la punctul 16 se utilizează ca martor de mediu pe fiecare placă din cadrul fiecărei serii de repetiții. În plus, celulele trebuie expuse, de asemenea, la eșantioane de martor tratat cu solvent pe fiecare placă din cadrul fiecărei serii de repetiții. Pentru solvenții prezentați la punctul 18 s-a obținut confirmarea că nu prezintă efecte adverse asupra viabilității celulelor SIRC.
|
|
21.
|
În metoda de testare STE, va fi utilizat lauril sulfat de sodiu (SLS) cu un conținut de 0,01 % în soluție salină ca martor pozitiv pe fiecare placă din cadrul fiecărei serii de repetiții. Pentru a calcula viabilitatea celulară a martorului pozitiv, fiecare repetare placă din cadrul fiecărei repetiții trebuie să includă, de asemenea, un martor tratat cu solvent salin.
|
|
22.
|
Este necesară o probă în vederea determinării compensației pentru densitatea optică și ar trebui să se recolteze din godeuri care conțin numai o soluție salină tamponată cu fosfat, dar fără calciu și magneziu (PBS-) sau celule.
|
|
23.
|
Fiecare eșantion (substanță chimică de testat la 5 % și 0,05 %, martor de mediu, martor tratat cu solvent și martor pozitiv) ar trebui să fie testat în trei replici la fiecare repetiție prin expunerea celulelor la 200 μl din substanța chimică de testat sau martor corespunzătoare timp de cinci minute la temperatura camerei.
|
|
24.
|
Substanțele de referință sunt utile pentru evaluarea potențialului iritant pentru ochi al substanțelor chimice necunoscute din cadrul unei anumite clase de substanțe sau produse chimice, sau pentru evaluarea potențialului iritant relativ al unei substanțe iritante pentru ochi într-un interval specific de reacții iritante.
|
Măsurarea viabilității celulare
|
25.
|
După expunere, celulele sunt spălate de două ori cu 200 μl de PBS și se adaugă 200 μl de soluție de MTT (0,5 mg de MTT/ml de mediu de cultură). După un timp de reacție de două ore într-un incubator (37 °C, 5 % CO2), soluția MTT este decantată, se extrage formazanul MTT prin adăugarea a 200 μl de 0,04 N acid hidroclorhidric-izopropanol timp de 60 minute la întuneric la temperatura ambiantă și se măsoară absorbanța soluției de formazan MTT la 570 nm cu un cititor de placă. Interferența substanțelor chimice de testat cu analiza MTT (prin coloranți sau reductori direcți ai MTT) se produce numai atunci când în sistemul de testare se păstrează o cantitate semnificativă de substanță chimică de testat după clătire în urma expunerii, așa cum este cazul țesutului de cornee umană reconstruit sau al țesutului de epidermă umană reconstruit 3D, dar nu este relevantă pentru culturile de celule 2D utilizate pentru metoda de testare STE.
|
Interpretarea rezultatelor și modelul predictiv
|
26.
|
Valorile densității optice (DO) obținute pentru fiecare substanță chimică de testat sunt apoi utilizate pentru a calcula viabilitatea celulară raportată la martorul tratat cu solvent, care este stabilită la 100 %. Viabilitatea celulară relativă este exprimată în procente și se obține împărțind DO a substanței chimice de testat la DO a martorului tratat cu solvent, după scăderea valorii DO a soluției sărace din ambele valori.

În mod similar, viabilitatea celulară relativă a fiecărui martor tratat cu solvent este exprimată în procente și se obține împărțind DO a fiecărui martor tratat cu solvent la DO a martorului de mediu, după scăderea valorii DO a soluției sărace din ambele valori.
|
|
27.
|
Ar trebui să se efectueze trei repetiții independente, fiecare conținând trei godeuri replicate (și anume, n = 9). Media aritmetică a celor trei godeuri pentru fiecare substanță chimică de testat și martor tratat cu solvent, în cadrul fiecărei repetiții independente, este utilizată pentru a calcula media aritmetică a viabilității celulare relative. Media aritmetică finală a viabilității celulare este calculată pe baza celor trei repetiții independente.
|
|
28.
|
În continuare sunt prezentate valorile-limită ale viabilității celulare pentru identificarea substanțelor chimice de testat care provoacă leziunea oculară gravă (categoria 1 din GHS al ONU/Regulamentul CLP) și substanțele chimice de testat care nu necesită clasificare pentru iritația oculară sau leziunea oculară gravă (fără încadrare în GHS al ONU/Regulamentul CLP).
|
Tabelul 2
Model predictiv al metodei de testare STE
|
Viabilitate celulară
|
Clasificare în GHS al ONU/CLP
|
Aplicabilitate
|
|
La 5 %
|
La 0,05 %
|
|
> 70 %
|
> 70 %
|
Neclasificată
|
Substanțe și amestecuri, cu excepția: i) substanțe foarte volatile, cu o presiune a vaporilor de peste 6 kPa (75) și ii) substanțe chimice solide (substanțe și amestecuri), cu excepția surfactanților și a amestecurilor compuse exclusiv din surfactanți
|
|
≤ 70 %
|
> 70 %
|
Nu se poate estima
|
Nu este cazul
|
|
≤ 70 %
|
≤ 70 %
|
Categoria 1
|
Substanțe și amestecuri (76)
|
Criterii de acceptare
|
29.
|
Rezultatele testului sunt considerate acceptabile în cazul în care sunt îndeplinite toate criteriile următoare:
|
a)
|
Densitatea optică a martorului de mediu (expunere la mediu de cultură) ar trebui să fie de cel puțin 0,3, după scăderea densității optice a soluției sărace.
|
|
b)
|
Viabilitatea martorului tratat cu solvent ar trebui să fie de cel puțin 80 % în raport cu martorul de mediu. În cazul în care, la fiecare repetiție, se utilizează mai mulți martori tratați cu solvent, fiecare dintre acești martori ar trebui să prezinte o viabilitate celulară mai mare de 80 % pentru a califica substanțele chimice de testat testate cu acești solvenți.
|
|
c)
|
Viabilitatea celulară obținută cu martorul pozitiv (0,01 % SLS) ar trebui să se încadreze în două abateri standard ale mediei istorice. Limitele superioare și inferioare de acceptabilitate pentru martorul pozitiv ar trebui să fie actualizate frecvent, adică o dată la trei luni sau de fiecare dată când este efectuat un test acceptabil în laboratoare în care testele sunt desfășurate rar (și anume, mai puțin de o dată pe lună). În cazul în care un laborator nu completează un număr suficient de experimente pentru a stabili o distribuție a martorilor pozitivi solidă din punct de vedere statistic, se acceptă utilizarea limitelor superioare și inferioare de acceptare stabilite de dezvoltatorul metodei, și anume între 21,1 % și 62,3 %, în conformitate cu datele sale istorice de laborator ale acestuia, în același timp realizându-se o distribuție internă în timpul primelor teste de rutină.
|
|
d)
|
Abaterea standard a viabilității celulare finale derivată în urma a trei repetiții independente ar trebui să fie mai mică de 15 % pentru ambele concentrații de 5 % și 0,05 % pentru substanța chimică de testat.
În cazul în care nu este îndeplinit unul sau mai multe dintre aceste criterii, rezultatele ar trebui să fie eliminate și ar trebui să se efectueze alte trei repetiții independente.
|
|
DATE ȘI RAPORTARE
Date
|
30.
|
Trebuis să se consemneze datele pentru fiecare godeu (de exemplu, valorile viabilității celulare) din cadrul fiecărei repetiții, precum și media globală, SD și clasificarea.
|
Raport testului
|
31.
|
Raportul testului ar trebui să includă următoarele informații:
|
|
Substanțe chimice de testat și substanțe martor
|
—
|
Substanța monocomponentă: identificarea chimică, precum denumirea (denumirile) IUPAC sau CAS, numărul (numerele) de registru CAS, codul SMILES sau InChI, formula structurală și/sau alți identificatori;
|
|
—
|
Substanța multicomponentă, UVCB și amestec: caracterizarea, dacă este posibil, de exemplu prin identitatea chimică (a se vedea mai sus), puritatea, apariția cantitativă și proprietățile fizico-chimice relevante (a se vedea mai sus) ale componentelor, dacă există;
|
|
—
|
starea fizică, volatilitatea, pH, LogP, masa moleculară, clasa de substanțe chimice și alte proprietăți fizico-chimice relevante pentru desfășurarea studiului, în măsura în care este posibil;
|
|
—
|
puritatea, identitatea chimică a impurităților, după caz și dacă este fezabil din punct de vedere practic etc.;
|
|
—
|
tratamentul înainte de efectuarea testului, dacă este cazul (de exemplu, încălzire, măcinare);
|
|
—
|
condițiile de depozitare și stabilitatea, dacă există.
|
|
|
|
Condițiile și procedurile metodei de testare
|
—
|
numele și adresa sponsorului, ale instalației de testare și ale directorului de studiu;
|
|
—
|
descrierea metodei de testare utilizate;
|
|
—
|
linia celulară utilizată, sursa sa, numărul de reînsămânțare și confluența celulelor utilizate pentru testare;
|
|
—
|
detalii privind procedura de testare utilizată;
|
|
—
|
numărul de repetiții și replici utilizate;
|
|
—
|
concentrațiile substanței chimice de testat utilizate (dacă acestea diferă de cele recomandate);
|
|
—
|
justificarea alegerii solventului pentru fiecare substanță chimică de testat;
|
|
—
|
durata expunerii la substanța chimică de testat (dacă este diferită de cea recomandată);
|
|
—
|
descrierea oricăror modificări ale procedurii de testare;
|
|
—
|
descrierea criteriilor de evaluare și de decizie utilizate;
|
|
—
|
trimiterea la media martorilor pozitivi testați anterior și la abaterea standard (SD):
|
|
—
|
Demonstrarea performanțelor de laborator în aplicarea metodei de testare (de exemplu, prin testarea unor substanțe de verificare) sau demonstrarea performanței reproductibile a metodei de testare în timp.
|
|
|
|
Rezultate
|
—
|
Pentru fiecare substanță chimică de testat și fiecare substanță martor, precum și pentru fiecare concentrație testată, ar trebui să se prezinte sub formă de tabel valorile DO individuale pentru fiecare godeu replicat, media aritmetică a valorilor DO pentru fiecare repetiție independentă, viabilitatea celulară % pentru fiecare repetiție independentă și media aritmetică finală % a viabilității celulare și SD în cadrul celor trei repetiții;
|
|
—
|
Rezultatele pentru mediu, solvent și martorul pozitiv care demonstrează criteriile adecvate de acceptare a studiului;
|
|
—
|
descrierea altor efecte observate;
|
|
—
|
clasificarea generală derivată, raportată la modelul predictiv/criteriile de decizie utilizate.
|
|
|
REFERINȚE BIBLIOGRAFICE
|
(1)
|
Organizația Națiunilor Unite (ONU) (2013). Sistemul global armonizat de clasificare și etichetare a substanțelor chimice (GHS). A cincea ediție revizuită. New York & Geneva: United Nations Publications. ISBN: 978-92-1-117006-1. Publicație disponibilă la adresa: http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html.
|
|
(2)
|
Scott L, et al. (2010). A proposed Eye Irritation Testing Strategy to Reduce and Replace in vivo Studies Using Bottom-Up and Top-Down Approaches. Toxicol. In Vitro 24, 1-9.
|
|
(3)
|
Mosmann T. (1983). Rapid Colorimetric Assay for Cellular Growth and Survival: Application to 7 Proliferation and Cytotoxicity Assays. J. Immunol. Methods 65, 55-63.
|
|
(4)
|
Takahashi Y, et al. (2008). Development of the Short Time Exposure (STE) Test: an In Vitro Eye Irritation Test Using SIRC Cells. Toxicol. In Vitro 22,760-770.
|
|
(5)
|
Sakaguchi H, et al. (2011). Validation Study of the Short Time Exposure (STE) Test to Assess the Eye Irritation Potential of Chemicals. Toxicol. In Vitro 25,796-809.
|
|
(6)
|
Kojima H, et al. (2013). Second-Phase Validation of Short Time Exposure Tests for Assessment of Eye Irritation Potency of Chemicals. Toxicol. In Vitro 27, pp.1855-1869.
|
|
(7)
|
ICCVAM (2013). Short Time Exposure (STE) Test Method Summary Review Document, NIH. Publicație disponibilă la adresa: [http://www.ntp.niehs.nih.gov/iccvam/docs/ocutox_docs/STE-SRD-NICEATM-508.pdf].
|
|
(8)
|
Capitolul B.5 din prezenta anexă, Iritație/coroziune oculară acută.
|
|
(9)
|
Saito K, et al. (2015). Predictive Performance of the Short Time Exposure Test for Identifying Eye Irritation Potential of Chemical Mixtures.
|
|
(10)
|
Mikkelson TJ, Chrai SS și Robinson JR. (1973). Altered Bioavailability of Drugs in the Eye Due to Drug-Protein Interaction. J. Pharm. Sci.1648-1653.
|
|
(11)
|
ECETOC (1998). Eye Irritation Reference Chemicals Data Bank. Technical Report [nr. 48. (2)], Bruxelles, Belgia.
|
|
(12)
|
Gautheron P, et al. (1992). Bovine Corneal Opacity and Permeability Test: an In Vitro Assay of Ocular Irritancy. Fundam Appl Toxicol. 18, 442-449.
|
|
(13)
|
OCDE (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environmental, Health and Safety Publications, Series on Testing and Assessment (nr. 34). Organizația pentru Cooperare și Dezvoltare Economică, Paris.
|
|
(14)
|
OCDE (2017). Guidance Document on an Integrated Approaches on Testing and Assessment for Serious Eye Damage and Eye irritation. Environmental, Health and Safety Publications, Series on Testing and Assessment (nr. 263). Organizația pentru Cooperare și Dezvoltare Economică, Paris.
|
Apendice
DEFINIȚII
Precizie
: gradul de apropiere dintre rezultatele metodei de testare și valorile de referință acceptate. Aceasta constituie una dintre caracteristicile de performanță ale metodei de testare și unul dintre aspectele relevanței acesteia. Termenul este adesea utilizat interschimbabil cu termenul de „concordanță” pentru a desemna proporția de rezultate corecte ale unei metode de testare (13).
Substanță de referință
: o substanță utilizată în calitate de standard pentru comparația cu o substanță chimică de testat. O substanță de referință ar trebui să aibă următoarele proprietăți: (i) o sursă (surse) compatibilă (compatibile) și sigură (sigure); (ii) similitudini structurale și funcționale cu clasa substanțelor care sunt testate; (iii) caracteristici fizico-chimice cunoscute; (iv) date justificative privind efecte cunoscute; și (v) puterea cunoscută în intervalul de răspuns dorit.
Abordare ascendentă
: abordare etapizată utilizată pentru o substanță chimică de testat susceptibilă de a nu necesita clasificare pentru iritația oculară sau leziunea oculară gravă, care începe cu determinarea substanțelor chimice care nu necesită clasificare (rezultat negativ) din alte substanțe chimice (rezultat pozitiv).
Substanță chimică
: o substanță sau un amestec.
Iritație oculară
: producerea de modificări la nivel ocular în urma aplicării unei substanțe chimice de testat pe suprafața anterioară a ochiului, care sunt complet reversibile în termen de 21 de zile de la aplicare. Termen utilizat în paralel cu „efecte reversibile asupra ochiului” și cu „categoria 2 din GHS al ONU”
Rată de rezultate fals negative
: proporția tuturor substanțelor chimice pozitive identificate în mod fals de o metodă de testare ca fiind negative. Acesta este unul dintre indicatorii de performanță ai metodei de testare.
Rată de rezultate fals pozitive
: proporția tuturor substanțelor chimice negative identificate în mod fals de o metodă de testare ca fiind pozitive. Acesta este unul dintre indicatorii de performanță ai metodei de testare.
Pericol
: proprietate intrinsecă a unui agent sau a unei situații care are potențialul de a produce efecte adverse atunci când un organism, un sistem sau o (sub)populație este expusă la agentul respectiv.
Martor pentru mediu
: o probă replică netratată care conține toate componentele unui sistem de testare. Această probă este analizată împreună cu probele tratate cu substanța chimică de testat și cu alte probe martor pentru a determina dacă solventul interacționează cu sistemul de testare.
Amestec
: un amestec sau o soluție compus/ă din două sau mai multe substanțe.
Substanță mono-constituent
: O substanță definită prin compoziția sa cantitativă, în care un singur component principal este prezent în proporție de cel puțin 80 % (g/g).
MTT
: 3-(4,5-dimetiltiazol-2-il)-2,5-bromură de difeniltetrazoliu; Bromură de tetrazoliu, albastru de tiazolil.
Substanță multicomponentă
: O substanță definită prin compoziția sa cantitativă, în care mai mult de un singur component principal este prezent într-o concentrație ≥ 10 % (g/g) și < de 80 % (g/g). O substanță multicomponentă este rezultatul unui proces de fabricație. Diferența dintre un amestec și o substanță multicomponentă este faptul că un amestec este obținut prin combinarea a două sau mai multe substanțe chimice fără o reacție chimică. O substanță multicomponentă este rezultatul unei reacții chimice.
DO
: Densitate optică.
Martor pozitiv
: o replică ce conține toate componentele sistemului de testare și care a fost tratată cu o substanță cunoscută pentru faptul că provoacă un răspuns pozitiv. Pentru a asigura posibilitatea evaluării variabilității în timp a reacției martorului pozitiv, amplitudinea reacției severe nu ar trebui să fie excesiv de mare.
Relevanță
: Descrierea relației dintre test și efectul de interes și dacă aceasta este relevantă și utilă pentru un anumit scop. Aceasta reprezintă măsura în care testul măsoară sau prezice în mod corect efectul biologic de interes. Relevanța include luarea în considerare a acurateței (conformității) unei metode de testare (10).
Fiabilitate
: măsuri în care o metodă de testare poate fi reprodusă în mod reproductibil, în același laborator sau în laboratoare diferite în timp, atunci când sunt efectuate utilizând același protocol. Aceasta se evaluează prin calcularea reproductibilității în același laborator sau între laboratoare și a repetabilității în același laborator (13).
Sensibilitate
: proporția tuturor substanțelor chimice pozitive/active clasificate în mod corect în urma testului. Acesta permite măsurarea acurateței unei metode de testare care conduce la rezultate clasificabile în categorii și constituie un aspect important în evaluarea relevanței unei metode de testare (10).
Leziune oculară gravă
: producerea de leziuni tisulare la nivel ocular sau deteriorarea fizică gravă a vederii în urma aplicării unei substanțe chimice de testat pe suprafața anterioară a ochiului, care nu este complet reversibilă într-un interval de 21 de zile de la aplicare. Termen utilizat în paralel cu „efecte ireversibile asupra ochiului” și cu „categoria 1 din GHS al ONU”
Martor solvent/vehicul
: o probă netratată care conține toate componentele unui sistem de testare, inclusiv solventul sau vehiculul analizat împreună cu substanța chimică de testat tratată și cu alte probe martor pentru a stabili reacția de bază pentru probele tratate cu substanța chimică de testat dizolvată în același solvent sau vehicul. Atunci când este testată concomitent cu un martor de mediu, această probă indică, de asemenea, dacă solventul/vehiculul interacționează cu sistemul testat.
Specificitate
: proporția tuturor substanțelor chimice negative/inactive clasificate în mod corect în urma testului. Acesta permite măsurarea acurateței unei metode de testare care conduce la rezultate clasificabile în categorii și constituie un aspect important în evaluarea relevanței unei metode de testare (13).
Substanță
: un element chimic și compușii săi în stare naturală sau obținuți prin orice proces de producție, inclusiv orice aditiv necesar pentru a-i păstra stabilitatea și orice impuritate care derivă din procesul utilizat, însă excluzând orice solvent care poate fi separat fără a afecta stabilitatea substanței sau fără a-i schimba compoziția.
Surfactant
: numit și agent tensioactiv, aceasta este o substanță chimică, cum ar fi un detergent, care poate să reducă tensiunea la suprafață a unui lichid și, astfel, să îi permită acesteia spumarea sau penetrarea solidelor; este cunoscut, de asemenea, ca agent de umectare.
Substanță chimică de testat
: orice substanță sau amestec testat utilizând această metodă de testare.
Strategie de testare etapizată
: o strategie de testare pe etape în care toate informațiile existente cu privire la o substanță chimică de testat sunt analizate, într-o anumită ordine, utilizând un proces de identificare a dovezilor în fiecare etapă pentru a determina dacă sunt disponibile suficiente informații pentru o decizie privind clasificarea pericolelor înainte de a trece la etapa următoare. În cazul în care potențialul iritant al unei substanțe chimice de testat poate fi apreciat pe baza informațiilor existente, nu este necesară o testare suplimentară. În cazul în care potențialul iritant al unei substanțe chimice de testat nu poate fi apreciat pe baza informațiilor existente, se efectuează o procedură de testare secvențială pe animale până în momentul în care se poate realiza o clasificare lipsită de echivoc.
Abordare descendentă
: o abordare pe etape utilizată pentru o substanță chimică susceptibilă de a provoca leziunea oculară gravă, care începe cu determinarea substanțelor chimice care provoacă leziunea oculară gravă (rezultat pozitiv) din alte substanțe chimice (rezultat negativ).
Sistemul global armonizat de clasificare și etichetare a substanțelor chimice al Organizației Națiunilor Unite (GHS al ONU)
: Un sistem care propune clasificarea substanțelor chimice (substanțe și amestecuri) pe tipuri și niveluri standardizate de pericole fizice, pentru sănătate și pentru mediu, și care vizează elemente de comunicare corespunzătoare, de exemplu pictograme, cuvinte de semnalizare, fraze de pericol, fraze de precauție și fișe cu date de securitate, pentru a transmite informații privind efectele adverse ale acestora, în scopul protejării populației (inclusiv a angajatorilor, a lucrătorilor, a transportatorilor, a consumatorilor și a serviciilor de urgență) și a mediului (1).
Categoria 1 din GHS al ONU/Regulamentul CLP
: A se vedea „Leziune oculară gravă”.
Categoria 2 din GHS al ONU/Regulamentul CLP
: A se vedea „iritația oculară”.
Neîncadrat în GHS al ONU/Regulamentul CLP
: Substanțe chimice care nu sunt clasificate drept substanțe chimice din categoria 1 sau 2 din GHS al ONU/Regulamentul CLP (sau în categoria 2 A sau 2B din GHS al ONU).
UVCB
: Substanțe cu compoziție necunoscută sau variabilă, produși de reacție complexă sau materiale biologice.
B.69. METODA DE TESTARE PE EPITELIU SIMILAR CORNEEI UMANE RECONSTRUIT (RhCE) PENTRU IDENTIFICAREA SUBSTANȚELOR CHIMICE CARE NU NECESITĂ CLASIFICARE PENTRU IRITAȚIA OCULARĂ SAU LEZIUNEA OCULARĂ GRAVĂ
INTRODUCERE
|
1.
|
Această metodă de testare (MT) este echivalentă cu Orientarea OCDE privind testarea (TG) nr. 492 (2017). Iritația cutanată gravă înseamnă producerea de leziuni ale țesutului ocular sau deteriorarea fizică gravă a vederii în urma aplicării unei substanțe chimice de testat pe suprafața anterioară a ochiului, care nu este pe deplin reversibilă în termen de 21 de zile de la aplicare, astfel cum este definită de Sistemul global armonizat de clasificare și etichetare a substanțelor chimice al Organizației Națiunilor Unite (GHS al ONU) (1) și în Regulamentul (CE) nr. 1272/2008 privind clasificarea, etichetarea și ambalarea substanțelor și a amestecurilor (CLP) (77). De asemenea, conform GHS al ONU și CLP, iritația oculară se referă la producerea de modificări la nivel ocular în urma aplicării unei substanțe chimice de testat pe suprafața anterioară a ochiului, care sunt complet reversibile în termen de 21 de zile de la aplicare. Substanțele chimice de testat care provoacă leziunea oculară gravă sunt încadrate în categoria 1 din GHS al ONU și Regulamentul CLP, iar cele care provoacă iritația oculară sunt încadrate în categoria 2 din GHS al ONU și Regulamentul CLP. Substanțele chimice de testat care nu sunt clasificate în ceea ce privește iritația oculară sau leziunea oculară gravă sunt definite ca fiind cele care nu îndeplinesc cerințele pentru încadrarea în categoria 1 sau 2 (2A sau 2B) din GHS al ONU, și anume, cele menționate ca nefiind încadrate într-o categorie din GHS al ONU sau din Regulamentul CLP.
|
|
2.
|
Evaluarea leziunii oculare grave/iritației oculare a implicat, de regulă, utilizarea animalelor de laborator [MT B.5 (2)]. Alegerea celei mai adecvate metode de testare și utilizarea acestei metode de testare ar trebui să fie avută în vedere în contextul documentului de orientare al OCDE referitor la o strategie integrată privind testarea și evaluarea pentru leziunea oculară gravă și iritația oculară (39).
|
|
3.
|
Prezenta metodă de testare descrie o procedură in vitro care permite identificarea substanțelor chimice (substanțe și amestecuri) care nu necesită clasificare și etichetare pentru iritația oculară sau leziunea oculară gravă în conformitate cu GHS al ONU și Regulamentul CLP. Aceasta folosește epiteliu similar corneei umane (RhCE), care imită îndeaproape proprietățile histologice, morfologice, biochimice și fiziologice ale epiteliului cornean uman. Alte patru metode de testare in vitro au fost validate, considerate valabile din punct de vedere științific și adoptate ca fiind TM B.47 (3), B.48 (4), B.61 (5) și B.68 (6) pentru a aborda leziunea oculară gravă/iritația oculară ca punct final pentru sănătatea umană.
|
|
4.
|
În această metodă de testare sunt incluse două teste validate care utilizează modele RhCE disponibile în comerț. Au fost efectuate studii de validare pentru evaluarea iritației oculare/leziunii oculare grave (7) (8) (9) (10) (11) (12) (13) utilizând testul EpiOcular™ Eye Irritation Test (EIT) și testul SkinEthic™ Human Corneal Epithelium (HCE) Eye Irritation Test (EIT). Fiecare dintre aceste teste utilizează structuri de țesuturi RhCE disponibile în comerț ca sistem de testare, care sunt menționate în textul de mai jos ca fiind metodele de referință validate – VRM 1 și, respectiv, VRM 2. Ca urmare a acestor studii de validare și a evaluării inter pares independentă a acestora (9) (12), s-a ajuns la concluzia că EpiOcular™ EIT și SkinEthic™ HCE EIT pot identifica în mod corect substanțe chimice (atât substanțe, cât și amestecuri) care nu necesită clasificare și etichetare pentru iritația oculară sau leziunea oculară gravă, în conformitate cu GHS al ONU, iar testele au fost recomandate ca fiind valabile din punct de vedere științific în acest scop (13).
|
|
5.
|
Este general acceptat în prezent faptul că, în viitorul apropiat, nicio metodă de testare in vitro nu va putea înlocui în totalitate testul in vivo pe ochi Draize (2) (14) pentru a estima întreaga gamă de răspunsuri privind leziunea oculară gravă/iritația oculară pentru diferite clase de substanțe chimice. Însă există combinații strategice de mai multe metode de testare alternative în cadrul unor strategii de testare (etapizate), precum abordarea ascendentă/descendentă, care ar putea înlocui pe deplin testul pe ochi Draize (15). Abordarea ascendentă (15) este concepută pentru a fi utilizată atunci când, pe baza informațiilor existente, se preconizează că o substanță chimică nu provoacă o iritație oculară suficientă pentru a impune o clasificare, iar abordarea descendentă (15) este concepută pentru a fi utilizată atunci când, pe baza informațiilor existente, se preconizează că o substanță chimică va provoca leziunea oculară gravă. Testele EpiOcular™ EIT și SkinEthic™ HCE EIT sunt recomandate pentru a identifica substanțele chimice care nu impun clasificarea pentru iritația oculară sau leziunea oculară gravă conform GHS al ONU/Regulamentului CLP (neîncadrată într-o categorie) fără teste suplimentare, în cadrul unei strategii de testare precum abordarea ascendentă/descendentă sugerată de Scott et al., de exemplu, ca o etapă inițială într-o abordare ascendentă sau ca una dintre etapele finale dintr-o abordare descendentă (15). Însă, testele EpiOcular™ EIT și SkinEthic™ HCE EIT nu sunt prevăzute a face diferența între categoria 1 din GHS al ONU/Regulamentul CLP (leziunea oculară gravă) și categoria 2 din GHS al ONU/Regulamentul CLP (iritația oculară). Această diferențiere va trebui să fie abordată la un alt nivel al unei strategii de testare (15). O substanță chimică de testat care este identificată ca necesitând o clasificare pentru iritația oculară/leziunea oculară gravă cu EpiOcular™ EIT sau SkinEthic™ HCE EIT va presupune astfel teste suplimentare (in vitro și/sau in vivo) pentru a ajunge la o concluzie definitivă (neîncadrarea într-o categorie din GHS al ONU/Regulamentul CLP, încadrarea în categoria 2 sau 1), utilizând, de exemplu, MT B.47, B.48, B.61 sau B.68.
|
|
6.
|
Scopul acestei metode de testare este de a descrie procedura utilizată pentru a evalua potențialul de pericol pentru ochi al unei substanțe chimice de testat, pe baza capacității acesteia de a induce citotoxicitatea într-o structură de țesut RhCE, astfel cum este măsurat prin analiza MTT (16) (a se vedea punctul 21). Viabilitatea țesutului RhCE în urma expunerii la o substanță chimică de testat este determinată în comparație cu țesuturile tratate cu substanța martor negativă (viabilitate %) și este apoi utilizată pentru a prezice potențialul de pericol pentru ochi al substanței chimice de testat.
|
|
7.
|
Standardele de performanță (17) sunt disponibile pentru a facilita validarea unor teste bazate pe RhCE in vitro noi sau modificate similare testelor EpiOcular™ EIT și SkinEthic™ HCE EIT, în conformitate cu principiile din documentul de orientare nr. 34 al OCDE (18), și permit modificarea oportună a TG nr. 492 a OCDE privind includerea acestora. Acceptarea reciprocă a datelor (MAD) conform acordului OCDE va fi garantată doar pentru testele validate în conformitate cu standardele de performanță, în cazul în care respectivele teste au fost revizuite și incluse în orientarea OCDE privind testarea corespunzătoare.
|
DEFINIȚII
|
8.
|
Definițiile sunt prevăzute în apendicele 1.
|
CONSIDERAȚII PRELIMINARE ȘI LIMITĂRI
|
9.
|
Această metodă de testare se bazează pe structuri de țesut comerciale tridimensionale RhCE care sunt produse utilizând fie cheratinocite epidermice primare umane (și anume, EpiOcular™ OCL-200), fie celule epiteliale corneene umane imortalizate (și anume, SkinEthic™ HCE/S). Structurile de țesut RhCE EpiOcular™ OCL-200 și SkinEthic™ HCE/S sunt similare structurii tridimensionale a epiteliului cornean in vivo și sunt produse cu ajutorul celulelor din specia de interes (19) (20). În plus, testele măsoară în mod direct citotoxicitatea care rezultă ca urmare a pătrunderii substanței chimice prin cornee și a producții de leziuni la nivelul celulelor și al țesuturilor; răspunsul citotoxic determină apoi rezultatul general de leziune oculară gravă/iritație oculară in vivo. Leziunile celulare se pot produce prin mai multe moduri de acțiune (a se vedea punctul 20), dar citotoxicitatea joacă un rol important, dacă nu rolul mecanic principal, în determinarea răspunsului general de leziune oculară gravă/iritație oculară al unei substanțe chimice, manifestându-se in vivo în principal prin opacitate corneană, inflamația irisului, înroșirea conjunctivei și/sau chemoza conjunctivei, indiferent de procesele fizico-chimice care determină lezarea tisulară.
|
|
10.
|
O gamă largă de substanțe chimice, care acoperă o varietate amplă de tipuri de substanțe chimice, clase de substanțe chimice, mase moleculare, valori LogP, structuri chimice etc., au fost testate în cadrul studiului de validare care stă la baza acestei metode de testare. Baza de date de validare EpiOcular™ EIT a cuprins în total 113 substanțe chimice din 95 de grupe funcționale organice diferite conform unui set de instrumente de analiză QSAR al OCDE (8). Majoritatea acestor substanțe chimice au reprezentat substanțe monocomponente, însă în studiu au fost incluse, de asemenea, mai multe substanțe multicomponente (inclusiv 3 homopolimeri, 5 copolimeri și 10 cvasipolimeri). În ceea ce privește starea fizică și categoriile din GHS al ONU/Regulamentul CLP, cele 113 substanțe chimice testate au fost distribuite după cum urmează: 13 lichide în categoria 1, 15 substanțe solide în categoria 1, 6 lichide în categoria 2A, 10 substanțe solide în categoria 2A, 7 lichide în categoria 2B, 7 substanțe solide în categoria 2B, 27 de lichide fără clasificare și 28 de substanțe solide fără clasificare (8). Baza de date de validare SkinEthic™ HCE EIT a cuprins în total 200 de substanțe chimice din 165 de grupe funcționale organice diferite (8) (10) (11). Majoritatea acestor substanțe chimice au reprezentat substanțe monocomponente, însă în studiu au fost incluse, de asemenea, mai multe substanțe multicomponente (inclusiv 10 polimeri). În ceea ce privește starea fizică și categoriile din GHS al ONU/Regulamentul CLP, cele 200 substanțe chimice testate au fost distribuite după cum urmează: 27 de lichide în categoria 1, 24 de substanțe solide în categoria 1, 19 lichide în categoria 2A, 10 substanțe solide în categoria 2A, 9 lichide în categoria 2B, 8 substanțe solide în categoria 2B, 50 de lichide fără clasificare și 53 de substanțe solide fără clasificare (10) (11).
|
|
11.
|
Această metodă de testare este aplicabilă substanțelor și amestecurilor, precum și substanțelor solide, substanțelor lichide, substanțelor semisolide și cerurilor. Lichidele pot fi apoase sau neapoase; solidele pot fi solubile sau insolubile în apă. În măsura în care este posibil, solidele trebuie transformate în pulbere fină înainte de aplicare; nu este necesar alt tratament prealabil al eșantionului. Gazele și aerosolii nu au fost evaluați în cadrul unui studiu de validare. Deși se recunoaște faptul că gazele și aerosolii pot fi testați cu ajutorul tehnologiei bazate pe RhCE, actuala metodă de testare nu permite testarea produselor de acest tip.
|
|
12.
|
Substanțele chimice de testat, care absorb lumina în același interval ca și formazanul MTT (în mod natural sau după tratament), și substanțele chimice de testat care pot să reducă direct colorantul MTT esențial (la formazan MTT) pot să interfereze cu măsurătorile de viabilitate tisulară și să necesite utilizarea unor martori adaptați pentru corecții. Tipul de martori adaptați care pot fi necesari vor varia în funcție de tipul de interferență produsă de substanța chimică de testat și de procedura utilizată pentru a măsura formazanul MTT (a se vedea punctele 36-42).
|
|
13.
|
Rezultatele generate în cadrul validării prealabile (21) (22) și al studiilor de validare complete (8) (10) (11) au demonstrat că atât EpiOcular™, EIT cât și SkinEthic™ HCE EIT sunt transferabile în laboratoarele considerate a avea puțină experiență în realizarea de analize și, de asemenea, sunt reproductibile în cadrul aceluiași laborator și între laboratoare. Pe baza acestor studii, nivelul de reproductibilitate în ceea ce privește concordanța dintre predicțiile care pot fi așteptate în urma testului EpiOcular™ EIT pornind de la date privind 113 substanțe chimice este de ordinul a 95 % în cadrul laboratoarelor și de 93 % între laboratoare. Nivelul de reproductibilitate în ceea ce privește concordanța dintre predicțiile care pot fi așteptate în urma testului SkinEthic™ HCE EIT pornind de la date privind 120 de substanțe chimice este de ordinul a 92 % în cadrul laboratoarelor și de 95 % între laboratoare.
|
|
14.
|
Testul EpiOcular™ EIT poate fi utilizat pentru a identifica substanțele chimice care nu necesită clasificare pentru iritația oculară sau leziunea oculară gravă conform sistemului de clasificare GHS al ONU/Regulamentului CLP. Având în vedere datele obținute în cadrul studiului de validare (8), EpiOcular™ EIT are o precizie totală de 80 % (pe baza a 112 substanțe chimice), o rată de sensibilitate de 96 % (pe baza a 57 de substanțe chimice), o rată de rezultate fals negative de 4 % (pe baza a 57 de substanțe chimice), o rată de specificitate de 63 % (pe baza a 55 de substanțe chimice) și o rată de rezultate fals pozitive de 37 % (pe baza a 55 de substanțe chimice), comparativ cu datele generate de testul in vivo pe ochi de iepure de referință (MT B.5) (2) (14) conform sistemului de clasificare GHS al ONU/Regulamentului CLP. Un studiu în care au fost testate 97 de formule agrochimice lichide au fost testate cu EpiOcular™ EIT a demonstrat o performanță similară a metodei de testare pentru acest tip de amestecuri, astfel cum a rezultat din studiul de validare (23). Cele 97 de formule au fost distribuite după cum urmează: 21 în categoria 1, 19 în categoria 2A, 14 în categoria 2B și 43 fără încadrare, clasificate conform sistemului de clasificare GHS al ONU pe baza testului in vivo pe ochi de iepure de referință (MT B.5) (2) (14). S-a obținut o precizie generală de 82 % (pe baza a 97 de formule), o sensibilitate de 91 % (pe baza a 54 de formule), o rată de rezultate fals negative de 9 % (pe baza a 54 de formule), o rată de specificitate de 72 % (pe baza a 43 de formule) și o rată de rezultate fals pozitive de 28 % (pe baza a 43 de formule) (23).
|
|
15.
|
Testul SkinEthic™ HCE EIT poate fi utilizat pentru a identifica substanțele chimice care nu necesită clasificare pentru iritația oculară sau leziunea oculară gravă conform sistemului de clasificare GHS al ONU/Regulamentului CLP. Având în vedere datele obținute în cadrul studiului de validare (10) (11), SkinEthic™ HCE EIT are o precizie totală de 84 % (pe baza a 200 de substanțe chimice), o rată de sensibilitate de 95 % (pe baza a 97 de substanțe chimice), o rată de rezultate fals negative de 5 % (pe baza a 97 de substanțe chimice), o rată de specificitate de 72 % (pe baza a 103 substanțe chimice) și o rată de rezultate fals pozitive de 28 % (pe baza a 103 de substanțe chimice), comparativ cu datele generate de testul in vivo pe ochi de iepure de referință (MT B.5) (2) (14) conform sistemului de clasificare GHS al ONU/Regulamentului CLP.
|
|
16.
|
Ratele de rezultate fals negative obținute cu ambele teste RhCE, cu substanțe sau amestecuri de substanțe, se încadrează în rata generală de probabilitate de 12 % ca substanțele chimice să fie identificate fie ca substanțe încadrate în categoria 2 din GHS al ONU și CLP, fie ca substanțe neîncadrate în nicio categorie din GHS al ONU și CLP prin testul ocular Draize in vivo în cadrul unor teste repetate; acest lucru se datorează viabilității inerente a metodei în cadrul testului (24). Ratele de rezultate fals pozitive obținute cu ambele metode de testare RhCE, cu substanțe sau amestecuri, nu sunt esențiale în contextul acestei metode de testare, întrucât toate substanțele chimice de testat care determină o viabilitate celulară egală cu sau mai mică decât valorile-limită stabilite (a se vedea punctul 44) vor trebui să fie supuse altor teste efectuate cu alte metode de testare in vitro sau, ca o ultimă opțiune, pe iepuri, în funcție de cerințele de reglementare și aplicând o strategie de testare secvențială în cadrul unei metode bazate pe dovezi. Aceste metode de testare pot fi utilizate pentru toate tipurile de substanțe chimice, iar un rezultat negativ al acestora ar trebui să fie acceptat pentru neclasificarea unei substanțe chimice pentru iritația oculară și leziunea oculară gravă (fără încadrare în GHS al ONU și Regulamentul CLP). Ar trebui să se consulte autoritățile de reglementare competente înainte de utilizarea EpiOcular™ EIT și SkinEthic™ HCE EIT în cadrul altor scheme de clasificare decât GHS al ONU/Regulamentul CLP.
|
|
17.
|
O limitare a acestei metode de testare este faptul că nu permite diferențierea între efectele iritante pentru ochi/reversibile asupra ochilor (categoria 2) și efectele de lezare oculară gravă/ireversibile asupra ochilor (categoria 1), astfel cum sunt definite în GHS al ONU și Regulamentul CLP, nici între substanțele iritante pentru ochi (categoria 2A opțională) și substanțele ușor iritante pentru ochi (categoria 2B opțională), astfel cum sunt definite în GHS al ONU (1). În acest sens, sunt necesare teste suplimentare cu alte metode de testare in vitro.
|
|
18.
|
Termenul „substanță chimică de testat” este utilizat în prezenta metodă de testare pentru a face referire la ceea ce este supus testării (78) și nu are legătură cu aplicabilitatea metodei de testare RhCE pentru testarea substanțelor și/sau a amestecurilor.
|
PRINCIPIUL TESTULUI
|
19.
|
Substanța chimică de testat se aplică local pe o suprafață minimă a două structuri de țesut RhCE tridimensionale și se măsoară viabilitatea țesutului în urma expunerii și după o perioadă de incubare post-tratament. Țesuturile RhCE sunt reconstituite din cheratinocite de epidermă umană primare sau din celule epiteliale corneene umane imortalizate care au fost cultivate timp de mai multe zile pentru a forma un epiteliu scuamos stratificat și foarte diferențiat, având o morfologie similară celui din corneea umană. Structura de țesut EpiOcular™ RhCE este formată din cel puțin trei straturi viabile de celule și o suprafață necheratinizată, care prezintă o structură similară corneei, analogă celei identificate in vivo. Structura de țesut SkinEthic™ HCE RhCE este formată din cel puțin patru straturi viabile de celule, incluzând celule columnare bazale, celule tranziționale poliedrice și celule superficiale scuamoase, similară celei de țesut epitelial cornean uman normal (20) (26).
|
|
20.
|
Iritația oculară gravă/iritația oculară indusă de o substanță chimică, manifestată in vivo în principal prin opacitate corneană, inflamația irisului, înroșirea conjunctivei și/sau chemoza conjunctivei, este rezultatul unei cascade de evenimente care încep cu pătrunderea substanței chimice prin cornee și/sau conjunctivă și producerea de leziuni la nivelul celulelor. Leziunea celulelor se poate produce prin mai multe moduri de acțiune, inclusiv: lizarea membranelor celulare (de exemplu, prin surfactanți, solvenți organici); coagularea macromoleculelor (în special a proteinelor) (de exemplu, prin surfactanți, solvenți organici, alcali și acizi); saponificarea lipidelor (de exemplu, prin alcali); și alchilarea sau alte interacțiuni covalente cu macromolecule (de exemplu, prin înălbitori, peroxizi și agenți de alchilare) (15) (27) (28). Însă, s-a demonstrat că citotoxicitatea joacă un rol important, dacă nu rolul mecanicist principal, în determinarea răspunsului general de leziune oculară gravă/iritație oculară al unei substanțe chimice, indiferent de caracteristicile fizico-chimice care stau la baza lezării țesuturilor (29) (30). Mai mult, potențialul de leziune oculară gravă/iritație oculară al unei substanțe chimice este determinat în principal de gravitatea leziunii inițiale (31), care este corelată cu gradul de moarte a celulelor (29) și cu amploarea răspunsurilor ulterioare și a eventualelor rezultate (32). Astfel, substanțele ușor iritante afectează, în general, doar epiteliul cornean superficial, substanțele ușor și moderat iritante afectează în principal epiteliul și stroma superficială, iar substanțele grav iritante deteriorează epiteliul, pătrund adânc în stromă și, uneori, în endoteliul cornean (30) (33). Măsurarea viabilității structurii țesutului RhCE după expunere topică la o substanță chimică de testat pentru a identifica substanțele chimice care nu necesită clasificare pentru leziune oculară gravă/iritație oculară (fără încadrare în GHS al ONU și Regulamentul CLP) se bazează pe ipoteza că toate substanțele chimice care provoacă leziunea oculară gravă sau iritația oculară vor induce citotoxicitate în epiteliul cornean și/sau în conjunctivă.
|
|
21.
|
Viabilitatea tisulară RhCE se măsoară, de regulă, prin conversia enzimatică a colorantului esențial MTT [bromură de 3-(4,5-dimetiltiazol-2-il)-2,5-difeniltetrazoliu; bromură de tetrazoliu, albastru de tiazolil; număr CAS 298-93-1], prin celulele viabile ale țesutului, într-o sare de formazan MTT de culoare albastră care este măsurată cantitativ după ce a fost extrasă din țesuturi (16). Substanțele chimice care nu necesită clasificare și etichetare în conformitate cu GHS al ONU/Regulamentul CLP (neclasificate) sunt identificate ca fiind acelea care nu reduc viabilitatea tisulară sub un prag definit [și anume, viabilitate tisulară > 60 %, la EpiOcular™ EIT și SkinEthic™ HCE EITL (79) sau > 50 %, la SkinEthic™ HCE EITS (80)) (a se vedea alineatul 44).
|
DEMONSTRAREA PERFORMANȚEI
|
22.
|
Înainte de utilizarea sistematică a testelor RhCE în scopuri de reglementare, laboratoarele ar trebui să demonstreze performanțele tehnice prin estimarea corectă a celor cincisprezece substanțe chimice de verificare enumerate în tabelul 1. Aceste substanțe chimice au fost selectate dintre cele utilizate în studiile de validare a VRM (8) (10) (11). Selecția include, în măsura posibilității, substanțe chimice care: (i) prezintă diferite stări fizice; (ii) acoperă întreaga gamă de răspunsuri de leziune oculară gravă/iritație oculară in vivo pe baza rezultatelor de înaltă calitate obținute prin testul in vivo pe ochi de iepure de referință (MT B.5) (2) (14), precum și a sistemului de clasificare GHS al ONU (și anume, categoriile 1, 2A, 2B sau fără clasificare) (1) și a sistemului de clasificare CLP (și anume, categoriile 1, 2 sau fără clasificare); (iii) se referă la diferiții factori determinanți de clasificare in vivo (24) (25); (iv) sunt reprezentative pentru clasele de substanțe chimice utilizate în studiul de validare (8) (10) (11); (V) prezintă o rată de reprezentare bună și la nivel amplu a grupurilor funcționale ecologice (8) (10) (11); (vi) prezintă structuri chimice bine definite (8) (10) (11); (vii) sunt colorate și/sau reductori direcți ai MTT; (viii) au produs rezultate reproductibile în cadrul metodelor de testare RhCE în cursul validărilor lor; (ix) au fost corect estimate prin metodele de testare RhCE în timpul studiilor de validare a acestora; (x) acoperă întreaga gamă de răspunsuri in vitro pe baza datelor de înaltă calitate generate prin metodele de testare RhCE (viabilitate între 0 și 100 %); (xi) sunt disponibile în comerț; și (xii) nu sunt asociate cu costuri de achiziție și/sau eliminare prohibitive. În situațiile în care o substanță chimică menționată în listă nu este disponibilă sau nu poate fi utilizată din alte motive întemeiate, ar putea fi utilizată o altă substanță chimică care îndeplinește criteriile descrise mai sus, de exemplu, dintre substanțele chimice utilizate în procesul de validare a VRM. Însă astfel de abateri ar trebui să fie justificate.
Tabelul 1
Lista substanțelor de verificare
|
Denumire chimică
|
NR CAS
|
Grup organic funcțional (81)
|
Stare fizică
|
Viabilitate VRM1 (%) (82)
|
Viabilitate VRM2 (%) (83) Predicția VRM
|
Reductor MTT
|
Interf. culorii
|
Categoria 1 in vivo
(84)
|
|
metil tioglicolat
|
|
|
2365-48-2
|
ester acid carboxilic; tioalcool
|
L
|
10,9 ± 6,4
|
5,5 ± 7,4
|
Nu se poate estima
|
Y (puternic)
|
N
|
acrilat de hidroxietil
|
|
818-61-1
|
acrilat;
|
alcool
|
L
|
7,5 ± 4,7 (85)
|
1,6 ± 1,0
|
Nu se poate estima
|
N
|
N
|
|
2,5-dimetil-2,5-hexanediol
|
110-03-2
|
alcool
|
S
|
2,3 ± 0,2
|
0,2 ± 0,1
|
Nu se poate estima
|
N
|
N
|
|
oxalat de sodiu
|
62-76-0
|
acid oxocarboxilic
|
S
|
29,0 ± 1,2
|
5,3 ± 4,1
|
Nu se poate estima
|
N
|
N
|
|
Categoria 2A in vivo (84)
|
|
|
2,4,11,13-tetraazatetradecan-diimidamidă, N,N″-bis(4-clorofenil)-3,12-diimino-, di-D-gluconat (20 %, soluție apoasă) (86)
|
18472-51-0
|
halidă aromatică heterocilică; aril halidă; grup dihidroxil; guanidină
|
L
|
4,0 ± 1,1
|
1,3 ± 0,6
|
Nu se poate estima
|
N
|
Y (slab)
|
|
benzoat de sodiu
|
532-32-1
|
aril; acid carboxilic
|
S
|
3,5 ± 2,6
|
0,6 ± 0,1
|
Nu se poate estima
|
N
|
N
|
|
Categoria 2B in vivo
(84)
|
|
|
dietiltoluamid
|
134-62-3
|
benzamidă
|
L
|
15,6 ± 6,3
|
2,8 ± 0,9
|
Nu se poate estima
|
N
|
N
|
|
2,2-dimetil-3-metilenbiciclo [2.2.1]heptan
|
79-92-5
|
alcan, ramificat cu carbon terțiar; alchenă; bicicloheptan; carbocicluri cu structură inelară; cicloalcan
|
S
|
4,7 ± 1,5
|
15,8 ± 1,1
|
Nu se poate estima
|
N
|
N
|
|
Fără clasificare in vivo
(84)
|
|
|
1-etil-3-sulfat de etil metilimidazoliu
|
342573-75-5
|
alcoxi; sare de amoniu; aril; imidazol; sulfat
|
L
|
79,9 ± 6,4
|
79,4 ± 6,2
|
Neclasificat
|
N
|
N
|
|
dicaprilil eter
|
629-82-3
|
alcoxi; Eter
|
L
|
97,8 ± 4,3
|
95,2 ± 3,0
|
Neclasificat
|
N
|
N
|
|
piperonil butoxid
|
51-03-6
|
alcoxi; benzodioxol; benzil; Eter
|
L
|
104,2 ± 4,2
|
96,5 ± 3,5
|
Neclasificat
|
N
|
N
|
|
ulei de ricin hidrogenat polietilen glicol (PEG-40)
|
61788-85-0
|
acilal; Alcool; alil; Eter
|
Vâscoasă
|
77,6 ± 5,4
|
89,1 ± 2,9
|
Neclasificat
|
N
|
N
|
|
1-(4-clorfenil)-3-(3,4-diclorfenil) uree
|
101-20-2
|
halidă aromatică heterocilică; aril halidă; derivați din uree
|
S
|
106,7 ± 5,3
|
101,9 ± 6,6
|
Neclasificat
|
N
|
N
|
|
2,2′-metilen-bis(6-(2H-benzotriazol-2-il)-4-(1,1,3,3-tetrametilbutil)fenol)
|
103597-45-1
|
alcan ramificat cu carbon terțiar; carbocicluri aromatice fuzionate; heterocicluri saturate fuzionate; compuși precursori ai chinoidului; terț-butil
|
S
|
102,7 ± 13,4
|
97,7 ± 5,6
|
Neclasificat
|
N
|
N
|
|
tetrafluorborat de potasiu
|
14075-53-7
|
sare anorganică
|
S
|
88,6 ± 3,3
|
92,9 ± 5,1
|
Neclasificat
|
N
|
N
|
|
Abrevieri:
Nr. CAS = Număr de înregistrare al Chemical Abstracts Service; GHS al ONU = Sistemul global armonizat de clasificare și etichetare a substanțelor chimice al Organizației Națiunilor Unite (1); VRM1 = metodă de referință validată, EpiOcular™ EIT; VRM2 = metodă de referință validată, SkinEthic™ HCE EIT; Interf. culorii = interferența culorii cu absorbanța standard [măsurarea densității optice (OD)] a formazanului MTT.
|
|
|
23.
|
În cadrul verificării performanței, se recomandă ca utilizatorii să verifice proprietățile de barieră ale țesuturilor după primire, astfel cum sunt specificate de către producătorul structurii de țesut RhCE (a se vedea punctele 25, 27 și 30). Acest lucru este deosebit de important în cazul în care țesuturile sunt transportate pe distanțe/perioade îndelungate de timp. Odată ce s-a stabilit cu succes un test, iar performanța acestuia în utilizare a fost asigurată și demonstrată, nu este necesar ca astfel de verificări să fie efectuate în mod sistematic. Cu toate acestea, atunci când se utilizează în mod sistematic un test, se recomandă evaluarea continuă a proprietăților de barieră la intervale regulate.
|
PROCEDURA
|
24.
|
Testele care sunt vizate în prezent de această metodă de testare sunt cele valabile din punct de vedere științific, EpiOcular™ EIT și SkinEthic™ HCE EIT (9) (12) (13), denumită în continuare metoda de referință validată (VRM1 și, respectiv, VRM2). Sunt disponibile proceduri standard de operare (PSO) pentru metodele de testare RhCE, care ar trebui să fie utilizate la punerea în aplicare și utilizarea metodelor de testare într-un laborator (34) (35). Punctele următoare și apendicele 2 descriu componentele și procedurile principale ale testelor RhCE.
|
COMPONENTELE METODEI DE TESTARE RHCE
Condiții generale
|
25.
|
Ar trebui să se utilizeze celule umane relevante pentru a reconstitui țesutul epitelial tridimensional, similar corneei, care ar trebui să fie compus din celule treptat stratificate, dar nu cornificate. Structura de țesut RhCE se prepară în inserții, cu o membrană sintetică poroasă, prin care nutrienții pot trece în celule. În epiteliul reconstruit similar corneei ar trebui să existe straturi multiple de celule epiteliale viabile necheratinizate. Structura de țesut RhCE ar trebui să aibă suprafața epitelială în contact direct cu aerul, astfel încât să permită expunerea locală directă a substanțelor chimice de testat într-un mod similar modului în care ar fi expus epiteliul corneean in vivo. Structura țesutului RhCE ar trebui să formeze o barieră funcțională suficient de robustă pentru a rezista la pătrunderea rapidă a substanțelor de referință citotoxice, de exemplu, e.g. Triton X-100 sau dodecilsulfat de sodiu (SDS). Funcția de barieră ar trebui să fie demonstrată și poate fi evaluată prin determinarea timpului de expunere necesar pentru a reduce viabilitatea tisulară cu 50 % (ET50) la aplicarea unei substanțe de referință la o concentrație specifică fixă (de exemplu, 100 μl de soluție 0,3 % (v/v) Triton X-100) sau la o concentrație la care o substanță de referință reduce viabilitatea tisulară cu 50 % (IC50) în urma unui timp de expunere fix (de exemplu, un tratament de 30 de minute cu 50 μl SDS) (a se vedea punctul 30). Proprietățile de etanșeitate ale structurii de țesut RhCE ar trebui să prevină trecerea substanței chimice de testat peste marginea țesutului viabil, ceea ce ar conduce la o slabă modelare a expunerii corneene. Celulele umane utilizate pentru a stabili structura țesutului RhCE nu ar trebui să fie contaminate cu bacterii, virusuri, micoplasmă și fungi. Sterilitatea structurii țesutului ar trebui să fie verificată de furnizor pentru absența contaminării cu fungi și bacterii.
|
Condiții funcționale
Viabilitate
|
26.
|
Analiza utilizată pentru cuantificarea viabilității tisulare este analiza MTT (16). Celulele viabile ale structurii țesutului RhCE reduc colorantul vital MTT într-un precipitat de formazan MTT de culoare albastră, care este apoi extras din țesut utilizând izopropanol (sau un solvent similar). Formazanul MTT extras poate fi cuantificat folosind fie o măsurare standard a absorbanței [densitatea optică - DO], fie o procedură de spectrofotometrie HPLC/UPLC (36). DO a solventului de extracție trebuie să fie suficient de mică, și anume DO < 0,1. Utilizatorii structurii țesutului RhCE ar trebui să se asigure de faptul că fiecare lot de structură de țesut RhCE utilizat îndeplinește criteriile stabilite pentru substanța martor negativă. Intervalele de acceptabilitate pentru valorile DO ale martorului negativ pentru VRM sunt date în tabelul 2. Un utilizator al spectrofotometriei HPLC/UPLC ar trebui să utilizeze intervalele DO ale martorului negativ din tabelul 2 ca și criteriu de acceptare pentru martorul negativ. Ar trebui documentat în raportul testului faptul că țesuturile tratate cu substanța martor negativă sunt stabile în cultură (prezintă măsurători ale viabilității tisulare similare) în perioada de expunere prin testare. O procedură similară ar trebui urmată de către producătorul țesuturilor în cadrul procedurii de eliberare a loturilor de țesuturi cu asigurarea controlui calității, dar, în acest caz, pot fi aplicate criterii de acceptare diferite de cele specificate în tabelul 2. Producătorul/furnizorul structurii de țesut RhCE ar trebui să stabilească un interval de acceptabilitate (limita superioară și inferioară) pentru valorile DO ale martorului negativ (în condițiile metodei de testare pentru controlul calității).
|
Tabelul 2
Intervalele de acceptabilitate pentru valorile DO ale martorului negativ (pentru utilizatorii testului)
|
Test
|
Limita inferioară de acceptabilitate
|
Limita superioară de acceptabilitate
|
|
EpiOcular™ EIT (OCL-200) – VRM1 (pentru protocoale privind substanțe lichide și solide)
|
> 0,8 (87)
|
< 2,5
|
|
SkinEthic™ HCE EIT (HCE/S) – VRM2 (pentru protocoale privind substanțe lichide și solide)
|
> 1,0
|
≤ 2,5
|
Funcția de barieră
|
27.
|
Structura de țesut RhCE ar trebui să fie suficient de groasă și solidă pentru a rezista la pătrunderea rapidă a substanțelor de referință citotoxice, astfel cum este estimat, de exemplu, prin ET50 (Triton X-100) sau IC50 (SDS) (tabelul 3). Funcția de barieră a fiecărui lot de structură de țesut RhCE utilizat ar trebui să fie demonstrată de către producătorul/furnizorul structurii de țesut RhCE la momentul livrării țesuturilor la utilizatorul final (a se vedea punctul 30).
|
Morfologie
|
28.
|
Examinarea histologică a structurii de țesut RhCE ar trebui să demonstreze faptul că aceasta este o structură de epiteliu similară corneei umane (incluzând cel puțin trei straturi de celule epiteliale viabile și o suprafață necheratinizată). Pentru VRM, morfologia corespunzătoare a fost stabilită de către producător/furnizor și, prin urmare, nu este necesar să fie demonstrată din nou de către un utilizator al metodei de testare pentru fiecare lot de țesuturi utilizat.
|
Reproductibilitate
|
29.
|
Rezultatele martorilor pozitivi și negativi ai metodei de testare ar trebui să demonstreze reproductibilitatea în timp.
|
Controlul calității (CC)
|
30.
|
Structura de țesut RhCE ar trebui să fie utilizată doar dacă producătorul/furnizorul demonstrează că fiecare lot de structură de țesut RhCE îndeplinește criteriile definite pentru introducere în procesul de fabricație, printre care cele mai relevante sunt criteriile privind viabilitatea (punctul 26) și funcția de barieră (punctul 27). Producătorul/furnizorul structurii de țesut ar trebui să se stabilească un interval de acceptabilitate (limite superioare și inferioare) pentru funcțiile de barieră, astfel cum este măsurat prin ET50 sau IC50 (a se vedea punctele 25 și 26). Intervalul de acceptabilitate ET50 și IC50 utilizat ca și criteriu de eliberare a lotului cu asigurarea CC de către producătorul/furnizorul structurilor de țesut RhCE (utilizate în VRM) este prezentat în tabelul 3. Datele care demonstrează conformitatea cu toate criteriile de eliberare din producție ar trebui furnizate de către producătorul/furnizorul structurii de țesut RhCE utilizatorilor metodei de testare, pentru ca aceștia să poată include aceste informații în raportul testului. Numai rezultatele obținute pentru țesuturi care îndeplinesc toate aceste criterii de eliberare din producție pot fi acceptate în vederea estimării fiabile a substanțelor chimice care nu necesită clasificare și etichetare pentru iritația oculară sau leziunea oculară gravă în conformitate cu GHS al ONU/Regulamentul CLP.
|
Tabelul 3
Criteriu de eliberare a lotului cu asigurarea CC
|
Test
|
Limita inferioară de acceptabilitate
|
Limita superioară de acceptabilitate
|
|
EpiOcular™ EIT (OCL-200) – VRM1 (100 μl de soluție 0,3 % (v/v) Triton X-100)
|
ET50 = 12,2 min
|
ET50 = 37,5 min
|
|
SkinEthic™ HCE EIT (HCE/S) – VRM2 (un tratament de 30 de minute cu 50 μl SDS)
|
IC50 = 1 mg/ml
|
IC50 = 3,2 mg/ml
|
Aplicarea substanțelor chimice de testat și a substanțelor martor
|
31.
|
Ar trebui să fie utilizate cel puțin două replicate de țesuturi pentru fiecare substanță chimică de testat și fiecare substanță martor din cadrul fiecărui test. Sunt utilizate două protocoale de tratament diferite, unul pentru substanțe chimice de testat lichide și unul pentru substanțe chimice de testat solide (34) (35). Pentru ambele metode și protocoale, suprafața structurii de țesut ar trebui să fie umezită cu soluție salină tamponată cu fosfat Dulbecco, fără calciu și fără magneziu, (DPBS fără Ca2+/Mg2+) înainte de aplicarea substanțelor chimice de testat, pentru a reproduce condițiile de umiditate a ochiului uman. Tratamentul aplicat țesuturilor este inițiat cu expunerea la substanța (substanțele) chimică (chimice) de testat și la substanțele martor. În cazul ambelor protocoale de tratament din ambele VRM, ar trebui să se aplice o cantitate suficientă de substanță chimică de testat sau martor pentru a acoperi uniform suprafața epitelială, evitându-se în același timp aplicarea unei doze infinite (a se vedea punctele 32 și 33) (apendicele 2).
|
|
32.
|
Substanțele chimice de testat care pot fi introduse cu pipeta la 37 °C sau la temperaturi mai scăzute (folosind o pipetă cu piston, dacă este necesar) sunt tratate ca substanțe lichide în VRM; în caz contrar, acestea ar trebui să fie tratate ca substanțe solide (a se vedea punctul 33). În VRM, substanța chimică de testat lichidă este distribuită uniform pe suprafața țesutului (adică o aplicare minimă de 60 μl/cm2) [a se vedea apendicele 2, (33) (34)]. Efectele capilare (efectele de tensiune la suprafață) care pot apărea din cauza volumelor mici aplicate pe locul de inserție (pe suprafața țesutului) ar trebui evitate în măsura în care este posibil pentru a garanta dozarea corectă a țesutului. Țesuturile tratate cu substanțe chimice de testat lichide se incubează timp de 30 de minute în condiții standard de cultură (37±2 °C, 5±1 % CO2, ≥ 95 % RH). La sfârșitul perioadei de expunere, substanța chimică de testat lichidă și substanțele martor r trebui să se elimine cu grijă de pe suprafața țesutului prin spălare intensă cu DPBS fără Ca2+/Mg2+ la temperatura camerei. Această etapă de spălare este urmată de o imersie după expunere în mediu proaspăt, la temperatura camerei (pentru a îndepărta orice substanță chimică de testat absorbită în țesut) pentru o perioadă predefinită de timp care variază în funcție de VRM utilizată. Numai în cazul VMR1 se aplică o incubare post-expunere în mediu proaspăt în condiții standard de cultură înainte de efectuarea analizei MTT [a se vedea apendicele 2, punctele (34) și (35)].
|
|
33.
|
Substanțele chimice de testat care nu pot fi introduse cu pipeta la temperaturi de până la 37 °C sunt tratate ca substanțe solide în cadrul VRM. Cantitatea de substanță chimică de testat aplicată ar trebui să fie suficientă pentru a acoperi întreaga suprafață a țesutului, adică ar trebui să se utilizeze o aplicare de minim 60 mg/cm2 (apendicele 2). În măsura în care este posibil, substanțele solide ar trebui să fie testate sub formă de pulbere fină. Țesuturile tratate cu substanțe chimice de testat solide sunt incubate pentru o perioadă predefinită de timp (în funcție de VRM utilizată) în condiții standard de cultură [a se vedea apendicele 2, (34) (35)]. La sfârșitul perioadei de expunere, substanța chimică de testat solidă și substanțele martor r trebui să se elimine cu grijă de pe suprafața țesutului prin spălare intensă cu DPBS fără Ca2+/Mg2+ la temperatura camerei. Această etapă de spălare este urmată de o imersie după expunere în mediu proaspăt, la temperatura camerei (pentru a îndepărta orice substanță chimică de testat absorbită în țesut) pentru o perioadă predefinită de timp care variază în funcție de VRM utilizată, precum și de o incubație post-expunere în mediu proaspăt în condiții de cultură standard înainte de efectuarea analizei MTT [a se vedea apendicele 2, (34) (35)].
|
|
34.
|
În fiecare etapă ar trebui să se includă martori negativi și pozitivi testați în paralel, pentru a demonstra că viabilitatea (determinată cu martorul negativ) și sensibilitatea (determinată prin martorul pozitiv) țesuturilor se situează în intervalele de acceptabilitate definite pe baza datelor istorice. Martorul negativ testat în paralel oferă, de asemenea, valoarea de referință (viabilitate tisulară de 100 %) pentru calcularea viabilității procentuale relative a țesuturilor tratate cu substanța chimică de testat (% viabilitatetest). Substanța martor pozitivă recomandată a fi utilizată cu metodele VRM este acetat de metil pur (nr. CAS 79-20-9, disponibil în comerț, de exemplu, de la Sigma-Aldrich, categ. nr. 45997; lichid). Substanțele martor negative recomandate pentru a fi utilizate împreună cu VRM1 și VRM2 sunt DPBS fără H2O și fără Ca2+/Mg2+ ultrapure. Acestea au fost substanțele de control utilizate în studiile de validare a metodelor VRM și sunt cele pentru care există cele mai multe date istorice. Utilizarea unor substanțe martor pozitive sau negative alternative adecvate ar trebui să fie justificată din punct de vedere științific și în mod adecvat. Martorii negativi și pozitivi ar trebui să fie testați cu același protocol/aceleași protocoale ca și cel(e) utilizat(e) pentru substanțele chimice de testat incluse în testare (și anume, pentru lichide și/sau solide). Această aplicare ar trebui să fie urmată de expunerea la tratament, spălare, o imersie post-expunere și o incubare post-expunere, după caz, astfel cum este descris în cazul substanțelor martor testate în paralel cu substanțele chimice de testat lichide (a se vedea punctul 32) sau, în cazul martorilor testați în paralel cu substanțele chimice de testat solide (a se vedea punctul 33), înainte de analiza MTT (a se vedea punctul 35) (34) (35). Un set unic de martori negativi și pozitivi este suficient pentru toate substanțele chimice de testat care au aceeași stare fizică (lichide sau solide) incluse în aceeași testare.
|
Măsurarea viabilității tisulare
|
35.
|
Testul MTT este o metodă cantitativă standardizată care ar trebui să fie utilizată pentru măsurarea viabilității tisulare în cadrul prezentei metode de testare. Acesta este compatibil cu o arhitectură tridimensională a țesutului. Analiza MTT este efectuată imediat după perioada de incubație post-expunere. În VRM, proba de structură de țesut RhCE este introdusă în soluție de 0,3 ml de MTT la 1 mg/ml timp de 180 ± 15 minute în condiții standard de cultură. Colorantul esențial MTT este redus într-un precipitat de formazan albastru prin celulele viabile ale structurii de țesut RhCE. Produsul precipitat de formazan albastru MTT este apoi extras din țesut utilizând un volum corespunzător de izopropanol (sau solvent similar) (34) (35). Țesuturile testate cu substanțe chimice de testat lichide ar trebui să fie extrase atât din partea superioară, cât și din partea inferioară a țesuturilor, în timp ce țesuturile testate cu substanțe chimice de testat solide și lichide colorate ar trebui să fie extrase numai din partea inferioară a țesutului (pentru a reduce la minimum orice posibilă contaminare a soluției de extracție izopropanol cu orice substanță chimică de testat care ar fi putut rămâne pe țesut). Țesuturile testate cu substanțe chimice de testat lichide care nu sunt spălate imediat pot fi extrase numai din partea inferioară a țesutului. Substanțele martor negative pozitive testate în paralel ar trebui să fie tratate în același mod ca și substanța chimică testată. Formazanul MTT extras poate fi cuantificat fie printr-o măsurare a absorbanței standard (DO) la 570 nm, cu ajutorul unei filtru de tip band pass de maximum ± 30 nm, fie utilizând o procedură de spectrofotometrie HPLC/UPLC (a se vedea punctul 42) (11) (36).
|
|
36.
|
Proprietățile optice ale substanței chimice de testat sau acțiunea sa chimică asupra MTT pot interfera cu măsurarea formazanului MTT, conducând la o estimare falsă a viabilității tisulare. Substanțele chimice de testat pot influența măsurarea formazanului MTT prin reducere directă a MTT în formazan albastru și/sau prin interferența culorii în cazul în care substanța chimică de testat absoarbe, în mod natural sau datorită procedurilor de tratament, în aceleași limite DO ca și pentru formazan MTT (și anume, 570 nm). Ar trebui să se efectueze verificări prealabile, înainte de testare, pentru a permite identificarea unor potențiali reductori direcți MTT și/sau a substanțelor chimice care influențează culoarea și ar trebui utilizați martori suplimentari pentru a detecta și a corecta posibilele interferențe din aceste acestor substanțe chimice de testat (a se vedea punctele 37-41). Acest lucru este deosebit de important atunci când o anumită substanță chimică de testat nu este îndepărtată complet din țesut prin spălare sau atunci când aceasta pătrunde în epiteliul similar corneei și, prin urmare, există în structurile de țesut RhCE atunci când este efectuată analiza MTT. În cazul substanțelor chimice de testat care absorb lumina în aceeași gamă ca și formazanul MTT (pe cale naturală sau după tratare), care nu sunt compatibile cu măsurarea absorbanței standard (DO) a formazanului MTT ca urmare a unei interferențe prea puternice, adică o absorbție puternică la 570 ± 30 nm, poate fi utilizată o procedură de spectrofotometrie HPLC/UPLC pentru măsurarea formazanului MTT (a se vedea punctele 41 și 42) (11) (36). O descriere detaliată a modului în care poate fi detectată și corectată direct reducerea MTT și interferențele cu ajutorul agenților de colorare este disponibilă în PSO ale VRM (34) (35) (36) (37). De asemenea, în apendicele III și IV sunt prezentate grafice de flux ilustrative care oferă indicații cu privire la modul de identificare și tratare a reductorilor MTT direcți și/sau a substanțelor chimice care influențează culoarea în cazul VRM1 și VRM2.
|
|
37.
|
Pentru a identifica o posibilă interferență din partea substanțelor chimice de testat care absorb lumina în același interval ca și formazanul MTT (în mod natural sau după tratament) și a decide cu privire la necesitatea unor martori suplimentari, substanța chimică de testat se adaugă în apă și/sau izopropanol și se lasă la incubat pentru un timp adecvat la temperatura camerei (a se vedea apendicele 2) (34) (35). În cazul în care substanța chimică de testat din apă și/sau izopropanol absoarbe lumină suficientă în intervalul 570 ± 20 nm în cazul VRM1 (a se vedea apendicele 3) sau dacă se obține o soluție colorată atunci când se amestecă substanța chimică de testat cu apă în cazul VRM2 (a se vedea apendicele 4), se presupune că substanța chimică de testat interferează cu măsurarea absorbanței standard (DO) a formazanului MTT și ar trebui să se obțină martori suplimentari de coloranți sau, în mod alternativ, să se utilizeze o procedură de spectrofotometrie HPLC/UPLC, caz în care nu sunt necesari acești martori (a se vedea punctele 41 și 42 și apendicele III și IV) (34) (35). La măsurarea absorbanței standard (DO), fiecare substanță chimică de testat care interferează ar trebui să fie aplicată pe cel puțin două replicate de țesut viabile, acestea fiind supuse întregii proceduri de testare, dar sunt incubate într-un mediu în locul soluției MTT în etapa de incubare a MTT pentru a genera un martor de o culoare nespecifică (NSCliving) (34) (35). Testul pentru martorul NSCliving trebuie efectuat concomitent cu testul substanței chimice de testat colorate și, în cazul testării multiple, trebuie efectuat un test pentru un martor independent NSCliving la fiecare test efectuat (în cadrul fiecărei secvențe de testare), din cauza variabilității biologice inerente a țesuturilor vii. Viabilitatea tisulară reală se calculează după cum urmează: procentul de viabilitate tisulară obținut la țesuturile vii expuse la substanța chimică de testat care interferează și incubate în soluția cu MTT (% viabilitatetest), minus procentul de culoare nespecifică obținut la țesuturi vii expuse la substanța chimică de testat care interferează și incubate în mediu fără MTT, testul fiind efectuat concomitent cu testul supus corectării (% NSCliving), și anume, viabilitate tisulară reală = [% Viabilitytest] - [% NSCliving].
|
|
38.
|
Pentru identificarea reductorilor direcți ai MTT, ar trebui să se adauge fiecare substanță chimică de testat la o soluție de MTT proaspăt preparată. O cantitate adecvată de substanță chimică de testat se adaugă la o soluție de MTT, iar amestecul este incubat timp de aproximativ 3 ore în condiții standard de cultură (a se vedea apendicele III și IV) (34) (35). Dacă amestecul cu MTT care conține substanța chimică de testat (sau suspensia pentru substanțe chimice insolubile) devine albastru/violet, se presupune că substanța chimică de testat reduce direct MTT și ar trebui să se efectueze o verificare funcțională suplimentară a structurilor de țesut RhCE neviabile, independent de utilizarea măsurării standard a absorbanței (DO) sau a unei proceduri HPLC/UPLC de spectrofotometrie. Această verificare funcțională suplimentară utilizează țesuturi moarte care au doar o activitate metabolică reziduală, dar care absorb și rețin substanța chimică de testat în mod similar țesuturilor viabile. Țesuturile ucise în cadrul VRM1 sunt preparate prin expunere la temperaturi scăzute (ucise prin congelare). Țesuturile ucise în cadrul VRM2 sunt preparate prin incubare prelungită (de exemplu, de cel puțin 24 ± 1 ore) în apă, urmată de o depozitare la temperatură joasă (ucise cu apă). Fiecare substanță chimică de testat care reduce MTT se aplică pe cel puțin două țesuturi moarte duplicate care sunt supuse întregii proceduri de testare pentru a genera un martor de reducere nespecifică a MTT (NSMTT) (34) (35). Un singur martor NSMTT este suficient pentru fiecare substanță chimică de testat, indiferent de numărul de teste/serii de teste independente efectuate. Viabilitatea tisulară reală se calculează după cum urmează: viabilitatea tisulară procentuală obținută pe țesuturi vii expuse la reductorul MTT (% viabilitatetest) minus procentul de reducere a MTT nespecific obținut pe țesuturi moarte expuse la același reductor MTT, calculat în raport cu efectuarea testului pe martorul negativ, concomitent cu testul corectat (% NSMTT), și anume, viabilitate tisulară reală = [% viabilitatetest] - [% NSMTT].
|
|
39.
|
Substanțele chimice de testat care sunt identificate ca producând atât interferența culorii (a se vedea punctul 37), cât și reducerea directă a MTT (a se vedea punctul 38) vor necesita, de asemenea, un al treilea set de martori atunci când se efectuează măsurarea absorbanței standard (DO), în afară de martorii NSMTT și NSCliving descriși la punctele anterioare. Acest lucru este valabil, de obicei, în cazul substanțelor chimice de testat care absorb lumina în intervalul 570±30 nm (de exemplu, de culoare albastră, violet, neagră), deoarece culoarea lor intrinsecă împiedică evaluarea capacității acestora de a reduce direct MTT, conform descrierii de la punctul 38. Acest lucru forțează utilizarea de martori NSMTT, în mod implicit, împreună cu martori NSCliving. Substanțele chimice de testat pentru care se produc atât martori NSMTT, cât și NSCliving, pot fi absorbite și reținute atât de țesuturile vii, cât și de cele moarte. Prin urmare, în acest caz, martorul NSMTT se poate corecta nu doar pentru o posibilă reducere directă a MTT prin substanța chimică de testat, ci și pentru interferența culorii ca urmare a aderării și reținerii substanței chimice de testat în țesuturile moarte. Aceasta ar putea conduce la o dublă corecție pentru interferența culorii, întrucât martorul NSCliving corectează deja interferența culorii determinată de absorbția și reținerea substanței chimice de testat în țesuturile vii. Pentru a evita o posibilă corecție dublă pentru interferența culorii, ar trebui efectuat un test pentru un al treilea martor de culoare nespecifică la țesuturi moarte (NSCkilled) (a se vedea apendicele III și IV) (34) (35). La acest martor suplimentar, substanța chimică de testat se aplică pe cel puțin două țesuturi duplicate moarte, care sunt supuse întregii proceduri de testare, dar se incubează în mediu în loc de MTT în etapa de incubare MTT. O singură verificare pentru martorul NSCkilled este suficientă pentru fiecare substanță chimică de testat, indiferent de numărul de teste independente, dar ar trebui să fie efectuată concomitent cu verificarea pentru martorul NSMTT și pe același lot de țesuturi. Viabilitatea tisulară reală se calculează după cum urmează: procentul de viabilitate tisulară obținut la țesuturile vii expuse la substanța chimică de testat (% viabilitatetest) % NSMTT minus % NSCliving
plus procentul de culoare nespecifică obținut la țesuturi moarte expuse la substanța chimică de testat care interferează și incubate în mediu fără MTT, cu calculare raportat la testul pentru martor negativ efectuat în paralel cu testul supus corectării (% NSCkilled), adică, viabilitate tisulară reală = [% viabilitatetest] - [% NSMTT] - [% NSCliving] + [% NSCkilled].
|
|
40.
|
Este important de remarcat faptul că reducerea MTT nespecifică și interferențele culorilor nespecifice pot crește densitatea optică (atunci când se efectuează măsurători ale absorbanței standard) a extractului de țesut peste intervalul de liniaritate a spectrofotometrului și că reducerea MTT nespecifică poate să crească, de asemenea, valoarea de vârf a formazanului MTT (atunci când sunt efectuate măsurători prin spectrometrie HPLC/UPLC) la extractul de țesut peste intervalul de liniaritate al spectrofotometrului. Pe această bază, este important ca fiecare laborator să stabilească DO/intervalul de liniaritate pentru valoarea de vârf al spectrofotometrului lor, de exemplu, cu formazan MTT (CAS nr. 57360-69-7), disponibil în comerț, de exemplu, de la Sigma-Aldrich (categ. nr. M2003) înainte de a începe testarea substanțelor chimice de testat în scopuri de reglementare.
|
|
41.
|
Măsurarea absorbanței standard (DO) cu ajutorul unui spectrofotometru este adecvată pentru evaluarea reductorilor MTT direcți și a substanțelor chimice de testat care influențează culoarea, în cazul în care interferența constatată la măsurarea formazanului MTT nu este prea puternică (și anume, valorile OD ale extractelor de țesuturi obținute cu substanța chimică de testat fără nicio corecție pentru reducerea directă a MTT și/sau interferență a culorii se află în intervalul liniar al spectrofotometrului). Cu toate acestea, rezultatele pentru substanțele chimice de testat care produc % NSMTT și/sau % NSCliving ≥ 60 % (VRM1 și VRM2 pentru protocolul substanțelor lichide) sau 50 % (pentru protocolul substanțelor solide) din martorul negativ ar trebui să fie tratate cu precauție, deoarece aceasta este valoarea-limită stabilită utilizată în cadrul VRM-urilor pentru a distinge substanțele chimice clasificate de cele neclasificate (a se vedea punctul 44). Însă absorbanța standard (DO) nu poate fi măsurată atunci când interferența cu măsurarea formazanului MTT este prea puternică (și anume, determinând valori ale DO necorectate ale extractelor de țesut de testare care nu se încadrează în intervalul linear al spectrofotometrului). Substanțele chimice de testat colorate sau substanțele chimice de testat care se colorează în contact cu apă sau izopropanol care interferează prea mult cu măsurarea absorbanței standard (DO) a formazanului MTT poate fi totuși evaluată cu ajutorul spectrofotometriei HPLC/UPLC (a se vedea apendicele III și IV). Aceasta se datorează faptului că sistemul HPLC/UPLC permite separarea formazanului MTT de substanța chimică înainte de cuantificarea sa (36). Din acest motiv, martorii NSCliving sau NSCkilled, nu sunt niciodată necesari atunci când se utilizează spectrofotometria HPLC/UPLC, independent de substanța chimică de testat. Însă ar trebui să fie utilizați martorii NSMTT în cazul în care substanța chimică de testat este suspectată a reduce MTT în mod direct (în conformitate cu procedura descrisă la punctul 38). Martorii NSMTT ar trebui să fie utilizați, de asemenea, cu substanțe chimice de testat care au o culoare (intrinsecă sau care apare în apă) ce împiedică evaluarea capacității acestora de a reduce MTT în mod direct, astfel cum este descris la punctul 38. Atunci când se utilizează spectrofotometria HPLC/UPLC pentru a măsura formazanul MTT, procentul de viabilitate tisulară se calculează ca procent al formazanului MTT de vârf, care este obținut la țesuturi vii expuse la substanța chimică de testat raportat la valoarea de vârf a formazanului MTT obținută pe martorul negativ testat în paralel. În cazul substanțelor chimice de testat care pot reduce MTT în mod direct, viabilitatea tisulară reală se calculează după cum urmează: % viabilitatetest
minus % NSMTT, astfel cum este descris în ultima teză de la punctul 38. În cele din urmă, ar trebui remarcat faptul că reductorii direcți ai MTT sau reductorii direcți ai MTT care pot interfera, de asemenea, cu culorile și care sunt reținuți în țesuturi după tratament și reduc MTT atât de mult încât conduc în mod semnificativ la valori ale DO (prin măsurarea standard a DO) sau la valori de vârf (folosind spectrofotometrul UPLC/HPLC) ale extractelor de țesuturi testate care nu se încadrează în intervalul de linearitate al spectrofotometrului, nu pot fi evaluați prin metodele de testare RhCE, deși se apariția acestora este preconizată doar în situații foarte rare.
|
|
42.
|
Spectrofotometria HPLC/UPLC poate fi utilizată, pe toate tipurile de substanțe chimice de testat (colorate, necolorate, reductori ai MTT și nereductori ai MTT) pentru măsurarea formazanului MTT (11) (36). Datorită diversității sistemelor de spectrophotometrie HPLC/UPLC, nu este fezabil pentru fiecare utilizator să stabilească exact același condiții ale sistemului. Ca atare, calificarea sistemului de spectrofotometrie HPLC/UPLC ar trebui să fie demonstrată înainte ca acesta să fie utilizat pentru a cuantifica formazanul MTT din extractele de țesuturi prin îndeplinirea criteriilor de acceptare pentru un set de parametri standard de calificare, pe baza celor descrise în orientarea U.S. Food and Drug Administration pentru industrie cu privire la validarea metodei bioanalitice (36) (38). Acești parametri-cheie și criteriile de acceptare aferente sunt prezentate în apendicele 5. Odată ce criteriile de acceptare definite în apendicele 5 au fost îndeplinite, sistemul de spectrofotometrie prin HPLC/UPLC este considerat a fi calificat și pregătit pentru a măsura formazanul MTT în condițiile experimentale descrise în prezenta metodă de testare.
|
Criterii de acceptare
|
43.
|
Pentru fiecare testare în care sunt utilizate loturi de țesuturi RhCE care au îndeplinit cerințele de control al calității (a se vedea punctul 30), țesuturile tratate cu substanța martor negativă ar trebui să prezinte o valoare DO care să reflecte calitatea țesuturilor în urma transportului, a etapelor de recepție și a tuturor proceselor de protocol și nu ar trebui să fie în afara limitelor stabilite în mod tradițional, care sunt descrise în tabelul 2 (a se vedea punctul 26). În mod similar, țesuturile tratate cu substanță martor pozitivă, și anume acetat de metil, ar trebui să prezinte o viabilitate tisulară medie < 50 % față de martorul negativ în cadrul VRM1, în protocoalele privind substanțele lichide sau cele solide, și ≤ 30 % (protocolul privind substanțele lichide) față de martorul negativ în cadrul VRM2, reflectând astfel capacitatea țesuturilor de a reacționa la o substanță chimică de testat iritantă în condițiile metodei de testare (34) (35). Variabilitatea între replicile de țesuturi ale substanțelor chimice de testat și cele ale substanțelor martor ar trebui să se încadreze în limitele acceptate (și anume, diferența de viabilitate dintre două replici de țesut ar trebui să fie mai mică de 20 % sau abaterea standard (SD) între trei replici de țesut nu ar trebui să depășească 18 %). În cazul în care martorul negativ sau martorul pozitiv inclus într-o testare nu se încadrează în intervalele acceptate, se consideră că testarea nu este calificată și ar trebui să fie repetată. Dacă variabilitatea dintre replicile de țesut ale unei substanțe chimice de testat se situează în afara intervalului acceptat, testul trebuie considerat a fi „necalificat”, iar substanța chimică de testat ar trebui să fie testată din nou.
|
Interpretarea rezultatelor și modelul predictiv
|
44.
|
Valorile densității optice/de vârf obținute cu replicile de extracte de țesut pentru fiecare substanță chimică de testat ar trebui să fie utilizate pentru a calcula media procentuală a viabilității tisulare (media între replicile de țesut) normalizată la martorul negativ, care este stabilită la 100 %. Valoarea-limită a viabilității tisulare procentuale pentru identificarea substanțelor chimice de testat care nu necesită clasificare pentru iritația oculară sau leziunea oculară gravă (fără clasificare conform GHS al ONU și Regulamentul CLP) este dată în tabelul 4. Rezultatele ar trebui să fie, așadar, interpretate după cum urmează:
|
—
|
Substanța chimică de testat este identificată ca nefiind necesar să fie clasificată și etichetată în conformitate cu GHS al ONU și cu Regulamentul CLP (neclasificată), în cazul în care procentul mediu de viabilitate tisulară după expunere și incubația după expunere este mai mare decât (>) valoarea-limită stabilită a procentului de viabilitate tisulară, astfel cum este indicat în tabelul 4. În acest caz, nu este necesară testarea suplimentară în cadrul altor metode de testare.
|
|
—
|
Dacă procentul mediu de viabilitate tisulară după expunere și incubația după expunere este mai mic decât sau egal cu (≤) valoarea-limită stabilită a procentului de viabilitate tisulară, nu se poate face o estimare, astfel cum se arată în tabelul 4. În acest caz, testarea suplimentară prin alte metode de testare va fi necesară deoarece metodele de testare RhCE indică un anumit număr de rezultate fals pozitive (a se vedea punctele 14-15) și nu pot face diferența între categoriile 1 și 2 din GHS al ONU și Regulamentul CLP (a se vedea punctul 17).
|
Tabelul 4
Modele predictive conform clasificării GHS al ONU și CLP
|
MRV
|
Neclasificată
|
Nu se poate estima
|
|
VRM 1 - EpiOcular™ EIT (pentru ambele protocoale)
|
Viabilitate tisulară medie > 60 %
|
Viabilitate tisulară medie ≤ 60 %
|
|
VRM 2 - SkinEthic™ HCE EIT (pentru protocolul lichidelor)
|
Viabilitate tisulară medie > 60 %
|
Viabilitate tisulară medie ≤ 60 %
|
|
VRM2 - SkinEthic™ HCE EIT (pentru protocolul solidelor)
|
Viabilitate tisulară medie > 50 %
|
Viabilitate tisulară medie ≤ 50 %
|
|
|
45.
|
Un singur test compus din cel puțin două duplicate de țesuturi ar trebui să fie suficient pentru o substanță chimică de testat atunci când rezultatul este fără echivoc. În schimb, în cazul rezultatelor ambigue, precum măsurători neconcordante ale replicilor și/sau un procent mediu de viabilitate tisulară egal cu 60 ± 5 % (VRM1 și VRM2 pentru protocolul lichidelor) sau 50 ± 5 % (VRM2 pentru protocolul solidelor), ar trebui avut în vedere un al doilea test, precum și un al treilea test în cazul unor rezultate discordante între primele două teste.
|
|
46.
|
Pentru anumite tipuri de amestecuri ar putea fi avute în vedere valori-limită procentuale diferite pentru viabilitatea tisulară care diferențiază substanțele chimice de testat clasificate de cele neclasificate, după caz și dacă acest lucru este justificat, pentru a crește nivelul general de performanță a metodei de testare pentru respectivele tipuri de amestecuri (a se vedea punctul 14). Substanțele chimice de referință pot fi utile pentru evaluarea potențialului de leziune oculară gravă/iritație oculară al substanțelor chimice de testat necunoscute sau din clasa de produse sau pentru evaluarea potențialului de toxicitate oculară relativă al unei substanțe chimice clasificate în limitele unui anumit interval de răspunsuri pozitive.
|
DATE ȘI RAPORTARE
Date
|
47.
|
Ar trebui să se consemneze în format tabelar, pentru fiecare substanță chimică de testat, datele fiecărei replici de țesut din cadrul unei testări (de exemplu, valorile DO/valorile de vârf ale formazanului MTT și datele procentuale calculate pentru viabilitatea tisulară pentru substanța chimică de testat și pentru substanțele martor, precum și estimarea finală a metodei de testare RhCE), inclusiv date provenind din testele repetate, după caz. În plus, ar trebui să se consemneze media procentuală de viabilitate a țesutului și diferența de viabilitate dintre două replici de țesut (dacă n = 2 replici de țesut) sau SD (dacă n ≥ 3 replici de țesut) pentru fiecare substanță chimică de testat și martor. Orice interferențe observate ale unei substanțe chimice de testat cu măsurarea formazan MTT prin reducerea directă a MTT și/sau interferența prin culoare ar trebui să fie consemnate pentru fiecare substanță chimică de testat.
|
Raport testului
|
48.
|
Raportul testului ar trebui să includă următoarele informații:
Substanța chimică de testat
|
|
Substanța monocomponentă
|
—
|
identificarea chimică, precum denumirea (denumirile) IUPAC sau CAS, numărul (numerele) de registru CAS, codul SMILES sau InChI, formula structurală și/sau alți identificatori;
|
|
—
|
starea fizică, volatilitatea, pH, LogP, masa moleculară, clasa de substanțe chimice și alte proprietăți fizico-chimice relevante pentru desfășurarea studiului, în măsura în care este posibil;
|
|
—
|
puritatea, identitatea chimică a impurităților, după caz și dacă este fezabil din punct de vedere practic etc.;
|
|
—
|
tratamentul înainte de efectuarea testului, dacă este cazul (de exemplu, încălzire, măcinare);
|
|
—
|
condițiile de depozitare și stabilitatea, dacă există.
|
|
|
|
Substanța multicomponentă, UVCB și amestec
|
—
|
Caracterizarea, dacă este posibil, de exemplu prin identitatea chimică (a se vedea mai sus), puritatea, apariția cantitativă și proprietățile fizico-chimice relevante (a se vedea mai sus) ale componentelor, dacă există;
|
|
—
|
starea fizică și alte proprietăți fizico-chimice relevante pentru desfășurarea studiului, în măsura în care este posibil;
|
|
—
|
puritatea, identitatea chimică a impurităților, după caz și dacă este fezabil din punct de vedere practic etc.;
|
|
—
|
tratamentul înainte de efectuarea testului, dacă este cazul (de exemplu, încălzire, măcinare);
|
|
—
|
condițiile de depozitare și stabilitatea, dacă există.
|
|
Substanțe martor pozitive și negative
|
—
|
identificarea chimică, precum denumirea (denumirile) IUPAC sau CAS, numărul (numerele) de registru CAS, codul SMILES sau InChI, formula structurală și/sau alți identificatori;
|
|
—
|
starea fizică, volatilitatea, masa moleculară, clasa de substanțe chimice și alte proprietăți fizico-chimice relevante pentru desfășurarea studiului, în măsura în care este posibil;
|
|
—
|
puritatea, identitatea chimică a impurităților, după caz și dacă este fezabil din punct de vedere practic etc.;
|
|
—
|
tratamentul înainte de efectuarea testului, dacă este cazul (de exemplu, încălzire, măcinare);
|
|
—
|
condițiile de depozitare și stabilitatea, dacă există;
|
|
—
|
justificarea utilizării unui tip de martor negativ diferit de H2O ultrapură sau DPBS fără Ca 2+/Mg2+, dacă este cazul;
|
|
—
|
justificarea utilizării unui martor pozitiv diferit de acetat de metil pur, dacă este cazul;
|
|
—
|
trimitere la rezultatele anterioare ale martorilor pozitivi și negativi care demonstrează criterii adecvate de acceptare a testelor.
|
Informații referitoare la sponsor și la instalația de testare
|
—
|
Numele și adresa sponsorului, ale instalației de testare și ale directorului de studiu.
|
|
—
|
Structura de țesut RhCE și protocolul utilizat (cu justificarea alegerii lor, dacă este cazul)
|
Condițiile metodei de testare
|
—
|
structura de țesut RhCE utilizată, inclusiv numărul de lot;
|
|
—
|
lungimea de undă și filtrul de tip band pass (dacă este cazul) utilizate pentru cuantificarea formazanului MTT, precum și intervalul de linearitate al dispozitivului de măsurare (de exemplu, spectrofotometru);
|
|
—
|
descrierea metodei utilizate pentru cuantificarea formazanului MTT;
|
|
—
|
descrierea sistemului de spectrofotometrie HPLC/UPLC utilizat, dacă este cazul;
|
|
—
|
informații de asistență complete privind structura de țesut RhE specifică utilizată, inclusiv performanța acestuia. Aceasta trebuie să includă, printre altele:
|
i)
|
controlul calității pentru viabilitate (furnizor)
|
|
ii)
|
viabilitate în condițiile metodei de testare (utilizator);
|
|
iii)
|
controlul calității funcției de barieră;
|
|
iv)
|
morfologia, dacă este disponibilă;
|
|
v)
|
Reproductibilitatea și capacitatea de estimare;
|
|
vi)
|
alte controale ale calității (CC) asupra structurii de țesut RhCE, dacă există;
|
|
|
—
|
trimiteri la datele istorice ale structurii de țesut RhCE. Aceasta trebuie să includă, printre altele: acceptabilitatea datelor CC cu trimitere la datele istorice privind lotul;
|
|
—
|
o declarație din care să reiasă că instalația de testare și-a demonstrat competența în utilizarea metodei de testare înainte de utilizarea sistematică prin testarea substanțelor chimice de verificare.
|
Durata și criteriile de acceptare a testului:
|
—
|
valorile medii ale martorilor pozitivi și negativi și intervale de acceptare pe baza datelor istorice;
|
|
—
|
variabilitatea acceptabilă dintre țesuturile duplicate pentru martorii pozitivi și negativi;
|
|
—
|
variabilitatea acceptabilă dintre replicile de țesut pentru substanța chimică de testat.
|
Procedura de testare
|
—
|
detalii cu privire la procedura de testare utilizată;
|
|
—
|
dozele de substanțe chimice de testat și de substanțe martor utilizate;
|
|
—
|
durata și temperatura expunerii, perioadele de imersie post-expunere și de incubație post-expunere (dacă este cazul);
|
|
—
|
descrierea oricăror modificări ale procedurii de testare;
|
|
—
|
indicarea martorilor utilizați pentru agenții direcți de reducere a MTT și/sau pentru substanțele testate pentru colorare, dacă este cazul;
|
|
—
|
numărul de replici de țesut utilizate pentru fiecare substanță chimică de testat și martori (martor pozitiv, martor negativ, NSMTT, NSCliving și NSCkilled, dacă este cazul);
|
Rezultate
|
—
|
prezentarea tabelară a datelor privind fiecare substanță chimică de testat și martor pe testări (inclusiv experimente repetate, dacă este cazul) și fiecare măsurare a replicilor, inclusiv valoarea DO sau valoarea de vârf pentru formazan MTT, procentul de viabilitate tisulară, procentul mediu de viabilitate tisulară, diferențele între replicile de țesut sau DS și estimarea finală;
|
|
—
|
dacă este cazul, rezultatele martorilor utilizați pentru reductorii direcți ai MTT și/sau substanțele chimice colorate, inclusiv valorile de vârf ale DO sau formazanului MTT, % NSMTT, % NSCliving, % NSCkilled, diferențe între replici de țesut sau DS, procentul final corect de viabilitate tisulară și estimarea finală;
|
|
—
|
rezultatele obținute cu substanța (substanțele) chimică(e) de testat și substanțele martor în legătură cu criteriile de acceptare ale verificării și testării specifice;
|
|
—
|
descrierea altor efecte observate, de exemplu, colorarea țesuturilor cu o substanță chimică de testat colorată;
|
Discutarea rezultatelor
Concluzii
|
REFERINȚE BIBLIOGRAFICE
|
(1)
|
ONU (2015). Sistemul global armonizat de clasificare și etichetare a substanțelor chimice al Organizației Națiunilor Unite (GHS). ST/SG/AC.10/30/Rev.6, a șasea ediție revizuită, New York și Geneva: Organizația Națiunilor Unite. Publicație disponibilă la adresa: http://www.unece.org/fileadmin/DAM/trans/danger/publi/ghs/ghs_rev06/English/ST-SG-AC10-30-Rev6e.pdf.
|
|
(2)
|
Capitolul B.5 din prezenta anexă, Iritație/coroziune oculară acută.
|
|
(3)
|
Capitolul B.47 din prezenta anexă, Metoda de testare a opacității și permeabilității corneene la bovine pentru identificarea i) substanțelor chimice care produc lezarea oculară gravă și a ii) substanțelor chimice care nu necesită clasificare pentru iritația oculară gravă sau lezarea oculară gravă.
|
|
(4)
|
Capitolul B.48 din prezenta anexă, Metoda de testare a ochiului izolat de pui pentru identificarea i) substanțelor chimice care produc lezarea oculară gravă și a ii) substanțelor chimice care nu necesită clasificare.
|
|
(5)
|
Capitolul B.61 din prezenta anexă, Metoda de testare pentru scăpări de fluoresceină în vederea identificării substanțelor corozive și iritante grave pentru ochi.
|
|
(6)
|
Capitolul B.68 din prezenta anexă, Metoda de testare in vitro prin expunere pe termen scurt pentru identificarea i) substanțelor chimice care produc lezarea oculară gravă și a ii) substanțelor chimice care nu necesită clasificare pentru iritația oculară sau leziunea oculară gravă.
|
|
(7)
|
Freeman, S.J., Alépée N., Barroso, J., Cole, T., Compagnoni, A., Rubingh, C., Eskes, C., Lammers, J., McNamee, P., Pfannenbecker, U., Zuang, V. (2010). Prospective Validation Study of Reconstructed Human Tissue Models for Eye Irritation Testing. ALTEX 27, ediție specială 2010, 261-266.
|
|
(8)
|
EC EURL ECVAM (2014). The EURL ECVAM - Cosmetics Europe prospective validation study of Reconstructed human Cornea-like Epithelium (RhCE)-based test methods for identifying chemicals not requiring classification and labelling for serious eye damage/eye irritation: Validation Study Report. EUR 28125 EN; doi:10.2787/41680. Publicație disponibilă la adresa: http://publications.jrc.ec.europa.eu/repository/handle/JRC100280.
|
|
(9)
|
EURL ECVAM Science Advisory Committee (2014). ESAC Opinion on the EURL ECVAM Eye Irritation Validation Study (EIVS) on EpiOcular™ EIT and SkinEthic™ HCE and a related Cosmetics Europe study on HPLC/UPLC-spectrophotometry as an alternative endpoint detection system for MTT-formazan. Avizul nr. 2014-03 al ESAC din 17 noiembrie 2014; EUR 28173 EN; doi: 10.2787/043697. Publicație disponibilă la adresa: http://publications.jrc.ec.europa.eu/repository/handle/JRC103702.
|
|
(10)
|
Alépée, N., Leblanc, V., Adriaens, E., Grandidier, M.H., Lelièvre, D, Meloni, M., Nardelli, L., Roper, C.S, Santirocco, E., Toner, F., Van Rompay, A., Vinall, J., Cotovio, J. (2016). Multi-laboratory validation of SkinEthic HCE test method for testing serious eye damage/eye irritation using liquid chemicals. Toxicol. In Vitro 31, 43-53.
|
|
(11)
|
Alépée, N., Adriaens, E., Grandidier, M.H., Meloni, M., Nardelli, L., Vinall, C.J., Toner, F., Roper, C.S, Van Rompay, A.R., Leblanc, V., Cotovio, J. (2016). Multi-laboratory evaluation of SkinEthic HCE test method for testing serious eye damage/eye irritation using solid chemicals and overall performance of the test method with regard to solid and liquid chemicals testing. Toxicol. In Vitro 34, 55-70.
|
|
(12)
|
EURL ECVAM Science Advisory Committee (2016). ESAC Opinion on the SkinEthic™ Human Corneal Epithelium (HCE) Eye Irritation Test (EIT). Avizul nr. 2016-02 al ESAC din 24 iunie 2016; EUR 28175 EN; doi: 10.2787/390390. Publicație disponibilă la adresa: http://publications.jrc.ec.europa.eu/repository/handle/JRC103704.
|
|
(13)
|
EC EURL ECVAM (2016). Recommendation on the Use of the Reconstructed human Cornea-like Epithelium (RhCE) Test Methods for Identifying Chemicals not Requiring Classification and Labelling for Serious Eye Damage/Eye Irritation According to UN GHS. (Manuscris în curs de elaborare).
|
|
(14)
|
Draize, J.H., Woodard, G., Calvery, H.O. (1944). Methods for the Study of Irritation and Toxicity of Substances Applied Topically to the Skin and Mucous Membranes. Journal of Pharmacol. and Exp. Therapeutics 82, 377-390.
|
|
(15)
|
Scott, L., Eskes, C., Hoffmann, S., Adriaens, E., Alépée, N., Bufo, M., Clothier, R., Facchini, D., Faller, C., Guest, R., Harbell, J., Hartung, T., Kamp, H., Le Varlet, B., Meloni, M., McNamee, P., Osborne, R., Pape, W., Pfannenbecker, U., Prinsen, M., Seaman, C., Spielman, H., Stokes, W., Trouba, K., Van den Berghe, C., Van Goethem, F., Vassallo, M., Vinardell, P., Zuang, V. (2010). A Proposed Eye Irritation Testing Strategy to Reduce and Replace In Vivo Studies Using Bottom-Up and Top-Down Approaches. Toxicol. In Vitro 24, 1-9.
|
|
(16)
|
Mosmann, T. (1983). Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 65, 55-63.
|
|
(17)
|
OCDE (2016). Series on Testing and Assessment No 216: Performance Standards for the Assessment of Proposed Similar or Modified In Vitro Reconstructed Human Cornea-Like Epithelium (RhCE) Test Methods for Identifying Chemicals not Requiring Classification and Labelling for Eye Irritation or Serious Eye Damage, Based on the Validated Reference Methods EpiOcular™ EIT and SkinEthic™ HCE EIT described in TG 492. Organizația pentru Cooperare și Dezvoltare Economică, Paris.
|
|
(18)
|
OCDE (2005). Series on Testing and Assessment No 34: Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Organizația pentru Cooperare și Dezvoltare Economică, Paris.
|
|
(19)
|
Kaluzhny, Y., Kandárová, H., Hayden, P., Kubilus, J., d’Argembeau-Thornton, L., Klausner, M. (2011). Development of the EpiOcular™ Eye Irritation Test for Hazard Identification and Labelling of Eye Irritating Chemicals in Response to the Requirements of the EU Cosmetics Directive and REACH Legislation. Altern. Lab. Anim. 39, 339-364.
|
|
(20)
|
Nguyen, D.H., Beuerman, R.W., De Wever, B., Rosdy, M. (2003). Three-dimensional construct of the human corneal epithelium for in vitro toxicology. În: Salem, H., Katz, S.A. (Eds), Alternative Toxicological Methods, CRC Press, pp. 147-159.
|
|
(21)
|
Pfannenbecker, U., Bessou-Touya, S., Faller, C., Harbell, J., Jacob, T., Raabe, H., Tailhardat, M., Alépée, N., De Smedt, A., De Wever, B., Jones, P., Kaluzhny, Y., Le Varlet, B., McNamee, P., Marrec-Fairley, M., Van Goethem, F. (2013). Cosmetics Europe multi-laboratory pre-validation of the EpiOcular™ reconstituted Human Tissue Test Method for the Prediction of Eye Irritation. Toxicol. In Vitro 27, 619-626.
|
|
(22)
|
Alépée, N., Bessou-Touya, S., Cotovio, J., de Smedt, A., de Wever, B., Faller, C., Jones, P., Le Varlet, B., Marrec-Fairley, M., Pfannenbecker, U., Tailhardat, M., van Goethem, F., McNamee, P. (2013). Cosmetics Europe Multi-Laboratory Pre-Validation of the SkinEthic™ Reconstituted Human Corneal Epithelium Test Method for the Prediction of Eye Irritation. Toxicol. In Vitro 27, 1476-1488.
|
|
(23)
|
Kolle, S.N., Moreno, M.C.R., Mayer, W., van Cott, A., van Ravenzwaay, B., Landsiedel, R. (2015). The EpiOcular™ Eye Irritation Test is the Method of Choice for In Vitro Eye Irritation Testing of Agrochemical Formulations: Correlation Analysis of EpiOcular™ Eye Irritation Test and BCOP Test Data to UN GHS, US EPA and Brazil ANIVSA Classifications. Altern. Lab. Anim. 43, 1-18.
|
|
(24)
|
Adriaens, E., Barroso, J., Eskes, C., Hoffmann, S., McNamee, P., Alépée, N., Bessou-Touya, S., De Smedt, A., De Wever, B., Pfannenbecker, U., Tailhardat, M., Zuang, V. (2014). Retrospective Analysis of the Draize Test for Serious Eye Damage/Eye Irritation: Importance of Understanding the In Vivo Endpoints Under UN GHS/EU CLP for the Development and Evaluation of In Vitro Test Methods. Arch. Toxicol. 88, 701-723.
|
|
(25)
|
Barroso, J., Pfannenbecker, U., Adriaens, E., Alépée, N., Cluzel, M., De Smedt, A., Hibatallah, J., Klaric, M., Mewes, K.R., Millet, M., Templier, M., McNamee, P. (2017). Cosmetics Europe compilation of historical serious eye damage/eye irritation in vivo data analysed by drivers of classification to support the selection of chemicals for development and evaluation of alternative methods/strategies: the Draize eye test Reference Database (DRD). Arch. Toxicol. 91, 521-547.
|
|
(26)
|
Meloni, M., De Servi, B., Marasco, D., Del Prete, S. (2011). Molecular mechanism of ocular surface damage: Application to an in vitro dry eye model on human corneal epithelium. Molecular Vision 17, 113-126.
|
|
(27)
|
Hackett, R.B., McDonald, T.O. (1991). Eye Irritation. In Advances in Modern Toxicology: Dermatoxicology Marzulli F.N.și Maibach H.I. (editori), a patra ediție, pp. 749–815. Washington, DC, SUA: Hemisphere Publishing Corporation.
|
|
(28)
|
Fox, D.A., Boyes, W.K. (2008). Toxic Responses of the Ocular and Visual System. In Cassaret and Doull’s Toxicology: The Basic Science of Poisons Klaassen C.D.(Ed.), a șaptea ediție, pp. 665–697. Withby, ON, Canada: McGraw-Hill Ryerson.
|
|
(29)
|
Jester, J.V., Li, H.F., Petroll, W.M., Parker, R.D., Cavanagh, H.D., Carr, G.J., Smith, B., Maurer, J.K. (1998). Area and Depth of Surfactant Induced Corneal Injury Correlates with Cell Death. Invest. Ophthalmol. Vis. Sci. 39, 922-936.
|
|
(30)
|
Maurer, J.K., Parker, R.D., Jester, J.V. (2002). Extent of Corneal Injury as the Mechanistic Basis for Ocular Irritation: Key Findings and Recommendations for the Development of Alternative Assays. Reg. Tox. Pharmacol. 36, 106-117.
|
|
(31)
|
Jester, J.V., Li, L., Molai, A., Maurer, J.K. (2001). Extent of Corneal Injury as a Mechanistic Basis for Alternative Eye Irritation Tests. Toxicol. In Vitro 15, 115-130.
|
|
(32)
|
Jester, J.V., Petroll, W.M., Bean, J., Parker, R.D., Carr, G.J., Cavanagh, H.D., Maurer, J.K. (1998). Area and Depth of Surfactant-Induced Corneal Injury Predicts Extent of Subsequent Ocular Responses. Invest. Ophthalmol. Vis. Sci. 39, 2610-2625.
|
|
(33)
|
Jester, J.V. (2006). Extent of Corneal Injury as a Biomarker for Hazard Assessment and the Development of Alternative Models to the Draize Rabbit Eye Test. Cutan. Ocul. Toxicol. 25, 41-54.
|
|
(34)
|
EpiOcular™ EIT SOP, versiunea a 8-a (5 martie 2013). EpiOcular™ EIT for the Prediction of Acute Ocular Irritation of Chemicals. Publicație disponibilă la adresa: [https://ecvam-dbalm.jrc.ec.europa.eu/beta/index.cfm/methodsAndProtocols/index].
|
|
(35)
|
SkinEthic™ HCE EIT SOP, versiunea 1. (20 iulie 2015). SkinEthic™ HCE Eye Irritation Test (EITL for Liquids, EITS for Solids) for the Prediction of Acute Ocular Irritation of Chemicals. Publicație disponibilă la adresa: https://ecvam-dbalm.jrc.ec.europa.eu/beta/index.cfm/methodsAndProtocols/index.
|
|
(36)
|
Alépée, N., Barroso, J., De Smedt, A., De Wever, B., Hibatallah, J., Klaric, M., Mewes, K.R., Millet, M., Pfannenbecker, U., Tailhardat, M., Templier, M., McNamee, P. (2015). Use of HPLC/UPLC-Spectrophotometry for Detection of Formazan in In Vitro Reconstructed Human Tissue (RhT)-Based Test Methods Employing the MTT-Reduction Assay to Expand their Applicability to Strongly Coloured Test Chemicals. Toxicol. In Vitro 29, 741-761.
|
|
(37)
|
Kaluzhny, Y., Kandárová, H., Handa, Y., DeLuca, J., Truong, T., Hunter, A., Kearney, P., d’Argembeau-Thornton, L., Klausner, M. (2015). EpiOcular™ Eye Irritation Test (EIT) for Hazard Identification and Labeling of Eye Irritating Chemicals: Protocol Optimization for Solid Materials and Extended Shipment Times. Altern. Lab Anim. 43, 101-127.
|
|
(38)
|
US FDA (2001). Guidance for Industry: Bioanalytical Method Validation. U.S. Department of Health and Human Services, Food and Drug Administration. Mai 2001. Publicație disponibilă la adresa: http://www.fda.gov/downloads/Drugs/Guidances/ucm070107.pdf.
|
|
(39)
|
OCDE (2017). Guidance Document on an Integrated Approaches on Testing and Assessment for Serious Eye Damage and Eye irritation. Series on Testing and Assessment, nr. 263. ENV Publications, Organizația pentru Cooperare și Dezvoltare Economică, Paris.
|
Apendicele 1
DEFINIȚII
Precizie
: gradul de apropiere dintre rezultatele metodei de testare și valorile de referință acceptate. Aceasta măsoară performanța metodei de testare și un aspect al „relevanței”. Termenul este adesea utilizat în paralel cu termenul de „concordanță” pentru a desemna proporția de rezultate corecte ale unei metode de testare (18).
Substanță chimică de referință
: o substanță chimică utilizată în calitate de standard pentru comparația cu o substanță chimică de testat. O substanță de referință ar trebui să aibă următoarele proprietăți: (i) o sursă (surse) consecventă (consecvente) și sigură (sigure) pentru identificarea și caracterizarea sa; (ii) asemănări structurale, funcționale și/sau chimice sau la nivelul clasei de produse cu substanța (substanțele) chimică (chimice) testată (testate); (iii) caracteristici fizico-chimice cunoscute; (iv) date de susținere privind efectele cunoscute; și (v) putere cunoscută în intervalul de răspuns dorit.
Abordarea ascendentă
: abordarea etapizată utilizată pentru o substanță chimică de testat susceptibilă de a nu necesita clasificare și etichetare pentru iritația oculară sau leziunea oculară gravă, care începe cu determinarea substanțelor chimice care nu necesită clasificare și etichetare (rezultat negativ) din alte substanțe chimice (rezultat pozitiv).
Substanță chimică
: o substanță sau un amestec.
Concordanță
: a se vedea punctul „Precizie”.
Cornee
: partea transparentă a părții frontale a globului ocular care acoperă irisul și pupila și care permite pătrunderea luminii în interior.
CV
: coeficient de variație.
Dev
: abatere.
EIT
: test de iritație cutanată.
EURL ECVAM
: Laboratorul de referință al UE pentru alternative la testarea pe animale.
Iritație oculară
: producerea de modificări la nivel ocular în urma aplicării unei substanțe chimice de testat pe suprafața anterioară a ochiului, care sunt complet reversibile în termen de 21 de zile de la aplicare. Termen utilizat în paralel cu „efecte reversibile asupra ochiului” și cu „categoria 2 din GHS al ONU”
ET50
: perioada de expunere necesară pentru a reduce viabilitatea tisulară cu 50 % la aplicarea unei substanțe chimice de referință cu o concentrație specifică fixă.
Rată de rezultate fals negative
: proporția tuturor substanțelor pozitive identificate în mod fals de o metodă de testare ca fiind negative. Acesta este unul dintre indicatorii de performanță ai metodei de testare.
Rată de rezultate fals pozitive
: proporția tuturor substanțelor negative identificate în mod fals de o metodă de testare ca fiind pozitive. Acesta este unul dintre indicatorii de performanță ai metodei de testare.
Pericol
: proprietate intrinsecă a unui agent sau a unei situații care are potențialul de a produce efecte adverse atunci când un organism, un sistem sau o (sub)populație este expusă la agentul respectiv.
HCE
: SkinEthic™ Human Corneal Epithelium.
HPLC
: cromatografie lichidă de înaltă performanță.
IC50
: concentrație la care o substanță chimică de referință reduce viabilitatea țesutului cu 50 % ca urmare a unui timp de expunere fix (de exemplu, un tratamen tde 30 de minute cu SDS).
Doză infinită
: cantitate de substanță chimică de testat aplicată pe structura de țesut RhCE care depășește cantitatea necesară pentru acoperirea completă și uniformă a suprafeței epiteliale.
Efecte ireversibile asupra ochilor
: A se vedea „Leziune oculară gravă”.
LLOQ
: limita de cuantificare inferioară.
LogP
: logaritmul coeficientului de partiționare octanol-apă
Amestec
: un amestec sau o soluție compus/ă din două sau mai multe substanțe.
Substanță mono-constituent
: O substanță definită prin compoziția sa cantitativă, în care un singur component principal este prezent în proporție de cel puțin 80 % (g/g).
Substanță multicomponentă
: O substanță definită prin compoziția sa cantitativă, în care mai mult de un singur component principal este prezent într-o concentrație ≥ 10 % (g/g) și < de 80 % (g/g). O substanță multicomponentă este rezultatul unui proces de fabricație. Diferența dintre un amestec și o substanță multicomponentă este faptul că un amestec este obținut prin combinarea a două sau mai multe substanțe chimice fără o reacție chimică. O substanță multicomponentă este rezultatul unei reacții chimice.
MTT
: 3-(4,5-dimetiltiazol-2-il)-2,5-bromură de difeniltetrazoliu; Bromură de tetrazoliu, albastru de tiazolil.
Martor negativ
: o probă ce conține toate componentele sistemului de testare și care a fost tratată cu o substanță cunoscută pentru faptul că nu provoacă un răspuns pozitiv în sistemul de testare. Această probă este prelucrată împreună cu probele tratate cu substanța chimică de testat și cu alte probe martor și este utilizată pentru a determina o viabilitate tisulară de 100 %.
Neclasificat
: Substanțe chimice care nu sunt clasificate pentru iritație oculară (categoria 2 din GHS al ONU/Regulamentul CLP, categoria 2A sau 2B din GHS al ONU) sau leziune oculară gravă (categoria 1 din GHS al ONU/Regulamentul CLP). Termen utilizat în paralel cu „neîncadrat într-o categorie din GHS al ONU/Regulamentul CLP”.
NSCkilled
: Culoare nespecifică la țesuturi moarte.
NSCliving
: Culoare nespecifică la țesuturi vii.
NSMTT
: Reducere nespecifică a MTT.
DO
: Densitate optică.
Standarde de performanță
: Standarde bazate pe o metodă de testare validată, care a fost considerată a fi valabilă din punct de vedere științific, care permit evaluarea comparabilității unei metode de testare propuse similare din punct de vedere al mecanismului și din punct de vedere funcțional. Se includ: (i) componentele esențiale ale metodei de testare; (ii) o listă minimală de substanțe chimice de referință selectate dintre substanțele chimice utilizate pentru a demonstra performanțele acceptabile ale metodei de referință validate; și (iii) nivelurile comparabile de precizie și fiabilitate, pe baza rezultatelor obținute în cazul metodei de testare validate, care ar trebui să fie demonstrate prin metoda de testare propusă la evaluarea acesteia pe baza listei minimale de substanțe chimice de referință (18).
Martor pozitiv
: o probă ce conține toate componentele sistemului de testare și care a fost tratată cu o substanță cunoscută pentru faptul că provoacă un răspuns pozitiv în sistemul de testare. Această probă este prelucrată împreună cu probele tratate cu substanța chimică de testat și cu alte probe martor. Pentru a asigura posibilitatea evaluării variabilității în timp a reacției martorului pozitiv, amplitudinea reacției severe nu ar trebui să fie excesiv de mare.
Relevanță
: Descrierea relației dintre test și efectul de interes și dacă aceasta este relevantă și utilă pentru un anumit scop. Aceasta reprezintă măsura în care testul măsoară sau prezice în mod corect efectul biologic de interes. Relevanța include luarea în considerare a acurateței (conformității) unei metode de testare (18).
Fiabilitate
: măsuri în care o metodă de testare poate fi reprodusă în mod reproductibil, în același laborator sau în laboratoare diferite în timp, atunci când sunt efectuate utilizând același protocol. Aceasta se evaluează prin calcularea reproductibilității în același laborator sau între laboratoare și a repetabilității în același laborator (18).
Test substitutiv
: Un test conceput să substituie un test utilizat în mod sistematic și acceptat pentru identificarea pericolelor și/sau evaluarea riscurilor și care a fost conceput pentru a asigura o protecție echivalentă sau îmbunătățită a sănătății umane sau a animalelor sau a mediului, după caz, comparativ cu testul acceptat pentru toate situațiile de testare posibile și toate substanțele chimice (18).
Reproductibilitate
: acordul dintre rezultatele obținute în urma testării repetate a aceleiași substanțe chimice de testat, utilizând același protocol de testare (a se vedea punctul „Fiabilitate”) (18).
Efecte reversibile asupra ochilor
: A se vedea „iritația oculară”.
RhCE
: epiteliu similar corneei umane reconstruit
Test
: un test este compus din unul sau mai multe substanțe chimice de testat, care sunt testate concomitent cu un martor negativ și un martor pozitiv.
SD
: abatere standard.
Sensibilitate
: Proporția tuturor substanțelor chimice pozitive/active de testat clasificate corect în urma testului. Acesta permite măsurarea acurateței unei metode de testare care conduce la rezultate clasificabile în categorii și constituie un aspect important în evaluarea relevanței unei metode de testare (18).
Leziune oculară gravă
: producerea de leziuni tisulare la nivel ocular sau deteriorarea fizică gravă a vederii în urma aplicării unei substanțe de testat pe suprafața anterioară a ochiului, care nu este complet reversibilă într-un interval de 21 de zile de la aplicare. Termen utilizat în paralel cu „efecte ireversibile asupra ochiului” și cu „categoria 1 din GHS al ONU”.
Proceduri standard de operare (PSO)
: proceduri formale scrise care descriu în detaliu modul în care ar trebui să se desfășoare fiecare operațiune de laborator de rutină și specifică testelor. Acestea sunt prevăzute în GLP.
Specificitate
: proporția tuturor substanțelor chimice de testat negative/inactive clasificate corect în urma testului. Acesta permite măsurarea acurateței unei metode de testare care conduce la rezultate clasificabile în categorii și constituie un aspect important în evaluarea relevanței unei metode de testare (18).
Substanță
: un element chimic și compușii săi în stare naturală sau obținuți prin orice proces de producție, inclusiv orice aditiv necesar pentru a-i păstra stabilitatea și orice impuritate care derivă din procesul utilizat, însă excluzând orice solvent care poate fi separat fără a afecta stabilitatea substanței sau fără a-i schimba compoziția.
Test
: o singură substanță chimică de testat testată concomitent pe minim două replici de țesut, astfel cum este definit în POS corespunzător.
Viabilitate tisulară
: parametru care măsoară activitatea totală a unei populații celulare dintr-un țesut reconstruit ca fiind capacitatea acestora de a reduce colorantul esențial MTT, care, în funcție de punctul final măsurat și structura testului utilizat, se corelează cu numărul total și/sau vitalitatea celulelor vii.
Abordare descendentă
: abordare pe etape utilizată pentru o substanță chimică susceptibilă de a provoca lezarea oculară gravă, care începe cu determinarea substanțelor chimice care provoacă lezarea oculară gravă (rezultat pozitiv) din alte substanțe chimice (rezultat negativ).
Substanța chimică de testat
: Orice substanță sau amestec testat utilizând prezenta metodă de testare.
Strategie de testare etapizată
: o strategie de testare pe etape, care utilizează metode de testare în mod secvențial. Toate informațiile existente cu privire la o substanță chimică de testat sunt analizate în fiecare etapă, utilizând un proces de identificare a dovezilor pentru a determina dacă sunt disponibile suficiente informații pentru o decizie privind clasificarea pericolelor înainte de a trece la etapa următoare a strategiei. În cazul în care potențialul/puterea de pericol a unei substanțe chimice de testat poate fi apreciat(ă) pe baza informațiilor existente într-o anumită etapă, nu este necesară o testare suplimentară (18).
ULOQ
: limita de cuantificare superioară.
Sistemul global armonizat de clasificare și etichetare a substanțelor chimice al Organizației Națiunilor Unite (GHS al ONU)
: Un sistem care propune clasificarea substanțelor chimice (substanțe și amestecuri) în funcție de tipuri și niveluri standardizate de pericole fizice, pericole pentru sănătate și pericole pentru mediu și care elaborează elemente de comunicare corespunzătoare, precum pictograme, cuvinte de avertizare, declarații cu privire la pericole, declarații de securitate și fișe tehnice de securitate pentru a transmite informații privind efectele adverse ale acestora în scopul protejării populației (inclusiv angajatorii, lucrătorii, transportatorii, consumatorii și cei care se ocupă de serviciile de urgență) și a mediului (1).
Categoria 1 din GHS al ONU și Regulamentul CLP
: A se vedea „Leziune oculară gravă”.
Categoria 2 din GHS al ONU și Regulamentul CLP
: A se vedea „iritația oculară”.
Neîncadrat în GHS al ONU și Regulamentul CLP
: substanțe chimice care nu îndeplinesc cerințele pentru a fi clasificate drept substanțe din categoria 1 sau 2 din GHS al ONU/Regulamentul CLP (sau din categoria 2A sau 2B din GHS al ONU). Termen utilizat în paralel cu „neclasificat”.
UPLC
: Cromatografie lichidă de performanță ultra-înaltă.
UVCB
: Substanțe cu compoziție necunoscută sau variabilă, produși de reacție complexă sau materiale biologice.
Metodă de testare valabilă
: o metodă de testare considerată a avea suficientă relevanță și fiabilitate pentru un scop specific și care se bazează pe principii solide din punct de vedere științific. o metodă de testare nu este niciodată valabilă într-un sens absolut, ci doar în legătură cu un scop definit (18).
Metodă de testare validată
: o metodă de testare pentru care au fost finalizate studii de validare pentru a determina relevanța (inclusiv precizia) și fiabilitatea pentru un anumit scop. Este important de precizat faptul că este posibil ca performanțele unei metode de testare validate să nu fie suficiente din punct de vedere al preciziei și fiabilității astfel încât aceasta să fie considerată acceptabilă pentru scopul propus (18).
VRM
: metodă de referință validată.
VRM1
: testul EpiOcular™ EIT este denumit metoda de referință validată 1.
VRM2
: testul SkinEthic™ EIT este denumit metoda de referință validată 2.
Identificarea dovezilor
: procesul prin care se analizează punctele tari și punctele slabe ale unor informații diverse pentru a formula și a susține o concluzie în ceea ce privește potențialul de pericol al unei substanțe chimice de testat.
Apendicele 2
COMPONENTELE PRINCIPALE ALE TESTELOR RHCE VALIDATE PENTRU IDENTIFICAREA SUBSTANȚELOR CHIMICE CARE NU NECESITĂ CLASIFICARE ȘI ETICHETARE PENTRU IRITAȚIA OCULARĂ SAU LEZIUNEA OCULARĂ GRAVĂ
|
Componentele testului
|
EpiOcular™ EIT
(VRM 1)
|
SkinEthic™ HCE EIT
(VRM 2)
|
|
Protocoale
|
Substanțe lichide
(pot fi introduse cu pipeta la 37 ± 1 °C sau la temperaturi mai mici timp de 15 minute)
|
Substanțe solide
(nu pot fi introduse cu pipeta)
|
Substanțe lichide și vâscoase
(pot fi introduse cu pipeta)
|
Substanțe solide
(nu pot fi introduse cu pipeta)
|
|
Suprafața modelelor
|
0,6 cm2
|
0,6 cm2
|
0,5 cm2
|
0,5 cm2
|
|
Numărul de țesuturi duplicate
|
Cel puțin două
|
Cel puțin două
|
Cel puțin două
|
Cel puțin două
|
|
Pre-verificareapentru interferența culorii
|
50 μl + 1 ml H2O timp de 60 de minute la 37 ± 2 oC, 5 ± 1 % CO2, ≥ 95 % RH (substanțe chimice de testat necolorate) sau o cantitate de 50 μl + 2 ml izopropanol amestecată timp de 2-3 ore la temperatura ambiantă (substanțe chimice de testat colorate)
|
 dacă DO a substanței chimice de testat la 570 ± 20 nm, după scăderea DO pentru izopropanol sau apă este > 0,08 (ceea ce corespunde unui procent de aproximativ 5 % din media DO a martorului negativ), ar trebui să fie obținuți martori vii adaptați. dacă DO a substanței chimice de testat la 570 ± 20 nm, după scăderea DO pentru izopropanol sau apă este > 0,08 (ceea ce corespunde unui procent de aproximativ 5 % din media DO a martorului negativ), ar trebui să fie obținuți martori vii adaptați.
|
|
50 mg + 1 ml H2O timp de 60 de minute la 37 ± 2 oC, 5 ± 1 % CO2, ≥ 95 % RH (substanțe chimice de testat necolorate)
și/sau
o cantitate de 50 mg + 2 ml izopropanol amestecată timp de 2-3 ore la temperatura ambiantă (substanțe chimice de testat colorate și necolorate)
|
 dacă DO a substanței chimice de testat la 570 ± 20 nm, după scăderea DO pentru izopropanol sau apă este > 0,08 (ceea ce corespunde unui procent de aproximativ 5 % din media DO a martorului negativ), ar trebui să fie obținuți martori vii adaptați. dacă DO a substanței chimice de testat la 570 ± 20 nm, după scăderea DO pentru izopropanol sau apă este > 0,08 (ceea ce corespunde unui procent de aproximativ 5 % din media DO a martorului negativ), ar trebui să fie obținuți martori vii adaptați.
|
|
o cantitate de 10 μl + 90 μl H2O amestecată timp de 30 ± 2 minute la temperatura camerei (TC, 18-28oC)
|
 în cazul în care substanța chimică de testat se colorează, ar trebui să se efectueze teste pe martori vii adaptați în cazul în care substanța chimică de testat se colorează, ar trebui să se efectueze teste pe martori vii adaptați
|
|
o cantitate de 10 mg + 90 μl H2O amestecată timp de 30 ± 2 minute la TC
|
 în cazul în care substanța chimică de testat se colorează, ar trebui să se efectueze teste pe martori vii adaptați în cazul în care substanța chimică de testat se colorează, ar trebui să se efectueze teste pe martori vii adaptați
|
|
|
Pre-verificare pentru reducția directă a MTT
|
o cantitate de 50 μl + 1 ml MTT 1 mg/ml soluție timp de 180 ± 15 minute
|
la 37 ± 2oC, 5 ± 1 % CO2, ≥ 95 % RH
 în cazul în care soluția capătă o culoare albastră/purpurie, ar trebui să fie efectueze teste pe martori adaptați eutanasiați prin congelare în cazul în care soluția capătă o culoare albastră/purpurie, ar trebui să fie efectueze teste pe martori adaptați eutanasiați prin congelare
|
|
(se utilizează 50 μl de apă deionizată sterilă în soluție de MTT ca martor negativ)
|
o cantitate de 50 μl + 1 ml MTT 1 mg/ml soluție timp de 180 ± 15 minute la 37 ± 2oC, 5 ± 1 % CO2, ≥ 95 % RH
 în cazul în care soluția capătă o culoare albastră/purpurie, ar trebui să fie efectueze teste pe martori adaptați eutanasiați prin congelare (se utilizează 50 μl de apă deionizată sterilă în soluție de MTT ca martor negativ) în cazul în care soluția capătă o culoare albastră/purpurie, ar trebui să fie efectueze teste pe martori adaptați eutanasiați prin congelare (se utilizează 50 μl de apă deionizată sterilă în soluție de MTT ca martor negativ)
|
|
30 μl + 300 μl MTT 1 mg/ml soluție pentru 180 ± 15 minute la 37 ± 2 oC, 5 ± 1 % CO2, ≥ 95 % RH
|
 în cazul în care soluția capătă o culoare albastră/purpurie, ar trebui să fie efectueze teste pe martori adaptați eutanasiați în apă în cazul în care soluția capătă o culoare albastră/purpurie, ar trebui să fie efectueze teste pe martori adaptați eutanasiați în apă
(se utilizează 30 μl de apă deionizată sterilă în soluție de MTT ca martor negativ)
|
|
30 mg + 300 μl MTT 1 mg/ml soluție pentru 180 ± 15 minute la 37 ± 2 oC, 5 ± 1 % CO2, ≥ 95 % RH
|
 în cazul în care soluția capătă o culoare albastră/purpurie, ar trebui să fie efectueze teste pe martori adaptați eutanasiați în apă în cazul în care soluția capătă o culoare albastră/purpurie, ar trebui să fie efectueze teste pe martori adaptați eutanasiați în apă
(se utilizează 30 μl de apă deionizată sterilă în soluție de MTT ca martor negativ)
|
|
|
Pre-tratament
|
20 μl DPBS fără Ca2+/Mg2+
timp de 30 ± 2 minute la 37 ± 2 oC, 5 ± 1 % CO2, ≥ 95 % RH, ferit de lumină.
|
20 μl DPBS fără Ca2+/Mg2+
timp de 30 ± 2 minute la 37 ± 2 oC, 5 ± 1 % CO2, ≥ 95 % RH, ferit de lumină.
|
—
|
—
|
|
Doze de tratament și aplicare
|
50 μl (83,3 μl/cm2)
|
50 mg (83,3 mg/cm2) cu ajutorul unui instrument calibrat (de exemplu, o nivelă cu lingură calibrată pentru a ține 50 mg de clorură de sodiu).
|
10 μl DPBS fără Ca2+/Mg2++ 30 ± 2 μl (60 μl/cm2)
Pentru substanțele vâscoase, utilizați o plasă din nailon
|
30 μl DPBS fără Ca2+/Mg2++ 30 ± 2 mg (60 mg/cm2)
|
|
Expunereincubației MTT
|
30 min (± 2 min)
În mediu de cultură
la 37 ± 2oC, 5 ± 1 % CO2, ≥ 95 % RH
|
6 ore (± 0,25 h)
În mediu de cultură
la 37 ± 2oC, 5 ± 1 % CO2, ≥ 95 % RH
|
30 min (± 2 min)
În mediu de cultură
la 37 ± 2oC, 5 ± 1 % CO2, ≥ 95 % RH
|
4 ore (± 0,1 h)
În mediu de cultură
la 37 ± 2oC, 5 ± 1 % CO2, ≥ 95 % RH
|
|
Spălare la temperatura camerei
|
de trei ori în 100 ml de DPBS fără Ca2+/Mg2+
|
de trei ori în 100 mlde DPBS fără Ca2+/Mg2+
|
20 ml DPBS fără Ca2+/Mg2+
|
25 ml DPBS fără Ca2+/Mg2+
|
|
Imersie post-expunere
|
12 min. (± 2 min.) la temperatura camerei în mediu de cultură
|
25 min. (± 2 min.) la temperatura camerei în mediu de cultură
|
30 min. (± 2 min.) la 37oC, 5 % CO2, 95 % RH în mediu de cultură
|
30 min. (± 2 min.) la temperatura camerei în mediu de cultură
|
|
Incubare post-expunere
|
120 min. (± 15 min.) în mediu de cultură la 37 ± 2 oC, 5 ± 1 % CO2, ≥ 95 % RH
|
18 min. (± 0,25 ore) în mediu de cultură la 37 ± 2 oC, 5 ± 1 % CO2, ≥ 95 % RH
|
nu există
|
18 min. (± 0,5 ore) în mediu de cultură la 37 ± 2 oC, 5 ± 1 % CO2, ≥ 95 % RH
|
|
Martor negativ
|
50 μl H2O
Testat în paralel
|
50 μl H2O
Testat în paralel
|
30 ± 2μl DPBS fără Ca2+/Mg2+
Testat în paralel
|
30 ± 2μl DPBS fără Ca2+/Mg2+
Testat în paralel
|
|
Martor pozitiv
|
50 μl acetat de metil
Testat în paralel
|
50 μl acetat de metil
Testat în paralel
|
30 ± 2μl acetat de metil
Testat în paralel
|
30 ± 2μl acetat de metil
Testat în paralel
|
|
Soluția MTT
|
300 μl 1 mg/ml
|
300 μl 1 mg/ml
|
300 μl 1 mg/ml
|
300 μl 1 mg/ml
|
|
incubației MTT Timpul și temperatura
|
180 min (± 15 min) la 37 ± 2 oC, 5 ± 1 % CO2, ≥ 95 % RH
|
180 min (± 15 min) la 37 ± 2 oC, 5 ± 1 % CO2, ≥ 95 % RH
|
180 min (± 15 min) la 37 ± 2 oC, 5 ± 1 % CO2, ≥ 95 % RH
|
180 min (± 15 min) la 37 ± 2 oC, 5 ± 1 % CO2, ≥ 95 % RH
|
|
Solvent de extracție
|
2 ml izopropanol
(extracție din partea superioară și inferioară a elementului inserat prin perforarea țesutului)
|
2 ml izopropanol
(extracție din partea inferioară a elementului inserat prin perforarea țesutului)
|
1,5 ml izopropanol
(extracție din partea superioară și inferioară a elementului inserat)
|
1,5 ml izopropanol
(extracție din partea inferioară a elementului inserat)
|
|
Timpul și temperatura de extracție
|
2-3 ore cu agitare (~ 120 rpm) la temperatura camerei sau peste noapte la 4-10 °C
|
2-3 ore cu agitare (~ 120 rpm) la temperatura camerei sau peste noapte la 4-10 °C
|
4 ore cu agitare (~ 120 rpm) la temperatura camerei sau cel puțin peste noapte fără agitare la 4-10 °C
|
Cel puțin 2 ore cu agitare (~ 120 rpm) la temperatura camerei
|
|
Citirea DO
|
570 nm (550 - 590 nm)
fără filtru de referință
|
570 nm (550-590 nm)
fără filtru de referință
|
570 nm (540 - 600 nm)
fără filtru de referință
|
570 nm (540 - 600 nm)
fără filtru de referință
|
|
Controlul calității țesutului
|
Tratament cu 100 μl de soluție 0,3 % (v/v) Triton X-100)
12,2 min ≤ ET50 ≤ 37,5 min
|
Tratament cu 100 μl de soluție 0,3 % (v/v) Triton X-100)
12,2 min ≤ ET50 ≤ 37,5 min
|
Tratament de 30 de min. cu SDS (50 μl)
1,0 mg/ml ≤ IC50 ≤ 3,5 mg/ml
|
Tratament de 30 de min. cu SDS (50 μl)
1,0 mg/ml ≤ IC50 ≤ 3,2 mg/ml
|
|
Criterii de acceptare
|
|
1.
|
DO medie a replicilor de țesut tratate cu martorul negativ ar trebui să fie > 0,8 și < 2,5
|
|
2.
|
Viabilitatea medie a țesuturilor duplicate expuse timp de 30 de minute cu martorul pozitiv, exprimată ca % din martorul negativ, ar trebui să fie < 50 %
|
|
3.
|
Diferența de viabilitate dintre două replici de țesut ar trebui să fie mai mică de 20 %.
|
|
|
1.
|
DO medie a replicilor de țesut tratate cu martorul negativ ar trebui să fie > 0,8 și < 2,5
|
|
2.
|
Viabilitatea medie a replicilor de țesut expuse timp de 6 ore cu martorul pozitiv, exprimată ca % din martorul negativ, ar trebui să fie < 50 %
|
|
3.
|
Diferența de viabilitate dintre două replici de țesut ar trebui să fie mai mică de 20 %.
|
|
|
1.
|
DO medie a replicilor de țesut tratate cu martorul negativ ar trebui să fie > 1,0 și ≤ 2,5
|
|
2.
|
Viabilitatea medie a replicilor de țesut expuse timp de 30 de minute cu martorul pozitiv, exprimată ca % din martorul negativ, ar trebui să fie ≤ 30 %
|
|
3.
|
Diferența de viabilitate dintre două replici de țesut ar trebui să fie mai mică de 20 %.
|
|
|
1.
|
DO medie a replicilor de țesut tratate cu martorul negativ ar trebui să fie > 1,0 și ≤ 2,5
|
|
2.
|
Viabilitatea medie a replicilor de țesut expuse timp de 4 ore cu martorul pozitiv, exprimată ca % din martorul negativ, ar trebui să fie ≤ 20 %
|
|
3.
|
Diferența de viabilitate dintre două replici de țesut ar trebui să fie mai mică de 20 %.
|
|
Apendicele 3
DIAGRAMĂ ILUSTRATIVĂ CARE OFERĂ ÎNDRUMĂRI PRIVIND MODUL DE IDENTIFICARE ȘI TRATARE A REDUCTORILOR MTT DIRECȚI ȘI/SAU A SUBSTANȚELOR CHIMICE CARE INTERFEREAZĂ PRIN CULOARE, PE BAZA PSO AFERENTE VRM1
Apendicele 4
DIAGRAMĂ ILUSTRATIVĂ CARE OFERĂ ÎNDRUMĂRI PRIVIND MODUL DE IDENTIFICARE ȘI TRATARE A REDUCTORILOR MTT DIRECțI ȘI/SAU A SUBSTANțELOR CHIMICE CARE INTERFEREAZĂ PRIN CULOARE, PE BAZA PSO AFERENTE VRM2

Apendicele 5
PARAMETRII-CHEIE ȘI CRITERIILE DE ACCEPTABILITATE PENTRU CALIFICAREA UNUI SISTEM DE SPECTROFOTOMETRIE HPLC/UPLC PENTRU MĂSURAREA FORMAZANULUI MTT EXTRAS DIN STRUCTURILE DE ȚESUT RHCE
|
Parametru
|
Protocol derivat din Orientarea FDA (36) (38)
|
Criterii de acceptare
|
|
Selectivitate
|
Analiza izopropanolului, a soluției sărace vii (extras de izopropanol din structuri de țesut RhCE vii fără tratament), a soluției sărace moarte (extras de izopropanol din structurile de țesut RhCE moarte fără tratament) și a unui colorant (de exemplu, albastru de metilen)
|
Zonăinterferență ≤ 20 % din ZonaLLOQ
(88)
|
|
Precizie
|
Controale ale calității (și anume, formazan MTT la 1,6 μg/ml, 16 μg/ml și 160 μg/ml) în izopropanol (n = 5)
|
CV ≤ 15 % sau ≤ 20 % pentru LLOQ
|
|
Precizie
|
Controlul calității la izopropanol (n = 5)
|
% Dev ≤ 15 % sau ≤ 20 % pentru LLOQ
|
|
Efectul de matrice
|
Controlul calității la proba vie (n = 5)
|
85 % ≤ % efect de matrice ≤ 115 %
|
|
Reportare
|
Analiza izopropanolului conform unui standard ULOQ (89)
|
Zonainterferență ≤ 20 % din ZonaLLOQ
|
|
Reproductibilitate (în aceeași zi)
|
3 curbe de calibrare independente (pe baza a 6 diluții 1/3 consecutive de formazan MTT în izopropanol începând de la valoarea ULOQ, și anume, 200 μg/ml);
Controlul calității la izopropanol (n = 5)
|
Curbe de calibrare: % Dev ≤ 15 % sau ≤ 20 % pentru LLOQ
Controale ale calității: % Dev ≤ 15 % și CV ≤ 15 %
|
|
Reproductibilitate (între zile diferite)
|
Ziua 1: 1 curbă de calibrare și controalele ale calității în izopropanol (n = 3)
Ziua 2: 1 curbă de calibrare și controalele ale calității în izopropanol (n = 3)
Ziua 3: 1 curbă de calibrare și controalele ale calității în izopropanol (n = 3)
|
|
Stabilitate pe termen scurt a formazanului MTT în extractul de țesut RhCE
|
Controalele calității pe proba vie (n = 3) sunt analizate în ziua preparării și după 24 de ore de la stocare la temperatura camerei
|
% Dev ≤ 15 %
|
|
Stabilitate pe termen lung a formazanului MTT în extractul de țesut RhCE, dacă este necesar
|
Controalele calității pe proba vie (n = 3) sunt analizate în ziua preparării și după mai multe zile de stocare la -20°C
|
% Dev ≤ 15 %
|
B.70 ANALIZE IN VITRO CU RECEPTOR DE ESTROGEN RECOMBINANT UMAN (hrER) PENTRU DETECTAREA SUBSTANȚELOR CHIMICE CU AFINITATE DE LEGARE DE ER
INTRODUCERE GENERALĂ
Orientarea OCDE privind testarea pe baza performanței
|
1.
|
Această metodă de testare este echivalentă cu Orientarea OCDE privind testarea (TG) nr. 493 (2015). TG nr. 493 este o orientare privind testarea bazată pe performanță (PBTG), în care este descrisă metodologia pentru analizele in vitro asupra recombinantului uman pentru detectarea substanțelor cu afinitate de legare de receptorul de estrogen (analize de legare hrER). Aceasta cuprinde două analize similare din punct de vedere mecanicist și funcțional pentru identificarea agenților de legare de receptorii de estrogen (și anume, ERα) și ar trebui să faciliteze dezvoltarea unor analize similare noi sau modificate în conformitate cu principiile de validare prevăzute în documentul de orientare al OCDE privind validarea și acceptarea internațională a metode de testare noi și actualizate pentru evaluarea riscurilor (1). Metodele de testare de referință validate integral (apendicele 2 și 3), care servesc drept bază pentru această PBTG, sunt:
|
—
|
Analiza legării receptorului de estrogen (ER) in vitro Freyberger-Wilson (FW) care utilizează un recombinant uman de lungime completă ERα (2) și
|
|
—
|
analiza legării receptorului de estrogen in vitro a Institutului de Evaluare și Cercetare în Chimie (Chemical Evaluation and Research Institute - CERI) care utilizează o proteină din domeniul de legare prin ligand recombinant uman (2).
|
Există standarde de performanță (SP) (3) pentru a facilita elaborarea și validarea unor metode de testare similare pentru același punct final de pericol și pentru a permite modificarea oportună a PBTG nr. 493 pentru a se putea adăuga analize similare unei PBTG actualizate. Însă analizele de testare similare vor fi adăugate doar după revizuirea și confirmarea de către OCDE a respectării standardelor de performanță. Testele incluse în TG nr. 493 pot fi utilizate fără nicio deosebire pentru a răspunde cerințelor statelor membre ale OCDE pentru rezultatele testelor privind legarea de receptori de estrogen, beneficiind în același timp de sistemul de acceptare reciprocă a datelor al OCDE.
|
Context și principii ale testelor incluse prezenta metodă de testare
|
2.
|
În 1998, OCDE a inițiat o activitate cu un nivel ridicat de prioritate pentru a revizui orientările existente privind testarea și a elabora noi orientări pentru depistarea și testarea substanțelor chimice cu potențial de perturbare a sistemului endocrin. Cadrul conceptual al OCDE (CC) pentru testarea și evaluarea substanțelor chimice cu potențial de perturbare a sistemului endocrin a fost revizuit în 2012. Cadrele conceptuale inițiale și revizuite sunt incluse ca anexe la documentul de orientare al privind orientările de testare standardizate pentru evaluarea substanțelor chimice în vederea identificării perturbatorilor endocrini (4). CC cuprinde cinci niveluri, fiecare nivel corespunzând unui nivel diferit de complexitate biologică. Testele de legare de ER descrise în această metodă de testare sunt de nivelul 2, care include „teste in vitro care furnizează date cu privire la (un) anumit(e) mecanism(e)/o cale endocrină/anumite căi endocrine.” Această metodă de testare este utilizată pentru analizele privind legarea de receptor in vitro, prevăzute a identifica liganzii pentru receptorul de estrogen uman alfa (ERα).
|
|
3.
|
A fost demonstrată în mod clar relevanța analizei de legare de ER in vitro pentru funcțiile biologice. Analizele de legare de ER sunt concepute pentru a identifica substanțele chimice care au potențialul de a perturba calea hormonilor estrogeni și au fost utilizate pe scară largă în ultimele două decenii pentru a caracteriza distribuția țesuturilor ER, precum și pentru a identifica agoniști/antagoniști ai ER. Aceste analize reflectă interacțiunea dintre receptori și liganzi care este treapta inițială a căii de semnalizare estrogenică și esențială pentru funcția de reproducere la toate vertebratele.
|
|
4.
|
Interacțiunea estrogenilor cu ER poate să afecteze transcrierea genelor controlate de estrogen și să inducă efecte non-genomice, ceea ce poate conduce la inducerea sau inhibarea proceselor celulare, inclusiv a celor necesare pentru proliferare celulară, dezvoltare normală a fătului și funcția de reproducere (5) (6) (7). Perturbarea sistemelor estrogenice normale poate avea potențialul de a declanșa efecte adverse asupra dezvoltării normale (ontogeneză), a sănătății reproductive și a integrității sistemului de reproducere. Semnalizarea ER inadecvată poate conduce la efecte cum ar fi un risc crescut de cancer hormono-dependent, afectarea fertilității și modificări ale creșterii și dezvoltării fetale (8).
|
|
5.
|
Analizele in vitro de legare se bazează pe o interacțiune directă a unei substanțe cu un anumit loc de legare a receptorului de ligand care reglează transcrierea genei. Componenta esențială a analizei de legare de receptorul de estrogen recombinant uman alpha (hrERα) măsoară capacitatea unui ligand marcat radioactiv ([3H]17-β-estradiol) de a se lega de ER în prezența concentrațiilor din ce în ce mai mari ale unei substanțe chimice de testat (și anume, concurent). Substanțele chimice de testat care prezintă o afinitate mare pentru ER concurează cu ligandul marcat radioactiv la o concentrație mai mică în comparație cu substanțele chimice care prezintă o afinitate mai redusă pentru receptor. Această analiză cuprinde două componente principale: un experiment de legare prin saturație pentru a caracteriza parametrii de interacțiune receptor-ligand și pentru documentarea specificității ER, urmat de un experiment de legare competitiv care caracterizează concurența dintre o substanță chimică de testat și un ligand marcat radioactiv pentru legarea de ER.
|
|
6.
|
Studiile de validare a analizelor de legare CERI și FW și-au demonstrat relevanța și fiabilitatea acestora pentru scopul prevăzut (2).
|
|
7.
|
Definițiile și abrevierile utilizate în prezenta metodă de testare sunt descrise în apendicele 1.
|
Domeniul de aplicare și limitările aferente analizelor de legare de receptori
|
8.
|
Aceste teste sunt propuse pentru depistare și stabilirea priorităților, dar pot furniza, de asemenea, informații pentru un eveniment de inițiere moleculară (MIE) care poate fi utilizat în cadrul unei abordări bazate pe dovezi. Acestea abordează legarea chimică de domeniul de legare a ligandului ERα în cadrul unui sistem in vitro. Astfel, rezultatele nu ar trebui să fie extrapolate în mod direct cu semnalizarea și reglarea complexă a sistemului endocrin in vivo.
|
|
9.
|
Legarea ligandului natural, 17β-estradiol, reprezintă prima etapă a unei serii de evenimente moleculare care activează transcrierea genelor țintă și, în cele din urmă, culminează cu o modificare fiziologică (9). Astfel legarea de domeniul de legare a ligandului de ERα este considerată drept unul dintre principalele mecanisme ale perturbării endocrine mediate de ER (ED), deși există și alte mecanisme prin care pot apărea ED, inclusiv interacțiunile cu (i) locuri ale ERα, altele decât cavitatea de legare a liganzilor, (ii) interacțiuni cu alți receptorii relevanți pentru semnalizarea estrogenului, receptorul de estrogen cuplat cu proteina G și ERβ, alți receptori și sisteme enzimatice din sistemul endocrin, (iii) sinteza hormonilor, (iv) activarea metabolică și/sau inactivarea hormonilor, (v) distribuția hormonilor către țesuturile țintă și (vi) eliminarea hormonilor din organism. Niciuna dintre analizele efectuate în cadrul acestei metode de testare nu abordează aceste moduri de acțiune.
|
|
10.
|
Această metodă de testare abordează capacitatea substanțelor de a se lega de ERα uman și nu face distincție între agoniști sau antagoniști ai ERα. Aceste analize nu abordează nici alte evenimente din aval precum transcrierea genelor, nici modificările fiziologice. Având în vedere că, în timpul validării, au fost utilizate numai substanțe monocomponente unice, aplicabilitatea pentru amestecuri de testare nu a fost abordată. Cu toate acestea, analizele sunt aplicabile teoretic pentru testarea substanțelor și amestecurilor multicomponente. Înainte de utilizarea metodei de testare pe un amestec pentru a genera date pentru un obiectiv de reglementare prevăzut, se ia în considerare dacă și, în caz afirmativ, din ce cauză aceasta poate oferi rezultate adecvate pentru respectivul scop. Astfel de considerații nu sunt necesare atunci când există o cerință normativă pentru testarea amestecului.
|
|
11.
|
Sistemele cu receptor fără celule nu au o capacitate metabolică intrinsecă și nu au fost validate în combinație cu sisteme enzimatice metabolice. Cu toate acestea, ar putea fi posibil să se integreze activitatea metabolică într-un proiect de studiu, dar acest lucru ar necesita eforturi suplimentare de validare.
|
|
12.
|
Substanțele chimice care pot denatura proteina (și anume, proteina receptoare), cum ar fi surfactantul sau substanțele chimice care pot schimba pH-ul soluției tampon a analizei, nu pot fi testate sau pot fi testate doar în concentrații lipsite de astfel de interacțiuni. În caz contrar, intervalul de concentrație care poate fi testat în cadrul analizelor pentru o substanță chimică de testat este limitat de solubilitatea acesteia în soluția tampon a analizei.
|
|
13.
|
În scop informativ, tabelul 1 prezintă rezultatele testului pentru cele 24 de substanțe care au fost testate în cadrul ambelor analize validate complet descrise în prezenta metodă de testare. Dintre aceste substanțe, 17 sunt clasificate ca fiind lianți de ER și 6 ca fiind non-lianți pe baza rapoartelor publicate, inclusiv a analizelor in vitro pentru activarea transcripțională a ER și sau a analizei uterotropice (9) (10) (11) (12) (13) (14) (15). În ceea ce privește datele sintetizate în tabelul 1, s-a ajuns la un acord de aproape 100 % între cele două analize privind clasificările tuturor substanțelor, până la 10-4M, și fiecare substanță a fost clasificată în mod corect ca fiind liant/non-liant de ER. Informații suplimentare privind acest grup de substanțe, precum și alte substanțe testate în cadrul analizelor de legare de ER în timpul studiilor de validare sunt prezentate în standardele de performanță pentru hrER (3), apendicele 2 (tabelele 1, 2 și 3).
Tabelul 1
Clasificarea substanțelor ca lianți sau non-lianți de ER la testarea în cadrul analizelor FW și CERI hrER, comparativ cu răspunsul preconizat
|
|
Denumirea substanței
|
NR. CAS
|
Răspuns preconizat
|
Analiza FW
|
Analiza CERI
|
Substanță chimică MESH Clasă
|
Clasa de produse
|
|
|
Interval de concentrații (M)
|
Clasificare
|
Interval de concentrații (M)
|
Clasificare
|
|
1
|
17β-Estradiol
|
50-28-2
|
Liant
|
1 × 10-11-1 × 10-6
|
Liant
|
1 × 10-11-1 × 10-6
|
Liant
|
Steroid
|
Produs farmaceutic, agent veterinar
|
|
2
|
Noretinodrel
|
68-23-5
|
Liant
|
3 × 10-9-30 × 10-4
|
Liant
|
3 × 10-9-30 × 10-4
|
Liant
|
Steroid
|
Produs farmaceutic, agent veterinar
|
|
3
|
Noretindronă
|
68-22-4
|
Liant
|
3 × 10-9-30 × 10-4
|
Liant
|
3 × 10-9-30 × 10-4
|
Liant
|
Steroid
|
Produs farmaceutic, agent veterinar
|
|
4
|
Ftalat de di-n-butil
|
84-74-2
|
Non-liant (*5)
|
1 × 10-10-1 × 10-4
|
Non-liant (*6)
(1)
|
1 × 10-10-1 × 10-4
|
Non-liant (*6)
(1)
|
Hidrocarbură (ciclică), ester
|
Plastifiant, substanță chimică intermediară
|
|
5
|
DES
|
56-53-1
|
Liant
|
1 × 10-10-1 × 10-3
|
Liant
|
1 × 10-10-1 × 10-3
|
Liant
|
Hidrocarbură (ciclică), fenol
|
Produs farmaceutic, agent veterinar
|
|
6
|
17α-etinilestradiol
|
57-63-6
|
Liant
|
1 × 10-10-1 × 10-3
|
Liant
|
1 × 10-10-1 × 10-3
|
Liant
|
Steroid
|
Produs farmaceutic, agent veterinar
|
|
7
|
Mezo-hexestrol
|
84-16-2
|
Liant
|
1 × 10-10-1 × 10-3
|
Liant
|
1 × 10-10-1 × 10-3
|
Liant
|
Hidrocarbură (ciclică), fenol
|
Produs farmaceutic, agent veterinar
|
|
8
|
Genisteină
|
446-72-0
|
Liant
|
1 × 10-10-1 × 10-3
|
Liant
|
1 × 10-10-1 × 10-3
|
Liant
|
Hidrocarbură (heterociclică), flavonoid
|
Produs natural
|
|
9
|
Ecvol
|
531-95-3
|
Liant
|
1 × 10-10-1 × 10-3
|
Liant
|
1 × 10-10-1 × 10-3
|
Liant
|
Metabolit fitoestrogen
|
Produs natural
|
|
10
|
Butilparaben (n 4-hidroxibenzoat de butil)
|
94-26-8
|
Liant
|
1 × 10-10-1 × 10-3
|
Liant
|
1 × 10-10-1 × 10-3
|
Liant
|
Paraben
|
Conservant
|
|
11
|
Nonilfenol (amestec)
|
84852-15-3
|
Liant
|
1 × 10-10-1 × 10-3
|
Liant
|
1 × 10-10-1 × 10-3
|
Liant
|
Alchilfenol
|
Compus intermediar
|
|
12
|
o,p’-DDT
|
789-02-6
|
Liant
|
1 × 10-10-1 × 10-3
|
Liant
|
1 × 10-10-1 × 10-3
|
Liant
|
Organoclorură
|
Insecticid
|
|
13
|
Corticosteron
|
50-22-6
|
Non-liant (*5)
|
1 × 10-10-1 × 10-4
|
Non-liant
|
1 × 10-10-1 × 10-4
|
Non-liant
|
Steroid
|
Produs natural
|
|
14
|
Zearalenonă
|
17924-92-4
|
Liant
|
1 × 10-10-1 × 10-3
|
Liant
|
1 × 10-10-1 × 10-3
|
Liant
|
Hidrocarbură (heterociclică), lactonă
|
Produs natural
|
|
15
|
Tamoxifen
|
10540-29-1
|
Liant
|
1 × 10-10-1 × 10-3
|
Liant
|
1 × 10-10-1 × 10-3
|
Liant
|
Hidrocarbură, (ciclică)
|
Produs farmaceutic, agent veterinar
|
|
16
|
5α-dihidrotestosteron
|
521-18-6
|
Liant
|
1 × 10-10-1 × 10-3
|
Liant
|
1 × 10-10-1 × 10-3
|
Liant
|
Steroid, nonfenolic
|
Produs natural
|
|
17
|
Bisfenol A
|
80-05-7
|
Liant
|
1 × 10-10-1 × 10-3
|
Liant
|
1 × 10-10-1 × 10-3
|
Liant
|
Fenol
|
Substanță chimică intermediară
|
|
18
|
4-n-heptilfenol
|
1987-50-4
|
Liant
|
1 × 10-10-1 × 10-3
|
Echivoc (5)
|
1 × 10-10-1 × 10-3
|
Liant
|
Alchilfenol
|
Intermediar
|
|
19
|
Kepone (clordeconă)
|
143-50-0
|
Liant
|
1 × 10-10-1 × 10-3
|
Liant
|
1 × 10-10-1 × 10-3
|
Liant
|
Hidrocarbură (halogenat)
|
Pesticid
|
|
20
|
Benzo(a)antracen
|
56-55-3
|
Non-liant
|
1 × 10-10-1 × 10-3
|
Non-liant (6)
|
1 × 10-10-1 × 10-3
|
Non-liant (6)
|
Hidrocarbură aromatică
|
Intermediar
|
|
21
|
Enterolactonă
|
78473-71-9
|
Liant
|
1 × 10-10-1 × 10-3
|
Liant
|
1 × 10-10-1 × 10-3
|
Liant
|
Fitoestrogen
|
Produs natural
|
|
22
|
Progesteron
|
57-83-0
|
Non-liant (*5)
|
1 × 10-10-1 × 10-4
|
Non-liant
|
1 × 10-10-1 × 10-4
|
Non-liant
|
Steroid
|
Produs natural
|
|
23
|
Octiltrietoxisilan
|
2943-75-1
|
Non-liant
|
1 × 10-10-1 × 10-3
|
Non-liant
|
1 × 10-10-1 × 10-3
|
Non-liant
|
Silan
|
Modificator de suprafață
|
|
24
|
Atrazină
|
1912-24-9
|
Non-liant (*5)
|
1 × 10-10-1 × 10-4
|
Non-liant
|
1 × 10-10-1 × 10-4
|
Non-liant
|
Compus heterociclic
|
Erbicid
|
|
COMPONENTELE ANALIZEI DE LEGARE DE hrER
Componentele esențiale ale testului
|
14.
|
Această metodă de testare se aplică pentru analizele care utilizează un receptor ER și un ligand suficient de puternic de receptor, care poate fi utilizat ca marker/detector pentru analiză și poate fi desprins la concentrații în creștere ale unei substanțe chimice de testat. Analizele de legare conțin următoarele două componente principale: 1) legarea prin saturație și 2) legarea competitivă. Analiza legării prin saturație este utilizată pentru a confirma specificitatea și activitatea preparatelor cu receptor, în timp ce experimentul de legare competitivă este utilizat pentru a evalua capacitatea unei substanțe chimice de testat de a se lega de hrER.
|
Martori
|
15.
|
Ar trebui să se descrie baza pentru estrogenul de referință propus a fi testat în paralel și martorii. Martorii testați în paralel [tratați cu solvent (vehicul), pozitivi (liant de ER; afinitate puternică și slabă), negativi (non-liant)], după caz, servesc drept indicatori pentru operativitatea testului în condițiile de testare și furnizează o bază pentru comparații între experimente; acestea fac parte, de obicei, din criteriile de acceptabilitate pentru un anumit experiment) (1). Pe fiecare placă, în timpul fiecărei testări, ar trebui să se utilizeze curbe complete de concentrații pentru estrogenul de referință și martori (și anume, liant slab și non-liant). Toate celelalte plăci ar trebui să conțină: 1) o concentrație ridicată (aproximativ desfacerea totală a ligandului marcat radioactiv) și o concentrație medie (aproximativ IC50) fiecare de liant E2 și slab în trei replici; 2) martor tratat cu solvent și legare nespecifică, fiecare în trei replici.
|
Proceduri standard de control al calității
|
16.
|
Ar trebui să fie desfășurate proceduri standard de control al calității, astfel cum sunt descrise pentru fiecare analiză pentru a asigura faptul că receptorii activi, concentrațiile chimice corecte și limitele de toleranță rămân stabile pe parcursul unor replici multiple și că își păstrează capacitatea de a asigura răspunsurile de legare de ER preconizate în timp.
|
Demonstrarea competenței laboratorului
|
17.
|
Înainte de a testa substanțele chimice necunoscute prin oricare dintre analizele din cadrul acestei metode de testare, fiecare laborator ar trebui să își demonstreze competența în utilizarea analizei prin efectuarea de analize de saturație pentru a confirma specificitatea și activitatea preparatului cu ER, precum și analize de legare competitivă cu estrogenul de referință și martori (liant slab și non-liant). Laboratorul ar trebui să dezvolte o bază de date istorice cu rezultate pentru estrogenul de referință și martori, care au fost generate în urma a 3-5 experimente independente desfășurate în zile diferite. Aceste experimente vor constitui baza pentru estrogenul de referință și martorii testați anterior pentru laborator și vor fi utilizate ca evaluare parțială a acceptabilității analizei în testele viitoare.
|
|
18.
|
Capacitatea de răspuns a sistemului de testare va fi, de asemenea, confirmată prin testarea substanțelor de verificare enumerate în tabelul 2. Lista substanțelor de verificare constituie un subset al substanțelor de referință prevăzute în standardele de performanță pentru analizele de legare de ER (3). Aceste substanțe sunt disponibile în comerț, reprezintă clasele de substanțe chimice asociate în mod obișnuit cu activitatea de legare de ER, prezintă un interval adecvat de putere preconizată pentru lianții de ER (și anume, puternice sau slabe) și non-lianți (și anume, negative). Pentru fiecare substanță de verificare, concentrațiile testate ar trebui să acopere intervalul indicat în tabelul 2. Ar trebui să fie efectuate cel puțin trei experimente pentru fiecare substanță, iar rezultatele ar trebui să fie în concordanță cu activitatea chimică preconizată. Fiecare experiment ar trebui să fie desfășurat în mod independent (și anume, cu diluții proaspete de receptor, substanțe chimice și reactiv), cu câte trei replici pentru fiecare concentrație. Performanța este demonstrată prin clasificarea corectă (pozitiv/negativ) a fiecărei substanțe de verificare. Competența ar trebui să fie testată de către fiecare tehnician atunci când învață analizele.
Tabelul 2
Lista martorilor și a substanțelor de verificare pentru analizele de legare competitivă de hrER
(90)
|
Nr.
|
Denumirea substanței
|
Nr. CAS (91)
|
Răspuns preconizat (92)
(93)
|
Interval de concentrații de testare (M)
|
Clasa de substanțe chimice MeSH (94)
|
Clasă de produse (95)
|
|
Martori (estrogen de referință, liant slab, non-liant)
|
|
1
|
17-εστραδιολ
|
50-28-2
|
Liant
|
1 × 10-11-1 × 10-6
|
Steroid
|
Farmaceutic, agent veterinar
|
|
2
|
Noretinodrel (sau) Noretindronă
|
68-23-5 (sau) 68-22-4
|
Liant
|
3 × 10-9-30 × 10-6
|
Steroid
|
Farmaceutic, agent veterinar
|
|
3
|
Octiltrietoxisilan
|
2943-75-1
|
Non-liant
|
1 × 10-10-1 × 10-3
|
Silan
|
Modificator de suprafață
|
|
Substanțe de verificare (95)
|
|
4
|
Dietilstilbestrol
|
56-53-1
|
Liant
|
1 × 10-11-1 × 10-6
|
Hidrocarbură (ciclică), fenol
|
Farmaceutic, agent veterinar
|
|
5
|
17α-etinilestradiol
|
57-63-6
|
Liant
|
1 × 10-11-1 × 10-6
|
Steroid
|
Farmaceutic, agent veterinar
|
|
6
|
Mezo-hexestrol
|
84-16-2
|
Liant
|
1 × 10-11-1 × 10-6
|
Hidrocarbură (ciclică), fenol
|
Farmaceutic, agent veterinar
|
|
7
|
Tamoxifen
|
10540-29-1
|
Liant
|
1 × 10-11-1 × 10-6
|
Hidrocarbură (ciclică)
|
Farmaceutic, agent veterinar
|
|
8
|
Genisteină
|
446-72-0
|
Liant
|
1 × 10-10-1 × 10-3
|
Compus heterociclic, flavonoid,
|
Produs natural
|
|
9
|
Bisfenol A
|
80-05-7
|
Liant
|
1 × 10-10-1 × 10-3
|
Fenol
|
Substanță chimică intermediară
|
|
10
|
Zaralonionă
|
17924-92-4
|
Liant
|
1 × 10-11-1 × 10-3
|
Compus heterociclic, lactonă
|
Produs natural
|
|
11
|
Butilparaben
|
94-26-8
|
Liant
|
1 × 10-11-1 × 10-3
|
Acid carboxilic, fenol
|
Conservant
|
|
12
|
Atrazină
|
1912-24-9
|
Non-liant
|
1 × 10-11-1 × 10-6
|
Compus heterociclic
|
Erbicid
|
|
13
|
Ftalat de di-n-butil (DBP) (96)
|
84-74-2
|
Non-liant (97)
|
1 × 10-10-1 × 10-4
|
Hidrocarbură (ciclică), ester
|
Plastifiant, substanță chimică intermediară
|
|
14
|
Corticosteron
|
50-22-6
|
Non-liant
|
1 × 10-11-1 × 10-4
|
Steroid
|
Produs natural
|
|
Teste de solubilitate și identificarea intervalului de concentrații pentru substanțele chimice de testat
|
19.
|
Ar trebui efectuat un test preliminar pentru a determina limita de solubilitate pentru fiecare substanță chimică de testat și pentru a identifica intervalul de concentrații adecvat care să fie utilizat la efectuarea testului. Limita de solubilitate a fiecărei substanțe chimice de testat va fi determinată inițial în solvent și confirmată ulterior în condiții de testare. Concentrația finală testată în cadrul analizei nu ar trebui să depășească 1 mM. Testul pentru identificarea intervalului cuprinde un martor tratat cu solvent și diluții în serie de 8 log, pornind de la concentrația maximă acceptabilă (de exemplu, cel mult 1 mM, în funcție de limita de solubilitate), și se consemnează gradul de tulburare sau precipitatul. Concentrațiile din cel de-al doilea și cel de-al treilea experiment ar trebui să fie ajustate după caz pentru o mai bună caracterizare a curbei concentrație-răspuns.
|
Criteriile de acceptabilitate a unui test
|
20.
|
Acceptarea sau respingerea unui test se bazează pe evaluarea rezultatelor obținute pentru estrogenul și martorul de referință utilizați pentru fiecare experiment. În primul rând, pentru placa 1, curbele complete de concentrații pentru martorii de referință, în cadrul fiecărui experiment, ar trebui să fie în concordanță cu valorile de măsurare ale performanței cu parametrii de formare a curbei (de exemplu, IC50 și Hillslope) pe baza rezultatelor raportate pentru protocoalele respective pentru analizele CERI și FW (apendicele 2 și 3) și a datelor privind martorii testați anterior din laboratorul care efectuează testul. Toți martorii (estrogenul de referință, liantul slab și non-liantul) ar trebui să fie clasificați în mod corect pentru fiecare experiment. În al doilea rând, martorii de pe toate plăcile ulterioare trebuie să fie evaluați pentru asigurarea consecvenței cu placa 1. Ar trebui să se utilizeze un interval suficient de concentrații ale substanței chimice de testat pentru a defini cu claritate partea superioară a curbei de legare competitivă. Variabilitatea între replici la fiecare concentrație a substanței chimice de testat, precum și între cele trei teste independente ar trebui să fie rezonabilă și justificată din punct de vedere științific. Capacitatea de a efectua în mod consecvent testul ar trebui să fie demonstrată prin dezvoltarea și întreținerea unei baze de date istorice pentru estrogenul și martorii de referință. Ca o măsură a reproductibilității în cadrul laboratorului, pot fi utilizate abateri standard (SD) sau coeficienți de variație (CV) pentru mediile de pe curbele estrogenului și martorului de referință ca lianți slabi care corelează parametrii din cadrul mai multor experimente. La analizarea rezultatelor privind martorii de pe placă din cadrul fiecărei testări, precum și pentru fiecare substanță chimică de testat, ar trebui să se aplice o apreciere profesională.
În plus, ar trebui să fie îndeplinite următoarele principii referitoare la criteriile de acceptabilitate:
|
—
|
Datele ar trebui să fie suficiente pentru evaluarea cantitativă a legării de ER.
|
|
—
|
Concentrațiile testate ar trebui să rămână în intervalul de solubilitate al substanței chimice de testat.
|
|
Analiza datelor
|
21.
|
Procedura specifică de analiză a datelor pentru datele privind legarea prin saturație și legarea competitivă ar trebui să respecte principiile esențiale pentru caracterizarea interacțiunilor receptor-ligand. De regulă, datele privind legarea prin saturație sunt analizate cu ajutorul unui model de regresie neliniară care determină legarea totală și legarea nespecifică. Este posibil să fie necesară o corecție pentru sărăcirea ligandului [de exemplu, Swillens, 1995, (19)] atunci când se stabilește Bmax și Kd. Datele provenite din analizele privind legarea competitivă sunt transformate, de regulă [de exemplu, legarea și concentrația procentuală a substanței chimice de testat (log M)]. Estimările valorii log (IC50) pentru fiecare substanță chimică de testat ar trebui să fie determinate utilizând un software de formare a curbelor neliniare adecvat pentru a se încadra într-o ecuație Hill cu patru parametri. În urma unei analize inițiale, ar trebui să se stabilească parametrii curbei și să se verifice vizual cât de bine sunt încadrate datele privind legarea pe curba de legare competitivă generată. În unele cazuri, poate fi necesară o analiză suplimentară pentru a obține cea mai bună curbă [de exemplu, prin stabilirea de limite pentru partea superioară și/sau cea inferioară a curbei, utilizarea regulii de 10 %, a se vedea apendicele 4 și referința 2 (secțiunea III.A.2).
|
|
22.
|
Îndeplinirea criteriilor de acceptabilitate (punctul 20) indică faptul că sistemul de analiză funcționează în mod corespunzător, dar nu asigură faptul că orice test specific va genera date precise. Reproducerea rezultatelor corecte ale primului test indică cel mai bine faptul că au fost generate date precise.
|
Criterii generale de interpretare a datelor
|
23.
|
În prezent, nu există nicio metodă convenită în mod universal pentru interpretarea datelor privind legarea de ER. Însă evaluările calitative (de exemplu, liant/non-liant) și/sau cantitative [de exemplu, log IC50, afinitatea de legare relativă (RBA)] ale activității mediate de hrER ar trebui să fie bazate pe date empirice și un raționament științific solid.
|
Raport testului
|
24.
|
Raportul testului ar trebui să includă următoarele informații:
|
|
Martor/substanță de referință/substanță chimică de testat
|
—
|
sursa, numărul lotului, data limită de utilizare, dacă sunt disponibile
|
|
—
|
stabilitatea substanței chimice de testat, dacă se cunoaște;
|
|
—
|
solubilitatea și stabilitatea substanței chimice de testat în solvent, dacă se cunosc;
|
|
—
|
măsurarea pH-ului, a osmolalității și a precipitatului în mediul de cultură în care s-a adăugat substanța chimică de testat, după caz.
|
|
|
|
Substanță mono-constituent:
|
—
|
aspectul fizic, solubilitatea în apă și alte proprietăți fizico-chimice relevante;
|
|
—
|
identificarea chimică, precum denumirea IUPAC sau CAS, numărul CAS, codul SMILES sau InChI, formula structurală, puritate, identitatea chimică a impurităților, după caz și dacă este practic fezabil etc.
|
|
|
|
Substanță multicomponentă, UVCB și amestecuri:
|
—
|
în măsura în care este posibil, caracterizată prin identitatea chimică (a se vedea mai sus), apariția cantitativă și proprietățile fizico-chimice relevante ale componentelor.
|
|
|
|
Solvent/vehicul:
|
—
|
caracterizare (natură, furnizor și lot);
|
|
—
|
justificarea alegerii solventului/vehiculului;
|
|
—
|
solubilitatea și stabilitatea substanței chimice de testat în solvent/vehicul, dacă se cunoaște;
|
|
|
|
Receptori:
|
—
|
sursa receptorilor (furnizor, nr. catalog, lot, specia receptorului, concentrația receptorului activ pusă la dispoziție de furnizor, certificarea din partea furnizorului)
|
|
—
|
caracterizarea receptorilor (inclusiv rezultatele privind legarea prin saturație): Kd, Bmax,
|
|
—
|
ligand marcat radioactiv:
|
|
—
|
furnizor, nr. catalog, lot, activitate specifică
|
|
|
|
Condiții de testare:
|
—
|
limitele de solubilitate în condiții de analiză;
|
|
—
|
componența soluției tampon de legare;
|
|
—
|
concentrația receptorului;
|
|
—
|
concentrația detectorului (și anume, ligand marcat radioactiv);
|
|
—
|
concentrațiile substanței chimice de testat;
|
|
—
|
procent de vehicul în analiza finală;
|
|
—
|
temperatura și timpul de incubație;
|
|
—
|
metoda legării/separării;
|
|
—
|
martori pozitivi și negativi/substanțe de referință;
|
|
—
|
criterii pentru caracterizarea testelor ca fiind pozitive, negative sau echivoce;
|
|
|
|
Verificarea de acceptabilitate:
|
—
|
valorile efective IC50 și Hillslope pentru martorii pozitivi/substanțele de referință testate în paralel;
|
|
|
|
Rezultate:
|
—
|
date brute și integrate/libere;
|
|
—
|
verificarea confirmării de denaturare, dacă este cazul;
|
|
—
|
dacă există, cea mai redusă valoare a concentrației efective (LEC);
|
|
—
|
valorile RBA și/sau IC50, după caz;
|
|
—
|
relația concentrație-răspuns, dacă este posibil;
|
|
—
|
analize statistice, dacă este cazul, împreună cu o măsură de eroare și de încredere (de exemplu, SEM, SD, CV sau 95 % CI) și o descriere a modului în care au fost obținute aceste valori;
|
|
|
|
Discutarea rezultatelor:
|
—
|
aplicarea regulii de 10 %
|
|
Concluzii
|
REFERINȚE BIBLIOGRAFICE
|
(1)
|
OCDE (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environmental Health and Safety Publications, Series on Testing and Assessment, (nr. 34), Organizația pentru Cooperare și Dezvoltare Economică, Paris.
|
|
(2)
|
OCDE (2015). Integrated Summary Report: Validation of Two Binding Assays Using Human Recombinant Estrogen Receptor Alpha (hrERα), Health and Safety Publications, Series on Testing and Assessment (nr. 226), Organizația pentru Cooperare și Dezvoltare Economică, Paris.
|
|
(3)
|
OCDE (2015). Performance Standards for Binding Assays Using Human Recombinant Estrogen Receptor Alpha (hrERα), Health and Safety Publications, Series on Testing and Assessment (nr. 222), Organizația pentru Cooperare și Dezvoltare Economică, Paris.
|
|
(4)
|
OCDE (2012). Guidance Document on Standardized Test Guidelines for Evaluating Chemicals for Endocrine Disruption. Environmental Health and Safety Publications, Series on Testing and Assessment, (nr. 150), Organizația pentru Cooperare și Dezvoltare Economică, Paris.
|
|
(5)
|
Cavailles V. (2002). Estrogens and Receptors: an Evolving Concept, Climacteric, 5 Suppl 2: p. 20-6.
|
|
(6)
|
Welboren W.J., et al. (2009). Genomic Actions of Estrogen Receptor Alpha: What are the Targets and How are they Regulated? Endocr. Relat. Cancer., 16(4): p. 1073-89.
|
|
(7)
|
Younes M. și Honma N. (2011). Estrogen Receptor Beta, Arch. Pathol. Lab. Med., 135(1): p. 63-6.
|
|
(8)
|
Diamanti-Kandarakis et al. (2009). Endocrine-Disrupting Chemicals: an Endocrine Society Sci. Statement, Endo Rev 30(4):293-342.
|
|
(9)
|
ICCVAM (2002). Background Review Document. Current Status of Test Methods for Detecting Endocrine Disruptors: In Vitro Estrogen Receptor Binding Assays. (NIH Publication nr. 03-4504). National Institute of Environmental Health Sciences, Research Triangle Park, NC.
|
|
(10)
|
ICCVAM (2003). ICCVAM Evaluation of In Vitro Test Methods for Detecting Potential Endocrine Disruptors: Estrogen Receptor and Androgen Receptor Binding and Transcriptional Activation Assays.
|
|
(11)
|
ICCVAM (2006). ICCVAM Evaluation of In Vitro Test Methods for Detecting Potential Endocrine Disruptors: Estrogen Receptor and Androgen Receptor Binding and Transcriptional Activation Assays.
|
|
(12)
|
Akahori Y. et al. (2008). Relationship Between the Results of In Vitro Receptor Binding Assay to Human Estrogen Receptor Alpha and In Vivo Uterotrophic Assay: Comparative Study with 65 Selected Chemicals, Toxicol. In Vitro, 22(1): 225-231.
|
|
(13)
|
OCDE (2007). Additional Data Supporting the Test Guideline on the Uterotrophic Bioassay in Rodents, Environment, Health and Safety Publications, Series on Testing and Assessment (nr. 67), Organizația pentru Cooperare și Dezvoltare Economică, Paris.
|
|
(14)
|
Takeyoshi, M. (2006). Draft Report of Pre-validation and Inter-laboratory Validation For Stably Transfected Transcriptional Activation (TA) Assay to Detect Estrogenic Activity - The Human Estrogen Receptor Alpha Mediated Reporter Gene Assay Using hER-HeLa-9903 Cell Line, Chemicals Evaluation and Research Institute (CERI): Japonia. p. 1-188.
|
|
(15)
|
Yamasaki, K; Noda, S; Imatanaka, N; Yakabe, Y. (2004). Comparative Study of the Uterotrophic Potency of 14 Chemicals in a Uterotrophic Assay and their Receptor-Binding Affinity, Toxicol. Letters, 146: 111-120.
|
|
(16)
|
Kummer V; Maskova, J; Zraly, Z; Neca, J; Simeckova, P; Vondracek, J; Machala, M. (2008). Estrogenic Activity of Environmental Polycyclic Aromatic Hydrocarbons in Uterus of Immature Wistar Rats. Toxicol. Letters, 180: 213-221.
|
|
(17)
|
Gozgit, JM; Nestor, KM; Fasco, MJ; Pentecost, BT; Arcaro, KF. (2004). Differential Action of Polycyclic Aromatic Hydrocarbons on Endogenous Estrogen-Responsive Genes and on a Transfected Estrogen-Responsive Reporter in MCF-7 Cells. Toxicol. and Applied Pharmacol., 196: 58-67.
|
|
(18)
|
Santodonato, J. (1997). Review of the Estrogenic and Antiestrogenic Activity of Polycyclic Aromatic Hydrocarbons: Relationship to Carcinogenicity. Chemosphere, 34: 835-848.
|
|
(19)
|
Swillens S (1995). Interpretation of Binding Curves Obtained with High Receptor Concentrations: Practical Aid for Computer Analysis, Mol Pharmacol 47(6):1197-1203.
|
Apendicele 1
Definiții și Abrevieri
Regula de 10 %
: O opțiune de excludere de la analiză a punctelor de date în cazul în care media replicilor pentru valoarea procentuală a legării specifice de [3H]17β-estradiol este de cel puțin 10 %, peste nivelul observat pentru valoarea medie la o concentrație mai mică (a se vedea apendicele 4).
Criterii de acceptabilitate
: Standarde minime pentru efectuarea de controale experimentale și standarde de referință. Toate criteriile de acceptabilitate ar trebui să fie îndeplinite pentru ca un experiment să fie considerat valabil.
Precizie (concordanță)
: Gradul de concordanță între rezultatele testului și valorile de referință acceptate. Aceasta măsoară performanța testului și unul dintre aspectele relevanței acestuia. Termenul este adesea utilizat în mod interschimbabil, iar concordanța înseamnă proporția de rezultate corecte ale unui test (1).
CF
: Cadrul conceptual al OCDE pentru testarea și evaluarea disruptorilor endocrini.
Substanță chimică
: O substanță sau un amestec.
CV
: Coeficient de variație
E2
: 17β-estradiol
ED
: Perturbare endocrină
hERα
: Receptor alfa de estrogen uman
ER
: Receptor de estrogen
Activitate estrogenă
: Capacitatea unei substanțe chimice de a imita 17β-estradiol în capacitatea sa de a lega receptori de estrogen. Legarea de hERα poate fi detectată prin această metodă de testare.
IC
50
: Jumătate din concentrația maximă efectivă a unei substanțe chimice de testat inhibitoare.
ICCVAM
: Comitetul de coordonare între agenții cu privire la validarea metodelor alternative (The Interagency Coordinating Committee on the Validation of Alternative Methods).
Reproductibilitatea între laboratoare
: Un indicator al măsurii în care diferite laboratoare calificate, utilizând același protocol și testând aceleași substanțe, pot produce rezultate similare din punct de vedere calitativ și cantitativ. Reproductibilitatea între laboratoare este determinată în timpul proceselor de pre-validare și de validare și indică măsura în care un test poate fi transferat cu succes între laboratoare, denumită de asemenea, reproductibilitate între laboratoare (1).
Reproductibilitatea în cadrul laboratorului
: O determinare a măsurii în care persoane calificate din cadrul aceluiași laborator pot repeta cu succes rezultatele la momente diferite, utilizând un protocol specific. Aceasta este denumită, de asemenea, reproductibilitate în cadrul laboratorului (1).
LEC
: Cea mai scăzută concentrație efectivă este cea mai scăzută concentrație a substanței chimice de testat care produce un răspuns (și anume, cea mai scăzută concentrație a substanței chimice de testat la care coeficientul de inducție multiplicată este diferit din punct de vedere statistic de martorul tratat cu vehicul testat în paralel).
Testul „me-too”
: O expresie colocvială utilizată pentru un test care este, din punct de vedere structural și funcțional, similar cu o metodă de testare de referință validată și acceptată. Utilizată alternativ cu metoda de testare similară.
PBTG
: Orientarea privind testarea pe baza performanței
Standarde de performanță
: Standarde bazate pe o metodă de testare validată, care asigură baza pentru evaluarea comparabilității unui test propus similar din punct de vedere al mecanismului și funcțional. Sunt incluse (1) componente esențiale ale testului; (2) o listă minimală de substanțe chimice de referință selectate dintre substanțele chimice utilizate pentru a demonstra performanța acceptabilă a metodei de testare validate; și (3) nivelurile comparabile de precizie și fiabilitate, pe baza rezultatelor obținute în cazul metodei de testare validate, care ar trebui să fie demonstrate prin testul propus la evaluarea acesteia pe baza listei minimale de substanțe chimice de referință (1).
Substanțe de verificare
: Un subset de substanțe de referință incluse în standardele de performanță, care pot fi utilizate de către laboratoare pentru a demonstra competența tehnică cu o analiză standardizată. Criteriile de selecție pentru aceste substanțe includ, de regulă, faptul că acestea reprezintă intervalul de răspunsuri, sunt disponibile în comerț și pentru acestea există date de referință de înaltă calitate.
Competență
: Capacitatea demonstrată de a efectua în mod corespunzător un test înainte de a testa substanțe necunoscute.
Estrogen de referință
: 17ß-estradiol (E2, CAS 50-28-2).
Metode de testare de referință
: Testele pe care se bazează PBTG 493.
RBA:
: Afinitate de legare relativă. Valoarea RBA a unei substanțe se calculează ca procent din log (IC50) pentru substanță cu raportare la log (IC50) pentru 17β-estradiol
Relevanță
: Descrierea relației dintre un test și efectul de interes și dacă aceasta este relevantă și utilă pentru un anumit scop. Aceasta reprezintă măsura în care testul măsoară sau prezice în mod corect efectul biologic de interes. Relevanța include analiza preciziei (a concordanței) unui test (1).
Fiabilitate
: Un indicator al faptului că un test poate fi efectuat în mod reproductibil, în același laborator și în laboratoare diferite în timp, atunci când este efectuat utilizând același protocol. Aceasta se evaluează prin calcularea reproductibilității în cadrul aceluiași laborator și între laboratoare.
SD
: Abatere standard.
Substanța chimică de testat
: Orice substanță sau amestec testat utilizând prezenta metodă de testare.
Metodă de testare validată
: Un test pentru care au fost realizate studii de validare pentru determinarea relevanței (inclusiv a preciziei) și a fiabilității pentru un anumit scop. Este important de precizat faptul că este posibil ca performanțele unei metode de testare validate să nu fie suficiente din punct de vedere al preciziei și fiabilității astfel încât aceasta să fie considerată acceptabilă pentru scopul propus (1).
Validare
: Procesul prin care se stabilește fiabilitatea și relevanța unei anumite abordări, metode, test, proces sau evaluări specifice pentru un scop definit (1).
Apendicele 2
ANALIZELE DE LEGARE PRIN SATURAȚIE ȘI LEGARE COMPETITIVĂ DE RECEPTORUL DE ESTROGEN (ERΑ) IN VITRO FREYBERGER-WILSON UTILIZÂND RECOMBINANT ERΑ DE LUNGIME TOTALĂ
CONSIDERAȚII ȘI LIMITĂRI INIȚIALE (A SE VEDEA, DE ASEMENEA, INTRODUCERE GENERALĂ)
|
1.
|
Această analiză de legare prin saturație și legare competitivă in vitro de receptorul de estrogen (ERα) utilizează un receptor uman de lungime totală ERα (hrERα) care este produs în și izolat de celulele unor insecte infectate cu baculovirus. Protocolul, care a fost dezvoltat de Freyberger și Wilson, a fost supus unui studiu internațional de validare în mai multe laboratoare (2) care a demonstrat relevanța și fiabilitatea acestuia în scopul prevăzut al analizei.
|
|
2.
|
Această analiză este o procedură de depistare pentru identificarea substanțelor care pot lega hrERα în lungime totală. Aceasta este utilizată pentru a stabili capacitatea unei substanțe chimice de testat de a concura cu 17β-estradiol pentru legarea de hrERα. Rezultatele cantitative ale analizei pot include valoarea IC50 (un indicator al concentrației unei substanțe chimice de testat care este necesară pentru a desprinde jumătate din [3H]-17β-estradiol de hrERα) și afinitățile de legare relative ale substanțelor chimice de testat pentru hrERα comparativ cu 17β-estradiol. În scopul depistării substanțelor chimice, rezultatele acceptabile ale analizei calitative pot include clasificări ale substanțelor chimice de testat ca lianți ai hrERα, non-lianți sau substanțe echivoce pe baza criteriilor descrise pentru curbele pentru legare.
|
|
3.
|
Analiza folosește un ligand radioactiv care necesită obținerea de către laborator a unei licențe pentru materiale radioactive. Toate procedurile cu radioizotopi și substanțe chimice periculoase ar trebui să respecte regulamentele și procedurile descrise în legislația națională.
|
|
4.
|
Ar trebui să se parcurgă secțiunile „INTRODUCERE GENERALĂ” și „COMPONENTE ALE ANALIZEI DE LEGARE DE hrER” înainte de utilizarea acestei analize în scopuri de reglementare. Definițiile și abrevierile utilizate în prezenta orientare privind testarea sunt descrise în apendicele 1.
|
PRINCIPIILE TESTULUI (A SE VEDEA, DE ASEMENEA, INTRODUCERE GENERALĂ)
|
5.
|
Analiza legării hrERα măsoară capacitatea unui ligand marcat radioactiv ([3H]17-β-estradiol) de a se lega de ER în prezența concentrațiilor din ce în ce mai mari ale unei substanțe chimice de testat (și anume, concurent). Substanțele chimice de testat care prezintă o afinitate mare pentru ER concurează cu ligandul marcat radioactiv la o concentrație mai mică în comparație cu substanțele chimice care prezintă o afinitate mai redusă pentru receptor.
|
|
6.
|
Această analiză cuprinde două componente principale: un experiment de legare prin saturație pentru a caracteriza parametrii de interacțiune receptor-ligand, urmat de un experiment de legare competitiv care caracterizează concurența dintre o substanță chimică de testat și un ligand marcat radioactiv pentru legarea de ER.
|
|
7.
|
Scopul experimentului de legare prin saturație este de a caracteriza un anumit lot de receptori în ceea ce privește afinitatea de legare și numărul în pregătirea pentru experimentul de legare competitivă. Experimentul de legare prin saturație măsoară, în condiții de echilibru, afinitatea unei concentrații fixe a receptorului de estrogen pentru ligandul său natural (reprezentată de constanta de disociere, Kd) și concentrația locurilor cu receptor activ (Bmax).
|
|
8.
|
Experimentul de legare competitivă măsoară afinitatea unei substanțe de a concura cu [3H]17β-estradiol pentru legarea de ER. Afinitatea este cuantificată prin concentrația substanței chimice de testat care, în stare de echilibru, inhibă 50 % din legarea specifică a [3H]17β-estradiol (denumită „concentrație inhibitoare de 50 %” sau IC50). Aceasta poate fi evaluată, de asemenea, utilizând afinitatea de legare relativă (RBA, în raport cu valoarea IC50 a estradiolului, măsurată separat în cadrul aceluiași test). Experimentul de legare competitivă măsoară legarea [3H]17β-estradiol la o concentrație fixă în prezența unui interval mare (opt ordine de mărime) de concentrații ale substanței chimice de testat. Datele sunt apoi corelate, dacă este posibil, într-o formă de ecuație Hill (Hill, 1910) care descrie desprinderea ligandului radioactiv de către un liant competitiv dintr-un loc. Gradul de desprindere a estradiolului marcat radioactiv, în stare de echilibru, este folosit pentru a caracteriza substanța chimică de testat ca liant, non-liant sau ca o substanță care generează un răspuns echivoc.
|
PROCEDURA
Demonstrarea performanței acceptabile a proteinei hrERα
|
9.
|
Înainte de efectuarea sistematică a analizelor de legare prin saturație și de legare competitivă, ar trebui să se demonstreze performanța corespunzătoare a fiecărui nou lot de hrERα în laboratorul în care acesta va fi utilizat. Pentru a demonstra performanța ar trebui să se utilizeze un proces în două etape. Aceste etape sunt următoarele:
|
—
|
Se desfășoară o analiză de legare a [3H]-17β-estradiol prin saturație pentru a demonstra specificitatea și saturația hrERα. Analiza regresiei non-liniare a acestor date (de exemplu, BioSoft; McPherson, 1985; Motulsky, 1995) și schița Scatchard ulterioară ar trebui să documenteze afinitățile de legare de hrERα ale [3H]-17β-estradiol (Kd) și numărul receptorilor (Bmax) pentru fiecare lot de hrERα.
|
|
—
|
Se desfășoară o analiză de legare competitivă cu ajutorul substanțelor martor [estrogenul de referință (17β-estradiol)], al unui liant slab (de exemplu, noretinodrel sau noredronă) și al unui non-liant (octiltrietoxisilan, OTES). Fiecare laborator ar trebui să stabilească o bază de date istorică pentru a documenta consecvența IC50 și alte valori relevante pentru estrogenul de referință și liantul slab obținute în urma experimentelor și pentru diferite loturi de hrERα. Parametrii curbelor privind legarea competitivă pentru substanțele martor ar trebui să se încadreze în limitele intervalului de încredere de 95 % (a se vedea tabelul 1), acestea fiind dezvoltate pe baza datelor din laboratoarele care au participat la studiul de validare pentru această analiză (2).
|
|
Tabelul 1
Criterii de performanță elaborate pentru estrogenul de referință și liantul slab, analiza FW privind legarea hrER.
|
Substanță
|
Parametru
|
Media (7)
|
Abaterea standard (n)
|
Interval de încredere de 95 % (8)
|
|
Limita inferioară
|
Limita superioară
|
|
17β-estradiol
|
Partea superioară (%)
|
100,44
|
10,84 (67)
|
97,8
|
103,1
|
|
Partea inferioară (%)
|
0,29
|
1,25 (67)
|
-0,01
|
0,60
|
|
Panta Hill
|
-1,06
|
0,20 (67)
|
-1,11
|
-1,02
|
|
LogIC50 (M)
|
-8,92 (9)
|
0,18 (67)
|
-8,97
|
-8,88
|
|
Noretinodrel
|
Partea superioară (%)
|
99,42
|
8,90 (68)
|
97,27
|
101,60
|
|
Partea inferioară (%)
|
2,02
|
3,42 (68)
|
1,19
|
2,84
|
|
Panta Hill
|
-1,01
|
0,38 (68)
|
-1,10
|
-0,92
|
|
Log IC50 (M)
|
-6,39
|
0,27 (68)
|
-6,46
|
-6,33
|
|
Noretindronă (9)
|
Partea superioară (%)
|
96,14
|
8,44 (27)
|
92,80
|
99,48
|
|
Partea inferioară (%)
|
2,38
|
5,02 (27)
|
0,40
|
4,37
|
|
Panta Hill
|
-1,41
|
0,32 (27)
|
-1,53
|
-1,28
|
|
LogIC50(M)
|
-5,73
|
0,27 (27)
|
-5,84
|
-5,62
|
Demonstrarea competenței laboratorului
|
10.
|
A se vedea punctele 17 și 18, precum și tabelul 2 din „COMPONENTE ALE ANALIZEI PRIVIND LEGAREA DE hrER” ale acestei metode de testare. Fiecare analiză (legare prin analiză și legare competitivă) ar trebui să conste în trei testări independente (și anume, cu diluții proaspete de receptor, substanțe chimice și reactivi) în zile diferite și fiecare testare ar trebui să cuprindă trei replici.
|
Determinarea concentrației receptorului (hrERα)
|
11.
|
Concentrația receptorului activ variază ușor în funcție de lot și de condițiile de păstrare. Din acest motiv, ar trebui să se determine concentrația receptorului activ, astfel cum a fost primită de la furnizor. Aceasta va determina concentrația corespunzătoare a receptorului activ la momentul testării.
|
|
12.
|
În condiții corespunzătoare legării competitive (și anume, 1 nM [3H]-estradiol), ar trebui să se incubeze concentrații nominale de 0,25, 0,5, 0,75 și 1 nM ale receptorului în absența (legare totală) și prezența (legare nespecifică) unei cantități de 1 μM estradiol neetichetat. Legarea specifică, calculată ca diferență între legarea totală și legarea nespecifică, este schițată în comparație cu concentrația nominală a receptorului. Concentrația receptorului, care determină valorile specifice de legare, reprezentând 20 % din marcajul radioactiv adăugat, este corelată cu concentrația nominală a receptorului și această concentrație a receptorului ar trebui utilizate pentru experimente de legare prin saturație și legare competitivă. În mod frecvent, o concentrație hrER finală de 0,5 nM va respecta această condiție.
|
|
13.
|
În cazul în care criteriul de 20 % nu poate fi respectat în mod repetat, ar trebui verificat setul experimental pentru identificarea unor posibile erori. Nerespectarea criteriului de 20 % poate indica faptul că există foarte puțini receptori activi în lotul recombinant și ar trebui atunci să se aibă în vedere utilizarea altui lot de receptori.
|
Analiza saturației
|
14.
|
Ar trebui să se evalueze opt concentrații din ce în ce mai mari de [3H]17β-estradiol în trei replici, în următoarele trei condiții (a se vedea tabelul 2):
|
—
|
în absența substanței 17β-estradiol nemarcate și în prezența ER. Astfel se determină legarea totală prin măsurarea radioactivității în godeurile care au doar [3H]17β-estradiol.
|
|
—
|
În prezența unei concentrații multiplicate de 1 000 de ori de 17β-estradiol nemarcat peste 17β-estradiol marcat și în prezența ER. Intenția acestei condiții este de a satura locurile cu legare activă cu 17β-estradiol nemarcat și, prin măsurarea radioactivității în godeuri, de a determina legarea nespecifică. Orice cantitate de estradiol rămasă la cald, care se poate lega de receptor, se consideră că se leagă într-un loc nespecific, întrucât cantitatea de estradiol rece ar trebui să fie într-o concentrație atât de mare încât să se lege de toate locurile specifice disponibile de pe receptor.
|
|
—
|
În absența substanței 17β-estradiol nemarcate și în absența ER (determinarea radioactivității totale)
|
|
Preparatul de [3H]-17β-estradiol și soluții 17β-estradiol nemarcate
|
15.
|
Diluțiile de [3H]-17β-estradiol ar trebui să fie preparate prin adăugarea de soluție tampon pentru analiză la o soluție mamă de 12 nM de [3H]-17β-estradiol pentru a obține concentrații inițiale de la 0,12 nM la 12 nM. Concentrațiile finale de analiză, de la 0,03 la 3,0 nM, se vor obține prin adăugarea a 40 μl din aceste soluții în godeurile de analiză respective de pe o placă de microtitrare cu 96 de godeuri (într-un volum final de 160 μl). Prepararea soluției tampon de analiză, a soluției mamă [3H]-17β-estradiol și a diluțiilor, precum și determinarea concentrațiilor sunt descrise în detaliu în protocolul FW (2).
|
|
16.
|
Diluțiile soluțiilor etanolice cu 17β-estradiol ar trebui să se prepare prin adăugarea de soluție tampon de analiză pentru a obține opt concentrații în creștere, cu valori inițiale de la 0,06 μΜ la 6 μΜ. Prin adăugarea a 80 μl din aceste soluții în godeurile de testare respective de pe o placă de microtitrare cu 96 de godeuri (într-un volum final de 160 μl), se vor obține concentrațiile finale de analiză, de la 0,03 μM la 3 μM. Concentrația finală de 17β-estradiol nemarcat în fiecare godeu din cadrul analizei privind legarea nespecifică ar trebui să fie de 1 000 de ori concentrația marcată a [3H]-17β- estradiol. Prepararea diluțiilor 17β-estradiol nemarcate este descrisă în detaliu în protocolul FW (2).
|
|
17.
|
Ar trebui să fie utilizată concentrația nominală a receptorului care prezintă o legare specifică de 20 ± 5 % (a se vedea punctele 12-13). Soluția hrERα ar trebui să fie preparată imediat înainte de utilizare.
|
|
18.
|
Plăcile de microtitrare cu 96 de godeuri sunt pregătite astfel cum este ilustrat în tabelul 2, cu 3 replici per concentrație. În apendicele 2.2 este prezentat un exemplu de distribuire a concentrației și volumului de [3H]-17β-estradiol, de 17β-estradiol nemarcat, de soluție tampon și de receptor.
|
Tabelul 2
Distribuția pe placa de microtitrare în cadrul analizei privind legarea prin saturație
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
|
A
|
0,03 nM [3H] E2 + ER
|
0,06 nM [3H] E2 + ER
|
0,08 nM [3H] E2 + ER
|
0,10 nM [3H] E2 + ER
|
Legare totală (solvent)
|
|
B
|
0,30 nM [3H] E2 + ER
|
0,60 nM [3H] E2 + ER
|
1,0 nM [3H] E2 + ER
|
3,0 nM [3H] E2 + ER
|
|
|
C
|
|
|
|
|
|
|
D
|
0,03 nM [3H] E2 + ER + 0,03 μM E2
|
0,06 nM [3H] E2 + ER + 0,06 μM E2
|
0,08 nM [3H] E2 + ER + 0,08 μM E2
|
0,10 nM [3H] E2 + ER
+ 0,10 μM E2
|
Legare nespecifică
|
|
E
|
0,30 nM [3H] E2 + ER + 0,30 μM E2
|
0,60 nM [3H] E2 + ER + 0,60 μM E2
|
1,0 nM [
3
H] E2 + ER
+ 1,0 μM E2
|
3,0 nM [3H] E2 + ER + 3,0 μM E2
|
|
|
F
|
|
|
|
|
|
|
G
|
|
|
|
|
|
|
H
|
|
|
|
|
|
|
[3H] E2:: [3H]-17β-estradiol
ER:: receptor de estrogen
E2:: 17β-estradiol nemarcat
|
|
19.
|
Plăcile de microtitrare pentru analiză ar trebui să fie incubate de la 2° până la 8 °C timp de 16-20 de ore și așezate pe un rotator în perioada de incubație.
|
Măsurarea [3H]-17β-estradiol legat de hrERα
|
20.
|
[3H]-17β-estradiol legat de hrERα ar trebui să fie separat de [3H]-17β-estradiol liber prin adăugarea a 80 μl de suspensie rece DCC în fiecare godeu, agitându-se plăcile de microtitrare timp de 10 minute și apoi supuse centrifugării timp de 10 minute la aproximativ 2 500 RPM. Pentru a reduce la minim desprinderea legăturii [3H]-17β-estradiol de hrERα în cursul acestui proces, este extrem de important ca soluțiile tampon și godeurile de analiză să fie păstrate la temperaturi între 2 și 8 °C și ca fiecare etapă să fie parcursă rapid. Este necesar un agitator pentru plăcile de microtitrare pentru ca acestea să fie prelucrate în mod eficient și rapid.
|
|
21.
|
Apoi ar trebui să se extragă cu multă atenție o cantitate de 50 μl de supernatant care conține [3H]-17β-estradiol legat de hrERα pentru a evita orice contaminare a godeurilor prin atingerea DCC și care ar trebui să fie așezată pe o a doua placă de microtitrare.
|
|
22.
|
Apoi se adaugă în fiecare godeu (A1-B12 și D1-E12) 200 μl de fluid de scintilație, care poate să convertească energia cinetică a emisiilor nucleare în energie luminoasă. Godeurile G1-H12 (identificate ca valoare DPMS totală) reprezintă diluții în serie de [3H]-17β-estradiol (40 μl) care ar trebui introduse direct în lichidul de scintilație din godeurile plăcii de măsurare, astfel cum se indică în tabelul 3, și anume, aceste godeuri conțin doar 200 μl de fluid de scintilație și diluția corespunzătoare de [3H]-17β-estradiol. Aceste măsuri demonstrează cât de mult [3H]-17β-estradiol, în DPMS, a fost adăugat în fiecare set de godeuri pentru legarea totală și legarea nespecifică.
|
Tabelul 3
Distribuția pe placa de microtitrare în cadrul analizei privind legarea prin saturație, măsurarea radioactivității
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
|
A
|
0,03 nM [3H] E2 + ER
|
0,06 nM [3H] E2 + ER
|
0,08 nM [3H] E2 + ER
|
0,10 nM [3 H] E2 + ER
|
Legare totală (solvent)
|
|
B
|
0,30 nM [3H] E2 + ER
|
0,60 nM [3H] E2 + ER
|
1,0 nM [3H] E2 + ER
|
3,0 nM [3H] E2 + ER
|
|
C
|
|
|
|
|
|
|
D
|
0,03 nM [3H] E2 + ER + 0,03 μM E2
|
0,06 nM [3H] E2 + ER + 0,06 μM E2
|
0,08 nM [3H] E2 + ER + 0,08 μM E2
|
0,10 nM [3H] E2 + ER
+ 0,10 μM E
2
|
Legare nespecifică
|
|
E
|
0,30 nM [3H] E2 + ER + 0,30 μM E2
|
0,60 nM [3H] E2 + ER + 0,60 μM E2
|
1,0 nM [
3
H] E2 + ER
+ 1,0 μM E2
|
3,0 nM [3H] E2 + ER + 3,0 μM E2
|
|
F
|
|
|
|
|
|
|
G
|
0,03 nM [3H] E2 (total dpms)
|
0,06 nM [3H] E2
|
0,08 nM [3H] E2
|
0,10 nM [3H] E2
|
Total dpms (*7)
|
|
H
|
0,30 nM [3H] E2
|
0,60 nM [3H] E2
|
1,0 nM [3H] E2
|
3,0 nM [3H] E2
|
|
[3H] E2:: [3H]-17β-estradiol
ER:: receptor de estrogen
E2:: 17β-estradiol nemarcat
dpms:: dezintegrări pe minut
|
|
23.
|
Măsurarea ar trebui să înceapă cu un decalaj de cel puțin 2 ore, iar timpul de numărare ar trebui să fie de 40 de minute per godeu. Ar trebui să se utilizeze un contor de scintilație pe placa de microtitrare pentru determinarea dpm/godeu cu o corecție de atenuare. În mod alternativ, în cazul în care nu există un contor de scintilație pentru placa de microtitrare, eșantioanele pot fi măsurate într-un contor convențional. În aceste condiții, se poate avea în vedere o reducere a timpului de numărare.
|
Analiza privind legarea competitivă
|
24.
|
Analiza privind legarea competitivă măsoară gradul de legare al unei singure concentrații de [3H]-17β-estradiol în prezența unor concentrații în creștere ale unei substanțe chimice de testat. La fiecare concentrație din cadrul unei testări ar trebui să se utilizeze trei replici concomitent. În plus, pentru fiecare substanță chimică de testat ar trebui să se efectueze trei teste la momente diferite. Substanțele pentru analiză ar trebui să fie configurate pe una sau mai multe plăci de microtitrare cu 96 de godeuri.
|
Martori
|
25.
|
Atunci când se efectuează analiza, ar trebui să se includă în fiecare experiment solventul și martorii tratați în paralel (și anume, estrogenul de referință, liantul slab și non-liantul). Pe fiecare placă, în timpul fiecărei testări, ar trebui să se utilizeze curbe complete de concentrații pentru estrogenul de referință și martori (și anume, liant slab și non-liant). Toate celelalte plăci ar trebui să conțină fiecare (i) E2 și un liant slab în trei replici la o concentrație ridicată (desprindere maximă) și medie (aproximativ IC50); (ii) martor tratat cu solvent și legare nespecifică, fiecare în cel puțin trei replici. Procedurile pentru prepararea soluției tampon de analiză, a martorilor, [3H]-17β-estradiol, hrERα și a soluțiilor cu substanță chimică de testat sunt descrise la referința 2 (anexa K, a se vedea Protocolul privind testarea FW).
|
Martor tratat cu solvent:
|
26.
|
Martorul tratat cu solvent indică faptul că solventul nu interacționează cu sistemul de testare și, de asemenea, măsoară legarea totală (TB). Etanolul este solventul preferat. În mod alternativ, în cazul în care cea mai mare concentrație a substanței chimice de testat nu este solubilă în etanol, poate fi utilizat DMSO. Concentrația de etanol sau DMSO, dacă este utilizată, în godeurile analizei finale este de 1,5 % și nu poate să depășească 2 %.
|
Martor tampon:
|
27.
|
Martorul tampon (BC) nu ar trebui să conțină nici solvent, nici substanță chimică de testat, ci toate celelalte componente ale analizei. Rezultatele martorului tampon sunt comparate cu martorul tratat cu solvent pentru a verifica dacă solventul utilizat nu afectează sistemul de analiză.
|
Liant puternic (estrogen de referință)
|
28.
|
17β-estradiol (CAS 50-28-2) este ligandul endogen și se leagă cu un grad de afinitate mare de ER, subtipul alfa. Pentru fiecare analiză privind legarea competitivă hrERα ar trebui să se pregătească o curbă standard utilizând 17β-estradiol nemarcat pentru a permite o evaluare a variabilității atunci când analiza este efectuată în timp, în același laborator. Ar trebui să se prepare opt soluții cu 17β-estradiol nemarcate în etanol, având concentrații în godeurile de analiză cuprinse în intervalul 100 nM-10 pM (de la -7 [logM] la -11 [logM]), repartizate după cum urmează: (-7 [logM], -8 [logM], -8,5 [logM], -9 [logM], - 9,5 [logM], -10 [logM], -11 [logM]). Cea mai mare concentrație de 17β-estradiol nemarcată (1 μM) servește, de asemenea, drept indicator de legare nespecifică. Această concentrație se distinge prin eticheta „NSB” în tabelul 4, chiar dacă aceasta este, de asemenea, integrată în curba standard.
|
Liant slab
|
29.
|
Un liant slab [noretinodrel (CAS 68-23-5) sau noretindronă (CAS 68-22-40] ar trebui să fie inclus pentru a demonstra sensibilitatea fiecărui experiment și a permite o evaluare a variabilității atunci când analiza este efectuată în timp. Ar trebui să se prepare opt soluții de liant slab în etanol, având concentrații în godeurile de analiză cuprinse între 3 nM și 30 μM (de la -8,5 [logM] la -4,5 [logM]), repartizate după cum urmează: -4,5 [logM], -5 [logM], -5,5 [logM], -6 [logM], -6,5 [logM], -7 [logM],-7,5 [logM], -8,5 [logM].
|
Non-liant
|
30.
|
Ca martor negativ ar trebui să se utilizeze octiltrietoxisilan (OTES, CAS 2 943-75-1) (non-liant). Acesta asigură faptul că, așa cum este efectuată analiza, va detecta momentul în care substanțele chimice de testat nu se leagă de hrERα. Ar trebui să se prepare opt soluții de non-liant în etanol, având concentrații în godeurile de analiză cuprinse între 0,1 nM și 1 000 μM (de la -10 [logM] la -3 [logM]), în creșteri treptate ale valorii log. Ftalatul de di-n-butil (DBP) poate fi utilizat ca și non-liant martor alternativ. S-a demonstrat că solubilitatea sa maximă este -4 [logM].
|
Concentrația hrERα
|
31.
|
Ar trebui să fie utilizată cantitatea de receptori care prezintă o legare specifică de 20 ± 5 % de 1 nm de ligant radioactiv (a se vedea punctele 12-13 din apendicele 2). Soluția hrERα ar trebui să fie preparată imediat înainte de utilizare.
|
[3H]-17β-estradiol
|
32.
|
Concentrația [3H]-17β-estradiol în godeurile de analiză ar trebui să fie de 1,0 nM.
|
Substanțe chimice de testat
|
33.
|
În primul rând, trebuie să se efectueze un test al solubilității pentru a determina limita de solubilitate pentru fiecare substanță chimică de testat și pentru a identifica intervalul de concentrații adecvat care să fie utilizat la efectuarea protocolului de testare. Limita de solubilitate a fiecărei substanțe chimice de testat va fi determinată inițial în solvent și confirmată ulterior în condiții de testare. Concentrația finală testată în cadrul analizei nu ar trebui să depășească 1 mM. Testul pentru identificarea intervalului cuprinde un martor tratat cu solvent și diluții în serie de 8 log, pornind de la concentrația maximă acceptabilă (de exemplu, cel mult 1 mM, în funcție de limita de solubilitate), și se consemnează gradul de tulburare sau precipitatul (a se vedea, de asemenea, punctul 35). Substanța chimică de testat ar trebui să fie testată folosind curbe ale concentrației repartizate la 8 log, astfel cum sunt definite în testul anterior de stabilire a intervalului. Concentrațiile din cel de-al doilea și cel de-al treilea experiment ar trebui să fie ajustate după caz pentru o mai bună caracterizare a curbei concentrație-răspuns.
|
|
34.
|
Diluțiile substanței chimice de testat ar trebui să fie preparate în solvent adecvat (a se vedea punctul 26 din apendicele 2). În cazul în care concentrația maximă a substanței chimice de testat nu este solubilă nici în etanol, nici în DMSO, iar dacă se adaugă mai mult solvent, concentrația solventului în eprubeta finală ar fi mai mare decât limita acceptabilă, concentrația maximă poate fi redusă la următoarea concentrație inferioară. În acest caz, se poate adăuga o concentrație suplimentară la limita inferioară a seriei de concentrații. Alte concentrări din cadrul seriei ar trebui să rămână neschimbate.
|
|
35.
|
Soluțiile cu substanță chimică de testat ar trebui să fie monitorizate atent atunci când se adaugă la godeul de analiză, deoarece substanța chimică testată se poate precipita atunci când este adăugată în acesta. Datele pentru toate godeurile care conțin precipitat ar trebui să fie excluse de la formarea curbei, iar motivul excluderii datelor ar trebui să fie consemnat.
|
|
36.
|
În cazul în care există informații anterioare din alte surse care furnizează o valoare log(IC50) pentru o substanță chimică de testat, ar putea fi adecvat ca diluțiile să se repartizate geometric [și anume, 0,5 unități log în jurul valorii preconizate log(IC50). Rezultatul final ar trebui să reflecte o marjă suficientă de concentrații pe fiecare parte a valorii log(IC50), inclusiv pe partea superioară și cea inferioară, astfel încât curba pentru legătură să poată fi caracterizată în mod corespunzător.
|
Organizara plăcii de analiză
|
37.
|
Ar trebui să se pregătească plăci de microtitrare etichetate luând în considerare incubațiile în șase replici cu coduri pentru martorul tratat cu solvent, cea mai mare concentrație a estrogenului de referință, care constituie, de asemenea, un indicator al legăturii nespecifice (NSB), și soluția tampon martor, precum și luând în considerare incubațiile în trei replici cu coduri pentru fiecare dintre cele opt concentrații ale martorului non-liant (octiltrietoxisilan), cele șapte concentrații inferioare pentru estrogenul de referință, cele opt niveluri ale dozelor de concentrații ale liantului slab și cele opt concentrații ale fiecărei substanțe chimice de testat (TC). În tabelul 4 de mai jos este prezentat un exemplu de model de diagramă a plăcii pentru curbele de concentrație complete pentru estrogenul de referință și martor. Se utilizează plăci de microtitrare suplimentare pentru substanțele chimice de testat, care ar trebui să includă martori pe placă (și anume, 1) E2 și un liant slab în trei replici având o concentrație ridicată (desprindere maximă) și medie (de aproximativ IC50); 2) martor tratat cu solvent și soluție de legare nespecifică, fiecare în șase replici (tabelul 5). În apendicele 2.3 este prezentat un exemplu de fișă de configurare a plăcilor de microtitrare pentru analiză competitivă, care utilizează trei substanțe chimice de testat necunoscute. Concentrațiile indicate în tabelele 4 și 5 reprezintă concentrațiile finale ale analizei. Concentrația maximă pentru E2 ar trebui să fie 1 × 10-7 M, iar pentru liantul slab, ar trebui să se utilizeze cea mai mare concentrație utilizată pentru liantul slab pe placa 1. Concentrația IC50 trebuie să fie stabilită de laborator pe baza bazei sale de date cu martori testați anterior. Se preconizează că această valoare va fi similară cu cea observată în studiile de validare (a se vedea tabelul 1).
|
Tabelul 4
Configurația pentru analiza privind legarea competitivă pe placa de microtitrare, curbe de concentrație complete pentru estrogenul de referință și martori (placa 1)
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
TB (doar solvent)
|
TB (doar solvent)
|
NSB
|
NSB
|
|
B
|
E2 (1×10-7)
|
E2 (1×10-8)
|
E2 (1×10-8,5)
|
E2 (1×10-9)
|
|
C
|
E2 (1×10-9,5)
|
E2 (1×10-10)
|
E2 (1×10-11)
|
Soluție săracă (*8)
|
|
D
|
NE (1×10-4,5)
|
NE (1×10-5)
|
NE (1×10-5,5)
|
NE (1×10-6)
|
|
E
|
NE (1×10-6,5)
|
NE (1×10-7)
|
NE (1×10-7,5)
|
NE (1×10-8,5)
|
|
F
|
OTES (1×10-3)
|
OTES (1×10-4)
|
OTES (1×10-5)
|
OTES (1×10-6)
|
|
G
|
OTES (1×10-7)
|
OTES (1×10-8)
|
OTES (1×10-9)
|
OTES (1×10-10)
|
|
H
|
Soluție săracă (pentru mediu la cald) (*9)
|
Soluție săracă (pentru mediu la cald) (*9)
|
Martor tampon
|
Martor tampon
|
|
În acest exemplu, liantul slab este noretinodrel (NE)
|
Tabelul 5
Configurația pentru analiza privind legarea competitivă pe placa de microtitrare, curbe de concentrație complete pentru substanțele chimice de testat și martorii de pe placă.
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
TB (doar solvent)
|
TB (doar solvent)
|
NSB
|
NSB
|
|
B
|
TC1 (1×10-3)
|
TC1 (1×10-4)
|
TC1 (1×10-5)
|
TC1 (1×10-6)
|
|
C
|
TC1 (1×10-7)
|
TC1 (1×10-8)
|
TC1 (1×10-9)
|
TC1 (1×10-10)
|
|
D
|
TC2 (1×10-3)
|
TC2 (1×10-4)
|
TC2 (1×10-5)
|
TC2 (1×10-6)
|
|
E
|
TC2 (1×10-7)
|
TC2 (1×10-8)
|
TC2 (1×10-9)
|
TC2 (1×10-10)
|
|
F
|
TC3 (1×10-3)
|
TC3 (1×10-4)
|
TC3 (1×10-5)
|
TC3 (1×10-6)
|
|
G
|
TC3 (1×10-7)
|
TC3 (1×10-8)
|
TC3 (1×10-9)
|
TC3 (1×10-10)
|
|
H
|
NE (IC50)
|
NE (1×10-4,5)
|
E2 (IC50)
|
E2 (1×10-7)
|
|
În acest exemplu, liantul slab este noretinodrel (NE)
|
Încheierea analizei privind legarea competitivă
|
38.
|
Astfel cum se arată în tabelul 6, în fiecare godeu ar trebui să se adauge 80 μl de martor tratat cu solvent, martor tampon, estrogen de referință, liant slab, non-liant și substanțe chimice de testat preparate în soluția-tampon a analizei. Apoi se adaugă în fiecare godeu 40 μl dintr-o soluție 4 nM [3H]-17β-estradiol. După o rotire ușoară timp de 10-15 minute la temperaturi între 2° și 8 °C, ar trebui să se adauge 40 μl din soluția hrERα. Plăcile de microtitrare pentru analiză ar trebui să fie incubate de la 2° până la 8 °C timp de 16-20 de ore și așezate pe un rotator în perioada de incubație.
|
Tabelul 6
Volumul componentelor de analiză pentru testul privind legarea competitivă hrER, plăci de microtitrare
|
Volum (μl)
|
Componentă
|
|
80
|
1/7-β-estradiol nemarcat, noretinodrel, OTES, substanțe chimice de testat, solvent sau soluție tampon
|
|
40
|
4 nM de soluție [3H]-17β-estradiol
|
|
40
|
Soluție hrERα având concentrația determinată
|
|
160
|
Volumul total în fiecare godeu de analiză
|
|
39.
|
Cuantificarea legării [3H]-17β-estradiol de hrERα, în urma separării [3H]-17β-estradiol legat de hrERα de [3H]-17β-estradiol liber, prin adăugarea, în fiecare godeu, a 80 μl de suspensie DCC rece, ar trebui să fie efectuată astfel cum este descris la punctele 20-23 pentru analiza privind legarea prin saturație.
|
|
40.
|
Godeurile H1-6 [identificate ca având soluție săracă (pentru mediu cald) în tabelul 4] reprezintă DPMS al [3H]-estradiol marcat în 40 μl. Partea alicotă de 40 μl ar trebui să fie introdusă direct în lichidul de scintilație din godeurile H1-H6.
|
Criterii de acceptabilitate
Analiza privind legarea prin saturație
|
41.
|
Curba specifică privind legarea ar trebui să ajungă la un platou, întrucât au fost utilizate concentrații crescătoare de [3H]-17β-estradiol, care indică saturația hrERα cu ligandul.
|
|
42.
|
Legarea specifică la 1 nM de [3H]-17β-estestradiol ar trebui să fie situată în intervalul acceptabil 15 %-25 % din radioactivitatea totală măsurată medie adăugată de la o testare la alta. Abaterile ocazionale ușoare de la acest interval sunt acceptabile, însă, dacă testele se situează în mod constant în afara acestui interval sau un anumit test este semnificativ în afara acestui interval, ar trebui să se ajusteze concentrația de proteine, iar analiza saturației ar trebui să fie repetată.
|
|
43.
|
Datele ar trebui să producă o reprezentare grafică liniară Scatchard.
|
|
44.
|
Legarea nespecifică nu ar trebui să fie excesivă. Valoarea pentru legarea nespecifică ar trebui, de regulă, să fie < 35 % din legarea totală. Însă, raportul poate depăși ocazional această limită atunci când se măsoară valoarea dpm foarte scăzută pentru concentrația minimă a substanței 17β-estradiol marcat radioactiv testate.
|
Analiza privind legarea competitivă
|
45.
|
Concentrațiile din ce în ce mai mari de 17β-estradiol nemarcat ar trebui să desprindă [3H]-17β- estradiol de receptor într-un mod compatibil cu o legarea competitivă dintr-un singur punct.
|
|
46.
|
Valoarea IC50 pentru estrogenul de referință (și anume, 17β-estradiol) ar trebui să fie aproximativ egal cu concentrația molară a [3H]-17β-estradiol, plus valorile Kd determinate din analiza privind legarea prin saturație.
|
|
47.
|
Legarea specifică totală ar trebui să se situeze în mod consecvent în intervalul acceptabil de 20 ± 5 % atunci când concentrația medie măsurată a radioactivității totale adăugate în fiecare godeu a fost de 1 nM în toate testele. Abaterile ocazionale ușoare de la acest interval sunt acceptabile, însă, dacă testele se situează în mod constant în afara acestui interval sau un anumit test este semnificativ în afara acestui interval, ar trebui să se ajusteze concentrația de proteine.
|
|
48.
|
Solventul nu ar trebui să modifice sensibilitatea sau reproductibilitatea analizei. Rezultatele martorului tratat cu solvent (godeuri TB) sunt comparate cu martorul tampon pentru a verifica dacă solventul utilizat nu afectează sistemul de analiză. Rezultatele pentru TB și martorul tampon ar trebui să fie comparabile în cazul în care nu există niciun efect al solventului asupra analizei.
|
|
49.
|
Non-liantul nu ar trebui să desprindă mai mult de 25 % din [3H]-17β-estradiol de hrERα atunci când se testează maxim 10-3 M (OTES) sau 10-4 M (DBP).
|
|
50.
|
Au fost elaborate criterii de performanță pentru estrogenul de referință și doi lianți slabi (de exemplu, noretinodrel, noretindronă), utilizând date din studiul de validare a analizei FW privind legarea hrER (anexa N la referința 2). Sunt prezentate intervale de încredere de 95 % pentru media (n) +/- SD pentru toate testele pe martori din laboratoarele participante la studiul de validare. Au fost calculate intervale de încredere de 95 % pentru parametrii de formare a curbei (și anume, partea superioară, partea inferioară, Hillslope, logIC50) pentru estrogenul de referință și lianții slabi, precum și pentru valoarea log10RBA a lianților slabi în raport cu estrogenul de referință, fiind furnizate ca și criterii de performanță pentru martorii pozitivi. Tabelul 1 furnizează intervalele preconizate pentru parametrii de formare a curbei care pot fi utilizați ca și criterii de performanță. În practică, intervalul IC50 poate varia ușor, în funcție de valoarea Kd a preparatul cu receptori și de concentrația ligandului.
|
|
51.
|
Nu au fost elaborate criterii de performanță pentru parametrii de formare a curbei în cazul substanțelor chimice de testat din cauza gamei largi de substanțe chimice de testat posibile și a variației posibilelor afinități și rezultatele (de exemplu, curbă completă, curbă parțială, fără formă de curbă). Însă, la analizarea rezultatelor din cadrul fiecărei testări, în cazul unei substanțe chimice de testat, ar trebui să se aplice o apreciere profesională. Ar trebui să se utilizeze un interval suficient de concentrații ale substanței chimice de testat pentru a defini cu claritate partea superioară (de exemplu, o legare de 90-100 %) a curbei de legare competitivă. Variabilitatea între replici la fiecare concentrație a substanței chimice de testat, precum și între cele trei teste independente ar trebui să fie rezonabilă și justificată din punct de vedere științific. Martorii din cadrul fiecărei serii, pentru o substanță chimică de testat, ar trebui să se apropie de măsurile de performanță raportate pentru această analiză FW și să fie în concordanță cu datele privind martorii testați anterior din fiecare laborator.
|
ANALIZA DATELOR
Analiza privind legarea prin saturație
|
52.
|
Se măsoară atât legarea totală, cât și cea nespecifică. Pornind de la aceste valori, se calculează legarea specifică a [3H]-17β-estradiol, în condiții de echilibru, la concentrații în creștere, scăzând valoarea legării nespecifice din total. Un grafic al legării specifice versus concentrația [3H]-17β-estradiol ar trebui să ajungă al un platou pentru o legare specifică maximă care indică o saturație a hrERα cu [3H]-17β-estradiol. În plus, analiza datelor ar trebui să documenteze legarea [3H]-17β- estradiol de un singur loc de legare cu un grad ridicat de afinitate. Legarea nespecifică, cea totală și cea specifică ar trebui să fie ilustrate pe o curbă de legare prin saturație. În cadrul unei analize ulterioare a acestor date ar trebui să se utilizeze o analiză de regresie nelineară (de exemplu, BioSoft; McPherson, 1985; Motulsky, 1995) cu o reprezentare finală a datelor sub forma unui grafic Scatchard.
|
|
53.
|
Prin analiza datelor ar trebui să se determine valorile Bmax și Kd doar din datele privind legarea totală, pe baza ipotezei că legarea nespecifică este liniară, cu excepția cazului în care se justifică utilizarea unei metode diferite. În plus, ar trebui să se utilizeze o regresie robustă atunci când se determină cea mai bună corelare, cu excepția cazului în care este furnizată o justificare. Ar trebui să se precizeze metoda aleasă pentru regresia robustă. Ar trebui să se recurgă întotdeauna la corecția pentru sărăcirea ligandului (de exemplu, utilizând metoda lui Swillens, 1995) atunci când se determină valorile Bmax și Kd din datele privind legarea prin saturație.
|
Analiza privind legarea competitivă
|
54.
|
Curba de legare competitivă este reprezentată grafic ca fiind o legare specifică [3H]-17β-estradiol versus concentrația (unități de log10) soluției testate în paralel. Concentrația substanței chimice de testat care inhibă 50 % din legarea specifică maximă a [3H]-17β-estradiol constituie valoarea IC50.
|
|
55.
|
Estimările valorilor log(IC50) pentru martorii pozitivi (de exemplu, estrogenul de referință și liantul slab) ar trebui să fie determinate utilizând un software de formare a curbelor neliniare adecvat pentru a se încadra într-o ecuație Hill cu patru parametri (de exemplu, BioSoft; McPherson, 1985; Motulsky, 1995). Partea superioară, cea inferioară, panta și log(IC50) ar trebui, în general, să rămână necondiționate atunci când se formează aceste curbe. Ar trebui să se utilizeze o regresie robustă atunci când se determină cea mai bună corelare, cu excepția cazului în care este furnizată o justificare. Nu ar trebui să se recurgă la corecția pentru sărăcirea ligandului. În urma analizei inițiale, ar trebui să se examineze fiecare curbă de legare pentru a asigura o corelare corespunzătoare cu modelul. Afinitatea de legare relativă (RBA) a unui liant slab ar trebui să se calculeze ca procent din log (IC50) pentru liantul slab raportat la valoarea log (IC50) pentru 17β-estradiol. Rezultatele martorilor pozitivi și ale martorului non-liant ar trebui să fie evaluate utilizând valorile de măsurare a performanței analizei de la punctele 45-50 din prezentul apendice 2.
|
|
56.
|
Datele pentru toate substanțele chimice de testat ar trebui să fie analizate printr-o abordare etapizată pentru a se asigura analizarea corespunzătoare a datelor și clasificarea corectă a fiecărei curbe de legare competitivă. Se recomandă ca fiecare testare pentru o substanță chimică de testat să fie supusă inițial unei analize a datelor standardizată care este identică cu cea utilizată pentru estrogenul de referință și lianții slabi martori (a se vedea punctul 55 de mai sus). După încheierea analizei, ar trebui să fie efectuată o analiză tehnică a parametrilor curbei și o verificare vizuală a modului în care datele se încadrează pe curba de legare competitivă generată pentru fiecare testare. În timpul acestei analize tehnice, observațiile privind o scădere dependentă de concentrație în procentul [3H]-17β-estradiol legat în mod specific, variabilitatea scăzută între replicile tehnice la fiecare concentrație chimică și consecvența parametrilor de corelare între cele trei teste constituie indicații bune pentru faptul că analiza și analizele datelor au fost efectuate în mod corespunzător.
|
Interpretarea datelor
|
57.
|
Cu condiția ca toate criteriile de acceptabilitate să fie îndeplinite, o substanță chimică de testat este considerată a fi un liant pentru hrERα în cazul în care poate fi formată o curbă de legare, iar punctul cel mai de jos de pe curba de răspuns în intervalul de date este sub 50 % (figura 1).
|
|
58.
|
Cu condiția îndeplinirii tuturor criteriilor de acceptabilitate, o substanță chimică de testat este considerată a fi un non-liant pentru hrERα dacă:
|
—
|
poate fi formată o curbă de legare, iar punctul cel mai de jos pe curba de răspuns formată, situat în intervalul de date, este mai mare de 75 % sau
|
|
—
|
nu poate fi formată o curbă de legare, iar media cea mai scăzută a procentului de legare neuniformizată între grupurile de concentrații în ceea ce privește datele este mai mare de 75 %.
|
|
|
59.
|
Substanțele chimice de testat sunt considerate echivoce dacă nu este îndeplinită niciuna dintre condițiile de mai sus (de exemplu, punctul cel mai de jos de pe curba de răspuns formată este între 76 și 51 %).
|
Tabelul 7
Criterii pentru clasificare pe baza curbe de legare competitive pentru o substanță chimică de testat.
|
Clasificare
|
Criterii
|
|
Lianta
|
Se poate forma o curbă de legare.
Punctul cel mai de jos al curbei de răspuns în intervalul de date este mai mic de 50 %.
|
|
Non-liantb
|
Dacă se poate forma o curbă de legare,
punctul cel mai de jos al curbei de răspuns în intervalul de date este peste 75 %.
Dacă nu se poate forma o curbă de legare,
media cea mai scăzută a procentului de legare neuniformizată între grupurile de concentrații în ceea ce privește datele este mai mare de 75 %.
|
|
Substanță echivocăc
|
Orice substanță care poate fi testată și care nu este nici liant, nici non-liant
(de exemplu, punctul cel mai de jos pe curba de răspuns formată este între 76 și 51 %).
|
Figura 1
Exemple de clasificare a unei substanțe chimice de testat utilizând curba de legare competitivă.
|
60.
|
Se combină mai multe testări realizate într-un laborator pentru o substanță chimică de testat prin atribuirea de valori numerice fiecărei testări și calcularea mediei testelor, astfel cum se arată în tabelul 8. Rezultatele pentru testele combinate din fiecare laborator sunt comparate cu clasificarea preconizată pentru fiecare substanță chimică de testat.
|
Tabelul 8
Metoda de clasificare a substanței chimice de testat utilizând teste multiple în cadrul unui laborator
|
Pentru atribuirea unei valori fiecărei testări:
|
|
|
Clasificare
|
Valoare numerică
|
|
Liant
|
2
|
|
Substanță echivocă
|
1
|
|
Non-liant
|
0
|
|
Pentru clasificarea mediei valorilor numerice ale testelor:
|
|
|
Clasificare
|
Valoare numerică
|
|
Liant
|
Media ≥ 1,5
|
|
Substanță echivocă
|
0,5 ≤ media < 1,5
|
|
Non-liant
|
Media < 0,5
|
RAPORTUL TESTULUI
|
61.
|
A se vedea punctul 24 din „COMPONENTELE ANALIZEI PRIVIND LEGAREA hrER” din prezenta metodă de testare.
|
Apendicele 2.1
LISTĂ DE TERMENI
[3H]E2
: 17β-estradiol marcat radioactiv cu tritiu
DCC
: Cărbune acoperit cu dextran
E2
: 17β-estradiol nemarcat (inert)
Soluția-tampon de analiză
: 10 mM Tris, 10 mg albumină serică bovină /ml, 2 mM DTT, 10 % glicerol, 0,2 mM leupeptin, pH 7,5
hrERα
: Receptor de estrogen recombinant uman alfa
Replică
: Unul dintre multiplele godeuri care conțin același conținut la aceleași concentrații și sunt analizate în paralel în cadrul unei singure testări. În acest protocol, fiecare concentrație a substanței chimice de testat este testată în trei replici; cu alte cuvinte, există trei replici care sunt testate simultan la fiecare concentrație a substanței chimice de testat.
Testare
: Un set complet de godeuri de pe o placă de microtitrare testate în paralel, care oferă toate informațiile necesare pentru a caracteriza legarea unei substanțe chimice de testat de hrERα (și anume, [3H]-17β-estradiol total adăugat în godeul de analiză, legarea maximă a [3H]-17β-estradiol de hrERα, legarea nespecifică și legarea totală la diferite concentrații ale substanței chimice de testat). O testare ar putea consta dintr-un singur godeu de analiză (și anume, replică) per concentrație, dar, deoarece acest protocol impune efectuarea analizei pe trei replici, o testare constă din trei godeuri de analiză per concentrație. În plus, acest protocol necesită trei teste independente (și anume, neefectuate în paralel) pe substanță chimică.
Apendicele 2.2
ANALIZĂ TIPICĂ A SATURAȚIEI [3H]-17Β-ESTRADIOL CU TREI REPLICI DE GODEURI
|
Analiză tipică a saturației [3H]-17β-estradiol cu trei replici de godeuri
|
|
Poziție
|
Replică
|
Cod tip godeu
|
Concentrație inițială E2 la cald (nM)
|
Volum E2 la cald (μl)
|
Concentrație finală E2 la cald (nM)
|
Concentrație inițială E2 la rece (μM)
|
Volum E2 la rece (μl)
|
Concentrație finală E2 la rece (μM)
|
Volum soluție-tampon (μl)
|
Volum receptor (μl)
|
Total volum în godeuri
|
|
A1
|
1
|
H
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A2
|
2
|
H
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A3
|
3
|
H
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A4
|
1
|
H
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A5
|
2
|
H
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A6
|
3
|
H
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A7
|
1
|
H
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A8
|
2
|
H
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A9
|
3
|
H
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A10
|
1
|
H
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A11
|
2
|
H
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A12
|
3
|
H
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B1
|
1
|
H
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B2
|
2
|
H
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B3
|
3
|
H
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B4
|
1
|
H
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B5
|
2
|
H
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B6
|
3
|
H
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B7
|
1
|
H
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B8
|
2
|
H
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B9
|
3
|
H
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B10
|
1
|
H
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B11
|
2
|
H
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B12
|
3
|
H
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
D1
|
1
|
HC
|
0,12
|
40
|
0,03
|
0,06
|
80
|
0,03
|
—
|
40
|
160
|
|
D2
|
2
|
HC
|
0,12
|
40
|
0,03
|
0,06
|
80
|
0,03
|
—
|
40
|
160
|
|
D3
|
3
|
HC
|
0,12
|
40
|
0,03
|
0,06
|
80
|
0,03
|
—
|
40
|
160
|
|
D4
|
1
|
HC
|
0,24
|
40
|
0,06
|
0,12
|
80
|
0,06
|
—
|
40
|
160
|
|
D5
|
2
|
HC
|
0,24
|
40
|
0,06
|
0,12
|
80
|
0,06
|
—
|
40
|
160
|
|
D6
|
3
|
HC
|
0,24
|
40
|
0,06
|
0,12
|
80
|
0,06
|
—
|
40
|
160
|
|
D7
|
1
|
HC
|
0,32
|
40
|
0,08
|
0,16
|
80
|
0,08
|
—
|
40
|
160
|
|
D8
|
2
|
HC
|
0,32
|
40
|
0,08
|
0,16
|
80
|
0,08
|
—
|
40
|
160
|
|
D9
|
3
|
HC
|
0,32
|
40
|
0,08
|
0,16
|
80
|
0,08
|
—
|
40
|
160
|
|
D10
|
1
|
HC
|
0,40
|
40
|
0,10
|
0,2
|
80
|
0,1
|
—
|
40
|
160
|
|
D11
|
2
|
HC
|
0,40
|
40
|
0,10
|
0,2
|
80
|
0,1
|
—
|
40
|
160
|
|
D12
|
3
|
HC
|
0,40
|
40
|
0,10
|
0,2
|
80
|
0,1
|
—
|
40
|
160
|
|
E1
|
1
|
HC
|
1,20
|
40
|
0,30
|
0,6
|
80
|
0,3
|
—
|
40
|
160
|
|
E2
|
2
|
HC
|
1,20
|
40
|
0,30
|
0,6
|
80
|
0,3
|
—
|
40
|
160
|
|
E3
|
3
|
HC
|
1,20
|
40
|
0,30
|
0,6
|
80
|
0,3
|
—
|
40
|
160
|
|
E4
|
1
|
HC
|
2,40
|
40
|
0,60
|
1,2
|
80
|
0,6
|
—
|
40
|
160
|
|
E5
|
2
|
HC
|
2,40
|
40
|
0,60
|
1,2
|
80
|
0,6
|
—
|
40
|
160
|
|
E6
|
3
|
HC
|
2,40
|
40
|
0,60
|
1,2
|
80
|
0,6
|
—
|
40
|
160
|
|
E7
|
1
|
HC
|
4,00
|
40
|
1,00
|
2
|
80
|
1
|
—
|
40
|
160
|
|
E8
|
2
|
HC
|
4,00
|
40
|
1,00
|
2
|
80
|
1
|
—
|
40
|
160
|
|
E9
|
3
|
HC
|
4,00
|
40
|
1,00
|
2
|
80
|
1
|
—
|
40
|
160
|
|
E10
|
1
|
HC
|
12,00
|
40
|
3,00
|
6
|
80
|
3
|
—
|
40
|
160
|
|
E11
|
2
|
HC
|
12,00
|
40
|
3,00
|
6
|
80
|
3
|
—
|
40
|
160
|
|
E12
|
3
|
HC
|
12,00
|
40
|
3,00
|
6
|
80
|
3
|
—
|
40
|
160
|
|
G1
|
1
|
La cald
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G2
|
2
|
La cald
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G3
|
3
|
La cald
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G4
|
1
|
La cald
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G5
|
2
|
La cald
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G6
|
3
|
La cald
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G7
|
1
|
La cald
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G8
|
2
|
La cald
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G9
|
3
|
La cald
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G10
|
1
|
La cald
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G11
|
2
|
La cald
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G12
|
3
|
La cald
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H1
|
1
|
La cald
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H2
|
2
|
La cald
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H3
|
3
|
La cald
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H4
|
1
|
La cald
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H5
|
2
|
La cald
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H6
|
3
|
La cald
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H7
|
1
|
La cald
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H8
|
2
|
La cald
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H9
|
3
|
La cald
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H10
|
1
|
La cald
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H11
|
2
|
La cald
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H12
|
3
|
La cald
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
Este de precizat că godeurile „la cald” sunt goale în timpul incubației. Cantitatea de 40 μl se adaugă doar pentru numărarea scintilației.
|
Apendicele 2.3:
CONFIGURAțIA GODEURILOR ÎN ANALIZA LEGĂRII COMPETITIVE
|
Placă
|
Poziție
|
Replică
|
Tip godeu
|
Cod godeu
|
Codul concentrației
|
Concentrația inițială a soluției testate în paralel (M)
|
Stocul hrER (μl)
|
Volum soluție-tampon (μl)
|
Volum detector (E2 la cald) (μL)
|
Volum din placa de diluție (μL)
|
Volum final (μl)
|
Concentrația finală a soluției testate în paralel (M)
|
|
S
|
A1
|
1
|
legare totală
|
TB
|
TB1
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A2
|
2
|
legare totală
|
TB
|
TB2
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A3
|
3
|
legare totală
|
TB
|
TB3
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A4
|
1
|
legare totală
|
TB
|
TB4
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A5
|
2
|
legare totală
|
TB
|
TB5
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A6
|
3
|
legare totală
|
TB
|
TB6
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A7
|
1
|
E2 la rece (ridicat)
|
NSB
|
S0
|
2,00 E-06
|
40
|
—
|
40
|
80
|
160
|
1,0 E-06
|
|
S
|
A8
|
2
|
E2 la rece (ridicat)
|
NSB
|
S0
|
2,00 E-06
|
40
|
—
|
40
|
80
|
160
|
1,0 E-06
|
|
S
|
A9
|
3
|
E2 la rece (ridicat)
|
NSB
|
S0
|
2,00 E-06
|
40
|
—
|
40
|
80
|
160
|
1,0 E-06
|
|
S
|
A10
|
1
|
E2 la rece (ridicat)
|
NSB
|
S0
|
2,00 E-06
|
40
|
—
|
40
|
80
|
160
|
1,0 E-06
|
|
S
|
A11
|
2
|
E2 la rece (ridicat)
|
NSB
|
S0
|
2,00 E-06
|
40
|
—
|
40
|
80
|
160
|
1,0 E-06
|
|
S
|
A12
|
3
|
E2 la rece (ridicat)
|
NSB
|
S0
|
2,00 E-06
|
40
|
—
|
40
|
80
|
160
|
1,0 E-06
|
|
S
|
B1
|
1
|
E2 la rece
|
S
|
S1
|
2,00 E-07
|
40
|
—
|
40
|
80
|
160
|
1,0 E-07
|
|
S
|
B2
|
2
|
E2 la rece
|
S
|
S1
|
2,00 E-07
|
40
|
—
|
40
|
80
|
160
|
1,0 E-07
|
|
S
|
B3
|
3
|
E2 la rece
|
S
|
S1
|
2,00 E-07
|
40
|
—
|
40
|
80
|
160
|
1,0 E-07
|
|
S
|
B4
|
1
|
E2 la rece
|
S
|
S2
|
2,00 E-08
|
40
|
—
|
40
|
80
|
160
|
1,0 E-08
|
|
S
|
B5
|
2
|
E2 la rece
|
S
|
S2
|
2,00 E-08
|
40
|
—
|
40
|
80
|
160
|
1,0 E-08
|
|
S
|
B6
|
3
|
E2 la rece
|
S
|
S2
|
2,00 E-08
|
40
|
—
|
40
|
80
|
160
|
1,0 E-08
|
|
S
|
B7
|
1
|
E2 la rece
|
S
|
S3
|
6.00E-09
|
40
|
—
|
40
|
80
|
160
|
3,0 E-09
|
|
S
|
B8
|
2
|
E2 la rece
|
S
|
S3
|
6.00E-09
|
40
|
—
|
40
|
80
|
160
|
3,0 E-09
|
|
S
|
B9
|
3
|
E2 la rece
|
S
|
S3
|
6.00E-09
|
40
|
—
|
40
|
80
|
160
|
3,0 E-09
|
|
S
|
B10
|
1
|
E2 la rece
|
S
|
S4
|
2,00 E-09
|
40
|
—
|
40
|
80
|
160
|
1.0E-09
|
|
S
|
B11
|
2
|
E2 la rece
|
S
|
S4
|
2,00 E-09
|
40
|
—
|
40
|
80
|
160
|
1.0E-09
|
|
S
|
B12
|
3
|
E2 la rece
|
S
|
S4
|
2,00 E-09
|
40
|
—
|
40
|
80
|
160
|
1.0E-09
|
|
S
|
C1
|
1
|
E2 la rece
|
S
|
S5
|
6.00E-10
|
40
|
—
|
40
|
80
|
160
|
3,0 E-10
|
|
S
|
C2
|
2
|
E2 la rece
|
S
|
S5
|
6.00E-10
|
40
|
—
|
40
|
80
|
160
|
3,0 E-10
|
|
S
|
C3
|
3
|
E2 la rece
|
S
|
S5
|
6.00E-10
|
40
|
—
|
40
|
80
|
160
|
3,0 E-10
|
|
S
|
C4
|
1
|
E2 la rece
|
S
|
S6
|
2,00 E-10
|
40
|
—
|
40
|
80
|
160
|
1.0E-10
|
|
S
|
C5
|
2
|
E2 la rece
|
S
|
S6
|
2,00 E-10
|
40
|
—
|
40
|
80
|
160
|
1.0E-10
|
|
S
|
C6
|
3
|
E2 la rece
|
S
|
S6
|
2,00 E-10
|
40
|
—
|
40
|
80
|
160
|
1.0E-10
|
|
S
|
C7
|
1
|
E2 la rece
|
S
|
S7
|
2,00 E-11
|
40
|
—
|
40
|
80
|
160
|
1.0E-11
|
|
S
|
C8
|
2
|
E2 la rece
|
S
|
S7
|
2,00 E-11
|
40
|
—
|
40
|
80
|
160
|
1.0E-11
|
|
S
|
C9
|
3
|
E2 la rece
|
S
|
S7
|
2,00 E-11
|
40
|
—
|
40
|
80
|
160
|
1.0E-11
|
|
S
|
C10
|
1
|
soluție săracă
|
soluție săracă
|
B1
|
—
|
—
|
160
|
—
|
—
|
160
|
—
|
|
S
|
C11
|
2
|
soluție săracă
|
soluție săracă
|
B2
|
—
|
—
|
160
|
—
|
—
|
160
|
—
|
|
S
|
C12
|
3
|
soluție săracă
|
soluție săracă
|
B3
|
—
|
—
|
160
|
—
|
—
|
160
|
—
|
|
S
|
D1
|
1
|
noretinodrel
|
NE
|
WP1
|
6.00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0 E-05
|
|
S
|
D2
|
1
|
noretinodrel
|
NE
|
WP1
|
6.00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0 E-05
|
|
S
|
D3
|
1
|
noretinodrel
|
NE
|
WP1
|
6.00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0 E-05
|
|
S
|
D4
|
1
|
noretinodrel
|
NE
|
WP2
|
2,00 E-05
|
40
|
—
|
40
|
80
|
160
|
1.0E-05
|
|
S
|
D5
|
1
|
noretinodrel
|
NE
|
WP2
|
2,00 E-05
|
40
|
—
|
40
|
80
|
160
|
1.0E-05
|
|
S
|
D6
|
1
|
noretinodrel
|
NE
|
WP2
|
2,00 E-05
|
40
|
—
|
40
|
80
|
160
|
1.0E-05
|
|
S
|
D7
|
1
|
noretinodrel
|
NE
|
WP3
|
6.00E-06
|
40
|
—
|
40
|
80
|
160
|
3,0 E-06
|
|
S
|
D8
|
1
|
noretinodrel
|
NE
|
WP3
|
6.00E-06
|
40
|
—
|
40
|
80
|
160
|
3,0 E-06
|
|
S
|
D9
|
1
|
noretinodrel
|
NE
|
WP3
|
6.00E-06
|
40
|
—
|
40
|
80
|
160
|
3,0 E-06
|
|
S
|
D10
|
1
|
noretinodrel
|
NE
|
WP4
|
2,00 E-06
|
40
|
—
|
40
|
80
|
160
|
1.0E-06
|
|
S
|
D11
|
1
|
noretinodrel
|
NE
|
WP4
|
2,00 E-06
|
40
|
—
|
40
|
80
|
160
|
1.0E-06
|
|
S
|
D12
|
1
|
noretinodrel
|
NE
|
WP4
|
2,00 E-06
|
40
|
—
|
40
|
80
|
160
|
1.0E-06
|
|
S
|
E1
|
1
|
noretinodrel
|
NE
|
WP
|
6,00 E-07
|
40
|
|
40
|
80
|
160
|
3,0 E-07
|
|
S
|
E2
|
2
|
noretinodrel
|
NE
|
WP
|
6,00 E-07
|
40
|
|
40
|
80
|
160
|
3,0 E-07
|
|
S
|
E3
|
3
|
noretinodrel
|
NE
|
WP
|
6,00 E-07
|
40
|
|
40
|
80
|
160
|
3,0 E-07
|
|
S
|
E4
|
1
|
noretinodrel
|
NE
|
WP
|
2,00 E-07
|
40
|
|
40
|
80
|
160
|
1.0E-07
|
|
S
|
E5
|
2
|
noretinodrel
|
NE
|
WP
|
2,00 E-07
|
40
|
|
40
|
80
|
160
|
1.0E-07
|
|
S
|
E6
|
3
|
noretinodrel
|
NE
|
WP
|
2,00 E-07
|
40
|
|
40
|
80
|
160
|
1.0E-07
|
|
S
|
E7
|
1
|
noretinodrel
|
NE
|
WP
|
6,00 E-08
|
40
|
—
|
40
|
80
|
160
|
3,0 E-08
|
|
S
|
E8
|
2
|
noretinodrel
|
NE
|
WP
|
6,00 E-08
|
40
|
—
|
40
|
80
|
160
|
3,0 E-08
|
|
S
|
E9
|
3
|
noretinodrel
|
NE
|
WP
|
6,00 E-08
|
40
|
—
|
40
|
80
|
160
|
3,0 E-08
|
|
S
|
E10
|
1
|
noretinodrel
|
NE
|
WP
|
6,00 E-09
|
40
|
—
|
40
|
80
|
160
|
3,0 E-09
|
|
S
|
E11
|
2
|
noretinodrel
|
NE
|
WP
|
6,00 E-09
|
40
|
—
|
40
|
80
|
160
|
3,0 E-09
|
|
S
|
E12
|
3
|
noretinodrel
|
NE
|
WP
|
6,00 E-09
|
40
|
—
|
40
|
80
|
160
|
3,0 E-09
|
|
S
|
F1
|
1
|
OTES
|
N
|
OTES
|
2,00 E-03
|
40
|
—
|
40
|
80
|
160
|
1.0E-03
|
|
S
|
F2
|
2
|
OTES
|
N
|
OTES
|
2,00 E-03
|
40
|
—
|
40
|
80
|
160
|
1.0E-03
|
|
S
|
F3
|
3
|
OTES
|
N
|
OTES
|
2,00 E-03
|
40
|
—
|
40
|
80
|
160
|
1.0E-03
|
|
S
|
F4
|
1
|
OTES
|
N
|
OTES
|
2,00 E-04
|
40
|
—
|
40
|
80
|
160
|
1.0E-04
|
|
S
|
F5
|
2
|
OTES
|
N
|
OTES
|
2,00 E-04
|
40
|
—
|
40
|
80
|
160
|
1.0E-04
|
|
S
|
F6
|
3
|
OTES
|
N
|
OTES
|
2,00 E-04
|
40
|
—
|
40
|
80
|
160
|
1.0E-04
|
|
S
|
F7
|
1
|
OTES
|
N
|
OTES
|
2,00 E-05
|
40
|
—
|
40
|
80
|
160
|
3,0 E-05
|
|
S
|
F8
|
2
|
OTES
|
N
|
OTES
|
2,00 E-05
|
40
|
—
|
40
|
80
|
160
|
3,0 E-05
|
|
S
|
F9
|
3
|
OTES
|
N
|
OTES
|
2,00 E-05
|
40
|
—
|
40
|
80
|
160
|
3,0 E-05
|
|
S
|
F10
|
1
|
OTES
|
N
|
OTES
|
2,00 E-06
|
40
|
—
|
40
|
80
|
160
|
1.0E-06
|
|
S
|
F11
|
2
|
OTES
|
N
|
OTES
|
2,00 E-06
|
40
|
—
|
40
|
80
|
160
|
1.0E-06
|
|
S
|
F12
|
3
|
OTES
|
N
|
OTES
|
2,00 E-06
|
40
|
—
|
40
|
80
|
160
|
1.0E-06
|
|
S
|
G1
|
1
|
OTES
|
N
|
OTES
|
2,00 E-07
|
40
|
—
|
40
|
80
|
160
|
3,0 E-07
|
|
S
|
G2
|
2
|
OTES
|
N
|
OTES
|
2,00 E-07
|
40
|
—
|
40
|
80
|
160
|
3,0 E-07
|
|
S
|
G3
|
3
|
OTES
|
N
|
OTES
|
2,00 E-07
|
40
|
—
|
40
|
80
|
160
|
3,0 E-07
|
|
S
|
G4
|
1
|
OTES
|
N
|
OTES
|
2,00 E-08
|
40
|
—
|
40
|
80
|
160
|
1.0E-08
|
|
S
|
G5
|
2
|
OTES
|
N
|
OTES
|
2,00 E-08
|
40
|
—
|
40
|
80
|
160
|
1.0E-08
|
|
S
|
G6
|
3
|
OTES
|
N
|
OTES
|
2,00 E-08
|
40
|
—
|
40
|
80
|
160
|
1.0E-08
|
|
S
|
G7
|
1
|
OTES
|
N
|
OTES
|
2,00 E-09
|
40
|
—
|
40
|
80
|
160
|
1.0E-09
|
|
S
|
G8
|
2
|
OTES
|
N
|
OTES
|
2,00 E-09
|
40
|
—
|
40
|
80
|
160
|
1.0E-09
|
|
S
|
G9
|
3
|
OTES
|
N
|
OTES
|
2,00 E-09
|
40
|
—
|
40
|
80
|
160
|
1.0E-09
|
|
S
|
G10
|
1
|
OTES
|
N
|
OTES
|
2,00 E-10
|
40
|
—
|
40
|
—
|
160
|
1.0E-10
|
|
S
|
G11
|
2
|
OTES
|
N
|
OTES
|
2,00 E-10
|
40
|
—
|
40
|
—
|
160
|
1.0E-10
|
|
S
|
G12
|
3
|
OTES
|
N
|
OTES
|
2,00 E-10
|
40
|
—
|
40
|
—
|
160
|
1.0E-10
|
|
S
|
H1
|
1
|
la cald
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H2
|
1
|
la cald
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H3
|
1
|
la cald
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H4
|
1
|
la cald
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H5
|
1
|
la cald
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H6
|
1
|
la cald
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H7
|
1
|
martor tampon
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H8
|
1
|
martor tampon
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H9
|
1
|
martor tampon
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H10
|
1
|
martor tampon
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H11
|
1
|
martor tampon
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H12
|
1
|
martor tampon
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
Este de precizat că godeurile „la cald” sunt goale în timpul incubației. Cantitatea de 40 μl se adaugă doar pentru numărarea scintilației.
|
Configurația godeurilor în analiza legării competitive
|
|
Placă
|
Poziție
|
Replică
|
Tip godeu
|
Cod godeu
|
Cod concentrație
|
Concentrația inițială a soluției testate în paralel (M)
|
Stocul hrER (μL)
|
Volum tampon (μL)
|
Volum detector (E2 la cald) (μL)
|
Volum din placa de diluție (μL)
|
Volum final (μl)
|
Concentrația finală a soluției testate în paralel (M)
|
|
P1
|
A1
|
1
|
legare totală
|
TB
|
TBB1B1
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A2
|
2
|
legare totală
|
TB
|
TB2
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A3
|
3
|
legare totală
|
TB
|
TB3
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A4
|
1
|
legare totală
|
TB
|
TB4
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A5
|
2
|
legare totală
|
TB
|
TB5
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A6
|
3
|
legare totală
|
TB
|
TB6
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A7
|
1
|
E2 la rece (ridicat)
|
NSB
|
S0
|
2,00 E-06
|
40
|
—
|
40
|
80
|
160
|
1.0E-06
|
|
P1
|
A8
|
2
|
E2 la rece (ridicat)
|
NSB
|
S0
|
2,00 E-06
|
40
|
—
|
40
|
80
|
160
|
1.0E-06
|
|
P1
|
A9
|
3
|
E2 la rece (ridicat)
|
NSB
|
S0
|
2,00 E-06
|
40
|
—
|
40
|
80
|
160
|
1.0E-06
|
|
P1
|
A10
|
1
|
E2 la rece (ridicat)
|
NSB
|
S0
|
2,00 E-06
|
40
|
—
|
40
|
80
|
160
|
1.0E-06
|
|
P1
|
A11
|
2
|
E2 la rece (ridicat)
|
NSB
|
S0
|
2,00 E-06
|
40
|
—
|
40
|
80
|
160
|
1.0E-06
|
|
P1
|
A12
|
3
|
E2 la rece (ridicat)
|
NSB
|
S0
|
2,00 E-06
|
40
|
—
|
40
|
80
|
160
|
1.0E-06
|
|
P1
|
B1
|
1
|
Substanța chimică de testat 1
|
TC1
|
1
|
2,00 E-03
|
40
|
0
|
40
|
80
|
160
|
1.0E-03
|
|
P1
|
B2
|
2
|
Substanța chimică de testat 1
|
TC1
|
1
|
2,00 E-03
|
40
|
0
|
40
|
80
|
160
|
1.0E-03
|
|
P1
|
B3
|
3
|
Substanța chimică de testat 1
|
TC1
|
1
|
2,00 E-03
|
40
|
0
|
40
|
80
|
160
|
1.0E-03
|
|
P1
|
B4
|
1
|
Substanța chimică de testat 1
|
TC1
|
2
|
2,00 E-04
|
40
|
0
|
40
|
80
|
160
|
1.0E-04
|
|
P1
|
B5
|
2
|
Substanța chimică de testat 1
|
TC1
|
2
|
2,00 E-04
|
40
|
0
|
40
|
80
|
160
|
1.0E-04
|
|
P1
|
B6
|
3
|
Substanța chimică de testat 1
|
TC1
|
2
|
2,00 E-04
|
40
|
0
|
40
|
80
|
160
|
1.0E-04
|
|
P1
|
B7
|
1
|
Substanța chimică de testat 1
|
TC1
|
3
|
2,00 E-05
|
40
|
0
|
40
|
80
|
160
|
1.0E-05
|
|
P1
|
B8
|
2
|
Substanța chimică de testat 1
|
TC1
|
3
|
2,00 E-05
|
40
|
0
|
40
|
80
|
160
|
1.0E-05
|
|
P1
|
B9
|
3
|
Substanța chimică de testat 1
|
TC1
|
3
|
2,00 E-05
|
40
|
0
|
40
|
80
|
160
|
1.0E-05
|
|
P1
|
B10
|
1
|
Substanța chimică de testat 1
|
TC1
|
4
|
2,00 E-06
|
40
|
0
|
40
|
80
|
160
|
1.0E-06
|
|
P1
|
B11
|
2
|
Substanța chimică de testat 1
|
TC1
|
4
|
2,00 E-06
|
40
|
0
|
40
|
80
|
160
|
1.0E-06
|
|
P1
|
B12
|
3
|
Substanța chimică de testat 1
|
TC1
|
4
|
2,00 E-06
|
40
|
0
|
40
|
80
|
160
|
1.0E-06
|
|
P1
|
C1
|
1
|
Substanța chimică de testat 1
|
TC1
|
5
|
2,00 E-07
|
40
|
0
|
40
|
80
|
160
|
1.0E-07
|
|
P1
|
C2
|
2
|
Substanța chimică de testat 1
|
TC1
|
5
|
2,00 E-07
|
40
|
0
|
40
|
80
|
160
|
1.0E-07
|
|
P1
|
C3
|
3
|
Substanța chimică de testat 1
|
TC1
|
5
|
2,00 E-07
|
40
|
0
|
40
|
80
|
160
|
1.0E-07
|
|
P1
|
C4
|
1
|
Substanța chimică de testat 1
|
TC1
|
6
|
2,00 E-08
|
40
|
0
|
40
|
80
|
160
|
1.0E-08
|
|
P1
|
C5
|
2
|
Substanța chimică de testat 1
|
TC1
|
6
|
2,00 E-08
|
40
|
0
|
40
|
80
|
160
|
1.0E-08
|
|
P1
|
C6
|
3
|
Substanța chimică de testat 1
|
TC1
|
6
|
2,00 E-08
|
40
|
0
|
40
|
80
|
160
|
1.0E-08
|
|
P1
|
C7
|
1
|
Substanța chimică de testat 1
|
TC1
|
7
|
2,00 E-09
|
40
|
0
|
40
|
80
|
160
|
1.0E-09
|
|
P1
|
C8
|
2
|
Substanța chimică de testat 1
|
TC1
|
7
|
2,00 E-09
|
40
|
0
|
40
|
80
|
160
|
1.0E-09
|
|
P1
|
C9
|
3
|
Substanța chimică de testat 1
|
TC1
|
7
|
2,00 E-09
|
40
|
0
|
40
|
80
|
160
|
1.0E-09
|
|
P1
|
C10
|
1
|
Substanța chimică de testat 1
|
TC1
|
8
|
2,00 E-10
|
40
|
0
|
40
|
80
|
160
|
1.0E-10
|
|
P1
|
C11
|
2
|
Substanța chimică de testat 1
|
TC1
|
8
|
2,00 E-10
|
40
|
0
|
40
|
80
|
160
|
1.0E-10
|
|
P1
|
C12
|
3
|
Substanța chimică de testat 1
|
TC1
|
8
|
2,00 E-10
|
40
|
0
|
40
|
80
|
160
|
1.0E-10
|
|
P1
|
D1
|
1
|
Substanța chimică de testat 2
|
TC2
|
1
|
2,00 E-03
|
40
|
0
|
40
|
80
|
160
|
1.0E-03
|
|
P1
|
D2
|
2
|
Substanța chimică de testat 2
|
TC2
|
1
|
2,00 E-03
|
40
|
0
|
40
|
80
|
160
|
1.0E-03
|
|
P1
|
D3
|
3
|
Substanța chimică de testat 2
|
TC2
|
1
|
2,00 E-03
|
40
|
0
|
40
|
80
|
160
|
1.0E-03
|
|
P1
|
D4
|
1
|
Substanța chimică de testat 2
|
TC2
|
2
|
2,00 E-04
|
40
|
0
|
40
|
80
|
160
|
1.0E-04
|
|
P1
|
D5
|
2
|
Substanța chimică de testat 2
|
TC2
|
2
|
2,00 E-04
|
40
|
0
|
40
|
80
|
160
|
1.0E-04
|
|
P1
|
D6
|
3
|
Substanța chimică de testat 2
|
TC2
|
2
|
2,00 E-04
|
40
|
0
|
40
|
80
|
160
|
1.0E-04
|
|
P1
|
D7
|
1
|
Substanța chimică de testat 2
|
TC2
|
3
|
2,00 E-05
|
40
|
0
|
40
|
80
|
160
|
1.0E-05
|
|
P1
|
D8
|
2
|
Substanța chimică de testat 2
|
TC2
|
3
|
2,00 E-05
|
40
|
0
|
40
|
80
|
160
|
1.0E-05
|
|
P1
|
D9
|
3
|
Substanța chimică de testat 2
|
TC2
|
3
|
2,00 E-05
|
40
|
0
|
40
|
80
|
160
|
1.0E-05
|
|
P1
|
D10
|
1
|
Substanța chimică de testat 2
|
TC2
|
4
|
2,00 E-06
|
40
|
0
|
40
|
80
|
160
|
1.0E-06
|
|
P1
|
D11
|
2
|
Substanța chimică de testat 2
|
TC2
|
4
|
2,00 E-06
|
40
|
0
|
40
|
80
|
160
|
1.0E-06
|
|
P1
|
D12
|
3
|
Substanța chimică de testat 2
|
TC2
|
4
|
2,00 E-06
|
40
|
0
|
40
|
80
|
160
|
1.0E-06
|
|
P1
|
E1
|
1
|
Substanța chimică de testat 2
|
TC2
|
5
|
2,00 E-07
|
40
|
0
|
40
|
80
|
160
|
1.0E-07
|
|
P1
|
E2
|
2
|
Substanța chimică de testat 2
|
TC2
|
5
|
2,00 E-07
|
40
|
0
|
40
|
80
|
160
|
1.0E-07
|
|
P1
|
E3
|
3
|
Substanța chimică de testat 2
|
TC2
|
5
|
2,00 E-07
|
40
|
0
|
40
|
80
|
160
|
1.0E-07
|
|
P1
|
E4
|
1
|
Substanța chimică de testat 2
|
TC2
|
6
|
—
|
40
|
0
|
40
|
80
|
160
|
1.0E-08
|
|
P1
|
E5
|
2
|
Substanța chimică de testat 2
|
TC2
|
6
|
—
|
40
|
0
|
40
|
80
|
160
|
1.0E-08
|
|
P1
|
E6
|
3
|
Substanța chimică de testat 2
|
TC2
|
6
|
—
|
40
|
0
|
40
|
80
|
160
|
1.0E-08
|
|
P1
|
E7
|
1
|
Substanța chimică de testat 2
|
TC2
|
7
|
2,00 E-06
|
40
|
0
|
40
|
80
|
160
|
1.0E-09
|
|
P1
|
E8
|
2
|
Substanța chimică de testat 2
|
TC2
|
7
|
2,00 E-06
|
40
|
0
|
40
|
80
|
160
|
1.0E-09
|
|
P1
|
E9
|
3
|
Substanța chimică de testat 2
|
TC2
|
7
|
2,00 E-06
|
40
|
0
|
40
|
80
|
160
|
1.0E-09
|
|
P1
|
E10
|
1
|
Substanța chimică de testat 2
|
TC2
|
8
|
2,00 E-06
|
40
|
0
|
40
|
80
|
160
|
1.0E-10
|
|
P1
|
E11
|
2
|
Substanța chimică de testat 2
|
TC2
|
8
|
2,00 E-06
|
40
|
0
|
40
|
80
|
160
|
1.0E-10
|
|
P1
|
E12
|
3
|
Substanța chimică de testat 2
|
TC2
|
8
|
2,00 E-06
|
40
|
0
|
40
|
80
|
160
|
1.0E-10
|
|
P1
|
F1
|
1
|
Substanța chimică de testat 3
|
TC3
|
1
|
2,00 E-03
|
40
|
0
|
40
|
80
|
160
|
1.0E-03
|
|
P1
|
F2
|
2
|
Substanța chimică de testat 3
|
TC3
|
1
|
2,00 E-03
|
40
|
0
|
40
|
80
|
160
|
1.0E-03
|
|
P1
|
F3
|
3
|
Substanța chimică de testat 3
|
TC3
|
1
|
2,00 E-03
|
40
|
0
|
40
|
80
|
160
|
1.0E-03
|
|
P1
|
F4
|
1
|
Substanța chimică de testat 3
|
TC3
|
2
|
2,00 E-04
|
40
|
0
|
40
|
80
|
160
|
1.0E-04
|
|
P1
|
F5
|
2
|
Substanța chimică de testat 3
|
TC3
|
2
|
2,00 E-04
|
40
|
0
|
40
|
80
|
160
|
1.0E-04
|
|
P1
|
F6
|
3
|
Substanța chimică de testat 3
|
TC3
|
2
|
2,00 E-04
|
40
|
0
|
40
|
80
|
160
|
1.0E-04
|
|
P1
|
F7
|
1
|
Substanța chimică de testat 3
|
TC3
|
3
|
2,00 E-05
|
40
|
0
|
40
|
80
|
160
|
1.0E-05
|
|
P1
|
F8
|
2
|
Substanța chimică de testat 3
|
TC3
|
3
|
2,00 E-05
|
40
|
0
|
40
|
80
|
160
|
1.0E-05
|
|
P1
|
F9
|
3
|
Substanța chimică de testat 3
|
TC3
|
3
|
2,00 E-05
|
40
|
0
|
40
|
80
|
160
|
1.0E-05
|
|
P1
|
F10
|
1
|
Substanța chimică de testat 3
|
TC3
|
4
|
2,00 E-06
|
40
|
0
|
40
|
80
|
160
|
1.0E-06
|
|
P1
|
F11
|
2
|
Substanța chimică de testat 3
|
TC3
|
4
|
2,00 E-06
|
40
|
0
|
40
|
80
|
160
|
1.0E-06
|
|
P1
|
F12
|
3
|
Substanța chimică de testat 3
|
TC3
|
4
|
2,00 E-06
|
40
|
0
|
40
|
80
|
160
|
1.0E-06
|
|
P1
|
G1
|
1
|
Substanța chimică de testat 3
|
TC3
|
5
|
2,00 E-07
|
40
|
0
|
40
|
80
|
160
|
1.0E-07
|
|
P1
|
G2
|
2
|
Substanța chimică de testat 3
|
TC3
|
5
|
2,00 E-07
|
40
|
0
|
40
|
80
|
160
|
1.0E-07
|
|
P1
|
G3
|
3
|
Substanța chimică de testat 3
|
TC3
|
5
|
2,00 E-07
|
40
|
0
|
40
|
80
|
160
|
1.0E-07
|
|
P1
|
G4
|
1
|
Substanța chimică de testat 3
|
TC3
|
6
|
2,00 E-08
|
40
|
0
|
40
|
80
|
160
|
1.0E-08
|
|
P1
|
G5
|
2
|
Substanța chimică de testat 3
|
TC3
|
6
|
2,00 E-08
|
40
|
0
|
40
|
80
|
160
|
1.0E-08
|
|
P1
|
G6
|
3
|
Substanța chimică de testat 3
|
TC3
|
6
|
2,00 E-08
|
40
|
0
|
40
|
80
|
160
|
1.0E-08
|
|
P1
|
G7
|
1
|
Substanța chimică de testat 3
|
TC3
|
7
|
2,00 E-09
|
40
|
0
|
40
|
80
|
160
|
1.0E-09
|
|
P1
|
G8
|
2
|
Substanța chimică de testat 3
|
TC3
|
7
|
2,00 E-09
|
40
|
0
|
40
|
80
|
160
|
1.0E-09
|
|
P1
|
G9
|
3
|
Substanța chimică de testat 3
|
TC3
|
7
|
2,00 E-09
|
40
|
0
|
40
|
80
|
160
|
1.0E-09
|
|
P1
|
G10
|
1
|
Substanța chimică de testat 3
|
TC3
|
8
|
2,00 E-10
|
40
|
0
|
40
|
80
|
160
|
1.0E-10
|
|
P1
|
G11
|
2
|
Substanța chimică de testat 3
|
TC3
|
8
|
2,00 E-10
|
40
|
0
|
40
|
80
|
160
|
1.0E-10
|
|
P1
|
G12
|
3
|
Substanța chimică de testat 3
|
TC3
|
8
|
2,00 E-10
|
40
|
0
|
40
|
80
|
160
|
1.0E-10
|
|
P1
|
H1
|
1
|
noretinodrel
|
NE
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H2
|
2
|
noretinodrel
|
NE
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H3
|
3
|
noretinodrel
|
NE
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H4
|
1
|
noretinodrel
|
NE
|
|
1.00E-4,5
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H5
|
2
|
noretinodrel
|
NE
|
|
1.00E-4,5
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H6
|
3
|
noretinodrel
|
NE
|
|
1.00E-4,5
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H7
|
1
|
E2 la rece S
|
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H8
|
2
|
E2 la rece S
|
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H9
|
3
|
E2 la rece S
|
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H10
|
1
|
E2 la rece S
|
|
|
1,00 E-7
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H11
|
2
|
E2 la rece S
|
|
|
1,00 E-7
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H12
|
3
|
E2 la rece S
|
|
|
1,00 E-7
|
40
|
0
|
40
|
80
|
160
|
|
Apendicele 3
ANALIZA LEGĂRII RECEPTORULUI DE ESTROGEN IN VITRO A INSTITUTULUI DE EVALUARE ȘI CERCETARE ÎN CHIMIE (CHEMICAL EVALUATION AND RESEARCH INSTITUTE - CERI) CARE UTILIZEAZĂ O PROTEINĂ DIN DOMENIUL DE LEGARE PRIN LIGAND RECOMBINANT UMAN
CONSIDERAȚII ȘI LIMITĂRI INIȚIALE (A SE VEDEA, DE ASEMENEA, INTRODUCERE GENERALĂ)
|
1.
|
Această analiză a legării prin saturație și competitive a receptorului de estrogen ERα in vitro utilizează un domeniu de legare a ligandului (LBD) al unui ERα uman (hrERα). Această structură proteică a fost realizată de Institutul de cercetare pentru evaluarea substanțelor chimice (Chemicals Evaluation Research Institute - CERI) din Japonia și există sub formă de proteină de fuziune glutation-S-transferază (GST) și este exprimată în E. coli. Protocolul CERI a fost supus unui studiu internațional de validare în mai multe laboratoare (2) care a demonstrat relevanța și fiabilitatea acestuia în scopul prevăzut al analizei.
|
|
2.
|
Această analiză este o procedură de depistare pentru identificarea substanțelor care pot lega hrERα. Aceasta este utilizată pentru a stabili capacitatea unei substanțe chimice de testat de a concura cu 17β-estradiol pentru legarea de hrERα-LBD. Rezultatele cantitative ale analizei pot include valoarea IC50 (un indicator al concentrației unei substanțe chimice de testat care este necesară pentru a desprinde jumătate din [3H]-17β-estradiol de hrERα) și afinitățile de legare relative ale substanțelor chimice de testat pentru hrERα comparativ cu 17β-estradiol. În scopul depistării substanțelor chimice, rezultatele acceptabile ale analizei calitative pot include clasificări ale substanțelor chimice de testat ca lianți ai hrERα, non-lianți sau substanțe echivoce pe baza criteriilor descrise pentru curbele pentru legare.
|
|
3.
|
Analiza folosește un ligand radioactiv care necesită obținerea de către laborator a unei licențe pentru materiale radioactive. Toate procedurile cu radioizotopi și substanțe chimice periculoase ar trebui să respecte regulamentele și procedurile descrise în legislația națională.
|
|
4.
|
Ar trebui să se parcurgă secțiunile „INTRODUCERE GENERALĂ” și „COMPONENTE ALE ANALIZEI DE LEGARE DE hrER” înainte de utilizarea acestei analize în scopuri de reglementare. Definițiile și abrevierile utilizate în prezenta orientare privind testarea sunt descrise în apendicele 1.
|
PRINCIPIILE TESTULUI (A SE VEDEA, DE ASEMENEA, INTRODUCERE GENERALĂ)
|
5.
|
Analiza legării hrERα măsoară capacitatea unui ligand marcat radioactiv ([3H]17-β-estradiol) de a se lega de ER în prezența concentrațiilor din ce în ce mai mari ale unei substanțe chimice de testat (și anume, concurent). Substanțele chimice de testat care prezintă o afinitate mare pentru ER concurează cu ligandul marcat radioactiv la o concentrație mai mică în comparație cu substanțele chimice care prezintă o afinitate mai redusă pentru receptor.
|
|
6.
|
Această analiză cuprinde două componente principale: un experiment de legare prin saturație pentru a caracteriza parametrii de interacțiune receptor-ligand, urmat de un experiment de legare competitiv care caracterizează concurența dintre o substanță chimică de testat și un ligand marcat radioactiv pentru legarea de ER.
|
|
7.
|
Scopul experimentului de legare prin saturație este de a caracteriza un anumit lot de receptori în ceea ce privește afinitatea de legare și numărul în pregătirea pentru experimentul de legare competitivă. Experimentul de legare prin saturație măsoară, în condiții de echilibru, afinitatea unei concentrații fixe a receptorului de estrogen pentru ligandul său natural (reprezentată de constanta de disociere, Kd) și concentrația locurilor cu receptor activ (Bmax).
|
|
8.
|
Experimentul de legare competitivă măsoară afinitatea unei substanțe de a concura cu [3H]17β-estradiol pentru legarea de ER. Afinitatea este cuantificată prin concentrația substanței chimice de testat care, în stare de echilibru, inhibă 50 % din legarea specifică a [3H]17β-estradiol (denumită „concentrație inhibitoare de 50 %” sau IC50). Aceasta poate fi evaluată, de asemenea, utilizând afinitatea de legare relativă (RBA, în raport cu valoarea IC50 a estradiolului, măsurată separat în cadrul aceluiași test). Experimentul de legare competitivă măsoară legarea [3H]17β-estradiol la o concentrație fixă în prezența unui interval mare (opt ordine de mărime) de concentrații ale substanței chimice de testat. Datele sunt apoi corelate, dacă este posibil, într-o formă de ecuație Hill (Hill, 1910) care descrie desprinderea ligandului radioactiv de către un liant competitiv dintr-un loc. Gradul de desprindere a estradiolului marcat radioactiv, în stare de echilibru, este folosit pentru a caracteriza substanța chimică de testat ca liant, non-liant sau ca o substanță care generează un răspuns echivoc.
|
PROCEDURA
Demonstrarea performanței acceptabile a proteinei hrERα
|
9.
|
Înainte de efectuarea sistematică a analizelor de legare prin saturație și de legare competitivă, ar trebui să se demonstreze performanța corespunzătoare a fiecărui nou lot de hrERα în laboratorul în care acesta va fi utilizat. Pentru a demonstra performanța ar trebui să se utilizeze un proces în două etape. Aceste etape sunt următoarele:
|
—
|
Se desfășoară o analiză de legare a [3H]-17β-estradiol prin saturație pentru a demonstra specificitatea și saturația hrERα. Analiza regresiei non-liniare a acestor date (de exemplu, BioSoft; McPherson, 1985; Motulsky, 1995) și schița Scatchard ulterioară ar trebui să documenteze afinitățile de legare de hrERα ale [3H]-17β-estradiol (Kd) și numărul receptorilor (Bmax) pentru un anumit lot de hrERα.
|
|
—
|
Se desfășoară o analiză de legare competitivă cu ajutorul substanțelor martor [estrogenul de referință (17β-estradiol), un liant slab (de exemplu, noretinodrel sau noredronă) și un non-liant (octiltrietoxisilan, OTES). Fiecare laborator ar trebui să stabilească o bază de date istorică pentru a documenta consecvența IC50 și valorile relevante pentru estrogenul de referință și liantul slab obținute în urma experimentelor și pentru diferite loturi de hrERα. În plus, parametrii curbelor privind legarea competitivă pentru substanțele martor ar trebui să se încadreze în limitele intervalului de încredere de 95 % (a se vedea tabelul 1), acestea fiind dezvoltate pe baza datelor din laboratoarele care au participat la studiul de validare pentru această analiză (2).
|
Tabelul 1
Criterii de performanță elaborate pentru estrogenul de referință și liantul slab, analiza CERI privind legarea hrER.
|
Substanță
|
Parametru
|
Media (10)
|
Abaterea standard (n)
|
Interval de încredere de 95 % (11)
|
|
Limita inferioară
|
Limita superioară
|
|
17β-estradiol
|
Partea superioară
|
104,74
|
13,12 (70)
|
101,6
|
107,9
|
|
Partea inferioară
|
0,85
|
2,41 (70)
|
0,28
|
1,43
|
|
HillSlope
|
-1,22
|
0,20 (70)
|
-1,27
|
-1,17
|
|
LogIC50
|
-8,93
|
0,23 (70)
|
-8,98
|
-8,87
|
|
Noretinodrel
|
Partea superioară
|
101,31
|
10,55 (68)
|
98,76
|
103,90
|
|
Partea inferioară
|
2,39
|
5,01 (68)
|
1,18
|
3,60
|
|
HillSlope
|
-1,04
|
0,21 (68)
|
-1,09
|
-0,99
|
|
LogIC50
|
-6,19
|
0,40 (68)
|
-6,29
|
-6,10
|
|
Noretindronă (12)
|
Partea superioară
|
92,27
|
7,79 (23)
|
88,90
|
95,63
|
|
Partea inferioară
|
16,52
|
10,59 (23)
|
11,94
|
21,10
|
|
Panta Hill
|
-1,18
|
0,32 (23)
|
-1,31
|
-1,04
|
|
LogIC50
|
-6,01
|
0,54 (23)
|
-6,25
|
-5,78
|
|
Demonstrarea competenței laboratorului
|
10.
|
A se vedea punctele 17 și 18, precum și tabelul 2 din „COMPONENTE ALE ANALIZEI PRIVIND LEGAREA DE hrER” ale acestei metode de testare. Fiecare analiză (legare prin analiză și legare competitivă) ar trebui să conste în trei testări independente (și anume, cu diluții proaspete de receptor, substanțe chimice și reactivi) în zile diferite și fiecare testare ar trebui să cuprindă trei replici.
|
Determinarea concentrației receptorului (hrERα)
|
11.
|
Concentrația receptorului activ variază ușor în funcție de lot și de condițiile de păstrare. Din acest motiv, ar trebui să se determine concentrația receptorului activ, astfel cum a fost primită de la furnizor. Aceasta va determina concentrația corespunzătoare a receptorului activ la momentul testării.
|
|
12.
|
În condiții corespunzătoare legării competitive (și anume, 0,5 nM [3H]-estradiol), ar trebui să se incubeze concentrații nominale de 0,1, 0,2, 0,4 și 0,6 nM ale receptorului în absența (legare totală) și prezența (legare nespecifică) unei cantități de 1 μM estradiol neetichetat. Legarea specifică, calculată ca diferență între legarea totală și legarea nespecifică, este schițată în comparație cu concentrația nominală a receptorului. Concentrația receptorului, care determină valorile specifice de legare, reprezentând 40 % din marcajul radioactiv adăugat, este corelată cu concentrația receptorului și această concentrație a receptorului ar trebui utilizate pentru experimente de legare prin saturație și legare competitivă. În mod frecvent, o concentrație hrER finală de 0,2 nM va respecta această condiție.
|
|
13.
|
În cazul în care criteriul de 40 % nu poate fi respectat în mod repetat, ar trebui verificat setul experimental pentru identificarea unor posibile erori. Nerespectarea criteriului de 40 % poate indica faptul că există foarte puțini receptori activi în lotul recombinant și ar trebui atunci să se aibă în vedere utilizarea altui lot de receptori.
|
Analiza saturației
|
14.
|
Ar trebui să se evalueze opt concentrații din ce în ce mai mari de [3H]17β-estradiol în trei replici, în următoarele trei condiții (a se vedea tabelul 2):
|
a.
|
în absența substanței 17β-estradiol nemarcate și în prezența ER. Astfel se determină legarea totală prin măsurarea radioactivității în godeurile care au doar [3H]17β-estradiol.
|
|
b.
|
În prezența unei concentrații multiplicate de 2000 de ori de 17β-estradiol nemarcat peste 17β-estradiol marcat și în prezența ER. Intenția acestei condiții este de a satura locurile cu legare activă cu 17β-estradiol nemarcat și, prin măsurarea radioactivității în godeuri, de a determina legarea nespecifică. Orice cantitate de estradiol rămasă la cald, care se poate lega de receptor, se consideră că se leagă într-un loc nespecific, întrucât cantitatea de estradiol rece ar trebui să fie într-o concentrație atât de mare încât să se lege de toate locurile specifice disponibile de pe receptor.
|
|
c.
|
În absența substanței 17β-estradiol nemarcate și în absența ER (determinarea radioactivității totale)
|
Preparatul de [3H]-17β-estradiol, soluții 17β-estradiol nemarcate și hrERα
|
15.
|
Ar trebui să se prepare o soluție 40 nM de [3H] -17β-estradiol dintr-o soluție mamă de 1 μM de [3H]-17β-estradiol în DMSO, prin adăugarea DMSO (pentru prepararea a 200 nM) și soluție-tampon de analiză la temperatura camerei (pentru a prepara 40 nM). Folosind această soluție de 40 nM, se prepară seria de diluții [3H]-17β-estradiol, variind între 0,313 nM și 40 nM cu soluție-tampon de analiză la temperatura camerei (astfel cum a fost reprezentat pe rândul 12 din tabelul 2). Concentrațiile finale de analiză, cuprinse între 0,0313 și 4,0 nM, se vor obține prin adăugarea a 10 μl din aceste soluții la godeurile de analiză respective de pe o placă de microtitrare cu 96 de godeuri (a se vedea tabelele 2 și 3). Prepararea soluției tampon de analiză, calcularea soluției mamă inițiale de [3H]-17β-estradiol pe baza activității sale specifice, prepararea diluțiilor și determinarea concentrațiilor sunt descrise în detaliu în protocolul CERI (2).
|
|
16.
|
Diluțiile soluțiilor cu 17β-estradiol nemarcate ar trebui să se prepare dintr-o soluție mamă de 1 μM 17β-estradiol prin adăugarea de soluție-tampon de analiză pentru a obține opt concentrații în creștere, cu valori inițiale de la 0,625 μΜ la 80 μΜ. Concentrațiile finale de analiză, cuprinse între 0,0625 și 8 μM, se vor obține prin adăugarea a 10 μl din aceste soluții la godeurile de analiză respective de pe o placă de microtitrare cu 96 de godeuri prevăzută pentru măsurarea legării nespecifice (a se vedea tabelele 2 și 3). Prepararea diluțiilor 17β-estradiol nemarcate este descrisă în detaliu în protocolul CERI (2).
|
|
17.
|
Ar trebui să fie utilizată concentrația receptorului care prezintă o legare specifică de 40 ± 10 % (a se vedea punctele 12-13). Soluția hrERα ar trebui să fie preparată cu soluție-tampon de analiză foarte rece, imediat înainte de utilizare, și anume, după ce au fost preparate toate godeurile pentru legare totală, legare nespecifică și doar ligandul la cald.
|
|
18.
|
Plăcile de microtitrare cu 96 de godeuri sunt pregătite astfel cum este ilustrat în tabelul 2, cu trei replici per concentrație de [3H]-17β-estradiol. Distribuirea volumului de [3H]-17β-estradiol, de 17β-estradiol nemarcat, de soluție tampon și de receptor este prezentată în tabelul 3.
Tabelul 2
Distribuția pe placa de microtitrare în cadrul analizei privind legarea prin saturație
|
|
1 (*10)
|
2 (*10)
|
3 (*10)
|
4 (*10)
|
5 (*10)
|
6 (*10)
|
7 (*10)
|
8 (*10)
|
9 (*10)
|
10
|
11 (*11)
|
12 (*11)
|
|
Pentru măsurarea TB
|
Pentru măsurarea NSB
|
Doar pentru determinarea ligandului la cald
|
|
Diluții E2 nemarcate pentru coloanele 4-6 de pe placă
|
[3H] Diluții E2 pentru coloanele 1-9 de pe placă
|
|
A
|
0,0313 nM [ 3H] E2+ ER
|
0,0313 nM [3H] E2+ 0,0625 μM E2+ ER
|
0,0313 nM
|
|
0,625 μM
|
0,313 nM
|
|
B
|
0,0625 nM [3H] E2+ ER
|
0,0625 nM [3H] E2+ 0,125 μM E2+ ER
|
0,0625 nM
|
|
1,25 μM
|
0,625 nM
|
|
C
|
0,125 nM [3H] E2+ ER
|
0,125 nM [3H] E2+ 0,25 μM E2+ ER
|
0,125 nM
|
|
2,5 μM
|
1,25 nM
|
|
D
|
0,250 nM [3H] E2+ ER
|
0,250 nM [3H] E2+ 0,5 μM E2+ ER
|
0,250 nM
|
|
5 μM
|
2,5 nM
|
|
E
|
0,50 nM [ H] E2+ ER
|
0,50 nM [3H] E2+ 1 μM E2+ ER
|
0,50 nM
|
|
10 μM
|
5 nM
|
|
F
|
1,00 nM [3H] E2+ ER
|
1,00 nM [3H] E2+ 2 μM E2+ ER
|
1,00 nM
|
|
20 μM
|
10 nM
|
|
G
|
2,00 nM [3H] E2+ ER
|
2,00 nM [3H] E2+ 4 μM E2+ ER
|
2,00 nM
|
|
40 μM
|
20 nM
|
|
H
|
4,00 nM [3H] E2+ ER
|
4,00 nM [3H] E2+ 8 μM E2+ ER
|
4,00 nM
|
|
80 μM
|
40 nM
|
|
TB
|
:
|
legare totală,
|
|
NSB
|
:
|
legare nespecifică
|
|
[3H] E2
|
:
|
[3H]17β-estradiol
|
|
E2
|
:
|
17β-estradiol nemarcat
|
|
Tabelul 3
Volume ale reactivilor pentru placa de microtitrare cu saturație
|
Număr rând
|
1
|
2
|
3
|
4
|
5
|
6
|
7 (*12)
|
8 (*12)
|
9 (*12)
|
|
Etape de preparare
|
Godeuri TB
|
Godeuri NSB
|
Doar ligand la cald
|
|
Volumul componentelor pentru godeurile de reacție de mai sus și ordinea adăugării
|
Soluție-tampon
|
60 μl
|
50 μl
|
90 μl
|
|
E2 nemarcat din rândul 11 din tabelul 2
|
—
|
10 μl
|
—
|
|
[3H]E2 de pe rândul 12 din tabelul 2
|
10 μl
|
10 μl
|
10 μl
|
|
hrERα
|
30 μl
|
30 μl
|
—
|
|
Volumul total al reacției
|
100 μl
|
100 μl
|
100 μl
|
|
Incubație
|
REACȚIE DUPĂ INCUBAȚIE DE DOUĂ ORE
|
Cuantificarea radioactivității imediat după preparare. Fără incubație
|
|
Tratament cu 0,4 % DCC
|
Da
|
Da
|
Nr.
|
|
Volumul DCC 0,4 %
|
100 μl
|
100 μl
|
—
|
|
Filtrare
|
Da
|
Da
|
Nr.
|
|
MĂSURAREA DPMS
|
|
Volumul de cuantificare adăugat la cocteilul de scintilație
|
100 μl (*13)
|
100 μl (*13)
|
50 μl
|
|
|
19.
|
Plăcile de microtitrare pentru analiză pentru determinarea legării totale și a legării nespecifice ar trebui să fie incubate la temperatura camerei (22 °C-28 °C) timp de două ore.
|
|
Măsurarea [3H]-17β-estradiol legat de hrERα
|
20.
|
După perioada de incubație de două ore, [3H]-17β-estradiol legat de hrERα ar trebui să fie separat de [3H]-17β-estradiol liber prin adăugarea a 100 μl de suspensie DCC 0,4 % rece ca gheața în godeuri. Plăcile ar trebui apoi să fie plasate pe gheață timp de 10 minute, iar amestecul de reacție și suspensia DCC ar trebui să fie filtrate, prin transferarea într-un filtru al plăcii de microtitrare, pentru eliminarea DCC. Apoi ar trebui să se adauge 100 μl de soluție filtrată în lichidul de scintilație din fiole cu LSC pentru determinarea dezintegrării pe minut (dpms) pe fiolă prin numărare în scintilație lichidă.
|
|
21.
|
Alternativ, dacă nu este disponibilă un filtru pe microplacă, suspensia DCC poate fi îndepărtată prin centrifugare. Apoi ar trebui să se extragă cu multă atenție o cantitate de 50 μl de supernatant care conține [3H]-17β-estradiol legat de hrERα pentru a evita orice contaminare a godeurilor prin atingerea DCC, și care ar trebui să fie utilizată pentru numărare în scintilație.
|
|
22.
|
Condiția de unic ligand cald se utilizează pentru determinarea dezintegrării pe minut (dpm) a [3H]-17β-estradiol adăugat în godeurile de analiză. Radioactivitatea ar trebui să fie cuantificată imediat după preparare. Aceste godeuri nu ar trebui să fie incubate și nu ar trebui să fie tratate cu suspensie DCC, dar conținutul lor ar trebui să fie golit direct în lichidul de scintilație. Aceste măsuri demonstrează cât de mult [3H]-17β-estradiol, în DPMS, a fost adăugat în fiecare set de godeuri pentru legarea totală și legarea nespecifică.
|
Analiza privind legarea competitivă
|
23.
|
Analiza privind legarea competitivă măsoară gradul de legare al unei singure concentrații de [3H]-17β-estradiol în prezența unor concentrații în creștere ale unei substanțe chimice de testat. La fiecare concentrație din cadrul unei testări ar trebui să se utilizeze trei replici concomitent. În plus, pentru fiecare substanță chimică de testat ar trebui să se efectueze trei teste la momente diferite. Substanțele pentru analiză ar trebui să fie configurate pe una sau mai multe plăci de microtitrare cu 96 de godeuri.
|
Martori
|
24.
|
Atunci când se efectuează analiza, ar trebui să se includă în fiecare experiment solventul și martorii tratați în paralel (și anume, estrogenul de referință, liantul slab și non-liantul). Pe fiecare placă, în timpul fiecărei testări, ar trebui să se utilizeze curbe complete de concentrații pentru estrogenul de referință și martori (și anume, liant slab și non-liant). Toate celelalte plăci ar trebui să conțină (i) E2 și un liant slab în trei replici la o concentrație ridicată (desprindere maximă, și anume, desprindere aproximativ integrală a ligandului marcat radioactiv) și medie (aproximativ IC50); (ii) martor tratat cu solvent și legare nespecifică, fiecare în trei replici. Procedurile pentru prepararea soluției-tampon de analiză, [3H]-17β-estradiol, hrERα și a soluțiilor cu substanță chimică de testat sunt descrise detaliat în protocolul CERI (2).
|
Martor tratat cu solvent:
|
25.
|
Martorul tratat cu solvent indică faptul că solventul nu interacționează cu sistemul de testare și, de asemenea, măsoară legarea totală (TB). Solventul preferat este DMSO. În mod alternativ, în cazul în care cea mai mare concentrație a substanței chimice de testat nu este solubilă în DMSO, poate fi utilizat etanol. Concentrația DMSO în godeurile de analiză finale ar trebui să fie de 2,05 % și ar putea fi mărită până la 2,5 % în cazul în care substanța chimică de testat este lipsită de solubilitate. Nu ar trebui să se utilizeze concentrații DMSO peste 2,5 % din cauza interferenței unor concentrații mai mari de solvenți cu analiza. Pentru substanțele chimice de testat care nu sunt solubile în DMSO, dar sunt solubile în etanol, se poate utiliza o soluție de maxim 2 % etanol în analiză, fără interferențe.
|
Martor tampon:
|
26.
|
Martorul tampon (BC) nu ar trebui să conțină nici solvent, nici substanță chimică de testat, ci toate celelalte componente ale analizei. Rezultatele martorului tampon sunt comparate cu martorul tratat cu solvent pentru a verifica dacă solventul utilizat nu afectează sistemul de analiză.
|
Liant puternic (estrogen de referință)
|
27.
|
17β-estradiol (CAS 50-28-2) este ligandul endogen și se leagă cu un grad de afinitate mare de ER, subtipul alfa. Pentru fiecare analiză privind legarea competitivă hrERα ar trebui să se pregătească o curbă standard utilizând 17β-estradiol nemarcat pentru a permite o evaluare a variabilității atunci când analiza este efectuată în timp, în același laborator. Ar trebui să se prepare opt soluții de 17β-estradiol nemarcat în DMSO și în soluția-tampon de analiză, iar concentrațiile finale din godeurile de analiză vor fi utilizate pentru curba standard , cu următoarea repartizare: 10–6, 10–7, 10–8, 10–8,5, 10–9, 10–9,5, 10–10, 10–11 M. Cea mai mare concentrație de 17β-estradiol nemarcat (1 μM) ar trebui să servească drept indicator al legării nespecifice. Această concentrație se distinge prin eticheta „NSB” în tabelul 4, chiar dacă aceasta este, de asemenea, integrată în curba standard.
|
Liant slab
|
28.
|
Un liant slab [noretinodrel (CAS 68-23-5) sau, ca alternativă, noretindronă (CAS 68-22-40] ar trebui să fie inclus pentru a demonstra sensibilitatea fiecărui experiment și a permite o evaluare a variabilității atunci când analiza este efectuată în timp. Ar trebui să se prepare opt soluții de liant slab în DMSO și în soluția-tampon de analiză, iar concentrațiile finale din godeurile de analiză fiind următoarele: 10–4,5, 10–5,5, 10–6, 10–6,5, 10–7, 10–7,5, 10–8 și 10–9 M.
|
Non-liant
|
29.
|
Ca martor negativ ar trebui să se utilizeze octiltrietoxisilan (OTES, CAS 2943-75-1) (non-liant). Acesta asigură faptul că, așa cum este efectuată analiza, aceasta va detecta substanțele chimice de testat care nu se leagă de hrERα. Ar trebui să se prepare opt soluții de non-liant în DMSO și în soluția-tampon de analiză, iar concentrațiile finale din godeurile de analiză fiind următoarele: 10–3,10–4, 10–5, 10–6, 10–7, 10–8, 10–9, 10–10 M. Ftalatul de di-n-butil (DBP, CAS 84-72-2) poate fi utilizat ca non-liant alternativ, însă poate fi testat doar la maxim 10–4M. S-a demonstrat că solubilitatea maximă a DBP în cadrul analizei este 10–4M.
|
Concentrația hrERα
|
30.
|
Ar trebui să fie utilizată cantitatea de receptori care prezintă o legare specifică de 40 ± 10 % (a se vedea punctele 12-13 din apendicele 3). Soluția hrERα ar trebui să fie preparată prin diluția hrERα funcțional în soluție-tampon de analiză rece ca gheața, imediat înainte de utilizare.
|
[3H]-17β-estradiol
|
31.
|
Concentrația [3H]-17β-estradiol finală în godeurile de analiză ar trebui să fie de 0,5 nM.
|
Substanțe chimice de testat
|
32.
|
În primul rând, trebuie să se efectueze un test al solubilității pentru a determina limita de solubilitate pentru fiecare substanță chimică de testat și pentru a identifica intervalul de concentrații adecvat care să fie utilizat la efectuarea protocolului de testare. Limita de solubilitate a fiecărei substanțe chimice de testat va fi determinată inițial în solvent și confirmată ulterior în condiții de testare. Concentrația finală testată în cadrul analizei nu ar trebui să depășească 1 mM. Testul pentru identificarea intervalului cuprinde un martor tratat cu solvent și diluții în serie de cel puțin 8 log, pornind de la concentrația maximă acceptabilă (de exemplu, cel mult 1 mM, în funcție de limita de solubilitate), și se consemnează gradul de tulburare sau precipitatul (a se vedea, de asemenea, punctul 35 din apendicele 3). După ce se determină intervalul de concentrații pentru testare, o substanță chimică de testat ar trebui să fie testată folosind concentrații de 8 log repartizate în mod corespunzător, astfel cum este definit în testul preliminar de stabilire a intervalului. Concentrațiile testate în cel de-al doilea și cel de-al treilea experiment ar trebui să fie apoi ajustate după caz pentru o mai bună caracterizare a curbei concentrație-răspuns, dacă este necesar.
|
|
33.
|
Diluțiile substanței chimice de testat ar trebui să fie preparate în solvent adecvat (a se vedea punctul 25 din apendicele 3). În cazul în care concentrația maximă a substanței chimice de testat nu este solubilă nici în DMSO, nici în etanol, iar dacă se adaugă mai mult solvent, concentrația solventului în eprubeta finală ar fi mai mare decât limita acceptabilă, concentrația maximă poate fi redusă la următoarea concentrație inferioară. În acest caz, se poate adăuga o concentrație suplimentară la limita inferioară a seriei de concentrații. Alte concentrări din cadrul seriei ar trebui să rămână neschimbate.
|
|
34.
|
Soluțiile cu substanță chimică de testat ar trebui să fie monitorizate atent atunci când se adaugă la godeul de analiză, deoarece substanța chimică testată se poate precipita atunci când este adăugată în acesta. Datele pentru toate godeurile care conțin precipitat ar trebui să fie excluse de la formarea curbei, iar motivul excluderii datelor ar trebui să fie consemnat.
|
|
35.
|
În cazul în care există informații anterioare din alte surse care furnizează o valoare log(IC50) pentru o substanță chimică de testat, ar putea fi adecvat ca diluțiile să se repartizate geometric mai apropiate, în jurul valorii preconizate log(IC50) (și anume, 0,5 unități de log). Rezultatele finale ar trebui să reflecte o marjă suficientă de concentrații pe fiecare parte a valorii log(IC50), inclusiv pe partea superioară și cea inferioară, astfel încât curba pentru legătură să poată fi caracterizată în mod corespunzător.
|
Organizara plăcii de analiză
|
36.
|
Ar trebui să se pregătească plăci de microtitrare etichetate utilizând incubații în șase replici pentru martorul tratat cu solvent, cea mai mare concentrație a estrogenului de referință (E2), care constituie, de asemenea, un indicator al legăturii nespecifice (NSB), soluția-tampon martor și incubații în trei replici pentru fiecare dintre cele opt concentrații ale martorului non-liant (octiltrietoxisilan), cele șapte concentrații inferioare pentru estrogenul de referință (E2), cele opt concentrații ale liantului slab (noretinodrel sau noretindronă) și cele opt concentrații ale fiecărei substanțe chimice de testat (TC). În tabelul 4 de mai jos este prezentat un exemplu de model de diagramă a plăcii pentru curbele de concentrație complete pentru estrogenul de referință și martori. Se utilizează plăci de microtitrare suplimentare pentru substanța chimică de testat, care ar trebui să includă martori pe placă [și anume, (i) E2 și un liant slab în trei replici având o concentrație ridicată (desprindere maximă) și medie (de aproximativ IC50); (ii) martor tratat cu solvent (ca legare totală) și soluție de legare nespecifică, fiecare în șase replici (tabelul 5). În apendicele 3.3 este prezentat un exemplu de fișă de configurare a plăcilor de microtitrare pentru analiză competitivă, care utilizează trei substanțe chimice de testat necunoscute. Concentrațiile indicate în fișa de lucru, precum și în tabelele 4 și 5 se referă la concentrațiile finale utilizate în fiecare godeu de analiză. Concentrația maximă pentru E2 ar trebui să fie 1 × 10-7 M, iar pentru liantul slab, ar trebui să se utilizeze cea mai mare concentrație utilizată pentru liantul slab pe placa 1. Concentrația IC50 trebuie să fie stabilită de laborator pe baza bazei sale de date cu martori testați anterior. Se preconizează că această valoare va fi similară cu cea observată în studiile de validare (a se vedea tabelul 1).
|
Tabelul 4
Configurația pentru analiza privind legarea competitivă pe placa de microtitrare (98), (99)
curbe de concentrație complete pentru estrogenul de referință și martori (placa 1)
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
Martor tampon și martor pozitiv (E2)
|
Slab pozitiv (noretinodrel)
|
Martor negativ (OTES)
|
TB și NSB
|
|
A
|
Soluție săracă (*14)
|
1 × 10–9 M
|
1 × 10–10 M
|
TB (martor tratat cu solvent) (2,05 % DMSO)
|
|
B
|
E2 (1 × 10–11 M)
|
1 × 10–8 M
|
1 × 10–9 M
|
|
C
|
E2 (1 × 10–10 M)
|
1 × 10–7,5 M
|
1 × 10–8 M
|
NSB (10–6 M E2)
|
|
D
|
E2 (1 × 10–9,5 M)
|
1 × 10–7 M
|
1 × 10–7 M
|
|
E
|
E2 (1 × 10–9 M)
|
1 × 10–6,5 M
|
1 × 10–6 M
|
Martor tampon
|
|
F
|
E2 (1 × 10–8,5 M)
|
1 × 10–6 M
|
1 × 10–5 M
|
|
G
|
E2 (1 × 10–8 M)
|
1 × 10–5,5 M
|
1 × 10–4 M
|
Soluție săracă (pentru mediu la cald) (*15)
|
|
H
|
E2 (1 × 10–7 M)
|
1 × 10–4,5 M
|
1 × 10–3 M
|
Tabelul 5
Configurația pentru analiza privind legarea competitivă pe placa de microtitrare, plăci suplimentare pentru substanțele chimice de testat (TC) și martorii de pe placă.
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
|
Substanța chimică de testat-1 (TC-1)
|
Substanța chimică de testat-2 (TC-2)
|
Substanța chimică de testat-3 (TC-3)
|
Martori
|
|
A
|
TC-1 (1 × 10–10 M)
|
TC-2 (1 × 10–10 M)
|
TC-3 (1 × 10–10 M)
|
E2 (1 × 10
–7
M)
|
|
B
|
TC-1 (1 × 10–9 M)
|
TC-2 (1 × 10–9 M)
|
TC-3 (1 × 10–9 M)
|
E2 (IC50)
|
|
C
|
TC-1 (1 × 10–8 M)
|
TC-2 (1 × 10–8 M)
|
TC-3 (1 × 10–8 M)
|
NE (1 × 10
–4,5
M)
|
|
D
|
TC-1 (1 × 10–7 M)
|
TC-2 (1 × 10–7 M)
|
TC-3 (1 × 10–7 M)
|
NE (IC50)
|
|
E
|
TC-1 (1 × 10–6 M)
|
TC-2 (1 × 10–6 M)
|
TC-3 (1 × 10–6 M)
|
NSB (10–6 M E2)
|
|
F
|
TC-1 (1 × 10–5 M)
|
TC-2 (1 × 10–5 M)
|
TC-3 (1 × 10–5 M)
|
|
G
|
TC-1 (1 × 10–4 M)
|
TC-2 (1 × 10–4 M)
|
TC-3 (1 × 10–4 M)
|
TB (Martor tratat cu solvent)
|
|
H
|
TC-1 (1 × 10–3 M)
|
TC-2 (1 × 10–3 M)
|
TC-3 (1 × 10–3 M)
|
|
În acest exemplu, liantul slab este noretinodrel (NE)
|
Încheierea analizei privind legarea competitivă
|
37.
|
Cu excepția godeurilor pentru legare totală și soluții sărace (pentru medii calde), astfel cum se arată în tabelul 6, 50 μl de soluție-tampon de analiză ar trebui să se introducă în fiecare godeu și să se amestece cu 10 μl de martor tratat cu solvent, estrogen de referință (E2), liant slab, non-liant și, respectiv, substanțe chimice de testat, respectiv 10 μl de soluție [3H]-17β-estradiol de 5 nM. Apoi o cantitate de 30 μl de soluție receptoare rece ca gheața a fost adăugată în fiecare placă și amestecată ușor. Soluția hrERα ar trebui să fie ultimul reactiv de adăugat. Plăcile de microtitrare de analiză ar trebui să fie incubate la temperatura camerei (22°-28 °C) timp de 2 ore.
Tabelul 6
Volumul componentelor de analiză pentru testul privind legarea competitivă hrER, plăci de microtitrare
|
Etape de preparare
|
Altele decât godeurile TB
|
Godeuri TB
|
Soluție săracă (pentru mediu cald)
|
|
Volumul componentelor pentru godeurile de reacție de mai sus și ordinea adăugării
|
Soluție-tampon de analiză la temperatura camerei
|
50 μl
|
60 μl
|
90 μl
|
|
E2 nemarcat, liant slab, non-liant, solvent și substanțe chimice de testat (*16)
|
10 μl
|
–
|
–
|
|
[3H]-17β-estradiol pentru a produce o concentrație finală de 0,5 nM (adică 5 nM)
|
10 μl
|
10 μl
|
10 μl
|
|
concentrația hrERα astfel cum este determinată (a se vedea punctele 12-13)
|
30 μl
|
30 μl
|
—
|
|
Volumul total în fiecare godeu de analiză
|
100 μl
|
100 μl
|
100 μl
|
|
|
38.
|
Cuantificarea legării [3H]-17β-estradiol de hrERα, în urma separării [3H]-17β-estradiol legat de hrERα de [3H]-17β-estradiol liber, prin adăugarea, în fiecare godeu, a 100 μl de suspensie DCC rece ca gheața, ar trebui să fie efectuată astfel cum este descris la punctele 21-23 din apendicele 3 pentru analiza privind legarea prin saturație.
|
|
39.
|
Godeurile G10-12 și H10-12 [identificate ca având soluție săracă (pentru mediu cald) în tabelul 4] reprezintă DPMS al [3H]-estradiol marcat în 10 μl. Partea alicotă de 10 μl ar trebui să fie introdusă direct în lichidul de scintilație.
|
Criterii de acceptabilitate
Analiza privind legarea prin saturație
|
40.
|
Curba specifică privind legarea ar trebui să ajungă la un platou, întrucât au fost utilizate concentrații crescătoare de [3H]-17β-estradiol, care indică saturația hrERα cu ligandul.
|
|
41.
|
Legarea specifică la 0,5 nM de [3H]-17β-estestradiol ar trebui să fie situată în intervalul acceptabil 30 %-50 % din radioactivitatea totală măsurată medie adăugată de la o testare la alta. Abaterile ocazionale ușoare de la acest interval sunt acceptabile, însă, dacă testele se situează în mod constant în afara acestui interval sau un anumit test este semnificativ în afara acestui interval, ar trebui să se ajusteze concentrația de proteine, iar analiza saturației ar trebui să fie repetată.
|
|
42.
|
Datele ar trebui să producă o reprezentare grafică liniară Scatchard.
|
|
43.
|
Legarea nespecifică nu ar trebui să fie excesivă. Valoarea pentru legarea nespecifică ar trebui, de regulă, să fie < 35 % din legarea totală. Însă, raportul poate depăși ocazional această limită atunci când se măsoară valoarea dpm foarte scăzută pentru concentrația minimă a substanței 17β-estradiol marcat radioactiv testate.
|
Analiza privind legarea competitivă
|
44.
|
Concentrațiile din ce în ce mai mari de 17β-estradiol nemarcat ar trebui să desprindă [3H]-17β-estradiol de receptor într-un mod compatibil cu o legarea competitivă dintr-un singur punct.
|
|
45.
|
Valoarea IC50 pentru estrogenul de referință (și anume, 17β-estradiol) ar trebui să fie aproximativ egal cu concentrația molară a [3H]-17β-estradiol, plus valorile Kd determinate din analiza privind legarea prin saturație.
|
|
46.
|
Legarea specifică totală ar trebui să se situeze în mod consecvent în intervalul acceptabil de 40 ± 10 % atunci când concentrația medie măsurată a radioactivității totale adăugate în fiecare godeu a fost de 0,5 nM în toate testele. Abaterile ocazionale ușoare de la acest interval sunt acceptabile, însă, dacă testele se situează în mod constant în afara acestui interval sau un anumit test este semnificativ în afara acestui interval, ar trebui să se ajusteze concentrația de proteine.
|
|
47.
|
Solventul nu ar trebui să modifice sensibilitatea sau reproductibilitatea analizei. Rezultatele martorului tratat cu solvent (godeuri TB) sunt comparate cu martorul tampon pentru a verifica dacă solventul utilizat nu afectează sistemul de analiză. Rezultatele pentru TB și martorul tampon ar trebui să fie comparabile în cazul în care nu există niciun efect al solventului asupra analizei.
|
|
48.
|
Non-liantul nu ar trebui să desprindă mai mult de 25 % din [3H]-17β-estradiol de hrERα atunci când se testează maxim 10–3 M (OTES) sau 10–4 M (DBP).
|
|
49.
|
Au fost elaborate criterii de performanță pentru estrogenul de referință și doi lianți slabi (de exemplu, noretinodrel, noretindronă), utilizând date din studiul de validare a analizei CERI privind legarea hrER (anexa N la referința 2). Sunt prezentate intervale de încredere de 95 % pentru medie +/- SD (n) pentru toate testele pe martori din cadrul celor patru laboratoare care au participat la studiul de validare. Au fost calculate intervale de încredere de 95 % pentru parametrii de formare a curbei (și anume, partea superioară, partea inferioară, Hillslope și logIC50) pentru estrogenul de referință și lianții slabi, precum și pentru valoarea log10RBA a lianților slabi în raport cu estrogenul de referință. Tabelul 1 furnizează intervalele preconizate pentru parametrii de formare a curbei care pot fi utilizați ca și criterii de performanță. În practică, intervalul IC50 poate varia ușor, în funcție de valoarea Kd derivată experimental a preparatul cu receptori și de concentrația ligandului utilizaztă pentru analiză.
|
|
50.
|
Nu au fost elaborate criterii de performanță pentru parametrii de formare a curbei în cazul substanțelor chimice de testat din cauza gamei largi de substanțe chimice de testat posibile și a variației posibilelor afinități și rezultatele (de exemplu, curbă completă, curbă parțială, fără formă de curbă). Însă, la analizarea rezultatelor din cadrul fiecărei testări, în cazul unei substanțe chimice de testat, ar trebui să se aplice o apreciere profesională. Ar trebui să se utilizeze un interval suficient de concentrații ale substanței chimice de testat pentru a defini cu claritate partea superioară (de exemplu, o legare de 90-100 %) a curbei de legare competitivă. Variabilitatea între replici la fiecare concentrație a substanței chimice de testat, precum și între cele trei teste independente ar trebui să fie rezonabilă și justificată din punct de vedere științific. Martorii din cadrul fiecărei serii, pentru o substanță chimică de testat, ar trebui să se apropie de măsurile de performanță raportate pentru această analiză FW și să fie în concordanță cu datele privind martorii testați anterior din fiecare laborator.
|
ANALIZA DATELOR
Analiza privind legarea prin saturație
|
51.
|
Se măsoară atât legarea totală, cât și cea nespecifică. Pornind de la aceste valori, se calculează legarea specifică a [3H]-17β-estradiol, în condiții de echilibru, la concentrații în creștere, scăzând valoarea legării nespecifice din total. Un grafic al legării specifice versus concentrația [3H]-17β-estradiol ar trebui să ajungă al un platou pentru o legare specifică maximă care indică o saturație a hrERα cu [3H]-17β-estradiol. În plus, analiza datelor ar trebui să documenteze legarea [3H]-17β- estradiol de un singur loc de legare cu un grad ridicat de afinitate. Legarea nespecifică, cea totală și cea specifică ar trebui să fie ilustrate pe o curbă de legare prin saturație. În cadrul unei analize ulterioare a acestor date ar trebui să se utilizeze o analiză de regresie nelineară (de exemplu, BioSoft; McPherson, 1985; Motulsky, 1995) cu o reprezentare finală a datelor sub forma unui grafic Scatchard.
|
|
52.
|
Prin analiza datelor ar trebui să se determine valorile Bmax și Kd doar din datele privind legarea totală, pe baza ipotezei că legarea nespecifică este liniară, cu excepția cazului în care se justifică utilizarea unei metode diferite. În plus, ar trebui să se utilizeze o regresie robustă atunci când se determină cea mai bună corelare, cu excepția cazului în care este furnizată o justificare. Ar trebui să se precizeze metoda aleasă pentru regresia robustă. Ar trebui să se recurgă întotdeauna la corecția pentru sărăcirea ligandului (de exemplu, utilizând metoda lui Swillens, 1995) atunci când se determină valorile Bmax și Kd din datele privind legarea prin saturație.
|
Analiza privind legarea competitivă
|
53.
|
Curba de legare competitivă este reprezentată grafic ca fiind o legare specifică [3H]-17β-estradiol versus concentrația (unități de log10) soluției testate în paralel. Concentrația substanței chimice de testat care inhibă 50 % din legarea specifică maximă a [3H]-17β-estradiol constituie valoarea IC50.
|
|
54.
|
Estimările valorilor log(IC50) pentru martorii pozitivi (de exemplu, estrogenul de referință și liantul slab) ar trebui să fie determinate utilizând un software de formare a curbelor neliniare adecvat pentru a se încadra într-o ecuație Hill cu patru parametri (de exemplu, BioSoft; McPherson, 1985; Motulsky, 1995). Partea superioară, cea inferioară, panta și log(IC50) ar trebui, în general, să rămână necondiționate atunci când se formează aceste curbe. Ar trebui să se utilizeze o regresie robustă atunci când se determină cea mai bună corelare, cu excepția cazului în care este furnizată o justificare. Nu ar trebui să se recurgă la corecția pentru sărăcirea ligandului. În urma analizei inițiale, ar trebui să se examineze fiecare curbă de legare pentru a asigura o corelare corespunzătoare cu modelul. Afinitatea de legare relativă (RBA) a unui liant slab ar trebui să se calculeze ca procent din log (IC50) pentru liantul slab raportat la valoarea log (IC50) pentru 17β-estradiol. Rezultatele martorilor pozitivi și ale martorului non-liant ar trebui să fie evaluate utilizând valorile de măsurare a performanței analizei de la punctele 44-49 din prezentul apendice 3.
|
|
55.
|
Datele pentru toate substanțele chimice de testat ar trebui să fie analizate printr-o abordare etapizată pentru a se asigura analizarea corespunzătoare a datelor și clasificarea corectă a fiecărei curbe de legare competitivă. Se recomandă ca fiecare testare pentru o substanță chimică de testat să fie supusă inițial unei analize a datelor standardizată care este identică cu cea utilizată pentru estrogenul de referință și lianții slabi martori (a se vedea punctul 54 din prezentul apendice 3). După încheierea analizei, ar trebui să fie efectuată o analiză tehnică a parametrilor curbei și o verificare vizuală a modului în care datele se încadrează pe curba de legare competitivă generată pentru fiecare testare. În timpul acestei analize tehnice, observațiile privind o scădere dependentă de concentrație în procentul [3H]-17β-estradiol legat în mod specific, variabilitatea scăzută între replicile tehnice la fiecare concentrație chimică și consecvența parametrilor de corelare între cele trei teste constituie indicații bune pentru faptul că analiza și analizele datelor au fost efectuate în mod corespunzător.
|
Interpretarea datelor
|
56.
|
Cu condiția ca toate criteriile de acceptabilitate să fie îndeplinite, o substanță chimică de testat este considerată a fi un liant pentru hrERα în cazul în care poate fi formată o curbă de legare, iar punctul cel mai de jos de pe curba de răspuns în intervalul de date este sub 50 % (figura 1).
|
|
57.
|
Cu condiția îndeplinirii tuturor criteriilor de acceptabilitate, o substanță chimică de testat este considerată a fi un non-liant pentru hrERα dacă:
|
—
|
poate fi formată o curbă de legare, iar punctul cel mai de jos pe curba de răspuns formată, situat în intervalul de date, este mai mare de 75 % sau
|
|
—
|
nu poate fi formată o curbă de legare, iar media cea mai scăzută a procentului de legare neuniformizată între grupurile de concentrații în ceea ce privește datele este mai mare de 75 %.
|
|
|
58.
|
Substanțele chimice de testat sunt considerate echivoce dacă nu este îndeplinită niciuna dintre condițiile de mai sus (de exemplu, punctul cel mai de jos de pe curba de răspuns formată este între 76 și 51 %).
|
Tabelul 7
Criterii pentru clasificare pe baza curbei de legare competitive pentru o substanță chimică de testat.
|
Clasificare
|
Criterii
|
|
Lianta
|
Se poate forma o curbă de legare.
Punctul cel mai de jos al curbei de răspuns în intervalul de date este mai mic de 50 %.
|
|
Non-liantb
|
Dacă se poate forma o curbă de legare,
punctul cel mai de jos al curbei de răspuns în intervalul de date este peste 75 %.
Dacă nu se poate forma o curbă de legare,
media cea mai scăzută a procentului de legare neuniformizată între grupurile de concentrații în ceea ce privește datele este mai mare de 75 %.
|
|
Substanță echivocăc
|
Orice substanță care poate fi testată și care nu este nici liant, nici non-liant
de exemplu, punctul cel mai de jos pe curba de răspuns formată este între 76 și 51 %).
|
Figura 1
Exemple de clasificare a unei substanțe chimice de testat utilizând curba de legare competitivă.
|
59.
|
Se combină mai multe testări realizate într-un laborator pentru o substanță chimică de testat prin atribuirea de valori numerice fiecărei testări și calcularea mediei testelor, astfel cum se arată în tabelul 8. Rezultatele pentru testele combinate din fiecare laborator sunt comparate cu clasificarea preconizată pentru fiecare substanță chimică de testat.
|
Tabelul 8
Metoda de clasificare a substanței chimice de testat utilizând teste multiple în cadrul unui laborator
|
Pentru atribuirea unei valori fiecărei testări:
|
|
Clasificare
|
Valoare numerică
|
|
Liant
|
2
|
|
Substanță echivocă
|
1
|
|
Non-liant
|
0
|
|
Pentru clasificarea mediei valorilor numerice ale testelor:
|
|
Clasificare
|
Valoare numerică
|
|
Liant
|
Media ≥ 1,5
|
|
Substanță echivocă
|
0,5 ≤ media < 1,5
|
|
Non-liant
|
Media < 0,5
|
RAPORTUL TESTULUI
|
60.
|
A se vedea punctul 24 din „COMPONENTELE ANALIZEI PRIVIND LEGAREA hrER” din prezenta metodă de testare.
|
Apendicele 3.1
Listă de Termeni
[3H]E2
: 17β-estradiol marcat radioactiv cu tritiu
DCC
: Cărbune acoperit cu dextran
E2
: 17β-estradiol nemarcat (inert)
Soluția-tampon de analiză
: 10 mM Tris-HCl, pH 7,4, conținând 1 mM EDTA, 1mM EGTA, 1 mM NaVO3, 10 % Glycerol, 0,2 mM Leupeptin, 1 mM Dithiothreitol și 10 mg/ml albumină serică bovină
hrERα
: Receptor de estrogen recombinant uman alfa (domeniu de legare cu ligand)
Replică
: Unul dintre multiplele godeuri care conțin același conținut la aceleași concentrații și sunt analizate în paralel în cadrul unei singure testări. În acest protocol, fiecare concentrație a substanței chimice de testat este testată în trei replici; cu alte cuvinte, există trei replici care sunt testate simultan la fiecare concentrație a substanței chimice de testat.
Testare
: Un set complet de godeuri de pe o placă de microtitrare testate în paralel, care oferă toate informațiile necesare pentru a caracteriza legarea unei substanțe chimice de testat de hrERα (și anume, [3H]-17β-estradiol total adăugat în godeul de analiză, legarea maximă a [3H]-17β-estradiol de hrERα, legarea nespecifică și legarea totală la diferite concentrații ale substanței chimice de testat). O testare ar putea consta dintr-un singur godeu de analiză (și anume, replică) per concentrație, dar, deoarece acest protocol impune efectuarea analizei pe trei replici, o testare constă din trei godeuri de analiză per concentrație. În plus, acest protocol necesită trei teste independente (și anume, neefectuate în paralel) pe substanță chimică.
Apendicele 3.2
CONFIGURAȚIA GODEURILOR ÎN ANALIZA LEGĂRII COMPETITIVE
|
Placă
|
Poziție
|
Replică
|
Tip godeu
|
Cod godeu
|
Cod concentrație
|
Concentrația inițială a soluției testate în paralel (M)
|
Soluție mamă hrER (μl)
|
Volum soluție-tampon (μl)
|
Volum detector (E2 la cald) (μL)
|
Volum din placa de diluție (μL)
|
Volum final (μl)
|
Concentrația finală a soluției testate în paralel (M)
|
|
S
|
A1
|
1
|
Soluție săracă
|
BK
|
BK1
|
—
|
—
|
—
|
—
|
—
|
—
|
—
|
|
S
|
A2
|
2
|
Soluție săracă
|
BK
|
BK2
|
—
|
—
|
—
|
—
|
—
|
—
|
—
|
|
S
|
A3
|
3
|
Soluție săracă
|
BK
|
BK3
|
—
|
—
|
—
|
—
|
—
|
—
|
—
|
|
S
|
B1
|
1
|
E2 la rece
|
S
|
S1
|
1.00E-10
|
30
|
50
|
10
|
10
|
100
|
1.0E-11
|
|
S
|
B2
|
2
|
E2 la rece
|
S
|
S1
|
1.00E-10
|
30
|
50
|
10
|
10
|
100
|
1.0E-11
|
|
S
|
B3
|
3
|
E2 la rece
|
S
|
S1
|
1.00E-10
|
30
|
50
|
10
|
10
|
100
|
1.0E-11
|
|
S
|
C1
|
1
|
E2 la rece
|
S
|
S2
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1.0E-10
|
|
S
|
C2
|
2
|
E2 la rece
|
S
|
S2
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1.0E-10
|
|
S
|
C3
|
3
|
E2 la rece
|
S
|
S2
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1.0E-10
|
|
S
|
D1
|
1
|
E2 la rece
|
S
|
S3
|
3,16 E-09
|
30
|
50
|
10
|
10
|
100
|
3,2 E-10
|
|
S
|
D2
|
2
|
E2 la rece
|
S
|
S3
|
3,16 E-09
|
30
|
50
|
10
|
10
|
100
|
3,2 E-10
|
|
S
|
D3
|
3
|
E2 la rece
|
S
|
S3
|
3,16 E-09
|
30
|
50
|
10
|
10
|
100
|
3,2 E-10
|
|
S
|
E1
|
1
|
E2 la rece
|
S
|
S4
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
S
|
E2
|
2
|
E2 la rece
|
S
|
S4
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
S
|
E3
|
3
|
E2 la rece
|
S
|
S4
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
S
|
F1
|
1
|
E2 la rece
|
S
|
S5
|
3,16 E-08
|
30
|
50
|
10
|
10
|
100
|
3,2 E-09
|
|
S
|
F2
|
2
|
E2 la rece
|
S
|
S5
|
3,16 E-08
|
30
|
50
|
10
|
10
|
100
|
3,2 E-09
|
|
S
|
F3
|
3
|
E2 la rece
|
S
|
S5
|
3,16 E-08
|
30
|
50
|
10
|
10
|
100
|
3,2 E-09
|
|
S
|
G1
|
1
|
E2 la rece
|
S
|
S6
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
S
|
G2
|
2
|
E2 la rece
|
S
|
S6
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
S
|
G3
|
3
|
E2 la rece
|
S
|
S6
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
S
|
H1
|
1
|
E2 la rece
|
S
|
S7
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
S
|
H2
|
2
|
E2 la rece
|
S
|
S7
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
S
|
H3
|
3
|
E2 la rece
|
S
|
S7
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
S
|
A4
|
1
|
noretinodrel
|
NE
|
WP1
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
S
|
A5
|
2
|
noretinodrel
|
NE
|
WP1
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
S
|
A6
|
3
|
noretinodrel
|
NE
|
WP1
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
S
|
B4
|
1
|
noretinodrel
|
NE
|
WP2
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
S
|
B5
|
2
|
noretinodrel
|
NE
|
WP2
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
S
|
B6
|
3
|
noretinodrel
|
NE
|
WP2
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
S
|
C4
|
1
|
noretinodrel
|
NE
|
WP3
|
3,16 E-07
|
30
|
50
|
10
|
10
|
100
|
3,2 E-08
|
|
S
|
C5
|
2
|
noretinodrel
|
NE
|
WP3
|
3,16 E-07
|
30
|
50
|
10
|
10
|
100
|
3,2 E-08
|
|
S
|
C6
|
3
|
noretinodrel
|
NE
|
WP3
|
3,16 E-07
|
30
|
50
|
10
|
10
|
100
|
3,2 E-08
|
|
S
|
D4
|
1
|
noretinodrel
|
NE
|
WP4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
S
|
D5
|
2
|
noretinodrel
|
NE
|
WP4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
S
|
D6
|
3
|
noretinodrel
|
NE
|
WP4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
S
|
E4
|
1
|
noretinodrel
|
NE
|
WP5
|
3,16 E-06
|
30
|
50
|
10
|
10
|
100
|
3,2 E-07
|
|
S
|
E5
|
2
|
noretinodrel
|
NE
|
WP5
|
3,16 E-06
|
30
|
50
|
10
|
10
|
100
|
3,2 E-07
|
|
S
|
E6
|
3
|
noretinodrel
|
NE
|
WP5
|
3,16 E-06
|
30
|
50
|
10
|
10
|
100
|
3,2 E-07
|
|
S
|
F4
|
1
|
noretinodrel
|
NE
|
WP6
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
S
|
F5
|
2
|
noretinodrel
|
NE
|
WP6
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
S
|
F6
|
3
|
noretinodrel
|
NE
|
WP6
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
S
|
G4
|
1
|
noretinodrel
|
NE
|
WP7
|
3,16 E-05
|
30
|
50
|
10
|
10
|
100
|
3,2 E-06
|
|
S
|
G5
|
2
|
noretinodrel
|
NE
|
WP7
|
3,16 E-05
|
30
|
50
|
10
|
10
|
100
|
3,2 E-06
|
|
S
|
G6
|
3
|
noretinodrel
|
NE
|
WP7
|
3,16 E-05
|
30
|
50
|
10
|
10
|
100
|
3,2 E-06
|
|
S
|
H4
|
1
|
noretinodrel
|
NE
|
WP8
|
3,16 E-04
|
30
|
50
|
10
|
10
|
100
|
3,2 E-05
|
|
S
|
H5
|
2
|
noretinodrel
|
NE
|
WP8
|
3,16 E-04
|
30
|
50
|
10
|
10
|
100
|
3,2 E-05
|
|
S
|
H6
|
3
|
noretinodrel
|
NE
|
WP8
|
3,16 E-04
|
30
|
50
|
10
|
10
|
100
|
3,2 E-05
|
|
S
|
A7
|
1
|
OTES
|
N
|
OTES1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1.0E-10
|
|
S
|
A8
|
2
|
OTES
|
N
|
OTES1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1.0E-10
|
|
S
|
A9
|
3
|
OTES
|
N
|
OTES1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1.0E-10
|
|
S
|
B7
|
1
|
OTES
|
N
|
OTES2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
S
|
B8
|
2
|
OTES
|
N
|
OTES2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
S
|
B9
|
3
|
OTES
|
N
|
OTES2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
S
|
C7
|
1
|
OTES
|
N
|
OTES3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
S
|
C8
|
2
|
OTES
|
N
|
OTES3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
S
|
C9
|
3
|
OTES
|
N
|
OTES3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
S
|
D7
|
1
|
OTES
|
N
|
OTES4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
S
|
D8
|
2
|
OTES
|
N
|
OTES4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
S
|
D9
|
3
|
OTES
|
N
|
OTES4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
S
|
E7
|
1
|
OTES
|
N
|
OTES5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
S
|
E8
|
2
|
OTES
|
N
|
OTES5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
S
|
E9
|
3
|
OTES
|
N
|
OTES5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
S
|
F7
|
1
|
OTES
|
N
|
OTES6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1.0E-05
|
|
S
|
F8
|
2
|
OTES
|
N
|
OTES6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1.0E-05
|
|
S
|
F9
|
3
|
OTES
|
N
|
OTES6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1.0E-05
|
|
S
|
G7
|
1
|
OTES
|
N
|
OTES7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1.0E-04
|
|
S
|
G8
|
2
|
OTES
|
N
|
OTES7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1.0E-04
|
|
S
|
G9
|
3
|
OTES
|
N
|
OTES7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1.0E-04
|
|
S
|
H7
|
1
|
OTES
|
N
|
OTES8 DBP7
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1.0E-03
|
|
S
|
H8
|
2
|
OTES
|
N
|
OTES88
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1.0E-03
|
|
S
|
H9
|
3
|
OTES
|
N
|
OTES8
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1.0E-03
|
|
S
|
A10
|
1
|
legare totală
|
TB
|
TB1
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
A11
|
2
|
legare totală
|
TB
|
TB2
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
A12
|
3
|
legare totală
|
TB
|
TB3
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
B10
|
4
|
legare totală
|
TB
|
TB4
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
B11
|
5
|
legare totală
|
TB
|
TB5
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
B12
|
6
|
legare totală
|
TB
|
TB6
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
C10
|
1
|
E2 la rece (ridicat)
|
NSB
|
S1
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
S
|
C11
|
2
|
E2 la rece (ridicat)
|
NSB
|
S2
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
S
|
C12
|
3
|
E2 la rece (ridicat)
|
NSB
|
S3
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
S
|
D10
|
4
|
E2 la rece (ridicat)
|
NSB
|
S4
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
S
|
D11
|
5
|
E2 la rece (ridicat)
|
NSB
|
S5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
S
|
D12
|
6
|
E2 la rece (ridicat)
|
NSB
|
S6
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
S
|
E10
|
1
|
Martor tampon
|
BC
|
BC1
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
E11
|
2
|
Martor tampon
|
BC
|
BC2
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
E12
|
3
|
Martor tampon
|
BC
|
BC3
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
F10
|
4
|
Martor tampon
|
BC
|
BC4
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
F11
|
5
|
Martor tampon
|
BC
|
BC5
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
G10 (*17)
|
1
|
Soluție săracă (pentru mediu cald)
|
La cald
|
H1
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
S
|
G11 (*17)
|
2
|
Soluție săracă (pentru mediu cald)
|
La cald
|
H2
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
S
|
G12 (*17)
|
3
|
Soluție săracă (pentru mediu cald)
|
La cald
|
H3
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
S
|
H10 (*17)
|
4
|
Soluție săracă (pentru mediu cald)
|
La cald
|
H4
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
S
|
H11 (*17)
|
5
|
Soluție săracă (pentru mediu cald)
|
La cald
|
H5
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
S
|
H12
|
6
|
Soluție săracă (pentru mediu cald)
|
La cald
|
H6
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
P1
|
A1
|
1
|
Necunoscut 1
|
U1
|
1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1.0E-10
|
|
P1
|
A2
|
2
|
Necunoscut 1
|
U1
|
1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1.0E-10
|
|
P1
|
A3
|
3
|
Necunoscut 1
|
U1
|
1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1.0E-10
|
|
P1
|
B1
|
1
|
Necunoscut 1
|
U1
|
2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
P1
|
B2
|
2
|
Necunoscut 1
|
U1
|
2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
P1
|
B3
|
3
|
Necunoscut 1
|
U1
|
2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
P1
|
C1
|
1
|
Necunoscut 1
|
U1
|
3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
P1
|
C2
|
2
|
Necunoscut 1
|
U1
|
3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
P1
|
C3
|
3
|
Necunoscut 1
|
U1
|
3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
P1
|
D1
|
1
|
Necunoscut 1
|
U1
|
4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
P1
|
D2
|
2
|
Necunoscut 1
|
U1
|
4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
P1
|
D3
|
3
|
Necunoscut 1
|
U1
|
4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
P1
|
E1
|
1
|
Necunoscut 1
|
U1
|
5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
P1
|
E2
|
2
|
Necunoscut 1
|
U1
|
5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
P1
|
E3
|
3
|
Necunoscut 1
|
U1
|
5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
P1
|
F1
|
1
|
Necunoscut 1
|
U1
|
6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1.0E-05
|
|
P1
|
F2
|
2
|
Necunoscut 1
|
U1
|
6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1.0E-05
|
|
P1
|
F3
|
3
|
Necunoscut 1
|
U1
|
6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1.0E-05
|
|
P1
|
G1
|
1
|
Necunoscut 1
|
U1
|
7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1.0E-04
|
|
P1
|
G2
|
2
|
Necunoscut 1
|
U1
|
7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1.0E-04
|
|
P1
|
G3
|
3
|
Necunoscut 1
|
U1
|
7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1.0E-04
|
|
P1
|
H1
|
1
|
Necunoscut 1
|
U1
|
8
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1.0E-03
|
|
P1
|
H2
|
2
|
Necunoscut 1
|
U1
|
8
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1.0E-03
|
|
P1
|
H3
|
3
|
Necunoscut 1
|
U1
|
8
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1.0E-03
|
|
P1
|
A4
|
1
|
Necunoscut 2
|
U2
|
1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1.0E-10
|
|
P1
|
A5
|
2
|
Necunoscut 2
|
U2
|
1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1.0E-10
|
|
P1
|
A6
|
3
|
Necunoscut 2
|
U2
|
1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1.0E-10
|
|
P1
|
B4
|
1
|
Necunoscut 2
|
U2
|
2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
P1
|
B5
|
2
|
Necunoscut 2
|
U2
|
2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
P1
|
B6
|
3
|
Necunoscut 2
|
U2
|
2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
P1
|
C4
|
1
|
Necunoscut 2
|
U2
|
3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
P1
|
C5
|
2
|
Necunoscut 2
|
U2
|
3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
P1
|
C6
|
3
|
Necunoscut 2
|
U2
|
3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
P1
|
D4
|
1
|
Necunoscut 2
|
U2
|
4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
P1
|
D5
|
2
|
Necunoscut 2
|
U2
|
4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
P1
|
D6
|
3
|
Necunoscut 2
|
U2
|
4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
P1
|
E4
|
1
|
Necunoscut 2
|
U2
|
5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
P1
|
E5
|
2
|
Necunoscut 2
|
U2
|
5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
P1
|
E6
|
3
|
Necunoscut 2
|
U2
|
5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
P1
|
F4
|
1
|
Necunoscut 2
|
U2
|
6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1.0E-05
|
|
P1
|
F5
|
2
|
Necunoscut 2
|
U2
|
6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1.0E-05
|
|
P1
|
F6
|
3
|
Necunoscut 2
|
U2
|
6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1.0E-05
|
|
P1
|
G4
|
1
|
Necunoscut 2
|
U2
|
7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1.0E-04
|
|
P1
|
G5
|
2
|
Necunoscut 2
|
U2
|
7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1.0E-04
|
|
P1
|
G6
|
3
|
Necunoscut 2
|
U2
|
7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1.0E-04
|
|
P1
|
H4
|
1
|
Necunoscut 2
|
U2
|
8
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1.0E-03
|
|
P1
|
H5
|
2
|
Necunoscut 2
|
U2
|
8
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1.0E-03
|
|
P1
|
H6
|
3
|
Necunoscut 2
|
U2
|
8
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1.0E-03
|
|
P1
|
A7
|
1
|
Necunoscut 3
|
U3
|
1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1.0E-10
|
|
P1
|
A8
|
2
|
Necunoscut 3
|
U3
|
1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1.0E-10
|
|
P1
|
A9
|
3
|
Necunoscut 3
|
U3
|
1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1.0E-10
|
|
P1
|
B7
|
1
|
Necunoscut 3
|
U3
|
2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
P1
|
B8
|
2
|
Necunoscut 3
|
U3
|
2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
P1
|
B9
|
3
|
Necunoscut 3
|
U3
|
2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
P1
|
C7
|
1
|
Necunoscut 3
|
U3
|
3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
P1
|
C8
|
2
|
Necunoscut 3
|
U3
|
3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
P1
|
C9
|
3
|
Necunoscut 3
|
U3
|
3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
P1
|
D7
|
1
|
Necunoscut 3
|
U3
|
4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
P1
|
D8
|
2
|
Necunoscut 3
|
U3
|
4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
P1
|
D9
|
3
|
Necunoscut 3
|
U3
|
4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
P1
|
E7
|
1
|
Necunoscut 3
|
U3
|
5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
P1
|
E8
|
2
|
Necunoscut 3
|
U3
|
5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
P1
|
E9
|
3
|
Necunoscut 3
|
U3
|
5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
P1
|
F7
|
1
|
Necunoscut 3
|
U3
|
6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1.0E-05
|
|
P1
|
F8
|
2
|
Necunoscut 3
|
U3
|
6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1.0E-05
|
|
P1
|
F9
|
3
|
Necunoscut 3
|
U3
|
6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1.0E-05
|
|
P1
|
G7
|
1
|
Necunoscut 3
|
U3
|
7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1.0E-04
|
|
P1
|
G8
|
2
|
Necunoscut 3
|
U3
|
7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1.0E-04
|
|
P1
|
G9
|
3
|
Necunoscut 3
|
U3
|
7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1.0E-04
|
|
P1
|
H7
|
1
|
Necunoscut 3
|
U3
|
8
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1.0E-03
|
|
P1
|
H8
|
2
|
Necunoscut 3
|
U3
|
8
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1.0E-03
|
|
P1
|
H9
|
3
|
Necunoscut 3
|
U3
|
8
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1.0E-03
|
|
P1
|
A10
|
1
|
Martor E2 (max)
|
S
|
E2 max1
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.00E-07
|
|
P1
|
A11
|
2
|
Martor E2 (max)
|
S
|
E2 max2
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.00E-07
|
|
P1
|
A12
|
3
|
Martor E2 (max)
|
S
|
E2 max3
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.00E-07
|
|
P1
|
B10
|
1
|
Martor E2 (IC50)
|
S
|
E2IC501
|
E2IC50x10
|
30
|
50
|
10
|
10
|
100
|
E2IC50
|
|
P1
|
B11
|
2
|
Martor E2 (IC50)
|
S
|
E2IC502
|
E2IC50x10
|
30
|
50
|
10
|
10
|
100
|
E2IC50
|
|
P1
|
B12
|
3
|
Martor E2 (IC50)
|
S
|
E2IC503
|
E2IC50x10
|
30
|
50
|
10
|
10
|
100
|
E2IC50
|
|
P1
|
C10
|
1
|
Martor NE (max)
|
S
|
Nemax1
|
1.00E-3,5
|
30
|
50
|
10
|
10
|
100
|
1.00E-4,5
|
|
P1
|
C11
|
2
|
Martor NE (max)
|
S
|
Nemax2
|
1.00E-3,5
|
30
|
50
|
10
|
10
|
100
|
1.00E-4,5
|
|
P1
|
C12
|
3
|
Martor NE (max)
|
S
|
Nemax3
|
1.00E-3,5
|
30
|
50
|
10
|
10
|
100
|
1.00E-4,5
|
|
P1
|
D10
|
1
|
Martor NE (IC50)
|
S
|
NEIC501
|
NEIC50 x10
|
30
|
50
|
10
|
10
|
100
|
NEIC50
|
|
P1
|
D11
|
2
|
Martor NE (IC50)
|
S
|
NEIC502
|
NEIC50 x10
|
30
|
50
|
10
|
10
|
100
|
NEIC50
|
|
P1
|
D12
|
3
|
Martor NE (IC50)
|
S
|
NEIC503
|
NEIC50 x10
|
30
|
50
|
10
|
10
|
100
|
NEIC50
|
|
P1
|
E10
|
1
|
E2 la rece (ridicat)
|
NSB
|
S1
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
P1
|
E11
|
2
|
E2 la rece (ridicat)
|
NSB
|
S2
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
P1
|
E12
|
3
|
E2 la rece (ridicat)
|
NSB
|
S3
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
P1
|
F10
|
4
|
E2 la rece (ridicat)
|
NSB
|
S4
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
P1
|
F11
|
5
|
E2 la rece (ridicat)
|
NSB
|
S5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
P1
|
F12
|
6
|
E2 la rece (ridicat)
|
NSB
|
S6
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
P1
|
G10
|
1
|
legare totală
|
TB
|
TB1
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
G11
|
2
|
legare totală
|
TB
|
TB2
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
G12
|
3
|
legare totală
|
TB
|
TB3
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
H10
|
4
|
legare totală
|
TB
|
TB4
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
H11
|
5
|
legare totală
|
TB
|
TB5
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
H12
|
6
|
legare totală
|
TB
|
TB6
|
—
|
30
|
60
|
10
|
—
|
100
|
-
|
Apendicele 4
CONSIDERAȚII PENTRU ANALIZA DATELOR DIN ANALIZA PRIVIND LEGAREA COMPETITIVĂ HREH
|
1.
|
Analiza privind legarea competitivă hrERα măsoară gradul de legare al unei singure concentrații de [3H]-17β-estradiol în prezența unor concentrații în creștere ale unei substanțe chimice de testat. Curba de legare competitivă este reprezentată grafic ca fiind o legare specifică [3H]-17β-estradiol versus concentrația (unități de log10) soluției testate în paralel. Concentrația substanței chimice de testat care inhibă 50 % din legarea specifică maximă a [3H]-17β-estradiol constituie valoarea IC50.
|
Analiza datelor pentru estrogenul de referință și liantul slab (1)
|
2.
|
Datele din testele pe martori se transformă (adică procentul de legare specifică [3H]-17β-estradiol și concentrația log a substanței chimice martor) în vederea unei analize ulterioare. Estimările valorilor log(IC50) pentru martorii pozitivi (de exemplu, estrogenul de referință și liantul slab) ar trebui să fie determinate utilizând un software de formare a curbelor neliniare adecvat pentru a se încadra într-o ecuație Hill cu patru parametri (de exemplu, BioSoft; GraphPad Prism) (2). Partea superioară, cea inferioară, panta și log(IC50) poate să rămână, de regulă, necondiționate atunci când se formează aceste curbe. Ar trebui să se utilizeze o regresie robustă atunci când se determină cea mai bună corelare, cu excepția cazului în care este furnizată o justificare. Ar trebui să se precizeze metoda aleasă pentru regresia robustă. Corecția pentru sărăcirea ligandului nu a fost necesară pentru analizele FW sau CERI hrER, dar poate fi avută în vedere dacă este necesar. În urma analizei inițiale, ar trebui să se examineze fiecare curbă de legare pentru a asigura o corelare corespunzătoare cu modelul. Afinitatea de legare relativă (RBA) a unui liant slab poate fi calculată ca procent din log (IC50) pentru liantul slab raportat la valoarea log (IC50) pentru 17β-estradiol. Rezultatele pentru martorii pozitivi și pentru martorul non-liant ar trebui să fie evaluate utilizând măsuri ale performanței analizei și criterii de acceptabilitate, astfel cum sunt descrise în prezenta metodă de testare (punctul 20), apendicele 2 (analiza FW, punctele 41-51) și apendicele 3 (analiza CERI, punctele 41-51). În figura 1 sunt prezentate exemple de trei teste pentru estrogenul de referință și liantul slab.
|
Figura 1
Exemple de curbe de legare competitivă pentru estrogenul de referință și liantul slab martor

Analiza datelor pentru substanțele chimice de testat
|
3.
|
Datele pentru toate substanțele chimice de testat ar trebui să fie analizate printr-o abordare etapizată pentru a se asigura analizarea corespunzătoare a datelor și clasificarea corectă a fiecărei curbe de legare competitivă. Fiecare testare pentru o substanță chimică de testat să fie supusă inițial unei analize a datelor standardizată care este identică cu cea utilizată pentru estrogenul de referință și lianții slabi martori. După încheierea analizei, ar trebui să fie efectuată o analiză tehnică a parametrilor curbei și o verificare vizuală a modului în care datele se încadrează pe curba de legare competitivă generată pentru fiecare testare. În timpul acestei analize tehnice, observațiile privind o scădere dependentă de concentrație în procentul [3H]-17β-estradiol legat în mod specific, variabilitatea scăzută între replicile tehnice la fiecare concentrație chimică și consecvența parametrilor de corelare între cele trei teste constituie indicații bune pentru faptul că analiza și analizele datelor au fost efectuate în mod corespunzător. La examinarea rezultatelor fiecărui test, pentru o substanță chimică de testat, ar trebui să se aplice o apreciere profesională, iar datele utilizate pentru a clasifica fiecare substanță chimică de testat ca liant sau non-liant ar trebui să fie justificabile din punct de vedere științific.
|
|
4.
|
Ocazional, ar putea exista exemple de date care necesită o atenție suplimentară pentru a analiza și a interpreta în mod corespunzător datele privind legarea hrER. Există studii anterioare care au arătat cazuri în care analiza și interpretarea datelor privind legarea competitivă de receptori pot fi complicate de o creștere a valorii procentuale a legării specifice atunci când se testează substanțe chimice la cele mai mari concentrații (figura 2). Aceasta este o problemă bine-cunoscută care a fost întâlnită atunci când au fost utilizate protocoale pentru o serie de analize privind legarea competitivă de receptori (3). În aceste cazuri, se observă un răspuns dependent de concentrație la concentrații mai mici, dar deoarece concentrația substanței chimice de testat se apropie de limita solubilității, gradul de desprindere a [3H]17β-estradiol nu mai scade. În aceste cazuri, datele pentru concentrațiile mai mari indică faptul că s-a atins limita biologică a analizei. De exemplu, acest fenomen este asociat de multe ori cu insolubilitatea substanței chimice și precipitarea la concentrații ridicate sau poate fi, de asemenea, o reflectare a depășirii capacității cărbunelui acoperit cu dextran de a prinde ligandul marcat radioactiv nelegat în timpul procedurii de separare la cele mai mari concentrații chimice. Includerea unor astfel de puncte de date atunci când se înregistrează datele privind legarea competitivă pe o curbă sigmoidă poate conduce uneori la o clasificare eronată a potențialului de legătură de ER în cazul unei substanțe chimice de testat (figura 2). Pentru a evita acest lucru, protocolul pentru analizele FW și CERI privind legarea de hrER include o opțiune de a exclude din analize punctele de date în cazul în care media replicilor pentru legarea procentuală specifică a [3H]17β-estradiol este de cel puțin 10 %, peste media observată la o concentrație mai mică (și anume, aceasta este denumită regula de 10 %). Această regulă poate fi folosită o singură dată pentru o anumită curbă și trebuie să existe date care rămân pentru cel puțin 6 concentrații, astfel încât curba să poată fi corect clasificată.
|
Figura 2
Exemple, curbe privind legarea competitivă cu și fără utilizarea regulii de 10 %.
|
5.
|
Utilizarea adecvată a regulii de 10 % pentru a corecta aceste curbe ar trebui să fie analizată cu atenție și rezervată pentru cazurile în care există o indicație clară a unui liant al hrER. În cursul desfășurării experimentelor pentru studiul de validare a analizei FW privind legarea de hrER, s-a observat că, uneori, regula de 10 % a avut o consecință neintenționată și neprevăzută. Substanțele chimice care nu au interacționat cu receptorul (și anume, non-lianți reali) au prezentat adesea o variabilitate de aproximativ 100 % în legarea ligandului radioactiv, care a depășit 10 % în intervalul de concentrații testate. În cazul în care valoarea cea mai scăzută s-a întâmplat să fie la un nivel redus de concentrare, datele privind toate concentrațiile mai mari ar putea fi eliminate din analiză prin utilizarea regulii de 10 %, chiar dacă aceste concentrări ar putea fi utile în stabilirea statutului de non-liant al substanței chimice. Figura 3 prezintă exemple în care utilizarea regulii de 10 % nu este adecvată.
|
Figura 3
Exemple, date privind legarea competitivă în cazul cărora utilizarea regulii de 10 % nu este adecvată.

Referințe
|
(1)
|
OCDE (2015). Integrated Summary Report: Validation of Two Binding Assays Using Human Recombinant Estrogen Receptor Alpha (hrERα), Health and Safety Publications, Series on Testing and Assessment (nr. 226), Organizația pentru Cooperare și Dezvoltare Economică, Paris.
|
|
(2)
|
Motulsky H. și Christopoulos A. (2003). The law of mass action, In Fitting Models to Biological Data Using Linear and Non-linear Regression. GraphPad Software Inc., San Diego, CA, pp 187-191. Www.graphpad.com/manuals/Prism4/RegressionBook.pdf
|
|
(3)
|
Laws SC, Yavanhxay S, Cooper RL, Eldridge JC. (2006). Nature of the Binding Interaction for 50 Structurally Diverse Chemicals with Rat Estrogen Receptors. Toxicological Sci. 94(1):46-56.
|
B.71 TESTELE IN VITRO PRIVIND SENSIBILIZAREA CUTANATĂ, CARE ABORDEAZĂ EVENIMENTUL IMPORTANT PRIVIND ACTIVAREA CELULELOR DENDRITICE ASUPRA PARCURSULUI RĂSPUNSURILOR ADVERSE (ADVERSE OUTCOME PATHWAY - AOP) PENTRU SENSIBILIZAREA CUTANATĂ
INTRODUCERE GENERALĂ
Metodă de testare bazată pe evenimentul important privind activarea celulelor dendritice
|
1.
|
O substanță sensibilizantă pentru piele înseamnă o substanță care conduce la o reacție alergică în urma contactului cu pielea, astfel cum este definit în Sistemul global armonizat de clasificare și etichetare a substanțelor chimice al Organizației Națiunilor Unite (GHS al ONU) (1) și în Regulamentul (CE) nr. 1272/2008 al Parlamentului European și al Consiliului privind clasificarea, etichetarea și ambalarea substanțelor și a amestecurilor (CLP) (100). Există un acord general cu privire la principalele evenimentele biologice care stau la baza sensibilizării cutanate. Cunoștințele existente cu privire la mecanismele biologice și chimice asociate cu sensibilizarea cutanată au fost sintetizate sub forma unui parcurs al răspunsurilor adverse (AOP) în cadrul programului OCDE AOP (2), începând cu evenimentul declanșator molecular, prin evenimentele intermediare, până la efectele adverse, și anume dermatita alergică de contact. În acest caz, evenimentul declanșator molecular (mai exact, primul eveniment important) este reprezentat de legarea covalentă a substanțelor electrofile de centrele nucleofile din proteinele din piele. Al doilea eveniment important în acest AOP se desfășoară în cheratinocite și include răspunsuri inflamatorii, precum și modificări ale expresiei genice asociate cu parcursurile de semnalizare de celule specifice, precum parcursurile care depind de elemente de răspuns antioxidant/electrofil (ERA). Al treilea eveniment important este activarea celulelor dendritice (CD), de regulă evaluate prin exprimarea markerilor celulari de suprafață specifici, chemochine și citocine. Al patrulea eveniment important este activarea și proliferarea celulară T, care este evaluată în mod indirect în testul local asupra ganglionilor limfatici murini (LLNA) (3).
|
|
2.
|
Această metodă de testare (MT) este echivalentă cu Orientarea OCDE (TG) privind testarea nr. 442E (2017). Aceasta descrie teste in vitro care abordează mecanismele descrise în cadrul evenimentului important privind activarea celulelor dendritice ale AOP pentru sensibilizarea cutanată (2). MT cuprinde teste care urmează să fie utilizate pentru susținerea diferențierii între substanțele sensibilizante pentru piele și cele nesensibilizante în conformitate cu GHS al ONU și Regulamentul CLP.
|
|
Testele descrise în prezenta MT sunt:
|
—
|
Testul de activare a liniilor celulare umane (h-CLAT)
|
|
—
|
Testul U937 de activare a liniilor celulare (U-SENS ™)
|
|
—
|
Testul Interleuk-8 Reporter Gene Assay (testul IL-8 Luc)
|
|
|
|
3.
|
Testele incluse în această metodă de testare și în Orientarea TG corespunzătoare a OCDE pot fi diferite în ceea ce privește procedura utilizată pentru a genera datele și valorile măsurate, dar pot fi utilizate în mod nediscriminatoriu pentru a aborda cerințele țărilor pentru rezultatele testelor privind evenimentul cheie cu privire la activarea celulelor dendritice ale AOP pentru sensibilizarea cutanată, beneficiind în același timp de acceptarea reciprocă a datelor a OCDE.
|
Context și principii ale testelor incluse în metoda de testare bazată pe evenimentul important
|
4.
|
Evaluarea sensibilizării cutanate a implicat, de regulă, utilizarea de animale de laborator. Metodele clasice care utilizează cobai, testul de maximizare pe cobai (GPMT) al lui Magnusson și Kligman și testul Buehler (MT B.6) (4) evaluează atât fazele de inducție, cât și cele de reacție de hipersensibilitate a sensibilizării cutanate. Testele pe murini, LLNA (MT B.42) (3) și cele două variante ale sale care nu sunt radioactive, LLNA: DA (MT B.50) (5) și LLNA: BrdU-ELISA (MT B.51) (6), toate evaluează exclusiv răspunsul de inducție și, de asemenea, au fost acceptate, întrucât acestea oferă un avantaj față de testele pe cobai din punctul de vedere al bunăstării animalelor, alături de o măsurare obiectivă a fazei de inducție a sensibilizării cutanate.
|
|
5.
|
Au fost adoptate metode de testare in chemico și in vitro recente, care sunt bazate pe mecanică și care abordează primul eveniment important [(MT B.59; Testul reactivității directe a peptidelor (7)] și al doilea eveniment important [(MT B.60; metoda de testare ARE-Nrf2 ale luciferazei (8)] al sensibilizării cutanate AOP, pentru a contribui la evaluarea potențialului de pericol de sensibilizare cutanată pe care îl prezintă substanțele chimice.
|
|
6.
|
Încercările descrise în prezenta metodă de testare măsoară fie modificarea în exprimarea markerului (markerilor) celular(i) de suprafață asociat (asociați) procesului de activare a monocitelor și a CD în urma expunerii la substanțe sensibilizante (de exemplu: CD54, CD86), fie modificările în exprimarea IL-8, o citocină asociată cu activarea CD. Au fost raportate substanțe sensibilizante pentru piele pentru a induce exprimarea markerilor cu membrană celulară, precum CD40, CD54, CD80, CD83 și CD86, în plus față de inducerea citocinelor proinflamatorii, precum IL-1β și TNF-α, precum și a mai multor chemochine, inclusiv IL-8 (CXCL8) și CCL3 (9) (10) (11) (12), asociate cu activarea CD (2).
|
|
7.
|
Însă, întrucât activarea CD reprezintă doar un singur eveniment important al AOP privind sensibilizarea cutanată (2) (13), este posibil ca informațiile obținute în urma testelor care măsoară doar markerii activării CD să nu fie suficiente pentru a concluziona cu privire la prezența sau absența potențialului de sensibilizare cutanată al substanțelor chimice. Prin urmare, datele obținute în urma testelor descrise în cadrul acestei metode de testare sunt propuse pentru susținerea diferențierii dintre substanțele sensibilizante pentru piele (mai exact, categoria 1 din GHS al ONU/Regulamentul CLP) și substanțele nesensibilizante atunci când sunt utilizate în cadrul strategiilor integrate pentru testare și evaluare (IATA), împreună cu alte informații suplimentare relevante, de exemplu, cele rezultate din teste in vitro care abordează alte evenimente importante ale AOP privind sensibilizarea cutanată, precum și metode care nu implică testarea, inclusiv extrapolări ale datelor referitoare la substanțe chimice analoage (13). Exemple de utilizare de date generate în urma acestor teste în cadrul abordărilor definite, mai exact, al abordărilor standardizate atât în legătură cu setul de surse informaționale utilizate, cât și în cadrul procedurii aplicate datelor pentru obținerea de previziuni, au fost publicate (13) și pot fi folosite ca elemente utile în cadrul IATA.
|
|
8.
|
Testele descrise în prezenta metodă de testare nu pot fi folosite pe cont propriu, nici pentru a subclasifica substanțele sensibilizante pentru piele în subcategoriile 1A și 1B, astfel cum sunt definite în GHS al ONU/Regulamentul CLP, pentru autoritățile care pun în aplicare aceste două subcategorii opționale, și nici pentru a prevedea potența pentru deciziile în materie de evaluare a siguranței. Cu toate acestea, în funcție de cadrul de reglementare, rezultatele pozitive obținute cu aceste metode pot fi utilizate în mod independent pentru a clasifica o substanță chimică în categoria 1 din GHS al ONU/Regulamentul CLP.
|
|
9.
|
Termenul „substanță chimică de testat” este utilizat în această metodă de testare pentru a se referi la ceea ce este supus testării (101) și nu are legătură cu aplicabilitatea testelor la testarea substanțelor monocomponente și a substanțelor și/sau amestecurilor multicomponente. În prezent sunt disponibile informații limitate privind aplicabilitatea testelor la substanțe/amestecuri multicomponente (14) (15). Cu toate acestea, testele sunt aplicabile din punct de vedere tehnic pentru testarea substanțelor și amestecurilor multicomponente. Cu toate acestea, înainte de utilizarea acestei metode de testare pe un amestec pentru a genera date pentru un obiectiv de reglementare specific, se ia în considerare dacă și, în caz afirmativ, din ce cauză aceasta poate oferi rezultate adecvate în scopul respectiv (102). Astfel de considerații nu sunt necesare atunci când există o cerință normativă pentru testarea amestecului. În plus, atunci când se testează substanțe sau amestecuri multicomponente, se ia în considerare posibila interferență a componentelor citotoxice cu răspunsurile observate.
|
REFERINȚE BIBLIOGRAFICE
|
(1)
|
Organizația Națiunilor Unite ONU (2015). Sistemul global armonizat de clasificare și etichetare a substanțelor chimice (GHS). A șasea ediție revizuită. New York & Geneva: United Nations Publications. ISBN: 978-92-1-117006-1. Publicație disponibilă la adresa: https://www.unece.org/trans/danger/publi/ghs/ghs_rev06/06files_e.html.
|
|
(2)
|
OCDE (2012). The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins. Part 1: Scientific Evidence. Series on Testing and Assessment No. 168. Publicație disponibilă la adresa: http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=ENV/JM/MONO(2012)10/PART1&docLanguage=En.
|
|
(3)
|
Capitolul B.42 din prezenta anexă: Studiu pe ganglionul limfatic local.
|
|
(4)
|
Capitolul B.6 din prezenta anexă: Sensibilizare cutanată.
|
|
(5)
|
Capitolul B.50 din prezenta anexă: Sensibilizare cutanată: Studiu pe ganglionul limfatic local: DA.
|
|
(6)
|
Capitolul B.51 din prezenta anexă: Sensibilizare cutanată: Studiu pe ganglionul limfatic local: BrdU-ELISA.
|
|
(7)
|
Capitolul B.59 din prezenta anexă: Sensibilizarea cutanată in chemico: Testul reactivității directe a peptidelor (Direct Peptide Reactivity Assay – DPRA).
|
|
(8)
|
Capitolul B.60 din prezenta anexă: Sensibilizarea cutanată in vitro: Metoda de testare a ERA-Nrf2 ale luciferazei.
|
|
(9)
|
Steinman RM. (1991). The dendritic cell system and its role in immunogenicity. Annu Rev Immunol 9:271-96.
|
|
(10)
|
Caux C, Vanbervliet B, Massacrier C, Azuma M, Okumura K, Lanier LL și Banchereau J. (1994). B70/B7-2 is identical to CD86 and is the major functional ligand for CD28 expressed on human dendritic cells. J Exp Med 180:1841-7.
|
|
(11)
|
Aiba S, Terunuma A, Manome H și Tagami H. (1997). Dendritic cells differently respond to haptens and irritants by their production of cytokines and expression of co-stimulatory molecules. Eur J Immunol 27:3031-8.
|
|
(12)
|
Aiba S, Manome H, Nakagawa S, Mollah ZU, Mizuashi M, Ohtani T, Yoshino Y și Tagami. H. (2003). p38 mitogen-activated protein kinase and extracellular signal-regulated kinases play distinct roles in the activation of dendritic cells by two representative haptens, NiCl2 and DNCB. J Invest Dermatol 120:390-8.
|
|
(13)
|
OCDE (2016). Series on Testing & Assessment No 256: Guidance Document On The Reporting Of Defined Approaches And Individual Information Sources To Be Used Within Integrated Approaches To Testing And Assessment (IATA) For Skin Sensitisation, anexele 1 și 2. ENV/JM/HA(2016)29. Organizația pentru Cooperare și Dezvoltare Economică, Paris. Publicație disponibilă la adresa: https://community.oecd.org/community/iatass.
|
|
(14)
|
Ashikaga T, Sakaguchi H, Sono S, Kosaka N, Ishikawa M, Nukada Y, Miyazawa M, Ito Y, Nishiyama N, Itagaki H. (2010). A comparative evaluation of in vitro skin sensitisation tests: the human cell-line activation test (h-CLAT) versus the local lymph node assay (LLNA). Altern. Lab. Anim. 38, 275-284.
|
|
(15)
|
Piroird, C., Ovigne, J.M., Rousset, F., Martinozzi-Teissier, S., Gomes, C., Cotovio, J., Alépée, N. (2015). The Myeloid U937 Skin Sensitization Test (U-SENS) addresses the activation of dendritic cell event in the adverse outcome pathway for skin sensitization. Toxicol. In Vitro 29, 901-916.
|
Apendicele 1
Sensibilizarea Cutanată in vitro: Testul de Activare a Liniilor Celulare Umane (h-clat)
CONSIDERAȚII PRELIMINARE ȘI LIMITĂRI
|
1.
|
Testul h-CLAT cuantifică modificările produse la nivelul exprimării markerilor celulari de suprafață asociați cu procesul de activare a monocitelor și celulelor dendritice (CD) (și anume, CD86 și CD54), la nivelul liniei celulare monocitice umane THP-1 afectate de leucemie în urma expunerii la substanțe sensibilizante (1)(2). Nivelurile de exprimare măsurate ale markerilor celulari de suprafață CD86 și CD54 sunt apoi utilizate pentru susținerea diferențierii între substanțele sensibilizante pentru piele și cele nesensibilizante.
|
|
2.
|
Testul h-CLAT a fost evaluat într-un laborator de referință al Uniunii Europene pentru metode alternative la testarea pe animale (EURL ECVAM) - un studiu de validare coordonat și o evaluare inter pares independentă ulterioară desfășurată de către Comitetul științific consultativ al EURL ECVAM (ESAC). Având în vedere toate dovezile disponibile și contribuțiile autorităților de reglementare și ale părților interesate, testul h-CLAT a fost recomandat de EURL ECVAM (3) pentru a fi utilizat în cadrul unei IATA în vederea sprijinirii diferențierii între substanțele sensibilizante și cele nesensibilizante în scopul clasificării și etichetării pericolelor. Exemple privind utilizarea datelor h-CLAT în combinație cu alte informații sunt prezentate în referințele bibliografice (4)(5)(6)(7)(8)(9)(10)(11).
|
|
3.
|
Testul h-CLAT s-a dovedit a fi transferabil către laboratoare cu experiență în tehnici de cultură celulară și analiza citometriei de flux. Nivelul de reproductibilitate în estimări, care poate fi preconizat în urma testării, este de ordinul a 80 % în cadrul aceluiași laborator și între laboratoare (3) (12). Rezultatele generate și incluse în studiul de validare (13), precum și în alte studii publicate (14), indică, în general, faptul că, în comparație cu rezultatele LLNA, precizia de deosebire a substanțelor sensibilizante pentru piele (și anume, categoria 1 din GHS al ONU/Regulamentul CLP) de cele nesensibilizante este de 85 % (N = 142) cu o sensibilitate de 93 % (94/101) și o specificitate de 66 % (27/41) (pe baza unei analize repetate efectuate de EURL ECVAM (12), având în vedere toate datele existente și neluând în calcul rezultatele negative pentru substanțele chimice cu o valoare a Log Kow mai mare de 3,5, astfel cum este descris la alineatul (4). Există o probabilitate mai mare ca estimările negative false realizate prin testul h-CLAT să vizeze substanțe chimice care prezintă un nivel scăzut până la moderat de potență de sensibilizare cutanată (și anume, subcategoria 1B din GHS al ONU/Regulamentul CLP) și nu substanțe chimice care prezintă un nivel ridicat de potență de sensibilizare cutanată (și anume, subcategoria 1A din GHS al ONU/Regulamentul CLP) (4) (13) (15). Coroborate, aceste informații indică utilitatea metodei h-CLAT în ceea ce privește contribuirea la identificarea pericolelor de sensibilizare cutanată. Cu toate acestea, valorile de precizie furnizate în prezentul document pentru testul h-CLAT ca test independent sunt doar orientative, întrucât testul ar trebui luat în considerare în combinație cu alte surse de informații în contextul unei IATA și în conformitate cu dispozițiile de la alineatele (7) și (8) din Introducerea generală. În plus, la evaluarea metodelor care nu se bazează pe animale pentru sensibilizarea cutanată, ar trebui reținut faptul că este posibil ca testul LLNA și alte teste pe animale să nu reflecte pe deplin situația oamenilor.
|
|
4.
|
Pe baza datelor disponibile, metoda h-CLAT s-a dovedit a fi aplicabilă substanțelor chimice de testat care acoperă o varietate de grupe organice funcționale, mecanisme de reacție, potență de sensibilizare cutanată (astfel cum a fost stabilit în cadrul studiilor in vivo), precum și proprietăți fizico-chimice (3) (14) (15). Metoda h-CLAT se aplică substanțelor chimice de testat solubile sau care formează dispersii stabile (și anume, un coloid sau o suspensie în care substanța chimică de testat nu se sedimentează sau se separă de solvent/vehicul în diferite faze) într-un solvent/vehicul adecvat [a se vedea alineatul (14)]. Substanțele chimice de testat cu un Log Kow mai mare de 3,5 tind să producă rezultate negative false (14). Prin urmare, nu ar trebui să fie luate în considerare rezultatele negative obținute în cazul substanțelor chimice de testat având o valoare Log Kow mai mare de 3,5. Cu toate acestea, rezultatele pozitive obținute în cazul substanțelor chimice de testat având o valoare Log Kow mai mare de 3,5 ar putea fi totuși utilizate pentru a sprijini identificarea substanței chimice de testat ca substanță sensibilizantă pentru piele. În plus, din cauza capacității metabolice limitate a liniei celulare utilizate (16), cât și din cauza condițiilor experimentale, prohaptenele (și anume, substanțele care necesită activare enzimatică, de exemplu prin intermediul enzimelor P 450) și prehaptenele (și anume, substanțe activate prin autooxidare), în special cele cu o rată de oxidare lentă, pot furniza, de asemenea, rezultate negative la testul h-CLAT (15). Substanțele chimice de testat fluorescente pot fi analizate prin testul h-CLAT (17); cu toate acestea, substanțele chimice de testat fluorescente puternice care emit la aceeași lungime de undă ca și izotiocianatul de fluoresceină (FITC) sau iodura de propidiu (PI) vor interfera cu detecția prin citometria de flux și, astfel, nu pot fi evaluate în mod corect folosind anticorpi conjugați cu FITC sau PI. Într-un astfel de caz, se pot folosi alți anticorpi marcați cu fluorocrom sau alți markeri de citotoxicitate, cu condiția să se poată demonstra că acestea oferă rezultate similare celor generate de anticorpii marcați cu FITC [a se vedea alineatul (24)] sau PI [a se vedea alineatul (18)], de exemplu prin testarea substanțelor de verificare din apendicele 1-2. Având în vedere cele de mai sus, rezultatele negative ar trebui interpretate în contextul limitărilor menționate și împreună cu alte surse de informații în cadrul IATA. În cazul în care există dovezi care demonstrează lipsa de aplicabilitate a metodei h-CLAT la alte categorii specifice de substanțe chimice de testat, acestea nu ar trebui utilizate pentru respectivele categorii specifice.
|
|
5.
|
Conform descrierii de mai sus, metoda h-CLAT susține diferențierea între substanțele sensibilizante pentru piele și cele nesensibilizante. Însă aceasta poate să contribuie, de asemenea, la evaluarea potenței de sensibilizare (4)(5)(9) atunci când este utilizată în abordări integrate precum IATA. Cu toate acestea, se impun studii suplimentare, de preferință pe baza datelor cu privire la oameni, pentru a determina în ce mod ar putea rezultatele h-CLAT să fundamenteze evaluarea potenței.
|
|
6.
|
Definițiile sunt prevăzute în apendicele 1.1.
|
PRINCIPIUL TESTULUI
|
7.
|
Metoda h-CLAT reprezintă un test in vitro care cuantifică modificările expresiei markerului celular de suprafață (și anume, CD86 și CD54) pe o linie celulară monocitică umană afectată de leucemie, celule THP-1, în urma expunerii timp de 24 de ore la substanța chimică de testat. Aceste molecule de suprafață constituie markeri tipici de activare THP-1 monocitică și pot imita activarea DC, care joacă un rol esențial în pregătirea de celule de tip T. Modificările expresiei markerului de suprafață se măsoară prin citometria de flux în urma colorării celulelor cu anticorpi marcați cu fluorocrom. De asemenea, măsurarea citotoxicității este efectuată în același timp cu scopul de a se evalua dacă se produce o reglare în sus a expresiei markerului de suprafață în cazul concentrațiilor subcitotoxice. Intensitatea relativă a fluorescenței markerilor de suprafață, în comparație cu martorul tratat cu solvent/vehicul, se calculează și se utilizează în modelul predictiv [a se vedea alineatul (26)], pentru a susține diferențierea între substanțe sensibilizante și nesensibilizante
|
DEMONSTRAREA PERFORMANȚEI
|
8.
|
Înainte de a introduce testul descris în prezentul apendice la metoda de testare B.71 pentru utilizare de rutină, laboratoarele ar trebui să demonstreze performanțele tehnice utilizând cele zece substanțe de verificare menționate în apendicele 1.2. În plus, utilizatorii testului ar trebui să întrețină o bază de date istorică incluzând date generate în urma verificărilor de reactivitate [a se vedea alineatul (11)] și a martorilor pozitivi și a celor tratați cu solvenți/vehicule [a se vedea alineatele (20)-(22)] și să utilizeze aceste date pentru a confirma faptul că reproductibilitatea testului în laboratorul lor este menținută în timp.
|
PROCEDURA
|
9.
|
Acest test este bazat pe protocolul nr. 158 (18) h-CLAT DataBase service on ALternative Methods to animal experimentation (DB-ALM), care reprezintă protocolul utilizat pentru studiul de validare coordonat de EURL ECVAM. Se recomandă utilizarea acestui protocol atunci când se pune în aplicare și se utilizează metoda h-CLAT în laborator. Componentele și procedurile principale pentru metoda h-CLAT sunt descrise în continuare, în două etape: testul de determinare a dozei și măsurarea expresiei CD86/CD54.
|
Pregătirea celulelor
|
10.
|
Linia celulară monocitică umană afectată de leucemie, și anume THP-1, ar trebui să fie utilizată pentru desfășurarea metodei h-CLAT. Se recomandă ca celulele (TIB-202™) să fie obținute de la o bancă de celule calificată, cum ar fi American Type Culture Collection.
|
|
11.
|
Celulele THP-1 sunt cultivate la 37oC în atmosferă de 5 % CO2 și de umiditate, în mediu RPMI-1640 completat cu 10 % ser fetal bovin (FBS), 0,05 mM 2-mercaptoetanol, 100 de unități/ml de penicilină și 100 μg/ml de streptomicină. Poate fi evitată utilizarea penicilinei și a streptomicinei în mediul de cultură. Însă, într-un astfel de caz, utilizatorii ar trebui să verifice dacă absența antibioticelor din mediul de cultură nu are niciun impact asupra rezultatelor, de exemplu prin testarea substanțelor de verificare enumerate în apendicele 1.2. În orice caz, pentru a reduce la minimum riscul de contaminare, bunele practici de cultură celulară ar trebui să fie urmate independent de prezența sau absența antibioticelor în mediul de cultură celulară. Celulele THP-1 sunt însămânțate în mod obișnuit la 2-3 zile la densitatea de 0,1-0,2 × 106 celule/ml. Acestea ar trebui să fie menținute la densități cuprinse între 0,1 și 1,0 × 106 celule/ml. Înainte de a le utiliza pentru testare, celulele ar trebui să fie calificate prin efectuarea unui control de reactivitate. Controlul de reactivitate ar trebui să fie efectuat asupra celulelor folosindu-se martori pozitivi, 2,4-dinitroclorbenzen (DNCB) (CAS nr. 97-00-7, puritate ≥ 99 %) și sulfat de nichel (NiSO4) (CAS nr. 10101-97-0, puritate ≥ 99 %) și martorul negativ, acidul lactic (AL) (CAS nr. 50-21-5, puritate ≥ 85 %), la două săptămâni după decongelare. Atât DNCB, cât și NiSO4 ar trebui să genereze un răspuns pozitiv al markerilor celulari de suprafață CD86 și CD54, iar AL ar trebui să genereze un răspuns negativ al markerilor celulari de suprafață CD86 și CD54. Sunt utilizate pentru analiză numai celulele pentru care verificarea de reactivitate a fost efectuată cu succes. Celulele pot fi reproduse până la două luni după decongelare. Numărul de trecere nu ar trebui să depășească 30. Verificarea de reactivitate ar trebui să fie efectuată conform procedurilor descrise la punctele 20-24.
|
|
12.
|
În scopul testării, celulele THP-1 sunt însămânțate la o densitate de 0,1 × 106 celule/ml sau de 0,2 × 106 celule/ml și cultivate în prealabil în baloane de cultură timp de 72 de ore sau, respectiv, 48 de ore. Este important ca densitatea celulară din balonul de cultură, imediat după perioada de cultivare prealabilă, să fie cât mai consecventă posibil în fiecare experiment (aplicându-se una dintre cele două condiții de cultivare prealabilă descrise mai sus), deoarece densitatea celulară din balonul de cultură, imediat după cultivarea prealabilă, ar putea afecta expresia CD86/CD54 indusă de alergeni (19). În ziua testării, celulele recoltate din balonul de cultură sunt suspendate din nou în mediu de cultură proaspăt, la 2 × 106 celule/ml. Apoi, celulele sunt distribuite pe o tavă cu fund plat cu 24 de godeuri (500 μl, 1 × 106 celule/godeu) sau pe o tavă cu fund plat cu 96 de godeuri (80 μl, 1,6 × 105 celule/godeu).
|
Testul de determinare a dozei
|
13.
|
Se efectuează un test de determinare a dozei pentru determinarea CV75, care este concentrația substanței chimice de testat, în urma căruia să se ajungă la o viabilitate celulară (VC) de 75 % în comparație cu martorul tratat cu solvent/vehicul. Valoarea CV75 este utilizată pentru a determina concentrația substanțelor chimice de testat pentru măsurarea expresiei CD86/CD54 [a se vedea alineatele (20)-(24)].
|
Pregătirea substanțelor chimice de testat și a substanțelor martor
|
14.
|
Substanțele chimice de testat și substanțele martor se pregătesc în ziua testării. Pentru metoda h-CLAT, substanțele chimice de testat sunt dizolvate sau dispersate în mod stabil [a se vedea, de asemenea, alineatul (4)] în soluție salină sau în mediu ca prime opțiuni de solvent/vehicul sau în dimetil sulfoxid (DMSO, puritate ≥99 %) ca a doua opțiune de solvent/vehicul dacă substanța chimică de testat nu este solubilă sau nu formează dispersii stabile în cei doi solvenți/vehicule anterioare, în concentrații finale de 100 mg/ml (în soluție salină sau în mediu) sau de 500 mg/ml (în DMSO). Se pot utiliza și alți solvenți/vehicule decât cele descrise mai sus în cazul în care se prezintă o justificare științifică suficientă. Ar trebui să se țină seama de stabilitatea substanței chimice de testat în solventul/vehiculul final.
|
|
15.
|
Pornind de la soluțiile mamă ale substanțelor de testat de 100 mg/ml (în soluție salină sau medie) sau de 500 mg/ml (în DMSO), ar trebui să fie parcurse următoarele etape de diluare:
|
—
|
Pentru soluția salină sau mediu ca solvent/vehicul: Se prepară opt soluții mamă (opt concentrații) cu ajutorul unor diluții în serie duble utilizând solventul/vehiculul aferent. Aceste soluții mamă sunt apoi diluate în continuare de 50 de ori în mediu de cultură (soluții de lucru). În cazul în care concentrația finală în partea superioară, în tava de 1 000 μg/ml, nu este toxică, concentrația maximă ar trebui să fie determinată din nou prin efectuarea unui nou test de citotoxicitate. Concentrația finală în placă nu ar trebui să depășească 5 000 μg/ml în cazul substanțelor chimice de testat dizolvate sau dispersate în mod stabil în soluție salină sau în mediu salin.
|
|
—
|
În cazul DMSO ca solvent/vehicul: Se prepară opt soluții mamă (opt concentrații) cu ajutorul unor diluții în serie duble utilizând solventul/vehiculul aferent. Aceste soluții mamă sunt apoi diluate în continuare de 250 de ori în mediu de cultură (soluții de lucru). Concentrația finală în plăci nu ar trebui să depășească 1 000 μg/ml, chiar dacă această concentrație nu este toxică.
|
Soluțiile de lucru sunt utilizate, în cele din urmă, pentru expunere prin adăugarea unui volum egal de soluție de lucru la volumul suspensiei celulare THP-1 din placă (a se vedea, de asemenea, punctul 17) pentru a obține încă o diluare dublă (de regulă, plaja finală de concentrații de pe placă este de 7,81-1 000 μg/ml).
|
|
16.
|
Martorul tratat cu solvent/vehicul utilizat în metoda h-CLAT este mediu de cultură [pentru substanțele chimice de testat solubilizate sau dispersate în mod stabil (a se vedea punctul 4 fie în mediu, fie în soluție salină) sau DMSO (pentru substanțe chimice de testat solubilizate sau dispersate în mod stabil în DMSO) testat la o singură concentrație finală în placă de 0,2 %. Acesta este supus aceluiași proces de diluție descris pentru soluțiile de lucru de la punctul 15.
|
Aplicarea substanțelor chimice de testat și a substanțelor martor
|
17.
|
Mediul de cultură sau soluțiile de lucru descrise la punctele 15 și 16 sunt amestecate 1: 1 (v/v) cu suspensiile de celule preparate pe placa cu fund plat de 24 sau 96 de godeuri (a se vedea punctul 12). Plăcile tratate sunt apoi incubate timp de 24 ± 0,5 ore la 37oC în soluție cu 5 % CO2. Ar trebui să se acorde o atenție deosebită pentru a se evita evaporarea substanțelor chimice de testat volatile și contaminarea încrucișată între godeuri a substanțelor chimice de testat, de exemplu, prin acoperirea etanșă a plăcilor înainte de incubația cu substanțele chimice de testat (20).
|
Colorarea cu iodură de propidiu (IP)
|
18.
|
După 24 ± 0,5 ore de expunere, celulele sunt transferate în eprubete de eșantioane și colectate prin centrifugare. Se îndepărtează supernatantele și se suspendă din nou celulele rămase în 200 μl (în cazul plăcii cu 96 de godeuri) sau în 600 μl (în cazul plăcii cu 24 de godeuri) de soluție salină tamponată cu fosfat conținând 0,1 % albumină din ser bovin (soluție tampon de colorare). Se transferă 200 μl de suspensie celulară pe o placă cu fund rotund de 96 de godeuri (în cazul plăcii cu 96 de godeuri) sau în micro-eprubetă (în cazul unei plăci cu 24 de godeuri) și se spală de două ori într-o cantitate de 200 μl (în cazul plăcii cu 96 de godeuri) sau de 600 μl (în cazul plăcii cu 24 de godeuri) de soluție tampon de colorare. În cele din urmă, celulele sunt suspendate din nou în soluție tampon de colorare (de exemplu, 400 μl) și se adaugă soluție IP (de exemplu, 20 μl) (spre exemplu, concentrația finală a IP este de 0,625 μg/ml). Se pot utiliza alți markeri de citotoxicitate, cum ar fi 7-aminoactinomicina D (7-AAD), albastru de tripan sau alții, în cazul în care se poate demonstra că acțiunile de colorare alterative generează rezultate similare, de exemplu, prin testarea substanțelor de verificare din apendicele 1.2.
|
Măsurarea citotoxicității prin citometria de flux și estimarea valorii CV75
|
19.
|
Absorbția IP este analizată utilizând citometria de flux la canalul de acumulare FL-3. Sunt acumulate în total 10 000 de celule vii (IP negativă). Viabilitatea celulară poate fi calculată folosind următoarea ecuație prin programul de analiză citometrică. Atunci când viabilitatea celulară este scăzută, trebuie să se acumuleze până la 30 000 de celule, inclusiv celule moarte. În mod alternativ, pot fi acumulate date timp de un minut după inițierea analizei.

Valoarea CV75 (a se vedea punctul 13), adică o concentrație care indică o rată de 75 % a THP-1 de supraviețuire a celulelor (25 % citotoxicitate), este calculată prin interpolare log-lineară folosind următoarea ecuație:

Unde:
a reprezintă valoarea minimă a viabilității celulare peste 75 %
c reprezintă valoarea maximă a viabilității celulare sub 75 %
b și d sunt concentrații care arată valoarea viabilității celulare a și, respectiv, c

Se pot utiliza alte abordări pentru derivarea valorii CV75, cu condiția să se demonstreze că acest lucru nu are niciun impact asupra rezultatelor (de exemplu, prin testarea substanțelor de verificare).
|
Măsurarea expresiei CD86/CD54
Pregătirea substanțelor chimice de testat și a substanțelor martor
|
20.
|
Solventul/vehiculul corespunzător (soluție salină, medie sau DMSO; a se vedea punctul 14) este utilizat pentru a dizolva sau a dispersa în mod stabil substanțele chimice de testat. Substanțele chimice de testat sunt diluate mai întâi la concentrația corespunzătoare valorii de 100 de ori (pentru soluția salină sau mediu) sau de 500 de ori (pentru DMSO) din 1,2 × CV75, determinate în testul de determinare a dozei (a se vedea punctul 19). În cazul în care valoarea CV75 nu poate fi determinată (și anume, dacă nu se observă un grad suficient de citotoxicitate în testul de determinare a dozei), ar trebui utilizată ca și concentrație inițială cea mai mare concentrație solubilă sau dispersată în mod stabil a substanței chimice de testat preparată împreună cu fiecare solvent/vehicul. Concentrația finală în placă nu ar trebui să depășească valoarea de 5 000 μg/ml (în cazul soluției saline sau al mediului) sau de 1 000 μg/ml (în cazul DMSO). Apoi sunt efectuate diluții în serie de 1,2 ori folosind solventul/vehiculul aferent pentru a obține soluțiile mamă [opt concentrații cuprinse între 100×1,2 × CV75 și 100×0,335 × CV75 (în cazul soluției saline sau al mediului) sau între 500×1,2 × CV75 și 500×0,335 × CV75 (în cazul DMSO)] pentru a fi testate prin metoda h-CLAT (a se vedea protocolul DB-ALM nr. 158 pentru un exemplu al schemei de dozare). Soluțiile mamă sunt apoi diluate în continuare de 50 de ori (în cazul soluției saline sau al mediului) sau de 250 de ori (în cazul DMSO) în mediul de cultură (soluțiile de lucru). Aceste soluții de lucru sunt utilizate, în cele din urmă, pentru expunerea la un alt factor final de diluare dublă în placă. În cazul în care rezultatele nu îndeplinesc criteriile de acceptare descrise la punctele 29 și 30 cu privire la viabilitatea celulară, testul de determinare a dozei poate fi repetat pentru a determina o valoare CV75 mai exactă. Este de precizat faptul că pot fi utilizate doar plăci cu 24 de godeuri pentru măsurarea expresiei CD86/CD54.
|
|
21.
|
Martorul tratat cu solvent/vehicul este preparat astfel cum este descris la punctul 16. Martorul pozitiv utilizat în metoda h-CLAT este DNCB (a se vedea punctul 11), pentru care soluțiile mamă sunt preparate în DMSO și diluate astfel cum este descris în cazul soluțiilor mamă de la punctul 20. DNCB ar trebui utilizat ca martor pozitiv pentru măsurarea expresiei CD86/CD54 la o concentrație unică finală în placă (de regulă, 4,0 μg/ml). Pentru a obține o concentrație de 4,0 μg/ml a DNCB în placă, se prepară o soluție mamă de 2 mg/ml de DNCB, care este apoi diluată în continuare de 250 de ori în mediu de cultură până la o soluție de lucru de 8 μg/ml. În mod alternativ, valoarea CV75 a DNCB, care este determinată în fiecare instalație de testare, ar putea fi utilizată, de asemenea, ca o concentrație a martorului pozitiv. Ar putea fi utilizați și alți martori pozitivi adecvați în cazul în care sunt disponibile date anterioare pentru a obține criterii comparabile de acceptare a testelor. În cazul martorilor pozitivi, concentrația finală unică în placă nu ar trebui să depășească valoarea de 5 000 μg/ml (în cazul soluției saline sau al mediului) sau de 1 000 μg/ml (în cazul DMSO). Criteriile de acceptare a testelor sunt aceleași cu cele descrise pentru substanța chimică testată (a se vedea punctul 29), cu excepția ultimului criteriu de acceptare, deoarece martorul pozitiv este testat într-o singură concentrație.
|
Aplicarea substanțelor chimice de testat și a substanțelor martor
|
22.
|
Pentru fiecare substanță chimică de testat și substanță martor este necesar un singur experiment pentru a obține o predicție. Fiecare experiment constă în cel puțin două testări independente pentru măsurarea expresiei CD86/CD54 (a se vedea punctele 26-28). Fiecare test independent este efectuat într-o altă zi sau în aceeași zi, cu condiția ca pentru fiecare test: a) să se prepare soluții mamă și soluții de lucru ale substanțelor chimice de testat și ale soluțiilor de anticorpi independente și proaspete și b) să se utilizeze celule recoltate în mod independent (de exemplu, se colectează celule din flacoane de cultură diferite); însă celulele pot proveni din aceeași trecere. Substanțele chimice de testat și substanțele martor preparate ca soluții de lucru (500 μl) sunt amestecate cu 500 μl de celule suspendate (1 x 106 celule) la un raport de 1:1, iar celulele sunt incubate timp de 24 ± 0,5 ore, conform descrierii de la punctele 20 și 21. În cadrul fiecărui test, o singură replică pentru fiecare concentrație a substanței chimice de testat și a substanței martor este suficientă, deoarece se obține o predicție din cel puțin două teste independente.
|
Colorarea și analiza celulelor
|
23.
|
După 24 ± 0,5 ore de expunere, celulele sunt transferate din placa de 24 de godeuri în eprubete pentru probe, colectate prin centrifugare și apoi spălate de două ori cu 1 ml de soluție tampon de colorare (dacă este necesar, se pot lua măsuri suplimentare de spălare). După spălare, celulele sunt blocate cu 600 μl de soluție de blocare [soluție tampon de colorare care conține globulină 0,01 % (g/v) (fracția Cohn fracțiune II, III, umană; SIGMA, #G2388-10G sau echivalentul)] și incubate la 4°C timp de 15 minute. După blocare, celulele sunt împărțite în trei părți alicote de 180 μl pe o placă de 96 de godeuri cu fund rotund sau în micro-eprubetă.
|
|
24.
|
După centrifugare, celulele sunt colorate cu 50 μl de soluție de anticorpi anti-CD86, anti-CD54 sau de șoarece IgG1 (izotip) etichetată FITC la 4oC timp de 30 de minute. Anticorpii descriși în protocolul DB-ALM nr. 158 al testului h-CLAT (18) ar trebui să fie utilizați prin diluarea în proporție de 3:25 v/v [pentru CD86 (BD-PharMingen, #555657; Clone: Fun-1)] sau de 3:50 v/v [pentru CD54 (DAKO, #F7143; Clone: 6,5B5) și IgG1 (DAKO, #X0927)] cu soluție tampon de colorare. Acești factori de diluție a anticorpilor au fost definiți de către dezvoltatorii de teste ca fiind cei care asigură cel mai bun raport semnal/zgomot. Pe baza experienței dezvoltatorilor de teste, intensitatea fluorescenței anticorpilor este, de obicei, consecventă între diferite loturi. Cu toate acestea, utilizatorii ar putea avea în vedere titrarea anticorpilor în condițiile din laboratorul propriu pentru a defini cele mai bune concentrații de utilizare. Se pot utiliza și alți anticorpi anti-CD86 marcați cu fluorocrom și/sau anti-CD54 în cazul în care se poate demonstra că aceștia generează rezultate similare cu cele ale anticorpilor conjugați FITC, de exemplu prin testarea substanțelor de verificare din apendicele 1.2. Trebuie remarcat faptul că modificarea clonei sau a furnizorului anticorpilor descriși în protocolul h-CLAT DB-ALM nr. 158 (18) poate afecta rezultatele. După ce sunt spălate de două sau de mai multe ori în 150 μl de soluție-tampon de colorare, celulele sunt resuspendate în soluție-tampon de colorare (de exemplu, 400 μl) și se adaugă soluție IP (de exemplu, 20 μl pentru a obține o concentrație finală de 0,625 μg/ml) sau o altă soluție a markerului de citotoxicitate (a se vedea punctul 18). Nivelurile de expresie ale CD86 și CD54 și viabilitatea celulară sunt analizate folosind citometria de flux.
|
DATE ȘI RAPORTARE
Evaluarea datelor
|
25.
|
Expresia CD86 și CD54 este analizată utilizând citometria de flux la canalul de acumulare FL-1. Pe baza intensității medii geometrice a fluorescenței (MFI), intensitatea relativă a fluorescenței (RFI) a CD86 și CD54 pentru celule și celule tratate chimic cu martor (ctrl) pozitiv se calculează conform ecuației următoare:
Viabilitatea celulară de la celulele cu martor izotip (ctrl) (care sunt colorate cu anticorpi IgG1 (izotip) de șoarece) este calculată, de asemenea, conform ecuației descrise la punctul 19.
|
Model predictiv
|
26.
|
Pentru măsurarea expresiei CD86/CD54, fiecare substanță chimică de testat este testată prin cel puțin două teste independente pentru a se obține o singură estimare (POZITIV sau NEGATIV). O estimare h-CLAT este considerată POZITIVĂ dacă este îndeplinită cel puțin una dintre următoarele condiții în cadrul derulării a 2 sau a cel puțin 2 din 3 teste independente; în caz contrar, estimarea h-CLAT este considerată NEGATIVĂ (figura 1):
|
—
|
Valoarea RFI a CD86 este mai mare sau egală cu 150 % la orice concentrație testată (cu viabilitate celulară ≥ 50 %);
|
|
—
|
Valoarea RFI a CD54 este mai mare sau egală cu 200 % la orice concentrație testată (cu viabilitate celulară ≥ 50 %).
|
|
|
27.
|
Pe baza celor de mai sus, dacă primele două teste generează rezultate pozitive pentru CD86 și/sau rezultate pozitive pentru CD54, estimarea h-CLAT este considerată a fi POZITIVĂ și nu este necesară derularea unui al treilea test. În mod similar, în cazul în care primele două teste sunt negative pentru ambii markeri, estimarea h-CLAT este considerată a fi NEGATIVĂ (ținând seama în mod corespunzător de dispozițiile de la punctul 30), fără a fi necesară derularea unui al treilea test. Însă, dacă rezultatele primelor două teste nu sunt concordante pentru cel puțin unul dintre markeri (CD54 sau CD86), este necesară derularea unui al treilea test, iar estimarea finală va fi bazată pe rezultatul majoritar al celor trei teste individuale (și anume, două din trei). În acest sens, ar trebui remarcat faptul că, dacă sunt derulate două teste independente, iar unul este pozitiv doar pentru CD86 (denumit în continuare „P1”) și celălalt este pozitiv doar pentru CD54 (denumit în continuare „P2”), este necesară derularea unui al treilea test. În cazul în care acest al treilea test este negativ pentru ambii markeri (denumiți în continuare „N”), estimarea h-CLAT este considerată ca fiind NEGATIVĂ. Pe de altă parte, în cazul în care al treilea test este pozitiv pentru oricare dintre markeri (P1 sau P2) sau pentru ambii markeri (denumit în continuare „P12”), estimarea h-CLAT este considerată POZITIVĂ.
Figura 1: Model predictiv utilizat în metoda h-CLAT. O estimare h-CLAT ar trebui să fie analizată în cadrul unei IATA și în conformitate cu dispozițiile de la punctele 7 și 8 din Introducerea generală.
P1: test cu estimare pozitivă doar pentru CD86; P2; test cu estimare pozitivă doar pentru CD54; P12: test cu estimare pozitivă pentru CD86 și CD54; N: test cu estimare pozitivă pentru CD86 sau CD54.
*Casetele prezintă combinațiile relevante ale rezultatelor obținute în cadrul primelor două teste, independent de ordinea în care acestea ar putea fi obținute.
#Casetele prezintă combinațiile relevante ale rezultatelor obținute în cadrul cele trei teste, pe baza rezultatelor obținute în cadrul primelor două teste prezentate în caseta de mai sus, însă nu reflectă ordinea în care acestea ar putea fi obținute.
|
|
28.
|
Pentru substanțele chimice de testat estimate ca fiind POZITIVE cu testul h-CLAT, ar putea fi determinate opțional două valori ale concentrațiilor efectivă (CE), CE150 pentru CD86 și CE200 pentru CD54, și anume concentrația la care substanțele chimice de testat au indus o valoare RFI de 150 sau 200. Aceste valori CE ar putea contribui la evaluarea potenței de sensibilizare (9) atunci când sunt utilizate în abordări integrate precum IATA (4) (5) (6) (7) (8). Acestea sunt calculate pe baza ecuațiilor următoare:
EC150 (for CD86) = Bconc
+ [(150 - BRFI
)/ARFI - BRFI
) × (Aconc - Bconc
)]
EC200 (for CD54) = Bconc
+ [(200 - BRFI
)/ARFI - BRFI
) × (Aconc - Bconc
)]
unde
Aconc este concentrația cea mai scăzută în μg/ml cu RFI > 150 (CD86) sau 200 (CD54)
Bconc este concentrația cea mai ridicată în μg/ml cu RFI < 150 (CD86) sau 200 (CD54)
ARFI este valoarea RFI la concentrația cea mai scăzută cu RFI > 150 (CD86) sau 200 (CD54)
BRFI este valoarea RFI la concentrația cea mai ridicată cu RFI < 150 (CD86) sau 200 (CD54)
Cu scopul de a deriva cu o precizie mai mare valorile CE150 și CE200, se pot solicita trei teste independente pentru măsurarea expresiei CD86/CD54. Valorile finale ale CE150 și CE200 sunt determinate apoi ca valoarea mediană a valorilor CE calculate în cadrul celor trei teste independente. Atunci când numai două din trei teste independente îndeplinesc criteriile de pozitivitate (a se vedea punctele 26-27), se adoptă valoarea cea mai mare CE150 sau CE200 dintre cele două valori calculate.
|
Criterii de acceptare
|
29.
|
Atunci când este utilizată metoda h-CLAT (22) (27), trebuie să fie îndeplinite următoarele criterii de acceptare.
|
—
|
Viabilitatea celulară a mediului și a martorilor tratați cu solvenți/vehicule ar trebui să fie mai mare de 90 %.
|
|
—
|
La martorul tratat cu solvent/vehicul, valorile RFI ale CD86 și CD54 nu ar trebui să fie peste criteriile pozitive (RFI ≥150 % pentru CD86 și RFI ≥ 200 % pentru CD54). Valorile RFI ale martorului tratat cu solvent/vehicul se calculează cu ajutorul formulei descrise la punctul 25 [„valoarea IMF a substanței chimice” ar trebui să fie înlocuită cu „valoarea IMF a solventului/vehiculului”, iar „valoarea IMF a solventului/vehiculului” ar trebui să fie înlocuită cu „valoarea IMF a martorului (mediului)”].
|
|
—
|
În cazul martorilor pentru mediu și a celor tratați cu solvent/vehicul, raportul IMF al CD86 și CD54 pe martor izotop ar trebui să fie > 105 %.
|
|
—
|
În cazul martorului pozitiv (DNCB), valorile RFI pentru CD86 și CD54 ar trebui să respecte criteriile pozitive (RFI ≥ 150 pentru CD86 și RFI ≥ 200 pentru CD54) și viabilitatea celulară ar trebui să fie mai mare de 50 %.
|
|
—
|
Pentru substanța chimică de testat, viabilitatea celulară ar trebui să fie peste 50 % în cel puțin patru concentrații verificate în cadrul fiecărui test.
|
|
|
30.
|
Rezultatele negative sunt acceptabile numai pentru substanțele chimice de testat care prezintă o viabilitate celulară mai mică de 90 % la cea mai mare concentrație testată (și anume, 1,2 x CV75 conform schemei de diluție în serie descrise la punctul 20). În cazul în care viabilitatea celulară la 1,2 × CV75 este mai mare sau egală cu 90 %, rezultatul negativ ar trebui să fie eliminat. Într-un astfel de caz, se recomandă să se încerce reluarea procesului de selecție a dozei prin repetarea testului pentru determinarea CV75. Ar trebui remarcat faptul că, atunci când se utilizează 5 000 μg/ml în soluție salină (sau mediu ori alți solvenți/vehicule), 1 000 μg/ml în DMSO sau concentrația solubilă maximă ca și concentrație de testare maximă a unei substanțe chimice de testat, un rezultat negativ este acceptabil chiar dacă viabilitatea celulară este mai mare de 90 %.
|
Raportul testului
|
31.
|
Raportul testului ar trebui să includă următoarele informații:
|
Substanța chimică de testat
|
Substanța monocomponentă
|
Identificarea chimică, precum denumirea (denumirile) IUPAC sau CAS, numărul (numerele) SMILES sau codul InChI, formula structurală și/sau alți identificatori;
|
|
Aspectul fizic, Log Kow, solubilitatea în apă, solubilitatea DMSO, masa moleculară și alte proprietăți fizico-chimice relevante, dacă există;
|
|
Puritatea, identitatea chimică a impurităților, după caz și dacă este fezabil din punct de vedere practic etc.;
|
|
Tratamentul înainte de efectuarea testului, dacă este cazul (de exemplu, încălzire, măcinare);
|
|
Concentrația (concentrațiile) testată(e);
|
|
Condițiile de depozitare și stabilitatea, dacă există;
|
|
Justificarea alegerii solventului/vehiculului pentru fiecare substanță chimică de testat.
|
|
|
Substanța multicomponentă, UVCB și amestec
|
Caracterizarea, dacă este posibil, de exemplu prin identitatea chimică (a se vedea mai sus), puritatea, apariția cantitativă și proprietățile fizico-chimice relevante (a se vedea mai sus) ale componentelor, dacă există;
|
|
Aspectul fizic, solubilitatea în apă, solubilitatea DMSO și alte proprietăți fizico-chimice relevante, dacă există;
|
|
Masa moleculară sau masa moleculară aparentă în cazul amestecurilor/polimerilor cu compoziții cunoscute sau alte informații relevante pentru efectuarea studiului;
|
|
Tratamentul înainte de efectuarea testului, dacă este cazul (de exemplu, încălzire, măcinare);
|
|
Concentrația (concentrațiile) testată(e);
|
|
Condițiile de depozitare și stabilitatea, dacă există;
|
|
Justificarea alegerii solventului/vehiculului pentru fiecare substanță chimică de testat.
|
|
|
|
Martori
|
Martor pozitiv
|
Identificarea chimică, precum denumirea (denumirile) IUPAC sau CAS, numărul (numerele) SMILES sau codul InChI, formula structurală și/sau alți identificatori;
|
|
Aspectul fizic, Log Kow, solubilitatea în apă, solubilitatea DMSO, masa moleculară și alte proprietăți fizico-chimice relevante, dacă există și dacă este cazul;
|
|
Puritatea, identitatea chimică a impurităților, după caz și dacă este fezabil din punct de vedere practic etc.;
|
|
Tratamentul înainte de efectuarea testului, dacă este cazul (de exemplu, încălzire, măcinare);
|
|
Concentrația (concentrațiile) testată(e);
|
|
Condițiile de depozitare și stabilitatea, dacă există;
|
|
Trimitere la rezultatele anterioare ale martorilor pozitivi care demonstrează criterii adecvate de acceptare a testelor, dacă este cazul.
|
|
|
|
Martor negativ și tratat cu solvent/vehicul
|
Identificarea chimică, precum denumirea (denumirile) IUPAC sau CAS, numărul (numerele) SMILES sau codul InChI, formula structurală și/sau alți identificatori;
|
|
Puritatea, identitatea chimică a impurităților, după caz și dacă este fezabil din punct de vedere practic etc.;
|
|
Aspectul fizic, masa moleculară și alte proprietăți fizico-chimice relevante în cazul în care sunt utilizați alți martori tratați cu solvent/vehicul decât cei menționați în Ghidul pentru teste și dacă aceștia există;
|
|
Condițiile de depozitare și stabilitatea, dacă există;
|
|
Justificarea alegerii solventului/vehiculului pentru fiecare substanță chimică de testat.
|
|
|
Condiții de testare
|
Numele și adresa sponsorului, ale instalației de testare și ale directorului de studiu;
|
|
Descrierea testului utilizat:
|
|
Linia celulară utilizată, condițiile de depozitare și sursa acesteia (de exemplu, instalația de la au fost obținute acestea);
|
|
Citometria de flux utilizată (de exemplu, modelul), inclusiv reglajele instrumentelor, globulina, anticorpii și markerul de citotoxicitate utilizat;
|
|
Procedura utilizată pentru a demonstra capabilitatea laboratorului de a efectua testul prin testarea unor substanțe de verificare și procedura utilizată pentru a demonstra performanța reproductibilă a testului în timp, de exemplu, datele anterioare privind martorii și/sau datele anterioare privind verificările asupra reactivității.
|
|
|
Criterii de acceptare a testului
|
Viabilitatea celulară, valorile IMF și RFI obținute cu martorul solventului/vehiculului în comparație cu intervalele de acceptare;
|
|
Viabilitatea celulară și valorile RFI obținute cu martorul pozitiv în comparație cu intervalele de acceptare;
|
|
Viabilitatea celulară a tuturor concentrațiilor testate ale substanței chimice de testat.
|
|
|
Procedura de testare
|
Numărul de teste utilizate;
|
|
Concentrațiile substanței chimice de testat, timpul de aplicare și de expunere utilizat (dacă acestea diferă de cele recomandate)
|
|
Descrierea criteriilor de evaluare și de decizie utilizate;
|
|
Descrierea oricăror modificări ale procedurii de testare.
|
|
|
Rezultate
|
Prezentarea tabelară a datelor, inclusiv a valorilor CV75 (dacă este cazul), a valorilor IMF geometrice individuale, a valorilor RFI, a valorilor de viabilitate celulară, a valorilor CE150/CE200 (dacă este cazul) obținute pentru substanța chimică de testat și pentru martorul pozitiv în cadrul fiecărui test, precum și o indicație privind clasificarea substanței chimice de testat în funcție de modelul predictiv;
|
|
O descriere a oricăror alte observații relevante, dacă este cazul.
|
|
|
Discutarea rezultatelor
|
Discutarea rezultatelor obținute cu ajutorul metodei h-CLAT;
|
|
Analiza rezultatelor testului în contextul unei IATA în cazul în care sunt disponibile alte informații relevante.
|
|
|
REFERINȚE BIBLIOGRAFICE
|
(1)
|
Ashikaga T, Yoshida Y, Hirota M, Yoneyama K, Itagaki H, Sakaguchi H, Miyazawa M, Ito Y, Suzuki H, Toyoda H. (2006). Development of an in vitro skin sensitization test using human cell lines: The human Cell Line Activation Test (h-CLAT) I. Optimization of the h-CLAT protocol. Toxicol. In Vitro 20, 767-773.
|
|
(2)
|
Miyazawa M, Ito Y, Yoshida Y, Sakaguchi H, Suzuki H. (2007). Phenotypic alterations and cytokine production in THP-1 cells in response to allergens. Toxicol. In Vitro 21, 428-437.
|
|
(3)
|
EC EURL-ECVAM (2013). Recommendation on the human Cell Line Activation Test (h-CLAT) for skin sensitisation testing. Material accesibil la adresa: https://eurl-ecvam.jrc.ec.europa.eu/eurl-ecvam-recommendations
|
|
(4)
|
Takenouchi O, Fukui S, Okamoto K, Kurotani S, Imai N, Fujishiro M, Kyotani D, Kato Y, Kasahara T, Fujita M, Toyoda A, Sekiya D, Watanabe S, Seto H, Hirota M, Ashikaga T, Miyazawa M. (2015). Test battery with the human cell line activation test, direct peptide reactivity assay and DEREK based on a 139 chemical data set for predicting skin sensitizing potential and potency of chemicals. J Appl Toxicol. 35, 1318-1332.
|
|
(5)
|
Hirota M, Fukui S, Okamoto K, Kurotani S, Imai N, Fujishiro M, Kyotani D, Kato Y, Kasahara T, Fujita M, Toyoda A, Sekiya D, Watanabe S, Seto H, Takenouchi O, Ashikaga T, Miyazawa M. (2015). Evaluation of combinations of in vitro sensitization test descriptors for the artificial neural network-based risk assessment model of skin sensitization. J Appl Toxicol. 35, 1333-1347.
|
|
(6)
|
Bauch C, Kolle SN, Ramirez T, Fabian E, Mehling A, Teubner W, van Ravenzwaay B, Landsiedel R. (2012). Putting the parts together: combining in vitro methods to test for skin sensitizing potencials. Regul Toxicol Parmacol. 63, 489-504.
|
|
(7)
|
Van der Veen JW, Rorije E, Emter R, Natch A, van Loveren H, Ezendam J. (2014). Evaluating the performance of integrated approaches for hazard identification of skin sensitizing chemicals. Regul Toxicol Pharmacol. 69, 371-379.
|
|
(8)
|
Urbisch D, Mehling A, Guth K, Ramirez T, Honarvar N, Kolle S, Landsiedel R, Jaworska J, Kern PS, Gerberick F, Natsch A, Emter R, Ashikaga T, Miyazawa M, Sakaguchi H. (2015). Assessing skin sensitization hazard in mice and men using non-animal test methods. Regul Toxicol Parmacol. 71, 337-351.
|
|
(9)
|
Jaworska JS, Natsch A, Ryan C, Strickland J, Ashikaga T, Miyazawa M. (2015). Bayesian integrated testing strategy (ITS) for skin sensitization potency assessment: a decision support system for quantitative weight of evidence and adaptive testing strategy. Arch Toxicol. 89, 2355-2383.
|
|
(10)
|
Strickland J, Zang Q, Kleinstreuer N, Paris M, Lehmann DM, Choksi N, Matheson J, Jacobs A, Lowit A, Allen D, Casey W. (2016). Integrated decision strategies for skin sensitization hazard. J Appl Toxicol. DOI 10.1002/jat.3281.
|
|
(11)
|
Nukada Y, Ashikaga T, Miyazawa M, Hirota M, Sakaguchi H, Sasa H, Nishiyama N. (2012). Prediction of skin sensitization potency of chemicals by human Cell Line Activation Test (h-CLAT) and an attempt at classifying skin sensitization potency. Toxicol. In Vitro 26, 1150-60.
|
|
(12)
|
EC EURL ECVAM (2015). Re-analysis of the within and between laboratory reproducibility of the human Cell Line Activation Test (h-CLAT). Material accesibil la adresa: https://eurl-ecvam.jrc.ec.europa.eu/eurl-ecvam-recommendations/eurl-ecvam-recommendation-on-the-human-cell-line-activation-test-h-clat-for-skin-sensitisation-testing
|
|
(13)
|
EC EURL ECVAM (2012). human Cell Line Activation Test (h-CLAT) Validation Study Report Accessible at: https://eurl-ecvam.jrc.ec.europa.eu/eurl-ecvam-recommendations
|
|
(14)
|
Takenouchi O, Miyazawa M, Saito K, Ashikaga T, Sakaguchi H. (2013). Predictive performance of the human Cell Line Activation Test (h-CLAT) for lipophilic with high octanol-water partition coefficients. J. Toxicol. Sci. 38, 599-609.
|
|
(15)
|
Ashikaga T, Sakaguchi H, Sono S, Kosaka N, Ishikawa M, Nukada Y, Miyazawa M, Ito Y, Nishiyama N, Itagaki H. (2010). A comparative evaluation of in vitro skin sensitisation tests: the human cell-line activation test (h-CLAT) versus the local lymph node assay (LLNA). Altern. Lab. Anim. 38, 275-284.
|
|
(16)
|
Fabian E., Vogel D., Blatz V., Ramirez T., Kolle S., Eltze T., van Ravenzwaay B., Oesch F., Landsiedel R. (2013). Xenobiotic metabolizin enzyme activities in cells used for testing skin sensitization in vitro. Arch Toxicol 87, 1683-1969.
|
|
(17)
|
Okamoto K, Kato Y, Kosaka N, Mizuno M, Inaba H, Sono S, Ashikaga T, Nakamura T, Okamoto Y, Sakaguchi H, Kishi M, Kuwahara H, Ohno Y. (2010). The Japanese ring study of a human Cell Line Activation Test (h-CLAT) for predicting skin sensitization potential (6th report): A study for evaluating oxidative hair dye sensitization potential using h-CLAT. AATEX 15, 81-88.
|
|
(18)
|
DB-ALM (INVITTOX) (2014). Protocol 158: human Cell Line Activation Test (h-CLAT), 23pp. Material accesibil la adresa: http://ecvam-dbalm.jrc.ec.europa.eu/
|
|
(19)
|
Mizuno M, Yoshida M, Kodama T, Kosaka N, Okamato K, Sono S, Yamada T, Hasegawa S, Ashikaga T, Kuwahara H, Sakaguchi H, Sato J, Ota N, Okamoto Y, Ohno Y. (2008). Effects of pre-culture conditions on the human Cell Line Activation Test (h-CLAT) results; Results of the 4th Japanese inter-laboratory study. AATEX 13, 70-82.
|
|
(20)
|
Sono S, Mizuno M, Kosaka N, Okamoto K, Kato Y, Inaba H, , Nakamura T, Kishi M, Kuwahara H, Sakaguchi H, Okamoto Y, Ashikaga T, Ohno Y. (2010). The Japanese ring study of a human Cell Line Activation Test (h-CLAT) for predicting skin sensitization potential (7th report): Evaluation of volatile, poorly soluble fragrance materials. AATEX 15, 89-96.
|
|
(21)
|
OCDE (2005). Guidance Document No 34 on The Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. OECD Series on Testing and Assessment. Organizația pentru Cooperare și Dezvoltare Economică, Paris, Franța, 2005, 96 pp.
|
|
(22)
|
OCDE (2012). The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins. Part 1: Scientific Evidence. Series on Testing and Assessment No 168. Publicație disponibilă la adresa: http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=ENV/JM/MONO(2012)10/PART1&docLanguage=En
|
|
(23)
|
Organizația Națiunilor Unite ONU (2013). Sistemul global armonizat de clasificare și etichetare a substanțelor chimice (GHS). A cincea ediție revizuită. New York & Geneva: United Nations Publications. ISBN: 978-92-1-117006-1. Publicație disponibilă la adresa: http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html
|
|
(24)
|
ECETOC (2003). Contact sensitization: Classification according to potency. European Centre for Ecotoxicology & Toxicology of Chemicals (Technical Report No 87).
|
|
(25)
|
Ashikaga T, Sakaguchi H, Okamoto K, Mizuno M, Sato J, Yamada T, Yoshida M, Ota N, Hasegawa S, Kodama T, Okamoto Y, Kuwahara H, Kosaka N, Sono S, Ohno Y. (2008). Assessment of the human Cell Line Activation Test (h-CLAT) for Skin Sensitization; Results of the First Japanese Inter-laboratory Study. AATEX 13, 27-35.
|
Apendicele 1.1
DEFINIțII
Precizie
: Gradul de concordanță între rezultatele testului și valorile de referință acceptate. Aceasta măsoară performanța testului și unul dintre aspectele relevanței acestuia. Termenul este adesea utilizat în mod interschimbabil, iar concordanța înseamnă proporția de rezultate corecte ale unui test (21).
AOP (parcursul răspunsurilor adverse)
: o serie de evenimente din structura chimică a unei substanțe chimice țintă sau un grup de substanțe chimice similare, de la evenimentul molecular declanșator până la un răspuns in vivo de interes (22).
Substanță chimică
: O substanță sau un amestec.
CV75
: Concentrația estimată care indică o viabilitate celulară de 75 %.
EC150
: concentrațiile care indică valorile RFI de 150 în expresia CD86
EC200
: concentrațiile care indică valorile RFI de 200 în expresia CD54
Citometria de flux
: o tehnică citometrică prin care celulele au fost suspendate într-un flux fluid una câte una, printr-o focalizare a luminii care excită și care este difuzată în modele caracteristice asupra celulelor și a componentelor acestora; celulele sunt etichetate frecvent cu markeri fluorescenți, astfel încât lumina să fie absorbită mai întâi și apoi emisă la frecvențe modificate.
Pericol
: Proprietate intrinsecă a unui agent sau a unei situații care are potențialul de a produce efecte adverse atunci când un organism, un sistem sau o (sub)populație este expusă la agentul respectiv.
IATA (Strategie integrată pentru testare și evaluare)
: O abordare structurată utilizată pentru identificarea pericolelor (potențial), caracterizarea pericolelor (potența) și/sau evaluarea siguranței (potențial/potență și expunere) pentru o substanță chimică sau un grup de substanțe chimice, care se integrează în mod strategic și măsoară toate datele relevante pentru a furniza informații cu privire la decizii de reglementare în ceea ce privește potențialele pericole și/sau riscuri și/sau necesitatea unor teste suplimentare specifice și, prin urmare, minime.
Martor pentru mediu
: O probă replică netratată care conține toate componentele unui sistem de testare. Această probă este analizată împreună cu probele tratate cu substanța chimică de testat și cu alte probe martor pentru a determina dacă solventul/vehiculul interacționează cu sistemul de testare.
Amestec
: un amestec sau o soluție compus/ă din două sau mai multe substanțe.
Substanță monocomponentă
: O substanță definită prin compoziția sa cantitativă, în care un singur component principal este prezent în proporție de cel puțin 80 % (g/g).
Substanță multicomponentă
: O substanță definită prin compoziția sa cantitativă, în care mai mult de un singur component principal este prezent într-o concentrație ≥ 10 % (g/g) și < de 80 % (g/g). O substanță multicomponentă este rezultatul unui proces de fabricație. Diferența dintre un amestec și o substanță multicomponentă este faptul că un amestec este obținut prin combinarea a două sau mai multe substanțe chimice fără o reacție chimică. O substanță multicomponentă este rezultatul unei reacții chimice.
Martor pozitiv
: O replică ce conține toate componentele sistemului de testare și care a fost tratată cu o substanță cunoscută pentru faptul că provoacă o reacție pozitivă. Pentru a asigura posibilitatea evaluării variabilității în timp a reacției martorului pozitiv, amplitudinea reacției severe nu trebuie să fie excesiv de mare.
Prehaptene
: substanțe chimice care devin sensibilizante prin transformări abiotice
Prohaptene
: substanțe chimice care necesită activare enzimatică pentru a exercita potențialul de sensibilizare cutanată
Intensitatea relativă a fluorescenței (RFI)
: Valorile relative ale intensității medii a fluorescenței (MFI) la celulele tratate chimic comparativ cu MFI din celulele tratate cu solvent/vehicul.
Relevanță
: Descrierea relației dintre test și efectul de interes și dacă aceasta este relevantă și utilă pentru un anumit scop. Aceasta reprezintă măsura în care testul măsoară sau prezice în mod corect efectul biologic de interes. Relevanța include analiza preciziei (a concordanței) unui test (21).
Fiabilitate
: Măsuri ale faptului că un test poate fi efectuat în mod reproductibil, în același laborator și în laboratoare diferite în timp, atunci când este efectuat utilizând același protocol. Aceasta se evaluează prin calcularea reproductibilității în același laborator sau între laboratoare și a repetabilității în același laborator (21).
Test
: Un test este compus din unul sau mai multe substanțe chimice de testat, care sunt testate concomitent cu un martor solvent/vehicul și un martor pozitiv.
Sensibilitate
: Proporția tuturor substanțelor chimice pozitive/active clasificate în mod corect în urma testului. Aceasta permite măsurarea preciziei în cazul unui test care generează rezultate clasificabile în categorii și constituie un aspect important în evaluarea relevanței unui test (21).
Soluție tampon de colorare
: O soluție salină tamponată cu fosfat conținând o cantitate de 0,1 % albumină din ser bovin.
Martor solvent/vehicul
: O probă netratată care conține toate componentele unui sistem de testare, cu excepția celor ale substanței chimice de testat, dar incluzând solventul/vehiculul utilizat. Acesta este utilizat pentru a stabili reacția de bază pentru probele tratate cu substanța chimică de testat dizolvată sau dispersată stabil în același solvent/vehicul. Atunci când este testată concomitent cu un martor de mediu, această probă indică, de asemenea, dacă solventul/vehiculul interacționează cu sistemul testat.
Specificitate
: Proporția tuturor substanțelor chimice negative/inactive clasificate în mod corect în urma testului. Aceasta permite măsurarea preciziei în cazul unui test care generează rezultate clasificabile în categorii și constituie un aspect important în evaluarea relevanței unui test (21).
Substanță
: un element chimic și compușii săi în stare naturală sau obținuți prin orice proces de producție, inclusiv orice aditiv necesar pentru a-i păstra stabilitatea și orice impuritate care derivă din procesul utilizat, însă excluzând orice solvent care poate fi separat fără a afecta stabilitatea substanței sau fără a-i schimba compoziția.
Substanță chimică de testat
: orice substanță sau amestec testat(ă) în cadrul acestei metode.
Sistemul global armonizat de clasificare și etichetare a substanțelor chimice al Organizației Națiunilor Unite (GHS al ONU)
: Un sistem care propune clasificarea substanțelor chimice (substanțe și amestecuri) în funcție de tipuri și niveluri standardizate de pericole fizice, pericole pentru sănătate și pericole pentru mediu și care se referă la elemente de comunicare corespunzătoare, precum pictograme, cuvinte de avertizare, declarații cu privire la pericole, declarații de securitate și fișe tehnice de siguranță pentru a transmite informații privind efectele adverse ale acestora în scopul protejării populației (inclusiv angajatorii, lucrătorii, transportatorii, consumatorii și cei care se ocupă de serviciile de urgență) și a mediului (23).
UVCB
: Substanțe cu compoziție necunoscută sau variabilă, produși de reacție complexă sau materiale biologice.
Test valabil
: Un test considerat a avea suficientă relevanță și fiabilitate pentru un scop specific și care se bazează pe principii solide din punct de vedere științific. Un test nu este niciodată valabil într-un sens absolut, ci doar în legătură cu un scop definit (21).
Apendicele 1.2
SUBSTANțE DE VERIFICARE
Înainte de utilizarea de rutină a testului descris în prezentul apendice la metoda de testare B.71, laboratoarele ar trebui să demonstreze performanțele tehnice prin obținerea în mod corect a estimării h-CLAT pentru cele 10 substanțe de verificare recomandate în tabelul 1 și prin obținerea de valori pentru CV75, EC150 și EC200 care să se încadreze în plaja de valori de referință corespunzătoare pentru cel puțin 8 dintre cele 10 substanțe de verificare. Substanțele de verificare au fost selectate pentru a reprezenta intervalul de reacții pentru pericolele sensibilizării cutanate. Alte criterii de selecție au fost ca substanțele să fie disponibile în comerț și să fie disponibile date de referință in vivo de înaltă calitate, precum și date in vitro de înaltă calitate generate cu metoda h-CLAT. De asemenea, sunt disponibile date de referință publicate pentru metoda h-CLAT (3) (14).
Tabelul 1
Substanțe recomandate pentru a demonstra performanțele tehnice prin metoda h-CLAT
|
Substanțe de verificare
|
NR CAS
|
Stare fizică
|
Estimare in vivo
(103)
|
CV75 Interval de referință în μg/ml (104)
|
Rezultate h-CLAT pentru CD86 (Interval de referință pentru EC150 în μg/ml) (104)
|
Rezultate h-CLAT pentru CD54 (Interval de referință pentru EC200 în μg/ml) (104)
|
|
2,4-dinitroclorbenzen
|
97-00-7
|
Solid
|
Sensibilizant (extrem)
|
2-12
|
Pozitiv (0,5-10)
|
Pozitiv (0,5-15)
|
|
4-fenilendiamină
|
106-50-3
|
Solid
|
Sensibilizant (puternic)
|
5-95
|
Pozitiv (<40)
|
Negativ (>1,5) (105)
|
|
Sulfat de nichel
|
10101-97-0
|
Solid
|
Sensibilizant (moderat)
|
30-500
|
Pozitiv (<100)
|
Pozitiv (10-100)
|
|
2-Mercaptobenzotiazol
|
149-30-4
|
Solid
|
Sensibilizant (moderat)
|
30-400
|
Negativ (>10) (105)
|
Pozitiv (10-140)
|
|
R(+)-Limonen
|
5989-27-5
|
Lichid
|
Sensibilizant (slab)
|
>20
|
Negativ (>5) (105)
|
Pozitiv (<250)
|
|
Imidazolidinil uree
|
39236-46-9
|
Solid
|
Sensibilizant (slab)
|
25-100
|
Pozitiv (20-90)
|
Pozitiv (20-75)
|
|
Izopropanol
|
67-63-0
|
Lichid
|
Non-sensibilizant
|
>5 000
|
Negativ (>5 000 )
|
Negativ (>5 000 )
|
|
Glicerină
|
56-81-5
|
Lichid
|
Non-sensibilizant
|
>5 000
|
Negativ (>5 000 )
|
Negativ (>5 000 )
|
|
Acid lactic
|
50-21-5
|
Lichid
|
Non-sensibilizant
|
1500-5 000
|
Negativ (>5 000 )
|
Negativ (>5 000 )
|
|
Acid 4-aminobenzoic
|
150-13-0
|
Solid
|
Non-sensibilizant
|
>1 000
|
Negativ (>1 000 )
|
Negativ (>1 000 )
|
|
Abrevieri: NR CAS = Număr de înregistrare al Chemical Abstracts Service
|
Apendicele 2
SENSIBILIZAREA CUTANATĂ IN VITRO: TESTUL U937 DE ACTIVARE A LINIEI CELULARE (U-SENS™)
CONSIDERAțII PRELIMINARE ȘI LIMITĂRI
|
1.
|
Testul U-SENS™ cuantifică modificarea produsă la nivelul exprimării unui marker celular de suprafață asociat cu procesul de activare a monocitelor și celulelor dendritice (CD) (și anume, CD86), la nivelul liniei celulare umane U937 afectate de limfomul histiocitar în urma expunerii la substanțe sensibilizante (1). Nivelurile de exprimare măsurate ale markerului celular de suprafață CD86 de la nivelul liniei celulare U937 sunt apoi utilizate pentru susținerea diferențierii între sensibilizantele și nesensibilizantele pentru piele.
|
|
2.
|
Testul U-SENS™ a fost evaluat în cadrul unui studiu de validare (2) coordonat de L’Oreal și ulterior supus unei evaluări independente inter pares efectuate de Comitetul Consultativ Științific (ESAC) al laboratorului de referință al Uniunii Europene pentru metode alternative la testarea pe animale (European Union Reference Laboratory for Alternatives to Animal Testing - EURL ECVAM) (3). Având în vedere toate dovezile disponibile și contribuțiile autorităților de reglementare și ale părților interesate, U-SENS™ a fost recomandat de EURL ECVAM (4) pentru a fi utilizat în cadrul unei IATA în vederea sprijinirii diferențierii între substanțele sensibilizante și cele nesensibilizante în scopul clasificării și etichetării pericolelor. În documentul său de orientare privind raportarea abordărilor structurate ale integrării datelor și ale surselor de informații individuale utilizate în cadrul IATA pentru sensibilizarea pielii, OCDE abordează în prezent o serie de studii de caz care descriu diferite strategii de testare și modele de predicție. Una dintre diferitele abordări definite este bazată pe testul U-SENS (5). În literatura de specialitate (4) (5) (7) sunt prezentate, de asemenea, exemple de utilizare a datelor U-SENS™, în combinație cu alte informații, inclusiv date istorice și actuale valabile despre oameni (6).
|
|
3.
|
Testul U-SENS™ s-a dovedit a fi transferabil către laboratoare cu experiență în tehnici de cultură celulară și analiza citometriei de flux. Nivelul de reproductibilitate în estimări, care poate fi preconizat în urma testării, este de ordinul a 90 % și 84 % în același laborator și între laboratoare (8). Rezultatele generate și incluse în studiul de validare (8), precum și în alte studii publicate (1), indică, în general, faptul că, în comparație cu rezultatele LLNA, precizia de deosebire a sensibilizantelor pentru piele (și anume, categoria 1 din GHS al ONU/Regulamentul CLP) de substanțele nesensibilizante este de 86 % (N = 166) cu o sensibilitate de 91 % (118/129) și o specificitate de 65 % (24/37). În comparație cu rezultatele umane, precizia de deosebire a sensibilizantelor pentru piele (și anume, categoria 1 din GHS al ONU/Regulamentul CLP) de substanțele nesensibilizante este de 77 % (N = 101) cu o sensibilitate de 100 % (58/58) și o specificitate de 47 % (20/43). Există o probabilitate mai mare ca estimările negative false realizate prin U-SENS™, comparativ cu LLNA, să vizeze substanțe chimice care prezintă un nivel scăzut până la moderat de potență de sensibilizare cutanată (și anume, subcategoria 1B din GHS al ONU/Regulamentul CLP) și nu substanțe chimice care prezintă un nivel ridicat de potență de sensibilizare cutanată (și anume, subcategoria 1A din GHS al ONU/Regulamentul CLP) (1) (8) (9). Coroborate, aceste informații indică utilitatea testului U-SENS™ în ceea ce privește contribuirea la identificarea pericolelor de sensibilizare cutanată. Cu toate acestea, valorile de precizie furnizate în prezentul document pentru U-SENS™ ca test independent sunt doar orientative, întrucât testul ar trebui luat în considerare în combinație cu alte surse de informații în contextul unei IATA și în conformitate cu dispozițiile de la alineatele (7) și (8) din Introducerea generală. În plus, la evaluarea metodelor care nu se bazează pe animale pentru sensibilizarea cutanată, ar trebui reținut faptul că este posibil ca testul LLNA și alte teste pe animale să nu reflecte pe deplin situația oamenilor.
|
|
4.
|
Pe baza datelor disponibile în prezent, testul U-SENS ™ s-a dovedit a fi aplicabil substanțelor chimice de testat (inclusiv ingrediente cosmetice, de exemplu, conservanți, surfactanți, agenți activi, coloranți) care acoperă o varietate de grupe funcționale organice, de proprietăți fizico-chimice, de grade de potență de sensibilizare cutanată (determinate în studii in vivo), precum și spectrul de mecanisme de reacție cunoscute a fi asociate cu sensibilizarea cutanată (și anume, acceptorul Michael, formarea bazei Schiff, agentul de transfer pe bază acrilică, reacția de substituție nucleofilă bimoleculară [SN2] sau reacția de substituție nucleofilă aromatică [SNAr]) (1) (8) (9) (10). Testul U-SENS™ se aplică substanțelor chimice de testat care sunt solubile sau care formează dispersii stabile (și anume, un coloid sau o suspensie în care substanța chimică de testat nu se sedimentează sau se separă de solvent/vehicul în diferite faze) într-un solvent/vehicul adecvat [a se vedea alineatul (13)]. Substanțele chimice din setul de date, careu au fost raportate ca fiind prehaptene (și anume, substanțe activate prin oxidare) sau prohaptene (și anume, substanțe care necesită activare enzimatică, de exemplu prin intermediul enzimelor P450), au fost corect estimate prin testul U-SENS™ (1) (10). Substanțele care perturbă membrana pot conduce la rezultate fals pozitive datorită unei creșteri nespecifice a expresiei CD86, întrucât 3 din 7 rezultate fals pozitive aferente clasificării de referință in vivo au fost surfactanți (1). Ca atare, rezultatele pozitive obținute cu surfactanți ar trebui să fie luate în considerare cu prudență, iar rezultatele negative cu surfactanți ar putea fi totuși utilizate pentru a sprijini identificarea substanței chimice de testat ca nefiind nesensibilizantă. Substanțele chimice de testat fluorescente pot fi analizate prin testul U-SENS™ (1); cu toate acestea, substanțele chimice de testat fluorescente puternice care emit la aceeași lungime de undă ca și izotiocianatul de fluoresceină (FITC) sau iodura de propidiu (PI) vor interfera cu detecția prin citometria de flux și, astfel, nu pot fi evaluate în mod corect folosind anticorpi conjugați cu FITC (rezultate potențial fals pozitive) sau PI (viabilitatea nu este măsurabilă). Într-un astfel de caz, se pot folosi alți anticorpi marcați cu fluorocrom sau alți markeri de citotoxicitate, cu condiția să se poată demonstra că acestea oferă rezultate similare celor generate de anticorpii marcați cu FITC sau PI [a se vedea alineatul (18)], de exemplu prin testarea substanțelor de verificare din apendicele 2.2. Având în vedere cele de mai sus, rezultatele pozitive obținute cu surfactanți și rezultatele negative obținute cu substanțele chimice de testat fluorescente puternic ar trebui interpretate în contextul limitărilor menționate și împreună cu alte surse de informații în cadrul IATA. În cazul în care există dovezi care demonstrează lipsa de aplicabilitate a testului U-SENS™ la alte categorii specifice de substanțe chimice de testat, acestea nu ar trebui utilizate pentru respectivele categorii specifice.
|
|
5.
|
Conform descrierii de mai sus, testul U-SENS™ susține diferențierea între sensibilizantele și nesensibilizantele pentru piele. Însă acesta poate contribui, de asemenea, la evaluarea potenței de sensibilizare atunci când este utilizat în abordări integrate precum IATA. Cu toate acestea, se impun studii suplimentare, de preferință pe baza datelor cu privire la oameni, pentru a determina în ce mod ar putea rezultatele U-SENS™ să fundamenteze evaluarea potenței.
|
|
6.
|
Definițiile sunt prevăzute în apendicele 2.1.
|
Principiul Testului
|
7.
|
Testul U-SENS™ reprezintă un test in vitro care cuantifică modificările expresiei markerului celular de suprafață la nivelul unei linii celulare umane, celule U937, afectate de limfomul histiocitar în urma expunerii timp de 45±3 ore la substanța chimică de testat. Markerul de suprafață CD86 este un marker tipic de activare U937. CD86 este cunoscut ca fiind o moleculă co-stimulatorie care poate să imite activarea monocitică, jucând un rol esențial în pregătirea de celule de tip T. Modificările expresiei markerului celular de suprafață CD86 se măsoară prin citometria de flux în urma colorării celulelor cu anticorpi etichetați cu izotiocianat de fluoresceină (FITC). De asemenea, măsurarea citotoxicității este efectuată (de exemplu, utilizând PI) în același timp cu scopul de a se evalua dacă se produce o reglare în sus a expresiei markerului celular de suprafață CD86 în cazul concentrațiilor subcitotoxice. Indicele de simulare (IS) al markerului celular de suprafață CD86, în comparație cu martorul tratat cu solvent/vehicul, se calculează și se utilizează în modelul predictiv (a se vedea punctul 19), pentru a susține diferențierea între sensibilizante și nesensibilizante.
|
Demonstrarea Performanței
|
8.
|
Înainte de utilizarea de rutină a testului descris în prezentul apendice pentru metoda de testare B.71, laboratoarele ar trebui să demonstreze performanțele tehnice, utilizând cele 10 substanțe de verificare enumerate în apendicele 2.2, în conformitate cu bunele practici privind metodele in vitro (11). În plus, utilizatorii testelor ar trebui să mențină o bază de date istorice generate în urma verificărilor de reactivitate (a se vedea punctul 11) și cu martori pozitivi și tratați cu solvent/vehicul (a se vedea punctele 15-16) și să utilizeze aceste date pentru a confirma faptul că reproductibilitatea testului în laboratorul propriu este asigurată în timp.
|
Procedura
|
9.
|
Acest test se bazează pe protocolul nr. 183 al U-SENS™ DataBase service on ALternative Methods to animal experimentation (DB-ALM) (12). Procedurile standard de operare (PSO) ar trebui să fie utilizate la punerea în aplicare și utilizarea testului U-SENS™ în laborator. Se poate utiliza un sistem automat de derulare a testului U-SENS ™ dacă se poate demonstra că acesta generează rezultate similare, de exemplu, prin testarea substanțelor de verificare din apendicele 2.2. Componentele și procedurile principale pentru testul U-SENS™ sunt descrise în continuare.
|
Pregătirea celulelor
|
10.
|
Linia celulară umană afectată de limfomul histiocitic, și anume U937 (13), ar trebui să fie utilizată pentru efectuarea testului U-SENS™. Celulele (clona CRL1593.2) ar trebui să fie obținute de la o bancă de celule calificată, cum ar fi American Type Culture Collection.
|
|
11.
|
Celulele U937 sunt cultivate la 37 °C în atmosferă de 5 % CO2 și de umiditate, în mediu RPMI-1 640 completat cu 10 % ser fetal de vițel (SFV), 2 mM L-glutamină, 100 de unități/ml de penicilină și 100 μg/ml de streptomicină (mediu complet). Celulele U937 sunt trecute, de regulă, la 2-3 zile la densitatea de 1,5 sau 3 × 105 celule/ml. Densitatea celulară nu ar trebui să depășească 2 × 106 celule/ml, iar viabilitatea celulară măsurată prin excluderea albastrului de tripan ar trebui să fie ≥ 90 % (nu se aplică la prima trecere după decongelare). Înainte de a le utiliza pentru testare, fiecare lot de celule, SFV sau anticorpi ar trebui să fie calificat prin efectuarea unui control de reactivitate. Verificarea de reactivitate a celulelor ar trebui să fie efectuat folosind martorul pozitiv, acidul picril-sulfonic (acid 2,4,6-trinitrobenzensulfonic: TNBS) (CASRN 2 508-19-2, puritate de ≥ 99 %) și acidul lactic ca martor negativ (AL) (CASRN 50-21-5, puritate de ≥ 85 %), după cel puțin o săptămână de la decongelare. Pentru verificarea de reactivitate, ar trebui să fie testate șase concentrații finale pentru fiecare dintre cei doi martori (TNBS: 1, 12.5, 25, 50, 75, 100μg/ml și AL: 1, 10, 20, 50, 100, 200μg/ml). TNBS solubilizat în mediu complet ar trebui să producă o reacție a CD86 pozitivă și legată de concentrație (de exemplu, în cazul în care o concentrație pozitivă, CD86 S.I. ≥ 150, este urmată de o concentrare cu o valoare a CD86 S.I crescută) și AL solubilizat în mediu complet ar trebui să producă o reacție negativă a CD86 (a se vedea punctul 21). Ar trebui utilizat pentru analiză numai lotul de celule pentru care verificarea de reactivitate a fost efectuată cu succes de două ori. Celulele pot fi reproduse până la șapte săptămâni după decongelare. Numărul de trecere nu ar trebui să depășească 21. Verificarea de reactivitate ar trebui să fie efectuată conform procedurilor descrise la punctele 18-22.
|
|
12.
|
În scopul testării, celulele U937 sunt însămânțate la o densitate de 3 x 105 celule/ml sau de 6 x 105 celule/ml și cultivate în prealabil în baloane de cultură pentru două sau, respectiv, o zi. Ar putea fi utilizate alte condiții de cultivare prealabilă decât cele descrise mai sus dacă este asigurată o argumentare științifică suficientă și dacă se poate demonstra că aceasta generează rezultate similare, de exemplu, prin testarea substanțelor de verificare din apendicele 2.2. În ziua testării, celulele recoltate din balonul de cultură sunt suspendate din nou în mediu de cultură proaspăt, la 5 × 105 celule/ml. Apoi, celulele sunt distribuite pe o placă de 96 de godeuri de 100 μl cu fund plat (densitate celulară finală de 0,5 × 105 celule/godeu).
|
Pregătirea substanțelor chimice de testat și a substanțelor martor
|
13.
|
Evaluarea solubilității este efectuată înainte de testare. În acest scop, substanțele chimice de testat se dizolvă sau se dispersează în mod stabil la o concentrație de 50 mg/ml în mediu complet ca primă opțiune de solvent sau în dimetilsulfoxid (DMSO, puritate de ≥ 99 %) ca al doilea solvent/vehicul dacă substanța chimică de testat nu este solubilă în solventul/vehiculul ca mediu complet. Pentru testare, substanța chimică de testat este dizolvată într-o concentrație finală de 0,4 mg/ml în mediu complet dacă substanța chimică este solubilă în acest solvent/vehicul. Dacă substanța chimică este solubilă numai în DMSO, aceasta se dizolvă la o concentrație de 50 mg/ml. Se pot utiliza și alți solvenți/vehicule decât cele descrise mai sus în cazul în care se prezintă o justificare științifică suficientă. Ar trebui să se țină seama de stabilitatea substanței chimice de testat în solventul/vehiculul final.
|
|
14.
|
Substanțele chimice de testat și substanțele martor se pregătesc în ziua testării. Deoarece nu este efectuat un test de determinare a dozelor, prima dată ar trebui să fie testate șase concentrații finale (1, 10, 20, 50, 100 și 200 μg/ml) în solventul/vehiculul corespunzător, fie în mediu complet, fie în mediu de 0,4 % DMSO. Pentru testele ulterioare, începând cu o cantitate de 0,4 mg/ml într-un mediu complet sau 50 mg/ml în DMSO, se prepară soluții de substanțe chimice de testat, cel puțin patru soluții de lucru (și anume, cel puțin patru concentrații) folosind solvent/vehicul corespunzător. Soluțiile de lucru sunt, în final, utilizate pentru tratare prin adăugarea unui volum egal de suspensie celulară U937 (a se vedea punctul 11 de mai sus) la volumul de soluție de lucru din placă pentru a obține o diluție suplimentară dublă (12). Concentrațiile (cel puțin patru concentrații) pentru orice test ulterior sunt alese pe baza rezultatelor individuale ale tuturor testelor anterioare (8). Concentrațiile finale utilizabile sunt de 1, 2, 3, 4, 5, 7.5, 10, 12.5, 15, 20, 25, 30, 35, 40, 45, 50, 60, 70, 80, 90, 100, 120, 140, 160, 180 și 200 μg/ml. Concentrația finală maximă este de 200 μg/ml. În cazul ]n care se observă o valoare pozitivă a CD86 la 1 μg/ml, se evaluează o cantitate de 0,1 μg/ml pentru a se afla concentrația substanței chimice de testat care nu induce CD86 deasupra pragului pozitiv. Pentru fiecare test, EC150 (concentrația la care o substanță chimică atinge pragul pozitiv al CD86 de 150 %, a se vedea punctul 19) se calculează dacă se observă o reacție sub forma unei concentrații pozitive de tip CD86. În cazul în care substanța chimică de testat induce o reacție pozitivă a CD86 nelegată de concentrație, calcularea EC150 ar putea să nu fie relevantă, astfel cum este descris în protocolul DB-ALM nr. 183 privind U-SENS ™ (12). Pentru fiecare test, CV70 (concentrația la care o substanță chimică atinge pragul de citotoxicitate de 70 %, a se vedea punctul 19) se calculează ori de câte ori este posibil (12). Pentru a analiza efectul reacției concentrației în urma creșterii CD86, orice concentrații dintre concentrațiile utilizabile ar trebui alese ca fiind repartizate în mod egal între valoarea EC150 (sau cea mai mare concentrație necitotoxică negativă CD86) și CV70 (sau cea mai mare concentrație admisă, și anume, 200 μg/ml). Ar trebui să fie testate minimum patru concentrații pe test și să existe cel puțin două concentrații comune cu testul (testele) anterior (anterioare) pentru comparație.
|
|
15.
|
Martorul tratat cu solvent/vehicul și utilizat în testul U-SENS ™ este un mediu complet (pentru substanțe chimice de testat solubilizate sau dispersate în mod stabil în mediu complet) (a se vedea punctul 4) sau o soluție de 0,4 % DMSO în mediu complet (pentru substanțe chimice de testat solubilizate sau dispersate în mod stabil în DMSO).
|
|
16.
|
Martorul pozitiv utilizat în testul U-SENS ™ este TNBS (a se vedea punctul 11), preparat în mediu complet. TNBS ar trebui utilizat ca martor pozitiv pentru măsurarea expresiei CD86 la o concentrație unică finală în placă (50 μg/ml) determinând o viabilitate celulară de > 70 %. Pentru a obține o concentrație de 50 μg/ml de TNBS în placă, se prepară o soluție mamă de 1 M (și anume, 293 mg/ml) de TNBS în mediu complet, care este apoi diluată de 2 930 de ori în mediu complet până la o soluție de lucru de 100 μg/ml. Acidul lactic (AL, CAS 50-21-5) ar trebui utilizat ca martor negativ în cantitate de 200 μg/ml solubilizat în mediu complet (dintr-o soluție mamă de 0,4 mg/ml). În fiecare placă din cadrul fiecărui test sunt preparate trei replici de martor netratat în mediu complet, martor tratat cu solvent/vehicul, precum și martori negativi și pozitivi (12). Ar putea fi utilizați și alți martori pozitivi adecvați în cazul în care sunt disponibile date anterioare pentru a obține criterii comparabile de acceptare a testelor. Criteriile de acceptare a testului sunt aceleași cu cele descrise pentru substanța chimică de testat (a se vedea punctul 12).
|
Aplicarea substanțelor chimice de testat și a substanțelor martor
|
17.
|
Martorul tratat cu solvent/vehicul sau soluțiile de lucru descrise la punctele 14-16 sunt amestecate 1:1 (v/v) cu suspensiile de celule preparate pe placa cu fund plat de 96 de godeuri (a se vedea punctul 12). Plăcile tratate sunt apoi incubate timp de 45 ± 3 ore la 37 °C în soluție cu 5 % CO2. Înainte de incubație, plăcile sunt închise etanș cu membrană semipermeabilă pentru a se evita evaporarea substanțelor chimice de testat volatile și contaminarea încrucișată între celulele tratate cu substanțe chimice de testat (12).
|
Colorarea celulelor
|
18.
|
După 45±3 ore de expunere, celulele sunt transferate într-o placă de microtitrare în formă de V și colectate prin centrifugare. Interferența în solubilitate este definită ca fiind cristale sau picături observate la microscop la 45±3 ore după tratament (înainte de colorarea celulelor). Se îndepărtează supernatantele, iar restul celulelor sunt spălate o singură dată cu o soluție salină cu tampon de fosfat (PBS) de 100 μl rece ca gheața conținând 5 % ser fetal de vițel (soluție tampon de colorare). După centrifugare, celulele sunt resuspendate în 100 μl de soluție tampon de colorare și colorate cu 5 μl (de exemplu, 0,25 μg) de anticorpi anti-CD86 etichetați FITC sau IgG1 (izotip) de șoarece la 4 °C timp de 30 de minute, departe de lumină. Ar trebui să se utilizeze anticorpii descriși în Protocolul DB-ALM nr. 183 al U-SENS ™ (pentru CD86: clona BD-PharMingen #5556 57: Fun-1 sau clona Caltag/Invitrogen # MHCD8601: BU63; și pentru IgG1: BD-PharMingen #5557 48 sau Caltag/Invitrogen # GM4992). Pe baza experienței dezvoltatorilor de teste, intensitatea fluorescenței anticorpilor este, de obicei, consecventă între diferite loturi. Pentru test ar putea fi utilizate alte clone sau alt furnizor de anticorpi pentru care a fost efectuată cu succes verificarea de reactivitate (a se vedea punctul 11). Cu toate acestea, utilizatorii ar putea avea în vedere titrarea anticorpilor în condițiile din laboratorul propriu pentru a defini cea mai bună concentrație de utilizare. Se poate utiliza și un alt sistem de detecție, de exemplu, anticorpi anti-CD86 marcați cu fluorocrom în cazul în care se poate demonstra că aceștia generează rezultate similare cu cele ale anticorpilor conjugați FITC, de exemplu prin testarea substanțelor de verificare din apendicele 2.2. După spălare de două ori în 100 μl de soluție tampon de colorare și o dată în 100 μl PBS rece ca gheața, celulele sunt resuspendate în PBS rece ca gheața (de exemplu, 125 μl pentru probe analizate manual eprubetă cu eprubetă sau 50 μl utilizând o placă cu auto-eșantionare) și se adaugă soluție IP (concentrație finală de 3 μg/ml). Se pot utiliza alți markeri de citotoxicitate, cum ar fi 7-aminoactinomicina D (7-AAD) sau albastru de tripan, în cazul în care se poate demonstra că acțiunile de colorare alternative generează rezultate similare cu IP, de exemplu, prin testarea substanțelor de verificare din apendicele 2.2.
|
Analiza prin citometria de flux
|
19.
|
Nivelurile de expresie ale CD86 și viabilitatea celulară sunt analizate folosind citometria de flux. Celulele sunt afișate în interiorul unui segment cu puncte având o dimensiune (FSC) și o granularitate (SSC) stabilite la scara logaritmică pentru a identifica în mod clar populația dintr-un prim grup R1 și a elimina resturile. Pentru fiecare godeu se impune un număr total vizat de 10 000 de celule în grupul (gate) R1. Celulele din același grup R1 sunt afișate pe un segment cu puncte FL3 sau FL4/SSC. Celulele viabile sunt delimitate prin plasarea unui al doilea grup R2 care selectează populația de celule negative de iodură de propidiu (canalul FL3 sau FL4). Viabilitatea celulară poate fi calculată folosind următoarea ecuație prin programul de analiză citometrică. Atunci când viabilitatea celulară este scăzută, ar putea fi acumulate până la 20 000 de celule, inclusiv celule moarte. În mod alternativ, pot fi acumulate date timp de un minut după inițierea analizei.
Apoi se măsoară procentul de celule pozitive FL1 se măsoară între aceste celule viabile grupate sub R2 (în cadrul R1). Exprimarea CD86 la suprafața celulei este analizată pe un segment cu puncte FL1/SSC care formează un grup de celule viabile (R2).
Pentru godeurile cu mediu complet/IgG1, markerul de analiză este stabilit în apropierea populației principale, astfel încât martorii tratați cu mediu complet să aibă IgG1 în intervalul-țintă 0,6-0,9 %.
Interferența culorii este definită ca o schimbare a segmentului cu puncte etichetat cu FITC IgG1 (media geometrică IgG1 FL1 S.I. ≥ 150 %).
Indicele de stimulare (S.I.) al CD86 pentru celulele martorilor (netratate sau în soluție de 0,4 % DMSO) și celulele tratate chimic se calculează conform ecuației următoare:.

% din celulele netratate ale martorilor IgG1+: denumit drept procent din celulele IgG1 pozitive FL1 definite cu markerul de analiză (interval acceptat de ≥ 0,6 % și < 1,5 %, a se vedea punctul 22) în celulele viabile netratate.
% din celulele tratate/ale martorilor IgG1+/CD86+: denumit drept procent din celulele IgG1 pozitive FL1/CD86 măsurate fără deplasarea markerului de analiză între celulele viabile ale martorilor/tratate.
|
Date Și Raportare
Evaluarea datelor
|
20.
|
În testul U-SENS ™ se calculează următorii parametri: valoarea CV70, adică o concentrație care indică 70 % din supraviețuirea celulelor U937 (30 % citotoxicitate) și valoarea EC150, și anume, concentrația la care substanțele chimice de testat au indus un indice de stimulare (S.I.) de 150 % pentru CD86.
CV70 se calculează prin interpolare logliniară folosind următoarea ecuație:
CV70 = C1 + [(V1 - 70) / (V1 – V2) * (C2 – C1)]
Unde:
V1 reprezintă valoarea minimă a viabilității celulare peste 70 %
V2 reprezintă valoarea maximă a viabilității celulare sub 70 %
C1 și C2 sunt concentrații care arată valoarea viabilității celulare V1 și, respectiv, V2
Se pot utiliza alte abordări pentru derivarea valorii CV70, cu condiția să se demonstreze că acest lucru nu are niciun impact asupra rezultatelor (de exemplu, prin testarea substanțelor de verificare).
EC150 se calculează prin interpolare logliniară folosind următoarea ecuație:
EC150 = C1 + [(150 – S.I.1) / (S.I.2 – S.I.1) * (C2 – C1)]
Unde:
C2 este cea mai mare concentrație în μg/ml cu S.I. al CD86 < 150 % (S.I. 1)
C2 este cea mai redusă concentrație în μg/ml cu S.I. al CD86 ≥ 150 % (S.I. 2).
Se calculează valorile EC150 și CV70
|
—
|
pentru fiecare test: valorile EC150 și CV70 individuale sunt utilizate ca instrumente de investigare a efectului reacției concentrației generate de creșterea CD86 (a se vedea punctul 14),
|
|
—
|
valoarea totală a CV70 este determinată pe baza valorilor viabilității medii (12),
|
|
—
|
valoarea totală a EC150 este determinată pe baza valorii medii a S.I. al valorilor CD86 pentru substanța chimică de testat prevăzută ca fiind POZITIVĂ în urma testului U-SENS ™ (a se vedea punctul 21) (12).
|
|
Model predictiv
|
21.
|
Pentru măsurarea expresiei CD86, fiecare substanță chimică de testat este testată în cel puțin patru concentrații și prin cel puțin două teste independente (efectuate în zile diferite) pentru a se obține o singură estimare (NEGATIV sau POZITIV).
|
—
|
Concluzia individuală a unui test U-SENS ™ este considerată negativă (denumită în continuare „N”) dacă S.I. al CD86 este mai mic de 150 % la toate concentrațiile care nu sunt citotoxice (viabilitate celulară ≥ 70 %) și dacă nu se observă interferențe (citotoxicitate, solubilitate: a se vedea punctul 18 sau culoarea: a se vedea punctul 19 indiferent de concentrațiile care nu sunt citotoxice la care este detectată interferența). În toate celelalte cazuri: atunci când S.I. al CD86 este mai mare sau egal cu 150 % și/sau se observă interferențe, concluzia individuală a unui test U-SENS ™ este considerată pozitivă (denumită în continuare „P”).
|
|
—
|
O estimare în urma unui test U-SENS™ este considerată NEGATIVĂ dacă cel puțin două teste independente sunt negative (N) (figura 1). Dacă primele două teste sunt ambele negative (N), estimarea U-SENS™ este considerată NEGATIVĂ și nu trebuie să fie efectuat un al treilea test.
|
|
—
|
O estimare în urma unui test U-SENS™ este considerată POZITIVĂ dacă cel puțin două teste independente sunt pozitive (P) (figura 1). Dacă primele două teste sunt ambele pozitive (P), estimarea U-SENS™ este considerată POZITIVĂ și nu trebuie să fie efectuat un al treilea test.
|
|
—
|
Deoarece nu este efectuată o analiză pentru determinarea dozelor, există o excepție în cazul în care, în cadrul primului test, S.I. al CD86 este mai mare sau egal cu 150 % doar la cea mai mare concentrație care nu este citotoxică. Atunci testul este considerat a fi NECONCLUDENT (NC) și ar trebui să se testeze concentrații suplimentare (între cea mai mare concentrație fără citotoxicitate și cea mai scăzută concentrația cu citotoxicitate - a se vedea punctul 20) în cadrul unor teste suplimentare. În cazul în care un test este identificat ca fiind NC, ar trebui să fie efectuate cel puțin încă două teste, precum și un al patrulea test în cazul în care al doilea și al treilea test nu sunt concordante (N și/sau P independent) (figura 1). Testele efectuate în continuare vor fi considerate pozitive chiar dacă numai în cazul unei concentrații care nu este citotoxică este generată o valoare a CD86 egală cu sau peste 150 %, deoarece valoarea concentrației a fost ajustată pentru substanța chimică de testat specifică. Estimarea finală va fi bazată pe rezultatul majoritar al celor trei sau patru teste individuale (și anume: două din trei sau două din patru) (figura 1).
|

Figura 1: Model predictiv utilizat în testul U-SENS™. O estimare U-SENS™ ar trebui să fie analizată în cadrul unei IATA și în conformitate cu dispozițiile de la punctul 4 și de la punctele 7, 8 și 9 din Introducerea generală.
N: Test fără o valoare pozitivă a CD86 sau observarea unei interferențe;
P: Test cu o valoare pozitivă a CD86 și/sau cu observarea unei (unor) interferențe;
NC: Neconcludent. Primul test fără concluzii atunci când valoarea CD86 este pozitivă numai la cea mai mare concentrație necitotoxică;
#: O concluzie individuală de tipul Neconcludent (NC), care este atribuită numai primului test efectuat determină în mod automat necesitatea efectuării unui al treilea test pentru a ajunge la o majoritate a concluziilor de tip Pozitiv (P) sau Negativ (N) în cadrul a cel puțin două din trei teste independente.
$: Casetele prezintă combinațiile relevante ale rezultatelor obținute în cadrul cele trei teste, pe baza rezultatelor obținute în cadrul primelor două teste prezentate în caseta de mai sus.
°: Casetele prezintă combinațiile relevante ale rezultatelor obținute în cadrul cele patru teste, pe baza rezultatelor obținute în cadrul primelor trei teste prezentate în caseta de mai sus.
|
Criterii de acceptare
|
22.
|
Atunci când este utilizat testul U-SENS™ (12), ar trebui să fie îndeplinite următoarele criterii de acceptare.
|
—
|
La sfârșitul perioadei de expunere de 45±3 ore, viabilitatea medie a celulelor U937 netratate triple trebuia să fie > 90 % și nu se observă nicio deviere a expresiei CD86. Expresia de bază a valorii CD86 a celulelor U937 netratate a trebuit să fie cuprinsă între ≥ 2 % și ≤ 25 %.
|
|
—
|
În cazul în care DMSO este utilizat ca solvent, valabilitatea martorului tratat cu vehicul DMSO este evaluată prin calcularea unui S.I. al DMSO în raport cu celulele netratate, iar viabilitatea medie a celulelor triple a trebuit să fie > 90 %. Martorul tratat cu vehicul DMSO este valabil dacă valoarea medie a S.I. triplu al CD86 a fost sub 250 % din valoarea medie a S.I. al valorii CD86 a celulelor U937 netratate.
|
|
—
|
Testele sunt considerate valabile dacă cel puțin două din trei valori IgG1 ale celulelor U937 netratate s-au încadrat între ≥ 0,6 % și < 1,5 %.
|
|
—
|
Martorul negativ testat în paralel (acidul lactic) este considerat valabil dacă cel puțin două din cele trei replici au fost negative (CD86 S.I. < 150 %) și fără citotoxicitate (viabilitate celulară ≥ 70 %).
|
|
—
|
Martorul pozitiv (TNBS) a fost considerat valabil dacă cel puțin două din cele trei replici au fost pozitive (CD86 S.I. < 150 %) și fără citotoxicitate (viabilitate celulară ≥ 70 %).
|
|
Raportul testului
|
23.
|
Raportul testului ar trebui să includă următoarele informații:
|
Substanță chimică de testat
Substanța monocomponentă
|
Identificarea chimică, precum denumirea (denumirile) IUPAC sau CAS, numărul (numerele) SMILES sau codul InChI, formula structurală și/sau alți identificatori;
|
|
aspectul fizic, solubilitate în mediu complet, solubilitate în DMSO, masa moleculară și alte proprietăți fizico-chimice relevante, dacă există;
|
|
Puritatea, identitatea chimică a impurităților, după caz și dacă este fezabil din punct de vedere practic etc.;
|
|
Tratamentul înainte de efectuarea testului, dacă este cazul (de exemplu, încălzire, măcinare);
|
|
Concentrația (concentrațiile) testată(e);
|
|
Condițiile de depozitare și stabilitatea, dacă există;
|
|
Justificarea alegerii solventului/vehiculului pentru fiecare substanță chimică de testat.
|
Substanța multicomponentă, UVCB și amestec:
|
Caracterizarea, dacă este posibil, de exemplu prin identitatea chimică (a se vedea mai sus), puritatea, apariția cantitativă și proprietățile fizico-chimice relevante (a se vedea mai sus) ale componentelor, dacă există;
|
|
aspectul fizic, solubilitate în mediu complet, solubilitate în DMSO și alte proprietăți fizico-chimice relevante, dacă există;
|
|
Masa moleculară sau masa moleculară aparentă în cazul amestecurilor/polimerilor cu compoziții cunoscute sau alte informații relevante pentru efectuarea studiului;
|
|
Tratamentul înainte de efectuarea testului, dacă este cazul (de exemplu, încălzire, măcinare);
|
|
Concentrația (concentrațiile) testată(e);
|
|
Condițiile de depozitare și stabilitatea, dacă există;
|
|
Justificarea alegerii solventului/vehiculului pentru fiecare substanță chimică de testat.
|
|
|
Martori
Martor pozitiv
|
Identificarea chimică, precum denumirea (denumirile) IUPAC sau CAS, numărul (numerele) SMILES sau codul InChI, formula structurală și/sau alți identificatori;
|
|
aspectul fizic, solubilitatea DMSO, masa moleculară și alte proprietăți fizico-chimice relevante, în măsura în care este posibil și dacă este cazul;
|
|
Puritatea, identitatea chimică a impurităților, după caz și dacă este fezabil din punct de vedere practic etc.;
|
|
Tratamentul înainte de efectuarea testului, dacă este cazul (de exemplu, încălzire, măcinare);
|
|
Concentrația (concentrațiile) testată(e);
|
|
Condițiile de depozitare și stabilitatea, dacă există;
|
|
Trimitere la rezultatele anterioare ale martorilor pozitivi care demonstrează criterii adecvate de acceptare a testelor, dacă este cazul.
|
Martor negativ și tratat cu solvent/vehicul
|
Identificarea chimică, precum denumirea (denumirile) IUPAC sau CAS, numărul (numerele) SMILES sau codul InChI, formula structurală și/sau alți identificatori;
|
|
Puritatea, identitatea chimică a impurităților, după caz și dacă este fezabil din punct de vedere practic etc.;
|
|
Aspectul fizic, masa moleculară și alte proprietăți fizico-chimice relevante în cazul în care sunt utilizați alți martori tratați cu solvent/vehicul decât cei menționați în Ghidul pentru teste și dacă aceștia există;
|
|
Condițiile de depozitare și stabilitatea, dacă există;
|
|
Justificarea alegerii solventului/vehiculului pentru fiecare substanță chimică de testat.
|
|
|
Condiții de testare
|
Numele și adresa sponsorului, ale instalației de testare și ale directorului de studiu;
|
|
descrierea testului utilizat;
|
|
Linia celulară utilizată, condițiile de depozitare și sursa acesteia (de exemplu, instalația de la au fost obținute acestea);
|
|
citometria de flux utilizată (de exemplu, modelul), inclusiv reglajele instrumentelor, anticorpii și markerul de citotoxicitate utilizat;
|
|
Procedura utilizată pentru a demonstra capabilitatea laboratorului de a efectua testul prin testarea unor substanțe de verificare și procedura utilizată pentru a demonstra performanța reproductibilă a testului în timp, de exemplu, datele anterioare privind martorii și/sau datele anterioare privind verificările asupra reactivității.
|
|
|
Criterii de acceptare a testului
|
viabilitatea celulară valorile S.I. ale CD86 obținute cu martorul tratat cu solvent/vehicul în comparație cu intervalele de acceptare;
|
|
viabilitatea celulară și valorile S.I. obținute cu martorul pozitiv în comparație cu intervalele de acceptare;
|
|
Viabilitatea celulară a tuturor concentrațiilor testate ale substanței chimice de testat.
|
|
|
Procedura de testare
|
Numărul de teste utilizate;
|
|
Concentrațiile substanței chimice de testat, timpul de aplicare și de expunere utilizat (dacă acestea diferă de cele recomandate)
|
|
Descrierea criteriilor de evaluare și de decizie utilizate;
|
|
Descrierea oricăror modificări ale procedurii de testare.
|
|
|
Rezultate
|
Prezentarea tabelară a datelor, inclusiv a valorilor CV70 (dacă este cazul), a valorilor de viabilitate celulară, a valorilor CE150 (dacă este cazul) obținute pentru substanța chimică de testat și pentru martorul pozitiv în cadrul fiecărui test, precum și o indicație privind clasificarea substanței chimice de testat în funcție de modelul predictiv;
|
|
O descriere a oricăror alte observații relevante, dacă este cazul.
|
|
|
Discutarea rezultatelor
|
Discutarea rezultatelor obținute în urma testului U-SENS™;
|
|
Analiza rezultatelor testului în contextul unei IATA în cazul în care sunt disponibile alte informații relevante.
|
|
Concluzii
|
REFERINȚE BIBLIOGRAFICE
|
(1)
|
Piroird, C., Ovigne, J.M., Rousset, F., Martinozzi-Teissier, S., Gomes, C., Cotovio, J., Alépée, N. (2015). The Myeloid U937 Skin Sensitization Test (U-SENS) addresses the activation of dendritic cell event in the adverse outcome pathway for skin sensitization. Toxicol. In Vitro 29, 901-916.
|
|
(2)
|
EURL ECVAM (2017). The U-SENS™ test method Validation Study Report. Material accesibil la adresa: http://ihcp.jrc.ec.europa.eu/our_labs/eurl-ecvam/eurl-ecvam-recommendations
|
|
(3)
|
EC EURL ECVAM (2016). ESAC Opinion No 2016-03 on the L'Oréal-coordinated study on the transferability and reliability of the U-SENS™ test method for skin sensitisation testing. EUR 28 178 EN; doi 10.2 787/8157 37. Publicație disponibilă la adresa: [http://publications.jrc.ec.europa.eu/repository/handle/JRC103705].
|
|
(4)
|
EC EURL ECVAM (2017). EURL ECVAM Recommendation on the use of non-animal approaches for skin sensitisation testing. EUR 28 553 EN; doi 10.2 760/5889 55. Publicație disponibilă la adresa: https://ec.europa.eu/jrc/en/publication/eur-scientific-and-technical-research-reports/eurl-ecvam-recommendation-use-non-animal-approaches-skin-sensitisation-testing.
|
|
(5)
|
Steiling, W. (2016). Safety Evaluation of Cosmetic Ingredients Regarding their Skin Sensitization Potential. doi:10.3390/cosmetics3020014.Cosmetics 3, 14.
|
|
(6)
|
OCDE (2016). Guidance Document on The Reporting of Defined Approaches and Individual Information Sources to be Used Within Integrated Approaches to Testing and Assessment (IATA) For Skin Sensitisation, Series on Testing & Assessment No 256, ENV/JM/MONO(2016)29. Organizația pentru Cooperare și Dezvoltare Economică, Paris. Publicație disponibilă la adresa: [http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(7)
|
Urbisch, D., Mehling, A., Guth, K., Ramirez, T., Honarvar, N., Kolle, S., Landsiedel, R., Jaworska, J., Kern, P.S., Gerberick, F., Natsch, A., Emter, R., Ashikaga, T., Miyazawa, M., Sakaguchi, H. (2015). Assessing skin sensitization hazard in mice and men using non-animal test methods. Regul. Toxicol. Pharmacol. 71, 337-351.
|
|
(8)
|
Alépée, N., Piroird, C., Aujoulat, M., Dreyfuss, S., Hoffmann, S., Hohenstein, A., Meloni, M., Nardelli, L., Gerbeix, C., Cotovio, J. (2015). Prospective multicentre study of the U-SENS test method for skin sensitization testing. Toxicol In Vitro 30, 373-382.
|
|
(9)
|
Reisinger, K., Hoffmann, S., Alépée, N., Ashikaga, T., Barroso, J., Elcombe, C., Gellatly, N., Galbiati, V., Gibbs, S., Groux, H., Hibatallah, J., Keller, D., Kern, P., Klaric, M., Kolle, S., Kuehnl, J., Lambrechts, N., Lindstedt, M., Millet, M., Martinozzi-Teissier, S., Natsch, A., Petersohn, D., Pike, I., Sakaguchi, H., Schepky, A., Tailhardat, M., Templier, M., van Vliet, E., Maxwell, G. (2014). Systematic evaluation of non-animal test methods for skin sensitisation safety assessment. Toxicol. In Vitro 29, 259-270.
|
|
(10)
|
Fabian, E., Vogel, D., Blatz, V., Ramirez, T., Kolle, S., Eltze, T., van Ravenzwaay, B., Oesch, F., Landsiedel, R. (2013). Xenobiotic metabolizin enzyme activities in cells used for testing skin sensitization in vitro. Arch. Toxicol. 87, 1 683-1 696.
|
|
(11)
|
OCDE. (2018). Draft Guidance document: Good In Vitro Method Practices (GIVIMP) for the Development and Implementation of In Vitro Methods for Regulatory Use in Human Safety Assessment. Organizația pentru Cooperare și Dezvoltare Economică, Paris. Publicație disponibilă la adresa: http://www.oecd.org/env/ehs/testing/OECD Final Draft GIVIMP.pdf.
|
|
(12)
|
DB-ALM (2016). Protocol no 183: Myeloid U937 Skin Sensitization Test (U-SENS™), 33pp. Material accesibil la adresa: [http://ecvam-dbalm.jrc.ec.europa.eu/].
|
|
(13)
|
Sundström, C., Nilsson, K. (1976). Establishment and characterization of a human histiocytic lymphoma cell line (U-937). Int. J. Cancer 17, 565-577.
|
|
(14)
|
OCDE (2005). Series on Testing and Assessment No. 34: Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Organizația pentru Cooperare și Dezvoltare Economică, Paris. Publicație disponibilă la adresa: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(15)
|
Organizația Națiunilor Unite ONU (2015). Sistemul global armonizat de clasificare și etichetare a substanțelor chimice (GHS). ST/SG/AC.10/30/Rev.6, a șasea ediție revizuită, New York & Geneva: United Nations Publications. Publicație disponibilă la adresa: http://www.unece.org/fileadmin/DAM/trans/danger/publi/ghs/ghs_rev06/English/ST-SG-AC10-30-Rev6e.pdf.
|
|
(16)
|
OCDE (2012). Series on Testing and Assessment No 168: The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins. Part 1: Scientific Evidence. Organizația pentru Cooperare și Dezvoltare Economică, Paris. Publicație disponibilă la adresa: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(17)
|
ECETOC (2003). Technical Report No 87: Contact sensitization: Classification according to potency. European Centre for Ecotoxicology & Toxicology of Chemicals, Bruxelles. Publicație disponibilă la adresa: https://ftp.cdc.gov/pub/Documents/OEL/06.%20Dotson/References/ECETOC_2003-TR87.pdf.
|
Apendicele 2.1
DEFINIțII
Precizie
: Gradul de concordanță între rezultatele testului și valorile de referință acceptate. Aceasta măsoară performanța testului și unul dintre aspectele relevanței acestuia. Termenul este adesea utilizat în mod interschimbabil, iar concordanța înseamnă proporția de rezultate corecte ale unui test (14).
AOP (parcursul răspunsurilor adverse)
: o serie de evenimente din structura chimică a unei substanțe chimice țintă sau un grup de substanțe chimice similare, de la evenimentul molecular declanșator până la un răspuns in vivo de interes (15).
Reacția concentrației CD86
: Există o dependență de concentrație (sau o reacție a concentrației) atunci când o concentrație pozitivă (CD86 S.I. ≥ 150) este urmată de o concentrație cu o valoare S.I. a CD86 în creștere.
Substanță chimică
: O substanță sau un amestec.
CV70
: Concentrația estimată care indică o viabilitate celulară de 70 %.
Deviere
: O deviere este definită prin următoarele situații i) valoarea % corectată a CD86+ a celei de a treia replici a martorului netratat este mai mică decât 50 % din media valorii % corectate a CD86+ a primei și celei de a doua replici ale martorului netratat; și ii) valoarea % corectată a CD86+ a celei de a treia replici a martorului negativ este mai mică decât 50 % din media valorii % corectate a CD86+ a primei și celei de a doua replici ale martorului negativ.
EC150
: concentrațiile estimate care arată S.I. de 150 % al expresiei CD86.
Citometria de flux
: o tehnică citometrică prin care celulele au fost suspendate într-un flux fluid una câte una, printr-o focalizare a luminii care excită și care este difuzată în modele caracteristice asupra celulelor și a componentelor acestora; celulele sunt etichetate frecvent cu markeri fluorescenți, astfel încât lumina să fie absorbită mai întâi și apoi emisă la frecvențe modificate.
Pericol
: Proprietate intrinsecă a unui agent sau a unei situații care are potențialul de a produce efecte adverse atunci când un organism, un sistem sau o (sub)populație este expusă la agentul respectiv.
IATA (Strategie integrată pentru testare și evaluare)
: O abordare structurată utilizată pentru identificarea pericolelor (potențial), caracterizarea pericolelor (potența) și/sau evaluarea siguranței (potențial/potență și expunere) pentru o substanță chimică sau un grup de substanțe chimice, care se integrează în mod strategic și măsoară toate datele relevante pentru a furniza informații cu privire la decizii de reglementare în ceea ce privește potențialele pericole și/sau riscuri și/sau necesitatea unor teste suplimentare specifice și, prin urmare, minime.
Amestec
: un amestec sau o soluție compus/ă din două sau mai multe substanțe.
Substanță monocomponentă
: O substanță definită prin compoziția sa cantitativă, în care un singur component principal este prezent în proporție de cel puțin 80 % (g/g).
Substanță multicomponentă
: O substanță definită prin compoziția sa cantitativă, în care mai mult de un singur component principal este prezent într-o concentrație ≥ 10 % (g/g) și < de 80 % (g/g). O substanță multicomponentă este rezultatul unui proces de fabricație. Diferența dintre un amestec și o substanță multicomponentă este faptul că un amestec este obținut prin combinarea a două sau mai multe substanțe chimice fără o reacție chimică. O substanță multicomponentă este rezultatul unei reacții chimice.
Martor pozitiv
: O replică ce conține toate componentele sistemului de testare și care a fost tratată cu o substanță cunoscută pentru faptul că provoacă o reacție pozitivă. Pentru a asigura posibilitatea evaluării variabilității în timp a reacției martorului pozitiv, amplitudinea reacției severe nu trebuie să fie excesiv de mare.
Prehaptene
: substanțe chimice care devin sensibilizante prin transformări abiotice, de exemplu, prin oxidare.
Prohaptene
: substanțe chimice care necesită activare enzimatică pentru a exercita potențialul de sensibilizare cutanată.
Relevanță
: Descrierea relației dintre test și efectul de interes și dacă aceasta este relevantă și utilă pentru un anumit scop. Aceasta reprezintă măsura în care testul măsoară sau prezice în mod corect efectul biologic de interes. Relevanța include analiza preciziei (a concordanței) unui test (14).
Fiabilitate
: Măsuri ale faptului că un test poate fi efectuat în mod reproductibil, în același laborator și în laboratoare diferite în timp, atunci când este efectuat utilizând același protocol. Aceasta se evaluează prin calcularea reproductibilității în același laborator sau între laboratoare și a repetabilității în același laborator (14).
Test
: Un test este compus din unul sau mai multe substanțe chimice de testat, care sunt testate concomitent cu un martor solvent/vehicul și un martor pozitiv.
Sensibilitate
: Proporția tuturor substanțelor chimice pozitive/active clasificate în mod corect în urma testului. Aceasta permite măsurarea preciziei în cazul unui test care generează rezultate clasificabile în categorii și constituie un aspect important în evaluarea relevanței unui test (14).
S.I.
: Indice de stimulare. Valorile relative ale intensității medii a fluorescenței la celulele tratate chimic comparativ cu celulele tratate cu solvent/vehicul.
Martor solvent/vehicul
: O probă netratată care conține toate componentele unui sistem de testare, cu excepția celor ale substanței chimice de testat, dar incluzând solventul/vehiculul utilizat. Acesta este utilizat pentru a stabili reacția de bază pentru probele tratate cu substanța chimică de testat dizolvată sau dispersată stabil în același solvent/vehicul. Atunci când este testată concomitent cu un martor de mediu, această probă indică, de asemenea, dacă solventul/vehiculul interacționează cu sistemul testat.
Specificitate
: Proporția tuturor substanțelor chimice negative/inactive clasificate în mod corect în urma testului. Aceasta permite măsurarea preciziei în cazul unui test care generează rezultate clasificabile în categorii și constituie un aspect important în evaluarea relevanței unui test (14).
Soluție tampon de colorare
: O soluție salină tamponată cu fosfat conținând o cantitate de 5 % ser fetal de vițel.
Substanță
: un element chimic și compușii săi în stare naturală sau obținuți prin orice proces de producție, inclusiv orice aditiv necesar pentru a-i păstra stabilitatea și orice impuritate care derivă din procesul utilizat, însă excluzând orice solvent care poate fi separat fără a afecta stabilitatea substanței sau fără a-i schimba compoziția.
Substanță chimică de testat
: orice substanță sau amestec testat(ă) în cadrul acestui test.
Sistemul global armonizat de clasificare și etichetare a substanțelor chimice al Organizației Națiunilor Unite (GHS al ONU)
: Un sistem care propune clasificarea substanțelor chimice (substanțe și amestecuri) pe tipuri și niveluri standardizate de pericole fizice, pentru sănătate și pentru mediu, și care vizează elemente de comunicare corespunzătoare, de exemplu pictograme, cuvinte de semnalizare, fraze de pericol, fraze de precauție și fișe cu date de securitate, pentru a transmite informații privind efectele adverse ale acestora, în scopul protejării populației (inclusiv a angajatorilor, a lucrătorilor, a transportatorilor, a consumatorilor și a serviciilor de urgență) și a mediului (16).
UVCB
: Substanțe cu compoziție necunoscută sau variabilă, produși de reacție complexă sau materiale biologice.
Test valabil
: Un test considerat a avea suficientă relevanță și fiabilitate pentru un scop specific și care se bazează pe principii solide din punct de vedere științific. Un test nu este niciodată valabil într-un sens absolut, ci doar în legătură cu un scop definit (14).
Apendicele 2.2
SUBSTANțE DE VERIFICARE
Înainte de utilizarea de rutină a testului descris în prezentul apendice la metoda de testare B.71, laboratoarele ar trebui să demonstreze performanțele tehnice prin obținerea în mod corect a estimării U-SENS™ pentru cele 10 substanțe de verificare recomandate în tabelul 1 și prin obținerea de valori pentru CV70 și EC150 care să se încadreze în plaja de valori de referință corespunzătoare pentru cel puțin 8 dintre cele 10 substanțe de verificare. Substanțele de verificare au fost selectate pentru a reprezenta intervalul de reacții pentru pericolele sensibilizării cutanate. Alte criterii de selecție au fost ca substanțele să fie disponibile în comerț și să fie disponibile date de referință in vivo de înaltă calitate, precum și date in vitro de înaltă calitate generate cu testul U-SENS™. De asemenea, sunt disponibile date de referință publicate pentru testul U-SENS™ (1) (8).
Tabelul 1
Substanțe recomandate pentru a demonstra performanțele tehnice cu testul U-SENS™
|
Substanțe de verificare
|
NR CAS
|
Stare fizică
|
Estimare in vivo
(106)
|
U-SENS Solvent/Vehicul
|
U-SENS CV70 Interval de referință în μg/ml (107)
|
U-SENS Interval de referință pentru EC150 în μg/ml (107)
|
|
4-fenilendiamină
|
106-50-3
|
Solid
|
Sensibilizant (puternic)
|
Mediu complet (108)
|
< 30
|
Pozitiv (≤ 10)
|
|
Acid picril-sulfonic
|
2508-19-2
|
Lichid
|
Sensibilizant (puternic)
|
Mediu complet
|
>50
|
Pozitiv (≤ 50)
|
|
Maleat de dietil
|
141-05-9
|
Lichid
|
Sensibilizant (moderat)
|
DMSO
|
10-100
|
Pozitiv (≤ 20)
|
|
Rezorcinol
|
108-46-3
|
Solid
|
Sensibilizant (moderat)
|
Mediu complet
|
>100
|
Pozitiv (≤ 50)
|
|
Alcool cinamic
|
104-54-1
|
Solid
|
Sensibilizant (slab)
|
DMSO
|
>100
|
Pozitiv (10-100)
|
|
4-alilanisol
|
140-67-0
|
Lichid
|
Sensibilizant (slab)
|
DMSO
|
>100
|
Pozitiv (<200)
|
|
Zaharină
|
81-07-2
|
Solid
|
Non-sensibilizant
|
DMSO
|
>200
|
Negativ (> 200)
|
|
Glicerină
|
56-81-5
|
Lichid
|
Non-sensibilizant
|
Mediu complet
|
>200
|
Negativ (> 200)
|
|
Acid lactic
|
50-21-5
|
Lichid
|
Non-sensibilizant
|
Mediu complet
|
>200
|
Negativ (> 200)
|
|
Acid salicilic
|
69-72-7
|
Solid
|
Non-sensibilizant
|
DMSO
|
>200
|
Negativ (> 200)
|
|
Abrevieri: NR CAS = Număr de înregistrare al Chemical Abstracts Service
|
Apendicele 3
SENSIBILIZARE CUTANATĂ IN VITRO: TESTUL IL-8 LUC
CONSIDERAȚII PRELIMINARE ȘI LIMITĂRI
|
1.
|
Spre deosebire de testele care analizează expresia markerilor celulari de suprafață, testul IL8-Luc cuantifică modificările expresiei IL-8, o citocină asociată activării celulelor dendritice (CD). În linia celulară raportoare de tip IL-8 derivată din THP-1 (THP-G8, determinată pornind de la linia celulară monocitică acută la om afectată de leucemie THP-1), expresia IL-8 se măsoară în urma expunerii la substanțe sensibilizante (1). Expresia luciferazei este utilizată apoi pentru a contribui la diferențierea între sensibilizantele și nesensibilizantele pentru piele.
|
|
2.
|
Testul IL-8 Luc a fost evaluat în cadrul unui studiu de validare (2) realizat de Centrul Japonez pentru Validarea Metodelor Alternative (Japanese Centre for the Validation of Alternative Methods - JaCVAM), Ministerul economiei, comerțului și industriei (METI) și Societatea Japoneză pentru Metode Alternative la Testarea pe Animale (JSAAE) și, ulterior, supus unei evaluări inter pares independente desfășurate sub auspiciile JaCVAM (3) și ale Ministerului Sănătății, Muncii și Bunăstării (MHLW) cu sprijinul International Cooperation on Alternative Test Methods ( ICATM). Având în vedere toate dovezile disponibile și contribuțiile autorităților de reglementare și ale părților interesate, testul IL-8 Luc este considerat util în cadrul unei IATA pentru a diferenția substanțele sensibilizante de cele nesensibilizante în scopul clasificării și etichetării pericolelor. Exemple privind utilizarea datelor din cadrul testului IL-8 Luc în combinație cu alte informații sunt prezentate în referințe bibliografice (4) (5) (6).
|
|
3.
|
Testul IL-8 Luc s-a dovedit a fi transmisibil la laboratoare cu experiență în cultura celulelor și măsurarea luciferazei. În cadrul aceluiași laborator și între laboratoare ratele de reproductibilitate au fost de 87,7 % și, respectiv, 87,5 % (2). Datele generate în cadrul studiului de validare (2) și alte lucrări publicate (1) (6) arată că, în comparație cu LLNA, prin testul IL-8 Luc un număr de 118 din 143 de substanțe chimice au fost estimate ca fiind pozitive sau negative, iar 25 de substanțe chimice au fost estimate ca fiind neconcludente; precizia testului IL-8 Luc privind distincția între sensibilizantele pentru piele (categoria 1 din GHS al ONU/Regulamentul CLP) și nesensibilizantele pentru piele (neîncadrate într-o categorie din GHS al ONU/Regulamentului CLP) este de 86 % (101/118) cu o sensibilitate de 96 % (92/96) și o specificitate de 41 % (9/22). Cu excepția substanțelor din afara domeniului de aplicabilitate descris mai jos (punctul 5), un număr de 113 din 136 de substanțe chimice au fost estimate ca fiind pozitive sau negative prin testul IL-8 Luc, iar 23 de substanțe chimice au fost estimate ca fiind neconcludente; precizia testului IL-8 Luc este de 89 % (101/113), sensibilitatea de 96 % (92/96) și specificitatea de 53 % (9/17). Folosind datele despre oameni citate în Urbisch et al. (7), prin testul IL-8 Luc un număr de 76 din 90 de substanțe chimice au fost estimate ca fiind pozitive sau negative, iar 14 substanțe chimice au fost estimate ca fiind neconcludente; precizia este de 80 % (61/76), sensibilitatea de 93 % (54/58) și specificitatea de 39 % (7/18). Cu excepția substanțelor din afara domeniului de aplicabilitate, un număr de 71 din 84 de substanțe chimice au fost estimate ca fiind pozitive sau negative prin testul IL-8 Luc, iar 13 de substanțe chimice au fost estimate ca fiind neconcludente; precizia este de 86 % (61/71), sensibilitatea de 93 % (54/58) și specificitatea de 54 % (7/13). Există o probabilitate mai mare să apară estimări negative false în urma testului IL-8 Luc în cazul substanțelor chimice care prezintă un nivel scăzut/moderat de potență de sensibilizare cutanată (subcategoria 1A din GHS al ONU/Regulamentul CLP) (6). Împreună, informațiile sprijină rolul testului IL-8 Luc în ceea ce privește identificarea pericolelor de sensibilizare a pielii. Valoarea preciziei furnizate pentru testul IL-8 Luc ca și test independent este doar orientativă, întrucât testul ar trebui luat în considerare în combinație cu alte surse de informații în contextul unei IATA și în conformitate cu dispozițiile de la alineatele (7) și (8) din Introducerea generală. În plus, la evaluarea testelor care nu se bazează pe animale pentru sensibilizarea cutanată, ar trebui reținut faptul că este posibil ca testul LLNA și alte teste pe animale să nu reflecte pe deplin situația oamenilor.
|
|
4.
|
Pe baza datelor disponibile, testul IL-8 Luc s-a dovedit a fi aplicabil substanțelor chimice de testat care acoperă o varietate de grupe organice funcționale, mecanisme de reacție, potență de sensibilizare cutanată (astfel cum a fost stabilit în cadrul studiilor in vivo), precum și proprietăți fizico-chimice (2) (6).
|
|
5.
|
Deși testul IL-8 Luc utilizează X-VIVOTM 15 ca și solvent, prin acesta au fost evaluate în mod corect substanțele chimice având Log Kow >3,5, precum și cele având o solubilitate în apă de aproximativ 100 μg/ml, astfel cum a fost calculată prin EPI SuiteTM, iar performanța acestuia de a detecta sensibilizantele cu o solubilitate mai scăzută în apă este mai bună decât a testului IL-8 Luc în care se utilizează dimetilsulfoxid (DMSO) ca și solvent (2). Însă rezultatele negative obținute în cazul substanțelor chimice de testat care nu se dizolvă în 20 mg/ml pot produce rezultate negative false din cauza incapacității acestora de a se dizolva în X-VIVOTM 15. Prin urmare, nu ar trebui să se ia în considerare rezultatele negative în cazul acestor substanțe chimice. În cadrul studiului de validare a fost observată o rată ridicată de rezultate negative false în cazul anhidridelor. În plus, din cauza capacității metabolice limitate a liniei celulare (8) și a condițiilor experimentale, este posibil ca prohaptenele (substanțe care necesită activare metabolică) și prehaptenele (substanțe activate prin oxidare cu aer) să genereze rezultate negative în cadrul testului. Cu toate acestea, deși rezultatele negative pentru prehaptene/prohaptene ar trebui să fie interpretate cu prudență, prin testul IL-8 Luc 11 din 11 prehaptene, 6/6 prohaptene și 6/8 prehaptene/prohaptene au fost estimate corect în cadrul setului de date pentru IL-8 Luc (2). Pe baza revizuirii cuprinzătoare efectuate recent asupra a trei teste care nu se bazează pe animale (DPRA, KeratinoSens™ și h-CLAT) pentru a detecta prehaptene și prohaptene (9) și pe baza faptului că celulele THP-G8 utilizate în testul IL-8 Luc constituie o linie celulară derivată din THP-1 care este utilizată în testul h-CLAT, testul IL-8 Luc poate contribui, de asemenea, la creșterea sensibilității testelor care nu se bazează pe animale pentru a detecta prehaptene și prohaptene în combinația cu alte teste. Surfactanții testați până la acest moment au generat rezultate pozitive (false) indiferent de tipul lor (de exemplu, cationic, anionic sau non-ionic). În cele din urmă, substanțele chimice care interferează cu luciferaza pot avea efecte asupra activității/măsurării acesteia, cauzând o inhibiție aparentă sau o creștere a luminiscenței (10). De exemplu, concentrațiile de fitoestrogen mai mari de 1 μM au fost raportate a interfera cu semnalele de luminiscență ale altor teste asupra genei raportoare bazate pe luciferază din cauza supraactivării genei raportoare pentru luciferază. În consecință, trebuie să se examineze cu atenție expresia luciferazei obținută la concentrații ridicate de fitoestrogeni sau compuși, care este suspectată de a produce o activare similară fitoestrogenilor a genei raportoare pentru luciferază (11). Pe baza considerațiilor de mai sus, surfactanții, anhidridele și substanțele chimice care interferează cu luciferaza sunt în afara domeniului de aplicabilitate al acestui test. În cazul în care există dovezi care demonstrează lipsa de aplicabilitate a testului IL-8 Luc la alte categorii specifice de substanțe chimice de testat, testul nu ar trebui utilizat pentru respectivele categorii specifice.
|
|
6.
|
Conform descrierii de mai sus, testul IL-8 Luc susține diferențierea între sensibilizantele și nesensibilizantele pentru piele. Sunt necesare studii suplimentare, de preferat pe baza datelor umane, pentru a stabili dacă rezultatele testului IL-8 Luc pot contribui la evaluarea potenței atunci când sunt analizate împreună cu alte surse de informații.
|
|
7.
|
Definițiile sunt prevăzute în apendicele 3.1.
|
PRINCIPIUL TESTULUI
|
8.
|
Testul IL-8 Luc utilizează o linie celulară monocitică umană afectată de leucemie THP-1 obținută de la American Type Culture Collection (Manassas, VA, SUA). Utilizând această linie celulară, Departamentul de Dermatologie din cadrul Universității School of Medicine din Tohoku a stabilit o linie celulară raportoare IL-8 derivată din THP-1, denumită THP-G8, care poartă gene de luciferază Stable Luciferase Orange (SLO) și Stable Luciferase Red (SLR) sub controlul promotorilor IL-8 și, respectiv, ai gliceraldehid-3-fosfat dehidrogenazei (GAPDH) (1). Acest lucru permite măsurarea cantitativă a inducției genei luciferază prin detectarea luminiscenței separat de lumina bine stabilită care produce substraturi de luciferază ca indicator al activității IL-8 și GAPDH la celule în urma expunerii la substanțe chimice sensibilizante.
|
|
9.
|
Sistemul de testare cu două culori cuprinde o luciferază care emite culoarea portocalie (SLO; λmax = 580 nm) (12) pentru expresia genică a promotorului IL-8 și o luciferază care emite culoarea roșie (SLR; λmax = 630 nm) (13) pentru expresia genică a promotorului martorului intern, GAPDH. Cele două luciferaze care emit culori diferite la reacția cu firefly d-luciferin și luminiscența lor se măsoară simultan în cadrul unei reacții într-o singură etapă prin detașarea emisiilor de amestecul de testare cu ajutorul unui filtru optic (14) (apendicele 3.2).
|
|
10.
|
Celulele THP-G8 sunt tratate timp de 16 ore cu substanța chimică de testat, după care se măsoară activitatea luciferazei SLO (SLOA-LA), care reflectă activitatea promotorului IL-8 și activitatea luciferazei SLR (SLR-LA), care reflectă activitatea promotorului GAPDH. Pentru a facilita înțelegerea abrevierilor, SLO-LA și SLR-LA sunt desemnate ca fiind IL8LA și, respectiv, GAPLA. Tabelul 1 prezintă o descriere a termenilor asociați activității luciferazei în cadrul testului IL-8 Luc. Valorile măsurate sunt utilizate pentru a calcula valoarea IL8LA normalizată (nIL8LA), care este raportul dintre IL8LA și GAPLA; inducția nIL8LA (Ind-IL8LA), care este raportul dintre mediile aritmetice ale valorilor măsurate de patru ori ale nIL8LA ale celulelor THP-G8 tratate cu o substanță chimică de testat și ale valorilor nIL8LA ale celulelor THP-G8 netratate; și inhibiția GAPLA (Inh-GAPLA), care este raportul dintre mediile aritmetice ale valorilor măsurate de patru ori ale GAPLA ale celulelor THP-G8 tratate cu o substanță chimică de testat și ale valorilor GAPLA ale celulelor THP-G8 netratate, utilizat ca indicator pentru citotoxicitate.
Tabelul 1
Descrierea termenilor asociați activității luciferazei în testul IL-8 Luc
|
Abrevieri
|
Definiție
|
|
GAPLA
|
Activitatea luciferazei SLR care reflectă activitatea promotorului GAPDH
|
|
IL8LA
|
Activitatea luciferazei SLO care reflectă activitatea promotorului IL-8
|
|
nIL8LA
|
IL8LA/GAPLA
|
|
Ind-IL8LA
|
nIL8LA a celulelor THP-G8 tratate cu substanțe chimice/nIL8LA a celulelor netratate
|
|
Inh-GAPLA
|
GAPLA a celulelor THP-G8 tratate cu substanțe chimice/GAPLA a celulelor netratate
|
|
CV05
|
Concentrația cea mai scăzută a substanței chimice la care Inh-GAPLA devine < 0,05.
|
|
|
11.
|
Există standarde de performanță (PS) (15) pentru a facilita validarea testelor in vitro IL-8 ale luciferazei modificate, care sunt similare cu testul IL-8 Luc, și a permite modificarea promptă a Ghidului 442E al OCDE privind testele pentru includerea acestora. Sistemul de acceptare reciprocă a datelor (MAD) al OCDE va fi garantat doar pentru testele validate în conformitate cu PS, în cazul în care aceste teste au fost revizuite și incluse în Ghidul 442E al OCDE privind testele (16).
|
DEMONSTRAREA PERFORMANȚEI
|
12.
|
Înainte de utilizarea de rutină a testului descris în prezentul apendice pentru metoda de testare B.71, laboratoarele ar trebui să demonstreze performanțele tehnice, utilizând cele 10 substanțe de verificare enumerate în apendicele 3.3, în conformitate cu bunele practici privind metodele in vitro (17). În plus, utilizatorii testelor ar trebui să mențină o bază de date istorice generate în urma verificărilor de reactivitate (a se vedea punctul 15) și cu martori pozitivi și tratați cu solvent/vehicul (a se vedea punctele 21-24) și să utilizeze aceste date pentru a confirma faptul că reproductibilitatea testului în laboratorul propriu este asigurată în timp.
|
PROCEDURA
|
13.
|
Există procedura standard de operare (PSO) în cazul testului IL-8 Luc, care ar trebui utilizată la efectuarea testului (18). Laboratoarele care doresc să efectueze testul pot obține linia celulară recombinantă THP-G8 de la laboratorul GPC Lab. Co. Ltd., Tottori, Japonia, la semnarea unui acord de transfer de material (ATM) în conformitate cu condițiile modelului OCDE. În paragrafele următoare sunt descrise componentele și procedurile principale ale testului.
|
Pregătirea celulelor
|
14.
|
Linia celulară THP-G8 de la GPC Lab. Co. Ltd., Tottori, Japonia ar trebui să fie utilizată pentru efectuarea testului IL-8 Luc (a se vedea punctele 8 și 13). La primire, celulele sunt multiplicate (2-4 pasaje) și păstrate înghețate ca stoc omogen. Celulele din acest stoc pot fi multiplicate până la maximum 12 treceri sau maximum 6 săptămâni. Mediul utilizat pentru multiplicare este mediul de cultură RPMI-1640 care conține 10 % ser fetal bovin (FBS), soluție antibiotică/antimicotică (100 U/ml de penicilină G, 100μg/ml de streptomicină și 0,25μg/ml de amfotericină B în 0,85 % soluție salină) (de exemplu, GIBCO Cat#15240-062), 0,15μg/ml Puromycin (de exemplu, CAS:58-58-2) și 300 μg/ml G418 (de exemplu, CAS:108321-42-2).
|
|
15.
|
Înainte de a fi utilizate pentru testare, celulele ar trebui să fie calificate prin efectuarea unei verificări de reactivitate. Această verificare ar trebui efectuată la 1-2 săptămâni sau după 2-4 de pasaje de la decongelare, cu ajutorul martorului pozitiv, bromura 4-nitrobenzil (4-NBB), (CAS:100-11-8, cu puritate ≥ 99 %) și al martorului negativ, acidul lactic (AL) (CAS:50-21-5, cu o puritate ≥ 85 %). 4-NBB ar trebui să producă un răspuns pozitiv la Ind-IL8LA (≥ 1,4), iar AL ar trebui să producă un răspuns negativ la Ind-IL8LA (< 1,4). Sunt utilizate pentru test numai celulele pentru care verificarea de reactivitate a fost efectuată cu succes. Verificarea ar trebui să fie efectuată conform procedurilor descrise la punctele 22-24.
|
|
16.
|
În scopul testării, celulele THP-G8 sunt însămânțate la o densitate de 5 × 105 celule/ml și cultivate în prealabil în baloane de cultură timp de 48-96 de ore. În ziua testului, celule recoltate din balonul de cultură sunt spălate cu RPMI-1640 conținând 10 % FBS fără antibiotice, și apoi resuspendate în RPMI-1640 conținând 10 % FBS fără antibiotice la 1 × 106 celule/ml. Apoi, celulele sunt distribuite pe o placă de 96 de godeuri cu fund plat de culoare neagră (de exemplu, Costar Cat#3603) de 50 μl (5 × 104 celule/godeu).
|
Pregătirea substanței chimice de testat și a substanțelor martor
|
17.
|
Substanța chimică de testat și substanțele martor se pregătesc în ziua testării. Pentru testul IL-8 Luc, substanțele chimice de testat se dizolvă în X-VIVOTM 15, un mediu fără ser disponibil în comerț (Lonza, 04-418Q), până la concentrația finală de 20 mg/ml. X-VIVOTM 15 se adaugă la 20 mg de substanță chimică de testat (indiferent de solubilitatea substanței chimice) într-o eprubetă de microcentrifugare și se aduce la un volum de 1 ml, apoi se rotește viguros și se agită pe un rotor la o viteză maximă de 8 rpm timp de 30 de minute la o temperatură ambiantă de aproximativ 20 ,°C. Apoi, dacă substanțele chimice solide sunt încă solide, eprubeta este supusă procesului sonic până când substanța chimică este dizolvată complet sau dispersată în mod stabil. Pentru substanțele chimice de testat solubile din X-VIVOTM 15, soluția este diluată cu un factor de 5 cu X-VIVOTM 15 și utilizată ca soluție mamă X-VIVOTM 15 a substanței chimice de testat (4 mg/ml). În cazul substanțelor chimice de testat care nu sunt solubile în X-VIVOTM 15, amestecul este rotit din nou timp de cel puțin 30 de minute, apoi centrifugat la 15000 rpm (≈ 20000g) timp de 5 minute; supernatantul rezultat este utilizat ca soluție mamă X-VIVOTM 15 a substanței chimice de testat. Utilizarea altor solvenți, cum ar fi DMSO, apă, sau mediu de cultură, ar trebui să fie justificată științific. Procedura detaliată pentru dizolvarea substanțelor chimice este prezentată în apendicele 3.5. Soluțiile X-VIVOTM 15 descrise la punctele 18-23 sunt amestecate 1:1 (v/v) cu suspensiile de celule preparate pe o placă de 96 de godeuri cu fund plat de culoare neagră (a se vedea punctul 16).
|
|
18.
|
Primul test are drept scop determinarea concentrației citotoxice și examinarea potențialului de sensibilizare cutanată al substanțelor chimice. Folosind X-VIVOTM 15, diluțiile în serie ale soluțiilor mamă X-VIVOTM 15 ale substanțelor chimice de testat sunt efectuate la factorul doi de diluție (a se vedea apendicele 3.5) folosind un bloc de analiză cu 96 de godeuri (de exemplu: Costar Cat#EW-01729-03). În continuare, se adaugă 50 μl de soluție diluată în fiecare godeu la 50 μl de suspensie celulară pe o placă de 96 de godeuri cu fund plat de culoare neagră. Astfel, pentru substanțele chimice de testat care sunt solubile în X-VIVOTM 15, concentrațiile finale ale substanțelor chimice de testat variază între 0,002 și 2 mg/ml (apendicele 3.5). Pentru substanțele chimice de testat care nu sunt solubile în X-VIVOTM 15 la 20 mg/ml, se determină numai factorii de diluare care se încadrează între 2 și 210, deși concentrațiile finale reale ale substanțelor chimice de testat rămân incerte și depind de concentrația saturată a substanțelor chimice de testat din soluția mamă X-VIVOTM 15.
|
|
19.
|
În cadrul testelor ulterioare (și anume, a doua, a treia și a patra replică), soluția mamă X-VIVOTM 15 este produsă la o concentrație de patru ori mai mare decât concentrația de viabilitate celulară 05 (CV05; cea mai scăzută concentrație la care Inh-GAPLA devine < 0,05) în primul experiment. Dacă Inh-GAPLA nu scade sub 0,05 la concentrația cea mai mare în cadrul primului test, soluția mamă X-VIVOTM 15 este preparată la cea mai ridicată concentrație a primului test. Concentrația CV05 se calculează împărțind concentrația soluției mamă din primul test la factorul de diluare pentru CV05 (X) [factorul de diluție CV05 (X); factorul de diluție necesar pentru diluarea soluției mamă la CV05) (a se vedea apendicele 3.5). În cazul substanțelor de testat care nu sunt solubile în X-VIVO la 20 mg/ml, CV05 este determinată prin concentrația soluției mamă × 1/X. Pentru testele 2-4, este preparată a doua soluție mamă ca 4 × CV05 (apendicele 3.5).
|
|
20.
|
Diluțiile în serie ale soluțiilor mamă de gradul doi X-VIVOTM 15 sunt realizate cu un factor de diluare de 1,5 utilizând un bloc de analiză cu 96 de godeuri. În continuare, se adaugă 50 μl de soluție diluată în fiecare godeu la 50 μl de suspensie celulară în godeurile unei plăci de 96 de godeuri cu fund plat de culoare neagră. Fiecare concentrație a fiecărei substanțe chimice de testat ar trebui să fie testată în patru godeuri. Eșantioanele sunt apoi amestecate pe un agitator cu placă și incubate timp de 16 ore la 37 °C și în soluție de 5 % CO2, după care se măsoară activitatea luciferazei conform descrierii de mai jos.
|
|
21.
|
Martorul tratat cu solvent este amestecul de 50 μl/godeu de X-VIVOTM 15 și de 50 μl/godeu de suspensie celulară în RPMI-1640 conținând 10 % FBS.
|
|
22.
|
Martorul pozitiv recomandat este 4-NBB. Într-o eprubetă de microcentrifugare se prepară 20 mg de 4-NBB, la care se adaugă X-VIVOTM 15 până la 1 ml. Eprubeta se rotește viguros și se agită pe un rotor la o viteză maximă de 8 rpm timp de 30 de minute. După centrifugare la 20000 g timp de 5 minute, supernatantul este diluat cu un factor de 4 în X-VIVOTM 15 și o cantitate de 500 μl de supernatant diluat se transferă într-un godeu din blocul de analiză cu 96 de godeuri. Supernatantul diluat este diluat în continuare în X-VIVOTM 15 la factori de 2 și 4 și se adaugă 50 μl de soluție la 50 μl de suspensie celulară THP-G8 în godeurile unei plăci cu 96 de godeuri cu fund plat de culoare neagră (apendicele 3.6). Fiecare concentrație a martorului pozitiv ar trebui să fie testată în patru godeuri. Placa se agită pentru un agitator cu placă și se incubează într-un incubator cu CO2 timp de 16 ore (37 °C, 5 % CO2), după care se măsoară activitatea luciferazei conform descrierii de la punctul 29.
|
|
23.
|
Martorul negativ recomandat este AL. Se prepară 20 mg de AL într-o eprubetă de microcentrifugare de 1,5 ml, la care se adaugă X-VIVOTM 15 până la 1 ml (20 mg/ml). Se diluează 20 mg/ml de soluție AL cu un factor de 5 în X-VIVOTM 15 (4 mg/ml); 500 μl din această soluție AL de 4 mg/ml AL se transferă într-un godeu dintr-un bloc de analiză cu 96 de godeuri. Această soluție este diluată cu un factor de 2 în X-VIVOTM 15 și apoi diluată din nou cu un factor de 2 pentru a produce soluții de 2 mg/ml și 1 mg/ml. 50 μl din aceste trei soluții și martorul tratat cu vehicul (X-VIVOTM 15) se adaugă la 50 μl de suspensie celulară THP-G8 în godeurile unei plăci de 96 de godeuri cu fund plat de culoare neagră. Fiecare concentrație a martorului negativ este testată în patru godeuri. Placa se agită pentru un agitator cu placă și se incubează într-un incubator cu CO2 timp de 16 ore (37 °C, 5 % CO2), după care se măsoară activitatea luciferazei conform descrierii de la punctul 29.
|
|
24.
|
Ar putea fi utilizați și alți martori pozitivi sau negativi adecvați în cazul în care sunt disponibile date anterioare pentru a obține criterii comparabile de acceptare a testelor.
|
|
25.
|
Ar trebui să se acorde o atenție deosebită pentru a se evita evaporarea substanțelor chimice de testat volatile și contaminarea încrucișată între godeuri a substanțelor chimice de testat, de exemplu, prin acoperirea etanșă a plăcilor înainte de incubația cu substanțele chimice de testat.
|
|
26.
|
Substanțele chimice de testat și martorul tratat cu solvent necesită efectuarea a 2-4 teste pentru obținerea unei estimări pozitive sau negative (a se vedea tabelul 2). Fiecare test independent este efectuat într-o altă zi cu soluții mamă X-VIVOTM 15 proaspete ale substanțelor chimice de testat și celule recoltate în mod independent. Celulele pot proveni din cadrul aceleiași treceri.
|
Măsurători de activitate ale luciferazei
|
27.
|
Luminiscența este măsurată cu ajutorul unui luminometru cu o microplacă de 96 de godeuri dotat cu filtre optice, de exemplu, Phelios (ATTO, Tokyo, Japonia), Tristan 941 (Berthold, Bad Wildbad, Germania) și seria ARVO (PerkinElmer, Waltham, MA, USA). Luminometrul trebuie calibrat pentru fiecare test pentru a se asigura reproductibilitatea (19). Pentru această calibrare sunt disponibile luciferaze recombinante de culoarea portocalie și roșie.
|
|
28.
|
Se transferă 100 μl de reactiv preîncălzit Tripluc® Luciferase (Tripluc) prevăzut pentru test în fiecare godeu al plăcii care conține suspensia celulară tratată cu sau fără substanță chimică. Placa se agită timp de 10 minute la o temperatură ambiantă de aproximativ 20 °C. Placa se plasează în luminometru pentru măsurarea activității luciferazei. Bioluminiscența se măsoară timp de 3 secunde fiecare în absența (F0) și prezența (F1) filtrului optic. Ar trebui să se furnizeze o justificare pentru utilizarea de valori alternative, de exemplu, în funcție de modelul de luminometru utilizat.
|
|
29.
|
Parametrii pentru fiecare concentrație sunt calculați de la valorile măsurate, de exemplu, IL8LA, GAPLA, nIL8LA, Ind-IL8LA, Inh-GAPLA, media ±SD a IL8LA, media ±SD a GAPLA, media ±SD a nIL8LA, media ±SD a Ind-IL8LA, media ±SD a Inh-GAPLA și intervalul de încredere de 95 % al Ind-IL8LA. Definițiile parametrilor utilizați în prezentul punct sunt prevăzute în apendicele 3.1 și 3.4.
|
|
30.
|
Înainte de măsurare, diferențierea culorilor în cadrul testelor de raportare a mai multor culori este realizată, în general, cu ajutorul detectoarelor (luminometrul și cititorul plăcii) prevăzute cu filtre optice, precum filtrele de tip „sharp-cut” (cu trecere lungă sau cu trecere scurtă) sau filtrele „trece banda”. Coeficienții de transmitere ai filtrelor pentru fiecare culoare a semnalului bioluminiscenței ar trebui calibrați înainte de testare, conform apendicelui 3.2.
|
DATE ȘI RAPORTARE
Evaluarea datelor
|
31.
|
Criteriile pentru o decizie pozitivă/negativă impun ca în cadrul fiecărui test:
|
—
|
o estimare a unui test IL-8 Luc să fie apreciată ca fiind pozitivă dacă o substanță chimică de testat are o valoare Ind-IL8LA ≥ 1,4 și limita inferioară a intervalului de încredere de 95 % al Ind-IL8LA ≥ 1,0
|
|
—
|
o estimare a unui test IL-8 Luc să fie apreciată ca fiind negativă dacă o substanță chimică de testat are o valoare Ind-IL8LA < 1,4 și limita inferioară a intervalului de încredere de 95 % al Ind-IL8LA < 1,0
|
|
Model predictiv
|
32.
|
Substanțele chimice de testat care generează două rezultate pozitive la testele 1, 2, 3 sau 4 sunt identificate ca fiind pozitive, iar cele care generează trei rezultate negative la testele 1, 2, 3 sau 4 sunt identificate și presupuse ca fiind negative (tabelul 2). Între presupusele substanțe chimice negative, substanțele chimice care sunt dizolvate la cantitatea de 20 mg/ml de X-VIVOTM 15 sunt estimate ca fiind negative, iar substanțele chimice care nu sunt dizolvate la 20 mg/ml de X-VIVOTM 15 nu ar trebui să fie luate în considerare (figura 1).
Tabelul 2
Criterii de identificare a rezultatelor pozitive și a celor presupuse drept negative
|
testul 1
|
testul 2
|
testul 3
|
testul 4
|
Estimare finală
|
|
Pozitiv
|
Pozitiv
|
–
|
–
|
Pozitiv
|
|
Negativ
|
Pozitiv
|
–
|
Pozitiv
|
|
Negativ
|
Pozitiv
|
Pozitiv
|
|
Negativ
|
Presupus negativ
|
|
Negativ
|
Pozitiv
|
Pozitiv
|
–
|
Pozitiv
|
|
Negativ
|
Pozitiv
|
Pozitiv
|
|
Negativ
|
Presupus negativ
|
|
Negativ
|
Pozitiv
|
Pozitiv
|
Pozitiv
|
|
Negativ
|
Presupus negativ
|
|
Negativ
|
–
|
Presupus negativ
|
|
Figura 1
Model predictiv pentru estimarea finală
Criterii de acceptare
|
33.
|
Atunci când se utilizează testul IL-8, ar trebui să fie îndeplinite următoarele criterii de acceptare:
|
—
|
Ind-IL8LA trebuie să fie peste 5,0 cel puțin într-o concentrație a martorului pozitiv, 4-NBB, în cadrul fiecărui test.
|
|
—
|
Ind-IL8LA ar trebui să fie sub 1,4 în orice concentrație a martorului negativ, acidul lactic, în cadrul fiecărui test.
|
|
—
|
Datele provenind din plăcile pentru care valoarea GAPLA din godeurile cu martor cu celule și Tripluc, dar fără substanțe chimice, este mai mică decât valoarea echivalentă a de cinci ori valoarea godeului conținând doar mediul de testare (50 μl/godeu de RPMI-1640 conținând 10 % FBS și 50 μl/godeu X-VIVOTM 15) ar trebui să fie respinse.
|
|
—
|
Datele provenind din plăcile pentru care valoarea Inh-GAPLA a tuturor concentrațiilor substanțelor chimice de testat sau martor este mai mică de 0,05 ar trebui să fie respinse. În acest caz, primul test ar trebui repetat astfel încât cea mai mare concentrație finală a testului repetat să fie cea mai scăzută concentrație finală a testului anterior.
|
|
Raportul testului
|
34.
|
Raportul testului ar trebui să includă următoarele informații:
|
Substanțele chimice de testat
Substanță monocomponentă:
|
Identificarea chimică, precum denumirea (denumirile) IUPAC sau CAS, numărul (numerele) SMILES sau codul InChI, formula structurală și/sau alți identificatori;
|
|
Aspectul fizic, solubilitatea în apă, masa moleculară și alte proprietăți fizico-chimice relevante, dacă există;
|
|
Puritatea, identitatea chimică a impurităților, după caz și dacă este fezabil din punct de vedere practic etc.;
|
|
Tratamentul înainte de efectuarea testului, dacă este cazul (de exemplu, încălzire, măcinare);
|
|
Solubilitatea în X-VIVOTM 15. În cazul substanțelor chimice insolubile în X-VIVOTM 15, dacă este respectată precipitarea sau flotația după centrifugare;
|
|
Concentrația (concentrațiile) testată(e);
|
|
Condițiile de depozitare și stabilitatea, dacă există;
|
|
Justificarea alegerii solventului/vehiculului pentru fiecare substanță chimică de testat dacă nu a fost utilizat X VIVOTM 15.
|
Substanța multicomponentă, UVCB și amestec:
|
Caracterizarea, dacă este posibil, de exemplu prin identitatea chimică (a se vedea mai sus), puritatea, apariția cantitativă și proprietățile fizico-chimice relevante (a se vedea mai sus) ale componentelor, dacă există;
|
|
Aspectul fizic, solubilitatea în apă și alte proprietăți fizico-chimice relevante, dacă există;
|
|
Masa moleculară sau masa moleculară aparentă în cazul amestecurilor/polimerilor cu compoziții cunoscute sau alte informații relevante pentru efectuarea studiului;
|
|
Tratamentul înainte de efectuarea testului, dacă este cazul (de exemplu, încălzire, măcinare);
|
|
Solubilitatea în X-VIVOTM 15. În cazul substanțelor chimice insolubile în X-VIVOTM 15, dacă este respectată precipitarea sau flotația după centrifugare;
|
|
Concentrația (concentrațiile) testată(e);
|
|
Condițiile de depozitare și stabilitatea, dacă există.
|
|
Justificarea alegerii solventului/vehiculului pentru fiecare substanță chimică de testat dacă nu a fost utilizat X VIVOTM 15.
|
|
|
Martori
Martor pozitiv:
|
Identificarea chimică, precum denumirea (denumirile) IUPAC sau CAS, numărul (numerele) SMILES sau codul InChI, formula structurală și/sau alți identificatori;
|
|
aspectul fizic, solubilitatea în apă, masa moleculară și alte proprietăți fizico-chimice relevante, dacă există și dacă este cazul;
|
|
puritatea, identitatea chimică a impurităților, după caz și dacă este fezabil din punct de vedere practic etc.;
|
|
Tratamentul înainte de efectuarea testului, dacă este cazul (de exemplu, încălzire, măcinare);
|
|
Concentrația (concentrațiile) testată(e);
|
|
Condițiile de depozitare și stabilitatea, dacă există;
|
|
Trimitere la rezultatele anterioare ale martorilor pozitivi care demonstrează criterii adecvate de acceptare, dacă este cazul.
|
Martor negativ:
|
Identificarea chimică, precum denumirea (denumirile) IUPAC sau CAS, numărul (numerele) CAS și/sau alți identificatori;
|
|
puritatea, identitatea chimică a impurităților, după caz și dacă este fezabil din punct de vedere practic etc.;
|
|
aspectul fizic, masa moleculară și alte proprietăți fizico-chimice relevante în cazul în care sunt utilizați alți martori negativi decât cei menționați în Ghidul pentru teste și dacă aceștia există;
|
|
Condițiile de depozitare și stabilitatea, dacă există;
|
|
Justificarea alegerii solventului pentru fiecare substanță chimică de testat.
|
|
|
Condiții de testare
|
Numele și adresa sponsorului, ale instalației de testare și ale directorului de studiu;
|
|
descrierea testului utilizat;
|
|
linia celulară utilizată, condițiile de depozitare și sursa acesteia (de exemplu, instalația de la a fost obținută aceasta);
|
|
numărul lotului și originea FBS, denumirea furnizorului, numărul lotului de pe placa de 96 de godeuri cu fund plat de culoare neagră și numărul lotului reactivului Tripluc;
|
|
numărul de trecere și densitatea celulară utilizate pentru testare;
|
|
metoda utilizată pentru numărarea celulelor pentru însămânțare înainte de testare și măsurile luate pentru a asigura distribuția omogenă a numărului de celule;
|
|
luminometrul utilizat (de exemplu, modelul), inclusiv reglajele aparatului, substratul de luciferază utilizat, precum și demonstrarea măsurătorilor corespunzătoare ale luminiscenței pe baza testului de control descris la apendicele 3.2;
|
|
procedura utilizată pentru a demonstra performanțele de laborator la efectuarea testului (de exemplu, prin testarea unor substanțe de verificare) sau pentru a demonstra performanța reproductibilă a testului în timp.
|
|
|
Procedura de testare
|
numărul de replici și teste efectuate;
|
|
concentrațiile substanței chimice de testat, procedura de aplicare și timpul de expunere utilizate (în cazul în care acestea diferă de cele recomandate);
|
|
Descrierea criteriilor de evaluare și de decizie utilizate;
|
|
descrierea criteriilor de acceptare a studiului utilizate;
|
|
Descrierea oricăror modificări ale procedurii de testare.
|
|
|
Rezultate
|
măsurătorile IL8LA și GAPLA;
|
|
calculele pentru nIL8LA, Ind-IL8LA și Inh-GAPLA;
|
|
intervalul de încredere de 95 % al Ind-IL8LA;
|
|
un grafic care descrie curbele de răspuns în funcție de doză pentru inducția activității luciferazei și viabilitate;
|
|
O descriere a oricăror alte observații relevante, dacă este cazul.
|
|
|
Discutarea rezultatelor
|
Discutarea rezultatelor obținute cu ajutorul testului IL-8 Luc;
|
|
Analiza rezultatelor testului în contextul unei IATA în cazul în care sunt disponibile alte informații relevante.
|
|
Concluzii
|
REFERINȚE BIBLIOGRAFICE
|
(1)
|
Takahashi T, Kimura Y, Saito R, Nakajima Y, Ohmiya Y, Yamasaki K și Aiba S. (2011). An in vitro test to screen skin sensitizers using a stable THP-1-derived IL-8 reporter cell line, THP-G8. Toxicol Sci 124:359-69.
|
|
(2)
|
OCDE (2017). Validation report for the international validation study on the IL-8 Luc assay as a test evaluating the skin sensitizing potential of chemicals conducted by the IL-8 Luc Assay. Series on Testing and Assessment No 267, ENV/JM/MONO(2017)19. Organizația pentru Cooperare și Dezvoltare Economică, Paris. Publicație disponibilă la adresa: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(3)
|
OCDE (2017). Report of the Peer Review Panel for the IL-8 Luciferase (IL-8 Luc) Assay for in vitro skin sensitisation. Series on Testing and Assessment No 258, ENV/JM/MONO(2017)20. Organizația pentru Cooperare și Dezvoltare Economică, Paris. Publicație disponibilă la adresa: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(4)
|
OCDE (2016) Guidance Document On The Reporting Of Defined Approaches And Individual Information Sources To Be Used Within Integrated Approaches To Testing And Assessment (IATA) For Skin Sensitisation, Series on Testing & Assessment No 256, ENV/JM/MONO(2016)29. Organizația pentru Cooperare și Dezvoltare Economică, Paris. Publicație disponibilă la adresa: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(5)
|
van der Veen JW, Rorije E, Emter R, Natsch A, van Loveren H și Ezendam J. (2014). Evaluating the performance of integrated approaches for hazard identification of skin sensitizing chemicals. Regul Toxicol Pharmacol 69:371-9.
|
|
(6)
|
Kimura Y, Fujimura C, Ito Y, Takahashi T, Nakajima Y, Ohmiya Y și Aiba S. (2015). Optimization of the IL-8 Luc assay as an in vitro test for skin sensitization. Toxicol In Vitro 29:1816-30.
|
|
(7)
|
Urbisch D, Mehling A, Guth K, Ramirez T, Honarvar N, Kolle S, Landsiedel R, Jaworska J, Kern PS, Gerberick F, et al. (2015). Assessing skin sensitization hazard in mice and men using non-animal test methods. Regul Toxicol Pharmacol 71:337-51.
|
|
(8)
|
Ashikaga T, Sakaguchi H, Sono S, Kosaka N, Ishikawa M, Nukada Y, Miyazawa M, Ito Y, Nishiyama N și Itagaki H. (2010). A comparative evaluation of in vitro skin sensitisation tests: the human cell-line activation test (h-CLAT) versus the local lymph node assay (LLNA). Alternatives to laboratory animals: ATLA 38:275-84.
|
|
(9)
|
Patlewicz G, Casati S, Basketter DA, Asturiol D, Roberts DW, Lepoittevin J-P, Worth A și Aschberger K (2016) Can currently available non-animal methods detect pre and pro haptens relevant for skin sensitisation? Regul Toxicol Pharmacol, 82:147-155.
|
|
(10)
|
Thorne N, Inglese J și Auld DS. (2010). Illuminating insights into firefly luciferase and other bioluminescent reporters used in chemical biology. Chem Biol 17:646-57.
|
|
(11)
|
OCDE (2016). Testul nr. 455: Performance-Based Test Guideline for Stably Transfected Transactivation In Vitro Assays to Detect Estrogen Receptor Agonists and Antagonists, OECD Publishing, Paris. http://dx.doi.org/10.1787/9789264265295-en.
|
|
(12)
|
Viviani V, Uchida A, Suenaga N, Ryufuku M și Ohmiya Y. (2001). Thr226 is a key residue for bioluminescence spectra determination in beetle luciferases. Biochem Biophys Res Commun 280:1286-91.
|
|
(13)
|
Viviani VR, Bechara EJ și Ohmiya Y. (1999). Cloning, sequence analysis, and expression of active Phrixothrix railroad-worms luciferases: relationship between bioluminescence spectra and primary structures. Biochemistry 38:8271-9.
|
|
(14)
|
Nakajima Y, Kimura T, Sugata K, Enomoto T, Asakawa A, Kubota H, Ikeda M și Ohmiya Y. (2005). Multicolor luciferase assay system: one-step monitoring of multiple gene expressions with a single substrate. Biotechniques 38:891-4.
|
|
(15)
|
OCDE (2017). Urmează a se publica - Performance Standards for the assessment of proposed similar or modified in vitro skin sensitisation IL-8 luc test methods. OECD Environment, Health and Safety Publications, Series on Testing and Assessment. OCDE, Paris, Franța
|
|
(16)
|
OCDE (2005). Guidance Document the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. OECD Environment, Health and Safety publications, OECD Series on Testing and Assessment No 34. OCDE, Paris, Franța.
|
|
(17)
|
OCDE (2018). Draft Guidance document: Good In Vitro Method Practices (GIVIMP) for the Development and Implementation of In Vitro Methods for Regulatory Use in Human Safety Assessment. Organizația pentru Cooperare și Dezvoltare Economică, Paris. Publicație disponibilă la adresa: http://www.oecd.org/env/ehs/testing/OECD Final Draft GIVIMP.pdf.
|
|
(18)
|
JaCVAM (2016). IL-8 Luc assay protocol, Publicație disponibilă la adresa: http://www.jacvam.jp/en_effort/effort02.html.
|
|
(19)
|
Niwa K, Ichino Y, Kumata S, Nakajima Y, Hiraishi Y, Kato D, Viviani VR și Ohmiya Y. (2010). Quantum yields and kinetics of the firefly bioluminescence reaction of beetle luciferases. Photochem Photobiol 86:1046-9.
|
|
(20)
|
OCDE (2012). The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins, Part 1: Scientific Evidence. OECD Environment, Health and Safety Publications, Series on Testing and Assessment No 168. OCDE, Paris, Franța.
|
|
(21)
|
Organizația Națiunilor Unite (2015). Sistemul global armonizat de clasificare și etichetare a substanțelor chimice (GHS). A șasea ediție revizuită. New York & Geneva: United Nations Publications. ISBN: 978-92-1-117006-1. Material disponibil la adresa: http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html.
|
Apendicele 3.1
DEFINIțII
Precizie
: Gradul de concordanță între rezultatele testului și valorile de referință acceptate. Aceasta măsoară performanța testului și unul dintre aspectele relevanței acestuia. Termenul este adesea utilizat în mod interschimbabil, iar concordanța înseamnă proporția de rezultate corecte ale unui test (16).
AOP (parcursul răspunsurilor adverse)
: o serie de evenimente din structura chimică a unei substanțe chimice țintă sau un grup de substanțe chimice similare, de la evenimentul molecular declanșator până la un răspuns in vivo de interes (20).
Substanță chimică
: O substanță sau un amestec.
CV05
: Viabilitatea celulară 05, și anume concentrația minimă la care substanțele chimice prezintă o valoare Inh-GAPLA mai mică de 0,05.
FInSLO-LA
: Abrevierea utilizată în raportul de validare și în publicațiile anterioare privind testul IL-8 Luc referitor la Ind-IL8LA. Pentru definiție, a se vedea Ind-IL8LA.
GAPLA
: Activitatea luciferazei Stable Luciferase Red (SLR) (λ max = 630 nm), reglementată de promotorul GAPDH, care demonstrează viabilitatea celulară și numărul de celule viabile.
Pericol
: Proprietate intrinsecă a unui agent sau a unei situații care are potențialul de a produce efecte adverse atunci când un organism, un sistem sau o (sub)populație este expusă la agentul respectiv.
IATA (Strategie integrată pentru testare și evaluare)
: O abordare structurată utilizată pentru identificarea pericolelor (potențial), caracterizarea pericolelor (potența) și/sau evaluarea siguranței (potențial/potență și expunere) pentru o substanță chimică sau un grup de substanțe chimice, care se integrează în mod strategic și măsoară toate datele relevante pentru a furniza informații cu privire la decizii de reglementare în ceea ce privește potențialele pericole și/sau riscuri și/sau necesitatea unor teste suplimentare specifice și, prin urmare, minime.
II-SLR-LA
: Abrevierea utilizată în raportul de validare și în publicațiile anterioare privind testul IL-8 Luc referitor la Inh-GAPLA. Pentru definiție, a se vedea Inh-GAPLA.
IL-8 (Interleukin-8)
: O citokină derivată din celulele endoteliale, fibroblaste, cheratinocite, macrofage și monocite care cauzează chemotaxia neutrofilelor și a și a limfocitelor T.
IL8LA
: Activitate luciferazei Stable Luciferase Orange (SLO) (λmax = 580 nm), reglementată de promotorul IL-8.
Ind-IL8LA
: Inducția în multipli a nIL8LA. Se obține prin împărțirea valorii nIL8LA a celulelor THP-G8 tratate cu substanțe chimice la valoarea celulelor THP-G8 nestimulate și reprezintă inducția activității promotorului IL-8 prin intermediul substanțelor chimice.
Inh-GAPLA
: Inhibarea GAPLA. Se obține prin împărțirea valorii GAPLA a celulelor THP-G8 tratate cu substanțe chimice la valoarea celulelor THP-G8 netratate și reprezintă citotoxicitatea substanțelor chimice.
Prag minim de inducție (PMI)
: concentrația cea mai scăzută la care o substanță chimică îndeplinește criteriile pozitive
Amestec
: un amestec sau o soluție compus/ă din două sau mai multe substanțe.
Substanță monocomponentă
: O substanță definită prin compoziția sa cantitativă, în care un singur component principal este prezent în proporție de cel puțin 80 % (g/g).
Substanță multicomponentă
: O substanță definită prin compoziția sa cantitativă, în care există mai multe componente principale într-o concentrație ≥ 10 % (g/g) și < 80 % (g/g). O substanță multicomponentă este rezultatul unui proces de fabricație. Diferența dintre un amestec și o substanță multicomponentă este faptul că un amestec este obținut prin combinarea a două sau mai multe substanțe chimice fără o reacție chimică. O substanță multicomponentă este rezultatul unei reacții chimice.
nIL8LA
: Activitatea luciferazei SLO care reflectă activitatea promotorului IL-8 (IL8LA), normalizată prin activitatea luciferazei SLR care reflectă activitatea promotorului GAPDH (GAPLA). Aceasta reprezintă o activitate a promotorului IL-8 după analizarea viabilității celulare sau a numărului de celule.
nSLO-LA
: Abrevierea utilizată în raportul de validare și în publicațiile anterioare privind testul IL-8 Luc referitor la nIL8LA. Pentru definiție, a se vedea nIL8LA.
Martor pozitiv
: O replică ce conține toate componentele sistemului de testare și care a fost tratată cu o substanță cunoscută pentru faptul că provoacă o reacție pozitivă. Pentru a asigura posibilitatea evaluării variabilității în timp a reacției martorului pozitiv, amplitudinea reacției severe nu trebuie să fie excesiv de mare.
Prehaptene
: Substanțe chimice care devin sensibilizante prin transformări abiotice.
Prohaptene
: Substanțe chimice care necesită activare enzimatică pentru a exercita potențialul de sensibilizare cutanată.
Relevanță
: Descrierea relației dintre test și efectul de interes și dacă aceasta este relevantă și utilă pentru un anumit scop. Aceasta reprezintă măsura în care testul măsoară sau prezice în mod corect efectul biologic de interes. Relevanța include analiza preciziei (a concordanței) unui test (16).
Fiabilitate
: Măsuri ale faptului că un test poate fi efectuat în mod reproductibil, în același laborator și în laboratoare diferite în timp, atunci când este efectuat utilizând același protocol. Aceasta se evaluează prin calcularea reproductibilității în același laborator sau între laboratoare și a repetabilității în același laborator (16).
Test
: Un test este compus din unul sau mai multe substanțe chimice de testat, care sunt testate concomitent cu un martor solvent/vehicul și un martor pozitiv.
Sensibilitate
: Proporția tuturor substanțelor chimice pozitive/active clasificate în mod corect în urma testului. Aceasta permite măsurarea preciziei în cazul unui test care generează rezultate clasificabile în categorii și constituie un aspect important în evaluarea relevanței unui test (16).
SLO-LA
: Abrevierea utilizată în raportul de validare și în publicațiile anterioare privind testul IL-8 Luc referitor la IL8LA. Pentru definiție, a se vedea IL8LA.
SLR-LA
: Abrevierea utilizată în raportul de validare și în publicațiile anterioare privind testul IL-8 Luc referitor la GAPLA. Pentru definiție, a se vedea GAPLA.
Martor solvent/vehicul
: O probă netratată care conține toate componentele unui sistem de testare, cu excepția celor ale substanței chimice de testat, dar incluzând solventul/vehiculul utilizat. Acesta este utilizat pentru a stabili reacția de bază pentru probele tratate cu substanța chimică de testat dizolvată sau dispersată stabil în același solvent/vehicul. Atunci când este testată concomitent cu un martor de mediu, această probă indică, de asemenea, dacă solventul/vehiculul interacționează cu sistemul testat.
Specificitate
: Proporția tuturor substanțelor chimice negative/inactive clasificate în mod corect în urma testului. Aceasta permite măsurarea preciziei în cazul unui test care generează rezultate clasificabile în categorii și constituie un aspect important în evaluarea relevanței unui test (16).
Substanță
: Un element chimic și compușii acestuia în stare naturală sau obținut prin orice proces de producție, inclusiv orice aditiv necesar pentru a menține stabilitatea produsului și orice impuritate rezultată din procesul utilizat, dar cu excepția oricărui solvent care poate fi separat fără a influența stabilitatea substanței sau fără a schimba compoziția acesteia.
Surfactant
: Numit și agent tensioactiv, aceasta este o substanță, cum ar fi un detergent, care poate să reducă tensiunea la suprafață a unui lichid și, astfel, să îi permită acesteia spumarea sau penetrarea solidelor; este cunoscut, de asemenea, ca agent de umectare. (TG437)
Substanță chimică de testat
: orice substanță sau amestec testat(ă) în cadrul acestei metode.
THP-G8
: O linie celulară raportoare IL-8 utilizată în cadrul testului IL-8 Luc. Linia celulară THP-1 similară macrofagului uman a fost transfectată în genele pentru luciferază SLO și SLR sub controlul promotorilor IL-8 și, respectiv, GAPDH.
Sistemul global armonizat de clasificare și etichetare a substanțelor chimice al Organizației Națiunilor Unite (GHS al ONU)
: Un sistem care propune clasificarea substanțelor chimice (substanțe și amestecuri) în funcție de tipuri și niveluri standardizate de pericole fizice, pericole pentru sănătate și pericole pentru mediu și care se referă la elemente de comunicare corespunzătoare, precum pictograme, cuvinte de avertizare, declarații cu privire la pericole, declarații de securitate și fișe tehnice de siguranță pentru a transmite informații privind efectele adverse ale acestora în scopul protejării populației (inclusiv angajatorii, lucrătorii, transportatorii, consumatorii și cei care se ocupă de serviciile de urgență) și a mediului (21).
UVCB
: Substanțe cu compoziție necunoscută sau variabilă, produși de reacție complexă sau materiale biologice.
Metodă de testare valabilă
: Un test considerat a avea suficientă relevanță și fiabilitate pentru un scop specific și care se bazează pe principii solide din punct de vedere științific. Un test nu este niciodată valabil într-un sens absolut, ci doar în legătură cu un scop definit.
Apendicele 3.2
PRINCIPIUL MĂSURĂRII ACTIVITĂțII LUCIFERAZEI șI AL DETERMINĂRII COEFICIENțILOR DE TRANSMISIE AI FILTRULUI OPTIC PENTRU SLO ȘI SLR
Se poate utiliza MultiReporter Assay System - Tripluc - cu un luminometru de tip microplacă prevăzut cu un sistem de detectare a mai multor culori, care poate fi montat la un filtru optic [de exemplu, Phelios AB-2350 (ATTO), ARVO (PerkinElmer), Tristar LB941 (Berthold)]. Filtrul optic utilizat pentru măsurare este un filtru de 600-620 nm cu trecere lungă sau scurtă, sau un filtru de 600-700 nm de tip band pass.
Măsurarea luciferazelor în două culori cu un filtru optic.
Acesta este un exemplu folosind filtrul Phelios AB-2350 (ATTO). Acest luminometru este prevăzut cu un filtru de 600 nm cu trecere lungă (R60 HOYA Co., de 600 nm LP, filtrul nr. 1) pentru scindarea luminiscenței SLO (λmax = 580 nm) și SLR (λmax = 630 nm).
Pentru a determina coeficienții de transmitere ai filtrului LP de 600 nm, folosind enzime purificate pentru luciferază SLO și SLR, măsurați mai întâi i) intensitatea bioluminiscenței SLO și SLR fără filtru (F0) și ii) intensitatea bioluminiscenței SLO și SLR care a trecut prin filtrul LP de 600 nm (filtrul 1) și iii) calculați coeficienții de transmitere ai filtrului LP de 600 nm pentru SLO și SLR care sunt menționați în continuare.
|
Transmission coefficients
|
Abbreviation
|
Definition
|
|
SLO
|
Filter 1 Transmission coefficients
|
=κOR60
|
The filter’s transmission coefficient for the SLO
|
|
SLR
|
Filter 1 Transmission coefficients
|
κRR60
|
The filter’s transmission coefficient for the SLR
|
Atunci când intensitatea SLO și SLR în eșantion este definită ca O și, respectiv, R, i) intensitatea luminii fără filtru (complet optic) F0 și ii) intensitatea luminii care transmite prin filtrul LP de 600 nm (filtrul 1) F1 sunt descrise mai jos.
F0=O+R
F1=κOR60 x O + κRR60 x R
Aceste formule ar putea fi reformulate, după cum urmează:

Apoi, folosind factorii de transmitanță calculați (κOR60 și κRR60) și având valorile F0 și F1 măsurate, puteți calcula valoarea O și R, după cum urmează:
Materiale și metode pentru determinarea factorului de transmitanță
|
(1)
|
Reactivi
Enzime simple purificate luciferaze:
|
Enzima SLO purificată liofilizată
|
|
Enzima SLR purificată liofilizată
|
|
(care, pentru activitatea de validare, au fost obținute de la laboratorul LMP Lab. Co. Ltd., Tottori, Japonia cu linia celulară THP-G8)
|
Reactiv pentru testare:
|
Reactivul pentru testare Tripluc® Luciferase (spre exemplu, de la TOYOBO Cat#MRA-301)
|
Mediu: pentru testul luciferazei (30 ml, stocat la 2-8 °C)
|
|
Concentr.
|
reactivului
|
Concentrație finală în mediu
|
Cantitate necesară
|
|
RPMI-1640
|
—
|
—
|
27 ml
|
|
FBS
|
—
|
10 %
|
3 ml
|
|
(2)
|
Prepararea soluției enzimatice
Se dizolvă o enzimă luciferază purificată lipofilizată în eprubetă, adăugând 200 μl de 10 ~ 100 mM Tris/HCl sau Hepes/HCl (pH 7,5 ~ 8,0), se completează cu glicerol în proporție de 10 % (g/v), se împarte soluția enzimei în alicote de 10 μl în eprubete de unică folosință de 1,5 ml și se depozitează într-un congelator la -80 °C. Soluția enzimatică înghețată poate fi utilizată timp de maxim șase luni. La utilizare, se adaugă 1 ml de mediu pentru testul luciferazei (RPMI-1640 cu 10 % FBS) în fiecare eprubetă care conține soluțiile enzimatice (soluție enzimatică diluată) și se păstrează pe gheață pentru a preveni dezactivarea.
|
|
(3)
|
Măsurarea bioluminiscenței
Reactivul pentru testare Tripluc® Luciferase (Tripluc) se decongelează și se păstrează la temperatura camerei, fie într-o baie de apă, fie la temperatura ambiantă. Se activează luminometrul cu 30 de minute înainte de începerea măsurătorii pentru a permite stabilizarea fotomultiplicatorului. Se transferă 100 μl de soluție enzimatică diluată pe o placă cu 96 de godeuri de culoare neagră (cu fund plat) (eșantion de referință SLO pentru #B1, #B2 și #B3 și eșantionul de referință SLR pentru #D1, #D2, #D3). Se transferă apoi 100 μl de Tripluc preîncălzit în fiecare godeu al plăcii care conține soluția enzimatică diluată, folosind o pipetă. Se agită placa timp de 10 minute la temperatura camerei (aproximativ 25 °C) folosind un agitator pentru placă. Dacă apar bule în soluțiile din godeuri acestea sunt eliminate. Se așază placa în luminometru pentru a măsura activitatea luciferazei. Bioluminiscența se măsoară timp de 3 secunde fiecare în absența (F0) și prezența (F1) filtrului optic.
Coeficientul de transmisie al filtrului optic a fost calculat după cum urmează:
Coeficientul de transmisie [SLO (κOR60)] = (#B1 al F1+ #B2 al F1+ #B3 al F1) / (#B1 al F0+ #B2 al F0+ #B3 al F0)
Coeficientul de transmisie [SLR (κRR60)] = (#D1 al F1+ #D2 al F1+ #D3 al F1) / (#D1 al F0+ #D2 al F0+ #D3 al F0)
Factorii de transmitanță calculați se utilizează pentru toate măsurătorile efectuate folosind același luminometru.
|
Controlul calității echipamentului
Ar trebui să fie utilizate procedurile descrise în protocolul IL-8 Luc (18).
Apendicele 3.3
SUBSTANțE DE VERIFICARE
Înainte de utilizarea de rutină a testului descris în prezentul apendice la metoda de testare B.71, laboratoarele ar trebui să demonstreze performanțele tehnice prin obținerea estimării în urma testului IL-8 Luc pentru cele nouă substanțe recomandate în tabelul 1 și prin obținerea de valori care să se încadreze în plaja de valori de referință corespunzătoare pentru cel puțin opt dintre cele nouă substanțe de verificare (selectate pentru a reprezenta intervalul de răspunsuri pentru pericolele de sensibilizare cutanată). Alte criterii de selecție au fost ca substanțele să fie disponibile în comerț și să fie disponibile date de referință in vivo de înaltă calitate, precum și date in vitro de înaltă calitate generate cu testul IL-8 Luc. De asemenea, sunt disponibile date de referință publicate pentru testul IL-8 Luc (6) (1).
Tabelul 1
Substanțe recomandate pentru demonstrarea performanțelor tehnice cu testul IL-8 Luc
|
Substanțe de verificare
|
Nr. CAS
|
Stare
|
Solubilitate în X-VIVO15 la 20 mg/ml
|
Estimarea in vivo
(109)
|
Estimarea IL-8 Luc (110)
|
Interval de referință (μg/ml) (111)
|
|
|
CV05
(112)
|
IL-8 Luc MIT (113)
|
|
2,4-dinitroclorbenzen
|
97-00-7
|
Solid
|
Insolubil
|
Sensibilizant (Extrem)
|
Pozitiv
|
2,3-3,9
|
0,5-2,3
|
|
Formaldehidă
|
50-00-0
|
Lichid
|
Solubil
|
Sensibilizant (Puternic)
|
Pozitiv
|
9-30
|
4-9
|
|
2-mercaptobenzotiazol
|
149-30-4
|
Solid
|
Insolubil
|
Sensibilizant (Moderat)
|
Pozitiv
|
250-290
|
60-250
|
|
Etilendiamină
|
107-15-3
|
Lichid
|
Solubil
|
Sensibilizant (Moderat)
|
Pozitiv
|
500-700
|
0,1-0,4
|
|
Etilen glicol dimetacrilat
|
97-90-5
|
Lichid
|
Insolubil
|
Sensibilizant (Slab)
|
Pozitiv
|
>2000
|
0,04-0,1
|
|
4-alilanisol (Estragol)
|
140-67-0
|
Lichid
|
Insolubil
|
Sensibilizant (Slab)
|
Pozitiv
|
>2000
|
0,01-0,07
|
|
Streptomicină sulfat
|
3810-74-0
|
Solid
|
Solubil
|
Non-sensibilizant
|
Negativ
|
>2000
|
>2000
|
|
Glicerină
|
56-81-5
|
Lichid
|
Solubil
|
Non-sensibilizant
|
Negativ
|
>2000
|
>2000
|
|
Izopropanol
|
67-63-0
|
Lichid
|
Solubil
|
Non-sensibilizant
|
Negativ
|
>2000
|
>2000
|
|
Abrevieri: Nr. CAS = Număr de înregistrare al Chemical Abstracts Service
|
Apendicele 3.4
INDICI șI CRITERII DE APRECIERE
nIL8LA (nSLO-LA)
Repetarea j-th (j = 1-4) a concentrației i-th (i = 0-11) se măsoară pentru IL8LA (SLO-LA) și, respectiv, GAPLA (SLR-LA). Valoarea IL8LA normalizată, denumită nIL8LA (nSLO-LA) și definită ca:
nIL8LAij = IL8LAij/GAPLAij
Aceasta este unitatea de măsură de bază în cadrul acestui test.
Ind-IL8LA (FInSLO-LA)
Creșterea în multipli a mediei nIL8LA (nSLO-LA) pentru repetiția la concentrația i-th, în comparație cu aceasta la concentrația 0, Ind-IL8LA, reprezintă principala măsură a acestui test. Acest coeficient este scris prin următoarea formulă:
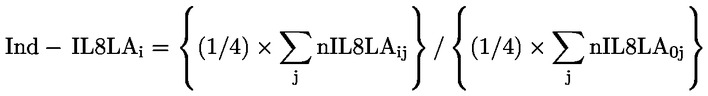
Laboratorul principal a propus ca o valoare de 1,4 să corespundă unui rezultat pozitiv pentru substanța chimică testată. Această valoare este bazată pe examinarea datelor istorice ale laboratorului principal. Echipa de gestionare a datelor a utilizat apoi această valoare în toate etapele studiului de validare. Rezultatul principal, Ind-IL8LA, constituie raportul dintre două medii aritmetice, astfel cum se arată în ecuație.
Interval de încredere de 95 % (CI 95 %)
Intervalul de încredere de 95 % (CI 95 %), care este bazat pe coeficient, poate fi estimat pentru a indica precizia măsurării acestui rezultat principal. Limita inferioară a CI de 95 % ≥ 1 indică faptul că valoarea nIL8LA cu o concentrație i-th este semnificativ mai mare decât cea cu martor tratat cu solvent. Există mai multe modalități de a construi CI de 95 %. În acest studiu am utilizat metoda cunoscută drept teorema lui Fieller. Această teoremă privind intervalul de încredere de 95 % este obținută din următoarea formulă:

Unde .


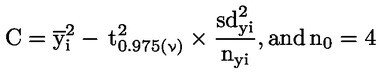
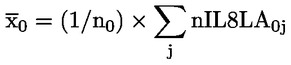
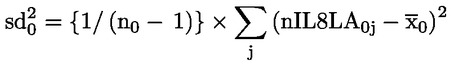

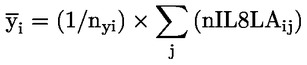
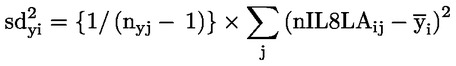
t0.975(ν)is 97.5 percentile din distribuția t centrală cu ν grade de libertate, unde
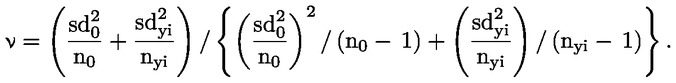
Inh-GAPLA (II-SLR-LA)
Valoarea Inh-GAPLA reprezintă un coeficient al mediei GAPLA (SLR-LA) pentru repetarea concentrației i-th în comparație cu cea cu martor tratat cu solvent, iar aceasta este scrisă prin
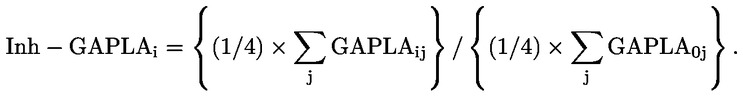
Întrucât valoarea GAPLA este numitorul nIL8LA, o valoare extrem de mică determină o variație mare a valorii nIL8LA. Prin urmare, valorile Ind-IL8LA având o valoare extrem de mică a Inh-GAPLA (sub 0,05) ar putea fi considerate a reprezenta un nivel scăzut de precizie.
Apendicele 3.5
SCHEMA METODELOR DE DIZOLVARE A SUBSTANțELOR CHIMICE PENTRU TESTUL IL-8 LUC.
|
(a)
|
Pentru substanțele chimice dizolvate în X-VIVOTM 15 la 20 mg/ml
|
|
(b)
|
Pentru substanțele chimice insolubile în X-VIVOTM 15 la 20 mg/ml

|
Apendicele 3.6
SCHEMA METODEI DE DIZOLVARE A 4-NBB PENTRU MARTORUL POZITIV AL TESTULUI IL-8 LUC.

” |