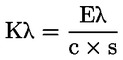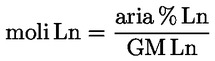1991R2568 — RO — 04.08.2016 — 029.001
Acest document are doar scop informativ și nu produce efecte juridice. Instituțiile Uniunii nu își asumă răspunderea pentru conținutul său. Versiunile autentice ale actelor relevante, inclusiv preambulul acestora, sunt cele publicate în Jurnalul Oficial al Uniunii Europene și disponibile pe site-ul EUR-Lex. Aceste texte oficiale pot fi consultate accesând linkurile integrate în prezentul document.
|
REGULAMENTUL (CEE) NR. 2568/91 AL COMISIEI din 11 iulie 1991 (JO L 248 5.9.1991, p. 1) |
Astfel cum a fost modificat prin:
|
(*) |
Acest act nu a fost publicat niciodată în limba română. |
REGULAMENTUL (CEE) NR. 2568/91 AL COMISIEI
din 11 iulie 1991
privind caracteristicile uleiurilor de măsline și ale uleiurilor din resturi de măsline, precum și metodele de analiză a acestora
Articolul 1
(1) Sunt considerate uleiuri de măsline virgine, în sensul punctului 1 literele (a) și (b) din anexa la Regulamentul nr. 136/66/CEE, uleiurile ale căror caracteristici sunt conforme celor indicate la anexa I punctele 1 și 2 la prezentul regulament.
(2) Este considerat ulei de măsline lampant, în sensul punctului 1 litera (c) din anexa la Regulamentul nr. 136/66/CEE, uleiul ale cărui caracteristici sunt conforme celor indicate la anexa I punctul 3 la prezentul regulament.
(3) Este considerat ulei de măsline rafinat, în sensul punctului 2 din anexa la Regulamentul nr. 136/66/CEE, uleiul ale cărui caracteristici sunt conforme celor indicate în anexa I punctul 4 la prezentul regulament.
(4) Este considerat ulei de măsline compus din ulei rafinat și din uleiuri de măsline virgine, în sensul punctului 3 din anexa la Regulamentul nr. 136/66/CEE, uleiul ale cărui caracteristici sunt conforme celor indicate la anexa I punctul 5 la prezentul regulament.
(5) Este considerat ulei din reziduuri de măsline brut, în sensul punctului 4 din anexa la Regulamentul nr. 136/66/CEE, uleiul ale cărui caracteristici sunt conforme celor indicate la anexa I punctul 6 la prezentul regulament.
(6) Este considerat ulei din reziduuri de măsline rafinat, în sensul punctului 5 din anexa la Regulamentul nr. 136/66/CEE, uleiul ale cărui caracteristici sunt conforme celor indicate la anexa I punctul 7 la prezentul regulament.
(7) Este considerat ulei din reziduuri de măsline, în sensul punctului 6 din anexa la Regulamentul nr. 136/66/CEE, uleiul ale cărui caracteristici sunt conforme celor indicate la anexa I punctul 8 la prezentul regulament.
Articolul 2
(1) Caracteristicile uleiurilor prevăzute în anexa I se determină în conformitate cu următoarele metodele de analiză:
(a) pentru determinarea acizilor grași liberi, exprimați în procente de acid oleic, metoda prevăzută în anexa II;
(b) pentru determinarea indicelui de peroxid, metoda prevăzută în anexa III;
(c) pentru determinarea conținutului de ceară, metoda prevăzută în anexa IV;
(d) pentru determinarea compoziției și a conținutului de steroli și de dialcoolii triterpenici prin cromatografie în fază gazoasă cu coloană capilară, metoda prevăzută în anexa V;
(e) pentru determinarea procentului de monopalmitat de 2-gliceril, metoda prevăzută în anexa VII;
(f) pentru analiza spectrofotometrică, metoda prevăzută în anexa IX;
(g) pentru determinarea compoziției de acizi grași, metoda prevăzută în anexa X;
(h) pentru determinarea solvenților halogenați volatili, metoda prevăzută în anexa XI;
(i) pentru evaluarea caracteristicilor organoleptice ale uleiului de măsline virgin, metoda prevăzută în anexa XII;
(j) pentru determinarea stigmastadienelor, metoda prevăzută în anexa XVII;
(k) pentru determinarea conținutului de trigliceride cu ECN42, metoda prevăzută în anexa XVIII;
(l) pentru determinarea conținutului de alcooli alifatici și triterpenici, metoda prevăzută în anexa XIX;
(m) pentru determinarea conținutului de ceruri, esteri metilici ai acizilor grași și esteri etilici ai acizilor grași, metoda descrisă în anexa XX.
▼M28 —————
(2) Verificarea de către autoritățile naționale sau de către reprezentanții acestora a caracteristicilor organoleptice ale uleiurilor virgine se efectuează în cadrul comisiilor de degustători aprobate de statele membre.
Caracteristicile organoleptice ale uleiurilor menționate la primul paragraf sunt considerate a fi conforme cu categoria declarată dacă o comisie aprobată de statul membru în cauză confirmă respectiva clasificare.
În cazul în care comisia nu confirmă categoria declarată în ceea ce privește caracteristicile organoleptice, autoritățile naționale sau reprezentanții acestora dispun, la cererea părții interesate, să se efectueze fără întârziere două contraanalize de către alte comisii aprobate, din care cel puțin una este aprobată de statul membru producător în cauză. Se consideră că respectivele caracteristici sunt conforme cu caracteristicile declarate dacă cel puțin două contraanalize confirmă clasificarea declarată. În caz contrar, costurile contraanalizelor sunt suportate de partea interesată.
(3) Atunci când autoritățile naționale sau reprezentanții acestora verifică caracteristicile uleiului după cum se prevede la alineatul (1), prelevarea probelor se efectuează în conformitate cu standardele internaționale EN ISO 661 privind pregătirea probei pentru analiză și EN ISO 5555 privind eșantionarea. Cu toate acestea, fără a aduce atingere punctului 6.8 din standardul EN ISO 5555, în cazul loturilor de astfel de uleiuri în ambalaje imediate, probele se prelevă în conformitate cu anexa Ia la prezentul regulament. În cazul uleiurilor în vrac pentru care eșantionarea nu se poate realiza în conformitate cu EN ISO 5555, eșantionarea se efectuează în conformitate cu instrucțiunile furnizate de autoritatea competentă a statului membru.
Fără a aduce atingere standardului EN ISO 5555 și capitolului 6 din standardul EN ISO 661, probele prelevate se păstrează cât mai repede posibil într-un loc întunecos și ferit de căldură puternică și se trimit la laborator pentru analiză cel mai târziu în a cincea zi lucrătoare de la prelevarea lor, iar în caz contrar probele se păstrează astfel încât să nu fie degradate sau deteriorate în timpul transportului sau al depozitării, înainte de a fi trimise la laborator.
(4) Pentru verificarea prevăzută la alineatul (3), în cazul produselor ambalate, analizele menționate la anexele II, III, IX, XII și XX, precum și, după caz, contraanalizele prevăzute de legislațiile naționale se efectuează înainte de data minimă de valabilitate. În cazul eșantionării de uleiuri în vrac, aceste analize se efectuează cel târziu în a șasea lună care urmează lunii în care a fost prelevată proba.
Pentru celelalte analize prevăzute de prezentul regulament nu se prevede nici o limită temporală.
Cu excepția cazului în care proba a fost prelevată la mai puțin de două luni de la data minimă de valabilitate, dacă rezultatele analizelor nu corespund caracteristicilor categoriei declarate de ulei de măsline sau de ulei din resturi de măsline, partea în cauză este înștiințată cel mai târziu cu o lună înainte de expirarea perioadei prevăzute la primul paragraf.
(5) În scopul determinării caracteristicilor uleiurilor de măsline prin metodele prevăzute la alineatul (1) primul paragraf, rezultatele analizelor se compară direct cu limitele prevăzute de prezentul regulament.
Articolul 2a
(1) În sensul prezentului articol, „ulei de măsline comercializat” înseamnă cantitatea totală de ulei de măsline și ulei din resturi de măsline a unui stat membru care este consumată în respectivul stat membru sau exportată din respectivul stat membru.
(2) Statele membre se asigură că verificările conformității sunt efectuate în mod selectiv, pe baza unei analize a riscurilor, și cu o frecvență corespunzătoare, astfel încât să se asigure conformitatea uleiului de măsline comercializat cu categoria declarată.
(3) Criteriile de evaluare a riscurilor pot include:
(a) categoria de ulei, perioada de producție, prețul uleiurilor în raport cu alte uleiuri vegetale, operațiunile de amestecare și ambalare, instalațiile și condițiile de depozitare, țara de origine, țara de destinație, mijloacele de transport sau volumul lotului;
(b) poziția operatorilor în lanțul de comercializare, volumul și/sau valoarea produselor comercializate de aceștia, gama de categorii de ulei pe care le comercializează, tipul de operațiune economică desfășurată, cum ar fi presarea, depozitarea, rafinarea, amestecarea, ambalarea sau vânzarea cu amănuntul;
(c) constatările din timpul verificărilor precedente, inclusiv numărul și tipul defectelor constatate, calitatea obișnuită a uleiurilor comercializate, performanța echipamentelor tehnice utilizate;
(d) fiabilitatea sistemelor de asigurare a calității sau a sistemelor de autocontrol ale operatorilor legate de conformitatea cu standardele de comercializare;
(e) locul de desfășurare a verificării, în special dacă este vorba de primul punct de intrare în Uniune, ultimul punct de ieșire din Uniune sau locul în care uleiurile sunt produse, ambalate, încărcate ori vândute către consumatorul final;
(f) orice alte informații care ar putea indica un risc de neconformitate.
(4) Statele membre stabilesc în prealabil:
(a) criteriile de evaluare a riscurilor de neconformitate a loturilor;
(b) pe baza unei analize a riscurilor pentru fiecare categorie de risc, numărul minim de operatori sau loturi și/sau cantități care vor fi supuse unei verificări a conformității.
O dată pe an se efectuează cel puțin o verificare a conformității la o mie de tone de ulei de măsline comercializat în statul membru.
(5) Statele membre verifică conformitatea:
(a) efectuând în ordine aleatorie analizele prevăzute în anexa I; sau
(b) respectând ordinea prevăzută în anexa Ib privind programul decizional, până se ajunge la una dintre deciziile menționate de programul respectiv.
▼M19 —————
Articolul 3
În cazul în care se constată că un ulei nu corespunde descrierii categoriei sale și fără a aduce atingere niciunei alte sancțiuni, statul membru în cauză aplică sancțiuni eficace, proporționale și disuasive, care se vor stabili în funcție de gravitatea neregulii detectate.
Atunci când, în urma verificărilor, se constată nereguli semnificative, statele membre măresc frecvența verificărilor cu privire la etapa de comercializare, categoria de ulei, originea sa ori alte criterii.
Articolul 4
1. The Member States may approve assessment panels so that national authorities or their representatives can assess and verify organoleptic characteristics.
The terms of approval shall be set by Member States and ensure that:
— the requirements of Annex XII.4 are met,
— the panel head is given training recognised for this purpose by the Member State,
— continued approval depends on performance in annual checks arranged by the Member State.
Member States shall notify to the Commission a list of approved panels and the action taken under this paragraph.
(2) Atunci când statele membre au dificultăți în înființarea comisiilor de degustători pe teritoriul lor, acestea pot solicita o comisie de degustători autorizată în alt stat membru.
(3) Fiecare stat membru elaborează o listă cu comisiile de degustători înființate de organizații profesionale sau inter-filiale în conformitate cu condițiile prevăzute la alineatul (1) și asigură respectarea acestor condiții.
▼M19 —————
Articolul 6
(1) Conținutul de ulei din resturile și alte reziduuri rezultate din extragerea uleiului cu codurile NC 2306 90 11 și 2306 90 19 se determină în conformitate cu metoda prezentată în anexa XV.
(2) Conținutul de ulei la care se face referire în alineatul (1) se exprimă ca procent din greutatea de ulei raportată la greutatea materiei uscate.
Articolul 7
Se aplică dispozițiile comunitare privind prezența contaminanților.
În ceea ce privește concentrația de solvenți halogenați, limitele pentru toate categoriile de ulei de măsline sunt următoarele:
— concentrația maximă pentru fiecare solvent halogenat detectat: 0,1 mg/kg;
— concentrația maximă pentru suma solvenților halogenați detectați: 0,2 mg/kg.
Articolul 7a
Persoanele fizice sau juridice, precum și grupurile de persoane care dețin, în scopuri profesionale sau comerciale, ulei de măsline și ulei din resturi de măsline, începând cu etapa extracției în concasor până la etapa de îmbuteliere, inclusiv, sunt obligate să țină registre cu intrările și ieșirile pentru fiecare categorie de astfel de uleiuri.
Statul membru se asigură că obligația prevăzută la primul paragraf este respectată în mod corespunzător.
Articolul 8
(1) Statele membre informează Comisia cu privire la măsurile de punere în aplicare a prezentului regulament. Acestea informează Comisia cu privire la orice modificări ulterioare.
(2) Până cel târziu la data de 31 mai a fiecărui an, statele membre transmit Comisiei un raport privind punerea în aplicare a prezentului regulament în cursul anului calendaristic anterior. Raportul conține cel puțin rezultatele verificărilor conformității efectuate în cazul uleiurilor de măsline, utilizând modelele prevăzute în anexa XXI.
(3) Notificările menționate în prezentul regulament se efectuează în conformitate cu Regulamentul (CE) nr. 792/2009 al Comisiei ( 1 ).
Articolul 9
Regulamentul (CEE) nr. 1058/77 se abrogă.
Articolul 10
(1) Prezentul regulament intră în vigoare în a treia zi de la data publicării în Jurnalul Oficial al Comunităților Europene.
Cu toate acestea, metoda prezentată în anexa XII se aplică de la ►M1 1 noiembrie 1992 ◄ , cu excepția operațiunilor privind sistemul de intervenție.
Metoda respectivă nu se aplică pentru uleiul de măsline virgin pregătit în vederea comercializării până la 1 noiembrie 1992.
(2) Prezentul regulament nu se aplică uleiurilor de măsline și uleiurilor din resturi de măsline ambalate înainte de intrarea în vigoare a prezentului regulament și comercializate până la 31 octombrie 1992.
Prezentul regulament este obligatoriu în toate elementele sale și se aplică direct în toate statele membre.
ANEXE
Cuprins
|
Anexa I: |
Caracteristicile uleiurilor de măsline |
|
Anexa Ia: |
Eșantionarea uleiului de măsline sau a uleiului din resturi de măsline livrat în ambalaje imediate |
|
Anexa Ib: |
Programul decizional pentru verificarea conformității unei probe de ulei de măsline cu categoria declarată |
|
Anexa II: |
Determinarea acizilor grași liberi, metoda la rece |
|
Anexa III: |
Determinarea indicelui de peroxid |
|
Anexa IV: |
Determinarea conținutului de ceară cu ajutorul cromatografiei în fază gazoasă cu coloană capilară |
|
Anexa V: |
Determinarea compoziției și a conținutului de steroli și de dialcooli triterpenici prin cromatografie în fază gazoasă cu coloană capilară |
|
Anexa VII: |
►M21 Determinarea procentului de monopalmitat de 2-gliceril ◄ |
|
▼M20 ————— |
|
|
Anexa IX: |
Analiză spectrofotometrică în ultraviolet |
|
Anexa X: |
Determinarea esterilor metilici ai acizilor grași prin cromatografie în fază gazoasă |
|
Anexa XI: |
Determinarea conținutului de solvenți halogenați volatili din uleiul de măsline |
|
Anexa XII: |
Metoda consiliului oleicol internațional pentru evaluarea organoleptică a uleiurilor de măsline virgine |
|
▼M20 ————— |
|
|
▼M19 ————— |
|
|
Anexa XV: |
Conținutul de ulei al resturilor de măsline |
|
Anexa XVI: |
Determinarea indicelui de iod |
|
Anexa XVII: |
Metodă de determinare a conținutului de stigmastadiene din uleiurile vegetale |
|
Anexa XVIII: |
Determinarea diferenței dintre conținutul real și conținutul teoretic de trigliceride cu NEC 42 |
|
Annex XIX: |
►M28 Determinarea conținutului de alcooli alifatici și triterpenici prin cromatografie în fază gazoasă cu coloană capilară ◄ |
|
Anexa XX: |
Metoda pentru determinarea conținutului de ceruri, esteri metilici ai acizilor grași și esteri etilici ai acizilor grași prin cromatografie în fază gazoasă cu coloană capilară |
|
▼M28 ————— |
|
|
Anexa XXI: |
Rezultatele verificărilor conformității efectuate în cazul uleiurilor de măsline menționate la articolul 8 alineatul (2) |
ANEXA I
CARACTERISTICILE ULEIURILOR DE MĂSLINE
|
Categorie |
Esteri etilici ai acizilor grași (EEAG) (*) |
Aciditate (%) (*) |
Indice de peroxid mEq O2/kg (*) |
Ceruri mg/kg (**) |
Monopalmitat de 2-gliceril (%) |
Stigmastadiene mg/kg (1) |
Diferență: ECN42 (HPLC) și ECN42 (calcul teoretic) |
K232 (*) |
K268 sau K270 (*) |
Delta-K (*) |
Evaluare organoleptică Mediana defectului (Md) (*) |
Evaluare organoleptică Mediana atributului fructat (Mf) (*) |
|
1. Ulei de măsline extravirgin |
EEAG ≤ 40 mg/kg (anul agricol 2013-2014) (2) EEAG ≤ 35 mg/kg (anii agricoli 2014-2016) EEAG ≤ 30 mg/kg (anii agricoli ulteriori anului 2016) |
≤ 0,8 |
≤ 20 |
C42 + C44 + C46 ≤ 150 |
≤ 0,9 dacă procentul de acid palmitic total ≤ 14 % |
≤ 0,05 |
≤ |0,2| |
≤ 2,50 |
≤ 0,22 |
≤ 0,01 |
Md = 0 |
Mf > 0 |
|
≤ 1,0 dacă procentul de acid palmitic total > 14 % |
||||||||||||
|
2. Ulei de măsline virgin |
— |
≤ 2,0 |
≤ 20 |
C42 + C44 + C46 ≤ 150 |
≤ 0,9 dacă procentul de acid palmitic total ≤ 14 % |
≤ 0,05 |
≤ |0,2| |
≤ 2,60 |
≤ 0,25 |
≤ 0,01 |
Md ≤ 3,5 |
Mf > 0 |
|
≤ 1,0 dacă procentul de acid palmitic total > 14 % |
||||||||||||
|
3. Ulei de măsline lampant |
— |
> 2,0 |
— |
C40 + C42 + C44 + C46 ≤ 300 (3) |
≤ 0,9 dacă procentul de acid palmitic total ≤ 14 % |
≤ 0,50 |
≤ |0,3| |
— |
— |
— |
Md > 3,5 (4) |
— |
|
≤ 1,1 dacă procentul de acid palmitic total > 14 % |
||||||||||||
|
4. Ulei de măsline rafinat |
— |
≤ 0,3 |
≤ 5 |
C40 + C42 + C44 + C46 ≤ 350 |
≤ 0,9 dacă procentul de acid palmitic total ≤ 14 % |
— |
≤ |0,3| |
— |
≤ 1,10 |
≤ 0,16 |
— |
— |
|
≤ 1,1 dacă procentul de acid palmitic total > 14 % |
||||||||||||
|
5. Ulei de măsline compus din uleiuri de măsline rafinate și uleiuri de măsline virgine |
— |
≤ 1,0 |
≤ 15 |
C40 + C42 + C44 + C46 ≤ 350 |
≤ 0,9 dacă procentul de acid palmitic total ≤ 14 % |
— |
≤ |0,3| |
— |
≤ 0,90 |
≤ 0,15 |
— |
— |
|
≤ 1,0 dacă procentul de acid palmitic total > 14 % |
||||||||||||
|
6. Ulei brut din resturi de măsline |
— |
— |
— |
C40 + C42 + C44 + C46 > 350 (5) |
≤ 1,4 |
— |
≤ |0,6| |
— |
— |
— |
— |
— |
|
7. Ulei rafinat din resturi de măsline |
— |
≤ 0,3 |
≤ 5 |
C40 + C42 + C44 + C46 > 350 |
≤ 1,4 |
— |
≤ |0,5| |
— |
≤ 2,00 |
≤ 0,20 |
— |
— |
|
8. Ulei din resturi de măsline |
— |
≤ 1,0 |
≤ 15 |
C40 + C42 + C44 + C46 > 350 |
≤ 1,2 |
— |
≤ |0,5| |
— |
≤ 1,70 |
≤ 0,18 |
— |
— |
|
(1) Suma izomerilor care ar putea să fie (sau să nu fie) separați prin coloana capilară. (2) Limita se aplică uleiurilor de măsline produse începând cu data de 1 martie 2014. (3) Uleiurile cu un conținut de ceară cuprins între 300 mg/kg și 350 mg/kg sunt considerate ulei de măsline lampant în cazul în care cantitatea totală de alcooli alifatici este mai mică sau egală cu 350 mg/kg sau în cazul în care procentul de eritrodiol și uvaol este mai mic sau egal cu 3,5. (4) Mediana defectului poate fi mai mică sau egală cu 3,5 și mediana atributului fructat este egală cu 0. (5) Uleiurile cu un conținut de ceară cuprins între 300 mg/kg și 350 mg/kg sunt considerate uleiuri brute din resturi de măsline dacă conținutul total de alcooli alifatici depășește 350 mg/kg și dacă conținutul de eritrodiol și uvaol depășește 3,5 %. |
||||||||||||
|
Categorie |
Compoziția de acizi grași (1) |
Suma izomerilor trans ai acidului oleic (%) |
Suma izomerilor trans ai acidului linoleic și ai acidului linolenic (%) |
Conținutul de steroli |
Steroli totali (mg/kg) |
Eritrodiol și uvaol (%) (**) |
||||||||||
|
Miristic (%) |
Linolenic (%) |
Arahidic (%) |
Eicosenoic (%) |
Behenic (%) |
Lignoceric (%) |
Colesterol (%) |
Brasicasterol (%) |
Campesterol (2) (%) |
Stigmasterol (%) |
β-Sitosterol ap (3) (%) (**) |
Delta-7-Stigmastenol (2) (%) |
|||||
|
1. Ulei de măsline extravirgin |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,40 |
≤ 0,20 |
≤ 0,20 |
≤ 0,05 |
≤ 0,05 |
≤ 0,5 |
≤ 0,1 |
≤ 4,0 |
< Camp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 000 |
≤ 4,5 |
|
2. Ulei de măsline virgin |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,40 |
≤ 0,20 |
≤ 0,20 |
≤ 0,05 |
≤ 0,05 |
≤ 0,5 |
≤ 0,1 |
≤ 4,0 |
< Camp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 000 |
≤ 4,5 |
|
3. Ulei de măsline lampant |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,40 |
≤ 0,20 |
≤ 0,20 |
≤ 0,10 |
≤ 0,10 |
≤ 0,5 |
≤ 0,1 |
≤ 4,0 |
— |
≥ 93,0 |
≤ 0,5 |
≥ 1 000 |
≤ 4,5 (4) |
|
4. Ulei de măsline rafinat |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,40 |
≤ 0,20 |
≤ 0,20 |
≤ 0,20 |
≤ 0,30 |
≤ 0,5 |
≤ 0,1 |
≤ 4,0 |
< Camp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 000 |
≤ 4,5 |
|
5. Ulei de măsline compus din uleiuri de măsline rafinate și uleiuri de măsline virgine |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,40 |
≤ 0,20 |
≤ 0,20 |
≤ 0,20 |
≤ 0,30 |
≤ 0,5 |
≤ 0,1 |
≤ 4,0 |
< Camp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 000 |
≤ 4,5 |
|
6. Ulei brut din resturi de măsline |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,40 |
≤ 0,30 |
≤ 0,20 |
≤ 0,20 |
≤ 0,10 |
≤ 0,5 |
≤ 0,2 |
≤ 4,0 |
— |
≥ 93,0 |
≤ 0,5 |
≥ 2 500 |
> 4,5 (5) |
|
7. Ulei rafinat din resturi de măsline |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,40 |
≤ 0,30 |
≤ 0,20 |
≤ 0,40 |
≤ 0,35 |
≤ 0,5 |
≤ 0,2 |
≤ 4,0 |
< Camp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 800 |
> 4,5 |
|
8. Ulei din resturi de măsline |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,40 |
≤ 0,30 |
≤ 0,20 |
≤ 0,40 |
≤ 0,35 |
≤ 0,5 |
≤ 0,2 |
≤ 4,0 |
< Camp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 600 |
> 4,5 |
|
(1) Conținutul de alți acizi grași (%): palmitic: 7,50-20,00; palmitoleic: 0,30-3,50; heptadecanoic: ≤ 0,30; heptadecenoic: ≤ 0,30; stearic: 0,50-5,00; oleic: 55,00-83,00; linoleic: 2,50-21,00. (2) A se vedea apendicele la prezenta anexă. (3) β-Sitosterol ap: delta-5,23-stigmastadienol + colesterol + betasitosterol + sitostanol + delta-5-avenasterol + delta-5,24-stigmastadienol. (4) Uleiurile cu un conținut de ceară cuprins între 300 mg/kg și 350 mg/kg sunt considerate ulei de măsline lampant în cazul în care cantitatea totală de alcooli alifatici este mai mică sau egală cu 350 mg/kg sau în cazul în care procentul de eritrodiol și uvaol este mai mic sau egal cu 3,5. (5) Uleiurile cu un conținut de ceară cuprins între 300 mg/kg și 350 mg/kg sunt considerate uleiuri brute din resturi de măsline dacă conținutul total de alcooli alifatici depășește 350 mg/kg sau dacă conținutul de eritrodiol și uvaol depășește 3,5 %. |
||||||||||||||||
Note:
(a) Rezultatele analizelor trebuie exprimate cu același număr de zecimale ca cele indicate pentru fiecare caracteristică. Ultima cifră trebuie mărită cu o unitate în cazul în care cifra următoare este mai mare de 4.
(b) Este de ajuns ca o singură caracteristică să nu fie conformă cu valorile indicate pentru ca uleiul să fie încadrat într-o altă categorie sau să fie declarat neconform din punct de vedere al purității, în sensul prezentului regulament.
(c) Caracteristicile indicate cu asterisc (*), care fac referire la calitatea uleiului, semnifică următoarele: – pentru uleiul de măsline lampant, limitele aferente pot să nu fie respectate în mod simultan; – pentru uleiurile de măsline virgine, nerespectarea cel puțin a uneia dintre aceste limite implică schimbarea categoriei, cu toate că uleiul rămâne clasificat într-una din categoriile de uleiuri de măsline virgine.
(d) Caracteristicile marcate cu două asteriscuri (**) implică faptul că, pentru toate uleiurile din resturi de măsline, limitele aferente pot să nu fie respectate în mod simultan.
Apendice
ARBORELE DECIZIONAL
Arborele decizional referitor la campesterol pentru uleiurile de măsline virgine și extravirgine:
Ceilalți parametri respectă limitele stabilite în prezentul regulament.
Arborele decizional referitor la Delta-7-stigmastenol:
— Uleiuri de măsline extravirgine și virgine
—
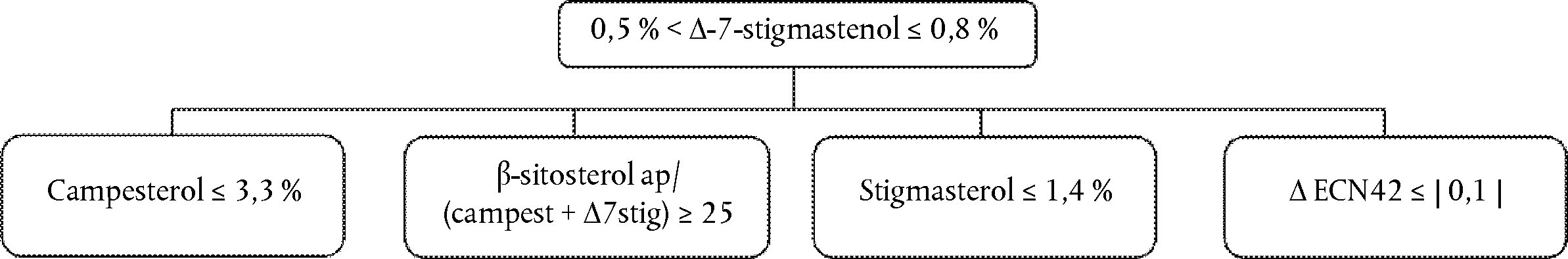
Ceilalți parametri respectă limitele stabilite în prezentul regulament.
— Uleiuri din resturi de măsline (brute și rafinate)
—

ANEXA Ia
EȘANTIONAREA ULEIULUI DE MĂSLINE SAU A ULEIULUI DIN RESTURI DE MĂSLINE LIVRAT ÎN AMBALAJE IMEDIATE
Această metodă de eșantionare se aplică loturilor de ulei de măsline sau de ulei din resturi de măsline în ambalaje imediate. Se aplică diferite metode de eșantionare, în funcție de volumul ambalajului imediat (mai mic sau mai mare de 5 litri).
Prin „lot” se înțelege un ansamblu de unități de vânzare produse, fabricate și ambalate astfel încât uleiul conținut în fiecare din aceste unități de vânzare este considerat omogen din punct de vedere al tuturor caracteristicilor analitice. Individualizarea unui lot trebuie să se efectueze în conformitate cu Directiva 2011/91/UE a Parlamentului European și a Consiliului ( 2 ).
„Prelevare” înseamnă cantitatea de ulei conținută într-un ambalaj imediat și extrasă dintr-un punct aleatoriu al lotului.
1. CONȚINUTUL UNEI PROBE PRIMARE
1.1. Ambalaje imediate care nu depășesc 5 litri
În cazul ambalajelor imediate care nu depășesc 5 litri, „probă primară” înseamnă numărul prelevărilor extrase dintr-un lot și conforme tabelului 1.
Tabelul 1
Dimensiunea minimă a fiecărei probe primare trebuie să fie alcătuită din
|
În cazul în care ambalajele imediate au o capacitate |
Proba primară trebuie să conțină ulei din |
|
(a) mai mare sau egală cu 1 litru |
(a) 1 ambalaj imediat; |
|
(b) mai mică de 1 litru |
(b) numărul minim de ambalaje cu o capacitate totală de cel puțin 1,0 litru |
Numărul de ambalaje menționat în tabelul 1 și care constituie o probă primară poate fi mărit de către fiecare stat membru, în conformitate cu propriile nevoi (de exemplu, evaluarea organoleptică de către un alt laborator decât cel care a efectuat analizele chimice, contraanaliză etc.).
1.2. Ambalaje imediate care depășesc 5 litri
În cazul ambalajelor imediate care depășesc 5 litri, „probă primară” înseamnă o parte reprezentativă a prelevărilor totale, obținute printr-un proces de reducere și conforme tabelului 2. Probele primare trebuie să fie compuse din diferite specimene.
„Specimen” de probă primară înseamnă fiecare dintre ambalajele care compun proba primară.
Tabelul 2
Numărul minim de prelevări care trebuie selectate
|
Număr de ambalaje pe lot |
Numărul minim de prelevări care trebuie selectate |
|
Până la 10 |
1 |
|
De la … 11 la 150 |
2 |
|
De la … 151 la 500 |
3 |
|
De la … 501 la 1 500 |
4 |
|
De la … 1 501 la 2 500 |
5 |
|
> 2 500 pentru 1 000 de ambalaje |
1 prelevare suplimentară |
Pentru a reduce volumul de ambalaje imediate la eșantionare, conținutul prelevărilor de eșantionare este omogenizat pentru pregătirea probei primare. Fracțiunile din diferitele prelevări sunt vărsate și amestecate într-un recipient comun în vederea omogenizării, într-un mod care să ofere o protecție optimală împotriva aerului.
Conținutul probei primare trebuie să fie turnat într-o serie de ambalaje cu o capacitate minimă de 1,0 litru, astfel încât fiecare dintre acestea să constituie un specimen din proba primară.
Numărul de probe primare poate fi mărit de către fiecare stat membru, în conformitate cu propriile nevoi (de exemplu, evaluarea organoleptică de către un alt laborator decât cel care a efectuat analizele chimice, contraanaliză etc.).
Fiecare ambalaj trebuie să fie umplut în așa fel încât să se reducă la minimum stratul superior de aer, după care este închis și sigilat în mod corespunzător pentru a se asigura protejarea produsului împotriva falsificării.
Specimenele trebuie să fie etichetate pentru a se garanta identificarea corectă a acestora.
2. ANALIZE ȘI REZULTATE
2.1. Fiecare probă primară trebuie să se împartă în probe de laborator, în conformitate cu punctul 2.5 din standardul EN ISO 5555 și trebuie să se analizeze urmându-se ordinea prezentată în programul decizional prevăzut în anexa IB sau în orice altă ordine aleatorie.
2.2. În cazul în care toate rezultatele analizelor sunt conforme caracteristicilor categoriei de ulei declarate, întregul lot este declarat conform.
Dacă un singur rezultat al analizelor nu este conform caracteristicilor categoriei de ulei declarate, întregul lot este declarat neconform.
3. VERIFICAREA CATEGORIEI LOTULUI
3.1. În vederea verificării categoriei lotului, autoritatea competentă poate mări numărul de probe primare extrase din diferite puncte ale lotului, în conformitate cu următorul tabel:
Tabelul 3
Numărul de probe primare stabilite în funcție de mărimea lotului
|
Mărimea lotului (în litri) |
Numărul de probe primare |
|
mai puțin de 7 500 |
2 |
|
de la 7 500 la mai puțin de 25 000 |
3 |
|
de la 25 000 la mai puțin de 75 000 |
4 |
|
de la 75 000 la mai puțin de 125 000 |
5 |
|
egală cu și mai mare de 125 000 |
6 + 1 pentru fiecare 50 000 de litri suplimentari |
Fiecare prelevare care constituie o probă primară trebuie să fie extrasă din același loc al lotului, fiind necesar să se noteze amplasarea fiecărei probe primare, care să fie identificată în mod inechivoc.
Alcătuirea fiecărei probe primare trebuie să se efectueze în conformitate cu procedurile menționate punctele 1.1 și 1.2.
Fiecare probă primară este apoi supusă analizelor menționate la articolul 2 alineatul (1).
3.2. În cazul în care unul dintre rezultatele analizelor menționate la articolul 2 alineatul (1) pentru cel puțin o probă primară nu este conform caracteristicilor categoriei de ulei declarate, se declară neconform întregul lot de eșantionare.
ANEXA Ib
PROGRAMUL DECIZIONAL PENTRU VERIFICAREA CONFORMITĂȚII UNEI PROBE DE ULEI DE MĂSLINE CU CATEGORIA DECLARATĂ
Tabelul 1
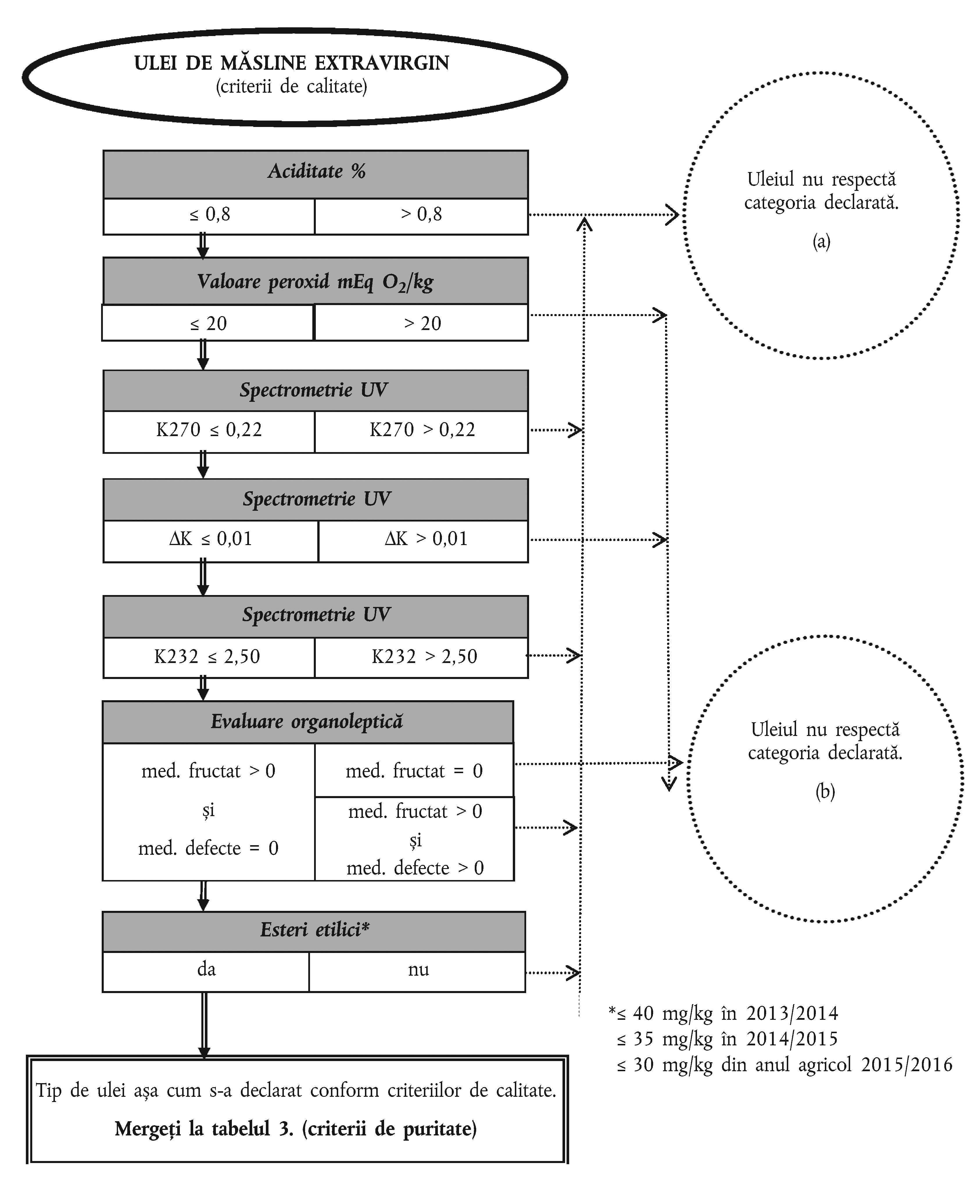
Tabelul 2

Tabelul 3
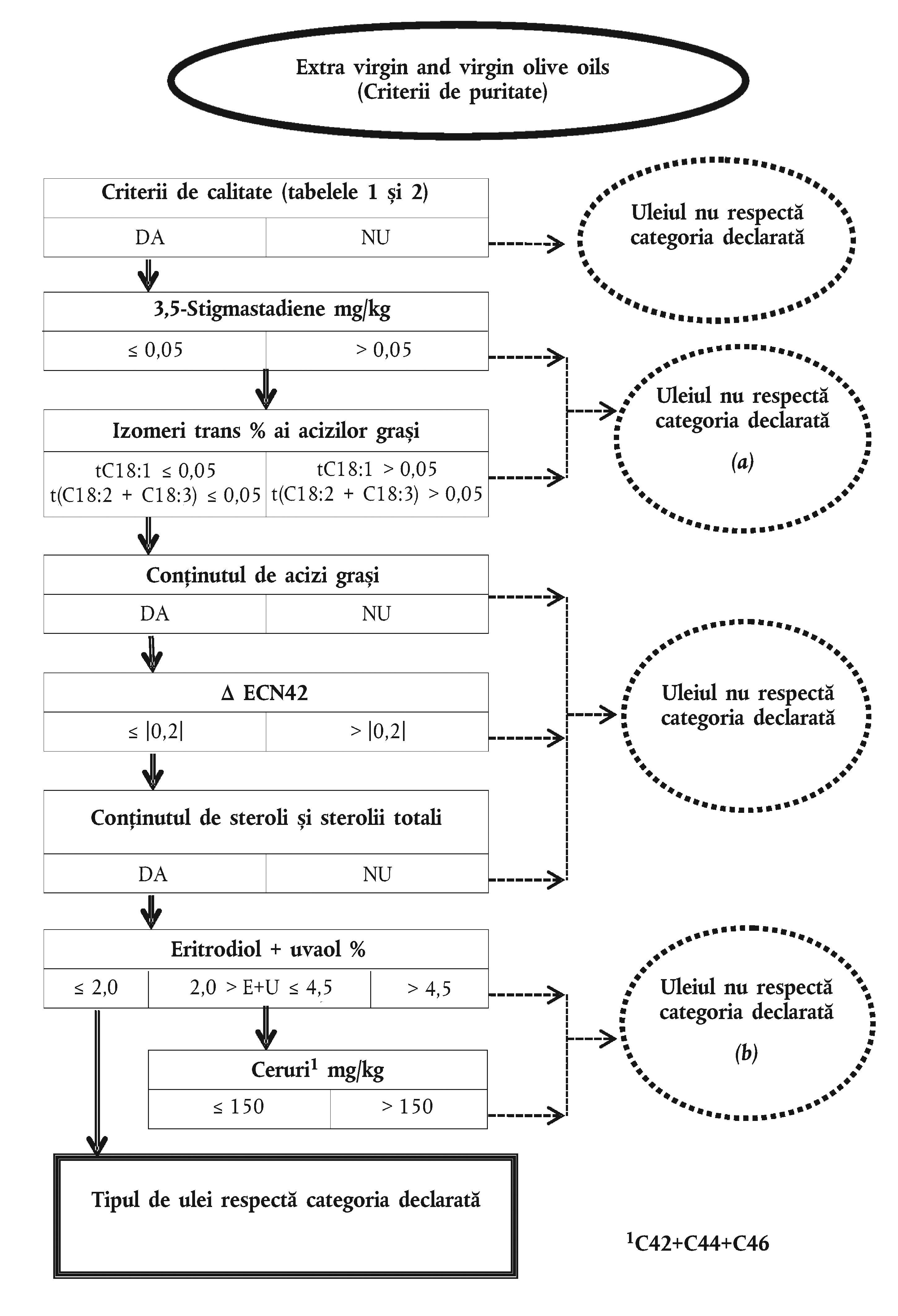
Apendicele 1
Tabel de corespondență între anexele la prezentul regulament și analizele menționate în programul decizional
|
— Aciditate |
Anexa II |
Determinarea acizilor grași liberi, metoda la rece |
|
— Indicele de peroxid |
Anexa III |
Determinarea indicelui de peroxid |
|
— Spectrometrie UV |
Anexa IX |
Analiza spectrofotometrică |
|
— Evaluarea organoleptică |
Anexa XII |
Evaluarea organoleptică a uleiului de măsline virgin |
|
— Esteri etilici |
Anexa XX |
Metoda pentru determinarea conținutului de ceruri, esteri metilici ai acizilor grași și esteri etilici ai acizilor grași prin cromatografie în fază gazoasă cu coloană capilară |
|
— Stigmasta-3,5-diene |
Anexa XVII |
Metodă de determinare a conținutului de stigmastadiene din uleiurile vegetale |
|
— Izomeri trans ai acizilor grași |
Anexa X |
Analiza prin cromatografie în fază gazoasă a esterilor metilici ai acizilor grași |
|
— Conținutul de acizi grași |
Anexa X |
Analiza prin cromatografie în fază gazoasă a esterilor metilici ai acizilor grași |
|
— ΔECN42 |
Anexa XVIII |
Determinarea compoziției trigliceridelor cu ECN42 (diferența dintre datele HPLC și conținutul teoretic) |
|
— Compoziția de steroli și steroli totali — Eritrodiol și uvaol |
Anexa V |
Determinarea compoziției și a conținutului de steroli și de dialcooli triterpenici prin cromatografie în fază gazoasă cu coloană capilară |
|
— Ceruri |
Anexa IV |
Determinarea conținutului de ceară cu ajutorul cromatografiei în fază gazoasă cu coloană capilară |
|
— Alcooli alifatici și triterpenici |
Anexa XIX |
Determinarea conținutului de alcooli alifatici și triterpenici prin cromatografie în fază gazoasă cu coloană capilară |
|
— Acizi grași saturați în poziția 2 |
Anexa VII |
Determinarea procentului de monopalmitat de 2-gliceril |
ANEXA II
DETERMINAREA ACIZILOR GRAȘI LIBERI, METODA LA RECE
1. OBIECTUL ȘI DOMENIUL DE APLICARE
Această metodă descrie determinarea acizilor grași liberi din uleiurile de măsline și din uleiurile din resturi de măsline. Conținutul de acizi grași liberi se exprimă prin aciditatea calculată ca procentaj de acid oleic.
2. PRINCIPIU
Se dizolvă o probă într-un amestec de solvenți, apoi se titrează acizii grași liberi prezenți cu ajutorul unei soluții de hidroxid de potasiu sau de hidroxid de sodiu.
3. REACTIVI
Toți reactivii trebuie să aibă o calitate analitică recunoscută, iar apa utilizată trebuie să fie distilată sau cu un grad de puritate echivalent.
|
3.1. |
Eter dietilic; etanol în concentrație de 95 % (V/V), amestec în volum de 1 la 1. Se neutralizează exact în momentul folosirii cu soluția de hidroxid de potasiu (3.2) în prezența prin adăugarea a 0,3 ml din soluția de fenolftaleină (3.3) la 100 ml de amestec. Nota 1: Eterul dietilic este foarte inflamabil și poate forma peroxizi explozivi. A se utiliza cu precauții speciale. Nota 2: În cazul în care nu se poate utiliza eter dietilic, se poate folosi un amestec de solvenți conținând etanol și toluen. Dacă este necesar, etanolul se poate înlocui cu propanol-2. |
|
3.2. |
Hidroxid de potasiu sau hidroxid de sodiu, soluție etanolică titrată sau soluție apoasă c(KOH) [sau c(NaOH)] aproximativ 0,1 mol/l sau, dacă este necesar, c(KOH) [sau c(NaOH)] aproximativ 0,5 moli la litru. Sunt disponibile soluții în comerț. Concentrația exactă a soluției de hidroxid de potasiu (sau a soluției de hidroxid de sodiu) trebuie să fie cunoscută și verificată înainte de folosire. Se utilizează o soluție preparată cu cel puțin cinci zile înainte de folosire și decantată într-un flacon de sticlă maro cu un dop de cauciuc. Soluția trebuie să fie incoloră sau de nuanța galben pai. Dacă se observă separarea fazelor atunci când se folosește soluție apoasă de hidroxid de potasiu (sau de hidroxid de sodiu), se înlocuiește soluția apoasă cu o soluție etanolică. Nota 3: O soluție incoloră stabilă de hidroxid de potasiu (sau de hidroxid de sodiu) se poate prepara în modul următor. Se pun la fiert 1 000 ml de etanol sau de apă cu 8 g de hidroxid de potasiu (sau de hidroxid de sodiu) și 0,5 g de pilitură de aluminiu și se continuă fierberea la reflux timp de o oră. Se distilează imediat. Se dizolvă în distilat cantitatea de hidroxid de potasiu (sau de hidroxid de sodiu) necesară. Se lasă în repaus timp de mai multe zile și se decantează lichidul limpede supernatant de precipitatul de carbonat de potasiu (sau de carbonat de sodiu). Soluția poate fi preparată și fără distilare, astfel: la 1 000 ml de etanol (sau de apă) se adaugă 4 ml de butilat de aluminiu și se lasă amestecul în repaus timp de câteva zile. Se decantează lichidul supernatant și se dizolvă cantitatea necesară de hidroxid de potasiu (sau de hidroxid de sodiu). Soluția se poate utiliza ca atare. |
|
3.3. |
Fenolftaleină, soluție de 10 g/l în etanol în concentrație de 95-96 % (V/V) sau albastru alcalin 6B sau timolftaleină, soluție de 20 g/l în etanol în concentrație de 95-96 % (V/V). În cazul uleiurilor foarte colorate, se folosește albastru alcalin sau timolftaleină. |
4. APARATURĂ
Materiale obișnuite de laborator, în special:
4.1. balanță analitică;
4.2. balon conic cu capacitatea de 250 ml;
4.3. biuretă cu capacitatea de 10 ml, clasa A, gradată la 0,05 ml, sau o biuretă automată echivalentă.
5. MOD DE LUCRU
5.1. Prepararea probei
Când este tulbure, proba trebuie filtrată.
5.2. Mostra
Se prelevează o probă, în funcție de aciditatea presupusă, după indicațiile din tabelul următor:
|
Aciditatea presupusă (aciditate oleică g/100g) |
Masa probei (în g) |
Precizia de cântărire a mostrei (în g) |
|
0-2 |
10 |
0,02 |
|
> 2-7,5 |
2,5 |
0,01 |
|
> 7,5 |
0,5 |
0,001 |
Se cântărește proba în balonul conic (4.2).
5.3. Determinare
Se dizolvă proba (5.2) într-o cantitate de 50-100 ml de amestec de eter dietilic și etanol (3.1), neutralizat în prealabil.
Se titrează în timp ce se agită, cu soluția de hidroxid de potasiu (sau de hidroxid de sodiu) de 0,1 mol/l (3.2) (a se vedea nota 4) până când se modifică indicatorul (culoarea indicatorului colorat persistă cel puțin 10 secunde).
Nota 4: În cazul în care cantitatea necesară de soluție de hidroxid de potasiu (sau de hidroxid de sodiu) de 0,1 mol/l depășește 10 ml, se folosește soluția de 0,5 mol/l sau se modifică masa probei în funcție de aciditatea liberă presupusă și de tabelul propus.
Nota 5: În cazul în care soluția devine tulbure în timpul titrării, se adaugă o cantitate suficientă de amestec de solvenți (3.1) pentru a se obține o soluție limpede.
O a doua determinare se efectuează doar dacă primul rezultat depășește limita specificată pentru categoria uleiului.
6. EXPRIMAREA REZULTATELOR
Aciditatea, exprimată în procente de acid oleic, în greutate, este egală cu:

unde:
|
V |
= |
volumul soluției titrate de hidroxid de potasiu (sau de hidroxid de sodiu) utilizate, în mililitri; |
|
c |
= |
concentrația exactă, în mol/l, a soluției titrate de hidroxid de potasiu (sau de hidroxid de sodiu) utilizate; |
|
M |
= |
282 g/mol, masa molară în grame la mol de acid oleic; |
|
m |
= |
masa în grame a probei. |
Aciditatea oleică se raportează după cum urmează:
(a) cu două zecimale pentru valorile de la 0 la 1 inclusiv;
(b) cu o zecimală pentru valorile de la 1 la 100 inclusiv.
ANEXA III
DETERMINAREA INDICELUI DE PEROXID
1. OBIECT
Prezenta normă descrie o metodă de determinare a indicelui de peroxid în uleiuri și materii grase.
2. DOMENIUL DE APLICARE
Prezenta normă se aplică uleiurilor și materiilor grase animale și vegetale.
3. DEFINIȚIE
Indicele de peroxid reprezintă cantitatea de substanțe din mostră (exprimată în miliechivalenți de oxigen activ la kilogram) care oxidează iodura de potasiu în condițiile de lucru descrise.
4. PRINCIPIU
Mostra sub formă de soluție într-un amestec de acid acetic și cloroform se tratează cu o soluție de iodură de potasiu. Iodul eliberat se titrează cu o soluție de tiosulfat de sodiu.
5. APARATURĂ
Echipamentele utilizate trebuie să fie lipsite de orice urmă de substanțe oxidante sau dezoxidante.
Nota bene: nu se gresează suprafețele din sticlă rodată.
|
5.1. |
Lingură de sticlă de 3 ml. |
|
5.2. |
Baloane de aproximativ 250 ml, cu gâtul și dopul din sticlă rodată, uscate în prealabil și umplute cu un gaz inert, uscat și pur (azot sau, de preferință, bioxid de carbon). |
|
5.3. |
Biuretă de 25 sau 50 ml cu gradare la 0,1 ml. |
6. REACTIVI
|
6.1. |
Cloroform de calitate analitică, fără oxigen (acesta din urmă fiind eliminat prin barbotarea unui curent de gaz inert, uscat și pur). |
|
6.2. |
Acid acetic glacial de calitate analitică, fără oxigen (acesta din urmă fiind eliminat prin barbotarea unui curent de gaz uscat și pur). |
|
6.3. |
Iodură de potasiu în soluție apoasă saturată în urma unei preparări recente, fără de iod și iodați. |
|
6.4. |
Soluție apoasă de tiosulfat de sodiu 0,01 sau 0,002 N, standardizată cu atenție chiar înainte de folosire. |
|
6.5. |
Soluție de amidon (dispersare apoasă de 10 g/l) preparată recent din amidon natural solubil. |
7. MOSTRA
Mostra trebuie să fie prelevată și păstrată la adăpost de lumină, conservată la rece și închisă în recipiente de sticlă umplute în întregime și închise ermetic cu dopuri de plută sau de sticlă rodată.
8. MOD DE LUCRU
Testul trebuie să fie realizat la o lumină difuză (lumina zilei) sau artificială. Într-o lingură de sticlă (5.1) sau, la nevoie, într-un balon (5.2), se cântărește, cu o aproximare de 0,001 grame, una dintre masele de mostră menționate în tabelul următor, în funcție de indicele de peroxid prevăzut.
|
Indicele prevăzut de peroxid (în meq O2/kg) |
Masa mostrei (în g) |
|
0–12 |
5,0–2,0 |
|
12–20 |
2,0–1,2 |
|
20–30 |
1,2–0,8 |
|
30–50 |
0,8–0,5 |
|
50–90 |
0,5–0,3 |
Se scoate dopul unui balon (5.2) și se introduce lingura de sticlă cu mostra. Se adaugă 10 ml de cloroform (6.1). Se dizolvă rapid mostra prin agitare. Se adaugă 15 ml de acid acetic (6.2), apoi 1 ml de soluție de iodură de potasiu (6.3). Se pune rapid dopul la loc, se agită timp de un minut și se lasă în repaus exact 5 minute, la adăpost de lumină și la o temperatură de la 15–25 °C.
Se adaugă aproximativ 75 ml de apă distilată. Se titrează iodul eliberat cu soluție de tiosulfat de sodiu (6.4) (soluție de 0,002 N, în cazul în care indicii prevăzuți sunt mai mici de 12, și de 0,001 N, în cazul în care sunt mai mari de 12) prin agitare puternică și prin folosirea soluției de amidon (6,5) ca indicator.
Se efectuează două analize pe aceeași mostră.
Simultan, se efectuează un test etalon. În cazul în care rezultatul acestuia din urmă este mai mare de 0,05 ml de soluție de tiosulfat de sodiu (6,4) 0,001 N, se înlocuiesc reactivii impuri.
9. EXPRIMAREA REZULTATELOR
Indicele de peroxid (IP), exprimat în miliechivalenți de oxigen activ la kilogram, este dat de formula:

,
în care:
|
V |
= |
numărul ml de soluție de tiosulfat de sodiu standardizată (6.4) utilizat pentru test, modificat în funcție de rezultatele mostrei etalon; |
|
T |
= |
factorul exact de standardizare al soluției de tiosulfat de sodiu (6,4) utilizate; |
|
m |
= |
masa (în grame) a mostrei. |
Se ia drept rezultat media aritmetică a celor două determinări.
ANEXA IV
DETERMINAREA CONȚINUTULUI DE CEARĂ CU AJUTORUL CROMATOGRAFIEI ÎN FAZĂ GAZOASĂ CU COLOANĂ CAPILARĂ
1. OBIECT
Metoda descrie un procedeu de determinare a conținutului de ceară din uleiurile de măsline. Tipurile de ceară sunt definite în funcție de numărul de atomi de carbon. Metoda poate fi folosită în special pentru a deosebi uleiul de măsline obținut prin presare de cel obținut prin extracție (ulei din resturi de măsline).
2. PRINCIPIU
Materia grasă, la care s-a adăugat un standard intern adecvat, este fracționată prin cromatografie pe o coloană de silicagel hidratat; fracțiunea eluată prima în condițiile de testare (a cărei polaritate este mai mică decât cea a trigliceridelor) este recuperată, apoi analizată direct prin cromatografie în fază gazoasă cu coloană capilară.
3. APARATURĂ
|
3.1. |
Pahar Erlenmeyer de 25 ml. |
|
3.2. |
Coloană de sticlă pentru cromatografie în fază gazoasă, cu un diametru interior de 15 mm, o înălțime de 30-40 cm, echipată cu un robinet. |
|
3.3. |
Cromatograf în fază gazoasă adaptat la funcționarea cu coloană capilară, echipat cu un sistem de introducere directă în coloană, alcătuit din:
|
|
3.4. |
Microseringă de 10 μl pentru introducerea directă în coloană, echipată cu un ac cementat. |
|
3.5. |
Agitator electric. |
|
3.6. |
Evaporator rotativ. |
|
3.7. |
Cuptor închis. |
|
3.8. |
Balanță analitică cu o precizie garantată a măsurii de + 0,1 mg. |
|
3.9. |
Sticlărie standard de laborator. |
4. REACTIVI
|
4.1. |
Silicagel cu o granulometrie cuprinsă între 60 și 200 μm. Silicagelul se păstrează în cuptor la 500 °C timp de cel puțin patru ore. După răcire, se adaugă 2 % apă în raport cu cantitatea de silicagel prelevată. Se agită bine pentru a omogeniza cantitatea. Se păstrează la întuneric timp de cel puțin 12 ore înainte de utilizare. |
|
4.2. |
n-hexan, pentru cromatografie. |
|
4.3. |
Eter etilic, pentru cromatografie. |
|
4.4. |
n-heptan, pentru cromatografie. |
|
4.5. |
Soluție standard de lauril arahidat, la 0,1 % (m/V) în hexan (standard intern) (se poate utiliza, de asemenea, palmitat de palmitil sau stearat de miristil).
|
|
4.6. |
Gaz transportor: hidrogen sau heliu pur, pentru cromatografie în fază gazoasă. |
|
4.7. |
Gaze auxiliare: — hidrogen pur, pentru cromatografie în fază gazoasă; — aer pur, pentru cromatografie în fază gazoasă. |
5. MOD DE LUCRU
5.1. Pregătirea coloanei cromatografice
Se prepară o suspensie de 15 g de silicagel (4.1) în n-hexan (4.2) și se introduce pe coloană (3.2). Se lasă să se clarifice spontan și se definitivează clarificarea cu ajutorul unui agitator electric (3.5) pentru omogenizarea stratului de cromatografie. Se percolează 30 ml de n-hexan pentru a înlătura orice impurități. Se cântăresc cu exactitate cu ajutorul balanței (3.8) 500 mg din mostră în paharul Erlenmeyer de 25 ml (3.1), se adaugă cantitatea adecvată de standard intern (4.5), în funcție de conținutul de ceară estimat.
Se adaugă, de exemplu, 0,1 mg de lauril arahidat în cazul uleiului de măsline și între 0,25 și 0,5 mg pentru uleiul din resturi de măsline. Se transferă mostra astfel preparată în coloana cromatografică cu ajutorul a două doze de câte 2 ml de n-hexan (4.2).
Se lasă să scadă nivelul solventului până la 1 mm deasupra marginii superioare a absorbantului, apoi se percolează o cantitate suplimentară de 70 ml de n-hexan, pentru a elimina n-alcanii prezenți în mod natural. Se începe eluarea cromatografică prin colectarea a 180 ml de amestec de n-hexan/eter etilic, în raport de 99:1, la un debit de aproximativ 15 picături la fiecare 10 secunde. Eluarea mostrei se efectuează la o temperatură ambiantă de 22 ± 4 °C.
Note:
— Amestecul de n-hexan/eter etilic (99:1) trebuie să fie pregătit zilnic.
— Pentru a controla vizual eluarea corectă a cerii, se pot adăuga la mostra de soluție 100 μl de sudan de 1 % în amestecul de eluant. Deoarece colorantul prezintă o retenție intermediară între ceruri și trigliceride, eluarea trebuie întreruptă în momentul în care baza coloanei cromatografice se colorează, întrucât toate tipurile de ceară au fost eluate.
Fracțiunea astfel obținută este uscată într-un evaporator rotativ (3.6) până la eliminarea aproape totală a solventului. Ultimii 2 ml de solvent se elimină cu ajutorul unui ușor curent de azot; se adaugă apoi 2-4 ml de n-heptan.
5.2. Analiza prin cromatografie în fază gazoasă
5.2.1. Operațiuni preliminare
Se instalează coloana în cromatograful în fază gazoasă (3.3), conectând extremitatea de intrare la sistemul coloanei și extremitatea de ieșire la detector. Se efectuează controalele generale ale complexului de cromatografie în faza gazoasă (etanșeitatea circuitului de gaz, eficacitatea detectorului și a sistemului de înregistrare etc.).
În cazul în care coloana este folosită pentru prima dată, se recomandă ca aceasta să fie condiționată. Se trece un ușor flux de gaz prin coloană, apoi se conectează complexul de cromatografie în fază gazoasă. Se încălzește treptat până când se ajunge, după aproximativ 4 ore, la o temperatură de 350 °C. Se menține această temperatură timp de cel puțin două ore, apoi se reglează complexul la condițiile de funcționare [se reglează debitul de gaz, se aprinde flacăra, se conectează la aparatul de înregistrare electronic (3.3.4), se reglează temperatura camerei pentru coloană, detectorul etc.] și se înregistrează semnalul la o sensibilitate cel puțin de două ori mai mare decât cea prevăzută pentru efectuarea analizei. Traseul liniei de bază obținute trebuie să fie liniar, fără valori de vârf, de orice natură, și nu trebuie să prezinte abateri.
O abatere rectilinie negativă indică o etanșeitate imperfectă a conexiunilor coloanei; o abatere pozitivă indică o condiționare insuficientă a coloanei.
5.2.2. Alegerea condițiilor de funcționare
Condițiile de funcționare care trebuie respectate sunt, în general, următoarele:
— temperatura coloanei:
—
|
|
20 °C/minut |
|
5 °C/minut |
|
20 °C/minut |
|
|
la început 80 °C (1′) |
→ |
240 °C |
→ |
325 °C (6′) |
→ |
340 °C (10′) |
— temperatura detectorului: 350 °C;
— cantitatea de substanță injectată: 1 μl de soluție (2-4 ml) de n-heptan;
— gaz transportor: heliu sau hidrogen cu viteză lineară optimă pentru gazul selectat (a se vedea apendicele);
— sensibilitatea instrumentelor: adaptată condițiilor de mai jos:
Aceste condiții pot fi modificate în funcție de caracteristicile coloanei și ale cromatografului în fază gazoasă, astfel încât să se obțină o separare a tuturor tipurilor de ceară, o rezoluție convenabilă a valorilor de vârf (a se vedea figura) și un timp de retenție al standardului intern C32, care trebuie să fie de 18 ± 3 minute. Valoarea de vârf cea mai reprezentativă a cerii trebuie să se situeze la cel puțin 60 % din scara totală.
Parametrii de integrare ai valorilor de vârf trebuie să fie astfel impuși încât să se obțină o evaluare corectă a ariilor valorilor de vârf care sunt luate în considerare.
Notă: Având în vedere temperatura finală ridicată, se admite o variație pozitivă, care nu trebuie să depășească 10 % din scara totală.
5.3. Efectuarea analizei
Se prelevează 1 μl de soluție cu microseringa de 10 μl; se trage înapoi pistonul seringii în așa fel încât acul să fie gol. Se introduce acul în complexul de injecție și după 1-2 secunde se injectează rapid; după aproximativ 5 secunde, acul se scoate lent.
Se procedează la înregistrare până la eluarea completă a cerii.
Linia de bază trebuie să corespundă întotdeauna condițiilor impuse.
5.4. Identificarea valorilor de vârf
Identificarea valorilor de vârf unice se efectuează pe baza timpilor de retenție și prin comparație cu amestecurile de ceară cu timpi de retenție cunoscuți, analizate în aceleași condiții.
Figura prezintă cromatograma cerii pentru un ulei de măsline virgin.
5.5. Evaluare cantitativă
Cu ajutorul integratorului, se procedează la calculul ariilor valorilor de vârf corespunzătoare standardului intern și esterilor alifatici de la C40 la C46.
Se calculează conținutul de ceară al fiecărui ester, în mg/kg de materie grasă, după formula:
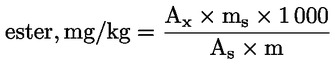
unde:
|
Ax |
= |
aria valorii de vârf a fiecărui ester, exprimată în milimetri pătrați; |
|
As |
= |
aria valorii de vârf a standardului intern, exprimată în milimetri pătrați; |
|
ms |
= |
masa standardului intern adăugată, exprimată în miligrame; |
|
m |
= |
masa mostrei prevalate pentru determinare, exprimată în grame. |
6. EXPRIMAREA REZULTATELOR
Se indică suma conținuturilor diferitelor tipuri de ceară de la C40 la C46 în mg/kg de substanță grasă (ppm).
Notă: Componentele care trebuie cuantificate se referă la valorile de vârf cu număr par de atomi de carbon, cuprinse între esterii C40 și C46, după modelul cromatogramei cerii uleiului de măsline, prezentate în figura următoare. Pentru a identifica esterul C46, în cazul în care apare de două ori, se recomandă analizarea fracțiunii de ceară a unui ulei din resturi de măsline, a cărei valoare de vârf C46, prezentă în cantitate majoritară, este ușor de identificat.
Rezultatele se exprimă cu o zecimală.
Figură
Cromatograma tipurilor de ceară ale unui ulei de măsline ( 3 )

Legendă:
|
I.S. |
= |
Lauril arahidat |
|
1 |
= |
Esteri diterpenici |
|
2 + 2′ |
= |
Esteri C40 |
|
3 + 3′ |
= |
Esteri C42 |
|
4 + 4′ |
= |
Esteri C44 |
|
5 |
= |
Esteri C46 |
|
6 |
= |
Esteri steroli și alcooli triterpenici. |
Apendice
Determinarea vitezei liniare a gazului
În cromatograful în fază gazoasă, reglat la condiții de funcționare normale, se injectează 1-3 μl de metan (sau propan). Se măsoară timpul în care gazul trece prin coloană, din momentul în care a fost injectat până când apare vârful (tM).
Viteza liniară în cm/sec se obține prin formula L/tM, unde L este lungimea coloanei în centimetri, iar tM este timpul măsurat în secunde.
ANEXA V
DETERMINAREA COMPOZIȚIEI ȘI A CONȚINUTULUI DE STEROLI ȘI DE DIALCOOLI TRITERPENICI PRIN CROMATOGRAFIE ÎN FAZĂ GAZOASĂ CU COLOANĂ CAPILARĂ
1. DOMENIUL DE APLICARE
Metoda descrie procedura de determinare a conținutului de steroli, simpli și totali, și de dialcooli triterpenici în uleiurile de măsline și în uleiurile din resturi de măsline.
2. PRINCIPIU
Uleiul, la care s-a adăugat α-colestanol ca etalon intern, este saponificat cu hidroxid de potasiu în soluție etanolică, apoi se extrage substanța nesaponificabilă cu eter etilic.
Fracțiunea de steroli și de dialcooli triterpenici se separă de substanța nesaponificabilă prin cromatografie în strat subțire pe placă de silicagel bazic. Fracțiunile recuperate din silicagel se transformă în trimetilsilileteri și apoi se analizează prin cromatografie în fază gazoasă cu coloană capilară.
3. APARATURA
Aparatura obișnuită de laborator și, în special, următoarele:
3.1. Balon de 250 ml echipat cu un condensator cu reflux cu racordurile din sticlă rodată.
3.2. Pâlnie de separare de 500 ml.
3.3. Baloane de 250 ml.
3.4. Echipament complet pentru analiză prin cromatografie în strat subțire cu plăci de sticlă de 20 × 20 cm.
3.5. Lampă cu lumină ultravioletă cu o lungime de undă de 254 sau 366 nm.
3.6. Microseringi de 100 μl și de 500 μl.
3.7. Pâlnie cilindrică de filtrare cu membrană poroasă G 3 (porozitate 15-40 μm) cu un diametru de aproximativ 2 cm și o înălțime de 5 cm, adaptată pentru filtrarea în vid cu un racord tată din sticlă rodată.
3.8. Balon conic cu vid de 50 ml cu un racord mamă din sticlă rodată, care este adaptabil la pâlnia de filtrare (punctul 3.7).
3.9. Eprubetă cu fund conic de 10 ml, prevăzută cu dop ermetic din sticlă.
3.10. Cromatograf cu gaz care poate fi utilizat cu coloană capilară, prevăzut cu un dispozitiv de injectare cu splitare, format din următoarele:
3.10.1. Etuvă cu termostat pentru coloane, care poate menține temperatura dorită cu o precizie de ± 1 °C;
3.10.2. Unitate de injectare termoreglabilă cu element vaporizator din sticlă tratată cu persilan și cu dispozitiv de splitare;
3.10.3. Detector cu ionizare în flacără (FID);
3.10.4. Sistem de culegere a datelor care poate fi utilizat cu detectorul FID (punctul 3.10.3.) și care poate efectua integrare manuală.
3.11. Coloană capilară de sticlă de siliciu cu o lungime de 20–30 m și un diametru interior de 0,25-0,32 mm, acoperită în interior cu 5 % difenil - 95 % dimetilpolisiloxan (faza staționară SE-52 sau SE-54 sau echivalent), cu o grosime uniformă cuprinsă între 0,10 și 0,30 μm.
3.12. Microseringă cu o capacitate de 10 μl, pentru cromatografie în fază gazoasă, cu ac cimentat, adecvată pentru injectarea cu splitare.
3.13. Exsicator cu diclorură de calciu
4. REACTIVI
4.1. Hidroxid de potasiu, titru minim 85 %.
4.2. Hidroxid de potasiu în soluție etanolică, la aproximativ 2 N.
Se dizolvă, prin răcire, 130 g de hidroxid de potasiu (punctul 4.1) în 200 ml de apă distilată, apoi se completează până la un litru cu etanol (punctul 4.10). Soluția se păstrează în vase de sticlă opacă bine închise și se depozitează maximum două zile.
4.3. Eter etilic, de calitate analitică.
4.4. Hidroxid de potasiu în soluție etanolică, la aproximativ 0,2 N.
Se dizolvă 13 g de hidroxid de potasiu (punctul 4.1) în 20 ml de apă distilată și se completează până la un litru cu etanol (punctul 4.10).
4.5. Sulfat de sodiu anhidru, de calitate analitică.
4.6. Plăci de sticlă (20 × 20 cm) acoperite cu silicagel fără indicator de fluorescență, cu o grosime de 0,25 mm (acestea sunt disponibile în comerț gata pentru utilizare).
4.7. Toluen, de calitate cromatografică.
4.8. Acetonă, de calitate cromatografică.
4.9. n-Hexan, de calitate cromatografică.
4.10. Eter etilic, de calitate cromatografică.
4.11. Etanol, de calitate analitică.
4.12. Acetat de etil, de calitate analitică.
4.13. Soluție de referință pentru cromatografie în strat subțire: colesterol sau fitosteroli și soluție de eritrodiol de 5 % în acetat de etil (punctul 4.11).
4.14. 2′,7′ Diclorfluoresceină, soluție etanolică de 0,2 %. Aceasta este făcută ușor bazică prin adăugarea câtorva picături de soluție alcoolică de hidroxid de potasiu la 2 N (punctul 4.2).
4.15. Piridină anhidră, de calitate cromatografică (a se vedea nota 5).
4.16. Hexametil-disilazan, de calitate analitică.
4.17. Trimetilclorosilan, de calitate analitică.
4.18. Soluție de probă de trimetilsilileter de steroli.
Se prepară în momentul folosirii din steroli și eritrodiol extrași din uleiurile care le conțin.
4.19. α-colestanol, cu puritate de peste 99 % (puritatea trebuie să fie controlată prin analiză GC).
4.20. Soluție etalon intern de α-colestanol, soluție de 0,2 % (m/V) în acetat de etil (punctul 4.11).
4.21. Soluție de fenolftaleină, 10 g/l în etanol (punctul 4.10).
4.22. Gaz purtător: hidrogen sau heliu pur, puritate pentru cromatografie în fază gazoasă.
4.23. Gaze auxiliare: hidrogen, heliu, azot și aer, puritate pentru cromatografie în fază gazoasă.
4.24. n-Hexan (punctul 4.9)/eter etilic (punctul 4.10) amestec de 65:35 (V/V).
4.25. Reactiv de sililare, constituit dintr-un amestec de 9:3:1 (V/V/V) de piridină/hexametil-disilazan/trimetilclorosilan.
5. PROCEDURA
|
5.1. |
Prepararea substanței nesaponificabile
|
|
5.2. |
Separarea fracțiunii de steroli și de dialcooli triterpenici (eritrodiol + uvaol)
|
|
5.3. |
Prepararea trimetilsilileterilor
|
|
5.4. |
Analiza prin cromatografie în faza gazoasă.
|
6. EXPRIMAREA REZULTATELOR
6.1. Se raportează concentrația fiecărui sterol în mg/kg de substanță grasă și suma lor ca „steroli totali”.
Compoziția pentru fiecare sterol și eritrodiol și uvaol se exprimă cu o zecimală.
Compoziția de steroli totali trebuie să fie exprimată fără zecimale.
6.2. Se calculează procentul fiecărui sterol pornind de la raportul dintre aria valorii de vârf corespondente și suma ariilor valorilor de vârf pentru steroli:

unde:
|
Ax |
= |
aria valorii de vârf pentru x; |
|
ΣA |
= |
suma ariilor valorilor de vârf pentru steroli. |
6.3. β-Sitosterol aparent: Δ5-23-stigmastadienol + clerosterol + β-sitosterol + sitostanol + Δ5-avenasterol + Δ5-24-stigmastadienol.
6.4. Se calculează procentul de eritrodiol și uvaol:

unde
|
ΣA |
= |
suma ariei pentru sterol, în unități ale sistemului de culegere a datelor; |
|
ER |
= |
aria pentru eritrodiol, în unități ale sistemului de culegere a datelor; |
|
Uv |
= |
aria pentru uvaol, în unități ale sistemului de culegere a datelor. |
Apendice
Determinarea vitezei liniare a gazului
În cromatograful în fază gazoasă, reglat în condiții de operare normale, se injectează 1–3 μl de metan (sau propan) și se cronometrează timpul în care gazul parcurge coloana, între momentul de injecție și cel al ieșirii valorii de vârf (tM).
Viteza liniară în cm/s este dată de L/tM, unde L este lungimea coloanei în centimetri și tM este timpul cronometrat în secunde.
Tabelul 1
Timpii de retenție relativi ai sterolilor
|
Vârf |
Identificare |
Timpi de retenție relativi |
||
|
SE 54 coloană |
SE 52 coloană |
|||
|
1 |
Colesterol |
Δ-5-cholesten-3ß-ol |
0,67 |
0,63 |
|
2 |
Colestanol |
5α-colestan-3ß-ol |
0,68 |
0,64 |
|
3 |
Brasicasterol |
[24S]-24-metil-Δ-5,22-colestadien-3ß-ol |
0,73 |
0,71 |
|
* |
Ergosterol |
[24S] 24 meti Δ5-7-22 colestatrien 3β-ol |
0,78 |
0,76 |
|
4 |
24-metilen-colesterol |
24-metilen-Δ-5,24-colestadien-3ß-o1 |
0,82 |
0,80 |
|
5 |
Campesterol |
(24R)-24-metil-Δ-5-colesten-3ß-ol |
0,83 |
0,81 |
|
6 |
Campestanol |
(24R)-24-metil-colestan-3ß-ol |
0,85 |
0,82 |
|
7 |
Stigmasterol |
(24S)-24-etill-Δ-5,22-colestadien-3ß-ol |
0,88 |
0,87 |
|
8 |
Δ-7-campesterol |
(24R)-24-metil-Δ-7-colesten-3ß-ol |
0,93 |
0,92 |
|
9 |
Δ-5,23-stigmastadienol |
(24R,S)-24-etil-Δ-5,23-coIestadien-3ß-ol |
0,95 |
0,95 |
|
10 |
Clerosterol |
(24S)-24-etil-Δ-5,25-colestadien-3ß-ol |
0,96 |
0,96 |
|
11 |
ß-sitosterol |
(24R)-24-etil-Δ-5-colesten-3ß-ol |
1,00 |
1,00 |
|
12 |
Sitostanol |
24-etil-colestan-3ß-ol |
1,02 |
1,02 |
|
13 |
Δ-5-avenasterol |
(24Z)-24-etiliden-Δ-colesten-3ß-ol |
1,03 |
1,03 |
|
14 |
Δ-5-24-stigmastadienol |
(24R,S)-24-etil-Δ-5,24-colestadien-3ß-ol |
1,08 |
1,08 |
|
15 |
Δ-7-stigmastenol |
(24R,S)-24-etil-Δ-7-colesten-3ß-ol |
1,12 |
1,12 |
|
16 |
Δ-7-avenasterol |
(24Z)-24-etiliden-Δ-7-colesten-3ß-ol |
1,16 |
1,16 |
|
17 |
Eritrodiol |
5α olean-12en-3β28 diol |
1,41 |
1,41 |
|
18 |
Uvaol |
Δ12-ursen-3β28 diol |
1,52 |
1,52 |
Figura 1
Cromatograma în fază gazoasă a fracțiunii de steroli și de dialcooli triterpenici a unui ulei de măsline lampant (eluat cu etalon intern)

Figura 2
Cromatograma în fază gazoasă a fracțiunii de steroli și de dialcooli triterpenici a unui ulei de măsline rafinat (eluat cu etalon intern)

Figura 3
Placa de cromatografie în strat subțire pentru uleiul din resturi de măsline, cu zona care trebuie să fie curățată pentru determinarea sterolilor și a dialcoolilor triterpenici

1 – Scualen
2 – Alcooli triterpenici și alifatici
3 – Steroli și dialcooli triterpenici
4 – Acizi grași inițiali și acizi grași liberi
▼M26 —————
ANEXA VII
DETERMINAREA PROCENTULUI DE MONOPALMITAT DE 2-GLICERIL
1. OBIECTUL ȘI DOMENIUL DE APLICARE
Prezenta metodă descrie procedura analitică de determinare a procentului de acid palmitic în poziția 2 a trigliceridelor cu ajutorul evaluării monopalmitatului de 2-gliceril.
Prezenta metodă se aplică uleiurilor vegetale lichide la temperatură ambiantă (20 °C).
2. PRINCIPIU
După preparare, mostra de ulei este supusă acțiunii lipazei pancreatice: o hidroliză parțială și specifică în pozițiile 1 și 3 ale moleculei de trigliceridă determină apariția monogliceridelor în poziția 2. Procentul de monopalmitat de 2-gliceril în fracțiunea de monogliceridă este determinat, după sililare, prin cromatografie în fază gazoasă cu coloană capilară.
3. APARATURĂ ȘI MATERIALE
|
3.1. |
Pahar Erlenmeyer de 25 ml. |
|
3.2. |
Pahare de laborator de 100, 250 și 300 ml. |
|
3.3. |
Coloană de sticlă pentru cromatografie, cu diametru interior de 21-23 mm, lungime de 400 mm, echipată cu un disc din sticlă sinterizată și un robinet. |
|
3.4. |
Eprubete gradate de 10, 50, 100 și 200 ml. |
|
3.5. |
Baloane de 100 și 250 ml. |
|
3.6. |
Evaporator rotativ. |
|
3.7. |
Tuburi de centrifugare, cu capăt conic de 10 ml, cu dop din sticlă rodată. |
|
3.8. |
Eprubetă de centrifugare pentru tuburi de 10 și 100 ml. |
|
3.9. |
Termostat pentru menținerea temperaturii la 40 °C ± 0,5 °C. |
|
3.10. |
Pipete gradate de 1 și 2 ml. |
|
3.11. |
Seringă hipodermică de 1 ml. |
|
3.12. |
Microseringă de 100 μl. |
|
3.13. |
Pâlnie, 1 000 ml. |
|
3.14. |
Cromatograf în fază gazoasă pentru coloane capilare, echipat cu un dispozitiv de injecție „on-column” la rece pentru introducerea directă a mostrei în coloană și cu un cuptor capabil să mențină temperatura aleasă în limita a 1 °C. |
|
3.15. |
Injector „on-column” la rece pentru introducerea directă a mostrei în coloană. |
|
3.16. |
Detector cu ionizare în flacără și electrometru. |
|
3.17. |
Aparat de înregistrare-integrator adaptat la electrometru cu un timp de răspuns sub 1 secundă și cu o viteză a hârtiei variabilă. |
|
3.18. |
Coloană capilară de sticlă sau silice topită cu o lungime de 8-12 metri și un diametru interior de la 0,25-0,32 mm, acoperită cu metil-polisiloxan sau 5 % fenil-metil-polisiloxan, într-un strat cu o grosime cuprinsă între 0,10 și 0,30 μm, care poate fi utilizată la 370 °C. |
|
3.19. |
Microseringă de 10 μl echipată cu un ac cementat, cu o lungime de cel puțin 7,5 cm pentru introducerea directă în coloană. |
4. REACTIVI
|
4.1. |
Silicagel cu o granulometrie cuprinsă între 0,063 și 0,200 mm (70/280 Mesh), preparat astfel: se introduce silicagelul într-o capsulă de porțelan, se lasă la uscat în etuvă la 160 °C timp de 4 ore, apoi se lasă la răcit într-un desicator, la temperatura ambiantă. Se adaugă un volum de apă echivalent cu 5 % din greutatea silicagelului, astfel: într-un pahar Erlenmeyer de 500 ml, se cântăresc 152 g de silicagel și se adaugă 8 g de apă distilată, se pune dopul și se agită ușor pentru a obține o repartizare uniformă a apei. Se lasă în repaus cel puțin 12 ore înainte de utilizare. |
|
4.2. |
n-hexan (pentru cromatografie). |
|
4.3. |
Izopropanol. |
|
4.4. |
Izopropanol, soluție apoasă 1/1 (V/V). |
|
4.5. |
Lipază pancreatică. Lipaza utilizată trebuie să aibă o activitate cuprinsă între 2-10 unități de lipază/mg. (Există în comerț lipaze pancreatice cu activitate cuprinsă între 2-10 unități/mg de enzimă). |
|
4.6. |
Soluție tampon de tri-hidroximetilaminometan soluție apoasă 1 M ajustată până la un pH de 8 (control cu potențiometrul) prin adăugarea de acid clorhidric concentrat (1/1 V/V). |
|
4.7. |
Colat de sodiu, calitate enzimatică, soluție apoasă de 0,1 % (această soluție trebuie utilizată în decurs de 15 zile de la preparare). |
|
4.8. |
Clorură de calciu, soluție apoasă de 22 %. |
|
4.9. |
Eter dietilic pentru cromatografie. |
|
4.10. |
Solvent de developare: amestec de n-hexan/eter dietilic (87/13 V/V). |
|
4.11. |
Hidroxid de sodiu, soluție de 12 % din greutate. |
|
4.12. |
Fenolftaleină, soluție de 1 % în etanol. |
|
4.13. |
Gaz transportor: hidrogen sau heliu, pentru cromatografie în fază gazoasă. |
|
4.14. |
Gaze auxiliare: hidrogen cu puritate de minimum 99 %, fără umiditate și impurități organice, și aer de puritate echivalentă, pentru cromatografie în fază gazoasă. |
|
4.15. |
Reactiv silanizant: amestec de piridină/hexametildisilazan, trimetilclorosilan 9/3/1 (V/V/V) (în comerț există soluții gata preparate. Pot fi utilizați alți reactivi silanizanți, cum ar fi bi-trimetilsililtrifluorescentamida + 1 % trimetilclorosilan, diluați cu un volum egal de piridină anhidră). |
|
4.16. |
Mostre de referință: monogliceride pure sau amestecuri de monogliceride cu o compoziție în procent cunoscută apropiată de cea a mostrei. |
5. PROCEDURĂ
5.1. Prepararea mostrei
|
5.1.1. |
În cazul uleiurilor cu o aciditate liberă mai mică de 3 %, nu este necesară neutralizarea acestora înainte de cromatografia în coloană de silicagel. Uleiurile cu o aciditate liberă mai mare de 3 % se neutralizează în conformitate cu indicațiile de la punctul 5.1.1.1.
|
|
5.1.2. |
Se introduce 1 g de ulei preparat conform indicațiilor anterioare într-un pahar Erlenmeyer de 25 ml (3.1.) și se dizolvă în 10 ml de amestec de developare (4.10). Soluția se lasă în repaus timp de cel puțin 15 minute înainte de cromatografia în coloană de silicagel. În cazul în care soluția este tulbure, se centrifughează pentru a asigura condiții optime pentru cromatografie. (Pot fi utilizate cartușe de silicagel SPE de 500 mg, gata pregătite.) |
|
5.1.3. |
Pregătirea coloanei cromatografice Se toarnă în coloană (3.3) aproximativ 30 ml de solvent de developare (4.10), se introduce o bucată de vată în partea de jos a coloanei cu ajutorul unei baghete de sticlă; se exercită presiune pentru a elimina aerul. Într-un pahar de laborator, se prepară o suspensie de 25 g de silicagel (4.1) în aproximativ 80 ml de solvent de developare și se toarnă în coloană cu ajutorul unei pâlnii. Se verifică dacă întreaga cantitate de silicagel a fost introdusă în coloană; se spală cu solvent de developare (4.10), se deschide robinetul și se lasă ca nivelul lichidului să ajungă la circa 2 mm deasupra nivelului superior al silicagelului. |
|
5.1.4. |
Cromatografie în coloană Într-un pahar Erlenmeyer de 25 ml (3.1), se cântărește exact 1 g din mostra preparată în conformitate cu indicațiile de la punctul 5.1. Se dizolvă mostra în 10 ml de solvent de developare (4.10). Soluția se toarnă în coloana cromatografică pregătită în conformitate cu indicațiile de la punctul 5.1.3. Se evită balansarea suprafeței coloanei. Se deschide robinetul și se lasă să se scurgă soluția mostrei până la atingerea nivelului de silicagel. Se developează cu ajutorul a 150 ml de solvent de developare. Se ajustează debitul la 2 ml/min. (astfel încât 150 ml să se scurgă în coloană în circa 60-70 de minute). Se recuperează soluția eluată într-un balon de 250 ml, cântărit în prealabil. Se evaporă solventul sub vid și se înlătură ultimele urme ale acestuia cu ajutorul unui curent de azot. Se cântărește balonul și se calculează materialul extras recuperat. [În cazul utilizării unor cartușe SPE de silice gata preparate, se procedează astfel: se introduce 1 ml de soluție (5.1.2) în cartușele pregătite în prealabil cu 3 ml de n-hexan. După percolarea soluției, se developează cu 4 ml de n-hexan/eter dietilic 9/1 (V/V). Se recuperează soluția eluată într-o eprubetă de 10 ml și se utilizează un curent de azot pentru evaporarea până la uscare. Reziduul uscat este supus acțiunii lipazei pancreatice (5.2). Este esențială verificarea compoziției de acizi grași înainte și după trecerea prin cartușul SPE]. |
5.2. Hidroliză cu lipază pancreatică
|
5.2.1. |
În eprubeta de centrifugare se cântăresc aproximativ 0,1 g de ulei preparat conform indicațiilor de la punctul 5.1. Se adaugă 2 ml de soluție tampon (4.6), 0,5 ml de soluție de colat de sodiu (4.7) și 0,2 ml de soluție de clorură de calciu, agitând bine după fiecare operațiune. Se închide eprubeta cu dopul din sticlă rodată și se pune în termostat la 40 ± 0,5 °C. |
|
5.2.2. |
Se adaugă 20 mg de lipază, se agită cu grijă (se va evita umezirea dopului) și se pune eprubeta în termostat timp de exact 2 minute, apoi se scoate din termostat și se agită energic timp de exact 1 minut, după care se lasă la răcit. |
|
5.2.3. |
Se adaugă 1 ml de eter dietilic, se închide și se agită energic, apoi se centrifughează și se transferă soluția de eter într-o eprubetă curată și uscată cu ajutorul unei microseringi. |
5.3. Prepararea derivaților silanizați și a cromatografiei în fază gazoasă
|
5.3.1. |
Cu ajutorul unei microseringi, se introduc 100 μl de soluție (5.2.3) într-o eprubetă cu fund conic de 10 ml. |
|
5.3.2. |
Se elimină solventul într-un ușor curent de azot, se adaugă 200 μl de reactiv silanizant (4.15), se astupă eprubeta și se lasă în repaus timp de 20 de minute. |
|
5.3.3. |
După 20 de minute se adaugă între 1-5 ml de n-hexan (în funcție de condițiile cromatografice): soluția obținută este gata pentru cromatografia în fază gazoasă. |
5.4. Cromatografia în fază gazoasă
Condițiile de funcționare sunt următoarele:
— temperatura injectorului (injector „on-column”) mai mică decât temperatura de fierbere a solventului (68 °C);
— temperatura detectorului: 350 °C;
— temperatura coloanei: programarea temperaturii cuptorului: 60 °C timp de 1 minut, crescând cu 15 °C pe minut până la 180 °C, apoi cu 5 °C pe minut până la 340 °C, apoi 340 °C timp de 13 minute;
— gaz transportor: hidrogen sau heliu, reglat la viteza lineară adecvată pentru a obține rezoluția indicată în figura 1. Timpul de retenție a trigliceridei C54 trebuie să fie de 40 ± 5 minute (a se vedea figura 2); (Condițiile de funcționare descrise anterior sunt propuse cu titlu indicativ. Fiecare operator va trebui să le optimizeze pentru a obține rezoluția dorită. Valoarea de vârf a monopalmitatului de 2-gliceril trebuie să atingă un nivel minim de 10 % din scara aparatului de înregistrare.);
— cantitatea de substanță injectată: 0,5-1 μl de soluție (5 ml) de n-hexan (5.3.3).
5.4.1. Identificarea valorilor de vârf
Monogliceridele individuale sunt identificate în funcție de timpii de retenție obținuți și în raport cu cei obținuți prin amestecurile standard de mopogliceride analizate în aceleași condiții.
5.4.2. Evaluare cantitativă
Aria fiecărei valori de vârf se calculează cu ajutorul unui integrator electronic.
6. EXPRIMAREA REZULTATELOR
Procentul de monopalmitat de gliceril se calculează pe baza raportului dintre aria valorii de vârf corespunzătoare și suma ariilor valorilor de vârf ale tuturor monogliceridelor (a se vedea figura 2), după formula:
Monopalmitat de gliceril(%):
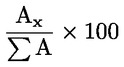
unde:
|
Ax |
= |
aria valorii de vârf corespunzătoare monopalmitatului de gliceril; |
|
ΣA |
= |
suma ariilor tuturor valorilor de vârf ale monogliceridelor. |
Rezultatul trebuie exprimat cu o zecimală.
7. RAPORTUL ANALIZEI
Raportul analizei va trebui să precizeze:
— trimiterea la această metodă;
— orice informație necesară identificării integrale a mostrei;
— rezultatul analizei;
— orice abatere de la această metodă, fie că este vorba de o decizie a părților implicate, fie dintr-un alt motiv;
— detaliile de identificare a laboratorului, data efectuării analizei și semnătura persoanelor responsabile de aceasta.
Figura 1
Cromatogramă a produșilor de silanizare, obținuți prin acțiunea lipazei asupra unui ulei de măsline rafinat cu adaos de 20 % ulei esterificat (100 %)

[Legendă: „acides gras libres” = acizi grași liberi; „Huile d'olive raffinée + 20 % huile estérifiée” = ulei de măsline rafinat + 20 % ulei esterificat; „1-2 monopalmitoléine” = 1-2 monopalmitolein; „1-2 mono C18 insat” = unsaturated 1-2 mono C18]
Figura 2
Cromatogramă a unui:
|
A. |
ulei de măsline neesterificat, după lipază; după silanizare; în aceste condiții (coloană capilară 8-12 mm), fracțiunea de ceară se eluează în același timp cu fracțiunea de digliceridă sau la puțin timp după aceasta. După lipază, conținutul de trigliceride nu ar trebui să depășească 15 %. 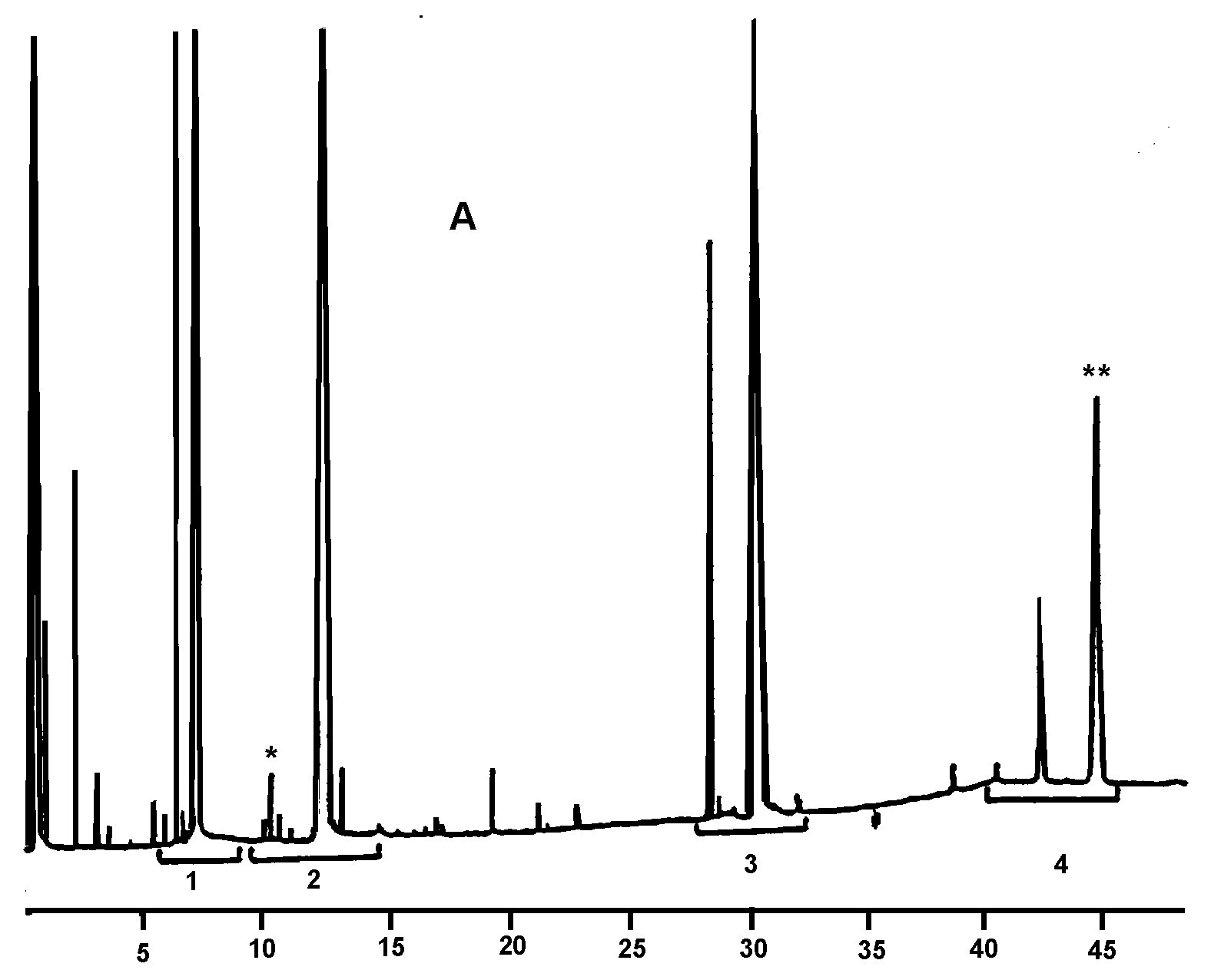
Legendă:
|
Cromatogramă a unui:
|
B. |
ulei esterificat după lipază; după silanizare; în aceste condiții (coloană capilară 8-12 mm), fracțiunea de ceară se eluează în același timp cu fracțiunea de digliceridă sau la puțin timp după aceasta. După lipază, conținutul de trigliceride nu ar trebui să depășească 15 %. 
Legendă:
|
8. NOTE
PREPARAREA LIPAZEI
Lipaze cu un nivel de activitate satisfăcător sunt disponibile în comerț. Acestea pot fi, de asemenea, preparate în laborator după cum urmează:
Se răcesc 5 kg de pancreas proaspăt de porc la 0 °C. Se elimină grăsimea solidă și țesutul conjunctiv din jur, apoi se triturează într-un malaxor până la obținerea unei paste fluide. Se agită această pastă într-un agitator cu 2,5 litri de acetonă anhidră timp de 4-6 ore, apoi se centrifughează. Se efectuează alte trei extracții de reziduu cu același volum de acetonă anhidră, apoi două extracții cu un amestec 1/1 (V/V) de acetonă și de eter dietilic și două extracții cu eter dietilic.
Se usucă reziduul sub vid timp de 48 ore pentru a obține o pudră stabilă, care se păstrează timp îndelungat în frigider și ferită de umiditate.
CONTROLUL ACTIVITĂȚII LIPAZICE
Se prepară o emulsie de ulei de măsline astfel:
Se agită timp de 10 minute într-un agitator adecvat un amestec compus din 165 ml de soluție de gumă arabică de 100 g/l, din 15 g de gheață pisată și 20 ml dintr-un ulei neutralizat în prealabil.
Într-un pahar de laborator de 50 ml se toarnă succesiv câte 10 ml din această emulsie, apoi 0,3 ml de soluție de colat de sodiu de 0,2 g/ml și 20 ml de apă distilată.
Se plasează paharul de laborator într-un termostat menținut la 37 °C; se introduc electrozii unui pH-metru și un agitator cu spirală.
Cu ajutorul unei biurete, se adaugă soluția de hidroxid de sodiu 0,1 N picătură cu picătură, până când pH-ul atinge valoarea de 8,3.
Se adaugă o cantitate suficientă de suspensie apoasă de lipază (0,1 g/ml de lipază). Din momentul în care pH-metrul indică un pH de 8,3, se declanșează cronometrul și se începe adăugarea soluției de hidroxid de sodiu picătură cu picătură cu viteza necesară pentru a menține constant pH-ul de 8,3. Se notează la fiecare minut volumul de soluție consumat.
Se raportează observațiile pe o diagramă ce indică pe abscisă timpii și pe ordonată mililitrii de soluție alcalină de 0,1 N consumați pentru menținerea pH-ului constant. Rezultatul obținut trebuie să fie un grafic liniar.
Activitatea lipazei, exprimată în unități lipazice pe mg, este calculată cu ajutorul formulei următoare:
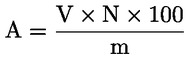
unde:
|
A |
reprezintă activitatea în unități lipazice/mg; |
|
V |
reprezintă numărul de mililitri de soluție de hidroxid de sodiu 0,1 N pe minut (calculat pe baza graficului); |
|
N |
reprezintă valoarea normală a soluției de hidroxid de sodiu; |
|
m |
reprezintă masa în mg a mostrei de lipază. |
Unitatea lipazică se definește ca fiind cantitatea de enzimă care produce 10 microechivalenți de acid pe minut.
▼M20 —————
ANEXA IX
ANALIZĂ SPECTROFOTOMETRICĂ ÎN ULTRAVIOLET
INTRODUCERE
Examinarea spectrofotometrică în ultraviolet poate furniza indicații privind calitatea unei materii grase, starea de conservare a acesteia și modificările datorate proceselor tehnologice. Absorbțiile cu lungimi de undă prevăzute în metodă sunt datorate prezenței sistemelor de diene și triene conjugate rezultând din procese de oxidare și/sau din practici de rafinare. Valorile acestor absorbții sunt exprimate ca extincția specifică
![]()
(extincția unei soluții de substanță grasă de 1 % greutate volumetrică în solventul prescris, într-o cuvă de 10 mm) notată în mod convențional cu K (denumit, de asemenea, „coeficient de extincție”).
1. DOMENIUL DE APLICARE
Prezenta anexă descrie procedeul executării examinării spectrofotometrice în ultraviolet a uleiurilor de măsline.
2. PRINCIPIUL METODEI
Mostra se dizolvă în solventul necesar, apoi se măsoară absorbanța soluției la lungimile de undă prevăzute, în raport cu solventul pur.
Extincțiile specifice la 232 nm și 268 nm în izooctan sau la 232 nm și 270 nm în ciclohexan se calculează pentru o concentrație de 1 % greutate volumetrică într-o cuvă de 10 mm.
3. APARATURĂ
|
3.1. |
Un spectofotometru adecvat pentru măsurători la lungimi de undă ultravioletă (între 220 nm și 360 nm), cu posibilitate de citire pentru fiecare unitate nanometrică. Se recomandă o verificare regulată a preciziei și reproductibilității scărilor de absorbanță și de lungime de undă, precum și a luminii difuze.
|
|
3.2. |
Cuve de cuarț dreptunghiulare, cu capac, adecvate pentru măsurători la lungimi de undă ultravioletă (între 220 și 360 nm) cu o lungime a parcursului optic de 10 mm. Cuvele, umplute cu apă sau cu alt solvent adecvat, nu trebuie să prezinte diferențe între ele mai mari de 0,01 unități de extincție. |
|
3.3. |
Baloane volumetrice gradate, cu o capacitate de 25 ml, clasa A. |
|
3.4. |
Balanță analitică, ce să poată fi citită cu o precizie de 0,0001 g. |
4. REACTIVI
Pe durata analizei, dacă nu se specifică altfel, se utilizează numai reactivi de puritate analitică recunoscută și apă distilată sau demineralizată sau apă de puritate echivalentă.
Solventul: Izooctan (2,2,4-trimetilpentan) pentru măsurătorile la 232 nm și 268 nm și ciclohexan pentru măsurători la 232 nm și 270 nm, având o absorbanță mai mică de 0,12 la 232 nm și mai mică de 0,05 la 270 nm în raport cu apa distilată, măsurată într-o cuvă de 10 mm.
5. MOD DE LUCRU
|
5.1. |
Proba trebuie să fie perfect omogenă și lipsită de impurități în suspensie. În caz contrar, aceasta trebuie să fie filtrată printr-un filtru de hârtie la o temperatură de aproximativ 30 °C. |
|
5.2. |
Se cântăresc aproximativ 0,25 g (cu o precizie de 1 mg) din proba astfel preparată într-un balon gradat de 25 ml, se completează până la gradație cu solventul prescris și se omogenizează. Soluția obținută trebuie să fie perfect limpede. În cazul în care soluția ar prezenta opalescență sau turbiditate, se filtrează rapid printr-un filtru de hârtie. NOTĂ: în general, o masă de 0,25-0,30 g este suficientă pentru măsurarea absorbanței uleiului de măsline virgin și extravirgin la 268 nm și la 270 nm. Pentru măsurători la 232 nm, este necesară o probă de 0,05 g, fiind preparate de obicei două soluții diferite. Pentru măsurători ale absorbanței uleiurilor din resturi de măsline, a uleiurilor rafinate de măsline și a uleiurilor modificate de măsline, este de obicei necesară o proporție mai redusă din probă, de exemplu 0,1 g, ca urmare a absorbanței mai ridicate. |
|
5.3. |
Dacă este necesar, se aplică o corecție a referinței (între 220 și 290 nm) cu solvent din ambele cuve de cuarț (probă și referință), apoi se umple cuva de cuarț a probei cu soluția de test și se măsoară extincțiile la 232, 268 sau 270 nm în raport cu solventul utilizat ca referință. Valorile de extincție înregistrate trebuie să fie cuprinse în intervalul 0,1-0,8 sau în intervalul liniarității specrofotometrului care ar trebui verificat. În caz contrar, este necesară repetarea măsurătorilor folosindu-se soluții mai concentrate sau mai diluate, după caz. |
|
5.4. |
După măsurarea absorbanței la 268 sau 270 nm, se măsoară absorbanța la λmax, λmax + 4 și λmax – 4. Aceste valori ale absorbanței sunt utilizate pentru a determina variația extincției specifice (ΔΚ). NOTĂ: se consideră că λmax este 268 nm pentru izooctanul utilizat ca solvent și 270 nm pentru ciclohexan. |
6. EXPRIMAREA REZULTATELOR
|
6.1. |
Se înregistrează extincțiile specifice (coeficienții de extincție) la diferitele lungimi de undă, calculate după cum urmează:
unde:
Rezultatele sunt exprimate cu două decimale. |
|
6.2. |
Variația extincției specifice (ΔΚ) Variația valorii absolute a extincției (ΔΚ) este dată de:
unde Km reprezintă extincția specifică la lungimea de undă corespunzătoare absorbției maxime de 270 nm pentru ciclohexan și de 268 nm, în funcție de solventul utilizat. Rezultatele sunt exprimate cu două decimale. |
ANEXA X
DETERMINAREA ESTERILOR METILICI AI ACIZILOR GRAȘI PRIN CROMATOGRAFIE ÎN FAZĂ GAZOASĂ
1. DOMENIUL DE APLICARE
Prezenta anexă oferă orientări privind determinarea prin cromatografie în fază gazoasă a acizilor grași liberi și legați din grăsimile și uleiurile vegetale în urma conversiei lor în esteri metilici ai acizilor grași (EMAG).
Acizii grași legați ai triacilglicerolilor (TAG) și, în funcție de metoda de esterificare, acizii grași liberi sunt convertiți în esteri metilici ai acizilor grași (EMAG), care sunt determinați prin cromatografie în fază gazoasă cu coloană capilară.
Metoda descrisă în prezenta anexă permite determinarea esterilor metilici ai acizilor grași de la C12 la C24, inclusiv a esterilor metilici ai acizilor grași saturați, cis și trans mononesaturați și cis și trans polinesaturați.
2. PRINCIPIU
Cromatografia în fază gazoasă este utilizată pentru analiza cantitativă a esterilor metilici ai acizilor grași. Esterii metilici ai acizilor grași se prepară în conformitate cu partea A. Aceștia sunt injectați și vaporizați în injector. Separarea esterilor metilici ai acizilor grași se realizează pe coloane analitice cu polaritate și lungime specifice. Un detector cu ionizare în flacără (FID) este utilizat pentru detectarea esterilor metilici ai acizilor grași. Condițiile pentru analiză sunt indicate în partea B.
Hidrogenul sau heliul se poate utiliza ca gaz purtător (faza mobilă) în cromatografia în fază gazoasă a esterilor metilici ai acizilor grași cu detector cu ionizare în flacără. Hidrogenul accelerează separarea și oferă vârfuri mai clare. Faza staționară este un strat microscopic al unei pelicule subțiri de lichid pe o suprafață solidă inertă din silice topită.
Pe măsură ce trec prin coloana capilară, compușii volatilizați analizați interacționează cu filmul fazei staționare de pe suprafața interioară a coloanei. Ca urmare a acestei interacțiuni între diferiți compuși, ei eluează la momente diferite, un astfel de moment purtând denumirea de timp de retenție a compusului pentru un set dat de parametri de analiză. Comparația timpilor de retenție este utilizată pentru identificarea diferiților compuși.
PARTEA A
PREPARAREA ESTERILOR METILICI AI ACIZILOR GRAȘI DIN ULEIUL DE MĂSLINE ȘI DIN ULEIUL DIN RESTURI DE MĂSLINE
1. DOMENIUL DE APLICARE
Această parte descrie prepararea esterilor metilici ai acizilor grași. Ea include metode pentru prepararea esterilor metilici ai acizilor grași din uleiurile de măsline și din uleiurile din resturi de măsline.
2. SFERA DE APLICARE
Prepararea esterilor metilici ai acizilor grași din uleiurile de măsline și din uleiurile din resturi de măsline se realizează prin transesterificarea cu soluție metanolică de hidroxid de potasiu la temperatura camerei. Necesitatea de purificare a probei anterior transesterificării depinde de conținutul de acizi grași liberi al probei și de parametrul analitic care trebuie determinat; se poate alege în funcție de tabelul următor:
|
Categoria uleiului |
Metoda |
|
Ulei de măsline virgin cu o aciditate ≤ 2,0 % |
1. Acizi grași 2. Acizi grași trans 3. ΔECN42 (după purificare cu silicagel SPE) |
|
Ulei de măsline rafinat |
|
|
Ulei de măsline compus din uleiuri de măsline rafinate și uleiuri de măsline virgine |
|
|
Ulei rafinat din resturi de măsline |
|
|
Ulei din resturi de măsline |
|
|
Ulei de măsline virgin cu o aciditate > 2,0 % Ulei brut din resturi de măsline |
1. Acizi grași (după purificare cu silicagel SPE) 2. Acizi grași trans (după purificare cu silicagel SPE) 3. ΔECN42 (după purificare cu silicagel SPE) |
3. METODOLOGIE
3.1. Transesterificarea cu soluție metanolică de hidroxid de potasiu la temperatura camerei.
3.1.1. Principiu
Esterii metilici se formează prin transesterificare cu hidroxid de potasiu metanolic ca o etapă intermediară înainte de a avea loc saponificarea.
3.1.2. Reactivi
|
3.1.2.1. |
Metanol cu un conținut maxim de apă de 0,5 % (m/m). |
|
3.1.2.2. |
Hexan, de calitate cromatografică. |
|
3.1.2.3. |
Heptan, de calitate cromatografică. |
|
3.1.2.4. |
Dietil eter, stabilizat pentru analiză. |
|
3.1.2.5. |
Acetonă, de calitate cromatografică. |
|
3.1.2.6. |
Solvent de eluare pentru purificarea uleiului prin cromatografie în coloană/extracție în fază solidă (SPE), amestec hexan/dietil eter 87/13 (v/v). |
|
3.1.2.7. |
Hidroxid de potasiu, soluție metanolică de aproximativ 2M: se dizolvă 11,2 g de hidroxid de potasiu în 100 ml de metanol. |
|
3.1.2.8. |
Cartușe de silicagel, 1 g (6 ml), pentru extracție în fază solidă. |
3.1.3. Aparatură
|
3.1.3.1. |
Eprubete cu dop filetat (5 ml volum), cu capac prevăzut cu o îmbinare cu PTFE. |
|
3.1.3.2. |
Pipete gradate sau automate, 2 ml și 0,2 ml. |
3.1.4. Purificarea probelor de ulei
Când este necesar, probele vor fi purificate prin trecerea uleiului printr-un cartuș de silicagel pentru extracție în fază solidă. Un cartuș de silicagel (3.1.2.8) este plasat într-un aparat de eluare sub vid și se spală cu 6 ml de hexan (3.1.2.2); spălarea se realizează fără vid. Apoi se încărcă pe coloană o soluție de ulei (aproximativ 0,12 g) în 0,5 ml de hexan (3.1.2.2). Soluția este trasă în jos și eluată apoi cu 10 ml de hexan/dietil eter (87:13 v/v) (3.1.2.6). Eluatele combinate sunt omogenizate și împărțite în două volume similare. O parte alicotă se evaporă până la uscare într-un evaporator rotativ la presiune scăzută și la temperatura camerei. Se dizolvă reziduul în 1 ml de heptan și soluția este pregătită pentru analiza acizilor grași prin cromatografie în fază gazoasă. A doua parte alicotă se evaporă, iar reziduul se dizolvă în 1 ml de acetonă pentru analiza trigliceridelor prin HPLC, dacă este necesar.
3.1.5. Procedură
Într-o eprubetă cu dop filetat de 5 ml (3.1.3.1), se cântărește o probă de ulei de aproximativ 0,1 g. Se adaugă 2 ml de heptan (3.1.2.2) și se agită. Se adaugă 0,2 ml din soluția de hidroxid de potasiu metanolic (3.1.2.7), se așază capacul prevăzut cu o îmbinare cu PTFE, se strânge capacul și se agită energic timp de 30 de secunde. Se lasă să se stratifice până când devine limpede soluția de deasupra. Se decantează stratul superior care conține esterii metilici. Soluția de heptan este pregătită pentru injectare în cromatograful în fază gazoasă. Se recomandă ca soluția să fie păstrată la frigider până la analiza cromatografică în fază gazoasă. Nu se recomandă depozitarea soluției pentru o perioadă mai mare de 12 ore.
PARTEA B
ANALIZA ESTERILOR METILICI AI ACIZILOR GRAȘI PRIN CROMATOGRAFIE ÎN FAZĂ GAZOASĂ
1. DOMENIUL DE APLICARE
Această parte oferă orientări generale pentru aplicarea cromatografiei în fază gazoasă cu coloană capilară pentru a se determina compoziția calitativă și cantitativă a unui amestec de esteri metilici ai acizilor grași obținut conform metodei specificate în partea A.
Această parte nu este aplicabilă acizilor grași polimerizați.
2. REACTIVI
2.1. Gaz purtător
Gaz inert (heliu sau hidrogen), complet uscat și cu un conținut de oxigen mai mic 10 mg/kg.
Nota 1: Hidrogenul poate dubla viteza de analiză, însă este periculos. Există dispozitive de securitate.
2.2. Gaze auxiliare
|
2.2.1. |
Hidrogen (cu puritate ≥ 99,9 %) care nu conține impurități organice. |
|
2.2.2. |
Aer sau oxigen care nu conține impurități organice. |
|
2.2.3. |
Azot (cu puritate > 99 %). |
2.3. Produse de etalonare
Amestec de esteri metilici ai acizilor grași puri sau de esteri metilici ai unei substanțe grase cu compoziție cunoscută, de preferat similară cu cea a substanței grase de analizat. Izomerii cis și trans ai esterilor metilici octadecenoici, octadecadienoici și octadecatrienoici sunt utili pentru identificarea izomerilor trans ai acizilor nesaturați.
Ar trebui să se facă eforturi pentru a se preveni oxidarea acizilor grași polinesaturați.
3. APARATURĂ
Instrucțiunile se referă la aparatele obișnuite de cromatografie în fază gazoasă care utilizează coloane capilare și un detector cu ionizare prin flacără.
3.1. Cromatograful în fază gazoasă
Cromatograful în fază gazoasă trebuie să cuprindă următoarele elemente.
3.1.1. Dispozitiv de injecție
Se utilizează un dispozitiv de injecție cu coloane capilare, caz în care dispozitivul de injecție trebuie să fie conceput special pentru utilizarea cu astfel de coloane. Poate fi cu divizare sau fără, cu injector pe coloană rece.
3.1.2. Cuptor
Cuptorul trebuie să fie în măsură să aducă coloana capilară la o temperatură de minimum 260 °C și să mențină temperatura dorită în limita a 0,1 °C. Această ultimă cerință este importantă îndeosebi atunci când se utilizează un tub de silice topită.
Utilizarea unui aparat de încălzit echipat cu un programator de temperatură este recomandată în toate cazurile, în special în prezența acizilor grași cu mai puțin de 16 atomi de carbon.
3.1.3. Coloană capilară
|
3.1.3.1. |
Tub din material inert față de substanțele de analizat (în general de sticlă sau de silice topită). Diametrul intern trebuie să fie cuprins între 0,20 și 0,32 mm. În interior, trebuie să suporte tratamente adecvate (prepararea stării de suprafață, inactivarea) înainte de a primi filmul fazei staționare. O lungime de 60 m este suficientă pentru acizii grași și pentru izomerii cis și trans ai acizilor grași. |
|
3.1.3.2. |
Fază staționară, coloanele de tip polisiloxani polari (cianopropil-silicon) grefate (reticulate) sunt adecvate. Nota 2: Polisiloxanii polari riscă să creeze dificultăți în identificarea și separarea acidului linolenic și a acizilor C20. Grosimile filmului trebuie să fie reduse: 0,1-0,2 μm. |
|
3.1.3.3. |
Montajul și condiționarea coloanei Se respectă precauțiile obișnuite de montare a coloanelor capilare, adică dispunerea coloanei în cuptor (suport), alegerea și montarea îmbinărilor (etanșeitate), poziționarea extremităților coloanei în injector și detectorul (reducerea volumelor moarte). Se pune coloana sub un flux de gaz purtător [de exemplu, 0,3 bar (30 kPa) pentru o coloană de 25 de metri lungime și cu un diametru interior de 0,3 mm]. Se condiționează coloana prin programarea temperaturii cuptorului la 3 °C/minut de la temperatura ambiantă până la o temperatură cu 10 °C sub limita de descompunere a fazei staționare. Se menține cuptorul la această temperatură timp de o oră, până la stabilizarea liniei de bază. Se revine la 180 °C pentru a se lucra în condiții izoterme. Nota 3: În comerț sunt disponibile coloane precondiționate adecvate. |
3.1.4. Detector cu ionizare în flacără și convertor-amplificator
3.2. Seringa
Seringa trebuie să aibă o capacitate de maximum 10 μl și să fie gradată la 0,1 μl.
3.3. Sistem de culegere a datelor
Sistemul de culegere a datelor, conectat online cu detectoare și utilizat cu un program software adecvat pentru integrarea și normalizarea vârfurilor.
4. MOD DE LUCRU
Operațiunile descrise la punctele 4.1-4.3 vizează utilizarea unui detector cu ionizare în flacără.
4.1. Condiții de testare
4.1.1. Alegerea condițiilor optime de operare pentru coloane capilare
Datorită eficienței și permeabilității coloanelor capilare, separarea constituenților și durata analizei sunt dependente în mare măsură de debitul gazului purtător în coloană. Prin urmare, va fi necesar să se optimizeze condițiile de operare prin ajustarea acestui parametru (sau pur și simplu pierderea încărcăturii din capul coloanei), în funcție de ceea ce se dorește, adică îmbunătățirea separărilor sau realizarea rapidă a analizei.
Următoarele condiții s-au dovedit adecvate pentru separarea esterilor metilici ai acizilor grași (C4-C26). Exemple de cromatograme sunt prezentate în apendicele B:
|
Temperatura injectorului: |
250 °C |
|
Temperatura detectorului: |
250 °C |
|
Temperatura cuptorului: |
165 °C (8 min) la 210 °C la 2 °C/min |
|
Gaz purtător: hidrogen: |
presiune în capul coloanei: 179 kPa |
|
Flux total: |
154,0 ml/min; |
|
Raportul de splitare: |
1:100 |
|
Volumul de injectare: |
1 μl |
4.1.2. Determinarea rezoluției (a se vedea apendicele A)
Se calculează rezoluția, R, a două vârfuri vecine I și II, cu ajutorul formulei:
R = 2 × [(dr(II) – d r(I))/(ω(I) + ω(II))] or R = 2 × [(tr(II) – t r(I))/(ω(I) + ω(II))] (USP) (United States Pharmacopeia),
sau
R = 1,18 × [(tr(II) – tr(I))/(ω0,5(I) + ω0,5(II))] (EP, BP, JP, DAB) [JP (Japanese Pharmacopeia), EP (Pharmacopée Européenne), BP (British Pharmacopeia)],
unde:
d r(I) este distanța de retenție a vârfului I;
d r(II) este distanța de retenție a vârfului II;
t r(I) este timpul de retenție al vârfului;
t r(II) este timpul de retenție al vârfului II;
ω(I) este lățimea bazei vârfului I;
ω(II) este lățimea bazei vârfului II;
ω0,5 este lățimea vârfului compusului specificat, la jumătatea înălțimii vârfului;
Dacă ω(I) ≈ ω(II), R se calculează cu ajutorul următoarelor formule:
R = (dr(II) – d r(I))/ω = (dr(II) – d r(I))/4σ
unde:
σ este deviația standard (a se vedea apendicele A, figura 1).
Dacă distanța dr dintre cele două vârfuri d r(II) – d r(I) este egală cu 4σ, factorul de rezoluție R = 1.
Dacă două vârfuri nu sunt separate complet, tangentele la punctele de inflexiune ale celor două vârfuri se intersectează în punctul C. Pentru a se separa complet cele două vârfuri, distanța dintre cele două vârfuri trebuie să fie egală cu:
d r(II) – d r(I) = 6 σ de unde R = 1,5 (a se vedea apendicele A, figura 3).
5. EXPRIMAREA REZULTATELOR
5.1. Analiza cantitativă
Se identifică vârfurile esterului metilic al probei din cromatograma din apendicele B figura 1, dacă este necesar prin interpolare, sau prin comparație cu cele ale amestecurilor etalon de esteri metilici (în conformitate cu punctul 2.3).
5.2. Analiza cantitativă
5.2.1. Determinarea compoziției
Se calculează fracțiunea de masă w i a esterilor metilici ai acizilor grași individuali, exprimată în procentaj de masă de esteri metilici, după cum urmează:
5.2.2. Mod de calcul
5.2.2.1. Caz general
Se calculează conținutul unui constituent dat i, exprimat în procentaj de masă de esteri metilici, prin determinarea procentajului reprezentat de aria vârfului corespunzător în raport cu suma ariilor tuturor vârfurilor, cu ajutorul următoarei formule:
wi = (Ai/ΣA) × 100,
unde:
Ai este aria valorii de vârf a esterilor metilici ai acizilor grași individuali i;
ΣA este suma ariilor totalității valorilor de vârf ale tuturor esterilor metilici ai acizilor grași individuali.
Rezultatele sunt exprimate cu două decimale.
Nota 4: Pentru grăsimi și uleiuri, fracția masică a esterilor metilici ai acizilor grași este egală cu fracția masică a triacilglicerolilor în grame per 100 g. Pentru cazurile în care această ipoteză nu este permisă, a se vedea 5.2.2.2.
5.2.2.2. Utilizarea factorilor de corecție
În anumite cazuri, de exemplu în prezența acizilor grași cu mai puțin de opt atomi de carbon sau a acizilor cu funcții secundare, ariile se corectează cu factori de corecție specifici (Fci). Acești factori se determină pentru fiecare instrument. În acest scop, se utilizează materiale de referință adecvate, cu compoziție de acizi grași certificată în intervalul corespunzător.
Nota 5: Acești factori de corecție nu sunt identici cu factorii de corecție teoretici FID, care sunt specificați în apendicele A, întrucât includ, de asemenea, performanța sistemului de injecție etc. Cu toate acestea, în cazul unor diferențe mai mari, întregul sistem ar trebui verificat din punctul de vedere al performanței.
Pentru acest amestec etalon, procentajul în masă al esterilor metilici ai acizilor grași i este dat de formula:
wi = (mi /Σm) × 100,
unde
m i este masa esterilor metilici ai acizilor grași i în amestecul etalon;
Σm este suma maselor diverșilor constituenți, precum esterii metilici ai acizilor grași, ai amestecului etalon.
Pornind de la cromatograma amestecului etalon, se calculează procentajul ca arie al esterilor metilici ai acizilor grași i, astfel:
wi = (Ai/ΣA) × 100,
unde:
Ai este aria esterilor metilici ai acizilor grași i în amestecul etalon;
ΣA este suma tuturor ariilor ale tuturor esterilor metilici ai acizilor grași din amestecul etalon.
Factorul de corecție Fc este deci:
Fc = (mi × ΣA)/(Ai/Σm).
Pentru probă, procentajul ca masă al fiecărui ester metilic al acizilor grași i este:
wi = (Fi × Ai)/Σ (Fi × Ai).
Rezultatele sunt exprimate cu două decimale.
Nota 6: Valoarea calculată corespunde procentajului în masă al acidului gras individual calculat ca triacilgliceroli per 100 g de substanță grasă.
5.2.2.3. Utilizarea unui etalon intern
În anumite analize (de exemplu, când nu sunt cuantificați toți acizii grași, precum atunci când acizii în C4 și în C6 sunt prezenți alături de acizii în C16 și C18, sau când este necesară determinarea cantității absolute de acizi grași dintr-o probă), este necesară utilizarea unui etalon intern. Acizii grași în C5, C15 sau C17 sunt utilizați în mod frecvent. Se determină factorul de corecție al etalonului intern (dacă este necesar).
Procentajul în masă al compusului i, exprimat în esteri metilici, este dat în continuare de formula:
wi = (mIS × Fi × Ai)/(m × FIS × AIS),
unde:
A i este aria esterilor metilici ai acizilor grași i;
A IS este aria etalonului intern;
F i este factorul de corecție al acidului gras i, exprimat ca EMAG;
F IS este factorul de corecție al etalonului intern;
m este masa probei de testare, în miligrame
m IS este masa etalonului intern, în miligrame.
Rezultatele sunt exprimate cu două decimale.
6. PROCESUL-VERBAL DE TESTARE
Procesul-verbal de testare trebuie să specifice metodele utilizate pentru prepararea esterilor metilici și pentru analiza prin cromatografie în fază gazoasă. Acesta trebuie să menționeze, de asemenea, toate detaliile de operare nespecificate în prezenta metodă standard sau considerate opționale, împreună cu detalii privind eventualele incidente care ar fi putut influența rezultatele.
Procesul-verbal de testare trebuie să includă toate informațiile necesare pentru identificarea completă a probei.
7. PRECIZIA
7.1. Rezultatele testului între laboratoare
Detaliile unui test între laboratoare privind precizia metodei sunt prezentate în anexa C la standardul IOC/T.20/Doc. nr. 33. Valorile derivate din acest test între laboratoare pot să nu fie aplicabile intervalelor de concentrație și matricelor, altele decât cele prezentate.
7.2. Repetabilitatea
Diferența absolută dintre două rezultate independente ale unui singur test, obținute prin aceeași metodă pe o substanță de testare identică, în același laborator, de către același operator, folosind același echipament la un interval scurt de timp, va fi mai mare în cel mult 5 % dintre cazuri decât valoarea R indicată în tabelele 1-14 din anexa C la standardul IOC/T.20/Doc. nr. 33.
7.3. Reproductibilitatea
Diferența absolută dintre două rezultate ale unui singur test, obținute prin aceeași metodă pe o substanță de testare identică, în laboratoare diferite, cu operatori diferiți, folosind echipamente diferite, va fi mai mare în cel mult 5 % dintre cazuri decât valoarea R indicată în tabelele 1-14 din anexa C la standardul IOC/T.20/Doc. nr. 33.
Apendicele A
Figura 1
ω0,5 reprezintă lățimea la jumătatea înălțimii triunghiului (ABC) și b reprezintă lățimea la jumătatea înălțimii triunghiului (NPM).
|
Figura 2 |
Figura 3 |
|
|
|
Apendicele B
Figura 1
Profilul cromatografic în fază gazoasă obținut prin metoda de metilare la rece din ulei din resturi de măsline
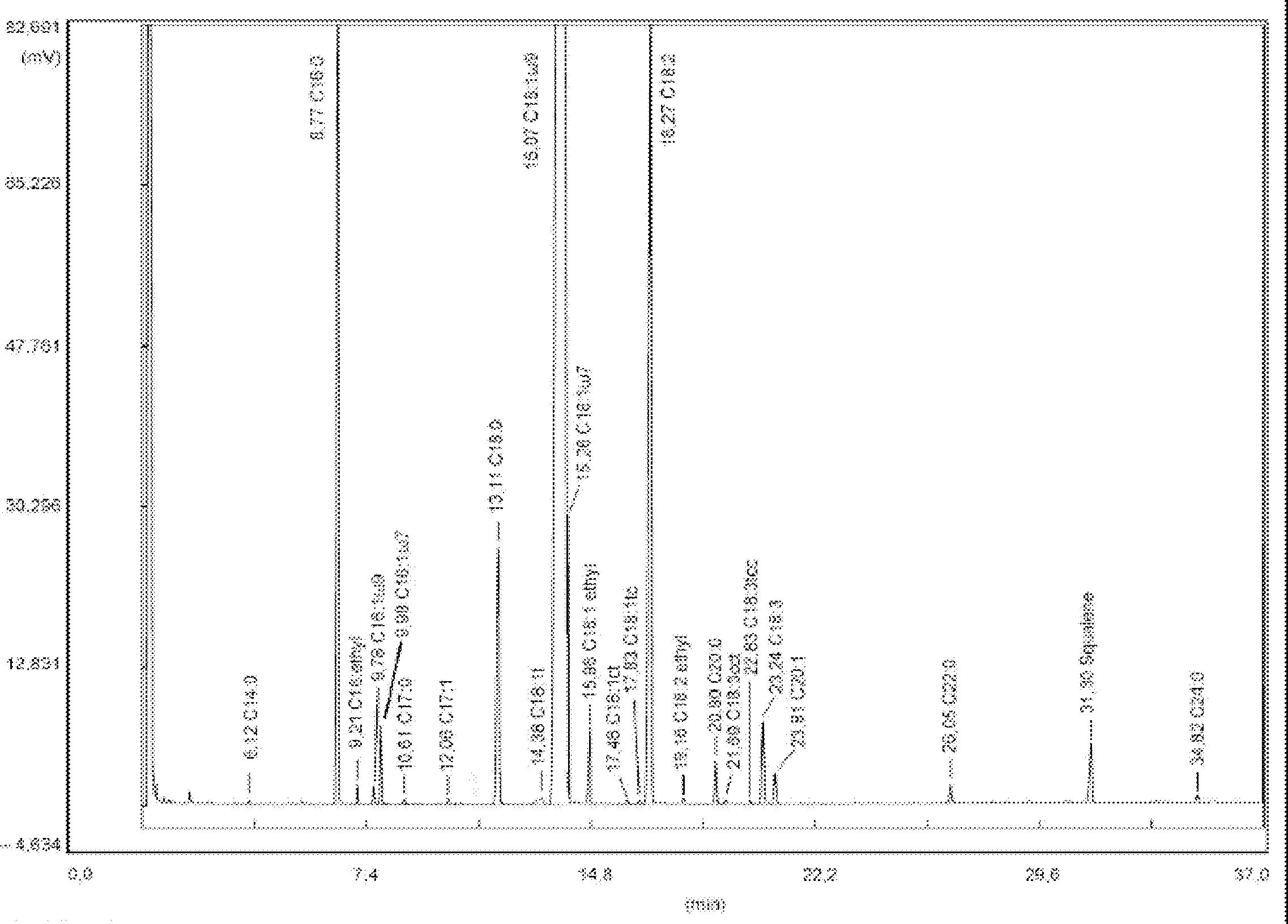
Vârfurile cromatografice corespund esterilor metilici și etilici, cu excepția cazurilor în care există indicații contrare.
ANEXA XI
DETERMINAREA CONȚINUTULUI DE SOLVENȚI HALOGENAȚI VOLATILI ÎN ULEIUL DE MĂSLINE
1. PRINCIPIU
Analiză prin cromatografie în fază gazoasă cu tehnica spațiului de cap (tehnica head space).
2. APARATURĂ
|
2.1. |
Aparat de cromatografie în fază gazoasă, dotat cu un detector cu captură de electroni (ECD). |
|
2.2. |
Aparatură pentru spațiul de cap (tehnica head space). |
|
2.3. |
Coloană de cromatografie în fază gazoasă din sticlă cu o lungime de 2 metri și un diametru de 2 mm, fază staționară. OV 101 la 10 % sau echivalent, impregnând un pământ cu diatomit calcinat, spălat cu acizi și silanizat, cu granulometrie de 80–100 Mesh. |
|
2.4. |
Gaz vector și gaz auxiliar: azot pentru cromatografie în fază gazoasă, adaptată la detectarea prin captură de electroni. |
|
2.5. |
Flacoane de sticlă de 10–15 mm dotate cu o garnitură de teflon și cu un dop de aluminiu și un orificiu care permite prelevarea cu seringa. |
|
2.6. |
Pensă cu închidere ermetică. |
|
2.7. |
Seringă pentru gaz de 0,5–2 ml. |
3. REACTIVI
Standard: solvenți halogenați volatili cu un grad de puritate adecvat utilizării cromatografiei în fază gazoasă.
4. PROCEDURA DE ANALIZĂ
|
4.1. |
Se cântăresc cu grijă aproximativ 3 g de ulei într-un flacon de sticlă (a nu se reutiliza), se astupă flaconul până la închiderea ermetică. Se introduce flaconul într-un termostat la 70 °C timp de 1 oră. Se prelevează cu precizie cu ajutorul seringii un volum de 0,2–0,5 ml din tehnica head space. Se injectează în coloana aparatului de cromatografie în fază gazoasă reglat după cum urmează: — temperatura injectorului: 150 °C; — temperatura coloanei: 70–80 °C; — temperatura detectorului: 200–250 °C. Pot fi utilizate și alte temperaturi, cu condiția ca rezultațele să fie echivalente. |
|
4.2. |
Soluții etalon. Se prepară soluții etalon, utilizând ulei rafinat fără urmă de solvenți, cu concentrații variabile între 0,05 și 1 miligram pe kilogram și în raport cu conținutul presupus al mostrei. Diluarea eventuală trebuie să se efectueze cu pentan. |
|
4.3. |
Evaluarea cantitativă. Se face raportul între suprafețele sau înălțimile valorilor de vârf ale mostrei și ale soluției etalon cu concentrația presupusă cea mai apropiată. În cazul în care abaterea relativă este mai mare de 10 %, trebuie să se refacă analiza prin compararea cu o nouă soluție etalon până când concentrația va respecta abaterea relativă menționată anterior. Conținutul este stabilit pe baza unei medii de injecții elementare. |
|
4.4. |
Exprimarea rezultatelor. Rezultatele sunt exprimate în miligrame pe kilogram (ppm). Limita de detectare a metodei este de 0,01 mg pe kilogram. |
ANEXA XII
METODA CONSILIULUI OLEICOL INTERNAȚIONAL PENTRU EVALUAREA ORGANOLEPTICĂ A ULEIURILOR DE MĂSLINE VIRGINE
1. SCOPUL ȘI DOMENIUL DE APLICARE
Scopul metodei internaționale descrise în prezenta anexă este de a determina procedura de evaluare a caracteristicilor organoleptice ale uleiului de măsline virgin în sensul punctului 1 din partea VIII a anexei VII la Regulamentul (UE) nr. 1308/2013 al Parlamentului European și a Consiliului ( 4 ) și de a stabili metoda de clasare a uleiului de măsline virgin pe baza acestor caracteristici. Metoda conține și indicații pentru etichetarea opțională.
Metoda prezentată nu se aplică decât uleiurilor de măsline virgine și clasificării sau etichetării acestora în funcție de intensitatea defectelor percepute și a gustului fructat, determinate de un grup de degustători selecționați, formați și monitorizați, constituiți într-o comisie.
Standardele COI menționate în prezenta anexă se utilizează în ultima versiune disponibilă.
2. VOCABULARUL GENERAL DE BAZĂ AL ANALIZEI SENZORIALE
A se vedea standardul IOC/T.20/Doc. nr. 4 „Analiza senzorială: Vocabular general de bază”
3. VOCABULAR SPECIFIC
3.1. Atribute negative
Stătut/de drojdie: aromă caracteristică uleiului extras din măsline strânse sau depozitate în condiții de natură să determine un stadiu avansat de fermentare anaerobă sau uleiului rămas în contact cu depunerile rezultate în urma decantării, care au suferit, de asemenea, un proces de fermentare anaerobă în containere și cuve.
Mucegăit/umed/de pământ: Aromă caracteristică uleiului de măsline obținut din fructe atacate de mucegaiuri și de drojdii, ca rezultat al depozitării acestora timp de mai multe zile în condiții de umiditate sau uleiului obținut din măsline care au fost culese cu pământ sau acoperite cu noroi și care nu au fost spălate.
De vin/oțetit/acid/acru: aromă caracteristică a anumitor uleiuri care amintește de vin sau de oțet. Această aromă se datorează în principal unui proces de fermentare aerobă a măslinelor sau a resturilor de pastă de măsline de pe rogojini care nu au fost probabil spălate corespunzător, ceea ce a condus la formarea de acid acetic, de acetat de etil și de etanol.
Rânced: aromă a uleiurilor care au fost supuse unui proces intens de oxidare.
Măsline înghețate (lemn umed): aromă caracteristică uleiurilor extrase din măsline care au suferit de pe urma înghețului în copaci.
3.1.1. Alte atribute negative
|
Încălzit sau ars |
Aromă caracteristică a uleiurilor, care provine din încălzirea excesivă și/sau prelungită în cursul obținerii acestora și, în special, în timpul malaxării termice a pastei, dacă aceasta se realizează în condiții termice necorespunzătoare. |
|
De fân-lemnos |
Aromă caracteristică anumitor uleiuri care provin din măsline uscate. |
|
Grosier |
Senzație bucotactilă densă și păstoasă produsă de anumite uleiuri vechi. |
|
De lubrifianți |
Aromă a uleiului care amintește de motorină, de grăsime sau de uleiul mineral. |
|
De ape de vegetație |
Aromă dobândită de ulei în urma unui contact prelungit cu apele de vegetație care au suferit procese de fermentare. |
|
De saramură |
Aroma uleiului obținut din măsline conservate în saramură. |
|
Metalică |
Aromă care amintește de cea a metalelor. Aceasta este caracteristică uleiului care a rămas în contact îndelungat cu suprafețe metalice, pe durata procesului de măcinare, de malaxare, de presare sau de stocare. |
|
Rogoz |
Aromă caracteristică uleiului obținut din măsline presate între rogojini împletite din rogoz nou. Aroma poate fi diferită în funcție de materialul folosit la fabricarea rogojinilor, și anume rogoz verde sau rogoz uscat. |
|
Mâncat de viermi |
Aroma uleiului provenit din măsline care au fost supuse unui atac puternic al larvelor muștei măslinului (Bactrocera oleae). |
|
Castravete |
Aromă care apare ca urmare a ambalării ermetice a uleiului, prelungite în mod excesiv, în special în recipiente de tinichea, și care este atribuită formării de 2,6 nonadienal. |
3.2. Atribute pozitive
|
Fructat |
Ansamblul senzațiilor olfactive caracteristice ale uleiului, în funcție de soiul măslinelor, provenind de la fructe sănătoase și proaspete, verzi sau coapte. Senzațiile sunt percepute direct și/sau retronazal. |
|
Amar |
Gust elementar caracteristic al uleiului obținut din măsline verzi sau în stadiul de coacere. Este perceput de papilele caliciforme care formează „V”-ul lingual. |
|
Picant |
Senzație tactilă de pișcătură, caracteristică uleiurilor produse la începutul sezonului, în principal pe bază de măsline încă verzi. Senzația poate fi percepută în toată cavitatea bucală, în special în gât. |
3.3. Terminologie opțională în scopul etichetării
La cerere, președintele comisiei poate atesta faptul că uleiurile evaluate respectă definițiile și intervalele care corespund doar termenilor următori, în funcție de intensitatea și de percepția atributelor.
Atributele pozitive (fructat, amar și picant): în funcție de intensitatea percepției:
— robust, atunci când mediana atributului este mai mare de 6;
— mediu, atunci când mediana atributului este cuprinsă între 3 și 6;
— delicat, atunci când mediana atributului este mai mică de 3.
|
Fructat |
Ansamblul senzațiilor olfactive caracteristice ale uleiului, care depinde de soiul măslinelor și care provine de la fructe sănătoase și proaspete, în care nu predomină nici atributul fructat verde și nici atributul fructat copt. Senzațiile sunt percepute direct și/sau retronazal. |
|
Fructat verde |
Set de senzații olfactive caracteristice uleiului, amintind de cele ale fructelor verzi, care depind de soiul măslinelor și provin de la fructe verzi, sănătoase și proaspete. Senzațiile sunt percepute direct și/sau retronazal. |
|
Fructat copt |
Set de senzații olfactive caracteristice uleiului, amintind de cele ale fructelor coapte, care depind de soiul măslinelor și provin de la fructe sănătoase și proaspete. Senzațiile sunt percepute direct și/sau retronazal. |
|
Echilibrat |
Un ulei care nu este dezechilibrat, adică senzația olfactivă, gustativă și tactilă generată de un ulei în cazul căruia mediana atributului amar și mediana atributului picant nu depășesc cu mai mult de două puncte mediana atributului fructat. |
|
Ulei dulce |
Un ulei în cazul căruia mediana atributului amar și cea a atributului picant sunt mai mici sau egale cu 2. |
Lista termenilor în funcție de intensitatea percepției:
|
Termeni care fac obiectul prezentării unui certificat de test organoleptic |
Mediana atributului |
|
Fructat |
— |
|
Fructat copt |
— |
|
Fructat verde |
— |
|
Fructat delicat |
Mai mică de 3 |
|
Fructat mediu |
Între 3 și 6 |
|
Fructat robust |
Mai mare de 6 |
|
Fructat copt delicat |
Mai mică de 3 |
|
Fructat copt mediu |
Între 3 și 6 |
|
Fructat copt robust |
Mai mare de 6 |
|
Fructat verde delicat |
Mai mică de 3 |
|
Fructat verde mediu |
Între 3 și 6 |
|
Fructat verde robust |
Mai mare de 6 |
|
Amar delicat |
Mai mică de 3 |
|
Amar mediu |
Între 3 și 6 |
|
Amar robust |
Mai mare de 6 |
|
Picant delicat |
Mai mică de 3 |
|
Picant mediu |
Între 3 și 6 |
|
Picant robust |
Mai mare de 6 |
|
Ulei echilibrat |
Mediana atributului amar și mediana atributului picant nu depășesc cu mai mult de 2 puncte mediana atributului fructat |
|
Ulei dulce |
Mediana atributului amar și mediana atributului picant nu sunt mai mari de 2 |
4. PAHAR PENTRU DEGUSTAREA ULEIURILOR
A se vedea standardul IOC/T.20/Doc. nr. 5, „Pahar pentru degustarea uleiurilor”.
5. SALA DE DEGUSTARE
A se vedea standardul IOC/T.20/Doc. nr. 6, „Ghid pentru instalarea unei săli de degustare”.
6. ACCESORII
Fiecare cabină trebuie să fie dotată cu următoarele accesorii, care să se afle la îndemâna degustătorului pentru a-i permite să-și îndeplinească sarcina în mod corespunzător:
— pahare (standardizate) care conțin probele, identificate cu un număr de cod, acoperite cu o sticlă de ceas și păstrate la 28 °C ± 2 °C;
— foaia de profil (a se vedea figura 1), pe suport de hârtie sau în format electronic, care îndeplinește condițiile privind foaia de profil, împreună cu instrucțiunile pentru utilizarea sa în cazul în care este necesar;
— pix sau cerneală indelebilă;
— tăvi cu felii de mere și/sau apă, apă gazoasă și/sau pesmeți;
— un pahar cu apă la temperatura ambiantă;
— o foaie care reamintește normele generale enumerate la punctele 8.4 și 9.1.1;
— scuipători.
7. PREȘEDINTELE COMISIEI ȘI DEGUSTĂTORII
7.1. Președintele comisiei
Președintele comisiei trebuie să aibă o pregătire corespunzătoare și să fie un expert în toate tipurile de uleiuri pe care le va degusta în timpul testării. El este personalitatea de bază din comisie și are responsabilitatea organizării și funcționării acesteia.
Activitățile președintelui comisiei necesită ca acesta să aibă o formare de bază în ceea ce privește instrumentele de analiză senzorială, aptitudini senzoriale, meticulozitate la pregătirea, organizarea și desfășurarea testelor, precum și capacitățile și răbdarea necesare pentru a planifica și efectua testele în mod științific.
El este singura persoană responsabilă cu selectarea, formarea și monitorizarea degustătorilor, pentru a garanta nivelul lor de aptitudini. El este, prin urmare, responsabil cu evaluarea degustătorilor, care trebuie întotdeauna să fie obiectivă și pentru care trebuie să dezvolte proceduri specifice bazate pe teste și pe criterii solide de acceptare și de respingere. A se vedea standardul IOC/T.20/Doc. nr. 14, „Ghid pentru selectarea, formarea și monitorizarea degustătorilor calificați de ulei de măsline virgin”.
Președintele comisiei este responsabil de performanța comisiei și, prin urmare, de evaluarea sa, în legătură cu care trebuie să furnizeze dovezi fiabile și obiective. În orice caz, trebuie să dovedească, în orice moment, că metoda și degustătorii fac obiectul unor controale. Se recomandă calibrarea periodică a comisiei (IOC/T.20/Doc. nr. 14, punctul 5).
El deține responsabilitatea finală pentru ținerea evidențelor comisiei. Evidențele trebuie să fie întotdeauna trasabile, să respecte cerințele de certitudine și de calitate stabilite în standardele internaționale privind analiza senzorială și să asigure întotdeauna anonimitatea probelor.
El este responsabil cu inventarierea aparaturii și a echipamentelor și trebuie să se asigure că acestea, care sunt necesare pentru a respecta specificațiile prezentei metode, sunt curățate și întreținute în mod corespunzător, păstrând dovezi scrise în această privință. De asemenea, el este responsabil cu respectarea condițiilor de testare.
El are responsabilitatea recepționării și depozitării probelor în momentul în care ajung la laborator, precum și pe cea a depozitării lor după testare. În acest context, el se asigură în permanență că probele rămân anonime și că sunt depozitate corespunzător, iar în acest scop trebuie să elaboreze proceduri scrise pentru a garanta că întregul proces este trasabil și sigur.
În plus, este responsabil cu pregătirea, alocarea unui cod și prezentarea probelor către degustători în conformitate cu un protocol experimental corespunzător care respectă protocoalele prestabilite și, de asemenea, răspunde de colectarea și prelucrarea statistică a datelor obținute de degustători.
El este responsabil cu elaborarea și redactarea oricăror alte proceduri care ar putea fi necesare pentru a completa prezentul standard și pentru a garanta funcționarea corespunzătoare a comisiei.
El trebuie să caute modalități de comparare a rezultatelor comisiei cu cele obținute de alte comisii care efectuează analiza uleiului de măsline virgin, pentru a stabili dacă comisia funcționează în mod corespunzător.
Președintele comisiei are datoria de a-i motiva pe membrii comisiei de degustători prin promovarea interesului, a curiozității și a spiritului competitiv în rândurile acestora. În acest sens, li se recomandă să asigure un bun flux bidirecțional de informații cu membrii comisiei, care trebuie să fie informați în privința tuturor sarcinilor pe care le efectuează, precum și a rezultatelor obținute. În plus, el nu trebuie să-și facă publică opinia și va evita situația în care eventualii lideri își impun propriile criterii celorlalți degustători.
El îi convoacă din timp pe degustători și răspunde întrebărilor acestora cu privire la desfășurarea testelor, dar nu le sugerează opinii referitoare la probă.
7.1.1. Președintele adjunct al comisiei
Din motive întemeiate, președintele comisiei poate fi înlocuit cu un președinte al comisiei adjunct care poate prelua sarcinile legate de efectuarea testelor. Acest înlocuitor trebuie să dețină toate competențele necesare solicitate unui președinte al comisiei.
7.2. Degustătorii
Persoanele care au calitatea de degustători în testarea organoleptică a uleiurilor de măsline trebuie să facă acest lucru în mod voluntar. Prin urmare, se recomandă depunerea unei cereri scrise de către candidați. Candidații sunt selecționați, formați și monitorizați de către președintele comisiei, în conformitate cu capacitatea lor de a face distincția între probe similare, ținând cont de faptul că precizia lor se va îmbunătăți ca urmare a formării.
Degustătorii trebuie să se comporte ca adevărați observatori senzoriali, lăsându-și deoparte gusturile personale și raportând doar senzațiile pe care le percep. În acest sens, ei trebuie să-și desfășoare întotdeauna activitatea în tăcere, într-un mod relaxat și lipsit de grabă, acordând o atenție senzorială maximă probei pe care o degustă.
Pentru fiecare test sunt necesari între 8 și 12 degustători, dar trebuie să se prevadă câțiva degustători suplimentari care să poată fi folosiți în cazul unei eventuale absențe.
8. CONDIȚIILE DE TESTARE
8.1. Prezentarea probei
Proba de ulei pentru analiză este prezentată în pahare de testare standardizate care respectă standardul IOC/T.20/Doc. nr. 5, „Pahar pentru degustarea uleiurilor”.
Paharul conține 14-16 ml de ulei, sau între 12,8 și 14,6 g dacă probele sunt cântărite, și se acoperă cu o sticlă de ceas.
Fiecare pahar trebuie să fie marcat cu un cod format din cifre sau o combinație de litere și de cifre alese la întâmplare. Marcarea codului se face cu ajutorul unui sistem nemirositor.
8.2. Temperatura de testare și cea a probelor
Probele de ulei destinate degustării trebuie să fie menținute în pahare la 28 °C ± 2 °C pe toată durata testării. Această temperatură a fost reținută pentru că ușurează observarea diferențelor organoleptice prin comparație cu temperatura ambiantă și pentru că la temperaturi mai scăzute compușii aromatici caracteristici acestor uleiuri sunt slab volatili, în timp ce temperaturile mai ridicate duc la formarea de compuși volatili caracteristici uleiurilor încălzite. A se vedea standardul IOC/T.20/Doc. nr. 5 „Pahar pentru degustarea uleiurilor” pentru metoda care trebuie utilizată pentru încălzirea probelor aflate în pahar.
Sala de degustare trebuie să aibă o temperatură de 20 ° - 25 ° C (a se vedea standardul IOC/T.20/Doc. nr. 6).
8.3. Orarul de degustare
Pentru degustarea uleiurilor, orele optime de lucru sunt cele de dimineață: s-a dovedit că există perioade optime de percepție a gustului și a mirosului în timpul zilei. Înainte de mese sensibilitatea gustativ-olfactivă crește, în timp ce după mese percepția gustativ-olfactivă scade.
Cu toate acestea, criteriul menționat nu trebuie absolutizat în cazul în care foamea poate să distragă atenția degustătorilor, ceea ce diminuează capacitatea lor de departajare, prin urmare se recomandă ca degustarea să aibă loc dimineața între orele 10 și 12.
8.4. Degustători: norme generale de conduită
Următoarele recomandări se referă la comportamentul degustătorilor în timpul degustării.
Atunci când sunt chemați de către președintele comisiei să participe la testul organoleptic, degustătorii trebuie să respecte orarul stabilit, precum și următoarele reguli:
— Să se abțină de la fumat și să nu bea cafea cu cel puțin 30 minute înainte de ora stabilită pentru test.
— Trebuie să nu folosească parfumuri, produse cosmetice sau un săpunuri al căror miros ar putea să persiste în momentul testului. Trebuie să-și spele mâinile cu un săpun fără miros, apoi să le clătească și să le usuce ori de câte ori este necesar pentru a elimina orice miros.
— Să nu mănânce nimic cu cel puțin o oră înaintea degustării.
— În cazul unui disconfort fizic, în special dacă le este afectat simțul mirosului sau al gustului sau dacă se află sub efectul unui impact psihologic care îi poate împiedica să se concentreze, degustătorii îl informează pe președintele comisiei pentru ca acesta să îi retragă din testare.
— După îndeplinirea celor menționate, degustătorii își ocupă locul în cabina care le-a fost atribuită, într-un mod ordonat și silențios.
— Ei trebuie să citească atent instrucțiunile care figurează pe foaia de profil și să înceapă examinarea probei numai după ce sunt pe deplin pregătiți pentru sarcina de îndeplinit (în mod relaxat și lipsit de grabă). În cazul unor nelămuriri, trebuie să se consulte în particular cu președintele comisiei.
— Atunci când își îndeplinesc sarcinile, degustătorii păstrează tăcerea.
— Telefoanele lor mobile trebuie să fie oprite pe toată durata degustării, pentru a nu afecta concentrația și activitatea colegilor.
9. PROCEDURA PENTRU EVALUAREA ORGANOLEPTICĂ ȘI CLASAREA ULEIULUI DE MĂSLINE VIRGIN
9.1. Tehnica de degustare
9.1.1. Degustătorii iau paharul, ținându-l acoperit cu sticla de ceas, apoi îl înclină ușor și în această poziție îl rotesc complet pentru a umezi interiorul acestuia pe o suprafață cât mai mare. După această operațiune, degustătorii îndepărtează sticla de ceas și miros proba inspirând lent și profund, pentru a evalua uleiul. Durata testului olfactiv nu trebuie să depășească 30 de secunde. Dacă nu au ajuns la nici o concluzie în acest timp, degustătorii fac o scurtă pauză înainte de a trece la o nouă testare.
După efectuarea testului olfactiv, degustătorii evaluează senzațiile bucale (senzații retronazale globale olfactive, gustative și tactile). În acest scop, se ia în gură o cantitate mică de ulei, de aproximativ 3 ml. Este foarte importantă distribuirea uleiului în toată cavitatea bucală, de la partea anterioară a gurii și limbii, trecând prin părțile laterale și partea posterioară, până la vălul palatului și gât, fiind bine cunoscut că gustul și senzațiile tactile sunt percepute cu o intensitate variabilă în funcție de diferitele zone ale limbii, ale palatului și ale gâtului.
Trebuie să se insiste asupra necesității de a răspândi uleiul în cantități suficiente și foarte lent în partea posterioară a limbii până la vălul palatului și gât, atenția degustătorului fiind dirijată spre ordinea apariției stimulilor amar și picant. Dacă nu se procedează astfel, la anumite uleiuri cei doi stimuli pot trece neobservați sau stimulul amar poate fi mascat de stimulul picant.
Inspirațiile scurte și succesive, care lasă aerul să pătrundă prin gură, permit nu numai buna răspândire a probei în toată cavitatea bucală, ci și perceperea pe cale retronazală a compușilor aromatici volatili, prin utilizarea forțată a acestui canal.
N.B. Când degustătorii nu percep atributul fructat dintr-o probă, iar intensitatea atributului de clasare negativ este de 3,5 sau mai mică, președintele comisiei poate decide să ia măsuri pentru ca degustătorii să analizeze proba din nou la temperatura ambiantă (COI/T.20/Doc. Nr. 6/Rev. 1, septembrie 2007, secțiunea 3 – Specificații generale pentru instalarea unei săli de degustare), specificând contextul și conceptul de temperatură ambiantă. Când proba ajunge la temperatura camerei, degustătorii trebuie să o evalueze din nou pentru a verifica doar dacă se percepe atributul fructat. Dacă da, degustătorii trebuie să marcheze intensitatea pe scară.
Senzația tactilă de picant trebuie luată în considerare. În acest scop, se recomandă ingerarea uleiului.
9.1.2. Când se efectuează evaluarea organoleptică a uleiului de măsline virgin, se recomandă evaluarea a maximum patru probe în fiecare sesiune, având cel mult trei sesiuni pe zi, pentru a evita efectul de contrast care ar putea apărea prin degustarea imediată a altor probe.
Având în vedere că degustările succesive sunt afectate de oboseală sau de pierderea acuității, cauzate de degustările precedente, trebuie să se utilizeze un produs capabil să elimine din gură resturile de ulei de la degustarea tocmai efectuată.
Se recomandă utilizarea unei bucățele de măr care, după mestecare, poate fi aruncată în scuipătoare. În continuare, se clătește gura cu puțină apă la temperatura ambiantă. Se lasă să treacă cel puțin 15 minute între sfârșitul unei sesiuni și începutul următoarei.
9.2. Utilizarea foii de profil de către degustători
Foaia de profil care trebuie utilizată de către degustători este reprodusă în figura 1 din prezenta anexă.
Fiecare degustător din comisie miroase și pe urmă gustă ( 5 ) uleiul în cauză. Pe urmă, introduce intensitatea cu care percepe fiecare dintre atributele negative și pozitive pe scara de 10 cm de pe foaia de profil pe care o are la dispoziție.
În cazul în care degustătorii percep atribute negative neindicate în secțiunea 4, acestea se înregistrează la rubrica „altele”, folosindu-se termenul sau termenii care descriu cel mai precis atributele respective.
9.3. Utilizarea datelor de către președintele comisiei
Președintele comisiei strânge foile de profil completate de fiecare degustător și trece în revistă intensitățile consemnate pentru diferitele atribute. Dacă identifică vreo anomalie, invită degustătorul să își revizuiască foaia de profil și, dacă este necesar, să repete testul.
Președintele comisiei introduce datele referitoare la evaluarea fiecărui membru al comisiei într-un program informatic de tipul celor prevăzute de standardul IOC/T.20/Doc. nr. 15, în vederea calculării din punct de vedere statistic a rezultatelor analizei, pe baza calculării medianei acestora. A se vedea punctul 9.4 și apendicele la prezenta anexă. Datele pentru o anumită probă se introduc cu ajutorul unei matrice care conține 9 coloane, reprezentând cele 9 atributele senzoriale, și n linii corespunzătoare celor n membri care fac parte din comisie.
Atunci când se percepe un defect care se înregistrează la rubrica „altele” de către cel puțin 50 % din comisie, președintele comisiei calculează mediana defectului și obține clasarea corespunzătoare.
Valoarea coeficientului de variație robust care definește clasarea (defectul cu cea mai mare intensitate și atributul fructat) trebuie să nu fie mai mare de 20 %.
În caz contrar, președintele comisiei trebuie să repete evaluarea probei respective într-o altă sesiune de degustare.
Dacă această situație este frecventă, se recomandă ca președintele comisiei să ofere degustătorilor o formare specifică suplimentară (IOC/T.20/Doc. nr. 14, punctul 5) și să utilizeze indicele de repetabilitate și indicele de deviație pentru a verifica performanțele degustătorilor (IOC/T.20/Doc. nr. 14, punctul 6).
9.4. Clasarea uleiului
Uleiul se clasează în categoriile de mai jos, în funcție de mediana defectelor și de mediana atributului fructat. Mediana defectelor se definește ca mediana defectului perceput cu cea mai mare intensitate. Mediana defectelor și mediana atributului fructat sunt exprimate cu o precizie de o zecimală.
Clasarea uleiului se efectuează prin compararea valorii medianei defectelor și a medianei atributului fructat cu intervalele de referință prezentate în cele ce urmează. Întrucât limitele acestor intervale s-au stabilit ținându-se cont de eroarea metodei, acestea sunt considerate absolute. Pachetele de software permit să se afișeze clasarea într-un tabel de date statistice sau ca grafic.
(a) În cazul uleiului de măsline extravirgin, mediana defectelor este egală cu 0, iar mediana atributului fructat este mai mare de 0.
(b) În cazul uleiului de măsline virgin, mediana defectelor este mai mare de 0 și mai mică sau egală cu 3,5, iar mediana atributului fructat este mai mare de 0.
(c) În cazul uleiului de măsline virgin lampant, mediana defectelor depășește 3,5 sau mediana defectelor este mai mică sau egală cu 3,5, iar mediana atributului fructat este egală cu 0.
Nota 1: Atunci când mediana atributului amar și/sau a atributului picant depășește 5,0, președintele comisiei trebuie să declare acest lucru pe certificatul de test.
Pentru evaluările care au ca scop monitorizarea conformității, trebuie efectuat un singur test. În cazul contraevaluărilor, analiza trebuie să se desfășoare în duplicat, în sesiuni de testare diferite. Rezultatele analizei duplicat trebuie să fie omogene din punct de vedere statistic. (A se vedea punctul 9.5). În caz contrar, proba trebuie analizată din nou de încă două ori. Valoarea finală a medianei atributelor de clasare se va calcula pe baza valorii medii a ambelor mediane.
9.5 Criterii pentru acceptarea și respingerea duplicatelor
Pentru a se stabili dacă cele două rezultate ale unei analize în duplicat sunt omogene sau acceptabile din punct de vedere statistic, se utilizează eroarea normalizată, definită în cele ce urmează:
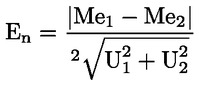
unde Me1 și Me2 sunt medianele celor două duplicate (mai exact, prima și a doua analiză), iar U1 și U2 sunt incertitudinile extinse obținute pentru cele două valori, calculate, după cum se specifică în apendice, astfel:
U
1 = c × s* and

În cazul incertitudinii extinse, c = 1,96; așadar:
U1 = 0,0196 × CVr × Me1
unde CVr este coeficientul de variație robust.
Pentru a se declara că cele două valori obținute nu sunt diferite din punct de vedere statistic, En trebuie să fie egală cu 1,0 sau mai mică.
Apendice
Metoda de calcul al medianei și al intervalelor de încredere
Mediana
![]()
Mediana se definește ca numărul real Xm caracterizat de faptul că probabilitatea (p) ca valorile distribuției (X) să fie inferioare acestui număr (Xm) este mai mică sau egală cu 0,5 și că, simultan, probabilitatea (p) ca valorile distribuției (X) să fie inferioare sau egale cu Xm este mai mare sau egală cu 0,5. O definiție mai practică consideră mediana ca fiind a 50-a percentilă a unei distribuții de numere ordonate în ordine crescătoare. În termeni mai simpli, mediana este punctul de mijloc al unui ansamblu ordonat de numere impare, sau media a două puncte de mijloc ale unui ansamblu ordonat de numere pare.
Deviația standard robustă
Pentru a se obține o estimare fiabilă a variabilității în jurul mediei, este necesar să se țină cont de deviația standard robustă, astfel cum a fost estimată conform Stuart și Kendall (4). Formula oferă deviația standard asimptotică robustă, adică estimarea robustă a variabilității datelor luate în considerare, în care N este numărul de observații și IQR este intervalul interquartil, care înglobează exact 50% din cazurile unei anumite distribuții de probabilitate:

Calculul intervalului interquartil se efectuează calculând magnitudinea diferenței dintre a 75-a și a 25-a percentilă.
![]()
unde percentila este valoarea Xpc caracterizată de faptul că probabilitatea (p) ca valorile distribuției să fie inferioare Xpc este mai mică sau egală cu o sutime specifică și că, simultan, probabilitatea (p) ca valorile distribuției să fie mai mici sau egale cu Xpc este mai mare sau egală cu acea sutime specifică. Sutimea indică cuantila de distribuție reținută. În cazul medianei, aceasta este egală cu 50/100.

În scopuri practice, percentila este valoarea de distribuție care corespunde unei arii specifice trasate dintr-o curbă de distribuție sau de densitate. De exemplu, a 25-a percentilă reprezintă valoarea de distribuție care corespunde unei arii egale cu 0,25 sau 25/100.
În prezenta metodă, percentilele sunt calculate pe baza valorilor reale care apar în datele din matrice (procedura de calculare a percentilelor).
Coeficientul de variație robust (%)
CVr% reprezintă un număr pur care indică procentajul de variabilitate al seriei de numere analizate. Din acest motiv, coeficientul este foarte util pentru verificarea fiabilității evaluatorilor comisiei.

Intervalele de încredere pentru mediană la 95 %
Intervalele de încredere la 95 % (valoarea erorii de tip I egale cu 0,05 sau 5 %) reprezintă intervalul în care valoarea medianei ar putea varia în ipoteza în care ar fi posibilă repetarea experienței de un număr infinit de ori. În practică, acesta indică intervalul de variabilitate al testului în condițiile de operare reținute, dacă se pleacă de la ipoteza că s-ar putea repeta de mai multe ori. Intervalul ajută la evaluarea fiabilității testului, ca și în cazul CVr%.


unde C = 1,96 pentru intervalul de încredere la nivelul de 95 %.
Un exemplu al fișei de calcul este prezentat în anexa I la standardul IOC/T 20/Doc. nr. 15.
Bibliografie
(1) Wilkinson, L. 1990. Systat: The system for statistics. Evanston, IL.SYSTAT Inc.
(2) Cicchitelli, G. 1984. Probabilità e Statistica. Maggioli Editore, Rimini.
(3) Massart, D.L.; Vandeginste, B.G.M.; Deming, Y.; Michotte, L. 1988. Chemometrics. A textbook. Elsevier. Amsterdam.
(4) Kendall, M.G.; Stuart, A. 1967. The advanced theory of statistics. Vol. 1. Hafner Publishing Co.
(5) McGill, R.; Tukey, J.W.; Larsen, W.A. 1978. Variation of Box Plots. The American Statistician, 32, (2), 12-16.
(6) IOC/T.28/Doc. nr. 1 septembrie 2007, Orientări pentru acreditarea laboratoarelor de teste senzoriale cu referire specială la uleiul de măsline virgin în conformitate cu standardul ISO/IEC 17025:2005.
(7) IOC/T.20/Doc. nr. 14.
(8) IOC/T.20/Doc. nr. 15.
(9) ISO/IEC 17025:05.
▼M20 —————
▼M19 —————
ANEXA XV
1. CONȚINUTUL DE ULEI AL RESTURILOR DE MĂSLINE
1.1. Material
— aparat de extracție adecvat, echipat cu un balon de 200–250 ml;
— baie cu încălzire electrică (baie de nisip, baie de apă etc.) sau placă de încălzire;
— balanță analitică;
— etuvă reglată la maxim 80 °C;
— etuvă cu încălzire electrică echipată cu un dispozitiv de termoreglare reglat la 103 °C ± 2 °C care permite realizarea unei insuflări de aer sau a unei presiuni reduse;
— concasor mecanic ușor de curățat care permite concasarea fără încălzire și fără diminuarea sensibilă a conținutului lor de apă și de ulei;
— cartuș de extracție și vată hidrofilă sau hârtie de filtru, fără produse care se extrag cu hexan;
— exsicator;
— sită cu găuri cu diametru de 1 mm;
— piatră ponce cu granule mici, uscată în prealabil.
1.2. Reactiv
n-hexan tehnic al cărui reziduu în urma evaporării complete trebuie să fie mai mic de 0,002 g pentru 100 de ml.
2. MOD DE LUCRU
2.1. Prepararea mostrei
Se sfărâmă mostra pentru laborator, dacă este necesar, în concasorul mecanic bine curățat în prealabil cu scopul de a-l mărunți în particule ce pot trece prin sită.
Se folosește aproximativ a douăzecea parte din mostră pentru a finaliza curățarea concasorului, se aruncă mostra măcinată, se macină restul, se colectează, se amestecă cu grijă și se analizează imediat.
2.2. Mostra
Se cântăresc, cu o precizie de 0,01 grame, după încheierea concasării, aproximativ 10 g de mostră pentru test.
2.3. Prepararea cartușului de extracție
Se plasează mostra în cartuș și se astupă cu tamponul de vată hidrofilă. În cazul în care se folosește hârtie de filtru, se ambalează mostra măcinată în această hârtie.
2.4. Uscarea prealabilă
În cazul în care resturile sunt foarte umede (conținutul de apă și de materii volatile este mai mare de 10 %), se efectuează o uscare prealabilă prin introducerea, pentru o perioadă de timp suficientă, a cartușului umplut (sau a hârtiei de filtru) în etuva încălzită la maximum 80 °C, pentru a reduce conținutul de apă și de materii volatile la mai puțin de 10 %.
2.5. Prepararea balonului
Se cântărește, cu o precizie de 1 miligram, balonul ce conține 1 sau 2 granule de piatră ponce, uscat în prealabil în etuvă la 103 °C ± 2 °C, apoi se răcește timp de cel puțin o oră într-un exsicator.
2.6. Prima extracție
Se pune în aparatul de extracție cartușul (sau hârtia de filtru) ce conține mostra. Se toarnă în balon cantitatea necesară de hexan. Se adaptează balonul la aparatul de extracție și se plasează totul pe baia cu încălzire electrică. Încălzirea se efectuează în așa fel încât debitul refluxului să fie de cel puțin trei picături pe secundă (fierbere moderată, fără clocote). După 4 ore de extracție, se lasă să se răcească. Se scoate cartușul aparatului de extracție și se plasează într-un curent de aer astfel încât să se elimine cea mai mare parte a solventului impregnat.
2.7. A doua extracție
Se golește cartușul în microconcasor și se concasează cât mai fin posibil. Se plasează din nou, cantitativ, amestecul în cartuș, și acesta se pune în aparatul de extracție.
Se începe din nou extracția timp de încă două ore folosindu-se același balon ce conține prima extracție.
Soluția obținută în balonul de extracție trebuie să fie limpede. În caz contrar, aceasta se filtrează printr-o hârtie de filtru, spălându-se de mai multe ori primul balon și hârtia de filtru cu hexan. Se colectează filtratul și solventul de spălare într-un al doilea balon, uscat în prealabil și tarat cu o precizie de 1 miligram.
2.8. Eliminarea solventului și cântărirea extractului
Se înlătură, prin distilare pe baia cu încălzire electrică, cea mai mare parte a solventului. Se elimină ultimele urme de solvent prin încălzirea balonului în etuvă la 103 °C ± 2 °C timp de 20 de minute. Se facilitează această eliminare fie prin insuflarea de aer, din când în când, sau, de preferință, a unui gaz inert, fie operând sub presiune redusă.
Se lasă să se răcească balonul într-un exsicator timp de cel puțin o oră, și se cântărește cu o precizie de 1 mg.
Se încălzește din nou 10 minute în aceleași condiții, se răcește în exsicator și se cântărește.
Diferența dintre rezultatele celor două cântăriri trebuie să fie mai mică sau egală cu 10 mg. În caz contrar, se încălzește din nou timp de câte 10 minute, perioade urmate de răciri și de cântăriri, până când diferența de masă este cel mult egală cu 10 mg. Se reține ultima cântărire a balonului.
Se efectuează două determinări pe aceeași mostră pentru testare.
3. EXPRIMAREA REZULTATELOR
3.1. Mod de calcul și formula
(a) Extractul exprimat în procente de masă a produsului ca atare, este egal cu:
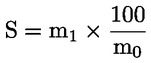
|
în care: |
S este procentul de masă al extractului produsului ca atare; m0 este masa, în grame, a mostrei; m1 este masa, în grame, a extractului după uscare. |
Se ia ca rezultat media aritmetică a celor două determinări, în cazul în care sunt îndeplinite condițiile lor de repetabilitate.
Rezultatul se exprimă cu o singură zecimală.
(b) Extractul este raportat la materia uscată folosind-se următoarea formulă:

în care:
S = este procentul de masă al extractului produsului ca atare [a se vedea litera (a)],
U = conținutul acestuia de apă și de materii volatile.
3.2. Repetabilitatea
Diferența dintre rezultatele celor două determinări, efectuate simultan sau rapid una după alta de către același analist, nu trebuie să fie mai mare de 0,2 g de extract cu hexan pentru 100 g de mostră.
În caz contrar, se repetă analiza pe alte două mostre pentru test. În cazul în care și de această dată diferența depășește 0,2 grame, se ia ca rezultat media aritmetică a celor patru determinări efectuate.
ANEXA XVI
DETERMINAREA INDICELUI DE IOD
1. OBIECT
Prezenta normă internațională descrie o metodă destinată determinării indicele de iod în substanțele grase de origine animală și vegetală.
2. DEFINIȚIE
În sensul prezentei norme internaționale, se aplică următoarele definiții.
|
2.1. |
Indice de iod: masa de iod absorbită de mostră în condiții de operare specificate de prezenta normă internațională. Indicele de iod se exprimă în număr de grame de iod pe 100 g de mostră. |
3. PRINCIPIU
Punerea în soluție a mostrei într-un solvent și adăugarea reactivului Wijs. După un interval de timp determinat, adăugarea unei soluții de iodură de potasiu și de apă; titrarea iodului eliberat cu ajutorul unei soluții de tiosulfat de sodiu.
4. REACTIVI
Toți reactivii sunt de calitate analitică recunoscută.
|
4.1. |
Apa, care să conformă cu cerințele ISO 3696, categoria a 3-a. |
|
4.2. |
Iodură de potasiu, soluție de 100 g/l, fără iod sau iodat. |
|
4.3. |
Soluție de amidon. Se amestecă 5 g de amidon solubil în 30 ml de apă, se adaugă acestui amestec 1 000 ml de apă fiartă, se fierbe 3 minute și se lasă să se răcească. |
|
4.4. |
Soluție volumetrică etalon de tiosulfat de sodiu. c(Na2S3O3.5H2O) = 0,1 mol pe litru, standardizat timp de cel mult șapte zile înainte de folosire. |
|
4.5. |
Solvent preparat prin amestecul de volume egale de ciclohexan și de acid acetic. |
|
4.6. |
Reactiv Wijs care conține monoclorură de iod în acid acetic. Se utilizează reactivul de Wijs care se găsește în comerț. |
5. APARATURĂ
Materialul de laborator obișnuit, în special:
|
5.1. |
Nacele de sticlă pentru cântărit, adecvate mostrei și care pot fi introduse în baloane (5.2.). |
|
5.2. |
Baloane conice, cu o capacitate de 500 ml, prevăzute cu capace de sticlă și complet uscate. |
6. PREPARAREA MOSTREI DE ANALIZAT
Mostra omogenizată este uscată pe sulfat de sodiu și filtrată.
7. MOD DE LUCRU
|
7.1. |
Mostra Masa mostrei variază conform indicelui de iod presupus, așa cum indică tabelul 1.
Tabelul 1
Se cântărește mostra cu o precizie de 0,1 mg într-o nacelă de sticlă pentru cântărit (5.1). |
|
7.2. |
Determinare Se introduce mostra într-un balon de 500 ml (5.2). Se adaugă 20 ml de solvent (4.4) pentru a dizolva substanțele grase. Se adaugă exact 25 ml de reactiv Wijs (4.5), se astupă, se agită conținutul și se pune balonul într-un loc întunecos. Pentru reactivul de Wijs nu se utilizează pipeta. Se prepară o mostră etalon cu solventul și cu reactivul, dar fără mostra pentru testare. Pentru mostrele care au un indice de iod mai mic de 150, se lasă baloanele într-un spațiu întunecos timp de o oră; pentru cele care au un indice de iod mai mare de 150 și pentru produsele polimerizate sau produse oxidate într-o măsură considerabilă, se lasă timp de două ore. După ce a trecut acest interval de timp, se adaugă 20 ml din soluția de iodură de potasiu (4.1) și 150 ml de apă în fiecare balon. Se titrează cu soluția de tiosulfat de sodiu volumetrică etalon (4.3) până când dispare culoarea galbenă datorată iodului. Se adaugă câteva picături din soluția de amidon (4.2) și se continuă titrarea până în momentul în care dispare culoarea albastră, după ce s-a agitat energic conținutul. Notă: Determinarea potențiometrică a punctului final este tolerată. |
|
7.3. |
Numărul de determinări Se efectuează două determinări pe aceeași mostră. |
8. EXPRIMAREA REZULTATELOR
Indicele de iod este egal cu:

,
în care:
c = valoarea numerică a concentrației exacte, în moli pe litru, a soluției etalon de tiosulfat de sodiu volumetric (4.3) utilizate;
V1 = valoarea numerică a volumului, în ml, a soluției etalon de tiosulfat de sodiu volumetric (4.3) utilizate pentru mostra etalon;
V2 = valoarea numerică a volumului, în ml, a soluției etalon de tiosulfat de sodiu volumetric (4.3) utilizate pentru determinare;
m = valoarea numerică a masei, în grame, a mostrei (7.1).
Se ia ca rezultat media aritmetică a celor două determinări, cu condiția să fie îndeplinit criteriul repetabilității.
ANEXA XVII
METODĂ DE DETERMINARE A CONȚINUTULUI DE STIGMASTADIENE DIN ULEIURILE VEGETALE
1. OBIECT
Determinarea conținutului de stigmastadiene din uleiurile vegetale conținând concentrații scăzute din aceste hidrocarburi, în special în uleiul virgin de măsline și în uleiul brut din resturi de măsline.
2. DOMENIU DE APLICARE
Metoda se utilizează pentru toate uleiurile vegetale, dar măsurătorile sunt fiabile doar în cazul în care conținutul de hidrocarburi este cuprins între 0,01 și 4,0 mg/kg. Această metodă este adecvată în special pentru detectarea prezenței uleiurilor vegetale rafinate (de măsline, resturi de măsline, floarea soarelui, palmier etc.) în ulei virgin de măsline, deoarece uleiurile rafinate conțin stigmastadiene, iar uleiurile virgine nu conțin.
3. PRINCIPIU
Izolarea substanței nesaponificabile. Separarea fracțiunii de hidrocarburi steroidale prin cromatografie în coloană pe silicagel și analiza prin cromatografie în fază gazoasă în coloană capilară.
4. APARATURĂ
|
4.1. |
Baloane adecvate de 250 ml cu un condensator cu reflux. |
|
4.2. |
Pâlnii de decantare de 500 ml. |
|
4.3. |
Baloane cu fund rotund de 100 ml. |
|
4.4. |
Evaporator rotativ. |
|
4.5. |
Coloană cromatografică de sticlă (diametru intern între 1,5 și 2,0 cm și lungime de 50 cm) cu robinet de teflon și un dop din fibră din vată de sticlă sau un disc din sticlă sinterizată la fund. Pentru a prepara coloana de silicagel, a se turna hexan în coloana cromatografică până la înălțimea de aproximativ 5 cm și apoi a se completa cu o suspensie de silicagel în hexan (15 g în 40 ml) cu ajutorul unor fracțiuni de hexan. A se lăsa să se depună și a se termina depunerea prin aplicarea unei ușoare vibrații. A se adăuga sulfat de sodiu anhidru până la o înălțime de aproximativ 0,5 cm și la sfârșit a se elua hexanul în exces. |
|
4.6. |
Cromatograf în fază gazoasă cu detector de ionizare a flăcării, injector cu splitare sau injector pe coloană rece „on-column” și cuptor programabil între ± 1 °C. |
|
4.7. |
Coloană capilară din silice topită pentru cromatografia în fază gazoasă (diametru intern de 0,25 sau 0,5 mm și lungime de 25 m), acoperită cu fază de fenilmetilsilicon 5 %, cu grosime a peliculei de 0,25 mm. Nota 1: Pot fi utilizate alte coloane cu polaritate similară sau mai scăzută. |
|
4.8. |
Integrator-înregistrator care permite o integrare vale-vale. |
|
4.9. |
Microseringă de 5-10 ml pentru cromatografie în fază gazoasă cu ac cimentat. |
|
4.10. |
Manta de încălzire electrică sau placă de încălzire. |
5. REACTIVI
Toți reactivii trebuie să fie puri, în lipsa unor indicații contrare. Apa utilizată trebuie să fie apă distilată sau apă cu o puritate cel puțin echivalentă.
|
5.1. |
Hexan sau amestec de alcani cu intervalul de fierbere între 65 și 70 °C, distilat cu coloană de rectificare. Nota 2: Solventul trebuie distilat pentru a îndepărta impuritățile. |
|
5.2. |
Etanol v/v 96. |
|
5.3. |
Sulfat de sodiu anhidru. |
|
5.4. |
Soluție de 10 % de hidroxid de potasiu alcoolic. A se adăuga 10 ml de apă la 50 g de hidroxid de potasiu, a se amesteca și apoi a se dizolva amestecul în etanol până la obținerea a 500 ml de soluție. Nota 3: Potasa alcoolică devine cafenie în repaus. Trebuie preparată proaspăt în fiecare zi și păstrată în vase de sticlă neagră bine astupate. |
|
5.5. |
Silicagel 60 pentru cromatografie în coloană, 70 - 230 mesh (Merck, referința 7734 sau similară). Nota 4: De obicei, silicagelul poate fi utilizat direct din recipient fără vreun tratament prealabil. Cu toate acestea, unele loturi de silicagel pot avea o activitate scăzută din cauza unei insuficiente separări cromatografice. În acest caz, silicagelul ar trebui tratat în modul următor: a se dezactiva prin încălzire timp de cel puțin 4 ore la 550 °C. După încălzire, a se plasa silicagelul într-un exsicator până la răcire și apoi a se transfera silicagelul într-un balon închis. A se adăuga 2 % apă și a se agita până la dispariția bulgărilor și obținerea unei pulberi care plutește liber. În cazul în care pentru unele loturi de silicagel rezultă cromatograme cu valori de vârf care interferează, silicagelul trebuie tratat după metoda indicată mai sus. O alternativă ar putea fi utilizarea unui alt silicagel extrapur 60 (Merck, referința 7754). |
|
5.6. |
Soluție-mamă (200 ppm) de colesta-3,5-dienă (Sigma, puritate 99 %) în hexan (10 mg în 50 ml). |
|
5.7. |
Soluție standard de colesta-3,5-dienă în hexan la o concentrație de 20 ppm, obținută prin diluarea soluției de mai sus. Nota 5: Soluțiile de la punctele 5.6 și 5.7 sunt stabile pentru o perioadă de cel puțin patru luni în cazul în care sunt păstrate la mai puțin de 4 °C. |
|
5.8. |
Soluție de n-nonacosan în hexan la o concentrație de aproximativ 100 ppm. |
|
5.9. |
Gaz purtător pentru cromatografie: heliu sau hidrogen cu puritate de de 99,9990 %. |
|
5.10. |
Gaze auxiliare pentru detectorul de ionizare cu flacără: hidrogen cu puritate 99,9990 % și aer purificat. |
6. MOD DE LUCRU
6.1. Prepararea substanței nesaponificabile
|
6.1.1. |
A se cântări 20 ± 0,1 g de ulei într-un balon de 250 ml (punctul 4.1), a se adăuga 1 ml din soluția standard de colesta-3,5-dienă (20 μg) și 75 ml de potasă alcoolică la 10 %, a se instala condensatorul cu reflux și a se încălzi menținând o fierbere moderată timp de 30 de minute. A se îndepărta de sursa de căldură balonul care conține eșantionul și a se permite răcirea ușoară a soluției (a nu se permite răcirea completă, întrucât eșantionul se va depune). A se adăuga 100 ml de apă și a se transfera soluția într-o pâlnie separatoare (punctul 4.2) cu ajutorul a 100 ml de hexan. A se agita puternic amestecul timp de 30 de secunde și a se permite formarea diferitelor straturi. Nota 6: În cazul în care se formează o emulsie care nu dispare rapid, a se adăuga mici cantități de etanol. |
|
6.1.2. |
A se transfera faza apoasă de dedesubt într-o a doua pâlnie separatoare și a se extrage din nou cu 100 ml de hexan. A se recupera din nou faza inferioară și a se spăla extractele de hexan (regrupate într-o altă pâlnie de separare) de trei ori, de fiecare dată cu 100 ml dintr-un amestec etanol-apă (1: 1) până se ajunge la un pH neutru. |
|
6.1.3. |
A se trece soluția de hexan prin sulfat de sodiu anhidru (50 g), a se spăla cu 20 ml hexan și a se usca într-un evaporator rotativ la 30 °C la presiune redusă. |
6.2. Separarea fracțiunii de hidrocarbură steroidală
|
6.2.1. |
A se așeza reziduul în coloana de fracționare cu ajutorul a două fracțiuni de 1 ml de hexan, a se scurge eșantionul de-a lungul coloanei permițând scăderea nivelului soluției până deasupra sulfatului de sodiu și a se începe eluția cromatografică cu hexan la un debit de aproximativ 1 ml/min. A se elimina primii 25-30 ml de eluat și apoi a se colecta următoarea fracțiune de 40 ml. După colectare, a se transfera această fracțiune într-un balon cu fundul rotund de 100 ml (punctul 4.3). Nota 7: Prima fracțiune conține hidrocarburi saturate (figura 1 a) și a doua fracțiune pe cele steroidale. Continuând eluția, se obțin squalen și compuși înrudiți. Pentru a obține o bună separare între hidrocarburile saturate și cele steroidale, este necesară optimizarea volumelor fracțiunilor. Pentru aceasta, volumul primei fracțiuni ar trebui ajustat astfel încât, atunci când a doua fracțiune este analizată, valorile de vârf care reprezintă hidrocarburile saturate să fie scăzute (a se vedea figura 1 c); în cazul în care acestea nu apar, dar intensitatea valorii de vârf standard este scăzută, trebuie redus volumul. Oricum, nu este necesară o separare completă între componenții primei și celei de a doua fracțiuni, dat fiind că nu există o suprapunere a valorilor de vârf în timpul analizei prin cromatografie în faza gazoasă, dacă condițiile CG sunt adaptate în conformitate cu punctul 6.3.1. Optimizarea volumului celei de-a doua fracțiuni este, în general, inutilă, întrucât se obține o bună separare cu ajutorul componentelor următoare. Cu toate acestea, prezența unei valori de vârf ridicate la un timp de retenție cu aproximativ 1,5 minute mai redus decât cel standard se datorează squalenului și indică o separare insuficientă. |
|
6.2.2. |
A se evapora a doua fracțiune într-un evaporator rotativ la 30 °C și la o presiune redusă până la uscare și a se dizolva imediat reziduul în 0,2 ml de hexan. A se păstra soluția la frigider până la analiză. Nota 8: Reziduurile menționate la punctele 6.1.3 și 6.2.2 nu trebuie păstrate uscate și la temperatura camerei. Imediat ce sunt obținute, ar trebui adăugat solventul și soluțiile ar trebui păstrate la frigider. |
6.3. Cromatografie în fază gazoasă
|
6.3.1. |
Condiții de lucru pentru injectare cu splitare: — temperatura injectorului: 300 °C; — temperatura detectorului: 320 °C; — integrator-înregistrator: parametrii pentru integrare trebuie fixați astfel încât să se permită o evaluare corectă a suprafețelor. Este recomandat modul de integrare vale-vale; — sensibilitate: aproximativ de 16 ori atenuarea minimă; — cantitatea de soluție injectată: 1 μl; — temperaturi de programare a cuptorului: temperatura inițială 235 °C timp de șase minute și apoi crescând cu 2 °C/minut până la 285 °C; — injector cu divizor de debit 1: 15; — purtător: heliu sau hidrogen la o presiune de aproximativ 120 kPa. Aceste condiții pot fi modificate în funcție de caracteristicile cromatografului și ale coloanei, pentru ca cromatogramele să îndeplinească următoarele cerințe: formarea valorii de vârf standard interne în aproximativ cinci minute din timpul indicat la punctul 6.3.2; valoarea de vârf standard internă trebuie să se întindă pe cel puțin 80 % din scara totală. Sistemul de cromatografie în fază gazoasă ar trebui să fie controlat prin injectarea unui amestec din soluția-mamă de colestadienă (punctul 5.6) și din soluția de n-nonacosan (punctul 5.8). Valoarea de vârf a colesta-3,5-dienei trebuie să apară înainte de cea a n-nonacosan-ului (figura 1c); în cazul în care aceasta nu apare, se pot lua două măsuri: reducerea temperaturii cuptorului și/sau utilizarea unei coloane mai puțin polare. |
|
6.3.2. |
Identificarea valorilor de vârf Valoarea de vârf standard internă apare la aproximativ 19 minute și 3,5-stigmastadiena la un timp de retenție relativ de aproximativ 1,29 (a se vedea figura 1b). 3,5-Stigmastadiena este însoțită de cantități mici de izomer și, în general, ambele produc o valoare de vârf cromatografică unică. Cu toate acestea, în cazul în care coloana este prea polară sau prezintă o putere de rezoluție prea mare, izomerul poate apărea ca o valoare de vârf scăzută înaintea și apropiată de cea a stigmasta-3,5-dienei (figura 2). Pentru a se asigura faptul că stigmastadienele produc o valoare de vârf unică, este recomandabil să se înlocuiască coloana cu una care este mai puțin polară sau are un diametru intern mai mare. Nota 9: Stigmastadienele de referință pot fi obținute din analiza unui ulei vegetal rafinat prin utilizarea unei cantități mai reduse din eșantion (1-2 g). Stigmastadienele produc o valoare de vârf proeminentă și ușor de identificat. |
|
6.3.3. |
Analiză cantitativă Conținutul de stigmastadiene este determinat în conformitate cu formula: mg/kg de stigmastadiene =
Limita de detecție: aproximativ 0,01 mg/kg. |


Figura 1
Cromatograme (cromatografii în fază gazoasă) obținute prin analiza eșantioanelor de ulei de măsline pe o coloană capilară cu silice topită (diametru intern de 0,25 mm și lungime de 25 m), acoperită cu fenilmetilsilicon 5 %, cu o grosime a peliculei de 0,25 μm.
(a) Prima fracțiune (30 ml) dintr-un ulei virgin, eluat cu etalon.
(b) A doua fracțiune (40 ml) dintr-un ulei de măsline conținând 0,10 mg/kg de stigmastadiene.
(c) A doua fracțiune (40 ml) conținând o mică proporție din prima fracțiune.

Figura 2
Cromatogramă în fază gazoasă obținută dintr-un eșantion de ulei de măsline rafinat analizat pe o coloană DB-5 pe care figurează izomerul de 3,5-stigmastadienă.
ANEXA XVIII
DETERMINAREA DIFERENȚEI DINTRE CONȚINUTUL REAL ȘI CONȚINUTUL TEORETIC DE TRIGLICERIDE CU NEC 42
1. DOMENIUL DE APLICARE
Determinarea diferenței absolute dintre valorile experimentale ale trigliceridelor (TG) cu număr echivalent de atomi de carbon egal cu 42 (NEC42HPLC) obținute prin determinarea în ulei prin cromatografie lichidă de înaltă performanță (HPLC) și valoarea teoretică a TG cu număr echivalent de atomi de carbon egal cu 42 (NEC 42teoretic) calculată pe baza compoziției de acizi grași.
2. SFERA DE APLICARE
Metoda se aplică uleiurilor de măsline. Metoda se aplică detectării prezenței unor cantități mici de uleiuri din semințe (bogate în acid linoleic) în fiecare categorie de uleiuri de măsline.
3. PRINCIPIU
În cazul uleiurilor de măsline pure, conținutul de trigliceride cu NEC 42 determinat prin analiza HPLC este, într-o oarecare măsură, echivalent cu conținutul teoretic de trigliceride cu NEC 42 (calculat pe baza compoziției de acizi grași determinate prin cromatografie în fază gazoasă-lichidă - GLC). O diferență mai mare decât valorile adoptate pentru fiecare categorie de ulei indică faptul că uleiul conține uleiuri din semințe.
4. METODĂ
Metoda de calcul pentru conținutul teoretic de trigliceride cu NEC 42 și pentru diferența în raport cu datele HPLC constă în principal în coordonarea datelor analitice obținute cu ajutorul altor metode. Se pot distinge trei faze: determinarea compoziției de acizi grași prin cromatografie în fază gazoasă cu coloană capilară, calculul compoziției teoretice a trigliceridelor cu NEC 42 și determinarea trigliceridelor cu NEC 42 prin HPLC.
4.1. Aparatură
|
4.1.1. |
Baloane de 250 și 500 ml |
|
4.1.2. |
Pahare Berzelius de 100 ml |
|
4.1.3. |
Coloană cromatografică din sticlă (diametru intern de 21 mm, lungime de 450 mm) cu robinet și șlif (mamă) în partea de sus |
|
4.1.4. |
Pâlnii de separare de 250 ml, cu șlif (tată) în partea de jos, adecvate pentru conectarea la partea superioară a coloanei |
|
4.1.5. |
Baghetă de sticlă de 600 mm lungime |
|
4.1.6. |
Pâlnie de sticlă de 80 mm diametru |
|
4.1.7. |
Baloane gradate de 50 ml |
|
4.1.8. |
Baloane gradate de 20 ml |
|
4.1.9. |
Evaporator rotativ |
|
4.1.10. |
Aparat de cromatografie în fază lichidă de înaltă performanță care permite un control termostatic al temperaturii coloanei |
|
4.1.11. |
Injectoare pentru 10 μl |
|
4.1.12. |
Detector: refractometru diferențial capabil să determine indicele de refracție cu aproximație de 10–4 |
|
4.1.13. |
Coloană: tub din oțel inoxidabil de 250 mm lungime × 4,5 mm diametru interior, prevăzut cu particule de siliciu cu diametru de 5 μm conținând 22-23 % carbon sub formă de octadecilsilan |
|
4.1.14. |
Software de prelucrare a datelor |
|
4.1.15. |
Flacoane, de aproximativ 2 ml, cu membrane din teflon și capace cu filet |
4.2. Reactivi
Reactivii trebuie să fie de puritate analitică, iar solvenții de eluare trebuie să fie degazați (ei pot fi reciclați de mai multe ori, fără ca acest lucru să aibă repercusiuni asupra separărilor).
|
4.2.1. |
Eter de petrol, 40-60 °C, de puritate pentru cromatografie sau hexan |
|
4.2.2. |
Eter etilic, fără peroxid, proaspăt distilat |
|
4.2.3. |
Solvent de eluare pentru purificarea uleiului prin cromatografie în coloană: amestec eter de petrol/eter etilic 87/13 (v/v) |
|
4.2.4. |
Silicagel, granulometrie 70-230, tipul Merck 7734, cu un conținut de apă standardizat de 5 % (g/g) |
|
4.2.5. |
Vată de sticlă |
|
4.2.6. |
Acetonă pentru HPLC |
|
4.2.7. |
Acetonitril sau propionitril pentru HPLC |
|
4.2.8. |
Solvent de eluare HPLC: acetonitril + acetonă (proporțiile trebuie să fie ajustate în funcție de separarea dorită; se începe cu un amestec 50:50) sau propionitril |
|
4.2.9. |
Solvent de solubilizare: acetonă |
|
4.2.10. |
Trigliceride de referință: pot fi utilizate trigliceride comerciale (tripalmitină, trioleină etc.), iar timpii de retenție se reprezintă grafic după numărul echivalent de atomi de carbon, sau, alternativ, cromatograme de referință obținute din ulei de soia, amestec 30:70 ulei de soia - ulei de măsline, și ulei de măsline pur (a se vedea notele 1 și 2 și figurile 1-4) |
|
4.2.11. |
Coloană de extracție în fază solidă, cu fază de silice 1 g, 6 ml |
4.3. Pregătirea probelor
Întrucât existența unui anumit număr de substanțe interferente poate conduce la rezultate fals pozitive, proba trebuie întotdeauna să fie purificată în conformitate cu metoda IUPAC 2.507, utilizată pentru determinarea conținutului de compuși polari în grăsimi de prăjit.
4.3.1. Pregătirea coloanei cromatografice
Se umple coloana (4.1.3) cu aproximativ 30 ml de solvent de eluare (4.2.3), apoi se introduce niște vată de sticlă (4.2.5) în interiorul coloanei și se împinge până la fundul coloanei cu ajutorul baghetei de sticlă (4.1.5).
Într-un pahar Berzelius de 100 ml, se prepară o suspensie cu 25 g de silicagel (4.2.4) în 80 ml de amestec de eluare (4.2.3), apoi se transferă în coloană cu ajutorul unei pâlnii de sticlă (4.1.6).
Pentru a asigura transferul complet al silicagelului în coloană, se spală paharul Berzelius cu amestecul de eluare și se transferă în coloană și lichidul de spălare.
Se deschide robinetul și se permite eluțiunea solventului din coloană până când nivelul său ajunge la aproximativ 1 cm deasupra silicagelului.
4.3.2. Cromatografia în coloană
Cu o precizie de 0,001 g, se cântăresc 2,5 ± 0,1 g de ulei, filtrat anterior, omogenizat și, dacă este necesar, complet deshidratat, într-un balon gradat de 50 ml (4.1.7).
Se dizolvă în aproximativ 20 ml de solvent de eluare (4.2.3). În cazul în care este necesar, se încălzește ușor pentru ca dizolvarea să se facă ușor. Se răcește la temperatura camerei și se aduce la volum cu solvent de eluare.
Cu ajutorul unei pipete gradate, se introduc 20 ml de soluție în interiorul coloanei pregătite în conformitate cu punctul 4.3.1, se deschide robinetul și se permite eluțiunea solventului până la nivelul stratului de silicagel.
Apoi se realizează eluțiunea cu 150 ml de solvent de eluare (4.2.3), reglând debitul solventului la aproximativ 2 ml/min (astfel încât să dureze aproximativ 60-70 de minute pentru ca 150 ml să treacă prin coloană).
Eluatul este recuperat într-un balon cu fund rotund de 250 ml (4.1.1) tarat în prealabil în cuptor și cântărit cu precizie. Se elimină solventul la presiune redusă într-un evaporator rotativ (4.1.9) și se cântărește reziduul care va fi utilizat pentru prepararea soluției pentru analiza HPLC și pentru prepararea metilesterilor.
Recuperarea probei în coloană trebuie să fie în proporție de cel puțin 90 % în cazul categoriilor de ulei extra virgin, virgin și rafinat și în proporție de minimum 80 % în cazul uleiului lampant și al celui din resturi de măsline.
4.3.3. Purificarea SPE (extracție în fază solidă)
Se activează coloana SPE de silice prin trecerea a 6 ml de hexan (4.2.3) sub vid, evitându-se uscarea.
Cu o precizie de 0,001 g, se cântăresc 0,12 g într-un flacon de 2 ml (4.1.15) și se dizolvă în 0,5 ml de hexan (4.2.3).
Se încarcă soluția în coloana SPE și se face eluțiunea cu 10 ml de hexan-dietil eter (87:13 v/v) (4.2.3) sub vid.
Fracțiunea colectată se evaporă până la uscare într-un evaporator rotativ (4.1.9) la presiune scăzută și la temperatura camerei. Se dizolvă reziduul în 2 ml de acetonă (4.2.6) pentru analiza trigliceridelor (TG).
4.4. Analiza HPLC
4.4.1. Pregătirea probelor pentru analiza cromatografică
Se prepară o soluție la 5 % din probele de analizat prin cântărirea a 0,5 ± 0,001 g de probă într-un balon gradat de 10 ml și se completează până la 10 ml cu solventul de solubilizare (4.2.9).
4.4.2. Procedură
Se reglează sistemul cromatografic. Se pompează solvent de eluare (4.2.8) la un debit de 1,5 ml/minut pentru a purja întregul sistem. Se așteaptă până la obținerea unei linii de bază stabile.
Se injectează 10 μl din proba preparată conform indicațiilor de la punctul 4.3.
4.4.3. Calcularea și exprimarea rezultatelor
Se utilizează metoda normalizării ariilor, altfel spus se presupune că suma ariilor picurilor corespunzând trigliceridelor (TG) de la NEC 42 până la NEC 52 este egală cu 100 %.
Se calculează procentajul relativ al fiecărei trigliceride, utilizând formula:
![]()
.
Rezultatele se exprimă cu cel puțin două zecimale.
A se vedea notele 1-4.
4.5. Calculul compoziției trigliceridelor (% moli) pe baza datelor privind compoziția de acizi grași (% arie)
4.5.1. Determinarea compoziției de acizi grași
Compoziția de acizi grași se determină în conformitate cu ISO 5508 cu ajutorul unei coloane capilare. Esterii metilici se prepară conform COI/T.20/Doc. nr. 24.
4.5.2. Acizi grași pentru calcul
Gliceridele sunt grupate după numărul lor echivalent de atomi de carbon (NEC), ținând seama de următoarele echivalențe dintre NEC și acizii grași. Numai acizii grași cu 16 și 18 atomi de carbon au fost luați în considerare, deoarece numai aceștia sunt importanți pentru uleiul de măsline. Acizii grași trebuie să fie normalizați la 100 %.
|
Acid gras (AG) |
Abreviere |
Greutate moleculară (GM) |
NEC |
|
Acid palmitic |
P |
256,4 |
16 |
|
Acid palmitoleic |
Po |
254,4 |
14 |
|
Acid stearic |
S |
284,5 |
18 |
|
Acid oleic |
O |
282,5 |
16 |
|
Acid linoleic |
L |
280,4 |
14 |
|
Acid linolenic |
Ln |
278,4 |
12 |
4.5.3. Conversia în moli a % din arie pentru toți acizii grași (1):
|
|
|
|
|
|
|
|
4.5.4. Normalizarea la 100 % a molilor de acizi grași (2):


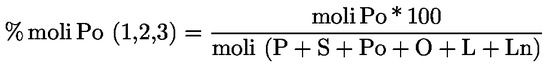


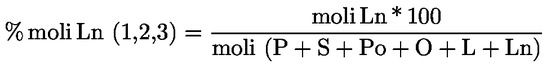
Rezultatul indică procentul din fiecare acid gras, în moli, în poziția globală (1, 2, 3) a TG.
Apoi se calculează suma acizilor grași saturați P și S (AGS) și a acizilor grași nesaturați Po, O, L și Ln (AGN) (3):
![]()
![]()
4.5.5. Calculul compoziției de acizi grași în pozițiile 2 și 1, 3 ale TG
Acizii grași sunt distribuiți în trei grupe, după cum urmează: una pentru poziția 2 și două identice pentru pozițiile 1 și 3, cu coeficienți diferiți pentru acizii saturați (P și S) și acizii nesaturați (PO, O, L și LN).
4.5.5.1. Acizi grași saturați în poziția 2 [P(2) și S(2)] (4):
![]()
![]()
4.5.5.2. Acizi grași nesaturați în poziția 2 [Po(2), O(2), L(2) și Ln(2)] (5):




4.5.5.3. Acizi grași în pozițiile 1,3 [P(1,3), S(1,3), Po(1,3), O(1,3), L(1,3) și Ln(1,3)] (6):
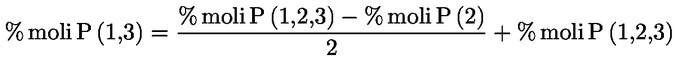

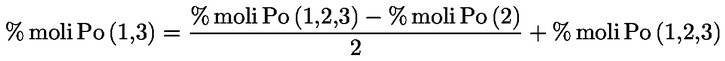



4.5.6. Calculul trigliceridelor
4.5.6.1. TG cu un acid gras (AAA, aici LLL, PoPoPo) (7)
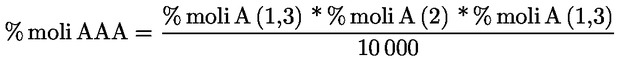
4.5.6.2. TG cu doi acizi grași (AAB, aici PoPoL, PoLL) (8)

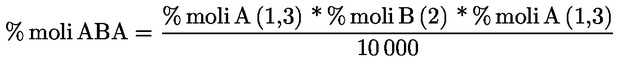
4.5.6.3. TG cu trei acizi grași diferiți (ABC, aici OLLn, PLLn, PoOLn, PPoLn) (9)



4.5.6.4. Trigliceride cu NEC42
Trigliceridele cu NEC42 se calculează cu ajutorul ecuațiilor 7, 8 și 9 și apoi se indică în ordinea eluării preconizate în HPLC (în mod normal, numai trei picuri).
LLL
PoLL și izomerul de poziție LPoL
OLLn și izomerii de poziție OLnL și LnOL
PoPoL și izomerul de poziție PoLPo
PoOLn și izomerii de poziție OPoLn și OLnPo
PLLn și izomerii de poziție LLnP și LnPL
PoPoPo
SLnLn și izomerul de poziție LnSLn
PPoLn și izomerii de poziție PLnPo și PoPLn
Trigliceridele cu NEC42 se obțin însumând cele nouă trigliceride, inclusiv izomerii de poziție ai acestora. Rezultatele se exprimă cu cel puțin două zecimale.
5. EVALUAREA REZULTATELOR
Se compară conținutul teoretic calculat cu cel determinat prin analiza HPLC. Dacă, în valoare absolută, diferența dintre datele HPLC și datele teoretice este mai mare decât valorile specificate pentru categoria corespunzătoare de ulei în standardul de comercializare, proba conține ulei din semințe.
Rezultatele se exprimă cu două zecimale.
6. EXEMPLU (NUMERELE SE REFERĂ LA SECȚIUNILE TEXTULUI METODEI)
— 4.5.1. Calculul % moli de acizi grași pe baza datelor CGL (% arie normalizată)
Următoarele date se obțin prin GLC pentru compoziția de acizi grași:
|
AG |
P |
S |
Po |
O |
L |
Ln |
|
GM |
256,4 |
284,5 |
254,4 |
282,5 |
280,4 |
278,4 |
|
% arie |
10,0 |
3,0 |
1,0 |
75,0 |
10,0 |
1,0 |
— 4.5.3. Conversia în moli a % din arie pentru toți acizii grași [a se vedea formula (1)]:
|
moli P |
= |
|
|
moli S |
= |
|
|
moli Po |
= |
|
|
moli O |
= |
|
|
moli L |
= |
|
|
moli Ln |
= |
|
|
Total |
= |
0,35821 moli TG |
— 4.5.4. Normalizarea la 100 % a molilor de acizi grași [a se vedea formula (2)]:
|
% moli P(1,2,3) |
= |
|
|
% moli S(1,2,3) |
= |
|
|
% moli Po(1,2,3) |
= |
|
|
% moli O(1,2,3) |
= |
|
|
% moli L(1,2,3) |
= |
|
|
% moli Ln(1,2,3) |
= |
|
|
Total % moli |
= |
100 % |
Suma acizilor grași saturați și nesaturați în pozițiile 1,2,3 ale TG [a se vedea formula (3)]:
![]()
![]()
— 4.5.5. Calculul compoziției de acizi grași în pozițiile 2 și 1,3 ale TG
— 4.5.5.1. Acizi grași saturați în poziția 2 [P(2) și S(2)] [a se vedea formula (4)]:
![]()
![]()
— 4.5.5.2. Acizi grași nesaturați în poziția 2 [Po(1,3), O(1,3), L(1,3) și Ln(1,3)] [a se vedea formula (5)]:


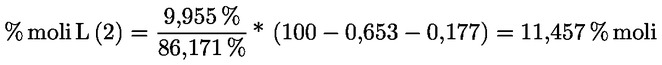

— 4.5.5.3. Acizi grași în pozițiile 1,3 [P(1,3), S(1,3), Po(1,3), O(1,3), L(1,3) și Ln(1,3)] [a se vedea formula (6)]:

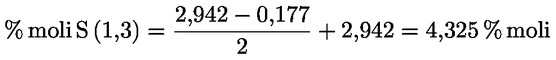
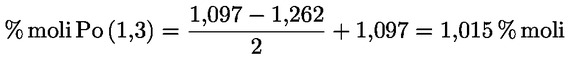



— 4.5.6. Calculul trigliceridelor
Pe baza compoziției de acizi grași în pozițiile 2 și 1,3:
|
AG în |
pozițiile 1,3 |
poziția 2 |
|
P |
16,004 % |
0,653 % |
|
S |
4,325 % |
0,177 % |
|
Po |
1,015 % |
1,262 % |
|
O |
68,526 % |
85,296 % |
|
L |
9,204 % |
11,457 % |
|
Ln |
0,927 % |
1,153 % |
|
Suma |
100,0 % |
100,0 % |
Se calculează trigliceridele următoare:
LLL
PoPoPo
PoLL cu un izomer de poziție
SLnLn cu un izomer de poziție
PoPoL cu un izomer de poziție
PPoLn cu doi izomeri de poziție
OLLn cu doi izomeri de poziție
PLLn cu doi izomeri de poziție
PoOLn cu doi izomeri de poziție
— 4.5.6.1. TG cu un acid gras (LLL, PoPoPo) [a se vedea formula (7)]:
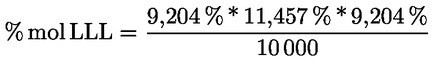 = 0,09706 mol LLL
= 0,09706 mol LLL

— 4.5.6.2. TG cu doi acizi grași (PoLL, SLnLn, PoPoL) [a se vedea formula (8)]:
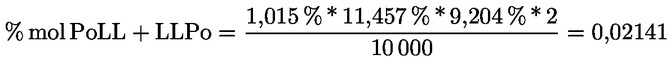

0,03210 mol PoLL

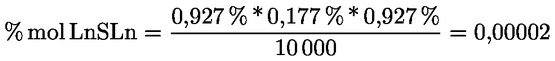
0,00094 mol SLnLn

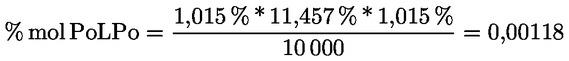
0,00354 mol PoPoL
— 4.5.6.3. TG cu trei acizi grași diferiți (PoPLn, OLLn, PLLn, PoOLn) [a se vedea formula (9)]:



0,00761 mol PPoLn


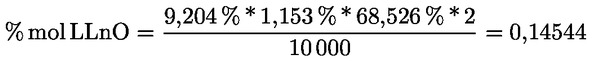
0,43655 mol OLLn



0,06907 mol PLLn

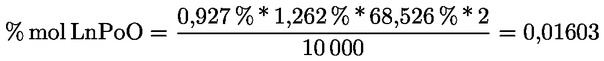

0,04812 mol PoOLn
NEC42 = 0,69512 mol TG
Nota 1: Este posibil să se determine ordinea de eluare prin calcularea numerelor echivalente de atomi de carbon, definite adeseori prin raportul ![]() , unde NC este numărul atomilor de carbon și n numărul de legături duble. Calculul se poate face cu o precizie mai mare ținând seama de originea legăturilor duble. Dacă no, nl și nln reprezintă numărul de legături duble ce pot fi atribuite acizilor oleic, linoleic și linolenic, numărul echivalent de atomi de carbon poate fi calculat după formula:
, unde NC este numărul atomilor de carbon și n numărul de legături duble. Calculul se poate face cu o precizie mai mare ținând seama de originea legăturilor duble. Dacă no, nl și nln reprezintă numărul de legături duble ce pot fi atribuite acizilor oleic, linoleic și linolenic, numărul echivalent de atomi de carbon poate fi calculat după formula:
![]()
unde coeficienții do, dl și dln pot fi calculați cu ajutorul trigliceridelor de referință. În condițiile specificate în prezenta metodă, rezultatul obținut este comparabil cu formula:
![]()
Nota 2: Cu mai multe trigliceride de referință, este posibil, de asemenea, să se calculeze rezoluția pentru trioleină:
![]()
trioleinăutilizând timpii de retenție corectați
![]()
.
Reprezentarea grafică a log α în funcție de f (număr de legături duble) permite determinarea indicilor de retenție pentru toate trigliceridele acizilor grași conținuți în trigliceridele de referință (a se vedea figura 1).
Nota 3: Eficacitatea coloanei trebuie să permită separarea netă a picului trilinoleinei de picurile trigliceridelor care au timpi de retenție apropiați. Eluarea se face până la picul NEC 52.
Nota 4: O măsurătoare corectă a ariilor tuturor picurilor de interes pentru prezenta determinare este asigurată dacă al doilea pic corespunzător NEC 50 este de 50 % din maximul scalei înregistratorului.
Figura 1
Reprezentarea grafică a log α în funcție de f (număr de legături duble)
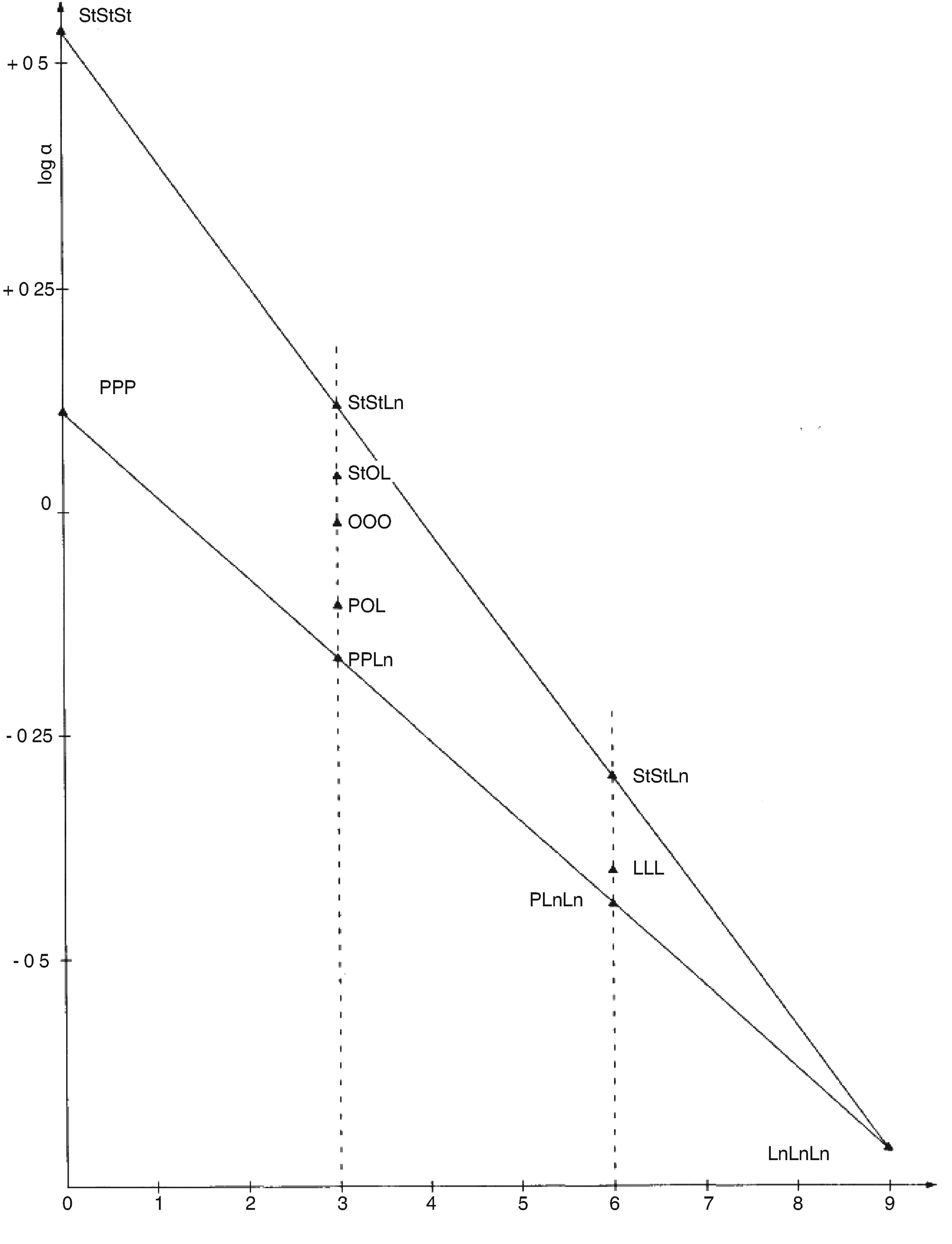
Numărul de legături duble
La: acid lauric; My: acid miristic; P: acid palmitic; S: acid stearic; O: acid oleic; L: acid linoleic; Ln: acid linolenic.
Figura 2
Ulei de măsline cu conținut scăzut de acid linoleic
(a)

Solvent: Acetonă/acetonitril.
Profilul a: componentele principale ale picurilor cromatografice: NEC42: (1) LLL + PoLL; (2) OLLn + PoOLn; (3) PLLn; NEC44: (4) OLL + PoOL; (5) OOLn + PLL; (6) POLn + PPoPo; (7) OOL + PoOO; NEC46: (8) OOL + LnPP; (9) PoOO; (10) SLL + PLO; (11) PoOP + SPoL + SOLn + SPoPo; (12) PLP; NEC48: (13) OOO + PoPP; (14 + 15) SOL + POO; (16) POP; NEC50: (17) SOO; (18) POS + SLS.
(b)

Solvent: Propionitril
Profilul b: componentele principale ale picurilor cromatografice: NEC42: (1) LLL; (2) OLLn + PoLL; (3) PLLn; NEC44: (4) OLL; (5) OOLn + PoOL; (6) PLL + PoPoO; (7) POLn + PPoPo + PPoL; NEC46: (8) OOL + LnPP; (9) PoOO; (10) SLL + PLO; (11) PoOP + SPoL + SOLn + SPoPo; (12) PLP; NEC48: (13) OOO + PoPP; (14) SOL; (15) POO; (16) POP; NEC50: (17) SOO; (18) POS + SLS.
Figura 3
Ulei de măsline cu conținut ridicat de acid linoleic
(a)
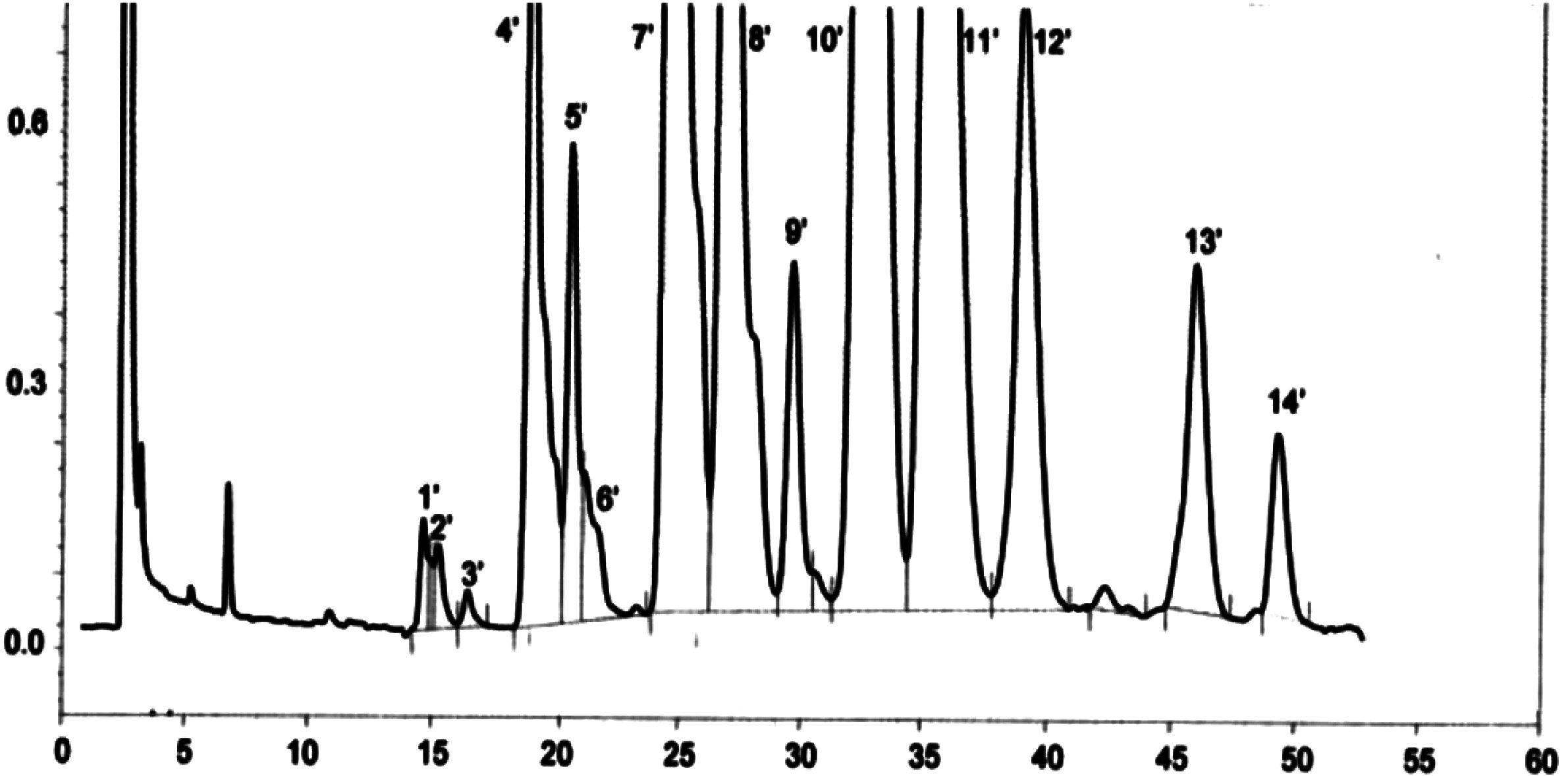
Solvent: Acetonă/acetonitril (50:50).
Profil a: componentele principale ale picurilor cromatografice: NEC42: (1’) LLL + PoLL; (2’) OLLn + PoOLn; (3’) PLLn; NEC44: (4’) OLL + PoOL; (5’) OOLn + PLL; (6’) POLn + PPoPo; NEC46: (7’) OOL + PoOO; (8’) PLO + SLL + PoOP; (9’) PLP + PoPP; NEC48: (10’) OOO; (11’) POO + SLL + PPoO; (12’) POP + PLS; NEC50: (13’) SOO; (14’) POS + SLS.
(b)
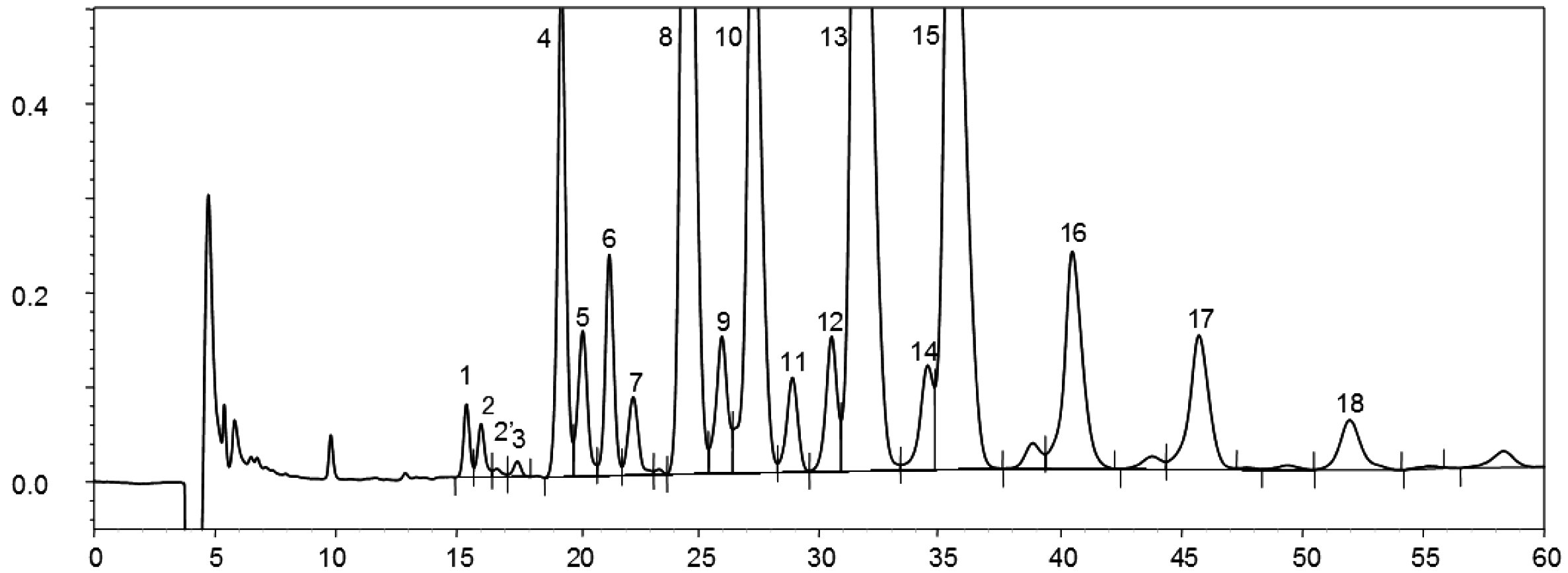
Solvent: Propionitril.
Profilul b: componentele principale ale picurilor cromatografice: NEC42: (1) LLL; (2 + 2’) OLLn + PoLL; (3) PLLn; NEC44: (4) OLL; (5) OOLn + PoOL; (6) PLL + PoPoO; (7) POLn + PPoPo + PPoL; NEC46: (8) OOL + LnPP; (9) PoOO; (10) SLL + PLO; (11) PoOP + SPoL + SOLn + SPoPo; NEC48: (12) PLP; (13) OOO + PoPP; (14) SOL; (15) POO; (16) POP; NEC50: (17) SOO; (18) POS + SLS; NEC52: (19) AOO.
ANNEX XIX
DETERMINAREA CONȚINUTULUI DE ALCOOLI ALIFATICI ȘI TRITERPENICI PRIN CROMATOGRAFIE ÎN FAZĂ GAZOASĂ CU COLOANĂ CAPILARĂ
1. OBIECT
Prezenta anexă descrie o metodă de determinare a conținutului de alcooli alifatici și triterpenici din uleiuri și grăsimi.
2. PRINCIPLE OF THE METHOD
The fatty substance, with 1-eicosanol added as internal standard, is saponified with ethanolic potassium hydroxide and then the unsaponifiable matter extracted with ethyl ether. The alcoholic fraction is separated from the unsaponifiable matter by chromatography on a basic silica gel plate; the alcohols recovered from the silica gel are transformed into trimethylsilyl ethers and analysed by capillary gas chromatography.
3. EQUIPMENT
3.1. 250 ml round-bottomed flask fitted with a reflux condenser having ground-glass joints.
3.2. 500 ml separating funnel.
3.3. 250 ml round-bottomed flasks.
3.4. Chromatographic tank for thin-layer chromatographic analysis, for glass plates of dimensions 20 x 20 cm.
3.5. Ultraviolet lamp having a wavelength of 366 or 254 nm.
3.6. 100 μl and 500 μl microsyringes.
3.7. A cylindrical filter funnel with a G3 porous septum (porosity 15 to 40 μm) of diameter approximately 2 cm and a depth of some 5 cm, with an attachment suitable for filtration under vacuum and a 12/21 male ground glass joint.
3.8. 50 ml vacuum conical flask with a 12/21 ground-glass female joint which can be fitted to the filter funnel (3.7).
3.9. A 10 ml test tube with a tapering bottom and a sealing stopper.
3.10. Gas chromatograph for use with a capillary column, and provided with a splitting system composed of:
3.10.1. Thermostatic chamber for columns (column oven) to hold the temperature desired with a precision of ± 1 °C.
3.10.2. A temperature-adjustable injection unit with a persilanised glass vaporising element.
3.10.3. A flame ionisation detector and converter-amplifier.
3.10.4. Recorder-integrator for operation with the converter-amplifier (3.10.3), with response time not exceeding one second and with variable paper-speed.
3.11. Glass or fused silica capillary column, of length 20 to 30 m, internal diameter 0,25 to 0,32 mm, with SE-52 or SE-54 liquid phase or equivalent, with a film thickness between 0,10 and 0,30 μm.
3.12. Microsyringe for gas chromatography, of 10 μl capacity with hardened needle.
3.13. Analytical balance sensitive to 1 mg (with 0,1 mg display).
4. REAGENTS
4.1. Potassium hydroxide, approximately 2 N ethanolic solution: 130 g potassium hydroxide (minimum concentration 85 %) is dissolved, with cooling, in 200 ml distilled water and then made up to one litre with ethanol. The solution should be stored in a well-stoppered opaque glass bottle.
4.2. Ethyl ether, pure for analysis.
4.3. Anhydrous sodium sulphate, analytical purity.
4.4. Glass plates coated with silica gel, without fluorescence indicator, thickness 0,25 mm (commercially available ready for use).
4.5. Potassium hydroxide, approximately 0,2 N ethanolic solution; 13 g of potassium hydroxide are dissolved in 20 ml of distilled water and made up to one litre with ethanol.
4.6. Benzene, for chromatography (see 5.2.2).
4.7. Acetone, for chromatography (See 5.2.2).
4.8. Hexane, for chromatography (see 5.2.2).
4.9. Ethyl ether, for chromatography (see 5.2.2).
4.10. Chloroform, for chromatography.
4.11. Soluție de referință pentru cromatografie în strat subțire: alcooli C20-C28 de 0,5 % în cloroform, sau o fracțiune de alcooli obținută conform indicațiilor de la punctul 5.2 din substanța nesaponificabilă a unui ulei din resturi de măsline.
4.12. 0,2 % solution of 2', 7'-dichlorofluorescein in ethanol. Make slightly basic by adding a few drops of 2 N alcoholic potassium hydroxide solution.
4.13. Anhydrous pyridine, for chromatography.
4.14. Hexamethyl disilazane.
4.15. Trimethylchlorosilane.
4.16. Standard solutions of trimethylsilyl ethers of aliphatic alcohols from C20 to C28. They may be prepared from mixtures of pure alcohols at the time they are required for use.
4.17. A 0,1 % (m/v) solution of 1-eicosanol in chloroform (internal standard).
4.18. Carrier gas: hydrogen or helium, gas-chromatographic purity.
4.19. Auxiliary gas: nitrogen, gas-chromatographic purity.
5. PROCEDURE
5.1. Preparation of the unsaponifiables
|
5.1.1. |
Using a 500 μl microsyringe place, into a 250 ml round-bottom flask, a volume of 0,1 % 1-eicosanol solution in chloroform (4.17) containing a quantity of 1-eicosanol approximately equal to 10 % of the aliphatic alcohols content in that portion of sample to be taken for analysis. For example, to 5 g of sample add 250 μl of the 0,1 % 1-eicosanol solution if olive oil and 1 500 μl if olive pomace oil. Evaporate to dryness in current of nitrogen and then weigh accurately 5 g of the dry filtered sample into the same flask. |
|
5.1.2. |
Add 50 ml of 2 N potassium hydroxide ethanolic solution, fit the reflux condenser and heat the apparatus to slight boiling on a steam bath, stirring continuously throughout the heating process until saponification has taken place (the solution becomes clear). Continue heating for a further 20 minutes and then add 50 ml of distilled water through the condenser. The condenser is then disconnected and the flask cooled to approximately 30 °C. |
|
5.1.3. |
The contents of the flask are quantitatively transferred to a separating funnel of 500 ml capacity by adding distilled water several times, using a total of around 50 ml distilled water. Add approximately 80 ml of ethyl ether, shake vigorously for approximately 30 seconds and allow to settle (Note 1). Separate off the lower aqueous phase collecting it in a second separating funnel. Two further extractions are effected on the aqueous phase, in the same manner, using each time 60 to 70 ml ethyl ether. Note 1: Emulsions may be eliminated by adding, using as a spray, small quantities of ethyl alcohol or methyl alcohol. |
|
5.1.4. |
The ethyl ether extracts are combined in a separating funnel and washed with distilled water (50 ml at a time) until the washing water gives a neutral reaction. Discard the washing water, dry with anhydrous sodium sulphate and filter, into a flask of 250 ml capacity which has been weighed beforehand, the funnel and filter being washed with small quantities of ethyl ether which are added to the total. |
|
5.1.5. |
Distil the ether down to a few ml, then bring to dryness under a slight vacuum or in a current of nitrogen, completing drying in an oven at 100 °C for approximately a quarter of an hour, and then weigh after cooling in a desiccator. |
5.2. Separation of alcoholic fractions
|
5.2.1. |
Preparation of basic TLC plates: the silica gel plates (4.4) are immersed completely, in 0,2 N potassium hydroxide solution (4.5) for 10 seconds, and then left to dry for two hours under an extractor hood and finally placed in an oven at 100 °C for one hour. Remove from the oven and keep in a calcium chloride desiccator until required for use (plates treated in this way must be used within 15 days). Note 2: When basic silica gel plates are used to separate the alcoholic fraction there is no need to treat the unsaponifiables with alumina. It follows that all acid compounds (fatty acids and others) are retained at the origin thereby obtaining both aliphatic alcohol and terpenic alcohol bands which are both separated distinctly from the sterol band. |
|
5.2.2. |
Place a 65/35 by volume hexane/ethyl ether mixture in the plate-developing chamber to a depth of approximately 1 cm ( 6 ). Close the chamber using an appropriate cover and leave for half an hour to allow equilibration between vapour and liquid. Strips of filter paper dipping into the eluent may be affixed to the inside surfaces of the tank to reduce the development time by approximately one third and obtain more uniform, regular elution of the components. Note 3: The developing solution must be replaced for each analysis in order to obtain reproducible developing conditions. |
|
5.2.3. |
An approximately 5 % solution of unsaponifiable matter (5.1.5) in chloroform is prepared and 0,3 ml of the solution is streaked as a uniform strip of minimum thickness, using the 100 μl microsyringe, on a TLC plate at approximately 2 cm from the bottom of the TLC plate. Aligned with the origin, 2 to 3 μl of the aliphatic alcohols reference solution (4.11) are spotted for the identification of the aliphatic alcohols band after development has been completed. |
|
5.2.4. |
Place the plate inside the development tank as stated in 5.2.2. The ambient temperature should be maintained between 15 and 20 °C. Immediately close the chamber with the cover and allow to elute until the solvent front reaches approximately 1 cm from the upper edge of the plate. The plate is then removed from the development chamber and the solvent evaporated under a hot air current or the plate is left for a while under the extractor hood. |
|
5.2.5. |
Placa se pulverizează ușor și uniform cu soluție de 2′, 7′-diclorofluoresceină atunci când placa este observată în lumină ultravioletă. Banda de alcooli alifatici poate fi identificată prin faptul că este aliniată cu pata obținută din soluția de referință: se marchează limitele benzii cu un creion negru; evidențiind banda de alcooli alifatici și banda imediat superioară, care este banda de alcooli terpenici, împreună (nota 4). Nota 4: Banda alcoolilor alifatici și banda alcoolilor terpenici trebuie grupate împreună în vederea posibilei migrări a unor alcooli alifatici în banda alcoolilor triterpenici. Un exemplu de separare TLC este oferit în figura 1 din apendice. |
|
5.2.6. |
Se răzuiește silicagelul cu o spatulă metalică în zona delimitată. Materialul extras, sfărâmat fin, se introduce în pâlnia de filtrare (3.7). Se adaugă 10 ml de cloroform cald, se amestecă cu grijă cu o spatulă metalică și se filtrează cu ajutorul vidului, apoi se colectează filtratul în balonul conic (3.8) conectat la pâlnia de filtrare. Se spală silicagelul din balon de trei ori cu eter etilic (aproximativ 10 ml de fiecare dată) și se colectează filtratul în același balon conectat la pâlnie. Se evaporă filtratul până ajunge la un volum de 4-5 ml, se transvazează soluția reziduală în eprubeta de 10 ml (3.9) cântărită în prealabil, se evaporă până la uscare prin încălzire ușoară într-un curent ușor de azot, se completează din nou folosind câteva picături de acetonă, se evaporă din nou până la uscare, se introduce într-un cuptor la 105 °C timp de aproximativ 10 minute și apoi se lasă să se răcească în exsicator și se cântărește. Reziduul din interiorul eprubetei este compus din fracțiunea alcoolilor. |
5.3. Preparation of the trimethylsilyl ethers
|
5.3.1. |
The reagent for silylation, consisting of a mixture of 9:3:1 by volume (Note 5) of pyridine-hexamethyldisilazane-trimethylchlorosilane in the proportion of 50 μl for each milligram of aliphatic alcohols, is added to the test tube containing the alcoholic fraction, avoiding all absorption of moisture (Note 6). Note 5: Solutions which are ready for use are available commercially. Other silanising reagents such as, for example, bis-trimethylsilyl, trifluor acetamide + 1 % trimethyl chlorosilane, which has to be diluted with an equal volume of anhydrous pyridine, are also available. Note 6: The slight opalescence which may form is normal and does not cause any interference. The formation of a white floc or the appearance of a pink colour are indicative of the presence of moisture or deterioration of the reagent. If these occur the test must be repeated. |
|
5.3.2. |
Stopper the test tube, shake carefully (without overturning) until the aliphatic alcohols are completely dissolved. Stand for at least 15 minutes at ambient temperature and then centrifuge for a few minutes. The clear solution is ready for gas chromatographic analysis. |
5.4. Gas chromatography analysis
5.4.1. Preliminary operations, column packing
|
5.4.1.1. |
Fit the column in the gas chromatograph, attaching the inlet end to the injector connected to the splitting system and the outlet end to the detector. Carry out a general check of the gas chromatography assembly (tightness of gas fittings, efficiency of the detector, efficiency of the splitting system and of the recording system, etc.). |
|
5.4.1.2. |
If the column is being used for the first time it is recommended that it should be subjected to conditioning. A little carrier gas is passed through the capillary column and then the gas chromatography assembly is switched on and gradually heated until a temperature not less than 20 °C above the operating temperature (see Note 7) is attained. That temperature is held for not less than two hours and then the assembly is brought to the operating conditions (regulation of gas flow, split flame ignition, connection to the electronic recorder, adjustment of the temperature of the capillary column oven, the detector and the injector, etc.) and the signal is adjusted to a sensitivity not less than twice the highest level contemplated for the execution of the analysis. The course of the base line must be linear, without peaks of any kind, and must not drift. A negative straight-line drift indicates leakage from the column connections; a positive drift indicates inadequate conditioning of the column. Note 7: The conditioning temperature shall be at least 20 °C less than the maximum temperature contemplated for the liquid phase employed. |
5.4.2. Choice of operating conditions
5.4.2.1. The guideline operating conditions are as follows:
— column temperature: the initial isotherm is set at 180 °C for eight minutes and then programmed at 5 °C/minute to 260 °C and a further 15 minutes at 260 °C,
— temperature of evaporator: 280 °C,
— temperature of detector: 290 °C,
— linear velocity of carrier gas: helium 20 to 35 cm/s, hydrogen 30 to 50 cm/s,
— splitting ratio: 1:50 to 1:100,
— sensitivity of instrument: 4 to 16 times the minimum attenuation,
— sensitivity of recording: 1 to 2 mV fs,
— paper speed: 30 to 60 cm/h,
— quantity of substance injected: 0,5 to 1 μl of TMSE solution.
The above conditions may be modified according to the characteristics of the column and of the gas chromatograph to obtain chromatograms satisfying the following conditions:
— alcohol C26 retention time shall be 18 ± 5 minutes,
— the alcohol C22 peak shall be 80 ± 20 % of the full scale value for olive oil and 40 ± 20 % of the full scale value for seed oil.
5.4.2.2. The above requirements are checked by repeated injection of the standard TMSE mixture of alcohols and the operating conditions are adjusted to yield the best possible results.
5.4.2.3. The parameters for the integration of peaks shall be set so that a correct appraisal of the areas of the peaks considered is obtained.
5.4.3. Analytical procedure
5.4.3.1. Using the microsyringe of 10 μl capacity draw in 1 μl of hexane followed by 0,5 μl of air and subsequently 0,5 to 1 μl of the sample solution; raise the plunger of the syringe further so the needle is emptied. Push the needle through the membrane of the injection unit and after one to two seconds inject rapidly, then slowly remove the needle after some five seconds.
5.4.3.2. Recording is effected until the TMSE of the aliphatic alcohols present have been eluted completely. The base line shall always correspond to the requirements of 5.4.1.2.
5.4.4. Identificarea valorilor de vârf
Identificarea valorilor de vârf individuale se efectuează pe baza timpilor de retenție și prin comparație cu amestecul etalon TMSE, analizați în aceleași condiții.
Exemple de cromatogramă pentru fracțiunea alcoolilor unui ulei de măsline rafinat sunt prezentate în figurile 2 și 3 din apendice.
5.4.5. Quantitative evaluation
5.4.5.1. The peak areas of 1-eicosanol and of the aliphatic alcohols C22, C24, C26 and C28 are calculated by electronic integration.
5.4.5.2. The contents of each aliphatic alcohol, expressed in mg/
1 000
g fatty substance, are calculated as follows:
![]()
where:
Ax = area of the alcohol peak x
As = area of 1-eicosanol
ms = mass of 1-eicosanol in milligrams
m = mass of sample drawn for determination, in grams.
6. EXPRESSION OF THE RESULTS
The contents of the individual aliphatic alcohols in mg/1 000 g of fatty substance and the sum of the „total aliphatic alcohols” are reported.
Apendice
Exemplu de separare TLC și exemple de cromatogramă
Figura 1
Placă pentru cromatografie pe strat subțire a fracțiunii nesaponificabile din uleiul de măsline eluat cu hexan/eter etilic (65/35)

|
1 |
Alcool C26 |
A |
Steroli |
|
2 |
Alcool C30 |
B |
Alcooli alifatici |
|
3 |
Alcool C20 |
C |
Alcooli triterpenici |
|
4 |
Amestec de alcooli C20-22-26-30 |
D |
Scualen |
|
5 |
Ulei extravirgin nesaponificabil |
|
|
Figura 2
Cromatograma fracțiunii alcoolice a unui ulei rafinat
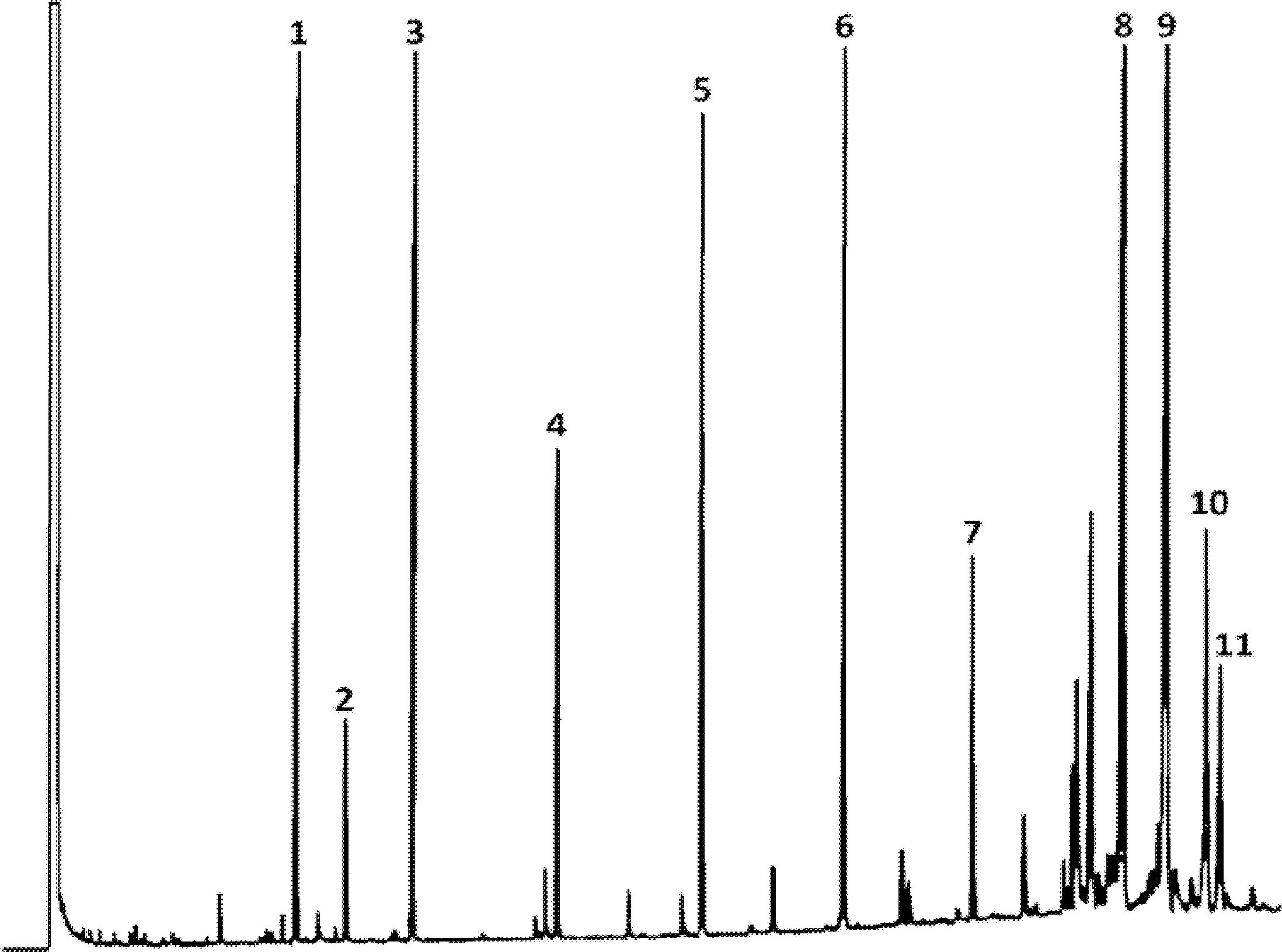
|
1 = Fitol |
5 = Alcool C24 |
9 = 24-Metilen-cicloartenol |
|
2 = Geranil geraniol |
6 = Alcool C26 |
10 = Citrostadienol |
|
3 = Alcool C20 (IS) |
7 = Alcool C28 |
11 = Ciclobranol |
|
4 = Alcool C22 |
8 = Cicloartenol |
|
Figura 3
Alcooli alifatici și triterpenici ai uleiului de măsline rafinat și ai unui ulei de măsline obținut la a doua centrifugare
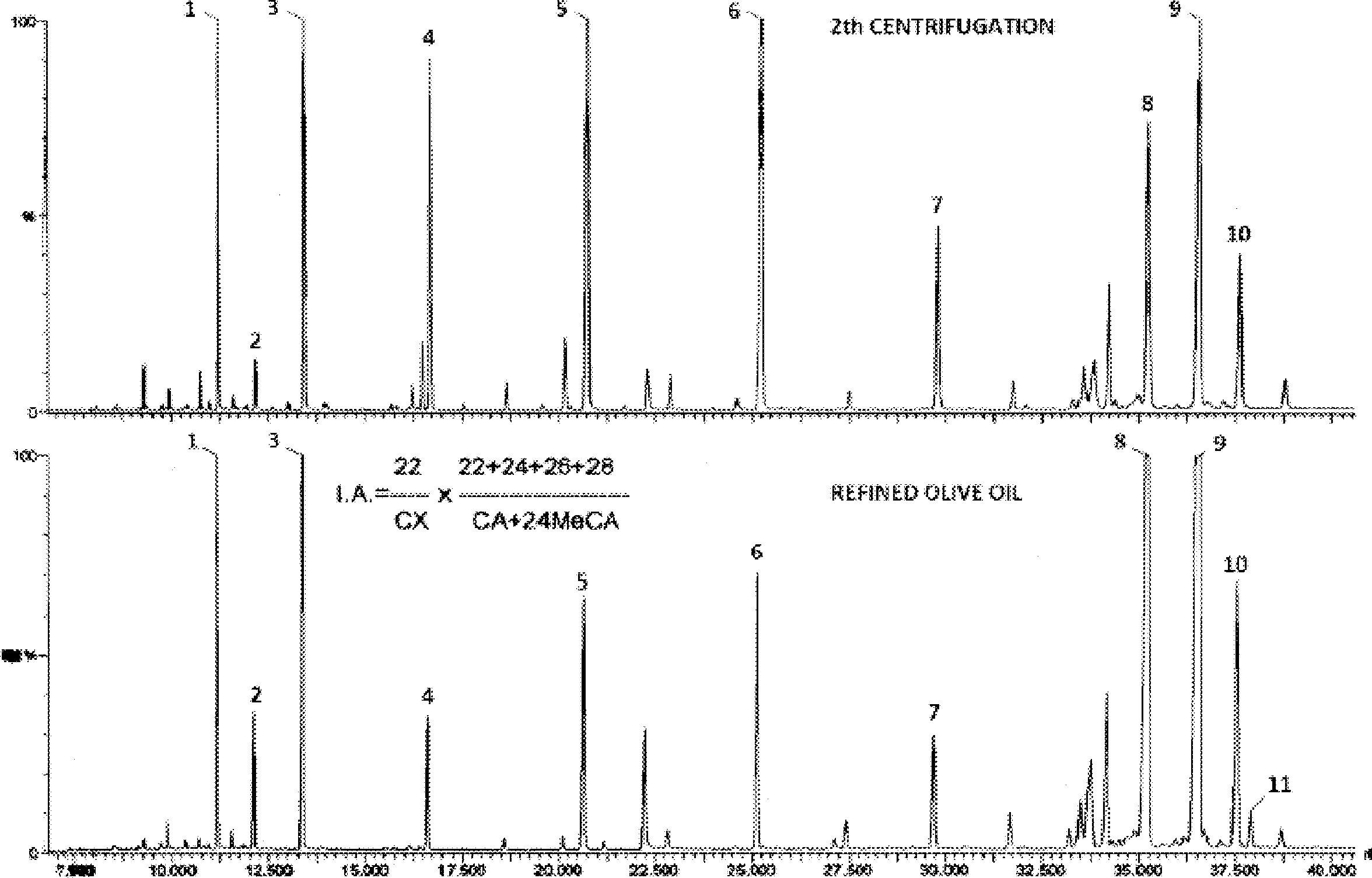
|
1 = Fitol |
5 = Alcool C24 |
9 = 24-Metilen-cicloartenol (24MeCA) |
|
2 = Geranil geraniol (CX) |
6 = Alcool C26 |
10 = Citrostadienol |
|
3 = Alcool C20 |
7 = Alcool C28 |
11 = Ciclobranol |
|
4 = Alcool C22 |
8 = Cicloartenol (CA) |
|
ANEXA XX
Metoda pentru determinarea conținutului de ceruri, esteri metilici ai acizilor grași și esteri etilici ai acizilor grași prin cromatografie în fază gazoasă cu coloană capilară
1. SCOP
Această metodă se utilizează pentru determinarea conținutului de ceruri, esteri metilici ai acizilor grași și esteri etilici ai acizilor grași în uleiurile de măsline. Cerurile și esterii alchilici se separă în funcție de numărul atomilor de carbon. Metoda se recomandă ca instrument pentru a face distincția între uleiul de măsline și uleiul din turte de măsline, precum și ca parametru de calitate pentru uleiurile de măsline extravirgine, permițând detectarea amestecurilor frauduloase de uleiuri de măsline extravirgine cu uleiuri de calitate inferioară, indiferent dacă este vorba despre uleiuri de măsline virgine, lampante sau unele uleiuri deodorizate.
2. PRINCIPIU
Adăugarea unor standarde interne adecvate pentru ulei, apoi fracționarea prin cromatografie pe o coloană cu silicagel hidratat. Recuperarea fracției eluate în condițiile de testare (cu o polaritate mai mică decât cea a triacilglicerolilor), apoi analiza directă prin cromatografie în fază gazoasă cu coloană capilară.
3. APARATURĂ
|
3.1. |
Pahar Erlenmayer, 25 ml. |
|
3.2. |
Coloană de sticlă pentru cromatografie în fază lichidă, diametru interior 15 mm, înălțime 30-40 cm, echipată cu un robinet adecvat. |
|
3.3. |
Cromatograf în fază gazoasă adecvat pentru funcționarea cu coloană capilară, echipat cu un sistem de introducere directă în coloană, alcătuit din:
|
|
3.4. |
Microseringă, 10 μl, cu ac călit, pentru injecție directă pe coloană. |
|
3.5. |
Agitator electric. |
|
3.6. |
Evaporator rotativ. |
|
3.7. |
Cuptor cu muflă. |
|
3.8. |
Balanță analitică pentru cântărirea cu o acuratețe de ± 0,1 mg. |
|
3.9. |
Sticlărie de laborator obișnuită. |
4. REACTIVI
|
4.1. |
Silicagel, 60-200 μm. Silicagelul se păstrează în cuptorul cu muflă la 500 °C timp de cel puțin patru ore. Se lasă să se răcească, apoi se adaugă 2% apă în raport cu cantitatea de silicagel utilizată. Se agită bine pentru omogenizarea suspensiei și se păstrează în desicator timp de cel puțin 12 ore înainte de utilizare. |
|
4.2. |
n-hexan, pentru cromatografie sau analiza reziduurilor (puritatea trebuie verificată). AVERTISMENT – Gazele de evacuare se pot aprinde. A se ține la distanță de surse de căldură, scântei sau flăcări deschise. A se verifica întotdeauna etanșeitatea recipientelor. A se asigura ventilarea corespunzătoare în timpul utilizării. A se evita acumularea gazelor de evacuare și a se lua măsuri de eliminarea a oricăror surse de incendiu, precum radiatoarele sau aparatele electrice care nu sunt fabricate din materiale neinflamabile. Toxic la inhalare, poate cauza deteriorarea celulelor sistemului nervos. A se evita inspirarea gazelor de evacuare. A se utiliza, dacă este necesar, un aparat de respirat adecvat. A se evita contactul cu ochii și cu pielea. |
|
4.3. |
Eter etilic, pentru cromatografie.
AVERTISMENT – Foarte inflamabil și cu toxicitate moderată. Irită pielea. Toxic la inhalare. Poate provoca leziuni ale ochilor. Efectele pot apărea cu întârziere. Poate da naștere unor peroxizi explozivi. Gazele de evacuare se pot aprinde. A se ține la distanță de surse de căldură, scântei sau flăcări deschise. A se verifica întotdeauna etanșeitatea recipientelor. A se asigura ventilarea corespunzătoare în timpul utilizării. A se evita acumularea gazelor de evacuare și a se lua măsuri de eliminarea a oricăror surse de incendiu, precum radiatoarele sau aparatele electrice care nu sunt fabricate din materiale neinflamabile. A nu se evapora până la desicare totală sau cvasitotală. Adăugarea de apă sau alt agent reducător adecvat poate diminua formarea de peroxizi. A nu se ingera. A se evita inspirarea gazelor de evacuare A se evita contactul prelungit sau repetat cu pielea. |
|
4.4. |
n-heptan, pentru cromatografie, sau izooctan. AVERTISMENT – Inflamabil. Toxic la inhalare. A se ține la distanță de surse de căldură, scântei sau flăcări deschise. A se verifica întotdeauna etanșeitatea recipientelor. A se asigura ventilarea corespunzătoare în timpul utilizării. A se evita inspirarea gazelor de evacuare A se evita contactul prelungit sau repetat cu pielea. |
|
4.5. |
Soluție standard de lauril arahidat (nota 3), la 0,05 % (m/V) în heptan (standard intern pentru ceruri). Nota 3: Se poate utiliza, de asemenea, palmitat de palmitil, stearat de miristil sau laureat de arahidil. |
|
4.6. |
Soluție standard de metil heptadecanoat, la 0,02 % (m/V) în heptan (standard intern pentru esteri metilici și etilici). |
|
4.7. |
Sudan 1 (1-fenilazo-2-naftol). |
|
4.8. |
Gaz transportor: hidrogen sau heliu, pur, pentru cromatografie în fază gazoasă. AVERTISMENT Hidrogen. Foarte inflamabil, sub presiune. A se ține la distanță de surse de căldură, scântei, flăcări deschise sau aparate electrice care nu sunt fabricate din materiale neinflamabile. A se verifica dacă supapa buteliei este bine închisă atunci când aceasta nu este utilizată. A se utiliza întotdeauna cu reductor de presiune. A se slăbi resortul reductorului înainte de a deschide supapa buteliei. Nu stați în fața punctului de ieșire a gazului din butelie la deschiderea supapei. A se asigura ventilarea corespunzătoare în timpul utilizării. A nu se transfera hidrogen dintr-o butelie în alta. A nu se amesteca gazul din butelie. Asigurați-vă că buteliile nu pot fi răsturnate. A se păstra la adăpost de lumina soarelui și de surse de căldură. A nu se depozita în mediu corosiv. A nu se utiliza butelii deteriorate sau neetichetate. Heliu. Gaz comprimat sub presiune ridicată. Reduce cantitatea de oxigen disponibilă pentru respirație. A se păstra recipientul închis. A se asigura ventilarea corespunzătoare în timpul utilizării. A nu se intra în spațiile de depozitare dacă nu sunt ventilate adecvat. A se utiliza întotdeauna cu reductor de presiune. A se slăbi resortul reductorului înainte de a deschide supapa buteliei. A nu se transfera gaz dintr-o butelie în alta. Asigurați-vă că buteliile nu pot fi răsturnate. Nu stați în fața punctului de ieșire a gazului din butelie la deschiderea supapei. A se păstra la adăpost de lumina soarelui și de surse de căldură. A nu se depozita în mediu corosiv. A nu se utiliza butelii deteriorate sau neetichetate. A nu se inhala. A se utiliza exclusiv pentru aplicații tehnice. |
|
4.9. |
Gaze auxiliare: — hidrogen, pur, pentru cromatografie în fază gazoasă. — aer, pur, pentru cromatografie în fază gazoasă. AVERTISMENT Aer. Gaz comprimat sub presiune ridicată. A se utiliza cu atenție în prezența substanțelor combustibile, deoarece temperatura de autoaprindere pentru majoritatea compușilor organici în aer se reduce substanțial la presiune mare. A se verifica dacă supapa buteliei este bine închisă atunci când aceasta nu este utilizată. A se utiliza întotdeauna un reductor de presiune. A se slăbi resortul reductorului înainte de a deschide supapa buteliei. Nu stați în fața punctului de ieșire a gazului din butelie la deschiderea supapei. A nu se transfera gaz dintr-o butelie în alta. A nu se amesteca gazul din butelie. Asigurați-vă că buteliile nu pot fi răsturnate. A se păstra la adăpost de lumina soarelui și de surse de căldură. A nu se depozita în mediu corosiv. A nu se utiliza butelii deteriorate sau neetichetate. Aerul destinat utilizării în aplicații tehnice nu trebuie inhalat sau utilizat pentru aparatele de respirat. |
5. PROCEDURA
5.1. Pregătirea coloanei cromatografice
Se prepară o suspensie de 15 g de silicagel (punctul 4.1) în n-hexan (punctul 4.2) și se introduce pe coloană (punctul 3.2). Se lasă să se clarifice spontan. Se definitivează clarificarea cu ajutorul unui agitator electric pentru a face banda de cromatografie mai omogenă. Se percolează 30 ml de n-hexan pentru a înlătura orice impurități. Cu ajutorul balanței analitice (punctul 3.8), se cântăresc exact 500 mg de probă într-o fiolă de 25 ml (punctul 3.1) și se adaugă o cantitate adecvată de soluție standard intern (punctul 4.5), în funcție de conținutul de ceară estimat, de exemplu se adaugă 0,1 mg de lauril arahidat în cazul uleiului de măsline, 0,25-0,50 mg în cazul uleiului din turte de măsline și 0,05 mg de metil heptadecanoat în cazul uleiurilor de măsline (punctul 4.6).
Se transferă proba preparată în coloana cromatografică cu ajutorul dozelor de 2 ml de n-hexan (punctul 4.2).
Se lasă solventul să curgă până la 1 mm deasupra nivelului superior al absorbantului. Se percolează un amestec de n-hexan/eter etilic (99:1) și se colectează 220 ml la un debit de aproximativ 15 picături la fiecare 10 secunde. (Această fracțiune conține esterii metilici, esterii etilici și cerurile). (Nota 4) (Nota 5).
Nota 4: Amestecul de n-hexan/eter etilic (99:1) trebuie să fie proaspăt pregătit în fiecare zi.
Nota 5: Pentru a verifica vizual eluția corectă a cerurilor, se poate adăuga la soluția probei 100 μl de colorant Sudan I diluat la 1 % în amestecul de eluție.
Colorantul are un timp de retenție cuprins între cel al cerurilor și cel al triacilglicerolilor. Prin urmare, atunci când colorantul atinge fundul coloanei cromatografice, eluția trebuie suspendată deoarece toate cerurile au fost eluate.
Se evaporă fracțiunile rezultate într-un evaporator rotativ până când aproape tot solventul este eliminat. Se elimină ultimii 2 ml de solvent cu ajutorul unui curent slab de azot. Se colectează fracțiunea care conține esterii metilici și etilici, diluată cu 2-4 ml de n-heptan sau izooctan.
5.2. Analiza prin cromatografie în fază gazoasă
5.2.1. Procedura preliminară
Se instalează coloana în cromatograful în fază gazoasă (punctul 3.3), conectând extremitatea de intrare la sistemul coloanei și extremitatea de ieșire la detector. Se verifică aparatura pentru cromatografia în fază gazoasă (funcționarea buclelor cu gaz, eficiența detectorului și a aparatului înregistrator etc.).
În cazul în care coloana este folosită pentru prima dată, se recomandă condiționarea sa. Se trece un debit slab de gaz prin coloană, apoi se deschide aparatul de cromatografie în fază gazoasă. Se încălzește treptat până la atingerea unei temperaturi de 350 °C, după aproximativ patru ore.
Se menține această temperatură timp de cel puțin două ore, apoi se reglează aparatul la condițiile de funcționare [se reglează debitul de gaz, se aprinde flacăra, se conectează la aparatul înregistrator electronic (punctul 3.3.4), se reglează temperatura cuptorului pentru coloană, detectorul etc.]. Se înregistrează semnalul la o sensibilitate cel puțin de două ori mai mare decât cea necesară pentru analiză. Traseul liniei de bază obținute trebuie să fie liniar, fără valori de vârf, de orice natură, și să nu prezinte abateri.
O abatere negativă în linie dreaptă indică faptul că nu sunt efectuate corect conexiunile coloanei, iar o abatere pozitivă indică faptul că nu a fost condiționată corect coloana.
5.2.2. Alegerea condițiilor de funcționare pentru ceruri, esteri metilici și esteri etilici (Nota 6)
Condițiile de funcționare care trebuie respectate sunt, în general, următoarele:
|
— |
Temperatura coloanei : 20 °C/min 5 °C/min 80 °C inițial (1′) |
|
— |
Temperatura detectorului : 350 °C. |
|
— |
Cantitatea injectată : 1 μl de soluție (2-4 ml) de n-heptan. |
|
— |
Gaz transportor : heliu sau hidrogen cu viteză lineară optimă pentru gazul ales (a se vedea apendicele A). |
|
— |
Sensibilitatea instrumentelor : adecvată pentru îndeplinirea condițiilor de mai sus. |
Nota 6: Datorită temperaturii finale ridicate, este permisă o abatere pozitivă care să nu depășească însă 10 % din valoarea maximă admisibilă la citire.
Aceste condiții se pot modifica pentru a se adapta caracteristicilor coloanei și ale cromatografului în fază gazoasă, în scopul separării tuturor cerurilor și esterilor metilici și etilici ai acizilor grași, precum și în scopul obținerii unei separări a vârfurilor satisfăcătoare (a se vedea figurile 2, 3 și 4) și a unui timp de retenție de 18 ± 3 minute pentru standardul intern de lauril arahidat. Vârful cel mai reprezentativ al cerurilor trebuie să fie peste 60 % din valoarea maximă admisibilă la citire, în timp ce standardul intern de metil heptadecanoat pentru esterii metilici și etilici trebuie să atingă valoarea maximă admisibilă la citire.
Se determină parametrii de integrare a vârfurilor în așa fel încât să se obțină o evaluare corectă a suprafeței vârfurilor luate în considerare.
5.3. Realizarea analizei
Se iau 10 μl de soluție cu ajutorul unei microseringi de 10 μl, retrăgând pistonul până când acul este gol. Se introduce acul în sistemul de injecție și se injectează repede, după una sau două secunde. După aproximativ 5 secunde, acul se scoate ușor.
Se înregistrează până la eluarea completă a cerurilor sau a stigmastadienelor, în funcție de fracțiunea analizată.
Linia de bază trebuie să corespundă întotdeauna condițiilor impuse.
5.4. Identificarea vârfurilor
Se identifică vârfurile timpilor de retenție, comparându-le cu amestecuri de ceruri cu timpi de retenție cunoscuți, analizate în aceleași condiții. Esterii alchilici se identifică din amestecuri de esteri metilici și etilici din principalii acizi grași conținuți în uleiurile de măsline (palmitic și oleic).
Figura 1 prezintă cromatograma cerurilor pentru un ulei de măsline virgin. Figurile 2 și 3 prezintă cromatogramele a două uleiuri de măsline extravirgine din comerț, unul cu esteri metilici și etilici, iar celălalt fără. Figura 4 prezintă cromatograma unui ulei de măsline extravirgin de bună calitate și cea a aceluiași ulei conținând în proporție de 20 % un ulei deodorizat.
5.5. Analiza cantitativă a cerurilor
Se determină suprafața vârfurilor corespunzătoare standardului intern de lauril arahidat și esterilor alifatici de la C40 la C46 cu ajutorul integratorului.
Se determină conținutul total de ceruri adunând fiecare tip de ceară, în mg/kg de grăsime, după cum urmează:

unde:
|
Ax |
= |
aria corespunzătoare vârfului pentru fiecare ester, calculată sub formă numerică |
|
As |
= |
aria corespunzătoare vârfului pentru standardul intern de lauril arahidat, calculată sub formă numerică |
|
ms |
= |
masa de standard intern de lauril arahidat adăugat, în miligrame; |
|
m |
= |
masa probei prelevate pentru determinare, în grame. |
5.5.1. Analiza cantitativă a esterilor metilici și etilici
Cu ajutorul integratorului, se determină ariile vârfurilor corespunzătoare standardului intern de metil heptadecanoat, esterilor metilici ai acizilor grași C16 și C18, precum și esterilor etilici ai acizilor grași C16 și C18.
Se determină conținutul pentru fiecare ester alchilic, în mg/kg de grăsime, după cum urmează:
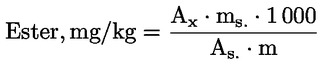
unde:
|
Ax |
= |
aria corespunzătoare vârfului pentru fiecare ester C16 și C18, calculată sub formă numerică |
|
As |
= |
aria corespunzătoare vârfului pentru standardul intern de metil heptadecanoat, calculată sub formă numerică |
|
ms |
= |
masa de standard intern de metil heptadecanoat adăugat, în miligrame; |
|
m |
= |
masa probei prelevate pentru determinare, în grame. |
6. EXPRIMAREA REZULTATELOR
Se indică suma de ceruri conținute de la C40 la C46 (Nota 7) în miligrame per kilogram de grăsime.
Se indică suma esterilor metilici și a esterilor etilici conținuți de la C16 la C18 și totalul celor două.
Rezultatele se aproximează la cea mai apropiată valoare în mg/kg.
Nota 7: Componentele de cuantificat se referă la vârfurile esterilor C40 - C46 cu un număr par de atomi de carbon, conform cromatogramei cerurilor din uleiul de măsline reproduse în figura atașată. În scopul identificării, dacă esterul C46 este scindat, se recomandă să se analizeze fracțiunea de ceruri a unui ulei din turte de măsline, unde vârful pentru C46 se poate distinge, deoarece acesta este clar predominant.
Se indică raportul dintre esterii etilici și esterii metilici.
Figura 1
Exemplu de cromatogramă în fază gazoasă a fracțiunii cerurilor dintr-un ulei de măsline ( 7 )
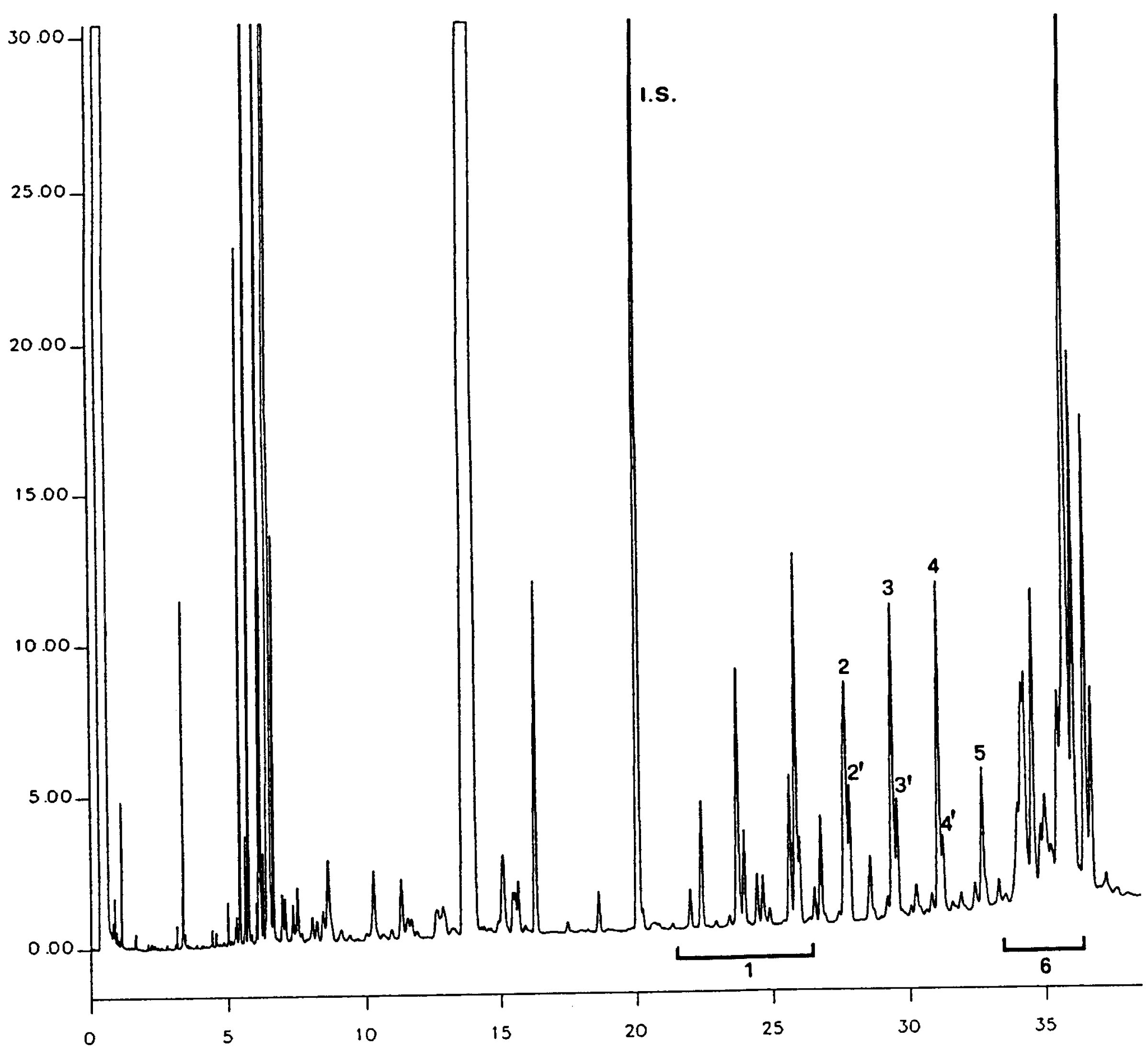
Vârfuri cu timp de retenție cuprins între cinci și opt minute a esterilor metilici și etilici ai acizilor grași
Explicații:
|
S.I. |
= |
Lauril arahidat |
|
1 |
= |
Esteri diterpenici |
|
2+2′ |
= |
Esteri C40 |
|
3+3′ |
= |
Esteri C42 |
|
4+4′ |
= |
Esteri C44 |
|
5 |
= |
Esteri C46 |
|
6 |
= |
Esteri steroli și alcooli triterpenici. |
Figura 2
Esteri metilici, esteri etilici și ceruri într-un ulei de măsline virgin

Explicații:
|
1 |
– |
Metil C16 |
|
2 |
– |
Etil C16 |
|
3 |
– |
S.I Metil heptadecanoat |
|
4 |
– |
Metil C18 |
|
5 |
– |
Etil C18 |
|
6 |
– |
Scualen |
|
7 |
– |
S.I Lauril arahidat |
|
A |
– |
Esteri diterpenici |
|
B |
– |
Ceruri |
|
C |
– |
Esteri steroli și esteri triterpenici |
Figura 3
Esteri metilici, esteri etilici și ceruri într-un ulei de măsline extravirgin

Explicații:
|
1 |
– |
S.I Metil heptadecanoat |
|
2 |
– |
Metil C18 |
|
3 |
– |
Etil C18 |
|
4 |
– |
Scualen |
|
5 |
– |
S.I Lauril arahidat |
|
A |
– |
Esteri diterpenici |
|
B |
– |
Ceruri |
|
C |
– |
Esteri steroli și esteri triterpenici |
Figura 4
Cromatograma parțială a unui ulei de măsline extravirgin și cea a aceluiași ulei conținând un ulei deodorizat

Explicații:
|
1 |
– |
S.I Miristat de metil |
|
2 |
– |
Palmitat de metil |
|
3 |
– |
Palmitat de etil |
|
4 |
– |
S.I Metil heptadecanoat |
|
5 |
– |
Linoleat de metil |
|
6 |
– |
Oleat de metil |
|
7 |
– |
Stearat de metil |
|
8 |
– |
Linoleat de etil |
|
9 |
– |
Oleat de etil |
|
10 |
– |
Stearat de etil |
Apendicele A
Determinarea vitezei liniare a gazului
Se injectează 1:3 μl de metan (sau propan) în cromatograful în fază gazoasă, după ce acesta a fost în prealabil reglat la condițiile normale de funcționare. Se măsoară timpul în care gazul trece prin coloană, din momentul în care a fost injectat până când apare vârful (tM).
Viteza liniară în cm/s se obține prin formula L/tM, unde L este lungimea coloanei în centimetri, iar tM este timpul măsurat în secunde.
▼M28 —————
ANEXA XXI
Rezultatele verificărilor conformității efectuate în cazul uleiurilor de măsline menționate la articolul 8 alineatul (2)
|
|
Etichetare |
Parametri chimici |
Caracteristici organoleptice (4) |
Concluzie finală |
|||||||||||||
|
Probă |
Categorie |
Țară de origine |
Locul de inspecție (1) |
Denumirea juridică |
Desemnarea originii |
Condiții de depozitare |
Informații eronate |
Lizibilitate |
C/NC (3) |
Parametri în afara limitelor DA/NU |
Dacă da, vă rugăm să precizați care (2) |
C/NC (3) |
Mediana defectului |
Mediana atributului fructat |
C/NC (3) |
Acțiune necesară |
Sancțiune |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(1) Piața internă (concasor, îmbuteliatori, etapa de vânzare cu amănuntul), exportul, importul. (2) Fiecare caracteristică a uleiului de măsline indicată în anexa I trebuie să aibă un cod. (3) Conform/neconform. (4) Nu sunt cerute pentru uleiul de măsline și pentru uleiul din resturi de măsline. |
|||||||||||||||||
( 1 ) JO L 228, 1.9.2009, p. 3.
( 2 ) Directiva 2011/91/UE a Parlamentului European și a Consiliului din 13 decembrie 2011 privind indicarea sau marcarea care permite identificarea lotului din care face parte un produs alimentar (JO L 334, 16.12.2011, p. 1).
( 3 ) După eluarea esterilor de steroli, traseul cromatografic nu trebuie să prezinte valori de vârf semnificative (trigliceride).
( 4 ) Regulamentul (UE) nr. 1308/2013 al Parlamentului European și al Consiliului din 17 decembrie 2013 de instituire a unei organizări comune a piețelor produselor agricole și de abrogare a Regulamentelor (CEE) nr. 922/72, (CEE) nr. 234/79, (CE) nr. 1037/2001 și (CE) nr. 1234/2007 ale Consiliului (JO L 347, 20.12.2013, p. 671).
( 5 ) Degustătorul se poate abține să deguste un ulei atunci când constată pe cale olfactivă directă vreun atribut negativ extrem de intens, notând pe foaia de profil această situație excepțională.
( 6 ) In these cases in particular, a 95/5 by volume benzene/acetone eluent mixture must be used to obtain distinct band separation.
( 7 ) După eluarea esterilor steroli, traseul cromatografic nu trebuie să prezinte vârfuri semnificative (triacilgliceroli).