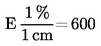ISSN 1725-2601
Jornal Oficial
da União Europeia
L 54

Edição em língua portuguesa
Legislação
52.o ano
26 de Fevreiro de 2009
|
ISSN 1725-2601 |
||
|
Jornal Oficial da União Europeia |
L 54 |
|
|
|
||
|
Edição em língua portuguesa |
Legislação |
52.o ano |
|
Índice |
|
I Actos aprovados ao abrigo dos Tratados CE/Euratom cuja publicação é obrigatória |
Página |
|
|
|
REGULAMENTOS |
|
|
|
* |
Regulamento (CE) n.o 152/2009 da Comissão, de 27 de Janeiro de 2009, que estabelece os métodos de amostragem e análise para o controlo oficial dos alimentos para animais ( 1 ) |
|
|
|
||
|
|
* |
|
|
|
|
|
(1) Texto relevante para efeitos do EEE |
|
PT |
Os actos cujos títulos são impressos em tipo fino são actos de gestão corrente adoptados no âmbito da política agrícola e que têm, em geral, um período de validade limitado. Os actos cujos títulos são impressos em tipo negro e precedidos de um asterisco são todos os restantes. |
I Actos aprovados ao abrigo dos Tratados CE/Euratom cuja publicação é obrigatória
REGULAMENTOS
|
26.2.2009 |
PT |
Jornal Oficial da União Europeia |
L 54/1 |
REGULAMENTO (CE) N. o 152/2009 DA COMISSÃO
de 27 de Janeiro de 2009
que estabelece os métodos de amostragem e análise para o controlo oficial dos alimentos para animais
(Texto relevante para efeitos do EEE)
A COMISSÃO DAS COMUNIDADES EUROPEIAS,
Tendo em conta o Tratado que institui a Comunidade Europeia,
Tendo em conta o Regulamento (CE) n.o 882/2004 do Parlamento Europeu e do Conselho, de 29 de Abril de 2004, relativo aos controlos oficiais realizados para assegurar a verificação do cumprimento da legislação relativa aos alimentos para animais e aos géneros alimentícios e das normas relativas à saúde e ao bem-estar dos animais (1), nomeadamente o n.o 4, alíneas a), b) e c), do artigo 11.o,
Considerando o seguinte:
|
(1) |
Para efeitos da execução da Directiva 70/373/CEE, foram adoptados os seguintes diplomas, que permanecem em vigor nos termos do n.o 2 do artigo 61.o do Regulamento (CE) n.o 882/2004:
|
|
(2) |
Uma vez que a Directiva 70/373/CEE foi substituída pelo Regulamento (CE) n.o 882/2004, afigura-se adequado substituir os actos de execução daquela directiva por um único regulamento. Em simultâneo, os métodos devem ser adaptados à luz da evolução dos conhecimentos científicos e técnicos. Deve ainda suprimir-se os métodos que deixaram de ser válidos para a finalidade pretendida. Embora esteja prevista a actualização, em tempo útil, das disposições relativas à amostragem a fim de ter em conta os progressos recentes em matéria de produção, armazenagem, transporte e comercialização de alimentos para animais, convém manter, por enquanto, as disposições existentes aplicáveis à amostragem. |
|
(3) |
As Directivas 71/250/CEE, 71/393/CEE, 72/199/CEE, 73/46/CEE, 76/371/CEE, 76/372/CEE, 78/633/CEE, 81/715/CEE, 84/425/CEE, 86/174/CEE, 93/70/CEE, 93/117/CE, 98/64/CE, 1999/27/CE, 1999/76/CE, 2000/45/CE, 2002/70/CE e 2003/126/CE devem, pois, ser revogadas. |
|
(4) |
As medidas previstas no presente regulamento estão em conformidade com o parecer do Comité Permanente da Cadeia Alimentar e da Saúde Animal, |
ADOPTOU O PRESENTE REGULAMENTO:
Artigo 1.o
A amostragem, para efeitos de controlo oficial, de alimentos para animais, no que se refere à determinação de constituintes, aditivos e substâncias indesejáveis, à excepção de resíduos de pesticidas e microrganismos, efectua-se de acordo com os métodos descritos no anexo I.
Artigo 2.o
A preparação das amostras para análise e a expressão dos resultados efectuam-se de acordo com os métodos descritos no anexo II.
Artigo 3.o
As análises para efeitos de controlo oficial dos alimentos para animais efectuam-se de acordo com os métodos descritos no anexo III (Métodos de análise para o controlo da composição das matérias-primas para a alimentação animal e dos alimentos compostos para animais), no anexo IV (Métodos de análise para o controlo do teor dos aditivos autorizados nos alimentos para animais), no anexo V (Métodos de análise para o controlo das substâncias indesejáveis nos alimentos para animais) e no anexo VI (Métodos de análise para a determinação dos constituintes de origem animal para o controlo oficial dos alimentos para animais).
Artigo 4.o
O valor energético dos alimentos compostos destinados a aves de capoeira é calculado em conformidade com o disposto no anexo VII.
Artigo 5.o
Usam-se, para efeitos de confirmação, os métodos de análise descritos no anexo VIII para o controlo da presença ilegal de aditivos que já não estão autorizados nos alimentos para animais.
Artigo 6.o
São revogadas as Directivas 71/250/CEE, 71/393/CEE, 72/199/CEE, 73/46/CEE, 76/371/CEE, 76/372/CEE, 78/633/CEE, 81/715/CEE, 84/425/CEE, 86/174/CEE, 93/70/CEE, 93/117/CE, 98/64/CE, 1999/27/CE, 1999/76/CE, 2000/45/CE, 2002/70/CE e 2003/126/CE.
As referências às directivas revogadas devem entender-se como sendo feitas para o presente regulamento e devem ser lidas de acordo com os quadros de correspondência constantes do anexo IX.
Artigo 7.o
O presente regulamento entra em vigor no vigésimo dia seguinte ao da sua publicação no Jornal Oficial da União Europeia.
É aplicável a partir de 26 de Agasto de 2009.
O presente regulamento é obrigatório em todos os seus elementos e directamente aplicável em todos os Estados-Membros.
Feito em Bruxelas, em 27 de Janeiro de 2009.
Pela Comissão
Androulla VASSILIOU
Membro da Comissão
(1) JO L 165 de 30.4.2004, p. 1. Rectificação no JO L 191 de 28.5.2004, p. 1.
(2) JO L 155 de 12.7.1971, p. 13.
(3) JO L 279 de 20.12.1971, p. 7.
(4) JO L 123 de 29.5.1972, p. 6.
(5) JO L 83 de 30.3.1973, p. 21.
(6) JO L 102 de 15.4.1976, p. 1.
(7) JO L 102 de 15.4.1976, p. 8.
(8) JO L 206 de 29.7.1978, p. 43.
(9) JO L 257 de 10.9.1981, p. 38.
(10) JO L 238 de 6.9.1984, p. 34.
(11) JO L 130 de 16.5.1986, p. 53.
(12) JO L 234 de 17.9.1993, p. 17.
(13) JO L 329 de 30.12.1993, p. 54.
(14) JO L 257 de 19.9.1998, p. 14.
(15) JO L 118 de 6.5.1999, p. 36.
(16) JO L 207 de 6.8.1999, p. 13.
(17) JO L 174 de 13.7.2000, p. 32.
(18) JO L 209 de 6.8.2002, p. 15.
(19) JO L 339 de 24.12.2003, p. 78.
ANEXO I
MÉTODOS DE AMOSTRAGEM
1. OBJECTO E ÂMBITO DE APLICAÇÃO
As amostras destinadas aos controlos oficiais de alimentos para animais são colhidas de acordo com os métodos seguidamente indicados. As amostras assim obtidas serão consideradas representativas dos lotes.
2. PESSOAL QUE EFECTUA A AMOSTRAGEM
As amostras são colhidas por pessoas autorizadas para o efeito pelos Estados-Membros.
3. DEFINIÇÕES
Lote: quantidade de produto constituindo uma unidade e que se presume ter características uniformes.
Amostra elementar: quantidade colhida num ponto do lote.
Amostra global: conjunto de amostras elementares colhidas no mesmo lote.
Amostra reduzida: parte representativa da amostra global, obtida através da sua redução.
Amostra final: parte de amostra reduzida ou da amostra global homogeneizada.
4. EQUIPAMENTO
|
4.1. |
O equipamento destinado à colheita de amostras deve ser fabricado com materiais que não contaminem os produtos a colher. Estes equipamentos podem ser aprovados oficialmente pelos Estados-Membros. |
4.2. Equipamento recomendado para a amostragem dos alimentos sólidos
4.2.1. Amostragem manual
|
4.2.1.1. |
Cápsula de fundo plano e bordos verticais. |
|
4.2.1.2. |
Sonda de fenda longa ou compartimentada. As dimensões da sonda devem ser adaptadas às características do lote (profundidade do recipiente, dimensões do saco, etc.) e ao tamanho das partículas que compõem o alimento. |
4.2.2. Amostragem mecânica
Podem ser utilizados aparelhos mecânicos aprovados para amostragem dos alimentos em movimento.
4.2.3. Divisor
Na colheita de amostras elementares, assim como na preparação das amostras reduzidas e amostras finais, podem ser utilizados aparelhos destinados a dividir a amostra em partes aproximadamente iguais.
5. EXIGÊNCIAS QUANTITATIVAS
|
5.A. |
Respeitantes ao controlo das substâncias ou produtos repartidos uniformemente nos alimentos |
|
|
5.A.1. |
Lote A dimensão do lote deve ser tal que todas as partes que o compõem possam ser amostradas. |
|
|
5.A.2. |
Amostras elementares |
|
|
5.A.2.1. |
Alimentos a granel: |
Número mínimo de amostras elementares: |
|
5.A.2.1.1. |
Lotes não excedendo 2,5 toneladas |
Sete |
|
5.A.2.1.2. |
Lotes de mais de 2,5 toneladas |
√ 20 vezes o número de toneladas que constituem o lote (1), limitado a um número máximo de 40 amostras elementares |
|
5.A.2.2. |
Alimentos embalados: |
Número mínimo de embalagens a amostrar (2): |
|
5.A.2.2.1. |
Embalagens de conteúdo superior a 1 kg: |
|
|
5.A.2.2.1.1. |
Lotes compostos por uma a quatro embalagens |
Todas as embalagens |
|
5.A.2.2.1.2. |
Lotes compostos por cinco a 16 embalagens |
Quatro |
|
5.A.2.2.1.3. |
Lotes compostos por mais de 16 embalagens |
√ número de embalagens que constituem o lote (1), limitado a um número máximo de 20 embalagens |
|
5.A.2.2.2. |
Embalagens de conteúdo não superior a 1 kg |
Quatro |
|
5.A.2.3. |
Alimentos líquidos ou semilíquidos: |
Número mínimo de recipientes a amostrar (2): |
|
5.A.2.3.1. |
Recipientes de conteúdo superior a 1 litro: |
|
|
5.A.2.3.1.1. |
Lotes compostos por um a quatro recipientes |
Todos os recipientes |
|
5.A.2.3.1.2. |
Lotes compostos por cinco a 16 recipientes |
Quatro |
|
5.A.2.3.1.3. |
Lotes compostos por mais de 16 recipientes |
√ número de recipientes que constituem o lote (1), limitado a um número máximo de 20 recipientes |
|
5.A.2.3.2. |
Recipientes de conteúdo não superior a 1 litro |
Quatro |
|
5.A.2.4. |
Alimentos em blocos e pedras minerais para lamber |
Número mínimo de blocos ou pedras a amostrar (2): um bloco ou pedra por cada lote de 25 unidades, limitado a um número máximo de quatro blocos ou pedras |
|
5.A.3. |
Amostra global É necessária uma só amostra global por lote. A dimensão total das amostras elementares que constituem a amostra global não pode ser inferior às quantidades seguintes: |
|
|
5.A.3.1. |
Alimentos a granel |
4 kg |
|
5.A.3.2. |
Alimentos embalados: |
|
|
5.A.3.2.1. |
Embalagens de conteúdo superior a 1 kg |
4 kg |
|
5.A.3.2.2. |
Embalagens de conteúdo não superior a 1 kg |
Massa do conteúdo de quatro embalagens de origem |
|
5.A.3.3. |
Alimentos líquidos ou semilíquidos: |
|
|
5.A.3.3.1. |
Recipientes de conteúdo superior a 1 litro: |
Quatro litros |
|
5.A.3.3.2. |
Recipientes de conteúdo não superior a 1 litro |
Volume do conteúdo de quatro recipientes de origem |
|
5.A.3.4. |
Alimentos em blocos e pedras minerais para lamber: |
|
|
5.A.3.4.1. |
Peso unitário superior a 1 kg |
4 kg |
|
5.A.3.4.2. |
Peso unitário não superior a 1 kg |
Quatro blocos ou pedras de origem |
|
5.A.4. |
Amostras finais A amostra global permitirá obter após redução, se necessário, as amostras finais. É exigida a análise de pelos menos uma amostra final. A dimensão da amostra final destinada a análise não pode ser inferior às quantidades seguintes: |
|
|
|
Alimentos sólidos |
500 g |
|
|
Alimentos líquidos ou semilíquidos |
500 ml |
|
5.B. |
Respeitantes ao controlo de substâncias ou produtos indesejáveis susceptíveis de não se encontrarem uniformemente repartidos nos alimentos, tais como aflatoxinas, cravagem do centeio, rícino e crotalária nas matérias-primas para a alimentação animal (3) |
|
|
5.B.1. |
Lote: ver ponto 5.A.1 |
|
|
5.B.2. |
Amostras elementares |
|
|
5.B.2.1. |
Alimentos a granel: ver ponto 5.A.2.1 |
|
|
5.B.2.2. |
Alimentos embalados: |
Número mínimo de embalagens a amostrar: |
|
5.B.2.2.1. |
Lotes compostos por uma a quatro embalagens |
Todas as embalagens |
|
5.B.2.2.2. |
Lotes compostos por cinco a 16 embalagens |
Quatro |
|
5.B.2.2.3. |
Lotes compostos por mais de 16 embalagens |
√ número de embalagens que constituem o lote (1), limitado a um número máximo de 40 embalagens |
|
5.B.3. |
Amostras globais O número de amostras globais varia em função da dimensão do lote. O número mínimo de amostras globais por lote é o que a seguir se indica. A massa total das amostras elementares que constituem cada amostra global não pode ser inferior a 4 kg. |
|
|
5.B.3.1. |
Alimentos a granel |
|
|
|
Massa do lote, em toneladas: |
Número mínimo de amostras globais por lote: |
|
|
Até 1 |
1 |
|
|
mais de 1 e até 10 |
2 |
|
|
mais de 10 e até 40 |
3 |
|
|
mais de 40 |
4 |
|
5.B.3.2. |
Alimentos embalados |
|
|
|
Dimensão do lote, em número de embalagens: |
Número mínimo de amostras globais por lote: |
|
|
1 a 16 |
1 |
|
|
17 a 200 |
2 |
|
|
201 a 800 |
3 |
|
|
mais de 800 |
4 |
|
5.B.4. |
Amostras finais Cada amostra global dá origem às amostras finais, após redução. É exigida a análise de pelos menos uma amostra final por cada amostra global. A massa da amostra final destinada a análise não pode ser inferior a 500 g. |
|
6. INSTRUÇÕES RELATIVAS À COLHEITA, À PREPARAÇÃO E AO ACONDICIONAMENTO DAS AMOSTRAS
6.1. Generalidades
Recolher e preparar as amostras tão rapidamente quanto possível tendo em conta as precauções requeridas para evitar que o produto seja alterado ou contaminado. Os instrumentos assim como as superfícies e os recipientes destinados a receber as amostras devem estar limpos e secos.
6.2. Amostras elementares
6.2.A. Respeitantes ao controlo das substâncias ou produtos repartidos uniformemente nos alimentos
As amostras elementares devem ser colhidas aleatoriamente no conjunto do lote e ser de dimensão aproximadamente equivalente.
6.2.A.1.
Dividir simbolicamente o lote em partes aproximadamente iguais. Escolher aleatoriamente um número de partes que correspondam ao número de amostras elementares previstas no ponto 5.A.2 e colher pelo menos uma amostra em cada uma dessas partes.
Sempre que adequado, a colheita das amostras pode ser efectuada aquando da movimentação do lote (carga ou descarga).
6.2.A.2.
Após a selecção do número necessário de embalagens a amostrar, tal como indicado no ponto 5.A.2, procede-se à colheita de uma parte do conteúdo de cada embalagem com o auxílio de uma sonda ou de uma pá. Sempre que necessário, colher as amostras após ter esvaziado separadamente as embalagens. Para cada amostra global, esmagar os aglomerados eventualmente presentes, se necessário separando-os da massa e reconstituindo em seguida o todo.
6.2.A.3.
Após a selecção do número necessário de recipientes a amostrar, tal como indicado no ponto 5.A.2, procede-se à colheita de uma quantidade em cada recipiente após ter homogeneizado o conteúdo, se necessário.
As amostras elementares podem eventualmente ser colhidas aquando da trasfega do produto.
6.2.A.4.
Após a selecção do número necessário de recipientes a amostrar, tal como indicado no ponto 5.A.2, procede-se à colheita das amostras a diferentes níveis.
As amostras podem igualmente ser colhidas aquando da trasfega do produto, mas rejeitando as primeiras fracções.
Nos dois casos, o volume total colhido não deve ser inferior a 10 litros.
6.2.A.5.
Após a selecção do número necessário de blocos ou pedras a amostrar, tal como indicado no ponto 5.A.2, retira-se uma parte de cada bloco ou pedra.
6.2.B. Respeitantes ao controlo de substâncias ou produtos indesejáveis susceptíveis de não se encontrarem uniformemente repartidos nos alimentos, tais como aflatoxinas, cravagem do centeio, rícino e crotalária nas matérias-primas para a alimentação animal
Dividir simbolicamente o lote num número de partes aproximadamente iguais, correspondendo ao número de amostras globais previstas no ponto 5.B.3. Se esse número for superior a um, repartir o número total de amostras elementares previsto no ponto 5.B.2 de maneira aproximadamente igual pelas diferentes partes. Efectuar seguidamente a colheita de amostras, com massas aproximadamente iguais (4) e de maneira a que a massa total das amostras, no que diz respeito a cada parte, não seja inferior à quantidade mínima de 4 quilogramas, requerida para cada amostra global. Não juntar as amostras elementares provindas de partes diferentes.
6.3. Preparação das amostras globais
6.3.A. Respeitantes ao controlo das substâncias ou produtos repartidos uniformemente nos alimentos
As amostras elementares devem ser misturadas para formar uma só amostra global.
6.3.B. Respeitantes ao controlo de substâncias ou produtos indesejáveis susceptíveis de não se encontrarem uniformemente repartidos nos alimentos, tais como aflatoxinas, cravagem do centeio, rícino e crotalária nas matérias-primas para a alimentação animal
Misturar as amostras elementares relativas a cada parte do lote e constituir o número de amostras globais previsto no ponto 5.B.3., tendo o cuidado de assinalar a proveniência de cada amostra global.
6.4. Preparação das amostras finais
Misturar cuidadosamente cada amostra global para obter uma amostra homogénea (5). Se necessário, reduzir para este efeito a amostra global até 2 quilogramas ou 2 litros pelo menos (amostra reduzida) quer com a ajuda dum divisor mecânico ou automático, quer pelo método dos quartos.
Preparar seguidamente pelo menos três amostras finais de dimensão aproximadamente equivalente e respondendo às exigências quantitativas requeridas no ponto 5.A.4 ou 5.B.4. Introduzir cada amostra num recipiente apropriado. Tomar todas as precauções necessárias para evitar qualquer modificação da composição da amostra ou qualquer contaminação ou adulteração que possam ocorrer no decurso do transporte ou armazenagem.
6.5. Acondicionamento das amostras finais
Selar e rotular os recipientes ou as embalagens (todo o rótulo deve estar incorporado no selo) de forma a que seja impossível abri-las sem deteriorar o selo.
7. REGISTO DE AMOSTRAGEM
Para cada colheita de amostras, deve ser estabelecido um registo que permita identificar sem ambiguidades o lote amostrado.
8. DESTINO DAS AMOSTRAS
Por cada amostra global, enviar pelo menos uma amostra final, o mais rapidamente possível, ao laboratório de análises autorizado, com as indicações necessárias a essa análise.
(1) Sempre que o número obtido for fraccionário, deve ser arredondado para o inteiro imediatamente superior.
(2) Para as embalagens ou os recipientes cujo conteúdo não exceda 1 quilograma ou 1 litro, assim como para os blocos ou pedras para lamber cujo peso unitário não exceda 1 quilograma, a amostra elementar é constituída pelo conteúdo de uma embalagem ou um recipiente de origem, um bloco ou uma pedra.
(3) Os métodos previstos no ponto 5.A são de aplicação no controlo das aflatoxinas, da cravagem do centeio, do rícino e da crotalária nos alimentos completos e complementares.
(4) Para alimentos embalados, recolher uma parte do conteúdo de cada embalagem a amostrar com o auxílio de uma sonda ou de uma pá após ter esvaziado separadamente as embalagens, caso necessário.
(5) Para cada amostra global, esmagar os aglomerados eventualmente presentes (se necessário separando-os da massa e reconstituindo em seguida o todo).
ANEXO II
DISPOSIÇÕES GERAIS RELATIVAS AOS MÉTODOS DE ANÁLISE DOS ALIMENTOS PARA ANIMAIS
A. PREPARAÇÃO DAS AMOSTRAS PARA ANÁLISE
1. Objecto
Os procedimentos descritos infra referem-se à preparação para análise das amostras finais enviadas aos laboratórios de controlo após amostragem efectuada segundo o disposto no anexo I.
A preparação destas amostras deverá permitir que as amostras de ensaio previstas pelos métodos de análise sejam homogéneas e representativas das amostras finais.
2. Precauções a tomar
O procedimento de preparação da amostra a seguir depende dos métodos de análise a utilizar. Importa pois garantir que o procedimento aplicado se adequa ao método de análise.
Todas as operações devem ser efectuadas de maneira a evitar tanto quanto possível a contaminação da amostra ou a alteração da sua composição.
Todas as triturações, misturas e peneiramentos devem fazer-se o mais rapidamente possível, evitando ao máximo expor a amostra ao ar e à luz. Não se devem usar moinhos ou trituradores susceptíveis de aquecer demasiado a amostra.
Recomenda-se a trituração manual para os alimentos particularmente sensíveis ao calor. Deve ainda ter-se o máximo cuidado em verificar que o equipamento não seja uma fonte de contaminação por oligoelementos.
Caso a amostra não possa ser preparada sem que se dê uma sensível variação no seu teor em humidade, determinar este teor, antes e depois da preparação, pelo método estabelecido na parte A do anexo III.
3. Procedimento
Dividir a amostra em subamostras adequadas para efeitos de análise e de procedimentos de arbitragem mediante a aplicação de técnicas de divisão adequadas, como sejam a formação de porções alternativas com a ajuda de uma pá ou o recurso a divisores mecânicos estacionários ou rotativos. Não se recomenda o uso do método dos quartos, dado que pode dar origem a subamostras com uma elevada margem de erro de divisão. A amostra destinada a procedimentos de arbitragem deve ser conservada num recipiente adequado, limpo, seco e munido de fecho hermético, e as subamostras para análise, com pelo menos 100 g, são preparadas como seguidamente se indica.
3.1. Alimentos que podem ser moídos tal como se encontram
Salvo indicação específica nos métodos de análise, peneirar a totalidade da amostra numa peneira de malha quadrada de 1 mm de lado (de harmonia com a recomendação ISO R565) depois de triturada, se necessário. Evitar triturar em demasia.
Misturar a amostra peneirada e recolhê-la para um recipiente adequado, limpo, seco e com fecho hermético. Voltar a misturá-la imediatamente antes de recolher a amostra de ensaio.
3.2. Alimentos que podem ser moídos após secagem
Salvo indicação específica nos métodos de análise, secar a amostra de maneira a baixar o seu teor de humidade para 8-12 %, utilizando o processo indicado no ponto 4.3 do método de determinação do teor de humidade referido na parte A do anexo III. Proceder em seguida como indicado no ponto 3.1.
3.3. Alimentos líquidos ou semilíquidos
Recolher a amostra para um recipiente adequado, limpo, seco e com fecho hermético. Misturar intimamente, imediatamente antes de colher a amostra de ensaio.
3.4. Outros alimentos
Caso a amostra não possa ser preparada por qualquer dos processos acima indicados, utilizar outro processo de preparação apropriado que permita a obtenção de amostras de ensaio homogéneas e representativas das amostras finais.
4. Conservação das amostras
Conservar as amostras a uma temperatura que não altere a sua composição. Utilizar recipientes de vidro castanho para as amostras destinadas a análise das vitaminas ou de substâncias particularmente sensíveis à luz.
B. DISPOSIÇÕES RELATIVAS AOS REAGENTES E EQUIPAMENTOS REQUERIDOS PELOS MÉTODOS DE ANÁLISE
|
1. |
Salvo indicação específica nos métodos de análise, todos os reagentes deverão ser de qualidade «para análise» (p.a.). Para a análise dos oligoelementos, a pureza dos reagentes deverá ser controlada mediante um ensaio em branco. Consoante o resultado obtido, poderá ou não vir a ser necessária a sua purificação suplementar. |
|
2. |
Todas as operações de preparação de soluções, diluição e lavagem mencionadas nos métodos de análise relativamente às quais não sejam fornecidas indicações quanto à natureza do solvente ou diluente a utilizar implicarão que se deverá utilizar água, em regra desmineralizada ou destilada. Em certos casos especiais, indicados nos métodos de análise, essa água deverá ser submetida a processos de purificação específicos. |
|
3. |
Atendendo ao equipamento corrente normalmente presente nos laboratórios de controlo, o método de análise indica apenas o material especial ou os instrumentos e aparelhos que devam responder a determinadas condições. Este material deverá estar bem limpo, particularmente para todas as determinações de quantidades vestigiais. |
C. APLICAÇÃO DOS MÉTODOS DE ANÁLISE E EXPRESSÃO DOS RESULTADOS
1. Procedimento de extracção
Vários métodos indicam um procedimento de extracção específico. Como regra geral, podem ser usados métodos de extracção diferentes do indicado, desde que o procedimento usado tenha dado provas de que proporciona, para a matriz em causa, a mesma eficiência de extracção que o procedimento mencionado no método de análise.
2. Procedimento de purificação
Vários métodos indicam um procedimento de purificação específico. Como regra geral, podem ser usados métodos de purificação diferentes do indicado, desde que o procedimento usado tenha dado provas de que proporciona, para a matriz em causa, resultados analíticos equivalentes aos do procedimento mencionado no método de análise.
3. Notificação do método de análise usado
Em geral, estabelece-se apenas um método de análise para cada uma das substâncias a determinar nos alimentos para animais. Sempre que se indicar mais de um método, o boletim de análise deverá mencionar qual deles foi utilizado pelo laboratório de controlo.
4. Número de determinações
O resultado indicado no boletim de análise será o valor médio obtido a partir de, pelo menos, duas determinações de boa repetibilidade, efectuadas com amostras de ensaio distintas.
Todavia, em caso de análise de substâncias indesejáveis, se o resultado da primeira determinação for significativamente (> 50 %) inferior à especificação a controlar, não são necessárias mais determinações, desde que se tenham aplicado os procedimentos de qualidade adequados.
Nos casos em que o controlo incide sobre o teor declarado de uma substância ou ingrediente, se o resultado da primeira determinação confirmar o teor declarado, ou seja, se o resultado analítico se situar dentro da gama de variação aceitável desse teor, não são necessárias mais determinações, desde que se tenham aplicado os procedimentos de qualidade adequados.
Nalguns casos, esta gama de variação aceitável encontra-se definida na legislação, como por exemplo a Directiva 79/373/CEE do Conselho (1).
5. Notificação do resultado analítico
O resultado analítico, que deverá ser expresso em conformidade com as indicações dadas no método de análise e conter o devido número de algarismos significativos, poderá, caso necessário, ser corrigido em função do teor de humidade apresentado pela amostra final antes da sua preparação para análise.
6. Incerteza da medição e taxa de recuperação no caso da análise de substâncias indesejáveis
No que diz respeito às substâncias indesejáveis na acepção da Directiva 2002/32/CE, incluindo dioxinas e PCB sob a forma de dioxina, um produto destinado à alimentação animal será considerado não conforme com o limite máximo fixado se se considerar que o resultado analítico ultrapassa o limite máximo tendo em conta a incerteza expandida da medição e a correcção em função da recuperação. Para a avaliação da conformidade, usa-se a concentração analisada, corrigida em função da recuperação, e subtrai-se a incerteza expandida da medição. Este procedimento só é aplicável nos casos em que o método de análise permita estimar a incerteza da medição e a correcção em função da recuperação (por exemplo, não é possível no caso da análise microscópica).
O resultado analítico será apresentado do seguinte modo (na medida em que o método de análise utilizado permita estimar a incerteza da medição e a taxa de recuperação):
|
a) |
Corrigido em função da recuperação, indicando o nível de recuperação. Se a taxa de recuperação se situar entre 90 e 110 % não é necessário efectuar a correcção; |
|
b) |
Como «x +/– U», em que x é o resultado analítico e U é a incerteza expandida da medição, utilizando um factor de expansão de 2, que permite obter um nível de confiança de cerca de 95 %. |
Todavia, se o resultado da análise for significativamente (> 50 %) inferior à especificação a controlar e se análise se destinar exclusivamente a verificar a observância das disposições legislativas, desde que se tenham aplicado os procedimentos de qualidade adequados, o resultado analítico pode ser notificado sem correcção em função da recuperação e, nestes casos, não é necessário transmitir os valores da taxa de recuperação nem da incerteza da medição.
ANEXO III
MÉTODOS DE ANÁLISE PARA O CONTROLO DA COMPOSIÇÃO DAS MATÉRIAS-PRIMAS PARA A ALIMENTAÇÃO ANIMAL E DOS ALIMENTOS COMPOSTOS PARA ANIMAIS
A. DETERMINAÇÃO DA HUMIDADE
1. Objecto e âmbito de aplicação
Este método permite determinar o teor de humidade dos alimentos para animais. Se os alimentos contiverem substâncias voláteis, como por exemplo ácidos orgânicos, deve ter-se em conta que, a par do teor de humidade, também se mede uma quantidade significativa de substâncias voláteis.
O método não abrange a análise dos produtos lácteos enquanto matérias-primas para a alimentação animal, a análise das substâncias minerais e das misturas essencialmente compostas de substâncias minerais, a análise das gorduras e óleos animais e vegetais nem a análise das sementes e frutos oleaginosos.
2. Princípio
A amostra é dessecada em condições definidas que variam em função da natureza do alimento. A perda de massa é determinada por pesagem. É necessário proceder a uma pré-dessecação sempre que se trate de alimentos sólidos com um elevado teor de humidade.
3. Equipamento
|
3.1. |
Triturador construído num material que não absorva a humidade, fácil de limpar e que permita uma trituração rápida e uniforme sem provocar aquecimento sensível, que evite o mais possível o contacto com o ar exterior, e que satisfaça as exigências indicadas nos pontos 4.1.1 e 4.1.2 (por exemplo, microtrituradores de martelos ou com arrefecimento por água, moinhos de cones desmontáveis, trituradores de movimento lento ou de discos dentados). |
|
3.2. |
Balança analítica com uma sensibilidade de 0,1 miligrama. |
|
3.3. |
Recipientes secos de metal inoxidável ou de vidro, munidos de uma tampa hermética; superfície útil que permita obter uma repartição da amostra da ordem dos 0,3 g/cm2. |
|
3.4. |
Estufa isotérmica (± 2 oC) de aquecimento eléctrico, com uma regulação rápida da temperatura e convenientemente ventilada (1). |
|
3.5. |
Estufa de vácuo, com aquecimento eléctrico regulável, munida de uma bomba de óleo e quer de um dispositivo de introdução de ar quente seco, quer de um agente desidratante (por ex., óxido de cálcio). |
|
3.6. |
Exsicador com placa de metal ou de porcelana espessa, perfurada, contendo um agente desidratante eficaz. |
4. Procedimento
|
N.B. |
: |
As operações descritas nesta secção devem ser efectuadas imediatamente após a abertura das embalagens que contêm as amostras. As análises devem ser efectuadas pelo menos em duplicado. |
4.1. Preparação
4.1.1.
Tomar pelo menos 50 g da amostra. Se for necessário, triturar ou dividir de maneira apropriada para evitar qualquer variação do teor de humidade (ver ponto 6).
4.1.2.
Tomar pelo menos 50 g da amostra. Moer em partículas, das quais pelo menos 50 % passem por uma peneira de malhas de 0,5 mm e não deixem mais de 10 % de resíduos sobre outro crivo de malhas redondas de 1 mm.
4.1.3.
Tomar e pesar, com uma aproximação de 10 mg, cerca de 25 g da amostra, adicionar-lhe uma quantidade apropriada de areia anidra, com uma aproximação de 10 mg, misturar até à obtenção de um produto homogéneo.
4.2. Dessecação
4.2.1.
Tarar, com uma aproximação de 1 mg, um recipiente (3.3) com a tampa. Pesar dentro dele, com uma aproximação de 1 mg, cerca de 5 g da amostra repartindo-a uniformemente. Colocar o recipiente destapado na estufa previamente aquecida a 103 oC. Introduzir o recipiente o mais rapidamente possível, para evitar que a temperatura da estufa desça demasiado. Deixar secar durante quatro horas, a partir do momento em que a estufa tiver atingido de novo a temperatura de 103 oC. Colocar a tampa no recipiente, retirar este da estufa, deixar arrefecer 30 a 45 minutos no exsicador (3.6) e pesar com uma aproximação de 1 mg.
No caso de alimentos constituídos essencialmente de matérias gordas, efectuar uma dessecação suplementar de 30 minutos na estufa a 130 oC. A diferença entre as duas pesagens não deve exceder o valor correspondente a 0,1 % de humidade.
4.2.2.
Tarar, com uma aproximação de 0,5 mg, um recipiente (3.3) com a tampa. Pesar dentro dele, com uma aproximação de 1 mg, cerca de 5 g da amostra triturada, repartindo-a uniformemente. Colocar o recipiente destapado na estufa previamente aquecida a 130 oC. Introduzir o recipiente o mais rapidamente possível, para evitar que a temperatura da estufa desça demasiado. Deixar secar durante duas horas, a partir do momento em que a estufa tiver atingido de novo a temperatura de 130 oC. Colocar a tampa no recipiente, retirar este da estufa, deixar arrefecer 30 a 45 minutos no exsicador (3.6) e pesar com uma aproximação de 1 mg.
|
4.2.3. |
Alimentos compostos contendo mais de 4 % de sacarose ou de lactose: matérias-primas para a alimentação animal tais como alfarroba, produtos cereais hidrolisados, germes de malte, rodelas de beterraba, solúveis de peixe e açúcares; alimentos compostos com mais de 25 % de sais minerais contendo água de cristalização Tarar, com uma aproximação de 0,5 mg, um recipiente (3.3) com a tampa. Pesar dentro dele, com uma aproximação de 1 mg, cerca de 5 g da amostra repartindo-a uniformemente. Colocar o recipiente destapado na estufa de vácuo (3.5) previamente aquecida a uma temperatura entre 80 e 85 oC. Introduzir o recipiente o mais rapidamente possível, para evitar que a temperatura da estufa desça demasiado. Elevar a pressão a 100 Torr e deixar secar a esta pressão durante quatro horas, quer sob uma corrente de ar seco e quente, quer por meio de um agente desidratante (cerca de 300 g para 20 amostras). Neste último caso, desligar a bomba de vácuo quando a pressão indicada tiver sido atingida. Contar o tempo de secagem a partir do momento em que a estufa tiver atingido de novo a temperatura de 80 a 85 oC. Reduzir cuidadosamente a pressão no interior da estufa até à pressão atmosférica. Abrir a estufa, tapar imediatamente o recipiente, retirá-lo da estufa, deixar arrefecer durante 30 a 45 minutos no exsicador (3.6) e pesar com uma aproximação de 1 mg. Proceder a uma dessecação complementar de 30 minutos na estufa de vácuo a uma temperatura entre 80 e 85 oC e pesar novamente. A diferença entre as duas pesagens não deve exceder o valor correspondente a 0,1 % de humidade. |
4.3. Pré-dessecação
4.3.1.
Os alimentos sólidos cujo teor de humidade é elevado e torna a trituração difícil devem ser pré-dessecados como se segue:
Pesar, com uma aproximação de 10 mg, 50 g de amostra não triturada (pode efectuar-se uma divisão grosseira, se necessário, no caso dos alimentos comprimidos ou aglomerados) num recipiente apropriado (por exemplo, numa placa de alumínio de 20 × 12 cm com um bordo de 0,5 cm). Deixar secar numa estufa a uma temperatura entre 60 e 70 oC até que o teor de humidade seja reduzido para um valor entre 8 e 12 %. Retirar da estufa, deixar arrefecer a descoberto no laboratório durante uma hora e pesar com uma aproximação de 10 mg. Triturar imediatamente, como indicado no ponto 4.1.1 e efectuar a dessecação como indicado nos pontos 4.2.1 ou 4.2.3 segundo a natureza do alimento.
4.3.2.
Os grãos com uma taxa de humidade superior a 17 % devem ser pré-dessecados como se segue:
Pesar, com uma aproximação de 10 mg, 50 g de grãos não moídos num recipiente apropriado (por exemplo, numa placa de alumínio de 20 × 12 cm com bordo de 0,5 cm). Deixar secar numa estufa durante 5 a 7 minutos, à temperatura de 130 oC. Retirar da estufa, deixar arrefecer a descoberto no laboratório durante duas horas e pesar, com uma aproximação de 10 mg. Moer imediatamente, como indicado no ponto 4.1.2 e efectuar a dessecação, como indicado no ponto 4.2.2.
5. Cálculo dos resultados
O teor de humidade (X), em percentagem da amostra, é dado pelas seguintes fórmulas:
5.1. Dessecação sem pré-dessecação

em que:
|
m |
= |
massa inicial da amostra, em gramas, |
|
m0 |
= |
massa da amostra seca, em gramas. |
5.2. Dessecação com pré-dessecação
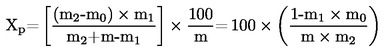
em que:
|
m |
= |
massa inicial da amostra, em gramas, |
|
m1 |
= |
massa da amostra, depois da pré-dessecação, em gramas, |
|
m2 |
= |
massa da amostra depois da trituração ou moagem, em gramas, |
|
m0 |
= |
massa da amostra seca, em gramas. |
5.3. Repetibilidade
A diferença entre os resultados de duas determinações paralelas efectuadas com a mesma amostra não deve exceder 0,2 % do valor absoluto da humidade.
6. Observações
Se for necessária uma trituração e dela resultar uma variação do teor de humidade do produto, os resultados da análise que dizem respeito aos componentes do alimento devem ser corrigidos de acordo com o teor de humidade da amostra inicial.
B. DETERMINAÇÃO DA HUMIDADE NAS MATÉRIAS GORDAS ANIMAIS E VEGETAIS
1. Objecto e âmbito de aplicação
O método permite determinar o teor em humidade (água e outras matérias voláteis) das gorduras e dos óleos animais e vegetais.
2. Princípio
A amostra é seca a 103 oC até peso constante (a perda de peso entre duas pesagens sucessivas deve ser inferior a 1 mg). A perda de peso é determinada por pesagem.
3. Equipamento
|
3.1. |
Cápsula de fundo plano, em material resistente à corrosão, diâmetro: 8 a 9 cm, altura: cerca de 3 cm. |
|
3.2. |
Termómetro com globo reforçado e câmara de dilatação na extremidade superior, graduado de cerca de 80 oC a pelo menos 110 oC, comprimento: cerca de 10 cm. |
|
3.3. |
Banho de areia ou placa de aquecimento eléctrica. |
|
3.4. |
Exsicador, contendo um agente desidratante eficaz. |
|
3.5. |
Balança analítica. |
4. Procedimento
Pesar, com uma aproximação de 1 mg, cerca de 20 g da amostra homogeneizada na cápsula (3.1) seca e com tara calculada, contendo o termómetro (3.2). Aquecer em banho de areia ou placa de aquecimento (3.3), agitando constantemente com a ajuda do termómetro, de modo a que a temperatura atinja 90 oC em cerca de 7 minutos.
Reduzir a intensidade do aquecimento, atendendo à frequência com que as bolhas sobem do fundo do recipiente. A temperatura não deverá ultrapassar 105 oC. Continuar a agitar, raspando o fundo da cápsula até que cesse a formação de bolhas.
A fim de assegurar a eliminação total da humidade, repetir várias vezes o aquecimento a 103 oC ± 2oC, arrefecendo até 93 oC entre aquecimentos sucessivos. Em seguida deixar arrefecer no exsicador (3.4) até que atinja a temperatura ambiente e pesar. Repetir esta operação até que a perda de massa entre duas pesagens sucessivas não exceda 2 mg.
|
N.B. |
: |
Um aumento de massa da amostra após aquecimento repetido indica uma oxidação da gordura. Neste caso, calcular o resultado a partir da pesagem efectuada imediatamente antes do começo do aumento de massa. |
5. Cálculo dos resultados
O teor de humidade (X) em percentagem da amostra é dado pela fórmula:

em que:
|
m |
= |
massa da amostra em ensaio, em gramas, |
|
m1 |
= |
massa da cápsula com o respectivo conteúdo, antes do aquecimento, em gramas, |
|
m2 |
= |
massa da cápsula com o respectivo conteúdo, após aquecimento, em gramas. |
Os resultados inferiores a 0,05 % devem ser registados com a referência «inferior a 0,05 %».
Repetibilidade
A diferença entre os resultados obtidos em duas determinações paralelas efectuadas com a mesma amostra não deve exceder 0,05 % em valor absoluto.
C. DETERMINAÇÃO DO TEOR DE PROTEÍNA BRUTA
1. Objecto e âmbito de aplicação
O presente método permite determinar o teor de proteína bruta dos alimentos para animais com base no teor de azoto, determinado pelo método de Kjeldahl.
2. Princípio
A amostra é mineralizada com ácido sulfúrico, na presença de um catalisador. A solução ácida é alcalinizada por adição de uma solução de hidróxido de sódio. O amoníaco é destilado e recolhido numa quantidade determinada de ácido sulfúrico, cujo excesso é titulado com uma solução-padrão de hidróxido de sódio.
Alternativamente, o amoníaco libertado é destilado numa solução de ácido bórico em excesso, seguido de titulação com uma solução de ácido clorídrico ou sulfúrico.
3. Reagentes
|
3.1. |
Sulfato de potássio. |
|
3.2. |
Catalisador: óxido de cobre (II) CuO ou sulfato de cobre (II) penta-hidratado CuSO4 · 5H2O. |
|
3.3. |
Zinco granulado. |
|
3.4. |
Ácido sulfúrico, ρ20 = 1,84 g/ml. |
|
3.5. |
Ácido sulfúrico, solução volumétrica padrão, c(H2SO4) = 0,25 mol/l. |
|
3.6. |
Ácido sulfúrico, solução volumétrica padrão, c(H2SO4) = 0,10 mol/l. |
|
3.7. |
Ácido sulfúrico, solução volumétrica padrão, c(H2SO4) = 0,05 mol/l. |
|
3.8. |
Indicador vermelho de metilo; dissolver 300 mg de vermelho de metilo em 100 ml de etanol, σ = 95-96 % (v/v). |
|
3.9. |
Solução de hidróxido de sódio (pode utilizar-se uma solução de pureza técnica), β = 40 g/100 ml (m/v: 40 %). |
|
3.10. |
Hidróxido de sódio, solução volumétrica padrão, c(NaOH) = 0,25 mol/l. |
|
3.11. |
Hidróxido de sódio, solução volumétrica padrão, c(NaOH) = 0,10 mol/l. |
|
3.12. |
Pedra-pomes granulada, lavada em ácido clorídrico e inflamada. |
|
3.13. |
Acetanilida (p.f.=114 oC, teor de N = 10,36 %). |
|
3.14. |
Sacarose (isenta de azoto). |
|
3.15. |
Ácido bórico (H3BO3). |
|
3.16. |
Solução de indicador vermelho de metilo: dissolver 100 mg de vermelho de metilo em 100 ml de etanol ou metanol. |
|
3.17. |
Solução de verde de bromocresol: dissolver 100 mg de verde de bromocresol em 100 ml de etanol ou metanol. |
|
3.18. |
Solução de ácido bórico (10 g/l a 40 g/l, em função do equipamento utilizado). Se se aplicar a detecção colorimétrica do ponto final, deve adicionar-se os indicadores vermelho de metilo e bromocresol às soluções de ácido bórico. Se se preparar um litro de solução de ácido bórico, adicionam-se, antes de ajustar o volume, 7 ml de solução de indicador vermelho de metilo (3.16) e 10 ml de solução de verde de bromocresol (3.17). O pH das soluções de ácido bórico preparadas pode diferir em função da água usada. Por vezes, é necessário efectuar um ajuste com um pequeno volume de uma solução alcalina a fim de obter um resultado positivo no ensaio em branco.
|
|
3.19. |
Ácido clorídrico, solução volumétrica padrão, c(HCl) = 0,10 mol/l.
|
4. Equipamento
Equipamento adequado para efectuar a mineralização, a destilação e a titulação pelo método de Kjeldahl.
5. Procedimento
5.1. Mineralização
Pesar, com uma aproximação de 0,001 g, 1 g de amostra e transferi-la para o recipiente do aparelho de mineralização. Adicionar 15 g de sulfato de potássio (3.1), uma quantidade adequada de catalisador (3.2) [0,3 a 0,4 g de óxido de cobre (II) ou 0,9 a 1,2 g de sulfato de cobre (II) penta-hidratado], 25 ml de ácido sulfúrico (3.4) e, se necessário, alguns grânulos de pedra-pomes (3.12), misturando.
Começar por aquecer moderadamente a mistura, agitando ocasionalmente se necessário, até à carbonização completa da massa e o desaparecimento da espuma. Aquecer seguidamente de um modo mais vigoroso até à ebulição; o aquecimento é adequado quando se observar a condensação do ácido nas paredes do recipiente. Evitar o sobreaquecimento lateral e a adesão de partículas orgânicas às paredes.
Quando a solução ficar límpida e assumir um tom verde claro, continuar a ebulição durante mais duas horas, arrefecendo de seguida.
5.2. Destilação
Adicionar cuidadosamente uma quantidade de água adequada para a dissolução completa dos sulfatos. Deixar arrefecer e adicionar, se necessário, alguns grânulos de zinco (3.3). Proceder de acordo com o disposto nos pontos 5.2.1 ou 5.2.2.
5.2.1.
Colocar no balão de recolha do dispositivo de destilação 25 ml, rigorosamente medidos, de ácido sulfúrico (3.5) ou (3.7), em função do teor presumido de azoto. Adicionar algumas gotas de vermelho de metilo (3.8).
Adaptar o recipiente de mineralização ao refrigerante do dispositivo de destilação, imergindo a extremidade do refrigerante no líquido contido no balão de recolha, até uma profundidade mínima de 1 cm (ver a observação 8.3). Juntar cuidadosamente 100 ml de solução de hidróxido de sódio (3.9) ao recipiente de mineralização, evitando perdas de amoníaco (ver a observação 8.1). Aquecer o recipiente até à destilação completa do amoníaco.
5.2.2.
Sempre que a titulação do teor de amoníaco no destilado for realizada manualmente, aplica-se o procedimento mencionado a seguir. Se a unidade de destilação for totalmente automatizada e incluir a titulação do teor de amoníaco no destilado, seguir as instruções do fabricante.
Colocar um balão de recolha com 25 a 30 ml da solução de ácido bórico (3.18) sob a saída do refrigerante de forma que o tubo de saída se situe sob a superfície da solução de ácido bórico em excesso. Regular a unidade de destilação de forma a adicionar 50 ml da solução de hidróxido de sódio (3.9). Colocar a unidade de destilação em funcionamento de acordo com as instruções do fabricante e destilar o amoníaco libertado com a adição da solução de hidróxido de sódio. Recolher o destilado na solução de ácido bórico. A quantidade de destilado (duração da destilação por arrastamento de vapor) depende da quantidade de azoto na amostra. Seguir as instruções do fabricante.
|
Nota |
: |
Numa unidade de destilação semiautomática, a adição de hidróxido de sódio em excesso e a destilação por arrastamento de vapor são realizadas automaticamente. |
5.3. Titulação
Proceder de acordo com o disposto nos pontos 5.3.1 ou 5.3.2.
5.3.1.
Titular o excesso de ácido sulfúrico existente no balão de recolha com a solução de hidróxido de sódio (3.10) ou (3.11), em função da concentração de ácido sulfúrico utilizada, até atingir o ponto final.
5.3.2.
Com o auxílio de uma bureta, titular o conteúdo do balão de recolha com a solução volumétrica padrão de ácido clorídrico (3.19) ou com a solução volumétrica padrão de ácido sulfúrico (3.6) e registar o volume de solução titulante usada.
Se se aplicar a detecção colorimétrica do ponto final, este é atingido assim que a solução começa a mudar de cor para rosa. A leitura da bureta é feita com uma aproximação de 0,05 ml. A visualização do ponto final pode ser facilitada pelo uso de uma placa de agitação magnética dotada de iluminação ou de um detector fotométrico.
Esta operação pode ser feita automaticamente se se usar um dispositivo de destilação por arrastamento de vapor com titulação automática.
Para o funcionamento do destilador ou destilador/titulador, seguir as instruções do fabricante.
|
Nota: |
Quando se usa um sistema de titulação automática, a titulação começa assim que se dá início à destilação e usa-se a solução de ácido bórico a 1 % (3.18). Se se recorrer a uma unidade de destilação totalmente automatizada, a titulação do amoníaco também se pode fazer com a detecção do ponto final através de um sistema potenciométrico de medição do pH. Neste caso, usa-se um titulador automático com um medidor de pH. O medidor de pH deve estar devidamente calibrado no intervalo de pH 4 a 7, segundo procedimentos laboratoriais normais. O ponto final da titulação é atingido a um pH de 4,6, correspondente ao ponto de inflexão da curva de titulação (ponto de maior declive). |
5.4. Ensaio em branco
Com vista a confirmar a ausência de azoto nos reagentes, efectuar um ensaio em branco (mineralização, destilação e titulação) utilizando 1 g de sacarose (3.14) em vez da amostra.
6. Cálculo dos resultados
Os cálculos efectuam-se em conformidade com o referido nos pontos 6.1 ou 6.2.
6.1. Cálculo para a titulação efectuada segundo o método exposto em 5.3.1
O teor de proteína bruta, expresso em percentagem em massa, é dado pela fórmula seguinte:

em que:
|
V0 |
= |
Volume, expresso em ml, de NaOH (3.10 ou 3.11) utilizado no ensaio em branco. |
|
V1 |
= |
Volume, expresso em ml, de NaOH (3.10 ou 3.11) utilizado na titulação da amostra. |
|
c |
= |
Concentração, expressa em mol/l, da solução de hidróxido de sódio (3.10 ou 3.11). |
|
m |
= |
Massa da amostra, expressa em gramas. |
6.2. Cálculo para a titulação efectuada segundo o método exposto em 5.3.2
6.2.1.
O teor de proteína bruta, expresso em percentagem em massa, é dado pela fórmula seguinte:

em que:
|
m |
= |
Massa da amostra, expressa em gramas. |
|
c |
= |
Concentração, expressa em mol/l, da solução volumétrica padrão de ácido clorídrico (3.19). |
|
V0 |
= |
Volume, em ml, de ácido clorídrico utilizado no ensaio em branco. |
|
V1 |
= |
Volume, em ml, de ácido clorídrico utilizado na titulação da amostra. |
6.2.2.
O teor de proteína bruta, expresso em percentagem em massa, é dado pela fórmula seguinte:

em que:
|
m |
= |
Massa da amostra, expressa em gramas. |
|
c |
= |
Concentração, expressa em mol/l, da solução volumétrica padrão de ácido sulfúrico (3.6). |
|
V0 |
= |
Volume, em ml, de ácido sulfúrico (3.6) utilizado no ensaio em branco. |
|
V1 |
= |
Volume, em ml, de ácido sulfúrico (3.6) utilizado na titulação da amostra. |
7. Verificação do método
7.1. Repetibilidade
A diferença entre os resultados de duas determinações paralelas efectuadas com a mesma amostra não deve exceder:
|
— |
0,2 %, em valor absoluto, para teores de proteína bruta inferiores a 20 %, |
|
— |
1,0 % do maior dos valores, para teores de proteína bruta compreendidos entre 20 % e 40 %, |
|
— |
0,4 %, em valor absoluto, para teores superiores a 40 %. |
7.2. Exactidão
Efectuar a análise (mineralização, destilação e titulação) de 1,5 a 2,0 g de acetanilida (3.13), na presença de 1 g de sacarose (3.14); 1 g de acetanilida deverá consumir 14,80 ml de ácido sulfúrico (3.5). A taxa de recuperação deve ser de, pelo menos, 99 %.
8. Observações
|
8.1. |
O equipamento utilizado pode ser de tipo manual, semiautomático ou automático. Se for necessário efectuar uma operação de transferência entre os processos de mineralização e destilação, não deverão observar-se perdas. Se o recipiente do dispositivo de destilação não se encontrar munido de uma ampola de carga, adicionar o hidróxido de sódio imediatamente antes de adaptar o recipiente ao refrigerante, vertendo o líquido cuidadosamente ao longo das paredes. |
|
8.2. |
Se o resíduo da mineralização solidificar, recomeçar o processo, utilizando um volume de ácido sulfúrico (3.4) superior ao especificado. |
|
8.3. |
No caso de produtos com baixo teor de azoto, o volume de ácido sulfúrico (3.7) a adicionar ao balão de recolha pode ser reduzido, se necessário, para 10 ou 15 ml e diluído para 25 ml com água. |
|
8.4. |
Em análises de rotina, pode recorrer-se a métodos alternativos para a determinação da proteína bruta, mas o método Kjeldahl descrito na presente parte C é o método de referência. Deve demonstrar-se, para cada matriz individualmente, a equivalência dos resultados obtidos com o método alternativo (por exemplo, DUMAS) em comparação com o método de referência. Dado que os resultados obtidos com métodos alternativos, mesmo depois de verificada a sua equivalência, podem apresentar um pequeno desvio em relação aos resultados derivados do método de referência, é necessário mencionar no relatório analítico o método de análise usado na determinação da proteína bruta. |
D. DETERMINAÇÃO DA UREIA
1. Objecto e âmbito de aplicação
Esta técnica permite determinar o teor de ureia em alimentos para animais.
2. Princípio
Suspende-se a amostra em água juntamente com um agente clarificante e filtra-se. Determina-se o teor de ureia do filtrado, após a adição de 4-dimetilaminobenzaldeído (4-DMAB), medindo a densidade óptica a um comprimento de onda de 420 nm.
3. Reagentes
|
3.1. |
Solução de 4-dimetilaminobenzaldeído: dissolver 1,6 g de 4-DMAB em 100 ml de etanol a 96 % e juntar 10 ml de ácido clorídrico (ρ20: 1,19 g/ml). O tempo de conservação máximo deste reagente é de duas semanas. |
|
3.2. |
Solução de Carrez I: dissolver em água 21,9 g de acetato de zinco, Zn(CH3COO)2 · 2H2O, e 3 g de ácido acético glacial. Perfazer o volume de 100 ml com água. |
|
3.3. |
Solução de Carrez II: dissolver em água 10,6 g de ferrocianeto de potássio K4Fe(CN)6 · 3H2O. Perfazer o volume de 100 ml com água. |
|
3.4. |
Carvão activado, não absorvente da ureia (a verificar). |
|
3.5. |
Solução de ureia a 0,1 % (m/v). |
4. Equipamento
|
4.1. |
Misturador (agitador): cerca de 35 a 40 rotações por minuto. |
|
4.2. |
Tubos de ensaio: 160 × 16 mm, com tampa esmerilada. |
|
4.3. |
Espectrofotómetro. |
5. Procedimento
5.1. Análise da amostra
Pesar, com uma aproximação de 1 mg, 2 g de amostra, introduzi-la com 1 g de carvão activado (3.4) num balão volumétrico de 500 ml. Juntar 400 ml de água e 5 ml de solução de Carrez I (3.2), agitar durante cerca de 30 segundos e juntar em seguida 5 ml de solução de Carrez II (3.3). Misturar durante 30 minutos no misturador. Completar o volume com água, agitar e filtrar.
Recolher 5 ml de filtrado límpido e incolor e transferi-los para os tubos de ensaio de tampa esmerilada, juntar 5 ml de solução de 4-DMAB (3.1) e misturar. Colocar os tubos em banho-maria a 20 oC (± 4 oC). Decorridos 15 minutos, medir a densidade óptica da solução de amostra, com o espectrofotómetro a 420 nm. Comparar com a solução de reagentes feita no ensaio em branco.
5.2. Curva de calibração
Recolher volumes de 1, 2, 4, 5 e 10 ml de solução de ureia (3.5), introduzi-los em balões volumétricos de 100 ml, completando os volumes com água. De cada um dos balões, retirar 5 ml de solução e juntar 5 ml de solução de 4-DMAB, homogeneizar e medir a densidade óptica, como referido, por comparação com uma solução de referência com 5 ml de 4-DMAB e 5 ml de água, isenta de ureia. Traçar a curva de calibração.
6. Cálculo dos resultados
A partir da curva de calibração, determinar a quantidade de ureia na amostra.
Exprimir o resultado em percentagem da amostra.
7. Observações
|
7.1. |
Para teores de ureia superiores a 3 %, reduzir a amostra de ensaio para 1 g, ou diluir a solução inicial, para que o teor de ureia por cada 500 ml não seja superior a 50 mg. |
|
7.2. |
Para teores de ureia pouco elevados, aumentar o peso da amostra de ensaio, sem deixar no entanto que a quantidade de amostra para análise impeça o filtrado de ficar límpido e incolor. |
|
7.3. |
Se a amostra contiver compostos azotados simples, tais como aminoácidos, é aconselhável efectuar a medição da densidade óptica ao comprimento de onda de 435 nm. |
E. DETERMINAÇÃO DAS BASES AZOTADAS VOLÁTEIS
I. POR MICRODIFUSÃO
1. Objecto e âmbito de aplicação
O método permite determinar o teor de bases azotadas voláteis, expressas em amoníaco, dos alimentos para animais.
2. Princípio
A amostra é extraída com água, a solução é clarificada e filtrada. As bases azotadas voláteis são separadas por microdifusão com uma solução de carbonato de potássio, recolhidas numa solução de ácido bórico e tituladas com ácido sulfúrico.
3. Reagentes
|
3.1. |
Ácido tricloroacético, solução a 20 % (m/v). |
|
3.2. |
Indicador: dissolver 33 mg de verde de bromocresol e 65 mg de vermelho de metilo em 100 ml de etanol a 95-96 % (v/v). |
|
3.3. |
Solução de ácido bórico: num balão volumétrico de 1 litro, dissolver 10 g de ácido bórico em 200 ml de etanol a 95-96 % (v/v) e 700 ml de água. Juntar 10 ml de indicador (3.2). Misturar e, se necessário, ajustar a coloração da solução a vermelho claro, por adição de uma solução de hidróxido de sódio. 1 ml desta solução permite fixar, no máximo, 300 μg de NH3. |
|
3.4. |
Solução saturada de carbonato de potássio: dissolver 100 g de carbonato de potássio em 100 ml de água em ebulição. Deixar arrefecer e filtrar. |
|
3.5. |
Ácido sulfúrico 0,01 mol/l. |
4. Equipamento
|
4.1. |
Misturador basculante: cerca de 35 a 40 rotações por minuto. |
|
4.2. |
Células de Conway (v. esquema) de vidro ou matéria plástica. |
|
4.3. |
Microburetas, com graduação de 1/100 ml. |
5. Procedimento
Pesar, com uma aproximação de 1 mg, 10 g de amostra e introduzir num balão volumétrico de 200 ml com 100 ml de água. Misturar ou agitar durante 30 minutos no misturador basculante. Adicionar 50 ml de solução de ácido tricloroacético (3.1), completar o volume com água, agitar vigorosamente e filtrar num filtro de pregas.
Introduzir com a pipeta, na parte central da célula de Conway, 1 ml de solução de ácido bórico (3.3) e, na parte periférica, 1 ml do filtrado da amostra. Cobrir parcialmente com a tampa com silicone. Introduzir rapidamente na parte periférica 1 ml de solução saturada de carbonato de potássio (3.4) e fechar hermeticamente a tampa. Agitar suavemente a célula dando-lhe um movimento de rotação num plano horizontal, para misturar os dois reagentes. Deixar incubar durante quatro horas, pelo menos, à temperatura ambiente, ou, então, durante uma hora a 40 oC.
Titular as bases voláteis na solução de ácido bórico por meio de ácido sulfúrico (3.5), utilizando uma microbureta (4.3).
Efectuar um ensaio em branco aplicando o mesmo método, na ausência de amostra a analisar.
6. Cálculo dos resultados
1 ml de H2SO40,01 mol/l corresponde a 0,34 mg de amoníaco.
Exprimir o resultado em percentagem da amostra.
Repetibilidade
A diferença entre os resultados de duas determinações paralelas efectuadas com a mesma amostra não deve exceder:
|
— |
10 % em valor relativo, para os teores de amoníaco inferiores a 1,0 %, |
|
— |
0,1 % em valor absoluto, para os teores de amoníaco iguais ou superiores a 1,0 %. |
7. Observações
Se o teor de amoníaco da amostra for superior a 0,6 %, diluir o filtrado inicial.
CONWAY CELL
Scale 1/1

II. POR DESTILAÇÃO
1. Objecto e âmbito de aplicação
Este método permite determinar o teor de bases azotadas voláteis, expressas em amoníaco, das farinhas de peixe que praticamente não contêm ureia. Só pode ser utilizado para os teores de amoníaco inferiores a 0,25 %.
2. Princípio
A amostra é extraída com água, a solução é clarificada e filtrada. As bases azotadas voláteis são separadas, no ponto de ebulição, por adição de óxido de magnésio e recolhidas numa quantidade determinada de ácido sulfúrico cujo excesso é titulado por meio de uma solução de hidróxido de sódio.
3. Reagentes
|
3.1. |
Ácido tricloroacético, solução a 20 % (m/v). |
|
3.2. |
Óxido de magnésio. |
|
3.3. |
Emulsão antiespuma (silicone por ex.). |
|
3.4. |
Ácido sulfúrico 0,05 mol/l. |
|
3.5. |
Solução de hidróxido de sódio 0,1 mol/l. |
|
3.6. |
Solução de vermelho de metilo a 0,3 % em etanol a 95-96 % (v/v). |
4. Equipamento
|
4.1. |
Misturador basculante: cerca de 35 a 40 rotações por minuto. |
|
4.2. |
Aparelho de destilação do tipo Kjeldahl. |
5. Procedimento
Pesar, com uma aproximação de 1 mg, 10 g da amostra e introduzir num balão volumétrico de 200 ml com 100 ml de água. Misturar ou agitar durante 30 minutos no misturador basculante. Adicionar 50 ml de solução de ácido tricloroacético (3.1), completar o volume com água, agitar vigorosamente e filtrar num filtro de pregas.
Recolher uma quantidade do filtrado límpido que seja adequada em função da concentração presumida de bases azotadas voláteis (geralmente 100 ml). Diluir para 200 ml e adicionar 2 g de óxido de magnésio (3.2) e algumas gotas de emulsão antiespuma (3.3). O pH da solução deve ser alcalino quando verificado com papel tornesol; se não o for, adicionar mais óxido de magnésio (3.2). Proceder de acordo com o disposto nos pontos 5.2 e 5.3 da parte C do presente anexo relativa ao método de determinação do teor de proteína bruta.
Efectuar um ensaio em branco aplicando o mesmo método, na ausência de amostra a analisar.
6. Cálculo dos resultados
1 ml de H2SO40,05 mol/l corresponde a 1,7 mg de amoníaco.
Exprimir o resultado em percentagem da amostra.
Repetibilidade
A diferença entre os resultados de duas determinações paralelas efectuadas com a mesma amostra não deve exceder, em valor relativo, 10 % de amoníaco.
F. DETERMINAÇÃO DOS AMINOÁCIDOS (COM EXCEPÇÃO DO TRIPTOFANO)
1. Objecto e âmbito de aplicação
O presente método destina-se à determinação do teor de aminoácidos livres (naturais e de síntese) e aminoácidos totais (ligados e livres) em alimentos para animais, utilizando um analisador de aminoácidos. O método é aplicável ao doseamento dos seguintes aminoácidos: cistina/cisteína, metionina, lisina, treonina, alanina, arginina, ácido aspártico, ácido glutâmico, glicina, histidina, isoleucina, leucina, fenilalanina, prolina, serina, tirosina e valina.
O método não permite distinguir as formas D e L, nem os sais de aminoácidos. Não é aplicável ao doseamento do triptofano nem de derivados hidroxilados de aminoácidos.
2. Princípio
2.1. Aminoácidos livres
Os aminoácidos livres são extraídos com ácido clorídrico diluído. As macromoléculas azotadas co-extraídas são precipitadas com ácido sulfossalicílico e removidas por filtração. O pH do filtrado é ajustado a 2,20. Os aminoácidos são separados por cromatografia de troca iónica e determinados fotometricamente a 570 nm, após reacção com ninidrina.
2.2. Aminoácidos totais
O procedimento a adoptar depende dos aminoácidos a dosear. A cistina/cisteína e a metionina necessitam de oxidação a ácido cisteico e a metionina sulfona, respectivamente, antes da hidrólise. A tirosina é determinada nos hidrolisados de amostras não oxidadas. Todos os restantes aminoácidos referidos no ponto 1 podem ser determinados a partir de amostras oxidadas ou não oxidadas.
A oxidação é realizada a 0 oC, com uma mistura ácido perfórmico/fenol. O excesso de reagente de oxidação é decomposto com bissulfito de sódio. A amostra, oxidada ou não, é hidrolisada com ácido clorídrico (3.20) durante 23 horas. O pH do hidrolisado é ajustado a 2,20. Os aminoácidos são separados por cromatografia de troca iónica e determinados fotometricamente a 570 nm (440 nm no caso da prolina), após reacção com ninidrina.
3. Reagentes
Deve utilizar-se água bidestilada ou de qualidade equivalente (condutividade inferior a 10 μS).
|
3.1. |
Peróxido de hidrogénio, 30 % (m/m). |
|
3.2. |
Ácido fórmico, 98-100 % (m/m). |
|
3.3. |
Fenol. |
|
3.4. |
Bissulfito de sódio. |
|
3.5. |
Hidróxido de sódio. |
|
3.6. |
Ácido 5-sulfossalicílico di-hidratado. |
|
3.7. |
Ácido clorídrico (densidade aproximada 1,18 g/ml). |
|
3.8. |
Citrato trissódico di-hidratado. |
|
3.9. |
2,2'-Tiodietanol (tiodiglicol). |
|
3.10. |
Cloreto de sódio. |
|
3.11. |
Ninidrina. |
|
3.12. |
Éter de petróleo, intervalo de ebulição: 40 a 60 oC. |
|
3.13. |
Norleucina ou outro composto adequado, para ser usado como padrão interno. |
|
3.14. |
Azoto gasoso (teor de oxigénio inferior a 10 ppm). |
|
3.15. |
1-Octanol. |
|
3.16. |
Aminoácidos. |
|
3.16.1. |
Substâncias-padrão referidas no ponto 1. Os produtos devem ser puros e não conter água de cristalização. Secar sob vácuo, com P2O5 ou H2SO4, durante uma semana, antes de usar. |
|
3.16.2. |
Ácido cisteico. |
|
3.16.3. |
Metionina sulfona. |
|
3.17. |
Solução de hidróxido de sódio 7,5 mol/l. Dissolver 300 g de NaOH (3.5) em água, e perfazer o volume de 1 litro. |
|
3.18. |
Solução de hidróxido de sódio 1 mol/l. Dissolver 40 g de NaOH (3.5) em água e perfazer o volume de 1 litro. |
|
3.19. |
Solução de ácido fórmico/fenol. Misturar 889 g de ácido fórmico (3.2) com 111 g de água e adicionar 4,73 g de fenol (3.3). |
|
3.20. |
Mistura de hidrólise (HCl 6 mol/l contendo 1 g de fenol/l). Adicionar 1 g de fenol (3.3) a 492 ml de HCl (3.7) e perfazer com água o volume de 1 litro. |
|
3.21. |
Mistura de extracção (HCl 0,1 mol/l contendo 2 % de tiodiglicol). Diluir 8,2 ml de HCl (3.7) em cerca de 900 ml de água, adicionar 20 ml de tiodiglicol (3.9) e perfazer com água o volume de 1 litro (não misturar directamente os reagentes 3.7 e 3.9). |
|
3.22. |
Ácido 5-sulfossalicílico, 6 % (m/v). Dissolver 60 g de ácido 5-sulfossalicílico (3.6) em água, e perfazer o volume de 1 litro. |
|
3.23. |
Mistura oxidante (ácido perfórmico/fenol). Misturar, num pequeno matraz, 0,5 ml de peróxido de hidrogénio (3.1) com 4,5 ml de solução de ácido fórmico/fenol (3.19). Tapar e manter a 20-30 oC durante uma hora, de modo a obter ácido perfórmico. Arrefecer num banho de gelo (15 minutos) e adicionar à amostra. Cuidado: evitar o contacto com a pele e utilizar vestuário de protecção. |
|
3.24. |
Tampão citrato (0,2 mol Na+/l; pH = 2,20). Dissolver 19,61 g de citrato de sódio (3.8), 5 ml de tiodiglicol (3.9), 1 g de fenol (3.3) e 16,50 ml de HCl (3.7) em cerca de 800 ml de água. Ajustar o pH a 2,20. Perfazer o volume de 1 l com água. |
|
3.25. |
Tampões de eluição, preparados em função do aparelho utilizado (4.9). |
|
3.26. |
Reagente de ninidrina, preparado em função do aparelho utilizado (4.9). |
|
3.27. |
Soluções-padrão de aminoácidos. Estas soluções devem ser armazenadas a uma temperatura inferior a 5 oC. |
|
3.27.1. |
Solução-mãe padrão de aminoácidos (3.16.1). c = 2,5 μmol/ml de cada aminoácido em ácido clorídrico. Disponíveis no comércio. |
|
3.27.2. |
Solução-mãe padrão de ácido cisteico e metionina sulfona, c = 1,25 μmol/ml. Dissolver, num balão volumétrico de 1 litro, 0,2115 g de ácido cisteico (3.16.2) e 0,2265 g de metionina sulfona (3.16.3) em tampão citrato (3.24), completando o volume com o mesmo tampão. Armazenar a uma temperatura inferior a 5 oC, por um período não superior a 12 meses. Esta solução não é necessária no caso de a solução-mãe padrão (3.27.1) conter ácido cisteico e metionina sulfona. |
|
3.27.3. |
Solução-mãe de padrão interno, por exemplo norleucina (c = 20 μmol/ml). Dissolver, num balão volumétrico de 250 ml, 0,6560 g de norleucina (3.13) em tampão citrato (3.24), completando o volume com o mesmo tampão. Armazenar a uma temperatura inferior a 5 oC, por um período não superior a 6 meses. |
|
3.27.4. |
Solução de calibração de aminoácidos-padrão para uso com os hidrolisados (c = 5 nmol/50 μl de ácido cisteico e metionina sulfona; c = 10 nmol/50 μl dos restantes aminoácidos). Dissolver, num matraz de 100 ml, 2,2 g de cloreto de sódio (3.10) em 30 ml de tampão citrato (3.24). Adicionar 4,00 ml de solução-mãe padrão de aminoácidos (3.27.1), 4,00 ml de solução-mãe padrão de ácido cisteico e metionina sulfona (3.27.2) e 0,50 ml de solução-mãe de padrão interno (3.27.3), se for caso disso. Ajustar o pH a 2,20 com solução de hidróxido de sódio (3.18). Transferir quantitativamente para um balão volumétrico de 50 ml, completar o volume com tampão citrato (3.24) e homegeneizar. Armazenar a uma temperatura inferior a 5 oC, por um período não superior a 3 meses. Ver igualmente a observação do ponto 9.1. |
|
3.27.5. |
Solução de calibração de aminoácidos-padrão para uso com os hidrolisados obtidos de acordo com o ponto 5.3.3.1, bem como com os extractos referidos no ponto 5.2. A solução de calibração é preparada em conformidade com o ponto 3.27.4, suprimindo o cloreto de sódio. Armazenar a uma temperatura inferior a 5 oC, por um período não superior a 3 meses. |
4. Equipamento
|
4.1. |
Balão de fundo redondo de 100 ml ou 250 ml equipado com um condensador de refluxo. |
|
4.2. |
Frasco de vidro borossilicato de 100 ml munido de tampa de rosca com revestimento de borracha ou teflon (por exemplo, Duran, Schott) para uso em estufa. |
|
4.3. |
Estufa munida de um sistema de ventilação e de um sistema de regulação de temperatura com exactidão superior a ± 2 oC. |
|
4.4. |
Medidor de pH (com leitura às milésimas). |
|
4.5. |
Filtro de membrana (porosidade 0,22 μm). |
|
4.6. |
Centrífuga. |
|
4.7. |
Evaporador rotativo de vácuo. |
|
4.8. |
Agitador mecânico ou magnético. |
|
4.9. |
Analisador de aminoácidos ou aparelho para HPLC munido de uma coluna de troca iónica, derivatização pós-coluna com ninidrina e detector fotométrico. O enchimento da coluna é de resinas de poliestireno sulfonado que permitem separar os aminoácidos entre si, bem como de outros produtos com reacção positiva à ninidrina. A estabilidade do fluxo das bombas que controlam o caudal do tampão de eluição e do reagente de ninidrina deve ser de cerca de ±0,5 %, quer durante o ensaio de calibração, quer durante a execução da análise. Em alguns analisadores de aminoácidos, é possível utilizar procedimentos de hidrólise cujos hidrolisados contenham uma concentração de sódio superior a 0,8 mol/l, bem como a totalidade do ácido fórmico residual do processo de oxidação. Noutros analisadores, a separação de determinados aminoácidos não é satisfatória caso os produtos de hidrólise contenham um excesso de ácido fórmico e/ou uma concentração elevada de ião sódio. Nestes casos, o volume de ácido é reduzido, por evaporação, a cerca de 5 ml, após a hidrólise e antes do ajuste do pH. A evaporação deve ser realizada sob vácuo, a uma temperatura não superior a 40 oC. |
5. Procedimento
5.1. Preparação da amostra
A amostra é moída a uma granulometria inferior a 0,5 mm. As amostras com elevado teor de humidade devem ser secas ao ar, a uma temperatura não superior a 50 oC, ou liofilizadas, antes da moagem. Se o seu teor de matérias gordas for elevado, as amostras terão de ser submetidas a uma extracção com éter de petróleo (3.12) antes da moagem.
5.2. Determinação de aminoácidos livres em alimentos para animais e pré-misturas
Pesar num erlenmeyer, com uma aproximação de 0,2 mg, uma quantidade adequada (1-5 g) de amostra preparada de acordo com o ponto 5.1 e adicionar 100,0 ml de mistura de extracção (3.21). Agitar durante 60 minutos em agitador mecânico ou magnético (4.8). Deixar sedimentar e pipetar 10,0 ml de solução sobrenadante para um matraz de 100 ml.
Adicionar, sob agitação, 5,0 ml de solução de ácido sulfossalicílico (3.22), mantendo a agitação durante 5 minutos. Filtrar ou centrifugar o sobrenadante, de modo a remover qualquer precipitado. Colocar 10,0 ml da solução resultante num matraz de 100 ml e ajustar o pH a 2,20 com solução de hidróxido de sódio (3.18). Transferir para um balão volumétrico de volume adequado e completar até à marca com tampão citrato (3.24).
Caso se utilize um padrão interno, adicionar 1,00 ml (3.27.3) por cada 100 ml de solução final, antes de completar até à marca com tampão (3.24).
Efectuar a análise cromatográfica de acordo com o ponto 5.4.
Se os extractos não forem sujeitos a cromatografia no mesmo dia, devem ser armazenados a uma temperatura inferior a 5 oC.
5.3. Determinação dos aminoácidos totais
5.3.1.
Pesar, com uma aproximação de 0,2 mg, entre 0,1 e 1 g de amostra preparada (5.1):
|
— |
num balão de fundo redondo de 100 ml (4.1), no caso de hidrólise em sistema aberto (5.3.2.3), |
|
— |
num balão de fundo redondo de 250 ml (4.1), caso seja necessário um teor de sódio reduzido (5.3.3.1), |
|
— |
num frasco de 100 ml munido de tampa de rosca (4.2), no caso de hidrólise em sistema fechado (5.3.2.4). |
A toma de amostra para ensaio deve ter um teor aproximado de azoto de 10 mg e um teor de humidade não superior a 100 mg.
Colocar o balão ou o frasco num banho de gelo, arrefecer a 0 oC, adicionar 5 ml de mistura oxidante (3.23) e misturar com uma vareta de vidro de ponta recurva. Tapar o balão ou o frasco contendo a vareta com película impermeável ao ar, e colocar o conjunto constituído pelo recipiente tapado e o banho de gelo no frigorífico, a 0 oC, durante 16 horas. Após este período, retirar do frigorífico e decompor o excesso de oxidante por adição de 0,84 g de bissulfito de sódio (3.4).
Continuar de acordo com o descrito no ponto 5.3.2.1.
5.3.2.
5.3.2.1.
À amostra oxidada como descrito no ponto 5.3.1, adicionar 25 ml de mistura de hidrólise (3.20) tendo o cuidado de arrastar quaisquer resíduos de amostra das paredes do recipiente e da vareta.
Proceder em conformidade com o ponto 5.3.2.3 ou 5.3.2.4, em função do tipo de hidrólise.
5.3.2.2.
Pesar num balão de fundo redondo de 100 ml ou 250 ml (4.1) ou num frasco munido de tampa de rosca (4.2), com uma aproximação de 0,2 mg, 0,1 a 1 g de amostra preparada (5.1). A toma de amostra para ensaio deve ter um teor aproximado de azoto de 10 mg. Adicionar cuidadosamente 25 ml da mistura de hidrólise (3.20) e misturar. Proceder de acordo com o descrito no ponto 5.3.2.3 ou 5.3.2.4.
5.3.2.3.
Juntar 3 pérolas de vidro como reguladores de ebulição à mistura preparada de acordo com o ponto 5.3.2.1 ou 5.3.2.2 e manter em ebulição contínua, sob refluxo, durante 23 horas. Após a hidrólise, lavar o condensador pelo topo, com 5 ml de tampão citrato (3.24). Desmontar o conjunto e arrefecer o balão num banho de gelo.
Proceder de acordo com o ponto 5.3.3.
5.3.2.4.
Colocar o frasco que contém a mistura preparada de acordo com o ponto 5.3.2.1 ou 5.3.2.2 numa estufa (4.3) a 110 oC. Durante a primeira hora, colocar a tampa de rosca no topo do frasco sem contudo o vedar, de modo a evitar um aumento da pressão devido aos gases libertados e o consequente risco de explosão. Em seguida, vedar o frasco com a tampa de rosca e manter em estufa (4.3) durante 23 horas. Após a hidrólise, remover o frasco da estufa, abri-lo cuidadosamente e colocá-lo num banho de gelo, até arrefecer.
Com o auxílio de tampão citrato (3.24), transferir quantitativamente o conteúdo do frasco para um matraz ou um balão de fundo redondo de 250 ml, em função do procedimento de ajuste do pH a utilizar (ponto 5.3.3).
Proceder de acordo com o ponto 5.3.3.
5.3.3.
Para o ajuste do pH, proceder de acordo com o ponto 5.3.3.1 ou 5.3.3.2, em função da tolerância do analisador de aminoácidos (4.9) ao sódio.
5.3.3.1.
É aconselhável utilizar uma solução-mãe de padrão interno (3.27.3), no caso de analisadores de aminoácidos que necessitem de um teor reduzido de sódio (uma vez que o volume de ácido deve ser reduzido).
Neste caso, adicionar 2,00 ml de solução-mãe de padrão interno (3.27.3) ao hidrolisado, antes da evaporação.
Adicionar 2 gotas de 1-octanol (3.15) ao hidrolisado obtido de acordo com os pontos 5.3.2.3 ou 5.3.2.4.
Em evaporador rotativo (4.7), reduzir o volume a 5-10 ml, sob vácuo, a 40 oC. Se, acidentalmente, o volume for reduzido a menos de 5 ml, o produto deve ser rejeitado e a análise recomeçada.
Ajustar o pH a 2,20 com solução de hidróxido de sódio (3.18), e proceder de acordo com o referido no ponto 5.3.4.
5.3.3.2.
Neutralizar parcialmente o hidrolisado obtido de acordo com 5.3.2.3 ou 5.3.2.4 mediante a adição cuidadosa, sob agitação, de 17 ml de solução de hidróxido de sódio (3.17), mantendo a temperatura inferior a 40 oC.
Ajustar o pH a 2,20, à temperatura ambiente, com as soluções de hidróxido de sódio (3.17) e (3.18). Proceder de acordo com o ponto 5.3.4.
5.3.4.
Com o auxílio de tampão citrato (3.24), transferir quantitativamente o produto de hidrólise com o pH ajustado (5.3.3.1 ou 5.3.3.2) para um balão volumétrico de 200 ml, completando até à marca com o mesmo tampão.
No caso de não se ter adicionado ainda um padrão interno, juntar 2,00 ml da solução-mãe de padrão interno (3.27.3) e completar até à marca com tampão citrato (3.24). Homogeneizar.
Efectuar a análise cromatográfica de acordo com o ponto 5.4.
Se as soluções de amostra não forem analisadas no mesmo dia, devem ser conservadas a uma temperatura inferior a 5 oC.
5.4. Cromatografia
Antes de iniciar a análise cromatográfica, deixar estabilizar o extracto (5.2) ou o produto de hidrólise (5.3.4) à temperatura ambiente. Agitar a mistura e filtrar uma quantidade adequada com filtro de membrana de 0,22 μm (4.5). A solução límpida resultante é sujeita a cromatografia de troca iónica no analisador de aminoácidos (4.9).
A injecção pode ser manual ou automática. A quantidade de solução utilizada na análise dos padrões e das amostras deve ser constante, com uma aproximação de ±0,5 %, excepto no caso de se utilizar um padrão interno; a razão entre as concentrações de sódio e de aminoácidos na solução-padrão e na amostra deve ser tão próxima quanto possível.
Em geral, a frequência dos ensaios de calibração depende da estabilidade do reagente de ninidrina e do sistema analítico. O padrão ou a amostra são diluídos com tampão citrato (3.24), de modo a que a área dos picos dos aminoácidos produzidos pelo padrão seja 30-200 % da área dos correspondentes picos dos aminoácidos da amostra.
A cromatografia dos aminoácidos varia ligeiramente em função do tipo de analisador e da resina utilizada. O sistema escolhido deve permitir a separação dos aminoácidos entre si, bem como dos restantes produtos com reacção positiva à ninidrina. Além disso, o sistema deverá dar uma resposta linear à alteração das quantidades de aminoácidos injectadas, na gama das concentrações utilizadas.
Durante a cromatografia, as razões entre a altura do vale e a altura do pico referidas infra aplicam-se na análise de uma solução equimolar (dos aminoácidos a determinar). A solução equimolar de aminoácidos deve conter, pelo menos, 30 % da quantidade máxima de cada aminoácido que pode ser determinada com exactidão no analisador de aminoácidos (4.9).
Para a separação treonina-serina, se os respectivos picos estiverem parcialmente sobrepostos, a razão entre a altura do vale e a altura do menor dos dois picos sobrepostos no cromatograma não deve exceder 2:10. Caso se pretenda determinar apenas a cistina/cisteína, a metionina, a treonina e a lisina, a má resolução de dois picos adjacentes afecta a quantificação. No que respeita aos restantes aminoácidos, a separação deve ser superior a 1:10.
O sistema deve assegurar a separação da lisina das substâncias afins e da ornitina.
6. Cálculo dos resultados
Procede-se à determinação da área do pico correspondente a cada aminoácido presente na amostra e no padrão, sendo o respectivo teor (X), expresso em g por kg de amostra, calculado pela fórmula:

Caso tenha sido utilizado um padrão interno, multiplicar pelo factor: 
|
A |
= |
área do pico do hidrolisado ou do extracto |
|
B |
= |
área do pico da solução-padrão de calibração |
|
C |
= |
área do pico do padrão interno, no hidrolisado ou no extracto |
|
D |
= |
área do pico do padrão interno, na solução-padrão de calibração |
|
M |
= |
massa molecular do aminoácido doseado |
|
c |
= |
concentração da solução-padrão, expressa em μmol/ml |
|
m |
= |
massa da amostra, corrigida em caso de secagem ou desengorduramento, expressa em g |
|
V |
= |
volume do hidrolisado (5.3.4) ou volume de diluição total do extracto (6.1), expressos em ml |
A cistina e a cisteína são determinadas na forma de ácido cisteico, no hidrolisado da amostra oxidada, sendo expressas em cistina (C6H12N2O4S2, M = 240,30 g/mol), considerando M = 120,15 g/mol (0,5 × 240,30 g/mol).
A metionina é determinada na forma de metionina sulfona, no hidrolisado da amostra oxidada, sendo expressa em metionina, considerando M = 149,21 g/mol.
A metionina livre adicionada é determinada na forma de metionina, após extracção, utilizando o mesmo valor de massa molecular.
|
6.1. |
O volume total de diluição dos extractos (F), para a determinação dos aminoácidos livres (5.2), é calculado do seguinte modo: 
|
7. Avaliação do método
O método foi testado num estudo internacional de comparação interlaboratorial realizado em 1990 com base em quatro tipos de alimentos para animais (alimento misto para suínos, alimento composto para frangos, concentrado proteico e pré-mistura). Após eliminação dos valores anómalos, obtiveram-se para a média e o desvio-padrão os valores que se apresentam nos quadros seguintes:
Médias, expressas em g/kg
|
Material de referência |
Aminoácidos |
||||||
|
Treonina |
Cistina/Cisteína |
Metionina |
Lisina |
||||
|
Alimento misto para suínos |
6,94 n = 15 |
3,01 n = 17 |
3,27 n = 17 |
9,55 n = 13 |
|||
|
Alimento composto para frangos |
9,31 n = 16 |
3,92 n = 18 |
5,08 n = 18 |
13,93 n = 16 |
|||
|
Concentrado proteico |
22,32 n = 16 |
5,06 n = 17 |
12,01 n = 17 |
47,74 n = 15 |
|||
|
Pré-mistura |
58,42 n = 16 |
— |
90,21 n = 16 |
98,03 n = 16 |
|||
|
|||||||
7.1. Repetibilidade
Apresentam-se de seguida os valores relativos à repetibilidade, expressos em termos de desvio-padrão intralaboratorial, obtidos na comparação interlaboratorial mencionada, para os aminoácidos em estudo:
Desvio-padrão intralaboratorial (Sr), expresso em g/kg
|
Material de referência |
Aminoácidos |
||||||
|
Treonina |
Cistina/Cisteína |
Metionina |
Lisina |
||||
|
Alimento misto para suínos |
0,13 n = 15 |
0,10 n = 17 |
0,11 n = 17 |
0,26 n = 13 |
|||
|
Alimento composto para frangos |
0,20 n = 16 |
0,11 n = 18 |
0,16 n = 18 |
0,28 n = 16 |
|||
|
Concentrado proteico |
0,48 n = 16 |
0,13 n = 17 |
0,27 n = 17 |
0,99 n = 15 |
|||
|
Pré-mistura |
1,30 n = 16 |
— |
2,19 n = 16 |
2,06 n = 16 |
|||
|
|||||||
Coeficiente de variação, expresso em percentagem, do desvio-padrão intralaboratorial (Sr)
|
Material de referência |
Aminoácidos |
||||||
|
Treonina |
Cistina/Cisteína |
Metionina |
Lisina |
||||
|
Alimento misto para suínos |
1,9 n = 15 |
3,3 n = 17 |
3,4 n = 17 |
2,8 n = 13 |
|||
|
Alimento composto para frangos |
2,1 n = 16 |
2,8 n = 18 |
3,1 n = 18 |
2,1 n = 16 |
|||
|
Concentrado proteico |
2,7 n = 16 |
2,6 n = 17 |
2,2 n = 17 |
2,4 n = 15 |
|||
|
Pré-mistura |
2,2 n = 16 |
— |
2,4 n = 16 |
2,1 n = 16 |
|||
|
|||||||
7.2. Reprodutibilidade
Apresentam-se no quadro seguinte os valores do desvio-padrão interlaboratorial obtidos na comparação interlaboratorial mencionada:
Desvio-padrão interlaboratorial (SR), expresso em g/kg
|
Material de referência |
Aminoácidos |
||||||
|
Treonina |
Cistina/Cisteína |
Metionina |
Lisina |
||||
|
Alimento misto para suínos |
0,28 n = 15 |
0,30 n = 17 |
0,23 n = 17 |
0,30 n = 13 |
|||
|
Alimento composto para frangos |
0,48 n = 16 |
0,34 n = 18 |
0,55 n = 18 |
0,75 n = 16 |
|||
|
Concentrado proteico |
0,85 n = 16 |
0,62 n = 17 |
1,57 n = 17 |
1,24 n = 15 |
|||
|
Pré-mistura |
2,49 n = 16 |
— |
6,20 n = 16 |
6,62 n = 16 |
|||
|
|||||||
Coeficiente de variação, expresso em percentagem, do desvio-padrão interlaboratorial (SR)
|
Material de referência |
Aminoácidos |
||||||
|
Treonina |
Cistina/Cisteína |
Metionina |
Lisina |
||||
|
Alimento misto para suínos |
4,1 n = 15 |
9,9 n = 17 |
7,0 n = 17 |
3,2 n = 13 |
|||
|
Alimento composto para frangos |
5,2 n = 16 |
8,8 n = 18 |
10,9 n = 18 |
5,4 n = 16 |
|||
|
Concentrado proteico |
3,8 n = 16 |
12,3 n = 17 |
13,0 n = 17 |
3,0 n = 15 |
|||
|
Pré-mistura |
4,3 n = 16 |
— |
6,9 n = 16 |
6,7 n = 16 |
|||
|
|||||||
8. Utilização de materiais de referência
Deve verificar-se a aplicação correcta do método mediante a realização de ensaios em duplicado com materiais de referência certificados, quando disponíveis. Recomenda-se a calibração com soluções de calibração de aminoácidos certificadas.
9. Observações
|
9.1. |
Em virtude das diferenças entre os analisadores de aminoácidos, deve tomar-se como orientação a concentração final das soluções de calibração dos aminoácidos-padrão (3.27.4 e 3.27.5) e do hidrolisado (5.3.4). A zona de resposta linear do aparelho deve ser verificada para todos os aminoácidos. A solução-padrão é diluída com tampão citrato de modo que os picos surjam no meio dessa zona. |
|
9.2. |
Quando se utilizar um aparelho de HPLC para análise dos hidrolisados, as condições experimentais devem ser optimizadas de acordo com as recomendações do fabricante. |
|
9.3. |
A aplicação do método a alimentos para animais com mais de 1 % de cloretos (concentrados, minerais, suplementos) pode originar uma subestimação da metionina, sendo por isso necessário um tratamento especial. |
G. DETERMINAÇÃO DO TRIPTOFANO
1. Objecto e âmbito de aplicação
O método permite determinar o triptofano livre e total em alimentos para animais. As formas D e L não são distinguidas.
2. Princípio
Para a determinação do triptofano total, procede-se à hidrólise da amostra em condições alcalinas com uma solução saturada de hidróxido de bário que se aquece em seguida a 110 oC durante 20 horas. Depois de concluída a hidrólise, adiciona-se um padrão interno.
Para a determinação do triptofano livre, efectua-se uma extracção da amostra em condições moderadamente ácidas na presença de um padrão interno.
O triptofano e o padrão interno no hidrolisado ou no extracto são determinados por cromatografia líquida de alta resolução (HPLC) com detecção por fluorescência.
3. Reagentes
|
3.1. |
Água bidestilada ou de qualidade equivalente (condutividade inferior a 10 μS/cm). |
|
3.2. |
Substância-padrão: triptofano (grau de pureza/teor ≥ 99 %) seco sob vácuo na presença de pentóxido de fósforo. |
|
3.3. |
Padrão interno: α-metiltriptofano (grau de pureza/teor ≥ 99 %) seco sob vácuo na presença de pentóxido de fósforo. |
|
3.4. |
Hidróxido de bário octa-hidratado (tomar as devidas precauções para não expor excessivamente o Ba(OH)2.8H2O ao ar, para evitar a formação de BaCO3, que poderia perturbar a determinação) (ver as observações do ponto 9.3). |
|
3.5. |
Hidróxido de sódio. |
|
3.6. |
Ácido ortofosfórico, w = 85 %. |
|
3.7. |
Ácido clorídrico, ρ20 = 1,19 g/ml. |
|
3.8. |
Metanol de qualidade equivalente para HPLC. |
|
3.9. |
Éter de petróleo, intervalo de ebulição: 40 a 60 oC. |
|
3.10. |
Solução de hidróxido de sódio 1 mol/l: Dissolver 40,0 g de NaOH (3.5) em água e completar o volume com água (3.1) até perfazer 1 litro. |
|
3.11. |
Ácido clorídrico 6 mol/l: Misturar 492 ml de HCl (3.7) com água (3.1) até perfazer 1 litro. |
|
3.12. |
Ácido clorídrico 1 mol/l: Misturar 82 ml de HCl (3.7) com água (3.1) até perfazer 1 litro. |
|
3.13. |
Ácido clorídrico 0,1 mol/l: Misturar 8,2 ml de HCl (3.7) com água (3.1) até perfazer 1 litro. |
|
3.14. |
Ácido ortofosfórico 0,5 mol/l: Misturar 34 ml de ácido ortofosfórico (3.6) com água (3.1) até perfazer 1 litro. |
|
3.15. |
Solução concentrada de triptofano (3.2) 2,50 μmol/ml: Em balão volumétrico de 500 ml, dissolver 0,2553 g de triptofano (3.2) em ácido clorídrico (3.13) e completar o volume com o mesmo ácido até ao traço. Conservar a -18 oC no máximo durante quatro semanas. |
|
3.16. |
Solução concentrada de padrão interno 2,50 μmol/ml: Em balão volumétrico de 500 ml, dissolver 0,2728 g de α-metiltriptofano (3.3) em ácido clorídrico (3.13) e completar o volume com o mesmo ácido até ao traço. Conservar a -18 oC no máximo durante quatro semanas. |
|
3.17. |
Solução-padrão de calibração de triptofano e de padrão interno: Tomar 2,00 ml da solução concentrada de triptofano (3.15) e 2,00 ml da solução concentrada de padrão interno (α-metiltriptofano) (3.16). Diluir com água (3.1) e metanol (3.8) até obter aproximadamente o mesmo volume e aproximadamente a mesma concentração de metanol (10-30 %) que o hidrolisado pronto. Esta solução é preparada imediatamente antes da sua utilização. Proteger da luz solar directa durante a preparação. |
|
3.18. |
Ácido acético. |
|
3.19. |
1,1,1-Tricloro-2-metil-2-propanol. |
|
3.20. |
Etanolamina w > 98 %. |
|
3.21. |
Solução de 1 g de 1,1,1-tricloro-2-metil-2-propanol (3.19) em 100 ml de metanol (3.8). |
|
3.22. |
Fase móvel para HPLC: 3,00 g de ácido acético (3.18) + 900 ml de água (3.1) +50,0 ml da solução 1 g/100 ml (3.21) de 1,1,1-tricloro-2-metil-2-propanol (3.19) em metanol (3.8). Ajustar o pH a 5,00 com etanolamina (3.20). Completar o volume com água (3.1) até perfazer 1 000 ml. |
4. Equipamento
|
4.1. |
Aparelho para HPLC com espectrofluorímetro. |
|
4.2. |
Coluna para cromatografia líquida com 125 mm x 4 mm e enchimento C18 de 3 μm, ou equivalente. |
|
4.3. |
Medidor de pH. |
|
4.4. |
Frasco de polipropileno de 125 ml, com colo largo e tampa de rosca. |
|
4.5. |
Filtro de membrana, porosidade 0,45 μm. |
|
4.6. |
Autoclave, 110 (± 2)oC, 1,4 (±0,1) bar. |
|
4.7. |
Agitador mecânico ou magnético. |
|
4.8. |
Agitador vortex. |
5. Procedimento
5.1. Preparação da amostra
A amostra é moída a uma granulometria inferior a 0,5 mm. As amostras com elevado teor de humidade devem ser secas ao ar, a uma temperatura não superior a 50 oC, ou liofilizadas, antes da moagem. Se o seu teor de matérias gordas for elevado, as amostras terão de ser submetidas a uma extracção com éter de petróleo (3.9) antes da moagem.
5.2. Determinação do triptofano livre (extracção)
Pesar num erlenmeyer, com uma aproximação de 1 mg, uma quantidade adequada (1-5 g) de amostra preparada de acordo com o ponto 5.1. Adicionar 100,0 ml de ácido clorídrico (3.13) e 5,00 ml da solução concentrada de padrão interno (3.16). Agitar durante 60 minutos em agitador mecânico ou magnético (4.7). Deixar sedimentar e pipetar 10,0 ml da solução sobrenadante para um matraz. Adicionar 5 ml de ácido ortofosfórico (3.14). Ajustar o pH a 3 com a solução de hidróxido de sódio (3.10). Adicionar metanol (3.8) em quantidade suficiente para obter uma concentração de metanol no volume final compreendida entre 10 % e 30 %. Transferir para um balão volumétrico de capacidade adequada e diluir com água até ao volume necessário para a cromatografia [aproximadamente o mesmo volume que a solução-padrão de calibração (3.17).
Filtrar alguns mililitros da solução por filtro de membrana de 0,45 μm (4.5) antes de proceder à injecção na coluna de HPLC. Efectuar a análise cromatográfica de acordo com o ponto 5.4.
Proteger a solução-padrão e os extractos da luz solar directa. Se os extractos não puderem ser analisados no mesmo dia, podem ser conservados a 5 oC durante um máximo de três dias.
5.3. Determinação do triptofano total (hidrólise)
Pesar em frasco de polipropileno (4.4), com uma aproximação de 0,2 mg, 0,1 g a 1 g da amostra preparada de acordo com o ponto 5.1. A toma de amostra para ensaio deve ter um teor aproximado de azoto de 10 mg. Adicionar 8,4 g de hidróxido de bário octa-hidratado (3.4) e 10 ml de água. Agitar em agitador vortex (4.8) ou magnético (4.7) (deixar o magnete revestido a teflon no frasco). Lavar as paredes do frasco com 4 ml de água. Colocar a tampa de rosca e fechar o frasco sem enroscar muito a tampa. Colocar na autoclave (4.6) com água em ebulição e manter sob a acção do vapor durante 30-60 minutos. Fechar a autoclave e manter a 110 (± 2)oC durante 20 horas.
Antes de abrir a autoclave, reduzir a temperatura a um pouco menos de 100 oC. Para evitar a cristalização do Ba(OH)2·8H2O, adicionar à mistura quente 30 ml de água à temperatura ambiente. Agitar com cuidado. Adicionar 2,00 ml da solução concentrada de padrão interno (α-metiltriptofano) (3.16). Arrefecer o recipiente num banho de gelo durante 15 minutos.
Adicionar, em seguida, 5 ml de ácido ortofosfórico (3.14). Manter o recipiente no banho de arrefecimento e neutralizar com HCl (3.11), sob agitação; ajustar o pH a 3,0 com HCl (3.12). Adicionar metanol em quantidade suficiente para obter uma concentração de metanol no volume final compreendida entre 10 % e 30 %. Transferir para um balão volumétrico de capacidade adequada e diluir com água até ao volume necessário para a cromatografia (por exemplo, 100 ml). A adição de metanol não deve produzir precipitação.
Filtrar alguns mililitros da solução por um filtro de membrana de 0,45 μm (4.5) antes de proceder à injecção na coluna de HPLC. Efectuar a análise cromatográfica de acordo com o ponto 5.4.
Proteger a solução-padrão e o hidrolisado da luz solar directa. Se o hidrolisado não puder ser analisado no mesmo dia, pode ser conservado a 5 oC durante um máximo de três dias.
5.4. Análise por HPLC
As condições a seguir especificadas para a eluição isocrática são-no a título indicativo. Poderão escolher-se outras condições, desde que produzam resultados equivalentes (ver igualmente as observações dos pontos 9.1 e 9.2):
|
Coluna para cromatografia líquida (4.2): |
125 mm × 4 mm, enchimento C18 de 3 μm, ou equivalente |
|
Temperatura da coluna: |
Temperatura ambiente |
|
Fase móvel (3.22): |
3,00 g de ácido acético (3.18) + 900 ml de água (3.1) +50,0 ml da solução 1 g/100 ml (3.21) de 1,1,1-tricloro-2-metil-2-propanol (3.19) em metanol (3.8). Ajustar o pH a 5,00 com etanolamina (3.20). Completar o volume com água (3.1) até perfazer 1 000 ml. |
|
Caudal: |
1 ml/minuto |
|
Tempo total de eluição: |
cerca de 34 minutos |
|
Comprimento de onda de detecção: |
excitação: 280 nm; emissão: 356 nm |
|
Volume injectado: |
20 μl |
6. Cálculo dos resultados
A quantidade de triptofano (X), expressa em gramas, por 100 gramas de amostra, é calculada através da fórmula seguinte:

|
A |
= |
área do pico do padrão interno na solução-padrão de calibração (3.17) |
|
B |
= |
área do pico do triptofano no extracto (5.2) ou no hidrolisado (5.3) |
|
V1 |
= |
volume em ml (2 ml) de solução concentrada de triptofano (3.15) adicionado à solução de calibração (3.17) |
|
c |
= |
concentração, em μmol/ml (= 2,50), da solução concentrada de triptofano (3.15) adicionada à solução de calibração (3.17) |
|
V2 |
= |
volume, em ml, da solução concentrada de padrão interno (3.16) adicionado na extracção (5.2) (= 5,00 ml) ou na hidrólise (5.3) (= 2,00 ml) |
|
C |
= |
área do pico do padrão interno no extracto (5.2) ou no hidrolisado (5.3) |
|
D |
= |
área do pico do triptofano na solução-padrão de calibração (3.17) |
|
V3 |
= |
volume em ml (= 2,00 ml) de solução concentrada de padrão interno (3.16) adicionado à solução-padrão de calibração (3.17) |
|
m |
= |
massa, em g, da amostra (corrigida para a massa original se tiver sido submetida a secagem e/ou desengorduramento) |
|
M |
= |
massa molecular do triptofano (= 204,23 g/mol) |
7. Repetibilidade
A diferença entre os resultados de duas determinações paralelas efectuadas com a mesma amostra não deve exceder 10 % do maior dos valores.
8. Resultados de um estudo interlaboratorial
Tendo em vista a certificação do método de hidrólise, foi organizado um estudo interlaboratorial comunitário (quarta comparação interlaboratorial), no âmbito do qual foram analisadas três amostras em 12 laboratórios. Cada amostra foi analisada em quintuplicado. Os resultados obtidos figuram no quadro seguinte:
|
|
Amostra 1 Alimento para suínos |
Amostra 2 Alimento para suínos com suplemento de L- -triptofano |
Amostra 3 Alimento concentrado para suínos |
|
L |
12 |
12 |
12 |
|
n |
50 |
55 |
50 |
|
Média [g/kg] |
2,42 |
3,40 |
4,22 |
|
sr [g/kg] |
0,05 |
0,05 |
0,08 |
|
r [g/kg] |
0,14 |
0,14 |
0,22 |
|
CVr [ %] |
1,9 |
1,6 |
1,9 |
|
SR [g/kg] |
0,15 |
0,20 |
0,09 |
|
R [g/kg] |
0,42 |
0,56 |
0,25 |
|
CVR [ %] |
6,3 |
6,0 |
2,2 |
|
L |
= |
número de laboratórios que apresentaram resultados |
|
n |
= |
número de resultados individuais considerado (com excepção dos valores anómalos, identificados pelo teste de Cochran, Dixon) |
|
sr |
= |
desvio-padrão da repetibilidade |
|
SR |
= |
desvio-padrão da reprodutibilidade |
|
r |
= |
repetibilidade |
|
R |
= |
reprodutibilidade |
|
CVr |
= |
coeficiente de variação da repetibilidade (%) |
|
CVR |
= |
coeficiente de variação da reprodutibilidade (%) |
Tendo em vista a certificação do método de extracção do triptofano livre, foi organizado um outro estudo interlaboratorial comunitário (terceira comparação interlaboratorial), no âmbito do qual foram analisadas duas amostras em 13 laboratórios. Cada amostra foi analisada em quintuplicado. Os resultados obtidos figuram no quadro seguinte:
|
|
Amostra 4 Mistura de trigo e soja |
Amostra 5 Mistura de trigo e soja (= amostra 4) com triptofano adicionado (0,457 g/kg) |
|
L |
12 |
12 |
|
n |
55 |
60 |
|
Média [g/kg] |
0,391 |
0,931 |
|
sr [g/kg] |
0,005 |
0,012 |
|
r [g/kg] |
0,014 |
0,034 |
|
CVr [ %] |
1,34 |
1,34 |
|
SR [g/kg] |
0,018 |
0,048 |
|
R [g/kg] |
0,050 |
0,134 |
|
CVR [ %] |
4,71 |
5,11 |
|
L |
= |
número de laboratórios que apresentaram resultados |
|
n |
= |
número de resultados individuais considerado (com excepção dos valores anómalos, identificados pelo teste de Cochran, Dixon) |
|
sr |
= |
desvio-padrão da repetibilidade |
|
SR |
= |
desvio-padrão da reprodutibilidade |
|
r |
= |
repetibilidade |
|
R |
= |
reprodutibilidade |
|
CVr |
= |
coeficiente de variação da repetibilidade (%) |
|
CVR |
= |
coeficiente de variação da reprodutibilidade (%) |
Tendo em vista a certificação do triptofano para a hidrólise, foi organizado um outro estudo interlaboratorial comunitário, no âmbito do qual foram analisadas quatro amostras em sete laboratórios. Os resultados obtidos figuram no quadro seguinte. Cada amostra foi analisada em quintuplicado.
|
|
Amostra 1 Alimento misto para suínos (CRM 117) |
Amostra 2 Farinha de peixe com baixo teor de matérias gordas (CRM 118) |
Amostra 3 Farinha de soja (CRM 119) |
Amostra 4 Leite em pó desnatado (CRM 120) |
|
L |
7 |
7 |
7 |
7 |
|
n |
25 |
30 |
30 |
30 |
|
Média [g/kg] |
2,064 |
8,801 |
6,882 |
5,236 |
|
sr [g/kg] |
0,021 |
0,101 |
0,089 |
0,040 |
|
r [g/kg] |
0,059 |
0,283 |
0,249 |
0,112 |
|
CVr [ %] |
1,04 |
1,15 |
1,30 |
0,76 |
|
SR [g/kg] |
0,031 |
0,413 |
0,283 |
0,221 |
|
R [g/kg] |
0,087 |
1,156 |
0,792 |
0,619 |
|
CVR [ %] |
1,48 |
4,69 |
4,11 |
4,22 |
|
L |
= |
número de laboratórios que apresentaram resultados |
|
n |
= |
número de resultados individuais considerado (com excepção dos valores anómalos, identificados pelo teste de Cochran, Dixon) |
|
sr |
= |
desvio-padrão da repetibilidade |
|
SR |
= |
desvio-padrão da reprodutibilidade |
|
r |
= |
repetibilidade |
|
R |
= |
reprodutibilidade |
|
CVr |
= |
coeficiente de variação da repetibilidade (%) |
|
CVR |
= |
coeficiente de variação da reprodutibilidade (%) |
9. Observações
|
9.1. |
A aplicação das condições cromatográficas a seguir especificadas pode permitir uma melhor separação entre o triptofano e o α-metiltriptofano. Eluição isocrática seguida de lavagem da coluna com aplicação de um gradiente:
|
||||||||||||||||||||||||||||||||||||||||||||
|
9.2. |
A cromatografia está dependente do tipo de aparelho para HPLC e do enchimento da coluna. O sistema seleccionado deve permitir separar o triptofano do padrão interno com retorno à linha de base. É também importante que os produtos de degradação sejam bem separados do triptofano e do padrão interno. Devem efectuar-se algumas passagens cromatográficas de hidrolisados sem padrão interno para avaliar da presença de impurezas na zona da linha de base correspondente ao padrão interno. É importante que o tempo total de eluição seja suficientemente longo para eluir todos os produtos de degradação; caso contrário, pode haver interferência de picos eluídos tardiamente em passagens cromatográficas posteriores. O sistema cromatográfico deve produzir uma resposta linear nas condições de trabalho. A linearidade da resposta deve ser determinada com uma concentração constante (normal) do padrão interno e concentrações variáveis de triptofano. É importante que o tamanho dos picos do triptofano e do padrão interno esteja dentro da zona de linearidade do sistema de HPLC/fluorescência. Se o(s) pico(s) do triptofano e/ou do padrão interno for(em) muito grande(s) ou muito pequeno(s), haverá que repetir a análise com outra dimensão de amostra e/ou um volume final devidamente ajustado. |
|
9.3. |
Hidróxido de bário
Com o tempo, a dissolução do hidróxido de bário torna-se mais difícil. Daí resulta uma solução turva para a análise por HPLC, a qual pode produzir resultados por defeito para o triptofano. |
H. DETERMINAÇÃO DAS MATÉRIAS GORDAS BRUTAS
1. Objecto e âmbito de aplicação
Este método tem por objectivo o doseamento das matérias gordas brutas nos alimentos para animais. Não diz respeito à análise das sementes e frutos oleaginosos.
A utilização dos dois processos a seguir descritos depende da natureza e da composição do alimento e da razão pela qual a análise é realizada.
1.1. Processo A — Matérias gordas brutas extraíveis directamente
Este método é aplicável a matérias-primas para a alimentação animal de origem vegetal, com excepção daquelas a que é aplicável o processo B.
1.2. Processo B — Matérias gordas brutas totais
Este método é aplicável a matérias-primas para a alimentação animal de origem animal e a todos os alimentos compostos para animais. Deve ser utilizado para todas as matérias-primas das quais não se possam extrair completamente as matérias gordas sem hidrólise prévia, como, por exemplo, glúten, leveduras, proteínas de batata e produtos submetidos a processos como a extrusão, transformação em flocos e aquecimento.
1.3. Interpretação dos resultados
Sempre que pelo processo B for obtido um resultado de valor mais elevado do que o obtido pelo processo A, o resultado obtido com o processo B será considerado como o verdadeiro valor.
2. Princípio
2.1. Processo A
A amostra é extraída com éter de petróleo. O solvente é destilado e o resíduo é seco e pesado.
2.2. Processo B
A amostra é tratada a quente com ácido clorídrico. A mistura é arrefecida e filtrada. Depois de ter sido lavado e seco, o resíduo é submetido a análise segundo o processo A.
3. Reagentes
|
3.1. |
Éter de petróleo, intervalo de ebulição: 40 a 60 oC O índice de bromo deve ser inferior a 1 e o resíduo após evaporação inferior a 2 mg/100 ml. |
|
3.2. |
Sulfato de sódio, anidro. |
|
3.3. |
Ácido clorídrico 3 mol/l. |
|
3.4. |
Adjuvante de filtração, por exemplo, terra de diatomáceas, Hyflo-supercel. |
4. Equipamento
|
4.1. |
Extractor. Se o dispositivo for munido de um sifão (aparelho Soxhlet), a taxa de refluxo deve ser regulada de maneira a obter cerca de 10 ciclos por hora; se se tratar de um aparelho sem sifão, a taxa de refluxo deve ser de cerca de 10 ml por minuto. |
|
4.2. |
Cartuchos de extracção, isentos de matérias solúveis em éter de petróleo, com uma porosidade compatível com as exigências do ponto 4.1. |
|
4.3. |
Estufa, quer de vácuo, a 75 oC ± 3 oC, quer de ar, a 100 oC ± 3 oC. |
5. Procedimento
5.1. Processo A (ver ponto 8.1)
Pesar 5 g de amostra, com uma aproximação de 1 mg, introduzi-los num cartucho de extracção (4.2) e cobri-los com um tampão de algodão isento de matéria gorda.
Colocar o cartucho num extractor (4.1) e proceder à extracção durante 6 horas com éter de petróleo (3.1). Recolher o extracto de éter de petróleo num balão seco, contendo fragmentos de pedra-pomes (2) e pesado.
Eliminar o solvente por destilação. Secar o resíduo, colocando o balão durante uma hora e meia numa estufa (4.3). Deixar arrefecer num exsicador e pesar. Secar de novo durante 30 minutos para ter a certeza de que o peso da matéria gorda permanece constante (a perda de peso entre duas pesagens sucessivas deve ser igual ou inferior a 1 mg).
5.2. Processo B
Pesar, com uma aproximação de 1 mg, 2,5 g de amostra (ver ponto 8.2), introduzi-los num copo de 400 ml ou num erlenmeyer de 300 ml e juntar 100 ml de ácido clorídrico (3.3) e alguns fragmentos de pedra-pomes. Cobrir o copo com um vidro de relógio ou munir o erlenmeyer de um condensador de refluxo. Deixar ferver suavemente durante uma hora, à chama fraca ou sobre uma placa de aquecimento. Evitar que o produto adira às paredes do recipiente.
Arrefecer e adicionar uma quantidade de adjuvante de filtração (3.4) suficiente para evitar qualquer perda de matéria gorda na filtração. Filtrar num filtro de papel duplo, humidificado e isento de matérias gordas. Lavar o resíduo com água fria até à neutralização do filtrado. Verificar que o filtrado não contém matérias gordas. A presença destas indica que, antes da hidrólise, deve ser efectuada uma extracção da amostra com éter de petróleo, segundo o processo A.
Colocar o papel de filtro duplo contendo o resíduo num vidro de relógio e secar durante uma hora e meia na estufa (4.3) a 100 oC ± 3 oC.
Introduzir o papel de filtro duplo contendo o resíduo seco num cartucho de extracção (4.2) e cobrir com um tampão de algodão isento de matéria gorda. Colocar o cartucho num extractor (4.1) e prosseguir como indicado nos segundo e terceiro parágrafos do ponto 5.1.
6. Expressão dos resultados
Exprimir o peso do resíduo em percentagem da amostra.
7. Repetibilidade
A diferença entre os resultados de duas determinações paralelas efectuadas com a mesma amostra pelo mesmo analista não deve exceder:
|
— |
0,2 %, em valor absoluto, para teores de matérias gordas brutas inferiores a 5 %, |
|
— |
4,0 % do maior dos valores para teores entre 5 e 10 %, |
|
— |
0,4 %, em valor absoluto, para teores superiores a 10 %. |
8. Observações
|
8.1. |
Para produtos de elevado teor de matérias gordas, difíceis de triturar ou inadequados para a colheita de uma amostra reduzida e homogénea, proceder como se segue: Pesar, com uma aproximação de 1 mg, 20 g de amostra e misturá-los com 10 g ou mais de sulfato de sódio anidro (3.2). Extrair com éter de petróleo (3.1) como indicado no ponto 5.1. Completar o extracto obtido com éter de petróleo (3.1) até perfazer 500 ml e misturar. Introduzir 50 ml de solução num pequeno matraz seco, contendo alguns fragmentos de pedra-pomes e pesado. Eliminar o solvente por destilação, secar e prosseguir como indicado no último parágrafo do ponto 5.1. Eliminar o solvente do resíduo de extracção que se encontra no cartucho de extracção, triturar o resíduo até atingir uma granulometria de 1 mm, colocá-lo novamente no cartucho de extracção (não adicionar sulfato de sódio) e prosseguir como indicado nos segundo e terceiro parágrafos do ponto 5.1. O teor de matérias gordas brutas em percentagem da amostra é dado pela fórmula: (10m1 + m2) × 5 em que:
|
|
8.2. |
A amostra de ensaio de produtos pobres em matérias gordas pode ser elevada a 5 g. |
|
8.3. |
Os alimentos para animais de companhia com um elevado teor de água podem ter que ser misturados com sulfato de sódio anidro antes da hidrólise e da extracção, como para o processo B. |
|
8.4. |
No ponto 5.2, pode ser mais eficaz utilizar água quente em vez de fria para lavar o resíduo após filtração. |
|
8.5. |
Para alguns alimentos para animais, o tempo de secagem de uma hora e meia pode ter que ser aumentado. A secagem excessiva deve ser evitada, pois pode levar a resultados por defeito. Pode também ser utilizado um forno de microondas. |
|
8.6. |
A pré-extracção pelo processo A antes da hidrólise e a reextracção pelo processo B são recomendadas quando o teor de matéria gorda bruta for superior a 15 %. Em certa medida, isto depende da natureza do alimento e da matéria gorda nele presente. |
I. DETERMINAÇÃO DA FIBRA BRUTA
1. Objecto e âmbito de aplicação
O presente método permite efectuar o doseamento, em alimentos para animais, de substâncias orgânicas isentas de gordura e insolúveis em meio ácido e em meio alcalino, convencionalmente designadas por fibra bruta.
2. Princípio
A amostra, eventualmente desengordurada, é tratada sucessivamente com soluções de ácido sulfúrico e hidróxido de potássio em ebulição, de concentrações determinadas. O resíduo é separado por filtração num cadinho de vidro sinterizado, lavado, seco, pesado e calcinado a uma temperatura compreendida entre 475 e 500 oC. A perda de massa resultante da calcinação corresponde à fibra bruta presente na amostra em estudo.
3. Reagentes
|
3.1. |
Ácido sulfúrico 0,13 mol/l. |
|
3.2. |
Agente antiespuma (por exemplo, n-octanol). |
|
3.3. |
Adjuvante de filtração (Celite 545 ou equivalente), aquecido a 500 oC durante quatro horas (8.6). |
|
3.4. |
Acetona. |
|
3.5. |
Éter de petróleo, intervalo de ebulição: 40 a 60 oC. |
|
3.6. |
Ácido clorídrico 0,5 mol/l. |
|
3.7. |
Solução de hidróxido de potássio 0,23 mol/l. |
4. Equipamento
|
4.1. |
Unidade de aquecimento para a mineralização com ácido sulfúrico e hidróxido de potássio, equipada com um suporte para o cadinho de filtração (4.2) e com um tubo, munido de uma torneira, para a saída do líquido e para efectuar o vácuo, por recurso eventual a ar comprimido. Diariamente, antes do uso, proceder ao pré-aquecimento da unidade com água em ebulição, durante cinco minutos. |
|
4.2. |
Cadinho de filtração de vidro, com uma placa filtrante de vidro fundido sinterizado de porosidade 40-90 μm. Antes da primeira utilização, aquecer a 500 oC durante alguns minutos, arrefecendo em seguida (8.6). |
|
4.3. |
Balão, próprio para ebulição, com uma capacidade mínima de 270 ml, equipado com um condensador de refluxo. |
|
4.4. |
Estufa com termóstato. |
|
4.5. |
Mufla com termóstato. |
|
4.6. |
Unidade de extracção a frio, constituída por um suporte para o cadinho de filtração (4.2) e por um tubo de descarga equipado com torneira, para efectuar o vácuo e para a saída do líquido. |
|
4.7. |
Juntas para a montagem da unidade de aquecimento (4.1), do cadinho de filtração (4.2) e do balão (4.3) assim como para a ligação da unidade de extracção a frio (4.6) com o cadinho. |
5. Procedimento
Pesar, com uma aproximação de 1 mg, 1 g de amostra, previamente preparada, transferindo-a para o cadinho de filtração (4.2) (ver observações nos pontos 8.1, 8.2 e 8.3) e adicionar 1 g de adjuvante de filtração (3.3).
Montar a unidade de aquecimento (4.1) e o cadinho de filtração (4.2), adaptando em seguida o balão (4.3) ao cadinho e adicionar 150 ml de ácido sulfúrico (3.1), em ebulição, juntando algumas gotas de agente antiespuma (3.2), se necessário.
Levar o líquido à ebulição em 5 ± 2 minutos, deixando ferver vigorosamente durante exactamente 30 minutos.
Abrir a torneira do tubo de descarga (4.1) e filtrar, sob vácuo, o ácido sulfúrico através do cadinho de filtração. Ainda sob vácuo, lavar o resíduo no filtro com três porções sucessivas, de 30 ml cada, de água em ebulição. Assegurar que o resíduo fica seco por aspiração, após cada lavagem.
Fechar a torneira e adicionar mais 150 ml de hidróxido de potássio (3.7) em ebulição. Juntar algumas gotas de agente antiespuma (3.2). Levar o líquido à ebulição em 5 ± 2 minutos, deixando ferver vigorosamente durante exactamente 30 minutos. Filtrar o resíduo, lavando-o de modo idêntico ao descrito no tratamento com ácido sulfúrico.
Após a última lavagem e secagem, retirar o cadinho com o seu conteúdo e adaptá-lo à unidade de extracção a frio (4.6). Ligar o vácuo e lavar o resíduo com três porções sucessivas, de 25 ml cada, de acetona (3.4), secando o resíduo por aspiração, após cada lavagem.
Secar o cadinho de filtração até massa constante, na estufa, a 130 oC. Após cada secagem, arrefecer no exsicador e pesar de imediato. Colocar o cadinho na mufla, calcinando entre 475 e 500 oC, durante pelo menos 30 minutos, até peso constante (a perda de peso entre duas pesagens sucessivas deve ser igual ou inferior a 2 mg).
Após cada calcinação, proceder ao arrefecimento na mufla e, em seguida no exsicador, antes de pesar.
Efectuar um ensaio em branco. A perda de massa resultante da calcinação não deverá, neste caso, exceder 4 mg.
6. Cálculo dos resultados
O teor de fibra bruta, expresso em percentagem da amostra, é dado pela expressão:
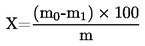
em que:
|
m |
= |
massa da amostra, em g. |
|
m0 |
= |
perda de massa após calcinação, durante o doseamento, em g. |
|
m1 |
= |
perda de massa após calcinação, durante o ensaio em branco, em g. |
7. Repetibilidade
A diferença entre os resultados de duas determinações paralelas efectuadas com a mesma amostra não deve exceder:
|
— |
0,6 % em valor absoluto, para os teores de fibra bruta inferiores a 10 %, |
|
— |
6 %, em valor relativo, em relação ao maior dos valores, para os teores de fibra bruta iguais ou superiores a 10 %. |
8. Observações
|
8.1. |
Os alimentos para animais cujo teor de gordura bruta seja superior a 10 % deverão ser previamente desengordurados, por recurso a éter de petróleo (3.5). Adaptar o cadinho de filtração (4.2) com o seu conteúdo à unidade de extracção a frio (4.6) e lavar, sob vácuo, com três porções sucessivas, de 30 ml cada, de éter de petróleo (3.5). Secar a amostra por aspiração. Adaptar o cadinho com o seu conteúdo à unidade de aquecimento (4.1), continuando de acordo com a descrição efectuada no ponto 5. |
|
8.2. |
Os alimentos para animais que contenham gorduras que não possam ser extraídas directamente com éter de petróleo (3.5) deverão ser desengordurados de acordo com o método descrito no ponto 8.1 e sujeitos a um novo processo de desengorduramento, precedido do tratamento com ácido em ebulição. Após tratamento com ácido e lavagem subsequente, adaptar o cadinho com o seu conteúdo à unidade de extracção a frio (4.6), lavando com três porções sucessivas, de 30 ml cada, de acetona, seguidas de três porções sucessivas, também de 30 ml, de éter de petróleo. Secar por aspiração e continuar o processo de acordo com a descrição efectuada no ponto 5, começando pelo tratamento com hidróxido de potássio. |
|
8.3. |
Se o teor em carbonatos dos alimentos para animais exceder 5 %, expressos em carbonato de cálcio, adaptar o cadinho (4.2) com a amostra pesada à unidade de aquecimento (4.1). Lavar a amostra com três porções sucessivas, de 30 ml cada, de ácido clorídrico (3.6). Após cada adição, deixar a amostra repousar durante cerca de um minuto, antes de filtrar. Lavar uma vez com 30 ml de água e continuar de acordo com a descrição efectuada no ponto 5. |
|
8.4. |
No caso da utilização de uma bateria de aparelhos (vários cadinhos ligados à mesma unidade de aquecimento), não se deverá proceder, na mesma série, a dois doseamentos diferentes com a mesma amostra. |
|
8.5. |
Se, após a ebulição, a filtração das soluções ácidas e alcalinas se revelar difícil, proceder à introdução de ar comprimido pelo tubo de descarga da unidade de aquecimento, continuando em seguida a filtração. |
|
8.6. |
Com vista a prolongar a vida média dos cadinhos de filtração de vidro, é conveniente não utilizar temperaturas de calcinação superiores a 500 oC. Dever-se-ão igualmente evitar quaisquer choques térmicos excessivos no decurso dos ciclos de aquecimento e arrefecimento. |
J. DETERMINAÇÃO DOS AÇÚCARES
1. Objecto e âmbito de aplicação
Esta técnica permite dosear os açúcares redutores e os açúcares totais após inversão, expressos sob a forma de glucose ou eventualmente sob a forma de sacarose, utilizando 0,95 como factor de conversão. Este doseamento é aplicável aos alimentos compostos. Existem técnicas especiais para outros alimentos para animais. Se for necessário, poderá dosear-se separadamente a lactose, não sem ter em conta o respectivo valor no cálculo dos resultados.
2. Princípio
Os açúcares são extraídos com etanol diluído e a solução é clarificada com soluções de Carrez I e II. Uma vez eliminado o etanol, efectuam-se os doseamentos, antes e depois da inversão, aplicando o método de Luff-Schoorl.
3. Reagentes
|
3.1. |
Etanol a 40 % (v/v), d: 0,948 g/ml a 20 oC, ajustado para o ponto de viragem da fenolftaleína. |
|
3.2. |
Solução de Carrez I: dissolver em água 21,9 g de acetato de zinco, Zn(CH3COO)2 · 2H2O, e 3 g de ácido acético glacial. Perfazer o volume de 100 ml com água. |
|
3.3. |
Solução de Carrez II: dissolver em água 10,6 g de ferrocianeto de potássio K4Fe(CN)6 · 3H2O. Perfazer o volume de 100 ml com água. |
|
3.4. |
Alaranjado de metilo, solução a 0,1 % (m/v). |
|
3.5. |
Ácido clorídrico 4 mol/l. |
|
3.6. |
Ácido clorídrico 0,1 mol/l. |
|
3.7. |
Solução de hidróxido de sódio 0,1 mol/l. |
|
3.8. |
Reagente de Luff-Schoorl: Deitar, agitando lentamente, a solução de ácido cítrico (3.8.2) na solução de carbonato de sódio (3.8.3). Juntar em seguida a solução de sulfato de cobre (3.8.1) e perfazer o volume de 1 litro com água. Deixar repousar uma noite e filtrar. Verificar a concentração do reagente assim obtido (Cu 0,05 mol/l; Na2CO3 1 mol/l) (ver o último parágrafo do ponto 5.4). O pH da solução deverá ser cerca de 9,4. |
|
3.8.1. |
Solução de sulfato de cobre: dissolver 25 g de sulfato de cobre, CuSO4 · 5H2O, isento de ferro, em 100 ml de água. |
|
3.8.2. |
Solução de ácido cítrico: dissolver 50 g de ácido cítrico, C6H8O7 H2O, em 50 ml de água. |
|
3.8.3. |
Solução de carbonato de sódio: dissolver 143,8 g de carbonato de sódio anidro em cerca de 300 ml de água quente. Deixar arrefecer. |
|
3.9. |
Solução de tiossulfato de sódio 0,1 mol/l. |
|
3.10. |
Solução de amido: juntar uma mistura de 5 g de amido solúvel e 30 ml de água a 1 l de água a ferver. Manter em ebulição durante três minutos, deixar arrefecer e juntar, se necessário, 10 mg de iodeto de mercúrio como conservante. |
|
3.11. |
Ácido sulfúrico 3 mol/l. |
|
3.12. |
Solução de iodeto de potássio a 30 % (m/v). |
|
3.13. |
Pedra-pomes granulada fervida em ácido clorídrico, lavada com água e seca. |
|
3.14. |
3-Metilbutan-l-ol. |
4. Equipamento
Misturador basculante: cerca de 35 a 40 rotações por minuto.
5. Procedimento
5.1. Extracção da amostra
Pesar, com uma aproximação de 1 mg, 2,5 g de amostra e introduzi-la num balão volumétrico de 250 ml. Juntar 200 ml de etanol (3.1) e misturar durante uma hora no misturador. Juntar 5 ml de solução de Carrez I (3.2) e agitar durante cerca de 30 segundos. Juntar em seguida 5 ml de solução de Carrez II (3.3) e tornar a agitar, durante um minuto. Completar o volume com etanol (3.1), homogeneizar e filtrar. Recolher 200 ml de filtrado e evaporá-lo até cerca de metade do volume a fim de eliminar a maior parte do etanol. Transferir quantitativamente o resíduo da evaporação com água quente para um balão volumétrico de 200 ml, deixar arrefecer, completar o volume com água, homogeneizar e, se necessário, filtrar. Esta será a solução utilizada para o doseamento dos açúcares redutores e, após inversão, para o doseamento dos açúcares totais.
5.2. Determinação dos açúcares redutores
Com uma pipeta, recolher uma quantidade de solução, não superior a 25 ml, que contenha menos de 60 mg de açúcares redutores, expressos em glucose. Se necessário, perfazer o volume de 25 ml com água destilada e determinar o teor de açúcares redutores pelo método de Luff-Schoorl. O resultado é expresso em percentagem de glucose na amostra.
5.3. Determinação dos açúcares totais após a inversão
Com a pipeta, recolher 50 ml de solução e deitá-la num balão volumétrico de 100 ml. Juntar algumas gotas de solução de alaranjado de metilo (3.4) e em seguida ácido clorídrico (3.5) lentamente e mexendo sempre, até que haja uma viragem nítida para vermelho. Juntar 15 ml de ácido clorídrico (3.6) e mergulhar o balão durante 30 minutos em água fervendo abundantemente. Arrefecer rapidamente até cerca de 20 oC e juntar 15 ml de solução de hidróxido de sódio (3.7). Perfazer o volume de 100 ml com água e homogeneizar. Recolher uma quantidade não superior a 25 ml e que contenha menos de 60 mg de açúcares redutores expressos em glucose. Se necessário, perfazer o volume de 25 ml com água destilada e determinar o teor de açúcares redutores pelo método de Luff-Schoorl. O resultado deverá ser expresso em percentagem de glucose, ou eventualmente sacarose, multiplicando pelo factor 0,95.
5.4. Titulação pelo método de Luff-Schoorl
Recolher com uma pipeta 25 ml de reagente de Luff-Schoorl (3.8) e deitá-lo num erlenmeyer de 300 ml; juntar 25 ml exactos da solução de açúcares já clarificada. Juntar dois pedaços de pedra-pomes granulada (3.13), aquecer, agitando manualmente sobre uma chama aberta de altura média, e levar o líquido à ebulição em cerca de dois minutos. Colocar imediatamente o erlenmeyer sobre uma placa com rede de amianto com uma abertura de cerca de 6 cm de diâmetro, sob a qual terá sido previamente acendida uma chama, regulada de forma a aquecer apenas o fundo do erlenmeyer. Ajustar ao erlenmeyer um condensador de refluxo. Deixar em ebulição durante exactamente 10 minutos. Arrefecer imediatamente em água fria durante cerca de cinco minutos e titular como segue:
Juntar 10 ml de solução de iodeto de potássio (3.12) e, imediatamente depois e com cuidado (dado o risco de formação de espuma abundante), adicionar 25 ml de ácido sulfúrico (3.11). Titular seguidamente com a solução de tiossulfato de sódio (3.9) até ao aparecimento de uma coloração amarelada, juntar o indicador de amido (3.10) e completar a titulação.
Efectuar a mesma titulação numa mistura, medida com exactidão, de 25 ml de reagente de Luff-Schoorl (3.8) e 25 ml de água, a que se juntam 10 ml de solução de iodeto de potássio (3.12) e 25 ml de ácido sulfúrico (3.11), sem levar à ebulição.
6. Cálculo dos resultados
Calcular por meio da tabela a quantidade de glucose, expressa em mg, correspondente à diferença entre os valores das duas titulações, expressos em ml de tiossulfato de sódio 0,1 mol/l. Exprimir o resultado em percentagem da amostra.
7. Procedimentos especiais
|
7.1. |
No caso de alimentos muito ricos em melaço ou pouco homogéneos, pesar uma amostra de 20 g e introduzi-la com 500 ml de água num balão volumétrico de 1 l. Misturar durante uma hora no misturador. Clarificar com soluções de Carrez I (3.2) e II (3.3) tal como se descreve no ponto 5.1, mas utilizando uma quantidade quatro vezes superior de cada solução. Completar o volume até à marca com etanol a 80 % (v/v). Homogeneizar e filtrar. Eliminar o etanol como se descreve no ponto 5.1. Na ausência de amido dextrinizado, completar o volume até à marca com água destilada. |
|
7.2. |
No caso dos melaços e das matérias-primas para a alimentação animal ricas em açúcares e praticamente isentas de amido (alfarroba, cascas secas de beterraba, etc.), pesar 5 g de amostra e introduzi-los num balão volumétrico de 250 ml, juntar 200 ml de água destilada e misturar durante uma hora ou mais, se necessário, no misturador. Clarificar em seguida com soluções de Carrez I (3.2) e II (3.3) tal como se descreve no ponto 5.1. Completar o volume com água, homogeneizar e filtrar. Para dosear os açúcares totais, proceder como descrito no ponto 5.3. |
8. Observações
|
8.1. |
Para evitar a formação de espuma, recomenda-se a adição (independentemente do volume) de cerca de 1 ml de 3-metilbutan-l-ol (3.14) antes da ebulição com o reagente de Luff-Schoorl. |
|
8.2. |
O teor de sacarose, em percentagem, será igual à diferença entre o teor de açúcares totais após a inversão, expressos em glucose, e o teor de açúcares redutores (igualmente expressos em glucose), multiplicada por 0,95. |
|
8.3. |
Para determinar o teor de açúcares redutores, com exclusão da lactose, podem utilizar-se dois processos: |
|
8.3.1. |
Para um cálculo aproximado, multiplica-se o teor de lactose, determinado num ensaio em separado, por 0,675 e subtrai-se o resultado obtido do teor de açúcares redutores. |
|
8.3.2. |
Para um cálculo mais rigoroso dos açúcares redutores, com exclusão da lactose, é necessário utilizar a mesma amostra nas duas determinações finais, efectuando-se uma análise com parte da solução obtida no ponto 5.1 e outra com parte da solução que se utiliza no método de doseamento da lactose (após a fermentação dos outros tipos de açúcares seguida de clarificação). Em ambos os casos, a quantidade de açúcar presente determina-se pelo método de Luff-Schoorl e o respectivo cálculo faz-se em mg de glucose, subtraindo um valor do outro e exprimindo o resultado em termos percentuais relativamente à amostra. Exemplo Os volumes recolhidos correspondem ambos, em cada um dos doseamentos, a uma amostra de 250 mg. No primeiro caso, são gastos 17 ml de solução de tiossulfato de sódio 0,1 mol/l, o que corresponde a 44,2 mg de glucose, e no segundo são gastos 11 ml, o que corresponde a 27,6 mg de glucose. A diferença é de 16,6 mg de glucose. O teor de açúcares redutores (excepto a lactose), calculado como glucose, será portanto de:
|
Tabela de valores para 25 ml de reagente de Luff-Schoorl
ml de Na2S2O30,1 mol/l, dois minutos de aquecimento e 10 minutos de ebulição
|
Na2 S2 O3 0,1 mol/l |
Glucose, frutose açúcares invertidos C6 H12 O6 |
Lactose C12 H22 O11 |
Maltose C12 H22 O11 |
Na2 S2 O3 0,1 mol/l |
|||
|
ml |
mg |
diferença |
mg |
diferença |
mg |
diferença |
ml |
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
2,4 4,8 7,2 9,7 12,2 14,7 17,2 19,8 22,4 25,0 27,6 30,3 33,0 35,7 38,5 41,3 44,2 47,1 50,0 53,0 56,0 59,1 62,2 |
2,4 2,4 2,5 2,5 2,5 2,5 2,6 2,6 2,6 2,6 2,7 2,7 2,7 2,8 2,8 2,9 2,9 2,9 3,0 3,0 3,1 3,1 |
3,6 7,3 11,0 14,7 18,4 22,1 25,8 29,5 33,2 37,0 40,8 44,6 48,4 52,2 56,0 59,9 63,8 67,7 71,7 75,7 79,8 83,9 88,0 |
3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,8 3,8 3,8 3,8 3,8 3,8 3,9 3,9 3,9 4,0 4,0 4,1 4,1 4,1 |
3,9 7,8 11,7 15,6 19,6 23,5 27,5 31,5 35,5 39,5 43,5 47,5 51,6 55,7 59,8 63,9 68,0 72,2 76,5 80,9 85,4 90,0 94,6 |
3,9 3,9 3,9 4,0 3,9 4,0 4,0 4,0 4,0 4,0 4,0 4,1 4,1 4,1 4,1 4,1 4,2 4,3 4,4 4,5 4,6 4,6 |
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
K. DETERMINAÇÃO DA LACTOSE
1. Objecto e âmbito de aplicação
Esta técnica permite determinar o teor de lactose em alimentos para animais que a contenham em mais de 0,5 %.
2. Princípio
Dissolvem-se os açúcares em água. Submete-se a solução a uma fermentação pela levedura Saccharomyces cerevisiae, que não modifica a lactose. Após clarificação e filtração, determina-se o teor de lactose do filtrado pelo método Luff-Schoorl.
3. Reagentes
|
3.1. |
Suspensão de Saccharomyces cerevisiae: suspender 25 g de levedura fresca em 100 ml de água. O tempo máximo de conservação desta suspensão em frigorífico é de uma semana. |
|
3.2. |
Solução de Carrez I: dissolver em água 21,9 g de acetato de zinco, Zn(CH3COO)2 · 2H2O e 3 g de ácido acético glacial. Perfazer o volume de 100 ml com água. |
|
3.3. |
Solução de Carrez II: dissolver em água 10,6 g de ferrocianeto de potássio K4Fe(CN)6 · 3H2O. Perfazer o volume de 100 ml com água. |
|
3.4. |
Reagente de Luff-Schoorl: Deitar a solução de ácido cítrico (3.4.2) na de carbonato de sódio (3.4.3), agitando com precaução. Juntar em seguida a solução de sulfato de cobre (3.4.1) e perfazer o volume de 1 litro com água. Deixar repousar uma noite e filtrar. Verificar a concentração do reagente assim obtido (Cu 0,05 mol/l; Na2CO3 1 mol/l). O pH da solução deverá ser cerca de 9,4. |
|
3.4.1. |
Solução de sulfato de cobre: dissolver 25 g de sulfato de cobre, Cu SO4 · 5H2O, isento de ferro, em 100 ml de água. |
|
3.4.2. |
Solução de ácido cítrico: dissolver 50 g de ácido cítrico, C6H8O7 H2O, em 50 ml de água. |
|
3.4.3. |
Solução de carbonato de sódio: dissolver 143,8 g de carbonato de sódio anidro em cerca de 300 ml de água quente. Deixar arrefecer. |
|
3.5. |
Pedra-pomes granulada fervida em ácido clorídrico, lavada com água e seca. |
|
3.6. |
Solução de iodeto de potássio a 30 % (m/v). |
|
3.7. |
Ácido sulfúrico 3 mol/l. |
|
3.8. |
Solução de tiossulfato de sódio 0,1 mol/l. |
|
3.9. |
Solução de amido: juntar uma mistura de 5 g de amido solúvel e 30 ml de água a 1 l de água a ferver. Manter em ebulição durante três minutos, deixar arrefecer e juntar, se necessário, 10 mg de iodeto de mercúrio como conservante. |
4. Equipamento
Tina de banho-maria, com termóstato regulado para 38-40 oC.
5. Procedimento
Pesar, com uma aproximação de 1 mg, 1 g de amostra e introduzi-la num balão volumétrico de 100 ml. Juntar 25 a 30 ml de água, colocar o balão durante trinta minutos em banho-maria a ferver, deixando arrefecer em seguida até cerca de 35 oC. Juntar 5 ml da suspensão de levedura (3.1) e homogeneizar. Deixar o balão durante duas horas em banho-maria a uma temperatura de 38 a 40oC e deixar arrefecer em seguida até cerca de 20 oC.
Juntar 2,5 ml de solução de Carrez I (3.2) e agitar durante 30 segundos; adicionar seguidamente 2,5 ml de solução de Carrez II (3.3), agitando novamente durante 30 segundos. Perfazer o volume de 100 ml com água, misturar e filtrar. Com uma pipeta, recolher uma quantidade de filtrado não superior a 25 ml, que contenha, de preferência, entre 40 e 80 mg de lactose, e introduzi-lo num erlenmeyer de 300 ml. Se necessário, perfazer o volume de 25 ml com água.
Efectuar segundo o mesmo processo um ensaio em branco com 5 ml de suspensão de levedura (3.1). Determinar o teor de lactose em conformidade com o método de Luff-Schoorl, como se indica a seguir: juntar a 25 ml exactos de reagente de Luff-Schoorl (3.4) dois grânulos de pedra-pomes (3.5). Aquecer, agitando manualmente sobre uma chama aberta de altura média e levar o líquido à ebulição em cerca de dois minutos. Colocar imediatamente o erlenmeyer sobre uma placa com rede de amianto com uma abertura de cerca de 6 cm de diâmetro, sob a qual terá sido previamente acendida uma chama, regulada de forma a aquecer apenas o fundo do erlenmeyer. Ajustar ao erlenmeyer um condensador de refluxo. Deixar em ebulição durante exactamente 10 minutos. Arrefecer imediatamente em água fria durante cerca de cinco minutos e titular como segue:
Juntar 10 ml de solução de iodeto de potássio (3.6) e, imediatamente depois e com cuidado (dado o risco de formação de espuma abundante), adicionar 25 ml de ácido sulfúrico (3.7). Titular seguidamente com a solução de tiossulfato de sódio (3.8) até ao aparecimento de uma coloração amarelada, juntar o indicador de amido (3.9) e completar a titulação.
Efectuar a mesma titulação numa mistura, medida com exactidão, de 25 ml de reagente de Luff-Schoorl (3.4) e 25 ml de água, a que se juntam 10 ml de solução de iodeto de potássio (3.6) e 25 ml de ácido sulfúrico (3.7), sem levar à ebulição.
6. Cálculo dos resultados
Utilizando a tabela, determinar a quantidade de lactose, em mg, correspondente à diferença entre os resultados das duas titulações, expressos em ml de tiossulfato de sódio 0,1 mol/l.
Exprimir o resultado obtido para a lactose anidra em percentagem da amostra.
7. Observações
Para os produtos que contenham mais de 40 % de açúcares fermentáveis, utilizar mais de 5 ml de suspensão de levedura (3.1).
Tabela de valores para 25 ml de reagente de Luff-Schoorl
ml de Na2S2O30,1 mol/l, dois minutos de aquecimento e 10 minutos de ebulição
|
Na2 S2 O3 0,1 mol/l |
Glucose, frutose açúcares invertidos C6 H12 O6 |
Lactose C12 H22 O11 |
Maltose C12 H22 O11 |
Na2 S2 O3 0,1 mol/l |
|||
|
ml |
mg |
diferença |
mg |
diferença |
mg |
diferença |
ml |
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
2,4 4,8 7,2 9,7 12,2 14,7 17,2 19,8 22,4 25,0 27,6 30,3 33,0 35,7 38,5 41,3 44,2 47,1 50,0 53,0 56,0 59,1 62,2 |
2,4 2,4 2,5 2,5 2,5 2,5 2,6 2,6 2,6 2,6 2,7 2,7 2,7 2,8 2,8 2,9 2,9 2,9 3,0 3,0 3,1 3,1 |
3,6 7,3 11,0 14,7 18,4 22,1 25,8 29,5 33,2 37,0 40,8 44,6 48,4 52,2 56,0 59,9 63,8 67,7 71,7 75,7 79,8 83,9 88,0 |
3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,8 3,8 3,8 3,8 3,8 3,8 3,9 3,9 3,9 4,0 4,0 4,1 4,1 4,1 |
3,9 7,8 11,7 15,6 19,6 23,5 27,5 31,5 35,5 39,5 43,5 47,5 51,6 55,7 59,8 63,9 68,0 72,2 76,5 80,9 85,4 90,0 94,6 |
3,9 3,9 3,9 4,0 3,9 4,0 4,0 4,0 4,0 4,0 4,0 4,1 4,1 4,1 4,1 4,1 4,2 4,3 4,4 4,5 4,6 4,6 |
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
L. DETERMINAÇÃO DO AMIDO
MÉTODO POLARIMÉTRICO
1. Objecto e âmbito de aplicação
O presente método permite determinar o teor de amido e dos seus produtos de degradação de elevado peso molecular em alimentos para animais, tendo em vista verificar o cumprimento do valor energético declarado (disposições do anexo VII) e da Directiva 96/25/CE do Conselho (3).
2. Princípio
O método inclui duas determinações. Na primeira, a amostra é tratada com ácido clorídrico diluído. Depois de clarificada e filtrada, mede-se o poder rotatório da solução por polarimetria.
Na segunda, a amostra é submetida a extracção com etanol a 40 %. Depois de acidificado o filtrado com ácido clorídrico, clarificação e filtração, mede-se o poder rotatório tal como na primeira determinação.
A diferença entre as duas medições, multiplicada por um factor conhecido, dá o teor de amido da amostra.
3. Reagentes
|
3.1. |
Ácido clorídrico, solução a 25 % (m/m), d: 1,126 g/ml. |
|
3.2. |
Ácido clorídrico, solução a 1,13 % (m/v). A concentração é verificada por titulação com uma solução 0,1 mol/l de hidróxido de sódio, na presença de vermelho de metilo a 0,1 % (m/v) em etanol a 94 % (v/v). Para a neutralização de 10 ml, são necessários 30,94 ml de NaOH 0,1 mol/l. |
|
3.3. |
Solução de Carrez I: dissolver em água 21,9 g de acetato de zinco, Zn(CH3COO)2 · 2H2O e 3 g de ácido acético glacial. Perfazer o volume de 100 ml com água. |
|
3.4. |
Solução de Carrez II: dissolver em água 10,6 g de ferrocianeto de potássio K4Fe(CN)6 · 3H2O. Perfazer o volume de 100 ml com água. |
|
3.5. |
Etanol a 40 % (v/v), d: 0,948 a 20 oC. |
4. Equipamento
|
4.1. |
Erlenmeyer de 250 ml com esmerilado normalizado e condensador de refluxo. |
|
4.2. |
Polarímetro ou sacarímetro. |
5. Procedimento
5.1. Preparação da amostra
Triturar a amostra de maneira a passar a totalidade da mesma por um crivo de malha redonda de 0,5 mm de diâmetro.
5.2. Determinação do poder rotatório total (P ou S) (ver observação 7.1)
Pesar, com uma aproximação de 1 mg, 2,5 g da amostra triturada para um balão volumétrico de 100 ml. Adicionar 25 ml de ácido clorídrico (3.2), agitar, para homogeneizar a amostra, e adicionar mais 25 ml de ácido clorídrico (3.2). Introduzir o balão num banho de água em ebulição e agitar enérgica e regularmente durante três minutos, para evitar a formação de aglomerados. A quantidade de água no banho deve ser suficiente para que este se mantenha em ebulição quando o balão for introduzido. O balão não pode ser retirado do banho durante a agitação. Decorridos 15 minutos exactos, retirar o balão do banho, adicionar 30 ml de água fria e arrefecer imediatamente a 20 oC.
Juntar 5 ml de solução de Carrez I (3.3) e agitar durante cerca de 30 segundos. Adicionar em seguida 5 ml da solução de Carrez II (3.4) e agitar de novo durante cerca de 30 segundos. Completar o volume com água, homogeneizar e filtrar. Se o filtrado não ficar perfeitamente límpido (o que é pouco frequente), recomeçar a análise utilizando uma quantidade maior das soluções de Carrez I e II, por exemplo, 10 ml.
Medir, em seguida, o poder rotatório da solução num tubo de 200 mm com o polarímetro ou o sacarímetro.
5.3. Determinação do poder rotatório (P' ou S') de substâncias solúveis em etanol a 40 %
Introduzir, num balão volumétrico de 100 ml, 5 g de amostra, pesada com uma aproximação de 1 mg, e adicionar cerca de 80 ml de etanol (3.5) (ver a observação 7.2). Deixar o balão em repouso à temperatura ambiente durante uma hora; durante esse lapso de tempo, agitar energicamente por seis vezes, para que a amostra fique bem misturada com o etanol. Perfazer o volume de 100 ml com etanol (3.5), homogeneizar e filtrar.
Pipetar 50 ml de filtrado (correspondentes a 2,5 g de amostra) para um erlenmeyer de 250 ml, adicionar 2,1 ml de ácido clorídrico (3.1) e agitar energicamente. Adaptar um condensador de refluxo ao erlenmeyer e mergulhar este num banho de água em ebulição. Decorridos exactamente 15 minutos, retirar o erlenmeyer do banho, transferir o conteúdo para um balão volumétrico de 100 ml, lavar com um pouco de água fria e arrefecer a 20 oC.
Clarificar, em seguida, com as soluções de Carrez I (3.3) e II (3.4), completar o volume com água, homogeneizar, filtrar e medir o poder rotatório como indicado nos segundo e terceiro parágrafos do ponto 5.2.
6. Cálculo dos resultados
O teor percentual de amido é calculado como segue:
6.1. Medições polarimétricas

|
P |
= |
poder rotatório total, em graus de arco |
||||||||||||||
|
P' |
= |
poder rotatório, em graus de arco, das substâncias solúveis em etanol a 40 % |
||||||||||||||
|
|
= |
poder rotatório específico do amido puro. Os valores convencionalmente admitidos para este factor são os seguintes:
|
6.2. Medições sacarimétricas

|
S |
= |
poder rotatório total, em graus sacarimétricos |
|
S' |
= |
poder rotatório, em graus sacarimétricos, das substâncias solúveis em etanol a 40 % |
|
N |
= |
peso, em gramas, de sacarose dissolvida em 100 ml de água, correspondente a um poder rotatório de 100o sacarimétricos, quando medido com um tubo de 200 mm 16,29 g para os sacarímetros franceses 26,00 g para os sacarímetros alemães 20,00 g para os sacarímetros mistos.
|
6.3. Repetibilidade
A diferença entre os resultados de duas determinações paralelas efectuadas com a mesma amostra não deve exceder 0,4, em valor absoluto, para teores de amido inferiores a 40 %, ou 1 %, em valor relativo, para teores de amido iguais ou superiores a 40 %.
7. Observações
|
7.1. |
Se a amostra contiver mais de 6 % de carbonatos, expressos em carbonato de cálcio, estes devem ser destruídos por tratamento com a quantidade apropriada, exacta, de ácido sulfúrico diluído antes da determinação do poder rotatório total. |
|
7.2. |
No caso dos produtos com teor de lactose elevado, como o soro de leite em pó ou o leite em pó desnatado, proceder como segue depois da adição de 80 ml de etanol (3.5): adaptar um condensador de refluxo ao balão e mergulhar este durante 30 minutos num banho de água a 50 oC. Em seguida, deixar arrefecer e prosseguir a análise como indicado no ponto 5.3. |
|
7.3. |
Caso estejam presentes em quantidades significativas, as seguintes matérias-primas para a alimentação animal produzem comprovadamente interferências na determinação do teor de amido por polarimetria, podendo originar resultados incorrectos:
|
M. DETERMINAÇÃO DAS CINZAS BRUTAS
1. Objecto e âmbito de aplicação
Esta técnica permite determinar o teor de cinzas brutas em alimentos para animais.
2. Princípio
Incinera-se amostra a 550 oC e pesa-se o resíduo.
3. Reagentes
Nitrato de amónio, solução a 20 % (m/v).
4. Equipamento
|
4.1. |
Placa de aquecimento. |
|
4.2. |
Mufla eléctrica com termóstato. |
|
4.3. |
Cadinhos para incineração em quartzo, porcelana ou platina, rectangulares (aprox. 60 × 40 × 25 mm) ou circulares (diâmetro: 60 a 75 mm, altura: 20 a 40 mm). |
5. Procedimento
Pesar, com uma aproximação de 1 mg, cerca de 5 g de amostra (2,5 g para os produtos com tendência para aumentar de volume) num cadinho para incineração previamente aquecido a 550 oC, arrefecido e tarado. Colocar o cadinho na placa de aquecimento e aquecer progressivamente até à carbonização da matéria. Proceder à incineração de acordo com o disposto no ponto 5.1 ou 5.2.
|
5.1. |
Introduzir o cadinho na mufla calibrada e regulada para 550 oC. Manter esta temperatura até à obtenção de cinzas brancas, de cor cinzenta clara ou avermelhada, e sem partículas de carvão visíveis. Colocar o cadinho num exsicador, deixar arrefecer e pesar imediatamente. |
|
5.2. |
Introduzir o cadinho na mufla calibrada e regulada para 550 oC. Incinerar durante 3 horas. Colocar o cadinho num exsicador, deixar arrefecer e pesar imediatamente. Incinerar de novo por 30 minutos por forma a garantir que o peso das cinzas permanece constante (a perda de peso entre duas pesagens sucessivas deve ser igual ou inferior a 1 mg). |
6. Cálculo dos resultados
Calcular o peso do resíduo subtraindo-lhe o peso da tara.
Exprimir o resultado em percentagem da amostra.
7. Observações
|
7.1. |
As cinzas de substâncias de difícil incineração devem ser submetidas a uma primeira incineração de pelo menos três horas e arrefecidas, adicionando-se-lhes em seguida algumas gotas de solução de nitrato de amónio a 20 % ou de água (com extremo cuidado, para evitar a dispersão das cinzas ou a formação de aglomerados). Prosseguir com a calcinação, após secagem em estufa. Repetir eventualmente a operação até à incineração total. |
|
7.2. |
Para as substâncias que resistam ao tratamento indicado no ponto 7.1, proceder do modo seguinte: depois de uma incineração de três horas, recolher as cinzas em água quente e filtrá-las num pequeno filtro, isento de cinzas. Incinerar o filtro e o seu conteúdo no primeiro cadinho. Colocar o filtrado no cadinho arrefecido, evaporar até à secura, incinerar e pesar. |
|
7.3. |
No caso das matérias gordas, pesar rigorosamente cerca de 25 g de amostra num cadinho de capacidade adequada. Carbonizar, inflamando a substância com uma torcida de papel de filtro isento de cinzas. Terminada a combustão, humedecer com o mínimo de água possível. Secar e incinerar como indicado no ponto 5. |
N. DETERMINAÇÃO DAS CINZAS INSOLÚVEIS EM ÁCIDO CLORÍDRICO
1. Objecto e âmbito de aplicação
Esta técnica permite determinar o teor de substâncias minerais insolúveis em ácido clorídrico existentes nos alimentos para animais. Podem utilizar-se dois processos, em função do tipo de amostra:
|
1.1. |
Processo A: aplicável às matérias-primas orgânicas e à maior parte dos alimentos compostos. |
|
1.2. |
Processo B: aplicável aos compostos e misturas minerais, assim como aos alimentos compostos cujo teor de substâncias minerais insolúveis em ácido clorídrico, determinado segundo o método A, seja superior a 1 %. |
2. Princípio
|
2.1. |
Processo A: incinera-se a amostra, tratam-se as cinzas em ácido clorídrico em ebulição, filtra-se o resíduo insolúvel e pesa-se. |
|
2.2. |
Processo B: trata-se a amostra com ácido clorídrico. Filtra-se a solução, incinera-se o resíduo e tratam-se as cinzas assim obtidas como no Processo A. |
3. Reagentes
|
3.1. |
Ácido clorídrico 3 mol/l. |
|
3.2. |
Ácido tricloroacético, solução a 20 % (m/v). |
|
3.3. |
Ácido tricloroacético, solução a 1 % (m/v). |
4. Equipamento
|
4.1. |
Placa de aquecimento. |
|
4.2. |
Mufla eléctrica com termóstato. |
|
4.3. |
Cadinhos para incineração em quartzo, porcelana ou platina, rectangulares (aprox. 60 × 40 × 25 mm) ou circulares (diâmetro: 60 a 75 mm, altura: 20 a 40 mm). |
5. Procedimento
5.1. Processo A
Incinerar a amostra de acordo com o método indicado para a determinação do teor de cinzas brutas. Podem igualmente utilizar-se as cinzas obtidas nessa determinação.
Introduzir as cinzas num copo de 250 a 400 ml com 75 ml de ácido clorídrico (3.1). Levar cuidadosamente o líquido a uma ebulição suave mantendo-a durante 15 minutos. Filtrar a solução quente com papel de filtro isento de cinzas e lavar o resíduo com água quente até desaparecer a reacção ácida. Secar o filtro que contém o resíduo e incinerá-lo, num cadinho tarado, a uma temperatura entre 550 oC (min.) e 700 oC (máx.). Arrefecer num exsicador e pesar.
5.2. Processo B
Pesar, com uma aproximação de 1 mg, 5 g de amostra e introduzi-la num copo de 250 a 400 ml. Juntar sucessivamente 25 ml de água e 25 ml de ácido clorídrico (3.1), misturar e deixar que acabe a efervescência. Juntar mais 50 ml de ácido clorídrico (3.1). Caso se verifique libertação de gás, aguardar que esta cesse completamente, colocar em seguida o copo num banho-maria em ebulição durante 30 minutos ou mais, se necessário, para hidrolisar completamente o amido eventualmente presente. Filtrar a quente com um filtro isento de cinzas e lavar o filtro com 50 ml de água quente (ver observação 7). Colocar o filtro com o resíduo num cadinho para incineração, secar e incinerar a uma temperatura entre 550 oC (mín.) e 700 oC (máx.). Introduzir as cinzas num copo de 250 a 400 ml com 75 ml de ácido clorídrico (3.1); prosseguir como indicado no segundo parágrafo do ponto 5.1.
6. Cálculo dos resultados
Calcular o peso do resíduo subtraindo-lhe o peso da tara. Exprimir o resultado em percentagem da amostra.
7. Observações
Se a filtração for difícil, repetir o doseamento substituindo os 50 ml de ácido clorídrico (3.1) por 50 ml de ácido tricloracético a 20 % (3.2), lavando o filtro com uma solução quente de ácido tricloracético a 1 % (3.3).
O. DETERMINAÇÃO DOS CARBONATOS
1. Objecto e âmbito de aplicação
Esta técnica permite dosear os carbonatos, convencionalmente expressos em carbonato de cálcio, na maior parte dos alimentos para animais.
No entanto, para certos casos (carbonato de ferro, por exemplo) é necessário utilizar uma técnica especial.
2. Princípio
Decompõe-se os carbonatos com ácido clorídrico e recolhe-se o dióxido de carbono que se liberta da reacção para um tubo graduado, comparando-se o seu volume com o do dióxido de carbono que se libertar, nas mesmas condições, a partir de uma quantidade conhecida de carbonato de cálcio.
3. Reagentes
|
3.1. |
Ácido clorídrico, de 1,10 g/ml de densidade. |
|
3.2. |
Carbonato de cálcio. |
|
3.3. |
Ácido sulfúrico cerca de 0,05 mol/l, corado com vermelho de metilo. |
4. Equipamento
Aparelho de Scheibler-Dietrich (v. esquema) ou aparelho equivalente.
5. Procedimento
Em função do teor de carbonatos da amostra, pesar uma porção da amostra para ensaio como a seguir se indica:
|
— |
0,5 g para os produtos com 50 a 100 % de carbonatos, expressos em carbonato de cálcio, |
|
— |
1 g para os produtos com 40 a 50 % de carbonatos, expressos em carbonato de cálcio, |
|
— |
2 g a 3 g para os outros produtos. |
Introduzir a amostra de ensaio num frasco especial (4) equipado com um pequeno tubo de matéria inquebrável, contendo 10 ml de ácido clorídrico (3.1), e ligar o frasco ao aparelho. Rodar a torneira de três vias (5) de maneira a que o tubo (1) comunique com o exterior. Com o tubo móvel (2) cheio de ácido sulfúrico corado (3.3) e ligado ao tubo graduado (1), acertar o nível de líquido para a graduação zero. Rodar a torneira (5) de maneira a estabelecer a comunicação entre os tubos (1) e (3) e certificar-se de que o nível está em zero.
Deixar cair lentamente o ácido clorídrico (3.1) sobre a amostra de ensaio, inclinando o frasco (4). Nivelar a pressão, baixando o tubo (2). Agitar o frasco (4) até à cessação completa da libertação de dióxido de carbono.
Tornar a nivelar a pressão colocando o líquido ao mesmo nível em ambos os tubos (1) e (2). Esperar alguns minutos até que o volume do gás se estabilize, fazendo então a leitura.
Efectuar nas mesmas condições um ensaio comparativo com 0,5 g de carbonato de cálcio (3.2).
6. Cálculo dos resultados
O teor de carbonatos, expresso em carbonato de cálcio, é dado pela seguinte fracção:
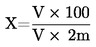
em que:
|
X |
= |
% (m/m) de carbonatos na amostra, expresso em carbonato de cálcio |
|
V |
= |
ml de CO2 libertados pela amostra de ensaio |
|
V1 |
= |
ml de CO2 libertados por 0,5 g de CaCO3 |
|
m |
= |
massa, em gramas, da amostra de ensaio. |
7. Observações
|
7.1. |
Sempre que a amostra de ensaio pesar mais de 2 g, introduzir 15 ml de água destilada no frasco (4) e misturar, previamente ao ensaio. Utilizar o mesmo volume de água para o ensaio comparativo. |
|
7.2. |
Se o aparelho utilizado tiver um volume diferente do de Scheibler-Dietrich, será necessário modificar consequentemente a quantidade de amostra de ensaio e substância de comparação, assim como o cálculo dos resultados.
|
P. DETERMINAÇÃO DO FÓSFORO TOTAL
MÉTODO FOTOMÉTRICO
1. Objecto e âmbito de aplicação
Este método permite determinar o teor de fósforo total dos alimentos para animais. É particularmente indicado para a análise de produtos pobres em fósforo. Nalguns casos (produtos ricos em fósforo), pode ser aplicado um método gravimétrico.
2. Princípio
A amostra é mineralizada, quer por via seca (para os alimentos orgânicos) quer por via húmida (para os compostos minerais e os alimentos líquidos) e colocada em solução ácida. A solução é tratada pelo reagente vanadomolíbdico. A densidade óptica da solução amarela assim obtida é medida em espectrofotómetro a 430 nm.
3. Reagentes
|
3.1. |
Carbonato de cálcio. |
|
3.2. |
Ácido clorídrico, ρ20 = 1,10 g/ml (aprox. 6 mol/l). |
|
3.3. |
Ácido nítrico, ρ20 = 1,045 g/ml. |
|
3.4. |
Ácido nítrico, ρ20 = 1,38 a 1,42 g/ml. |
|
3.5. |
Ácido sulfúrico, ρ20 = 1,84 g/ml. |
|
3.6. |
Reagente vanadomolíbdico: misturar 200 ml de solução de heptamolibdato de amónio (3.6.1), 200 ml de solução de monovanadato de amónio (3.6.2) e 134 ml de ácido nítrico (3.4) num balão volumétrico de 1 litro. Perfazer o volume com água. |
|
3.6.1. |
Solução de heptamolibdato de amónio: dissolver em água quente 100 g de heptamolibdato de amónio (NH4)6Mo7O24 · 4H2O. Juntar 10 ml de amónia (d: 0,91 g/ml) e perfazer 1 l com água. |
|
3.6.2. |
Solução de monovanadato de amónio: em 400 ml de água quente, dissolver 2,35 g de monovanadato de amónio NH4VO3. Adicionar lentamente, e agitando sempre, 20 ml de ácido nítrico diluído (7 ml de HNO3 (3.4) + 13 ml de H2O) e perfazer o volume de 1 l com água. |
|
3.7. |
Solução-padrão a 1 mg/ml de fósforo: dissolver em água 4,387 g de di-hidrogenofosfato de potássio KH2PO4. Perfazer o volume de 1 l com água. |
4. Equipamento
|
4.1. |
Cadinhos para incineração em quartzo, porcelana ou platina. |
|
4.2. |
Mufla eléctrica com termóstato regulado para 550 oC. |
|
4.3. |
Balão Kjeldahl, 250 ml. |
|
4.4. |
Balões volumétricos e pipetas de precisão. |
|
4.5. |
Espectrofotómetro. |
|
4.6. |
Tubos de ensaio, diâmetro: cerca de 16 mm, de rosca normalizada de 14,5 mm; capacidade: 25 a 30 ml. |
5. Procedimento
5.1. Preparação da solução
Consoante a natureza da amostra, preparar uma solução tal como indicado nos pontos 5.1.1 ou 5.1.2.
5.1.1.
Pesar 1 g ou mais de amostra, com uma aproximação de 1 mg. Introduzir a amostra num balão Kjeldahl, adicionar 20 ml de ácido sulfúrico (3.5) agitar para impregnar completamente a substância com ácido e evitar que adira às paredes do balão, aquecer e manter em ebulição durante 10 minutos. Deixar arrefecer ligeiramente, adicionar 2 ml de ácido nítrico (3.4), aquecer lentamente, deixar arrefecer ligeiramente, juntar de novo um pouco de ácido nítrico (3.4) e levar à ebulição. Repetir estas operações até à obtenção de uma solução incolor. Arrefecer, juntar um pouco de água, transferir o líquido para um balão volumétrico de 500 ml, enxaguando o balão de Kjeldahl com água quente. Deixar arrefecer, completar o volume com água, homogeneizar e filtrar.
5.1.2.
Pesar, com uma aproximação de 1 mg, cerca de 2,5 g de amostra num cadinho de incineração. Misturar intimamente a amostra com 1 g de carbonato de cálcio (3.1). Calcinar na mufla a 550oC até à obtenção de cinzas brancas ou cinzentas (uma pequena quantidade de carvão não prejudica). Transferir as cinzas para um copo de precipitação de 250 ml. Juntar 20 ml de água e ácido clorídrico (3.2) até que cesse a efervescência. Juntar mais 10 ml de ácido clorídrico (3.2). Colocar o copo num banho de areia e evaporar até à secura para insolubilizar a sílica. Redissolver o resíduo em 10 ml de ácido nítrico (3.3) e levar a ebulição, durante 5 minutos num banho de areia, sem evaporar até à secura. Transferir o líquido para um balão volumétrico de 500 ml, enxaguando o copo de precipitação várias vezes com água quente. Deixar arrefecer, completar o volume com água, homogeneizar e filtrar.
5.2. Desenvolvimento da coloração e medição da densidade óptica
Diluir uma alíquota do filtrado obtido no ponto 5.1.1 ou 5.1.2 de modo a obter uma concentração em fósforo não superior a 40 μg/ml. Introduzir 10 ml desta solução num tubo de ensaio (4.6) e adicionar 10 ml do reagente vanadomolíbdico (3.6). Homogeneizar e deixar repousar 10 minutos, pelo menos, à temperatura de 20 oC. Medir a densidade óptica em espectrofotómetro a 430 nm por comparação com uma solução obtida por adição de 10 ml do reagente vanadomolíbdico (3.6) a 10 ml de água.
5.3. Curva de calibração
A partir da solução-padrão (3.7) preparar soluções contendo respectivamente 5, 10, 20, 30 e 40 μg de fósforo por ml. Tomar 10 ml de cada uma destas soluções e adicionar 10 ml do reagente vanadomolíbdico (3.6). Homogeneizar e deixar repousar 10 minutos, pelo menos, à temperatura de 20 oC. Medir a densidade óptica como indicado no ponto 5.2. Traçar a curva de calibração representado em ordenadas os valores da densidade óptica e, em abcissas, as quantidades correspondentes de fósforo. A curva é linear para concentrações compreendidas entre 0 e 40 μg/ml.
6. Cálculo dos resultados
Recorrendo à curva de calibração, determinar a quantidade de fósforo na amostra.
Exprimir o resultado em percentagem da amostra.
Repetibilidade
A diferença entre os resultados de duas determinações paralelas efectuadas com a mesma amostra não deve exceder:
|
— |
3 %, em valor relativo, em relação ao maior dos valores, para os teores de fósforo inferiores a 5 %, |
|
— |
0,15 % em valor absoluto para os teores de fósforo iguais ou superiores a 5 %. |
Q. DETERMINAÇÃO DO CLORO SOB A FORMA DE CLORETOS
1. Objecto e âmbito de aplicação
Esta técnica permite dosear o cloro dos cloretos solúveis em água, convencionalmente expressos como cloreto de sódio, e é aplicável a todos os alimentos para animais.
2. Princípio
Dissolvem-se os cloretos em água. Se o produto contiver matérias orgânicas, procede-se à sua clarificação. Acidifica-se ligeiramente a solução com ácido nítrico e os cloretos precipitam sob a forma de cloreto de prata com uma solução de nitrato de prata. Titula-se o excesso de nitrato de prata com uma solução de tiocianato de amónio segundo o método de Volhard.
3. Reagentes
|
3.1. |
Solução de tiocianato de amónio 0,1 mol/l. |
|
3.2. |
Solução de nitrato de prata 0,1 mol/l. |
|
3.3. |
Solução saturada de sulfato férrico de amónio (NH4)Fe(SO4)2. |
|
3.4. |
Ácido nítrico, d: 1,38 g/mol. |
|
3.5. |
Éter dietílico. |
|
3.6. |
Acetona. |
|
3.7. |
Solução de Carrez I: dissolver em água 21,9 g de acetato de zinco, Zn(CH3COO)2 · 2H2O, e 3 g de ácido acético glacial. Perfazer o volume de 100 ml com água. |
|
3.8. |
Solução de Carrez II: dissolver em água 10,6 g de ferrocianeto de potássio K4Fe(CN)6 · 3H2O. Perfazer o volume de 100 ml com água. |
|
3.9. |
Carvão activado, isento de cloretos e não absorvente. |
4. Equipamento
Misturador basculante: cerca de 35 a 40 rotações por minuto.
5. Procedimento
5.1. Preparação da solução
Conforme o tipo de amostra, preparar uma solução como se indica no ponto 5.1.1, 5.1.2 ou 5.1.3.
Efectuar ao mesmo tempo, sem a amostra, um «ensaio em branco».
5.1.1.
Pesar, com uma aproximação de 1 mg, uma amostra de ensaio não superior a 10 g, que não contenha mais de 3 g de cloro sob a forma de cloretos, e introduzi-la, com 400 ml de água, num balão volumétrico de 500 ml, a cerca de 20 oC. Misturar durante 30 minutos no misturador, completar o volume, homogeneizar e filtrar.
5.1.2.
Pesar, com uma aproximação de 1 mg, cerca de 5 g de amostra e introduzi-la com 1 g de carvão activado num balão volumétrico de 500 ml. Adicionar 400 ml de água a cerca de 20 oC e 5 ml de solução de Carrez I (3.7), agitar durante 30 segundos e juntar em seguida 5 ml de solução de Carrez II (3.8). Misturar durante 30 minutos no misturador, completar o volume, homogeneizar e filtrar.
5.1.3.
Preparar a solução como se indica no ponto 5.1.2 mas sem filtrar. Decantar (se necessário, centrifugar), recolher 100 ml do líquido sobrenadante e introduzi-lo num balão volumétrico de 200 ml. Adicionar acetona (3.6), completando o volume, homogeneizar e filtrar.
5.2. Titulação
Com uma pipeta, transferir para um erlenmeyer 25 a 100 ml do filtrado (dependendo do teor presumido de cloro) obtido no ponto 5.1.1, 5.1.2 ou 5.1.3. A alíquota não deverá conter mais de 150 mg de cloro (Cl). Se for necessário, diluir com água, para obter, pelo menos, 50 ml de filtrado, juntar 5 ml de ácido nítrico (3.4), 20 ml de solução saturada de sulfato férrico de amónio (3.3) e duas gotas de solução de tiocianato de amónio (3.1), que se deitam com uma bureta cheia até à marca zero. Em seguida, juntar-lhe a solução de nitrato de prata (3.2) com uma bureta até obter um excesso de 5 ml. Adicionar 5 ml de éter dietílico (3.5) e agitar fortemente para formar o precipitado. Titular o excesso de nitrato de prata com a solução de tiocianato de amónio (3.1) até a viragem para vermelho acastanhado se manter durante um minuto.
6. Cálculo dos resultados
O teor de cloro (X), expresso em percentagem de cloreto de sódio, é dado pela seguinte fórmula:

em que:
|
V1 |
= |
ml de solução de nitrato de prata 0,1 mol/l adicionada |
|
V2 |
= |
ml da solução de tiocianato de amónio 0,1 mol/l gasta na titulação. |
|
m |
= |
massa da amostra. |
Se o ensaio em branco revelar que houve consumo de solução de nitrato de prata 0,1 mol/l, subtrair o valor da quantidade consumida ao volume (V1 - V2).
7. Observações
|
7.1. |
A titulação pode igualmente fazer-se por potenciometria. |
|
7.2. |
Para os produtos muito ricos em matérias gordas, proceder a um desengorduramento prévio com éter dietílico ou éter de petróleo. |
|
7.3. |
Para as farinhas de peixe, a titulação pode ser efectuada pelo método de Mohr. |
(1) Para a dessecação dos cereais, assim como das farinhas e sêmolas grossas e finas, a estufa deve ter uma capacidade térmica tal que, regulada previamente à temperatura de 131 oC, possa atingir de novo esta temperatura menos de 45 minutos após a colocação do número máximo de amostras a secar simultaneamente. A estufa deverá ainda ter uma ventilação tal que, secando durante 2 horas todas as amostras de trigo mole que pode conter, os resultados apresentem uma diferença inferior a 0,15 % relativamente aos resultados obtidos após quatro horas de dessecação.
(2) Substituir os fragmentos de pedra-pomes por pérolas de vidro sempre que a matéria gorda deva ser objecto de exames qualitativos posteriores.
ANEXO IV
MÉTODOS DE ANÁLISE PARA O CONTROLO DO TEOR DE ADITIVOS AUTORIZADOS NOS ALIMENTOS PARA ANIMAIS
A. DETERMINAÇÃO DA VITAMINA A
1. Objecto e âmbito de aplicação
Este método permite determinar o teor de vitamina A (retinol) em alimentos para animais e pré-misturas. A vitamina A determinada pelo presente método abrange o álcool retinílico totalmente trans e os seus isómeros cis. O teor de vitamina A é expresso em unidades internacionais (UI) por quilograma. Uma unidade internacional corresponde à actividade de 0,300 μg de vitamina A totalmente trans na forma de álcool ou 0,344 μg de vitamina A totalmente trans na forma de acetato ou 0,550 μg de vitamina A totalmente trans na forma de palmitato.
O limite de quantificação é 2 000 UI de vitamina A por kg.
2. Princípio
Hidrólise da amostra com solução etanólica de hidróxido de potássio e extracção da vitamina A com éter de petróleo. Remoção do solvente por evaporação e dissolução do resíduo em metanol; se necessário, diluição até à concentração requerida. Determinação do teor de vitamina A por cromatografia líquida de alta resolução (HPLC) de fase reversa, com um detector de UV ou de fluorescência. Os parâmetros cromatográficos são escolhidos de forma a não haver separação entre a vitamina A na forma de álcool totalmente trans e os seus isómeros cis.
3. Reagentes
|
3.1. |
Etanol, σ = 96 %. |
|
3.2. |
Éter de petróleo, intervalo de ebulição: 40 a 60 oC. |
|
3.3. |
Metanol. |
|
3.4. |
Solução de hidróxido de potássio, 50 g/100 ml. |
|
3.5. |
Solução de ascorbato de sódio, 10 g/100 ml (ver as observações do ponto 7.7) |
|
3.6. |
Sulfureto de sódio, Na2S· xH2O (x = 7–9). |
|
3.6.1. |
Solução de sulfureto de sódio, 0,5 mol/l em glicerol, 120 g/l (para x = 9) (ver as observações do ponto 7.8). |
|
3.7. |
Solução de fenolftaleína, 2 g/100 ml em etanol (3.1). |
|
3.8. |
2-Propanol. |
|
3.9. |
Fase móvel para HPLC: mistura de metanol (3.3) e água, por exemplo 980 + 20 (v + v). A proporção exacta dependerá das características da coluna utilizada. |
|
3.10. |
Azoto, isento de oxigénio. |
|
3.11. |
Vitamina A totalmente trans na forma de acetato, extrapura, de actividade certificada, por exemplo 2,80 x 106 UI/g. |
|
3.11.1. |
Solução-mãe de vitamina A totalmente trans na forma de acetato: num balão volumétrico de 100 ml, pesar, com uma aproximação de 0,1 mg, 50 mg de vitamina A na forma de acetato (3.11). Dissolver com 2-propanol (3.8) e completar o volume com o mesmo solvente até ao traço. A concentração nominal desta solução é de 1 400 UI de vitamina A por ml. A concentração exacta é determinada conforme descrito no ponto 5.6.3.1. |
|
3.12. |
Vitamina A totalmente trans na forma de palmitato, extrapura, de actividade certificada, por exemplo 1,80 x 106 UI/g. |
|
3.12.1. |
Solução-mãe de vitamina A totalmente trans na forma de palmitato: num balão volumétrico de 100 ml, pesar, com uma aproximação de 0,1 mg, 80 mg de vitamina A na forma de palmitato (3.12). Dissolver com 2-propanol (3.8) e completar o volume com o mesmo solvente até ao traço. A concentração nominal desta solução é de 1 400 UI de vitamina A por ml. A concentração exacta é determinada conforme descrito no ponto 5.6.3.2. |
|
3.13. |
2,6-Di-terc-butil-4-metilfenol (BHT) (ver as observações do ponto 7.5). |
4. Equipamento
|
4.1. |
Evaporador rotativo de vácuo. |
|
4.2. |
Material de vidro ambarizado. |
|
4.2.1. |
Balões de 500 ml de fundo plano ou erlenmeyer, com colo esmerilado. |
|
4.2.2. |
Balões volumétricos de 10, 25, 100 e 500 ml, com colo estreito e tampa esmerilada. |
|
4.2.3. |
Ampolas de decantação, de 1 000 ml, com tampa esmerilada. |
|
4.2.4. |
Balões em forma de pêra, de 250 ml, com colo esmerilado. |
|
4.3. |
Condensador de Allihn, com camisa de 300 mm, com junta esmerilada e adaptador para alimentação de gás. |
|
4.4. |
Papel de filtro de pregas, com 185 mm de diâmetro, para a separação de fases (por exemplo, Schleicher & Schuell 597 HY 1/2). |
|
4.5. |
Aparelho para HPLC com sistema de injecção. |
|
4.5.1. |
Coluna para cromatografia líquida, com 250 mm x 4 mm, enchimento C18 de 5 ou 10 μm, ou equivalente (critério de eficiência: pico único para todos os isómeros do retinol nas condições da HPLC). |
|
4.5.2. |
Detector de UV ou de fluorescência, com comprimento de onda regulável. |
|
4.6. |
Espectrofotómetro com células de quartzo de 10 mm. |
|
4.7. |
Banho de água equipado com um agitador magnético. |
|
4.8. |
Aparelho de extracção, constituído por (ver figura 1): |
|
4.8.1. |
Cilindro de vidro de 1 litro com colo esmerilado e tampa. |
|
4.8.2. |
Dispositivo de vidro com colo esmerilado, adaptável ao precedente, com uma tubuladura lateral e um tubo axial ajustável. O tubo ajustável termina em U na extremidade inferior e é afilado na extremidade oposta, para permitir a transferência da fase líquida superior do cilindro para uma ampola de decantação. |
5. Procedimento
|
Nota: |
A vitamina A é sensível à luz (ultravioleta) e à oxidação. Efectuar todas as operações na ausência de luz (utilizando material de vidro ambarizado ou protegido com folha de alumínio) e de oxigénio (sob corrente de azoto). Durante a extracção, o ar acima do líquido deve ser substituído por azoto (para evitar excessos de pressão, soltar a tampa de vez em quando). |
5.1. Preparação da amostra
Moer a amostra de forma a poder passá-la por um crivo de 1 mm, tomando as devidas precauções para evitar a geração de calor. Efectuar a moagem imediatamente antes da pesagem e saponificação; caso contrário, pode haver perdas de vitamina A.
5.2. Saponificação
Em função do teor de vitamina A, pesar, com uma aproximação de 1 mg, 2 g a 25 g de amostra num balão de 500 ml de fundo plano ou erlenmeyer (4.2.1). Adicionar sucessivamente, sob agitação, 130 ml de etanol (3.1), aproximadamente 100 mg de BHT (3.13), 2 ml da solução de ascorbato de sódio (3.5) e 2 ml da solução de sulfureto de sódio (3.6). Adaptar o condensador (4.3) ao balão e mergulhar este último no banho de água equipado com um agitador magnético (4.7). Aquecer até à ebulição e deixar em refluxo durante 5 minutos. Adicionar em seguida, pelo condensador (4.3), 25 ml da solução de hidróxido de potássio (3.4) e deixar em refluxo durante mais 25 minutos, com agitação e sob uma corrente ligeira de azoto. Lavar o condensador com cerca de 20 ml de água e arrefecer o conteúdo do balão até à temperatura ambiente.
5.3. Extracção
Transferir quantitativamente a solução de saponificação para uma ampola de decantação de 1 000 ml (4.2.3) ou para o aparelho de extracção (4.8), lavando com um volume total de 250 ml de água. Lavar o balão de saponificação sucessivamente com 25 ml de etanol (3.1) e 100 ml de éter de petróleo (3.2) e transferir os líquidos de lavagem para a ampola de decantação ou o aparelho de extracção. A proporção de água e etanol na solução combinada assim constituída deve ser aproximadamente 2:1. Agitar vigorosamente durante 2 minutos e deixar em repouso durante 2 minutos.
5.3.1.
Depois da separação das fases (ver as observações do ponto 7.3), transferir a fase etérea para outra ampola de decantação (4.2.3). Repetir duas vezes a operação de extracção com 100 ml de éter de petróleo (3.2) e mais duas vezes com 50 ml de éter de petróleo (3.2).
Lavar os extractos combinados na ampola de decantação por duas vezes, com volumes de 100 ml de água cada, agitando suavemente por rotação (para evitar a formação de emulsões), e, em seguida, agitar vigorosamente com mais volumes de 100 ml de água até esta permanecer incolor à solução de fenolftaleína (3.7) (quatro lavagens são normalmente suficientes). Filtrar o extracto lavado com um filtro de pregas de separação de fases (4.4) seco para um balão volumétrico de 500 ml (4.2.2), para remover alguma água ainda em suspensão. Lavar a ampola de decantação e o filtro com 50 ml de éter de petróleo (3.2), completar o volume com éter de petróleo (3.2) até ao traço e homogeneizar.
5.3.2.
Depois da separação das fases (ver as observações do ponto 7.3), substituir a tampa do cilindro de vidro (4.8.1) pelo dispositivo de vidro com esmerilado (4.8.2) e colocar a extremidade inferior em U do tubo ajustável de modo a que fique imediatamente acima do nível da interface. Aumentar a pressão por alimentação de azoto através da tubuladura lateral e transferir a fase etérea superior para uma ampola de decantação de 1 000 ml (4.2.3). Adicionar 100 ml de éter de petróleo (3.2) ao cilindro, tapar e agitar vigorosamente. Depois da separação das fases, transferir a fase superior para a ampola de decantação conforme descrito acima. Repetir a operação de extracção com mais 100 ml de éter de petróleo (3.2) e duas vezes com volumes de 50 ml de éter de petróleo (3.2), transferindo as fases etéreas para a ampola de decantação.
Lavar os extractos etéreos combinados conforme descrito no ponto 5.3.1 e proceder conforme descrito no mesmo ponto.
5.4. Preparação da solução de amostra para a HPLC
Pipetar uma alíquota da solução de éter de petróleo (obtida no ponto 5.3.1 ou 5.3.2) para um balão em forma de pêra de 250 ml (4.2.4). Evaporar o solvente quase até à secura no evaporador rotativo (4.1), sob pressão reduzida, num banho a uma temperatura não superior a 40 oC. Restabelecer a pressão atmosférica com uma alimentação de azoto (3.10) e retirar o balão do evaporador rotativo. Remover o solvente restante sob uma corrente de azoto (3.10) e dissolver imediatamente o resíduo com um volume conhecido (10 ml-100 ml) de metanol (3.3) (a concentração de vitamina A deve estar compreendida entre 5 UI/ml e 30 UI/ml).
5.5. Determinação por HPLC
Procede-se à separação da vitamina A numa coluna de fase reversa C18 (4.5.1) e determina-se a sua concentração com um detector de UV (325 nm) ou de fluorescência (excitação: 325 nm; emissão: 475 nm) (4.5.2).
Injectar uma alíquota (por exemplo, 20 μl) da solução metanólica obtida no ponto 5.4 e eluir com a fase móvel (3.9). Calcular a altura ou área média dos picos correspondentes a várias injecções da mesma solução de amostra e a altura ou área média dos picos correspondentes a várias injecções das soluções de calibração (5.6.2).
As condições a seguir especificadas são-no a título indicativo. Poderão escolher-se outras condições, desde que produzam resultados equivalentes.
|
Coluna para cromatografia líquida (4.5.1): |
250 mm x 4 mm, enchimento C18 de 5 ou 10 μm, ou equivalente |
|
Fase móvel (3.9): |
Mistura de metanol (3.3) e água, por exemplo 980 + 20 (v + v) |
|
Caudal: |
1-2 ml/minuto |
|
Detector (4.5.2): |
Detector de UV (325 nm) ou de fluorescência (excitação: 325 nm; emissão: 475 nm) |
5.6. Calibração
5.6.1.
Pipetar 20 ml da solução-mãe de vitamina A na forma de acetato (3.11.1) ou 20 ml da solução-mãe de vitamina A na forma de palmitato (3.12.1) para um balão de 500 ml de fundo plano ou erlenmeyer (4.2.1) e efectuar a hidrólise conforme descrito no ponto 5.2, mas sem adicionar BHT. Proceder, em seguida, à extracção com éter de petróleo (3.2) conforme descrito no ponto 5.3 e completar o volume com éter de petróleo (3.2) até perfazer 500 ml. Evaporar 100 ml do extracto obtido no evaporador rotativo (ver ponto 5.4) quase até à secura, remover o solvente restante sob uma corrente de azoto (3.10) e redissolver o resíduo com 10,0 ml de metanol (3.3). A concentração nominal desta solução é de 560 UI de vitamina A por ml. A concentração exacta é determinada conforme descrito no ponto 5.6.3.3. Preparar a solução-padrão de trabalho imediatamente antes da sua utilização.
Pipetar 2,0 ml desta solução-padrão de trabalho para um balão volumétrico de 20 ml, completar o volume com metanol (3.3) até ao traço e homogeneizar. A concentração nominal desta solução-padrão de trabalho diluída é de 56 UI de vitamina A por ml.
5.6.2.
Pipetar 1,0, 2,0, 5,0 e 10,0 ml da solução-padrão de trabalho diluída para uma série de balões volumétricos de 20 ml, completar o volume com metanol (3.3) até ao traço e homogeneizar. A concentração nominal destas soluções é de 2,8, 5,6, 14,0 e 28,0 UI de vitamina A por ml.
Injectar vários volumes de 20 μl de cada solução de calibração e determinar as alturas (ou áreas) médias dos picos. Traçar uma curva de calibração com base nas alturas (ou áreas) médias dos picos e nos resultados obtidos na padronização no ultravioleta (5.6.3.3).
5.6.3.
5.6.3.1.
Pipetar 2,0 ml da solução-mãe de vitamina A na forma de acetato (3.11.1) para um balão volumétrico de 50 ml (4.2.2) e completar o volume com 2-propanol (3.8) até ao traço. A concentração nominal desta solução é de 56 UI de vitamina A por ml. Pipetar 3,0 ml desta solução diluída de vitamina A na forma de acetato para um balão volumétrico de 25 ml e completar o volume com 2-propanol (3.8) até ao traço. A concentração nominal desta solução é de 6,72 UI de vitamina A por ml. Traçar o espectro de UV desta solução no espectrofotómetro (4.6), entre 300 nm e 400 nm, em relação ao 2-propanol (3.8). O máximo de extinção deve estar compreendido entre 325 nm e 327 nm.
Cálculo do teor de vitamina A:
UI de vitamina A/ml = E326 × 19,0
(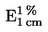 da vitamina A na forma de acetato = 1 530 a 326 nm, em 2-propanol)
da vitamina A na forma de acetato = 1 530 a 326 nm, em 2-propanol)
5.6.3.2.
Pipetar 2,0 ml da solução-mãe de vitamina A na forma de palmitato (3.12.1) para um balão volumétrico de 50 ml (4.2.2) e completar o volume com 2-propanol (3.8) até ao traço. A concentração nominal desta solução é de 56 UI de vitamina A por ml. Pipetar 3,0 ml desta solução diluída de vitamina A na forma de palmitato para um balão volumétrico de 25 ml e completar o volume com 2-propanol (3.8) até ao traço. A concentração nominal desta solução é de 6,72 UI de vitamina A por ml. Traçar o espectro de UV desta solução no espectrofotómetro (4.6), entre 300 nm e 400 nm, em relação ao 2-propanol (3.8). O máximo de extinção deve estar compreendido entre 325 nm e 327 nm.
Cálculo do teor de vitamina A:
UI de vitamina A/ml = E326 × 19,0
( da vitamina A na forma de palmitato = 957 a 326 nm, em 2-propanol)
da vitamina A na forma de palmitato = 957 a 326 nm, em 2-propanol)
5.6.3.3.
Pipetar 3,0 ml da solução-padrão de trabalho não diluída de vitamina A, preparada conforme descrito no ponto 5.6.1, para um balão volumétrico de 50 ml (4.2.2) e completar o volume com 2-propanol (3.8) até ao traço. Pipetar 5,0 ml desta solução para um balão volumétrico de 25 ml e completar o volume com 2-propanol (3.8) até ao traço. A concentração nominal desta solução é de 6,72 UI de vitamina A por ml. Traçar o espectro de UV desta solução no espectrofotómetro (4.6), entre 300 nm e 400 nm, em relação ao 2-propanol (3.8). O máximo de extinção deve estar compreendido entre 325 nm e 327 nm.
Cálculo do teor de vitamina A:
UI de vitamina A/ml = E325 × 18,3
( da vitamina A na forma de álcool = 1 821 a 325 nm, em 2-propanol)
da vitamina A na forma de álcool = 1 821 a 325 nm, em 2-propanol)
6. Cálculo dos resultados
A partir da altura ou área média dos picos da vitamina A da solução de amostra, determinar a concentração desta em UI/ml, com base na curva de calibração (5.6.2).
O teor de vitamina A, w, da amostra, expresso em UI/kg, é dado pela seguinte fórmula:
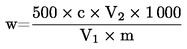 [UI/kg]
[UI/kg]
em que:
|
c |
= |
concentração, em UI/ml, de vitamina A da solução de amostra (5.4) |
|
V1 |
= |
volume, em ml, de solução de amostra (5.4) |
|
V2 |
= |
volume, em ml, da alíquota tomada no ponto 5.4 |
|
m |
= |
massa, em g, da toma analisada |
7. Observações
|
7.1. |
No caso das amostras com baixas concentrações de vitamina A, pode ser útil combinar os extractos etéreos de duas saponificações (quantidade pesada: 25 g) numa única solução de amostra para a determinação por HPLC. |
|
7.2. |
A toma de amostra utilizada na análise não deve conter mais de 2 g de matérias gordas. |
|
7.3. |
Se não se der a separação de fases, adicionar aproximadamente 10 ml de etanol (3.1), para desfazer a emulsão. |
|
7.4. |
No caso do óleo de fígado de bacalhau e outras matérias gordas puras, o tempo de saponificação deve ser aumentado para 45-60 minutos. |
|
7.5. |
Em vez de BHT, pode utilizar-se hidroquinona. |
|
7.6. |
É possível separar os isómeros do retinol com uma coluna de fase normal. Mas, neste caso, para a realização dos cálculos, devem somar-se as alturas (ou áreas) dos picos correspondentes a todos os isómeros cis e trans. |
|
7.7. |
Em vez da solução de ascorbato de sódio, podem utilizar-se aproximadamente 150 mg de ácido ascórbico. |
|
7.8. |
Em vez da solução de sulfureto de sódio, podem utilizar-se aproximadamente 50 mg de EDTA. |
|
7.9. |
No caso das análises da vitamina A nos substitutos do leite, deve dedicar-se uma atenção especial aos seguintes aspectos:
Para verificar se o método de análise utilizado produz resultados fiáveis nesta matriz específica (substituto do leite) deve realizar-se um ensaio de recuperação com uma toma de ensaio adicional. Se a taxa de recuperação for inferior a 80 %, o resultado analítico tem de ser corrigido em função da recuperação. |
8. Repetibilidade
A diferença entre os resultados de duas determinações paralelas efectuadas com a mesma amostra não deve exceder 15 % do maior dos valores.
9. Resultados de um estudo interlaboratorial (1)
|
|
Pré-mistura |
Alimento pré-misturado |
Concentrado de minerais |
Alimento proteico |
Alimento para leitões |
|
L |
13 |
12 |
13 |
12 |
13 |
|
n |
48 |
45 |
47 |
46 |
49 |
|
média [UI/kg] |
17,02 x 106 |
1,21 x 106 |
537 100 |
151 800 |
18 070 |
|
sr [UI/kg] |
0,51 x 106 |
0,039 x 106 |
22 080 |
12 280 |
682 |
|
r [UI/kg] |
1,43 x 106 |
0,109 x 106 |
61 824 |
34 384 |
1 910 |
|
CVr [ %] |
3,0 |
3,5 |
4,1 |
8,1 |
3,8 |
|
sR [IU/kg] |
1,36 x 106 |
0,069 x 106 |
46 300 |
23 060 |
3 614 |
|
R [UI/kg] |
3,81 x 106 |
0,193 x 106 |
129 640 |
64 568 |
10 119 |
|
CVR [ %] |
8,0 |
6,2 |
8,6 |
15 |
20 |
|
L |
= |
número de laboratórios |
|
n |
= |
número de valores individuais |
|
sr |
= |
desvio-padrão da repetibilidade |
|
sR |
= |
desvio-padrão da reprodutibilidade |
|
r |
= |
repetibilidade |
|
R |
= |
reprodutibilidade |
|
CVr |
= |
coeficiente de variação da repetibilidade |
|
CVR |
= |
coeficiente de variação da reprodutibilidade |

B. DETERMINAÇÃO DA VITAMINA E
1. Objecto e âmbito de aplicação
Este método permite determinar o teor de vitamina E em alimentos para animais e pré-misturas. O teor de vitamina E é expresso em mg de acetato de DL-α-tocoferol por quilograma. 1 mg de acetato de DL-α-tocoferol corresponde a 0,91 mg de DL-α- -tocoferol (vitamina E).
O limite de quantificação é de 2 mg de vitamina E por kg e só pode ser alcançado com um detector de fluorescência. Com um detector de UV, o limite de quantificação é de 10 mg/kg.
2. Princípio
Hidrólise da amostra com solução etanólica de hidróxido de potássio e extracção da vitamina E com éter de petróleo. Remoção do solvente por evaporação e dissolução do resíduo em metanol; se necessário, diluição até à concentração requerida. Determinação do teor de vitamina E por cromatografia líquida de alta resolução (HPLC) de fase reversa, com um detector de fluorescência ou de UV.
3. Reagentes
|
3.1. |
Etanol, σ = 96 %. |
|
3.2. |
Éter de petróleo, intervalo de ebulição: 40 a 60 oC. |
|
3.3. |
Metanol. |
|
3.4. |
Solução de hidróxido de potássio, 50 g/100 ml. |
|
3.5. |
Solução de ascorbato de sódio, 10 g/100 ml (ver as observações do ponto 7.7). |
|
3.6. |
Sulfureto de sódio, Na2S· xH2O (x = 7–9). |
|
3.6.1. |
Solução de sulfureto de sódio, 0,5 mol/l em glicerol, 120 g/l (para x = 9) (ver as observações do ponto 7.8). |
|
3.7. |
Solução de fenolftaleína, 2 g/100 ml em etanol (3.1). |
|
3.8. |
Fase móvel para HPLC: mistura de metanol (3.3) e água, por exemplo 980 + 20 (v + v). A proporção exacta dependerá das características da coluna utilizada. |
|
3.9. |
Azoto, isento de oxigénio. |
|
3.10. |
Acetato de DL-α-tocoferol, extrapuro, de actividade certificada. |
|
3.10.1. |
Solução-mãe de acetato de DL-α-tocoferol: num balão volumétrico de 100 ml, pesar, com uma aproximação de 0,1 mg, 100 mg de acetato de DL-α-tocoferol 3.11. Dissolver com etanol (3.1) e completar o volume com o mesmo solvente até ao traço. 1 ml desta solução contém 1 mg de acetato de DL-α-tocoferol. (Padronização no UV: ver o ponto 5.6.1.3; estabilização: ver as observações do ponto 7.4). |
|
3.11. |
DL-α-tocoferol, extrapuro, de actividade certificada. |
|
3.11.1. |
Solução-mãe de DL-α-tocoferol: num balão volumétrico de 100 ml, pesar, com uma aproximação de 0,1 mg, 100 mg de DL-α-tocoferol (3.11). Dissolver com etanol (3.1) e completar o volume com o mesmo solvente até ao traço. 1 ml desta solução contém 1 mg de DL-α-tocoferol. (padronização no UV: ver o ponto 5.6.2.3; estabilização: ver as observações do ponto 7.4). |
|
3.12. |
2,6-Di-terc-butil-4-metilfenol (BHT) (ver as observações do ponto 7.5). |
4. Equipamento
|
4.1. |
Evaporador rotativo. |
|
4.2. |
Material de vidro ambarizado. |
|
4.2.1. |
Balões de 500 ml de fundo plano ou erlenmeyer, com colo esmerilado. |
|
4.2.2. |
Balões volumétricos de 10, 25, 100 e 500 ml, com colo estreito e tampa esmerilada. |
|
4.2.3. |
Ampolas de decantação, de 1 000 ml, com colo esmerilado e tampa. |
|
4.2.4. |
Balões em forma de pêra, de 250 ml, com colo esmerilado. |
|
4.3. |
Condensador de Allihn, com camisa de 300 mm, junta esmerilada e adaptador para alimentação de gás. |
|
4.4. |
Papel de filtro de pregas, com 185 mm de diâmetro, para a separação de fases (por exemplo, Schleicher & Schuell 597 HY 1/2). |
|
4.5. |
Aparelho para HPLC com sistema de injecção. |
|
4.5.1. |
Coluna para cromatografia líquida com 250 mm x 4 mm e enchimento C18 de 5 ou 10 μm, ou equivalente. |
|
4.5.2. |
Detector de fluorescência ou de UV, com comprimento de onda regulável. |
|
4.6. |
Espectrofotómetro com células de quartzo de 10 mm. |
|
4.7. |
Banho de água equipado com um agitador magnético. |
|
4.8. |
Aparelho de extracção, constituído por (ver figura 1): |
|
4.8.1. |
Cilindro de vidro de 1 litro com colo esmerilado e tampa. |
|
4.8.2. |
Dispositivo de vidro com colo esmerilado, adaptável ao precedente, com uma tubuladura lateral e um tubo axial ajustável. O tubo ajustável termina em U na extremidade inferior e é afilado na extremidade oposta, para permitir a transferência da fase líquida superior do cilindro para uma ampola de decantação. |
5. Procedimento
|
Nota |
A vitamina E é sensível à luz (ultravioleta) e à oxidação. Efectuar todas as operações na ausência de luz (utilizando material de vidro ambarizado ou protegido com folha de alumínio) e de oxigénio (sob corrente de azoto). Durante a extracção, o ar acima do líquido deve ser substituído por azoto (para evitar excessos de pressão, soltar a tampa de vez em quando). |
5.1. Preparação da amostra
Moer a amostra de forma a poder passá-la por um crivo de 1 mm, tomando as devidas precauções para evitar a geração de calor. Efectuar a moagem imediatamente antes da pesagem e da saponificação; caso contrário, pode haver perdas de vitamina E.
5.2. Saponificação
Em função do teor de vitamina E, pesar com uma aproximação de 0,01 g, 2 g a 25 g de amostra num balão de fundo plano ou erlenmeyer de 500 ml (4.2.1). Adicionar sucessivamente, sob agitação, 130 ml de etanol (3.1), aproximadamente 100 mg de BHT (3.12), 2 ml da solução de ascorbato de sódio (3.5) e 2 ml da solução de sulfureto de sódio (3.6). Adaptar o condensador (4.3) ao balão e mergulhar este último no banho de água equipado com um agitador magnético (4.7). Aquecer até à ebulição e deixar em refluxo durante 5 minutos. Adicionar em seguida, pelo condensador (4.3), 25 ml da solução de hidróxido de potássio (3.4) e deixar em refluxo durante mais 25 minutos, com agitação e sob uma corrente ligeira de azoto. Lavar o condensador com cerca de 20 ml de água e arrefecer o conteúdo do balão até à temperatura ambiente.
5.3. Extracção
Transferir quantitativamente a solução de saponificação para uma ampola de decantação de 1 000 ml (4.2.3) ou para o aparelho de extracção (4.8), lavando com um volume total de 250 ml de água. Lavar o balão de saponificação sucessivamente com 25 ml de etanol (3.1) e 100 ml de éter de petróleo (3.2) e transferir os líquidos de lavagem para a ampola de decantação ou o aparelho de extracção. A proporção de água e etanol na solução combinada assim constituída deve ser aproximadamente 2:1. Agitar vigorosamente durante 2 minutos e deixar em repouso durante 2 minutos.
5.3.1.
Depois da separação das fases (ver as observações do ponto 7.3), transferir a fase etérea para outra ampola de decantação (4.2.3). Repetir duas vezes a operação de extracção com 100 ml de éter de petróleo (3.2) e mais duas vezes com 50 ml de éter de petróleo (3.2).
Lavar os extractos combinados na ampola de decantação por duas vezes, com volumes de 100 ml de água cada, agitando suavemente por rotação (para evitar a formação de emulsões), e, em seguida, agitar vigorosamente com mais volumes de 100 ml de água até esta permanecer incolor à solução de fenolftaleína (3.7) (quatro lavagens são normalmente suficientes). Filtrar o extracto lavado com um filtro de pregas de separação de fases (4.4) seco para um balão volumétrico de 500 ml (4.2.2), para remover alguma água ainda em suspensão. Lavar a ampola de decantação e o filtro com 50 ml de éter de petróleo (3.2), completar o volume com éter de petróleo (3.2) até ao traço e homogeneizar.
5.3.2.
Depois da separação das fases (ver as observações do ponto 7.3), substituir a tampa do cilindro de vidro (4.8.1) pelo dispositivo de vidro com esmerilado (4.8.2) e colocar a extremidade inferior em U do tubo ajustável de modo a que fique imediatamente acima do nível da interface. Aumentar a pressão por alimentação de azoto através da tubuladura lateral e transferir a fase etérea superior para uma ampola de decantação de 1 000 ml (4.2.3). Adicionar 100 ml de éter de petróleo (3.2) ao cilindro, tapar e agitar vigorosamente. Depois da separação das fases, transferir a fase superior para a ampola de decantação conforme descrito acima. Repetir a operação de extracção com mais 100 ml de éter de petróleo (3.2) e duas vezes com volumes de 50 ml de éter de petróleo (3.2), transferindo as fases etéreas para a ampola de decantação.
Lavar os extractos etéreos combinados conforme descrito no ponto 5.3.1 e proceder conforme descrito no mesmo ponto.
5.4. Preparação da solução de amostra para a HPLC
Pipetar uma alíquota da solução de éter de petróleo (obtida no ponto 5.3.1 ou 5.3.2) para um balão em forma de pêra de 250 ml (4.2.4). Evaporar o solvente quase até à secura no evaporador rotativo (4.1), sob pressão reduzida, num banho a uma temperatura não superior a 40 oC. Restabelecer a pressão atmosférica com uma alimentação de azoto (3.9) e retirar o balão do evaporador rotativo. Remover o solvente restante sob uma corrente de azoto (3.9) e dissolver imediatamente o resíduo com um volume conhecido (10 ml — 100 ml) de metanol (3.3) (a concentração de DL-α-tocoferol deve estar compreendida entre 5 μg/ml e 30 μg/ml).
5.5. Determinação por HPLC
Procede-se à separação da vitamina E numa coluna de fase reversa C18 (4.5.1) e determina-se a sua concentração com um detector de fluorescência (excitação: 295 nm; emissão: 330 nm) ou de UV (292 nm) (4.5.2).
Injectar uma alíquota (por exemplo, 20 μl) da solução metanólica obtida no ponto 5.4 e eluir com a fase móvel (3.8). Calcular a altura ou área média dos picos correspondentes a várias injecções da mesma solução de amostra e a altura ou área média dos picos correspondentes a várias injecções das soluções de calibração (5.6.2).
As condições a seguir especificadas são-no a título indicativo. Poderão escolher-se outras condições, desde que produzam resultados equivalentes.
|
Coluna para cromatografia líquida (4.5.1): |
250 mm x 4 mm, enchimento C18 de 5 ou 10 μm, ou equivalente |
|
Fase móvel (3.8): |
Mistura de metanol (3.3) e água, por exemplo 980 + 20 (v + v) |
|
Caudal: |
1-2 ml/minuto |
|
Detector (4.5.2): |
Detector de fluorescência (excitação: 295 nm; emissão: 330 nm) ou detector de UV (292 nm) |
5.6. Calibração (acetato de DL-α-tocoferol ou DL-α-tocoferol)
5.6.1.
5.6.1.1.
Pipetar 25 ml da solução-mãe de acetato de DL-α-tocoferol (3.10.1) para um balão de 500 ml de fundo plano ou erlenmeyer (4.2.1) e efectuar a hidrólise conforme descrito no ponto 5.2. Proceder, em seguida, à extracção com éter de petróleo (3.2) conforme descrito no ponto 5.3 e completar o volume com éter de petróleo até perfazer 500 ml. Evaporar 25 ml do extracto obtido no evaporador rotativo (ver ponto 5.4) quase até à secura, remover o solvente restante sob uma corrente de azoto (3.9) e redissolver o resíduo com 25,0 ml de metanol (3.3). A concentração nominal desta solução é 45,5 μg de DL-α-tocoferol/ml, equivalente a 50 μg de acetato de DL-α-tocoferol/ml. Preparar a solução-padrão de trabalho imediatamente antes da sua utilização.
5.6.1.2.
Pipetar 1,0, 2,0, 4,0 e 10,0 ml da solução-padrão de trabalho para uma série de balões volumétricos de 20 ml, completar o volume com metanol (3.3) até ao traço e homogeneizar. A concentração nominal destas soluções é de 2,5, 5,0, 10,0 e 25,0 μg de acetato de DL-α-tocoferol/ml, equivalentes a 2,28, 4,55, 9,10 e 22,8 μg de DL-α-tocoferol/ml.
Injectar vários volumes de 20 μl de cada solução de calibração e determinar as alturas (ou áreas) médias dos picos. Traçar uma curva de calibração com base nas alturas (ou áreas) médias dos picos.
5.6.1.3.
Diluir, com etanol, 5,0 ml da solução-mãe de acetato de DL-α-tocoferol (3.10.1) até perfazer 25,0 ml e traçar o espectro de UV desta solução no espectrofotómetro (4.6), entre 250 nm e 320 nm, em relação ao etanol (3.1).
O máximo de absorção deve situar-se nos 284 nm:
 = 43,6 a 284 nm, em etanol
= 43,6 a 284 nm, em etanol
Para esta diluição, deve obter-se uma extinção compreendida entre 0,84 e 0,88.
5.6.2.
5.6.2.1.
Pipetar 2 ml da solução-mãe de DL-α-tocoferol (3.11.1) para um balão volumétrico de 50 ml, dissolver com metanol (3.3) e completar o volume com metanol até ao traço. A concentração nominal desta solução é 40 μg de DL-α-tocoferol/ml, equivalente a 44,0 μg de acetato de DL-α-tocoferol/ml. Preparar a solução-padrão de trabalho imediatamente antes da sua utilização.
5.6.2.2.
Pipetar 1,0, 2,0, 4,0 e 10,0 ml da solução-padrão de trabalho para uma série de balões volumétricos de 20 ml, completar o volume com metanol (3.3) até ao traço e homogeneizar. A concentração nominal destas soluções é de 2,0, 4,0, 8,0 e 20,0 μg de DL-α- -tocoferol/ml, equivalentes a 2,20, 4,40, 8,79 e 22,0 μg de acetato de DL-α-tocoferol/ml.
Injectar vários volumes de 20 μl de cada solução de calibração e determinar as alturas (ou áreas) médias dos picos. Traçar uma curva de calibração com base nas alturas (ou áreas) médias dos picos.
5.6.2.3.
Diluir, com etanol, 2,0 ml da solução-mãe de DL-α-tocoferol (3.11.1) até perfazer 25,0 ml e traçar o espectro de UV desta solução no espectrofotómetro (4.6), entre 250 nm e 320 nm, em relação ao etanol (3.1). O máximo de absorção deve situar-se nos 292 nm:
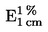 = 75,8 a 292 nm, em etanol
= 75,8 a 292 nm, em etanol
Para esta diluição, deve obter-se uma extinção de 0,6.
6. Cálculo dos resultados
A partir da altura ou área média dos picos da vitamina E da solução de amostra, determinar a concentração desta em μg/ml (expressa em acetato de α-tocoferol) com base na curva de calibração (5.6.1.2 ou 5.6.2.2).
O teor de vitamina E, w, da amostra, expresso em mg/kg, é dado pela seguinte fórmula:
 [mg/kg]
[mg/kg]
em que:
|
c |
= |
concentração, em μg/ml, de vitamina E (expressa em acetato de α-tocoferol) da solução de amostra (5.4) |
|
V1 |
= |
volume, em ml, de solução de amostra (5.4) |
|
V2 |
= |
volume, em ml, da alíquota tomada no ponto 5.4 |
|
m |
= |
massa, em g, da toma analisada |
7. Observações
|
7.1. |
No caso das amostras com baixas concentrações de vitamina E, pode ser útil combinar os extractos etéreos de duas saponificações (quantidade pesada: 25 g) numa única solução de amostra para a determinação por HPLC. |
|
7.2. |
A toma de amostra utilizada na análise não deve conter mais de 2 g de matérias gordas. |
|
7.3. |
Se não se der a separação de fases, adicionar aproximadamente 10 ml de etanol (3.1), para desfazer a emulsão. |
|
7.4. |
Depois das medições espectrofotométricas efectuadas com a solução de acetato de DL-α-tocoferol ou de DL-α-tocoferol de acordo com os pontos 5.6.1.3 ou 5.6.2.3, respectivamente, adicionar à solução (3.10.1 ou 3.10.2) cerca de 10 mg de BHT (3.12) e conservar a mesma em frigorífico (prazo de validade: máximo quatro semanas). |
|
7.5. |
Em vez de BHT, pode utilizar-se hidroquinona. |
|
7.6. |
É possível separar os isómeros α-, β-, γ- e δ-tocoferol com uma coluna de fase normal. |
|
7.7. |
Em vez da solução de ascorbato de sódio, podem utilizar-se aproximadamente 150 mg de ácido ascórbico. |
|
7.8. |
Em vez da solução de sulfureto de sódio, podem utilizar-se aproximadamente 50 mg de EDTA. |
|
7.9. |
Em condições alcalinas, o acetato de vitamina E hidrolisa muito rapidamente, pelo que é muito sensível à oxidação, especialmente na presença de oligoelementos como o ferro ou o cobre. A determinação da vitamina E em pré-misturas com teores superiores a 5 000 mg/kg pode ter por consequência a degradação da vitamina E. Assim, recomenda-se a confirmação através de um ensaio por HPLC que inclua uma digestão enzimática da formulação com vitamina E sem a etapa de saponificação em meio alcalino. |
8. Repetibilidade
A diferença entre os resultados de duas determinações paralelas efectuadas com a mesma amostra não deve exceder 15 % do maior dos valores.
9. Resultados de um estudo interlaboratorial (2)
|
|
Pré-mistura |
Alimento pré-misturado |
Concentrado de minerais |
Alimento proteico |
Alimento para leitões |
|
L |
12 |
12 |
12 |
12 |
12 |
|
n |
48 |
48 |
48 |
48 |
48 |
|
média [mg/kg] |
17 380 |
1 187 |
926 |
315 |
61,3 |
|
sr [mg/kg] |
384 |
45,3 |
25,2 |
13,0 |
2,3 |
|
r [mg/kg] |
1 075 |
126,8 |
70,6 |
36,4 |
6,4 |
|
CVr [ %] |
2,2 |
3,8 |
2,7 |
4,1 |
3,8 |
|
SR mg/kg] |
830 |
65,0 |
55,5 |
18,9 |
7,8 |
|
R [mg/kg] |
2 324 |
182,0 |
155,4 |
52,9 |
21,8 |
|
CVR [ %] |
4,8 |
5,5 |
6,0 |
6,0 |
12,7 |
|
L |
= |
número de laboratórios |
|
n |
= |
número de valores individuais |
|
sr |
= |
desvio-padrão da repetibilidade |
|
SR |
= |
desvio-padrão da reprodutibilidade |
|
r |
= |
repetibilidade |
|
R |
= |
reprodutibilidade |
|
CVr |
= |
coeficiente de variação da repetibilidade |
|
CVR |
= |
coeficiente de variação da reprodutibilidade |

(C) DETERMINAÇÃO DOS OLIGOELEMENTOS FERRO, COBRE, MANGANÊS E ZINCO
1. Objecto e âmbito de aplicação
Esta técnica permite dosear os oligoelementos ferro, cobre, manganês e zinco nos alimentos para animais. Os limites de quantificação são os seguintes:
|
— |
Ferro (Fe): 20 mg/kg |
|
— |
Cobre (Cu): 10 mg/kg |
|
— |
Manganês (Mn): 20 mg/kg |
|
— |
Zinco (Zn): 20 mg/kg |
2. Princípio
A amostra é colocada em solução em ácido clorídrico, após a destruição da matéria orgânica eventualmente presente. Os elementos ferro, cobre, manganês e zinco são determinados, após uma diluição apropriada, por espectrometria de absorção atómica.
3. Reagentes
Observações prévias
A água utilizada na preparação dos reagentes e das soluções necessárias ao processo de análise deverá estar isenta dos catiões a determinar, devendo obter-se mediante dupla destilação num aparelho em borossilicato ou quartzo, ou por tratamento duplo com uma resina de permuta iónica.
Os reagentes devem ser, pelo menos, da qualidade «para análise». A ausência do elemento a determinar deve ser verificada por um ensaio em branco. Os reagentes podem, se necessário, ser submetidos a uma maior purificação.
As soluções-padrão adiante descritas poderão ser substituídas por soluções-padrão comerciais, desde que venham garantidas e sejam testadas antes da sua utilização.
|
3.1. |
Ácido clorídrico (d: 1,19 g/ml). |
|
3.2. |
Ácido clorídrico 6 mol/l. |
|
3.3. |
Ácido clorídrico 0,5 mol/l. |
|
3.4. |
Ácido fluorídrico a 38-40 % (v/v), com um teor de ferro inferior a 1 mg/l e resíduo de evaporação inferior a 10 mg (expressos em sulfato)/l. |
|
3.5. |
Ácido sulfúrico (d: 1,84 g/ml). |
|
3.6. |
Água oxigenada a aproximadamente 100 vol. de oxigénio (30 % em peso). |
|
3.7. |
Solução-padrão de ferro (1 000 μg Fe/ml), preparada da seguinte forma, ou solução comercial equivalente: dissolver 1 g de arame de ferro em 200 ml de ácido clorídrico 6 mol/l (3.2), adicionar 16 ml de água oxigenada (3.6) e perfazer o volume de 1 litro com água. |
|
3.7.1. |
Solução-padrão de trabalho de ferro (100 μg Fe/ml): diluir a solução-padrão (3.7) em água, na proporção 1:9. |
|
3.8. |
Solução-padrão de cobre (1 000 μg Cu/ml), preparada da seguinte forma, ou solução comercial equivalente:
|
|
3.8.1. |
Solução-padrão de trabalho de cobre (10 μg Cu/ml): diluir a solução-padrão (3.8) em água na proporção de 1:9; diluir em seguida esta solução em água, novamente na proporção de 1:9. |
|
3.9. |
Solução-padrão de manganês (1 000 μg Mn/ml), preparada da seguinte forma, ou solução comercial equivalente:
|
|
3.9.1. |
Solução-padrão de trabalho de manganês (10 μg Mn/ml): diluir a solução-padrão (3.9) em água na proporção de 1:9; diluir em seguida esta solução em água, novamente na proporção de 1:9. |
|
3.10. |
Solução-padrão de zinco (1 000 μg Zn/ml), preparada da seguinte forma, ou solução comercial equivalente:
|
|
3.10.1. |
Solução-padrão de trabalho de zinco (10 μg Zn/ml): diluir a solução-padrão (3.10) em água na proporção de 1:9; diluir em seguida esta solução em água, novamente na proporção de 1:9. |
|
3.11. |
Solução de cloreto de lantânio preparada da seguinte forma: dissolver 12 g de óxido de lantânio em 150 ml de água, adicionar 100 ml de ácido clorídrico 6 mol/l (3.2) e perfazer o volume de 1 litro com água. |
4. Equipamento
|
4.1. |
Mufla de temperatura regulável e, de preferência, com registador. |
|
4.2. |
Material de vidro em borossilicato resistente. Recomenda-se a utilização de material que sirva exclusivamente para a dosagem dos oligoelementos. |
|
4.3. |
Espectrofotómetro de absorção atómica, com características de sensibilidade e precisão compatíveis com as exigências desta técnica. |
5. ProcedimentO (3)
5.1. Amostras com matéria orgânica
5.1.1. (4)
|
5.1.1.1. |
Colocar entre 5 e 10 g de amostra, pesado com uma aproximação de 0,2 mg, num cadinho de quartzo ou platina [ver nota b)], secar em estufa a 105 oC e introduzir o cadinho na mufla (4.1) a frio. Fechar a mufla [ver nota c)] e elevar progressivamente a sua temperatura de forma a que atinja os 450 a 475 oC em 90 minutos. Manter esta temperatura durante 4 a 16 horas (durante a noite, por exemplo) para eliminar a substância carbonosa, abrir em seguida a mufla e deixar arrefecer [ver nota d)]. Humedecer as cinzas com água a transferi-las em seguida para um copo de 250 ml. Lavar o cadinho com cerca de 5 ml de ácido clorídrico (3.1) e transferir, lentamente e com precaução, a solução de lavagem para o copo (poderá produzir-se uma reacção violenta pela formação de CO2). Em seguida adicionar ácido clorídrico (3.1) gota a gota, com agitação, até que cesse a efervescência. Evaporar até à secura, mexendo regularmente com uma vareta de vidro. Juntar ao resíduo 15 ml de ácido clorídrico 6 mol/l (3.2) e, em seguida, aproximadamente 120 ml de água. Agitar com vareta de vidro, que deve ser deixada no copo, e tapar com um vidro de relógio. Levar lentamente a ebulição e ferver em lume brando até que a dissolução das cinzas deixe de ser visível. Filtrar com papel de filtro, isento de cinzas, e recolher o filtrado num balão volumétrico de 250 ml. Lavar o copo e o filtro com 5 ml de ácido clorídrico 6 mol/l (3.2) quente e duas vezes com água a ferver. Completar o volume com água (a concentração em HCl é aproximadamente de 0,5 mol/l). |
|
5.1.1.2. |
Se o resíduo que ficou no filtro for de cor negra (carbonosa) colocá-lo de novo na mufla e incinerá-lo a 450-475 oC. Esta incineração, que requer apenas algumas horas (três a cinco horas, aproximadamente), considerar-se-á terminada quando as cinzas se apresentarem de cor branca ou quase branca. Dissolver o resíduo em aproximadamente 2 ml de ácido clorídrico (3.1), evaporá-lo até à secura e adicionar 5 ml de ácido clorídrico 6 mol/l (3.2). Aquecer, filtrar a solução para o balão volumétrico e completar o volume com água (a concentração em HCl é de aproximadamente 0,5 mol/l). Notas:
|
5.1.2.
5.1.2.1.
A partir das soluções-padrão de trabalho 3.7.1, 3.8.1, 3.9.1 e 3.10.1, preparar, para cada oligoelemento a dosear, uma gama de soluções de calibração, apresentando cada uma delas uma concentração em HC1 de aproximadamente 0,5 mol/l e, no caso do ferro, do manganês e do zinco, uma concentração em cloreto de lantânio correspondente a 0,1 % de lantânio (m/v).
As concentrações de oligoelementos escolhidas deverão situar-se na zona de sensibilidade do espetrofotómetro utilizado. Os quadros adiante apresentados indicam, a título de exemplo, as composições de gamas típicas de soluções de calibração; poderá ser necessário, conforme o tipo e a sensibilidade do espectrofotómetro utilizado, optar por outras concentrações.
Ferro
|
μg Fe/ml |
0 |
0,5 |
1 |
2 |
3 |
4 |
5 |
|
ml de solução-padrão de trabalho (3.7.1) (1 ml = 100 μg Fe) |
0 |
0,5 |
1 |
2 |
3 |
4 |
5 |
|
ml HCl (3.2) |
7 |
7 |
7 |
7 |
7 |
7 |
7 |
|
+ 10 ml de solução de cloreto de lantânio (3.11); perfazer o volume de 100 ml com água. |
|||||||
Cobre
|
μg Cu/ml |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
ml de solução-padrão de trabalho (3.8.1) (1 ml = 10 μg Cu) |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
|
ml HCl (3.2) |
8 |
8 |
8 |
8 |
8 |
8 |
8 |
Manganês
|
μg Mn/ml |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
ml de solução-padrão de trabalho (3.9.1) (1 ml = 10 μg Mn) |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
|
ml HCl (3.2) |
7 |
7 |
7 |
7 |
7 |
7 |
7 |
|
+ 10 ml de solução de cloreto de lantânio (3.11); perfazer o volume de 100 ml com água. |
|||||||
Zinco
|
μg Zn/ml |
0 |
0,05 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
|
ml de solução-padrão de trabalho (3.10.1) (1 ml = 10 μg Zn) |
0 |
0,5 |
1 |
2 |
4 |
6 |
8 |
|
ml HCl (3.2) |
7 |
7 |
7 |
7 |
7 |
7 |
7 |
|
+ 10 ml de solução de cloreto de lantânio (3.11); perfazer o volume de 100 ml com água. |
|||||||
5.1.2.2.
Para o doseamento do cobre, a solução preparada como indicado no ponto 5.1.1 poderá, regra geral, ser utilizada directamente. Se for necessário diluir a solução de forma a que a sua concentração se situe na gama de concentrações das soluções de calibração, poderá pipetar-se uma alíquota para um balão volumétrico de 100 ml e completar o volume com ácido clorídrico 0,5 mol/l (3.3).
Para a determinação do ferro, manganês e zinco, pipetar uma alíquota da solução preparada como indicado no ponto 5.1.1 para um balão volumétrico de 100 ml, adicionar 10 ml de solução de cloreto de lantânio (3.11) e completar o volume com ácido clorídrico 0,5 mol/l (3.3) (ver também o ponto 8 «Observações»).
5.1.2.3.
Efectuar sem a amostra um ensaio em branco que inclua todas as etapas constantes do método. A solução de calibração «0» não deve ser utilizada como «branco».
5.1.2.4.
Medir a absorção atómica das soluções de calibração e da solução a analisar utilizando uma chama oxidante ar-acetileno aos seguintes comprimentos de onda:
|
|
Fe: 248,3 nm |
|
|
Cu: 324,8 nm |
|
|
Mn: 279,5 nm |
|
|
Zn: 213,8 nm |
Repetir quatro vezes cada medição.
5.2. Alimentos minerais
Na ausência de matérias orgânicas é inútil a incineração prévia. Seguir o método a partir do segundo parágrafo do ponto 5.1.1.1. Pode ser dispensada a evaporação em presença de ácido fluorídrico.
6. Cálculo dos resultados
Calcular a concentração do oligoelemento na solução a analisar, por meio de uma curva de calibração, e exprimir o resultado em mg de oligoelemento por kg de amostra (ppm).
7. Repetibilidade
A diferença entre os resultados de duas determinações paralelas efectuadas com a mesma amostra pelo mesmo analista não deve exceder:
|
— |
5 mg/kg, em valor absoluto, para teores do oligoelemento em causa inferiores a 50 mg/kg, |
|
— |
10 % do maior dos valores, para teores entre 50 e 100 mg/kg, |
|
— |
10 mg/kg, em valor absoluto, para teores entre 100 e 200 mg/kg, |
|
— |
5 % do maior dos valores, para teores superiores a 200 mg/kg. |
8. Observações
A presença de grandes quantidades de fosfatos poderá interferir com a determinação do ferro, manganês e zinco. Esta interferência deve ser corrigida por adição de uma solução de cloreto de lantânio (3.11). Se, no entanto, a amostra apresentar uma razão ponderal [(Ca + Mg)/P] > 2, pode ser dispensada a adição da solução de cloreto de lantânio (3.11) à solução a analisar e às soluções de calibração.
(D) DETERMINAÇÃO DA HALOFUGINONA
Bromidrato de DL-trans-7-bromo-6-cloro-3-[3-(3-hidroxi-2-piperidil)acetonil]-quinazolin-4(3H)-ona
1. Objecto e âmbito de aplicação
O presente método destina-se à determinação da halofuginona em alimentos para animais. O limite de quantificação é 1 mg/kg.
2. Princípio
Após tratamento com água quente, a halofuginona é extraída com acetato de etilo sob a forma de base livre e posteriormente transferida por partição, sob a forma de cloridrato, para uma solução aquosa de ácido acético. O extracto é purificado por cromatografia de troca iónica, sendo o teor de halofuginona determinado por cromatografia líquida de alta resolução (HPLC) de fase reversa, com um detector de ultravioleta.
3. Reagentes
|
3.1. |
Acetonitrilo de qualidade equivalente para HPLC. |
|
3.2. |
Resina Amberlite XAD-2. |
|
3.3. |
Acetato de amónio. |
|
3.4. |
Acetato de etilo. |
|
3.5. |
Ácido acético glacial. |
|
3.6. |
Halofuginona, substância-padrão (bromidrato de DL-trans-7-bromo-6-cloro-3-[3-(3-hidroxi-2-piperidil)acetonil]-quinazolin-4-(3H)-ona, E 764). |
|
3.6.1. |
. Pesar, com uma aproximação de 0,1 mg, 50 mg de halofuginona (3.6) num balão volumétrico de 500 ml. Dissolver com solução-tampão de acetato de amónio (3.18), perfazer o volume com a mesma solução e agitar. Esta solução mantém-se estável durante três semanas, a 5 oC, na ausência de luz. |
|
3.6.2. |
Transferir, respectivamente, 1,0, 2,0, 3,0, 4,0 e 6,0 ml de solução-mãe padrão de halofuginona (3.6.1) para uma série de balões volumétricos de 100 ml. Completar até ao traço com a fase móvel (3.21) e agitar. As soluções resultantes possuem concentrações de halofuginona de 1,0, 2,0, 3,0, 4,0 e 6,0 μg/ml, respectivamente. Estas soluções têm que ser preparadas antes de cada utilização. |
|
3.7. |
Ácido clorídrico, ρ20 = 1,16 g/ml, aproximadamente |
|
3.8. |
Metanol. |
|
3.9. |
Nitrato de prata. |
|
3.10. |
Ascorbato de sódio. |
|
3.11. |
Carbonato de sódio. |
|
3.12. |
Cloreto de sódio. |
|
3.13. |
EDTA (sal dissódico do ácido etilenodiaminotetracético). |
|
3.14. |
Água de qualidade equivalente para HPLC. |
|
3.15. |
Solução de carbonato de sódio 10 g/100 ml. |
|
3.16. |
Solução de carbonato de sódio saturada com cloreto de sódio, 5 g/100 ml Dissolver 50 g de carbonato de sódio (3.11) em água, diluir para 1 litro e adicionar cloreto de sódio (3.12) até obter uma solução saturada. |
|
3.17. |
Ácido clorídrico, aprox. 0,1 mol/l Diluir 10 ml de HCl (3.7) em água e perfazer o volume de 1 litro. |
|
3.18. |
Solução-tampão de acetato de amónio, aprox. 0,25 mol/l. Dissolver 19,3 g de acetato de amónio (3.3) e 30 ml de ácido acético (3.5) em água (3.14), perfazendo o volume de 1 litro. |
|
3.19. |
Preparação da resina Amberlite XAD-2 Lavar com água uma quantidade adequada de Amberlite (3.2), até à remoção completa dos iões cloreto, verificada através da realização do teste com nitrato de prata (3.20) com a fase aquosa recolhida. Seguidamente, lavar a resina com 50 ml de metanol (3.8), rejeitar o metanol e armazenar a resina numa nova porção de metanol fresco. |
|
3.20. |
Solução de nitrato de prata, aprox. 0,1 mol/l. Dissolver 0,17 g de nitrato de prata (3.9) em 10 ml de água. |
|
3.21. |
Fase móvel para HPLC Misturar 500 ml de acetonitrilo (3.1) com 300 ml de solução-tampão de acetato de amónio (3.18) e 1 200 ml de água (3.14). Ajustar o pH a 4,3 com ácido acético (3.5). Filtrar com um filtro de 0,22 μm (4.8) e desgaseificar a solução (por exemplo por aplicação de ultra-sons durante 10 minutos). A solução permanece estável durante um mês, se for armazenada na ausência de luz e num recipiente fechado. |
4. Equipamento
|
4.1. |
Banho de ultra-sons. |
|
4.2. |
Evaporador rotativo. |
|
4.3. |
Centrífuga. |
|
4.4. |
Aparelho para HPLC com detector de ultravioleta de comprimento de onda variável ou detector de díodos. |
|
4.4.1. |
Coluna para cromatografia líquida, de 300 mm × 4 mm, com enchimento C18 de 10 μm, ou equivalente. |
|
4.5. |
Coluna de vidro (300 mm × 10 mm), equipada com um filtro de vidro sinterizado e torneira. |
|
4.6. |
Filtros de fibra de vidro de 150 mm de diâmetro. |
|
4.7. |
Filtros de membrana, porosidade 0,45 μm. |
|
4.8. |
Filtros de membrana, porosidade 0,22 μm. |
5. Procedimento
|
Nota: |
Sob a forma de base livre, a halofuginona é instável em soluções alcalinas e em acetato de etilo, não devendo permanecer neste último durante mais de 30 minutos. |
5.1. Generalidades
|
5.1.1. |
De modo a comprovar a ausência de halofuginona e outras substâncias susceptíveis de interferir no processo, deve proceder-se à análise de uma amostra em branco de um alimento para animais (ensaio em branco). |
|
5.1.2. |
Deve efectuar-se um ensaio de recuperação utilizando a amostra do ensaio em branco, fortificada com uma quantidade conhecida de halofuginona semelhante à quantidade presente na amostra. Para obter uma fortificação a um nível de 3 mg/kg, adicionam-se 300 μl de solução-mãe padrão (3.6.1) a 10 g de amostra para o ensaio em branco. Misturar, deixar em repouso 10 minutos e proceder à extracção (5.2).
|
5.2. Extracção
Pesar, com uma aproximação de 0,1 g, 10 g da amostra previamente preparada para um tubo de centrífuga de 200 ml. Adicionar 0,5 g de ascorbato de sódio (3.10), 0,5 g de EDTA (3.13) e 20 ml de água, agitando. Colocar o tubo num banho de água a 80 oC, durante 5 minutos. Após arrefecimento à temperatura ambiente, adicionar 20 ml de solução de carbonato de sódio (3.15), agitando. Adicionar imediatamente 100 ml de acetato de etilo (3.4) e agitar manualmente com vigor durante 15 segundos. Colocar o tubo durante três minutos num banho de ultra-sons (4.1), desenroscando a rolha. Centrifugar durante dois minutos e decantar a fase de acetato de etilo, através de um filtro de fibra de vidro (4.6), para uma ampola de decantação de 500 ml. Repetir a extracção da amostra com uma segunda porção de 100 ml de acetato de etilo. Lavar os extractos combinados durante um minuto com 50 ml de solução de carbonato de sódio saturada com cloreto de sódio (3.16), rejeitando a fase aquosa.
Extrair a fase orgânica durante um minuto com 50 ml de ácido clorídrico (3.17). Transferir a camada inferior de ácido para uma ampola de decantação de 250 ml. Extrair novamente a fase orgânica durante 1,5 minutos com uma nova porção de 50 ml de ácido clorídrico, combinando este extracto com o primeiro. Lavar os extractos ácidos combinados por agitação com 10 ml de acetato de etilo (3.4), durante cerca de 10 segundos.
Transferir quantitativamente a fase aquosa para um balão de fundo redondo de 250 ml, rejeitando a fase orgânica. Evaporar o acetato de etilo remanescente da solução ácida, por recurso a um evaporador rotativo (4.2). A temperatura do banho de água não deve exceder 40 oC. A uma pressão aproximada de 25 mbar, todo o acetato de etilo residual deverá ser removido em 5 minutos, a 38 oC.
5.3. Purificação
5.3.1.
Deve preparar-se uma coluna de XAD-2 para cada extracto de amostra. Com o auxílio de metanol (3.8), transferir para uma coluna de vidro (4.5) 10 g de Amberlite preparada (3.19). Colocar um pequeno tampão de lã de vidro no topo do leito de resina. Remover o metanol da coluna e lavar a resina com 100 ml de água, suspendendo o fluxo quando o nível de líquido atingir o topo do leito de resina. Deixar a coluna estabilizar durante 10 minutos antes da utilização. Não permitir, em caso algum, a secagem da coluna.
5.3.2.
Transferir quantitativamente o extracto (5.2) para o topo da coluna de Amberlite preparada (5.3.1) e eluir, desprezando o eluato. O caudal de eluição não deverá exceder 20 ml/min. Lavar o balão de fundo redondo com 20 ml de ácido clorídrico (3.17), aproveitando a solução residual para lavar a coluna de resina. Remover quaisquer resíduos de solução ácida por recurso a uma corrente de ar. Rejeitar os resíduos de lavagem. Adicionar à coluna 100 ml de metanol (3.8) e deixar eluir 5 a 10 ml, recolhendo o eluato num balão de fundo redondo de 250 ml. Estabilizar a resina com o metanol remanescente durante 10 minutos e continuar a eluição a um caudal não superior a 20 ml/min, recolhendo o eluato no mesmo balão de fundo redondo. Evaporar o metanol no evaporador rotativo (4.2); a temperatura do banho de água não deverá exceder 40 oC. Com o auxílio da fase móvel (3.21), transferir quantitativamente o resíduo para um balão volumétrico de 10 ml. Perfazer o volume com fase móvel e agitar. Filtrar uma alíquota num filtro de membrana (4.7), destinando esta solução à análise por HPLC (5.4).
5.4. Análise por HPLC
5.4.1.
As condições a seguir especificadas são-no a titulo indicativo. Poderão escolher-se outras condições, desde que produzam resultados equivalentes.
Coluna para cromatografia líquida (4.4.1)
Fase móvel para HPLC (3.21)
Caudal: 1,5 a 2 ml/minuto
Comprimento de onda de detecção: 243 nm
Volume injectado: 40 to 100 μl.
Verificar a estabilidade do sistema cromatográfico mediante a injecção repetida da solução de calibração (3.6.2), que contém 3,0 μg/ml de halofuginona, até obter picos com alturas (ou áreas) e tempos de retenção reprodutíveis.
5.4.2.
Injectar várias vezes cada solução de calibração (3.6.2) e medir a altura (ou área) dos picos correspondentes a cada concentração. Traçar uma curva de calibração, colocando a altura (ou área) média dos picos das soluções de calibração em ordenadas e as concentrações correspondentes, expressas em μg/ml, em abcissas.
5.4.3.
Injectar várias vezes o extracto de amostra (5.3.2), utilizando o mesmo volume utilizado para as soluções de calibração, e determinar a altura (ou área) média dos picos correspondentes à halofuginona.
6. Cálculo dos resultados
A partir da altura (ou área) média dos picos da halofuginona da solução de amostra, determinar a concentração desta em μg/ml, com base na curva de calibração (5.4.2).
O teor de halofuginona da amostra, w, expresso em mg/kg, é dado pela fórmula:

em que:
|
c |
= |
concentração de halofuginona da solução de amostra, expressa em μg/ml, |
|
m |
= |
massa da toma de ensaio, expressa em gramas. |
7. Validação dos resultados
7.1. Identidade
A identidade do analito pode ser confirmada por co-cromatografia ou por recurso a um detector de díodos, comparando os espectros do extracto de amostra e da solução de calibração (3.6.2) que contém 6,0 μg/ml de halofuginona.
7.1.1.
Fortificar um extracto de amostra com a adição de uma quantidade adequada de uma solução de calibração (3.6.2). A quantidade de halofuginona adicionada deve ser idêntica à quantidade de halofuginona presumida no extracto de amostra.
Apenas se deverá verificar um aumento da altura do pico correspondente à halofuginona, atendendo à quantidade adicionada e à diluição do extracto. A largura do pico a meia altura não poderá diferir mais de 10 % da largura inicial.
7.1.2.
Os resultados são avaliados de acordo com os seguintes critérios:
|
a) |
O comprimento de onda de absorção máxima dos espectros da amostra e do padrão, registado no máximo do pico cromatográfico, deve ser o mesmo, com uma margem de variação dependente da resolução do sistema de detecção. No caso de um detector de díodos, esta margem é, em geral, de ± 2 nm; |
|
b) |
Entre 225 e 300 nm, os espectros da amostra e do padrão, registados no máximo dos picos cromatográficos, não devem diferir entre si no que respeita às zonas do espectro que apresentem uma absorvância relativa compreendida entre 10 e 100 %. Este critério considera-se satisfeito se estiverem presentes os mesmos máximos e a diferença entre ambos os espectros não exceder, em caso algum, 15 % da absorvância do analito-padrão; |
|
c) |
Entre 225 e 300 nm, os espectros na curva ascendente, no máximo e na curva descendente do pico do extracto da amostra não devem diferir entre si nas partes do espectro que apresentem uma absorvância relativa compreendida entre 10 % e 100 %. Este critério considera-se satisfeito se estiverem presentes os mesmos máximos e a diferença entre os espectros não exceder, em caso algum, 15 % da absorvância do espectro no máximo do pico. |
Se algum destes critérios não for satisfeito, a presença do analito não pode ser confirmada.
7.2. Repetibilidade
A diferença entre os resultados de duas determinações paralelas efectuadas com a mesma amostra não deve exceder 0,5 mg/kg, para teores de halofuginona até 3 mg/kg.
7.3. Recuperação
A taxa de recuperação da amostra em branco fortificada deve ser de, pelo menos, 80 %.
8. Resultados de um estudo interlaboratorial
Num estudo interlaboratorial (5), procedeu-se à análise de três amostras por oito laboratórios.
Resultados
|
|
Amostra A (branco) aquando da recepção |
Amostra B (farinha) |
Amostra C (granulado) |
||
|
|
|
aquando da recepção |
decorridos dois meses |
aquando da recepção |
decorridos dois meses |
|
Média [mg/kg] |
ND |
2,80 |
2,42 |
2,89 |
2,45 |
|
SR [mg/kg] |
— |
0,45 |
0,43 |
0,40 |
0,42 |
|
CVR [ %] |
— |
16 |
18 |
14 |
17 |
|
Rec. [ %] |
|
86 |
74 |
88 |
75 |
|
ND |
= |
não detectado |
|
SR |
= |
desvio-padrão da reprodutibilidade |
|
CVR |
= |
coeficiente de variação da reprodutibilidade ( %) |
|
Rec. |
= |
recuperação ( %) |
E. DETERMINAÇÃO DA ROBENIDINA
Cloridrato de 1,3-bis[(4-clorobenzilideno)amino]guanidina
1. Objecto e âmbito de aplicação
Este método destina-se à determinação da robenidina em alimentos para animais. O limite de quantificação é 5 mg/kg.
2. Princípio
A amostra é extraída com metanol acidificado. O extracto é seco, sendo uma alíquota submetida a purificação numa coluna de óxido de alumínio. A robenidina é eluída da coluna com metanol, concentrada e posteriormente diluída com fase móvel para um volume adequado. O teor de robenidina é determinado por cromatografia líquida de alta resolução (HPLC) de fase reversa, utilizando um detector de ultravioleta.
3. Reagentes
|
3.1. |
Metanol |
|
3.2. |
Metanol acidificado Transferir 4,0 ml de ácido clorídrico (ρ 20 = 1,18 g/ml) para um balão volumétrico de 500 ml, completar até ao traço com metanol (3.1) e agitar. A solução deve ser preparada antes de cada uso. |
|
3.3. |
Acetonitrilo de qualidade equivalente para HPLC |
|
3.4. |
Peneiro molecular Tipo 3A, partículas 8-12 mesh (partículas de aluminossilicato cristalino com 1,6-2,5 mm de diâmetro e poros com 0,3 mm de diâmetro). |
|
3.5. |
Óxido de alumínio ácido, de grau de actividade I, para cromatografia em coluna Transferir 100 g de óxido de alumínio para um recipiente adequado e adicionar 2,0 ml de água. Tapar e agitar durante cerca de 20 minutos. Guardar num recipiente bem rolhado. |
|
3.6. |
Solução de di-hidrogenofosfato de potássio 0,025 mol/l Dissolver, num balão volumétrico de 1 000 ml, 3,40 g de di-hidrogenofosfato de potássio em água de qualidade para HPLC, perfazer o volume e agitar. |
|
3.7. |
Solução de hidrogenofosfato dissódico, c= 0,025 mol/l Dissolver, num balão volumétrico de 1 000 ml, 3,55 g de hidrogenofosfato dissódico anidro (ou 4,45 g de hidrogenofosfato dissódico di-hidratado, ou ainda 8,95 g de hidrogenofosfato dissódico dodeca-hidratado) em água de qualidade equivalente para HPLC, completar até ao traço e agitar. |
|
3.8. |
Fase móvel para HPLC Misturar os seguintes reagentes:
Filtrar com um filtro de 0,22 μm (4.6) e desgaseificar a solução (por exemplo por aplicação de ultra-sons durante 10 minutos). |
|
3.9. |
Substância-padrão Robenidina pura: cloridrato de 1,3-bis[(4-clorobenzilideno)amino]guanidina. |
|
3.9.1. |
Pesar, com uma aproximação de 0,1 mg, 30 mg de robenidina padrão (3.9). Dissolver em metanol acidificado (3.2) num balão volumétrico de 100 ml, perfazer o volume com o mesmo solvente e agitar. Envolver o balão em folha de alumínio e armazenar ao abrigo da luz. |
|
3.9.2. |
Transferir 10,0 ml de solução-mãe padrão (3.9.1) para um balão volumétrico de 250 ml, completar o volume com fase móvel (3.8) e agitar. Envolver o balão em folha de alumínio e armazenar ao abrigo da luz. |
|
3.9.3. |
Transferir, respectivamente, 5,0, 10,0, 15,0, 20,0 e 25,0 ml de solução-padrão intermédia (3.9.2) para balões volumétricos de 50 ml. Perfazer o volume com a fase móvel (3.8) e agitar. As soluções obtidas contêm, respectivamente, 1,2, 2,4, 3,6, 4,8 e 6,0 μg/ml de robenidina e devem ser preparadas imediatamente antes da utilização. |
|
3.10. |
Água de qualidade equivalente para HPLC. |
4. Equipamento
|
4.1. |
Coluna de vidro Coluna de vidro ambarizado, equipada com torneira e reservatório de cerca de 150 ml, possuindo 10-15 mm de diâmetro interno e 250 mm de comprimento. |
|
4.2. |
Agitador mecânico ou magnético. |
|
4.3. |
Evaporador rotativo. |
|
4.4. |
Aparelho para HPLC com detector de ultravioleta de comprimento de onda variável ou detector de díodos adequado para determinações na gama de comprimentos de onda de 250 a 400 nm. |
|
4.4.1. |
Coluna para cromatografia líquida: 300 mm x 4 mm, com enchimento C18 de 10 μm ou equivalente. |
|
4.5. |
Filtro de fibra de vidro (Whatman GF/A ou equivalente) |
|
4.6. |
Filtros de membrana, porosidade 0,22 μm. |
|
4.7. |
Filtros de membrana, porosidade 0,45 μm. |
5. Procedimento
|
Nota: |
A robenidina é fotossensível, devendo utilizar-se material de vidro ambarizado para todas as operações. |
5.1. Generalidades
|
5.1.1. |
De modo a comprovar a ausência de robenidina e outras substâncias susceptíveis de interferir no processo, deve proceder-se à análise de uma amostra em branco de um alimento para animais (ensaio em branco). |
|
5.1.2. |
Deve efectuar-se um ensaio de recuperação utilizando a amostra do ensaio em branco (5.1.1), fortificada com uma quantidade conhecida de robenidina semelhante à quantidade presente na amostra. Para obter uma fortificação a um nível de 60 mg/kg, transferir 3,0 ml de solução-mãe padrão (3.9.1) para um erlenmeyer de 250 ml. Evaporar a solução sob uma corrente de azoto até obter um volume aproximado de 0,5 ml. Adicionar 15 g de amostra para o ensaio em branco, misturar e esperar 10 minutos antes de iniciar a extracção (5.2).
|
5.2. Extracção
Pesar, com uma aproximação de 0,01 g, cerca de 15 g de amostra preparada. Transferi-la para um erlenmeyer de 250 ml e adicionar 100,0 ml de metanol acidificado (3.2). Tapar e agitar durante uma hora no agitador (4.2). Filtrar a solução num filtro de fibra de vidro (4.5) e recolher a totalidade do filtrado num erlenmeyer de 150 ml. Adicionar 7,5 g de peneiro molecular (3.4), tapar e agitar durante cinco minutos. Filtrar imediatamente num filtro de fibra de vidro. Guardar esta solução para as operações de purificação (5.3).
5.3. Purificação
5.3.1.
Colocar um pequeno tampão de fibra de vidro na extremidade inferior da coluna de vidro (4.1), comprimindo-o com o auxílio de uma vareta de vidro. Pesar 11,0 g de óxido de alumínio preparado de acordo com o descrito no ponto 3.5 e transferi-lo para a coluna. Devem tomar-se precauções no sentido de minimizar a exposição ao ar no decurso destas operações. Bater levemente na extremidade inferior da coluna para que o óxido de alumínio assente.
5.3.2.
Pipetar para a coluna 5,0 ml de extracto de amostra preparado de acordo com o ponto 5.2. Colocar a ponta da pipeta junto da parede da coluna e esperar que a solução seja absorvida pelo óxido de alumínio. Eluir a robenidina da coluna por recurso a 100 ml de metanol (3.1), a um caudal de 2-3 ml/minuto, recolhendo o eluato num balão de fundo redondo de 250 ml. Evaporar até à secura a solução de metanol, sob pressão reduzida, a 40oC, com o auxílio de um evaporador rotativo (4.3). Dissolver o resíduo em 3-4 ml de fase móvel (3.8) e transferir quantitativamente para um balão volumétrico de 10 ml. Lavar o balão de fundo redondo com diversas porções de 1-2 ml de fase móvel, transferindo as mesmas para o balão volumétrico. Perfazer o volume com o mesmo solvente e agitar. Filtrar uma alíquota com um filtro de membrana de 0,45 μm (4.7). Guardar esta solução para a análise por HPLC (5.4).
5.4. Análise por HPLC
5.4.1.
As condições a seguir especificadas são-no a titulo indicativo. Poderão escolher-se outras condições, desde que produzam resultados equivalentes.
Coluna para cromatografia líquida (4.4.1)
Fase móvel para HPLC (3.8)
Caudal: 1,5 a 2 ml/minuto
Comprimento de onda de detecção: 317 nm
Volume injectado: 20 a 50 μl
Verificar a estabilidade do sistema cromatográfico mediante a injecção repetida da solução de calibração (3.9.3), que contém 3,6 μg/ml de robenidina, até obter picos com alturas (ou áreas) e tempos de retenção reprodutíveis.
5.4.2.
Injectar várias vezes cada solução de calibração (3.9.3) e medir a altura (ou área) dos picos correspondentes a cada concentração. Traçar uma curva de calibração, colocando a altura (ou área) média dos picos das soluções de calibração em ordenadas e as concentrações correspondentes, expressas em μg/ml, em abcissas.
5.4.3.
Injectar várias vezes o extracto de amostra (5.3.2), utilizando o mesmo volume utilizado para as soluções de calibração, e determinar a altura (ou área) média dos picos correspondentes à robenidina.
6. Cálculo dos resultados
A partir da altura (ou área) média dos picos da robenidina da solução de amostra, determinar a concentração desta em μg/ml, com base na curva de calibração (5.4.2).
O teor de robenidina da amostra, w, expresso em mg/kg, é dado pela fórmula:
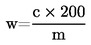
em que:
|
c |
= |
concentração de robenidina da solução de amostra, expressa em μg/ml |
|
m |
= |
massa da toma de ensaio, expressa em gramas. |
7. Validação dos resultados
7.1. Identidade
A identidade do analito pode ser confirmada por co-cromatografia ou por recurso a um detector de díodos, comparando os espectros do extracto de amostra e da solução de calibração (3.9.3) que contém 6 μg/ml de robenidina.
7.1.1.
Fortificar um extracto de amostra com a adição de uma quantidade adequada de solução de calibração (3.9.3). A quantidade de robenidina adicionada deve ser idêntica à quantidade de robenidina presumida no extracto de amostra.
Apenas se deverá verificar um aumento da altura do pico correspondente à robenidina, atendendo à quantidade adicionada e à diluição do extracto. A largura do pico a meia altura não poderá diferir mais de 10 % da largura inicial.
7.1.2.
Os resultados são avaliados de acordo com os seguintes critérios:
|
a) |
O comprimento de onda de absorção máxima dos espectros da amostra e do padrão, registado no máximo do pico cromatográfico, deve ser o mesmo, com uma margem de variação dependente da resolução do sistema de detecção. No caso de um detector de díodos, esta margem é, em geral, de ± 2 nm. |
|
b) |
Entre 250 e 400 nm, os espectros da amostra e do padrão, registados no máximo dos picos cromatográficos, não devem diferir entre si no que respeita às zonas do espectro que apresentem uma absorvância relativa compreendida entre 10 % e 100 %. Este critério considera-se satisfeito se estiverem presentes os mesmos máximos e a diferença entre ambos os espectros não exceder, em caso algum, 15 % da absorvância do analito-padrão; |
|
c) |
Entre 250 e 400 nm, os espectros na curva ascendente, no máximo e na curva descendente do pico do extracto da amostra não diferem entre si nas partes do espectro compreendidas entre 10 % e 100 % de absorvância relativa. Este critério considera-se satisfeito se estiverem presentes os mesmos máximos e a diferença entre os espectros não exceder, em caso algum, 15 % da absorvância do espectro no máximo do pico. |
Se algum destes critérios não for satisfeito, a presença do analito não pode ser confirmada.
7.2. Repetibilidade
A diferença entre os resultados de duas determinações paralelas efectuadas com a mesma amostra não deve exceder 10 % do maior dos valores, para teores de robenidina superiores a 15 mg/kg.
7.3. Recuperação
A taxa de recuperação de uma amostra em branco fortificada deve ser de, pelo menos, 85 %.
8. Resultados de um estudo interlaboratorial
Num estudo interlaboratorial promovido a nível comunitário, procedeu-se à análise por 12 laboratórios de quatro amostras de alimentos para aves de capoeira e coelhos, sob a forma de farinha e granulado. Para cada amostra, foram efectuadas análises em duplicado. Os resultados obtidos foram os seguintes:
|
|
Aves de capoeira |
Coelhos |
||
|
|
Farinha |
Granulado |
Farinha |
Granulado |
|
Média [mg/kg] |
27,00 |
27,99 |
43,6 |
40,1 |
|
sr [mg/kg] |
1,46 |
1,26 |
1,44 |
1,66 |
|
CVr [ %] |
5,4 |
4,5 |
3,3 |
4,1 |
|
SR [mg/kg] |
4,36 |
3,36 |
4,61 |
3,91 |
|
CVR [ %] |
16,1 |
12,0 |
10,6 |
9,7 |
|
Recuperação [ %] |
90,0 |
93,3 |
87,2 |
80,2 |
|
sr |
= |
desvio-padrão da repetibilidade |
|
CVr |
= |
coeficiente de variação da repetibilidade ( %) |
|
SR |
= |
desvio-padrão da reprodutibilidade |
|
CVR |
= |
coeficiente de variação da reprodutibilidade ( %) |
(F) DETERMINAÇÃO DO DICLAZURIL
(+)-4-clorofenil[2,6-dicloro-4-(2,3,4,5-tetra-hidro-3,5-dioxo-1,2,4-triazin-2-il)fenil]-acetonitrilo
1. Objecto e âmbito de aplicação
Este método permite determinar o teor de diclazuril em alimentos para animais e pré-misturas. O limite de detecção é de 0,1 mg/kg; o limite de quantificação é de 0,5 mg/kg.
2. Princípio
Depois de adicionado um padrão interno, submete-se a amostra a uma operação de extracção com metanol acidificado. No caso dos alimentos para animais, purifica-se uma alíquota do extracto num cartucho de extracção com fase sólida C18, elui-se o diclazuril do cartucho com uma mistura de metanol acidificado e água, evapora-se e dissolve-se o resíduo em DMF/água. No caso das pré-misturas, evapora-se o extracto e dissolve-se o resíduo em DMF/água. O teor de diclazuril é determinado por cromatografia líquida de alta resolução (HPLC) de fase reversa com gradiente ternário, com um detector de UV.
3. Reagentes
|
3.1. |
Água de qualidade equivalente para HPLC. |
|
3.2. |
Acetato de amónio. |
|
3.3. |
Hidrogenossulfato de tetrabutilamónio (TBHS). |
|
3.4. |
Acetonitrilo de qualidade equivalente para HPLC. |
|
3.5. |
Metanol de qualidade equivalente para HPLC. |
|
3.6. |
N, N-Dimetilformamida (DMF). |
|
3.7. |
Ácido clorídrico, ρ20 = 1,19 g/ml. |
|
3.8. |
Substância padrão: diclazuril II-24, (+)-4-clorofenil[2,6-dicloro-4-(2,3,4,5-tetra-hidro-3,5-dioxo-1,2,4-triazin-2-il)fenil]-acetonitrilo de grau de pureza garantido, E 771. |
|
3.8.1. |
. Num balão volumétrico de 50 ml, pesar, com uma aproximação de 0,1 mg, 25 mg de diclazuril-padrão (3.8). Dissolver em DMF (3.6), completar o volume com DMF (3.6) e homogeneizar. Envolver o balão com folha de alumínio ou utilizar material de vidro ambarizado e guardar no frigorífico. A temperaturas não superiores a 4 oC, a solução mantém- -se estável durante um mês. |
|
3.8.2. |
. Transferir 5,00 ml da solução-mãe padrão (3.8.1) para um balão volumétrico de 50 ml, completar o volume com DMF (3.6) e homogeneizar. Envolver o balão com folha de alumínio ou utilizar material de vidro ambarizado e guardar no frigorífico. A temperaturas não superiores a 4 oC a solução mantém-se estável durante um mês. |
|
3.9. |
Padrão interno: a substância utilizada é o 2,6-dicloro-α-(4-clorofenil)-4-[4,5-di-hidro-3,5-dioxo1,2,4-triazin-2(3H)-il]-α-metilbenzenoacetonitrilo. |
|
3.9.1. |
. Num balão volumétrico de 50 ml, pesar, com uma aproximação de 0,1 mg, 25 mg de padrão interno (3.9). Dissolver em DMF (3.6), completar o volume com DMF (3.6) e homogeneizar. Envolver o balão com folha de alumínio ou utilizar material de vidro ambarizado e guardar no frigorífico. A temperaturas não superiores a 4 oC, a solução mantém-se estável durante um mês. |
|
3.9.2. |
. Transferir 5,00 ml da solução-mãe de padrão interno (3.9.1) para um balão volumétrico de 50 ml, completar o volume com DMF (3.6) e homogeneizar. Envolver o balão com folha de alumínio ou utilizar material de vidro ambarizado e guardar no frigorífico. A temperaturas não superiores a 4 oC, a solução mantém-se estável durante um mês. |
|
3.9.3. |
(p = teor nominal, em mg/kg, de diclazuril na pré-mistura) Num balão volumétrico de 100 ml, pesar, com uma aproximação de 0,1 mg, p/10 mg do padrão interno, dissolver em DMF (3.6) num banho de ultra-sons (4.6), completar o volume com DMF e homogeneizar. Envolver o balão com folha de alumínio ou utilizar material de vidro ambarizado e guardar no frigorífico. A temperaturas não superiores a 4 oC, a solução mantém-se estável durante um mês. |
|
3.10. |
Solução de calibração, 2 μg/ml. Pipetar 2,00 ml da solução-padrão de diclazuril (3.8.2) e 2,00 ml da solução de padrão interno (3.9.2) para um balão volumétrico de 50 ml. Adicionar 16 ml de DMF (3.6), completar o volume com água e homogeneizar. Esta solução é preparada imediatamente antes da sua utilização. |
|
3.11. |
Cartucho de extracção com fase sólida C18, por exemplo Bond Elut; volume: 1 cm3, massa de substância sorvente: 100 mg. |
|
3.12. |
Solvente de extracção: metanol acidificado. Pipetar 5,0 ml de ácido clorídrico (3.7) para um recipiente com 1 000 ml de metanol (3.5) e homogeneizar. |
|
3.13. |
Fase móvel para HPLC. |
|
3.13.1. |
Eluente A: solução de acetato de amónio e hidrogenossulfato de tetrabutilamónio. Dissolver 5 g de acetato de amónio (3.2) e 3,4 g de TBHS (3.3) em 1 000 ml de água (3.1) e homogeneizar. |
|
3.13.2. |
Eluente B: acetonitrilo (3.4). |
|
3.13.3. |
Eluente C: metanol (3.5). |
4. Equipamento
|
4.1. |
Agitador mecânico. |
|
4.2. |
Equipamento para HPLC com gradiente ternário. |
|
4.2.1. |
Coluna para cromatografia líquida com 100 mm x 4,6 mm e enchimento Hypersil ODS de 3 μm, ou equivalente. |
|
4.2.2. |
Detector de UV com regulação do comprimento de onda ou detector de díodos. |
|
4.3. |
Evaporador de vácuo. |
|
4.4. |
Filtros de membrana, porosidade 0,45 μm. |
|
4.5. |
Sistema de vácuo. |
|
4.6. |
Banho de ultra-sons. |
5. Procedimento
5.1. Generalidades
5.1.1.
De modo a comprovar a ausência de diclazuril e outras substâncias susceptíveis de interferir no processo, deve proceder-se à análise de uma amostra em branco de um alimento para animais (ensaio em branco). Essa amostra em branco deve ser de tipo similar ao da amostra em análise e não devem ser detectados diclazuril nem substâncias susceptíveis de interferir no processo.
5.1.2.
Deve efectuar-se um ensaio de recuperação utilizando a amostra do ensaio em branco, fortificada com uma quantidade conhecida de diclazuril semelhante à quantidade presente na amostra. Para obter uma fortificação a um nível de 1 mg/kg, adicionar 0,1 ml da solução-mãe padrão (3.8.1) a 50 g da amostra para o ensaio em branco, misturar bem e deixar em repouso durante 10 minutos, misturando de novo várias vezes antes de prosseguir (5.2).
Caso não se disponha de um alimento para animais para o ensaio em branco de tipo similar ao da amostra (ver 5.1.1), pode determinar-se a recuperação pelo método da adição de padrão. Nesse caso, adiciona-se à amostra a analisar uma quantidade de diclazuril semelhante à que já se encontra presente e analisa-se a amostra assim obtida juntamente com a amostra sem adição. A recuperação é calculada por subtracção.
5.2. Extracção
5.2.1.
Pesar, com uma aproximação de 0,01 g, cerca de 50 g de amostra. Transferir a quantidade pesada para um erlenmeyer de 500 ml, adicionar 1,00 ml da solução de padrão interno (3.9.2) e 200 ml do solvente de extracção (3.12) e tapar. Colocar o erlenmeyer no agitador (4.1) e deixar a agitar de um dia para o outro. Deixar assentar durante 10 minutos. Transferir uma alíquota de 20 ml do líquido sobrenadante para um recipiente de vidro adequado e diluir com 20 ml de água. Verter esta solução num cartucho de extracção (3.11) e forçar a passagem por aplicação de vácuo (4.5). Lavar o cartucho com 25 ml de uma mistura 65+35 (v+v) de solvente de extracção (3.12) e água. Rejeitar as fracções recolhidas e eluir os compostos retidos com 25 ml de uma mistura 80+20 (v+v) de solvente de extracção (3.12) e água. Evaporar esta fracção a 60 oC num evaporador rotativo (4.3), suspendendo a operação logo que seja atingida a secura. Dissolver o resíduo em 1,0 ml de DMF (3.6), adicionar 1,5 ml de água (3.1) e homogeneizar. Filtrar por um filtro de membrana (4.4). Efectuar em seguida a análise por HPLC (5.3).
5.2.2.
Pesar, com uma aproximação de 0,001 g, cerca de 1 g de amostra. Transferir a quantidade pesada para um erlenmeyer de 500 ml, adicionar 1,00 ml da solução de padrão interno (3.9.3) e 200 ml do solvente de extracção (3.12) e tapar. Colocar o erlenmeyer no agitador (4.1) e deixar a agitar de um dia para o outro. Deixar assentar durante 10 minutos. Transferir uma aliquota de 10 000/p ml (p = teor nominal, em mg/kg, de diclazuril na pré-mistura) do líquido sobrenadante para um balão de vidro de fundo redondo de volume adequado. Evaporar a 60 oC, sob pressão reduzida, num evaporador rotativo (4.3), suspendendo a operação logo que seja atingida a secura. Redissolver o resíduo em 10,0 ml de DMF (3.6), adicionar 15,0 ml de água (3.1) e homogeneizar. Efectuar em seguida a análise por HPLC (5.3).
5.3. Análise por HPLC
5.3.1.
As condições a seguir especificadas são-no a título indicativo. Poderão escolher-se outras condições, desde que produzam resultados equivalentes.
|
Coluna para cromatografia líquida (4.21): |
100 mm × 4,6 mm e enchimento Hypersil ODS de 3 μm, ou equivalente |
|
||||||
|
Fase móvel: |
Eluente A (3.13.1): |
solução aquosa de acetato de amónio e hidrogenossulfato de tetrabutilamónio |
||||||
|
|
Eluente B (3.13.2): |
acetonitrilo |
||||||
|
|
Eluente C (3.13.3): |
metanol |
||||||
|
Modo de eluição: |
Lavagem com B durante 10 minutos |
|||||||
|
Caudal: |
1,5-2 ml/minuto |
|
||||||
|
Volume injectado: |
20 μl |
|
||||||
|
Comprimento de onda de detecção: |
280 nm |
|
||||||
Para verificar a estabilidade do sistema cromatográfico, injectar várias vezes a solução de calibração (3.10), de concentração 2,0 μg/ml, até se obterem alturas de pico e tempos de retenção constantes.
5.3.2.
Injectar várias vezes 20 μl da solução de calibração (3.10) e determinar a altura ou área média dos picos do diclazuril e do padrão interno.
5.3.3.
Injectar várias vezes 20 μl da solução da amostra (5.2.1 ou 5.2.2) e determinar a altura ou área média dos picos do diclazuril e do padrão interno.
6. Cálculo dos resultados
6.1. Alimentos para animais
O teor de diclazuril, w, da amostra, expresso em mg/kg, é dado pela seguinte fórmula:
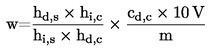 [mg/kg]
[mg/kg]
em que:
|
hd,s |
= |
altura ou área do pico do diclazuril da solução de amostra (5.2.1) |
|
hi,s |
= |
altura ou área do pico do padrão interno da solução de amostra (5.2.1) |
|
hd,c |
= |
altura ou área do pico do diclazuril da solução de calibração (3.10) |
|
hi,c |
= |
altura ou área do pico do padrão interno da solução de calibração (3.10) |
|
cd,c |
= |
concentração de diclazuril, em μg/ml, da solução de calibração (3.10) |
|
m |
= |
massa, em g, da toma analisada |
|
V |
= |
volume, em ml, do extracto de amostra, de acordo com 5.2.1 (ou seja, 2,5 ml). |
6.2. Pré-misturas
O teor de diclazuril, w, da amostra, expresso em mg/kg, é dado pela seguinte fórmula:
 [mg/kg]
[mg/kg]
em que:
|
hd,c |
= |
altura ou área do pico do diclazuril da solução de calibração (3.10) |
|
hi,c |
= |
altura ou área do pico do padrão interno da solução de calibração (3.10) |
|
hd,s |
= |
altura ou área do pico do diclazuril da solução de amostra (5.2.1) |
|
hi,s |
= |
altura ou área do pico do padrão interno da solução de amostra (5.2.1) |
|
cd,c |
= |
concentração de diclazuril, em μg/ml, da solução de calibração (3.10) |
|
m |
= |
massa, em g, da toma analisada |
|
V |
= |
volume, em ml, do extracto de amostra, de acordo com 5.2.2 (isto é, 25 ml) |
|
p |
= |
teor nominal, em mg/kg, de diclazuril na pré-mistura. |
7. Validação dos resultados
7.1. Identidade
A identidade do analito pode ser confirmada por co-cromatografia ou com um detector de díodos que permita comparar os espectros do extracto de amostra (5.2.1 ou 5.2.2) e da solução de calibração (3.10).
7.1.1.
Adiciona-se uma quantidade apropriada da solução de calibração (3.10) a um extracto da amostra (5.2.1 ou 5.2.2). A quantidade de diclazuril adicionada deve ser semelhante à existente no extracto de amostra.
Apenas se deverá verificar um aumento da altura dos picos correspondentes ao diclazuril e ao padrão interno, atendendo à quantidade adicionada e à diluição do extracto. A largura desses picos a meia altura não poderá diferir mais de 10 % da largura inicial do pico do diclazuril e do pico do padrão interno, respectivamente, do extracto de amostra não fortificado.
7.1.2.
Os resultados são avaliados de acordo com os seguintes critérios:
|
a) |
O comprimento de onda de absorção máxima dos espectros da amostra e do padrão, registado no máximo do pico cromatográfico, deve ser o mesmo, com uma margem de variação dependente da resolução do sistema de detecção. No caso de um detector de díodos, essa margem é, em geral, de ± 2 nm; |
|
b) |
Entre 230 e 320 nm, os espectros da amostra e do padrão, registados no máximo dos picos cromatográficos, não devem diferir entre si no que respeita às zonas do espectro que apresentem uma absorvância relativa compreendida entre 10 % e 100 %. Este critério considera-se satisfeito se estiverem presentes os mesmos máximos e a diferença entre ambos os espectros não exceder, em caso algum, 15 % da absorvância do analito-padrão; |
|
c) |
Entre 230 e 320 nm, os espectros na curva ascendente, no máximo e na curva descendente do pico do extracto da amostra não devem diferir entre si nas partes do espectro que apresentem uma absorvância relativa compreendida entre 10 % e 100 %. Este critério considera-se satisfeito se estiverem presentes os mesmos máximos e a diferença entre os espectros não exceder, em caso algum, 15 % da absorvância do espectro no máximo do pico. |
Se algum destes critérios não for satisfeito, a presença do analito não pode ser confirmada.
7.2. Repetibilidade
A diferença entre os resultados de duas determinações paralelas efectuadas com a mesma amostra não deve exceder:
|
— |
para teores de diclazuril compreendidos entre 0,5 mg/kg e 2,5 mg/kg: 30 % do maior dos valores, |
|
— |
para teores de diclazuril compreendidos entre 2,5 mg/kg e 5 mg/kg: 0,75 mg/kg, |
|
— |
para teores de diclazuril superiores a 5 mg/kg: 15 % do maior dos valores. |
7.3. Recuperação
Utilizando uma amostra (em branco) fortificada, a recuperação deve ser, pelo menos, de 80 %.
8. Resultados de um estudo interlaboratorial
Num estudo interlaboratorial, procedeu-se à análise de cinco amostras em 11 laboratórios. Foram analisadas duas amostras de pré-misturas (uma, O 100, misturada com uma matriz orgânica; a outra, A 100, com uma matriz inorgânica) com um teor teórico de diclazuril de 100 mg/kg, e três amostras (L1, Z1 e K1) de misturas alimentares para aves de capoeira de três produtores distintos dos Países Baixos, com um teor teórico de diclazuril de 1 mg/kg. Os laboratórios receberam instruções para analisarem cada amostra uma vez ou em duplicado. (No Journal of AOAC International, Volume 77, n.o 6, 1994, p. 1359-1361 podem ser encontrados elementos mais pormenorizados sobre o estudo interlaboratorial efectuado.) Os resultados obtidos figuram no quadro seguinte:
|
|
Amostra 1 A 100 |
Amostra 2 O 100 |
Amostra 3 L1 |
Amostra 4 Z1 |
Amostra 5 K1 |
|
L |
11 |
11 |
11 |
11 |
6 |
|
n |
19 |
18 |
19 |
19 |
12 |
|
Média |
100,8 |
103,5 |
0,89 |
1,15 |
0,89 |
|
Sr (mg/kg) |
5,88 |
7,64 |
0,15 |
0,02 |
0,03 |
|
CVr ( %) |
5,83 |
7,38 |
17,32 |
1,92 |
3,34 |
|
SR (mg/kg) |
7,59 |
7,64 |
0,17 |
0,11 |
0,12 |
|
CVR ( %) |
7,53 |
7,38 |
18,61 |
9,67 |
13,65 |
|
Teor nominal (mg/kg) |
100 |
100 |
1 |
1 |
1 |
|
L |
= |
número de laboratórios |
|
n |
= |
número de valores individuais |
|
Sr |
= |
desvio-padrão da repetibilidade |
|
CVr |
= |
coeficiente de variação da repetibilidade |
|
SR |
= |
desvio-padrão da reprodutibilidade |
|
CVR |
= |
coeficiente de variação da reprodutibilidade |
9. Observações
É necessário comprovar previamente a linearidade da resposta do diclazuril em toda a gama de concentrações objecto das determinações.
(G) DETERMINAÇÃO DO LASALOCIDO DE SÓDIO
Sal sódico de um ácido monocarboxílico polieterificado produzido por Streptomyces lasaliensis
1. Objecto e âmbito de aplicação
Este método permite determinar o teor de lasalocido de sódio em alimentos para animais e pré-misturas. O limite de detecção é de 5 mg/kg; o limite de quantificação é de 10 mg/kg.
2. Princípio
O lasalocido de sódio é extraído da amostra com metanol acidificado e doseado por cromatografia líquida de alta resolução (HPLC) de fase reversa, com um detector espectrofluorimétrico.
3. Reagentes
|
3.1. |
Di-hidrogenofosfato de potássio (KH2PO4). |
|
3.2. |
Ácido ortofosfórico, w = 85 %. |
|
3.3. |
Solução de ácido ortofosfórico, c = 20 %. Diluir 23,5 ml de ácido ortofosfórico (3.2) para 100 ml, com água. |
|
3.4. |
6-Metil-2-heptilamina (1,5-dimetil-hexilamina), w = 99 %. |
|
3.5. |
Metanol de qualidade equivalente para HPLC. |
|
3.6. |
Ácido clorídrico, d: 1,19 g/ml. |
|
3.7. |
Solução de tampão fosfato, c = 0,01 mol/l. Dissolver 1,36 g de KH2PO4 (3.1) em 500 ml de água (3.11); adicionar 3,5 ml de ácido ortofosfórico (3.2) e 10,0 ml de 6-metil-2-heptilamina (3.4). Ajustar o pH para 4,0 com solução de ácido ortofosfórico (3.3) e diluir para 1 000 ml com água (3.11). |
|
3.8. |
Metanol acidificado Transferir 5,0 ml de ácido clorídrico (3.6) para um balão volumétrico de 1 000 ml, perfazer o volume com metanol (3.5) e agitar. Esta solução deve ser preparada imediatamente antes da sua utilização. |
|
3.9. |
Fase móvel para HPLC: solução de tampão fosfato-metanol 5 + 95 (v/v). Misturar 5 ml de solução de tampão fosfato (3.7) com 95 ml de metanol (3.5). |
|
3.10. |
Lasalocido de sódio-padrão de pureza garantida (C34H53O8Na, sal sódico de um ácido monocarboxílico polieterificado produzido por Streptomyces lasaliensis), E 763. |
|
3.10.1. |
. Num balão volumétrico de 100 ml, pesar, com uma aproximação de 0,1 mg, 50 mg de lasalocido de sódio (3.10) e dissolver em metanol acidificado (3.8). Completar o volume com o mesmo solvente e homogeneizar. A solução deve ser preparada imediatamente antes da utilização. |
|
3.10.2. |
. Pipetar 10,0 ml da solução-mãe padrão (3.10.1) para um balão volumétrico de 100 ml, completar o volume com metanol acidificado (3.8) e homogeneizar. A solução deve ser preparada imediatamente antes da utilização. |
|
3.10.3. |
Transferir 1,0, 2,0, 4,0, 5,0 e 10,0 ml da solução-padrão intermédia (3.10.2) para uma série de balões volumetricos de 50 ml. Perfazer o volume com metanol acidificado (3.8) e homogeneizar. As soluções obtidas contêm 1,0, 2,0, 4,0, 5,0 e 10,0 μg de lasalocido de sódio por ml, respectivamente, e são preparadas imediatamente antes da utilização. |
|
3.11. |
Água de qualidade equivalente para HPLC. |
4. Equipamento
|
4.1. |
Banho de ultra-sons (ou banho de água com agitação) com temperatura regulável. |
|
4.2. |
Filtros de membrana, porosidade 0,45 μm. |
|
4.3. |
Aparelho para HPLC com sistema de injecção com capacidade de injecção de volumes de 20 μl. |
|
4.3.1. |
Coluna para cromatografia líquida de fase reversa com 125 mm x 4 mm e enchimento C18 com granulometria de 5 μm, ou equivalente. |
|
4.3.2. |
Espectrofluorímetro com possibilidade de regulação dos comprimentos de onda de emissão e de excitação. |
5. Procedimento
5.1. Generalidades
5.1.1.
Para a realização do ensaio de recuperação (5.1.2) analisa-se uma amostra em branco de um alimento para animais para comprovar a ausência de lasalocido de sódio e de substâncias susceptíveis de interferir no processo. Essa amostra em branco deve ser de tipo similar ao da amostra em análise e não devem ser detectados lasalocido de sódio nem substâncias susceptíveis de interferir no processo.
5.1.2.
Deve efectuar-se um ensaio de recuperação utilizando a amostra do ensaio em branco, fortificada com uma quantidade conhecida de lasalocido de sódio semelhante à quantidade presente na amostra. Para obter uma fortificação a um nível de 100 mg/kg, transferir 10,0 ml da solução-mãe padrão (3.10.1) para um erlenmeyer de 250 ml e concentrar por evaporação até cerca de 0,5 ml. Adicionar 50 g da amostra em branco, misturar bem e deixar em repouso durante 10 minutos, misturando de novo várias vezes antes de proceder à extracção (5.2).
Caso não se disponha de um alimento para animais para o ensaio em branco de tipo similar ao da amostra (ver 5.1.1), pode determinar-se a recuperação pelo método da adição de padrão. Nesse caso, adiciona-se à amostra a analisar uma quantidade de lasalocido de sódio semelhante à que já se encontra presente e analisa-se a amostra assim obtida juntamente com a amostra sem adição. A recuperação é calculada por subtracção.
5.2. Extracção
5.2.1.
Num erlenmeyer de 250 ml, pesar, com uma aproximação de 0,01 g, 5 a 10 g de amostra. Com uma pipeta, adicionar 100,0 ml de metanol acidificado (3.8). Tapar o erlenmeyer, homogeneizar e colocar no banho de ultra-sons (4.1) a cerca de 40 oC, durante 20 minutos. Retirar o erlenmeyer do banho e arrefecer à temperatura ambiente. Deixar repousar durante cerca de uma hora, até à deposição das matérias em suspensão, e filtrar uma alíquota com um filtro de membrana de 0,45 μm (4.2) para um recipiente adequado. Efectuar em seguida a análise por HPLC (5.3).
5.2.2.
Num erlenmeyer de 250 ml, pesar, com uma aproximação de 0,001 g, cerca de 2 g da pré-mistura. Adicionar 100,0 ml de metanol acidificado (3.8), homogeneizando por agitação. Colocar no banho de ultra-sons (4.1) a cerca de 40 oC, durante 20 minutos. Retirar o erlenmeyer do banho e arrefecer à temperatura ambiente. Diluir até ao traço com metanol acidificado (3.8) e agitar. Deixar repousar durante cerca de uma hora, até à deposição das matérias em suspensão, e filtrar uma alíquota com um filtro de membrana de 0,45 μm (4.2). Diluir um volume adequado do filtrado com metanol acidificado (3.8), de modo a obter uma solução de ensaio com cerca de 4 μg/ml de lasalocido de sódio. Efectuar em seguida a análise por HPLC (5.3).
5.3. Análise por HPLC
5.3.1.
As condições a seguir especificadas são-no a título indicativo. Poderão escolher-se outras condições, desde que produzam resultados equivalentes.
|
Coluna para cromatografia líquida (4.3.1): |
125 mm x 4 mm, enchimento de fase reversa C18 de 5 μm, ou equivalente |
|
Fase móvel (3.9): |
mistura de solução de tampão fosfato (3.7) com metanol (3.5), 5 + 95 (v + v) |
|
Caudal: |
1,2 ml/minuto |
|
Comprimentos de onda de detecção: |
|
|
Excitação: |
310 nm |
|
Emissão: |
419 nm |
|
Volume injectado: |
20 μl |
Para verificar a estabilidade do sistema cromatográfico, injectar várias vezes a solução de calibração (3.10.3) com 4,0 μg/ml até se obterem alturas (ou áreas) de pico e tempos de retenção constantes.
5.3.2.
Injectar várias vezes cada solução de calibração (3.10.3) e determinar a altura ou área média dos picos para cada concentração. Traçar uma curva de calibração, colocando a altura (ou área) média dos picos das soluções de calibração em ordenadas e as concentrações correspondentes, expressas em μg/ml, em abcissas.
5.3.3.
Injectar várias vezes volumes dos extractos de amostra obtidos em 5.2.1 e 5.2.2, idênticos ao volume utilizado para as soluções de calibração e determinar a altura ou a área média dos picos do lasalocido de sódio.
6. Cálculo dos resultados
A partir da altura ou área média dos picos obtidos por injecção da solução da amostra (5.3.3), determinar a concentração de lasalocido de sódio em μg/ml com base na curva de calibração.
6.1. Alimentos para animais
O teor de lasalocido de sódio, w, da amostra, expresso em mg/kg, é dado pela seguinte fórmula:
 [mg/kg]
[mg/kg]
em que:
|
c |
= |
concentração de lasalocido de sódio, em μg/ml, do extracto da amostra (5.2.1) |
|
V1 |
= |
volume, em ml, do extracto, de acordo com 5.2.1 (isto é, 100 ml) |
|
m |
= |
massa, em g, da toma analisada. |
6.2. Pré-misturas
O teor de lasalocido de sódio, w, da amostra, expresso em mg/kg, é dado pela seguinte fórmula:
 [mg/kg]
[mg/kg]
em que:
|
c |
= |
concentração de lasalocido de sódio, em μg/ml, do extracto da amostra (5.2.2) |
|
V2 |
= |
volume, em ml, do extracto, de acordo com 5.2.2 (isto é, 250 ml) |
|
f |
= |
factor de diluição, de acordo com 5.2.2 |
|
m |
= |
massa, em g, da toma analisada. |
7. Validação dos resultados
7.1. Identidade
Os métodos baseados na espectrofluorimetria estão menos sujeitos a interferências que os métodos que utilizam um detector de UV. A identidade do analito pode ser confirmada por co-cromatografia.
7.1.1.
Adiciona-se uma quantidade apropriada da solução de calibração (3.10.3) a um extracto da amostra (5.2.1 ou 5.2.2). A quantidade de lasalocido de sódio adicionada deve ser semelhante à existente no extracto da amostra. Apenas se deverá verificar um aumento da altura do pico correspondente ao lasalocido de sódio, atendendo à quantidade adicionada e à diluição do extracto. A largura do pico a meia altura não poderá diferir mais de 10 % da largura inicial do pico do lasalocido de sódio do extracto da amostra não fortificado.
7.2. Repetibilidade
A diferença entre os resultados de duas determinações paralelas efectuadas com a mesma amostra não deve exceder:
|
— |
para teores de lasalocido de sódio compreendidos entre 30 mg/kg e 100 mg/kg: 15 % do maior dos valores, |
|
— |
para teores de lasalocido de sódio compreendidos entre 100 mg/kg e 200 mg/kg: 15 mg/kg, |
|
— |
para teores de lasalocido de sódio superiores a 200 mg/kg: 7,5 % do maior dos valores. |
7.3. Recuperação
Utilizando uma amostra (em branco) fortificada, a recuperação deve ser pelo menos de 80 %. No respeitante às amostras de pré-misturas fortificadas, a recuperação deve ser pelo menos de 90 %.
8. Resultados de um estudo interlaboratorial
Apresentam-se no quadro seguinte os resultados das análises efectuadas a duas pré-misturas (amostras 1 e 2) e cinco alimentos para animais (amostras 3 a 7) no âmbito de um estudo interlaboratorial (6) com 12 laboratórios. Para cada amostra, foram efectuadas análises em duplicado. Os resultados obtidos figuram no quadro seguinte:
|
|
Amostra 1 Prémistura para frangos |
Amostra 2 Prémistura para perus |
Amostra 3 Granulado para perus |
Amostra 4 Farelos para frangos |
Amostra 5 Alimento para perus |
Amostra 6 Alimento para aves de capoeira A |
Amostra 7 Alimento para aves de capoeira B |
|
L |
12 |
12 |
12 |
12 |
12 |
12 |
12 |
|
N |
23 |
23 |
23 |
23 |
23 |
23 |
23 |
|
Média [mg/kg] |
5 050 |
16 200 |
76,5 |
78,4 |
92,9 |
48,3 |
32,6 |
|
sr [mg/kg] |
107 |
408 |
1,71 |
2,23 |
2,27 |
1,93 |
1,75 |
|
Cvr [ %] |
2,12 |
2,52 |
2,24 |
2,84 |
2,44 |
4,00 |
5,37 |
|
SR [mg/kg] |
286 |
883 |
3,85 |
7,32 |
5,29 |
3,47 |
3,49 |
|
CVR [ %] |
5,66 |
5,45 |
5,03 |
9,34 |
5,69 |
7,18 |
10,70 |
|
Teor nominal [mg/kg] |
5 000 (7) |
16 000 (7) |
80 (7) |
105 (7) |
120 (7) |
50 (8) |
35 (8) |
|
L |
= |
número de laboratórios |
|
n |
= |
número de valores individuais |
|
sr |
= |
desvio-padrão da repetibilidade |
|
SR |
= |
desvio-padrão da reprodutibilidade |
|
CVr |
= |
coeficiente de variação da repetibilidade ( %) |
|
CVR |
= |
coeficiente de variação da reprodutibilidade ( %) |
(1) Organizado pelo grupo de trabalho «Alimentos para animais» do Verband Deutscher Landwirtschaftlicher Untersuchungs-und Forschungsanstalten (VDLUFA).
(2) Organizado pelo grupo de trabalho «Alimentos para animais» do Verband Deutscher Landwirtschaftlicher Untersuchungs-und Forschungsanstalten (VDLUFA).
(3) Poderão ser utilizados outros métodos de mineralização desde que se tenha demonstrado terem resultados semelhantes (por exemplo, a mineralização por microondas sob pressão).
(4) As forragens verdes (frescas ou desidratadas) são susceptíveis de conter grandes quantidades de sílica vegetal que pode reter oligoelementos e deve ser eliminada. As amostras destes alimentos devem ser submetidas ao seguinte procedimento modificado: efectuar a operação do ponto 5.1.1.1 até ao estádio da filtração. Lavar duas vezes em água a ferver o papel de filtro que contém o resíduo insolúvel e colocá-lo num cadinho de quartzo ou platina. Incinerar na mufla (4.1) a uma temperatura inferior a 550 oC até desaparecer completamente toda a substância carbonosa. Deixar arrefecer, juntar algumas gotas de água, deitar em seguida 10 a 15 ml de ácido fluorídrico (3.4) e evaporar até à secura, a aproximadamente 150 oC. Se o resíduo ainda contiver sílica, dissolvê-la em alguns ml de ácido fluorídrico (3.4) e evaporar até à secura. Juntar 5 gotas de ácido sulfúrico (3.5) e aquecer até ao desaparecimento do fumo branco. Juntar 5 ml de ácido clorídrico 6 mol/l (3.2) e aproximadamente 30 ml de água, aquecer, filtrar a solução para um balão volumétrico de 250 ml e completar o volume com água (a concentração em HC1 é aproximadamente de 0,5 mol/l). Prosseguir a operação a partir do ponto 5.1.2.
(5) The Analyst 108, 1983, pp. 1 252-1 256.
(6) Analyst, 1995, 120, 2 175-2 180.
(7) teor declarado pelo fabricante
(8) alimento preparado no laboratório
ANEXO V
MÉTODOS DE ANÁLISE PARA O CONTROLO DE SUBSTÂNCIAS INDESEJÁVEIS NOS ALIMENTOS PARA ANIMAIS
A. DETERMINAÇÃO DO GOSSIPOL LIVRE E TOTAL
1. Objecto e âmbito de aplicação
Este método permite dosear o gossipol livre, o gossipol total e as substâncias quimicamente aparentadas, nas sementes, farinhas e bagaços de algodão, assim como nos alimentos compostos que os contêm em teor superior a 20 mg/kg.
2. Princípio
O gossipol é extraído em presença de 3-aminopropan-1-ol, quer com uma mistura de propan-2-ol e hexano para o doseamento do gossipol livre, quer com dimetilformamida, para o doseamento do gossipol total. Por meio de anilina, procede-se à conversão do gossipol em gossipol-dianilina, cuja densidade óptica é medida a 440 nm.
3. Reagentes
|
3.1. |
Mistura propan-2-ol-hexano: misturar 60 partes de propan-2-ol com 40 partes de n-hexano (v+v). |
|
3.2. |
Solvente A: num balão volumétrico de 1 l, colocar cerca de 500 ml da mistura de propan-2-ol-hexano (3.1), 2 ml de 3-aminopropan-1-ol, 8 ml de ácido acético glacial e 50 ml de água. Perfazer o volume com a mistura de propan-2- -ol-hexano (3.1). Este reagente é estável durante uma semana. |
|
3.3. |
Solvente B: para um balão volumétrico de 100 ml, pipetar 2 ml de 3-aminopropan-1-ol e 10 ml de ácido acético glacial. Deixar arrefecer até à tempertura ambiente e completar o volume com N, N-dimetilformamida. Este reagente é estável durante uma semana. |
|
3.4. |
Anilina: se a densidade óptica do ensaio em branco exceder 0,022, destilar a anilina em pó de zinco, eliminando as primeiras e últimas fracções de 10 % do destilado. Em frasco bem rolhado de vidro escuro e no frigorífico, este reagente conserva-se vários meses. |
|
3.5. |
Solução-padrão A de gossipol: num balão volumérico de 250 ml, colocar 27,9 mg de acetato de gossipol. Dissolver e completar o volume com o solvente A (3.2). Pipetar 50 ml desta solução para um balão volumétrico de 250 ml e completar o volume com o solvente A. A concentração em gossipol desta solução é de 0,02 mg/ml. Deixar repousar durante uma hora à temperatura ambiente, antes de utilizar. |
|
3.6. |
Solução-padrão B de gossipol: num balão volumétrico de 50 ml, colocar 27,9 mg de acetato de gossipol. Dissolver e completar o volume com o solvente B (3.3). A concentração em gossipol desta solução é de 0,5 mg/ml. Conservadas ao abrigo da luz, as soluções-padrão A e B de gossipol são estáveis durante 24 horas. |
4. Equipamento
|
4.1. |
Misturador basculante: cerca de 35 rotações por minuto. |
|
4.2. |
Espectrofotómetro. |
5. Procedimento
5.1. Amostra para ensaio
A dimensão da amostra para ensaio depende do teor presumido de gossipol na amostra. É preferível trabalhar com uma pequena amostra para ensaio e com uma alíquota do filtrado relativamente importante, de maneira a obter uma quantidade de gossipol suficiente para efectuar uma medida fotométrica precisa. Para o doseamento do gossipol livre nas sementes, nas farinhas e nos bagaços de algodão, a amostra para ensaio não deve exceder 1 g; para os alimentos compostos poderá atingir 5 g. Na maioria dos casos, uma alíquota de 10 ml do filtrado é conveniente: deverá conter 50 a 100 μg de gossipol. Para o doseamento do gossipol total, a amostra para ensaio poderá variar de 0,5 a 5 g para que uma alíquota de 2 ml do filtrado contenha 40 a 200 μg de gossipol.
A análise deve ser feita a uma temperatura ambiente próxima de 20 oC.
5.2. Doseamento do gossipol livre
Introduzir a amostra para ensaio num balão de colo esmerilado de 250 ml, com o fundo coberto de vidro moído. Com uma pipeta, adicionar 50 ml de solvente A (3.2) rolhar o balão e misturar durante uma hora no misturador basculante. Filtrar num filtro seco e recolher o filtrado num pequeno balão de colo esmerilado. Durante a filtração, cobrir o funil com um vidro de relógio.
Pipetar alíquotas idênticas de filtrado contendo 50 a 100 μg de gossipol para dois balões volumétricos de 25 ml (A e B). Completar eventualmente o volume para 10 ml com o solvente A (3.2). Em seguida, perfazer o volume de 25 ml do balão A com a mistura de propan-2-ol-hexano (3.1). Esta solução será utilizada como solução de referência para a medição da solução da amostra.
Para dois outros balões volumétricos de 25 ml (balões C e D) pipetar 10 ml do solvente A (3.2). Completar o volume do balão C com a mistura de propan-2-ol-hexano (3.1). Esta solução será utilizada como solução de referência para a medição do ensaio em branco.
A cada um dos balões B e D, adicionar 2 ml de anilina (3.4). Aquecer durante 30 minutos num banho-maria em ebulição para fazer desenvolver a coloração. Arrefecer até à temperatura ambiente, perfazer o volume de 25 ml com a mistura de propan-2-ol-hexano (3.1), homogeneizar e deixar repousar durante uma hora.
Determinar em espectrofotómetro a 440 nm, em células de vidro de 1 cm, a densidade óptica da solução do ensaio em branco (D), por comparação com a solução de referência (C) e a densidade óptica da solução da amostra (B) por comparação com a solução de referência (A).
Subtrair a densidade óptica da solução do ensaio em branco da densidade óptica da solução da amostra (= densidade óptica corrigida). Calcular, a partir deste valor, o teor de gossipol livre, com indicado no ponto 6.
5.3. Doseamento do gossipol total
Introduzir uma amostra para ensaio contendo de 1 a 5 mg de gossipol num balão volumétrico de 50 ml e adicionar 10 ml de solvente B (3.3). Preparar simultaneamente um ensaio em branco, introduzindo 10 ml de solvente B (3.3) noutro balão volumétrico de 50 ml. Aquecer os dois balões durante 30 minutos num banho-maria em ebulição. Arrefecer até à temperatura ambiente e perfazer o volume de cada balão com a mistura de propan-2-ol-hexano (3.1). Homogeneizar e deixar assentar durante 10 a 15 minutos, filtrar em seguida e recolher os filtrados em balões de colo esmerilado.
Para dois balões volumétricos de 25 ml, pipetar 2 ml do filtrado da amostra e para dois outros balões de 25 ml, pipetar 2 ml do filtrado do ensaio em branco. Para um balão de cada série, perfazer o volume de 25 ml com a mistura de propan-2-ol-hexano (3.1). Estas soluções serão utilizadas como soluções de referência.
A cada um dos dois outros balões, adicionar 2 ml de anilina (3.4). Aquecer durante 30 minutos num banho-maria em ebulição para fazer desenvolver a coloração. Arrefecer até à temperatura ambiente, perfazer o volume de 25 ml com a mistura de propan-2-ol-hexano (3.1), homogeneizar e deixar repousar durante uma hora.
Determinar a densidade óptica, como indicado no ponto 5.2 para o gossipol livre. Calcular, a partir deste valor, o teor de gossipol total, como indicado no ponto 6.
6. Cálculo dos resultados
O cálculo dos resultados pode fazer-se quer a partir da densidade óptica específica (6.1) quer por referência a uma curva de calibração (6.2).
6.1. A partir da densidade óptica específica
Nas condições descritas, as densidades ópticas específicas são as seguintes:
|
Gossipol livre: |
|
|
Gossipol total: |
|
O teor de gossipol livre ou total da amostra é dado pela seguinte fórmula:

em que:
|
E |
= |
densidade óptica corrigida, determinada como indicado no ponto 5.2 |
|
p |
= |
massa, em g, da amostra para ensaio |
|
a |
= |
alíquota do filtrado, em ml. |
6.2. A partir de uma curva de calibração
6.2.1.
Preparar duas séries de cinco balões volumétricos de 25 ml. Em cada série, pipetar para os balões, respectivamente, 2,0, 4,0, 6,0, 8,0 e 10,0 ml da solução-padrão A de gossipol (3.5). Completar o volume de 10 ml com o solvente A (3.2). Completar cada série com um balão volumétrico de 25 ml contendo unicamente 10 ml de solvente A (3.2) (ensaio em branco).
Nos balões da primeira série (incluindo o do ensaio em branco), perfazer o volume de 25 ml com a mistura propan-2-ol-hexano (3.1) (série de referência).
A cada balão da segunda série (incluindo o do ensaio em branco) adicionar 2 ml de anilina (3.4). Aquecer durante 30 minutos num banho-maria em ebulição para fazer desenvolver a coloração. Arrefecer até à temperatura ambiente, perfazer o volume de 25 ml com a mistura de propan-2-ol-hexano (3.1), homogeneizar e deixar repousar durante uma hora (série-padrão).
Tal como se indica no ponto 5.2, determinar a densidade óptica das soluções da série-padrão, por comparação com as soluções correspondentes da série de referência. Traçar a curva de calibração representado os valores da densidade óptica em função das quantidades correspondentes de gossipol (em μg).
6.2.2.
Preparar 6 balões volumétricos de 50 ml. No primeiro balão, introduzir 10 ml de solvente B (3.3) e nos outros, respectivamente, 2,0, 4,0, 6,0, 8,0 e 10,0 ml da solução-padrão B de gossipol (3.6). Completar o conteúdo de cada balão para 10 ml com solvente B (3.3). Aquecer durante 30 minutos num banho-maria em ebulição. Arrefecer até à temperatura ambiente, completar o volume com a mistura de propan-2-ol-hexano (3.1) e homogeneizar.
Introduzir 2,0 ml de cada uma destas soluções em, respectivamente, duas séries de seis balões volumétricos de 25 ml. Nos balões da primeira série, perfazer o volume de 25 ml com a mistura propan-2-ol-hexano (3.1) (série de referência).
A cada balão da segunda série adicionar 2 ml de anilina (3.4). Aquecer durante 30 minutos num banho-maria em ebulição. Arrefecer até à temperatura ambiente, perfazer o volume de 25 ml com a mistura de propan-2-ol-hexano (3.1), homogeneizar e deixar repousar durante uma hora (série-padrão).
Tal como se indica no ponto 5.2, determinar a densidade óptica das soluções da série-padrão, por comparação com as soluções correspondentes da série de referência. Traçar a curva de calibração representado os valores da densidade óptica em função das quantidades correspondentes de gossipol (em μg).
6.3. Repetibilidade
A diferença entre os resultados de duas determinações paralelas efectuadas com a mesma amostra não deve exceder:
|
— |
15 % do maior dos valores, para os teores de gossipol inferiores a 500 mg/kg, |
|
— |
75 mg/kg, em valor absoluto, para os teores compreendidos entre 500 e 750 mg/kg, |
|
— |
10 % do maior dos valores, para os teores superiores a 750 mg/kg. |
B. DETERMINAÇÃO DOS NÍVEIS DE DIOXINAS (PCDD/PCDF) E DE PCB SOB A FORMA DE DIOXINA PARA EFEITOS DE CONTROLO OFICIAL
I. MÉTODOS DE AMOSTRAGEM E INTERPRETAÇÃO DOS RESULTADOS ANALÍTICOS
1. Objecto e âmbito de aplicação
As amostras destinadas ao controlo oficial do teor de dioxinas (PCDD/PCDF) e do teor de PCB sob a forma de dioxina (1) nos alimentos para animais devem ser colhidas em conformidade com os métodos descritos no anexo I. Devem aplicar-se as exigências quantitativas respeitantes ao controlo de substâncias ou produtos repartidos uniformemente nos alimentos, tal como se prevê no ponto 5.A do anexo I. As amostras globais assim obtidas são consideradas representativas dos lotes ou sublotes dos quais foram colhidas. A observância dos limites máximos previstos na Directiva 2002/32/CE do Parlamento Europeu e do Conselho (2) será estabelecida em função dos níveis detectados nas amostras de laboratório.
2. Conformidade do lote ou do sublote com a especificação
O lote é aceite se o resultado analítico de uma única análise não for superior ao respectivo limite máximo, tal como estabelecido na Directiva 2002/32/CE, tomando em consideração a incerteza da medição.
O lote é considerado não-conforme relativamente ao limite máximo estabelecido na Directiva 2002/32/CE se o limite superior do resultado analítico (3), confirmado por uma análise em duplicado (4), for superior ao limite máximo, com um grau de confiança elevado, tendo em conta a incerteza da medição.
A incerteza da medição pode ser tomada em consideração de acordo com uma das seguintes abordagens:
|
— |
calculando a incerteza expandida, utilizando um factor de expansão de 2, que permite obter um nível de confiança de cerca de 95 %. Um lote não é conforme se o valor medido menos U for superior ao limite máximo. No caso de uma determinação em separado das dioxinas e dos PCB sob a forma de dioxina, na soma dos resultados correspondentes às dioxinas e aos PCB sob a forma de dioxina usa-se a soma da incerteza expandida estimada dos resultados analíticos separados de cada determinação. |
|
— |
estabelecendo o limite de decisão (CCα) em conformidade com a Decisão 2002/657/CE da Comissão (5) (ponto 3.1.2.5. do anexo — caso das substâncias com um limite permitido definido). Um lote é considerado não-conforme se o valor medido for igual ou superior ao CCα. |
As presentes regras de interpretação são aplicáveis ao resultado analítico obtido na amostra para o controlo oficial. Não afectam o direito de os Estados-Membros aplicarem normas nacionais às análises para efeitos de direito de recurso ou procedimentos de arbitragem.
II. PREPARAÇÃO DAS AMOSTRAS E REQUISITOS RESPEITANTES AOS MÉTODOS DE ANÁLISE UTILIZADOS NO CONTROLO OFICIAL DOS TEORES DE DIOXINAS (PCDD/PCDF) E DE PCB SOB A FORMA DE DIOXINA
1. Objecto e âmbito de aplicação
Estes requisitos devem ser aplicados quando se analisam alimentos para animais e matérias-primas para a alimentação animal tendo em vista a determinação das dioxinas [dibenzo-p-dioxinas policloradas (PCDD) e dibenzofuranos policlorados (PCDF)] e dos bifenilos policlorados (PCB) sob a forma de dioxina.
A monitorização da presença de dioxinas em alimentos para animais pode ser realizada mediante uma estratégia que englobe um método de pré-selecção, a fim de escolher as amostras com teores de dioxinas e de PCB sob a forma de dioxina que excedam o teor requerido ou lhe sejam inferiores em menos de 25 %. É necessário que a concentração de dioxinas nas amostras com teores significativos seja determinada/confirmada por um método de confirmação.
Os métodos de pré-selecção são métodos utilizados para a detecção da presença de dioxinas e de PCB sob a forma de dioxina com o teor requerido. Estes métodos têm capacidade para processar um elevado número de amostras e são utilizados para seleccionar grandes números de amostras potencialmente positivas. São concebidos especificamente para evitar a obtenção de falsos negativos.
Os métodos de confirmação são métodos que fornecem informação completa ou complementar que permite a identificação e a quantificação inequívocas ao teor requerido de dioxinas e de PCB sob a forma de dioxina.
2. Antecedentes
Uma vez que as amostras ambientais e biológicas (incluindo as amostras de alimentos para animais/matérias-primas para a alimentação animal) contêm, regra geral, misturas complexas de diferentes congéneres de dioxinas, introduziu-se o conceito de factores de equivalência de toxicidade (FET) para facilitar a avaliação dos riscos. Estes FET foram estabelecidos para exprimir em equivalente tóxico (TEQ) de 2,3,7,8-TCDD as concentrações de misturas de PCDD e PCDF substituídos nas posições 2,3,7 e 8 e alguns PCB não-orto e mono-orto cloro-substituídos que possuem uma actividade semelhante às dioxinas. As concentrações de cada substância numa determinada amostra são multiplicadas pelo respectivo FET e, subsequentemente, somadas para se obter a concentração total de compostos sob a forma de dioxina expressa em TEQ.
Exclusivamente para efeitos do presente regulamento, o limite específico de quantificação aceite para um congénere individual será a concentração de um analito no extracto de uma amostra que produza uma resposta instrumental a dois iões diferentes, a qual será controlada com um rácio sinal/ruído (S/R) de 3:1 para o sinal menos sensível e o cumprimento de requisitos básicos, tais como, por exemplo, o tempo de retenção e o rácio isotópico, de acordo com o procedimento de determinação descrito no método EPA 1613, revisão B.
3. Requisitos de garantia da qualidade a cumprir na preparação da amostra
Aplicam-se as disposições gerais relativas à preparação de amostras para análise estabelecidas no anexo II.
Devem ainda ser cumpridos os seguintes requisitos:
|
— |
As amostras devem ser conservadas e transportadas em contentores de vidro, alumínio, polipropileno ou polietileno. Devem ser removidos do recipiente da amostra os vestígios de poeiras de papel. O material de vidro deve ser enxaguado com solventes previamente submetidos a um controlo destinado a detectar a presença de dioxinas. |
|
— |
Efectuar uma análise em branco através da realização de todo o procedimento analítico, omitindo apenas a amostra. |
|
— |
O peso da amostra utilizada para extracção deve ser suficiente para respeitar os requisitos no tocante à sensibilidade. |
4. Requisitos aplicáveis aos laboratórios
|
— |
Os laboratórios devem demonstrar o desempenho de um método na gama do teor requerido (por exemplo, 0,5 vezes, 1 vez e 2 vezes o teor requerido) com um coeficiente de variação aceitável para análises repetidas. No que se refere aos critérios de aceitação, ver o ponto 5. |
|
— |
O limite de quantificação de um método de confirmação deve situar-se na gama de cerca de um quinto do teor requerido, para garantir que, na gama do teor requerido, são atingidos coeficientes de variação aceitáveis. |
|
— |
Os controlos regulares com ensaios em branco, com amostras enriquecidas ou análises de amostras de controlo (de preferência, se disponível, material de referência certificado) devem ser realizados como medidas internas de controlo da qualidade. |
|
— |
O êxito da participação em estudos interlaboratoriais que avaliam a competência do laboratório é a melhor forma de provar competência em análises específicas. No entanto, o êxito da participação em estudos interlaboratoriais relativos, por exemplo, a amostras de solo ou de águas residuais não prova necessariamente competência no domínio das amostras de alimentos para consumo humano ou animal, que apresentam níveis de contaminação inferiores. Consequentemente, é obrigatória a participação constante em estudos interlaboratoriais respeitantes à determinação de dioxinas e de PCB sob a forma de dioxina nas matrizes relevantes de alimentos para animais/géneros alimentícios. |
|
— |
Os laboratórios devem ser acreditados por um organismo reconhecido que opere em conformidade com o Guia ISO 58, a fim de assegurar que aplicam a garantia da qualidade analítica. Os laboratórios devem ser acreditados em conformidade com a norma ISO/IEC/17025. |
5. Requisitos aplicáveis aos procedimentos analíticos para a determinação de dioxinas e de PCB sob a forma de dioxina
Requisitos básicos de aceitação dos procedimentos analíticos:
|
— |
Sensibilidade elevada e limites de detecção baixos. Para os PCDD e PCDF, as quantidades detectáveis devem ser da ordem do picograma (10-12 g) de TEQ devido à extrema toxicidade de alguns destes compostos. Sabe-se que os PCB ocorrem em teores mais elevados do que os PCDD e PCDF. Para bastantes congéneres de PCB, uma sensibilidade na gama do nanograma (10–9 g) já é suficiente. No entanto, para a medição dos congéneres de PCB sob a forma de dioxina mais tóxicos (designadamente, os congéneres não-orto substituídos), deve ser conseguida a mesma sensibilidade que para os PCDD e PCDF. |
|
— |
Selectividade (especificidade) elevada. É necessário estabelecer uma distinção entre PCDD, PCDF, PCB sob a forma de dioxina e inúmeros compostos co-extraídos e eventualmente interferentes, presentes em concentrações que podem atingir várias ordens de grandeza superiores às dos analitos em causa. Nos métodos de cromatografia gasosa/espectroscopia de massa (CG/EM), é necessária uma diferenciação entre vários congéneres, nomeadamente entre isómeros tóxicos (por exemplo, os dezassete PCDD e PCDF substituídos nas posições 2,3,7 e 8 e os PCB sob a forma de dioxina) e outros congéneres. Os bioensaios deveriam ser capazes de determinar valores de TEQ de forma selectiva como a soma de PCDD, PCDF e PCB sob a forma de dioxina. |
|
— |
Exactidão elevada (rigor e precisão). A determinação deve fornecer uma estimativa válida e fiável da verdadeira concentração numa amostra. É necessária uma exactidão elevada (exactidão da medição: proximidade de concordância entre o resultado de uma medição e o valor verdadeiro ou que se presume verdadeiro da grandeza medida) por forma a evitar a rejeição do resultado da análise de uma amostra com base na reduzida fiabilidade da estimativa de TEQ. A exactidão é expressa em termos de rigor (diferença entre o valor médio medido para um analito num material certificado e o respectivo valor certificado, expresso em percentagem deste valor) e de precisão (RSDR, desvio-padrão relativo, calculado a partir de resultados obtidos em condições de reprodutibilidade). |
Os métodos de pré-selecção podem incluir bioensaios e métodos CG-EM; os métodos de confirmação são a cromatografia gasosa de elevada resolução/espectroscopia de massa de elevada resolução (CGER/EMER).
O valor de TEQ total tem de respeitar os seguintes critérios:
|
|
Métodos de pré-selecção |
Métodos de confirmação |
|
Taxa de falsos negativos |
< 1 % |
|
|
Rigor |
|
- 20 % a + 20 % |
|
Precisão RSDR |
< 30 % |
< 15 % |
6. Requisitos específicos aplicáveis aos métodos CG/EM usados para fins de pré-selecção ou de confirmação
|
— |
Logo no início do método analítico, por exemplo antes da extracção, deve proceder-se à adição de padrões internos de PCDD/F, cloro-substituídos nas posições 2,3,7 e 8, marcados com 13C e de padrões internos de PCB sob a forma de dioxina marcados com 13C, por forma a validar o procedimento analítico. Deve ser adicionado, pelo menos, um congénere para cada grupo homólogo de PCDD/F tetra a octo-clorado e, pelo menos, um congénere para cada grupo homólogo de PCB sob a forma de dioxina (alternativamente, deve ser utilizado para o controlo de PCDD/F e de PCB sob a forma de dioxina, pelo menos, um congénere para cada função de registo de iões seleccionados pela espectrometria de massa). Deve haver uma clara preferência, sobretudo no caso dos métodos de confirmação, pela utilização de padrões internos da totalidade dos 17 PCDD/F substituídos nas posições 2,3,7 e 8 e marcados com 13C e de todos os padrões internos dos 12 PCB sob a forma de dioxina marcados com 13C. |
|
— |
Também devem ser determinados factores de resposta relativos dos congéneres para os quais não se adiciona um composto análogo marcado com 13C, através da utilização de soluções de calibração adequadas. |
|
— |
Em relação aos alimentos para animais de origem vegetal e aos alimentos para animais de origem animal que contenham menos de 10 % de gorduras, a adição de padrões internos é obrigatória antes da extracção. Em relação aos alimentos para animais de origem animal que contenham mais de 10 % de gorduras, os padrões internos podem ser adicionados antes ou após a extracção de gorduras. Deve ser efectuada uma validação adequada da eficiência da extracção, dependendo da fase em que são introduzidos os padrões internos e de os resultados serem notificados em função da quantidade de produto ou de gordura. |
|
— |
Antes da análise por CG/EM, devem ser adicionados 1 ou 2 padrões de recuperação (substitutos). |
|
— |
É necessário efectuar o controlo da recuperação. Para os métodos de confirmação, as recuperações de cada padrão interno deverão situar-se na gama de 60 a 120 %. São aceitáveis recuperações inferiores ou superiores para congéneres individuais, nomeadamente para algumas dibenzodioxinas e dibenzofuranos hepta e octo-clorados, desde que a sua contribuição para o valor TEQ não exceda 10 % do valor TEQ total (com base na soma de PCDD/F e PCB sob a forma de dioxina). Para os métodos de pré-selecção, as recuperações deverão situar-se na gama de 30 a 140 %. |
|
— |
Deve proceder-se à separação entre dioxinas e compostos clorados interferentes, tais como PCB não semelhantes a dioxina e éteres difenílicos clorados através de técnicas cromatográficas adequadas (de preferência, com uma coluna de florisil, alumina e/ou carbono). |
|
— |
Devia ser suficiente a separação de isómeros por cromatografia gasosa (< 25 % de pico a pico entre 1,2,3,4,7,8-HxCDF e 1,2,3,6,7,8-HxCDF). |
|
— |
A determinação deve realizar-se de acordo com o Método EPA 1613 revisão B: Dioxinas e furanos tetra a octo-clorados por CGER/EMER e diluição isotópica ou outro método com critérios de desempenho equivalentes. |
|
— |
A diferença entre o limite superior e o limite inferior de detecção não deve exceder 20 % no caso de alimentos para animais com uma contaminação por dioxinas na gama ou acima do limite máximo. No tocante aos alimentos para animais com níveis de contaminação bastante inferiores ao limite máximo, a diferença pode situar-se na gama de 25 a 40 %. |
7. Métodos de análise de pré-selecção
7.1. Introdução
Num método de pré-selecção podem utilizar-se diferentes abordagens analíticas: uma abordagem puramente de pré-selecção e uma abordagem quantitativa.
A resposta das amostras é comparada com a de uma amostra de referência com o teor requerido. As amostras com uma resposta inferior à da referência são declaradas negativas, as amostras com uma resposta superior são suspeitas de serem positivas. Requisitos:
|
— |
Em cada série de testes tem de se incluir uma amostra em branco e uma de referência, que são extraídas e testadas ao mesmo tempo e em condições idênticas. A amostra de referência deve apresentar uma resposta claramente elevada em comparação com a amostra em branco. |
|
— |
Devem incluir-se outras amostras de referência com 0,5x e 2x o teor requerido para demonstrar o desempenho correcto do teste dentro da gama requerida para o controlo do teor requerido. |
|
— |
Quando se testam outras matrizes, tem de se demonstrar a adequação das amostras de referência, preferencialmente incluindo amostras cuja CGER/EMER revelou conterem um teor TEQ próximo do da amostra de referência ou então uma amostra em branco enriquecida para este teor. |
|
— |
Uma vez que não se podem utilizar padrões internos nos bioensaios, são muito importantes os testes de repetibilidade para se obter informações sobre o desvio-padrão numa série de testes. O coeficiente de variação deve ser inferior a 30 %. |
|
— |
Para os bioensaios deverão ser definidos os compostos-alvos, as possíveis interferências e os valores máximos toleráveis para a amostra em branco. |
A abordagem quantitativa exige várias séries de diluições do padrão, purificação e medições em duplicado ou triplicado, bem como controlos em branco e da recuperação. O resultado pode ser expresso em TEQ, partindo-se assim do princípio de que os compostos responsáveis pelo sinal correspondem ao princípio subjacente ao TEQ. Para o efeito, pode usar-se TCDD (ou uma mistura-padrão de dioxina/furano/PCB sob a forma de dioxina) para produzir uma curva de calibração a fim de calcular o teor de TEQ no extracto e, consequentemente, na amostra. Posteriormente, este resultado é corrigido em função do valor de TEQ calculado para uma amostra em branco (para ter em conta as impurezas provenientes de solventes e produtos químicos utilizados) e da recuperação (calculada a partir do valor TEQ numa amostra de controlo de qualidade com um teor próximo do teor requerido). É essencial ter em conta que parte da perda de recuperação aparente pode dever-se a efeitos de matriz e/ou a diferenças entre os valores dos FET nos bioensaios e os valores oficiais dos FET fixados pela OMS.
7.2. Requisitos aplicáveis aos métodos de análise utilizados na pré-selecção
|
— |
Na pré-selecção, podem usar-se os métodos de análise CG/EM e os bioensaios. Para os métodos CG/EM, são aplicáveis os requisitos descritos no ponto 6. Relativamente aos bioensaios com células, os requisitos específicos estão fixados no ponto 7.3 e, relativamente aos bioensaios com kits, no ponto 7.4. |
|
— |
É necessária informação sobre o número de resultados falsos positivos e falsos negativos de um conjunto grande de amostras abaixo e acima do teor máximo ou do nível de acção, em comparação com o teor de TEQ conforme determinado por um método de análise de confirmação. As taxas efectivas de falsos negativos devem ser inferiores a 1 %. A taxa de amostras com resultados falsos positivos deve ser suficientemente baixa para se poder recorrer vantajosamente ao instrumento de pré-selecção. |
|
— |
Os resultados positivos têm sempre de ser confirmados por um método de análise de confirmação (CGER/EMER). Além disso, devem confirmar-se por CGER/EMER as amostras de uma ampla gama de TEQ (aproximadamente 2-10 % das amostras negativas). Deverá ser disponibilizada informação sobre a correspondência entre os resultados com bioensaios e com CGER/EMER. |
7.3. Requisitos específicos aplicáveis aos bioensaios com células
|
— |
Quando se procede a um bioensaio, para cada teste é necessário uma série de concentrações de referência de TCDD ou de uma mistura de dioxina/furano (curva de dose-resposta completa com um R2 > 0,95). No entanto, para efeitos de pré-selecção, pode usar-se uma curva expandida na zona de baixas concentrações para analisar as amostras com teores reduzidos. |
|
— |
Para o resultado do bioensaio durante um período de tempo constante, deve usar-se uma concentração de referência de TCDD (cerca de 3 x o limite de quantificação) constante de uma ficha de controlo de qualidade. Como alternativa, pode utilizar-se a resposta relativa de uma amostra de referência por comparação com a recta de calibração de TCDD, uma vez que a resposta das células pode depender de muitos factores. |
|
— |
Devem registar-se e verificar-se os gráficos de controlo de qualidade (CQ) para cada tipo de material de referência, a fim de garantir que o resultado está conforme com as directrizes definidas. |
|
— |
Em especial para os cálculos quantitativos, a indução da diluição da amostra utilizada deve encontrar-se dentro da porção linear da curva de resposta. As amostras acima da porção linear da curva de resposta devem ser diluídas e testadas de novo. Assim, recomenda-se a realização simultânea de testes com, pelo menos, três ou mais diluições. |
|
— |
O desvio-padrão percentual não deve ser superior a 15 % numa determinação em triplicado para cada diluição da amostra e não superior a 30 % entre três experiências independentes. |
|
— |
O limite de detecção pode ser fixado como 3x o desvio-padrão da solução em branco de solvente ou da resposta de base. Outra abordagem consiste em aplicar uma resposta que seja superior à resposta de base (factor de indução 5x superior ao da solução em branco de solvente) calculada a partir da curva de calibração do dia. O limite de quantificação pode ser fixado como 5 a 6x o desvio-padrão da solução em branco de solvente ou da resposta de base ou aplicar uma resposta que seja nitidamente superior à resposta de base (factor de indução 10x superior ao da solução em branco de solvente) calculada a partir da curva de calibração do dia. |
7.4. Requisitos específicos aplicáveis aos bioensaios com kits
|
— |
Deve garantir-se que os bioensaios com kits são suficientemente sensíveis e fidedignos para serem aplicados aos alimentos para animais. |
|
— |
Devem ser respeitadas as instruções do fabricante no que se refere à preparação da amostra e às análises. |
|
— |
Os kits não devem ser utilizados depois do prazo de validade. |
|
— |
Não se devem usar materiais ou componentes concebidos para serem usados com outros kits. |
|
— |
Os kits de ensaio devem ser conservados a uma temperatura dentro dos limites definidos e ser usados à temperatura de utilização especificada. |
|
— |
O limite de detecção para os imunoensaios é determinado como a soma da média e de 3x o desvio-padrão, com base numa análise de 10 réplicas da solução em branco, a dividir pelo valor do declive da equação de regressão linear. |
|
— |
Devem ser utilizados padrões de referência para os testes no laboratório, a fim de se garantir que a capacidade de resposta ao padrão se encontra numa gama aceitável. |
8. Notificação dos resultados
Na medida em que o procedimento analítico utilizado o permita, os resultados analíticos devem conter os teores de cada congénere de PCDD/F e PCB e serem indicados em termos de limite inferior, limite superior e limite médio, a fim de incluir o máximo de informações possível na notificação dos resultados e, deste modo, permitir a interpretação dos resultados de acordo com requisitos específicos.
Deve também notificar-se o teor de lípidos da amostra bem como o método utilizado para a respectiva extracção.
As recuperações de cada padrão interno devem ser disponibilizadas se se situarem fora da gama mencionada no ponto 6, no caso de o limite máximo ser excedido e noutros casos mediante pedido.
Como a incerteza da medição deve ser tida em conta ao decidir sobre a conformidade de uma amostra, este parâmetro deve igualmente ser indicado. Assim, o resultado analítico deve ser notificado enquanto x +/- U, em que x é o resultado analítico e U é a incerteza da medição expandida, utilizando um factor de expansão de 2, que permite obter um nível de confiança de cerca de 95 %. No caso de uma determinação em separado das dioxinas e dos PCB sob a forma de dioxina, na soma dos resultados correspondentes às dioxinas e aos PCB sob a forma de dioxina usa-se a soma da incerteza expandida estimada dos resultados analíticos separados de cada determinação.
Se a incerteza da medição for tida em conta mediante a aplicação de um CCα (tal como descrito no ponto 2 da secção I), este parâmetro deve ser mencionado.
(1) Factores de equivalência tóxica (FET) para as dioxinas, furanos e PCB sob a forma de dioxina
|
Congénere |
Valor do FET |
Congénere |
Valor do FET |
|
Dibenzo-p-dioxinas («PCDD») |
|
PCB «sob a forma de dioxina»: |
|
|
2,3,7,8-TCDD |
1 |
|
|
|
1,2,3,7,8-PeCDD |
1 |
PCB não-orto |
|
|
1,2,3,4,7,8-HxCDD |
0,1 |
PCB 77 |
0,0001 |
|
1,2,3,6,7,8-HxCDD |
0,1 |
PCB 81 |
0,0001 |
|
1,2,3,7,8,9-HxCDD |
0,1 |
PCB 126 |
0,1 |
|
1,2,3,4,6,7,8-HpCDD |
0,01 |
PCB 169 |
0,01 |
|
OCDD |
0,0001 |
PCB mono-orto |
|
|
|
|
PCB 105 |
0,0001 |
|
Dibenzofuranos («PCDF») |
|
PCB 114 |
0,0005 |
|
2,3,7,8-TCDF |
0,1 |
PCB 118 |
0,0001 |
|
1,2,3,7,8-PeCDF |
0,05 |
PCB 123 |
0,0001 |
|
2,3,4,7,8-PeCDF |
0,5 |
PCB 156 |
0,0005 |
|
1,2,3,4,7,8-HxCDF |
0,1 |
PCB 157 |
0,0005 |
|
1,2,3,6,7,8-HxCDF |
0,1 |
PCB 167 |
0,00001 |
|
1,2,3,7,8,9-HxCDF |
0,1 |
PCB 189 |
0,0001 |
|
2,3,4,6,7,8-HxCDF |
0,1 |
|
|
|
1,2,3,4,6,7,8-HpCDF |
0,01 |
|
|
|
1,2,3,4,7,8,9-HpCDF |
0,01 |
|
|
|
OCDF |
0,0001 |
|
|
|
Abreviaturas utilizadas: T = tetra; Pe = penta; Hx = hexa; Hp = hepta; O = octo; CDD = clorodibenzo-p-dioxina; CDF = clorodibenzofurano; CB = clorobifenilo. |
|||
(2) JO L 140 de 30.5.2002, p. 10.
(3) O conceito de «limite superior» preconiza que o contributo para a concentração tóxica equivalente (TEQ) de cada congénere não quantificado seja igual ao limite de quantificação.
O conceito de «limite inferior» preconiza que o contributo para a TEQ de cada congénere não quantificado seja igual a zero.
O conceito de «limite médio» preconiza que o contributo para a TEQ de cada congénere não quantificado seja igual a metade do limite de quantificação.
(4) A análise em duplicado é necessária para se excluir a possibilidade de contaminação cruzada interna ou de uma troca acidental de amostras. A primeira análise, tendo em devida conta a incerteza da medição, é utilizada para verificar a conformidade.
No caso de a análise ser realizada no contexto de um incidente de contaminação por dioxinas, poderá dispensar-se a confirmação através de uma análise em duplicado, se as amostras seleccionadas para análise estiverem associadas, por medidas de rastreabilidade, a esse incidente de contaminação por dioxinas.
ANEXO VI
MÉTODOS DE ANÁLISE PARA A DETERMINAÇÃO DE CONSTITUINTES DE ORIGEM ANIMAL NO QUADRO DO CONTROLO OFICIAL DOS ALIMENTOS PARA ANIMAIS
Condições para a detecção, identificação e quantificação por estimativa, por exame microscópico, de constituintes de origem animal em alimentos para animais
1. Objecto e âmbito de aplicação
As presentes condições são aplicáveis sempre que a detecção de constituintes de origem animal (definidos como produtos do processamento de carcaças e partes de carcaça de mamíferos, aves de capoeira e peixes) em alimentos para animais seja efectuada por exame microscópico no quadro do programa coordenado de controlo no domínio da alimentação animal previsto no Regulamento (CE) n.o 882/2004 (1). Desde que os métodos do presente anexo sejam utilizados em todos os exames oficiais, poderá ser igualmente efectuado um segundo exame, com base em variantes dos métodos ou métodos alternativos, para melhorar a detecção de determinados tipos de constituintes de origem animal ou melhor especificar a origem desses constituintes. Além disso, poderá fazer-se uso de uma variante do protocolo no exame de determinados constituintes específicos de origem animal, como o plasma ou ossos presentes no sebo (ver o ponto 9), desde que essas análises sejam efectuadas em complemento das previstas no programa coordenado de controlo.
2. Sensibilidade
Podem ser detectadas quantidades muito pequenas (inferiores a 0,1 %) de constituintes de origem animal em alimentos para animais, dependendo da natureza desses constituintes.
3. Princípio
Utiliza-se na identificação uma amostra representativa, colhida de acordo com o disposto no anexo I e preparada de modo adequado. O protocolo a seguir descrito adequa-se a alimentos para animais com baixo teor de humidade. Os alimentos para animais com teor de humidade superior a 14 % terão de ser previamente secos (ou condensados). Determinados alimentos para animais ou matérias-primas para a alimentação animal (por exemplo, óleos ou gorduras) exigem um tratamento específico (ver o ponto 9). Os constituintes de origem animal são identificados com base em características típicas detectáveis por exame microscópico (por exemplo, fibras musculares e outras partículas de carne, cartilagens, ossos, chifres, pêlos, cerdas, sangue, penas, cascas de ovos, espinhas ou escamas). Procede-se à identificação na fracção peneirada (6.1) e no sedimento concentrado (6.2) da amostra.
4. Reagentes
4.1. Meios de montagem
|
4.1.1. |
Hidrato de cloral (solução aquosa a 60 %, m/v). |
|
4.1.2. |
Solução alcalina (solução a 2,5 %, m/v, de NaOH ou solução a 2,5 %, m/v, de KOH) para as fracções peneiradas. |
|
4.1.3. |
Óleo parafínico ou glicerol (viscosidade: 68-81) para as observações microscópicas no sedimento. |
4.2. Agentes de lavagem
|
4.2.1. |
Álcool a 96 %. |
|
4.2.2. |
Acetona. |
4.3. Agente de concentração
|
4.3.1. |
Tetracloroetileno (densidade: 1,62). |
4.4. Reagentes de coloração
|
4.4.1. |
Solução de iodo/iodeto de potássio (dissolver 2 g de iodeto de potássio em 100 ml de água e adicionar 1 g de iodo, agitando com frequência). |
|
4.4.2. |
Vermelho de alizarina (diluir 2,5 ml de ácido clorídrico 1 M em 100 ml de água e adicionar a esta solução 200 mg de vermelho de alizarina). |
|
4.4.3. |
Reagente da cistina (2 g de acetato de chumbo, 10 g de NaOH/100 ml H2O). |
|
4.4.4. |
Solução de iodo/iodeto de potássio (dissolvida em etanol a 70 %). |
4.5. Reagente descolorante
|
4.5.1 |
Solução comercial de hipoclorito de sódio (9,6 % de cloro activo). |
5. Equipamento e acessórios
|
5.1. |
Balança analítica (precisão de 0,01 g; excepto no caso do sedimento concentrado: 0,001 g). |
|
5.2. |
Meios de moagem (moinho ou almofariz, em especial no caso dos alimentos para animais com teor de matéria gorda em análise superior a 15 %). |
|
5.3. |
Peneira com rede de orifícios quadrados de lado não superior a 0,50 mm. |
|
5.4. |
Ampola de decantação ou vaso de decantação de fundo cónico. |
|
5.5. |
Microscópio estereoscópico (ampliação mínima: 40 vezes). |
|
5.6. |
Microscópio composto (ampliação mínima: 400 vezes), de luz transmitida ou luz polarizada. |
|
5.7. |
Material de vidro de uso laboratorial corrente. O equipamento deve apresentar-se perfeitamente limpo. As ampolas de decantação e o restante material de vidro devem ser lavados em máquina de lavar. As peneiras devem ser limpas com uma escova rija. |
6. Procedimento
Os alimentos para animais em granulado podem ser previamente peneirados, se ambas as fracções forem analisadas como amostras distintas.
Deve tratar-se pelo menos 50 g de amostra [moer com precaução, utilizando, se necessário, os meios de moagem adequados (5.2) para obter uma estrutura apropriada]. Tomar duas partes representativas da matéria moída, uma para a peneiração (pelo menos 5 g) (6.1) e outra para a concentração do sedimento (pelo menos 5 g) (6.2). Para facilitar a identificação, podem utilizar-se ainda reagentes de coloração (6.3).
A fim de detectar a natureza das proteínas animais e a origem das partículas, pode recorrer-se a um sistema de apoio à decisão (como o ARIES) e a amostras de referência.
6.1. Identificação de constituintes de origem animal nas fracções peneiradas
Peneirar (5.3), em duas fracções, uma quantidade mínima de 5 g da amostra.
Depositar a fracção (ou as várias partes em que seja dividida a fracção) de maior granulometria, ou uma porção representativa da mesma, em camada fina, num suporte adequado e pesquisar, de modo sistemático, ao microscópio estereoscópico (5.5), a diversas ampliações, a presença de constituintes de origem animal.
Pesquisar, de modo sistemático, ao microscópio composto (5.6), a diversas ampliações, a presença de constituintes de origem animal em lâminas preparadas com a fracção peneirada de menor granulometria.
6.2. Identificação de constituintes de origem animal no sedimento concentrado
Depositar uma quantidade mínima de 5 g (com uma aproximação de 0,01 g) da amostra numa ampola de decantação ou vaso de decantação de fundo cónico e adicionar pelo menos 50 ml de tetracloroetileno (4.3.1). Agitar a mistura diversas vezes.
|
— |
Se se utilizar uma ampola de decantação fechada, deixar assentar o sedimento durante tempo suficiente (pelo menos 3 minutos) antes de o separar. Agitar de novo e voltar a deixar assentar o sedimento durante pelo menos 3 minutos. Voltar a separar o sedimento. |
|
— |
Se se utilizar um vaso aberto, deixar assentar o sedimento durante pelo menos 5 minutos antes de o separar. |
Secar e depois pesar (com uma aproximação de 0,001 g) o sedimento total. A pesagem só é necessária caso se pretenda efectuar uma quantificação por estimativa. Se o sedimento for constituído por muitas partículas grandes poderá ser separado em duas fracções por peneiração (5.3). Pesquisa-se a presença de constituintes ósseos no sedimento seco ao microscópio estereoscópico (5.5) e ao microscópio composto (5.6).
6.3. Utilização de meios de montagem e reagentes de coloração
A identificação microscópica dos constituintes de origem animal pode ser facilitada pelo recurso a meios de montagem e reagentes de coloração especiais.
Hidrato de cloral (4.1.1): Aquecendo cuidadosamente, é possível observar mais claramente as estruturas celulares, pois os grãos de amido sofrem gelatinização e os conteúdos indesejados são removidos das células.
Solução alcalina (4.1.2): O hidróxido de sódio e o hidróxido de potássio clarificam as matérias constituintes do alimento para animais, ajudando na detecção de fibras musculares e de pêlos e outras estruturas queratínicas.
Óleo parafínico e glicerol (4.1.3): Os constituintes ósseos podem ser bem identificados neste meio de montagem, pois, na sua maioria, as lacunas permanecem cheias de ar e surgem como buracos negros de 5-15 μm.
Solução de iodo/iodeto de potássio (4.4.1): É utilizada na detecção de amido (cor azul violáceo) e de proteínas (cor amarelo alaranjado). Se necessário, as soluções podem ser diluídas.
Solução de vermelho de alizarina (4.4.2): Coloração vermelha/rosa dos ossos, espinhas e escamas. Antes de secar o sedimento (ver ponto 6.2), transferir o sedimento total para um tubo de ensaio de vidro e lavar duas vezes com aproximadamente 5 ml de álcool (4.2.1) (utilizar um agitador de vórtex em cada lavagem e deixar o solvente em repouso durante cerca de um minuto antes de o decantar). Antes de utilizar este reagente de coloração, descorar o sedimento com pelo menos 1 ml de solução de hipoclorito de sódio (4.5.1). Deixar reagir durante 10 minutos. Encher o tubo com água, deixar depositar o sedimento durante 2-3 minutos e decantar a água e as partículas em suspensão. Lavar mais duas vezes o sedimento com cerca de 10 ml de água (utilizar um agitador de vórtex, deixar em repouso e decantar a água de cada vez). Adicionar duas a 10 ou mais gotas (dependendo da quantidade de resíduo) de solução de vermelho de alizarina. Agitar a mistura e deixar reagir durante alguns segundos. Lavar duas vezes o sedimento corado com aproximadamente 5 ml de álcool (4.2.1) e, em seguida, uma vez com acetona (4.2.2) (utilizar um agitador de vórtex em cada lavagem e deixar o solvente em repouso durante cerca de um minuto antes de o decantar). O sedimento estará pronto para a secagem.
Reagente da cistina (4.4.3): Aquecendo cuidadosamente, os constituintes com cistina (pêlos, penas, etc.) ficam castanho escuro.
6.4. Exame de alimentos para animais que eventualmente contenham farinhas de peixe
Examinar ao microscópio composto pelo menos uma lâmina da fracção peneirada de menor granulometria e da fracção fina do sedimento (ver pontos 6.1 e 6.2).
Se a rotulagem incluir farinhas de peixe nos ingredientes ou se suspeitar da presença de farinhas de peixe ou esta tiver sido detectada no exame inicial, devem examinar-se pelo menos mais duas lâminas da fracção de menor granulometria da amostra inicial e também a fracção sedimento total.
7. Cálculos e avaliação
Os Estados-Membros devem assegurar a aplicação dos procedimentos descritos no presente ponto quando for efectuada uma análise oficial com o intuito de estimar a quantidade (e não apenas a presença) de constituintes de origem animal.
Os cálculos só podem ser efectuados se os constituintes de origem animal contiverem fragmentos ósseos.
Com base nas lacunas características que apresentam, os fragmentos de ossos de animais terrestres de sangue quente (mamíferos e aves) são distinguíveis dos diversos tipos de espinhas de peixe na lâmina de observação microscópica. A proporção de constituintes de origem animal na amostra pode ser estimada tendo em conta:
|
— |
a proporção estimada (percentagem ponderal) de fragmentos ósseos no sedimento concentrado, |
|
— |
a proporção (percentagem ponderal) de ossos nos constituintes de origem animal. |
A estimativa deve basear-se na observação de pelo menos (se possível) três lâminas e de um mínimo de cinco campos por lâmina. Nos alimentos compostos para animais, o sedimento concentrado contém, em geral, não apenas fragmentos de ossos de animais terrestres e de espinhas de peixes, mas também outras partículas de massa específica elevada, nomeadamente minerais, areia, fragmentos lenhificados de plantas, etc.
7.1. Estimativa da percentagem de fragmentos ósseos
Percentagem de fragmentos de ossos de animais terrestres = (S × c)/W
Percentagem de fragmentos de espinhas e escamas de peixes = (S × d)/W
|
(S = |
massa de sedimento (mg); c = factor de correcção ( %) correspondente à estimativa de fragmentos de ossos de animais terrestres no sedimento; d = factor de correcção ( %) correspondente à estimativa de fragmentos de espinhas e escamas de peixes no sedimento; W = massa da amostra para sedimentação (mg)]. |
7.2. Estimativa da percentagem de constituintes de origem animal
A proporção óssea pode variar grandemente nos produtos de origem animal. A percentagem de ossos é de 50-60 % no caso das farinhas de ossos e de 20-30 % no caso das farinhas de carne; no caso das farinhas de peixe, a proporção de espinhas e escamas varia em função da categoria e origem da farinha, sendo, normalmente, da ordem de 10-20 %).
Se o tipo de farinha de origem animal presente na amostra for conhecido, será possível efectuar as seguintes estimativas:
Proporção estimada de constituintes originários de animais terrestres ( %) = (S × c)/(W × f) × 100
Proporção estimada de constituintes originários de peixe ( %) = (S × d)/(W × f) × 100
|
(S = |
massa de sedimento (mg); c = factor de correcção ( %) correspondente à estimativa de fragmentos de ossos de animais terrestres no sedimento; d = factor de correcção ( %) correspondente à estimativa de fragmentos de espinhas e escamas de peixes no sedimento; f = factor de correcção correspondente à proporção de ossos nos constituintes de origem animal da amostra examinada; W = massa da amostra para sedimentação (mg)). |
8. Expressão dos resultados do exame
O relatório deve conter, no mínimo, informações sobre a presença de constituintes originários de animais terrestres e de farinhas de peixe. Os diversos casos possíveis serão descritos do seguinte modo:
|
8.1. |
Quanto à presença de constituintes originários de animais terrestres:
|
|
8.2. |
Quanto à presença de farinhas de peixe:
Se forem detectados constituintes originários de peixe ou de animais terrestres, o relatório do resultado do exame pode, se necessário, apresentar ainda uma estimativa da quantidade de constituintes detectada (x %, < 0,1 %, 0,1-0,5 %, 0,5-5 % ou > 5 %) e especificar o tipo de animal terrestre, se possível, e os constituintes de origem animal identificados (fibras musculares, cartilagem, ossos, chifres, pêlos, cerdas, penas, sangue, cascas de ovos, espinhas, escamas). Se for estimada uma quantidade de ingredientes de origem animal, deve ser indicado o factor de correcção f utilizado. Se forem identificados constituintes ósseos provenientes de animais terrestres, o relatório deverá incluir a seguinte frase: «Não é de excluir a possibilidade de estes constituintes serem originários de mamíferos.» Esta frase suplementar não será necessária se tiver sido especificado tratar-se de fragmentos ósseos de animais terrestres provenientes de aves de capoeira ou de mamíferos. |
9. Protocolo facultativo para a análise de óleos ou gorduras
Na análise de óleos ou gorduras pode recorrer-se ao seguinte protocolo:
|
— |
Se a gordura se apresentar no estado sólido, deve ser aquecida (por exemplo, num forno de microondas) até fundir. |
|
— |
Com uma pipeta, transferir 40 ml de gordura da base da amostra para um tubo de centrifugação. |
|
— |
Centrifugar a 4 000 rpm durante 10 minutos. |
|
— |
Se a gordura se apresentar no estado sólido depois da centrifugação, deve ser aquecida de novo num forno até fundir. Voltar a centrifugar a 4 000 rpm durante 5 minutos. |
|
— |
Com uma pequena colher ou espátula, transferir metade das impurezas decantadas para uma pequena placa de Petri ou uma lâmina de microscópio, para a identificação ao microscópio da eventual presença de constituintes de origem animal (fibras de carne, penas, fragmentos ósseos, etc.). Para a observação ao microscópio, é recomendada a utilização de óleo parafínico ou de glicerol como meio de montagem. |
|
— |
As restantes impurezas serão utilizadas na sedimentação descrita no ponto 6.2. |
(1) JO L 165 de 30.4.2004, p. 1. Rectificação no JO L 191 de 28.5.2004, p. 1.
ANEXO VII
MÉTODO DE CÁLCULO DO VALOR ENERGÉTICO DOS ALIMENTOS COMPOSTOS DESTINADOS ÀS AVES DE CAPOEIRA
1. Modo de cálculo e expressão do valor energético
O valor energético dos alimentos compostos destinados às aves de capoeira é calculado segundo a fórmula adiante indicada, a partir da percentagem de determinados componentes analíticos dos alimentos. Esse valor é expresso em megajoules (MJ), de energia metabolizável (EM), corrigida para o azoto, por quilograma de alimento composto tal que:
MJ/kg de EM = 0,1551 × % de proteína bruta +0,3431 × % de matérias gordas brutas +0,1669 × % de amido +0,1301 × % de açúcares totais (expressos em sacarose).
2. Tolerâncias aplicáveis aos valores declarados
Se, na sequência de um controlo oficial, for constatada uma diferença entre o resultado do controlo e o valor declarado (valor energético do alimento superior ou inferior) é aplicada uma tolerância mínima de 0,4 MJ/kg de EM.
3. Expressão do resultado
Após a aplicação da fórmula pré-citada, o resultado obtido é expresso com uma casa decimal.
4. Métodos de amostragem e de análise
A colheita da amostra do alimento composto e a determinação do teor dos componentes analíticos indicados no método de cálculo são realizados segundo os métodos comunitários de amostragem e análise para o controlo oficial dos alimentos para animais.
São aplicáveis:
|
— |
para a determinação do teor de proteína bruta: o procedimento B do método de determinação do teor de matérias gordas brutas, estabelecido na parte H do anexo III, |
|
— |
para a determinação do teor de amido: o método polarimétrico estabelecido na parte L do anexo III. |
ANEXO VIII
MÉTODOS DE ANÁLISE PARA O CONTROLO DA PRESENÇA ILEGAL DE ADITIVOS QUE JÁ NÃO ESTÃO AUTORIZADOS NOS ALIMENTOS PARA ANIMAIS
Observação importante:Pode recorrer-se a métodos de análise mais sensíveis que os descritos no presente anexo para detectar a presença ilegal de aditivos que deixaram de estar autorizados nos alimentos para animais.Os métodos de análise referidos no presente anexo podem ser usados para efeitos de confirmação.
(A) DETERMINAÇÃO DO METILBENZOQUATO
7-benziloxi-6-butil-3-metoxicarbonil-4-quinolona
1. Objecto e âmbito de aplicação
O presente método destina-se à determinação do teor de metilbenzoquato em alimentos para animais. O limite de quantificação é 1 mg/kg.
2. Princípio
O metilbenzoquato é extraído da amostra por recurso a uma solução metanólica de ácido metanossulfónico. O extracto é purificado com diclorometano e por cromatografia de permuta iónica, seguindo-se nova purificação com diclorometano. O teor de metilbenzoquato é determinado por cromatografia líquida de alta resolução (HPLC) de fase reversa, utilizando um detector de ultravioleta.
3. Reagentes
|
3.1. |
Diclorometano |
|
3.2. |
Metanol de qualidade equivalente para HPLC. |
|
3.3. |
Fase móvel para HPLC Mistura de metanol (3.2) e água (qualidade equivalente para HPLC) 75 + 25 (v + v). Filtrar com um filtro de 0,22 μm (4.5) e desgaseificar a solução (por exemplo por aplicação de ultra-sons durante 10 minutos). |
|
3.4. |
Solução de ácido metanossulfónico a 2 % Diluir 20,0 ml de ácido metanossulfónico em metanol (3.2) de modo a perfazer o volume de 1 000 ml. |
|
3.5. |
Solução d ácido clorídrico a 10 %: Diluir 100 ml de ácido clorídrico (ρ20 = 1,18 g/ml) em água de modo a perfazer o volume de 1 000 ml. |
|
3.6. |
Resina de permuta catiónica Amberlite CG-120 (Na), 100-200 mesh Tratamento da resina antes do uso: misturar 100 g de resina em 500 ml de solução de ácido clorídrico (3.5) e aquecer em placa até à ebulição, agitando continuamente. Deixar arrefecer e decantar o ácido. Filtrar num filtro de papel, sob vácuo. Lavar a resina com duas porções de 500 ml de água, seguidas de 250 ml de metanol (3.2). Lavar a resina com uma nova porção de 250 ml de metanol e secar, mediante a passagem de uma corrente de ar através do bolo de filtração. Guardar a resina seca num recipiente rolhado. |
|
3.7. |
Substância-padrão: metilbenzoquato puro (7-benziloxi-6-butil-3-metoxicarbonil-4-quinolona). |
|
3.7.1. |
Pesar, com uma aproximação de 0,1 mg, 50 mg de substância-padrão (3.7). Dissolver em solução de ácido metanossulfónico (3.4), num balão volumétrico de 100 ml, perfazer o volume e agitar. |
|
3.7.2. |
Transferir 5,0 ml de solução-mãe padrão de metilbenzoquato (3.7.1) para um balão volumétrico de 50 ml. Perfazer o volume com metanol (3.2) e agitar. |
|
3.7.3. |
Transferir, respectivamente, 1,0, 2,0, 3,0, 4,0 e 5,0 ml de solução-padrão intermédia de metilbenzoquato (3.7.2) para uma série de balões volumétricos de 25 ml. Perfazer o volume com fase móvel (3.3) e agitar. As soluções obtidas contêm, respectivamente, 2,0, 4,0, 6,0, 8,0 e 10,0 μg/ml de metilbenzoquato e devem ser preparadas imediatamente antes da utilização. |
4. Equipamento
|
4.1. |
Agitador mecânico. |
|
4.2. |
Evaporador rotativo. |
|
4.3. |
Coluna de vidro (250 mm × 15 mm) munida de torneira e reservatório de cerca de 200 ml. |
|
4.4. |
Aparelho para HPLC com detector de ultravioleta de comprimento de onda variável ou detector de díodos. |
|
4.4.1. |
Coluna para cromatografia líquida: 300 mm × 4 mm, com enchimento C18 de 10 μm ou equivalente. |
|
4.5. |
Filtros de membrana, porosidade 0,22 μm. |
|
4.6. |
Filtros de membrana, porosidade 0,45 μm. |
5. Procedimento
5.1. Generalidades
|
5.1.1. |
De modo a comprovar a ausência de metilbenzoquato e outras substâncias susceptíveis de interferir no processo, deve-se proceder à análise de uma amostra em branco de um alimento para animais (ensaio em branco). |
|
5.1.2. |
Deve-se efectuar um ensaio de recuperação utilizando a amostra do ensaio em branco, fortificada com uma quantidade conhecida de metilbenzoquato semelhante à quantidade presente na amostra. Para obter uma fortificação a um nível de 15 mg/kg, adicionam-se 600 μl de solução-mãe padrão (3.7.1) a 20 g de amostra para o ensaio em branco. Misturar, deixar em repouso 10 minutos e proceder à extracção (5.2). Nota: Para os fins do presente método, a amostra para o ensaio em branco deve ter composição semelhante à da amostra em análise e não deve acusar a presença de benzoquato de metilo. |
5.2. Extracção
Pesar, com uma aproximação de 0,01 g, cerca de 20 g de amostra preparada e transferir para um erlenmeyer de 250 ml. Adicionar 100,0 ml de solução de ácido metanossulfónico (3.4) e agitar durante 30 minutos com o agitador mecânico (4.1). Filtrar a solução num filtro de papel e guardar o filtrado para a operação de partição líquido-líquido (5.3).
5.3. Partição líquido-líquido
Transferir 25,0 ml do filtrado obtido no ponto 5.2 para uma ampola de decantação de 500 ml contendo 100 ml de solução de ácido clorídrico (3.5). Adicionar 100 ml de diclorometano (3.1) e agitar a ampola durante 1 minuto. Esperar a separação das fases e transferir a camada inferior (diclorometano) para um balão de fundo redondo de 500 ml. Repetir a extracção da fase aquosa com duas novas porções de 40 ml de diclorometano e combinar os extractos resultantes com o primeiro extracto, no balão de fundo redondo. Evaporar o extracto de diclorometano até à secura, no evaporador rotativo (4.2), a 40 oC, sob pressão reduzida. Dissolver o resíduo em 20-25 ml de metanol (3.2), tapar o balão e guardar a totalidade do extracto para a cromatografia de permuta iónica (5.4).
5.4. Cromatografia de permuta iónica
5.4.1.
Colocar um tampão de fibra de vidro na extremidade inferior de uma coluna de vidro (4.3). Preparar uma suspensão de 5,0 g de resina de permuta catiónica tratada (3.6) em 50 ml de ácido clorídrico (3.5), verter na coluna de vidro e deixar assentar. Escoar o excesso de ácido, até o nível deste último se situar ligeiramente acima da superfície da resina, e lavar a coluna com água até o efluente apresentar reacção neutra ao papel de tornesol. Verter na coluna 50 ml de metanol (3.2) e deixar escoar até à superfície da resina.
5.4.2.
Com o auxílio de uma pipeta, transferir cuidadosamente para a coluna o extracto obtido no ponto 5.3. Lavar o balão de fundo redondo com duas porções de 5-10 ml de metanol (3.2), transferindo as mesmas para a coluna. Deixar impregnar o extracto até à superfície da resina. Lavar a coluna com 50 ml de metanol, tomando precauções para que o caudal não exceda 5 ml por minuto. Rejeitar o efluente. Eluir o metilbenzoquato com 150 ml de solução de ácido metanossulfónico (3.4), recolhendo o eluato num erlenmeyer de 250 ml.
5.5. Partição líquido-líquido
Transferir para uma ampola de decantação de 1 litro o eluato obtido no ponto 5.4.2. Lavar o erlenmeyer com 5-10 ml de metanol (3.2) que se adicionam ao conteúdo da ampola de decantação. Adicionar 300 ml de solução de ácido clorídrico (3.5) e 130 ml de diclorometano (3.1). Agitar durante um minuto e esperar a separação das fases. Escoar a camada inferior (diclorometano) para um balão de fundo redondo de 500 ml. Repetir a extracção da fase aquosa com duas novas porções de 70 ml de diclorometano, combinando estes extractos com o primeiro, no balão de fundo redondo.
Evaporar até à secura o extracto de diclorometano, no evaporador rotativo (4.2), a 40 oC, sob pressão reduzida. Dissolver o resíduo do balão em cerca de 5 ml de metanol (3.2) e transferir quantitativamente esta solução para um balão volumétrico de 10 ml. Lavar o balão de fundo redondo com duas novas porções de 1-2 ml de metanol e transferir para o balão volumétrico. Perfazer o volume com metanol e agitar. Filtrar uma alíquota através de um filtro de membrana (4.6). Guardar esta solução para a análise por HPLC (5.6).
5.6. Análise por HPLC
5.6.1.
As condições a seguir especificadas são-no a título indicativo. Poderão escolher-se outras condições, desde que produzam resultados equivalentes.
|
— |
coluna para cromatografia líquida (4.4.1), |
|
— |
fase móvel para HPLC: mistura metanol-água (3.3), |
|
— |
caudal: 1 a 1,5 ml/minuto, |
|
— |
comprimento de onda de detecção: 265 nm, |
|
— |
Volume injectado: 20 a 50 μl. |
Verificar a estabilidade do sistema cromatográfico mediante a injecção repetida da solução de calibração (3.7.3), que contém 4 μg/ml de metilbenzoquato, até obter picos com alturas (ou áreas) e tempos de retenção reprodutíveis.
5.6.2.
Injectar várias vezes cada solução de calibração (3.7.3) e medir a altura (ou área) dos picos correspondentes a cada concentração. Traçar uma curva de calibração, colocando a altura (ou área) média dos picos das soluções de calibração em ordenadas e as concentrações correspondentes, expressas em μg/ml, em abcissas.
5.6.3.
Injectar várias vezes o extracto de amostra (5.5), utilizando o mesmo volume utilizado para as soluções de calibração e determinar a altura (ou área) média dos picos correspondentes ao metilbenzoquato.
6. Cálculo dos resultados
A partir da altura (ou área) média dos picos do metilbenzoquato da solução de amostra, determinar a concentração desta em μg/ml, com base na curva de calibração (5.6.2).
O teor de metilbenzoquato da amostra, w, expresso em mg/kg, é dado pela fórmula:

em que:
|
c |
= |
concentração de metilbenzoquato da solução da amostra, expressa em μg/ml, |
|
m |
= |
massa da toma de ensaio, expressa em gramas. |
7. Validação dos resultados
7.1. Identidade
A identidade do analito pode ser confirmada por co-cromatografia ou por recurso a um detector de díodos, comparando os espectros do extracto de amostra e da solução de calibração (3.9.3) que contém 10 μg/ml de metilbenzoquato.
7.1.1.
Fortificar um extracto da amostra com a adição de uma quantidade adequada de solução-padrão intermédia (3.7.2). A quantidade de metilbenzoquato adicionada deve ser idêntica à quantidade de metilbenzoquato presumida no extracto de amostra.
Apenas a altura do pico correspondente ao metilbenzoquato deverá exibir um aumento adequado, tendo em conta a quantidade adicionada e a diluição do extracto. A largura do pico a meia altura não poderá diferir mais de 10 % da largura inicial.
7.1.2.
Os resultados são avaliados de acordo com os seguintes critérios:
|
a) |
O comprimento de onda de absorção máxima dos espectros da amostra e do padrão, registado no máximo do pico cromatográfico, deve ser o mesmo, com uma margem de variação dependente da resolução do sistema de detecção. No caso de um detector de díodos, esta margem é, em geral, de cerca de 2 nm; |
|
b) |
Entre 220 e 350 nm, os espectros da amostra e do padrão, registados no máximo dos picos cromatográficos, não devem diferir entre si no que respeita às zonas do espectro que apresentem uma absorvância relativa compreendida entre 10 % e 100 %. Este critério considera-se satisfeito se estiverem presentes os mesmos máximos e a diferença entre ambos os espectros não exceder, em caso algum, 15 % da absorvância do analito-padrão; |
|
c) |
Entre 220 e 350 nm, os espectros na curva ascendente, no máximo e na curva descendente do pico do extracto da amostra não devem diferir entre si nas partes do espectro que apresentem uma absorvância relativa compreendida entre 10 e 100 %. Este critério considera-se satisfeito se estiverem presentes os mesmos máximos e a diferença entre os espectros não exceder, em caso algum, 15 % da absorvância do espectro no máximo do pico. |
Se algum destes critérios não for satisfeito, a presença do analito não pode ser confirmada.
7.2. Repetibilidade
A diferença entre os resultados de duas determinações paralelas efectuadas com a mesma amostra não deve exceder: 10 %, em valor relativo, em relação ao maior dos valoress, para os teores de metilbenzoquato compreendidos entre 4 e 20 mg/kg.
7.3. Recuperação
A taxa de recuperação de uma amostra em branco fortificada deve ser de, pelo menos, 90 %.
8. Resultados de um estudo interlaboratorial
Procedeu-se à análise de cinco amostras por 10 laboratórios. Para cada amostra, foram efectuadas análises em duplicado.
|
|
Branco |
Farinha 1 |
Granulado 1 |
Farinha 2 |
Granulado 2 |
|
Média [mg/kg] |
n.d. |
4,50 |
4,50 |
8,90 |
8,70 |
|
sr [mg/kg] |
— |
0,30 |
0,20 |
0,60 |
0,50 |
|
Cvr [ %] |
— |
6,70 |
4,40 |
6,70 |
5,70 |
|
SR [mg/kg] |
— |
0,40 |
0,50 |
0,90 |
1,00 |
|
CVR [ %] |
— |
8,90 |
11,10 |
10,10 |
11,50 |
|
Recuperação [ %] |
— |
92,00 |
93,00 |
92,00 |
89,00 |
|
n.d. |
= |
não detectado |
|
sr |
= |
desvio-padrão da repetibilidade |
|
CVr |
= |
coeficiente de variação da repetibilidade ( %) |
|
SR |
= |
desvio-padrão da reprodutibilidade |
|
CVR |
= |
coeficiente de variação da reprodutibilidade ( %) |
(B) DETERMINAÇÃO DO OLAQUINDOX
N1,N4-dióxido de 2-(N-2'-(hidroxietil)carbamoíl)-3-metilquinoxalina
1. Objecto e âmbito de aplicação
Esta técnica permite determinar o teor de olaquindox em alimentos para animais. O limite de quantificação é 5 mg/kg.
2. Princípio
A amostra é submetida a extracção com uma mistura metanol-água. O teor de olaquindox é determinado por cromatografia líquida de alta resolução (HPLC) de fase reversa, utilizando um detector de ultravioleta.
3. Reagentes
|
3.1. |
Metanol |
|
3.2. |
Metanol de qualidade equivalente para HPLC. |
|
3.3. |
Água de qualidade equivalente para HPLC. |
|
3.4. |
Fase móvel para HPLC. Mistura água (3.3)-metanol (3.2), 900 + 100 (v + v). |
|
3.5. |
Substância-padrão: olaquindox puro. [N1,N4-dióxido de 2-(N-2'-(hidroxietil)carbamoíl)-3-metilquinoxalina], E 851. |
|
3.5.1. |
Pesar, com uma aproximação de 0,1 mg, 50 mg de olaquindox (3.5) num balão volumétrico de 200 ml e acrescentar aproximadamente 190 ml de água. Colocar o balão num banho de ultra-sons (4.1) durante 20 minutos. Depois do tratamento ultra-sónico, levar a solução à temperatura ambiente, perfazer o volume com água e agitar. Envolver o balão com uma folha de alumínio e colocá-lo no frigorífico. Esta solução deve ser renovada mensalmente. |
|
3.5.2. |
Transferir 10,0 ml de solução-mãe padrão (3.5.1) para um balão volumétrico de 100 ml, perfazer o volume com a fase móvel e agitar. Envolver o balão com uma folha de alumínio e colocá-lo no frigorífico. Esta solução deve ser renovada diariamente. |
|
3.5.3. |
Transferir para uma série de balões volumétricos de 50 ml: 1,0, 2,0, 5,0, 10,0, 15,0 e 20,0 ml da solução-padrão intermédia (3.5.2). Perfazer o volume com fase móvel (3.4) e agitar. Envolver os balões com folha de alumínio. Estas soluções contêm, respectivamente, 0,5, 1,0, 2,5, 5,0, 7,5 e 10,0 μg de olaquindox por ml. Devem ser renovadas diariamente. |
4. Equipamento
|
4.1. |
Banho de ultra-sons. |
|
4.2. |
Agitador mecânico. |
|
4.3. |
Aparelho para HPLC com detector de ultravioleta de comprimento de onda variável ou detector de díodos. |
|
4.3.1. |
Coluna para cromatografia líquida com 250 mm × 4 mm e enchimento C18 de 10 μm, ou equivalente. |
|
4.4. |
Filtros de membrana, porosidade 0,45 μm. |
5. Procedimento
|
Nota: |
o olaquindox é sensível à luz. Efectuar todas as operações sob luz difusa ou utilizar material de vidro ambarizado. |
5.1. Generalidades
|
5.1.1. |
De modo a comprovar a ausência de olaquindox e outras substâncias susceptíveis de interferir no processo, deve proceder-se à análise de uma amostra em branco de um alimento para animais (ensaio em branco). |
|
5.1.2. |
Deve efectuar-se um ensaio de recuperação utilizando a amostra do ensaio em branco, fortificada com uma quantidade conhecida de olaquindox semelhante à quantidade presente na amostra. Para obter uma fortificação a um nível de 50 mg/kg, transferir 10,0 ml da solução-mãe padrão (3.5.1) para um erlenmeyer de 250 ml e concentrar a solução por evaporação até cerca de 0,5 ml. Acrescentar 50 g da amostra em branco, misturar cuidadosamente e esperar 10 minutos misturando de novo várias vezes antes de proceder à extracção (5.2).
|
5.2. Extracção
Pesar, com uma aproximação de 0,01 g, cerca de 50 g da amostra. Transferir para um erlenmeyer de 1 000 ml, acrescentar 100 ml de metanol (3.1) e colocar o balão durante cinco minutos num banho de ultra-sons (4.1). Acrescentar 410 ml de água e deixar no banho de ultra-sons durante mais 15 minutos. Retirar o erlenmeyer do banho de ultra-sons, agitá-lo durante 30 minutos no agitador (4.2) e filtrar através de um filtro dobrado. Transferir 10,0 ml do filtrado para um balão volumétrico de 20 ml, perfazer o volume com água e agitar. Filtrar uma alíquota através de um filtro de membrana (4.4) (ver ponto 9). Efectuar em seguida a análise por HPLC (5.3).
5.3. Análise por HPLC
5.3.1.
As condições a seguir especificadas são-no a título indicativo. Poderão escolher-se outras condições, desde que produzam resultados equivalentes.
|
Coluna analítica (4.3.1) |
|
|
Fase móvel (3.4): |
mistura metanol (3.2)-água (3.3),900 + 100 (v + v) |
|
Caudal: |
1,5-2 ml/minuto |
|
Comprimento de onda de detecção: |
380 nm |
|
Volume injectado: |
20 μl-100 μl |
Verificar a estabilidade do sistema cromatográfico mediante a injecção repetida da solução de calibração (3.5.3) que contém 2,5 μg/ml, até obter picos com alturas (ou áreas) e tempos de retenção reprodutíveis.
5.3.2.
Injectar várias vezes cada solução de calibração (3.5.3) e determinar a altura ou área média dos picos para cada concentração. Traçar uma curva de calibração, colocando a altura (ou área) média dos picos das soluções de calibração em ordenadas e as concentrações correspondentes, expressas em μg/ml, em abcissas.
5.3.3.
Injectar o extracto de amostra (5.2) várias vezes, utilizando o mesmo volume usado para as soluções de calibração, e determinar a altura (ou área) média dos picos do olaquindox.
6. Cálculo dos resultados
A partir da altura (ou área) média dos picos do olaquindox da solução de amostra, determinar a concentração desta em μg/ml, com base na curva de calibração (5.3.2).
O teor de olaquindox, w, expresso em mg/kg de amostra, é dado pela fórmula seguinte:

em que:
|
c |
= |
concentração de olaquindox do extracto da amostra (5.2) em μg/ml |
|
m |
= |
massa da toma para análise em g. |
7. Validação dos resultados
7.1. Identidade
A identidade do analito pode ser confirmada por co-cromatografia ou pela utilização de um detector de díodos que permita comparar os espectros do extracto de amostra (5.2) e da solução de calibração (3.5.3) que contém 5,0 μg/ml.
7.1.1.
Fortificar um extracto de amostra (5.2) com a adição de uma quantidade adequada de uma solução de calibração (3.5.3). A quantidade de olaquindox adicionada deve ser semelhante à quantidade de olaquindox presente no extracto de amostra.
Apenas se deverá verificar um aumento da altura do pico correspondente ao olaquindox, atendendo à quantidade adicionada e à diluição do extracto. A largura do pico a meia altura não poderá diferir mais de 10 % da largura inicial do pico do olaquindox do extracto de amostra não fortificado.
7.1.2.
Os resultados são avaliados de acordo com os seguintes critérios:
|
a) |
O comprimento de onda de absorção máxima dos espectros da amostra e do padrão, registado no máximo do pico cromatográfico, deve ser o mesmo, com uma margem de variação dependente da resolução do sistema de detecção. No caso de um detector de díodos, essa margem é, em geral, de ± 2 nm; |
|
b) |
Entre 220 e 400 nm, os espectros da amostra e do padrão, registados no máximo dos picos cromatográficos, não devem diferir entre si no que respeita às zonas do espectro que apresentem uma absorvância relativa compreendida entre 10 % e 100 %. Este critério considera-se satisfeito se estiverem presentes os mesmos máximos e a diferença entre ambos os espectros não exceder, em caso algum, 15 % da absorvância do analito-padrão; |
|
c) |
Entre 220 e 400 nm, os espectros na curva ascendente, no máximo e na curva descendente do pico do extracto da amostra não devem diferir entre si nas partes do espectro que apresentem uma absorvância relativa compreendida entre 10 e 100 %. Este critério considera-se satisfeito se estiverem presentes os mesmos máximos e a diferença entre os espectros não exceder, em caso algum, 15 % da absorvância do espectro no máximo do pico. |
Se algum destes critérios não for satisfeito, a presença do analito não pode ser confirmada.
7.2. Repetibilidade
A diferença entre os resultados de duas determinações paralelas efectuadas com a mesma amostra não deve exceder 15 % do maior dos valores, para teores de olaquindox entre 10 e 200 mg/kg.
7.3. Recuperação
A taxa de recuperação de uma amostra em branco fortificada deve ser de, pelo menos, 90 %.
8. Resultados de um estudo interlaboratorial
Foi organizado um estudo interlaboratorial a nível comunitário, no âmbito do qual foram analisadas, em 13 laboratórios, quatro amostras de alimento para leitões, incluindo uma amostra em branco. Os resultados desse estudo são apresentados em seguida:
|
|
Amostra 1 |
Amostra 2 |
Amostra 3 |
Amostra 4 |
|
L |
13 |
10 |
11 |
11 |
|
n |
40 |
40 |
44 |
44 |
|
média [mg/kg] |
— |
14,6 |
48,0 |
95,4 |
|
Sr [mg/kg] |
— |
0,82 |
2,05 |
6,36 |
|
SR [mg/kg] |
— |
1,62 |
4,28 |
8,42 |
|
CVr [ %] |
— |
5,6 |
4,3 |
6,7 |
|
CVR [ %] |
— |
11,1 |
8,9 |
8,8 |
|
Teor nominal |
|
|
|
|
|
[mg/kg] |
— |
15 |
50 |
100 |
|
Recuperação % |
— |
97,3 |
96,0 |
95,4 |
|
L |
= |
número de laboratórios |
|
n |
= |
número de valores individuais |
|
sr |
= |
desvio-padrão da repetibilidade |
|
SR |
= |
desvio-padrão da reprodutibilidade |
|
CVr |
= |
coeficiente de variação da repetibilidade |
|
CVR |
= |
coeficiente de variação da reprodutibilidade |
9. Observações
Embora não tenha sido validado em alimentos para animais com teores de olaquindox superiores a 100 mg/kg, o método pode ser utilizado satisfatoriamente mediante uma redução da toma de ensaio e/ou diluição do extracto da amostra (5.2), de forma a obter uma concentração no intervalo da curva de calibração (5.3.2).
(C) DETERMINAÇÃO DO AMPROLIUM
Cloridrato de cloreto de 1-[(4-amino-2-propil-5-pirimidinil)metil]-2-picolínio
1. Objecto e âmbito de aplicação
Este método permite determinar o teor de amprolium em alimentos para animais e pré-misturas. O limite de detecção é de 1 mg/kg; o limite de quantificação de 5 mg/kg.
2. Princípio
Extracção da amostra com uma mistura metanol-água. Diluição com a fase móvel, filtração com um filtro de membrana e determinação do teor de amprolium por cromatografia líquida de alta resolução (HPLC) de permuta catiónica, com um detector de UV.
3. Reagentes
|
3.1. |
Metanol. |
|
3.2. |
Acetonitrilo de qualidade equivalente para HPLC. |
|
3.3. |
Água de qualidade equivalente para HPLC. |
|
3.4. |
Solução de di-hidrogenofosfato de sódio 0,1 mol/l. Num balão volumétrico de 1 000 ml, dissolver em água (3.3) 13,80 g de di-hidrogenofosfato de sódio mono-hidratado, completar o volume com água (3.3) e homogeneizar. |
|
3.5. |
Solução de perclorato de sódio 1,6 mol/l Num balão volumétrico de 1 000 ml, dissolver em água (3.3) 224,74 g de perclorato de sódio mono-hidratado, completar o volume com água (3.3) e homogeneizar. |
|
3.6. |
Fase móvel para HPLC (ver ponto 9.1). Mistura 450 + 450 + 100 (v + v + v) de acetonitrilo (3.2), solução de di-hidrogenofosfato de sódio (3.4) e solução de perclorato de sódio (3.5). Antes de utilizar a fase móvel, filtrar por um filtro de membrana de 0,22 μm (4.3) e desgaseificar a solução [por exemplo, num banho de ultra-sons (4.4) durante pelo menos 15 minutos]. |
|
3.7. |
Substância-padrão: amprolium puro. Cloreto de 1-[(4-amino-2-propilpirimidin-5-il)metil]-2-metilpiridínio, na forma cloridrato, E 750 (ver ponto 9.2). |
|
3.7.1. |
Num balão volumétrico de 100 ml, pesar, com uma aproximação de 0,1 mg, 50 mg de amprolium (3.7) e dissolver em 80 ml de metanol (3.1), colocando depois o balão durante 10 minutos no banho de ultra-sons (4.4). Depois do tratamento por ultra-sons, levar a solução à temperatura ambiente, perfazer o volume com água e homogeneizar. A temperaturas não superiores a 4 oC, a solução mantém-se estável durante um mês. |
|
3.7.2. |
Pipetar 5,0 ml da solução-mãe padrão (3.7.1) para um balão volumétrico de 50 ml, completar o volume com solvente de extracção (3.8) e homogeneizar. A temperaturas não superiores a 4 oC, a solução mantém-se estável durante um mês. |
|
3.7.3. |
Transferir 0,5, 1,0 e 2,0 ml da solução-padrão intermédia (3.7.2) para uma série de balões volumétricos de 50 ml. Perfazer o volume com fase móvel (3.6) e agitar. As soluções obtidas contêm 0,5, 1,0 e 2,0 μg de amprolium por ml, respectivamente, e são preparadas imediatamente antes da utilização. |
|
3.8. |
Solvente de extracção. Mistura 2:1 (v+v) de metanol (3.1) e água. |
4. Equipamento
|
4.1. |
Aparelho para HPLC com sistema de injecção com capacidade de injecção de volumes de 100 μl. |
|
4.1.1. |
Coluna para cromatografia líquida com 125 mm × 4 mm e enchimento de permuta catiónica Nucleosil 10 SA de 5 ou 10 μm, ou equivalente. |
|
4.1.2. |
Detector de UV com regulação do comprimento de onda ou detector de díodos. |
|
4.2. |
Filtro de membrana de PTFE, porosidade 0,45 μm. |
|
4.3. |
Filtro de membrana, porosidade 0,22 μm. |
|
4.4. |
Banho de ultra-sons. |
|
4.5. |
Agitador mecânico ou magnético. |
5. Procedimento
5.1. Generalidades
5.1.1.
Para a realização do ensaio de recuperação (5.1.2), analisa-se uma amostra em branco de um alimento para animais, para comprovar a ausência de amprolium e de substâncias susceptíveis de interferir no processo. Essa amostra em branco deve ser de tipo similar ao da amostra em análise e não devem ser detectados amprolium nem substâncias susceptíveis de interferir no processo.
5.1.2
Deve efectuar-se um ensaio de recuperação utilizando a amostra do ensaio em branco, fortificada com uma quantidade conhecida de amprolium semelhante à quantidade presente na amostra. Para obter uma fortificação a um nível de 100 mg/kg, transferir 10,0 ml da solução-mãe padrão de amprolium (3.7.1) para um erlenmeyer de 250 ml e concentrar por evaporação até cerca de 0,5 ml. Acrescentar 50 g da amostra em branco, misturar cuidadosamente e esperar 10 minutos misturando de novo várias vezes antes de proceder à extracção (5.2).
Caso não se disponha de um alimento para animais para o ensaio em branco de tipo similar ao da amostra (ver ponto 5.1.1), pode determinar-se a recuperação pelo método da adição de padrão. Nesse caso, adiciona-se à amostra a analisar uma quantidade de amprolium semelhante à que já se encontra presente na amostra. A amostra assim obtida é analisada juntamente como a amostra não fortificada e a recuperação é calculada por subtracção.
5.2. Extracção
5.2.1.
Num erlenmeyer de 500 ml, pesar, com uma aproximação de 0,01 g, 5 a 40 g da amostra, em função do teor de amprolium, e adicionar 200 ml do solvente de extracção (3.8). Colocar o erlenmeyer no banho de ultra-sons (4.4) e deixar actuar durante 15 minutos. Retirar o erlenmeyer do banho e agitar durante uma hora em agitador mecânico ou magnético (4.5). Tomar uma alíquota do extracto e diluir com a fase móvel (3.6), de modo a obter um teor de amprolium de 0,5 a 2 μg/ml, e homogeneizar (ver a observação 9.3). Filtrar 5 a 10 ml desta solução diluída por um filtro de membrana (4.2). Efectuar em seguida a análise por HPLC (5.3).
5.2.2.
Num erlenmeyer de 500 ml, pesar, com uma aproximação de 0,001 g, 1 a 4 g da pré-mistura, em função do teor de amprolium, e adicionar 200 ml do solvente de extracção (3.8). Colocar o erlenmeyer no banho de ultra-sons (4.4) e deixar actuar durante 15 minutos. Retirar o erlenmeyer do banho e agitar durante uma hora em agitador mecânico ou magnético (4.5). Tomar uma alíquota do extracto e dilui-la com a fase móvel (3.6), de modo a obter um teor de amprolium de 0,5 a 2 μg/ml, e homogeneizar. Filtrar 5 a 10 ml desta solução diluída por um filtro de membrana (4.2). Efectuar em seguida a análise por HPLC (5.3).
5.3. Análise por HPLC
5.3.1.
As condições a seguir especificadas são-no a título indicativo. Poderão escolher-se outras condições, desde que produzam resultados equivalentes.
|
Coluna para cromatografia |
|
|
líquida (4.1.1): |
125 mm × 4 mm e enchimento de permuta catiónica Nucleosil 10 SA de 10 μm, ou equivalente |
|
Fase móvel (3.6): |
Mistura 450 + 450 + 100 (v + v + v) de acetonitrilo (3.2), solução de di-hidrogenofosfato de sódio (3.4) e solução de perclorato de sódio (3.5) |
|
Caudal: |
0,7-1 ml/minuto |
|
Comprimento de onda de detecção: |
264 nm |
|
Volume injectado: |
100 μl |
Para verificar a estabilidade do sistema cromatográfico, injectar várias vezes a solução de calibração (3.7.3) com 1,0 μg/ml até se obterem alturas de pico e tempos de retenção constantes.
5.3.2.
Injectar várias vezes cada solução de calibração (3.7.3) e determinar a altura (ou área) média dos picos para cada concentração. Traçar uma curva de calibração, colocando a altura (ou área) média dos picos das soluções de calibração em ordenadas e as concentrações correspondentes, expressas em μg/ml, em abcissas.
5.3.3.
Injectar várias vezes um volume de extracto de amostra (5.2) idêntico ao utilizado para as soluções de calibração e determinar a altura (ou área) média dos picos do amprolium.
6. Cálculo dos resultados
A partir da altura (ou área) média dos picos do amprolium da solução de amostra, determinar a concentração desta em μg/ml com base na curva de calibração (5.3.2).
O teor de amprolium, w, da amostra, expresso em mg/kg, é dado pela seguinte fórmula:
 [mg/kg]
[mg/kg]
em que:
|
V |
= |
volume, em ml, do solvente de extracção (3.8), de acordo com o ponto 5.2 (isto é, 200 ml) |
|
c |
= |
concentração de amprolium, em μg/ml, do extracto de amostra (5.2) |
|
f |
= |
factor de diluição, de acordo com o ponto 5.2 |
|
m |
= |
massa, em g, da toma analisada. |
7. Validação dos resultados
7.1. Identidade
A identidade do analito pode ser confirmada por co-cromatografia ou com um detector de díodos que permita comparar os espectros do extracto de amostra (5.2) e da solução de calibração (3.7.3) com 2,0 μg/ml.
7.1.1.
Fortificar um extracto de amostra (5.2) com a adição de uma quantidade adequada de uma solução de calibração (3.7.3). A quantidade de amprolium adicionada deve ser semelhante à existente no extracto de amostra.
Apenas se deverá verificar um aumento da altura do pico correspondente ao amprolium, atendendo à quantidade adicionada e à diluição do extracto. A largura do pico a meia altura não poderá diferir mais de 10 % da largura inicial do pico do amprolium do extracto de amostra não fortificado.
7.1.2.
Os resultados são avaliados de acordo com os seguintes critérios:
|
a) |
O comprimento de onda de absorção máxima dos espectros da amostra e do padrão, registado no máximo do pico cromatográfico, deve ser o mesmo, com uma margem de variação dependente da resolução do sistema de detecção. No caso de um detector de díodos, essa margem é, em geral, de ± 2 nm; |
|
b) |
Entre 210 e 320 nm, os espectros da amostra e do padrão, registados no máximo dos picos cromatográficos, não devem diferir entre si no que respeita às zonas do espectro que apresentem uma absorvância relativa compreendida entre 10 % e 100 %. Este critério considera-se satisfeito se estiverem presentes os mesmos máximos e a diferença entre ambos os espectros não exceder, em caso algum, 15 % da absorvância do analito-padrão; |
|
c) |
Entre 210 e 320 nm, os espectros na curva ascendente, no máximo e na curva descendente do pico do extracto da amostra não devem diferir entre si nas partes do espectro que apresentem uma absorvância relativa compreendida entre 10 e 100 %. Este critério considera-se satisfeito se estiverem presentes os mesmos máximos e a diferença entre os espectros não exceder, em caso algum, 15 % da absorvância do espectro no máximo do pico. |
Se algum destes critérios não for satisfeito, a presença do analito não pode ser confirmada.
7.2. Repetibilidade
A diferença entre os resultados de duas determinações paralelas efectuadas com a mesma amostra não deve exceder:
|
— |
15 % do maior dos valores, para teores de amprolium compreendidos entre 25 mg/kg e 500 mg/kg, |
|
— |
75 mg/kg, para teores de amprolium compreendidos entre 500 mg/kg e 1 000 mg/kg, |
|
— |
7,5 % do maior dos valores, para teores de amprolium superiores a 1 000 mg/kg. |
7.3. Recuperação
Utilizando uma amostra (em branco) fortificada, a recuperação deve ser, pelo menos, de 90 %.
8. Resultados de um estudo interlaboratorial
Apresentam-se no quadro seguinte os resultados das análises efectuadas a três alimentos para aves de capoeira (amostras 1 a 3), um suplemento mineral (amostra 4) e uma pré-mistura (amostra 5) no âmbito de um estudo interlaboratorial.
|
|
Amostra 1 (amostra em branco) |
Amostra 2 |
Amostra 3 |
Amostra 4 |
Amostra 5 |
|
L |
14 |
14 |
14 |
14 |
15 |
|
n |
56 |
56 |
56 |
56 |
60 |
|
média [mg/kg] |
— |
45,5 |
188 |
5 129 |
25 140 |
|
sr [mg/kg] |
— |
2,26 |
3,57 |
178 |
550 |
|
CVr [ %] |
— |
4,95 |
1,90 |
3,46 |
2,20 |
|
SR [mg/kg] |
— |
2,95 |
11,8 |
266 |
760 |
|
CVR [ %] |
— |
6,47 |
6,27 |
5,19 |
3,00 |
|
teor nominal [mg/kg] |
— |
50 |
200 |
5 000 |
25 000 |
|
L |
= |
número de laboratórios |
|
n |
= |
número de valores individuais |
|
sr |
= |
desvio-padrão da repetibilidade |
|
CVr |
= |
coeficiente de variação da repetibilidade |
|
SR |
= |
desvio-padrão da reprodutibilidade |
|
CVR |
= |
coeficiente de variação da reprodutibilidade |
9. Observações
|
9.1. |
Se a amostra contiver tiamina, o pico da tiamina aparecerá no cromatograma imediatamente antes do pico do amprolium. Como o presente método exige a separação do amprolium da tiamina, se a coluna utilizada (4.1.1) não efectuar essa separação, substituir, na fase móvel (3.6) até 50 % do acetonitrilo por metanol. |
|
9.2. |
De acordo com a British Pharmacopoeia, o espectro de uma solução 0,02 mol/l de amprolium em ácido clorídrico 0,1 mol/l apresenta máximos a 246 nm e 262 nm. As absorvâncias serão, respectivamente, 0,84 e 0,80. |
|
9.3. |
O extracto deve ser sempre diluído com fase móvel. Caso contrário, devido a variações da força iónica, o tempo de retenção do pico do amprolium pode apresentar desvios significativos. |
(D) DETERMINAÇÃO DO CARBADOX
N1,N4-Dióxido-3-(2-quinoxalinilmetileno)carbazato de metilo
1. Objecto e âmbito de aplicação
Este método permite determinar o teor de carbadox em alimentos para animais, pré-misturas e preparações. O limite de detecção é de 1 mg/kg; o limite de quantificação de 5 mg/kg.
2. Princípio
Adição de água à amostra, estabilização e extracção com uma mistura metanol-acetonitrilo. No caso dos alimentos para animais, limpeza de uma alíquota do extracto filtrado numa coluna de óxido de alumínio. No caso das pré-misturas e preparações, diluição apropriada de uma alíquota do extracto filtrado com água, metanol e acetonitrilo. Determinação do teor de carbadox por cromatografia líquida de alta resolução (HPLC) de fase reversa, com um detector de UV.
3. Reagentes
|
3.1. |
Metanol. |
|
3.2. |
Acetonitrilo de qualidade equivalente para HPLC. |
|
3.3. |
Ácido acético a 100 %. |
|
3.4. |
Óxido de alumínio (neutro, grau de actividade I). |
|
3.5. |
Mistura metanol-acetonitrilo 1:1 (v + v). Misturar 500 ml de metanol (3.1) com 500 ml de acetonitrilo (3.2). |
|
3.6. |
Ácido acético a 10 %. Diluir 10 ml de ácido acético (3.3) para 100 ml com água. |
|
3.7. |
Acetato de sódio. |
|
3.8. |
Água de qualidade equivalente para HPLC. |
|
3.9. |
Tampão acetato (0,01 mol/l, pH 6,0). Dissolver 0,82 g de acetato de sódio (3.7) em 700 ml de água (3.8) e ajustar o pH a 6,0 com ácido acético (3.6). Transferir para um balão volumétrico de 1 000 ml, perfazer o volume com água (3.8) e homogeneizar. |
|
3.10. |
Fase móvel para HPLC. Misturar 825 ml de tampão acetato (3.9) com 175 ml de acetonitrilo (3.2). Filtrar com um filtro de 0,22 μm (4.5) e desgaseificar a solução (por exemplo por aplicação de ultra-sons durante 10 minutos). |
|
3.11. |
Substância-padrão. Carbadox puro: N1,N4-dióxido-3-(2-quinoxalinilmetileno)carbazato de metilo, E 850. |
|
3.11.1. |
Num balão volumétrico de 250 ml, pesar, com uma aproximação de 0,1 mg, 25 mg de carbadox-padrão (3.11). Dissolver em metanol-acetonitrilo (3.5) por aplicação de ultra-sons (4.7). Depois do tratamento ultrassónico, levar a solução à temperatura ambiente, perfazer o volume com metanol-acetonitrilo (3.5) e homogeneizar. Envolver o balão com folha de alumínio ou utilizar material de vidro ambarizado e guardar no frigorífico. A temperaturas não superiores a 4 oC, a solução mantém-se estável durante um mês. |
|
3.11.2. |
Transferir 2,0, 5,0, 10,0 e 20,0 ml da solução-mãe padrão (3.11.1) para uma série de balões volumétricos de 100 ml. Adicionar 30 ml de água, perfazer o volume com a mistura metanol-acetonitrilo (3.5) e homogeneizar. Envolver os balões com folha de alumínio. As soluções obtidas contêm, respectivamente, 2,0, 5,0, 10,0 e 20,0 μg de carbadox por ml. Estas soluções de calibração devem ser preparadas antes de cada utilização.
|
|
3.12. |
Mistura 300 + 700 (v + v) de água e mistura metanol-acetonitrilo (3.5). Misturar 300 ml de água com 700 ml da mistura metanol-acetonitrilo (3.5). |
4. Equipamento
|
4.1. |
Agitador mecânico ou magnético. |
|
4.2. |
Filtro de fibra de vidro (Whatman GF/A ou equivalente). |
|
4.3. |
Coluna de vidro sinterizado (300 a 400 mm de comprimento e diâmetro interno aproximado de 10 mm) com válvula de escoamento.
|
|
4.4. |
Aparelho para HPLC com sistema de injecção com capacidade de injecção de volumes de 20 μl. |
|
4.4.1. |
Coluna para cromatografia líquida: 300 mm x 4 mm, com enchimento C18 de 10 μm ou equivalente. |
|
4.4.2. |
Detector de UV com regulação do comprimento de onda ou detector de díodos para comprimentos de onda compreendidos entre 225 nm e 400 nm. |
|
4.5. |
Filtro de membrana, porosidade 0,22 μm. |
|
4.6. |
Filtro de membrana, porosidade 0,45 μm. |
|
4.7. |
Banho de ultra-sons. |
5. Procedimento
|
Nota: |
O carbadox é sensível à luz. Trabalhar com luz difusa ou utilizar material de vidro ambarizado ou envolvido em folha de alumínio. |
5.1. Generalidades
5.1.1.
Na determinação da recuperação (5.1.2), analisa-se uma amostra em branco de um alimento para animais, para comprovar a ausência de carbadox e de substâncias susceptíveis de interferir no processo. Essa amostra em branco deve ser de tipo similar ao da amostra em análise e não devem ser detectados carbadox nem substâncias susceptíveis de interferir no processo.
5.1.2.
Deve efectuar-se um ensaio de recuperação utilizando a amostra do ensaio em branco (5.1.1), fortificada com uma quantidade conhecida de carbadox semelhante à quantidade presente na amostra. Para obter uma fortificação a um nível de 50 mg/kg, transferir 5,0 ml de solução-mãe padrão (3.11.1) para um erlenmeyer de 200 ml. Evaporar a solução sob uma corrente de azoto até obter um volume aproximado de 0,5 ml. Adicionar 10 g de amostra para o ensaio em branco, misturar e esperar 10 minutos antes de iniciar a extracção (5.2).
Caso não se disponha de um alimento para animais para o ensaio em branco de tipo similar ao da amostra (ver ponto 5.1.1), pode determinar-se a recuperação pelo método da adição de padrão. Nesse caso, adiciona-se à amostra a analisar uma quantidade de carbadox semelhante à que já se encontra presente e analisa-se a amostra assim obtida juntamente com a amostra sem adição. A recuperação é calculada por subtracção.
5.2. Extracção
5.2.1.
Pesar, com uma aproximação de 0,01 g, cerca de 10 g de amostra. Transferir a quantidade pesada para um erlenmeyer de 200 ml, adicionar 15,0 ml de água, homogeneizar e deixar estabilizar durante cinco minutos. Adicionar 35,0 ml da mistura metanol-acetonitrilo (3.5), tapar e agitar ou misturar durante 30 minutos no agitador mecânico ou magnético (4.1). Filtrar a solução obtida com um papel de filtro de fibra de vidro (4.2). Guardar esta solução para as operações de purificação (5.3).
5.2.2.
Pesar, com uma aproximação de 0,001 g, cerca de 1 g de amostra não triturada. Transferir a quantidade pesada para um erlenmeyer de 200 ml, adicionar 15,0 ml de água, homogeneizar e deixar estabilizar durante cinco minutos. Adicionar 35,0 ml da mistura metanol-acetonitrilo (3.5), tapar e agitar ou misturar durante 30 minutos no agitador mecânico ou magnético (4.1). Filtrar a solução obtida com um papel de filtro de fibra de vidro (4.2).
Pipetar uma alíquota do filtrado para um balão volumétrico de 50 ml. Adicionar 15,0 ml de água, perfazer o volume com a mistura metanol-acetonitrilo (3.5) e homogeneizar. A concentração de carbadox na solução final deve ser próxima de 10 μg/ml. Filtrar uma alíquota com um filtro de 0,45 μm (4.6).
Efectuar em seguida a análise por HPLC (5.4).
5.2.3.
Pesar, com uma aproximação de 0,001 g, cerca de 0,2 g de amostra não triturada. Transferir a quantidade pesada para um erlenmeyer de 200 ml, adicionar 45,0 ml de água, homogeneizar e deixar estabilizar durante cinco minutos. Adicionar 105,0 ml da mistura metanol-acetonitrilo (3.5), tapar e homogeneizar. Aplicar ultra-sons (4.7) à amostra durante 15 minutos e agitar ou misturar durante mais 15 minutos (4.1). Filtrar a solução obtida com um papel de filtro de fibra de vidro (4.2).
Diluir uma alíquota do filtrado com a mistura água-metanol-acetonitrilo (3.12) de modo a obter uma concentração final de carbadox de 10-15 μg/ml (para uma preparação a 10 %, o factor de diluição é 10). Filtrar uma alíquota com um filtro de 0,45 μm (4.6).
Efectuar em seguida a análise por HPLC (5.4).
5.3. Purificação
5.3.1.
Pesar 4 g de óxido de alumínio (3.4) e transferir a quantidade pesada para a coluna de vidro (4.3).
5.3.2.
Transferir 15 ml do extracto filtrado (5.2.1) para a coluna de óxido de alumínio e rejeitar os primeiros 2 ml de eluato. Recolher os 5 ml seguintes e filtrar uma alíquota através de um filtro de 0,45 μm (4.6).
Efectuar em seguida a análise por HPLC (5.4).
5.4. Análise por HPLC
5.4.1.
As condições a seguir especificadas são-no a titulo indicativo. Poderão escolher-se outras condições, desde que produzam resultados equivalentes.
|
Coluna para cromatografia líquida (4.4.1): |
300 mm × 4 mm, com enchimento C18 de 10 μm ou equivalente |
|
Fase móvel (3.10): |
mistura 825 + 175 (v+v) de tampão acetato (3.9) e acetonitrilo (3.2) |
|
Caudal: |
1,5-2 ml/minuto |
|
Comprimento de onda de detecção: |
365 nm |
|
Volume injectado: |
20 μl |
Para verificar a estabilidade do sistema cromatográfico, injectar várias vezes a solução de calibração (3.11.2) com 5,0 μg/ml até se obterem alturas (ou áreas) de pico e tempos de retenção constantes.
5.4.2.
Injectar várias vezes cada solução de calibração (3.11.2) e determinar as alturas (ou áreas) dos picos para cada concentração. Traçar uma curva de calibração, colocando a altura (ou área) média dos picos das soluções de calibração em ordenadas e as concentrações correspondentes, expressas em μg/ml, em abcissas.
5.4.3.
Injectar várias vezes o extracto de amostra [(5.3.2) no caso dos alimentos para animais, (5.2.2) no caso das pré-misturas e (5.2.3) no caso das preparações] e determinar a altura (ou área) média dos picos do carbadox.
6. Cálculo dos resultados
A partir da altura (ou área) média dos picos do carbadox da solução de amostra, determinar a concentração desta em μg/ml com base na curva de calibração (5.4.2).
6.1. Alimentos para animais
O teor de carbadox, w, da amostra, expresso em mg/kg, é dado pela seguinte fórmula:
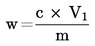 [mg/kg]
[mg/kg]
em que:
|
c |
= |
concentração de carbadox, em μg/ml, no extracto de amostra (5.3.2), |
|
V1 |
= |
volume de extracção, em ml (isto é, 50 ml), |
|
m |
= |
massa da toma analisada, em gramas. |
6.2. Pré-misturas e preparações
O teor de carbadox, w, da amostra, expresso em mg/kg, é dado pela seguinte fórmula:
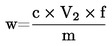 [mg/kg]
[mg/kg]
em que:
|
c |
= |
concentração de carbadox, em μg/ml, no extracto de amostra (5.2.2 ou 5.2.3), |
|
V2 |
= |
volume de extracção, em ml (isto é, 50 ml no caso das pré-misturas e 150 ml no caso das preparações), |
|
f |
= |
factor de diluição, de acordo com 5.2.2 (pré-misturas) ou 5.2.3 (preparações), |
|
m |
= |
massa da toma analisada, em gramas. |
7. Validação dos resultados
7.1. Identidade
A identidade do analito pode ser confirmada por co-cromatografia ou com um detector de díodos que permita comparar os espectros do extracto da amostra e da solução de calibração (3.11.2) com 10,0 μg/ml.
7.1.1.
Adiciona-se uma quantidade apropriada da solução de calibração (3.11.2) a um extracto de amostra. A quantidade de carbadox adicionada deve ser semelhante à que se prevê existir no extracto de amostra.
Apenas se deverá verificar um aumento da altura do pico correspondente ao carbadox, atendendo à quantidade adicionada e à diluição do extracto. A largura do pico a meia altura não poderá diferir mais de 10 % da largura inicial.
7.1.2.
Os resultados são avaliados de acordo com os seguintes critérios:
|
a) |
O comprimento de onda de absorção máxima dos espectros da amostra e do padrão, registado no máximo do pico cromatográfico, deve ser o mesmo, com uma margem de variação dependente da resolução do sistema de detecção. No caso de um detector de díodos, essa margem é, em geral, de ± 2 nm; |
|
b) |
Entre 225 e 400 nm, os espectros da amostra e do padrão, registados no máximo dos picos cromatográficos, não devem diferir entre si no que respeita às zonas do espectro que apresentem uma absorvância relativa compreendida entre 10 % e 100 %. Este critério considera-se satisfeito se estiverem presentes os mesmos máximos e a diferença entre ambos os espectros não exceder, em caso algum, 15 % da absorvância do analito-padrão; |
|
c) |
Entre 225 e 400 nm, os espectros na curva ascendente, no máximo e na curva descendente do pico do extracto da amostra, não devem diferir entre si nas partes do espectro que apresentem uma absorvância relativa compreendida entre 10 e 100 %. Este critério considera-se satisfeito se estiverem presentes os mesmos máximos e a diferença entre os espectros não exceder, em caso algum, 15 % da absorvância do espectro no máximo do pico. |
Se algum destes critérios não for satisfeito, a presença do analito não pode ser confirmada.
7.2. Repetibilidade
Para teores iguais ou superiores a 10 mg/kg, a diferença entre os resultados de duas determinações paralelas efectuadas com a mesma amostra não deve exceder 15 % do maior dos valores.
7.3. Recuperação
Utilizando uma amostra (em branco) fortificada, a recuperação deve ser, pelo menos, de 90 %.
8. Resultados de um estudo interlaboratorial
Foi organizado um estudo interlaboratorial, que envolveu a análise de seis alimentos para animais, quatro pré-misturas e três preparações em oito laboratórios. Para cada amostra, foram efectuadas análises em duplicado. (No Journal of the AOAC, Volume 71, 1988, p. 484-490, podem ser encontrados elementos mais pormenorizados sobre o estudo interlaboratorial efectuado). Os resultados obtidos (com excepção dos valores anómalos) são apresentados no quadro seguinte:
Quadro 1:
Resultados do esrtudo interlaboratorial com alimentos para animais
|
|
Amostra 1 |
Amostra 2 |
Amostra 3 |
Amostra 4 |
Amostra 5 |
Amostra 6 |
|
L |
8 |
8 |
8 |
8 |
8 |
8 |
|
n |
15 |
14 |
15 |
15 |
15 |
15 |
|
Média (mg/kg) |
50,0 |
47,6 |
48,2 |
49,7 |
46,9 |
49,7 |
|
Sr (mg/kg) |
2,90 |
2,69 |
1,38 |
1,55 |
1,52 |
2,12 |
|
CVr ( %) |
5,8 |
5,6 |
2,9 |
3,1 |
3,2 |
4,3 |
|
SR (mg/kg) |
3,92 |
4,13 |
2,23 |
2,58 |
2,26 |
2,44 |
|
CVR ( %) |
7,8 |
8,7 |
4,6 |
5,2 |
4,8 |
4,9 |
|
Teor nominal (mg/kg) |
50,0 |
50,0 |
50,0 |
50,0 |
50,0 |
50,0 |
Quadro 2:
Resultados do teste interlaboratorial com pré-misturas e preparações
|
|
Pré-misturas |
Preparação |
|||||
|
A |
B |
C |
D |
A |
B |
C |
|
|
L |
7 |
7 |
7 |
7 |
8 |
8 |
8 |
|
n |
14 |
14 |
14 |
14 |
16 |
16 |
16 |
|
Média (g/kg) |
8,89 |
9,29 |
9,21 |
8,76 |
94,6 |
98,1 |
104 |
|
Sr (g/kg) |
0,37 |
0,28 |
0,28 |
0,44 |
4,1 |
5,1 |
7,7 |
|
CVr ( %) |
4,2 |
3,0 |
3,0 |
5,0 |
4,3 |
5,2 |
7,4 |
|
SR (g/kg) |
0,37 |
0,28 |
0,40 |
0,55 |
5,4 |
6,4 |
7,7 |
|
CVR ( %) |
4,2 |
3,0 |
4,3 |
6,3 |
5,7 |
6,5 |
7,4 |
|
Teor nominal (g/kg) |
10,0 |
10,0 |
10,0 |
10,0 |
100 |
100 |
100 |
|
L |
= |
número de laboratórios |
|
n |
= |
número de valores individuais |
|
Sr |
= |
desvio-padrão da repetibilidade |
|
CVr |
= |
coeficiente de variação da repetibilidade |
|
SR |
= |
desvio-padrão da reprodutibilidade |
|
CVR |
= |
coeficiente de variação da reprodutibilidade |
ANEXO IX
QUADROS DE CORRESPONDÊNCIA REFERIDOS NO ARTIGO 6.o
1. Directiva 71/250/CEE
|
Directiva 71/250/CEE |
Presente regulamento |
|
Artigo 1.o, primeiro parágrafo |
Artigo 3.o |
|
Artigo 1.o, segundo parágrafo |
Artigo 2.o |
|
Artigo 2.o |
— |
|
Artigo 3.o |
— |
|
Anexo, parte 1 |
Anexo II |
|
Anexo, parte 2 |
— |
|
Anexo, parte 3 |
— |
|
Anexo, parte 4 |
Anexo III, parte O |
|
Anexo, parte 5 |
Anexo III, parte M |
|
Anexo, parte 6 |
Anexo III, parte N |
|
Anexo, parte 7 |
Anexo III, parte Q |
|
Anexo, parte 9 |
Anexo III, parte K |
|
Anexo, parte 10 |
— |
|
Anexo, parte 11 |
— |
|
Anexo, parte 12 |
Anexo III, parte J |
|
Anexo, parte 14 |
Anexo III, parte D |
|
Anexo, parte 16 |
— |
2. Directiva 71/393/CEE
|
Directiva 71/393/CEE |
Presente regulamento |
|
Artigo 1.o |
Artigo 3.o |
|
Artigo 2.o |
— |
|
Artigo 3.o |
— |
|
Anexo, parte I |
Anexo III, parte A |
|
Anexo, parte II |
Anexo III, parte E |
|
Anexo, parte III |
Anexo III, parte P |
|
Anexo, parte IV |
Anexo III, parte H |
3. Directiva 72/199/CEE
|
Directiva 72/199/CEE |
Presente regulamento |
|
Artigo 1.o |
Artigo 3.o |
|
Artigo 2.o |
— |
|
Artigo 3.o |
— |
|
Artigo 4.o |
— |
|
Anexo I, parte 1 |
Anexo III, parte L |
|
Anexo I, parte 2 |
Anexo III, parte C |
|
Anexo I, parte 3 |
— |
|
Anexo I, parte 4 |
— |
|
Anexo I, parte 5 |
Anexo V, parte A |
|
Anexo II |
— |
4. Directiva 73/46/CEE
|
Directiva 73/46/CEE |
Presente regulamento |
|
Artigo 1.o |
Artigo 3.o |
|
Artigo 3.o |
— |
|
Artigo 4.o |
— |
|
Anexo I, parte 1 |
Anexo III, parte B |
|
Anexo I, parte 2 |
— |
|
Anexo I, parte 3 |
Anexo III, parte I |
5. Directiva 76/371/CEE
|
Directiva 76/371/CEE |
Presente regulamento |
|
Artigo 1.o |
Artigo 1.o |
|
Artigo 2.o |
— |
|
Artigo 3.o |
— |
|
Anexo |
Anexo I |
6. Directiva 76/372/CEE
|
Directiva 76/372/CEE |
Presente regulamento |
|
Artigo 1.o |
— |
|
Artigo 2.o |
— |
|
Artigo 3.o |
— |
|
Anexo |
— |
7. Directiva 78/633/CEE
|
Directiva 78/633/CEE |
Presente regulamento |
|
Artigo 1.o |
Artigo 3.o |
|
Artigo 2.o |
— |
|
Artigo 3.o |
— |
|
Anexo, parte 1 |
— |
|
Anexo, parte 2 |
— |
|
Anexo, parte 3 |
Anexo IV, parte C |
8. Directiva 81/715/CEE
|
Directiva 81/715/CEE |
Presente regulamento |
|
Artigo 1.o |
— |
|
Artigo 2.o |
— |
|
Artigo 3.o |
— |
|
Anexo |
— |
9. Directiva 84/425/CEE
|
Directiva 84/425/CEE |
Presente regulamento |
|
Artigo 1.o |
— |
|
Artigo 2.o |
— |
|
Artigo 3.o |
— |
|
Anexo |
— |
10. Directiva 86/174/CEE
|
Directiva 86/174/CEE |
Presente regulamento |
|
Artigo 1.o |
Artigo 4.o |
|
Artigo 2.o |
— |
|
Artigo 3.o |
— |
|
Anexo |
Anexo VII |
11. Directiva 93/70/CEE
|
Directiva 93/70/CEE |
Presente regulamento |
|
Artigo 1.o |
Artigo 3.o |
|
Artigo 2.o |
— |
|
Artigo 3.o |
— |
|
Anexo |
Anexo IV, parte D |
12. Directiva 93/117/CE
|
Directiva 93/117/CE |
Presente regulamento |
|
Artigo 1.o |
Artigos 3.o e 5.o |
|
Artigo 2.o |
— |
|
Artigo 3.o |
— |
|
Anexo, parte 1 |
Anexo IV, parte E |
|
Anexo, parte 2 |
Anexo VIII, parte A |
13. Directiva 98/64/CE
|
Directiva 98/64/CE |
Presente regulamento |
|
Artigo 1.o |
Artigos 3.o e 5.o |
|
Artigo 2.o |
— |
|
Artigo 3.o |
— |
|
Artigo 4.o |
— |
|
Anexo, parte A |
Anexo III, parte F |
|
Anexo, parte B |
Anexo VIII, parte B |
14. Directiva 1999/27/CE
|
Directiva 1999/27/CE |
Presente regulamento |
|
Artigo 1.o |
Artigos 3.o e 5.o |
|
Artigo 2.o |
— |
|
Artigo 3.o |
— |
|
Artigo 4.o |
— |
|
Artigo 5.o |
— |
|
Artigo 6.o |
— |
|
Artigo 7.o |
— |
|
Anexo, parte A |
Anexo VIII, parte C |
|
Anexo, parte B |
Anexo IV, parte F |
|
Anexo, parte C |
Anexo VIII, parte D |
15. Directiva 1999/76/CE
|
Directiva 1999/76/CE |
Presente regulamento |
|
Artigo 1.o |
Artigo 3.o |
|
Artigo 2.o |
— |
|
Artigo 3.o |
— |
|
Artigo 4.o |
— |
|
Anexo |
Anexo IV, parte G |
16. Directiva 2000/45/CE
|
Directiva 2000/45/CE |
Presente regulamento |
|
Artigo 1.o |
Artigo 3.o |
|
Artigo 2.o |
— |
|
Artigo 3.o |
— |
|
Artigo 4.o |
— |
|
Anexo, parte A |
Anexo IV, parte A |
|
Anexo, parte B |
Anexo IV, parte B |
|
Anexo, parte C |
Anexo III, parte G |
17. Directiva 2002/70/CE
|
Directiva 2002/70/CE |
Presente regulamento |
|
Artigo 1.o |
Artigo 1.o |
|
Artigo 2.o |
Artigos 2.o e 3.o |
|
Artigo 3.o |
— |
|
Artigo 4.o |
— |
|
Artigo 5.o |
— |
|
Anexo I |
Anexo I e anexo V, parte B (I) |
|
Anexo II |
Anexo II e anexo V, parte B (II) |
18. Directiva 2003/126/CE
|
Directiva 2003/126/CE |
Presente regulamento |
|
Artigo 1.o |
Artigo 3.o |
|
Artigo 2.o |
— |
|
Artigo 3.o |
— |
|
Artigo 4.o |
— |
|
Artigo 5.o |
— |
|
Artigo 6.o |
— |
|
Anexo |
Anexo VI |
|
26.2.2009 |
PT |
Jornal Oficial da União Europeia |
L 54/s3 |
AVISO AO LEITORAs instituições europeias decidiram deixar de referir, nos seus textos, a última redacção dos actos citados.Salvo indicação em contrário, entende-se que os actos aos quais é feita referência nos textos aqui publicados correspondem aos actos com a redacção em vigor.


 = Poder rotatório específico do amido puro (ver ponto 6.1).
= Poder rotatório específico do amido puro (ver ponto 6.1).