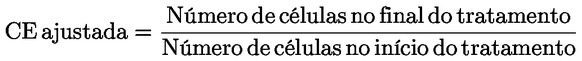|
(8)
|
Na parte B, são aditados os seguintes capítulos:
«B.63 ENSAIO DE TOXICIDADE PARA A REPRODUÇÃO/O DESENVOLVIMENTO
INTRODUÇÃO
|
1.
|
O presente método de ensaio é equivalente à Test Guideline 421 (2016) da OCDE. As diretrizes da OCDE para o ensaio de produtos químicos são revistas periodicamente à luz do progresso científico. A diretriz n.o 421 para os testes de rastreio inicial foi adotada em 1995, com base num protocolo «ensaio preliminar de despistagem da toxicidade para a reprodução/o desenvolvimento», debatido em duas reuniões de peritos, realizadas em Londres, em 1990, (1) e em Tóquio, em 1992 (2).
|
|
2.
|
O presente método de ensaio foi atualizado com parâmetros relativos a perturbadores do sistema endócrino, na sequência da atividade da OCDE altamente prioritária, iniciada em 1998, de revisão das diretrizes de ensaio existentes e elaboração de novas orientações para a análise e o ensaio de possíveis perturbadores do sistema endócrino (3). Neste contexto, a TG 407 da OCDE (estudo de toxicidade pela repetição de uma dose oral durante 28 dias em roedores – capítulo B.7 do presente anexo) foi enriquecida em 2008 com parâmetros adequados para detetar a atividade endócrina de produtos químicos em estudo. O objetivo da atualização da Test Guideline 421 era incluir alguns parâmetros importantes no rastreio dos perturbadores do sistema endócrino nas Test Guideline cujos períodos de exposição abrangem alguns dos períodos sensíveis do desenvolvimento (período pré-natal e primeira fase do período pós-natal).
|
|
3.
|
Os restantes parâmetros aplicáveis aos perturbadores do sistema endócrino selecionados, igualmente incluídos na Test Guideline 443 (estudo alargado da toxicidade reprodutiva numa geração – capítulo B.56 deste anexo) foram incluídos na Test Guideline 421 com base num estudo de viabilidade sobre as questões científicas e técnicas, bem como em eventuais adaptações da conceção do ensaio necessárias à sua inclusão (4).
|
|
4.
|
O presente método de ensaio destina-se a gerar informações limitadas sobre os efeitos de um produto químico em estudo no desempenho reprodutor masculino e feminino, como a função gonadal, o comportamento de acasalamento, a fecundação e o desenvolvimento do feto e o parto. Não constitui uma alternativa aos métodos de ensaio B.31, B.34, B.35 ou B.56, nem os substitui.
|
CONSIDERAÇÕES INICIAIS
|
5.
|
O presente método de ensaio pode ser utilizado para fornecer informações preliminares sobre possíveis efeitos na reprodução e/ou no desenvolvimento, quer numa fase inicial da avaliação das propriedades toxicológicas dos produtos químicos, quer sobre produtos químicos que suscitem preocupação. Pode também ser utilizado no âmbito de um conjunto de ensaios preliminares com produtos químicos existentes sobre os quais se disponha de pouca ou nenhuma informação toxicológica, como estudo exploratório de determinação da gama de dosagens para estudos mais exaustivos sobre a reprodução/o desenvolvimento, ou sempre que for considerado importante. Na realização do estudo, deverão seguir-se os princípios de orientação e as considerações descritas no documento de orientação 19 da OCDE quanto ao reconhecimento, avaliação e utilização dos sinais clínicos como parâmetros extrapoláveis aos seres humanos para experiências com animais utilizados em avaliações de segurança (5).
|
|
6.
|
O presente método de ensaio não fornece informações completas sobre todos os aspetos da reprodução e do desenvolvimento. Faculta apenas, nomeadamente, meios limitados de deteção de manifestações pós-natais de exposição pré-natal ou de efeitos que possam ser induzidos durante a exposição pós-natal. Devido, entre outros motivos, ao número relativamente reduzido de animais dos grupos de dosagem, à seletividade dos parâmetros e à curta duração do estudo, o método não fornece dados que apoiem a declaração definitiva de inexistência de efeitos. Além disso, na ausência de dados de outros ensaios de toxicidade para a reprodução/o desenvolvimento, os resultados positivos são úteis para a avaliação preliminar dos perigos e contribuem para informar as decisões relativas à necessidade e à oportunidade de ensaios adicionais.
|
|
7.
|
Os resultados correspondentes aos parâmetros relacionados com o sistema endócrino devem interpretar-se com base no documento Conceptual Framework for Testing and Assessment of Endocrine Disrupting Chemicals (6), elaborado pela OCDE, que estabelece um quadro conceptual para o ensaio e a avaliação de produtos químicos perturbadores do sistema endócrino. Nesse quadro, o método correspondente à Test Guideline 421 da OCDE melhorada consta do nível 4, como ensaio in vivo que fornece dados sobre efeitos nocivos nos parâmetros pertinentes do sistema endócrino. Contudo, um sinal do sistema endócrino não pode, por si só, ser considerado prova suficiente de que o produto em causa é perturbador do sistema endócrino.
|
|
8.
|
O presente método prevê a administração por via oral do produto químico em estudo. Poderá ser necessário introduzir alterações se forem utilizadas outras vias de exposição.
|
|
9.
|
Antes da aplicação do método de ensaio a uma mistura para obter dados com fins normativos, importa ponderar se – e, em caso afirmativo, por que motivo – o método pode proporcionar resultados adequados para o efeito. Essas considerações não são necessárias se existir um requisito normativo para o ensaio da mistura.
|
|
10.
|
Definem-se no apêndice 1 alguns conceitos utilizados.
|
PRINCÍPIO DO ENSAIO
|
11.
|
O produto químico em estudo é administrado, em doses escalonadas, a vários grupos de machos e fêmeas. Os machos devem ser tratados durante, pelo menos, quatro semanas – até ao dia anterior ao da eutanásia, inclusive –, o que abrange um mínimo de duas semanas antes do acasalamento, o período de acasalamento e aproximadamente duas semanas após este. Tendo em conta o período limitado de dosagem antes do acasalamento dos machos, a fertilidade não pode constituir um indicador particularmente sensível da toxicidade testicular. Por conseguinte, é essencial um exame histológico pormenorizado dos testículos. A combinação de um período de dosagem pré-acasalamento de duas semanas e de observações subsequentes de acasalamento/fertilidade com um período global de dosagem de, pelo menos, quatro semanas, seguido de histopatologia pormenorizada das gónadas do macho, é considerada suficiente para permitir a deteção da maioria dos efeitos na fertilidade masculina e na espermatogénese.
|
|
12.
|
As fêmeas devem ser tratadas ao longo de todo o estudo – ou seja, duas semanas antes do acasalamento –, com o objetivo de abranger, pelo menos, dois ciclos éstricos completos, o período variável até à fecundação, a duração da gravidez e, pelo menos, treze dias após o parto, até ao dia anterior ao da eutanásia, inclusive.
|
|
13.
|
A duração do estudo após a aclimatação, com uma avaliação do ciclo éstrico antes do início do tratamento, depende do desempenho das fêmeas, e é de cerca de 63 dias [pelo menos 14 dias antes do acasalamento, até 14 dias de acasalamento, 22 dias de gestação, 13 dias de aleitamento].
|
|
14.
|
No período de administração, deve verificar-se atentamente, todos os dias, se os animais evidenciam sinais de toxicidade. Os animais que morrem ou são eutanasiados durante o ensaio são autopsiados; no final do ensaio, são eutanasiados e autopsiados os animais sobreviventes.
|
DESCRIÇÃO DO MÉTODO
Seleção de espécies animais
|
15.
|
O presente método de ensaio foi concebido para ratazanas. Se os parâmetros que nele se especificam forem estudados noutra espécie de roedor, essa opção deve ser justificada pormenorizadamente. No programa de validação internacional para a deteção de perturbadores do sistema endócrino com base na TG 407 da OECD (correspondente ao capítulo B.7 do presente anexo), as ratazanas foram a única espécie utilizada. Deve evitar-se o recurso a estirpes de fecundidade baixa ou com uma incidência elevada de deficiências de desenvolvimento. Devem utilizar-se animais virgens saudáveis, não sujeitos a experiências anteriores, e especificar-se a espécie, a estirpe, o sexo, o peso e/ou a idade dos animais. No início do estudo, a diferença de peso entre os animais deve ser mínima, não devendo exceder 20 % do peso médio dos animais de cada sexo. Nos casos em que for realizado como estudo preliminar de um estudo a longo prazo ou de geração, é preferível utilizar animais da mesma estirpe e proveniência em ambos os estudos.
|
Condições de alojamento e de alimentação
|
16.
|
Todos os procedimentos devem respeitar as normas locais de manipulação de animais de laboratório. A temperatura do compartimento experimental dos animais deve ser de 22 °C (±3). A humidade relativa deve estar compreendida entre 50 % e 60 %, embora sejam aceitáveis valores compreendidos entre 30 %, no mínimo, e um valor máximo que, preferencialmente, não deve exceder 70 %, salvo durante os períodos de limpeza do biotério. A iluminação deve ser artificial, com uma sequência de 12 horas de luz seguidas de 12 horas de escuridão. Para a alimentação, podem ser usadas dietas convencionais de laboratório, com acesso ilimitado a água potável. A escolha da dieta pode depender da necessidade de garantir uma mistura adequada do produto químico em estudo, quando administrado por essa via.
|
|
17.
|
Os animais devem ser alojados em pequenos grupos do mesmo sexo. Podem ser alojados individualmente, se tal se justificar do ponto de vista científico. Em caso de alojamento coletivo, cada gaiola não deve alojar mais de cinco animais. O acasalamento deve ocorrer em gaiolas adequadas para o efeito. As fêmeas prenhes devem ser colocadas em gaiolas individuais e dispor de materiais de nidificação. As fêmeas em lactação devem ser colocadas em gaiolas individuais com as crias.
|
|
18.
|
Deve efetuar-se com regularidade uma pesquisa de contaminantes nos alimentos fornecidos e conservar-se uma amostra da dieta até o relatório estar concluído.
|
Preparação dos animais
|
19.
|
Distribuem-se aleatoriamente animais adultos, jovens e saudáveis pelos grupos de controlo e pelos grupos expostos. As gaiolas devem estar dispostas de forma a minimizar possíveis efeitos derivados do seu posicionamento. Os animais são identificados de forma inequívoca, devendo ser aclimatados às condições de laboratório durante, pelo menos, cinco dias antes do início do estudo.
|
Preparação das doses
|
20.
|
Recomenda-se que a consulta do PON pertinente aquando da implementação e utilização de um desses modelos no laboratório. Se for selecionada a via oral, o produto químico em estudo é geralmente administrado por gavagem; no entanto, os produtos químicos em estudo podem ser administrados através dos alimentos ou da água de beber.
|
|
21.
|
Quando necessário, o produto químico em estudo pode ser dissolvido ou suspenso num veículo adequado. Sempre que possível, recomenda-se, como primeira opção, uma solução ou suspensão aquosa; caso isso não seja viável, pode optar-se por uma solução ou emulsão num óleo (por exemplo, em óleo de milho); em último caso, pode recorrer-se a uma solução noutro veículo. Se este não for aquoso, devem conhecer-se as suas características tóxicas. Importa determinar a estabilidade e a homogeneidade do produto químico em estudo no veículo.
|
PROCEDIMENTO
Número e sexo dos animais
|
22.
|
Recomenda-se que cada grupo seja iniciado com, pelo menos, 10 machos e 12-13 fêmeas. As fêmeas serão avaliadas para pré-exposição do ciclo éstrico; os animais que não apresentem ciclos típicos de 4-5 dias não são incluídos no estudo. Assim, é recomendável utilizar um número mais elevado de fêmeas, a fim de assegurar a presença de 10 fêmeas reprodutoras em cada grupo. Exceto no caso de efeitos tóxicos marcados, prevê-se que o número de fêmeas prenhes por grupo seja, no mínimo, igual a 8, que, normalmente, é o número mínimo aceitável de fêmeas prenhes por grupo. O objetivo é produzir gravidezes e crias em número suficiente para assegurar uma avaliação significativa do potencial do produto químico em estudo para afetar a fertilidade, a gravidez, o comportamento materno e de aleitamento, o crescimento e o desenvolvimento da geração F1, desde a conceção até ao dia 13 pós-parto.
|
Dosagem
|
23.
|
De modo geral, devem utilizar-se, no mínimo, três lotes de ensaio e um lote de controlo. As doses podem basear-se em informações dos ensaios de toxicidade aguda ou em resultados de estudos de dose repetida. Salvo no que respeita à exposição ao produto químico em estudo, os animais dos grupos de controlo devem ser tratados do mesmo modo que os animais dos grupos ensaiados. Se for utilizado um veículo para administrar o produto químico em estudo, o grupo de controlo deve receber o volume máximo de veículo utilizado.
|
|
24.
|
Na seleção das doses devem ter-se em conta os dados eventualmente disponíveis em matéria de toxicidade e toxicocinética. Deve também ter-se em conta o facto de poder haver diferenças de sensibilidade entre animais prenhes e não prenhes. Importa escolher como dose mais elevada uma dose que induza efeitos tóxicos, mas não cause mortalidade nem sofrimento intenso. Deve selecionar-se uma sequência descendente de doses que permita detetar qualquer resposta relacionada com estas e determinar o nível sem efeito nocivo observável (NSEAO) à dose mais baixa. A inclusão de um quarto grupo de ensaio é muitas vezes preferível ao uso de intervalos muito grandes (fatores superiores a 10) entre as dosagens.
|
|
25.
|
Caso se observem sinais de toxicidade generalizada (por exemplo, redução do peso corporal, efeitos ao nível hepático, cardíaco, pulmonar ou renal, etc.) ou outras alterações que possam não ser reações tóxicas (por exemplo, diminuição da quantidade de alimentos ingerida, dilatação hepática, etc.), deverá interpretar-se com cautela qualquer efeito ao nível dos parâmetros endócrinos.
|
Ensaio no limite
|
26.
|
Se um estudo oral com uma dose de, pelo menos, 1 000 mg/kg de peso corporal/dia – ou, no caso da administração por via alimentar ou pela água de beber, de uma percentagem equivalente na alimentação ou na água de beber –, utilizando os procedimentos descritos para o estudo, não produzir efeitos tóxicos observáveis e, com base nos dados relativos a substâncias estruturalmente afins, não for de esperar efeitos tóxicos, poderá não ser necessário efetuar um estudo completo com várias doses. O ensaio-limite é sempre válido, exceto nos casos em que se preveja exposição humana e em que seja necessário testar um nível de dose oral mais elevado. Quando se recorre a outras formas de administração, como a inalação ou a aplicação cutânea, as propriedades físico-químicas do produto químico de ensaio são muitas vezes indicativas e limitativas do nível máximo de exposição praticável.
|
Administração das doses
|
27.
|
Os animais são tratados diariamente com o produto químico em estudo durante 7 dias por semana. A administração forçada por meio de uma sonda esofágica deve efetuar-se numa dose única, utilizando um tubo estomacal ou uma cânula de intubação adequada. O volume máximo de líquido administrado em cada operação depende das dimensões do animal, não devendo exceder 1 ml/100 g de massa corporal, exceto no caso de soluções aquosas, em que podem administrar-se 2 ml/100 g de massa corporal. Exceto para produtos químicos irritantes ou corrosivos, que normalmente produzem efeitos exacerbados em concentrações superiores, a variabilidade no volume de ensaio deve ser minimizada, ajustando a concentração de modo a garantir um volume constante em todos os níveis de dosagem.
|
|
28.
|
No caso de produtos químicos em estudo administrados através da alimentação ou da água de beber, é importante assegurar que as quantidades do produto não interferem com a nutrição normal ou com a composição da água. Se o produto químico em estudo for administrado na alimentação, pode optar-se por concentrações constantes nesta (da ordem das ppm) ou por doses constantes em relação ao peso corporal de cada animal. Caso o produto químico em estudo seja administrado por gavagem, a dose deve ser administrada todos os dias à mesma hora, devendo ajustar-se, pelo menos, uma vez por semana, a fim de se manter uma dose constante em relação ao peso corporal do animal.
|
Calendário da experiência
|
29.
|
O tratamento de ambos os sexos deve ter início 2 semanas antes do acasalamento, depois de os animais serem aclimatados durante pelo menos 5 dias e de as fêmeas serem sujeitas a exames de deteção de ciclos éstricos normais (num período de 2 semanas anterior ao tratamento). O estudo deve ser programado de forma a que a avaliação do ciclo éstrico comece pouco depois de os animais terem atingido a plena maturidade sexual, o que pode variar ligeiramente nas diferentes linhagens de ratazanas em laboratórios diferentes, sendo, por exemplo, às 10 semanas nas ratazanas Sprague Dawley e cerca das 12 semanas nas ratazanas Wistar. As mães com crias devem ser eutanasiadas no dia 13 após o parto, ou pouco depois. O dia de nascimento (ou seja, quando o parto é concluído) é definido como o dia 0 após o parto. As fêmeas que não apresentem qualquer indício de copulação são mortas 24 a 26 dias após o último dia do período de acasalamento. O nível de dosagem é mantido em ambos os sexos durante o período de acasalamento. Os machos devem continuar a ser tratados após o período de acasalamento, pelo menos até ter decorrido o período total mínimo de dosagem, de 28 dias. Em seguida, são eutanasiados ou, em alternativa, são mantidos e continuam a ser tratados, para um eventual segundo acasalamento, se isso for considerado adequado.
|
|
30.
|
As fêmeas prenhes devem continuar a ser tratadas durante a gravidez e, pelo menos, até ao dia 13 após o parto ou ao dia anterior ao abate. No caso de o produto químico em estudo ser administrado por inalação ou por via cutânea, o tratamento deve continuar a ser administrado pelo menos até ao dia 19 de gestação, inclusive, e retomado o mais rapidamente possível, o mais tardar no dia 4 após o parto.
|
|
31.
|
O apêndice 2 apresenta um diagrama do calendário da experiência.
|
Processo de acasalamento
|
32.
|
Normalmente, devem ser utilizados neste estudo acasalamentos de 1:1 (um macho e uma fêmea). Pode haver exceções em caso de morte ocasional de machos. A fêmea deve permanecer com o mesmo macho até se observarem indícios de copulação ou terem decorrido duas semanas. Deverão examinar-se as fêmeas todas as manhãs, para verificar a presença de esperma ou de rolhão vaginal. O dia 0 da gravidez é definido como o dia em que houver provas de acasalamento (deteção de rolhão vaginal ou esperma). Se a tentativa de acasalamento for mal sucedida, poderá tentar-se um novo acasalamento das fêmeas com machos do mesmo grupo comprovadamente aptos a procriar.
|
Número de animais por ninhada
|
33.
|
No dia 4 após o parto, o número de animais de cada ninhada pode ser ajustado eliminando as crias excessivas por seleção aleatória, a fim de se obter, na medida do possível, quatro ou cinco crias por sexo e por ninhada, consoante o tamanho normal das ninhadas das estirpes de ratazanas utilizadas. Devem ser colher-se amostras de sangue de duas das crias excedentárias, que serão agrupadas e utilizadas para a determinação dos níveis do soro T4. Não é adequada a eliminação seletiva das crias, por exemplo, com base no peso corporal ou na distância anogenital (DAG). Sempre que o número de crias do sexo masculino ou feminino impeça a obtenção de quatro ou cinco animais de cada sexo por ninhada, é admissível um ajustamento parcial (por exemplo, seis machos e quatro fêmeas). Não devem ser eliminadas crias se as ninhadas ficarem aquém da meta de abate (8 ou 10 crias/ninhada). Se existir uma única cria além da meta de abate, apenas uma cria deve ser eliminada e utilizada para a colheita de sangue para possíveis avaliações séricas de T4.
|
|
34.
|
Se o tamanho da ninhada não for ajustado, eutanasiam-se duas crias por ninhada no dia 4 após o nascimento e são colhidas amostras de sangue para medição das concentrações de hormonas da tiroide. Se possível, essas duas crias de cada ninhada devem ser fêmeas, a fim de reservar as crias do sexo masculino para as avaliações de retenção de mamilo, exceto se essas crias não deixarem outras fêmeas para avaliação no termo da experiência. Não devem ser eliminadas crias se a ninhada ficar com menos de 8 ou 10 crias/ninhada (em função do tamanho normal das ninhadas das estirpes de ratazanas utilizadas). Se existir apenas uma cria além do tamanho normal das ninhadas, apenas uma cria deve ser eliminada e utilizada para a colheita de sangue destinada a possíveis avaliações séricas de T4.
|
Observações in vivo
Observações clínicas
|
35.
|
Ao longo de todo o período de ensaio, devem efetuar-se exames clínicos gerais, pelo menos, uma vez por dia, frequência que pode aumentar em caso de observação de sinais de toxicidade. Devem ser feitas, de preferência, à(s) mesma(s) hora(s) todos os dias e tendo em conta o período de pico dos efeitos previstos após a administração da dose. Devem registar-se modificações de comportamento significativas, sinais de parto difícil ou prolongado, bem como qualquer sinal de toxicidade, incluindo a mortalidade. Estes registos devem indicar o momento da ocorrência, o grau e a duração dos sinais de toxicidade.
|
Peso corporal e consumo de alimentos/água
|
36.
|
Os machos e as fêmeas devem ser pesados no primeiro dia de tratamento, pelo menos uma vez por semana a partir de então e no final. Durante a gestação, as fêmeas devem ser pesadas nos dias 0, 7, 14 e 20 e nas 24 horas seguintes ao parto (dia 0 ou 1 pós-parto) e, pelo menos, nos dias 4 e 13 pós-parto. As observações devem ser registadas individualmente para cada animal adulto.
|
|
37.
|
Durante o pré-acasalamento, a gestação e o aleitamento, o consumo de alimentos deve ser medido pelo menos uma vez por semana. A medição do consumo de alimentos durante o acasalamento é facultativa. Se o produto químico em estudo for administrado através da água para beber, o consumo de água durante estes períodos também deve ser determinado.
|
Ciclos éstricos
|
38.
|
Os ciclos éstricos devem ser monitorizados antes do início do tratamento, de forma a selecionar para o estudo fêmeas com um ciclo regular (ver ponto 22). Os esfregaços vaginais também devem ser monitorizados diariamente desde o início do período de tratamento até haver indicações de acasalamento. Se houver preocupações quanto a efeitos agudos causados pelo stress que possam alterar os ciclos éstricos no início do tratamento, os laboratórios podem expor os animais durante 2 semanas, e em seguida colher esfregaço vaginal diariamente, a fim de monitorizar o ciclo éstrico durante, pelo menos, duas semanas no período de pré-acasalamento e igualmente durante o período de acasalamento, até que haja provas de copulação. Aquando da colheita de células vaginais/cervicais, deve ter-se o cuidado de evitar perturbar a mucosa, o que pode induzir uma pseudogravidez (7) (8).
|
Parâmetros relativos à descendência
|
39.
|
A duração da gestação deve ser registada e calculada a partir do dia 0. Cada ninhada deve ser examinada o mais rapidamente possível após o nascimento, a fim de determinar o número e o sexo das crias, os nados-mortos, os nados-vivos e as crias que são significativamente menores do que as crias de controlo correspondentes, bem como a presença de anomalias relevantes.
|
|
40.
|
As crias vivas devem ser contadas e determinado o sexo de cada uma; as ninhadas devem ser pesadas nas 24 horas seguintes ao parto (dia 0 ou 1 após o parto) e, pelo menos, nos dias 4 e 13 pós-parto. Para além das observações descritas no ponto 35, deve registar-se qualquer comportamento anormal das crias.
|
|
41.
|
A DAG de cada cria deve ser medida no mesmo dia pós-natal, entre o dia 0 e o dia 4. O peso corporal da cria deve ser medido no dia em que a DAG for determinada; as DAG devem ser normalizadas de acordo com uma medida do tamanho da cria, preferencialmente a raiz cúbica do peso corporal (9). O número de mamilos/aréolas em crias do sexo masculino deve ser contado no dia 12 ou 13 após o nascimento, como recomenda a GD 151 (10) da OCDE.
|
Bioquímica clínica
|
42.
|
As amostras de sangue são colhidas num ponto específico, de acordo com o seguinte calendário:
|
—
|
pelo menos, de duas crias por ninhada, no dia 4 após o nascimento, se o número de crias o permitir (ver pontos 33 a 34)
|
|
—
|
de todas as mães e, pelo menos, de duas crias por ninhada, no final (dia 13), e
|
|
—
|
de todos os machos adultos, no final.
|
|
Todas as amostras de sangue são armazenadas em condições adequadas. As amostras de sangue do dia 13 das crias e dos machos adultos são analisadas para determinação dos níveis séricos de hormonas da tiroide (T4). Se necessário, procede-se a uma avaliação mais aprofundada de T4 nas amostras de sangue das mães e do dia 4 das crias. Em alternativa, podem ser medidas outras hormonas, se necessário. O sangue das crias pode ser agrupado por ninhada para a realização de análises das hormonas da tiroide. As hormonas da tiroide (T4 e TSH) devem ser, de preferência, medidas como «totais».
|
43.
|
Os fatores a seguir indicados podem afetar a variabilidade dos resultados das análises hormonais e as concentrações absolutas nelas determinadas:
|
—
|
momento da eutanásia, devido à variação das concentrações hormonais ao longo do dia,
|
|
—
|
método utilizado para eutanasiar os animais sem lhes causar tensões desnecessárias, que poderiam afetar as concentrações hormonais,
|
|
—
|
diferenças ao nível das curvas de calibração dos conjuntos para as determinações hormonais.
|
|
|
44.
|
As amostras de plasma especificamente destinadas a determinações hormonais devem colher-se à mesma hora do dia. Os conjuntos existentes no comércio para determinar concentrações hormonais podem dar valores diferentes.
|
Patologia
Autópsia macroscópica
|
45.
|
No momento da eutanásia ou da morte durante o estudo, os animais adultos são objeto de um exame macroscópico, a fim de se detetarem quaisquer anomalias estruturais ou modificações patológicas. Deve prestar-se especial atenção aos órgãos do sistema reprodutivo. O número de locais de implantação deve ser registado. Os esfregaços vaginais devem ser examinados na manhã do dia da autópsia, para determinar a fase do ciclo éstrico e permitir a correlação com a histopatologia dos ovários.
|
|
46.
|
Os testículos e os epidídimos, bem como a próstata e as vesículas seminais, com as glândulas coagulantes, de todos os machos adultos devem ser limpos de qualquer tecido aderente, conforme adequado, e o seu peso líquido deve ser determinado logo que possível após a dissecação, para evitar a secagem. Além disso, os pesos de órgãos facultativos podem incluir o complexo formado pelos músculos elevatórios do ânus e bulbocavernosos, as glândulas de Cowper e a glande peniana, nos machos, e os ovários (em peso húmido) e o útero (incluindo o colo do útero), nas fêmeas;
|
|
47.
|
As crias mortas e as crias eutanasiadas até ao dia 13 após o parto ou pouco depois, devem, pelo menos, ser cuidadosamente examinadas externamente para efeitos de deteção de anomalias macroscópicas. O aparelho reprodutor externo deve ser objeto de especial atenção, devendo ser inspecionado para a deteção de sinais de alterações no desenvolvimento. No dia 13, deve conservar-se a tiroide de uma cria macho e de uma cria fêmea por ninhada.
|
|
48.
|
Devem conservar-se os ovários, os testículos, os órgãos sexuais secundários (útero e colo do útero, epidídimos, próstata, vesículas seminais e glândulas de coagulação), a tiroide e todos os órgãos com lesões macroscópicas de todos os animais adultos. A fixação em formalina não é recomendada para o exame de rotina dos testículos e dos epidídimos. A utilização de fixador de Bouin ou de Davidatos modificado constitui um método aceitável para estes tecidos (11). Para que o fixador penetre rapidamente, deve puncionar-se superficialmente a túnica albugínea com uma agulha, com cuidado, em ambos os polos do órgão.
|
Histopatologia
|
49.
|
Devem ser efetuados exames histológicos pormenorizados aos ovários, aos testículos e aos epidídimos (com especial ênfase nas fases da espermatogénese e na histopatologia da estrutura celular testicular intersticial) dos animais do grupo de dosagem mais elevada e do grupo de controlo. Os outros órgãos conservados, incluindo a tiroide das crias e dos animais adultos, podem ser examinados, se necessário. A pesagem da tiroide pode realizar-se após fixação. A remoção dos tecidos aderentes à tiroide deve efetuar-se com muito cuidado e também só depois da fixação, para evitar danificar tecidos, o que, a ocorrer, poderia comprometer a análise histopatológica. Os exames devem ser alargados aos animais de outros grupos de dosagens, quando forem observadas alterações no grupo de dosagem mais elevada. O documento com a referência 11, que contém orientações no domínio da histopatologia, dá mais informações sobre a dissecação, a fixação, a colheita de amostras e a histopatologia de tecidos do sistema endócrino.
|
DADOS E RELATÓRIOS
Dados
|
50.
|
Devem apresentar-se os dados individuais para cada animal. Além disso, todos os dados devem ser resumidos num quadro, indicando, para cada grupo de ensaio, o número de animais no início deste, o número de animais encontrados mortos durante o ensaio ou abatidos por intervenção humana, a hora da morte de cada animal, a descrição e evolução temporal dos efeitos tóxicos, o número de animais férteis, o número de fêmeas prenhes, o número de animais que apresentam sinais de toxicidade, uma descrição dos sinais de toxicidade observados, incluindo o momento em que surgiram, a duração e a gravidade dos eventuais efeitos tóxicos, os tipos de alterações histopatológicas e quaisquer dados relevantes sobre a ninhada. No apêndice 3 figura um modelo de relatório sob a forma de tabela, que se tem revelado muito útil para a avaliação de efeitos na reprodução/no desenvolvimento.
|
|
51.
|
Devido às dimensões limitadas do estudo, as análises estatísticas, sob a forma de ensaios de «significância», têm um valor limitado para muitos parâmetros, em especial os parâmetros de reprodução. Se se utilizarem análises estatísticas, o método escolhido deve ser adequado à distribuição da variável examinada, que importa selecionar antes do início do estudo. A análise estatística da DAG e da retenção de mamilo deve basear-se em dados individuais das crias, tendo em conta os efeitos da ninhada. Se for adequado, a ninhada é escolhida como unidade de análise. A análise estatística do peso corporal das crias deve basear-se nos dados relativos a cada indivíduo, tendo em conta o tamanho da ninhada. Devido ao pequeno tamanho do grupo, a utilização de dados históricos de controlo (por exemplo, para o tamanho das ninhadas), quando disponíveis, pode também ser útil para auxiliar a interpretação do estudo.
|
Avaliação dos resultados
|
52.
|
As conclusões do estudo de toxicidade devem ser ponderadas com base nos efeitos observados, na autópsia e nos resultados microscópicos. A análise deverá considerar a relação (ou a ausência de relação) entre a dose do produto químico em estudo e a presença ou ausência, incidência e gravidade de anomalias, incluindo lesões macroscópicas, órgãos identificados como alvos, infertilidade, alterações na reprodução e na procriação, alterações do peso corporal, efeitos na mortalidade e quaisquer outros efeitos tóxicos.
|
|
53.
|
Devido ao período reduzido de tratamento dos machos, a histopatologia dos testículos e dos epidídimos deve ser analisada juntamente com os dados de fertilidade, aquando da avaliação dos efeitos para a reprodução masculina. A utilização de dados históricos de controlo sobre a reprodução/o desenvolvimento (por exemplo, para o tamanho da ninhada, a DAG, a retenção de mamilo, os níveis séricos de T4), quando disponíveis, pode também ser útil para auxiliar a interpretação do estudo.
|
|
54.
|
Para fins de controlo de qualidade, propõe-se a compilação de dados de controlo históricos e o cálculo de coeficientes de variação dos dados numéricos, especialmente no caso dos parâmetros relacionados com a deteção de perturbadores do sistema endócrino. Estes dados podem ser utilizados para fins comparativos na avaliação de estudos reais.
|
Relatório de ensaio
|
55.
|
O relatório de ensaio deve conter as seguintes informações:
|
|
Produto químico em estudo:
|
—
|
origem, número de lote e data-limite de utilização, se conhecida
|
|
—
|
estabilidade do produto químico em estudo, se conhecida.
|
|
|
|
Substância monocomponente:
|
—
|
aspeto físico, hidrossolubilidade e outras propriedades físico-químicas pertinentes;
|
|
—
|
dados de identificação química, como as denominações IUPAC ou CAS, número CAS, código SMILES ou InChI, fórmula estrutural, pureza, identidade química de impurezas, caso se justifique e seja exequível, etc.
|
|
|
|
Substância multicomponentes, UVCB e misturas:
|
—
|
caracterizada, na medida do possível, pela identidade química (ver acima), pela ocorrência quantitativa e pelas propriedades físico-químicas pertinentes dos componentes.
|
|
|
|
Veículo (se adequado):
|
—
|
Justificação da escolha do veículo, se não for aquoso;
|
|
|
|
Animais utilizados no ensaio:
|
—
|
espécie e estirpe utilizadas;
|
|
—
|
número, idade e sexo dos animais;
|
|
—
|
proveniência, condições de alojamento, alimentação, etc.;
|
|
—
|
peso individual dos animais no início do ensaio
|
|
—
|
caso não sejam utilizadas ratazanas, justificação do recurso a outra espécie.
|
|
|
|
Condições de realização do ensaio:
|
—
|
fundamentação da escolha das doses;
|
|
—
|
elementos relativos à formulação do produto químico em estudo e/ou à incorporação do mesmo na alimentação dos animais; concentração atingida, estabilidade e homogeneidade da preparação;
|
|
—
|
elementos relativos à administração do produto químico em estudo;
|
|
—
|
se pertinente, equivalência entre a concentração do produto químico em estudo na dieta ou na água de beber, expressa em ppm, e a dose real, expressa em mg/kg de peso corporal/dia,
|
|
—
|
elementos relativos à qualidade dos alimentos e da água;
|
|
—
|
descrição pormenorizada do processo de aleatorização para selecionar as crias para abate, se o abate for seletivo.
|
|
|
|
Resultados:
|
—
|
pesos corporais e alterações de peso corporal;
|
|
—
|
consumo de alimentos e consumo de água, se disponíveis;
|
|
—
|
reações tóxicas em função do sexo e da dose administrada, nomeadamente fertilidade, gestação e outros sinais de toxicidade;
|
|
—
|
duração do período de gestação;
|
|
—
|
efeitos tóxicos (ou outros) na reprodução, descendência, crescimento pós-natal, etc.;
|
|
—
|
natureza, intensidade e duração dos sinais clínicos observados (reversíveis ou irreversíveis),
|
|
—
|
número de fêmeas adultas com ciclo éstrico normal ou anormal e duração do ciclo;
|
|
—
|
número de nados-vivos e de perdas pós-implantação;
|
|
—
|
dados relativos à massa corporal da cria;
|
|
—
|
DAG de todas as crias (e peso corporal no dia da medição da DAG);
|
|
—
|
retenção de mamilo em crias do sexo masculino,
|
|
—
|
níveis das hormonas da tiroide no dia 13, para as crias e os machos adultos (bem como para as mães e as crias no dia 4, caso tenham sido medidos)
|
|
—
|
número de crias com anomalias macroscópicas visíveis, avaliação bruta dos órgãos genitais externos, número de crias significativamente menores do que as crias de controlo correspondentes;
|
|
—
|
momento do óbito durante o estudo ou indicação de que os animais sobreviveram até ao término do estudo;
|
|
—
|
número de implantações, dimensão da ninhada e pesos da ninhada no momento do registo;
|
|
—
|
peso corporal no momento da eutanásia e peso dos órgãos dos animais progenitores;
|
|
—
|
resultados das autópsias;
|
|
—
|
descrição pormenorizada das observações histopatológicas;
|
|
—
|
dados de absorção (se disponíveis);
|
|
—
|
tratamento estatístico dos resultados, se for o caso.
|
|
Discussão dos resultados
Conclusões
|
Interpretação dos resultados
|
56.
|
O estudo fornece avaliações da toxicidade para a reprodução/o desenvolvimento associada à administração de doses repetidas (ver pontos 5 e 6). Pode dar uma indicação da necessidade de realizar mais estudos e fornece orientações para a conceção dos estudos subsequentes. Deve consultar-se o documento de orientações 43 da OCDE para a interpretação dos resultados sobre a reprodução e o desenvolvimento (12). O documento de orientações 106 da OCDE sobre a avaliação histológica dos ensaios endócrinos e de reprodução em roedores (11) fornece informações sobre a preparação e a avaliação de órgãos endócrinos e de esfregaços vaginais, que podem ser úteis no contexto da presente Test Guideline.
|
REFERÊNCIAS BIBLIOGRÁFICAS
|
(1)
|
OCDE (1990). Room Document No 1 for the 14th Joint Meeting of the Chemicals Group and Management Committee. Available upon request at Organisation for Economic and Cooperation and Development, Paris.
|
|
(2)
|
OCDE (1992). Chairman’s Report of the ad hoc Expert Meeting on Reproductive Toxicity Screening Methods, Tokyo, 27th-29th October, 1992. Disponível mediante pedido na Organização de Cooperação e de Desenvolvimento Económicos, Paris.
|
|
(3)
|
OCDE (1998). Report of the First Meeting of the OECD Endocrine Disrupter Testing and Assessment (EDTA) Task Force, 10th-11th March 1998. Disponível mediante pedido na Organização de Cooperação e de Desenvolvimento Económicos, Paris.
|
|
(4)
|
OCDE (2015). Feasibility Study for Minor Enhancements of TG 421/422 with ED Relevant Endpoints. Environment, Health and Safety Publications, Series on Testing and Assessment (No 217), Organisation for Economic Cooperation and Development, Paris.
|
|
(5)
|
OCDE (2000). Guidance Document on the Recognition, Assessment, and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluations. Series on Testing and Assessment, (No 19), Organisation for Economic Cooperation and Development,.Paris.
|
|
(6)
|
OCDE (2011). Guidance Document on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption. Environment, Health and Safety Publications, Series on Testing and Assessment(No 150), Organisation for Economic Cooperation and Development, Paris.
|
|
(7)
|
Goldman, J.M., Murr A.S., Buckalew A.R., Ferrell J.M. and Cooper R.L. (2007). The Rodent Estrous Cycle: Characterization of Vaginal Cytology and its Utility in Toxicological Studies, Birth Defects Research, Part B, 80 (2), 84-97.
|
|
(8)
|
Sadleir R.M.F.S (1979). Cycles and Seasons, in Auston C.R. and Short R.V. (eds.), Reproduction in Mammals: I. Germ Cells and Fertilization, Cambridge, New York.
|
|
(9)
|
Gallavan R.H. Jr, Holson J.F., Stump D.G., Knapp J.F. and Reynolds V.L.(1999). Interpreting the Toxicologic Significance of Alterations in Anogenital Distance: Potential for Confounding Effects of Progeny Body Weights, Reproductive Toxicology, 13: 383-390.
|
|
(10)
|
OCDE (2013). Guidance Document in Support of the Test Guideline on the Extended One Generation Reproductive Toxicity Study. Environment, Health and Safety Publications, Series on Testing and Assessment (No 151), Organisation for Economic Cooperation and Development, Paris.
|
|
(11)
|
OCDE (2009). Guidance Document for Histologic Evaluation of Endocrine and Reproductive Tests in Rodents. Environment, Health and Safety Publications, Series on Testing and Assessment (No106), Organisation for Economic Cooperation and Development, Paris.
|
|
(12)
|
OCDE (2008). Guidance Document on Mammalian Reproductive Toxicity Testing and Assessment. Environment, Health and Safety Publications, Series on Testing and Assessment (No 43), Organisation for Economic Cooperation and Development, Paris.
|
Apêndice 1
DEFINIÇÕES (VER TAMBÉM A TEST GUIDELINE 150 DA OCDE) (6)
Atividade anfrogénica: capacidade de um produto químico de agir como uma hormona androgénica natural (por exemplo, a testosterona), num mamífero.
Atividade antiandrogénica: capacidade de um produto químico de suprimir a ação de uma hormona androgénica natural (por exemplo, a testosterona), num mamífero.
Atividade antiestrogénica: capacidade de um produto químico de suprimir a ação de uma hormona estrogénica natural (por exemplo, o 17ß-estradiol), num mamífero.
Atividade antitiroideia: capacidade de um produto químico de suprimir a ação de uma hormona da tiroide natural (por exemplo, a T3), num mamífero.
Atividade estrogénica: capacidade de um produto químico de agir como hormona estrogénica natural (por exemplo o 17ß-estradiol), num mamífero.
Atividade tiroideia: capacidade de um produto químico de agir como uma hormona da tiroide natural (por exemplo a T3) num mamífero.
Diminuição da fertilidade: reflete perturbações de funções ou capacidades reprodutoras masculinas ou femininas.
Dosagem: termo geral que abrange a dose, a frequência desta e o tempo de aplicação da mesma.
Dose: quantidade de produto químico em estudo administrada. Exprime-se em peso diário do produto químico por unidade de peso corporal do animal em estudo (por exemplo, mg/kg de peso corporal/dia), ou sob a forma de uma concentração constante na dieta.
NOAEL: abreviatura de «No Observed Adverse Effect Level» («nível sem observação de efeitos nocivos»); constitui a dose máxima cuja exposição não produz efeitos nocivos observáveis.
Produto químico: uma substância ou mistura.
Produto químico em estudo: qualquer substância ou mistura à qual seja aplicado o presente método de ensaio.
Toxicidade para o desenvolvimento: manifestação de toxicidade reprodutiva, sob a forma de perturbações estruturais pré-natal, peri-natal e pós-natal ou perturbações funcionais nos descendentes.
Toxicidade evidente: termo geral que descreve a existência de sinais claros de toxicidade após a administração do produto químico em estudo. Os sinais em causa devem ser suficientes para a avaliação dos perigos e devem permitir prever que o aumento da dose administrada provoque o aparecimento de sinais intensos de toxicidade e provável mortalidade.
Toxicidade materna: efeitos nocivos em fêmeas grávidas, quer ocorram especificamente (efeitos diretos) ou não especificamente (efeitos indiretos).
Toxicidade para a reprodução: designa efeitos prejudiciais na descendência e/ou perturbações de funções ou capacidades reprodutoras masculinas ou femininas.
Validação: processo científico concebido para caracterizar as limitações e os imperativos operacionais de um método de ensaio e demonstrar a fiabilidade e pertinência do mesmo para um determinado fim.
Apêndice 2
DIAGRAMA DO CALENDÁRIO EXPERIMENTAL, COM INDICAÇÃO DA DURAÇÃO MÁXIMA DO ESTUDO – PERÍODO TOTAL DE ACASALAMENTO DE 14 DIAS
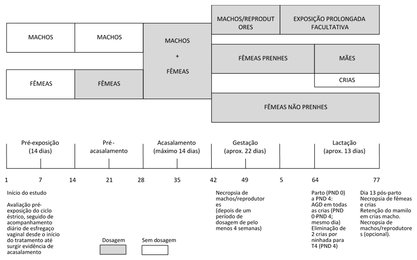
Apêndice 3
RELATÓRIO DE SÍNTESE SOBRE OS EFEITOS NA REPRODUÇÃO/NO DESENVOLVIMENTO
|
OBSERVAÇÕES
|
VALORES
|
|
|
|
Dosagem (unidades)
|
0 (controlo)
|
…
|
…
|
…
|
…
|
|
Pares iniciais (N)
|
|
|
|
|
|
|
Ciclo éstrico (pelo menos, a duração média e a frequência de ciclos irregulares)
|
|
|
|
|
|
|
Fêmeas que apresentam indícios de copulação (N)
|
|
|
|
|
|
|
Fêmeas que engravidaram (N)
|
|
|
|
|
|
|
Conceção nos dias 1-5 (N)
|
|
|
|
|
|
|
Conceção nos dias 6 -... (21) (N)
|
|
|
|
|
|
|
Gestação ≤ 21 dias (N)
|
|
|
|
|
|
|
Gestação = 22 dias (N)
|
|
|
|
|
|
|
Gestação ≥ 23 dias (N)
|
|
|
|
|
|
|
Mães com nados-vivos (N)
|
|
|
|
|
|
|
Mães com nados-vivos no dia 4 pp (N)
|
|
|
|
|
|
|
Implantes/mãe (média)
|
|
|
|
|
|
|
Crias vivas/mãe à nascença (média)
|
|
|
|
|
|
|
Crias vivas/mãe no dia 4 (média)
|
|
|
|
|
|
|
Rácio sexual (m/f) à nascença (média)
|
|
|
|
|
|
|
Rácio sexual (m/f), no dia 4 (média)
|
|
|
|
|
|
|
Peso da ninhada à nascença (média)
|
|
|
|
|
|
|
Peso da ninhada no dia 4 (média)
|
|
|
|
|
|
|
Peso da cria à nascença (média)
|
|
|
|
|
|
|
Peso da cria no momento da medição da DAG (média dos machos, média das fêmeas).
|
|
|
|
|
|
|
DAG da cria no mesmo dia, dia 4 após o nascimento (média dos machos, média das fêmeas, nota PND)
|
|
|
|
|
|
|
Peso das crias no dia 4 (média)
|
|
|
|
|
|
|
Retenção de mamilo das crias do sexo masculino no dia 13 (média)
|
|
|
|
|
|
|
Peso das crias no dia 13 (média)
|
|
|
|
|
|
|
|
|
CRIAS ANORMAIS
|
|
Mães com 0
|
|
|
|
|
|
|
Mães com 1
|
|
|
|
|
|
|
Mães com ≥ 2
|
|
|
|
|
|
|
|
|
PERDA DE CRIAS
|
|
|
|
Implantações pré-natais/pós-natais (implantações menos nados-vivos)
|
|
Fêmeas com 0
|
|
|
|
|
|
|
Fêmeas com 1
|
|
|
|
|
|
|
Fêmeas com 2
|
|
|
|
|
|
|
Fêmeas com ≥ 3
|
|
|
|
|
|
|
|
|
Pós-natal (nados-vivos menos vivos no dia 13 pós-natal)
|
|
Fêmeas com 0
|
|
|
|
|
|
|
Fêmeas com 1
|
|
|
|
|
|
|
Fêmeas com 2
|
|
|
|
|
|
|
Fêmeas com ≥ 3
|
|
|
|
|
|
B.64 ESTUDO DA TOXICIDADE DE DOSE REPETIDA COMBINADO COM O ENSAIO DE RASTREIO DA TOXICIDADE PARA A REPRODUÇÃO/O DESENVOLVIMENTO
INTRODUÇÃO
|
1.
|
O presente método de ensaio é equivalente à Test Guideline 422 (2016) da OCDE. As diretrizes da OCDE para o ensaio de produtos químicos são revistas periodicamente à luz do progresso científico. A diretriz original para o ensaio, n.o 422, foi adotada em 1996, com base num protocolo denominado Combined Repeat Dose and Reproductive/Developmental Screening Test, debatido em duas reuniões de peritos, realizadas em Londres, em 1990 (1), e em Tóquio, em 1992 (2).
|
|
2.
|
O presente método combina uma parte de verificação da toxicidade para a reprodução/o desenvolvimento que se baseia na experiência adquirida nos Estados-Membros com a utilização do método original para os produtos químicos existentes com elevado volume de produção e com ensaios exploratórios com substâncias de controlo positivo (3) (4), com uma parte relativa à toxicidade de dose repetida, em conformidade com a Test Guideline 407 da OCDE (Repeated Dose 28-Day Oral Toxicity Study in Rodents, correspondente ao capítulo B.7 deste anexo).
|
|
3.
|
O presente método de ensaio foi atualizado com parâmetros relativos a perturbadores do sistema endócrino, na sequência da atividade altamente prioritária da OCDE, iniciada em 1998, de revisão das diretrizes de ensaio existentes e elaboração de novas orientações para a análise e o ensaio de potenciais perturbadores do sistema endócrino (5). Neste contexto, a Test Guideline 407 (correspondente ao capítulo B.7 deste anexo) foi melhorada em 2008 com parâmetros adequados para detetar a atividade endócrina de produtos químicos em estudo. O objetivo da atualização da Test Guideline 422 era incluir alguns parâmetros relevantes no rastreio dos perturbadores do sistema endócrino nas Test Guidelines cujos períodos de exposição abrangem alguns dos períodos sensíveis do desenvolvimento (período pré-natal e primeira fase do período pós-natal).
|
|
4.
|
A Test Guideline 443 (estudo alargado de toxicidade reprodutiva uma geração, correspondente ao capítulo B.56 deste anexo) foi incluída na Test Guideline 422 com base num estudo de viabilidade relativo a questões científicas e técnicas relacionadas com a inclusão, bem como eventuais adaptações da conceção do ensaio necessárias para a mesma (6).
|
|
5.
|
O presente método de ensaio destina-se a produzir informações limitadas sobre os efeitos de um produto químico em estudo no desempenho reprodutor masculino e feminino, como a função gonadal, o comportamento de acasalamento, a fecundação e o desenvolvimento do feto e o parto. Não constitui uma alternativa aos métodos de ensaio B.31, B.34, B.35 ou B.56, nem os substitui.
|
CONSIDERAÇÕES INICIAIS
|
6.
|
Na apreciação e avaliação das características de toxicidade de produtos químicos em estudo pode determinar-se a toxicidade oral por dose repetida depois de se obterem informações sobre a toxicidade aguda a partir de ensaios realizados para o efeito. O presente estudo fornece informações sobre os possíveis riscos para a saúde de uma exposição repetida num período relativamente limitado. O método compreende um estudo básico de toxicidade por dose repetida, que pode utilizar-se para produtos químicos que não justifiquem um estudo a 90 dias (por exemplo, se o volume de produção não exceder determinados limites) ou como estudo exploratório com vista a um estudo a longo prazo. Na realização do estudo, devem seguir-se os princípios de orientação e as considerações descritas no documento de orientações n.o 19 da OCDE quanto ao reconhecimento, avaliação e utilização dos sinais clínicos como parâmetros de tratamento humano para experiências com animais utilizados em avaliações de segurança (7).
|
|
7.
|
O estudo inclui ainda um ensaio de despistagem da toxicidade para a reprodução/o desenvolvimento; por conseguinte, pode também ser utilizado para fornecer informações preliminares sobre possíveis efeitos no desempenho reprodutor dos machos e das fêmeas, como a função gonadal, o comportamento de acasalamento, a fecundação e o desenvolvimento do feto e o parto, quer numa fase inicial de avaliação das propriedades toxicológicas dos produtos químicos em estudo, quer no ensaio de produtos químicos que suscitam preocupação. O método de ensaio não fornece informações completas sobre todos os aspetos da reprodução e do desenvolvimento. Proporciona apenas, nomeadamente, meios limitados para a deteção de manifestações pós-natais de exposição pré-natal ou de efeitos que possam ser induzidos durante a exposição pós-natal. Devido, entre outros motivos, ao número relativamente reduzido de animais dos grupos de dosagem, à seletividade dos parâmetros e à curta duração do estudo, o método não fornece dados que apoiem a declaração definitiva de inexistência de efeitos na reprodução/no desenvolvimento. Além disso, na ausência de dados de outros ensaios de toxicidade para a reprodução/o desenvolvimento, os resultados positivos são úteis para a avaliação preliminar dos perigos e contribuem para fundamentar as decisões quanto à necessidade e à oportunidade de realizar ensaios adicionais.
|
|
8.
|
Os resultados correspondentes aos parâmetros relacionados com o sistema endócrino devem interpretar-se com base no documento Conceptual Framework for Testing and Assessment of Endocrine Disrupting Chemicals (8), elaborado pela OCDE, que estabelece um quadro concetual para o ensaio e a avaliação de produtos químicos perturbadores do sistema endócrino. Nesse quadro, o método correspondente à Test Guideline 422 da OCDE melhorada consta do nível 4, constituindo um ensaio in vivo que fornece dados sobre efeitos nocivos em determinados parâmetros do sistema endócrino. Contudo, um sinal do sistema endócrino não pode, por si só, ser considerado prova suficiente de que o produto químico em estudo é um perturbador desse sistema.
|
|
9.
|
O método confere especial importância aos efeitos neurológicos, devendo efetuar-se um exame clínico pormenorizado dos animais, de modo a obter o máximo possível de informações. Deve permitir identificar substâncias com potencial neurotóxico passíveis de necessitarem de uma investigação mais aprofundada. Além disso, pode também fornecer uma indicação básica dos efeitos imunológicos.
|
|
10.
|
Na ausência de dados de outros estudos de toxicidade sistémica, de toxicidade para a reprodução/o desenvolvimento, de neurotoxicidade e/ou de imunotoxicidade, os resultados positivos são úteis para a avaliação preliminar dos perigos e contribuem para fundamentar as decisões relativas à necessidade e à oportunidade de ensaios adicionais. O ensaio pode ser particularmente útil no âmbito do Screening Information Data Set (conjunto de dados de informação de despistagem) da OCDE para a avaliação dos produtos químicos existentes relativamente aos quais existe pouca ou nenhuma informação toxicológica, podendo também servir de alternativa à realização de dois ensaios separados de toxicidade de dose repetida (Test Guideline 407 da OCDE, que corresponde ao capítulo B.7 deste anexo) e de toxicidade para a reprodução/o desenvolvimento (Test Guideline 421 da OCDE, que corresponde ao capítulo B.63 deste anexo), respetivamente. Pode também ser utilizado como estudo exploratório de determinação da gama de dosagens para estudos mais exaustivos sobre a reprodução/o desenvolvimento, ou sempre que isso for considerado relevante.
|
|
11.
|
Em geral, parte-se do princípio de que existem diferenças de sensibilidade entre os animais prenhes e não prenhes. Por isso, pode ser mais complexo determinar no ensaio combinado dosagens adequadas para avaliar tanto a toxicidade sistémica generalizada como a toxicidade específica para a reprodução/o desenvolvimento do que em ensaios realizados separadamente. Além disso, a interpretação dos resultados dos ensaios, no que respeita à toxicidade sistémica geral, pode ser mais difícil do que quando é realizado um estudo separado de doses repetidas, sobretudo se os parâmetros séricos e histopatológicos não forem avaliados em simultâneo no estudo. Devido a estas complexidades técnicas, é necessária uma experiência considerável em ensaios de toxicidade para realizar o ensaio combinado. Por outro lado, além do recurso a um número inferior de animais, o ensaio combinado pode constituir um meio mais adequado para distinguir os efeitos diretos na reprodução/no desenvolvimento dos efeitos secundários em relação a outros efeitos (sistémicos).
|
|
12.
|
No presente ensaio, o período de dosagem é mais longo do que num estudo convencional de dose repetida com 28 dias. No entanto, utiliza menos animais de cada sexo por grupo do que os estudos convencionais de dose repetida a 28 dias realizados em complemento a ensaios da toxicidade na reprodução/no desenvolvimento.
|
|
13.
|
O presente método prevê a administração por via oral do produto químico em estudo. Pode ser necessário introduzir alterações se forem utilizadas outras vias de exposição.
|
|
14.
|
Antes da aplicação do método de ensaio a uma mistura para obter dados com fins normativos, importa ponderar se – e, em caso afirmativo, por que motivo – o método pode proporcionar resultados adequados para o efeito. Essas considerações não são necessárias se existir um requisito normativo para o ensaio da mistura.
|
|
15.
|
Definem-se no apêndice 1 alguns conceitos utilizados.
|
PRINCÍPIO DO ENSAIO
|
16.
|
O produto químico em estudo é administrado, em doses escalonadas, a vários grupos de machos e fêmeas. Os machos devem ser tratados durante, pelo menos, quatro semanas, até ao dia anterior ao da eutanásia, inclusive, o que abrange, no mínimo, duas semanas antes do acasalamento, o período de acasalamento e aproximadamente duas semanas após o acasalamento. Tendo em conta o período limitado de dosagem pré-acasalamento dos machos, a fertilidade pode não constituir um indicador particularmente sensível da toxicidade testicular. Por conseguinte, é essencial um exame histológico pormenorizado dos testículos. A combinação de um período de dosagem pré-acasalamento de duas semanas e de observações subsequentes de acasalamento/fertilidade com um período global de dosagem de, pelo menos, quatro semanas, seguido de histopatologia pormenorizada das gónadas do macho, é considerada suficiente para permitir a deteção da maioria dos efeitos na fertilidade masculina e na espermatogénese.
|
|
17.
|
As fêmeas devem ser tratadas ao longo de todo o estudo, ou seja, duas semanas antes do acasalamento – com o objetivo de abranger, pelo menos, dois ciclos éstricos completos –, o período variável até à conceção, a gravidez e, pelo menos, treze dias após o parto, até ao dia anterior ao da eutanásia, inclusive.
|
|
18.
|
A duração do estudo, após a aclimatização, com uma avaliação do ciclo éstrico efetuada antes do início do tratamento, depende do desempenho das fêmeas, e é de cerca de 63 dias, [pelo menos 14 dias antes do acasalamento, até 14 dias de acasalamento, 22 dias de gestação, 13 dias de aleitamento].
|
|
19.
|
No decurso do período de administração, verifica-se atentamente, todos os dias, se os animais evidenciam sinais de toxicidade. Os que morrem ou são eutanasiados no período de ensaio são autopsiados; no final do ensaio, são eutanasiados e autopsiados os animais sobreviventes.
|
DESCRIÇÃO DO MÉTODO
Seleção de espécies animais
|
20.
|
O presente método de ensaio foi concebido para ratazanas. Se os parâmetros que nele se especificam forem estudados noutra espécie de roedor, essa opção deve ser justificada pormenorizadamente. A ratazana foi a única espécie utilizada no programa internacional de validação da deteção de perturbadores do sistema endócrino (TG 407). Deve evitar-se a utilização de estirpes de fecundidade baixa ou nas quais se verifique uma incidência elevada de deficiências de desenvolvimento. Devem utilizar-se animais virgens saudáveis, não sujeitos a experiências anteriores. Importa especificar a espécie, a estirpe, o sexo, o peso e/ou a idade dos animais utilizados. No início do estudo, as diferenças de peso entre os animais devem ser mínimas, não se desviando mais de 20 % do peso médio dos animais de cada sexo. Nos casos em que for realizado como estudo preliminar de um estudo a longo prazo ou numa geração, é preferível utilizar animais da mesma estirpe e proveniência em ambos os estudos.
|
Condições de alojamento e de alimentação
|
21.
|
Todos os procedimentos devem respeitar as normas locais de manipulação de animais de laboratório. A temperatura do biotério deve ser de 22 °C ± 3 °C. A humidade relativa deve ser, no mínimo, de 30 % e, de preferência, não deve exceder 70 %, exceto durante a limpeza do biotério. A iluminação deve ser artificial, com uma sequência de 12 horas de luz seguidas de 12 horas de escuridão. Para a alimentação, podem ser usadas dietas convencionais de laboratório, com acesso ilimitado a água potável. A escolha da dieta pode ser influenciada pela necessidade de garantir uma mistura adequada do produto químico em estudo, quando administrado por essa via.
|
|
22.
|
Os animais devem ser alojados em pequenos grupos do mesmo sexo. Os animais podem ser alojados individualmente, se tal se justificar do ponto de vista científico. Em caso de alojamento coletivo, cada gaiola não deve alojar mais de cinco animais. O acasalamento deve ocorrer em gaiolas adequadas para o efeito. As fêmeas prenhes devem ser colocadas em gaiolas individuais e dispor de materiais de nidificação. As fêmeas em lactação devem ser colocadas em gaiolas individuais com as crias.
|
|
23.
|
Deve efetuar-se com regularidade uma pesquisa de contaminantes nos alimentos fornecidos e conservar-se uma amostra da dieta até o relatório estar concluído.
|
Preparação dos animais
|
24.
|
Os animais adultos jovens e saudáveis são repartidos ao acaso pelos grupos de tratamento e por gaiolas. As gaiolas devem estar dispostas de forma a minimizar possíveis efeitos decorrentes do seu posicionamento. Os animais são identificados de forma inequívoca, devendo ser aclimatados às condições de laboratório durante, pelo menos, cinco dias antes de se iniciar o estudo.
|
Preparação das doses
|
25.
|
Recomenda-se a consulta do PON aquando da implementação e utilização de um desses modelos no laboratório. Se for selecionada a via oral, o produto químico em estudo é geralmente administrado por gavagem; no entanto, os produtos podem ser administrados através dos alimentos ou da água de beber.
|
|
26.
|
Se necessário, o produto químico em estudo pode ser dissolvido ou suspenso num veículo adequado. Recomenda-se que, sempre que possível, se opte por uma solução ou suspensão aquosa; caso isso não seja viável, pode optar-se por uma solução ou suspensão num óleo (por exemplo, em óleo de milho); em último caso, pode recorrer-se a uma solução noutro veículo. Se este não for aquoso, devem conhecer-se as suas características de toxicidade. Importa determinar a estabilidade e a homogeneidade do produto químico em estudo no veículo.
|
PROCEDIMENTO
Número e sexo dos animais
|
27.
|
Recomenda-se que cada grupo seja iniciado com, pelo menos, 10 machos e 12-13 fêmeas. As fêmeas serão avaliadas para pré-exposição do ciclo éstrico; os animais que não apresentem ciclos típicos de 4-5 dias não são incluídos no estudo. Assim, é recomendável utilizar um número mais elevado de fêmeas, a fim de assegurar a presença de 10 fêmeas reprodutoras por grupo. Exceto no caso de efeitos tóxicos marcados, prevê-se que o número de fêmeas prenhes por grupo seja, no mínimo, igual a 8, que, normalmente, é o número mínimo aceitável de fêmeas prenhes por grupo. O objetivo é produzir gravidezes e crias em número suficiente para assegurar uma avaliação significativa do potencial do produto químico em estudo para afetar a fertilidade, a gravidez, o comportamento materno e de aleitamento, o crescimento e o desenvolvimento da geração F1, desde a fecundação até ao dia 13 pós-parto. Caso se preveja a eutanásia de alguns animais durante o ensaio, o referido número deve ser acrescido do número de animais a sacrificar. Deve ponderar-se a possibilidade de constituir um grupo-satélite adicional de cinco animais de cada sexo nos grupos de controlo e de dose máxima, para observar a reversibilidade, a persistência ou a ocorrência tardia de efeitos tóxicos sistémicos, durante, pelo menos, 14 dias após o tratamento. Os animais dos grupos-satélite não devem acasalar, pelo que não são utilizados na avaliação da toxicidade para a reprodução/o desenvolvimento.
|
Dosagem
|
28.
|
De modo geral, devem utilizar-se, no mínimo, três lotes de ensaio e um lote de controlo. Se não se dispuser de dados gerais de toxicidade adequados, pode efetuar-se um estudo exploratório, com animais da mesma estirpe e proveniência, para facilitar a determinação das doses a utilizar. Salvo no que respeita à exposição ao produto químico em estudo, os animais dos grupos de controlo devem ser tratados do mesmo modo que os animais dos grupos de ensaio. Se for utilizado um veículo para administrar o produto químico em estudo, o grupo de controlo deve receber o volume máximo de veículo utilizado.
|
|
29.
|
Na seleção das doses devem ter-se em conta os dados eventualmente disponíveis em matéria de toxicidade e toxicocinética. Importa também ter em conta o facto de poder haver diferenças de sensibilidade entre animais prenhes e não prenhes. Deve escolher-se como dose mais elevada uma dose que induza efeitos tóxicos, mas não cause mortalidade nem sofrimento evidente. Posteriormente, deve selecionar-se uma sequência decrescente de doses, com o objetivo de evidenciar uma correlação entre a dose administrada e a reação, bem como a ausência de efeitos nocivos associados à administração da dose mais reduzida. O intervalo ótimo entre doses consecutivas é frequentemente definido por um fator de 2 a 4. A inclusão de um quarto grupo de ensaio é muitas vezes preferível ao uso de intervalos muito grandes entre as dosagens (fatores superiores a 10).
|
|
30.
|
Caso se observem sinais de toxicidade generalizada (por exemplo, redução do peso corporal, efeitos ao nível hepático, cardíaco, pulmonar ou renal, etc.) ou outras alterações que possam não ser reações tóxicas (por exemplo, diminuição da quantidade de alimentos ingerida, dilatação hepática, etc.), deverá interpretar-se com cautela qualquer efeito observado ao nível dos parâmetros endócrinos.
|
Ensaio no limite
|
31.
|
Sempre que um ensaio, realizado de acordo com o presente método, que utilize uma dose de, pelo menos, 1 000 mg/kg de massa corporal/dia ou – no caso da administração através dos alimentos ou da água para beber – uma percentagem equivalente em relação aos mesmos (com base na determinação da massa corporal), não produza efeitos tóxicos observáveis, ou se, tendo em conta dados referentes a substâncias estruturalmente afins, não se prever a ocorrência de efeitos tóxicos, pode não ser necessário efetuar um estudo completo com várias doses. Nesses casos, justifica-se a realização de um ensaio no limite, exceto se os dados relativos à exposição humana aconselharem o ensaio de doses superiores. Caso se usem outras formas de administração, como a inalação ou a aplicação cutânea, as propriedades físico-químicas do produtos químico de ensaio são muitas vezes indicativas e limitativas do nível máximo de exposição praticável.
|
Administração das doses
|
32.
|
Os animais são tratados diariamente com o produto químico em estudo, durante uma semana. A administração forçada por meio de uma sonda esofágica deve efetuar-se numa dose única, utilizando um tubo estomacal ou uma cânula de intubação adequada. O volume máximo de líquido que pode ser administrado de cada vez depende do tamanho do animal. O volume máximo de líquido que pode ser administrado numa toma depende também do tamanho do animal de ensaio, não devendo exceder 1 ml/100 g de peso corporal; excetuam-se as soluções aquosas, que podem ser administradas na proporção de 2 ml/100 g de peso corporal. Exceto no caso de produtos químicos irritantes ou corrosivos, que normalmente revelam efeitos exacerbados em concentrações superiores, a variabilidade no volume de ensaio deve ser minimizada ajustando a concentração de modo a garantir um volume constante em todos os níveis de dosagem.
|
|
33.
|
No caso de produtos administrados através da alimentação ou da água de beber, é importante assegurar que as quantidades do produto não interferem com a nutrição normal ou com a composição da água. Se o produto for administrado na alimentação, pode optar-se por concentrações constantes desta (da ordem das ppm) ou por doses constantes em relação ao peso corporal de cada animal. Se o produto químico em estudo for administrado por gavagem, a dose deve ser administrada todos os dias à mesma hora, devendo ajustar-se, pelo menos, uma vez por semana a fim de se manter uma dose constante em relação ao peso corporal do animal; caso o estudo combinado de toxicidade oral da dose repetida preceda um estudo a longo prazo, é conveniente utilizar uma dieta semelhante em ambos os ensaios.
|
Calendário da experiência
|
34.
|
O tratamento de ambos os sexos deve ter início 2 semanas antes do acasalamento, depois de os animais serem aclimatados durante, pelo menos, 5 dias e as fêmeas serem sujeitas a exames de deteção de ciclos éstricos normais (num período de 2 semanas anterior ao tratamento). O estudo deve ser programado de forma a que a avaliação do ciclo éstrico comece pouco depois de os animais terem atingido a plena maturidade sexual, o que pode variar ligeiramente nas diferentes linhagens de ratazanas em laboratórios diferentes, sendo, por exemplo, às 10 semanas nas ratazanas Sprague Dawley e cerca das 12 semanas nas ratazanas Wistar. As mães com crias devem ser eutanasiadas no dia 13 após o parto, ou pouco depois. Para permitir que os animais jejuem desde a véspera da colheita de sangue (caso seja preferível esta opção), as mães e as crias não têm necessariamente de ser abatidas no mesmo dia. O dia de nascimento (ou seja, quando o parto é concluído) é definido como o dia 0 após o parto. As fêmeas que não apresentem qualquer indício de copulação são mortas 24 a 26 dias após o último dia do período de acasalamento. O nível de dosagem é mantido em ambos os sexos durante o período de acasalamento. Os machos devem continuar a ser tratados após o período de acasalamento, pelo menos até ter decorrido o período total mínimo de dosagem, de 28 dias. Em seguida, são eutanasiados ou, em alternativa, são mantidos e continuam a ser tratados, para um eventual segundo acasalamento, se isso for considerado adequado.
|
|
35.
|
As fêmeas prenhes devem continuar a ser tratadas durante a gravidez e, pelo menos, até ao dia 13 após o parto ou ao dia anterior ao abate. No caso de o produto químico em estudo ser administrado por inalação ou por via cutânea, o tratamento deve continuar a ser administrado pelo menos até ao dia 19 de gestação, inclusive, e ser retomado o mais rapidamente possível, o mais tardar no dia 4 após o parto.
|
|
36.
|
Os animais de grupos-satélite com observações subsequentes, caso sejam utilizados, não acasalam. Devem ser mantidos durante, pelo menos, 14 dias após o primeiro abate programado de mães, sem tratamento que permita detetar a ocorrência tardia, a persistência ou a superação dos efeitos tóxicos.
|
|
37.
|
O apêndice 2 apresenta um diagrama do calendário da experiência.
|
Ciclos éstricos
|
38.
|
Os ciclos éstricos devem ser monitorizados antes do início do tratamento, de forma a selecionar para o estudo fêmeas com um ciclo regular (ver ponto 27). Os esfregaços vaginais também devem ser monitorizados diariamente desde o início do período de tratamento, até haver indicações de acasalamento. Se houver preocupações quanto a efeitos agudos ligados ao stress que possam alterar os ciclos éstricos com o início do tratamento, os laboratórios podem expor os animais durante 2 semanas, e em seguida colher esfregaços vaginal diariamente, a fim de monitorizar o ciclo éstrico durante, pelo menos, duas semanas durante o período de pré-acasalamento e igualmente durante o período de acasalamento, até que haja provas de copulação. Aquando da colheita de células vaginais/cervicais, deve ter-se o cuidado de evitar perturbar a mucosa, o que pode induzir uma pseudogravidez (8) (9).
|
Processo de acasalamento
|
39.
|
Normalmente, devem ser utilizados neste estudo acasalamentos de 1:1 (um macho e uma fêmea). Pode haver exceções em caso de morte ocasional de machos. A fêmea deve ser colocada com o mesmo macho até se observarem indícios de copulação ou terem decorrido duas semanas. Devem examinar-se as fêmeas todas as manhãs, para verificar a presença de esperma ou de rolhão vaginal. O dia 0 da gravidez é definido como o dia em que é confirmada a evidência de acasalamento (deteção de rolhão vaginal ou esperma). Se a tentativa de acasalamento for mal sucedida, poderá tentar-se um novo acasalamento das fêmeas com machos do mesmo grupo comprovadamente aptos a procriar.
|
Número de animais por ninhada
|
40.
|
No dia 4 após o parto, o número de animais de cada ninhada pode ser ajustado, eliminando as crias excessivas por seleção aleatória, a fim de se obter, tão perto quanto possível, quatro ou cinco crias por sexo e por ninhada, consoante o tamanho normal das ninhadas das estirpes de ratazanas utilizadas. Devem ser colhidas amostras de sangue de duas das crias excedentárias, que serão agrupadas e utilizadas para a determinação dos níveis do soro T4. Não é adequada a eliminação seletiva das crias, por exemplo, com base no peso corporal ou na distância anogenital (DAG). Sempre que o número de crias do sexo masculino ou feminino impeça a obtenção de quatro ou cinco animais de cada sexo por ninhada, é admissível um ajustamento parcial – por exemplo, seis machos e quatro fêmeas. Não devem ser eliminadas crias se as ninhadas ficarem aquém da meta de abate (8 ou 10 crias/ninhada). Se existir uma única cria além da meta de abate, apenas uma cria deve ser eliminada e utilizada para a colheita de sangue com vista a possíveis avaliações séricas de T4.
|
|
41.
|
Se o tamanho da ninhada não for ajustado, eutanasiam-se duas crias por ninhada no dia 4 após o nascimento e colhem-se amostras de sangue para medição das concentrações de hormonas da tiroide. Se possível, essas duas crias de cada ninhada devem ser fêmeas, a fim de reservar as crias do sexo masculino para as avaliações de retenção de mamilo, exceto se essas crias não deixarem outras fêmeas para avaliação no termo da experiência. Não devem ser eliminadas crias se a ninhada ficar com menos de 8 ou 10 crias (em função do tamanho normal das ninhadas das estirpes de ratazanas em causa). Se existir uma única cria além do tamanho normal das ninhadas, apenas uma cria deve ser eliminada e utilizada para a colheita de sangue destinada a possíveis avaliações séricas de T4.
|
Observações
|
42.
|
Devem ser feitas observações clínicas gerais pelo menos uma vez por dia, de preferência à(s) mesma(s) hora(s), tendo em conta o período de pico de efeitos antecipados após a administração da dose. Deve registar-se o estado de saúde dos animais. Pelo menos, duas vezes por dia verificam-se os casos de morbidez ou mortalidade no conjunto dos animais.
|
|
43.
|
Deve efetuar-se um exame clínico aprofundado de cada animal progenitor antes da primeira exposição (para permitir comparações subsequentes com o estado inicial do animal) e, posteriormente, pelo menos uma vez por semana. O exame deve decorrer no exterior da gaiola, num recinto adequado e, de preferência, todos os dias à mesma hora. As observações devem ser cuidadosamente registadas, de preferência por recurso a um sistema definido em pormenor pelo laboratório em causa. Deve zelar-se por que as condições de ensaio sejam o mais constantes possível; os exames devem ser efetuados, de preferência, por pessoal que não esteja a par do ensaio realizado. Entre os sinais a registar contam-se alterações da pele, da pelagem, dos olhos e das mucosas e a ocorrência de secreções, excreções ou reações neurovegetativas (por exemplo lacrimação, horripilação, alterações da dimensão pupilar ou respiração anormal). Deve também registar-se qualquer alteração da forma de o animal se mover, da postura e da reação à manipulação, bem como a ocorrência de movimentos clónicos ou tónicos e de comportamentos estereotipados (por exemplo, atos de higiene repetitivos ou movimentação repetitiva em círculo), partos difíceis ou prolongados ou comportamentos estranhos (automutilação, locomoção para trás, etc.) (10).
|
|
44.
|
Num dado momento do estudo, deve proceder-se à avaliação da reação sensorial a diferentes estímulos (por exemplo, estímulos auditivos, visuais e propriocetivos) (8) (9) (11), da força de preensão (12) e da atividade motora (13), em cinco machos e cinco fêmeas, selecionados aleatoriamente de cada grupo. As referências bibliográficas contêm mais informações sobre a forma de proceder em cada caso, embora possam adotar-se procedimentos distintos dos nelas descritos. Nos machos, as observações funcionais devem ser feitas perto do final do período de dosagem, pouco antes do abate programado, mas antes da colheita de amostras de sangue para hematologia ou química clínica (ver pontos 53-56, incluindo a nota de rodapé 1). As fêmeas devem encontrar-se num estado fisiológico semelhante durante estes ensaios funcionais e devem, de preferência, ser testadas uma vez durante a última semana de aleitamento (por exemplo, dias de aleitamento 6-13), pouco antes do seu abate programado. Na medida do possível, deve minimizar-se o tempo de separação das mães e das crias.
|
|
45.
|
As observações funcionais efetuadas perto do final do estudo podem ser omitidas se o mesmo for realizado a título de estudo preliminar a um estudo posterior de toxicidade subcrónica (90 dias) ou a um estudo de longo prazo. Nesse caso, os exames funcionais devem realizar-se no estudo subsequente. Não obstante, os dados das observações funcionais efetuadas durante o estudo de dose repetida podem facilitar a escolha do nível de doses a utilizar no estudo de toxicidade subcrónica ou do estudo de longo prazo posterior.
|
|
46.
|
A título excecional, podem também omitir-se as observações funcionais no caso dos grupos de animais que apresentem sinais de toxicidade cuja intensidade interferiria de modo significativo com o desempenho do ensaio.
|
|
47.
|
A duração da gestação deve ser registada e calculada a partir do dia 0. Deve examinar-se cada ninhada o mais rapidamente possível após o parto, a fim de determinar o número e o sexo das crias, os nados-mortos, os nados-vivos, as crias que são significativamente inferiores às crias de controlo correspondentes e a presença de anomalias macroscópicas.
|
|
48.
|
As crias vivas devem ser contadas e determinado o sexo de cada uma; as ninhadas devem ser pesadas nas 24 horas seguintes ao parto (dia 0 ou 1 após o parto) e, pelo menos, nos dias 4 e dia 13 pós-parto. Além das observações sobre os animais progenitores (ver pontos 43 e 44), deve registar-se qualquer comportamento anormal das crias.
|
|
49.
|
A DAG de cada cria deve ser medida no mesmo dia pós-natal, entre o dia 0 e o dia 4. O peso corporal da cria deve ser medido no dia em que a DAG for medida e as DAG devem ser normalizadas de acordo com uma medida do tamanho da cria, preferencialmente a raiz cúbica do peso corporal (14). O número de mamilos/auréolas em crias do sexo masculino deve ser contado no dia 12 ou 13 após o nascimento, tal como recomendado na GD 151 (15) da OCDE.
|
Peso corporal e consumo de alimentos/água
|
50.
|
Os machos e as fêmeas devem ser pesados no primeiro dia de tratamento, pelo menos uma vez por semana a partir de então e no final. Durante a gestação, as fêmeas devem ser pesadas nos dias 0, 7, 14 e 20 e nas 24 horas seguintes ao parto (dia 0 ou 1 pós-parto) e, pelo menos, nos dias 4 e 13 pós-parto. As observações devem ser registadas individualmente para cada animal adulto.
|
|
51.
|
Durante o pré-acasalamento, a gestação e o aleitamento, o consumo de alimentos deve ser medido pelo menos uma vez por semana. A medição do consumo de alimentos durante o acasalamento é facultativa. Se o produto químico em estudo for administrado através da água de beber, o consumo de água deve também ser medido nestes períodos.
|
Hematologia
|
52.
|
Durante o ensaio, devem determinar-se os seguintes parâmetros hematológicos de cinco machos e cinco fêmeas escolhidos aleatoriamente de cada grupo: hematócrito, concentração de hemoglobina, contagem de eritrócitos, reticulócitos, contagem total de leucócitos e fórmula leucocitária, contagem de plaquetas e tempo e potencial de coagulação. Caso o produto químico em estudo ou os seus metabolitos potenciais tenham, ou se suspeite que tenham, propriedades oxidantes, devem efetuar-se outras análises, como a determinação da concentração de meta-hemoglobina e de corpos de Heinz.
|
|
53.
|
As amostras de sangue devem ser colhidas num determinado ponto. As fêmeas devem estar em condições fisiológicas semelhantes durante a colheita das amostras. A fim de evitar dificuldades práticas relacionadas com a variabilidade do início da gestação, a colheita de sangue das fêmeas pode ser feita no final do período de pré-acasalamento, e não imediatamente antes ou no âmbito do processo de eutanásia. De preferência, as amostras de sangue dos machos devem ser colhidas imediatamente antes ou como parte integrante do processo de eutanásia. Em alternativa, a colheita de sangue nos machos pode também ser feita no final do período de pré-acasalamento, se se optar por este momento para as fêmeas.
|
|
54.
|
As amostras de sangue devem ser armazenadas em condições adequadas.
|
Bioquímica clínica
|
55.
|
Devem efetuar-se determinações bioquímicas destinadas a investigar os principais efeitos tóxicos sobre os tecidos, nomeadamente renal e hepático, com amostras de sangue dos cinco machos e cinco fêmeas escolhidos aleatoriamente em todos os grupos. Recomenda-se que os animais sejam jejuados desde a véspera da colheita de sangue (22). Os parâmetros a determinar no plasma ou no soro são os seguintes: sódio, potássio, glucose, colesterol total, ureia, creatinina, proteínas totais e albumina, além de, pelo menos, duas enzimas indicadoras dos efeitos hepatocelulares (como a alanina-aminotransferase, a aspartato-aminotransferase e o sorbitol-desidrogenase). Em certos casos, a determinação de outras enzimas (de origem hepática ou outra origem) e da bilirrubina pode fornecer informações úteis.
|
|
56.
|
Em cada local, as amostras de sangue são colhidas com base no seguinte calendário:
|
—
|
pelo menos, de duas crias por ninhada no dia 4 após o nascimento, se o número de crias o permitir (ver pontos 40 e 41)
|
|
—
|
de todas as mães e, pelo menos, duas crias por ninhada, no final (dia 13), e
|
|
—
|
de todos os machos adultos, no final.
|
Todas as amostras de sangue são armazenadas em condições adequadas. As amostras de sangue do dia 13 das crias e dos machos adultos são analisadas para a determinação dos níveis séricos de hormonas da tiroide (T4). Se necessário, procede-se a uma avaliação mais aprofundada de T4 nas amostras de sangue das mães e do dia 4 das crias. Em alternativa, podem ser medidas outras hormonas, se necessário. O sangue das crias pode ser agrupado por ninhada para a realização de análises das hormonas da tiroide. As hormonas da tiroide (T4 e TSH) devem ser, de preferência, medidas como «totais».
|
|
57.
|
Como opção, podem realizar-se na última semana do ensaio as seguintes análises de urina em cinco machos de cada grupo escolhidos aleatoriamente na última semana do estudo, recolhida de acordo com um programa previamente estabelecido: aparência, volume, osmolalidade ou gravidade específica, pH, proteínas, glucose e sangue/hematócitos.
|
|
58.
|
Além disso, deve prever-se a pesquisa de marcadores séricos que forneçam indicações gerais sobre a lesão de tecidos. Caso as propriedades conhecidas do produto químico em estudo afetem, ou se preveja que possam afetar, o perfil metabólico da mesma, devem realizar-se outras determinações, nomeadamente de cálcio, fosfatos, triglicéridos em jejum e glucose em jejum, hormonas específicas, meta-hemoglobina e colinesterase. Estas determinações devem ser identificadas caso a caso.
|
|
59.
|
Os fatores a seguir indicados podem afetar a variabilidade dos resultados das análises hormonais e as concentrações absolutas nelas determinadas:
|
—
|
momento da eutanásia, devido à variação das concentrações hormonais ao longo do dia,
|
|
—
|
método utilizado para eutanasiar os animais sem lhes causar tensões desnecessárias, que poderiam afetar as concentrações hormonais,
|
|
—
|
diferenças ao nível das curvas de calibração dos conjuntos para as determinações hormonais.
|
|
|
60.
|
As amostras de plasma especificamente destinadas a determinações hormonais devem colher-se à mesma hora do dia. Os conjuntos existentes no comércio para determinar concentrações hormonais podem dar valores diferentes.
|
|
61.
|
Se os dados históricos de base forem inadequados, deve ponderar-se a determinação da variabilidade dos parâmetros hematológicos e de bioquímica clínica antes de iniciar a exposição dos animais às doses previstas ou – o que será preferível – num conjunto de animais não incluídos nos grupos ensaiados. No caso das fêmeas, os dados devem dizer respeito a animais em lactação.
|
PATOLOGIA
Autópsia macroscópica
|
62.
|
Deve ser realizada a todos os animais adultos estudados uma autópsia macroscópica completa e pormenorizada, através do exame cuidadoso da superfície exterior do corpo, dos orifícios, das cavidades craniana, torácica e abdominal e do conteúdo de cada uma destas. Deve prestar-se especial atenção aos órgãos do sistema reprodutivo. Deve registar-se o número de locais de implantação. Os esfregaços vaginais devem ser examinados na manhã do dia da autópsia para determinar a fase do ciclo éstrico e permitir a correlação com a histopatologia dos órgãos reprodutores femininos.
|
|
63.
|
Os testículos e os epidídimos, bem como a próstata e as vesículas seminais, com as glândulas coagulantes, de todos os machos adultos devem ser limpos de qualquer tecido aderente, conforme adequado, e o seu peso húmido determinado logo que possível após a dissecação, para evitar a secagem. Além disso, os pesos de órgãos facultativos podem incluir o complexo formado pelos músculos elevatórios do ânus e bulbocavernosos, as glândulas de Cowper e a glande peniana, nos machos, e os ovários (peso húmido) e o útero (incluindo o colo), nas fêmeas; devem conservar-se os ovários, os testículos, os epidídimos, os órgãos sexuais acessórios e todos os órgãos com lesões macroscópicas de todos os animais adultos.
|
|
64.
|
Devem conservar-se, no meio de fixação mais adequado ao exame histopatológico subsequente previsto, as glândulas tiroideias de todos os machos e fêmeas adultos, bem como de uma cria de sexo masculino e de outra de sexo feminino, de cada ninhada, colhidas no 13.o dia de vida. A pesagem da tiroide pode realizar-se após fixação. A remoção dos tecidos aderentes à tiroide deve efetuar-se com muito cuidado e também só depois da fixação, para evitar danificar tecidos, o que, a ocorrer, poderia comprometer a análise histopatológica. As colheitas de sangue devem ser efetuadas num determinado ponto a indicar, imediatamente antes da eutanásia dos animais ou integradas nesta, e as amostras devem ser armazenadas em condições adequadas (ver ponto 56).
|
|
65.
|
Além disso, o fígado, os rins, as glândulas suprarrenais, o timo, o baço, o cérebro e o coração de, pelo menos, cinco machos e fêmeas adultos escolhidos aleatoriamente em cada grupo (excluindo os animais moribundos e/ou eutanasiados antes do termo do estudo) devem ser limpos de qualquer tecido aderente, de forma adequada, e o seu peso húmido deve ser determinado logo que possível após a dissecação, para evitar a secagem. Os órgãos e tecidos que se seguem devem ser conservados num meio de fixação adequado aos mesmos, bem como ao tipo de exame histopatológico subsequente: todas as lesões macroscópicas, o encéfalo (regiões representativas, incluindo os hemisférios cerebrais, o cerebelo e a protuberância anelar), a medula espinal, os olhos, o estômago, o intestino delgado e o intestino grosso (incluindo as placas de Peyer), o fígado, os rins, as glândulas suprarrenais, o baço, o coração, o timo, a traqueia e os pulmões (conservados por injeção de fixador, seguida de imersão), as gónadas (testículos e ovários), os órgãos sexuais secundários (útero e respetivo colo, epidídimos, próstata + vesículas seminais e glândulas de coagulação), a vagina, a bexiga urinária, os gânglios linfáticos – o gânglio linfático mais próximo e outro gânglio linfático, a selecionar em função da experiência anterior do laboratório (16), um nervo periférico (ciático ou tibial), de preferência na vizinhança do músculo, músculo esquelético e osso com medula óssea (corte ou, em alternativa, uma punção recente de medula óssea). Recomenda-se a fixação dos testículos por imersão em fixador de Bouin ou em fixador de Davidson modificado (16)(17)(18). A fixação em formalina não é recomendada para estes tecidos. Para que o fixador penetre rapidamente, deve puncionar-se superficialmente a túnica albugínea com uma agulha, com cuidado, em ambos os polos do órgão. Os resultados clínicos, ou outros, podem aconselhar o exame de outros tecidos. Devem ser conservados também todos os órgãos que as propriedades conhecidas do produto químico em estudo indiciem que serão provavelmente afetados.
|
|
66.
|
Os seguintes tecidos podem dar indicações úteis sobre efeitos ao nível do sistema endócrino: gónadas (ovários e testículos), órgãos sexuais secundários (útero – incluindo o colo –, epidídimos, vesículas seminais e glândulas de coagulação, bem como a próstata dorsolateral e ventral), vagina, hipófise, glândulas mamárias masculinas e glândulas suprarrenais. Não existe documentação suficiente sobre alterações das glândulas mamárias masculinas, mas este parâmetro pode ser muito sensível às substâncias com atividade estrogénica. A observação dos órgãos e tecidos não enumerados no ponto 65 é facultativa.
|
|
67.
|
As crias mortas e as crias abatidas até ao dia 13 seguinte ao parto, ou pouco depois, devem, pelo menos, ser cuidadosamente examinadas externamente para a pesquisa de anomalias macroscópicas. O aparelho reprodutor externo deve ser objeto de especial atenção, devendo ser inspecionado para a pesquisa de sinais de alterações no desenvolvimento.
|
Histopatologia
|
68.
|
Deve efetuar-se uma histopatologia completa dos órgãos e tecidos conservados dos animais selecionados dos grupos de controlo e de dose elevada (com especial destaque para as fases da espermatogénese nos machos e a histopatologia da célula testicular intersticial). A tiroide das crias e dos restantes animais adultos poderá ser examinada quando necessário. Caso o exame destes últimos revele alterações atribuíveis à substância em estudo, devem examinar-se também os animais de todos os lotes restantes. O documento com a referência 10, que contém orientações no domínio da histopatologia, dá mais informações sobre a dissecação, a fixação, a colheita de amostras e a histopatologia de tecidos do sistema endócrino.
|
|
69.
|
Devem examinar-se todas as lesões importantes. Para ajudar na elucidação dos NOAEL, devem ser examinados os órgãos-alvo para outros grupos de dose, em especial nos grupos que apresentem um pedido para indicar um NOAEL.
|
|
70.
|
Caso se tenha constituído um grupo satélite de animais, deve efetuar-se o exame histopatológico dos tecidos e órgãos que tenham revelado alterações nos animais dos grupos expostos a uma dose do produto químico em estudo.
|
DADOS E RELATÓRIOS
Dados
|
71.
|
Devem ser apresentar-se os dados individuais de cada animal. Além disso, todos os dados devem ser resumidos num quadro, indicando, para cada grupo de ensaio, o número de animais no início deste, o número de animais encontrados mortos durante o ensaio ou eutanasiados por intervenção humana, a hora da morte de cada animal, a descrição e evolução temporal dos efeitos tóxicos, o número de animais férteis, o número de fêmeas prenhes, o número de animais que apresentam sinais de toxicidade, uma descrição dos sinais de toxicidade observados, incluindo o momento em que surgiram, a duração e a gravidade dos eventuais efeitos tóxicos, os tipos de alterações histopatológicas e quaisquer dados relevantes sobre a ninhada. No apêndice 3 figura um modelo de relatório sob a forma de tabela, que se revelou muito útil para a avaliação de efeitos na reprodução/no desenvolvimento.
|
|
72.
|
Se possível, os resultados numéricos devem ser avaliados por um método estatístico corrente adequado. Na comparação de um efeito numa gama de doses, deve evitar-se o recurso a testes t múltiplos. Os métodos estatísticos devem ser escolhidos ao planear o estudo. A análise estatística da DAG e da retenção de mamilo deve basear-se em dados individuais das crias, tendo em conta os efeitos da ninhada. Se for adequado, a ninhada é escolhida como unidade de análise. A análise estatística do peso corporal das crias deve basear-se em dados relativos a cada indivíduo, tendo em conta o tamanho da ninhada. Devido às dimensões limitadas do estudo, as análises estatísticas, sob a forma de ensaios de significância, têm um valor limitado para muitos parâmetros, em especial os parâmetros de reprodução. Alguns dos métodos mais utilizados, especialmente os ensaios paramétricos para medição da tendência central, são inadequados. Se se utilizarem análises estatísticas, o método escolhido deve adequar-se à distribuição da variável examinada, que deve ser selecionada antes do início do estudo.
|
Avaliação dos resultados
|
73.
|
As conclusões do presente estudo de toxicidade devem ser ponderadas com base nos efeitos observados, na autópsia e nos resultados microscópicos. A análise deverá considerar a relação (ou a ausência desta) entre a dose do produto químico de ensaio e a presença ou ausência, incidência e gravidade das anomalias, incluindo lesões macroscópicas, órgãos identificados como alvos, infertilidade, alterações da reprodução e procriação, alterações do peso corporal, efeitos na mortalidade e quaisquer outros efeitos tóxicos.
|
|
74.
|
Devido ao curto período de tratamento dos machos, a histopatologia dos testículos e dos epidídimos deve ser tida em conta juntamente com os dados de fertilidade, aquando da avaliação dos efeitos para a reprodução masculina. A utilização de dados históricos de controlo sobre a reprodução/o desenvolvimento (por exemplo, para as dimensões da ninhada, a DAG, a retenção de mamilo, os níveis séricos de T4), se disponíveis, pode também ser útil para auxiliar a interpretação do estudo.
|
|
75.
|
Para fins de controlo de qualidade, propõe-se a compilação de dados de controlo históricos e o cálculo de coeficientes de variação dos dados numéricos, especialmente no caso dos parâmetros relacionados com a deteção de perturbadores do sistema endócrino. Estes dados podem ser utilizados para fins comparativos na avaliação de estudos reais.
|
Relatório de ensaio
|
76.
|
O relatório de ensaio deve conter as seguintes informações:
|
|
Produto químico em estudo:
|
—
|
origem, número de lote e data-limite de utilização, se conhecida
|
|
—
|
estabilidade do produto químico em estudo, se conhecida.
|
|
|
|
Substância monocomponente:
|
—
|
aspeto físico, hidrossolubilidade e outras propriedades físico-químicas pertinentes;
|
|
—
|
dados de identificação química, como as denominações IUPAC ou CAS, número CAS, código SMILES ou InChI, fórmula estrutural, pureza, identidade química das impurezas, caso se justifique e seja exequível, etc.
|
|
|
|
Substância multicomponentes, UVCB e misturas:
|
—
|
caracterizada, na medida do possível, pela identidade química (ver acima), pela ocorrência quantitativa e pelas principais propriedades físico-químicas dos componentes
|
. |
|
|
Veículo (se adequado):
|
—
|
justificação da escolha do veículo, se não for água.
|
|
|
|
Animais utilizados no ensaio:
|
—
|
espécie e estirpe utilizadas;
|
|
—
|
número, idade e sexo dos animais;
|
|
—
|
proveniência, condições de alojamento, alimentação, etc.;
|
|
—
|
peso individual dos animais no início do ensaio
|
|
—
|
caso não sejam utilizados ratazanas, justificação do recurso a outra espécie.
|
|
|
|
Condições de realização do ensaio:
|
—
|
fundamentação da escolha das doses;
|
|
—
|
elementos relativos à formulação do produto químico em estudo/à incorporação do mesmo na dieta dos animais; concentração atingida, estabilidade e homogeneidade da preparação;
|
|
—
|
elementos relativos à administração do produto químico em estudo;
|
|
—
|
se pertinente, equivalência entre a concentração do produto químico em estudo na dieta ou na água de beber, expressa em ppm, e a dose real, expressa em mg/kg de peso corporal/dia,
|
|
—
|
elementos relativos à qualidade dos alimentos e da água;
|
|
—
|
descrição pormenorizada do processo de aleatorização para selecionar as crias para abate, se este for seletivo.
|
|
|
|
Resultados:
|
—
|
peso corporal e suas alterações;
|
|
—
|
consumo de alimentos e de água, se pertinente,
|
|
—
|
reações tóxicas em função do sexo e da dose administrada, nomeadamente na fertilidade, na gestação e outros sinais de toxicidade;
|
|
—
|
efeitos nocivos, ou outros, na reprodução, na descendência, no crescimento pós-natal, etc.;
|
|
—
|
natureza, intensidade e duração dos sinais clínicos observados (reversíveis ou irreversíveis),
|
|
—
|
avaliações da atividade sensorial, da força de preensão e da atividade motora;
|
|
—
|
análises hematológicas com valores de base relevantes;
|
|
—
|
análises bioquímicas com valores de base relevantes;
|
|
—
|
número de fêmeas adultas com ciclo éstrico normal ou anormal, e duração do ciclo;
|
|
—
|
número de nados-vivos e de perdas pós-implantação;
|
|
—
|
número de crias com anomalias macroscópicas visíveis, avaliação bruta dos órgãos genitais externos, número de crias são significativamente menores do que as crias de controlo correspondentes;
|
|
—
|
momento do óbito durante o estudo, ou indicação de que os animais sobreviveram até ao final do mesmo;
|
|
—
|
número de implantações, dimensão da ninhada e pesos da ninhada no momento do registo;
|
|
—
|
dados relativos à massa corporal das crias;
|
|
—
|
DAG de todas as crias (e peso corporal no dia da medição da DAG);
|
|
—
|
retenção de mamilo em crias do sexo masculino,
|
|
—
|
níveis das hormonas da tiroide no dia 13, para as crias e os machos adultos (bem como para as mães e as crias no dia 4, caso tenham sido medidos)
|
|
—
|
peso corporal no momento do abate e peso dos órgãos dos animais progenitores;
|
|
—
|
resultados das autópsias;
|
|
—
|
descrição pormenorizada dos resultados histopatológicos;
|
|
—
|
dados de absorção (se disponíveis);
|
|
—
|
tratamento estatístico dos resultados, se for o caso.
|
|
|
|
Discussão dos resultados.
|
|
Interpretação dos resultados
|
77.
|
O estudo proporciona avaliações da toxicidade para a reprodução/o desenvolvimento associada à administração de doses repetidas. Uma vez que o estudo incide tanto nos parâmetros de toxicidade geral como de toxicidade para a reprodução/o desenvolvimento, os resultados obtidos permitem estabelecer uma distinção entre os efeitos para a reprodução/o desenvolvimento não associados a uma toxicidade geral e os efeitos nocivos apenas induzidos a níveis igualmente tóxicos para os progenitores (ver pontos 7-11). Pode fornecer uma indicação da necessidade de realizar mais estudos, bem como orientações para a conceção de estudos subsequentes. Para a interpretação dos resultados sobre a reprodução e o desenvolvimento, deve consultar-se o documento de orientações 43 da OCDE (19). O documento de orientação 106 da OCDE sobre a avaliação histológica dos ensaios endócrinos e de reprodução em roedores (16) fornece informações sobre a preparação e a avaliação de órgãos (endócrinos) e de esfregaços vaginais que podem ser úteis para a presente Test Guideline.
|
REFERÊNCIAS BIBLIOGRÁFICAS
|
(1)
|
OCDE (1990). Room Document No 1 for the 14th Joint Meeting of the Chemicals Group and Management Committee. Disponível mediante pedido junto da Organização de Cooperação e de Desenvolvimento Económicos, Paris
|
|
(2)
|
OCDE (1992). Chairman’s Report of the ad hoc Expert Meeting on Reproductive Toxicity Screening Methods, Tokyo, 27th-29th October, 1992. Disponível mediante pedido junto da Organização de Cooperação e de Desenvolvimento Económicos, Paris
|
|
(3)
|
Mitsumori K., Kodama Y., Uchida O., Takada K., Saito M. Naito K., Tanaka S., Kurokawa Y., Usami, M., Kawashima K., Yasuhara K., Toyoda K., Onodera H., Furukawa F., Takahashi M. and Hayashi Y. (1994). Confirmation Study, Using Nitro-Benzene, of the Combined Repeat Dose and Reproductive/ Developmental Toxicity Test Protocol Proposed by the Organization for Economic Cooperation and Development (OECD). J. Toxicol, Sci., 19, 141-149.
|
|
(4)
|
Tanaka S., Kawashima K., Naito K., Usami M., Nakadate M., Imaida K., Takahashi M., Hayashi Y., Kurokawa Y. and Tobe M. (1992). Combined Repeat Dose and Reproductive/Developmental Toxicity Screening Test (OECD): Familiarization Using Cyclophosphamide. Fundam. Appl. Toxicol., 18, 89-95.
|
|
(5)
|
OCDE (1998). Report of the First Meeting of the OECD Endocrine Disrupter Testing and Assessment (EDTA) Task Force, 10th-11th March 1998, Available upon request at Organisation for Economic Cooperation and Development, Paris
|
|
(6)
|
OCDE (2015). Feasibility Study for Minor Enhancements of TG 421/422 with ED Relevant Endpoints. Environment, Health and Safety Publications, Series on Testing and Assessment (No 217), Organisation for Economic Cooperation and Development, Paris.
|
|
(7)
|
OCDE (2000). Guidance Document on the Recognition, Assessment, and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluations, Environment, Health and Safety Publications, Series on Testing and Assessment, (No 19), Organisation for Economic Cooperation and Development, Paris.
|
|
(8)
|
Goldman J.M., Murr A.S., Buckalew A.R., Ferrell J.M.and Cooper R.L. (2007). The Rodent Estrous Cycle: Characterization of Vaginal Cytology and its Utility in Toxicological Studies, Birth Defects Research, Part B, 80 (2), 84-97.
|
|
(9)
|
Sadleir R.M.F.S. (1979). Cycles and Seasons, in Auston C.R. and Short R.V. (Eds.), Reproduction in Mammals: I. Germ Cells and Fertilization, Cambridge, New York.
|
|
(10)
|
IPCS (1986). Principles and Methods for the Assessment of Neurotoxicity Associated with Exposure to Chemicals. Environmental Health Criteria Document (No 60).
|
|
(11)
|
Moser V.C., McDaniel K.M. and Phillips P.M. (1991). Rat Strain and Stock Comparisons Using a Functional Observational Battery: Baseline Values and Effects of Amitraz. Toxicol. Appl. Pharmacol., 108, 267-283.
|
|
(12)
|
Meyer O.A., Tilson H.A., Byrd W.C. and Riley M.T. (1979). A Method for the Routine Assessment of Fore- and Hindlimb Grip Strength of Rats and Mice. Neurobehav. Toxicol., 1, 233-236.
|
|
(13)
|
Crofton K.M., Howard J.L., Moser V.C., Gill M.W., Reiter L.W., Tilson H.A., MacPhail R.C. (1991). Interlaboratory Comparison of Motor Activity Experiments: Implication for Neurotoxicological Assessments. Neurotoxicol. Teratol. 13, 599-609.
|
|
(14)
|
Gallavan R.H. Jr, J.F. Holson, D.G. Stump, J.F. Knapp and V.L. Reynolds. (1999). «Interpreting the Toxicologic Significance of Alterations in Anogenital Distance: Potential for Confounding Effects of Progeny Body Weights», Reproductive Toxicology, 13: 383-390.
|
|
(15)
|
OCDE (2013). Guidance Document in Support of the Test Guideline on the Extended One Generation Reproductive Toxicity Study. Environment, Health and Safety Publications, Series on Testing and Assessment (No 151). Organisation for Economic Cooperation and Development, Paris.
|
|
(16)
|
OECD (2009).Guidance Document for Histologic Evaluation of Endocrine and Reproductive Tests in Rodents. Environment, Health and Safety Publications, Series on Testing and Assessment (No. 106) Organisation for Economic Cooperation and Development, Paris.
|
|
(17)
|
Hess RA e Moore B.J. (1993). Histological Methods for the Evaluation of the Testis. In: Methods in Reproductive Toxicology, Chapin RE and Heindel JJ (Eds.). Academic Press: San Diego, CA, pp. 52-85.
|
|
(18)
|
Latendresse JR, Warbrittion AR, Jonassen H, Creasy DM. (2002). Fixation of Testes and Eyes Using a Modified Davidson’s Fluid: Comparison with Bouin’s Fluid and Conventional Davidson’s fluid. Toxicol. Pathol. 30, 524-533.
|
|
(19)
|
OCDE (2008). Guidance Document on Mammalian Reproductive Toxicity Testing and Assessment. Environment, Health and Safety Publications, Series on Testing and Assessment (No 43), Organisation for Economic Cooperation and Development, Paris.
|
|
(20)
|
OECD (2011), Guidance Document on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption (No 150), Organisation for Economic Cooperation and Development, Paris.
|
Apêndice 1
DEFINIÇÕES (VER TAMBÉM O DOCUMENTO GD 150 DA OCDE) (29)
Atividade anfrogénica capacidade de um produto químico de agir como uma hormona androgénica natural (por exemplo, a testosterona) num mamífero.
Atividade antiandrogénica capacidade de um produto químico de suprimir a ação de uma hormona androgénica natural (por exemplo a testosterona) num mamífero.
Atividade antiestrogénica capacidade de um produto químico de suprimir a ação de uma hormona estrogénica natural (por exemplo, o 17ß-estradiol) num mamífero.
Atividade antitiroideia capacidade de um produto químico de suprimir a ação de uma hormona da tiroide natural (por exemplo, a T3) num mamífero.
Atividade estrogénica capacidade de um produto químico de agir como hormona estrogénica natural (por exemplo o 17ß-estradiol) num mamífero.
Atividade tiroideia capacidade de um produto químico de agir como uma hormona da tiroide natural (por exemplo, a T3) num mamífero.
Diminuição da fertilidade reflete perturbações de funções ou capacidades reprodutoras masculinas ou femininas.
Dosagem termo geral que inclui a dose, a sua frequência e a duração da aplicação da dose.
Dose quantidade de produto químico em estudo administrada. Exprime-se em peso diário do produto químico em estudo por unidade de peso corporal do animal de ensaio (por exemplo mg/kg de peso corporal/dia) ou sob a forma de uma concentração constante nos alimentos.
NOAEL abreviatura de «No Observed Adverse Effect Level» («nível sem observação de efeitos nocivos»), que constitui a dose máxima que não produz efeitos nocivos observáveis da exposição à mesma.
Produto químico uma substância ou mistura.
Produto químico em estudo qualquer substância ou mistura à qual seja aplicado o presente método de ensaio.
Toxicidade evidente termo geral que descreve a existência de sinais claros de toxicidade após a administração do produto químico em estudo. Os sinais em causa devem ser suficientes para a avaliação dos perigos e devem ser tais que seja de prever que o aumento da dose administrada provoque o aparecimento de sinais intensos de toxicidade e provável mortalidade.
Toxicidade materna efeitos nocivos nas fêmeas prenhes, que se apresentam especificamente (efeito direto) ou não especificamente (efeito indireto) e estão relacionados com a gestação.
Toxicidade para a reprodução designa efeitos prejudiciais na descendência e/ou perturbações de funções ou capacidades reprodutoras masculinas ou femininas.
Toxicidade para o desenvolvimento a manifestação de toxicidade reprodutiva, sob a forma de perturbações estruturais pré-natal, peri-natal e pós-natal ou perturbações funcionais nos descendentes.
Validação processo científico concebido para caracterizar as limitações e os imperativos operacionais de um método de ensaio e demonstrar a fiabilidade e pertinência do mesmo para um determinado fim.
Apêndice 2
DIAGRAMA DO CALENDÁRIO EXPERIMENTAL, COM INDICAÇÃO DA DURAÇÃO MÁXIMA DO ESTUDO, COM BASE NUM PERÍODO TOTAL DE ACASALAMENTO DE 14 DIAS


Apêndice 3
RELATÓRIO DE SÍNTESE SOBRE OS EFEITOS NA REPRODUÇÃO/NO DESENVOLVIMENTO
|
OBSERVAÇÕES
|
VALORES
|
|
Dosagem (unidades).......
|
0 (controlo)
|
…
|
…
|
…
|
…
|
|
Pares iniciais (N)
|
|
|
|
|
|
|
Ciclo éstrico (pelo menos, o comprimento médio e a frequência de ciclos irregulares)
|
|
|
|
|
|
|
Fêmeas que apresentam indícios de copulação (N)
|
|
|
|
|
|
|
Fêmeas que engravidaram (N)
|
|
|
|
|
|
|
Conceção nos dias 1-5 (N)
|
|
|
|
|
|
|
Conceção nos dias 6 -... (23) (N)
|
|
|
|
|
|
|
Gestação ≤ 21 dias (N)
|
|
|
|
|
|
|
Gestação = 22 dias (N)
|
|
|
|
|
|
|
Gravidez ≥ 23 dias (N)
|
|
|
|
|
|
|
Mães com nados-vivos (N)
|
|
|
|
|
|
|
Mães com nados-vivos no dia 4 pp (N)
|
|
|
|
|
|
|
Implantes/mãe (média)
|
|
|
|
|
|
|
Crias vivas/mãe à nascença (média)
|
|
|
|
|
|
|
Crias vivas/mãe no dia 4 (média)
|
|
|
|
|
|
|
Rácio sexual (m/f) à nascença (média)
|
|
|
|
|
|
|
Rácio sexual (m/f), no dia 4 (média)
|
|
|
|
|
|
|
Peso da ninhada à nascença (média)
|
|
|
|
|
|
|
Peso da ninhada no dia 4 (média)
|
|
|
|
|
|
|
Peso da cria à nascença (média)
|
|
|
|
|
|
|
Peso da cria no momento da medição da DAG (média dos machos, média das fêmeas).
|
|
|
|
|
|
|
DAG da cria no mesmo dia pós-natal, dia 4 após o nascimento (média dos machos, média das fêmeas, nota PND)
|
|
|
|
|
|
|
Peso das crias no dia 4 (média)
|
|
|
|
|
|
|
Peso das crias no dia 13 (média)
|
|
|
|
|
|
|
Retenção de mamilo das crias do sexo masculino no dia 13 (média)
|
|
|
|
|
|
|
CRIAS ANORMAIS
|
|
Mães com 0
|
|
|
|
|
|
|
Mães com 1
|
|
|
|
|
|
|
Mães com ≥ 2 duas crias
|
|
|
|
|
|
|
PERDA DE CRIAS
|
|
Pré-natal (implantações menos nados-vivos)
|
|
Fêmeas com 0
|
|
|
|
|
|
|
Fêmeas com 1
|
|
|
|
|
|
|
Fêmeas com 2
|
|
|
|
|
|
|
Fêmeas com ≥ 3
|
|
|
|
|
|
|
Pós-natal (nados-vivos menos vivos no dia 13 pós-natal)
|
|
Fêmeas com 0
|
|
|
|
|
|
|
Fêmeas com 1
|
|
|
|
|
|
|
Fêmeas com 2
|
|
|
|
|
|
|
Fêmeas com ≥ 3
|
|
|
|
|
|
B.65 MÉTODO DE ENSAIO IN VITRO DE MEMBRANA DE ESTANQUIDADE PARA A CORROSÃO CUTÂNEA
INTRODUÇÃO
|
1.
|
O presente método de ensaio é equivalente à Test Guideline 435 da OCDE (2015). Entende-se por corrosão da pele a produção de danos irreversíveis nos tecidos cutâneos, que se manifestam na forma de necrose visível em toda a epiderme e atingindo a derme, por aplicação de um produto químico em estudo, definido pelo Sistema Mundial Harmonizado de Classificação e Rotulagem de Produtos Químicos da ONU (GHS) (1) e pelo Regulamento (UE) n.o 1272/2008 da União Europeia relativo à classificação, rotulagem e embalagem de substâncias e misturas (CRE) (24). Trata-se de um método de membrana de estanquidade in vitro que pode ser utilizado para identificar produtos químicos corrosivos. Utiliza uma membrana artificial concebida para reagir a produtos químicos corrosivos de forma similar à pele animal in situ.
|
|
2.
|
A corrosividade cutânea tem sido tradicionalmente avaliada pela aplicação do produto químico em estudo à pele de animais vivos, seguida da avaliação dos danos causados aos tecidos após um determinado período (2). Além do presente, foi adotada uma série de outros métodos de ensaio in vitro alternativos (3)(4) ao procedimento normalizado da pele de coelho in vivo (capítulo B.4 do presente anexo, equivalente à Test Guideline 404 da OCDE), para identificar produtos químicos corrosivos (2). A estratégia de ensaio sequencial por etapas do GHS da ONU e o documento de orientação da OCDE sobre abordagens integradas de ensaio e avaliação (IATA) para a corrosão e irritação da pele recomendam o uso de métodos de ensaio in vitro validados e aceites nos módulos 3 e 4 (1) (5). A IATA descreve vários módulos que contêm fontes de informação e ferramentas de análise: (i) fornece orientações sobre como integrar e utilizar os ensaios existentes e os dados não provenientes de ensaios para a avaliação dos potenciais de irritação cutânea e de corrosão cutânea dos produtos químicos e (ii) propõe uma abordagem em caso de necessidade de ensaios complementares, mesmo quando se obtêm resultados negativos (5). Nesta abordagem modular, os resultados positivos dos métodos de ensaio in vitro podem ser utilizados para classificar um produto químico como corrosivo, sem a necessidade de experimentação animal, reduzindo e tornando mais seletiva a utilização de animais em ensaios e evitando a dor e o sofrimento que podem ocorrer quando são usados animais para esta finalidade.
|
|
3.
|
Foram realizados estudos de validação do modelo in vitro de membrana de estanquidade disponível comercialmente com a denominação Corrositex ® (6) (7) (8), estudos esses que apresentaram uma exatidão global de previsão de corrosividade cutânea de 79 % (128/163), uma sensibilidade de 85 % (76/89) e uma especificidade de 70 % (52/74) para 163 substâncias e misturas constantes de uma base de dados (7). Atendendo à sua validade reconhecida, este método de referência validado (MRV) foi recomendado para utilização no âmbito de uma estratégia de ensaio sequencial destinada a avaliar o potencial de corrosão dérmica de produtos químicos (5) (7). Antes de se poder utilizar um método proposto de ensaio in vitro da membrana de estanquidade para a corrosão cutânea para efeitos normativos, é necessário determinar a sua fiabilidade e adequação (precisão), bem como os limites da utilização proposta, para garantir a similaridade com o MRV (9), de acordo com os requisitos das normas de desempenho (10). A aceitação mútua de dados da OCDE só será garantida após a revisão e inclusão de qualquer método novo ou atualizado proposto na sequência das substâncias prioritárias, para inclusão nas diretrizes de ensaio correspondentes da OCDE. Atualmente, apenas um método in vitro é abrangido pela Test Guideline 435 da OCDE (o presente método de ensaio, que utiliza o já referido modelo Corrositex®).
|
|
4.
|
Outros métodos de ensaio para determinação da corrosividade cutânea baseiam-se no uso de pele humana reconstruída (TG 431 OCDE) (3) e em pele de ratazana isolada (TG 430 da OCDE) (4). A mesma diretriz prevê a subcategorização dos produtos químicos corrosivos nas três subcategorias de corrosividade do sistema GHS da ONU e nos três grupos de embalagem da ONU para o risco de corrosividade. Esta Test Guideline foi adotada em 2006 e atualizada em 2015, para ter em conta o documento de orientação da IATA e para atualizar a lista de substâncias de referência.
|
DEFINIÇÕES
|
5.
|
As definições utilizadas constam do apêndice.
|
CONSIDERAÇÕES INICIAIS E LIMITAÇÕES
|
6.
|
O ensaio descrito no presente método permite a identificação de produtos químicos corrosivos, bem como a subcategorização de produtos químicos em estudo corrosivos de acordo com o GHS/CRE (quadro 1). Além disso, pode ser usado para fundamentar decisões sobre a corrosividade e a não corrosividade de determinadas classes de produtos químicos, por exemplo, ácidos orgânicos e inorgânicos, derivados de ácidos (25) e bases para fins de ensaio (7)(11)(12). Descreve um procedimento genérico semelhante ao método de ensaio de referência validado (7). Embora o presente método não forneça informações adequadas sobre a irritação da pele, deve notar-se que o método de ensaio B.46 (equivalente à Test Guideline 439 da OCDE) incide especificamente no efeito de irritação cutânea da pele in vitro (13). Para uma avaliação exaustiva dos efeitos locais na pele após uma exposição única por via dérmica, deve consultar-se o documento de orientação da OCDE relativo a abordagens integradas de ensaio e avaliação (5).
Quadro 1
Categoria e subcategorias de corrosividade cutânea do sistema GHS da ONU (26)
|
Categoria de corrosividade (categoria 1) (para entidades que não utilizam subcategorias)
|
Subcategorias de corrosividade potencial (26) (para entidades que utilizam subcategorias, incluindo o Regulamento CRE)
|
Corrosivo em ≥ 1 de 3 animais
|
|
Exposição
|
Observação
|
|
Corrosivo
|
Subcategoria de corrosividade 1A
|
≤ 3 minutos
|
≤ 1 hora
|
|
Subcategoria de corrosividade 1B
|
> 3 minutos/ ≤ 1 hora
|
≤ 14 dias
|
|
Subcategoria de corrosividade 1C
|
> 1 hora/ ≤ 4 horas
|
≤ 14 dias
|
|
|
7.
|
Uma limitação do método de referência validado (7) reside em que muitos produtos químicos não corrosivos e alguns produtos químicos corrosivos podem não ser adequados para ensaio, com base nos resultados do ensaio de compatibilidade inicial (ver ponto 13). Muitas substâncias químicas aquosas com pH compreendido entre 4,5 e 8,5 não são adequadas para o ensaio; contudo, 85 % dos produtos químicos deste intervalo de pH estudados não se revelaram corrosivos em ensaios com animais (7). O método da membrana de estanquidade in vitro pode ser utilizado para estudar sólidos (solúveis ou insolúveis na água), líquidos (aquosos ou não aquosos) e emulsões. No entanto, os produtos químicos em estudo que não provocam uma alteração detetável no ensaio de compatibilidade (ou seja, uma mudança de cor no CDS do sistema de deteção do método de ensaio de referência validado) não podem ser ensaiados com o método da membrana de estanquidade, devendo-lhes ser aplicados outros métodos de ensaio.
|
PRINCÍPIO DO ENSAIO
|
8.
|
O sistema do ensaio tem dois componentes: uma biobarreira macromolecular sintética e um sistema de deteção de produtos químicos (CDS); o método deteta, através da barreira da membrana CDS, os danos causados por produtos químicos em estudo corrosivos após a aplicação do produto em causa na superfície da barreira da membrana macromolecular sintética (7), provavelmente pelo(s) mesmo(s) mecanismo(s) de corrosão que age(m) na pele viva.
|
|
9.
|
A penetração da barreira da membrana (ou rutura) pode ser medida por um conjunto de procedimentos ou CDS, incluindo a alteração da cor de um indicador de pH ou de qualquer outra propriedade da solução do indicador sob a barreira.
|
|
10.
|
A barreira da membrana deve ser avaliada para ser válida, ou seja, adequada e fiável para o uso pretendido. Contudo, é necessário garantir que as diferentes preparações são compatíveis com as propriedades da barreira, por exemplo, que são capazes de constituir uma barreira a produtos químicos corrosivos e de permitir classificar as propriedades dos produtos químicos corrosivos nas várias subcategorias de corrosividade do GHS da ONU (1). A classificação atribuída baseia-se no tempo que o produto demora a penetrar na membrana até atingir a solução do indicador.
|
DEMONSTRAÇÃO DE COMPETÊNCIA
|
11.
|
Antes de utilizarem de forma rotineira o método da barreira de membrana in vitro em conformidade com o presente método de ensaio, os laboratórios devem demonstrar a sua competência técnica, classificando corretamente as doze substâncias recomendadas no quadro 2. No caso de uma substância incluída na lista estar indisponível ou sempre que se justifique, pode ser utilizada outra substância para a qual estejam disponíveis dados adequados de referência in vivo e in vitro (por exemplo, da lista de produtos químicos de referência (10)), desde que sejam aplicados os critérios de seleção descritos no quadro 1.
Quadro 2
Substâncias para a demonstração de competência
(27)
|
Substância (28)
|
N.o CAS
|
Classe química
|
Subcategoria in vivo do sistema GHS da ONU (29)
|
Subcategoria in vitro do sistema GHS da ONU (29)
|
|
Trifluoreto de boro di-hidratado
|
13319-75-0
|
Ácido inorgânico
|
1A
|
1A
|
|
Ácido nítrico
|
7697-37-2
|
Ácido inorgânico
|
1A
|
1A
|
|
Pentacloreto de fósforo
|
10026-13-8
|
Precursor de ácido inorgânico
|
1A
|
1A
|
|
Cloreto de valerilo
|
638-29-9
|
Cloreto de acilo
|
1B
|
1B
|
|
Hidróxido de sódio
|
1310-73-2
|
Base inorgânica
|
1B
|
1B
|
|
1-(2-Aminoetil)piperazina
|
140-31-8
|
Amina alifática
|
1B
|
1B
|
|
Cloreto de benzenossulfonilo
|
98-09-9
|
Cloreto de acilo
|
1 C
|
1 C
|
|
N,N-Dimetilbenzilamina
|
103-83-3
|
Anilina
|
1 C
|
1 C
|
|
Tetraetilenopentamina
|
112-57-2
|
Amina alifática
|
1 C
|
1 C
|
|
Eugenol
|
97-53-0
|
Fenol
|
NC
|
NC
|
|
Acrilato de nonilo
|
2664-55-3
|
Acrilato/metacrilato
|
NC
|
NC
|
|
Bicarbonato de sódio
|
144-55-8
|
Sal inorgânico
|
NC
|
NC
|
|
PROCEDIMENTO
|
12.
|
Os pontos seguintes descrevem os componentes e os procedimentos de um método de ensaio de membrana de estanquidade artificial, para avaliação da corrosão (7)(15), com base no MRV, designadamente o MRV Corrositex® disponível no comércio. A membrana de estanquidade e o indicador de compatibilidade, bem como as soluções de categorização podem ser construídos, elaborados ou obtidos comercialmente, como é o caso do MRV Corrositex®. Existe uma amostra do protocolo do método de ensaio para o método de ensaio de referência validado (7). Os ensaios devem ser realizados à temperatura ambiente (17-25 °C) e os componentes devem cumprir as condições que se descrevem de seguida.
|
Ensaio de compatibilidade do produto químico em estudo
|
13.
|
Antes da realização do ensaio de membrana de estanquidade, efetua-se um ensaio de compatibilidade para determinar se o produto químico em estudo é detetável pelo CDS. Se o CDS não detetar o produto em causa, o método de ensaio de membrana de estanquidade não é adequado para avaliar a potencial corrosividade desse produto, devendo utilizar-se outro método de ensaio. O CDS e as condições de exposição utilizadas para o ensaio de compatibilidade devem refletir a exposição no ensaio de membrana de estanquidade subsequente.
|
Ensaio de classificação dos produtos químicos em estudo em função do tempo
|
14.
|
Se for adequado no contexto do método de ensaio, um produto químico em estudo que tenha sido qualificado pelo teste de compatibilidade deve ser sujeito a um ensaio de classificação em função do tempo (ensaio de rastreio para distinguir os ácidos ou bases fracos dos fortes). Por exemplo, no método de referência validado, utiliza-se um ensaio de classificação em função do tempo para indicar qual de dois períodos se deve utilizar para a deteção de um potencial de acidez ou alcalinidade significativo. Devem utilizar-se dois períodos diferentes para determinar a corrosividade para a pele e a respetiva subcategoria no sistema GHS da ONU, com base no potencial de acidez ou alcalinidade do produto químico em estudo.
|
COMPONENTES DO MÉTODO DE ENSAIO DE MEMBRANA DE ESTANQUIDADE
Membrana de estanquidade
|
15.
|
A membrana de estanquidade tem dois componentes: um gel aquoso macromolecular proteico e uma membrana permeável de apoio. O gel proteico deve ser impermeável a líquidos e sólidos, mas pode ser corroído e tornar-se permeável. A membrana de estanquidade deve ser armazenada inteira, em condições previamente determinadas e que impeçam a deterioração do gel, por exemplo, por secagem, crescimento microbiano, deslocação, fendas, para que o seu desempenho não seja afetado. Deve determinar-se o período de armazenagem aceitável, não devendo as preparações da membrana de estanquidade ser utilizadas após esse período.
|
|
16.
|
A membrana de apoio permeável confere apoio mecânico ao gel proteico durante o processo de gelificação e de exposição ao produto químico em estudo. Deve impedir o amolecimento ou a deslocação do gel e ser facilmente permeável a todos os produtos químicos em estudo.
|
|
17.
|
O gel proteico, constituído, por exemplo, por queratina, colagénio ou misturas de proteínas que formam uma matriz de gel serve de alvo para o produto químico em estudo. A matéria proteica é colocada sobre a superfície da membrana de apoio, onde gelifica antes de a membrana de estanquidade ser colocada sobre a solução do indicador. O gel proteico deve manter sempre a mesma espessura e densidade e não exibir bolhas de ar ou defeitos que possam afetar a sua integridade funcional.
|
Sistema de deteção química (CDS)
|
18.
|
A solução do indicador, que é a mesma utilizada para o ensaio de compatibilidade, deve reagir à presença do produto químico em estudo. Deve utilizar-se um indicador de pH corante ou com uma combinação de corantes – por exemplo, vermelho de cresol e alaranjado de metilo – que mude de cor devido à presença do produto químico. O sistema de medição pode ser visual ou eletrónico.
|
|
19.
|
Os sistemas de deteção desenvolvidos para detetar a passagem do produto químico em estudo através da membrana de estanquidade devem ser avaliados quanto à sua adequação e fiabilidade, a fim de definir a gama de produtos químicos que podem ser detetados e os limites quantitativos de deteção.
|
REALIZAÇÃO DO ENSAIO
Montagem dos componentes do método de ensaio
|
20.
|
A membrana é colocada num frasco (ou tubo) que contém a solução do indicador, de modo a que a membrana de apoio esteja totalmente em contacto com a solução do indicador e que não se observem bolhas de ar. Deve ter-se o cuidado de assegurar a integridade da membrana.
|
Aplicação do produto químico em estudo
|
21.
|
É cuidadosamente espalhada e uniformemente distribuída pela superfície superior da membrana uma quantidade adequada do produto químico em estudo, como, por exemplo, 500 μl de um líquido ou 500 mg de um sólido finamente pulverizado (7). Prepara-se para cada substância, e para os despectivos controlos, um número adequado de replicados – por exemplo, quatro (7) (ver pontos 23 a 25). Regista-se o momento da aplicação do produto químico em estudo à membrana. Para assegurar o registo exato dos tempos de corrosão curtos, procede-se ao escalonamento dos tempos de aplicação do produto químico nos frascos replicados.
|
Medição das penetrações na membrana
|
22.
|
Cada frasco é adequadamente monitorizado e é registado o momento da primeira mudança na solução do indicador, ou seja, a penetração da membrana, determinando-se o tempo decorrido entre a aplicação e a penetração da membrana.
|
Controlos
|
23.
|
Nos ensaios que utilizem um veículo ou solvente com o produto químico de estudo, o veículo ou solvente deve ser compatível com o sistema da membrana de estanquidade, ou seja, não deve alterar a integridade sistema da membrana nem a corrosividade do produto químico. Se for caso disso, o controlo do solvente (ou do veículo) deve ser ensaiado em simultâneo com o produto químico em estudo, a fim de demonstrar a compatibilidade do solvente com o sistema de membrana.
|
|
24.
|
Deve ensaiar-se em simultâneo com o produto químico em estudo um controlo positivo (corrosivo) com corrosividade intermédia – por exemplo, atividade de 110 ±15 mg de hidróxido de sódio (subcategoria de corrosividade 1B do GHS da ONU) (7) –, para avaliar se o sistema está a funcionar de forma aceitável. Um segundo controlo positivo com a mesma classe química do produto químico em estudo pode ser útil para avaliar o potencial de corrosividade relativo de um produto químico corrosivo. Devem selecionar-se controlo(s) positivo(s) de corrosividade intermédia (por exemplo, da subcategoria 1B do GHS da ONU), a fim de detetar mudanças no tempo de penetração, que pode ser inaceitavelmente maior ou menor do que o valor de referência estabelecido, indicando assim que o sistema de teste não funciona corretamente. Para o efeito, os produtos químicos extremamente corrosivos (subcategoria 1A do sistema GHS da ONU) ou não corrosivos são de utilidade limitada. Um produto químico corrosivo da subcategoria 1B do sistema GHS da ONU permitirá detetar um período demasiado rápido ou demasiado lento. Uma substância fracamente corrosiva (subcategoria 1C do sistema GHS da ONU) pode ser utilizada como controlo positivo, com vista a medir a capacidade do método de ensaio para estabelecer uma distinção sistemática entre substâncias químicas fracamente corrosivas e não corrosivas. Independentemente da abordagem utilizada, deve definir-se uma gama aceitável de respostas ao controlo positivo com base na gama histórica de momentos de avanço para o(s) controlo(s) positivo(s) utilizado(s), como, por exemplo, os desvios-padrão médios 2-3. Em cada estudo, deve determinar-se o tempo de penetração exato para o controlo positivo, para que possam ser detetados desvios fora da gama aceitável.
|
|
25.
|
Um controlo negativo (não corrosivo) – por exemplo, 10 % de ácido cítrico e 6 % de ácido propiónico (7) – também devem ser ensaiados simultaneamente com o produto químico em estudo, constituindo outra medida de controlo de qualidade para demonstrar a integridade funcional da membrana.
|
Critérios de aceitabilidade do estudo
|
26.
|
Atendendo aos parâmetros temporais estabelecidos para cada subcategoria de corrosividade do sistema GHS da ONU, o tempo (em minutos) decorrido entre a aplicação do produto químico em estudo na membrana e a penetração nesta é utilizado para prever a corrosividade do produto em causa. Para que um estudo possa ser considerado aceitável, o controlo positivo paralelo deve produzir o tempo de penetração esperado (por exemplo, 8-16 min até atingir o hidróxido de sódio, se usado como controlo positivo); o controlo negativo simultâneo não deve ser corrosivo. Se for utilizado, o controlo de solvente simultâneo não deve ser corrosivo nem deve alterar a corrosividade potencial do produto químico. Antes de utilizarem um método de ensaio de rotina conforme com o presente método de ensaio, os laboratórios devem demonstrar a sua competência técnica utilizando as doze substâncias recomendadas no quadro 2. No caso dos novos métodos «me-too», desenvolvidos no âmbito deste método de ensaio, que são estruturalmente semelhantes ao método de referência validado (14), deve recorrer-se às normas de desempenho predefinidas para demonstrar a fiabilidade e a exatidão de um novo método antes de ser utilizado para ensaios regulamentares (10).
|
Interpretação dos resultados e classificação das substâncias químicas em estudo
|
27.
|
Utiliza-se o tempo (em minutos) decorrido entre a aplicação do produto químico em estudo na membrana e a penetração nesta para classificar o produto em causa em termos das subcategorias de corrosividade do sistema GHS da ONU (1) e, se for caso disso, do grupo de embalagens da ONU (16). Para cada método de ensaio proposto, são estabelecidos valores-limite para cada uma das três subcategorias. As decisões finais sobre tempos-limite devem ter em conta a necessidade de minimizar a subclassificação do perigo de corrosão (ou seja, os falsos negativos). No presente ensaio, devem utilizar-se os tempos-limite do Corrositex®, descritos no quadro 3, uma vez que este constitui o único método de ensaio atualmente abrangido pela Test Guideline em vigor (7).
Quadro 3
Modelo de previsão do Corrositex®
|
Tempo médio de penetração (min)
|
Previsão do GHS da ONU (32)
|
|
Produtos químicos em estudo da categoria (30) (determinados pelo ensaio de classificação do método)
|
Produtos químicos em estudo da categoria (31) (determinados pelo método de classificação do método)
|
|
0-3 min
|
0-3 min
|
Subcategoria facultativa de corrosividade 1A
|
|
> 3-60 min
|
> 3-30 min
|
Subcategoria facultativa de corrosividade 1B
|
|
> 60-240 min
|
> 30-60 min
|
Subcategoria facultativa de corrosividade 1C
|
|
> 240 min
|
> 60 min
|
Não corrosivo
|
|
DADOS E RELATÓRIOS
Dados
|
28.
|
O tempo (em minutos) decorrido entre a aplicação e a penetração da barreira pelo produto químico em estudo e pelo(s) controlo(s)positivo(s) deve ser indicado num quadro que inclua os dados correspondentes a cada replicado, bem como o desvio-padrão aproximado para cada ensaio.
|
Relatório de ensaio
|
29.
|
O relatório de ensaio deve conter as seguintes informações:
|
|
Produto químico em estudo e substâncias de controlo:
|
—
|
Substância monocomponente: dados de identificação química, como as denominações IUPAC ou CAS, número CAS, código SMILES ou InChI, fórmula estrutural, pureza, identidade química das impurezas, caso se justifique e seja exequível, etc.;
|
|
—
|
Substância multicomponentes, UVCB e mistura: caracterizada, na medida do possível, pela identidade química (ver acima), ocorrência quantitativa e principais propriedades físico-químicas dos componentes;
|
|
—
|
Aspeto físico, solubilidade em água e outras propriedades físico-químicas pertinentes;
|
|
—
|
Proveniência, número do lote, se disponíveis;
|
|
—
|
Tratamento do produto químico em estudo/substância de controlo antes do ensaio, se for o caso;
|
|
—
|
Estabilidade do produto químico em estudo, data-limite de utilização ou data de reanálise, se conhecidas.
|
|
—
|
Condições de armazenagem.
|
|
|
|
Veículo:
|
—
|
Identificação, concentração (se pertinente), volume utilizado;
|
|
—
|
Justificação do excipiente escolhido.
|
|
|
|
Modelo de membrana de estanquidade in vitro e protocolo utilizado, incluindo a exatidão e fiabilidade demonstradas
|
|
|
Condições de realização do ensaio:
|
—
|
Descrição da montagem e dos processos de preparação utilizados;
|
|
—
|
Origem e composição da membrana de estanquidade in vitro;
|
|
—
|
Composição e propriedades da solução do indicador;
|
|
—
|
Quantidades do produto químico em estudo e da substância de controlo;
|
|
—
|
Descrição e justificação do ensaio de classificação temporal realizado;
|
|
—
|
Descrição dos critérios de avaliação e de classificação aplicados;
|
|
—
|
Demonstração da competência na execução do método de ensaio antes da sua utilização regular, através do ensaio das substâncias químicas de demonstração de competência técnica.
|
|
|
|
Resultados:
|
—
|
Quadro com os dados brutos obtidos a partir de amostras individuais de ensaio e de controlo, para cada replicado;
|
|
—
|
Descrição de outros efeitos observados;
|
|
—
|
Classificação atribuída, mencionando o modelo de previsão ou os critérios de decisão utilizados.
|
|
Discussão dos resultados
Conclusões
|
REFERÊNCIAS BIBLIOGRÁFICAS
|
(1)
|
Nações Unidas (ONU) (2013). Globally Harmonized System of Classification and Labelling of Chemicals (GHS), Fifth Revised Edition, UN New York and Geneva, 2013. Disponível em: http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html.
|
|
(2)
|
Capítulo B.4 deste anexo, «Toxicidade aguda: irritação/corrosão dérmica»
|
|
(3)
|
Capítulo B.40.A deste anexo, «Corrosão da pele in vitro: Método de ensaio da epiderme humana reconstruída (RHE)».
|
|
(4)
|
Capítulo B.40 deste anexo, «Corrosão da pele in vitro: Ensaio da resistência elétrica transcutânea (RET)».
|
|
(5)
|
OCDE (2015). Guidance Document on Integrated Approaches to Testing and Assessment of Skin Irritation/Corrosion. Environment, Health and Safety Publications, Series on Testing and Assessment, (No 203). Organisation for Economic Cooperation and Development, Paris.
|
|
(6)
|
Fentem, J.H., Archer, G.E.B., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Esdaile, D.J., Holzhutter, H.-G. and Liebsch, M. (1998). The ECVAM International Validation Study on In Vitro Tests for Skin Corrosivity. 2. Results and Evaluation by the Management Team. Toxicology In Vitro 12, 483-524.
|
|
(7)
|
ICCVAM (1999). Corrositex®. An In Vitro Test Method for Assessing Dermal Corrosivity Potential of Chemicals. The Results of an Independent Peer Review Evaluation Coordinated by ICCVAM, NTP and NICEATM. NIEHS, NIH Publication (No 99-4495.)
|
|
(8)
|
Gordon V.C., Harvell J.D. and Maibach H.I. (1994). Dermal Corrosion, the Corrositex® System: A DOT Accepted Method to Predict Corrosivity Potential of Test Materials. In vitro Skin Toxicology-Irritation, Phototoxicity, Sensitization. Alternative Methods in Toxicology 10, 37-45.
|
|
(9)
|
OCDE (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environmental, Health and Safety Publications. Series on testing and Assessment (No 34).
|
|
(10)
|
OCDE (2014). Performance Standards for the Assessment of Proposed Similar or Modified In Vitro Membrane Barrier Test Method for Skin Corrosion in Relation to TG 435. Organisation for Economic Cooperation and Development, Paris. Disponível em: http://www.oecd.org/chemicalsafety/testing/PerfStand-TG430-June14.pdf.
|
|
(11)
|
ECVAM (2001). Statement on the Application of the CORROSITEX® Assay for Skin Corrosivity Testing. 15th Meeting of ECVAM Scientific Advisory Committee (ESAC), Ispra, Italy. ATLA 29, 96-97.
|
|
(12)
|
U.S. DOT (2002). Exemption DOT-E-10904 (Fifth Revision). (September 20, 2002). Washington, D.C., U.S. DOT.
|
|
(13)
|
Capítulo B.46 deste anexo, «Corrosão da pele in vitro: Método de ensaio em epiderme humana reconstruída». ICCVAM (2004). ICCVAM Recommended Performance Standards for In Vitro Test Methods for Skin Corrosion. NIEHS, NIH Publication No 04-4510. Disponível em: http://www.ntp.niehs.nih.gov/iccvam/docs/dermal_docs/ps/ps044510.pdf.
|
|
(14)
|
U.S. EPA (1996). Method 1120, Dermal Corrosion. Disponível em: http://www.epa.gov/osw/hazard/testmethods/sw846/pdfs/1120.pdf.
|
|
(15)
|
Nações Unidas (ONU) (2013). UN Recommendations on the Transport of Dangerous Goods, Model Regulations, 18th Revised Edition (Part, Chapter 2.8), UN, 2013. Disponível em: http://www.unece.org/fileadmin/DAM/trans/danger/publi/unrec/rev18/English/Rev18_Volume1_Part2.pdf.
|
Apêndice
DEFINIÇÕES
Adequação
: Relação do método de ensaio com o efeito em causa; pertinência e utilidade do ensaio para o fim em vista. Traduz a medida em que o método de ensaio mede ou prevê corretamente o efeito biológico em causa. A adequação compreende a exatidão (concordância) do método de ensaio (9).
Concordância
: Mede o desempenho do método de ensaio no caso dos métodos cujos resultados sejam estabelecidos em função de categorias e constitui um dos aspetos da adequação. Este termo e o termo «exatidão» são muitas vezes utilizados indistintamente para indicar a proporção de produtos químicos testados que são corretamente classificados como positivos ou negativos. A concordância é altamente dependente da prevalência do produto químico em estudo positivo no tipo de produto químico em estudo (9).
Corrosão da pele in vivo
: Produção de danos irreversíveis à pele, nomeadamente a necrose visível da epiderme, prolongando-se para a derme, após a aplicação de um produto químico em estudo durante um máximo de quatro horas. São exemplos típicos de reações corrosivas as úlceras, hemorragias e escaras sanguinolentas e, no final do período de observação de 14 dias, a descoloração, devido à perda de pigmentação da pele, e a formação de zonas de alopécia total e de cicatrizes. As lesões duvidosas poderão ser elucidadas por métodos histopatológicos.
Especificidade
: Proporção dos produtos químicos negativos/inativos que são corretamente classificados pelo método de ensaio. Constitui uma medida da exatidão de um método de ensaio cujos resultados sejam estabelecidos em função de categorias e é um aspeto importante a ter em conta na avaliação da adequação do método em causa (9).
Exatidão
: Grau de acordo entre os resultados do método de ensaio e os valores de referência aceites. Constitui uma medida do desempenho do método e um dos aspetos da adequação. Este termo e o termo «concordância» são muitas vezes utilizados indistintamente para indicar a proporção de resultados corretos de um método de ensaio (9).
Fiabilidade
: Medida em que, utilizando o mesmo protocolo, um método de ensaio pode ser continuadamente reproduzido no mesmo laboratório e em laboratórios diferentes. A fiabilidade é avaliada com base nos valores calculados das reprodutibilidades intralaboratorial e interlaboratorial (9).
GHS(Sistema Mundial Harmonizado de Classificação e Rotulagem de Produtos Químicos)
: Sistema que propõe a classificação dos produtos químicos (substâncias e misturas) em função de tipos e níveis normalizados de perigos físicos, sanitários e ambientais e trata ainda dos correspondentes elementos de comunicação, como pictogramas, palavras-sinal, advertências de perigo, recomendações de prudência e fichas de dados de segurança, de modo a transmitir informações sobre os efeitos indesejáveis do produto em causa, com vista à proteção das pessoas (empregadores, trabalhadores, transportadores, consumidores, pessoal dos serviços de emergência, etc.) e do ambiente (1).
IATA
: Abordagem integrada de ensaio e avaliação.
Mistura
: Uma mistura ou solução composta por duas ou mais substâncias.
NC
: Não corrosivo.
Normas de desempenho
: Normas associadas a um método de ensaio validado com base nas quais pode ser avaliada a comparabilidade de um método de ensaio proposto que lhe seja funcional e mecanisticamente similar. Incluem: (i) componentes essenciais do método de ensaio; (ii) uma lista mínima de produtos químicos de referência, escolhidos de entre os produtos químicos utilizados para demonstrar o desempenho aceitável do método de ensaio validado; e (iii) níveis de exatidão e fiabilidade comparáveis, baseados no que foi obtido para o método de ensaio validado, que o método de ensaio proposto deve demonstrar quando avaliado pela utilização da lista mínima de produtos químicos de referência (9).
Produto químico
: uma substância ou mistura.
Produto químico de ensaio
: qualquer substância ou mistura à qual seja aplicado o presente método de ensaio.
Sensibilidade
: Proporção dos produtos químicos positivos/ativos que são corretamente classificados pelo método de ensaio. Constitui uma medida da exatidão de um método de ensaio cujos resultados são estabelecidos em função de categorias e é um aspeto importante a ter em conta para avaliar a adequação do método em causa (9).
Sistema de deteção química (CDS)
: Sistema de medição visual ou eletrónico com uma solução indicadora que reage à presença de um produto químico em estudo, por exemplo, com a alteração do corante, ou combinação de corantes, do indicador de pH que mostre uma alteração da cor em reação à presença do produto químico em estudo ou com outros tipos de reações químicas ou eletroquímicas.
Substância
: um elemento químico e os seus compostos no estado natural ou obtido por qualquer processo de produção, incluindo qualquer aditivo necessário para preservar a sua estabilidade e qualquer impureza derivada do processo utilizado, mas excluindo qualquer solvente que possa ser separado sem afetar a estabilidade da substância nem modificar a sua composição.
Substância monocomponente
: Substância, definida pela sua composição quantitativa, na qual um dos principais componentes está presente num teor de, pelo menos, 80 % (m/m).
Substância multicomponentes
: Substância, definida pela sua composição quantitativa, na qual mais de um dos principais componentes está presente numa concentração ≥ 10 % (m/m) e < 80 % (m/m). As substâncias multicomponentes resultam de processos de fabrico. A diferença entre uma mistura e uma substância multicomponentes reside em que a primeira se obtém misturando duas ou mais substâncias, sem reação química. As substâncias multicomponentes resultam de reações químicas.
UVCB
: Substâncias de composição desconhecida ou variável, produtos de reação complexos ou materiais biológicos.
B.66 ENSAIOS DE TRANSATIVAÇÃO TRANSFETADA COM ESTABILIDADE PARA DETEÇÃO DE RECETORES DE ESTROGÉNIO AGONISTAS E ANTAGONISTAS E ANTAGONISTAS
INTRODUÇÃO GERAL
Método de ensaio baseado na Test Guideline da OCDE
|
1.
|
O presente método de ensaio é equivalente à Test Guideline 455 (2016) da OCDE. Esta última é uma diretriz de ensaio baseada no desempenho (PBTG), que descreve a metodologia de ensaios de transativação transfetada in vitro para deteção de recetores de estrogénio agonistas e antigonistas (ensaios ER TA). É composto por vários métodos de ensaio, mecânica e funcionalmente afins, para a identificação de recetores de estrogénio (ERα e/ou ERβ) agonistas e antagonistas, e deve facilitar o desenvolvimento de novos métodos de ensaio semelhantes ou modificados de acordo com os princípios de validação estabelecidos no Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment (1) da OCDE. Os métodos de ensaio de referência inteiramente validados (apêndice 2 e apêndice 3) que constituem a base deste PBTG são os seguintes:
|
—
|
The Stably Transfected TA (STTA) assay (2) using the (h) ERα-HeLa-9903 cell line; and
|
|
—
|
The VM7Luc ER TA assay (3) using the VM7Luc4E2 cell line (33) which predominately expresses hERα with some contribution from hERβe(4)(5).
|
Para o desenvolvimento e a validação de ensaios semelhantes para o mesmo parâmetro de risco, existem normas de desempenho (PS) (6) (7), que importa utilizar. Permitem a alteração atempada do PBTG 455, para que se possam acrescentar novos ensaios semelhantes a um PBTG atualizado. No entanto, só serão adicionados ensaios semelhantes após análise e aprovação pela OCDE do cumprimento das normas de desempenho. Os ensaios incluídos no método TG 455 podem ser utilizados indiscriminadamente para satisfazer as necessidades dos países membros da OCDE relativamente a resultados de ensaios de transativação de recetores de estrogénio, beneficiando simultaneamente da aceitação mútua de dados da OCDE.
|
Antecedentes e princípios dos ensaios incluídos no presente método
|
2.
|
A OCDE iniciou em 1998 uma atividade altamente prioritária com o objetivo de rever as diretrizes em vigor para a pesquisa e o ensaio de produtos químicos com possíveis efeitos de perturbação endócrina, bem como elaborar novas diretrizes. O quadro concetual da OCDE para ensaio e avaliação de produtos químicos potencialmente perturbadores do sistema endócrino do sistema endócrino foi revisto em 2012. Os quadros concetuais original e revisto constam, como anexo, do Guidance Document on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption da OCDE (8). O quadro concetual contém cinco níveis, cada um dos quais corresponde a um nível diferente de complexidade biológica. O ensaio de transativação de ER (TA) descrito no presente método de ensaio é do nível 2, que abrange os ensaios in vitro que facultam dados sobre mecanismos/vias endócrinos selecionados. O método destina-se a ensaios de transativação (TA) in vitro, concebidos para identificar recetores de estrogénio (RE) agonistas e antagonistas.
|
|
3.
|
A interação dos estrogénios com os RE pode afetar a transcrição de genes estrogénicos, que podem conduzir à indução ou inibição de processos celulares, incluindo os necessários para a proliferação celular, para o desenvolvimento fetal normal e para a função reprodutora (9) (10) (11). A perturbação de sistemas estrogénicos normais pode provocar efeitos nocivos no desenvolvimento normal (ontogénese), na saúde reprodutiva e na integridade do aparelho reprodutor.
|
|
4.
|
Os ensaios de TA in vitro baseiam-se numa interação direta ou indireta das substâncias com um recetor específico que regula a transcrição de um produto de gene repórter. Estes ensaios foram amplamente utilizados para avaliar a expressão dos genes regulados por recetores nucleares específicos, como os ER (12) (13) (14) (15) (16). Foram propostos para deteção da transativação estrogénica regulada pelo ER (17) (18) (19). Há, pelo menos, dois grandes subtipos de RE nucleares, α e β, que são codificados por genes distintos. As proteínas respetivas têm funções biológicas diferentes, bem como distribuições de tecidos e afinidades com ligandos diferentes (20) (21) (22) (23) (24) (25) (26). O ERα nuclear determina a reação estrogénica clássica (27) (28) (29) (30), pelo que a maioria dos modelos atualmente em desenvolvimento para medir a ativação ou a inibição dos ER são específicos do ERα. Os ensaios são utilizados para identificar produtos químicos que ativem (ou inibam) o ER na sequência de ligações de ligandos, após os quais o complexo recetor-ligando se liga a elementos de resposta de ADN específicos e transativa um gene repórter, que provoca o aumento da expressão celular de um marcador proteico. Nestes ensaios podem ser utilizadas diferentes respostas do repórter. Em sistemas à base de luciferase, a enzima luciferase transforma o substrato de luciferina num produto bioluminescente que pode ser medido quantitativamente com um luminómetro. Outros exemplos de repórteres comuns são a proteína fluorescente e o gene lacZ, que codificam a β-galactosidase, uma enzima que pode transformar o substrato incolor X-Gal (5-bromo-4-cloro-indolil-galactopiranosídeo azul) num produto quantificável com um espectrofotómetro. Estes repórteres podem ser avaliados de forma rápida e pouco onerosa com os conjuntos de ensaio existentes no mercado.
|
|
5.
|
Os estudos de validação do STTA e dos ensaios VM7Luc demonstraram a sua adequação e fiabilidade para o fim a que se destinam (3) (4) (5) (30). As normas de desempenho para ensaios ER TA baseados na luminescência com linhas celulares mamárias constam do ICCVAM Test Method Evaluation Report on the LUMI-CELL® ER (VM7Luc ER TA) Test Method: An In Vitro Assay for Identifying Human Estrogen Recetor Agonist and Antagonist Activity of Chemicals (3). Estas normas de desempenho foram alteradas para serem aplicáveis tanto aos ensaios STTA como aos ensaios VM7Luc (2).
|
|
6.
|
As definições e abreviaturas utilizadas no presente método de ensaio são descritas no apêndice 1.
|
Âmbito e limitações relacionados com os ensaios de TA
|
7.
|
Os ensaios são propostos para efeitos de rastreio e de definição de prioridades, mas podem também fornecer informações mecanísticas passíveis de serem utilizadas numa abordagem de ponderação da suficiência da prova. Centram-se na TA induzida por um produto químico que se liga ao ER num sistema in vitro. Assim, os resultados não devem ser extrapolados diretamente para o complexo de sinalização e regulação do sistema endócrino intacto in vivo.
|
|
8.
|
A TA mediada pelos ER é considerada um dos principais mecanismos de desregulação endócrina, embora existam outros mecanismos através dos quais o ED pode ocorrer, incluindo (i) interações com outros recetores e sistemas enzimáticos no sistema endócrino, (ii) síntese das hormonas, (iii) ativação e/ou inativação metabólica de hormonas, (iv) distribuição de hormonas por tecidos-alvo e (v) eliminação de hormonas do organismo. Nenhum dos ensaios no âmbito do presente método aborda estes modos de ação.
|
|
9.
|
O presente método de ensaio incide na capacidade dos produtos químicos de ativarem (ou seja, agirem como agonistas) e eliminarem (ou seja, agirem como antagonistas) a transcrição dependente do ER. Alguns produtos químicos podem, em função do tipo de células, apresentar atividades de agonista e antagonista, sendo conhecidos como moduladores seletivos de recetores de estrogénio (SERMS). Os produtos químicos que são negativos nestes ensaios podem ser avaliados num ensaio de ligação de ER antes de se concluir que não se ligam ao recetor. Além disso, os ensaios só são suscetíveis de informar sobre a atividade da molécula-mãe tendo em conta a limitada capacidade de metabolização dos sistemas de células in vitro. Considerando que, na validação, apenas foram utilizadas substâncias estremes, a aplicabilidade a misturas de ensaio não foi tida em conta. No entanto, o método é teoricamente aplicável ao ensaio de substâncias multicomponentes, UVCB e misturas. Antes da sua aplicação a uma substância multicomponentes, UVCB ou mistura para obter dados com uma finalidade normativa, deve ponderar-se se e, em caso afirmativo, por que razão o método pode proporcionar resultados adequados para o efeito. Essas considerações não são necessárias se existir um requisito normativo para o ensaio da mistura.
|
|
10.
|
Para efeitos informativos, o quadro 1 fornece os resultados do ensaio de agonistas para as 34 substâncias que foram ensaiadas com ambos os métodos de ensaio de referência plenamente validados descritos no presente. Destas substâncias, 26 são classificadas como ER agonistas e 8 negativas, com base em relatórios publicados, incluindo ensaios in vitro de ligação de ER e TA e/ou o ensaio uterotrófico (2) (3) (18) (31) (32) (33) (34). O quadro 2 fornece os resultados do ensaio de antagonistas relativos às 15 substâncias ensaiadas em ambos os métodos de ensaio de referência plenamente validados descritos no presente método de ensaio. Entre estas substâncias, 4 são classificadas como antagonistas ER definitivas/presumidas e 10 negativas, com base em relatórios publicados, incluindo os ensaios in vitro de ligação ER e TA (2) (3) (18) (31). Relativamente aos dados resumidos nos quadros 1 e 2, houve concordância total entre os dois métodos de ensaio de referência para as classificações de todas as substâncias, com exceção de uma (mifepristona) para o ensaio antagonista, e cada substância foi classificada corretamente como agonista/antagonista de ER ou negativa. Constam das normas de desempenho para o AETR (6) (7), apêndice 2 (quadros 1, 2 e 3), informações suplementares sobre este grupo de produtos químicos, assim como ensaios de produtos químicos adicionais no STTA e no VM7Luc ER TA realizados durante os estudos de validação.
|
Quadro 1
Panorâmica dos resultados de ensaios STTA e VM7Luc ER TA para substâncias ensaiadas em ambos os ensaios agonistas e classificadas como ER agonistas (POS) ou negativas (NEG
|
|
Substância
|
N.o CAS
|
Ensaio STTA (35)
|
Ensaio VM7Luc ER TA (36)
|
Fonte de dados para classificação (38)
|
|
ER TA Atividade
|
Valor PC10 (M)
|
Valor PC50
(2) (M)
|
Atividade ER TA
|
Valor EC50
(2), (37) (M)
|
Outros ER TAs (34)
|
ER Ligante
|
Uterotrófico
|
|
1
|
17ß-Estradiol (1)
|
50-28-2
|
POS
|
< 1,00 × 10–11
|
< 1,00 × 10–11
|
POS
|
5,63 × 10–12
|
POS (227/227)
|
POS
|
POS
|
|
2
|
17α-Estradiol (1)
|
57-91-0
|
POS
|
7,24 × 10–11
|
6,44 × 10–10
|
POS
|
1,40 × 10–9
|
POS(11/11)
|
POS
|
POS
|
|
3
|
17α-Etinilestradiol (1)
|
57-63-6
|
POS
|
< 1,00 × 10–11
|
< 1,00 × 10–11
|
POS
|
7,31 × 10–12
|
POS(22/22)
|
POS
|
POS
|
|
4
|
17β-Trembolona
|
10161-33-8
|
POS
|
1,78 × 10–8
|
2,73 × 10–7
|
POS
|
4,20 × 10–8
|
POS (2/2)
|
NT
|
NT
|
|
5
|
19-Nortestosteron (1)
|
434-22-0
|
POS
|
9,64 × 10–9
|
2,71 × 10–7
|
POS
|
1,80 × 10–6
|
POS(4/4)
|
POS
|
POS
|
|
6
|
4-Cumilfenol (1)
|
599-64-4
|
POS
|
1,49 × 10–7
|
1,60 × 10–6
|
POS
|
3,20 × 10–7
|
POS(5/5)
|
POS
|
NT
|
|
7
|
4-terc-Octilfenol (1)
|
140-66-9
|
POS
|
1,85 × 10–9
|
7,37 × 10–8
|
POS
|
3,19 × 10–8
|
POS(21/24)
|
POS
|
POS
|
|
8
|
Apigenin (1)
|
520-36-5
|
POS
|
1,31 × 10–7
|
5,71 × 10–7
|
POS
|
1,60 × 10–6
|
POS(26/26)
|
POS
|
NT
|
|
9
|
Atrazin (1)
|
1912-24-9
|
NEG.
|
—
|
—
|
NEG.
|
—
|
NEG (30/30)
|
NEG.
|
NT
|
|
10
|
Bisfenol A (1)
|
80-05-7
|
POS
|
2,02 × 10–8
|
2,94 × 10–7
|
POS
|
5,33 × 10–7
|
POS (65/65)
|
POS
|
POS
|
|
11
|
Bisfenol B (1)
|
77-40-7
|
POS
|
2,36 × 10–8
|
2,11 × 10–7
|
POS
|
1,95 × 10–7
|
POS(6/6)
|
POS
|
POS
|
|
12
|
Ftalato de benzilo e butilo (1)
|
85-68-7
|
POS
|
1,14 × 10–6
|
4,11 × 10–6
|
POS
|
1,98 × 10–6
|
POS(12/14)
|
POS
|
NEG.
|
|
13
|
Corticosteron (1)
|
50-22-6
|
NEG.
|
—
|
—
|
NEG.
|
—
|
NEG (6/6)
|
NEG.
|
NT
|
|
14
|
Cumesterol (1)
|
479-13-0
|
POS
|
1,23 × 10–9
|
2,00 × 10–8
|
POS
|
1,32 × 10–7
|
POS(30/30)
|
POS
|
NT
|
|
15
|
Daidzeín (1)
|
486-66-8
|
POS
|
1,76 × 10–8
|
1,51 × 10–7
|
POS
|
7,95 × 10–7
|
POS(39/39)
|
POS
|
POS
|
|
16
|
Dietilestilbesterol (1)
|
56-53-1
|
POS
|
< 1,00 × 10–11
|
2,04 × 10–11
|
POS
|
3,34 × 10–11
|
POS(42/42)
|
POS
|
NT
|
|
17
|
Ftalato de di-n-butilo
|
84-74-2
|
POS
|
4,09 × 10–6
|
|
POS
|
4,09 × 10–6
|
POS(6/11)
|
POS
|
NEG.
|
|
18
|
Etilparabeno
|
120-47-8
|
POS
|
5,00 × 10–6
|
(no PC50)
|
POS
|
2,48 × 10–5
|
POS
|
|
NT
|
|
19
|
Estron (1)
|
53-16-7
|
POS
|
3,02 × 10–11
|
5,88 × 10–10
|
POS
|
2,34 × 10–10
|
POS(26/28)
|
POS
|
POS
|
|
20
|
Genisteín (1)
|
446-72-0
|
POS
|
2,24 × 10–9
|
2,45 × 10–8
|
POS
|
2,71 × 10–7
|
POS(100/102)
|
POS
|
POS
|
|
21
|
Haloperidol
|
52-86-8
|
NEG.
|
—
|
—
|
NEG.
|
—
|
NEG (2/2)
|
NEG.
|
NT
|
|
22
|
Campferol (1)
|
520-18-3
|
POS
|
1,36 × 10–7
|
1,21 × 10–6
|
POS
|
3,99 × 10–6
|
POS(23/23)
|
POS
|
NT
|
|
23
|
Clordecon (1)
|
143-50-0
|
POS
|
7,11 × 10–7
|
7,68 × 10–6
|
POS
|
4,91 × 10–7
|
POS(14/18)
|
POS
|
NT
|
|
24
|
Cetoconazol
|
65277-42-1
|
NEG.
|
—
|
—
|
NEG.
|
—
|
NEG (2/2)
|
NEG.
|
NT
|
|
25
|
Linurão (1)
|
330-55-2
|
NEG.
|
—
|
—
|
NEG.
|
—
|
NEG (8/8)
|
NEG.
|
NT
|
|
26
|
meso-Hexesterol (1)
|
84-16-2
|
POS
|
< 1,00 × 10–11
|
2,75 × 10–11
|
POS
|
1,65 × 10–11
|
POS(4/4)
|
POS
|
NT
|
|
27
|
Metiltestosteron (1)
|
58-18-4
|
POS
|
1,73 × 10–7
|
4,11 × 10–6
|
POS
|
2,68 × 10–6
|
POS(5/6)
|
POS
|
NT
|
|
28
|
Morina
|
480-16-0
|
POS
|
5,43 × 10–7
|
4,16 × 10–6
|
POS
|
2,37 × 10–6
|
POS(2/2)
|
POS
|
NT
|
|
29
|
Noretinodrel (1)
|
68-23-5
|
POS
|
1,11 × 10–11
|
1,50 × 10–9
|
POS
|
9,39 × 10–10
|
POS(5/5)
|
POS
|
NT
|
|
30
|
p,p’-Metoxicloro (1)
|
72-43-5
|
POS
|
1,23 × 10–6
|
(no PC50) (2)
|
POS
|
1,92 × 10–6
|
POS(24/27)
|
POS
|
POS
|
|
31
|
Fenobarbital (1)
|
57-30-7
|
NEG.
|
—
|
—
|
NEG.
|
—
|
NEG (2/2)
|
NEG.
|
NT
|
|
32
|
Reserpina
|
50-55-5
|
NEG.
|
—
|
—
|
NEG.
|
—
|
NEG (4/4)
|
NEG.
|
NT
|
|
33
|
Espironolacton (1)
|
52-01-7
|
NEG.
|
—
|
—
|
NEG.
|
—
|
NEG (4/4)
|
NEG.
|
NT
|
|
34
|
Testosterona
|
58-22-0
|
POS
|
2,82 × 10–8
|
9,78 × 10–6
|
POS
|
1,75 × 10–5
|
POS(5/10)
|
POS
|
NT
|
|
Abreviaturas: N.o CAS = Número de registo do Chemical Abstracts Service; M = molar; EC50 = Concentração semimáxima eficaz de um produto químico em estudo; NEG = negativo; POS= positivo; NT = não ensaiado; PC10 (e PC50) = concentração um produto químico em estudo em que a resposta é 10 % (ou 50 % para PC50) da resposta induzida pelo controlo positivo (E2, 1nM) em cada placa.
|
Quadro 2
Comparação dos resultados dos ensaios SSTTA e VM7Luc ER TA para substâncias ensaiadas em ambos os ensaios agonistas e classificadas como ER antagonistas (POS) ou negativas (NEG)
|
|
Substância (3)
|
N.o CAS
|
Ensaio ER STTA (39)
|
Ensaio VM7Luc ER TA (40)
|
Efeitos candidatos ER STTA (42)
|
Classificação consensual do ICCVAM (43) Classe de produtos químicos
|
MeSH (44)
|
Classe de produtos (45)
|
|
Atividade ER TA
|
Valor IC50
(4) (M)
|
Atividade ER TA
|
Valor IC50
(4), (41) (M)
|
|
1
|
4-Hidroxitamoxifeno
|
68047-06-3
|
POS
|
3,97 × 10–9
|
POS
|
2,08 × 10–7
|
POS moderado
|
POS
|
Hidrocarboneto cíclico
|
Produtos farmacêuticos
|
|
2
|
Dibenzo[a.h]antraceno
|
53-70-3
|
POS
|
N.o IC50
|
POS
|
N.o IC50
|
POS
|
PP
|
Composto policíclico
|
Produto químico de laboratório, produto natural
|
|
3
|
Mifepristona
|
84371-65-3
|
POS
|
5,61 × 10–6
|
NEG.
|
—
|
POS moderado
|
NEG.
|
Esteroide
|
Produtos farmacêuticos
|
|
4
|
Raloxifeno-HCl
|
82640-04-8
|
POS
|
7,86 × 10–10
|
POS
|
1,19 × 10–9
|
POS moderado
|
POS
|
Hidrocarboneto cíclico
|
Produtos farmacêuticos
|
|
5
|
Tamoxifeno
|
10540-29-1
|
POS
|
4,91 × 10–7
|
POS
|
8,17 × 10–7
|
POS
|
POS
|
Hidrocarboneto cíclico
|
Produto farmacêutico
|
|
6
|
17β-Estradiol
|
50-28-2
|
NEG.
|
—
|
NEG.
|
—
|
PN
|
PN
|
Esteroide
|
Produto farmacêutico, agente veterinário
|
|
7
|
Apigenina
|
520-36-5
|
NEG.
|
—
|
NEG.
|
—
|
NEG.
|
NEG.
|
Composto heterocíclico
|
Corante, produto natural, produto farmacêutico intermédio
|
|
8
|
Atrazina
|
1912-24-9
|
NEG.
|
—
|
NEG.
|
—
|
NEG.
|
PN
|
Composto heterocíclico
|
Herbicida
|
|
9
|
Ftalato de di-n-butilo
|
84-74-2
|
NEG.
|
—
|
NEG.
|
—
|
NEG.
|
NEG.
|
Éster de ácido ftálico
|
Ingrediente cosmético, produto químico industrial, plastificante
|
|
10
|
Fenarimol
|
60168-88-9
|
NEG.
|
—
|
NEG.
|
—
|
não ensaiado
|
PN
|
Composto heterocíclico, pirimidina
|
Fungicida
|
|
11
|
Flavona
|
525-82-6
|
NEG.
|
—
|
NEG.
|
—
|
PN
|
PN
|
Flavonoide, composto heterocíclico
|
Produto natural, produto farmacêutico
|
|
12
|
Flutamida
|
13311-84-7
|
NEG.
|
—
|
NEG.
|
—
|
NEG.
|
PN
|
Amida
|
Produto farmacêutico, agente veterinário
|
|
13
|
Genisteína
|
446-72-0
|
NEG.
|
—
|
NEG.
|
—
|
PN
|
NEG.
|
Flavonoide, composto heterocíclico
|
Produto natural, produto farmacêutico
|
|
14
|
p-n-Nonilfenol
|
104-40-5
|
NEG.
|
—
|
NEG.
|
—
|
não ensaiado
|
NEG.
|
Fenol
|
Produto químico intermédio
|
|
15
|
Resveratrol
|
501-36-0
|
NEG.
|
—
|
NEG.
|
—
|
PN
|
NEG.
|
Hidrocarboneto cíclico
|
Produto natural
|
|
Abreviaturas: N.o CAS = Número de registo do Chemical Abstracts Service; M = molar; IC50 = Concentração semimáxima inibitória de uma substância química em estudo; NEG = negativo; PN = negativo presumível; POS= positivo; PP = presumível positivo.
|
COMPONENTES DOS ENSAIOS ER TA
Componentes de ensaio essenciais
|
11.
|
O presente método de ensaio aplica-se aos ensaios que utilizam um recetor ERα transfetado estavelmente ou endógeno e um constituinte de gene repórter transfetado estavelmente sob o controlo de um ou mais elementos de resposta estrogénicos; no entanto, podem estar presentes outros recetores, como o ERβ. Estes são componentes essenciais do ensaio.
|
Controlos
|
12.
|
Deve ser descrita a base dos padrões de referência concorrentes propostos para cada ensaio de agonistas e antagonistas. Os controlos paralelos (negativo, solvente e positivo), se necessário, servem como indicação de que o método de ensaio está a funcionar nas condições do ensaio e fornecem uma base para comparações entre experiências; geralmente, são parte dos critérios de aceitabilidade de uma experiência (1).
|
Procedimentos normalizados de controlo de qualidade
|
13.
|
Devem ser aplicados os procedimentos normalizados de controlo da qualidade descritos para cada ensaio, a fim de garantir que a linha celular se mantém estável através de passagens múltiplas, continua isenta de micoplasma (isto é, sem contaminação bacteriana) e mantém a capacidade de fornecer as respostas mediadas pelo ER previstas ao longo do tempo. As linhas celulares devem ser sujeitas a um controlo suplementar para verificar se a sua identidade está correta, bem como para identificar outros contaminantes (por exemplo, fungos, leveduras e vírus).
|
Demonstração da competência técnica do laboratório
|
14.
|
Antes de ensaiar produtos químicos desconhecidos com qualquer um dos ensaios no âmbito do presente método de ensaio, cada laboratório deve demonstrar a sua competência na utilização do ensaio. Para demonstrar a competência, cada laboratório deve ensaiar as 14 substâncias enumeradas no quadro 3 no que respeita ao ensaio de agonistas e as 10 substâncias do quadro 4 para o ensaio antagonista. Este ensaio de competência deve também confirmar a capacidade de resposta do sistema de ensaio. A lista de substâncias de competência é um subconjunto das substâncias de referência constantes das normas de desempenho para os ensaios ER TA (6). Estas substâncias estão comercialmente disponíveis, representam as classes de produtos químicos normalmente associadas à atividade agonista ou antagonista de ER e apresentam uma gama de potência adequada e esperada para agonistas/antagonistas de ER (ou seja, de fortes a fracos) e incluem negativos. Os ensaios das substâncias de competência devem ser reproduzidos pelo menos duas vezes, em dias diferentes. A competência é demonstrada por uma classificação correta (positiva/negativa) de cada uma das substâncias. O ensaio de competência deve ser repetido por todos os técnicos quando aprendem os ensaios. Em função do tipo de célula, algumas destas substâncias de competência podem comportar-se como SERMS e exibir atividades agonistas e antagonistas. No entanto, as substâncias para a demonstração de competência são classificadas nos quadros 3 e 4 de acordo com a sua atividade predominante conhecida, que deve ser utilizada na avaliação da competência.
|
|
15.
|
Para demonstrar o desempenho e para fins de controlo de qualidade, cada laboratório deve elaborar bases de dados históricos agonistas e antagonistas com padrões de referência (p. ex., 17β-estradiol e tamoxifeno), produtos químicos de controlo positivo, negativo e de solvente (p.e., DMSO). Inicialmente, a base de dados deve ser gerada a partir de uma série de, pelo menos, 10 ensaios de agonistas independentes (por ex., 17β-estradiol) e 10 ensaios de antagonistas independentes (por exemplo, tamoxifeno). Os resultados de futuras análises destes padrões de referência e dos controlos de solventes devem ser incluídos para aumentar a base de dados, a fim de garantir a coerência e o desempenho do bioensaio por parte do laboratório ao longo do tempo.
|
Quadro 3
Lista de (14) substâncias para o ensaio de competência do agonistas
(53)
|
No
(52)
|
Substância
|
N.o CAS
|
Resposta prevista (46)
|
Ensaio STTA
|
Ensaio VM7Luc ER TA
|
Classe de produtos químicos MESH (50)
|
Classe de produtos (51)
|
|
Valor PC10 (M) (47)
|
Valor PC50 (M) (47)
|
Ensaio Conc. Gama (M)
|
Valor VM7Luc EC50 (M) (48)
|
Conc. mais elevada na procura de gamas (M) (49)
|
|
14
|
Dietilestilbesterol
|
56-53-1
|
POS
|
< 1,00 × 10–11
|
2,04 × 10–11
|
10–14 – 10–8
|
3,34 × 10–11
|
3,73 × 10–4
|
Hidrocarboneto cíclico
|
Agente farmacêutico de uso veterinário
|
|
12
|
17α-Estradiol
|
57-91-0
|
POS
|
4,27 × 10–11
|
6,44 × 10–10
|
10–11 – 10–5
|
1,40 × 10–9
|
3,67 × 10–3
|
Esteroide
|
Produto farmacêutico, agente veterinário
|
|
15
|
meso-Hexesterol
|
84-16-2
|
POS
|
< 1,00 × 10–11
|
2,75 × 10–11
|
10–11 – 10–5
|
1,65 × 10–11
|
3,70 × 10–3
|
Hidrocarboneto cíclico, fenol
|
Produto farmacêutico, agente veterinário
|
|
11
|
4-terc-Octilfenol
|
140-66-9
|
POS
|
1,85 × 10–9
|
7,37 × 10–8
|
10–11 – 10–5
|
3,19 × 10–8
|
4,85 × 10–3
|
Fenol
|
Produto químico intermédio
|
|
9
|
Genisteína
|
446-72-0
|
POS
|
2,24 × 10–9
|
2,45 × 10–8
|
10–11 – 10–5
|
2,71 × 10–7
|
3,70 × 10–4
|
Flavonoide, composto heterocíclico
|
Produto natural, produto farmacêutico
|
|
6
|
Bisfenol A
|
80-05-7
|
POS
|
2,02 × 10–8
|
2,94 × 10–7
|
10–11 – 10–5
|
5,33 × 10–7
|
4,38 × 10–3
|
Fenol
|
Produto químico intermédio
|
|
2
|
Campferol
|
520-18-3
|
POS
|
1,36 × 10–7
|
1,21 × 10–6
|
10–11 – 10–5
|
3,99 × 10–6
|
3,49 × 10–3
|
Flavonoide, composto heterocíclico
|
Produto natural
|
|
3
|
Ftalato de butilo
|
85-68-7
|
POS
|
1,14 × 10–6
|
4,11 × 10–6
|
10–11 – 10–5
|
1,98 × 10–6
|
3,20 × 10–4
|
Ácido carboxílico, éster, ácido ftálico
|
Plastificante, Produtos Químicos Industriais
|
|
4
|
p,p’-Metoxicloro
|
72-43-5
|
POS
|
1,23 × 10–6
|
—
|
10–11 – 10–5
|
1,92 × 10–6
|
2,89 × 10–3
|
Hidrocarboneto halogenado
|
Pesticida, Agente Veterinário
|
|
1
|
Etilparabeno
|
120-47-8
|
POS
|
5,00 × 10–6
|
—
|
10–11 – 10–5
|
2,48 × 10–5
|
6,02 × 10–3
|
Ácido carboxílico, fenol
|
Produto farmacêutico, conservante
|
|
17
|
Atrazina
|
1912-24-9
|
NEG.
|
—
|
—
|
10–10 – 10–4
|
—
|
4,64 × 10–4
|
Composto heterocíclico
|
Herbicida
|
|
20
|
Espironolactona
|
52-01-7
|
NEG.
|
—
|
—
|
10–11 – 10–5
|
—
|
2,40 × 10–3
|
Lactona, esteroide
|
Produto farmacêutico
|
|
21
|
Cetoconazol
|
65277-42-1
|
NEG.
|
—
|
—
|
10–11 – 10–5
|
—
|
9,41 × 10–5
|
Composto heterocíclico
|
Produto farmacêutico
|
|
22
|
Reserpina
|
50-55-5
|
NEG.
|
—
|
—
|
10–11 – 10–5
|
—
|
1,64 × 10–3
|
Composto heterocíclico, indol
|
Produto farmacêutico, agente veterinário
|
|
Abreviaturas: N.o CAS = Número de registo do Chemical Abstracts Service; EC50 = Concentração semimáxima eficaz de uma substância química em estudo; NEG = negativo; POS= positivo; PC10 (e PC50) = concentração de uma substância de ensaio em que a resposta é 10 % (ou 50 % para PC50) da resposta induzida pelo controlo positivo (E2, 1nM) em cada placa.
|
Quadro 4
Lista de (10) substâncias para o ensaio de competência de antagonistas
|
|
Substância (5)
|
N.o CAS
|
Ensaio ER STTA (54)
|
Ensaio VM7Luc ER TA (55)
|
Efeitos candidatos ER STTA (54)
|
CCVAM (58) Consensus Classification Classe de produtos químicos
|
MeSH (59)
|
Classe de produtos (60)
|
|
Atividade ER TA
|
IC50 (M)
|
Gama de concentrações de ensaio (M)
|
Atividade ER TA
|
IC50
(56) (M)
|
Conc. mais elevada na procura de gamas (M) (57)
|
|
1
|
4-Hidroxioxitamoxifeno
|
68047-06-3
|
POS
|
3,97 × 10–9
|
10–12 – 10–7
|
POS
|
2,08 × 10–7
|
2,58 × 10–4
|
POS moderado
|
POS
|
Hidrocarboneto cíclico
|
Produto farmacêutico
|
|
2
|
Raloxifeno-HCl
|
82640-04-8
|
POS
|
7,86 × 10–10
|
10–12 – 10–7
|
POS
|
1,19 × 10–9
|
1,96 × 10–4
|
POS moderado
|
POS
|
Hidrocarboneto cíclico
|
Produto farmacêutico
|
|
3
|
Tamoxifeno
|
10540-29-1
|
POS
|
4,91 × 10–7
|
10–10 – 10–5
|
POS
|
8,17 × 10–7
|
2,69 × 10–4
|
POS
|
POS
|
Hidrocarboneto cíclico
|
Produto farmacêutico
|
|
4
|
17β-Estradiol
|
50-28-2
|
NEG.
|
—
|
10–9 – 10–4
|
NEG.
|
—
|
3,67 × 10–3
|
para ser negativa (*4)
|
PN
|
Esteroide
|
Produto farmacêutico, agente veterinário
|
|
5
|
Apigenina
|
520-36-5
|
NEG.
|
—
|
10–9 – 10–4
|
NEG.
|
—
|
3,70 × 10–4
|
NEG.
|
NEG.
|
Composto heterocíclico
|
Corante, produto natural, produto farmacêutico intermédio
|
|
6
|
Ftalato de di-n-butilo
|
84-74-2
|
NEG.
|
—
|
10–8 – 10–3
|
NEG.
|
—
|
3,59 × 10–3
|
NEG.
|
NEG.
|
Éster, ácido ftálico
|
Ingrediente cosmético, produto químico industrial, plastificante
|
|
7
|
Flavona
|
525-82-6
|
NEG.
|
—
|
10–8 – 10–3
|
NEG.
|
—
|
4,50 × 10–4
|
para ser negativa (*4)
|
PN
|
Flavonoide, composto heterocíclico
|
Produto natural, produto farmacêutico
|
|
8
|
Genisteína
|
446-72-0
|
NEG.
|
—
|
10–9 – 10–4
|
NEG.
|
—
|
3,70 × 10–4
|
para ser negativa (*4)
|
NEG.
|
Flavonoide, composto heterocíclico
|
Produto natural, produto farmacêutico
|
|
9
|
p-n-Nonilfenol
|
104-40-5
|
NEG.
|
—
|
10–9 – 10–4
|
NEG.
|
—
|
4,54 × 10–4
|
não ensaiado
|
NEG.
|
Fenol
|
Produto químico intermédio
|
|
10
|
Resveratrol
|
501-36-0
|
NEG.
|
—
|
10–8 – 10–3
|
NEG.
|
—
|
4,38 × 10–4
|
para ser negativa (*4)
|
NEG.
|
Hidrocarboneto cíclico
|
Produto natural
|
|
Abreviaturas: N.o CAS = Número de registo do Chemical Abstracts Service; M = molar; IC50 = Concentração semimáxima inibitória de uma substância química em estudo; NEG = negativo; PN = negativo presumível; POS= positivo;
|
Critérios de aceitabilidade dos ensaios
|
16.
|
A aceitação ou rejeição de uma série de ensaios baseia-se na avaliação dos resultados obtidos para os padrões de referência e dos controlos utilizados para cada experiência. Os valores para PC50 (EC50) ou IC50 (padrões de referência) devem satisfazer os critérios de aceitabilidade previstos para o ensaio selecionado (para STTA ver apêndice 2, para VM7Luc ER TA ver apêndice 3), e todos os controlos positivos/negativos devem ser corretamente classificados para cada experiência aceite. A capacidade de efetuar o ensaio de forma consistente deve ser demonstrada através da criação e da manutenção de uma base de dados histórica das normas e controlos de referência (ver ponto 15). Os desvios-padrão (desvio-padrão) ou os coeficientes de variação (CV) das médias dos padrões de referência para os parâmetros das curvas de várias experiências podem ser utilizados como indicadores da reprodutibilidade interna do laboratório. Além disso, devem cumprir-se os seguintes princípios relativos aos critérios de aceitabilidade:
|
—
|
Os dados devem ser suficientes para efetuar uma avaliação quantitativa da ativação ER (ensaio de agonistas) ou supressão (ensaio antagonistas) (isto é, eficácia e potência).
|
|
—
|
A atividade média do repórter para a concentração dos estrogénios de referência deve ser, pelo menos, a mínima especificada nos ensaios relativamente ao controlo do veículo (solvente), a fim de garantir a devida sensibilidade. Para os ensaios STTA e VM7Luc ER, esta é quatro vezes a da média do controlo do veículo (solvente) de cada placa.
|
|
—
|
As concentrações ensaiadas devem manter-se no intervalo de solubilidade dos produtos químicos em estudo e não demonstrar citotoxicidade.
|
|
Análise dos dados
|
17.
|
O procedimento de interpretação dos dados definido para cada ensaio deve ser utilizado para classificar uma resposta positiva e negativa.
|
|
18.
|
O cumprimento dos critérios de aceitação (ponto 16) indica que o ensaio está a funcionar corretamente, mas não assegura que um determinado ensaio específico produza dados exatos. A replicação dos resultados da primeira série de ensaios é a melhor indicação de que foram produzidos dados exatos. Se duas séries de ensaios derem resultados reprodutíveis (por exemplo, os resultados dos dois ensaios indicarem que um produto químico em estudo é positivo), não é necessário realizar uma terceira série de ensaios.
|
|
19.
|
Se duas séries de ensaios não permitirem obter resultados reprodutíveis (por exemplo, um produto químico em estudo der positivo numa série e negativo na outra) ou se for necessário um grau de certeza mais elevado quanto ao resultado deste ensaio, devem realizar-se pelo menos três séries de ensaios independentes. Neste caso, a classificação baseia-se em dois resultados concordantes em três.
|
Critérios gerais para a interpretação dos dados
|
20.
|
Não existe atualmente um método universalmente aceite para a interpretação dos dados ER TA. No entanto, a avaliação qualitativa (por exemplo positivo/negativo) e/ou quantitativa (p. ex., EC50, PC50, IC50) da atividade mediada pelo ER deve basear-se em dados empíricos e em pareceres científicos sólidos. Sempre que possível, os resultados positivos devem ser caracterizados pela magnitude do efeito em comparação com o do controlo do veículo (solvente) ou do estrogénio de referência, bem como pela concentração a que o efeito se produz (por exemplo, EC50, PC50, RPCmáx, IC50, etc.).
|
Relatório de ensaio
|
21.
|
O relatório de ensaio deve conter as seguintes informações:
|
|
Método de ensaio:
|
—
|
Método de ensaio utilizado;
|
|
—
|
Padrões de controlo/referência/produto químico em estudo
|
|
—
|
Origem, número de lote e data-limite de utilização, se conhecida
|
|
—
|
Estabilidade do produto químico em estudo, se conhecida;
|
|
—
|
Solubilidade e estabilidade do produto químico em estudo no solvente, se conhecida;
|
|
—
|
Medição do pH, da pressão osmótica e da quantidade de precipitado no meio de cultura ao qual foi adicionado o produto químico em estudo, consoante o que for adequado.
|
|
|
|
Substância monocomponente:
|
—
|
Aspeto físico, hidrossolubilidade e outras propriedades físico-químicas pertinentes;
|
|
—
|
Dados de identificação química, como as denominações IUPAC ou CAS, número CAS, código SMILES ou InChI, fórmula estrutural, pureza, identidade química das impurezas, se justificado e exequível, etc.
|
|
|
|
Substância multicomponentes, UVCB e misturas:
|
—
|
Caracterizada, na medida do possível, pela identidade química (ver acima), ocorrência quantitativa e principais propriedades físico-químicas dos componentes.
|
|
|
|
Solvente/veículo:
|
—
|
Caracterização (natureza, fornecedor e lote);
|
|
—
|
Justificação para a escolha do solvente/veículo;
|
|
—
|
Solubilidade e estabilidade do produto químico em estudo no solvente/veículo, se conhecida.
|
|
|
|
Células:
|
—
|
Tipo e origem das células:
|
—
|
É o ER expresso endogenamente? Na negativa, que recetor(es) foi/foram transfetado(s)?
|
|
—
|
Constituinte(s) do(s) repórter(es) utilizado(s) (incluindo as espécies de origem);
|
|
—
|
Método de seleção para a manutenção da estabilidade da transfeção (quando aplicável);
|
|
—
|
O método de transfeção é relevante para linhas estáveis?
|
|
|
—
|
Número de passagens celulares (desde a descongelação);
|
|
—
|
Número de passagens celulares aquando da descongelação;
|
|
—
|
Métodos de manutenção das culturas celulares.
|
|
|
|
Condições de realização do ensaio:
|
—
|
Limitações da solubilidade;
|
|
—
|
Descrição dos métodos de avaliação da viabilidade aplicados;
|
|
—
|
Composição dos meios, concentração de CO2;
|
|
—
|
Concentrações do produto químico em estudo;
|
|
—
|
Volumes adicionados do veículo e produto químico em estudo;
|
|
—
|
Temperatura e humidade de incubação;
|
|
—
|
Densidade celular no início e no decurso do tratamento;
|
|
—
|
Padrões de referência positivos e negativos;
|
|
—
|
Reagentes do repórter (nome do produto, fornecedor e lote);
|
|
—
|
Critérios definidos para que o ensaio seja considerado positivo, negativo ou inconclusivo.
|
|
|
|
Verificação da aceitabilidade:
|
—
|
Fator de indução de cada placa de ensaio e se cumprem o mínimo exigido pelo ensaio com base em controlos históricos;
|
|
—
|
Valores reais para critérios de aceitabilidade, por exemplo, log10(EC50), log10(PC50), log10(IC50) e valores da curva de Hill para os controlos positivos/padrões de referência;
|
|
|
|
Resultados:
|
—
|
Dados brutos e normalizados;
|
|
—
|
Nível do fator de indução máximo;
|
|
—
|
Dados relativos à citotoxicidade;
|
|
—
|
Se existir, a concentração mínima eficaz (LEC);
|
|
—
|
Valores RPCmáx, PCmáx, PC50, IC50 e/ou EC50, consoante o caso;
|
|
—
|
Relação concentração-resposta, quando for possível determiná-la;
|
|
—
|
Análise estatística, se for caso disso, associada a uma medida de erro e confiança (por exemplo, SEM, DP, CV ou IC 95 %) e descrição da forma como os valores foram obtidos.
|
|
|
REFERÊNCIAS BIBLIOGRÁFICAS
|
(1)
|
OCDE (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environment, Health and Safety Publications, Series on Testing and Assessment (No 34.), Organisation for Economic Cooperation and Development, Paris.
|
|
(2)
|
OCDE (2015). Report of the Inter-Laboratory Validation for Stably Transfected Transactivation Assay to detect Estrogenic and Anti-estrogenic Activity. Environment, Health and Safety Publications, Series on Testing and Assessment (No 225), Organisation for Economic Cooperation and Development, Paris.
|
|
(3)
|
ICCVAM (2011). ICCVAM Test Method Evaluation Report on the LUMI-CELL® ER (BG1Luc ER TA) Test Method, an In Vitro Method for Identifying ER Agonists and Antagonists, National Institute of Environmental Health Sciences: Research Triangle Park, NC.
|
|
(4)
|
Pujol P. et al. (1998). Differential Expression of Estrogen Recetor-Alpha and -Beta Messenger RNAs as a Potential Marker of Ovarian Carcinogenesis, Cancer. Res., 58(23): p. 5367-73.
|
|
(5)
|
Rogers J.M. and Denison M.S. (2000). Recombinant Cell Bioassays for Endocrine Disruptors: Development of a Stably Transfected Human Ovarian Cell Line for the Detection of Estrogenic and Anti-Estrogenic Chemicals, In Vitro and Molecular Toxicology: Journal of Basic and Applied Research, 13(1): p. 67-82.
|
|
(6)
|
OCDE (2012). Performance Standards For Stably Transfected Transactivation In Vitro Assay to Detect Estrogen Recetor Agonists (for TG 455). Environment, Health and Safety Publications, Series on Testing and Assessment (No 173.), Organisation for Economic Cooperation and Development, Paris.
|
|
(7)
|
OCDE (2015). Performance Standards For Stably Transfected Transactivation In Vitro Assay to Detect Estrogen Recetor Antagonists. Environment, Health and Safety Publications, Series on Testing and Assessment (No 174.), Organisation for Economic Cooperation and Development, Paris.
|
|
(8)
|
OCDE (2012). Guidance Document on Standardized Test Guidelines for Evaluating Chemicals for Endocrine Disruption. Environment, Health and Safety Publications, Series on Testing and Assessment (No 150.), Organisation for Economic Cooperation and Development, Paris.
|
|
(9)
|
Cavailles V. (2002). Estrogens and Recetors: an Evolving Concept. Climacteric, 5 Suppl 2: p. 20- 6.
|
|
(10)
|
Welboren W.J. et al. (2009). Genomic Actions of Estrogen Recetor Alpha: What are the Targets and how are they Regulated? Endocr. Relat. Cancer, 16(4): p. 1073-89.
|
|
(11)
|
Younes M. and Honma N. (2011). Estrogen Recetor Beta, Arch. Pathol. Lab. Med., 135(1): p. 63- 6.
|
|
(12)
|
Jefferson W.N., et al. (2002). Assessing Estrogenic Activity of Phytochemicals Using Transcriptional Activation and Immature Mouse Uterotrophic Responses, Journal of Chromatography B, 777(1-2): p. 179-189.
|
|
(13)
|
Sonneveld E. et al. (2006). Comparison of In Vitro and In Vivo Screening Models for Androgenic and Estrogenic Activities, Toxicol. Sci., 89(1): p. 173-187.
|
|
(14)
|
Takeyoshi M. et al. (2002). The Efficacy of Endocrine Disruptor Screening Tests in Detecting Anti- Estrogenic Effects Downstream of Recetor-Ligand Interactions, Toxicology Letters, 126(2): p. 91- 98.
|
|
(15)
|
Combes R.D. (2000). Endocrine Disruptors: a Critical Review of In Vitro and In Vivo Testing Strategies for Assessing their Toxic Hazard to Humans, ATLA Alternatives to Laboratory Animals,28(1): p. 81-118.
|
|
(16)
|
Escande A. et al. (2006). Evaluation of Ligand Selectivity Using Reporter Cell Lines Stably Expressing Estrogen Recetor Alpha or Beta, Biochem. Pharmacol,71(10): p. 1459-69.
|
|
(17)
|
Gray L.E. Jr. (1998). Tiered Screening and Testing Strategy for Xenoestrogens and Antiandrogens, Toxicol. Lett, 102-103, 677-680.
|
|
(18)
|
EDSTAC (1998). Endocrine Disruptor Screening and Testing Advisory Committee (EDSTAC) Final Report.
|
|
(19)
|
ICCVAM (2003). ICCVAM Evaluation of In Vitro Test Methods for Detecting Potential Endocrine Disruptors: Estrogen Recetor and Androgen Recetor Binding and Transcriptional Activation Assays.
|
|
(20)
|
Gustafsson J.Ö. (1999). Estrogen Recetor ß - A New Dimension in Estrogen Mechanism of Action, Journal of Endocrinology, 163(3): p. 379-383.
|
|
(21)
|
Ogawa S. et al. (1998). The Complete Primary Structure of Human Estrogen Recetor ß (hERß) and its Heterodimerization with ERα In Vivo and In Vitro, Biochemical and Biophysical Research Communications, 243(1): p. 122-126.
|
|
(22)
|
Enmark E. et al. (1997). Human Estrogen Recetor ß-Gene Structure, Chromosomal Localization, and Expression Pattern, Journal of Clinical Endocrinology and Metabolism,82(12): p. 4258-4265.
|
|
(23)
|
Ball L.J. et al. (2009). Cell Type- and Estrogen Recetor-Subtype Specific Regulation of Selective Estrogen Recetor Modulator Regulatory Elements, Molecular and Cellular Endocrinology, 299(2): p. 204-211.
|
|
(24)
|
Barkhem T. et al. (1998). Differential Response of Estrogen Recetor Alpha and Estrogen Recetor Beta to Partial Estrogen Agonists/Antagonists, Mol. Pharmacol, 54(1): p. 105-12.
|
|
(25)
|
Deroo B.J. and Buensuceso A.V. (2010). Minireview: Estrogen Recetor-ß: Mechanistic Insights from Recent Studies, Molecular Endocrinology, 24(9): p. 1703-1714.
|
|
(26)
|
Harris D.M. et al. (2005). Phytoestrogens Induce Differential Estrogen Recetor Alpha- or Beta- Mediated Responses in Transfected Breast Cancer Cells, Experimental Biology and Medicine, 230(8): p. 558-568.
|
|
(27)
|
Anderson J.N. Clark J.H. and Peck E.J.Jr. (1972). The Relationship Between Nuclear Recetor- Estrogen Binding and Uterotrophic Responses, Biochemical and Biophysical Research Communications, 48(6): p. 1460-1468.
|
|
(28)
|
Toft D. (1972). The Interaction of Uterine Estrogen Recetors with DNA, Journal of Steroid Biochemistry, 3(3): p. 515-522.
|
|
(29)
|
Gorski J. et al. (1968), Hormone Recetors: Studies on the Interaction of Estrogen with the Uterus, Recent Progress in Hormone Research, 24: p. 45-80.
|
|
(30)
|
Jensen E.V. et al. (1967), Estrogen-Recetor Interactions in Target Tissues, Archives d’Anatomie Microscopique et de Morphologie Experimentale, 56(3):p. 547-569.
|
|
(31)
|
ICCVAM (2002). Background Review Document: Estrogen Recetor Transcriptional Activation (TA) Assay. Appendix D, Substances Tested in the ER TA Assay, NIH Publication Report (No 03-4505).
|
|
(32)
|
Kanno J. et al. (2001). The OECD Program to Validate the Rat Uterotrophic Bioassay to Screen Compounds for In Vivo Estrogenic Responses: Phase 1, Environ. Health Persp., 109:785-94.
|
|
(33)
|
Kanno J. et al. (2003). The OECD Program to Validate the Rat Uterotrophic Bioassay: Phase Two Dose -Response Studies, Environ. Health Persp., 111:1530-1549.
|
|
(34)
|
Kanno J. et al. (2003), The OECD Program to Validate the Rat Uterotrophic Bioassay: Phase Two – Coded Single-Dose Studies, Environ. Health Persp., 111:1550-1558.
|
|
(35)
|
Geisinger et al. (1989) Characterization of a human ovarian carcinoma cell line with estrogen and progesterone recetors, Cancer 63, 280-288.
|
|
(36)
|
Baldwin et al. (1998) BG-1 ovarian cell line: an alternative model for examining estrogen-dependent growth in vitro, In Vitro Cell. Dev. Biol. – Animal, 34, 649-654.
|
|
(37)
|
Li, Y. et al. (2014) Research resource: STR DNA profile and gene expression comparisons of human BG-1 cells and a BG-1/MCF-7 clonal variant, Mol. Endo. 28, 2072-2081.
|
|
(38)
|
Rogers, J.M. and Denison, M.S. (2000) Recombinant cell bioassays for endocrine disruptors: development of a stably transfected human ovarian cell line for the detection of estrogenic and anti-estrogenic chemicals, In Vitro & Molec. Toxicol. 13, 67-82.
|
Apêndice 1
DEFINIÇÕES E ABREVIATURAS
Adequação
: descrição da relação de um ensaio com o objeto de interesse e da sua importância e utilidade para um determinado fim. Traduz a medida em que o ensaio mede ou prevê corretamente o efeito biológico em causa. A adequação comporta a exatidão (concordância) de um ensaio (1).
Agonista
: substância que produz uma resposta, por exemplo, transcrição, quando se liga a um recetor específico.
Antagonista
: tipo de ligando a recetor ou produto químico que não provoca uma resposta biológica com a ligação a um recetor, mas bloqueia ou amortece respostas mediadas por agonistas.
ARN
: ácido ribonucleico.
Atividade antiestrogénica
: capacidade de um produto químico de suprimir a ação do 17β-estradiol mediada por recetores de estrogénios.
Atividade estrogénica
: a capacidade de um produto químico reproduzir o 17β-estradiol na sua capacidade de se fixar em recetores de estrogénio e de os ativar. O presente método de ensaio permite detetar atividade estrogénia mediada por hERα.
CF
: quadro conceptual da OCDE para ensaio e avaliação de desreguladores do sistema endócrino.
Citotoxicidade
: efeitos nocivos para a estrutura ou a função celular que podem causar a morte celular e se podem traduzir numa redução do número de células presentes no poço no final do período de exposição ou numa redução da capacidade para uma medida da função celular, quando comparada com o controlo do veículo simultâneo.
Critérios de aceitabilidade
: normas mínimas para a realização de controlos experimentais e padrões de referência. Para que uma experiência seja considerada válida, devem ser satisfeitos todos os critérios de aceitabilidade.
CP (Amostra de controlo positivo)
: substância fortemente ativa, de preferência 17ß-estradiol, que é incluída em todos os ensaios para ajudar a garantir o bom funcionamento do método.
CP
10
: concentração do produto químico em estudo a que a atividade medida num ensaio de agonistas é igual a 10 % da atividade máxima induzida pelo controlo positivo (E2 a 1nM para o ensaio STTA) em cada placa.
CP
50
: concentração do produto químico em estudo em que a atividade medida num ensaio de agonistas é igual a 50 % da atividade máxima induzida pelo PC (E2 com a concentração de referência indicada no método de ensaio) em cada placa.
CP
máx
: concentração do produto químico em estudo que induz a RPC Máx.
CV
: coeficiente de variação.
Competência técnica
: capacidade comprovada para efetuar um ensaio de forma adequada antes de ensaiar substâncias desconhecidas.
Controlo positivo fraco
: substância fracamente ativa, selecionada a partir da lista de substâncias químicas de referência que está incluída em todos os ensaios a fim de assegurar o correto funcionamento do ensaio.
DCC-FBS
: soro fetal bovino tratado com carvão revestido com dextrano.
DMEM
: meio de Eagle modificado por Dulbecco.
DMSO
: dimetilsulfóxido.
DP
: desvio-padrão.
E2
: 17β-estradiol.
EC50
: concentração semimáxima efetiva de um produto químico em estudo.
ED
: desregulação do sistema endócrino.
Ensaio STTA
: ensaio de transativação transfetada de forma estável, o ensaio de ativação transcricional ERα com recurso à linha celular HeLa 9 903.
Ensaio «me-too»
: expressão coloquial para designar um método que é, estrutural e funcionalmente, semelhante a um método de ensaio de referência validado e aceite. O termo é sinónimo de «método de ensaio similar».
Estrogénio de referência (controlo positivo, PC)
: 17β-estradiol (E2, CAS 50-28-2).
Estudo
: todo o trabalho experimental realizado para avaliar uma substância única e específica através de um ensaio específico. Um estudo compreende todas as etapas, incluindo ensaios de diluição da substância em estudo nos meios de ensaio, séries de ensaios preliminares para avaliações da gama, todas as séries de ensaios exaustivos necessárias, análises de dados, controlo de qualidade, avaliações de citotoxicidade, etc. A realização de um estudo permite a classificação da atividade do produto químico em estudo no alvo de toxicidade (ou seja, ativo, inativo ou inconclusivo), que é avaliada pelo método usado e por uma estimativa de potência em relação ao produto químico de referência positiva.
Exatidão (concordância)
: grau de concordância entre os resultados do ensaio e um valor de referência aceite. Constitui uma medida da eficiência do método e um aspeto da adequação. Este termo e o termo «concordância» são muitas vezes utilizados indistintamente para indicar a proporção de resultados corretos do método de ensaio (1).
ERE
: elemento de resposta do estrogénio.
ERTA
: transativação do recetor de estrogénio.
Especificidade
: proporção de substâncias negativas/inativa que é corretamente classificada pelo ensaio. Constitui uma medida da exatidão de um método cujos resultados sejam estabelecidos em função de categorias e é um aspeto importante a ter em conta na avaliação da adequação de um método (1).
FBS
: soro fetal bovino.
Fiabilidade
: indica em que medida um ensaio pode ser reproduzido ao longo do tempo num mesmo laboratório e entre laboratórios utilizando o mesmo protocolo. A fiabilidade é avaliada com base nos valores calculados das reprodutibilidades intralaboratorial e interlaboratorial.
hERα
: recetor alfa de estrogénio humano.
hERß
: recetor beta de estrogénio humano.
HeLa
: linha celular imortal do colo do útero humano.
HeLa9903
: subclone da célula HeLa para a qual foram transfetados de forma estável hERαs e um gene repórter da luciferase.
IC
50
: concentração semimáxima efetiva de um produto químico em estudo inibidor.
ICCVAM
: Comité de Coordenação Interagências na Validação de Métodos Alternativos.
LEC
: menor concentração eficaz é a menor concentração do produto químico em estudo que produz uma resposta (ou seja, a menor concentração do produto químico em estudo em que o fator de indução é estatisticamente diferente do controlo do veículo concorrente).
Morfologia celular
: forma e aspeto das células cultivadas numa única camada num único poço de uma placa de cultura de tecidos. As células que estão a morrer apresentam muitas vezes uma morfologia celular anormal.
MSE
: meio sem estrogénio. Meio de Eagle modificado por Dulbecco (DMEM), complementado com 4,5 % de FBS tratado com carvão/dextrano, 1,9 % de L-glutamina e 0,9 % de «pen-strep».
MCE
: Menor Concentração Eficaz. É a menor concentração do produto químico em estudo que produz uma resposta (ou seja, a menor concentração do produto químico em estudo em que o fator de indução é estatisticamente diferente do controlo do veículo concorrente).
Método de ensaio
: no contexto do presente método de ensaio, um ensaio é uma das metodologias aceites como válidas para cumprir os critérios de desempenho indicados. Os componentes do ensaio incluem, por exemplo, a linha celular específica com as condições de crescimento associadas, o meio específico em que o ensaio é efetuado, as condições de configuração das placas, disposições e diluições dos produtos químicos em estudo, a par de outras medidas de controlo da qualidade exigidas e de etapas de avaliação dos dados conexas.
Método de ensaio validado
: método de ensaio relativamente ao qual foram realizados estudos de validação com vista a determinar a sua adequação (incluindo a exatidão) e fiabilidade para um determinado fim. Importa referir que um método de ensaio validado pode não ser suficientemente exato e fiável para ser considerado aceitável para o fim pretendido (1).
Métodos de ensaio de referência
: ensaios em que se baseia o PBTG 455.
MMTV
: vírus do tumor mamário do rato.
MT
: metalotioneína.
Normas de desempenho
: normas assentes num método de ensaio validado que constituam uma base para avaliar a comparabilidade de um ensaio proposto que seja mecanística e funcionalmente similar. Incluem: (1) componentes essenciais do método de ensaio; (2) uma lista mínima de produtos químicos de referência, escolhidos de entre os produtos químicos utilizados para demonstrar o desempenho aceitável do método de ensaio validado; e (3) níveis de exatidão e fiabilidade comparáveis, baseados no que foi obtido para o método de ensaio validado, que o método de ensaio proposto deve demonstrar quando avaliado pela utilização da lista mínima de produtos químicos de referência (1).
OHT
: 4-hidroxitamoxifeno.
Padrão de referência
: substância de referência utilizada para demonstrar a adequação de um ensaio. O 17β-estradiol é o padrão de referência para os ensaios STTA e VM7Luc ER TA.
PBTG
: diretriz de ensaio baseada no desempenho.
Produto químico
: uma substância ou mistura.
Produto químico em estudo
: qualquer substância ou mistura à qual seja aplicado o presente método de ensaio.
RCP
máx
: nível máximo de resposta induzido pelo produto químico em estudo, expresso em percentagem da resposta induzida por 1 nM E2 na mesma placa.
RE
: recetor de estrogénio.
Reprodutibilidade interlaboratorial
: medida do grau em que diferentes laboratórios qualificados que utilizam o mesmo protocolo e testam as mesmas substâncias podem produzir resultados qualitativa e quantitativamente similares. A reprodutibilidade interlaboratorial é determinada durante os processos de pré-validação e validação e indica em que medida um método pode ser transferido com êxito entre laboratórios, igualmente designada «reprodutibilidade entre laboratórios» (1).
Reprodutibilidade intralaboratorial
: medida do grau em que pessoal qualificado do mesmo laboratório consegue replicar resultados com êxito utilizando o mesmo protocolo, em momentos diferentes. Igualmente designada «reprodutibilidade intralaboratorial» (1).
RLU
: unidades de luz relativas.
RPMI
: meio RPMI 1 640 complementado com 0,9 % de «pen-strep» e 8,0 % de soro fetal bovino (FBS).
Sensibilidade
: proporção de todas as substâncias positivas/ativas que é corretamente classificada pelo método. Constitui uma medida da exatidão de um método cujos resultados sejam estabelecidos em função de categorias e é um aspeto importante a ter em conta na avaliação da adequação de um método (1).
Série de ensaios
: experiência individual que avalia a ação química sobre o resultado biológico do ensaio. Cada série de ensaios constitui uma experiência completa realizada em poços de células retiradas de um mesmo agregado de células ao mesmo tempo.
Série de ensaios independente
: experiência realizada independente que avalia a ação química sobre o resultado biológico do ensaio, utilizando células de um conjunto diferente de produtos químicos recém-diluídos, realizada em dias diferentes ou no mesmo dia por pessoas diferentes.
Substância
: nos termos do REACH, uma substância é definida como um elemento químico e seus compostos, no estado natural ou obtidos por qualquer processo de fabrico, incluindo qualquer aditivo necessário para preservar a sua estabilidade e qualquer impureza que derive do processo utilizado, mas excluindo qualquer solvente que possa ser separado sem afetar a estabilidade da substância nem modificar a sua composição. No contexto do sistema GHS da ONU é utilizada uma definição muito semelhante (61).
Substâncias para demonstração de competência
: subconjunto das substâncias de referência incluídas nas normas de desempenho que podem ser utilizadas pelos laboratórios para demonstrar a sua competência técnica com um método de ensaio normalizado. Os critérios de seleção destas substâncias incluem normalmente o facto de representarem a gama de respostas, estarem comercialmente disponíveis e disporem de dados de referência de elevada qualidade.
TA (Transativação)
: início da síntese do mARN em resposta a um sinal químico específico, como uma ligação de um estrogénio no recetor de estrogénios.
Tratamento por carvão vegetal/dextrano
: tratamento de soro utilizado na cultura celular. O tratamento com carvão vegetal/dextrano (frequentemente designado «separação») retira hormonas endógenas e proteínas de ligação de hormonas.
Transfeção estável
: quando o ADN é transferido para células de cultura de modo a ser estavelmente integrado no genoma celular, o que resulta na expressão estável dos genes transfetados. Os clones de células transfetadas de forma estável são selecionados por marcadores estáveis (por exemplo, resistência ao G418).
Transcrição
: síntese do mARN.
UVCB
: substâncias químicas de composição desconhecida ou variável, produtos de reação complexos e materiais biológicos.
Validação
: processo de estabelecimento da fiabilidade e adequação de uma determinada abordagem, método, processo ou avaliação para um determinado fim (1).
VC (controlo do veículo)
: solvente que é utilizado para dissolver os produtos químicos em estudo e de controlo; é testado exclusivamente na qualidade de veículo, sem adição de produtos químicos dissolvidos.
VM7
: célula de adenocarcinoma imortalizada que endogenamente expressa o recetor de estrogénio.
VM7Luc4E2
: linha celular VM7Luc4E2, obtida a partir de células VM7 imortalizadas de adenocarcinoma de origem humana, que expressam endogenamente ambas as formas do recetor de estrogénios (ERα e ERβ) e que foram transfetadas de forma estável, com o plasmídeo pGudLuc7.ERE. O plasmídeo contém quatro cópias de um oligonucleótido sintético que contém o elemento de resposta estrogénica a montante do promotor do tumor mamário viral do rato (MMTV) e o gene da luciferase do pirilampo
Apêndice 2
RECETOR Α DE ESTROGÉNIO HUMANO TRANSFETADO DE FORMA ESTÁVEL. ENSAIO DE TRANSATIVAÇÃO PARA DETEÇÃO DE ATIVIDADE ESTROGÉNICA AGONISTA E ANTAGONISTA NOS PRODUTOS QUÍMICOS COM RECURSO À LINHA CELULAR HERΑ-HELA-9903
CONSIDERAÇÕES INICIAIS E LIMITAÇÕES (VER TAMBÉM INTRODUÇÃO GERAL)
|
1.
|
O presente ensaio de transativação (TA) utiliza a linha celular hERα-HeLa-9 903 para detetar atividade estrogénica agonista mediada por um recetor de estrogénio humano alfa (hERα). O estudo de validação do ensaio de transativação transfetada de forma estável, realizado pelo Japanese Chemicals Evaluation and Research Institute (CERI), utilizando a linha celular hERα-HeLa-9 903 para detetar atividade estrogénica agonista e antagonista mediada pelo recetor alfa de estrogénio humano (hERα), demonstrou a adequação e a fiabilidade do ensaio para o fim a que se destina (1).
|
|
2.
|
O ensaio foi concebido especificamente para a deteção de TA mediada pelo hERα, tendo como parâmetro a medição por quimioluminescência. No entanto, foram referidos sinais de luminescência não mediada pelo recetor em concentrações de fitoestrogénios superiores a 1 μM devido à sobreativação do gene repórter da luciferase (2) (3). Embora a curva de respostas à dose indique que a ativação real do sistema do ER ocorre a concentrações inferiores, é necessário examinar cuidadosamente a expressão da luciferase obtida em concentrações elevadas de fitoestrogénios ou de compostos similares suspeitos de produzirem o mesmo tipo de sobreativação de fitoestrogénios do gene repórter da luciferase nos sistemas de ensaio de ER TA transfetados de modo estável (apêndice 1).
|
|
3.
|
Devem ler-se as secções «INTRODUÇÃO GERAL» e «COMPONENTES DO ENSAIO DE ER TA» antes de utilizar este ensaio para fins normativos. As definições e abreviaturas utilizadas no presente método de ensaio são descritas no apêndice 2.1.
|
PRINCÍPIO DO ENSAIO (VER TAMBÉM INTRODUÇÃO GERAL)
|
4.
|
O presente ensaio é utilizado para fixar um sinal no recetor de estrogénio com um ligando. Após a ligação do ligando, o complexo recetor-ligando transloca-se para o núcleo, onde se fixa a elementos de resposta de ADN específicos e transativa um gene repórter da luciferase do pirilampo que provoca um aumento da expressão celular da enzima luciferase. A luciferina é um substrato que é transformado pela enzima luciferase num produto bioluminescente mensurável quantitativamente com um luminómetro. A atividade da luciferase pode ser avaliada de forma rápida e pouco onerosa, com vários conjuntos de ensaio existentes no mercado.
|
|
5.
|
O sistema de ensaio utiliza a linha celular hERα-HeLa-9 903, que é derivada de um tumor do colo do útero humano, com dois elementos estavelmente inseridos: (i) o elemento de expressão hERαs (que codifica o recetor humano) e (ii) um elemento do repórter da luciferase pirilampo com cinco repetições de um elemento de vitelogenina responsivo ao estrogénio (ERE), conduzidos por um elemento TATA promotor de metalotioneína (MT) do rato. Foi demonstrado que o constituinte TATA MT do gene do rato tem o melhor desempenho, pelo que é frequentemente utilizado. Por conseguinte, esta linha celular hERIα-HeLa-9 903 pode medir a capacidade de um produto químico em estudo de induzir a transativação da expressão do gene da luciferase mediada pelo hERα.
|
|
6.
|
No caso do ensaio do agonista do ER, a interpretação dos dados baseia-se no facto de o nível máximo de resposta induzido por um produto químico em estudo ser ou não igual ou superior a uma resposta agonista igual a 10 % da resposta induzida por uma concentração indutora maximizada (1 nM) do agente de controlo positivo (PC) 17β-estradiol (E2) (ou seja, o PC10). No caso do ensaio de antagonistas do ER, a interpretação dos dados baseia-se no facto de a resposta revelar, ou não, uma redução de pelo menos 30 % da atividade da resposta induzida pelo pico do controlo (25 pM de E2) sem citotoxicidade. A análise e a interpretação dos dados são abordadas em pormenor nos pontos 34 a 48.
|
PROCEDIMENTO
Linhas celulares
|
7.
|
Para o ensaio, deve utilizar-se a linha celular HeLa-9 903 transfetada de modo estável. A linha celular pode ser obtida na Japanese Collection of Research Bioresources (JCRB) Cell Bank (62), mediante a assinatura de um acordo de transferência de material (MTA).
|
|
8.
|
Só devem ser utilizadas nos ensaios células caracterizadas como isentas de micoplasmas. O RT-PCR (reação em cadeia da polimerase em tempo real) é o método ideal para uma deteção sensível de infeção por micoplasmas (4) (5) (6).
|
Estabilidade da linha celular
|
9.
|
Para monitorizar a estabilidade da linha celular, devem utilizar-se E2, 17α-estradiol, 17α-metiltestosterona e corticosterona como padrões de referência para ensaios de agonista, devendo ser medida uma curva completa de respostas à concentração da gama concentrações de ensaio constantes do quadro 1, pelo menos, uma vez sempre que o ensaio for realizado, devendo os resultados estar de acordo com os apresentados no quadro 1.
|
|
10.
|
No caso do ensaio antagonista, devem ser medidas em paralelo, em cada série de ensaios, as curvas de concentração completas para dois padrões de referência – taxifeno e flutamida. A classificação qualitativa correta como positiva ou negativa para os dois produtos químicos deve ser monitorizada.
|
Cultura celular e condições de incubação em placas
|
11.
|
As células devem ser mantidas em meio mínimo essencial de Eagle (EMEM) sem vermelho de fenol, complementado com 60 mg/l do antibiótico canamicina e 10 % de soro fetal bovino tratado com carvão revestido de dextrano (DCC-SFB), numa incubadora de CO2 (5 % CO2) a 37±1 oC. Quando atingem uma confluência de 75-90 %, as células podem ser subcultivadas em 10 ml de 0,4 × 105 – 1 × 105 células/ml para recipientes de cultura de 100 mm. As células devem ser suspensas com 10 % de SFB-EMEM (que é o mesmo que EMEM com DCC-FBS) e, em seguida, colocadas em poços de uma microplaca a uma densidade de 1 × 104 células/(100 μl × poço). Em seguida, as células devem ser pré-incubadas numa incubadora com 5 % de CO2 a 37±1 oC durante 3 horas antes da exposição ao produto químico. Os artigos de plástico não devem ter atividade estrogénica.
|
|
12.
|
Para manter a integridade da resposta, as células devem ser cultivadas por mais do que uma passagem da cultura congelada nos meios condicionados e não devem ser cultivadas por mais de 40 passagens. No caso da linha celular hERα-HeLa-9 903, este período deve ser inferior a três meses. Contudo, o desempenho das células pode ser reduzido se forem cultivadas em condições de cultura inadequadas.
|
|
13.
|
O DCC-FBS pode ser preparado como descrito no apêndice 2.2, ou obtido no comércio.
|
Critérios de aceitabilidade
Padrões de referência positivos e negativos para o ensaio de agonistas do ER
|
14.
|
Antes e durante o estudo, deve verificar-se a capacidade de resposta do sistema de ensaio utilizando as concentrações adequadas de um estrogénio forte (E2), um estrogénio fraco (17α-estradiol), um agonista muito fraco (17α-metiltestosterona) e uma substância negativa (corticosterona). Os valores de gama aceitáveis derivados do estudo de validação (1) são indicados no quadro 1. Estes 4 padrões de referência concomitantes devem ser incluídos em todas as experiências e os resultados devem situar-se dentro dos limites admissíveis. Se tal não for o caso, deve averiguar-se a causa da impossibilidade de cumprir os critérios de aceitabilidade (por exemplo, manipulação das células, soro e antibióticos para a qualidade e concentração) e o ensaio deve ser repetido. Uma vez cumpridos os critérios de aceitabilidade, é essencial uma utilização consistente dos materiais de cultura celular para assegurar a variabilidade mínima dos valores de EC50, PC50 and PC10. Os quatro padrões de referência concorrentes, que devem ser incluídos em cada ensaio (realizado nas mesmas condições, incluindo os materiais, o nível de passagem das células e os técnicos), podem garantir a sensibilidade do ensaio, na medida em que os PC10 dos três padrões de referência positivos devem situar-se dentro da gama aceitável, tal como os PC50 e EC50, quando puderem ser calculados (ver quadro 1).
|
Quadro 1
Gama de valores aceitáveis dos quatro padrões de referência para o ensaio de ER agonista
|
Nome
|
log(PC50)
|
log(PC10)
|
log(EC50)
|
Curva de Hill
|
Gama de ensaio
|
|
17β-Estradiol (E2) N.o CAS: 50-28-2
|
-11,4~-10,1
|
<-11
|
-11,3~-10,1
|
0,7~1,5
|
10-14~10-8M
|
|
17α-Estradiol N.o CAS: 57-91-0
|
-9,6~-8,1
|
-10,7~-9,3
|
-9,6~-8,4
|
0,9~2,0
|
10-12~10-6M
|
|
Corticosterona N.o CAS: 50-22-6
|
—
|
—
|
—
|
—
|
10-10~10-4M
|
|
17α-Metiltestosterona N.o CAS: 58-18-4
|
-6,0~-5,1
|
-8,0~-6,2
|
—
|
—
|
10-11~10-5M
|
Padrão de referência positivo e negativo para o ensaio de antagonistas do ER
|
15.
|
Antes e durante o estudo, deve verificar-se a capacidade de resposta do sistema de ensaio utilizando as concentrações adequadas de uma substância positiva (tamoxifeno) e de uma substância negativa (flutamida). Os valores de gama aceitáveis derivados do estudo de validação (1) são indicados no quadro 2. Estes dois padrões de referência concorrentes devem ser tidos em conta em todas as experiências, devendo os resultados ser considerados corretos, conforme indicado nos critérios. Se tal não for o caso, há que determinar a causa do incumprimento dos critérios (por exemplo, manipulação de células, soro e antibióticos para a qualidade e a concentração) e o ensaio deve ser repetido. Além disso, devem calcular-se os valores IC50 para uma substância positiva (tamoxifeno), devendo os resultados situar-se dentro dos limites aceitáveis indicados. Quando os critérios de aceitabilidade tiverem sido alcançados, é essencial a utilização coerente dos materiais de cultura de células para assegurar a variabilidade mínima dos valores IC50. Os dois padrões de referência concorrentes, que devem ser utilizados em cada experiência (realizadas nas mesmas condições, incluindo os materiais, o nível de passagem das células e os técnicos), podem garantir a sensibilidade do ensaio (ver quadro 2).
|
Quadro 2
Critérios e gama de valores aceitáveis dos dois padrões de referência para o ensaio de antagonistas do ER
|
Nome
|
Critérios
|
log ( IC50)
|
Gama de ensaio
|
|
Tamoxifeno N.o CAS: 10 540 -29-1
|
Positivos: O IC50 deve ser calculado
|
-5,942~-7,596
|
10-10~10-5M
|
|
Flutamida N.o CAS: 13 311 -84-7
|
Negativos: O IC30 não deve ser calculado
|
—
|
10-10~10-5M
|
Controlos positivos e controlo do veículo
|
16.
|
O controlo positivo (PC) do ensaio de agonistas do ER (1 nM de E2) e do ensaio de antagonistas do ER (10 μM TAM) deve ser efetuado, pelo menos, em triplicado em cada placa. O veículo utilizado para dissolver um produto químico em estudo deve ser ensaiado como controlo do veículo (VC), pelo menos, em triplicado em cada placa. Além deste CV, se o PC utilizar um veículo diferente do produto químico em estudo, deve ensaiar-se outro CV, pelo menos em triplicado, na mesma placa com o PC.
|
Critérios de qualidade para o ensaio de agonistas do ER
|
17.
|
A atividade da luciferase do controlo positivo (1 nM E2) deve ser pelo menos 4 vezes superior à do CV médio em cada placa. Este critério baseia-se na fiabilidade dos valores dos parâmetros do estudo de validação (historicamente um fator entre 4 e 30).
|
|
18.
|
Em relação ao controlo de qualidade do ensaio, o fator de indução correspondente ao valor PC10 do PC simultâneo (1 nM E2 deve ser superior a 1+2DP do valor do fator de indução (=1) do VC simultâneo. Para efeitos de definição de prioridades, o valor PC10 pode ser útil para simplificar a análise de dados necessária comparativamente a uma análise estatística. Embora uma análise estatística forneça informações sobre a significância, essa análise não é um parâmetro quantitativo no que diz respeito ao potencial com base na concentração, pelo que é menos útil para efeitos de definição de prioridades.
|
Critérios de qualidade para o ensaio de antagonistas do ER
|
19.
|
A atividade média da luciferase no pico do controlo (25 pM E2) deve ser pelo menos 4 vezes superior à atividade média do CV em todas as placas. Este critério é estabelecido com base na fiabilidade dos valores dos parâmetros do estudo de validação.
|
|
20.
|
Em relação ao controlo de qualidade do ensaio, a ativação transcricional relativa (RTA) de 1 nM E2 deve ser superior a 100 %, a RTA de 1μM 4-hidroxitamoxifeno (OHT) deve ser inferior a 40,6 % e a RTA de 100 μM de digitonina (Dig) deve ser inferior a 0 %.
Demonstração da competência técnica do laboratório (ver os pontos 14 e os quadros 3 e 4 em «COMPONENTES DO ENSAIO DE ER TA» do presente método de ensaio).
|
Veículo
|
21.
|
Deve utilizar-se como VC, ensaiado em paralelo, dimetilsulfóxido (DMSO) ou um solvente adequado, na concentração utilizada para os diferentes controlos positivos e negativos, bem como os produtos químicos em estudo. Estes devem ser dissolvidos num solvente que os dissolva e seja miscível no meio celular. A água, o etanol (95 % a 100 %de pureza) e o DMSO são veículos adequados. Caso se utilize DMSO, o seu teor não deve exceder 0,1 % (v/v). Para qualquer veículo, deve demonstrar-se que o volume máximo utilizado não é citotóxico e não interfere com o desempenho do ensaio.
|
Preparação dos produtos químicos em estudo
|
22.
|
Em geral, os produtos químicos em estudo devem ser dissolvidos em DMSO ou noutro solvente adequado e diluídos sequencialmente com o mesmo solvente, à razão de 1:10, a fim de preparar soluções para diluição no meio.
|
Solubilidade e citotoxicidade: Considerações para a determinação da gama de dosagem
|
23.
|
Deve ser efetuado um ensaio preliminar para determinar a gama de concentrações adequada do produto químico em estudo e se este pode apresentar problemas de solubilidade ou citotoxicidade. Inicialmente, os produtos químicos são ensaiados até à concentração máxima de 1 μg/ml, 1 mg/ml ou 1 mM, consoante a que for mais baixa. Com base no grau de citotoxicidade ou na falta de solubilidade observados no ensaio preliminar, a primeira série de ensaios definitiva deve utilizar o produto químico em algoritmos sequenciais de diluições a partir da concentração máxima aceitável (por exemplo, 1 mM, 100 μM, 10 μM, etc.), observando a presença de turbidez, precipitado ou citotoxicidade. As concentrações da segunda e, se necessário, terceira séries de ensaios devem ser ajustadas conforme adequado para melhor caracterizar a curva de respostas à concentração e evitar concentrações que se verifique serem insolúveis ou produzam citotoxicidade excessiva.
|
|
24.
|
No caso dos agonistas e antagonistas do ER, a presença de níveis crescentes de citotoxicidade pode alterar significativamente ou eliminar a resposta sigmoidal característica e deve ser tida em conta na interpretação dos dados. Devem utilizar-se métodos de ensaio de citotoxicidade que possam fornecer informações sobre a viabilidade de 80 % das células, com recurso a um ensaio adequado baseado na experiência adquirida no laboratório.
|
|
25.
|
Caso os resultados do ensaio de citotoxicidade revelem que a concentração do produto químico em estudo reduziu o número de células em 20 % ou mais, esta concentração deve ser considerada citotóxica e devem excluir-se da avaliação as concentrações iguais ou superiores à de concentração citotóxica.
|
Exposição ao produto químico e organização das placas do ensaio
|
26.
|
O procedimento relativo às diluições do produto químico (etapas 1 e 2) e à exposição a células (etapa ) é o seguinte:
Etapa 1: Cada produto químico deve ser diluído sequencialmente em DMSO, ou num solvente apropriado, e adicionado aos poços de uma placa de microtitulação para atingir as concentrações sequenciais finais determinadas pelo ensaio preliminar de determinação das gamas de dosagem [geralmente, numa série de, por exemplo, 1 mM, 100 μM, 10 μM, 1 μM, 100 nM, 10 nM, 1 nM, 100 pM e 10 pM (10-3-10-11 M)] para ensaio em triplicado.
Etapa 2: diluição do produto químico: Em primeiro lugar, diluir 1,5 μl do produto químico em estudo no solvente até um volume de 500 μl no meio.
Etapa 3: exposição das células ao produto químico: Adicionar 50 μl da diluição com o meio (preparada na etapa 2) a um poço que contenha 104 células/100 μl/poço.
O volume final recomendado de meio para cada poço é de 150 μl. As amostras para ensaio e os padrões de referência podem ser afetados como indicado no quadro 3 e no quadro 4.
|
Quadro 3
Exemplo da atribuição de concentração por placa dos padrões de referência na placa de ensaio no ensaio de agonistas do ER
|
Linha
|
17α-metiltestosterona
|
Corticosterona
|
17α-estradiol
|
E2
|
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
Conc. 1 (10 μM)
|
→
|
→
|
100 μM
|
→
|
→
|
1 μM
|
→
|
→
|
10 nM
|
→
|
→
|
|
B
|
Conc. 2 (1 μM)
|
→
|
→
|
10 μM
|
→
|
→
|
100 nM
|
→
|
→
|
1 nM
|
→
|
→
|
|
C
|
Conc. 3 (100 nM)
|
→
|
→
|
1 μM
|
→
|
→
|
10 nM
|
→
|
→
|
100 pM
|
→
|
→
|
|
D
|
Conc. 4 (10 nM)
|
→
|
→
|
100 nM
|
→
|
→
|
1 nM
|
→
|
→
|
10 pM
|
→
|
→
|
|
E
|
Conc. 5 (1 nM)
|
→
|
→
|
10 nM
|
→
|
→
|
100 pM
|
→
|
→
|
1 pM
|
→
|
→
|
|
F
|
Conc. 6 (100 pM)
|
→
|
→
|
1 nM
|
→
|
→
|
10 pM
|
→
|
→
|
0,1 pM
|
→
|
→
|
|
G
|
Conc. 7 (10 pM)
|
→
|
→
|
100 pM
|
→
|
→
|
1 pM
|
→
|
→
|
0,01 pM
|
→
|
→
|
|
H
|
VC
|
→
|
→
|
→
|
→
|
→
|
PC
|
→
|
→
|
→
|
→
|
→
|
|
VC: Controlo do veículo (0,1 % DMSO), PC: Controlo positivo (1 nM E2)
|
|
27.
|
Os padrões de referência (E2, (E2, 17α-estradiol, 17α-metiltestosterona devem ser ensaiados em todas as séries de ensaios (quadro 3). Apenas os poços do PC tratados com 1 nM de E2 que podem produzir a indução máxima de E2 e os poços de VC tratados com DMSO (ou solvente apropriado) devem ser incluídos em todas as placas de ensaio (quadro 4). Se forem utilizadas células de diferentes origens (p. ex. números de passagem diferentes, lotes diferentes, etc.) na mesma experiência, os padrões de referência devem ser ensaiados para cada origem de células.
|
Quadro 4
Exemplo de atribuição de concentrações a placas de ensaio e placas dos produtos químicos em estudo e de controlo das placas no ensaio de agonistas do ER
|
Linha
|
Produto químico em estudo 1
|
Produto químico em estudo 2
|
Produto químico em estudo 3
|
Produto químico em estudo 4
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
Conc. 1 (10 μM)
|
→
|
→
|
1 mM
|
→
|
→
|
1 μM
|
→
|
→
|
10 nM
|
→
|
→
|
|
B
|
Conc. 2 (1 μM)
|
→
|
→
|
100 μM
|
→
|
→
|
100 nM
|
→
|
→
|
1 nM
|
→
|
→
|
|
C
|
Conc. 3 (100 nM)
|
→
|
→
|
10 μM
|
→
|
→
|
10 nM
|
→
|
→
|
100 pM
|
→
|
→
|
|
D
|
Conc. 4 (10 nM)
|
→
|
→
|
1 μM
|
→
|
→
|
1 nM
|
→
|
→
|
10 pM
|
→
|
→
|
|
E
|
Conc. 5 (1 nM)
|
→
|
→
|
100 nM
|
→
|
→
|
100 pM
|
→
|
→
|
1 pM
|
→
|
→
|
|
F
|
Conc. 6 (100 pM)
|
→
|
→
|
10 nM
|
→
|
→
|
10 pM
|
→
|
→
|
0,1 pM
|
→
|
→
|
|
G
|
Cconc. 7 (10 pM)
|
→
|
→
|
1 nM
|
→
|
→
|
1 pM
|
→
|
→
|
0,01 pM
|
→
|
→
|
|
H
|
VC
|
→
|
→
|
→
|
→
|
→
|
PC
|
→
|
→
|
→
|
→
|
→
|
|
VC: Controlo do veículo (0,1 % DMSO), PC: Controlo positivo (1 nM E2)
|
Quadro 5
Exemplo de atribuição das concentrações por placa dos padrões de referência na placa de ensaio no ensaio de antagonistas do ER

|
28.
|
Para avaliar a atividade antagonista dos produtos químicos, os poços de ensaio situados nas linhas A a G devem ser enriquecidos com 25pM E2. Os padrões de referência (tamoxifeno e flutamida) devem ser ensaiados em todas as séries de ensaios. Os poços de PC tratados com 1 nM de E2 que podem ser utilizados para o controlo da qualidade da linha celular hERα-HeLa-9 903, os poços VC tratados com DMSO (ou com um solvente adequado), os poços de 0,1 % de DMSO, tratados com adição de DMSO ao E2 enriquecido correspondente ao pico no controlo, os poços tratados com a concentração final 1 μM OHT e os poços tratados com 100 μM Dig devem ser incluídos em todas as placas de ensaio (quadro 5). A placa de ensaio subsequente deve ter a mesma configuração, sem poços de padrões de referência (quadro 6). Se forem utilizadas células de diferentes origens (p. ex,. números de passagem diferentes, lotes diferentes, etc.) na mesma experiência, os padrões de referência devem ser ensaiados para cada origem de células.
|
Quadro 6
Exemplo de atribuição de concentrações de placas ao produto químico em estudo e de controlo na placa do ensaio de antagonistas do ER
|
29.
|
A ausência de efeitos de borda deve ser confirmada, conforme adequado; se houver suspeitas desses efeitos, a configuração das placas deve ser alterada para evitá-los. Pode utilizar-se, por exemplo, uma configuração que exclua os poços de borda.
|
|
30.
|
Após a adição dos produtos químicos, as placas de ensaio devem ser incubadas numa incubadora com 5 % de CO2, a 37 °C ±1 oC, durante 20 a 24 horas, para induzir os produtos génicos repórteres.
|
|
31.
|
Os compostos altamente voláteis requerem considerações específicas. Nesses casos, os poços de controlo próximos podem gerar falsos positivos, o que deve ser apreciado à luz dos valores esperados e dos valores de controlo históricos. Nos poucos casos em que a volatilidade seja preocupante, a utilização de «selantes de placas» pode contribuir para isolar eficazmente os poços individuais durante os ensaios, sendo, por conseguinte, recomendada.
|
|
32.
|
A repetição dos ensaios definitivos para o mesmo produto químico deve ser feita em dias diferentes, a fim de garantir a sua independência.
|
Ensaio da luciferase
|
33.
|
Pode utilizar-se um reagente comercial de ensaio da luciferase [p. ex., Steady-Glo® Luciferase Assay System (Promega, E2510, ou equivalente)], desde que sejam respeitados os critérios de aceitação. Os reagentes do ensaio devem ser escolhidos com base na sensibilidade do luminómetro a utilizar. Caso se recorra ao sistema de ensaio padrão da luciferase – Cell Culture Lysis Reagent (p.e., Promega, E1531 ou equivalente) –, este deve ser utilizado antes da adição do substrato. O reagente de luciferase deve ser aplicado de acordo com as instruções do fabricante.
|
ANÁLISE DOS DADOS
Ensaio de agonistas do ER
|
34.
|
No caso do ensaio de agonistas do ER, para obter a atividade transcricional relativa para o PC (1 nM de E2), os sinais de luminescência da mesma placa podem ser analisados de acordo com os seguintes passos (também são aceitáveis outros processos matemáticos equivalentes):
Etapa 1: Calcular o valor médio para o VC.
Etapa 2: Subtrair o valor médio do VC de todos os poços, para normalizar os dados.
Etapa 3: Calcular a média para o PC normalizado.
Etapa 4: Dividir o valor normalizado de cada poço da placa pelo valor médio do PC normalizado (PC = 100 %).
O valor final de cada poço corresponde à atividade transcricional relativa desse poço em comparação com a resposta do PC.
Etapa 5: Calcular o valor médio da atividade transcricional relativa para cada grupo de concentração do produto químico em estudo. A resposta tem duas vertentes: a média da atividade transcricional (resposta) e a concentração a que ocorre a resposta (ver secção seguinte).
|
Considerações sobre a indução de EC50, PC50 e PC10
|
35.
|
É necessária uma curva completa concentrações-resposta para o cálculo do EC50, mas isso pode nem sempre ser possível ou praticável devido às limitações da gama de concentrações do ensaio – por exemplo, devido a problemas de citotoxicidade ou de solubilidade. No entanto, como o EC50 e o nível máximo de indução (correspondente ao valor mais elevado da equação de Hill) são parâmetros informativos, estes parâmetros devem ser comunicados sempre que possível. Para o cálculo do EC50 e do nível máximo de indução, deve utilizar-se software estatístico adequado (por exemplo, o software Graphpad Prism). Se a equação logística de Hill for aplicável aos dados das respostas às concentrações, o EC50 deverá ser calculado com recurso à seguinte equação (7):
Y = Inferior + (Superior-Inferior) / {1+10 [exp (log(EC50)-X) × curva de Hill]}, em que:
X é o logaritmo da concentração;
Y é a resposta e o Y começa na «inferior» e vai até à superior numa curva sigmoide. A inferior é fixada em zero na equação logística de Hill.
|
|
36.
|
Para cada produto químico em estudo, devem apresentar-se:
O RPCmáx, que corresponde ao nível máximo de resposta induzido pelo produto químico em estudo, expresso em percentagem da resposta induzida por 1 nM E2 na mesma placa, bem como o PCmáx (concentração associada ao RPCmáx); e
No caso de produtos químicos positivos, as concentrações que induzem o PC10 e, se for caso disso, o PC50.
|
|
37.
|
O valor PCx pode ser calculado através da interpolação entre 2 pontos na coordenada X-Y, um imediatamente acima e um imediatamente abaixo de um valor PCx. Se os pontos de dados situados imediatamente acima e abaixo do valor PCx tiverem as coordenadas (a, b) e (c, d), respetivamente, o valor PCx pode ser calculado pela seguinte equação:
log[PCx] = log[c]+(x-d)/(d-b).
|
|
38.
|
As descrições dos valores dos PC constam da figura 1.
Figura 1
Exemplo de como calcular valores do PC. O PC (1 nM de E2) é incluído em todas as placas de ensaio
|
Ensaio de antagonistas do ER
|
39.
|
No caso do ensaio de antagonistas do ER, para obter a atividade transcricional relativa (RTA) do pico do controlo (25 pM de E2), os sinais de luminescência da mesma placa podem ser analisados de acordo com as seguintes etapas (também são aceitáveis outros processos matemáticos equivalentes):
Etapa 1: Calcular o valor médio para o VC.
Etapa 2: Subtrair o valor médio do VC de todos os poços, para normalizar os dados. Etapa 3: Calcular a média dos picos de controlo normalizados.
Etapa 4: Dividir o valor normalizado de cada poço da placa pelo valor médio do pico do controlo normalizado (pico de controlo = 100 %).
O valor final de cada poço corresponde à atividade transcricional relativa desse poço em comparação com a resposta do pico de controlo.
Etapa 5: Calcular o valor médio da atividade transcricional relativa para cada tratamento.
|
Considerações de indução de IC30 e IC50
|
40.
|
No caso de produtos químicos positivos, devem indicar-se as concentrações que induzem o IC30 e, se for caso disso, o IC50.
|
|
41.
|
O valor ICx pode ser calculado através da interpolação entre 2 pontos na coordenada X-Y, um imediatamente acima e um imediatamente abaixo de um valor ICx. Se os pontos situados imediatamente acima e abaixo do valor ICx tiverem as coordenadas (c, d) e (a, b), respetivamente, o valor ICx pode ser calculado pela seguinte equação:
lin ICx = a-[b-(100-x)] (a-c) /(b-d)
Figura 2
Exemplo de como calcular valores IC. O pico de controlo (25 pM de E2) está incluído em todas as placas de ensaio

RTA: atividade transcricional relativa
|
|
42.
|
Os resultados devem basear-se em duas (ou três) séries de ensaios independentes. Se duas séries derem resultados compráveis e, por conseguinte, reprodutíveis, não é necessário realizar uma terceira série de ensaios. Para serem aceitáveis, os resultados devem:
|
—
|
Cumprir os critérios de aceitabilidade (ver critérios de aceitabilidade, pontos 14-20),
|
|
Critérios de interpretação dos dados
Quadro 7
Critérios de decisão positiva e negativa no ensaio de agonistas do ER
|
Positiva
|
Se o RPCmáx obtido for igual ou superior a 10 % da resposta do controlo positivo em, pelo menos, duas de duas ou duas de três séries de ensaios.
|
|
Negativa
|
Se a RPCmáx não atingir, pelo menos, 10 % da resposta do controlo positivo em duas de duas ou duas de três séries de ensaios.
|
Quadro 8
Critérios de decisão positiva e negativa no ensaio de antagonistas do ER
|
Positiva
|
Se o IC30 for calculado em, pelo menos, duas de duas ou duas de três séries de ensaios.
|
|
Negativa
|
Se o IC30 não pode ser calculado em duas de duas ou duas de três séries de ensaios.
|
|
43.
|
Os critérios de interpretação dos dados constam dos quadros 7 e 8. Os resultados positivos serão caracterizados pela magnitude do efeito e pela concentração a que o efeito ocorre. Se os resultados forem expressos como uma concentração à qual se obtêm os valores de 50 % (PC50) ou 10 % (PC10) de PC, para o ensaio de agonistas, e 50 % (IC50) ou 30 % (IC30) dos valores de inibição do pico de controlo, para o ensaio de antagonistas, estão cumpridos ambos os objetivos. No entanto, um produto químico em estudo é considerado positivo se a resposta máxima induzida pelo produto químico em estudo (RPCmáx) for igual ou superior a 10 % da resposta do PC em, pelo menos, duas de duas ou duas de três séries de ensaios, sendo considerado negativo se a RCPmáx não corresponder a, pelo menos, 10 % da resposta do controlo positivo em duas de duas ou duas de três séries de ensaios.
|
|
44.
|
Os cálculos de PC10, PC50 e PCmáx no ensaio de agonistas do ER e de IC30 e IC50 no ensaio de antagonistas do ER podem ser efetuados com recurso a uma folha de cálculo disponibilizada com a Test Guideline no sítio Web público da OCDE (63).
|
|
45.
|
Essa folha de cálculo deve ser suficiente para obter os valores de PC10 ou PC50 e de IC30 ou IC50 pelo menos duas vezes. Porém, se os dados de base resultantes para o mesmo intervalo de concentração mostrarem variabilidade com um coeficiente de variação (CV %) – inaceitavelmente elevado –, os dados não podem considerar-se fiáveis e importa identificar a origem da elevada variabilidade. O CV dos triplicados dos dados brutos (ou seja, os dados relativos à intensidade de luminescência) dos pontos utilizados para o cálculo do PC10 deve ser inferior a 20 %.
|
|
46.
|
O cumprimento dos critérios de aceitabilidade indica que o sistema de ensaio está a funcionar corretamente, mas não assegura que uma determinada operação produza dados exatos. A duplicação dos resultados da primeira série de ensaios é a melhor garantia de que foram produzidos dados exatos.
|
|
47.
|
No caso do ensaio de agonistas do ER, em que são necessárias mais informações para além dos fins de monitorização e priorização da presente Test Guideline para produtos químicos positivos, especialmente para produtos químicos PC10-PC49, bem como para produtos químicos suspeitos de estimular excessivamente a luciferase, pode confirmar-se que a atividade da luciferase observada é unicamente uma resposta específica do antagonista do ERα, utilizando um antagonista do ERα (ver apêndice 2.1).
|
RELATÓRIO DO ENSAIO
|
48.
|
Ver ponto 20 dos «COMPONENTES DO ENSAIO ER TA».
|
REFERÊNCIAS BIBLIOGRÁFICAS
|
(1)
|
OCDE (2015). Report of the Inter-Laboratory Validation for Stably Transfected Transactivation Assay to detect Estrogenic and Anti-estrogenic Activity. Environment, Health and Safety Publications, Series on Testing and Assessment (No 225), Organisation for Economic Cooperation and Development, Paris.
|
|
(2)
|
Escande A., et al. (2006). Evaluation of Ligand Selectivity Using Reporter Cell Lines Stably Expressing Estrogen Recetor Alpha or Beta, Biochem. Pharmacol., 71, 1 459-1 469.
|
|
(3)
|
Kuiper G.G., et al. (1998). Interaction of Estrogenic Chemicals and Phytoestrogens with Estrogen Recetor Beta, Endocrinol., 139, 4 252-4 263.
|
|
(4)
|
Spaepen M., et al. (1992). Detection of Bacterial and Mycoplasma Contamination in Cell Cultures by Polymerase Chain Reaction, FEMS Microbiol. Lett., 78(1), 89-94.
|
|
(5)
|
Kobayashi H., et al. (1995). Rapid Detection of Mycoplasma Contamination in Cell Cultures by Enzymatic Detection of Polymerase Chain Reaction (PCR) Products, J. Vet. Med. Sci., 57(4), 769- 71.
|
|
(6)
|
Dussurget O. and Roulland-Dussoix D. (1994). Rapid, Sensitive PCR-Based Detection of Mycoplasmas in Simulated Samples of Animal Sera, Appl. Environ. Microbiol., 60(3), 953-9.
|
|
(7)
|
De Lean A., Munson P.J. and Rodbard D. (1978). Simultaneous Analysis of Families of Sigmoidal Curves: Application to Bioassay, Radioligand Assay, and Physiological Dose-Response Curves, Am. J. Physiol., 235, E97-El02.
|
Apêndice 2.1
FALSOS POSITIVOS: AVALIAÇÃO DE SINAIS DE LUMINESCÊNCIA NÃO MEDIADOS PELO RECETOR
|
1.
|
No ensaio de agonistas do ER, podem ser gerados falsos positivos pela ativação do gene da luciferase não mediada pelo ER, pela ativação direta do produto génico ou por fluorescência independente. Esses efeitos são indicados por uma curva incompleta ou invulgar de respostas às dosagens. Se tais efeitos forem suspeitos, deve examinar-se o efeito na resposta de um antagonista do ER [por exemplo, 2-hidroxitamoxifeno (OHT) numa concentração não tóxica]. O antagonista ICI 1827 80 pode não ser adequado para este efeito, uma vez que uma concentração suficiente de ICI 1827 80 pode diminuir o valor do VC, o que irá afetar a análise dos dados.
|
|
2.
|
Para assegurar a validade desta abordagem, é necessário ensaiar o seguinte na mesma placa:
|
—
|
Atividade agonística do produto químico desconhecido, com /sem 10 μM de OHT
|
|
—
|
1 nM de E2 (em triplicado) como PC agonista
|
|
—
|
1 nM de E2 + OHT (em triplicado)
|
|
Critérios de interpretação dos dados
Nota: Todos os poços devem ser tratados com a mesma concentração do veículo.
|
—
|
Se a atividade agonística do produto químico desconhecido NÃO for afetada pelo tratamento com o antagonista do ER, este é classificado como «negativo».
|
|
—
|
Se a atividade agonística do produto químico desconhecido for completamente inibida, devem aplicar-se os critérios de decisão.
|
|
—
|
Se a atividade agonística na menor concentração for igual ou superior à resposta PC10, o produto químico desconhecido é inibido de forma igual ou superior à resposta PC10. Calcula-se a diferença das respostas dos poços não tratados e tratados com o antagonista do ER, devendo esta diferença ser considerada a resposta verdadeira, utilizada no cálculo dos parâmetros adequados para permitir uma decisão de classificação.
|
Análise dos dados
Verificar a norma de desempenho.
Verificar o CV entre os poços tratados nas mesmas condições.
|
1.
|
Calcular a média do VC
|
|
2.
|
Subtrair a média do VC de cada poço não tratado com OHT
|
|
3.
|
Calcular a média do OHT
|
|
4.
|
Subtrair a média do VC de cada poço tratado com OHT
|
|
5.
|
Calcular a média do PC
|
|
6.
|
Calcular a atividade transcricional relativa de todos os outros poços em relação ao PC.
|
Apêndice 2.2
PREPARAÇÃO DE SORO TRATADO COM CARVÃO REVESTIDO DE DEXTRANO (DCC)
|
1.
|
O tratamento do soro com carvão revestido de dextrano (DCC) é um método geral para a remoção de compostos estrogénicos do soro que é adicionado ao meio celular, a fim de excluir a resposta distorcida associada aos estrogénios residuais no soro. Podem ser tratados com este procedimento 500 ml de soro fetal bovino (FBS).
|
Componentes
|
2.
|
São necessários os seguintes materiais e equipamentos:
|
Materiais
|
Cloreto de magnésio hexa-hidratado (MgCl2 6H2O)
|
|
1 M de solução-tampão HEPES (pH 7,4)
|
|
Água ultrapura produzida a partir de um sistema de filtros
|
Equipamento
Recipiente de vidro autoclavado (a dimensão deve ser adaptada); centrifugadora geral de laboratório (que pode ser regulada para uma temperatura de 4 °C)
Procedimento
|
3.
|
O seguinte procedimento é ajustado para a utilização de tubos de centrifugação de 50 ml:
[Dia 1] Preparar uma suspensão de carvão revestido de dextrano com 1 l de água ultrapura contendo 1,5 mM de MgCl2, 0,25 M de sacarose, 2,5 g de carvão, 0,25 g de dextrano e 5 mM de HEPES; agitá-la a 4 °C, de um dia para o outro.
[Dia 2] Distribuir a suspensão por tubos de centrifugação de 50 ml e centrifugar a 10 000 rpm, a 4 °C, durante 10 minutos. Remover o sobrenadante e armazenar metade do sedimento de carvão a 4 °C para ser utilizado no dia 3. Suspender a outra metade do carvão vegetal com FBS descongelado suavemente para evitar precipitação e inativado pelo calor a 56 °C durante 30 minutos e, em seguida, transferir para um recipiente de vidro autoclavado, como um erlenmeyer. Agitar suavemente esta suspensão a 4 °C de um dia para o outro.
[Dia 3] Distribuir a suspensão com o FBS por tubos de centrifugação para centrifugação a 10 000 rpm a 4 °C, durante 10 minutos. Recolher o FBS e transferi-lo para os novos sedimentos de carvão preparados e armazenados no dia 2. Suspender os sedimentos de carvão e agitar suavemente esta suspensão a 4 °C num recipiente de vidro autoclavado, de um dia para o outro.
[Dia 4] Distribuir a suspensão para centrifugação a 10 000 rpm a 4 °C por 10 minutos e esterilizar o sobrenadante por filtração através de um filtro estéril de 0,2 μm. O FBS tratado com DCC deve ser armazenado a –20 °C e pode ser utilizado durante um ano.
|
Apêndice 3
ENSAIO DE TRANSATIVAÇÃO DO RECETOR DE ESTROGÉNIO VM7LUC PARA IDENTIFICAR OS AGONISTAS E OS ANTAGONISTAS DO RECETOR DE ESTROGÉNIO
CONSIDERAÇÕES INICIAIS E LIMITAÇÕES (VER TAMBÉM INTRODUÇÃO GERAL)
|
1.
|
O presente ensaio utiliza a linha celular VM7Luc4E2 (64). Foi validado pelo National Toxicology Program Interagency Center for the Evaluation of Alternative Toxicological Methods (ICVAM), e pelo Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM) (1). As linhas celulares VM7Luc expressam predominantemente ERα e uma pequena quantidade de ERβ endógenos (2) (3) (4).
|
|
2.
|
O presente ensaio é aplicável a uma vasta gama de substâncias, desde que possam ser dissolvidas em dimetilsulfóxido (DMSO; N.o CAS 67-68-5), não reajam com o DMSO ou com o meio de cultura das células e não sejam citotóxicas nas concentrações ensaiadas. Se não for possível utilizar o DMSO, pode utilizar-se outro veículo, como o etanol ou a água (ver ponto 12). O desempenho demonstrado pelo ensaio de (ant)agonistas VM7Luc ER TA sugere que os dados gerados com este ensaio podem informar sobre os mecanismos de ação mediados pelo ER e podem ser tidos em conta para a priorização de substâncias para a realização de ensaios complementares.
|
|
3.
|
O presente ensaio foi especificamente concebido para a deteção de TA mediada por hERα e hERß, por medição da quimioluminescência como parâmetro final. A utilização de quimioluminescência em bioensaios é generalizada, em virtude da relação sinal/origem elevada. No entanto, a atividade de luciferase do pirilampo em ensaios baseados em células pode ser confundida por substâncias que inibem a enzima luciferase, causando tanto inibição aparente ou o aumento da luminescência devido à estabilização de proteínas. Além disso, em alguns ensaios de genes repórteres do ER baseados na luciferase, foram comunicados sinais de luminescência não mediados por recetores em concentrações de fitoestrogénios superiores a 1 μM, devido à sobreativação do gene repórter da luciferase. Embora a curva dose-resposta indique que a ativação real do sistema ER ocorre a concentrações inferiores, a expressão da luciferase obtida a concentrações elevadas de fitoestrogénios ou de compostos semelhantes suspeitos de produzir uma sobreativação do gene repórter da luciferase idêntica à dos fitoestrogénios deve ser cuidadosamente examinada nos sistemas de ensaio de ER TA transfetados de forma estável.
|
|
4.
|
Devem ler-se a «INTRODUÇÃO GERAL» e os «COMPONENTES DO ENSAIO ER TA» antes de utilizar o presente ensaio para fins normativos. As definições e abreviaturas utilizadas no presente método de ensaio constam do apêndice 1.
|
PRINCÍPIO DO ENSAIO (VER TAMBÉM INTRODUÇÃO GERAL)
|
5.
|
O presente ensaio é utilizado para indicar uma ligação ao ER por ligando, seguida de uma translocação do complexo recetor-ligando ao núcleo. No núcleo, o complexo recetor-ligando fixa-se a elementos específicos de resposta do ADN e transativa o gene resposta (luc), o que resulta na produção da luciferase e na subsequente emissão de luz, que pode ser quantificada com o auxílio de um luminómetro. A atividade da luciferase pode ser rapidamente avaliada, de forma pouco onerosa, com uma série de conjuntos disponíveis no comércio. O VM7Luc ER TA utiliza uma linha celular do adenocarcinoma da mama humano, VM7, respondendo ao ER, que foi transfetada estavelmente com um constituinte do repórter luc do pirilampo sob o controlo de quatro elementos de resposta estrogénicos localizados a montante do promotor do vírus do tumor mamário do rato (MMTV), para detetar substâncias com atividade agonista ou antagonista do ER in vitro. Este promotor de MMTV apresenta apenas uma fraca reatividade cruzada com outras hormonas esteroides e não esteroides (8). Os critérios para a interpretação dos dados são descritos em pormenor no ponto 41. Rapidamente, é identificada uma resposta positiva por uma curva concentração-resposta com, pelo menos, três pontos com barras de erro não sobrepostas (±DP médio), bem como uma mudança de amplitude (unidade relativa de luz [RLU] normalizada) de, no mínimo, 20 % do valor máximo para o padrão de referência (17β-estradiol [E2; N.o CAS 50-28-2] para o ensaio de agonista, raloxifeno HCl [Ral; No CAS 84449-90-1]/E2 para o ensaio de antagonistas).
|
PROCEDIMENTO
Linha celular
|
6.
|
Para o ensaio, deve utilizar-se a linha celular VM7Luc4E2 estavelmente transfetada. Na atualidade, esta linha celular apenas está disponível mediante acordo de licença técnica da Universidade da Califórnia, Davis, Califórnia, EUA (65), e da Xenobiotic Detection Systems Inc., Durham, North Carolina, EUA (66).
|
Estabilidade da linha celular
|
7.
|
Para manter a estabilidade e a integridade da linha celular, as células devem ser cultivadas durante mais de uma passagem a partir da cultura-mãe congelada em meios de manutenção de células (ver ponto 9). Estas não devem ser cultivadas por mais de 30 passagens. Para a linha celular VM7Lu4E2, 30 passagens correspondem a cerca de três meses.
|
Cultura celular e condições de incubação em placas
|
8.
|
Devem ser seguidos os procedimentos especificados na Guidance on Good Cell Culture Practice [Orientações de boas práticas de cultura celular] (5) (6), para garantir a qualidade de todos os materiais e métodos, a fim de manter a integridade, a validade e a reprodutibilidade dos trabalhos realizados.
|
|
9.
|
As células VM7Luc4E2, são mantidas num meio RPMI 1640 suplementado com 0,9 % de «pen-strep» e 8,0 % de soro fetal bovino (FBS), numa incubadora de cultura de tecidos específica, a 37 oC ±1 oC, 90 % ±5 % de humidade e 5,0 % ±1 % CO2/ar.
|
|
10.
|
Quando atingirem uma confluência de cerca de 80 %, as células VM7Luc4E2 são subcultivadas e condicionadas durante 48 horas num ambiente sem estrogénios, antes de serem colocadas em placas com 96 poços para exposição aos produtos químicos em estudo e para análise da indução dependente de estrogénios da atividade da luciferase. O meio isento de estrogénios (EFM) contém meio de Eagle modificado por Dulbecco (DMEM) sem vermelho de fenol, complementado com 4,5 % de FBS tratado com carvão/dextrano, 1,9 % de L-glutamina e 0,9 % de «pen-strep». Todo o material de plástico deve ser isento de atividade estrogénica [ver protocolo pormenorizado (7)].
|
Critérios de aceitabilidade
|
11.
|
A aceitação ou rejeição de um ensaio baseia-se na avaliação do padrão de referência e dos resultados do controlo de todas as experiências realizadas numa placa de 96 poços. Cada padrão de referência é ensaiado em várias concentrações, havendo várias amostras de cada concentração de referência e de controlo. Os resultados são comparados com os controlos de qualidade (QC) para os parâmetros derivados das bases de dados agonistas e antagonistas históricos geradas por cada laboratório durante a demonstração de competência técnica. As bases de dados históricos são continuamente atualizadas com os padrões de referência e os valores de controlo. A mudança de equipamento ou das condições laboratoriais pode exigir a criação de bases de dados históricos atualizadas.
|
Ensaio de agonistas
Ensaio de determinação da gama
|
•
|
Indução: A indução nas placas deve ser medida dividindo o valor médio mais elevado da unidade de luz relativa (RLU) – padrão de referência E2 – pelo valor médio RLU do controlo com DMSO. Normalmente, é atingido um nível de indução cinco vezes superior, mas, para efeitos de aceitação, a indução deve ser superior ou igual a quatro vezes.
|
|
•
|
Resultados do controlo com DMSO: Os valores RLU de controlo do solvente devem ser 2,5 vezes superiores ao desvio-padrão do valor histórico do valor médio RLU do controlo do solvente histórico.
|
|
•
|
Uma experiência que não cumpra um critério de aceitação deve ser rejeitada e repetida.
|
Ensaio global
Inclui critérios de aceitabilidade do ensaio de determinação da gama dos agonistas e o seguinte:
|
•
|
Resultados do padrão de referência: A curva E2 de concentração-resposta do padrão de referência (curva concentração-resposta) deve ter uma forma sigmoidal e, no mínimo, três valores dentro da parte linear da curva concentração-resposta.
|
|
•
|
Resultados do controlo positivo: Os valores RLU do controlo do metoxicloro devem ser superiores ao DMSO médio mais três vezes o desvio-padrão médio do DMSO.
|
|
•
|
Uma experiência que não cumpra um critério de aceitação único deve ser rejeitada e repetida.
|
Ensaio de antagonistas
Ensaio de determinação da gama
|
•
|
Redução: A redução na placa é medida dividindo a média do valor RLU do padrão de referência Ral/E2 mais elevado pelo valor médio RLU de controlo com DMSO. Normalmente, é atingido um nível de redução de fator cinco, mas, para efeitos de aceitação, a redução deve ser superior ou igual a um fator três.
|
|
•
|
Resultados do controlo E2: Os valores RLU do controlo E2 devem situar-se no intervalo de 2,5 vezes o desvio-padrão do valor RLU histórico médio do controlo E2.
|
|
•
|
Resultados do controlo com DMSO: Os valores RLU do controlo com DMSO devem situar-se no intervalo de 2,5 vezes o desvio-padrão do valor RLU histórico médio do controlo do solvente.
|
|
•
|
Uma experiência que não cumpra um dado critério de aceitação será rejeitada e repetida.
|
Ensaio global
Inclui os critérios de aceitação do ensaio de determinação da gama dos antagonistas e o seguinte:
|
•
|
Resultados do padrão de referência: A curva concentração-resposta do padrão de referência Ral/E2 (curva concentração-resposta) deve ter uma forma sigmoidal e, no mínimo, três valores dentro da parte linear da curva concentração-resposta.
|
|
•
|
Resultados do controlo positivo: Os valores RLU do controlo tamoxifeno/E2 devem ser inferiores ao controlo médio E2 menos três vezes o desvio-padrão do controlo médio E2.
|
|
•
|
Uma experiência que não cumpra um qualquer critério de aceitação será rejeitada e repetida.
|
Padrões de referência, controlos positivos e controlos de veículo
Controlo do veículo (ensaios de agonistas e antagonistas)
|
12.
|
O veículo utilizado para dissolver os produtos químicos em estudo deve ser sujeito a ensaio de controlo. O veículo utilizado durante a validação do ensaio VM7Luc ER TA foi 1 % (v/v) de dimetilsulfóxido (DMSO, N.o CAS 67-68-5) (ver ponto 24). Se for utilizado um veículo diferente do DMSO, todos os padrões de referência, controlos e produtos químicos em estudo devem ser ensaiados no mesmo veículo, se pertinente.
|
Padrão de referência (determinação de gama para agonistas)
|
13.
|
O padrão de referência é o E2 (N.o CAS 50-28-2). Para os ensaios de determinação de gama, o padrão de referência é constituído por uma diluição sequencial de quatro concentrações de E2 (1,84 × 10-10, 4,59 × 10-11, 1,15 × 10-11 and 2,87 × 10-12 M), sendo cada concentração ensaiada em dois poços.
|
Padrão de referência (completo para agonistas)
|
14.
|
O E2 para um ensaio completo resulta de uma diluição em série de 1:2 constituída por 11 concentrações (de 3,67 × 10-10 a 3,59 × 10-13 M) de E2 em dois poços.
|
Padrão de referência (determinação de gama para antagonistas)
|
15.
|
O padrão de referência é uma combinação de Ral (N.o CAS 84449-90-1) e E2 (N.o CAS 50-28-2). O RAL/E2 para a realização de ensaios de determinação de gama resulta de uma diluição em série de três concentrações de Ral (3,06 x 10-9, 7.67 x 10-10 e 1,92 x 10-10M) mais uma concentração fixa (9,18 × 10-11 M) de E2 em dois poços.
|
Padrão de referência (completo para antagonistas)
|
16.
|
O RAL/E2 de ensaio completo consiste numa diluição em série de 1:2 de Ral (de 2,45x 10-8 a 9,57 x 10-11M) mais uma concentração fixa (9,18 × 10-11 M) de Ral/E2 constituída por nove concentrações de Ral/E2 em dois poços.
|
Controlo positivo fraco (agonistas)
|
17.
|
O controlo positivo fraco é uma solução 9,06 x 10-6 M p,p’-metoxicloro (metoxicloro; N.o CAS 72-43-5) em EFM.
|
Controlo positivo fraco (antagonista)
|
18.
|
O controlo positivo fraco é tamoxifeno (N.o CAS 10540-29-1) 3,36 × 10-6M com 9,18 × 10-11 M E2 em EFM.
|
Controlo E2 (ensaio para antagonistas)
|
19.
|
O controlo E2 é 9,18 × 10-11 M E2 em EFM, sendo utilizado como controlo negativo de base.
|
Fator de indução (agonistas)
|
20.
|
A indução da atividade da luciferase do padrão de referência (E2) é medida dividindo o valor RLU médio mais elevado do padrão de referência E2 pelo valor RLU do controlo com DMSO médio, devendo o resultado ter um fator superior a quatro.
|
Fator de redução (antagonistas)
|
21.
|
A atividade média da luciferase do padrão de referência (Ral/E2) é medida dividindo o valor RLU médio mais elevado do padrão de referência Ral/E2 pelo valor médio do DMSO, devendo ser superior ao triplo.
Demonstração de competência técnica do laboratório (ver ponto 14 e quadros 3 e 4 da secção «COMPONENTES DO ENSAIO ER TA» do presente método de ensaio)
|
Veículo
|
22.
|
Os produtos químicos em estudo devem ser dissolvidos num solvente que os solubilize e ser miscíveis com o meio celular. A água, o etanol (95 % a 100 % de pureza) e o DMSO são veículos adequados. Caso seja utilizado DMSO, o seu teor não deve exceder 1 % (v/v). Para qualquer veículo, deve demonstrar-se que o volume máximo utilizado não é citotóxico e não interfere com a eficácia do ensaio. Os padrões de referência e os controlos são dissolvidos em 100 % de solvente e, em seguida, diluídos em concentrações adequadas em EFM.
|
Preparação dos produtos químicos em estudo
|
23.
|
Os produtos químicos em estudo são dissolvidos em DMSO a 100 % (ou solvente adequado) e, em seguida, diluídos em concentrações adequadas. Antes de serem dissolvidos e diluídos, todos os produtos químicos em estudo devem estar à temperatura ambiente. As soluções do produto químico em estudo devem ser preparadas de novo para cada experiência. As soluções não devem ter precipitados ou turbidez percetíveis. Os padrões de referência e as culturas de controlo podem preparar-se a granel; contudo, o padrão de referência, as diluições de controlo e os produtos químicos em estudo finais devem ser preparados para cada experiência e utilizados nas 24 horas seguintes à preparação.
|
Solubilidade e citotoxicidade: considerações para a determinação da gama
|
24.
|
O ensaio de determinação da gama é composto por sete pontos — diluições em série de 1:10 realizadas em duplicado. Inicialmente, os produtos químicos são ensaiados até à concentração máxima de 1 mg/ml (~1 mM), para o ensaio de agonistas, e 20 μg/ml (~10 M), para o ensaio de antagonistas. As experiências de determinação da gama são utilizadas para determinar o seguinte:
|
—
|
Concentrações iniciais do produto químico em estudo a utilizar durante os ensaios completos
|
|
—
|
Diluições do produto químico em estudo (1:2 ou 1:5) a utilizar durante os ensaios completos
|
|
|
25.
|
Os protocolos dos ensaios de agonistas e antagonistas (7) incluem uma avaliação da viabilidade/citotoxicidade celular, integrada nas determinações da gama e nos ensaios completos. O método de citotoxicidade que foi usado para avaliar a viabilidade celular durante a validação do VM7Luc ER TA (1) é um método qualitativo de observação visual graduado; no entanto, é possível utilizar um método quantitativo para a determinação da citotoxicidade - ver protocolo (7). Não podem utilizar-se dados de concentrações de produtos químicos em estudo que causem uma redução da viabilidade superior a 20 %.
|
Exposição ao produto químico em estudo e organização das placas do ensaio
|
26.
|
As células são contadas e colocadas em placas de cultura de tecidos de 96 poços (2 × 105 células por poço) em EFM e incubadas durante 24 horas, para permitir que as células se fixem à placa. O EFM é removido e substituído pelos produtos químicos em estudo e de referência em EFM, incubados durante 19-24 horas. É necessário prestar especial atenção às substâncias altamente voláteis, porquanto os poços de controlo mais próximos podem gerar resultados falsos positivos. Nesses casos, a utilização de «selantes de placas» permite isolar eficazmente os poços individuais durante os ensaios, sendo, por conseguinte, recomendada.
|
Ensaios de determinação da gama
|
27.
|
O ensaio de determinação da gama utiliza todos os poços da placa de 96 poços para ensaiar até seis produtos químicos em estudo em sete diluições em série 1:10 em duplicado (ver figuras 1 e 2).
|
—
|
O ensaio de determinação da gama para agonistas utiliza quatro concentrações de E2 em duplicado como padrão de referência e quatro poços replicados para o controlo com DMSO.
|
|
—
|
O ensaio de determinação da gama para agonistas utiliza três concentrações de Ral/E2 com 9,18 × 10-11 M E2 em duplicado como padrão de referência, com três poços replicados para os controlos com E2 e DMSO.
|
|
Figura 1
Configuração da placa de 96 poços para o ensaio de determinação da gama para agonistas

Abreviaturas: E2-1 a E2-4 = concentrações (da mais alta para a mais baixa) do padrão de referência E2; TC1-1 a TC1-7 = concentrações (da mais alta para a mais baixa) do produto químico em estudo 1 (TC1); TC2-1 a TC2-7 = concentrações (da mais alta para a mais baixa) do produto químico em estudo 2 (TC2); TC3-1 a TC3-7 = concentrações (da mais alta para a mais baixa) do produto químico em estudo 3 (TC3); TC4-1 a TC4-7 = concentrações (da mais alta para a mais baixa) do produto químico em estudo 4 (TC4); TC5-1 a TC5-7 = concentrações (da mais alta para a mais baixa) do produto químico em estudo 5 (TC5); TC6-1 a TC6-7 = concentrações (da mais alta para a mais baixa) do produto químico em estudo 6 (TC6); VC = controlo do veículo (DMSO [1 % v/v EFM.]).
Figura 2
Configuração da placa de 96 poços para o ensaio de determinação da gama para antagonistas
Abreviaturas: E2= controlo E2; Ral-1 a Ral-3 = concentrações do padrão de referência raloxifeno/E2 (da mais alta para a mais baixa); TC1-1 a TC1-7 = concentrações (da mais alta para a mais baixa) do produto químico em estudo 1 (TC1); TC2-1 a TC2-7 = concentrações (da mais alta para a mais baixa) do produto químico em estudo 2 (TC2); TC3-1 a TC3-7 = concentrações (da mais alta para a mais baixa) do produto químico em estudo 3 (TC3); TC4-1 a TC4-7 = concentrações (da mais alta para a mais baixa) do produto químico em estudo 4 (TC4); TC5-1 a TC5-7 = concentrações (da mais alta para a mais baixa) do produto químico em estudo 5 (TC5); TC6-1 a TC6-7 = concentrações (da mais alta para a mais baixa) do produto químico em estudo 6 (TC6); VC = controlo do veículo = (DMSO [1 % v/v EFM.]).
Nota: Todos os produtos químicos em estudo são ensaiados na presença de 9,18 × 10-11 M E2.
|
28.
|
O volume final recomendado de meio necessário para cada poço é de 200 μl. Utilizar apenas as placas de ensaio em que as células de todos os poços tiverem uma viabilidade igual ou superior a 80 %.
|
|
29.
|
A determinação das concentrações iniciais do ensaio completo para agonistas é descrita pormenorizadamente no respetivo protocolo (7). Em síntese, utilizam-se os seguintes critérios:
|
—
|
Se não houver pontos na curva de concentração do produto químico em estudo superiores à média mais três vezes o desvio-padrão do controlo com DMSO, os ensaios completos devem ser efetuados com uma diluição em série de 1:2 em 11 pontos a partir da concentração máxima solúvel.
|
|
—
|
Se existirem pontos na curva de concentração do produto químico em estudo superiores à média mais três vezes o desvio-padrão do controlo com DMSO, a concentração inicial a utilizar para diluição em 11 pontos em ensaios completos deve ser uma unidade logarítmica mais elevada do que a concentração com que se obtém o valor RLU ajustado mais elevado na determinação da gama. O esquema de diluição de 11 pontos deve basear-se nas diluições de 1:2 ou de 1:5, de acordo com os seguintes critérios:
|
Utilizar uma diluição em série de 1:2 em 11 pontos se a gama de concentrações resultante abranger toda a gama de respostas com base na curva concentração-resposta gerada no ensaio de determinação da gama. Caso contrário, utilizar uma diluição de 1:5.
|
|
|
—
|
Se o produto químico em estudo apresentar uma curva de resposta à concentração bifásica no ensaio de determinação da gama de concentrações, ambas as fases devem também ser resolvidas no ensaio completo.
|
|
|
30.
|
A determinação das concentrações iniciais para os ensaios completos para antagonistas é descrita pormenorizadamente no respetivo protocolo (7). Em síntese, utilizam-se os seguintes critérios:
|
—
|
Se não existirem pontos da curva de concentração do produto químico em estudo inferiores à média menos o triplo do desvio-padrão do E2, deve efetuar-se um ensaio completo do controlo com uma diluição em série de 1:2 em 11 pontos com início na concentração máxima solúvel.
|
|
—
|
Se existirem pontos na curva de concentrações do produto químico em estudo à média menos o triplo do desvio-padrão do E2, a concentração inicial a utilizar no esquema de diluição em 11 pontos em ensaios completos deve ser uma das seguintes:
|
—
|
A concentração com que se obtém o mais baixo valor RLU ajustado na determinação da gama;
|
|
—
|
A máxima concentração solúvel [ver protocolo relativo aos antagonistas (7), figura 14-2];
|
|
—
|
A mais baixa concentração citotóxica [ver protocolo relativo aos antagonistas (7), figura 14-3 para um exemplo conexo].
|
|
|
—
|
O esquema de diluição em 11 pontos terá como base uma série de diluições de 1:2 ou1:5, ou uma diluição de acordo com os seguintes critérios:
|
Utilizar uma diluição em série de 1:2 em 11 pontos se a gama de concentrações resultante abranger toda a gama de respostas, com base na curva concentração-resposta gerada no ensaio de determinação da gama. Caso contrário, utilizar uma diluição de 1:5.
|
|
|
Ensaios completos
|
31.
|
Os ensaios completos consistem em diluições em série em 11 pontos (diluições em série de 1:2 ou 1:5 com base na concentração inicial dos critérios para ensaios completos), sendo cada concentração ensaiada em três poços da placa de 96 poços (ver figuras 3 e 4).
|
—
|
Os ensaios completos para agonistas utilizam 11 concentrações de E2 em duplicado como padrão de referência. São incluídos em cada placa quatro poços replicados para o controlo com DMSO e quatro poços replicados para o controlo com metoxicloro (9,06 × 10-6 M);
|
|
—
|
Os ensaios completos para antagonistas utilizam 9 concentrações de Ral/E2 com 9,18 × 10-11 M E2 em duplicado como padrão de referência com quatro poços replicados para o controlo E2 9,18 x 10-11 M, quatro poços replicados para os controlos com DMSO e quatro poços replicados para tamoxifeno 3,36 × 10-6M.
|
|
Figura 3
Configuração da placa de 96 poços para o ensaio completo para agonistas

Abreviaturas: TC11-1 a TC1-11 = concentrações (da mais alta para a mais baixa) do produto químico em estudo 1; 1 TC2-1 a TC2-11 = concentrações (da mais alta para a mais baixa) do produto químico em estudo 2; E2-1 a E2-11 = concentrações do padrão de referência E2 (da mais alta para a mais baixa); Met = controlo positivo fraco do p,p’- metoxicloro; VC = DMSO (1 % v/v) do controlo do veículo EFM
Figura 4
Configuração da placa de 96 poços para o ensaio completo para antagonistas

Abreviaturas: E2 = controlo E2; Ral-1 a Ral-9 = concentrações do padrão de referência raloxifeno/E2 (da mais alta para a mais baixa); Tam = controlo positivo fraco tamoxifeno/E2; TC1-1 a TC1-11 = concentrações (da mais alta para a mais baixa) do produto químico 1 (TC1); TC2-1 a TC2-11 = concentrações (da mais alta para a mais baixa) do produto químico em estudo 2 (TC2); VC = controlo do veículo (DMSO [1 % v/v EFM.]).
Nota: Tal como referido, todos os poços de referência e de ensaio contêm uma concentração fixa de E2 (9,18 × 10-11M)
|
32.
|
As repetições dos ensaios completos para o mesmo produto químico devem ser realizadas em dias diferentes, a fim de garantir a sua independência. Devem efetuar-se, pelo menos, dois ensaios completos. Se os seus resultados forem contraditórios (por exemplo, um ensaio é positivo, o outro negativo) ou se um dos ensaios for inadequado, deve realizar-se um terceiro ensaio.
|
Medição da luminescência
|
33.
|
A luminescência é medida na gama de 300 a 650 nm, com um luminómetro de injeção e software de controlo do volume de injeção e do intervalo de medição (7). A emissão de luz de cada poço é expressa em RLU por poço.
|
ANÁLISE DOS DADOS
Determinação de EC50 /IC50
|
34.
|
Os valores EC50 (metade da concentração efetiva máxima de um produto químico em estudo [agonistas]) e IC50 (metade da concentração inibitória de um produto químico em estudo [antagonistas]) são determinados a partir dos dados concentração-resposta. Para produtos químicos em estudo positivos em uma ou mais concentrações, a concentração que provoca uma resposta semimáxima (IC50 ou EC50) é calculada com recurso a uma análise funcional de Hill ou a uma alternativa adequada. A análise de Hill é um modelo matemático logístico de quatro parâmetros, que relaciona a concentração do produto químico em estudo com a resposta (geralmente após uma curva sigmoidal), utilizando a equação seguinte:
|

em que:
Y= resposta (i.e., RLU);
X= o logaritmo de concentração;
Base= a resposta mínima;
Topo= a resposta máxima;
Log(EC50) [ou log(IC50)]= o logaritmo de X como resposta intermédia entre o topo e a base;
Curva de Hill= o declive da curva.
O modelo calcula o valor mais adequado para o topo, a base, a curva de Hill e os parâmetros IC50 e EC50. Para o cálculo dos valores de EC50 e IC50, deve utilizar-se um software estatístico adequado (por exemplo, Graphpad Prism®).
Determinação de anomalias
|
35.
|
Uma boa avaliação estatística pode ser facilitada pela inclusão (sem caráter limitativo) do ensaio-Q – ver protocolos relativos aos agonistas e antagonistas (7) –, para determinar os poços «inutilizáveis», a excluir da análise de dados.
|
|
36.
|
Para os replicados do padrão de referência E2 (amostra de dois), qualquer valor RLU ajustado para um replicado a uma dada concentração de E2 é considerado anormal se o seu valor for superior ou inferior em mais de 20 % ao valor RLU ajustado para essa concentração, que consta da base de dados históricos.
|
Recolha e ajustamento dos dados do luminómetro para determinação da gama
|
37.
|
Os dados brutos do luminómetro devem ser transferidos para um modelo de folha de cálculo concebida para o ensaio. Deve determinar-se se há pontos aberrantes que necessitem de ser excluídos (ver critérios de aceitação do ensaio para parâmetros determinados nas análises). Devem efetuar-se os cálculos a seguir indicados:
|
Agonistas
|
Etapa 1
|
Calcular o valor médio para o controlo do veículo (VC) DMSO
|
|
Etapa 2
|
Subtrair o valor médio do VC DMSO do valor de cada poço, a fim de normalizar os dados.
|
|
Etapa 3
|
Calcular o fator de indução médio para o padrão de referência (E2).
|
|
Etapa 4
|
Calcular o valor médio de EC50 para os produtos químicos em estudo.
|
Antagonistas
|
Etapa 1
|
Calcular o valor médio do VC DMSO.
|
|
Etapa 2
|
Subtrair o valor médio do VC DMSO do valor de cada poço, a fim de normalizar os dados.
|
|
Etapa 3
|
Calcular o fator de redução médio para o padrão de referência (Ral/E2).
|
|
Etapa 4
|
Calcular o valor médio do padrão de referência E2.
|
|
Etapa 5
|
Calcular o valor médio de IC50 para os produtos químicos em estudo.
|
Recolha e ajustamento dos dados do luminómetro para o ensaio global
|
38.
|
Os dados brutos do luminómetro devem ser transferidos para um modelo de folha de cálculo concebida para o ensaio. Deve determinar-se se há pontos aberrantes que necessitem de ser excluídos (ver critérios de aceitação do ensaio para parâmetros determinados nas análises). Efetuam-se os seguintes cálculos:
|
Agonistas
|
Etapa 1
|
Calcular o valor médio do VC DMSO.
|
|
Etapa 2
|
Subtrair o valor médio do VC DMSO do valor de cada poço, a fim de normalizar os dados.
|
|
Etapa 3
|
Calcular o fator de indução médio para o padrão de referência (E2).
|
|
Etapa 4
|
Calcular o valor médio de EC50 para E2 e para os produtos químicos em estudo.
|
|
Etapa 5
|
Calcular o valor RLU ajustado médio para o metoxicloro.
|
Antagonistas
|
Etapa 1
|
Calcular o valor médio do VC DMSO.
|
|
Etapa 2
|
Subtrair o valor médio do VC DMSO do valor de cada poço, a fim de normalizar os dados.
|
|
Etapa 3
|
Calcular o fator de indução médio para o padrão de referência (Ral/E2).
|
|
Etapa 4
|
Calcular o valor médio de IC50 para Ral/E2 e para os produtos químicos em estudo.
|
|
Etapa 5
|
Calcular o valor RLU ajustado médio para o tamoxifeno.
|
|
Etapa 6
|
Calcular o valor médio do padrão de referência E2.
|
Critérios de interpretação dos dados
|
39.
|
O VM7Luc ER TA destina-se a fazer parte de uma abordagem de ponderação das provas que contribuirá para priorizar substâncias para ensaios de ED in vivo. Uma parte deste procedimento de priorização consiste na classificação do produto químico em estudo como positivo ou negativo em termos de atividade agonista ou de atividade antagonista. Os critérios de decisão positiva e negativa utilizados no estudo de validação da VM7Luc ER TA são descritos no quadro 1.
|
Quadro 1
Critérios de decisão positivos e negativos
|
|
|
ATIVIDADE AGONISTA
|
|
Positiva
|
|
—
|
Todos os produtos químicos em estudo classificados como positivo para a atividade agonista do ER devem apresentar uma curva concentração-resposta que consista numa linha de base, seguida de uma inclinação positiva que termina num plano ou num pico. Em alguns casos, apenas duas destas características (linha de base–inclinação ou inclinação–pico) podem ser definidas.
|
|
—
|
A linha que define a inclinação positiva deve conter, pelo menos, três pontos com barras de erro não sobrepostas (DP ±média). Excluem-se os pontos que constituem a linha de base, mas a parte linear da curva pode incluir o pico ou o primeiro ponto do plano.
|
|
—
|
Uma classificação positiva requer uma amplitude de resposta, ou seja, a diferença entre a linha de base e o pico, de, pelo menos, 20 % do valor máximo para o padrão de referência E2 (i.e., 2000 RLU ou mais, se o valor máximo de resposta dos padrões de referência [E2] for ajustado para 10000 RLU).
|
|
—
|
Se possível, deve calcular-se um valor EC50 para cada produto químico em estudo positivo.
|
|
|
Negativa
|
O valor RLU ajustado médio para uma dada concentração é igual ou inferior ao valor RLU médio do controlo com DMSO mais três vezes o desvio-padrão do RLU do DMSO.
|
|
Inadequada
|
Os dados que não possam ser interpretados como válidos para demonstrar a presença ou ausência de atividade, devido a limitações qualitativas ou quantitativas importantes, são considerados inadequados e não podem ser utilizados para determinar se o produto químico é positivo ou negativo. Os produtos químicos em causa devem ser objeto de novo ensaio.
|
|
|
|
ATIVIDADE ANTAGONISTA
|
|
Positiva
|
|
—
|
Os dados do produto químico em estudo produzem uma curva concentração-resposta que consiste numa linha de base seguida de uma inclinação negativa.
|
|
—
|
A linha que define a inclinação negativa deve ter, pelo menos, três pontos com barras de erro não sobrepostas; os pontos que formam a linha de base são excluídos, mas a parte linear da curva pode incluir o primeiro ponto do plano.
|
|
—
|
Deve registar-se, pelo menos, uma redução de 20 % na atividade do valor máximo do padrão de referência Ral/E2 (i.e., 8000 RLU ou menos, quando o valor máximo da resposta do padrão de referência [Ral/E2] é ajustado para 10000 RLU).
|
|
—
|
As concentrações não citotóxicas mais elevadas do produto químico em estudo devem ser iguais ou inferiores a 1 x 10-5 M.
|
|
—
|
Se possível, deve calcular-se um valor IC50 para cada produto químico em estudo positivo.
|
|
|
Negativa
|
Todos os pontos de dados são acima do valor EC80 (80 % da resposta de E2 ou 8000 RLU), em concentrações inferiores a 1,0 x 10-5 M.
|
|
Inadequada
|
Os dados que não possam ser interpretados como válidos para demonstrar a presença ou ausência de atividade devido a limitações qualitativas ou quantitativas importantes são considerados inadequados e não podem ser utilizados para determinar se o produto químico é positivo ou negativo. O produto químico em causa deve ser objeto de novo ensaio.
|
|
40.
|
Sempre que possível, os resultados positivos serão caracterizados pela magnitude do efeito e pela concentração a que este ocorre. Nas figuras 5 e 6, apresentam-se exemplos de dados positivos, negativos e inadequados.
|
Figura 5
Exemplos de agonistas: Dados positivos, negativos e inadequados

A linha a tracejado indica 20 % da resposta do E2, 2000 RLU ajustados e normalizados.
Figura 6
Exemplos de antagonistas: Dados positivos, negativos e inadequados

A linha a tracejado indica 80 % da resposta de Ral/E2, 8000 RLU ajustados e normalizados.
A linha contínua indica 1,00 x 10-5 M. Para uma resposta ser considerada positiva, deve ser inferior à linha de 8000 RLU e em concentrações inferiores a 1,00 x 10-5 M.
As concentrações marcadas com asterisco no gráfico do meso-hexesterol indicam pontuações de viabilidade iguais ou superiores a «2».
Os resultados do ensaio para o meso-hexesterol são considerados inadequados porque a única resposta que está abaixo de 8000 RLU ocorre a 1,00 x 10-5 M.
|
41.
|
Os cálculos de EC50 e IC50 podem ser feitos por recurso a uma função de Hill de quatro parâmetros (para mais informações, ver protocolo relativo aos agonistas e antagonistas) (7). O cumprimento dos critérios de aceitabilidade indica que o sistema está a funcionar corretamente, mas não assegura que uma determinada série de ensaios produz dados exatos. A duplicação dos resultados da primeira série de ensaios é a melhor garantia de que foram produzidos dados exatos (ver ponto 19 da secção «COMPONENTES DO ENSAIO ER TA»).
|
RELATÓRIO DO ENSAIO
|
42.
|
Ver ponto 20 dos «COMPONENTES DO ENSAIO ER TA».
|
REFERÊNCIAS BIBLIOGRÁFICAS
|
(1)
|
ICCVAM. (2011). ICCVAM Test Method Evaluation Report on the LUMI-CELL® ER (BG1Luc ER TA) Test Method: An In Vitro Method for Identifying ER Agonists and Antagonists, National Institute of Environmental Health Sciences: Research Triangle Park, NC.
|
|
(2)
|
Monje P., Boland R. (2001). Subcellular Distribution of Native Estrogen Recetor α and β Isoforms in Rabbit Uterus and Ovary, J. Cell Biochem., 82(3): 467-479.
|
|
(3)
|
Pujol P., et al. (1998). Differential Expression of Estrogen Recetor-Alpha and -Beta Messenger RNAs as a Potential Marker of Ovarian Carcinogenesis, Cancer Res., 58(23): 5367-5373.
|
|
(4)
|
Weihua Z., et al. (2000). Estrogen Recetor (ER) β, a Modulator of ERα in the Uterus, Proceedings of the National Academy of Sciences of the United States of America 97(11): 936-5941.
|
|
(5)
|
Balls M., et al. (2006). The Importance of Good Cell Culture Practice (GCCP), ALTEX, 23(Suppl): p. 270-273.
|
|
(6)
|
Coecke S., et al. (2005). Guidance on Good Cell Culture Practice: a Report of the Second ECVAM Task Force on Good Cell Culture Practice, Alternatives to Laboratory Animals, 33: p. 261-287.
|
|
(7)
|
ICCVAM (2011). ICCVAM Test Method Evaluation Report, The LUMI-CELL® ER (BG1Luc ER TA) Test Method: An In Vitro Assay for Identifying Human Estrogen Recetor Agonist and Antagonist Activity of Chemicals, NIH Publication No 11-7850.
|
|
(8)
|
Rogers J.M., Denison M.S. (2000). Recombinant Cell Bioassays for Endocrine Disruptors: Development of a Stably Transfected Human Ovarian Cell Line for the Detection of Estrogenic and Anti-Estrogenic Chemicals, In Vitro Mol. Toxicol.,13(1):67-82.
|
|
(9)
|
Escande A., et al. (2006). Evaluation of Ligand Selectivity Using Reporter Cell Lines Stably Expressing Estrogen Recetor Alpha or Beta, Biochem. Pharmacol., 71(10):1459-69.
|
|
(10)
|
Thorne N., Inglese J., Auld D.S. (2010). Illuminating Insights into Firefly Luciferase and Other Bioluminescent Reporters Used in Chemical Biology, Chemistry and Biology,17(6):646-57.
|
|
(11)
|
Kuiper G.G, et al. (1998). Interaction of Estrogenic Chemicals and Phytoestrogens with Estrogen Recetor Beta, Endocrinology,139(10):4252-63.
|
|
(12)
|
Geisinger, et al. (1989). Characterization of a human ovarian carcinoma cell line with estrogen and progesterone recetors, Cancer 63, 280-288.
|
|
(13)
|
Baldwin, et al. (1998). BG-1 ovarian cell line: an alternative model for examining estrogen-dependent growth in vitro, In Vitro Cell. Dev. Biol. – Animal, 34, 649-654.
|
|
(14)
|
Li, Y., et al. (2014). Research resource: STR DNA profile and gene expression comparisons of human BG-1 cells and a BG-1/MCF-7 clonal variant, Mol. Endo. 28, 2072-2081.
|
|
(15)
|
Rogers, J.M. and Denison, M.S. (2000). Recombinant cell bioassays for endocrine disruptors:development of a stably transfected human ovarian cell line for the detection of estrogenicand anti-estrogenic chemicals, In Vitro & Molec. Toxicol. 13, 67-82.
|
Apêndice 4
ENSAIO DE TRANSATIVAÇÃO DE RECETOR-Α DE ESTROGÉNIO HUMANO ESTAVELMENTE TRANSFETADO PARA DETEÇÃO DE ATIVIDADE ESTROGÉNICA AGONISTA E ANTAGONISTA NOS PRODUTOS QUÍMICOS COM RECURSO À LINHA CELULAR ERΑ CALUX
CONSIDERAÇÕES INICIAIS E LIMITAÇÕES (VER TAMBÉM INTRODUÇÃO GERAL)
|
1.
|
O ensaio de transativação do ERα CASX utiliza a linha celular humana U2OS para detetar atividade estrogénica agonista e antagonista mediada através do recetor alfa de estrogénio humano (hERα). O estudo de validação do bioensaio do ERα CALUX estavelmente transfetado pela BioDetection Systems BV (Amesterdão, Países Baixos) demonstrou a adequação e a fiabilidade do ensaio para o fim a que se destina (1). A linha celular ERá CALUX expressa apenas ERá humano transfetado de forma estável (2) (3).
|
|
2.
|
Este ensaio foi especificamente concebido para detetar o transativação mediada pelo hERα tendo a medição da bioluminescência como parâmetro. A utilização da bioluminescência é habitualmente utilizada nos bioensaios devido à elevada relação sinal/ruído (4).
|
|
3.
|
Foram comunicadas concentrações de fitoestrogénios superiores a 1 μM que sobreativam o gene repórter da luciferase, o que resultou em luminescência não mediada pelo recetor (5) (6) (7). Por conseguinte, concentrações mais elevadas de fitoestrogénios ou de outros compostos semelhantes que podem sobreativar a expressão da luciferase devem ser cuidadosamente examinadas nos ensaios de transativação em ER transfetados de forma estável (ver apêndice 2).
|
|
4.
|
Devem ler-se a «INTRODUÇÃO GERAL» e os «COMPONENTES DO ENSAIO ER TA» antes de utilizar este ensaio para fins normativos. As definições e abreviaturas utilizadas no presente método de ensaio são descritas no apêndice 1.
|
PRINCÍPIO DO ENSAIO (VER TAMBÉM INTRODUÇÃO GERAL)
|
5.
|
O bioensaio é utilizado para avaliar a ligação do ligando ao ER e subsequente translocação do complexo recetor-ligando para o núcleo. No núcleo, o complexo recetor-ligando fixa-se a elementos de resposta específicos do ADN e transativa um gene repórter da luciferase do pirilampo, que provoca um aumento da expressão celular da enzima luciferase. Após a adição do substrato luciferina da luciferase, a luciferina é transformada num produto bioluminescente. A luz produzida pode ser facilmente detetada e quantificada com o auxílio de um luminómetro.
|
|
6.
|
O sistema de ensaio utiliza células ERα CALUX transfetadas de forma estável. As células ERα CALUX são provenientes da linha celular U2OS do osteossarcoma osteoblástico humano. As células humanas U2OS foram estavelmente transfetadas com 3xHRE-TATA-Luc e pSG5-neo-hERα pelo método de coprecipitação com fosfato de cálcio. A linha celular U2OS foi selecionada como a melhor candidata a funcionar como linha celular repórter que responde a estrogénios (e a outras hormonas esteroides), com base na observação de que revelou pouca ou nenhuma atividade recetora endógena. A ausência de recetores endógenos foi avaliada com recurso unicamente a repórteres da luciferase plasmídeos, que não revelaram nenhuma atividade após a adição de ligandos aos recetores. Além disso, esta linha celular suportou respostas fortes mediadas por hormonas quando foram transitoriamente introduzidos recetores cognatos (2) (3) (8).
|
|
7.
|
O ensaio de produtos químicos para atividade estrogénica ou antiestrogénica com recurso à linha celular ERα CALUX inclui ume várias séries de ensaios completas. Durante a série de ensaios de avaliação prévia, são determinadas a solubilidade, a citotoxicidade e uma gama de concentrações mais pormenorizada dos produtos químicos em estudo, com vista à realização de ensaios completos. Durante as séries completas, as gamas de concentração mais pormenorizada dos produtos químicos em estudo são ensaiadas nos bioensaios ERα CALUX, seguidas da classificação dos produtos químicos em estudo para agonismo ou antagonismo.
|
|
8.
|
Os critérios para a interpretação dos dados são descritos em pormenor no ponto 59. Em suma, um produto químico em estudo é considerado positivo para agonismo se, pelo menos, duas concentrações consecutivas do produto apresentarem uma resposta igual ou superior a 10 % da resposta máxima do padrão de referência, 17β-estradiol (PC10). Um produto químico em estudo é considerado positivo para antagonismo se, pelo menos, duas concentrações consecutivas do produto químico em estudo apresentarem uma resposta igual ou inferior a 80 % da resposta máxima do padrão de referência, tamoxifeno (PC80).
|
PROCEDIMENTO
Linhas celulares
|
9.
|
Para o ensaio, deve ser utilizada a linha celular U2OS ERα CALUX transfetada de forma estável. A linha celular pode ser obtida na BioDetection Systems BV, Amesterdão, Países Baixos, através de um acordo de licença técnica.
|
|
10.
|
Devem utilizar-se apenas culturas de células livres de micoplasmas. Os lotes de células utilizados devem ser certificados como negativos para contaminação por micoplasmas ou deve proceder-se à realização de um ensaio de micoplasmas antes da sua utilização. Deve usar-se a RT-PCR (reação em cadeia da polimerase em tempo real) para deteção sensível de infeção por micoplasmas (9).
|
Estabilidade da linha celular
|
11.
|
Para manter a sua estabilidade e integridade, as células CALUX devem ser armazenadas em azoto líquido (-80 oC). Após o descongelamento para iniciar uma nova cultura, as células devem ser subcultivadas, pelo menos, duas vezes antes de serem utilizadas para avaliar a atividade estrogénica agonista e antagonista dos produtos químicos. As células não devem ser subcultivadas por mais de 30 passagens.
|
|
12.
|
Para monitorizar a estabilidade da linha celular ao longo do tempo, deve verificar-se a capacidade de resposta do sistema de ensaio agonístico e antagonístico, através da avaliação do EC50 ou do IC50 do padrão de referência. Além disso, é necessário monitorizar a indução relativa da amostra de controlo positivo (PC) e da amostra de controlo negativo (NC). Os resultados devem estar de acordo com os critérios de aceitação para o bioensaio ERα CALUX antagonístico (quadro 3C) ou antagonístico (quadro 4C). Os padrões de referência e os controlos positivo e negativo são apresentados no quadros 1 e no quadro 2 para os modos agonístico e antagonístico, respetivamente.
|
Cultura celular e condições de colocação em placa
|
13.
|
As células U2OS são cultivadas em meio de crescimento (DMEM/F12 (1:1) com vermelho de fenol (indicador de pH), complementado com soro fetal bovino (7,5 %), aminoácidos não essenciais (1 %), 10 unidades/ml de penicilina, estreptomicina e geneticina (G-418), marcadores de seleção. As células devem ser colocadas numa incubadora de CO2 (5 % de CO2) a 37 oC e com 100 % de humidade. Quando atingirem uma confluência de 85-95 %, devem ser subcultivadas ou preparadas para sementeira em placas de microtitulação com 96 poços. Neste último caso, devem ser ressuspensas em 1 × 105 células/ml em meio de ensaio isento de estrogénio – DMEM/F12 (1:1) sem vermelho de fenol, complementado com soro fetal bovino tratado com carvão revestido de dextrano (5 % v/v), aminoácidos não essenciais (1 % v/v) e 10 unidades/ml de penicilina e estreptomicina) e colocadas nos poços das placas de microtitulação de 96 poços (100 μl de suspensão celular homogeneizada). As células devem ser pré-incubadas numa incubadora de CO2 (5 % CO2, 37 oC, 100 % de humidade) durante 24 horas antes da exposição. O material de plástico deve ser isento de estrogénio.
|
Critérios de aceitabilidade
|
14.
|
As atividades agonísticas e antagonístas do(s) produto(s) químico(s) em estudo são ensaiadas em série de ensaios. Uma série de ensaios é constituída por um máximo de 6 placas de microtitulação. Cada série de ensaios contém pelo menos 1 série completa de diluições de um padrão de referência, uma amostra de controlo positivo, uma amostra de controlo negativo e solvente. As figuras 1 e 2 apresentam a configuração das placas para as séries de ensaios agonísticas e antagonísticas.
|
|
15.
|
Todas as diluições dos padrões de referência, dos produtos químicos em estudo, dos controlos do solvente e dos controlos positivo e negativo devem ser analisadas em triplicado. Todas as análises em triplicado devem satisfazer os requisitos indicados nos quadros 3A e 4A.
|
|
16.
|
Em cada série de ensaios, é medida, na primeira placa, uma série completa de diluições do padrão de referência (17β-estradiol para agonismo; tamoxifeno para antagonismo). Para ser possível comparar os resultados da análise das restantes 5 microplacas com a primeira placa de microtitulação – que tem a curva concentração-resposta completa do padrão de referência –, todas as placas deverão conter 3 amostras de controlo: controlo do solvente, a mais elevada concentração do padrão de referência ensaiado e a concentração aproximada de EC50 (agonismo) ou IC50 (antagonismo) do padrão de referência. O rácio da média das amostras de controlo na primeira placa e das 5 restantes deve cumprir os requisitos indicados no quadro 3C (agonismo) ou no quadro 4C (antagonismo).
|
|
17.
|
Para cada placa de microtitulação de série de ensaios, calcula-se o fator z (10) utilizando as respostas à concentração mais elevada e mais baixa do padrão de referência. Uma placa de microtitulação é considerada válida se preencher os requisitos constantes do quadro 3C (agonismo) ou do quadro 4C (antagonismo).
|
|
18.
|
O padrão de referência deve produzir uma curva sigmoidal dose-resposta. Os EC50 ou IC50 derivados da resposta da série de diluições do padrão de referência devem cumprir os requisitos indicados no quadro 3C (agonismo) ou no quadro 4C (antagonismo).
|
|
19.
|
Cada série de ensaios deve abranger uma amostra de controlo positivo e uma amostra de controlo negativo. A indução relativa calculada da amostra de controlo positivo e da amostra de controlo negativo deve satisfazer os requisitos indicados no quadro 3C (agonismo) ou no quadro 4C (antagonismo).
|
|
20.
|
Durante todas as medições, o fator de indução da mais elevada concentração do padrão de referência deve ser medido dividindo a resposta média da unidade de luz relativa (RLU) mais elevada do padrão de referência, 17β-estradiol, pela resposta de controlo RLU média do solvente de referência. Este fator de indução deve cumprir os requisitos mínimos relativos ao fator de indução prescrito no quadro 3C (agonismo) ou no quadro 4C (antagonismo).
|
|
21.
|
Só são consideradas válidas as placas de microtitulação que satisfaçam todos os critérios de aceitação acima mencionados e possam ser utilizadas para avaliar a resposta do produto químico em estudo.
|
|
22.
|
Os critérios de aceitação são aplicáveis tanto a séries de ensaios prévios como a séries de ensaios completas.
|
Quadro 1
Concentrações do padrão de referência, controlo positivo (PC) e controlo negativo (NC) para o bioensaio agonístico CALUX
|
|
Substância
|
N.o CAS
|
Intervalo de ensaio (M)
|
|
Padrão de referência
|
17β-Estradiol
|
50-28-2
|
1*10-13 - 1*10-10
|
|
Controlo positivo (PC)
|
17α-Metiltestosterona
|
58-18-4
|
3*10-06
|
|
Controlo negativo (NC)
|
Corticosterona
|
50-22-6
|
1*10-08
|
Quadro 2
Concentrações do padrão de referência, controlo positivo (PC) e controlo negativo (NC) para o bioensaio antagonístico CALUX
|
|
Substância
|
N.o CAS
|
Intervalo de ensaio (M)
|
|
Padrão de referência
|
Tamoxifeno
|
10540-29-1
|
3*10-9 - 1*10-5
|
|
Controlo positivo (PC)
|
4-Hidroxitamoxifeno
|
68047-06-3
|
1*10-9
|
|
Controlo negativo (NC)
|
Resveratrol
|
501-36-0
|
1*10-5
|
Quadro 3
Critérios de aceitação para o bioensaio agonístico ERα CALUX
|
A - amostras individuais numa placa
|
Critério
|
|
1
|
% DP máximo dos poços triplicados (para NC, PC, cada diluição do produto químico em estudo e o padrão de referência, exceto C0)
|
< 15 %
|
|
2
|
% DP máximo dos poços triplicados [para o padrão de referência e para os controlos do solvente do produto químico em estudo (C0, SC)]
|
< 30 %
|
|
3
|
Fuga máxima de LDH, como medida de citotoxicidade.
|
< 120 %
|
|
B - numa única placa de microtitulação
|
|
|
4
|
Relação entre o controlo do solvente do padrão de referência (C0; placa 1) e o controlo do solvente do produto químico em estudo (SC; placas 2 a x),
|
0,5 a 2,0
|
|
5
|
Relação entre as concentrações mais elevadas aprox. de EC50 e do padrão de referência na placa 1 e as concentrações mais elevadas aprox. de EC50 e do padrão de referência nas placas 2 a x (C4, C8)
|
0,70 a 1,30
|
|
6
|
Fator Z de cada placa
|
> 0,6
|
|
C - numa única série de análises (todas as placas de uma série)
|
|
|
7
|
Curva sigmoidal dos padrões de referência
|
Sim (17ß-estradiol)
|
|
8
|
Gama EC50 para o padrão de referência (17ß-estradiol)
|
4*10-12 – 4*10-11 M
|
|
9
|
Fator de indução mínimo da concentração mais elevada de 17ß-estradiol, em relação ao controlo de solvente do padrão de referência.
|
5
|
|
10
|
(%) PC indução relativa.
|
> 30 %
|
|
11
|
(%) NC indução relativa
|
< 10 %
|
Aprox.: aproximado; PC: controlo positivo; NC: controlo negativo; SC: controlo de solvente do produto químico em estudo; C0: controlo de solvente do padrão de referência; DP: desvio-padrão; DHL: desidrogenase láctica
Quadro 4
Critérios de aceitação para o bioensaio antagonístico ERα CALUX
|
A - amostras individuais numa placa
|
Critério
|
|
1
|
% DP máximo dos poços triplicados (para NC, PC, cada diluição do produto químico em estudo e o controlo do solvente (C0) do padrão de referência)
|
< 15 %
|
|
2
|
% DP máximo dos poços triplicados (para o controlo do veículo (VC) e para a concentração máxima do padrão de referência (C8))
|
< 30 %
|
|
3
|
Fuga máxima de LDH, como medida de citotoxicidade.
|
< 120 %
|
|
B - numa única placa de microtitulação
|
|
|
4
|
Relação entre o controlo de solvente do padrão de referência mais elevadas e o controlo de solvente do produto químico de ensaio (SC; placas 2 a x)
|
0,70 a 1,30
|
|
5
|
Relação entre as concentrações aprox. de IC50 e do padrão de referência na placa 1 e as concentrações aprox. de IC50 e do padrão de referência nas placas 2 a x (C4)
|
0,70 a 1,30
|
|
6
|
Relação entre as concentrações mais elevadas do padrão de referência na placa 1 e as concentrações mais elevadas do padrão de referência nas placas 2 a x (C8)
|
0,50 a 2,0
|
|
7
|
Fator Z de cada placa
|
> 0,6
|
|
C - numa única série de análises (todas as placas de uma série)
|
|
|
8
|
Curva sigmoidal dos padrões de referência
|
Sim (tamoxifeno)
|
|
9
|
Gama IC50 do padrão de referência (tamoxifeno)
|
1*10-8 - 1*10-7 M
|
|
10
|
Fator de indução mínimo do controlo de solvente do padrão de referência em relação à concentração mais elevada de tamoxifeno.
|
2,5
|
|
11
|
(%) PC indução relativa.
|
< 70 %
|
|
12
|
(%) NC indução relativa
|
> 85 %
|
Aprox.: aproximativo; PC: controlo positivo; NC: controlo negativo; VC: controlo do veículo (controlo do solvente sem concentração fixa do padrão de referência agonista); SC: controlo de solvente do produto químico em estudo; C0: controlo de solvente do padrão de referência; DP: desvio padrão; DHL: desidrogenase lática
Controlo do solvente/veículo, padrões de referência, controlos positivos, controlos negativos
|
23.
|
Tanto na série de ensaios prévios como na série de ensaios completas deve utilizar-se os mesmos controlos do solvente/veículo, padrões de referência, controlos positivos e controlos negativos. Além disso, a concentração dos padrões de referência, dos controlos positivos e dos controlos negativos deve ser a mesma.
|
Controlo do solvente
|
24.
|
O solvente utilizado para dissolver os produtos químicos em estudo deve ser ensaiado como solvente de controlo. O dimetilsulfóxido (DMSO 1 % (v/v); N.o CAS 67-68-5) foi utilizado como veículo durante a validação do bioensaio ERα CALUX. Se for utilizado um solvente que não seja o DMSO, devem ser ensaiados no mesmo veículo todos os padrões de referência, controlos e produtos químicos em estudo. Note-se que o controlo do solvente para estudos antagonistas contém uma concentração fixa do padrão de referência agonista, 17β-estradiol (aproximadamente a concentração EC50). Para ensaiar o solvente utilizado nos estudos antagonísticos, deve ser preparado e ensaiado um controlo do veículo.
|
Controlo do veículo (antagonismo)
|
25.
|
Para o ensaio do antagonismo, completa-se o meio com uma concentração fixa do padrão de referência agonista, 17β-estradiol (aproximadamente a concentração EC50). Para ensaiar o solvente utilizado para dissolver o produto químico em estudo, deve preparar-se um meio de ensaio sem uma concentração fixa do padrão de referência agonista (17β-estradiol). Esta amostra de controlo é indicada como o controlo do veículo. O dimetilsulfóxido [DMSO 1 % (v/v); n.o CAS 67-68-5] foi utilizado como veículo durante a validação do bioensaio ERα CALUX. Se for utilizado um solvente que não seja o DMSO, devem ser ensaiados no mesmo veículo todos os padrões de referência, controlos e produtos químicos em estudo.
|
Padrões de referência
|
26.
|
O padrão de referência agonístico é o 17β-estradiol (quadro 1). Os padrões de referência incluem uma série de diluições de oito concentrações de 17β-estradiol (1*10-13, 3*10-13, 1*10-12, 3*10-12, 6*10-12, 1*10-11, 3*10-11, 1*10-10 M).
|
|
27.
|
O padrão de referência antagonístico é o tamoxifeno (quadro 2). Os padrões de referência incluem uma série de diluições de oito concentrações de tamoxifeno (3*10-09, 1*10-08, 3*10-08, 1*10-07, 3*10-07, 1*10-06, 3*10-06, 1*10-05 M). Cada uma das concentrações do padrão de referência antagonístico é coincubada com uma concentração fixa do padrão de referência agonístico 17β-estradiol (3*10-12 M).
|
Controlo positivo
|
28.
|
No caso de estudos agonísticos, o controlo positivo é a 17α-metiltestosterona (quadro 1).
|
|
29.
|
O controlo positivo para os estudos antagonísticos é o 4-hidroxitamoxifeno (quadro 2). O controlo positivo antagonístico é coincubado com uma concentração fixa do padrão de referência agonístico, 17β-estradiol (3*10-12 M).
|
Controlo negativo
|
30.
|
O controlo negativo para os estudos agonísticos é a corticosterona (quadro 1).
|
|
31.
|
O controlo negativo para os estudos antagonísticos é o resveratrol (quadro 2). O controlo negativo antagonístico é coincubado com uma concentração fixa do padrão de referência agonístico, 17β-estradiol (3*10-12 M).
|
Demonstração da competência técnica do laboratório (ver pontos 14 e os quadros 3 e 4 da secção «COMPONENTES DO ENSAIO ER TA» do presente método)
Veículo
|
32.
|
O solvente utilizado para dissolver o produto químico em estudo deve solubilizá-lo completamente e ser miscível com o meio celular. O DMSO, a água e o etanol (pureza 95 % a 100 %) são solventes adequados. Caso seja utilizado DMSO como solvente, a sua concentração máxima durante a incubação não deve exceder 1 % (v/v). Antes da utilização, o solvente deve ser ensaiado para verificar a ausência de citotoxicidade e de interferência com o desempenho do ensaio.
|
Preparação de padrões de referência, dos controlos positivos, dos controlos negativos e dos produtos químicos em estudo
|
33.
|
Os padrões de referência, os controlos positivos, os controlos negativos e o produto químico em estudo são dissolvidos em DMSO a 100 %, ou num solvente adequado. Devem em seguida ser preparadas diluições adequadas (em série) no mesmo solvente. Antes da dissolução, todas as substâncias devem poder estabilizar à temperatura ambiente. As soluções recentemente preparadas dos padrões de referência, controlos positivos, controlos negativos e produtos químicos em estudo não devem apresentar precipitados nem turbidez percetíveis. O padrão de referência e os controlos podem ser preparados a granel. As soluções de produtos químicos em estudo devem ser preparadas antes de cada ensaio. As diluições finais dos padrões de referência, dos controlos positivos, dos controlos negativos e do produto químico em estudo devem ser preparadas para cada experiência e utilizadas nas 24 horas seguintes à preparação.
|
Solubilidade, citotoxicidade e determinação da gama.
|
34.
|
Durante a série de ensaios de avaliação prévia, é determinada a solubilidade do produto químico em estudo no solvente escolhido. Prepara-se uma concentração máxima de 0,1 M. No caso de esta concentração mostrar problemas de solubilidade, devem preparar-se soluções de concentração mais baixa até os produtos químicos serem completamente solubilizados. Durante a série de ensaios de avaliação prévia, são ensaiadas diluições em série de 1:10 do produto químico em estudo. A concentração máxima de ensaio agonistas ou antagonistas é de 1 mM. Após a série de ensaios de avaliação prévia, é derivada uma gama de concentração pormenorizada para produtos químicos em estudo que deve ser ensaiada durante as séries de ensaios completos. As diluições utilizadas para os ensaios completos devem ser 1×, 3×, 10×, 30×, 100×, 300×, 1000× e 3000×.
|
|
35.
|
O ensaio de citotoxicidade está incluído no protocolo de ensaio agonista e antagonista (11). O ensaio de citotoxicidade é integrado tanto na série de ensaios de avaliação prévia como na série de ensaios completos. O método utilizado para avaliar a citotoxicidade durante a validação do bioensaio ERα CALUX foi o ensaio de fugas da desidrogenase láctica (LDH) em combinação com a inspeção visual qualitativa das células (ver apêndice 4.1) após a exposição aos produtos químicos em estudo. No entanto, podem utilizar-se outros métodos quantitativos para a determinação da citotoxicidade (por exemplo, ensaio colorimétrico à base de tetrazólio – MTT – ou bioensaio de citotoxicidade CALUX). Em geral, as concentrações do produto químico em estudo que apresentem uma redução da viabilidade celular superior a 20 % são consideradas citotóxicas e não podem, portanto, ser utilizadas na avaliação dos dados. No que respeita ao ensaio de fuga de LDH, a concentração do produto químico em estudo é considerada citotóxica se a percentagem de LDH for superior a 120 %.
|
Exposição ao produto químico em estudo e a organização da placa do ensaio
|
36.
|
Após a tripsinização de um balão confluente de células de cultura, as células voltam a ser suspensas em 1 × 105 células/ml num meio de ensaio isento de estrogénio. Colocam-se 100 μl de células ressuspensas nos poços interiores de uma placa de microtitulação com 96 poços. Os poços exteriores são preenchidos com 200 μl de solução salina tamponada de fosfato (PBS) (ver figuras 1 e 2). As células colocadas nas placas são pré-incubadas durante 24 horas numa incubadora de CO2 (5 % de CO2, 37 oC, 100 % de humidade).
|
|
37.
|
Após a pré-incubação, as placas são inspecionadas para verificação da citotoxicidade visual (ver apêndice 4.1), da contaminação e da confluência. Para o ensaio, apenas são utilizadas placas que não apresentem citotoxicidade ou contaminação e tenham uma confluência mínima de 85 %. O meio dos poços interiores é cuidadosamente removido e substituído por 200 μl de meio de ensaio sem estrogénio com as diluições adequadas dos padrões de referência, produtos químicos em estudo, controlos positivos, controlos negativos e controlos de solvente (quadro 5: estudos de agonistas; quadro 6: estudos de antagonista). Todos os padrões de referência, produtos químicos em estudo, controlos positivos, controlos negativos e controlos dos solventes são ensaiados em triplicado. Na figura 1, é apresentada a configuração das placas para os ensaios de agonistas. Na figura 2, é apresentada a configuração da placa para os ensaios de antagonistas. A configuração da placa para os ensaios de avaliação prévia e para os ensaios completos é idêntica. No caso dos ensaios antagonísticos, todos os poços interiores, exceto os poços do controlo do veículo (VC), contêm igualmente uma concentração fixa do padrão de referência agonista, 17β-estradiol (3*10-12 M). Note-se que os padrões de referência C8 e C4 devem ser adicionados a todas as placas de TC.
|
|
38.
|
Após a exposição das células a todos os produtos químicos, as placas de microtitulação de 96 poços devem ser incubadas por mais 24 horas numa incubadora de CO2 (5 % CO2, 37 oC, 100 % de humidade).
|
Figura 1
Configuração das placas de microtitulação de 96 poços para avaliação prévia e avaliação do efeito agonístico

C0 = Solvente padrão de referência.
C(1-8) = Série de diluições (1-8, da concentração mais baixa para a mais alta) do padrão de referência.
PC = Controlo positivo.
NC = Controlo negativo.
TCx-(1-8) = Diluições (1-8, da concentração mais baixa para a mais alta) do produto químico em estudo para a série de ensaios de avaliação prévia e para avaliação do efeito agonístico do produto químico x.
SC = Controlo do solvente do produto químico em estudo (idealmente, o mesmo solvente de C0, mas eventualmente de outro lote).
Células cinzentas: = Poços exteriores, preenchidos com 200 μl de PBS.
Figura 2
Configuração das placas de microtitulação de 96 poços para avaliação antagonística prévia e avaliação do efeito antagonístico

C0 = Solvente padrão de referência.
C(1-8) = Série de diluições (1-8, da concentração mais baixa para a mais alta) do padrão de referência.
NC = Controlo negativo.
PC = Controlo positivo.
TCx-(1-8) = Diluições (1-8, da concentração mais baixa para a mais alta) do produto químico em estudo para a série de ensaios de avaliação prévia e para avaliação do efeito antagonístico do produto químico x.
SC = Controlo do solvente do produto químico em estudo (idealmente, o mesmo solvente de C0, mas eventualmente de outro lote).
VC = Controlo do veículo (controlo do solvente sem concentração fixa do padrão de referência agonista 17β-estradiol).
Células cinzentas: = Poços exteriores, preenchidos com 200 μl de PBS.
Nota: todos os poços interiores, exceto os poços do controlo do veículo (VC), contêm igualmente uma concentração fixa do padrão de referência agonista, 17β-estradiol (3,0*10-12 M)
Medição da luminescência
|
39.
|
A medição da luminescência é descrita em pormenor no protocolo de ensaio relativo ao agonista e ao antagonista (10). O meio dos poços deve ser removido e as células devem ser lisadas após 24 horas de incubação, de modo a abrirem a membrana celular e permitirem a medição da atividade da luciferase.
|
|
40.
|
Para medir a luminescência, este procedimento exige um luminómetro equipado com dois injetores. A reação da luciferase é desencadeada por injeção do substrato luciferino. A reação é interrompida por adição de 0,2 M NaOH, por forma a impedir a transferência de luminescência de um poço para outro.
|
|
41.
|
A luz emitida por cada poço é expressa em unidades de luz relativa (RLU) por poço.
|
Série de ensaios de avaliação prévia
|
42.
|
Os resultados das análises prévias são utilizados para determinar um intervalo de concentração mais preciso de produtos químicos em estudo, com vista à realização dos ensaios completos. A avaliação dos resultados das análises prévias e a determinação da gama de concentrações mais precisa de produtos químicos em estudo para ensaios completos é descrita de forma aprofundada no protocolo de ensaio de agonistas e antagonistas (10). Apresenta-se aqui um breve resumo dos procedimentos de determinação da gama de concentrações dos produtos químicos em estudo para os ensaios agonista e antagonista. Ver os quadros 5 e 6 para diretrizes para a conceção da diluição em série.
|
Seleção das concentrações para avaliação de efeitos agonísticos
|
43.
|
Durante a série de ensaios de avaliação prévia, os produtos químicos em estudo devem ser ensaiados com a série de diluições indicada nos quadros 5 (agonismo) e 6 (antagonismo). Todas as concentrações devem ser ensaiadas em triplicado, de acordo com a configuração da placa indicada nas figuras 1 (agonismo) ou 2 (antagonismo).
|
|
44.
|
Apenas os resultados de análises que satisfaçam os critérios de aceitação (quadro 3) são considerados válidos e podem ser utilizados para avaliar a resposta dos produtos químicos em estudo. Se uma ou mais placas de microtitulação de uma série de análises não cumprirem os critérios de aceitação, as placas de microtitulação correspondentes devem ser reanalisadas. Caso a primeira placa com a série completa de diluições do padrão de referência não cumpra os critérios de aceitação, a série completa de ensaios (6 placas) tem de ser repetida.
|
|
45.
|
As gamas de concentração inicial dos produtos químicos em estudo devem ser ajustadas e a série de ensaios de avaliação prévia repetida no caso de:
|
—
|
se observar citotoxicidade. O procedimento de avaliação prévia deve ser repetido com concentrações não citotóxicas mais baixas do produto químico em estudo;
|
|
—
|
a avaliação prévia do produto químico em estudo não produzir uma curva dose-resposta completa, porque as concentrações ensaiadas geram indução máxima. Repete-se a série de ensaios de avaliação prévia com concentrações menores do produto químico em estudo.
|
|
|
46.
|
Quando se observa uma resposta válida relacionada com a dose, deve selecionar-se a menor concentração à qual é observada a indução máxima sem revelar citotoxicidade. A concentração mais elevada do produto químico em estudo nas séries de ensaios completos deve ser 3 vezes superior a esta concentração selecionada.
|
|
47.
|
Deve preparar-se uma série completa de diluições mais precisas do produto químico em estudo com as etapas de diluição indicadas no quadro 5, começando pela concentração mais alta determinada supra.
|
|
48.
|
Um produto químico em estudo que não provoque qualquer efeito antagonista deve ser ensaiado nas séries de ensaio completas, começando com a concentração não citotóxica mais alta identificada na série de ensaios de avaliação prévia.
|
Seleção das concentrações para avaliação dos efeitos antagónicos
|
49.
|
Apenas os resultados de análises que satisfaçam os critérios de aceitação (quadro 4) são considerados válidos e podem ser utilizados para avaliar a resposta dos produtos químicos em estudo. Se uma ou mais placas de microtitulação de uma série de análises não cumprirem os critérios de aceitação, devem ser reanalisadas. Caso a primeira placa com a série completa de diluições do padrão de referência não cumpra os critérios de aceitação, a série completa de ensaios (6 placas) tem de ser repetida.
|
|
50.
|
As gamas de concentração inicial dos produtos químicos em estudo devem ser ajustadas e a série de ensaios de avaliação prévia repetida no caso de:
|
—
|
se observar citotoxicidade. O procedimento de avaliação prévia deve ser repetido com concentrações não citotóxicas mais baixas do produto químico em estudo;
|
|
—
|
a avaliação prévia do produto químico em estudo não produzir uma curva dose-resposta completa, porque as concentrações ensaiadas geram inibição máxima. Deve repetir-se a série de ensaios de avaliação prévia com concentrações menores do produto químico em estudo.
|
|
|
51.
|
Quando se observa uma resposta válida relacionada com a dose, deve selecionar-se a menor concentração à qual é observada a inibição máxima sem revelar citotoxicidade. A concentração mais elevada do produto químico em estudo nas séries de ensaios completos deve ser 3 vezes superior a esta concentração selecionada.
|
|
52.
|
Deve preparar-se uma série completa de diluições mais precisas do produto químico em estudo com as etapas de diluição indicadas no quadro 6, começando pela concentração mais alta determinada supra.
|
|
53.
|
Um produto químico em estudo que não provoque qualquer efeito antagonista deve ser ensaiado nas séries de ensaio completas, começando com a concentração não citotóxica mais alta ensaiada na série de ensaios de avaliação prévia.
|
Séries de ensaios completos
|
54.
|
Após a seleção das gamas de concentração aperfeiçoadas, os produtos químicos em estudo devem ser submetidos a um ensaio completo, utilizando a série de diluições indicada no quadro 5 (agonismo) e 6 (antagonismo). Todas as concentrações devem ser ensaiadas em triplicado, de acordo com a configuração da placa indicada nas figuras 1 (agonismo) ou 2 (antagonismo).
|
|
55.
|
Apenas os resultados de análises que satisfaçam os critérios de aceitação (quadros 3 e 4) são considerados válidos e podem ser utilizados para avaliar a resposta dos produtos químicos em estudo. Se uma ou mais placas de microtitulação de uma série de análises não cumprirem os critérios de aceitação, devem ser reanalisadas. Caso a primeira placa contendo a série completa de diluições do padrão de referência não cumpra os critérios de aceitação, a série completa de ensaios (6 placas) tem de ser repetida.
|
Quadro 5
Concentração e diluições dos padrões de referência, controlos e produtos químicos em estudo utilizados nos ensaios agonistas
|
17β-estradiol de referência
|
TCx - série de ensaios de avaliação prévia
|
TCx - série de ensaios completos
|
Controlos
|
|
conc. (M)
|
diluição
|
diluição
|
conc. (M)
|
|
C0
|
0
|
TCx-1
|
10 000 000 x
|
TCx-1
|
3 000 x
|
PC
|
3*10-6
|
|
C1
|
1*10-13
|
TCx-2
|
1 000 000 x
|
TCx-2
|
1 000 x
|
NC
|
1*10-8
|
|
C2
|
3*10-13
|
TCx-3
|
100 000 x
|
TCx-3
|
300 x
|
C0
|
0
|
|
C3
|
1*10-12
|
TCx-4
|
10 000 x
|
TCx-4
|
100 x
|
SC
|
0
|
|
C4
|
3*10-12
|
TCx-5
|
1 000 x
|
TCx-5
|
30 x
|
|
|
|
C5
|
6*10-12
|
TCx-6
|
100 x
|
TCx-6
|
10 x
|
|
|
|
C6
|
1*10-11
|
TCx-7
|
10 x
|
TCx-7
|
3 x
|
|
|
|
C7
|
3*10-11
|
TCx-8
|
1 x
|
TCx-8
|
1 x
|
|
|
|
C8
|
1*10-10
|
|
|
|
|
|
|
|
TCx - Produto químico em estudo x
PC - Controlo positivo (17α-metiltestosterona)
NC - Controlo negativo (corticosterona)
C0 - Controlo do solvente padrão de referência
SC - Controlo do solvente do produto químico em estudo
|
Quadro 6
Concentração e diluições dos padrões de referência, controlos e produtos químicos em estudo utilizados nos ensaios antagonistas
|
Tamoxifeno de referência
|
TCx - série de ensaios de avaliação prévia
|
TCx - série de ensaios completos
|
Controlos
|
|
conc. (M)
|
diluição
|
diluição
|
conc. (M)
|
|
C0
|
0
|
TCx-1
|
10 000 000 x
|
TCx-1
|
3 000 x
|
PC
|
1*10-9
|
|
C1
|
3*10-9
|
TCx-2
|
1 000 000 x
|
TCx-2
|
1 000 x
|
NC
|
1*10-5
|
|
C2
|
1*10-8
|
TCx-3
|
100 000 x
|
TCx-3
|
300 x
|
C0
|
0
|
|
C3
|
3*10-8
|
TCx-4
|
10 000 x
|
TCx-4
|
100 x
|
SC
|
0
|
|
C4
|
1*10-7
|
TCx-5
|
1 000 x
|
TCx-5
|
30 x
|
|
|
|
C5
|
3*10-7
|
TCx-6
|
100 x
|
TCx-6
|
10 x
|
Agonista complementado
|
|
C6
|
1*10-6
|
TCx-7
|
10 x
|
TCx-7
|
3 x
|
conc. (M)
|
|
C7
|
3*10-6
|
TCx-8
|
1 x
|
TCx-8
|
1 x
|
17β-estradiol
|
3*10-12
|
|
C8
|
1*10-5
|
|
|
|
|
|
|
|
TCx - Produto químico em estudo x
PC - Controlo positivo (4-hidroxitamoxafeno)
NC - Controlo negativo (resveratrol)
C0 - Controlo do solvente padrão de referência
SC - Controlo do solvente do produto químico em estudo
VC - Controlo do veículo (não contém uma concentração fixa do padrão de referência agonístico, 17β-estradiol (3*10-12 M).
|
Recolha e análise de dados
|
56.
|
Após a série de ensaios de avaliação prévia e a série de ensaios completos, devem ser determinados os valores EC10, EC50, PC10, PC50 e de indução máxima (TCxmáx) de um produto químico em estudo para o ensaio agonístico. Para este último, devem ser calculados os valores de IC20, IC50, PC80, PC50 e de indução mínima (TCxmin). Nas figuras 3 (agonismo) e 4 (antagonismo), apresenta-se uma representação gráfica destes parâmetros. Os parâmetros exigidos são calculados com base na indução relativa de cada produto químico em estudo [relativa à indução máxima do padrão de referência (= 100 %)]. Deve utilizar-se a regressão não linear (declive variável, 4 parâmetros) para a avaliação dos dados, de acordo com a seguinte equação:

em que:
X = Log de dose ou concentração
Y = Resposta [(%) indução relativa]
Topo = (%) indução máxima
Base = (%) indução mínima
Log(EC50) = Log da concentração à qual se observam 50 % da resposta máxima
Declive = Declive da curva de Hill
|
|
57.
|
Os dados brutos do luminómetro, expressos em unidades de luz relativa (RLU), devem ser transferidos para a folha de cálculo da análise de dados para a série de ensaios de avaliação prévia e a série de ensaios completos. Os dados em bruto devem satisfazer os critérios de aceitação indicados nos quadros 3A e 3B (agonismo) ou 4A e 4B (antagonismo). Caso os dados em bruto satisfaçam os critérios de aceitação, efetuam-se as seguintes etapas de cálculo para determinar os parâmetros exigidos:
|
Agonistismo
|
—
|
Subtrair a RLU média do controlo do solvente do padrão referência de cada um dos dados brutos de análise dos padrões de referência.
|
|
—
|
Subtrair a RLU média para o controlo do solvente do produto químico em estudo de cada um dos dados de análise brutos dos produtos químicos em estudo.
|
|
—
|
Calcular a indução relativa de cada concentração do padrão de referência. Fixar a indução da mais elevada concentração do padrão de referência em 100 %.
|
|
—
|
Calcular a indução relativa de cada concentração do produto químico em estudo em relação à concentração mais elevada do padrão de referência e estabelecê-la como 100 %.
|
|
—
|
Avaliar os resultados da análise após a regressão não linear (inclinação variável, 4 parâmetros).
|
|
—
|
Determinar os valores EC50 e EC10 do padrão de referência.
|
|
—
|
Determinar os valores EC50 e EC10 dos produtos químicos em estudo.
|
|
—
|
Determinar a indução relativa máxima do produto químico em estudo (TCmáx).
|
|
—
|
Determinar os valores PC10 e PC50 dos produtos químicos em estudo.
|
No caso dos produtos químicos em estudo, pode nem ser sempre obtida uma curva dose-resposta completa devido, por exemplo, a problemas de citotoxicidade ou de solubilidade. Assim, não podem ser determinados os valores EC50, EC10 e PC50. Nesse caso, apenas podem ser determinados o PC10 e TCmáx
Antagonismo
|
—
|
Subtrair a RLU média da concentração mais elevada do padrão de referência de cada um dos dados brutos de análise dos padrões de referência.
|
|
—
|
Subtrair a RLU média da concentração mais elevada do padrão de referência de cada um dos dados brutos de análise dos produtos químicos em estudo.
|
|
—
|
Calcular a indução relativa de cada concentração do padrão de referência. Fixar a indução da mais baixa concentração do padrão de referência em 100 %.
|
|
—
|
Calcular a indução relativa de cada concentração do produto químico em estudo em relação à concentração mais baixa do padrão de referência e estabelecê-la como 100 %.
|
|
—
|
Avaliar os resultados da análise após a regressão não linear (inclinação variável, 4 parâmetros).
|
|
—
|
Determinar os valores IC50 e IC20 do padrão de referência.
|
|
—
|
Determinar os valores IC50 e IC20 dos produtos químicos em estudo.
|
|
—
|
Determinar a indução relativa mínima do produto químico em estudo (TCmín).
|
|
—
|
Determinar os valores PC80 e PC50 dos produtos químicos em estudo.
|
Figura 3
Panorâmica dos parâmetros determinados no ensaio de agonistas

EC10 = Concentração de uma substância em que se observam 10 % da sua resposta máxima.
EC50 = Concentração de uma substância em que se observam 50 % da sua resposta máxima.
PC10 = Concentração de um produto químico em estudo na qual a sua resposta é igual a EC10 do padrão de referência.
PC50 = Concentração de um produto químico em estudo na qual a sua resposta é igual à EC50 do padrão de referência.
TCxmáx = Indução máxima relativa do produto químico em estudo.
Figura 4
Panorâmica dos parâmetros determinados no ensaio de antagonistas

IC20 = Concentração de uma substância em que se observa 80 % da sua resposta máxima (20 % de inibição).
IC50 = Concentração de uma substância em que se observa 50 % da sua resposta máxima (50 % de inibição).
PC80 = Concentração de um produto químico em estudo na qual a sua resposta é igual à IC20 do padrão de referência.
PC50 = Concentração de um produto químico em estudo na qual a sua resposta é igual à IC50 do padrão de referência.
TCxmin = Indução relativa mínima do produto químico em estudo.
No caso dos produtos químicos em estudo, pode nem ser sempre obtida uma curva dose-resposta completa devido, por exemplo, a problemas de citotoxicidade ou de solubilidade. Assim, não é possível determinar o IC50, o IC20 e o PC50. Nesse caso, só o PC20 e a TCmín podem ser determinados.
|
58.
|
Os resultados devem basear-se em duas (ou três) séries de ensaios independentes. Se duas séries derem resultados comparáveis e, por conseguinte, reprodutíveis, não é necessário realizar uma terceira série de ensaios. Para serem aceitáveis, os resultados devem:
|
—
|
satisfazer os critérios de aceitabilidade (ver critérios de aceitabilidade, pontos 14-22),
|
|
Critérios de interpretação dos dados
|
59.
|
Para a interpretação dos dados e a decisão sobre se um produto químico é considerado positivo ou negativo, devem ser utilizados os seguintes critérios:
|
Para cada série completa de ensaios, considera-se positivo o produto químico em estudo nos seguintes casos:
|
1.
|
A TCmáx é igual ou superior a 10 % da resposta máxima do padrão de referência (REF10).
|
|
2.
|
Pelo menos, duas concentrações consecutivas do produto químico em estudo são iguais ou superiores ao REF10.
|
|
|
Para cada série de ensaios completos, um produto químico em estudo é considerado negativo no caso de:
|
1.
|
A TCmáx não exceder 10 % da resposta máxima do padrão de referência (REF10).
|
|
2.
|
Menos de duas concentrações do produto químico em estudo serem iguais ou superiores à REF10.
|
|
|
Para cada série completa de ensaios, considera-se positivo o produto químico em estudo nos seguintes casos:
|
1.
|
A TCmín é igual ou superior a 80 % da resposta máxima do padrão de referência (REF80 = 20 % de inibição).
|
|
2.
|
Pelo menos, duas concentrações consecutivas do produto químico em estudo são iguais ou superiores à REF10.
|
|
|
Para cada série de ensaios completos, um produto químico em estudo é considerado negativo no caso de:
|
1
|
A TCmín exceder 80 % da resposta máxima do padrão de referência (inibição RF80 = 20 % de inibição).
|
|
2
|
Menos de duas concentrações do produto químico em estudo são inferiores ou iguais à REF80.
|
|
|
|
60.
|
Para caracterizar a potência da resposta positiva de um produto químico em estudo, a magnitude do efeito (agonismo: TCmáx; antagonismo: TCmín) e a concentração em que o efeito ocorre (agonismo: EC10, EC50, PC10, PC50; antagonismo: IC20, IC50, PC80, PC50) devem ser comunicadas.
|
RELATÓRIO DO ENSAIO
|
61.
|
Ver ponto 20 dos «COMPONENTES DO ENSAIO ER TA»
|
REFERÊNCIAS BIBLIOGRÁFICAS
|
(1)
|
OCDE (2016). Draft Validation report of the (anti-) ERα CALUX bioassay - transactivation bioassay for the detection of compounds with (anti)estrogenic potential. Environmental Health and Safety Publications, Series on Testing and Assessment (No 240). Organisation for Economic Cooperation and Development, Paris
|
|
(2)
|
Sonneveld E, Jansen HJ, Riteco JA, Brouwer A, van der Burg B. (2005). Development of androgen- and estrogen-responsive bioassays, members of a panel of human cell line-based highly selective steroid-responsive bioassays. Toxicol Sci. 83(1), 136-148.
|
|
(3)
|
Quaedackers ME, van den Brink CE, Wissink S, Schreurs RHMM, Gustafsson JA, van der Saag PT, and van der Burg B. (2001). 4-Hydroxytamoxifen trans-represses nuclear factor-kB Activity in human osteoblastic U2OS cells through estrogen recetor (ER)α and not through ERβ. Endocrinology 142(3), 1156-1166.
|
|
(4)
|
Thorne N, Inglese J and Auld DS. (2010). Illuminating Insights into Firefly Luciferase and Other Bioluminescent Reporters Used in Chemical Biology, Chemistry and Biology17(6):646-57.
|
|
(5)
|
Escande A, Pillon A, Servant N, Cravedi JP, Larrea F, Muhn P, Nicolas JC, Cavaillès V and Balaguer P. (2006). Evaluation of ligand selectivity using reporter cell lines stably expressing estrogen recetor alpha or beta. Biochem. Pharmacol., 71, 1459-1469.
|
|
(6)
|
Kuiper GG, Lemmen JG, Carlsson B, Corton JC, Safe SH, van der Saag PT, van der Burg B and Gustafsson JA. (1998). Interaction of estrogenic chemicals and phytoestrogens with estrogen recetor beta. Endocrinol., 139, 4252-4263.
|
|
(7)
|
Sotoca AM, Bovee TFH, Brand W, Velikova N, Boeren S, Murk AJ, Vervoort J, Rietjens IMCM. (2010). Superinduction of estrogen recetor mediated gene expression in luciferase based reporter gene assays is mediated by a post-transcriptional mechanism. J. Steroid. Biochem. Mol. Biol., 122, 204–211.
|
|
(8)
|
Sonneveld E, Riteco JAC, Jansen HJ, Pieterse B, Brouwer A, Schoonen WG, and van der Burg B. (2006). Comparison of in vitro and in vivo screening models for androgenic and estrogenic activities. Toxicol. Sci., 89(1), 173–187.
|
|
(9)
|
Kobayashi H, Yamamoto K, Eguchi M, Kubo M, Nakagami S, Wakisaka S, Kaizuka M and Ishii H. (1995). Rapid detection of mycoplasma contamination in cell cultures by enzymatic detection of polymerase chain reaction (PCR) products. J. Vet. Med. Sci., 57(4), 769-771.
|
|
(10)
|
Zhang J-H, Chung TDY, and Oldenburg KR. (1999). A simple statistical parameter for use in evaluation and validation of high throuphut screening assays. J. Biomol. Scr., 4, 67-73
|
|
(11)
|
Besselink H, Middelhof I, and Felzel, E. (2014). Transactivation assay for the detection of compounds with (anti)estrogenic potential using ERα CALUX cells. BioDetection Systems BV (BDS). Amsterdam, the Netherlands.
|
Apêndice 4.1
INSPEÇÃO VISUAL DA VIABILIDADE CELULAR
B.67 ENSAIO DE MUTAÇÃO GENÉTICA EM CÉLULAS DE MAMÍFEROS IN VITRO COM O GENE DA TIMIDINA-CINASE
INTRODUÇÃO
|
1.
|
O presente método de ensaio é equivalente à Test Guideline 490 (2016) da OCDE. Os métodos de ensaio são analisados e revistos periodicamente à luz do progresso científico, das necessidades normativas e do bem-estar dos animais. O ensaio do linfoma do rato (MLA) e o ensaio TK6 com o lócus da timidina-cinase (TK) estavam originalmente incluídos no método de ensaio B.17. Posteriormente, o grupo de trabalho de peritos do AML do International Workshop for Genotoxicity Testing (IWGT) desenvolveu recomendações harmonizadas a nível internacional de critérios de aceitação e interpretação dos dados para o MLA (1)(2)(3)(4)(5), tendo estas recomendações sido incorporadas no novo método de ensaio B.67. O presente método de ensaio foi concebido para o ensaio MLA e, dado que utiliza também o lócus TK, para o ensaio TK6. Embora o MLA tenha sido amplamente utilizado para fins normativos, o TK6 tem sido utilizado com muito menos frequência. Note-se que, apesar da semelhança entre os parâmetros, as duas linhas celulares não são permutáveis e os programas de regulamentação podem manifestar preferência no respeitante a uma determinada utilização regulamentar. Por exemplo, a validação do MLA demonstrou que o método é adequado para detetar não só as mutações genéticas, mas também a capacidade de um produto químico em estudo para induzir danos cromossómicos estruturais. O presente método faz parte de uma série de métodos de ensaio no domínio da toxicologia genética. Está disponível um documento da OCDE que fornece informações sucintas sobre ensaios de toxicologia genética e resume as alterações recentemente introduzidas nas orientações da OCDE neste domínio (6).
|
|
2.
|
O objetivo dos ensaios de mutação genética de células de mamíferos in vitro é detetar mutações genéticas induzidas por produtos químicos. As linhas celulares utilizadas nestes ensaios indicam mutações em genes repórteres, nomeadamente no gene da timidina-cinase endógeno (TK para células humanas e Tk para células de roedores, coletivamente designadas TK). O presente método de ensaio destina-se a ser utilizado com duas linhas celulares: a linha celular L5178Y TK+/––3.7.2C do linfoma do rato (geralmente designada L5178Y) e a linha celular linfoblastoide humana TK6 (geralmente designada TK6). Embora as duas linhas variem devido à sua origem, crescimento celular, estatuto p53, etc., os ensaios da mutação do gene Tk podem ser realizados de forma semelhante em ambos os tipos de células, conforme descrito no presente método de ensaio.
|
|
3.
|
A natureza autossómica e heterozigótica do gene da timidina-cinase permite detetar colónias viáveis cujas células são deficientes em enzima timidina-cinase após a mutação de TK+/–
para TK–/–
. Essa deficiência pode resultar de eventos genéticos que afetam o gene Tk, incluindo mutações genéticas (mutações pontuais, mutações que deslocam o quadro de leitura, supressão de pequenas sequências, etc.) e eventos cromossómicos (supressão de grandes sequências e rearranjos cromossómicos, recombinação mitótica). Estes últimos são expressos em perdas de heterozigosidade, que é uma mudança genética comum de genes supressores tumorais na tumorigénese humana. Teoricamente, a perda de todo o cromossoma que comporta o gene Tk na sequência de perturbações do fuso mitótico e/ou não disjunção mitótica pode ser detetada no MLA. Com efeito, uma combinação de análises citogenéticas e moleculares mostra claramente que alguns TK mutantes do MLA resultam da ausência de disjunção. No entanto, a análise das provas mostra que os ensaios de mutação do gene Tk não permitem detetar de forma fiável a aneugénese aplicando critérios normais de citotoxicidade (descritos no presente método de ensaio), pelo que não é adequado utilizar estes ensaios para detetar a aneugénese (7)(8)(9).
|
|
4.
|
A mutação do gene Tk gera duas classes fenotípicas distintas de mutantes TK: os mutantes de crescimento normal, que crescem ao mesmo ritmo que as células heterozigóticas TK, e os mutantes de crescimento lento, que crescem com períodos de duplicação longos. Os mutantes de crescimento normal e de crescimento lento são reconhecidos como mutantes de colónia grande e mutantes de colónia pequena no MLA e como mutantes de colónia rápida e mutantes de colónia tardia no TK6. Foi explorada aprofundadamente a natureza molecular e citogenética dos mutantes do MLA de colónia grande e colónia pequena (8) (10) (11) (12) (13). A natureza molecular e citogenética dos mutantes TK6 de colónia rápida e de colónia tardia foram também objeto de estudo exaustivo (14) (15) (16) (17). Os mutantes de crescimento lento de ambos os tipos de células sofreram danos genéticos que envolvem genes putativos reguladores do crescimento perto do lócus do TK e que resultam em tempos de duplicação prolongados e na formação de colónias tardias ou pequenas (18). A indução de mutantes de crescimento lento foi associada à ação de produtos químicos que induzem grandes variações estruturais ao nível do cromossoma. As células cujo dano não implica o(s) gene(s) putativo(s) regulador(es) do crescimento perto do lócus do TK crescem a ritmos similares aos das células parentais e tornam-se mutantes de crescimento normal. A indução de mutantes de crescimento normal está associada a produtos químicos que funcionam principalmente como mutagénicos pontuais. Por conseguinte, é essencial contar tanto os mutantes de crescimento lento como os mutantes de crescimento normal para recuperar todos os mutantes e fornecer informações sobre o(s) tipo(s) de danos (mutagénicos/clastogénicos) induzidos pelo produto químico em estudo (10) (12) (18) (19).
|
|
5.
|
O método de ensaio está estruturado de modo a fornecer informações gerais aplicáveis tanto ao MLA como ao TK6, e orientações específicas para os ensaios individuais.
|
|
6.
|
As definições utilizadas constam do apêndice 1.
|
CONSIDERAÇÕES INICIAIS E LIMITAÇÕES
|
7.
|
Os ensaios realizados in vitro exigem geralmente a utilização de uma fonte exógena de ativação metabólica. Os sistemas exógenos de ativação metabólica não reproduzem totalmente as condições in vivo.
|
|
8.
|
Deve ter-se o cuidado de evitar condições suscetíveis de conduzir a resultados falsos positivos (ou seja, possível interação com o sistema de ensaio) não causados pela interação direta entre o produto químico em estudo e o material genético da célula; essas condições incluem variações do pH ou da pressão osmótica, a interação com os componentes do meio (20) (21) ou níveis excessivos de citotoxicidade (22)(23)(24). No MLA e no TK6, é considerada excessiva uma citotoxicidade que exceda os níveis de citotoxicidade máxima recomendados, estabelecidos no ponto 28. Além disso, é de notar que os produtos químicos em estudo análogos da timidina, ou com comportamento idêntico aos produtos análogos da timidina, podem aumentar a frequência de mutação por via do crescimento seletivo dos mutantes espontâneos durante o tratamento celular, exigindo métodos de ensaio complementares para uma avaliação adequada (25).
|
|
9.
|
No caso dos nanomateriais fabricados, podem ser necessárias adaptações específicas ao presente método de ensaio, que não são descritas no mesmo.
|
|
10.
|
Antes da aplicação do método de ensaio a uma mistura para obter dados com fins normativos, importa ponderar se – e, em caso afirmativo, por que motivo – o método pode proporcionar resultados adequados para o efeito. Essas considerações não são necessárias se existir um requisito regulamentar para o ensaio da mistura.
|
|
11.
|
As células mutantes deficientes em atividade enzimática da timidina-cinase devido a uma mutação TK
+/– para TK
–/– são resistentes aos efeitos citostáticos do análogo de pirimidina da trifluorotimidina (TFT). As células que dispõem de timidina-cinase são sensíveis ao TFT, que causa inibição do metabolismo celular e impede a divisão da célula. Logo, as células mutantes podem proliferar na presença de TFT, o que não acontece com as células que contêm a enzima timidina-cinase.
|
PRINCÍPIO DO ENSAIO
|
12.
|
As células em suspensão são expostas ao produto químico em estudo, com e sem uma fonte exógena de ativação metabólica (ver ponto 19), por um período adequado (ver ponto 33), e, em seguida, subcultivadas para determinar a citotoxicidade e permitir a expressão fenotípica antes da seleção de mutantes. A citotoxicidade é determinada pelo crescimento relativo total (RTG, ver ponto 25), para o MLA, e pela sobrevivência relativa (RS, ver ponto 26), para o TK6. As culturas expostas são mantidas em meio de crescimento durante um período suficiente, específico de cada tipo de célula (ver ponto 37), por forma a permitir uma expressão fenotípica tão boa quanto possível das mutações induzidas. Após a expressão fenotípica, determina-se a frequência de mutação mediante a inoculação de um número conhecido de células num meio que contenha o agente seletivo, para a deteção de colónias mutantes, e num meio sem agente seletivo, para determinar as respetivas eficiências de clonagem (viabilidade). Após um período de incubação apropriado, procede-se à contagem das colónias. A frequência de mutação é calculada com base no número de colónias mutantes corrigido pela eficiência de clonagem no momento da seleção dos mutantes.
|
DESCRIÇÃO DO MÉTODO
Preparações
Células
|
13.
|
Para o MLA: Dado o MLA ter sido foi desenvolvido e caracterizado com a sublinha TK
+/– –3.7.2C das células L5178Y, tem de se utilizar esta sublinha específica. A linha celular L5178Y foi derivada de um linfoma num timo de rato induzido por metilcolantreno DBA2 (26). Clive et al. trataram células L5178Y (designadas por Clive como TK+/+ –3) com metanossulfonato de etilo e isolaram um clone TK–/– (designado TK–/– –3.7), usando bromodesoxiuridina como agente seletivo. A partir do clone TK–/–, foram isolados e caracterizados para uso na AML um clone TK+/– espontâneo (designado TK+/– –3.7.2.) e um subclone (designado TK+/––3.7.2C) (27). O cariótipo da linha celular encontra-se publicado (28) (29) (30) (31). O número modal de cromossomas é 40. Existe um cromossoma metacêntrico (t12; 13) que deve ser contabilizado como cromossoma. O lócus da TK do rato está localizado na extremidade distal do cromossoma 11. A linha celular L5178Y TK
+/– –3,7.2C tem mutações em ambos os alelos p53 e produz a proteína mutante p53 (32) (33). O estatuto p53 da linha celular TK+/––3,7.2C é provavelmente responsável pela capacidade do ensaio para detetar danos em grande escala (17).
|
|
14.
|
Para o TK6: O TK6 é uma linha celular linfoblastoide humana. A linha celular-mãe é uma linha transformada pelo vírus de Epstein-Barr, WI-L2, originalmente derivada de um indivíduo do sexo masculino de 5 anos com esferocitose hereditária. O primeiro clone isolado, HH4, foi mutagenisado com ICR191, tendo sido gerada uma linha celular TK heterozigótica – TK6 (34). As células TK6 são quase diploides e o cariotipo representativo é 47, XY, 13+, t(14; 20), t(3; 21) (35). O lócus da TK humana está situado no braço longo do cromossoma 17. O TK6 é uma linha celular competente em p53, pois possui uma sequência de p53 de tipo selvagem em ambos os alelos e exprime apenas proteínas p53 do tipo selvagem (36).
|
|
15.
|
Tanto para o MLA como para o TK6, quando se estabelece ou reconstitui uma solução de reserva celular primária, é aconselhável que o laboratório de ensaio possa garantir a ausência de contaminação por micoplasma, identifique o cariótipo das células ou marque os cromossomas com o lócus da TK, e verifique o tempo de duplicação da população. Deve estabelecer-se o tempo normal do ciclo celular para as células utilizadas no laboratório de ensaio, tempo esse que deve ser coerente com as características celulares publicadas (16) (19) (37). Esta solução de reserva celular primária deve ser armazenada a uma temperatura não superior a -150 °C e usada para preparar todas as soluções de reserva de células de trabalho.
|
|
16.
|
Quer antes do estabelecimento de um grande número de soluções de reserva de trabalho criopreservadas, quer imediatamente antes da sua utilização numa experiência, a cultura pode ter de ser limpa de células mutantes pré-existentes [a menos que a frequência de mutação (MF) do controlo do solvente já se encontre dentro da margem admissível – ver o quadro 2 para o MLA]. Para tal, utiliza-se metotrexato (aminopterina) para selecionar células deficientes em TK e adicionar à cultura timidina, hipoxantina e glicina (L5178Y) ou 2’-desoxicitidina (TK6), de forma a garantir o crescimento ótimo das células competentes em TK (19)(38)(39) e (40), para TK6. Em (19) (31) (37) (39) (41) podem encontrar-se orientações gerais sobre boas práticas de manutenção de culturas celulares, bem como aconselhamento específico para as células L5178Y e TK6. Relativamente aos laboratórios que necessitam de soluções de reserva celulares primárias para iniciar o MLA ou o TK6 ou para obter soluções de reserva de células principais, está disponível um repositório de células bem caracterizadas (37).
|
Meios e condições de cultura
|
17.
|
Em ambos os ensaios, devem utilizar-se meios de cultura e condições de incubação adequados para a conservação das culturas (por exemplo, recipientes de cultura, atmosfera humidificada com 5 % de CO2, temperatura de incubação de 37 °C). As culturas celulares devem ser sempre mantidas em condições que assegurem o seu crescimento exponencial. É particularmente importante escolher meios e condições de cultura que garantam um crescimento ótimo das células durante o período de expressão e clonagem das células mutantes e não mutantes. Para o MLA e o TK6, é também importante que as condições de cultura garantam um crescimento ótimo dos mutantes TK, tanto de grande colónia ou colónia rápida como de pequena colónia ou colónia tardia. Para mais informações sobre a cultura, incluindo a necessidade de aquecer adequadamente o soro de cavalo inativado, caso se utilize o meio RPMI durante a seleção de mutantes, consultar (19) (31) (38) (39) (40) (42).
|
Preparação das culturas
|
18.
|
As células são propagadas a partir de soluções de reserva de culturas, inoculadas num meio de cultura a uma densidade que permita que as culturas em suspensão continuem a crescer exponencialmente durante o tratamento e os períodos de expressão.
|
Ativação metabólica
|
19.
|
Quando se utilizam as células L5178Y e TK6, deve recorrer-se a sistemas metabolizantes exógenos, dado a sua capacidade metabólica endógena ser inadequada. O sistema que se utiliza com maior frequência, recomendado por defeito – salvo justificação em contrário – é uma fração pós-mitocondrial reforçada com co-fator (S9) preparada a partir de fígados de roedores (em geral, ratazanas) tratados com agentes de indução enzimática, como, por exemplo, Aroclor 1254 (43) (44) (45) ou uma mistura de fenobarbital e β-naftoflavona (46) (47) (48) (49) (50) (51). Esta última combinação não é contrária à Convenção de Estocolmo sobre Poluentes Orgânicos Persistentes (52) e comprovou-se ser tão eficaz na indução de oxidases de função mista como o Aroclor 1254 (45)(46)(47)(48)(49). A fração S9 é habitualmente utilizada em concentrações na gama de 1 % a 2 %, que podem ser aumentadas para a 10 % (v/v) no meio de ensaio final. A escolha do tipo e da concentração de sistema exógeno de ativação metabólica ou do indutor metabólico utilizado pode ser influenciada pela classe dos produtos químicos em estudo.
|
Preparação do produto químico em estudo
|
20.
|
Os produtos químicos em estudo sólidos devem ser dissolvidos em solventes adequados e, se necessário, diluídos antes do tratamento das células (ver ponto 21). Os produtos químicos es estudo líquidos podem ser adicionados diretamente ao sistema de ensaio e/ou ser diluídos antes de serem utilizados no tratamento. Para o ensaio de produtos químicos gasosos ou voláteis, devem efetuar-se alterações adequadas aos protocolos normalizados, optando, por exemplo, pelo tratamento em recipientes de cultura fechados (53)(54)(55). A preparação do produto químico em estudo deve ser feita imediatamente antes do tratamento, a menos que os dados de estabilidade demonstrem que pode ser armazenado.
|
CONDIÇÕES DE REALIZAÇÃO DO ENSAIO
Solventes
|
21.
|
O solvente deve ser escolhido de modo a otimizar a solubilidade dos produtos químicos em estudo sem afetar negativamente a realização do ensaio – por exemplo, alterando o crescimento celular, afetando a integridade do produto químico em estudo, reagindo com os recipientes de cultura ou alterando o sistema de ativação metabólica. Sempre que possível, recomenda-se a utilização de um solvente (ou meio de cultura) aquoso. A água e o dimetilsulfóxido, por exemplo, são solventes cujo desempenho é bem conhecido. No meio de tratamento final, os solventes orgânicos não devem exceder, em geral, 1 % (v/v) e os solventes aquosos (salinos ou água) não devem exceder 10 % (v/v). Se forem utilizados solventes cujo desempenho não é bem conhecido (p. ex., etanol ou acetona), devem fornecer-se dados que comprovem a respetiva compatibilidade com os produtos químicos e o sistema em estudo, bem como a inexistência de genotoxicidade à concentração utilizada. Na ausência de dados comprovativos, é importante incluir amostras de controlo não tratadas (ver apêndice 1, «Definições») para demonstrar que o solvente escolhido não tem efeitos deletérios ou mutagénicos.
|
MEDIÇÃO DA CITOTOXICIDADE E ESCOLHA DAS CONCENTRAÇÕES DE TRATAMENTO
|
22.
|
Ao determinar a concentração máxima a ensaiar do produto químico em estudo, devem evitar-se concentrações passíveis de gerar respostas positivas falsas, como as que produzam citotoxicidade excessiva (ver ponto 28), precipitação no meio de cultura (ver ponto 20) ou alterações pronunciadas do pH ou da pressão osmótica (ver ponto 8). Se, ao ser adicionado, o produto químico em estudo causar uma alteração pronunciada do pH do meio, este pode ser ajustado por tamponamento do meio de tratamento final, de modo a evitar falsos resultados positivos e manter condições de cultura adequadas.
|
|
23.
|
A seleção da concentração baseia-se na citotoxicidade e noutras considerações (ver pontos 27-30). Embora a avaliação da citotoxicidade num ensaio preliminar possa ser útil para definir melhor as concentrações a utilizar no ensaio principal, não é indispensável realizar esse ensaio. Mesmo no caso de se proceder a uma avaliação preliminar da citotoxicidade, continua a ser necessária a determinação da citotoxicidade em cada cultura no ensaio principal. Se for realizado um ensaio de determinação da gama de concentrações, este deve abranger uma vasta gama e pode terminar no dia 1 após o tratamento ou prolongar-se durante os dois dias de expressão, até à seleção de mutantes (caso se verifique que as concentrações utilizadas são adequadas).
|
|
24.
|
Deve determinar-se a citotoxicidade para cada cultura de ensaio e para cada cultura de controlo: os métodos para MLA (2) e TK6 (15) são definidos por práticas acordadas a nível internacional.
|
|
25.
|
Tanto para o ágar como para as variantes micropoços do MLA: deve avaliar-se a citotoxicidade utilizando o crescimento relativo total (RTG) originalmente definido por Clive e Spector em 1975 (2). Esta medida inclui a suspensão relativa do crescimento durante o tratamento das células (RSG: cultura de ensaio versus controlo do solvente), o tempo de expressão e a eficiência relativa de clonagem no momento em que os mutantes são selecionados (RCE: cultura de ensaio versus controlo do solvente) (2). Note-se que a RSG inclui todas as perdas de células que ocorram na cultura de ensaio durante o tratamento (ver fórmulas no apêndice 2).
|
|
26.
|
Para o TK6: A citotoxidade deve ser avaliada utilizando a taxa de sobrevivência relativa (RS), ou seja, a eficiência de clonagem das células colocadas em placas imediatamente após o tratamento, ajustada para ter em conta qualquer perda de células durante o tratamento, com base na contagem de células em comparação com o controlo negativo (taxa de sobrevivência de 100 %) (ver a fórmula no apêndice 2).
|
|
27.
|
Devem avaliar-se pelo menos quatro concentrações de ensaio (não incluindo os controlos de solvente e positivos) que satisfaçam os critérios de aceitabilidade (citotoxicidade adequada, número de células, etc.). Embora seja aconselhável a utilização de culturas em duplicado, são igualmente aceitáveis culturas únicas em todas as concentrações a ensaiar. Os resultados obtidos com culturas replicadas independentes, a uma dada concentração, devem ser comunicados separadamente, mas podem ser agrupados para a análise de dados (55). No caso dos produtos químicos que demonstrem pouca ou nenhuma citotoxicidade, são normalmente adequadas concentrações espaçadas por um fator de 2 a 3. Quando se observa citotoxicidade, as concentrações escolhidas devem abranger uma gama com início na concentração que produz citotoxicidade, conforme descrito no ponto 28, e que inclua as concentrações às quais se observa pouca ou nenhuma citotoxicidade. Muitos produtos químicos em estudo apresentam curvas concentração-resposta com declive acentuado, pelo que, para abranger toda a gama de citotoxicidade ou para estudar em pormenor a relação dose-resposta, será necessário recorrer a concentrações menos espaçadas e a mais de quatro concentrações, em especial nos casos em que for necessário repetir o ensaio (ver ponto 70). A utilização de mais de quatro concentrações pode ser particularmente importante quando se utilizam culturas únicas.
|
|
28.
|
Se a concentração máxima se basear na citotoxicidade, a concentração mais elevada deverá visar entre 20 % e 10 % de RTG para o MLA e entre 20 % e 10 % de RS para o TK6 (ponto 67).
|
|
29.
|
No caso de produtos químicos pouco solúveis que não sejam citotóxicos a concentrações inferiores à concentração insolúvel mínima, a maior concentração analisada deve produzir, no final do tratamento com o produto químico em estudo, turbidez ou um precipitado visível a olho nu ou com o auxílio de um microscópio invertido. Mesmo no caso de se observar citotoxicidade acima da menor concentração insolúvel, é conveniente ensaiar uma única concentração que produza turbidez ou um precipitado visível, devido aos efeitos falsos que possam ser induzidos pelo precipitado. Dado que os ensaios MLA e TK6 utilizam culturas em suspensão, deve ter-se o cuidado de assegurar que os precipitados não interferem com a realização do ensaio. Neste contexto, pode igualmente ser útil determinar a solubilidade no meio de cultura antes do ensaio.
|
|
30.
|
Se não se observar precipitado nem citotoxicidade condicionante, a maior concentração ensaiada deve corresponder à menor das seguintes concentrações: 10 mM, 2 mg/ml ou 2 μl/ml (57)(58). Se o produto químico em estudo não tiver composição definida (caso, p. ex., de uma substância de composição desconhecida ou variável, de produtos de reação complexos ou de materiais biológicos [ou seja, substâncias químicas de composição desconhecida ou variável (UVCB)], de extratos ambientais, etc.), a concentração de topo poderá ter de ser mais elevada (p. ex., 5 mg/ml) na ausência de citotoxicidade, para aumentar a concentração de cada um dos componentes. Importa, contudo, notar que estes requisitos podem diferir no caso de medicamentos para uso humano (59).
|
Controlos
|
31.
|
Para cada uma das condições experimentais, devem também realizar-se controlos negativos paralelos (ver ponto 21), em que as células são expostas apenas ao solvente e ao meio de tratamento, procedendo-se do mesmo modo que num ensaio normal.
|
|
32.
|
São necessários controlos positivos para demonstrar a capacidade do laboratório para identificar mutagénios nas condições do protocolo de ensaio, a eficácia do sistema exógeno de ativação metabólica – se pertinente – e para detetar de forma adequada mutantes TK de colónias pequenas/tardias e de colónias grandes/rápidas. O quadro 1 apresenta exemplos de controlos positivos. Se tal se justificar, podem utilizar-se outras substâncias de controlo positivo. Dado que os ensaios de toxicidade genética in vitro em células de mamíferos estão suficientemente normalizados no que respeita a tratamentos paralelos de curta duração (3-4 horas) com e sem ativação metabólica por período idêntico, o recurso a produtos químicos de controlo positivo pode limitar-se a um mutagénico que necessite de ativação metabólica. Neste caso, a resposta de controlo positivo única demonstrará a atividade do sistema de ativação metabólica e a capacidade de resposta do sistema de ensaio. No entanto, devem efetuar-se controlos positivos para os tratamentos de longa duração (24 horas sem S9), dado que a duração do tratamento diferirá da do ensaio com ativação metabólica. Cada amostra de controlo positiva deverá ser utilizada numa ou mais das concentrações a que se prevê um aumento detetável e reprodutível relativamente à base, a fim de demonstrar a sensibilidade do sistema de ensaio; a resposta não deve ser comprometida pela citotoxicidade, de modo a exceder os limites especificados no método de ensaio (ver ponto 28).
|
Quadro 1
Substâncias de referência recomendadas para avaliação da competência técnica de um laboratório e para a seleção dos controlos positivos
|
Categoria
|
Substância
|
N.o CAS
|
|
1. Mutagénios ativos sem ativação metabólica
|
|
Metanossulfonato de metilo
|
66-27-3
|
|
Mitomicina C
|
50-07-7
|
|
N-óxido de 4-nitroquinolina
|
56-57-5
|
|
2. Mutagénios que necessitam de ativação metabólica
|
|
Benzo[a]pireno
|
50-32-8
|
|
Ciclofosfamida mono-hidratada
|
50-18-0 (6055-19-2)
|
|
7,12-Dimetilbenzoantraceno
|
57-97-6
|
|
3-Metilcolantreno
|
56-49-5
|
PROCEDIMENTO
Tratamento com o produto químico em estudo
|
33.
|
As células em proliferação são expostas ao produto químico em estudo na presença e na ausência de um sistema de ativação metabólica. A exposição deve ter uma duração adequada (normalmente, de 3 a 4 horas). Importa, contudo, notar que estes requisitos podem diferir no caso de medicamentos para uso humano (59). No caso do MLA, se o tratamento de curta duração produzir resultados negativos e existirem dados que sugiram a necessidade de um tratamento mais longo [por exemplo, dispositivos análogos dos nucleósidos, produtos químicos pouco solúveis, (5) (59)], deve ponderar-se a possibilidade de prolongar o ensaio – por exemplo, 24 horas sem S9.
|
|
34.
|
O número mínimo de células utilizado para cada cultura (controlada e tratada), em cada fase do ensaio, deve basear-se na frequência de mutação espontânea. Uma orientação genérica consiste em tratar e passar células suficientes em cada cultura experimental para manter, no mínimo, 100 mutantes espontâneos em todas as fases do ensaio (tratamento, expressão fenotípica e seleção de mutantes) (56).
|
|
35.
|
Para o MLA, a frequência de mutação espontânea recomendada aceitável situa-se entre 35-140 × 10–6 (versão ágar) e 50-170 × 10–6 (versão micropoço) (ver quadro 2). Para ter pelo menos 10 e, idealmente, 100 mutantes espontâneos sobreviventes ao tratamento em cada cultura de ensaio, é necessário tratar pelo menos 6 × 106 células. O tratamento deste número de células e a manutenção de um número suficiente de células durante a fase de expressão e de clonagem para seleção de mutantes assegura um número suficiente de mutantes espontâneos (10 ou mais) em todas as fases da experiência, mesmo para as culturas tratadas em concentrações que resultam numa citotoxicidade de 90 % (medida por um RTG de 10 %) (19) (38) (39).
|
|
36.
|
Para o TK6, a frequência de mutação espontânea é, em geral, entre 2 e 10 × 10–6. Para ter, pelo menos, 10 mutantes espontâneos sobreviventes ao tratamento em cada cultura, é necessário tratar, pelo menos, 20 × 106 células. O tratamento deste número de células assegura um número suficiente de mutantes espontâneos (10 ou mais), mesmo nas culturas tratadas com concentrações que provoquem 90 % de citotoxicidade durante o tratamento (10 % RS). Além disso, deve cultivar-se um número suficiente de células durante o período de expressão, colocadas em placas para seleção de mutantes (60).
|
Período de expressão fenotípica e determinação da citotoxicidade e da frequência de mutação
|
37.
|
No final do período de exposição, as células são cultivadas por um determinado período, para permitir a expressão fenotípica quase ótima das mutações recentemente induzidas, específicas de cada linha celular. Para o MLA, o período de expressão fenotípica é de 2 dias. Para o TK6, é de 3-4 dias. Se for utilizado um tratamento de 24 horas, o período de expressão tem início após o final do tratamento.
|
|
38.
|
Durante o período de expressão fenotípica, as células são contadas diariamente. Para o MLA, as contagens de células diárias são utilizadas para calcular o crescimento diário da suspensão (SG). Após o período de expressão de 2 dias, as células são suspensas num meio com e sem agente seletivo, para a determinação do número de mutantes (placas de seleção) e da eficiência de clonagem (placas de viabilidade), respetivamente. No caso do MLA, existem dois métodos igualmente aceitáveis de clonagem para seleção de mutantes: um que utiliza ágar macio e outro que utiliza meio líquido em placas de 96 poços (19) (38) (39). A clonagem no TK6 é efetuada com um meio líquido e placas de 96 poços (16).
|
|
39.
|
A trifluorotimidina (TFT) é o único agente seletivo recomendado para mutantes TK (61).
|
|
40.
|
Para o MLA, as placas de ágar e as placas de micropoços são contadas após 10 a 12 dias de incubação. No que diz respeito ao TK6, as colónias em placas de micropoços são contadas após 10 a 14 dias, para a deteção de mutantes precoces. A fim de recuperar os mutantes TK6 de crescimento lento (aparecimento tardio), é necessário voltar a expor as células a um meio de crescimento e a TFT após contagem dos mutantes precoces, e incubar as placas durante mais 7-10 dias (62). Ver pontos 42 e 44 para uma reflexão sobre a contagem dos mutantes TK de crescimento lento e normal.
|
|
41.
|
O apêndice 2 especifica os cálculos adequados para os dois ensaios, incluindo os dois métodos (ágar e micropoço) para o MLA. No respeitante ao método ágar do MLA, as colónias são contadas e o número de colónias mutantes ajustado pela eficiência de clonagem para calcular uma MF. Quanto à versão de micropoços do MLA e do TK6, a eficiência de clonagem das placas de seleção e de clonagem é determinada de acordo com a distribuição de Poisson (63). Calcula-se a MF é calculada a partir destas duas eficiências de clonagem.
|
Caracterização de colónias mutantes
|
42.
|
No MLA, se o produto químico em estudo for positivo (ver pontos 62 e 63), deve proceder-se à caracterização das colónias por dimensão ou por crescimento em, pelo menos, uma das culturas de ensaio (geralmente a concentração positiva mais elevada aceitável) e nos controlos negativos e positivos. Se o produto químico em estudo for negativo (ver ponto 64), a caracterização das colónias mutantes deve basear-se nos controlos negativos e positivos. No método dos micropoços do MLA, as colónias de mutantes pequenas são definidas como as que cobrem menos de 25 % do diâmetro do poço e as colónias de mutantes grandes como as que cobrem mais de 25 % do diâmetro do poço. No método do ágar, é utilizado um contador de colónias automático para contar as colónias mutantes e para medição do tamanho das colónias. As abordagens relativas à medição do tamanho das colónias constam das referências bibliográficas (19) (38) (40). É necessário caracterizar as colónias nos controlos negativos e positivos para demonstrar que os estudos são realizados de modo adequado.
|
|
43.
|
O produto químico em estudo não pode ser considerado negativo se não se detetarem adequadamente colónias de mutantes grandes e colónias de mutantes pequenas no controlo positivo. A caracterização das colónias pode ser usada para obter informações gerais sobre a capacidade do produto químico em estudo de provocar mutações pontuais e/ou eventos cromossómicos (ponto 4).
|
|
44.
|
TK6: Os mutantes de crescimento normal e de crescimento lento são diferenciados pela diferença do tempo de incubação (ver ponto 40). Para o TK6, em geral, os mutantes precoces e tardios são contados em todas as culturas, incluindo os controlos negativos e positivos. A caracterização das colónias dos controlos negativos e positivos é necessária para demonstrar que os estudos são realizados de modo adequado. O produto químico em estudo não pode ser considerado negativo se não for adequadamente detetada no controlo positivo a presença de mutantes precoces e tardios. A caracterização das colónias pode ser usada para fornecer informações gerais sobre a capacidade do produto químico em estudo de provocar mutações pontuais e/ou eventos cromossómicos (ponto 4).
|
Competência técnica do laboratório
|
45.
|
A fim de demonstrar que possui experiência suficiente com o ensaio antes de o realizar de forma rotineira, o laboratório deve ter realizado uma série de experiências com substâncias de referência positivas, que atuem através de diferentes mecanismos (pelo menos uma substância ativa com ativação metabólica e uma substância ativa sem ativação metabólica, selecionadas a partir das substâncias enumeradas no quadro 1), e vários controlos negativos (culturas não tratadas e vários solventes/veículos). As respostas observadas aos controlos positivos e negativos devem ser coerentes com as referências bibliográficas. Este requisito não é aplicável a laboratórios com experiência, isto é, que disponham de uma base de dados históricos, tal como se define nos pontos 47-50. No caso do MLA, os valores obtidos para os controlos positivos e negativos devem ser coerentes com as recomendações do IWGT (ver quadro 2).
|
|
46.
|
Deve estudar-se uma seleção de substâncias de controlo positivo (ver quadro 1), com tratamentos curtos e longos (se forem utilizados tratamentos longos) na ausência de ativação metabólica, bem como com tratamentos curtos na presença de ativação metabólica, com vista a demonstrar competência técnica para detetar produtos químicos mutagénicos, determinar a eficácia do sistema de ativação metabólica e demonstrar a adequação das condições de crescimento das células durante o tratamento, a expressão fenotípica, a seleção de mutantes e os procedimentos de contagem. A gama de concentrações das substâncias selecionadas deve ser escolhida de forma a proporcionar aumentos reprodutíveis e dependentes da concentração relativamente ao nível de base, que permitam demonstrar a sensibilidade e a gama dinâmica do sistema de ensaio.
|
Dados históricos de controlo
|
47.
|
O laboratório deve elaborar:
|
—
|
um historial da gama e da distribuição dos controlos positivos;
|
|
—
|
um historial da gama e da distribuição dos controlos negativos (amostras não tratadas, solvente).
|
|
|
48.
|
Quando se obtêm os primeiros dados para uma distribuição histórica do controlo negativo, os controlos negativos paralelos devem ser coerentes com os dados de controlo negativo publicados. À medida que forem sendo adicionados mais dados experimentais à distribuição dos controlos, os controlos negativos em paralelo devem, de preferência, situar-se dentro dos limites de controlo de 95 % daquela distribuição (64) (65).
|
|
49.
|
A base de dados históricos de controlo negativo deve ser inicialmente constituída por um mínimo de 10 experiências, embora de preferência, seja constituída por, pelo menos, 20 experiências efetuadas em condições comparáveis. Os laboratórios devem utilizar métodos de controlo de qualidade, como gráficos de controlo [p. ex., gráficos C ou gráficos de barras (65)], para identificar a variabilidade dos seus dados de controlo positivo e negativo e demonstrar que, no seu laboratório, a metodologia está sob controlo (66). A referência bibliográfica (64) contém mais informações e recomendações sobre a forma de obter e utilizar os dados históricos.
|
|
50.
|
Os dados de controlo negativo devem consistir em frequências de mutação em culturas únicas ou, de preferência, replicadas, conforme descrito no ponto 27. Os controlos negativos em paralelo devem, de preferência, situar-se dentro dos limites de controlo de 95 % da distribuição da base de dados históricos do laboratório sobre o controlo negativo. Os dados do controlo negativo que se situem fora dos limites de controlo de 95 % podem ser aceitáveis para inclusão no historial de distribuição de controlo se não forem valores extremos, se houver indícios de que o sistema de ensaio está sob controlo (ver ponto 49) e se não houver indícios de erros humanos ou técnicos.
|
|
51.
|
As eventuais alterações ao protocolo experimental devem ser ponderadas em função da coerência dos dados com as bases de dados históricos de controlo do laboratório. As incoerências de monta devem conduzir à constituição de uma nova base de dados históricos de controlo.
|
DADOS E RELATÓRIOS
Apresentação dos resultados
|
52.
|
A apresentação de dados para o MLA e para o TK6 deve incluir, no respeitante às culturas tratadas e de controlo, os dados necessários para o cálculo da citotoxidade (RTG ou RS, respetivamente) e das frequências de mutação, conforme a seguir se descreve.
|
|
53.
|
No caso do MLA, devem ser fornecidos dados de cultura individuais para a RSG, o RTG, a eficiência de clonagem no momento da seleção de mutantes e o número de colónias mutantes (na versão ágar) ou o número de poços vazios (na versão micropoços). A MF deve ser expressa em número de células mutantes por milhão de células sobreviventes. Se a resposta for positiva, devem fornecer-se as MF das pequenas e grandes colónias (e/ou a percentagem da MF total) para, pelo menos, uma concentração do produto químico em estudo (geralmente a concentração positiva mais elevada) e para os controlos negativos e positivos. Em caso de resposta negativa, devem indicar-se as MF das colónias grande e pequena para o controlo negativo e o controlo positivo.
|
|
54.
|
No que diz respeito ao TK6, devem fornecer-se dados de cultura individuais para a RS, a eficiência de clonagem no momento da seleção de mutantes e o número de poços vazios de mutantes precoces e tardios. A MF deve ser expressa em número de células mutantes por número de células sobreviventes, incluindo a MF total e a MF (e/ou a percentagem da MF total) dos mutantes precoces e tardios.
|
Critérios de aceitabilidade
|
55.
|
Antes da determinação dos resultados globais relativos a um dado produto químico em estudo, devem estar cumpridos os seguintes critérios, tanto para o MLA como para o TK6:
|
—
|
Foram ensaiadas duas condições experimentais (tratamento curto com e sem ativação metabólica – ver ponto 33), salvo se uma tiver dado resultados positivos.
|
|
—
|
Deve ser possível analisar um número adequado de células e concentrações (ver pontos 27 e 34-36).
|
|
—
|
Os critérios de seleção da concentração máxima são coerentes com os descritos nos pontos 28 a 30.
|
|
Critérios de aceitabilidade dos controlos negativos e positivos
|
56.
|
A análise de um volume importante de dados pelo grupo de trabalho de peritos do IWGT sobre o MLA resultou num consenso internacional quanto aos critérios de aceitabilidade específicos do MLA (1) (2) (3) (4) (5). Por conseguinte, o presente método de ensaio formula recomendações específicas para determinar a aceitabilidade dos controlos negativos e positivos e para a avaliação dos resultados das várias substâncias no MLA. Para o TK6, a base de dados é muito mais reduzida e não foi avaliada por um grupo de trabalho.
|
|
57.
|
Em relação ao MLA, cada experiência deve ser avaliada de modo a verificar se as amostras de controlo não tratadas e com solvente cumprem os critérios de aceitação do grupo de trabalho do IWGT sobre o MLA [(4) e quadro 2 infra] no respeitante ao seguinte: (1) as MF (note-se que as MF aceitáveis para o IWGT diferem para as versões ágar e de micropoços do MLA), (2) a eficiência de clonagem (CE) no momento do processo da seleção de mutantes e (3) o crescimento das suspensões (SG) para o controlo do solvente (ver fórmulas no apêndice 2).
Quadro 2
Critérios de aceitabilidade para o MLA
|
Parâmetro
|
Método do ágar macio
|
Método de micropoços
|
|
Frequência de mutação
|
35 – 140 × 10–6
|
50 – 170 × 10–6
|
|
Eficiência de clonagem
|
65 – 120 %
|
65 – 120 %
|
|
Crescimento da suspensão
|
8-32 vezes (tratamento de 3 a 4 horas) 32-180 vezes (tratamento de 24 horas, se realizado)
|
8-32 vezes (tratamento de 3 a 4 horas) 32-180 vezes (tratamento de 24 horas, se realizado)
|
|
|
58.
|
No respeitante ao MLA, cada ensaio deve também ser avaliado quanto à questão de saber se o(s) controlo(s) positivo(s) satisfaz(em), pelo menos, um dos dois critérios de aceitação seguintes, definidos pelo grupo de trabalho do IWGT:
|
—
|
O controlo positivo mostra um aumento absoluto da MF total, ou seja, um aumento em relação à MF espontânea [uma MF induzida (IMF)] de, pelo menos, 300 × 10–6. Pelo menos 40 % da IMF deve refletir-se na MF da colónia.
|
|
—
|
O controlo positivo mostra um aumento da MF da colónia pequena de, pelo menos, 150 × 10–6, em relação à observada na amostra de controlo não tratada ou do solvente paralela (uma pequena colónia IMF de 150 × 10–6).
|
|
|
59.
|
No caso do TK6, um ensaio é aceitável se o controlo negativo paralelo for considerado aceitável para adição à base de dados históricos de controlo negativo do laboratório, conforme descrito nos pontos 48 e 49. Além disso, os controlos positivos paralelos (ver ponto 32) devem induzir respostas compatíveis com as geradas na base de dados históricos de controlo positivo e produzir um aumento estatisticamente significativo em relação ao controlo negativo paralelo.
|
|
60.
|
Para ambos os ensaios, o limite superior de citotoxicidade observada na cultura de controlo positivo deve ser idêntico ao das culturas experimentais, ou seja, o RTG/RS não deve ser inferior a 10 %. Basta utilizar uma única concentração (ou uma das concentrações das culturas de controlo positivo, se se utilizar mais do que uma concentração) para demonstrar que os critérios de aceitação para o controlo positivo foram satisfeitos. Além disso, a MF do controlo positivo deve situar-se dentro da gama aceitável estabelecida para o laboratório.
|
Avaliação e interpretação dos resultados
|
61.
|
No que se refere ao MLA, o Mouse Lymphoma Expert Workgroup do IWGT realizou um trabalho significativo sobre a importância biológica e os critérios de uma resposta positiva (4). Por conseguinte, o presente método de ensaio apresenta recomendações específicas para a interpretação dos resultados do produto químico em estudo do MLA (ver pontos 62-64). A base de dados do TK6 é muito mais reduzida e não foi avaliada por um grupo de trabalho. Por conseguinte, as recomendações para a interpretação dos dados para o TK6 são apresentadas em termos mais genéricos (ver pontos 65-66). Existem recomendações adicionais para ambos os ensaios (ver pontos 67-71).
|
MLA
|
62.
|
Recomenda-se a adoção de uma abordagem para definir as respostas positivas e negativas, a fim de assegurar que o número acrescido de MF é biologicamente relevante. Em vez da análise estatística geralmente usada para outros ensaios, o presente baseia-se no recurso a uma frequência de mutação induzida predefinida (ou seja, um aumento da MF acima do controlo paralelo), designada «Fator de Avaliação Global» (GEF), baseada na análise da distribuição do controlo negativo dos dados sobre a MF dos laboratórios participantes (4). Para a versão do ágar do MLA, o GEF é de 90 × 10–6 e para a versão dos micropoços do MLA o GEF é de 126 × 10–6.
|
|
63.
|
Se se verificar que são cumpridos todos os critérios de aceitabilidade, considera-se o produto químico em estudo inequivocamente positivo se, numa das condições de ensaio examinadas (ver ponto 33), o aumento da MF acima dos valores paralelos exceder o GEF e estiver relacionado com a concentração (por exemplo, utilizando uma análise de tendências). O produto químico em estudo é então considerado apto a induzir mutações, no contexto do sistema de ensaio.
|
|
64.
|
Se forem cumpridos todos os critérios de aceitabilidade, considera-se o produto químico em estudo inequivocamente negativo se, em todas as condições de ensaio examinadas (ver ponto 33), não se verificar uma resposta relacionada com a concentração ou, caso se verifique um aumento da MF, este não exceder o GEF. Assim, o produto químico não é considerado passível de induzir mutações, no contexto do sistema de ensaio.
|
TK6
|
65.
|
Se forem cumpridos todos os critérios de aceitabilidade, considera-se o produto químico em estudo inequivocamente positivo se, em qualquer das condições experimentais examinadas (ver ponto 33):
|
—
|
pelo menos uma das concentrações de ensaio apresentar um aumento estatisticamente significativo em relação ao controlo negativo,
|
|
—
|
o aumento, avaliado com base numa análise de tendências adequada, depender da concentração (ver ponto 33),
|
|
—
|
nenhum dos resultados estiver fora da distribuição dos dados históricos de controlo negativo (p. ex., limites de controlo de 95 % com base numa distribuição de Poisson; ver ponto 48).
|
Se todos estes critérios forem preenchidos, considera-se o produto químico em estudo passível de induzir mutações no âmbito do presente sistema de ensaio. As referências bibliográficas (66) (67) contêm recomendações sobre os métodos estatísticos mais adequados.
|
|
66.
|
Se forem cumpridos todos os critérios de aceitabilidade, o produto químico em estudo é considerado inequivocamente negativo se, em todas as condições experimentais examinadas (ver ponto 33):
|
—
|
nenhuma das concentrações de ensaio apresentar um aumento estatisticamente significativo em relação ao controlo negativo,
|
|
—
|
não se observar qualquer aumento dependente da dose, numa avaliação feita com base numa análise de tendências adequada.
|
|
—
|
todos os resultados se situarem dentro da distribuição dos dados históricos de controlo negativo (p. ex., limites de controlo de 95 % com base numa distribuição de Poisson; ver ponto 48).
|
Nesse caso, considera-se que o produto químico não é passível de induzir mutações, no contexto do sistema de ensaio.
|
Para o MLA e o TK6:
|
67.
|
Se a concentração máxima se basear na citotoxicidade, a concentração mais elevada deverá estar entre 20 % e 10 % do RTG/RS. É consensual que se deve ter cuidado ao interpretar os resultados positivos apenas entre 20 % e 10 % do RTG/RS e que um resultado não deve ser considerado positivo se o aumento da MF ocorrer apenas a 10 % do RTG/RS ou abaixo deste valor (se avaliado) (2) (59).
|
|
68.
|
Em alguns casos, certas informações adicionais podem ajudar a determinar que um produto químico em estudo não é mutagénico, se não existir nenhuma cultura com um valor de RTG compreendido entre 10 e 20 % do RTG/RS. Estas situações definem-se do seguinte modo: (1) não há provas de mutagenicidade (por exemplo, inexistência de resposta às doses, de frequências de mutação acima das observadas nas gamas de controlo negativo paralelas ou históricas, etc.) numa série de pontos de dados compreendida entre 100 % e 20 % do RTG/RS, observando-se, pelo menos, um ponto de dados entre 20 % e 25 % do RTG/RS; (2) não há provas de mutagenicidade (por exemplo, iexistência de resposta às doses, de frequências de mutação acima das observadas nas gamas de controlo negativo paralelas ou históricas, etc.) numa série de pontos de dados entre 100 % e 25 % do RTG/RS, observando-se um ponto negativo ligeiramente inferior a 10 % do RTG/RS. Em ambas as situações, o produto químico em estudo pode ser considerado negativo.
|
|
69.
|
Não há nenhuma exigência concreta para a verificação de uma resposta inequivocamente positiva ou negativa.
|
|
70.
|
No caso de a resposta não ser inequivocamente positiva nem inequivocamente negativa, como se descreveu, e/ou para se tirarem conclusões quanto à importância biológica do resultado, os dados devem ser avaliados por peritos e/ou ser objeto de estudos complementares. Pode ser útil proceder a um ensaio de repetição, eventualmente alterando as condições experimentais (intervalo diferente entre as concentrações – para aumentar a probabilidade de atingir pontos de dados entre 10 e 20 % do RTG/RS –, condições de ativação metabólica diferentes – p. ex., concentração ou origem do S9 – e duração diferente do tratamento).
|
|
71.
|
Em casos raros, mesmo após a realização de estudos complementares, os dados obtidos não permitem concluir por um resultado positivo ou negativo. Nessas circunstâncias, deve concluir-se que a resposta do produto químico em estudo é ambígua (interpretada como igualmente suscetível de ser positiva ou negativa).
|
RELATÓRIO DE ENSAIO
|
72.
|
O relatório do ensaio deve incluir as seguintes informações:
|
|
Produto químico em estudo:
|
—
|
origem, número de lote e data-limite de utilização, se conhecidos;
|
|
—
|
estabilidade do produto químico em estudo, se conhecida;
|
|
—
|
solubilidade e estabilidade do produto químico em estudo no solvente, se conhecidas;
|
|
—
|
medição do pH, da pressão osmótica e da quantidade de precipitado no meio de cultura ao qual foi adicionado o produto químico em estudo, consoante o que for adequado.
|
|
|
Substância monocomponente:
|
—
|
aspeto físico, hidrossolubilidade e outras propriedades físico-químicas pertinentes;
|
|
—
|
dados de identificação química, como as denominações IUPAC ou CAS, número CAS, código SMILES ou InChI, fórmula estrutural, pureza, identidade química das impurezas, se justificado e exequível, etc.
|
|
|
|
Substâncias multicomponentes, UVCB e misturas:
|
—
|
caracterizada, na medida do possível, pela identidade química (ver acima), ocorrência quantitativa e principais propriedades físico-químicas dos componentes.
|
|
|
|
|
Solvente:
|
—
|
justificação da escolha do solvente;
|
|
—
|
percentagem de solvente no meio de cultura final.
|
|
|
|
Células:
|
|
Para as culturas principais de laboratório:
|
—
|
tipo e origem das células e antecedentes no laboratório de ensaio;
|
|
—
|
características do cariótipo e/ou número modal de cromossomas.
|
|
—
|
métodos de manutenção das culturas celulares;
|
|
—
|
ausência de micoplasma;
|
|
—
|
tempo de duplicação celular.
|
|
|
|
|
Condições de realização do ensaio:
|
—
|
justificação da escolha das concentrações e do número de culturas de células, incluindo, por exemplo, dados sobre a citotoxicidade e sobre as limitações de solubilidade;
|
|
—
|
composição dos meios, concentração de CO2, nível de humidade;
|
|
—
|
concentração do produto químico em estudo, expressa em concentração final no meio de cultura (p. ex., μg ou mg/ml ou mM do meio de cultura);
|
|
—
|
concentração (e/ou volume) de solvente e de produto químico em estudo adicionada ao meio de cultura;
|
|
—
|
temperatura de incubação;
|
|
—
|
densidade celular durante a exposição;
|
|
—
|
tipo e composição do sistema de ativação metabólica (fonte de S9, método de preparação da mistura de S9, concentração ou volume da mistura de S9 e de S9 no meio de cultura final, controlos de qualidade do S9);
|
|
—
|
substâncias de controlo positivo e negativo, concentrações finais para cada condição de tratamento;
|
|
—
|
duração do período de expressão (incluindo o número de células inoculadas e os calendários de subcultura e de alimentação, quando aplicável),
|
|
—
|
identidade do agente seletivo e respetiva concentração;
|
|
—
|
para o MLA, indicar a versão utilizada (ágar ou micropoço).
|
|
—
|
critérios de aceitabilidade dos ensaios;
|
|
—
|
métodos utilizados para a contagem das células mutantes e viáveis,
|
|
—
|
métodos de medição da citotoxicidade;
|
|
—
|
outros dados pertinentes relativos à citotoxicidade e ao método utilizado;
|
|
—
|
duração dos períodos de incubação após a colocação em placas;
|
|
—
|
definição das colónias cuja dimensão e tipo são caracterizados (incluindo os critérios para a definição de colónias «pequenas» e «grandes», conforme apropriado).
|
|
—
|
critérios para que o estudo seja considerado positivo, negativo ou inconclusivo;
|
|
—
|
métodos utilizados para determinar o pH, a pressão osmótica e a precipitação, se pertinente.
|
|
|
|
Resultados:
|
—
|
número de células expostas e número de células subcultivadas para cada cultura;
|
|
—
|
parâmetros de toxicidade (RTG para o MLA e RS para o TK6);
|
|
—
|
sinais de precipitação e instante da determinação;
|
|
—
|
número de células colocadas em placas em meio seletivo e não seletivo;
|
|
—
|
número de colónias em meio não seletivo, número de colónias resistentes em meio seletivo e frequências de mutação conexas;
|
|
—
|
definição do tamanho das colónias para os controlos negativos e positivos e, se o produto químico em estudo for positivo, pelo menos uma concentração e as frequências de mutação afins;
|
|
—
|
relação concentração-resposta, quando for possível determiná-la;
|
|
—
|
dados relativos aos controlos negativos (solvente) e controlos positivos (concentrações e solventes) realizados em paralelo;
|
|
—
|
dados históricos sobre o controlo negativo (solvente) e positivo (concentrações e solventes), com as correspondentes gamas, médias e desvios-padrão; número de ensaios em que se baseiam os controlos históricos;
|
|
—
|
análises estatísticas (para culturas individuais e replicados agrupados, se for caso disso) e valores p, caso existam; para o MLA, a avaliação GEF.
|
|
|
REFERÊNCIAS BIBLIOGRÁFICAS
|
(1)
|
Moore, M.M., Honma, M. Clements, J. (Rapporteur), Awogi, T., Bolcsfoldi, G., Cole, J., Gollapudi, B., Harrington-Brock, K., Mitchell, A., Muster, W., Myhr, B., O'Donovan, M., Ouldelhkim, M-C., San, R., Shimada, H. and Stankowski, L.F. Jr. (2000). Mouse Lymphoma Thymidine Kinase Locus (TK) Gene Mutation Assay: International Workshop on Genotoxicity Test Procedures (IWGTP) Workgroup Report, Environ. Mol. Mutagen., 35 (3): 185-190.
|
|
(2)
|
Moore, M.M., Honma, M., Clements, J., Harrington-Brock, K., Awogi, T., Bolcsfoldi, G., Cifone, M., Collard, D., Fellows, M., Flanders, K., Gollapudi, B., Jenkinson, P., Kirby, P., Kirchner, S., Kraycer, J., McEnaney, S., Muster, W., Myhr, B., O’Donovan, M., Oliver, Ouldelhkim, M-C., Pant, K., Preston, R., Riach, C., San, R., Shimada, H. and Stankowski, L.F. Jr. (2002). Mouse Lymphoma Thymidine Kinase Locus Gene Mutation Assay: Follow-Up International Workshop on Genotoxicity Test Procedures, New Orleans, Louisiana, (April 2000), Environ. Mol. Mutagen., 40 (4): 292-299.
|
|
(3)
|
Moore, M.M., Honma, M., Clements, J., Bolcsfoldi, G., Cifone, M., Delongchamp, R., Fellows, M., Gollapudi, B., Jenkinson, P., Kirby, P., Kirchner, S., Muster, W., Myhr, B., O’Donovan, M., Ouldelhkim, M-C., Pant, K., Preston, R., Riach, C., San, R., Stankowski, L.F. Jr., Thakur, A., Wakuri, S. and Yoshimura, I. (2003). Mouse Lymphoma Thymidine Kinase Locus Gene Mutation Assay: International Workshop (Plymouth, UK) on Genotoxicity Test Procedures Workgroup Report, Mutation Res., 540: 127-140.
|
|
(4)
|
Moore, M.M., Honma, M., Clements, J., Bolcsfoldi, G., Burlinson, B., Cifone, M., Clarke, J., Delongchamp, R., Durward, R., Fellows, M., Gollapudi, B., Hou, S., Jenkinson, P., Lloyd, M., Majeska, J., Myhr, B., O’Donovan, M., Omori, T., Riach, C., San, R., Stankowski, L.F. Jr., Thakur, A.K., Van Goethem, F., Wakuri, S. and Yoshimura, I. (2006). Mouse Lymphoma Thymidine Kinase Gene Mutation Assay: Follow-Up Meeting of the International Workshop on Genotoxicity Tests – Aberdeen, Scotland, 2003 – Assay Acceptance Criteria, Positive Controls, and Data Evaluation, Environ. Mol. Mutagen., 47 (1): 1-5.
|
|
(5)
|
Moore, M.M., Honma, M., Clements, J., Bolcsfoldi, G., Burlinson, B., Cifone, M., Clarke, J., Clay, P., Doppalapudi, R., Fellows, M., Gollapudi, B., Hou, S., Jenkinson, P., Muster, W., Pant, K., Kidd, D.A., Lorge, E., Lloyd, M., Myhr, B., O’Donovan, M., Riach, C., Stankowski, L.F. Jr., Thakur A.K. and Van Goethem, F. (2007). Mouse Lymphoma Thymidine Kinase Mutation Assay: Meeting of the International Workshop on Genotoxicity Testing, San Francisco, 2005, Recommendations for 24-h Treatment, Mutation. Res., 627 (1): 36-40.
|
|
(6)
|
OCDE (2016). Overview of the set of OECD Genetic Toxicology Test Guidelines and updates performed in 2014-2015. ENV Publications. Series on Testing and Assessment, No 234, OECD, Paris.
|
|
(7)
|
Fellows M.D., Luker, T., Cooper, A. and O'Donovan, M.R. (2012). Unusual Structure-Genotoxicity Relationship in Mouse Lymphoma Cells Observed with a Series of Kinase Inhibitors. Mutation, Res., 746 (1): 21-28.
|
|
(8)
|
Honma, M., Momose, M., Sakamoto, H., Sofuni, T. and Hayashi, M. (2001). Spindol Poisons Induce Allelic Loss in Mouse Lymphoma Cells Through Mitotic Non-Disjunction. Mutation Res., 493 (1-2): 101-114.
|
|
(9)
|
Wang, J., Sawyer, J.R., Chen, L., Chen, T., Honma, M., Mei, N. and Moore, M.M. (2009). The Mouse Lymphoma Assay Detects Recombination, Deletion, and Aneuploidy, Toxicol. Sci., 109 (1): 96-105.
|
|
(10)
|
Applegate, M.L., Moore, M.M., Broder, C.B., Burrell, A., and Hozier, J.C. (1990). Molecular Dissection of Mutations at the Heterozygous Thymidine Kinase Locus in Mouse Lymphoma Cells. Proc. National. Academy. J. Environ. Sci. USA, 87 (1): 51-55.
|
|
(11)
|
Hozier, J., Sawyer, J., Moore, M., Howard, B. and Clive, D. (1981). Cytogenetic Analysis of the L5178Y/TK+/– Leads to TK–/– Mouse Lymphoma Mutagenesis Assay System, Mutation Res., 84 (1): 169-181.
|
|
(12)
|
Hozier, J., Sawyer, J., Clive, D. and Moore, M.M. (1985). Chromosome 11 Aberrations in Small Colony L5178Y TK–/– Mutants Early in their Clonal History, Mutation Res., 147 (5): 237-242.
|
|
(13)
|
Moore, M.M., Clive, D., Hozier, J.C., Howard, B.E., Batson, A.G., Turner, N.T. and Sawyer, J. (1985). Analysis of Trifluorothymidine-Resistant (TFTr) Mutants of L5178Y/TK+/– Mouse Lymphoma Cells. Mutation Res., 151 (1): 161-174.
|
|
(14)
|
Liber H.L., Call K.M. and Little J.B. (1987). Molecular and Biochemical Analyses of Spontaneous and X-Ray-Induced Mutants in Human Lymphoblastoid Cells. Mutation Res., 178 (1): 143-153.
|
|
(15)
|
Li C.Y., Yandell D.W. and Little J.B. (1992). Molecular Mechanisms of Spontaneous and Induced Loss of Heterozygosity in Human Cells In Vitro. Somat. Cell Mol. Genet., 18 (1): 77-87.
|
|
(16)
|
Honma M., Hayashi M. and Sofuni T. (1997). Cytotoxic and Mutagenic Responses to X-Rays and Chemical Mutagens in Normal and P53-Mutated Human Lymphoblastoid Cells. Mutation. Res., 374 (1): 89-98.
|
|
(17)
|
Honma, M., Momose, M., Tanabe, H., Sakamoto, H., Yu, Y., Little, J.B., Sofuni, T. and Hayashi, M. (2000). Requirement of Wild-Type P53 Protein for Maintenance of Chromosomal Integrity. Mol. Carcinogen., 28 (4): 203-14.
|
|
(18)
|
Amundson S.A. and Liber H.L. (1992). A Comparison of Induced Mutation at Homologous Alleles of the TK Locus in Human Cells. II. Molecular Analysis of Mutants. Mutation Res., 267 (1): 89-95.
|
|
(19)
|
Schisler M.R., Moore M.M. and Gollapudi B.B. (2013). In Vitro Mouse Lymphoma (L5178Y TK+/– –3.7.2C) Forward Mutation Assay. In Protocols in Genotoxicity Assessment A. Dhawan and M. Bajpayee (Eds.), Springer Protocols, Humana Press: 27-50.
|
|
(20)
|
Long, L.H., Kirkland, D., Whitwell, J. and Halliwell, B. (2007). Different Cytotoxic and Clastogenic Effects of Epigallocatechin Gallate in Various Cell-Culture Media Due to Variable Rates of its Oxidation in the Culture Medium, Mutation Res., 634 (1-2): 177-183.
|
|
(21)
|
Nesslany, F., Simar-Meintieres, S., Watzinger, M., Talahari, I. and Marzin, D. (2008). Characterization of the Genotoxicity of Nitrilotriacetic Acid. Environ. Mol. Mutagen., 49 (6): 439-452.
|
|
(22)
|
Brusick D. (1986). Genotoxic Effects in Cultured Mammalian Cells Produced by Low pH Treatment Conditions and Increased Ion Concentrations. Environ. Mutagen., 8 (6): 879-886.
|
|
(23)
|
Morita, T., Nagaki, T., Fukuda, I. and Okumura, K. (1992). Clastogenicity of Low pH to Various Cultured Mammalian Cells. Mutation Res., 268 (2): 297-305.
|
|
(24)
|
Scott, D., Galloway, S.M., Marshall, R.R., Ishidate, M.Jr, Brusick, D., Ashby, J. and Myhr, B.C. (1991). Genotoxicity under Extreme Culture Conditions. A report from ICPEMC Task Group 9. Mutation Res., 257: 147-204.
|
|
(25)
|
Wang J., Heflich R.H. and Moore M.M. (2007). A Method to Distinguish Between the De Novo Induction of Thymidine Kinase Mutants and the Selection of Pre-Existing Thymidine Kinase Mutants in the Mouse Lymphoma Assay. Mutation Res., 626 (1-2): 185-190.
|
|
(26)
|
Fischer, G.A. (1958). Studies on the Culture of Leukemic Cells In Vitro. Ann. N.Y. Academy Sci., 76: 673-680.
|
|
(27)
|
Clive, D., Johnson, K.O., Spector, J.F.S., Batson, A.G. and Brown, M.M.M. (1979). Validation and Characterization of the L5178Y/TK+/– Mouse Lymphoma Mutagen Assay System. Mutation Res., 59(1): 61-108.
|
|
(28)
|
Sawyer, J., Moore, M.M., Clive, D. and Hozier, J. (1985). Cytogenetic Characterization of the L5178Y TK+/– 3.7.2C Mouse Lymphoma Cell Line, Mutation Res., 147 (5): 243-253.
|
|
(29)
|
Sawyer J.R., Moore M.M. and Hozier J.C. (1989). High-Resolution Cytogenetic Characterization of the L5178Y TK+/– Mouse Lymphoma Cell Line, Mutation Res., 214 (2): 181-193.
|
|
(30)
|
Sawyer, J.R., Binz, R.L., Wang, J. and Moore, M.M. (2006). Multicolor Spectral Karyotyping of the L5178Y TK+/––3.7.2C Mouse Lymphoma Cell Line, Environ. Mol. Mutagen., 47 (2): 127-131.
|
|
(31)
|
Fellows, M.D., McDermott, A., Clare, K.R., Doherty, A. and Aardema, M.J. (2014). The Spectral Karyotype of L5178Y TK+/– Mouse Lymphoma Cells Clone 3.7.2C and Factors Affecting Mutant Frequency at the Thymidine Kinase (TK) Locus in the Microtitre Mouse Lymphoma Assay, Environ. Mol. Mutagen., 55 (1): 35-42.
|
|
(32)
|
Storer, R.D., Jraynak, A.R., McKelvey, T.W., Elia, M.C., Goodrow, T.L. and DeLuca, J.G. (1997). The Mouse Lymphoma L5178Y TK+/– Cell Line is Heterozygous for a Codon 170 Mutation in the P53 Tumor Suppressor Gene. Mutation. Res., 373 (2): 157-165.
|
|
(33)
|
Clark L.S., Harrington-Brock, K., Wang, J., Sargent, L., Lowry, D., Reynolds, S.H. and Moore, M.M. (2004). Loss of P53 Heterozygosity is not Responsible for the Small Colony Thymidine Kinase Mutant Phenotype in L5178Y Mouse Lymphoma Cells. Mutagen., 19 (4): 263-268.
|
|
(34)
|
Skopek T.R., Liber, H.L., Penman, B.W. and Thilly, W.G. (1978). Isolation of a Human Lymphoblastoid Line Heterozygous at the Thymidine Kinase Locus: Possibility for a Rapid Human Cell Mutation Assay. Biochem. Biophys. Res. Commun., 84 (2): 411-416.
|
|
(35)
|
Honma M. (2005). Generation of Loss of Heterozygosity and its Dependency on P53 Status in Human Lymphoblastoid Cells. Environ. Mol. Mutagen., 45 (2-3): 162-176.
|
|
(36)
|
Xia, F., Wang, X., Wang, Y.H., Tsang, N.M., Yandell, D.W., Kelsey, K.T. and Liber, H.L. (1995). Altered P53 Status Correlates with Differences in Sensitivity to Radiation-Induced Mutation and Apoptosis in Two Closely Related Human Lymphoblast Lines. Cancer. Res., 55 (1): 12-15.
|
|
(37)
|
Lorge, E., M. Moore, J. Clements, M. O Donovan, M. Honma, A. Kohara, J. van Benthem, S. Galloway, M.J. Armstrong, V. Thybaud, B. Gollapudi, M. Aardema, J. Kim, A. Sutter, D.J. Kirkland (2015). Standardized Cell Sources and Recommendations for Good Cell Culture Practices in Genotoxicity Testing. (Manuscrito em preparação).
|
|
(38)
|
Lloyd M. and Kidd D. (2012). The Mouse Lymphoma Assay. Springer Protocols: Methods in Molecular Biology 817, Genetic Toxicology Principles and Methods, ed. Parry and Parry, Humana Press. ISBN, 978-1-61779-420-9, 35-54.
|
|
(39)
|
Mei N., Guo X. and Moore M.M. (2014). Methods for Using the Mouse Lymphoma Assay to Screen for Chemical Mutagenicity and Photo-Mutagenicity. In: Optimization in Drug Discover: In Vitro Methods: Yan Z and Caldwell(Eds), 2nd Edition, GW; Humana Press, Totowa, NJ.
|
|
(40)
|
Liber H.L. and Thilly W.G. (1982). Mutation Assay at the Thymidine Kinase Locus in Diploidhuman Lymphoblasts. Mutation Res., 94 (2): 467-485.
|
|
(41)
|
Coecke, S., Balls, M., Bowe, G., Davis, J., Gstraunthaler, G., Hartung, T., Hay, R., Merten, OW., Price, A., Schechtman, L., Stacey, G. and Stokes, W. (2005). Guidance on Good Cell Culture Practice. A Report of the Second ECVAM Task Force on Good Cell Culture Practice. ATLA, 33 (3): 261-287.
|
|
(42)
|
Moore M.M. and Howard B.E. (1982). Quantitation of Small Colony Trifluorothymidine-Resistant Mutants of L5178Y/TK+/- Mouse Lymphoma Cells in RPMI-1640 Medium, Mutation Res., 104 (4-5): 287-294.
|
|
(43)
|
Ames B.N., McCann J. and Yamasaki E. (1975). Methods for Detecting Carcinogens and Mutagens with the Salmonella/Mammalian Microsome Mutagenicity Test. Mutation Res., 31 (6): 347-364.
|
|
(44)
|
Maron D.M. and Ames B.N. (1983). Revised Methods for the Salmonella Mutagenicity Test. Mutation Res., 113 (3-4): 173-215.
|
|
(45)
|
Natarajan, A.T., Tates, A.D, Van Buul, P.P.W., Meijers, M. and De Vogel, N. (1976). Cytogenetic Effects of Mutagens/Carcinogens After Activation in a Microsomal System In Vitro, I. Induction of Chromosomal Aberrations and Sister Chromatid Exchanges by Diethylnitrosamine (DEN) and Dimethylnitrosamine (DMN) in CHO Cells in the Presence of Rat-Liver Microsomes. Mutation Res., 37 (1): 83-90.
|
|
(46)
|
Matsuoka A., Hayashi M. and Ishidate M. Jr. (1979). Chromosomal Aberration Tests on 29 Chemicals Combined with S9 Mix In Vitro. Mutation Res., 66 (3): 277-290.
|
|
(47)
|
Ong T.M., et al. (1980). Differential Effects of Cytochrome P450-Inducers on Promutagen Activation Capabilities and Enzymatic Activities of S-9 from Rat Liver, J. Environ. Pathol. Toxicol., 4 (1): 55-65
|
|
(48)
|
Elliott, B.M., Combes, R.D., Elcombe, C.R., Gatehouse, D.G., Gibson, G.G., Mackay, J.M. and Wolf, R.C. (1992). Report of UK Environmental Mutagen Society Working Party. Alternatives to Aroclor 1254-Induced S9 in In Vitro Genotoxicity Assays. Mutagen., 7 (3): 175-177.
|
|
(49)
|
Matsushima, T., Sawamura, M., Hara, K. and Sugimura, T. (1976). A Safe Substitute for Polychlorinated Biphenyls as an Inducer of Metabolic Activation Systems. In: In Vitro Metabolic Activation in Mutagenesis Testing. de Serres F.J., et al. (Eds, Elsevier, North-Holland, pp. 85-88.
|
|
(50)
|
Galloway S.M., et al. (1994). Report from Working Group on In Vitro Tests for Chromosomal Aberrations. Mutation Res., 312 (3): 241-261.
|
|
(51)
|
Johnson T.E., Umbenhauer D.R. and Galloway S.M. (1996). Human Liver S-9 Metabolic Activation: Proficiency in Cytogenetic Assays and Comparison with Phenobarbital/Beta-Naphthoflavone or Aroclor 1254 Induced Rat S-9, Environ. Mol. Mutagen., 28 (1): 51-59.
|
|
(52)
|
UNEP (2001). Stockholm Convention on Persistent Organic Pollutants, United Nations Environment Programme (UNEP).
|
|
(53)
|
Krahn D.F., Barsky F.C. and McCooey K.T. (1982). CHO/HGPRT Mutation Assay: Evaluation of Gases and Volatile Liquids. In: Genotoxic Effects of Airborne Agents Tice R.R., Costa D.L.and Schaich K.M. (Eds.). New York, Plenum, pp. 91-103.
|
|
(54)
|
Zamora, P.O., Benson, J.M., Li, A.P. and Brooks, A.L. (1983). Evaluation of an Exposure System Using Cells Grown on Collagen Gels for Detecting Highly Volatile Mutagens in the CHO/HGPRT Mutation Assay. Environ. Mutagen., 5 (6): 795-801.
|
|
(55)
|
Asakura M., Sasaki T., Sugiyama T., Arito H., Fukushima, S. and Matsushima, T. (2008). An Improved System for Exposure of Cultured Mammalian Cells to Gaseous Compounds in the Chromosomal Aberration Assay. Mutation Res., 652 (2): 122-130.
|
|
(56)
|
Arlett C.F., et al. (1989). Mammalian Cell Gene Mutation Assays Based upon Colony Formation. In: Statistical Evaluation of Mutagenicity Test Data, Kirkland, D.J. (Ed.), CambridgeUniversity Press, pp. 66-101.
|
|
(57)
|
Morita T., Honma M. and Morikawa K. (2012). Effect of Reducing the Top Concentration Used in the In Vitro Chromosomal Aberration Test in CHL Cells on the Evaluation of Industrial Chemical Genotoxicity. Mutation Res., 741 (1-2): 32-56.
|
|
(58)
|
Brookmire L., Chen J.J. and Levy D.D. (2013). Evaluation of the Highest Concentrations Used in the In Vitro Chromosome Aberrations Assay. Environ. Mol. Mutagen., 54 (1): 36-43.
|
|
(59)
|
USFDA (2012). International Conference on Harmonisation (ICH) Guidance S2 (R1) on Genotoxicity Testing and Data Interpretation for Pharmaceuticals Intended For Human Use. Disponível em: [https://www.federalregister.gov/a/2012-13774].
|
|
(60)
|
Honma M. and Hayashi M. (2011). Comparison of In Vitro Micronucleus and Gene Mutation Assay Results for P53-Competent Versus P53-Deficient Human Lymphoblastoid Cells. Environ. Mol. Mutagen., 52 (5): 373-384.
|
|
(61)
|
Moore-Brown, M.M., Clive, D., Howard, B.E., Batson, A.G. and Johnson, K.O. (1981). The Utilization of Trifluorothymidine (TFT) to Select for Thymidine Kinase-Deficient (TK–/–) Mutants from L5178Y/TK+/– Mouse Lymphoma Cells, Mutation Res., 85 (5): 363-378.
|
|
(62)
|
Liber H.L., Yandell D.W. and Little J.B. (1989). A Comparison of Mutation Induction at the TK and HRPT Loci in Human Lymphoblastoid Cells; Quantitative Differences are Due to an Additional Class of Mutations at the Autosomal TK locus. Mutation Res., 216 (1): 9-17.
|
|
(63)
|
Furth E.E., Thilly, W.G., Penman, B.W., Liber, H.L. and Rand, W.M. (1981). Quantitative Assay for Mutation in Diploid Human Lymphoblasts Using Microtiter Plates. Anal. Biochem., 110 (1): 1-8.
|
|
(64)
|
Hayashi, M, Dearfield, K., Kasper, P., Lovell, D., Martus, H. J. and Thybaud, V. (2011). Compilation and Use of Genetic Toxicity Historical Control Data, Mutation Res., 723 (2): 87-90.
|
|
(65)
|
Ryan T.P. (2000). Statistical Methods for Quality Improvement. John Wiley and Sons, New York 2nd Edition.
|
|
(66)
|
OCDE (2014). Statistical analysis supporting the revision of the genotoxicity Test Guidelines. Environmental, Health and Safety Publications, Series on Testing and Assessment (No 199.), Organisation for Economic Cooperation and Development, Paris.
|
|
(67)
|
Fleiss J.L., Levin B. and Paik M.C. (2003). Statistical Methods for Rates and Proportions, Third Edition, New York: John Wiley & Sons.
|
Apêndice 1
DEFINIÇÕES
Amostra de controlo não tratada
: Cultura não sujeita a qualquer tratamento (com o produto químico em estudo ou solvente), mas processada do mesmo modo que as culturas tratadas com o produto químico em estudo.
Aneugénio
: Produto ou processo químico que, por interação com os componentes do aparelho do ciclo mitótico e meiótico de divisão celular, origina aneuploidia em células ou organismos.
Aneuploidia
: Desvio, num único ou em mais cromossomas, mas não por séries completas de cromossomas (poliploidia), do número diploide (ou haploide) normal de cromossomas.
Citotoxicidade
: No âmbito dos ensaios abrangidos pelo presente método de ensaio, a citotoxicidade é identificada como uma redução do crescimento relativo total (RTG) ou da sobrevivência relativa (RS) para, respetivamente, o MLA e o TK6.
Clastogénio
: Produto ou processo químico que provoca aberrações cromossómicas estruturais em populações de células ou organismos.
Crescimento da suspensão (SG)
: Aumento do número de células durante as fases de tratamento e expressão do MLA. O SG é calculado multiplicando o aumento registado no dia 1, pelo aumento do dia 2 para o tratamento curto (3-4 horas). Se for utilizado um tratamento de 24 horas, o SG é o aumento durante o tratamento de 24 horas multiplicado pelos aumentos dos dias 1 e 2.
Crescimento relativo em suspensão (RSG)
: No caso do MLA, crescimento total relativo da cultura após dois dias em suspensão em comparação com o crescimento total do controlo negativo/de solvente em dois dias (Clive e Spector, 1975). O RSG deve incluir o crescimento relativo da cultura de ensaio em relação ao controlo negativo/de solvente durante o período de exposição.
Crescimento total relativo (RTG)
: Utilizado como medida da citotoxicidade do MLA decorrente do tratamento. É uma medida do crescimento relativo (em relação ao controlo do veículo) das culturas de ensaio durante as fases de tratamento, dois dias de expressão, seleção e clonagem de mutantes do ensaio. O RSG de cada cultura de ensaio é multiplicado pela eficiência relativa de clonagem da cultura de ensaio no momento da seleção de mutantes e expresso em relação à eficiência de clonagem da amostra de controlo negativa/solvente (Clive e Spector, 1975).
Controlo do solvente
: Termo geral que define as culturas de controlo tratadas apenas com o solvente utilizado para dissolver o produto químico em estudo.
Eficiência de clonagem
: Percentagem de células incubadas em placas em baixa densidade que consegue originar uma colónia que pode ser contada.
Frações de fígado S9
: Sobrenadante após centrifugação a 9 000 g do homogeneizado hepático (extrato de fígado em bruto).
Frequência de mutação (MF)
: Relação entre o número de células mutantes observadas e o número de células viáveis.
Genotóxico
: Termo geral que abrange todos os tipos de danos ao ADN ou aos cromossomas, incluindo quebra do ADN, aduções, rearranjos, mutações, aberrações cromossómicas e aneuploidia. Nem todos os tipos de efeitos genotóxicos originam mutações ou danos cromossómicos estáveis.
Mistura S9
: Mistura da fração de fígado S9 e dos cofatores necessários à atividade enzimática metabólica.
Mutação para diante
: Uma mutação genética do tipo parental para a forma mutante que causa uma alteração ou perda de atividade enzimática da função da proteína codificada.
Mutagéneos por deslocação do quadro de leitura
: Produtos químicos que causam a adição ou supressão de um par de bases ou de uma sequência de pares de bases do ADN.
Mutagéneos por substituição de um par de bases
: Produtos químicos que causam a substituição de pares de bases do ADN.
Mutagénico
: Que produz uma alteração hereditária de sequências de pares de bases do ADN em genes ou da estrutura dos cromossomas (aberrações cromossómicas).
Período de expressão fenotípica
: Período após o tratamento durante o qual a modificação genética é fixada no genoma e os produtos dos genes inalterados se esgotam ao ponto de alterar o caráter fenotípico.
Produto químico
: uma substância ou mistura.
Produto químico em estudo
: Qualquer substância ou mistura à qual seja aplicado o presente método de ensaio.
Recombinação mitótica
: Recombinação entre cromatídeos homólogos durante a mitose, eventualmente resultante na indução de quebras em cadeias duplas do ADN ou numa perda de heterozigotia.
Sobrevivência relativa (RS)
: Utilizada para medir a citotoxicidade do TK6 decorrente da exposição. Consiste na eficiência de clonagem (CE) relativa de células colocadas em placas imediatamente após o tratamento, ajustada por qualquer perda de células durante o tratamento, em comparação com a eficiência de clonagem do controlo negativo.
Apêndice 2
FÓRMULAS
Citotoxicidade
Para ambas as versões do MLA (ágar e micropoço)
A citotoxicidade é definida como o crescimento total relativo (RTG), que abrange o crescimento relativo em suspensão (RSG) durante o período de expressão de 2 dias e a eficiência de clonagem relativa (RCE) obtida no momento da seleção de mutantes. O RTG, a RSG e a RCE são expressos em percentagem.
Cálculo da RSG: O crescimento em suspensão 1 (SG1) é a taxa de crescimento entre o dia 0 e o dia 1 (concentração celular no dia 1 / concentração celular no dia 0); o crescimento em suspensão dois (SG2) é a taxa de crescimento entre o dia 1 e o dia 2 (concentração celular no dia 2 / concentração celular no dia 1). O RSG é o SG total (SG1 × SG2) da cultura tratada em relação ao controlo sem tratamento/do solvente. Ou seja: RSG = [SG1(ensaio) × SG2(ensaio)] / [SG1(controlo) × SG2(controlo)] O SG1 deve ser calculado a partir da concentração inicial de células utilizada no início do tratamento celular. Quantifica qualquer citotoxicidade diferencial que ocorra na(s) cultura(s) de ensaio durante o tratamento das células.
A RCE é a eficiência relativa de clonagem da cultura de ensaio comparada com a eficiência relativa de clonagem da amostra de controlo não tratada/de solvente obtida no momento da seleção de mutantes.
Crescimento total relativo (RTG):
RTG=RSG × RCE
TK6
Sobrevivência relativa (RS):
A citotoxicidade é avaliada pela sobrevivência relativa, ou seja, a eficiência de clonagem (CE) de células colocadas em placas imediatamente após o tratamento, ajustada por eventuais perdas de células durante este, em comparação com a eficiência de clonagem nos controlos negativos (sobrevivência de 100 %). O ajustamento pela perda de células durante o tratamento é calculado do seguinte modo:
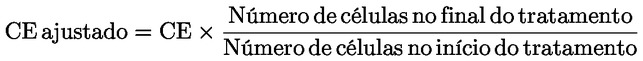
A RS de uma cultura tratada pelo produto químico em estudo é calculada do seguinte modo:

Frequência de mutação para o MLA e o ML6
A frequência de mutação (MF) é a eficiência de clonagem de colónias mutantes em meio seletivo (CEM) ajustada pela eficiência de clonagem em meio não seletivo no momento da seleção de mutantes (CEV). Ou seja, MF=CEM/CEV. O cálculo destas duas eficiências de clonagem para os métodos de clonagem do ágar e dos micropoços é descrito a seguir.
MLA versão ágar:
Na versão de ágar macio do MLA, o número de colónias na placa de seleção de mutantes (CM) e o número de colónias na placa de não seleção ou de eficiência da clonagem (contagem viável) (CV) são obtidos através da contagem direta dos clones. Quando houver 600 células em placas para eficiência de clonagem (CE) para a seleção de mutantes (CEM) e as placas não selecionadas ou de eficiência de clonagem (contagem viável) (CEV) e 3 × 106 células forem utilizadas para a seleção de mutantes,
CEM = CM / (3 × 106) = (CM / 3) × 10-6
CEV = CV / 600
MLA e TK6 versão micropoços:
Na versão micropoços dos MLA, a CM e a CV são definidas como o produto do número total de micropoços (TW) e o número provável de colónias por poço (P) nas placas de micropoços.
CM = PM × TWM
CV = PV × TWV
A partir do termo zero da distribuição de Poisson (Furth et al., 1981), o P é dado por
P = - ln (EW / TW)
Em que EW são os poços vazios e TW são os poços totais. Portanto,
CEM = CM / TM = (PM × TWM) / TM
CEV = CV / TV = (PV × TWV) / TV
Para a versão micropoços do MLA, as frequências de mutação das colónias pequenas e grandes serão calculadas de modo idêntico, utilizando o número de poços vazios tanto para as pequenas como para as grandes colónias.
No caso do TK6, as frequências de mutação das pequenas e grandes colónias baseiam-se nos mutantes precoces e tardios.
B.68 MÉTODO DE ENSAIO IN VITRO DE EXPOSIÇÃO DE CURTA DURAÇÃO PARA IDENTIFICAR i) PRODUTOS QUÍMICOS INDUTORES DE LESÕES OCULARES GRAVES E ii) PRODUTOS QUÍMICOS QUE NÃO NECESSITAM DE SER CLASSIFICADOS EM TERMOS DE IRRITAÇÃO OCULAR NEM DE LESÕES OCULARES GRAVES
INTRODUÇÃO
|
1.
|
O presente método de ensaio é equivalente à Test Guideline (TG) 491 (2017) da OCDE. O método de ensaio de exposição de curta duração (STE) é um método in vitro que pode ser utilizado, em determinadas circunstâncias e com limitações específicas, para a classificação e rotulagem de perigo de produtos químicos (substâncias e misturas) que provoquem danos oculares graves, bem de produtos químicos que não necessitem de classificação no que diz respeito quer a lesões oculares graves quer a irritação ocular, tal como definido no Sistema Mundial Harmonizado de Classificação e Rotulagem de Produtos Químicos da ONU (GHS) (1) e no Regulamento (UE) n.o 1272/2008 da União Europeia relativo à classificação, rotulagem e embalagem de substâncias e misturas (CRE) (67).
|
|
2.
|
Durante muitos anos, o potencial de perigosidade ocular dos produtos químicos foi avaliado essencialmente com recurso a um ensaio ocular in vivo no coelho (método de ensaio B.5 (8), equivalente à Test Guideline 405 da OCDE). É geralmente aceite que, num futuro previsível, nenhum ensaio in vitro alternativo isolado poderá substituir integralmente o ensaio ocular in vivo no coelho por forma a prever toda a gama de reações de lesões oculares graves ou irritação ocular das diferentes classes de produtos químicos. No entanto, combinações estratégicas de métodos de ensaio alternativos utilizados numa estratégia de ensaio (sequencial) podem conseguir substituir integralmente o ensaio ocular no coelho (2). A abordagem descendente foi concebida para o ensaio de produtos químicos que, previsivelmente, com base nas informações existentes, têm um elevado potencial de irritação ou de indução de lesões oculares graves. Por outro lado, a abordagem ascendente foi concebida para o ensaio de produtos químicos que, previsivelmente, com base nas informações existentes, não causam irritação ocular suficiente para deverem ser classificadas. Embora não se considere um substituto integral do ensaio ocular in vivo no coelho, o método de ensaio STE é adequado para ser utilizado, no âmbito de uma estratégia de ensaio sequencial para a classificação e rotulagem regulamentares, como a abordagem descendente/ascendente, para identificar, sem ensaios complementares: i) produtos químicos indutores de lesões oculares graves (categoria 1 do sistema GHS da ONU/CRE); ii) produtos químicos (excluindo substâncias altamente voláteis e todos os sólidos não tensioativos) que não necessitem de ser classificados em termos de irritação ocular ou de lesões oculares graves (nenhuma categoria do sistema GHS da ONU/CRE) (1) (2). No entanto, para se estabelecer uma classificação definitiva de um produto químico que, previsivelmente, não provoque lesões oculares graves (categoria 1 do sistema GHS da ONU/CRE) ou não pertença a nenhuma categoria GHS da ONU/CRE (não induz lesões oculares graves nem irritação ocular) de acordo com o método de ensaio STE, será necessário realizar ensaios complementares. Além disso, as entidades reguladoras competentes devem ser consultadas antes de se utilizar a STE numa abordagem ascendente ao abrigo de outros sistemas de classificação que não o GHS da ONU/CRE. A escolha do método de ensaio mais adequado e a utilização do presente método de ensaio devem ser vistos no contexto do documento de orientação da OCDE sobre as abordagens integrada de ensaio e avaliação de lesões oculares graves e de irritação ocular (14).
|
|
3.
|
O presente método de ensaio tem por objetivo descrever os procedimentos utilizados para avaliar o potencial de perigosidade ocular de um produto químico em estudo, com base na sua capacidade de induzir a citotoxicidade no método de ensaio de exposição de curta duração. Os efeitos citotóxicos dos produtos químicos nas células epiteliais da córnea são modos de ação (MOA) importantes que conduzem à deterioração do epitélio da córnea e à irritação ocular. No método STE, a viabilidade celular é avaliada através da medição quantitativa, após extração a partir de células, de sal azul de formazano produzido pelas células vivas através da conversão enzimática do corante vital MTT [brometo de 3-(4,5-dimetiltiazol-2-il)-2,5-difeniltetrazólio], também conhecido como brometo de tiazolilo e azul de tetrazólio (3). A viabilidade celular obtida é comparada com o controlo do solvente (viabilidade relativa) e utilizada para estimar o risco ocular potencial do produto químico em estudo. Este é classificado na categoria 1 do sistema GHS da ONU/CRE quando concentrações de 5 % e 0,05 % resultam numa uma viabilidade celular igual ou inferior a 70 %. Inversamente, um produto químico é classificado como pertencente a «nenhuma categoria» no sistema GHS da ONU/CRE quando concentrações de 5 % e 0,05 % resultam numa uma viabilidade celular superior a 70 %.
|
|
4.
|
A expressão «produto químico em estudo» é utilizada no presente método de ensaio para referir o produto que está a ser estudado, não remetendo para a aplicabilidade do método de ensaio STE ao estudo de substâncias e/ou misturas. Para definições, consultar o apêndice.
|
CONSIDERAÇÕES INICIAIS E LIMITAÇÕES
|
5.
|
O presente método de ensaio baseia-se num protocolo desenvolvido pela Kao Corporation (4) que foi objeto de dois estudos de validação diferentes: um do Validation Committee of the Japanese Society for Alternative to Animal Experiments (JSAAE) (5) e outro do Japanese Center for the Validation of Alternative Methods (JaCVAM) (6). Com base nos relatórios dos estudos de validação e nos documentos-base de recensão sobre o método de ensaio (7), o NICEATM/ICCVAM realizou uma avaliação por pares.
|
|
6.
|
Quando utilizados para identificar produtos químicos (substâncias e misturas) indutores de lesões oculares graves (categoria 1 do sistema GHS da ONU/CRE) (1), os dados obtidos com o método de ensaio STE em 125 produtos químicos (incluindo substâncias e misturas) apresentaram uma exatidão global de 83 % (104/125), uma taxa de falsos positivos de 1 % (1/86) e uma taxa de falsos negativos de 51 % (20/39) em relação ao ensaio ocular in vivo no coelho (7). A taxa de falsos negativos obtida não é crítica no atual contexto, uma vez que todos os produtos químicos que induzem uma viabilidade celular igual ou inferior a 70 % a uma concentração de 5 % e uma viabilidade celular superior a 70 % a uma concentração de 0,05 % são depois estudados por outros métodos de ensaio in vitro devidamente validados ou, como última opção, no ensaio ocular in vivo no coelho, consoante os requisitos regulamentares e em conformidade com a estratégia de ensaio sequencial e com as abordagens de ponderação da suficiência da prova atualmente recomendadas (1) (8). Foram ensaiadas, principalmente, substâncias monocomponentes, embora exista também uma quantidade limitada de dados sobre o ensaio de misturas. Considera-se, porém, que o método é tecnicamente aplicável ao ensaio de substâncias e misturas multicomponentes. Contudo, antes da sua aplicação a uma mistura para obter dados com fins regulamentares, importa ponderar se — e, em caso afirmativo, por que motivo — o método pode proporcionar resultados adequados para o efeito. Tais considerações não são necessárias se existir um requisito regulamentar para o ensaio da mistura. O método de ensaio STE não revelou quaisquer outras deficiências específicas quando utilizado para identificar produtos químicos em estudo como pertencentes à categoria 1 do sistema GHS da ONU/CRE. Os investigadores podem ponderar a utilização do presente método de ensaio com produtos químicos em estudo em que uma viabilidade celular ≤ 70 %, tanto numa concentração de 5 % como numa concentração de 0,05 %, deve ser aceite como indicativa de uma resposta que induz danos oculares graves, que devem ser classificados na categoria 1 do sistema GHS da ONU/CRE, sem mais ensaios.
|
|
7.
|
Quando utilizados para identificar produtos químicos (substâncias e misturas) indutores de lesões oculares graves (nenhuma categoria do sistema GHS da ONU/CRE), os dados obtidos com o método de ensaio STE em 130 produtos químicos (incluindo substâncias e misturas) apresentaram uma exatidão global de 85 % (110/130), uma taxa de falsos positivos de 12 % (9/73) e uma taxa de falsos negativos de 19 % (11/57) em relação ao ensaio ocular in vivo no coelho (7). Se se excluírem do conjunto de dados as substâncias altamente voláteis e as substâncias sólidas não tensioativas, a exatidão global sobe para 90 % (92/102), a taxa de falsos negativos desce para 2 % (1/54) e a de falsos positivos para 19 % (9/48) (7). Assim, as possíveis deficiências do método de ensaio STE, quando utilizado para identificar produtos químicos em estudo que não necessitem de classificação em termos de irritação ocular nem de lesões oculares graves (nenhuma categoria no sistema GHS da ONU/CRE), consistem numa elevada taxa de falsos negativos para: i) substâncias altamente voláteis com uma pressão de vapor superior a 6 kPa; ii) produtos químicos sólidos (substâncias e misturas), com exceção dos tensioativos e das misturas constituídas apenas por tensioativos. Estes produtos químicos estão excluídos do domínio de aplicabilidade do método de ensaio STE (7).
|
|
8.
|
Além dos produtos químicos referidos nos pontos 6 e 7, o conjunto de dados gerado pelo método de ensaio STE contém também dados obtidos internamente relativos a 40 misturas, que, em comparação com o ensaio ocular de Draize in vivo, revelaram, na avaliação de misturas que não necessitam de classificação nos sistemas de classificação GHS da ONU/CRE (9), uma exatidão de 88 % (35/40), uma taxa de falsos positivos de 50 % (5/10) e uma taxa de falsos negativos de 0 % (0/30). Por conseguinte, o método de ensaio STE pode ser aplicado para identificar misturas como não pertencentes a nenhuma categoria no sistema GHS da ONU/CRE, numa abordagem ascendente, excetuando-se as misturas sólidas que não sejam constituídas apenas por tensioativos, na esteira da limitação relativa a substâncias sólidas. Além disso, as misturas que contenham substâncias com uma pressão de vapor superior a 6kPa devem ser avaliadas com precaução, a fim de evitar eventuais subavaliações, devendo a sua avaliação ser justificada caso a caso.
|
|
9.
|
O método de ensaio STE não pode ser utilizado para identificar produtos químicos em estudo como pertencentes à categoria 2, à categoria 2A (irritação ocular) ou à categoria 2B (irritação ocular suave) do sistema GHS da ONU/CRE, devido ao número considerável de produtos químicos da categoria 1 do sistema GHS da ONU/CRE subavaliados como pertencentes às categorias 2, 2A ou 2B e de produtos químicos não pertencentes a nenhuma categoria do GHS da ONU/CRE sobreavaliados como pertencentes às categorias 2, 2A ou 2B (7). Nesses casos, podem ser necessários ensaios complementares por outro método que seja adequado.
|
|
10.
|
O método de ensaio STE é adequado para o ensaio de produtos químicos dissolvidos ou suspensos uniformemente durante, pelo menos, 5 minutos em soro fisiológico, de solução salina de dimetilsulfóxido (DMSO) a 5 %ou em óleo mineral. Não é adequado para o ensaio de produtos químicos insolúveis ou que não possam ser suspensos uniformemente durante, pelo menos, 5 minutos em soro fisiológico, solução salina de DMSO a 5 % ou em óleo mineral. A utilização de óleo mineral no método de ensaio STE é possível devido à exposição de curta duração. Por conseguinte, o método é adequado para prever o potencial de perigosidade ocular de produtos químicos em estudo insolúveis na água (p. ex., álcoois gordos de cadeia longa ou cetonas), desde que sejam miscíveis em, pelo menos, um dos três solventes supramencionados (4).
|
|
11.
|
A expressão «produto químico em estudo» utilizada no presente método de ensaio designa o produto que está a ser estudado (68), não abrangendo a aplicabilidade do método de ensaio STE ao ensaio de substâncias e/ou misturas.
|
PRINCÍPIO DO ENSAIO
|
12.
|
O método de ensaio STE é um ensaio de toxicidade in vitro realizado numa única camada confluente de células Statens Seruminstitut Cornea (SIRC) cultivadas numa microplaca de policarbonato com 96 poços (4). Após cinco minutos de exposição ao produto químico em estudo, a citotoxicidade é medida quantitativamente como a viabilidade relativa das células SIRC, por recurso ao ensaio do MTT (4). A viabilidade celular reduzida é utilizada para prever potenciais efeitos nocivos indutores de danos oculares.
|
|
13.
|
Encontra-se referenciado que 80 % de uma solução que caia no olho de um coelho são excretados através do saco conjuntival nos três-quatro minutos seguintes, enquanto mais de 80 % de uma solução que caia no olho humano são excretados em 1 a 2 minutos (10). O método de ensaio STE procura aproximar estes tempos de exposição e utiliza a citotoxicidade como parâmetro para avaliar a extensão dos danos causados às células SIRC após cinco minutos de exposição ao produto químico em estudo.
|
DEMONSTRAÇÃO DE COMPETÊNCIA TÉCNICA
|
14.
|
Antes de utilizarem de forma rotineira o método de ensaio STE descrito, os laboratórios devem demonstrar a sua competência técnica, classificando corretamente as onze substâncias recomendadas no quadro 1. Estas substâncias foram selecionadas para representar a gama completa de reações de lesão ocular grave ou irritação ocular com base nos resultados de ensaios oculares in vivo no coelho (TG 405) e no sistema de classificação GHS da ONU/CRE (1). Outros critérios de seleção incluíam a disponibilidade comercial das substâncias, a disponibilidade de dados de referência in vivo de elevada qualidade e a existência de dados in vitro de alta qualidade relativos ao método de ensaio STE (3). Nas situações em que uma substância incluída na lista esteja indisponível ou sempre que se justifique, podem utilizar-se outras substâncias para as quais estejam disponíveis dados de referência in vivo e in vitro adequados, desde que se apliquem os critérios descritos.
Quadro 1
Lista de substâncias para demonstração de competência técnica
|
Substância
|
N.o CAS
|
Classe química (69)
|
Estado físico
|
Cat. in vivo do GHS da ONU/CRE (70)
|
Solvente no ensaio STE
|
Categoria STE do GHS da ONU/CRE
|
|
Cloreto de benzalcónio (sol. aquosa a 10 %)
|
8001-54-5
|
Sal de amónio quaternário
|
Líquido
|
Categoria 1
|
Salino
|
Categoria 1
|
|
Triton X-100 (100 %)
|
9002-93-1
|
Éter
|
Líquido
|
Categoria 1
|
Salino
|
Categoria 1
|
|
Vermelho ácido 92
|
18472-87-2
|
Composto heterocíclico; composto de bromo; composto de cloro
|
Sólido
|
Categoria 1
|
Salino
|
Categoria 1
|
|
Hidróxido de sódio
|
1310-73-2
|
Alcalinos; produtos químicos inorgânicos
|
Sólido
|
Categoria 1 (71)
|
Salino
|
Categoria 1
|
|
Butirolactona
|
96-48-0
|
Lactona; composto heterocíclico
|
Líquido
|
Categoria 2A (categoria 2 do CRE)
|
Salino
|
Previsão impossível
|
|
1-Octanol
|
111-87-5
|
Álcool
|
Líquido
|
Categoria 2A/B (72)(categoria 2 do CRE)
|
Óleos minerais
|
Previsão impossível
|
|
Ciclopentanol
|
96-41-3
|
Álcool; Hidrocarboneto cíclico
|
Líquido
|
Categoria 2A/B (73) (categoria 2 do CLP)
|
Salino
|
Previsão impossível
|
|
Acetato de 2-etoxietilo
|
111-15-9
|
Álcool; éter
|
Líquido
|
Nenhuma categoria
|
Salino
|
Nenhuma categoria
|
|
Dodecano
|
112-40-3
|
Hidrocarboneto acíclico
|
Líquido
|
Nenhuma categoria
|
Óleos minerais
|
Nenhuma categoria
|
|
Metilisobutilcetona
|
108-10-1
|
Cetona
|
Líquido
|
Nenhuma categoria
|
Óleos minerais
|
Nenhuma categoria
|
|
Sulfato de 1,1-dimetilguanidina
|
598-65-2
|
Amidina; composto de enxofre
|
Sólido
|
Nenhuma categoria
|
Salino
|
Nenhuma categoria
|
|
Abreviaturas: N.o de registo CAS = Número de registo do Chemical Abstracts Service
|
|
PROCEDIMENTO
Preparação da monocamada celular
|
15.
|
Para a realização do ensaio STE, deve utilizar-se a linha celular SIRC da córnea do coelho. Recomenda-se que as células SIRC provenham de um banco de células devidamente qualificado, como o American Type Culture Collection CCl60.
|
|
16.
|
As células SIRC são cultivadas a 37 °C, numa atmosfera humidificada com 5 % de CO2, num balão de cultura com um meio de cultura constituído por meio mínimo essencial de Eagle (MEM) complementado com 10 % de soro fetal bovino (FBS), 2 mM de L-glutamina, 50-100 unidades/ml de penicilina e 50-100 μg/ml de estreptomicina. As células que se tenham tornado confluentes no frasco de cultura devem ser separadas por meio da solução de tripsina-ácido etilenodiaminotetracético, com ou sem recurso a um raspador. As células são propagadas (2 a 3 passagens) num recipiente de cultura antes de serem utilizadas em ensaios de rotina, não devendo ser objeto de mais de 25 passagens desde a descongelação.
|
|
17.
|
As células prontas a utilizar no ensaio STE são preparadas à densidade adequada e inoculadas em placas de 96 poços. A densidade de inoculação das células recomendada é de 6,0 × 103 células por poço, se as células forem utilizadas quatro dias após a inoculação, ou de 3,0 × 103 células por poço, se forem utilizadas cinco dias após a inoculação, num volume de cultura de 200 μl. As células utilizadas para o ensaio STE que sejam inoculadas num meio de cultura com a densidade adequada atingirão uma confluência superior a 80 % no momento do ensaio, isto é, quatro ou cinco dias após a inoculação.
|
Aplicação dos produtos químicos em estudo e de controlo
|
18.
|
A primeira escolha do solvente para dissolução ou suspensão dos produtos químicos em estudo é uma solução de soro fisiológico. Se o produto apresentar uma baixa solubilidade ou não puder ser dissolvido ou suspenso uniformemente durante, pelo menos, cinco minutos em soro fisiológico, utiliza-se, como segunda opção, DMSO (n.o CAS 67-68-5) a 5 % em soro fisiológico. Caso os produtos químicos em estudo não possam ser dissolvidos ou suspensos de modo uniforme durante, pelo menos, cinco minutos soro fisiológico ou em DMSO a 5 % em soro fisiológico, utiliza-se óleo mineral (n.o CAS 8042-47-5) como terceira opção.
|
|
19.
|
Os produtos químicos em estudo são dissolvidos ou suspensos uniformemente no solvente selecionado, a uma concentração de 5 % (m/m), e novamente diluídos numa diluição sequencial de fator 10 até 0,5 % e uma concentração de 0,05 %. Cada produto químico em estudo deve ser ensaiado a concentrações de 5 % e 0,05 %. As células cultivadas na placa de 96 poços são expostas a 200 μl/poço de uma concentração da solução (ou suspensão) do produto químico em estudo de 5 % ou de 0,05 %, durante 5 minutos à temperatura ambiente. Os produtos químicos em estudo (substâncias monocomponentes ou substâncias ou misturas multicomponentes) são considerados substâncias puras, sendo diluídos ou suspensos de acordo com o método, independentemente do seu grau de pureza.
|
|
20.
|
Utiliza-se como meio de controlo, em cada placa de cada réplica, o meio de cultura descrito no ponto 16. Além disso, as células devem ser expostas também a amostras de controlo de solvente em cada uma das placas de cada réplica. Está confirmado que os solventes enumerados no ponto 18 não têm efeitos nocivos na viabilidade das células SIRC.
|
|
21.
|
No método de ensaio STE, utiliza-se uma solução salina a 0,01 % de laurilsulfato de sódio como controlo positivo em cada réplica. A fim de calcular a viabilidade celular do controlo positivo, cada placa de cada réplica deve incluir também um controlo do solvente salino.
|
|
22.
|
É necessário um ensaio em branco para determinar a compensação por densidade ótica, ensaio esse que deve ser realizado em poços contendo apenas solução salina tamponada de fosfatos (PBS), mas sem cálcio e magnésio (PBS-) nem células.
|
|
23.
|
Cada amostra (produto químico em estudo a 5 % e 0,05 %, controlo de meio, controlo do solvente e controlo positivo) deve ser ensaiada em triplicado em cada réplica, expondo as células a 200 μl do produto químico em estudo ou de controlo, durante 5 minutos, à temperatura ambiente.
|
|
24.
|
As substâncias de referência são úteis para avaliar o potencial de irritação ocular de produtos químicos desconhecidos de uma determinada classe química – ou classe de produtos – e para avaliar o potencial de irritação relativo de uma substância irritante ocular numa determinada gama de reações de irritação.
|
Medição da viabilidade celular
|
25.
|
Após a exposição, as células são lavadas duas vezes com 200 μl de PBS e adicionam-se 200 μl de solução de MTT (0,5 mg de MTT/ml de meio de cultura). Após um período de reação de duas horas numa incubadora (37 °C, 5 % de CO2), a solução de MTT é decantada, o MTT formazano é extraído durante 60 minutos através da adição de 200 μl de 0,04 N ácido clorídrico-isopropanol, ao abrigo da luz e à temperatura ambiente, medindo-se a absorvância da solução de MTT formazano a 570 nm, com um leitor de placas. A interferência de produtos químicos em estudo com o ensaio do MTT (por corantes ou redutores diretos do MTT) só ocorre se uma quantidade significativa do produto químico em estudo ficar retida no sistema de ensaio após a lavagem subsequente à exposição, o que acontece com córnea humana reconstruída em 3D ou com tecidos da epiderme humana reconstruídos, mas não é relevante para culturas de células 2D utilizadas para o método de teste STE.
|
Interpretação dos resultados e modelo de previsão
|
26.
|
Utilizam-se os valores da densidade ótica (DO) obtidos para cada produto químico em estudo para calcular a viabilidade celular em relação ao controlo do solvente, que é fixada em 100 %. A viabilidade celular relativa é expressa em percentagem e obtida dividindo a densidade ótica do produto químico em estudo pela densidade ótica do controlo do solvente depois de subtrair a densidade ótica do produto em branco de ambos os valores.

Do mesmo modo, a viabilidade celular relativa de cada controlo de solvente é expressa em percentagem e obtida dividindo a densidade ótica do controlo do solvente pela densidade ótica do controlo do meio, após subtração da densidade ótica do branco a ambos os valores.
|
|
27.
|
Devem realizar-se três repetições independentes, cada uma das quais com três poços replicados (ou seja, n = 9). A média aritmética dos três poços de cada produto químico em estudo e do controlo do solvente em cada réplica independente é utilizada para calcular a média aritmética da viabilidade celular relativa. A média aritmética final da viabilidade celular é calculada a partir das três repetições independentes.
|
|
28.
|
Apresentam-se a seguir os valores-limite de viabilidade celular para a identificação de produtos químicos indutores de lesões oculares graves (categoria 1 do sistema GHS da ONU/CRE) e de produtos químicos que não necessitem de ser classificados em termos de irritação ocular nem de lesões oculares graves (nenhuma categoria no sistema GHS da ONU/CRE).
|
Quadro 2
Modelo de previsão do método de ensaio STE
|
Viabilidade celular
|
Classificação GHS da ONU/CRE
|
Aplicabilidade
|
|
A 5 %
|
A 0,05 %
|
|
> 70 %
|
> 70 %
|
Nenhuma categoria
|
Substâncias e misturas, com exceção de: i) Substâncias altamente voláteis com uma pressão de vapor superior a 6 kPa (74) e ii) Produtos químicos sólidos (substâncias e misturas), com exceção dos tensioativos e das misturas constituídas apenas por tensioativos
|
|
≤ 70 %
|
> 70 %
|
Previsão impossível
|
Não aplicável
|
|
≤ 70 %
|
≤ 70 %
|
Categoria 1
|
Substâncias e misturas (75)
|
Critérios de aceitação
|
29.
|
Os resultados dos ensaios são considerados aceitáveis se forem cumpridos os seguintes critérios:
|
a)
|
A densidade ótica do controlo do meio (exposto ao meio de cultura) deve ser igual ou superior a 0,3 após dedução da densidade ótica do branco.
|
|
b)
|
A viabilidade do controlo do solvente deve ser igual ou superior a 80 % do controlo do meio. Caso se utilizem vários controlos de solvente em cada repetição, cada um desses controlos deve indicar uma viabilidade celular superior a 80 % para qualificar os produtos químicos em estudo ensaiados com esses solventes.
|
|
c)
|
A viabilidade celular obtida com o controlo positivo (0,01 % SLS) deve situar-se no intervalo de dois desvios-padrão da média histórica. Os limites superior e inferior de aceitação do controlo positivo devem ser atualizados com frequência, ou seja, de três em três meses ou sempre que se realize um ensaio aceitável num laboratório que efetue ensaios com pouca frequência (ou seja, menos de uma vez por mês). Caso um laboratório não realize um número de experiências suficiente para estabelecer uma distribuição de controlo positiva estatisticamente sólida, é aceitável que se utilizem os limites superior e inferior de aceitação estabelecidos pelo criador do método, ou seja, entre 21,1 % e 62,3 %, de acordo com os seus dados históricos de laboratório, enquanto uma distribuição interna é construída durante os primeiros ensaios de rotina.
|
|
d)
|
O desvio-padrão da viabilidade celular final derivado de três repetições independentes deve ser inferior a 15 % para as concentrações de 5 % e 0,05 % do produto químico em estudo.
Se um ou mais destes critérios não forem cumpridos, os resultados devem ser rejeitados e deve proceder-se a três novas repetições independentes.
|
|
DADOS E RELATÓRIOS
Dados
|
30.
|
Devem comunicar-se os dados relativos a cada poço individual (por exemplo, valores de viabilidade celular) de cada repetição, bem como a média global, o DP e a classificação.
|
Relatório de ensaio
|
31.
|
O relatório do ensaio deve incluir as seguintes informações:
|
|
Produto químico em estudo e substâncias de controlo
|
—
|
Substâncias monocomponentes: dados de identificação química, como as denominações IUPAC ou CAS, o(s) número(s) de registo CAS, o código SMILES ou InChI, a fórmula estrutural e/ou outros elementos de identificação;
|
|
—
|
Substância multicomponentes, UVCB e mistura: Caracterização, tanto quanto possível — por exemplo, por identidade química (ver supra), pureza, ocorrência quantitativa e principais propriedades físico-químicas (ver supra) dos componentes, na medida em que estejam disponíveis;
|
|
—
|
Estado físico, volatilidade, pH, log(P), peso molecular, classe química e outras propriedades físico-químicas pertinentes para a realização do estudo, na medida em que estejam disponíveis;
|
|
—
|
Grau de pureza, identidade química das impurezas, conforme adequado e exequível, etc.;
|
|
—
|
Pré-tratamento, se for o caso (aquecimento ou moagem, por exemplo);
|
|
—
|
Condições de armazenagem e de estabilidade, se conhecidas.
|
|
|
|
Condições e procedimentos do método de ensaio
|
—
|
Nome e endereço do patrocinador, do laboratório e do diretor do estudo;
|
|
—
|
Descrição do método de ensaio utilizado;
|
|
—
|
Linha celular utilizada, origem, número de passagens e confluência das células utilizadas para o ensaio;
|
|
—
|
Pormenores sobre o protocolo de ensaio seguido;
|
|
—
|
Número de réplicas e replicados utilizados;
|
|
—
|
Concentrações do produto químico em estudo utilizadas (se diferentes das recomendadas);
|
|
—
|
Condições de armazenagem e de estabilidade, se conhecidas;
|
|
—
|
Tempo de exposição ao produto químico em estudo (se diferente do recomendado);
|
|
—
|
Descrição de eventuais modificações do protocolo do ensaio;
|
|
—
|
Descrição dos critérios de avaliação e de decisão utilizados;
|
|
—
|
Referência à média histórica do controlo positivo e ao desvio-padrão (DP);
|
|
—
|
Demonstração da competência técnica do laboratório para executar o método de ensaio (p. ex., ensaio de substâncias utilizadas para demonstrar a competência técnica) ou demonstração da reprodutibilidade do método de ensaio ao longo do tempo.
|
|
|
|
Resultados
|
—
|
Para cada produto químico em estudo, cada substância de controlo e cada concentração estudada, deve apresentar-se uma tabela dos valores da densidade ótica por poço replicado, os valores da média aritmética da densidade ótica para cada repetição independente, a percentagem de viabilidade celular para cada repetição independente e a média aritmética final da percentagem de viabilidade celular das três repetições, bem como o desvio-padrão das três repetições;
|
|
—
|
Resultados relativos ao meio, ao controlo do solvente e ao controlo positivo que demonstrem o cumprimento dos critérios pertinentes de aceitação do estudo;
|
|
—
|
Descrição de outros efeitos eventualmente observados;
|
|
—
|
Classificação global atribuída, com menção do modelo de previsão ou dos critérios de decisão utilizados.
|
|
|
REFERÊNCIAS BIBLIOGRÁFICAS
|
(1)
|
United Nations UN (2013). Globally Harmonized System of Classification and Labelling of Chemicals (GHS). Fifth revised edition. New York & Geneva: United Nations Publications. ISBN: 978-92-1-117006-1. Disponível em: http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html.
|
|
(2)
|
Scott L, et al. (2010). A proposed Eye Irritation Testing Strategy to Reduce and Replace in vivo Studies Using Bottom-Up and Top-Down Approaches. Toxicol. In Vitro 24, 1-9.
|
|
(3)
|
Mosmann T. (1983). Rapid Colorimetric Assay for Cellular Growth and Survival: Application to 7 Proliferation and Cytotoxicity Assays. J. Immunol. Methods 65, 55-63.
|
|
(4)
|
Takahashi Y, et al. (2008). Development of the Short Time Exposure (STE) Test: an In Vitro Eye Irritation Test Using SIRC Cells. Toxicol. In Vitro 22,760-770.
|
|
(5)
|
Sakaguchi H, et al. (2011). Validation Study of the Short Time Exposure (STE) Test to Assess the Eye Irritation Potential of Chemicals. Toxicol. In Vitro 25,796-809.
|
|
(6)
|
Kojima H, et al. (2013). Second-Phase Validation of Short Time Exposure Tests for Assessment of Eye Irritation Potency of Chemicals. Toxicol. In Vitro 27, pp.1 855-1 869.
|
|
(7)
|
ICCVAM (2013). Short Time Exposure (STE) Test Method Summary Review Document, NIH. Disponível em: [http://www.ntp.niehs.nih.gov/iccvam/docs/ocutox_docs/STE-SRD-NICEATM-508.pdf].
|
|
(8)
|
Chapter B.5 of this Annex, Acute Eye Irritation/Corrosion.
|
|
(9)
|
Saito K, et al. (2015). Predictive Performance of the Short Time Exposure Test for Identifying Eye Irritation Potential of Chemical Mixtures.
|
|
(10)
|
Mikkelson TJ, Chrai SS and Robinson JR. (1973). Altered Bioavailability of Drugs in the Eye Due to Drug-Protein Interaction. J. Pharm. Sci.1 648-1 653.
|
|
(11)
|
ECETOC (1998). Eye Irritation Reference Chemicals Data Bank. Technical Report (No 48. (2)), Brussels, Belgium.
|
|
(12)
|
Gautheron P, et al. (1992). Bovine Corneal Opacity and Permeability Test: an In Vitro Assay of Ocular Irritancy. Fundam Appl Toxicol. 18, 442-449.
|
|
(13)
|
OCDE (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environmental, Health and Safety Publications, Series on Testing and Assessment (No 34). Organisation for Economic Cooperation and Development, Paris.
|
|
(14)
|
OCDE (2017). Guidance Document on an Integrated Approaches on Testing and Assessment for Serious Eye Damage and Eye irritation. Environmental, Health and Safety Publications, Series on Testing and Assessment (No 263). Organisation for Economic Cooperation and Development, Paris.
|
Apêndice
DEFINIÇÕES
Abordagem ascendente
: Metodologia por etapas utilizada para produtos químicos em estudo que se pense não necessitarem de ser classificados em termos de irritação ocular ou de lesões oculares graves; inicia-se com a determinação de produtos químicos que não careçam de classificação (resultado negativo) a partir de outros produtos químicos (resultado positivo).
Abordagem descendente
: Metodologia por etapas utilizada para produtos químicos em estudo que se suspeite causarem lesões oculares graves; inicia-se com a determinação de produtos químicos que induzam lesões oculares graves (resultado positivo) a partir de outros produtos químicos (resultado negativo).
Adequação
: Relação do ensaio com o efeito em causa; pertinência e utilidade do ensaio para o fim em vista. Traduz a medida em que o ensaio mede ou prevê corretamente o efeito biológico em apreciação. A adequação compreende a exatidão (concordância) do método de ensaio (10).
Agente tensioativo
: Produto químico, como um detergente, capaz de reduzir a tensão superficial dos líquidos, permitindo-lhes formar espumas ou penetrar em sólidos; igualmente designado por «agente molhante».
Amostra de controlo do solvente/veículo
: Amostra não tratada que contém todos os componentes do sistema de ensaio, incluindo o solvente ou excipiente, e é ensaiada, juntamente com as amostras tratadas com o produto químico em estudo e as outras amostras de controlo, para estabelecer a linha de base de reação para as amostras tratadas com o produto químico em estudo, dissolvido no mesmo solvente ou excipiente. Quando ensaiada com uma amostra de controlo do meio correspondente, esta amostra também permite determinar se o solvente ou o veículo interage com o sistema de ensaio.
Categoria 1 do sistema GHS da ONU/CRE
: Ver «lesão ocular grave».
Categoria 2 do sistema GHS da ONU/CRE
: Ver «irritação ocular».
Controlo do meio
: Replicado não tratado que contém todos os componentes do sistema de ensaio. Esta amostra é ensaiada juntamente com as amostras tratadas com o produto químico em estudo e as outras amostras de controlo, para determinar se o solvente interage com o sistema de ensaio.
Controlo positivo
: Replicado que contém todos os componentes do sistema de ensaio e foi tratado com uma substância que comprovadamente induz reação positiva. Para que possa determinar-se a variabilidade no tempo da reação a esta amostra de controlo, essa reação não deve ser excessivamente positiva.
DO
: Densidade ótica.
Especificidade
: Proporção dos produtos químicos negativos/inativos que são corretamente classificados pelo ensaio. Constitui uma medida da exatidão de um método de ensaio cujos resultados sejam estabelecidos em função de categorias e é um aspeto importante a ter em conta na avaliação da adequação do método de ensaio (13).
Estratégia de ensaio sequencial por etapas
: Estratégia sequencial de ensaio em que, seguindo uma ordem estabelecida, se avalia toda a informação disponível sobre o produto químico em estudo, por um processo baseado na ponderação da suficiência da prova em cada etapa, com o objetivo de determinar se há dados suficientes para uma decisão de classificação de perigosidade antes de passar à etapa seguinte. Se os dados existentes possibilitarem a atribuição de um potencial de irritação ao produto químico em estudo, não serão necessários mais ensaios. No caso contrário, procede-se a uma série de ensaios sequenciais em animais até ser possível atribuir uma classificação inequívoca.
Exatidão
: Grau de acordo entre os resultados do método de ensaio e os valores de referência aceites. Constitui uma medida do desempenho do método e um dos aspetos da sua adequação. Este termo e o termo «concordância» são muitas vezes utilizados indistintamente para indicar a proporção de resultados corretos do método de ensaio (13).
Fiabilidade
: Medida em que, utilizando o mesmo protocolo, um método de ensaio pode ser continuadamente reproduzido no mesmo laboratório e em laboratórios diferentes. A fiabilidade é avaliada com base nos valores calculados das reprodutibilidades intralaboratorial e interlaboratorial, bem como da repetibilidade intralaboratorial (13).
Irritação ocular
: Alteração ocular em consequência da aplicação do produto químico em estudo na superfície anterior do olho, totalmente reversíveis nos 21 dias após a aplicação. Esta expressão e as expressões «efeitos oculares reversíveis» ou «categoria 2 do sistema GHS da ONU/CRE» são utilizados indistintamente.
Lesão ocular grave
: Lesão do tecido ocular ou degradação grave da visão devido à aplicação do produto químico em estudo na superfície anterior do olho, não totalmente reversível nos 21 dias após a aplicação. Esta expressão e as expressões «efeitos oculares irreversíveis» ou «categoria 1 do sistema GHS da ONU/CRE» são utilizadas indistintamente.
Mistura
: Uma mistura ou solução composta por duas ou mais substâncias.
MTT
: Brometo de [3-(4,5-dimetiltiazol-2-il)-2,5-difeniltetrazólio]; brometo de tiazolilo; azul de tetrazólio.
Nenhuma categoria no sistema GHS da ONU/CRE
: Produtos químicos não classificados nas categorias 1 ou 2 (ou categorias 2A ou 2B) do sistema GHS da ONU/CRE.
Perigo
: Propriedade intrínseca de um agente, ou de uma situação, suscetível de causar efeitos nocivos num organismo, sistema, população ou subpopulação que seja exposto ao agente em causa.
Produto químico
: Uma substância ou mistura.
Produto químico em estudo
: Qualquer substância ou mistura à qual seja aplicado o presente método de ensaio.
Sensibilidade
: Proporção dos produtos químicos positivos/ativos que são corretamente classificados pelo ensaio. Constitui uma medida da exatidão de um método de ensaio cujos resultados sejam classificados em função de categorias e é um aspeto importante a ter em conta na avaliação da adequação do método de ensaio (10).
Sistema Mundial Harmonizado de Classificação e Rotulagem de Produtos Químicos das Nações Unidas (em inglês: Globally Harmonized System of Classification and Labelling of Chemicals – UN GHS)
: Sistema que propõe a classificação dos produtos químicos (substâncias e misturas) em função de tipos e níveis normalizados de perigos físicos, sanitários e ambientais e trata ainda dos correspondentes elementos de comunicação, como pictogramas, palavras-sinal, advertências de perigo, recomendações de prudência e fichas de dados de segurança, de modo a transmitir informações sobre os efeitos indesejáveis do produto em causa, com vista à proteção das pessoas (empregadores, trabalhadores, transportadores, consumidores, pessoal dos serviços de emergência, etc.) e do ambiente (1).
Substância
: Um elemento químico e os seus compostos, no estado natural ou obtidos por qualquer processo de produção, incluindo qualquer aditivo necessário para preservar a sua estabilidade e qualquer impureza derivada do processo utilizado, mas excluindo qualquer solvente que possa ser separado sem afetar a estabilidade da substância nem modificar a sua composição.
Substância de referência
: Substância utilizada como padrão de comparação com o produto químico em estudo. Uma substância de referência deve ter as seguintes características: (i) origem ou origens uniformes e fiáveis; (ii) similaridade estrutural e funcional com a classe de substâncias em estudo; (iii) propriedades físico-químicas conhecidas; (iv) disponibilidade de dados sobre os efeitos conhecidos; e v) potência conhecida, situada na gama de reação pretendida.
Substância monocomponente
: Substância, definida pela sua composição quantitativa, na qual um dos principais componentes está presente num teor de, pelo menos, 80 % (m/m).
Substância multicomponentes
: Substância, definida pela sua composição quantitativa, da qual mais de um dos principais componentes está presente numa concentração ≥ 10 % (m/m) e < 80 % (m/m). As substâncias multicomponentes resultam de processos de fabrico. A diferença entre uma mistura e uma substância multicomponentes reside no facto de a primeira ser obtida misturando duas ou mais substâncias, sem reação química. As substâncias multicomponentes resultam de reações químicas.
Taxa de falsos negativos
: Proporção dos produtos químicos positivos que o método de ensaio considera, erradamente, negativos. É um dos indicadores da eficiência dos métodos de ensaio.
Taxa de falsos positivos
: Proporção dos produtos químicos negativos que o método de ensaio considera, erradamente, positivos. É um dos indicadores da eficiência dos métodos de ensaio.
UVCB
: Substâncias de composição desconhecida ou variável, produtos de reação complexos ou matérias biológicas.
B.69 MÉTODO DE ENSAIO DO EPITÉLIO HUMANO SIMILAR À CÓRNEA RECONSTRUÍDO (RhCE), PARA IDENTIFICAÇÃO DE PRODUTOS QUÍMICOS QUE NÃO NECESSITAM DE CLASSIFICAÇÃO E ROTULAGEM EM MATÉRIA DE IRRITAÇÃO OCULAR E LESÕES OCULARES GRAVES
INTRODUÇÃO
|
1.
|
O presente método de ensaio é equivalente à Test Guideline (TG) 492 (2017) da OCDE. Por lesões oculares graves entende-se a produção de lesões oculares graves ou uma degradação grave da visão, na sequência da aplicação de um produto químico na superfície anterior do olho, que não sejam totalmente reversíveis nos 21 dias após a aplicação, de acordo com o Sistema Mundial Harmonizado de Classificação e Rotulagem de Produtos Químicos das Nações Unidas (GHS) (1), e pelo Regulamento (UE) n.o 1272/2008 da União Europeia, relativo à classificação, rotulagem e embalagem de substâncias e misturas (CRE) (76). Ainda de acordo com o sistema GHS da ONU e com o CRE, entende-se por irritação ocular a produção de alterações no olho, após a aplicação do produto químico em estudo na sua superfície anterior, que são totalmente reversíveis nos 21 dias seguintes à aplicação. Os produtos químicos em estudo que induzem lesões oculares graves são classificados na categoria 1 do sistema GHS da ONU e na categoria 1 do CRE, enquanto os que induzem irritação ocular são classificados na categoria 2 do sistema GHS da ONU e na categoria 2 do CRE. Os produtos químicos em estudo não classificados em termos de irritação ocular nem de lesões oculares graves são definidos como produtos químicos que não preenchem os requisitos para classificação nas categorias 1 ou 2 (2A ou 2B) do sistema de classificação GHS da ONU/CRE, ou seja, são referidos como não pertencendo a «nenhuma categoria» no sistema GHS da ONU/CRE.
|
|
2.
|
A avaliação das lesões oculares graves/irritação ocular implica, normalmente, a utilização de animais de laboratório (método de ensaio B.5) (2). A escolha do método de ensaio mais adequado e a utilização do presente método de ensaio devem ser ponderadas no contexto do documento de orientação da OCDE sobre abordagens integradas de ensaio e avaliação de lesões oculares graves e de irritação ocular (39).
|
|
3.
|
O presente método de ensaio descreve um procedimento in vitro que permite a identificação de produtos químicos (substâncias e misturas) que não necessitam de classificação e rotulagem em termos de irritação ocular nem de lesões oculares graves, em conformidade com o GHS da ONU e com o CRE. Utiliza epitélio humano reconstruído similar à córnea (RhCE), o qual reproduz com rigor as propriedades histológicas, morfológicas, bioquímicas e fisiológicas do epitélio da córnea humana. Quatro outros métodos de ensaio in vitro foram validados, considerados cientificamente válidos e adotados como métodos de ensaio B.47 (3), B.48 (4), B.61 (5) e B.68 (6), a fim de resolver a questão do parâmetro «lesões oculares graves/irritação ocular».
|
|
4.
|
O presente método inclui dois ensaios validados, comercialmente disponíveis, de RhCE. Realizaram-se estudos de validação para avaliar irritação ocular/lesões oculares graves (7) (8) (9) (10) (11) (12) (13) utilizando os ensaios de irritação ocular EpiOcular™ Eye Irritation Test (EIT) e SkinEthic™ Human Corneal Epithelium (HCE) Eye Irritation Test (EIT). Cada um destes testes utiliza, como sistema de ensaio, construções de tecido RhCE disponíveis no comércio, a seguir referidas como métodos de referência validados – VRM1 e VRM2, respetivamente. A partir destes estudos de validação e da sua avaliação independente por pares (9)(12), concluiu-se que o IET EpiOcular™ e o HCE IET SkinEthic™ permitem identificar corretamente os produtos químicos (substâncias e misturas) que, de acordo com o GHS da ONU, não necessitam de classificação e rotulagem em matéria de irritação ocular ou de lesões oculares graves, tendo os ensaios sido recomendados como cientificamente válidos para o efeito (13).
|
|
5.
|
É geralmente aceite que, num futuro previsível, nenhum ensaio in vitro alternativo isolado poderá substituir integralmente o ensaio ocular pelo método de Draize in vivo (2) (14) para prever toda a gama de reações de lesões oculares graves/irritação ocular das diferentes classes de produtos químicos. Porém, a combinação de vários métodos de ensaio alternativos no âmbito de uma estratégia de ensaio ascendente/descendente pode substituir-se ao ensaio ocular pelo método de Draize (15). A abordagem ascendente (15) foi concebida para ser utilizada quando, com base nos dados existentes, se preveja que um produto químico não cause irritação ocular suficiente para necessitar de ser classificado, enquanto a abordagem descendente (15) é concebida para ser utilizada quando, com base nos dados existentes, se preveja que um produto químico cause lesões oculares graves. O EIT EpiOcular™ e o EIT HCE SkinEthic™ são recomendados para a identificação de produtos químicos que não necessitam de classificação para irritação ou lesões oculares graves de acordo com o GHS da ONU/CRE(nenhuma categoria) sem qualquer outro ensaio, no âmbito de uma estratégia como a abordagem ascendente/descendente sugerida por Scott et al., por exemplo, como uma etapa inicial de uma abordagem ascendente ou como uma das últimas etapas de uma abordagem descendente (15). No entanto, o EIT EpiOcular™ e o EIT HCE SkinEthic™ não se destinam a estabelecer uma distinção entre a categoria 1 do sistema GHS da ONU/CRE (lesões oculares graves) e a categoria 2 do sistema GHS da ONU/CRE (irritação ocular). Esta diferenciação terá de ser feita a outro nível da estratégia de ensaio (15). No caso de produtos químicos em estudo identificados como necessitando de classificação para a irritação ocular/lesões oculares graves com o EIT EpiOcular™ ou o EIT HCE SkinEthic™ será, por conseguinte, necessário realizar ensaios complementares (in vitro e/ou in vivo) para se chegar a uma conclusão definitiva (nenhuma categoria, categoria 2 ou categoria 1 no sistema GHS da ONU/CRE), utilizando, por exemplo, os métodos B.47, B.48, B.61 ou B.68.
|
|
6.
|
O presente método de ensaio tem por objetivo descrever o procedimento utilizado para avaliar o potencial de perigosidade ocular de um produto químico em estudo, com base na sua capacidade de induzir a citotoxicidade num tecido RhCE, medido pelo ensaio do MTT (16) (ver ponto 21). A viabilidade do tecido RhCE na sequência da exposição a um produto químico em estudo é determinada em relação aos tecidos tratados com a substância de controlo negativa (taxa de viabilidade em %) e é utilizada para prever o potencial de perigosidade ocular do produto em causa.
|
|
7.
|
Existem normas de desempenho (17) para facilitar a validação de ensaios in vitro, novos ou modificados, baseados no RhCE, semelhantes ao EIT EpiOcular™ e ao EIT HCE SkinEthic™, em conformidade com os princípios do documento de orientação n.o 34 da OCDE (18), e permitir a alteração oportuna da Test Guideline 492 da OCDE, para que possa abranger os referidos ensaios. A aceitação mútua de dados (MAD) nos termos do acordo da OCDE só poderá ser garantida para métodos de ensaio validados de acordo com as normas de desempenho, se tiverem sido revistos e incluídos na correspondente diretriz de ensaio da OCDE.
|
DEFINIÇÕES
|
8.
|
O apêndice 1 contém definições.
|
CONSIDERAÇÕES INICIAIS E LIMITAÇÕES
|
9.
|
O presente método de ensaio baseia-se em tecidos RhCE tridimensionais disponíveis no comércio que são produzidos utilizando queratinócitos primários de epiderme humana (OCL-200 da EpiOcular™) ou células epiteliais da córnea humana imortalizadas (HCE/S da SkinEthic ™). Os tecidos RhCE OCL-200 da EpiOcular™ e HCE/S da SkinEthic™ são similares à estrutura tridimensional do epitélio corneano in vivo e são produzidos utilizando células da espécie-alvo (19)(20). Além disso, os ensaios medem diretamente a citotoxicidade resultante da penetração do produto químico através da córnea e a produção de danos nas células e nos tecidos; a resposta citotóxica determina in vivo as lesões oculares graves/a irritação ocular totais resultantes. Os danos nas células podem ocorrer através de vários modos de ação (ver ponto 20), mas a citotoxicidade desempenha um papel mecanístico importante – se não essencial – na determinação da resposta global, em termos de lesões oculares graves/irritação ocular, a um produto químico, expressa in vivo principalmente por opacidade da córnea, irite, vermelhidão da conjuntiva e/ou equimose conjuntival, independentemente dos processos físico-químicos subjacentes aos danos dos tecidos.
|
|
10.
|
Uma vasta gama de produtos químicos, que abrange uma grande diversidade de produtos químicos, classes químicas, pesos moleculares, log(P), estruturas químicas, etc., foi ensaiada no estudo de validação subjacente ao presente método de ensaio. A base de dados de validação do EIT EpiOcular™ contém um total de 113 produtos químicos, que abrangem 95 grupos funcionais orgânicos diferentes, de acordo com uma análise da caixa de ferramentas da QSAR Toolbax da OCDE (8). A maioria destes produtos químicos representa substâncias monocomponentes, mas várias substâncias multicomponentes (incluindo 3 homopolímeros, 5 copolímeros e 10 quase-polímeros) foram igualmente incluídas no estudo. No que respeita ao estado físico e às categorias do sistema GHS da ONU, os 113 produtos químicos objeto de ensaio foram distribuídos do seguinte modo: 13 líquidos da categoria 1, 15 sólidos da categoria 1, 6 líquidos da categoria 2A, 10 sólidos da categoria 2A, 7 líquidos da categoria 2B, 7 sólidos da categoria 2B, 27 líquidos de nenhuma categoria e 28 sólidos de nenhuma categoria (8). A base de dados do EIT HCE da SkinEthic™ contém um total de 200 produtos químicos, que abrangem 165 grupos funcionais orgânicos diferentes (8) (10) (11). A maioria destes produtos químicos representava substâncias monocomponentes, mas várias substâncias multicomponentes (incluindo 10 polímeros) foram igualmente incluídas no estudo. No que respeita ao estado físico e às categorias do sistema GHS da ONU, os 200 produtos químicos objeto de ensaio foram distribuídos do seguinte modo: 27 líquidos da categoria 1, 24 sólidos da categoria 1, 19 líquidos da categoria 2A, 10 sólidos da categoria 2A, 7 líquidos da categoria 2B, 8 sólidos da categoria 2B, 50 líquidos de nenhuma categoria e 53 sólidos de nenhuma categoria (10) (11).
|
|
11.
|
O método é aplicável a substâncias e misturas, sólidos, líquidos, semissólidos e ceras. Os líquidos podem ser aquosos ou não e os sólidos podem ser solúveis ou insolúveis em água. Sempre que possível, os sólidos devem moídos em pó fino antes da aplicação; não é necessário nenhum outro tratamento prévio da amostra. O estudo de validação não incidiu em gases e aerossóis. Embora se admita que uns e outros possam ser ensaiados recorrendo à tecnologia RhCE, a versão atual do método não está vocacionada para ensaios de gases nem de aerossóis.
|
|
12.
|
Os produtos químicos em estudo que absorvem a luz na gama do MTT formazano (naturalmente ou após tratamento) e os produtos químicos em estudo capazes de reduzir diretamente o corante vital MTT (a MTT formazano) podem interferir com as medições da viabilidade dos tecidos e requerer a realização de controlos adaptados para correção. O tipo de controlos adaptados que podem ser requeridos varia em função do tipo de interferência produzida pelo produto químico em estudo e do procedimento utilizado para quantificar o MTT formazano (ver pontos 36 a 42).
|
|
13.
|
Os resultados obtidos nos estudos de pré-validação (21) (22) e de validação (8) (10) (11) demonstraram que tanto o EIT EpiOcular™ como o EIT HCE SkinEthic™ são transferíveis para laboratórios considerados pouco experientes na realização dos ensaios e são igualmente reprodutíveis, quer no mesmo laboratório quer em laboratórios diferentes. Com base nestes estudos, o nível de reprodutibilidade das previsões expectáveis para o EIT da EpiOcular™ a partir de dados relativos a 113 produtos químicos é da ordem de 95 % a nível intralaboratorial e de 93 % a nível interlaboratorial. O nível de reprodutibilidade em termos de concordância das previsões expectáveis para o EIT HCE da SkinEthic™ a partir de dados relativos a 120 produtos químicos é da ordem dos 92 % a nível intralaboratorial e de 95 % a nível interlaboratorial.
|
|
14.
|
O EIT da EpiOcular™ pode ser utilizado para identificar produtos químicos que não necessitem de ser classificados em termos de irritação ocular nem de lesões oculares graves de acordo com o sistema de classificação GHS da ONU e com o CRE. Tendo em conta os dados obtidos no estudo de validação (8), o EIT da EpiOcular™ tem uma exatidão global de 80 % (com base em 112 produtos químicos), uma sensibilidade de 96 % (57 produtos químicos) uma taxa de falsos negativos de 4 % (com base em 57 produtos químicos), uma especificidade de 63 % (55 produtos químicos) e uma taxa de falsos positivos de 37 % (55 produtos químicos) comparativamente com dados do ensaio ocular in vivo no coelho – método de ensaio B.5 (2)(14) classificado de acordo com o sistema de classificação GHS da ONU e com o CRE. Um estudo em que foram ensaiados com o EIT da EpiOcular™ 97 formulações de produtos agroquímicos líquidos demonstrou um desempenho do método de ensaio para este tipo de misturas similar ao obtido no estudo de validação (23). As 97 formulações distribuíram-se do seguinte modo: 21 da categoria 1, 19 da categoria 2A, 14 da categoria 2B e 43 sem categoria, classificadas de acordo com o sistema de classificação GHS da ONU com base em dados de referência do ensaio ocular in vivo do coelho (método de ensaio B.5) (2)(14). Foi obtida uma exatidão global de 82 % (com base em 97 formulações), uma sensibilidade de 91 % (54 formulações), uma taxa de falsos negativos de 9 % (54 formulações), uma especificidade de 72 % (43 formulações) e uma taxa de falsos positivos de 28 % (43 formulações) (23).
|
|
15.
|
O IET HCE da SkinEthic™ pode ser utilizado para identificar produtos químicos que não necessitem de ser classificados em termos de irritação ocular nem de lesões oculares graves de acordo com o sistema de classificação GHS da ONU e com o CRE. Tendo em conta os dados obtidos no estudo de validação (10) (11), o EIT HCE da SkinEthic™ tem uma exatidão global de 84 % (com base em 200 produtos químicos), uma sensibilidade de 95 % (97 produtos químicos) uma taxa de falsos negativos de 5 % (97 produtos químicos), uma especificidade de 72 % (103 produtos químicos) e uma taxa de falsos positivos de 28 % (103 produtos químicos) comparativamente com dados do ensaio ocular in vivo no coelho (método de ensaio B.5 (2)(14) classificado de acordo com o sistema de classificação GHS da ONU e com o CRE.
|
|
16.
|
As taxas de falsos negativos obtida com ambos os ensaios RhCE, tanto para as substâncias como para as misturas, estão dentro dos 12 % de probabilidade de os produtos químicos serem identificados como pertencendo à categoria 2 ou a nenhuma do GHS da ONU e do CRE pelo ensaio ocular de Draize in vivo, em testes repetidos; ou seja, são devidas à variabilidade intrínseca do método de ensaio (24). As taxas de falsos positivos obtidas com ambos os métodos de ensaio RhCE tanto com substâncias como com misturas não são críticas no contexto do presente método de ensaio, uma vez que todos os produtos químicos que produzem uma viabilidade dos tecidos igual ou inferior aos limites estabelecidos (ver ponto 44) requerem ensaios complementares por recurso a outros métodos de ensaio in vitro, ou, em último recurso, em coelhos, consoante os requisitos regulamentares, utilizando uma estratégia de ensaios sequenciais, numa abordagem de ponderação da suficiência da prova. Estes métodos podem ser usados para todos os tipos de produtos químicos, devendo um resultado negativo ser aceite para que não se classifique um produto químico em termos de irritação ocular e de lesões oculares graves (nenhuma categoria do GHS da ONU/CRE). Devem consultar-se as entidades reguladoras competentes antes da utilização do EIT da EpiOcular™ e do EIT HCE da SkinEthic™ quanto à aplicação de sistemas de classificação diferentes do GHS da ONU/CRE.
|
|
17.
|
Uma limitação do presente método de ensaio reside no facto de não permitir distinguir irritação ocular/efeitos oculares reversíveis (categoria 2) e lesões oculares graves/efeitos oculares irreversíveis (categoria 1), definidos no sistema GHS da ONU e no CRE, nem entre irritantes oculares (categoria 2A facultativa) e irritantes oculares ligeiros (categoria 2B facultativa), definidos no sistema GHS da ONU (1). Para o efeito, são necessários ensaios complementares por recurso a outros métodos de ensaio in vitro.
|
|
18.
|
A expressão «produto químico em estudo» é utilizada no presente método de ensaio refere-se ao objeto do estudo (77), não pressupondo a aplicabilidade do método de ensaio RhCE ao ensaio de substâncias e/ou misturas.
|
PRINCÍPIO DO ENSAIO
|
19.
|
O produto químico em estudo é aplicado localmente a um mínimo de dois sistemas tridimensionais de tecido RhCE, sendo a viabilidade dos tecidos medida após a exposição e um período de incubação subsequente ao tratamento. Os tecidos RhCE são reconstruídos a partir de queratinócitos primários da epiderme humana ou de células epiteliais da córnea humana imortalizadas, cultivadas durante vários dias para formar um epitélio escamoso, estratificado, altamente diferenciado e morfologicamente semelhante ao encontrado na córnea humana. O tecido RhCE da EpiOcular™ consiste em, pelo menos, três camadas viáveis de células e uma superfície não queratinizada, apresentando uma estrutura análoga à estrutura in vivo. O tecido HCE RhCE da SkinEthic™ consiste em, pelo menos, quatro camadas viáveis de células, incluindo células colunares basais, células transicionais aladas e células escamosas superficiais similares às do epitélio da córnea humana normal. (20) (26).
|
|
20.
|
As lesões oculares graves, ou irritação ocular grave, induzidas por produtos químicos, que se manifestam in vivo, principalmente, por opacidade da córnea, irite, vermelhidão da conjuntiva e/ou equimose conjuntival, resultam de uma sucessão de eventos que se inicia com a penetração do produto químico na córnea e/ou na conjuntiva e a produção de danos nas células. Estes podem ocorrer por vários modos de ação, nomeadamente lise da membrana celular (caso, por exemplo, dos tensioativos e solventes orgânicos), coagulação macromolecular (sobretudo proteínas) (por exemplo, tensioativos, solventes orgânicos, ácidos e compostos alcalinos), saponificação de lípidos (por exemplo, por compostos alcalinos), interações de alquilação ou outras interações covalentes com macromoléculas (por exemplo, por descolorantes, peróxidos e alquiladores) (15) (27) (28). No entanto, ficou demonstrado que a citotoxicidade desempenha um papel mecanístico importante, se não essencial, na determinação da resposta global a um produto químico, expressa em lesões oculares graves/irritação ocular, independentemente dos processos físico-químicos subjacentes aos danos nos tecidos (29) (30). Além disso, o potencial de lesões oculares graves/irritação ocular de um produto químico é principalmente determinado pela gravidade da lesão inicial (31), relacionada com a extensão da morte celular (29) e com a intensidade das respostas e eventuais resultados posteriores (32). Assim, de um modo geral, os irritantes leves apenas afetam o epitélio superficial da córnea, os irritantes ligeiros e moderados afetam, principalmente, o epitélio e o estroma superficial e os irritantes fortes afetam o epitélio, o estroma profundo e, por vezes, o endotélio corneano (30) (33). A medição da viabilidade do tecido RhCE, após exposição tópica a um produto químico em estudo, para identificar produtos químicos que não necessitem de ser classificados em caso de lesões oculares graves/irritação ocular (nenhuma categoria no sistema GHS da ONU e no CRE) baseia-se no pressuposto de que todos os produtos químicos indutores de lesões oculares ou irritação ocular graves induzirão citotoxicidade no epitélio da córnea e/ou na conjuntiva.
|
|
21.
|
A viabilidade dos tecidos RhCE é normalmente medida pela conversão enzimática do corante vital MTT [3-(4,5-dimetiltiazol-2-il)-2,5-difeniltetrazólio, azul de tiazolilo, n.o CAS 298-93-1] pelas células viáveis do tecido num sal azul de MTT formazano, que é determinado quantitativamente depois de extraído dos tecidos (16). Os produtos químicos que não necessitam de classificação e rotulagem de acordo com o sistema GHS da ONU/CRE (nenhuma categoria) são identificados como aqueles relativamente aos quais a viabilidade dos tecidos não diminui aquém de um limiar definido (ou seja, viabilidade dos tecidos não diminui > 60 %, no caso do EITL da EpiOcular™ e do HCE EITL da SkinEthic™ (78), e > 50 %, no caso do HCE EITS da SkinEthic™ (79)) (ver ponto 44).
|
DEMONSTRAÇÃO DE COMPETÊNCIA TÉCNICA
|
22.
|
Antes de utilizarem de forma rotineira os ensaios RhCE para fins normativos, os laboratórios devem demonstrar a sua competência técnica através da previsão correta dos quinze produtos químicos para a demonstração de competência técnica que constam do quadro 1. Estes produtos químicos foram selecionados a partir dos produtos utilizados nos estudos de validação dos VRM (8) (10) (11). A seleção inclui, na medida do possível, produtos químicos que: (i) abrangem diferentes estados físicos; (ii) cobrem toda a gama de respostas in vivo expressas em lesões oculares graves/irritação ocular, com base em resultados de alta qualidade obtidos no ensaio ocular in vivo de referência no coelho (método de ensaio B.5) (2)(14), no sistema de classificação GHS da ONU (categorias 1, 2A, 2B ou nenhuma categoria) (1) e no sistema de classificação CRE (categorias 1, 2 ou nenhuma categoria); (iii) cobrem os vários fatores de classificação in vivo (24)(25); (iv) são representativos das classes de produtos químicos utilizadas no estudo de validação (8)(10)(11); (v) cobrem uma boa e ampla representação dos grupos funcionais orgânicos (8)(10)(11); (vi) possuem estruturas químicas bem definidas (8)(10)(11); (vii) são coloridos e/ou redutores diretos do MTT; (viii) produziram resultados reprodutíveis em métodos de ensaio RhCE durante as respetivas validações; (ix) foram corretamente preditos por métodos de ensaio RhCE durante os respetivos estudos de validação; (x) cobrem toda a gama de respostas in vitro baseadas em dados de alta qualidade relativos aos métodos de ensaio RhCE (viabilidade de 0 a 100 %); (xi) estão comercialmente disponíveis; e (xii) não têm custos de aquisição e/ou de eliminação proibitivos. Caso um produto químico constante da lista não esteja disponível ou não possa ser utilizado por outros motivos justificados, pode utilizar-se outro produto que satisfaça os critérios acima descritos, por exemplo, com base nos produtos químicos utilizados na validação do método de referência. Qualquer alteração deve, no entanto, ser justificada.
Quadro 1
Lista de produtos químicos recomendados para demonstração de competência técnica
|
Denominação química
|
N.o CAS
|
Grupo funcional orgânico (80)
|
Estado físico
|
VRM1 — (%) viabilidade (81)
|
VRM2 — (%) viabilidade (82)
|
Previsão do VRM
|
Redutor do MTT
|
Interf. de cor
|
|
Categoria in vivo1 (83)
|
|
|
Tioglicolato de metilo
|
2365-48-2
|
Éster de ácido carboxílico; tioálcool
|
L
|
10,9 ±6,4
|
5,5 ±7,4
|
Previsão impossível
|
Y (forte)
|
N
|
|
Acrilato de hidroexitilo
|
818-61-1
|
Acrilato; Álcool
|
L
|
7,5±4,7 (84)
|
1,6 ±1,0
|
Previsão impossível
|
N
|
N
|
|
2,5-Dimetil-2,5-hexanodiol
|
110-03-2
|
Álcool
|
S
|
2,3 ±0,2
|
0,2 ±0,1
|
Previsão impossível
|
N
|
N
|
|
Oxalato de sódio
|
62-76-0
|
Ácido dicarboxílico
|
S
|
29,0 ±1,2
|
5,3 ±4,1
|
Previsão impossível
|
N
|
N
|
|
Categoria in vivo2A (83)
|
|
|
Di-D-gluconato de N,N″-bis(4-clorofenil)-3,12-diimino- 2,4,11,13-tetradecano-diimidamida (20 %, aquoso) (85)
|
18472-51-0
|
Halogeneto de heterocíclicos aromáticos; halogeneto de arilo; grupo di-hidroxilo; guanidina;
|
L
|
4,0 ±1,1
|
1,3 ±0,6
|
Previsão impossível
|
N
|
Y (fraco)
|
|
Benzoato de sódio
|
532-32-1
|
Arilo; ácido carboxílico
|
S
|
3,5 ±2,6
|
0,6 ±0,1
|
Previsão impossível
|
N
|
N
|
|
Categoria in vivo 2B (83)
|
|
|
Dietiltoluamida
|
134-62-3
|
Benzamida
|
L
|
15,6 ±6,3
|
2,8 ±0,9
|
Previsão impossível
|
N
|
N
|
|
2,2-Dimetil-3-metilenobiciclo [2.2.1]heptano
|
79-92-5
|
Alcano ramificado com carbono terciário; alceno; biciclo-heptano; carbociclo de anel em ponte; carbociclo cicloalcano
|
S
|
4,7 ±1,5
|
15,8 ±1,1
|
Previsão impossível
|
N
|
N
|
|
Nenhuma categoria in vivo
(83)
|
|
|
Etilsulfato de 1-etil-3-metilimidazólio
|
342573-75-5
|
Alcóxido; sal de amónio; arilo; imidazolo; sulfato
|
L
|
79,9 ±6,4
|
79,4 ±6,2
|
Nenhuma cat.
|
N
|
N
|
|
Éter dicaprílico
|
629-82-3
|
Alcóxido;éter
|
L
|
97,8 ±4,3
|
95,2 ±3,0
|
Nenhuma cat.
|
N
|
N
|
|
Butóxido de piperonilo
|
51-03-6
|
Alcoxi; benzodioxole; benzilo; éter
|
L
|
104,2 ±4,2
|
96,5 ±3,5
|
Nenhuma cat.
|
N
|
N
|
|
Polietilenoglicol (PEG-40) com óleo de rícino hidrogenado
|
61788-85-0
|
Acila; álcool; alilo; éter
|
Viscosidade
|
77,6 ±5,4
|
89,1 ±2,9
|
Nenhuma cat.
|
N
|
N
|
|
1-(4-Clorofenil)-3-(3,4-diclorofenil)ureia
|
101-20-2
|
Heterocíclicos aromáticos; halogeneto de arilo; derivados de ureia
|
S
|
106,7 ±5,3
|
101,9 ±6,6
|
Nenhuma cat.
|
N
|
N
|
|
2,2′-Metileno-bis[6-(2H-benzotriazol-2-il)-4-(1,1,3,3-tetrametilbutil)fenol]
|
103597-45-1
|
Alcano ramificado com carbono quaternário; carbocíclicos fundidos aromáticos; heterocíclicos fundidos saturados; compostos quinóides precursores; terc-butilo
|
S
|
102,7 ±13,4
|
97,7 ±5,6
|
Nenhuma cat.
|
N
|
N
|
|
Tetrafluoroborato de potássio
|
14075-53-7
|
Sal inorgânico
|
S
|
88,6 ±3,3
|
92,9 ±5,1
|
Nenhuma cat.
|
N
|
N
|
|
Abreviaturas:
N.o CAS = Número de registo do Chemical Abstracts Service; GHS da ONU = Sistema Mundial Harmonizado de Classificação e Rotulagem de Produtos Químicos das Nações Unidas (em inglês: Globally Harmonized System of Classification and Labelling of Chemicals – UN GHS); (1); VRM1 = Método de referência validado, IET da EpiOcular™; VRM2 = Método de referência validado, HCE IET da SkinEthic™; Interf. de cor = interferência de cor com a medição da absorvância padrão (densidade ótica – DO) do MTT formazano.
|
|
|
23.
|
No âmbito do ensaio de competência técnica, recomenda-se que o utilizador verifique, após a receção dos tecidos, as propriedades de barreira dos mesmos, especificadas pelo produtor do modelo de tecido RhCE. Este aspeto é especialmente importante se os tecidos forem expedidos a longas distâncias ou demorarem muito tempo a chegar. A partir do momento em que um método de ensaio esteja implantado e tenha sido adquirida e demonstrada competência técnica para o utilizar, não é necessário efetuar por rotina a referida verificação. Recomenda-se, contudo, que, ao utilizar com regularidade um método de ensaio, se continue a avaliar periodicamente as propriedades de barreira.
|
PROCEDIMENTO
|
24.
|
Os ensaios atualmente abrangidos por este método de ensaio são os cientificamente válidos EIT da EpiOcular™ e HCE EIT da SkinEthic™ (9) (12) (13), referidos como métodos de referência validados (VRM1 e VRM2, respetivamente). Existem procedimentos operacionais normalizados (PON) para os métodos de ensaio RhCE, que devem ser utilizados nas fases de implementação e utilização dos métodos de ensaio no laboratório (34) (35). Os pontos seguintes e o apêndice 2 descrevem os principais componentes e procedimentos dos ensaios RhCE.
|
COMPONENTES DO MÉTODO DE ENSAIO RHCE
Condições gerais
|
25.
|
Devem utilizar-se células de origem humana adequadas para reconstruir o tecido tridimensional do epitélio corneano, composto por células progressivamente estratificadas, mas não cornificadas. O modelo do tecido RhCE é preparado em inserções com uma membrana sintética porosa, através da qual os nutrientes podem passar para as células. Devem estar presentes no epitélio corneano reconstruído várias camadas de células epiteliais viáveis, não queratinizadas. O modelo de tecido RhCE deve ter a superfície epitelial em contato direto com o ar, de modo a permitir a exposição tópica direta aos produtos químicos em estudo numa forma semelhante àquela como o epitélio corneano seria exposto in vivo. O modelo de tecido RhCE deve formar uma barreira funcional com robustez suficiente para resistir a uma penetração rápida de substâncias de referência citotóxicas, por exemplo, o Triton X-100 ou SDS. Importa demonstrar a eficácia da barreira, que pode ser avaliada pela determinação do tempo de exposição necessário para reduzir a viabilidade tecidual em 50 % (ET50), por aplicação de uma substância de referência a uma concentração específica fixa – p.ex., 100 μl de Triton X-100 a 0,3 % (v/v) – ou à concentração na qual a substância de referência reduz a viabilidade dos tecidos em 50 % (IC50) após um tempo de exposição determinado (p.ex., 30 minutos de exposição com 50 μl de SDS) – ver ponto 30. As propriedades de contenção do tecido RhCE reconstruído devem impedir a passagem do produto químico em estudo na extremidade do tecido viável, o que redundaria numa modelação deficiente da exposição da córnea. As células de origem humana utilizadas para reconstruir o tecido RhCE devem estar isentas de contaminação por bactérias, vírus, micoplasmas e fungos. A esterilidade do tecido deve ser verificada pelo fornecedor, para assegurar que não está contaminado por fungos e bactérias.
|
Condições funcionais
Viabilidade
|
26.
|
O ensaio utilizado para quantificar a viabilidade dos tecidos é o ensaio MTT (16). As células viáveis do tecido RhCE reconstruído reduzem o corante vital MTT a um precipitado azul de MTT formazano, que é depois extraído do tecido com isopropanol (ou solvente similar). O MTT formazano extraído pode ser quantificado por um método normalizado de medição da absorvância (densidade ótica – DO) ou um procedimento de espetrofotometria HPLC/UPLC (36). A densidade ótica do solvente de extração isolado deve ser suficientemente baixa (< 0,1). Os utilizadores do tecido RhCE reconstruído devem garantir que cada lote utilizado preenche os critérios definidos para o controlo negativo. Os intervalos de aceitabilidade para os valores de densidade ótica dos controlos negativos para o VRMS são indicados no quadro 2. Os utilizadores de espetrofotometria HPLC/UPLC devem utilizar as gamas de controlo negativo da densidade ótica indicadas no quadro 2 como critério de aceitação para o controlo negativo. O relatório do ensaio deve documentar a estabilidade em cultura dos tecidos tratados com a substância de controlo negativo (fornecer medições da viabilidade de tecidos similares) no período de exposição do ensaio. O procedimento deve ser idêntico ao seguido pelo produtor de tecidos no âmbito do controlo de qualidade da liberação dos lotes de tecido; neste caso, podem ser aplicáveis critérios de aceitação diferentes dos especificados no quadro 2. O fabricante de tecidos RHCE deve estabelecer um intervalo de aceitabilidade (limite superior e inferior) para os valores de densidade ótica dos controlos negativos (nas condições do método de ensaio do controlo da qualidade) do concetor/fornecedor.
|
Quadro 2
Intervalos de aceitabilidade dos valores de densidade ótica dos controlos negativos (para os utilizadores do ensaio)
|
Ensaio
|
Limite inferior de aceitabilidade
|
Limite superior de aceitabilidade
|
|
EIT da EpiOcular™ (OCL-200) – VRM1 (para protocolos de líquidos e sólidos)
|
> 0,8 (86)
|
< 2,5
|
|
HCE EIT (HCE/S) da SkinEthic™ – VRM2 (para protocolos de líquidos e sólidos)
|
> 1,0
|
≤ 2,5
|
Função de barreira
|
27.
|
O tecido RhCE deve ser suficientemente espesso e robusto para resistir à rápida penetração de substâncias de referência citotóxicas, estimada, por exemplo, pelo EF50 (Triton X-100) ou pela IC50 (SDS) (quadro 3). A função de barreira de cada lote do tecido RhCE utilizado deve ser demonstrada pelo concetor/fornecedor do RhCE, aquando do fornecimento dos tecidos ao utilizador final (ver ponto 30).
|
Morfologia
|
28.
|
O exame histológico do tecido RhCE deve mostrar uma estrutura de epitélio corneano humano (incluindo pelo menos 3 camadas de células epiteliais viáveis e uma superfície não queratinizada). Para os VRM, a morfologia adequada foi estabelecida pelo criador/fornecedor, pelo que não precisa de ser novamente demonstrada por um utilizador do método de ensaio para cada lote de tecidos utilizado.
|
Reprodutibilidade
|
29.
|
Os resultados dos controlos positivos e negativos do método de ensaio devem mostrar reprodutibilidade ao longo do tempo.
|
Controlo de qualidade
|
30.
|
O tecido RhCE só deve ser utilizado se o concetor/fornecedor demonstrar que cada lote obedece a critérios definidos de liberação da produção, dos quais o de viabilidade (ponto 26) e o de função de barreira (ver ponto 27) são os mais relevantes. O concetor/fornecedor do tecido RhCE deve estabelecer uma gama de aceitabilidade (limites superior e inferior) para as funções de barreira, medidas por ET50 ou IC50 (ver pontos 25 e 26). A gama de aceitabilidade do ET50 e IC50, utilizada como critério de controlo da qualidade para a liberação dos lotes de RhCE pelo seu concetor/fornecedor (utilizado nos VRM), figura no quadro 3. O concetor/fornecedor do tecido RhCE deve fornecer aos utilizadores do método de ensaio dados que demonstrem o cumprimento de todos os critérios de liberação da produção, para que os utilizadores possam incluir essas informações no relatório de ensaio. Apenas os resultados obtidos com tecidos que satisfaçam todos os critérios de liberação da produção podem ser aceites para a previsão fiável de produtos químicos que, em conformidade com o GHS da ONU/CRE, não necessitem de classificação e rotulagem em termos de irritação ocular ou de lesões oculares graves.
|
Quadro 3
Critério de QC para a liberação de lotes
|
Ensaio
|
Limite inferior de aceitabilidade
|
Limite superior de aceitabilidade
|
|
EIT da EpiOcular™ (OCL-200) – VRM1 [100 μl de Triton X-100 a 0,3 % (v/v)]
|
ET50 = 12,2 min
|
ET50 = 37,5 min
|
|
HCE IET (HCE/S) da SkinEthic™ – VRM2 (tratamento de 30 minutos, com 50 μl de SDS)
|
IC50 = 1 mg/ml
|
IC50 = 3,2 mg/ml
|
Aplicação dos produtos químicos em estudo e das substâncias de controlo
|
31.
|
Em cada ensaio de cada série, devem utilizar-se, pelo menos, dois replicados de tecido para cada produto químico em estudo e para cada substância de controlo. Utilizam-se dois protocolos de tratamento diferentes, um para produtos químicos em estudo no estado líquido e outro para produtos químicos no estado sólido (34) (35). Para ambos os métodos e protocolos, a superfície do modelo de tecido deve ser humedecida com solução salina tamponada de fosfatos de Dulbecco, sem cálcio e sem magnésio (DPBS sem Ca2+/Mg2+), antes da aplicação dos produtos químicos em estudo, para simular as condições de humidade do olho humano. O tratamento dos tecidos é iniciado com a exposição ao(s) produto(s) químico(s) em estudo e às substâncias de controlo. Para ambos os protocolos de tratamento de ambos os VRM, aplica-se uma quantidade suficiente do produto químico em estudo ou da substância de controlo para cobrir uniformemente a superfície epitelial, evitando-se uma dose infinita (ver pontos 32 e 33) (apêndice 2).
|
|
32.
|
Os produtos químicos em estudo que possam ser pipetados a 37 °C ou a temperaturas inferiores – utilizando uma pipeta de deslocamento positivo, se necessário – são tratados como líquidos nos VRM; caso contrário, devem ser tratados como sólidos (ver ponto 33). Nos VRM, o produto químico líquido em estudo está uniformemente espalhado pela superfície do tecido – ou seja, um mínimo de 60 μl/cm de aplicação; ver apêndice 2, (33) (34). Devem evitar-se, na medida do possível, efeitos capilares (efeitos de tensão superficial) que possam ocorrer devido aos baixos volumes aplicados na inserção (na superfície do tecido), para garantir a dosagem correta do tecido. Os tecidos tratados com produtos químicos líquidos em estudo são incubados durante 30 minutos em condições de cultura normais (37±2 °C, 5±1 % CO2, ≥ 95 % HR). No final do período de exposição, devem remover-se cuidadosamente da superfície do tecido o produto químico líquido em estudo e as substâncias de controlo, através de uma lavagem extensiva com DPBS sem Ca2+/Mg2+, à temperatura ambiente. Esta fase de lavagem é seguida por uma imersão pós-exposição em meio fresco à temperatura ambiente – para retirar o produto químico em estudo absorvido pelo tecido – por um período predefinido, que varia em função do VRM utilizado. Unicamente para o VRM1, antes da realização do ensaio com MTT, é praticada uma incubação pós-exposição em meio fresco, em condições de cultura normais – ver apêndice 2, (34) (35).
|
|
33.
|
Os produtos químicos em estudo que não possam ser pipetados a temperaturas até 37 °C são tratados como sólidos no VRM. A quantidade de produto aplicada deve ser suficiente para cobrir toda a superfície do tecido, ou seja, deve utilizar-se um mínimo de 60 mg/cm2 (apêndice 2). Sempre que possível, os sólidos devem ser ensaiados na forma de pó fino. Os tecidos tratados com produtos químicos sólidos são incubados por um período predefinido, consoante o VRM utilizado, em condições de cultura normais – ver apêndice 2, (34) (35). No final do período de exposição, devem remover-se cuidadosamente da superfície do tecido o produto químico em estudo e as substâncias de controlo, através de uma lavagem extensiva com DPBS sem Ca2+/Mg2+, à temperatura ambiente. Esta fase de lavagem é seguida de uma imersão pós-exposição em meio fresco, à temperatura ambiente – para remoção do produto químico em estudo absorvido pelo tecido –, por um período predefinido que varia em função do VRM utilizado, e de uma incubação após exposição em meio fresco em condições de cultura normais, antes da realização do ensaio com MTT – ver apêndice 2, (34) (35).
|
|
34.
|
Devem prever-se controlos negativos e positivos paralelos, para demonstrar que a viabilidade (determinada com o controlo negativo) e a sensibilidade (determinada com o controlo positivo) dos tecidos se encontram dentro dos intervalos de aceitação definidos com base em dados históricos. O controlo negativo paralelo proporciona ainda a linha de base (100 % de viabilidade dos tecidos) para calcular a percentagem de viabilidade relativa dos tecidos tratados com o produto químico em estudo (ensaio de % de viabilidade). A substância de controlo positivo recomendada para o VRM é o acetato de metilo puro (n.o CAS 79-20-9, comercializado, por exemplo, pela Sigma-Aldrich, Cat n.o 45997; líquido). As substâncias de controlo negativo recomendadas para os VRM1 e VRM2 são água ultrapura e DPBS sem Ca2+/Mg2+, respetivamente. Foram estas as substâncias de controlo utilizadas nos estudos de validação dos VRM e aquelas para as quais existem mais dados. A utilização de outras substâncias de controlo, positivas ou negativas, adequadas deverá justificar-se científica e adequadamente. Os controlos negativos e positivos devem ser ensaiados com o(s) protocolo(s) utilizado(s) para os produtos químicos em estudo incluídos na série de ensaios (líquidos e/ou sólidos). A aplicação deve ser seguida da exposição ao produto químico em estudo, da lavagem, da imersão pós-exposição e da incubação pós-exposição, quando pertinente, conforme descrito para os controlos paralelos aos ensaios dos produtos químicos líquidos (ver ponto 32) ou para os controlos paralelos aos ensaios dos produtos químicos sólidos (ver ponto 33), antes da realização do ensaio com MTT (ver ponto 35) (34) (35). Um único conjunto de controlos negativos e positivos é suficiente para todos os produtos químicos no mesmo estado físico (líquidos ou sólidos) incluídos na mesma série de ensaios.
|
Medições de viabilidade dos tecidos
|
35.
|
Para medir a viabilidade celular no âmbito do presente método, deve utilizar-se o método quantitativo validado do MTT, que é compatível com a arquitetura tridimensional dos tecidos. O ensaio do MTT é efetuado imediatamente após o período de incubação pós-exposição. Nos VRM, a amostra de tecido RhCE é colocada em 0,3 ml de solução de MTT a 1 mg/ml durante 180±15 minutos, em condições de cultura normais. O corante vital MTT é reduzido a um precipitado azul de MTT formazano pelas células viáveis do tecido RhCE. O precipitado é em seguida extraído do tecido com recurso a um volume adequado de isopropanol, ou a um solvente similar (34) (35). No caso dos tecidos ensaiados com produtos químicos líquidos em estudo, a extração deve ser feita da parte superior e inferior, enquanto no caso dos tecidos ensaiados com produtos químicos sólidos e líquidos coloridos, a extração devem ser efetuada unicamente da parte inferior, para minimizar uma eventual contaminação da solução de extração de isopropanol com produto químico em estudo que possa ter permanecido no tecido. No caso dos tecidos que tenham sido ensaiados com produtos químicos líquidos e que não tenham sido imediatamente lavados, a extração também deve ser feita apenas da parte inferior. As substâncias de controlo positivo e positivo paralelo devem ser tratadas de forma similar à do produto químico em estudo. O MTT formazano extraído pode ser quantificado quer por medição da absorvância-padrão (DO) a 570 nm, utilizando um filtro de banda passante de ±30 nm, no máximo, quer por um procedimento de espetrofotometria HPLC/UPLC –ver ponto 42 (11) (36).
|
|
36.
|
As propriedades óticas do produto químico em estudo ou a sua ação química sobre o MTT podem interferir na medição do MTT formazano, conduzindo a uma estimativa falsa da viabilidade dos tecidos. Os produtos químicos em estudo podem interferir com a medição do MTT formazano por redução direta do MTT a azul de MTT formazano e/ou por interferência cromática, se a sua absorvância, quer natural, quer devida a procedimentos de tratamento, se situar na mesma gama de densidade ótica que o MTT formazano (cerca de 570 nm). Devem realizar-se controlos prévios para permitir a identificação de potenciais redutores diretos do MTT e/ou de produtos químicos com interferência cromática; devem ainda efetuar-se controlos adicionais para detetar e corrigir possíveis interferências desses produtos químicos em estudo (ver os pontos 37 a 41). Este aspeto é especialmente importante quando um produto químico em estudo não é completamente removido do tecido RhCE por lavagem ou quando penetra no epitélio corneano propriamente dito e está, portanto, presente no tecido RhCE quando é realizado o ensaio do MTT. No caso dos produtos químicos que absorvem a luz na mesma gama do MTT formazano (naturalmente ou após tratamento) e que não são compatíveis com a medição da absorvância-padrão (DO) do MTT formazano devido a uma interferência demasiado forte – ou seja, uma forte absorvância a 570 ±30 nm –, pode recorrer-se a um procedimento de espetrofotometria-HPLC/UPLC para medir o MTT formazano (ver pontos 41 e 42) (11) (36). Nos PON dos VRM está disponível uma descrição pormenorizada do modo de deteção e correção da redução direta do MTT e das interferências dos agentes corantes (34) (35). Os apêndices III e IV contêm fluxogramas com orientações sobre o modo de identificar e manusear os redutores diretos do MTT e/ou os produtos químicos suscetíveis de interferirem cromaticamente no VRM1 e VRM2, respetivamente.
|
|
37.
|
Para identificar eventuais interferências provocadas por produtos químicos em estudo com a mesma gama de absorvância da luz que o MTT (naturalmente ou após tratamento) e determinar a necessidade de proceder a controlos adicionais, o produto químico em estudo é adicionado a água e/ou isopropanol e incubado, por um período adequado, à temperatura ambiente – ver apêndice 2, (34) (35). Se o produto químico em estudo, dissolvido em água e/ou em isopropanol, mostrar uma absorvância suficiente na gama de 570 ±20 nm para o VRM1 (ver apêndice 3), ou caso se obtenha uma solução colorida quando se mistura o produto com água, para o VRM2 (ver apêndice 4), presume-se que o produto químico em causa interfere com a medição da absorvância-padrão do MTT formazano, devendo realizar-se controlos de corante complementares ou, em alternativa, utilizar um procedimento de espetrofotometria HPLC/UPLC, caso em que estes controlos não são exigidos (ver pontos 41 e 42 e apêndices III e IV) (34) (35). Ao efetuar a medição da absorvância-padrão, deve-se aplicar cada um dos produtos químicos em estudo interferentes em, pelo menos, dois replicados viáveis do tecido, que são submetidos à totalidade do processo de ensaio, mas são incubados em meio e não na solução de MTT durante a fase de incubação com MTT, de modo a produzir uma cor não específica no controlo dos tecidos vivos (NSCvivos) (34) (35). O controlo NSCvivos tem de ser realizado em paralelo ao ensaio do produto químico corado em estudo; no caso de ensaios múltiplos, é necessário efetuar, em cada série de ensaios, um NSCvivos independente para cada ensaio, devido à variabilidade biológica intrínseca dos tecidos vivos. A viabilidade real do tecido é calculada como a percentagem de viabilidade dos tecidos obtida com tecidos vivos expostos ao produto químico em estudo interferente e incubados com a solução de MTT (ensaio de % de viabilidade) menos a percentagem de cor não específica obtida com tecidos vivos expostos ao produto químico em estudo interferente e incubados em meio sem MTT paralelamente à correção do ensaio (% NSCvivos), ou seja: viabilidade real do tecido = [ensaio de % de viabilidade] - [% NSCvivos].
|
|
38.
|
Para identificar os redutores diretos do MTT, os produtos químicos de ensaio devem ser adicionados a soluções de MTT recentemente preparadas. Adiciona-se uma quantidade adequada do produto químico em estudo a uma solução de MTT, sendo a mistura incubada durante cerca de 3 horas em condições de cultura normais (ver apêndices III e IV) (34) (35). Se a mistura de MTT que contém o produto químico em estudo (ou a suspensão, no caso de produtos químicos insolúveis) se tornar azul/púrpura, considera-se que o produto reduz diretamente o MTT, devendo proceder-se a um controlo funcional complementar nos tecidos RhCE não viáveis, independentemente de se recorrer à medição da absorvância padrão ou a um procedimento de espetrofotometria HPLC/UPLC. Este controlo funcional complementar utiliza tecidos mortos, que apenas possuem atividade metabólica residual, mas absorvem e retêm o produto químico em estudo de forma semelhante aos tecidos viáveis. Os tecidos do VRM1 mortos «por congelamento» são preparados por exposição a baixas temperaturas. Os tecidos do VRM2 mortos «em água» são preparados por incubação prolongada (pelo menos, 24 ±1 h) em água, seguida de armazenagem a baixa temperatura. Cada produto químico em estudo redutor do MTT é aplicado em, pelo menos, dois tecidos replicados mortos, que são submetidos a todo o processo de ensaio a fim de gerarem um controlo não específico da redução do MTT (NSMTT) (32) (33) (34) (35). Basta um único controlo do NSMTT para cada produto químico em estudo, independentemente do número de ensaios ou de ensaios/séries de ensaios realizados. A viabilidade real do tecido é calculada como a percentagem de viabilidade do tecido obtida com os tecidos vivos expostos ao redutor do MTT (ensaio de % de viabilidade), menos a percentagem de redução não específica do MTT obtida com os tecidos mortos expostos ao mesmo redutor do MTT, calculada em relação ao controlo negativo concomitante ao ensaio a corrigir, ou seja: viabilidade real do tecido = [ensaio de % de viabilidade] - [(% NSMTT].
|
|
39.
|
Os produtos químicos em estudo identificados como produzindo tanto interferências cromáticas (ver ponto 37) como uma redução direta do MTT (ver ponto 38) deverão também ser objeto de um terceiro conjunto de controlos no âmbito da medição da absorvância-padrão (DO), para além dos controlos de NSMTT e NSCvivos a que se referem os números anteriores. Trata-se, em geral, de produtos químicos de cor escura que absorvem luz na gama 570 ±30 nm (por exemplo, azuis, púrpura, pretos), porque a sua cor intrínseca impede a avaliação da capacidade de reduzirem diretamente o MTT, conforme descrito no ponto 38. Esta situação força o recurso, por defeito, a controlos de NSMTT em conjunto com os controlos NSCvivos. Os produtos químicos em estudo para os quais se realizem ambos os controlos NSMTT e NSCvivos podem ser absorvidos e retidos por tecidos vivos e mortos. Por conseguinte, neste caso, o controlo NSMTT permite não só corrigir uma potencial redução direta do MTT pelo produto químico em estudo, como também evitar as interferências cromáticas resultantes da absorção e da retenção do produto pelos tecidos mortos. Pode, assim observar-se a uma dupla correção das interferências cromáticas, uma vez que o controlo NCSvivos corrige já as interferências que resultam da absorção e retenção do produto químico em estudo pelos tecidos vivos. Para evitar uma eventual dupla correção da interferência cromática, importa realizar um terceiro controlo cromático não específico nos tecidos mortos (NSCmortos) – ver apêndices III e IV (34) (35). Neste controlo adicional, aplica-se o produto químico em estudo em, pelo menos, dois replicados dos tecidos mortos, que são submetidos à totalidade do procedimento de ensaio mas são incubados com meio durante a etapa de incubação com MTT, em vez da solução de MTT. Basta um único controlo NSCmortos por produto químico em estudo, independentemente do número de ensaios ou séries de ensaios, mas deve ser realizado concomitantemente com o controlo NSMTT e com o mesmo lote de tecidos. A viabilidade real do tecido é calculada como a percentagem de viabilidade dos tecidos obtida com tecidos vivos expostos ao produto químico (ensaio de % de viabilidade) menos %NSMTT menos % NSCvivos
mais a percentagem de cor não específica obtida com tecidos mortos expostos ao produto químico em estudo interferente e incubados em meio sem MTT, calculada em relação à amostra do controlo negativo realizado em simultâneo com o ensaio a corrigir (% NSCmortos), ou seja: viabilidade real do tecido = [ensaio de % de viabilidade] - [%NSMTT] - [%NSCvivos] + [%NSCmortos].
|
|
40.
|
É importante assinalar que a redução não específica do MTT e as interferências cromáticas não específicas podem aumentar a densidade ótica (nas medições da absorvância-padrão) do extrato de tecido acima da gama de linearidade do espetrofotómetro e que, na medição por espetrofotometria HPLC/UPLC, a redução não específica do MTT pode também aumentar a área do pico de formazano do extrato de tecido acima da gama de linearidade do espetrofotómetro. Neste contexto, cada laboratório deve determinar a densidade ótica/área de pico da gama de linearidade do respetivo espetrofotómetro, por exemplo, com MTT formazano (n.o CAS 57360-69-7), comercializado, por exemplo, pela Sigma-Aldrich (n.o Cat M2003), antes de iniciar o ensaio de produtos químicos em estudo para fins normativos.
|
|
41.
|
A medição da absorvância-padrão (DO) com espetrofotómetro é adequada para avaliar produtos químicos em estudo redutores diretos do MTT e com interferência cromática, se a interferência observada com a medição do MTT formazano não for muito forte (ou seja, se as DO dos extratos de tecidos obtidos com o produto químico em estudo, sem qualquer correção para a redução direta do MTT e/ou para a interferência cromática, se situarem na gama linear do espetrofotómetro). No entanto, os resultados para produtos químicos em estudo que produzam % NSMTT e/ou % NSCvivos iguais ou superiores a 60 % (VRM1 e protocolo de líquidos do VRM2) ou 50 % (protocolo de sólidos do VRM2) do controlo negativo devem ser interpretados com precaução, dado tratar-se do valor-limite estabelecido no VRMS para distinguir os produtos químicos classificados dos não classificados (ver ponto 44). Contudo, não é possível medir a absorvância-padrão (DO) se a interferência com a medição de MTT formazano for muito intensa (ou seja, de forma a que as DO não corrigidas dos extratos de tecidos se situem fora do intervalo linear do espectrofotómetro). Os produtos químicos em estudo corados ou que adquiram coloração em contacto com a água ou com o isopropanol e que interfiram muito fortemente com a medição da absorvância-padrão (DO) do MTT formazano podem também ser avaliados por recurso à espetrofotometria HPLC/UPLC (ver apêndices III e IV), pelo facto de o sistema HPLC/UPLC permitir a separação do MTT formazano do produto químico antes da sua quantificação (36). Por este motivo, nunca são necessários controlos NCSvivos ou NCSmortos quando se utiliza este método, independentemente do produto químico em estudo. Contudo, devem realizar-se controlos NSMTT se se suspeitar que o produto em causa reduz diretamente o MTT (cf. procedimento descrito no ponto 38). Devem também realizar-se estes controlos no caso de produtos químicos com uma coloração (intrínseca ou aparente quando dissolvidos em água) que impeça a avaliação da sua capacidade de reduzirem diretamente o MTT, conforme descrito no ponto 38. Caso se recorra à espetrofotometria HPLC/UPLC para medir o MTT formazano, a percentagem de viabilidade do tecido é calculada como a percentagem de todos os picos do MTT formazano obtidos com tecidos vivos expostos ao produto químico em estudo em relação a um pico obtido em paralelo com o controlo negativo. No caso de produtos químicos suscetíveis de reduzirem diretamente o MTT, a viabilidade real dos tecidos é calculada do seguinte modo: ensaio da % de viabilidade menos % NSMTT, tal como descrito no último período do ponto 38. Por último, importa referir que os redutores diretos do MTT, nomeadamente os que são também interferentes cromáticos e que são retidos nos tecidos após o tratamento e reduzem tão fortemente o MTT que produzem DO (pelo método de medição da densidade ótica padrão) ou áreas de picos (por espetrofotometria UPLC/HPLC) não abrangidas pela gama de linearidade do espetrofotómetro, não permitem que os extratos de tecidos sejam avaliados pelos métodos do ensaio RhCE, embora se preveja que tal ocorra apenas em casos muito raros.
|
|
42.
|
A espetrofotometria HPLC/UPLC pode ser utilizada para a medição do MTT formazano com todos os tipos de produtos químicos em estudo (corados ou não, redutores de MTT ou não) (36). Atendendo à diversidade dos sistemas de espetrofotometria HPLC/UPLC, não é exequível que cada utilizador adote exatamente as mesmas condições sistémicas. Pelo mesmo motivo, a qualificação dos sistemas de espetrofotometria HPLC/UPLC deve ser demonstrada antes de serem utilizados para quantificar o MTT formazano de extratos de tecidos, através do cumprimento dos critérios de aceitação de um conjunto de parâmetros de qualificação normalizados conformes com os descritos na U.S. Food and Drug Administration guidance for industry on bio-analytical method validation. Estes parâmetros-chave e os respetivos critérios de aceitação constam do apêndice 5. Se os critérios de aceitação definidos no apêndice 5 forem cumpridos, considera-se o sistema de espetrofotometria HPLC/UPLC qualificado para determinar o MTT formazano nas condições experimentais descritas no presente método de ensaio.
|
Critérios de aceitação
|
43.
|
Para cada série de ensaios que utilize lotes de tecidos RhCE conformes com o controlo de qualidade (ver ponto 30), os tecidos tratados com a substância de controlo negativa devem apresentar uma densidade ótica que reflita a qualidade dos tecidos subsequente à expedição, à receção e a todos os processos do protocolo, não devendo situar-se fora dos limites históricos descritos no quadro 2 (ver ponto 26). Da mesma forma, os tecidos tratados com a substância de controlo positivo (acetato de metilo) devem apresentar uma viabilidade tecidual média < 50 %, em relação ao controlo negativo no VRM1, no caso dos protocolos para líquidos ou sólidos, e ≤ 30 % (protocolo para líquidos) ou ≤ 20 % (protocolo para sólidos), em relação ao controlo negativo do VRM2, refletindo a sua capacidade de reação a um produto químico irritante, nas condições do método de ensaio (34)(35). A variabilidade entre os tecidos replicados dos produtos químicos em estudo e/ou dos produtos químicos de controlo deve situar-se dentro dos limites aceites (a diferença de viabilidade entre os dois tecidos replicados não deve exceder 20 % ou o desvio-padrão entre os três tecidos replicados não deve ser superior a 18 %). Se o controlo negativo ou positivo de uma série de ensaios se situar fora das gamas aceites, considera-se a série «não qualificada», pelo que deve ser repetida. Se a variabilidade entre os tecidos replicados do produto químico em estudo se situar fora da gama aceite, o ensaio é considerado «não qualificado», devendo o produto químico em estudo ser submetido a novo ensaio.
|
Interpretação dos resultados e modelo de previsão
|
44.
|
Os valores da densidade ótica/das áreas dos picos obtidos com extratos de tecidos replicados, para cada produto químico em estudo, são utilizados para calcular a percentagem média de viabilidade dos tecidos (média entre os replicados dos tecidos) normalizada para o controlo negativo, fixada em 100 %. O valor-limite da percentagem de viabilidade dos tecidos para identificar produtos químicos em estudo que não necessitem de ser classificados em termos de irritação ocular nem de lesões oculares graves (nenhuma categoria no sistema GHS da ONU e na categoria CRE) consta do quadro 4. Os resultados devem ser interpretados do seguinte modo:
|
—
|
Considera-se que o produto químico em estudo não necessita de classificação e rotulagem em conformidade com o sistema GHS da ONU e com o CRE (nenhuma categoria) se a percentagem média de viabilidade dos tecidos após exposição e incubação pós-exposição for superior ao valor-limite percentual estabelecido para a viabilidade dos tecidos, indicado no quadro 4. Neste caso, não são necessários ensaios complementares por outros métodos de ensaio.
|
|
—
|
Se a percentagem média de viabilidade dos tecidos após a exposição e a incubação pós-exposição for inferior ou igual ao valor-limite estabelecido para a percentagem de viabilidade dos tecidos, não é possível efetuar qualquer previsão, como se pode comprovar no quadro 4. Neste caso, serão necessários ensaios complementares por outros métodos de ensaio, dado que os métodos RhCE apresentam um certo número de resultados falsos positivos (ver pontos 14 a 15) e não permitem distinguir entre as categorias 1 e 2 do sistema GHS da ONU e do CRE (ver ponto 17).
|
Quadro 4
Modelos de previsão de acordo com a classificação GHS da ONU e da CRE
|
VRM
|
Nenhuma categoria
|
Previsão impossível
|
|
VRM 1 - EpiOcular™ EIT (para ambos os protocolos)
|
Viabilidade média dos tecidos > 60 %
|
Viabilidade média dos tecidos ≤ 60 %
|
|
VRM 2 - SkinEthic™ HCE EIT (para protocolos de líquidos)
|
Viabilidade média dos tecidos > 60 %
|
Viabilidade média dos tecidos ≤ 60 %
|
|
VRM2 - SkinEthic™ HCE EIT (para protocolos de sólidos)
|
Viabilidade média dos tecidos > 50 %
|
Viabilidade média dos tecidos ≤ 50 %
|
|
|
45.
|
Se a classificação do produto químico em estudo for inequívoca, basta normalmente um ensaio constituído por, pelo menos, dos replicados de tecidos. Porém, nos casos inconclusivos (por exemplo, medições discordantes dos replicados e/ou percentagem média de viabilidade do tecido igual a 60±5 % (VRM1 e VRM2, no caso do protocolo para líquidos) ou 50 ±5 % (VRM2, no caso do protocolo para sólidos), é de ponderar a realização de um segundo ensaio, bem como de um terceiro se os resultados dos dois primeiros forem discordantes.
|
|
46.
|
Para aumentar o desempenho global do método de ensaio para certos tipos de misturas, podem ponderar-se, quando adequado e justificável, valores-limite diferentes, no que respeita à percentagem de viabilidade dos tecidos, para distinguir produtos químicos em estudo classificados e não classificados (ver ponto 14). Os produtos químicos de referência podem ser úteis para avaliar o potencial de lesões oculares graves/irritação ocular dos produtos químicos em estudo ou de produtos desconhecidos de determinadas classes, bem como para avaliar o potencial de toxicidade ocular relativa de um produto químico classificado numa gama específica de respostas positivas.
|
DADOS E RELATÓRIOS
Dados
|
47.
|
Para cada produto químico em estudo, devem apresentar-se num quadro os dados relativos aos tecidos individuais replicados numa série de ensaios (por exemplo, valores de DO/áreas de pico do MTT formazano e dados relativos à percentagem calculada de viabilidade dos tecidos e a previsão final do método de ensaio RhCE), incluindo os dados dos ensaios repetidos, se pertinente. Além disso, deve comunicar-se a percentagem média de viabilidade dos tecidos e a diferença de viabilidade entre dois tecidos replicados (se n = 2 tecidos replicados) ou o DP (se n ≥ 3 tecidos replicados), para cada produto químico em estudo e para cada amostra de controlo. Devem também comunicar-se as eventuais interferências do produto químico em estudo com a medição do MTT formazano, expressas na redução direta do MTT e/ou em interferências cromáticas.
|
Relatório de ensaio
|
48.
|
O relatório do ensaio deve incluir as seguintes informações:
Produto químico em estudo
|
|
Substância monocomponente:
|
—
|
Identificação química, como as denominações IUPAC ou CAS, o(s) número(s) de registo CAS, o código SMILES ou InChI, a fórmula estrutural e/ou outros identificadores;
|
|
—
|
Estado físico, volatilidade, pH, log(P), peso molecular, classe química e outras propriedades físico-químicas pertinentes para a realização do estudo, na medida em que estejam disponíveis;
|
|
—
|
Grau de pureza, identidade química das impurezas, conforme adequado e exequível, etc.;
|
|
—
|
Pré-tratamento, se for o caso (aquecimento ou moagem, por exemplo);
|
|
—
|
Condições de armazenagem e de estabilidade, se conhecidas.
|
|
|
|
Substância multicomponentes, UVCB e mistura:
|
—
|
Caracterização, tanto quanto possível, por exemplo, por identidade química (ver supra), pureza, ocorrência quantitativa e principais propriedades físico-químicas (ver supra) dos componentes, na medida em que estejam disponíveis;
|
|
—
|
Estado físico e outras propriedades físico-químicas pertinentes para a realização do estudo, na medida em que estejam disponíveis;
|
|
—
|
Grau de pureza, identidade química das impurezas, conforme adequado e exequível, etc.;
|
|
—
|
Pré-tratamento, se for o caso (aquecimento ou moagem, por exemplo);
|
|
—
|
Condições de armazenagem e de estabilidade, se conhecidas.
|
|
Substâncias de controlo positivas e negativas
|
—
|
Identificação química, como as denominações IUPAC ou CAS, o(s) número(s) de registo CAS, o código SMILES ou InChI, a fórmula estrutural e/ou outros identificadores;
|
|
—
|
Estado físico, volatilidade, peso molecular, classe química e outras propriedades físico-químicas pertinentes para a realização do estudo, na medida em que estejam disponíveis;
|
|
—
|
Grau de pureza, identidade química das impurezas, conforme adequado e exequível, etc.;
|
|
—
|
Pré-tratamento, se for o caso (aquecimento ou moagem, por exemplo);
|
|
—
|
Condições de armazenagem e de estabilidade, se conhecidas;
|
|
—
|
Se for caso disso, justificação do recurso a um controlo negativo diferente da água ultrapura ou do DPBS sem Ca2+/Mg2+;
|
|
—
|
Se for caso disso, justificação do recurso a um controlo positivo diferente do acetato de metilo puro;
|
|
—
|
Referência a resultados históricos de controlo positivo e negativo que demonstrem critérios de aceitação válidos.
|
Informações relativas ao patrocinador e ao laboratório
|
—
|
Nome e endereço do patrocinador, do laboratório e do diretor do estudo.
|
|
—
|
Modelo de tecido RhCE e protocolo utilizados (e fundamentação das escolhas, se for caso disso)
|
Condições do método de ensaio
|
—
|
O modelo de tecido RhCE utilizado, incluindo o número do lote;
|
|
—
|
Comprimento de onda e gama de comprimentos de onda (se pertinente) para quantificar o MTT formazano e gama de linearidade do dispositivo de medição (espetrofotómetro, por exemplo).
|
|
—
|
Descrição do método utilizado para quantificar o MTT formazano;
|
|
—
|
Descrição do sistema de sistema de espetrofotometria HPLC/UPLC utilizado, se pertinente;
|
|
—
|
Informações completas que substanciem o modelo de tecido RhCE utilizado, incluindo dados sobre o respetivo desempenho. Devem incluir, nomeadamente, o seguinte:
|
i)
|
Controlo de qualidade da viabilidade (fornecedor)
|
|
ii)
|
Viabilidade nas condições do método de ensaio (utilizador);
|
|
iii)
|
Controlo da qualidade da função de barreira;
|
|
iv)
|
Morfologia, se conhecida;
|
|
v)
|
Capacidade de reprodutibilidade e de previsão;
|
|
vi)
|
Outros controlos de qualidade (QC) do modelo de tecidos RhCE, se pertinente;
|
|
|
—
|
Referência a dados históricos sobre o modelo do tecido RhCE. Deve incluir, nomeadamente, a aceitabilidade dos dados de controlo de qualidade em relação aos dados históricos dos lotes,
|
|
—
|
Declaração de que o laboratório de ensaio demonstrou competência técnica para a aplicação do método de ensaio antes da sua utilização regular, tendo procedido ao ensaio dos produtos químicos recomendados para demonstração de competência técnica;
|
Critérios de aceitação de séries de ensaios e de ensaios
|
—
|
Meios de controlo positivo e negativo e gamas de aceitação baseadas em dados históricos;
|
|
—
|
Variabilidade aceitável entre os tecidos replicados, para os controlos positivos e negativos;
|
|
—
|
Variabilidade aceitável entre os tecidos replicados, para o produto químico em estudo;
|
Procedimento de ensaio
|
—
|
Elementos sobre o procedimento de ensaio adotado;
|
|
—
|
Doses do produto químico em estudo e das substâncias de controlo utilizadas;
|
|
—
|
Duração e temperatura da exposição, da imersão pós-exposição e dos períodos de incubação pós-exposição (se pertinente);
|
|
—
|
Descrição de eventuais modificações do protocolo do ensaio;
|
|
—
|
Indicação dos controlos utilizados nos redutores diretos do MTT e/ou dos produtos químicos em estudo que apresentem coloração, se pertinente;
|
|
—
|
Número de replicados de tecidos utilizados por cada produto químico em estudo e de controlo (NSMTT, NSCvivos e NSCmortos, se pertinente);
|
Resultados
|
—
|
Quadro com os dados relativos a todos os produtos químicos e substâncias de controlo de cada série de ensaios (incluindo ensaios repetidos, se pertinente), a todas as medições de replicados – incluindo a densidade ótica (DO) ou a área de pico do MTT formazano –, percentagem de viabilidade dos tecidos, percentagem média de viabilidade dos tecidos, diferenças entre os tecidos replicados, ou DP, e predição final;
|
|
—
|
Se for caso disso, os resultados dos controlos utilizados para os redutores diretos do MTT e/ou para os produtos químicos em estudo corados, incluindo o valor da DO ou a área do pico do MTT formazano, % NSMTT, % NSCvivos, % NSCmortos, diferença entre os tecidos replicados ou DP, percentagem final correta de viabilidade dos tecidos e previsão final;
|
|
—
|
Resultados obtidos com o(s) produto(s) químico(s) em estudo e as substâncias de controlo no que diz respeito ao(s) critério(s) de definição e de aceitação do ensaio;
|
|
—
|
Descrição de quaisquer outros efeitos observados, por exemplo, coloração dos tecidos por um produto químico corado;
|
Discussão dos resultados
Conclusão
|
REFERÊNCIAS BIBLIOGRÁFICAS
|
(1)
|
ONU (2015). United Nations Globally Harmonized System of Classification and Labelling of Chemicals (GHS). ST/SG/AC.10/30/Rev.6, Sixth Revised Edition, New York and Geneva: United Nations. Disponível em: http://www.unece.org/fileadmin/DAM/trans/danger/publi/ghs/ghs_rev06/English/ST-SG-AC10-30-Rev6e.pdf.
|
|
(2)
|
Capítulo B.5 do presente anexo, «Irritação/corrosão ocular agudas».
|
|
(3)
|
Capítulo B.47 do presente anexo, «Método de ensaio de opacidade e permeabilidade da córnea em bovinos para identificação de produtos químicos indutores de lesões oculares graves e de produtos químicos que não necessitem de ser classificados em termos de irritação ocular nem de lesões oculares graves».
|
|
(4)
|
Capítulo B.48 do presente anexo, «Método de ensaio em olhos de frango isolados para identificação de produtos químicos indutores de lesões oculares graves e de produtos químicos que não necessitem de ser classificados em termos de irritação ocular nem de lesões oculares graves».
|
|
(5)
|
Capítulo B.61 do presente anexo, «Método de ensaio de difusão de fluoresceína para identificação de corrosivos oculares e de irritantes oculares severos».
|
|
(6)
|
Capítulo B.68 do presente anexo, «Método de ensaio de exposição in vitro de curta duração para identificação i) de produtos químicos indutores de lesões oculares graves e ii) de produtos químicos que não necessitam de ser classificados em termos de irritação ocular nem de lesões oculares graves».
|
|
(7)
|
Freeman, S.J., Alépée N., Barroso, J., Cole, T., Compagnoni, A., Rubingh, C., Eskes, C., Lammers, J., McNamee, P., Pfannenbecker, U., Zuang, V. (2010). Prospective Validation Study of Reconstructed Human Tissue Models for Eye Irritation Testing. ALTEX 27, Special Issue 2010,261-266.
|
|
(8)
|
EC EURL ECVAM (2014). The EURL ECVAM - Cosmetics Europe prospective validation study of Reconstructed human Cornea-like Epithelium (RhCE)-based test methods for identifying chemicals not requiring classification and labelling for serious eye damage/eye irritation: Validation Study Report. EUR 28125 EN; doi:10.2787/41680. Disponível em: http://publications.jrc.ec.europa.eu/repository/handle/JRC100280.
|
|
(9)
|
EURL ECVAM Science Advisory Committee (2014). ESAC Opinion on the EURL ECVAM Eye Irritation Validation Study (EIVS) on EpiOcular™ EIT and SkinEthic™ HCE and a related Cosmetics Europe study on HPLC/UPLC-spectrophotometry as an alternative endpoint detection system for MTT-formazan. ESAC Opinion No 2014-03 of 17 November 2014; EUR 28173 EN; doi: 10.2787/043697. Disponível em: http://publications.jrc.ec.europa.eu/repository/handle/JRC103702.
|
|
(10)
|
Alépée, N., Leblanc, V., Adriaens, E., Grandidier, M.H., Lelièvre, D, Meloni, M., Nardelli, L., Roper, C.S, Santirocco, E., Toner, F., Van Rompay, A., Vinall, J., Cotovio, J. (2016). Multi-laboratory validation of SkinEthic HCE test method for testing serious eye damage/eye irritation using liquid chemicals. Toxicol. In Vitro 31, 43-53.
|
|
(11)
|
Alépée, N., Adriaens, E., Grandidier, M.H., Meloni, M., Nardelli, L., Vinall, C.J., Toner, F., Roper, C.S, Van Rompay, A.R., Leblanc, V., Cotovio, J. (2016). Multi-laboratory evaluation of SkinEthic HCE test method for testing serious eye damage/eye irritation using solid chemicals and overall performance of the test method with regard to solid and liquid chemicals testing. Toxicol. In Vitro 34, 55-70.
|
|
(12)
|
EURL ECVAM Science Advisory Committee (2016). ESAC Opinion on the SkinEthic™ Human Corneal Epithelium (HCE) Eye Irritation Test (EIT). ESAC Opinion No 2016-02 of 24 June 2016; EUR 28175 EN; doi: 10.2787/390390. Disponível em: http://publications.jrc.ec.europa.eu/repository/handle/JRC103704.
|
|
(13)
|
EC EURL ECVAM (2016). Recommendation on the Use of the Reconstructed human Cornea-like Epithelium (RhCE) Test Methods for Identifying Chemicals not Requiring Classification and Labelling for Serious Eye Damage/Eye Irritation According to UN GHS. (Manuscrito em preparação).
|
|
(14)
|
Draize, J.H., Woodard, G., Calvery, H.O. (1944). Methods for the Study of Irritation and Toxicity of Substances Applied Topically to the Skin and Mucous Membranes. Journal of Pharmacol. and Exp. Therapeutics 82, 377-390.
|
|
(15)
|
Scott, L., Eskes, C., Hoffmann, S., Adriaens, E., Alépée, N., Bufo, M., Clothier, R., Facchini, D., Faller, C., Guest, R., Harbell, J., Hartung, T., Kamp, H., Le Varlet, B., Meloni, M., McNamee, P., Osborne, R., Pape, W., Pfannenbecker, U., Prinsen, M., Seaman, C., Spielman, H., Stokes, W., Trouba, K., Van den Berghe, C., Van Goethem, F., Vassallo, M., Vinardell, P., Zuang, V. (2010). A Proposed Eye Irritation Testing Strategy to Reduce and Replace In Vivo Studies Using Bottom-Up and Top-Down Approaches. Toxicol. In Vitro 24, 1-9.
|
|
(16)
|
Mosmann, T. (1983). Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 65, 55-63.
|
|
(17)
|
OCDE (2016). Series on Testing and Assessment No 216: Performance Standards for the Assessment of Proposed Similar or Modified In Vitro Reconstructed Human Cornea-Like Epithelium (RhCE) Test Methods for Identifying Chemicals not Requiring Classification and Labelling for Eye Irritation or Serious Eye Damage, Based on the Validated Reference Methods EpiOcular™ EIT and SkinEthic™ HCE EIT described in TG 492. Organisation for Economic Cooperation and Development, Paris.
|
|
(18)
|
OCDE (2005). Series on Testing and Assessment No 34: Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Organisation for Economic Cooperation and Development, Paris.
|
|
(19)
|
Kaluzhny, Y., Kandárová, H., Hayden, P., Kubilus, J., d’Argembeau-Thornton, L., Klausner, M. (2011). Development of the EpiOcular™ Eye Irritation Test for Hazard Identification and Labelling of Eye Irritating Chemicals in Response to the Requirements of the EU Cosmetics Directive and REACH Legislation. Altern. Lab. Anim. 39, 339-364.
|
|
(20)
|
Nguyen, D.H., Beuerman, R.W., De Wever, B., Rosdy, M. (2003). Three-dimensional construct of the human corneal epithelium for in vitro toxicology. In: Salem, H., Katz, S.A. (Eds), Alternative Toxicological Methods, CRC Press, pp. 147-159.
|
|
(21)
|
Pfannenbecker, U., Bessou-Touya, S., Faller, C., Harbell, J., Jacob, T., Raabe, H., Tailhardat, M., Alépée, N., De Smedt, A., De Wever, B., Jones, P., Kaluzhny, Y., Le Varlet, B., McNamee, P., Marrec-Fairley, M., Van Goethem, F. (2013). Cosmetics Europe multi-laboratory pre-validation of the EpiOcular™ reconstituted Human Tissue Test Method for the Prediction of Eye Irritation. Toxicol. In Vitro 27, 619-626.
|
|
(22)
|
Alépée, N., Bessou-Touya, S., Cotovio, J., de Smedt, A., de Wever, B., Faller, C., Jones, P., Le Varlet, B., Marrec-Fairley, M., Pfannenbecker, U., Tailhardat, M., van Goethem, F., McNamee, P. (2013). Cosmetics Europe Multi-Laboratory Pre-Validation of the SkinEthic™ Reconstituted Human Corneal Epithelium Test Method for the Prediction of Eye Irritation. Toxicol. In Vitro 27, 1476-1488.
|
|
(23)
|
Kolle, S.N., Moreno, M.C.R., Mayer, W., van Cott, A., van Ravenzwaay, B., Landsiedel, R. (2015). The EpiOcular™ Eye Irritation Test is the Method of Choice for In Vitro Eye Irritation Testing of Agrochemical Formulations: Correlation Analysis of EpiOcular™ Eye Irritation Test and BCOP Test Data to UN GHS, US EPA and Brazil ANIVSA Classifications. Altern. Lab. Anim. 43, 1-18.
|
|
(24)
|
Adriaens, E., Barroso, J., Eskes, C., Hoffmann, S., McNamee, P., Alépée, N., Bessou-Touya, S., De Smedt, A., De Wever, B., Pfannenbecker, U., Tailhardat, M., Zuang, V. (2014). Retrospective Analysis of the Draize Test for Serious Eye Damage/Eye Irritation: Importance of Understanding the In Vivo Endpoints Under UN GHS/EU CLP for the Development and Evaluation of In Vitro Test Methods. Arch. Toxicol. 88, 701-723.
|
|
(25)
|
Barroso, J., Pfannenbecker, U., Adriaens, E., Alépée, N., Cluzel, M., De Smedt, A., Hibatallah, J., Klaric, M., Mewes, K.R., Millet, M., Templier, M., McNamee, P. (2017). Cosmetics Europe compilation of historical serious eye damage/eye irritation in vivo data analysed by drivers of classification to support the selection of chemicals for development and evaluation of alternative methods/strategies: the Draize eye test Reference Database (DRD). Arch. Toxicol. 91, 521-547.
|
|
(26)
|
Meloni, M., De Servi, B., Marasco, D., Del Prete, S. (2011). Molecular mechanism of ocular surface damage: Application to an in vitro dry eye model on human corneal epithelium. Molecular Vision 17, 113-126.
|
|
(27)
|
Hackett, R.B., McDonald, T.O. (1991). Eye Irritation. In Advances in Modern Toxicology: Dermatoxicology Marzulli F.N.and Maibach H.I. (Eds.), 4th Edition, pp. 749–815. Washington, DC, USA: Hemisphere Publishing Corporation.
|
|
(28)
|
Fox, D.A., Boyes, W.K. (2008). Toxic Responses of the Ocular and Visual System. In Cassaret and Doull’s Toxicology: The Basic Science of Poisons Klaassen C.D.(Ed.), 7th Edition, pp. 665–697. Withby, ON, Canada: McGraw-Hill Ryerson.
|
|
(29)
|
Jester, J.V., Li, H.F., Petroll, W.M., Parker, R.D., Cavanagh, H.D., Carr, G.J., Smith, B., Maurer, J.K. (1998). Area and Depth of Surfactant Induced Corneal Injury Correlates with Cell Death. Invest. Ophthalmol. Vis. Sci. 39, 922-936.
|
|
(30)
|
Maurer, J.K., Parker, R.D., Jester, J.V. (2002). Extent of Corneal Injury as the Mechanistic Basis for Ocular Irritation: Key Findings and Recommendations for the Development of Alternative Assays. Reg. Tox. Pharmacol. 36, 106-117.
|
|
(31)
|
Jester, J.V., Li, L., Molai, A., Maurer, J.K. (2001). Extent of Corneal Injury as a Mechanistic Basis for Alternative Eye Irritation Tests. Toxicol. In Vitro 15, 115-130.
|
|
(32)
|
Jester, J.V., Petroll, W.M., Bean, J., Parker, R.D., Carr, G.J., Cavanagh, H.D., Maurer, J.K. (1998). Area and Depth of Surfactant-Induced Corneal Injury Predicts Extent of Subsequent Ocular Responses. Invest. Ophthalmol. 39, 2610-2625.
|
|
(33)
|
Jester, J.V. (2006). Extent of Corneal Injury as a Biomarker for Hazard Assessment and the Development of Alternative Models to the Draize Rabbit Eye Test. Cutan. Ocul. Toxicol. 25, 41-54.
|
|
(34)
|
EpiOcular™ EIT SOP, Version 8 (March 05, 2013). EpiOcular™ EIT for the Prediction of Acute Ocular Irritation of Chemicals. Disponível em: [https://ecvam-dbalm.jrc.ec.europa.eu/beta/index.cfm/methodsAndProtocols/index].
|
|
(35)
|
SkinEthic™ HCE EIT SOP, Version 1. (July 20, 2015). SkinEthic™ HCE Eye Irritation Test (EITL for Liquids, EITS for Solids) for the Prediction of Acute Ocular Irritation of Chemicals. Disponível em: https://ecvam-dbalm.jrc.ec.europa.eu/beta/index.cfm/methodsAndProtocols/index.
|
|
(36)
|
Alépée, N., Barroso, J., De Smedt, A., De Wever, B., Hibatallah, J., Klaric, M., Mewes, K.R., Millet, M., Pfannenbecker, U., Tailhardat, M., Templier, M., McNamee, P. (2015). Use of HPLC/UPLC-Spectrophotometry for Detection of Formazan in In Vitro Reconstructed Human Tissue (RhT)-Based Test Methods Employing the MTT-Reduction Assay to Expand their Applicability to Strongly Coloured Test Chemicals. Toxicol. In Vitro 29, 741-761.
|
|
(37)
|
Kaluzhny, Y., Kandárová, H., Handa, Y., DeLuca, J., Truong, T., Hunter, A., Kearney, P., d’Argembeau-Thornton, L., Klausner, M. (2015). EpiOcular™ Eye Irritation Test (EIT) for Hazard Identification and Labeling of Eye Irritating Chemicals: Protocol Optimization for Solid Materials and Extended Shipment Times. Altern. Lab Anim. 43, 101-127.
|
|
(38)
|
US FDA (2001). Guidance for Industry: Bioanalytical Method Validation. U.S. Department of Health and Human Services, Food and Drug Administration. May 2001. Disponível em: http://www.fda.gov/downloads/Drugs/Guidances/ucm070107.pdf.
|
|
(39)
|
OCDE (2017). Guidance Document on an Integrated Approaches on Testing and Assessment for Serious Eye Damage and Eye irritation. Series on Testing and Assessment No 263. ENV Publications, Organisation for Economic Cooperation and Development, Paris.
|
Apêndice 1
DEFINIÇÕES
Abordagem ascendente
: Metodologia por etapas utilizada para produtos químicos que se pense não necessitarem de ser classificados e rotulados em termos de irritação ocular ou de lesões oculares graves; inicia-se com a determinação de produtos químicos que não careçam de classificação e rotulagem (resultado negativo) a partir de outros produtos químicos (resultado positivo).
Abordagem descendente
: Metodologia por etapas utilizada para produtos químicos que se suspeite causarem lesões oculares graves; inicia-se com a determinação dos produtos químicos que induzam lesões oculares graves (resultado positivo) a partir de outros produtos químicos (resultado negativo).
Adequação
: Relação do ensaio com o efeito em causa; pertinência e utilidade do ensaio para o fim em vista. Traduz a medida em que o ensaio mede ou prevê corretamente o efeito biológico em estudo. A adequação compreende a exatidão (concordância) do método de ensaio (18).
Amostra de controlo negativo
: Amostra que contém todos os componentes de um sistema de ensaio e é tratada com uma substância conhecida por induzir uma reação positiva no sistema de ensaio. Esta amostra é preparada juntamente com as amostras tratadas com o produto químico em estudo e as outras amostras de controlo, sendo utilizada para determinar a viabilidade dos tecidos a 100 %.
Amostra de controlo positivo
: Amostra que contém todos os componentes de um sistema de ensaio e é tratada com uma substância conhecida por induzir uma reação positiva no sistema de ensaio. Esta amostra tratada juntamente com as amostras tratadas com o produto químico em estudo e as outras amostras de controlo. Para que possa determinar-se a variabilidade no tempo da reação a esta amostra de controlo, essa reação não deve ser excessivamente positiva.
Categoria 1 do sistema GHS da ONU/CRE
: Ver «lesões oculares graves».
Categoria 2 do sistema GHS da ONU/CRE
: Ver «irritação ocular».
Concordância
: Ver «exatidão».
Córnea
: Parte anterior transparente do globo ocular que recobre a íris e a pupila e deixa passar a luz para o interior.
CV
: Coeficiente de variação.
Dev
: Desvio.
DO
: Densidade ótica.
DP
: Desvio-padrão.
Dose infinita
: Quantidade de produto químico em estudo aplicada no tecido RhCE que excede a quantidade necessária para cobrir completa e uniformemente a superfície epitelial.
Efeitos oculares irreversíveis
: Ver «lesões oculares graves».
Efeitos oculares reversíveis
: Ver «irritação ocular».
EIT
: Ensaio de irritação ocular.
Ensaio
: Um único produto químico em estudo ensaiado em paralelo com, pelo menos, dois tecidos replicados, como definido no PON correspondente.
Ensaio alternativo
: Ensaio destinado a substituir um ensaio de rotina aceite para identificação de perigos e/ou avaliação de riscos, que se concluiu proporcionar proteção equivalente, ou melhorar a proteção, da saúde pública e animal ou do ambiente – consoante o caso – comparativamente ao ensaio de rotina aceite, no que respeita a todos os produtos químicos e situações de ensaio possíveis (18).
Especificidade
: Proporção dos produtos químicos em estudo negativos/inativos que são corretamente classificados pelo ensaio. Constitui uma medida da exatidão de um método de ensaio cujos resultados são estabelecidos em função de categorias e é um aspeto importante a ter em conta na avaliação da adequação do método (18).
Estratégia de ensaio sequencial por etapas
: Estratégia de ensaios por etapas que usa métodos de ensaio de forma sequencial. As informações disponíveis sobre o produto químico em estudo são avaliadas a cada etapa, por um processo baseado na ponderação da suficiência da prova, com o objetivo de determinar se são suficientes para a classificação de perigosidade, antes de passar à etapa seguinte. Se as informações existentes possibilitarem a atribuição de um potencial de perigosidade ao produto químico em estudo numa dada etapa, não serão necessários mais ensaios (18).
EURL ECVAM
: Laboratório de referência da União Europeia para as alternativas à experimentação animal.
Exatidão
: Grau de acordo entre os resultados do método de ensaio e os valores de referência aceites. Constitui uma medida da eficiência do método e um dos aspetos da «adequação». Este termo e o termo «concordância» são muitas vezes utilizados indistintamente para indicar a proporção de resultados corretos do método de ensaio (18).
ET50
: Tempo de exposição necessário para reduzir a viabilidade do tecido em 50 % após aplicação de uma concentração específica e fixa de um produto químico de referência.
Fiabilidade
: Medida em que, utilizando o mesmo protocolo, um método de ensaio pode ser continuadamente reproduzido no mesmo laboratório e em laboratórios diferentes. A fiabilidade é avaliada com base nos valores calculados das reprodutibilidades intralaboratorial e interlaboratorial, bem como da repetibilidade intralaboratorial (18).
Irritação ocular
: Alterações oculares decorrentes da aplicação do produto químico em estudo na superfície anterior do olho, totalmente reversíveis nos 21 dias após a aplicação. Esta expressão, bem como as expressões «efeitos oculares reversíveis» ou «categoria 2 do sistema GHS da ONU/CRE», são utilizadas indistintamente.
Lesões oculares graves
: Lesão do tecido ocular ou degradação grave da visão decorrente da aplicação de um produto químico em estudo na superfície anterior do olho, não totalmente reversível nos 21 dias após a aplicação. Esta expressão, bem como as expressões «efeitos oculares irreversíveis» ou «categoria 1 do sistema GHS da ONU/CRE», são utilizadas indistintamente.
LLOQ
: Limite de quantificação inferior.
Log(P)
: Logaritmo do coeficiente de repartição octanol/água.
Método de ensaio válido
: Método de ensaio cuja importância e cuja fiabilidade são consideradas suficientes para um objetivo específico e que se baseia em princípios cientificamente sólidos. Um método de ensaio nunca é válido em sentido absoluto, mas apenas em relação a um determinado objetivo (18).
Método de ensaio validado
: Método de ensaio relativamente ao qual foram concluídos estudos de validação com vista a determinar a sua adequação (incluída a exatidão) e a sua fiabilidade para um determinado fim. Importa referir que um método de ensaio validado pode não ser suficientemente exato e fiável para ser considerado aceitável para o fim pretendido (18).
Mistura
: Uma mistura ou solução composta por duas ou mais substâncias.
MTT
: Brometo de [3-(4,5-dimetiltiazol-2-il)-2,5-difeniltetrazólio]; brometo de tiazolilo; azul de tetrazólio.
Não classificado
: Produtos químicos não classificados em termos de irritação ocular (categoria 2 do sistema GHS da ONU/CRE, categorias 2A ou 2B do sistema GHS da ONU) ou de lesões oculares graves (categoria 1 do sistema GHS da ONU/CRE). Designação utilizada indistintamente com a classificação «nenhuma categoria» no sistema GHS da ONU/CRE.
«Nenhuma categoria» do sistema GHS da ONU/CRE
: Produtos químicos que não satisfaçam os requisitos para classificação nas categorias 1 ou 2 do sistema GHS da ONU/CRE (ou na categoria 2A ou 2B do sistema GHS da ONU). Esta expressão e a expressão «não classificado» são utilizadas indistintamente.
Normas de desempenho
: Normas baseadas um método de referência validado que tenha sido considerado cientificamente válido e que constituem a base para a avaliação da comparabilidade de um método de ensaio proposto mecanística e funcionalmente similar. Incluem-se: i) componentes essenciais do método de ensaio; ii) uma lista mínima de produtos químicos de referência, escolhidos de entre os produtos químicos utilizados para demonstrar o desempenho aceitável do método de ensaio validado; iii) níveis de exatidão e fiabilidade comparáveis, baseados no que foi obtido para o método de ensaio validado, que o método de ensaio proposto deve demonstrar quando avaliado pela utilização da lista mínima de produtos químicos de referência (18).
NSMTT
: Redução não específica do MTT.
NSCmortos
: Cor não específica em tecidos mortos.
NSCvivos
: Cor não específica em tecidos vivos.
Perigo
: Propriedade intrínseca de um agente, ou de uma situação, suscetível de causar efeitos nocivos num organismo, sistema, população ou subpopulação exposto ao agente em causa.
Ponderação da suficiência da prova
: Processo que consiste em ponderar os pontos fortes e fracos dos vários elementos de informação para tirar conclusões fundamentadas sobre o potencial de perigosidade do produto químico em estudo.
Procedimentos operacionais normalizados (PON)
: Procedimentos formais e escritos que descrevem em pormenor o modo como devem ser realizadas as operações laboratoriais de rotina e as específicas do ensaio. São previstos pelo CRE.
Produto químico
: Uma substância ou mistura.
Produto químico de referência
: Produto químico utilizado como padrão de comparação com o produto químico em estudo. Um produto químico de referência deve ter as seguintes propriedades: (i) origem ou origens uniformes e fiáveis para a sua identificação e caracterização; (ii) similaridade estrutural, funcional e/ou química com a classe de produtos químicos em estudo; (iii) características físico-químicas conhecidas; (iv) existência de dados sobre os efeitos conhecidos; e v) representatividade conhecida, situada na gama de reação pretendida.
Produto químico em estudo
: Qualquer substância ou mistura à qual seja aplicado o presente método de ensaio.
Reprodutibilidade
: Acordo entre resultados obtidos em ensaios com o mesmo produto químico, utilizando o mesmo protocolo de ensaio (ver «fiabilidade») (18).
RhCE
: Epitélio corneano humano reconstruído.
Sensibilidade
: Proporção dos produtos químicos em estudo positivos/ativos que são corretamente classificados pelo ensaio. Constitui uma medida da exatidão de um método de ensaio cujos resultados sejam estabelecidos em função de categorias e é um aspeto importante a ter em conta na avaliação da adequação do método (18).
Série de ensaios
: Uma série de ensaios consiste no ensaio de um ou mais produtos químicos em estudo em paralelo com um controlo negativo e um controlo positivo.
Sistema Mundial Harmonizado de Classificação e Rotulagem de Produtos Químicos das Nações Unidas (em inglês: Globally Harmonized System of Classification and Labelling of Chemicals – UN GHS)
: Sistema que propõe a classificação dos produtos químicos (substâncias e misturas) em função de tipos e níveis normalizados de perigos físicos, sanitários e ambientais e trata ainda dos correspondentes elementos de comunicação, como pictogramas, palavras-sinal, advertências de perigo, recomendações de prudência e fichas de dados de segurança, de modo a transmitir informações sobre os efeitos indesejáveis do produto em causa, com vista à proteção das pessoas (empregadores, trabalhadores, transportadores, consumidores, pessoal dos serviços de emergência, etc.) e do ambiente (1).
Substância
: Um elemento químico e os seus compostos, no estado natural ou obtidos por qualquer processo de produção, incluindo os aditivos necessários para preservar a sua estabilidade e qualquer impureza derivada do processo utilizado, mas excluindo os solventes que possam ser separados sem afetar a estabilidade da substância nem modificar a sua composição.
Substância monocomponentes
: Substância, definida pela sua composição quantitativa, na qual um dos principais componentes está presente num teor de, pelo menos, 80 % (m/m).
Substância multicomponentes
: Substância, definida pela sua composição quantitativa, da qual mais de um dos principais componentes está presente numa concentração ≥ 10 % (m/m) e < 80 % (m/m). As substâncias multicomponentes resultam de processos de fabrico. A diferença entre uma mistura e uma substância multicomponentes reside no facto de a primeira ser obtida misturando duas ou mais substâncias, sem reação química. As substâncias multicomponentes resultam de reações químicas.
Taxa de falsos negativos
: Proporção das substâncias positivas que o método de ensaio considera erradamente negativas. É um dos indicadores da eficiência dos métodos.
Taxa de falsos positivos
: Proporção das substâncias negativas que o método de ensaio considera erradamente positivas. É um dos indicadores da eficiência dos métodos.
ULOQ
: Limite de quantificação superior.
UPLC
: Cromatografia líquida de muito alta resolução.
UVCB
: Substâncias de composição desconhecida ou variável, produtos de reação complexos ou matérias biológicas.
Viabilidade dos tecidos
: Parâmetro que mede a atividade total de uma população celular num tecido reconstruído e a sua capacidade para reduzir o corante vital MTT, capacidade esse que, consoante o parâmetro determinado e do tipo de ensaio realizado, está correlacionada com o número total de células vivas e/ou com a vitalidade dessas células.
VRM
: Método de referência validado.
VRM1
: O EIT da EpiOcular™ é referido como «método de referência validado 1».
VRM2
: O EIT HCE da SkinEthic™ é referido como «método de referência validado 2».
Apêndice 2
PRINCIPAIS COMPONENTES DOS ENSAIOS RHCE VALIDADOS PARA IDENTIFICAÇÃO DE PRODUTOS QUÍMICOS QUE NÃO NECESSITAM DE CLASSIFICAÇÃO E ROTULAGEM EM MATÉRIA DE IRRITAÇÃO OCULAR OU LESÕES OCULARES GRAVES
|
Componentes do ensaio
|
EIT da EpiOcular™
(VRM 1)
|
EIT HCE da SkinEthic™
(VRM 2)
|
|
Protocolos
|
Líquidos
(pipetáveis a 37 oC ±1 oC ou a temperaturas mais baixas durante 15 min)
|
Sólidos
(não pipetáveis)
|
Líquidos e viscosos
(pipetáveis)
|
Sólidos
(não pipetáveis)
|
|
Superfície do modelo
|
0,6 cm2
|
0,6 cm2
|
0,5 cm2
|
0,5 cm2
|
|
Número de tecidos replicados
|
Pelo menos 2
|
Pelo menos 2
|
Pelo menos 2
|
Pelo menos 2
|
|
Controlo prévio de interferência cromática
|
50 μl + 1 ml H2O durante 60 min a 37±2oC, 5±1 % CO2, ≥ 95 % RH (produtos químicos em estudo não corados), ou 50 μl +2 ml de mistura de isopropanol durante 2-3 horas à temperatura ambiente (produtos químicos corados)
|
 Se a densidade ótica do produto químico em estudo a 570±20 nm, após subtração da densidade ótica do isopropanol ou da água, for superior a 0,08 (o que corresponde a cerca de 5 % da DO total do controlo negativo), devem realizar-se controlos adaptados in vivo.
Se a densidade ótica do produto químico em estudo a 570±20 nm, após subtração da densidade ótica do isopropanol ou da água, for superior a 0,08 (o que corresponde a cerca de 5 % da DO total do controlo negativo), devem realizar-se controlos adaptados in vivo.
|
|
50 mg + 1 ml H2O durante 60 min a 37±2oC, 5±1 % CO2, ≥ 95 % à temperatura ambiente (produtos químicos não corados)
e/ou
50 mg +2 ml de mistura de isopropanol durante 2 a 3 horas à temperatura ambiente (produtos químicos de ensaio corados e não corados)
|
 Se a densidade ótica do produto químico em estudo a 570±20 nm, após subtração da densidade ótica do isopropanol ou da água, for superior a 0,08 (o que corresponde a cerca de 5 % do total da DO de controlo negativo), devem realizar-se controlos adaptados in vivo.
Se a densidade ótica do produto químico em estudo a 570±20 nm, após subtração da densidade ótica do isopropanol ou da água, for superior a 0,08 (o que corresponde a cerca de 5 % do total da DO de controlo negativo), devem realizar-se controlos adaptados in vivo.
|
|
10 μl + 90 μl H2O misturados durante 30 ±2 min à temperatura ambiente (temperatura ambiente: 18-28 oC)
|
 se o produto químico em estudo for corado, devem realizar-se controlos adaptados in vivo
se o produto químico em estudo for corado, devem realizar-se controlos adaptados in vivo
|
|
10 mg + 90 μl H2O misturados durante 30±2 min à temperatura ambiente
|
 se o produto químico em estudo for corado, devem realizar-se controlos adaptados in vivo
se o produto químico em estudo for corado, devem realizar-se controlos adaptados in vivo
|
|
|
Controlo prévio da redução direta do MTT
|
50 μl+1 ml de MTT 1 mg/ml de solução durante 180±15 min a 37±2 oC, 5±1 % de CO2, ≥ 95 % de humidade relativa
|
 se a solução ficar de cor azul/púrpura, devem realizar-se controlos adaptados «mortos por congelamento»
se a solução ficar de cor azul/púrpura, devem realizar-se controlos adaptados «mortos por congelamento»
(utilizam-se como controlo negativo 50 μl de água desionizada estéril numa solução de MTT)
|
|
50 mg + 1 ml MTT 1 mg/ml de solução durante 180 ±15 min a 37±2 oC, 5±1 % de CO2, ≥ 95 % de humidade relativa
|
 se a solução ficar azul/púrpura, devem realizar-se controlos adaptados «mortos por congelamento».
se a solução ficar azul/púrpura, devem realizar-se controlos adaptados «mortos por congelamento».
(utilizam-se como controlo negativo 50 μl de água desionizada estéril numa solução de MTT).
|
|
30 μl + 300 μl MTT 1 mg/ml de solução durante 180±15 min a 37±2 oC, 5±1 % CO2, ≥95 % de humidade relativa
|
 se a solução ficar azul/púrpura, devem realizar-se controlos adaptados à água
se a solução ficar azul/púrpura, devem realizar-se controlos adaptados à água
(utilizam-se como controlo negativo 30 μl de água desionizada estéril numa solução de MTT).
|
|
30 mg +300 μl de MTT
|
 S1 mg/ml de solução durante 180±15 min a 37±2 oC, 5±1 % de CO2, ≥95 % de humidade relativa se a solução ficar azul/púrpura, devem realizar-se controlos adaptados à água
S1 mg/ml de solução durante 180±15 min a 37±2 oC, 5±1 % de CO2, ≥95 % de humidade relativa se a solução ficar azul/púrpura, devem realizar-se controlos adaptados à água
(utilizam-se como controlo negativo 30 μl de água desionizada estéril numa solução de MTT).
|
|
|
Pré-tratamento
|
20 μl de DPSB sem Ca2+/Mg2+
durante 30 ± 2 min a 37±2 oC, 5±1 % CO2, ≥ 95 % de humidade relativa, ao abrigo da luz.
|
20 μl de DPSB sem Ca2+/Mg2+
durante 30 ± 2 min a 37±2 oC, 5±1 % CO2, ≥ 95 % de humidade relativa, ao abrigo da luz.
|
-
|
-
|
|
Doses e aplicação do tratamento
|
50 μl (83,3 μl/cm2)
|
50 mg (83,3 mg/cm2) utilizando um aparelho calibrado (por exemplo, uma colher nivelada calibrada para conter 50 mg de cloreto de sódio).
|
10 μl de DPSB sem Ca2+/Mg2++ 30 ± 2 μl (60 μl/cm2)
Para produtos viscosos, utilizar uma malha de nylon
|
30 μl de DPSB sem Ca2+/Mg2++ 30 ± 2 mg (60 mg/cm2)
|
|
Tempo e temperaturade exposição
|
30 min (± 2 min)
em meio de cultura
a 37±2 oC, 5±1 % de CO2, ≥ 95 % de humidade relativa
|
6 horas (± 0,25 h)
em meio de cultura
a 37±2 oC, 5±1 % de CO2, ≥ 95 % de humidade relativa
|
30 min (± 2 min)
em meio de cultura
a 37±2 oC, 5±1 % de CO2, ≥ 95 % de humidade relativa
|
4 horas (± 0,1 h)
em meio de cultura
a 37±2 oC, 5±1 % de CO2, ≥ 95 % de humidade relativa
|
|
Lavagem à temperatura ambiente
|
3 vezes em 100 ml de DPSB sem Ca2+/Mg2+
|
3 vezes em 100 ml de DPSB sem Ca2+/Mg2+
|
20 ml de DPSB sem Ca2+/Mg2+
|
25 ml de DPSB sem Ca2+/Mg2+
|
|
Imersão pós-exposição
|
12 min (± 2 min) à temperatura ambiente em meio de cultura
|
25 min (± 2 min) à temperatura ambiente em meio de cultura
|
30 min (± 2 min) a 37 oC, 5 % CO2, 95 % de humidade relativa em meio de cultura
|
30 min (± 2 min) à temperatura ambiente em meio de cultura
|
|
Incubação pós-exposição
|
120 min (± 15 min) em meio de cultura a 37±2 oC, 5±1 % CO2, ≥ 95 % de humidade relativa
|
18 h (± 0,25 h) em meio de cultura a 37±2 oC, 5±1 % CO2, ≥ 95 % de humidade relativa
|
nenhuma
|
18 h (± 0,5 h) em meio de cultura a 37±2 oC, 5±1 % CO2, ≥ 95 % de humidade relativa
|
|
Controlo negativo
|
50 μl H2O
Ensaio paralelo
|
50 μl H2O
Ensaio paralelo
|
30 ±2μl de DPBS sem Ca2+/Mg2+
Ensaio paralelo
|
30 ±2μl de DPBS sem Ca2+/Mg2+
Ensaio paralelo
|
|
Controlo positivo
|
50 μl de acetato de metilo
Ensaio paralelo
|
50 μl de acetato de metilo
Ensaio paralelo
|
30±2 μl de acetato de metilo
Ensaio paralelo
|
30±2 μl de acetato de metilo
Ensaio paralelo
|
|
Solução de MTT
|
300 μl 1 mg/ml
|
300 μl 1 mg/ml
|
300 μl 1 mg/ml
|
300 μl 1 mg/ml
|
|
Tempo e temperaturade incubação do MTT
|
180 min (± 15 min) a 37±2 oC, 5±1 % CO2, ≥ 95 % de humidade relativa
|
180 min (± 15 min) a 37±2 oC, 5±1 % CO2, ≥ 95 % de humidade relativa
|
180 min (± 15 min) a 37±2 oC, 5±1 % CO2, ≥ 95 % de humidade relativa
|
180 min (± 15 min) a 37±2 oC, 5±1 % CO2, ≥ 95 % de humidade relativa
|
|
Extração do solvente
|
2 ml de isopropanol
(extração da superfície e do fundo da inserção por perfuração do tecido)
|
2 ml de isopropanol
(extração do fundo da inserção por perfuração do tecido)
|
1,5 ml de isopropanol
(extração da superfície e do fundo da inserção)
|
1,5 ml de isopropanol
(extração do fundo da inserção)
|
|
Tempo e temperatura de extração
|
2-3 horas com agitação (~20 rpm) à temperatura ambiente ou de um dia para o outro a 4-10 oC
|
2-3 horas com agitação (~20 rpm) à temperatura ambiente ou de um dia para o outro a 4-10 oC
|
4 h com agitação (~120 rpm) à temperatura ambiente ou, pelo menos, de um dia para o outro, sem agitação, a 4-10 oC
|
Pelo menos 2 horas com agitação (~120 rpm), à temperatura ambiente
|
|
Leitura da densidade ótica
|
570 nm (550 - 590 nm)
sem filtro de referência
|
570 nm (550 - 590 nm)
sem filtro de referência
|
570 nm (540 - 600 nm)
sem filtro de referência
|
570 nm (540 - 600 nm)
sem filtro de referência
|
|
Controlo da qualidade dos tecidos
|
Tratamento com 100 μl de Triton X-100 a 0,3 %
12,2 min ≤ ET50 ≤ 37,5 min
|
Tratamento com 100 μl de Triton X-100 a 0,3 %
12,2 min ≤ ET50 ≤37,5 min
|
30 minutos de tratamento com SDS (50 μl)
1,0 mg/ml ≤ IC50 ≤3,5 mg/ml
|
30 minutos de tratamento com SDS (50 μl)
1,0 mg/ml ≤ IC50 ≤ 3,2 mg/ml
|
|
Critérios de aceitação
|
|
1.
|
A densidade média dos replicados tratados com o controlo negativo deve ser > 0,8 e < 2,5
|
|
2.
|
A viabilidade média dos tecidos replicados expostos durante 30 minutos com o controlo positivo, expressa em % da amostra de controlo negativa, deve ser < 50 %.
|
|
3.
|
A diferença de viabilidade entre dois tecidos replicados deve ser inferior a 20 %.
|
|
|
1.
|
A densidade média dos replicados tratados com o controlo negativo deve ser > 0,8 e < 2,5
|
|
2.
|
A viabilidade média dos tecidos replicados expostos durante 6 horas com o controlo positivo, expressa em % da amostra de controlo negativa, deve ser < 50 %.
|
|
3.
|
A diferença de viabilidade entre dois tecidos replicados deve ser inferior a 20 %.
|
|
|
1.
|
A densidade média dos replicados tratados com o controlo negativo deve ser > 1,0 e ≤ 2,5
|
|
2.
|
A viabilidade média dos replicados de tecidos expostos durante 30 minutos com o controlo positivo, expressa em % do controlo negativo, deve ser ≤ 30 %
|
|
3.
|
A diferença de viabilidade entre dois tecidos replicados deve ser inferior a 20 %.
|
|
|
1.
|
A densidade média dos tecidos replicados tratados com o controlo negativo deve ser > 1,0 e ≤ 2,5
|
|
2.
|
A viabilidade média dos tecidos replicados expostos durante 4 horas com o controlo positivo, expressa em % do controlo negativo, deve ser ≤ 20 %
|
|
3.
|
A diferença de viabilidade entre dois tecidos replicados deve ser inferior a 20 %.
|
|
Apêndice 3
FLUXOGRAMA ILUSTRATIVO DE ORIENTAÇÃO PARA IDENTIFICAR E MANUSEAR OS REDUTORES DIRETOS DO MTT E/OU OS PRODUTOS QUÍMICOS INTERFERENTES CROMÁTICOS COM BASE NOS PON DO VRM1

Apêndice 4
FLUXOGRAMA ILUSTRATIVO DE ORIENTAÇÃO PARA IDENTIFICAR E MANUSEAR OS REDUTORES DIRETOS DO MTT E/OU OS PRODUTOS QUÍMICOS INTERFERENTES CROMÁTICOS COM BASE NOS PON DO VRM2
Apêndice 5
PRINCIPAIS PARÂMETROS E CRITÉRIOS DE ACEITAÇÃO PARA A QUALIFICAÇÃO DE UM SISTEMA DE ESPETROFOTOMETRIA HPLC/UPLC PARA MEDIÇÃO DO MTT FORMAZANO EXTRAÍDO DE TECIDOS RHCE
|
Parâmetro
|
Protocolo derivado do documento de orientação da FDA (36) (38)
|
Critérios de aceitação
|
|
Seletividade
|
Análise do isopropanol, em branco vivo (extrato de isopropanol extraído de tecido RhCE vivo sem qualquer tratamento), em branco morto (extrato de isopropanol sem qualquer tratamento extraído de tecido RhCE morto sem qualquer tratamento) e de um corante (por exemplo, azul de metileno)
|
Áreainterferência ≤20 % da ÁreaLLOQ
(87)
|
|
Precisão
|
Controlos de qualidade (MTT formazano a 1,6 μg/ml, 16 μg/ml e 160 μg/ml) em isopropanol (n = 5)
|
CV ≤ 15 % or ≤ 20 % para o LLOQ
|
|
Exatidão
|
Controlos de qualidade em isopropanol (n = 5)
|
% Dev ≤ 15 % ou ≤ 20 % para LLOQ
|
|
Efeito matriz
|
Controlos de qualidade em branco vivo (n = 5)
|
85 % ≤ % efeito de matriz ≤ 115 %
|
|
Passagem
|
Análise de isopropanol após um padrão ULOQ (88)
|
Áreainterferência ≤20 % da ÁreaLLOQ
|
|
Reprodutibilidade (intradiária)
|
3 curvas de calibração independentes (com base em 6 diluições consecutivas de 1/3 de MTT formazano em isopropanol, com início em ULOQ, isto é, 200 μg/ml);
Controlos de qualidade em isopropanol (n = 5)
|
Curvas de calibração: % Dev ≤ 15 % ou ≤ 20 % para LLOQ
Controlos de qualidade: % Dev ≤ 15 % e CV ≤ 15 %
|
|
Reprodutibilidade (intradiária)
|
Dia 1: 1 curva de calibração e controlos de qualidade no isopropanol (n = 3)
Dia 2: 1 curva de calibração e controlos de qualidade no isopropanol (n = 3)
Dia 3: 1 curva de calibração e controlos de qualidade no isopropanol (n = 3)
|
|
Estabilidade a curto prazo do MTT formazano em extratos de tecidos RhCE
|
Controlos de qualidade em branco vivo (n = 3), analisados no dia da preparação e após 24 horas de armazenagem à temperatura ambiente.
|
% Dev ≤ 15 %
|
|
Estabilidade a longo prazo do MTT formazano em extratos de tecidos RhCE, se necessário
|
Controlos de qualidade em branco vivo (n = 3), analisados no dia da preparação e após vários dias de armazenagem a -20 oC
|
% Dev ≤ 15 %
|
B.70 ENSAIOS IN VITRO DE RECETOR DE ESTROGÉNIO HUMANO RECOMBINANTE PARA DETEÇÃO DE PRODUTOS QUÍMICOS COM AFINIDADE LIGANTE COM O ER
INTRODUÇÃO GERAL
Método de ensaio baseado na Test Guideline da OCDE
|
1.
|
O presente método de ensaio é equivalente à Test Guideline 493 (2015) da OCDE. A Test Guideline 493 é uma diretriz de ensaio baseada no desempenho (PBTG), que descreve a metodologia para os ensaios in vitro de recetor de estrogénio humano recombinante para a deteção de produtos químicos com afinidade ligante com recetores de estrogénios (ensaios de ligação do hrER). É composto por vários métodos de ensaio mecânica e funcionalmente semelhantes para a identificação de ligantes do recetor de estrogénio (i.e., ERα) e deve facilitar o desenvolvimento de métodos de ensaio novos ou modificados de acordo com os princípios de validação estabelecidos no Guidance Document on the Validation and International Acceptance of New or Updated Composição for Hazard Assessment (1) da OCDE. Os métodos de ensaio de referência inteiramente validados (apêndice 2 e apêndice 3) que constituem a base deste PBTG são:
|
—
|
O In Vitro Estrogen Recetor (ER) Binding Assay Using a Full Length Human Recombinant ERα [ensaio in vitro de ligação do recetor de estrogénio com um ERα recombinante inteiro humano] de Freyberger-Wilson (FW) (2) e
|
|
—
|
O In Vitro Estrogen Recetor Binding Assay Using a Human Recombinant Ligand Binding Domain Protein (ensaio in vitro de ligação do recetor de estrogénio através de uma proteína ligando recombinante de ligação do Chemical Evaluation and Research Institute (CERI) (2).
|
Existem normas de desempenho (PS) (3) para facilitar o desenvolvimento e a validação de métodos de ensaio similares para o mesmo parâmetro de perigo e permitir a alteração oportuna do PBTG 493, por forma a que possam ser adicionados novos ensaios similares a um PBTG atualizado. No entanto, só serão adicionados ensaios semelhantes após análise e aprovação pela OCDE do cumprimento das normas de desempenho. Os ensaios incluídos no método TG 493 podem ser utilizados indiscriminadamente para satisfazer as necessidades dos países membros da OCDE quanto a resultados de ensaios de ligação de recetores de estrogénio, beneficiando, em simultâneo, da aceitação mútua de dados da OCDE.
|
Antecedentes e princípios dos ensaios incluídos no presente método de ensaio
|
2.
|
A OCDE iniciou em 1998 uma atividade altamente prioritária com o objetivo de rever as diretrizes em vigor para a pesquisa e o ensaio de produtos químicos com possíveis efeitos de perturbação endócrina, bem como de estabelecer novas diretrizes. O quadro conceptual da OCDE para ensaio e avaliação de produtos químicos potencialmente perturbadores do sistema endócrino foi revisto em 2012. O quadro conceptual original e o quadro revisto constam, como anexo, do Guidance Document on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption (4). O quadro conceptual inclui cinco níveis, correspondendo cada nível a um nível diferente de complexidade biológica. Os ensaios de ligação do RE descritos no presente método de ensaio são de nível 2, que abrange ensaios in vitro que proporcionam dados sobre um ou vários mecanismos/vias endócrinos selecionados. O método refere-se a ensaios de ligação de recetores in vitro, concebidos para identificar ligandos para o recetor de estrogénios humanos alfa (ERα).
|
|
3.
|
A relevância do ensaio de ligação do ER in vitro para funções biológicas já foi claramente demonstrada. Os ensaios de ligação do ER são concebidos para identificar produtos químicos suscetíveis de perturbar a via hormonal do estrogénio e têm sido amplamente utilizados nas duas últimas décadas para caracterizar a distribuição dos tecidos do ER, bem como para identificar agonistas/antagonistas do ER. Estes ensaios refletem a interação ligando-recetor, que constitui a primeira etapa da via de sinalização do estrogénio e é essencial para a função reprodutora em todos os vertebrados.
|
|
4.
|
A interação dos estrogénios com os ER pode afetar a transcrição dos genes controlados pelo estrogénio e induzir efeitos não genómicos passíveis de conduzir à indução ou inibição de processos celulares, incluindo os necessários para a proliferação celular, o desenvolvimento fetal normal e a função reprodutora (5) (6) (7). A perturbação dos sistemas estrogénicos normais pode provocar efeitos nocivos no desenvolvimento normal (ontogénese), na saúde reprodutiva e na integridade do aparelho reprodutor. Uma sinalização inadequada do ER pode provocar efeitos como o aumento de risco de cancro hormono-dependente, perturbações da fertilidade e alterações no crescimento e desenvolvimento fetais (8).
|
|
5.
|
Os ensaios obrigatórios in vitro baseiam-se numa interação direta de uma substância com um ligando de ligação do recetor específico que regula a transcrição génica. O principal componente do ensaio de ligação do recetor de estrogénio alfa recombinante humano (hrERα) mede a capacidade de um ligando com marcação radioativa ([3H]17β-estradiol) para se fixar ao ER na presença de concentrações crescentes de um produto químico concorrente Os produtos químicos em estudo que possuem uma elevada afinidade para o ER competem com o ligando com marcação radioativa a uma concentração mais baixa comparativamente com os produtos químicos com menor afinidade para o recetor. O presente ensaio é composto por dois componentes principais: uma experiência de ligantes por saturação com vista a caracterizar os parâmetros da interação recetor-ligando e a documentação da especificidade do ER, seguida de uma experiência de ligantes competitivos que caracteriza a concorrência entre um produto químico em estudo e um ligando com marcação radioativa para ligação ao ER.
|
|
6.
|
Os estudos de validação do CERI e os ensaios de ligação de FW demonstraram a sua adequação e fiabilidade para o fim a que se destinam (2).
|
|
7.
|
As definições e abreviaturas utilizadas no presente método de ensaio constam do apêndice 1.
|
Âmbito de aplicação e limitações relacionadas com os ensaios de ligação do recetor
|
8.
|
Estes ensaios são propostos para efeitos de análise e de definição de prioridades, mas podem também fornecer informações para um evento de iniciação molecular (MIE) passível de ser utilizado numa abordagem de ponderação da suficiência da prova. Abordam a ligação do produto químico ao domínio de ligação do ligando do ERα num sistema in vitro. Os resultados não devem, pois, ser extrapolados diretamente para o complexo de sinalização e regulação do sistema endócrino intacto in vivo.
|
|
9.
|
A ligação do ligando natural (17β-estradiol) constitui a primeira etapa de uma série de eventos moleculares que ativam a transcrição dos genes-alvo e, em última análise, culminam numa mudança fisiológica (9). Assim, a ligação ao domínio de ligação do ligando do ERα é considerada um dos principais mecanismos da desregulação do sistema endócrino (ED) mediada pelo ER, embora haja outros mecanismos através dos quais pode ocorrer a desregulação do sistema endócrino, incluindo: i) interações com pontos de ERα diferentes da bolsa de ligação do ligando; ii) interações com outros recetores relevantes para a sinalização do estrogénio, recetores de estrogénios emparelhados com o ERα e proteína-G e outros recetores e sistemas enzimáticos do sistema endócrino; iii) a síntese hormonal; iv) ativação metabólica e/ou a inativação hormonal; v) distribuição de hormonas aos tecidos-alvo; vi) eliminação de hormonas do organismo. Nenhum dos ensaios segundo o presente método aborda estes modos de ação.
|
|
10.
|
O presente método de ensaio incide na capacidade das substâncias de se fixarem ao ERα humano e não estabelece qualquer distinção entre agonistas ou antagonistas do ERα. Os ensaios não abordam outros eventos a jusante, como a transcrição génica ou as alterações fisiológicas. Tendo em conta que, durante a validação, apenas foram utilizadas substâncias monocomponentes, não foi abordada a aplicabilidade ao estudo de misturas. Considera-se, porém, que os ensaios são tecnicamente aplicáveis a substâncias e misturas multicomponentes. Antes da aplicação do método de ensaio a uma mistura para obter dados com fins normativos, importa ponderar se – e, em caso afirmativo, por que motivo – o método pode proporcionar resultados adequados para o efeito. Essas considerações não são necessárias se existir um requisito normativo para o ensaio da mistura.
|
|
11.
|
Os sistemas recetores sem células não têm capacidade metabólica intrínseca e não foram validados em combinação com sistemas enzimáticos metabólicos. No entanto, poderá ser possível integrar a atividade metabólica numa conceção do estudo, mas isso exigirá novos esforços de validação.
|
|
12.
|
Os produtos químicos suscetíveis de desnaturarem as proteínas do recetor, como os tensioativos ou as substâncias químicas que possam alterar o pH do tampão de ensaio, não podem ser ensaiados, ou apenas o podem ser em concentrações que não provoquem tais interações. Caso contrário, a gama de concentrações que pode ser estudada nos ensaios de um produto químico em estudo é limitada pela sua solubilidade no tampão.
|
|
13.
|
A título informativo, o quadro 1 fornece os resultados do ensaio para as 24 substâncias que foram ensaiadas em ambos os ensaios plenamente validados descritos no presente método. Destas substâncias, 17 são classificadas como ligantes do ER e 6 como não ligantes, com base em relatórios publicados, incluindo ensaios in vitro para a ativação transcricional do ER e/ou o ensaio uterotrófico (9) (10) (11) (12) (13) (14) (15). Em referência aos dados resumidos no quadro 1, houve um acordo de quase 100 % entre os dois ensaios sobre as classificações de todas as substâncias até 10–4 M, e cada substância foi corretamente classificada como ligante ou não ligante. Durante os estudos de validação, são fornecidas informações suplementares sobre este grupo de substâncias, bem como sobre outras substâncias ensaiadas com os ensaios de ligação para efeitos de ER nos estudos de validação, de acordo com as normas de desempenho para o ensaio vinculativo (3), apêndice 2 (quadros 1, 2 e 3).
Quadro 1
Classificação de substâncias como ligantes ou não ligantes quando ensaiados com os ensaios de ligação de FW e do de hrER do CERI hrER, em comparação com a resposta esperada
|
|
Denominação da substância
|
N.o CAS
|
Resposta esperada
|
Ensaio de FW
|
Ensaio do CERI
|
Produto químico MESH Classe
|
Classe de produto
|
|
|
Concentração Gama (M)
|
Classificação
|
Concentração Gama (M)
|
Classificação
|
|
1
|
17β-Estradiol
|
50-28-2
|
Ligante
|
1×10–11 – 1×10–6
|
Ligante
|
1×10–11 – 1×10–6
|
Ligante
|
Esteroide
|
Produto farmacêutico, agente veterinário
|
|
2
|
Noretinodrel
|
68-23-5
|
Ligante
|
3×10–9 – 30×10–4
|
Ligante
|
3×10–9 – 30×10–4
|
Ligante
|
Esteroide
|
Produto farmacêutico, agente veterinário
|
|
3
|
Norentindrona
|
68-22-4
|
Ligante
|
3×10–9 – 30×10–4
|
Ligante
|
3×10–9 – 30×10–4
|
Ligante
|
Esteroide
|
Produto farmacêutico, agente veterinário
|
|
4
|
Ftalato de di-n-butilo
|
84-74-2
|
Não ligante (*5)
|
1×10–10 – 1×10–4
|
Não ligando (*6)
(1)
|
1×10–10 – 1×10–4
|
Não ligando (*6)
(1)
|
Hidrocarboneto cíclico, éster
|
Plastificante, Produto químico intermédio
|
|
5
|
DES
|
56-53-1
|
Ligante
|
1×10–10 – 1×10–3
|
Ligante
|
1×10–10 – 1×10–3
|
Ligante
|
Hidrocarboneto cíclico, fenol
|
Produto farmacêutico, agente veterinário
|
|
6
|
17α-Etinilestradiol
|
57-63-6
|
Ligante
|
1×10–10 – 1×10–3
|
Ligante
|
1×10–10 – 1×10–3
|
Ligante
|
Esteroide
|
Produto farmacêutico, agente veterinário
|
|
7
|
Meso-hexesterol
|
84-16-2
|
Ligante
|
1×10–10 – 1×10–3
|
Ligante
|
1×10–10 – 1×10–3
|
Ligante
|
Hidrocarboneto cíclico, fenol
|
Produto farmacêutico, agente veterinário
|
|
8
|
Genisteína
|
446-72-0
|
Ligante
|
1×10–10 – 1×10–3
|
Ligante
|
1×10–10 – 1×10–3
|
Ligante
|
Hidrocarboneto heterocíclico, flavonoide
|
Produto natural
|
|
9
|
Equol
|
531-95-3
|
Ligante
|
1×10–10 – 1×10–3
|
Ligante
|
1×10–10 – 1×10–3
|
Ligante
|
Fitoestrogénio Metabolito
|
Produto natural
|
|
10
|
Butilparabeno (4-hidroxibenzoato de n–butilo)
|
94-26-8
|
Ligante
|
1×10–10 – 1×10–3
|
Ligante
|
1×10–10 – 1×10–3
|
Ligante
|
Parabeno
|
Conservante
|
|
11
|
Nonilfenol (mistura)
|
84852-15-3
|
Ligante
|
1×10–10 – 1×10–3
|
Ligante
|
1×10–10 – 1×10–3
|
Ligante
|
Alquilfenol
|
Composto intermédio
|
|
12
|
o,p’-DDT
|
789-02-6
|
Ligante
|
1×10–10 – 1×10–3
|
Ligante
|
1×10–10 – 1×10–3
|
Ligante
|
Organoclorado
|
Inseticida
|
|
13
|
Corticosterona
|
50-22-6
|
Não ligante (*5)
|
1×10–10 – 1×10–4
|
Não ligante
|
1×10–10 – 1×10–4
|
Não ligante
|
Esteroide
|
Produto natural
|
|
14
|
Zearalenona
|
17924-92-4
|
Ligante
|
1×10–10 – 1×10–3
|
Ligante
|
1×10–10 – 1×10–3
|
Ligante
|
Hidrocarboneto heterocíclico, lactona
|
Produto natural
|
|
15
|
Tamoxifeno
|
10540-29-1
|
Ligante
|
1×10–10 – 1×10–3
|
Ligante
|
1×10–10 – 1×10–3
|
Ligante
|
Hidrocarboneto cíclico
|
Produto farmacêutico, agente veterinário
|
|
16
|
5α-Di-hidrotestosterona
|
521-18-6
|
Ligante
|
1×10–10 – 1×10–3
|
Ligante
|
1×10–10 – 1×10–3
|
Ligante
|
Esteroide não fenólico
|
Produto natural
|
|
17
|
Bisfenol A
|
80-05-7
|
Ligante
|
1×10–10 – 1×10–3
|
Ligante
|
1×10–10 – 1×10–3
|
Ligante
|
Fenol
|
Produto químico intermédio
|
|
18
|
4-n-Heptilfenol
|
1987-50-4
|
Ligante
|
1×10–10 – 1×10–3
|
Equívoco (6)
|
1×10–10 – 1×10–3
|
Ligante
|
Alquilfenol
|
Intermédio
|
|
19
|
Kepone (Clordecona)
|
143-50-0
|
Ligante
|
1×10–10 – 1×10–3
|
Ligante
|
1×10–10 – 1×10–3
|
Ligante
|
Hidrocarboneto halogenado
|
Pesticida
|
|
20
|
Benzo(a)antraceno
|
56-55-3
|
Não ligante
|
1×10–10 – 1×10–3
|
Não ligante (7)
|
1×10–10 – 1×10–3
|
Não ligante (7)
|
Hidrocarboneto aromático
|
Intermédio
|
|
21
|
Enterolactona
|
78473-71-9
|
Ligante
|
1×10–10 – 1×10–3
|
Ligante
|
1×10–10 – 1×10–3
|
Ligante
|
Fitoestrogénio
|
Produto natural
|
|
22
|
Progesterona
|
57-83-0
|
Não ligante (*5)
|
1×10–10 – 1×10–4
|
Não ligante
|
1×10–10 – 1×10–4
|
Não ligante
|
Esteroide
|
Produto natural
|
|
23
|
Octiltrietoxissilano
|
2943-75-1
|
Não ligante
|
1×10–10 – 1×10–3
|
Não ligante
|
1×10–10 – 1×10–3
|
Não ligante
|
Silano
|
Modificador de superfície
|
|
24
|
Atrazina
|
1912-24-9
|
Não ligante (*5)
|
1×10–10 – 1×10–4
|
Não ligante
|
1×10–10 – 1×10–4
|
Não ligante
|
Composto heterocíclico
|
Herbicida
|
|
COMPONENTES DO ENSAIO DE FIXAÇÃO HRER
Componentes de ensaio essenciais
|
14.
|
O presente método de ensaio aplica-se a ensaios que utilizam um recetor ER e um ligando suficientemente forte ao recetor, que pode ser utilizado como marcador/rastreador para o ensaio e pode ser deslocado com concentrações crescentes de um produto químico em estudo. Os ensaios de ligação contêm os dois componentes principais seguintes: 1) ligantes por saturação; 2) ligantes competitivos. O ensaio de saturação é utilizado para confirmar a especificidade e a atividade das preparações do recetor, enquanto a experiência de ligantes competitivos é utilizada para avaliar a capacidade de um produto químico para se fixar ao hrER.
|
Controlos
|
15.
|
Deve ser descrever-se a base dos estrogénios e dos controlos de referência paralelos propostos. Os controlos paralelos [de solvente (veículo), positivos (ER, ligante, afinidade forte e fraca), negativo (não ligante)], consoante o caso, servem como indicação de que o ensaio está a progredir em condições adequadas e fornecem uma base para comparações entre experiências; geralmente, fazem parte dos critérios de aceitabilidade de uma experiência (1). As curvas de concentração inteiras para os estrogénios e os controlos de referência (por exemplo, ligante fraco e não ligante) devem ser utilizadas numa única placa em cada série de ensaios. Todas as outras placas devem conter: 1) uma concentração elevada (aproximadamente o deslocamento total do ligando com marcação radioativa) e média (aproximadamente IC50), cada um dos E2 e do ligante fraco em triplicado; 2) controlo do solvente e ligante não específico, ambos em triplicado.
|
Procedimentos normalizados de controlo de qualidade
|
16.
|
Devem ser aplicados os procedimentos normalizados de controlo da qualidade descritos para cada ensaio, a fim de garantir recetores ativos, concentrações corretas de produtos químicos, estabilidade dos limites de segurança ao longo de múltiplas replicações e manutenção da capacidade de fornecer as respostas ER-ligante esperadas ao longo do tempo.
|
Demonstração da competência técnica do laboratório
|
17.
|
Antes de ensaiarem produtos químicos desconhecidos com qualquer dos ensaios do presente método, os laboratórios devem demonstrar competência técnica na realização do ensaio através da realização de ensaios de saturação para confirmar a especificidade e a atividade de preparação do ER e ensaios de ligação competitivos com os estrogénios e os controlos de referência (ligante fraco e não ligante). O laboratório deve estabelecer uma base de dados históricos com resultados relativos aos estrogénios e aos controlos de referência gerados no decurso de 3-5 experiências independentes realizadas em dias diferentes. Estas experiências serão a base dos estrogénios e dos controlos de referência históricos do laboratório e serão utilizadas para avaliação parcial da aceitabilidade do ensaio para futuras séries.
|
|
18.
|
A capacidade de resposta do sistema de ensaio será também confirmada através do ensaio das substâncias indicadas no quadro 2. A lista de substâncias para demonstração de competência técnica é um subconjunto das substâncias de referência constantes das normas de desempenho para os ensaios de ligação do ER (3). Estas substâncias estão disponíveis no comércio, representam as classes de produtos químicos geralmente associadas à atividade de ligantes do ER e apresentam uma gama de potência adequada e previsível para ligantes de ER (ou seja, de forte a fraco) e não ligantes (ou seja, negativo). Para cada substância de demonstração de competência técnica, as concentrações ensaiadas devem abranger o intervalo de variação indicado no quadro 2. Devem efetuar-se pelo menos três experiências relativamente a cada substância, e os resultados devem ser conformes com a atividade química esperada. Cada experiência deve ser realizada de forma independente (ou seja, com diluições frescas de recetores, produtos químicos e reagentes), com três replicados para cada concentração. A competência é demonstrada por uma classificação correta (positiva/negativa) de cada uma das substâncias. Os ensaios de demonstração da competência técnica devem ser realizados por todos os técnicos que aprendem a realizar os ensaios.
Quadro 2
Lista de substância de controlo e de demonstração da competência técnica para ensaios de ligação competitivos do hrER (89)
|
N.o
|
Denominação da substância
|
N.o CAS (90)
|
Resposta prevista (91)
(92)
|
Gama de concentrações de ensaio (M)
|
Classe química MeSH (93)
|
Classe de produtos (94)
|
|
Controlos (estrogénios de referência, ligante fraco, não ligante)
|
|
1
|
17β-Estradiol
|
50-28-2
|
Ligante
|
1×10–11 – 1×10–6
|
Esteroide
|
Produto farmacêutico, agente veterinário
|
|
2
|
Noretinodrel (ou) Norentinodrona
|
68-23-5 (ou) 68-22-4
|
Ligante
|
3×10–9 – 30×10–6
|
Esteroide
|
Produto farmacêutico, agente veterinário
|
|
3
|
Octiltrietoxissilano
|
2943-75-1
|
Não ligante
|
1×10–10 – 1×10–3
|
Silano
|
Modificador de superfície
|
|
Substâncias para a demonstração de competência técnica (94)
|
|
4
|
Dietilestilbesterol
|
56-53-1
|
Ligante
|
1×10–11 – 1×10–6
|
Hidrocarboneto cíclico, fenol
|
Produto farmacêutico, agente veterinário
|
|
5
|
17α-Etinilestradiol
|
57-63-6
|
Ligante
|
1×10–11 – 1×10–6
|
Esteroide
|
Produto farmacêutico, agente veterinário
|
|
6
|
Meso-hexesterol
|
84-16-2
|
Ligante
|
1×10–11 – 1×10–6
|
Hidrocarboneto cíclico, fenol
|
Produto farmacêutico, agente veterinário
|
|
7
|
Tamoxifeno
|
10540-29-1
|
Ligante
|
1×10–11 – 1×10–6
|
Hidrocarboneto cíclico
|
Produto farmacêutico, agente veterinário
|
|
8
|
Genisteína
|
446-72-0
|
Ligante
|
1×10–10 – 1×10–3
|
Composto heterocíclico, flavonoide
|
Produto natural
|
|
9
|
Bisfenol A
|
80-05-7
|
Ligante
|
1×10–10 – 1×10–3
|
Fenol
|
Produto químico intermédio
|
|
10
|
Zearalenona
|
17924-92-4
|
Ligante
|
1×10–11 – 1×10–3
|
Composto heterocíclico, lactona
|
Produto natural
|
|
11
|
Butilparabeno
|
94-26-8
|
Ligante
|
1×10–11 – 1×10–3
|
Ácido carboxílico, fenol
|
Conservante
|
|
12
|
Atrazina
|
1912-24-9
|
Não ligante
|
1×10–11 – 1×10–6
|
Composto heterocíclico
|
Herbicida
|
|
13
|
Ftalato de di-n-butilo (DBP) (95)
|
84-74-2
|
Não ligante (96)
|
1×10–10 – 1×10–4
|
Hidrocarboneto cíclico, éster
|
Plastificante, Produto químico intermédio
|
|
14
|
Corticosterona
|
50-22-6
|
Não ligante
|
1×10–11 – 1×10–4
|
Esteroide
|
Produto natural
|
|
Ensaio de solubilidade e determinação da gama de concentrações de produtos químicos em estudo
|
19.
|
Deve efetuar-se um ensaio preliminar para determinar o limite de solubilidade de cada produto químico em estudo e identificar a gama de concentrações adequadas a utilizar no ensaio. O limite de solubilidade de cada produto químico em estudo deve ser determinado inicialmente no solvente e confirmado em condições de ensaio. A concentração final estudada no ensaio não deve exceder 1 mM. O ensaio de determinação da gama de concentrações consiste num controlo de solvente, a par de oito diluições sequenciais, a partir da concentração máxima aceitável (por ex., 1 mM ou inferior, com base no limite de solubilidade), e da presença de turbidez ou precipitado percetível. As concentrações na segunda e na terceira experiência devem ser ajustadas na medida do necessário para caracterizar melhor a curva concentração-resposta.
|
Critérios de aceitabilidade dos ensaios
|
20.
|
A aceitação ou rejeição de uma série de ensaios baseia-se na avaliação dos resultados obtidos para os estrogénios e controlos de referência utilizados para cada experiência. Em primeiro lugar, para a placa 1, as curvas de concentração inteiras para os controlos de referência de cada experiência devem cumprir as medidas de desempenho relativas aos parâmetros das curvas (p. ex., IC50 e curva de Hill), com base nos resultados relatados para os protocolos correspondentes relativos aos ensaios do CERI e ensaios de FW de (apêndices 2 e 3) e nos dados históricos de controlo do laboratório que realiza o ensaio. Todos os controlos (estrogénios de referência, ligante fraco e não ligante) devem ser corretamente classificados para cada experiência. Em segundo lugar, os controlos de todas as placas subsequentes têm de ser avaliados em termos de coerência com a placa 1. Deve utilizar-se uma gama de concentrações do produto químico em estudo suficiente para definir com clareza o topo da curva do ligante competitivo. A variabilidade dos replicados em cada concentração do produto químico em estudo, bem como entre os três ensaios independentes, deve ser razoável e cientificamente aceitável. A capacidade para efetuar o ensaio de forma consistente deve ser demonstrada através da constituição e da manutenção de uma base de dados histórica para os estrogénios de referência. Os desvios-padrão (DP) ou os coeficientes de variação (CV) para as médias dos estrogénios de referência e para os parâmetros das curvas do ligante fraco de controlo de várias experiências podem ser utilizados como indicadores da reprodutibilidade interna do laboratório. Devem utilizar-se critérios profissionais para a avaliação dos resultados do controlo das placas de cada série de ensaios, bem como para cada produto químico em estudo.
Além disso, devem ser respeitados os seguintes princípios relativos aos critérios de aceitabilidade:
|
—
|
Os dados devem ser suficientes para uma avaliação quantitativa do ligante do ER,
|
|
—
|
As concentrações ensaiadas devem manter-se dentro da gama de solubilidade dos produtos químicos em estudo.
|
|
Análise dos dados
|
21.
|
O procedimento definido para a análise dos dados relativos aos ligantes por saturação e competitivo devem respeitar os princípios fundamentais da caracterização das interações recetor-ligando. Normalmente, os dados relativos aos ligantes por saturação são analisados por recurso a um modelo de regressão não linear que contabiliza a ligação total e não específica. Pode ser necessária uma correção por depleção de ligantes [ex. Swillens, 1995 (19)] aquando da determinação de Bmáx e Kd. Os dados provenientes de ensaios de ligação competitivos são geralmente software transformados [por exemplo, percentagem de ligação específica e concentração do produto químico em estudo (logM)]. Para cada produto químico em estudo, as estimativas de log(IC50) devem ser determinadas com recurso a um software de ajustamento de curva não linear adequado, a fim de a adaptar à equação de quatro parâmetros de Hill. Na sequência de uma análise preliminar, devem ser definidos os parâmetros de ajustamento da curva e realizada uma análise visual da forma como os dados do ligante se adaptam à curva de ligação competitiva gerada. Em alguns casos, pode ser necessária uma análise adicional para obter o melhor ajustamento da curva (por exemplo, restrição do topo e/ou da base da curva, utilização da regra dos 10 %, ver apêndice 4 e referência 2 (secção III.A.2).
|
|
22.
|
O cumprimento dos critérios de aceitabilidade (ponto 20) indica que o ensaio está a funcionar corretamente, mas não garante que produz dados exatos. A replicação dos resultados corretos do primeiro ensaio constitui a melhor indicação de que foram produzidos dados exatos.
|
Critérios gerais de interpretação dos dados
|
23.
|
Não existe na atualidade um método universalmente aceite para a interpretação dos dados de ligantes de ER. No entanto, tanto a avaliação qualitativa (por exemplo ligante/não ligante) como a quantitativa [p. ex., log(IC50), afinidade de ligação relativa (RBA), etc.] da atividade mediada pelo ER devem basear-se em dados empíricos e em pareceres científicos sólidos.
|
Relatório de ensaio
|
24.
|
O relatório do ensaio deve incluir as seguintes informações:
|
|
Controlo/referência/produto químico em estudo
|
—
|
origem, número de lote e data-limite de utilização, se conhecida;
|
|
—
|
estabilidade do produto químico em estudo, se conhecida;
|
|
—
|
solubilidade e estabilidade do produto químico em estudo no solvente, se conhecida;
|
|
—
|
medição do pH, da pressão osmótica e da quantidade de precipitado no meio de cultura ao qual foi adicionado o produto químico em estudo, consoante o que for adequado.
|
|
|
|
Substância monocomponente:
|
—
|
aspeto físico, hidrossolubilidade e outras propriedades físico-químicas pertinentes;
|
|
—
|
dados de identificação química, como as denominações IUPAC ou CAS, número CAS, código SMILES ou InChI, fórmula estrutural, pureza, identidade química de impurezas, caso se justifique e seja exequível, etc.
|
|
|
|
Substância multicomponentes, UVCB e misturas:
|
—
|
caracterizada, na medida do possível, pela identidade química (ver acima), ocorrência quantitativa e principais propriedades físico-químicas dos componentes.
|
|
|
|
Solvente/veículo:
|
—
|
caracterização (natureza, fornecedor e lote);
|
|
—
|
justificação para a escolha do solvente/veículo;
|
|
—
|
solubilidade e estabilidade do produto químico em estudo no solvente/veículo, se conhecida;
|
|
|
|
Recetores:
|
—
|
origem dos recetores (fornecedor, número de catálogo, lote, espécie do recetor, concentração ativa do recetor fornecido pelo fornecedor, certificação do fornecedor)
|
|
—
|
caracterização de recetores (incluindo os resultados dos ligantes por saturação): Kd, Bmáx,
|
|
—
|
armazenamento dos recetores
|
|
—
|
ligando com marcação radioativa:
|
|
—
|
fornecedor, número de catálogo, lote, atividade específica
|
|
|
|
Condições de realização do ensaio:
|
—
|
limitações da solubilidade nas condições de ensaio,
|
|
—
|
composição do tampão do ligante;
|
|
—
|
concentração do recetor;
|
|
—
|
concentração do traçador (ou seja, ligante com marcação radioativa);
|
|
—
|
concentrações do produto químico em estudo;
|
|
—
|
percentagem do veículo no ensaio final;
|
|
—
|
tempo e temperatura de incubação;
|
|
—
|
método de separação agrupado/isolado;
|
|
—
|
controlos positivos e negativos/substâncias de referência;
|
|
—
|
critérios definidos para que os ensaios sejam considerados positivos, negativos ou inconclusivos,
|
|
|
|
Verificação da aceitabilidade:
|
—
|
valores reais de IC50 e da curva de Hill para controlos positivos/substâncias de referência,
|
|
|
|
Resultados:
|
—
|
dados brutos agrupados e isolados;
|
|
—
|
controlo de confirmação da desnaturação, se for caso disso;
|
|
—
|
se existir, a concentração mínima efetiva (LEC);
|
|
—
|
valores RBA e/ou IC50, consoante o caso;
|
|
—
|
relação concentração-resposta, quando for possível determiná-la;
|
|
—
|
análise estatística, se for caso disso, associada a uma medida de erro e confiança (por exemplo, SEM, DP, CV ou IC 95 %) e descrição da forma como os valores foram obtidos;
|
|
|
|
Discussão dos resultados:
|
—
|
aplicação da regra dos 10 %
|
|
Conclusão
|
REFERÊNCIAS BIBLIOGRÁFICAS
|
(1)
|
OCDE (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environmental, Health and Safety Publications, Series on Testing and Assessment (No 34), Organisation for Economic Cooperation and Development, Paris.
|
|
(2)
|
OCDE (2015). Integrated Summary Report: Validation of Two Binding Assays Using Human Recombinant Estrogen Recetor Alpha (hrERα), Health and Safety Publications, Series on Testing and Assessment (No 226), Organisation for Economic Cooperation and Development, Paris.
|
|
(3)
|
OCDE (2015). Performance Standards for Binding Assays Using Human Recombinant Estrogen Recetor Alpha (hrERα), Health and Safety Publications, Series on Testing and Assessment (No 222), Organisation for Economic Cooperation and Development, Paris.
|
|
(4)
|
OCDE (2012). Guidance Document on Standardized Test Guidelines for Evaluating Chemicals for Endocrine Disruption. Environmental, Health and Safety Publications, Series on Testing and Assessment (No 150), Organisation for Economic Cooperation and Development, Paris.
|
|
(5)
|
Cavailles V. (2002). Estrogens and Recetors: an Evolving Concept, Climacteric, 5 Suppl 2: p.20-6.
|
|
(6)
|
Welboren W.J., et al. (2009). Genomic Actions of Estrogen Recetor Alpha: What are the Targets and How are they Regulated? Endocr. Relat. Cancer., 16(4): p. 1073-89.
|
|
(7)
|
Younes M. and Honma N. (2011). Estrogen Recetor Beta, Arch. Pathol. Lab. Med., 135(1): p. 63-6.
|
|
(8)
|
Diamanti-Kandarakis et al. (2009). Endocrine-Disrupting Chemicals: an Endocrine Society Sci. Statement, Endo Rev 30(4):293-342.
|
|
(9)
|
ICCVAM (2002). Background Review Document. Current Status of Test Methods for Detecting Endocrine Disruptors: In Vitro Estrogen Recetor Binding Assays. (NIH Publication No 03-4504). National Institute of Environmental Health Sciences, Research Triangle Park, NC.
|
|
(10)
|
ICCVAM (2003). ICCVAM Evaluation of In Vitro Test Methods for Detecting Potential Endocrine Disruptors: Estrogen Recetor and Androgen Recetor Binding and Transcriptional Activation Assays.
|
|
(11)
|
ICCVAM (2006). ICCVAM Evaluation of In Vitro Test Methods for Detecting Potential Endocrine Disruptors: Estrogen Recetor and Androgen Recetor Binding and Transcriptional Activation Assays.
|
|
(12)
|
Akahori Y. et al. (2008). Relationship Between the Results of In Vitro Recetor Binding Assay to Human Estrogen Recetor Alpha and In Vivo Uterotrophic Assay: Comparative Study with 65 Selected Chemicals, Toxicol. In Vitro, 22(1): 225-231.
|
|
(13)
|
OCDE (2007). Additional Data Supporting the Test Guideline on the Uterotrophic Bioassay in Rodents, Environment, Health and Safety Publications, Series on Testing and Assessment (No 67), Organisation for Economic Cooperation and Development, Paris.
|
|
(14)
|
Takeyoshi, M. (2006). Draft Report of Pre-validation and Inter-laboratory Validation For Stably Transfected Transcriptional Activation (TA) Assay to Detect Estrogenic Activity - The Human Estrogen Recetor Alpha Mediated Reporter Gene Assay Using hER-HeLa-9903 Cell Line, Chemicals Evaluation and Research Institute (CERI): Japan. p. 1-188.
|
|
(15)
|
Yamasaki, K; Noda, S; Imatanaka, N; Yakabe, Y. (2004). Comparative Study of the Uterotrophic Potency of 14 Chemicals in a Uterotrophic Assay and their Recetor-Binding Affinity, Toxicol. Letters, 146: 111-120.
|
|
(16)
|
Kummer V; Maskova, J; Zraly, Z; Neca, J; Simeckova, P; Vondracek, J; Machala, M. (2008). Estrogenic Activity of Environmental Polycyclic Aromatic Hydrocarbons in Uterus of Immature Wistar Rats. Toxicol. Letters, 180: 213-221.
|
|
(17)
|
Gozgit, JM; Nestor, KM; Fasco, MJ; Pentecost, BT; Arcaro, KF. (2004). Differential Action of Polycyclic Aromatic Hydrocarbons on Endogenous Estrogen-Responsive Genes and on a Transfected Estrogen-Responsive Reporter in MCF-7 Cells. Toxicol. and Applied Pharmacol., 196: 58-67.
|
|
(18)
|
Santodonato, J. (1997). Review of the Estrogenic and Antiestrogenic Activity of Polycyclic Aromatic Hydrocarbons: Relationship to Carcinogenicity. Chemosphere, 34: 835-848.
|
|
(19)
|
Swillens S (1995). Interpretation of Binding Curves Obtained with High Recetor Concentrations: Practical Aid for Computer Analysis, Mol Pharmacol 47(6):1197-1203.
|
Apêndice 1
DEFINIÇÕES E ABREVIATURAS
Adequação: descrição da relação de um ensaio com o objeto de interesse e da sua importância e utilidade para um determinado fim. Traduz a medida em que o ensaio mede ou prevê corretamente o efeito biológico em causa. A adequação comporta a exatidão (concordância) de um ensaio (1).
Atividade estrogénica: capacidade de um produto químico reproduzir o 17β-estradiol na sua capacidade de ligação a recetores de estrogénios. O presente método de ensaio permite detetar a ligação ao hERα.
CF: quadro conceptual da OCDE para ensaio e avaliação de perturbadores do sistema endócrino.
Competência técnica: capacidade comprovada para efetuar um ensaio de forma adequada antes de ensaiar substâncias desconhecidas.
Critérios de aceitabilidade: normas mínimas para a realização de controlos experimentais e padrões de referência. Para que uma experiência seja considerada válida, devem ser satisfeitos todos os critérios de aceitabilidade.
CV: coeficiente de variação.
DP: desvio-padrão.
E2: 17β-estradiol.
ED: desregulação do sistema endócrino.
Ensaio «me-too»: expressão coloquial para designar um método que é, estrutural e funcionalmente, semelhante a um método de ensaio de referência validado e aceite. O termo é sinónimo de «método de ensaio similar».
ER: Recetor de estrogénio.
Estrogénio de referência: 17ß-estradiol (E2, CAS 50-28-2).
Exatidão (concordância): grau de concordância entre os resultados do ensaio e um valor de referência aceite. Constitui uma medida da eficiência do método e um aspeto da adequação. Este termo e o termo «concordância» são muitas vezes utilizados indistintamente para indicar uma proporção de resultados corretos do método de ensaio (1).
Fiabilidade: indica em que medida um ensaio pode ser reproduzido ao longo do tempo num mesmo laboratório e entre laboratórios utilizando o mesmo protocolo. A fiabilidade é avaliada com base nos valores calculados das reprodutibilidades intralaboratorial e interlaboratorial.
hERα: recetor alfa de estrogénio humano.
IC50
: concentração semimáxima efetiva de um produto químico em estudo inibidor.
ICCVAM: Interagency Coordinating Committee on the Validation of Alternative Methods [Comissão de Coordenação da Validação de Métodos Alternativos].
LEC: menor concentração eficaz é a menor concentração do produto químico em estudo que produz uma resposta (ou seja, a menor concentração do produto químico em estudo em que o fator de indução é estatisticamente diferente do controlo do veículo concorrente).
Método de ensaio validado: método de ensaio relativamente ao qual foram realizados estudos de validação com vista a determinar a sua adequação (incluindo a exatidão) e fiabilidade para um determinado fim. Importa referir que um método de ensaio validado pode não ser suficientemente exato e fiável para ser considerado aceitável para o fim pretendido (1).
Métodos de ensaio de referência: ensaios em que se baseia o PBTG 493.
Normas de desempenho: normas, associadas a um método de ensaio validado, com base nas quais pode ser avaliada a comparabilidade de um ensaio proposto que seja mecanística e funcionalmente similar. Incluem: (1) componentes essenciais do método de ensaio; (2) uma lista mínima de produtos químicos de referência, escolhidos de entre os produtos químicos utilizados para demonstrar o desempenho aceitável do método de ensaio validado; e (3) níveis de exatidão e fiabilidade comparáveis, baseados no que foi obtido para o método de ensaio validado, que o método de ensaio proposto deve demonstrar quando avaliado pela utilização da lista mínima de produtos químicos de referência (1).
PBTG: diretriz de ensaio baseada no desempenho.
Produto químico: uma substância ou mistura.
Produto químico em estudo: qualquer substância ou mistura à qual seja aplicado o presente método de ensaio.
RBA: afinidade de ligação relativa. A RBA de uma substância é calculada como uma percentagem do log(IC50) para a substância relativamente ao log(IC50) para o 17β-estradiol.
Regra dos 10 %: opção de excluir da análise dados em que a média dos replicados para a percentagem de ligação específica do [3H]17β-estradiol tenha superior em 10 % ou mais à observada para o valor médio a uma concentração inferior (ver apêndice 4).
Reprodutibilidade interlaboratorial: uma medida do grau em que diferentes laboratórios qualificados que utilizam o mesmo protocolo e testam as mesmas substâncias podem produzir resultados qualitativa e quantitativamente similares. A reprodutibilidade interlaboratorial é determinada durante os processos de pré-validação e validação e indica em que medida um método pode ser transferido com êxito entre laboratórios, igualmente designada «reprodutibilidade entre laboratórios» (1).
Reprodutibilidade intralaboratorial: uma medida do grau em que pessoal qualificado do mesmo laboratório consegue replicar resultados com êxito utilizando o mesmo protocolo, em momentos diferentes. Igualmente designada «reprodutibilidade intralaboratorial» (1).
Substâncias para a demonstração de competência técnica: um subconjunto das substâncias de referência incluídas nas normas de desempenho que pode ser utilizado pelos laboratórios para demonstrar competência técnica com um ensaio normalizado. Os critérios de seleção destas substâncias incluem normalmente o facto de representarem a gama de respostas, estarem comercialmente disponíveis e disporem de dados de referência de elevada qualidade.
Validação: o processo de estabelecimento da fiabilidade e adequação de uma determinada abordagem, método, processo ou avaliação para um determinado fim (1).
Apêndice 2
ENSAIOS IN VITRO DE LIGAÇÃO POR SATURAÇÃO E COMPETITIVA AO RECETOR DE ESTROGÉNIO (ERΑ) COM RECURSO A ERΑ RECOMBINANTE INTEIRO DE FREYBERGER-WILSON
CONSIDERAÇÕES INICIAIS E LIMITAÇÕES (VER TAMBÉM INTRODUÇÃO GERAL)
|
1.
|
Este ensaio in vitro de ligação por saturação e competitiva ao recetor de estrogénio (ER) utiliza o recetor humano inteiro ERαα(hrERα), que é produzido em células de insetos infetadas com baculovírus e destas isolado. O protocolo, desenvolvido por Freiberger e Wilson, foi submetido a um estudo de validação internacional em vários laboratórios (2), que demonstrou a sua pertinência e fiabilidade para o fim a que se destina.
|
|
2.
|
Este ensaio é um procedimento de análise para identificação de substâncias suscetíveis de se ligarem a toda a extensão do hrERα. É utilizado para determinar a capacidade de um produto químico em estudo para competir com o 17β-estradiol pela ligação ao hrERα. Os resultados quantitativos do ensaio podem incluir o IC50 (uma medição da concentração do produto químico em estudo necessária para deslocar metade do [3H]17β-estradiol do hrERα) e as afinidades de ligação relativas dos produtos químicos em estudo para o hrERα em comparação com o 17β-estradiol. Para fins de análise química, os resultados qualitativos de ensaios aceitáveis podem incluir classificações produtos químicos em estudo como ligantes ou não ligantes do hrERα, ou como inconclusivos com base nos critérios descritos para as curvas de ligação.
|
|
3.
|
O ensaio utiliza ligandos radioativos, sendo necessário que o laboratório disponha de uma licença para o manuseamento desses materiais. Todos os procedimentos com radioisótopos e produtos químicos perigosos devem seguir a regulamentação e os procedimentos previstos na legislação nacional.
|
|
4.
|
As secções «INTRODUÇÃO GERAL» e «COMPONENTES DO ENSAIO DE LIGAÇÃO DO hrER» devem ser lidas antes da utilização do presente ensaio para fins normativos. As definições e abreviaturas utilizadas são descritas no apêndice 1.
|
PRINCÍPIOS DO ENSAIO (VER TAMBÉM INTRODUÇÃO GERAL)
|
5.
|
O ensaio de ligação ao hrERα mede a capacidade de um ligando com marcação radioativa – [3H]17β-estradiol – para se ligar ao ER na presença de concentrações crescentes de um produto químico em estudo (concorrente). Os produtos químicos em estudo que possuem uma elevada afinidade para o ER competem com o ligando com marcação radioativa a uma concentração mais baixa comparativamente com os produtos químicos com menor afinidade para o recetor.
|
|
6.
|
O presente ensaio é composto por dois componentes principais: uma experiência de ligantes por saturação com vista a caracterizar os parâmetros da interação recetor-ligando, seguida de uma experiência de ligantes competitivos que caracteriza a concorrência entre um produto químico em estudo e um ligando com marcação radioativa para ligação ao ER.
|
|
7.
|
O objetivo da experiência de ligação por saturação é caracterizar um determinado lote de recetores para afinidade de ligação e número de recetores em preparação para a realização da experiência de ligação competitiva. A experiência de ligação por saturação mede, em condições de equilíbrio, a afinidade de uma concentração fixa do recetor de estrogénio com o seu ligando natural (representado pela constante de dissociação, Kd) e a concentração de pontos de recetores ativos (Bmáx).
|
|
8.
|
A experiência de ligação competitiva mede a afinidade de uma substância para competir com o [3H]17β-estradiol pela ligação ao ER. A afinidade é quantificada pela concentração do produto químico em estudo que, em equilíbrio, inibe 50 % do ligante específico do [3H]17β-estradiol (designada «concentração inibidora 50 %» ou «IC50»). A afinidade pode igualmente ser avaliada através da afinidade de ligação relativa (RBA, em relação à IC50 do estradiol, medida separadamente na mesma série de ensaios). A experiência de ligação competitiva mede a ligação do [3H]17β-estradiol a uma concentração fixa na presença de uma vasta gama de concentrações (oito ordens de grandeza) do produto químico em estudo. Os dados são, então, ajustados, na medida do possível, a uma forma da equação de Hill (Hill, 1910) que descreve o deslocamento do ligando radioativo através de um ligante concorrente num ponto. A extensão da deslocação do estradiol com marcação radioativa em equilíbrio é utilizada para caracterizar o produto químico em estudo como ligante, não ligante ou inconclusivo.
|
PROCEDIMENTO
Demonstração do desempenho proteico aceitável do hrERα
|
9.
|
Antes de realizar rotineiramente os ensaios de ligação por saturação e de ligação competitiva, deve verificar-se se cada novo lote de hrERα tem um desempenho correto no laboratório em que será usado. Para demonstrar esse desempenho, deve utilizar-se um processo em duas etapas:
|
—
|
Realização de um ensaio de ligação por saturação [3H]17β-estradiol para demonstrar a especificidade e a saturação do hrERα. A análise de regressão não linear dos dados (por exemplo, BioSoft; McPherson, 1985; Motulsky, 1995) e a subsequente aplicação da equação de Scatchard devem comprovar a afinidade de ligação do hrERα com o [3H]17β-estradiol (Kd) e o número de recetores (Bmáx) de cada lote de hrERα.
|
|
—
|
Realizar um ensaio de ligação competitiva com recurso às substâncias de controlo [estrogénio de referência (17β-estradiol], um ligante fraco (por exemplo, noretinodrel ou norentinodrona), e um não ligante (octiltrietoxissilano, OTES). Cada laboratório deve estabelecer uma base de dados histórica para documentar a coerência do IC50 e outros valores relevantes para os estrogénios de referência e de um ligante fraco, entre as experiências e os diferentes lotes de hrERα. Os parâmetros das curvas de ligação competitiva para as substâncias de controlo devem situar-se dentro dos limites do intervalo de confiança de 95 % (ver quadro 1) que foram estabelecidos com recurso a dados de laboratórios que participaram no estudo de validação deste ensaio (2).
|
|
Quadro 1
Critérios de desempenho estabelecidos para os estrogénios de referência e para o ligante fraco, ensaio de ligação de hrER de FW
|
Substância
|
Parâmetro
|
Média (8)
|
Desvio-padrão (n)
|
Intervalos de confiança de 95 % (9)
|
|
Limite inferior
|
Limite superior
|
|
17β-Estradiol
|
Topo (%)
|
100,44
|
10,84 (67)
|
97,8
|
103,1
|
|
Base (%)
|
0,29
|
1,25 (67)
|
-0,01
|
0,60
|
|
Curva de Hill
|
-1,06
|
0,20 (67)
|
-1,11
|
-1,02
|
|
log(IC50) (M)
|
-8,92 (10)
|
0,18 (67)
|
-8,97
|
-8,88
|
|
Noretinodrel
|
Topo (%)
|
99,42
|
8,90 (68)
|
97,27
|
101,60
|
|
Base (%)
|
2,02
|
3,42 (68)
|
1,19
|
2,84
|
|
Curva de Hill
|
-1,01
|
0,38 (68)
|
-1,10
|
-0,92
|
|
log(IC50) (M)
|
-6,39
|
0,27 (68)
|
-6,46
|
-6,33
|
|
Noretinedrona (10)
|
Topo (%)
|
96,14
|
8,44 (27)
|
92,80
|
99,48
|
|
Base (%)
|
2,38
|
5,02 (27)
|
0,40
|
4,37
|
|
Curva de Hill
|
-1,41
|
0,32 (27)
|
-1,53
|
-1,28
|
|
log(IC50) (M)
|
-5,73
|
0,27 (27)
|
-5,84
|
-5,62
|
Demonstração da competência técnica do laboratório
|
10.
|
Ver os pontos 17 e 18 e o quadro 2 da secção «COMPONENTES DA ENSAIO DE LIGAÇÃO DO hrER» do presente método de ensaio. Cada ensaio (ligação por saturação e ligação competitiva) deve ser constituído por três séries de ensaios independentes (ou seja, diluições de recetor, produtos químicos e reagentes) em dias diferentes, devendo cada série conter três replicados.
|
Determinação do da concentração do recetor (hrERα)
|
11.
|
A concentração do recetor ativo varia ligeiramente em função do lote e das condições de armazenagem. Por esta razão, deve determinar-se a concentração do recetor ativo recebida do fornecedor. Obtém-se assim a concentração adequada do recetor ativo no momento da série de ensaios.
|
|
12.
|
Em condições correspondentes à ligação competitiva (1 nM [3H]-estradiol), as concentrações nominais de recetor de 0,25, 0,5, 0,75 e 1 nM devem ser incubadas na ausência (ligação total) e na presença (ligação não específica) de 1 μM de estradiol não marcado. À ligação específica, calculada como a diferença entre a ligação total e a ligação não específica, faz-se corresponder a concentração nominal do recetor. A concentração do recetor que der valores de ligação específicos correspondentes a 20 % da marcação radioativa adicionada está relacionada com a concentração nominal de recetor correspondente, devendo esta ser utilizada para as experiências de saturação e de ligação competitiva. Frequentemente, uma concentração final de hrER de 0,5 nM satisfaz esta condição.
|
|
13.
|
Se o critério dos 20 % não puder, repetidamente, ser satisfeito, importa verificar o procedimento experimental para deteção de eventuais erros. O incumprimento do critério dos 20 % pode indicar que há muito pouco recetor ativo no lote recombinante, caso em que se deve ponderar a utilização de outro lote de recetores.
|
Ensaio de saturação
|
14.
|
Devem ser avaliadas em triplicado oito concentrações crescentes de [3H]17β-estradiol, nas três condições seguintes (ver quadro 2):
|
—
|
Ausência de 17β-estradiol não marcado e presença de ER. Trata-se de determinar a ligação total por medida da radioatividade nos poços que apenas têm [3H]17β-estradiol.
|
|
—
|
Presença de uma concentração excessiva (1 000 vezes mais) de β-estradiol não marcado em relação ao 17β-estradiol marcado e presença de ER. O objetivo desta condição é saturar os pontos de ligação ativos com 17β-estradiol não marcado e, mediante a medição da radioatividade nos poços, determinar a ligação não específica. Qualquer estradiol quente remanescente que possa ligar-se ao recetor é considerado ligante a um ponto não específico, dado que o estradiol frio deve estar numa concentração tão elevada que se liga a todos os pontos disponíveis no recetor.
|
|
—
|
Ausência de 17β-estradiol não marcado e ausência de ER (determinação da radioatividade total)
|
|
Preparação de soluções de [3H]17β-estradiol e de 17β-estradiol não marcado
|
15.
|
As diluições de [3H]17β-estradiol devem ser preparadas por adição de tampão de ensaio a uma solução-mãe de [3H]17β-estradiol 12 nM para obter concentrações inicialmente compreendidas entre 0,12 nM e 12 nM. Adicionando 40 μl destas soluções aos respetivos poços de uma placa de microtitulação com 96 poços (para um volume final de 160 μl), obtêm-se concentrações finais compreendidas entre 0,03 e 3,0 nM. A preparação do tampão de ensaio, da solução e diluições de [3H]17β-estradiol e a determinação das concentrações são descritas em pormenor no protocolo de FW (2).
|
|
16.
|
As diluições de soluções de β-estradiol devem ser preparadas por adição de tampão de ensaio, a fim de se obter oito concentrações crescentes, compreendidas, inicialmente, entre 0,06 μM e 6 μM. Adicionando 80 μl destas soluções aos respetivos poços de uma placa de microtitulação com 96 poços (para um volume final de 160 μl), obtêm-se as concentrações finais do ensaio, compreendidas entre 0,03 μM e 3 μM. A concentração final do 17β-estradiol não marcado nos poços individuais não específicos deve corresponder a 1 000 vezes a concentração do [3H]17β-estradiol não marcado. A preparação das diluições não marcadas de 17β-estradiol é descrita em pormenor no protocolo de FW (2).
|
|
17.
|
Deve usar-se a concentração nominal do recetor que dê uma ligação específica de 20±5 % (ver os pontos 12 a 13). A solução de hrERIα deve ser preparada imediatamente antes de ser utilizada.
|
|
18.
|
As placas de microtitulação de 96 poços são preparadas de acordo com o quadro 2, com 3 replicados por concentração. O apêndice 2.2 apresenta exemplos de concentrações da placa e distribuição de volumes do [3H]17β-estradiol, 17β-estradiol não marcado, tampão e recetor.
|
Quadro 2
Configuração da placa de microtitulação no ensaio de ligação por saturação
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
|
A
|
0,03 nM [3H] E2 + ER
|
0,06 nM [3H] E2 + ER
|
0,08 nM [3H] E2 + ER
|
0,10 nM [3H] E2 + ER
|
Ligante total (solvente)
|
|
B
|
0,30 nM [3H] E2 + ER
|
0,60 nM [3H] E2 + ER
|
1,0 nM [3H] E2 + ER
|
3,0 nM [3H] E2 + ER
|
|
C
|
|
|
|
|
|
|
D
|
0,03 nM [3H] E2 + ER + 0,03 μM E2
|
0,06 nM [3H] E2 + ER + 0,06 μM E2
|
0,08 nM [3H] E2 + ER + 0,08 μM E2
|
0,10 nM [3H] E2 + ER + 0,10 μM E2
|
Ligante não específico
|
|
E
|
0,30 nM [3H] E2 + ER + 0,30 μM E2
|
0,60 nM [3H] E2 + ER + 0,60 μM E2
|
1,0 nM [3H] E2 + ER + 1,0 μM E2
|
3,0 nM [3H] E2 + ER + 3,0 μM E2
|
|
F
|
|
|
|
|
|
|
G
|
|
|
|
|
|
|
H
|
|
|
|
|
|
[3H] E2: [3H]17β-estradiol
ER: recetor de estrogénio
E2: 17β-estradiol não marcado
|
|
19.
|
As placas de microtitulação devem ser incubadas a 2-8o C, durante 16-20 horas, e colocadas num mecanismo de rotação durante o período de incubação.
|
Medição de [3H]17β-estradiol ligado à hrERα
|
20.
|
O [3H]17β-estradiol ligado ao hrERα deve ser separado do [3H]17β-estradiol livre por adição de 80 μl de suspensão de DCC fria a cada poço, agitando as placas de microtitulação durante 10 minutos e centrifugando-as, durante 10 minutos, a cerca de 2 500 rpm. Para minimizar a dissociação de [3H]17β-estradiol ligado ao hrERα durante este processo, é extremamente importante que os tampões e os poços sejam mantidos a 2-8 °C e que cada etapa seja realizada rapidamente. Para utilizar as placas de microtitulação de forma eficiente e rápida, é necessário um agitador.
|
|
21.
|
Para evitar qualquer contaminação dos poços por contacto com o DCC, devem retirar-se com extremo cuidado 50 μl do sobrenadante contendo [3H]17β-estradiol ligado ao hrERα.
|
|
22.
|
Adicionam-se então a cada poço 200 μl de fluido de cintilação, capaz de converter a energia cinética das emissões nucleares em energia luminosa (A1-B12 e D1-E12). Os poços G1-H12 (identificados como dpms total) representam diluições sequenciais do [3H]17β-estradiol (40 μl),) que devem ser adicionadas diretamente ao líquido de cintilação nos poços da placa de medição conforme indicado no quadro 3; assim, esses poços contêm apenas 200 μl de líquido de cintilação e a diluição adequada do [3H]17β-estradiol. Estas medidas demonstram quanto [3H]17β-estradiol foi adicionado a cada conjunto de poços para a ligação total e para a ligação não específica.
|
Quadro 3
Configuração da placa de microtitulação no ensaio de ligação por saturação Medição da radioatividade
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
|
A
|
0,03 nM [3H] E2 + ER
|
0,06 nM [3H] E2 + ER
|
0,08 nM [3H] E2 + ER
|
0,10 nM [3H] E2 + ER
|
Ligante total (solvente)
|
|
B
|
0,30 nM [3H] E2 + ER
|
0,60 nM [3H] E2 + ER
|
1,0 nM [3H] E2 + ER
|
3,0 nM [3H] E2 + ER
|
|
C
|
|
|
|
|
|
|
D
|
0,03 nM [3H] E2 + ER + 0,03 μM E2
|
0,06 nM [3H] E2 + ER + 0,06 μM E2
|
0,08 nM [3H] E2 + ER + 0,08 μM E2
|
0,10 nM [3H] E2 + ER + 0,10 μM E2
|
Ligante não específico
|
|
E
|
0,30 nM [3H] E2 + ER + 0,30 μM E2
|
0,60 nM [3H] E2 + ER + 0,60 μM E2
|
1,0 nM [3H] E2+ ER + 1,0 μM E2
|
3,0 nM [3H] E2 + ER + 3,0 μM E2
|
|
F
|
|
|
|
|
|
|
G
|
0,03 nM [3H] E2 (dpms total)
|
0,06 nM [3H] E2
|
0,08 nM [3H] E2
|
0,10 nM [3H] E2
|
Dpms total (*7)
|
|
H
|
0,30 nM [3H] E2
|
0,60 nM [3H] E2
|
1,0 nM [3H] E2
|
3,0 nM [3H] E2
|
|
[3H] E2: [3H]17β-estradiol
ER: recetor de estrogénio
E2: 17β-estradiol não marcado
Dpms: desintegrações por minuto
|
|
23.
|
As medições devem iniciar-se com um desfasamento de, pelo menos, 2 horas, devendo o tempo de contagem ser de 40 minutos por poço. Para a determinação de dpm/poço, com correção do efeito de atenuação, deve utilizar-se um contador de cintilação em placas de microtitulação. Em alternativa, se não se dispuser de um contador de cintilação deste tipo, as amostras podem ser medidas num contador convencional. Nestas condições, pode ponderar-se uma redução do tempo de contagem.
|
Ensaio de ligação competitiva
|
24.
|
O ensaio de ligação competitiva mede a ligação de uma única concentração de [3H17β-estradiol na presença de concentrações crescentes de um produto químico em estudo. Devem utilizar-se três replicados paralelos de cada concentração numa mesma série de ensaios. Além disso, devem efetuar-se três ensaios não paralelos para cada produto químico ensaiado. O ensaio deve utilizar uma ou mais placas de microtitulação de 96 poços.
|
Controlos
|
25.
|
experiência do ensaio deve incluir o solvente e os controlos paralelos (isto é, estrogénios de referência, ligante fraco e não ligante). As curvas de concentração inteiras para os estrogénios e os controlos de referência (por exemplo, ligante fraco e não ligante) devem corresponder a uma única placa em cada série de ensaios. Todas as outras placas devem conter: i) uma concentração elevada (deslocamento máximo) e média (aproximadamente a IC50) de cada um dos E2 e ligante fraco em triplicado; ii) controlo do solvente e ligação não específica, cada um, no mínimo, em triplicado. Os procedimentos para a preparação das soluções-tampão de ensaio, controlos, [3H]17β-estradiol, hrERα e produtos químicos em estudo estão descritos na referência bibliográfica 2 (anexo K, ver «FW Assay Protocol»).
|
Controlo do solvente
|
26.
|
O controlo do solvente indica que este não interage com o sistema de ensaio e mede a ligação total (TB). O etanol é o solvente preferencial. Em alternativa, se a concentração mais elevada do produto químico em estudo não for solúvel em etanol, pode utilizar-se DMSO. A concentração de etanol – ou DMSO, se for o caso – nos poços do ensaio final é de 1,5 %, não podendo exceder 2 %.
|
Controlo do tampão
|
27.
|
O controlo do tampão (BC) não deve conter solvente nem produto químico em estudo, mas conter todos os outros componentes do ensaio. Os resultados do controlo do tampão são comparados com o controlo do solvente, para verificar se o solvente utilizado não afeta o sistema de ensaio.
|
Ligante forte (estrogénio de referência)
|
28.
|
O 17β-estradiol (n.o CAS 50-28-2) é o ligando endógeno e liga-se com elevada afinidade ao ER de subtipo alfa. Deve preparar-se uma curva padrão com 17β-estradiol não marcado para cada ensaio de ligação competitiva do hrERα, a fim de se proceder a uma avaliação da variabilidade entre ensaios realizados no mesmo laboratório. Devem preparar-se, em etanol, oito soluções de 17β-estradiol não marcado, com concentrações nos poços compreendidas entre 100 nM e 10 PM -7[logM] a -11[logM]), espaçadas do seguinte modo: (-7[logM], -8[logM], -8,5[logM], -9[logM], - 9.5[logM], -10[logM], -11[logM]). A maior concentração de 17β-estradiol (1 μM) não marcado serve também de indicador de ligação inespecífica. Esta concentração distingue-se pelo rótulo «NSB» no quadro 4, embora também faça parte da curva-padrão.
|
Ligante fraco
|
29.
|
Para demonstrar a sensibilidade de cada experiência e permitir uma avaliação da variabilidade ao longo do tempo, deve ser incluído um ligante fraco [noretinodrel (n.o CAS 68-23-5) ou noretinodrona (n.o CAS 68-22-4)]. Devem preparar-se, em etanol, oito soluções de ligante fraco, com concentrações nos poços compreendidas entre 3 nM e 30 μM (-8,5[logM] to -4,5[logM]), espaçadas do seguinte modo: -4,5[logM], -5[logM], -5,5[logM], -6[logM], -6,5[logM], -7[logM],-7,5[logM], -8,5[logM].
|
Não ligante
|
30.
|
Deve ser usado octiltrietoxissilano (OTES, n.o CAS 2 943-75-1) como controlo negativo (não ligante), que garante que o ensaio permite detetar os produtos químicos em estudo que não se ligam ao hrERα. Devem preparar-se em etanol oito soluções do não ligante, com concentrações nos poços do ensaio compreendidas entre 0,1 nM e 1 000 μM (-10[logM] a -3[logM]) e escalonadas. Pode utilizar-se ftalato de di-n-butilo (DBP), como não ligante de controlo alternativo. A sua solubilidade máxima demonstrada é de -4 [logM].
|
Concentração de hrERα
|
31.
|
Deve utilizar-se a quantidade de recetor que dá um valor específico de ligação de 20±5 % de 1 nM de ligando radioativo (ver pontos 12-13 do apêndice 2). A solução de hrERIα deve ser preparada imediatamente antes de ser utilizada.
|
[3H]17β-estradiol
|
32.
|
A concentração de [3H]17β-estradiol nos poços deve ser de 1,0 nM.
|
Produtos químicos em estudo
|
33.
|
Em primeiro lugar, é necessário realizar um ensaio para determinar o limite de solubilidade de cada produto químico em estudo e identificar a gama de concentrações a utilizar na condução do protocolo de ensaio. O limite de solubilidade de cada produto químico em estudo no solvente deve ser objeto de determinação preliminar e confirmado em condições de ensaio. A concentração final estudada no ensaio não deve exceder 1 mM. O ensaio de determinação da gama inclui um controlo do solvente, juntamente com, pelo menos, 8 diluições sequenciais, com início na concentração máxima aceitável (por exemplo: 1 mM ou menos, com base no limite de solubilidade) e a presença de ligeira turbidez ou precipitado (ver também o ponto 35). O produto químico em estudo deve ser ensaiado utilizando curvas de intervalos de concentrações de 8 log, tal como definido no ensaio de determinação de gama anterior. As concentrações na segunda e na terceira experiência devem ser ajustadas na medida do necessário para caracterizar melhor a curva concentração-resposta.
|
|
34.
|
As diluições do produto químico em estudo devem ser preparadas no solvente adequado (ver ponto 26 do apêndice 2). Se a concentração mais elevada do produto químico em estudo não for solúvel nem em etanol nem em DMSO e a introdução de um novo solvente implicar uma concentração de solvente no tubo final superior ao limite aceitável, a concentração mais elevada pode ser reduzida para a concentração imediatamente inferior. Neste caso, pode acrescentar-se uma concentração adicional na extremidade inferior da série de concentrações. As outras concentrações das séries devem permanecer inalteradas.
|
|
35.
|
As soluções do produto químico em estudo devem ser monitorizadas de perto quando adicionadas ao poço de ensaio, uma vez que o produto químico em estudo precipitar para fora do poço. Os dados relativos a todos os poços que contenham precipitado não devem ser tidos em conta e os respetivos motivos de exclusão devem ser anotados.
|
|
36.
|
Se houver informação prévia, de outras fontes, que forneça o log(IC50) de um produto químico em estudo, pode ser conveniente espaçar as diluições de forma geométrica, mais perto do log(IC50) esperado, ou seja, 0,5 unidades logarítmicas em torno do log(IC50) esperado. O resultado final deve refletir a extensão suficiente das concentrações para os dois lados do log(IC50), incluindo o «topo» e o «base», de modo a que a curva de ligação possa ser adequadamente caracterizada.
|
Organização da placa de ensaio
|
37.
|
Devem preparar-se placas de microtitulação marcadas – tendo em conta as incubações sêxtuplas – com códigos para o controlo do solvente, a concentração mais elevada do estrogénio de referência (que serve igualmente como indicador de ligação não específica – NSB) e o controlo do tampão, tendo em mente as incubações triplas com códigos para cada uma das oito concentrações do controlo não ligante (octiltrietoxissilano), as sete concentrações mais baixas do estrogénio de referência, as oito concentrações das doses do ligante fraco e as oito concentrações de cada produto químico em estudo (TC). No quadro 4 figura um exemplo do diagrama da placa para as curvas de concentração inteiras dos estrogénios de referência e do controlo. São utilizadas placas de microtitulação adicionais para os produtos químicos em estudo, placas essas que devem incluir controlos de placa, nomeadamente 1) uma concentração elevada (deslocamento máximo) e média (aproximadamente a IC50) de cada um dos E2 e ligante fraco em triplicado; 2) controlo do solvente e ligação não específica, cada uma sextuplicada (quadro 5). O apêndice 2.3 apresenta um exemplo de folha de cálculo com um esquema de configuração das placas de microtitulação com três produtos químicos em estudo desconhecidos para o ensaio de ligação competitiva. As concentrações indicadas nos quadros 4 e 5 são as concentrações finais do ensaio. A concentração máxima para E2 deve ser 1×10-7 M; para o ligante fraco, deve ser usada a concentração mais elevada utilizada para o mesmo na placa 1. A concentração IC50 deve ser determinada pelo laboratório com base na sua base de dados históricos de controlo. Espera-se que esse valor seja semelhante ao observado nos estudos de validação (ver quadro 1).
|
Quadro 4
Configuração das placas de microtitulação do ensaio de ligação competitiva, curvas de concentração inteiras para o estrogénio e os controlos de referência (placa 1)
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
TB (apenas solvente)
|
TB (apenas solvente)
|
NSB
|
NSB
|
|
B
|
E2 (1×10-7)
|
E2 (1×10-8)
|
E2 (1×10-8,5)
|
E2 (1×10-9)
|
|
C
|
E2 (1×10-9,5)
|
E2 (1×10-10)
|
E2 (1×10-11)
|
Branco (*8)
|
|
D
|
NE (1×10-4,5)
|
NE (1×10-5)
|
NE (1×10-5,5)
|
NE (1×10-6)
|
|
E
|
NE (1×10-6,5)
|
NE (1×10-7)
|
NE (1×10-7,5)
|
NE (1×10-8,5)
|
|
F
|
OTES (1×10-3)
|
OTES (1×10-4)
|
OTES (1×10-5)
|
OTES (1×10-6)
|
|
G
|
OTES (1×10-7)
|
OTES (1×10-8)
|
OTES (1×10-9)
|
OTES (1×10-10)
|
|
H
|
Vazio (para quente) (*9)
|
Vazio (para quente) (*9)
|
Tampão de controlo
|
Tampão de controlo
|
|
Neste exemplo, o ligando fraco é o noretinodrel (NE)
|
Quadro 5
Configuração das placas de microtitulação do ensaio de ligação competitiva, curvas de concentração para os produtos químicos em estudo e controlos da placa
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
TB (apenas solvente)
|
TB (apenas solvente)
|
NSB
|
NSB
|
|
B
|
TC1 (1×10-3)
|
TC1 (1×10-4)
|
TC1 (1×10-5)
|
TC1 (1×10-6)
|
|
C
|
TC1 (1×10-7)
|
TC1 (1×10-8)
|
TC1 (1×10-9)
|
TC1 (1×10-10)
|
|
D
|
TC2 (1×10-3)
|
TC2 (1×10-4)
|
TC2 (1×10-5)
|
TC2 (1×10-6)
|
|
E
|
TC2 (1×10-7)
|
TC2 (1×10-8)
|
TC2 (1×10-9)
|
TC2 (1×10-10)
|
|
F
|
TC3 (1×10-3)
|
TC3 (1×10-4)
|
TC3 (1×10-5)
|
TC3 (1×10-6)
|
|
G
|
TC3 (1×10-7)
|
TC3 (1×10-8)
|
TC3 (1×10-9)
|
TC3 (1×10-10)
|
|
H
|
NE (IC50)
|
NE (1×10-4,5)
|
E2 (IC50)
|
E2 (1×10-7)
|
|
Neste exemplo, o ligando fraco é o noretinodrel (NE)
|
Realização do ensaio de ligação competitiva
|
38.
|
Conforme se mostra no quadro 6, devem ser adicionados aos poços 80 μl de solvente de controlo do estrogénio de referência, ligante fraco, não ligante e produtos químicos em estudo, preparados em tampão de ensaio. Em seguida, deve adicionar-se a cada poço 40 μl de uma solução de 4 nM [3H]17β-estradiol. Após uma rotação delicada durante 10 a 15 minutos, entre 2 e 15 oC, adicionam-se 40 μl da solução de hrERα. As placas de microtitulação do ensaio devem ser incubadas a uma temperatura de 2 a 8 oC, durante 16-20 horas, e colocadas num mecanismo de rotação durante o período de incubação.
|
Quadro 6
Quantidade de componentes de ensaio para o ensaio de ligação competitiva nos hrER, placas microtituladoras
|
Volume (μl)
|
Componente
|
|
80
|
17β-Estradiol não marcado, noretinodrel, OTES, produtos químicos em estudo, solvente ou solução-tampão
|
|
40
|
Solução de 4 nM [3H]17β-estradiol
|
|
40
|
Solução de hrERα com a concentração determinada
|
|
160
|
Volume total em cada poço
|
|
39.
|
Deve proceder-se à quantificação do valor da ligação do [3H]17β-estradiol ao hrERα, após a separação do [3H]17β-estradiol ligado ao hrERα do [3H]17β-estradiol livre mediante a adição de 80 μl de suspensão de DCC fria a cada poço, conforme descrito nos pontos 20 a 23, para o ensaio de ligações por saturação.
|
|
40.
|
Os poços H1-6 [identificados como «vazio (para quente)» no quadro 4] representam o dpms do [3H]17β-estradiol marcado em 40 μl. A alíquota de 40 μl deve ser diretamente depositada no fluido de cintilação, nos poços H1-H6.
|
Critérios de aceitabilidade
Ensaio de ligação por saturação
|
41.
|
A curva de ligação específica deve atingir um patamar elevado à medida em que forem sendo utilizadas concentrações crescentes de [3H]17β-estradiol, indicando saturação do hrERα com o ligando.
|
|
42.
|
A ligação específica a 1 nM de [3H]17β-estradiol deve situar-se no intervalo aceitável, entre 15 % e 25 % da radioatividade total média medida adicionada entre séries de ensaios. São aceitáveis pequenos desvios a este intervalo, mas se as séries de ensaios se situarem permanentemente fora deste intervalo de variação, ou se uma séries de ensaios estiver consideravelmente fora deste intervalo, a concentração de proteínas deve ser ajustada, repetindo-se o ensaio de saturação.
|
|
43.
|
Os dados devem produzir um diagrama linear de Scatchard.
|
|
44.
|
A ligação não específica não deve ser excessiva. O seu valor deve, normalmente, ser < 35 % da ligação total. No entanto, o rácio pode, ocasionalmente, ultrapassar este limite se for medido um dpm muito baixo para a concentração mais baixa de 17β-estradiol com marcação radioativa.
|
Ensaio de ligação competitiva
|
45.
|
O aumento das concentrações do 17β-estradiol não marcado deve deslocar o [3H]17β-estradiol do recetor de forma compatível com uma ligação competitiva num ponto.
|
|
46.
|
O valor de IC50 para o estrogénio de referência (17β-estradiol) deve ser aproximadamente igual à concentração molar de [3H]17β-estradiol mais o Kd determinado a partir do ensaio de saturação da ligação.
|
|
47.
|
Nas séries de ensaios, a ligação específica total deve estar sistematicamente dentro de um intervalo aceitável de 20±5 % se a concentração média de radioatividade total medida adicionada a cada poço for de 1 nM. São aceitáveis pequenos desvios, mas, se as séries de ensaios se situarem permanentemente fora deste intervalo de variação, ou se uma série de ensaios se situar consideravelmente fora do mesmo, a concentração de proteínas deve ser ajustada.
|
|
48.
|
O solvente não deve alterar a sensibilidade ou a reprodutibilidade do ensaio. Os resultados do controlo do solvente (poços de TB) são comparados com o controlo de tampão, a fim de verificar se o solvente utilizado não afeta o sistema de ensaio. Os resultados do TB e do controlo da solução-tampão devem ser comparáveis se não houver nenhum efeito do solvente no ensaio.
|
|
49.
|
O não ligante não deve deslocar mais de 25 % do [3H]17β-estradiol do hrERα quando ensaiado a até 10-3 M (OTES) ou 10-4 M (DBP).
|
|
50.
|
Foram estabelecidos critérios de desempenho para os estrogénios de referência e dois ligantes fracos (noretinodrel, noretindrona), com base nos dados do estudo de validação do ensaio de ligação FW hrER FW (anexo N da referência 2). Estão previstos intervalos de confiança de 95 % para DPDP (n) ±±DPDP médio em todos os controlos em todos os laboratórios que participam no estudo de validação. Foram calculados intervalos de confiança de 95 % para os parâmetros de ajustamento da curva [ou seja, topo, base, curva de Hill, log(IC50)]logIC50) para os estrogénios de referência e ligantes fracos, e para o log10(RBA)log10RBA com base nos estrogénios de referência e são fornecidos como critérios de desempenho para os controlos positivos. O quadro 1 apresenta os intervalos previstos para os parâmetros de ajustamento da curva que podem ser utilizados como critérios de desempenho. Na prática, o intervalo de variação do IC50 pode variar ligeiramente com base no Kd da preparação do recetor e na concentração do ligando.
|
|
51.
|
Não foram definidos critérios de desempenho para os parâmetros de ajustamento da curva em relação aos produtos químicos em estudo, devido à grande variedade de produtos químicos em estudo existentes e à variação das principais afinidades e dos resultados (p. ex., curva inteira, curva parcial, sem ajustamento de curva). Contudo, devem ser utilizados critérios profissionais aquando da avaliação dos resultados de cada série de ensaios para um produto químico em estudo. Deve utilizar-se uma gama de concentrações do produto suficiente para definir claramente o topo (por exemplo, 90-100 % de ligação) da curva de ligação competitiva. A variabilidade dos replicados para cada concentração do produto químico em estudo, bem como entre as 3 séries não paralelas, deve ser razoável e cientificamente aceitável. Os controlos de cada série de ensaios em relação a um produto químico em estudo devem aproximar-se das medições de desempenho comunicadas para este ensaio do FW e ser coerentes com os dados históricos de controlo de cada laboratório.
|
ANÁLISE DOS DADOS
Ensaio de ligação por saturação
|
52.
|
São medidas as ligações totais e as ligações não específicas. A partir destes valores, a ligação específica de concentrações crescentes de [3H]17β-estradiol em condições de equilíbrio é calculada subtraindo-se a ligação não específica do total. Um gráfico que apresente a ligação específica versus a concentração de [3H]17β-estradiol deve atingir um plano para o indicativo máximo específico da ligação por saturação do hrERα [3H-17H]β-estradiol. Além disso, a análise dos dados deve documentar a ligação do [3H]17β-estradiol a um único ponto de ligação de alta afinidade. As ligações não específicas, totais e específicas devem ser visíveis numa curva de ligação por saturação. Uma análise mais aprofundada destes dados deve utilizar uma análise de regressão não linear (ex., BioSoft; McPherson, 1985; Motulsky, 1995) com uma apresentação final dos dados de acordo com Scatchard.
|
|
53.
|
A análise de dados deve determinar a Bmáx e Kd a partir, unicamente dos dados relativos à ligação total, partindo do pressuposto de que a ligação não específica é de linear, salvo se for justificada a utilização de outro método. Ao determinar a melhor adequação, importa efetuar uma regressão sólida, salvo justificação. Deve indicar-se o método escolhido para uma regressão sólida. A correção da depleção de ligando (por exemplo, utilizando o método de Swillens 1995) deve ser sempre utilizada na determinação de Bmáx e Kd a partir dos dados da ligação por saturação.
|
Ensaio de ligação competitiva
|
54.
|
A curva de ligação competitiva é representada como a ligação específica do [3H]17β-estradiol versus a concentração (log 10 unidades) do concorrente. A concentração do produto químico em estudo que inibe 50 % da ligação máxima específica do [3H]17β-estradiol é IC50.
|
|
55.
|
As estimativas de log(IC50) para os controlos positivos (por exemplo, estrogénios de referência e ligantes fracos) devem ser determinadas com recurso a um software de ajustamento não linear de ajustamento adequado, a fim de se adaptar à equação de quatro parâmetros de Hill (p. ex., BioSoft; McPherson, 1985; Motulsky, 1995). Em geral, o topo, a base, a curva e o log(IC50) devem ser mantidos inalterados quando se ajustam as curvas. Quando se determinar a melhor adequação, deve utilizar-se uma regressão sólida, a menos que seja apresentada uma justificação. Não devem ser feitas correções para a depleção do ligando. Após a análise inicial, cada curva de ligação deve ser avaliada de modo a garantir a devida adequação ao modelo. A afinidade de ligação relativa (RBA) do ligando fraco deve ser calculada como percentagem do log(IC50) para o ligante fraco em relação ao log(IC50) para o 17β-estradiol. Os resultados dos controlos positivos e do controlo não ligante devem ser avaliados de acordo com as medidas do desempenho do ensaio constantes dos pontos 45 a 50 do presente apêndice 2.
|
|
56.
|
Os dados relativos a todos os produtos químicos em estudo devem ser analisados através de uma abordagem por etapas, para garantir que os dados são devidamente analisados e que cada ligação competitiva é devidamente classificada. Recomenda-se que cada série de ensaios para um produto químico em estudo seja inicialmente submetida a uma análise normalizada de dados idêntica à utilizada para os estrogénios de referência e para controlos de ligantes fracos (ponto 55). Uma vez concluída, deve ser efetuada uma avaliação técnica dos parâmetros de ajustamento da curva, bem como uma análise visual do grau de adequação dos dados à curva de ligação competitiva gerada para cada série de ensaios. Durante esta avaliação técnica, as observações de uma diminuição dependente da concentração do [3H]17β-estradiol especificamente ligado, da baixa variabilidade entre os replicados técnicos em cada concentração do produto químico em estudo e da coerência dos parâmetros de ajustamento entre as três séries de ensaios constituem uma boa indicação de que a análise de ensaios e de dados foi realizada de forma adequada.
|
Interpretação dos dados
|
57.
|
Caso se cumpram todos os critérios de aceitabilidade, o produto químico em estudo é considerado um ligante para o hrERα se for possível ajustar uma curva ligação e o ponto mais baixo da curva de resposta dentro da gama de dados for inferior a 50 % (figura 1).
|
|
58.
|
O produto químico em estudo é considerado como não ligante se, cumpridos todos os critérios de aceitabilidade:
|
—
|
for possível ajustar uma curva de ligação e o ponto mais baixo da curva de resposta dentro da gama de dados for superior a 75 %, ou
|
|
—
|
não for possível ajustar uma curva de ligação e a média percentual mais baixa não ajustada entre os grupos de concentração dos dados for superior a 75 %.
|
|
|
59.
|
Os produtos químicos em estudo são considerados equívocos se nenhuma das condições acima referidas estiver preenchida (por exemplo, o ponto mais baixo da curva de resposta ajustada situa-se entre 76 e 51 %).
|
Quadro 7
Critérios para classificação com base numa curva de ligação competitiva para um produto químico em estudo
|
Classificação
|
Critérios
|
|
Ligantea
|
Uma curva de ligação pode ser ajustada.
O ponto mais baixo na curva de resposta dentro da gama de dados é inferior a 50 %.
|
|
Não liganteb
|
Se for possível ajustar a curva de ligação,
o ponto mais baixo da curva de resposta dentro da gama de dados é superior a 75 %.
Se não for possível ajustar a curva de ligação,
a média percentual mais baixa não ajustada entre os grupos de concentração nos dados é superior a 75 %.
|
|
Inconclusivoc
|
Qualquer produto químico em estudo que não seja ligante nem não ligante.
(Por exemplo, o ponto mais baixo da curva de resposta ajustada situa-se entre 76 e 51 %).
|
Figura 1
Exemplos de classificação de produtos químicos em estudo por meio de uma curva de ligação competitiva
|
60.
|
As várias séries de ensaios realizados num laboratório para um produto químico em estudo são combinadas através da atribuição de valores numéricos a cada série de ensaios e fazendo a média das diferentes séries, como se indica no quadro 8. Os resultados para as séries combinadas em cada laboratório são comparados com a classificação prevista para cada produto químico em estudo.
|
Quadro 8
Método de classificação do produto químico em estudo utilizando múltiplas séries de ensaios
|
Para atribuir valor a cada série de ensaios
|
|
|
Classificação
|
Valor numérico
|
|
Ligante
|
2
|
|
Inconclusivo
|
1
|
|
Não ligante
|
0
|
|
Para classificar a média dos valores numéricos das diferentes séries de ensaios
|
|
|
Classificação
|
Valor numérico
|
|
Ligante
|
Média ≥ 1,5
|
|
Inconclusivo
|
0,5 ≤ média < 1,5
|
|
Não ligante
|
Média < 0,5
|
RELATÓRIO DO ENSAIO
|
61.
|
Ver ponto 24 da secção «COMPONENTES DO ENSAIO DE LIGAÇÃO DO hrER»
|
Apêndice 2.1
LISTA DE TERMOS
DCC: carvão revestido com dextrose.
[3H]E2: 17β-estradiol com marcação radioativa por trítio.
E2: 17β-estradiol não marcado (inerte).
hrERα: recetor alfa de estrogénio recombinante humano.
Replicado: um ou vários poços que contêm o mesmo conteúdo com as mesmas concentrações e que são objeto de ensaios paralelos numa única série de ensaios. Neste protocolo, cada concentração do produto químico em estudo é ensaiada em triplicado; isto é, existem três replicados que são ensaiados simultaneamente em cada concentração do produtos químicos em estudo.
Série de ensaios: conjunto completo de poços de placas de microtitulação que fornece todas as informações necessárias para caracterizar a ligação de um produto químico em estudo ao hrERα (viz., total [3H]17β-estradiol adicionado ao poço, ligação máxima do [3H]17β-estradiol ao hrERα ligação não específica e ligação total em diferentes concentrações do produto químico em estudo). Uma série de ensaio pode consistir em apenas um poço bem (ou seja, replicado) por concentração; contudo, dado que este protocolo requer que os ensaios sejam realizados em triplicado, uma série de ensaios consiste em três poços por concentração. Além disso, este protocolo requer três séries de ensaio independentes (ou seja, não paralelas) por produto químico.
Tampão de ensaio: 10 mM Tris, 10 mg de albumina de soro bovino/ml, 2 mM TDT, 10 % de glicerol, 0,2 mM leupeptina, pH 7,5.
Apêndice 2.2
ENSAIO DE SATURAÇÃO TÍPICA DO [3H]17Β-ESTRADIOL COM TRÊS POÇOS REPLICADOS
|
Ensaio de saturação típica do [3H]17β-estradiol com três poços replicados
|
|
Posição
|
Replicado
|
Código do tipo do poço
|
Concentração inicial a quente de E2 (nM)
|
Volume a quente do E2 (μl)
|
Concentração final a quente do E2 (nM)
|
Concentração inicial a frio do E2 (μM)
|
Volume a frio do E2 (μl)
|
Concentração final a frio do E2 (μM)
|
Volume do tampão (μl)
|
Volume do recetor (μl)
|
Volume total em poços
|
|
A1
|
1
|
H
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A2
|
2
|
H
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A3
|
3
|
H
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A4
|
1
|
H
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A5
|
2
|
H
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A6
|
3
|
H
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A7
|
1
|
H
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A8
|
2
|
H
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A9
|
3
|
H
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A10
|
1
|
H
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A11
|
2
|
H
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A12
|
3
|
H
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B1
|
1
|
H
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B2
|
2
|
H
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B3
|
3
|
H
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B4
|
1
|
H
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B5
|
2
|
H
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B6
|
3
|
H
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B7
|
1
|
H
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B8
|
2
|
H
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B9
|
3
|
H
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B10
|
1
|
H
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B11
|
2
|
H
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B12
|
3
|
H
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
D1
|
1
|
HC
|
0,12
|
40
|
0,03
|
0,06
|
80
|
0,03
|
—
|
40
|
160
|
|
D2
|
2
|
HC
|
0,12
|
40
|
0,03
|
0,06
|
80
|
0,03
|
—
|
40
|
160
|
|
D3
|
3
|
HC
|
0,12
|
40
|
0,03
|
0,06
|
80
|
0,03
|
—
|
40
|
160
|
|
D4
|
1
|
HC
|
0,24
|
40
|
0,06
|
0,12
|
80
|
0,06
|
—
|
40
|
160
|
|
D5
|
2
|
HC
|
0,24
|
40
|
0,06
|
0,12
|
80
|
0,06
|
—
|
40
|
160
|
|
D6
|
3
|
HC
|
0,24
|
40
|
0,06
|
0,12
|
80
|
0,06
|
—
|
40
|
160
|
|
D7
|
1
|
HC
|
0,32
|
40
|
0,08
|
0,16
|
80
|
0,08
|
—
|
40
|
160
|
|
D8
|
2
|
HC
|
0,32
|
40
|
0,08
|
0,16
|
80
|
0,08
|
—
|
40
|
160
|
|
D9
|
3
|
HC
|
0,32
|
40
|
0,08
|
0,16
|
80
|
0,08
|
—
|
40
|
160
|
|
D10
|
1
|
HC
|
0,40
|
40
|
0,10
|
0,2
|
80
|
0,1
|
—
|
40
|
160
|
|
D11
|
2
|
HC
|
0,40
|
40
|
0,10
|
0,2
|
80
|
0,1
|
—
|
40
|
160
|
|
D12
|
3
|
HC
|
0,40
|
40
|
0,10
|
0,2
|
80
|
0,1
|
—
|
40
|
160
|
|
E1
|
1
|
HC
|
1,20
|
40
|
0,30
|
0,6
|
80
|
0,3
|
—
|
40
|
160
|
|
E2
|
2
|
HC
|
1,20
|
40
|
0,30
|
0,6
|
80
|
0,3
|
—
|
40
|
160
|
|
E3
|
3
|
HC
|
1,20
|
40
|
0,30
|
0,6
|
80
|
0,3
|
—
|
40
|
160
|
|
E4
|
1
|
HC
|
2,40
|
40
|
0,60
|
1,2
|
80
|
0,6
|
—
|
40
|
160
|
|
E5
|
2
|
HC
|
2,40
|
40
|
0,60
|
1,2
|
80
|
0,6
|
—
|
40
|
160
|
|
E6
|
3
|
HC
|
2,40
|
40
|
0,60
|
1,2
|
80
|
0,6
|
—
|
40
|
160
|
|
E7
|
1
|
HC
|
4,00
|
40
|
1,00
|
2
|
80
|
1
|
—
|
40
|
160
|
|
E8
|
2
|
HC
|
4,00
|
40
|
1,00
|
2
|
80
|
1
|
—
|
40
|
160
|
|
E9
|
3
|
HC
|
4,00
|
40
|
1,00
|
2
|
80
|
1
|
—
|
40
|
160
|
|
E10
|
1
|
HC
|
12,00
|
40
|
3,00
|
6
|
80
|
3
|
—
|
40
|
160
|
|
E11
|
2
|
HC
|
12,00
|
40
|
3,00
|
6
|
80
|
3
|
—
|
40
|
160
|
|
E12
|
3
|
HC
|
12,00
|
40
|
3,00
|
6
|
80
|
3
|
—
|
40
|
160
|
|
G1
|
1
|
Quente
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G2
|
2
|
Quente
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G3
|
3
|
Quente
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G4
|
1
|
Quente
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G5
|
2
|
Quente
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G6
|
3
|
Quente
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G7
|
1
|
Quente
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G8
|
2
|
Quente
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G9
|
3
|
Quente
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G10
|
1
|
Quente
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G11
|
2
|
Quente
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G12
|
3
|
Quente
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H1
|
1
|
Quente
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H2
|
2
|
Quente
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H3
|
3
|
Quente
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H4
|
1
|
Quente
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H5
|
2
|
Quente
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H6
|
3
|
Quente
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H7
|
1
|
Quente
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H8
|
2
|
Quente
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H9
|
3
|
Quente
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H10
|
1
|
Quente
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H11
|
2
|
Quente
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H12
|
3
|
Quente
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
Note-se que os poços «quentes» estão vazios durante a incubação. Os 40 μl são adicionados apenas para contagem de cintilação.
|
Apêndice 2.3
CONFIGURAÇÃO DO ENSAIO DE LIGAÇÃO COMPETITIVA
|
Placa
|
Posição
|
Replicado
|
Tipo de poço
|
Código do poço
|
Código de concentração
|
Concentração inicial do concorrente (M)
|
Solução de reserva de hrER (μl)
|
Volume do tampão (μl)
|
Volume (μl) do traçador (E2 quente)
|
Volume a partir da placa de diluição (μl)
|
Volume final (μl)
|
Concentração final concorrente (M)
|
|
S
|
A1
|
1
|
Ligação total
|
TB
|
TB1
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A2
|
2
|
Ligação total
|
TB
|
TB2
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A3
|
3
|
Ligação total
|
TB
|
TB3
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A4
|
1
|
Ligação total
|
TB
|
TB4
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A5
|
2
|
Ligação total
|
TB
|
TB5
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A6
|
3
|
Ligação total
|
TB
|
TB6
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A7
|
1
|
E2 frio (elevado)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
A8
|
2
|
E2 frio (elevado)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
A9
|
3
|
E2 frio (elevado)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
A10
|
1
|
E2 frio (elevado)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
A11
|
2
|
E2 frio (elevado)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
A12
|
3
|
E2 frio (elevado)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
B1
|
1
|
E2 frio
|
S
|
S1
|
2,00E-07
|
40
|
—
|
40
|
80
|
160
|
1,0E-07
|
|
S
|
B2
|
2
|
E2 frio
|
S
|
S1
|
2,00E-07
|
40
|
—
|
40
|
80
|
160
|
1,0E-07
|
|
S
|
B3
|
3
|
E2 frio
|
S
|
S1
|
2,00E-07
|
40
|
—
|
40
|
80
|
160
|
1,0E-07
|
|
S
|
B4
|
1
|
E2 frio
|
S
|
S2
|
2,00E-08
|
40
|
—
|
40
|
80
|
160
|
1,0E-08
|
|
S
|
B5
|
2
|
E2 frio
|
S
|
S2
|
2,00E-08
|
40
|
—
|
40
|
80
|
160
|
1,0E-08
|
|
S
|
B6
|
3
|
E2 frio
|
S
|
S2
|
2,00E-08
|
40
|
—
|
40
|
80
|
160
|
1,0E-08
|
|
S
|
B7
|
1
|
E2 frio
|
S
|
S3
|
6,00E-09
|
40
|
—
|
40
|
80
|
160
|
3,0E-09
|
|
S
|
B8
|
2
|
E2 frio
|
S
|
S3
|
6,00E-09
|
40
|
—
|
40
|
80
|
160
|
3,0E-09
|
|
S
|
B9
|
3
|
E2 frio
|
S
|
S3
|
6,00E-09
|
40
|
—
|
40
|
80
|
160
|
3,0E-09
|
|
S
|
B10
|
1
|
E2 frio
|
S
|
S4
|
2,00E-09
|
40
|
—
|
40
|
80
|
160
|
1,0E-09
|
|
S
|
B11
|
2
|
E2 frio
|
S
|
S4
|
2,00E-09
|
40
|
—
|
40
|
80
|
160
|
1,0E-09
|
|
S
|
B12
|
3
|
E2 frio
|
S
|
S4
|
2,00E-09
|
40
|
—
|
40
|
80
|
160
|
1,0E-09
|
|
S
|
C1
|
1
|
E2 frio
|
S
|
S5
|
6,00E-10
|
40
|
—
|
40
|
80
|
160
|
3,0E-10
|
|
S
|
C2
|
2
|
E2 frio
|
S
|
S5
|
6,00E-10
|
40
|
—
|
40
|
80
|
160
|
3,0E-10
|
|
S
|
C3
|
3
|
E2 frio
|
S
|
S5
|
6,00E-10
|
40
|
—
|
40
|
80
|
160
|
3,0E-10
|
|
S
|
C4
|
1
|
E2 frio
|
S
|
S6
|
2,00E-10
|
40
|
—
|
40
|
80
|
160
|
1,0E-10
|
|
S
|
C5
|
2
|
E2 frio
|
S
|
S6
|
2,00E-10
|
40
|
—
|
40
|
80
|
160
|
1,0E-10
|
|
S
|
C6
|
3
|
E2 frio
|
S
|
S6
|
2,00E-10
|
40
|
—
|
40
|
80
|
160
|
1,0E-10
|
|
S
|
C7
|
1
|
E2 frio
|
S
|
S7
|
2,00E-11
|
40
|
—
|
40
|
80
|
160
|
1,0E-11
|
|
S
|
C8
|
2
|
E2 frio
|
S
|
S7
|
2,00E-11
|
40
|
—
|
40
|
80
|
160
|
1,0E-11
|
|
S
|
C9
|
3
|
E2 frio
|
S
|
S7
|
2,00E-11
|
40
|
—
|
40
|
80
|
160
|
1,0E-11
|
|
S
|
C10
|
1
|
Vazio
|
Vazio
|
B1
|
—
|
—
|
160
|
—
|
—
|
160
|
—
|
|
S
|
C11
|
2
|
Vazio
|
Vazio
|
B2
|
—
|
—
|
160
|
—
|
—
|
160
|
—
|
|
S
|
C12
|
3
|
Vazio
|
Vazio
|
B3
|
—
|
—
|
160
|
—
|
—
|
160
|
—
|
|
S
|
D1
|
1
|
Noretinodrel
|
NE
|
WP1
|
6,00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0E-05
|
|
S
|
D2
|
1
|
Noretinodrel
|
NE
|
WP1
|
6,00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0E-05
|
|
S
|
D3
|
1
|
Noretinodrel
|
NE
|
WP1
|
6,00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0E-05
|
|
S
|
D4
|
1
|
Noretinodrel
|
NE
|
WP2
|
2,00E-05
|
40
|
—
|
40
|
80
|
160
|
1,0E-05
|
|
S
|
D5
|
1
|
Noretinodrel
|
NE
|
WP2
|
2,00E-05
|
40
|
—
|
40
|
80
|
160
|
1,0E-05
|
|
S
|
D6
|
1
|
Noretinodrel
|
NE
|
WP2
|
2,00E-05
|
40
|
—
|
40
|
80
|
160
|
1,0E-05
|
|
S
|
D7
|
1
|
Noretinodrel
|
NE
|
WP3
|
6,00E-06
|
40
|
—
|
40
|
80
|
160
|
3,0E-06
|
|
S
|
D8
|
1
|
Noretinodrel
|
NE
|
WP3
|
6,00E-06
|
40
|
—
|
40
|
80
|
160
|
3,0E-06
|
|
S
|
D9
|
1
|
Noretinodrel
|
NE
|
WP3
|
6,00E-06
|
40
|
—
|
40
|
80
|
160
|
3,0E-06
|
|
S
|
D10
|
1
|
Noretinodrel
|
NE
|
WP4
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
D11
|
1
|
Noretinodrel
|
NE
|
WP4
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
D12
|
1
|
Noretinodrel
|
NE
|
WP4
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
E1
|
1
|
Noretinodrel
|
NE
|
WP
|
6,00E-07-
|
40
|
|
40
|
80
|
160
|
3,0E-07
|
|
S
|
E2
|
2
|
Noretinodrel
|
NE
|
WP
|
6,00E-07-
|
40
|
|
40
|
80
|
160
|
3,0E-07
|
|
S
|
E3
|
3
|
Noretinodrel
|
NE
|
WP
|
6,00E-07-
|
40
|
|
40
|
80
|
160
|
3,0E-07
|
|
S
|
E4
|
1
|
Noretinodrel
|
NE
|
WP
|
2,00E-07-
|
40
|
|
40
|
80
|
160
|
1,0E-07
|
|
S
|
E5
|
2
|
Noretinodrel
|
NE
|
WP
|
2,00E-07-
|
40
|
|
40
|
80
|
160
|
1,0E-07
|
|
S
|
E6
|
3
|
Noretinodrel
|
NE
|
WP
|
2,00E-07-
|
40
|
|
40
|
80
|
160
|
1,0E-07
|
|
S
|
E7
|
1
|
Noretinodrel
|
NE
|
WP
|
6,00E-08-
|
40
|
—
|
40
|
80
|
160
|
3,0E-08
|
|
S
|
E8
|
2
|
Noretinodrel
|
NE
|
WP
|
6,00E-08-
|
40
|
—
|
40
|
80
|
160
|
3,0E-08
|
|
S
|
E9
|
3
|
Noretinodrel
|
NE
|
WP
|
6,00E-08-
|
40
|
—
|
40
|
80
|
160
|
3,0E-08
|
|
S
|
E10
|
1
|
Noretinodrel
|
NE
|
WP
|
6,00E-09-
|
40
|
—
|
40
|
80
|
160
|
3,0E-09
|
|
S
|
E11
|
2
|
Noretinodrel
|
NE
|
WP
|
6,00E-09-
|
40
|
—
|
40
|
80
|
160
|
3,0E-09
|
|
S
|
E12
|
3
|
Noretinodrel
|
NE
|
WP
|
6,00E-09-
|
40
|
—
|
40
|
80
|
160
|
3,0E-09
|
|
S
|
F1
|
1
|
OTES
|
N
|
OTES
|
2,00E-03
|
40
|
—
|
40
|
80
|
160
|
1,0E-03
|
|
S
|
F2
|
2
|
OTES
|
N
|
OTES
|
2,00E-03
|
40
|
—
|
40
|
80
|
160
|
1,0E-03
|
|
S
|
F3
|
3
|
OTES
|
N
|
OTES
|
2,00E-03
|
40
|
—
|
40
|
80
|
160
|
1,0E-03
|
|
S
|
F4
|
1
|
OTES
|
N
|
OTES
|
2,00E-04
|
40
|
—
|
40
|
80
|
160
|
1,0E-04
|
|
S
|
F5
|
2
|
OTES
|
N
|
OTES
|
2,00E-04
|
40
|
—
|
40
|
80
|
160
|
1,0E-04
|
|
S
|
F6
|
3
|
OTES
|
N
|
OTES
|
2,00E-04
|
40
|
—
|
40
|
80
|
160
|
1,0E-04
|
|
S
|
F7
|
1
|
OTES
|
N
|
OTES
|
2,00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0E-05
|
|
S
|
F8
|
2
|
OTES
|
N
|
OTES
|
2,00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0E-05
|
|
S
|
F9
|
3
|
OTES
|
N
|
OTES
|
2,00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0E-05
|
|
S
|
F10
|
1
|
OTES
|
N
|
OTES
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
F11
|
2
|
OTES
|
N
|
OTES
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
F12
|
3
|
OTES
|
N
|
OTES
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
G1
|
1
|
OTES
|
N
|
OTES
|
2,00E-07
|
40
|
—
|
40
|
80
|
160
|
3,0E-07
|
|
S
|
G2
|
2
|
OTES
|
N
|
OTES
|
2,00E-07
|
40
|
—
|
40
|
80
|
160
|
3,0E-07
|
|
S
|
G3
|
3
|
OTES
|
N
|
OTES
|
2,00E-07
|
40
|
—
|
40
|
80
|
160
|
3,0E-07
|
|
S
|
G4
|
1
|
OTES
|
N
|
OTES
|
2,00E-08
|
40
|
—
|
40
|
80
|
160
|
1,0E-08
|
|
S
|
G5
|
2
|
OTES
|
N
|
OTES
|
2,00E-08
|
40
|
—
|
40
|
80
|
160
|
1,0E-08
|
|
S
|
G6
|
3
|
OTES
|
N
|
OTES
|
2,00E-08
|
40
|
—
|
40
|
80
|
160
|
1,0E-08
|
|
S
|
G7
|
1
|
OTES
|
N
|
OTES
|
2,00E-09
|
40
|
—
|
40
|
80
|
160
|
1,0E-09
|
|
S
|
G8
|
2
|
OTES
|
N
|
OTES
|
2,00E-09
|
40
|
—
|
40
|
80
|
160
|
1,0E-09
|
|
S
|
G9
|
3
|
OTES
|
N
|
OTES
|
2,00E-09
|
40
|
—
|
40
|
80
|
160
|
1,0E-09
|
|
S
|
G10
|
1
|
OTES
|
N
|
OTES
|
2,00E-10
|
40
|
—
|
40
|
—
|
160
|
1,0E-10
|
|
S
|
G11
|
2
|
OTES
|
N
|
OTES
|
2,00E-10
|
40
|
—
|
40
|
—
|
160
|
1,0E-10
|
|
S
|
G12
|
3
|
OTES
|
N
|
OTES
|
2,00E-10
|
40
|
—
|
40
|
—
|
160
|
1,0E-10
|
|
S
|
H1
|
1
|
Quente
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H2
|
1
|
Quente
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H3
|
1
|
Quente
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H4
|
1
|
Quente
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H5
|
1
|
Quente
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H6
|
1
|
Quente
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H7
|
1
|
Tampão de controlo
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H8
|
1
|
Tampão de controlo
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H9
|
1
|
Tampão de controlo
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H10
|
1
|
Tampão de controlo
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H11
|
1
|
Tampão de controlo
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H12
|
1
|
Tampão de controlo
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
Note-se que os poços «quentes» estão vazios durante a incubação. Os 40 μl são adicionados apenas para contagem de cintilação.
|
Configuração do ensaio de ligação competitiva
|
|
Placa
|
Posição
|
Replicado
|
Tipo de poço
|
Código do poço
|
Código de concentração
|
Concentração inicial do concorrente (M)
|
Solução de reserva de hrER (μl)
|
Volume de tampões (μl)
|
Volume (μl) do traçador (E2 quente)
|
Volume da placa de diluição (μl)
|
Volume final (μl)
|
Concentração final do concorrente (M)
|
|
P1
|
A1
|
1
|
Ligação total
|
TB
|
TBB1B1
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A2
|
2
|
Ligação total
|
TB
|
TB2
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A3
|
3
|
Ligação total
|
TB
|
TB3
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A4
|
1
|
Ligação total
|
TB
|
TB4
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A5
|
2
|
Ligação total
|
TB
|
TB5
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A6
|
3
|
Ligação total
|
TB
|
TB6
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A7
|
1
|
E2 frio (elevado)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
A8
|
2
|
E2 frio (elevado)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
A9
|
3
|
E2 frio (elevado)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
A10
|
1
|
E2 frio (elevado)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
A11
|
2
|
E2 frio (elevado)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
A12
|
3
|
E2 frio (elevado)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
B1
|
1
|
Produto químico em estudo 1
|
TC1
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
B2
|
2
|
Produto químico em estudo 1
|
TC1
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
B3
|
3
|
Produto químico em estudo 1
|
TC1
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
B4
|
1
|
Produto químico em estudo 1
|
TC1
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
B5
|
2
|
Produto químico em estudo 1
|
TC1
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
B6
|
3
|
Produto químico em estudo 1
|
TC1
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
B7
|
1
|
Produto químico em estudo 1
|
TC1
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
B8
|
2
|
Produto químico em estudo 1
|
TC1
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
B9
|
3
|
Produto químico em estudo 1
|
TC1
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
B10
|
1
|
Produto químico em estudo 1
|
TC1
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
B11
|
2
|
Produto químico em estudo 1
|
TC1
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
B12
|
3
|
Produto químico em estudo 1
|
TC1
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
C1
|
1
|
Produto químico em estudo 1
|
TC1
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
C2
|
2
|
Produto químico em estudo 1
|
TC1
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
C3
|
3
|
Produto químico em estudo 1
|
TC1
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
C4
|
1
|
Produto químico em estudo 1
|
TC1
|
6
|
2,00E-08
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
C5
|
2
|
Produto químico em estudo 1
|
TC1
|
6
|
2,00E-08
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
C6
|
3
|
Produto químico em estudo 1
|
TC1
|
6
|
2,00E-08
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
C7
|
1
|
Produto químico em estudo 1
|
TC1
|
7
|
2,00E-09
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
C8
|
2
|
Produto químico em estudo 1
|
TC1
|
7
|
2,00E-09
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
C9
|
3
|
Produto químico em estudo 1
|
TC1
|
7
|
2,00E-09
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
C10
|
1
|
Produto químico em estudo 1
|
TC1
|
8
|
2,00E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
C11
|
2
|
Produto químico em estudo 1
|
TC1
|
8
|
2,00E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
C12
|
3
|
Produto químico em estudo 1
|
TC1
|
8
|
2,00E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
D1
|
1
|
Produto químico em estudo 2
|
TC2
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
D2
|
2
|
Produto químico em estudo 2
|
TC2
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
D3
|
3
|
Produto químico em estudo 2
|
TC2
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
D4
|
1
|
Produto químico em estudo 2
|
TC2
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
D5
|
2
|
Produto químico em estudo 2
|
TC2
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
D6
|
3
|
Produto químico em estudo 2
|
TC2
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
D7
|
1
|
Produto químico em estudo 2
|
TC2
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
D8
|
2
|
Produto químico em estudo 2
|
TC2
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
D9
|
3
|
Produto químico em estudo 2
|
TC2
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
D10
|
1
|
Produto químico em estudo 2
|
TC2
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
D11
|
2
|
Produto químico em estudo 2
|
TC2
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
D12
|
3
|
Produto químico em estudo 2
|
TC2
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
E1
|
1
|
Produto químico em estudo 2
|
TC2
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
E2
|
2
|
Produto químico em estudo 2
|
TC2
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
E3
|
3
|
Produto químico em estudo 2
|
TC2
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
E4
|
1
|
Produto químico em estudo 2
|
TC2
|
6
|
—
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
E5
|
2
|
Produto químico em estudo 2
|
TC2
|
6
|
—
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
E6
|
3
|
Produto químico em estudo 2
|
TC2
|
6
|
—
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
E7
|
1
|
Produto químico em estudo 2
|
TC2
|
7
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
E8
|
2
|
Produto químico em estudo 2
|
TC2
|
7
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
E9
|
3
|
Produto químico em estudo 2
|
TC2
|
7
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
E10
|
1
|
Produto químico em estudo 2
|
TC2
|
8
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
E11
|
2
|
Produto químico em estudo 2
|
TC2
|
8
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
E12
|
3
|
Produto químico em estudo 2
|
TC2
|
8
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
F1
|
1
|
Produto químico em estudo 3
|
TC3
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
F2
|
2
|
Produto químico em estudo 3
|
TC3
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
F3
|
3
|
Produto químico em estudo 3
|
TC3
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
F4
|
1
|
Produto químico em estudo 3
|
TC3
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
F5
|
2
|
Produto químico em estudo 3
|
TC3
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
F6
|
3
|
Produto químico em estudo 3
|
TC3
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
F7
|
1
|
Produto químico em estudo 3
|
TC3
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
F8
|
2
|
Produto químico em estudo 3
|
TC3
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
F9
|
3
|
Produto químico em estudo 3
|
TC3
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
F10
|
1
|
Produto químico em estudo 3
|
TC3
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
F11
|
2
|
Produto químico em estudo 3
|
TC3
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
F12
|
3
|
Produto químico em estudo 3
|
TC3
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
G1
|
1
|
Produto químico em estudo 3
|
TC3
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
G2
|
2
|
Produto químico em estudo 3
|
TC3
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
G3
|
3
|
Produto químico em estudo 3
|
TC3
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
G4
|
1
|
Produto químico em estudo 3
|
TC3
|
6
|
2,00E-08
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
G5
|
2
|
Produto químico em estudo 3
|
TC3
|
6
|
2,00E-08
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
G6
|
3
|
Produto químico em estudo 3
|
TC3
|
6
|
2,00E-08
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
G7
|
1
|
Produto químico em estudo 3
|
TC3
|
7
|
2,00E-09
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
G8
|
2
|
Produto químico em estudo 3
|
TC3
|
7
|
2,00E-09
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
G9
|
3
|
Produto químico em estudo 3
|
TC3
|
7
|
2,00E-09
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
G10
|
1
|
Produto químico em estudo 3
|
TC3
|
8
|
2,00E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
G11
|
2
|
Produto químico em estudo 3
|
TC3
|
8
|
2,00E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
G12
|
3
|
Produto químico em estudo 3
|
TC3
|
8
|
2,00E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
H1
|
1
|
Noretinodrel
|
NE
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H2
|
2
|
Noretinodrel
|
NE
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H3
|
3
|
Noretinodrel
|
NE
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H4
|
1
|
Noretinodrel
|
NE
|
|
1,00E-4,5
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H5
|
2
|
Noretinodrel
|
NE
|
|
1,00E-4,5
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H6
|
3
|
Noretinodrel
|
NE
|
|
1,00E-4,5
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H7
|
1
|
E2 S frio
|
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H8
|
2
|
E2 S frio
|
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H9
|
3
|
E2 S frio
|
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H10
|
1
|
E2 S frio
|
|
|
1,00 E-7
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H11
|
2
|
E2 S frio
|
|
|
1,00 E-7
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H12
|
3
|
E2 S frio
|
|
|
1,00 E-7
|
40
|
0
|
40
|
80
|
160
|
|
Apêndice 3
ENSAIO IN VITRO DO LIGAÇÃO AO RECETOR DO ESTROGÉNIO COM RECURSO A UMA PROTEÍNA DE DOMÍNIO DE LIGAÇÃO DO LIGANDO ERα RECOMBINANTE HUMANA DO CHEMICAL EVALUATION AND RESEARCH INSTITUTE (CERI)
CONSIDERAÇÕES INICIAIS E LIMITAÇÕES (VER TAMBÉM INTRODUÇÃO GERAL)
|
1.
|
Este ensaio in vitro de ligação por saturação e competitiva ao recetor estrogénico utiliza um domínio de ligação do ligando (LBD) do ERα humano (hrERα). Este modelo de proteína foi produzido pelo Chemicals Evaluation Research Institute (CERI), no Japão, existe como proteína de fusão da glutationa-S-transferase (GST) e é expressa em Escherichia coli. O protocolo CERI foi objeto de um estudo internacional de validação em vários laboratórios (2) que demonstrou a sua adequação e fiabilidade para o fim a que se destina o ensaio.
|
|
2.
|
Este ensaio é um procedimento de análise para identificação de substâncias suscetíveis de se ligar ao hrERα. É utilizado para determinar a capacidade de um produto químico em estudo para competir com o 17β-estradiol pela ligação ao hrERα-LBD. Os resultados quantitativos do ensaio podem incluir o IC50 (uma medição da concentração do produto químico em estudo necessária para deslocar metade do [3H]17β-estradiol do hrERα) e as afinidades de ligação relativas dos produtos químicos em estudo para o hrERα em comparação com o 17β-estradiol. Para fins de análise química, os resultados qualitativos de ensaios aceitáveis podem incluir classificações produtos químicos em estudo como ligantes ou não ligantes do hrERα, ou como inconclusivos com base nos critérios descritos para as curvas de ligação.
|
|
3.
|
O ensaio utiliza ligandos radioativos que requerem que o laboratório possua uma licença para materiais radioativos. Todos os procedimentos com radioisótopos e produtos químicos perigosos devem seguir a regulamentação e os procedimentos previstos na legislação nacional.
|
|
4.
|
As secções «INTRODUÇÃO GERAL» e «COMPONENTES DO ENSAIO DE LIGAÇÃO DO hrER» devem ser lidas antes de o presente ensaio ser utilizado para fins normativos. As definições e abreviaturas utilizadas no presente método de ensaio são descritas no apêndice 1.
|
PRINCÍPIOS DO ENSAIO (VER TAMBÉM INTRODUÇÃO GERAL)
|
5.
|
O ensaio de ligação ao hrERα mede a capacidade de um ligando com marcação radioativa ([3H]17β-estradiol) para se ligar com o ER na presença de concentrações crescentes de um produto químico em estudo (concorrente). Os produtos químicos em estudo que possuem uma elevada afinidade para o ER competem com o ligando com marcação radioativa a uma concentração mais baixa comparativamente com os produtos químicos com menor afinidade para o recetor.
|
|
6.
|
O presente ensaio é composto por dois componentes principais: uma experiência de ligantes por saturação com vista a caracterizar os parâmetros da interação recetor-ligando, seguida de uma experiência de ligantes competitivos que caracteriza a concorrência entre um produto químico em estudo e um ligando com marcação radioativa para ligação ao ER.
|
|
7.
|
O objetivo da experiência de ligação por saturação é caracterizar um determinado lote de recetores relativamente à sua afinidade de ligação e número de recetores em preparação para a realização da experiência de ligação competitiva. A experiência de ligação por saturação mede, em condições de equilíbrio, a afinidade de uma concentração fixa do recetor de estrogénio com o seu ligando natural (representado pela constante de dissociação, Kd) e a concentração de pontos de recetores ativos (Bmáx).
|
|
8.
|
A experiência de ligação competitiva mede a afinidade de uma substância para competir com o ([3H]17β-estradiol) pela ligação ao ER. A afinidade é quantificada pela concentração do produto químico em estudo que, em equilíbrio, inibe 50 % do ligante específico do ([3H]17β-estradiol (designada «concentração inibidora 50 %» ou «IC50»). A afinidade pode igualmente ser avaliada através da afinidade de ligação relativa (RBA, em relação à IC50 do estradiol, medida separadamente na mesma série de ensaios). A experiência de ligação competitiva mede a ligação do ([3H]17β-estradiol) a uma concentração fixa na presença de uma vasta gama de concentrações (oito ordens de grandeza) do produto químico em estudo. Os dados são, então, ajustados, na medida do possível, a uma forma da equação de Hill (Hill, 1910) que descreve o deslocamento do ligando radioativo através de um ligante concorrente num ponto. A extensão do deslocação do estradiol com marcação radioativa em equilíbrio é utilizada para caracterizar o produto químico em estudo como ligante, não ligante ou inconclusivo.
|
PROCEDIMENTO
Demonstração do desempenho proteico aceitável do hrERα
|
9.
|
Antes de realizar rotineiramente os ensaios de ligação por saturação e de ligação competitiva, deve verificar-se se cada novo lote de hrERα tem um desempenho correto no laboratório em que será usado. Para demonstrar esse desempenho, deve ser utilizado um processo em duas etapas:
|
—
|
Realização de um ensaio de ligação por saturação [3H]17β-estradiol para demonstrar a especificidade e a saturação do hrERα. A análise de regressão não linear de tais dados (por exemplo, BioSoft; McPherson, 1985; Motulsky, 1995) e a subsequente equação de Scatchard devem documentar a afinidade de ligação do hrERα com o [3H]17β-estradiol (Kd) e o número de recetores (Bmáx) de cada lote de hrERα.
|
|
—
|
Realizar um ensaio de ligação competitiva com recurso às substâncias de controlo [estrogénio de referência (17β-estradiol)], um ligante fraco (por exemplo, noretinodrel ou norentinodrona), e um não ligante (octiltrietoxissilano, OTES). Cada laboratório deve estabelecer uma base de dados histórica para documentar a coerência do IC50 e os valores relevantes para os estrogénios de referência e de um ligante fraco entre as experiências e os diferentes lotes de hrERα. Além disso, os parâmetros das curvas de ligação competitiva para as substâncias de controlo devem situar-se dentro dos limites do intervalo de confiança de 95 % (ver quadro 1) que foram desenvolvidos com recurso a dados de laboratórios que participaram no estudo de validação deste ensaio (2).
|
Quadro 1
Critérios de desempenho desenvolvidos para os estrogénios de referência e para o ligante fraco, ensaio de ligação de hrER do CERI
|
Substância
|
Parâmetro
|
Média (11):
|
Desvio-padrão (n)
|
Intervalos de confiança de 95 % (12)
|
|
Limite inferior
|
Limite superior
|
|
17β-estradiol
|
Topo
|
104,74
|
13,12 (70)
|
101,6
|
107,9
|
|
Base
|
0,85
|
2,41 (70)
|
0,28
|
1,43
|
|
Curva de Hill
|
–1,22
|
0,20 (70)
|
–1,27
|
–1,17
|
|
log(IC50)
|
–8,93
|
0,23 (70)
|
–8,98
|
–8,87
|
|
Noretinodrel
|
Topo
|
101,31
|
10,55 (68)
|
98,76
|
103,90
|
|
Base
|
2,39
|
5,01 (68)
|
1,18
|
3,60
|
|
Curva de Hill
|
–1,04
|
0,21 (68)
|
–1,09
|
–0,99
|
|
log(IC50)
|
–6,19
|
0,40 (68)
|
–6,29
|
–6,10
|
|
Noretindrona (13)
|
Topo
|
92,27
|
7,79 (23)
|
88,90
|
95,63
|
|
Base
|
16,52
|
10,59 (23)
|
11,94
|
21,10
|
|
Curva de Hill
|
–1,18
|
0,32 (23)
|
–1,31
|
–1,04
|
|
log(IC50)
|
–6,01
|
0,54 (23)
|
–6,25
|
–5,78
|
|
Demonstração da competência técnica do laboratório
|
10.
|
Os pontos 17 e 18 e o quadro 2 da secção «COMPONENTES DA ENSAIO DE LIGAÇÃO DO hrER» do presente método de ensaio. Cada ensaio (ligação por saturação e ligação competitiva) deve ser constituído por três séries de ensaios independentes (ou seja, diluições de recetor, produtos químicos e reagentes) em dias diferentes, devendo cada série conter três replicados.
|
Determinação do da concentração do recetor (hrERα)
|
11.
|
A concentração do recetor ativo varia ligeiramente em função do lote e das condições de armazenagem. Por esta razão, deve determinar-se a concentração do recetor ativo recebida do fornecedor. Obtém-se, assim, a concentração adequada do recetor ativo no momento da série de ensaios.
|
|
12.
|
Em condições correspondentes à ligação competitiva (0,5 nM [3H]-estradiol), as concentrações nominais de recetor de 0,1, 0,2, 0,4 e 1 nM devem ser incubadas na ausência (ligação total) e na presença (ligação não específica) de 1 μM de estradiol não marcado. À ligação específica, calculada como a diferença entre a ligação total e a ligação inespecífica, faz-se corresponder a concentração nominal do recetor. A concentração do recetor que der valores de ligação específicos correspondentes a 40 % da marcação radioativa adicionada está relacionada com a concentração de recetor correspondente, devendo esta concentração ser utilizada para as experiências de saturação e de ligação competitiva. Frequentemente, uma concentração final de hrER de 0,2 nM satisfaz esta condição.
|
|
13.
|
Se o critério dos 40 % não puder, repetidamente, ser satisfeito, o procedimento experimental deve ser verificado para deteção de eventuais erros. O incumprimento do critério dos 40 % pode indicar que há muito pouco recetor ativo no lote recombinante, caso em que deve ser considerada a utilização de outro lote de recetores.
|
Ensaio de saturação
|
14.
|
Devem ser avaliadas em triplicado oito concentrações crescentes de [3H]17β-estradiol, nas três condições seguintes (ver quadro 2):
|
a.
|
Ausência de 17β-estradiol não marcado e presença de ER. Esta é a determinação da ligação total por medida da radioatividade nos poços que apenas têm 0,5 nM [3H]17β-estradiol.
|
|
b.
|
Presença de uma concentração excessiva (2000 vezes superior) de 17β-estradiol não marcado em relação ao 17β-estradiol marcado e presença de ER. O objetivo desta condição é saturar os pontos de ligação ativos com 17β-estradiol não marcado e, mediante a medição da radioatividade nos poços, determinar a ligação não específica. Qualquer estradiol quente remanescente que possa ligar-se ao recetor é considerado ligante a um ponto não específico, dado que o estradiol frio deve estar numa concentração tão elevada que se liga a todos os pontos disponíveis no recetor.
|
|
c.
|
Ausência de 17β-estradiol não marcado e ausência de ER (determinação da radioatividade total)
|
Preparação de soluções de [3H]17β-estradiol, de 17β-estradiol não marcado e de hrERα
|
15.
|
Deve preparar-se uma solução de 40 nM de [3H]17β-estradiol a partir de uma solução de 1 μM de [3H]17β-estradiol em DMSO, adicionando DMSO (para preparar 200 nM) e tampão à temperatura ambiente (para preparar 40 nM). Usando esta solução de 40 nM, a série de diluições preparadas do [3H]17β-estradiol, que variam entre 0,313 nM e 40 nM, com tampão de ensaio à temperatura ambiente (conforme representado na coluna 12 do quadro 2). As concentrações finais do ensaio, de 0,0313 a 4,0 nM, serão obtidas mediante a adição de 10 μl destas soluções aos respetivos poços de uma placa de microtitulação com 96 poços (ver quadros 2 e 3). A preparação do tampão de ensaio, cálculo da solução original do [3H]17β-estradiol baseada na sua atividade específica, a preparação de diluições e a determinação das concentrações estão descritas em pormenor no protocolo de CERI (2).
|
|
16.
|
As diluições de soluções de 17β-estradiol devem ser preparadas a partir de uma solução de 1 nM 17β-estradiol por adição de tampão de ensaio, a fim de se obter oito concentrações crescentes, compreendidas, inicialmente, entre 0,625 μM e 80 μM. As concentrações finais do ensaio, de 0,0625 to 8 nM, serão obtidas mediante a adição de 10 μl destas soluções aos respetivos poços de uma placa de microtitulação com 96 poços específica para a medição da ligação não específica (ver quadros 2 e 3). A preparação das diluições não marcadas de 17β-estradiol é descrita em pormenor no protocolo do CERI (2).
|
|
17.
|
Deve usar-se a concentração do recetor que dê uma ligação específica de 40±10 % (ver os pontos 12 e 13). A solução de hrERα deve ser preparada com tampão de ensaio gelado imediatamente antes do uso, ou seja, depois de terem sido preparados todos os poços para ligação total, ligação não específica e ligandos.
|
|
18.
|
As placas de microtitulação de 96 poços são preparadas de acordo com o quadro 2, com 3 replicados por concentração de [3H]17β-estradiol. O quadro 3 apresenta exemplos da distribuição de volumes do [3H]17β-estradiol, 17β-estradiol não marcado, tampão e recetor.
Quadro 2
Configuração da placa de microtitulação no ensaio de ligação por saturação
|
|
1 (*10)
|
2 (*10)
|
3 (*10)
|
4 (*10)
|
5 (*10)
|
6 (*10)
|
7 (*10)
|
8 (*10)
|
9 (*10)
|
10
|
11 (*11)
|
12 (*11)
|
|
Para medição de TB
|
Para medição de NSB
|
Apenas para determinação do ligando quente
|
|
Diluições de E2 não rotuladas para as colunas de placas 4-6
|
Diluições de [3H]E2 para as colunas de placas 1-9
|
|
A
|
0,0313 nM [3H] E2+ ER
|
0,0313 nM [3H] E2+ 0,0625 μM E2+ ER
|
0,0313 nM
|
|
0,625 μM
|
0,313 nM
|
|
B
|
0,0625 nM [3H] E2+ ER
|
0,0625 nM [3H] E2+ 0,125 μM E2+ ER
|
0,0625 nM
|
|
1,25 μM
|
0,625 nM
|
|
C
|
30,125 nM [3H] E2+ ER
|
0,125 nM [3H] E2+ 0,25 μM E2+ ER
|
0,125 nM
|
|
2,5 μM
|
1,25 nM
|
|
D
|
0,250 nM [3H] E2+ ER
|
0,250 nM [3H] E2+ 0,5 μM E2+ ER
|
0,250 nM
|
|
5 μM
|
2,5 nM
|
|
E
|
0,50 nM [3H] E2+ ER
|
0,50 nM [3H] E2+ 1 μM E2+ ER
|
0,50 nM
|
|
10 μM
|
5 nM
|
|
F
|
1,00 nM [3H] E2+ ER
|
1,00 nM [3H] E2+ 2 μM E2+ ER
|
1,00 nM
|
|
20 μM
|
10 nM
|
|
G
|
2,00 nM [3H] E2+ ER
|
2,00 nM [3H] E2+ 4 μM E2+ ER
|
2,00 nM
|
|
40 μM
|
20 nM
|
|
H
|
4,00 nM [3H] E2+ ER
|
4,00 nM [3H] E2+ 8 μM E2+ ER
|
4,00 nM
|
|
80 μM
|
40 nM
|
|
TB
|
:
|
ligação total
|
|
NSB
|
:
|
ligação não específica
|
|
[3H] E2
|
:
|
[3H]17β-estradiol
|
|
E2
|
:
|
17β-estradiol não marcado
|
|
Quadro 3
Volumes de reagente para a placa de microtitulação para saturação
|
Número da coluna
|
1
|
2
|
3
|
4
|
5
|
6
|
7 (*12)
|
8 (*12)
|
9 (*12)
|
|
Etapas de preparação
|
Poços TB
|
Poços NSB
|
Apenas ligando
|
|
Volume dos componentes para os poços de reação acima e ordem de adição
|
Tampão
|
60 μl
|
50 μl
|
90 μl
|
|
E2 não marcado da coluna 11 do quadro 2
|
—
|
10 μl
|
—
|
|
[3H] E2 da coluna 12 do quadro 2
|
10 μl
|
10 μl
|
10 μl
|
|
hrERα
|
30 μl
|
30 μl
|
-
|
|
Volume total de reação
|
100 μl
|
100 μl
|
100 μl
|
|
Incubação
|
REAÇÃO APÓS 2 HORAS DE INCUBAÇÃO
|
Quantificação da radioatividade imediatamente após a preparação. Sem incubação
|
|
Tratamento com 0,4 % DCC
|
Sim
|
Sim
|
N.o
|
|
Volume de 0,4 % DCC
|
100 μl
|
100 μl
|
-
|
|
Filtração
|
Sim
|
Sim
|
N.o
|
|
MEDIÇÃO DO DPMS
|
|
Quantificação do volume adicionado ao cocktail de cintilação
|
100 μl (*13)
|
100 μl (*13)
|
50 μl
|
|
|
19.
|
As placas de microtitulação para determinação da ligação total e não específicas devem ser incubadas à temperatura ambiente (22 °C a 28 °C) durante duas horas.
|
|
Medição da ligação do [3H]17β-estradiol à hrERα
|
20.
|
Após o período de incubação de duas horas o [3H]17β-estradiol ligado ao hrERα deve ser separada do [3H]17β-estradiol livre pela adição de 100 μl de uma suspensão em gelo frio de 0,4 % DCC aos poços. As placas devem então ser colocadas em gelo durante 10 minutos, devendo a mistura de reação e a suspensão de DCC ser filtradas por meio de transferência para um filtro de placas de microtitulação, para remover o DCC. Adicionar depois 100 μl do filtrado ao fluido de cintilação em frascos de LSC para determinação da desintegração por minuto (dpms) e por frasco por contagem da cintilação em meio líquido.
|
|
21.
|
Em alternativa, se não estiver disponível um filtro de microplacas, a extração do DCC pode ser feita por centrifugação. Para evitar qualquer contaminação dos poços por contacto com o DCC, 50 μl do sobrenadante contendo [3H]17β-estradiol ligado ao hrERα devem ser retirados com extremo cuidado.
|
|
22.
|
O ligando quente isolado é usado para determinar a desintegração por minuto (dpm) do [3H]17β-estradiol adicionado aos poços. A radioatividade deve ser quantificada imediatamente após a preparação. Estes poços não devem ser incubados e não devem ser tratados com a suspensão DCC, mas o seu conteúdo deve ser diretamente adicionado ao fluido de cintilação. Estas medidas demonstram quanto [3H]17β-estradiol em foi adicionado a cada conjunto de poços para a ligação total e para a ligação não específica.
|
Ensaio de ligação competitiva
|
23.
|
O ensaio de ligação competitiva mede a ligação de uma única concentração de [3H]17β-estradiol na presença de concentrações crescentes de um produto químico em estudo. Devem utilizar-se três replicados paralelos de cada concentração numa mesma série de ensaios. Além disso, devem efetuar-se três ensaios não paralelos para cada produto químico ensaiado. O ensaio deve ser configurado com uma ou várias placas de microtitulação de 96 poços.
|
Controlos
|
24.
|
Durante a realização do ensaio, devem ser incluídos em cada experiência o solvente e os controlos paralelos (isto é, estrogénios de referência, ligante fraco e não ligante). As curvas de concentração inteiras para os estrogénios e os controlos de referência (por exemplo, ligante fraco e não ligante) devem ser utilizadas numa única placa em cada série de ensaios. Todas as outras placas devem conter: (i) uma concentração elevada (deslocamento máximo, ou seja, deslocamento quase total do ligando com marcação radioativa) e média (aproximadamente a IC50) de cada um dos E2 e ligante fraco em triplicado; (ii) controlo do solvente e ligação não específica, cada um, no mínimo, em triplicado. Os procedimentos para a preparação do tampão de ensaio [3H]17β-estradiol, do hrERα e das soluções do produto químico em estudo são descritos de forma aprofundada no protocolo CERI (2).
|
Controlo do solvente
|
25.
|
O controlo do solvente indica que o solvente não interage com o sistema de ensaio e mede a ligação total (TB). O DMSO é o solvente preferencial. Em alternativa, se a concentração mais elevada do produto químico em estudo não for solúvel em DMSO, pode ser utilizado etanol. A concentração de DMSO nos poços finais deve ser de 2,05 % e poderá ser aumentada para 2,5 % em caso de ausência de solubilidade do produto químico em estudo. As concentrações de DMSO acima de 2,5 % não devem ser utilizadas devido à interferência das concentrações mais elevadas de solvente no ensaio. Para ensaiar os produtos químicos que não são solúveis em DMSO, mas são solúveis em etanol, pode ser utilizado um máximo de 2 % de etanol no ensaio sem interferência.
|
Controlo do tampão
|
26.
|
O controlo do tampão (BC) não deve conter solvente nem produto químico em estudo, mas deve conter todos os outros componentes do ensaio. Os resultados do controlo do tampão são comparados com o controlo do solvente, para verificar se o solvente utilizado não afeta o sistema de ensaio.
|
Ligante forte (estrogénio de referência)
|
27.
|
O 17β-estradiol (n.o CAS 50-28-2) é o ligando endógeno e liga-se com elevada afinidade ao ER de subtipo alfa. Deve preparar-se uma curva padrão com 17β-estradiol não marcado para cada ensaio de ligação competitiva do hrERα, a fim de se proceder a uma avaliação da variabilidade entre ensaios realizados no mesmo laboratório. Devem ser preparadas oito soluções do 17β-estradiol não marcado em DMSO e no tampão de ensaio, com concentrações finais dos poços a utilizar na curva normal espaçada do seguinte modo: 10–6, 10–7, 10–8, 10–8,5, 10–9, 10–9,5, 10–10, 10–11M. A maior concentração de 17β–estradiol (1 μM) não marcado deve ser o indicador de ligação não específico. Esta concentração distingue-se pelo rótulo «NSB» no quadro 4, embora também faça parte da curva-padrão.
|
Ligante fraco
|
28.
|
Para demonstrar a sensibilidade de cada experiência e permitir uma avaliação da variabilidade ao longo do tempo, deve ser incluído um ligante fraco [noretinodrel (n.o CAS 68-23-5) ou noretinodrona (n.o CAS 68-22-4)]. Devem ser preparadas oito soluções de ligante fraco em DMSO e em tampão de ensaio, com as seguintes concentrações finais nos poços: 10–4,5, 10–5,5, 10–6, 10–6,5, 10–7, 10–7,5, 10–8 e 10–9 M.
|
Não ligante
|
29.
|
O octiltrietoxissilano (OTES, n.o CAS 2943-75-1) deve ser utilizado como controlo negativo (não ligante), que garante que o ensaio permite detetar os produtos químicos em estudo que não se ligam ao hrERα. Devem ser preparadas oito soluções do não ligante em DMSO e tampão de ensaio, com as seguintes concentrações finais nos poços: 10-3, 10310–4, 10–5, 10–6, 10–7, 10–8, 10-9, 10-10 M. O ftalato de di-n-butilo (DBP, n.o CAS 84-72-2) pode ser usado como uma alternativa não ligante, mas apenas ensaiado até 10-4 M. Demonstrou-se que a solubilidade máxima do DBP no ensaio é de 10-4 M4M.
|
Concentração do hrERα
|
30.
|
Deve ser utilizada a quantidade de recetor que dá um valor específico de ligação de 40±10 % (ver pontos 12-13 do apêndice 3). A solução hrERα deve ser preparada por diluição do hrERα funcional em tampão gelo frio imediatamente antes do uso.
|
[3H]17β-estradiol
|
31.
|
A concentração final de [3H]17β-estradiol nos poços deve ser de 0,5 nM.
|
Produtos químicos em estudo
|
32.
|
Em primeiro lugar, é necessário realizar um ensaio de solubilidade para determinar o limite de solubilidade de cada produto químico em estudo e identificar a gama de concentrações a utilizar na condução do protocolo de ensaio. O limite de solubilidade de cada substância química de ensaio deve ser determinado inicialmente no solvente e posteriormente confirmado em condições de ensaio. A concentração final estudada no ensaio não deve exceder 1 mM. O ensaio de determinação da gama inclui um controlo do solvente, juntamente com, pelo menos, 8 diluições sequenciais, com início na concentração máxima aceitável (por exemplo: 1 mM ou menos, com base no limite de solubilidade) e a presença de ligeira turbidez ou precipitado (ver também o ponto Apêndice 35). Uma vez determinada a gama de concentrações a ensaiar, o produto químico em estudo deve ser ensaiado utilizando curvas de intervalos de concentrações de 8 log, espaçadas conforme definido no ensaio de determinação de gama anterior. As concentrações ensaiadas na segunda e na terceira experiências devem ser novamente ajustadas, de modo a caracterizar melhor a curva concentração-resposta, se necessário.
|
|
33.
|
As diluições do produto químico em estudo devem ser preparadas no solvente adequado (ver ponto 25 do apêndice 3). Se a concentração mais elevada do produto químico em estudo não for solúvel nem em DMSO nem em etanol e se a introdução de um novo solvente implicar uma concentração de solvente no tubo final superior ao limite aceitável, a concentração mais elevada pode ser reduzida para a concentração imediatamente inferior. Neste caso, pode ser acrescentada uma concentração adicional na extremidade inferior da série de concentrações. As outras concentrações das séries devem permanecer inalteradas.
|
|
34.
|
As soluções do produto químico em estudo devem ser monitorizadas de perto quando adicionadas ao poço de ensaio, uma vez que o produto químico em estudo precipitar para fora do poço. Os dados relativos a todos os poços que contenham precipitado não devem ser tidos em conta e os respetivos motivos de exclusão devem ser anotados.
|
|
35.
|
Se houver informação prévia, de outras fontes, que forneçam um log(IC50) de um produto químico em estudo, pode ser conveniente espaçar as diluições de forma geométrica, mais perto do log(IC50) esperado (ou seja, 0,5 unidades logarítmicas em torno do log(IC50) esperado. Os resultados finais deve refletir a extensão suficiente das concentrações para os dois lados do log(IC50), incluindo o «topo» e a «base», de modo a que a curva de ligação possa ser adequadamente caracterizada.
|
Organização da placa de ensaio
|
36.
|
Devem ser preparadas placas de microtitulação marcadas, tendo em conta as incubações sêxtuplas, com códigos para o controlo do solvente, a concentração mais elevada do estrogénio de referência, que serve igualmente como indicador de ligação não específica (NSB), o controlo do tampão e incubações em triplicado de cada uma das oito concentrações do controlo não ligante (octiltrietoxissilano), as sete concentrações mais baixas do estrogénio de referência, as oito concentrações das doses do ligante fraco (noretinodrel ou noretinodrina) e as oito concentrações de cada produto químico em estudo (TC). No quadro 4 figura um exemplo do diagrama da placa para as curvas de concentração inteiras dos estrogénios de referência e do controlo. São utilizadas placas de microtitulação adicionais para os produtos químicos em estudo e devem incluir controlos da placa, nomeadamente 1), uma concentração elevada (deslocamento máximo) e média (aproximadamente a IC50) de cada um dos E2 e ligante fraco em triplicado; 2) controlo do solvente (como ligante total) e ligação não específica, cada uma sextuplicada (quadro 5). O apêndice 3.3 apresenta um exemplo de folha de cálculo com um esquema de configuração das placas de microtitulação com três produtos químicos em estudo desconhecidos para o ensaio de ligação competitiva. As concentrações indicadas na folha de cálculo, bem como nos quadros 4 e 5, referem-se às concentrações finais utilizadas em cada ensaio. A concentração máxima para E2 deve ser 1×10-7 M e, para o ligante fraco, deve ser usada a concentração mais elevada utilizada para o ligante fraco na placa 1. A concentração IC50 deve ser determinada pelo laboratório com base na sua base de dados históricos de controlo. Prevê-se que este valor seja semelhante ao observado nos estudos de validação (ver quadro 1).
|
Quadro 4
Configuração das placas de microtitulação para o ensaio de ligação competitiva
(97)
(98)
, curvas de concentração inteiras para os estrogénios e os controlos de referência (placa 1)
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
Controlo do tampão e controlo positivo (E2)
|
Fracamente positivo Noretinodrel
|
Controlo negativo (OTES)
|
TB e NSB
|
|
A
|
Vazio (*14)
|
1×10–9 M
|
1×10–10 M
|
TB (controlo de solvente) (2,05 % DMSO)
|
|
B
|
E2 (1×10–11 M)
|
1×10–8 M
|
1×10–9 M
|
|
C
|
E2 (1×10–10 M)
|
1×10–7,5 M
|
1×10–8 M
|
NSB (E2 10–6 M)
|
|
D
|
E2 (1×10–9,5 M)
|
1×10–7 M
|
1×10–7 M
|
|
E
|
E2 (1×10–9 M)
|
1×10–6,5 M
|
1×10–6 M
|
Tampão de controlo
|
|
F
|
E2 (1×10–8,5 M)
|
1×10–6 M
|
1×10–5 M
|
|
G
|
E2 (1×10–8 M)
|
1×10–5,5 M
|
1×10–4 M
|
Vazio (para quente) (*15)
|
|
H
|
E2 (1×10–7 M)
|
1×10–4,5 M
|
1×10–3 M
|
Quadro 5
Materiais de simulação para a produção de placas vinculativas, placas adicionais vinculativas, placas adicionais para produtos químicos de ensaio (TC) e placas de controlo
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
|
Produto químico em estudo-1 (TC-1)
|
Produto químico em estudo-2 (TC-2)
|
Produto químico em estudo-3 (TE-3)
|
Controlos
|
|
A
|
TC-1 (1×10–10 M)
|
TC-2 (1×10–10 M)
|
TC-3 (1×10–10 M)
|
E2 (1×10–7M)
|
|
B
|
TC-1 (1×10–9 M)
|
TC-2 (1×10–9 M)
|
TC-3 (1×10–9 M)
|
E2 (IC50)
|
|
C
|
TC-1 (1×10–8 M)
|
TC-2 (1×10–8 M)
|
TC-3 (1×10–8 M)
|
NE (1×10–4,5M)
|
|
D
|
TC-1 (1×10–7 M)
|
TC-2 (1×10–7 M)
|
TC-3 (1×10–7 M)
|
NE (IC50)
|
|
E
|
TC-1 (1×10–6 M)
|
TC-2 (1×10–6 M)
|
TC-3 (1×10–6 M)
|
NSB (E2 10–6 M)
|
|
F
|
TC-1 (1×10–5 M)
|
TC-2 (1×10–5 M)
|
TC-3 (1×10–5 M)
|
|
G
|
TC-1 (1×10–4 M)
|
TC-2 (1×10–4 M)
|
TC-3 (1×10–4 M)
|
TB (controlo de solvente)
|
|
H
|
TC-1 (1×10–3 M)
|
TC-2 (1×10–3 M)
|
TC-3 (1×10–3 M)
|
|
Neste exemplo, o ligando fraco é o noretinodrel (NE)
|
Realização do ensaio de ligação competitiva
|
37.
|
Com exceção dos poços destinados a ligação total e dos vazios (para quente), conforme ao quadro 6, introduzir em cada poço 50 μl de tampão de ensaio e misturar com 10 μl do controlo do solvente, estrogénio de referência (E2), ligante fraco, não ligante e produto químico em estudo, respetivamente, 10 μl de uma solução a 5 nM de [3H]17β-estradiol. Em seguida, adicionar 30 μl de solução do recetor gelo frio a cada placa e misturar delicadamente. A solução de hrERIα deve ser o último reagente a ser adicionado. As placas de microtitulação do ensaio devem ser incubadas à temperatura ambiente (22-28 °C) durante 2 horas.
Quadro 6
Quantidade de componentes de ensaio para o ensaio de ligação competitiva nos hrER, placas microtituladoras
|
Etapas de preparação
|
Outros que não os poços de TB
|
Poços de TB
|
Vazio (para quente)
|
|
Volume dos componentes para os poços de reação acima e ordem de adição
|
Tampão de ensaio à temperatura ambiente
|
50 μl
|
60 μl
|
90 μl
|
|
E2 não marcado, ligante fraco não ligante, solventes e produtos químicos de ensaio (*16)
|
10 μl
|
—
|
—
|
|
[3H]17β-estradiol para concentração final de 0,5 nM (i.e. 5 nM)
|
10 μl
|
10 μl
|
10 μl
|
|
Concentração de hrERα conforme determinada (ver pontos 12-13)
|
30 μl
|
30 μl
|
—
|
|
Volume total em cada poço
|
100 μl
|
100 μl
|
100 μl
|
|
|
38.
|
Deve proceder-se à quantificação do valor da ligação do [3H]17β-estradiol ao hrERα, após a separação do [3H]17β-estradiol ligado ao hrERα do [3H]17β-estradiol livre mediante a adição de 100 μl de suspensão de DCC gelo frio a cada poço, conforme descrito nos pontos 21 a 23 do apêndice 3 para o ensaio de ligação por saturação.
|
|
39.
|
Os poços G10-12 e H10-12, identificados como vazios (para quente) no quadro 4, representam o dpms do [3H] ]17β-estradiol marcado em 10 μl. A alíquota de 10 μl deve ser diretamente adicionada ao fluido de cintilação.
|
Critérios de aceitabilidade
Ensaio de ligação por saturação
|
40.
|
A curva de ligação específica deve atingir um patamar elevado à medida em que forem sendo utilizadas concentrações crescentes de [3H]17β-estradiol, indicando saturação do hrERα com o ligando.
|
|
41.
|
A ligação específica a 0,5 nM de [3H]17β-estradiol deve situar-se na gama aceitável, entre 30 % e 50 % da média da radioatividade total medida adicionada ao longo nas diferentes séries de ensaio. São aceitáveis pequenos desvios a este intervalo, mas se as séries de ensaios se situarem permanentemente fora deste intervalo de variação, ou se uma séries de ensaios estiver consideravelmente fora deste intervalo, a concentração de proteínas deve ser ajustada, repetindo-se o ensaio de saturação.
|
|
42.
|
Os dados devem produzir um diagrama linear de Scatchard.
|
|
43.
|
A ligação não específica não deve ser excessiva. O valor da ligação não específica deve, normalmente, ser < 35 % da ligação total. No entanto, o rácio pode, ocasionalmente, ultrapassar este limite quando é medido um dpm muito baixo para a concentração mais baixa de e 17β-estradiol com marcação radioativa do β-estradiol.
|
Ensaio de ligação competitiva
|
44.
|
O aumento das concentrações do 17β-estradiol não marcado deve deslocar o [3H]17β-estradiol do recetor de forma compatível com uma ligação competitiva num sítio.
|
|
45.
|
O valor de IC50 para o estrogénio de referência (i.e., 17β-estradiol) deve ser aproximadamente igual à concentração molar de [3H]17β-estradiol mais o Kd determinado a partir do ensaio de saturação da ligação.
|
|
46.
|
A ligação específica total deve estar consistentemente dentro de um intervalo aceitável de 40±10 % quando a concentração média de radioatividade total medida adicionada a cada poço for de 0,5 nM nas séries de ensaios. São aceitáveis pequenos desvios a este intervalo, mas se as séries de ensaios se situarem permanentemente fora deste intervalo de variação, ou se uma séries de ensaios estiver consideravelmente fora deste intervalo, a concentração de proteínas deve ser ajustada.
|
|
47.
|
O solvente não deve alterar a sensibilidade ou a reprodutibilidade do ensaio. Os resultados do controlo do solvente (poços de TB) são comparados com o controlo de tampão, a fim de verificar se o solvente utilizado não afeta o sistema de ensaio. Os resultados do TB e do controlo da solução-tampão devem ser comparáveis se não houver nenhum efeito do solvente no ensaio.
|
|
48.
|
O não ligante não deve deslocar mais de 25 % do [3H]17β-estradiol do hrERα quando ensaiado até 10–3 M (OTES) ou 10–4 M (DBP).
|
|
49.
|
Foram desenvolvidos critérios de desempenho para os estrogénios de referência e dois ligantes fracos (por exemplo, noretinodrel, noretindrona) com base nos dados do estudo de validação do ensaio de ligação CERIhrER (anexo N da referência 2). A média de 95 % de intervalos de confiança é fornecida para o DP médio (n) de todos os controlos em quatro laboratórios que participaram no estudo de validação. Foram calculados intervalos de confiança de 95 % para os parâmetros de ajustamento da curva [isto é, topo, base, curva de Hill e log( IC50)]) para os estrogénios de referência e os ligantes fracos, e o log10(RBA)Log10RBA dos ligantes fracos em relação aos estrogénios de referência. O quadro 1 apresenta os intervalos previstos para os parâmetros de ajustamento da curva que podem ser utilizados como critérios de desempenho. Na prática, o IC50 pode variar ligeiramente em função dos valores de Kd obtidos experimentalmente a partir da preparação de recetor e da concentração de ligando utilizados para o ensaio.
|
|
50.
|
Não foram definidos critérios de desempenho para os parâmetros de ajustamento da curva em relação aos produtos químicos em estudo, devido à grande variedade de produtos químicos em estudo existentes e à variação das principais afinidades e dos resultados (p. ex., curva inteira, curva parcial, sem ajustamento de curva). Contudo, devem ser utilizados critérios profissionais aquando da avaliação dos resultados de cada série de ensaios para um produto químico em estudo. Deve ser utilizada uma gama de concentrações do produto químico em estudo suficiente para definir claramente o topo (por exemplo, 90-100 % de ligação) da curva de ligação competitiva. A variabilidade dos replicados para cada concentração do produto químico em estudo, bem como entre as 3 séries não paralelas, deve ser razoável e cientificamente aceitável. Os controlos de cada ciclo para um produto químico em estudo devem abordar as medidas de desempenho comunicadas para este ensaio CERI e ser coerentes com os dados históricos de controlo de cada laboratório.
|
ANÁLISE DOS DADOS
Ensaio de ligação por saturação
|
51.
|
São medidas as ligações totais e as ligações não específicas. A partir destes valores, a ligação específica de concentrações crescentes de [3H]17β-estradiol em condições de equilíbrio é calculada subtraindo-se a ligação não específica do total. Um gráfico que apresente a ligação específica versus a concentração de [3H]17β-estradiol deve atingir um plano para o indicativo máximo específico da ligação por saturação do hrERα [3-17H]β-estradiol. Além disso, a análise dos dados deve documentar a ligação do [3H]17β-estradiol a um único ponto de ligação de alta afinidade. As ligações não específicas, totais e específicas devem ser visíveis numa curva de ligação por saturação. Uma análise mais aprofundada destes dados deve utilizar uma análise de regressão não linear (ex., BioSoft; McPherson, 1985; Motulsky, 1995) com uma apresentação final dos dados de acordo com Scatchard.
|
|
52.
|
A análise de dados deve determinar o Bmáx e a Kd a partir, unicamente dos dados relativos à ligação total, partindo do pressuposto de que a ligação não específica é de linear, salvo se for justificada a utilização de outro método. Quando se determinar a melhor adequação, deve utilizar-se uma regressão sólida, a menos que seja apresentada uma justificação. Deve ser indicado método escolhido para uma regressão sólida. A correção da depleção de ligando (por exemplo, utilizando o método de Swillens 1995) deve ser sempre utilizada na determinação de Bmáx e Kd a partir dos dados da ligação por saturação.
|
Ensaio de ligação competitiva
|
53.
|
A curva de ligação competitiva é representada como a ligação específica do [3H]17β-estradiol versus a concentração [log10(unidades)] do concorrente. A concentração do produto químico em estudo que inibe 50 % da ligação máxima específica do [3H]17β-estradiol é IC50.
|
|
54.
|
As estimativas de log(IC50) para os controlos positivos (por exemplo, estrogénios de referência e ligantes fracos) devem ser determinadas com recurso a um software de ajustamento não linear de ajustamento adequado, a fim de se adaptar à equação de quatro parâmetros de Hill (p. ex., BioSoft; McPherson, 1985; Motulsky, 1995). Em geral, o topo, a base, a curva e o log(IC50) devem ser mantidos inalterados quando se ajustam as curvas. Quando se determinar a melhor adequação, deve utilizar-se uma regressão sólida, a menos que seja apresentada uma justificação. Não devem ser feitas correções para a depleção do ligando. Após a análise inicial, cada curva de ligação deve ser avaliada de modo a garantir a devida adequação ao modelo. A afinidade de ligação relativa (RBA) do ligando fraco deve ser calculada como percentagem do log(IC50) para o ligante fraco em relação ao log(IC50) para o 17β-estradiol. Os resultados dos controlos positivos e do controlo do não ligante devem ser avaliados de acordo com as medidas do desempenho do ensaio constantes dos pontos 44 a 49 do presente apêndice 3.
|
|
55.
|
Os dados relativos a todos os produtos químicos em estudo devem ser analisados através de uma abordagem por etapas, para garantir que os dados são devidamente analisados e que cada ligação competitiva é devidamente classificada. Recomenda-se que cada série de ensaios para um produto químico em estudo seja inicialmente submetida a uma análise normalizada de dados idêntica à utilizada para os estrogénios de referência e para controlos de ligantes fracos (ponto 54 do presente apêndice 3). Uma vez concluída, deve ser efetuada uma avaliação técnica dos parâmetros de ajustamento da curva, bem como uma análise visual do grau de adequação dos dados à curva de ligação competitiva gerada para cada série de ensaios. Durante esta avaliação técnica, as observações de uma diminuição dependente da concentração do [3H]17β-estradiol especificamente ligado, da baixa variabilidade entre os replicados técnicos em cada concentração do produto químico em estudo e da coerência dos parâmetros de ajustamento entre as três séries de ensaios constituem uma boa indicação de que a análise de ensaios e de dados foi realizada de forma adequada.
|
Interpretação dos dados
|
56.
|
Caso se cumpram todos os critérios de aceitabilidade, o produto químico em estudo é considerado um ligante para o hrERα se for possível ajustar uma curva ligação e o ponto mais baixo da curva de resposta dentro da gama de dados for inferior a 50 % (figura 1).
|
|
57.
|
O produto químico em estudo é considerado como não ligante se, cumpridos todos os critérios de aceitabilidade:
|
—
|
For possível ajustar uma curva de ligação e o ponto mais baixo da curva de resposta dentro da gama de dados for superior a 75 %, ou
|
|
—
|
Não for possível ajustar uma curva de ligação e a média percentual mais baixa não ajustada entre os grupos de concentração dos dados for superior a 75 %.
|
|
|
58.
|
Os produtos químicos em estudo são considerados equívocos se nenhuma das condições acima referidas estiver preenchida (por exemplo, o ponto mais baixo da curva de resposta ajustada situa-se entre 76 e 51 %).
|
Quadro 7
Critérios para classificação com base numa curva de ligação competitiva para um produto químico em estudo
|
Classificação
|
Critérios
|
|
Ligantea
|
Uma curva de ligação pode ser ajustada.
O ponto mais baixo na curva de resposta dentro da gama de dados é inferior a 50 %.
|
|
Não liganteb
|
Se for possível ajustar a curva de ligação,
o ponto mais baixo da curva de resposta dentro da gama de dados é superior a 75 %.
Se não for possível ajustar a curva de ligação,
a média percentual mais baixa não ajustada entre os grupos de concentração nos dados é superior a 75 %.
|
|
Inconclusivoc
|
Qualquer produto químico em estudo que não seja ligante nem não ligante.
(Por exemplo, o ponto mais baixo da curva de resposta ajustada situa-se entre 76 e 51 %).
|
Figura 1
Exemplos de classificação de produtos químicos em estudo por meio de uma curva de ligação competitiva

|
59.
|
As várias séries de ensaios realizados num laboratório para um produto químico em estudo são combinadas através da atribuição de valores numéricos a cada série de ensaios e fazendo a média das diferentes séries, como se indica no quadro 8. Os resultados para as séries combinadas em cada laboratório são comparados com a classificação prevista para cada produto químico em estudo.
|
Quadro 8
Método de classificação do produto químico em estudo utilizando múltiplas séries de ensaios
|
Para atribuir valor a cada série de ensaios:
|
|
Classificação
|
Valor numérico
|
|
Ligante
|
2
|
|
Inconclusivo
|
1
|
|
Não ligante
|
0
|
|
Para classificar a média dos valores numéricos das diferentes séries de ensaios:
|
|
Classificação
|
Valor numérico
|
|
Ligante
|
Média ≥ 1,5
|
|
Inconclusivo
|
0,5 ≤ média < 1,5
|
|
Não ligante
|
Média < 0,5
|
RELATÓRIO DO ENSAIO
|
60.
|
Ver ponto 24 de «COMPONENTES DO ENSAIO DE LIGAÇÃO hrER» do presente método de ensaio.
|
Apêndice 3.1
LISTA DE TERMOS
DCC
: carvão revestido com dextrose.
[3H]E2
: 17β-estradiol com marcação radioativa por trítio.
E2
: 17β-estradiol não marcado (inerte).
hrERα
: recetor alfa de estrogénio recombinante humano (domínio de ligação do ligando).
Replicado
: um ou vários poços que contêm o mesmo conteúdo com as mesmas concentrações e que são objeto de ensaios paralelos numa única série de ensaios. Neste protocolo, cada concentração do produto químico em estudo é ensaiada em triplicado; isto é, existem três replicados que são ensaiados simultaneamente em cada concentração do produtos químico em estudo.
Série de ensaios
: conjunto completo de poços de placas de microtitulação que fornece todas as informações necessárias para caracterizar a ligação de um produto químico em estudo ao hrERα (viz., total [3H]17β-estradiol adicionado ao poço, ligação máxima do [3H]17β-estradiol ao hrERα ligação não específica e ligação total em diferentes concentrações do produto químico em estudo). Uma série de ensaio pode consistir em apenas um poço de ensaio (ou seja, replicado) por concentração; contudo, dado que este protocolo requer que os ensaios sejam realizados em triplicado, uma série de ensaios consiste em três poços por concentração. Além disso, este protocolo requer três séries de ensaio independentes (ou seja, não paralelas) por produto químico.
Tampão de ensaio
: 10 mM de Tris-HCl, pH 7,4, contendo 1 mM de EDTA, EGTA 1 mM, 1 mM NaVO3, 10 % de glicerol, 0,2 mM de leupeptina, 1 mM ditiotritol e 10 mg/ml de albumina sérica bovina.
Apêndice 3.2
CONFIGURAÇÃO DO ENSAIO DE LIGAÇÃO COMPETITIVA
|
Placa
|
Posição
|
Replicado
|
Tipo de poço
|
Código do poço
|
Código de concentração
|
Concentração inicial do concorrente (M)
|
Solução de reserva de hrER (μl)
|
Volume do tampão (μl)
|
Volume (μl) do traçador (E2 quente)
|
Volume a partir da placa de diluição (μl)
|
Volume final (μl)
|
Concentração final do concorrente (M)
|
|
S
|
A1
|
1
|
Em branco
|
BK
|
BK1
|
—
|
—
|
—
|
—
|
—
|
—
|
—
|
|
S
|
A2
|
2
|
Em branco
|
BK
|
BK2
|
—
|
—
|
—
|
—
|
—
|
—
|
—
|
|
S
|
A3
|
3
|
Em branco
|
BK
|
BK3
|
—
|
—
|
—
|
—
|
—
|
—
|
—
|
|
S
|
B1
|
1
|
E2 frio
|
S
|
S1
|
1,,00E-10
|
30
|
50
|
10
|
10
|
100
|
1,,0E-11
|
|
S
|
B2
|
2
|
E2 frio
|
S
|
S1
|
1,,00E-10
|
30
|
50
|
10
|
10
|
100
|
1,,0E-11
|
|
S
|
B3
|
3
|
E2 frio
|
S
|
S1
|
1,,00E-10
|
30
|
50
|
10
|
10
|
100
|
1,,0E-11
|
|
S
|
C1
|
1
|
E2 frio
|
S
|
S2
|
1,,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,,0E-10
|
|
S
|
C2
|
2
|
E2 frio
|
S
|
S2
|
1,,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,,0E-10
|
|
S
|
C3
|
3
|
E2 frio
|
S
|
S2
|
1,,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,,0E-10
|
|
S
|
D1
|
1
|
E2 frio
|
S
|
S3
|
3,16E-09
|
30
|
50
|
10
|
10
|
100
|
3,,2E-10
|
|
S
|
D2
|
2
|
E2 frio
|
S
|
S3
|
3,16E-09
|
30
|
50
|
10
|
10
|
100
|
3,,2E-10
|
|
S
|
D3
|
3
|
E2 frio
|
S
|
S3
|
3,16E-09
|
30
|
50
|
10
|
10
|
100
|
3,,2E-10
|
|
S
|
E1
|
1
|
E2 frio
|
S
|
S4
|
1,,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,,0E-09
|
|
S
|
E2
|
2
|
E2 frio
|
S
|
S4
|
1,,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,,0E-09
|
|
S
|
E3
|
3
|
E2 frio
|
S
|
S4
|
1,,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,,0E-09
|
|
S
|
F1
|
1
|
E2 frio
|
S
|
S5
|
3,16E-08
|
30
|
50
|
10
|
10
|
100
|
3,,2E-09
|
|
S
|
F2
|
2
|
E2 frio
|
S
|
S5
|
3,16E-08
|
30
|
50
|
10
|
10
|
100
|
3,,2E-09
|
|
S
|
F3
|
3
|
E2 frio
|
S
|
S5
|
3,16E-08
|
30
|
50
|
10
|
10
|
100
|
3,,2E-09
|
|
S
|
G1
|
1
|
E2 frio
|
S
|
S6
|
1,,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,,0E-08
|
|
S
|
G2
|
2
|
E2 frio
|
S
|
S6
|
1,,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,,0E-08
|
|
S
|
G3
|
3
|
E2 frio
|
S
|
S6
|
1,,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,,0E-08
|
|
S
|
H1
|
1
|
E2 frio
|
S
|
S7
|
1,,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,,0E-07
|
|
S
|
H2
|
2
|
E2 frio
|
S
|
S7
|
1,,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,,0E-07
|
|
S
|
H3
|
3
|
E2 frio
|
S
|
S7
|
1,,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,,0E-07
|
|
S
|
A4
|
1
|
Noretinodrel
|
NE
|
WP1
|
1,,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,,0E-09
|
|
S
|
A5
|
2
|
Noretinodrel
|
NE
|
WP1
|
1,,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,,0E-09
|
|
S
|
A6
|
3
|
Noretinodrel
|
NE
|
WP1
|
1,,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,,0E-09
|
|
S
|
B4
|
1
|
Noretinodrel
|
NE
|
WP2
|
1,,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,,0E-08
|
|
S
|
B5
|
2
|
Noretinodrel
|
NE
|
WP2
|
1,,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,,0E-08
|
|
S
|
B6
|
3
|
Noretinodrel
|
NE
|
WP2
|
1,,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,,0E-08
|
|
S
|
C4
|
1
|
Noretinodrel
|
NE
|
WP3
|
3,16E-07
|
30
|
50
|
10
|
10
|
100
|
3,,2E-08
|
|
S
|
C5
|
2
|
Noretinodrel
|
NE
|
WP3
|
3,16E-07
|
30
|
50
|
10
|
10
|
100
|
3,,2E-08
|
|
S
|
C6
|
3
|
Noretinodrel
|
NE
|
WP3
|
3,16E-07
|
30
|
50
|
10
|
10
|
100
|
3,,2E-08
|
|
S
|
D4
|
1
|
Noretinodrel
|
NE
|
WP4
|
1,,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,,0E-07
|
|
S
|
D5
|
2
|
Noretinodrel
|
NE
|
WP4
|
1,,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,,0E-07
|
|
S
|
D6
|
3
|
Noretinodrel
|
NE
|
WP4
|
1,,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,,0E-07
|
|
S
|
E4
|
1
|
Noretinodrel
|
NE
|
WP5
|
3,16E-06
|
30
|
50
|
10
|
10
|
100
|
3,,2E-07
|
|
S
|
E5
|
2
|
Noretinodrel
|
NE
|
WP5
|
3,16E-06
|
30
|
50
|
10
|
10
|
100
|
3,,2E-07
|
|
S
|
E6
|
3
|
Noretinodrel
|
NE
|
WP5
|
3,16E-06
|
30
|
50
|
10
|
10
|
100
|
3,,2E-07
|
|
S
|
F4
|
1
|
Noretinodrel
|
NE
|
WP6
|
1,,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,,0E-06
|
|
S
|
F5
|
2
|
Noretinodrel
|
NE
|
WP6
|
1,,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,,0E-06
|
|
S
|
F6
|
3
|
Noretinodrel
|
NE
|
WP6
|
1,,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,,0E-06
|
|
S
|
G4
|
1
|
Noretinodrel
|
NE
|
WP7
|
3,16E-05
|
30
|
50
|
10
|
10
|
100
|
3,,2E-06
|
|
S
|
G5
|
2
|
Noretinodrel
|
NE
|
WP7
|
3,16E-05
|
30
|
50
|
10
|
10
|
100
|
3,,2E-06
|
|
S
|
G6
|
3
|
Noretinodrel
|
NE
|
WP7
|
3,16E-05
|
30
|
50
|
10
|
10
|
100
|
3,,2E-06
|
|
S
|
H4
|
1
|
Noretinodrel
|
NE
|
WP8
|
3,16E-04
|
30
|
50
|
10
|
10
|
100
|
3,,2E-05
|
|
S
|
H5
|
2
|
Noretinodrel
|
NE
|
WP8
|
3,16E-04
|
30
|
50
|
10
|
10
|
100
|
3,,2E-05
|
|
S
|
H6
|
3
|
Noretinodrel
|
NE
|
WP8
|
3,16E-04
|
30
|
50
|
10
|
10
|
100
|
3,,2E-05
|
|
S
|
A7
|
1
|
OTES
|
N
|
OTES1
|
1,,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,,0E-10
|
|
S
|
A8
|
2
|
OTES
|
N
|
OTES1
|
1,,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,,0E-10
|
|
S
|
A9
|
3
|
OTES
|
N
|
OTES1
|
1,,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,,0E-10
|
|
S
|
B7
|
1
|
OTES
|
N
|
OTES2
|
1,,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,,0E-09
|
|
S
|
B8
|
2
|
OTES
|
N
|
OTES2
|
1,,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,,0E-09
|
|
S
|
B9
|
3
|
OTES
|
N
|
OTES2
|
1,,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,,0E-09
|
|
S
|
C7
|
1
|
OTES
|
N
|
OTES3
|
1,,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,,0E-08
|
|
S
|
C8
|
2
|
OTES
|
N
|
OTES3
|
1,,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,,0E-08
|
|
S
|
C9
|
3
|
OTES
|
N
|
OTES3
|
1,,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,,0E-08
|
|
S
|
D7
|
1
|
OTES
|
N
|
OTES4
|
1,,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,,0E-07
|
|
S
|
D8
|
2
|
OTES
|
N
|
OTES4
|
1,,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,,0E-07
|
|
S
|
D9
|
3
|
OTES
|
N
|
OTES4
|
1,,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,,0E-07
|
|
S
|
E7
|
1
|
OTES
|
N
|
OTES5
|
1,,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,,0E-06
|
|
S
|
E8
|
2
|
OTES
|
N
|
OTES5
|
1,,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,,0E-06
|
|
S
|
E9
|
3
|
OTES
|
N
|
OTES5
|
1,,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,,0E-06
|
|
S
|
F7
|
1
|
OTES
|
N
|
OTES6
|
1,,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,,0E-05
|
|
S
|
F8
|
2
|
OTES
|
N
|
OTES6
|
1,,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,,0E-05
|
|
S
|
F9
|
3
|
OTES
|
N
|
OTES6
|
1,,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,,0E-05
|
|
S
|
G7
|
1
|
OTES
|
N
|
OTES7
|
1,,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,,0E-04
|
|
S
|
G8
|
2
|
OTES
|
N
|
OTES7
|
1,,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,,0E-04
|
|
S
|
G9
|
3
|
OTES
|
N
|
OTES7
|
1,,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,,0E-04
|
|
S
|
H7
|
1
|
OTES
|
N
|
OTES8DBP7
|
1,,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,,0E-03
|
|
S
|
H8
|
2
|
OTES
|
N
|
OTES88
|
1,,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,,0E-03
|
|
S
|
H9
|
3
|
OTES
|
N
|
OTES8
|
1,,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,,0E-03
|
|
S
|
A10
|
1
|
Ligação total
|
TB
|
TB1
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
A11
|
2
|
Ligação total
|
TB
|
TB2
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
A12
|
3
|
Ligação total
|
TB
|
TB3
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
B10
|
4
|
Ligação total
|
TB
|
TB4
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
B11
|
5
|
Ligação total
|
TB
|
TB5
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
B12
|
6
|
Ligação total
|
TB
|
TB6
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
C10
|
1
|
E2 frio (elevado)
|
NSB
|
S1
|
1,,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,,0E-06
|
|
S
|
C11
|
2
|
E2 frio (elevado)
|
NSB
|
S2
|
1,,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,,0E-06
|
|
S
|
C12
|
3
|
E2 frio (elevado)
|
NSB
|
S3
|
1,,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,,0E-06
|
|
S
|
D10
|
4
|
E2 frio (elevado)
|
NSB
|
S4
|
1,,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,,0E-06
|
|
S
|
D11
|
5
|
E2 frio (elevado)
|
NSB
|
S5
|
1,,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,,0E-06
|
|
S
|
D12
|
6
|
E2 frio (elevado)
|
NSB
|
S6
|
1,,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,,0E-06
|
|
S
|
E10
|
1
|
Tampão de controlo
|
BC
|
BC1
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
E11
|
2
|
Tampão de controlo
|
BC
|
BC2
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
E12
|
3
|
Tampão de controlo
|
BC
|
BC3
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
F10
|
4
|
Tampão de controlo
|
BC
|
BC4
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
F11
|
5
|
Tampão de controlo
|
BC
|
BC5
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
F12
|
6
|
Tampão de controlo
|
BC
|
BC6
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
G10 (*17)
|
1
|
Vazio (para quente)
|
Quente
|
H1
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
S
|
G11 (*17)
|
2
|
Vazio (para quente)
|
Quente
|
H2
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
S
|
G12 (*17)
|
3
|
Vazio (para quente)
|
Quente
|
H3
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
S
|
H10 (*17)
|
4
|
Vazio (para quente)
|
Quente
|
H4
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
S
|
H11 (*17)
|
5
|
Vazio (para quente)
|
Quente
|
H5
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
S
|
H12
|
6
|
Vazio (para quente)
|
Quente
|
H6
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
P1
|
A1
|
1
|
Desconhecido 1
|
U1
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A2
|
2
|
Desconhecido 1
|
U1
|
1
|
1,,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A3
|
3
|
Desconhecido 1
|
U1
|
1
|
1,,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
B1
|
1
|
Desconhecido 1
|
U1
|
2
|
1,,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
B2
|
2
|
Desconhecido 1
|
U1
|
2
|
1,,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
B3
|
3
|
Desconhecido 1
|
U1
|
2
|
1,,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
C1
|
1
|
Desconhecido 1
|
U1
|
3
|
1,,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
C2
|
2
|
Desconhecido 1
|
U1
|
3
|
1,,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
C3
|
3
|
Desconhecido 1
|
U1
|
3
|
1,,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
D1
|
1
|
Desconhecido 1
|
U1
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
D2
|
2
|
Desconhecido 1
|
U1
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
D3
|
3
|
Desconhecido 1
|
U1
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
E1
|
1
|
Desconhecido 1
|
U1
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E2
|
2
|
Desconhecido 1
|
U1
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E3
|
3
|
Desconhecido 1
|
U1
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
F1
|
1
|
Desconhecido 1
|
U1
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
F2
|
2
|
Desconhecido 1
|
U1
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
F3
|
3
|
Desconhecido 1
|
U1
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
G1
|
1
|
Desconhecido 1
|
U1
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
G2
|
2
|
Desconhecido 1
|
U1
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
G3
|
3
|
Desconhecido 1
|
U1
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
H1
|
1
|
Desconhecido 1
|
U1
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
H2
|
2
|
Desconhecido 1
|
U1
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
H3
|
3
|
Desconhecido 1
|
U1
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
A4
|
1
|
Desconhecido 2
|
U2
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A5
|
2
|
Desconhecido 2
|
U2
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A6
|
3
|
Desconhecido 2
|
U2
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
B4
|
1
|
Desconhecido 2
|
U2
|
2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
B5
|
2
|
Desconhecido 2
|
U2
|
2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
B6
|
3
|
Desconhecido 2
|
U2
|
2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
C4
|
1
|
Desconhecido 2
|
U2
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
C5
|
2
|
Desconhecido 2
|
U2
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
C6
|
3
|
Desconhecido 2
|
U2
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
D4
|
1
|
Desconhecido 2
|
U2
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
D5
|
2
|
Desconhecido 2
|
U2
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
D6
|
3
|
Desconhecido 2
|
U2
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
E4
|
1
|
Desconhecido 2
|
U2
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E5
|
2
|
Desconhecido 2
|
U2
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E6
|
3
|
Desconhecido 2
|
U2
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
F4
|
1
|
Desconhecido 2
|
U2
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
F5
|
2
|
Desconhecido 2
|
U2
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
F6
|
3
|
Desconhecido 2
|
U2
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
G4
|
1
|
Desconhecido 2
|
U2
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
G5
|
2
|
Desconhecido 2
|
U2
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
G6
|
3
|
Desconhecido 2
|
U2
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
H4
|
1
|
Desconhecido 2
|
U2
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
H5
|
2
|
Desconhecido 2
|
U2
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
H6
|
3
|
Desconhecido 2
|
U2
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
A7
|
1
|
Desconhecido 3
|
U3
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A8
|
2
|
Desconhecido 3
|
U3
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A9
|
3
|
Desconhecido 3
|
U3
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
B7
|
1
|
Desconhecido 3
|
U3
|
2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
B8
|
2
|
Desconhecido 3
|
U3
|
2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
B9
|
3
|
Desconhecido 3
|
U3
|
2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
C7
|
1
|
Desconhecido 3
|
U3
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
C8
|
2
|
Desconhecido 3
|
U3
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
C9
|
3
|
Desconhecido 3
|
U3
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
D7
|
1
|
Desconhecido 3
|
U3
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
D8
|
2
|
Desconhecido 3
|
U3
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
D9
|
3
|
Desconhecido 3
|
U3
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
E7
|
1
|
Desconhecido 3
|
U3
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E8
|
2
|
Desconhecido 3
|
U3
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E9
|
3
|
Desconhecido 3
|
U3
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
F7
|
1
|
Desconhecido 3
|
U3
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
F8
|
2
|
Desconhecido 3
|
U3
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
F9
|
3
|
Desconhecido 3
|
U3
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
G7
|
1
|
Desconhecido 3
|
U3
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
G8
|
2
|
Desconhecido 3
|
U3
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
G9
|
3
|
Desconhecido 3
|
U3
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
H7
|
1
|
Desconhecido 3
|
U3
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
H8
|
2
|
Desconhecido 3
|
U3
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
H9
|
3
|
Desconhecido 3
|
U3
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
A10
|
1
|
Controlo E2 (máx.)
|
S
|
E2máx1
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,00E-07
|
|
P1
|
A11
|
2
|
Controlo E2 (máx.)
|
S
|
E2máx2
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,00E-07
|
|
P1
|
A12
|
3
|
Controlo E2 (máx.)
|
S
|
E2máx3
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,00E-07
|
|
P1
|
B10
|
1
|
Controlo E2 (IC50)
|
S
|
E2IC501
|
E2IC50 × 10
|
30
|
50
|
10
|
10
|
100
|
E2IC50
|
|
P1
|
B11
|
2
|
Controlo E2 (IC50)
|
S
|
E2IC502
|
E2IC50 × 10
|
30
|
50
|
10
|
10
|
100
|
E2IC50
|
|
P1
|
B12
|
3
|
Controlo E2 (IC50)
|
S
|
E2IC503
|
E2IC50 × 10
|
30
|
50
|
10
|
10
|
100
|
E2IC50
|
|
P1
|
C10
|
1
|
Controlo NE (máx.)
|
S
|
Nemáx1
|
1,00E-3,5
|
30
|
50
|
10
|
10
|
100
|
1,00E-4,5
|
|
P1
|
C11
|
2
|
Controlo NE (máx.)
|
S
|
Nemáx2
|
1,00E-3,5
|
30
|
50
|
10
|
10
|
100
|
1,00E-4,5
|
|
P1
|
C12
|
3
|
Controlo NE (máx.)
|
S
|
Nemáx3
|
1,00E-3,5
|
30
|
50
|
10
|
10
|
100
|
1,00E-4,5
|
|
P1
|
D10
|
1
|
Controlo NE (IC50)
|
S
|
NEIC501
|
NEIC50 × 10
|
30
|
50
|
10
|
10
|
100
|
NEIC50
|
|
P1
|
D11
|
2
|
Controlo NE (IC50)
|
S
|
NEIC502
|
NEIC50 × 10
|
30
|
50
|
10
|
10
|
100
|
NEIC50
|
|
P1
|
D12
|
3
|
Controlo NE (IC50)
|
S
|
NEIC503
|
NEIC50 × 10
|
30
|
50
|
10
|
10
|
100
|
NEIC50
|
|
P1
|
E10
|
1
|
E2 frio (elevado)
|
NSB
|
S1
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E11
|
2
|
E2 frio (elevado)
|
NSB
|
S2
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E12
|
3
|
E2 frio (elevado)
|
NSB
|
S3
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
F10
|
4
|
E2 frio (elevado)
|
NSB
|
S4
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
F11
|
5
|
E2 frio (elevado)
|
NSB
|
S5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
F12
|
6
|
E2 frio (elevado)
|
NSB
|
S6
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
G10
|
1
|
Ligação total
|
TB
|
TB1
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
G11
|
2
|
Ligação total
|
TB
|
TB2
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
G12
|
3
|
Ligação total
|
TB
|
TB3
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
H10
|
4
|
Ligação total
|
TB
|
TB4
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
H11
|
5
|
Ligação total
|
TB
|
TB5
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
H12
|
6
|
Ligação total
|
TB
|
TB6
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
Apêndice 4
CONSIDERAÇÕES PARA A ANÁLISE DE DADOS DO ENSAIO DE LIGAÇÃO COMPETITIVA HRER
|
1.
|
O ensaio de ligação competitiva hrERα mede a ligação de uma única concentração de [3H]17β-estradiol na presença de concentrações crescentes de um produto químico em estudo. A curva de ligação competitiva é representada como a ligação específica do [3H]17β-estradiol versus a concentração [log10(unidades)] do concorrente. A concentração do produto químico em estudo que inibe 50 % da ligação máxima específica do [3H]17β-estradiol é IC50.
|
Análise de dados para estrogénios de referência e ligante fraco (1)
|
2.
|
Os dados do controlo (isto é, a percentagem de ligação específica do [3H]17β-estradiol e o logaritmo da concentração do produto químico de controlo) são transformados para análise. As estimativas de log(IC50) para os controlos positivos (por exemplo, estrogénios de referência e ligantes fracos) devem ser determinadas com recurso a um software de ajustamento não linear de ajustamento adequado, a fim de se adaptar à equação de quatro parâmetros de Hill (p. ex., BioSoft, GraphPad Prism). Em geral, o topo, a base, a curva e o log(IC50) devem ser mantidos inalterados quando se ajustam as curvas. Quando se determinar a melhor adequação, deve utilizar-se uma regressão sólida, a menos que seja apresentada uma justificação. Deve ser indicado método escolhido para uma regressão sólida. Não foi necessária correção da depleção do ligando para os ensaios de FW ou hrER do CERI, mas tal pode ser considerado, se necessário. Após a análise inicial, cada curva de ligação deve ser avaliada de modo a assegurar a adequação ao modelo. A afinidade de ligação relativa (RBA) do ligando fraco pode ser calculada como percentagem do log(IC50) para o ligante fraco em relação ao log(IC50) para o 17β-estradiol. Os resultados dos controlos positivos e do controlo do não ligante devem ser avaliados utilizando medidas de desempenho do ensaio e critérios de aceitabilidade conforme descritos no presente método de ensaio (ponto 20), no apêndice 2 (ensaio FW, pontos 41-51) e no apêndice 3 (ensaio CERI, pontos 41 a 51). A figura 1 apresenta exemplos de 3 séries de ensaios para os estrogénios de referência e os ligantes fracos.
|
Figura 1
Exemplos de curvas de ligação competitiva para os estrogénios de referência e para o controlo do ligante fraco

Análise de dados relativos aos produtos químicos em estudo
|
3.
|
Os dados relativos a todos os produtos químicos em estudo devem ser analisados através de uma abordagem por etapas, para garantir que os dados são devidamente analisados e que cada ligação competitiva é devidamente classificada. Cada série de ensaios para um produto químico em estudo é inicialmente submetida a uma análise de dados normalizada idêntica à utilizada para os estrogénios de referência e para os controlos de ligantes fracos (ponto 55). Uma vez concluída, deve ser efetuada uma avaliação técnica dos parâmetros de ajustamento da curva, bem como uma análise visual do grau de adequação dos dados à curva de ligação competitiva gerada para cada série de ensaios. Durante esta avaliação técnica, as observações de uma diminuição dependente da concentração do [3H]17β-estradiol especificamente ligado, da baixa variabilidade entre os replicados técnicos em cada concentração do produto químico em estudo e da coerência dos parâmetros de ajustamento entre as três séries de ensaios constituem uma boa indicação de que a análise de ensaios e de dados foi realizada de forma adequada. Aquando da análise dos resultados de cada produto químico em estudo, devem ser utilizados critérios profissionais e os dados utilizados para classificar cada produto químico em estudo como ligante ou não ligante devem ser cientificamente justificados.
|
|
4.
|
Ocasionalmente, podem existir exemplos de dados que exijam uma atenção adicional na análise e interpretação adequada dos dados relativos à ligação do hrER. Estudos anteriores revelaram casos em que a análise e a interpretação de dados relativos à ligação competitiva ao recetor podem ser complicadas por uma mudança da percentagem específica de ligação no ensaio de produtos químicos com as concentrações mais elevadas (figura 2). Este é um problema bem conhecido, que ocorre quando se utilizam protocolos para uma série de ensaios de ligação competitiva ao recetor (3). Nestes casos, a resposta é dependente da concentração observada em concentrações mais baixas, mas, como a concentração do produto químico em estudo se aproxima do limite de solubilidade, o deslocamento do [3H]17β-estradiol não diminui. Nestes casos, os dados relativos às concentrações mais elevadas indicam que o limite biológico do ensaio foi atingido. Por exemplo, este fenómeno está muitas vezes associado à insolubilidade e à precipitação dos produtos químicos em concentrações elevadas, embora também possa ser um reflexo da capacidade excessiva do carvão revestido de dextrano para capturar o ligando com marcação radioativa livre durante o procedimento de separação em concentrações químicas mais elevadas. Manter os pontos de dados em que os dados sobre o ajustamento da ligação competitiva a uma curva sigmoide pode, por vezes, conduzir a uma classificação incorreta do potencial de ligação do ER a um produto químico em estudo (figura 2). Para evitar tal facto, o protocolo relativo aos ensaios de ligação de FW e de hrER do CERI prevê a opção de excluir da análise pontos de dados em que a média dos replicados para a percentagem de ligação específica do [3H]17β-estradiol seja superior em 10 % ou mais à média observada a uma concentração inferior (conhecida por «regra dos 10 %»). Esta regra apenas pode ser utilizada uma vez para uma curva determinada e deve haver dados remanescentes relativos a, pelo menos, 6 concentrações, de modo a que a curva possa ser corretamente classificada.
|
Figura 2
Exemplos de curvas de ligação competitiva com e sem recurso à regra dos 10 %
|
5.
|
A utilização adequada da regra dos 10 % para corrigir estas curvas deve ser cuidadosamente considerada e reservada para os casos em que exista uma forte indicação de uma ligante hrER. Durante a realização de experiências para o estudo de validação do ensaio de ligação hrER de FW, observou-se que a regra dos 10 % tinha por vezes uma consequência imprevista e inesperada. Os produtos químicos que não interagiam com o recetor (ou seja, os verdadeiros não ligantes) apresentavam frequentemente uma variabilidade de cerca de 100 % para a ligação radioativa que era superior a 10 % da gama de concentrações ensaiadas. Se o valor mais baixo correspondesse a uma concentração baixa, os dados de todas as concentrações mais elevadas poderiam potencialmente ser eliminados da análise, através da regra dos 10 %, mesmo que essas concentrações pudessem ser úteis para determinar se o produto químico era não ligante. A figura 3 mostra exemplos de situações em que a utilização da regra dos 10 % não é adequada.
|
Figura3
Exemplos, dados sobre a ligação competitiva em que não é adequada a aplicação da regra dos 10 %


Referências bibliográficas
|
(1)
|
OCDE (2015). Integrated Summary Report: Validation of Two Binding Assays Using Human Recombinant Estrogen Recetor Alpha (hrERα), Health and Safety Publications, Series on Testing and Assessment (No 226), Organisation for Economic Cooperation and Development, Paris.
|
|
(2)
|
Motulsky H. and Christopoulos A. (2003). The law of mass action, In Fitting Models to Biological Data Using Linear and Non-linear Regression. GraphPad Software Inc., San Diego, CA, pp 187-191. Www.graphpad.com/manuals/Prism4/RegressionBook.pdf
|
|
(3)
|
Laws SC, Yavanhxay S, Cooper RL, Eldridge JC. (2006). Nature of the Binding Interaction for 50 Structurally Diverse Chemicals with Rat Estrogen Recetors. Toxicological Sci. 94(1):46-56.
|
B.71 ENSAIOS DE SENSIBILIZAÇÃO CUTÂNEA IN VITRO RESPEITANTES A EVENTOS ESSENCIAIS DE ATIVAÇÃO DAS CÉLULAS DENDRÍTICAS NA VIA DETERMINANTE DOS EFEITOS NOCIVOS PARA A SENSIBILIZAÇÃO CUTÂNEA
INTRODUÇÃO GERAL
Método de ensaio baseado no evento essencial relativo à ativação das células dendríticas
|
1.
|
Um sensibilizante cutâneo é uma substância que provoca uma reação alérgica após contacto com a pele, conforme definido pelo Sistema Mundial Harmonizado de Classificação e Rotulagem de Produtos Químicos das Nações Unidas (GHS da ONU) (1) e o Regulamento (UE) n.o 1272/2008 relativo à classificação, rotulagem e embalagem de substâncias e misturas (CLP) (99). Existe consenso quanto aos principais eventos biológicos subjacentes à sensibilização cutânea. Os conhecimentos atuais sobre os mecanismos químicos e biológicos associados à sensibilização cutânea foram sintetizados na forma de uma via determinante dos efeitos nocivos (AOP – Adverse Outcome Pathway) no âmbito do programa AOP da OCDE (2), desde o evento molecular iniciador, passando pelos eventos intermédios, até ao efeito nocivo, designadamente a dermatite alérgica de contacto. Neste caso, o evento molecular iniciador (isto é, o primeiro evento essencial) é a ligação covalente das substâncias eletrófilas aos centros nucleófilos das proteínas da pele. O segundo evento essencial nesta AOP tem lugar nos queratinócitos e inclui respostas inflamatórias, bem como variações na expressão de genes associadas a vias de sinalização celular específicas, como as vias dependentes do elemento de resposta antioxidante/eletrófilo (ARE – Antioxidant Response Element). O terceiro evento essencial é a ativação das células dendríticas, normalmente avaliada com base na expressão de marcadores específicos da superfície celular (quimocinas e citocinas). O quarto evento essencial consiste na ativação e na proliferação de células T, que é avaliada indiretamente no ensaio de gânglios linfáticos locais com murídeos (LLNA – Local Lymph Node Assay) (3).
|
|
2.
|
O presente método de ensaio é equivalente à orientação de ensaio (TG — Test Guideline) 442E (2017) da OCDE. Descreve os ensaios in vitro respeitantes aos mecanismos descritos no evento essencial relativo à ativação das células dendríticas na AOP para a sensibilização cutânea (2). O método de ensaio inclui ensaios para apoiar a discriminação entre sensibilizantes e não sensibilizantes cutâneos, em conformidade com o GHS da ONU e com o CLP.
|
|
Os ensaios descritos no presente método de ensaio são os seguintes:
|
—
|
Teste de ativação da linha celular humana (h-CLAT)
|
|
—
|
Teste de ativação da linha celular U937 (U-SENS™)
|
|
—
|
Ensaio por gene repórter da interleucina-8 (ensaio IL-8 Luc)
|
|
|
|
3.
|
Os ensaios incluídos no presente método e as orientações de ensaio da OCDE correspondentes podem diferir em relação ao procedimento utilizado para gerar os dados e efetuar as medições, mas podem ser utilizados indiscriminadamente para satisfazer as exigências dos países quanto aos resultados dos ensaios sobre o evento essencial de ativação das células dendríticas na AOP para a sensibilização cutânea, beneficiando simultaneamente da aceitação mútua de dados da OCDE.
|
Antecedentes e princípios dos ensaios incluídos no método de ensaio baseado nos eventos essenciais
|
4.
|
Tradicionalmente, a determinação da sensibilização cutânea implica o recurso a animais de laboratório. Os métodos clássicos que utilizam porquinhos-da-índia — ensaio de maximização com porquinhos-da-índia (GMPT) de Magnusson e Kligman e ensaio de Buehler (TM B.6) (4) — avaliam as fases de indução e de desencadeamento da sensibilização cutânea. Vulgarizaram-se também os ensaios com murídeos, o LLNA (TM B.42) (3) e duas alterações deste sem recurso a isótopos radioativos — LLNA: DA (TM B.50) (5) e LLNA: BrdU-ELISA (TM B.51) (6) —, que determinam exclusivamente a resposta indutiva, uma vez que apresentam vantagem em relação aos ensaios com porquinhos-da-índia no tocante ao bem-estar animal e proporcionam uma medição objetiva da fase de indução da sensibilização cutânea.
|
|
5.
|
Mais recentemente, foram adotados métodos de ensaio de tipo mecanístico in chemico e in vitro respeitantes ao primeiro evento essencial (TM B.59; ensaio de reatividade direta de péptidos (7)) e ao segundo evento essencial [TM B.60; método ARE-Nrf2 luciferase (8)] da AOP de sensibilização cutânea, a fim de contribuir para a avaliação do potencial de perigo de sensibilização cutânea dos produtos químicos.
|
|
6.
|
Os ensaios descritos no presente método de ensaio permitem quantificar quer as variações na expressão do(s) marcador(es) da superfície celular associada(s) ao processo de ativação dos monócitos e das células dendríticas após a exposição a um sensibilizante (por exemplo, CD54, CD86), quer as variações na expressão da IL-8, uma citocina associada à ativação das células dendríticas. Existem referências de que os sensibilizantes cutâneos induzem a expressão de marcadores da membrana celular, nomeadamente CD40, CD54, CD80, CD83 e CD86, para além de induzirem citocinas pró-inflamatórias, tais como a IL-1β e a TNF-α, e várias quimocinas, entre as quais a IL-8 (CXCL8) e a CCL3 (9) (10) (11) (12), associados à ativação das células dendríticas (2).
|
|
7.
|
No entanto, como a ativação das células dendríticas representa apenas um evento essencial da AOP de sensibilização cutânea (2) (13), as informações geradas pelos ensaios que medem unicamente os marcadores da ativação das células dendríticas podem não bastar para concluir a existência ou ausência de potencial de sensibilização cutânea dos produtos químicos. Por conseguinte, propõem-se os dados gerados com os ensaios descritos no presente método de ensaio para ajudar a distinguir os sensibilizantes (ou seja, categoria 1 do GHS da ONU/CLP) dos não sensibilizantes cutâneos utilizados no âmbito das abordagens integradas de ensaio e avaliação (IATA — Integrated Approaches to Testing and Assessment), juntamente com outras informações complementares decorrentes, por exemplo, de ensaios in vitro respeitantes a outros eventos essenciais da AOP de sensibilização cutânea, bem como métodos sem ensaios, nomeadamente a interpolação com base em dados relativos a produtos químicos análogos (13). Encontram-se na literatura exemplos de utilização dos dados obtidos com estes ensaios no âmbito de abordagens definidas – ou seja, abordagens normalizadas no que diz respeito ao conjunto de fontes de informação utilizadas e aos procedimentos de análise dos dados para efeitos de previsão (13) –, que podem ser elementos úteis no âmbito das IATA.
|
|
8.
|
Os ensaios descritos no presente método não podem ser utilizados isoladamente, nem para classificar os sensibilizantes cutâneos nas subcategorias 1A e 1B do GHS da ONU/CLP pelas autoridades responsáveis pela aplicação destas duas subcategorias facultativas, nem para prever o potencial de sensibilização no contexto de avaliações de segurança. Todavia, consoante o quadro regulamentar, os resultados positivos obtidos com estes métodos podem ser utilizados isoladamente para classificar um produto químico na categoria 1 do GHS da ONU/CLP.
|
|
9.
|
No presente método de ensaio, o termo «produto químico em estudo» designa o produto que é objeto do ensaio (100) e não as substâncias monocomponentes, substâncias multicomponentes e/ou misturas às quais os métodos sejam aplicáveis. Atualmente, dispõe-se de informações limitadas sobre a aplicabilidade dos ensaios a substâncias multicomponentes/misturas (14) (15). Os ensaios são, porém, tecnicamente aplicáveis ao ensaio de substâncias multicomponentes e misturas. Contudo, antes da aplicação do presente método de ensaio a uma mistura para se obterem dados para efeitos regulamentares, importa ponderar se — e, em caso afirmativo, por que motivo — o método pode proporcionar resultados adequados para o efeito (101). Tais considerações não são necessárias se houver um requisito regulamentar para o ensaio da mistura. Acresce que, no caso de ensaios de substâncias multicomponentes ou de misturas, deve ser tida em conta a possível interferência de componentes citotóxicos com as respostas observadas.
|
REFERÊNCIAS BIBLIOGRÁFICAS
|
(1)
|
United Nations UN (2015). Globally Harmonized System of Classification and Labelling of Chemicals (GHS). Sixth revised edition. Nova Iorque e Genebra: United Nations Publications. ISBN: 978-92-1-117006-1. Disponível em: https://www.unece.org/trans/danger/publi/ghs/ghs_rev06/06files_e.html.
|
|
(2)
|
OECD (2012). The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins. Part 1: Scientific Evidence. Series on Testing and Assessment No. 168. Disponível em: http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=ENV/JM/MONO(2012)10/PART1&docLanguage=En.
|
|
(3)
|
Capítulo B.42 do presente anexo: Ensaio de gânglios linfáticos locais.
|
|
(4)
|
Capítulo B.6 do presente anexo: Sensibilização cutânea.
|
|
(5)
|
Capítulo B.50 do presente anexo: Sensibilização cutânea: ensaio de gânglios linfáticos locais: DA.
|
|
(6)
|
Capítulo B.51 do presente anexo: Sensibilização cutânea: ensaio de gânglios linfáticos locais: BrdU-ELISA.
|
|
(7)
|
Capítulo B.59 do presente anexo: Sensibilização cutânea in chemico: ensaio de reatividade direta de péptidos (DPRA).
|
|
(8)
|
Capítulo B.60 do presente anexo: Sensibilização cutânea in vitro: método ARE-Nrf2 luciferase.
|
|
(9)
|
Steinhman RM. (1991). The dendritic cell system and its role in immunogenicity. Annu Rev Immunol 9:271-96.
|
|
(10)
|
Caux C, Vanbervliet B, Massacrier C, Azuma M, Okumura K, Lanier LL, and Banchereau J. (1994). B70/B7-2 is identical to CD86 and is the major functional ligand for CD28 expressed on human dendritic cells. J Exp Med 180:1841-7.
|
|
(11)
|
Aiba S, Terunuma A, Manome H, and Tagami H. (1997). Dendritic cells differently respond to haptens and irritants by their production of cytokines and expression of co-stimulatory molecules. Eur J Immunol 27:3031-8.
|
|
(12)
|
Aiba S, Manome H, Nakagawa S, Mollah ZU, Mizuashi M, Ohtani T, Yoshino Y, and Tagami. H. (2003). p38 mitogen-activated protein kinase and extracellular signal-regulated kinases play distinct roles in the activation of dendritic cells by two representative haptens, NiCl2 and DNCB. J Invest Dermatol 120:390-8.
|
|
(13)
|
OECD (2016). Series on Testing & Assessment No 256: Guidance Document On The Reporting Of Defined Approaches And Individual Information Sources To Be Used Within Integrated Approaches To Testing And Assessment (IATA) For Skin Sensitisation, Annex 1 and Annex 2. ENV/JM/HA(2016)29. Organisation for Economic Cooperation and Development, Paris. Disponível em: https://community.oecd.org/community/iatass.
|
|
(14)
|
Ashikaga T, Sakaguchi H, Sono S, Kosaka N, Ishikawa M, Nukada Y, Miyazawa M, Ito Y, NishiyamaN, Itagaki H. (2010). A comparative evaluation of in vitro skin sensitisation tests: the human cell-line activation test (h-CLAT) versus the local lymph node assay (LLNA). Altern. Lab. Anim. 38, 275-284.
|
|
(15)
|
Piroird, C., Ovigne, J.M., Rousset, F., Martinozzi-Teissier, S., Gomes, C., Cotovio, J., Alépée, N. (2015). The Myeloid U937 Skin Sensitization Test (U-SENS) addresses the activation of dendritic cell event in the adverse outcome pathway for skin sensitization. Toxicol. In Vitro 29, 901-916.
|
Apêndice 1
SENSIBILIZAÇÃO CUTÂNEA IN VITRO: TESTE DE ATIVAÇÃO DA LINHA CELULAR HUMANA (H-CLAT)
CONSIDERAÇÕES INICIAIS E LIMITAÇÕES
|
1.
|
O método de ensaio h-CLAT permite quantificar as variações na expressão dos marcadores da superfície celular associadas ao processo de ativação dos monócitos e das células dendríticas (ou seja, CD86 e CD54), na linha celular da leucemia monocítica humana THP-1, após a exposição a sensibilizantes (1) (2). Os níveis de expressão medidos para os marcadores da superfície celular CD86 e CD54 são, em seguida, utilizados para ajudar a distinguir os sensibilizantes dos não sensibilizantes cutâneos.
|
|
2.
|
O método de ensaio h-CLAT foi avaliado num estudo de validação coordenado pelo Laboratório de Referência da União Europeia para as Alternativas à Experimentação em Animais (EURL ECVAM), seguido de uma análise independente por pares do Comité Científico Consultivo do EURL ECVAM (ESAC). Tendo em conta todos os dados disponíveis e os contributos das entidades reguladoras e das partes interessadas, o método foi recomendado pelo EURL ECVAM (3) para utilização no âmbito de uma IATA, com o objetivo de ajudar a distinguir os sensibilizantes dos não sensibilizantes para efeitos de classificação e rotulagem de perigos. As referências bibliográficas contêm exemplos da utilização de dados h-CLAT combinados com outras informações (4) (5) (6) (7) (8) (9) (10) (11).
|
|
3.
|
O método de ensaio h-CLAT demonstrou ser transferível para laboratórios com experiência em técnicas de cultura celular e análise por citometria de fluxo. O nível de reprodutibilidade esperado das previsões decorrentes do ensaio é da ordem dos 80 %, tanto intralaboratorial como interlaboratorial (3) (12). Os resultados do estudo de validação (13) e de outros estudos publicados (14) indicam, em termos gerais, que, comparativamente com os resultados obtidos pelo método LLNA, a precisão na distinção entre sensibilizantes cutâneos (cat. 1 do GHS da ONU/CLP) e não sensibilizantes é de 85 % (N=142), com uma sensibilidade de 93 % (94/101) e uma especificidade de 66 % (27/41), a partir de uma nova análise do EURL ECVAM (12), tendo em conta todos os dados existentes, mas excluindo os resultados negativos obtidos para produtos químicos com coeficiente de partição octanol-água (log P) superior a 3,5, conforme descrito no ponto 4. É mais provável que os falsos resultados negativos nas previsões efetuadas com o método h-CLAT digam respeito a produtos químicos com potencial de sensibilização cutânea baixo a moderado (subcategoria 1B do GHS da ONU/CLP) do que a produtos químicos com um potencial de sensibilização cutânea elevado (subcategoria 1A do GHS da ONU/CLP) (4) (13) (15). No seu conjunto, estas informações demonstram a utilidade do método h-CLAT para a identificação dos perigos de sensibilização cutânea. No entanto, os valores relativos à precisão do método de ensaio h-CLAT utilizado isoladamente são meramente indicativos, uma vez que o presente método deve ser combinado com outras fontes de informação no contexto de uma IATA e em conformidade com o disposto nos pontos 7 e 8 da Introdução Geral. Além disso, na avaliação de métodos de estudo da sensibilização cutânea que não utilizem animais, deve ter-se em mente que o método LLNA, tal como outros ensaios que utilizam animais, pode não refletir de forma adequada a situação no ser humano.
|
|
4.
|
Os dados atualmente disponíveis demonstram que o método h-CLAT é aplicável a produtos químicos em estudo que abrangem uma grande diversidade de grupos funcionais orgânicos, mecanismos de reação, potências de sensibilização cutânea (determinadas em estudos in vivo) e propriedades físico-químicas (3) (14) (15). O método é aplicável a produtos químicos solúveis ou que formem uma dispersão estável – ou seja, um coloide ou uma suspensão em que o produto em estudo não se deposite nem se separe do solvente/veículo formando diferentes fases – num solvente/veículo adequado (ver ponto 14). Os produtos químicos com log P superior a 3,5 tendem a produzir resultados negativos falsos (14). Por conseguinte, os resultados negativos obtidos com produtos químicos em estudo que apresentem um log P superior a 3,5 não devem ser tidos em conta. No entanto, os resultados positivos obtidos com produtos químicos em estudo que apresentem um log P superior a 3,5 poderão ser utilizados para ajudar a identificar o produto químico em estudo como sensibilizante cutâneo. Além disso, devido à limitada capacidade metabólica da linha celular utilizada (16) e às condições experimentais, os pró-haptenos (substâncias que requerem ativação enzimática, por exemplo através das enzimas P450) e os pré-haptenos (substâncias ativadas por oxidação), em particular os que apresentem uma taxa de oxidação lenta, podem também produzir resultados negativos no h-CLAT (15). É possível avaliar produtos químicos fluorescentes com o h-CLAT (17). No entanto, os produtos fortemente fluorescentes que emitem no mesmo comprimento de onda que o isotiocianato de fluoresceína (FITC) ou que o iodeto de propídio (IP) interferem com a deteção por citometria de fluxo, pelo que não podem ser corretamente avaliados utilizando anticorpos conjugados com FITC ou IP. Neste caso, é possível recorrer, respetivamente, a outros anticorpos marcados com fluorocromos ou a outros marcadores de citotoxicidade, desde que seja possível demonstrar que produzem resultados semelhantes aos dos anticorpos marcados com FITC (ver ponto 24) ou com IP (ver ponto 18), por exemplo testando as substâncias para demonstração de competência indicadas nos apêndices 1-2. À luz do que precede, os resultados negativos devem ser interpretados no contexto das limitações indicadas e em conjunto com outras fontes de informação no âmbito da IATA. Nos casos em que se demonstre que o método h-CLAT não é aplicável a outras categorias específicas de produtos químicos em estudo, o mesmo não deve ser utilizado para essas categorias específicas.
|
|
5.
|
Tal como descrito acima, o método h-CLAT ajuda a distinguir os sensibilizantes cutâneos dos não sensibilizantes. Porém, quando utilizado no âmbito de abordagens integradas do tipo IATA, pode também contribuir para a avaliação do potencial de sensibilização (4) (5) (9). Contudo, é necessário continuar a trabalhar, de preferência com base em dados humanos, para determinar o modo como os resultados do h-CLAT poderão vir a ser úteis para este tipo de avaliação.
|
|
6.
|
São apresentadas definições no apêndice 1.1.
|
PRINCÍPIO DO ENSAIO
|
7.
|
O método h-CLAT é um ensaio in vitro que permite quantificar as variações da expressão de marcadores (CD86 e CD54) da superfície das células da linha celular da leucemia monocítica humana THP-1, após uma exposição de 24 horas ao produto químico em estudo. Estas moléculas de superfície são marcadores típicos da ativação dos monócitos THP-1 e podem imitar a ativação das células dendríticas, o que desempenha um papel crítico na ativação das células T. As variações na expressão dos marcadores de superfície são medidas por citometria de fluxo após coloração das células com anticorpos marcados com fluorocromo. A citotoxicidade é medida em paralelo, a fim de determinar se a ativação da expressão dos marcadores de superfície ocorre a concentrações subcitotóxicas. A intensidade relativa de fluorescência dos marcadores de superfície, em comparação com a do controlo do solvente/veículo, é calculada e utilizada no modelo preditivo (ver ponto 26), para ajudar a distinguir os sensibilizantes dos não sensibilizantes.
|
DEMONSTRAÇÃO DE COMPETÊNCIA
|
8.
|
Antes de utilizarem, por rotina, o ensaio descrito no presente apêndice do método de ensaio B.71, os laboratórios devem demonstrar a sua competência utilizando as dez substâncias enumeradas no apêndice 1.2. Além disso, os utilizadores do ensaio devem manter uma base de dados históricos resultantes dos controlos de reatividade (ver ponto 11) e obtidos com o controlo positivo e com o controlo do solvente/veículo (ver pontos 20 a 22) e devem utilizar esses dados para confirmar que a reprodutibilidade do método de ensaio no seu laboratório se mantém ao longo do tempo.
|
PROCEDIMENTO
|
9.
|
O presente método de ensaio baseia-se no protocolo n.o 158 do serviço de base de dados sobre os métodos alternativos à experimentação em animais (DB-ALM — DataBase service on ALternative Methods to animal experimentation) h-CLAT (18), que é o protocolo utilizado para o estudo de validação coordenado pelo EURL ECVAM. Recomenda-se a utilização deste protocolo aquando da implementação e utilização, no laboratório, do método h-CLAT. Segue-se uma descrição dos principais elementos e procedimentos do método h-CLAT, que compreende duas etapas: um ensaio de determinação da dose e uma medição da expressão de CD86/CD54.
|
Preparação das células
|
10.
|
Para a execução do método h-CLAT, deve utilizar-se a linha celular da leucemia monocítica humana, THP-1. Recomenda-se que as células (TIB-202™) sejam obtidas junto de um banco de células reconhecido, como a American Type Culture Collection.
|
|
11.
|
As células THP-1 são cultivadas a 37°C numa atmosfera humidificada a 5 % CO2, em meio RPMI-1640 complementado com 10 % de soro bovino fetal (SBF), 0,05 mM de 2-mercaptoetanol, 100 unidades/ml de penicilina e 100 μg/ml de estreptomicina. A utilização da penicilina e da estreptomicina no meio de cultura pode ser evitada. No entanto, nesse caso, os utilizadores devem verificar se a ausência de antibióticos no meio de cultura não tem qualquer impacto nos resultados, por exemplo testando as substâncias enumeradas no apêndice 1.2. De qualquer modo, para minimizar o risco de contaminação, devem seguir-se as boas práticas de cultura celular independentemente da presença ou não de antibióticos no meio de cultura celular. As células THP-1 são inoculadas regularmente a cada 2-3 dias, a uma densidade compreendida entre 0,1 e 0,2 × 106 células/ml, e devem ser mantidas a uma densidade compreendida entre 0,1 e 1,0 × 106 células/ml. Antes da sua utilização para o ensaio, as células devem ser qualificadas através de um controlo de reatividade. Este controlo deve ser realizado duas semanas após a descongelação, utilizando como controlos positivos o 2,4-dinitroclorobenzeno (DNCB) (n.o CAS 97-00-7, pureza ≥ 99 %) e o sulfato de níquel (NiSO4) (n.o CAS 10101-97-0, pureza ≥ 99 %) e como controlo negativo o ácido láctico (AL) (n.o CAS 50-21-5, pureza ≥ 85 %). Tanto o DNCB como o NiSO4 devem dar respostas positivas para os marcadores de superfície celular CD86 e CD54; o ácido láctico deve dar uma resposta negativa para os marcadores de superfície celular CD86 e CD54. Apenas serão utilizadas para o ensaio as células que tenham passado o controlo de reatividade. As células podem ser propagadas até dois meses após a descongelação, sem ultrapassar as 30 passagens. O controlo de reatividade deve ser efetuado de acordo com os procedimentos descritos nos pontos 20 a 24.
|
|
12.
|
Para o ensaio, as células THP-1 são inoculadas a uma densidade de 0,1 × 106 células/ml ou 0,2 × 106 células/ml e pré-cultivadas em frascos de cultura durante 72 horas ou 48 horas, respetivamente. É importante que a densidade celular no frasco de cultura imediatamente após o período de pré-cultura seja o mais coerente possível em cada experiência (utilizando uma das duas condições de pré-cultura acima descritas), uma vez que a densidade celular no frasco de cultura imediatamente após a pré-cultura pode influenciar a expressão de CD86/CD54 induzida por alergénios (19). No dia do ensaio, as células colhidas do frasco de cultura são novamente colocadas em suspensão num meio de cultura fresco a uma densidade de 2 × 106 células/ml. Em seguida, distribuem-se as células numa placa de 24 poços de fundo plano (500 μl, 1 × 106 células/poço) ou numa placa de 96 poços de fundo plano (80 μl, 1,6 × 105 células/poço).
|
Ensaio de determinação da dose
|
13.
|
Efetua-se um ensaio de determinação da dose para apurar o valor CV75, ou seja, a concentração do produto químico em estudo que resulta numa viabilidade celular (CV) de 75 %, em comparação com o controlo do solvente/veículo. O valor CV75 é utilizado para determinar a concentração de produtos químicos em estudo a utilizar para a medição da expressão de CD86/CD54 (ver pontos 20 a 24).
|
Preparação dos produtos químicos em estudo e das substâncias de controlo
|
14.
|
Os produtos químicos em estudo e as substâncias de controlo são preparados no dia do ensaio. No caso do método h-CLAT, os produtos químicos em estudo são dissolvidos ou dispersos de forma estável (ver também o ponto 4) escolhendo de preferência como solvente/veículo uma solução salina ou o meio ou, como segunda opção, dimetilsulfóxido (DMSO, pureza ≥ 99 %), se o produto químico não for solúvel ou não formar uma dispersão estável nos dois solventes/veículos anteriores, até concentrações finais de 100 mg/ml (solução salina ou meio) ou de 500 mg/ml (DMSO). É possível utilizar solventes/veículos diferentes dos acima descritos, desde que a escolha seja suficientemente fundamentada do ponto de vista científico. Deve ter-se em conta a estabilidade do produto químico em estudo no solvente/veículo final.
|
|
15.
|
A partir das soluções-mãe dos produtos químicos em estudo a 100 mg/ml (em solução salina ou meio) ou a 500 mg/ml (em DMSO), devem efetuar-se as diluições seguintes:
|
—
|
Se o solvente/veículo for a solução salina ou o meio: preparam-se oito soluções-mãe (oito concentrações) através de uma série de diluições de fator 2 utilizando o solvente/veículo correspondente. Estas soluções-mãe são depois novamente diluídas por um fator de 50 no meio de cultura (soluções de trabalho). Se a concentração final máxima de 1 000 μg/ml na placa não for tóxica, a concentração máxima deve ser novamente determinada através da realização de um novo ensaio de citotoxicidade. A concentração final na placa não deve exceder 5 000 μg/ml para os produtos químicos em estudo dissolvidos ou em dispersão estável na solução salina ou no meio.
|
|
—
|
Se o solvente/veículo for o DMSO: preparam-se oito soluções-mãe (oito concentrações) através de uma série de diluições de fator 2 utilizando o solvente/veículo correspondente. Estas soluções-mãe são depois novamente diluídas por um fator de 250 no meio de cultura (soluções de trabalho). A concentração final na placa, mesmo que não seja tóxica, não deve exceder 1 000 μg/ml.
|
Para a exposição, utilizam-se, por fim, as soluções de trabalho, adicionando o mesmo volume de cada uma destas ao volume de células THP-1 em suspensão na placa (ver também o ponto 17), a fim de obter uma diluição suplementar de fator 2 (em geral, as concentrações finais na placa encontram-se no intervalo entre 7,81-1 000 μg/ml).
|
|
16.
|
O controlo do solvente/veículo utilizado no método h-CLAT é o meio de cultura (para os produtos químicos em estudo dissolvidos ou em dispersão estável (ver ponto 4) no meio ou na solução salina) ou o DMSO (para os produtos químicos em estudo dissolvidos ou em dispersão estável no DMSO) a uma concentração final única de 0,2 % na placa. Efetua-se uma diluição idêntica à descrita para as soluções de trabalho no ponto 15.
|
Aplicação dos produtos químicos em estudo e das substâncias de controlo
|
17.
|
O meio de cultura ou as soluções de trabalho, que se descrevem nos pontos 15 e 16, misturam-se na proporção 1:1 (v/v) com as suspensões celulares preparadas na placa de 24 poços ou de 96 poços de fundo plano (ver ponto 12). As placas tratadas são, em seguida, incubadas durante 24 ± 0,5 horas a 37°C, 5 % CO2. Deve ter-se o cuidado de evitar a evaporação dos produtos químicos em estudo, caso sejam voláteis, e a contaminação cruzada entre poços pelos referidos produtos, por exemplo selando a placa antes da incubação com os produtos químicos em estudo (20).
|
Coloração com iodeto de propídio (IP)
|
18.
|
Após 24 ± 0,5 horas de exposição, as células são transferidas para tubos de ensaio e recolhidas por centrifugação. Rejeitam-se os sobrenadantes e as restantes células são novamente colocadas em suspensão em 200 μl (no caso da placa de 96 poços) ou 600 μl (no caso da placa de 24 poços) de um tampão fosfato salino contendo 0,1 % de albumina de soro bovino (tampão de coloração). Transferem-se 200 μl da suspensão de células para uma placa de 96 poços de fundo redondo (no caso da placa de 96 poços) ou para um microtubo (no caso da placa de 24 poços); em seguida, as células são lavadas duas vezes com 200 μl (no caso da placa de 96 poços) ou 600 μl (no caso da placa de 24 poços) de tampão de coloração. Por último, as células são novamente suspensas no tampão de coloração (por exemplo, 400 μl), sendo acrescentada uma solução de IP (por exemplo, 20 μl) (para obter, por exemplo, uma concentração final de IP de 0,625 μl/ml). Podem utilizar-se outros marcadores de citotoxicidade, tais como a 7-aminoactinomicina D (7-AAD), o azul de Trypan ou outros, desde que se possa demonstrar que produzem resultados semelhantes ao IP, por exemplo testando as substâncias enumeradas no apêndice 1.2.
|
Medição da citotoxicidade por citometria de fluxo e estimativa do valor CV75
|
19.
|
O nível de fixação do IP é analisado por citometria de fluxo no canal FL-3. No total, são analisadas 10 000 células vivas (IP negativo). A viabilidade celular pode ser calculada pelo programa de análise do citómetro utilizando a equação indicada em baixo. Se a viabilidade celular for baixa, devem ser adquiridas, no máximo, 30 000 células, incluindo células mortas. Uma outra opção consiste em adquirir os dados durante um minuto após o início da análise.
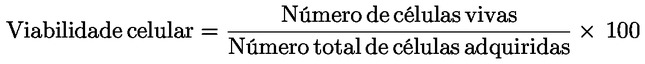
O valor CV75 (ver ponto 13), isto é, a concentração que provoca 75 % de sobrevivência das células THP-1 (25 % de citotoxicidade), é calculado por interpolação linear logarítmica utilizando a seguinte equação:

em que:
a é o valor mínimo de viabilidade celular superior a 75 %
c é o valor máximo de viabilidade celular inferior a 75 %
b e d são as concentrações que provocam os valores de viabilidade celular a e c, respetivamente
Podem utilizar-se outras abordagens para calcular o CV75, desde que fique demonstrado que tal não tem qualquer impacto nos resultados (por exemplo, testando as substâncias para demonstração de competência).
|
Medição da expressão de CD86/CD54
Preparação dos produtos químicos em estudo e das substâncias de controlo
|
20.
|
Utiliza-se o solvente/veículo adequado (solução salina, meio ou DMSO; ver o ponto 14) para dissolver os produtos químicos em estudo ou para criar uma dispersão estável. Os produtos químicos em estudo são primeiro diluídos até uma concentração correspondente a 100 vezes (na solução salina ou no meio) ou 500 vezes (no DMSO) o valor CV75 × 1,2 determinado no ensaio de determinação da dose (ver ponto 19). Se o CV75 não puder ser determinado (ou seja, se a citotoxicidade observada no ensaio de determinação da dose for insuficiente), deve utilizar-se como concentração inicial a concentração solúvel ou em dispersão estável mais elevada do produto químico em estudo preparado com cada solvente/veículo. Note-se que a concentração final na placa não deve exceder 5 000 μg/ml (no caso da solução salina ou do meio) ou 1 000 μg/ml (no caso do DMSO). Seguidamente, são realizadas diluições em série de fator 1:2 utilizando o solvente/veículo correspondente para obter as soluções-mãe (oito concentrações entre 100 × 1,2 × CV75 e 100×0,335 × CV75 (na solução salina ou no meio) ou entre 500×1,2 × CV75 e 500×0,335 × CV75 (no DMSO)), que serão testadas de acordo com o método h-CLAT (ver o protocolo DB-ALM n.o 158 para um exemplo do esquema de dosagem). As soluções-mãe são depois novamente diluídas por um fator de 50 (solução salina ou meio) ou de 250 (DMSO) no meio de cultura (soluções de trabalho). Estas soluções de trabalho são finalmente utilizadas para exposição, após uma diluição final de fator 2 na placa. Se os resultados não satisfizerem os critérios de aceitação descritos nos pontos 29 e 30 quanto à viabilidade celular, pode repetir-se o ensaio de determinação da dose para deduzir um valor CV75 mais preciso. Note-se que apenas podem ser utilizadas placas de 24 poços para a medição da expressão de CD86/CD54.
|
|
21.
|
O controlo do solvente/veículo é preparado conforme descrito no ponto 16. O controlo positivo utilizado no método h-CLAT é o DNCB (ver ponto 11), para o qual são preparadas soluções-mãe em DMSO e diluídas tal como descrito para as soluções-mãe no ponto 20. O DNCB deve ser utilizado como controlo positivo para a medição da expressão de CD86/CD54 a uma concentração final única na placa (geralmente 4,0 μg/ml). Para obter uma concentração de 4,0 μg/ml de DNCB na placa, prepara-se uma solução-mãe de 2 mg/ml de DNCB em DMSO, que é depois novamente diluída por um fator de 250 no meio de cultura, até se obter uma solução de trabalho a 8 μg/ml. Em alternativa, pode utilizar-se o valor CV75 do DNCB, determinado em cada instalação de ensaio, como concentração do controlo positivo. Podem utilizar-se outros controlos positivos adequados se houver dados históricos que permitam definir critérios de aceitação comparáveis para a execução. Para os controlos positivos, a concentração final única na placa não deve exceder 5 000 μg/ml (no caso da solução salina ou meio) ou 1 000 μg/ml (no caso do DMSO). Os critérios de aceitação para a execução são idênticos aos descritos para o produto químico em estudo (ver ponto 29), exceto no que diz respeito ao último critério, uma vez que o controlo positivo é testado a uma concentração única.
|
Aplicação dos produtos químicos em estudo e das substâncias de controlo
|
22.
|
Para obter uma previsão, é necessária uma experiência para cada produto químico em estudo e para cada substância de controlo. Cada experiência consiste em pelo menos duas execuções independentes para a medição de expressão de CD86/CD54 (ver pontos 26 a 28). Cada execução independente é efetuada num dia diferente ou no mesmo dia, desde que, para cada execução: a) sejam preparadas soluções-mãe e de trabalho frescas, independentes, do produto químico em estudo e soluções de anticorpos, e b) sejam utilizadas células colhidas em dois tempos independentes (provenientes de frascos de cultura diferentes); no entanto, as células podem ser provenientes da mesma passagem. Os produtos químicos em estudo e as substâncias de controlo preparados como soluções de trabalho (500 μl) são misturados com 500 μl de suspensão de células (1x106 células) a um rácio de 1:1 e as células são incubadas durante 24 ± 0,5 horas, conforme descrito nos pontos 20 e 21. Em cada execução, basta um único replicado para cada concentração do produto química em estudo e da substância de controlo, dado que a previsão é obtida a partir de, pelo menos, duas execuções independentes.
|
Coloração celular e análise
|
23.
|
Após 24 ± 0,5 horas de exposição, as células são transferidas da placa de 24 poços para tubos de ensaio e recolhidas por centrifugação, antes de serem lavadas duas vezes com 1 ml de tampão de coloração (se necessário, podem ser efetuadas etapas de lavagem suplementares). Após a lavagem, as células são bloqueadas com 600 μl de solução de bloqueio [tampão de coloração com 0,01 % (m/v) de globulina (fração Cohn II, III, humana; SIGMA, #G2388-10G ou equivalente)] e incubadas a 4oC durante 15 minutos. Após o bloqueio, as células são divididas em três alíquotas de 180 μl numa placa de 96 poços de fundo redondo ou num microtubo.
|
|
24.
|
Após centrifugação, as células são coradas a 4oC durante 30 minutos com 50 μl de anticorpos marcados com FITC anti-CD86, anti-CD54 ou de ratinho (isótipo) IgG1. Os anticorpos descritos no protocolo DB-ALM n.o 158 h-CLAT (18) devem ser diluídos no tampão de coloração a um rácio de 3:25 v/v [para CD86 (BD-PharMingen, #555657; Clone: Fun-1) ou de 3:50 v/v (para CD54 (DAKO, #F7143; Clone: 6.5B5) e IgG1 (DAKO, #X0927)]. Estes rácios de diluição dos anticorpos foram definidos aquando do desenvolvimento do ensaio como aqueles que apresentam a melhor relação sinal/ruído. A experiência adquirida aquando do desenvolvimento do ensaio indica que a intensidade da fluorescência dos anticorpos é geralmente coerente entre diferentes lotes. No entanto, os utilizadores podem ponderar a possibilidade de proceder à titulação dos anticorpos em condições laboratoriais próprias, a fim de definir as concentrações mais adequadas. Podem ser utilizados anticorpos anti-CD86 e/ou anti-CD54 marcados por outros fluorocromos, desde que seja possível demonstrar que produzem resultados semelhantes aos anticorpos conjugados por FITC, por exemplo testando as substâncias enumeradas no apêndice 1.2. Note-se que a alteração do clone ou do fornecedor dos anticorpos, tal como descrito no protocolo DB-ALM n.o 158 h-CLAT (18), pode afetar os resultados. Após terem sido lavadas duas ou mais vezes com 150 μl de tampão de coloração, as células são novamente colocadas em suspensão no tampão de coloração (por exemplo, 400 μl), sendo acrescentada a solução de IP (por exemplo, 20 μl para obter uma concentração final de 0,625 μg/ml) ou um outro marcador de citotoxicidade (ver ponto 18). Os níveis de expressão de CD86 e de CD54 e a viabilidade celular são analisados por citometria de fluxo.
|
DADOS E RELATÓRIOS
Avaliação dos dados
|
25.
|
O nível de expressão de CD86 e CD54 é analisado por citometria de fluxo com o canal de aquisição FL-1. Com base na média geométrica da intensidade de fluorescência (IMF, intensidade média de fluorescência), a intensidade relativa de fluorescência (IRF) de CD86 e de CD54 para as células com controlo (ctrl) positivo e para as células tratadas com o produto químico é calculada utilizando a seguinte equação:

A viabilidade celular das células de controlo (ctrl) do isótipo [coradas com anticorpos de ratinho IgG1 (isótipo)] também é calculada de acordo com a equação descrita no ponto 19.
|
Modelo preditivo
|
26.
|
Para determinar a expressão de CD86/CD54, cada produto químico em estudo é testado em, pelo menos, duas execuções independentes para obter uma previsão única (POSITIVA ou NEGATIVA). Uma previsão por h-CLAT é considerada POSITIVA se for preenchida pelo menos uma das seguintes condições em duas de duas ou em pelo menos duas de três execuções independentes; caso contrário, a previsão por h-CLAT é considerada NEGATIVA (figura 1):
|
—
|
A IRF de CD86 é igual ou superior a 150 % a qualquer concentração testada (com viabilidade celular ≥ 50 %);
|
|
—
|
A IRF de CD54 é igual ou superior a 200 % a qualquer concentração testada (com viabilidade celular ≥ 50 %).
|
|
|
27.
|
Atendendo ao que precede, se as duas primeiras execuções forem ambas positivas para CD86 e/ou forem ambas positivas para CD54, a previsão por h-CLAT é considerada POSITIVA e não é necessário efetuar uma terceira execução. Do mesmo modo, se as duas primeiras execuções forem negativas para ambos os marcadores, a previsão por h-CLAT é considerada NEGATIVA (tendo devidamente em conta o disposto no ponto 30), não sendo necessário efetuar uma terceira execução. Se, no entanto, as duas primeiras execuções não forem concordantes em relação a pelo menos um dos marcadores (CD54 ou CD86), é necessário realizar uma terceira execução e a previsão final basear-se-á no resultado obtido na maioria das três execuções individuais (ou seja, dois de três). A este respeito, importa notar que, se forem efetuadas duas execuções independentes e uma só for positiva para CD86 (a seguir designada P1) e a outra só for positiva para CD54 (a seguir designada P2), é necessário realizar uma terceira execução. Se esta terceira execução for negativa para ambos os marcadores (a seguir designada N), a previsão por h-CLAT é considerada NEGATIVA. Em contrapartida, se a terceira execução for positiva para um dos marcadores (P1 ou P2) ou para ambos os marcadores (a seguir designada P12), a previsão por h-CLAT é considerada POSITIVA.

Figura 1: Modelo preditivo usado no método h-CLAT. Uma previsão por h-CLAT deve ser analisada no âmbito de uma IATA e em conformidade com o disposto nos pontos 7 e 8 da Introdução Geral.
P1: execução positiva apenas para CD86; P2; execução positiva apenas para CD54; P12: execução positiva para CD86 e CD54; N: execução não positiva para CD86 nem para CD54.
* As caixas mostram as combinações pertinentes de resultados das duas primeiras execuções, independentemente da ordem de obtenção dos resultados.
# As caixas mostram as combinações pertinentes de resultados das três execuções com base nos resultados obtidos nas duas primeiras execuções indicados na caixa acima, mas não refletem a ordem de obtenção dos resultados.
|
|
28.
|
No caso dos produtos químicos em estudo que se prevê sejam POSITIVOS com o h-CLAT, podem determinar-se dois valores de concentração efetiva (CE), CE150 para CD86 e CE200 para CD54, ou seja, a concentração à qual os produtos químicos em estudo produzem uma IRF de 150 ou 200. Estes valores CE podem contribuir para a avaliação do potencial de sensibilização (9) no âmbito de uma abordagem integrada do tipo IATA (4) (5) (6) (7) (8). Podem ser calculados utilizando as seguintes equações:
EC 150 (for CD86) = Bconc
+ [(150 – BRFI
)/ARFI – BRFI
) × (Aconc – Bconc
)]
EC 200 (for CD86) = Bconc
+ [(200 – BRFI
)/ARFI – BRFI
) × (Aconc – Bconc
)]
em que
Aconc é a concentração mais baixa em μg/ml com IRF > 150 (CD86) ou 200 (CD54)
Bconc é a concentração mais elevada em μg/ml com IRF < 150 (CD86) ou 200 (CD54)
AIRF é a IRF à concentração mais baixa com IRF > 150 (CD86) ou 200 (CD54)
BIRF é a IRF à concentração mais elevada com IRF < 150 (CD86) ou 200 (CD54)
Para determinar de modo mais preciso os valores CE150 e CE200, podem ser necessárias três execuções independentes para a medição da expressão de CD86/CD54. Os valores CE150 e CE200 finais são, então, determinados como o valor mediano das CE calculado a partir das três execuções independentes. Se apenas duas das três execuções independentes satisfizerem os critérios de positividade (ver pontos 26 e 27), é adotado o valor CE150 ou CE200 mais elevado.
|
Critérios de aceitação
|
29.
|
Os critérios de aceitação a seguir indicados devem ser respeitados ao utilizar o método h-CLAT (22) (27).
|
—
|
Os valores de viabilidade celular dos controlos do meio e do solvente/veículo devem ser superiores a 90 %.
|
|
—
|
Para o controlo do solvente/veículo, as IRF de CD86 e de CD54 não devem exceder os critérios positivos (CD86 IRF ≥ 150 % e CD54 IRF ≥ 200 %). Os valores de IRF para o controlo do solvente/veículo são calculados utilizando a fórmula indicada no ponto 25 (substituindo a menção «IMF do produto químico» por «IMF do solvente/veículo» e «IMF do solvente/veículo» por «IMF do controlo (meio)»).
|
|
—
|
Para o controlo do meio e do solvente/veículo, os rácios da IMF CD86 e CD54 para o controlo de isótipo devem ser > 105 %.
|
|
—
|
Para o controlo positivo (DNCB), os valores IRF de CD86 e de CD54 devem cumprir os critérios positivos (CD86 IRF ≥ 150 e CD54 IRF ≥ 200) e a viabilidade celular deve ser superior a 50 %.
|
|
—
|
Para o produto químico em estudo, a viabilidade celular deve ser superior a 50 % em, pelo menos, quatro concentrações testadas em cada execução.
|
|
|
30.
|
Os resultados negativos só são aceitáveis no caso de produtos químicos em estudo que apresentem uma viabilidade celular inferior a 90 % à concentração mais elevada testada (ou seja, 1,2 × CV75 de acordo com o esquema de diluições em série descrito no ponto 20). Se a viabilidade celular a 1,2 × CV75 for igual ou superior a 90 %, o resultado negativo não deve ser tido em conta. Neste caso, é recomendável tentar aperfeiçoar a seleção das doses repetindo a determinação do valor CV75. Note-se que, quando uma concentração de 5 000 μg/ml na solução salina (ou no meio ou outros solventes/veículos), uma concentração de 1 000 μg/ml em DMSO ou a concentração solúvel mais elevada é utilizada como concentração máxima de ensaio para um produto químico em estudo, pode aceitar-se um resultado negativo mesmo que a viabilidade celular seja superior a 90 %.
|
Relatório de ensaio
|
31.
|
O relatório de ensaio deve incluir as informações que a seguir se indicam.
|
Produto químico em estudo
|
Substância monocomponente
|
Identificação química, como o(s) nome(s) IUPAC ou CAS, o(s) número(s) CAS, o código SMILES ou InChI, a fórmula estrutural e/ou outros identificadores;
|
|
Aspeto físico, log P, solubilidade na água, solubilidade em DMSO, massa molecular e outras propriedades físico-químicas relevantes, se disponíveis;
|
|
Grau de pureza, identidade química das impurezas, conforme adequado e exequível, etc.;
|
|
Pré-tratamento, se for o caso (aquecimento ou moagem, por exemplo);
|
|
Concentração(ões) testada(s);
|
|
Condições de armazenagem e de estabilidade, se conhecidas;
|
|
Justificação da escolha do solvente/veículo para cada produto químico em estudo.
|
|
|
Substância multicomponentes, UVCB e mistura
|
Caracterização, tanto quanto possível, por exemplo, por identidade química (ver supra), pureza, ocorrência quantitativa e propriedades físico-químicas relevantes (ver supra) dos componentes, na medida em que estejam disponíveis;
|
|
Aspeto físico, solubilidade na água, solubilidade em DMSO e outras propriedades físico-químicas relevantes, na medida em que estejam disponíveis;
|
|
Massa molecular, ou massa molecular aparente no caso de misturas/polímeros de composições conhecidas, ou outras informações relevantes para a realização do estudo;
|
|
Pré-tratamento, se for o caso (aquecimento ou moagem, por exemplo);
|
|
Concentração(ões) testada(s);
|
|
Condições de armazenagem e de estabilidade, se conhecidas;
|
|
Justificação da escolha do solvente/veículo para cada produto químico em estudo.
|
|
|
|
Controlos
|
Controlo positivo
|
Identificação química, como o(s) nome(s) IUPAC ou CAS, o(s) número(s) CAS, o código SMILES ou InChI, a fórmula estrutural e/ou outros identificadores;
|
|
Aspeto físico, log P, água, solubilidade na água, solubilidade em DMSO, massa molecular e outras propriedades físico-químicas relevantes, se conhecidas e se aplicável;
|
|
Grau de pureza, identidade química das impurezas, conforme adequado e exequível, etc.;
|
|
Pré-tratamento, se for o caso (aquecimento ou moagem, por exemplo);
|
|
Concentração(ões) testada(s);
|
|
Condições de armazenagem e de estabilidade, se conhecidas;
|
|
Referência ao historial dos resultados dos controlos positivos que demonstrem o cumprimento dos critérios de aceitação da execução, se aplicável.
|
|
|
|
Controlo negativo e controlo do solvente/veículo
|
Identificação química, como o(s) nome(s) IUPAC ou CAS, o(s) número(s) CAS, o código SMILES ou InChI, a fórmula estrutural e/ou outros identificadores;
|
|
Grau de pureza, identidade química das impurezas, conforme adequado e exequível, etc.;
|
|
Aspeto físico, massa molecular e outras propriedades físico-químicas relevantes caso seja utilizado um solvente/veículo diferente dos mencionados nas orientações de ensaio, na medida em que estejam disponíveis;
|
|
Condições de armazenagem e de estabilidade, se conhecidas;
|
|
Justificação da escolha do solvente/veículo para cada produto químico em estudo.
|
|
|
Condições de ensaio
|
Nome e endereço do promotor, do laboratório e do diretor do estudo;
|
|
Descrição do ensaio utilizado;
|
|
Linha celular utilizada, respetivas condições de armazenamento e origem (por exemplo, instalação de onde provêm as células);
|
|
Citometria de fluxo utilizada (por exemplo, modelo), incluindo parâmetros dos instrumentos, globulina, anticorpos e marcador de citotoxicidade utilizados;
|
|
O procedimento utilizado para demonstrar a competência do laboratório na realização do ensaio, mediante o teste de substâncias para demonstração de competência, e o procedimento utilizado para demonstrar a reprodutibilidade do desempenho do ensaio ao longo do tempo, por exemplo dados históricos do controlo e/ou dados históricos dos controlos de reatividade.
|
|
|
Critérios de aceitação do ensaio
|
Viabilidade celular, valores IMF e IRF obtidos com o controlo do solvente/veículo comparativamente com os intervalos de aceitação;
|
|
Viabilidade celular e valores IRF obtidos com o controlo positivo comparativamente com os intervalos de aceitação;
|
|
Viabilidade celular de todas as concentrações testadas do produto químico em estudo.
|
|
|
Procedimento de ensaio
|
Número de execuções realizadas;
|
|
Concentrações do produto químico em estudo, aplicação e tempo de exposição utilizado (se diferente do recomendado);
|
|
Descrição dos critérios de avaliação e de decisão utilizados;
|
|
Descrição de eventuais modificações do procedimento de ensaio.
|
|
|
Resultados
|
Quadro com os dados, incluindo o valor CV75 (se aplicável), a IMF geométrica individual, a IRF, valores de viabilidade celular, valores CE150/CE200 (se aplicável) obtidos para o produto químico em estudo e para o controlo positivo em cada execução, bem como uma indicação da classificação do produto químico em estudo segundo o modelo preditivo;
|
|
Descrição de quaisquer outras observações pertinentes, se for caso disso.
|
|
|
Discussão dos resultados
|
Discussão dos resultados obtidos com o método h-CLAT;
|
|
Análise dos resultados do ensaio no contexto de uma IATA, caso estejam disponíveis outras informações pertinentes.
|
|
|
REFERÊNCIAS BIBLIOGRÁFICAS
|
(1)
|
Ashikaga T, Yoshida Y, Hirota M, Yoneyama K, Itagaki H, Sakaguchi H, Miyazawa M, Ito Y, Suzuki H, Toyoda H. (2006). Development of an in vitro skin sensitization test using human cell lines: The human Cell Line Activation Test (h-CLAT) I. Optimization of the h-CLAT protocol. Toxicol. In Vitro 20, 767–773.
|
|
(2)
|
Miyazawa M, Ito Y, Yoshida Y, Sakaguchi H, Suzuki H. (2007). Phenotypic alterations and cytokine production in THP-1 cells in response to allergens. Toxicol. In Vitro 21, 428-437.
|
|
(3)
|
EC EURL-ECVAM (2013). Recommendation on the human Cell Line Activation Test (h-CLAT) for skin sensitisation testing. Disponível em: https://eurl-ecvam.jrc.ec.europa.eu/eurl-ecvam-recommendations
|
|
(4)
|
Takenouchi O, Fukui S, Okamoto K, Kurotani S, Imai N, Fujishiro M, Kyotani D, Kato Y, Kasahara T, Fujita M, Toyoda A, Sekiya D, Watanabe S, Seto H, Hirota M, Ashikaga T, Miyazawa M. (2015). Test battery with the human cell line activation test, direct peptide reactivity assay and DEREK based on a 139 chemical data set for predicting skin sensitizing potential and potency of chemicals. J Appl Toxicol. 35, 1318-1332.
|
|
(5)
|
Hirota M, Fukui S, Okamoto K, Kurotani S, Imai N, Fujishiro M, Kyotani D, Kato Y, Kasahara T, Fujita M, Toyoda A, Sekiya D, Watanabe S, Seto H, Takenouchi O, Ashikaga T, Miyazawa M. (2015). Evaluation of combinations of in vitro sensitization test descriptors for the artificial neural network-based risk assessment model of skin sensitization. J Appl Toxicol. 35, 1333-1347.
|
|
(6)
|
Bauch C, Kolle SN, Ramirez T, Fabian E, Mehling A, Teubner W, van Ravenzwaay B, Landsiedel R. (2012). Putting the parts together: combining in vitro methods to test for skin sensitizing potencials. Regul Toxicol Parmacol. 63, 489-504.
|
|
(7)
|
Van der Veen JW, Rorije E, Emter R, Natch A, van Loveren H, Ezendam J. (2014). Evaluating the performance of integrated approaches for hazard identification of skin sensitizing chemicals. Regul Toxicol Pharmacol. 69, 371-379.
|
|
(8)
|
Urbisch D, Mehling A, Guth K, Ramirez T, Honarvar N, Kolle S, Landsiedel R, Jaworska J, Kern PS, Gerberick F, Natsch A, Emter R, Ashikaga T, Miyazawa M, Sakaguchi H. (2015). Assessing skin sensitization hazard in mice and men using non-animal test methods. Regul Toxicol Parmacol. 71, 337-351.
|
|
(9)
|
Jaworska JS, Natsch A, Ryan C, Strickland J, Ashikaga T, Miyazawa M. (2015). Bayesian integrated testing strategy (ITS) for skin sensitization potency assessment: a decision support system for quantitative weight of evidence and adaptive testing strategy. Arch Toxicol. 89, 2355-2383.
|
|
(10)
|
Strickland J, Zang Q, Kleinstreuer N, Paris M, Lehmann DM, Choksi N, Matheson J, Jacobs A, Lowit A, Allen D, Casey W. (2016). Integrated decision strategies for skin sensitization hazard. J Appl Toxicol. DOI 10.1002/jat.3281.
|
|
(11)
|
Nukada Y, Ashikaga T, Miyazawa M, Hirota M, Sakaguchi H, Sasa H, Nishiyama N. (2012). Prediction of skin sensitization potency of chemicals by human Cell Line Activation Test (h-CLAT) and an attempt at classifying skin sensitization potency. Toxicol. In Vitro 26, 1150-60.
|
|
(12)
|
EC EURL ECVAM (2015). Re-analysis of the within and between laboratory reproducibility of the human Cell Line Activation Test (h-CLAT). Disponível em: https://eurl-ecvam.jrc.ec.europa.eu/eurl-ecvam-recommendations/eurl-ecvam-recommendation-on-the-human-cell-line-activation-test-h-clat-for-skin-sensitisation-testing
|
|
(13)
|
EC EURL ECVAM (2012). human Cell Line Activation Test (h-CLAT) Validation Study Report. Disponível em: https://eurl-ecvam.jrc.ec.europa.eu/eurl-ecvam-recommendations
|
|
(14)
|
Takenouchi O, Miyazawa M, Saito K, Ashikaga T, Sakaguchi H. (2013). Predictive performance of the human Cell Line Activation Test (h-CLAT) for lipophilic with high octanol-water partition coefficients. J. Toxicol. Sci. 38, 599-609.
|
|
(15)
|
Ashikaga T, Sakaguchi H, Sono S, Kosaka N, Ishikawa M, Nukada Y, Miyazawa M, Ito Y, NishiyamaN, Itagaki H. (2010). A comparative evaluation of in vitro skin sensitisation tests: the human cell-line activation test (h-CLAT) versus the local lymph node assay (LLNA). Altern. Lab. Anim. 38, 275-284.
|
|
(16)
|
Fabian E., Vogel D., Blatz V., Ramirez T., Kolle S., Eltze T., van Ravenzwaay B., Oesch F., Landsiedel R. (2013). Xenobiotic metabolizin enzyme activities in cells used for testing skin sensitization in vitro. Arch Toxicol 87, 1683-1969.
|
|
(17)
|
Okamoto K, Kato Y, Kosaka N, Mizuno M, Inaba H, Sono S, Ashikaga T, Nakamura T, Okamoto Y, Sakaguchi H, Kishi M, Kuwahara H, Ohno Y. (2010). The Japanese ring study of a human Cell Line Activation Test (h-CLAT) for predicting skin sensitization potential (6th report): A study for evaluating oxidative hair dye sensitization potential using h-CLAT. AATEX 15, 81-88.
|
|
(18)
|
DB-ALM (INVITTOX) (2014). Protocol 158: human Cell Line Activation Test (h-CLAT), 23pp. Disponível em: http://ecvam-dbalm.jrc.ec.europa.eu/
|
|
(19)
|
Mizuno M, Yoshida M, Kodama T, Kosaka N, Okamato K, Sono S, Yamada T, Hasegawa S, Ashikaga T, Kuwahara H, Sakaguchi H, Sato J, Ota N, Okamoto Y, Ohno Y. (2008). Effects of pre-culture conditions on the human Cell Line Activation Test (h-CLAT) results; Results of the 4th Japanese inter-laboratory study. AATEX 13, 70-82.
|
|
(20)
|
Sono S, Mizuno M, Kosaka N, Okamoto K, Kato Y, Inaba H, Nakamura T, Kishi M, Kuwahara H, Sakaguchi H, Okamoto Y, Ashikaga T, Ohno Y. (2010). The Japanese ring study of a human Cell Line Activation Test (h-CLAT) for predicting skin sensitization potential (7th report): Evaluation of volatile, poorly soluble fragrance materials. AATEX 15, 89-96.
|
|
(21)
|
OECD (2005). Guidance Document No 34 on The Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. OECD Series on Testing and Assessment. Organization for Economic Cooperation and Development, Paris, França, 2005, 96 pp.
|
|
(22)
|
OECD (2012). The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins. Part 1: Scientific Evidence. Series on Testing and Assessment No 168. Disponível em: http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=ENV/JM/MONO(2012)10/PART1&docLanguage=En
|
|
(23)
|
United Nations UN (2013). Globally Harmonized System of Classification and Labelling of Chemicals (GHS). Fifth revised edition. Nova Iorque e Genebra: United Nations Publications. ISBN: 978-92-1-117006-1. Disponível em: http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html
|
|
(24)
|
ECETOC (2003). Contact sensitization: Classification according to potency. European Centre for Ecotoxicology & Toxicology of Chemicals (Technical Report No 87).
|
|
(25)
|
Ashikaga T, Sakaguchi H, Okamoto K, Mizuno M, Sato J, Yamada T, Yoshida M, Ota N, Hasegawa S, Kodama T, Okamoto Y, Kuwahara H, Kosaka N, Sono S, Ohno Y. (2008). Assessment of the human Cell Line Activation Test (h-CLAT) for Skin Sensitization; Results of the First Japanese Inter-laboratory Study. AATEX 13, 27-35.
|
Apêndice 1.1
DEFINIÇÕES
AOP (Adverse Outcome Pathway – via determinante dos efeitos nocivos)
: Sequência de eventos que parte da estrutura química de um produto químico alvo ou de um grupo de produtos químicos análogos, passa pelo evento molecular iniciador, e resulta num resultado com consequências in vivo (22).
CE150
: Concentrações que apresentam IRF de 150 para a expressão de CD86.
CE200
: Concentrações que apresentam IRF de 200 para a expressão de CD54.
Citometria de fluxo
: Técnica citométrica em que células em suspensão num fluido passam, uma a uma, por um foco de excitação luminosa, disperso em padrões característicos das células e dos seus componentes; as células são frequentemente marcadas com marcadores fluorescentes para que a luz seja primeiro absorvida e depois emitida a uma frequência diferente.
Controlo do meio
: Replicado não tratado que contém todos os componentes de um sistema de ensaio. Esta amostra é tratada com amostras tratadas com o produto químico em estudo e outras amostras de controlo, para determinar se o solvente/veículo interage com o sistema de ensaio.
Controlo positivo
: Replicado que contém todos os componentes de um sistema de ensaio e foi tratado com uma substância conhecida por induzir uma resposta positiva. Para que seja possível determinar a variabilidade no tempo da resposta do controlo positivo, a magnitude da resposta positiva não deve ser excessiva.
Controlo do solvente/veículo
: Amostra não tratada que contém todos os componentes de um sistema de ensaio, com exceção do produto químico em estudo, mas incluindo o solvente/veículo utilizado. Serve para determinar a resposta de referência para as amostras tratadas com o produto químico em estudo dissolvido ou em dispersão estável no mesmo solvente/veículo. Quando testada simultaneamente com um controlo de meio, esta amostra também indica se o solvente/veículo interage com o sistema de ensaio.
CV75
: Concentração estimada que apresenta uma viabilidade celular de 75 %.
Ensaio válido
: Um ensaio considerado suficientemente pertinente e fiável para um fim específico e que se baseia em princípios cientificamente sólidos. Um ensaio nunca é válido em termos absolutos, mas apenas em relação a um objetivo definido (21).
Especificidade
: Proporção dos produtos químicos negativos/inativos que são corretamente classificados pelo ensaio. Permite medir a precisão de um ensaio que produz resultados categóricos e é um aspeto importante a ter em conta na avaliação da pertinência de um ensaio (21).
Execução
: Consiste em testar um ou mais produtos químicos em estudo em simultâneo com um controlo do solvente/veículo e com um controlo positivo.
Fiabilidade
: Indica em que medida um ensaio pode ser reproduzido ao longo do tempo num mesmo laboratório e entre laboratórios utilizando o mesmo protocolo. Para a avaliar, calcula-se a reprodutibilidade intralaboratorial e interlaboratorial, bem como a repetibilidade intralaboratorial (21).
IATA (Integrated Approach to Testing and Assessment – abordagem integrada de ensaio e avaliação)
: Abordagem estruturada utilizada para a identificação (potencial) e caracterização (potência) do perigo e/ou para a avaliação da segurança (potencial/potência e exposição) de um produto químico ou grupo de produtos químicos, que integra e pondera, de forma estratégica, todos os dados pertinentes com o objetivo de ser tida em conta numa decisão regulamentar sobre o potencial perigo e/ou risco e/ou a necessidade de realizar ensaios complementares específicos.
Intensidade relativa de fluorescência (IRF)
: Valores relativos da média geométrica da intensidade de fluorescência (IMF) das células tratadas com o produto químico em comparação com a IMF das células tratadas com o solvente/veículo.
Mistura
: Uma mistura ou solução composta por duas ou mais substâncias.
Perigo
: Propriedade intrínseca de um agente, ou de uma situação, suscetível de causar efeitos nocivos quando um organismo, sistema ou (sub)população é exposto ao agente em causa.
Pertinência
: Descrição da relação do ensaio com o efeito em estudo e da adequação e utilidade do ensaio para um fim específico. Traduz a medida em que o ensaio mede ou prevê corretamente o efeito biológico em causa. A pertinência tem em conta a precisão (concordância) de um ensaio (21).
Precisão
: Grau de acordo entre os resultados do ensaio e os valores de referência aceites. Trata-se de uma medida do desempenho do ensaio e de um dos aspetos da sua pertinência. Este termo e o termo «concordância» são muitas vezes utilizados indistintamente para indicar a proporção de resultados corretos de um ensaio (21).
Pré-haptenos
: Produtos químicos que se tornam sensibilizantes por transformação abiótica.
Pró-haptenos
: Produtos químicos que requerem ativação enzimática para exercerem potencial de sensibilização cutânea.
Produto químico
: Uma substância ou mistura.
Produto químico em estudo
: Qualquer substância ou mistura à qual seja aplicado o presente método.
Sensibilidade
: Proporção de todos os produtos químicos positivos/ativos que são corretamente classificados pelo ensaio. Permite medir a precisão de um ensaio que produz resultados categóricos e é um aspeto importante a ter em conta na avaliação da pertinência de um ensaio (21).
Sistema Mundial Harmonizado de Classificação e Rotulagem de Produtos Químicos das Nações Unidas – GHS da ONU (em inglês: United Nations Globally Harmonized System of Classification and Labelling of Chemicals – UN GHS)
: Sistema que propõe a classificação dos produtos químicos (substâncias e misturas) em função de tipos e níveis normalizados de perigos físicos, sanitários e ambientais e trata dos elementos de comunicação correspondentes, nomeadamente pictogramas, palavras-sinal, advertências de perigo, recomendações de prudência e fichas de dados de segurança, de modo a transmitir informações sobre os respetivos efeitos nocivos, tendo em vista a proteção das pessoas (incluindo empregadores, trabalhadores, transportadores, consumidores e pessoal dos serviços de emergência) e do ambiente (23).
Substância
: Um elemento químico e seus compostos, no estado natural ou obtidos por qualquer processo de produção, incluindo qualquer aditivo necessário para preservar a sua estabilidade e quaisquer impurezas resultantes do processo utilizado, mas excluindo qualquer solvente que possa ser separado sem afetar a estabilidade da substância nem alterar a sua composição.
Substância monocomponente
: Uma substância, definida pela sua composição quantitativa, da qual um dos principais componentes está presente num teor de, pelo menos, 80 % (m/m).
Substância multicomponentes
: Uma substância, definida pela sua composição quantitativa, da qual mais do que um dos principais componentes está presente numa concentração ≥ 10 % (m/m) e < 80 % (m/m). As substâncias multicomponentes resultam de processos de fabrico. A diferença entre uma mistura e uma substância multicomponentes é que a primeira se obtém misturando duas ou mais substâncias, sem reação química. As substâncias multicomponentes resultam de reações químicas.
Tampão de coloração
: Uma solução salina de tampão fosfato com 0,1 % de albumina de soro bovino.
UVCB
: Substâncias de composição desconhecida ou variável, produtos de reação complexos ou materiais biológicos.
Apêndice 1.2
SUBSTÂNCIAS PARA A DEMONSTRAÇÃO DE COMPETÊNCIA
Antes de utilizarem, por rotina, o ensaio descrito no presente apêndice do método de ensaio B.71, os laboratórios devem demonstrar a sua competência técnica obtendo corretamente a previsão esperada com o método h-CLAT para as dez substâncias recomendadas no quadro 1 e obtendo valores CV75, CE150 e CE200 que se encontrem dentro do intervalo de referência correspondente para, pelo menos, oito das dez substâncias de demonstração de competência. Estas substâncias foram selecionadas de modo a representarem o conjunto de respostas possíveis no que diz respeito aos perigos de sensibilização cutânea. Outros critérios de seleção foram a disponibilidade comercial das substâncias e a disponibilidade de dados de referência in vivo de elevada qualidade, bem como de dados in vitro de alta qualidade produzidos com o método h-CLAT. Além disso, estão disponíveis dados de referência publicados para o método h-CLAT (3) (14).
Quadro 1
Substâncias recomendadas para a demonstração de competência técnica com o método h-CLAT
|
Substâncias para a demonstração de competência
|
N.o CAS
|
Estado físico
|
Previsão in vivo
(102)
|
Intervalo de referência CV75 em μg/ml (103)
|
Resultados h-CLAT para CD86 (intervalo de referência CE150 em μg/ml) (103)
|
Resultados h-CLAT para CD54 (intervalo de referência CE200 em μg/ml) (103)
|
|
2,4-Dinitroclorobenzeno
|
97-00-7
|
Sólido
|
Sensibilizante (extremo)
|
2-12
|
Positivo (0,5-10)
|
Positivo (0,5-15)
|
|
4-Fenilenodiamina
|
106-50-3
|
Sólido
|
Sensibilizante (forte)
|
5-95
|
Positivo (< 40)
|
Negativo (> 1,5) (104)
|
|
Sulfato de níquel
|
10101-97-0
|
Sólido
|
Sensibilizante (moderado)
|
30-500
|
Positivo (< 100)
|
Positivo (10-100)
|
|
2-Mercaptobenzotiazol
|
149-30-4
|
Sólido
|
Sensibilizante (moderado)
|
30-400
|
Negativo (> 10) (104)
|
Positivo (10-140)
|
|
R(+)-Limoneno
|
5989-27-5
|
Líquido
|
Sensibilizante (fraco)
|
> 20
|
Negativo (> 5) (104)
|
Positivo (< 250)
|
|
Imidazolidinilureia
|
39236-46-9
|
Sólido
|
Sensibilizante (fraco)
|
25-100
|
Positivo (20-90)
|
Positivo (20-75)
|
|
Isopropanol
|
67-63-0
|
Líquido
|
Não sensibilizante
|
> 5 000
|
Negativo (> 5 000 )
|
Negativo (> 5 000 )
|
|
Glicerol
|
56-81-5
|
Líquido
|
Não sensibilizante
|
> 5 000
|
Negativo (> 5 000 )
|
Negativo (> 5 000 )
|
|
Ácido láctico
|
50-21-5
|
Líquido
|
Não sensibilizante
|
1500-5 000
|
Negativo (> 5 000 )
|
Negativo (> 5 000 )
|
|
Ácido 4-aminobenzoico
|
150-13-0
|
Sólido
|
Não sensibilizante
|
> 1 000
|
Negativo (> 1 000 )
|
Negativo (> 1 000 )
|
|
Abreviaturas: N.o CAS = Número de registo do Chemical Abstracts Service
|
Apêndice 2
SENSIBILIZAÇÃO CUTÂNEA IN VITRO: TESTE DE ATIVAÇÃO DA LINHA CELULAR U937 (U-SENS™)
CONSIDERAÇÕES INICIAIS E LIMITAÇÕES
|
1.
|
O método de ensaio U-SENS™ permite quantificar as variações na expressão de um marcador de superfície celular associadas ao processo de ativação dos monócitos e das células dendríticas (ou seja, CD86), na linha celular do linfoma histiocítico humano U937, após a exposição a sensibilizantes (1). Os níveis de expressão medidos para o marcador de superfície celular CD86 na linha celular U937 são, em seguida, utilizados para ajudar a distinguir os sensibilizantes dos não sensibilizantes cutâneos.
|
|
2.
|
O método de ensaio U-SENS™ foi avaliado num estudo de validação (2) coordenado pela L’Oréal, seguido de uma análise independente pelos pares conduzida pelo Comité Científico Consultivo (ESAC) do Laboratório de Referência da União Europeia para as Alternativas à Experimentação em Animais (EURL ECVAM) (3). Tendo em conta todos os dados disponíveis e os contributos das entidades reguladoras e das partes interessadas, o método de ensaio U-SENS™ foi recomendado pelo EURL ECVAM (4) para ser utilizado no âmbito de uma IATA para ajudar a distinguir os sensibilizantes dos não sensibilizantes para efeitos de classificação e rotulagem de perigos. No seu documento de orientação relativo à elaboração de relatórios sobre as abordagens estruturadas de integração de dados e de utilização de fontes individuais de informação no âmbito de uma IATA para a sensibilização cutânea, a OCDE analisa atualmente vários estudos de casos que descrevem diferentes estratégias de ensaio e modelos preditivos. Uma das diferentes abordagens definidas baseia-se no ensaio U-SENS (5). Encontram-se na literatura (4) (5) (7) exemplos da utilização de dados U-SENS™ combinados com outras informações, incluindo dados históricos e dados humanos válidos (6).
|
|
3.
|
O método de ensaio U-SENS™ demonstrou ser transferível para laboratórios com experiência em técnicas de cultura celular e análise por citometria de fluxo. O nível de reprodutibilidade esperado das previsões efetuadas pelo ensaio é da ordem dos 90 % intralaboratorial e 84 % interlaboratorial (8). Os resultados do estudo de validação (8) e de outros estudos publicados (1) indicam, em termos gerais, que, em comparação com os resultados obtidos pelo método LLNA, a precisão na distinção entre sensibilizantes cutâneos (cat. 1 do GHS da ONU/CLP) e não sensibilizantes é de 86 % (N=166), com uma sensibilidade de 91 % (118/129) e uma especificidade de 65 % (24/37). Em comparação com os resultados obtidos no ser humano, a precisão na distinção entre sensibilizantes (cat. 1 do GHS da ONU/CLP) e não sensibilizantes é de 77 % (N=101), com uma sensibilidade de 100 % (58/58) e uma especificidade de 47 % (20/43). É mais provável que os falsos negativos nas previsões efetuadas com o método U-SENS™ em comparação com o LLNA digam respeito a produtos químicos com potencial de sensibilização cutânea baixo a moderado (subcategoria 1B do GHS da ONU/CLP) do que a produtos químicos com um potencial de sensibilização cutânea elevado (subcategoria 1A do GHS da ONU/CLP) (1) (8) (9). No seu conjunto, estas informações demonstram a utilidade do método U-SENS™ para a identificação dos perigos de sensibilização cutânea. No entanto, os valores relativos à precisão do ensaio U-SENS™ utilizado isoladamente são meramente indicativos, uma vez que o presente método deve ser combinado com outras fontes de informação no contexto de uma IATA e em conformidade com o disposto nos pontos 7 e 8 da Introdução Geral. Além disso, na avaliação de métodos de estudo da sensibilização cutânea que não utilizem animais, deve ter-se em mente que o método LLNA e outros ensaios com animais podem não refletir de forma adequada a situação no ser humano.
|
|
4.
|
Os dados atualmente disponíveis demonstram que o método U-SENS™ é aplicável a produtos químicos em estudo (incluindo ingredientes cosméticos, tais como conservantes, tensioativos, substâncias ativas, corantes) abrangendo diversos grupos funcionais orgânicos, propriedades físico-químicas, potência de sensibilização cutânea (tal como determinadas por estudos in vivo) e o espetro de mecanismos de reação que se sabe estarem associados à sensibilização cutânea (ou seja, aceitador de Michael, formação de base de Schiff, agente acilante, substituição nucleófila bimolecular [SN2] ou substituição nucleófila aromática [SNAr]) (1) (8) (9) (10). O método de ensaio U-SENS™ é aplicável a produtos químicos que sejam solúveis ou que formem uma dispersão estável (ou seja, um coloide ou uma suspensão em que o produto químico não se deposite nem se separe do solvente/veículo formando diferentes fases) num solvente/veículo adequado (ver ponto 13). Os produtos químicos constantes da base de dados assinalados como pré-haptenos (substâncias ativadas por oxidação) ou pró-haptenos (substâncias que requerem ativação enzimática, por exemplo através de enzimas P450) foram corretamente previstos pelo ensaio U-SENS™ (1) (10). As substâncias capazes de provocar a rutura de membranas podem produzir resultados falsos positivos, devido a um aumento não específico da expressão de CD86. Com efeito, três dos sete falsos positivos em relação à classificação de referência in vivo eram tensioativos (1). Por isso, os resultados positivos com tensioativos devem ser tratados com prudência, enquanto os resultados negativos com tensioativos podem continuar a ser utilizados para apoiar a identificação do produto químico em estudo como não sensibilizante. É possível avaliar produtos químicos em estudo fluorescentes com o U-SENS™ (1). No entanto, os produtos fortemente fluorescentes que emitem no mesmo comprimento de onda que o isotiocianato de fluoresceína (FITC) ou que o iodeto de propídio (IP) provocam interferência com a deteção por citometria de fluxo, pelo que não podem ser corretamente avaliados utilizando anticorpos conjugados com FITC (potenciais falsos negativos) ou IP (viabilidade não mensurável). Neste caso, é possível recorrer, respetivamente, a outros anticorpos marcados com fluorocromos ou a outros marcadores de citotoxicidade, desde que seja possível demonstrar que produzem resultados semelhantes aos dos anticorpos marcados com FITC ou com IP (ver ponto 18), por exemplo testando as substâncias para demonstração de competência indicadas no apêndice 2.2. À luz do que precede, os resultados positivos com tensioativos e os resultados negativos com produtos químicos em estudo fortemente fluorescentes devem ser interpretados no contexto das limitações indicadas e em conjunto com outras fontes de informação no âmbito da IATA. Nos casos em que seja demonstrado que o ensaio U-SENS™ não é aplicável a outras categorias específicas de produtos químicos em estudo, este ensaio não deve ser utilizado para essas categorias específicas.
|
|
5.
|
Tal como descrito acima, o ensaio U-SENS™ ajuda a distinguir os sensibilizantes cutâneos dos não sensibilizantes. No entanto, quando utilizado no âmbito de abordagens integradas do tipo IATA, pode também contribuir para a avaliação do potencial de sensibilização. Contudo, é necessário continuar a trabalhar, de preferência com base em dados humanos, para determinar o modo como os resultados do U-SENS™ poderão vir a ser úteis para este tipo de avaliação.
|
|
6.
|
Apresentam-se definições no apêndice 2.1.
|
PRINCÍPIO DO ENSAIO
|
7.
|
O método U-SENS™ é um ensaio in vitro que permite quantificar as variações da expressão do marcador de superfície celular CD86 numa linha celular do linfoma histiocítico humano – as células U937– após uma exposição de 45 ± 3 horas ao produto químico em estudo. O marcador de superfície CD86 é um marcador típico de ativação das células U937. Trata-se de uma molécula de coestimulação conhecida por ser capaz de simular a ativação monocitária, o que desempenha um papel crítico na ativação das células T. As variações na expressão do marcador de superfície celular CD86 são medidas por citometria de fluxo após coloração das células, geralmente com anticorpos marcados com isotiocianato de fluoresceína (FITC). A citotoxicidade é medida em paralelo (por exemplo, utilizando IP), a fim de determinar se a ativação da expressão do marcador de superfície celular CD86 ocorre a concentrações subcitotóxicas. O índice de estimulação (IE) do marcador de superfície celular CD86, em comparação com o do controlo do solvente/veículo, é calculado e utilizado no modelo preditivo (ver ponto 19) para ajudar a distinguir os sensibilizantes dos não sensibilizantes.
|
DEMONSTRAÇÃO DE COMPETÊNCIA
|
8.
|
Antes de utilizarem, por rotina, o ensaio descrito no presente apêndice do método de ensaio B.71, os laboratórios devem demonstrar a sua competência técnica utilizando as dez substâncias enumeradas no apêndice 2.2, em conformidade com as boas práticas para os métodos in vitro (11). Além disso, os utilizadores do ensaio devem manter uma base de dados históricos dos controlos de reatividade (ver ponto 11), obtidos com o controlo positivo e com o controlo do solvente/veículo (ver pontos 15 e 16), e utilizar esses dados para confirmar que a reprodutibilidade do método de ensaio no seu laboratório é mantida ao longo do tempo.
|
PROCEDIMENTO
|
9.
|
O presente método de ensaio baseia-se no protocolo n.o 183 do serviço de base de dados sobre os métodos alternativos à experimentação em animais (DB-ALM — DataBase service on ALternative Methods to animal experimentation) U-SENS™ (12). Devem utilizar-se procedimentos operacionais normalizados (PON) aquando da implementação e utilização do método de ensaio U-SENS™ em laboratório. Pode utilizar-se um sistema automatizado para executar o método de ensaio U-SENS™ se for possível demonstrar que produz resultados semelhantes, por exemplo testando as substâncias enumeradas no apêndice 2.2. Segue-se uma descrição dos principais elementos e procedimentos do método U-SENS™.
|
Preparação das células
|
10.
|
Para a realização do ensaio U-SENS™, deve utilizar-se a linha celular do linfoma histiocítico humano, U937 (13). As células (clone CRL1593.2) devem ser obtidas junto de um banco de células reconhecido, como a American Type Culture Collection.
|
|
11.
|
As células U937 são cultivadas a 37 °C numa atmosfera humidificada a 5 % CO2, em meio RPMI-1 640 complementado com 10 % de soro fetal de vitelo (SFV), 2 mM de L-glutamina, 100 unidades/ml de penicilina e 100 μg/ml de estreptomicina (meio completo). As células U937 são passadas regularmente a cada 2-3 dias a uma densidade de 1,5 ou 3 × 105 células/ml, respetivamente. A densidade celular não deve exceder 2 × 106 células/ml e a viabilidade celular medida pela exclusão do azul de Trypan deve ser ≥ 90 % (não deve aplicar-se na primeira passagem após descongelação). Antes de ser utilizado para o ensaio, cada lote de células, de SFV ou de anticorpos deve ser qualificado mediante a realização de um controlo de reatividade. O controlo de reatividade das células deve ser efetuado com o controlo positivo, o ácido picrilsulfónico (ácido 2,4,6-trinitrobenzenossulfónico: TNBS) (n.o CAS 2 508-19-2, pureza ≥ 99 %) e com o controlo negativo, o ácido láctico (n.o CAS 50-21-5, pureza ≥ 85 %), pelo menos uma semana após a descongelação. O controlo de reatividade deve ser efetuado com seis concentrações finais para cada um dos dois controlos (TNBS: 1, 12.5, 25, 50, 75, 100 μg/ml e ácido láctico: 1, 10, 20, 50, 100, 200 μg/ml). O TNBS dissolvido no meio completo deve produzir uma resposta positiva para o CD86, relacionada com a concentração (por exemplo, quando uma concentração positiva, IE CD86 ≥ 150, é seguida por uma concentração com um IE CD86 crescente); o ácido láctico dissolvido no meio completo deve produzir uma resposta negativa para o CD86 (ver ponto 21). Apenas devem ser utilizados para o ensaio os lotes de células que tenham passado duas vezes o controlo de reatividade. As células podem ser propagadas até sete semanas após a descongelação. O número de passagens não deve exceder 21. O controlo de reatividade deve ser efetuado de acordo com os procedimentos descritos nos pontos 18 a 22.
|
|
12.
|
Para o ensaio, as células U937 são inoculadas a uma densidade de 3 × 105 células/ml ou 6 × 105 células/ml e pré-cultivadas em frascos de cultura durante dois dias ou um dia, respetivamente. Podem utilizar-se condições de pré-cultura diferentes das descritas acima, desde que a escolha seja suficientemente fundamentada do ponto de vista científico e seja possível demonstrar que produz resultados semelhantes, por exemplo testando as substâncias enumeradas no apêndice 2.2. No dia do ensaio, as células colhidas do frasco de cultura são novamente colocadas em suspensão num meio de cultura fresco a uma densidade de 5 × 105 células/ml. Em seguida, distribuem-se as células numa placa de 96 poços de fundo plano com 100 μl (densidade celular final de 0,5 × 105 células/poço).
|
Preparação dos produtos químicos em estudo e das substâncias de controlo
|
13.
|
A avaliação da solubilidade deve ser efetuada antes do ensaio. Para este efeito, os produtos químicos em estudo são dissolvidos ou dispersos de forma estável a uma concentração de 50 mg/ml, escolhendo de preferência como solvente o meio completo ou, como segunda opção de solvente/veículo, o dimetilsulfóxido (DMSO, pureza ≥ 99 %), se o produto químico em estudo não for solúvel no meio completo. Para o ensaio, dissolve-se o produto químico em estudo até uma concentração final de 0,4 mg/ml no meio completo, se o produto químico for solúvel neste solvente/veículo. Se apenas for solúvel em DMSO, é dissolvido a uma concentração de 50 mg/ml. É possível utilizar solventes/veículos diferentes dos descritos, desde que a escolha seja fundamentada do ponto de vista científico. Deve ser tida em conta a estabilidade do produto químico em estudo no solvente/veículo final.
|
|
14.
|
Os produtos químicos em estudo e as substâncias de controlo são preparados no dia do ensaio. Uma vez que não é realizado um ensaio de determinação da dose, devem testar-se, para a primeira execução, seis concentrações finais (1, 10, 20, 50, 100 e 200 μg/ml) no solvente/veículo correspondente, quer no meio completo, quer em DMSO a 0,4 % no meio. Para as execuções seguintes, partindo das soluções do produto químico em estudo a uma concentração de 0,4 mg/ml em meio completo, ou de 50 mg/ml em DMSO, preparam-se pelo menos quatro soluções de trabalho (ou seja, pelo menos quatro concentrações) utilizando o solvente/veículo correspondente. Para terminar, as soluções de trabalho são utilizadas para o tratamento, acrescentando um volume igual de células U937 em suspensão (ver ponto 11 supra) ao volume da solução de trabalho na placa para obter uma diluição suplementar de fator 2 (12). As concentrações (pelo menos quatro) para eventuais execuções suplementares são selecionadas com base nos resultados individuais de todas as execuções anteriores (8). As concentrações finais utilizáveis são 1, 2, 3, 4, 5, 7.5, 10, 12.5, 15, 20, 25, 30, 35, 40, 45, 50, 60, 70, 80, 90, 100, 120, 140, 160, 180 e 200 μg/ml. A concentração final máxima é de 200 μg/ml. Se for obtido um valor positivo para CD86 a 1 μg/ml, convém avaliar 0,1 μg/ml para encontrar a concentração do produto químico em estudo que não transpõe o limiar de positividade de CD86. Para cada execução, calcula-se o valor CE150 (concentração à qual um produto químico atinge o limiar de positividade de 150 % para CD86, ver ponto 19) se for observada uma resposta positiva para CD86 dependente da concentração. Se o produto químico em estudo induzir uma resposta positiva para CD86 não relacionada com a concentração, o cálculo do valor CE150 pode não ser pertinente, tal como descrito no protocolo DB-ALM n.o 183 U-SENS™ (12). Para cada execução, é calculado, sempre que possível, o valor CV70 (concentração à qual um produto químico atinge o limiar de citotoxicidade de 70 %, ver ponto 19) (12). Para avaliar o efeito de resposta ligada à concentração do aumento de CD86, quaisquer concentrações escolhidas entre as concentrações utilizáveis devem ser repartidas uniformemente entre a CE150 (ou a concentração mais elevada não citotóxica negativa para CD86) e a CV70 (ou a concentração mais elevada permitida, ou seja, de 200 μg/ml). Para efeitos de comparação, devem testar-se, no mínimo, quatro concentrações por execução sendo, pelo menos, duas concentrações comuns à execução ou execuções anteriores.
|
|
15.
|
O controlo do solvente/veículo utilizado no ensaio U-SENS™ é o meio completo – para os produtos químicos em estudo dissolvidos ou em dispersão estável no meio completo, ver ponto 4 – ou o DMSO a 0,4 % em meio completo – para os produtos químicos em estudo dissolvidos ou em dispersão estável em DMSO.
|
|
16.
|
O controlo positivo utilizado no ensaio U-SENS™ é o TNBS (ver ponto 11), preparado em meio completo. O TNBS deve ser utilizado como controlo positivo para a medição da expressão de CD86 a uma concentração final única na placa (50 μg/ml) que produza uma viabilidade celular > 70 %. Para obter uma concentração de 50 μg/ml de TNBS na placa, prepara-se uma solução-mãe de TNBS a 1 M (ou seja, 293 mg/ml) em meio completo, antes de proceder a uma diluição por um fator de 2 930 no meio completo até se obter uma solução de trabalho a 100 μg/ml. O ácido láctico (AL, CAS 50-21-5) deve ser utilizado como controlo negativo, dissolvido a 200 μg/ml no meio completo, a partir de uma solução-mãe a 0,4 mg/ml. Em cada placa de cada execução, preparam-se três replicados de meio completo controlo não tratado, o controlo do solvente/veículo e os controlos negativo e positivo (12). Podem utilizar-se outros controlos positivos adequados se houver dados históricos que permitam definir critérios de aceitação comparáveis para a execução. Os critérios de aceitação da execução são idênticos aos descritos para o produto químico em estudo (ver ponto 12).
|
Aplicação dos produtos químicos em estudo e das substâncias de controlo
|
17.
|
O controlo do solvente/veículo ou as soluções de trabalho descritas nos pontos 14 a 16 são misturados na proporção 1:1 (v/v) com as suspensões de células preparadas na placa de 96 poços de fundo plano (ver ponto 12). As placas tratadas são, em seguida, incubadas durante 45 ± 3 horas a 37 °C, 5 % CO2. Antes da incubação, as placas são seladas com uma membrana semipermeável, para evitar a evaporação de produtos químicos em estudo voláteis e a contaminação cruzada entre células tratadas com os produtos químicos em estudo (12).
|
Coloração celular
|
18.
|
Após 45 ± 3 horas de exposição, as células são transferidas para uma placa de microtitulação de fundo cónico e recolhidas por centrifugação. A interferência de solubilidade é definida pela presença de cristais ou gotas visíveis ao microscópio 45 ± 3 horas após o tratamento (antes da coloração celular). Os sobrenadantes são rejeitados e as restantes células são lavadas uma vez com 100 μl de um tampão fosfato salino (TFS) gelado contendo 5 % de soro fetal de vitelo (tampão de coloração). Após centrifugação, as células são novamente colocadas em suspensão em 100 μl de tampão de coloração e coradas com 5 μl (ou seja, 0,25 μg) de anticorpos anti-CD86 ou anticorpos de ratinho (isótipo) IgG1 marcados com FITC a 4 °C durante 30 minutos, ao abrigo da luz. Devem utilizar-se os anticorpos descritos no protocolo DB-ALM n.o 183 U-SENS™ (12) (para CD86: BD-PharMingen #5556 57 Clone: Fun-1, ou Caltag/Invitrogen # MHCD8601 Clone: BU63; e para IgG1: BD-PharMingen #5557 48, ou Caltag/Invitrogen # GM4992). A experiência adquirida aquando do desenvolvimento do ensaio indica que a intensidade da fluorescência dos anticorpos é geralmente coerente entre diferentes lotes. É possível utilizar para o ensaio outros clones ou fornecedor dos anticorpos que tenham passado o controlo de reatividade (ver o ponto 11). No entanto, os utilizadores podem ponderar a possibilidade de proceder à titulação dos anticorpos em condições laboratoriais próprias, a fim de definir a concentração mais adequada. Podem utilizar-se outros sistemas de deteção, por exemplo anticorpos anti-CD86 marcados por outros fluorocromos, desde que seja possível demonstrar que produzem resultados semelhantes aos anticorpos conjugados por FITC, por exemplo testando as substâncias enumeradas no apêndice 2.2. Após duas lavagens com 100 μl de tampão de coloração e uma lavagem com 100 μl de TFS gelado, as células são novamente colocadas em suspensão em TFS gelado (por exemplo, 125 μl para as amostras a analisar manualmente tubo a tubo, ou 50 μl para uma placa de auto-amostragem) e acrescenta-se a solução de IP (concentração final: 3 μg/ml). Podem utilizar-se outros marcadores de citotoxicidade, tais como a 7-aminoactinomicina D (7-AAD) ou o azul de Trypan, desde que se possa demonstrar que produzem resultados semelhantes ao IP, por exemplo testando as substâncias enumeradas no apêndice 2.2.
|
Análise por citometria de fluxo
|
19.
|
O nível de expressão de CD86 e a viabilidade celular são analisados por citometria de fluxo. As células são dispostas num gráfico de pontos à escala logarítmica de acordo com o seu tamanho (FSC) e granularidade (CSS), a fim de identificar claramente a população numa primeira janela R1 e eliminar os resíduos. O objetivo consiste em adquirir 10 000 células na janela R1 para cada poço. As células pertencentes a uma mesma janela R1 são representadas num gráfico de pontos FL3 ou FL4/CSS. As células viáveis são identificadas traçando uma segunda janela R2 de seleção da população celular negativa para o iodeto de propídio (canais FL3 ou FL4). Pode calcular-se a viabilidade celular pelo programa de análise do citómetro utilizando a equação indicada em baixo. Se a viabilidade celular for baixa, devem ser adquiridas, no máximo, 20 000 células, incluindo células mortas. Uma outra opção consiste em adquirir os dados durante um minuto após o início da análise.

A percentagem das células FL1-positivas é, em seguida, medida entre as células viáveis retidas em R2 (dentro de R1). A expressão de CD86 na superfície celular é analisada através de um gráfico de pontos FL1/CSS em células viáveis (R2).
No caso dos poços que contêm meio completo/IgG1, o marcador de análise é fixado próximo da população principal, de modo a que os controlos em meio completo apresentem um valor IgG1 na zona alvo de 0,6 a 0,9 %.
A interferência de cor é definida como uma transferência da dispersão correspondente aos IgG1 marcados com FITC (médio geométrica de IE IgG1 FL1 ≥ 150 %).
O índice de estimulação (IE) de CD86 para as células de controlo (não tratadas ou em DMSO a 0,4 %) e para as células tratadas com produtos químicos é calculado pela seguinte equação:.

% de células de controlo não tratadas IgG1+: percentagem de células FL1-positivas reconhecidas pelo IgG1 que ultrapassam o marcador de análise (intervalo de aceitação ≥ 0,6 % e < 1,5 %, ver ponto 22) entre as células viáveis não tratadas.
% de células de controlo IgG1+/células tratadas CD86+: percentagem de células FL1-positivas reconhecidas pelo IgG1/CD86 medida sem deslocar o marcador de análise, entre as células viáveis de controlo/tratadas.
|
DADOS E RELATÓRIOS
Avaliação dos dados
|
20.
|
Os parâmetros seguintes são calculados no ensaio U-SENS™: valor CV70, isto é, a concentração que produz uma sobrevivência de 70 % das células U937 (30 % de citotoxicidade) e o valor CE150, ou seja, a concentração à qual os produtos químicos em estudo induzem um índice de estimulação (IE) de CD86 de 150 %.
O valor CV70 é calculado por interpolação linear logarítmica utilizando a seguinte equação:
CV70 = C1 + [(V1 - 70) / (V1 – V2) * (C2 – C1)]
em que:
V1 é o valor mínimo de viabilidade celular superior a 70 %
V2 é o valor máximo de viabilidade celular inferior a 70 %
C1 e C2 são as concentrações que provocam os valores de viabilidade celular V1 e V2, respetivamente.
Podem ser utilizadas outras abordagens para calcular o valor CV70, desde que fique demonstrado que tal não tem qualquer impacto nos resultados – por exemplo, testando as substâncias para demonstração de competência.
O valor CE150 é calculado por interpolação linear logarítmica utilizando a seguinte equação:
CE150 = C1 + [(150 – IE 1) / (IE 2 – IE 1) * (C2 – C1)]
Em que:
C1 é a concentração mais elevada em μg/ml com um IE CD86 < 150 % (IE 1)
C2 é a concentração mais baixa em μg/ml com um IE CD86 ≥ 150 % (IE 2)

Os valores CE150 e CV70 são calculados
|
—
|
para cada execução: os valores CE150 e CV70 são utilizados como ferramentas para investigar o efeito de resposta ligada à concentração do aumento de CD86 (ver ponto 14);
|
|
—
|
o valor CV70 global é determinado em função da viabilidade média (12);
|
|
—
|
o valor CE150 global de um produto químico em estudo que se preveja POSITIVO com o ensaio U-SENS™ é determinado em função dos valores médios de IE CD86 (ver ponto 21) (12).
|
|
Modelo preditivo
|
21.
|
Para a medição de expressão CD86, cada produto químico em estudo é testado em, pelo menos, quatro concentrações e em, pelo menos, duas execuções independentes (efetuadas em dias diferentes) para chegar a uma previsão única (NEGATIVO ou POSITIVO).
|
—
|
A conclusão individual de uma execução do ensaio U-SENS™ é considerada negativa (a seguir designada por N) se o IE de CD86 for inferior a 150 % a todas as concentrações não citotóxicas (viabilidade celular ≥ 70 %) e se não for observada nenhuma interferência (citotoxicidade, solubilidade: ver ponto 18, ou cor: ver ponto 19, independentemente das concentrações não citotóxicas às quais a interferência é detetada). Em todos os outros casos (para um IE de CD86 superior ou igual a 150 % e/ou interferências observadas), a conclusão individual de uma execução do ensaio U-SENS™ é considerada positiva, sendo a seguir designada por P.
|
|
—
|
Uma previsão U-SENS™ é considerada NEGATIVA se, pelo menos, duas execuções independentes forem negativas (N) (figura 1). Se as duas primeiras execuções forem negativas (N), a previsão U-SENS™ é considerada NEGATIVA e não é necessário proceder a uma terceira execução.
|
|
—
|
Uma previsão U-SENS™ é considerada POSITIVA se, pelo menos, duas execuções independentes forem positivas (P) (figura 1). Se as duas primeiras execuções forem positivas (P), a previsão U-SENS™ é considerada POSITIVA e não é necessário proceder a uma terceira execução.
|
|
—
|
Uma vez que não é realizado um ensaio de determinação da dose, há uma exceção se, na primeira execução, o IE de CD86 for superior ou igual a 150 % apenas à concentração não citotóxica mais elevada. Neste caso, a execução é considerada INCONCLUSIVA (NC), devendo testar-se, em execuções suplementares, concentrações adicionais (entre a concentração mais elevada não citotóxica e a concentração mais baixa citotóxica — ver ponto 20). Se uma execução for identificada como NC, convém realizar pelo menos duas execuções suplementares, e uma quarta execução caso os resultados das execuções 2 e 3 não sejam concordantes (N e/ou P, a título independente) (figura 1). As execuções seguintes serão consideradas positivas mesmo que apenas uma das concentrações não citotóxicas dê um valor de CD86 superior ou igual a 150 %, uma vez que os parâmetros de concentração foram adaptados especificamente a este produto químico em estudo. A previsão final basear-se-á no resultado maioritário das três ou quatro execuções individuais (ou seja, 2 de 3 ou 2 de 4) (figura 1).
|
Figura 1: Modelo preditivo utilizado no ensaio U-SENS™. Uma previsão por U-SENS™ deve ser considerada no âmbito de uma IATA e em conformidade com o disposto no ponto 4 e nos pontos 7, 8 e 9 da Introdução Geral.
N: Execução sem resultado positivo para CD86 nem interferência observada.
P: Execução com resultado positivo para CD86 e/ou interferência(s) observada(s).
NC: Inconclusivo. Primeira execução inconclusiva se CD86 for positivo apenas à concentração não citotóxica mais elevada.
#: Um resultado individual inconclusivo (NC) atribuído apenas aquando da primeira execução requer automaticamente uma terceira execução para chegar a uma maioria de conclusões positivas (P) ou negativas (N) em pelo menos duas de três execuções independentes.
$: As caixas mostram as combinações pertinentes de resultados das três execuções com base nos resultados obtidos nas duas primeiras execuções indicados na caixa acima.
o: As caixas mostram as combinações pertinentes de resultados das quatro execuções com base nos resultados obtidos nas três primeiras execuções indicados na caixa acima.
|
Critérios de aceitação
|
22.
|
Ao utilizar o método de ensaio U-SENS™ devem respeitar-se os critérios de aceitação a seguir indicados (12).
|
—
|
Após uma exposição de 45 ± 3 horas, a viabilidade média dos três replicados de células U937 não tratadas deve ser > 90 % e não deve observar-se qualquer deriva da expressão de CD86. A expressão basal de CD86 nas células U937 não tratadas deve estar compreendida entre ≥ 2 % e ≤ 25 %.
|
|
—
|
Se o solvente utilizado for o DMSO, a sua validade como controlo do veículo é avaliada calculando o IE do DMSO em comparação com o das células não tratadas, devendo a viabilidade média dos três replicados de células ser > 90 %. O controlo de veículo DMSO é válido se o IE médio de CD86 nos três replicados de DMSO for inferior a 250 % do IE médio de CD86 nos três replicados de células U937 não tratadas.
|
|
—
|
As execuções são consideradas válidas se, pelo menos, dois dos três valores IgG1 das células U937 não tratadas forem ≥ 0,6 % e < 1,5 %.
|
|
—
|
O controlo negativo testado em paralelo (ácido láctico) é considerado válido se, pelo menos, dois dos três replicados forem negativos (IE CD86 < 150 %) e não citotóxicos (viabilidade celular ≥ 70 %).
|
|
—
|
O controlo positivo (TNBS) é considerado válido se pelo menos dois dos três replicados forem positivos (IE CD86 ≥ 150 %) e não citotóxicos (viabilidade celular ≥ 70 %).
|
|
Relatório de ensaio
|
23.
|
O relatório de ensaio deve incluir as informações indicadas a seguir.
|
Produto químico em estudo
Substância monocomponente
|
Identificação química, como o(s) nome(s) IUPAC ou CAS, o(s) número(s) CAS, o código SMILES ou InChI, a fórmula estrutural e/ou outros identificadores;
|
|
Aspeto físico, solubilidade no meio completo, solubilidade em DMSO, massa molecular e outras propriedades físico-químicas relevantes, se disponíveis;
|
|
Grau de pureza, identidade química das impurezas, conforme adequado e exequível, etc.;
|
|
Pré-tratamento, se for o caso (aquecimento ou moagem, por exemplo);
|
|
Concentração(ões) testada(s);
|
|
Condições de armazenagem e de estabilidade, se conhecidas;
|
|
Justificação da escolha do solvente/veículo para cada produto químico em estudo.
|
Substância multicomponentes, UVCB e mistura:
|
Caracterização, tanto quanto possível, por exemplo, por identidade química (ver supra), pureza, ocorrência quantitativa e propriedades físico-químicas relevantes (ver supra) dos componentes, na medida em que estejam disponíveis;
|
|
Aspeto físico, solubilidade no meio completo, solubilidade em DMSO e outras propriedades físico-químicas relevantes, na medida em que estejam disponíveis;
|
|
Massa molecular, ou massa molecular aparente no caso de misturas/polímeros de composições conhecidas, ou outras informações relevantes para a realização do estudo;
|
|
Pré-tratamento, se for o caso (aquecimento ou moagem, por exemplo);
|
|
Concentração(ões) testada(s);
|
|
Condições de armazenagem e de estabilidade, se conhecidas;
|
|
Justificação da escolha do solvente/veículo para cada produto químico em estudo.
|
|
|
Controlos
Controlo positivo
|
Identificação química, como o(s) nome(s) IUPAC ou CAS, o(s) número(s) CAS, o código SMILES ou InChI, a fórmula estrutural e/ou outros identificadores;
|
|
Aspeto físico, solubilidade em DMSO, massa molecular e outras propriedades físico-químicas, se conhecidas e se pertinente;
|
|
Grau de pureza, identidade química das impurezas, conforme adequado e exequível, etc.;
|
|
Pré-tratamento, se for o caso (aquecimento ou moagem, por exemplo);
|
|
Concentração(ões) testada(s);
|
|
Condições de armazenagem e de estabilidade, se conhecidas;
|
|
Referência ao historial dos resultados dos controlos positivos que demonstrem o cumprimento dos critérios de aceitação da execução, se aplicável.
|
Controlo negativo e controlo do solvente/veículo
|
Identificação química, como o(s) nome(s) IUPAC ou CAS, o(s) número(s) CAS, o código SMILES ou InChI, a fórmula estrutural e/ou outros identificadores;
|
|
Grau de pureza, identidade química das impurezas, conforme adequado e exequível, etc.;
|
|
Aspeto físico, massa molecular e outras propriedades físico-químicas pertinentes, caso seja utilizado um solvente/veículo diferente dos mencionados nas orientações de ensaio, na medida em que estejam disponíveis;
|
|
Condições de armazenagem e de estabilidade, se conhecidas;
|
|
Justificação da escolha do solvente/veículo para cada produto químico em estudo.
|
|
|
Condições de ensaio
|
Nome e endereço do promotor, do laboratório e do diretor do estudo;
|
|
Descrição do método de ensaio utilizado;
|
|
Linha celular utilizada, respetivas condições de armazenamento e origem (por exemplo, instalação de onde provêm as células);
|
|
Citometria de fluxo utilizada (por exemplo, modelo), incluindo parâmetros dos instrumentos, anticorpos e marcador de citotoxicidade utilizados;
|
|
Procedimento utilizado para demonstrar as competências do laboratório na realização do ensaio, mediante o teste de substâncias para demonstração de competência, e procedimento utilizado para demonstrar a reprodutibilidade do desempenho do ensaio ao longo do tempo, por exemplo dados históricos do controlo e/ou dados históricos dos controlos de reatividade.
|
|
|
Critérios de aceitação do ensaio
|
Viabilidade celular e valores IE CD86 obtidos com o controlo do solvente/veículo, em comparação com os intervalos de aceitação;
|
|
Viabilidade celular e valores IE obtidos com o controlo positivo, em comparação com os intervalos de aceitação;
|
|
Viabilidade celular de todas as concentrações testadas do produto químico em estudo.
|
|
|
Procedimento de ensaio
|
Número de execuções realizadas;
|
|
Concentrações do produto químico em estudo, aplicação e tempo de exposição utilizado (se diferente do recomendado);
|
|
Descrição dos critérios de avaliação e de decisão utilizados;
|
|
Descrição de eventuais modificações do procedimento de ensaio.
|
|
|
Resultados
|
Quadro com os dados, incluindo o valor CV70 (se pertinente), IE, valores de viabilidade celular, valores CE150 (se pertinente) obtidos para o produto químico em estudo e para o controlo positivo em cada execução, bem como uma indicação da classificação do produto químico em estudo segundo o modelo preditivo;
|
|
Descrição de quaisquer outras observações pertinentes, se for caso disso.
|
|
|
Discussão dos resultados
|
Discussão dos resultados obtidos com o método U-SENS™;
|
|
Análise dos resultados do ensaio no contexto de uma IATA, caso se disponha de outras informações pertinentes.
|
|
Conclusões
|
REFERÊNCIAS BIBLIOGRÁFICAS
|
(1)
|
Piroird, C., Ovigne, J.M., Rousset, F., Martinozzi-Teissier, S., Gomes, C., Cotovio, J., Alépée, N. (2015). The Myeloid U937 Skin Sensitization Test (U-SENS) addresses the activation of dendritic cell event in the adverse outcome pathway for skin sensitization. Toxicol. In Vitro 29, 901-916.
|
|
(2)
|
EURL ECVAM (2017). The U-SENS™ test method Validation Study Report. Disponível em: http://ihcp.jrc.ec.europa.eu/our_labs/eurl-ecvam/eurl-ecvam-recommendations
|
|
(3)
|
EC EURL ECVAM (2016). ESAC Opinion No 2016-03 on the L'Oréal-coordinated study on the transferability and reliability of the U-SENS™ test method for skin sensitisation testing. EUR 28 178 EN; doi 10.2 787/8157 37. Disponível em: [http://publications.jrc.ec.europa.eu/repository/handle/JRC103705].
|
|
(4)
|
EC EURL ECVAM (2017). EURL ECVAM Recommendation on the use of non-animal approaches for skin sensitisation testing. EUR 28 553 EN; doi 10.2 760/5889 55. Disponível em: https://ec.europa.eu/jrc/en/publication/eur-scientific-and-technical-research-reports/eurl-ecvam-recommendation-use-non-animal-approaches-skin-sensitisation-testing.
|
|
(5)
|
Steiling, W. (2016). Safety Evaluation of Cosmetic Ingredients Regarding their Skin Sensitization Potential. doi:10.3390/cosmetics3020014.Cosmetics 3, 14.
|
|
(6)
|
OECD (2016). Guidance Document on The Reporting of Defined Approaches and Individual Information Sources to be Used Within Integrated Approaches to Testing and Assessment (IATA) For Skin Sensitisation, Series on Testing & Assessment No 256, ENV/JM/MONO(2016)29. Organisation for Economic Cooperation and Development, Paris. Disponível em: [ http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(7)
|
Urbisch, D., Mehling, A., Guth, K., Ramirez, T., Honarvar, N., Kolle, S., Landsiedel, R., Jaworska, J., Kern, P.S., Gerberick, F., Natsch, A., Emter, R., Ashikaga, T., Miyazawa, M., Sakaguchi, H. (2015). Assessing skin sensitization hazard in mice and men using non-animal test methods. Regul. Toxicol. Pharmacol. 71, 337-351.
|
|
(8)
|
Alépée, N., Piroird, C., Aujoulat, M., Dreyfuss, S., Hoffmann, S., Hohenstein, A., Meloni, M., Nardelli, L., Gerbeix, C., Cotovio, J. (2015). Prospective multicentre study of the U-SENS test method for skin sensitization testing. Toxicol In Vitro 30, 373-382.
|
|
(9)
|
Reisinger, K., Hoffmann, S., Alépée, N., Ashikaga, T., Barroso, J., Elcombe, C., Gellatly, N., Galbiati, V., Gibbs, S., Groux, H., Hibatallah, J., Keller, D., Kern, P., Klaric, M., Kolle, S., Kuehnl, J., Lambrechts, N., Lindstedt, M., Millet, M., Martinozzi-Teissier, S., Natsch, A., Petersohn, D., Pike, I., Sakaguchi, H., Schepky, A., Tailhardat, M., Templier, M., van Vliet, E., Maxwell, G. (2014). Systematic evaluation of non-animal test methods for skin sensitisation safety assessment. Toxicol. In Vitro 29, 259-270.
|
|
(10)
|
Fabian, E., Vogel, D., Blatz, V., Ramirez, T., Kolle, S., Eltze, T., van Ravenzwaay, B., Oesch, F., Landsiedel, R. (2013). Xenobiotic metabolizin enzyme activities in cells used for testing skin sensitization in vitro. Arch. Toxicol. 87, 1683-1696.
|
|
(11)
|
OECD. (2018). Draft Guidance document: Good In Vitro Method Practices (GIVIMP) for the Development and Implementation of In Vitro Methods for Regulatory Use in Human Safety Assessment. Organisation for Economic Cooperation and Development, Paris. Disponível em: http://www.oecd.org/env/ehs/testing/OECD Final Draft GIVIMP.pdf.
|
|
(12)
|
DB-ALM (2016). Protocol no 183: Myeloid U937 Skin Sensitization Test (U-SENS™), 33pp. Disponível em: [http://ecvam-dbalm.jrc.ec.europa.eu/].
|
|
(13)
|
Sundström, C., Nilsson, K. (1976). Establishment and characterization of a human histiocytic lymphoma cell line (U-937). Int. J. Cancer 17, 565-577.
|
|
(14)
|
OECD (2005). Series on Testing and Assessment No. 34: Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Organisation for Economic Cooperation and Development, Paris. Disponível em: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(15)
|
United Nations UN (2015). Globally Harmonized System of Classification and Labelling of Chemicals (GHS). ST/SG/AC.10/30/Rev.6, Sixth Revised Edition, Nova Iorque e Genebra: United Nations Publications. Disponível em: http://www.unece.org/fileadmin/DAM/trans/danger/publi/ghs/ghs_rev06/English/ST-SG-AC10-30-Rev6e.pdf.
|
|
(16)
|
OECD (2012). Series on Testing and Assessment No 168: The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins. Part 1: Scientific Evidence. Organisation for Economic Cooperation and Development, Paris. Disponível em: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(17)
|
ECETOC (2003). Technical Report No 87: Contact sensitization: Classification according to potency. European Centre for Ecotoxicology & Toxicology of Chemicals, Bruxelas. Disponível em:https://ftp.cdc.gov/pub/Documents/OEL/06.%20Dotson/References/ECETOC_2003-TR87.pdf.
|
Apêndice 2.1
DEFINIÇÕES
AOP (Adverse Outcome Pathway – via determinante dos efeitos nocivos)
: Sequência de eventos que parte da estrutura química de um produto químico alvo ou de um grupo de produtos químicos análogos, passa pelo evento molecular iniciador, e resulta num resultado com consequências in vivo (15).
CV70
: Concentração estimada que apresenta uma viabilidade celular de 70 %.
Deriva
: A deriva é definida pelo seguinte i) valor corrigido %CD86+ do replicado 3 do controlo não tratado é inferior a 50 % da média corrigida do valor %CD86+ dos replicados 1 e 2 do controlo não tratado; e ii) valor corrigido %CD86+ do replicado 3 do controlo negativo é inferior a 50 % da média corrigida do valor %CD86+ dos replicados 1 e 2 do controlo negativo.
CE150
: Concentrações estimadas que apresentam um IE de 150 % para a expressão de CD86.
Citometria de fluxo
: Técnica citométrica na qual células em suspensão num fluido passam, uma a uma, por um foco de excitação luminosa, disperso em padrões característicos das células e dos seus componentes; as células são frequentemente marcadas com marcadores fluorescentes, para que a luz seja primeiro absorvida e depois emitida a uma frequência diferente.
Controlo positivo
: Replicado que contém todos os componentes de um sistema de ensaio e foi tratado com uma substância conhecida por induzir uma resposta positiva. Para que seja possível determinar a variabilidade no tempo da resposta do controlo positivo, a magnitude da resposta positiva não deve ser excessiva.
Controlo do solvente/veículo
: Amostra não tratada que contém todos os componentes de um sistema de ensaio, com exceção do produto químico em estudo, mas incluindo o solvente/veículo utilizado. Serve para determinar a resposta de referência para as amostras tratadas com o produto químico em estudo dissolvido ou em dispersão estável no mesmo solvente/veículo. Quando testada simultaneamente com um controlo de meio, esta amostra também indica se o solvente/veículo interage com o sistema de ensaio.
Ensaio válido
: Um ensaio considerado suficientemente pertinente e fiável para um fim específico e que se baseia em princípios cientificamente sólidos. Um ensaio nunca é válido em termos absolutos, mas apenas em relação a um objetivo definido (14).
Especificidade
: Proporção dos produtos químicos negativos/inativos que são corretamente classificados pelo ensaio. Permite medir a precisão de um ensaio que produz resultados categóricos e é um aspeto importante a ter em conta na avaliação da pertinência de um ensaio (14).
Execução
: Consiste em testar um ou mais produtos químicos em estudo simultaneamente com um controlo do solvente/veículo e com um controlo positivo.
Fiabilidade
: Indica em que medida um ensaio pode ser reproduzido ao longo do tempo num mesmo laboratório e entre laboratórios, utilizando o mesmo protocolo. Para a avaliar, calcula-se a reprodutibilidade intralaboratorial e interlaboratorial, bem como a repetibilidade intralaboratorial (14).
IATA (Integrated Approach to Testing and Assessment – abordagem integrada de ensaio e avaliação)
: Abordagem estruturada utilizada para a identificação (potencial) e caracterização (potência) do perigo e/ou para a avaliação da segurança (potencial/potência e exposição) de um produto químico ou grupo de produtos químicos, que integra e pondera, de forma estratégica, todos os dados pertinentes com o objetivo de ser tida em conta numa decisão regulamentar sobre o potencial perigo e/ou risco e/ou a necessidade de realizar ensaios complementares específicos.
IE
: Índice de estimulação. Valores relativos da média geométrica da intensidade de fluorescência das células tratadas com o produto químico em comparação com as células tratadas com o solvente.
Mistura
: Uma mistura ou solução composta por duas ou mais substâncias.
Perigo
: Propriedade intrínseca de um agente, ou de uma situação, suscetível de causar efeitos nocivos quando um organismo, sistema ou (sub)população é exposto ao agente em causa.
Pertinência
: Descrição da relação do ensaio com o efeito em estudo e da adequação e utilidade do ensaio para um fim específico. Traduz a medida em que o ensaio mede ou prevê corretamente o efeito biológico em causa. A pertinência tem em conta a precisão (concordância) de um ensaio (14).
Precisão
: Grau de acordo entre os resultados do ensaio e os valores de referência aceites. Trata-se de uma medida do desempenho do ensaio e de um dos aspetos da sua pertinência. Este termo e o termo «concordância» são muitas vezes utilizados indistintamente para indicar a proporção de resultados corretos de um ensaio (14).
Pré-haptenos
: Produtos químicos que se tornam sensibilizantes por transformação abiótica, por exemplo por oxidação.
Pró-haptenos
: Produtos químicos que requerem ativação enzimática para exercerem potencial de sensibilização cutânea.
Produto químico
: Uma substância ou mistura.
Produto químico em estudo
: Qualquer substância ou mistura à qual seja aplicado o presente método.
Resposta de CD86 ligada à concentração
: O efeito é dependente da concentração (ou resposta ligada à concentração) quando uma concentração positiva (IE CD86 ≥ 150) é seguida de uma concentração com um IE CD86 ainda maior.
Sensibilidade
: Proporção de todos os produtos químicos positivos/ativos que são corretamente classificados pelo ensaio. Permite medir a precisão de um ensaio que produz resultados categóricos e é um aspeto importante a ter em conta na avaliação da pertinência de um ensaio (14).
Sistema Mundial Harmonizado de Classificação e Rotulagem de Produtos Químicos das Nações Unidas – GHS da ONU (em inglês
: United Nations Globally Harmonized System of Classification and Labelling of Chemicals – UN GHS) Sistema que propõe a classificação dos produtos químicos (substâncias e misturas) em função de tipos e níveis normalizados de perigos físicos, sanitários e ambientais e trata dos elementos de comunicação correspondentes, nomeadamente pictogramas, palavras-sinal, advertências de perigo, recomendações de prudência e fichas de dados de segurança, de modo a transmitir informações sobre os respetivos efeitos nocivos, tendo em vista a proteção das pessoas (incluindo empregadores, trabalhadores, transportadores, consumidores e pessoal dos serviços de emergência) e do ambiente (16).
Substância
: Um elemento químico e seus compostos, no estado natural ou obtidos por qualquer processo de produção, incluindo qualquer aditivo necessário para preservar a sua estabilidade e quaisquer impurezas resultantes do processo utilizado, mas excluindo qualquer solvente que possa ser separado sem afetar a estabilidade da substância nem alterar a sua composição.
Substância monocomponente
: Uma substância, definida pela sua composição quantitativa, da qual um dos principais componentes está presente num teor de, pelo menos, 80 % (m/m).
Substância multicomponentes
: Uma substância, definida pela sua composição quantitativa, da qual mais do que um dos principais componentes está presente numa concentração ≥ 10 % (m/m) e < 80 % (m/m). As substâncias multicomponentes resultam de processos de fabrico. A diferença entre uma mistura e uma substância multicomponentes é que a primeira se obtém misturando duas ou mais substâncias, sem reação química. As substâncias multicomponentes resultam de reações químicas.
Tampão de coloração
: Uma solução salina de tampão fosfato com 5 % de soro fetal de vitelo.
UVCB
: Substâncias de composição desconhecida ou variável, produtos de reação complexos ou materiais biológicos.
Apêndice 2.2
SUBSTÂNCIAS PARA A DEMONSTRAÇÃO DE COMPETÊNCIA
Antes de utilizarem, por rotina, o ensaio descrito no presente apêndice do método de ensaio B.71, os laboratórios devem demonstrar a sua competência técnica obtendo corretamente a previsão esperada com o método U-SENS™ para as dez substâncias recomendadas no quadro 1 e obtendo valores CV70 e CE150 que se encontrem dentro do intervalo de referência correspondente para, pelo menos, oito das dez substâncias de demonstração de competência. Estas substâncias foram selecionadas de modo a representarem o conjunto de respostas possíveis no que diz respeito aos perigos de sensibilização cutânea. Outros critérios de seleção foram a disponibilidade comercial das substâncias e a disponibilidade de dados de referência in vivo de elevada qualidade, bem como de dados in vitro de alta qualidade produzidos com o método U-SENS™. Além disso, estão disponíveis dados de referência publicados para o método U-SENS™ (1) (8).
Quadro 1
Substâncias recomendadas para a demonstração de competência técnica com o método U-SENS™
|
Substâncias para a demonstração de competência
|
N.o CAS
|
Estado físico
|
Previsão in vivo
(105)
|
U-SENS Solvente/Veículo
|
U-SENS Intervalo de referência CV70 em μg/ml (106)
|
U-SENS Intervalo de referência CE150 em μg/ml (106)
|
|
4-Fenilenodiamina
|
106-50-3
|
Sólido
|
Sensibilizante (forte)
|
Meio completo (107)
|
< 30
|
Positivo (≤ 10)
|
|
Ácido picrilsulfónico
|
2508-19-2
|
Líquido
|
Sensibilizante (forte)
|
Meio completo
|
> 50
|
Positivo (≤ 50)
|
|
Maleato de dietilo
|
141-05-9
|
Líquido
|
Sensibilizante (moderado)
|
DMSO
|
10-100
|
Positivo (≤ 20)
|
|
Resorcinol
|
108-46-3
|
Sólido
|
Sensibilizante (moderado)
|
Meio completo
|
> 100
|
Positivo (≤ 50)
|
|
Álcool cinâmico
|
104-54-1
|
Sólido
|
Sensibilizante (fraco)
|
DMSO
|
> 100
|
Positivo (10-100)
|
|
4-Alilanisol
|
140-67-0
|
Líquido
|
Sensibilizante (fraco)
|
DMSO
|
> 100
|
Positivo (< 200)
|
|
Sacarina
|
81-07-2
|
Sólido
|
Não sensibilizante
|
DMSO
|
> 200
|
Negativo (> 200)
|
|
Glicerol
|
56-81-5
|
Líquido
|
Não sensibilizante
|
Meio completo
|
> 200
|
Negativo (> 200)
|
|
Ácido láctico
|
50-21-5
|
Líquido
|
Não sensibilizante
|
Meio completo
|
> 200
|
Negativo (> 200)
|
|
Ácido salicílico
|
69-72-7
|
Sólido
|
Não sensibilizante
|
DMSO
|
> 200
|
Negativo (> 200)
|
|
Abreviaturas: N.o CAS = Número de registo do Chemical Abstracts Service
|
Apêndice 3
SENSIBILIZAÇÃO CUTÂNEA IN VITRO: ENSAIO IL-8 LUC
CONSIDERAÇÕES INICIAIS E LIMITAÇÕES
|
1.
|
Ao contrário dos ensaios que visam analisar o nível de expressão de marcadores de superfície celular, o ensaio IL-8 Luc permite quantificar as variações da expressão da IL-8, uma citocina associada à ativação das células dendríticas. Na linha celular repórter IL-8 derivada da linha celular THP-1 (THP-G8, estabelecida a partir da linha celular da leucemia monocítica aguda humana THP-1), a expressão da IL-8 é medida após exposição a sensibilizantes (1). O nível de expressão da luciferase é, depois, utilizado para ajudar a distinguir os sensibilizantes dos não sensibilizantes cutâneos.
|
|
2.
|
O ensaio IL-8 Luc foi objeto de um estudo de validação (2) realizado pelo Centro Japonês para a Validação de Métodos Alternativos (JaCVAM), pelo Ministério da Economia, do Comércio e da Indústria (METI) e pela Sociedade Japonesa para Alternativas à Experimentação em Animais (JSAAE), seguido de uma análise independente pelos pares (3) sob os auspícios do JaCVAM e do Ministério da Saúde, do Trabalho e da Segurança Social (MHLW), com o apoio da Cooperação Internacional em matéria de Métodos de Ensaio Alternativos (ICATM — International Cooperation on Alternative Test Methods). Tendo em conta todos os dados disponíveis e os contributos das entidades reguladoras e das partes interessadas, o ensaio IL-8 Luc é considerado útil no âmbito de uma IATA para distinguir os sensibilizantes dos não sensibilizantes, para efeitos de classificação e rotulagem de perigos. As referências bibliográficas contêm exemplos da utilização de dados do ensaio IL-8 Luc combinados com outras informações (4) (5) (6).
|
|
3.
|
O ensaio IL-8 Luc demonstrou ser transferível para laboratórios experientes em cultura celular e na medição da luciferase. O nível de reprodutibilidade intralaboratorial e interlaboratorial foi de 87,7 % e 87,5 %, respetivamente. Os dados obtidos com o estudo de validação (2) e outros estudos publicados (1) (6) demonstram que, em comparação com o método LLNA, o ensaio IL-8 Luc permitiu efetuar uma previsão positiva ou negativa para 118 de 143 produtos químicos, produziu resultados inconclusivos para 25 produtos químicos e apresenta uma precisão de 86 % (101/118), uma sensibilidade de 96 % (92/96) e uma especificidade de 41 % (9/22) para a distinção entre os produtos químicos sensibilizantes cutâneos (categoria 1 do GHS da ONU/CLP) e não sensibilizantes (sem categoria, de acordo com o GHS da ONU/CLP). Excluindo as substâncias que não são abrangidas pelo âmbito de aplicação descrito infra (ponto 5), o ensaio IL-8 Luc permitiu classificar 113 de 136 produtos químicos como positivos ou negativos e 23 produtos químicos como inconclusivos. A precisão do ensaio IL-8 Luc é de 89 % (101/113), com uma sensibilidade de 96 % (92/96) e uma especificidade de 53 % (9/17). Utilizando os dados humanos citados em Urbisch et al. (7), o ensaio IL-8 Luc permitiu classificar 76 de 90 produtos químicos como positivos ou negativos e 14 produtos químicos como inconclusivos, com uma precisão de 80 % (61/76), uma sensibilidade de 93 % (54/58) e uma especificidade de 39 % (7/18). Excluindo as substâncias que não são abrangidas pelo seu âmbito de aplicação, o ensaio IL-8 Luc permitiu classificar 71 de 84 produtos químicos como positivos ou negativos e 13 produtos químicos como inconclusivos, com uma precisão de 86 % (61/71), uma sensibilidade de 93 % (54/58) e uma especificidade de 54 % (7/13). É mais provável que ocorram falsos negativos nas previsões efetuadas com o ensaio IL-8 Luc com produtos químicos com potencial de sensibilização cutânea baixo/moderado (subcategoria 1B do GHS da ONU/CLP) do que com produtos químicos com um potencial elevado (subcategoria 1A do GHS da ONU/CLP) (6). No seu conjunto, estas informações corroboram o papel do ensaio do IL-8 Luc na identificação dos perigos de sensibilização cutânea. Os valores relativos à precisão do método de ensaio IL-8 Luc utilizado isoladamente servem apenas de orientação, uma vez que o presente método deve ser combinado com outras fontes de informação no contexto de uma IATA e em conformidade com o disposto nos pontos 7 e 8 da Introdução Geral. Além disso, na avaliação de métodos de estudo da sensibilização cutânea que não utilizem animais, deve ter-se em mente que o método LLNA e outros ensaios com animais podem não refletir de forma adequada a situação no ser humano.
|
|
4.
|
Os dados atualmente disponíveis demonstram que o ensaio IL-8 Luc é aplicável a produtos químicos em estudo que abrangem uma grande diversidade de grupos funcionais orgânicos, mecanismos de reação, potências de sensibilização cutânea (determinadas em estudos in vivo) e propriedades físico-químicas (2) (6).
|
|
5.
|
Embora utilize o solvente X-VIVOTM 15, o ensaio IL-8 Luc permite avaliar corretamente os produtos químicos com log P > 3,5 e os produtos químicos com uma solubilidade na água de cerca de 100 μg/ml, tal como calculado com o programa EPI SuiteTM. Além disso, o seu desempenho para detetar sensibilizantes com fraca solubilidade na água é superior ao do ensaio IL-8 Luc utilizando dimetilsulfóxido (DMSO) como solvente (2). No entanto, os resultados negativos obtidos com produtos químicos em estudo não dissolvidos a 20 mg/ml podem ser falsos negativos, devido à sua incapacidade para se dissolverem no X-VIVOTM 15. Por conseguinte, para estes produtos químicos, os resultados negativos não devem ser tidos em conta. No estudo de validação, observou-se uma elevada taxa de falsos negativos para os anidridos. Além disso, devido à capacidade metabólica limitada da linha celular utilizada (8) e às condições experimentais, os pró-haptenos (substâncias que requerem ativação metabólica) e os pré-haptenos (substâncias ativadas por oxidação) podem produzir resultados negativos no ensaio. No entanto, embora os resultados negativos obtidos com potenciais pré-haptenos ou pró-haptenos devam ser interpretados com prudência, o método IL-8 Luc permitiu identificar corretamente 11 de 11 pré-haptenos, 6/6 pró-haptenos e 6/8, pré-/pró-haptenos, no conjunto de dados do ensaio IL-8 Luc (2). O exame completo recentemente efetuado de três métodos de ensaio sem experimentação animal (o DPRA, o KeratinoSens™ e o h-CLAT) para detetar pré-haptenos e pró-haptenos (9) e o facto de as células THP-G8 utilizadas no ensaio IL-8 Luc serem uma linha celular derivada da THP-1, utilizada no método h-CLAT, permitem concluir que o ensaio IL-8 Luc, combinado com outros métodos, pode contribuir igualmente para melhorar a sensibilidade dos métodos sem recurso a animais na deteção de pré-haptenos e pró-haptenos. Os tensioativos testados até à data deram resultados positivos falsos, independentemente da sua tipologia (catiónicos, aniónicos ou não iónicos). Por último, os produtos químicos que interferem com a luciferase podem alterar a sua atividade/medição, provocando uma inibição aparente ou um aumento da luminescência (10). Por exemplo, foi notificado que concentrações de fitoestrogénios superiores a 1 μM interferem com sinais de luminescência noutros ensaios por gene repórter da luciferase, devido à sobreativação do gene repórter da luciferase. Por conseguinte, a expressão da luciferase obtida na presença de concentrações elevadas de fitoestrogénios ou de compostos suspeitos de ativar o gene repórter da luciferase de modo similar deve ser examinada com prudência (11). Atendendo ao que precede, os tensioativos, os anidridos e os produtos químicos que interferem com a luciferase são excluídos do âmbito de aplicação do presente ensaio. Nos casos em que seja demonstrado que o ensaio IL-8 Luc não é aplicável a outras categorias específicas de produtos químicos em estudo, este ensaio não deve ser utilizado para essas categorias específicas.
|
|
6.
|
Tal como descrito acima, o ensaio IL-8 Luc ajuda a distinguir os sensibilizantes cutâneos dos não sensibilizantes. É necessário prosseguir os trabalhos, de preferência com base em dados humanos, para determinar se os resultados do IL-8 Luc poderão contribuir para a avaliação do potencial de sensibilização cutânea quando considerados em combinação com outras fontes de informação.
|
|
7.
|
São apresentadas definições no apêndice 3.1.
|
PRINCÍPIO DO ENSAIO
|
8.
|
O método de ensaio IL-8 Luc utiliza uma linha celular da leucemia monocítica humana, THP-1, obtida junto da American Type Culture Collection (Manassas, VA, EUA). A partir desta linha celular, o Departamento de Dermatologia da Faculdade de Medicina da Universidade de Tohoku desenvolveu a linha celular repórter IL-8, derivada da THP-1, a THP-G8, que contém os genes da luciferase laranja (Stable Luciferase Orange, SLO) e vermelha (Stable Luciferase Red, SLR), sob o controlo dos promotores da IL-8 e da gliceraldeído-3-fosfato desidrogenase (GAPDH), respetivamente (1). Este sistema permite quantificar a indução do gene da luciferase por deteção da luminescência produzida por substratos de luciferase conhecidos pela sua emissão luminosa como indicador da atividade da IL-8 e da GAPDH nas células após a exposição a produtos químicos sensibilizantes.
|
|
9.
|
O sistema de ensaio bicolor inclui uma luciferase emissora de laranja (SLO; λmáx. = 580 nm) (12) para a expressão genética do promotor da IL-8, bem como uma luciferase emissora de vermelho (SLR; λmáx. = 630 nm) (13) para a expressão genética do promotor do controlo interno, GAPDH. As duas luciferases emitem cores diferentes quando reagem à d-luciferina de pirilampo e a sua luminescência é medida simultaneamente aquando de uma reação numa só etapa, dividindo a emissão proveniente da mistura do ensaio com o auxílio de um filtro ótico (14) (apêndice 3.2).
|
|
10.
|
As células THP-G8 são tratadas durante 16 horas com o produto químico em estudo, após o que a atividade da luciferase SLO (SLO-LA), indicativa da atividade do promotor da IL-8, e da luciferase SLR (SLR-LA), indicativa da atividade do promotor da GAPDH, são medidas. Para facilitar a compreensão das abreviaturas, SLO-LA e SLR-LA são designadas, respetivamente, por IL8LA e GAPLA. O quadro 1 apresenta uma descrição dos termos associados à atividade da luciferase no ensaio IL-8 Luc. Os valores medidos são utilizados para calcular a IL8LA normalizado (nIL8LA), ou seja, o rácio IL8LA/GAPLA; a indução da nIL8LA (Ind-IL8LA), que é o rácio entre a média aritmética dos quatro valores medidos da nIL8LA das células THP-G8 tratadas com o produto químico em estudo e os valores da nIL8LA das células THP-G8 não tratadas; e a inibição da GAPLA (Inh-GAPLA), que é o rácio entre a média aritmética dos quatro valores medidos da GAPLA das células THP-G8 tratadas com um produto químico em estudo e os valores da GAPLA das células THP-G8 não tratadas, e é utilizada como indicador de citotoxicidade.
Quadro 1
Descrição dos termos associados à atividade da luciferase no ensaio IL-8 Luc
|
Abreviaturas
|
Definição
|
|
GAPLA
|
Atividade da luciferase SLR indicativa da atividade do promotor da GAPDH
|
|
IL8LA
|
Atividade da luciferase SLO indicativa da atividade do promotor da IL-8
|
|
nIL8LA
|
IL8LA/GAPLA
|
|
Ind-IL8LA
|
nIL8LA das células THP-G8 tratadas com produtos químicos/nIL8LA das células não tratadas
|
|
Inh-GAPLA
|
GAPLA das células THP-G8 tratadas com produtos químicos/GAPLA das células não tratadas
|
|
CV05
|
A concentração mais baixa do produto químico à qual a Inh-GAPLA passa a ser < 0,05.
|
|
|
11.
|
Estão disponíveis normas de desempenho (15) que permitem facilitar a validação de métodos de ensaio in vitro modificados utilizando ensaios IL-8 da luciferase semelhantes ao ensaio IL-8 Luc, bem como modificar rapidamente as orientações de ensaio 442E da OCDE para que passem a incluir os referidos métodos. A aceitação mútua de dados (AMD) da OCDE só será garantida para métodos de ensaio validados de acordo com as normas de desempenho, se esses métodos tiverem sido examinados e incluídos nas orientações de ensaio 442E pela OCDE (16).
|
DEMONSTRAÇÃO DE COMPETÊNCIA
|
12.
|
Antes de utilizarem, por rotina, o ensaio descrito no presente apêndice do método de ensaio B.71, os laboratórios devem demonstrar a sua competência técnica utilizando as dez substâncias enumeradas no apêndice 3.3, em conformidade com as boas práticas para os métodos in vitro (17). Além disso, os utilizadores do ensaio devem conservar uma base de dados históricos resultantes dos controlos de reatividade (ver ponto 15), obtidos com o controlo positivo e com o controlo do solvente/veículo (ver pontos 21 a 24), e utilizar esses dados para confirmar que a reprodutibilidade do método de ensaio no seu laboratório se mantém ao longo do tempo.
|
PROCEDIMENTO
|
13.
|
O procedimento operacional normalizado (PON) para o ensaio IL-8 Luc está disponível e deve ser utilizado para a realização do ensaio (18). Os laboratórios que desejem aplicar este ensaio podem obter a linha celular recombinante THP-G8 junto do laboratório GPC Lab. Co. Ltd., Tottori, Japão, após a assinatura de um acordo de transferência de material, em conformidade com as condições enunciadas no modelo de acordo da OCDE. Os pontos que se seguem apresentam uma descrição dos principais elementos e procedimentos do ensaio.
|
Preparação das células
|
14.
|
Convém utilizar a linha celular THP-G8 do laboratório GPC Lab. Co. Ltd., Tottori, Japão, para realizar o ensaio IL-8 Luc (ver pontos 8 e 13). Após a receção, as células são propagadas (2-4 passagens) e armazenadas congeladas, como uma reserva homogénea. As células desta reserva podem ser propagadas até um máximo de 12 passagens ou um máximo de seis semanas. O meio utilizado para a propagação é o meio de cultura RPMI-1640 com 10 % de soro bovino fetal (SBF), uma solução antibiótica/antimicótica (100 U/ml de penicilina G, 100 μg/ml de estreptomicina e 0,25 μg/ml de anfotericina B numa solução salina a 0,85 %) (por exemplo, GIBCO Cat#15240-062), 0,15 μg/ml de puromicina (por exemplo, n.o CAS 58-58-2) e 300 μg/ml de G418 (por exemplo, n.o CAS 108321-42-2).
|
|
15.
|
Antes da utilização para o ensaio, as células devem ser qualificadas através de um controlo de reatividade. Esta verificação deve ser efetuada uma a duas semanas ou duas a quatro passagens após descongelação, utilizando como controlo positivo o brometo de 4-nitrobenzilo (4-NBB) (n.o CAS: 100-11-8, pureza ≥ 99 %) e como controlo negativo o ácido láctico (AL) (n.o CAS: 50-21-5, pureza ≥ 85 %). O 4-NBB deve produzir uma resposta positiva para Ind-IL8LA (≥ 1,4), ao passo que o ácido láctico deve produzir uma resposta negativa para Ind-IL8LA (< 1,4). Apenas são utilizadas para o ensaio células que tenham passado o controlo de reatividade. O controlo de reatividade deve ser efetuado de acordo com os procedimentos descritos nos pontos 22 a 24.
|
|
16.
|
Para o ensaio, as células THP-G8 são inoculadas a uma densidade de 2 a 5 × 105 células/ml e pré-cultivadas em frascos de cultura durante 48 a 96 horas. No dia do ensaio, as células colhidas do frasco de cultura são lavadas com RPMI-1640 contendo 10 % de SBF sem antibióticos e, em seguida, colocadas novamente em suspensão com RPMI-1640 contendo 10 % de SBF sem antibióticos a 1 × 106 células/ml. Em seguida, as células são distribuídas por uma placa negra de 96 poços de fundo plano (por exemplo, Costar Cat#3603) com 50 μl (5 × 104 de células/poço).
|
Preparação do produto químico em estudo e das substâncias de controlo
|
17.
|
O produto químico em estudo e as substâncias de controlo são preparados no dia do ensaio. Para o ensaio IL-8 Luc, os produtos químicos em estudo são dissolvidos em X-VIVOTM 15, um meio isento de soro comercialmente disponível (Lonza, 04-418Q), até atingir uma concentração final de 20 mg/ml. O meio X-VIVOTM 15 é adicionado, num tubo de microcentrífuga, a 20 mg de produto químico em estudo (independentemente da solubilidade deste), até atingir um volume de 1 ml e, em seguida, é misturado vigorosamente num vórtex e agitado num rotor a uma velocidade máxima de 8 rpm durante 30 minutos a uma temperatura ambiente de cerca de 20 oC. Além disso, se os produtos químicos sólidos continuarem por dissolver, o tubo é sonicado até à dissolução completa do produto químico ou até à obtenção de uma dispersão estável. No caso de produtos químicos solúveis em X-VIVOTM 15, a solução é diluída por um fator de 5 no X-VIVOTM 15 e utilizada como solução-mãe do produto químico em estudo no X-VIVOTM 15 (4 mg/ml). No caso de produtos químicos não solúveis em X-VIVOTM 15, a mistura é novamente misturada por rotação durante pelo menos 30 minutos, e depois centrifugada a 15000 rpm (≈20000g) durante 5 minutos; o sobrenadante é utilizado como solução-mãe do produto químico em estudo no X-VIVOTM 15. Caso sejam utilizados outros solventes, como DMSO, água ou o meio de cultura, a escolha deve ser fundamentada cientificamente. O procedimento pormenorizado para a dissolução dos produtos químicos é descrito no apêndice 3.5. As soluções X-VIVOTM 15 descritas nos pontos 18 a 23 são misturadas na proporção 1:1 (v/v) com as suspensões de células preparadas numa placa negra de 96 poços de fundo plano (ver ponto 16).
|
|
18.
|
A primeira execução do ensaio destina-se a determinar a concentração citotóxica e a examinar o potencial de sensibilização cutânea dos produtos químicos. Utilizando o X-VIVOTM 15, são efetuadas diluições em série, de fator 2, das soluções-mãe dos produtos químicos em estudo no X-VIVOTM 15 (ver apêndice 3.5) utilizando um bloco de ensaio de 96 poços (por exemplo, Costar Cat#EW-01729-03). Em seguida, acrescentam-se 50 μl/poço de solução diluída a 50 μl das células em suspensão numa placa negra de 96 poços de fundo plano. Assim, no caso de produtos químicos solúveis em X-VIVOTM 15, as concentrações finais dos produtos químicos em estudo variam entre 0,002 e 2 mg/ml (apêndice 3.5). No caso dos produtos químicos em estudo que não são solúveis em X-VIVOTM 15 a 20 mg/ml, apenas são determinados fatores de diluição entre 2 e 210, apesar de as concentrações finais reais dos produtos químicos em estudo se manterem incertas e dependerem da concentração saturada dos produtos químicos em estudo na solução-mãe X-VIVOTM 15.
|
|
19.
|
Nas execuções seguintes (isto é, o segundo, o terceiro e o quarto replicados), a solução-mãe X-VIVOTM 15 é preparada a uma concentração quatro vezes superior à concentração de viabilidade celular 05 (CV05; a concentração mais baixa à qual a Inh-GAPLA passa a ser < 0,05) observada na primeira experiência. Se a Inh-GAPLA não descer abaixo dos 0,05 à concentração mais elevada na primeira execução, a solução-mãe X-VIVOTM 15 é preparada à concentração mais elevada da primeira execução. A concentração CV05 é calculada dividindo a concentração da solução-mãe da primeira execução pelo fator de diluição de CV05 (X) (fator de diluição CV05 (X); o fator de diluição necessário para diluir a solução-mãe até alcançar o valor CV05) (ver apêndice 3.5). Para as substâncias em estudo não solúveis em X-VIVO a 20 mg/ml, o valor CV05 é determinado pela concentração da solução-mãe × 1/X. Para as execuções 2 a 4, prepara-se uma segunda solução-mãe a uma concentração de 4 × CV05 (apêndice 3.5).
|
|
20.
|
Efetuam-se diluições em série das segundas soluções-mãe do X-VIVOTM 15 por um fator de 1,5 utilizando um bloco de ensaio de 96 poços. Em seguida, acrescentam-se 50 μl/poço de solução diluída a 50 μl da suspensão de células nos poços de uma placa negra de 96 poços de fundo plano. Cada concentração de cada produto químico em estudo deve ser testada em 4 poços. As amostras são, em seguida, misturadas num agitador de placas e incubadas durante 16 horas a 37 oC e 5 % CO2, após o que a atividade da luciferase é determinada, como se descreve em seguida.
|
|
21.
|
O controlo do solvente é a mistura de 50 μl/poço de X-VIVOTM 15 e de 50 μl/poço da suspensão de células no RPMI-1640 contendo 10 % de SBF.
|
|
22.
|
O controlo positivo recomendado é o 4-NBB. A 20 mg de 4-NBB preparados num tubo de microcentrífuga de 1,5 ml, é acrescentado X-VIVOTM 15 até atingir 1 ml. O tubo é misturado num vórtex vigorosamente e agitado num rotor a uma velocidade máxima de 8 rpm durante, pelo menos, 30 minutos. Após centrifugação a 20000 g durante 5 minutos, o sobrenadante é diluído por um fator de 4 no X-VIVOTM 15 e 500 μl do sobrenadante diluído são transferidos para um poço num bloco de ensaio de 96 poços. O sobrenadante diluído é novamente diluído em X-VIVOTM 15 por fatores de 2 e de 4 e, em seguida, são adicionados 50 μl da solução a 50 μl de suspensão de células THP-G8 nos poços de uma placa negra de 96 poços de fundo plano (apêndice 3.6). Cada concentração do controlo positivo deve ser testada em 4 poços. A placa é agitada num agitador de placas e incubada numa incubadora de CO2 durante 16 horas (37 oC, 5 % CO2), após o que a atividade da luciferase é medida, conforme descrito no ponto 29.
|
|
23.
|
O controlo negativo recomendado é o ácido láctico. A 20 mg deste, preparado num tubo de microcentrífuga de 1,5 ml, acrescenta-se X-VIVOTM 15 até atingir 1 ml (20 mg/ml). Diluem-se 20 mg/ml de solução de ácido láctico por um fator de 5 no X-VIVOTM 15 (4 mg/ml) e transferem-se 500 μl desta solução de ácido láctico a 4 mg/ml para um poço de um bloco de ensaio de 96 poços. Esta solução é diluída por um fator de 2 em X-VIVOTM 15 e, em seguida, diluída novamente por um fator de 2, de modo a obter soluções a 2 mg/ml e 1 mg/ml. Acrescentam-se 50 μl destas três soluções e de controlo do veículo (X-VIVOTM 15) a 50 μl de células THP-G8 em suspensão nos poços de uma placa negra de 96 poços de fundo plano. Cada concentração do controlo negativo é testada em 4 poços. A placa é agitada num agitador de placas e incubada numa incubadora de CO2 durante 16 horas (37 oC, 5 % CO2), após o que a atividade da luciferase é medida, conforme descrito no ponto 29.
|
|
24.
|
Podem utilizar-se outros controlos positivos ou negativos adequados, se houver dados históricos que permitam definir critérios de aceitação comparáveis para a execução.
|
|
25.
|
Deve ter-se o cuidado de evitar a evaporação de produtos químicos em estudo voláteis e a contaminação cruzada entre poços pelos produtos químicos em estudo, por exemplo selando a placa antes da incubação com os produtos químicos em estudo.
|
|
26.
|
Os produtos químicos em estudo e o controlo do solvente exigem 2 a 4 execuções para derivar uma previsão positiva ou negativa (ver quadro 2). Cada execução é efetuada num dia diferente, com uma solução-mãe dos produtos químicos em estudo no X-VIVOTM 15 fresca e células colhidas separadamente. As células podem ser provenientes da mesma passagem.
|
Medições da atividade da luciferase
|
27.
|
A luminescência é medida utilizando um luminómetro para microplacas de 96 poços equipado com filtros óticos, por exemplo das séries Phelios (ATTO, Tóquio, Japão), Tristan 941 (Berthold, Bad Wildbad, Alemanha) ou ARVO (PerkinElmer, Waltham, MA, EUA). Para garantir a reprodutibilidade, o luminómetro deve ser calibrado para cada ensaio (19). Estão disponíveis para esta calibração luciferases recombinantes de cor de laranja e vermelha.
|
|
28.
|
Transfere-se 100 μl de reagente pré-aquecido Tripluc® Luciferase (Tripluc) para cada poço da placa que contém a suspensão de células tratadas, com ou sem produto químico. A placa é agitada durante 10 minutos a uma temperatura ambiente de cerca de 20 oC antes de ser colocada no luminómetro para medir a atividade da luciferase. A bioluminescência é medida durante três segundos sem filtro ótico (F0) e, em seguida, durante três segundos com filtro ótico (F1). Deve justificar-se o recurso a parâmetros alternativos, por exemplo em função do modelo de luminómetro utilizado.
|
|
29.
|
Para cada concentração, os parâmetros são calculados a partir dos valores medidos, por exemplo IL8LA, GAPLA, nIL8LA, Ind-IL8LA, Inh-GAPLA, a média ± desvio padrão (DP) da IL8LA, a média ± DP da GAPLA, a média ± DP da nIL8LA, a média ± DP da Ind-IL8LA, a média ± DP da Inh-GAPLA e o intervalo de confiança de 95 % da Ind-IL8LA. As definições dos parâmetros utilizados no presente ponto são fornecidas nos apêndices 3.1 e 3.4, respetivamente.
|
|
30.
|
Antes de proceder à medição num ensaio de repórter multicolor, convém realizar uma discriminação das cores, geralmente com o auxílio de detetores (luminómetro e leitor de placas) equipados com filtros óticos, tais como filtros de rejeição de banda (passa-alta ou passa-baixa) ou filtros passa-banda. Os coeficientes de transmissão dos filtros para cada cor do sinal de bioluminescência devem ser calibrados antes do ensaio, em conformidade com o apêndice 3.2.
|
DADOS E RELATÓRIOS
Avaliação dos dados
|
31.
|
Os critérios a respeitar em cada execução para obter uma decisão positiva/negativa são os seguintes:
|
—
|
uma previsão com o ensaio IL-8 Luc é considerada positiva se o produto químico em estudo apresentar um valor Ind-IL8LA ≥ 1,4 e se o limite inferior do intervalo de confiança de 95 % para Ind-IL8LA for ≥ 1,0;
|
|
—
|
uma previsão com o ensaio IL-8 Luc é considerada negativa se o produto químico em estudo apresentar um valor Ind-IL8LA < 1,4 e/ou se o limite inferior do intervalo de confiança de 95 % para Ind-IL8LA for < 1,0.
|
|
Modelo preditivo
|
32.
|
Os produtos químicos em estudo que apresentam dois resultados positivos entre a primeira, a segunda, a terceira ou a quarta execuções são considerados positivos, ao passo que os que apresentam três resultados negativos entre a primeira, a segunda, a terceira ou a quarta execuções são considerados potencialmente negativos (quadro 2). Entre os produtos químicos potencialmente negativos, os produtos químicos dissolvidos a 20 mg/ml no X-VIVOTM 15 são considerados negativos, ao passo que os produtos químicos não dissolvidos a 20 mg/ml de X-VIVOTM 15 não devem ser tidos em conta (figura 1).
Quadro 2
Critérios de identificação dos produtos positivos e potencialmente negativos
|
1.a execução
|
2.a execução
|
3.a execução
|
4.a execução
|
Previsão final
|
|
Positivo
|
Positivo
|
–
|
–
|
Positivo
|
|
Negativo
|
Positivo
|
–
|
Positivo
|
|
Negativo
|
Positivo
|
Positivo
|
|
Negativo
|
Potencialmente negativo
|
|
Negativo
|
Positivo
|
Positivo
|
–
|
Positivo
|
|
Negativo
|
Positivo
|
Positivo
|
|
Negativo
|
Potencialmente negativo
|
|
Negativo
|
Positivo
|
Positivo
|
Positivo
|
|
Negativo
|
Potencialmente negativo
|
|
Negativo
|
–
|
Potencialmente negativo
|
|
Figura 1
Modelo preditivo para a decisão final

Critérios de aceitação
|
33.
|
Ao utilizar o método de ensaio IL-8 Luc, devem respeitar-se os critérios de aceitação a seguir indicados:
|
—
|
O valor Ind-IL8LA deve ser superior a 5,0, pelo menos, a uma concentração do controlo positivo, 4-NBB, em cada execução.
|
|
—
|
O valor Ind-IL8LA deve ser inferior a 1,4 a qualquer concentração do controlo negativo (ácido láctico), em cada execução.
|
|
—
|
Os dados resultantes de placas para as quais o valor GAPLA dos poços de controlo com células e Tripluc mas sem produtos químicos seja inferior a 5 vezes o valor dos poços que contêm apenas o meio de ensaio (50 μl/poço de RPMI-1640 contendo 10 % de SBF e 50 μl/poço de X-VIVOTM 15) não devem ser tidos em conta.
|
|
—
|
Os dados resultantes de placas para as quais o valor Inh-GAPLA a todas as concentrações dos produtos químicos em estudo ou de controlo seja inferior a 0,05 não devem ser tidos em conta. Neste caso, a primeira execução deve ser repetida, de modo a que a concentração final mais elevada no ensaio replicado seja a concentração final mais baixa da execução anterior.
|
|
Relatório de ensaio
|
34.
|
O relatório de ensaio deve conter as seguintes informações:
|
Produtos químicos em estudo
Substância monocomponente:
|
Identificação química, como o(s) nome(s) IUPAC ou CAS, o(s) número(s) CAS, o código SMILES ou InChI, a fórmula estrutural e/ou outros identificadores;
|
|
Aspeto físico, solubilidade na água, massa molecular e outras propriedades físico-químicas pertinentes, na medida em que estejam disponíveis;
|
|
Grau de pureza, identidade química das impurezas, conforme adequado e exequível, etc.;
|
|
Pré-tratamento, se for o caso (aquecimento ou moagem, por exemplo);
|
|
Solubilidade em X-VIVOTM 15. No caso de produtos químicos insolúveis em X-VIVOTM 15, se se observa precipitação ou flotação após centrifugação;
|
|
Concentração(ões) testada(s);
|
|
Condições de armazenagem e de estabilidade, se conhecidas;
|
|
Justificação da escolha do solvente/veículo para cada produto químico em estudo, se não tiver sido utilizado o X-VIVOTM 15.
|
Substância multicomponentes, UVCB e mistura:
|
Caracterização, tanto quanto possível, por exemplo, por identidade química (ver supra), pureza, ocorrência quantitativa e propriedades físico-químicas relevantes (ver supra) dos componentes, na medida em que estejam disponíveis;
|
|
Aspeto físico, solubilidade na água e outras propriedades físico-químicas relevantes, na medida em que estejam disponíveis;
|
|
Massa molecular, ou massa molecular aparente no caso de misturas/polímeros de composições conhecidas, ou outras informações relevantes para a realização do estudo;
|
|
Pré-tratamento, se for o caso (aquecimento ou moagem, por exemplo);
|
|
Solubilidade em X-VIVOTM 15. No caso de produtos químicos insolúveis em X-VIVOTM 15, se se observa precipitação ou flotação após centrifugação;
|
|
Concentração(ões) testada(s);
|
|
Condições de armazenagem e de estabilidade, se conhecidas.
|
|
Justificação da escolha do solvente/veículo para cada produto químico em estudo, se não tiver sido utilizado o X-VIVOTM 15.
|
|
|
Controlos
Controlo positivo:
|
Identificação química, como o(s) nome(s) IUPAC ou CAS, o(s) número(s) CAS, o código SMILES ou InChI, a fórmula estrutural e/ou outros identificadores;
|
|
Aspeto físico, solubilidade na água, massa molecular e outras propriedades físico-químicas, se conhecidas e se pertinente;
|
|
Grau de pureza, identidade química das impurezas, conforme adequado e exequível, etc.;
|
|
Pré-tratamento, se for o caso (aquecimento ou moagem, por exemplo);
|
|
Concentração(ões) testada(s);
|
|
Condições de armazenagem e de estabilidade, se conhecidas;
|
|
Referência a resultados históricos relativos ao controlo positivo que demonstrem a conformidade com os critérios de aceitação, se pertinente.
|
Controlo negativo:
|
Identificação química, como o(s) nome(s) IUPAC ou CAS, o(s) número(s) CAS e/ou outros identificadores;
|
|
Grau de pureza, identidade química das impurezas, conforme adequado e exequível, etc.;
|
|
Aspeto físico, massa molecular e outras propriedades físico-químicas relevantes, se forem utilizados controlos negativos diferentes dos mencionados nas orientações de ensaio e na medida em que se encontrem disponíveis;
|
|
Condições de armazenagem e de estabilidade, se conhecidas;
|
|
Justificação da escolha do solvente, para cada produto químico em estudo.
|
|
|
Condições de ensaio
|
Nome e endereço do promotor, do laboratório e do diretor do estudo;
|
|
Descrição do método de ensaio utilizado;
|
|
Linha celular utilizada, respetivas condições de armazenamento e origem (por exemplo, instalação de onde provêm as células);
|
|
Número de lote e origem do SBF, nome do fornecedor, número de lote da placa negra de 96 poços de fundo plano e número de lote do reagente Tripluc;
|
|
Número de passagens e densidade celular utilizadas para o ensaio;
|
|
Método de contagem celular utilizado para a inoculação antes do ensaio e medidas tomadas para assegurar a homogeneidade da distribuição do número de células;
|
|
Luminómetro utilizado (por exemplo, modelo), incluindo parâmetros dos instrumentos, substrato de luciferase utilizado e demonstração da adequação das medições de luminescência com base no ensaio de controlo descrito no apêndice 3.2;
|
|
Procedimento utilizado para demonstrar a competência do laboratório para a realização do ensaio (por exemplo, através testando as substâncias para a demonstração de competência) ou para demonstrar a reprodutibilidade do desempenho do ensaio ao longo do tempo.
|
|
|
Procedimento de ensaio
|
Número de replicados e de execuções efetuadas;
|
|
Concentrações do produto químico em estudo, procedimento de aplicação e tempo de exposição (se forem diferentes dos recomendados);
|
|
Descrição dos critérios de avaliação e de decisão utilizados;
|
|
Descrição dos critérios de aceitação do estudo utilizados;
|
|
Descrição de eventuais modificações do procedimento de ensaio.
|
|
|
Resultados
|
Medições de IL8LA e GAPLA;
|
|
Cálculos para nIL8LA, Ind-IL8LA e Inh-GAPLA;
|
|
O intervalo de confiança de 95 % de Ind-IL8LA;
|
|
Gráfico ilustrativo das curvas dose-resposta para a indução da atividade da luciferase e a viabilidade;
|
|
Descrição de quaisquer outras observações pertinentes, se for caso disso.
|
|
|
Discussão dos resultados
|
Discussão dos resultados obtidos com o ensaio IL-8 Luc;
|
|
Exame dos resultados do ensaio no contexto de uma IATA, caso estejam disponíveis outras informações pertinentes.
|
|
Conclusão
|
REFERÊNCIAS BIBLIOGRÁFICAS
|
(1)
|
Takahashi T, Kimura Y, Saito R, Nakajima Y, Ohmiya Y, Yamasaki K, and Aiba S. (2011). An in vitro test to screen skin sensitizers using a stable THP-1-derived IL-8 reporter cell line, THP-G8. Toxicol Sci 124:359-69.
|
|
(2)
|
OECD (2017). Validation report for the international validation study on the IL-8 Luc assay as a test evaluating the skin sensitizing potential of chemicals conducted by the IL-8 Luc Assay. Series on Testing and Assessment No 267, ENV/JM/MONO(2017)19. Organisation for Economic Cooperation and Development, Paris. Disponível em: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(3)
|
OECD (2017). Report of the Peer Review Panel for the IL-8 Luciferase (IL-8 Luc) Assay for in vitro skin sensitisation. Series on Testing and Assessment No 258, ENV/JM/MONO(2017)20. Organisation for Economic Cooperation and Development, Paris. Disponível em: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(4)
|
OECD (2016) Guidance Document On The Reporting Of Defined Approaches And Individual Information Sources To Be Used Within Integrated Approaches To Testing And Assessment (IATA) For Skin Sensitisation, Series on Testing & Assessment No 256, ENV/JM/MONO(2016)29. Organisation for Economic Cooperation and Development, Paris. Disponível em: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(5)
|
van der Veen JW, Rorije E, Emter R, Natsch A, van Loveren H, and Ezendam J. (2014). Evaluating the performance of integrated approaches for hazard identification of skin sensitizing chemicals. Regul Toxicol Pharmacol 69:371-9.
|
|
(6)
|
Kimura Y, Fujimura C, Ito Y, Takahashi T, Nakajima Y, Ohmiya Y, and Aiba S. (2015). Optimization of the IL-8 Luc assay as an in vitro test for skin sensitization. Toxicol In Vitro 29:1816-30.
|
|
(7)
|
Urbisch D, Mehling A, Guth K, Ramirez T, Honarvar N, Kolle S, Landsiedel R, Jaworska J, Kern PS, Gerberick F, et al. (2015). Assessing skin sensitization hazard in mice and men using non-animal test methods. Regul Toxicol Pharmacol 71:337-51.
|
|
(8)
|
Ashikaga T, Sakaguchi H, Sono S, Kosaka N, Ishikawa M, Nukada Y, Miyazawa M, Ito Y, Nishiyama N, and Itagaki H. (2010). A comparative evaluation of in vitro skin sensitisation tests: the human cell-line activation test (h-CLAT) versus the local lymph node assay (LLNA). Alternatives to laboratory animals: ATLA 38:275-84.
|
|
(9)
|
Patlewicz G, Casati S, Basketter DA, Asturiol D, Roberts DW, Lepoittevin J-P, Worth A and Aschberger K (2016) Can currently available non-animal methods detect pre and pro haptens relevant for skin sensitisation? Regul Toxicol Pharmacol, 82:147-155.
|
|
(10)
|
Thorne N, Inglese J, and Auld DS. (2010). Illuminating insights into firefly luciferase and other bioluminescent reporters used in chemical biology. Chem Biol 17:646-57.
|
|
(11)
|
OECD (2016). Test No 455: Performance-Based Test Guideline for Stably Transfected Transactivation In Vitro Assays to Detect Estrogen Receptor Agonists and Antagonists, OECD Publishing, Paris. http://dx.doi.org/10.1787/9789264265295-en.
|
|
(12)
|
Viviani V, Uchida A, Suenaga N, Ryufuku M, and Ohmiya Y. (2001). Thr226 is a key residue for bioluminescence spectra determination in beetle luciferases. Biochem Biophys Res Commun 280:1286-91.
|
|
(13)
|
Viviani VR, Bechara EJ, and Ohmiya Y. (1999). Cloning, sequence analysis, and expression of active Phrixothrix railroad-worms luciferases: relationship between bioluminescence spectra and primary structures. Biochemistry 38:8271-9.
|
|
(14)
|
Nakajima Y, Kimura T, Sugata K, Enomoto T, Asakawa A, Kubota H, Ikeda M, and Ohmiya Y. (2005). Multicolor luciferase assay system: one-step monitoring of multiple gene expressions with a single substrate. Biotechniques 38:891-4.
|
|
(15)
|
OECD (2017). A publicar - Performance Standards for the assessment of proposed similar or modified in vitro skin sensitisation IL-8 luc test methods. OECD Environment, Health and Safety Publications, Series on Testing and Assessment. OECD, Paris, França
|
|
(16)
|
OECD (2005). Guidance Document the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. OECD Environment, Health and Safety publications, OECD Series on Testing and Assessment No 34. OECD, Paris, França.
|
|
(17)
|
OECD (2018). Draft Guidance document: Good In Vitro Method Practices (GIVIMP) for the Development and Implementation of In Vitro Methods for Regulatory Use in Human Safety Assessment. Organisation for Economic Cooperation and Development, Paris. Disponível em: http://www.oecd.org/env/ehs/testing/OECD Final Draft GIVIMP.pdf.
|
|
(18)
|
JaCVAM (2016). IL-8 Luc assay protocol. Disponível em: http://www.jacvam.jp/en_effort/effort02.html.
|
|
(19)
|
Niwa K, Ichino Y, Kumata S, Nakajima Y, Hiraishi Y, Kato D, Viviani VR, and Ohmiya Y. (2010). Quantum yields and kinetics of the firefly bioluminescence reaction of beetle luciferases. Photochem Photobiol 86:1046-9.
|
|
(20)
|
OECD (2012). The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins, Part 1: Scientific Evidence. OECD Environment, Health and Safety Publications, Series on Testing and Assessment No 168. OECD, Paris, França.
|
|
(21)
|
United Nations (2015). Globally Harmonized System of Classification and Labelling of Chemicals (GHS). Sixth revised edition. Nova Iorque e Genebra: United Nations Publications. ISBN: 978-92-1-117006-1. Disponível em: http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html.
|
Apêndice 3.1
DEFINIÇÕES
AOP (Adverse Outcome Pathway – via determinante dos efeitos nocivos)
: Sequência de eventos que parte da estrutura química de um produto químico alvo ou de um grupo de produtos químicos análogos, passa pelo evento molecular iniciador, e resulta num resultado com consequências in vivo (20).
Controlo positivo
: Replicado que contém todos os componentes de um sistema de ensaio e foi tratado com uma substância conhecida por induzir uma resposta positiva. Para que seja possível determinar a variabilidade no tempo da resposta do controlo positivo, a magnitude da resposta positiva não deve ser excessiva.
Controlo do solvente/veículo
: Amostra não tratada que contém todos os componentes de um sistema de ensaio, com exceção do produto químico em estudo, mas incluindo o solvente/veículo utilizado. Serve para determinar a resposta de referência para as amostras tratadas com o produto químico em estudo dissolvido ou em dispersão estável no mesmo solvente/veículo. Quando testada simultaneamente com um controlo de meio, esta amostra também indica se o solvente/veículo interage com o sistema de ensaio.
CV05
: Viabilidade celular 05, ou seja, a concentração mínima à qual os produtos químicos produzem um valor Inh-GAPLA inferior a 0,05.
Especificidade
: Proporção dos produtos químicos negativos/inativos que são corretamente classificados pelo ensaio. Permite medir a precisão de um ensaio que produz resultados categóricos e é um aspeto importante a ter em conta na avaliação da pertinência de um ensaio (16).
Execução
: Consiste em testar um ou mais produtos químicos em estudo simultaneamente com um controlo do solvente/veículo e com um controlo positivo.
Fiabilidade
: Indica em que medida um ensaio pode ser reproduzido ao longo do tempo num mesmo laboratório e entre laboratórios, utilizando o mesmo protocolo. Para a avaliar, calcula-se a reprodutibilidade intralaboratorial e interlaboratorial, bem como a repetibilidade intralaboratorial (16).
FInSLO-LA
: Abreviatura utilizada no relatório de validação e em publicações anteriores relativas ao ensaio do IL-8 Luc para designar a Ind-IL8LA. Ver Ind-IL8LA para a definição.
GAPLA
: Atividade da luciferase vermelha Stable Luciferase Red (SLR) (λmáx. = 630 nm), regulada pelo promotor da GAPDH e que demonstra a viabilidade celular e o número de células viáveis.
IATA (Integrated Approach to Testing and Assessment – abordagem integrada de ensaio e avaliação)
: Abordagem estruturada utilizada para a identificação (potencial) e caracterização (potência) do perigo e/ou para a avaliação da segurança (potencial/potência e exposição) de um produto químico ou grupo de produtos químicos, que integra e pondera, de forma estratégica, todos os dados pertinentes com o objetivo de ser tida em conta numa decisão regulamentar sobre o potencial perigo e/ou risco e/ou a necessidade de realizar ensaios complementares específicos.
II-SLR-LA
: Abreviatura utilizada no relatório de validação e em publicações anteriores relativas ao ensaio do IL-8 Luc para designar a Inh-GAPLA. Ver Inh-GAPLA para a definição.
IL-8 (interleucina-8)
: Uma citocina derivada de células endoteliais, fibroblastos, queratinócitos, macrófagos e monócitos que provoca a quimiotaxia dos neutrófilos e dos linfócitos T.
IL8LA
: Atividade da luciferase laranja Stable Luciferase Orange (SLRO) (λmáx. = 580 nm), regulada pelo promotor da IL-8.
Ind-IL8LA
: Variação de indução da nIL8LA. Obtém-se dividindo o valor nIL8LA das células THP-G8 tratadas com produtos químicos pelo valor das células THP-G8 não estimuladas; representa a indução da atividade do promotor da IL-8 por produtos químicos.
Inh-GAPLA
: Inibição da GAPLA. Obtém-se dividindo o valor GAPLA das células THP-G8 tratadas com produtos químicos pelo valor GAPLA das células THP-G8 não tratadas; representa a citotoxicidade dos produtos químicos.
Limiar mínimo de indução (LMI)
: A concentração mais baixa à qual um produto químico satisfaz os critérios de positividade.
Método de ensaio válido
: Um ensaio considerado suficientemente pertinente e fiável para um fim específico e que se baseia em princípios cientificamente sólidos. Um ensaio nunca é válido em termos absolutos, mas apenas em relação a um objetivo definido.
Mistura
: Uma mistura ou solução composta por duas ou mais substâncias.
nIL8LA
: A atividade da SLO que reflete a atividade do promotor da IL-8 (IL8LA) normalizada pela atividade da SLR que reflete a atividade do promotor da GAPDH (GAPLA). Representa a atividade do promotor da IL-8 depois de considerar a viabilidade celular ou o número de células.
nSLO-LA
: Abreviatura utilizada no relatório de validação e em publicações anteriores relativas ao ensaio do IL-8 Luc para designar a nIL8LA. Ver nIL8LA para a definição
Perigo
: Propriedade intrínseca de um agente, ou de uma situação, suscetível de causar efeitos nocivos quando um organismo, sistema ou (sub)população é exposto ao agente em causa.
Pertinência
: Descrição da relação do ensaio com o efeito em estudo e da adequação e utilidade do ensaio para um fim específico. Traduz a medida em que o ensaio mede ou prevê corretamente o efeito biológico em causa. A pertinência tem em conta a precisão (concordância) de um ensaio (16).
Precisão
: Grau de acordo entre os resultados do ensaio e os valores de referência aceites. Trata-se de uma medida do desempenho do ensaio e de um dos aspetos da sua pertinência. Este termo e o termo «concordância» são muitas vezes utilizados indistintamente para indicar a proporção de resultados corretos de um ensaio (16).
Pré-haptenos
: Produtos químicos que se tornam sensibilizantes por transformação abiótica.
Pró-haptenos
: Produtos químicos que requerem ativação enzimática para exercerem potencial de sensibilização cutânea.
Produto químico
: Uma substância ou mistura.
Produto químico em estudo
: Qualquer substância ou mistura à qual seja aplicado o presente método.
Sensibilidade
: Proporção de todos os produtos químicos positivos/ativos que são corretamente classificados pelo ensaio. Permite medir a precisão de um ensaio que produz resultados categóricos e é um aspeto importante a ter em conta na avaliação da pertinência de um ensaio (16).
Sistema Mundial Harmonizado de Classificação e Rotulagem de Produtos Químicos das Nações Unidas – GHS da ONU (em inglês: United Nations Globally Harmonized System of Classification and Labelling of Chemicals – UN GHS)
: Sistema que propõe a classificação dos produtos químicos (substâncias e misturas) em função de tipos e níveis normalizados de perigos físicos, sanitários e ambientais e trata dos elementos de comunicação correspondentes, nomeadamente pictogramas, palavras-sinal, advertências de perigo, recomendações de prudência e fichas de dados de segurança, de modo a transmitir informações sobre os respetivos efeitos nocivos, tendo em vista a proteção das pessoas (incluindo empregadores, trabalhadores, transportadores, consumidores e pessoal dos serviços de emergência) e do ambiente (21).
SLO-LA
: Abreviatura utilizada no relatório de validação e em publicações anteriores relativas ao ensaio do IL-8 Luc para designar a IL8LA. Ver IL8LA para a definição.
SLR-LA
: Abreviatura utilizada no relatório de validação e em publicações anteriores relativas ao ensaio do IL-8 Luc para designar a GAPLA. Ver GAPLA para a definição.
Substância
: Um elemento químico e seus compostos, no estado natural ou obtidos por qualquer processo de produção, incluindo qualquer aditivo necessário para preservar a sua estabilidade e quaisquer impurezas resultantes do processo utilizado, mas excluindo qualquer solvente que possa ser separado sem afetar a estabilidade da substância nem alterar a sua composição.
Substância monocomponente
: Uma substância, definida pela sua composição quantitativa, da qual um dos principais componentes está presente num teor de, pelo menos, 80 % (m/m).
Substância multicomponentes
: Uma substância, definida pela sua composição quantitativa, da qual mais do que um dos principais componentes está presente numa concentração ≥ 10 % (m/m) e < 80 % (m/m). As substâncias multicomponentes resultam de processos de fabrico. A diferença entre uma mistura e uma substância multicomponentes é que a primeira se obtém misturando duas ou mais substâncias, sem reação química. As substâncias multicomponentes resultam de reações químicas.
Tensioativo
: Também designado agente de superfície, é uma substância, como um detergente, capaz de reduzir a tensão superficial de um líquido, permitindo-lhe formar espuma ou penetrar em sólidos; é igualmente designado por agente molhante. (TG437)
THP-G8
: Uma linha celular repórter da IL-8 utilizada no ensaio IL-8 Luc. A linha celular humana semelhante a macrófagos THP-1 foi transfetada com os genes da luciferase SLO e SLR sob o controlo dos promotores da IL-8 e da GAPDH, respetivamente.
UVCB
: Substâncias de composição desconhecida ou variável, produtos de reação complexos ou materiais biológicos.
Apêndice 3.2
PRINCÍPIO DE MEDIÇÃO DA ATIVIDADE DA LUCIFERASE E DETERMINAÇÃO DOS COEFICIENTES DE TRANSMISSÃO DE FILTRO ÓTICO PARA SLO E SLR
O sistema de ensaio multirrepórter – Tripluc – pode ser utilizado com luminómetro de microplacas equipado com um sistema de deteção multicolor com filtro ótico (por exemplo, Phelios AB-2350 (ATTO), ARVO (PerkinElmer), Tristar LB941 (Berthold)). O filtro ótico utilizado na medição é um filtro passa-alta ou passa-baixa de 600-620 nm ou um filtro passa-banda de 600-700 nm.
Medição de duas cores de luciferase com um filtro ótico
O exemplo seguinte utiliza o Phelios AB-2350 (ATTO). Este luminómetro está equipado com um filtro passa-alta de 600 nm (R60 HOYA Co., 600 nm LP, filtro 1) para separar a luminescência SLO (λmáx. = 580 nm) da luminescência SLR (λmáx. = 630 nm).
Para determinar os coeficientes de transmissão do filtro passa-alta de 600 nm, convém, em primeiro lugar, utilizando enzimas luciferases SLO e SLR purificadas: i) medir a intensidade da bioluminescência de SLO e SLR na ausência de filtro (F0), ii) medir a intensidade da bioluminescência de SLO e SLR que atravessou o filtro 1 (passa-alta 600 nm) e iii) calcular os coeficientes de transmissão do filtro 1 (600 nm) para SLO e SLR abaixo indicados.
|
Transmission coefficients
|
Abbreviation
|
Definition
|
|
SLO
|
Filter 1 Transmission coefficients
|
=κOR60
|
The filter’s transmission coefficient for the SLO
|
|
SLR
|
Filter 1 Transmission coefficients
|
κRR60
|
The filter’s transmission coefficient for the SLR
|
Se a intensidade de SLO e SLR na amostra de ensaio for definida como O e R, respetivamente, i) a intensidade da luz sem filtro (todas as óticas) F0 e ii) a intensidade da luz transmitida através do filtro 1 (600 nm) F1 são descritas como se segue.
F0 = O + R
F1 = κOR60 × O + κRR60 × R
Estas equações podem ser reformuladas do seguinte modo:
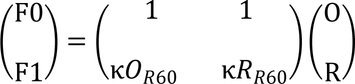
Em seguida, utilizando os fatores de transmissão calculados (κOR60 e κRR60) e os valores F0 e F1 medidos, é possível calcular os valores O e R do seguinte modo:
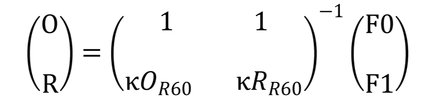
Materiais e métodos para determinação do fator de transmissão
|
(1)
|
Reagentes
Enzimas luciferases purificadas:
|
Enzima SLO purificada liofilizada
|
|
Enzima SLR purificada liofilizada
|
|
(que, para os trabalhos de validação, foram obtidas junto do GPC Lab. Co. Ltd., Tottori, Japão com a linha celular THP-G8)
|
Reagente do ensaio:
|
Reagente Tripluc® Luciferase (por exemplo, de TOYOBO Cat#MRA-301)
|
Meio: para o ensaio da luciferase (30 ml, armazenada a 2-8 °C)
|
|
Reagente
|
Conc.
|
Conc. final no meio
|
Volume necessário
|
|
RPMI-1640
|
—
|
—
|
27 ml
|
|
SBF
|
—
|
10 %
|
3 ml
|
|
(2)
|
Preparação da solução enzimática
Dissolver a enzima luciferase purificada liofilizada no tubo, adicionando 200 μl de 10 ~ 100 mM Tris/HCl ou Hepes/HCl (pH 7.5 ~ 8.0) e complementado com 10 % (m/v) de glicerol; dividir a solução enzimática em alíquotas de 10 μl em tubos descartáveis de 1,5 ml e armazená-los num congelador a -80 °C. A solução enzimática congelada pode ser utilizada no prazo de seis meses. Para utilizar as soluções, adicionar 1 ml de meio do ensaio da luciferase (RPMI-1640 com 10 % de SBF) a cada tubo de solução enzimática (solução enzimática diluída) e mantê-los em gelo, a fim de evitar a desativação.
|
|
(3)
|
Medição da bioluminescência
Descongelar o reagente do ensaio Tripluc® Luciferase e mantê-lo à temperatura ambiente, quer em banho-maria, quer à temperatura do ar. Ligar o luminómetro 30 minutos antes do início da medição para permitir a estabilização do fotomultiplicador. Transferir 100 μl da solução enzimática diluída para uma placa negra de 96 poços (fundo plano) (amostra de referência SLO para #B1, #B2, #B3 e amostra de referência SLR para #D1, #D2, #D3). Em seguida, transferir 100 μl de Tripluc pré-aquecido para cada poço da placa que contém a solução enzimática diluída, utilizando uma pipeta automática. Agitar a placa durante 10 minutos à temperatura ambiente (cerca de 25 °C) num agitador de placas. Retirar as bolhas que se formarem nas soluções nos poços. Colocar a placa no luminómetro para medir a atividade da luciferase. A bioluminescência é medida durante três segundos sem filtro ótico (F0) e, em seguida, durante três segundos com filtro ótico (F1).
O coeficiente de transmissão do filtro ótico foi calculado do seguinte modo:
Coeficiente de transmissão (SLO (κOR60))= (#B1 de F1+ #B2 de F1+ #B3 de F1) / (#B1 de F0+ #B2 de F0+ #B3 de F0)
Coeficiente de transmissão (SLR (κRR60))= (#D1 de F1+ #D2 de F1+ #D3 de F1) / (#D1 de F0+ #D2 de F0+ #D3 de F0)
Os fatores de transmissão calculados são utilizados para todas as medições executadas utilizando o mesmo luminómetro.
|
Controlo da qualidade do equipamento
Devem utilizar-se os procedimentos descritos no protocolo IL-8 Luc (18).
Apêndice 3.3
SUBSTÂNCIAS PARA A DEMONSTRAÇÃO DE COMPETÊNCIA
Antes de utilizarem, por rotina, o ensaio descrito no presente apêndice do método de ensaio B.71, os laboratórios devem demonstrar a sua competência técnica obtendo a previsão esperada com o ensaio IL-8 Luc para as dez substâncias recomendadas no quadro 1 e obtendo valores que se encontrem dentro do intervalo de referência correspondente para, pelo menos, oito das dez substâncias de demonstração de competência (selecionadas para representar o intervalo de respostas possíveis ao perigo de sensibilização cutânea). Outros critérios de seleção foram a disponibilidade comercial das substâncias e a disponibilidade de dados de referência in vivo de elevada qualidade, bem como de dados in vitro de alta qualidade produzidos com o ensaio IL-8 Luc. Além disso, existem dados de referência publicados para o ensaio IL-8 Luc (6) (1).
Quadro 1
Substâncias recomendadas para a demonstração de competência técnica com o ensaio IL-8 Luc
|
Substâncias para a demonstração de competência
|
N.o CAS
|
Estado
|
Solubilidade em X-VIVO15 a 20 mg/ml
|
Previsão in vivo
(108)
|
Previsão IL-8 Luc (109)
|
Intervalo de referência (μg/ml) (110)
|
|
|
CV05
(111)
|
MIT IL-8 Luc (112)
|
|
2,4-Dinitroclorobenzeno
|
97-00-7
|
Sólido
|
Insolúvel
|
Sensibilizante (extremo)
|
Positivo
|
2,3-3,9
|
0,5-2,3
|
|
Formaldeído
|
50-00-0
|
Líquido
|
Solúvel
|
Sensibilizante (forte)
|
Positivo
|
9-30
|
4-9
|
|
2-Mercaptobenzotiazol
|
149-30-4
|
Sólido
|
Insolúvel
|
Sensibilizante (moderado)
|
Positivo
|
250-290
|
60-250
|
|
Etilenodiamina
|
107-15-3
|
Líquido
|
Solúvel
|
Sensibilizante (moderado)
|
Positivo
|
500-700
|
0,1-0,4
|
|
Dimetacrilato de etilenoglicol
|
97-90-5
|
Líquido
|
Insolúvel
|
Sensibilizante (fraco)
|
Positivo
|
> 2000
|
0,04-0,1
|
|
4-Alilanisol (estragol)
|
140-67-0
|
Líquido
|
Insolúvel
|
Sensibilizante (fraco)
|
Positivo
|
> 2000
|
0,01-0,07
|
|
Sulfato de estreptomicina
|
3810-74-0
|
Sólido
|
Solúvel
|
Não sensibilizante
|
Negativo
|
> 2000
|
> 2000
|
|
Glicerol
|
56-81-5
|
Líquido
|
Solúvel
|
Não sensibilizante
|
Negativo
|
> 2000
|
> 2000
|
|
Isopropanol
|
67-63-0
|
Líquido
|
Solúvel
|
Não sensibilizante
|
Negativo
|
> 2000
|
> 2000
|
|
Abreviaturas: N.o CAS = Número de registo do Chemical Abstracts Service.
|
Apêndice 3.4
ÍNDICES E CRITÉRIOS DE APRECIAÇÃO
nIL8LA (nSLO-LA)
A j-ésima repetição (j = 1 a 4) da i-ésima concentração (i = 0 a 11) é medida para IL8LA (SLO-LA) e GAPLA (SLR-LA), respetivamente. A IL8LA normalizada, designada por nIL8LA (nSLO-LA), é definida como:
nIL8LAij = IL8LAij/GAPLAij
Trata-se da unidade de medida de base neste ensaio.
Ind-IL8LA (FInSLO-LA)
O aumento médio do valor nIL8LA (nSLO-LA) para a repetição à i-ésima concentração em comparação com a mesma à concentração 0, Ind-IL8LA, é a medida principal deste ensaio. Esta relação é escrita através da fórmula seguinte:
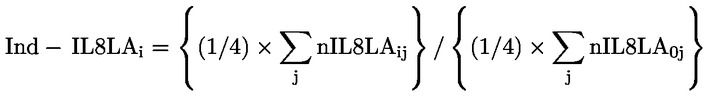
O laboratório principal propôs que um valor de 1,4 corresponda a um resultado positivo para o produto químico em estudo. Este valor baseia-se na investigação dos dados históricos do laboratório principal. A equipa de gestão dos dados utilizou, em seguida, este valor em todas as fases do estudo de validação. O principal resultado, Ind-IL8LA, é o rácio de duas médias aritméticas, tal como demonstrado na equação.
Intervalo de confiança de 95 % (IC 95 %)
A precisão desta medida do resultado principal é estimada graças ao intervalo de confiança de 95 % (IC 95 %) baseado no rácio. O limite inferior do IC 95 % ≥ 1 indica que o valor niL8LA à i-ésima concentração é significativamente superior ao valor obtido com o controlo do solvente. O IC 95 % pode ser calculado de várias formas. No presente estudo, foi utilizado o método conhecido como teorema de Fieller. Segundo este teorema, o intervalo de confiança de 95 % obtém-se por recurso à seguinte fórmula:

em que






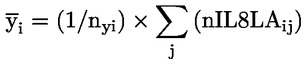

é o percentil 97,5 da distribuição central t com o ν do grau de liberdade, em que

Inh-GAPLA (II-SLR-LA)
O valor Inh-GAPLA é um rácio do valor GAPLA médio (SLR-LA) para a repetição da i-ésima concentração, comparativamente ao valor obtido com o controlo do solvente, escrito conforme se segue:

Dado que o valor GAPLA é o denominador da niL8LA, um valor extremamente baixo provoca uma grande variação no valor nIL8LA. Por conseguinte, os valores Ind-IL8LA com um valor extremamente baixo para Inh-GAPLA (inferior a 0,05), podem ser considerados pouco precisos.
Apêndice 3.5
ESQUEMA DOS MÉTODOS DE DISSOLUÇÃO DE PRODUTOS QUÍMICOS PARA O ENSAIO IL-8 LUC.
|
(a)
|
Para os produtos químicos dissolvidos em X-VIVOTM 15 a 20 mg/ml

|
|
(b)
|
Para produtos químicos insolúveis em X-VIVOTM 15 a 20 mg/ml

|
Apêndice 3.6
ESQUEMA DO MÉTODO DE DISSOLUÇÃO DE 4-NBB PARA O CONTROLO POSITIVO DO ENSAIO IL-8 LUC.
» |