ISSN 1977-0766
Dziennik Urzędowy
Unii Europejskiej
L 272

Wydanie polskie
Legislacja
Rocznik 64
30 lipca 2021
|
ISSN 1977-0766 |
||
|
Dziennik Urzędowy Unii Europejskiej |
L 272 |
|
|
|
||
|
Wydanie polskie |
Legislacja |
Rocznik 64 |
|
|
|
|
|
(1) Tekst mający znaczenie dla EOG. |
|
PL |
Akty, których tytuły wydrukowano zwykłą czcionką, odnoszą się do bieżącego zarządzania sprawami rolnictwa i generalnie zachowują ważność przez określony czas. Tytuły wszystkich innych aktów poprzedza gwiazdka, a drukuje się je czcionką pogrubioną. |
II Akty o charakterze nieustawodawczym
ROZPORZĄDZENIA
|
30.7.2021 |
PL |
Dziennik Urzędowy Unii Europejskiej |
L 272/1 |
ROZPORZĄDZENIE WYKONAWCZE RADY (UE) 2021/1241
z dnia 29 lipca 2021 r.
w sprawie wykonania art. 21 ust. 2 rozporządzenia (UE) 2016/44 w sprawie środków ograniczających w związku z sytuacją w Libii oraz uchylającego rozporządzenie (UE) nr 204/2011
RADA UNII EUROPEJSKIEJ,
uwzględniając Traktat o funkcjonowaniu Unii Europejskiej,
uwzględniając rozporządzenie Rady (UE) 2016/44 z dnia 18 stycznia 2016 r. w sprawie środków ograniczających w związku z sytuacją w Libii oraz uchylające rozporządzenie (UE) nr 204/2011 (1), w szczególności jego art. 21 ust. 2,
uwzględniając wniosek Wysokiego Przedstawiciela Unii do Spraw Zagranicznych i Polityki Bezpieczeństwa,
a także mając na uwadze, co następuje:
|
(1) |
W dniu 18 stycznia 2016 r. Rada przyjęła rozporządzenie (UE) 2016/44. |
|
(2) |
Zgodnie z art. 21 ust. 6 rozporządzenia (UE) 2016/44 Rada przeprowadziła przegląd wykazu wskazanych osób i podmiotów, zamieszczonego w załączniku III do tego rozporządzenia. |
|
(3) |
Rada stwierdziła, że wpis dotyczący jednej osoby powinien zostać skreślony, ponieważ osoba ta zmarła, oraz że należy utrzymać środki ograniczające skierowane przeciwko wszystkim osobom i podmiotom figurującym w wykazie zamieszczonym w załączniku III do rozporządzenia (UE) 2016/44. Ponadto należy zaktualizować informacje umożliwiające identyfikację dotyczące jednej osoby. |
|
(4) |
Należy zatem odpowiednio zmienić rozporządzenie (UE) 2016/44, |
PRZYJMUJE NINIEJSZE ROZPORZĄDZENIE:
Artykuł 1
W załączniku III do rozporządzenia (UE) 2016/44 wprowadza się zmiany zgodnie z załącznikiem do niniejszego rozporządzenia.
Artykuł 2
Niniejsze rozporządzenie wchodzi w życie w dniu jego opublikowania w Dzienniku Urzędowym Unii Europejskiej.
Niniejsze rozporządzenie wiąże w całości i jest bezpośrednio stosowane we wszystkich państwach członkowskich.
Sporządzono w Brukseli dnia 29 lipca 2021 r.
W imieniu Rady
G. DOVŽAN
Przewodniczący
ZAŁĄCZNIK
W części A (Osoby) załącznika III (Wykaz osób fizycznych i prawnych, podmiotów lub organów, o których mowa w art. 6 ust. 2) do rozporządzenia (UE) 2016/44 wprowadza się następujące zmiany:
|
1) |
skreśla się wpis 3 (dotyczący generała Khaleda TOHAMIEGO); |
|
2) |
wpis 6 (dotyczący Baghdadiego AL-MAHMOUDIEGO) otrzymuje brzmienie:
|
|
30.7.2021 |
PL |
Dziennik Urzędowy Unii Europejskiej |
L 272/4 |
ROZPORZĄDZENIE WYKONAWCZE RADY (UE) 2021/1242
z dnia 29 lipca 2021 r.
wykonujące rozporządzenie (UE) nr 267/2012 w sprawie środków ograniczających wobec Iranu
RADA UNII EUROPEJSKIEJ,
uwzględniając Traktat o funkcjonowaniu Unii Europejskiej,
uwzględniając rozporządzenie Rady (UE) nr 267/2012 z dnia 23 marca 2012 r. w sprawie środków ograniczających wobec Iranu i uchylające rozporządzenie (UE) nr 961/2010 (1), w szczególności jego art. 46 ust. 2,
uwzględniając wniosek Wysokiego Przedstawiciela Unii do Spraw Zagranicznych i Polityki Bezpieczeństwa,
a także mając na uwadze, co następuje:
|
(1) |
W dniu 23 marca 2012 r. Rada przyjęła rozporządzenie (UE) nr 267/2012. |
|
(2) |
W dniu 18 czerwca 2020 r. Rada przyjęła rozporządzenie (UE) 2020/847 (2) wykonujące rozporządzenie (UE) nr 267/2012. |
|
(3) |
W następstwie wyroku Sądu w sprawie T-580/19 (3) Sayed Shamsuddin Borborudi powinien zostać wykreślony z wykazu osób i podmiotów objętych środkami ograniczającymi zamieszczonego w załączniku IX do rozporządzenia (UE) nr 267/2012. |
|
(4) |
Ponadto z przeglądu załącznika II do decyzji Rady 2010/413/WPZiB (4) wynika, że należy utrzymać środki ograniczające wobec wszystkich osób i podmiotów znajdujących się w wykazie zamieszczonym w tym załączniku, w zakresie, w jakim ich imiona i nazwiska oraz nazwy nie są wymienione w załączniku VI do tej decyzji, oraz że należy zaktualizować 21 wpisów zamieszczonych w załączniku IX do rozporządzenia (UE) nr 267/2012. |
|
(5) |
Należy zatem odpowiednio zmienić rozporządzenie (UE) nr 267/2012, |
PRZYJMUJE NINIEJSZE ROZPORZĄDZENIE:
Artykuł 1
W załączniku IX do rozporządzenia (UE) nr 267/2012 wprowadza się zmiany zgodnie z załącznikiem do niniejszego rozporządzenia.
Artykuł 2
Niniejsze rozporządzenie wchodzi w życie następnego dnia po jego opublikowaniu w Dzienniku Urzędowym Unii Europejskiej.
Niniejsze rozporządzenie wiąże w całości i jest bezpośrednio stosowane we wszystkich państwach członkowskich.
Sporządzono w Brukseli dnia 29 lipca 2021 r.
W imieniu Rady
G. DOVŽAN
Przewodniczący
(1) Dz.U. L 88 z 24.3.2012, s. 1.
(2) Rozporządzenie wykonawcze Rady (UE) 2020/847 z dnia 18 czerwca 2020 r. wykonujące rozporządzenie (UE) nr 267/2012 w sprawie środków ograniczających wobec Iranu (Dz.U. L 196 z 19.6.2020, s. 1).
(3) Wyrok Sądu z dnia 9 czerwca 2021 r., Sayed Shamsuddin Borborudi przeciwko Radzie Unii Europejskiej, T-580/19, ECLI:EU:T:2021:330.
(4) Decyzja Rady 2010/413/WPZiB z dnia 26 lipca 2010 r. w sprawie środków ograniczających wobec Iranu i uchylająca wspólne stanowisko 2007/140/WPZiB (Dz.U. L 195 z 27.7.2010, s. 39).
ZAŁĄCZNIK
W załączniku IX do rozporządzenia (UE) nr 267/2012 wprowadza się następujące zmiany:
|
1) |
pod nagłówkiem „I. Osoby i podmioty zaangażowane w działania na rzecz broni jądrowej lub pocisków balistycznych oraz osoby i podmioty popierające rząd Iranu”, w sekcji „A. Osoby fizyczne” skreśla się następujący wpis: „25. Sayed Shamsuddin Borborudi”; |
|
2) |
pod nagłówkiem „I. Osoby i podmioty zaangażowane w działania na rzecz broni jądrowej lub pocisków balistycznych oraz osoby i podmioty popierające rząd Iranu”, w wykazie zamieszczonym w sekcji „A. Osoby fizyczne”, następujące wpisy zastępują odpowiadające im wpisy:
|
|
3) |
pod nagłówkiem „I. Osoby i podmioty zaangażowane w działania na rzecz broni jądrowej lub pocisków balistycznych oraz osoby i podmioty popierające rząd Iranu”, w wykazie zamieszczonym w sekcji „B. Podmioty”, następujące wpisy zastępują odpowiadające im wpisy:
|
|
4) |
pod nagłówkiem „II. Korpus Strażników Rewolucji Irańskiej”, w wykazie zamieszczonym w sekcji „A. Osoby fizyczne”, następujące wpisy zastępują odpowiadające im wpisy:
|
|
5) |
pod nagłówkiem „II. Korpus Strażników Rewolucji Irańskiej”, w wykazie zamieszczonym w sekcji „B. Podmioty”, następujące wpisy zastępują odpowiadające im wpisy:
|
|
30.7.2021 |
PL |
Dziennik Urzędowy Unii Europejskiej |
L 272/11 |
ROZPORZĄDZENIE DELEGOWANE KOMISJI (UE) 2021/1243
z dnia 19 kwietnia 2021 r.
uzupełniające rozporządzenie Parlamentu Europejskiego i Rady (UE) 2019/2144 przez ustanowienie szczegółowych przepisów dotyczących ułatwień w zakresie montażu alkomatów blokujących zapłon w pojazdach silnikowych oraz zmieniające załącznik II do tego rozporządzenia
(Tekst mający znaczenie dla EOG)
KOMISJA EUROPEJSKA,
uwzględniając Traktat o funkcjonowaniu Unii Europejskiej,
uwzględniając rozporządzenie Parlamentu Europejskiego i Rady (UE) 2019/2144 z dnia 27 listopada 2019 r. w sprawie wymogów dotyczących homologacji typu pojazdów silnikowych i ich przyczep oraz układów, komponentów i oddzielnych zespołów technicznych przeznaczonych do tych pojazdów, w odniesieniu do ich ogólnego bezpieczeństwa oraz ochrony osób znajdujących się w pojeździe i niechronionych uczestników ruchu drogowego, zmieniające rozporządzenie Parlamentu Europejskiego i Rady (UE) 2018/858 oraz uchylające rozporządzenia Parlamentu Europejskiego i Rady (WE) nr 78/2009, (WE) nr 79/2009 i (WE) nr 661/2009 oraz rozporządzenia Komisji (WE) nr 631/2009, (UE) nr 406/2010, (UE) nr 672/2010, (UE) nr 1003/2010, (UE) nr 1005/2010, (UE) nr 1008/2010, (UE) nr 1009/2010, (UE) nr 19/2011, (UE) nr 109/2011, (UE) nr 458/2011, (UE) nr 65/2012, (UE) nr 130/2012, (UE) nr 347/2012, (UE) nr 351/2012, (UE) nr 1230/2012 i (UE) 2015/166 (1), w szczególności jego art. 4 ust. 6 i art. 6 ust. 6,
a także mając na uwadze, co następuje:
|
(1) |
W art. 6 rozporządzenia (UE) 2019/2144 wprowadzono obowiązek wyposażenia pojazdów silnikowych kategorii M i N w pewne zaawansowane układy pojazdów, w tym ułatwienie w zakresie montażu alkomatów blokujących zapłon. W załączniku II do tego rozporządzenia określono podstawowe wymogi dotyczące homologacji typu pojazdów silnikowych w odniesieniu do ułatwień w zakresie montażu alkomatów blokujących zapłon w tych pojazdach. |
|
(2) |
Alkomaty blokujące zapłon zwiększają bezpieczeństwo ruchu drogowego, zapobiegając prowadzeniu pojazdu silnikowego przez osoby, u których stężenie alkoholu w organizmie przekracza ustaloną wartość dopuszczalną. |
|
(3) |
Niezbędne są szczegółowe przepisy dotyczące szczególnych wymogów homologacji pojazdów w odniesieniu do ułatwień w zakresie montażu alkomatów blokujących zapłon. |
|
(4) |
Seria norm europejskich EN 50436 określa metody badań i zasadnicze wymogi dotyczące parametrów alkomatów blokujących zapłon oraz zawiera wytyczne dla organów, decydentów, nabywców i użytkowników. Normy w tej serii obejmują również szczególne przepisy dotyczące pojazdów silnikowych w celu ułatwienia montażu alkomatów blokujących zapłon. |
|
(5) |
Alkomaty blokujące zapłon są głównie przeznaczone do montażu na rynku wtórnym. W tym celu są one podłączone do obwodów elektrycznych i sterujących pojazdu. Taka instalacja nie powinna zakłócać prawidłowego działania lub konserwacji pojazdu, nie powinna negatywnie wpływać na bezpieczeństwo i ochronę pojazdu, ale powinna być również jak najprostsza dla wyspecjalizowanych i wyszkolonych instalatorów. |
|
(6) |
Należy zatem zobowiązać producentów pojazdów do udostępniania na swoich stronach internetowych dokumentu zawierającego jasne instrukcje montażu alkomatów blokujących zapłon („dokument instalacji”), aby umożliwić technikom właściwe zainstalowanie alkomatu blokującego zapłon w danym modelu pojazdu. |
|
(7) |
Ponieważ niektóre informacje zawarte w dokumencie instalacji mogą dotyczyć usług w zakresie informacji dotyczących naprawy i konserwacji pojazdów, związanych z ochroną, powinny one być dostępne wyłącznie dla niezależnych podmiotów, które uzyskały zezwolenie od podmiotów akredytowanych zgodnie z dodatkiem 3 do załącznika X do rozporządzenia Parlamentu Europejskiego i Rady (UE) 2018/858 (2). |
|
(8) |
Tabela zawierająca wykaz wymogów w załączniku II do rozporządzenia (UE) 2019/2144 nie zawiera żadnego odniesienia do aktów prawnych dotyczących ułatwień w zakresie montażu alkomatów blokujących zapłon. Konieczne jest zatem wprowadzenie do tego załącznika odniesienia do niniejszego rozporządzenia. |
|
(9) |
Należy zatem odpowiednio zmienić rozporządzenie (UE) 2019/2144. |
|
(10) |
Ponieważ rozporządzenie (UE) 2019/2144 ma być stosowane od dnia 6 lipca 2022 r., niniejsze rozporządzenie należy także stosować od tej daty, |
PRZYJMUJE NINIEJSZE ROZPORZĄDZENIE:
Artykuł 1
Wymogi dotyczące ułatwień w zakresie montażu alkomatów blokujących zapłon
Homologacja typu pojazdów silnikowych w odniesieniu do ułatwień w zakresie montażu alkomatów blokujących zapłon podlega wymogom określonym w załączniku I.
Artykuł 2
Zmiana rozporządzenia (UE) 2019/2144
W załączniku II do rozporządzenia (UE) 2019/2144 wprowadza się zmiany określone w załączniku II do niniejszego rozporządzenia.
Artykuł 3
Wejście w życie i rozpoczęcie stosowania
Niniejsze rozporządzenie wchodzi w życie dwudziestego dnia po jego opublikowaniu w Dzienniku Urzędowym Unii Europejskiej.
Niniejsze rozporządzenie stosuje się od dnia 6 lipca 2022 r.
Niniejsze rozporządzenie wiąże w całości i jest bezpośrednio stosowane we wszystkich państwach członkowskich.
Sporządzono w Brukseli dnia 19 kwietnia 2021 r.
W imieniu Komisji
Ursula VON DER LEYEN
Przewodnicząca
(1) Dz.U. L 325 z 16.12.2019, s. 1.
(2) Rozporządzenie Parlamentu Europejskiego i Rady (UE) 2018/858 z dnia 30 maja 2018 r. w sprawie homologacji i nadzoru rynku pojazdów silnikowych i ich przyczep oraz układów, komponentów i oddzielnych zespołów technicznych przeznaczonych do tych pojazdów, zmieniające rozporządzenie (WE) nr 715/2007 i (WE) nr 595/2009 oraz uchylające dyrektywę 2007/46/WE (Dz.U. L 151 z 14.6.2018, s. 1).
ZAŁĄCZNIK I
Wymogi techniczne
1.
Ułatwienia w zakresie montażu alkomatów blokujących zapłon umożliwiają montaż lub modernizację alkomatów blokujących zapłon zgodnych z normami europejskimi EN 50436-1:2014 lub EN 50436-2:2014+A1:2015.
2.
Układ pojazdu w odniesieniu do ułatwień w zakresie montażu alkomatów blokujących zapłon w każdym pojeździe silnikowym kategorii M i N musi być zgodny z odpowiednim modelem pojazdu określonym w dokumencie instalacji alkomatów blokujących zapłon („dokument instalacji”), zgodnym z normą europejską EN 50436-7:2016. W tym celu dokument instalacji musi obejmować co najmniej jedną z opcji określonych w normie EN 50436-7:2016 załącznik C pkt 3a, 3b lub 3c. Producent pojazdu może, w porozumieniu z organem udzielającym homologacji i służbą techniczną, dostarczyć dokument instalacji zgodny z późniejszymi wersjami normy europejskiej.
3.
Dokument instalacji
3.1.
Dokument instalacji musi zawierać szczegółowy opis, schematy i rysunki wyjaśniające sposób instalacji alkomatu blokującego zapłon, obejmujące którykolwiek z następujących zestawów informacji:|
a) |
informacje dotyczące zasilania z akumulatora, uziemienia, stanu gotowości pojazdu i urządzenia umożliwiającego uruchomienie; |
|
b) |
informacje dotyczące zasilania z akumulatora, uziemienia, stanu gotowości pojazdu i ścieżki umożliwiającej uruchomienie lub unieruchomienie oraz opcjonalnej detekcji działania napędu (np. działanie silnika) lub sygnalizacji ruchu pojazdu; lub |
|
c) |
informacje dotyczące zasilania z akumulatora, uziemienia i łącza magistrali danych. |
3.2.
Wszelkie dodatkowe oprogramowanie, sprzęt komputerowy lub procedury niezbędne do zainstalowania alkomatu blokującego zapłon w standardowym pojeździe muszą być określone i wskazane w dokumencie instalacji.
3.3.
W normalnej sytuacji alkomat blokujący zapłon musi znajdować się w trybie dezaktywacji. Tryb dezaktywacji alkomatu blokującego zapłon uzyskuje się za pomocą otwartego przekaźnika wyjściowego, odnośnego sygnału wyjściowego lub odnośnego komunikatu magistrali cyfrowej. Zamknięcie tego przekaźnika lub zmiana blokującego sygnału wyjściowego na nieblokujący sygnał wyjściowy bądź przekazanie odnośnego komunikatu magistrali danych dotyczącego nieblokowania muszą mieć miejsce, gdy dostarczono próbkę oddechu, w której stężenie alkoholu jest niższe niż wcześniej ustalona wartość graniczna.
3.4.
Zainstalowany alkomat blokujący zapłon ingeruje jedynie w proces uruchamiania silnika lub w umożliwianie pojazdowi poruszania się z własnym napędem po uruchomieniu głównego wyłącznika pojazdu, i nie może wpływać na działający silnik lub poruszający się pojazd.
4.
Dostęp do informacji dotyczących ułatwienia w zakresie montażu alkomatów blokujących zapłon
4.1.
Producenci pojazdów wprowadzają niezbędne ustalenia i procedury w celu zapewnienia, aby informacje na temat ułatwienia w zakresie montażu alkomatów blokujących zapłon w formie odpowiednich szczegółowych informacji w znormalizowanym dokumencie instalacji były dostępne zgodnie z załącznikiem X do rozporządzenia (UE) 2018/858. Ponieważ niektóre informacje mogą dotyczyć usług w zakresie informacji dotyczących naprawy i konserwacji pojazdów, związanych z bezpieczeństwem, dostęp do informacji dotyczących ułatwienia w zakresie montażu alkomatów blokujących zapłon jest ograniczony do niezależnych podmiotów, które przestrzegają procedury określonej w dodatku 3 do tego załącznika.
5.
Producent pojazdu załącza do dokumentu informacyjnego deklarację sporządzoną przy użyciu wzoru określonego w dodatku do niniejszego załącznika.
Dodatek
Deklaracja producenta
(Producent):
…
(Adres producenta):
…
Zaświadcza, że
zapewnia dostęp do dokumentu instalacji alkomatów blokujących zapłon zgodnie z art. 1 rozporządzenia delegowanego Komisji (UE) 2021/1243 (1) w odniesieniu do następującej marki i typu pojazdu:
adresy głównych stron internetowych, na których można uzyskać dostęp do dokumentu instalacji alkomatu blokującego zapłon, są wymienione w załączniku A do niniejszej deklaracji. Dane kontaktowe odpowiedzialnego przedstawiciela producenta, który podpisał niniejszą deklarację, podano w załączniku B do niniejszej deklaracji.
Sporządzono w … [miejscowość]
W dniu … [data]
[podpis] [stanowisko][]
Załącznik A: adres strony internetowej (adresy stron internetowych)
Załącznik B: dane kontaktowe
(1) Rozporządzenie delegowane Komisji (UE) 2021/1243 z dnia 19 kwietnia 2021 r. uzupełniające rozporządzenie Parlamentu Europejskiego i Rady (UE) 2019/2144 przez ustanowienie szczegółowych przepisów dotyczących ułatwień w zakresie montażu alkomatów blokujących zapłon w pojazdach silnikowych oraz zmieniające załącznik II do tego rozporządzenia (Dz.U. L 272 z …..2021, s. 11).
ZAŁĄCZNIK II
Zmiana rozporządzenia (UE) 2019/2144
W załączniku II do rozporządzenia (UE) 2019/2144 wiersz dotyczący wymogu E1 otrzymuje brzmienie:
|
„E1 Ułatwienia w zakresie montażu alkomatów blokujących zapłon |
Rozporządzenie delegowane Komisji (UE) 2021/1243 (*1) |
|
B |
B |
B |
B |
B |
B |
|
|
|
|
|
|
(*1) Rozporządzenie delegowane Komisji (UE) 2021/1243 z dnia 19 kwietnia 2021 r. uzupełniające rozporządzenie Parlamentu Europejskiego i Rady (UE) 2019/2144 przez ustanowienie szczegółowych przepisów dotyczących ułatwień w zakresie montażu alkomatów blokujących zapłon w pojazdach silnikowych oraz zmieniające załącznik II do tego rozporządzenia (Dz.U. L 272 z ..2021, s. 11).”.
|
30.7.2021 |
PL |
Dziennik Urzędowy Unii Europejskiej |
L 272/16 |
ROZPORZĄDZENIE DELEGOWANE KOMISJI (UE) 2021/1244
z dnia 20 maja 2021 r.
zmieniające załącznik X do rozporządzenia Parlamentu Europejskiego i Rady (UE) 2018/858 w odniesieniu do znormalizowanego dostępu do informacji z pokładowego układu diagnostycznego oraz informacji dotyczących naprawy i konserwacji pojazdów, a także wymogów i procedur w zakresie dostępu do informacji dotyczących zabezpieczeń pojazdu
KOMISJA EUROPEJSKA,
uwzględniając Traktat o funkcjonowaniu Unii Europejskiej,
uwzględniając rozporządzenie Parlamentu Europejskiego i Rady (UE) 2018/858 z dnia 30 maja 2018 r. w sprawie homologacji i nadzoru rynku pojazdów silnikowych i ich przyczep oraz układów, komponentów i oddzielnych zespołów technicznych przeznaczonych do tych pojazdów, zmieniające rozporządzenie (WE) nr 715/2007 i (WE) nr 595/2009 oraz uchylające dyrektywę 2007/46/WE (1), w szczególności jego art. 61 ust. 11,
a także mając na uwadze, co następuje:
|
(1) |
W art. 61 ust. 2 rozporządzenia 2018/858 nałożono na producentów pojazdów obowiązek udostępniania na swoich stronach internetowych informacji z pokładowego układu diagnostycznego (OBD) pojazdu oraz informacji dotyczących naprawy i konserwacji pojazdów (RMI). Brakuje jednak zharmonizowanych kryteriów dotyczących sposobu udostępniania tych informacji, co wymaga od niezależnych podmiotów dostosowania się do licznych i zróżnicowanych usług internetowych i terminologii. |
|
(2) |
W sprawozdaniu Komisji dla Parlamentu Europejskiego i Rady z dnia 9 grudnia 2016 r. (2) w sprawie funkcjonowania systemu dostępu do informacji dotyczących naprawy i utrzymania pojazdów stwierdzono, że dzięki standaryzacji tych stron internetowych i odpowiedniej terminologii obciążenie niezależnych podmiotów mogłoby zostać zmniejszone. |
|
(3) |
Ponieważ dostęp do informacji z OBD pojazdu oraz RMI pojazdu powinien być możliwy niezależnie od typu mechanizmu napędowego danego pojazdu, konieczne jest wyjaśnienie, że taki dostęp jest obowiązkowy nie tylko w przypadku wymogów dotyczących emisji zanieczyszczeń. |
|
(4) |
W dniu 15 września 2014 r. Europejski Komitet Normalizacyjny (CEN) opublikował części 1–5 normy EN ISO 18541 „Pojazdy drogowe – Zharmonizowany dostęp do informacji dotyczących naprawy i utrzymania pojazdów (RMI)”. Części te mają na celu ułatwienie wymiany między producentami i niezależnymi podmiotami informacji z OBD pojazdu i RMI pojazdu poprzez ustanowienie wymogów technicznych i procedur ułatwiających dostęp do tych informacji. Należy zatem w załączniku X do rozporządzenia (UE) 2018/858 zawrzeć odniesienie do wymogów części 1–5 normy EN ISO 18541-2014. |
|
(5) |
Biorąc pod uwagę, że informacje z OBD pojazdu i RMI pojazdu obejmują informacje niezbędne do zapewnienia bezpieczeństwa pojazdu, dostęp do niektórych zabezpieczeń pojazdu należy zapewnić wyłącznie niezależnym podmiotom spełniającym wymogi określone w niniejszym załączniku. |
|
(6) |
Zgodnie z zaleceniami Forum ds. dostępu do informacji o pojazdach, o którym mowa w art. 66 ust. 1 rozporządzenia (UE) 2018/858, wymogi te powinny obejmować zatwierdzanie zainteresowanych niezależnych podmiotów oraz udzielanie autoryzacji ich pracownikom wykonującym stosowne czynności przez akredytowane podmioty. Konieczne jest zatem ustanowienie procedury zatwierdzania i autoryzacji niezależnych podmiotów w zakresie dostępu do zabezpieczeń pojazdów, która powinna się opierać na „Systemie akredytacji, zatwierdzania i autoryzacji w zakresie dostępu do związanych z zabezpieczeniami informacji dotyczących naprawy i konserwacji (RMI)”, który został zatwierdzony w dniu 19 maja 2016 r. przez organizację Europejska Współpraca w Dziedzinie Akredytacji. Należy również ocenić, czy podmioty te nie są zaangażowane w nielegalną działalność gospodarczą. |
|
(7) |
Ponadto konieczne jest określenie roli i zakresów odpowiedzialności organów zaangażowanych w zatwierdzanie i autoryzację niezależnych podmiotów i ich pracowników w zakresie uzyskania dostępu do związanych z zabezpieczeniami informacji dotyczących naprawy i konserwacji pojazdów. |
|
(8) |
Aby umożliwić państwom członkowskim, organom krajowym i podmiotom gospodarczym przygotowanie się do stosowania nowych przepisów wprowadzonych niniejszym rozporządzeniem, należy odroczyć datę rozpoczęcia jego stosowania. |
|
(9) |
Należy zatem odpowiednio zmienić załącznik X do rozporządzenia (UE) 2018/858, |
PRZYJMUJE NINIEJSZE ROZPORZĄDZENIE:
Artykuł 1
W załączniku X do rozporządzenia (UE) 2018/858 wprowadza się zmiany zgodnie z załącznikiem do niniejszego rozporządzenia.
Artykuł 2
Niniejsze rozporządzenie wchodzi w życie dwudziestego dnia po jego opublikowaniu w Dzienniku Urzędowym Unii Europejskiej.
Niniejsze rozporządzenie stosuje się od dnia 30 lipca 2023 r.
Niniejsze rozporządzenie wiąże w całości i jest bezpośrednio stosowane we wszystkich państwach członkowskich.
Sporządzono w Brukseli dnia 20 maja 2021 r.
W imieniu Komisji
Ursula VON DER LEYEN
Przewodnicząca
(1) Dz.U. L 151 z 14.6.2018, s. 1.
(2) Sprawozdanie Komisji dla Parlamentu Europejskiego i Rady w sprawie funkcjonowania systemu dostępu do informacji dotyczących naprawy i utrzymania pojazdów ustanowionego rozporządzeniem (WE) nr 715/2007 w sprawie homologacji typu pojazdów silnikowych w odniesieniu do emisji zanieczyszczeń pochodzących z lekkich pojazdów pasażerskich i użytkowych (Euro 5 i Euro 6) oraz w sprawie dostępu do informacji dotyczących naprawy i utrzymania pojazdów, COM(2016) 782 final.
ZAŁĄCZNIK
W załączniku X do rozporządzenia (UE) 2018/858 wprowadza się następujące zmiany:
|
1) |
pkt 2.1 otrzymuje brzmienie:
|
|
2) |
pkt 2.5.2 otrzymuje brzmienie:
|
|
3) |
pkt 2.9 akapit pierwszy otrzymuje brzmienie: „Do celów OBD, diagnostyki, naprawy i konserwacji, monitorowania i kontroli pojazdu bezpośredni strumień danych o pojeździe, w tym kody błędów, oraz funkcje diagnostyczne udostępnia się przez port szeregowy znormalizowanego łącza danych określony w pkt 6.5.1.4 oraz zgodnie ze specyfikacjami określonymi w pkt 6.5.3 dodatku 1 do załącznika 11 do regulaminu nr 83 Europejskiej Komisji Gospodarczej Organizacji Narodów Zjednoczonych (EKG ONZ) (*1) oraz zgodnie z pkt 4.7.3 załącznika 9B i wzorcowymi dokumentami standardowymi określonymi w dodatku 6 do załącznika do regulaminu nr 49 Europejskiej Komisji Gospodarczej Organizacji Narodów Zjednoczonych (EKG ONZ) (*2). (*1) Regulamin nr 83 Europejskiej Komisji Gospodarczej Organizacji Narodów Zjednoczonych (EKG ONZ) – Jednolite przepisy dotyczące homologacji pojazdów w zakresie emisji zanieczyszczeń w zależności od paliwa zasilającego silnik (Dz.U. L 42 z 15.2.2012, s. 1)." (*2) Regulamin nr 49 Europejskiej Komisji Gospodarczej Organizacji Narodów Zjednoczonych (EKG/ONZ) – Jednolite przepisy dotyczące działań, jakie mają zostać podjęte przeciwko emisji zanieczyszczeń gazowych i pyłowych przez silniki o zapłonie samoczynnym (ZS) stosowane w pojazdach oraz emisji zanieczyszczeń gazowych z silników o zapłonie iskrowym (ZI) napędzanych gazem ziemnym lub skroplonym gazem węglowodorowym stosowanych w pojazdach (Dz.U. L 180 z 8.7.2011, s. 53).”;" |
|
4) |
pkt 6.1 akapit pierwszy otrzymuje brzmienie: „Zgodność z częściami normy EN ISO 18541, o których mowa w pkt 2.1, stanowi podstawę domniemania spełnienia obowiązków producentów polegających na zapewnieniu informacji z OBD oraz informacji dotyczących naprawy i konserwacji pojazdów na ich stronach internetowych z wykorzystaniem znormalizowanego formatu.”; |
|
5) |
pkt 6.2 otrzymuje brzmienie: „Dostęp do zabezpieczeń pojazdu jest udostępniany niezależnym podmiotom z zastrzeżeniem ochrony technologii zabezpieczeń zgodnie z następującymi wymogami:”; |
|
6) |
w pkt 6.3 wprowadza się następujące zmiany:
|
|
7) |
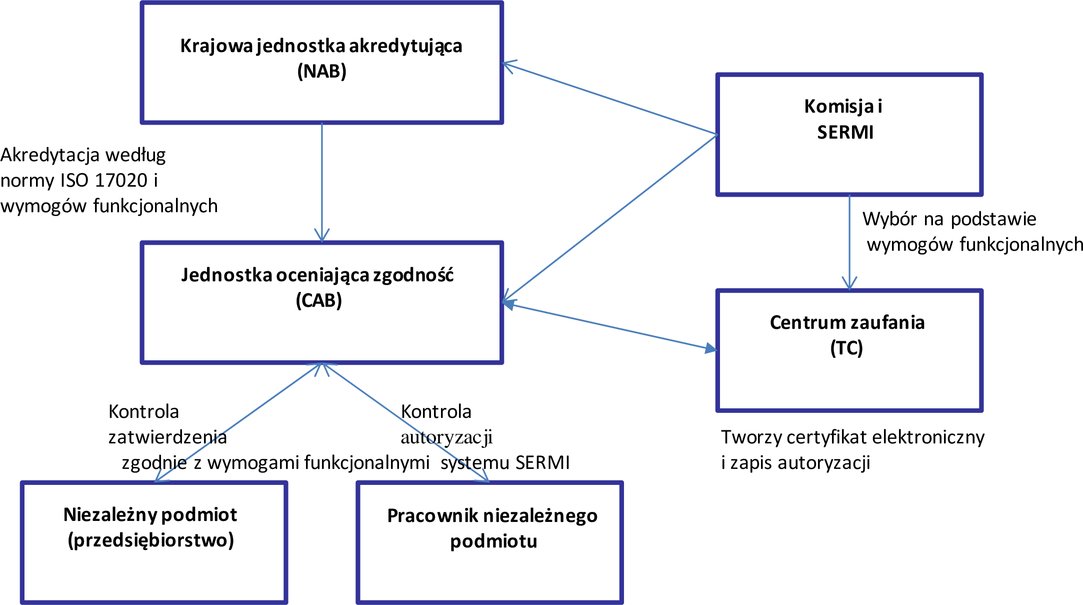
dodaje się dodatek 3 w brzmieniu: „Dodatek 3 Procedura zatwierdzania i autoryzacji niezależnych podmiotów w zakresie dostępu do zabezpieczeń pojazdu (*5) 1. Zakres Niniejszy dodatek zawiera wymogi do celów zatwierdzania i autoryzacji niezależnych podmiotów wymagających dostępu do związanych z zabezpieczeniami informacji dotyczących naprawy i konserwacji pojazdów (RMI). Określono w nim szczegółowo procedurę i organy wymagane do zatwierdzania i autoryzacji niezależnych podmiotów do uzyskania dostępu do związanych z zabezpieczeniami informacji dotyczących napraw i konserwacji lekkich pojazdów pasażerskich i użytkowych oraz pojazdów ciężarowych. 2. Definicje i skróty 2.1. Definicje Do celów niniejszego dodatku stosuje się następujące definicje: 2.1.1. »akredytacja« »akredytacja« oznacza akredytację zgodnie z definicją w art. 2 pkt 10 rozporządzenia (WE) nr 765/2008; 2.1.2. »pracownik niezależnego podmiotu« »pracownik niezależnego podmiotu« oznacza pracownika zatwierdzonego niezależnego podmiotu, który na podstawie autoryzacji od swojej jednostki oceniającej zgodność będzie miał dostęp do RMI związanych z zabezpieczeniami; 2.1.3. »związane z zabezpieczeniami informacje dotyczące naprawy i konserwacji« lub »RMI związane z zabezpieczeniami« »związane z zabezpieczeniami informacje dotyczące naprawy i konserwacji« lub »RMI związane z zabezpieczeniami« oznaczają informacje, oprogramowanie, funkcje i usługi wymagane do celów naprawy i utrzymania zabezpieczeń, które zostały zainstalowane w pojeździe przez producenta, aby zapobiec kradzieży pojazdu lub odjechaniu pojazdem oraz umożliwić śledzenie i odzyskiwanie pojazdu; 2.1.4. »świadectwo kontroli zatwierdzenia« »świadectwo kontroli zatwierdzenia« oznacza świadectwo wydawane przez jednostkę oceniającą zgodność niezależnym podmiotom spełniającym kryteria zatwierdzenia określone w niniejszym dodatku, które potwierdza, że te niezależne podmioty zostały zatwierdzone oraz że pracownicy niezależnego podmiotu mogą się ubiegać o autoryzację w zakresie dostępu do RMI związanych z zabezpieczeniami; 2.1.5. »świadectwo kontroli autoryzacji« »świadectwo kontroli autoryzacji« oznacza świadectwo wydane przez jednostkę oceniającą zgodność pracownikom niezależnego podmiotu spełniającym kryteria autoryzacji określone w niniejszym dodatku, które potwierdza, że pracownicy ci są autoryzowani w zakresie dostępu do RMI związanych z zabezpieczeniami na stronie internetowej producenta pojazdów; 2.1.6. »centrum zaufania« lub »TC« »centrum zaufania« lub »TC« oznacza organ wyznaczony przez SERMI i zatwierdzony przez Komisję, odpowiedzialny za:
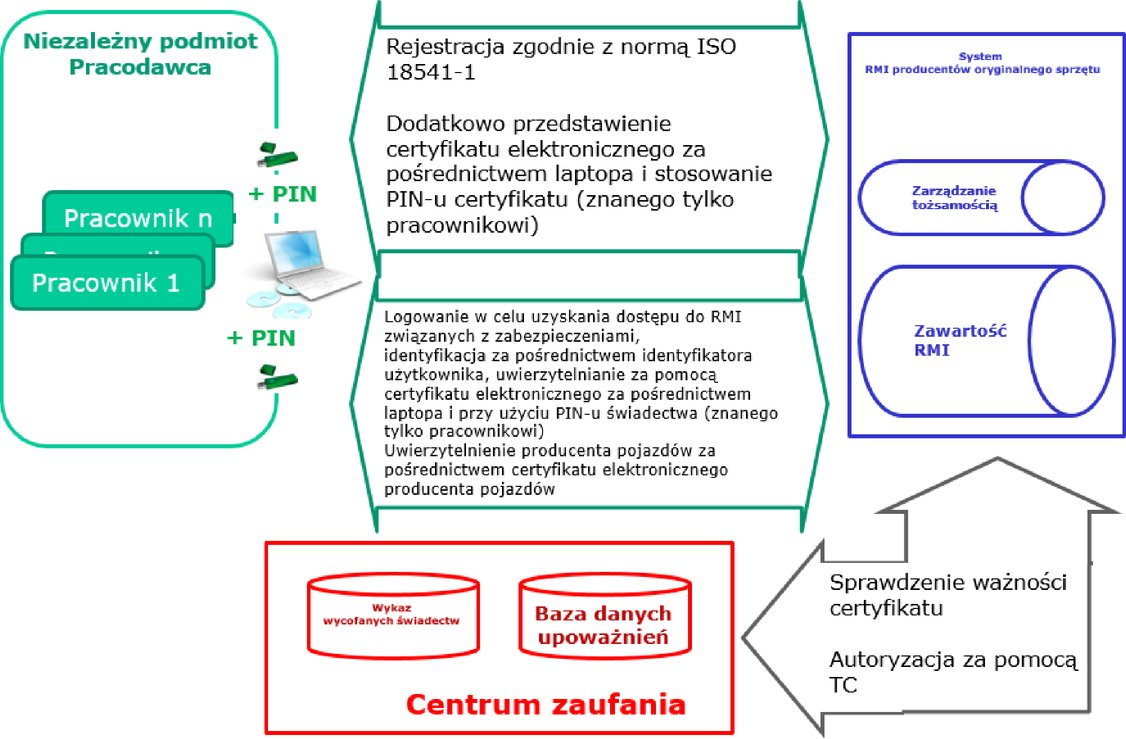
2.1.7. »token bezpieczeństwa« »token bezpieczeństwa« oznacza urządzenie, które umożliwia bezpieczne uwierzytelnienie niezależnego podmiotu; 2.1.8. »certyfikat cyfrowy« »certyfikat cyfrowy« oznacza certyfikat elektroniczny, który wymaga podpisu cyfrowego centrum zaufania wydającego certyfikat w celu powiązania klucza publicznego z tożsamością pracownika niezależnego podmiotu, zgodnie z normą ISO 9594; 2.1.9. »baza danych autoryzacji« »baza danych autoryzacji« oznacza bazę danych prowadzoną przez centrum zaufania, która zawiera szczegółowe informacje dotyczące autoryzacji zanonimizowanych autoryzowanych pracowników niezależnego podmiotu oraz rejestracji zatwierdzonych niezależnych podmiotów; 2.1.10. »baza danych certyfikacji« »baza danych certyfikacji« oznacza bazę danych prowadzoną przez centrum zaufania w celu zarządzania ważnością certyfikatów cyfrowych i identyfikatorami autoryzowanych pracowników niezależnych podmiotów; 2.1.11. »Europejska Współpraca w Dziedzinie Akredytacji« lub »EA« »Europejska Współpraca w Dziedzinie Akredytacji« lub »EA« oznacza jednostkę uznaną przez Komisję zgodnie z art. 14 rozporządzenia (WE) nr 765/2008, która jest odpowiedzialna za opracowanie, utrzymanie i wdrożenie akredytacji w Unii; 2.1.12. »Forum na rzecz dostępu do RMI pojazdu związanych z zabezpieczeniami« lub »SERMI« »Forum na rzecz dostępu do RMI pojazdu związanych z zabezpieczeniami« lub »SERMI« oznacza podmiot odpowiedzialny za koordynację i doradzanie Komisji w zakresie wdrażania procedur »akredytacji, zatwierdzania i autoryzacji do celów dostępu« do RMI związanych z zabezpieczeniami; 2.1.13. »odpowiednie organy« »odpowiednie organy« oznaczają te organy publiczne, które posiadają uprawnienia do działania w dziedzinie ochrony przed przestępstwami przeciwko bezpieczeństwu pojazdów, prowadzenia dochodzeń związanych z takimi przestępstwami i ścigania takich przestępstw. 3. Akredytacja jednostek oceniających zgodność, zatwierdzanie niezależnych podmiotów oraz autoryzację pracowników niezależnych podmiotów Wyłącznie jednostki oceniające zgodność akredytowane przez zdefiniowaną w art. 2 pkt 11 rozporządzenia (WE) nr 765/2008 krajową jednostkę akredytującą państwa członkowskiego, w którym mają siedzibę, wydają świadectwa kontroli zatwierdzenia poświadczające, że niezależny podmiot został zatwierdzony, oraz świadectwa kontroli autoryzacji poświadczające, że pracownik niezależnego podmiotu ma mieć dostęp do RMI związanych z zabezpieczeniami. Zatwierdzenia niezależnego podmiotu i autoryzacji pracownika niezależnego podmiotu udziela się na okres 60 miesięcy, począwszy od daty wydania odpowiednich świadectw kontroli. Niezależne podmioty, które chcą otrzymać RMI związane z zabezpieczeniami, muszą uzyskać świadectwo kontroli zatwierdzenia od jednostki oceniającej zgodność akredytowanej przez krajową jednostkę akredytującą państwa członkowskiego, w którym niezależny podmiot ma siedzibę. Pracownicy niezależnego podmiotu, którzy mają się posługiwać RMI związanymi z zabezpieczeniami, muszą uzyskać świadectwo kontroli autoryzacji od jednostki oceniającej zgodność akredytowanej przez krajową jednostkę akredytującą państwa członkowskiego, w którym zamieszkuje pracownik niezależnego podmiotu. Jednostki oceniające zgodność powiadamiają TC o wszelkich wydanych świadectwach kontroli zatwierdzenia lub świadectwach kontroli autoryzacji, na podstawie których TC tworzą rejestr autoryzacji i wydają token bezpieczeństwa oraz certyfikat cyfrowy zawierający szczegółowe dane umożliwiające niepowtarzalną identyfikację pracowników niezależnego podmiotu na stronie internetowej producenta pojazdów dotyczącej RMI. Jednostki oceniające zgodność wyposażają indywidualnych pracowników niezależnego podmiotu w token bezpieczeństwa i certyfikat cyfrowy. Producenci pojazdów mogą żądać opłaty za rejestrację pracowników niezależnego podmiotu na stronach internetowych producentów pojazdów dotyczących RMI producentów pojazdów oraz za dostęp do RMI związanych z zabezpieczeniami. Przedmiotowa opłata musi być proporcjonalna do kosztów takiej rejestracji i zapewnienia dostępu. Należne opłaty określa się na stronach internetowych producentów pojazdów dotyczących RMI. Wszystkie transfery danych cyfrowych między niezależnymi podmiotami, TC i jednostkami oceniającymi zgodność muszą być realizowane w drodze transakcji między przedsiębiorstwami (B2B) z wykorzystaniem bezpiecznych protokołów i w sposób terminowy.
Oświadczenie zaświadczające, że niezależny podmiot prowadzi legalną działalność gospodarczą, o której mowa w pkt 6.3 niniejszego załącznika, jest podpisywane przez niezależny podmiot wnioskujący o udzielenie autoryzacji przez jednostkę oceniającą zgodność. Niezależny podmiot zatwierdza się dopiero po przeprowadzeniu przez jednostkę oceniającą zgodność kontroli, w ramach której sprawdza się, czy deklaracja ta została podpisana i czy niezależny podmiot i jego poszczególni pracownicy spełniają wymogi określone w niniejszym dodatku. Poszczególnym pracownikom niezależnego podmiotu udziela się autoryzacji dopiero po kontroli ze strony jednostki oceniającej zgodność. Jednostki oceniające zgodność sprawdzają przedłożone dokumenty i weryfikują, czy dany pracownik niezależnego podmiotu złożył uprzednio wniosek o autoryzację, który został odrzucony przez zainteresowaną jednostkę oceniającą zgodność lub jakąkolwiek inną jednostkę oceniającą zgodność na szczeblu Unii. Jednostki oceniające zgodność przesyłają do TC wszystkie dane niezbędne do przygotowania certyfikatu cyfrowego i tokena bezpieczeństwa, które jednostka oceniająca zgodność przesyła pracownikom niezależnego podmiotu. Pracownicy niezależnego podmiotu, którzy uzyskali autoryzację, otrzymują od jednostek oceniających zgodność PIN powiązany z certyfikatem cyfrowym.
3.1. Dostęp do RMI związanych z zabezpieczeniami w zarysie Producenci pojazdów zapewniają dostęp do RMI związanych z zabezpieczeniami za pośrednictwem swojej strony internetowej dotyczącej RMI, pod warunkiem że pracownicy niezależnego podmiotu są autoryzowani i mogą przedstawić świadectwo kontroli autoryzacji oraz że niezależny podmiot, w imieniu którego wykonują pracę pracownicy niezależnego podmiotu, posiada świadectwo kontroli zatwierdzenia. Producenci mogą oferować autoryzowanym pracownikom niezależnego podmiotu pracującym dla zatwierdzonych niezależnych podmiotów dostęp do internetowego narzędzia do składania zamówień na części związane z zabezpieczeniami za pomocą specjalnej aplikacji powiązanej ze stroną internetową dotyczącą RMI. Po otrzymaniu wniosku o dostęp do strony internetowej dotyczącej RMI strony internetowe producentów pojazdów żądają identyfikacji za pomocą niepowtarzalnego identyfikatora pracownika niezależnego podmiotu oraz żądają uwierzytelnienia. Uwierzytelnianie pracowników niezależnego podmiotu odbywa się wyłącznie przy użyciu certyfikatów cyfrowych. Po otrzymaniu certyfikatu elektronicznego strony internetowe producenta pojazdów dotyczące RMI weryfikują niepowtarzalny identyfikator pracownika niezależnego podmiotu oraz aktualny status certyfikatu cyfrowego i autoryzacji, komunikując się z TC określonym w certyfikacie elektronicznym. Wszystkie transfery danych cyfrowych między niezależnymi podmiotami, producentami pojazdów, TC i jednostkami oceniającymi zgodność muszą być realizowane w drodze transakcji między przedsiębiorstwami (B2B) z wykorzystaniem bezpiecznych protokołów i w sposób terminowy. Po zweryfikowaniu niepowtarzalnego identyfikatora pracownika niezależnego podmiotu i statusu autoryzacji pracownika niezależnego podmiotu producent pojazdów zapewnia na swojej stronie internetowej dostęp do wymaganych RMI związanych z zabezpieczeniami.
4. Szczegółowe zasady dotyczące dostępu do RMI związanych z zabezpieczeniami 4.1. Rola SERMI 4.1.1. Zakresy odpowiedzialności i obowiązki SERMI monitoruje wdrażanie procesu akredytacji w państwach członkowskich i odpowiednio powiadamia o tym Komisję. SERMI doradza Komisji w sprawie wniosków o wprowadzenie zmian w procesie akredytacji.
4.1.2. Wybór centrum zaufania SERMI wybiera TC i zgłasza Komisji do zatwierdzenia. Wybrane TC musi być przestrzegać normy ETSI TS 319 411-3, spełniać wymogi dotyczące podpisów elektronicznych określone w rozporządzeniu Parlamentu Europejskiego i Rady (UE) nr 910/2014 (*6) oraz wymogi określone w pkt 4.6 niniejszego dodatku. Ponadto TC musi:
4.2. Rola krajowych jednostek akredytujących Krajowa jednostka akredytująca jest odpowiedzialna za akredytację jednostek oceniających zgodność do celów zatwierdzania niezależnych podmiotów i autoryzacji pracowników niezależnego podmiotu w zakresie dostępu do RMI związanych z zabezpieczeniami. 4.2.1. Zakresy odpowiedzialności i wymogi Zakresy odpowiedzialności i wymogi odnoszące się do krajowych jednostek akredytujących określono w art. 8–12 rozporządzenia (WE) nr 765/2008. 4.2.2. Kryteria akredytacji jednostek oceniających zgodność Jednostki oceniające zgodność akredytuje się jako organy kontrolne typu A zgodnie z normą ISO/IEC 17020:2012. Jednostki oceniające zgodność muszą spełniać wymogi dotyczące najwyższego poziomu niezależności. Ponadto krajowa jednostka akredytująca ocenia zdolność jednostek oceniających zgodność do spełnienia wymogów określonych w pkt 4.3.1–4.3.4. Personel odpowiedzialny za kontrole niezależnych podmiotów musi posiadać poziom wiedzy w zakresie naprawy i konserwacji pojazdów samochodowych oraz specyfiki motoryzacyjnego rynku wtórnego, który jest odpowiedni do wykonywanych zadań. 4.3. Rola jednostek oceniających zgodność Jednostka oceniająca zgodność jest odpowiedzialna za kontrole niezależnych podmiotów i ich pracowników oraz za wydawanie świadectw kontroli zatwierdzenia i autoryzacji zgodnie z niniejszym dodatkiem, a także za unieważnianie takich świadectw. 4.3.1. Zakresy odpowiedzialności i wymogi
4.3.2. Przedłużenie zatwierdzenia Na wniosek niezależnego podmiotu lub 6 miesięcy przed wygaśnięciem ważności zatwierdzenia, jednostki oceniające zgodność przeprowadzają kontrolę na miejscu, a w przypadku pozytywnego wyniku kontroli – przedłużają zatwierdzenie. Jednostki oceniające zgodność wydają nowe świadectwo kontroli zatwierdzenia dla niezależnego podmiotu, który spełnia kryteria zatwierdzenia. Jednostki oceniające zgodność dokonują oceny wniosków o przedłużenie autoryzacji i wydają świadectwo kontroli autoryzacji pracownikom niezależnego podmiotu spełniającym kryteria autoryzacji. 4.3.3. Kryteria zatwierdzania niezależnego podmiotu przez jednostkę oceniającą zgodność Przed zatwierdzeniem niezależnego podmiotu oraz podczas każdej kontroli na miejscu w okresie ważności zatwierdzenia jednostki oceniające zgodność sprawdzają:
4.3.4. Kryteria autoryzacji pracownika niezależnego podmiotu przez jednostkę oceniającą zgodność Przed autoryzacją pracownika niezależnego podmiotu oraz podczas każdej kontroli na miejscu w okresie ważności zatwierdzenia jednostka oceniająca zgodność weryfikuje, czy:
4.4. Rola niezależnego podmiotu 4.4.1. Zakresy odpowiedzialności i wymogi
4.5. Rola pracowników niezależnych podmiotów 4.5.1. Zakresy odpowiedzialności i wymogi
4.6. Rola centrum zaufania TC tworzą i przesyłają certyfikaty cyfrowe do niezależnych podmiotów i pracowników niezależnych podmiotów za pośrednictwem odpowiednich jednostek oceniających zgodność. TC prowadzą bazę danych wydanych świadectw kontroli autoryzacji. TC zapewniają producentom pojazdów dostęp do interfejsu w celu weryfikacji statusu certyfikatów cyfrowych i świadectw kontroli autoryzacji. TC przechowują informacje dotyczące pracowników niezależnego podmiotu w bazie danych autoryzacji przez dodatkowy okres nieprzekraczający 60 miesięcy. Okres ten nie może być dłuższy niż pozostały okres ważności zatwierdzenia udzielonego niezależnemu podmiotowi, w którym świadczy pracę dany pracownik niezależnego podmiotu. 4.6.1. Zakresy odpowiedzialności i wymogi
4.7. Rola producentów pojazdów Producenci pojazdów zapewniają wszystkim zatwierdzonym niezależnym podmiotom i autoryzowanym pracownikom niezależnego podmiotu dostęp do związanych z zabezpieczeniami informacji dotyczących naprawy i konserwacji. Producenci pojazdów komunikują się z TC w celu weryfikacji autoryzacji oraz statusu autoryzacji pracowników niezależnego podmiotu ubiegających się o dostęp do takich informacji. 4.7.1. Zakresy odpowiedzialności i wymogi
4.7.2. Wymogi proceduralne dla producentów pojazdów Producenci pojazdów nie udzielają dostępu do RMI związanych z zabezpieczeniami, o ile nie spełniono wszystkich następujących wymogów proceduralnych:
(*5) Wymogi określone w niniejszym dodatku opierają się na wymogach określonych w »Systemie akredytacji, zatwierdzania i autoryzacji w zakresie dostępu do związanych z zabezpieczeniami informacji dotyczących naprawy i konserwacji (RMI)«, zatwierdzonym w dniu 19 maja 2016 r. przez organizację Europejska Współpraca w Dziedzinie Akredytacji (https://www.vehiclesermi.eu/)." (*6) Rozporządzenie Parlamentu Europejskiego i Rady (UE) nr 910/2014 z dnia 23 lipca 2014 r. w sprawie identyfikacji elektronicznej i usług zaufania w odniesieniu do transakcji elektronicznych na rynku wewnętrznym oraz uchylające dyrektywę 1999/93/WE (Dz.U. L 257 z 28.8.2014, s. 73)." |
(*1) Regulamin nr 83 Europejskiej Komisji Gospodarczej Organizacji Narodów Zjednoczonych (EKG ONZ) – Jednolite przepisy dotyczące homologacji pojazdów w zakresie emisji zanieczyszczeń w zależności od paliwa zasilającego silnik (Dz.U. L 42 z 15.2.2012, s. 1).
(*2) Regulamin nr 49 Europejskiej Komisji Gospodarczej Organizacji Narodów Zjednoczonych (EKG/ONZ) – Jednolite przepisy dotyczące działań, jakie mają zostać podjęte przeciwko emisji zanieczyszczeń gazowych i pyłowych przez silniki o zapłonie samoczynnym (ZS) stosowane w pojazdach oraz emisji zanieczyszczeń gazowych z silników o zapłonie iskrowym (ZI) napędzanych gazem ziemnym lub skroplonym gazem węglowodorowym stosowanych w pojazdach (Dz.U. L 180 z 8.7.2011, s. 53).”;
(*3) Zgodnie z definicją w art. 3 pkt 10 rozporządzenia (WE) nr 715/2007.
(*4) Zgodnie z definicją w art. 3 pkt 8 rozporządzenia (WE) nr 595/2009.”;
(*5) Wymogi określone w niniejszym dodatku opierają się na wymogach określonych w »Systemie akredytacji, zatwierdzania i autoryzacji w zakresie dostępu do związanych z zabezpieczeniami informacji dotyczących naprawy i konserwacji (RMI)«, zatwierdzonym w dniu 19 maja 2016 r. przez organizację Europejska Współpraca w Dziedzinie Akredytacji (https://www.vehiclesermi.eu/).
(*6) Rozporządzenie Parlamentu Europejskiego i Rady (UE) nr 910/2014 z dnia 23 lipca 2014 r. w sprawie identyfikacji elektronicznej i usług zaufania w odniesieniu do transakcji elektronicznych na rynku wewnętrznym oraz uchylające dyrektywę 1999/93/WE (Dz.U. L 257 z 28.8.2014, s. 73).«
|
30.7.2021 |
PL |
Dziennik Urzędowy Unii Europejskiej |
L 272/29 |
ROZPORZĄDZENIE KOMISJI (UE) 2021/1245
z dnia 23 lipca 2021 r.
zatwierdzające zmianę w specyfikacji chronionej nazwy pochodzenia lub chronionego oznaczenia geograficznegoe [„Coteaux du Pont du Gard” (ChOG)]
KOMISJA EUROPEJSKA,
uwzględniając Traktat o funkcjonowaniu Unii Europejskiej,
uwzględniając rozporządzenie Parlamentu Europejskiego i Rady (UE) nr 1308/2013 z dnia 17 grudnia 2013 r. ustanawiające wspólną organizację rynków produktów rolnych oraz uchylające rozporządzenia Rady (EWG) nr 922/72, (EWG) nr 234/79, (WE) nr 1037/2001 i (WE) nr 1234/2007 (1), w szczególności jego art. 99,
a także mając na uwadze, co następuje,
|
(1) |
Komisja rozpatrzyła wniosek o zatwierdzenie zmiany w specyfikacji chronionego oznaczenia geograficznego „Coteaux du Pont du Gard” przesłany przez Francję zgodnie z art. 105 rozporządzenia (UE) nr 1308/2013. |
|
(2) |
Komisja opublikowała wniosek o zatwierdzenie zmiany w specyfikacji, w zastosowaniu art. 97 ust. 3 rozporządzenia (UE) nr 1308/2013, w Dzienniku Urzędowym Unii Europejskiej (2). |
|
(3) |
Komisja nie otrzymała żadnego oświadczenia o sprzeciwie na podstawie art. 98 rozporządzenia (UE) nr 1308/2013. |
|
(4) |
Należy zatem zatwierdzić zmianę w specyfikacji produktu zgodnie z art. 99 rozporządzenia (UE) nr 1308/2013. |
|
(5) |
Środki przewidziane w niniejszym rozporządzeniu są zgodne z opinią Komitetu ds. Wspólnej Organizacji Rynków Rolnych, |
PRZYJMUJE NINIEJSZE ROZPORZĄDZENIE:
Artykuł 1
Zatwierdza się zmianę w specyfikacji opublikowaną w Dzienniku Urzędowym Unii Europejskiej dotyczącą nazwy „Coteaux du Pont du Gard” (ChOG).
Artykuł 2
Niniejsze rozporządzenie wchodzi w życie dwudziestego dnia po jego opublikowaniu w Dzienniku Urzędowym Unii Europejskiej.
Niniejsze rozporządzenie wiąże w całości i jest bezpośrednio stosowane we wszystkich państwach członkowskich.
Sporządzono w Brukseli dnia 23 lipca 2021 r.
W imieniu Komisji,
za Przewodniczącą,
Janusz WOJCIECHOWSKI
Członek Komisji
|
30.7.2021 |
PL |
Dziennik Urzędowy Unii Europejskiej |
L 272/30 |
ROZPORZĄDZENIE WYKONAWCZE KOMISJI (UE) 2021/1246
z dnia 28 lipca 2021 r.
zmieniające rozporządzenie (WE) nr 1484/95 w odniesieniu do ustalania cen reprezentatywnych w sektorach mięsa drobiowego i jaj oraz w odniesieniu do albumin jaj
KOMISJA EUROPEJSKA,
uwzględniając Traktat o funkcjonowaniu Unii Europejskiej,
uwzględniając rozporządzenie Parlamentu Europejskiego i Rady (UE) nr 1308/2013 z dnia 17 grudnia 2013 r. ustanawiające wspólną organizację rynków produktów rolnych oraz uchylające rozporządzenia Rady (EWG) nr 922/72, (EWG) nr 234/79, (WE) nr 1037/2001 i (WE) nr 1234/2007 (1), w szczególności jego art. 183 lit. b),
uwzględniając rozporządzenie Parlamentu Europejskiego i Rady (UE) nr 510/2014 z dnia 16 kwietnia 2014 r. ustanawiające zasady handlu niektórymi towarami pochodzącymi z przetwórstwa produktów rolnych oraz uchylające rozporządzenia Rady (WE) nr 1216/2009 i (WE) nr 614/2009 (2), w szczególności jego art. 5 ust. 6 lit. a),
a także mając na uwadze, co następuje:
|
(1) |
Rozporządzeniem Komisji (WE) nr 1484/95 (3) ustanowiono szczegółowe zasady stosowania systemu dodatkowych należności przywozowych oraz ustalono ceny reprezentatywne w sektorach mięsa drobiowego i jaj oraz w odniesieniu do albumin jaj. |
|
(2) |
Z regularnych kontroli danych, na podstawie których są określane ceny reprezentatywne produktów w sektorach mięsa drobiowego i jaj oraz w odniesieniu do albumin jaj, wynika, że należy zmienić ceny reprezentatywne w przywozie niektórych produktów, uwzględniając wahania cen w zależności od pochodzenia tych produktów. |
|
(3) |
Należy zatem odpowiednio zmienić rozporządzenie (WE) nr 1484/95. |
|
(4) |
Ze względu na konieczność zagwarantowania, że środek ten będzie mieć zastosowanie możliwie jak najszybciej po udostępnieniu aktualnych danych, niniejsze rozporządzenie powinno wejść w życie z dniem jego opublikowania, |
PRZYJMUJE NINIEJSZE ROZPORZĄDZENIE:
Artykuł 1
Załącznik I do rozporządzenia (WE) nr 1484/95 zastępuje się tekstem znajdującym się w załączniku do niniejszego rozporządzenia.
Artykuł 2
Niniejsze rozporządzenie wchodzi w życie z dniem jego opublikowania w Dzienniku Urzędowym Unii Europejskiej.
Niniejsze rozporządzenie wiąże w całości i jest bezpośrednio stosowane we wszystkich państwach członkowskich.
Sporządzono w Brukseli dnia 28 lipca 2021 r.
W imieniu Komisji,
za Przewodniczącą,
Wolfgang BURTSCHER
Dyrektor Generalny
Dyrekcja Generalna ds. Rolnictwa i Rozwoju Obszarów Wiejskich
(1) Dz.U. L 347 z 20.12.2013, s. 671.
(2) Dz.U. L 150 z 20.5.2014, s. 1.
(3) Rozporządzenie Komisji (WE) nr 1484/95 z dnia 28 czerwca 1995 r. określające szczegółowe zasady wdrażania systemu dodatkowych należności przywozowych oraz ustalające ceny reprezentatywne w sektorach mięsa drobiowego i jaj oraz w odniesieniu do albumin jaj i uchylające rozporządzenie nr 163/67/EWG (Dz.U. L 145 z 29.6.1995, s. 47).
ZAŁĄCZNIK
„ZAŁĄCZNIK I
|
Kod CN |
Opis towarów |
Cena reprezentatywna (EUR/100 kg) |
Zabezpieczenie, o którym mowa w art. 3 (EUR/100 kg) |
Pochodzenie (1) |
|
0207 14 10 |
Kawałki z ptactwa z gatunku Gallus domesticus bez kości, zamrożone |
140,3 176,2 |
60 42 |
BR TH |
(1) Nomenklatura krajów ustalona w rozporządzeniu Komisji (UE) nr 1106/2012 z dnia 27 listopada 2012 r. w sprawie wykonania rozporządzenia Parlamentu Europejskiego i Rady (WE) nr 471/2009 w sprawie statystyk Wspólnoty dotyczących handlu zagranicznego z państwami trzecimi, w odniesieniu do aktualizacji nazewnictwa państw i terytoriów (Dz.U. L 328 z 28.11.2012, s. 7).”
|
30.7.2021 |
PL |
Dziennik Urzędowy Unii Europejskiej |
L 272/33 |
ROZPORZĄDZENIE KOMISJI (UE) 2021/1247
z dnia 29 lipca 2021 r.
zmieniające załącznik II do rozporządzenia Parlamentu Europejskiego i Rady (WE) nr 396/2005 w odniesieniu do najwyższych dopuszczalnych poziomów pozostałości mandestrobiny w winogronach i truskawkach
(Tekst mający znaczenie dla EOG)
KOMISJA EUROPEJSKA,
uwzględniając Traktat o funkcjonowaniu Unii Europejskiej,
uwzględniając rozporządzenie (WE) nr 396/2005 Parlamentu Europejskiego i Rady z dnia 23 lutego 2005 r. w sprawie najwyższych dopuszczalnych poziomów pozostałości pestycydów w żywności i paszy pochodzenia roślinnego i zwierzęcego oraz na ich powierzchni, zmieniające dyrektywę Rady 91/414/EWG (1), w szczególności jego art. 14 ust. 1 lit. a),
a także mając na uwadze, co następuje:
|
(1) |
Najwyższe dopuszczalne poziomy pozostałości (NDP) mandestrobiny określono w załączniku II do rozporządzenia (WE) nr 396/2005. |
|
(2) |
Zgodnie z art. 6 ust. 2 i 4 rozporządzenia (WE) nr 396/2005 złożono wniosek o tolerancję importową w odniesieniu do mandestrobiny stosowanej w Kanadzie na powierzchni truskawek i winogron. Wnioskodawca twierdzi, że dozwolone zastosowania tej substancji na wspomnianych uprawach w Kanadzie skutkują pozostałościami przekraczającymi NDP dozwolone na podstawie rozporządzenia (WE) nr 396/2005 oraz że konieczne jest ustanowienie wyższych NDP, aby uniknąć barier handlowych w odniesieniu do przywozu winogron i truskawek. |
|
(3) |
Zgodnie z art. 8 rozporządzenia (WE) nr 396/2005 wniosek ten został poddany ocenie przez państwo członkowskie, którego dotyczył, a sprawozdanie z oceny przekazano Komisji. |
|
(4) |
Europejski Urząd ds. Bezpieczeństwa Żywności („Urząd”) ocenił wniosek i sprawozdanie z oceny, analizując w szczególności ryzyko dla konsumentów oraz, w stosownych przypadkach, dla zwierząt, i wydał uzasadnioną opinię dotyczącą proponowanych NDP (2). Przekazał tę opinię wnioskodawcy, Komisji i państwom członkowskim oraz podał ją do publicznej wiadomości. |
|
(5) |
Urząd stwierdził, że spełniono wszystkie wymogi dotyczące danych oraz że zmiany NDP, o które wystąpił wnioskodawca, są dopuszczalne z punktu widzenia bezpieczeństwa konsumentów na podstawie oceny narażenia konsumentów dokonanej dla 27 określonych grup konsumentów europejskich. Urząd wziął pod uwagę najnowsze informacje na temat właściwości toksykologicznych przedmiotowej substancji. Długotrwałe narażenie na działanie substancji w wyniku spożywania wszystkich produktów spożywczych, które mogą ją zawierać, wykazało, że nie istnieje ryzyko przekroczenia dopuszczalnego dziennego spożycia. Ponadto Urząd stwierdził, że ustanowienie ostrej dawki referencyjnej nie jest konieczne ze względu na niski profil ostrej toksyczności substancji. |
|
(6) |
Na podstawie uzasadnionej opinii Urzędu oraz po uwzględnieniu czynników istotnych dla rozpatrywanej kwestii stwierdzono, że proponowane zmiany NDP spełniają wymogi art. 14 ust. 2 rozporządzenia (WE) nr 396/2005. |
|
(7) |
Należy zatem odpowiednio zmienić rozporządzenie (WE) nr 396/2005. |
|
(8) |
Środki przewidziane w niniejszym rozporządzeniu są zgodne z opinią Stałego Komitetu ds. Roślin, Zwierząt, Żywności i Pasz, |
PRZYJMUJE NINIEJSZE ROZPORZĄDZENIE:
Artykuł 1
W załączniku II do rozporządzenia (WE) nr 396/2005 wprowadza się zmiany zgodnie z załącznikiem do niniejszego rozporządzenia.
Artykuł 2
Niniejsze rozporządzenie wchodzi w życie dwudziestego dnia po jego opublikowaniu w Dzienniku Urzędowym Unii Europejskiej.
Niniejsze rozporządzenie wiąże w całości i jest bezpośrednio stosowane we wszystkich państwach członkowskich.
Sporządzono w Brukseli dnia 29 lipca 2021 r.
W imieniu Komisji
Ursula VON DER LEYEN
Przewodnicząca
(1) Dz.U. L 70 z 16.3.2005, s. 1.
(2) Sprawozdania naukowe EFSA są dostępne na stronie internetowej: http://www.efsa.europa.eu: „Reasoned opinion on the setting of import tolerances for mandestrobin in strawberries and table and wine grapes” (Uzasadniona opinia dotycząca określenia tolerancji importowych w odniesieniu do mandestrobiny w truskawkach oraz w winogronach stołowych i winogronach do produkcji wina). Dziennik EFSA 2018; 16(8): 5395.
ZAŁĄCZNIK
W załączniku II do rozporządzenia (WE) nr 396/2005 kolumna dotycząca mandestrobiny otrzymuje brzmienie:
„Pozostałości pestycydów i najwyższe dopuszczalne poziomy pozostałości (mg/kg)
|
Numer kodu |
Grupy i przykłady poszczególnych produktów, do których odnoszą się najwyższe dopuszczalne poziomy pozostałości (NDP) (1) |
Mandestrobina |
||
|
(1) |
(2) |
(3) |
||
|
0100000 |
OWOCE, ŚWIEŻE lub MROŻONE; ORZECHY Z DRZEW ORZECHOWYCH |
|
||
|
0110000 |
Owoce cytrusowe |
0,01 (*1) |
||
|
0110010 |
Grejpfruty |
|
||
|
0110020 |
Pomarańcze |
|
||
|
0110030 |
Cytryny |
|
||
|
0110040 |
Limy/limonki |
|
||
|
0110050 |
Mandarynki |
|
||
|
0110990 |
Pozostałe (2) |
|
||
|
0120000 |
Orzechy z drzew orzechowych |
0,01 (*1) |
||
|
0120010 |
Migdały |
|
||
|
0120020 |
Orzechy brazylijskie |
|
||
|
0120030 |
Orzechy nerkowca |
|
||
|
0120040 |
Kasztany jadalne |
|
||
|
0120050 |
Orzechy kokosowe |
|
||
|
0120060 |
Orzechy laskowe/Orzechy leszczyny pospolitej |
|
||
|
0120070 |
Orzechy makadamiowe/Orzechy makadamii trójlistnej |
|
||
|
0120080 |
Orzechy pekan |
|
||
|
0120090 |
Orzeszki piniowe |
|
||
|
0120100 |
Orzeszki pistacjowe |
|
||
|
0120110 |
Orzechy włoskie |
|
||
|
0120990 |
Pozostałe (2) |
|
||
|
0130000 |
Owoce ziarnkowe |
0,01 (*1) |
||
|
0130010 |
Jabłka |
|
||
|
0130020 |
Gruszki |
|
||
|
0130030 |
Pigwy |
|
||
|
0130040 |
Owoce nieszpułki zwyczajnej |
|
||
|
0130050 |
Owoc nieśplika japońskiego/Owoc miszpelnika japońskiego |
|
||
|
0130990 |
Pozostałe (2) |
|
||
|
0140000 |
Owoce pestkowe |
|
||
|
0140010 |
Morele |
2 |
||
|
0140020 |
Czereśnie |
3 |
||
|
0140030 |
Brzoskwinie |
2 |
||
|
0140040 |
Śliwki |
0,5 |
||
|
0140990 |
Pozostałe (2) |
0,01 (*1) |
||
|
0150000 |
Owoce jagodowe i drobne owoce |
|
||
|
0151000 |
|
5 |
||
|
0151010 |
Winogrona stołowe |
|
||
|
0151020 |
Winogrona do produkcji wina |
|
||
|
0152000 |
|
3 |
||
|
0153000 |
|
0,01 (*1) |
||
|
0153010 |
Jeżyny |
|
||
|
0153020 |
Jeżyny popielice |
|
||
|
0153030 |
Maliny (czerwone i żółte) |
|
||
|
0153990 |
Pozostałe (2) |
|
||
|
0154000 |
|
0,01 (*1) |
||
|
0154010 |
Borówki amerykańskie |
|
||
|
0154020 |
Żurawiny |
|
||
|
0154030 |
Porzeczki (czarne, czerwone i białe) |
|
||
|
0154040 |
Agrest (zielony, czerwony i żółty) |
|
||
|
0154050 |
Róża dzika |
|
||
|
0154060 |
Morwy (czarne i białe) |
|
||
|
0154070 |
Głóg włoski |
|
||
|
0154080 |
Bez czarny |
|
||
|
0154990 |
Pozostałe (2) |
|
||
|
0160000 |
Owoce różne z |
0,01 (*1) |
||
|
0161000 |
|
|
||
|
0161010 |
Daktyle |
|
||
|
0161020 |
Figi |
|
||
|
0161030 |
Oliwki stołowe |
|
||
|
0161040 |
Kumkwat |
|
||
|
0161050 |
Karambola |
|
||
|
0161060 |
Szaron/Persymona/Kaki/Huma wschodnia |
|
||
|
0161070 |
Czapetka kuminowa |
|
||
|
0161990 |
Pozostałe (2) |
|
||
|
0162000 |
|
|
||
|
0162010 |
Kiwi (zielone, czerwone, żółte) |
|
||
|
0162020 |
Liczi chińskie/Śliwka chińska |
|
||
|
0162030 |
Marakuja/Męczennica jadalna |
|
||
|
0162040 |
Owoc opuncji figowej/Figa indyjska |
|
||
|
0162050 |
Caimito |
|
||
|
0162060 |
Owoc hebanowca wirginijskiego/Owoc hurmy wirginijskiej |
|
||
|
0162990 |
Pozostałe (2) |
|
||
|
0163000 |
|
|
||
|
0163010 |
Awokado |
|
||
|
0163020 |
Banany |
|
||
|
0163030 |
Mango |
|
||
|
0163040 |
Papaja |
|
||
|
0163050 |
Granaty/Jabłka granatu |
|
||
|
0163060 |
Czerymoja |
|
||
|
0163070 |
Gujawa/Gruszla |
|
||
|
0163080 |
Ananasy |
|
||
|
0163090 |
Owoce chlebowca |
|
||
|
0163100 |
Owoce duriana właściwego |
|
||
|
0163110 |
Owoce flaszowca miękkociernistego/Owoce guanabany |
|
||
|
0163990 |
Pozostałe (2) |
|
||
|
0200000 |
WARZYWA, ŚWIEŻE lub MROŻONE |
0,01 (*1) |
||
|
0210000 |
Warzywa korzeniowe i bulwiaste |
|
||
|
0211000 |
|
|
||
|
0212000 |
|
|
||
|
0212010 |
Maniok jadalny/Podpłomycz najużyteczniejszy |
|
||
|
0212020 |
Kartofel słodki/Batat |
|
||
|
0212030 |
Pochrzyn |
|
||
|
0212040 |
Maranta trzcinowata |
|
||
|
0212990 |
Pozostałe (2) |
|
||
|
0213000 |
|
|
||
|
0213010 |
Buraki |
|
||
|
0213020 |
Marchew |
|
||
|
0213030 |
Seler zwyczajny |
|
||
|
0213040 |
Chrzan pospolity |
|
||
|
0213050 |
Słonecznik bulwiasty |
|
||
|
0213060 |
Pasternak |
|
||
|
0213070 |
Korzeń pietruszki zwyczajnej |
|
||
|
0213080 |
Rzodkiew zwyczajna |
|
||
|
0213090 |
Kozibród porolistny/Salsefia |
|
||
|
0213100 |
Brukiew/Karpiel |
|
||
|
0213110 |
Rzepa biała/Rzepa jadalna |
|
||
|
0213990 |
Pozostałe (2) |
|
||
|
0220000 |
Warzywa cebulowe |
|
||
|
0220010 |
Czosnek |
|
||
|
0220020 |
Cebula |
|
||
|
0220030 |
Szalotka |
|
||
|
0220040 |
Dymka/Cebula szczypiorowa i cebula siedmiolatka |
|
||
|
0220990 |
Pozostałe (2) |
|
||
|
0230000 |
Warzywa owocowe |
|
||
|
0231000 |
|
|
||
|
0231010 |
Pomidory |
|
||
|
0231020 |
Papryka roczna |
|
||
|
0231030 |
Oberżyny/Bakłażany |
|
||
|
0231040 |
Piżmian jadalny/Ketmia jadalna/Okra |
|
||
|
0231990 |
Pozostałe (2) |
|
||
|
0232000 |
|
|
||
|
0232010 |
Ogórki |
|
||
|
0232020 |
Korniszony |
|
||
|
0232030 |
Cukinie |
|
||
|
0232990 |
Pozostałe (2) |
|
||
|
0233000 |
|
|
||
|
0233010 |
Melony |
|
||
|
0233020 |
Dynie olbrzymie |
|
||
|
0233030 |
Arbuzy |
|
||
|
0233990 |
Pozostałe (2) |
|
||
|
0234000 |
|
|
||
|
0239000 |
|
|
||
|
0240000 |
Warzywa kapustne (oprócz korzeni warzyw kapustnych oraz oprócz kapustnych o młodych/drobnych liściach) |
|
||
|
0241000 |
|
|
||
|
0241010 |
Brokuły |
|
||
|
0241020 |
Kalafiory |
|
||
|
0241990 |
Pozostałe (2) |
|
||
|
0242000 |
|
|
||
|
0242010 |
Brukselka/Kapusta brukselska |
|
||
|
0242020 |
Kapusta głowiasta |
|
||
|
0242990 |
Pozostałe (2) |
|
||
|
0243000 |
|
|
||
|
0243010 |
Kapusta pekińska/Kapusta petsai |
|
||
|
0243020 |
Jarmuż |
|
||
|
0243990 |
Pozostałe (2) |
|
||
|
0244000 |
|
|
||
|
0250000 |
Warzywa liściowe, zioła, kwiaty jadalne |
|
||
|
0251000 |
|
|
||
|
0251010 |
Roszpunka warzywna |
|
||
|
0251020 |
Sałaty |
|
||
|
0251030 |
Endywia o liściach szerokich |
|
||
|
0251040 |
Rzeżucha i inne kiełki i pędy |
|
||
|
0251050 |
Gorczycznik wiosenny |
|
||
|
0251060 |
Rokietta siewna/rukola |
|
||
|
0251070 |
Gorczyca sarepska |
|
||
|
0251080 |
Warzywa o młodych/drobnych liściach (w tym gatunki warzyw kapustnych) |
|
||
|
0251990 |
Pozostałe (2) |
|
||
|
0252000 |
|
|
||
|
0252010 |
Szpinak |
|
||
|
0252020 |
Portulaka pospolita |
|
||
|
0252030 |
Boćwina |
|
||
|
0252990 |
Pozostałe (2) |
|
||
|
0253000 |
|
|
||
|
0254000 |
|
|
||
|
0255000 |
|
|
||
|
0256000 |
|
|
||
|
0256010 |
Trybula |
|
||
|
0256020 |
Szczypiorek |
|
||
|
0256030 |
Liście selera |
|
||
|
0256040 |
Pietruszka – nać |
|
||
|
0256050 |
Szałwia lekarska |
|
||
|
0256060 |
Rozmaryn lekarski |
|
||
|
0256070 |
Tymianek pospolity/Macierzanka tymianek |
|
||
|
0256080 |
Bazylia pospolita i kwiaty jadalne |
|
||
|
0256090 |
Liście laurowe/Wawrzyn szlachetny/Laur szlachetny |
|
||
|
0256100 |
Estragon |
|
||
|
0256990 |
Pozostałe (2) |
|
||
|
0260000 |
Warzywa strączkowe |
|
||
|
0260010 |
Fasola (w strąkach) |
|
||
|
0260020 |
Fasola (bez strąków) |
|
||
|
0260030 |
Groch (w strąkach) |
|
||
|
0260040 |
Groch (bez strąków) |
|
||
|
0260050 |
Soczewica |
|
||
|
0260990 |
Pozostałe (2) |
|
||
|
0270000 |
Warzywa łodygowe |
|
||
|
0270010 |
Szparagi |
|
||
|
0270020 |
Karczochy hiszpańskie |
|
||
|
0270030 |
Seler |
|
||
|
0270040 |
Fenkuł włoski/Koper włoski |
|
||
|
0270050 |
Karczochy zwyczajne |
|
||
|
0270060 |
Pory |
|
||
|
0270070 |
Rabarbar |
|
||
|
0270080 |
Pędy bambusa |
|
||
|
0270090 |
Rdzenie palmowe |
|
||
|
0270990 |
Pozostałe (2) |
|
||
|
0280000 |
Grzyby, mchy i porosty |
|
||
|
0280010 |
Grzyby uprawne |
|
||
|
0280020 |
Grzyby dzikie |
|
||
|
0280990 |
Mchy i porosty |
|
||
|
0290000 |
Algi i organizmy prokariotyczne |
|
||
|
0300000 |
JADALNE NASIONA ROŚLIN STRĄCZKOWYCH |
0,01 (*1) |
||
|
0300010 |
Fasola |
|
||
|
0300020 |
Soczewica |
|
||
|
0300030 |
Groch |
|
||
|
0300040 |
Łubin biały |
|
||
|
0300990 |
Pozostałe (2) |
|
||
|
0400000 |
NASIONA I OWOCE OLEISTE |
0,01 (*1) |
||
|
0401000 |
Nasiona oleiste |
|
||
|
0401010 |
Siemię lniane |
|
||
|
0401020 |
Orzeszki ziemne/Orzechy arachidowe |
|
||
|
0401030 |
Nasiona maku |
|
||
|
0401040 |
Ziarna sezamu |
|
||
|
0401050 |
Ziarna słonecznika |
|
||
|
0401060 |
Ziarna rzepaku |
|
||
|
0401070 |
Ziarna soi zwyczajnej |
|
||
|
0401080 |
Nasiona gorczycy |
|
||
|
0401090 |
Nasiona bawełny |
|
||
|
0401100 |
Nasiona dyni |
|
||
|
0401110 |
Nasiona krokoszu barwierskiego |
|
||
|
0401120 |
Nasiona ogórecznika lekarskiego |
|
||
|
0401130 |
Nasiona lnicznika siewnego |
|
||
|
0401140 |
Nasiona konopi siewnych |
|
||
|
0401150 |
Rącznik pospolity |
|
||
|
0401990 |
Pozostałe (2) |
|
||
|
0402000 |
Owoce oleiste |
|
||
|
0402010 |
Oliwki do produkcji oliwy |
|
||
|
0402020 |
Nasiona palm olejowych |
|
||
|
0402030 |
Owoce palm olejowych |
|
||
|
0402040 |
Puchowiec pięciopręcikowy/Drzewo kapokowe |
|
||
|
0402990 |
Pozostałe (2) |
|
||
|
0500000 |
ZBOŻA |
0,01 (*1) |
||
|
0500010 |
Jęczmień |
|
||
|
0500020 |
Gryka zwyczajna i pozostałe zboża rzekome |
|
||
|
0500030 |
Kukurydza |
|
||
|
0500040 |
Proso zwyczajne |
|
||
|
0500050 |
Owies zwyczajny |
|
||
|
0500060 |
Ryż siewny |
|
||
|
0500070 |
Żyto zwyczajne |
|
||
|
0500080 |
Sorgo japońskie |
|
||
|
0500090 |
Pszenica zwyczajna |
|
||
|
0500990 |
Pozostałe (2) |
|
||
|
0600000 |
HERBATY, KAWA, NAPARY ZIOŁOWE, KAKAO I SZARAŃCZYN STRĄKOWY |
0,05 (*1) |
||
|
0610000 |
Herbaty |
|
||
|
0620000 |
Ziarna kawy |
|
||
|
0630000 |
Napary ziołowe z |
|
||
|
0631000 |
|
|
||
|
0631010 |
Rumian szlachetny/Rumianek rzymski |
|
||
|
0631020 |
Ketmia szczawiowa |
|
||
|
0631030 |
Róża |
|
||
|
0631040 |
Jaśmin |
|
||
|
0631050 |
Lipa |
|
||
|
0631990 |
Pozostałe (2) |
|
||
|
0632000 |
|
|
||
|
0632010 |
Truskawka |
|
||
|
0632020 |
Aspalat prosty |
|
||
|
0632030 |
Ostrokrzew paragwajski |
|
||
|
0632990 |
Pozostałe (2) |
|
||
|
0633000 |
|
|
||
|
0633010 |
Kozłek lekarski |
|
||
|
0633020 |
Żeń-szeń |
|
||
|
0633990 |
Pozostałe (2) |
|
||
|
0639000 |
|
|
||
|
0640000 |
Ziarna kakaowe |
|
||
|
0650000 |
Szarańczyn strąkowy/Chleb świętojański |
|
||
|
0700000 |
CHMIEL |
0,05 (*1) |
||
|
0800000 |
PRZYPRAWY |
|
||
|
0810000 |
Przyprawy nasienne |
0,05 (*1) |
||
|
0810010 |
Anyż/Anyżek/Biedrzeniec anyż |
|
||
|
0810020 |
Kminek czarny/Czarny kmin |
|
||
|
0810030 |
Seler |
|
||
|
0810040 |
Kolendra siewna |
|
||
|
0810050 |
Kmin rzymski |
|
||
|
0810060 |
Koper ogrodowy |
|
||
|
0810070 |
Koper włoski/Fenkuł włoski |
|
||
|
0810080 |
Kozieradka pospolita |
|
||
|
0810090 |
Gałka muszkatołowa |
|
||
|
0810990 |
Pozostałe (2) |
|
||
|
0820000 |
Przyprawy owocowe |
0,05 (*1) |
||
|
0820010 |
Ziele angielskie/pieprz angielski |
|
||
|
0820020 |
Pieprz syczuański |
|
||
|
0820030 |
Kminek zwyczajny |
|
||
|
0820040 |
Kardamon malabarski |
|
||
|
0820050 |
Jagody jałowca |
|
||
|
0820060 |
Ziarna pieprzu (czarnego, zielonego i białego) |
|
||
|
0820070 |
Wanilia |
|
||
|
0820080 |
Tamarynd |
|
||
|
0820990 |
Pozostałe (2) |
|
||
|
0830000 |
Przyprawy korowe |
0,05 (*1) |
||
|
0830010 |
Cynamon |
|
||
|
0830990 |
Pozostałe (2) |
|
||
|
0840000 |
Przyprawy korzeniowe lub kłączowe |
|
||
|
0840010 |
Lukrecja |
0,05 (*1) |
||
|
0840020 |
Imbir lekarski (10) |
|
||
|
0840030 |
Kurkuma |
0,05 (*1) |
||
|
0840040 |
Chrzan pospolity (11) |
|
||
|
0840990 |
Pozostałe (2) |
0,05 (*1) |
||
|
0850000 |
Przyprawy pączkowe |
0,05 (*1) |
||
|
0850010 |
Goździki |
|
||
|
0850020 |
Kapary |
|
||
|
0850990 |
Pozostałe (2) |
|
||
|
0860000 |
Przyprawy ze słupków kwiatowych |
0,05 (*1) |
||
|
0860010 |
Szafran |
|
||
|
0860990 |
Pozostałe (2) |
|
||
|
0870000 |
Przyprawy z osnówki nasiona |
0,05 (*1) |
||
|
0870010 |
Kwiat muszkatołowy |
|
||
|
0870990 |
Pozostałe (2) |
|
||
|
0900000 |
ROŚLINY CUKRODAJNE |
0,01 (*1) |
||
|
0900010 |
Korzenie buraka cukrowego |
|
||
|
0900020 |
Trzcina cukrowa |
|
||
|
0900030 |
Cykoria podróżnik, korzenie |
|
||
|
0900990 |
Pozostałe (2) |
|
||
|
1000000 |
PRODUKTY POCHODZENIA ZWIERZĘCEGO - ZWIERZĘTA LĄDOWE |
|
||
|
1010000 |
Towary z |
0,01 (*1) |
||
|
1011000 |
|
|
||
|
1011010 |
Mięśnie |
|
||
|
1011020 |
Tłuszcz |
|
||
|
1011030 |
Wątroba |
|
||
|
1011040 |
Nerka |
|
||
|
1011050 |
Podroby jadalne (inne niż wątroba i nerki) |
|
||
|
1011990 |
Pozostałe (2) |
|
||
|
1012000 |
|
|
||
|
1012010 |
Mięśnie |
|
||
|
1012020 |
Tłuszcz |
|
||
|
1012030 |
Wątroba |
|
||
|
1012040 |
Nerka |
|
||
|
1012050 |
Podroby jadalne (inne niż wątroba i nerki) |
|
||
|
1012990 |
Pozostałe (2) |
|
||
|
1013000 |
|
|
||
|
1013010 |
Mięśnie |
|
||
|
1013020 |
Tłuszcz |
|
||
|
1013030 |
Wątroba |
|
||
|
1013040 |
Nerka |
|
||
|
1013050 |
Podroby jadalne (inne niż wątroba i nerki) |
|
||
|
1013990 |
Pozostałe (2) |
|
||
|
1014000 |
|
|
||
|
1014010 |
Mięśnie |
|
||
|
1014020 |
Tłuszcz |
|
||
|
1014030 |
Wątroba |
|
||
|
1014040 |
Nerka |
|
||
|
1014050 |
Podroby jadalne (inne niż wątroba i nerki) |
|
||
|
1014990 |
Pozostałe (2) |
|
||
|
1015000 |
|
|
||
|
1015010 |
Mięśnie |
|
||
|
1015020 |
Tłuszcz |
|
||
|
1015030 |
Wątroba |
|
||
|
1015040 |
Nerka |
|
||
|
1015050 |
Podroby jadalne (inne niż wątroba i nerki) |
|
||
|
1015990 |
Pozostałe (2) |
|
||
|
1016000 |
|
|
||
|
1016010 |
Mięśnie |
|
||
|
1016020 |
Tłuszcz |
|
||
|
1016030 |
Wątroba |
|
||
|
1016040 |
Nerka |
|
||
|
1016050 |
Podroby jadalne (inne niż wątroba i nerki) |
|
||
|
1016990 |
Pozostałe (2) |
|
||
|
1017000 |
|
|
||
|
1017010 |
Mięśnie |
|
||
|
1017020 |
Tłuszcz |
|
||
|
1017030 |
Wątroba |
|
||
|
1017040 |
Nerka |
|
||
|
1017050 |
Podroby jadalne (inne niż wątroba i nerki) |
|
||
|
1017990 |
Pozostałe (2) |
|
||
|
1020000 |
Mleko |
0,01 (*1) |
||
|
1020010 |
Bydło |
|
||
|
1020020 |
Owce |
|
||
|
1020030 |
Kozy |
|
||
|
1020040 |
Konie |
|
||
|
1020990 |
Pozostałe (2) |
|
||
|
1030000 |
Jaja ptasie |
0,01 (*1) |
||
|
1030010 |
Kury |
|
||
|
1030020 |
Kaczki |
|
||
|
1030030 |
Gęsi |
|
||
|
1030040 |
Przepiórki |
|
||
|
1030990 |
Pozostałe (2) |
|
||
|
1040000 |
Miód i pozostałe produkty pszczele (7) |
0,05 (*1) |
||
|
1050000 |
Płazy i gady |
0,01 (*1) |
||
|
1060000 |
Bezkręgowe zwierzęta lądowe |
0,01 (*1) |
||
|
1070000 |
Dzikie kręgowe zwierzęta lądowe |
0,01 (*1) |
||
|
1100000 |
PRODUKTY POCHODZENIA ZWIERZĘCEGO – RYBY, PRODUKTY RYBNE I POZOSTAŁE MORSKIE I SŁODKOWODNE PRODUKTY SPOŻYWCZE (8) |
|
||
|
1200000 |
PRODUKTY LUB CZĘŚCI PRODUKTÓW UŻYWANE WYŁĄCZNIE DO PRODUKCJI PASZ (8) |
|
||
|
1300000 |
PRZETWORZONE PRODUKTY SPOŻYWCZE (9) |
|
(*1) Wskazuje granicę oznaczalności.
(1) Pełny wykaz produktów pochodzenia roślinnego i zwierzęcego, do których stosują się NDP, można znaleźć w załączniku I.”
|
30.7.2021 |
PL |
Dziennik Urzędowy Unii Europejskiej |
L 272/46 |
ROZPORZĄDZENIE WYKONAWCZE KOMISJI (UE) 2021/1248
z dnia 29 lipca 2021 r.
w sprawie środków w zakresie dobrej praktyki dystrybucyjnej dotyczącej weterynaryjnych produktów leczniczych zgodnie z rozporządzeniem Parlamentu Europejskiego i Rady (UE) 2019/6
(Tekst mający znaczenie dla EOG)
KOMISJA EUROPEJSKA,
uwzględniając Traktat o funkcjonowaniu Unii Europejskiej,
uwzględniając rozporządzenie Parlamentu Europejskiego i Rady (UE) 2019/6 z dnia 11 grudnia 2018 r. w sprawie weterynaryjnych produktów leczniczych i uchylające dyrektywę 2001/82/WE (1), w szczególności jego art. 99 ust. 6,
a także mając na uwadze, co następuje:
|
(1) |
W art. 101 ust. 5 rozporządzenia (UE) 2019/6 zobowiązano hurtowników do stosowania dobrej praktyki dystrybucyjnej dotyczącej weterynaryjnych produktów leczniczych przyjętej przez Komisję. |
|
(2) |
Środki w zakresie dobrej praktyki dystrybucyjnej powinny zapewniać tożsamość, integralność, identyfikowalność i jakość weterynaryjnych produktów leczniczych w całym łańcuchu dostaw. Ponadto środki te powinny gwarantować odpowiednie przechowywanie i transport weterynaryjnych produktów leczniczych oraz postępowanie z nimi, a także zapewniać, by pozostawały one w legalnym łańcuchu dostaw podczas przechowywania i transportu. |
|
(3) |
W odniesieniu do produktów leczniczych stosowanych u ludzi istnieją normy międzynarodowe i wytyczne w sprawie dobrej praktyki dystrybucyjnej (2) (3) (4) (5). Na poziomie Unii przyjęto wytyczne w sprawie dobrej praktyki dystrybucyjnej wyłącznie w odniesieniu do produktów leczniczych stosowanych u ludzi (6). Analogiczne środki w dziedzinie weterynarii powinny uwzględniać doświadczenie zdobyte podczas stosowania obecnego systemu na mocy dyrektywy 2001/83/WE Parlamentu Europejskiego i Rady (7) w świetle podobieństw i potencjalnych różnic między wymogami dobrej praktyki dystrybucyjnej w odniesieniu do produktów leczniczych stosowanych u ludzi i w odniesieniu do weterynaryjnych produktów leczniczych. |
|
(4) |
Hurtownicy często zajmują się zarówno produktami leczniczymi stosowanymi u ludzi, jak i weterynaryjnymi produktami leczniczymi. Ponadto kontrole dobrej praktyki dystrybucyjnej w odniesieniu do obu rodzajów produktów leczniczych są często przeprowadzane przez tych samych ekspertów właściwych organów. W związku z tym, aby uniknąć niepotrzebnych obciążeń administracyjnych dla przemysłu i właściwych organów, praktyczne jest stosowanie w dziedzinie weterynarii podobnych środków jak w dziedzinie dotyczącej zdrowia ludzi, chyba że nie jest to wskazane z określonych względów. |
|
(5) |
Aby nie wpływać negatywnie na dostępność weterynaryjnych produktów leczniczych w Unii, wymogi dobrej praktyki dystrybucyjnej dotyczącej weterynaryjnych produktów leczniczych nie powinny być bardziej rygorystyczne niż odpowiadające im wymogi dotyczące produktów leczniczych stosowanych u ludzi. |
|
(6) |
Środki w zakresie dobrej praktyki dystrybucyjnej dotyczącej weterynaryjnych produktów leczniczych ustanowione w niniejszym rozporządzeniu powinny zapewniać spójność ze środkami wykonawczymi w zakresie dobrej praktyki wytwarzania weterynaryjnych produktów leczniczych i substancji czynnych stosowanych jako materiały wyjściowe, o których mowa w art. 93 ust. 2 rozporządzenia (UE) 2019/6, oraz środkami wykonawczymi w zakresie dobrej praktyki dystrybucyjnej dotyczącej substancji czynnych stosowanych jako materiały wyjściowe w weterynaryjnych produktach leczniczych, o których mowa w art. 95 ust. 8 tego rozporządzenia, oraz uzupełniać te środki wykonawcze. |
|
(7) |
Każda osoba działająca jako hurtownik weterynaryjnych produktów leczniczych musi posiadać pozwolenie na dystrybucję hurtową zgodnie z art. 99 ust. 1 rozporządzenia (UE) 2019/6 oraz przestrzegać dobrej praktyki dystrybucyjnej dotyczącej weterynaryjnych produktów leczniczych zgodnie z art. 101 ust. 5 tego rozporządzenia. Zgodnie z art. 99 ust. 5 tego rozporządzenia pozwolenie na wytwarzanie pozwala na dystrybucję hurtową weterynaryjnych produktów leczniczych objętych tym pozwoleniem na wytwarzanie. W związku z tym producenci zajmujący się dystrybucją własnych weterynaryjnych produktów leczniczych muszą również przestrzegać zasad dobrej praktyki dystrybucyjnej dotyczącej weterynaryjnych produktów leczniczych. |
|
(8) |
Definicja dystrybucji hurtowej określona w art. 4 pkt 36 rozporządzenia (UE) 2019/6 nie wyklucza hurtowników mających siedzibę lub działających w ramach szczególnych procedur celnych, takich jak wolne obszary celne lub składy celne. W związku z tym wszystkie obowiązki związane z działaniami w zakresie dystrybucji hurtowej (takimi jak wywóz, przechowywanie lub dostarczanie) mają również zastosowanie do tych hurtowników w odniesieniu do dobrej praktyki dystrybucyjnej dotyczącej weterynaryjnych produktów leczniczych. |
|
(9) |
Odpowiednie sekcje dobrej praktyki dystrybucyjnej dotyczącej weterynaryjnych produktów leczniczych powinny być również przestrzegane przez strony trzecie zaangażowane w dystrybucję hurtową weterynaryjnych produktów leczniczych i powinny być częścią ich zobowiązań umownych. Warunkiem skutecznego przeciwdziałania fałszowaniu weterynaryjnych produktów leczniczych jest stosowanie spójnego podejścia przez wszystkich partnerów w łańcuchu dostaw. |
|
(10) |
Aby zapewnić osiągnięcie celów dobrej praktyki dystrybucyjnej, niezbędny jest system jakości, który powinien jasno określać obowiązki, procesy i zasady zarządzania ryzykiem w odniesieniu do działań podejmowanych przez hurtownika. Za taki system jakości powinny odpowiadać osoby kierujące organizacją, które powinny przewodzić temu procesowi i aktywnie w nim uczestniczyć, przy wsparciu i zaangażowaniu ze strony pracowników. |
|
(11) |
Prawidłowa dystrybucja weterynaryjnych produktów leczniczych w znacznym stopniu zależy od liczby kompetentnych pracowników odpowiedniej do wykonywania wszystkich zadań, za które odpowiedzialny jest hurtownik. Pracownicy powinni mieć pełną świadomość swoich obowiązków, a obowiązki te powinny być udokumentowane. |
|
(12) |
Osoby zajmujące się dystrybucją weterynaryjnych produktów leczniczych powinny dysponować odpowiednimi pomieszczeniami, urządzeniami i sprzętem, tak aby zapewnić należyte przechowywanie i dystrybucję produktów leczniczych. |
|
(13) |
Dobra dokumentacja powinna być istotnym elementem każdego systemu jakości. Należy wprowadzić wymóg sporządzania pisemnej dokumentacji, aby zapobiec błędom wynikającym z komunikacji ustnej i umożliwić śledzenie poszczególnych czynności w trakcie hurtowej dystrybucji weterynaryjnych produktów leczniczych. Należy zdefiniować i stosować wszystkie rodzaje dokumentów. |
|
(14) |
Procedury powinny opisywać wszystkie działania związane z dystrybucją, które mają wpływ na tożsamość, identyfikowalność i jakość weterynaryjnych produktów leczniczych. |
|
(15) |
Należy prowadzić i przechowywać ewidencję wszystkich istotnych działań lub zdarzeń w celu zapewnienia identyfikowalności pochodzenia i przeznaczenia weterynaryjnych produktów leczniczych, a także identyfikacji wszystkich dostawców lub odbiorców takich weterynaryjnych produktów leczniczych. Ewidencja taka powinna ułatwiać, w razie potrzeby, wycofanie serii weterynaryjnego produktu leczniczego, jak również badanie sfałszowanych weterynaryjnych produktów leczniczych lub badanie weterynaryjnych produktów leczniczych podejrzewanych o to, że są sfałszowane. |
|
(16) |
Jeżeli chodzi o przetwarzanie danych osobowych pracowników, skarżących lub innych osób fizycznych, rozporządzenie Parlamentu Europejskiego i Rady (UE) 2016/679 (8) w sprawie ochrony osób fizycznych ma zastosowanie do przetwarzania danych osobowych i swobodnego przepływu takich danych. |
|
(17) |
System jakości powinien w pełni opisywać wszystkie kluczowe operacje w odpowiedniej dokumentacji. |
|
(18) |
Należy starannie ewidencjonować i rozpatrywać, zgodnie z ustanowionymi procedurami, skargi oraz sprawy dotyczące zwrotów, podejrzeń fałszowania weterynaryjnych produktów leczniczych oraz przypadki wycofania weterynaryjnych produktów leczniczych. Należy udostępniać ewidencję właściwym organom. Przed dopuszczeniem zwróconych weterynaryjnych produktów leczniczych do ponownej sprzedaży należy poddać je ocenie. |
|
(19) |
Każde działanie objęte dobrą praktyką dystrybucyjną dotyczącą weterynaryjnych produktów leczniczych, które jest zlecane podmiotom zewnętrznym, powinno być zdefiniowane, uzgodnione i kontrolowane, aby uniknąć nieporozumień, które mogłyby wpłynąć na integralność weterynaryjnego produktu leczniczego. Pisemna umowa między zleceniodawcą a zleceniobiorcą powinna wyraźnie określać obowiązki obu stron. |
|
(20) |
Niezależnie od środka transportu należy zapewnić możliwość wykazania, że weterynaryjne produkty lecznicze nie znajdowały się w warunkach, które mogłyby pogorszyć ich jakość i integralność. Przy planowaniu i podczas transportu weterynaryjnych produktów leczniczych należy stosować podejście oparte na analizie ryzyka. |
|
(21) |
Regularne kontrole wewnętrzne są niezbędne, aby monitorować wdrażanie i przestrzeganie dobrej praktyki dystrybucyjnej dotyczącej weterynaryjnych produktów leczniczych oraz proponować niezbędne środki naprawcze i zapobiegawcze. |
|
(22) |
Środki przewidziane w niniejszym rozporządzeniu są zgodne z opinią Stałego Komitetu ds. Weterynaryjnych Produktów Leczniczych, o którym mowa w art. 145 rozporządzenia (UE) 2019/6, |
PRZYJMUJE NINIEJSZE ROZPORZĄDZENIE:
ROZDZIAŁ I
PRZEPISY OGÓLNE
Artykuł 1
Przedmiot i zakres stosowania
1. W niniejszym rozporządzeniu ustanawia się środki w zakresie dobrej praktyki dystrybucyjnej dotyczącej weterynaryjnych produktów leczniczych.
2. Niniejsze rozporządzenie stosuje się do posiadaczy pozwolenia na wytwarzanie prowadzących dystrybucję hurtową weterynaryjnych produktów leczniczych objętych tym pozwoleniem na wytwarzanie oraz do posiadaczy pozwolenia na dystrybucję hurtową, w tym posiadaczy takiego pozwolenia, którzy mają siedzibę lub działają w ramach szczególnych procedur celnych, takich jak wolne obszary celne lub składy celne.
Artykuł 2
Definicje
Do celów niniejszego rozporządzenia stosuje się następujące definicje:
|
a) |
„dobra praktyka dystrybucyjna dotycząca weterynaryjnych produktów leczniczych” oznacza tę część zapewniania jakości w całym łańcuchu dostaw, która zapewnia utrzymanie jakości weterynaryjnych produktów leczniczych na wszystkich etapach łańcucha dostaw, od zakładu producenta do osób, o których mowa w art. 101 ust. 2 rozporządzenia (UE) 2019/6; |
|
b) |
„wolny obszar celny” oznacza wolny obszar celny wyznaczony przez państwa członkowskie zgodnie z art. 243 rozporządzenia Parlamentu Europejskiego i Rady (UE) nr 952/2013 (9); |
|
c) |
„skład celny” oznacza każdy ze składów, o których mowa w art. 240 ust. 1 rozporządzenia (UE) nr 952/2013; |
|
d) |
„system jakości” oznacza sumę wszystkich aspektów systemu wdrażającego politykę jakości i zapewniającego osiąganie celów w zakresie jakości; |
|
e) |
„zarządzanie ryzykiem w zakresie jakości” oznacza systematyczny proces oceny, kontroli, komunikowania i przeglądu ryzyka związanego z jakością weterynaryjnych produktów leczniczych w całym cyklu życia produktu, stosowany zarówno proaktywnie, jak i retrospektywnie; |
|
f) |
„walidacja” oznacza udokumentowany program gwarantujący wysoki stopień pewności, że dany proces, metoda lub system będą stale prowadziły do wyników spełniających z góry określone kryteria dopuszczalności; |
|
g) |
„procedura” oznacza udokumentowany opis czynności, które należy wykonać, oraz środków ostrożności i innych środków, które należy wprowadzić, pośrednio lub bezpośrednio, w odniesieniu do dystrybucji weterynaryjnych produktów leczniczych; |
|
h) |
„dokumentacja” oznacza pisemne procedury, instrukcje, umowy, ewidencje i dane, w formie papierowej lub elektronicznej; |
|
i) |
„pozyskiwanie” oznacza otrzymywanie, nabywanie lub zakup weterynaryjnych produktów leczniczych od producentów, importerów lub innych hurtowników; |
|
j) |
„przechowywanie” oznacza przechowywanie weterynaryjnych produktów leczniczych; |
|
k) |
„dostarczanie” oznacza wszelkie działania polegające na zapewnianiu, sprzedaży lub przekazywaniu w ramach darowizny weterynaryjnych produktów leczniczych osobom, o których mowa w art. 101 ust. 2 rozporządzenia (UE) 2019/6; |
|
l) |
„transport” oznacza przemieszczanie weterynaryjnych produktów leczniczych między dwoma miejscami bez przechowywania ich przez nieuzasadniony czas; |
|
m) |
„odstępstwo” oznacza odchylenie od zatwierdzonej dokumentacji lub ustalonej normy; |
|
n) |
„sfałszowany weterynaryjny produkt leczniczy” oznacza każdy weterynaryjny produkt leczniczy, który został fałszywie przedstawiony w zakresie któregokolwiek z poniższych elementów:
|
|
o) |
„zanieczyszczenie” oznacza niepożądane wprowadzenie zanieczyszczeń pochodzenia chemicznego lub mikrobiologicznego lub ciał obcych do weterynaryjnego produktu leczniczego lub na jego powierzchnię podczas produkcji, pobierania próbek, pakowania lub przepakowywania, przechowywania lub transportu; |
|
p) |
„kalibracja” oznacza zbiór czynności służących ustaleniu, w określonych warunkach, zależności między wartościami wskazywanymi przez przyrząd pomiarowy lub system pomiarowy bądź wartościami reprezentowanymi przez wzorzec miary, a odpowiadającymi im znanymi wartościami wzorca odniesienia; |
|
q) |
„kwalifikacja” oznacza dowiedzenie, że dany sprzęt działa właściwie i pozwala rzeczywiście osiągnąć spodziewane rezultaty; |
|
r) |
„podpisano” oznacza zapis dotyczący osoby, która wykonała określoną czynność lub dokonała określonego przeglądu. Zapis ten może mieć postać inicjałów, pełnego własnoręcznego podpisu, osobistej pieczęci lub zaawansowanego podpisu elektronicznego zdefiniowanego w art. 3 pkt 11 rozporządzenia Parlamentu Europejskiego i Rady (UE) nr 910/2014 (10); |
|
s) |
„seria” oznacza określoną ilość materiału wyjściowego, opakowania lub produktu przetworzonego w ramach jednego procesu lub szeregu procesów w taki sposób, że zakłada się jej jednorodność; |
|
t) |
„termin ważności” oznacza datę umieszczoną na opakowaniu weterynaryjnego produktu leczniczego określającą czas, w którym ten weterynaryjny produkt leczniczy ma utrzymywać zgodność ze specyfikacją ustaloną dla okresu trwałości, o ile jest przechowywany w określonych warunkach; jest to termin, po którym produkt nie powinien być używany; |
|
u) |
„numer serii” oznacza niepowtarzalną kombinację cyfr lub liter, która jednoznacznie identyfikuje daną serię. |
ROZDZIAŁ II
ZARZĄDZANIE JAKOŚCIĄ
Artykuł 3
Opracowanie i prowadzenie systemu jakości
1. Osoby, o których mowa w art. 1 ust. 2, opracowują i utrzymują system jakości.
2. System jakości uwzględnia wielkość, strukturę i złożoność działań tych osób oraz zmiany przewidziane wobec tych działań.
3. Osoby, o których mowa w art. 1 ust. 2, zapewniają kompetentnych pracowników do obsługi wszystkich komponentów systemu jakości, a także przeznaczają na jego potrzeby odpowiednie i wystarczające pomieszczenia, sprzęt i infrastrukturę.
Artykuł 4
Wymagania dotyczące systemu jakości
1. System jakości określa obowiązki, procesy i zasady zarządzania ryzykiem w zakresie jakości w odniesieniu do działań osób, o których mowa w art. 1 ust. 2. Wszystkie działania w zakresie dystrybucji hurtowej są jasno zdefiniowane i podlegają systematycznemu przeglądowi. Wszystkie kluczowe etapy dystrybucji hurtowej i ich istotne zmiany są uzasadniane, a w stosownych przypadkach podlegają walidacji.
2. System jakości obejmuje strukturę organizacyjną, procedury, procesy i zasoby, a także działania niezbędne do zapewnienia, by dostarczane weterynaryjne produkty lecznicze utrzymywały jakość i integralność oraz pozostawały w legalnym łańcuchu dostaw podczas przechowywania lub transportu.
3. System jakości jest w pełni udokumentowany. Jego skuteczność jest monitorowana. Wszystkie działania związane z systemem jakości są zdefiniowane i udokumentowane.
4. Opracowuje się podręcznik dotyczący jakości lub wprowadza się równoważne podejście oparte na dokumentacji, w ramach którego zawarto opis wszelkich różnic w systemie jakości pod względem postępowania z różnymi rodzajami weterynaryjnych produktów leczniczych.
5. Wprowadza się system kontroli zmian, który uwzględnia zasady zarządzania ryzykiem w zakresie jakości oraz jest proporcjonalny i skuteczny.
6. System jakości zapewnia wypełnienie następujących obowiązków:
|
a) |
pozyskiwanie, przechowywanie, dostarczanie, transport lub wywóz weterynaryjnych produktów leczniczych spełnia wymogi dobrej praktyki dystrybucyjnej dotyczącej weterynaryjnych produktów leczniczych ustanowione w niniejszym rozporządzeniu; |
|
b) |
obowiązki kierownictwa są jasno określone; |
|
c) |
weterynaryjne produkty lecznicze są dostarczane właściwym odbiorcom w odpowiednim terminie; |
|
d) |
działania są na bieżąco ewidencjonowane; |
|
e) |
odstępstwa są dokumentowane i badane; |
|
f) |
podejmowane są odpowiednie działania naprawcze i zapobiegawcze (corrective and preventive actions – CAPA) zgodnie z zasadami zarządzania ryzykiem w zakresie jakości; |
|
g) |
zmiany, które mogą mieć wpływ na przechowywanie i dystrybucję weterynaryjnych produktów leczniczych, poddawane są analizie. |
Artykuł 5
Zarządzanie działaniami zlecanymi podmiotom zewnętrznym
System jakości obejmuje kontrolę i przegląd wszystkich działań zlecanych podmiotom zewnętrznym, związanych z dystrybucją hurtową weterynaryjnych produktów leczniczych. Taka kontrola i przegląd uwzględniają zarządzanie ryzykiem w zakresie jakości i obejmują:
|
a) |
ocenę kompetencji zleceniobiorcy, ustalenie, czy nadaje się do przeprowadzenia danego działania, a także sprawdzenie, czy posiada pozwolenie, jeżeli jest ono wymagane; |
|
b) |
określenie obowiązków i procesów komunikacyjnych w odniesieniu do związanych z jakością działań zaangażowanych podmiotów; |
|
c) |
regularne monitorowanie i przegląd realizacji zlecenia przez zleceniobiorcę, regularne sprawdzanie, czy potrzebne są jakiekolwiek usprawnienia, oraz ich wprowadzanie. |
Artykuł 6
Przegląd zarządczy i monitorowanie
1. Kierownictwo osób, o których mowa w art. 1 ust. 2, ustanawia i wdraża formalny proces okresowego przeglądu systemu jakości.
2. Przegląd obejmuje:
|
a) |
pomiar realizacji celów systemu jakości; |
|
b) |
ocenę:
|
|
c) |
nowe przepisy, wytyczne i kwestie związane z jakością, które mogą wpływać na system jakości; |
|
d) |
innowacje, które mogą usprawniać system jakości; |
|
e) |
zmiany otoczenia biznesowego i celów. |
3. Wyniki każdego przeglądu zarządczego systemu jakości są dokumentowane w stosownym czasie, a informacje o nich są skutecznie przekazywane pracownikom.
Artykuł 7
Zarządzanie ryzykiem w zakresie jakości
1. Osoby, o których mowa w art. 1 ust. 2, stosują zarządzanie ryzykiem w zakresie jakości.
2. Zarządzanie ryzykiem w zakresie jakości zapewnia, aby ocena ryzyka dla jakości opierała się na wiedzy naukowej i doświadczeniach w stosowaniu danego procesu, a także by w ostatecznym rozrachunku była związana z ochroną leczonego zwierzęcia lub grupy zwierząt oraz ochroną osób odpowiedzialnych za zwierzę i leczenie, konsumenta zwierzęcia, od którego lub z którego pozyskuje się żywność, oraz środowiska.
3. Stopień szczegółowości i zakres udokumentowania procesu zarządzania ryzykiem w zakresie jakości jest proporcjonalny do poziomu ryzyka dla jakości.
ROZDZIAŁ III
WYMAGANIA DOTYCZĄCE PRACOWNIKÓW
Artykuł 8
Obowiązki osób odpowiedzialnych za dystrybucję hurtową
1. Osoby odpowiedzialne za dystrybucję hurtową, o których mowa w art. 101 ust. 3 rozporządzenia (UE) 2019/6 („osoby odpowiedzialne”), zapewniają zgodność z dobrą praktyką dystrybucyjną dotyczącą weterynaryjnych produktów leczniczych. Oprócz wymogu określonego w art. 100 ust. 2 lit. a) tego rozporządzenia osoby odpowiedzialne posiadają odpowiednie kompetencje i doświadczenie, a także wiedzę i przeszkolenie w zakresie zgodności z dobrą praktyką dystrybucyjną dotyczącą weterynaryjnych produktów leczniczych.
2. Osoby odpowiedzialne ponoszą osobistą odpowiedzialność za wypełnianie swoich obowiązków, a kontakt z nimi jest możliwy w każdej chwili.
3. Osoby odpowiedzialne mogą przekazywać zadania, ale nie obowiązki.
4. Jeżeli osoby odpowiedzialne nie są dostępne, osoby, o których mowa w art. 1 ust. 2, wyznaczają zastępcę na okres niezbędny do zapewnienia ciągłości działania.
5. Opis stanowiska pracy osoby odpowiedzialnej, sporządzony na piśmie, określa jej uprawnienia decyzyjne w zakresie jej obowiązków. Osoby, o których mowa w art. 1 ust. 2, przekazują osobom odpowiedzialnym określone uprawnienia, zasoby i zakres odpowiedzialności niezbędne do wypełniania ich obowiązków.
6. Osoby odpowiedzialne wykonują swoje zadania w sposób zapewniający, by odpowiednie osoby, o których mowa w art. 1 ust. 2, były w stanie wykazać zgodność z dobrą praktyką dystrybucyjną dotyczącą weterynaryjnych produktów leczniczych, oraz by spełnione były obowiązki, o których mowa w art. 101 ust. 4 rozporządzenia (UE) 2019/6.
7. Obowiązki osób odpowiedzialnych obejmują:
|
a) |
zapewnienie, aby wprowadzono i stosowano system jakości; |
|
b) |
koncentrację na zarządzaniu działalnością objętą pozwoleniem oraz na rzetelności i jakości ewidencji; |
|
c) |
zapewnienie, aby wprowadzono i realizowano programy kształcenia i doskonalenia zawodowego; |
|
d) |
koordynację i szybkie przeprowadzanie czynności związanych z wycofaniem weterynaryjnych produktów leczniczych; |
|
e) |
zapewnianie efektywnego rozpatrywania skarg klientów; |
|
f) |
zapewnianie, aby dostawcy i klienci byli zatwierdzeni; |
|
g) |
zatwierdzanie działań wykonywanych przez podwykonawców, które mogą mieć wpływ na dobrą praktykę dystrybucyjną dotyczącą weterynaryjnych produktów leczniczych; |
|
h) |
zapewnianie, aby w odpowiednich, regularnych odstępach były przeprowadzane kontrole wewnętrzne według ustalonego programu, a także dopilnowanie, by wprowadzono odpowiednie CAPA; |
|
i) |
właściwe ewidencjonowanie zadań zleconych innym osobom; |
|
j) |
podejmowanie decyzji w sprawie ostatecznego przeznaczenia zwróconych, odrzuconych, wycofanych lub sfałszowanych weterynaryjnych produktów leczniczych; |
|
k) |
zatwierdzanie zwrotów do zapasów przeznaczonych do sprzedaży; |
|
l) |
zapewnianie, aby przestrzegane były wszelkie dodatkowe wymogi dotyczące określonych weterynaryjnych produktów leczniczych przewidziane prawem krajowym; |
|
m) |
dokumentowanie odstępstw i podejmowanie decyzji w sprawie CAPA w celu skorygowania odstępstw i uniknięcia ich ponownego wystąpienia oraz monitorowanie skuteczności CAPA. |
Artykuł 9
Inny personel
1. We wszystkich etapach dystrybucji hurtowej weterynaryjnych produktów leczniczych bierze udział odpowiednia liczba kompetentnych pracowników. Liczba ta jest proporcjonalna do wielkości i zakresu działań.
2. Struktura organizacyjna osób, o których mowa w art. 1 ust. 2, jest zapisana w formie diagramu organizacyjnego. Diagram jasno określa poszczególne role, obowiązki wszystkich pracowników oraz wzajemne relacje między nimi. Każdy członek personelu rozumie swoją rolę i swoje obowiązki.
3. Role i obowiązki pracowników na najważniejszych stanowiskach, a także zasady ich zastępowania, są określone w opisach stanowisk.
Artykuł 10
Szkolenie pracowników
1. Wszyscy pracownicy zajmujący się dystrybucją hurtową są przeszkoleni w zakresie wymogów dobrej praktyki dystrybucyjnej dotyczącej weterynaryjnych produktów leczniczych. Ponadto mają oni odpowiednie kompetencje i doświadczenie przed rozpoczęciem wykonywania swoich zadań.
2. Pracownicy odbywają kształcenie i doskonalenie zawodowe stosowne do pełnionych ról, w oparciu o procedury i według pisemnego programu szkolenia. Osoby odpowiedzialne utrzymują swoje kompetencje w zakresie dobrej praktyki dystrybucyjnej dotyczącej weterynaryjnych produktów leczniczych poprzez regularne szkolenia.
3. Szkolenia obejmują identyfikację sfałszowanych weterynaryjnych produktów leczniczych i zapobieganie ich wprowadzaniu do łańcucha dostaw.
4. Pracownicy zajmujący się weterynaryjnymi produktami leczniczymi wymagającymi bardziej rygorystycznych warunków postępowania, takimi jak produkty niebezpieczne, produkty stwarzające szczególne ryzyko nadużyć, w tym substancje odurzające i psychotropowe, oraz produkty wrażliwe na temperaturę, przechodzą specjalne szkolenie.
5. Osoby, o których mowa w art. 1 ust. 2, prowadzą ewidencję wszystkich szkoleń oraz okresowo oceniają i dokumentują ich skuteczność.
Artykuł 11
Higiena
Osoby, o których mowa w art. 1 ust. 2, ustanawiają odpowiednie procedury dotyczące higieny pracowników, w tym ich zdrowia i odpowiedniej odzieży, związane z prowadzoną działalnością. Pracownicy przestrzegają tych procedur.
ROZDZIAŁ IV
POMIESZCZENIA I SPRZĘT
Artykuł 12
Pomieszczenia
1. Pomieszczenia są zaprojektowane w sposób zapewniający utrzymywanie wymaganych warunków przechowywania bądź zaadaptowane do tego celu. Pomieszczenia te są odpowiednio zabezpieczone, mają solidną konstrukcję i wystarczającą pojemność, tak by umożliwiały bezpieczne przechowywanie weterynaryjnych produktów leczniczych i postępowanie z nimi. Obszary magazynowe są wyposażone w odpowiednie oświetlenie, tak by wszystkie czynności mogły być wykonywane dokładnie i bezpiecznie. Weterynaryjne produkty lecznicze są przechowywane z zachowaniem odpowiedniej przestrzeni, aby umożliwić sprzątanie i inspekcję. Palety powinny być utrzymywane w dobrym stanie pod względem czystości i naprawy.
2. Jeżeli danymi pomieszczeniami nie zarządzają bezpośrednio osoby, o których mowa w art. 1 ust. 2, zawiera się stosowną umowę. Osoby, o których mowa w art. 1 ust. 2, mogą korzystać z pomieszczeń objętych umową tylko wtedy, gdy pomieszczenia te są objęte odrębnym pozwoleniem na dystrybucję hurtową.
3. Weterynaryjne produkty lecznicze są przechowywane na oddzielonych, jasno oznakowanych obszarach, do których dostęp ma wyłącznie upoważniony personel.
4. Każdy system zastępujący fizyczne oddzielenie, stosownie do przypadku, na przykład segregacja elektroniczna oparta na skomputeryzowanym systemie, zapewnia równoważne bezpieczeństwo i podlega stosownej walidacji.
5. Weterynaryjne produkty lecznicze oczekujące na decyzję o ich unieszkodliwieniu lub weterynaryjne produkty lecznicze, które usunięto z zapasów przeznaczonych do sprzedaży, są fizycznie oddzielone lub – jeżeli dostępny jest równoważny system elektroniczny – są oddzielone elektronicznie, co obejmuje zwrócone weterynaryjne produkty lecznicze.
6. Weterynaryjne produkty lecznicze otrzymane z państwa trzeciego, ale nieprzeznaczone na rynek Unii, są oddzielane fizycznie i elektronicznie, jeżeli dostępny jest system elektroniczny.
7. Wszelkie przeterminowane weterynaryjne produkty lecznicze, wycofane weterynaryjne produkty lecznicze i odrzucone weterynaryjne produkty lecznicze są natychmiast fizycznie oddzielane i przechowywane na przeznaczonym do tego obszarze z dala od wszystkich innych weterynaryjnych produktów leczniczych. Na tych obszarach stosuje się odpowiedni poziom bezpieczeństwa, aby zapewnić stałe oddzielenie tych produktów od zapasów przeznaczonych do sprzedaży. Obszary te są wyraźnie oznakowane.
8. Pomieszczenia są zaprojektowane lub zaadaptowane w sposób zapewniający przechowywanie weterynaryjnych produktów leczniczych podlegających specjalnym środkom przechowywania i postępowania z nimi, takich jak substancje odurzające i psychotropowe, zgodnie z pisemnymi instrukcjami i z zastrzeżeniem odpowiednich środków bezpieczeństwa.
9. Zapewnia się co najmniej jeden specjalny obszar oraz wprowadzić odpowiednie środki bezpieczeństwa i zabezpieczenia w odniesieniu do przechowywania niebezpiecznych weterynaryjnych produktów leczniczych, a także weterynaryjnych produktów leczniczych stwarzających szczególne zagrożenie pożarem lub wybuchem, takich jak gazy medyczne, substancje palne, łatwopalne ciecze i substancje stałe.
10. Doki do przyjmowania i wydawania towarów chronią weterynaryjne produkty lecznicze przed panującymi warunkami pogodowymi. Odpowiednio oddziela się od siebie obszary, w których przyjmuje się, wydaje oraz przechowuje produkty. Wprowadzone są procedury służące utrzymywaniu kontroli nad przyjmowanymi i wydawanymi towarami. Wyznacza się i odpowiednio wyposaża obszary przyjmowania towarów, w których sprawdza się dostawy po ich przyjęciu.
11. Nieuprawnionemu dostępowi do wszystkich obszarów objętych zezwoleniem zapobiega się za pomocą odpowiednich urządzeń, takich jak monitorowany system alarmowy sygnalizacji włamania oraz odpowiednia kontrola dostępu. Odwiedzającym przez cały czas towarzyszy pracownik.
12. Pomieszczenia i infrastruktura magazynowa są czyste oraz pozbawione śmieci i kurzu. Wprowadzone są harmonogramy, instrukcje i ewidencja dotyczące sprzątania. Dobiera się odpowiedni sprzęt do sprzątania i środki czystości oraz używa się ich w taki sposób, by nie stanowiły one źródła zanieczyszczeń.
13. Pomieszczenia są suche, a temperatura jest w nich utrzymywana w dopuszczalnych granicach.
14. Stosowane są odpowiednie procedury usuwania śladów ewentualnego rozlania, aby zapewnić całkowite wyeliminowanie wszelkiego ryzyka zanieczyszczenia.
15. Pojazdy są regularnie czyszczone. Sprzęt wybrany i używany do czyszczenia pojazdów nie może stanowić źródła zanieczyszczenia.
16. Pomieszczenia są zaprojektowane i wyposażone w taki sposób, aby zapewniały ochronę przed owadami, gryzoniami i innymi zwierzętami. Stosuje się zapobiegawczy program zwalczania szkodników.
17. Pomieszczenia służące pracownikom do odpoczynku, mycia się i spożywania posiłków są odpowiednio oddzielone od obszarów magazynowych. Zakazane jest wprowadzanie do obszarów magazynowych żywności, napojów, wyrobów tytoniowych i produktów leczniczych do użytku osobistego.
Artykuł 13
Kontrola temperatury i otoczenia
1. Stosuje się odpowiedni sprzęt i procedury w celu kontrolowania otoczenia, w którym przechowywane są weterynaryjne produkty lecznicze. Pod uwagę brane są następujące czynniki środowiskowe: temperatura, światło, wilgotność i czystość pomieszczeń.
2. Przed rozpoczęciem korzystania z obszaru magazynowego przeprowadza się wstępne mapowanie temperatury w reprezentatywnych warunkach. Sprzęt do monitorowania temperatury rozmieszcza się przy uwzględnieniu wyników mapowania, tak aby urządzenia monitorujące znalazły się na obszarach, gdzie występują skrajne wartości fluktuacji temperatur. Mapowanie zostaje powtórzone, jeżeli wymagają tego wyniki oceny ryzyka oraz w każdym przypadku, gdy są wprowadzane istotne zmiany infrastruktury lub sprzętu do regulacji temperatury. W przypadku małych pomieszczeń o powierzchni kilku metrów kwadratowych, w których panuje temperatura pokojowa, przeprowadza się ocenę potencjalnego ryzyka, na przykład związanego z grzejnikami, i na jej podstawie rozmieszcza się urządzenia monitorujące temperaturę.
Artykuł 14
Sprzęt
1. Każdy sprzęt mający wpływ na przechowywanie i dystrybucję weterynaryjnych produktów leczniczych jest zaprojektowany, umieszczony i poddawany konserwacji zgodnie z normą odpowiednią do celu, do którego jest przeznaczony. W przypadku najważniejszego sprzętu, niezbędnego dla funkcjonalności danej czynności, stosuje się plan konserwacji.
2. Sprzęt używany do kontrolowania lub monitorowania otoczenia, w którym przechowywane są weterynaryjne produkty lecznicze, jest kalibrowany w określonych odstępach czasu w oparciu o ocenę ryzyka i niezawodności.
3. Kalibracja sprzętu odbywa się zgodnie z krajową lub międzynarodową normą pomiaru. Wprowadza się odpowiednie systemy alarmowe ostrzegające o odchyleniach od ustalonych warunków przechowywania. Poziomy alarmowe są właściwie ustawione, a alarmy są poddawane regularnym testom w celu zapewnienia odpowiedniej funkcjonalności.
4. Naprawy, konserwacja i kalibracja sprzętu odbywają się w sposób nienaruszający integralności weterynaryjnych produktów leczniczych.
5. Uszkodzone pojazdy i sprzęt nie są używane i są oznakowane jako takie lub wycofane z eksploatacji.
6. Sprzęt nieistotny dla działalności hurtowej nie jest przechowywany na obszarze, gdzie przechowywane są weterynaryjne produkty lecznicze.
7. Prowadzi się odpowiednią ewidencję dotyczącą napraw, konserwacji i kalibracji najważniejszego sprzętu, takiego jak chłodnie, monitorowane systemy alarmowe sygnalizacji włamania i systemy kontroli dostępu, chłodziarki, termohigrometry lub inne urządzenia rejestrujące temperaturę i wilgotność, urządzenia wentylacyjne i wszelki sprzęt używany do celów dalszego łańcucha dostaw, i przechowuje się wyniki takich napraw, konserwacji i kalibracji.
Artykuł 15
Skomputeryzowane systemy
1. Przed rozpoczęciem korzystania ze skomputeryzowanego systemu wykazuje się, w formie odpowiednich badań walidujących lub weryfikujących, że system ten jest w stanie osiągać pożądane wyniki w sposób dokładny, konsekwentny i powtarzalny.
2. Sporządza się na piśmie szczegółowy opis skomputeryzowanego systemu, obejmujący w stosownych przypadkach diagramy. Opis ten jest aktualizowany. W dokumencie tym opisuje się zasady, cele, środki bezpieczeństwa, zakres systemu i jego główne cechy, przedstawia się sposób korzystania z niego i jego interakcje z innymi systemami.
3. Tylko upoważnione osoby wprowadzają dane do skomputeryzowanego systemu lub je zmieniają.
4. Dane są zabezpieczone środkami fizycznymi lub elektronicznymi oraz chronione przed przypadkowymi lub nieuprawnionymi zmianami. Okresowo sprawdzana jest dostępność przechowywanych danych. W regularnych odstępach czasu sporządza się kopie zapasowe danych. Dane zapasowe przechowuje się w oddzielnym i bezpiecznym miejscu przez co najmniej 5 lat lub przez okres wyznaczony w mającym zastosowanie prawie krajowym, jeżeli okres ten jest dłuższy niż 5 lat.
5. Określa się procedury na wypadek błędu lub awarii systemu. Procedury obejmują systemy odzyskiwania danych.
Artykuł 16
Kwalifikacja i walidacja
1. Osoby, o których mowa w art. 1 ust. 2, ustalają, w jakich przypadkach niezbędna jest kwalifikacja najważniejszego sprzętu i walidacja najważniejszych procesów, aby zapewnić prawidłową instalację i funkcjonowanie. Zakres i zasięg takich działań związanych z kwalifikacją i walidacją, dotyczących np. przechowywania, procesów pobierania i pakowania produktów, są określone zgodnie z podejściem opartym na udokumentowanej ocenie ryzyka.
2. Sprzęt i procesy podlegają odpowiednio kwalifikacji lub walidacji przed rozpoczęciem ich stosowania oraz po każdej istotnej zmianie, takiej jak naprawa lub konserwacja.
3. Sporządza się sprawozdania z kwalifikacji i walidacji, zawierające podsumowanie uzyskanych wyników i uwagi na temat wszystkich odnotowanych odstępstw. W razie potrzeby stosuje się zasady CAPA. Odpowiedni pracownicy sporządzają i zatwierdzają dokumenty poświadczające zadowalającą walidację i akceptację poszczególnych procesów lub sprzętów.
ROZDZIAŁ V
DOKUMENTACJA, PROCEDURY I PROWADZENIE EWIDENCJI
Artykuł 17
Wymagania dotyczące dokumentacji
1. Dokumentacja spełnia następujące wymagania:
|
a) |
jest łatwo dostępna lub możliwa do łatwego wyszukania; |
|
b) |
jest wystarczająco kompleksowa w odniesieniu do zakresu działań osób, o których mowa w art. 1 ust. 2; |
|
c) |
jest napisana w języku zrozumiałym dla pracowników; |
|
d) |
jest napisana jasnym i jednoznacznym językiem. |
2. Dokumentacja jest zatwierdzona, podpisana i opatrzona datą przez odpowiednie upoważnione osoby, zgodnie z wymaganiami. Nie jest sporządzana odręcznie, chyba że zapisy własnoręczne są uzasadnione względami praktycznymi. W takim przypadku należy zapewnić wystarczającą ilość miejsca na prowadzenie takiej ewidencji.
3. W przypadku stwierdzenia błędów w dokumentacji są one niezwłocznie korygowane, z wyraźnym wskazaniem, kto je poprawił i kiedy.
4. Każda zmiana w dokumentacji jest opatrzona podpisem i datą. Zmiany wprowadza się tak, by możliwe było odczytanie pierwotnej informacji. W stosownych przypadkach zapisuje się powód wprowadzenia zmiany.
5. Dokumenty przechowuje się przez co najmniej 5 lat lub przez okres wyznaczony w mającym zastosowanie prawie krajowym, jeżeli okres ten jest dłuższy niż 5 lat. Dane osobowe usuwa się, gdy tylko ich przechowywanie nie jest już potrzebne do celów działań związanych z dystrybucją.
6. Każdy pracownik ma łatwy dostęp do całej dokumentacji niezbędnej do wykonywania jego zadań.
7. W odniesieniu do wszystkich systemów papierowych, elektronicznych i hybrydowych określa się relacje i środki kontroli w odniesieniu do oryginalnych dokumentów i kopii urzędowych, przetwarzania danych i ewidencji.
Artykuł 18
Procedury
1. Procedury opisują działania w zakresie dystrybucji hurtowej wpływające na jakość weterynaryjnych produktów leczniczych. Działania te obejmują:
|
a) |
odbiór i sprawdzanie dostaw; kontrolę dostawców i klientów; |
|
b) |
przechowywanie; |
|
c) |
czyszczenie i konserwację pomieszczeń i sprzętu, w tym zwalczanie szkodników; |
|
d) |
sprawdzanie i rejestrowanie warunków przechowywania; |
|
e) |
ochronę weterynaryjnych produktów leczniczych podczas transportu; |
|
f) |
bezpieczeństwo zapasów na miejscu i przesyłek przewożonych w tranzycie; |
|
g) |
wycofywanie z zapasów przeznaczonych do sprzedaży; |
|
h) |
postępowanie ze zwróconymi weterynaryjnymi produktami leczniczymi; |
|
i) |
plany wycofania; |
|
j) |
kwalifikację i walidację; |
|
k) |
procedury i środki unieszkodliwiania nienadających się do użytku weterynaryjnych produktów leczniczych; |
|
l) |
procedury rozpatrywania i rozstrzygania skarg; |
|
m) |
procedury identyfikacji weterynaryjnych produktów leczniczych, w przypadku których podejrzewa się, że są sfałszowane. |
2. Procedury są zatwierdzone, podpisane i opatrzone datą przez osoby odpowiedzialne.
3. Stosuje się odpowiednie i zatwierdzone procedury. Dokumenty są jasne i odpowiednio szczegółowe. Podaje się tytuł, charakter i cel dokumentów. Dokumenty są poddawane regularnemu przeglądowi oraz aktualizacji. Prowadzona jest kontrola wersji procedur. Stosowany jest system, który w przypadku zmiany dokumentu zapobiega przypadkowemu użyciu jego nieaktualnej wersji. Nieaktualne lub przestarzałe procedury są usuwane ze stacji roboczych i poddawane archiwizacji.
Artykuł 19
Ewidencja
1. Każda transakcja dotycząca otrzymanych lub dostarczonych weterynaryjnych produktów leczniczych jest ewidencjonowana w postaci faktur zakupu lub sprzedaży lub kwitów dostawy, lub w formie elektronicznej.
2. Oprócz szczegółowej dokumentacji, o której mowa w art. 101 ust. 7 rozporządzenia (UE) 2019/6, ewidencja obejmuje w stosownych przypadkach wszelkie dodatkowe wymagania określone w prawie krajowym.
3. Każdy przypadek przeprowadzenia danej czynności jest na bieżąco ewidencjonowany. Jeżeli ewidencja sporządzana jest odręcznie, musi być napisana w sposób jasny, czytelny i nieusuwalny.
ROZDZIAŁ VI
CZYNNOŚCI
Artykuł 20
Wymagania dotyczące czynności
1. Osoby, o których mowa w art. 1 ust. 2, zapewniają zachowanie tożsamości weterynaryjnego produktu leczniczego podczas dystrybucji hurtowej i stosują wszelkie dostępne środki w celu zminimalizowania ryzyka wprowadzenia sfałszowanych weterynaryjnych produktów leczniczych do legalnego łańcucha dostaw.
2. Osoby, o których mowa w art. 1 ust. 2, zapewniają, aby dystrybucja hurtowa weterynaryjnych produktów leczniczych odbywała się zgodnie z informacjami na opakowaniu zewnętrznym.
3. Osoby, o których mowa w art. 1 ust. 2, zapewniają, aby wszystkie weterynaryjne produkty lecznicze, które dystrybuują w Unii, były:
|
a) |
objęte pozwoleniem na dopuszczenie do obrotu wydanym przez właściwy organ lub, w stosownych przypadkach, przez Komisję; |
|
b) |
objęte rejestracją dokonaną przez właściwy organ; |
|
c) |
objęte zwolnieniem, przyznanym przez właściwy organ, z wymogów pozwolenia na dopuszczenie do obrotu; |
|
d) |
objęte zatwierdzeniem do celów handlu równoległego wydanym przez właściwy organ państwa członkowskiego przeznaczenia; |
|
e) |
objęte zezwoleniem na stosowanie zgodnie z art. 110 ust. 2 i 3 rozporządzenia (UE) 2019/6; lub |
|
f) |
w przypadku produktów przeznaczonych do stosowania na podstawie art. 112 ust. 2, art. 113 ust. 2 lub art. 114 ust. 4 rozporządzenia (UE) 2019/6 – przywożone przez posiadaczy pozwolenia na wytwarzanie wydanego zgodnie z art. 90 tego rozporządzenia lub zgodnie z procedurami, o których mowa w art. 106 ust. 3 tego rozporządzenia, stosownie do przypadku. |
4. Wszystkie najważniejsze czynności osób, o których mowa w art. 1 ust. 2, są w pełni opisane w systemie jakości w odpowiedniej dokumentacji.
Artykuł 21
Weryfikacja kwalifikowalności i zatwierdzanie dostawców
1. W przypadku gdy weterynaryjne produkty lecznicze są otrzymywane od osoby, o której mowa w art. 1 ust. 2, odbierający je hurtownik sprawdza, czy dostawca przestrzega zasad dobrej praktyki dystrybucyjnej dotyczącej weterynaryjnych produktów leczniczych określonych w niniejszym rozporządzeniu i czy posiada pozwolenie. Informacje te uzyskuje się od właściwych organów krajowych lub z unijnej bazy danych dotyczącej wytwarzania, przywozu i dystrybucji hurtowej, o której mowa w art. 91 ust. 1 rozporządzenia (UE) 2019/6. Odpowiednia weryfikacja kwalifikowalności i zatwierdzanie dostawców odbywa się przed pozyskaniem weterynaryjnych produktów leczniczych. Proces ten jest kontrolowany za pomocą procedury, a wyniki są dokumentowane i okresowo sprawdzane w oparciu o zasady zarządzania ryzykiem w zakresie jakości.
2. Zawierając umowę z nowymi dostawcami, osoby, o których mowa w art. 1 ust. 2, przeprowadzają tzw. analizę due diligence, oceniając odpowiedniość, kompetencje i wiarygodność danego dostawcy. W ramach analizy due diligence uwzględnia się:
|
a) |
reputację lub wiarygodność dostawcy; |
|
b) |
oferty weterynaryjnych produktów leczniczych, w przypadku których istnieje większe prawdopodobieństwo fałszerstwa; |
|
c) |
duże oferty weterynaryjnych produktów leczniczych, które co do zasady dostępne są tylko w ograniczonych ilościach; |
|
d) |
nietypowo dużą różnorodność weterynaryjnych produktów leczniczych obsługiwanych przez dostawcę; |
|
e) |
rażąco niskie ceny. |
Artykuł 22
Weryfikacja kwalifikowalności i zatwierdzanie klientów
1. Osoby, o których mowa w art. 1 ust. 2, przeprowadzają wstępne i, w stosownych przypadkach, okresowe kontrole w celu ustalenia, czy ich klienci spełniają wymogi określone w art. 101 ust. 2 rozporządzenia (UE) 2019/6. Może to obejmować prośbę o przekazanie kopii pozwoleń klienta wydanych na podstawie prawa krajowego, sprawdzenie jego statusu na stronie internetowej właściwego organu oraz prośbę o przekazanie dowodów kwalifikacji lub uprawnienia na podstawie prawa krajowego.
2. Osoby, o których mowa w art. 1 ust. 2, monitorują swoje transakcje i badają wszystkie nieprawidłowości w strukturze sprzedaży substancji odurzających, substancji psychotropowych i innych niebezpiecznych substancji. Wszystkie przypadki nietypowej struktury sprzedaży, które mogą stanowić nielegalne lub niewłaściwe wykorzystywanie weterynaryjnych produktów leczniczych, są badane i w stosownych przypadkach zgłaszane właściwym organom.
Artykuł 23
Przyjmowanie weterynaryjnych produktów leczniczych
1. Osoby odpowiedzialne za przyjmowanie weterynaryjnych produktów leczniczych upewniają się co do prawidłowości przychodzącej przesyłki, sprawdzają, czy weterynaryjne produkty lecznicze pochodzą od zatwierdzonych dostawców oraz czy nie zostały one uszkodzone podczas transportu.
2. Pierwszeństwo daje się weterynaryjnym produktom leczniczym wymagającym specjalnych środków w zakresie przechowywania lub bezpieczeństwa, a po przeprowadzeniu odpowiednich kontroli produkty te kieruje się niezwłocznie do właściwej infrastruktury magazynowej.
3. Serie weterynaryjnych produktów leczniczych przeznaczone na rynek Unii są kierowane do zapasów przeznaczonych do sprzedaży dopiero po uzyskaniu pewności zgodnie z procedurami, że produkty te są dopuszczone do sprzedaży. W przypadku serii pochodzących z innego państwa członkowskiego odpowiednio przeszkoleni pracownicy powinni, przed skierowaniem ich do zapasów przeznaczonych do sprzedaży, uważnie sprawdzić sprawozdanie kontrolne, o którym mowa w art. 97 ust. 6 i 9 rozporządzenia (UE) 2019/6, wyniki niezbędnych badań, o których mowa w art. 97 ust. 7 tego rozporządzenia, stosownie do przypadku, lub inny dowód dopuszczenia produktów do obrotu w danym państwie, sporządzony na podstawie równoważnego systemu.
Artykuł 24
Przechowywanie
1. Weterynaryjne produkty lecznicze są przechowywane oddzielnie od innych produktów, które mogłyby zmienić ich właściwości, i chronione od szkodliwego wpływu światła, temperatury, wilgoci i innych czynników zewnętrznych. Ze szczególną uwagą traktuje się weterynaryjne produkty lecznicze wymagające specjalnych warunków przechowywania.
2. Przychodzące pojemniki z weterynaryjnymi produktami leczniczymi są w razie potrzeby czyszczone przed skierowaniem do magazynu. Wszelkie działania podejmowane w odniesieniu do przychodzących towarów nie mogą wpływać na jakość weterynaryjnych produktów leczniczych.
3. Czynności magazynowe wykonuje się w sposób zapewniający utrzymanie odpowiednich warunków przechowywania i pozwalający na odpowiednie zabezpieczenie zapasów.
4. Rotacja zapasów odbywa się w oparciu o zasadę, że w pierwszej kolejności wydawane są produkty o najbliższym terminie ważności. Wyjątki od tej zasady zapisuje się w dokumentacji.
5. Sposób postępowania z weterynaryjnymi produktami leczniczymi i ich przechowywania zapobiega ich rozlaniu, uszkodzeniu, zanieczyszczeniu lub zmieszaniu. Weterynaryjne produkty lecznicze nie są przechowywane bezpośrednio na podłodze, chyba że ich opakowanie jest zaprojektowane w sposób pozwalający na takie przechowywanie, na przykład w przypadku niektórych butli z gazami medycznymi.
6. Weterynaryjne produkty lecznicze, w przypadku których zbliża się termin ważności, są natychmiast oddzielane od zapasów przeznaczonych do sprzedaży fizycznie lub, jeżeli dostępny jest równoważny system elektroniczny, w formie elektronicznej.
7. Zapasy są regularnie inwentaryzowane z uwzględnieniem wymogów prawa krajowego. Nieprawidłowości dotyczące zapasów są badane i dokumentowane.
Artykuł 25
Niszczenie zdezaktualizowanych weterynaryjnych produktów leczniczych
1. Weterynaryjne produkty lecznicze przeznaczone do zniszczenia są odpowiednio identyfikowane, oraz trzymane oddzielnie i postępuje się z nimi zgodnie z procedurą.
2. Niszczenie weterynaryjnych produktów leczniczych odbywa się zgodnie z mającymi zastosowanie wymogami dotyczącymi postępowania z takimi produktami, ich transportu i unieszkodliwiania.
3. Ewidencję wszystkich zniszczonych weterynaryjnych produktów leczniczych przechowuje się przez czas określony w systemie jakości, o którym mowa w art. 3.
Artykuł 26
Pobieranie produktów
Wprowadzone są środki kontrolne zapewniające pobranie właściwego weterynaryjnego produktu leczniczego. Pobrany weterynaryjny produkt leczniczy ma odpowiedni pozostały okres trwałości i nie może być uszkodzony podczas przechowywania.
Artykuł 27
Dostawy
1. Do wszystkich dostaw dołącza się dokument elektroniczny lub fizyczny zawierający, oprócz informacji, o których mowa w art. 101 ust. 7 rozporządzenia (UE) 2019/6, niepowtarzalny numer umożliwiający identyfikację polecenia dostawy, mające zastosowanie warunki transportu i przechowywania oraz dodatkowe wymogi określone w prawie krajowym.
2. Należy prowadzić ewidencję elektroniczną lub fizyczną w taki sposób, aby znać lokalizację weterynaryjnego produktu leczniczego.
Artykuł 28
Wywóz
1. Przy wywozie weterynaryjnych produktów leczniczych, w odniesieniu do których właściwy organ krajowy ani Komisja, stosownie do przypadku, nie wydały pozwolenia na dopuszczenie do obrotu zgodnie z rozdziałem III rozporządzenia (UE) 2019/6, hurtownicy wprowadzają odpowiednie środki, aby zapobiec wejściu tych produktów leczniczych do obrotu w Unii Europejskiej.
2. Jeżeli osoby, o których mowa w art. 1 ust. 2, dostarczają weterynaryjne produkty lecznicze osobom w państwach trzecich, dostarczają te produkty wyłącznie osobom posiadającym pozwolenie lub uprawnienie do otrzymywania weterynaryjnych produktów leczniczych na potrzeby dystrybucji hurtowej lub na potrzeby dostaw dla ogółu społeczeństwa zgodnie z mającymi zastosowanie przepisami prawnymi i administracyjnymi w tych państwach trzecich.
ROZDZIAŁ VII
SKARGI, ZWROTY, PODEJRZENIA FAŁSZOWANIA WETERYNARYJNYCH PRODUKTÓW LECZNICZYCH ORAZ PRZYPADKI WYCOFANIA
Artykuł 29
Skargi
1. Skargi są ewidencjonowane wraz z wszystkimi zawartymi w nich danymi. Stosuje się rozróżnienie na skargi dotyczące jakości weterynaryjnego produktu leczniczego i skargi dotyczące dystrybucji hurtowej.
W razie otrzymania skargi dotyczącej jakości weterynaryjnego produktu leczniczego i potencjalnej wady produktu bezzwłocznie informuje się producenta lub posiadacza pozwolenia na dopuszczenie do obrotu.
Każda skarga dotycząca dystrybucji weterynaryjnego produktu leczniczego jest gruntownie rozpatrywana, aby ustalić podstawę lub przyczynę skargi.
2. Wyznacza się osobę odpowiedzialną za rozpatrywanie skarg, a do wsparcia tej osoby przydziela się wystarczającą liczbę pracowników.
3. W razie potrzeby po rozpatrzeniu i ocenie skargi podejmuje się odpowiednie działania następcze (w tym CAPA), w tym, gdy jest to wymagane, zgłasza się sprawę właściwym organom krajowym.
Artykuł 30
Zwroty
1. Ze zwróconymi weterynaryjnymi produktami leczniczymi postępuje się zgodnie z pisemną procedurą opartą na analizie ryzyka, z uwzględnieniem charakteru danego weterynaryjnego produktu leczniczego, wszelkich wymaganych specjalnych warunków przechowywania oraz czasu, jaki upłynął od jego dostarczenia. Zwroty przeprowadza się zgodnie z prawem krajowym i zgodnie z ustaleniami umownymi między stronami.
2. Weterynaryjne produkty lecznicze, które nie znajdują się już pod kontrolą osób, o których mowa w art. 1 ust. 2, są zwracane do zapasów przeznaczonych do sprzedaży tylko pod warunkiem spełnienia wszystkich poniższych warunków:
|
a) |
weterynaryjne produkty lecznicze znajdują się w swoich nieotwartych i nieuszkodzonych opakowaniach zbiorczych i są w dobrym stanie; |
|
b) |
nie upłynął termin ważności weterynaryjnych produktów leczniczych i nie zostały one wycofane; |
|
c) |
weterynaryjne produkty lecznicze zwrócone przez klienta nieposiadającego pozwolenia na dystrybucję hurtową lub przez apteki lub osoby uprawnione do dostarczania ogółowi społeczeństwa weterynaryjnych produktów leczniczych zgodnie z prawem krajowym danego państwa członkowskiego zostały zwrócone w określonym dopuszczalnym terminie wyznaczonym przy zastosowaniu zasad zarządzania ryzykiem w zakresie jakości; |
|
d) |
weterynaryjne produkty lecznicze nie zostały zwrócone przez właściciela zwierzęcia do apteki ani do innych osób uprawnionych do dostarczania ogółowi społeczeństwa weterynaryjnych produktów leczniczych zgodnie z prawem krajowym danego państwa członkowskiego, chyba że taki zwrot jest dozwolony na mocy prawa krajowego tego państwa członkowskiego; |
|
e) |
klient wykazał, że transport weterynaryjnych produktów leczniczych, ich przechowywanie i postępowanie z nimi odbywało się zgodnie z konkretnymi wymogami dotyczącymi ich przechowywania; |
|
f) |
weterynaryjne produkty lecznicze zostały sprawdzone i ocenione przez dostatecznie przeszkoloną, kompetentną osobę upoważnioną do takich czynności; |
|
g) |
osoby, o których mowa w art. 1 ust. 2, mają dostateczne dowody na to, że weterynaryjny produkt leczniczy został dostarczony klientowi zwracającemu weterynaryjny produkt leczniczy, co potwierdzają kopie oryginalnych specyfikacji wysyłkowych lub numery faktur, numery serii, termin ważności itp., zgodnie z wymogami prawa krajowego, oraz że nie ma powodów, aby przypuszczać, że weterynaryjny produkt leczniczy został sfałszowany. |
3. Ponadto weterynaryjne produkty lecznicze wymagające specjalnej temperatury przechowywania, na przykład niskiej temperatury, wracają do zapasów przeznaczonych do sprzedaży tylko pod warunkiem przedstawienia dokumentów poświadczających, że produkty te były przez cały czas przechowywane w dopuszczonych warunkach przechowywania w okresach, o których mowa w lit. a)–f). Jeżeli doszło do odstępstwa, przeprowadza się ocenę ryzyka, która musi wykazać integralność weterynaryjnego produktu leczniczego. Dowody obejmują wszystkie następujące etapy:
|
a) |
dostawa do klienta; |
|
b) |
sprawdzenie weterynaryjnego produktu leczniczego; |
|
c) |
otwarcie opakowania transportowego; |
|
d) |
ponowne umieszczenie weterynaryjnego produktu leczniczego w opakowaniu; |
|
e) |
pobranie i zwrot osobom, o których mowa w art. 1 ust. 2; |
|
f) |
powrót produktu do chłodziarki w miejscu dystrybucji hurtowej. |
4. Produkty zwrócone do zapasów przeznaczonych do sprzedaży są rozmieszczone w taki sposób, aby zapewnić efektywność systemu, w którym w pierwszej kolejności wydawane są produkty o najbliższym terminie ważności.
5. Skradzione weterynaryjne produkty lecznicze, które odzyskano, nie wracają do zapasów przeznaczonych do sprzedaży ani nie zostają sprzedane klientom.
Artykuł 31
Sfałszowane weterynaryjne produkty lecznicze
1. Oprócz powiadomienia, o którym mowa w art. 101 ust. 6 rozporządzenia (UE) 2019/6, hurtownicy niezwłocznie zaprzestają dystrybucji wszelkich weterynaryjnych produktów leczniczych, które uznają za sfałszowane lub co do których podejrzewają, że zostały sfałszowane, oraz działają zgodnie z instrukcjami określonymi przez właściwe organy. Wprowadza się procedurę dotyczącą takich incydentów. Są one ewidencjonowane wraz z wszystkimi danymi dotyczącymi danej sprawy oraz badane.
2. Wszelkie weterynaryjne produkty lecznicze wykryte w łańcuchu dostaw, co do których istnieje podejrzenie, że zostały sfałszowane, natychmiast oddziela się fizycznie lub, jeżeli dostępny jest równoważny system elektroniczny – elektronicznie. Wszelkie sfałszowane weterynaryjne produkty lecznicze wykryte w łańcuchu dostaw są natychmiast oddzielane fizycznie, przechowywane na specjalnym obszarze oddzielnie od wszystkich innych weterynaryjnych produktów leczniczych i odpowiednio oznakowane. Wszystkie stosowne czynności dotyczące takich produktów są dokumentowane, a ewidencja jest przechowywana.
Artykuł 32
Wycofanie
1. Stosuje się dokumentację i procedury w celu zapewnienia identyfikowalności otrzymanych i dystrybuowanych weterynaryjnych produktów leczniczych do celów ewentualnego wycofania produktu.
2. W przypadku wycofania weterynaryjnego produktu leczniczego osoby, o których mowa w art. 1 ust. 2, informują, z zastosowaniem odpowiedniego stopnia pilności i jasnych instrukcji do wykonania, wszystkich zainteresowanych klientów, wśród których produkt został rozprowadzony.
3. Osoby, o których mowa w art. 1 ust. 2, informują odpowiedni właściwy organ krajowy o każdym wycofaniu weterynaryjnego produktu leczniczego. W przypadku wywożonego weterynaryjnego produktu leczniczego osoby, o których mowa w art. 1 ust. 2, informują klientów z państwa trzeciego lub właściwe organy państwa trzeciego o wycofaniu produktu zgodnie z wymogami prawa krajowego.
4. Osoby, o których mowa w art. 1 ust. 2, regularnie oceniają skuteczność rozwiązań dotyczących wycofywania weterynaryjnych produktów leczniczych na podstawie zasad zarządzania ryzykiem w zakresie jakości.
5. Osoby, o których mowa w art. 1 ust. 2, zapewniają możliwość szybkiego rozpoczęcia czynności związanych z wycofaniem produktu w dowolnym momencie.
6. Osoby, o których mowa w art. 1 ust. 2, przestrzegają wskazówek zawartych w informacji o wycofaniu produktu, która jest zatwierdzona, jeżeli jest to wymagane, przez właściwe organy.
7. Wszystkie czynności związane z wycofaniem produktu są ewidencjonowane w momencie ich podjęcia. Ewidencję tę udostępnia się właściwym organom.
8. Ewidencja dotycząca dystrybucji jest łatwo dostępna dla osób odpowiedzialnych za wycofanie produktu i zawiera wystarczające informacje na temat dystrybutorów i klientów, którym bezpośrednio dostarczono produkt (wraz z adresami, numerami telefonów i środkami komunikacji elektronicznej w godzinach pracy i poza nimi, numerami serii zgodnie z wymogami prawa krajowego i dostarczonymi ilościami), i obejmuje tego rodzaju ewidencję dotyczącą wywożonych weterynaryjnych produktów leczniczych i próbek weterynaryjnych produktów leczniczych.
9. Postępy w procesie wycofywania produktu zapisuje się w sprawozdaniu końcowym obejmującym uzgodnienie dostarczonych i odzyskanych ilości wycofanego weterynaryjnego produktu leczniczego.
ROZDZIAŁ VIII
DZIAŁANIA ZLECANE PODMIOTOM ZEWNĘTRZNYM
Artykuł 33
Obowiązki zleceniodawcy
1. Zleceniodawca jest odpowiedzialny za zlecane działania.
2. Zleceniodawca jest odpowiedzialny za ocenę kompetencji zleceniobiorcy w zakresie prawidłowego przeprowadzenia wymaganych prac, a także za zapewnienie, w drodze umowy i audytów, przestrzegania zasad dobrej praktyki dystrybucyjnej dotyczącej weterynaryjnych produktów leczniczych. Zleceniodawca przeprowadza audyt zleceniobiorcy przed rozpoczęciem zleconych działań oraz monitoruje realizację zlecenia przez zleceniobiorcę i dokonuje przeglądu tej realizacji. Częstotliwość audytów jest określana na podstawie ryzyka, w zależności od charakteru zleconych działań. W przypadku zmiany zleconych działań zleceniodawca stosuje ocenę ryzyka w ramach kontroli zmian, aby ustalić, czy konieczny jest ponowny audyt. Zleceniobiorca zezwala zleceniodawcy na przeprowadzanie audytów zleconych działań.
3. Zleceniodawca przekazuje zleceniobiorcy wszystkie informacje niezbędne do wykonywania zleconych czynności zgodnie ze specjalnymi wymogami dotyczącymi weterynaryjnego produktu leczniczego i innymi stosownymi wymaganiami.
Artykuł 34
Obowiązki zleceniobiorcy
1. Zleceniobiorca posiada odpowiedni sprzęt, procedury, wiedzę i doświadczenie oraz kompetentnych pracowników, aby wykonywać prace zlecone przez zleceniodawcę, a także pomieszczenia, jeżeli jest to wymagane na potrzeby danej działalności.
2. Zleceniobiorca może zlecić stronom trzecim podwykonawstwo prac powierzonych mu na mocy umowy pod warunkiem że zleceniodawca dokona uprzedniej oceny takich rozwiązań i je zatwierdzi, a także pod warunkiem że zleceniodawca lub zleceniobiorca przeprowadzą audyt strony trzeciej. Ustalenia między zleceniobiorcą i stroną trzecią zapewniają udostępnianie informacji dotyczących dystrybucji hurtowej w ten sam sposób jak pomiędzy pierwotnym zleceniodawcą a zleceniobiorcą.
3. Zleceniobiorca powstrzymuje się od jakichkolwiek działań, które mogą mieć niekorzystny wpływ na jakość weterynaryjnych produktów leczniczych, którymi zajmuje się na zlecenie zleceniodawcy.
4. Zleceniobiorca przekazuje zleceniodawcy wszystkie informacje, które mogą wpływać na jakość weterynaryjnych produktów leczniczych, zgodnie z wymaganiami umowy.
ROZDZIAŁ IX
KONTROLE WEWNĘTRZNE
Artykuł 35
Program kontroli wewnętrznych
Wprowadza się program kontroli wewnętrznych o ustalonych ramach czasowych, obejmujący wszystkie aspekty dobrej praktyki dystrybucyjnej dotyczącej weterynaryjnych produktów leczniczych oraz przestrzeganie niniejszego rozporządzenia i procedur.
Artykuł 36
Przeprowadzanie i ewidencja kontroli wewnętrznych
1. Kontrole wewnętrzne można podzielić na kilka kontroli o ograniczonym zakresie.
2. Kontrole wewnętrzne są prowadzone bezstronnie i w szczegółowy sposób przez wyznaczonych kompetentnych pracowników. Audyty niezależnych ekspertów zewnętrznych nie mogą zastępować kontroli wewnętrznych.
3. Wszystkie kontrole wewnętrzne są ewidencjonowane. Sprawozdania zawierają wszelkie spostrzeżenia z kontroli. Egzemplarz sprawozdania przekazuje się kierownictwu i innym odpowiednim osobom.
4. W razie odnotowania nieprawidłowości lub niedociągnięć ustala się ich przyczynę, a także dokumentuje się CAPA i przeprowadza w związku z nimi działania następcze. Dokonuje się przeglądu skuteczności CAPA.
ROZDZIAŁ X
TRANSPORT
Artykuł 37
Wymagania dotyczące transportu
1. Osoby, o których mowa w art. 1 ust. 2, dostarczające weterynaryjne produkty lecznicze są odpowiedzialne za ochronę tych weterynaryjnych produktów leczniczych przed uszkodzeniem, sfałszowaniem i kradzieżą oraz za zapewnienie, by podczas transportu temperatura była utrzymywana w dopuszczalnych granicach oraz, w miarę możliwości, monitorują tę temperaturę.
2. Podczas transportu wymagane warunki przechowywania lub transportu weterynaryjnych produktów leczniczych, stosownie do przypadku, utrzymywane są w granicach określonych przez producentów i posiadaczy pozwolenia na dopuszczenie do obrotu lub podanych na opakowaniu zewnętrznym.
3. Jeżeli podczas transportu wystąpiło odstępstwo, takie jak odchylenia temperatury lub uszkodzenie weterynaryjnego produktu leczniczego, zgłasza się to osobom, o których mowa w art. 1 ust. 2, oraz odbiorcy odnośnych weterynaryjnych produktów leczniczych w celu dokonania przez nich oceny potencjalnego wpływu na jakość odnośnych weterynaryjnych produktów leczniczych. Wprowadzona jest procedura dotycząca badania odchyleń temperatury i postępowania w takich przypadkach.
4. Osoby, o których mowa w art. 1 ust. 2, zapewniają, aby pojazdy i sprzęt wykorzystywane do dystrybucji i przechowywania weterynaryjnych produktów leczniczych lub postępowania z nimi nadawały się do takiego zastosowania i były odpowiednio wyposażone, tak aby zapobiegać narażeniu weterynaryjnych produktów leczniczych na działanie warunków, które mogą wpłynąć na ich jakość i integralność opakowania.
5. Wprowadzone są pisemne procedury dotyczące korzystania z wszystkich pojazdów i sprzętu używanych w procesie dystrybucji oraz procedury ich konserwacji, które obejmują kwestie czyszczenia tych pojazdów i sprzętu oraz środki ostrożności.
6. Sprzęt wybrany i używany do czyszczenia pojazdów nie może stanowić źródła zanieczyszczenia.
7. Stosowana jest ocena ryzyka tras dostawy, aby ustalić, gdzie wymagana jest kontrola temperatury. Sprzęt wykorzystywany do monitorowania temperatury podczas transportu w pojazdach lub pojemnikach jest konserwowany i poddawany kalibracji w regularnych odstępach czasu ustalonych na podstawie zasad zarządzania ryzykiem w zakresie jakości.
8. Na potrzeby weterynaryjnych produktów leczniczych i produktów leczniczych stosowanych u ludzi używa się, o ile to możliwe, pojazdów i sprzętu specjalnie przeznaczonych do tego celu. Jeżeli używane są inne pojazdy lub sprzęt, stosowane są procedury zapewniające utrzymanie jakości weterynaryjnych produktów leczniczych.
9. Dostawy są kierowane na adres wskazany w specyfikacji wysyłkowej pod pieczę odbiorcy lub do jego pomieszczeń. Weterynaryjnych produktów leczniczych nie zostawia się w innych pomieszczeniach.
10. Wyznacza się osoby do obsługi dostaw nadzwyczajnych poza zwykłymi godzinami pracy przedsiębiorstwa oraz wprowadza się procedury dotyczące takich przypadków.
11. W przypadku transportu wykonywanego przez stronę trzecią obowiązująca umowa zawiera wymogi art. 33 i 34 oraz jasno określa obowiązki tej strony trzeciej w zakresie zapewnienia zgodności z dobrą praktyką dystrybucyjną dotyczącą weterynaryjnych produktów leczniczych. Osoby, o których mowa w art. 1 ust. 2, informują przewoźników o odpowiednich warunkach transportu mających zastosowanie do przesyłki.
12. Jeżeli trasa przewozu obejmuje rozładunek i ponowny załadunek lub składowanie tranzytowe w węźle transportowym, wszelkie obiekty, w których tymczasowo przechowywane są produkty lecznicze, są czyste i bezpieczne oraz umożliwiają monitorowanie temperatury, stosownie do przypadku.
13. Zapewnia się, aby okres tymczasowego przechowywania produktów oczekujących na kolejny etap trasy przewozu był ograniczony do minimum.
Artykuł 38
Pojemniki, opakowania i oznakowanie opakowania
1. Weterynaryjne produkty lecznicze są transportowane w pojemnikach, które nie mają niekorzystnego wpływu na ich jakość oraz zapewniają odpowiednią ochronę przed czynnikami zewnętrznymi, w tym przed zanieczyszczeniem.
2. Wybór pojemnika i opakowania opiera się na następujących elementach:
|
a) |
wymogi dotyczące przechowywania i transportu weterynaryjnych produktów leczniczych; |
|
b) |
przestrzeń wymagana dla danej ilości weterynaryjnych produktów leczniczych; |
|
c) |
postać farmaceutyczna, w tym premiksy lecznicze; |
|
d) |
przewidywane maksymalne i minimalne wartości temperatury zewnętrznej; |
|
e) |
szacowany maksymalny czas transportu, w tym czas składowania tranzytowego w składzie celnym; |
|
f) |
status kwalifikacji opakowania; |
|
g) |
status walidacji pojemników transportowych. |
3. Oznakowanie pojemników zapewnia dostateczne informacje o wymogach dotyczących postępowania z weterynaryjnymi produktami leczniczymi i ich przechowywania oraz o środkach ostrożności, które zapewnią ciągłe właściwe postępowanie z produktami i ich stałe zabezpieczenie. Pojemniki umożliwiają identyfikację ich zawartości i ustalenie ich pochodzenia.
Artykuł 39
Produkty wymagające specjalnych warunków
1. W przypadku dostaw weterynaryjnych produktów leczniczych wymagających specjalnych warunków, takich jak substancje odurzające lub substancje psychotropowe, osoby, o których mowa w art. 1 ust. 2, utrzymują bezpieczny i zabezpieczony łańcuch dostaw takich produktów, zgodnie z wymogami określonymi przez państwa członkowskie, których to dotyczy. W odniesieniu do dostaw takich produktów stosuje się dodatkowe systemy kontrolne. Wprowadzony jest protokół postępowania na wypadek kradzieży.
2. Weterynaryjne produkty lecznicze zawierające wysoce aktywne materiały są transportowane w bezpiecznych, tylko do tego przeznaczonych i zabezpieczonych pojemnikach i pojazdach zgodnie z mającymi zastosowanie środkami bezpieczeństwa.
3. W przypadku weterynaryjnych produktów leczniczych wrażliwych na temperaturę stosuje się sprzęt podlegający kwalifikacji, taki jak opakowania termiczne, pojemniki o regulowanej temperaturze lub pojazdy o regulowanej temperaturze, aby zapewnić utrzymanie prawidłowych warunków transportu między producentem, hurtownikiem i klientem, chyba że wykazano stabilność produktu w innych warunkach transportu.
4. Jeżeli stosowane są pojazdy o regulowanej temperaturze, sprzęt do monitorowania temperatury używany podczas transportu jest regularnie konserwowany i poddawany kalibracji. Przeprowadza się mapowanie temperatury w reprezentatywnych warunkach, biorąc pod uwagę wahania sezonowe.
5. Na żądanie klientów z odpowiednim uzasadnieniem, a w każdym razie w przypadku incydentu, osoby, o których mowa w art. 1 ust. 2, dostarczają klientom informacje w celu wykazania, że weterynaryjne produkty lecznicze spełniały warunki temperatury przechowywania lub transportu.
6. Jeżeli w izolowanych pojemnikach stosowane są wkłady chłodzące, umieszcza się je w sposób zapewniający, aby weterynaryjny produkt leczniczy nie stykał się z nimi bezpośrednio.
7. Pracownicy są przeszkoleni w zakresie obsługi izolowanych pojemników, w tym ustawień sezonowych, i ponownego wykorzystania wkładów chłodzących.
8. Osoby, o których mowa w art. 1 ust. 2, posiadają system kontroli ponownego wykorzystania wkładów chłodzących zapobiegający omyłkowemu użyciu nie w pełni schłodzonych wkładów. Osoby, o których mowa w art. 1 ust. 2, zapewniają odpowiednie fizyczne oddzielenie mrożących i chłodzących wkładów lodowych.
9. Osoby, o których mowa w art. 1 ust. 2, opisują w procedurze proces dostarczania wrażliwych weterynaryjnych produktów leczniczych oraz kontrolę sezonowych wahań temperatury.
ROZDZIAŁ XI
PRZEPISY KOŃCOWE
Artykuł 40
Wejście w życie i rozpoczęcie stosowania
Niniejsze rozporządzenie wchodzi w życie dwudziestego dnia po jego opublikowaniu w Dzienniku Urzędowym Unii Europejskiej.
Niniejsze rozporządzenie wiąże w całości i jest bezpośrednio stosowane we wszystkich państwach członkowskich.
Sporządzono w Brukseli dnia 29 lipca 2021 r.
W imieniu Komisji
Ursula VON DER LEYEN
Przewodnicząca
(1) Dz.U. L 4 z 7.1.2019, s. 43.
(2) Good storage and distribution practices for medical products. W: WHO Expert Committee on Specifications for Pharmaceutical Preparations: fifty-fourth report. Genewa: Światowa Organizacja Zdrowia; 2020: Annex 7 (WHO Technical Report Series, No. 1025).
(3) Guide to good storage practices for pharmaceuticals. W: WHO Expert Committee on Specifications for Pharmaceutical Preparations: thirty-seventh report. Genewa: Światowa Organizacja Zdrowia; 2003: Annex 9 (WHO Technical Report Series, No. 908).
(4) Model guidance for the storage and transport of time- and temperature-sensitive pharmaceutical products. W: WHO Expert Committee on Specifications for Pharmaceutical Preparations: forty-fifth report. Genewa: Światowa Organizacja Zdrowia; 2011: Annex 9 (WHO Technical Report Series, No. 961).
(5) PIC/S Guide to good distribution practice for medicinal products, PIC/S, PE 011-1, 1.6.2014.
(6) Wytyczne z dnia 5 listopada 2013 r. w sprawie dobrej praktyki dystrybucyjnej dotyczącej produktów leczniczych do stosowania u ludzi (2013/C 343/01) (Dz.U. C 343 z 23.11.2013, s. 1).
(7) Dyrektywa 2001/83/WE Parlamentu Europejskiego i Rady z dnia 6 listopada 2001 r. w sprawie wspólnotowego kodeksu odnoszącego się do produktów leczniczych stosowanych u ludzi (Dz.U. L 311 z 28.11.2001, s. 67).
(8) Rozporządzenie Parlamentu Europejskiego i Rady (UE) 2016/679z dnia 27 kwietnia 2016 r. w sprawie ochrony osób fizycznych w związku z przetwarzaniem danych osobowych i w sprawie swobodnego przepływu takich danych oraz uchylenia dyrektywy 95/46/WE (ogólne rozporządzenie o ochronie danych) (Dz.U. L 119 z 4.5.2016, s. 1).
(9) Rozporządzenie Parlamentu Europejskiego i Rady (UE) nr 952/2013 z dnia 9 października 2013 r. ustanawiające unijny kodeks celny (Dz.U. L 269 z 10.10.2013, s. 1).
(10) Rozporządzenie Parlamentu Europejskiego i Rady (UE) nr 910/2014 z dnia 23 lipca 2014 r. w sprawie identyfikacji elektronicznej i usług zaufania w odniesieniu do transakcji elektronicznych na rynku wewnętrznym oraz uchylające dyrektywę 1999/93/WE (Dz.U. L 257 z 28.8.2014, s. 73).
DECYZJE
|
30.7.2021 |
PL |
Dziennik Urzędowy Unii Europejskiej |
L 272/67 |
DECYZJA RADY (UE) 2021/1249
z dnia 26 lipca 2021 r.
w sprawie stanowiska, jakie ma być zajęte w imieniu Unii Europejskiej w ramach Wspólnego Komitetu EOG w odniesieniu do zmiany Protokołu 31 w sprawie współpracy w konkretnych dziedzinach poza czterema swobodami, załączonego do Porozumienia EOG (Linia budżetowa 07 20 03 01 – Zabezpieczenie społeczne)
(Tekst mający znaczenie dla EOG)
RADA UNII EUROPEJSKIEJ,
uwzględniając Traktat o funkcjonowaniu Unii Europejskiej, w szczególności jego art. 46 i 48 w związku z art. 218 ust. 9,
uwzględniając rozporządzenie Rady (WE) nr 2894/94 z dnia 28 listopada 1994 r. w sprawie uzgodnień dotyczących stosowania Porozumienia o Europejskim Obszarze Gospodarczym (1), w szczególności jego art. 1 ust. 3,
uwzględniając wniosek Komisji Europejskiej,
a także mając na uwadze, co następuje:
|
(1) |
Porozumienie o Europejskim Obszarze Gospodarczym (2) (zwane dalej „Porozumieniem EOG”) weszło w życie z dniem 1 stycznia 1994 r. |
|
(2) |
Zgodnie z art. 98 Porozumienia EOG Wspólny Komitet EOG może podjąć decyzję o zmianie, między innymi, Protokołu 31 w sprawie współpracy w konkretnych dziedzinach poza czterema swobodami (zwanego dalej „Protokołem 31”), załączonego do Porozumienia EOG. |
|
(3) |
Należy kontynuować współpracę pomiędzy Umawiającymi się Stronami Porozumienia EOG w ramach działań Unii finansowanych z budżetu ogólnego Unii w zakresie swobodnego przepływu pracowników, koordynacji systemów zabezpieczenia społecznego i działań na rzecz migrantów, w tym migrantów z państw trzecich. |
|
(4) |
Należy zatem zmienić protokół 31 do Porozumienia EOG, aby umożliwić kontynuowanie takiej rozszerzonej współpracy od dnia 1 stycznia 2021 r. |
|
(5) |
Stanowisko Unii w ramach Wspólnego Komitetu EOG powinno zatem opierać się na projekcie decyzji Wspólnego Komitetu EOG, |
PRZYJMUJE NINIEJSZĄ DECYZJĘ:
Artykuł 1
Stanowisko, jakie ma być zajęte w imieniu Unii w ramach Wspólnego Komitetu EOG, dotyczące proponowanej zmiany Protokołu 31 w sprawie współpracy w konkretnych dziedzinach poza czterema swobodami, załączonego do Porozumienia EOG, oparte jest na projekcie decyzji Wspólnego Komitetu EOG (3).
Artykuł 2
Niniejsza decyzja wchodzi w życie z dniem jej przyjęcia.
Sporządzono w Brukseli dnia 26 lipca 2021 r.
W imieniu Rady
G. DOVŽAN
Przewodniczący
(1) Dz.U. L 305 z 30.11.1994, s. 6.
(2) Dz.U. L 1 z 3.1.1994, s. 3.
(3) Zob. dokument ST 10507/21 na stronie http://register.consilium.europa.eu.
|
30.7.2021 |
PL |
Dziennik Urzędowy Unii Europejskiej |
L 272/69 |
DECYZJA RADY (UE) 2021/1250
z dnia 26 lipca 2021 r.
w sprawie stanowiska, jakie ma być zajęte w imieniu Unii Europejskiej w ramach Wspólnego Komitetu EOG w odniesieniu do zmiany Protokołu 31 w sprawie współpracy w konkretnych dziedzinach poza czterema swobodami, załączonego do Porozumienia EOG (Europejski Fundusz Obronny)
(Tekst mający znaczenie dla EOG)
RADA UNII EUROPEJSKIEJ,
uwzględniając Traktat o funkcjonowaniu Unii Europejskiej, w szczególności jego art. 173 ust. 3, art. 182 ust. 4, art. 183 i art. 188 akapit drugi, w związku z art. 218 ust. 9,
uwzględniając rozporządzenie Rady (WE) nr 2894/94 z dnia 28 listopada 1994 r. w sprawie uzgodnień dotyczących stosowania Porozumienia o Europejskim Obszarze Gospodarczym (1), w szczególności jego art. 1 ust. 3,
uwzględniając wniosek Komisji Europejskiej,
a także mając na uwadze, co następuje:
|
(1) |
Porozumienie o Europejskim Obszarze Gospodarczym (2) (zwane dalej „Porozumieniem EOG”) weszło w życie z dniem 1 stycznia 1994 r. |
|
(2) |
Zgodnie z art. 98 Porozumienia EOG Wspólny Komitet EOG może podjąć decyzję o zmianie, między innymi, protokołu 31 w sprawie współpracy w konkretnych dziedzinach poza czterema swobodami (zwanego dalej „protokołem 31”), załączonego do Porozumienia EOG. |
|
(3) |
W Porozumieniu EOG należy uwzględnić rozporządzenie Parlamentu Europejskiego i Rady (UE) 2021/697 (3). |
|
(4) |
Należy zatem odpowiednio zmienić protokół 31 do Porozumienia EOG. |
|
(5) |
Stanowisko Unii w ramach Wspólnego Komitetu EOG powinno zatem opierać się na projekcie decyzji Wspólnego Komitetu EOG, |
PRZYJMUJE NINIEJSZĄ DECYZJĘ:
Artykuł 1
Stanowisko, jakie ma być zajęte w imieniu Unii w ramach Wspólnego Komitetu EOG, dotyczące proponowanej zmiany protokołu 31 w sprawie współpracy w konkretnych dziedzinach poza czterema swobodami, załączonego do Porozumienia EOG, oparte jest na projekcie decyzji Wspólnego Komitetu EOG (4).
Artykuł 2
Niniejsza decyzja wchodzi w życie z dniem jej przyjęcia.
Sporządzono w Brukseli dnia 26 lipca 2021 r.
W imieniu Rady
G. DOVŽAN
Przewodniczący
(1) Dz.U. L 305 z 30.11.1994, s. 6.
(2) Dz.U. L 1 z 3.1.1994, s. 3.
(3) Rozporządzenie Parlamentu Europejskiego i Rady (UE) 2021/697 z dnia 29 kwietnia 2021 r. ustanawiające Europejski Fundusz Obronny i uchylające rozporządzenie (UE) 2018/1092 (Dz.U. L 170 z 12.5.2021, s. 149).
(4) Zob. dokument ST 10693/21 na stronie http://register.consilium.europa.eu.
|
30.7.2021 |
PL |
Dziennik Urzędowy Unii Europejskiej |
L 272/71 |
DECYZJA RADY (WPZiB) 2021/1251
z dnia 29 lipca 2021 r.
zmieniająca decyzję (WPZiB) 2015/1333 w sprawie środków ograniczających w związku z sytuacją w Libii
RADA UNII EUROPEJSKIEJ,
uwzględniając Traktat o Unii Europejskiej, w szczególności jego art. 29,
uwzględniając wniosek Wysokiego Przedstawiciela Unii do Spraw Zagranicznych i Polityki Bezpieczeństwa,
a także mając na uwadze, co następuje:
|
(1) |
W dniu 31 lipca 2015 r. Rada przyjęła decyzję (WPZiB) 2015/1333 (1). |
|
(2) |
Zgodnie z art. 17 ust. 2 decyzji (WPZiB) 2015/1333 Rada przeprowadziła przegląd wykazów wskazanych osób i podmiotów zamieszczonych w załącznikach II i IV do tej decyzji. |
|
(3) |
Rada stwierdziła, że wpis dotyczący jednej osoby, która zmarła, oraz wpis dotyczący innej osoby, wobec której środki ograniczające miały zastosowanie do dnia 2 kwietnia 2021 r., powinny zostać skreślone oraz że należy utrzymać środki ograniczające skierowane przeciwko wszystkim osobom i podmiotom figurującym w wykazach zamieszczonych w załącznikach II i IV do decyzji (WPZiB) 2015/1333. Ponadto należy zaktualizować informacje umożliwiające identyfikację dotyczące jednej osoby. |
|
(4) |
Należy zatem odpowiednio zmienić decyzję (WPZiB) 2015/1333, |
PRZYJMUJE NINIEJSZĄ DECYZJĘ:
Artykuł 1
W decyzji (WPZiB) 2015/1333 wprowadza się następujące zmiany:
|
1) |
w art. 17 uchyla się ust. 3 i 4; |
|
2) |
w załącznikach II i IV wprowadza się zmiany zgodnie z załącznikiem do niniejszej decyzji. |
Artykuł 2
Niniejsza decyzja wchodzi w życie następnego dnia po jej opublikowaniu w Dzienniku Urzędowym Unii Europejskiej.
Sporządzono w Brukseli dnia 29 lipca 2021 r.
W imieniu Rady
G. DOVŽAN
Przewodniczący
(1) Decyzja Rady (WPZiB) 2015/1333 z dnia 31 lipca 2015 r. w sprawie środków ograniczających w związku z sytuacją w Libii oraz uchylająca decyzję 2011/137/WPZiB (Dz.U. L 206 z 1.8.2015, s. 34).
ZAŁĄCZNIK
W decyzji (WPZiB) 2015/1333 wprowadza się następujące zmiany:
|
1) |
w części A (Osoby) załącznika II (Wykaz osób i podmiotów, o których mowa w art. 8 ust. 2) wprowadza się następujące zmiany:
|
|
2) |
w części A (Osoby) załącznika IV (Wykaz osób i podmiotów, o których mowa w art. 9 ust. 2) wprowadza się następujące zmiany:
|
|
30.7.2021 |
PL |
Dziennik Urzędowy Unii Europejskiej |
L 272/73 |
DECYZJA RADY (WPZiB) 2021/1252
z dnia 29 lipca 2021 r.
zmieniająca decyzję 2010/413/WPZiB w sprawie środków ograniczających wobec Iranu
RADA UNII EUROPEJSKIEJ,
uwzględniając Traktat o Unii Europejskiej, w szczególności jego art. 29,
uwzględniając wniosek Wysokiego Przedstawiciela Unii do Spraw Zagranicznych i Polityki Bezpieczeństwa,
a także mając na uwadze, co następuje:
|
(1) |
W dniu 26 lipca 2010 r. Rada przyjęła decyzję 2010/413/WPZiB (1) w sprawie środków ograniczających wobec Iranu. |
|
(2) |
W dniu 18 czerwca 2020 r. Rada przyjęła decyzję (WPZiB) 2020/849 (2) zmieniającą decyzję 2010/413/WPZiB. |
|
(3) |
W następstwie wyroku Sądu w sprawie T-580/19 (3) Sayed Shamsuddin Borborudi powinien zostać wykreślony z wykazu osób i podmiotów objętych środkami ograniczającymi zamieszczonego w załączniku II do decyzji 2010/413/WPZiB. |
|
(4) |
Zgodnie z art. 26 ust. 3 decyzji 2010/413/WPZiB Rada przeprowadziła przegląd wykazu wskazanych osób i podmiotów zamieszczonego w załączniku II do tej decyzji. |
|
(5) |
Z tego przeglądu wynika, że należy utrzymać środki ograniczające wobec wszystkich osób i podmiotów znajdujących się w wykazie zamieszczonym w załączniku II do decyzji 2010/413/WPZiB, w zakresie, w jakim ich imiona i nazwiska oraz nazwy nie są wymienione w załączniku VI do tej decyzji, oraz że należy zaktualizować 21 wpisów zamieszczonych w załączniku II. |
|
(6) |
Należy zatem odpowiednio zmienić decyzję 2010/413/WPZiB, |
PRZYJMUJE NINIEJSZĄ DECYZJĘ:
Artykuł 1
W załączniku II do decyzji 2010/413/WPZiB wprowadza się zmiany zgodnie z załącznikiem do niniejszej decyzji.
Artykuł 2
Niniejsza decyzja wchodzi w życie następnego dnia po jej opublikowaniu w Dzienniku Urzędowym Unii Europejskiej.
Sporządzono w Brukseli dnia 29 lipca 2021 r.
W imieniu Rady
G. DOVŽAN
Przewodniczący
(1) Decyzja Rady 2010/413/CFSP z dnia 26 lipca 2010 r. w sprawie środków ograniczających wobec Iranu i uchylająca wspólne stanowisko 2007/140/WPZiB (Dz.U. L 195 z 27.7.2010, s. 39).
(2) Decyzja Rady (WPZiB) 2020/849 z dnia 18 czerwca 2020 r. zmieniająca decyzję 2010/413/WPZiB w sprawie środków ograniczających wobec Iranu (Dz.U. L 196 z 19.6.2020, s. 8).
(3) Wyrok Sądu z dnia 9 czerwca 2021 r., Sayed Shamsuddin Borborudi przeciwko Radzie Unii Europejskiej, T-580/19, ECLI:EU:T:2021:330.
ZAŁĄCZNIK
Do załącznika II do decyzji 2010/413/WPZiB wprowadza się następujące zmiany:
|
1) |
pod nagłówkiem „I. Osoby i podmioty zaangażowane w działania na rzecz broni jądrowej lub pocisków balistycznych oraz osoby i podmioty popierające rząd Iranu”, w sekcji „A. Osoby fizyczne”, skreśla się następujący wpis: „25. Sayed Shamsuddin Borborudi”; |
|
2) |
pod nagłówkiem „I. Osoby i podmioty zaangażowane w działania na rzecz broni jądrowej lub pocisków balistycznych oraz osoby i podmioty popierające rząd Iranu”, w wykazie zamieszczonym w sekcji „A. Osoby fizyczne”, następujące wpisy zastępują odpowiadające im wpisy:
|
|
3) |
pod nagłówkiem „I. Osoby i podmioty zaangażowane w działania na rzecz broni jądrowej lub pocisków balistycznych oraz osoby i podmioty popierające rząd Iranu”, w wykazie zamieszczonym w sekcji „B. Podmioty”, następujące wpisy zastępują odpowiadające im wpisy:
|
|
4) |
pod nagłówkiem „II. Korpus Strażników Rewolucji Islamskiej”, w wykazie zamieszczonym w sekcji „A. Osoby fizyczne”, następujące wpisy zastępują odpowiadające im wpisy:
|
|
5) |
pod nagłówkiem „II. Korpus Strażników Rewolucji Islamskiej”, w wykazie zamieszczonym w sekcji „B. Podmioty”, następujące wpisy zastępują odpowiadające im wpisy:
|